
アゾ顔料の製造方法
【課題】易分散なアゾ顔料微粒子を、高効率(純度および生産性)かつ低コストで製造することのできるアゾ顔料の製造方法を提供することを目的とする。
【解決手段】非晶質な下記式(1)で表されるアゾ化合物、その塩、水和物又は溶媒和物を結晶変換することにより、下記式(1)で表されるアゾ顔料を製造する。

【解決手段】非晶質な下記式(1)で表されるアゾ化合物、その塩、水和物又は溶媒和物を結晶変換することにより、下記式(1)で表されるアゾ顔料を製造する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、アゾ顔料の製造方法に関する。
【背景技術】
【0002】
アゾ化合物の合成法については古くから種々の方法が知られており、酸化反応による合成、還元反応による合成、置換反応による合成、付加反応による合成、縮合反応による合成、その他の合成法などがあった(たとえば非特許文献1参照)。しかしながらアゾ色素化合物の工業的製造法として利用されているのは、原料の入手性、コスト、収率などの点からジアゾニウム化合物と、アニリン、フェノール類などのカップリング成分とをアゾカップリング反応させて合成する方法がほとんどであり、また、この方法もジアゾニウム化合物の爆発の危険性があったり、ジアゾニウム化合物やカップリング成分の種類によっては低収率であったりするなどの欠点を有していた。特に複素環ジアゾニウム化合物はジアゾニウム化合物が不安定である場合が多く、一般性の高い合成法は知られていない。
【0003】
また、色材として広く利用されている有機顔料には、特に液晶ディスプレー用カラーフィルターやインクジェット用インク等の用途において、鮮明性や透明性などをより向上させることが求められている。鮮明性や透明性を向上させるためには、有機顔料を微細に分散させることが効果的なことが知られている。有機顔料を微細分散させるには、それに適した分散剤や分散機械を用いることが必要であるが、さらには有機顔料自身が微細(微粒子)であるということも大前提として必要である。有機顔料が粗大な粒子であると、分散剤や分散方法を改良しても優れた微細分散体を得ることは困難である。したがって、顔料の製造にあっては、純度や収率等の通常の要求項目に加えて微粒子として製造することが求められている。
【0004】
有機顔料微粒子の製造方法としては、例えばアゾ顔料のように、合成時に適切な反応条件を選択することにより、微細で整粒された粒子を得ることができるものがある。その他、銅フタロシアニングリーン顔料のように、合成時に生成する極めて微細で凝集した粒子を、後工程で粒子成長、整粒させることにより顔料化するもの、銅フタロシアニンブルー顔料のように、合成時に生成する粗大で不揃いな粒子を後工程で微細化し、整粒させることにより顔料化を行うものもある。例えば、ジケトピロロピロール顔料は、一般的には、琥珀酸ジエステルと芳香族ニトリルとを有機溶媒中で反応させることにより粗製顔料として合成される(例えば、特許文献1参照)。そして、粗製ジケトピロロピロール顔料は、水又は有機溶剤中で熱処理し、次に湿式摩砕のごとき粉末化を行うことにより、使用に適する形態にされる(例えば、特許文献2参照)。
【0005】
さらに、有機顔料には多形性を示すものがあり、このような顔料は、同一の化学組成を有するにもかかわらず2つ以上の結晶形態をとりうることが知られている。
例えば、C.I.ピグメントレッド254は、α型とβ型の結晶形態が知られている(例えば、特許文献3参照)。また、アゾ顔料であるC.I.ピグメントイエロー181は、数種の結晶形態が知られている(例えば、特許文献4参照)。
上記のように、特定の結晶構造を持つことを特徴とする知見がある一方、非晶質であることに特徴を持たせた顔料も知られており、C.I.ピグメントイエロー181を非晶質とすることで分散性を発揮させるといった例もある(特許文献5)。
【0006】
微細な有機顔料を得る方法として、特許文献6には顔料をアミド系の有機溶媒に溶解した溶液を貧溶媒に注入してナノサイズの微粒子を得る方法が記載されており、貧溶媒種により得られる顔料の結晶形態が異なる旨の記述がある。しかしながら、この方法では顔料溶液の濃度が10mM程度と低く、顔料を得るのに大量の溶媒を使用するため経済的ではない。
【0007】
特許文献7には、特定のモノアゾ顔料を高純度で製造できる方法が記載されている。しかしながら、より不安定な複素環ジアゾニウム化合物にこの方法を適用して高収率及び高純度でアゾ顔料を得ることは一般的に困難である。
【0008】
特許文献8には、ピラゾールのジアゾニウム化合物を用いたアゾ化合物を、高収率及び高純度で得る製造方法が記載されている。この方法は染料の製造には有効であるが、この方法で製造される化合物の粒子の大きさについては記載がない。
【0009】
特許文献9にも、ピラゾールのジアゾニウム化合物を用いたアゾ化合物を、高収率及び高純度で得る製造方法が記載されている。この方法により均一な微粒子が得られる旨の記載があるが、分散性に更なる改良の余地を残していた。なお、顔料の結晶形態に関する記述はない。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】特開昭58−210084号公報
【特許文献2】特開平5−222314号公報
【特許文献3】特開平8−48908号公報
【特許文献4】米国特許出願公開第2008/0058531号明細書
【特許文献5】特開2003−261792号公報
【特許文献6】特開2004−91560号公報
【特許文献7】特開2008−63524号公報
【特許文献8】特開2007−217681号公報
【特許文献9】特開2011−74375号公報
【非特許文献】
【0011】
【非特許文献1】新実験化学講座(丸善株式会社)14−III巻、1516〜1534頁
【発明の概要】
【発明が解決しようとする課題】
【0012】
本発明は特定の置換基を有するピラゾール環をアゾ基及びトリアジン環を介して連結したアゾ顔料の製造方法に関するが、上記のように、アゾ顔料の性能と製造性の両立には検討の余地が残っていた。
本発明は、分散性が良好なアゾ顔料微粒子を高効率かつ低コストで製造することのできるアゾ顔料の製造方法を提供することを目的とする。
【課題を解決するための手段】
【0013】
本発明者等は前記した実情に鑑みて鋭意検討した結果、非晶質な化合物を結晶変換することで分散性が良好なアゾ顔料微粒子を得ることができ、さらにはジアゾニウム塩とカップリング成分とをアゾカップリング反応させたときに、反応液中においてアゾ化合物が完全に析出していない状態(アゾ化合物の一部が反応液に溶解している状態)とし、この反応液をアゾ化合物の貧溶媒に注入し、非晶質な化合物を得ることで、上記課題が達成できることを見出し、本発明を完成した。
【0014】
即ち、本発明は、以下のとおりである。
【0015】
〔1〕
非晶質な下記式(1)で表されるアゾ化合物、その塩、水和物又は溶媒和物を結晶変換することを特徴とする下記式(1)で表されるアゾ顔料の製造方法。
【0016】
【化1】
【0017】
〔2〕
非晶質の前記式(1)で表されるアゾ化合物、その塩、水和物又は溶媒和物を溶媒中で加熱攪拌することにより結晶変換することを特徴とする〔1〕に記載のアゾ顔料の製造方法。
〔3〕
非晶質な前記式(1)で表されるアゾ化合物を、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が4.8°、7.2°及び9.7°に特徴的X線回折ピークを有する結晶形に結晶変換することを特徴とする〔1〕又は〔2〕に記載のアゾ顔料の製造方法。
〔4〕
非晶質な前記式(1)で表されるアゾ化合物を、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が4.8°、7.2°、9.7°、20.1°、及び26.8°に特徴的X線回折ピークを有する結晶形に結晶変換することを特徴とする〔1〕〜〔3〕のいずれか一項に記載のアゾ顔料の製造方法。
〔5〕
非晶質な前記式(1)で表されるアゾ化合物を、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的X線回折ピークを有する結晶形に結晶変換することを特徴とする〔1〕〜〔4〕のいずれか一項に記載のアゾ顔料の製造方法。
〔6〕
(i)非晶質な前記式(1)で表されるアゾ化合物を、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が4.8°、7.2°及び9.7°に特徴的X線回折ピークを有する結晶形に結晶変換する工程、及び、(ii)前記工程(i)で得たアゾ顔料と、水溶性無機塩と、水溶性有機溶剤とを含む混合物を混練して、窒素吸着法によるBET比表面積を50m2/g以上の結晶にする工程、を含むことを特徴とする、〔3〕〜〔5〕のいずれか一項に記載のアゾ顔料、又はその互変異性体の製造方法。
〔7〕
(a)ジアゾ化剤と下記式(2)で表されるアミノ化合物とを混合させる工程、(b)前記工程(a)で得た反応生成物と下記式(3)で表されるカップリング成分とを混合することにより反応を行い、該反応により生成した下記式(1)で表されるアゾ化合物の少なくとも一部が溶解した溶液を得る工程、(c)前記工程(b)で得た溶液と、前記アゾ化合物に対する貧溶媒とを混合して、下記式(1)で表される非晶質なアゾ化合物を晶析させる工程、を含むことを特徴とする〔1〕〜〔6〕のいずれか一項に記載のアゾ顔料の製造方法。
【0018】
【化2】
【0019】
【化3】
【0020】
【化4】
【0021】
〔8〕
前記工程(b)において、得られた溶液が酸性溶液であることを特徴とする〔7〕に記載のアゾ顔料の製造方法。
〔9〕
前記酸性溶液が、酢酸及び硫酸の少なくとも一方を含むことを特徴とする〔8〕に記載のアゾ顔料の製造方法。
〔10〕
前記工程(b)において、得られた溶液が、カップリング反応により生成した前記式(1)で表されるアゾ化合物が完全に溶解した溶液であることを特徴とする〔7〕〜〔9〕のいずれか一項に記載のアゾ顔料の製造方法。
〔11〕
前記(c)工程において、前記工程(b)で得られた前記式(1)で表されるアゾ化合物の溶解度が1g/L以下である貧溶媒と混合することを特徴とする〔7〕〜〔10〕のいずれか一項に記載のアゾ顔料の製造方法。
〔12〕
前記貧溶媒が、水及び、炭素数1〜3のアルコール、炭素数1〜6のグリコールからなる群から選ばれる1種以上の溶媒を含むことを特徴とする〔7〕〜〔11〕のいずれか一項に記載のアゾ顔料の製造方法。
〔13〕
〔1〕〜〔12〕のいずれか一項に記載の製造方法により得られるアゾ顔料。
〔14〕
〔13〕に記載のアゾ顔料、分散剤、及び水を含む顔料分散物。
〔15〕
分散剤が水溶性高分子であることを特徴とする〔14〕に記載の水系顔料分散物。
〔16〕
前記水溶性高分子分散剤が少なくとも1つのカルボキシ基を有し、少なくとも50mgKOH/g以上の酸価を有することを特徴とする〔15〕に記載の水系顔料分散物。
〔17〕
前記水系顔料分散物が、架橋剤により架橋されていることを特徴とする〔15〕又は〔16〕に記載の水系顔料分散物。
【発明の効果】
【0022】
本発明によれば、易分散なアゾ顔料微粒子を、高効率(純度および生産性)かつ低コストで製造することのできるアゾ顔料の製造方法を提供することができる。
【図面の簡単な説明】
【0023】
【図1】実施例1に従って合成されたδ型結晶形態のアゾ顔料(1)−2のX線回折の図である。
【図2】実施例2に従って合成されたδ型結晶形態のアゾ顔料(1)−4のX線回折の図である。
【図3】実施例3に従って合成された中間生成物であるζ型結晶形態のアゾ顔料(1)−5のX線回折の図である。
【図4】実施例3に従って合成されたδ型結晶形態のアゾ顔料(1)−7のX線回折の図である。
【図5】実施例4に従って合成されたδ型結晶形態のアゾ顔料(1)−9のX線回折の図である。
【図6】実施例5に従って合成されたδ型結晶形態のアゾ顔料(1)−11のX線回折の図である。
【図7】実施例6に従って合成されたδ型結晶形態のアゾ顔料(1)−13のX線回折の図である。
【図8】実施例7に従って合成されたδ型結晶形態のアゾ顔料(1)−14のX線回折の図である。
【図9】実施例8に従って合成されたδ型結晶形態のアゾ顔料(1)−15のX線回折の図である。
【図10】実施例9に従って合成されたδ型結晶形態のアゾ顔料(1)−16のX線回折の図である。
【図11】実施例10に従って合成されたδ型結晶形態のアゾ顔料(1)−17のX線回折の図である。
【図12】実施例11に従って合成されたδ型結晶形態のアゾ顔料(1)−18のX線回折の図である。
【図13】実施例11−2に従って合成されたζ型結晶形態のアゾ顔料(1)−101のX線回折の図である。
【図14】実施例12に従って合成された中間生成物である非晶質なアゾ顔料(1)−19のX線回折の図である。
【図15】実施例12に従って合成されたδ型結晶形態のアゾ顔料(1)−20のX線回折の図である。
【図16】実施例12−2に従って合成されたδ型結晶形態のアゾ顔料(1)−103のX線回折の図である。
【図17】実施例13に従って合成されたδ型結晶形態のアゾ顔料(1)−21のX線回折の図である。
【図18】実施例14に従って合成されたδ型結晶形態のアゾ顔料(1)−22のX線回折の図である。
【図19】実施例15に従って合成されたδ型結晶形態のアゾ顔料(1)−23のX線回折の図である。
【図20】実施例16に従って合成されたδ型結晶形態のアゾ顔料(1)−24のX線回折の図である。
【図21】実施例17に従って合成されたδ型結晶形態のアゾ顔料(1)−25のX線回折の図である。
【図22】実施例18に従って合成されたδ型結晶形態のアゾ顔料(1)−27のX線回折の図である。
【図23】実施例18−2に従って合成されたδ型結晶形態のアゾ顔料(1)−104のX線回折の図である。
【図24】比較例1に従って合成されたδ型結晶形態のアゾ顔料(1)−26のX線回折の図である。
【図25】実施例20に従って合成されたδ型結晶形態のアゾ顔料(1)−2−AのX線回折の図である。
【図26】比較例3に従って合成されたδ型結晶形態のアゾ顔料(1)−32−AのX線回折の図である。
【図27】比較例4に従って合成されたδ型結晶形態のアゾ顔料(1)−29−AのX線回折の図である。
【図28】比較例5に従って合成されたδ型結晶形態のアゾ顔料(1)−31−AのX線回折の図である。
【図29】比較例6に従って合成されたζ型結晶形態のアゾ顔料(1)−30−AのX線回折の図である。
【0024】
本発明の製造方法により製造された顔料の分散性が良好となる理由に関して、以下に推察を述べる。X線回折において、特徴的X線回折ピークを示す結晶は、分子が一定の規則性を持って配列し、分子間の相互作用が強固に結ばれている。その結果、溶媒に対する溶解性が低くなる。そのため例えば溶媒中で、このような結晶から別の結晶形に変換すると、少しずつ溶解しつつ結晶変換が進行するため、核となる結晶が少なく、結晶は大きく成長してしまう。一方、本発明のようにX線回折において、特徴的なX線回折ピークを示さない非晶質な固体は、分子間の相互作用が弱く、溶媒に対する溶解性が高い。そのため、例えば溶媒中で結晶変換を行うと、非晶質は固体からは多くが溶け出すが、結晶は溶解性が低いため、即座に過飽和状態になり、核となる結晶が多数析出し、結晶は成長しにくい。結果、微細な顔料が得られ、分散性が良好となると考えている。
【発明を実施するための形態】
【0025】
以下、本発明について詳細に説明する。
【0026】
本発明において非晶質とは、結晶のような長距離秩序は無いが、短距離秩序はある物質の状態を示し、X線回折において、特徴的X線回折ピークを示さないことを表す。
【0027】
本明細書においては以下、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が4.8°、7.2°及び9.7°に特徴的X線回折ピークを有する下記式(1)で表されるアゾ顔料をδ型結晶形態アゾ顔料と称する。
また、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が6.5°、7.1°及び21.8°に特徴的X線回折ピークを有する式(1)で表されるアゾ顔料をα型結晶形態アゾ顔料、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が6.3°、6.4°及び22.3°に特徴的X線回折ピークを有する式(1)で表されるアゾ顔料をβ型結晶形態アゾ顔料、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が4.9°、8.8°及び13.1°に特徴的X線回折ピークを有する式(1)で表されるアゾ顔料をε型結晶形態アゾ顔料、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が6.6°、9.2°及び21.5°に特徴的X線回折ピークを有する式(1)で表されるアゾ顔料をζ型結晶形態アゾ顔料と称する。
【0028】
【化5】
【0029】
本発明において、上記式(1)で表されるアゾ化合物、及びアゾ顔料のX線回折の測定は、日本工業規格JISK0131(X線回析分析通則)に準じて、例えば、粉末X線回折測定装置RINT2500(株式会社リガク製)にて行うことができる。
【0030】
図2にX線回折図を示して、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が4.8°、7.2°及び9.7°に特徴的X線回折ピークを有する式(1)で表されるアゾ顔料について更に詳細に説明する。
【0031】
〔アゾ顔料〕
本発明のアゾ顔料の製造方法によって製造されるアゾ顔料は下記式(1)で表される。
【0032】
以下、本発明における式(1)で表されるアゾ化合物及びアゾ顔料について詳細に説明する。なお当該アゾ顔料は、その塩、水和物又は溶媒和物であってもよい。
【0033】
【化6】
【0034】
式(1)で表される顔料において、結晶中に水分子を含む水和物、あるいは、溶媒(例えば、メタノール,エタノール,2−プロパノール,t−ブチルアルコール等のアルコール類や、アセトン、メチルエチルケトン等のケトン類や、アセトニトリル、ジメチルスルホキシド,ジメチルホルムアミド,ジメチルアセトアミド,N−メチルピロリドン、トルエン等の非プロトン性溶媒など)を含む溶媒和物であっても良い。
【0035】
また、式(1)で示される顔料に関しては、その互変異性体(例えば、アゾ−ヒドラゾンの互変異性体や式(4)で表されるような幾何異性体)も、本発明においては、これらの一般式に含まれるものとする。
式(4):
【0036】
【化7】
【0037】
〔アゾ顔料の製造方法〕
以下に、本発明の製造方法に関して詳細に説明する。本発明のアゾ顔料の製造方法は、
非晶質な下記式(1)で表されるアゾ化合物、その塩、水和物又は溶媒和物を結晶変換する下記式(1)で表されるアゾ顔料の製造方法である。
【0038】
【化8】
【0039】
本発明における結晶変換とは、結晶形態を変換することを表し、非晶質である上記式(1)で表されるアゾ化合物、その塩、水和物又は溶媒和物を、特定の結晶形態を有するアゾ顔料に変換することを言う。結晶変換の方法としては、ソルベントソルトミリング、ソルトミリング、ドライミリング、ソルベントミリング、アシッドペースティング等の磨砕処理、溶媒加熱処理が挙げられ、好ましくは溶媒加熱処理である。
【0040】
(溶媒加熱処理)
本発明において溶媒加熱処理とは、具体的には、非晶質な式(1)で表されるアゾ化合物、その塩、水和物又は溶媒和物を溶媒中で加熱撹拌することをいう。
溶媒加熱処理によって、効率よく結晶変換をすることができる。例えば、非晶質なアゾ化合物の溶媒和物を加熱攪拌することでδ型結晶形態のアゾ顔料を得ることができる。
【0041】
本発明において、式(1)で表されるアゾ化合物をCuKα特性X線回折におけるブラッグ角(2θ±0.2°)が4.8°、7.2°及び9.7°に特徴的なX線回折ピークを有する結晶形態に結晶変換することが好ましい。
上記のような特徴的X線回折ピークを有するδ型結晶形態アゾ顔料とすることにより分散性が向上する、すなわち、短時間で目標の粒子径まで分散できるようになる。
CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が4.8°、7.2°及び9.7°に特徴的なX線回折ピークを有する結晶形態は、さらに4.8°、7.2°、9.7°、20.1°、及び26.8°に特徴的なX線回折ピークを有する結晶形態がより好ましい。その中でも特に、4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを有する結晶形態が最も好ましい。
【0042】
本発明の結晶変換に用いることのできる溶媒としては、水、有機酸、無機酸、有機溶媒を用いることができるが、水および有機溶媒が好ましい。また、結晶変換をする際に結晶成長を抑制させる点から、結晶変換した後の式(1)で表されるアゾ顔料の溶解性が低い溶媒が好ましい。より好ましい溶媒としては、水、メタノール、エタノール、イソプロパノール、イソブタノール、エチレングリコール、ジエチレングリコール、ジエチレングリコールジエチルエーテル、ジエチレングリコールモノメチルエーテル、ジプロピレングリコール、酢酸、プロピオン酸、硫酸、またはそれらの混合溶媒が挙げられ、エチレングリコール、水、酢酸、硫酸、あるいはそれらの混合溶媒である場合が更に好ましく、エチレングリコールであることが最も好ましい。
【0043】
溶媒加熱処理に使用する溶媒の量は、式(1)で表されるアゾ顔料に対して1〜100倍であることが好ましく、5〜50倍であることがさらに好ましく、8〜30倍であることがより好ましい。1倍以上であれば、攪拌性を確保できるため好ましい。また、100倍以下であれば、生産性が高くなり、経済的なため好ましい。
【0044】
溶媒加熱処理における加熱攪拌の温度は、所望する顔料の一次粒子径の大きさによって異なるが、15〜150℃が好ましく、20〜120℃であることがより好ましく、20〜100℃がさらに好ましい。15℃以上であれば、結晶変換が起こるために長時間を要することなく、効率的である。一方、150℃以下であれば、アゾ顔料(1)の一部が分解するのを抑制できるため好ましい。
【0045】
結晶変換のための撹拌時間は特に制限はないが、5〜1500分が好ましく、10〜600分が更に好ましく、30〜300分がより好ましい。5分以上であれば、部分的に非晶質な箇所が残存するのを抑制できるため好ましい。一方、1500分以下であれば、効率的であり好ましい。
【0046】
以下に、結晶変換工程に供する非晶質な下記式(1)で表されるアゾ化合物の製造方法に関して詳細に説明する。
本発明の結晶変換に用いる式(1)で表されるアゾ化合物の製造方法は、(a)ジアゾ化剤と式(2)で表されるアミノ化合物とを混合させる工程、(b)前記工程(a)で得た反応生成物と式(3)で表されるカップリング成分とを混合することにより反応を行い、該反応により生成した下記式(1)で表されるアゾ化合物の少なくとも一部が溶解した溶液を得る工程、(c)前記工程(b)で得た溶液と、前記アゾ化合物に対する貧溶媒とを混合して、下記一般式(1)で表される非晶質なアゾ化合物を晶析させる工程、を含むことが好ましい。
【0047】
【化9】
【0048】
【化10】
【0049】
【化11】
【0050】
本発明に係わる工程(a)について詳細を説明する。
工程(a)では、ジアゾ化剤とアミノ化合物とを混合させることで、アミノ化合物とジアゾ化剤との反応によりジアゾニウム化合物が誘導される。この反応は酸を含む媒質中で行うことが好ましい。本明細書では、このジアゾニウム化合物を含む液を「ジアゾニウム化合物調製液」と呼ぶ。アミノ化合物と酸とジアゾ化剤の混合の方法に特に限定はないが、アミノ化合物と酸の溶液中にジアゾ化剤を添加することが好ましい。工程(a)におけるジアゾ化剤とは、アミノ化合物をジアゾニウム化合物に誘導するために使用されるものであり、そのような作用を持つものであれば限定はされない。ジアゾ化剤として代表的なものには、亜硝酸エステル類(例えば亜硝酸イソペンチルが挙げられる)、亜硝酸塩(例えば亜硝酸ナトリウムや亜硝酸カリウムが挙げられる)、亜硝酸イソアミル、ニトロシル硫酸が挙げられ、更に好ましくは亜硝酸ナトリウム、亜硝酸カリウム、ニトロシル硫酸であり、その中でも、ジアゾニウム化合物を安定かつ効率的に調製できる観点から、ニトロシル硫酸が特に好ましい。
【0051】
工程(a)で使用する酸とは、式(2)で表されるアミノ化合物を完溶させないまでも、わずかでも溶解できる酸を意味し、好ましくはアミノ化合物を完溶させる酸である。酸には無機酸及び有機酸が使用でき、無機酸としては塩酸、リン酸、硫酸が挙げられ、好ましくはリン酸、硫酸であり、更に好ましくは硫酸である。有機酸には蟻酸、酢酸、プロピオン酸、メタンスルホン酸が挙げられ、好ましくは酢酸、プロピオン酸、メタンスルホン酸であり、更に好ましくは酢酸、プロピオン酸である。また、これらの酸は単独で用いても良いし、混合して用いても良い。混合酸としては、リン酸/酢酸、硫酸/酢酸、メタンスルホン酸/酢酸、酢酸/プロピオン酸が挙げられ、好ましくは、リン酸/酢酸、硫酸/酢酸、硫酸/酢酸/プロピオン酸、酢酸/プロピオン酸であり、その中でも、硫酸/酢酸、酢酸/プロピオン酸が特に好ましい。これら混合酸の質量比は1/(0.1〜20)が好ましく、1/(0.5〜10)がより好ましく、更に好ましくは1/(1〜10)である。
【0052】
工程(a)における、アミノ化合物に対する酸の添加量は質量比で1〜100倍であり、より好ましくは2〜50倍であり、3〜25倍が更に好ましい。質量比が1倍以上であると、攪拌性が良化し、より確実にジアゾニウム化合物を誘導できる。一方、質量比が100倍以下になると生産性が向上に経済的となる。
