
アセトイン産生細胞および当該細胞を用いたアセトインの製造方法
【課題】アセトインを効率的かつ簡便に産生することのできる細胞および、当該細胞を用いたアセトイン製造方法の提供。
【解決手段】アセトイン脱水素酵素の機能が欠損または低減しており、ピルビン酸からアセト乳酸、及び、アセト乳酸からアセトインの合成を司る酵素群が構成的に発現している、アセトイン産生細胞。アセトイン脱水素酵素、リン酸アセチル基転移酵素、および乳酸脱水素酵素の3種のタンパク質の機能が欠損または低減している、アセトイン産生細胞。これらアセトイン産生細胞を用いたアセトインの製造方法。
【解決手段】アセトイン脱水素酵素の機能が欠損または低減しており、ピルビン酸からアセト乳酸、及び、アセト乳酸からアセトインの合成を司る酵素群が構成的に発現している、アセトイン産生細胞。アセトイン脱水素酵素、リン酸アセチル基転移酵素、および乳酸脱水素酵素の3種のタンパク質の機能が欠損または低減している、アセトイン産生細胞。これらアセトイン産生細胞を用いたアセトインの製造方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、アセトイン産生細胞および当該細胞を用いたアセトインの製造方法に関する。
【背景技術】
【0002】
アセトイン(3−ヒドロキシ−2−ブタノン)は、ヨーグルト、バター様の香りを持つ化合物であり、食品添加物、化粧品原料として使用される。またアセトインに類似する化合物であるジアセチル(2,3−ブタンジオン)は、食品添加物として用いられるのみならず、医薬品製造においても利用されている。
【0003】
アセトインは微生物等の細胞を用いて産生することが可能である。細胞内においてアセトインは、例えばクエン酸回路で生じたピルビン酸の二量体化によって生成するアセト乳酸が脱炭酸されることにより生成される。しかしながら細胞では、酢酸や乳酸などの他の代謝物が生成されてしまうため、アセトインの分離精製が困難であり、また細胞によるアセトイン産生の効率が悪く経済的でないという問題点がある。
【0004】
微生物等の細胞を用いたアセトインの製造方法に関して、従来より乳酸菌を用いた手法が検討されている(特許文献1,2)。特許文献1では、鉄ポルフィリンや金属塩等を添加した培地にて乳酸菌を培養することによりアセトイン類を製造する方法が開示されている。特許文献2では、薬剤を用いて突然変異を誘発することにより、アセトイン類の生成能の高い乳酸菌株を得たことが開示されている。
【0005】
枯草菌は元来糖質に富む培地条件で培養すると、細胞増殖の過程でアセトインを生産し培地中に蓄積すること(非特許文献1)、その後に細胞増殖を停止すると培地中のアセトインを再度分解してエネルギーを得ることが知られている。非特許文献1では、枯草菌を培養する際に、培地成分等の培養条件を変更することにより、アセトインの産生が増加したことが開示されている。また、アセトイン還元酵素をコードするbdhA遺伝子を破壊した枯草菌では、定常期にアセトインを蓄積することが知られている(非特許文献2)。
【0006】
枯草菌のアセトイン合成経路を司る酵素群は、単一のオペロンを形成し、かかるオペロン(alsSD)の発現を制御する制御因子としてalsRが知られている。alsR遺伝子は、alsSDオペロンとは独立に転写されており、alsR遺伝子を破壊するとalsDが発現しなかったことが非特許文献3にて報告されている。また、alsSDの発現を促進するalsR変異株を得たことが非特許文献3にて報告されている。
【0007】
これまで微生物等の細胞を用いてアセトインを効率よく産生するために、単一の遺伝子のみではなく、代謝系に関連する複数の遺伝子に着目して改変を施した例は報告されていない。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】特開平3−219884号公報
【特許文献2】特開平4−99480号公報
【非特許文献】
【0009】
【非特許文献1】Appl Micorobiol Biotechonol 74(1), 61-67(2007)
【非特許文献2】Appl Environ Microbiol 74(22), 6832-6838(2008)
【非特許文献3】J Bacteriol 175(12),3863-3875(1993)
【発明の概要】
【発明が解決しようとする課題】
【0010】
本発明は、アセトインを効率的かつ簡便に産生することのできる細胞および、当該細胞を用いたアセトイン製造方法を提供することを課題とする。
【課題を解決するための手段】
【0011】
上記課題を解決するために本発明者らは、鋭意検討した結果、枯草菌においてアセトインの再分解系を削除するとともに、アセトイン合成酵素群が構成的に発現するように遺伝子制御を改変する、および/または、アセトイン合成への原料供給を増やすように代謝系を改変することにより、アセトインを効率よく産生可能な細胞を作製しうることを見出し、本発明を完成した。
【0012】
すなわち、本発明は以下よりなる。
1.アセトイン脱水素酵素の機能が欠損または低減しており、ピルビン酸からアセト乳酸、及び、アセト乳酸からアセトインの合成を司る酵素群が構成的に発現している、アセトイン産生細胞。
2.前記酵素群をコードする遺伝子が単一のオペロンを形成している、前項1に記載のアセトイン産生細胞。
3.アセトイン脱水素酵素の機能の欠損または低減が、アセトイン脱水素酵素をコードする遺伝子を人為的に破壊したことによるものであり、且つ、前記酵素群の構成的発現が、前記オペロンの発現制御系を改変するような変異を導入したことによるものである、前項2に記載のアセトイン産生細胞。
4.アセトイン脱水素酵素、リン酸アセチル基転移酵素、および乳酸脱水素酵素の3種のタンパク質の機能が欠損または低減している、アセトイン産生細胞。
5.アセトイン脱水素酵素、リン酸アセチル基転移酵素、および乳酸脱水素酵素の3種のタンパク質の機能の欠損または低減が、アセトイン脱水素酵素をコードする遺伝子、リン酸アセチル基転移酵素をコードする遺伝子、および乳酸脱水素酵素をコードする遺伝子の3種の遺伝子を人為的に破壊したことによるものである、前項4に記載のアセトイン産生細胞。
6.さらに、リン酸アセチル基転移酵素および乳酸脱水素酵素のうち、少なくとも1種のタンパク質の機能が欠損または低減している、前項1〜3のいずれか1に記載のアセトイン産生細胞。
7.リン酸アセチル基転移酵素および乳酸脱水素酵素のうち、少なくとも1種のタンパク質の機能の欠損または低減が、リン酸アセチル基転移酵素をコードする遺伝子、および乳酸脱水素酵素をコードする遺伝子のうち、少なくとも1種の遺伝子を人為的に破壊したことによるものである、前項6に記載のアセトイン産生細胞。
8.細胞がバチルス属に属する菌である前項1〜7のいずれか1に記載の細胞。
9.細胞が、受領番号FERM AP-22036(受領日:2010年10月28日)の枯草菌変異株である、前項8に記載の細胞。
10.前項1〜9のいずれか1に記載の細胞を、グルコースの存在下で培養する工程を含む、アセトイン製造方法。
11.前項1〜3、6〜9のいずれか1に記載の細胞を、1〜10w/v%グルコースの存在下で培養する工程を含む、アセトイン製造方法。
12.前項4、5,8のいずれか1に記載の細胞を、0.2〜2w/v%グルコースの存在下で培養する工程を含む、アセトイン製造方法。
13.前記の培養する工程で得られた培養ろ液からアセトインを単離する工程をさらに含む、前項10〜12のいずれか1に記載のアセトイン製造方法。
【発明の効果】
【0013】
本発明の細胞は、アセトインを、グルコース等の糖類を原料として、細胞内で安価にかつ、90%(モル比)という高効率で産生することができる。本発明の細胞は、細胞の増殖量や生存率は、野生型の細胞と同様であるため、培養の際に特別な操作や特別な添加物等を必要とすることもない。また、かかる細胞を用いた本発明のアセトインの製造方法は、アセトインの産生を1段階で行うことができ簡便である。
【図面の簡単な説明】
【0014】
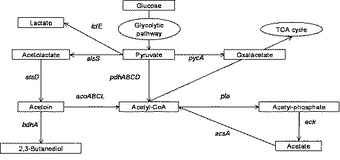
【図1】枯草菌の代謝系の概略を示す図である。
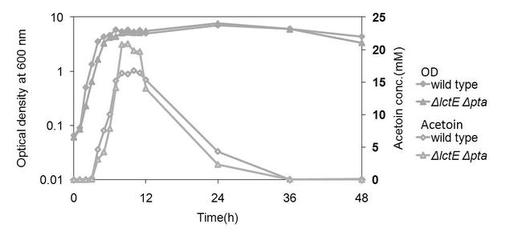
【図2】変異株1(ΔlctEΔpta)を、グルコースを含む培地にて培養した場合の、アセトイン産生量を示す図である。(参考例1)
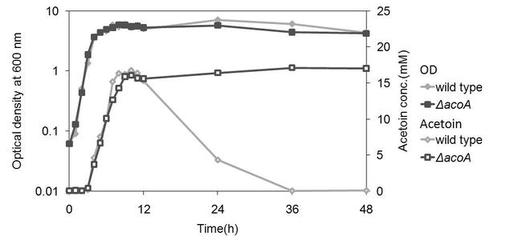
【図3】変異株2(ΔacoA)を、グルコースを含む培地にて培養した場合の、アセトイン産生量を示す図である。(参考例2)
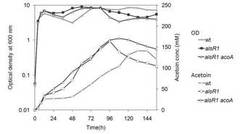
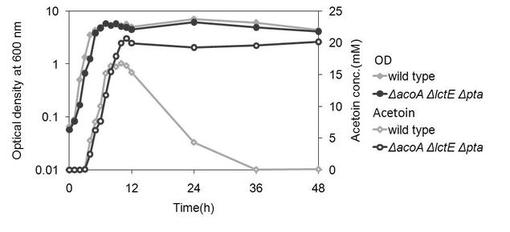
【図4】アセトイン産生細胞1(ΔacoAΔlctEΔpta)を、0.4w/v%グルコースを含む培地にて培養した場合の、アセトイン産生量を示す図である。(実施例2)
【図5】アセトイン産生細胞1(ΔacoAΔlctEΔpta)を、4w/v%グルコース含む培地にて培養した場合の、アセトイン産生量を示す図である。(実施例2)
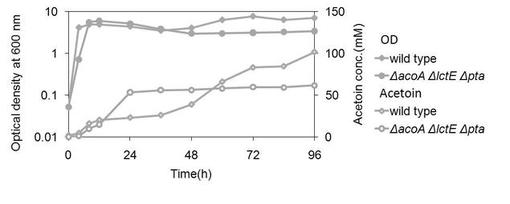
【図6】変異株3(ΔalsR1)を、グルコースを含む培地にて培養した場合の、アセトイン産生量を示す図である。(参考例3)
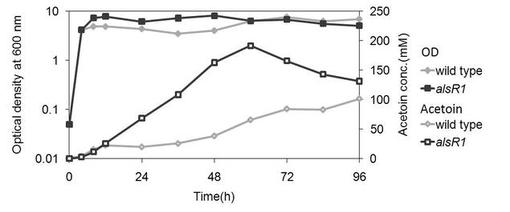
【図7】アセトイン産生細胞2(alsR1ΔacoA)を、4w/v%グルコース含む培地にて培養した場合の、アセトイン産生量を示す図である。(実施例4)
【発明を実施するための形態】
【0015】
本発明は、細胞内の代謝系が改変されたことによりアセトインを効率的に産生することのできる細胞を対象とする。本発明では、細胞内の代謝系の改変することにより、特定の酵素の機能が欠損または低減している、および/または特定の酵素群が構成的に発現する細胞を得ることができる。
【0016】
本発明において、「酵素の機能が欠損または低減している細胞」とは、野生型の細胞に比べて、対象となる酵素自体の機能が低下もしくは消失していたり、量が低減していたり、発現していない細胞などが例示される。具体的には、「酵素の機能が欠損または低減している細胞」とは、対象となる酵素をコードする遺伝子に変異が導入されていたり、該遺伝子が破壊されている細胞が例示される。本発明の細胞は、天然に生じたものであってもよいし、人為的に当該遺伝子を破壊することにより作製したものであってもよい。本発明の細胞は、好ましくは、人為的に対象となる酵素をコードする遺伝子を破壊することにより作製したものである。
【0017】
本発明において「構成的」とは本発明が属する技術分野において一般的に用いられている意味で用いられる。本発明において、「酵素群が構成的に発現している細胞」とは、環境条件による調節を受けずに常に一定レベルで目的の酵素群が発現している細胞であり、例えば、野生型の細胞では対象となる酵素群の発現が抑制されるような環境条件下であっても、定常的に目的の酵素群が発現し続けている細胞を意味する。
【0018】
本発明において「細胞」とは、アセトイン代謝系の酵素を有し、細胞内の代謝系が合理的に改変された結果アセトインを効率的に産生し得る細胞であればよく、微生物、動物細胞、植物細胞など、種類は制限されない。好ましくは、アセトイン代謝系を有する微生物を用いることができる。微生物は細菌であることが好ましく、グラム陽性菌であることがより好ましい。グラム陽性菌としては、バチルス属(Bacillus)、ジオバチルス属(Geobacillus)、ラクトバシラス属(Lactobacillus)、ラクトコッカス属(Lactococcus)、スタフィロコッカス属(Staphylococcus)、クレブシエラ属(Klebsiella)、ストレプトコッカス属(Streptococcus)に属する菌を用いることが可能である。特に、バチルス属に属する菌を用いることが好ましく、枯草菌(Bacillus subtilis)を用いることがより好ましい。
【0019】
(アセトイン産生細胞)
本発明のアセトイン産生細胞は、アセトイン脱水素酵素の機能が欠損または低減しており、ピルビン酸からアセト乳酸、及び、アセト乳酸からアセトインの合成を司る酵素群が構成的に発現していることを特徴とする。
【0020】
アセトイン脱水素酵素は、アセトインを再分解してアセチルCoAを生成する経路に関与する酵素である。本発明のアセトイン産生細胞内においては、アセトイン脱水素酵素の機能が欠損または低減しており、細胞外に蓄積するアセトインを消費する能力を欠失または低減している。アセトイン脱水素酵素の機能を欠損または低減させる手段は、いかなる手段によってもよいが、アセトイン脱水素酵素をコードする遺伝子を破壊する手段が好ましい。アセトイン脱水素酵素をコードする遺伝子の具体例としては、例えば、配列番号1で表される塩基配列からなる遺伝子(acoA遺伝子、NCBI-GI: 16077873、NC_000964.3参照)および当該遺伝子と同等の機能を有する遺伝子が挙げられる。配列番号1で表される塩基配列からなる遺伝子と同等の機能を有する遺伝子としては、配列番号1で表される塩基配列において1個以上(例えば2〜3個)のヌクレオチドが置換、欠失、挿入および/もしくは付加された塩基配列からなり、アセトイン脱水素酵素の活性を有するタンパク質をコードする遺伝子が挙げられる。
