
アスペルギルス変異体細胞におけるポリペプチドの生産方法
【課題】毒素欠失アスペルギルス変異体細胞における着目のポリペプチドの生産方法を提供する。
【解決手段】着目のポリペプチドの生産方法であって、(a) 親のアスペルギルス細胞の変異体を培養し、ここで(i) 前記変異体は前記ポリペプチドをコードする第一の核酸配列を含んでなり、そして(ii) 前記変異体は同一条件下で培養した時に少なくとも1つの着目の毒素を親のアスペルギルス細胞よりも少量生産し;そして(b) 培地から前記ポリペプチドを単離することを含んでなる方法を提供する。また、アスペルギルス細胞の変異体、並びに該変異体細胞の獲得方法も提供する。
【解決手段】着目のポリペプチドの生産方法であって、(a) 親のアスペルギルス細胞の変異体を培養し、ここで(i) 前記変異体は前記ポリペプチドをコードする第一の核酸配列を含んでなり、そして(ii) 前記変異体は同一条件下で培養した時に少なくとも1つの着目の毒素を親のアスペルギルス細胞よりも少量生産し;そして(b) 培地から前記ポリペプチドを単離することを含んでなる方法を提供する。また、アスペルギルス細胞の変異体、並びに該変異体細胞の獲得方法も提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、毒素欠失アスペルギルス変異体細胞における着目のポリペプチドの生産方法に関する。本発明はまた、アスペルギルス細胞の変異体および前記変異体細胞の獲得方法にも関する。
【背景技術】
【0002】
発明の背景
最近、非相同(異種)ポリペプチドの発現に組換え宿主細胞を利用することにより、他の方法では少量しか得られないかまたは天然源からの精製によってしか得られない商業的に価値あるポリペプチド、例えば工業的に重要な酵素や二次代謝産物、の大量生産が大幅に簡素化した。現在、ある特定のポリペプチドの生産のために選択される発現系には、真正細菌および真核宿主をはじめとする多様な選択肢がある。適当な発現系の選択は、しばしば所望の組成とコンホメーションを有するポリペプチドを生産する宿主細胞の能力に依存するだけでなく、大抵は、タンパク質の意図する最終用途によっても左右される。
【0003】
或る宿主系の使用に伴って遭遇する1つの問題はマイコトキシンの生産である。着目のポリペプチドの生産の際に宿主細胞として使われる多数の真菌は、様々な毒素の生合成に関与する酵素をコードする遺伝子を所有している。例えば、シクロピアゾン酸、コウジ酸、3−ニトロプロピオン酸およびアフラトキシンは、例えばアスペルギルス・フラーブス(Aspergillus flavus)中で生産される既知の毒素である。同様に、トリコテセン類も多数の真菌、例えばフザリウム種、例えばフザリウム・ベネナタム(Fusarium venenatum)およびトリコデルマ種において生産される。様々な真菌中での毒素の形成の詳細な概説はHandbook of Toxic Fungal Metabolites, Richard J. Cole & Richard H. Cox, Academic Press, 1981に見つけることができる。
【0004】
着目のポリペプチドの発酵の最中に起こるそのような毒素の形成は、それらが作業者と使用者の両方の健康および環境に対して有害となり得るので、非常に望ましくない。
【0005】
従って、関係する生産に使用する条件下で、健康に影響を及ぼすと考えられる量でそのような毒素が形成されないよう保証する多大な努力が費やされている。これは、主として、毒素を直接分析するかまたはバイオアッセイおよび/または食餌実験により分析することによる大規模な分析プログラムによって行われる。多くの場合、そのような大規模なプログラムは1回生産ごとに行われ、生産原価と製品を販売できるまでの時間の両方に影響を及ぼす。
【0006】
シクロピアゾン酸(以後“CPA”とも称する)は弱酸であり(pKa:3.5)酸性条件下で沈澱する。それは金属キレートを形成し、金属キレートは希酸によって分離される。CPAは特に多数の器官において変性病変と壊死を引き起こすので猛毒であり、そしてCa2+−ATPアーゼを選択的に阻害する。CPAはα形とβ形で生産され、β形はα形の前駆体である。CPAはアスペルギルス属により生産されるが、他の真菌、例えばペニシリウム属によっても生産される。
【0007】
コウジ酸(以後“KA”とも称する)は多数のアスペルギルス属により生産されるが他の真菌、例えばペニシリウム属および更に幾つかの細菌によっても生産される。それは弱アルカリ性(pKa:7.9;フェノール基)であり、多くの金属イオンと錯体を形成する。それは抗菌活性を有し、動物に対する毒性は弱い。それは殺虫剤や色素などといった多数の合成化合物の前駆体である。
【0008】
3−ニトロプロピオン酸(以後“3−NPA”とも称する)は天然ニトロ化合物である。それはある種の真菌、特にアスペルギルス属〔A.フラーブス(A. flavus)、A.ウエンティ(A. wentii)〕およびペニシリウム属〔P.アトロベンタム(P. atroventum)〕により生産される。数種の細菌における生産も報告されている。この酸およびそのエステルは幾つかの植物においても見つかっている。それは、例えば貧血を引き起こすのでそれ自体の方がむしろ毒性である。また、それは胃腸管において別の毒性化合物である亜硝酸塩に部分的に変換され得る。3−ニトロプロピオン酸は、コハク酸デヒドロゲナーゼを不可逆的に阻害しそしてイソクエン酸リアーゼ、フマラーゼおよびアスパルターゼを可逆的に阻害することにより、クレブス回路に影響を及ぼす。
【0009】
アフラトキシンはきわめて生物学的に活性であり、真菌アスペルギルス・フラーブス(Aspergillus flavus)Link ex. Friesおよびアスペルギルス・パラシチクス(Aspergillus parasiticus)Speareにより生産される二次代謝産物である;R.W. Detroy他、"Aflatoxin and related compounds", Microbial Toxins, 第6巻(A. Ciegler, S. KadisおよびS.J. Ajl編), Academic, New York, 1971, 3-178頁を参照のこと。主要なアフラトキシンはB1,B2,G1およびG2である。この代謝産物、特にアフラトキシンB1は動物とヒトに対して毒性であるだけでなく、全ての既知天然化合物のうち最も発ガン性でもある。
【0010】
マルフォルミンおよびオクラトキシンはA.ニガー(A. niger)により生産される。
【0011】
毒素を生産する宿主生物の能力を除去または低下させることにより、正式認可手続がずっと簡単になり、且つ分析プログラムを減少できるので生産段階における時間と費用を削減できるだろう。
【0012】
現在、着目のポリペプチドを効率的且つ経済的な方法で生産するのに適当である毒素欠失アスペルギルス変異体細胞(すなわち、好ましくはGRASとして分類される安全生物)が必要とされている。本発明は、毒素欠失アスペルギルス変異体宿主細胞を使って着目のポリペプチドの生産を提供することにより、およびそのような変異体宿主細胞の作製方法を提供することにより、この要望を満たす。発現されない、すなわちサイレント遺伝子である、1または複数の毒素遺伝子を有するアスペルギルス変異体細胞を提供することも必要とされている。
【発明の概要】
【0013】
本発明の一面によれば、アスペルギルス変異体宿主細胞により着目のポリペプチドを生産する方法であって、(a) 親のアスペルギルス細胞の変異体を培養し、ここで(i) 前記変異体は前記着目のポリペプチドをコードする第一の核酸配列と、少なくとも1つの毒素の生合成または分泌の原因となる少なくとも1つの遺伝子の変更を含んでなる第二の核酸配列とを含んでなり、そして(ii)前記変異体は同一条件下で培養した時に親のアスペルギルス細胞よりも少量の毒素を生産し;そして(b) 培地から前記ポリペプチドを単離することを含んでなる方法が提供される。
【0014】
本発明の好ましい態様では、変異体アスペルギルス細胞が同一条件下で培養した時に親細胞よりも少なくとも90%少なく毒素を生産する。好ましくは、変異体が同一条件下で培養した時に親のアスペルギルス細胞よりもシクロピアゾン酸、コウジ酸、3−ニトロプロピオン酸およびアフラトキシンのうちの1つまたは複数を少量生産する。
【0015】
本発明の別の態様によれば、着目の非相同ポリペプチドの生産に有用な毒素欠失アスペルギルス変異体宿主細胞であって、同一条件下で培養した時にアスペルギルス親細胞に比べて少なくとも1つの毒素を少量生産させるために遺伝子的に変更されている細胞が提供される。
【0016】
本発明のアスペルギルス細胞は、好ましくは、A.オリゼ(A. oryzae)、A.アキュレータス(A. aculeatus)、A.ニデュランス(A. nidulans)、A.フィキュウム(A. ficuum)、A.フラーブス(A. flavus)、A.フェチダス(A. foetidus)、A.ソヤ(A. soja)、A.サケ(A. sake)、A.ニガー(A. niger)、A.ジャポニカス(A. japonicus)、A.パラシチクス(A. parasiticus)およびA.フェニクス(A. phoeicus)からなる群より選ばれる。
【0017】
本発明の別の態様では、毒素欠失アスペルギルス変異体宿主細胞を獲得する方法であって、(a) アスペルギルス親宿主細胞中に、着目のポリペプチドをコードする第一の核酸配列と、少なくとも1つの毒素の生合成または分泌の原因である少なくとも1つの遺伝子の変更を含んでなる第二の核酸配列とを導入し;そして(b) 段階(a)から前記第一および第二の核酸配列を含んでなる変異体を同定することを含んでなる方法が提供される。
【0018】
本発明は、一面では、1または複数のサイレント毒性遺伝子が除去されている、非相同ポリペプチドの発現に適当なアスペルギルス変異体細胞に関する。
上記のおよび他の態様は下記の記載においてより詳細に説明されるだろう。
【図面の簡単な説明】
【0019】
【図1】図1は、プローブとしてDCAT-S遺伝子を含むpJaL499からの1 kb 32P標識DNA BglII断片を用いて、制限酵素EcoRI, SalI, BbuI, XhoIおよびXbaIを使って行った、アスペルギルス・オリゼ DCAT-S遺伝子のゲノム制限地図を示す。
【図2】図2は、破壊プラスミド p(DCAT-S-pyrG)の作製を示す。
【0020】
発明の具体的説明
定義
本願の目的上、本発明のより良い理解のために次の用語を定義する。
「ベクター」なる用語は、着目のポリペプチド(その前駆体形を含む)をコードする遺伝子またはDNA配列の挿入、増殖および発現を可能にするプラスミド、コスミド、ファージまたは他の任意媒介物を意味する。
【0021】
「宿主」なる用語は、着目のポリペプチド(その前駆体形を含む)の発現を可能にするであろう任意細胞を意味する。
【0022】
「形質転換」なる用語は、細胞による非相同DNA配列の発現を可能にする取り込みを意味する。
「変異体」宿主(または株)なる用語は、野生型株の変異体と形質転換体の両方を包含する、遺伝的に変更された株を意味する。
【0023】
「毒素」なる用語は、植物毒性、動物毒性および抗生物質活性を有する真菌の代謝産物を意味する。用語「マイコトキシン」は、暴露のレベルでヒトおよび動物に不利な健康障害を引き起こす可能性を有する、真菌により生産される二次代謝産物として一般に定義される。本明細書中では、「毒素」と「マイコトキシン」は互いに交換可能に用いることができる。
【0024】
本発明の変異体細胞について使われる「毒素欠失」なる用語は、その変異体がそれの親株に比較して不完全な毒素生産を有すること、すなわち、その変異体が少なくとも1つ、好ましくは複数の毒素を親細胞よりも少量生産することを意味する。
【0025】
本発明の方法の段階a)において使われる「同一条件」なる用語は、親細胞の毒素生産に対する変異体の毒素生産に関し、そして変異体と親株の発酵に例えばpH、温度、酸素などについて同様の条件が用いられることを示すために用いられる。着目のポリペプチドの生産を行う条件下で、または1もしくは複数の毒素の生産を行う条件下で、問題の毒素の生産に関して親細胞と変異体細胞を比較することができる。
【0026】
宿主細胞
本発明は、親細胞に比較して有意に減少したレベルの1または複数の毒素を発現させるために遺伝的に変更されている、着目のポリペプチドの発現に有用なアスペルギルス宿主細胞の変異体を提供する。宿主細胞は親細胞から誘導されるが、野生型細胞であってもよい。
【0027】
宿主株は、着目のポリペプチドの発現に従来使用されている任意のアスペルギルス宿主細胞であることができる。
【0028】
好ましい態様では、着目のポリペプチドの生産に有用であるアスペルギルス宿主細胞が、アスペルギルス亜群ユーロチウム(Eurotium)〔例えばA.レストリクツス(A. restrictus)種により代表される〕、ケトサルトリア(Chaetosartorya)〔例えばA.クレメウス(A. cremeus)種により代表される〕、スクレロクレイスタ(Sclerocleista)〔例えばA.オルナティ(A. ornati)種により代表される〕、サトイア(Satoia)〔例えばA.ニガー(A. niger)種により代表される〕、ネオサルトリア(Neosartorya)〔例えばA.フミガーツス(A. fumigatus)、A.セルビヌス(A. cervinus)種により代表される〕、ヘミカーペンテレス(Hemicarpenteles)〔例えばA.クラバツス(A. clavatus)種により代表される〕、ペトロミセス(Petromyces)〔例えばA.フラーブス(A. flavus)、A.カンジダス(A. candidus)、A.スパルサス(A. sparsus)種により代表される〕、エメリセラ(例えばA.ニデュランス(A. nidulans)、A.ベルシカラー(A. versicolor)、A.ウスツス(A. ustus)種により代表される〕、およびフェネリア(Fenellia)〔例えばA.テレウス(A. terreus)種により代表される〕からなる群より選ばれる。
【0029】
特に好ましい態様では、アスペルギルス宿主細胞が、A.オリゼ、A.アキュレータス、A.フィキュウム、A.フラーブス、A.フェチダス、A.ソヤ、A.サケ、A.ニガー、A.ニデュランスおよびA.ジャポニカスからなる群より選ばれる。それらのうち、アスペルギルス・オリゼ、アスペルギルス・ニガー、A.パラシチクスおよびA.フェニクスが最も好ましい。
本発明の宿主細胞の上記例は、現在受け入れられている生物分類学に従って命名される。
【0030】
毒素
本発明に従ってその生産を減少または除去しようとする毒素は、アスペルギルス属により生産されるか、またはアスペルギルス属が所有する遺伝子によりコードされるが必ずしも発現されなくてもよい、いずれの毒素であってもよい。例えば、アフラトキシン遺伝子はA.オリゼ中に存在するが、この種からは発現されないことが知られている。しかしながら、たとえそれらが発現されなくても、アフラトキシン経路遺伝子を除去することはまだ有利である場合がある。これは下記に説明され、実施例6に例示される。
【0031】
特に、減少または除去すべき毒素は、シクロピアゾン酸(CPA)、例えばそのα形またはβ形、コウジ酸(KA)、3−ニトロプロピオン酸(NPA)、エモジン、マルフォルミン(例えばマルホルミンAまたはB)、アフラトキシン、オクラトキシン、フラビオリンおよびセカロン酸(例えばセカロン酸D)からなる群より選ばれる。それらの毒素の説明については、本明細書中の発明の背景の項目とその中に言及したthe Handbook of Toxic Fungal Metabolitesを参照のこと。
【0032】
本発明のジメチルアリル−シクロアセトアセチル−L−トリプロファンシンターゼ(DCAT-S)
本発明は、(a) 配列番号2のアミノ酸配列を有するジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼ;(b) (a) の対立遺伝子変異体;および(c) ジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼ活性を有する(a)または(b)の断片からなる群より選ばれる、糸状菌から得られる単離されたジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼにも関する。
【0033】
好ましくは、本発明のジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼは配列番号2のアミノ酸配列またはそれの対立遺伝子変異体を含んでなる。より好ましい態様では、本発明のジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼは配列番号2のアミノ酸配列を含んでなる。別の好ましい態様では、本発明のジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼは配列番号2またはそれの断片のアミノ酸配列を有し、ここで前記断片はジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼ活性を有する。配列番号2の断片は、このアミノ酸配列のアミノ末端および/またはカルボキシ末端から1または複数のアミノ酸配列が削除されているポリペプチドである。最も好ましい態様では、ジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼが配列番号2のアミノ酸配列を有する。
【0034】
好ましくは、配列番号2の断片が少なくとも320アミノ酸残基、最も好ましくは少なくとも350アミノ酸残基を有する。
【0035】
対立遺伝子変異体は、同一染色体遺伝子座を占有する遺伝子の2以上の別形態のいずれかを意味する。対立遺伝子変異は突然変異を通じて自然に発生し、そして集団内に表現型多形を引き起こし得る。遺伝子変異はサイレント(コードされるポリペプチドに全く変化なし)であってもよく、変更されたアミノ酸配列を有するポリペプチドをコードしてもよい。ポリペプチドの対立遺伝子変異体という用語は、遺伝子の対立遺伝子変異体によりコードされるポリペプチドである。
【0036】
配列番号2のアミノ酸配列またはそれの部分配列を使って、オリゴヌクレオチドプローブ、すなわち本発明のジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼをコードする核酸配列をデザインすることができ、例えば、当業界で周知の方法に従って、配列番号1の核酸配列もしくはそれの部分配列を使って別の糸状菌株からジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼをコードするDNAを同定およびクローニングすることができる。特に、そのようなプローブは、対応する遺伝子を同定および単離するため、標準サザンブロッティング技術に従った着目の属または種のゲノムDNAまたはcDNAとのハイブリダイゼーションに使用することができる。そのようなプローブは、完全配列よりもかなり短鎖であることができるが、長さが少なくとも15、好ましくは少なくとも25、より好ましくは少なくとも40ヌクレオチドであるべきである。更に長いプローブを使用してもよい。DNAプローブとRNAプローブの両方を使用できる。プローブは典型的には対応する遺伝子を検出するために標識される(例えば32P,3H,35S,ビオチンまたはアビジンで)。
【0037】
ハイブリダイゼーションは、標準サザンブロッティング技術に従って、低〜高緊縮性条件下で(すなわち、5×SSPE,0.3%SDS,200μg/ml尖断変性サケ精子DNA、低、中および高緊縮性条件についてそれぞれ25%, 35%または50%のいずれかのホルムアミド中での42℃でのプレハイブリダイゼーションとハイブリダイゼーション)、配列番号1に示されるまたはpJaL499中に含まれる核酸配列のポリペプチドコード部分に相当するオリゴヌクレオチドプローブに核酸配列がハイブリダイズすることを示す。
【0038】
こうして、別の糸状菌株より調製したゲノムライブラリー、cDNAライブラリーまたはコンビナトリアル・ケミストリーライブラリーを、上記プローブとハイブリダイズし且つジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼをコードするDNAについてスクリーニングすることができる。別の糸状菌株からのゲノムDNAまたは他のDNAをアガロースもしくはポリアクリルアミドゲル電気泳動、または他の分離技術により分離することができる。ライブラリーからのDNAまたは分離したDNAは、ニトロセルロースまたは他の適当な担体材料上に移行せしめそして固定することができる。配列番号1と相同であるクローンまたはDNAを同定するために、サザンブロット法において担体材料を使用し、該担体材料を2×SSC,0.2%SDSを使って、好ましくは少なくとも50℃で、より好ましくは少なくとも55℃で、より好ましくは少なくとも60℃で、より好ましくは少なくとも65℃で、更により好ましくは少なくとも70℃で、最も好ましくは少なくとも75℃で、30分間ずつ3回最終洗浄する。それらの条件下でオリゴヌクレオチドプローブがハイブリダイズする分子を、X線フィルムを使って検出する。
【0039】
好ましい態様では、本発明のジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼはアスペルギルスの株、より好ましくはA.オリゼから得られ、例えばそのポリペプチドは配列番号2のアミノ酸配列を有する。
【0040】
本明細書中で定義する「単離された」ジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼは、本質的に他のポリペプチドを含まないポリペプチド、例えばSDS−PAGEで測定した時に少なくとも約20%純粋、好ましくは少なくとも約40%純粋、より好ましくは約60%純粋、更により好ましくは約80%純粋、最も好ましくは約90%純粋、更に最も好ましくは約95%純粋であるポリペプチドである。
【0041】
本発明は、糸状菌より得られるジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼをコードする単離された核酸配列にも関する。より好ましい態様では、該核酸配列がアスペルギルス種、例えばA.オリゼから得られ、特に配列番号1に記載の核酸配列を有する。別の好ましい態様では、該核酸配列がプラスミドpJaL499中に含まれる配列である。本発明は、遺伝暗号の縮重によって配列番号1とは異なる核酸配列も包含する。本発明は、ジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼ活性を有するポリペプチド断片をコードする、配列番号1の部分配列またはpJaL499のポリペプチドコード部分の部分配列にも関する。配列番号1の部分配列またはpJaL499のポリペプチドコード部分の部分配列は、5′および/または3′末端から1もしくは複数のヌクレオチドが欠失していること以外は、配列番号1またはpJaL499のポリペプチドコード部分により包含される核酸配列である。好ましくは、配列番号1の部分配列は少なくとも870ヌクレオチド、より好ましくは少なくとも960ヌクレオチド、最も好ましくは少なくとも1050ヌクレオチドを含む。
【0042】
該核酸配列はアスペルギルス・オリゼの分類学上の同等物である微生物より得ることができる。
【0043】