また、工程(a)における、アミノ化合物に対するジアゾ化剤の添加量は、モル比で1.0〜20倍であり、より好ましくは1.0〜10倍であり、1.0〜5倍が更に好ましい。ジアゾ化剤がアミノ化合物に対してモル比で1倍以上であることにより、ジアゾニウム化合物をより確実に誘導でき、20倍以下であることにより、副反応によりジアゾニウム化合物が分解することを抑制できる。
【0053】
工程(a)のジアゾ化剤とアミノ化合物の混合では、50℃以下で実施されることが好ましく、40℃以下で実施されることがより好ましく、更に好ましくは30℃以下で実施することが望ましい。50℃以上におけるジアゾ液の調製ではジアゾ化剤の分解が懸念される。ジアゾニウム化合物へ誘導する攪拌時間は0.3〜10時間が好ましく、0.5〜5時間がより好ましく、更に好ましくは1〜3時間である。上記攪拌時間が0.3時間以上であることにより、ジアゾニウム化合物に完全に誘導しやすく、10時間以下であることにより、ジアゾニウム化合物の分解が生じにくい。また、混合には通常の攪拌機が用いられ、特に限定はない。製造設備に依存することはあるが、好ましい攪拌の回転数は、30〜300rpmが好ましく、40〜200rpmがより好ましく、更に好ましくは50〜200rpmである。攪拌速度が回転数で30rpm以上であることにより、ジアゾニウム化合物調製液の攪拌効率が良好となるため、所望の反応の進行を確実に実施できる。
【0054】
工程(a)で混合することのできる溶媒は、誘導されるジアゾニウム化合物が分解を受けなければ特に限定はない。混合可能な溶媒として例えば、ヘキサン、ベンゼン、トルエン等の炭化水素系溶媒、ジエチルエーテル、テトラヒドロフラン等のエーテル系溶媒、アセトン、メチルエチルケトン等のケトン系溶媒、ジメチルホルムアミド、ジメチルアセトアミド、ピロリドン、N−メチル−2−ピロリドン等のアミド系溶媒、他ジメチルスルホキシド、スルホラン、アセトニトリル、水が挙げられる。
【0055】
工程(a)におけるジアゾニウム化合物調製液の好ましいpHは、7以下が好ましく、5以下がより好ましく、3以下が更に好ましい。工程(a)におけるジアゾニウム化合物調製液のpHが7超過になると、誘導されるジアゾニウム化合物の分解が懸念される。
【0056】
次に、本発明に係わる工程(b)について詳細を説明する。
工程(b)は、前記工程(a)で得た反応生成物とカップリング成分とを混合することにより反応を行い、該反応により生成した式(1)で表されるアゾ化合物の少なくとも一部が溶解した溶液を得る工程である。
本明細書では、このアゾ化合物の少なくとも一部が溶解した溶液を「アゾ化合物溶解液」と呼ぶ。
【0057】
アゾ化合物溶解液の調製方法としては、(i)工程(a)で得た反応生成物とカップリング成分とを混合することによりカップリング反応を行い、反応の結果、析出した式(1)で表されるアゾ顔料を、溶剤に溶解させて得る方法、及び、(ii)上記カップリング反応によって得られる式(1)で表される化合物の少なくとも一部が反応液に溶解するように、該カップリング反応を実施し、その反応液を、そのまま、アゾ化合物溶解液とする方法、又は、このようにして得られたアゾ化合物溶解液を下記に詳述する工程(c)に適用することにより得られた(晶析された)アゾ顔料を、更に溶剤に溶解させて得る方法、が挙げられる。
【0058】
上記形態(i)及び(ii)のいずれにおいても、工程(a)で得たジアゾニウム化合物調製液とカップリング成分との混合の方法に特に制限はないが、該カップリング成分を溶媒に一部または全部溶解させて添加すること、あるいは溶媒を用いずに固体で添加することが好ましく、工程(a)で得たジアゾニウム化合物調製液の中に、カップリング成分の溶液を添加すること、あるいは工程(a)で得たジアゾニウム化合物調製液の中に、カップリング成分を固体で添加することが更に好ましい。
【0059】
また、工程(b)におけるカップリング成分に対する前記工程(a)で得たジアゾニウム化合物調製液中のジアゾニウム化合物の量は、カップリング成分のカップリング位に対し0.8〜3当量が好ましく、より好ましくはカップリング位に対し0.9〜2当量であり、更に好ましくはカップリング位に対し0.95〜1.5当量である。0.8当量以上であることにより、未反応のカップリング位をもつカップリング成分の残存を抑制でき、また、3当量以下であることにより、未反応のジアゾニウム化合物の残存を抑制できるため、より経済的である。
【0060】
なお、上記形態(ii)においては、工程(b)において一般式(1)で表されるアゾ化合物の少なくとも一部が溶解しているため、カップリング反応がよりスムーズに進行してより高純度のアゾ化合物を製造することができる。この理由は以下のように推測される。前記式(3)はカップリング位が2個以上あるため、例えば1個のカップリング位のみが反応した反応中間体を経由する。この反応中間体が反応系で析出してしまうと、2個目以降のカップリング反応の反応速度が遅くなる。一方、ジアゾニウム化合物は不安定であるため、長時間経過すると分解が起こる懸念がある。したがって、カップリング反応は早く進行させてやることが重要であり、工程(b)において析出物を生成させない上記形態(ii)の製造方法は、結果として、高純度の顔料を製造するのに、より好適である。
【0061】
工程(b)においては溶媒を使用せずにカップリング成分を添加しても良いが、溶媒と混合して添加しても良いが、溶媒を使用せずに添加することが好ましい。工程(b)においてカップリング成分に溶媒を使用する場合、特に限定はされないが、上記形態(ii)となるように、すなわち、反応後に生成した前記一般式(1)で表されるアゾ化合物の少なくとも一部が溶解した溶液が得られるような溶媒であることが好ましい。
【0062】
上記形態(i)の場合、すなわち、顔料を析出させる場合、溶媒の例としては、水、有機酸、無機酸、有機溶媒を用いることができるが、特に水、有機溶媒が好ましく、反応時に液体分離現象を起こさず、溶媒と均一な溶液を呈する溶媒が好ましい。例えば、水、メタノール、エタノール、プロパノール、イソプロパノール、ブタノール、t−ブチルアルコール、アミルアルコール等のアルコール性有機溶媒、アセトン、メチルエチルケトン等のケトン系有機溶媒、エチレングリコール、ジエチレングリコール、トリエチレングリコール、プロピレングリコール、ジプロピレングリコール、1,3−プロパンジオール等のジオール系有機溶媒、エチレングリコールモノメチルエーテル、エチレングリコールモノエチルエーテル、エチレングリコールジエチルエーテル等のエーテル系有機溶媒、テトラヒドロフラン、ジオキサン、アセトニトリル等が挙げられる、これらの溶媒は2種類以上の混合液であってもよい。
【0063】
好ましくは、極性パラメータ(ET)の値が40以上の有機溶媒である。なかでも溶媒分子中に水酸基を2個以上有するグリコール系の溶媒、あるいは炭素原子数が3個以下のアルコール系溶媒、総炭素数5以下のケトン系溶媒、好ましくは炭素原子数が2以下のアルコール溶媒(例えば、メタノール、エチレングリコール)、総炭素数4以下のケトン系溶媒(例えばアセトン、メチルエチルケトン)が好ましい。またこれらの混合溶媒も含まれる。
【0064】
また、上記形態(ii)の場合、すなわち、式(1)で表される化合物の少なくとも一部が反応液に溶解するようにカップリング反応を行う場合、溶媒の例としては、水、メタノール、イソプロパノール、エチレングリコール等のアルコール系溶媒、アセトン、メチルエチルケトン等のケトン系溶媒、酢酸、プロピオン酸、メタンスルホン酸等の有機酸溶媒、硫酸、塩酸、リン酸等の無機酸溶媒、ジメチルホルムアミド、ジメチルアセトアミド、ピロリドン、N−メチル−2−ピロリドン等のアミド系溶媒、他ジメチルスルホキシド、スルホラン、アセトニトリル、が挙げられる。中でも、好ましくは、アセトン、メチルエチルケトン等のケトン系溶媒、酢酸、プロピオン酸、メタンスルホン酸等の有機酸溶媒、硫酸、塩酸、リン酸等の無機酸溶媒であり、更に好ましくは、有機酸又は無機酸の酸性溶媒であり、最も好ましくは、酢酸、メタンスルホン酸、リン酸、硫酸である。また、上記で示した溶媒の混合溶媒も好適である。
特に上記形態(ii)の場合、工程(b)においては、カップリング成分を酸性溶媒に溶解又は懸濁させた酸性溶液と、工程(a)で得た反応生成物とを混合する、あるいはカップリング成分を、溶媒を使用せずに、工程(a)で得た反応生成物に添加するのが好ましい。とりわけ、酸性溶媒は、酢酸及び硫酸の少なくとも一方を含む溶媒であることが好ましい。
【0065】
上記形態(i)及び(ii)のいずれにおいても、カップリング成分に対する好ましい溶媒の添加量は、質量比で0.5〜200倍が好ましく、1〜100倍がより好ましく、1〜50倍が更に好ましい。カップリング成分に対する好ましい溶媒量の添加量として、質量比で0.5倍未満ではカップリング成分と溶媒の製造機における攪拌が困難になり、所望の反応が進行しない。また、200倍超過では不経済となる。
【0066】
アゾ化合物溶解液の調製方法が、上記形態(i)である場合、あるいは、上記形態(ii)であって、かつ、式(1)で表される化合物の少なくとも一部が溶解されたカップリング反応液を工程(c)に適用して得られるアゾ顔料を、更に溶剤に溶解させてアゾ化合物溶解液を調製する場合において、得られたアゾ顔料を溶解するための溶剤としては、アゾ顔料の少なくとも一部を溶解できれば特に限定されないが、上記形態(ii)において好ましいとして前掲した溶媒の例を同様に挙げることができる。
【0067】
上記形態(i)、(ii)のいずれの工程をとるにせよ、工程(b)において最終的に得られるアゾ化合物溶解液としては、酸性溶液であることが好ましく、とりわけ、酢酸及び硫酸の少なくとも一方を含む溶液であることが好ましい。
【0068】
工程(b)において得られるアゾ化合物溶解液としては、工程(b)によって生成したアゾ化合物の全量(アゾ化合物溶解液に溶解している式(1)で表されるアゾ化合物と、アゾ化合物溶解液から析出した式(1)で表されるアゾ顔料との総和)に対する、アゾ化合物溶解液に溶解している式(1)で表されるアゾ化合物の割合が50質量%以上であることが好ましく、75質量%以上であることが好ましく、90質量%以上であることが好ましく、100質量%(工程(b)によって生成したアゾ化合物が反応液に完全に溶解している状態)であることが最も好ましく、これにより、顔料の粒子径をより低下できる傾向となる。
【0069】
工程(b)における工程(a)のジアゾニウム化合物調製液とカップリング成分の混合温度は50℃以下で実施されることが好ましく、30℃以下で実施されることがより好ましく、更に好ましくは25℃以下で実施することが望ましい。50℃超過では工程(a)で誘導されたジアゾニウム化合物、並びに生成した式(1)で表されるアゾ化合物の分解が懸念される。また、混合には通常の攪拌機が用いられ、特に限定はない。製造設備に依存することはあるが、好ましい攪拌の回転数は、30〜300rpmが好ましく、40〜200rpmがより好ましく、更に好ましくは50〜200rpmである。攪拌速度が回転数で30rpm未満となると混合液の攪拌効率が悪くなり所望の反応の進行が懸念される。工程(b)における攪拌時間は0.1〜10時間が好ましく、0.3〜5時間がより好ましく、更に好ましくは0.3〜3時間である。0.1時間未満では完全に顔料へ誘導することが難しく、10時間超過では式(1)で表されるアゾ化合物の分解が懸念される。
【0070】
次に本発明に係わる工程(c)について詳細を説明する。
工程(c)は、前記工程(b)で得たアゾ化合物溶解液を、該アゾ化合物の溶解性が低い貧溶媒と混合して、顔料を晶析させる工程である。工程(b)で得たアゾ化合物溶解液と貧溶媒との混合の方法に特に制限はないが、工程(b)で得たアゾ化合物溶解液を貧溶媒の中に添加することが好ましく、その際に貧溶媒が攪拌された状態であることが好ましい。
攪拌速度は100〜10000rpmとすることが好ましく、150〜8000rpmとすることがより好ましく、200〜6000rpmとすることが特に好ましい。添加にはポンプ等を用いることもできる。このとき、液中添加でも液外添加でもよいが、液中添加がより好ましい。更に供給管を介してポンプで液中に連続供給することが好ましい。
【0071】
貧溶媒は特に限定されないが、アゾ化合物の溶解度が1g/L以下であることが好ましく、0.1g/L以下であることがより好ましい。この溶解度は酸又はアルカリの存在下で溶解された場合の溶解度であってもよい。工程(b)で得たアゾ化合物溶解液と貧溶媒の相溶性若しくは均一混合性は、該アゾ化合物の良溶媒の貧溶媒に対する溶解量が30質量%以上であることが好ましく、50質量%以上であることがより好ましい。本明細書において、溶解度は25℃における溶解度を指す。
【0072】
貧溶媒としては、例えば、水、塩酸、アンモニア水、水酸化ナトリウム水溶液等の水性溶媒、メタノール、エタノール、イソプロピルアルコール、1−メトキシ−2−プロパノール等のアルコール系溶媒、エチレングリコール、ジエチレングリコール等のグリコール系溶媒、アセトン、メチルエチルケトン、メチルイソブチルケトン、シクロヘキサノン等のケトン化合物溶媒、ジエチルエーテル、テトラヒドロフラン等のエーテル系溶媒、ヘキサン、ベンゼン、トルエン等の炭化水素系溶媒、アセトニトリル等のニトリル系溶媒、ジクロロメタン、トリクロロエチレン等のハロゲン系溶媒、酢酸エチル、乳酸エチル、2−(1−メトキシ)プロピルアセテート等のエステル系溶媒等が挙げられ、好ましくは、水、塩酸、アンモニア水、水酸化ナトリウム水溶液等の水性溶媒、メタノール、エタノール、イソプロピルアルコール、1−メトキシ−2−プロパノール等のアルコール系溶媒、エチレングリコール、ジエチレングリコール等のグリコール系溶媒、アセトン、メチルエチルケトン、メチルイソブチルケトン、シクロヘキサノン等のケトン化合物溶媒であり、さらに好ましくは、水、アンモニア水等の水性溶媒、炭素数1〜3のアルコール溶媒、炭素数1〜6のグリコール系溶媒、である。また、上記で示した溶媒の混合溶媒も好適である。最も好ましくは、水及び炭素数1〜3のアルコール、炭素数1〜6のグリコールからなる群から選択される1種以上の溶媒である。
【0073】
工程(b)で得たアゾ化合物溶解液と貧溶媒との混合比は体積比で1/50〜2/3が好ましく、1/40〜1/2がより好ましく、1/20〜1/2が特に好ましい。体積比で2/3以下であると、顔料の晶析が充分に起こって反応収率が上がり、体積比が1/50以上であると、生産性が向上して経済的となる。
【0074】
工程(b)で得たアゾ化合物溶解液と貧溶媒との混合温度には特に制限はないが、−10〜50℃で実施されることが好ましく、−5〜30℃で実施されることがより好ましく、10〜25℃で実施されることが最も好ましい。
【0075】
工程(b)で得たアゾ化合物溶解液と貧溶媒との混合にあたり、レイノルズ数を調節することにより、析出生成させる有機ナノ粒子の粒子径を制御することができる。ここでレイノルズ数は流体の流れの状態を表す無次元数であり次式で表される。
【0076】
数式(1):Re=ρUL/μ
【0077】
(数式(1)中、Reはレイノルズ数を表し、ρは工程(b)で得たアゾ化合物溶解液の密度[kg/m3]を表し、Uはアゾ化合物溶解液と貧溶媒とが出会うときの相対速度[m/s]を表し、Lはアゾ化合物溶解液と貧溶媒が出会う部分の流路若しくは供給口の等価直径[m]を表し、μはアゾ化合物溶解液の粘性係数[Pa・s]を表す。)
【0078】
等価直径Lとは、任意断面形状の配管の開口径や流路に対し等価な円管を想定するとき、その等価円管の直径をいう。等価直径Lは、配管の断面積をA、配管のぬれぶち長さ(周長)又は流路の外周をpとすると下記数式(2)で表される。
【0079】
数式(2):L=4A/p
【0080】
アゾ化合物溶解液と貧溶媒とが出会うときの相対速度Uは、両者が出会う部分の面に対して垂直方向の相対速度で定義される。すなわち、例えば静止している貧溶媒中にアゾ化合物溶解液を注入して混合する場合は、供給口から注入する速度が相対速度Uに等しくなる。相対速度Uの値は特に制限されないが、例えば、0.5〜100m/sとすることが好ましく、1.0〜50m/sとすることがより好ましい。
【0081】
アゾ化合物溶解液の密度ρは、選択される材料の種類により定められる値であるが、例えば、0.8〜2.0kg/m3であることが実際的である。また、アゾ化合物溶解液の粘性係数μについても用いられる材料や環境温度等により定められる値であるが、例えば、0.5〜100mPa・sであることが好ましく、1.0〜50.0mPa・sであることがより好ましい。
【0082】
レイノルズ数の値は、小さいほど層流を形成しやすく、大きいほど乱流を形成しやすい。例えば、レイノルズ数を60以上で調節して顔料ナノ粒子の粒子径を制御して得ることができ、100以上とすることが好ましく、150以上とすることがより好ましい。レイノルズ数に特に上限はないが、例えば、100000以下の範囲で調節して制御することで所望の平均粒子径を持つ顔料粒子を制御して得ることができる。このとき、上記の範囲内においては、通常レイノルズ数を高めることで、より粒径の小さな顔料粒子を制御して得ることができる。
【0083】
本発明の製造方法により得られる顔料粒子の1次粒子を、透過型顕微鏡で観察した際の長軸方向の長さは、1nm〜1μmであることが好ましく、5〜500nmであることがより好ましく、10〜200nmであることが更に好ましく、10〜100nmであることが特に好ましい。
【0084】
なお、顔料粒子の粒子径に関しては、計測法により数値化して集団の平均の大きさを表現する方法があるが、よく使用されるものとして、分布の最大値を示すモード径、積分分布曲線の中央値に相当するメジアン径、各種の平均径(数平均、長さ平均、面積平均、質量平均、体積平均等)などがあり、本発明においては、特に断りのない限り、平均粒子径とは数平均粒子径をいう。
顔料粒子の粒径の測定方法としては、顕微鏡法、質量法、光散乱法、光遮断法、電気抵抗法、音響法、動的光散乱法が挙げられ、顕微鏡法、動的光散乱法が特に好ましい。顕微鏡法に用いられる顕微鏡としては、例えば、走査型電子顕微鏡、透過型電子顕微鏡などが挙げられる。動的光散乱法による粒子測定装置として、例えば、日機装社製ナノトラックUPA−EX150、大塚電子社製ダイナミック光散乱光度計DLS−7000シリーズなどが挙げられる。
【0085】
上記の顔料粒子の好ましい平均粒子径は、(1)工程(c)における温度、(2)貧溶媒に対するアゾ化合物の溶解度、及び、(3)攪拌速度(あるいは、レイノルズ数)を、適宜、調整することによって、達成される。
【0086】
顔料微粒子を析出させ分散液を調製するにあたり、アゾ化合物溶解液及び貧溶媒の少なくとも一方に分散剤を含有させてもよい。このとき、アゾ化合物溶解液に分散剤を含有させることが好ましい。分散剤は(1)析出した顔料表面に素早く吸着して、微細なナノ粒子を形成し、かつ(2)これらの粒子が再び凝集することを防ぐ作用を有するものである。
分散剤としては、例えば、アニオン性、カチオン性、両イオン性、ノニオン性の低分子又は高分子分散剤を使用することができる。
【0087】
高分子分散剤としては、その質量平均分子量が1000〜500000であることが好ましく、10000〜500000であることがより好ましく、10000〜100000であることが特に好ましい。
具体的には、ポリビニルピロリドン、ポリビニルアルコール、ポリビニルメチルエーテル、ポリエチレングリコール、ポリプロピレングリコール、ポリアクリルアミド、ビニルアルコール−酢酸ビニル共重合体、ポリビニルアルコール−ブブンホルマール化物、ポリビニルアルコール−部分ブチラール化物、ビニルピロリドン−酢酸ビニル共重合体、ポリエチレンオキシド/プロピレンオキシドブロック共重合体、ポリアクリル酸塩、ポリビニル硫酸塩、ポリ(4−ビニルピリジン)塩、ポリアミド、ポリアリルアミン塩、縮合ナフタレンスルホン酸塩、セルロース誘導体、澱粉誘導体などが挙げられる。その他、アルギン酸塩、ゼラチン、アルブミン、カゼイン、アラビアゴム、トンガントゴム、リグニンスルホン酸塩などの天然高分子類も使用できる。なかでも、ポリビニルピロリドンが好ましい。これら高分子化合物は、1種単独であるいは2種以上を組み合わせて用いることができ、また、低分子量の分散剤を組み合わせて用いてもよい。顔料の分散に用いる分散剤に関しては、「顔料分散安定化と表面処理技術・評価」(化学情報協会、2001年12月発行)の29〜46頁に詳しく記載されている。
【0088】
アニオン性分散剤(アニオン性界面活性剤)としては、N−アシル−N−アルキルタウリン塩、脂肪酸塩、アルキル硫酸エステル塩、アルキルベンゼンスルホン酸塩、アルキルナフタレンスルホン酸塩、ジアルキルスルホコハク酸塩、アルキルリン酸エステル塩、ナフタレンスルホン酸ホルマリン縮合物、ポリオキシエチレンアルキル硫酸エステル塩等を挙げることができる。なかでも、N−アシル−N−アルキルタウリン塩が好ましい。N−アシル−N−アルキルタウリン塩としては、特開平3−273067号明細書に記載されているものが好ましい。これらアニオン性分散剤は、単独であるいは2種以上を組み合わせて用いることができる。
【0089】
カチオン性分散剤(カチオン性界面活性剤)には、四級アンモニウム塩、アルコキシル化ポリアミン、脂肪族アミンポリグリコールエーテル、脂肪族アミン、脂肪族アミンと脂肪族アルコールから誘導されるジアミン及びポリアミン、脂肪酸から誘導されるイミダゾリン及びこれらのカチオン性物質の塩が含まれる。これらカチオン性分散剤は、単独であるいは2種以上を組み合わせて用いることができる。
【0090】
両イオン性分散剤は、前記アニオン性分散剤が分子内に有するアニオン基部分とカチオン性分散剤が分子内に有するカチオン基部分をともに分子内に有する分散剤である。
ノニオン性分散剤(ノニオン性界面活性剤)としては、ポリオキシエチレンアルキルエーテル、ポリオキシエチエレンアルキルアリールエーテル、ポリオキシエチレン脂肪酸エステル、ソルビタン脂肪酸エステル、ポリオキシエチレンソルビタン脂肪酸エステル、ポリオキシエチレンアルキルアミン、グリセリン脂肪酸エステルなどを挙げることができる。なかでも、ポリオキシエチレンアルキルアリールエーテルが好ましい。これらノニオン性分散剤は、単独であるいは2種以上を組み合わせて用いることができる。
【0091】
分散剤の含有量は、顔料100質量部に対して0.1〜1000質量部の範囲であることが好ましく、より好ましくは1〜500質量部の範囲であり、更に好ましくは5〜200質量部の範囲である。また、分散剤は、単独で用いても、複数のものを組み合わせて用いてもよい。
【0092】
本発明の顔料の製造方法においては、上記工程(a)〜(c)によって得られる生成物は通常の有機合成反応の後処理方法に従って処理した後、精製してあるいは精製せずに次の結晶変換工程に供することができる。
すなわち、例えば、反応系から遊離したものを精製せずに、あるいは再結晶、造塩等にて精製する操作を単独、あるいは組み合わせて行ない、供することができる。
【0093】
また、反応終了後、反応溶媒を留去して、あるいは留去せずに水、又は氷にあけ、中和してあるいは中和せずに、遊離したものをあるいは有機溶媒/水溶液にて抽出したものを、精製せずにあるいは再結晶、晶析、造塩等にて精製する操作を単独に又は組み合わせて行なった後、供することもできる。
【0094】
工程(c)で得られた非晶質アゾ化合物は、アゾ化合物の懸濁液からアゾ化合物を取り出さずに結晶変換をおこなっても良いし、取り出してから結晶変換を行ってもよい。
【0095】
工程(c)で得られた固体が非晶質ではない場合、アゾ化合物を良溶媒に溶解させて、非晶質な固体が得られる貧溶媒と混合することで非晶質なアゾ化合物を得てもよい。
【0096】
次に、本発明の製造方法により得られる顔料粒子、すなわち、以上の方法により非晶質なアゾ化合物から結晶変換して得られた式(1)で表されるアゾ顔料について述べる。
【0097】
本発明の製造方法により得られる顔料粒子の体積平均粒子径は、1nm〜10μmであることが好ましく、5nm〜5μmであることがより好ましく、10nm〜1μmであることがさらに好ましく、10〜500nmであることが特に好ましい。
【0098】
なお、顔料粒子の体積平均粒子径とは、顔料そのものの粒子径、又は色材に分散剤等の添加物が付着している場合には、添加物が付着した粒子径をいう。本発明において、顔料粒子の体積平均粒子径の測定装置には、ナノトラックUPA粒度分析計(UPA−EX150;日機装社製)を用いることができる。その測定は、顔料分散物3mlを測定セルに入れ、所定の測定方法に従って行うことができる。なお、測定時に入力するパラメーターとしては、粘度にはインク粘度を、分散粒子の密度には顔料の密度を用いる。
【0099】
上記の顔料粒子の好ましい体積平均粒子径は、既述の結晶変換した後の式(1)で表されるアゾ顔料の溶解性が低い溶媒を用いて結晶変換をする際に結晶成長を抑制させることや、結晶変換の温度、時間、溶媒量を、適宜、調整することで達成される。
【0100】
顔料の分散性を更に好ましくし、着色力を更に向上させる観点から、下記式(1)で表されるδ型結晶形態アゾ顔料の窒素吸着法によるBET比表面積は50m2/g以上であることが好ましく、60m2/g以上であることが特に好ましい。
ここで、窒素吸着法によるBET比表面積とは、窒素ガスを粉体粒子に吸着させ、吸着平衡状態における吸着平衡圧を求め、BETの関係式により単分子層吸着量を算出し求められた比表面積をいう。窒素吸着法によるBET比表面積は、例えば、日本工業規格JIS Z8830の付属書2に規定される「1点法による気体吸着量の測定方法」に従って測定することができる。具体的には、比表面積測定装置「MONOSORB MS−17」(ユアサアイオニクス(株)製)等を用いることにより測定することができる。