【0021】
例えば枯草菌では、炭素源が枯渇している場合にアセトインからアセチルCoAを生成するアセトイン再分解系として、acoABCLオペロンという一連の遺伝子群が知られている(図1、Huang, M., Oppermann-Sanio, .FB., and Steinbuchel, A., J Bacteriol. 1999 Jun;181(12):3837-3841)。acoABCLオペロンの遺伝子は、アセトイン脱水素酵素を構成するサブユニットを各々コードしている。枯草菌においては、アセトイン再分解系を抑制するために、acoABCLオペロンのいずれの遺伝子を破壊してもよいが、好ましくはacoA遺伝子を破壊した変異株を作製すればよい。アセトイン再分解系を抑制することにより、アセトイン産生細胞により産生されたアセトインが培地中から減少することを防ぐことができ、培地中のアセトイン量を維持することが可能となる。
【0022】
本発明のアセトイン産生細胞においては、アセトインの合成経路そのものを増強するために、ピルビン酸からアセト乳酸、及び、アセト乳酸からアセトインの合成を司る酵素群が構成的に発現している。ピルビン酸からアセト乳酸、及び、アセト乳酸からアセトインの合成を司る酵素群は、アセト乳酸合成酵素およびアセト乳酸脱炭酸酵素を含む酵素群である。これらの酵素群は、オペロンによりコードされていてもよく、当該オペロンとしては例えばalsSDオペロンが例示される。当該酵素群を構成的に発現させる手段は、いかなる手段によってもよいが、該酵素群がオペロンによりコードされる場合は、前記オペロンの発現制御系を改変するような変異を導入することが好ましい。前記オペロンの発現制御系の改変としては、オペロンの発現を制御する制御因子に変異を導入して恒常的活性化型にすることが例示され、好ましくは、当該制御因子の恒常的活性化型が細胞内に導入される。
【0023】
当該制御因子の恒常的活性化型をコードする遺伝子の具体例としては、例えば、配列番号2で表される塩基配列(alsR遺伝子、NC_000964.3参照)において、601番目のアデニン(A)がグアニン(G)に置換された塩基配列を有するalsR1遺伝子(すなわち、非特許文献3、FIG. 4の塩基配列における1028番目のチミン(T)がシトシン(C)に置換された塩基配列を有するalsR1遺伝子)、および当該alsR1遺伝子と同等の機能を有する遺伝子が挙げられる。alsR1遺伝子と同等の機能を有する遺伝子としては、配列番号2の塩基配列において1個以上(例えば2〜3個)のヌクレオチドが置換、欠失、挿入および/もしくは付加された塩基配列からなり、前記オペロンの制御因子の恒常的活性化型としての機能を有するタンパク質をコードする遺伝子が挙げられる。alsR1遺伝子と同等の機能を有する遺伝子の具体例としてalsR8遺伝子が例示される。alsR8遺伝子は、配列番号2で表される塩基配列において、700番目のアデニン(A)がシトシン(C)に置換された塩基配列を有する遺伝子(すなわち、非特許文献3、FIG. 4の塩基配列における929番目のチミン(T)がグアニン(G)に置換された塩基配列を有するalsR8遺伝子)である。
【0024】
例えば、枯草菌ではアセトインの合成経路に関連する酵素(アセト乳酸合成酵素およびアセト乳酸脱炭酸酵素)は、alsSDオペロンにより発現される。alsSDオペロンの転写は、制御因子のalsRが、alsSDオペロンの特定の配列に結合することにより促進される。alsRはリガンドに結合することにより活性化型となり、配列特異的DNA結合能を獲得する。alsRはリガンドとの結合が解除されることにより不活性化型となり、alsSDオペロンの発現が停止する。alsRの変異型タンパク質として、alsR1(alsR1遺伝子によりコードされるタンパク質であり、alsRのアミノ酸配列(NCBI-GI: 16080655)において201番目のスレオニンがアラニンにより置換されているタンパク質)やalsR8(alsR8遺伝子によりコードされるタンパク質であり、alsRのアミノ酸配列において234番目のセリンがアラニンにより置換されているタンパク質)などがあるが、これらはalsRの恒常的活性化型として知られている(非特許文献3)。枯草菌において、alsSDオペロンを構成的に発現させるためには、alsR1やalsR8等の恒常的活性化型の制御因子を細胞内に導入すればよい。恒常的活性化型の制御因子が導入された細胞は、アセトインの合成経路が増強されているため、高効率でアセトインを産生することができる。
【0025】
アセトイン脱水素酵素の機能が欠損または低減しており、ピルビン酸からアセト乳酸、及び、アセト乳酸からアセトインの合成を司る酵素群が構成的に発現していることを特徴とするアセトイン産生細胞では、90%(モル比)という高いアセトイン変換率を達成するとともに、長時間培養した場合でも、アセトインが再分解されることなく、培地中のアセトイン量が維持され続ける。また、かかるアセトイン産生細胞を培養する培地中におけるグルコース濃度は特に限定されないが、高濃度(2〜10w/v%、好ましくは2〜6w/v%、より好ましくは3〜5w/v%、さらに好ましくは4w/v%)でグルコースを含む培地にて培養した場合であっても、高いアセトイン変換率を有する。グルコースを高濃度で含む培地においては、浸透圧が上昇するため細胞の生存率自体が低下する場合もあるが、本発明のアセトイン産生細胞は、増殖、生存が可能である。
【0026】
本発明は、アセトイン脱水素酵素の機能が欠損または低減しており、ピルビン酸からアセト乳酸、及び、アセト乳酸からアセトインの合成を司る酵素群が構成的に発現していることを特徴とするアセトイン産生細胞は、さらにリン酸アセチル基転移酵素および乳酸脱水素酵素のうち、少なくとも1種のタンパク質の機能が欠損または低減しているアセトイン産生細胞を包含する。
【0027】
リン酸アセチル基転移酵素は、アセチルCoAに他の分子からリン酸基を転移・付加してアセチルリン酸を合成する反応を触媒する酵素であり、アセチルCoAの酢酸発酵に関与する酵素である。本発明のアセトイン産生細胞内においては、リン酸アセチル基転移酵素の機能が欠損または低減している。リン酸アセチル基転移酵素の機能を欠損または低減させる手段は、いかなる手段によってもよいが、リン酸アセチル基転移酵素をコードする遺伝子を破壊する手段が好ましい。リン酸アセチル基転移酵素をコードする遺伝子の具体例としては、配列番号3で表される塩基配列からなる遺伝子(pta(phosphotransacetylase)遺伝子、NCBI-GI: 16080818、NC_000964.3参照)および当該遺伝子と同等の機能を有する遺伝子が挙げられる。配列番号3で表される塩基配列からなる遺伝子と同等の機能を有する遺伝子としては、配列番号3で表される塩基配列において1個以上(例えば2〜3個)のヌクレオチドが置換、欠失、挿入および/もしくは付加された塩基配列からなり、リン酸アセチル基転移酵素の活性を有するタンパク質をコードする遺伝子が挙げられる。
【0028】
乳酸脱水素酵素は、ピルビン酸から乳酸を合成する反応を触媒する酵素であり、ピルビン酸の乳酸発酵に関与する酵素である。本発明のアセトイン産生細胞内においては、乳酸脱水素酵素の機能が欠損または低減している。乳酸脱水素酵素の機能を欠損または低減させる手段は、いかなる手段によってもよいが、乳酸脱水素酵素をコードする遺伝子を破壊する手段が好ましい。乳酸脱水素酵素をコードする遺伝子としての具体例は、例えば、配列番号4で表される塩基配列からなる遺伝子(lct(L-lactate dehydrogenase)遺伝子、NCBI-GI: 255767083、NC_000964.3参照)および当該遺伝子と同等の機能を有する遺伝子が挙げられる。配列番号4で表される塩基配列からなる遺伝子と同等の機能を有する遺伝子としては、配列番号4で表される塩基配列において1個以上(例えば2〜3個)のヌクレオチドが置換、欠失、挿入および/もしくは付加された塩基配列からなり、乳酸脱水素酵素の活性を有するタンパク質をコードする遺伝子が挙げられる。
【0029】
対象とするタンパク質が上記各種酵素や制御因子としての活性を有するか否か、対象とする遺伝子がこれらの活性を有するタンパク質をコードしているか否かは、自体公知の酵素学的アッセイなどの方法で確認可能である。
【0030】
本発明のアセトイン産生細胞として、具体的には、独立行政法人産業技術総合研究所特許生物寄託センター(〒305-8566 茨城県つくば市東1-1-1 つくばセンター 中央第6)に、受領番号FERM AP-22036(受領日:2010年10月28日)にて寄託した枯草菌変異株(名称 Bacillus subtilis MY05株)が例示される。
【0031】
また本発明は、アセトイン脱水素酵素、リン酸アセチル基転移酵素、および乳酸脱水素酵素の3種のタンパク質の機能が欠損または低減していることを特徴とするアセトイン産生細胞も包含する。
【0032】
例えば枯草菌では、グルコースなどの糖類を利用する代謝系として、TCA回路、乳酸発酵、アセトイン発酵、乳酸発酵等が知られている。例えば、lctE遺伝子(乳酸脱水素酵素をコードする遺伝子)および/またはpta遺伝子(リン酸アセチル基転移酵素をコードする遺伝子)を破壊した枯草菌変異株では、乳酸発酵および/または酢酸発酵が抑制され、原料であるピルビン酸のアセトイン合成への供給が高められており、アセトインを高効率で産生することができる。lctE遺伝子およびpta遺伝子に加えて、アセトイン脱水素酵素をコードする遺伝子(acoA遺伝子)の三種の遺伝子を破壊したアセトイン産生細胞を用いれば、アセトインを高効率で産生することができるとともに、培地中のアセトインは減少することなく、維持され続ける。
【0033】
すなわち、アセトイン脱水素酵素、リン酸アセチル基転移酵素、および乳酸脱水素酵素の3種のタンパク質の機能が欠損または低減していることを特徴とするアセトイン産生細胞では、90%(モル比)という高いアセトイン変換率を達成するとともに、長時間培養した場合でも、アセトインが再分解されることなく、培地中のアセトイン量が維持され続ける。また、かかるアセトイン産生細胞を培養する培地中におけるグルコース濃度は特に限定されないが、低濃度(0.2〜2w/v%、好ましくは0.2〜1w/v%、より好ましくは0.4w/v%)でグルコースを含む培地にて培養することが好ましい。
【0034】
(アセトイン産生細胞の作製方法)
本発明のアセトイン産生細胞は、目的の酵素をコードする遺伝子を破壊する方法および、目的の制御因子をコードする遺伝子を導入する方法を用いて作製することができる。
【0035】
目的の酵素の機能を欠損または低減させる方法としては、目的の酵素をコードする遺伝子を破壊する方法が例示され、通常の変異による方法を使用することができる。例えば、物理的および/または化学的変異誘発処理では、UV照射、放射線照射などの物理的変異方法の他、ニトロソグアニジン、メタンスルホン酸エチル、亜硝酸、メタンスルホン酸メチル、アクリジン色素、ベンゾピレン、硫酸ジメチルなどの変異剤の混合による化学的変異方法が例示される。これらの変異処理は、遺伝子上で塩基の挿入、欠失および/または置換が期待される方法である。変異処理においては、遺伝子が破壊される限り、一個の塩基を挿入、欠失および/または置換してもよく、複数個の塩基を挿入、欠失、および/または置換してもよい。
【0036】
さらに、目的の酵素をコードする遺伝子の別の人為的な破壊方法として、相同組換えによる塩基の挿入、欠失および/または置換を行なう方法がある。相同組換え法では、人為的に塩基の挿入、欠失および/または置換を施した目的の遺伝子と同じ配列を部分的に有する塩基配列を導入し、相同組換えによって変異を行なう方法である。例えば、薬剤耐性遺伝子であるクロラムフェニコール耐性遺伝子、カナマイシン耐性遺伝子、テトラサイクリン耐性遺伝子、エリスロマイシン耐性遺伝子、スペクチノマイシン耐性遺伝子、アンピシリン耐性遺伝子、ハイグロマイシン耐性遺伝子、ネオマイシン耐性遺伝子などを挿入した塩基配列を導入した後、相当する薬剤で選抜し、生存する細胞を得る方法である。さらに、望むべき部分に挿入されたことをPCRなどの機器を用いて確認することによって、目的の細胞を得ることが可能である。なお、「目的の酵素の機能もしくは目的の酵素をコードする遺伝子を人為的に破壊する」とは、例えば突然変異の導入、または相同組み換えにより、当該遺伝子の転写を抑制し、または当該遺伝子にコードされるタンパク質の機能を破壊することをいう。
【0037】
本発明の細胞の作製において、目的のタンパク質を構成的に発現させるために、目的のタンパク質の発現を制御する制御因子をコードする遺伝子を導入する方法が例示される。当該制御因子をコードする遺伝子を導入する方法としては、自体公知の方法を用いることができる。
【0038】