そのような核酸配列を単離またはクローニングするのに利用する技術については後述する。本明細書中で用いる「単離された核酸配列」という用語は他の核酸配列を本質的に含有しない核酸配列、例えば、アガロースゲル電気泳動により測定した時に、少なくとも約20%純粋、好ましくは少なくとも約40%純粋、より好ましくは少なくとも約60%純粋、更により好ましくは少なくとも約80%純粋、最も好ましくは少なくとも約90%純粋である核酸配列を言う。該核酸配列はゲノムDNA、cDNA、RNA、半合成、合成起源またはそれらの任意組合せのものであることができる。
【0044】
本発明のジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼをコードする核酸配列の変更が、該ポリペプチドと実質的に同じ酵素の合成に必要かもしれない。ジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼと「実質的に同じ」とは、該酵素の非天然形態を指して言う。それらのジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼは、何らかの操作方法によって、その生来の源より単離された酵素と異なっていてもよい。例えば部位特異的変異誘発法を用いて、比活性、熱安定性、最適pHなどの点が異なっている該酵素の変異体を合成することが重要となる場合がある。類似配列は、配列番号1のポリペプチドコード部分として与えられる核酸配列、例えばそれの部分配列に基づいて、および/または該核酸配列によりコードされるポリペプチドの別のアミノ酸配列を与えず且つ該ポリペプチドの生産用の宿主生物のコドン用法に対応するようなヌクレオチド置換の導入により、または異なるアミノ酸配列を与えるようなヌクレオチド置換の導入により、作製することができる。ヌクレオチド置換の一般記載については、例えば、Ford他, 1991, Protein Expression and Purification 2: 95-107を参照のこと。
【0045】
そのような置換は分子の機能にとって重大である領域の外側で行うことができ、更にまだ生物学的に活性なジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼをもたらすものであることは、当業者に明らかであろう。本発明の単離された核酸配列によりコードされるジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼの活性にとって必須であり、従って置換を受けないことが好ましいアミノ酸残基は、部位特異的変異誘発またはアラニンスキャニング変異誘発といった当業界で既知の手法に従って同定することができる(例えば、Cunningham & Wells, 1989, Science 244: 1081-1085参照)。後者の技術では、分子中の正電荷を有する残基ごとに突然変異を導入し、そして生じた変異体分子をジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼ活性について試験し、該分子の活性にとって重要であるアミノ酸残基を同定する。核磁気共鳴分析、結晶学または光親和性標識法のような技術により測定される三次元構造の解析によって、基質−酵素相互作用を調べることもできる(例えば、de Vos他, 1992, Science 255: 306-312; Smith他, 1992, Journal of Molecular Biology 224: 899-904; Wlodaver他, 1992, FEBS Letters 309: 59-64)。
【0046】
本発明の核酸配列またはそれの相同体もしくは断片の好ましい使用目的は、特定の宿主細胞、特にA.オリゼの細胞のジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼ活性を除去または減少させ、それにより前記細胞からCPAの生産を減少または排除することである。
【0047】
本発明は、配列番号1の核酸配列またはpJaL499のポリペプチドコード部分、それの部分配列もしくは相同体を含有する、該配列の発現のための構成物、組換え発現ベクターおよび宿主細胞にも関する。該構成物およびベクターは本明細書中に記載の通りに作製することができる。宿主細胞は核酸配列の発現に適当な任意の細胞であることができ、そして例えば本明細書中に記載の親細胞または変異体細胞より選択することができる。
【0048】
宿主細胞の遺伝的変更
1または複数の毒素の発現レベルを有意に減少させるために本発明の宿主細胞は遺伝的に変更されるが、それは当業者に既知の標準技術を使って達成することができる。毒素活性の生産の原因となる遺伝子配列を不活性化するかまたは部分的にもしくは完全に除去することができる。こうして、本発明のアスペルギルス変異体宿主細胞は、減少したレベルまたは検出不可能なレベルの1または複数の毒素を発現する。
【0049】
特定の態様では、不活性化は、特定の毒素の形成または分泌に関与する各々の構造領域もしくは調節領域(例えば遺伝子)の変更により得られる。既知の有用な技術としては、非限定的に、特異的もしくはランダム変異誘発、PCR生成変異誘発、部位特異的DNA欠失、挿入および/または置換、遺伝子破壊または遺伝子置換、アンチセンス技術、またはそれらの組合せ等が挙げられる。
【0050】
変異誘発は適当な物理的もしくは化学的変異誘発剤を使って行うことができる。本発明の目的上適当である物理的もしくは化学的変異誘発剤の例としては、非限定的に、UV照射、イオン化照射、例えばガンマ照射、ヒドロキシルアミン、N−メチル−N′−ニトロ−N−ニトロソグアニジン(MNNG)、O−メチルヒドロキシルアミン、亜硝酸、エチルメタンスルホネート(EMS)、亜硫酸水素ナトリウム、およびヌクレオチド類似体が挙げられる。そのような剤を用いる場合、典型的には、変異させようとする細胞を適当な条件下で特定の変異誘発剤の存在下でインキュベートし、そして有意に減少した標的毒素生産を示す細胞について選択することにより、変異誘発が行われる。
【0051】
宿主細胞による特定の毒素の生産の減少または除去は、該毒素の生産または分泌に関与するかまたは他の形で必要であるヌクレオチド配列を変更することによっても達成することができる。例えば、該ヌクレオチド配列が、毒素生産に至る経路の中の必要機能を有する遺伝子生成物であってもよい。ヌクレオチド配列は、例えば、配列番号1に示すものまたはpJaL499のポリペプチドコード部分であることができる。変更は、該ヌクレオチド配列中または該配列の転写もしくは翻訳に必要な調節要素中への1もしくは複数のヌクレオチドの導入、置換または除去によって達成することができる。例えば、該ヌクレオチド配列への終止コドンの導入、開始コドンの除去または転写解読枠の変更をもたらすようにヌクレオチドを挿入または除去してもよい。該配列またはその調節要素の変更または不活性化は、当業界で周知の方法に従った部位特異的もしくはランダム変異誘発またはPCR生成変異誘発により達成することができる。理論的には生体内で、すなわち毒素遺伝子を発現する細胞に直接、変更を行うことができるけれども、現在のところは下記に例示するように試験管内で変更を行うことが好ましい。
【0052】
特定の糸状菌細胞における着目の毒素、例えばCPAの生産を不活性化または減少させる便利な方法の一例は、遺伝子置換、遺伝子欠失または遺伝子破壊の技術に基づいている。例えば、遺伝子破壊法では、着目の遺伝子または遺伝子断片(例えば本発明のDCAT-S遺伝子)に相当する核酸配列を試験管内変異誘発せしめて欠陥核酸配列を生産させ、次いでその欠陥核酸配列を用いて親細胞を形質転換させて欠陥遺伝子を生成せしめる。相同組換えにより、欠陥遺伝子配列が内因性遺伝子または遺伝子断片と置き換わる。欠陥遺伝子または遺伝子断片が更にマーカーをコードすることが望ましいかもしれない。該マーカーは、核酸配列が変更または破壊されている形質転換体の選択に利用することができる。
【0053】
あるいは、遺伝子の変更または不活性化は、該遺伝子の核酸配列に相補的なヌクレオチド配列を使用する、確立されたアンチセンス技術によって行うこともできる。より詳しくは、細胞内で転写されることができ且つ細胞内で生産されたmRNAにハイブリダイズすることができる、該遺伝子の核酸配列に相補的であるヌクレオチド配列を導入することにより、糸状菌細胞による該遺伝子の発現を減少または除去することができる。こうして、相補的アンチセンスヌクレオチド配列をmRNAにハイブリダイズ可能にする条件下では、翻訳されるタンパク質の量が減少または除去される。
【0054】
毒素経路の遺伝子の変異誘発または別の変更の後、減少または除去された毒素生産について変異体がスクリーニングされる。毒素についてスクリーニングする方法の具体例は下記の実施例に与えられる。あるいは、有用なスクリーニングアッセイが"Handbook of Toxic Fungal Metabolites"中に記載されており、または例えば食品中のマイコトキシンのレベルを一般検査する機関で入手可能である。
【0055】
従って、遺伝的変更のため、本発明のアスペルギルス変異体宿主細胞は有意に減少したレベルの毒素を発現する。好ましい態様では、変異体宿主細胞により発現されるそれらの毒素のレベルは、個々に約50%以上、好ましくは約85%以上、より好ましくは約90%以上、最も好ましくは約95%以上、更により好ましくは99%以上減少されている。別の好ましい態様では、本発明の変異体宿主細胞中の複数毒素が任意の組合せで減少されてもよい。更に別の好ましい態様では、宿主細胞により発現される生成物がシクロピアゾン酸、コウジ酸、3−ニトロプロピオン酸、エモジン、マルフォルミン、アフラトキシン、オクラトキシンおよびセカロン酸のうちの少なくとも1つの毒素を本質的に含有しない。特に好ましい態様では、宿主細胞により発現される生成物が少なくともシクロピアゾン酸を本質的に含有せず、より好ましくは少なくともシクロピラゾン酸とコウジ酸またはアフラトキシンを本質的に含有せず、最も好ましくは少なくともシクロピアゾン酸、コウジ酸および3−ニトロプロピオン酸を含有しない。
【0056】
好ましい態様では、宿主細胞が、NPA,CPA,コウジ酸(KA)またはマルトリジンのうちの1つまたは複数;好ましくはそれらの毒素のうちの少なくとも2つ、例えばNPAとCPA;NPAとKA;CPAとKA;またはNPAとCPAとKA、の生産が減少または除去されているA.オリゼの株である。更に、それらの1または複数の毒素の除去または減少に加えて、好ましくは得られた変異体株がアフラトキシンを生産できないように、特定のアフラトキシン経路の遺伝子が不活性化される。A.フラーブス(A. flavus)からのアフラトキシン遺伝子は周知であるのでそれをA.オリゼ中の対応遺伝子の同定に利用することができ、次いでその対応遺伝子を当該技術分野で既知の方法により不活性化することができる。
【0057】
別の好ましい態様では、宿主細胞が、マルフォルミン(例えばマルフォルミンA1またはB)、オクラトキシン(例えばオクラトキシンA)およびフラビオリンのうちの1つまたは複数、好ましくはそれらの毒素のうちの少なくとも2つ、例えばマルフォルミンとオクラトキシン;マルフォルミンとフラビオリン;オクラトキシンとフラビオリン;およびマルフォルミンとオクラトキシンとフラビオリンの生産が減少または除去されているA.ニガー(A. niger)またはA.フィキュウム(A. ficuum)の株である。
【0058】
別の好ましい態様では、宿主細胞が、セカロン酸(例えばセカロン酸D)またはエモジン(セカロン酸Dの前駆体)、好ましくはそれらの毒素の両方の生産が減少または除去されているA.アキュレータス(A. aculeatus)の株である。
【0059】
ポリペプチドの生産方法
本発明の方法により、或る1または複数の標的毒素の量は有意に減少されるが、その一方で着目のポリペプチドをコードする遺伝的に変更された遺伝子の細胞内安定維持、細胞の生産能力、および着目のポリペプチドの収率の点からの変異体宿主細胞の特徴は実質的に保持される。より詳しくは、本発明の方法により、宿主細胞は1または複数の着目の毒素の生産または分泌に必要な構造領域および/または調節領域の内部で遺伝的に変更され、それにより前記毒素の生産または分泌が減少または除去される。
【0060】
従って、本発明の別の面は、本発明のアスペルギルス変異体宿主細胞におけるポリペプチドまたはタンパク質(非相同ポリペプチドまたはタンパク質を包含する)の生産方法であって、前記変異体宿主細胞中に着目のポリペプチドをコードする核酸配列を導入し、適当な増殖培地中で変異体宿主細胞を培養し、そして前記着目のポリペプチドを回収することを含んでなる方法を提供する。
【0061】
よって、本発明の変異体宿主細胞は、着目のポリペプチドの発現に必要な構造および調節遺伝子領域を含まなければならない。そのような構造および調節領域の性質は、大部分、目的とする生成物と特定のアスペルギルス宿主株により決まる。本発明の宿主細胞の遺伝子設計は、宿主細胞の形質転換またはトランスフェクションのための標準組換えDNA技術を使って当業者により成し遂げることができる(例えばSambrook他参照)。
【0062】
好ましくは、宿主細胞は、着目の所望のポリペプチドをコードするDNA断片を含んで成る適当なクローニングビヒクル、すなわちプラスミドまたはベクター、の導入のための周知技術により変更される。クローニングビヒクルは自己複製プラスミドとして宿主細胞中に導入されてもよく、または染色体中に組み込まれてもよい。好ましくは、クローニングビヒクルは1または複数の適当な調節領域に作用可能に連結された1または複数の構造領域を含んでなる。
【0063】
構造遺伝子は着目のポリペプチドをコードするヌクレオチド配列の領域である。調節領域は、転写および翻訳調節配列を含んでなるプロモーター領域、終止シグナルを含んでなるターミネーター領域、およびポリアデニル化領域を包含する。プロモーター、すなわち特定の宿主細胞において転写活性を示すヌクレオチド配列は、細胞外または細胞内タンパク質、好ましくは酵素、例えばアミラーゼ、グルコアミラーゼ、プロテアーゼ、リパーゼ、セルラーゼ、キシラナーゼ、オキシドレダクターゼ、ペクチナーゼ、クチナーゼまたは解糖系酵素をコードする遺伝子から誘導することができる。本発明の方法において核酸構成物の転写を指令するのに適当なプロモーターの例は、アスペルギルス・オリゼ(Aspergillus oryzae)TAKAアミラーゼ、リゾムーコル・ミーヘイ(Rhizomucor miehei)アスパラギン酸プロテイナーゼ、アスペルギルス・ニガー(Aspergillus niger)中性α−アミラーゼ、アスペルギルス・ニガー(Aspergillus niger)酸安定性α−アミラーゼ、アスペルギルス・ニガー(Aspergillus niger)もしくはアスペルギルス・アワモリ(Aspergillus awamori)グルコアミラーゼ(glaA)、リゾムーコル・ミーヘイ(Rhizomucor miehei)リパーゼ、アスペルギルス・オリゼ(Aspergillus oryzae)アルカリ性プロテアーゼ、アスペルギルス・オリゼ(Aspergillus oryzae)トリオースリン酸イソメラーゼ、アスペルギルス・ニデュランス(Aspergillus nidulans)アセトアミダーゼ(amdS)、フザリウム・オキシスポラム(Fusarium oxysporum)トリプシン様プロテアーゼ(米国特許第4,288,627号)をコードする遺伝子から得られるプロモーター、並びにそれらの変異体、短縮形およびハイブリッドプロモーターである。特に好ましいプロモーターは、NA2-tpiプロモーター(アスペルギルス・ニガー中性α−アミラーゼをコードする遺伝子由来のプロモーターとアスペルギルス・オリゼのトリオースリン酸イソメラーゼをコードする遺伝子由来のプロモーターとのハイブリッド)、グルコアミラーゼプロモーター、およびTAKAアミラーゼプロモーターである。
【0064】
クローニングビヒクルは選択マーカーを含んでもよい。選択マーカーは、その生成物が殺生剤耐性、ウイルス耐性、重金属耐性、原栄養株に対する栄養要求変異体、などに備える遺伝子である。糸状菌宿主細胞用の選択マーカーは、非限定的に、amdS(アセトアミダーゼ)、argB(オルニチンカルバモイルトランスフェラーゼ)、bar(ホスフィノスリシンアセチルトランスフェラーゼ)、hygB(ヒグロマイシンホスホトランスフェラーゼ)、niaD(硝酸レダクターゼ)、pyrG(オロチジン−5′−リン酸デカルボキシラーゼ)、sC(硫酸アデニルトランスフェラーゼ)およびtrpC(アントラニル酸シンターゼ)並びに別の種からの同等物からなる群より選ぶことができる。アスペルギルス細胞用に好ましいのは、アスペルギルス・ニデュランスまたはアスペルギルス・オリゼのamdSおよびpyrG遺伝子、並びにストレプトマイセス・ヒグロスコピクス(Streptomyces hygroscopics)のbar遺伝子である。
【0065】
更に、選択は同時形質転換によって行ってもよく、この場合は、2つのベクターの混合物を使って形質転換を行い、そして1つのベクターのみについて選択を行う。
【0066】
本発明のDNA構成物、プロモーター、ターミネーターおよび他の要素をそれぞれ連結せしめそしてそれらを複製に必要な情報を含む適当なクローニングビヒクル中に挿入するのに用いる手法は、当業者に周知である(例えばSambrook他, 1989, 前掲を参照のこと)。
【0067】
変異体糸状菌細胞は、当該技術分野で既知の方法を使って、着目のポリペプチドの生産に適した普通培地中で培養される。例えば、細胞は、適当な培地中でそして非相同ポリペプチドを発現および/または単離可能にする条件下で行われる、振盪フラスコ培養、実験室用もしくは工業用発酵槽中での小規模または大規模発酵(連続発酵、バッチ発酵、フェドバッチ発酵または固相発酵を含む)により培養することができる。培養は、当該技術分野で既知の手法を使って、炭素源、窒素源および無機塩類を含有する適当な普通培地中で行われる。適当な培地は民間供給業者から入手可能であり、または発表された組成〔例えばアメリカン・タイプ・カルチャー・コレクション(ATCC)のカタログ中〕に従って調製することができる。分泌されたポリペプチドは培地から直接回収することができる。
【0068】
ポリペプチドは該ポリペプチドに特有である当業界で既知の方法を使って検出することができる。それらの検出方法としては、特異抗体の使用、酵素生成物の形成、酵素基質の消失、またはSDS−PAGEが挙げられる。例えば、酵素アッセイを使ってポリペプチドの活性を測定してもよい。酵素活性の測定方法は様々な酵素について知られている。
【0069】
得られたポリペプチドは当該技術分野で既知の方法により単離することができる。例えば、非限定的に遠心分離、濾過、抽出、噴霧乾燥、蒸発または沈澱をはじめとする常用手順により、普通培地から単離することができる。次いで単離されたポリペプチドは、非限定的にクロマトグラフィー(例えばイオン交換、アフィニティー、疎水的、等電点およびサイズ排除)、電気泳動(例えば分取用等電点電気泳動)、分別溶解度(例えば硫酸アンモニウム沈澱)または抽出法をはじめとする既知の様々な方法により、更に精製することができる(例えばProtein Purification, J.-C. Janson & Lars Ryden編,VCH Publishers, New York, 1989を参照のこと)。
【0070】
生成物
所望の最終生成物、すなわちアスペルギルス変異体宿主細胞により発現される着目のポリペプチドは、いずれの相同または非相同タンパク質またはペプチドであってもよい。
【0071】
該ポリペプチドは、変異体糸状菌細胞にとって非相同である任意のポリペプチドであることができる。「ポリペプチド」なる用語は、コードされる生成物の特定の長さについて言及するものではなく、従ってペプチド、オリゴペプチドおよびタンパク質を包含するものである。非相同ポリペプチドは或るポリペプチドの改変変異体であってもよい。「非相同ポリペプチド」なる用語は、本明細書中では糸状菌細胞にとって生来でないポリペプチドとして定義される。変異体糸状菌細胞は非相同ポリペプチドをコードする核酸配列を1または複数コピー含んでもよい。
【0072】
本発明の方法では、変異体糸状菌細胞を該細胞にとって生来であるポリペプチドの組換え生産に使用してもよい。例えば、該ポリペプチドの発現を増強するため、シグナル配列の使用により細胞の外側への着目の生来のポリペプチドの輸送を促進するため、および細胞により通常に生産されるポリペプチドをコードする遺伝子のコピー数を増加させるために、別のプロモーターの調節下に該ポリペプチドをコードする遺伝子を配置することにより、生来のポリペプチドを組換え生産せしめることができる。本発明は、そのような発現が細胞にとって生来でない遺伝要素の使用を伴うかまたは宿主細胞で通常は起こらないような形で機能するように操作されている生来の要素の使用を伴う限り、「非相同ポリペプチド」の用語の範囲内に、そのような組換え生産も包含する。
【0073】
より具体的な態様では、生成物が療法上有効なペプチドまたはタンパク質、例えばホルモン、特にインスリン、成長ホルモン、グルカゴンまたはソマトスタチン;インターロイキン、特にインターフェロン;造血増殖因子、特にPDGE(血小板由来増殖因子)、EPO(エリスロポイエチン)またはTPO(トロンボポイエチン);プロテアーゼ、特に第VII因子、第VIII因子、ウロキナーゼ、キモシンまたはTPA;または血清アルブミンである。
【0074】
別の好ましい態様では、生成物が真菌または細菌起源の酵素である。酵素は好ましくはグリコシダーゼ酵素、例えば、アミラーゼ、特にα−アミラーゼ、β−アミラーゼまたはグルコアミラーゼ;グルカン1,4−α−グルコシダーゼ;アミノペプチダーゼ;カルボヒドラーゼ;カルボキシペプチダーゼ;カタラーゼ;セルラーゼ;特にエンド−1,4−β−グルカナーゼまたはエンド−1,3(4)−β−グルカナーゼ;セルロース−1,4−β−セロビオシダーゼ;キチナーゼ;クチナーゼ;シクロデキストリングリコシルトランスフェラーゼ;デオキシリボヌクレアーゼ;ガラクタナーゼ;ガラクトシダーゼ、特にα−ガラクトシダーゼまたはβ−ガラクトシダーゼ;エンドグルカナーゼ、特にエンド−1,3−β−グルカナーゼ、エンド−1,3−α−グルカナーゼ、エンド−1,2−β−グルカナーゼまたはエンド−1,6−β−グルカナーゼ;グルコシダーゼ;特にα−グルコシダーゼまたはβ−グルコシダーゼ;インベルターゼ;ラッカーゼ;脂質分解酵素、特にリパーゼ、エステラーゼ、ホスホリパーゼまたはリゾ−ホスホリパーゼ;リアーゼまたはペクテートリアーゼ;マンナーゼ;マンノシダーゼ;ポリガラクツロナーゼ;ムタナーゼ;オキシダーゼまたはオキシドレダクターゼ、例えばペルオキシダーゼまたはポリフェノールオキシダーゼ;オキシゲナーゼ;ペクチナーゼ、エンドペプチダーゼまたはエキソペプチダーゼ;フィターゼ;ポリガラクツロナーゼ;プロテアーゼ;リボヌクレアーゼ;トランスグルタミナーゼ;およびキシラナーゼ、特にエンド−1,4−β−キシラナーゼまたはキシラン−エンド−1,3−β−キシロシダーゼである。
【0075】
別の好ましい態様では、生成物がハイブリッドポリペプチド、例えばプロキモシンおよびプロトリプシン様プロテアーゼである。宿主細胞により発現される非相同ポリペプチドは、適当な条件下で、例えば実質的なプロテアーゼ活性の非存在下で、チモーゲンのような前駆体タンパク質、ハイブリッドタンパク質、プロ配列もしくはプレ−プロ配列として得られるタンパク質、または他の任意の未成熟形であってもよい。
【0076】
本発明を下記の実施例に関して更に説明するが、この実施例は特許請求の範囲において定義されるような本発明の範囲を限定すると解釈してはならない。