窒素吸着法によるBET比表面積を上記の範囲とすることにより顔料の一次粒子が十分微細化されるが、微細化された状態においても、式(1)で表されるδ型結晶形態アゾ顔料は耐光性を落とすことなく分散性及び着色力が更に向上する。
窒素吸着法によるBET比表面積が50m2/g以上である下記式(1)で表されるδ型結晶形態アゾ顔料は、好ましくは、後述するソルベントソルトミリングを含む工程により製造することができる。
【0101】
本発明の方法で製造された式(1)で表されるアゾ顔料は、必要に応じて後処理を行ってもよい。この後処理の方法としては、例えば、ソルベントソルトミリング、ソルトミリング、ドライミリング、ソルベントミリング、アシッドペースティング等の摩砕処理、溶媒加熱処理などによる顔料粒子制御工程、樹脂、界面活性剤及び分散剤等による表面処理工程が挙げられる。
【0102】
また、本発明の式(1)で表される化合物は後処理としてソルベントソルトミリングを行うことが好ましい。
【0103】
(ソルベントソルトミリング)
ソルベントソルトミリングとしては、例えば、アゾ顔料(以下、混練磨砕する前のアゾ顔料を「粗アゾ顔料」ということがある。)と、無機塩と、それを溶解しない有機溶剤とを含む混合物を混練機に仕込み、その中で混練磨砕を行うことが挙げられる。上記無機塩としては、水溶性無機塩が好適に使用でき、例えば塩化ナトリウム、塩化カリウム、硫酸ナトリウム、硫酸カリウム等の無機塩を用いることが好ましい。また、前記水溶性無機塩の粒子径としては特に制限はないが、アゾ顔料の2次凝集体の粒子径制御の観点から、水溶性無機塩の粒子径が体積基準のメディアン径で0.5〜50μmであることが好ましく、1〜20μmであることがより好ましく、1〜10μmであることがさらに好ましい。当該無機塩の使用量は、粗アゾ顔料に対して、1〜30質量倍とすることができ、生産性の観点から、3〜20質量倍とするのが好ましく、5〜15質量倍とするのがより好ましい。有機溶剤としては、水溶性有機溶剤が好適に使用でき、混練時の温度上昇により溶剤が蒸発し易い状態になるため、安全性の点から高沸点溶剤が好ましい。このような有機溶剤としては、例えばジエチレングリコール、グリセリン、エチレングリコール、プロピレングリコール、液体ポリエチレングルコール、液体ポリプロピレングリコール、2−(メトキシメトキシ)エタノール、2−ブトキシエタノール、2ー(イソペンチルオキシ)エタノール、2−(ヘキシルオキシ)エタノール、ジエチレングリコールモノメチルエーテル、ジエチレングルコールモノエチルエーテル、ジエチレングリコールモノブチルエーテル、トリエチレングリコール、トリエチレングリコールモノメチルエーテル、1−メトキシ−2−プロパノール、1−エトキシ−2−プロパノール、ジプロピレングリコール、ジプロピレングリコールモノメチルエーテル、ジプロピレングリコールモノメチルエーテル、ジプロピレングリコールまたはこれらの混合物が挙げられる。また、その他の水溶性有機溶剤として、プロピルアルコール、2−ブチルアルコール、tert−ブチルアルコールなどの1価アルコール系溶剤も好適に使用することができる。当該水溶性有機溶剤の使用量は、粗アゾ顔料に対して0.1〜5質量倍が好ましく、さらに好ましくは2〜3質量倍である。混練温度は、20〜130℃が好ましく、40〜110℃が特に好ましい。混練機としては、例えばニーダーやミックスマーラー等を使用することができ、具体的には、ニーダー等のバッチ式混練機、スーパーミキサー((株)カワタ製)やトリミックス((株)井上製作所製)等のバッチ式混練機、連続式1軸混練機KCKミル(浅田鉄工(株)製)等の連続式混練機を用いることができる。
【0104】
前記連続式混練機としては、例えば、その磨砕部分が混練分散に必要な要素である圧縮・せん断・混合(置換)の三つの作用を粗顔料に与えることができる固定ブレードと回転ブレードとを有していることが好ましい。また前記固定ブレードと回転ブレードとの山と山は隙間(ギャップ)を形成し、せん断作用はこのギャップにおいて発生し、また、前記回転ブレードと固定ブレードとの谷間の材料がお互いにキャビティースライスを受けていることが好ましい。
【0105】
前記固定ブレードと回転ブレードの形状には特に制限はないが、それぞれ、菊型、扇型および臼状型の3種類から選ばれることが好ましい。固定ブレードと回転ブレードとを交互に多段に重なっていることが好ましく、これにより各々のブレードの両面にキャビティーを放射状に形成することができる。また、回転ブレードと中間スクリューが回転軸上に交互に組み込まれ、固定ブレードはせん断室シリンダーと交互にタイロッドによってフィードシリンダーに固定されていることが好ましい、これにより固定ブレードと回転ブレードとスクリューの組合せにより混練物を押し出すことができる。
【0106】
さらに連続式混練機は、混合物の投入部、磨砕部および押出部に少なくとも6箇所の温度調節部を有していることが好ましい。これにより、粗アゾ顔料の磨砕工程における温度範囲を幅広く設定できる。
前記磨砕工程における処理温度としては、特に制限はなく、例えば5〜200℃とすることができるが、アゾ顔料粒子の変色、粒度分布の観点から、5〜50℃であることが好ましく、10〜35℃であることがより好ましい。
【0107】
また、前記連続混練機は、粗アゾ顔料、水溶性無機塩および水溶性有機溶剤の混合割合、または軸回転数により、吐出量を変えることが可能であることが好ましい。吐出量を変えることで、アゾ顔料の磨砕粒径を所望の粒径に容易に制御することができる。
【0108】
本発明のアゾ顔料磨砕物の製造方法は、前記磨砕工程に加え、必要に応じてその他の工程を備えることができる。その他の工程としては、例えば、磨砕工程後の前記混合物を、水等に投入して攪拌した後、アゾ顔料磨砕物をろ過等で分離することで水溶性無機塩および水溶性有機溶剤を除去する洗浄工程、前記洗浄工程で得られたアゾ顔料磨砕物を乾燥する乾燥工程等を挙げることができる。
これら洗浄工程および乾燥工程は、いわゆるソルベントソルトミリング法で通常用いられる方法を本発明においても特に制限なく適用することができる。
【0109】
本発明におけるアゾ顔料磨砕物の1次粒子の粒径としては、80nm以下であることが好ましく、30〜50nmであることがより好ましい。また1次粒子が凝集した2次粒子の粒径としては、120nm以下であることが好ましく、60〜100nmであることがより好ましい。
前記アゾ顔料磨砕物の1次粒子および2次粒子の粒径は、透過型電子顕微鏡(TEM)を用いて測定される。
【0110】
本発明は、非晶質な下記式(1)で表されるアゾ化合物、その塩、水和物又は溶媒和物を、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が(a)4.8°、7.2°及び9.7°、(b)4.8°、7.2°、9.7°、20.1°、及び26.8°又は(c)4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的X線回折ピークを有する結晶形に結晶変換することを特徴とする下記式(1)で表されるアゾ顔料の製造方法であって、(i)非晶質な前記式(1)で表されるアゾ化合物を、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が4.8°、7.2°及び9.7°に特徴的X線回折ピークを有する結晶形に結晶変換する工程、及び、(ii)前記工程(i)で得たアゾ顔料と、水溶性無機塩と、水溶性有機溶剤とを含む混合物を混練して、比表面積を50m2/g以上の結晶にする工程、を含むアゾ顔料、又はその互変異性体の製造方法にも関する。
上記水溶性無機塩及び水溶性有機溶剤としては上述のものと同様のものを使用することができ、好ましい範囲もまた同様である。混練に使用する混練機も、上述のものと同様のものを使用することができる。
【0111】
本発明において分散剤は低分子および高分子、さらに水溶性および水不溶性の中から任意で選択できるが、印画物の画質の観点から、高分子が好ましい。さらに、水系で分散を行うので、分散性、分散物安定性の観点から、水溶性であることが好ましい。本発明において分散剤は、水溶性高分子であることが特に好ましい。
また、本発明において「分散剤」は、架橋剤で架橋化された状態のものも意味する。本発明の顔料分散物では、この分散剤が顔料に吸着されたものが望ましい。
【0112】
分散剤は分子中に、電荷反発による効果があるため、分散物の保存安定性の観点から1以上、好ましくは10以上のカルボキシ基を持つことが好ましい。架橋剤が2つのエポキシ基をもつときは、架橋反応によりエポキシ基とカルボキシ基が架橋するために、カルボキシ基が減るので、ポリマーは10以上のカルボキシ基を持つことが好ましい。
ポリマー中にあるカルボキシ基は酸(−COOH)の形でも、塩の形でもよい。塩としては、例えば、金属イオン、アンモニウム、置換アンモニウム、4級アンモニウムまたはピリジニウム塩などが挙げられる。好ましくは、金属イオン、アンモニウムであり、さらに好ましくはカリウムイオン、ナトリウムイオンである。
【0113】
本発明の高分子分散剤はポリウレタン、ポリエステル、ポリビニルを含み、より好ましくはポリウレタン、ポリエステル、ポリビニルであり、最も好ましくはポリビニル(ビニルポリマー)である。本発明では2種類以上のポリマーを組み合わせてもよい。
ポリマーへのカルボキシ基の導入は少なくとも1つのカルボキシ基を含むモノマーの共重合によって得られる。好ましいポリビニルには、モノマーとしてイタコン酸、マレイン酸、フマール酸、クロトン酸、メタクリル酸、アクリル酸、β−カルボキシエチルアクリレートを用いるが、好ましくはメタクリル酸、アクリル酸、β−カルボキシエチルアクリレートを用いる。
【0114】
ポリマー中のカルボキシル基は、まず、架橋剤中の架橋性基と架橋する作用をもつ。架橋性基としては酸無水物、エポキシ基が挙げられ、エポキシ基が特に望ましい。反応性が高いので、温和な条件で架橋することができるからである。更に、未反応カルボキシル基は最終微粒子分散物の沈降及び凝集に対する安定性に有効である。カルボキシル基は極性溶媒特に水溶媒中で安定性基として有効である。カルボキシル基が顔料分散物中で安定性に寄与する唯一の基である場合、全てのカルボキシ基が架橋剤と架橋してしまうと、分散物の安定性が著しく低下する。そのため、架橋反応が完結した後に未反応カルボキシル基が残るように、エポキシ基に対してカルボキシル基のモル過剰とすることが好ましく、エポキシ基に対してカルボキシル基をモル比で30:1〜1.1:1、より好ましくは25:1〜1.1:1、特に好ましくは20:1〜2:1とすることが望ましい。
ポリマーは他の安定性基を持っていてもよい。安定性基の選択及びその量は溶媒の性質に大きく依存する。安定性基は実際、親水性(例えば、極性溶媒)であるか、疎水性(例えば、無極性溶媒)であるかに依存する。
好ましいポリマー分散剤は親水性モノマー、疎水性モノマーの両方から得られる。
【0115】
親水性モノマーはイオン性基または非イオン性基である親水性を含むモノマーである。イオン性基はカチオンでもよいが、好ましくはアニオンである。カチオン性基も、またアニオン性基も分散剤に両性的安定性(amphotericstabilisation)を与える。好ましいアニオン性基はフェノキシ、スルホン酸、硫酸、ホスホン酸、ポリ燐酸、燐酸の基(塩でもよい)である。好ましいカチオン性基は4級アンモニウム、ベンズアルコニウム、グアニジン、ビグアニジン、及びピリジニウムである。これらは水酸化物、硫酸塩、硝酸塩、塩化物、臭化物、沃化物及び弗化物のような塩の形でもよい。好ましい非イオン性基はグルコキシド、糖類、ピロリドン、アクリルアミド、及び特にヒドロキシル基及びポリ(アルキレンオキシド)基であり、より好ましくはポリ(エチレンオキシド)基またはポリ(プロピレンオキシド)基であり、特に−(CH2CH2O)nHまたは−(CH2CH2O)nC1−4−アルキルである。ここで、nは3〜200(好ましくは4〜20)を表す。これ以降、例えばC1−4−の表現は “炭素数1〜4の”を表す。ポリマーは非イオン性基のみを、ポリマー全体で複数の非イオン性基を、また非イオン性基を含む1以上のポリマー鎖を含んでいてもよい。ヒドロキシル基はポリビニルアルコール、ポリヒドロキシル機能のアクリリックス及びセルロースを用いて挿入される。エチレンオキシ基はポリエチレンオキシドのようなポリマー鎖を用いて挿入される。
疎水性モノマーは疎水性基を含むモノマーである。疎水性基を有する代表的なものは3以下で好ましくは0の親水性基を持つ、炭化水素類、フルオロカーボン類、ポリC3−4アルキレンオキシ類及びアルキルシロキサン類である。疎水性基は、好ましくはC3−50鎖であり、また疎水性モノマー中にプロピレンオキシドを側鎖または直鎖に有し得る。
ポリマーは、ホモポリマーでもよいが、好ましくは共重合体(コポリマー)である。ポリマーはランダムポリマー(統計上短いブロックまたはセグメント)を含むが、好ましくは、グラフトポリマー(長いブロックまたはセグメント)を含む。また、ポリマーは交互(alternating)ポリマーでもよい。ポリマーは分岐していてもよいが、好ましくは直鎖である。ポリマーは2以上のセグメント(例えば、ブロック及びグラフト、コポリマー)を持っていてもよいが好ましくはランダムである。
ポリマーが2以上のセグメントを持つ場合の態様では、少なくとも1つのセグメントは疎水性であり、少なくとも1つのセグメントは互いに関連性の親水性であることが好ましい。疎水性及び親水性セグメントをつくる好ましい方法はそれぞれ疎水性及び親水性モノマーの共重合による。ポリマーが少なくとも1つの疎水性セグメント及び少なくとも1つの親水性セグメントもつ場合、カルボキシル基は疎水性セグメントにあっても、また親水性セグメントにあっても、また両方のセグメントにあってもよい。
【0116】
ビニルポリマー(ポリビニル)はどのような適切な手段によって製造されてもよい。ビニルポリマーの好ましい製造方法は、特に(メタ)アクリレートとビニルナフタレン(特にスチレンモノマー)のようなビニルモノマーを用いるフリーラジカル重合である。適切なフリーラジカル重合は懸濁重合、溶液重合、分散重合、乳化重合に限定されないが、好ましくは溶液重合である。
ビニルポリマーは(メタ)アクリレートモノマーを用いる場合が好ましい。
ビニルポリマーは好ましくは共重合体(コポリマー)である。
疎水性モノマー及び親水性モノマーから導かれるコポリビニル分散剤は好ましくは実質的にセグメントをもたない。例えば、コポリビニルポリマーはセグメント長が非常に短いか存在しないようなフリーラジカル重合によって製造される。かかる場合はしばしば「ランダム」重合と呼ばれる。セグメントをもつコポリビニルポリマーはリビング重合、特に原子団転移(group transfer)重合、原子転移(atom transfer)重合、マクロモノマー(macromonomer)重合、グラフト重合、アニオンまたはカチオン重合のような重合方法によって製造される。好適な親水性ビニルモノマーは非イオン性及びイオン性モノマーである。好ましい非イオン性モノマーは糖類、グルコース、アミド、ピロリドンであり、特にヒドロキシ基及びエトキシ基をもつものである。好ましい非イオン性モノマーの例としては、ヒドロキシ エチルアクリレート、ヒドロキシ エチルメタアクリレート、ビニルピロリドン、エトキシ化された(メタ)アクリレート及び(メタ)アクリルアミドが挙げられる。好適なイオン性ビニルモノマーはカチオン性であってもよいが、好ましくはアニオン性である。
好ましいアニオン性ビニルモノマーはカルボキシ基および/または燐酸基および/またはスルホン酸基(これらの酸はフリーでも塩でもよい)を含むものである。好ましい例として、(メタ)アクリル酸、スチレンスルホン酸、ビニルベンジルスルホン酸、ビニルスルホン酸、(メタ)アクリロイルオキシアルキルスルホン酸(例えば、アクリロイルオキシメチルスルホン酸、アクリロイルオキシエチルスルホン酸、アクリロイルオキシプロピルスルホン酸、アクリロイルオキシブチルスルホン酸、メタクリロイルオキシメチルスルホン酸、メタクリロイルオキシエチルスルホン酸、メタクリロイルオキシプロピルスルホン酸、メタクリロイルオキシブチルスルホン酸)、2−アクリルアミド−2−アルキルアルカンスルホン酸(例えば、2−アクリルアミド−2−メチルエタンスルホン酸、2−アクリルアミド−2−メチルプロパンスルホン酸、2−アクリルアミド−2−メチルブタンスルホン酸)、2−メタクリルアミド−2−アルキルアルカンスルホン酸(例えば、2−メタクリルアミド−2−メチルエタンスルホン酸、2−メタクリルアミド−2−メチルプロパンスルホン酸、2−メタクリルアミド−2−メチルブタンスルホン酸)、モノ−(アクリロイルオキシアルキル)燐酸塩(例えば、モノ−(アクリロイルオキシエチル)燐酸塩、モノ−(3−アクリロイルオキシプロピル)燐酸塩)、モノ−(メタクリロイルオキシアルキル)燐酸塩(例えば、モノ−(メタクリロイルオキシエチル)燐酸塩、モノ−(3−メタクリロイルオキシプロピル)燐酸塩)が挙げられる。
好ましいカチオンビニルモノマーは4級アミン、ピリジン、グアニジン及びビグアニジン基を含むものである。
好ましい疎水性ビニルモノマーは親水性基を持たない。好ましい疎水性ビニルモノマーとしてはC1−20−ヒドロカルビル(メタ)アクリレート、ブタジエン、スチレン及びビニルナフタレンが挙げられ、C1−20−ヒドロカルビル(メタ)アクリレート(例、メチル(メタ)アクリレート、ブチル(メタ)アクリレート、オクチル(メタ)アクリレート、2−エチルヘキシル(メタ)アクリレート、イソボルニルアクリレート、ラウリルアクリレート、ステアリルアクリレート、ベンジル(メタ)アクリレート、フェノキシエチル(メタ)アクリレート)が好ましく、メチルメタクリレート、ベンジルメタクリレート、2−エチルヘキシルメタクリレート、フェノキシエチルメタクリレートが特に好ましい。これらのヒドロカルビル基は分岐でもよいが、好ましくは直鎖である。
【0117】
少なくとも1つのカルボキシル基を持つポリエステルはジオールモノマーと過剰量のジカルボン酸モノマーとの反応によっても生成される。カルボキシル基はカルボキシル基を持つジオールとジカルボン酸モノマーとの共重合によっても導入できる。
ポリエステルはジカルボン酸とジオールとのエステル化で製造されるのが典型的なものである。
【0118】
カルボキシル基を有するポリエステルは、例えば、カルボキシル基含有化合物と水酸基含有化合物とを、カルボキシル基が残存するように、溶融法、溶剤法などの公知の方法によって脱水縮合反応を行うことにより製造することができる。
【0119】
ポリエステルは、一塩基酸、多塩基酸の如きカルボキシル基を有する化合物と、ジオール、ポリオールの如き水酸基を有する化合物とを適宜選択して脱水縮合させて得られるもの等が挙げられ、さらに、油脂類または脂肪酸類を使用したものがアルキッド樹脂となる。
【0120】
本発明で使用するポリエステルが有するカルボキシル基は、主に、ポリエステルを構成する二塩基酸以上の多塩基酸に由来する未反応のカルボキシル基である。
【0121】
多塩基酸としては、例えば、アジピン酸、(無水)コハク酸、セバシン酸、ダイマー酸、(無水)マレイン酸、(無水)フタル酸、イソフタル酸、テレフタル酸、テトラヒドロ(無水)フタル酸、ヘキサヒドロ(無水)フタル酸、ヘキサヒドロテレフタル酸、2,6−ナフタレンジカルボン酸、(無水)トリメリット酸、(無水)ピロメリット酸などが挙げられる。
【0122】
多塩基酸以外に使用可能なカルボキシル基を有する化合物としては、例えば、テレフタル酸ジメチルの如き酸の低級アルキルエステル類;安息香酸、p−ターシャリブチル安息香酸、ロジン、水添ロジンの如き一塩基酸類;脂肪酸および油脂類;分子末端に1個又は2個のカルボキシル基を有するマクロモノマー類;5−ソジウムスルフォイソフタル酸およびそのジメチルエステル類などが挙げられる。
【0123】
水酸基を有する化合物としては、例えば、エチレングリコール、ネオペンチルグリコール、プロピレングリコール、ジエチレングリコール、ジプロピレングリコール、2−メチル−1,3−プロパンジオール、2,2−ジエチル−1,3−プロパンジオール、1,4−ブタンジオール、1,3−プロパンジオール、1,6−ヘキサンジオール、1,4−シクロヘキサンジメタノール、1,5−ペンタンジオール、ビスフェノールAのアルキレンオキサイド付加物、水添ビスフェノールA、水添ビスフェノールAのアルキレンオキサイド付加物、ポリエチレングリコール、ポリプロピレングリコール、ポリテトラメチレングリコールの如きジオール類;グリセリン、トリメチロールプロパン、トリメチロールエタン、ジグリセリン、ペンタエリスリトール、トリスヒドロキシエチルイソシアヌレートの如きポリオール類;「カージュラE−10」(シェル化学工業株式会社製の合成脂肪酸のグリシジルエステル)などのモノグリシジル化合物類、分子片末端に水酸基を2個有するマクロモノマー類などが挙げられる。
【0124】
また、ポリエステルを合成する際に、ひまし油、12−ヒドロキシステアリン酸などの水酸基含有脂肪酸または油脂類;ジメチロールプロピオン酸、p−ヒドロキシ安息香酸、ε−カプロラクトンの如きカルボキシル基と水酸基とを有する化合物なども使用できる。
【0125】
さらに、二塩基酸の一部をジイソシアネート化合物に代えることもできる。
【0126】
また、カルボキシル基を有するポリエステルは、水酸基を有するポリエステルに、無水マレイン酸、無水フタル酸、テトラヒドロ無水フタル酸、ヘキサヒドロ無水フタル酸、無水トリメリット酸などの無水酸を付加反応せしめる方法によっても製造することができる。
【0127】
水酸基とカルボキシル基とを有するポリエステルは、例えば、ポリエステル樹脂の脱水縮合反応において、公知の方法に従って、水酸基とカルボキシル基とが残存するように反応させることによって容易に製造することができる。
【0128】
第3級アミノ基とカルボキシル基とを有するポリエステルは、例えば、トリエタノールアミン、N−メチルジエタノールアミン、N,N−ジメチルエタノールアミン等の第3級アミノ基と水酸基とを有する化合物を、ポリエステル樹脂を製造する際のアルコール成分として使用することによって容易に製造することができる。
【0129】
ラジカル重合性不飽和基とカルボキシル基を有するポリエステルは、例えば、[1]水酸基とカルボキシル基とを有するポリエステルに、2−メタクリロイルオキシエチルイソシアネートなどのイソシアネート基を有するラジカル重合性不飽和基含有モノマー類、あるいは、無水マレイン酸などのラジカル重合性不飽和基を有する無水酸を付加反応せしめる方法、[2]カルボキシル基を有するポリエステル樹脂に、エポキシ基を有する重合性モノマー類を付加反応せしめる方法、[3]酸成分として無水マレイン酸などのラジカル重合性不飽和基含有モノマーを使用してポリエステル樹脂を合成する方法、等によって容易に製造することができる。
【0130】
ポリウレタンはポリオール成分(例えば、ジ−イソシアネート)とポリオール成分(例えば、ジオール)との縮合反応で好ましく製造される。
カルボキシル基を有するポリウレタンは、例えば、カルボキシル基を導入する成分としてのジメチロールプロピオン酸の如きカルボキシル基と水酸基とを有する化合物を含有するポリオール成分と、ポリイソシアネート成分とを反応させることによって、容易に製造することができる。
【0131】
ポリオール成分としては、ポリエステルの製造方法において掲げたジオール成分のほか、必要に応じて、3官能以上のポリオール化合物を使用することもできる。
【0132】
ポリイソシアネート成分としては、例えば、2,4−トリレンジイソシアネート、2,6−トリレンジイソシアネート、4,4’−ジフェニルメタンジイソシアネート、ヘキサメチレンジイソシアネート、フェニレンジイソシアネート、1,5−ナフタレンジイソシアネート、メタキシリレンジイソシアネート、イソホロンジイソシアネート、水添トリレンジイソシアネート、水添4,4’−ジフェニルメタンジイソシアネート、水添メタキシリレンジイソシアネート、粗製4,4’−ジフェニルメタンジイソシアネートの如きジイソシアネート化合物のほか、ポリメチレンポリフェニルイソシアネートの如きポリイソシアネート化合物も使用できる。
【0133】
ポリウレタンの製造は、常法に従えばよい。例えば、イソシアネート基と反応しない不活性な有機溶剤溶液中で、室温又は40〜100℃程度の温度で付加反応を行うのが好ましい。その際、ジブチル錫ジラウレート等の公知の触媒を使用しても良い。
【0134】
ポリウレタンを製造する際の反応系には、ジアミン、ポリアミン、N−メチルジエタノールアミンの如きN−アルキルジアルカノールアミン;ジヒドラジド化合物などの公知の鎖伸長剤も使用できる。
【0135】
水酸基とカルボキシル基とを有するポリウレタンは、例えば、ポリウレタンを製造する際に、イソシアネート基よりも水酸基が多くなる割合で反応させることにより容易に製造することができる。あるいは、カルボキシル基と末端イソシアネート基とを有するポリイソシアネートに、1分子中に水酸基を2個以上有する化合物を付加反応させることによっても容易に製造することができる。
【0136】
第3級アミノ基とカルボキシル基とを有するポリウレタンは、例えば、ポリオール成分の一部としてN−メチルジエタノールアミンなどのN−アルキルジアルカノールアミンを使用することにより容易に製造することができる。
【0137】
ブロック化イソシアネート基とカルボキシル基とを有するポリウレタンは、例えば、カルボキシル基と末端イソシアネート基とを有するポリイソシアネートに、公知のブロック剤を付加反応させることによって容易に製造することができる。
