
アセンブリ誤り検出のための方法およびシステム(アセンブリ誤り検出)
【課題】読取りデータの再アセンブリはアセンブリにおける配列誤りを含み得る。なぜならセグメントを正確な元の順序に戻すことは難しいことがあるからである。
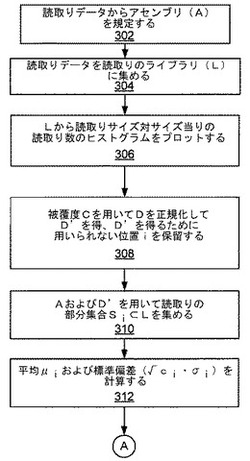
【解決手段】遺伝子配列アセンブリの誤りを検出するための方法は、遺伝子データの配列のアセンブリ(A)を規定するステップと、読取りデータを読取りのライブラリ(L)に集めるステップと、読取りのサイズ対サイズ当りの読取り数のヒストグラムをプロットするステップと、分布(D)を被覆度Cで正規化することによって、平均(μ)および標準偏差(σ)を有するD’を得て、D’を得るために用いられない位置(i)を保留するステップと、AおよびD’を用いて読取りの部分集合(Si⊂L)を集めるステップと、Siを用いて平均(μi)および標準偏差(√ci・σi)を計算するステップと、ユーザに対してディスプレイ上に結果を出力するステップとを含む。
【解決手段】遺伝子配列アセンブリの誤りを検出するための方法は、遺伝子データの配列のアセンブリ(A)を規定するステップと、読取りデータを読取りのライブラリ(L)に集めるステップと、読取りのサイズ対サイズ当りの読取り数のヒストグラムをプロットするステップと、分布(D)を被覆度Cで正規化することによって、平均(μ)および標準偏差(σ)を有するD’を得て、D’を得るために用いられない位置(i)を保留するステップと、AおよびD’を用いて読取りの部分集合(Si⊂L)を集めるステップと、Siを用いて平均(μi)および標準偏差(√ci・σi)を計算するステップと、ユーザに対してディスプレイ上に結果を出力するステップとを含む。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、デオキシリボ核酸(deoxyribonucleic acid:DNA)におけるアセンブリ誤り検出、ならびにリボ核酸(Ribonucleic acid:RNA)における過剰発現および低発現検出に関する。
【背景技術】
【0002】
デオキシリボ核酸(DNA)ゲノム配列は、DNAをいくつかの塩基の配列を有するいくつかのセグメントまたは小片に分割する方法を用いて決定されることがある。各セグメント内の塩基の配列決定と、セグメントの順序の決定とをともに用いて、DNA全体の配列を決定してもよい。セグメントの順序の決定は、バイオインフォマティクス・アセンブリ法を用いてイン・シリコ(in−silico)で行なわれてもよい。
【発明の概要】
【発明が解決しようとする課題】
【0003】
読取りデータの再アセンブリはアセンブリにおける配列誤りを含み得る。なぜならセグメントを正確な元の順序に戻すことは難しいことがあるからである。
【課題を解決するための手段】
【0004】
本発明の一局面において、遺伝子配列アセンブリの誤りを検出するための方法は、遺伝子データの配列のアセンブリ(A)を規定するステップと、読取りデータを読取りのライブラリ(L)に集めるステップと、読取りのサイズ対サイズ当りの読取り数のヒストグラムをプロットするステップと、分布(D)を被覆度(coverage)(C)で正規化することによって、平均(μ)および標準偏差(σ)を有するD’を得て、D’を得るために用いられない位置(i)を保留するステップと、AおよびD’を用いて読取りの部分集合(Si⊂L)を集めるステップと、Siを用いて平均(μi)および標準偏差(√ci・σi)を計算するステップと、ユーザに対してディスプレイ上に結果を出力するステップとを含む。
【0005】
本発明の別の局面において、遺伝子配列の誤りを検出するためのシステムは、メモリと、ディスプレイと、遺伝子データの配列のアセンブリ(A)を規定し、読取りデータを読取りのライブラリ(L)に集め、読取りのサイズ対サイズ当りの読取り数のヒストグラムをプロットし、分布(D)を被覆度(C)で正規化することによって、平均(μ)および標準偏差(σ)を有するD’を得て、D’を得るために用いられない位置(i)を保留し、AおよびD’を用いて読取りの部分集合(Si⊂L)を集め、Siを用いて平均(μi)および標準偏差(√ci・σi)を計算し、ユーザに対してディスプレイ上に結果を出力するために動作するプロセッサとを含む。
【0006】
本発明の技術によって、付加的な特徴および利点が実現される。本明細書には本発明の他の実施形態および局面が詳細に記載されており、請求される発明の一部とみなされる。利点および特徴を伴う本発明をより良く理解するために、説明および図面を参照されたい。
