
アセチルコリン受容体クラスター形成阻害活性を有するアグリンを特異的に認識する抗体並びに該抗体を含むアセチルコリン受容体クラスター形成能促進剤及び該抗体を充填したアセチルコリン受容体クラスター形成能を阻害するアグリン除去カラム
【課題】アセチルコリン受容体のクラスター形成能の障害により生じる病気の治療剤の開発を解決すべき課題とした。
【解決手段】アセチルコリン受容体クラスター形成能を阻害する機能を持つアグリンのスプライシング・バリアントが存在することを新規に見出した。
上記知見に基づいて、アセチルコリン受容体クラスター形成阻害活性を持つアグリンを特異的に認識する抗体が、アセチルコリン受容体クラスター形成能促進剤及びアセチルコリン受容体クラスター形成阻害活性を持つアグリン除去カラムに使用できることを見出した。
【解決手段】アセチルコリン受容体クラスター形成能を阻害する機能を持つアグリンのスプライシング・バリアントが存在することを新規に見出した。
上記知見に基づいて、アセチルコリン受容体クラスター形成阻害活性を持つアグリンを特異的に認識する抗体が、アセチルコリン受容体クラスター形成能促進剤及びアセチルコリン受容体クラスター形成阻害活性を持つアグリン除去カラムに使用できることを見出した。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、アセチルコリン受容体クラスター形成阻害活性を有するアグリンを特異的に認識する抗体並びに該抗体を含むアセチルコリン受容体クラスター形成能促進剤及び該抗体を充填したアセチルコリン受容体クラスター形成阻害活性を有するアグリン除去カラム、に関する。
【背景技術】
【0002】
運動ニューロンの軸索終末と骨格筋細胞との接合部は、神経筋接合部 (neuromuscular junction; NMJ) と呼ばれる高度に分化した生体構造である。活動電位が運動ニューロンの細胞体から軸索を伝わり神経終末に達すると、神経終末の電位依存性カルシウム (Ca2+) チャネル (voltage-gated calcium channel; VGCC) が開口し細胞外からCa2+ が流入し、シナプス小胞内のアセチルコリン (acetylcholine; ACh) がシナプス間隙に放出される。AChが終板に存在するニコチン性アセチルコリン受容体 (nicotinic acetylcholine receptor; nAChR) に結合すると、受容体に内蔵されるカチオンチャネルが開口し、興奮性シナプス後電位が生じる。この脱分極性膜刺激はさらに電位依存性Na+ チャネルを活性化し、活動電位が筋全体に広がる。活動電位は横行小管に存在するジヒドロピリジン受容体により感知され、筋小胞体膜に存在するリアノジン受容体が活性化されると、筋小胞体内の Ca2+ が細胞質に放出される。細胞内Ca2+ 濃度上昇によりアクチンやミオシン等の筋収縮関連タンパク質の相互作用が開始され、筋収縮が起こる。NMJはこれら一連の情報伝達の要となっている。
【0003】
NMJの分化の最も特徴的な現象は、nAChRがNMJ形成時にシナプス後膜に集合することである。この現象はクラスター形成と呼ばれ、nAChRの集合体はクラスターと呼ばれる。NMJの分化・形成には従来から様々な因子の関与が考えられている。なかでもシナプス後膜の分化が重要だと考えられ、それを裏付ける実験が多数報告されてきた。
【0004】
重症筋無力症 (Myasthenia gravis; MG) は、このNMJのシナプス後膜上に存在するタンパク質を標的とした自己免疫疾患である。その病態はシナプス機能障害による神経筋伝達阻害である。患者は骨格筋の易疲労性を訴え、休息後に軽快するのが臨床的特徴である。また、MG患者の80%で胸腺の腫瘍や過形成を伴うことから、胸腺の異常が発症に関与していると考えられている。MGは自己抗体の種類によって (1) 抗nAChR抗体陽性MG、(2) 抗筋特異的チロシンキナーゼ (muscle-specific tyrosine kinase; MuSK) 抗体陽性MG、および (3) 前記の抗体が検出されないdouble seronegative MGに分類される。MG患者の約80%が (1) に相当する。
【0005】
上記(1) における抗nAChR抗体の作用機序としては、AChとnAChRの結合阻害、nAChR崩壊促進、補体介在性シナプス後膜破壊などが考察されてきた。特に、補体介在性シナプス後膜破壊に伴うnAChR数の減少が病態の本質であると考えられている。
一方、上記(2)では抗nAChR抗体が検出されず、上記(1) と異なり抗MuSK抗体の大部分はIgG4サブクラスに属し、補体介在性シナプス後膜破壊を伴わない {Hoch, W., McConville, J., Helms, S., Newsom-Davis, J., Melms, A. and Vincent, A. (2001). Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat. Med. 7, 365−368.}。抗MuSK抗体がどのような機序でMG症状を引き起こしているのかは、現在不明である。
【0006】
MuSKは分子量110 kDaの膜貫通型リン酸化酵素で、筋膜上でnAChRと隣接している。このMuSKはアグリンの刺激によりリン酸化され、このリン酸化が下流に位置するラプシンを介してnAChRのクラスター形成を促進する。nAChRのクラスター形成を引き起こすアグリン-MuSK-ラプシン-nAChRシグナル伝達経路は様々なレベルの調節を受けていることが推測されているが、未だ解明されていない。
【0007】
また、アグリン又はMuSKに関する以下の特許文献が公開されている。
【0008】
特許文献1は、「ヒトアグリンの短縮型デルタ9部位が、MuSKレセプターのリン酸化を誘導すること」を開示している。
【0009】
特許文献2は、「アグリン由来物を投与することを特徴とするLAMA2遺伝子変異による先天的筋肉機能不全患者の治療方法」を開示している。
【0010】
特許文献3は、「アグリンの断片をマーカーとするニューロトリプシン関連疾患の診断方法」を開示している。
【0011】
しかしながら、上記文献では、アセチルコリン受容体のクラスター形成に関与するアグリン分子の構造と機能との関係は開示又は示唆されていない。
【先行技術文献】
【特許文献】
【0012】
【特許文献1】特表2000−504929号公報
【特許文献2】US7078379
【特許文献3】特開平5−304993号公報
【発明の概要】
【発明が解決しようとする課題】
【0013】
本発明の課題は、アセチルコリン受容体のクラスター形成能の障害により生じる病気の治療剤の開発を解決すべき課題とした。より詳しくは、アセチルコリン受容体のクラスター形成に関与するアグリン分子の構造の多様性と機能を解明し、アセチルコリン受容体のクラスター形成能の障害により生じる病気の新規な治療剤の開発を課題とした。
【課題を解決するための手段】
【0014】
本発明者らは、上記課題を解決するために、アグリンのスプライシング・バリアントのcDNAクローニングによる同定とその発現タンパク質の機能解析を行った。
該機能解析の結果、アセチルコリン受容体クラスター形成能を阻害する機能を持つアグリンのスプライシング・バリアントが存在することを新規に見出した。
そして、本発明者らは、上記知見に基づいて、アセチルコリン受容体クラスター形成阻害活性を持つアグリンを特異的に認識する抗体が、アセチルコリン受容体クラスター形成能促進剤及びアセチルコリン受容体クラスター形成阻害活性を持つアグリン除去カラムに使用できることを見出した。
【0015】
すなわち、本発明は以下の通りである。
1.SC2型アグリン及び/又はz0型アグリンを特異的に認識する抗体。
2.下記配列番号1で表されるアミノ酸配列を含むペプチドを免疫原として使用する前項1の抗体。
Glu-His-Trp-Gln-Pro-Arg-Ala-Glu-Thr(配列番号1)
3.下記の特徴を有する前項1又は2に記載の抗体。
(1)Glu-His-Trp-Gln-Pro-Arg-Ala-Glu-Thr(配列番号1)で表されるアミノ酸配列に1ないし数個のアミノ酸残基の欠失、置換、挿入もしくは付加、又は修飾を施すことにより得られるアミノ酸配列からなるペプチドを免疫原として使用する
(2)SC1型アグリン、z8型アグリン、z11型アグリン及び/又はz19型アグリンに結合しない
4.モノクローナル抗体である前項1〜3のいずれか1に記載の抗体。
5.前項1〜4のいずれか1に記載の抗体を含むアセチルコリン受容体クラスター形成能促進剤。
6.前記促進剤が、以下のいずれか1に記載の治療剤である前項5に記載のアセチルコリン受容体クラスター形成能促進剤。
(1)重症筋無力症
(2)筋萎縮性側索硬化症
(3)先天性筋ジストロフィー
(4)アルツハイマー病
(5)パーキンソン病
7.前項1〜4のいずれか1に記載の抗体を充填していることを特徴とするアセチルコリン受容体クラスター形成阻害活性を有するアグリン除去カラム。
8.前記カラムは、以下の治療用途であることを特徴とするカラム。
(1)重症筋無力症
(2)筋縮性側索硬化症
(3)先天性筋ジストロフィー
(4)アルツハイマー病
(5)パーキンソン病
9.以下の工程を含むz0型アグリン又はSC2型アグリンのアセチルコリン受容体クラスター形成能阻害活性を阻害する物質のスクリーニング方法;
(1)z0型アグリン及び/又はSC2型アグリン並びに候補阻害物質を細胞中に添加する工程、
(2)該細胞中のアセチルコリン受容体クラスター形成能を指標として、候補阻害物質の阻害活性を測定する工程。
【発明の効果】
【0016】
本発明者らは、従来知られていなかったアセチルコリン受容体クラスター形成阻害活性を持つアグリンの構造を基にして、該アグリンを特異的に認識できる抗体の提供を可能とした。さらに、該抗体を、アセチルコリン受容体クラスター形成能促進剤及びアセチルコリン受容体クラスター形成阻害活性を持つアグリン除去カラムに使用できる。
【図面の簡単な説明】
【0017】
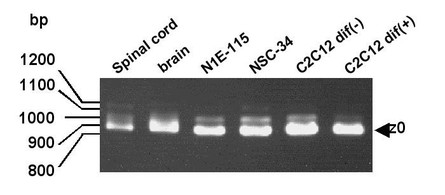
【図1】複数の試料についてのアグリンの(C)末端部位をPCRにより増幅した結果。図中の矢印は、通常のC末端長を有する典型的なz0型アグリンのバンドを示す。
【図2】各種のアグリン転写物のcDNA配列の比較図。
【図3】各種のアグリンのC末端アミノ酸配列の比較図。図中のY部位(KSRK)は二重の下線で示し、z8挿入部位(ELTNEIPA)は下線で示し、z11挿入部位(PETLDSRALFS)は、点下線で示す。各囲みは、各アグリン特有のアミノ酸配列を示す(例:アミノ酸配列EHWQPRAETは、SC2(short C-terminal variant-2)アグリンの特有の配列である)。
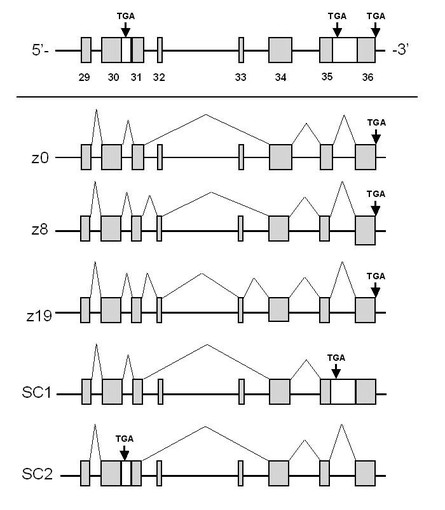
【図4】各種のアグリンmRNAの選択的スプライシングの模式図。図中の各ボックスは、エキソンを示す。
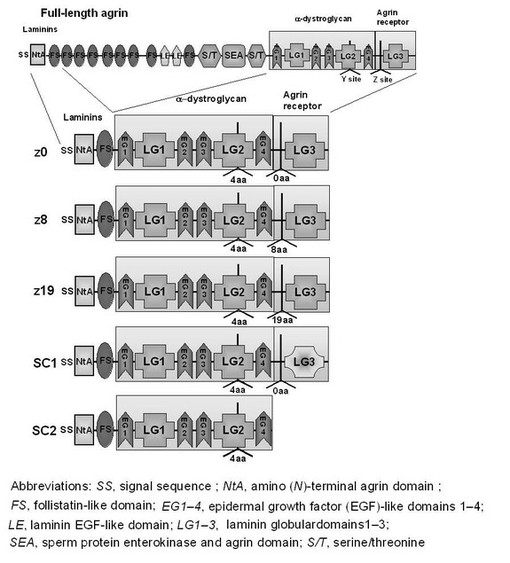
【図5】各種のアグリンのドメイン構造を示す模式図{最上位のアグリンは、フルアグリン(全長アグリン)であり、それ以外はミニアグリンを示す}。
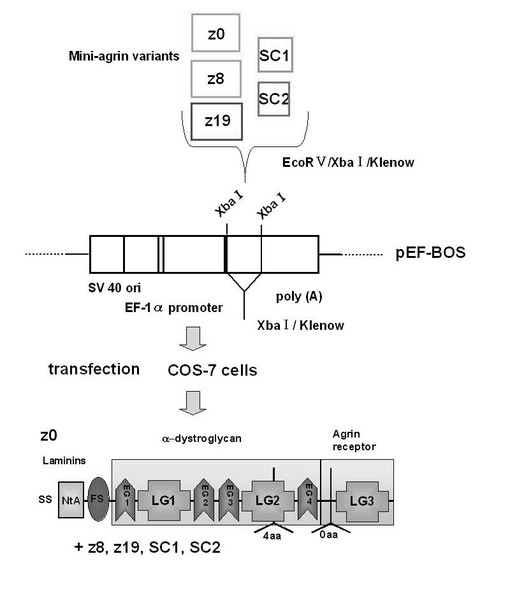
【図6】各種のミニアグリンの発現を示す模式図。
【図7】発現した各種のミニアグリンのイムノブロット解析の結果図。
【図8】nAChRクラスター形成の観察と評価結果。図中の矢印は、nAChRクラスターが形成されていることを示す。
【図9】z0型アグリン、SC2型アグリンのnAChRのクラスター形成阻害活性の評価。図中の矢印は、nAChRクラスターが形成されていることを示す。
【発明を実施するための形態】
【0018】
(アグリン)
アグリンは225 kDaのタンパク質をコアに持つヘパラン硫酸プロテオグリカンの一種であり、アミノ (N) 末端部と中央部に複数の推定糖鎖付加部位を持つ。アグリンは約40個のエクソンにコードされており、mRNAはオルタナティブ・スプライシングを受けて異なる翻訳開始点が生じる。このため49アミノ酸からなるshort N-terminal (SN) と、150アミノ酸からなるlong N-terminal (LN) の2種類のN末端を持つアグリンが産生される 。
さらにLNは、分泌に必須のシグナル配列 (signal sequence; SS) とラミニン結合領域であるN-terminal agrin domain (NtA) とを持つ分泌型、およびtransmembrane segment (TM) を持つ膜貫通型とに分けられる。
一方、C末端側にはX,Y,およびZのオルタナティブ・スプライシングによる可変部位が3箇所ある。Y部位への4個のアミノ酸挿入は、a-ジストログリカン (a-dystroglycan; a-DG) との親和性を高めると言われている。Z部位には、アミノ酸挿入がないz0 型に加えて、8、11、19個のアミノ酸が挿入されたz8,z11,z19型があり、z+型と総称されている (Ferns, M. J., Campanelli, J. T., Hoch, W., Scheller, R. H., and Hall, Z. (1993). The ability of agrin to cluster AChRs depends on alternative splicing and on cell surface)。
【0019】
本発明者らは、下記実施例1から明らかなように、上記既知の標準型C末端をもつz0,z8,z19型とは異なり、標準型よりも短いC末端をコードすることが推定される2種類のバリアントを新たに見出した。これらをSC1 (short C-terminal variant-1)、SC2 (short C-terminal variant-2) 型アグリンと命名した(参照:図3)。
なお、過去の報告と本実施例で明らかされた、オルタナティブ・スプライシングの様式を下記表1に示す。アグリン分子の多様性は,N末端,X部位,Y部位,Z部位,およびC末端の少なくとも5か所で生じる可変的な領域数の掛け算によって創出されていると考えられる。
さらに、本発明者らは、下記実施例2から明らかなように、SC2型アグリン及びz0型アグリンが、アセチルコリン受容体クラスター形成阻害活性を持つことを、新たに見出した。
【0020】
【表1】

【0021】
(アセチルコリン受容体クラスター形成阻害活性を持つアグリンを特異的に認識する抗体)
本発明の{アセチルコリン受容体クラスター形成阻害活性を持つアグリンを特異的に認識する抗体(以下、「本発明の抗体」と称する場合がある)}とは、SC2型アグリン及び/又はz0型アグリンを特異的に認識する抗体を意味し、特に限定されない。
さらに、前記抗体は、好ましくは、公知のz8型アグリン、z19型アグリン、及び/又は新規のSC1型アグリンには結合しない。
加えて、前記抗体は、ポリクローナル抗体、ポリクローナル抗体を含む抗血清又はモノクローナル抗体のいずれのタイプのものをも含み、また、これらの抗体のフラグメント{Fab、F(ab′)2又はFab′等}をも含むものである。なお、好ましくは、前記抗体は、モノクローナル抗体であることが好ましい。さらに、前記抗体は、自体公知の方法により、ヒト化抗体にすることが好ましい。
【0022】
{本発明の抗体作製に使用する免疫原(抗原)}
本発明の抗体を産出させるための免疫原として、哺乳動物由来、特に好ましくはヒト又はマウス由来のSC2型アグリン及び/又はz0型アグリンに特有のアミノ酸配列、より詳しくは、z8型アグリン、z11型アグリン、z19型アグリン、及び/又はSC1型アグリンには存在しないアミノ酸配列の全部又は一部を含むペプチドを用いることができる。
なお、下記実施例1から明らかなように、SC2型アグリンは、他のアグリンにない特有の以下のアミノ酸配列(配列番号1:参照図3)を有する。よって、好ましくは、以下のアミノ酸配列を含むペプチドを免疫原として使用することができる。
Glu-His-Trp-Gln-Pro-Arg-Ala-Glu-Thr(配列番号1)
さらに、Glu-His-Trp-Gln-Pro-Arg-Ala-Glu-Thr(配列番号1)で表されるアミノ酸配列に1ないし数個のアミノ酸残基の欠失、置換、挿入もしくは付加、又は修飾を施すことにより得られるアミノ酸配列からなるペプチドを免疫原として使用することもできる。
【0023】
(免疫原となるペプチドの合成)
ペプチドの製造は、遺伝子工学的手法、化学合成、および無細胞タンパク質合成により実施できる。ペプチドは、製造された後に、さらに精製して用いることができる。
【0024】
ペプチドの製造は、該ペプチドをコードする遺伝子の塩基配列情報又はアミノ酸配列に基づいて一般的な遺伝子工学的手法{サムブルック(Sambrook)ら編、「モレキュラークローニング,ア ラボラトリーマニュアル 第2版」、1989年、コールドスプリングハーバーラボラトリー;村松正實編、「ラボマニュアル遺伝子工学」、1988年、丸善株式会社;ウルマー(Ulmer, K.M.)、「サイエンス(Science)」、1983年、第219巻、p. 666-671;エールリッヒ(Ehrlich, H.A.)編、「PCRテクノロジー,DNA増幅の原理と応用」、1989年、ストックトンプレス}により実施できる。
【0025】
ペプチドの製造は、また、一般的な化学合成法により製造できる。ペプチドの化学合成方法として、例えば、固相合成方法や液相合成方法等が知られているがいずれも利用できる。
【0026】
ペプチドの精製および/または分離は、その物理的性質、化学的性質等を利用した各種分離操作方法により実施できる。分離操作方法として、硫酸アンモニウム沈殿、限外ろ過、ゲルクロマトグラフィー、イオン交換クロマトグラフィー、アフィニティークロマトグラフィー、高速液体クロマトグラフィーおよび透析法等の公知の方法を例示できる。これら方法は単独でまたは適宜組合せて使用できる。
【0027】
(免疫方法)
上記記載の免疫原となる精製したペプチド又は部分ペプチドを、リン酸緩衝液(PBS)などの適当な緩衝液中に溶解あるいは懸濁したものを抗原液として使用する。抗原液は通常抗原物質を50〜500μg/mL程度含む濃度に調製すればよい。また、ペプチド単独だけでは抗原性が低い場合には、アルブミンやキーホールリンペットヘモシアニン(KLH)などの適当なキャリアータンパク質に架橋して用いることができる。
当該抗原で免疫感作する動物(被免疫動物)は、マウス、ラット、ハムスター、ウマ、ヤギ、ウサギなどが例示される。好ましくはマウス、より好ましくはBALB/cマウスである。
【0028】
上記被免疫動物の抗原への応答性を高めるため、前記抗原溶液をアジュバントと混合して投与することができる。ここで使用可能なアジュバントは、フロイント完全アジュバント(FCA)、フロイント不完全アジュバント(FIA)、Ribi(MPL)、Ribi(TDM)、Ribi(MPL+TDM)。百日咳ワクチン(Boredetella pertussis vaccine)、ムラミルジペプチド(MDP)、アルミニウムアジュバント(ALUM)、およびこれらの組合せが例示されるが、初回免疫時にFCA、追加免疫時にFIAやRibiアジュバントを使用する組合せが特に好ましい。
【0029】