【0138】
エポキシ基とカルボキシル基とを有するポリウレタンは、例えば、カルボキシル基と末端イソシアネート基とを有するポリイソシアネートに、水酸基とエポキシ基とを有する化合物を付加反応させることによって容易に製造することができる。
【0139】
水酸基とエポキシ基とを有する化合物としては、例えば、グリシドール、グリセリンジグリシジルエーテル、トリメチロールプロパンジグリシジルエーテル、ビスフェノールAのジグリシジルエーテル等が挙げられる。
【0140】
ラジカル重合性不飽和基を、酸性基としてカルボキシル基を有するポリウレタンは、例えば、末端イソシアネート基を有するポリイソシアネートに、前述した如き水酸基を有する重合性モノマー類、及びグリセロールモノ(メタ)アクリレート、トリメチロールプロパンジ(メタ)アクリレート、ペンタエリスリトールトリアクリレートなどの水酸基とラジカル重合性不飽和基とを有する化合物を付加反応せしめる方法等によって容易に製造することができる。
【0141】
加水分解性アルコキシシラン基を、酸性基としてカルボキシル基を有するポリウレタンは、例えば、末端イソシアネート基を有するポリイソシアネートに、γ−メルカプトプロピルトリメトキシシラン、γ−メルカプトプロピルメチルジメトキシシラン、γ−アミノプロピルトリメトキシシラン、γ−アミノプロピルトリエトキシシランの如きイソシアネート基と反応しうる活性水素を有するシランカップリング剤を付加反応させる方法等により容易に製造することができる。
【0142】
ポリマーは微粒子分散物を製造する過程で用いる液体媒体に合うようにまた微粒子分散物に用いられる最終組成物(例えば、インク)中の液体展色剤(ベヒクル)に合うように選ばれる。例えば、微粒子分散物が水性のインクジェット記録用インクに用いられる場合には、好ましくはポリマーは親水性である。
〔分子量〕
分散剤の重量平均分子量は10000以上200000以下が好ましく、さらに15000以上150000以下であることが好ましく、中でも20000以上100000以下であることがより好ましい。10000以上では印画物の画質が優れ好ましい一方、200000以下では、粘度が高くなるのを抑制でき、さらに貯蔵安定性がの低下を防ぎ、好ましい。
【0143】
〔D/P値〕
分散剤の含有量は、顔料100質量部に対して20〜100質量部の範囲であることが好ましく、より好ましくは25〜90質量部の範囲であり、さらに好ましくは30〜70質量部の範囲である。また、分散剤は、単独で用いても、複数のものを組み合わせて用いてもよい。
分散剤の含有量が20質量部未満の場合、分散剤の量が顔料に対して不十分になり、貯蔵安定性が不十分になる。一方、100質量部超過の場合、粘度が高くなり、さらに貯蔵安定性が低下するため不適である。
前記顔料分散物中の着色剤の含有量をP、分散剤の含有量をDとし、含有量Dと含有量Pとの比をD/P値としたときに、D/P値が0.15以上1.0以下であることが好ましく、0.16以上0.8以下であることがより好ましく、0.17以上0.7以下であることが更に好ましい。。
【0144】
〔酸価〕
分散剤は架橋剤と架橋するために十分な酸価をもつ必要があり、少なくとも50mgKOH/g以上の酸価をもつものが好ましい。
全ての態様において、上記の酸価は好ましくは70〜200mgKOH/gであり、より好ましくは70〜160mgKOH/gである。係る酸価をもつ分散剤は改良された保存安定性を与える。
また、50mgKOH/gより低いと、水系溶媒への溶解性が低いため不適である。
【0145】
〔溶解性〕
分散剤は水不溶性、水溶性のどちらでも良いが、水への溶解性として、1g/100mL以上であることが好ましく、さらに好ましくは、3g/100mL以上であり、特に好ましくは5g/100mL以上である。
1g/(100m)L未満では、水への溶解性が低いために、顔料粒子に吸着しにくくなり、分散性が低下する場合がある。
【0146】
〔架橋〕
前記水系分散物が、架橋剤により架橋されていることが好ましい。
本発明のより好ましい形態は、分散剤は架橋する前に顔料表面に吸着し、相対的に安定な分散物が形成され、そしてこの分散工程に引き続き、架橋剤を用いて架橋する工程を実施することによりより高度な保存安定性を有し、印画物の画質に優れる分散物が得られる。
少なくとも50mg/KOH以上の酸価をもつ分散剤を用いる場合には、架橋剤はオリゴマー分散基を持っていても、持たなくてもよい。「オリゴマー」という言葉は分子量に上限はないし、また繰り返し単位の上限もない意味で用いる。1以上のオリゴマー分散基を持つ架橋剤は生じた微粒子分散物の安定性を増加させる。この増加された安定性はインクジェット記録に用いる液体展色剤(ビヒクル)において特に有用である。それは50mg/KOH未満の酸価をもつ分散剤では分散が困難であるからである。
【0147】
オリゴマー分散基は好ましくはポリアルキレンオキシドであり、より好ましくはポリC2−4−アルキレンオキシドであり、特に好ましくはポリエチレンオキシドである。ポリアルキレンオキシドは生じた微粒子分散物の安定性を改良する。ポリアルキレンオキシドは好ましくは3〜200、より好ましくは5〜50、特に好ましくは5〜20のアルキレンオキシド繰り返し単位を有する。
【0148】
架橋剤は2つ以上のエポキシ基を持つことが好ましい。少なくとも2つのエポキシ基を持つ好ましい架橋剤はエピクロロヒドリン誘導体である。2つ以上のエポキシ基を持ち、オリゴマー分散基を持たない架橋剤はエチレングリコールジグリシジルエーテル、レゾルシノールジグリシジルエーテル、ネオペンチルグリコールジグリシジルエーテル、1,6−ヘキサンジオールジグリシジルエーテル、ハロゲン化されたビスフェノールAジグリシジルエーテル、トリメチロールプロパンポリグリシジルエーテル、ポリグリセロールポリグリシジルエーテル、グリセロールポリグリシジルエーテル、ペンタエリスリトールポリグリシジルエーテル、ジグリセロールポリグリシジルエーテル、ソルビトールポリグリシジルエーテル、及びポリブタジエンジグリシジルエーテルである。2つのエポキシ基を持ち、かつ1以上のオリゴマー分散基をもつ好ましい架橋剤はジエチレングリコールジグリシジルエーテル、ポリエチレングリコールジグリシジルエーテル、またはジプロピレングリコールジグリシジルエーテルである。
また、無水フタル酸、無水コハク酸等の酸無水物も架橋剤として用いることができる。
【0149】
〔温度、pH〕
本発明では架橋反応は100℃以下、pH6以上で行うことが好ましい。更に好ましい架橋反応は30℃〜90℃、より好ましくは40℃〜85℃である。
架橋反応の好ましいpHは7〜10であり、より好ましくは8〜9.5である。
架橋剤がさらにカルボキシ基を含み、カルボキシ基とエポキシ基の間の架橋反応を100℃以下、pH6以上で行うことが好ましい。
【0150】
架橋反応は水系で行うため、100℃以下が好ましい。逆に、低温では架橋反応の進行が遅くなるため、好ましくなく、30℃以上が好ましく、40℃以上がさらに好ましい。
【0151】
pHが10超過では、架橋反応で熱を加えるとポリマーが加水分解してしまう可能性がある。一方、pHが6未満では、顔料分散物が凝集を起こしやすくなり不安定になってしまうので、好ましくない。
【0152】
〔膜精製〕
膜精製には逆浸透膜(NF膜)、限外ろ過膜(UF膜)を使用することができ、加圧してもしなくても良いが、加圧する場合の方が、精製に要する時間が短くなり、効率的である。UF膜としては、分画分子量10000以上150000以下が好ましく、20000以上100000以下がより好ましい。10000未満では精製するための時間が長くなってしまうため、非効率である。一方、150000超過では、分散剤が流出してしまう可能性があるため、好ましくない。
【0153】
本発明のアゾ顔料の用途としては、画像、特にカラー画像を形成するための画像記録材料が挙げられ、具体的には、以下に詳述するインクジェット方式記録材料を始めとして、感熱記録材料、感圧記録材料、電子写真方式を用いる記録材料、転写式ハロゲン化銀感光材料、印刷インク、記録ペン等があり、好ましくはインクジェット方式記録材料、感熱記録材料、電子写真方式を用いる記録材料であり、更に好ましくはインクジェット方式記録材料である。
【0154】
また、CCDなどの固体撮像素子やLCD、PDP等のディスプレーで用いられるカラー画像を記録・再現するためのカラーフィルター、各種繊維の染色の為の染色液にも適用できる。
【0155】
以上に説明した本発明の製造方法によれば、非晶質なアゾ化合物から結晶変換することで、結晶成長をコントロールし、易分散なアゾ顔料を製造することができる。
【0156】
工程(b)において式(1)で表されるアゾ化合物が反応液中の少なくとも一部が溶解しているため、特に、アゾ化合物溶解液の調整方法が上記形態(ii)である場合は、カップリング反応がよりスムーズに進行してより高純度のアゾ化合物を得ることができる。このことは、最終的に得られるアゾ顔料の高効率な製造に寄与する。
【0157】
ところで、上記したように、カップリング反応後の反応液中で析出した特許文献6の顔料(一度、粒子として得られた顔料)を有機溶媒に溶解させることは困難である。すなわち、顔料の粒子径をより微細なものにするべく、特許文献6の顔料を有機溶剤に溶解させようとする場合、大量の有機溶媒が使用する必要があり、製造コストが増大する上、大量の有機溶媒を使用したとしても溶解させられない場合も多く、ましてや完全に溶解させることは非常に難しい。
【0158】
一方、本発明によれば、工程(b)の終了後には、大量の有機溶媒を使用することなく(高濃度で)、目的とするアゾ化合物の少なくとも一部が溶解した溶解液を得ることができ、このことは、最終的に得られるアゾ顔料の高効率な製造に寄与する。また、工程(b)で得られたアゾ化合物溶解液と、このアゾ化合物に対する貧溶媒とを混合することにより、アゾ顔料を微粒子として、析出させることができる。
【0159】
以上のように、本発明のアゾ顔料の製造方法によれば、易分散なアゾ顔料微粒子を高効率かつ低コストで製造することができる。
【0160】
[着色組成物]
本発明の着色組成物は、上記した本発明のアゾ顔料、その塩、水和物又は溶媒和物を少なくとも1種含有する。本発明の着色組成物は、媒体を含有させることができるが、媒体として溶媒を用いた場合は特にインクジェット記録用インクとして好適である。本発明の着色組成物は、媒体として、親油性媒体や水性媒体を用いて、それらの中に、本発明の顔料を分散させることによって作製することができる。好ましくは、水性媒体を用いる場合である。本発明の着色組成物には、媒体を除いたインク用組成物も含まれる。本発明の着色組成物は、必要に応じてその他の添加剤を、本発明の効果を害しない範囲内において含有しうる。その他の添加剤としては、例えば、乾燥防止剤(湿潤剤)、褪色防止剤、乳化安定剤、浸透促進剤、紫外線吸収剤、防腐剤、防黴剤、pH調整剤、表面張力調整剤、消泡剤、粘度調整剤、分散剤、分散安定剤、防錆剤、キレート剤等の公知の添加剤(特開2003−306623号公報に記載)が挙げられる。これらの各種添加剤は、水溶性インクの場合にはインク液に直接添加する。油溶性インクの場合には、アゾ顔料分散物の調製後分散物に添加するのが一般的であるが、調製時に油相又は水相に添加してもよい。
【実施例】
【0161】
以下、本発明を実施例に基づきさらに詳細に説明するが、本発明はこれらの実施例に何ら限定されるものではない。なお、実施例中、「部」とは質量部を表す。
【0162】
<アゾ化合物、アゾ顔料の製造>
【0163】
以下の実施例にて得られたアゾ化合物の1次粒子径については、透過型顕微鏡(日本電子(株)製:JEM−1010電子顕微鏡)を用いて目視にて観察した。
【0164】
アゾ化合物、アゾ顔料のX線回折の測定は、日本工業規格JISK0131(X線回析分析通則)に準じて、粉末X線回折測定装置RINT2500(株式会社リガク製)にてCuKα線を用い、次の条件で行ったものである。
【0165】
使用測定器:Rigaku社製 自動X線回折装置RINT2500
X線管球:Cu
管電圧:55KV
管電流:280mA
スキャン方法:2θ/θスキャン
スキャン速度:6deg./min
サンプリング間隔:0.100deg.
スタート角度(2θ):5deg.
ストップ角度(2θ):55deg.
ダイバージェンススリット:2deg.
スキャッタリングスリット:2deg.
レシービングスリット:0.6mm
縦型ゴニオメータ使用
【0166】
[実施例1]
亜硝酸ナトリウム2.2gを水50mLに溶解させた。別に式(2)で表されるアミノ化合物5.8gを濃塩酸50mLに溶解させた後、内温−10℃まで冷却した。この中に内温が0℃以下になるように先述の亜硝酸ナトリウム水溶液を滴下した。内温−10℃〜0℃にて1時間攪拌した後、内温0℃以下にて尿素1.8gを加えた。添加終了後15分間同温度にて攪拌し、ジアゾニウム塩溶液を得た。別に式(3)の化合物5gをメタノール175mLに加えた後昇温し、還流下溶解させた。この溶液を内温0℃まで冷却し、先述のジアゾニウム塩溶液を内温が10℃以下になるように添加した。内温10℃にて1時間攪拌した後、析出している固体を濾別した。メタノール、水で十分にかけ洗いを行った後、水300mLに懸濁させ、28%アンモニア水溶液を添加してpHを6.0に調整した。析出している固体を濾別して水で十分にかけ洗いを行い、60℃にて乾燥後、非晶質なアゾ化合物(1)−1を9.8g得た。
得られた非晶質なアゾ化合物(1)−1の1次粒子の長軸方向の長さは、約0.5μmであった。
非晶質なアゾ化合物(1)−1のX線回折の測定を上記の条件により行ったところ、特徴的なX線回折ピークが見られなかった。
【0167】
得られた非晶質なアゾ化合物(1)−1 5gをエチレングリコール50mLに懸濁させた。内温100℃まで昇温した後、同温度にて2時間攪拌した。内温30℃まで冷却した後、析出している固体を濾別し、δ型結晶形態のアゾ顔料(1)−2を4.5g得た。
得られたδ型結晶形態のアゾ顔料(1)−2の1次粒子の長軸方向の長さは、約0.6μmであった。
得られたδ型結晶形態のアゾ顔料(1)−2のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図1に示す。
【0168】
[実施例2]
亜硝酸ナトリウム2.2gを水50mLに溶解させた。別に式(2)で表されるアミノ化合物5.8gを濃塩酸50mLに溶解させた後、内温−10℃まで冷却した。この中に内温が0℃以下になるように先述の亜硝酸ナトリウム水溶液を滴下した。内温−10℃〜0℃にて1時間攪拌した後、内温0℃以下にて尿素1.8gを加えた。添加終了後15分間同温度にて攪拌し、ジアゾニウム塩溶液を得た。このジアゾニウム塩溶液に式(3)の化合物5gを内温が5℃以下になるように少しずつ加えた。添加終了後、内温10℃まで昇温し、同温度にて3時間攪拌した後、析出している固体を濾別した。水で十分にかけ洗いを行った後、水200mLに懸濁させ、28%アンモニア水溶液を添加してpHを6.0に調整した。固体を濾別して水で十分にかけ洗いを行い、60℃にて乾燥後、非晶質なアゾ化合物(1)−3を9.9g得た。
得られたアゾ化合物(1)−3の1次粒子の長軸方向の長さは、約0.3μmであった。
アゾ化合物(1)−3のX線回折の測定を上記の条件により行ったところ、特徴的なX線回折ピークが見られなかった。
【0169】
得られた非晶質なアゾ化合物(1)−3 5gをエチレングリコール50mLに懸濁させた。内温120℃まで昇温した後、同温度にて2時間攪拌した。内温30℃まで冷却した後、固体を濾別し、δ型結晶形態のアゾ顔料(1)−4を4.5g得た。
得られたアゾ顔料(1)−4の1次粒子の長軸方向の長さは、約0.5μmであった。
得られたアゾ顔料(1)−4のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°及び9.7°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図2に示す。
【0170】
[実施例3]
式(2)の化合物34.6gを酢酸150gに懸濁し、硫酸24gを内温が20℃〜30℃になるように滴下した。さらに内温が20℃〜30℃になるように43%ニトロシル硫酸の硫酸溶液48.6gを滴下し、内温20℃にて1時間攪拌後、尿素0.28gを添加してジアゾニウム塩溶液を得た。このジアゾニウム塩溶液に式(3)の化合物30gを内温が20℃〜30℃になるように分割して添加し、内温25℃にて1時間攪拌し、アゾ化合物の均一反応液を得た。別に360gのメタノールを内温25℃にて用意し、上述のアゾ化合物の均一反応液を内温が30℃以下になるように添加し、10分間攪拌した後、析出している固体を濾別した。300mLのメタノールでかけ洗いした後、水900mLに懸濁させ、28%アンモニウム水溶液を添加してpHを6.0に調整した。固体を濾別し、ζ型結晶形態のアゾ顔料(1)−5を得た。
得られたアゾ顔料(1)−5の1次粒子の長軸方向の長さは、約2μmであった。
得られたアゾ顔料(1)−5のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が6.5°、6.7°、9.1°および21.3°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図3に示す。
【0171】
得られたアゾ顔料(1)−5 5gを硫酸50mLに溶解させ、水300mLに内温が15℃以下になるように添加した。析出している固体を濾別し、十分に水でかけ洗いした後、水300mLに懸濁させ、28%アンモニア水溶液を添加してpHを6.1に調整した。固体を濾別して、水で十分にかけ洗いを行い、60℃にて乾燥後、非晶質なアゾ化合物(1)−6を3.9g得た。
得られたアゾ化合物(1)−6の1次粒子の長軸方向の長さは、約0.2μmであった。
アゾ化合物(1)−6のX線回折の測定を上記の条件により行ったところ、特徴的なX線回折ピークが見られなかった。
【0172】
得られた非晶質なアゾ化合物(1)−6 3gをエチレングリコール30mLに懸濁させた。内温120℃まで昇温した後、同温度にて2時間攪拌した。内温30℃まで冷却した後、固体を濾別し、δ型結晶形態のアゾ顔料(1)−7を2.4g得た。
得られたアゾ顔料(1)−7の1次粒子の長軸方向の長さは、約0.3μmであった。
得られたアゾ顔料(1)−7のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図4に示す。
【0173】
[実施例4]
実施例3で得られたアゾ顔料(1)−5 5gをリン酸50mLに溶解させ、水300mLに内温が15℃以下になるように添加した。析出している固体を濾別し、十分に水でかけ洗いした後、水300mLに懸濁させ、28%アンモニア水溶液を添加してpHを7.2に調整した。固体を濾別して、水で十分にかけ洗いを行い、60℃にて乾燥後、非晶質なアゾ化合物(1)−8を4.2g得た。
得られたアゾ化合物(1)−8の1次粒子の長軸方向の長さは、約0.2μmであった。
アゾ化合物(1)−8のX線回折の測定を上記の条件により行ったところ、特徴的なX線回折ピークが見られなかった。
【0174】
得られた非晶質なアゾ化合物(1)−8 3gをエチレングリコール30mLに懸濁させた。内温120℃まで昇温した後、同温度にて2時間攪拌した。内温30℃まで冷却した後、固体を濾別し、δ型結晶形態のアゾ顔料(1)−9を2.4g得た。
得られたアゾ顔料(1)−9の1次粒子の長軸方向の長さは、約0.2μmであった。
得られたアゾ顔料(1)−9のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図5に示す。
【0175】
[実施例5]
式(2)の化合物11.5gを酢酸50gに懸濁させ、内温が20℃〜30℃になるようにニトロシル硫酸の43%硫酸溶液16.2gを滴下した。内温20℃にて1時間攪拌した後、尿素0.1gを添加してジアゾニウム塩溶液を得た。別に式(3)の化合物10gを酢酸100mLに溶解させ、上述のジアゾニウム塩溶液に内温が20℃〜25℃になるように滴下した。内温20℃にて1時間攪拌し、アゾ化合物(1)の均一反応液を得た。別に水150gを用意し、内温20℃〜25℃にて上述のアゾ化合物(1)の均一反応液を滴下した。析出している固体を濾別した後、水で十分にかけ洗いを行い、水200mLに懸濁させ、28%アンモニア水溶液を添加してpHを6.2に調整した。固体を濾別して、水で十分にかけ洗い、非晶質なアゾ化合物(1)−10を得た。
得られたアゾ化合物(1)−10の1次粒子の長軸方向の長さは、約0.2μmであった。
水分測定を行ったところ、水の含率が68%だった。
アゾ化合物(1)−10の一部を乾燥し、X線回折の測定を上記の条件により行ったところ、特徴的なX線回折ピークが見られなかった。
【0176】
得られた非晶質な含水のアゾ化合物(1)−10 10gをエチレングリコール30mLに懸濁させた。内温95℃まで昇温した後、同温度にて2時間攪拌した。内温30℃まで冷却した後、固体を濾別し、δ型結晶形態のアゾ顔料(1)−11を2.9g得た。
得られたアゾ顔料(1)−11の1次粒子の長軸方向の長さは、約0.15μmであった。
得られたアゾ顔料(1)−11のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図6に示す。
【0177】
[実施例6]
実施例5で得られた非晶質な含水のアゾ化合物(1)−10を乾燥し、アゾ化合物(1)−12を得た。アゾ化合物(1)−12、10gをエチレングリコール100mLに懸濁させ、内温120℃まで昇温した後、同温度にて2時間攪拌した。内温30℃まで冷却した後、固体を濾別し、δ型結晶形態のアゾ顔料(1)−13を9.1g得た。
得られたアゾ顔料(1)−13の1次粒子の長軸方向の長さは、約0.2μmであった。
得られたアゾ顔料(1)−13のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図7に示す。
【0178】
[実施例7]
アゾ化合物(1)−12、10gをエチレングリコール50mL、水50mLの混合溶媒に懸濁させ、内温95℃まで昇温した後、同温度にて2時間攪拌した。内温30℃まで冷却した後、固体を濾別し、δ型結晶形態のアゾ顔料(1)−14を9.3g得た。
得られたアゾ顔料(1)−14の1次粒子の長軸方向の長さは、約0.2μmであった。
得られたアゾ顔料(1)−14のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図8に示す。
【0179】
[実施例8]
アゾ化合物(1)−12、10gをエチレングリコール5mL、水95mLの混合溶媒に懸濁させ、内温85℃まで昇温した後、同温度にて2時間攪拌した。内温30℃まで冷却した後、固体を濾別し、δ型結晶形態のアゾ顔料(1)−15を9.5g得た。
得られたアゾ顔料(1)−15の1次粒子の長軸方向の長さは、約0.15μmであった。
得られたアゾ顔料(1)−15のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図9に示す。
【0180】
[実施例9]
アゾ化合物(1)−12、10gをイソプロパノール40mL、水60mLの混合溶媒に懸濁させ、内温80℃まで昇温した後、同温度にて2時間攪拌した。内温30℃まで冷却した後、固体を濾別し、δ型結晶形態のアゾ顔料(1)−16を8.2g得た。
得られたアゾ顔料(1)−16の1次粒子の長軸方向の長さは、約5μmであった。
得られたアゾ顔料(1)−16のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図10に示す。
【0181】
[実施例10]
アゾ化合物(1)−12、10gをイソブチルアルコール100mL、水10mLに懸濁させ、内温80℃まで昇温した後、同温度にて2時間攪拌した。内温30℃まで冷却した後、固体を濾別し、δ型結晶形態のアゾ顔料(1)−17を7.9g得た。
得られたアゾ顔料(1)−17の1次粒子の長軸方向の長さは、約15μmであった。
得られたアゾ顔料(1)−17のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図11に示す。
【0182】
[実施例11]
アゾ化合物(1)−12、10gを酢酸ブチル100mLに懸濁させ、内温90℃まで昇温した後、同温度にて2時間攪拌した。内温30℃まで冷却した後、固体を濾別し、δ型結晶形態のアゾ顔料(1)−18を8.5g得た。
得られたアゾ顔料(1)−18の1次粒子の長軸方向の長さは、約20μmであった。
得られたアゾ顔料(1)−18のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図12に示す。
【0183】
[実施例11−2]
アゾ化合物(1)−12、10gをメタノール100mLに懸濁させた。室温にて2時間攪拌した後に固体を濾別しζ型結晶形態のアゾ顔料(1)−101を9.4g得た。
得られたアゾ顔料(1)−101の1次粒子の長軸方向の長さは約10μmであった。
得られたアゾ顔料(1)−101のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が6.5°、6.7°、9.1°、及び21.3°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図13に示す。
【0184】
[実施例12]