【0077】
サイレント毒素遺伝子が除去されているアスペルギルス変異体
アフラトキシンの生合成経路はアフラトキシン産生種であるアスペルギルス・フラーブスおよびアスペルギルス・パラシチクスにおいて研究されている。どちらの種も、多数の遺伝子が同定されそして大クラスターとしてマッピングされることが示されている〔Woloshuk, C.P. & Prieto, R., FEMS Microbiology Letter (1998) 160:169-176〕。別経路遺伝子の発現調節遺伝子をコードする遺伝子であるaflR、およびO−メチルトランスフェラーゼをコードするomtAといった幾つかの遺伝子はクローニングされ、そして配列決定されている。
【0078】
アフラトキシン遺伝子はA.オリゼのゲノム中に存在するが、この種からは発現されない。アフラトキシンが全く発現されないくても、それらのサイレントアフラトキシン経路遺伝子の1つまたは複数を除去することは有利である。アフラトキシン遺伝子、例えばaflRおよび/またはomtA遺伝子が除去されているアスペルギルス属変異体、例えばA.オリゼ変異体は、問題の1または複数のアフラトキシンの生産について新規変異体株を試験する必要がないので、有利である。
【0079】
よって、本発明は、一面では、1または複数のサイレント毒素遺伝子が除去されている、非相同ポリペプチドの発現に適したアスペルギルス変異体細胞に関する。
【0080】
サイレント毒素遺伝子が「除去」されているとは、前記サイレント遺伝子が変異体細胞中に含まれなくなるような形で、例えば当該技術分野で周知の遺伝子置換技術または遺伝子破壊技術(例えばMiller他, 1985, Molecular and Cellular Biology, p.1714-1721参照)により、問題の(1または複数の)遺伝子が変更または削除されていることを意味する。
【0081】
「サイレント」毒素遺伝子なる用語は、該毒素が発現されないことを意味する。
【0082】
毒素遺伝子は該毒素遺伝子の全部または一部の不可逆的欠失または破壊により除去されてもよい。
【0083】
「毒素遺伝子の全部または一部の不可逆的欠失または破壊」なる用語は、前記遺伝子が毒素をコードせず、且つ毒素をコードする遺伝子へと自然に復帰変異する(例えば生産中に)ことができないような形で、問題の毒素遺伝子が除去または変更されていることを意味する。
【0084】
問題のアスペルギルス細胞は上記のものいずれでもよく、そしてアスペルギルス亜群のユーロチウム(Eurotium)、ケトサルトリア(Chaetosartorya)、スクレロクレイスタ(Sclerocleista)、サトイア(Satoia)、ネオサルトリア(Neosartorya)、ヘミカーペンテレス(Hemicarpenteles)、ペトロミセス(Petromyces)、エメリセラ(Emericella)およびフェネリア(Fenellia)からなる群より選択されてもよい。
【0085】
1または複数の毒素をコードする問題の毒素遺伝子としては、シクロピアゾン酸、コウジ酸、3−ニトロプロピオン酸、エモジン、マルフォルミン、アフラトキシン、オクラトキシンおよびセカロン酸からなる群より選ばれる毒素が挙げられる。
【0086】
特に期待されるのは、アフラトキシンをコードする毒素遺伝子、特にA.オリゼのアフラトキシンクラスターからの毒素遺伝子、特にomtA, aflR, pksA, Nor-1, fas-beta, fas-alpha, vber-1, avnA, ord-2から選ばれた毒素遺伝子である。
【0087】
親のアスペルギルス細胞はA.オリゼ細胞、特にA.オリゼ A1560 (IFO 0417)であることができる。
【実施例】
【0088】
実験
材料と方法
1.菌株
アスペルギルス・オリゼ(Aspergillus oryzae)A1560はIFO 04177と同一である(下記参照)。
アスペルギルス・オリゼ IFO 4177:Institute for Fermentation, Osaka(財団法人発酵研究所;大阪府淀川区十三本町2丁目17−25)より入手可能。WO 98/12300参照。
JaL228:中性メタロプロテアーゼ NpI遺伝子が破壊されているアスペルギルス・オリゼ株;この株の作製はWO 98/12300に記載されている。
【0089】
BECh 1:このCPA陰性アスペルギルス・オリゼ株の作製は実施例1に記載される。
BECh 2:このCPA陰性KA陰性アスペルギルス・オリゼ株の作製は実施例1に記載される。
BECh 3:このCPA陰性KA陰性アスペルギルス・オリゼ株の作製は実施例1に記載される。
【0090】
BZ14:WO 92/17573に記載の通りToC90とphD450により同時形質されたアスペルギルス・オリゼの株。
JaL 250:このアスペルギルス・オリゼ株の作製は実施例6に記載される。
【0091】
寄託:
プラスミドpJaL499を含有するE.コリ株は、1999年1月13日にDSMZ(Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH;Mascheroder Weg 1b, D-38124 Brauschweig)に寄託され、そして寄託番号DSM 12622を付与された。
【0092】
2.遺伝子
DMAT-S:この遺伝子は、麦角アルカロイドの生合成に関与する酵素であるジメチルアリル−L−トリプトファンシンターゼをコードする。
DCAT-S:この遺伝子は、シクロピアゾン酸(CPA)生合成に関与する酵素であるジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼをコードする。
pyrG:この遺伝子は、ウリジン生合成に関与する酵素であるオロチジン−5′−リン酸デカルボキシラーゼをコードする。
【0093】
3.プラスミド
pAHL:このプラスミドはWO 97/07202に記載されている。
pCaHj483:このプラスミドはWO 98/00529に記載されている。
pCaHj493:このプラスミドは実施例2に記載される。
pJaL335:このプラスミドはWO 98/12300に記載されている。
pJaL499:このプラスミドは実施例4に記載される。
【0094】
4.培地と溶液
緩衝液および基質として使用する薬品は少なくとも試薬用品質のものであった。
スクリーニング培地1(1リットルあたり)
マンニトール 30 g
グルコース 10 g
コハク酸 10 g
カザミノ酸 3 g
KH2PO4 1 g
MgSO4・7H2O 0.3 g
FeSO4・7H2O 0.2 g
2,6−ジクロロ−4−アニリン 2 ppm
寒天 20 g
14% NH4OHで最終pH 5.6に調整。
【0095】
Cove N
Cove塩溶液 50 ml
ソルビトール 218 g
デキストロース 10 g
硝酸カリウム 2.02 g
寒天 35 g
脱イオン水 1000 ml
【0096】
Cove塩溶液(1リットルあたり)
KCl 26 g
MgSO4 26 g
KH2PO4 76 g
微量金属溶液 50 ml
CHCl3 2 ml
【0097】
微量金属溶液(1リットルあたり)
Na2B4O7・10H2O 40 mg
CuSO4・5H2O 400 mg
FeSO4・7H2O 800 mg
MnSO4・2H2O 800 mg
Na2MoO4・2H2O 800 mg
ZnSO4・7H2O 8000 mg
【0098】
G1-gly
酵母エキス 18 g
グリセロール87% 24 ml
プルロニック PE6100 1 ml
水道水で 1000 mlに
【0099】
1/5 MDU-2BP
マルトース 9 g
MgSO4・7H2O 0.2 g
NaCl 0.2 g
K2SO4 0.4 g
KH2PO4 2.4 g
酵母エキス 1.4 g
AMG微量金属 0.1 ml
プルロニック PE6100 0.02 ml
脱イオン水 1000 mlに
最終pH 5.0;接種前に1.0 mlの50%尿素を添加する。
【0100】
MDU-IB(1リットルあたり)
マルトデキストリン MD01 45.0 g
MgSO4・7H2O 1.0 g
NaCl 1.0 g
K2SO4 2.0 g
KH2PO4 12.0 g
酵母エキス 7.0 g
AMG微量金属 0.5 ml
プルロニック PE6100 1 ml
最終pH 5.0;接種前に100 ml培地あたり1.3 mlの50%尿素を添加する。
【0101】
AMG微量金属溶液(1リットルあたり)
FeSO4・7H2O 13.9 g
MnSO4・H2O 8.45 g
ZnCl2 6.8 g
CuSO4・5H2O 2.5 g
NiCl2・6H2O 2.5 g
クエン酸 ≧3.0 g
微量金属溶液 1 ml
【0102】
KM2培地(1リットルあたり)
酵母エキス 2.5 g
KH2PO4 10 g
MgSO4・7H2O 0.5 g
KCl 0.5 g
FeSO4 0.01 g
グルコース 100 g
最終pHを6.0に調整
固形プレートで使用する前に、KMZ培地を20 g/l寒天で凝固させる。
コロニー増殖制限剤として300 μl/lのTriton X-100を添加する。
【0103】
ナカムラ培地(1リットルあたり)
ショ糖 50 g
ペプトン 20 g
KH2PO4 5 g
CaHPO4 2.5 g
MgSO4 2.5 g
最終pHを6.0に調整。
【0104】
5.アッセイ
A.HPキャピラリー電気泳動によるCPAについてのアッセイ方法
Supelclean LC-18 SPEチューブ(2 mlのメタノールと2 mlのMilli-Q水でコンディショニングした、カタログNo.5-7012のSupelco社製プレパック3 mlカラム)上での固相抽出により、キャピラリー電気泳動(CE)用に試料の1mlアリコートを調製する。溶液をカラムに強制的に流すために吸引マニホールドを使用する。3 mlのMilli-Q水で洗浄した後、3 mlのメタノールを用いて試料を溶離させる。更に後処理せずに、溶出液をCE分析にかける。場合により形成した沈澱を遠心により除去する。
【0105】
CE分析は、Hewlet-PackardフォトダイオードアレイCE装置(3D−CE)を使って行う。試料を10秒間かけて3400 Pa(34 mbar)の静水圧下で注入する。30℃で50 mmのキャピラリー(有効長さ56 cm)を使用し、それを0.1 M NaOHで1分間、次いで100 mMホウ酸/NaOH pH 9.1で5分間コンディショニングしておく。電圧を17 kVに設定する。ピークを同定するのにUVスペクトルを常時収集するが、280 nmで泳動を追跡する。標準試料として市販のシクロピアゾン酸を使用する(Sigma Co., St. Louis MO, USA, カタログNo. C1530, 最低98%純度)。この方法の感度の下限は約1ppmである。
【0106】
B.薄層クロマトグラフィー(TLC)によるCPAについてのアッセイ方法
1.平板培養物の分析
寒天プラグはFiltenborg O., Frisvad J.C. & Svendsen J.A.: "Simple Screening Method for Mold Producing Intracellular Mycotoxins in Pure Cultures", Applied and Environmental Microbiology (1983) 45:581-585に記載された通りに分析する。
【0107】
2.液体培養物の分析
上清の10μl試料をTLCプレート(Merck Silica Gel 60)の対角の縁に適用する。メタノール:クロロホルム(容量で1:2)混合物中に50 ppm, 25 ppm, 5 ppmおよび2.5 ppmに溶解希釈したシクロピアゾン酸(Sigma C 1530)を標準として使用する。まずプレートをCAP(クロロホルム:アセトン:プロパン−2−オール=容量で85:15:20)中で15分間展開し、乾燥し、次いで上下逆反転して、もう半分をTEF(トルエン:酢酸エチル:蟻酸=容量で5:4:1)中で15分間展開する。
【0108】
あるいは、プレートをEMA(酢酸エチル:メタノール:25%水酸化アンモニウム=容量で16:8:2)中とTEF中で各々15分間ずつ上記と同様に展開する。
プレートをヒュームフード中で完全に(1時間)乾燥させた後、エールリッヒ試薬(85 mlの96%エタノール中に溶かした2 gの4−ジメチルアミノベンズアルデヒドに、37%塩酸を添加したもの)を噴霧する。
【0109】
CPAは、CAP系(中性系)の場合は典型的な低い泳動度を有し、青紫色のマッシュルーム型スポットとして観察され、一方で酸性TEF系の場合は適用位置と展開剤の最前線の中間に細長く伸びた典型的なスミアを生じる。EMA系(塩基性/アルカリ性系)では、シクロピアゾン酸は小さい濃厚なスポットに集約される。
【0110】
プレートの直接外観検査によれば、≧2.5 ppmのCPA濃度は紫色の帯状スミアまたはスポットとして(展開系による)観察することができる。卓上フラットベッド・スキャナ−上でTLCプレートをスキャンし、次いで適当な画像処理プログラム(本件ではPaint Shop Pro 4)を使って電子画像を加工および強調処理することにより、感度が5〜10倍高められる。
この分析の全感度は(抽出なしで)約0.5〜1ppm CPAであり;抽出を使うと感度が少なくとも10倍高まる。
【0111】
C.キャピラリー電気泳動によるコウジ酸についてのアッセイ方法
メタノールと10 mMホウ酸/NaOH, 4 M KCl, pH 9.1でコンディショニングしたSupelclean LC-18 SPEチューブ(カタログNo.5-7012のSupelco社製プレパック3 mlカラム)上での固相抽出により、CE分析用に試料の1〜3mlアリコートを調製する。溶液をカラムに強制的に流すために吸引マニホールドを使用する。3 mlの10 mMホウ酸/NaOH, 4 M KCl pH 9.1と0.3 mlの10 mM ホウ酸/NaOH pH 9.1で洗浄した後、7.5 mlの10 mMホウ酸/NaOH pH 9.1を用いて試料を溶離させる。更に後処理せずに、シクロピアゾン酸について上述した手順に準じて溶出液をCE分析にかける。場合により形成した沈澱を遠心により除去する。この方法の感度の下限は約6ppmである。
【0112】
D.薄層クロマトグラフィーによるコウジ酸についてのアッセイ方法
試料のアリコートをCPAについて上述したのと同様にTLCプレートの対角の端に適用し、CPAと同じ溶媒系を使って展開する。乾燥したプレートに0.1 M HCl中の1%FeCl3を噴霧する。試料中のコウジ酸の存在は赤色スポットにより示され、対照として適用した純粋なコウジ酸によって生じた赤色スポットの強度と比較される。検出の下限は50 ppmである。
【0113】
E.キャピラリー電気泳動による3−NPAについてのアッセイ方法
キャピラリー電気泳動(CE)分析への準備としての試料精製の必要性は試料の伝導度に依存する。伝導度が10 mS未満である場合、溶出緩衝液として0.1 M KClを使ったVarian SAXアニオン交換体(Varian Instrumetns, Palo Alto CA)上でのイオン交換により試料を精製する。伝導度が100 mSより大きい場合、2−ブタノールを使って試料を抽出する。その場合、酸性化/高塩処理による沈澱形成と、10 mM Tris/HCl pH 7.0中への沈澱の再溶解の後、2mlの試料を6 mlのブタノールで抽出する。
【0114】
30℃の温度でおよび25 mMホウ酸/リン酸 pH 7.6のコンディショニング緩衝液を使う直径50μmで有効長さ56 cmの未コーティングシリカのキャピラリーを使用するHP−CE装置ダイオードアレイ検出を用いる。試料を20秒間かけて静水圧下で注入する。電圧を30 kVに設定する。この方法の検出の下限は6ppmである。
【0115】
F.薄層クロマトグラフィーによる3−NPAについてのアッセイ方法
CPAについて記載したのと同様にTLCプレートに発酵液のスポットを適用しそして展開する。次いでW. Majak & R.J. Bose, "Chromatographic methods for the isolation of miserotoxin and the detection of aliphatic nitro compounds", Phytochemistry (1974) 13:1005-1010により記載された通りに、それらにジアゾ化p−ニトロアニリンを噴霧する。対照物質に対比したスポットの強度と位置が3−NPA濃度の尺度である。TLCプレート上での検出レベルは25〜50 ppmである。
【0116】
あるいは、100μl試料に50μlの1 M NaOHと70μlのジアゾ化p−ニトロアニリンを添加することにより3−NPAを分光光度的に(λ=540 nm)分析する。検出レベルは5〜10 ppmである。
【0117】
実施例1
A.A.オリゼ BZ 14由来のCPA陰性株の作製
アスペルギルス・オリゼ BZ14株の凍結乾燥胞子に1000 Gy〜1250 Gyの最適線量でγ線を照射し、次いでその胞子を25〜50コロニー/9 cmプレートの密度においてスクリーニング培地1上に接種した。シクロピアゾン酸を生産するコロニーは、赤色の不溶性CPA−Fe錯体を形成するためスクリーニング培地1上に赤色の裏面(コロニーの下側)を生成する。
【0118】
照射した胞子から約50,000個のコロニーをスクリーニングし、そして乳白色(クリーム/白)外観により特徴づけられる154のCPA欠失コロニーを分離した。再分離後、64の株がスクリーニング培地1のプレート上で非赤色裏面を保持した。TLCプラグアッセイにより、52個の株でCPAが検出されなかった。次いでそれらの株を、毒素誘発(34℃、250 rpmで5日間)振盪フラスコ発酵条件下で、MDU-1B培地中で培養した。36個の株がTLCにより測定した時に上清中に検出可能レベルのCPAを示さなかった。
【0119】
B.A.オリゼ JaL228由来のCPA陰性株BECh 1の作製
JaL228の凍結乾燥胞子を上記と同様にγ線照射しそしてスクリーニングした。推定上のCPA欠失分離株を毒素誘発条件(34℃、250 rpmで5日間)下でMDU-1B培地上で振盪フラスコ培養し、そして上清をTLC法によりCPAについて分析した。上清はそのままでまたは抽出液として試験した。
【0120】
抽出用には、全試料のうちの50 mlを10 mlの0.1 M HClで酸性化した。次いでこの混合物を70 mlメタノール/クロロホルム(1:2)と共に3〜5分間激しく振盪した。相分離後(約3時間後)、下層(約25 ml)を300 mlビーカーに移し、クロロホルムを蒸発させた。残渣を5mlのクロロホルム中に再溶解し、25 mlビーカーに移し、そしてクロロホルムを蒸発させた。残渣を100μlのクロロホルム中に溶かした。
【0121】
上清またはクロロホルム抽出液10μlを20 cm×20 cmのTLCプレートの対角の縁に適用し、そしてアッセイに関する前掲の項目に記載した通りに処理した。
BECh 1を含む3つの株(分離株)がCPAを生産しなかった。
【0122】
C.CPA陰性KA陰性の株BECh 2およびBECh 3の作製
BECh 1をCove N斜面上で培養した。胞子を0.01%Tween 中に3-5×106の密度に懸濁し、短波UV照射(殺菌灯からの254 nm)にかけた。1〜5%生存率を生じるUV線量で照射した胞子をその後のスクリーニングに使用した。
【0123】
照射済胞子をKM2培地中に約0.7胞子/100μlの密度に希釈し、その100μlを96ウエルマイクロタイタープレートの各ウエルに接種した。培養物を湿潤チャンバーの中に入れ、静止状態で34℃にて5〜7日間インキュベートした。次いで、増殖徴候を有する各ウエルに40μlの1%FeCl3/0.1 M HClを添加した。
濃赤色の発色はコウジ酸(KA)生産を示し;発色がないのはコウジ酸生産を欠くコロニーを示す。
【0124】
あるいは、凝固させたKM2培地(Triton X-100により増殖制限)上に胞子をまき、成熟コロニーが観察されたら、プレートを0.1 M HCl中の1%FeCl3で潅水させた。コロニーの周囲に赤色域がないものは、推定上の非コウジ酸生産株を示す。
【0125】
約7000 のマイクロタイター培養物の中から、132個の推定KA陰性コロニー、すなわちFeCl3と発色反応を示さないものが単離された。しかしながら、一次KM2スクリーニング寒天プレート上で試験すると、該コロニーのうち1つもKA陰性であると確認されなかった。
【0126】
固形培地上と静止液体KM2培地中(30°)の両培地を使用したKA誘発条件下での陰性コロニーの再試験は、KA陰性変異体の数を11にまで絞り込んだ。それらの株を振盪フラスコ中で試験すると、8つの株でKAが生産された。残りの3つは、TLCにより調べたときにも直接上清をアッセイしたときにもどちらも発色反応を与えなかった。同様な並行培養において増殖させると、予想通り、対照株BECh 1によりKAは生産された。分離株のうちの1つは異常な形態を示した。MDU-1B上での長期増殖後、残りの2つの分離株をCPAとKAの両方について試験した。CPAもKAもどちらも検出されなかった。この2つの株をBECh 2およびBECh 3と命名した。
【0127】
D.既にCPA陰性で且つKA陰性である3−NPA陰性A.オリゼ株の作製
BECh2株とBECh3株それぞれを上述したようなUV変異誘発にかけた。照射した胞子を96ウエルマイクロタイタープレート中で0.7生存胞子/100μlナカムラ培地の密度に希釈した。湿潤チャンバー中で30℃にて5〜7日間インキュベートした。
【0128】
増殖を示すウエルからの発酵ブロス試料を新たなマイクロウエルプレートに移して分光光度分析するかまたはTLCプレートに適用した。3−NPA陰性である株をナカムラ培地の入った振盪フラスコ中で再培養し、そして3−NPAについて分析した。3−NPA陰性である株を実施例2に記載の通りにpCaHj 493により形質転換せしめ、そして形質転換体を実施例3に記載の通りに処理した。
【0129】
実施例2
A.オリゼ株 JAL228およびBECh 1中でのリパーセ遺伝子の発現
A.プラスミド pCaHj493の作製
リパーゼプラスミド pAHL (WO 97/07202)をBamHIとSalIで消化し、得られたリパーゼをコードする916 bp断片を単離した。
WO 98/00529に記載のpCaHj 483をBamHIとXhoIで消化し、6757 bpベクター断片を上記リパーゼ断片と連結せしめた。その連結混合物を用いてE.コリ DH5α細胞を形質転換せしめ、そして所望のプラスミドを含有する形質転換体を単離した。得られたプラスミドをpCaHj 493と命名した。
【0130】
B.JaL228およびBECh-1中へのpCaHj 493の形質転換
ヨーロッパ特許出願第0531372号に記載のようなアセトアミド上での選択を使って、pCaHj493によりアスペルギルス・オリゼ株 JaL228およびBECh1を形質転換せしめた。形質転換体を2回胞子再分離した。各形質転換体の2回目の再分離からの胞子を、振盪フラスコ中とマイクロタイター皿培養においてリパーゼ生産について試験した。
【0131】
実施例3
A.CPA陰性A.オリゼ株とCPA陽性A.オリゼ株におけるリパーゼ生産
実施例2に記載のように調製した18個のJaL228形質転換体と30個のBECh 1形質転換体を振盪フラスコ培養においてリパーゼ生産について調べた。
【0132】