免疫方法は、使用する抗原の種類やアジュバント混合の有無などにより、注射部位、スケジュールなどを適宜変化させることができるが、例えば、被免疫動物としてマウスを用いる場合は、アジュバント混合抗原液0.05〜1ml(抗原物質10〜200μg)を腹腔内、皮下、筋肉内または(尾)静脈内に注射し、初回免疫から約4〜21日毎に1〜4回追加免疫を行い、さらに約1〜4週間後に最終免疫を行う。抗原量を多くして腹腔内注射することで、当該抗原溶液をアジュバントを使用せずに投与することもできる。抗体価は追加免疫の約5〜10日後に採血して調べる。抗体価の測定は、後述の抗体価アッセイに準じ、通常行われる方法で行うことができる。最終免疫より約3〜5日後、該免疫動物から脾細胞を分離して抗体産生細胞を得る。
【0030】
(モノクローナル抗体の作製)
モノクローナル抗体(以下、「MoAb」と略する場合がある)は、自体公知の方法、例えばケーラーとミルシュタインの方法(Kohler G, Milstein C. (1975) Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256, 495−497.)にしたがって作製することができる。
例えば、免疫動物から抗体産出細胞を含む組織(例えば、脾臓又はリンパ節)を回収し、該抗体産出細胞と自体抗体の腫瘍細胞(例えば、骨腫瘍細胞)とを融合させることによってハイブリドーマを作製し、次いでハイブリドーマをクローン化した後、所望の抗体を産出しているハイブリドーマを選別し、このハイブリドーマの培養液から抗体を回収する。
骨髄腫細胞として、マウス、ラット、ヒトなど由来のものが使用され、例えばマウスミエローマP3X63-Ag8、P3X63-Ag8-U1、P3NS1-Ag4、SP2/o-Ag14、P3X63-Ag8・653などの株化骨髄腫細胞が例示される。骨髄腫細胞には免疫グロブリン軽鎖を産生しているものがあり、これを融合対象として用いると、抗体産生細胞が産生する免疫グロブリン重鎖とこの軽鎖とがランダムに結合することがあるので、特に免疫グロブリン軽鎖を産生しない骨髄腫細胞、例えばP3X63-Ag8・653やSP2/o-Ag14などを用いることが好ましい。
抗体産生細胞と骨髄腫細胞とは、同種動物、特に同系統の動物由来であることが好ましい。骨髄腫細胞の保存方法は自体公知の手法に従って行えばよく、例えばウマ、ウサギもしくはウシ胎児血清を添加した一般的な培地で継代培養したものについて凍結により保存される。また細胞融合には対数増殖期の細胞を用いるのが好ましい。
【0031】
(ハイブリドーマの作製)
抗体産生細胞と骨髄腫細胞とを融合させてハイブリドーマを作製する方法は、ポリエチレングリコール(PEG)を用いる方法、センダイウイルスを用いる方法、電気融合装置を用いる方法などが例示される。例えばPEG法の場合、約30〜60%のPEG(平均分子量1,000〜6,000)を含む適当な培地または緩衝液中に脾細胞と骨髄腫細胞を1〜10:1、好ましくは5〜10:1の混合比で懸濁し、温度約25〜37℃、pH6〜8の条件下で、約30秒〜3分間程度反応させればよい。反応終了後、細胞を洗浄しPEG溶液を除いて培地に再懸濁し、マイクロタイタープレート中に播種して培養を続ける。
融合操作後の細胞は選択培地で培養して、ハイブリドーマの選択を行う。選択培地は、親細胞株を死滅させ、融合細胞のみが増殖しえる培地であり、通常ヒポキサンチン−アミノプテリン−チミジン(HAT)培地が使用される。ハイブリドーマの選択は、通常融合操作の1〜7日後に、培地の一部、好ましくは約半量を選択培地と交換し、さらに2、3日毎に同様の培地交換を繰り返しながら培養することにより行う。顕微鏡観察によりハイブリドーマのコロニーが生育しているウエルを確認する。
【0032】
生育しているハイブリドーマが所望の抗体を産生しているかどうかを知るには、培養上清を採取して抗体価アッセイを自体公知の方法により行えばよい。
さらに限界希釈法、軟寒天法、蛍光励起セルソーターを用いた方法などにより単一クローンを分離する。
【0033】
ハイブリドーマが産生する抗体の免疫グロブリンサブクラスを調べるためには、該ハイブリドーマを一般的な条件で培養し、その培養上清中に分泌された抗体を市販の抗体クラス・サブクラス判定用キットなどを用いて分析することにより知ることができる。
【0034】
(モノクローナル抗体の取得方法)
ハイブリドーマからのMoAbの取得方法は、必要量やハイブリドーマの性状などによって適宜選択することができる。例えば、該ハイブリドーマを移植したマウス腹水から取得する方法、細胞培養により培養上清から取得する方法などが例示される。マウス腹腔内で増殖可能なハイブリドーマであれば、腹水から数mg/mLの高濃度のMoAbを得ることができる。インビボで増殖できないハイブリドーマは細胞培養の培養上清から取得する。
細胞培養によるMoAbの取得は、抗体産生量はインビボより低いが、マウス腹腔内に含まれる免疫グロブリンや他の夾雑物質の混入が少なく、精製が容易であるという利点がある。
抗体を、ハイブリドーマを移植したマウス腹腔内から取得する場合、例えば、予めプリスタン(2, 6, 10, 14-テトラメチルペンタデカン)などの免疫抑制作用を有する物質を投与したBALB/cマウスの腹腔内へハイブリドーマ(約106個以上)を移植し、約1〜3週間後に貯留した腹水を採取する。異種ハイブリドーマ(例えばマウスとラット)の場合には、ヌードマウス、放射線処理マウスを使用することが好ましい。
細胞培養上清から抗体を取得する場合、例えば、細胞維持に用いられる静置培養法の他に、高密度培養方法あるいはスピンナーフラスコ培養方法などの培養法を用い、当該ハイブリドーマを培養し抗体を含有する培養上清を得る。
腹水や培養上清からのMoAbの精製は、自体公知の方法により行うことができる。例えば、免疫グロブリンの精製法として従来既知の硫酸アンモニウムや硫酸ナトリウムを用いた塩析による分画法、ポリエチレングリコール分画法、エタノール分画法、DEAEイオン交換クロマトグラフィー法、ゲル濾過法などを応用することで、容易に達成される。
さらに、MoAbが、マウスIgGである場合には、プロテインA結合単体あるいは抗マウスイムノグロブリン結合単体を用いたアフィニティークロマトグラフィー法により精製することが可能であり、簡便である。
【0035】
{z0型アグリン又はSC2型アグリンのアセチルコリン受容体クラスター形成能阻害活性を阻害する物質のスクリーニング方法}
本発明のスクリーニング方法では、少なくとも以下の工程を含む。
(1)z0型アグリン及び/又はSC2型アグリン並びに候補阻害物質を培養した細胞中に添加する工程
細胞は、該細胞中のアセチルコリン受容体クラスター形成能を測定できる細胞であれば特に限定されない。
(2)該細胞中のアセチルコリン受容体クラスター形成能を指標として、候補阻害物質の阻害活性を測定する工程
候補阻害物質並びにz0型アグリン及び/又はSC2型アグリンを添加した細胞中のアセチルコリン受容体クラスター形成能が、z0型アグリン及び/又はSC2型アグリンを添加した細胞中のアセチルコリン受容体クラスター形成能と比較して、高い場合には、該候補阻害物質は、z0型アグリン又はSC2型アグリンのアセチルコリン受容体クラスター形成能阻害活性を阻害する物質、言い換えれば、アセチルコリン受容体クラスター形成を促進することができる物質(アセチルコリン受容体クラスター形成促進物質)である。
なお、アセチルコリン受容体クラスター形成能の測定方法は、下記実施例2を参照することができる。
【0036】
(候補阻害物質)
上記スクリーニングで使用する候補阻害物質としては任意の物質を使用することができる。候補阻害物質の種類は特に限定されず、個々の低分子合成化合物特にsiRNAでもよいし、天然物抽出物中に存在する化合物でもよく、合成ペプチドでもよい。あるいは、候補阻害物質はまた、化合物ライブラリー、ファージディスプレーライブラリーもしくはコンビナトリアルライブラリーでもよい。候補阻害物質は、好ましくは低分子化合物であり、低分子化合物の化合物ライブラリーが好ましい。化合物ライブラリーの構築は当業者に公知であり、また市販の化合物ライブラリーを使用することもできる。
【0037】
(アセチルコリン受容体クラスター形成能促進剤)
本発明の「アセチルコリン受容体クラスター形成能促進剤」とは、アセチルコリン受容体クラスター形成を抑制、阻害及び/又は低下させる機能を持つSC2型アグリン及び/又はz0型アグリンの作用を阻害若しくはこれらのアグリンと結合することにより、アセチルコリン受容体クラスター形成を促進させる。
本発明の促進剤の特徴は、従来のアセチルコリン産出量を増大させる薬剤若しくはアセチルコリン分解酵素阻害剤とは明らかにメカニズムが異なる。従来のアセチルコリン産出量を増大させる薬剤では、患者の体内中のアセチルコリン産出量を増大させる若しくはアセチルコリンの分解を阻害することができても、アセチルコリン受容体が少なければ(クラスターが形成されていなければ)、アセチルコリンがアセチルコリン受容体(ニコチン性アセチルコリン受容)に結合できず、受容体に内蔵されるカチオンチャネルが開口できず、興奮性シナプス後電位が生じなかった。
しかし、本発明の促進剤は、従来の上記薬剤とはメカニズムが異なり、アセチルコリン受容体クラスターの形成能を促進させるので、少ないアセチルコリン産出量の患者においても、少ないアセチルコリンが効率的にアセチルコリン受容体に結合することができ、受容体に内蔵されるカチオンチャネルが開口し、興奮性シナプス後電位を生じさせることができる。
さらに、本発明の促進剤は、従来のアセチルコリン産出量を増大させる薬剤若しくはアセチルコリン分解酵素阻害薬と併用又は組み合わせることにより、優れた効果、特に従来の薬剤では効果がない患者に有用であると考えられる。
【0038】
本発明の促進剤は、上記記載のアセチルコリン受容体クラスター形成阻害活性を持つアグリンを特異的に認識する抗体及び/又は上記記載のスクリーニング方法により得られたアセチルコリン受容体クラスター形成促進物質を有効成分として含む。
さらに、治療等(予防も含む)の目的に応じて、散剤、顆粒剤、錠剤、カプセル剤、腸溶剤、液剤、注射剤(液剤、懸濁剤)または遺伝子療法に用いる形態などの各種の形態に、常法にしたがって調製することができる。
加えて、充填剤、増量剤、結合剤、付湿剤、崩壊剤、滑沢剤、希釈剤および賦形剤も含むことができる。その他、安定化剤、殺菌剤、緩衝剤、等張化剤、キレート剤、界面活性剤、およびpH調整剤等を適宜使用することもできる。
【0039】
本発明の促進剤の投与量または摂取量については、本発明の効果が得られるものであれば特に限定されるものではなく、含有される成分の有効性、投与形態、投与経路、疾患の種類、対象の性質(体重、年齢、病状および他の医薬の使用の有無等)、および担当医師の判断等に応じて適宜選択される。本発明の促進剤は、1日1〜数回に分けて投与または摂取することができ、数日または数週間に1回の割合で間欠的に投与または摂取してもよい。
【0040】
(本発明の促進剤の用途)
本発明の促進剤は、アセチルコリンの減少、アセチルコリン受容体のクラスター形成能の低下が関与する疾患、例えば、以下の疾患の治療剤とすることができる。
(1)重症筋無力症
(2)筋萎縮性側索硬化症
(3)先天性筋ジストロフィー
(4)アルツハイマー病
(5)パーキンソン病
【0041】
(アセチルコリン受容体クラスター形成阻害活性を持つアグリン除去カラム)
本発明の「アセチルコリン受容体クラスター形成阻害活性を持つアグリン除去カラム」とは、上記記記載のアセチルコリン受容体クラスター形成阻害活性を持つアグリンを特異的に認識する抗体が充填されている。より詳しくは、SC2型アグリン及び/又はz0型アグリンを特異的に認識する抗体が充填されたカラムである。
上記抗体を吸着する担体としては、特に限定されないが、セルロース系ゲル、デキストラン系ゲル、アガロース系ゲル、ポリアクリルアミド系ゲル、多孔質ガラス、ビニルポリマーゲル等の有機または無機の多孔体が使用でき、通常のアフィニティクロマトグラフィーに用いられる担体を使用することができる。
加えて、上記抗体を担体に結合及び/又は吸着させる方法としては、自体公知の物理的吸着法、化学的結合法又はこれらの併用等の公知の方法により行うことができる。
さらに、必要に応じて担体と上記抗体との間に任意の長さの分子(スペーサー)を導入することもできる。
非特異的反応等を抑制するために、抗体を固相化させた担体の表面又は内壁面に、ウシ血清アルブミン等、界面活性剤又は脱脂粉乳等を接触させ被覆させること等の公知の方法により処理して、ブロッキング処理を行ってもよい。
【0042】
本発明のカラムを体外循環用カラムとして用いる場合には、大きく次の二通りの方法がある。一つには、体内から取り出した血液を遠心分離機もしくは膜型血漿分離器を使用して、血漿成分と血球成分とに分離した後、血漿成分を該カラムに通過させ、SC2型アグリン及び/又はz0型アグリンを除去した後、血球成分と合わせて体内にもどす方法であり、他の一つは、体内から取り出した血液を直接該カラムに通過させ、SC2型アグリン及び/又はz0型アグリンを除去する方法である。
なお、重症筋無力症の治療方法の一つとして、単純血漿交換療法により、血漿中の抗アセチルコリン受容体抗体を除去する療法がある。本発明のカラムを用いた療法では、該、重症筋無力症の単純血漿交換療法を参考にして行うこともできる。
【0043】
(本発明のカラムの用途)
本発明のカラムは、アセチルコリンの減少、アセチルコリン受容体のクラスター形成能の低下が関与する疾患、例えば、以下の疾患の治療に使用することができると考えられる。
(1)重症筋無力症
(2)筋萎縮性側索硬化症
(3)先天性筋ジストロフィー
(4)アルツハイマー病
(5)パーキンソン病
【0044】
{重症筋無力症(MG)}
MGでは抗nAChR抗体によって引き起こされる補体介在性シナプス後膜破壊と、それに伴うnAChR数の減少が病態の原因であり、これにより神経筋伝達障害が起きている。
すなわち、本発明の促進剤をMG患者に投与すること又は本発明のカラムによりMG患者からSC2型アグリン及び/又はz0型アグリンを除去することにより、少ないnAChRの発現量でも、アセチルコリンが効率的にnAChRに結合することができるので、重症筋無力症の治療効果があると考えられる。
【0045】
{筋萎縮性側索硬化症(ALS)}
筋萎縮性側索硬化症は、運動ニューロンの変性や萎縮、骨格筋の麻痺を主徴とする進行性神経変性病の一つである。また、ALSのモデルマウスであるSODマウスでは病態の末期においてアグリン発現量の低下が見られることが報告されている(Dobrowolny, G., Giacinti, C., Pelosi, L., Nicoletti, C., Winn, N., Barberi, L., Molinaro, M., Rosenthal, N., and Musaro, A. (2005). Muscle expression of a local Igf-1 isoform protects motor neurons in an ALS mouse model. J. Cell Biol. 168, 193-199)。
すなわち、本発明の促進剤をALS患者に投与すること又は本発明のカラムによるALS患者からSC2型アグリン及び/又はz0型アグリンを除去することにより、少ないアグリンの発現量でも、アセチルコリンが効率的にnAChRに結合することができるので、筋萎縮性側索硬化症の治療効果があると考えられる。
【0046】
{先天性筋ジストロフィー(congenital muscular dystrophy; CMD)}
先天性筋ジストロフィーは乳幼児期に筋力低下、関節拘縮、運動発達遅滞で発症し、筋病理で壊死と再生所見を認める筋ジストロフィーの総称で、中枢神経症状を合併する福山型 (Fukuyama CMD; FCMD)と、中枢神経症状を呈さない非福山型 (non-FCMD) に大別されている。non-FCMDの中には筋線維に発現するラミニンのα2鎖を指令する遺伝子LAMA2の変異によって起きるものがあり、CMDのモデルマウスはヒトのラミニン2欠損型先天性筋ジストロフィーと似た症状を呈する。この疾患での筋線維の変性は、基底膜構造に必要な1次ラミニン骨格の形成ができず、筋基底膜とジストロフィン-糖タンパク質複合体 (dystrophin-glycoprotein complex; DGC) との結合、または筋基底膜とインテグリン類との結合が失われるためと考えられている。このCMDモデルマウスにz-型のミニ・アグリンcDNAを組み込んだアデノ随伴ウイルスを腹腔内注射すると、ミニ・アグリンが基底膜およびDGCの一要素であるα-DGに結合し、ミニ・アグリンが仲介するα-DGとラミニンのα5鎖の安定化機構によって、筋病態を改善したという報告がある (Moll, J., Barzaghi, P., Lin, S., Bezakova, G., Lochmuller, H., Engvall, E., Muller, U., and Ruegg, M. A. (2001). An agrin minigene rescues dystrophic symptoms in a mouse model for congenital muscular dystrophy. Nature 413, 302-307.)。
すなわち、本発明の促進剤をCMD患者に投与すること又は本発明のカラムによるCMD患者からSC2型アグリン及び/又はz0型アグリンを除去することにより、少ないアグリンの発現量でも、アセチルコリンが効率的にnAChRに結合することができるので、先天性筋ジストロフィーの治療効果があると考えられる。
【0047】
アルツハイマー病は、中枢性アセチルコリン受容体伝達障害が原因であることが知られており。パーキンソン病は、中枢性ドーパミン伝達障害が原因であることが知られている。よって、本発明の促進剤をアルツハイマー病又はパーキンソン病患者に投与すること又は本発明のカラムによる該患者の血液からSC2型アグリン及び/又はz0型アグリンを該患者から除去することにより、治療効果があると考えられる。
【0048】
以下、本発明を実施例により具体的に説明する。しかし、本発明は下記実施例に限定されるものではない。
【実施例1】
【0049】
(アグリンのスプライシング・バリアントのcDNAクローニング及びそのタンパク質の発現)
本実施例では、哺乳動物の例としてマウス神経組織と神経筋組織に由来する種々の培養細胞株を用いてアグリンのスプライシング・バリアントのcDNAクローニングによる同定とそのタンパク質発現実験を行った。詳細は、以下の通りである。
【0050】
(1)細胞培養
マウス筋芽細胞由来培養細胞株C2C12は、American Type Culture Collection (Manassas, VA, USA) より購入した。マウス運動ニューロン由来細胞株NSC-34は、田平武博士 (国立長寿医療センター研究所) より供与を受けた。C2C12細胞、NSC-34細胞およびサル腎線維芽細胞由来COS-7細胞の維持培養液には、Dulbecco's Modified Eagle Medium (DMEM; GIBCO BRL, Grand Island, NY, USA) に10%ウシ胎児血清 (fetal bovine serum; FBS; GIBCO BRL)、ペニシリン100 units/ml (GIBCO BRL) 及びストレプトマイシン100 μg/ml (GIBCO BRL) を加えて使用した。継代時には培養液を吸引除去し、付着細胞をMg2+, Ca2+含有ハンクス平衡緩衝塩溶液 (Hanks' balanced salt solution; HBSS; GIBCO BRL) で洗浄し、0.25%トリプシン、1 mMエチレンジアミン四酢酸を含むMg2+, Ca2+不含HBSS (GIBCO BRL) を加え、CO2インキュベーター (37℃,5% CO2; BNA-111; TABAI ESPEC, 大阪) 内で10分間反応させた。