【0007】
本発明であるとみなされる主題は、請求項において特定的に示され明確に主張されている。本発明の前述およびその他の特徴および利点は、以下の詳細な説明および添付の図面から明らかである。
【図面の簡単な説明】
【0008】
【図1】複数のDNA配列およびそれらの配列のセグメントへの分割を示す図である。
【図2】配列中の誤りを定めるためのシステム200の例示的な実施形態を示す図である。
【図3】図2のシステムによって行なわれ得る例示的な処理方法を示すブロック図である。
【図4】図2のシステムによって行なわれ得る例示的な処理方法を示すブロック図である。
【図5】読取りの頻度のヒストグラムを示す図である。
【発明を実施するための形態】
【0009】
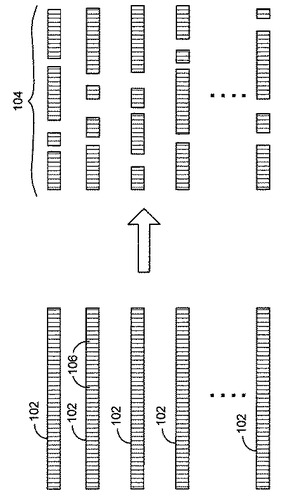
デオキシリボ核酸(DNA)ゲノム配列は、たとえば圧縮空気装置(噴霧器)または制限酵素などを用いることによって、DNAをいくつかの塩基の配列を有するいくつかのセグメントまたは小片に分割することによって決定されることがある。図1は、複数の類似のDNA配列、およびその配列のセグメントへの分割を示している。これに関して、いくつかの類似のDNA鎖102(例、50個またはそれ以上の鎖)が、たとえば50塩基から500塩基などの範囲のいくつかの塩基106を有する複数のセグメント104に分裂または切断されてもよい。セグメント104は必ずしも等しい長さに切断されるわけではない。セグメント104が切断されると、セグメント104を読取って塩基106が識別され、識別された塩基106の各セグメントにおける位置が定められることによって、各セグメント104に対する読取りデータがもたらされる。代替的には、セグメントの端部(例、各端部から100塩基)を読取って塩基を識別してもよい。セグメントの読取りは、たとえばヌクレオチドの蛍光ラベリングおよび高分解能レーザ・イメージングを含む、合成による配列決定(sequencing−by−synthesis)のプロセスなどによって行なわれてもよい。結果として生じるデータは複数の読取りを含み、各読取りは塩基106と、各セグメント104におけるその塩基106の位置とを識別する。読取りデータはグループ化されて、特定の長さにおける読取りの頻度(すなわち特定の長さの塩基を有する読取りの数)を含む読取りのライブラリ(L)に入れられる。被覆度(C)とは、配列決定されたDNAにおける位置が重複しているセグメント104の平均コピー数である。配列決定されたセグメント104の長さに加えてDNA配列の長さが既知であれば、被覆度Cが分かる。DNAゲノム配列の長さが未知であるときには、ユーザが推定の長さを与えてもよい。読取りデータの「再アセンブリ」を行なうことによって、DNAゲノム配列の一部または全体を表わすアセンブリ(A)データをもたらしてもよい。アセンブリは、たとえばアセンブラ(イン・シリコのバイオインフォマティクス・ツール)などを用いて、読取りにおける塩基間の重複を考慮し、可能なところで重複した読取りを連結することによって行なわれてもよい。アセンブリ・データは、所与の位置iにおける読取りカウントciおよび読取り長さlを含むベクトルV=<i,ci,l1,l2,…,lci>を含む。ベクトルの一例はV=<34,3,10,12,102>を含み、これは位置34がそれぞれ長さ10、12および102の3つの読取りによって重複することを示す。読取りデータの再アセンブリはアセンブリにおける配列誤りを含み得る。なぜならセグメントを正確な元の順序に戻すことは難しいことがあるからである。以下に説明される例示的な方法およびシステムは、アセンブリにおける誤りの検出を改善するものである。
【0010】
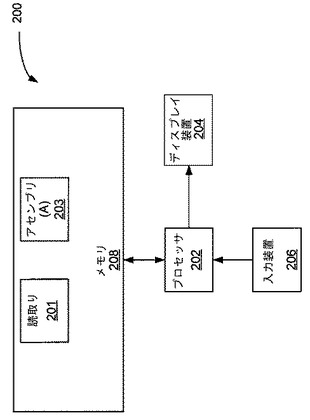
これに関して、図2は配列中の誤りを定めるためのシステム200の例示的な実施形態を示す。示される実施形態は、ディスプレイ装置204と、入力装置206と、読取りデータ201およびアセンブリ203を保存するメモリ208とに通信的に接続されたプロセッサ202を含む。
【0011】
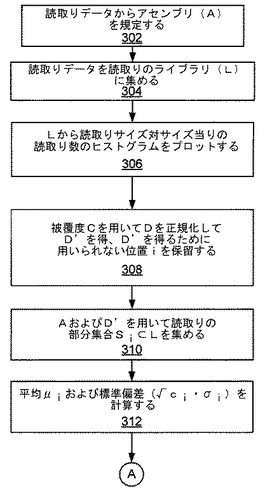
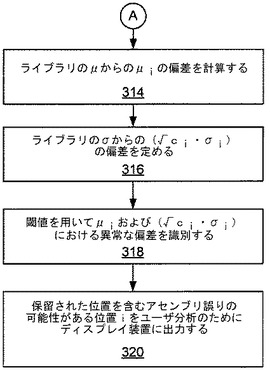