形質転換体のCove N斜面培養物を、10 mlの0.1%Tween溶液を使って収得し、胞子懸濁液を500 mlのツーバッフル(two-baffled)振盪フラスコに入れた100 mlのG1-Gly培地への接種材料として使用した。培養物を回転式振盪器上で34℃にて250 rpmで24時間インキュベートした。次いでG1-Gly培養物の10 mlを500 mlの振盪フラスコに入った100 mlの1/5 MDU-2BPに移し入れ、更に34℃にて250 rpmでインキュベートした。
【0133】
50時間後に試料を採取し、ミラクロスを通して濾過し、遠心分離した(4000×g)。単純放射状免疫拡散法〔Scand, J. Immunol. Vol.17, suppl. 10, 41-56,(1983), "Handbook of Immunoprecipitation-in-Gel Techniques", N.H. Axelsen編、Blackwell Scientific Publications, 1983〕を使って、上清中のリパーゼ濃度(LU/mlで表す)を検出した。
【0134】
30個のCPA陰性BECh 1形質転換体は、18個のCPA陽性JaL228形質転換体と同じかそれより高いリパーゼ収量を有した。表2はその分布の概観を与える。
【0135】
B.CPA陰性A.オリゼ株とCPA陽性A.オリゼ株によるキシラナーゼ生産
検出可能なCPA生産を示さない10個の株(実施例1Aで調製したもの)とCPAが検出可能である3個の株をキシラナーゼ生産について評価した。その結果を下記の表1に要約する。2列目の欄は、単純放射状免疫拡散法〔Scand, J. Immunol. Vol.17, suppl. 10, 41-56 (1983), "Handbook of Immunoprecipitation-in-Gel Techniques", N.H. Axelsen編、Blackwell Scientific Publications, 1983〕によりアッセイした時の真菌キシラナーゼ単位(FXU)で測定した振盪フラスコ培養で生産されたキシラナーゼの量を示す。
【0136】
この結果は、CPA陰性株がCPA陽性株に匹敵する量でキシラナーゼを生産できることを示す。
【0137】
【表1】

【0138】
C.CPA陰性KA陰性の株におけるリパーゼ生産
2つのCPA陰性KA陰性株、BECh 2とBECh 3を実施例2に記載の通りにプラスミド pCaHj 493により形質転換せしめた。得られた形質転換体を2回胞子分離した。各形質転換体の2回目の再分離からの胞子を実施例2に記載の通りにリパーゼ生産について試験した。
【0139】
表2は、それらの2つの株からの形質転換体のリパーゼ収量の度数分布を示す。その結果は、A.オリゼ株 BECh 1およびJaL228について与えられた値に比較して、得られる発現能力に何ら損傷がないことを示す。
【0140】
リパーゼ生産培養に使用したのと同じ胞子懸濁液から、CPA生産用のMDU1B振盪フラスコに接種を行い、そして上述したのと同様に5日間インキュベートした。項目5B2に従ってDPA分析を行った。BECh 1株はいずれもCPAを生産しなかったが、一方で18個のJaL228株のうちの17個は25 ppm超のCPAを生産し、大部分は100 ppm超のCPAを生産した。
【0141】
【表2】
【0142】
実施例4
A.オリゼDCAT-S遺伝子の同定およびゲノムクローニング
A.A.オリゼのジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼ(DCAT-S)遺伝子の同定
cDNAクローン(pJaL499)は配列番号1に示すDNA配列を含有する。それはクラビセプス・プルプレア(Claviceps purpurea)のジメチルアリルトリプトファンシンターゼ(DMAT-S)遺伝子に対する相同性により、CPA生合成に関連することが同定されている。A.オリゼcDNAクローンの配列決定は、それが長さ1393塩基対(配列番号1)であり、クラビセプス・プルプレア由来のDMAT-Sと42.1%同一である473アミノ酸ポリペプチド(配列番号2)をコードすることを示した。
【0143】
A.オリゼDCAT-Sポリペプチドは、シクロアセトアセチル−L−トリプトファンからのβ−CPA生合成に関与している(Nethling D.C. & McGrath, Can. J. Microbiol. (1977) 23:856-872)。
JaL228株とBECh 1株から染色体DNAを調製した。DNAをBglII, NcoI, XhoIおよびSpeIで消化し、そしてプローブとしてDCAT-S遺伝子を含むpJaL499由来の1kb 32P標識BglII DNA断片を使ってサザンブロッティングにより分析した。サザンブロット分析は、CPA生産株JaL228が1つのDCAT-S遺伝子を有し、一方でBECh 1ではそのDCAT-S遺伝子が染色体から欠失していることを示した。
【0144】
B.DCAT-S遺伝子のゲノムクローンのクローニング
次の制限酵素:EcoRI, SalI, BbuI, XhoIおよびXbaIを使って、プローブとしてDCAT-S遺伝子を含むpJaL499由来の1kb 32P標識BglII DNA断片を使って、A.オリゼ DCAT-S遺伝子のゲノム制限地図作成を行った(図1)。これは、DCAT-S遺伝子の唯一のコピーが存在することを示す。
【0145】
JaL228のゲノムDNAをTsp509Iで部分消化するかまたは0.7%アガロースゲル上で泳動した。7 kbと10 kbの間のサイズを有する断片を精製した。
【0146】
次いで精製済DNAを製造業者(StratageneR)により提供されたプロトコルを使ってLambda ZAP II中にクローニングした。製造業者により与えられる教示に従って、λベクター内に含まれる任意のクローン挿入断片を生体内切除し、再環化せしめて、DNAライブラリー用にクローン化挿入断片を含むファジミドを作製した。標準方法の参考書〔例えばJ. Sambrook, E.F. Fritsch & T. Maniatis編(1989) "Molecular Cloning: A Laboratory Manual", 第2版,Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York〕に概説されるように、プローブとしてDCAT-S遺伝子を含むpJaL499からの1kb 32P標識DNA BglII断片を使ったコロニーハイブリダイゼーションにより、DCAT-S遺伝子をコードするクローンについてスクリーニングを行った。
【0147】
実施例5
アスペルギルス・オリゼのジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼ(DCAT-S)の遺伝子破壊によるアスペルギルス・オリゼCPA陰性株の作製
選択マーカーとしてA.オリゼ pyrG遺伝子を使って、A.オリゼのpyrG陰性株中のDCAT-S遺伝子を一段階遺伝子置換法〔B.L. Miller他,Mol. Cell. Biol. (1985) 5:1714-1721;G. May, Applied Molecular Genetics of Filamentous Fungi, 1-25頁, J.R. Kinghorn & G. Turner編, Blakie Academic and Professional, 1992〕により破壊した。
【0148】
A.DCAT-S破壊プラスミドの作製
プラスミドpJaL499をSacIIで消化し、クレノウポリメラーゼで処理して平滑末端を作り、そして製造業者(Boehringer Mannheim)の教示に従って細菌アルカリ性ホスファターゼで処理して5′リン酸基を除去し、次いでフェノール抽出しそして沈澱させた。
【0149】
WO 98/12300に記載のプラスミド pJaL335をHindIIIで消化してA.オリゼpyrG遺伝子を含む3.5 kb断片を得、それをクレノウポリメラーゼで処理して平滑末端にし、ゲル電気泳動により単離し、そして精製した。次いで2つの断片を一緒に混合し、連結せしめた。E.コリ中への形質転換後、正しいプラスミドを担持しているコロニーをミニプラスミド調製物の制限酵素消化により同定した。破壊プラスミド(pDCAT-S-pyrG)の作製を図2に要約する。
【0150】
B.pyrG陰性A.オリゼ株 JaL250の単離
A.オリゼ株 JaL228を5−フルオロオロト酸に対する耐性についてスクリーニングし、自然pyrG変異体を同定した。JaL250と命名した1つの株がpyrG陰性であると同定された。この変異体はウリジン依存性であるので、それを野生型pyrG遺伝子を用いて形質転換し、そしてウリジンの非存在下での増殖能力により形質転換体を選択することができる。
【0151】
C.アスペルギルス・オリゼ DCAT-S欠失株の作製
プラスミド pDCAT-S-pyrGの4.9 kb NotI-EcoRI断片をゲル精製し、Christensen他、Biotechnology (1988) 6:1419-1422により記載された通りA.オリゼ株 JaL250を形質転換せしめるのに使った。次いでウリジン非存在下でのそれの増殖能力により形質転換体を選択した。2回再分離した後、実施例1に記載の通りにCPAを生産する能力について形質転換体をスクリーニングした。
【0152】
DCAT-S遺伝子が破壊されていることを確かめるために、CPAを生産しない形質転換体から染色体DNAを調製した。該DNAをEcoRIで消化し、DCAT-S遺伝子を含むpJaL499からの1kb 32P標識DNA BglII断片をプローブとして使ってサザンブロッティングにより分析した。DCAT-S遺伝子の破壊を有する形質転換体は、6.3 kb上の野生型EcoRIバンドが9.8 kb上のEcoRIバンドの方へ移動することにより識別される。
【0153】
実施例6
BECh1とBECh2がアフラトキシン生合成経路クラスターからの2つの遺伝子を欠くことの確証
A.オリゼ IFO 4177およびそれの多数の誘導体においてアフラトキシン生合成経路クラスター由来のaflRおよびomtAアフラトキシン遺伝子の存在を探求した。プライマー5956(5'-GGATCCAGGGCTCCCTGGAG-3')(配列番号3)と5955(5'-CCTGACCAGCCAGATCTCCT-3')(配列番号4)を使ったPCRにより、A.オリゼ IFO4177のゲノムDNAからaflR相同体を単離した。0.9 kb PCR断片を獲得し、それをInvitrogenから入手したベクター pCR2中にクローニングした。M13正(-40)プライマーと逆プライマーを使って、得られたプラスミド pToC280を配列分析することにより、クローン化断片の素性を確認した。また、プライマー 6120(5'-AGTGAGAGAACTCCCTCCTC-3')(配列番号5)と6121(5'-CCATATCTTCTCAGTCTCCA-3')(配列番号6)を使ったPCRにより、ゲノム IFO4177 DNAからomtA相同体を単離した。1.2 kb断片を得、それをInvirtogenからのベクターpCR2中にクローニングし、そして得られたプラスミド pToC276をM13正(-40)および逆プライマーを使って配列分析することにより、クローン化断片の素性を確認した。
【0154】
上記aflRおよびomtAのクローン化断片をハイブリダイゼーション実験における32P標識プローブとして使用した。IFO4177, JaL228, BECh1およびBECh2からのゲノムDNAを制限酵素EcoRIで消化し、生じた断片を0.7%アガロースゲル上で分離した。DNAを膜上にブロッティングし、上記2つの32P標識プローブを一つずつ用いて緊縮条件下でハイブリダイズさせた〔方法は. Sambrook, E.F. Fritsch, T. Maniatis編(1989) "Molecular Cloning: A Laboratory Manual", 第2版, Cold Spring Harbor, New Yorkに記載されている〕。ブロットは、IFO4177とJaL228からの両プローブを用いたとき陽性のハイブリダイゼーションシグナルを示し、一方でBECh1とBECh2 DNAを含むレーンでは全くバンドが検出されなかった。IFO4177とJaL228レーンでは、omtAプローブを使って1つの約3.8 kb断片が観察でき、aflRプローブを使った場合は約0.5 kbと4.3 kbの2つのバンドが観察できた。
【0155】
従って、A.オリゼ IFO4177は、アフラトキシン生合成経路からの少なくとも2つの遺伝子、すなわちaflR遺伝子とomtA遺伝子を含有する。A.フラーブスおよびA.パラシチクスでは、この2つの遺伝子は約32 kbだけ離れている〔Woloshuk, C.P. & Prieto, R., FEMS Microbiology Letter (1998) 160:169-176〕。IFO4177誘導体であるBECh1とBECh2には、それらの遺伝子のいずれも存在しない。
【0156】
本発明は、本明細書中に開示される特定の態様によりその範囲が限定されるものではなく、それらの態様は本発明の幾つかの面の例示として与えられるものである。任意の同等な態様も本発明の範囲内に含めることができる。実際、上記に例示および記載されたものに加えて、上記説明より本発明の様々な変更が当業者には明らかであろう。そのような変更は特許請求の範囲内に含まれるものである。
【0157】
様々な参考文献を本明細書中に引用したが、その開示の全内容が参考として組み込まれる。
【技術分野】
【0001】
本発明は、毒素欠失アスペルギルス変異体細胞における着目のポリペプチドの生産方法に関する。本発明はまた、アスペルギルス細胞の変異体および前記変異体細胞の獲得方法にも関する。
【背景技術】
【0002】
発明の背景
最近、非相同(異種)ポリペプチドの発現に組換え宿主細胞を利用することにより、他の方法では少量しか得られないかまたは天然源からの精製によってしか得られない商業的に価値あるポリペプチド、例えば工業的に重要な酵素や二次代謝産物、の大量生産が大幅に簡素化した。現在、ある特定のポリペプチドの生産のために選択される発現系には、真正細菌および真核宿主をはじめとする多様な選択肢がある。適当な発現系の選択は、しばしば所望の組成とコンホメーションを有するポリペプチドを生産する宿主細胞の能力に依存するだけでなく、大抵は、タンパク質の意図する最終用途によっても左右される。
【0003】
或る宿主系の使用に伴って遭遇する1つの問題はマイコトキシンの生産である。着目のポリペプチドの生産の際に宿主細胞として使われる多数の真菌は、様々な毒素の生合成に関与する酵素をコードする遺伝子を所有している。例えば、シクロピアゾン酸、コウジ酸、3−ニトロプロピオン酸およびアフラトキシンは、例えばアスペルギルス・フラーブス(Aspergillus flavus)中で生産される既知の毒素である。同様に、トリコテセン類も多数の真菌、例えばフザリウム種、例えばフザリウム・ベネナタム(Fusarium venenatum)およびトリコデルマ種において生産される。様々な真菌中での毒素の形成の詳細な概説はHandbook of Toxic Fungal Metabolites, Richard J. Cole & Richard H. Cox, Academic Press, 1981に見つけることができる。
【0004】
着目のポリペプチドの発酵の最中に起こるそのような毒素の形成は、それらが作業者と使用者の両方の健康および環境に対して有害となり得るので、非常に望ましくない。
【0005】
従って、関係する生産に使用する条件下で、健康に影響を及ぼすと考えられる量でそのような毒素が形成されないよう保証する多大な努力が費やされている。これは、主として、毒素を直接分析するかまたはバイオアッセイおよび/または食餌実験により分析することによる大規模な分析プログラムによって行われる。多くの場合、そのような大規模なプログラムは1回生産ごとに行われ、生産原価と製品を販売できるまでの時間の両方に影響を及ぼす。
【0006】
シクロピアゾン酸(以後“CPA”とも称する)は弱酸であり(pKa:3.5)酸性条件下で沈澱する。それは金属キレートを形成し、金属キレートは希酸によって分離される。CPAは特に多数の器官において変性病変と壊死を引き起こすので猛毒であり、そしてCa2+−ATPアーゼを選択的に阻害する。CPAはα形とβ形で生産され、β形はα形の前駆体である。CPAはアスペルギルス属により生産されるが、他の真菌、例えばペニシリウム属によっても生産される。
【0007】
コウジ酸(以後“KA”とも称する)は多数のアスペルギルス属により生産されるが他の真菌、例えばペニシリウム属および更に幾つかの細菌によっても生産される。それは弱アルカリ性(pKa:7.9;フェノール基)であり、多くの金属イオンと錯体を形成する。それは抗菌活性を有し、動物に対する毒性は弱い。それは殺虫剤や色素などといった多数の合成化合物の前駆体である。
【0008】
3−ニトロプロピオン酸(以後“3−NPA”とも称する)は天然ニトロ化合物である。それはある種の真菌、特にアスペルギルス属〔A.フラーブス(A. flavus)、A.ウエンティ(A. wentii)〕およびペニシリウム属〔P.アトロベンタム(P. atroventum)〕により生産される。数種の細菌における生産も報告されている。この酸およびそのエステルは幾つかの植物においても見つかっている。それは、例えば貧血を引き起こすのでそれ自体の方がむしろ毒性である。また、それは胃腸管において別の毒性化合物である亜硝酸塩に部分的に変換され得る。3−ニトロプロピオン酸は、コハク酸デヒドロゲナーゼを不可逆的に阻害しそしてイソクエン酸リアーゼ、フマラーゼおよびアスパルターゼを可逆的に阻害することにより、クレブス回路に影響を及ぼす。
【0009】
アフラトキシンはきわめて生物学的に活性であり、真菌アスペルギルス・フラーブス(Aspergillus flavus)Link ex. Friesおよびアスペルギルス・パラシチクス(Aspergillus parasiticus)Speareにより生産される二次代謝産物である;R.W. Detroy他、"Aflatoxin and related compounds", Microbial Toxins, 第6巻(A. Ciegler, S. KadisおよびS.J. Ajl編), Academic, New York, 1971, 3-178頁を参照のこと。主要なアフラトキシンはB1,B2,G1およびG2である。この代謝産物、特にアフラトキシンB1は動物とヒトに対して毒性であるだけでなく、全ての既知天然化合物のうち最も発ガン性でもある。
【0010】
マルフォルミンおよびオクラトキシンはA.ニガー(A. niger)により生産される。
【0011】
毒素を生産する宿主生物の能力を除去または低下させることにより、正式認可手続がずっと簡単になり、且つ分析プログラムを減少できるので生産段階における時間と費用を削減できるだろう。
【0012】
現在、着目のポリペプチドを効率的且つ経済的な方法で生産するのに適当である毒素欠失アスペルギルス変異体細胞(すなわち、好ましくはGRASとして分類される安全生物)が必要とされている。本発明は、毒素欠失アスペルギルス変異体宿主細胞を使って着目のポリペプチドの生産を提供することにより、およびそのような変異体宿主細胞の作製方法を提供することにより、この要望を満たす。発現されない、すなわちサイレント遺伝子である、1または複数の毒素遺伝子を有するアスペルギルス変異体細胞を提供することも必要とされている。
【発明の概要】
【0013】
本発明の一面によれば、アスペルギルス変異体宿主細胞により着目のポリペプチドを生産する方法であって、(a) 親のアスペルギルス細胞の変異体を培養し、ここで(i) 前記変異体は前記着目のポリペプチドをコードする第一の核酸配列と、少なくとも1つの毒素の生合成または分泌の原因となる少なくとも1つの遺伝子の変更を含んでなる第二の核酸配列とを含んでなり、そして(ii)前記変異体は同一条件下で培養した時に親のアスペルギルス細胞よりも少量の毒素を生産し;そして(b) 培地から前記ポリペプチドを単離することを含んでなる方法が提供される。
【0014】
本発明の好ましい態様では、変異体アスペルギルス細胞が同一条件下で培養した時に親細胞よりも少なくとも90%少なく毒素を生産する。好ましくは、変異体が同一条件下で培養した時に親のアスペルギルス細胞よりもシクロピアゾン酸、コウジ酸、3−ニトロプロピオン酸およびアフラトキシンのうちの1つまたは複数を少量生産する。
【0015】
本発明の別の態様によれば、着目の非相同ポリペプチドの生産に有用な毒素欠失アスペルギルス変異体宿主細胞であって、同一条件下で培養した時にアスペルギルス親細胞に比べて少なくとも1つの毒素を少量生産させるために遺伝子的に変更されている細胞が提供される。
【0016】
本発明のアスペルギルス細胞は、好ましくは、A.オリゼ(A. oryzae)、A.アキュレータス(A. aculeatus)、A.ニデュランス(A. nidulans)、A.フィキュウム(A. ficuum)、A.フラーブス(A. flavus)、A.フェチダス(A. foetidus)、A.ソヤ(A. soja)、A.サケ(A. sake)、A.ニガー(A. niger)、A.ジャポニカス(A. japonicus)、A.パラシチクス(A. parasiticus)およびA.フェニクス(A. phoeicus)からなる群より選ばれる。
【0017】
本発明の別の態様では、毒素欠失アスペルギルス変異体宿主細胞を獲得する方法であって、(a) アスペルギルス親宿主細胞中に、着目のポリペプチドをコードする第一の核酸配列と、少なくとも1つの毒素の生合成または分泌の原因である少なくとも1つの遺伝子の変更を含んでなる第二の核酸配列とを導入し;そして(b) 段階(a)から前記第一および第二の核酸配列を含んでなる変異体を同定することを含んでなる方法が提供される。
【0018】
本発明は、一面では、1または複数のサイレント毒性遺伝子が除去されている、非相同ポリペプチドの発現に適当なアスペルギルス変異体細胞に関する。
上記のおよび他の態様は下記の記載においてより詳細に説明されるだろう。
【図面の簡単な説明】
【0019】
【図1】図1は、プローブとしてDCAT-S遺伝子を含むpJaL499からの1 kb 32P標識DNA BglII断片を用いて、制限酵素EcoRI, SalI, BbuI, XhoIおよびXbaIを使って行った、アスペルギルス・オリゼ DCAT-S遺伝子のゲノム制限地図を示す。
【図2】図2は、破壊プラスミド p(DCAT-S-pyrG)の作製を示す。
【0020】
発明の具体的説明
定義
本願の目的上、本発明のより良い理解のために次の用語を定義する。
「ベクター」なる用語は、着目のポリペプチド(その前駆体形を含む)をコードする遺伝子またはDNA配列の挿入、増殖および発現を可能にするプラスミド、コスミド、ファージまたは他の任意媒介物を意味する。
【0021】
「宿主」なる用語は、着目のポリペプチド(その前駆体形を含む)の発現を可能にするであろう任意細胞を意味する。
【0022】
「形質転換」なる用語は、細胞による非相同DNA配列の発現を可能にする取り込みを意味する。
「変異体」宿主(または株)なる用語は、野生型株の変異体と形質転換体の両方を包含する、遺伝的に変更された株を意味する。
【0023】
「毒素」なる用語は、植物毒性、動物毒性および抗生物質活性を有する真菌の代謝産物を意味する。用語「マイコトキシン」は、暴露のレベルでヒトおよび動物に不利な健康障害を引き起こす可能性を有する、真菌により生産される二次代謝産物として一般に定義される。本明細書中では、「毒素」と「マイコトキシン」は互いに交換可能に用いることができる。