剥離した細胞を回収後、1500 rpm,20℃で5分遠心した。上清を吸引除去した後、新たな培養液に浮遊させ、細胞数を5×104cells/ml に調製し、そのうち5 mlを培養フラスコに播種し、CO2インキュベーター (37℃,5% CO2) で培養した。培養液は3日ごとに交換し、1週間で継代した。
マウス神経芽細胞腫×マウス線維芽細胞雑種細胞株N1E-115細胞 (Amano et al., 1972) は、DMEMに最終濃度が5%ウシ胎児血清 (fetal calf serum; FCS; GIBCO BRL)、1 mM ヒポキサンチン、0.16 mM d-チミジン、0.01 mM アミノプテリンになるように加えた培養液を用いて継代培養した。
上記の培養に加えて,C2C12細胞の分化誘導も行った。継代後2日目に培養液を吸引除去し、分化誘導用に調製した2%ウマ血清 (horse serum; HS; GIBCO BRL) を含むDMEMと置き換えた。さらに4日間、CO2インキュベーター (37℃,5% CO2) で培養した。
【0051】
(2)Total RNAの抽出
成体マウス (C57BL/6,BALB/c) をネンブタール腹腔内注射により深く麻酔し、頚椎脱臼の後、頚動脈をはさみで切り脱血させた。摘出した脳、脊髄をMg2+, Ca2+不含リン酸緩衝生理食塩水{phosphate-buffered saline (−); PBS(−)}}で洗浄し、組織片30 mgに600 μlのLysis Solutionを加え、テフロンホモジナイザーを用いて組織懸濁液を得た。 また、NSC-34およびN1E-115細胞は、60 mm培養皿 (IWAKI) にそれぞれ3×104cells/mlで細胞をまき、CO2インキュベーター (37℃,5% CO2)で4日間培養後にPBS (−) で洗浄し細胞を回収した。未分化型C2C12細胞は、60 mm培養皿で1×104cells/mlで細胞をまいて3日間培養し、同様に細胞を回収した。分化型C2C12細胞は、未分化型と同様に細胞をまき、3日間の通常培養と4日間の分化誘導後に回収した。いずれの細胞も、Lysis Solution 350 μlを加えて溶解した。
上記の組織懸濁液と細胞溶解液からtotal RNA 抽出/精製 Kit Mini (Agilent Technologies CA, USA) を使用して以下の通りにtotal RNAを抽出した。
Lysis Solution 350 μlを加えて溶解した組織懸濁液と細胞溶解液全量をMini Prefiltrationで遠心 (16000 × g,3分,20℃) して、上清 350 μlを70% エタノール (Amresco, OH, USA) 350 μlと混合した。エタノール/Lysis Solution 混合液700 μlをMini Isolation Columnで遠心 (16000 × g,30秒,20℃) して濾過液を廃棄し、Wash Solution 500 μlを用いて遠心 (16000 × g,30秒,20℃) してカラムの洗浄を2度行い、遠心 (16000 × g,2分,20℃) した。蒸留水 (distilled water; DW) 30 μlを加え1分間静置した。16000 × g,1分,20℃で遠心にかけRNAを溶出した。
得られたサンプルに混入したDNAを除くためにDNA分解処理を行った。反応液は全量50 μlで、各Total RNAサンプル0.4-1 μg、40 mM Tris-HCl (pH 7.5)、8 mM MgCl2、5 mM ジチオトレイトール、DNase I 2.5 unitを含んだ。37℃、10分間の反応後、フェノール・クロロホルム処理、ジエチルエーテル処理し、エタノール沈殿した。RNAは最終的にDW 11μlに溶解し、UV分光光度計 (Nano Drop Technologies) を用いて濃度を測定した。
【0052】
(3)逆転写酵素‐ポリメラーゼ連鎖反応
C57BL/6マウスの脊髄、BALB/cマウスの全脳、N1E-115細胞、NSC-34細胞、未分化型C2C12細胞および分化型C2C12細胞から抽出したtotal RNAから、Transcriptor First Strand cDNA Synthesis Kit (Roche Applied Science, Mannheim, Germany) を使用して相補DNA (complementary DNA; cDNA) を合成した。
反応液は全量20 μlで、各total RNA 0.4-1 μg,50 mM Tris-HCl (pH 8.5),30 mM KCl,8 mM MgCl2,60 μM Random Hexamer Primer,RNase Inhibitor 20 unit,10 mM dNTPs,AMV (Avian myeloblastosis virus) 逆転写酵素10 unitを混ぜて、25℃で10分、さらに50℃で1 時間反応させ、DW 180 μlを加えて使用時まで−80 °Cで保存した。
マウス・アグリン遺伝子の塩基配列に基づいて4種類のプライマー (AgAFw,AgBFw,AgCRv,及びAgDRv; 参照:表2) を合成した。調整した各cDNAを鋳型として、1 × 添付のbuffer,0.4 mM dNTPs,0.3 μM AgAFwプライマー,0.3 μM AgDRvプライマー,cDNAサンプル1 μl,KOD FX DNAポリメラーゼ1.0 unitを含む反応液でPCRを施行した。94℃で2分間熱変性の後、熱変性 (98℃,10秒)、アニーリング (60.4℃,30秒)、伸長 (68℃,1分) の反応サイクルを20回繰り返した。さらに,得られた反応産物1 μlを再度同じ組成の反応液に加えて同様の反応を25サイクル行った。得られたPCR産物は1%アガロースゲルの電気泳動で分離し、エチジウムブロマイド溶液 (1 μg/ml) にて染色後、UVサンプル撮影装置 (FAS-III; TOYOBO, 大阪) を用いて観察、記録した。
【0053】
【表2】
【0054】
(4)cDNAクローンの単離
上記(3)のRT-PCRの反応産物を電気泳動し、0.9-1.1 kbのDNA断片をGENECLEAN II Kit (MP Biomedicals LLC, Irvine, CA, USA) を用いてゲルから回収した。精製したDNA断片は、rTaq (TaKaRa, 滋賀) を含む溶液中{10 mM Tris-HCl (pH 8.3),50 mM KCl,1.5 mM MgCl2,0.25 mM dNTPs,0.25 μM A1 AgBFw,0.25 μM AgCRvプライマー,rTaq 0.25 unit,cDNAサンプル30 μl}で72℃、10分間反応させ、A付加反応を施した。この反応液からMin Elute PCR Purification Kit (QIAGEN) を用いてDNA断片を精製し、DW 10 μlに溶解した。回収したDNA断片1 μlにSalt Solution (Invitrogen, Carlsbad, CA, USA ) 0.5 μl、pCR 2.1-TOPO (Invitrogen) 0.5 μlを混ぜ、室温で30分、続いて4℃で6時間連結反応を行った。さらに、各反応液に大腸菌TOP10株コンピテントセル25 μlを加え氷上で30分間、続いて42℃で30秒反応させた後、SOC培地250 μlを加え37℃で1時間振とう培養した。
この溶液を5-bromo-4-chloro-3-indolyl-β-D-galactoside (X-gal; 8 mg/ml) を塗布したアンピシリン (50 μl/ml) 含有LB寒天培地に60 μlずつ播いて37℃で12時間培養した。
次に、出現したコロニーから白色のものを選び、cDNAが組込まれていることを確認するため、プラスミドを以下の通りに抽出した。白色コロニーをアンピシリン (50 μl/ml) 含有LB液体培地5 mlを加えたチューブに入れ、37℃,200 rpmで振とう培養した。12時間後、培養液を3000 rpm、10分遠心して集菌し、得られた菌体をRNase I (10 mg/ml) を含むBuffer P1 250 μlで懸濁し、別のチューブに移した。そこに、Buffer P2 250 μlを加えて転倒混和し、直後にBuffer N3 350 μlを加えて再度転倒混和した後、13000 rpmで10分間遠心した。得られた上清をQIAprep spin columnに移し13000 rpmで1分間遠心し、濾液を除去した。この後、Buffer PB 500 μlを加えて13000 rpmで1分間遠心し濾液を捨て、Buffer PE 750 μlを加えて13000 rpmで1分間遠心し濾液を捨て、さらに13000 rpmで1分間遠心し残液を除いた。QIAprep spin columnを別のチューブに移してDW 50 μl加え室温で1分間静置し、13000 rpm,1分間の遠心でプラスミド溶液を得た。DNA濃度はUV分光光度計 (Nano Drop Technologies) にて測定した。
【0055】
(5)塩基配列決定
抽出した各プラスミドDNA 300 ngをABI PRISM BigDye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) 添付の1 × Big Dye ver1.1 Reaction Mix, 1 × Big Dye Sequence Buffer, 0.8 μM 合成プライマー用いて、ダイデオキシ法により行った。反応産物をBigDye XTerminatorTM 精製キット (Applied Biosystems) により精製後、ABI PRISM 3130 Genetic Analyzer (Applied Biosystems) を用い、Sequence Scanner v1.0,DNA Sequencing AnalysisソフトウェアVersion 5.1を用いて塩基配列を決定した。
【0056】
(6)データベース解析
日本DNAデータバンク (DNA Data Bank of Japan; DDBJ) のホームページにアクセスし、BLAST version 2. 2. 18を用いた。
【0057】
(7)タンパク質発現用プラスミドの作製
(7−1)ミニ・アグリン (mini-agrin) cDNAの部分断片の調製
N1E-115細胞由来cDNA 1 μlを鋳型として、KOD FX DNAポリメラーゼ (TOYOBO) とそれぞれA1 AgEFw,AgGRvプライマーを用いて熱変性: 98℃,10秒、アニーリング: 62.2℃,30秒,伸長: 68℃,1分の各条件で35サイクル,B1 AgHFw,AgKRvプライマーを用いて熱変性: 98℃,10秒,アニーリング: 54.4℃,30秒,伸長: 68℃,1分の各条件で35サイクル,C1 AgBFw,AgNRvプライマーを用いて熱変性: 98℃,10秒,アニーリング: 57.3℃,30秒,伸長: 68℃,1分間の各条件で35サイクルPCRを行い、増幅したA1フラグメント 743 bp,B1フラグメント1275 bp,C1フラグメント879 bpをMin Elute PCR Purification Kit (QIAGEN, Hilden, Germany) を用いて精製した。
この精製したPCR産物全量についてrTaq (TaKaRa) を用いて調製した溶液 {10 mM Tris-HCl (pH 8.3),50 mM KCl,1.5 mM MgCl2,0.25 mM dNTPs,各0.25 μMの特異的プライマー,rTaq 0.25 unit,cDNAサンプル30 μl}}で72℃,10分間反応させ、各反応産物を再度Min Elute PCR Purification Kit (QIAGEN) を用いて精製し、DW 10 μlに溶解した。 AgEFw,AgGRvプライマー,AgHFw,AgKRvプライマー,AgBFw,AgNRvプライマーの各組合せで得られたDNA断片を本発明ではA1,B1,C1とした。
精製したA1,B1,C1の各DNA断片1 μlとSalt Solution (Invitrogen) 0.5 μl,pCR 2.1-TOPO (Invitrogen) 0.5 μlを混ぜ、室温で30分,4℃で6時間連結反応を行った。各連結反応産物に大腸菌TOP10株コンピテントセル25 μlを加え、氷上で30分間反応させた。その後42℃で30秒反応させ、SOC培地250 μlを加えて37℃で1時間振とう培養した。滅菌コンラージ棒でアンピシリン (50 μl/ml) 含有Luria-Bertani (LB) 寒天培地にX-gal (8 mg/ml) を塗りインキュベーターで乾かした。アンピシリン (50 μl/ml) 含有LB寒天培地に60 μlずつ播いて37 °Cで12時間培養した。プラスミドDNAは、上記(4)と同様に調製し (A2,B2,C2) とした。
【0058】
(7−2)ベクターの調製
pBluescript II KS (+) (pBSII KS(+); Stratagene) は、37℃、2時間EcoRVとXhoI,XhoIとBamHI,あるいはXbaIとSacIで消化した。切断されたプラスミドを、37℃,1時間バクテリア・アルカリホスファターゼ (bacterial alkaline phosphatase; BAP) で処理し、1%アガロースゲルで泳動後DEAEペーパーに吸着させて回収し、フェノール・クロロホルム処理、ジエチルエーテル処理、エタノール沈殿の後、50 μlのDWに溶解し、ベクターとして用いた。
pCR2.1-TOPO (Invitrogen) は、37℃、2時間XbaIとSacIで消化した。切断したプラスミドを、37℃、1時間BAP処理し、1%アガロースゲルで泳動した。DEAEペーパーに吸着させて回収し、フェノール・クロロホルム処理、ジエチルエーテル処理、エタノール沈殿の後、50 μlのDWに溶解し、これをベクターとして用いた。
pcDNA3.1 (+) (Invitrogen) は、37℃、2時間EcoRVとNotIで消化し、切断されたプラスミドを、37℃、1時間BAP処理し、1%アガロースゲルで泳動した。DEAEペーパーに吸着させ回収し、フェノール・クロロホルム処理、ジエチルエーテル処理、エタノール沈殿の後、50 μlのDWに溶解し、これをベクターとして用いた。
哺乳動物細胞発現用プラスミドpEF-BOS (京都大学大学院医学系研究科長田重一教授より供与された)は、37℃,1時間XbaIで消化し、その後37℃で1時間BAP処理した。フェノール・クロロホルム処理、ジエチルエーテル処理、エタノール沈殿の後、50 μlのDWに溶解した。このうちの20μlに、50 mM Tris-HCl (pH 7.5),100 mM NaCl,10 mM MgCl2,1 mM DTT,250 μM dNTPs,0.01 units/μl Klenow fragment of DNA polymerase Iとなるように加えて37℃、20分間反応させた。1%アガロースゲルで泳動した後、GENECLEAN II Kit (MP Biomedicals LLC) を用いて回収し、これをベクターとして用いた。
【0059】
(7−3)ミニ・アグリンcDNAの構築
A2,B2,C2の各プラスミドDNAを100℃で10分間反応させ、このうち1 μlを鋳型としてA2プラスミドをT7とAgGRvプライマーで、B2プラスミドをAgHFwとM13revプライマーあるいはT7とAgKRvプライマーで、C2プラスミドをAgBFwとM13revプライマーでアニーリング温度52.2℃,20サイクルの条件で KOD FX ポリメラーゼ (TOYOBO) を用いて増幅し、Min Elute PCR Purification Kit (QIAGEN) を用いて精製し、DW 15.4 μlに溶解した。得られたDNA断片を順にA3,B3,B3',C3とした。
A3,B3,B3',C3の末端リン酸化を、1×Protruding End Kinase Buffer、T4 polynucleotide kinase 0.5-2 units/μl,1 mM ATPを含む反応液中で37℃で30分間行い、65℃,5分間の加熱によって停止させた。ここで得られたDNA断片をA4,B4,B4',C4とした。A4 1 μlとB4 1 μl,B4' 1 μlとC4 1 μlの組み合わせで混合し、それぞれにMighty mix 2 μl (TaKaRa) を加えて16℃で30分間連結反応を行い、反応産物をD1,E1とした。
さらに、D1を鋳型にT7とAgJRvプライマーで、E1を鋳型にT7とM13revプライマーでPCRを行った。アニーリング温度52℃、35サイクルの条件でKOD FXポリメラーゼ(TOYOBO) を用いた。反応液を1%アガロースゲルで電気泳動し、D1由来の1741 bp付近のバンド、E1由来の2344 bp付近のバンドをGENECLEAN II Kit (MP Biomedicals LLC) を用いて回収し、それぞれD2,E2とした。D2は制限EcoRVとXhoIで、E2はXhoIとBamHIでそれぞれ消化し、1%アガロースゲルで電気泳動後、D2由来の1153 bp付近の断片、E2由来の1781 bp付近の断片をGENECLEAN II Kitで回収し、D3,E3とした。
D3断片1.5 μl、E3断片1.5 μlは、上記(7−2)で調製したEcoRVとXhoIで切断したpBSII-KS (+) 1 μl (25 ng)、XhoIとBamHIで切断したpBS2-KS (+) 1 μl (25 ng) とそれぞれ混ぜ、それぞれにMighty mix 2.5 μl (TaKaRa) を加えて連結反応を16℃、30分間行い、反応産物をそれぞれD4,E4とした。このD4,E4に50 mM MgCl2/10 mM CaCl2を100 μlと大腸菌SCS1株コンピテントセル100 μlを加えて、氷上で30分間反応させた。
その後42℃で1分間反応させ、SOC培地を250 μl加えて37℃で1時間反応させた。この溶液をアンピシリン (50 μl/ml) 含有LB寒天培地に60 μlずつ播いて、37℃で12時間培養した。得られたD4,E4の各コロニーからPlasmid DNA purification QIAprep spin Miniprep Kit (QIAGEN) を用いてプラスミド抽出し、このプラスミドをD5,E5とした。
D5をXbaIとXhoIで、E5をSacIとXhoIで消化し、1%アガロースゲルで電気泳動し、D5由来の1181 bp付近の断片、E5由来の947 bp付近の断片をGENECLEAN II Kit (MP Biomedicals LLC) で回収し、それぞれD6,E6とした。D6断片1 μl、E6断片1 μl、及び上記(7−2)で作製したXbaIとSacIで切断したpCR2.1 vector 1 μl (25 ng) を混ぜ、さらにMighty mix 3 μl (TaKaRa) を加え、16℃で30分間連結反応を行い、反応産物をF1とした。このF1サンプルに大腸菌DH5α株のコンピテントセル (TaKaRa) 20 μlを加えて、氷上で30分間反応させ、42℃で、45秒間反応後、SOC培地を200 μl加えて37℃、200 rpmで1時間振とう培養した。この溶液をX-gal (8 mg/ml) 塗布アンピシリン (50 μl/ml) 含有LB寒天培地に60 μlずつ播いて37℃で12時間培養した。得られたF1のコロニーをPlasmid DNA purification QIAprep spin Miniprep Kit (QIAGEN) を用いてプラスミド抽出し、これをF2 とした。