まず目的の制御因子をコードする遺伝子を、微生物等のゲノムDNAを鋳型にして変異を導入して取得するか、又は合成することにより取得する。微生物等のゲノムDNAから所望の遺伝子を取得する方法および遺伝子に変異を導入する方法は分子生物学の分野において周知である。例えば遺伝子の配列が既知の場合、制限エンドヌクレアーゼ消化により適したゲノムライブラリを作り、所望の遺伝子配列に相補的なプローブを用いてスクリーニングすることができる。配列が単離されたら、ポリメラーゼ連鎖反応(PCR)(米国特許第4,683,202号)のような標準的増幅法を用いてDNAを増幅し、形質転換に適した量のDNAを得ることができる。なお、クローニングに用いるゲノムDNAライブラリの作製、ハイブリダイゼーション、PCR、プラスミドDNAの調製、DNAの切断及び連結、形質転換等の方法は、Sambrook, J., Fritsch,E.F., Maniatis,T., Molecular Cloning, Cold Spring Harbor Laboratory Press, 1.21(1989)に記載されている。遺伝子への変異の導入方法は前述の通りである。
【0039】
本発明において、目的の制御因子をコードする遺伝子の導入はアセトイン合成する経路を増強させる。したがって、目的の制御因子をコードする遺伝子は、強力なプロモーターにより発現制御させることが好ましく、このためには、例えば、強力なプロモーター等を目的の制御因子をコードする遺伝子に連結させることが便利である。
【0040】
また、目的の制御因子をコードする遺伝子が導入された株を選別するため、マーカーとなる遺伝子がベクター内に組み込まれていてもよい。マーカー遺伝子としては、アミノ酸などの栄養要求性に関連する遺伝子や上述の薬剤耐性遺伝子などがあげられる。
【0041】
上記の目的の制御因子をコードする遺伝子を、宿主細胞で複製可能なベクターに挿入することにより本発明のアセトイン産生細胞を作製するための組み換えベクターが得られる。使用するベクターは宿主細胞で複製可能なものであれば限定されない。大腸菌や枯草菌のシャトルベクターであれば都合がよい。
【0042】
本発明の組み換えベクターを宿主細胞に導入することにより、目的の制御因子が発現する形質転換体が得られる。形質転換の方法としては、特に制限されないが、例えば、電気パルス法(Y. Kurusu et al., Agric. Biol. Chem. 54:443-447(1990))、カルシウムイオンを用いる方法(Proc. Natl. Acad. Sci. USA, 69, 2110(1972))、プロトプラスト法(特開昭63-2483942)、エレクトロポレーション法(Nucleic Acids Res., 16, 6127(1988))等を挙げることができる。エレクトロポレーション法が好ましい。
【0043】
目的の制御因子をコードする遺伝子を導入した細胞は、相同組み換えを利用したゲノムへの挿入による方法によって調製することもできる。相同組み換えによる方法は、ゲノム上の配列と相同な配列に目的の制御因子をコードする遺伝子を挿入し、このDNA断片をエレクトロポレーションによって細胞内に導入して相同組み換えを起こさせることにより実施できる。ゲノムへの導入の際には目的の制御因子をコードする遺伝子と薬剤耐性遺伝子を連結したDNA断片を用いると容易に相同組み換えが起こった株を選抜することができる。
【0044】
(アセトインの製造方法)
本発明はさらに、本発明のアセトイン産生細胞を用いたアセトインの製造方法を包含する。アセトインの製造方法は、本発明のアセトイン産生細胞を、グルコース等の炭素源を含有する培地で培養する工程を含む。さらに、本願のアセトインの製造方法は、好ましくは前記培養する工程で得られた培養ろ液から細胞を除去する工程と、細胞を除去した培養ろ液からアセトインを単離する工程とを含む。
【0045】
本発明において使用される培地は解糖系、ヘキソースリン酸経路、エントナー・ドゥドロフ経路等を経てピルビン酸を与える炭素源を含有していればよく、目的に達する限り何ら特別の制限はない。炭素源としては糖類や有機酸(グルコース、リンゴ酸、グリセロール、シュークロース、マルトース、澱粉など)が挙げられるが、糖類であることが好ましく、グルコースであることがさらに好ましい。さらに、培地は、炭素源に加えて、窒素源、有機栄養源、無機塩類等を含有する培地であればよく、合成培地または天然培地のいずれも使用できる。なお培地は、培養液(液体培地)であることが好ましい。
【0046】
培地は例えば以下のように配合されたものを用いることが好ましい。培地全体の重量に対し、炭素源としては糖類(好ましくはグルコース)を0.1w/v%〜10w/v%添加し、窒素源としては、ソイトン、カザミノ酸、ペプトン、トリプトン、酵母エキス、硫酸アンモニウム、塩化アンモニウム、硝酸アンモニウムおよび/または尿素等を0.01w/v%〜5.0w/v%、好ましくは0.5w/v%〜2.0w/v%添加するのが望ましい。
【0047】
その他、必要に応じ、ナトリウム、カリウム、カルシウム、マグネシウム、コバルト、マンガン、亜鉛、鉄、銅、モリブデン、リン酸、硫酸などのイオンを生成することができる無機塩類を培地中に添加することができる。無機塩類の添加量は、当業者が任意に決定することができる。
【0048】
培地中の水素イオン濃度は特に調整する必要は無いが、好ましくはpH5〜10、より好ましくはpH6〜9に調整し培養することができる。
【0049】
培養条件は、菌の種類によっても異なるが、培養温度は12〜45℃、好ましくは15〜37℃とすることができる。また、培養は培地(培養液)を振とうしたり、培地(培養液)に空気あるいは酸素ガスを吹き込むなどして好気的に行うこともできる。但し、通気量を大きく設定しすぎるとアセトイン生産量が減少する場合もあるので、生育が低下しない程度に通気量を制限することが望ましい。培養期間は、アセトインが最大または、必要量の蓄積量を示すまで行えば良く、通常1〜7日、好ましくは1〜2日である。
【0050】
細胞培養後の培地(培養液)には、アセトインのみならず、その他の生成物(例えば、2,3−ブタンジオール等)も含まれ得る。培地(培養液)からアセトインを分別する方法は、自体公知の方法または今後開発される方法を適用することができる。なお、アセトインの分別は、目的に応じた精製度でよく、アセトインと他の所望の生成物との混合物等として分別してもよい。
【0051】
例えば、培地からアセトインを採取する方法は、通常の水溶性中性物質を単離精製する一般的な方法を応用することができる。すなわち、培地から菌体を除去した後、培養上清液を活性炭やイオン交換樹脂等で処理することにより、アセトイン類以外の不純物をほとんど除くことができる。その後、再結晶等の方法を用いることにより、目的物質を単離することができる。
【0052】
具体的には後述する実施例の手法によりアセトインを採取することができるが、例えば、アセトインが蓄積した培養上清液を、望ましくない成分の除去の目的で強酸性陽イオン交換樹脂、例えばデュオライト(登録商標)C-20(H+型)を充填したカラムに通過させ通過液を集め、その後このカラムに脱イオン水を通過させ洗浄して洗浄液を集め、得られた通過液および洗浄液を合併する。こうして得られた溶液を強塩基性陰イオン交換樹脂、例えばデュオライト(登録商標)A116(OH-型)を充填したカラムに通過させ通過液を集め、その後このカラムに脱イオン水を通過させ洗浄して洗浄液を集め、得られた通過液および洗浄液を合併して、アセトイン類を含みそれ以外の不純物をほとんど含まない水溶液を取得する。この水溶液を濃縮して得られたアセトインの濃厚溶液に、エタノールの適当量を加え、室温または低温で一晩放置すると、純粋なアセトインの結晶を晶出できる。
【実施例】
【0053】
以下、本発明の理解を深めるために参考例および実施例により発明内容を具体的に説明するが、これらは本発明の範囲を限定するものではないことはいうまでもない。
【0054】
(参考例1) 変異株1(ΔlctEΔpta)の作製と培養
(1)変異株1(ΔlctEΔpta)として、遺伝子型がlctE::spec pta::neoである株を、以下のように段階的に作製した。
始めにlctE遺伝子破壊株を作製した。枯草菌168株の染色体を鋳型として、DNAプライマーlctE-F1(5'-ttggagccaggtaaatgctt-3'(配列番号5))とlctE-R1(5'-acaaaacccgctccgatta-3'(配列番号6))のペア、ならびにlctE-F2(5'-tcgcaggtatcactgagctg-3'(配列番号7))とlctE-R2(5'-gcaatgctggaccgaataat-3'(配列番号8))のペアによるPCR増幅によって、それぞれ約500 bpのDNA断片2種を得た。前者はlctE遺伝子上流側の隣接領域、また後者はlctE遺伝子下流側の隣接領域に相当する。一方、FU341株(Yoshida, K., Fujita, Y., & Ehrlich, S. D. (1999)., J. Bacteriol. 181, 6081-6091)の染色体を鋳型として、プライマーlctE-spec-F(5'-taatcggagcgggttttgtcaataacgctattgggag-3'(配列番号9))とlctE-spec-R(5'-cagctcagtgatacctgcgactatatgctccttctggc-3'(配列番号10))のペアによるPCR増幅によって、スペクチノマイシン耐性遺伝子を含むDNA断片を得た。以上3種のDNA断片を混合したものを鋳型とし、プライマーlctE-F1とlctE-R2のペアを用いてPCR増幅を行うと、lctE-spec-FとlctE-spec-Rには、それぞれlctE-R1とlctE -F2に相補的となる配列が5'側に付加されており、3つの断片が連結した長いDNA断片が得られる。このDNA断片を用いて枯草菌168株を形質転換して、50μg/mlスペクチノマイシンに耐性を示す変異体を選別しlctE遺伝子破壊株とした。
【0055】
次にpta遺伝子破壊株を作製した。枯草菌168株の染色体を鋳型として、DNAプライマーpta-F1(5'-atccattgtgcggaaatcat-3'(配列番号11))とpta-R1(5'-ccctatttatagacgctgtggctcgtctaagccttcagga-3'(配列番号12)) のペア、ならびにpta-F2(5'-gagccatcagcctaaagaagttcaagaggatgtaacgctgaa -3'(配列番号13))とpta-R2(5'-gccttggcttgttcgtagag-3'(配列番号14)) のペアによるPCR増幅によって、それぞれ約500 bpのDNA断片2種を得た。前者はpta遺伝子上流側の隣接領域、後者はpta遺伝子下流側の隣接領域に相当する。一方、FU340株(Yoshida, K., Fujita, Y., & Ehrlich, S. D. (1999)., J. Bacteriol. 181, 6081-6091)の染色体を鋳型として、プライマーpta-neo-F(5'-gttggctgtttgaatttgattggaattccggcacagcgtctataaataggg-3'(配列番号15))とpta-neo-R(5'-aatacaaaaaccccacccctggatccgcgcttctttaggctgatggctc-3'(配列番号16)) のペアによるPCR増幅によって、ネオマイシン耐性遺伝子を含むDNA断片を得た。以上3種のDNA断片を混合したものを鋳型とし、プライマーpta-F1とpta-R2のペアを用いてPCR増幅を行うと、pta-neo-Fとpta-R1、 pta-neo-Rとpta-F2には、それぞれに対して互いが相補的となる配列が5'側に付加されており、3つの断片が連結した長いDNA断片が得られる。このDNA断片を用いて枯草菌168株を形質転換して15μg/mlネオマイシンに耐性を示す変異体を選別し、pta遺伝子破壊株とした。
【0056】
pta遺伝子破壊株より抽出した染色体を用いて、lctE遺伝子破壊株を形質転換して、50 μg/mlスペクチノマイシンと15μg/mlネオマイシンの両方に耐性を獲得した変異株を選抜し変異株1とした。
【0057】
(2)変異株1について培養することにより、変異株1のアセトイン産生能を確認した。
変異株1を、5 mlのLB液体培地(1.0w/v% Bacto Tryptone、1.0w/v% Bacto Yeast Extract、1.0w/v% NaCl)にて、37℃、180 rpmで一晩振盪培養し、前培養とした。本培養は以下のようにして行った。前培養後の培地を、200 ml三角フラスコ内の50 mlの本培養培地(Soytone培地;1.0w/v% Bacto Soytone、0.5w/v% Bacto Yeast Extract、1.0w/v% NaCl、0.4w/v% グルコースを含む)に細胞濁度OD600 = 0.050となるように希釈して混合した。その後、37℃、150 rpmで振盪培養した。三角フラスコはシリコ栓でふたをして内外の空気の交換が最小限になるようにした。
【0058】
(3)培地中のアセトイン濃度測定を、既知の方法に従い行った(Grundy, F. J., Waters, D. A., Takova, T. Y. & Henkin, T. M. (1993)., Mol. Microbiol. 10(2). 259-271)。簡単に説明すると、本培養後の培地(培養液)500μlを1.5 ml容マイクロチューブに移し、遠心分離(4℃、21,900×g、1 min)して上清を回収、上清を蒸留水で200倍希釈したものをサンプル溶液とした。サンプル溶液1 mlに対して、5% α−ナフトールを含む2.5 N NaOH水溶液を0.2 ml、0.5% クレアチン水溶液を0.2 ml加え、十分に混合して室温で1時間インキュベートした後、OD540を測定した。濃度既知のアセトインサンプルによって検量線を作成し、アセトイン濃度を算出した。