【0024】
本発明の変異体細胞について使われる「毒素欠失」なる用語は、その変異体がそれの親株に比較して不完全な毒素生産を有すること、すなわち、その変異体が少なくとも1つ、好ましくは複数の毒素を親細胞よりも少量生産することを意味する。
【0025】
本発明の方法の段階a)において使われる「同一条件」なる用語は、親細胞の毒素生産に対する変異体の毒素生産に関し、そして変異体と親株の発酵に例えばpH、温度、酸素などについて同様の条件が用いられることを示すために用いられる。着目のポリペプチドの生産を行う条件下で、または1もしくは複数の毒素の生産を行う条件下で、問題の毒素の生産に関して親細胞と変異体細胞を比較することができる。
【0026】
宿主細胞
本発明は、親細胞に比較して有意に減少したレベルの1または複数の毒素を発現させるために遺伝的に変更されている、着目のポリペプチドの発現に有用なアスペルギルス宿主細胞の変異体を提供する。宿主細胞は親細胞から誘導されるが、野生型細胞であってもよい。
【0027】
宿主株は、着目のポリペプチドの発現に従来使用されている任意のアスペルギルス宿主細胞であることができる。
【0028】
好ましい態様では、着目のポリペプチドの生産に有用であるアスペルギルス宿主細胞が、アスペルギルス亜群ユーロチウム(Eurotium)〔例えばA.レストリクツス(A. restrictus)種により代表される〕、ケトサルトリア(Chaetosartorya)〔例えばA.クレメウス(A. cremeus)種により代表される〕、スクレロクレイスタ(Sclerocleista)〔例えばA.オルナティ(A. ornati)種により代表される〕、サトイア(Satoia)〔例えばA.ニガー(A. niger)種により代表される〕、ネオサルトリア(Neosartorya)〔例えばA.フミガーツス(A. fumigatus)、A.セルビヌス(A. cervinus)種により代表される〕、ヘミカーペンテレス(Hemicarpenteles)〔例えばA.クラバツス(A. clavatus)種により代表される〕、ペトロミセス(Petromyces)〔例えばA.フラーブス(A. flavus)、A.カンジダス(A. candidus)、A.スパルサス(A. sparsus)種により代表される〕、エメリセラ(例えばA.ニデュランス(A. nidulans)、A.ベルシカラー(A. versicolor)、A.ウスツス(A. ustus)種により代表される〕、およびフェネリア(Fenellia)〔例えばA.テレウス(A. terreus)種により代表される〕からなる群より選ばれる。
【0029】
特に好ましい態様では、アスペルギルス宿主細胞が、A.オリゼ、A.アキュレータス、A.フィキュウム、A.フラーブス、A.フェチダス、A.ソヤ、A.サケ、A.ニガー、A.ニデュランスおよびA.ジャポニカスからなる群より選ばれる。それらのうち、アスペルギルス・オリゼ、アスペルギルス・ニガー、A.パラシチクスおよびA.フェニクスが最も好ましい。
本発明の宿主細胞の上記例は、現在受け入れられている生物分類学に従って命名される。
【0030】
毒素
本発明に従ってその生産を減少または除去しようとする毒素は、アスペルギルス属により生産されるか、またはアスペルギルス属が所有する遺伝子によりコードされるが必ずしも発現されなくてもよい、いずれの毒素であってもよい。例えば、アフラトキシン遺伝子はA.オリゼ中に存在するが、この種からは発現されないことが知られている。しかしながら、たとえそれらが発現されなくても、アフラトキシン経路遺伝子を除去することはまだ有利である場合がある。これは下記に説明され、実施例6に例示される。
【0031】
特に、減少または除去すべき毒素は、シクロピアゾン酸(CPA)、例えばそのα形またはβ形、コウジ酸(KA)、3−ニトロプロピオン酸(NPA)、エモジン、マルフォルミン(例えばマルホルミンAまたはB)、アフラトキシン、オクラトキシン、フラビオリンおよびセカロン酸(例えばセカロン酸D)からなる群より選ばれる。それらの毒素の説明については、本明細書中の発明の背景の項目とその中に言及したthe Handbook of Toxic Fungal Metabolitesを参照のこと。
【0032】
本発明のジメチルアリル−シクロアセトアセチル−L−トリプロファンシンターゼ(DCAT-S)
本発明は、(a) 配列番号2のアミノ酸配列を有するジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼ;(b) (a) の対立遺伝子変異体;および(c) ジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼ活性を有する(a)または(b)の断片からなる群より選ばれる、糸状菌から得られる単離されたジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼにも関する。
【0033】
好ましくは、本発明のジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼは配列番号2のアミノ酸配列またはそれの対立遺伝子変異体を含んでなる。より好ましい態様では、本発明のジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼは配列番号2のアミノ酸配列を含んでなる。別の好ましい態様では、本発明のジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼは配列番号2またはそれの断片のアミノ酸配列を有し、ここで前記断片はジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼ活性を有する。配列番号2の断片は、このアミノ酸配列のアミノ末端および/またはカルボキシ末端から1または複数のアミノ酸配列が削除されているポリペプチドである。最も好ましい態様では、ジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼが配列番号2のアミノ酸配列を有する。
【0034】
好ましくは、配列番号2の断片が少なくとも320アミノ酸残基、最も好ましくは少なくとも350アミノ酸残基を有する。
【0035】
対立遺伝子変異体は、同一染色体遺伝子座を占有する遺伝子の2以上の別形態のいずれかを意味する。対立遺伝子変異は突然変異を通じて自然に発生し、そして集団内に表現型多形を引き起こし得る。遺伝子変異はサイレント(コードされるポリペプチドに全く変化なし)であってもよく、変更されたアミノ酸配列を有するポリペプチドをコードしてもよい。ポリペプチドの対立遺伝子変異体という用語は、遺伝子の対立遺伝子変異体によりコードされるポリペプチドである。
【0036】
配列番号2のアミノ酸配列またはそれの部分配列を使って、オリゴヌクレオチドプローブ、すなわち本発明のジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼをコードする核酸配列をデザインすることができ、例えば、当業界で周知の方法に従って、配列番号1の核酸配列もしくはそれの部分配列を使って別の糸状菌株からジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼをコードするDNAを同定およびクローニングすることができる。特に、そのようなプローブは、対応する遺伝子を同定および単離するため、標準サザンブロッティング技術に従った着目の属または種のゲノムDNAまたはcDNAとのハイブリダイゼーションに使用することができる。そのようなプローブは、完全配列よりもかなり短鎖であることができるが、長さが少なくとも15、好ましくは少なくとも25、より好ましくは少なくとも40ヌクレオチドであるべきである。更に長いプローブを使用してもよい。DNAプローブとRNAプローブの両方を使用できる。プローブは典型的には対応する遺伝子を検出するために標識される(例えば32P,3H,35S,ビオチンまたはアビジンで)。
【0037】
ハイブリダイゼーションは、標準サザンブロッティング技術に従って、低〜高緊縮性条件下で(すなわち、5×SSPE,0.3%SDS,200μg/ml尖断変性サケ精子DNA、低、中および高緊縮性条件についてそれぞれ25%, 35%または50%のいずれかのホルムアミド中での42℃でのプレハイブリダイゼーションとハイブリダイゼーション)、配列番号1に示されるまたはpJaL499中に含まれる核酸配列のポリペプチドコード部分に相当するオリゴヌクレオチドプローブに核酸配列がハイブリダイズすることを示す。
【0038】
こうして、別の糸状菌株より調製したゲノムライブラリー、cDNAライブラリーまたはコンビナトリアル・ケミストリーライブラリーを、上記プローブとハイブリダイズし且つジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼをコードするDNAについてスクリーニングすることができる。別の糸状菌株からのゲノムDNAまたは他のDNAをアガロースもしくはポリアクリルアミドゲル電気泳動、または他の分離技術により分離することができる。ライブラリーからのDNAまたは分離したDNAは、ニトロセルロースまたは他の適当な担体材料上に移行せしめそして固定することができる。配列番号1と相同であるクローンまたはDNAを同定するために、サザンブロット法において担体材料を使用し、該担体材料を2×SSC,0.2%SDSを使って、好ましくは少なくとも50℃で、より好ましくは少なくとも55℃で、より好ましくは少なくとも60℃で、より好ましくは少なくとも65℃で、更により好ましくは少なくとも70℃で、最も好ましくは少なくとも75℃で、30分間ずつ3回最終洗浄する。それらの条件下でオリゴヌクレオチドプローブがハイブリダイズする分子を、X線フィルムを使って検出する。
【0039】
好ましい態様では、本発明のジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼはアスペルギルスの株、より好ましくはA.オリゼから得られ、例えばそのポリペプチドは配列番号2のアミノ酸配列を有する。
【0040】
本明細書中で定義する「単離された」ジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼは、本質的に他のポリペプチドを含まないポリペプチド、例えばSDS−PAGEで測定した時に少なくとも約20%純粋、好ましくは少なくとも約40%純粋、より好ましくは約60%純粋、更により好ましくは約80%純粋、最も好ましくは約90%純粋、更に最も好ましくは約95%純粋であるポリペプチドである。
【0041】
本発明は、糸状菌より得られるジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼをコードする単離された核酸配列にも関する。より好ましい態様では、該核酸配列がアスペルギルス種、例えばA.オリゼから得られ、特に配列番号1に記載の核酸配列を有する。別の好ましい態様では、該核酸配列がプラスミドpJaL499中に含まれる配列である。本発明は、遺伝暗号の縮重によって配列番号1とは異なる核酸配列も包含する。本発明は、ジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼ活性を有するポリペプチド断片をコードする、配列番号1の部分配列またはpJaL499のポリペプチドコード部分の部分配列にも関する。配列番号1の部分配列またはpJaL499のポリペプチドコード部分の部分配列は、5′および/または3′末端から1もしくは複数のヌクレオチドが欠失していること以外は、配列番号1またはpJaL499のポリペプチドコード部分により包含される核酸配列である。好ましくは、配列番号1の部分配列は少なくとも870ヌクレオチド、より好ましくは少なくとも960ヌクレオチド、最も好ましくは少なくとも1050ヌクレオチドを含む。
【0042】
該核酸配列はアスペルギルス・オリゼの分類学上の同等物である微生物より得ることができる。
【0043】
そのような核酸配列を単離またはクローニングするのに利用する技術については後述する。本明細書中で用いる「単離された核酸配列」という用語は他の核酸配列を本質的に含有しない核酸配列、例えば、アガロースゲル電気泳動により測定した時に、少なくとも約20%純粋、好ましくは少なくとも約40%純粋、より好ましくは少なくとも約60%純粋、更により好ましくは少なくとも約80%純粋、最も好ましくは少なくとも約90%純粋である核酸配列を言う。該核酸配列はゲノムDNA、cDNA、RNA、半合成、合成起源またはそれらの任意組合せのものであることができる。
【0044】
本発明のジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼをコードする核酸配列の変更が、該ポリペプチドと実質的に同じ酵素の合成に必要かもしれない。ジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼと「実質的に同じ」とは、該酵素の非天然形態を指して言う。それらのジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼは、何らかの操作方法によって、その生来の源より単離された酵素と異なっていてもよい。例えば部位特異的変異誘発法を用いて、比活性、熱安定性、最適pHなどの点が異なっている該酵素の変異体を合成することが重要となる場合がある。類似配列は、配列番号1のポリペプチドコード部分として与えられる核酸配列、例えばそれの部分配列に基づいて、および/または該核酸配列によりコードされるポリペプチドの別のアミノ酸配列を与えず且つ該ポリペプチドの生産用の宿主生物のコドン用法に対応するようなヌクレオチド置換の導入により、または異なるアミノ酸配列を与えるようなヌクレオチド置換の導入により、作製することができる。ヌクレオチド置換の一般記載については、例えば、Ford他, 1991, Protein Expression and Purification 2: 95-107を参照のこと。
【0045】
そのような置換は分子の機能にとって重大である領域の外側で行うことができ、更にまだ生物学的に活性なジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼをもたらすものであることは、当業者に明らかであろう。本発明の単離された核酸配列によりコードされるジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼの活性にとって必須であり、従って置換を受けないことが好ましいアミノ酸残基は、部位特異的変異誘発またはアラニンスキャニング変異誘発といった当業界で既知の手法に従って同定することができる(例えば、Cunningham & Wells, 1989, Science 244: 1081-1085参照)。後者の技術では、分子中の正電荷を有する残基ごとに突然変異を導入し、そして生じた変異体分子をジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼ活性について試験し、該分子の活性にとって重要であるアミノ酸残基を同定する。核磁気共鳴分析、結晶学または光親和性標識法のような技術により測定される三次元構造の解析によって、基質−酵素相互作用を調べることもできる(例えば、de Vos他, 1992, Science 255: 306-312; Smith他, 1992, Journal of Molecular Biology 224: 899-904; Wlodaver他, 1992, FEBS Letters 309: 59-64)。
【0046】
本発明の核酸配列またはそれの相同体もしくは断片の好ましい使用目的は、特定の宿主細胞、特にA.オリゼの細胞のジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼ活性を除去または減少させ、それにより前記細胞からCPAの生産を減少または排除することである。
【0047】
本発明は、配列番号1の核酸配列またはpJaL499のポリペプチドコード部分、それの部分配列もしくは相同体を含有する、該配列の発現のための構成物、組換え発現ベクターおよび宿主細胞にも関する。該構成物およびベクターは本明細書中に記載の通りに作製することができる。宿主細胞は核酸配列の発現に適当な任意の細胞であることができ、そして例えば本明細書中に記載の親細胞または変異体細胞より選択することができる。
【0048】
宿主細胞の遺伝的変更
1または複数の毒素の発現レベルを有意に減少させるために本発明の宿主細胞は遺伝的に変更されるが、それは当業者に既知の標準技術を使って達成することができる。毒素活性の生産の原因となる遺伝子配列を不活性化するかまたは部分的にもしくは完全に除去することができる。こうして、本発明のアスペルギルス変異体宿主細胞は、減少したレベルまたは検出不可能なレベルの1または複数の毒素を発現する。
【0049】
特定の態様では、不活性化は、特定の毒素の形成または分泌に関与する各々の構造領域もしくは調節領域(例えば遺伝子)の変更により得られる。既知の有用な技術としては、非限定的に、特異的もしくはランダム変異誘発、PCR生成変異誘発、部位特異的DNA欠失、挿入および/または置換、遺伝子破壊または遺伝子置換、アンチセンス技術、またはそれらの組合せ等が挙げられる。
【0050】
変異誘発は適当な物理的もしくは化学的変異誘発剤を使って行うことができる。本発明の目的上適当である物理的もしくは化学的変異誘発剤の例としては、非限定的に、UV照射、イオン化照射、例えばガンマ照射、ヒドロキシルアミン、N−メチル−N′−ニトロ−N−ニトロソグアニジン(MNNG)、O−メチルヒドロキシルアミン、亜硝酸、エチルメタンスルホネート(EMS)、亜硫酸水素ナトリウム、およびヌクレオチド類似体が挙げられる。そのような剤を用いる場合、典型的には、変異させようとする細胞を適当な条件下で特定の変異誘発剤の存在下でインキュベートし、そして有意に減少した標的毒素生産を示す細胞について選択することにより、変異誘発が行われる。
【0051】
宿主細胞による特定の毒素の生産の減少または除去は、該毒素の生産または分泌に関与するかまたは他の形で必要であるヌクレオチド配列を変更することによっても達成することができる。例えば、該ヌクレオチド配列が、毒素生産に至る経路の中の必要機能を有する遺伝子生成物であってもよい。ヌクレオチド配列は、例えば、配列番号1に示すものまたはpJaL499のポリペプチドコード部分であることができる。変更は、該ヌクレオチド配列中または該配列の転写もしくは翻訳に必要な調節要素中への1もしくは複数のヌクレオチドの導入、置換または除去によって達成することができる。例えば、該ヌクレオチド配列への終止コドンの導入、開始コドンの除去または転写解読枠の変更をもたらすようにヌクレオチドを挿入または除去してもよい。該配列またはその調節要素の変更または不活性化は、当業界で周知の方法に従った部位特異的もしくはランダム変異誘発またはPCR生成変異誘発により達成することができる。理論的には生体内で、すなわち毒素遺伝子を発現する細胞に直接、変更を行うことができるけれども、現在のところは下記に例示するように試験管内で変更を行うことが好ましい。
【0052】
特定の糸状菌細胞における着目の毒素、例えばCPAの生産を不活性化または減少させる便利な方法の一例は、遺伝子置換、遺伝子欠失または遺伝子破壊の技術に基づいている。例えば、遺伝子破壊法では、着目の遺伝子または遺伝子断片(例えば本発明のDCAT-S遺伝子)に相当する核酸配列を試験管内変異誘発せしめて欠陥核酸配列を生産させ、次いでその欠陥核酸配列を用いて親細胞を形質転換させて欠陥遺伝子を生成せしめる。相同組換えにより、欠陥遺伝子配列が内因性遺伝子または遺伝子断片と置き換わる。欠陥遺伝子または遺伝子断片が更にマーカーをコードすることが望ましいかもしれない。該マーカーは、核酸配列が変更または破壊されている形質転換体の選択に利用することができる。
【0053】
あるいは、遺伝子の変更または不活性化は、該遺伝子の核酸配列に相補的なヌクレオチド配列を使用する、確立されたアンチセンス技術によって行うこともできる。より詳しくは、細胞内で転写されることができ且つ細胞内で生産されたmRNAにハイブリダイズすることができる、該遺伝子の核酸配列に相補的であるヌクレオチド配列を導入することにより、糸状菌細胞による該遺伝子の発現を減少または除去することができる。こうして、相補的アンチセンスヌクレオチド配列をmRNAにハイブリダイズ可能にする条件下では、翻訳されるタンパク質の量が減少または除去される。
【0054】
毒素経路の遺伝子の変異誘発または別の変更の後、減少または除去された毒素生産について変異体がスクリーニングされる。毒素についてスクリーニングする方法の具体例は下記の実施例に与えられる。あるいは、有用なスクリーニングアッセイが"Handbook of Toxic Fungal Metabolites"中に記載されており、または例えば食品中のマイコトキシンのレベルを一般検査する機関で入手可能である。
【0055】
従って、遺伝的変更のため、本発明のアスペルギルス変異体宿主細胞は有意に減少したレベルの毒素を発現する。好ましい態様では、変異体宿主細胞により発現されるそれらの毒素のレベルは、個々に約50%以上、好ましくは約85%以上、より好ましくは約90%以上、最も好ましくは約95%以上、更により好ましくは99%以上減少されている。別の好ましい態様では、本発明の変異体宿主細胞中の複数毒素が任意の組合せで減少されてもよい。更に別の好ましい態様では、宿主細胞により発現される生成物がシクロピアゾン酸、コウジ酸、3−ニトロプロピオン酸、エモジン、マルフォルミン、アフラトキシン、オクラトキシンおよびセカロン酸のうちの少なくとも1つの毒素を本質的に含有しない。特に好ましい態様では、宿主細胞により発現される生成物が少なくともシクロピアゾン酸を本質的に含有せず、より好ましくは少なくともシクロピラゾン酸とコウジ酸またはアフラトキシンを本質的に含有せず、最も好ましくは少なくともシクロピアゾン酸、コウジ酸および3−ニトロプロピオン酸を含有しない。
【0056】
好ましい態様では、宿主細胞が、NPA,CPA,コウジ酸(KA)またはマルトリジンのうちの1つまたは複数;好ましくはそれらの毒素のうちの少なくとも2つ、例えばNPAとCPA;NPAとKA;CPAとKA;またはNPAとCPAとKA、の生産が減少または除去されているA.オリゼの株である。更に、それらの1または複数の毒素の除去または減少に加えて、好ましくは得られた変異体株がアフラトキシンを生産できないように、特定のアフラトキシン経路の遺伝子が不活性化される。A.フラーブス(A. flavus)からのアフラトキシン遺伝子は周知であるのでそれをA.オリゼ中の対応遺伝子の同定に利用することができ、次いでその対応遺伝子を当該技術分野で既知の方法により不活性化することができる。
【0057】
別の好ましい態様では、宿主細胞が、マルフォルミン(例えばマルフォルミンA1またはB)、オクラトキシン(例えばオクラトキシンA)およびフラビオリンのうちの1つまたは複数、好ましくはそれらの毒素のうちの少なくとも2つ、例えばマルフォルミンとオクラトキシン;マルフォルミンとフラビオリン;オクラトキシンとフラビオリン;およびマルフォルミンとオクラトキシンとフラビオリンの生産が減少または除去されているA.ニガー(A. niger)またはA.フィキュウム(A. ficuum)の株である。
【0058】