図3および図4は、システム200によって行なわれ得る例示的な処理方法のブロック図を示す。図3を参照すると、ブロック302において読取りデータを含むアセンブリ(A)が規定される。ブロック304において、読取りデータが読取りのライブラリ(L)に集められる。ブロック306において、Lから読取りのサイズ対サイズ当りの読取り数のヒストグラムがプロットされる。ヒストグラムの一例を図5に示す。ブロック308において、被覆度Cを用いて分布Dを正規化して(D’)を得、ここでD’はLの期待標準分布であり、平均μおよび標準偏差σを有する。正規化は、Aに対する被覆度Cを用い、(ユーザによって与えられる上側および下側のカットオフを用いて)被覆度Cを表わすとは考えられないベクトルVをフィルタリングして除去することによって行なわれる。前のステップの出力を用いてライブラリが再び計算される。D’を得るために用いられない位置(i)が保留される。ブロック310において、アセンブリA内の各位置(i)に対して、位置iが重複している読取りの部分集合Si⊂LがベクトルViに集められる。ブロック312において、Siから平均(μi)および標準偏差(√ci・σi)が算出される。(図4の)ブロック314において、ライブラリのμからのμiの偏差が計算される。ブロック316において、ライブラリのσからの(√ci・σi)の偏差が定められる。ブロック318において、閾値を用いて、μiおよび(√ci・σi)における異常な偏差(すなわち閾値の外側の偏差)が定められる。
【0012】
ブロック320において、ユーザ分析のために結果がディスプレイ装置に出力されてもよい。アセンブリにおける各位置iに対して、平均(μi)が期待値から所与の閾値よりも多く逸脱しているか、または標準偏差(√ci・σi)が所与の閾値よりも大きいとき、その位置iはアセンブリの誤りの可能性があるとしてフラグを立てられる。次いでユーザは、別の方法によってデータを再アセンブリするか、追加の読取りを生成して再アセンブリするか、または配列情報の代替的供給源を用いることによって、これらのフラグを立てられた領域におけるアセンブリの間違いの可能性を修正することに集中できる。
【0013】
RNAデータに対しても同様のプロセスを用いることができるが、フラグを立てられる位置は過剰発現または低発現に関連付けられる。
【0014】
本明細書において用いられる用語は特定の実施形態を説明する目的のためのみのものであって、本発明を限定することは意図されない。本明細書において用いられる単数形「a」、「an」および「the」は、状況が明らかに別様を示していない限り、複数形をも含むことが意図される。さらに、「含む(comprises)」もしくは「含む(comprising)」という用語またはその両方が本明細書において用いられるとき、それは述べられる特徴、完全体、ステップ、動作、構成要素もしくは成分またはその組合わせの存在を特定するが、1つまたはそれ以上の他の特徴、完全体、ステップ、動作、構成要素成分もしくはそのグループまたはその組合わせの存在または追加を排除するものではないことが理解されるだろう。
【0015】
以下の請求項におけるすべての手段またはステップ・プラス機能(means or step plus function)要素に対応する構造、材料、動作、および同等物は、特定的に請求される他の請求要素と組合わせてその機能を行なうためのあらゆる構造、材料または動作を含むことが意図される。本発明の説明は例示および説明の目的のために提供されたものであるが、網羅的になったり、開示される形に本発明を制限したりすることは意図されない。本発明の範囲および趣旨から逸脱することなく、通常の当業者には多くの修正および変更が明らかになるだろう。実施形態は、本発明の原理および実際の適用を最も良く説明し、他の通常の当業者が予期される特定の使用に好適であるようなさまざまな修正を伴うさまざまな実施形態に対して本発明を理解できるようにするために選択されて記載されたものである。
【0016】
本明細書に示される図面は単なる一例である。本発明の趣旨から逸脱することなく、この図面または本明細書に記載されるステップ(もしくは動作)には多くの変更形が存在するだろう。たとえば、これらのステップが異なる順序で行なわれてもよいし、ステップが追加、削除または変更されてもよい。これらの変更形はすべて、請求される本発明の一部であるとみなされる。
【0017】
本発明に対する好ましい実施形態を説明したが、現在および将来にわたり、当業者が以下の請求項の範囲内のさまざまな改善および強化を行ない得ることが理解されるだろう。これらの請求項は、最初に記載された本発明に対する適切な保護を維持するものと解釈されるべきである。
【符号の説明】
【0018】
302 読取りデータからアセンブリ(A)を規定する
304 読取りデータを読取りのライブラリ(L)に集める