【0059】
結果を図2に示す。変異株1では、野生株に比較してアセトインの産生量が増加することがわかった。また、培地中のアセトイン量は一過的に増加するものの、培養後約12時間から徐々に減少することがわかった。変異株1では野生株と比較して、グルコースからアセトインの変換率(モル比)は、30%程度上昇することがわかった。
【0060】
(参考例2) 変異株2(ΔacoA)の作製
(1)変異株2(ΔacoA)として、遺伝子型がacoA::catである株を、以下のようにして作製した。
枯草菌168株の染色体を鋳型として、DNAプライマーacoA-F1(5'-gctgaaaaagcgcctatgag-3'(配列番号17))とacoA-R1(5'-ttgtgcgcctccttctattt-3'(配列番号18))のペア、ならびにacoA-F2(5'-gaaaaagccgtctcgttcag-3'(配列番号19))とacoA-R2(5'-cgccgaacatataacggaat-3'(配列番号20)) のペアによるPCR増幅によって、それぞれ約500 bpのDNA断片2種を得た。前者はacoA遺伝子上流側の隣接領域、後者はacoA遺伝子下流側の隣接下流側の隣接領域に相当する。一方、FU342株(Yoshida, K., Fujita, Y., & Ehrlich, S. D.(1999)., J. Bacteriol. 181,6081-6091.)の染色体を鋳型として、プライマーacoA-cat-F(5'-aaatagaaggaggcgcacaagattggagctgatgtcac-3'(配列番号21))とacoA-cat-R(5'-ctgaacgagacggctttttcgaacctacctctcctcaa-3'(配列番号22)) のペアによるPCR増幅によって、クロラムフェニコール耐性遺伝子を含むDNA断片を得た。以上3種のDNA断片を混合したものを鋳型とし、プライマーacoA-F1とacoA-R2を用いてPCR増幅を行うと、acoA-cat-FとacoA-cat-Rには、それぞれacoA-R1とacoA-F2に相補的となる配列が5'側に付加されており、3つの断片が連結した長いDNA断片が得られる。このDNA断片を用いて枯草菌168株を形質転換して10μg/mlクロラムフェニコールに耐性を示す変異体を選別し、その一つを変異株2とした。
【0061】
(2)変異株2を用いて、前培養および本培養を参考例1(2)と同様の方法によって行い、培地中のアセトイン濃度を参考例1(3)と同様の手法により測定した。
【0062】
結果を図3に示す。野生株と比較して変異株2では、12時間以上培養を続けても、培地中のアセトインが減少することはなかった。
【0063】
(実施例1) アセトイン産生細胞1(ΔacoAΔlctEΔpta)の作製
アセトイン産生細胞1(ΔacoAΔlctEΔpta)として、遺伝子型がacoA::cat lctE::spec pta::neoである株を作製した。アセトイン産生細胞1は、参考例2にて作製した変異株2(acoA::cat)の染色体DNAを用いて、参考例1にて作製した変異株1(lctE::spec pta::neo)の形質転換を行い、10μg/mlクロラムフェニコール、50μg/mlスペクチノマイシン、15μg/mlネオマイシンのすべてに対して耐性を獲得した変異株を選択することにより、作製した。
【0064】
(実施例2) アセトインの製造
実施例1にて得られたアセトイン産生細胞1を培養して、アセトインを産生させた。
まず、参考例1(2)と同様の手法により、前培養および、0.4w/v%グルコースを含む培地を用いて本培養を行った。また、参考例1(2)と同様の手法により前培養を行った後、本培養を高濃度グルコース添加Soytone培地(Soytone培地に3.6w/v% グルコースを添加したもの)を4w/v%グルコースを含む培地として用いた以外は、参考例1(2)と同様の手法により培養を行った。
【0065】
アセトイン濃度測定は、参考例1(3)と同様の操作により行った。また、本培養に高濃度グルコース添加Soytone培地を用いた場合は、培地(培養液)の上清を適宜希釈したものをアセトイン濃度測定のサンプル溶液として用いた。
【0066】
結果を図4および5に示す。0.4w/v%グルコースを含む培地の場合、野生型では培養後8時間で、アセトイン量がほぼ最大に達し、12時間以後は徐々に減少していくのに対し、アセトイン産生細胞1では、培養後約12時間までアセトイン量が増加し続け、その後、48時間まで減少しなかった。アセトイン産生細胞1のグルコースからのアセトイン変換率(モル比)は、90%を達成した。
4w/v%グルコースを含む培地の場合、野生型では徐々にアセトイン量が増加していくが、アセトイン産生細胞1により産生されたアセトイン量は、培養後24時間でほぼ最大に達し、その後96時間後まで変化が見られなかった。
【0067】
(参考例3) 変異株3(ΔalsR1)の作製
(1)変異株3(ΔalsR1)として、遺伝子型がamyE::cat, Pspac-alsR1である株を、枯草菌染色体のamyE領域に、大腸菌プラスミドpCRE-test-alsR1より得られたDNA鎖を相同組み換えにより導入することで作製した。具体的には以下のようにして作製した。
まず、pCRE-test-alsR1の作製では、168株の染色体を鋳型として、プライマーalsR1-F1(5'-cgcggatccatggagcttcgccatcttcaa-3'(配列番号23))とalsR1-R1(5'-catgtatagagcaggccatgc-3'(配列番号24))のペア、ならびにalsR1-F2(5'-gcatggcctgctctatacatg-3'(配列番号25))とalsR1-R2(5'-cgcggatcctgtacctgcatcactctcttt-3'(配列番号26))のペアを用いてPCR増幅を行い、alsRのORF内の5'側断片(約600 bp)と3'側断片(約300 bp)をそれぞれ増幅した(プライマーの塩基配列中の下線部は、制限酵素認識配列を示す)。それぞれのPCR断片を混合したものを鋳型とし、プライマーalsR1-F1とalsR1-R2のペアを用いてPCR増幅を行うと、alsR1-R1とalsR1-F2は互いに相補的であり、かつalsRのORFの592残基目から612残基目にかけての相補的な配列を持ち、さらに601残基目のアデニンがグアニンとなる点変異を持つようにデザインされているためalsRアレル(alsR1)が増幅される。このPCR産物をBamHIで制限酵素処理したものをインサートDNAとしてベクタープラスミドpCRE-test(Miwa, Y., Nakata, A., Ogiwara, A., Yamamoto, M. & Fujita, Y. (2000)., Nucleic. acids. Res. 28, 1206-1210.)のBamHIクローニングサイトに挿入した。alsR1のORFの601残基目の点変異を塩基配列の決定により確認し、pCRE-test-alsR1とした。
このpCRE-test-alsR1をPstIにより制限酵素処理することで得られたDNA鎖を用いて枯草菌168株を形質転換し、10μg/mlクロラムフェニコール耐性株の一つを変異株3とした。
【0068】
(2)変異株3の培養および、培地中のアセトイン濃度測定の測定は、実施例2と同様にして、本培養に4w/v%グルコースを含む培地を用いて行った。
【0069】
結果を図6に示す。野生株では、培養後96時間まで、徐々にアセトイン量が増加していた。一方変異株3では、培養後60時間におけるグルコースからのアセトイン変換率(モル比)は90%であったが、その後アセトイン量が減少することがわかった。
【0070】
(実施例3) アセトイン産生細胞2(alsR1ΔacoA)の作製
(1)アセトイン産生細胞2(alsR1ΔacoA)として、遺伝子型がamyE::tet,Pspac-alsR1 acoA::catである株を以下のように作製した。
pCm::Tc(Steinmetz, M. & Richter, R. (1994). Gene.142, 79-83)DNAを用いて、参考例3にて得られた変異株3(amyE::cat, Pspac-alsR1)を形質転換し、12.5μg/mlテトラサイクリン耐性かつ10μg/mlクロラムフェニコール感受性となった変異株(染色体上のクロラムフェニコール耐性遺伝子がテトラサイクリン耐性遺伝子に置き換えられている)を得た。得られたテトラサイクリン耐性株を、参考例2にて得た変異株2(acoA::cat)の染色体DNAを用いて形質転換し、12.5μg/mlテトラサイクリンと10μg/mlクロラムフェニコールの両方に耐性となった株のうち一つをアセトイン産生変異細胞2(名称 Bacillus subtilis MY05株)とした。
【0071】
(実施例4) アセトインの製造
参考例3(2)と同様にして、アセトイン産生細胞2を4w/v%グルコースを含む培地により培養し、アセトインを産生させた。培地中のアセトイン濃度測定の測定は参考例3(2)と同様にして行った。
【0072】
結果を図7に示す。アセトイン産生細胞2は、培養後96時間までアセトイン産生量が増加した。変異株3(ΔacoA)では96時間以後はアセトイン量が減少するのに対して、アセトイン産生細胞2ではアセトイン量の減少が抑制されることがわかった。
【0073】
(実施例5) アセトイン産生細胞3(alsR1ΔacoAΔlacE)および4(alsR1ΔacoAΔpta)の作製
アセトイン産生細胞3(alsR1ΔacoAΔlacE)として、遺伝子型がamyE::tet,Pspac-alsR1 acoA::cat lctE::specである株を、次のようにして作製した。参考例1にて作製したlctE遺伝子破壊株より抽出した染色体を用いて、アセトイン産生変異細胞2を形質転換し、12.5μg/mlテトラサイクリン、10μg/mlクロラムフェニコール、50μg/mlスペクチノマイシンのいずれに対しても耐性となった株のうち一つをアセトイン産生変異細胞3とした。
アセトイン産生細胞4(alsR1ΔacoAΔpta)として、遺伝子型がamyE::tet,Pspac-alsR1 acoA::cat pta::neoである株を、次のようにして作製した。参考例1にて作製したpta遺伝子破壊株より抽出した染色体を用いて、アセトイン産生変異細胞2を形質転換し、12.5μg/mlテトラサイクリン、10μg/mlクロラムフェニコール、10μg/mlネオマイシンのいずれに対しても耐性となった株のうち一つをアセトイン産生変異細胞4とした。
【産業上の利用可能性】
【0074】
本発明の細胞はアセトインを、グルコースを原料として、細胞内で安価にかつ効率よく産生することができる。また、かかる細胞を用いた本発明のアセトインの製造方法は、アセトインの産生を1段階で行うことができ簡便である。得られたアセトインは、食品添加物、化粧品原料として応用可能であるのに加えて、ジアセチルや2,3−ブタンジオールの中間体としても有用である。
【技術分野】
【0001】
本発明は、アセトイン産生細胞および当該細胞を用いたアセトインの製造方法に関する。
【背景技術】
【0002】
アセトイン(3−ヒドロキシ−2−ブタノン)は、ヨーグルト、バター様の香りを持つ化合物であり、食品添加物、化粧品原料として使用される。またアセトインに類似する化合物であるジアセチル(2,3−ブタンジオン)は、食品添加物として用いられるのみならず、医薬品製造においても利用されている。
【0003】
アセトインは微生物等の細胞を用いて産生することが可能である。細胞内においてアセトインは、例えばクエン酸回路で生じたピルビン酸の二量体化によって生成するアセト乳酸が脱炭酸されることにより生成される。しかしながら細胞では、酢酸や乳酸などの他の代謝物が生成されてしまうため、アセトインの分離精製が困難であり、また細胞によるアセトイン産生の効率が悪く経済的でないという問題点がある。
【0004】
微生物等の細胞を用いたアセトインの製造方法に関して、従来より乳酸菌を用いた手法が検討されている(特許文献1,2)。特許文献1では、鉄ポルフィリンや金属塩等を添加した培地にて乳酸菌を培養することによりアセトイン類を製造する方法が開示されている。特許文献2では、薬剤を用いて突然変異を誘発することにより、アセトイン類の生成能の高い乳酸菌株を得たことが開示されている。
【0005】
枯草菌は元来糖質に富む培地条件で培養すると、細胞増殖の過程でアセトインを生産し培地中に蓄積すること(非特許文献1)、その後に細胞増殖を停止すると培地中のアセトインを再度分解してエネルギーを得ることが知られている。非特許文献1では、枯草菌を培養する際に、培地成分等の培養条件を変更することにより、アセトインの産生が増加したことが開示されている。また、アセトイン還元酵素をコードするbdhA遺伝子を破壊した枯草菌では、定常期にアセトインを蓄積することが知られている(非特許文献2)。
【0006】
枯草菌のアセトイン合成経路を司る酵素群は、単一のオペロンを形成し、かかるオペロン(alsSD)の発現を制御する制御因子としてalsRが知られている。alsR遺伝子は、alsSDオペロンとは独立に転写されており、alsR遺伝子を破壊するとalsDが発現しなかったことが非特許文献3にて報告されている。また、alsSDの発現を促進するalsR変異株を得たことが非特許文献3にて報告されている。