式(2)の化合物11.5gを酢酸50gに懸濁させ、内温が20℃〜30℃になるようにニトロシル硫酸の43%硫酸溶液16.2gを滴下した。内温20℃にて1時間攪拌した後、尿素0.1gを添加してジアゾニウム塩溶液を得た。別に式(3)の化合物10gを酢酸100mLに溶解させ、上述のジアゾニウム塩溶液に内温が20℃〜25℃になるように滴下した。内温20℃にて1時間攪拌し、アゾ化合物(1)の均一反応液を得た。別に水150gを用意し、内温20℃〜25℃にて上述のアゾ化合物(1)の均一反応液を滴下した。析出している固体を濾別した後、水で十分にかけ洗いを行い、非晶質なアゾ化合物(1)−19を得た。
得られたアゾ化合物(1)−19の1次粒子の長軸方向の長さは、約0.2μmであった。
CuKα特性X線回折図を図14に示す。
アゾ化合物(1)−19のX線回折の測定を上記の条件により行ったところ、特徴的なX線回折ピークが見られなかった。
【0185】
得られた非晶質なアゾ化合物(1)−19を水120mL、エチレングリコール180mLの混合溶媒に懸濁させた。28%アンモニア水溶液でpHを6.28に調整した後、内温85℃まで昇温し、同温度にて2時間攪拌した。内温30℃まで冷却した後、結晶を濾別し、水で十分にかけ洗いを行い、δ型結晶形態のアゾ顔料(1)−20を19.5g得た。
得られたアゾ顔料(1)−20の1次粒子の長軸方向の長さは、約0.3μmであった。
得られたアゾ顔料(1)−20のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図15に示す。
【0186】
[実施例12−2]
実施例12において、アゾ化合物(1)の均一反応液をエチレングリコール150gに滴下すること以外は実施例12と同様にして、非晶質なアゾ化合物(1)−102を得た。得られたアゾ化合物(1)−102の1次粒子の長軸方向の長さは約0.7μmであった。
アゾ化合物(1)−102のX線回折の測定を上記の条件により行ったところ、特徴的なX線回折ピークが見られなかった。
続いて、実施例12と同様の操作で結晶変換を行い、δ型結晶形態のアゾ顔料(1)−103を19.1g得た。
得られたアゾ顔料(1)−103の1次粒子の長軸方向の長さは、約0.7μmであった。
得られたアゾ顔料(1)−103のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図16に示す。
【0187】
[実施例13]
式(2)の化合物11.5gを酢酸50gに懸濁させ、内温が20℃〜30℃になるようにニトロシル硫酸の43%硫酸溶液16.2gを滴下した。内温20℃にて1時間攪拌した後、尿素0.1gを添加してジアゾニウム塩溶液を得た。別に式(3)の化合物10gを酢酸100mLに溶解させ、上述のジアゾニウム塩溶液に内温が20℃〜25℃になるように滴下した。内温20℃にて1時間攪拌し、アゾ化合物(1)の均一反応液を得た。別に水150gを用意し、内温20℃〜25℃にて上述のアゾ化合物(1)の均一反応液を滴下した。固体(非晶質なアゾ化合物)が析出した懸濁液を同温度にて30分間攪拌した後、エチレングリコール20mLを添加し、内温85℃まで昇温し、同温度にて2時間攪拌した。内温30℃まで冷却した後、析出している結晶を濾別し、水で十分にかけ洗いを行い、δ型結晶形態のアゾ顔料(1)−21を19.9g得た。
得られたアゾ顔料(1)−21の1次粒子の長軸方向の長さは、約0.2μmであった。
得られたアゾ顔料(1)−21のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図17に示す。
【0188】
[実施例14]
式(2)の化合物11.5gを酢酸50gに懸濁させ、内温が20℃〜30℃になるようにニトロシル硫酸の43%硫酸溶液16.2gを滴下した。内温20℃にて1時間攪拌した後、尿素0.1gを添加してジアゾニウム塩溶液を得た。別に式(3)の化合物10gを酢酸100mLに溶解させ、上述のジアゾニウム塩溶液に内温が20℃〜25℃になるように滴下した。内温20℃にて1時間攪拌し、アゾ化合物(1)の均一反応液を得た。別に水150gを用意し、内温20℃〜25℃にて上述のアゾ化合物(1)の均一反応液を滴下した。固体(非晶質なアゾ化合物)が析出した懸濁液を同温度にて30分間攪拌した後、エチレングリコール20mLを添加した。内温30℃以下になるように28%アンモニア水溶液を添加してpHを4.01に調整した後、内温85℃まで昇温し、同温度にて2時間攪拌した。内温30℃まで冷却した後、析出している結晶を濾別し、水で十分にかけ洗いを行い、δ型結晶形態のアゾ顔料(1)−22を19.9g得た。
得られたアゾ顔料(1)−22の1次粒子の長軸方向の長さは、約0.5μmであった。
得られたアゾ顔料(1)−22のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図18に示す。
【0189】
[実施例15]
式(2)の化合物11.5gを酢酸50gに懸濁させ、内温が20℃〜30℃になるようにニトロシル硫酸の43%硫酸溶液16.2gを滴下した。内温20℃にて1時間攪拌した後、尿素0.1gを添加してジアゾニウム塩溶液を得た。別に式(3)の化合物10gを酢酸100mLに溶解させ、上述のジアゾニウム塩溶液に内温が20℃〜25℃になるように滴下した。内温20℃にて1時間攪拌し、アゾ化合物(1)の均一反応液を得た。別に水150gを用意し、内温20℃〜25℃にて上述のアゾ化合物(1)の均一反応液を滴下した。固体(非晶質なアゾ化合物)が析出した懸濁液を同温度にて30分間攪拌した後、エチレングリコール20mLを添加し、内温85℃まで昇温し、同温度にて2時間攪拌した。内温30℃まで冷却した後、内温が30℃以下になるように28%アンモニア水溶液を添加してpHを6.50に調整した。析出している結晶を濾別し、水で十分にかけ洗いを行い、δ型結晶形態のアゾ顔料(1)−23を19.9g得た。
得られたアゾ顔料(1)−23の1次粒子の長軸方向の長さは、約0.4μmであった。
得られたアゾ顔料(1)−23のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図19に示す。
【0190】
[実施例16]
式(2)の化合物11.5gを酢酸50gに懸濁させ、内温が20℃〜30℃になるようにニトロシル硫酸の43%硫酸溶液16.2gを滴下した。内温20℃にて1時間攪拌した後、尿素0.1gを添加してジアゾニウム塩溶液を得た。別に式(3)の化合物10gを酢酸100mLに溶解させ、上述のジアゾニウム塩溶液に内温が20℃〜25℃になるように滴下した。内温20℃にて1時間攪拌し、アゾ化合物(1)の均一反応液を得た。別に水150gを用意し、内温20℃〜25℃にて上述のアゾ化合物(1)の均一反応液を滴下した。固体(非晶質なアゾ化合物)が析出した懸濁液を同温度にて30分間攪拌した後、エチレングリコール20mLを添加し、内温85℃まで昇温し、同温度にて2時間攪拌した。内温30℃まで冷却した後、析出している結晶を濾別し、水で十分にかけ洗いを行い、1%炭酸水素ナトリウム水溶液100mLでかけ洗いを行った。さらに水で十分にかけ洗いを行い、δ型結晶形態のアゾ顔料(1)−24を19.8g得た。
得られたアゾ顔料(1)−24の1次粒子の長軸方向の長さは、約0.15μmであった。
得られたアゾ顔料(1)−24のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図20に示す。
【0191】
[実施例17]
実施例6で得られたアゾ化合物(1)−12、10gをエチレングリコール100mLに懸濁させ、室温にて24時間攪拌した。固体を濾別し、δ型結晶形態のアゾ顔料(1)−25を9.5g得た。
得られたアゾ顔料(1)−25の1次粒子の長軸方向の長さは、約0.2μmであった。
得られたアゾ顔料(1)−25のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図21に示す。
【0192】
[実施例18]
式(2)の化合物11.4gを90%酢酸50gに懸濁させ、内温が10〜20℃となるまで冷却した。この温度範囲で、硫酸4g、続いてニトロシル硫酸の43%硫酸溶液16.2gを滴下した。内温10〜20℃にて1時間攪拌した後、尿素0.1gを添加してジアゾニウム塩溶液を得た。このジアゾニウム塩溶液の中に、式(3)の化合物10gを内温が10〜20℃になるように分割添加した。内温10〜20℃にて1時間攪拌し、アゾ化合物(1)の均一反応液を得た。別に水150gを用意し、内温20℃〜25℃にて上述のアゾ化合物(1)の均一反応液を滴下した。固体(非晶質なアゾ化合物)が析出した懸濁液を同温度にて15分間攪拌した後、エチレングリコール20gを添加した。続いて、内温70℃まで昇温し、同温度にて1時間攪拌して結晶変換を行った。内温30℃まで冷却した後、析出している結晶を濾別し、水およびメタノールで十分にかけ洗いを行い、δ型結晶形態のアゾ顔料(1)−27を19.9g得た。
得られたアゾ顔料(1)−27の1次粒子の長軸方向の長さは、約0.5μmであった。
得られたアゾ顔料(1)−27のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図22に示す。
[実施例18−2]
式(2)の化合物11.4gを90%酢酸50gに懸濁させ、内温が10〜20℃となるまで冷却した。この温度範囲で、硫酸4g、続いてニトロシル硫酸の43%硫酸溶液16.2gを滴下した。内温10〜20℃にて1時間攪拌した後、尿素0.1gを添加してジアゾニウム塩溶液を得た。このジアゾニウム塩溶液の中に、式(3)の化合物10gを内温が10〜20℃になるように分割添加した。内温10〜20℃にて1時間攪拌し、アゾ化合物(1)の均一反応液を得た。別に水150gを用意し、内温40℃〜45℃にて上述のアゾ化合物(1)の均一反応液を滴下した。固体(非晶質なアゾ化合物)が析出した懸濁液を同温度にて15分間攪拌した後、エチレングリコール20gを添加した。同温度にて16時間攪拌して結晶変換を行った。内温30℃まで冷却した後、析出している結晶を濾別し、水およびメタノールで十分にかけ洗いを行い、δ型結晶形態のアゾ顔料(1)−104を19.9g得た。
得られたアゾ顔料(1)−104の1次粒子の長軸方向の長さは、約0.4μmであった。
得られたアゾ顔料(1)−104のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図23に示す。
【0193】
[比較例1]
実施例3で得られたアゾ顔料(1)−5 5gをエチレングリコール50mLに懸濁させ、内温120℃まで昇温した後、同温度にて2時間攪拌した。内温30℃まで冷却した後、析出している固体を濾別し、δ型結晶形態のアゾ顔料(1)−26を3.9g得た。
得られたアゾ顔料(1)−26を光学顕微鏡(ニコン(株)製:ECLIPSE LV150)で目視にて観察したところ、1次粒子の長軸方向の長さは、約80μmであった。
得られたアゾ顔料(1)−26のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図24に示す。
【0194】
【化12】
【0195】
【化13】
【0196】
【化14】
【0197】
<顔料分散物の作製>
【0198】
上記実施例1で合成したδ型結晶形態アゾ顔料(1)−2を2.5部、オレイン酸ナトリウム0.5部、グリセリン5部、水42部を混合し、直径0.1mmのジルコニアビーズ100部とともに遊星型ボールミルを用いて毎分300回転、3時間分散を行った。分散終了後、ジルコニアビーズを分離し、黄色の顔料分散物1を得た。
上記実施例2〜17で合成したδ型結晶形態アゾ顔料についてもそれぞれ同様の操作を行い、黄色の顔料分散物2〜17を得た。
【0199】
比較のために、上記比較例1で合成したδ型結晶形態アゾ顔料(1)−26を2.5部、オレイン酸ナトリウム0.5部、グリセリン5部、水42部を混合し、直径0.1mmのジルコニアビーズ100部とともに遊星型ボールミルを用いて毎分300回転、8時間分散を行った。分散終了後、ジルコニアビーズを分離し、黄色の比較顔料分散物1を得た。
さらに上記実施例1にて合成した非晶質なアゾ化合物(1)−1を2.5部、オレイン酸ナトリウム0.5部、グリセリン5部、水42部を混合し、直径0.1mmのジルコニアビーズ100部とともに遊星型ボールミルを用いて毎分300回転、7時間分散を行った。分散終了後、ジルコニアビーズを分離し、黄色の比較顔料分散物2を得た。
【0200】
顔料分散物中の顔料の体積平均粒子径を、日機装(株)製Nanotrac150(UPA−EX150)を用いて測定した。測定結果を表1に示す。
【0201】
【表1】
【0202】
以上の結果より、本発明の製造方法により得られる特定の結晶構造を有するアゾ顔料は、より短時間で体積平均粒子径(nm)の小さな分散物を調製することが可能であることが分かった。
【0203】
〔比表面積が50m2/g以上の顔料を含有する顔料分散物の製造方法と性能評価〕
<非晶質なアゾ顔料の結晶変換工程を含まないアゾ顔料(1)の製造>
【0204】
[合成例1]α型結晶形態アゾ顔料(1)−29の製造
前記式(2)で表される化合物67.5gをリン酸530mLに溶解させ、氷冷して内温を3℃まで冷却した。内温が4℃以下になるように15分間かけて亜硝酸ナトリウム26.9gを分割して添加した。添加終了後、同温度にて50分間攪拌し、尿素18.6gを分割して添加し、ジアゾニウム塩溶液を得た。別に前記式(3)で表される化合物47.9gをメタノール1680mLに加え、還流下完溶させた。氷冷して内温を0℃まで冷却し、ここに上述のジアゾニウム塩溶液を内温が10℃以下になるように30分かけて添加した。内温5℃にて1時間30分攪拌した後、水1.6Lに添加し、30分間攪拌した。析出している結晶を濾別し、水1Lでかけ洗った。得られた結晶を水2.5Lに懸濁させ、28%アンモニア水を加えてpHが6.1になるように調製した。結晶を濾別し、水で十分にかけ洗いを行い、γ型結晶形態アゾ顔料を得た。得られた結晶をアセトン1.5Lに懸濁させ、昇温して還流下2時間攪拌した。結晶を熱時にて濾別して、アセトンで十分かけ洗いを行い、β型結晶形態アゾ顔料(1)−28を103.5g得た。
得られたβ型結晶形態アゾ顔料(1)−28を60℃24時間乾燥させ、α型結晶形態アゾ顔料(1)−29を92.8g(収率88.8%)得た。
【0205】
[合成例2]ε型結晶形態アゾ顔料(1)−31の製造
50gの酢酸、8gの硫酸からなる混合溶媒にニトロシル硫酸の43%硫酸溶液16.2gを20分かけて加えた。この溶液を3℃まで冷却し、前記式(2)で表される化合物11.55gを粉末で添加しジアゾ化反応を行った。同温度で1時間攪拌後、余分のニトロシル硫酸を尿素0.094gで失活させ、ジアゾニウム化合物調製液を得た。
この上述のジアゾニウム化合物調製液の中に前記式(3)で表される化合物10gを粉体にて5℃以下で分割添加した。添加終了後、20℃へ昇温し2時間反応させてアゾ化合物溶解液を得た。なお、カップリング反応中、顔料の析出は見られず、アゾ化合物溶解液は、得られたアゾ化合物を完全に溶解している状態であった。
メタノール150mLからなる貧溶媒を用意し5℃、200rpmで攪拌した。この貧溶媒中に上述のアゾ化合物溶解液を滴下した。
そのまま15分攪拌後、生成した結晶を濾別し、前記式(1)で表されるζ型結晶形態のアゾ顔料(1)−30を得た。
【0206】
得られたζ型結晶形態のアゾ顔料(1)−30の結晶を水200mLに懸濁させ、28%アンモニア水を加えてpHを6.0に調製した。析出している結晶(ζ型)を濾別し、水で十分にかけ洗いを行い、60℃で24時間乾燥させた。得られた結晶(ζ型)をアセトン200mLに懸濁させ、昇温して還流下2時間攪拌した。室温まで冷却し、結晶を濾別し、前記式(1)で表されるη型結晶形態のアゾ顔料を得た。得られた結晶をアセトンで十分にかけ洗いを行い、60℃にて24時間乾燥させて前記式(1)で表されるε型結晶形態アゾ顔料(1)−31を18.9g得た。
【0207】
[合成例3]δ型結晶形態のアゾ顔料(1)−32の合成
前記式(2)の化合物34.6gを酢酸150gに懸濁し、硫酸24gを内温が20℃〜30℃になるように滴下した。さらに内温が20℃〜30℃になるように43%ニトロシル硫酸の硫酸溶液48.6gを滴下し、内温20℃にて1時間攪拌後、尿素0.28gを添加してジアゾニウム塩溶液を得た。このジアゾニウム塩溶液に前記式(3)の化合物30gを内温が20℃〜30℃になるように分割して添加し、内温25℃にて1時間攪拌し、アゾ化合物の均一反応液を得た。別に360gのメタノールを内温25℃にて用意し、上述のアゾ化合物の均一反応液を内温が30℃以下になるように添加し、10分間攪拌した後、析出している固体を濾別した。300mLのメタノールでかけ洗いした後、水900mLに懸濁させ、28%アンモニウム水溶液を添加してpHを6.0に調整した。析出している固体を濾別し、前記式(1)で表されるアゾ顔料を得た。
得られたアゾ顔料の1次粒子の長軸方向の長さは、約2μmであった。
得られたアゾ顔料のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が6.5°、6.7°、9.1°および21.3°に特徴的なX線回折ピークを示し、ζ型結晶形態を有することが判明した。
上記で得られたζ型結晶形態アゾ顔料 5gをエチレングリコール50mLに懸濁させ、内温120℃まで昇温した後、同温度にて2時間攪拌した。内温30℃まで冷却した後、析出している固体を濾別し、前記式(1)で表されるδ型結晶形態のアゾ顔料(1)−32を3.9g得た。
得られたδ型結晶形態のアゾ顔料(1)−32を光学顕微鏡(ニコン(株)製:ECLIPSE LV150)で目視にて観察したところ、1次粒子の長軸方向の長さは、約80μmであった。
得られたδ型結晶形態のアゾ顔料(1)−32のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
【0208】
<ミリング>
〔実施例19及び比較例2〕
実施例19のアゾ顔料は、実施例1で得られたアゾ顔料(1)−2をミリングすることなく用いた。同様に、比較例2のアゾ顔料は、合成例2の途中で生成するアゾ顔料(1)−30をミリングすることなくそのまま用いた。
【0209】
その他の実施例20及び比較例3〜6のアゾ顔料は、下記のとおりミリングしたものを用いた。
〔実施例20〕
以下の組成となるように、スーパーミキサーに粗アゾ顔料及び食塩を投入して混合した。スーパーミキサーを回転させながらジエチレングリコールを少しずつ添加して混合物(以下、「予備混合物」ということがある)を調製した。
・実施例1で得られたアゾ顔料(1)−2 ・・・150g
・食塩(ナイカイ塩業(株)製 ナクルUM−05) ・・・1500g
・ジエチレングリコール ・・・300g
続いて、連続式1軸混練機(浅田鉄工(株)製、ミラクルKCK−L)の磨砕部および押し出し部の5箇所の温度を15〜20℃に、軸回転数50rpmに設定し、上記で得られた予備混合物を投入し、混練物を得た。この時、電流値(負荷)は約4Aで、吐出量は50g/分、吐出物の温度は16℃であった。
こうして得られた混練物を1%希塩酸5000gへ投入して攪拌処理を行った後、濾過および十分に水洗をして食塩およびジエチレングリコールを除去し、乾燥した。得られたδ型結晶形態アゾ顔料(1)−2−AのX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.6°、10.7°、17.3°、18.9°、20.0°、及び26.7°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図25に示す。
【0210】
〔比較例3〜6〕
・比較例3:合成例3のアゾ顔料(1)−32
ブラッグ角(2θ±0.2°)が4.8°、7.1°、9.6°、10.7°、17.3°、18.9°、20.0°、及び26.7°に特徴的なX線回折ピークを示すδ型アゾ顔料(1)−32−Aを得た。
CuKα特性X線回折図を図26に示す。
【0211】
・比較例4:合成例1のアゾ顔料(1)−29
ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示すδ型アゾ顔料(1)−29−Aを得た。
CuKα特性X線回折図を図27に示す。
【0212】
・比較例5:合成例2のアゾ顔料(1)−31
ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示すδ型アゾ顔料(1)−31−Aを得た。
CuKα特性X線回折図を図28に示す。
【0213】
・比較例6:合成例2の途中で生成するアゾ顔料(1)−30
ブラッグ角(2θ±0.2°)が6.6°、9.2°、10.3°および21.4°に特徴的なX線回折ピークを示すζ型アゾ顔料(1)−30−Aを得た。
CuKα特性X線回折図を図29に示す。
【0214】
〔実施例21〕
実施例20において、食塩の使用量を750gに変えたこと以外は実施例6と同様にして、δ型結晶アゾ顔料(1)−2−Bを得た。δ型結晶形態アゾ顔料(1)−2−BのX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.1°、9.5°、10.7°、17.3°、18.9°、20.0°、及び26.7°に特徴的なX線回折ピークを示した。
【0215】
〔実施例22〕
実施例20において、ジエチレングリコールの使用量を400gに変えたこと以外は実施例6と同様にして、δ型結晶アゾ顔料(1)−2−Cを得た。δ型結晶形態アゾ顔料(1)−2−CのX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.1°、9.5°、10.7°、17.3°、18.9°、20.0°、及び26.7°に特徴的なX線回折ピークを示した。
【0216】
<BET比表面積の測定>
あらかじめ80℃において真空乾燥した顔料0.1gを試料セルに加え、比表面積測定装置「MONOSORB MS−17」(ユアサアイオニクス(株)製)を用いて測定を行った。なお、測定には、He:N2=7:3の混合ガスを用いた。
【0217】
<顔料分散物の製造>
上記実施例20で合成したδ型結晶形態アゾ顔料(1)−2−Aを10g、オレイン酸ナトリウム5g、グリセリン10g、水75gを混合し、直径0.1mmのジルコニアビーズ375gとともにサンドグラインダミルTSG1(アイメックス社製)を用いて毎分1500回転、45℃で分散を行った。体積平均粒子径Mvが100nm以下となるまで分散を行った後、ジルコニアビーズを分離し、顔料分散物を得た。
実施例19及び比較例2のミリングしなかったアゾ顔料、及び、比較例3〜6で合成した各アゾ顔料についても同様の方法により、顔料分散物を得た。
実施例20及び比較例2〜6の顔料分散物の下記評価は、実施例19の顔料分散物との相対的な評価により行った。
【0218】
<分散性評価>
分散性は、上記顔料分散物の製造において、体積平均粒子径Mvが100nm以下となるまでの時間で評価した(日機装(株)製Nanotrac150(UPA−EX150)を用いて測定)。ミリング前のδ型結晶系アゾ顔料より更に優れるものをA、ミリング前のδ型結晶系アゾ顔料と同程度のものをB、ミリング前のδ型結晶系アゾ顔料より劣るものをCとした。結果を表2に示す。
【0219】
<着色力評価>
上記実施例及び比較例で得られた顔料分散物を水で10質量倍に希釈してからNo.3のバーコーターを用いてエプソン社製フォトマット紙に塗布した。得られた塗布物の画像濃度を反射濃度計(X−Rite社製X−Rite938)を用いて測定し、「着色力(OD:Optical Density)」を以下の基準で評価した。ODがミリング前のδ型結晶系アゾ顔料より更に優れるものをA、ミリング前のδ型結晶系アゾ顔料と同程度のものをB、ミリング前のδ型結晶系アゾ顔料より劣るものをCとした。結果を表2に示す。
【0220】
【表2】
【0221】
[合成例4]
窒素雰囲気下、ジプロピレングリコール58.7gを内温70℃に昇温し、ここにメタクリル酸を10.8g、メタクリル酸ベンジルを39.4g、V−601を1.2g、ジプロピレングリコールを58.7gを混合した溶液を3時間かけて滴下した。同温度にてさらに1時間攪拌した後、V−601(重合開始剤:和光純薬社製)を0.6g添加し、同温度にてさらに2時間攪拌した。同温度にて50%水酸化カリウム水溶液を11.3g滴下した後、同温度で1時間攪拌した。室温にまで冷却し、メタクリル酸ベンジル(66.7モル%)、メタクリル酸(33.3モル%)の共重合体(Mw=83,000、酸価140mgKOH)のジプロピレングリコール溶液を得た。
【0222】
[合成例5]
合成例4のV−601の量を1.2gから2.5gに増量し、さらに温度を86℃にし、同様の操作を行うことで、メタクリル酸ベンジル(66.7モル%)、メタクリル酸(33.3モル%)の共重合体(Mw=25,000、酸価128mgKOH)のジプロピレングリコール溶液を得た。
【0223】
[合成例6]
窒素雰囲気下、ジプロピレングリコール41.1gを内温70℃に昇温し、ここにメタクリル酸を9.6g、メタクリル酸メチルを16.8g、メタクリル酸2−エチルヘキシルを8.9g、V-601を2.5g、ジプロピレングリコールを41.1gを混合した溶液を3時間かけて滴下した。他の操作は合成例4と同様に行うことで、メタクリル酸メチル(47.8モル%)、メタクリル酸(31.8モル%)、メタクリル酸2−エチルヘキシル(20.4モル%)の共重合体(Mw=83,000、酸価154mgKOH)のジプロピレングリコール溶液を得た。
【0224】
〔実施例23〕