306 Lから読取りサイズ対サイズ当りの読取り数のヒストグラムをプロットする
308 被覆度Cを用いてDを正規化してD’を得、D’を得るために用いられない位置iを保留する
310 AおよびD’を用いて読取りの部分集合Si⊂Lを集める
312 平均μiおよび標準偏差(√ci・σi)を計算する
【技術分野】
【0001】
本発明は、デオキシリボ核酸(deoxyribonucleic acid:DNA)におけるアセンブリ誤り検出、ならびにリボ核酸(Ribonucleic acid:RNA)における過剰発現および低発現検出に関する。
【背景技術】
【0002】
デオキシリボ核酸(DNA)ゲノム配列は、DNAをいくつかの塩基の配列を有するいくつかのセグメントまたは小片に分割する方法を用いて決定されることがある。各セグメント内の塩基の配列決定と、セグメントの順序の決定とをともに用いて、DNA全体の配列を決定してもよい。セグメントの順序の決定は、バイオインフォマティクス・アセンブリ法を用いてイン・シリコ(in−silico)で行なわれてもよい。
【発明の概要】
【発明が解決しようとする課題】
【0003】
読取りデータの再アセンブリはアセンブリにおける配列誤りを含み得る。なぜならセグメントを正確な元の順序に戻すことは難しいことがあるからである。
【課題を解決するための手段】
【0004】
本発明の一局面において、遺伝子配列アセンブリの誤りを検出するための方法は、遺伝子データの配列のアセンブリ(A)を規定するステップと、読取りデータを読取りのライブラリ(L)に集めるステップと、読取りのサイズ対サイズ当りの読取り数のヒストグラムをプロットするステップと、分布(D)を被覆度(coverage)(C)で正規化することによって、平均(μ)および標準偏差(σ)を有するD’を得て、D’を得るために用いられない位置(i)を保留するステップと、AおよびD’を用いて読取りの部分集合(Si⊂L)を集めるステップと、Siを用いて平均(μi)および標準偏差(√ci・σi)を計算するステップと、ユーザに対してディスプレイ上に結果を出力するステップとを含む。
【0005】
本発明の別の局面において、遺伝子配列の誤りを検出するためのシステムは、メモリと、ディスプレイと、遺伝子データの配列のアセンブリ(A)を規定し、読取りデータを読取りのライブラリ(L)に集め、読取りのサイズ対サイズ当りの読取り数のヒストグラムをプロットし、分布(D)を被覆度(C)で正規化することによって、平均(μ)および標準偏差(σ)を有するD’を得て、D’を得るために用いられない位置(i)を保留し、AおよびD’を用いて読取りの部分集合(Si⊂L)を集め、Siを用いて平均(μi)および標準偏差(√ci・σi)を計算し、ユーザに対してディスプレイ上に結果を出力するために動作するプロセッサとを含む。
【0006】
本発明の技術によって、付加的な特徴および利点が実現される。本明細書には本発明の他の実施形態および局面が詳細に記載されており、請求される発明の一部とみなされる。利点および特徴を伴う本発明をより良く理解するために、説明および図面を参照されたい。
【0007】
本発明であるとみなされる主題は、請求項において特定的に示され明確に主張されている。本発明の前述およびその他の特徴および利点は、以下の詳細な説明および添付の図面から明らかである。
【図面の簡単な説明】
【0008】
【図1】複数のDNA配列およびそれらの配列のセグメントへの分割を示す図である。
【図2】配列中の誤りを定めるためのシステム200の例示的な実施形態を示す図である。
【図3】図2のシステムによって行なわれ得る例示的な処理方法を示すブロック図である。
【図4】図2のシステムによって行なわれ得る例示的な処理方法を示すブロック図である。
【図5】読取りの頻度のヒストグラムを示す図である。
【発明を実施するための形態】
【0009】
デオキシリボ核酸(DNA)ゲノム配列は、たとえば圧縮空気装置(噴霧器)または制限酵素などを用いることによって、DNAをいくつかの塩基の配列を有するいくつかのセグメントまたは小片に分割することによって決定されることがある。図1は、複数の類似のDNA配列、およびその配列のセグメントへの分割を示している。これに関して、いくつかの類似のDNA鎖102(例、50個またはそれ以上の鎖)が、たとえば50塩基から500塩基などの範囲のいくつかの塩基106を有する複数のセグメント104に分裂または切断されてもよい。セグメント104は必ずしも等しい長さに切断されるわけではない。