F2プラスミドをEcoRVとSacIで消化し、1%アガロースゲルで電気泳動し、2092 bp付近の断片をDEAEペーパーに吸着した。これを1.5 M NaCl/10 mM Tris-HCl(pH 7.5)で溶出し、フェノール・クロロホルム抽出後、ジエチルエーテル処理、エタノール沈殿し、50 μlのDWに溶解した。得られたDNA断片をF3とした。
C末端側バリアントの各DNA断片をSacIとNotIで消化し、1%アガロースゲルで電気泳動した後、851-1108 bpの各バンドをGENECLEAN II Kit (MP Biomedicals LLC) で回収した。それぞれのフラグメント1 μl、事前に作製したF3 1 μl、及びEcoRVとNotIとで切断したpcDNA 3.1ベクター1 μl (25 ng) を混ぜ、そこにMighty mix 3 μl (TaKaRa) を加えて混ぜて合わせ、16℃で30分間連結反応を行い、この反応産物をG1とした。このG1サンプルに大腸菌DH5α株のコンピテントセル (TaKaRa) 20 μlを加えて、氷上で30分間反応させ、42℃で45秒間反応後、SOC培地を200 μl加えて37℃、200 rpmで1時間振とう培養した。これをX-gal (8 mg/ml) を塗布したアンピシリン (50 μl/ml) 含有LB寒天培地に60 μlずつ播いて37℃で12時間培養した。得られたG1のコロニーをQIAprep spin Miniprep Kit (QIAGEN) を用いてプラスミド抽出し、これ発現プラスミドとした。
さらに,これらpcDA3.1 (+) に組込まれたcDNAを、さらにEcoRVとXhoIで切り出し、Klenow fragment of DNA polymerase I で末端を平滑化し、上記(7−2)で調製したpEF-BOSベクターに組込み、発現用プラスミドとして併用した。
【0060】
(8)遺伝子導入
COS-7細胞を1×105cells/mlに調製し、そのうち2 mlを組織培養用6ウエルプレート (IWAKI) に播種し,CO2インキュベーター (37℃,5% CO2) で維持した。継代培養して1,2日後に培養液を吸引除去し、同量のDMEM-10% FBSに置き換えた。DMEM 97 μlとFuGENE 6 Transfection Reagent (Roche Applied Science) 3 μlを混ぜ、室温で5分間放置した後、上記(7−3)の各種ミニ・アグリンcDNAを組込んだ発現プラスミド 2 μgを混ぜ、室温で60分間反応させた。この混合液を1ウエルにつき100 μl加え、CO2インキュベーター (37℃,5% CO2) で培養した。
【0061】
(9)発現タンパク質のイムノブロット解析
遺伝子導入から24時間後に、培養液を同量のFCS不含DMEMに置き換えた。さらに24時間CO2インキュベーター (37℃,5% CO2) で培養した後、上清を回収し、1500 rpm、20℃で5分間遠心した。得られた上清をさらに Microcon YM-10 (Millipore, Billerica, MA, USA) を用いて濃縮し、イムノブロットのサンプルとした。これを等量の2 × サンプルBuffer (Tris Base 282 mM,Tris-HCl 212 mM,146 nM lithium dodecyl sulfate,2.18 M Glycerol,1.02 mM EDTA,100 mM DTT,0.44 mM Serva Blue G250,0.35 mM Phenol Red,pH 8.4; Invitrogen) と混合し、95℃で5分加熱した。各サンプル5-15 μl (50 ng-3 μg) を4-12%SDSポリアクリルゲル電気泳動 (NuPAGE 4-12% Bis-Tris GelおよびNuPAGE MOPS SDS Running Buffer; Invitrogen) にて、200 Vで50分泳動した。泳動後のタンパク質はPVDFメンブレン (Invitrogen) に30 Vで60分かけて転写した。転写後のメンブレンは、3% BSAを含むトリス緩衝生理食塩水 (Tris-buffered saline; TBS; 20 mM Tris,150 mM NaCl, pH 7.5) に浸して室温で1時間ブロッキングした。続いて、0.05% Tween 20 (Bio-Rad Laboratories, CA, USA) を含むTBS (TTBS) でメンブレンを10分間ずつ3回洗浄した後、ウサギ抗アグリンポリクローナル抗体 (sc-25528; SANTA CRUZ BIOTECHNOLOGY, Santa Cruz, CA, USA) を1% BSAを含むTTBS (1% BSA-TTBS) で500倍希釈した溶液を用いて1次抗体反応を行った。4℃で1晩反応させた後、メンブレンを10分ずつ3回洗浄した後、HRP標識ヤギ抗ウサギIgG抗体 (MP Biomdicals, Inc.-Cappel Products, Costa Mesa, CA, USA) を50,000倍希釈した溶液で2次抗体反応を行った。室温で1時間反応させた後、メンブレンを10分間ずつ3回洗浄し、ECL Plus (GE Healthcare UK Ltd., Buckinghamshire, England) 試薬と室温で5分反応させた。シグナルの検出には、LAS-4000UVmini (Fujifilm, 東京) を使用した。検出方法はchemiluminescence,露出方法はprecisionに設定した。
【0062】
(アグリン・バリアントのcDNAクローニングの結果)
C57BL/6脊髄、BALB/c全脳、N1E-115細胞、NSC-34細胞、未分化型C2C12細胞、分化型C2C12細胞からtotal RNAを抽出し、RT-PCRを行った。プライマーには、既知のアグリンmRNAの翻訳領域と3'側非翻訳領域の塩基配列に基づいて合成した。初回に使用したプライマーのセット (AgAFwとAgCRv;参照:表2) では非特異的増幅が高頻度で認められたため、さらに1組のプライマー(AgBFwとAgDRv; 参照:表2) を使用し、ネステッドPCRを行った。PCR産物を1%アガロースゲルで電気泳動したところ、全てのサンプルで800-1200 bpの範囲の複数のバンドが検出された (図1)。
増幅されたDNA断片の塩基配列を決定するために、PCR産物を組織、細胞ごとにアガロースゲルから回収し、pCR2.1-TOPOベクターに組込んだ。大腸菌コンピテントセルを形質転換し、X-galを塗布した寒天培地上で得られた白色コロニー6個を1つのプールとし、プラスミドDNAを抽出してPCRを行った。既知のz0型のアグリンから予想される916 bpよりも長い増幅産物を与えるプールを選択し、さらに個別のコロニーを同様に検索して目的とするcDNAクローンを得た。このような過程を経て最終的に、C57BL/6マウス脊髄から9個、BALB/cマウス全脳から7個、N1E-115細胞から6個、NSC-34細胞から2個、未分化型C2C12細胞から2個、分化型C2C12細胞から2個のcDNAクローンが得られた。挿入されたcDNAの塩基配列を決定し、BLAST version 2. 2. 18を用いてホモロジー検索を行った結果、いずれの配列も既知のマウス・アグリンと少なくとも部分的に一致した。
【0063】
(アグリンmRNAの多様性の確認)
C57BL/6脊髄由来のPCR産物には、Z部位にエクソン挿入の無いz0型、24 bpのエクソン挿入のあるz8型、57 bpのエクソン挿入のあるz19型のcDNAクローンが含まれていた。これらの塩基配列は、既にHochらの報告{Hoch, W., Ferns, M., Campanelli, J. T., Hall, Z. W., and Scheller, R. H. (1993). Developmental regulation of highly active alternatively spliced forms of agrin. Neuron 11, 479-490.}しているスプライシング・バリアントと一致した。これらに加えて、新たな2種類のバリアントを見出した。一方のバリアントでは、これまでに35番目のイントロンがスプライシングを受けずに残存していた。このmRNAの場合、異なる終止コドンが現れるため、翻訳領域が既知のz0型よりも102 bp短くなるものであった (図4)。もう一方のバリアントは、30番目のイントロンの残存により途中で終止コドンが現れ、C末端側の翻訳領域が既知のz0型より543 bp短くなるものであった。これらのmRNAから予想されるタンパク産物は既知のアグリンのC末端と比較して86アミノ酸、233アミノ酸ずつ短くなることが推定された。特にSC2では、LG3ドメイン全体が欠落することが予想された (図3及び5)。本発明では、これらのタイプをSC1 (short C-terminal variant-1)、SC2 (short C-terminal variant-2) 型と命名した。さらにこれ以外にも、Z部位に24 bpエクソン挿入があり、かつイントロン残存のあるz8-SC2型のcDNAクローンを単離した。
またBALB/c 全脳由来のPCR産物からは、Z部位にエクソン挿入の無いz0型、57 bpのエクソン挿入のあるz19型、イントロン残存のあるSC2型、Z部位に57 bpエクソン挿入があり、かつイントロン残存のあるz19-SC2型を同定した。N1E-115細胞由来のPCR産物からZ部位にエクソン挿入の無いz0型、イントロン残存のあるSC2型を同定した。NSC-34細胞由来のPCR産物からZ部位にエクソン挿入の無いz0型を同定した。未分型C2C12細胞由来のPCR産物からZ部位にエクソン挿入の無いz0型、エクソン挿入が無くかつイントロン残存のあるz0-SC1型を同定した。分化型C2C12由来のPCR産物からZ部位にエクソン挿入の無いz0型を同定した (図2及び4)。
【0064】
(タンパク質発現結果)
今回得られたアグリン分子のスプライシング・バリアントがどのような生理機能に関与するのかを調べるために、タンパク質発現系を確立した。従来の研究報告に基づいて、N末端がLN型になるようにすると同時に、神経細胞接着分子やヘパリン結合成長因子が結合する8つのFSドメイン (follistatin-like domain)、2つのLEドメイン (laminin EGF-like domain)、1つのSEAドメイン (sperm protein enterokinase and agrin domain)、2つのS/T部位 (serine/threonine sites) を除いたミニ・アグリン構造をとるように、発現プラスミドを構築した (図5)。N1E-115細胞から抽出したtotal RNAからRT-PCRによって得られた3つのDNA断片をつなぎ合わせて、原報通りのミニ・アグリンのN末端側cDNAを発現プラスミドpEF-BOS に組込んだ。さらに、このC末端側にあたる z0,z8,z19,SC1,SC2型のcDNAとをつなぎ合わせることにより、各バリアントの最終的な発現プラスミドを構築した (図6)。
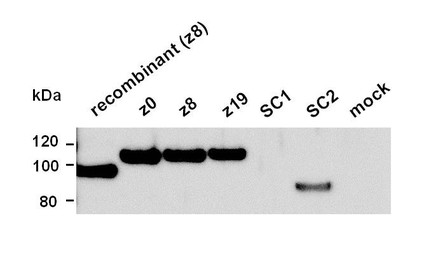
次に、ミニ・アグリンcDNAを組込んだプラスミドをCOS-7細胞に導入し、24-48時間後の培養上清を回収し、分泌型のミニ・アグリンが作られているか、イムノブロット法により検出を試みた。その結果、z0,z8,z19,SC2型のサンプルについて単一のバンドを検出した(図7)。z0,z8,z19,SC2型のミニ・アグリンの推定分子量は、それぞれ109 kDa,110 kDa,111 kDa,84 kDaであり、図7の各バンドから計測した分子量,113 kDa,113 kDa,116 kDa,88 kDaに近い値であった。
【実施例2】
【0065】
(各アグリンのアセチルコリン受容体クラスター形成能の評価)
上記実施例1で発現させた各アグリンのアセチルコリン受容体クラスター形成能又は阻害活性を確認した。詳細は、以下の通りである。
【0066】
(nACRクラスター形成の確認方法)
C2C12細胞を5×104cells/mlに調製し、組織培養用6ウエルプレート (IWAKI) の各ウエルに2 mlずつ播種し、CO2インキュベーター (37℃,5% CO2) で培養した。24時間後に2% HS-DMEMに交換し、4日間培養して分化させた。myotubeが確認されたウエルに0.2% BSA-PBS(−) に溶解した組換え型アグリン (Rat C-terminal Agrin; R&D SYSTEMS, Minneapolis, MN, USA) 単独、COS-7細胞の培養上清に分泌された各種ミニ・アグリン単独、又は組換え型アグリン (R&D SYSTEMS) とCOS-7細胞の培養上清に分泌された各種ミニ・アグリンとの混合物を添加した。陰性対照として、それぞれの溶解希釈液を添加した。
添加15時間後に培養上清を除去し、PBS(−)で洗浄後、3.7%ホルムアルデヒド (16% ultrapure formaldehyde methanol free; Polysciences, Eppelheim, Germany)-PBSを加え、細胞を室温で10分間固定した。1% BSA-PBS(−)で1分間ずつ3回洗浄した後、蛍光標識されたa-ブンガロトキシン (a-bungarotoxin; a-BuTX, Alexa Fluor 488 conjugate; Molecular Probes, Eugene, OR, USA) を最終濃度100 nMになるように添加し、遮光して室温で1時間反応させた。1% BSA-PBS(−)で5分間ずつ2回洗浄した後Propong Gold antifade regent (Molecular Probes) を添加して封入した。蛍光観察には、20倍対物レンズ、NIBAフィルターを装着した倒立型顕微鏡 (IX 71; OLYMPUS, 東京) を使用した。画像の取り込みには、DP71デジタルカメラ (OLYMPUS) と専用ソフトウェア (DP ControllerとDP Manager; OLYMPUS) を使用した。評価方法は、Alexa 488のシグナルが直径10 μm以上のもをnAChRクラスターと定義して数えた。各ウエルにつき5視野ずつ撮影し、視野毎にmyotube 1本あたりのnAChRクラスター数を計算し、1ウエルあたりの平均値を算出した。mock-trasfectionで得られた値を100%として、それに対する各サンプルの割合を百分率に換算した。有意差検定はTukey-Kramer検定により、有意水準を1%とした。
【0067】
(各アグリンのアセチルコリン受容体クラスター形成能の評価)
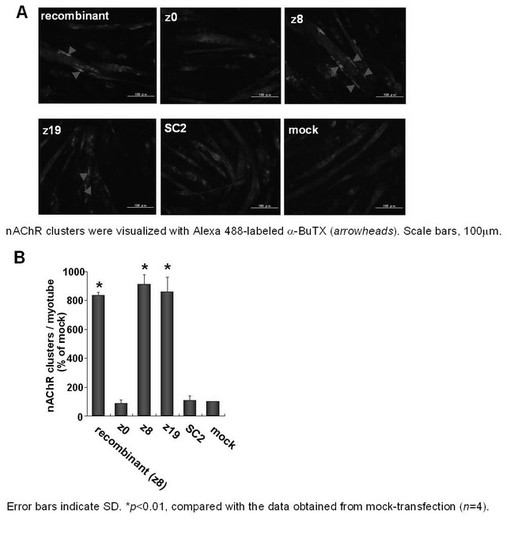
COS-7細胞培養上清から回収したミニ・アグリンバリアントを分化誘導したC2C12細胞に添加した。添加15時間後にAlexa 488で蛍光標識されたa-BuTX を用いてnAChRの視覚化を試みた。クラスターはnAChRが集合した直径10μm以上の集合体と定義した。組換え型アグリン、z8型、及びz19型のミニ・アグリンを添加したC2C12細胞では、nAChRのクラスター形成が明瞭に観察された(図8A)。myotubeあたりのクラスター数は、mock-transfectionの値を100%とした場合、それぞれ835±21% (n=4),911±64% (n=4),859±103% (n=4) の値を示した(図8B)。これに対して、z0,SC2型のミニ・アグリンを添加したC2C12細胞では、ベクターのみ導入 (mock-transfection) したCOS-7細胞の上清と同様、nAChRのクラスター形成はほとんど観察されなかった(図8A)。myotubeあたりのクラスター数は、mock-transfectionと比べて、それぞれ89±21% (n=4)、106±30% (n=4) であった(図8B)。
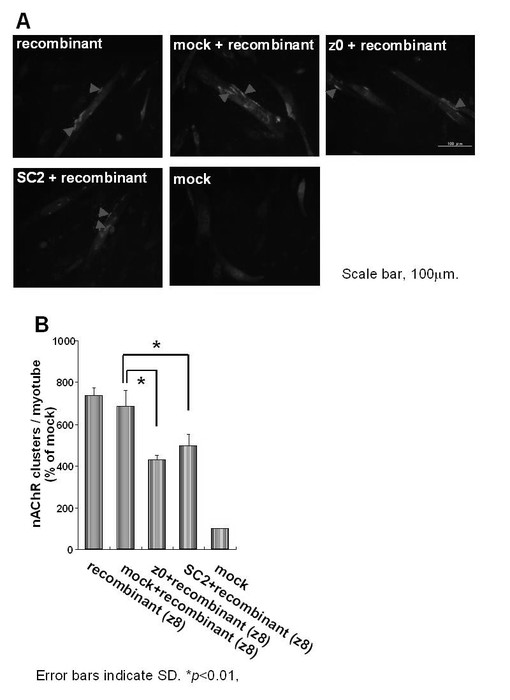
次に、z0,SC2型のnAChRのクラスター形成阻害活性を検討した。mock-transfectionと組換え型アグリンとの混合物を添加した値686±77% (n=4) と比較して、z0型と組換え型アグリンとの混合物、及びSC2型と組換え型アグリンとの混合物ではそれぞれ430±24% (n=4)、498±55% (n=4) の値を示し、nAChRクラスター形成の有意な減少を確認した (p<0.01; 図9)。
【0068】
(総論)
上記実施例の結果より、z0型アグリン及びSC2型アグリンは、アセチルコリン受容体クラスター形成阻害活性を有することを確認した。これにより、z0型アグリン及び/又はSC2型アグリンを特異的に認識する抗体は、z0型アグリン及び/又はSC2型アグリンのアセチルコリン受容体クラスター形成阻害活性を阻害できると考えられる。
【産業上の利用可能性】
【0069】
本発明者らは、アセチルコリン受容体クラスター形成阻害活性を持つアグリンを特異的に認識できる抗体の提供を可能とした。さらに、該抗体は、アセチルコリン受容体クラスター形成能促進剤及びアセチルコリン受容体クラスター形成能を阻害するアグリン除去カラムに使用できる可能性がある。
【技術分野】
【0001】
本発明は、アセチルコリン受容体クラスター形成阻害活性を有するアグリンを特異的に認識する抗体並びに該抗体を含むアセチルコリン受容体クラスター形成能促進剤及び該抗体を充填したアセチルコリン受容体クラスター形成阻害活性を有するアグリン除去カラム、に関する。
【背景技術】
【0002】
運動ニューロンの軸索終末と骨格筋細胞との接合部は、神経筋接合部 (neuromuscular junction; NMJ) と呼ばれる高度に分化した生体構造である。活動電位が運動ニューロンの細胞体から軸索を伝わり神経終末に達すると、神経終末の電位依存性カルシウム (Ca2+) チャネル (voltage-gated calcium channel; VGCC) が開口し細胞外からCa2+ が流入し、シナプス小胞内のアセチルコリン (acetylcholine; ACh) がシナプス間隙に放出される。AChが終板に存在するニコチン性アセチルコリン受容体 (nicotinic acetylcholine receptor; nAChR) に結合すると、受容体に内蔵されるカチオンチャネルが開口し、興奮性シナプス後電位が生じる。この脱分極性膜刺激はさらに電位依存性Na+ チャネルを活性化し、活動電位が筋全体に広がる。活動電位は横行小管に存在するジヒドロピリジン受容体により感知され、筋小胞体膜に存在するリアノジン受容体が活性化されると、筋小胞体内の Ca2+ が細胞質に放出される。細胞内Ca2+ 濃度上昇によりアクチンやミオシン等の筋収縮関連タンパク質の相互作用が開始され、筋収縮が起こる。NMJはこれら一連の情報伝達の要となっている。