【0007】
これまで微生物等の細胞を用いてアセトインを効率よく産生するために、単一の遺伝子のみではなく、代謝系に関連する複数の遺伝子に着目して改変を施した例は報告されていない。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】特開平3−219884号公報
【特許文献2】特開平4−99480号公報
【非特許文献】
【0009】
【非特許文献1】Appl Micorobiol Biotechonol 74(1), 61-67(2007)
【非特許文献2】Appl Environ Microbiol 74(22), 6832-6838(2008)
【非特許文献3】J Bacteriol 175(12),3863-3875(1993)
【発明の概要】
【発明が解決しようとする課題】
【0010】
本発明は、アセトインを効率的かつ簡便に産生することのできる細胞および、当該細胞を用いたアセトイン製造方法を提供することを課題とする。
【課題を解決するための手段】
【0011】
上記課題を解決するために本発明者らは、鋭意検討した結果、枯草菌においてアセトインの再分解系を削除するとともに、アセトイン合成酵素群が構成的に発現するように遺伝子制御を改変する、および/または、アセトイン合成への原料供給を増やすように代謝系を改変することにより、アセトインを効率よく産生可能な細胞を作製しうることを見出し、本発明を完成した。
【0012】
すなわち、本発明は以下よりなる。
1.アセトイン脱水素酵素の機能が欠損または低減しており、ピルビン酸からアセト乳酸、及び、アセト乳酸からアセトインの合成を司る酵素群が構成的に発現している、アセトイン産生細胞。
2.前記酵素群をコードする遺伝子が単一のオペロンを形成している、前項1に記載のアセトイン産生細胞。
3.アセトイン脱水素酵素の機能の欠損または低減が、アセトイン脱水素酵素をコードする遺伝子を人為的に破壊したことによるものであり、且つ、前記酵素群の構成的発現が、前記オペロンの発現制御系を改変するような変異を導入したことによるものである、前項2に記載のアセトイン産生細胞。
4.アセトイン脱水素酵素、リン酸アセチル基転移酵素、および乳酸脱水素酵素の3種のタンパク質の機能が欠損または低減している、アセトイン産生細胞。
5.アセトイン脱水素酵素、リン酸アセチル基転移酵素、および乳酸脱水素酵素の3種のタンパク質の機能の欠損または低減が、アセトイン脱水素酵素をコードする遺伝子、リン酸アセチル基転移酵素をコードする遺伝子、および乳酸脱水素酵素をコードする遺伝子の3種の遺伝子を人為的に破壊したことによるものである、前項4に記載のアセトイン産生細胞。
6.さらに、リン酸アセチル基転移酵素および乳酸脱水素酵素のうち、少なくとも1種のタンパク質の機能が欠損または低減している、前項1〜3のいずれか1に記載のアセトイン産生細胞。
7.リン酸アセチル基転移酵素および乳酸脱水素酵素のうち、少なくとも1種のタンパク質の機能の欠損または低減が、リン酸アセチル基転移酵素をコードする遺伝子、および乳酸脱水素酵素をコードする遺伝子のうち、少なくとも1種の遺伝子を人為的に破壊したことによるものである、前項6に記載のアセトイン産生細胞。
8.細胞がバチルス属に属する菌である前項1〜7のいずれか1に記載の細胞。
9.細胞が、受領番号FERM AP-22036(受領日:2010年10月28日)の枯草菌変異株である、前項8に記載の細胞。
10.前項1〜9のいずれか1に記載の細胞を、グルコースの存在下で培養する工程を含む、アセトイン製造方法。
11.前項1〜3、6〜9のいずれか1に記載の細胞を、1〜10w/v%グルコースの存在下で培養する工程を含む、アセトイン製造方法。
12.前項4、5,8のいずれか1に記載の細胞を、0.2〜2w/v%グルコースの存在下で培養する工程を含む、アセトイン製造方法。
13.前記の培養する工程で得られた培養ろ液からアセトインを単離する工程をさらに含む、前項10〜12のいずれか1に記載のアセトイン製造方法。
【発明の効果】
【0013】
本発明の細胞は、アセトインを、グルコース等の糖類を原料として、細胞内で安価にかつ、90%(モル比)という高効率で産生することができる。本発明の細胞は、細胞の増殖量や生存率は、野生型の細胞と同様であるため、培養の際に特別な操作や特別な添加物等を必要とすることもない。また、かかる細胞を用いた本発明のアセトインの製造方法は、アセトインの産生を1段階で行うことができ簡便である。
【図面の簡単な説明】
【0014】
【図1】枯草菌の代謝系の概略を示す図である。
【図2】変異株1(ΔlctEΔpta)を、グルコースを含む培地にて培養した場合の、アセトイン産生量を示す図である。(参考例1)
【図3】変異株2(ΔacoA)を、グルコースを含む培地にて培養した場合の、アセトイン産生量を示す図である。(参考例2)
【図4】アセトイン産生細胞1(ΔacoAΔlctEΔpta)を、0.4w/v%グルコースを含む培地にて培養した場合の、アセトイン産生量を示す図である。(実施例2)
【図5】アセトイン産生細胞1(ΔacoAΔlctEΔpta)を、4w/v%グルコース含む培地にて培養した場合の、アセトイン産生量を示す図である。(実施例2)
【図6】変異株3(ΔalsR1)を、グルコースを含む培地にて培養した場合の、アセトイン産生量を示す図である。(参考例3)
【図7】アセトイン産生細胞2(alsR1ΔacoA)を、4w/v%グルコース含む培地にて培養した場合の、アセトイン産生量を示す図である。(実施例4)
【発明を実施するための形態】
【0015】
本発明は、細胞内の代謝系が改変されたことによりアセトインを効率的に産生することのできる細胞を対象とする。本発明では、細胞内の代謝系の改変することにより、特定の酵素の機能が欠損または低減している、および/または特定の酵素群が構成的に発現する細胞を得ることができる。
【0016】
本発明において、「酵素の機能が欠損または低減している細胞」とは、野生型の細胞に比べて、対象となる酵素自体の機能が低下もしくは消失していたり、量が低減していたり、発現していない細胞などが例示される。具体的には、「酵素の機能が欠損または低減している細胞」とは、対象となる酵素をコードする遺伝子に変異が導入されていたり、該遺伝子が破壊されている細胞が例示される。本発明の細胞は、天然に生じたものであってもよいし、人為的に当該遺伝子を破壊することにより作製したものであってもよい。本発明の細胞は、好ましくは、人為的に対象となる酵素をコードする遺伝子を破壊することにより作製したものである。
【0017】
本発明において「構成的」とは本発明が属する技術分野において一般的に用いられている意味で用いられる。本発明において、「酵素群が構成的に発現している細胞」とは、環境条件による調節を受けずに常に一定レベルで目的の酵素群が発現している細胞であり、例えば、野生型の細胞では対象となる酵素群の発現が抑制されるような環境条件下であっても、定常的に目的の酵素群が発現し続けている細胞を意味する。
【0018】
本発明において「細胞」とは、アセトイン代謝系の酵素を有し、細胞内の代謝系が合理的に改変された結果アセトインを効率的に産生し得る細胞であればよく、微生物、動物細胞、植物細胞など、種類は制限されない。好ましくは、アセトイン代謝系を有する微生物を用いることができる。微生物は細菌であることが好ましく、グラム陽性菌であることがより好ましい。グラム陽性菌としては、バチルス属(Bacillus)、ジオバチルス属(Geobacillus)、ラクトバシラス属(Lactobacillus)、ラクトコッカス属(Lactococcus)、スタフィロコッカス属(Staphylococcus)、クレブシエラ属(Klebsiella)、ストレプトコッカス属(Streptococcus)に属する菌を用いることが可能である。特に、バチルス属に属する菌を用いることが好ましく、枯草菌(Bacillus subtilis)を用いることがより好ましい。
【0019】
(アセトイン産生細胞)
本発明のアセトイン産生細胞は、アセトイン脱水素酵素の機能が欠損または低減しており、ピルビン酸からアセト乳酸、及び、アセト乳酸からアセトインの合成を司る酵素群が構成的に発現していることを特徴とする。
【0020】
アセトイン脱水素酵素は、アセトインを再分解してアセチルCoAを生成する経路に関与する酵素である。本発明のアセトイン産生細胞内においては、アセトイン脱水素酵素の機能が欠損または低減しており、細胞外に蓄積するアセトインを消費する能力を欠失または低減している。アセトイン脱水素酵素の機能を欠損または低減させる手段は、いかなる手段によってもよいが、アセトイン脱水素酵素をコードする遺伝子を破壊する手段が好ましい。アセトイン脱水素酵素をコードする遺伝子の具体例としては、例えば、配列番号1で表される塩基配列からなる遺伝子(acoA遺伝子、NCBI-GI: 16077873、NC_000964.3参照)および当該遺伝子と同等の機能を有する遺伝子が挙げられる。配列番号1で表される塩基配列からなる遺伝子と同等の機能を有する遺伝子としては、配列番号1で表される塩基配列において1個以上(例えば2〜3個)のヌクレオチドが置換、欠失、挿入および/もしくは付加された塩基配列からなり、アセトイン脱水素酵素の活性を有するタンパク質をコードする遺伝子が挙げられる。
【0021】
例えば枯草菌では、炭素源が枯渇している場合にアセトインからアセチルCoAを生成するアセトイン再分解系として、acoABCLオペロンという一連の遺伝子群が知られている(図1、Huang, M., Oppermann-Sanio, .FB., and Steinbuchel, A., J Bacteriol. 1999 Jun;181(12):3837-3841)。acoABCLオペロンの遺伝子は、アセトイン脱水素酵素を構成するサブユニットを各々コードしている。枯草菌においては、アセトイン再分解系を抑制するために、acoABCLオペロンのいずれの遺伝子を破壊してもよいが、好ましくはacoA遺伝子を破壊した変異株を作製すればよい。アセトイン再分解系を抑制することにより、アセトイン産生細胞により産生されたアセトインが培地中から減少することを防ぐことができ、培地中のアセトイン量を維持することが可能となる。
【0022】
本発明のアセトイン産生細胞においては、アセトインの合成経路そのものを増強するために、ピルビン酸からアセト乳酸、及び、アセト乳酸からアセトインの合成を司る酵素群が構成的に発現している。ピルビン酸からアセト乳酸、及び、アセト乳酸からアセトインの合成を司る酵素群は、アセト乳酸合成酵素およびアセト乳酸脱炭酸酵素を含む酵素群である。これらの酵素群は、オペロンによりコードされていてもよく、当該オペロンとしては例えばalsSDオペロンが例示される。当該酵素群を構成的に発現させる手段は、いかなる手段によってもよいが、該酵素群がオペロンによりコードされる場合は、前記オペロンの発現制御系を改変するような変異を導入することが好ましい。前記オペロンの発現制御系の改変としては、オペロンの発現を制御する制御因子に変異を導入して恒常的活性化型にすることが例示され、好ましくは、当該制御因子の恒常的活性化型が細胞内に導入される。
【0023】
当該制御因子の恒常的活性化型をコードする遺伝子の具体例としては、例えば、配列番号2で表される塩基配列(alsR遺伝子、NC_000964.3参照)において、601番目のアデニン(A)がグアニン(G)に置換された塩基配列を有するalsR1遺伝子(すなわち、非特許文献3、FIG. 4の塩基配列における1028番目のチミン(T)がシトシン(C)に置換された塩基配列を有するalsR1遺伝子)、および当該alsR1遺伝子と同等の機能を有する遺伝子が挙げられる。alsR1遺伝子と同等の機能を有する遺伝子としては、配列番号2の塩基配列において1個以上(例えば2〜3個)のヌクレオチドが置換、欠失、挿入および/もしくは付加された塩基配列からなり、前記オペロンの制御因子の恒常的活性化型としての機能を有するタンパク質をコードする遺伝子が挙げられる。alsR1遺伝子と同等の機能を有する遺伝子の具体例としてalsR8遺伝子が例示される。alsR8遺伝子は、配列番号2で表される塩基配列において、700番目のアデニン(A)がシトシン(C)に置換された塩基配列を有する遺伝子(すなわち、非特許文献3、FIG. 4の塩基配列における929番目のチミン(T)がグアニン(G)に置換された塩基配列を有するalsR8遺伝子)である。
【0024】
例えば、枯草菌ではアセトインの合成経路に関連する酵素(アセト乳酸合成酵素およびアセト乳酸脱炭酸酵素)は、alsSDオペロンにより発現される。alsSDオペロンの転写は、制御因子のalsRが、alsSDオペロンの特定の配列に結合することにより促進される。alsRはリガンドに結合することにより活性化型となり、配列特異的DNA結合能を獲得する。alsRはリガンドとの結合が解除されることにより不活性化型となり、alsSDオペロンの発現が停止する。alsRの変異型タンパク質として、alsR1(alsR1遺伝子によりコードされるタンパク質であり、alsRのアミノ酸配列(NCBI-GI: 16080655)において201番目のスレオニンがアラニンにより置換されているタンパク質)やalsR8(alsR8遺伝子によりコードされるタンパク質であり、alsRのアミノ酸配列において234番目のセリンがアラニンにより置換されているタンパク質)などがあるが、これらはalsRの恒常的活性化型として知られている(非特許文献3)。枯草菌において、alsSDオペロンを構成的に発現させるためには、alsR1やalsR8等の恒常的活性化型の制御因子を細胞内に導入すればよい。恒常的活性化型の制御因子が導入された細胞は、アセトインの合成経路が増強されているため、高効率でアセトインを産生することができる。