上記実施例20で合成したδ型結晶形態アゾ顔料(1)−2−Aを20gに合成例4で得られた分散剤(メタクリル酸ベンジル(66.7モル%)、メタクリル酸(33.3モル%)の共重合体のジプロピレングリコール溶液、Mw=83,000、酸価140mgKOH)32.2g(固形分含率30.8%、固形分9.9g)、水58gを混合し、直径0.1mmφのジルコニアビーズ375gとともにサンドグラインダミルTSG1(アイメックス社製)を用いて毎分1500回転、45℃で分散を3時間行った後、ジルコニアビーズを分離し、水で洗浄して、顔料濃度15.4重量%の粗顔料分散液(1)を99g得た(平均体積粒子径Mv=91.7nm)。
得られた粗顔料分散液(1)99gにデナコールEX−321(ナガセケムテックス株式会社製)を0.43g、6.18%のホウ酸水溶液3.02g、水40gを加え、70℃にて5時間攪拌した。反応終了後、室温まで冷却し、孔径1.0μmのフィルターを通して粗大粒子を除去した後、遠心分離機でさらに粗大粒子を沈降させた(7000rpm、10分間)。沈降した固体を除去した後、分画分子量50,000のフィルターを用いて、水で十分に洗浄し、顔料濃度10.8%の顔料分散液(1)を121g得た。
【0225】
〔実施例24〕
上記実施例6で合成したδ型結晶形態アゾ顔料(1)−3−Aを20gに合成例5で得られた分散剤(メタクリル酸ベンジル(66.7モル%)、メタクリル酸(33.3モル%)の共重合体のジプロピレングリコール溶液、Mw=25,000、酸価128mgKOH)28.6g(固形分含率35%、固形分10.0g)、水58gを混合し、直径0.1mmφのジルコニアビーズ375gとともにサンドグラインダミルTSG1(アイメックス社製)を用いて毎分1500回転、45℃で分散を2時間行った後、ジルコニアビーズを分離し、水で洗浄して、顔料濃度14.6重量%の粗顔料分散液(2)を109g得た(平均体積粒子径Mv=89.6nm)。
得られた粗顔料分散液(2)109gにデナコールEX−321(ナガセケムテックス株式会社製)を0.17g、6.18%のホウ酸水溶液1.19g、水50gを加え、70℃にて5時間攪拌した。反応終了後、室温まで冷却し、孔径1.0μmのフィルターを通して粗大粒子を除去した後、遠心分離機でさらに粗大粒子を沈降させた(7000rpm、10分間)。沈降した固体を除去した後、分画分子量50,000のフィルターを用いて、水で十分に洗浄し、顔料濃度9.7%の顔料分散液(2)を149g得た。
【0226】
〔実施例25〕
上記実施例6で合成したδ型結晶形態アゾ顔料(1)−3−Aを20gに合成例6で得られた分散剤(メタクリル酸メチル(47.8モル%)、メタクリル酸(31.8モル%)、メタクリル酸2−エチルヘキシル(20.4モル%)の共重合体のジプロピレングリコール溶液、Mw=83,000、酸価154mgKOH)28.4g(固形分含率35.2%、固形分10.0g)、水62gを混合し、直径0.1mmφのジルコニアビーズ375gとともにサンドグラインダミルTSG1(アイメックス社製)を用いて毎分1500回転、45℃で分散を3時間行った後、ジルコニアビーズを分離し、水で洗浄して、顔料濃度13.9重量%の粗顔料分散液(3)を112g得た(平均体積粒子径Mv=96.7nm)。
得られた粗顔料分散液(3)112gにデナコールEX−321(ナガセケムテックス株式会社製)を0.77g、6.18%のホウ酸水溶液5.4g、水30gを加え、70℃にて5時間攪拌した。反応終了後、室温まで冷却し、孔径1.0μmのフィルターを通して粗大粒子を除去した後、遠心分離機でさらに粗大粒子を沈降させた(7000rpm、10分間)。沈降した固体を除去した後、分画分子量50,000のフィルターを用いて、水で十分に洗浄し、顔料濃度10.4%の顔料分散液(3)を127g得た。
【0227】
〔実施例26〕
上記合成例2で合成したδ型結晶形態アゾ顔料(1)−2を20gに合成例4で得られた分散剤(メタクリル酸ベンジル(66.7モル%)、メタクリル酸(33.3モル%)の共重合体のジプロピレングリコール溶液、Mw=83,000、酸価140mgKOH)32.4g(固形分含率30.8%、固形分10.0g)、水46gを混合し、直径0.1mmφのジルコニアビーズ375gとともにサンドグラインダミルTSG1(アイメックス社製)を用いて毎分1500回転、45℃で分散を9時間行った後、ジルコニアビーズを分離し、水で洗浄して、顔料濃度15.3重量%の粗顔料分散液(4)を98g得た(平均体積粒子径Mv=98.2nm)。
得られた粗顔料分散液(4)98gにデナコールEX−321(ナガセケムテックス株式会社製)を0.75g、6.18%のホウ酸水溶液5.3g、水50gを加え、70℃にて5時間攪拌した。反応終了後、室温まで冷却し、孔径1.0μmのフィルターを通して粗大粒子を除去した後、遠心分離機でさらに粗大粒子を沈降させた(7000rpm、10分間)。沈降した固体を除去した後、分画分子量50,000のフィルターを用いて、水で十分に洗浄し、顔料濃度9.4%の顔料分散液(4)を145g得た。
【0228】
〔比較例7〕
P.Y.128(Cromophtal Yellow 8GN、チバ社製)を20gに合成例5で得られた分散剤(メタクリル酸ベンジル(66.7モル%)、メタクリル酸(33.3モル%)の共重合体のジプロピレングリコール溶液、Mw=83,000、酸価140mgKOH)32.4g(固形分含率30.8%、固形分10.0g)、水46gを混合し、直径0.1mmφのジルコニアビーズ375gとともにサンドグラインダミルTSG1(アイメックス社製)を用いて毎分1500回転、45℃で分散を6時間行った後、ジルコニアビーズを分離し、水で洗浄して、顔料濃度16.3重量%の比較粗顔料分散液(1)を81g得た(平均体積粒子径Mv=93.4nm)。
得られた比較粗顔料分散液(1)81gにデナコールEX−321(ナガセケムテックス株式会社製)を0.66g、6.18%のホウ酸水溶液4.7g、水45gを加え、70℃にて5時間攪拌した。反応終了後、室温まで冷却し、孔径1.0μmのフィルターを通して粗大粒子を除去した後、遠心分離機でさらに粗大粒子を沈降させた(7000rpm、10分間)。沈降した固体を除去した後、分画分子量50,000のフィルターを用いて、水で十分に洗浄し、顔料濃度10.2%の比較顔料分散液(4)を115g得た。
【0229】
<着色力評価>
上記実施例23〜26で得られた顔料分散物を水で10質量倍に希釈してからNo.3のバーコーターを用いてエプソン社製フォトマット紙に塗布した。得られた塗布物の画像濃度を反射濃度計(X−Rite社製X−Rite938)を用いて測定し、「着色力(OD:Optical Density)」を以下の基準で評価した。ODが実施例20より更に優れるものをA、実施例20と同程度のものをB、実施例20より劣るが、実施例19より優れるものをC、実施例19と同程度のものをD、実施例19より劣るものをEとした。結果を表3に示す。
【0230】
【表3】
【0231】
〔実施例27〕
実施例23で得られた顔料分散液(1)を固形分で5質量%、グリセリン10質量%、2−ピロリドン5質量%、1,2−ヘキサンジオール2質量%、トリエチレングリコールモノブチルエーテル2質量%、プロピレングリコール0.5質量%、サーフィノール465を1質量%、イオン交換水74.5質量%になるように各成分を加えて、得られた混合液を1.2μmのフィルター(アセチルセルロース膜、外径:25mm、富士フイルム(株)社製)を取り付けた容量20mLのシリンジで濾過し、粗大粒子を除去することにより顔料インク(1)を得た。
【0232】
〔実施例28、29〕
上記実施例27における顔料分散液(1)を、実施例24で得られた顔料分散液(2)及び実施例25で得られた顔料分散液(3)に代えて、それぞれ顔料インク(2)及び顔料インク(3)を得た。
【0233】
〔比較例8〕
セイコーエプソン(株)社製ICY−42を用いて、下記評価を行った。
【0234】
<評価>
インク液をセイコーエプソン(株)社製インクジェットプリンターPX−V630のイエローインク液のカートリッジに装てんし、受像シートはセイコーエプソン(株)社製写真用紙クリスピア<高光沢>にカラー設定:色補正なし、印刷品質:フォトで、イエローのO.D.が1.0となるように印画したものおよびベタ印画したものを作製し、耐光性および濃度を評価した。
【0235】
<着色力評価>
ベタ印画したものの画像濃度を反射濃度計(X−Rite社製X−Rite938)を用いて測定し、「着色力(OD:Optical Density)」を以下の基準で評価した。ODが比較例8より優れるものをA、比較例8と同程度のものをB、比較例8より劣るものをCとした。結果を表4に示す。
【0236】
<耐光性評価>
O.D.が1.0となるように印画したものを、フェードメーターを用いてキセノン光(99000lux;TACフィルター存在下)を28日間照射し、キセノン照射前後の画像濃度を反射濃度計を用いて測定し、色素残存率[(照射後濃度/照射前濃度)×100%]が90%以上の場合をA、90%未満の場合をBとして評価した。結果を表4に示す。
【0237】
【表4】
【技術分野】
【0001】
本発明は、アゾ顔料の製造方法に関する。
【背景技術】
【0002】
アゾ化合物の合成法については古くから種々の方法が知られており、酸化反応による合成、還元反応による合成、置換反応による合成、付加反応による合成、縮合反応による合成、その他の合成法などがあった(たとえば非特許文献1参照)。しかしながらアゾ色素化合物の工業的製造法として利用されているのは、原料の入手性、コスト、収率などの点からジアゾニウム化合物と、アニリン、フェノール類などのカップリング成分とをアゾカップリング反応させて合成する方法がほとんどであり、また、この方法もジアゾニウム化合物の爆発の危険性があったり、ジアゾニウム化合物やカップリング成分の種類によっては低収率であったりするなどの欠点を有していた。特に複素環ジアゾニウム化合物はジアゾニウム化合物が不安定である場合が多く、一般性の高い合成法は知られていない。
【0003】
また、色材として広く利用されている有機顔料には、特に液晶ディスプレー用カラーフィルターやインクジェット用インク等の用途において、鮮明性や透明性などをより向上させることが求められている。鮮明性や透明性を向上させるためには、有機顔料を微細に分散させることが効果的なことが知られている。有機顔料を微細分散させるには、それに適した分散剤や分散機械を用いることが必要であるが、さらには有機顔料自身が微細(微粒子)であるということも大前提として必要である。有機顔料が粗大な粒子であると、分散剤や分散方法を改良しても優れた微細分散体を得ることは困難である。したがって、顔料の製造にあっては、純度や収率等の通常の要求項目に加えて微粒子として製造することが求められている。
【0004】
有機顔料微粒子の製造方法としては、例えばアゾ顔料のように、合成時に適切な反応条件を選択することにより、微細で整粒された粒子を得ることができるものがある。その他、銅フタロシアニングリーン顔料のように、合成時に生成する極めて微細で凝集した粒子を、後工程で粒子成長、整粒させることにより顔料化するもの、銅フタロシアニンブルー顔料のように、合成時に生成する粗大で不揃いな粒子を後工程で微細化し、整粒させることにより顔料化を行うものもある。例えば、ジケトピロロピロール顔料は、一般的には、琥珀酸ジエステルと芳香族ニトリルとを有機溶媒中で反応させることにより粗製顔料として合成される(例えば、特許文献1参照)。そして、粗製ジケトピロロピロール顔料は、水又は有機溶剤中で熱処理し、次に湿式摩砕のごとき粉末化を行うことにより、使用に適する形態にされる(例えば、特許文献2参照)。
【0005】
さらに、有機顔料には多形性を示すものがあり、このような顔料は、同一の化学組成を有するにもかかわらず2つ以上の結晶形態をとりうることが知られている。
例えば、C.I.ピグメントレッド254は、α型とβ型の結晶形態が知られている(例えば、特許文献3参照)。また、アゾ顔料であるC.I.ピグメントイエロー181は、数種の結晶形態が知られている(例えば、特許文献4参照)。
上記のように、特定の結晶構造を持つことを特徴とする知見がある一方、非晶質であることに特徴を持たせた顔料も知られており、C.I.ピグメントイエロー181を非晶質とすることで分散性を発揮させるといった例もある(特許文献5)。
【0006】
微細な有機顔料を得る方法として、特許文献6には顔料をアミド系の有機溶媒に溶解した溶液を貧溶媒に注入してナノサイズの微粒子を得る方法が記載されており、貧溶媒種により得られる顔料の結晶形態が異なる旨の記述がある。しかしながら、この方法では顔料溶液の濃度が10mM程度と低く、顔料を得るのに大量の溶媒を使用するため経済的ではない。
【0007】
特許文献7には、特定のモノアゾ顔料を高純度で製造できる方法が記載されている。しかしながら、より不安定な複素環ジアゾニウム化合物にこの方法を適用して高収率及び高純度でアゾ顔料を得ることは一般的に困難である。
【0008】
特許文献8には、ピラゾールのジアゾニウム化合物を用いたアゾ化合物を、高収率及び高純度で得る製造方法が記載されている。この方法は染料の製造には有効であるが、この方法で製造される化合物の粒子の大きさについては記載がない。
【0009】
特許文献9にも、ピラゾールのジアゾニウム化合物を用いたアゾ化合物を、高収率及び高純度で得る製造方法が記載されている。この方法により均一な微粒子が得られる旨の記載があるが、分散性に更なる改良の余地を残していた。なお、顔料の結晶形態に関する記述はない。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】特開昭58−210084号公報
【特許文献2】特開平5−222314号公報
【特許文献3】特開平8−48908号公報
【特許文献4】米国特許出願公開第2008/0058531号明細書
【特許文献5】特開2003−261792号公報
【特許文献6】特開2004−91560号公報
【特許文献7】特開2008−63524号公報
【特許文献8】特開2007−217681号公報
【特許文献9】特開2011−74375号公報
【非特許文献】
【0011】
【非特許文献1】新実験化学講座(丸善株式会社)14−III巻、1516〜1534頁
【発明の概要】
【発明が解決しようとする課題】
【0012】
本発明は特定の置換基を有するピラゾール環をアゾ基及びトリアジン環を介して連結したアゾ顔料の製造方法に関するが、上記のように、アゾ顔料の性能と製造性の両立には検討の余地が残っていた。
本発明は、分散性が良好なアゾ顔料微粒子を高効率かつ低コストで製造することのできるアゾ顔料の製造方法を提供することを目的とする。
【課題を解決するための手段】
【0013】
本発明者等は前記した実情に鑑みて鋭意検討した結果、非晶質な化合物を結晶変換することで分散性が良好なアゾ顔料微粒子を得ることができ、さらにはジアゾニウム塩とカップリング成分とをアゾカップリング反応させたときに、反応液中においてアゾ化合物が完全に析出していない状態(アゾ化合物の一部が反応液に溶解している状態)とし、この反応液をアゾ化合物の貧溶媒に注入し、非晶質な化合物を得ることで、上記課題が達成できることを見出し、本発明を完成した。
【0014】
即ち、本発明は、以下のとおりである。
【0015】
〔1〕
非晶質な下記式(1)で表されるアゾ化合物、その塩、水和物又は溶媒和物を結晶変換することを特徴とする下記式(1)で表されるアゾ顔料の製造方法。
【0016】
【化1】
【0017】
〔2〕
非晶質の前記式(1)で表されるアゾ化合物、その塩、水和物又は溶媒和物を溶媒中で加熱攪拌することにより結晶変換することを特徴とする〔1〕に記載のアゾ顔料の製造方法。
〔3〕
非晶質な前記式(1)で表されるアゾ化合物を、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が4.8°、7.2°及び9.7°に特徴的X線回折ピークを有する結晶形に結晶変換することを特徴とする〔1〕又は〔2〕に記載のアゾ顔料の製造方法。
〔4〕
非晶質な前記式(1)で表されるアゾ化合物を、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が4.8°、7.2°、9.7°、20.1°、及び26.8°に特徴的X線回折ピークを有する結晶形に結晶変換することを特徴とする〔1〕〜〔3〕のいずれか一項に記載のアゾ顔料の製造方法。
〔5〕
非晶質な前記式(1)で表されるアゾ化合物を、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的X線回折ピークを有する結晶形に結晶変換することを特徴とする〔1〕〜〔4〕のいずれか一項に記載のアゾ顔料の製造方法。
〔6〕
(i)非晶質な前記式(1)で表されるアゾ化合物を、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が4.8°、7.2°及び9.7°に特徴的X線回折ピークを有する結晶形に結晶変換する工程、及び、(ii)前記工程(i)で得たアゾ顔料と、水溶性無機塩と、水溶性有機溶剤とを含む混合物を混練して、窒素吸着法によるBET比表面積を50m2/g以上の結晶にする工程、を含むことを特徴とする、〔3〕〜〔5〕のいずれか一項に記載のアゾ顔料、又はその互変異性体の製造方法。
〔7〕
(a)ジアゾ化剤と下記式(2)で表されるアミノ化合物とを混合させる工程、(b)前記工程(a)で得た反応生成物と下記式(3)で表されるカップリング成分とを混合することにより反応を行い、該反応により生成した下記式(1)で表されるアゾ化合物の少なくとも一部が溶解した溶液を得る工程、(c)前記工程(b)で得た溶液と、前記アゾ化合物に対する貧溶媒とを混合して、下記式(1)で表される非晶質なアゾ化合物を晶析させる工程、を含むことを特徴とする〔1〕〜〔6〕のいずれか一項に記載のアゾ顔料の製造方法。
【0018】
【化2】
【0019】
【化3】
【0020】
【化4】
【0021】
〔8〕
前記工程(b)において、得られた溶液が酸性溶液であることを特徴とする〔7〕に記載のアゾ顔料の製造方法。
〔9〕
前記酸性溶液が、酢酸及び硫酸の少なくとも一方を含むことを特徴とする〔8〕に記載のアゾ顔料の製造方法。
〔10〕
前記工程(b)において、得られた溶液が、カップリング反応により生成した前記式(1)で表されるアゾ化合物が完全に溶解した溶液であることを特徴とする〔7〕〜〔9〕のいずれか一項に記載のアゾ顔料の製造方法。
〔11〕
前記(c)工程において、前記工程(b)で得られた前記式(1)で表されるアゾ化合物の溶解度が1g/L以下である貧溶媒と混合することを特徴とする〔7〕〜〔10〕のいずれか一項に記載のアゾ顔料の製造方法。
〔12〕
前記貧溶媒が、水及び、炭素数1〜3のアルコール、炭素数1〜6のグリコールからなる群から選ばれる1種以上の溶媒を含むことを特徴とする〔7〕〜〔11〕のいずれか一項に記載のアゾ顔料の製造方法。
〔13〕
〔1〕〜〔12〕のいずれか一項に記載の製造方法により得られるアゾ顔料。
〔14〕
〔13〕に記載のアゾ顔料、分散剤、及び水を含む顔料分散物。
〔15〕
分散剤が水溶性高分子であることを特徴とする〔14〕に記載の水系顔料分散物。
〔16〕
前記水溶性高分子分散剤が少なくとも1つのカルボキシ基を有し、少なくとも50mgKOH/g以上の酸価を有することを特徴とする〔15〕に記載の水系顔料分散物。
〔17〕
前記水系顔料分散物が、架橋剤により架橋されていることを特徴とする〔15〕又は〔16〕に記載の水系顔料分散物。
【発明の効果】
【0022】
本発明によれば、易分散なアゾ顔料微粒子を、高効率(純度および生産性)かつ低コストで製造することのできるアゾ顔料の製造方法を提供することができる。
【図面の簡単な説明】
【0023】
【図1】実施例1に従って合成されたδ型結晶形態のアゾ顔料(1)−2のX線回折の図である。
【図2】実施例2に従って合成されたδ型結晶形態のアゾ顔料(1)−4のX線回折の図である。
【図3】実施例3に従って合成された中間生成物であるζ型結晶形態のアゾ顔料(1)−5のX線回折の図である。
【図4】実施例3に従って合成されたδ型結晶形態のアゾ顔料(1)−7のX線回折の図である。
【図5】実施例4に従って合成されたδ型結晶形態のアゾ顔料(1)−9のX線回折の図である。
【図6】実施例5に従って合成されたδ型結晶形態のアゾ顔料(1)−11のX線回折の図である。
【図7】実施例6に従って合成されたδ型結晶形態のアゾ顔料(1)−13のX線回折の図である。
【図8】実施例7に従って合成されたδ型結晶形態のアゾ顔料(1)−14のX線回折の図である。
【図9】実施例8に従って合成されたδ型結晶形態のアゾ顔料(1)−15のX線回折の図である。
【図10】実施例9に従って合成されたδ型結晶形態のアゾ顔料(1)−16のX線回折の図である。
【図11】実施例10に従って合成されたδ型結晶形態のアゾ顔料(1)−17のX線回折の図である。
【図12】実施例11に従って合成されたδ型結晶形態のアゾ顔料(1)−18のX線回折の図である。
【図13】実施例11−2に従って合成されたζ型結晶形態のアゾ顔料(1)−101のX線回折の図である。
【図14】実施例12に従って合成された中間生成物である非晶質なアゾ顔料(1)−19のX線回折の図である。
【図15】実施例12に従って合成されたδ型結晶形態のアゾ顔料(1)−20のX線回折の図である。
【図16】実施例12−2に従って合成されたδ型結晶形態のアゾ顔料(1)−103のX線回折の図である。
【図17】実施例13に従って合成されたδ型結晶形態のアゾ顔料(1)−21のX線回折の図である。
【図18】実施例14に従って合成されたδ型結晶形態のアゾ顔料(1)−22のX線回折の図である。
【図19】実施例15に従って合成されたδ型結晶形態のアゾ顔料(1)−23のX線回折の図である。
【図20】実施例16に従って合成されたδ型結晶形態のアゾ顔料(1)−24のX線回折の図である。
【図21】実施例17に従って合成されたδ型結晶形態のアゾ顔料(1)−25のX線回折の図である。
【図22】実施例18に従って合成されたδ型結晶形態のアゾ顔料(1)−27のX線回折の図である。
【図23】実施例18−2に従って合成されたδ型結晶形態のアゾ顔料(1)−104のX線回折の図である。
【図24】比較例1に従って合成されたδ型結晶形態のアゾ顔料(1)−26のX線回折の図である。
【図25】実施例20に従って合成されたδ型結晶形態のアゾ顔料(1)−2−AのX線回折の図である。
【図26】比較例3に従って合成されたδ型結晶形態のアゾ顔料(1)−32−AのX線回折の図である。
【図27】比較例4に従って合成されたδ型結晶形態のアゾ顔料(1)−29−AのX線回折の図である。
【図28】比較例5に従って合成されたδ型結晶形態のアゾ顔料(1)−31−AのX線回折の図である。
【図29】比較例6に従って合成されたζ型結晶形態のアゾ顔料(1)−30−AのX線回折の図である。
【0024】
本発明の製造方法により製造された顔料の分散性が良好となる理由に関して、以下に推察を述べる。X線回折において、特徴的X線回折ピークを示す結晶は、分子が一定の規則性を持って配列し、分子間の相互作用が強固に結ばれている。その結果、溶媒に対する溶解性が低くなる。そのため例えば溶媒中で、このような結晶から別の結晶形に変換すると、少しずつ溶解しつつ結晶変換が進行するため、核となる結晶が少なく、結晶は大きく成長してしまう。一方、本発明のようにX線回折において、特徴的なX線回折ピークを示さない非晶質な固体は、分子間の相互作用が弱く、溶媒に対する溶解性が高い。そのため、例えば溶媒中で結晶変換を行うと、非晶質は固体からは多くが溶け出すが、結晶は溶解性が低いため、即座に過飽和状態になり、核となる結晶が多数析出し、結晶は成長しにくい。結果、微細な顔料が得られ、分散性が良好となると考えている。
【発明を実施するための形態】
【0025】
以下、本発明について詳細に説明する。
【0026】
本発明において非晶質とは、結晶のような長距離秩序は無いが、短距離秩序はある物質の状態を示し、X線回折において、特徴的X線回折ピークを示さないことを表す。
【0027】
本明細書においては以下、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が4.8°、7.2°及び9.7°に特徴的X線回折ピークを有する下記式(1)で表されるアゾ顔料をδ型結晶形態アゾ顔料と称する。
また、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が6.5°、7.1°及び21.8°に特徴的X線回折ピークを有する式(1)で表されるアゾ顔料をα型結晶形態アゾ顔料、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が6.3°、6.4°及び22.3°に特徴的X線回折ピークを有する式(1)で表されるアゾ顔料をβ型結晶形態アゾ顔料、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が4.9°、8.8°及び13.1°に特徴的X線回折ピークを有する式(1)で表されるアゾ顔料をε型結晶形態アゾ顔料、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が6.6°、9.2°及び21.5°に特徴的X線回折ピークを有する式(1)で表されるアゾ顔料をζ型結晶形態アゾ顔料と称する。
【0028】
【化5】
【0029】
本発明において、上記式(1)で表されるアゾ化合物、及びアゾ顔料のX線回折の測定は、日本工業規格JISK0131(X線回析分析通則)に準じて、例えば、粉末X線回折測定装置RINT2500(株式会社リガク製)にて行うことができる。
【0030】