【0003】
NMJの分化の最も特徴的な現象は、nAChRがNMJ形成時にシナプス後膜に集合することである。この現象はクラスター形成と呼ばれ、nAChRの集合体はクラスターと呼ばれる。NMJの分化・形成には従来から様々な因子の関与が考えられている。なかでもシナプス後膜の分化が重要だと考えられ、それを裏付ける実験が多数報告されてきた。
【0004】
重症筋無力症 (Myasthenia gravis; MG) は、このNMJのシナプス後膜上に存在するタンパク質を標的とした自己免疫疾患である。その病態はシナプス機能障害による神経筋伝達阻害である。患者は骨格筋の易疲労性を訴え、休息後に軽快するのが臨床的特徴である。また、MG患者の80%で胸腺の腫瘍や過形成を伴うことから、胸腺の異常が発症に関与していると考えられている。MGは自己抗体の種類によって (1) 抗nAChR抗体陽性MG、(2) 抗筋特異的チロシンキナーゼ (muscle-specific tyrosine kinase; MuSK) 抗体陽性MG、および (3) 前記の抗体が検出されないdouble seronegative MGに分類される。MG患者の約80%が (1) に相当する。
【0005】
上記(1) における抗nAChR抗体の作用機序としては、AChとnAChRの結合阻害、nAChR崩壊促進、補体介在性シナプス後膜破壊などが考察されてきた。特に、補体介在性シナプス後膜破壊に伴うnAChR数の減少が病態の本質であると考えられている。
一方、上記(2)では抗nAChR抗体が検出されず、上記(1) と異なり抗MuSK抗体の大部分はIgG4サブクラスに属し、補体介在性シナプス後膜破壊を伴わない {Hoch, W., McConville, J., Helms, S., Newsom-Davis, J., Melms, A. and Vincent, A. (2001). Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat. Med. 7, 365−368.}。抗MuSK抗体がどのような機序でMG症状を引き起こしているのかは、現在不明である。
【0006】
MuSKは分子量110 kDaの膜貫通型リン酸化酵素で、筋膜上でnAChRと隣接している。このMuSKはアグリンの刺激によりリン酸化され、このリン酸化が下流に位置するラプシンを介してnAChRのクラスター形成を促進する。nAChRのクラスター形成を引き起こすアグリン-MuSK-ラプシン-nAChRシグナル伝達経路は様々なレベルの調節を受けていることが推測されているが、未だ解明されていない。
【0007】
また、アグリン又はMuSKに関する以下の特許文献が公開されている。
【0008】
特許文献1は、「ヒトアグリンの短縮型デルタ9部位が、MuSKレセプターのリン酸化を誘導すること」を開示している。
【0009】
特許文献2は、「アグリン由来物を投与することを特徴とするLAMA2遺伝子変異による先天的筋肉機能不全患者の治療方法」を開示している。
【0010】
特許文献3は、「アグリンの断片をマーカーとするニューロトリプシン関連疾患の診断方法」を開示している。
【0011】
しかしながら、上記文献では、アセチルコリン受容体のクラスター形成に関与するアグリン分子の構造と機能との関係は開示又は示唆されていない。
【先行技術文献】
【特許文献】
【0012】
【特許文献1】特表2000−504929号公報
【特許文献2】US7078379
【特許文献3】特開平5−304993号公報
【発明の概要】
【発明が解決しようとする課題】
【0013】
本発明の課題は、アセチルコリン受容体のクラスター形成能の障害により生じる病気の治療剤の開発を解決すべき課題とした。より詳しくは、アセチルコリン受容体のクラスター形成に関与するアグリン分子の構造の多様性と機能を解明し、アセチルコリン受容体のクラスター形成能の障害により生じる病気の新規な治療剤の開発を課題とした。
【課題を解決するための手段】
【0014】
本発明者らは、上記課題を解決するために、アグリンのスプライシング・バリアントのcDNAクローニングによる同定とその発現タンパク質の機能解析を行った。
該機能解析の結果、アセチルコリン受容体クラスター形成能を阻害する機能を持つアグリンのスプライシング・バリアントが存在することを新規に見出した。
そして、本発明者らは、上記知見に基づいて、アセチルコリン受容体クラスター形成阻害活性を持つアグリンを特異的に認識する抗体が、アセチルコリン受容体クラスター形成能促進剤及びアセチルコリン受容体クラスター形成阻害活性を持つアグリン除去カラムに使用できることを見出した。
【0015】
すなわち、本発明は以下の通りである。
1.SC2型アグリン及び/又はz0型アグリンを特異的に認識する抗体。
2.下記配列番号1で表されるアミノ酸配列を含むペプチドを免疫原として使用する前項1の抗体。
Glu-His-Trp-Gln-Pro-Arg-Ala-Glu-Thr(配列番号1)
3.下記の特徴を有する前項1又は2に記載の抗体。
(1)Glu-His-Trp-Gln-Pro-Arg-Ala-Glu-Thr(配列番号1)で表されるアミノ酸配列に1ないし数個のアミノ酸残基の欠失、置換、挿入もしくは付加、又は修飾を施すことにより得られるアミノ酸配列からなるペプチドを免疫原として使用する
(2)SC1型アグリン、z8型アグリン、z11型アグリン及び/又はz19型アグリンに結合しない
4.モノクローナル抗体である前項1〜3のいずれか1に記載の抗体。
5.前項1〜4のいずれか1に記載の抗体を含むアセチルコリン受容体クラスター形成能促進剤。
6.前記促進剤が、以下のいずれか1に記載の治療剤である前項5に記載のアセチルコリン受容体クラスター形成能促進剤。
(1)重症筋無力症
(2)筋萎縮性側索硬化症
(3)先天性筋ジストロフィー
(4)アルツハイマー病
(5)パーキンソン病
7.前項1〜4のいずれか1に記載の抗体を充填していることを特徴とするアセチルコリン受容体クラスター形成阻害活性を有するアグリン除去カラム。
8.前記カラムは、以下の治療用途であることを特徴とするカラム。
(1)重症筋無力症
(2)筋縮性側索硬化症
(3)先天性筋ジストロフィー
(4)アルツハイマー病
(5)パーキンソン病
9.以下の工程を含むz0型アグリン又はSC2型アグリンのアセチルコリン受容体クラスター形成能阻害活性を阻害する物質のスクリーニング方法;
(1)z0型アグリン及び/又はSC2型アグリン並びに候補阻害物質を細胞中に添加する工程、
(2)該細胞中のアセチルコリン受容体クラスター形成能を指標として、候補阻害物質の阻害活性を測定する工程。
【発明の効果】
【0016】
本発明者らは、従来知られていなかったアセチルコリン受容体クラスター形成阻害活性を持つアグリンの構造を基にして、該アグリンを特異的に認識できる抗体の提供を可能とした。さらに、該抗体を、アセチルコリン受容体クラスター形成能促進剤及びアセチルコリン受容体クラスター形成阻害活性を持つアグリン除去カラムに使用できる。
【図面の簡単な説明】
【0017】
【図1】複数の試料についてのアグリンの(C)末端部位をPCRにより増幅した結果。図中の矢印は、通常のC末端長を有する典型的なz0型アグリンのバンドを示す。
【図2】各種のアグリン転写物のcDNA配列の比較図。
【図3】各種のアグリンのC末端アミノ酸配列の比較図。図中のY部位(KSRK)は二重の下線で示し、z8挿入部位(ELTNEIPA)は下線で示し、z11挿入部位(PETLDSRALFS)は、点下線で示す。各囲みは、各アグリン特有のアミノ酸配列を示す(例:アミノ酸配列EHWQPRAETは、SC2(short C-terminal variant-2)アグリンの特有の配列である)。
【図4】各種のアグリンmRNAの選択的スプライシングの模式図。図中の各ボックスは、エキソンを示す。
【図5】各種のアグリンのドメイン構造を示す模式図{最上位のアグリンは、フルアグリン(全長アグリン)であり、それ以外はミニアグリンを示す}。
【図6】各種のミニアグリンの発現を示す模式図。
【図7】発現した各種のミニアグリンのイムノブロット解析の結果図。
【図8】nAChRクラスター形成の観察と評価結果。図中の矢印は、nAChRクラスターが形成されていることを示す。
【図9】z0型アグリン、SC2型アグリンのnAChRのクラスター形成阻害活性の評価。図中の矢印は、nAChRクラスターが形成されていることを示す。
【発明を実施するための形態】
【0018】
(アグリン)
アグリンは225 kDaのタンパク質をコアに持つヘパラン硫酸プロテオグリカンの一種であり、アミノ (N) 末端部と中央部に複数の推定糖鎖付加部位を持つ。アグリンは約40個のエクソンにコードされており、mRNAはオルタナティブ・スプライシングを受けて異なる翻訳開始点が生じる。このため49アミノ酸からなるshort N-terminal (SN) と、150アミノ酸からなるlong N-terminal (LN) の2種類のN末端を持つアグリンが産生される 。
さらにLNは、分泌に必須のシグナル配列 (signal sequence; SS) とラミニン結合領域であるN-terminal agrin domain (NtA) とを持つ分泌型、およびtransmembrane segment (TM) を持つ膜貫通型とに分けられる。
一方、C末端側にはX,Y,およびZのオルタナティブ・スプライシングによる可変部位が3箇所ある。Y部位への4個のアミノ酸挿入は、a-ジストログリカン (a-dystroglycan; a-DG) との親和性を高めると言われている。Z部位には、アミノ酸挿入がないz0 型に加えて、8、11、19個のアミノ酸が挿入されたz8,z11,z19型があり、z+型と総称されている (Ferns, M. J., Campanelli, J. T., Hoch, W., Scheller, R. H., and Hall, Z. (1993). The ability of agrin to cluster AChRs depends on alternative splicing and on cell surface)。
【0019】
本発明者らは、下記実施例1から明らかなように、上記既知の標準型C末端をもつz0,z8,z19型とは異なり、標準型よりも短いC末端をコードすることが推定される2種類のバリアントを新たに見出した。これらをSC1 (short C-terminal variant-1)、SC2 (short C-terminal variant-2) 型アグリンと命名した(参照:図3)。
なお、過去の報告と本実施例で明らかされた、オルタナティブ・スプライシングの様式を下記表1に示す。アグリン分子の多様性は,N末端,X部位,Y部位,Z部位,およびC末端の少なくとも5か所で生じる可変的な領域数の掛け算によって創出されていると考えられる。
さらに、本発明者らは、下記実施例2から明らかなように、SC2型アグリン及びz0型アグリンが、アセチルコリン受容体クラスター形成阻害活性を持つことを、新たに見出した。
【0020】
【表1】
【0021】
(アセチルコリン受容体クラスター形成阻害活性を持つアグリンを特異的に認識する抗体)
本発明の{アセチルコリン受容体クラスター形成阻害活性を持つアグリンを特異的に認識する抗体(以下、「本発明の抗体」と称する場合がある)}とは、SC2型アグリン及び/又はz0型アグリンを特異的に認識する抗体を意味し、特に限定されない。
さらに、前記抗体は、好ましくは、公知のz8型アグリン、z19型アグリン、及び/又は新規のSC1型アグリンには結合しない。
加えて、前記抗体は、ポリクローナル抗体、ポリクローナル抗体を含む抗血清又はモノクローナル抗体のいずれのタイプのものをも含み、また、これらの抗体のフラグメント{Fab、F(ab′)2又はFab′等}をも含むものである。なお、好ましくは、前記抗体は、モノクローナル抗体であることが好ましい。さらに、前記抗体は、自体公知の方法により、ヒト化抗体にすることが好ましい。
【0022】
{本発明の抗体作製に使用する免疫原(抗原)}
本発明の抗体を産出させるための免疫原として、哺乳動物由来、特に好ましくはヒト又はマウス由来のSC2型アグリン及び/又はz0型アグリンに特有のアミノ酸配列、より詳しくは、z8型アグリン、z11型アグリン、z19型アグリン、及び/又はSC1型アグリンには存在しないアミノ酸配列の全部又は一部を含むペプチドを用いることができる。
なお、下記実施例1から明らかなように、SC2型アグリンは、他のアグリンにない特有の以下のアミノ酸配列(配列番号1:参照図3)を有する。よって、好ましくは、以下のアミノ酸配列を含むペプチドを免疫原として使用することができる。
Glu-His-Trp-Gln-Pro-Arg-Ala-Glu-Thr(配列番号1)
さらに、Glu-His-Trp-Gln-Pro-Arg-Ala-Glu-Thr(配列番号1)で表されるアミノ酸配列に1ないし数個のアミノ酸残基の欠失、置換、挿入もしくは付加、又は修飾を施すことにより得られるアミノ酸配列からなるペプチドを免疫原として使用することもできる。
【0023】
(免疫原となるペプチドの合成)
ペプチドの製造は、遺伝子工学的手法、化学合成、および無細胞タンパク質合成により実施できる。ペプチドは、製造された後に、さらに精製して用いることができる。
【0024】
ペプチドの製造は、該ペプチドをコードする遺伝子の塩基配列情報又はアミノ酸配列に基づいて一般的な遺伝子工学的手法{サムブルック(Sambrook)ら編、「モレキュラークローニング,ア ラボラトリーマニュアル 第2版」、1989年、コールドスプリングハーバーラボラトリー;村松正實編、「ラボマニュアル遺伝子工学」、1988年、丸善株式会社;ウルマー(Ulmer, K.M.)、「サイエンス(Science)」、1983年、第219巻、p. 666-671;エールリッヒ(Ehrlich, H.A.)編、「PCRテクノロジー,DNA増幅の原理と応用」、1989年、ストックトンプレス}により実施できる。
【0025】
ペプチドの製造は、また、一般的な化学合成法により製造できる。ペプチドの化学合成方法として、例えば、固相合成方法や液相合成方法等が知られているがいずれも利用できる。
【0026】
ペプチドの精製および/または分離は、その物理的性質、化学的性質等を利用した各種分離操作方法により実施できる。分離操作方法として、硫酸アンモニウム沈殿、限外ろ過、ゲルクロマトグラフィー、イオン交換クロマトグラフィー、アフィニティークロマトグラフィー、高速液体クロマトグラフィーおよび透析法等の公知の方法を例示できる。これら方法は単独でまたは適宜組合せて使用できる。
【0027】
(免疫方法)
上記記載の免疫原となる精製したペプチド又は部分ペプチドを、リン酸緩衝液(PBS)などの適当な緩衝液中に溶解あるいは懸濁したものを抗原液として使用する。抗原液は通常抗原物質を50〜500μg/mL程度含む濃度に調製すればよい。また、ペプチド単独だけでは抗原性が低い場合には、アルブミンやキーホールリンペットヘモシアニン(KLH)などの適当なキャリアータンパク質に架橋して用いることができる。
当該抗原で免疫感作する動物(被免疫動物)は、マウス、ラット、ハムスター、ウマ、ヤギ、ウサギなどが例示される。好ましくはマウス、より好ましくはBALB/cマウスである。
【0028】
上記被免疫動物の抗原への応答性を高めるため、前記抗原溶液をアジュバントと混合して投与することができる。ここで使用可能なアジュバントは、フロイント完全アジュバント(FCA)、フロイント不完全アジュバント(FIA)、Ribi(MPL)、Ribi(TDM)、Ribi(MPL+TDM)。百日咳ワクチン(Boredetella pertussis vaccine)、ムラミルジペプチド(MDP)、アルミニウムアジュバント(ALUM)、およびこれらの組合せが例示されるが、初回免疫時にFCA、追加免疫時にFIAやRibiアジュバントを使用する組合せが特に好ましい。
【0029】
免疫方法は、使用する抗原の種類やアジュバント混合の有無などにより、注射部位、スケジュールなどを適宜変化させることができるが、例えば、被免疫動物としてマウスを用いる場合は、アジュバント混合抗原液0.05〜1ml(抗原物質10〜200μg)を腹腔内、皮下、筋肉内または(尾)静脈内に注射し、初回免疫から約4〜21日毎に1〜4回追加免疫を行い、さらに約1〜4週間後に最終免疫を行う。抗原量を多くして腹腔内注射することで、当該抗原溶液をアジュバントを使用せずに投与することもできる。抗体価は追加免疫の約5〜10日後に採血して調べる。抗体価の測定は、後述の抗体価アッセイに準じ、通常行われる方法で行うことができる。最終免疫より約3〜5日後、該免疫動物から脾細胞を分離して抗体産生細胞を得る。
【0030】
(モノクローナル抗体の作製)
モノクローナル抗体(以下、「MoAb」と略する場合がある)は、自体公知の方法、例えばケーラーとミルシュタインの方法(Kohler G, Milstein C. (1975) Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256, 495−497.)にしたがって作製することができる。
例えば、免疫動物から抗体産出細胞を含む組織(例えば、脾臓又はリンパ節)を回収し、該抗体産出細胞と自体抗体の腫瘍細胞(例えば、骨腫瘍細胞)とを融合させることによってハイブリドーマを作製し、次いでハイブリドーマをクローン化した後、所望の抗体を産出しているハイブリドーマを選別し、このハイブリドーマの培養液から抗体を回収する。
骨髄腫細胞として、マウス、ラット、ヒトなど由来のものが使用され、例えばマウスミエローマP3X63-Ag8、P3X63-Ag8-U1、P3NS1-Ag4、SP2/o-Ag14、P3X63-Ag8・653などの株化骨髄腫細胞が例示される。骨髄腫細胞には免疫グロブリン軽鎖を産生しているものがあり、これを融合対象として用いると、抗体産生細胞が産生する免疫グロブリン重鎖とこの軽鎖とがランダムに結合することがあるので、特に免疫グロブリン軽鎖を産生しない骨髄腫細胞、例えばP3X63-Ag8・653やSP2/o-Ag14などを用いることが好ましい。
抗体産生細胞と骨髄腫細胞とは、同種動物、特に同系統の動物由来であることが好ましい。骨髄腫細胞の保存方法は自体公知の手法に従って行えばよく、例えばウマ、ウサギもしくはウシ胎児血清を添加した一般的な培地で継代培養したものについて凍結により保存される。また細胞融合には対数増殖期の細胞を用いるのが好ましい。
【0031】
(ハイブリドーマの作製)
抗体産生細胞と骨髄腫細胞とを融合させてハイブリドーマを作製する方法は、ポリエチレングリコール(PEG)を用いる方法、センダイウイルスを用いる方法、電気融合装置を用いる方法などが例示される。例えばPEG法の場合、約30〜60%のPEG(平均分子量1,000〜6,000)を含む適当な培地または緩衝液中に脾細胞と骨髄腫細胞を1〜10:1、好ましくは5〜10:1の混合比で懸濁し、温度約25〜37℃、pH6〜8の条件下で、約30秒〜3分間程度反応させればよい。反応終了後、細胞を洗浄しPEG溶液を除いて培地に再懸濁し、マイクロタイタープレート中に播種して培養を続ける。