セグメント104が切断されると、セグメント104を読取って塩基106が識別され、識別された塩基106の各セグメントにおける位置が定められることによって、各セグメント104に対する読取りデータがもたらされる。代替的には、セグメントの端部(例、各端部から100塩基)を読取って塩基を識別してもよい。セグメントの読取りは、たとえばヌクレオチドの蛍光ラベリングおよび高分解能レーザ・イメージングを含む、合成による配列決定(sequencing−by−synthesis)のプロセスなどによって行なわれてもよい。結果として生じるデータは複数の読取りを含み、各読取りは塩基106と、各セグメント104におけるその塩基106の位置とを識別する。読取りデータはグループ化されて、特定の長さにおける読取りの頻度(すなわち特定の長さの塩基を有する読取りの数)を含む読取りのライブラリ(L)に入れられる。被覆度(C)とは、配列決定されたDNAにおける位置が重複しているセグメント104の平均コピー数である。配列決定されたセグメント104の長さに加えてDNA配列の長さが既知であれば、被覆度Cが分かる。DNAゲノム配列の長さが未知であるときには、ユーザが推定の長さを与えてもよい。読取りデータの「再アセンブリ」を行なうことによって、DNAゲノム配列の一部または全体を表わすアセンブリ(A)データをもたらしてもよい。アセンブリは、たとえばアセンブラ(イン・シリコのバイオインフォマティクス・ツール)などを用いて、読取りにおける塩基間の重複を考慮し、可能なところで重複した読取りを連結することによって行なわれてもよい。アセンブリ・データは、所与の位置iにおける読取りカウントciおよび読取り長さlを含むベクトルV=<i,ci,l1,l2,…,lci>を含む。ベクトルの一例はV=<34,3,10,12,102>を含み、これは位置34がそれぞれ長さ10、12および102の3つの読取りによって重複することを示す。読取りデータの再アセンブリはアセンブリにおける配列誤りを含み得る。なぜならセグメントを正確な元の順序に戻すことは難しいことがあるからである。以下に説明される例示的な方法およびシステムは、アセンブリにおける誤りの検出を改善するものである。
【0010】
これに関して、図2は配列中の誤りを定めるためのシステム200の例示的な実施形態を示す。示される実施形態は、ディスプレイ装置204と、入力装置206と、読取りデータ201およびアセンブリ203を保存するメモリ208とに通信的に接続されたプロセッサ202を含む。
【0011】
図3および図4は、システム200によって行なわれ得る例示的な処理方法のブロック図を示す。図3を参照すると、ブロック302において読取りデータを含むアセンブリ(A)が規定される。ブロック304において、読取りデータが読取りのライブラリ(L)に集められる。ブロック306において、Lから読取りのサイズ対サイズ当りの読取り数のヒストグラムがプロットされる。ヒストグラムの一例を図5に示す。ブロック308において、被覆度Cを用いて分布Dを正規化して(D’)を得、ここでD’はLの期待標準分布であり、平均μおよび標準偏差σを有する。正規化は、Aに対する被覆度Cを用い、(ユーザによって与えられる上側および下側のカットオフを用いて)被覆度Cを表わすとは考えられないベクトルVをフィルタリングして除去することによって行なわれる。前のステップの出力を用いてライブラリが再び計算される。D’を得るために用いられない位置(i)が保留される。ブロック310において、アセンブリA内の各位置(i)に対して、位置iが重複している読取りの部分集合Si⊂LがベクトルViに集められる。ブロック312において、Siから平均(μi)および標準偏差(√ci・σi)が算出される。(図4の)ブロック314において、ライブラリのμからのμiの偏差が計算される。ブロック316において、ライブラリのσからの(√ci・σi)の偏差が定められる。ブロック318において、閾値を用いて、μiおよび(√ci・σi)における異常な偏差(すなわち閾値の外側の偏差)が定められる。
【0012】
ブロック320において、ユーザ分析のために結果がディスプレイ装置に出力されてもよい。アセンブリにおける各位置iに対して、平均(μi)が期待値から所与の閾値よりも多く逸脱しているか、または標準偏差(√ci・σi)が所与の閾値よりも大きいとき、その位置iはアセンブリの誤りの可能性があるとしてフラグを立てられる。次いでユーザは、別の方法によってデータを再アセンブリするか、追加の読取りを生成して再アセンブリするか、または配列情報の代替的供給源を用いることによって、これらのフラグを立てられた領域におけるアセンブリの間違いの可能性を修正することに集中できる。