別の好ましい態様では、宿主細胞が、セカロン酸(例えばセカロン酸D)またはエモジン(セカロン酸Dの前駆体)、好ましくはそれらの毒素の両方の生産が減少または除去されているA.アキュレータス(A. aculeatus)の株である。
【0059】
ポリペプチドの生産方法
本発明の方法により、或る1または複数の標的毒素の量は有意に減少されるが、その一方で着目のポリペプチドをコードする遺伝的に変更された遺伝子の細胞内安定維持、細胞の生産能力、および着目のポリペプチドの収率の点からの変異体宿主細胞の特徴は実質的に保持される。より詳しくは、本発明の方法により、宿主細胞は1または複数の着目の毒素の生産または分泌に必要な構造領域および/または調節領域の内部で遺伝的に変更され、それにより前記毒素の生産または分泌が減少または除去される。
【0060】
従って、本発明の別の面は、本発明のアスペルギルス変異体宿主細胞におけるポリペプチドまたはタンパク質(非相同ポリペプチドまたはタンパク質を包含する)の生産方法であって、前記変異体宿主細胞中に着目のポリペプチドをコードする核酸配列を導入し、適当な増殖培地中で変異体宿主細胞を培養し、そして前記着目のポリペプチドを回収することを含んでなる方法を提供する。
【0061】
よって、本発明の変異体宿主細胞は、着目のポリペプチドの発現に必要な構造および調節遺伝子領域を含まなければならない。そのような構造および調節領域の性質は、大部分、目的とする生成物と特定のアスペルギルス宿主株により決まる。本発明の宿主細胞の遺伝子設計は、宿主細胞の形質転換またはトランスフェクションのための標準組換えDNA技術を使って当業者により成し遂げることができる(例えばSambrook他参照)。
【0062】
好ましくは、宿主細胞は、着目の所望のポリペプチドをコードするDNA断片を含んで成る適当なクローニングビヒクル、すなわちプラスミドまたはベクター、の導入のための周知技術により変更される。クローニングビヒクルは自己複製プラスミドとして宿主細胞中に導入されてもよく、または染色体中に組み込まれてもよい。好ましくは、クローニングビヒクルは1または複数の適当な調節領域に作用可能に連結された1または複数の構造領域を含んでなる。
【0063】
構造遺伝子は着目のポリペプチドをコードするヌクレオチド配列の領域である。調節領域は、転写および翻訳調節配列を含んでなるプロモーター領域、終止シグナルを含んでなるターミネーター領域、およびポリアデニル化領域を包含する。プロモーター、すなわち特定の宿主細胞において転写活性を示すヌクレオチド配列は、細胞外または細胞内タンパク質、好ましくは酵素、例えばアミラーゼ、グルコアミラーゼ、プロテアーゼ、リパーゼ、セルラーゼ、キシラナーゼ、オキシドレダクターゼ、ペクチナーゼ、クチナーゼまたは解糖系酵素をコードする遺伝子から誘導することができる。本発明の方法において核酸構成物の転写を指令するのに適当なプロモーターの例は、アスペルギルス・オリゼ(Aspergillus oryzae)TAKAアミラーゼ、リゾムーコル・ミーヘイ(Rhizomucor miehei)アスパラギン酸プロテイナーゼ、アスペルギルス・ニガー(Aspergillus niger)中性α−アミラーゼ、アスペルギルス・ニガー(Aspergillus niger)酸安定性α−アミラーゼ、アスペルギルス・ニガー(Aspergillus niger)もしくはアスペルギルス・アワモリ(Aspergillus awamori)グルコアミラーゼ(glaA)、リゾムーコル・ミーヘイ(Rhizomucor miehei)リパーゼ、アスペルギルス・オリゼ(Aspergillus oryzae)アルカリ性プロテアーゼ、アスペルギルス・オリゼ(Aspergillus oryzae)トリオースリン酸イソメラーゼ、アスペルギルス・ニデュランス(Aspergillus nidulans)アセトアミダーゼ(amdS)、フザリウム・オキシスポラム(Fusarium oxysporum)トリプシン様プロテアーゼ(米国特許第4,288,627号)をコードする遺伝子から得られるプロモーター、並びにそれらの変異体、短縮形およびハイブリッドプロモーターである。特に好ましいプロモーターは、NA2-tpiプロモーター(アスペルギルス・ニガー中性α−アミラーゼをコードする遺伝子由来のプロモーターとアスペルギルス・オリゼのトリオースリン酸イソメラーゼをコードする遺伝子由来のプロモーターとのハイブリッド)、グルコアミラーゼプロモーター、およびTAKAアミラーゼプロモーターである。
【0064】
クローニングビヒクルは選択マーカーを含んでもよい。選択マーカーは、その生成物が殺生剤耐性、ウイルス耐性、重金属耐性、原栄養株に対する栄養要求変異体、などに備える遺伝子である。糸状菌宿主細胞用の選択マーカーは、非限定的に、amdS(アセトアミダーゼ)、argB(オルニチンカルバモイルトランスフェラーゼ)、bar(ホスフィノスリシンアセチルトランスフェラーゼ)、hygB(ヒグロマイシンホスホトランスフェラーゼ)、niaD(硝酸レダクターゼ)、pyrG(オロチジン−5′−リン酸デカルボキシラーゼ)、sC(硫酸アデニルトランスフェラーゼ)およびtrpC(アントラニル酸シンターゼ)並びに別の種からの同等物からなる群より選ぶことができる。アスペルギルス細胞用に好ましいのは、アスペルギルス・ニデュランスまたはアスペルギルス・オリゼのamdSおよびpyrG遺伝子、並びにストレプトマイセス・ヒグロスコピクス(Streptomyces hygroscopics)のbar遺伝子である。
【0065】
更に、選択は同時形質転換によって行ってもよく、この場合は、2つのベクターの混合物を使って形質転換を行い、そして1つのベクターのみについて選択を行う。
【0066】
本発明のDNA構成物、プロモーター、ターミネーターおよび他の要素をそれぞれ連結せしめそしてそれらを複製に必要な情報を含む適当なクローニングビヒクル中に挿入するのに用いる手法は、当業者に周知である(例えばSambrook他, 1989, 前掲を参照のこと)。
【0067】
変異体糸状菌細胞は、当該技術分野で既知の方法を使って、着目のポリペプチドの生産に適した普通培地中で培養される。例えば、細胞は、適当な培地中でそして非相同ポリペプチドを発現および/または単離可能にする条件下で行われる、振盪フラスコ培養、実験室用もしくは工業用発酵槽中での小規模または大規模発酵(連続発酵、バッチ発酵、フェドバッチ発酵または固相発酵を含む)により培養することができる。培養は、当該技術分野で既知の手法を使って、炭素源、窒素源および無機塩類を含有する適当な普通培地中で行われる。適当な培地は民間供給業者から入手可能であり、または発表された組成〔例えばアメリカン・タイプ・カルチャー・コレクション(ATCC)のカタログ中〕に従って調製することができる。分泌されたポリペプチドは培地から直接回収することができる。
【0068】
ポリペプチドは該ポリペプチドに特有である当業界で既知の方法を使って検出することができる。それらの検出方法としては、特異抗体の使用、酵素生成物の形成、酵素基質の消失、またはSDS−PAGEが挙げられる。例えば、酵素アッセイを使ってポリペプチドの活性を測定してもよい。酵素活性の測定方法は様々な酵素について知られている。
【0069】
得られたポリペプチドは当該技術分野で既知の方法により単離することができる。例えば、非限定的に遠心分離、濾過、抽出、噴霧乾燥、蒸発または沈澱をはじめとする常用手順により、普通培地から単離することができる。次いで単離されたポリペプチドは、非限定的にクロマトグラフィー(例えばイオン交換、アフィニティー、疎水的、等電点およびサイズ排除)、電気泳動(例えば分取用等電点電気泳動)、分別溶解度(例えば硫酸アンモニウム沈澱)または抽出法をはじめとする既知の様々な方法により、更に精製することができる(例えばProtein Purification, J.-C. Janson & Lars Ryden編,VCH Publishers, New York, 1989を参照のこと)。
【0070】
生成物
所望の最終生成物、すなわちアスペルギルス変異体宿主細胞により発現される着目のポリペプチドは、いずれの相同または非相同タンパク質またはペプチドであってもよい。
【0071】
該ポリペプチドは、変異体糸状菌細胞にとって非相同である任意のポリペプチドであることができる。「ポリペプチド」なる用語は、コードされる生成物の特定の長さについて言及するものではなく、従ってペプチド、オリゴペプチドおよびタンパク質を包含するものである。非相同ポリペプチドは或るポリペプチドの改変変異体であってもよい。「非相同ポリペプチド」なる用語は、本明細書中では糸状菌細胞にとって生来でないポリペプチドとして定義される。変異体糸状菌細胞は非相同ポリペプチドをコードする核酸配列を1または複数コピー含んでもよい。
【0072】
本発明の方法では、変異体糸状菌細胞を該細胞にとって生来であるポリペプチドの組換え生産に使用してもよい。例えば、該ポリペプチドの発現を増強するため、シグナル配列の使用により細胞の外側への着目の生来のポリペプチドの輸送を促進するため、および細胞により通常に生産されるポリペプチドをコードする遺伝子のコピー数を増加させるために、別のプロモーターの調節下に該ポリペプチドをコードする遺伝子を配置することにより、生来のポリペプチドを組換え生産せしめることができる。本発明は、そのような発現が細胞にとって生来でない遺伝要素の使用を伴うかまたは宿主細胞で通常は起こらないような形で機能するように操作されている生来の要素の使用を伴う限り、「非相同ポリペプチド」の用語の範囲内に、そのような組換え生産も包含する。
【0073】
より具体的な態様では、生成物が療法上有効なペプチドまたはタンパク質、例えばホルモン、特にインスリン、成長ホルモン、グルカゴンまたはソマトスタチン;インターロイキン、特にインターフェロン;造血増殖因子、特にPDGE(血小板由来増殖因子)、EPO(エリスロポイエチン)またはTPO(トロンボポイエチン);プロテアーゼ、特に第VII因子、第VIII因子、ウロキナーゼ、キモシンまたはTPA;または血清アルブミンである。
【0074】
別の好ましい態様では、生成物が真菌または細菌起源の酵素である。酵素は好ましくはグリコシダーゼ酵素、例えば、アミラーゼ、特にα−アミラーゼ、β−アミラーゼまたはグルコアミラーゼ;グルカン1,4−α−グルコシダーゼ;アミノペプチダーゼ;カルボヒドラーゼ;カルボキシペプチダーゼ;カタラーゼ;セルラーゼ;特にエンド−1,4−β−グルカナーゼまたはエンド−1,3(4)−β−グルカナーゼ;セルロース−1,4−β−セロビオシダーゼ;キチナーゼ;クチナーゼ;シクロデキストリングリコシルトランスフェラーゼ;デオキシリボヌクレアーゼ;ガラクタナーゼ;ガラクトシダーゼ、特にα−ガラクトシダーゼまたはβ−ガラクトシダーゼ;エンドグルカナーゼ、特にエンド−1,3−β−グルカナーゼ、エンド−1,3−α−グルカナーゼ、エンド−1,2−β−グルカナーゼまたはエンド−1,6−β−グルカナーゼ;グルコシダーゼ;特にα−グルコシダーゼまたはβ−グルコシダーゼ;インベルターゼ;ラッカーゼ;脂質分解酵素、特にリパーゼ、エステラーゼ、ホスホリパーゼまたはリゾ−ホスホリパーゼ;リアーゼまたはペクテートリアーゼ;マンナーゼ;マンノシダーゼ;ポリガラクツロナーゼ;ムタナーゼ;オキシダーゼまたはオキシドレダクターゼ、例えばペルオキシダーゼまたはポリフェノールオキシダーゼ;オキシゲナーゼ;ペクチナーゼ、エンドペプチダーゼまたはエキソペプチダーゼ;フィターゼ;ポリガラクツロナーゼ;プロテアーゼ;リボヌクレアーゼ;トランスグルタミナーゼ;およびキシラナーゼ、特にエンド−1,4−β−キシラナーゼまたはキシラン−エンド−1,3−β−キシロシダーゼである。
【0075】
別の好ましい態様では、生成物がハイブリッドポリペプチド、例えばプロキモシンおよびプロトリプシン様プロテアーゼである。宿主細胞により発現される非相同ポリペプチドは、適当な条件下で、例えば実質的なプロテアーゼ活性の非存在下で、チモーゲンのような前駆体タンパク質、ハイブリッドタンパク質、プロ配列もしくはプレ−プロ配列として得られるタンパク質、または他の任意の未成熟形であってもよい。
【0076】
本発明を下記の実施例に関して更に説明するが、この実施例は特許請求の範囲において定義されるような本発明の範囲を限定すると解釈してはならない。
【0077】
サイレント毒素遺伝子が除去されているアスペルギルス変異体
アフラトキシンの生合成経路はアフラトキシン産生種であるアスペルギルス・フラーブスおよびアスペルギルス・パラシチクスにおいて研究されている。どちらの種も、多数の遺伝子が同定されそして大クラスターとしてマッピングされることが示されている〔Woloshuk, C.P. & Prieto, R., FEMS Microbiology Letter (1998) 160:169-176〕。別経路遺伝子の発現調節遺伝子をコードする遺伝子であるaflR、およびO−メチルトランスフェラーゼをコードするomtAといった幾つかの遺伝子はクローニングされ、そして配列決定されている。
【0078】
アフラトキシン遺伝子はA.オリゼのゲノム中に存在するが、この種からは発現されない。アフラトキシンが全く発現されないくても、それらのサイレントアフラトキシン経路遺伝子の1つまたは複数を除去することは有利である。アフラトキシン遺伝子、例えばaflRおよび/またはomtA遺伝子が除去されているアスペルギルス属変異体、例えばA.オリゼ変異体は、問題の1または複数のアフラトキシンの生産について新規変異体株を試験する必要がないので、有利である。
【0079】
よって、本発明は、一面では、1または複数のサイレント毒素遺伝子が除去されている、非相同ポリペプチドの発現に適したアスペルギルス変異体細胞に関する。
【0080】
サイレント毒素遺伝子が「除去」されているとは、前記サイレント遺伝子が変異体細胞中に含まれなくなるような形で、例えば当該技術分野で周知の遺伝子置換技術または遺伝子破壊技術(例えばMiller他, 1985, Molecular and Cellular Biology, p.1714-1721参照)により、問題の(1または複数の)遺伝子が変更または削除されていることを意味する。
【0081】
「サイレント」毒素遺伝子なる用語は、該毒素が発現されないことを意味する。
【0082】
毒素遺伝子は該毒素遺伝子の全部または一部の不可逆的欠失または破壊により除去されてもよい。
【0083】
「毒素遺伝子の全部または一部の不可逆的欠失または破壊」なる用語は、前記遺伝子が毒素をコードせず、且つ毒素をコードする遺伝子へと自然に復帰変異する(例えば生産中に)ことができないような形で、問題の毒素遺伝子が除去または変更されていることを意味する。
【0084】
問題のアスペルギルス細胞は上記のものいずれでもよく、そしてアスペルギルス亜群のユーロチウム(Eurotium)、ケトサルトリア(Chaetosartorya)、スクレロクレイスタ(Sclerocleista)、サトイア(Satoia)、ネオサルトリア(Neosartorya)、ヘミカーペンテレス(Hemicarpenteles)、ペトロミセス(Petromyces)、エメリセラ(Emericella)およびフェネリア(Fenellia)からなる群より選択されてもよい。
【0085】
1または複数の毒素をコードする問題の毒素遺伝子としては、シクロピアゾン酸、コウジ酸、3−ニトロプロピオン酸、エモジン、マルフォルミン、アフラトキシン、オクラトキシンおよびセカロン酸からなる群より選ばれる毒素が挙げられる。
【0086】
特に期待されるのは、アフラトキシンをコードする毒素遺伝子、特にA.オリゼのアフラトキシンクラスターからの毒素遺伝子、特にomtA, aflR, pksA, Nor-1, fas-beta, fas-alpha, vber-1, avnA, ord-2から選ばれた毒素遺伝子である。
【0087】
親のアスペルギルス細胞はA.オリゼ細胞、特にA.オリゼ A1560 (IFO 0417)であることができる。
【実施例】
【0088】
実験
材料と方法
1.菌株
アスペルギルス・オリゼ(Aspergillus oryzae)A1560はIFO 04177と同一である(下記参照)。
アスペルギルス・オリゼ IFO 4177:Institute for Fermentation, Osaka(財団法人発酵研究所;大阪府淀川区十三本町2丁目17−25)より入手可能。WO 98/12300参照。
JaL228:中性メタロプロテアーゼ NpI遺伝子が破壊されているアスペルギルス・オリゼ株;この株の作製はWO 98/12300に記載されている。
【0089】
BECh 1:このCPA陰性アスペルギルス・オリゼ株の作製は実施例1に記載される。
BECh 2:このCPA陰性KA陰性アスペルギルス・オリゼ株の作製は実施例1に記載される。
BECh 3:このCPA陰性KA陰性アスペルギルス・オリゼ株の作製は実施例1に記載される。
【0090】
BZ14:WO 92/17573に記載の通りToC90とphD450により同時形質されたアスペルギルス・オリゼの株。
JaL 250:このアスペルギルス・オリゼ株の作製は実施例6に記載される。
【0091】
寄託:
プラスミドpJaL499を含有するE.コリ株は、1999年1月13日にDSMZ(Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH;Mascheroder Weg 1b, D-38124 Brauschweig)に寄託され、そして寄託番号DSM 12622を付与された。
【0092】
2.遺伝子
DMAT-S:この遺伝子は、麦角アルカロイドの生合成に関与する酵素であるジメチルアリル−L−トリプトファンシンターゼをコードする。
DCAT-S:この遺伝子は、シクロピアゾン酸(CPA)生合成に関与する酵素であるジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼをコードする。
pyrG:この遺伝子は、ウリジン生合成に関与する酵素であるオロチジン−5′−リン酸デカルボキシラーゼをコードする。
【0093】
3.プラスミド
pAHL:このプラスミドはWO 97/07202に記載されている。
pCaHj483:このプラスミドはWO 98/00529に記載されている。
pCaHj493:このプラスミドは実施例2に記載される。
pJaL335:このプラスミドはWO 98/12300に記載されている。
pJaL499:このプラスミドは実施例4に記載される。
【0094】
4.培地と溶液
緩衝液および基質として使用する薬品は少なくとも試薬用品質のものであった。
スクリーニング培地1(1リットルあたり)
マンニトール 30 g
グルコース 10 g
コハク酸 10 g
カザミノ酸 3 g
KH2PO4 1 g
MgSO4・7H2O 0.3 g
FeSO4・7H2O 0.2 g
2,6−ジクロロ−4−アニリン 2 ppm
寒天 20 g
14% NH4OHで最終pH 5.6に調整。
【0095】
Cove N
Cove塩溶液 50 ml
ソルビトール 218 g
デキストロース 10 g
硝酸カリウム 2.02 g
寒天 35 g
脱イオン水 1000 ml
【0096】
Cove塩溶液(1リットルあたり)
KCl 26 g
MgSO4 26 g
KH2PO4 76 g
微量金属溶液 50 ml
CHCl3 2 ml
【0097】
微量金属溶液(1リットルあたり)
Na2B4O7・10H2O 40 mg
CuSO4・5H2O 400 mg
FeSO4・7H2O 800 mg
MnSO4・2H2O 800 mg
Na2MoO4・2H2O 800 mg
ZnSO4・7H2O 8000 mg
【0098】
G1-gly
酵母エキス 18 g
グリセロール87% 24 ml
プルロニック PE6100 1 ml
水道水で 1000 mlに
【0099】
1/5 MDU-2BP
マルトース 9 g
MgSO4・7H2O 0.2 g
NaCl 0.2 g
K2SO4 0.4 g
KH2PO4 2.4 g
酵母エキス 1.4 g
AMG微量金属 0.1 ml
プルロニック PE6100 0.02 ml
脱イオン水 1000 mlに
最終pH 5.0;接種前に1.0 mlの50%尿素を添加する。
【0100】
MDU-IB(1リットルあたり)
マルトデキストリン MD01 45.0 g
MgSO4・7H2O 1.0 g
NaCl 1.0 g
K2SO4 2.0 g
KH2PO4 12.0 g
酵母エキス 7.0 g
AMG微量金属 0.5 ml
プルロニック PE6100 1 ml
最終pH 5.0;接種前に100 ml培地あたり1.3 mlの50%尿素を添加する。
【0101】
AMG微量金属溶液(1リットルあたり)
FeSO4・7H2O 13.9 g
MnSO4・H2O 8.45 g
ZnCl2 6.8 g
CuSO4・5H2O 2.5 g
NiCl2・6H2O 2.5 g
クエン酸 ≧3.0 g
微量金属溶液 1 ml
【0102】
KM2培地(1リットルあたり)
酵母エキス 2.5 g
KH2PO4 10 g
MgSO4・7H2O 0.5 g
KCl 0.5 g
FeSO4 0.01 g
グルコース 100 g
最終pHを6.0に調整
固形プレートで使用する前に、KMZ培地を20 g/l寒天で凝固させる。
コロニー増殖制限剤として300 μl/lのTriton X-100を添加する。
【0103】
ナカムラ培地(1リットルあたり)
ショ糖 50 g
ペプトン 20 g
KH2PO4 5 g
CaHPO4 2.5 g
MgSO4 2.5 g
最終pHを6.0に調整。
【0104】
5.アッセイ
A.HPキャピラリー電気泳動によるCPAについてのアッセイ方法
Supelclean LC-18 SPEチューブ(2 mlのメタノールと2 mlのMilli-Q水でコンディショニングした、カタログNo.5-7012のSupelco社製プレパック3 mlカラム)上での固相抽出により、キャピラリー電気泳動(CE)用に試料の1mlアリコートを調製する。溶液をカラムに強制的に流すために吸引マニホールドを使用する。3 mlのMilli-Q水で洗浄した後、3 mlのメタノールを用いて試料を溶離させる。更に後処理せずに、溶出液をCE分析にかける。場合により形成した沈澱を遠心により除去する。
【0105】
CE分析は、Hewlet-PackardフォトダイオードアレイCE装置(3D−CE)を使って行う。試料を10秒間かけて3400 Pa(34 mbar)の静水圧下で注入する。30℃で50 mmのキャピラリー(有効長さ56 cm)を使用し、それを0.1 M NaOHで1分間、次いで100 mMホウ酸/NaOH pH 9.1で5分間コンディショニングしておく。電圧を17 kVに設定する。ピークを同定するのにUVスペクトルを常時収集するが、280 nmで泳動を追跡する。標準試料として市販のシクロピアゾン酸を使用する(Sigma Co., St. Louis MO, USA, カタログNo. C1530, 最低98%純度)。この方法の感度の下限は約1ppmである。
【0106】
B.薄層クロマトグラフィー(TLC)によるCPAについてのアッセイ方法
1.平板培養物の分析
寒天プラグはFiltenborg O., Frisvad J.C. & Svendsen J.A.: "Simple Screening Method for Mold Producing Intracellular Mycotoxins in Pure Cultures", Applied and Environmental Microbiology (1983) 45:581-585に記載された通りに分析する。
【0107】
2.液体培養物の分析