融合操作後の細胞は選択培地で培養して、ハイブリドーマの選択を行う。選択培地は、親細胞株を死滅させ、融合細胞のみが増殖しえる培地であり、通常ヒポキサンチン−アミノプテリン−チミジン(HAT)培地が使用される。ハイブリドーマの選択は、通常融合操作の1〜7日後に、培地の一部、好ましくは約半量を選択培地と交換し、さらに2、3日毎に同様の培地交換を繰り返しながら培養することにより行う。顕微鏡観察によりハイブリドーマのコロニーが生育しているウエルを確認する。
【0032】
生育しているハイブリドーマが所望の抗体を産生しているかどうかを知るには、培養上清を採取して抗体価アッセイを自体公知の方法により行えばよい。
さらに限界希釈法、軟寒天法、蛍光励起セルソーターを用いた方法などにより単一クローンを分離する。
【0033】
ハイブリドーマが産生する抗体の免疫グロブリンサブクラスを調べるためには、該ハイブリドーマを一般的な条件で培養し、その培養上清中に分泌された抗体を市販の抗体クラス・サブクラス判定用キットなどを用いて分析することにより知ることができる。
【0034】
(モノクローナル抗体の取得方法)
ハイブリドーマからのMoAbの取得方法は、必要量やハイブリドーマの性状などによって適宜選択することができる。例えば、該ハイブリドーマを移植したマウス腹水から取得する方法、細胞培養により培養上清から取得する方法などが例示される。マウス腹腔内で増殖可能なハイブリドーマであれば、腹水から数mg/mLの高濃度のMoAbを得ることができる。インビボで増殖できないハイブリドーマは細胞培養の培養上清から取得する。
細胞培養によるMoAbの取得は、抗体産生量はインビボより低いが、マウス腹腔内に含まれる免疫グロブリンや他の夾雑物質の混入が少なく、精製が容易であるという利点がある。
抗体を、ハイブリドーマを移植したマウス腹腔内から取得する場合、例えば、予めプリスタン(2, 6, 10, 14-テトラメチルペンタデカン)などの免疫抑制作用を有する物質を投与したBALB/cマウスの腹腔内へハイブリドーマ(約106個以上)を移植し、約1〜3週間後に貯留した腹水を採取する。異種ハイブリドーマ(例えばマウスとラット)の場合には、ヌードマウス、放射線処理マウスを使用することが好ましい。
細胞培養上清から抗体を取得する場合、例えば、細胞維持に用いられる静置培養法の他に、高密度培養方法あるいはスピンナーフラスコ培養方法などの培養法を用い、当該ハイブリドーマを培養し抗体を含有する培養上清を得る。
腹水や培養上清からのMoAbの精製は、自体公知の方法により行うことができる。例えば、免疫グロブリンの精製法として従来既知の硫酸アンモニウムや硫酸ナトリウムを用いた塩析による分画法、ポリエチレングリコール分画法、エタノール分画法、DEAEイオン交換クロマトグラフィー法、ゲル濾過法などを応用することで、容易に達成される。
さらに、MoAbが、マウスIgGである場合には、プロテインA結合単体あるいは抗マウスイムノグロブリン結合単体を用いたアフィニティークロマトグラフィー法により精製することが可能であり、簡便である。
【0035】
{z0型アグリン又はSC2型アグリンのアセチルコリン受容体クラスター形成能阻害活性を阻害する物質のスクリーニング方法}
本発明のスクリーニング方法では、少なくとも以下の工程を含む。
(1)z0型アグリン及び/又はSC2型アグリン並びに候補阻害物質を培養した細胞中に添加する工程
細胞は、該細胞中のアセチルコリン受容体クラスター形成能を測定できる細胞であれば特に限定されない。
(2)該細胞中のアセチルコリン受容体クラスター形成能を指標として、候補阻害物質の阻害活性を測定する工程
候補阻害物質並びにz0型アグリン及び/又はSC2型アグリンを添加した細胞中のアセチルコリン受容体クラスター形成能が、z0型アグリン及び/又はSC2型アグリンを添加した細胞中のアセチルコリン受容体クラスター形成能と比較して、高い場合には、該候補阻害物質は、z0型アグリン又はSC2型アグリンのアセチルコリン受容体クラスター形成能阻害活性を阻害する物質、言い換えれば、アセチルコリン受容体クラスター形成を促進することができる物質(アセチルコリン受容体クラスター形成促進物質)である。
なお、アセチルコリン受容体クラスター形成能の測定方法は、下記実施例2を参照することができる。
【0036】
(候補阻害物質)
上記スクリーニングで使用する候補阻害物質としては任意の物質を使用することができる。候補阻害物質の種類は特に限定されず、個々の低分子合成化合物特にsiRNAでもよいし、天然物抽出物中に存在する化合物でもよく、合成ペプチドでもよい。あるいは、候補阻害物質はまた、化合物ライブラリー、ファージディスプレーライブラリーもしくはコンビナトリアルライブラリーでもよい。候補阻害物質は、好ましくは低分子化合物であり、低分子化合物の化合物ライブラリーが好ましい。化合物ライブラリーの構築は当業者に公知であり、また市販の化合物ライブラリーを使用することもできる。
【0037】
(アセチルコリン受容体クラスター形成能促進剤)
本発明の「アセチルコリン受容体クラスター形成能促進剤」とは、アセチルコリン受容体クラスター形成を抑制、阻害及び/又は低下させる機能を持つSC2型アグリン及び/又はz0型アグリンの作用を阻害若しくはこれらのアグリンと結合することにより、アセチルコリン受容体クラスター形成を促進させる。
本発明の促進剤の特徴は、従来のアセチルコリン産出量を増大させる薬剤若しくはアセチルコリン分解酵素阻害剤とは明らかにメカニズムが異なる。従来のアセチルコリン産出量を増大させる薬剤では、患者の体内中のアセチルコリン産出量を増大させる若しくはアセチルコリンの分解を阻害することができても、アセチルコリン受容体が少なければ(クラスターが形成されていなければ)、アセチルコリンがアセチルコリン受容体(ニコチン性アセチルコリン受容)に結合できず、受容体に内蔵されるカチオンチャネルが開口できず、興奮性シナプス後電位が生じなかった。
しかし、本発明の促進剤は、従来の上記薬剤とはメカニズムが異なり、アセチルコリン受容体クラスターの形成能を促進させるので、少ないアセチルコリン産出量の患者においても、少ないアセチルコリンが効率的にアセチルコリン受容体に結合することができ、受容体に内蔵されるカチオンチャネルが開口し、興奮性シナプス後電位を生じさせることができる。
さらに、本発明の促進剤は、従来のアセチルコリン産出量を増大させる薬剤若しくはアセチルコリン分解酵素阻害薬と併用又は組み合わせることにより、優れた効果、特に従来の薬剤では効果がない患者に有用であると考えられる。
【0038】
本発明の促進剤は、上記記載のアセチルコリン受容体クラスター形成阻害活性を持つアグリンを特異的に認識する抗体及び/又は上記記載のスクリーニング方法により得られたアセチルコリン受容体クラスター形成促進物質を有効成分として含む。
さらに、治療等(予防も含む)の目的に応じて、散剤、顆粒剤、錠剤、カプセル剤、腸溶剤、液剤、注射剤(液剤、懸濁剤)または遺伝子療法に用いる形態などの各種の形態に、常法にしたがって調製することができる。
加えて、充填剤、増量剤、結合剤、付湿剤、崩壊剤、滑沢剤、希釈剤および賦形剤も含むことができる。その他、安定化剤、殺菌剤、緩衝剤、等張化剤、キレート剤、界面活性剤、およびpH調整剤等を適宜使用することもできる。
【0039】
本発明の促進剤の投与量または摂取量については、本発明の効果が得られるものであれば特に限定されるものではなく、含有される成分の有効性、投与形態、投与経路、疾患の種類、対象の性質(体重、年齢、病状および他の医薬の使用の有無等)、および担当医師の判断等に応じて適宜選択される。本発明の促進剤は、1日1〜数回に分けて投与または摂取することができ、数日または数週間に1回の割合で間欠的に投与または摂取してもよい。
【0040】
(本発明の促進剤の用途)
本発明の促進剤は、アセチルコリンの減少、アセチルコリン受容体のクラスター形成能の低下が関与する疾患、例えば、以下の疾患の治療剤とすることができる。
(1)重症筋無力症
(2)筋萎縮性側索硬化症
(3)先天性筋ジストロフィー
(4)アルツハイマー病
(5)パーキンソン病
【0041】
(アセチルコリン受容体クラスター形成阻害活性を持つアグリン除去カラム)
本発明の「アセチルコリン受容体クラスター形成阻害活性を持つアグリン除去カラム」とは、上記記記載のアセチルコリン受容体クラスター形成阻害活性を持つアグリンを特異的に認識する抗体が充填されている。より詳しくは、SC2型アグリン及び/又はz0型アグリンを特異的に認識する抗体が充填されたカラムである。
上記抗体を吸着する担体としては、特に限定されないが、セルロース系ゲル、デキストラン系ゲル、アガロース系ゲル、ポリアクリルアミド系ゲル、多孔質ガラス、ビニルポリマーゲル等の有機または無機の多孔体が使用でき、通常のアフィニティクロマトグラフィーに用いられる担体を使用することができる。
加えて、上記抗体を担体に結合及び/又は吸着させる方法としては、自体公知の物理的吸着法、化学的結合法又はこれらの併用等の公知の方法により行うことができる。
さらに、必要に応じて担体と上記抗体との間に任意の長さの分子(スペーサー)を導入することもできる。
非特異的反応等を抑制するために、抗体を固相化させた担体の表面又は内壁面に、ウシ血清アルブミン等、界面活性剤又は脱脂粉乳等を接触させ被覆させること等の公知の方法により処理して、ブロッキング処理を行ってもよい。
【0042】
本発明のカラムを体外循環用カラムとして用いる場合には、大きく次の二通りの方法がある。一つには、体内から取り出した血液を遠心分離機もしくは膜型血漿分離器を使用して、血漿成分と血球成分とに分離した後、血漿成分を該カラムに通過させ、SC2型アグリン及び/又はz0型アグリンを除去した後、血球成分と合わせて体内にもどす方法であり、他の一つは、体内から取り出した血液を直接該カラムに通過させ、SC2型アグリン及び/又はz0型アグリンを除去する方法である。
なお、重症筋無力症の治療方法の一つとして、単純血漿交換療法により、血漿中の抗アセチルコリン受容体抗体を除去する療法がある。本発明のカラムを用いた療法では、該、重症筋無力症の単純血漿交換療法を参考にして行うこともできる。
【0043】
(本発明のカラムの用途)
本発明のカラムは、アセチルコリンの減少、アセチルコリン受容体のクラスター形成能の低下が関与する疾患、例えば、以下の疾患の治療に使用することができると考えられる。
(1)重症筋無力症
(2)筋萎縮性側索硬化症
(3)先天性筋ジストロフィー
(4)アルツハイマー病
(5)パーキンソン病
【0044】
{重症筋無力症(MG)}
MGでは抗nAChR抗体によって引き起こされる補体介在性シナプス後膜破壊と、それに伴うnAChR数の減少が病態の原因であり、これにより神経筋伝達障害が起きている。
すなわち、本発明の促進剤をMG患者に投与すること又は本発明のカラムによりMG患者からSC2型アグリン及び/又はz0型アグリンを除去することにより、少ないnAChRの発現量でも、アセチルコリンが効率的にnAChRに結合することができるので、重症筋無力症の治療効果があると考えられる。
【0045】
{筋萎縮性側索硬化症(ALS)}
筋萎縮性側索硬化症は、運動ニューロンの変性や萎縮、骨格筋の麻痺を主徴とする進行性神経変性病の一つである。また、ALSのモデルマウスであるSODマウスでは病態の末期においてアグリン発現量の低下が見られることが報告されている(Dobrowolny, G., Giacinti, C., Pelosi, L., Nicoletti, C., Winn, N., Barberi, L., Molinaro, M., Rosenthal, N., and Musaro, A. (2005). Muscle expression of a local Igf-1 isoform protects motor neurons in an ALS mouse model. J. Cell Biol. 168, 193-199)。
すなわち、本発明の促進剤をALS患者に投与すること又は本発明のカラムによるALS患者からSC2型アグリン及び/又はz0型アグリンを除去することにより、少ないアグリンの発現量でも、アセチルコリンが効率的にnAChRに結合することができるので、筋萎縮性側索硬化症の治療効果があると考えられる。
【0046】
{先天性筋ジストロフィー(congenital muscular dystrophy; CMD)}
先天性筋ジストロフィーは乳幼児期に筋力低下、関節拘縮、運動発達遅滞で発症し、筋病理で壊死と再生所見を認める筋ジストロフィーの総称で、中枢神経症状を合併する福山型 (Fukuyama CMD; FCMD)と、中枢神経症状を呈さない非福山型 (non-FCMD) に大別されている。non-FCMDの中には筋線維に発現するラミニンのα2鎖を指令する遺伝子LAMA2の変異によって起きるものがあり、CMDのモデルマウスはヒトのラミニン2欠損型先天性筋ジストロフィーと似た症状を呈する。この疾患での筋線維の変性は、基底膜構造に必要な1次ラミニン骨格の形成ができず、筋基底膜とジストロフィン-糖タンパク質複合体 (dystrophin-glycoprotein complex; DGC) との結合、または筋基底膜とインテグリン類との結合が失われるためと考えられている。このCMDモデルマウスにz-型のミニ・アグリンcDNAを組み込んだアデノ随伴ウイルスを腹腔内注射すると、ミニ・アグリンが基底膜およびDGCの一要素であるα-DGに結合し、ミニ・アグリンが仲介するα-DGとラミニンのα5鎖の安定化機構によって、筋病態を改善したという報告がある (Moll, J., Barzaghi, P., Lin, S., Bezakova, G., Lochmuller, H., Engvall, E., Muller, U., and Ruegg, M. A. (2001). An agrin minigene rescues dystrophic symptoms in a mouse model for congenital muscular dystrophy. Nature 413, 302-307.)。
すなわち、本発明の促進剤をCMD患者に投与すること又は本発明のカラムによるCMD患者からSC2型アグリン及び/又はz0型アグリンを除去することにより、少ないアグリンの発現量でも、アセチルコリンが効率的にnAChRに結合することができるので、先天性筋ジストロフィーの治療効果があると考えられる。
【0047】
アルツハイマー病は、中枢性アセチルコリン受容体伝達障害が原因であることが知られており。パーキンソン病は、中枢性ドーパミン伝達障害が原因であることが知られている。よって、本発明の促進剤をアルツハイマー病又はパーキンソン病患者に投与すること又は本発明のカラムによる該患者の血液からSC2型アグリン及び/又はz0型アグリンを該患者から除去することにより、治療効果があると考えられる。
【0048】
以下、本発明を実施例により具体的に説明する。しかし、本発明は下記実施例に限定されるものではない。
【実施例1】
【0049】
(アグリンのスプライシング・バリアントのcDNAクローニング及びそのタンパク質の発現)
本実施例では、哺乳動物の例としてマウス神経組織と神経筋組織に由来する種々の培養細胞株を用いてアグリンのスプライシング・バリアントのcDNAクローニングによる同定とそのタンパク質発現実験を行った。詳細は、以下の通りである。
【0050】
(1)細胞培養
マウス筋芽細胞由来培養細胞株C2C12は、American Type Culture Collection (Manassas, VA, USA) より購入した。マウス運動ニューロン由来細胞株NSC-34は、田平武博士 (国立長寿医療センター研究所) より供与を受けた。C2C12細胞、NSC-34細胞およびサル腎線維芽細胞由来COS-7細胞の維持培養液には、Dulbecco's Modified Eagle Medium (DMEM; GIBCO BRL, Grand Island, NY, USA) に10%ウシ胎児血清 (fetal bovine serum; FBS; GIBCO BRL)、ペニシリン100 units/ml (GIBCO BRL) 及びストレプトマイシン100 μg/ml (GIBCO BRL) を加えて使用した。継代時には培養液を吸引除去し、付着細胞をMg2+, Ca2+含有ハンクス平衡緩衝塩溶液 (Hanks' balanced salt solution; HBSS; GIBCO BRL) で洗浄し、0.25%トリプシン、1 mMエチレンジアミン四酢酸を含むMg2+, Ca2+不含HBSS (GIBCO BRL) を加え、CO2インキュベーター (37℃,5% CO2; BNA-111; TABAI ESPEC, 大阪) 内で10分間反応させた。
剥離した細胞を回収後、1500 rpm,20℃で5分遠心した。上清を吸引除去した後、新たな培養液に浮遊させ、細胞数を5×104cells/ml に調製し、そのうち5 mlを培養フラスコに播種し、CO2インキュベーター (37℃,5% CO2) で培養した。培養液は3日ごとに交換し、1週間で継代した。
マウス神経芽細胞腫×マウス線維芽細胞雑種細胞株N1E-115細胞 (Amano et al., 1972) は、DMEMに最終濃度が5%ウシ胎児血清 (fetal calf serum; FCS; GIBCO BRL)、1 mM ヒポキサンチン、0.16 mM d-チミジン、0.01 mM アミノプテリンになるように加えた培養液を用いて継代培養した。
上記の培養に加えて,C2C12細胞の分化誘導も行った。継代後2日目に培養液を吸引除去し、分化誘導用に調製した2%ウマ血清 (horse serum; HS; GIBCO BRL) を含むDMEMと置き換えた。さらに4日間、CO2インキュベーター (37℃,5% CO2) で培養した。
【0051】
(2)Total RNAの抽出
成体マウス (C57BL/6,BALB/c) をネンブタール腹腔内注射により深く麻酔し、頚椎脱臼の後、頚動脈をはさみで切り脱血させた。摘出した脳、脊髄をMg2+, Ca2+不含リン酸緩衝生理食塩水{phosphate-buffered saline (−); PBS(−)}}で洗浄し、組織片30 mgに600 μlのLysis Solutionを加え、テフロンホモジナイザーを用いて組織懸濁液を得た。 また、NSC-34およびN1E-115細胞は、60 mm培養皿 (IWAKI) にそれぞれ3×104cells/mlで細胞をまき、CO2インキュベーター (37℃,5% CO2)で4日間培養後にPBS (−) で洗浄し細胞を回収した。未分化型C2C12細胞は、60 mm培養皿で1×104cells/mlで細胞をまいて3日間培養し、同様に細胞を回収した。分化型C2C12細胞は、未分化型と同様に細胞をまき、3日間の通常培養と4日間の分化誘導後に回収した。いずれの細胞も、Lysis Solution 350 μlを加えて溶解した。
上記の組織懸濁液と細胞溶解液からtotal RNA 抽出/精製 Kit Mini (Agilent Technologies CA, USA) を使用して以下の通りにtotal RNAを抽出した。
Lysis Solution 350 μlを加えて溶解した組織懸濁液と細胞溶解液全量をMini Prefiltrationで遠心 (16000 × g,3分,20℃) して、上清 350 μlを70% エタノール (Amresco, OH, USA) 350 μlと混合した。エタノール/Lysis Solution 混合液700 μlをMini Isolation Columnで遠心 (16000 × g,30秒,20℃) して濾過液を廃棄し、Wash Solution 500 μlを用いて遠心 (16000 × g,30秒,20℃) してカラムの洗浄を2度行い、遠心 (16000 × g,2分,20℃) した。蒸留水 (distilled water; DW) 30 μlを加え1分間静置した。16000 × g,1分,20℃で遠心にかけRNAを溶出した。