【0025】
アセトイン脱水素酵素の機能が欠損または低減しており、ピルビン酸からアセト乳酸、及び、アセト乳酸からアセトインの合成を司る酵素群が構成的に発現していることを特徴とするアセトイン産生細胞では、90%(モル比)という高いアセトイン変換率を達成するとともに、長時間培養した場合でも、アセトインが再分解されることなく、培地中のアセトイン量が維持され続ける。また、かかるアセトイン産生細胞を培養する培地中におけるグルコース濃度は特に限定されないが、高濃度(2〜10w/v%、好ましくは2〜6w/v%、より好ましくは3〜5w/v%、さらに好ましくは4w/v%)でグルコースを含む培地にて培養した場合であっても、高いアセトイン変換率を有する。グルコースを高濃度で含む培地においては、浸透圧が上昇するため細胞の生存率自体が低下する場合もあるが、本発明のアセトイン産生細胞は、増殖、生存が可能である。
【0026】
本発明は、アセトイン脱水素酵素の機能が欠損または低減しており、ピルビン酸からアセト乳酸、及び、アセト乳酸からアセトインの合成を司る酵素群が構成的に発現していることを特徴とするアセトイン産生細胞は、さらにリン酸アセチル基転移酵素および乳酸脱水素酵素のうち、少なくとも1種のタンパク質の機能が欠損または低減しているアセトイン産生細胞を包含する。
【0027】
リン酸アセチル基転移酵素は、アセチルCoAに他の分子からリン酸基を転移・付加してアセチルリン酸を合成する反応を触媒する酵素であり、アセチルCoAの酢酸発酵に関与する酵素である。本発明のアセトイン産生細胞内においては、リン酸アセチル基転移酵素の機能が欠損または低減している。リン酸アセチル基転移酵素の機能を欠損または低減させる手段は、いかなる手段によってもよいが、リン酸アセチル基転移酵素をコードする遺伝子を破壊する手段が好ましい。リン酸アセチル基転移酵素をコードする遺伝子の具体例としては、配列番号3で表される塩基配列からなる遺伝子(pta(phosphotransacetylase)遺伝子、NCBI-GI: 16080818、NC_000964.3参照)および当該遺伝子と同等の機能を有する遺伝子が挙げられる。配列番号3で表される塩基配列からなる遺伝子と同等の機能を有する遺伝子としては、配列番号3で表される塩基配列において1個以上(例えば2〜3個)のヌクレオチドが置換、欠失、挿入および/もしくは付加された塩基配列からなり、リン酸アセチル基転移酵素の活性を有するタンパク質をコードする遺伝子が挙げられる。
【0028】
乳酸脱水素酵素は、ピルビン酸から乳酸を合成する反応を触媒する酵素であり、ピルビン酸の乳酸発酵に関与する酵素である。本発明のアセトイン産生細胞内においては、乳酸脱水素酵素の機能が欠損または低減している。乳酸脱水素酵素の機能を欠損または低減させる手段は、いかなる手段によってもよいが、乳酸脱水素酵素をコードする遺伝子を破壊する手段が好ましい。乳酸脱水素酵素をコードする遺伝子としての具体例は、例えば、配列番号4で表される塩基配列からなる遺伝子(lct(L-lactate dehydrogenase)遺伝子、NCBI-GI: 255767083、NC_000964.3参照)および当該遺伝子と同等の機能を有する遺伝子が挙げられる。配列番号4で表される塩基配列からなる遺伝子と同等の機能を有する遺伝子としては、配列番号4で表される塩基配列において1個以上(例えば2〜3個)のヌクレオチドが置換、欠失、挿入および/もしくは付加された塩基配列からなり、乳酸脱水素酵素の活性を有するタンパク質をコードする遺伝子が挙げられる。
【0029】
対象とするタンパク質が上記各種酵素や制御因子としての活性を有するか否か、対象とする遺伝子がこれらの活性を有するタンパク質をコードしているか否かは、自体公知の酵素学的アッセイなどの方法で確認可能である。
【0030】
本発明のアセトイン産生細胞として、具体的には、独立行政法人産業技術総合研究所特許生物寄託センター(〒305-8566 茨城県つくば市東1-1-1 つくばセンター 中央第6)に、受領番号FERM AP-22036(受領日:2010年10月28日)にて寄託した枯草菌変異株(名称 Bacillus subtilis MY05株)が例示される。
【0031】
また本発明は、アセトイン脱水素酵素、リン酸アセチル基転移酵素、および乳酸脱水素酵素の3種のタンパク質の機能が欠損または低減していることを特徴とするアセトイン産生細胞も包含する。
【0032】
例えば枯草菌では、グルコースなどの糖類を利用する代謝系として、TCA回路、乳酸発酵、アセトイン発酵、乳酸発酵等が知られている。例えば、lctE遺伝子(乳酸脱水素酵素をコードする遺伝子)および/またはpta遺伝子(リン酸アセチル基転移酵素をコードする遺伝子)を破壊した枯草菌変異株では、乳酸発酵および/または酢酸発酵が抑制され、原料であるピルビン酸のアセトイン合成への供給が高められており、アセトインを高効率で産生することができる。lctE遺伝子およびpta遺伝子に加えて、アセトイン脱水素酵素をコードする遺伝子(acoA遺伝子)の三種の遺伝子を破壊したアセトイン産生細胞を用いれば、アセトインを高効率で産生することができるとともに、培地中のアセトインは減少することなく、維持され続ける。
【0033】
すなわち、アセトイン脱水素酵素、リン酸アセチル基転移酵素、および乳酸脱水素酵素の3種のタンパク質の機能が欠損または低減していることを特徴とするアセトイン産生細胞では、90%(モル比)という高いアセトイン変換率を達成するとともに、長時間培養した場合でも、アセトインが再分解されることなく、培地中のアセトイン量が維持され続ける。また、かかるアセトイン産生細胞を培養する培地中におけるグルコース濃度は特に限定されないが、低濃度(0.2〜2w/v%、好ましくは0.2〜1w/v%、より好ましくは0.4w/v%)でグルコースを含む培地にて培養することが好ましい。
【0034】
(アセトイン産生細胞の作製方法)
本発明のアセトイン産生細胞は、目的の酵素をコードする遺伝子を破壊する方法および、目的の制御因子をコードする遺伝子を導入する方法を用いて作製することができる。
【0035】
目的の酵素の機能を欠損または低減させる方法としては、目的の酵素をコードする遺伝子を破壊する方法が例示され、通常の変異による方法を使用することができる。例えば、物理的および/または化学的変異誘発処理では、UV照射、放射線照射などの物理的変異方法の他、ニトロソグアニジン、メタンスルホン酸エチル、亜硝酸、メタンスルホン酸メチル、アクリジン色素、ベンゾピレン、硫酸ジメチルなどの変異剤の混合による化学的変異方法が例示される。これらの変異処理は、遺伝子上で塩基の挿入、欠失および/または置換が期待される方法である。変異処理においては、遺伝子が破壊される限り、一個の塩基を挿入、欠失および/または置換してもよく、複数個の塩基を挿入、欠失、および/または置換してもよい。
【0036】
さらに、目的の酵素をコードする遺伝子の別の人為的な破壊方法として、相同組換えによる塩基の挿入、欠失および/または置換を行なう方法がある。相同組換え法では、人為的に塩基の挿入、欠失および/または置換を施した目的の遺伝子と同じ配列を部分的に有する塩基配列を導入し、相同組換えによって変異を行なう方法である。例えば、薬剤耐性遺伝子であるクロラムフェニコール耐性遺伝子、カナマイシン耐性遺伝子、テトラサイクリン耐性遺伝子、エリスロマイシン耐性遺伝子、スペクチノマイシン耐性遺伝子、アンピシリン耐性遺伝子、ハイグロマイシン耐性遺伝子、ネオマイシン耐性遺伝子などを挿入した塩基配列を導入した後、相当する薬剤で選抜し、生存する細胞を得る方法である。さらに、望むべき部分に挿入されたことをPCRなどの機器を用いて確認することによって、目的の細胞を得ることが可能である。なお、「目的の酵素の機能もしくは目的の酵素をコードする遺伝子を人為的に破壊する」とは、例えば突然変異の導入、または相同組み換えにより、当該遺伝子の転写を抑制し、または当該遺伝子にコードされるタンパク質の機能を破壊することをいう。
【0037】
本発明の細胞の作製において、目的のタンパク質を構成的に発現させるために、目的のタンパク質の発現を制御する制御因子をコードする遺伝子を導入する方法が例示される。当該制御因子をコードする遺伝子を導入する方法としては、自体公知の方法を用いることができる。
【0038】
まず目的の制御因子をコードする遺伝子を、微生物等のゲノムDNAを鋳型にして変異を導入して取得するか、又は合成することにより取得する。微生物等のゲノムDNAから所望の遺伝子を取得する方法および遺伝子に変異を導入する方法は分子生物学の分野において周知である。例えば遺伝子の配列が既知の場合、制限エンドヌクレアーゼ消化により適したゲノムライブラリを作り、所望の遺伝子配列に相補的なプローブを用いてスクリーニングすることができる。配列が単離されたら、ポリメラーゼ連鎖反応(PCR)(米国特許第4,683,202号)のような標準的増幅法を用いてDNAを増幅し、形質転換に適した量のDNAを得ることができる。なお、クローニングに用いるゲノムDNAライブラリの作製、ハイブリダイゼーション、PCR、プラスミドDNAの調製、DNAの切断及び連結、形質転換等の方法は、Sambrook, J., Fritsch,E.F., Maniatis,T., Molecular Cloning, Cold Spring Harbor Laboratory Press, 1.21(1989)に記載されている。遺伝子への変異の導入方法は前述の通りである。
【0039】
本発明において、目的の制御因子をコードする遺伝子の導入はアセトイン合成する経路を増強させる。したがって、目的の制御因子をコードする遺伝子は、強力なプロモーターにより発現制御させることが好ましく、このためには、例えば、強力なプロモーター等を目的の制御因子をコードする遺伝子に連結させることが便利である。
【0040】
また、目的の制御因子をコードする遺伝子が導入された株を選別するため、マーカーとなる遺伝子がベクター内に組み込まれていてもよい。マーカー遺伝子としては、アミノ酸などの栄養要求性に関連する遺伝子や上述の薬剤耐性遺伝子などがあげられる。
【0041】
上記の目的の制御因子をコードする遺伝子を、宿主細胞で複製可能なベクターに挿入することにより本発明のアセトイン産生細胞を作製するための組み換えベクターが得られる。使用するベクターは宿主細胞で複製可能なものであれば限定されない。大腸菌や枯草菌のシャトルベクターであれば都合がよい。
【0042】
本発明の組み換えベクターを宿主細胞に導入することにより、目的の制御因子が発現する形質転換体が得られる。形質転換の方法としては、特に制限されないが、例えば、電気パルス法(Y. Kurusu et al., Agric. Biol. Chem. 54:443-447(1990))、カルシウムイオンを用いる方法(Proc. Natl. Acad. Sci. USA, 69, 2110(1972))、プロトプラスト法(特開昭63-2483942)、エレクトロポレーション法(Nucleic Acids Res., 16, 6127(1988))等を挙げることができる。エレクトロポレーション法が好ましい。
【0043】
目的の制御因子をコードする遺伝子を導入した細胞は、相同組み換えを利用したゲノムへの挿入による方法によって調製することもできる。相同組み換えによる方法は、ゲノム上の配列と相同な配列に目的の制御因子をコードする遺伝子を挿入し、このDNA断片をエレクトロポレーションによって細胞内に導入して相同組み換えを起こさせることにより実施できる。ゲノムへの導入の際には目的の制御因子をコードする遺伝子と薬剤耐性遺伝子を連結したDNA断片を用いると容易に相同組み換えが起こった株を選抜することができる。
【0044】
(アセトインの製造方法)
本発明はさらに、本発明のアセトイン産生細胞を用いたアセトインの製造方法を包含する。アセトインの製造方法は、本発明のアセトイン産生細胞を、グルコース等の炭素源を含有する培地で培養する工程を含む。さらに、本願のアセトインの製造方法は、好ましくは前記培養する工程で得られた培養ろ液から細胞を除去する工程と、細胞を除去した培養ろ液からアセトインを単離する工程とを含む。
【0045】
本発明において使用される培地は解糖系、ヘキソースリン酸経路、エントナー・ドゥドロフ経路等を経てピルビン酸を与える炭素源を含有していればよく、目的に達する限り何ら特別の制限はない。炭素源としては糖類や有機酸(グルコース、リンゴ酸、グリセロール、シュークロース、マルトース、澱粉など)が挙げられるが、糖類であることが好ましく、グルコースであることがさらに好ましい。さらに、培地は、炭素源に加えて、窒素源、有機栄養源、無機塩類等を含有する培地であればよく、合成培地または天然培地のいずれも使用できる。なお培地は、培養液(液体培地)であることが好ましい。