図2にX線回折図を示して、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が4.8°、7.2°及び9.7°に特徴的X線回折ピークを有する式(1)で表されるアゾ顔料について更に詳細に説明する。
【0031】
〔アゾ顔料〕
本発明のアゾ顔料の製造方法によって製造されるアゾ顔料は下記式(1)で表される。
【0032】
以下、本発明における式(1)で表されるアゾ化合物及びアゾ顔料について詳細に説明する。なお当該アゾ顔料は、その塩、水和物又は溶媒和物であってもよい。
【0033】
【化6】
【0034】
式(1)で表される顔料において、結晶中に水分子を含む水和物、あるいは、溶媒(例えば、メタノール,エタノール,2−プロパノール,t−ブチルアルコール等のアルコール類や、アセトン、メチルエチルケトン等のケトン類や、アセトニトリル、ジメチルスルホキシド,ジメチルホルムアミド,ジメチルアセトアミド,N−メチルピロリドン、トルエン等の非プロトン性溶媒など)を含む溶媒和物であっても良い。
【0035】
また、式(1)で示される顔料に関しては、その互変異性体(例えば、アゾ−ヒドラゾンの互変異性体や式(4)で表されるような幾何異性体)も、本発明においては、これらの一般式に含まれるものとする。
式(4):
【0036】
【化7】
【0037】
〔アゾ顔料の製造方法〕
以下に、本発明の製造方法に関して詳細に説明する。本発明のアゾ顔料の製造方法は、
非晶質な下記式(1)で表されるアゾ化合物、その塩、水和物又は溶媒和物を結晶変換する下記式(1)で表されるアゾ顔料の製造方法である。
【0038】
【化8】
【0039】
本発明における結晶変換とは、結晶形態を変換することを表し、非晶質である上記式(1)で表されるアゾ化合物、その塩、水和物又は溶媒和物を、特定の結晶形態を有するアゾ顔料に変換することを言う。結晶変換の方法としては、ソルベントソルトミリング、ソルトミリング、ドライミリング、ソルベントミリング、アシッドペースティング等の磨砕処理、溶媒加熱処理が挙げられ、好ましくは溶媒加熱処理である。
【0040】
(溶媒加熱処理)
本発明において溶媒加熱処理とは、具体的には、非晶質な式(1)で表されるアゾ化合物、その塩、水和物又は溶媒和物を溶媒中で加熱撹拌することをいう。
溶媒加熱処理によって、効率よく結晶変換をすることができる。例えば、非晶質なアゾ化合物の溶媒和物を加熱攪拌することでδ型結晶形態のアゾ顔料を得ることができる。
【0041】
本発明において、式(1)で表されるアゾ化合物をCuKα特性X線回折におけるブラッグ角(2θ±0.2°)が4.8°、7.2°及び9.7°に特徴的なX線回折ピークを有する結晶形態に結晶変換することが好ましい。
上記のような特徴的X線回折ピークを有するδ型結晶形態アゾ顔料とすることにより分散性が向上する、すなわち、短時間で目標の粒子径まで分散できるようになる。
CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が4.8°、7.2°及び9.7°に特徴的なX線回折ピークを有する結晶形態は、さらに4.8°、7.2°、9.7°、20.1°、及び26.8°に特徴的なX線回折ピークを有する結晶形態がより好ましい。その中でも特に、4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを有する結晶形態が最も好ましい。
【0042】
本発明の結晶変換に用いることのできる溶媒としては、水、有機酸、無機酸、有機溶媒を用いることができるが、水および有機溶媒が好ましい。また、結晶変換をする際に結晶成長を抑制させる点から、結晶変換した後の式(1)で表されるアゾ顔料の溶解性が低い溶媒が好ましい。より好ましい溶媒としては、水、メタノール、エタノール、イソプロパノール、イソブタノール、エチレングリコール、ジエチレングリコール、ジエチレングリコールジエチルエーテル、ジエチレングリコールモノメチルエーテル、ジプロピレングリコール、酢酸、プロピオン酸、硫酸、またはそれらの混合溶媒が挙げられ、エチレングリコール、水、酢酸、硫酸、あるいはそれらの混合溶媒である場合が更に好ましく、エチレングリコールであることが最も好ましい。
【0043】
溶媒加熱処理に使用する溶媒の量は、式(1)で表されるアゾ顔料に対して1〜100倍であることが好ましく、5〜50倍であることがさらに好ましく、8〜30倍であることがより好ましい。1倍以上であれば、攪拌性を確保できるため好ましい。また、100倍以下であれば、生産性が高くなり、経済的なため好ましい。
【0044】
溶媒加熱処理における加熱攪拌の温度は、所望する顔料の一次粒子径の大きさによって異なるが、15〜150℃が好ましく、20〜120℃であることがより好ましく、20〜100℃がさらに好ましい。15℃以上であれば、結晶変換が起こるために長時間を要することなく、効率的である。一方、150℃以下であれば、アゾ顔料(1)の一部が分解するのを抑制できるため好ましい。
【0045】
結晶変換のための撹拌時間は特に制限はないが、5〜1500分が好ましく、10〜600分が更に好ましく、30〜300分がより好ましい。5分以上であれば、部分的に非晶質な箇所が残存するのを抑制できるため好ましい。一方、1500分以下であれば、効率的であり好ましい。
【0046】
以下に、結晶変換工程に供する非晶質な下記式(1)で表されるアゾ化合物の製造方法に関して詳細に説明する。
本発明の結晶変換に用いる式(1)で表されるアゾ化合物の製造方法は、(a)ジアゾ化剤と式(2)で表されるアミノ化合物とを混合させる工程、(b)前記工程(a)で得た反応生成物と式(3)で表されるカップリング成分とを混合することにより反応を行い、該反応により生成した下記式(1)で表されるアゾ化合物の少なくとも一部が溶解した溶液を得る工程、(c)前記工程(b)で得た溶液と、前記アゾ化合物に対する貧溶媒とを混合して、下記一般式(1)で表される非晶質なアゾ化合物を晶析させる工程、を含むことが好ましい。
【0047】
【化9】
【0048】
【化10】
【0049】
【化11】
【0050】
本発明に係わる工程(a)について詳細を説明する。
工程(a)では、ジアゾ化剤とアミノ化合物とを混合させることで、アミノ化合物とジアゾ化剤との反応によりジアゾニウム化合物が誘導される。この反応は酸を含む媒質中で行うことが好ましい。本明細書では、このジアゾニウム化合物を含む液を「ジアゾニウム化合物調製液」と呼ぶ。アミノ化合物と酸とジアゾ化剤の混合の方法に特に限定はないが、アミノ化合物と酸の溶液中にジアゾ化剤を添加することが好ましい。工程(a)におけるジアゾ化剤とは、アミノ化合物をジアゾニウム化合物に誘導するために使用されるものであり、そのような作用を持つものであれば限定はされない。ジアゾ化剤として代表的なものには、亜硝酸エステル類(例えば亜硝酸イソペンチルが挙げられる)、亜硝酸塩(例えば亜硝酸ナトリウムや亜硝酸カリウムが挙げられる)、亜硝酸イソアミル、ニトロシル硫酸が挙げられ、更に好ましくは亜硝酸ナトリウム、亜硝酸カリウム、ニトロシル硫酸であり、その中でも、ジアゾニウム化合物を安定かつ効率的に調製できる観点から、ニトロシル硫酸が特に好ましい。
【0051】
工程(a)で使用する酸とは、式(2)で表されるアミノ化合物を完溶させないまでも、わずかでも溶解できる酸を意味し、好ましくはアミノ化合物を完溶させる酸である。酸には無機酸及び有機酸が使用でき、無機酸としては塩酸、リン酸、硫酸が挙げられ、好ましくはリン酸、硫酸であり、更に好ましくは硫酸である。有機酸には蟻酸、酢酸、プロピオン酸、メタンスルホン酸が挙げられ、好ましくは酢酸、プロピオン酸、メタンスルホン酸であり、更に好ましくは酢酸、プロピオン酸である。また、これらの酸は単独で用いても良いし、混合して用いても良い。混合酸としては、リン酸/酢酸、硫酸/酢酸、メタンスルホン酸/酢酸、酢酸/プロピオン酸が挙げられ、好ましくは、リン酸/酢酸、硫酸/酢酸、硫酸/酢酸/プロピオン酸、酢酸/プロピオン酸であり、その中でも、硫酸/酢酸、酢酸/プロピオン酸が特に好ましい。これら混合酸の質量比は1/(0.1〜20)が好ましく、1/(0.5〜10)がより好ましく、更に好ましくは1/(1〜10)である。
【0052】
工程(a)における、アミノ化合物に対する酸の添加量は質量比で1〜100倍であり、より好ましくは2〜50倍であり、3〜25倍が更に好ましい。質量比が1倍以上であると、攪拌性が良化し、より確実にジアゾニウム化合物を誘導できる。一方、質量比が100倍以下になると生産性が向上に経済的となる。
また、工程(a)における、アミノ化合物に対するジアゾ化剤の添加量は、モル比で1.0〜20倍であり、より好ましくは1.0〜10倍であり、1.0〜5倍が更に好ましい。ジアゾ化剤がアミノ化合物に対してモル比で1倍以上であることにより、ジアゾニウム化合物をより確実に誘導でき、20倍以下であることにより、副反応によりジアゾニウム化合物が分解することを抑制できる。
【0053】
工程(a)のジアゾ化剤とアミノ化合物の混合では、50℃以下で実施されることが好ましく、40℃以下で実施されることがより好ましく、更に好ましくは30℃以下で実施することが望ましい。50℃以上におけるジアゾ液の調製ではジアゾ化剤の分解が懸念される。ジアゾニウム化合物へ誘導する攪拌時間は0.3〜10時間が好ましく、0.5〜5時間がより好ましく、更に好ましくは1〜3時間である。上記攪拌時間が0.3時間以上であることにより、ジアゾニウム化合物に完全に誘導しやすく、10時間以下であることにより、ジアゾニウム化合物の分解が生じにくい。また、混合には通常の攪拌機が用いられ、特に限定はない。製造設備に依存することはあるが、好ましい攪拌の回転数は、30〜300rpmが好ましく、40〜200rpmがより好ましく、更に好ましくは50〜200rpmである。攪拌速度が回転数で30rpm以上であることにより、ジアゾニウム化合物調製液の攪拌効率が良好となるため、所望の反応の進行を確実に実施できる。
【0054】
工程(a)で混合することのできる溶媒は、誘導されるジアゾニウム化合物が分解を受けなければ特に限定はない。混合可能な溶媒として例えば、ヘキサン、ベンゼン、トルエン等の炭化水素系溶媒、ジエチルエーテル、テトラヒドロフラン等のエーテル系溶媒、アセトン、メチルエチルケトン等のケトン系溶媒、ジメチルホルムアミド、ジメチルアセトアミド、ピロリドン、N−メチル−2−ピロリドン等のアミド系溶媒、他ジメチルスルホキシド、スルホラン、アセトニトリル、水が挙げられる。
【0055】
工程(a)におけるジアゾニウム化合物調製液の好ましいpHは、7以下が好ましく、5以下がより好ましく、3以下が更に好ましい。工程(a)におけるジアゾニウム化合物調製液のpHが7超過になると、誘導されるジアゾニウム化合物の分解が懸念される。
【0056】
次に、本発明に係わる工程(b)について詳細を説明する。
工程(b)は、前記工程(a)で得た反応生成物とカップリング成分とを混合することにより反応を行い、該反応により生成した式(1)で表されるアゾ化合物の少なくとも一部が溶解した溶液を得る工程である。
本明細書では、このアゾ化合物の少なくとも一部が溶解した溶液を「アゾ化合物溶解液」と呼ぶ。
【0057】
アゾ化合物溶解液の調製方法としては、(i)工程(a)で得た反応生成物とカップリング成分とを混合することによりカップリング反応を行い、反応の結果、析出した式(1)で表されるアゾ顔料を、溶剤に溶解させて得る方法、及び、(ii)上記カップリング反応によって得られる式(1)で表される化合物の少なくとも一部が反応液に溶解するように、該カップリング反応を実施し、その反応液を、そのまま、アゾ化合物溶解液とする方法、又は、このようにして得られたアゾ化合物溶解液を下記に詳述する工程(c)に適用することにより得られた(晶析された)アゾ顔料を、更に溶剤に溶解させて得る方法、が挙げられる。
【0058】
上記形態(i)及び(ii)のいずれにおいても、工程(a)で得たジアゾニウム化合物調製液とカップリング成分との混合の方法に特に制限はないが、該カップリング成分を溶媒に一部または全部溶解させて添加すること、あるいは溶媒を用いずに固体で添加することが好ましく、工程(a)で得たジアゾニウム化合物調製液の中に、カップリング成分の溶液を添加すること、あるいは工程(a)で得たジアゾニウム化合物調製液の中に、カップリング成分を固体で添加することが更に好ましい。
【0059】
また、工程(b)におけるカップリング成分に対する前記工程(a)で得たジアゾニウム化合物調製液中のジアゾニウム化合物の量は、カップリング成分のカップリング位に対し0.8〜3当量が好ましく、より好ましくはカップリング位に対し0.9〜2当量であり、更に好ましくはカップリング位に対し0.95〜1.5当量である。0.8当量以上であることにより、未反応のカップリング位をもつカップリング成分の残存を抑制でき、また、3当量以下であることにより、未反応のジアゾニウム化合物の残存を抑制できるため、より経済的である。
【0060】
なお、上記形態(ii)においては、工程(b)において一般式(1)で表されるアゾ化合物の少なくとも一部が溶解しているため、カップリング反応がよりスムーズに進行してより高純度のアゾ化合物を製造することができる。この理由は以下のように推測される。前記式(3)はカップリング位が2個以上あるため、例えば1個のカップリング位のみが反応した反応中間体を経由する。この反応中間体が反応系で析出してしまうと、2個目以降のカップリング反応の反応速度が遅くなる。一方、ジアゾニウム化合物は不安定であるため、長時間経過すると分解が起こる懸念がある。したがって、カップリング反応は早く進行させてやることが重要であり、工程(b)において析出物を生成させない上記形態(ii)の製造方法は、結果として、高純度の顔料を製造するのに、より好適である。
【0061】
工程(b)においては溶媒を使用せずにカップリング成分を添加しても良いが、溶媒と混合して添加しても良いが、溶媒を使用せずに添加することが好ましい。工程(b)においてカップリング成分に溶媒を使用する場合、特に限定はされないが、上記形態(ii)となるように、すなわち、反応後に生成した前記一般式(1)で表されるアゾ化合物の少なくとも一部が溶解した溶液が得られるような溶媒であることが好ましい。
【0062】
上記形態(i)の場合、すなわち、顔料を析出させる場合、溶媒の例としては、水、有機酸、無機酸、有機溶媒を用いることができるが、特に水、有機溶媒が好ましく、反応時に液体分離現象を起こさず、溶媒と均一な溶液を呈する溶媒が好ましい。例えば、水、メタノール、エタノール、プロパノール、イソプロパノール、ブタノール、t−ブチルアルコール、アミルアルコール等のアルコール性有機溶媒、アセトン、メチルエチルケトン等のケトン系有機溶媒、エチレングリコール、ジエチレングリコール、トリエチレングリコール、プロピレングリコール、ジプロピレングリコール、1,3−プロパンジオール等のジオール系有機溶媒、エチレングリコールモノメチルエーテル、エチレングリコールモノエチルエーテル、エチレングリコールジエチルエーテル等のエーテル系有機溶媒、テトラヒドロフラン、ジオキサン、アセトニトリル等が挙げられる、これらの溶媒は2種類以上の混合液であってもよい。
【0063】
好ましくは、極性パラメータ(ET)の値が40以上の有機溶媒である。なかでも溶媒分子中に水酸基を2個以上有するグリコール系の溶媒、あるいは炭素原子数が3個以下のアルコール系溶媒、総炭素数5以下のケトン系溶媒、好ましくは炭素原子数が2以下のアルコール溶媒(例えば、メタノール、エチレングリコール)、総炭素数4以下のケトン系溶媒(例えばアセトン、メチルエチルケトン)が好ましい。またこれらの混合溶媒も含まれる。
【0064】
また、上記形態(ii)の場合、すなわち、式(1)で表される化合物の少なくとも一部が反応液に溶解するようにカップリング反応を行う場合、溶媒の例としては、水、メタノール、イソプロパノール、エチレングリコール等のアルコール系溶媒、アセトン、メチルエチルケトン等のケトン系溶媒、酢酸、プロピオン酸、メタンスルホン酸等の有機酸溶媒、硫酸、塩酸、リン酸等の無機酸溶媒、ジメチルホルムアミド、ジメチルアセトアミド、ピロリドン、N−メチル−2−ピロリドン等のアミド系溶媒、他ジメチルスルホキシド、スルホラン、アセトニトリル、が挙げられる。中でも、好ましくは、アセトン、メチルエチルケトン等のケトン系溶媒、酢酸、プロピオン酸、メタンスルホン酸等の有機酸溶媒、硫酸、塩酸、リン酸等の無機酸溶媒であり、更に好ましくは、有機酸又は無機酸の酸性溶媒であり、最も好ましくは、酢酸、メタンスルホン酸、リン酸、硫酸である。また、上記で示した溶媒の混合溶媒も好適である。
特に上記形態(ii)の場合、工程(b)においては、カップリング成分を酸性溶媒に溶解又は懸濁させた酸性溶液と、工程(a)で得た反応生成物とを混合する、あるいはカップリング成分を、溶媒を使用せずに、工程(a)で得た反応生成物に添加するのが好ましい。とりわけ、酸性溶媒は、酢酸及び硫酸の少なくとも一方を含む溶媒であることが好ましい。
【0065】
上記形態(i)及び(ii)のいずれにおいても、カップリング成分に対する好ましい溶媒の添加量は、質量比で0.5〜200倍が好ましく、1〜100倍がより好ましく、1〜50倍が更に好ましい。カップリング成分に対する好ましい溶媒量の添加量として、質量比で0.5倍未満ではカップリング成分と溶媒の製造機における攪拌が困難になり、所望の反応が進行しない。また、200倍超過では不経済となる。
【0066】
アゾ化合物溶解液の調製方法が、上記形態(i)である場合、あるいは、上記形態(ii)であって、かつ、式(1)で表される化合物の少なくとも一部が溶解されたカップリング反応液を工程(c)に適用して得られるアゾ顔料を、更に溶剤に溶解させてアゾ化合物溶解液を調製する場合において、得られたアゾ顔料を溶解するための溶剤としては、アゾ顔料の少なくとも一部を溶解できれば特に限定されないが、上記形態(ii)において好ましいとして前掲した溶媒の例を同様に挙げることができる。
【0067】
上記形態(i)、(ii)のいずれの工程をとるにせよ、工程(b)において最終的に得られるアゾ化合物溶解液としては、酸性溶液であることが好ましく、とりわけ、酢酸及び硫酸の少なくとも一方を含む溶液であることが好ましい。
【0068】
工程(b)において得られるアゾ化合物溶解液としては、工程(b)によって生成したアゾ化合物の全量(アゾ化合物溶解液に溶解している式(1)で表されるアゾ化合物と、アゾ化合物溶解液から析出した式(1)で表されるアゾ顔料との総和)に対する、アゾ化合物溶解液に溶解している式(1)で表されるアゾ化合物の割合が50質量%以上であることが好ましく、75質量%以上であることが好ましく、90質量%以上であることが好ましく、100質量%(工程(b)によって生成したアゾ化合物が反応液に完全に溶解している状態)であることが最も好ましく、これにより、顔料の粒子径をより低下できる傾向となる。
【0069】
工程(b)における工程(a)のジアゾニウム化合物調製液とカップリング成分の混合温度は50℃以下で実施されることが好ましく、30℃以下で実施されることがより好ましく、更に好ましくは25℃以下で実施することが望ましい。50℃超過では工程(a)で誘導されたジアゾニウム化合物、並びに生成した式(1)で表されるアゾ化合物の分解が懸念される。また、混合には通常の攪拌機が用いられ、特に限定はない。製造設備に依存することはあるが、好ましい攪拌の回転数は、30〜300rpmが好ましく、40〜200rpmがより好ましく、更に好ましくは50〜200rpmである。攪拌速度が回転数で30rpm未満となると混合液の攪拌効率が悪くなり所望の反応の進行が懸念される。工程(b)における攪拌時間は0.1〜10時間が好ましく、0.3〜5時間がより好ましく、更に好ましくは0.3〜3時間である。0.1時間未満では完全に顔料へ誘導することが難しく、10時間超過では式(1)で表されるアゾ化合物の分解が懸念される。
【0070】
次に本発明に係わる工程(c)について詳細を説明する。
工程(c)は、前記工程(b)で得たアゾ化合物溶解液を、該アゾ化合物の溶解性が低い貧溶媒と混合して、顔料を晶析させる工程である。工程(b)で得たアゾ化合物溶解液と貧溶媒との混合の方法に特に制限はないが、工程(b)で得たアゾ化合物溶解液を貧溶媒の中に添加することが好ましく、その際に貧溶媒が攪拌された状態であることが好ましい。
攪拌速度は100〜10000rpmとすることが好ましく、150〜8000rpmとすることがより好ましく、200〜6000rpmとすることが特に好ましい。添加にはポンプ等を用いることもできる。このとき、液中添加でも液外添加でもよいが、液中添加がより好ましい。更に供給管を介してポンプで液中に連続供給することが好ましい。
【0071】
貧溶媒は特に限定されないが、アゾ化合物の溶解度が1g/L以下であることが好ましく、0.1g/L以下であることがより好ましい。この溶解度は酸又はアルカリの存在下で溶解された場合の溶解度であってもよい。工程(b)で得たアゾ化合物溶解液と貧溶媒の相溶性若しくは均一混合性は、該アゾ化合物の良溶媒の貧溶媒に対する溶解量が30質量%以上であることが好ましく、50質量%以上であることがより好ましい。本明細書において、溶解度は25℃における溶解度を指す。
【0072】
貧溶媒としては、例えば、水、塩酸、アンモニア水、水酸化ナトリウム水溶液等の水性溶媒、メタノール、エタノール、イソプロピルアルコール、1−メトキシ−2−プロパノール等のアルコール系溶媒、エチレングリコール、ジエチレングリコール等のグリコール系溶媒、アセトン、メチルエチルケトン、メチルイソブチルケトン、シクロヘキサノン等のケトン化合物溶媒、ジエチルエーテル、テトラヒドロフラン等のエーテル系溶媒、ヘキサン、ベンゼン、トルエン等の炭化水素系溶媒、アセトニトリル等のニトリル系溶媒、ジクロロメタン、トリクロロエチレン等のハロゲン系溶媒、酢酸エチル、乳酸エチル、2−(1−メトキシ)プロピルアセテート等のエステル系溶媒等が挙げられ、好ましくは、水、塩酸、アンモニア水、水酸化ナトリウム水溶液等の水性溶媒、メタノール、エタノール、イソプロピルアルコール、1−メトキシ−2−プロパノール等のアルコール系溶媒、エチレングリコール、ジエチレングリコール等のグリコール系溶媒、アセトン、メチルエチルケトン、メチルイソブチルケトン、シクロヘキサノン等のケトン化合物溶媒であり、さらに好ましくは、水、アンモニア水等の水性溶媒、炭素数1〜3のアルコール溶媒、炭素数1〜6のグリコール系溶媒、である。また、上記で示した溶媒の混合溶媒も好適である。最も好ましくは、水及び炭素数1〜3のアルコール、炭素数1〜6のグリコールからなる群から選択される1種以上の溶媒である。
【0073】
工程(b)で得たアゾ化合物溶解液と貧溶媒との混合比は体積比で1/50〜2/3が好ましく、1/40〜1/2がより好ましく、1/20〜1/2が特に好ましい。体積比で2/3以下であると、顔料の晶析が充分に起こって反応収率が上がり、体積比が1/50以上であると、生産性が向上して経済的となる。
【0074】
工程(b)で得たアゾ化合物溶解液と貧溶媒との混合温度には特に制限はないが、−10〜50℃で実施されることが好ましく、−5〜30℃で実施されることがより好ましく、10〜25℃で実施されることが最も好ましい。
【0075】
工程(b)で得たアゾ化合物溶解液と貧溶媒との混合にあたり、レイノルズ数を調節することにより、析出生成させる有機ナノ粒子の粒子径を制御することができる。ここでレイノルズ数は流体の流れの状態を表す無次元数であり次式で表される。
【0076】
数式(1):Re=ρUL/μ
【0077】
(数式(1)中、Reはレイノルズ数を表し、ρは工程(b)で得たアゾ化合物溶解液の密度[kg/m3]を表し、Uはアゾ化合物溶解液と貧溶媒とが出会うときの相対速度[m/s]を表し、Lはアゾ化合物溶解液と貧溶媒が出会う部分の流路若しくは供給口の等価直径[m]を表し、μはアゾ化合物溶解液の粘性係数[Pa・s]を表す。)
【0078】
等価直径Lとは、任意断面形状の配管の開口径や流路に対し等価な円管を想定するとき、その等価円管の直径をいう。等価直径Lは、配管の断面積をA、配管のぬれぶち長さ(周長)又は流路の外周をpとすると下記数式(2)で表される。
【0079】
数式(2):L=4A/p
【0080】
アゾ化合物溶解液と貧溶媒とが出会うときの相対速度Uは、両者が出会う部分の面に対して垂直方向の相対速度で定義される。すなわち、例えば静止している貧溶媒中にアゾ化合物溶解液を注入して混合する場合は、供給口から注入する速度が相対速度Uに等しくなる。相対速度Uの値は特に制限されないが、例えば、0.5〜100m/sとすることが好ましく、1.0〜50m/sとすることがより好ましい。
【0081】
アゾ化合物溶解液の密度ρは、選択される材料の種類により定められる値であるが、例えば、0.8〜2.0kg/m3であることが実際的である。また、アゾ化合物溶解液の粘性係数μについても用いられる材料や環境温度等により定められる値であるが、例えば、0.5〜100mPa・sであることが好ましく、1.0〜50.0mPa・sであることがより好ましい。
【0082】
レイノルズ数の値は、小さいほど層流を形成しやすく、大きいほど乱流を形成しやすい。例えば、レイノルズ数を60以上で調節して顔料ナノ粒子の粒子径を制御して得ることができ、100以上とすることが好ましく、150以上とすることがより好ましい。レイノルズ数に特に上限はないが、例えば、100000以下の範囲で調節して制御することで所望の平均粒子径を持つ顔料粒子を制御して得ることができる。このとき、上記の範囲内においては、通常レイノルズ数を高めることで、より粒径の小さな顔料粒子を制御して得ることができる。
【0083】