上清の10μl試料をTLCプレート(Merck Silica Gel 60)の対角の縁に適用する。メタノール:クロロホルム(容量で1:2)混合物中に50 ppm, 25 ppm, 5 ppmおよび2.5 ppmに溶解希釈したシクロピアゾン酸(Sigma C 1530)を標準として使用する。まずプレートをCAP(クロロホルム:アセトン:プロパン−2−オール=容量で85:15:20)中で15分間展開し、乾燥し、次いで上下逆反転して、もう半分をTEF(トルエン:酢酸エチル:蟻酸=容量で5:4:1)中で15分間展開する。
【0108】
あるいは、プレートをEMA(酢酸エチル:メタノール:25%水酸化アンモニウム=容量で16:8:2)中とTEF中で各々15分間ずつ上記と同様に展開する。
プレートをヒュームフード中で完全に(1時間)乾燥させた後、エールリッヒ試薬(85 mlの96%エタノール中に溶かした2 gの4−ジメチルアミノベンズアルデヒドに、37%塩酸を添加したもの)を噴霧する。
【0109】
CPAは、CAP系(中性系)の場合は典型的な低い泳動度を有し、青紫色のマッシュルーム型スポットとして観察され、一方で酸性TEF系の場合は適用位置と展開剤の最前線の中間に細長く伸びた典型的なスミアを生じる。EMA系(塩基性/アルカリ性系)では、シクロピアゾン酸は小さい濃厚なスポットに集約される。
【0110】
プレートの直接外観検査によれば、≧2.5 ppmのCPA濃度は紫色の帯状スミアまたはスポットとして(展開系による)観察することができる。卓上フラットベッド・スキャナ−上でTLCプレートをスキャンし、次いで適当な画像処理プログラム(本件ではPaint Shop Pro 4)を使って電子画像を加工および強調処理することにより、感度が5〜10倍高められる。
この分析の全感度は(抽出なしで)約0.5〜1ppm CPAであり;抽出を使うと感度が少なくとも10倍高まる。
【0111】
C.キャピラリー電気泳動によるコウジ酸についてのアッセイ方法
メタノールと10 mMホウ酸/NaOH, 4 M KCl, pH 9.1でコンディショニングしたSupelclean LC-18 SPEチューブ(カタログNo.5-7012のSupelco社製プレパック3 mlカラム)上での固相抽出により、CE分析用に試料の1〜3mlアリコートを調製する。溶液をカラムに強制的に流すために吸引マニホールドを使用する。3 mlの10 mMホウ酸/NaOH, 4 M KCl pH 9.1と0.3 mlの10 mM ホウ酸/NaOH pH 9.1で洗浄した後、7.5 mlの10 mMホウ酸/NaOH pH 9.1を用いて試料を溶離させる。更に後処理せずに、シクロピアゾン酸について上述した手順に準じて溶出液をCE分析にかける。場合により形成した沈澱を遠心により除去する。この方法の感度の下限は約6ppmである。
【0112】
D.薄層クロマトグラフィーによるコウジ酸についてのアッセイ方法
試料のアリコートをCPAについて上述したのと同様にTLCプレートの対角の端に適用し、CPAと同じ溶媒系を使って展開する。乾燥したプレートに0.1 M HCl中の1%FeCl3を噴霧する。試料中のコウジ酸の存在は赤色スポットにより示され、対照として適用した純粋なコウジ酸によって生じた赤色スポットの強度と比較される。検出の下限は50 ppmである。
【0113】
E.キャピラリー電気泳動による3−NPAについてのアッセイ方法
キャピラリー電気泳動(CE)分析への準備としての試料精製の必要性は試料の伝導度に依存する。伝導度が10 mS未満である場合、溶出緩衝液として0.1 M KClを使ったVarian SAXアニオン交換体(Varian Instrumetns, Palo Alto CA)上でのイオン交換により試料を精製する。伝導度が100 mSより大きい場合、2−ブタノールを使って試料を抽出する。その場合、酸性化/高塩処理による沈澱形成と、10 mM Tris/HCl pH 7.0中への沈澱の再溶解の後、2mlの試料を6 mlのブタノールで抽出する。
【0114】
30℃の温度でおよび25 mMホウ酸/リン酸 pH 7.6のコンディショニング緩衝液を使う直径50μmで有効長さ56 cmの未コーティングシリカのキャピラリーを使用するHP−CE装置ダイオードアレイ検出を用いる。試料を20秒間かけて静水圧下で注入する。電圧を30 kVに設定する。この方法の検出の下限は6ppmである。
【0115】
F.薄層クロマトグラフィーによる3−NPAについてのアッセイ方法
CPAについて記載したのと同様にTLCプレートに発酵液のスポットを適用しそして展開する。次いでW. Majak & R.J. Bose, "Chromatographic methods for the isolation of miserotoxin and the detection of aliphatic nitro compounds", Phytochemistry (1974) 13:1005-1010により記載された通りに、それらにジアゾ化p−ニトロアニリンを噴霧する。対照物質に対比したスポットの強度と位置が3−NPA濃度の尺度である。TLCプレート上での検出レベルは25〜50 ppmである。
【0116】
あるいは、100μl試料に50μlの1 M NaOHと70μlのジアゾ化p−ニトロアニリンを添加することにより3−NPAを分光光度的に(λ=540 nm)分析する。検出レベルは5〜10 ppmである。
【0117】
実施例1
A.A.オリゼ BZ 14由来のCPA陰性株の作製
アスペルギルス・オリゼ BZ14株の凍結乾燥胞子に1000 Gy〜1250 Gyの最適線量でγ線を照射し、次いでその胞子を25〜50コロニー/9 cmプレートの密度においてスクリーニング培地1上に接種した。シクロピアゾン酸を生産するコロニーは、赤色の不溶性CPA−Fe錯体を形成するためスクリーニング培地1上に赤色の裏面(コロニーの下側)を生成する。
【0118】
照射した胞子から約50,000個のコロニーをスクリーニングし、そして乳白色(クリーム/白)外観により特徴づけられる154のCPA欠失コロニーを分離した。再分離後、64の株がスクリーニング培地1のプレート上で非赤色裏面を保持した。TLCプラグアッセイにより、52個の株でCPAが検出されなかった。次いでそれらの株を、毒素誘発(34℃、250 rpmで5日間)振盪フラスコ発酵条件下で、MDU-1B培地中で培養した。36個の株がTLCにより測定した時に上清中に検出可能レベルのCPAを示さなかった。
【0119】
B.A.オリゼ JaL228由来のCPA陰性株BECh 1の作製
JaL228の凍結乾燥胞子を上記と同様にγ線照射しそしてスクリーニングした。推定上のCPA欠失分離株を毒素誘発条件(34℃、250 rpmで5日間)下でMDU-1B培地上で振盪フラスコ培養し、そして上清をTLC法によりCPAについて分析した。上清はそのままでまたは抽出液として試験した。
【0120】
抽出用には、全試料のうちの50 mlを10 mlの0.1 M HClで酸性化した。次いでこの混合物を70 mlメタノール/クロロホルム(1:2)と共に3〜5分間激しく振盪した。相分離後(約3時間後)、下層(約25 ml)を300 mlビーカーに移し、クロロホルムを蒸発させた。残渣を5mlのクロロホルム中に再溶解し、25 mlビーカーに移し、そしてクロロホルムを蒸発させた。残渣を100μlのクロロホルム中に溶かした。
【0121】
上清またはクロロホルム抽出液10μlを20 cm×20 cmのTLCプレートの対角の縁に適用し、そしてアッセイに関する前掲の項目に記載した通りに処理した。
BECh 1を含む3つの株(分離株)がCPAを生産しなかった。
【0122】
C.CPA陰性KA陰性の株BECh 2およびBECh 3の作製
BECh 1をCove N斜面上で培養した。胞子を0.01%Tween 中に3-5×106の密度に懸濁し、短波UV照射(殺菌灯からの254 nm)にかけた。1〜5%生存率を生じるUV線量で照射した胞子をその後のスクリーニングに使用した。
【0123】
照射済胞子をKM2培地中に約0.7胞子/100μlの密度に希釈し、その100μlを96ウエルマイクロタイタープレートの各ウエルに接種した。培養物を湿潤チャンバーの中に入れ、静止状態で34℃にて5〜7日間インキュベートした。次いで、増殖徴候を有する各ウエルに40μlの1%FeCl3/0.1 M HClを添加した。
濃赤色の発色はコウジ酸(KA)生産を示し;発色がないのはコウジ酸生産を欠くコロニーを示す。
【0124】
あるいは、凝固させたKM2培地(Triton X-100により増殖制限)上に胞子をまき、成熟コロニーが観察されたら、プレートを0.1 M HCl中の1%FeCl3で潅水させた。コロニーの周囲に赤色域がないものは、推定上の非コウジ酸生産株を示す。
【0125】
約7000 のマイクロタイター培養物の中から、132個の推定KA陰性コロニー、すなわちFeCl3と発色反応を示さないものが単離された。しかしながら、一次KM2スクリーニング寒天プレート上で試験すると、該コロニーのうち1つもKA陰性であると確認されなかった。
【0126】
固形培地上と静止液体KM2培地中(30°)の両培地を使用したKA誘発条件下での陰性コロニーの再試験は、KA陰性変異体の数を11にまで絞り込んだ。それらの株を振盪フラスコ中で試験すると、8つの株でKAが生産された。残りの3つは、TLCにより調べたときにも直接上清をアッセイしたときにもどちらも発色反応を与えなかった。同様な並行培養において増殖させると、予想通り、対照株BECh 1によりKAは生産された。分離株のうちの1つは異常な形態を示した。MDU-1B上での長期増殖後、残りの2つの分離株をCPAとKAの両方について試験した。CPAもKAもどちらも検出されなかった。この2つの株をBECh 2およびBECh 3と命名した。
【0127】
D.既にCPA陰性で且つKA陰性である3−NPA陰性A.オリゼ株の作製
BECh2株とBECh3株それぞれを上述したようなUV変異誘発にかけた。照射した胞子を96ウエルマイクロタイタープレート中で0.7生存胞子/100μlナカムラ培地の密度に希釈した。湿潤チャンバー中で30℃にて5〜7日間インキュベートした。
【0128】
増殖を示すウエルからの発酵ブロス試料を新たなマイクロウエルプレートに移して分光光度分析するかまたはTLCプレートに適用した。3−NPA陰性である株をナカムラ培地の入った振盪フラスコ中で再培養し、そして3−NPAについて分析した。3−NPA陰性である株を実施例2に記載の通りにpCaHj 493により形質転換せしめ、そして形質転換体を実施例3に記載の通りに処理した。
【0129】
実施例2
A.オリゼ株 JAL228およびBECh 1中でのリパーセ遺伝子の発現
A.プラスミド pCaHj493の作製
リパーゼプラスミド pAHL (WO 97/07202)をBamHIとSalIで消化し、得られたリパーゼをコードする916 bp断片を単離した。
WO 98/00529に記載のpCaHj 483をBamHIとXhoIで消化し、6757 bpベクター断片を上記リパーゼ断片と連結せしめた。その連結混合物を用いてE.コリ DH5α細胞を形質転換せしめ、そして所望のプラスミドを含有する形質転換体を単離した。得られたプラスミドをpCaHj 493と命名した。
【0130】
B.JaL228およびBECh-1中へのpCaHj 493の形質転換
ヨーロッパ特許出願第0531372号に記載のようなアセトアミド上での選択を使って、pCaHj493によりアスペルギルス・オリゼ株 JaL228およびBECh1を形質転換せしめた。形質転換体を2回胞子再分離した。各形質転換体の2回目の再分離からの胞子を、振盪フラスコ中とマイクロタイター皿培養においてリパーゼ生産について試験した。
【0131】
実施例3
A.CPA陰性A.オリゼ株とCPA陽性A.オリゼ株におけるリパーゼ生産
実施例2に記載のように調製した18個のJaL228形質転換体と30個のBECh 1形質転換体を振盪フラスコ培養においてリパーゼ生産について調べた。
【0132】
形質転換体のCove N斜面培養物を、10 mlの0.1%Tween溶液を使って収得し、胞子懸濁液を500 mlのツーバッフル(two-baffled)振盪フラスコに入れた100 mlのG1-Gly培地への接種材料として使用した。培養物を回転式振盪器上で34℃にて250 rpmで24時間インキュベートした。次いでG1-Gly培養物の10 mlを500 mlの振盪フラスコに入った100 mlの1/5 MDU-2BPに移し入れ、更に34℃にて250 rpmでインキュベートした。
【0133】
50時間後に試料を採取し、ミラクロスを通して濾過し、遠心分離した(4000×g)。単純放射状免疫拡散法〔Scand, J. Immunol. Vol.17, suppl. 10, 41-56,(1983), "Handbook of Immunoprecipitation-in-Gel Techniques", N.H. Axelsen編、Blackwell Scientific Publications, 1983〕を使って、上清中のリパーゼ濃度(LU/mlで表す)を検出した。
【0134】
30個のCPA陰性BECh 1形質転換体は、18個のCPA陽性JaL228形質転換体と同じかそれより高いリパーゼ収量を有した。表2はその分布の概観を与える。
【0135】
B.CPA陰性A.オリゼ株とCPA陽性A.オリゼ株によるキシラナーゼ生産
検出可能なCPA生産を示さない10個の株(実施例1Aで調製したもの)とCPAが検出可能である3個の株をキシラナーゼ生産について評価した。その結果を下記の表1に要約する。2列目の欄は、単純放射状免疫拡散法〔Scand, J. Immunol. Vol.17, suppl. 10, 41-56 (1983), "Handbook of Immunoprecipitation-in-Gel Techniques", N.H. Axelsen編、Blackwell Scientific Publications, 1983〕によりアッセイした時の真菌キシラナーゼ単位(FXU)で測定した振盪フラスコ培養で生産されたキシラナーゼの量を示す。
【0136】
この結果は、CPA陰性株がCPA陽性株に匹敵する量でキシラナーゼを生産できることを示す。
【0137】
【表1】
【0138】
C.CPA陰性KA陰性の株におけるリパーゼ生産
2つのCPA陰性KA陰性株、BECh 2とBECh 3を実施例2に記載の通りにプラスミド pCaHj 493により形質転換せしめた。得られた形質転換体を2回胞子分離した。各形質転換体の2回目の再分離からの胞子を実施例2に記載の通りにリパーゼ生産について試験した。
【0139】
表2は、それらの2つの株からの形質転換体のリパーゼ収量の度数分布を示す。その結果は、A.オリゼ株 BECh 1およびJaL228について与えられた値に比較して、得られる発現能力に何ら損傷がないことを示す。
【0140】
リパーゼ生産培養に使用したのと同じ胞子懸濁液から、CPA生産用のMDU1B振盪フラスコに接種を行い、そして上述したのと同様に5日間インキュベートした。項目5B2に従ってDPA分析を行った。BECh 1株はいずれもCPAを生産しなかったが、一方で18個のJaL228株のうちの17個は25 ppm超のCPAを生産し、大部分は100 ppm超のCPAを生産した。
【0141】
【表2】
【0142】
実施例4
A.オリゼDCAT-S遺伝子の同定およびゲノムクローニング
A.A.オリゼのジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼ(DCAT-S)遺伝子の同定
cDNAクローン(pJaL499)は配列番号1に示すDNA配列を含有する。それはクラビセプス・プルプレア(Claviceps purpurea)のジメチルアリルトリプトファンシンターゼ(DMAT-S)遺伝子に対する相同性により、CPA生合成に関連することが同定されている。A.オリゼcDNAクローンの配列決定は、それが長さ1393塩基対(配列番号1)であり、クラビセプス・プルプレア由来のDMAT-Sと42.1%同一である473アミノ酸ポリペプチド(配列番号2)をコードすることを示した。
【0143】
A.オリゼDCAT-Sポリペプチドは、シクロアセトアセチル−L−トリプトファンからのβ−CPA生合成に関与している(Nethling D.C. & McGrath, Can. J. Microbiol. (1977) 23:856-872)。
JaL228株とBECh 1株から染色体DNAを調製した。DNAをBglII, NcoI, XhoIおよびSpeIで消化し、そしてプローブとしてDCAT-S遺伝子を含むpJaL499由来の1kb 32P標識BglII DNA断片を使ってサザンブロッティングにより分析した。サザンブロット分析は、CPA生産株JaL228が1つのDCAT-S遺伝子を有し、一方でBECh 1ではそのDCAT-S遺伝子が染色体から欠失していることを示した。
【0144】
B.DCAT-S遺伝子のゲノムクローンのクローニング
次の制限酵素:EcoRI, SalI, BbuI, XhoIおよびXbaIを使って、プローブとしてDCAT-S遺伝子を含むpJaL499由来の1kb 32P標識BglII DNA断片を使って、A.オリゼ DCAT-S遺伝子のゲノム制限地図作成を行った(図1)。これは、DCAT-S遺伝子の唯一のコピーが存在することを示す。
【0145】
JaL228のゲノムDNAをTsp509Iで部分消化するかまたは0.7%アガロースゲル上で泳動した。7 kbと10 kbの間のサイズを有する断片を精製した。
【0146】
次いで精製済DNAを製造業者(StratageneR)により提供されたプロトコルを使ってLambda ZAP II中にクローニングした。製造業者により与えられる教示に従って、λベクター内に含まれる任意のクローン挿入断片を生体内切除し、再環化せしめて、DNAライブラリー用にクローン化挿入断片を含むファジミドを作製した。標準方法の参考書〔例えばJ. Sambrook, E.F. Fritsch & T. Maniatis編(1989) "Molecular Cloning: A Laboratory Manual", 第2版,Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York〕に概説されるように、プローブとしてDCAT-S遺伝子を含むpJaL499からの1kb 32P標識DNA BglII断片を使ったコロニーハイブリダイゼーションにより、DCAT-S遺伝子をコードするクローンについてスクリーニングを行った。
【0147】
実施例5
アスペルギルス・オリゼのジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼ(DCAT-S)の遺伝子破壊によるアスペルギルス・オリゼCPA陰性株の作製
選択マーカーとしてA.オリゼ pyrG遺伝子を使って、A.オリゼのpyrG陰性株中のDCAT-S遺伝子を一段階遺伝子置換法〔B.L. Miller他,Mol. Cell. Biol. (1985) 5:1714-1721;G. May, Applied Molecular Genetics of Filamentous Fungi, 1-25頁, J.R. Kinghorn & G. Turner編, Blakie Academic and Professional, 1992〕により破壊した。
【0148】
A.DCAT-S破壊プラスミドの作製
プラスミドpJaL499をSacIIで消化し、クレノウポリメラーゼで処理して平滑末端を作り、そして製造業者(Boehringer Mannheim)の教示に従って細菌アルカリ性ホスファターゼで処理して5′リン酸基を除去し、次いでフェノール抽出しそして沈澱させた。
【0149】
WO 98/12300に記載のプラスミド pJaL335をHindIIIで消化してA.オリゼpyrG遺伝子を含む3.5 kb断片を得、それをクレノウポリメラーゼで処理して平滑末端にし、ゲル電気泳動により単離し、そして精製した。次いで2つの断片を一緒に混合し、連結せしめた。E.コリ中への形質転換後、正しいプラスミドを担持しているコロニーをミニプラスミド調製物の制限酵素消化により同定した。破壊プラスミド(pDCAT-S-pyrG)の作製を図2に要約する。
【0150】
B.pyrG陰性A.オリゼ株 JaL250の単離
A.オリゼ株 JaL228を5−フルオロオロト酸に対する耐性についてスクリーニングし、自然pyrG変異体を同定した。JaL250と命名した1つの株がpyrG陰性であると同定された。この変異体はウリジン依存性であるので、それを野生型pyrG遺伝子を用いて形質転換し、そしてウリジンの非存在下での増殖能力により形質転換体を選択することができる。
【0151】
C.アスペルギルス・オリゼ DCAT-S欠失株の作製
プラスミド pDCAT-S-pyrGの4.9 kb NotI-EcoRI断片をゲル精製し、Christensen他、Biotechnology (1988) 6:1419-1422により記載された通りA.オリゼ株 JaL250を形質転換せしめるのに使った。次いでウリジン非存在下でのそれの増殖能力により形質転換体を選択した。2回再分離した後、実施例1に記載の通りにCPAを生産する能力について形質転換体をスクリーニングした。
【0152】
DCAT-S遺伝子が破壊されていることを確かめるために、CPAを生産しない形質転換体から染色体DNAを調製した。該DNAをEcoRIで消化し、DCAT-S遺伝子を含むpJaL499からの1kb 32P標識DNA BglII断片をプローブとして使ってサザンブロッティングにより分析した。DCAT-S遺伝子の破壊を有する形質転換体は、6.3 kb上の野生型EcoRIバンドが9.8 kb上のEcoRIバンドの方へ移動することにより識別される。
【0153】
実施例6
BECh1とBECh2がアフラトキシン生合成経路クラスターからの2つの遺伝子を欠くことの確証
A.オリゼ IFO 4177およびそれの多数の誘導体においてアフラトキシン生合成経路クラスター由来のaflRおよびomtAアフラトキシン遺伝子の存在を探求した。プライマー5956(5'-GGATCCAGGGCTCCCTGGAG-3')(配列番号3)と5955(5'-CCTGACCAGCCAGATCTCCT-3')(配列番号4)を使ったPCRにより、A.オリゼ IFO4177のゲノムDNAからaflR相同体を単離した。0.9 kb PCR断片を獲得し、それをInvitrogenから入手したベクター pCR2中にクローニングした。M13正(-40)プライマーと逆プライマーを使って、得られたプラスミド pToC280を配列分析することにより、クローン化断片の素性を確認した。また、プライマー 6120(5'-AGTGAGAGAACTCCCTCCTC-3')(配列番号5)と6121(5'-CCATATCTTCTCAGTCTCCA-3')(配列番号6)を使ったPCRにより、ゲノム IFO4177 DNAからomtA相同体を単離した。1.2 kb断片を得、それをInvirtogenからのベクターpCR2中にクローニングし、そして得られたプラスミド pToC276をM13正(-40)および逆プライマーを使って配列分析することにより、クローン化断片の素性を確認した。
【0154】