【0046】
培地は例えば以下のように配合されたものを用いることが好ましい。培地全体の重量に対し、炭素源としては糖類(好ましくはグルコース)を0.1w/v%〜10w/v%添加し、窒素源としては、ソイトン、カザミノ酸、ペプトン、トリプトン、酵母エキス、硫酸アンモニウム、塩化アンモニウム、硝酸アンモニウムおよび/または尿素等を0.01w/v%〜5.0w/v%、好ましくは0.5w/v%〜2.0w/v%添加するのが望ましい。
【0047】
その他、必要に応じ、ナトリウム、カリウム、カルシウム、マグネシウム、コバルト、マンガン、亜鉛、鉄、銅、モリブデン、リン酸、硫酸などのイオンを生成することができる無機塩類を培地中に添加することができる。無機塩類の添加量は、当業者が任意に決定することができる。
【0048】
培地中の水素イオン濃度は特に調整する必要は無いが、好ましくはpH5〜10、より好ましくはpH6〜9に調整し培養することができる。
【0049】
培養条件は、菌の種類によっても異なるが、培養温度は12〜45℃、好ましくは15〜37℃とすることができる。また、培養は培地(培養液)を振とうしたり、培地(培養液)に空気あるいは酸素ガスを吹き込むなどして好気的に行うこともできる。但し、通気量を大きく設定しすぎるとアセトイン生産量が減少する場合もあるので、生育が低下しない程度に通気量を制限することが望ましい。培養期間は、アセトインが最大または、必要量の蓄積量を示すまで行えば良く、通常1〜7日、好ましくは1〜2日である。
【0050】
細胞培養後の培地(培養液)には、アセトインのみならず、その他の生成物(例えば、2,3−ブタンジオール等)も含まれ得る。培地(培養液)からアセトインを分別する方法は、自体公知の方法または今後開発される方法を適用することができる。なお、アセトインの分別は、目的に応じた精製度でよく、アセトインと他の所望の生成物との混合物等として分別してもよい。
【0051】
例えば、培地からアセトインを採取する方法は、通常の水溶性中性物質を単離精製する一般的な方法を応用することができる。すなわち、培地から菌体を除去した後、培養上清液を活性炭やイオン交換樹脂等で処理することにより、アセトイン類以外の不純物をほとんど除くことができる。その後、再結晶等の方法を用いることにより、目的物質を単離することができる。
【0052】
具体的には後述する実施例の手法によりアセトインを採取することができるが、例えば、アセトインが蓄積した培養上清液を、望ましくない成分の除去の目的で強酸性陽イオン交換樹脂、例えばデュオライト(登録商標)C-20(H+型)を充填したカラムに通過させ通過液を集め、その後このカラムに脱イオン水を通過させ洗浄して洗浄液を集め、得られた通過液および洗浄液を合併する。こうして得られた溶液を強塩基性陰イオン交換樹脂、例えばデュオライト(登録商標)A116(OH-型)を充填したカラムに通過させ通過液を集め、その後このカラムに脱イオン水を通過させ洗浄して洗浄液を集め、得られた通過液および洗浄液を合併して、アセトイン類を含みそれ以外の不純物をほとんど含まない水溶液を取得する。この水溶液を濃縮して得られたアセトインの濃厚溶液に、エタノールの適当量を加え、室温または低温で一晩放置すると、純粋なアセトインの結晶を晶出できる。
【実施例】
【0053】
以下、本発明の理解を深めるために参考例および実施例により発明内容を具体的に説明するが、これらは本発明の範囲を限定するものではないことはいうまでもない。
【0054】
(参考例1) 変異株1(ΔlctEΔpta)の作製と培養
(1)変異株1(ΔlctEΔpta)として、遺伝子型がlctE::spec pta::neoである株を、以下のように段階的に作製した。
始めにlctE遺伝子破壊株を作製した。枯草菌168株の染色体を鋳型として、DNAプライマーlctE-F1(5'-ttggagccaggtaaatgctt-3'(配列番号5))とlctE-R1(5'-acaaaacccgctccgatta-3'(配列番号6))のペア、ならびにlctE-F2(5'-tcgcaggtatcactgagctg-3'(配列番号7))とlctE-R2(5'-gcaatgctggaccgaataat-3'(配列番号8))のペアによるPCR増幅によって、それぞれ約500 bpのDNA断片2種を得た。前者はlctE遺伝子上流側の隣接領域、また後者はlctE遺伝子下流側の隣接領域に相当する。一方、FU341株(Yoshida, K., Fujita, Y., & Ehrlich, S. D. (1999)., J. Bacteriol. 181, 6081-6091)の染色体を鋳型として、プライマーlctE-spec-F(5'-taatcggagcgggttttgtcaataacgctattgggag-3'(配列番号9))とlctE-spec-R(5'-cagctcagtgatacctgcgactatatgctccttctggc-3'(配列番号10))のペアによるPCR増幅によって、スペクチノマイシン耐性遺伝子を含むDNA断片を得た。以上3種のDNA断片を混合したものを鋳型とし、プライマーlctE-F1とlctE-R2のペアを用いてPCR増幅を行うと、lctE-spec-FとlctE-spec-Rには、それぞれlctE-R1とlctE -F2に相補的となる配列が5'側に付加されており、3つの断片が連結した長いDNA断片が得られる。このDNA断片を用いて枯草菌168株を形質転換して、50μg/mlスペクチノマイシンに耐性を示す変異体を選別しlctE遺伝子破壊株とした。
【0055】
次にpta遺伝子破壊株を作製した。枯草菌168株の染色体を鋳型として、DNAプライマーpta-F1(5'-atccattgtgcggaaatcat-3'(配列番号11))とpta-R1(5'-ccctatttatagacgctgtggctcgtctaagccttcagga-3'(配列番号12)) のペア、ならびにpta-F2(5'-gagccatcagcctaaagaagttcaagaggatgtaacgctgaa -3'(配列番号13))とpta-R2(5'-gccttggcttgttcgtagag-3'(配列番号14)) のペアによるPCR増幅によって、それぞれ約500 bpのDNA断片2種を得た。前者はpta遺伝子上流側の隣接領域、後者はpta遺伝子下流側の隣接領域に相当する。一方、FU340株(Yoshida, K., Fujita, Y., & Ehrlich, S. D. (1999)., J. Bacteriol. 181, 6081-6091)の染色体を鋳型として、プライマーpta-neo-F(5'-gttggctgtttgaatttgattggaattccggcacagcgtctataaataggg-3'(配列番号15))とpta-neo-R(5'-aatacaaaaaccccacccctggatccgcgcttctttaggctgatggctc-3'(配列番号16)) のペアによるPCR増幅によって、ネオマイシン耐性遺伝子を含むDNA断片を得た。以上3種のDNA断片を混合したものを鋳型とし、プライマーpta-F1とpta-R2のペアを用いてPCR増幅を行うと、pta-neo-Fとpta-R1、 pta-neo-Rとpta-F2には、それぞれに対して互いが相補的となる配列が5'側に付加されており、3つの断片が連結した長いDNA断片が得られる。このDNA断片を用いて枯草菌168株を形質転換して15μg/mlネオマイシンに耐性を示す変異体を選別し、pta遺伝子破壊株とした。
【0056】
pta遺伝子破壊株より抽出した染色体を用いて、lctE遺伝子破壊株を形質転換して、50 μg/mlスペクチノマイシンと15μg/mlネオマイシンの両方に耐性を獲得した変異株を選抜し変異株1とした。
【0057】
(2)変異株1について培養することにより、変異株1のアセトイン産生能を確認した。
変異株1を、5 mlのLB液体培地(1.0w/v% Bacto Tryptone、1.0w/v% Bacto Yeast Extract、1.0w/v% NaCl)にて、37℃、180 rpmで一晩振盪培養し、前培養とした。本培養は以下のようにして行った。前培養後の培地を、200 ml三角フラスコ内の50 mlの本培養培地(Soytone培地;1.0w/v% Bacto Soytone、0.5w/v% Bacto Yeast Extract、1.0w/v% NaCl、0.4w/v% グルコースを含む)に細胞濁度OD600 = 0.050となるように希釈して混合した。その後、37℃、150 rpmで振盪培養した。三角フラスコはシリコ栓でふたをして内外の空気の交換が最小限になるようにした。
【0058】
(3)培地中のアセトイン濃度測定を、既知の方法に従い行った(Grundy, F. J., Waters, D. A., Takova, T. Y. & Henkin, T. M. (1993)., Mol. Microbiol. 10(2). 259-271)。簡単に説明すると、本培養後の培地(培養液)500μlを1.5 ml容マイクロチューブに移し、遠心分離(4℃、21,900×g、1 min)して上清を回収、上清を蒸留水で200倍希釈したものをサンプル溶液とした。サンプル溶液1 mlに対して、5% α−ナフトールを含む2.5 N NaOH水溶液を0.2 ml、0.5% クレアチン水溶液を0.2 ml加え、十分に混合して室温で1時間インキュベートした後、OD540を測定した。濃度既知のアセトインサンプルによって検量線を作成し、アセトイン濃度を算出した。
【0059】
結果を図2に示す。変異株1では、野生株に比較してアセトインの産生量が増加することがわかった。また、培地中のアセトイン量は一過的に増加するものの、培養後約12時間から徐々に減少することがわかった。変異株1では野生株と比較して、グルコースからアセトインの変換率(モル比)は、30%程度上昇することがわかった。
【0060】
(参考例2) 変異株2(ΔacoA)の作製
(1)変異株2(ΔacoA)として、遺伝子型がacoA::catである株を、以下のようにして作製した。
枯草菌168株の染色体を鋳型として、DNAプライマーacoA-F1(5'-gctgaaaaagcgcctatgag-3'(配列番号17))とacoA-R1(5'-ttgtgcgcctccttctattt-3'(配列番号18))のペア、ならびにacoA-F2(5'-gaaaaagccgtctcgttcag-3'(配列番号19))とacoA-R2(5'-cgccgaacatataacggaat-3'(配列番号20)) のペアによるPCR増幅によって、それぞれ約500 bpのDNA断片2種を得た。前者はacoA遺伝子上流側の隣接領域、後者はacoA遺伝子下流側の隣接下流側の隣接領域に相当する。一方、FU342株(Yoshida, K., Fujita, Y., & Ehrlich, S. D.(1999)., J. Bacteriol. 181,6081-6091.)の染色体を鋳型として、プライマーacoA-cat-F(5'-aaatagaaggaggcgcacaagattggagctgatgtcac-3'(配列番号21))とacoA-cat-R(5'-ctgaacgagacggctttttcgaacctacctctcctcaa-3'(配列番号22)) のペアによるPCR増幅によって、クロラムフェニコール耐性遺伝子を含むDNA断片を得た。以上3種のDNA断片を混合したものを鋳型とし、プライマーacoA-F1とacoA-R2を用いてPCR増幅を行うと、acoA-cat-FとacoA-cat-Rには、それぞれacoA-R1とacoA-F2に相補的となる配列が5'側に付加されており、3つの断片が連結した長いDNA断片が得られる。このDNA断片を用いて枯草菌168株を形質転換して10μg/mlクロラムフェニコールに耐性を示す変異体を選別し、その一つを変異株2とした。
【0061】
(2)変異株2を用いて、前培養および本培養を参考例1(2)と同様の方法によって行い、培地中のアセトイン濃度を参考例1(3)と同様の手法により測定した。
【0062】
結果を図3に示す。野生株と比較して変異株2では、12時間以上培養を続けても、培地中のアセトインが減少することはなかった。
【0063】
(実施例1) アセトイン産生細胞1(ΔacoAΔlctEΔpta)の作製
アセトイン産生細胞1(ΔacoAΔlctEΔpta)として、遺伝子型がacoA::cat lctE::spec pta::neoである株を作製した。アセトイン産生細胞1は、参考例2にて作製した変異株2(acoA::cat)の染色体DNAを用いて、参考例1にて作製した変異株1(lctE::spec pta::neo)の形質転換を行い、10μg/mlクロラムフェニコール、50μg/mlスペクチノマイシン、15μg/mlネオマイシンのすべてに対して耐性を獲得した変異株を選択することにより、作製した。
【0064】
(実施例2) アセトインの製造
実施例1にて得られたアセトイン産生細胞1を培養して、アセトインを産生させた。
まず、参考例1(2)と同様の手法により、前培養および、0.4w/v%グルコースを含む培地を用いて本培養を行った。また、参考例1(2)と同様の手法により前培養を行った後、本培養を高濃度グルコース添加Soytone培地(Soytone培地に3.6w/v% グルコースを添加したもの)を4w/v%グルコースを含む培地として用いた以外は、参考例1(2)と同様の手法により培養を行った。