得られたサンプルに混入したDNAを除くためにDNA分解処理を行った。反応液は全量50 μlで、各Total RNAサンプル0.4-1 μg、40 mM Tris-HCl (pH 7.5)、8 mM MgCl2、5 mM ジチオトレイトール、DNase I 2.5 unitを含んだ。37℃、10分間の反応後、フェノール・クロロホルム処理、ジエチルエーテル処理し、エタノール沈殿した。RNAは最終的にDW 11μlに溶解し、UV分光光度計 (Nano Drop Technologies) を用いて濃度を測定した。
【0052】
(3)逆転写酵素‐ポリメラーゼ連鎖反応
C57BL/6マウスの脊髄、BALB/cマウスの全脳、N1E-115細胞、NSC-34細胞、未分化型C2C12細胞および分化型C2C12細胞から抽出したtotal RNAから、Transcriptor First Strand cDNA Synthesis Kit (Roche Applied Science, Mannheim, Germany) を使用して相補DNA (complementary DNA; cDNA) を合成した。
反応液は全量20 μlで、各total RNA 0.4-1 μg,50 mM Tris-HCl (pH 8.5),30 mM KCl,8 mM MgCl2,60 μM Random Hexamer Primer,RNase Inhibitor 20 unit,10 mM dNTPs,AMV (Avian myeloblastosis virus) 逆転写酵素10 unitを混ぜて、25℃で10分、さらに50℃で1 時間反応させ、DW 180 μlを加えて使用時まで−80 °Cで保存した。
マウス・アグリン遺伝子の塩基配列に基づいて4種類のプライマー (AgAFw,AgBFw,AgCRv,及びAgDRv; 参照:表2) を合成した。調整した各cDNAを鋳型として、1 × 添付のbuffer,0.4 mM dNTPs,0.3 μM AgAFwプライマー,0.3 μM AgDRvプライマー,cDNAサンプル1 μl,KOD FX DNAポリメラーゼ1.0 unitを含む反応液でPCRを施行した。94℃で2分間熱変性の後、熱変性 (98℃,10秒)、アニーリング (60.4℃,30秒)、伸長 (68℃,1分) の反応サイクルを20回繰り返した。さらに,得られた反応産物1 μlを再度同じ組成の反応液に加えて同様の反応を25サイクル行った。得られたPCR産物は1%アガロースゲルの電気泳動で分離し、エチジウムブロマイド溶液 (1 μg/ml) にて染色後、UVサンプル撮影装置 (FAS-III; TOYOBO, 大阪) を用いて観察、記録した。
【0053】
【表2】
【0054】
(4)cDNAクローンの単離
上記(3)のRT-PCRの反応産物を電気泳動し、0.9-1.1 kbのDNA断片をGENECLEAN II Kit (MP Biomedicals LLC, Irvine, CA, USA) を用いてゲルから回収した。精製したDNA断片は、rTaq (TaKaRa, 滋賀) を含む溶液中{10 mM Tris-HCl (pH 8.3),50 mM KCl,1.5 mM MgCl2,0.25 mM dNTPs,0.25 μM A1 AgBFw,0.25 μM AgCRvプライマー,rTaq 0.25 unit,cDNAサンプル30 μl}で72℃、10分間反応させ、A付加反応を施した。この反応液からMin Elute PCR Purification Kit (QIAGEN) を用いてDNA断片を精製し、DW 10 μlに溶解した。回収したDNA断片1 μlにSalt Solution (Invitrogen, Carlsbad, CA, USA ) 0.5 μl、pCR 2.1-TOPO (Invitrogen) 0.5 μlを混ぜ、室温で30分、続いて4℃で6時間連結反応を行った。さらに、各反応液に大腸菌TOP10株コンピテントセル25 μlを加え氷上で30分間、続いて42℃で30秒反応させた後、SOC培地250 μlを加え37℃で1時間振とう培養した。
この溶液を5-bromo-4-chloro-3-indolyl-β-D-galactoside (X-gal; 8 mg/ml) を塗布したアンピシリン (50 μl/ml) 含有LB寒天培地に60 μlずつ播いて37℃で12時間培養した。
次に、出現したコロニーから白色のものを選び、cDNAが組込まれていることを確認するため、プラスミドを以下の通りに抽出した。白色コロニーをアンピシリン (50 μl/ml) 含有LB液体培地5 mlを加えたチューブに入れ、37℃,200 rpmで振とう培養した。12時間後、培養液を3000 rpm、10分遠心して集菌し、得られた菌体をRNase I (10 mg/ml) を含むBuffer P1 250 μlで懸濁し、別のチューブに移した。そこに、Buffer P2 250 μlを加えて転倒混和し、直後にBuffer N3 350 μlを加えて再度転倒混和した後、13000 rpmで10分間遠心した。得られた上清をQIAprep spin columnに移し13000 rpmで1分間遠心し、濾液を除去した。この後、Buffer PB 500 μlを加えて13000 rpmで1分間遠心し濾液を捨て、Buffer PE 750 μlを加えて13000 rpmで1分間遠心し濾液を捨て、さらに13000 rpmで1分間遠心し残液を除いた。QIAprep spin columnを別のチューブに移してDW 50 μl加え室温で1分間静置し、13000 rpm,1分間の遠心でプラスミド溶液を得た。DNA濃度はUV分光光度計 (Nano Drop Technologies) にて測定した。
【0055】
(5)塩基配列決定
抽出した各プラスミドDNA 300 ngをABI PRISM BigDye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) 添付の1 × Big Dye ver1.1 Reaction Mix, 1 × Big Dye Sequence Buffer, 0.8 μM 合成プライマー用いて、ダイデオキシ法により行った。反応産物をBigDye XTerminatorTM 精製キット (Applied Biosystems) により精製後、ABI PRISM 3130 Genetic Analyzer (Applied Biosystems) を用い、Sequence Scanner v1.0,DNA Sequencing AnalysisソフトウェアVersion 5.1を用いて塩基配列を決定した。
【0056】
(6)データベース解析
日本DNAデータバンク (DNA Data Bank of Japan; DDBJ) のホームページにアクセスし、BLAST version 2. 2. 18を用いた。
【0057】
(7)タンパク質発現用プラスミドの作製
(7−1)ミニ・アグリン (mini-agrin) cDNAの部分断片の調製
N1E-115細胞由来cDNA 1 μlを鋳型として、KOD FX DNAポリメラーゼ (TOYOBO) とそれぞれA1 AgEFw,AgGRvプライマーを用いて熱変性: 98℃,10秒、アニーリング: 62.2℃,30秒,伸長: 68℃,1分の各条件で35サイクル,B1 AgHFw,AgKRvプライマーを用いて熱変性: 98℃,10秒,アニーリング: 54.4℃,30秒,伸長: 68℃,1分の各条件で35サイクル,C1 AgBFw,AgNRvプライマーを用いて熱変性: 98℃,10秒,アニーリング: 57.3℃,30秒,伸長: 68℃,1分間の各条件で35サイクルPCRを行い、増幅したA1フラグメント 743 bp,B1フラグメント1275 bp,C1フラグメント879 bpをMin Elute PCR Purification Kit (QIAGEN, Hilden, Germany) を用いて精製した。
この精製したPCR産物全量についてrTaq (TaKaRa) を用いて調製した溶液 {10 mM Tris-HCl (pH 8.3),50 mM KCl,1.5 mM MgCl2,0.25 mM dNTPs,各0.25 μMの特異的プライマー,rTaq 0.25 unit,cDNAサンプル30 μl}}で72℃,10分間反応させ、各反応産物を再度Min Elute PCR Purification Kit (QIAGEN) を用いて精製し、DW 10 μlに溶解した。 AgEFw,AgGRvプライマー,AgHFw,AgKRvプライマー,AgBFw,AgNRvプライマーの各組合せで得られたDNA断片を本発明ではA1,B1,C1とした。
精製したA1,B1,C1の各DNA断片1 μlとSalt Solution (Invitrogen) 0.5 μl,pCR 2.1-TOPO (Invitrogen) 0.5 μlを混ぜ、室温で30分,4℃で6時間連結反応を行った。各連結反応産物に大腸菌TOP10株コンピテントセル25 μlを加え、氷上で30分間反応させた。その後42℃で30秒反応させ、SOC培地250 μlを加えて37℃で1時間振とう培養した。滅菌コンラージ棒でアンピシリン (50 μl/ml) 含有Luria-Bertani (LB) 寒天培地にX-gal (8 mg/ml) を塗りインキュベーターで乾かした。アンピシリン (50 μl/ml) 含有LB寒天培地に60 μlずつ播いて37 °Cで12時間培養した。プラスミドDNAは、上記(4)と同様に調製し (A2,B2,C2) とした。
【0058】
(7−2)ベクターの調製
pBluescript II KS (+) (pBSII KS(+); Stratagene) は、37℃、2時間EcoRVとXhoI,XhoIとBamHI,あるいはXbaIとSacIで消化した。切断されたプラスミドを、37℃,1時間バクテリア・アルカリホスファターゼ (bacterial alkaline phosphatase; BAP) で処理し、1%アガロースゲルで泳動後DEAEペーパーに吸着させて回収し、フェノール・クロロホルム処理、ジエチルエーテル処理、エタノール沈殿の後、50 μlのDWに溶解し、ベクターとして用いた。
pCR2.1-TOPO (Invitrogen) は、37℃、2時間XbaIとSacIで消化した。切断したプラスミドを、37℃、1時間BAP処理し、1%アガロースゲルで泳動した。DEAEペーパーに吸着させて回収し、フェノール・クロロホルム処理、ジエチルエーテル処理、エタノール沈殿の後、50 μlのDWに溶解し、これをベクターとして用いた。
pcDNA3.1 (+) (Invitrogen) は、37℃、2時間EcoRVとNotIで消化し、切断されたプラスミドを、37℃、1時間BAP処理し、1%アガロースゲルで泳動した。DEAEペーパーに吸着させ回収し、フェノール・クロロホルム処理、ジエチルエーテル処理、エタノール沈殿の後、50 μlのDWに溶解し、これをベクターとして用いた。
哺乳動物細胞発現用プラスミドpEF-BOS (京都大学大学院医学系研究科長田重一教授より供与された)は、37℃,1時間XbaIで消化し、その後37℃で1時間BAP処理した。フェノール・クロロホルム処理、ジエチルエーテル処理、エタノール沈殿の後、50 μlのDWに溶解した。このうちの20μlに、50 mM Tris-HCl (pH 7.5),100 mM NaCl,10 mM MgCl2,1 mM DTT,250 μM dNTPs,0.01 units/μl Klenow fragment of DNA polymerase Iとなるように加えて37℃、20分間反応させた。1%アガロースゲルで泳動した後、GENECLEAN II Kit (MP Biomedicals LLC) を用いて回収し、これをベクターとして用いた。
【0059】
(7−3)ミニ・アグリンcDNAの構築
A2,B2,C2の各プラスミドDNAを100℃で10分間反応させ、このうち1 μlを鋳型としてA2プラスミドをT7とAgGRvプライマーで、B2プラスミドをAgHFwとM13revプライマーあるいはT7とAgKRvプライマーで、C2プラスミドをAgBFwとM13revプライマーでアニーリング温度52.2℃,20サイクルの条件で KOD FX ポリメラーゼ (TOYOBO) を用いて増幅し、Min Elute PCR Purification Kit (QIAGEN) を用いて精製し、DW 15.4 μlに溶解した。得られたDNA断片を順にA3,B3,B3',C3とした。
A3,B3,B3',C3の末端リン酸化を、1×Protruding End Kinase Buffer、T4 polynucleotide kinase 0.5-2 units/μl,1 mM ATPを含む反応液中で37℃で30分間行い、65℃,5分間の加熱によって停止させた。ここで得られたDNA断片をA4,B4,B4',C4とした。A4 1 μlとB4 1 μl,B4' 1 μlとC4 1 μlの組み合わせで混合し、それぞれにMighty mix 2 μl (TaKaRa) を加えて16℃で30分間連結反応を行い、反応産物をD1,E1とした。
さらに、D1を鋳型にT7とAgJRvプライマーで、E1を鋳型にT7とM13revプライマーでPCRを行った。アニーリング温度52℃、35サイクルの条件でKOD FXポリメラーゼ(TOYOBO) を用いた。反応液を1%アガロースゲルで電気泳動し、D1由来の1741 bp付近のバンド、E1由来の2344 bp付近のバンドをGENECLEAN II Kit (MP Biomedicals LLC) を用いて回収し、それぞれD2,E2とした。D2は制限EcoRVとXhoIで、E2はXhoIとBamHIでそれぞれ消化し、1%アガロースゲルで電気泳動後、D2由来の1153 bp付近の断片、E2由来の1781 bp付近の断片をGENECLEAN II Kitで回収し、D3,E3とした。
D3断片1.5 μl、E3断片1.5 μlは、上記(7−2)で調製したEcoRVとXhoIで切断したpBSII-KS (+) 1 μl (25 ng)、XhoIとBamHIで切断したpBS2-KS (+) 1 μl (25 ng) とそれぞれ混ぜ、それぞれにMighty mix 2.5 μl (TaKaRa) を加えて連結反応を16℃、30分間行い、反応産物をそれぞれD4,E4とした。このD4,E4に50 mM MgCl2/10 mM CaCl2を100 μlと大腸菌SCS1株コンピテントセル100 μlを加えて、氷上で30分間反応させた。
その後42℃で1分間反応させ、SOC培地を250 μl加えて37℃で1時間反応させた。この溶液をアンピシリン (50 μl/ml) 含有LB寒天培地に60 μlずつ播いて、37℃で12時間培養した。得られたD4,E4の各コロニーからPlasmid DNA purification QIAprep spin Miniprep Kit (QIAGEN) を用いてプラスミド抽出し、このプラスミドをD5,E5とした。
D5をXbaIとXhoIで、E5をSacIとXhoIで消化し、1%アガロースゲルで電気泳動し、D5由来の1181 bp付近の断片、E5由来の947 bp付近の断片をGENECLEAN II Kit (MP Biomedicals LLC) で回収し、それぞれD6,E6とした。D6断片1 μl、E6断片1 μl、及び上記(7−2)で作製したXbaIとSacIで切断したpCR2.1 vector 1 μl (25 ng) を混ぜ、さらにMighty mix 3 μl (TaKaRa) を加え、16℃で30分間連結反応を行い、反応産物をF1とした。このF1サンプルに大腸菌DH5α株のコンピテントセル (TaKaRa) 20 μlを加えて、氷上で30分間反応させ、42℃で、45秒間反応後、SOC培地を200 μl加えて37℃、200 rpmで1時間振とう培養した。この溶液をX-gal (8 mg/ml) 塗布アンピシリン (50 μl/ml) 含有LB寒天培地に60 μlずつ播いて37℃で12時間培養した。得られたF1のコロニーをPlasmid DNA purification QIAprep spin Miniprep Kit (QIAGEN) を用いてプラスミド抽出し、これをF2 とした。
F2プラスミドをEcoRVとSacIで消化し、1%アガロースゲルで電気泳動し、2092 bp付近の断片をDEAEペーパーに吸着した。これを1.5 M NaCl/10 mM Tris-HCl(pH 7.5)で溶出し、フェノール・クロロホルム抽出後、ジエチルエーテル処理、エタノール沈殿し、50 μlのDWに溶解した。得られたDNA断片をF3とした。
C末端側バリアントの各DNA断片をSacIとNotIで消化し、1%アガロースゲルで電気泳動した後、851-1108 bpの各バンドをGENECLEAN II Kit (MP Biomedicals LLC) で回収した。それぞれのフラグメント1 μl、事前に作製したF3 1 μl、及びEcoRVとNotIとで切断したpcDNA 3.1ベクター1 μl (25 ng) を混ぜ、そこにMighty mix 3 μl (TaKaRa) を加えて混ぜて合わせ、16℃で30分間連結反応を行い、この反応産物をG1とした。このG1サンプルに大腸菌DH5α株のコンピテントセル (TaKaRa) 20 μlを加えて、氷上で30分間反応させ、42℃で45秒間反応後、SOC培地を200 μl加えて37℃、200 rpmで1時間振とう培養した。これをX-gal (8 mg/ml) を塗布したアンピシリン (50 μl/ml) 含有LB寒天培地に60 μlずつ播いて37℃で12時間培養した。得られたG1のコロニーをQIAprep spin Miniprep Kit (QIAGEN) を用いてプラスミド抽出し、これ発現プラスミドとした。
さらに,これらpcDA3.1 (+) に組込まれたcDNAを、さらにEcoRVとXhoIで切り出し、Klenow fragment of DNA polymerase I で末端を平滑化し、上記(7−2)で調製したpEF-BOSベクターに組込み、発現用プラスミドとして併用した。
【0060】
(8)遺伝子導入
COS-7細胞を1×105cells/mlに調製し、そのうち2 mlを組織培養用6ウエルプレート (IWAKI) に播種し,CO2インキュベーター (37℃,5% CO2) で維持した。継代培養して1,2日後に培養液を吸引除去し、同量のDMEM-10% FBSに置き換えた。DMEM 97 μlとFuGENE 6 Transfection Reagent (Roche Applied Science) 3 μlを混ぜ、室温で5分間放置した後、上記(7−3)の各種ミニ・アグリンcDNAを組込んだ発現プラスミド 2 μgを混ぜ、室温で60分間反応させた。この混合液を1ウエルにつき100 μl加え、CO2インキュベーター (37℃,5% CO2) で培養した。
【0061】
(9)発現タンパク質のイムノブロット解析