上記aflRおよびomtAのクローン化断片をハイブリダイゼーション実験における32P標識プローブとして使用した。IFO4177, JaL228, BECh1およびBECh2からのゲノムDNAを制限酵素EcoRIで消化し、生じた断片を0.7%アガロースゲル上で分離した。DNAを膜上にブロッティングし、上記2つの32P標識プローブを一つずつ用いて緊縮条件下でハイブリダイズさせた〔方法は. Sambrook, E.F. Fritsch, T. Maniatis編(1989) "Molecular Cloning: A Laboratory Manual", 第2版, Cold Spring Harbor, New Yorkに記載されている〕。ブロットは、IFO4177とJaL228からの両プローブを用いたとき陽性のハイブリダイゼーションシグナルを示し、一方でBECh1とBECh2 DNAを含むレーンでは全くバンドが検出されなかった。IFO4177とJaL228レーンでは、omtAプローブを使って1つの約3.8 kb断片が観察でき、aflRプローブを使った場合は約0.5 kbと4.3 kbの2つのバンドが観察できた。
【0155】
従って、A.オリゼ IFO4177は、アフラトキシン生合成経路からの少なくとも2つの遺伝子、すなわちaflR遺伝子とomtA遺伝子を含有する。A.フラーブスおよびA.パラシチクスでは、この2つの遺伝子は約32 kbだけ離れている〔Woloshuk, C.P. & Prieto, R., FEMS Microbiology Letter (1998) 160:169-176〕。IFO4177誘導体であるBECh1とBECh2には、それらの遺伝子のいずれも存在しない。
【0156】
本発明は、本明細書中に開示される特定の態様によりその範囲が限定されるものではなく、それらの態様は本発明の幾つかの面の例示として与えられるものである。任意の同等な態様も本発明の範囲内に含めることができる。実際、上記に例示および記載されたものに加えて、上記説明より本発明の様々な変更が当業者には明らかであろう。そのような変更は特許請求の範囲内に含まれるものである。
【0157】
様々な参考文献を本明細書中に引用したが、その開示の全内容が参考として組み込まれる。
【特許請求の範囲】
【請求項1】
着目のポリペプチドの生産方法であって、
(a) 親のアスペルギルス細胞の変異体を培養し、ここで(i) 前記変異体は前記ポリペプチドをコードする第一の核酸配列を含んでなり、そして(ii) 前記変異体は同一条件下で培養した時に少なくとも1つの着目の毒素を親のアスペルギルス細胞よりも少量生産し;そして
(b) 培地から前記ポリペプチドを単離する
ことを含んでなる方法。
【請求項2】
前記変異体が、1または複数の毒素の生合成または分泌の原因となる少なくとも1つの遺伝子の変更の結果として前記毒素を少量生産する、請求項1に記載の方法。
【請求項3】
前記変異体が同一条件下で培養した時に前記毒素を親細胞よりも少なくとも約90%少なく生産する、請求項1または2に記載の方法。
【請求項4】
前記毒素がシクロピアゾン酸、コウジ酸、3−ニトロプロピオン酸、エモジン、マルフォルミン、アフラトキシン、オクラトキシンおよびセカロン酸からなる群より選ばれる、請求項1〜3のいずれか一項に記載の方法。
【請求項5】
前記変異体が同一条件下で培養した時にシクロピアゾン酸と少なくとも第二の毒素を親のアスペルギルス細胞よりも少量生産する、上記請求項のいずれか一項に記載の方法。
【請求項6】
前記第二の毒素がコウジ酸またはアフラトキシンである、請求項5に記載の方法。
【請求項7】
前記変異体が同一条件下で培養した時に更に3−ニトロプロピオン酸を親のアスペルギルス細胞よりも少量生産する、請求項5または6に記載の方法。
【請求項8】
前記アスペルギルス細胞がアスペルギルス亜群ユーロチウム、ケトサルトリア、スクレロクレイスタ、サトイア、ネオサルトリア、ヘミカーペンテレス、ペトロミセス、エメリセラおよびフェネリアからなる群より選ばれる、上記請求項のいずれか一項に記載の方法。
【請求項9】
前記着目のポリペプチドがアスペルギルス宿主細胞にとって生来である、上記請求項のいずれか一項に記載の方法。
【請求項10】
前記着目のポリペプチドが、同一条件下で培養した時に、親のアスペルギルス宿主細胞による発現に比較してアスペルギルス変異体宿主細胞により過剰発現される、請求項9に記載の方法。
【請求項11】
前記着目のポリペプチドがアスペルギルス宿主細胞にとって非相同である、請求項10に記載の方法。
【請求項12】
前記ポリペプチドが、ホルモンまたはそれの前駆体形、酵素もしくは酵素変異体またはそれの前駆体形、抗体またはそれの機能性断片、レセプターまたはそれの機能性断片、およびレポーターからなる群より選ばれる、上記請求項のいずれか一項に記載の方法。
【請求項13】
前記酵素がアミノペプチダーゼ、アミラーゼ、カルボヒドラーゼ、カルボキシペプチダーゼ、カタラーゼ、セルラーゼ、キチナーゼ、クチナーゼ、シクロデキストリングルコシルトランスフェラーゼ、デオキシリボヌクレアーゼ、エステラーゼ、ガラクタナーゼ、α−ガラクトシダーゼ、β−ガラクトシダーゼ、グルコアミラーゼ、α−グルコシダーゼ、β−グルコシダーゼ、インベルターゼ、ラッカーゼ、リパーゼ、リアーゼ、ペクテートリアーゼ、マンナーゼ、マンノシダーゼ、ムタナーゼ、オキシダーゼ、オキシゲナーゼ、ペクチナーゼ、エンドペプチダーゼ、エキソペプチダーゼ、ペルオキシダーゼ、フィターゼ、ポリフェノールオキシダーゼ、プロテアーゼ、リボヌクレアーゼ、トランスグルタミナーゼおよびキシラナーゼからなる群より選ばれる、請求項12に記載の方法。
【請求項14】
着目の非相同ポリペプチドの生産に有用な毒素欠失アスペルギルス変異体宿主細胞であって、同一条件下で培養した時にアスペルギルス親細胞に比較して少なくとも1つの毒素をより少量生産させるために該細胞が遺伝的に変更されていることを特徴とする変異体細胞。
【請求項15】
着目のポリペプチドをコードする第一の核酸配列と、前記毒素の生合成または分泌の原因となる少なくとも1つの遺伝子の変更を含んでなる第二の核酸配列とを含んでなる、請求項14に記載の変異体細胞。
【請求項16】
前記細胞が前記第一の核酸配列の少なくとも2コピーを含んでなる、請求項14または15に記載の変異体細胞。
【請求項17】
前記着目のポリペプチドが前記変異体細胞にとって生来である、請求項14〜16のいずれか一項に記載の変異体細胞。
【請求項18】
前記着目のポリペプチドが前記変異体細胞にとって非相同である、請求項14〜16のいずれか一項に記載の変異体細胞。
【請求項19】
前記毒素がシクロピアゾン酸、コウジ酸、3−ニトロプロピオン酸、エモジン、マルフォルミン、アフラトキシン、オクラトキシンおよびセカロン酸からなる群より選ばれる、請求項13〜18のいずれか一項に記載の変異体細胞。
【請求項20】
前記毒素が、シクロピアゾン酸である第一の毒素と、コウジ酸、3−ニトロプロピオン酸およびアフラトキシンからなる群より選ばれる第二の毒素とを含んでなる、請求項13〜19のいずれか一項に記載の変異体細胞。
【請求項21】
請求項13〜20のいずれか一項に記載の毒素欠失アスペルギルス変異体宿主細胞を獲得する方法であって、前記細胞は少なくとも1つの着目の毒素の生産を欠いており、(a) 親細胞を突然変異誘発にかけそして(b) 着目の毒素の生産が減少または除去されている変異体細胞についてスクリーニングすることを含んでなる方法。
【請求項22】
前記突然変異誘発がランダムである、請求項21に記載の方法。
【請求項23】
前記突然変異誘発が特異的であり、そして前記着目の毒素の生産または分泌に関係しており且つそれに必要であるヌクレオチド配列を変更するように行われる、請求項22に記載の方法。
【請求項24】
前記毒素の生合成または分泌の原因である少なくとも1つの遺伝子の変更を含んでなる核酸配列を、アスペルギルス宿主細胞中の対応する生来の遺伝子との相同組換えを媒介する条件下で、前記遺伝子を不活性化するようにアスペルギルス宿主細胞中に導入し、それにより前記生来の遺伝子と不活性な変更遺伝子との置換を引き起こす、請求項23に記載の方法。
【請求項25】
着目のポリペプチドをコードする核酸配列をアスペルギルス親宿主細胞中に導入することを更に含んでなる、請求項21〜24のいずれか一項に記載の方法。
【請求項26】
前記着目のポリペプチドがアスペルギルス細胞にとって生来である、請求項25に記載の方法。
【請求項27】
前記着目のポリペプチドがアスペルギルス細胞にとって非相同である、請求項25に記載の方法。
【請求項28】
請求項13〜20のいずれか一項に記載の毒素欠失アスペルギルス変異体宿主細胞を獲得する方法であって、(a) 着目のポリペプチドをコードする第一の核酸配列と、少なくとも1つの毒素の生合成または分泌の原因である少なくとも1つの遺伝子の変更を含んでなる第二の核酸配列とを、アスペルギルス親宿主細胞中に導入し;そして(b) 段階(a)から前記第一および第二の核酸配列を含んでなる変異体を同定することを含んで成る方法。
【請求項29】
前記第一および第二の核酸配列の導入の前または後にアスペルギルス親宿主細胞を突然変異誘発にかけることを更に含んでなる、請求項28に記載の方法。
【請求項30】
前記突然変異誘発が、特異的もしくはランダム突然変異誘発、PCR作製突然変異誘発、部位特異的DNA欠失、挿入および/または置換、遺伝子破壊または置換技術、並びにアンチセンス技術からなる群より選ばれる1または複数の方法により達成される、請求項29に記載の方法。
【請求項31】
配列番号1に記載のものと実質的に同じ、ジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼをコードする核酸配列。
【請求項32】
糸状菌宿主株における類似遺伝子の破壊のための、請求項31に記載の核酸配列またはそれの活性断片の使用。
【請求項33】
前記糸状菌宿主株がアスペルギルス、トリコデルマ、ペニシリウムおよびフザリウム種からなる群より選ばれる、請求項32に記載の使用。
【請求項34】
アスペルギルス・オリゼ(Aspergillus oryzae)株より得られる単離されたジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼであって、
(a) 実質的に配列番号2のアミノ酸配列を有するジメチルアリル−シクロアセチル−L−トリプトファンシンターゼ;
(b) (a) の対立遺伝子変異体;および
(c) ジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼ活性を有する、(a)または(b)の断片
からなる群より選ばれる、単離されたジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼ。
【請求項35】
1または複数のサイレント毒素遺伝子が除去されている、非相同ポリペプチドの発現に適当な変異体アスペルギルス細胞。
【請求項36】
前記1または複数の毒素遺伝子の全部または一部の不可逆的欠失または破壊により1または複数の毒素遺伝子が除去されている、請求項35に記載の変異体。
【請求項37】
前記アスペルギルス細胞がアスペルギルス亜群ユーロチウム、ケトサルトリア、スクレロクレイスタ、サトイア、ネオサルトリア、ヘミカーペンテレス、ペトロミセス、エメリセラおよびフェネリアからなる群より選ばれる、請求項35または36のいずれか一項に記載の変異体。
【請求項38】
前記毒素遺伝子が、シクロピアゾン酸、コウジ酸、3−ニトロプロピオン酸、エモジン、マルフォルミン、アフラトキシン、オクラトキシンおよびセカロン酸からなる群より選ばれる1または複数の毒素をコードする、請求項35〜37のいずれか一項に記載の変異体。
【請求項39】
問題の前記毒素遺伝子がアフラトキシンをコードする、請求項35〜38のいずれか一項に記載の変異体。
【請求項40】
前記毒素遺伝子がA.オリゼのアフラトキシンクラスターからの遺伝子であり、特にomtA, aflR, pksA, Nor-1, fas-beta, fas-alpha, vber-1, avnA, ord-2からなる群より選ばれる、請求項35〜39のいずれか一項に記載の変異体。
【請求項41】
親のアスペルギルス細胞がA.オリゼ、特にA.オリゼ A1560 (IFO 0417)である、請求項40に記載の変異体。
【請求項42】
前記アフラトキシン遺伝子がomtAおよびaflRから成る群より選ばれる、請求項41に記載の変異体。
【請求項1】
着目のポリペプチドの生産方法であって、
(a) 親のアスペルギルス細胞の変異体を培養し、ここで(i) 前記変異体は前記ポリペプチドをコードする第一の核酸配列を含んでなり、そして(ii) 前記変異体は同一条件下で培養した時に少なくとも1つの着目の毒素を親のアスペルギルス細胞よりも少量生産し;そして
(b) 培地から前記ポリペプチドを単離する
ことを含んでなる方法。
【請求項2】
前記変異体が、1または複数の毒素の生合成または分泌の原因となる少なくとも1つの遺伝子の変更の結果として前記毒素を少量生産する、請求項1に記載の方法。
【請求項3】
前記変異体が同一条件下で培養した時に前記毒素を親細胞よりも少なくとも約90%少なく生産する、請求項1または2に記載の方法。
【請求項4】
前記毒素がシクロピアゾン酸、コウジ酸、3−ニトロプロピオン酸、エモジン、マルフォルミン、アフラトキシン、オクラトキシンおよびセカロン酸からなる群より選ばれる、請求項1〜3のいずれか一項に記載の方法。
【請求項5】
前記変異体が同一条件下で培養した時にシクロピアゾン酸と少なくとも第二の毒素を親のアスペルギルス細胞よりも少量生産する、上記請求項のいずれか一項に記載の方法。
【請求項6】
前記第二の毒素がコウジ酸またはアフラトキシンである、請求項5に記載の方法。
【請求項7】
前記変異体が同一条件下で培養した時に更に3−ニトロプロピオン酸を親のアスペルギルス細胞よりも少量生産する、請求項5または6に記載の方法。
【請求項8】
前記アスペルギルス細胞がアスペルギルス亜群ユーロチウム、ケトサルトリア、スクレロクレイスタ、サトイア、ネオサルトリア、ヘミカーペンテレス、ペトロミセス、エメリセラおよびフェネリアからなる群より選ばれる、上記請求項のいずれか一項に記載の方法。
【請求項9】
前記着目のポリペプチドがアスペルギルス宿主細胞にとって生来である、上記請求項のいずれか一項に記載の方法。
【請求項10】
前記着目のポリペプチドが、同一条件下で培養した時に、親のアスペルギルス宿主細胞による発現に比較してアスペルギルス変異体宿主細胞により過剰発現される、請求項9に記載の方法。
【請求項11】
前記着目のポリペプチドがアスペルギルス宿主細胞にとって非相同である、請求項10に記載の方法。
【請求項12】
前記ポリペプチドが、ホルモンまたはそれの前駆体形、酵素もしくは酵素変異体またはそれの前駆体形、抗体またはそれの機能性断片、レセプターまたはそれの機能性断片、およびレポーターからなる群より選ばれる、上記請求項のいずれか一項に記載の方法。
【請求項13】
前記酵素がアミノペプチダーゼ、アミラーゼ、カルボヒドラーゼ、カルボキシペプチダーゼ、カタラーゼ、セルラーゼ、キチナーゼ、クチナーゼ、シクロデキストリングルコシルトランスフェラーゼ、デオキシリボヌクレアーゼ、エステラーゼ、ガラクタナーゼ、α−ガラクトシダーゼ、β−ガラクトシダーゼ、グルコアミラーゼ、α−グルコシダーゼ、β−グルコシダーゼ、インベルターゼ、ラッカーゼ、リパーゼ、リアーゼ、ペクテートリアーゼ、マンナーゼ、マンノシダーゼ、ムタナーゼ、オキシダーゼ、オキシゲナーゼ、ペクチナーゼ、エンドペプチダーゼ、エキソペプチダーゼ、ペルオキシダーゼ、フィターゼ、ポリフェノールオキシダーゼ、プロテアーゼ、リボヌクレアーゼ、トランスグルタミナーゼおよびキシラナーゼからなる群より選ばれる、請求項12に記載の方法。
【請求項14】
着目の非相同ポリペプチドの生産に有用な毒素欠失アスペルギルス変異体宿主細胞であって、同一条件下で培養した時にアスペルギルス親細胞に比較して少なくとも1つの毒素をより少量生産させるために該細胞が遺伝的に変更されていることを特徴とする変異体細胞。
【請求項15】
着目のポリペプチドをコードする第一の核酸配列と、前記毒素の生合成または分泌の原因となる少なくとも1つの遺伝子の変更を含んでなる第二の核酸配列とを含んでなる、請求項14に記載の変異体細胞。
【請求項16】
前記細胞が前記第一の核酸配列の少なくとも2コピーを含んでなる、請求項14または15に記載の変異体細胞。
【請求項17】
前記着目のポリペプチドが前記変異体細胞にとって生来である、請求項14〜16のいずれか一項に記載の変異体細胞。
【請求項18】
前記着目のポリペプチドが前記変異体細胞にとって非相同である、請求項14〜16のいずれか一項に記載の変異体細胞。
【請求項19】
前記毒素がシクロピアゾン酸、コウジ酸、3−ニトロプロピオン酸、エモジン、マルフォルミン、アフラトキシン、オクラトキシンおよびセカロン酸からなる群より選ばれる、請求項13〜18のいずれか一項に記載の変異体細胞。
【請求項20】
前記毒素が、シクロピアゾン酸である第一の毒素と、コウジ酸、3−ニトロプロピオン酸およびアフラトキシンからなる群より選ばれる第二の毒素とを含んでなる、請求項13〜19のいずれか一項に記載の変異体細胞。
【請求項21】
請求項13〜20のいずれか一項に記載の毒素欠失アスペルギルス変異体宿主細胞を獲得する方法であって、前記細胞は少なくとも1つの着目の毒素の生産を欠いており、(a) 親細胞を突然変異誘発にかけそして(b) 着目の毒素の生産が減少または除去されている変異体細胞についてスクリーニングすることを含んでなる方法。
【請求項22】
前記突然変異誘発がランダムである、請求項21に記載の方法。
【請求項23】
前記突然変異誘発が特異的であり、そして前記着目の毒素の生産または分泌に関係しており且つそれに必要であるヌクレオチド配列を変更するように行われる、請求項22に記載の方法。
【請求項24】
前記毒素の生合成または分泌の原因である少なくとも1つの遺伝子の変更を含んでなる核酸配列を、アスペルギルス宿主細胞中の対応する生来の遺伝子との相同組換えを媒介する条件下で、前記遺伝子を不活性化するようにアスペルギルス宿主細胞中に導入し、それにより前記生来の遺伝子と不活性な変更遺伝子との置換を引き起こす、請求項23に記載の方法。
【請求項25】
着目のポリペプチドをコードする核酸配列をアスペルギルス親宿主細胞中に導入することを更に含んでなる、請求項21〜24のいずれか一項に記載の方法。
【請求項26】
前記着目のポリペプチドがアスペルギルス細胞にとって生来である、請求項25に記載の方法。
【請求項27】
前記着目のポリペプチドがアスペルギルス細胞にとって非相同である、請求項25に記載の方法。
【請求項28】
請求項13〜20のいずれか一項に記載の毒素欠失アスペルギルス変異体宿主細胞を獲得する方法であって、(a) 着目のポリペプチドをコードする第一の核酸配列と、少なくとも1つの毒素の生合成または分泌の原因である少なくとも1つの遺伝子の変更を含んでなる第二の核酸配列とを、アスペルギルス親宿主細胞中に導入し;そして(b) 段階(a)から前記第一および第二の核酸配列を含んでなる変異体を同定することを含んで成る方法。
【請求項29】
前記第一および第二の核酸配列の導入の前または後にアスペルギルス親宿主細胞を突然変異誘発にかけることを更に含んでなる、請求項28に記載の方法。
【請求項30】
前記突然変異誘発が、特異的もしくはランダム突然変異誘発、PCR作製突然変異誘発、部位特異的DNA欠失、挿入および/または置換、遺伝子破壊または置換技術、並びにアンチセンス技術からなる群より選ばれる1または複数の方法により達成される、請求項29に記載の方法。
【請求項31】
配列番号1に記載のものと実質的に同じ、ジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼをコードする核酸配列。
【請求項32】
糸状菌宿主株における類似遺伝子の破壊のための、請求項31に記載の核酸配列またはそれの活性断片の使用。
【請求項33】
前記糸状菌宿主株がアスペルギルス、トリコデルマ、ペニシリウムおよびフザリウム種からなる群より選ばれる、請求項32に記載の使用。
【請求項34】
アスペルギルス・オリゼ(Aspergillus oryzae)株より得られる単離されたジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼであって、
(a) 実質的に配列番号2のアミノ酸配列を有するジメチルアリル−シクロアセチル−L−トリプトファンシンターゼ;
(b) (a) の対立遺伝子変異体;および
(c) ジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼ活性を有する、(a)または(b)の断片
からなる群より選ばれる、単離されたジメチルアリル−シクロアセトアセチル−L−トリプトファンシンターゼ。
【請求項35】
1または複数のサイレント毒素遺伝子が除去されている、非相同ポリペプチドの発現に適当な変異体アスペルギルス細胞。
【請求項36】
前記1または複数の毒素遺伝子の全部または一部の不可逆的欠失または破壊により1または複数の毒素遺伝子が除去されている、請求項35に記載の変異体。
【請求項37】
前記アスペルギルス細胞がアスペルギルス亜群ユーロチウム、ケトサルトリア、スクレロクレイスタ、サトイア、ネオサルトリア、ヘミカーペンテレス、ペトロミセス、エメリセラおよびフェネリアからなる群より選ばれる、請求項35または36のいずれか一項に記載の変異体。
【請求項38】
前記毒素遺伝子が、シクロピアゾン酸、コウジ酸、3−ニトロプロピオン酸、エモジン、マルフォルミン、アフラトキシン、オクラトキシンおよびセカロン酸からなる群より選ばれる1または複数の毒素をコードする、請求項35〜37のいずれか一項に記載の変異体。
【請求項39】
問題の前記毒素遺伝子がアフラトキシンをコードする、請求項35〜38のいずれか一項に記載の変異体。
【請求項40】
前記毒素遺伝子がA.オリゼのアフラトキシンクラスターからの遺伝子であり、特にomtA, aflR, pksA, Nor-1, fas-beta, fas-alpha, vber-1, avnA, ord-2からなる群より選ばれる、請求項35〜39のいずれか一項に記載の変異体。
【請求項41】
親のアスペルギルス細胞がA.オリゼ、特にA.オリゼ A1560 (IFO 0417)である、請求項40に記載の変異体。
【請求項42】
前記アフラトキシン遺伝子がomtAおよびaflRから成る群より選ばれる、請求項41に記載の変異体。
【図1】

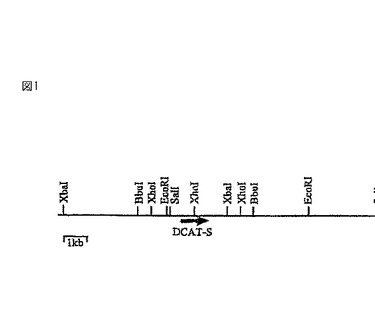
【図2】
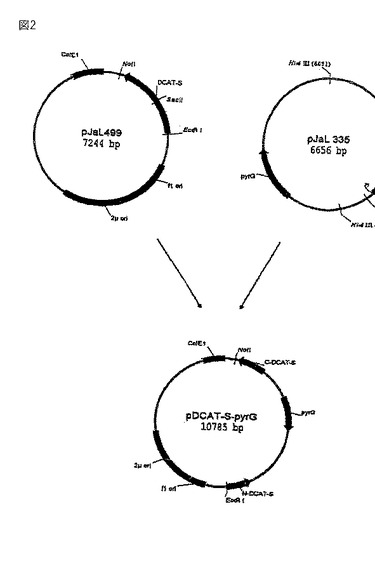
【図2】
【公開番号】特開2011−188863(P2011−188863A)
【公開日】平成23年9月29日(2011.9.29)
【国際特許分類】
【外国語出願】
【出願番号】特願2011−103808(P2011−103808)
【出願日】平成23年5月6日(2011.5.6)
【分割の表示】特願2000−591212(P2000−591212)の分割
【原出願日】平成11年12月22日(1999.12.22)
【出願人】(500586299)ノボザイムス アクティーゼルスカブ (164)
【Fターム(参考)】
【公開日】平成23年9月29日(2011.9.29)
【国際特許分類】
【出願番号】特願2011−103808(P2011−103808)
【出願日】平成23年5月6日(2011.5.6)
【分割の表示】特願2000−591212(P2000−591212)の分割
【原出願日】平成11年12月22日(1999.12.22)
【出願人】(500586299)ノボザイムス アクティーゼルスカブ (164)
【Fターム(参考)】
[ Back to top ]