【0065】
アセトイン濃度測定は、参考例1(3)と同様の操作により行った。また、本培養に高濃度グルコース添加Soytone培地を用いた場合は、培地(培養液)の上清を適宜希釈したものをアセトイン濃度測定のサンプル溶液として用いた。
【0066】
結果を図4および5に示す。0.4w/v%グルコースを含む培地の場合、野生型では培養後8時間で、アセトイン量がほぼ最大に達し、12時間以後は徐々に減少していくのに対し、アセトイン産生細胞1では、培養後約12時間までアセトイン量が増加し続け、その後、48時間まで減少しなかった。アセトイン産生細胞1のグルコースからのアセトイン変換率(モル比)は、90%を達成した。
4w/v%グルコースを含む培地の場合、野生型では徐々にアセトイン量が増加していくが、アセトイン産生細胞1により産生されたアセトイン量は、培養後24時間でほぼ最大に達し、その後96時間後まで変化が見られなかった。
【0067】
(参考例3) 変異株3(ΔalsR1)の作製
(1)変異株3(ΔalsR1)として、遺伝子型がamyE::cat, Pspac-alsR1である株を、枯草菌染色体のamyE領域に、大腸菌プラスミドpCRE-test-alsR1より得られたDNA鎖を相同組み換えにより導入することで作製した。具体的には以下のようにして作製した。
まず、pCRE-test-alsR1の作製では、168株の染色体を鋳型として、プライマーalsR1-F1(5'-cgcggatccatggagcttcgccatcttcaa-3'(配列番号23))とalsR1-R1(5'-catgtatagagcaggccatgc-3'(配列番号24))のペア、ならびにalsR1-F2(5'-gcatggcctgctctatacatg-3'(配列番号25))とalsR1-R2(5'-cgcggatcctgtacctgcatcactctcttt-3'(配列番号26))のペアを用いてPCR増幅を行い、alsRのORF内の5'側断片(約600 bp)と3'側断片(約300 bp)をそれぞれ増幅した(プライマーの塩基配列中の下線部は、制限酵素認識配列を示す)。それぞれのPCR断片を混合したものを鋳型とし、プライマーalsR1-F1とalsR1-R2のペアを用いてPCR増幅を行うと、alsR1-R1とalsR1-F2は互いに相補的であり、かつalsRのORFの592残基目から612残基目にかけての相補的な配列を持ち、さらに601残基目のアデニンがグアニンとなる点変異を持つようにデザインされているためalsRアレル(alsR1)が増幅される。このPCR産物をBamHIで制限酵素処理したものをインサートDNAとしてベクタープラスミドpCRE-test(Miwa, Y., Nakata, A., Ogiwara, A., Yamamoto, M. & Fujita, Y. (2000)., Nucleic. acids. Res. 28, 1206-1210.)のBamHIクローニングサイトに挿入した。alsR1のORFの601残基目の点変異を塩基配列の決定により確認し、pCRE-test-alsR1とした。
このpCRE-test-alsR1をPstIにより制限酵素処理することで得られたDNA鎖を用いて枯草菌168株を形質転換し、10μg/mlクロラムフェニコール耐性株の一つを変異株3とした。
【0068】
(2)変異株3の培養および、培地中のアセトイン濃度測定の測定は、実施例2と同様にして、本培養に4w/v%グルコースを含む培地を用いて行った。
【0069】
結果を図6に示す。野生株では、培養後96時間まで、徐々にアセトイン量が増加していた。一方変異株3では、培養後60時間におけるグルコースからのアセトイン変換率(モル比)は90%であったが、その後アセトイン量が減少することがわかった。
【0070】
(実施例3) アセトイン産生細胞2(alsR1ΔacoA)の作製
(1)アセトイン産生細胞2(alsR1ΔacoA)として、遺伝子型がamyE::tet,Pspac-alsR1 acoA::catである株を以下のように作製した。
pCm::Tc(Steinmetz, M. & Richter, R. (1994). Gene.142, 79-83)DNAを用いて、参考例3にて得られた変異株3(amyE::cat, Pspac-alsR1)を形質転換し、12.5μg/mlテトラサイクリン耐性かつ10μg/mlクロラムフェニコール感受性となった変異株(染色体上のクロラムフェニコール耐性遺伝子がテトラサイクリン耐性遺伝子に置き換えられている)を得た。得られたテトラサイクリン耐性株を、参考例2にて得た変異株2(acoA::cat)の染色体DNAを用いて形質転換し、12.5μg/mlテトラサイクリンと10μg/mlクロラムフェニコールの両方に耐性となった株のうち一つをアセトイン産生変異細胞2(名称 Bacillus subtilis MY05株)とした。
【0071】
(実施例4) アセトインの製造
参考例3(2)と同様にして、アセトイン産生細胞2を4w/v%グルコースを含む培地により培養し、アセトインを産生させた。培地中のアセトイン濃度測定の測定は参考例3(2)と同様にして行った。
【0072】
結果を図7に示す。アセトイン産生細胞2は、培養後96時間までアセトイン産生量が増加した。変異株3(ΔacoA)では96時間以後はアセトイン量が減少するのに対して、アセトイン産生細胞2ではアセトイン量の減少が抑制されることがわかった。
【0073】
(実施例5) アセトイン産生細胞3(alsR1ΔacoAΔlacE)および4(alsR1ΔacoAΔpta)の作製
アセトイン産生細胞3(alsR1ΔacoAΔlacE)として、遺伝子型がamyE::tet,Pspac-alsR1 acoA::cat lctE::specである株を、次のようにして作製した。参考例1にて作製したlctE遺伝子破壊株より抽出した染色体を用いて、アセトイン産生変異細胞2を形質転換し、12.5μg/mlテトラサイクリン、10μg/mlクロラムフェニコール、50μg/mlスペクチノマイシンのいずれに対しても耐性となった株のうち一つをアセトイン産生変異細胞3とした。
アセトイン産生細胞4(alsR1ΔacoAΔpta)として、遺伝子型がamyE::tet,Pspac-alsR1 acoA::cat pta::neoである株を、次のようにして作製した。参考例1にて作製したpta遺伝子破壊株より抽出した染色体を用いて、アセトイン産生変異細胞2を形質転換し、12.5μg/mlテトラサイクリン、10μg/mlクロラムフェニコール、10μg/mlネオマイシンのいずれに対しても耐性となった株のうち一つをアセトイン産生変異細胞4とした。
【産業上の利用可能性】
【0074】
本発明の細胞はアセトインを、グルコースを原料として、細胞内で安価にかつ効率よく産生することができる。また、かかる細胞を用いた本発明のアセトインの製造方法は、アセトインの産生を1段階で行うことができ簡便である。得られたアセトインは、食品添加物、化粧品原料として応用可能であるのに加えて、ジアセチルや2,3−ブタンジオールの中間体としても有用である。
【特許請求の範囲】
【請求項1】
アセトイン脱水素酵素の機能が欠損または低減しており、ピルビン酸からアセト乳酸、及び、アセト乳酸からアセトインの合成を司る酵素群が構成的に発現している、アセトイン産生細胞。
【請求項2】
前記酵素群をコードする遺伝子が単一のオペロンを形成している、請求項1に記載のアセトイン産生細胞。
【請求項3】
アセトイン脱水素酵素の機能の欠損または低減が、アセトイン脱水素酵素をコードする遺伝子を人為的に破壊したことによるものであり、且つ、前記酵素群の構成的発現が、前記オペロンの発現制御系を改変するような変異を導入したことによるものである、請求項2に記載のアセトイン産生細胞。
【請求項4】
アセトイン脱水素酵素、リン酸アセチル基転移酵素、および乳酸脱水素酵素の3種のタンパク質の機能が欠損または低減している、アセトイン産生細胞。
【請求項5】
アセトイン脱水素酵素、リン酸アセチル基転移酵素、および乳酸脱水素酵素の3種のタンパク質の機能の欠損または低減が、アセトイン脱水素酵素をコードする遺伝子、リン酸アセチル基転移酵素をコードする遺伝子、および乳酸脱水素酵素をコードする遺伝子の3種の遺伝子を人為的に破壊したことによるものである、請求項4に記載のアセトイン産生細胞。
【請求項6】
さらに、リン酸アセチル基転移酵素および乳酸脱水素酵素のうち、少なくとも1種のタンパク質の機能が欠損または低減している、請求項1〜3のいずれか1に記載のアセトイン産生細胞。
【請求項7】
リン酸アセチル基転移酵素および乳酸脱水素酵素のうち、少なくとも1種のタンパク質の機能の欠損または低減が、リン酸アセチル基転移酵素をコードする遺伝子、および乳酸脱水素酵素をコードする遺伝子のうち、少なくとも1種の遺伝子を人為的に破壊したことによるものである、請求項6に記載のアセトイン産生細胞。
【請求項8】
細胞がバチルス属に属する菌である請求項1〜7のいずれか1に記載の細胞。
【請求項9】
細胞が、受領番号FERM AP-22036(受領日:2010年10月28日)の枯草菌変異株である、請求項8に記載の細胞。
【請求項10】
請求項1〜9のいずれか1に記載の細胞を、グルコースの存在下で培養する工程を含む、アセトイン製造方法。
【請求項11】
請求項1〜3、6〜9のいずれか1に記載の細胞を、1〜10w/v%グルコースの存在下で培養する工程を含む、アセトイン製造方法。
【請求項12】
請求項4、5,8のいずれか1に記載の細胞を、0.2〜2w/v%グルコースの存在下で培養する工程を含む、アセトイン製造方法。
【請求項13】
前記の培養する工程で得られた培養ろ液からアセトインを単離する工程をさらに含む、請求項10〜12のいずれか1に記載のアセトイン製造方法。
【請求項1】
アセトイン脱水素酵素の機能が欠損または低減しており、ピルビン酸からアセト乳酸、及び、アセト乳酸からアセトインの合成を司る酵素群が構成的に発現している、アセトイン産生細胞。
【請求項2】
前記酵素群をコードする遺伝子が単一のオペロンを形成している、請求項1に記載のアセトイン産生細胞。
【請求項3】
アセトイン脱水素酵素の機能の欠損または低減が、アセトイン脱水素酵素をコードする遺伝子を人為的に破壊したことによるものであり、且つ、前記酵素群の構成的発現が、前記オペロンの発現制御系を改変するような変異を導入したことによるものである、請求項2に記載のアセトイン産生細胞。
【請求項4】
アセトイン脱水素酵素、リン酸アセチル基転移酵素、および乳酸脱水素酵素の3種のタンパク質の機能が欠損または低減している、アセトイン産生細胞。
【請求項5】
アセトイン脱水素酵素、リン酸アセチル基転移酵素、および乳酸脱水素酵素の3種のタンパク質の機能の欠損または低減が、アセトイン脱水素酵素をコードする遺伝子、リン酸アセチル基転移酵素をコードする遺伝子、および乳酸脱水素酵素をコードする遺伝子の3種の遺伝子を人為的に破壊したことによるものである、請求項4に記載のアセトイン産生細胞。
【請求項6】
さらに、リン酸アセチル基転移酵素および乳酸脱水素酵素のうち、少なくとも1種のタンパク質の機能が欠損または低減している、請求項1〜3のいずれか1に記載のアセトイン産生細胞。
【請求項7】
リン酸アセチル基転移酵素および乳酸脱水素酵素のうち、少なくとも1種のタンパク質の機能の欠損または低減が、リン酸アセチル基転移酵素をコードする遺伝子、および乳酸脱水素酵素をコードする遺伝子のうち、少なくとも1種の遺伝子を人為的に破壊したことによるものである、請求項6に記載のアセトイン産生細胞。
【請求項8】
細胞がバチルス属に属する菌である請求項1〜7のいずれか1に記載の細胞。
【請求項9】
細胞が、受領番号FERM AP-22036(受領日:2010年10月28日)の枯草菌変異株である、請求項8に記載の細胞。
【請求項10】
請求項1〜9のいずれか1に記載の細胞を、グルコースの存在下で培養する工程を含む、アセトイン製造方法。
【請求項11】
請求項1〜3、6〜9のいずれか1に記載の細胞を、1〜10w/v%グルコースの存在下で培養する工程を含む、アセトイン製造方法。
【請求項12】
請求項4、5,8のいずれか1に記載の細胞を、0.2〜2w/v%グルコースの存在下で培養する工程を含む、アセトイン製造方法。
【請求項13】
前記の培養する工程で得られた培養ろ液からアセトインを単離する工程をさらに含む、請求項10〜12のいずれか1に記載のアセトイン製造方法。
【図1】


【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2012−105582(P2012−105582A)
【公開日】平成24年6月7日(2012.6.7)
【国際特許分類】
【出願番号】特願2010−256714(P2010−256714)
【出願日】平成22年11月17日(2010.11.17)
【出願人】(504150450)国立大学法人神戸大学 (421)
【Fターム(参考)】
【公開日】平成24年6月7日(2012.6.7)
【国際特許分類】
【出願日】平成22年11月17日(2010.11.17)
【出願人】(504150450)国立大学法人神戸大学 (421)
【Fターム(参考)】
[ Back to top ]