【0013】
RNAデータに対しても同様のプロセスを用いることができるが、フラグを立てられる位置は過剰発現または低発現に関連付けられる。
【0014】
本明細書において用いられる用語は特定の実施形態を説明する目的のためのみのものであって、本発明を限定することは意図されない。本明細書において用いられる単数形「a」、「an」および「the」は、状況が明らかに別様を示していない限り、複数形をも含むことが意図される。さらに、「含む(comprises)」もしくは「含む(comprising)」という用語またはその両方が本明細書において用いられるとき、それは述べられる特徴、完全体、ステップ、動作、構成要素もしくは成分またはその組合わせの存在を特定するが、1つまたはそれ以上の他の特徴、完全体、ステップ、動作、構成要素成分もしくはそのグループまたはその組合わせの存在または追加を排除するものではないことが理解されるだろう。
【0015】
以下の請求項におけるすべての手段またはステップ・プラス機能(means or step plus function)要素に対応する構造、材料、動作、および同等物は、特定的に請求される他の請求要素と組合わせてその機能を行なうためのあらゆる構造、材料または動作を含むことが意図される。本発明の説明は例示および説明の目的のために提供されたものであるが、網羅的になったり、開示される形に本発明を制限したりすることは意図されない。本発明の範囲および趣旨から逸脱することなく、通常の当業者には多くの修正および変更が明らかになるだろう。実施形態は、本発明の原理および実際の適用を最も良く説明し、他の通常の当業者が予期される特定の使用に好適であるようなさまざまな修正を伴うさまざまな実施形態に対して本発明を理解できるようにするために選択されて記載されたものである。
【0016】
本明細書に示される図面は単なる一例である。本発明の趣旨から逸脱することなく、この図面または本明細書に記載されるステップ(もしくは動作)には多くの変更形が存在するだろう。たとえば、これらのステップが異なる順序で行なわれてもよいし、ステップが追加、削除または変更されてもよい。これらの変更形はすべて、請求される本発明の一部であるとみなされる。
【0017】
本発明に対する好ましい実施形態を説明したが、現在および将来にわたり、当業者が以下の請求項の範囲内のさまざまな改善および強化を行ない得ることが理解されるだろう。これらの請求項は、最初に記載された本発明に対する適切な保護を維持するものと解釈されるべきである。
【符号の説明】
【0018】
302 読取りデータからアセンブリ(A)を規定する
304 読取りデータを読取りのライブラリ(L)に集める
306 Lから読取りサイズ対サイズ当りの読取り数のヒストグラムをプロットする
308 被覆度Cを用いてDを正規化してD’を得、D’を得るために用いられない位置iを保留する
310 AおよびD’を用いて読取りの部分集合Si⊂Lを集める
312 平均μiおよび標準偏差(√ci・σi)を計算する
【特許請求の範囲】
【請求項1】
コンピュータ・システムにより遺伝子配列アセンブリの誤りを検出するための方法であって、前記コンピュータ・システムはメモリとディスプレイとプロセッサとを含み、
前記方法は、
前記プロセッサが前記メモリ上で遺伝子データの配列のアセンブリ(A)を規定するステップと、
前記プロセッサが前記メモリ上で読取りデータを読取りのライブラリ(L)に集めるステップと、
前記プロセッサが読取りのサイズ対サイズ当りの読取り数のヒストグラムをプロットするステップと、
前記プロセッサが分布(D)を被覆度Cで正規化することによって、平均(μ)および標準偏差(σ)を有するD’を得て、D’を得るために用いられない位置(i)を保留するステップと、
前記プロセッサがAおよびD’を用いて読取りの部分集合(Si⊂L)を集めるステップと、
前記プロセッサがSiを用いて平均(μi)および標準偏差(√ci・σi)を計算するステップと、
前記プロセッサがユーザに対して前記ディスプレイ上に結果を出力するステップと
を含む、方法。
【請求項2】
前記方法は、前記プロセッサが読取りの前記ライブラリから各位置(i)に対するμからのμiの偏差を計算するステップをさらに含む、請求項1に記載の方法。
【請求項3】
前記方法は、前記プロセッサが読取りの前記ライブラリから各位置(i)に対するσからの√ci・σiの偏差を定めるステップをさらに含む、請求項1に記載の方法。
【請求項4】
前記方法は、前記偏差を閾値と比較して、前記閾値よりも大きいかまたは小さい偏差を識別するステップをさらに含む、請求項2に記載の方法。