遺伝子導入から24時間後に、培養液を同量のFCS不含DMEMに置き換えた。さらに24時間CO2インキュベーター (37℃,5% CO2) で培養した後、上清を回収し、1500 rpm、20℃で5分間遠心した。得られた上清をさらに Microcon YM-10 (Millipore, Billerica, MA, USA) を用いて濃縮し、イムノブロットのサンプルとした。これを等量の2 × サンプルBuffer (Tris Base 282 mM,Tris-HCl 212 mM,146 nM lithium dodecyl sulfate,2.18 M Glycerol,1.02 mM EDTA,100 mM DTT,0.44 mM Serva Blue G250,0.35 mM Phenol Red,pH 8.4; Invitrogen) と混合し、95℃で5分加熱した。各サンプル5-15 μl (50 ng-3 μg) を4-12%SDSポリアクリルゲル電気泳動 (NuPAGE 4-12% Bis-Tris GelおよびNuPAGE MOPS SDS Running Buffer; Invitrogen) にて、200 Vで50分泳動した。泳動後のタンパク質はPVDFメンブレン (Invitrogen) に30 Vで60分かけて転写した。転写後のメンブレンは、3% BSAを含むトリス緩衝生理食塩水 (Tris-buffered saline; TBS; 20 mM Tris,150 mM NaCl, pH 7.5) に浸して室温で1時間ブロッキングした。続いて、0.05% Tween 20 (Bio-Rad Laboratories, CA, USA) を含むTBS (TTBS) でメンブレンを10分間ずつ3回洗浄した後、ウサギ抗アグリンポリクローナル抗体 (sc-25528; SANTA CRUZ BIOTECHNOLOGY, Santa Cruz, CA, USA) を1% BSAを含むTTBS (1% BSA-TTBS) で500倍希釈した溶液を用いて1次抗体反応を行った。4℃で1晩反応させた後、メンブレンを10分ずつ3回洗浄した後、HRP標識ヤギ抗ウサギIgG抗体 (MP Biomdicals, Inc.-Cappel Products, Costa Mesa, CA, USA) を50,000倍希釈した溶液で2次抗体反応を行った。室温で1時間反応させた後、メンブレンを10分間ずつ3回洗浄し、ECL Plus (GE Healthcare UK Ltd., Buckinghamshire, England) 試薬と室温で5分反応させた。シグナルの検出には、LAS-4000UVmini (Fujifilm, 東京) を使用した。検出方法はchemiluminescence,露出方法はprecisionに設定した。
【0062】
(アグリン・バリアントのcDNAクローニングの結果)
C57BL/6脊髄、BALB/c全脳、N1E-115細胞、NSC-34細胞、未分化型C2C12細胞、分化型C2C12細胞からtotal RNAを抽出し、RT-PCRを行った。プライマーには、既知のアグリンmRNAの翻訳領域と3'側非翻訳領域の塩基配列に基づいて合成した。初回に使用したプライマーのセット (AgAFwとAgCRv;参照:表2) では非特異的増幅が高頻度で認められたため、さらに1組のプライマー(AgBFwとAgDRv; 参照:表2) を使用し、ネステッドPCRを行った。PCR産物を1%アガロースゲルで電気泳動したところ、全てのサンプルで800-1200 bpの範囲の複数のバンドが検出された (図1)。
増幅されたDNA断片の塩基配列を決定するために、PCR産物を組織、細胞ごとにアガロースゲルから回収し、pCR2.1-TOPOベクターに組込んだ。大腸菌コンピテントセルを形質転換し、X-galを塗布した寒天培地上で得られた白色コロニー6個を1つのプールとし、プラスミドDNAを抽出してPCRを行った。既知のz0型のアグリンから予想される916 bpよりも長い増幅産物を与えるプールを選択し、さらに個別のコロニーを同様に検索して目的とするcDNAクローンを得た。このような過程を経て最終的に、C57BL/6マウス脊髄から9個、BALB/cマウス全脳から7個、N1E-115細胞から6個、NSC-34細胞から2個、未分化型C2C12細胞から2個、分化型C2C12細胞から2個のcDNAクローンが得られた。挿入されたcDNAの塩基配列を決定し、BLAST version 2. 2. 18を用いてホモロジー検索を行った結果、いずれの配列も既知のマウス・アグリンと少なくとも部分的に一致した。
【0063】
(アグリンmRNAの多様性の確認)
C57BL/6脊髄由来のPCR産物には、Z部位にエクソン挿入の無いz0型、24 bpのエクソン挿入のあるz8型、57 bpのエクソン挿入のあるz19型のcDNAクローンが含まれていた。これらの塩基配列は、既にHochらの報告{Hoch, W., Ferns, M., Campanelli, J. T., Hall, Z. W., and Scheller, R. H. (1993). Developmental regulation of highly active alternatively spliced forms of agrin. Neuron 11, 479-490.}しているスプライシング・バリアントと一致した。これらに加えて、新たな2種類のバリアントを見出した。一方のバリアントでは、これまでに35番目のイントロンがスプライシングを受けずに残存していた。このmRNAの場合、異なる終止コドンが現れるため、翻訳領域が既知のz0型よりも102 bp短くなるものであった (図4)。もう一方のバリアントは、30番目のイントロンの残存により途中で終止コドンが現れ、C末端側の翻訳領域が既知のz0型より543 bp短くなるものであった。これらのmRNAから予想されるタンパク産物は既知のアグリンのC末端と比較して86アミノ酸、233アミノ酸ずつ短くなることが推定された。特にSC2では、LG3ドメイン全体が欠落することが予想された (図3及び5)。本発明では、これらのタイプをSC1 (short C-terminal variant-1)、SC2 (short C-terminal variant-2) 型と命名した。さらにこれ以外にも、Z部位に24 bpエクソン挿入があり、かつイントロン残存のあるz8-SC2型のcDNAクローンを単離した。
またBALB/c 全脳由来のPCR産物からは、Z部位にエクソン挿入の無いz0型、57 bpのエクソン挿入のあるz19型、イントロン残存のあるSC2型、Z部位に57 bpエクソン挿入があり、かつイントロン残存のあるz19-SC2型を同定した。N1E-115細胞由来のPCR産物からZ部位にエクソン挿入の無いz0型、イントロン残存のあるSC2型を同定した。NSC-34細胞由来のPCR産物からZ部位にエクソン挿入の無いz0型を同定した。未分型C2C12細胞由来のPCR産物からZ部位にエクソン挿入の無いz0型、エクソン挿入が無くかつイントロン残存のあるz0-SC1型を同定した。分化型C2C12由来のPCR産物からZ部位にエクソン挿入の無いz0型を同定した (図2及び4)。
【0064】
(タンパク質発現結果)
今回得られたアグリン分子のスプライシング・バリアントがどのような生理機能に関与するのかを調べるために、タンパク質発現系を確立した。従来の研究報告に基づいて、N末端がLN型になるようにすると同時に、神経細胞接着分子やヘパリン結合成長因子が結合する8つのFSドメイン (follistatin-like domain)、2つのLEドメイン (laminin EGF-like domain)、1つのSEAドメイン (sperm protein enterokinase and agrin domain)、2つのS/T部位 (serine/threonine sites) を除いたミニ・アグリン構造をとるように、発現プラスミドを構築した (図5)。N1E-115細胞から抽出したtotal RNAからRT-PCRによって得られた3つのDNA断片をつなぎ合わせて、原報通りのミニ・アグリンのN末端側cDNAを発現プラスミドpEF-BOS に組込んだ。さらに、このC末端側にあたる z0,z8,z19,SC1,SC2型のcDNAとをつなぎ合わせることにより、各バリアントの最終的な発現プラスミドを構築した (図6)。
次に、ミニ・アグリンcDNAを組込んだプラスミドをCOS-7細胞に導入し、24-48時間後の培養上清を回収し、分泌型のミニ・アグリンが作られているか、イムノブロット法により検出を試みた。その結果、z0,z8,z19,SC2型のサンプルについて単一のバンドを検出した(図7)。z0,z8,z19,SC2型のミニ・アグリンの推定分子量は、それぞれ109 kDa,110 kDa,111 kDa,84 kDaであり、図7の各バンドから計測した分子量,113 kDa,113 kDa,116 kDa,88 kDaに近い値であった。
【実施例2】
【0065】
(各アグリンのアセチルコリン受容体クラスター形成能の評価)
上記実施例1で発現させた各アグリンのアセチルコリン受容体クラスター形成能又は阻害活性を確認した。詳細は、以下の通りである。
【0066】
(nACRクラスター形成の確認方法)
C2C12細胞を5×104cells/mlに調製し、組織培養用6ウエルプレート (IWAKI) の各ウエルに2 mlずつ播種し、CO2インキュベーター (37℃,5% CO2) で培養した。24時間後に2% HS-DMEMに交換し、4日間培養して分化させた。myotubeが確認されたウエルに0.2% BSA-PBS(−) に溶解した組換え型アグリン (Rat C-terminal Agrin; R&D SYSTEMS, Minneapolis, MN, USA) 単独、COS-7細胞の培養上清に分泌された各種ミニ・アグリン単独、又は組換え型アグリン (R&D SYSTEMS) とCOS-7細胞の培養上清に分泌された各種ミニ・アグリンとの混合物を添加した。陰性対照として、それぞれの溶解希釈液を添加した。
添加15時間後に培養上清を除去し、PBS(−)で洗浄後、3.7%ホルムアルデヒド (16% ultrapure formaldehyde methanol free; Polysciences, Eppelheim, Germany)-PBSを加え、細胞を室温で10分間固定した。1% BSA-PBS(−)で1分間ずつ3回洗浄した後、蛍光標識されたa-ブンガロトキシン (a-bungarotoxin; a-BuTX, Alexa Fluor 488 conjugate; Molecular Probes, Eugene, OR, USA) を最終濃度100 nMになるように添加し、遮光して室温で1時間反応させた。1% BSA-PBS(−)で5分間ずつ2回洗浄した後Propong Gold antifade regent (Molecular Probes) を添加して封入した。蛍光観察には、20倍対物レンズ、NIBAフィルターを装着した倒立型顕微鏡 (IX 71; OLYMPUS, 東京) を使用した。画像の取り込みには、DP71デジタルカメラ (OLYMPUS) と専用ソフトウェア (DP ControllerとDP Manager; OLYMPUS) を使用した。評価方法は、Alexa 488のシグナルが直径10 μm以上のもをnAChRクラスターと定義して数えた。各ウエルにつき5視野ずつ撮影し、視野毎にmyotube 1本あたりのnAChRクラスター数を計算し、1ウエルあたりの平均値を算出した。mock-trasfectionで得られた値を100%として、それに対する各サンプルの割合を百分率に換算した。有意差検定はTukey-Kramer検定により、有意水準を1%とした。
【0067】
(各アグリンのアセチルコリン受容体クラスター形成能の評価)
COS-7細胞培養上清から回収したミニ・アグリンバリアントを分化誘導したC2C12細胞に添加した。添加15時間後にAlexa 488で蛍光標識されたa-BuTX を用いてnAChRの視覚化を試みた。クラスターはnAChRが集合した直径10μm以上の集合体と定義した。組換え型アグリン、z8型、及びz19型のミニ・アグリンを添加したC2C12細胞では、nAChRのクラスター形成が明瞭に観察された(図8A)。myotubeあたりのクラスター数は、mock-transfectionの値を100%とした場合、それぞれ835±21% (n=4),911±64% (n=4),859±103% (n=4) の値を示した(図8B)。これに対して、z0,SC2型のミニ・アグリンを添加したC2C12細胞では、ベクターのみ導入 (mock-transfection) したCOS-7細胞の上清と同様、nAChRのクラスター形成はほとんど観察されなかった(図8A)。myotubeあたりのクラスター数は、mock-transfectionと比べて、それぞれ89±21% (n=4)、106±30% (n=4) であった(図8B)。
次に、z0,SC2型のnAChRのクラスター形成阻害活性を検討した。mock-transfectionと組換え型アグリンとの混合物を添加した値686±77% (n=4) と比較して、z0型と組換え型アグリンとの混合物、及びSC2型と組換え型アグリンとの混合物ではそれぞれ430±24% (n=4)、498±55% (n=4) の値を示し、nAChRクラスター形成の有意な減少を確認した (p<0.01; 図9)。
【0068】
(総論)
上記実施例の結果より、z0型アグリン及びSC2型アグリンは、アセチルコリン受容体クラスター形成阻害活性を有することを確認した。これにより、z0型アグリン及び/又はSC2型アグリンを特異的に認識する抗体は、z0型アグリン及び/又はSC2型アグリンのアセチルコリン受容体クラスター形成阻害活性を阻害できると考えられる。
【産業上の利用可能性】
【0069】
本発明者らは、アセチルコリン受容体クラスター形成阻害活性を持つアグリンを特異的に認識できる抗体の提供を可能とした。さらに、該抗体は、アセチルコリン受容体クラスター形成能促進剤及びアセチルコリン受容体クラスター形成能を阻害するアグリン除去カラムに使用できる可能性がある。
【特許請求の範囲】
【請求項1】
SC2型アグリン及び/又はz0型アグリンを特異的に認識する抗体。
【請求項2】
下記配列番号1で表されるアミノ酸配列を含むペプチドを免疫原として使用する請求項1の抗体。
Glu-His-Trp-Gln-Pro-Arg-Ala-Glu-Thr(配列番号1)
【請求項3】
下記の特徴を有する請求項1又は2に記載の抗体。
(1)Glu-His-Trp-Gln-Pro-Arg-Ala-Glu-Thr(配列番号1)で表されるアミノ酸配列に1ないし数個のアミノ酸残基の欠失、置換、挿入もしくは付加、又は修飾を施すことにより得られるアミノ酸配列からなるペプチドを免疫原として使用する
(2)SC1型アグリン、z8型アグリン、z11型アグリン及び/又はz19型アグリンに結合しない
【請求項4】
モノクローナル抗体である請求項1〜3のいずれか1に記載の抗体。
【請求項5】
請求項1〜4のいずれか1に記載の抗体を含むアセチルコリン受容体クラスター形成能促進剤。
【請求項6】
前記促進剤が、以下のいずれか1に記載の治療剤である請求項5に記載のアセチルコリン受容体クラスター形成能促進剤。
(1)重症筋無力症
(2)筋萎縮性側索硬化症
(3)先天性筋ジストロフィー
(4)アルツハイマー病
(5)パーキンソン病
【請求項7】
請求項1〜4のいずれか1に記載の抗体を充填していることを特徴とするアセチルコリン受容体クラスター形成阻害活性を有するアグリン除去カラム。
【請求項8】
前記カラムは、以下の治療用途であることを特徴とするカラム。
(1)重症筋無力症
(2)筋縮性側索硬化症
(3)先天性筋ジストロフィー
(4)アルツハイマー病
(5)パーキンソン病
【請求項9】
以下の工程を含むz0型アグリン又はSC2型アグリンのアセチルコリン受容体クラスター形成能阻害活性を阻害する物質のスクリーニング方法;
(1)z0型アグリン及び/又はSC2型アグリン並びに候補阻害物質を細胞中に添加する工程、
(2)該細胞中のアセチルコリン受容体クラスター形成能を指標として、候補阻害物質の阻害活性を測定する工程。
【請求項1】
SC2型アグリン及び/又はz0型アグリンを特異的に認識する抗体。
【請求項2】
下記配列番号1で表されるアミノ酸配列を含むペプチドを免疫原として使用する請求項1の抗体。
Glu-His-Trp-Gln-Pro-Arg-Ala-Glu-Thr(配列番号1)
【請求項3】
下記の特徴を有する請求項1又は2に記載の抗体。
(1)Glu-His-Trp-Gln-Pro-Arg-Ala-Glu-Thr(配列番号1)で表されるアミノ酸配列に1ないし数個のアミノ酸残基の欠失、置換、挿入もしくは付加、又は修飾を施すことにより得られるアミノ酸配列からなるペプチドを免疫原として使用する
(2)SC1型アグリン、z8型アグリン、z11型アグリン及び/又はz19型アグリンに結合しない
【請求項4】
モノクローナル抗体である請求項1〜3のいずれか1に記載の抗体。
【請求項5】
請求項1〜4のいずれか1に記載の抗体を含むアセチルコリン受容体クラスター形成能促進剤。
【請求項6】
前記促進剤が、以下のいずれか1に記載の治療剤である請求項5に記載のアセチルコリン受容体クラスター形成能促進剤。
(1)重症筋無力症
(2)筋萎縮性側索硬化症
(3)先天性筋ジストロフィー
(4)アルツハイマー病
(5)パーキンソン病
【請求項7】
請求項1〜4のいずれか1に記載の抗体を充填していることを特徴とするアセチルコリン受容体クラスター形成阻害活性を有するアグリン除去カラム。
【請求項8】
前記カラムは、以下の治療用途であることを特徴とするカラム。
(1)重症筋無力症
(2)筋縮性側索硬化症
(3)先天性筋ジストロフィー
(4)アルツハイマー病
(5)パーキンソン病
【請求項9】
以下の工程を含むz0型アグリン又はSC2型アグリンのアセチルコリン受容体クラスター形成能阻害活性を阻害する物質のスクリーニング方法;
(1)z0型アグリン及び/又はSC2型アグリン並びに候補阻害物質を細胞中に添加する工程、
(2)該細胞中のアセチルコリン受容体クラスター形成能を指標として、候補阻害物質の阻害活性を測定する工程。
【図1】

【図2】

【図3】

【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公開番号】特開2011−173793(P2011−173793A)
【公開日】平成23年9月8日(2011.9.8)
【国際特許分類】
【出願番号】特願2010−36739(P2010−36739)
【出願日】平成22年2月23日(2010.2.23)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.テフロン
【出願人】(504160781)国立大学法人金沢大学 (282)
【Fターム(参考)】
【公開日】平成23年9月8日(2011.9.8)
【国際特許分類】
【出願日】平成22年2月23日(2010.2.23)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.テフロン
【出願人】(504160781)国立大学法人金沢大学 (282)
【Fターム(参考)】
[ Back to top ]