【請求項5】
前記方法は、前記プロセッサが前記偏差を閾値と比較して、前記閾値よりも大きいかまたは小さい偏差を識別するステップをさらに含む、請求項3に記載の方法。
【請求項6】
前記方法は、前記プロセッサが前記識別された偏差の位置iをユーザに対して前記ディスプレイ上に出力させるステップを含む、請求項4に記載の方法。
【請求項7】
前記方法は、前記プロセッサが前記識別された偏差の位置iをユーザに対して前記ディスプレイ上に出力させるステップを含む、請求項5に記載の方法。
【請求項8】
前記アセンブリは、配列アセンブリのためのイン・シリコのバイオインフォマティクス法によって規定される、請求項1に記載の方法。
【請求項9】
前記読取りデータは、デオキシリボ核酸(DNA)のセグメント内の複数の塩基の位置および識別子を含む、請求項1に記載の方法。
【請求項10】
読取りの前記ライブラリは複数の読取りデータを含む、請求項1に記載の方法。
【請求項11】
遺伝子配列の誤りを検出するためのシステムであって、前記システムは、
メモリと、
ディスプレイと、
遺伝子データの配列のアセンブリ(A)を規定し、読取りデータを読取りのライブラリ(L)に集め、読取りのサイズ対サイズ当りの読取り数のヒストグラムをプロットし、分布(D)を被覆度Cで正規化することによって、平均(μ)および標準偏差(σ)を有するD’を得て、D’を得るために用いられない位置(i)を保留し、AおよびD’を用いて読取りの部分集合(Si⊂L)を集め、Siを用いて平均(μi)および標準偏差(√ci・σi)を計算し、ユーザに対して前記ディスプレイ上に結果を出力するために動作するプロセッサと
を含む、システム。
【請求項12】
前記プロセッサは、読取りの前記ライブラリから各位置(i)に対するμからのμiの偏差を計算するためにさらに動作する、請求項11に記載のシステム。
【請求項13】
前記プロセッサは、読取りの前記ライブラリから各位置(i)に対するσからの√ci・σiの偏差を定めるためにさらに動作する、請求項11に記載のシステム。
【請求項14】
前記プロセッサは、前記偏差を閾値と比較して、前記閾値よりも大きいかまたは小さい偏差を識別するためにさらに動作する、請求項12に記載のシステム。
【請求項15】
前記プロセッサは、前記偏差を閾値と比較して、前記閾値よりも大きいかまたは小さい偏差を識別するためにさらに動作する、請求項13に記載のシステム。
【請求項16】
前記プロセッサは、前記識別された偏差の位置iをユーザに対して前記ディスプレイ上に出力するためにさらに動作する、請求項14に記載のシステム。
【請求項17】
前記プロセッサは、前記識別された偏差の位置iをユーザに対して前記ディスプレイ上に出力するためにさらに動作する、請求項15に記載のシステム。
【請求項18】
前記アセンブリは、配列アセンブリのためのイン・シリコのバイオインフォマティクス法によって規定される、請求項11に記載のシステム。
【請求項19】
前記読取りデータは、デオキシリボ核酸(DNA)のセグメント内の複数の塩基の位置および識別子を含む、請求項11に記載のシステム。
【請求項20】
読取りの前記ライブラリは複数の読取りデータを含む、請求項11に記載のシステム。
【請求項1】
コンピュータ・システムにより遺伝子配列アセンブリの誤りを検出するための方法であって、前記コンピュータ・システムはメモリとディスプレイとプロセッサとを含み、
前記方法は、
前記プロセッサが前記メモリ上で遺伝子データの配列のアセンブリ(A)を規定するステップと、
前記プロセッサが前記メモリ上で読取りデータを読取りのライブラリ(L)に集めるステップと、
前記プロセッサが読取りのサイズ対サイズ当りの読取り数のヒストグラムをプロットするステップと、
前記プロセッサが分布(D)を被覆度Cで正規化することによって、平均(μ)および標準偏差(σ)を有するD’を得て、D’を得るために用いられない位置(i)を保留するステップと、
前記プロセッサがAおよびD’を用いて読取りの部分集合(Si⊂L)を集めるステップと、
前記プロセッサがSiを用いて平均(μi)および標準偏差(√ci・σi)を計算するステップと、
前記プロセッサがユーザに対して前記ディスプレイ上に結果を出力するステップと
を含む、方法。
【請求項2】
前記方法は、前記プロセッサが読取りの前記ライブラリから各位置(i)に対するμからのμiの偏差を計算するステップをさらに含む、請求項1に記載の方法。
【請求項3】
前記方法は、前記プロセッサが読取りの前記ライブラリから各位置(i)に対するσからの√ci・σiの偏差を定めるステップをさらに含む、請求項1に記載の方法。
【請求項4】
前記方法は、前記偏差を閾値と比較して、前記閾値よりも大きいかまたは小さい偏差を識別するステップをさらに含む、請求項2に記載の方法。
【請求項5】
前記方法は、前記プロセッサが前記偏差を閾値と比較して、前記閾値よりも大きいかまたは小さい偏差を識別するステップをさらに含む、請求項3に記載の方法。
【請求項6】
前記方法は、前記プロセッサが前記識別された偏差の位置iをユーザに対して前記ディスプレイ上に出力させるステップを含む、請求項4に記載の方法。
【請求項7】
前記方法は、前記プロセッサが前記識別された偏差の位置iをユーザに対して前記ディスプレイ上に出力させるステップを含む、請求項5に記載の方法。
【請求項8】
前記アセンブリは、配列アセンブリのためのイン・シリコのバイオインフォマティクス法によって規定される、請求項1に記載の方法。
【請求項9】
前記読取りデータは、デオキシリボ核酸(DNA)のセグメント内の複数の塩基の位置および識別子を含む、請求項1に記載の方法。
【請求項10】
読取りの前記ライブラリは複数の読取りデータを含む、請求項1に記載の方法。
【請求項11】
遺伝子配列の誤りを検出するためのシステムであって、前記システムは、
メモリと、
ディスプレイと、
遺伝子データの配列のアセンブリ(A)を規定し、読取りデータを読取りのライブラリ(L)に集め、読取りのサイズ対サイズ当りの読取り数のヒストグラムをプロットし、分布(D)を被覆度Cで正規化することによって、平均(μ)および標準偏差(σ)を有するD’を得て、D’を得るために用いられない位置(i)を保留し、AおよびD’を用いて読取りの部分集合(Si⊂L)を集め、Siを用いて平均(μi)および標準偏差(√ci・σi)を計算し、ユーザに対して前記ディスプレイ上に結果を出力するために動作するプロセッサと
を含む、システム。
【請求項12】
前記プロセッサは、読取りの前記ライブラリから各位置(i)に対するμからのμiの偏差を計算するためにさらに動作する、請求項11に記載のシステム。
【請求項13】
前記プロセッサは、読取りの前記ライブラリから各位置(i)に対するσからの√ci・σiの偏差を定めるためにさらに動作する、請求項11に記載のシステム。
【請求項14】
前記プロセッサは、前記偏差を閾値と比較して、前記閾値よりも大きいかまたは小さい偏差を識別するためにさらに動作する、請求項12に記載のシステム。
【請求項15】
前記プロセッサは、前記偏差を閾値と比較して、前記閾値よりも大きいかまたは小さい偏差を識別するためにさらに動作する、請求項13に記載のシステム。
【請求項16】
前記プロセッサは、前記識別された偏差の位置iをユーザに対して前記ディスプレイ上に出力するためにさらに動作する、請求項14に記載のシステム。
【請求項17】
前記プロセッサは、前記識別された偏差の位置iをユーザに対して前記ディスプレイ上に出力するためにさらに動作する、請求項15に記載のシステム。
【請求項18】
前記アセンブリは、配列アセンブリのためのイン・シリコのバイオインフォマティクス法によって規定される、請求項11に記載のシステム。
【請求項19】
前記読取りデータは、デオキシリボ核酸(DNA)のセグメント内の複数の塩基の位置および識別子を含む、請求項11に記載のシステム。
【請求項20】
読取りの前記ライブラリは複数の読取りデータを含む、請求項11に記載のシステム。
【図1】


【図2】
【図3】
【図4】
【図5】
【図2】
【図3】
【図4】
【図5】
【公開番号】特開2012−155715(P2012−155715A)
【公開日】平成24年8月16日(2012.8.16)
【国際特許分類】
【出願番号】特願2012−7764(P2012−7764)
【出願日】平成24年1月18日(2012.1.18)
【出願人】(390009531)インターナショナル・ビジネス・マシーンズ・コーポレーション (4,084)
【氏名又は名称原語表記】INTERNATIONAL BUSINESS MASCHINES CORPORATION
【Fターム(参考)】
【公開日】平成24年8月16日(2012.8.16)
【国際特許分類】
【出願日】平成24年1月18日(2012.1.18)
【出願人】(390009531)インターナショナル・ビジネス・マシーンズ・コーポレーション (4,084)
【氏名又は名称原語表記】INTERNATIONAL BUSINESS MASCHINES CORPORATION
【Fターム(参考)】
[ Back to top ]