本発明の製造方法により得られる顔料粒子の1次粒子を、透過型顕微鏡で観察した際の長軸方向の長さは、1nm〜1μmであることが好ましく、5〜500nmであることがより好ましく、10〜200nmであることが更に好ましく、10〜100nmであることが特に好ましい。
【0084】
なお、顔料粒子の粒子径に関しては、計測法により数値化して集団の平均の大きさを表現する方法があるが、よく使用されるものとして、分布の最大値を示すモード径、積分分布曲線の中央値に相当するメジアン径、各種の平均径(数平均、長さ平均、面積平均、質量平均、体積平均等)などがあり、本発明においては、特に断りのない限り、平均粒子径とは数平均粒子径をいう。
顔料粒子の粒径の測定方法としては、顕微鏡法、質量法、光散乱法、光遮断法、電気抵抗法、音響法、動的光散乱法が挙げられ、顕微鏡法、動的光散乱法が特に好ましい。顕微鏡法に用いられる顕微鏡としては、例えば、走査型電子顕微鏡、透過型電子顕微鏡などが挙げられる。動的光散乱法による粒子測定装置として、例えば、日機装社製ナノトラックUPA−EX150、大塚電子社製ダイナミック光散乱光度計DLS−7000シリーズなどが挙げられる。
【0085】
上記の顔料粒子の好ましい平均粒子径は、(1)工程(c)における温度、(2)貧溶媒に対するアゾ化合物の溶解度、及び、(3)攪拌速度(あるいは、レイノルズ数)を、適宜、調整することによって、達成される。
【0086】
顔料微粒子を析出させ分散液を調製するにあたり、アゾ化合物溶解液及び貧溶媒の少なくとも一方に分散剤を含有させてもよい。このとき、アゾ化合物溶解液に分散剤を含有させることが好ましい。分散剤は(1)析出した顔料表面に素早く吸着して、微細なナノ粒子を形成し、かつ(2)これらの粒子が再び凝集することを防ぐ作用を有するものである。
分散剤としては、例えば、アニオン性、カチオン性、両イオン性、ノニオン性の低分子又は高分子分散剤を使用することができる。
【0087】
高分子分散剤としては、その質量平均分子量が1000〜500000であることが好ましく、10000〜500000であることがより好ましく、10000〜100000であることが特に好ましい。
具体的には、ポリビニルピロリドン、ポリビニルアルコール、ポリビニルメチルエーテル、ポリエチレングリコール、ポリプロピレングリコール、ポリアクリルアミド、ビニルアルコール−酢酸ビニル共重合体、ポリビニルアルコール−ブブンホルマール化物、ポリビニルアルコール−部分ブチラール化物、ビニルピロリドン−酢酸ビニル共重合体、ポリエチレンオキシド/プロピレンオキシドブロック共重合体、ポリアクリル酸塩、ポリビニル硫酸塩、ポリ(4−ビニルピリジン)塩、ポリアミド、ポリアリルアミン塩、縮合ナフタレンスルホン酸塩、セルロース誘導体、澱粉誘導体などが挙げられる。その他、アルギン酸塩、ゼラチン、アルブミン、カゼイン、アラビアゴム、トンガントゴム、リグニンスルホン酸塩などの天然高分子類も使用できる。なかでも、ポリビニルピロリドンが好ましい。これら高分子化合物は、1種単独であるいは2種以上を組み合わせて用いることができ、また、低分子量の分散剤を組み合わせて用いてもよい。顔料の分散に用いる分散剤に関しては、「顔料分散安定化と表面処理技術・評価」(化学情報協会、2001年12月発行)の29〜46頁に詳しく記載されている。
【0088】
アニオン性分散剤(アニオン性界面活性剤)としては、N−アシル−N−アルキルタウリン塩、脂肪酸塩、アルキル硫酸エステル塩、アルキルベンゼンスルホン酸塩、アルキルナフタレンスルホン酸塩、ジアルキルスルホコハク酸塩、アルキルリン酸エステル塩、ナフタレンスルホン酸ホルマリン縮合物、ポリオキシエチレンアルキル硫酸エステル塩等を挙げることができる。なかでも、N−アシル−N−アルキルタウリン塩が好ましい。N−アシル−N−アルキルタウリン塩としては、特開平3−273067号明細書に記載されているものが好ましい。これらアニオン性分散剤は、単独であるいは2種以上を組み合わせて用いることができる。
【0089】
カチオン性分散剤(カチオン性界面活性剤)には、四級アンモニウム塩、アルコキシル化ポリアミン、脂肪族アミンポリグリコールエーテル、脂肪族アミン、脂肪族アミンと脂肪族アルコールから誘導されるジアミン及びポリアミン、脂肪酸から誘導されるイミダゾリン及びこれらのカチオン性物質の塩が含まれる。これらカチオン性分散剤は、単独であるいは2種以上を組み合わせて用いることができる。
【0090】
両イオン性分散剤は、前記アニオン性分散剤が分子内に有するアニオン基部分とカチオン性分散剤が分子内に有するカチオン基部分をともに分子内に有する分散剤である。
ノニオン性分散剤(ノニオン性界面活性剤)としては、ポリオキシエチレンアルキルエーテル、ポリオキシエチエレンアルキルアリールエーテル、ポリオキシエチレン脂肪酸エステル、ソルビタン脂肪酸エステル、ポリオキシエチレンソルビタン脂肪酸エステル、ポリオキシエチレンアルキルアミン、グリセリン脂肪酸エステルなどを挙げることができる。なかでも、ポリオキシエチレンアルキルアリールエーテルが好ましい。これらノニオン性分散剤は、単独であるいは2種以上を組み合わせて用いることができる。
【0091】
分散剤の含有量は、顔料100質量部に対して0.1〜1000質量部の範囲であることが好ましく、より好ましくは1〜500質量部の範囲であり、更に好ましくは5〜200質量部の範囲である。また、分散剤は、単独で用いても、複数のものを組み合わせて用いてもよい。
【0092】
本発明の顔料の製造方法においては、上記工程(a)〜(c)によって得られる生成物は通常の有機合成反応の後処理方法に従って処理した後、精製してあるいは精製せずに次の結晶変換工程に供することができる。
すなわち、例えば、反応系から遊離したものを精製せずに、あるいは再結晶、造塩等にて精製する操作を単独、あるいは組み合わせて行ない、供することができる。
【0093】
また、反応終了後、反応溶媒を留去して、あるいは留去せずに水、又は氷にあけ、中和してあるいは中和せずに、遊離したものをあるいは有機溶媒/水溶液にて抽出したものを、精製せずにあるいは再結晶、晶析、造塩等にて精製する操作を単独に又は組み合わせて行なった後、供することもできる。
【0094】
工程(c)で得られた非晶質アゾ化合物は、アゾ化合物の懸濁液からアゾ化合物を取り出さずに結晶変換をおこなっても良いし、取り出してから結晶変換を行ってもよい。
【0095】
工程(c)で得られた固体が非晶質ではない場合、アゾ化合物を良溶媒に溶解させて、非晶質な固体が得られる貧溶媒と混合することで非晶質なアゾ化合物を得てもよい。
【0096】
次に、本発明の製造方法により得られる顔料粒子、すなわち、以上の方法により非晶質なアゾ化合物から結晶変換して得られた式(1)で表されるアゾ顔料について述べる。
【0097】
本発明の製造方法により得られる顔料粒子の体積平均粒子径は、1nm〜10μmであることが好ましく、5nm〜5μmであることがより好ましく、10nm〜1μmであることがさらに好ましく、10〜500nmであることが特に好ましい。
【0098】
なお、顔料粒子の体積平均粒子径とは、顔料そのものの粒子径、又は色材に分散剤等の添加物が付着している場合には、添加物が付着した粒子径をいう。本発明において、顔料粒子の体積平均粒子径の測定装置には、ナノトラックUPA粒度分析計(UPA−EX150;日機装社製)を用いることができる。その測定は、顔料分散物3mlを測定セルに入れ、所定の測定方法に従って行うことができる。なお、測定時に入力するパラメーターとしては、粘度にはインク粘度を、分散粒子の密度には顔料の密度を用いる。
【0099】
上記の顔料粒子の好ましい体積平均粒子径は、既述の結晶変換した後の式(1)で表されるアゾ顔料の溶解性が低い溶媒を用いて結晶変換をする際に結晶成長を抑制させることや、結晶変換の温度、時間、溶媒量を、適宜、調整することで達成される。
【0100】
顔料の分散性を更に好ましくし、着色力を更に向上させる観点から、下記式(1)で表されるδ型結晶形態アゾ顔料の窒素吸着法によるBET比表面積は50m2/g以上であることが好ましく、60m2/g以上であることが特に好ましい。
ここで、窒素吸着法によるBET比表面積とは、窒素ガスを粉体粒子に吸着させ、吸着平衡状態における吸着平衡圧を求め、BETの関係式により単分子層吸着量を算出し求められた比表面積をいう。窒素吸着法によるBET比表面積は、例えば、日本工業規格JIS Z8830の付属書2に規定される「1点法による気体吸着量の測定方法」に従って測定することができる。具体的には、比表面積測定装置「MONOSORB MS−17」(ユアサアイオニクス(株)製)等を用いることにより測定することができる。
窒素吸着法によるBET比表面積を上記の範囲とすることにより顔料の一次粒子が十分微細化されるが、微細化された状態においても、式(1)で表されるδ型結晶形態アゾ顔料は耐光性を落とすことなく分散性及び着色力が更に向上する。
窒素吸着法によるBET比表面積が50m2/g以上である下記式(1)で表されるδ型結晶形態アゾ顔料は、好ましくは、後述するソルベントソルトミリングを含む工程により製造することができる。
【0101】
本発明の方法で製造された式(1)で表されるアゾ顔料は、必要に応じて後処理を行ってもよい。この後処理の方法としては、例えば、ソルベントソルトミリング、ソルトミリング、ドライミリング、ソルベントミリング、アシッドペースティング等の摩砕処理、溶媒加熱処理などによる顔料粒子制御工程、樹脂、界面活性剤及び分散剤等による表面処理工程が挙げられる。
【0102】
また、本発明の式(1)で表される化合物は後処理としてソルベントソルトミリングを行うことが好ましい。
【0103】
(ソルベントソルトミリング)
ソルベントソルトミリングとしては、例えば、アゾ顔料(以下、混練磨砕する前のアゾ顔料を「粗アゾ顔料」ということがある。)と、無機塩と、それを溶解しない有機溶剤とを含む混合物を混練機に仕込み、その中で混練磨砕を行うことが挙げられる。上記無機塩としては、水溶性無機塩が好適に使用でき、例えば塩化ナトリウム、塩化カリウム、硫酸ナトリウム、硫酸カリウム等の無機塩を用いることが好ましい。また、前記水溶性無機塩の粒子径としては特に制限はないが、アゾ顔料の2次凝集体の粒子径制御の観点から、水溶性無機塩の粒子径が体積基準のメディアン径で0.5〜50μmであることが好ましく、1〜20μmであることがより好ましく、1〜10μmであることがさらに好ましい。当該無機塩の使用量は、粗アゾ顔料に対して、1〜30質量倍とすることができ、生産性の観点から、3〜20質量倍とするのが好ましく、5〜15質量倍とするのがより好ましい。有機溶剤としては、水溶性有機溶剤が好適に使用でき、混練時の温度上昇により溶剤が蒸発し易い状態になるため、安全性の点から高沸点溶剤が好ましい。このような有機溶剤としては、例えばジエチレングリコール、グリセリン、エチレングリコール、プロピレングリコール、液体ポリエチレングルコール、液体ポリプロピレングリコール、2−(メトキシメトキシ)エタノール、2−ブトキシエタノール、2ー(イソペンチルオキシ)エタノール、2−(ヘキシルオキシ)エタノール、ジエチレングリコールモノメチルエーテル、ジエチレングルコールモノエチルエーテル、ジエチレングリコールモノブチルエーテル、トリエチレングリコール、トリエチレングリコールモノメチルエーテル、1−メトキシ−2−プロパノール、1−エトキシ−2−プロパノール、ジプロピレングリコール、ジプロピレングリコールモノメチルエーテル、ジプロピレングリコールモノメチルエーテル、ジプロピレングリコールまたはこれらの混合物が挙げられる。また、その他の水溶性有機溶剤として、プロピルアルコール、2−ブチルアルコール、tert−ブチルアルコールなどの1価アルコール系溶剤も好適に使用することができる。当該水溶性有機溶剤の使用量は、粗アゾ顔料に対して0.1〜5質量倍が好ましく、さらに好ましくは2〜3質量倍である。混練温度は、20〜130℃が好ましく、40〜110℃が特に好ましい。混練機としては、例えばニーダーやミックスマーラー等を使用することができ、具体的には、ニーダー等のバッチ式混練機、スーパーミキサー((株)カワタ製)やトリミックス((株)井上製作所製)等のバッチ式混練機、連続式1軸混練機KCKミル(浅田鉄工(株)製)等の連続式混練機を用いることができる。
【0104】
前記連続式混練機としては、例えば、その磨砕部分が混練分散に必要な要素である圧縮・せん断・混合(置換)の三つの作用を粗顔料に与えることができる固定ブレードと回転ブレードとを有していることが好ましい。また前記固定ブレードと回転ブレードとの山と山は隙間(ギャップ)を形成し、せん断作用はこのギャップにおいて発生し、また、前記回転ブレードと固定ブレードとの谷間の材料がお互いにキャビティースライスを受けていることが好ましい。
【0105】
前記固定ブレードと回転ブレードの形状には特に制限はないが、それぞれ、菊型、扇型および臼状型の3種類から選ばれることが好ましい。固定ブレードと回転ブレードとを交互に多段に重なっていることが好ましく、これにより各々のブレードの両面にキャビティーを放射状に形成することができる。また、回転ブレードと中間スクリューが回転軸上に交互に組み込まれ、固定ブレードはせん断室シリンダーと交互にタイロッドによってフィードシリンダーに固定されていることが好ましい、これにより固定ブレードと回転ブレードとスクリューの組合せにより混練物を押し出すことができる。
【0106】
さらに連続式混練機は、混合物の投入部、磨砕部および押出部に少なくとも6箇所の温度調節部を有していることが好ましい。これにより、粗アゾ顔料の磨砕工程における温度範囲を幅広く設定できる。
前記磨砕工程における処理温度としては、特に制限はなく、例えば5〜200℃とすることができるが、アゾ顔料粒子の変色、粒度分布の観点から、5〜50℃であることが好ましく、10〜35℃であることがより好ましい。
【0107】
また、前記連続混練機は、粗アゾ顔料、水溶性無機塩および水溶性有機溶剤の混合割合、または軸回転数により、吐出量を変えることが可能であることが好ましい。吐出量を変えることで、アゾ顔料の磨砕粒径を所望の粒径に容易に制御することができる。
【0108】
本発明のアゾ顔料磨砕物の製造方法は、前記磨砕工程に加え、必要に応じてその他の工程を備えることができる。その他の工程としては、例えば、磨砕工程後の前記混合物を、水等に投入して攪拌した後、アゾ顔料磨砕物をろ過等で分離することで水溶性無機塩および水溶性有機溶剤を除去する洗浄工程、前記洗浄工程で得られたアゾ顔料磨砕物を乾燥する乾燥工程等を挙げることができる。
これら洗浄工程および乾燥工程は、いわゆるソルベントソルトミリング法で通常用いられる方法を本発明においても特に制限なく適用することができる。
【0109】
本発明におけるアゾ顔料磨砕物の1次粒子の粒径としては、80nm以下であることが好ましく、30〜50nmであることがより好ましい。また1次粒子が凝集した2次粒子の粒径としては、120nm以下であることが好ましく、60〜100nmであることがより好ましい。
前記アゾ顔料磨砕物の1次粒子および2次粒子の粒径は、透過型電子顕微鏡(TEM)を用いて測定される。
【0110】
本発明は、非晶質な下記式(1)で表されるアゾ化合物、その塩、水和物又は溶媒和物を、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が(a)4.8°、7.2°及び9.7°、(b)4.8°、7.2°、9.7°、20.1°、及び26.8°又は(c)4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的X線回折ピークを有する結晶形に結晶変換することを特徴とする下記式(1)で表されるアゾ顔料の製造方法であって、(i)非晶質な前記式(1)で表されるアゾ化合物を、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が4.8°、7.2°及び9.7°に特徴的X線回折ピークを有する結晶形に結晶変換する工程、及び、(ii)前記工程(i)で得たアゾ顔料と、水溶性無機塩と、水溶性有機溶剤とを含む混合物を混練して、比表面積を50m2/g以上の結晶にする工程、を含むアゾ顔料、又はその互変異性体の製造方法にも関する。
上記水溶性無機塩及び水溶性有機溶剤としては上述のものと同様のものを使用することができ、好ましい範囲もまた同様である。混練に使用する混練機も、上述のものと同様のものを使用することができる。
【0111】
本発明において分散剤は低分子および高分子、さらに水溶性および水不溶性の中から任意で選択できるが、印画物の画質の観点から、高分子が好ましい。さらに、水系で分散を行うので、分散性、分散物安定性の観点から、水溶性であることが好ましい。本発明において分散剤は、水溶性高分子であることが特に好ましい。
また、本発明において「分散剤」は、架橋剤で架橋化された状態のものも意味する。本発明の顔料分散物では、この分散剤が顔料に吸着されたものが望ましい。
【0112】
分散剤は分子中に、電荷反発による効果があるため、分散物の保存安定性の観点から1以上、好ましくは10以上のカルボキシ基を持つことが好ましい。架橋剤が2つのエポキシ基をもつときは、架橋反応によりエポキシ基とカルボキシ基が架橋するために、カルボキシ基が減るので、ポリマーは10以上のカルボキシ基を持つことが好ましい。
ポリマー中にあるカルボキシ基は酸(−COOH)の形でも、塩の形でもよい。塩としては、例えば、金属イオン、アンモニウム、置換アンモニウム、4級アンモニウムまたはピリジニウム塩などが挙げられる。好ましくは、金属イオン、アンモニウムであり、さらに好ましくはカリウムイオン、ナトリウムイオンである。
【0113】
本発明の高分子分散剤はポリウレタン、ポリエステル、ポリビニルを含み、より好ましくはポリウレタン、ポリエステル、ポリビニルであり、最も好ましくはポリビニル(ビニルポリマー)である。本発明では2種類以上のポリマーを組み合わせてもよい。
ポリマーへのカルボキシ基の導入は少なくとも1つのカルボキシ基を含むモノマーの共重合によって得られる。好ましいポリビニルには、モノマーとしてイタコン酸、マレイン酸、フマール酸、クロトン酸、メタクリル酸、アクリル酸、β−カルボキシエチルアクリレートを用いるが、好ましくはメタクリル酸、アクリル酸、β−カルボキシエチルアクリレートを用いる。
【0114】
ポリマー中のカルボキシル基は、まず、架橋剤中の架橋性基と架橋する作用をもつ。架橋性基としては酸無水物、エポキシ基が挙げられ、エポキシ基が特に望ましい。反応性が高いので、温和な条件で架橋することができるからである。更に、未反応カルボキシル基は最終微粒子分散物の沈降及び凝集に対する安定性に有効である。カルボキシル基は極性溶媒特に水溶媒中で安定性基として有効である。カルボキシル基が顔料分散物中で安定性に寄与する唯一の基である場合、全てのカルボキシ基が架橋剤と架橋してしまうと、分散物の安定性が著しく低下する。そのため、架橋反応が完結した後に未反応カルボキシル基が残るように、エポキシ基に対してカルボキシル基のモル過剰とすることが好ましく、エポキシ基に対してカルボキシル基をモル比で30:1〜1.1:1、より好ましくは25:1〜1.1:1、特に好ましくは20:1〜2:1とすることが望ましい。
ポリマーは他の安定性基を持っていてもよい。安定性基の選択及びその量は溶媒の性質に大きく依存する。安定性基は実際、親水性(例えば、極性溶媒)であるか、疎水性(例えば、無極性溶媒)であるかに依存する。
好ましいポリマー分散剤は親水性モノマー、疎水性モノマーの両方から得られる。
【0115】
親水性モノマーはイオン性基または非イオン性基である親水性を含むモノマーである。イオン性基はカチオンでもよいが、好ましくはアニオンである。カチオン性基も、またアニオン性基も分散剤に両性的安定性(amphotericstabilisation)を与える。好ましいアニオン性基はフェノキシ、スルホン酸、硫酸、ホスホン酸、ポリ燐酸、燐酸の基(塩でもよい)である。好ましいカチオン性基は4級アンモニウム、ベンズアルコニウム、グアニジン、ビグアニジン、及びピリジニウムである。これらは水酸化物、硫酸塩、硝酸塩、塩化物、臭化物、沃化物及び弗化物のような塩の形でもよい。好ましい非イオン性基はグルコキシド、糖類、ピロリドン、アクリルアミド、及び特にヒドロキシル基及びポリ(アルキレンオキシド)基であり、より好ましくはポリ(エチレンオキシド)基またはポリ(プロピレンオキシド)基であり、特に−(CH2CH2O)nHまたは−(CH2CH2O)nC1−4−アルキルである。ここで、nは3〜200(好ましくは4〜20)を表す。これ以降、例えばC1−4−の表現は “炭素数1〜4の”を表す。ポリマーは非イオン性基のみを、ポリマー全体で複数の非イオン性基を、また非イオン性基を含む1以上のポリマー鎖を含んでいてもよい。ヒドロキシル基はポリビニルアルコール、ポリヒドロキシル機能のアクリリックス及びセルロースを用いて挿入される。エチレンオキシ基はポリエチレンオキシドのようなポリマー鎖を用いて挿入される。
疎水性モノマーは疎水性基を含むモノマーである。疎水性基を有する代表的なものは3以下で好ましくは0の親水性基を持つ、炭化水素類、フルオロカーボン類、ポリC3−4アルキレンオキシ類及びアルキルシロキサン類である。疎水性基は、好ましくはC3−50鎖であり、また疎水性モノマー中にプロピレンオキシドを側鎖または直鎖に有し得る。
ポリマーは、ホモポリマーでもよいが、好ましくは共重合体(コポリマー)である。ポリマーはランダムポリマー(統計上短いブロックまたはセグメント)を含むが、好ましくは、グラフトポリマー(長いブロックまたはセグメント)を含む。また、ポリマーは交互(alternating)ポリマーでもよい。ポリマーは分岐していてもよいが、好ましくは直鎖である。ポリマーは2以上のセグメント(例えば、ブロック及びグラフト、コポリマー)を持っていてもよいが好ましくはランダムである。
ポリマーが2以上のセグメントを持つ場合の態様では、少なくとも1つのセグメントは疎水性であり、少なくとも1つのセグメントは互いに関連性の親水性であることが好ましい。疎水性及び親水性セグメントをつくる好ましい方法はそれぞれ疎水性及び親水性モノマーの共重合による。ポリマーが少なくとも1つの疎水性セグメント及び少なくとも1つの親水性セグメントもつ場合、カルボキシル基は疎水性セグメントにあっても、また親水性セグメントにあっても、また両方のセグメントにあってもよい。
【0116】
ビニルポリマー(ポリビニル)はどのような適切な手段によって製造されてもよい。ビニルポリマーの好ましい製造方法は、特に(メタ)アクリレートとビニルナフタレン(特にスチレンモノマー)のようなビニルモノマーを用いるフリーラジカル重合である。適切なフリーラジカル重合は懸濁重合、溶液重合、分散重合、乳化重合に限定されないが、好ましくは溶液重合である。
ビニルポリマーは(メタ)アクリレートモノマーを用いる場合が好ましい。
ビニルポリマーは好ましくは共重合体(コポリマー)である。
疎水性モノマー及び親水性モノマーから導かれるコポリビニル分散剤は好ましくは実質的にセグメントをもたない。例えば、コポリビニルポリマーはセグメント長が非常に短いか存在しないようなフリーラジカル重合によって製造される。かかる場合はしばしば「ランダム」重合と呼ばれる。セグメントをもつコポリビニルポリマーはリビング重合、特に原子団転移(group transfer)重合、原子転移(atom transfer)重合、マクロモノマー(macromonomer)重合、グラフト重合、アニオンまたはカチオン重合のような重合方法によって製造される。好適な親水性ビニルモノマーは非イオン性及びイオン性モノマーである。好ましい非イオン性モノマーは糖類、グルコース、アミド、ピロリドンであり、特にヒドロキシ基及びエトキシ基をもつものである。好ましい非イオン性モノマーの例としては、ヒドロキシ エチルアクリレート、ヒドロキシ エチルメタアクリレート、ビニルピロリドン、エトキシ化された(メタ)アクリレート及び(メタ)アクリルアミドが挙げられる。好適なイオン性ビニルモノマーはカチオン性であってもよいが、好ましくはアニオン性である。
好ましいアニオン性ビニルモノマーはカルボキシ基および/または燐酸基および/またはスルホン酸基(これらの酸はフリーでも塩でもよい)を含むものである。好ましい例として、(メタ)アクリル酸、スチレンスルホン酸、ビニルベンジルスルホン酸、ビニルスルホン酸、(メタ)アクリロイルオキシアルキルスルホン酸(例えば、アクリロイルオキシメチルスルホン酸、アクリロイルオキシエチルスルホン酸、アクリロイルオキシプロピルスルホン酸、アクリロイルオキシブチルスルホン酸、メタクリロイルオキシメチルスルホン酸、メタクリロイルオキシエチルスルホン酸、メタクリロイルオキシプロピルスルホン酸、メタクリロイルオキシブチルスルホン酸)、2−アクリルアミド−2−アルキルアルカンスルホン酸(例えば、2−アクリルアミド−2−メチルエタンスルホン酸、2−アクリルアミド−2−メチルプロパンスルホン酸、2−アクリルアミド−2−メチルブタンスルホン酸)、2−メタクリルアミド−2−アルキルアルカンスルホン酸(例えば、2−メタクリルアミド−2−メチルエタンスルホン酸、2−メタクリルアミド−2−メチルプロパンスルホン酸、2−メタクリルアミド−2−メチルブタンスルホン酸)、モノ−(アクリロイルオキシアルキル)燐酸塩(例えば、モノ−(アクリロイルオキシエチル)燐酸塩、モノ−(3−アクリロイルオキシプロピル)燐酸塩)、モノ−(メタクリロイルオキシアルキル)燐酸塩(例えば、モノ−(メタクリロイルオキシエチル)燐酸塩、モノ−(3−メタクリロイルオキシプロピル)燐酸塩)が挙げられる。
好ましいカチオンビニルモノマーは4級アミン、ピリジン、グアニジン及びビグアニジン基を含むものである。
好ましい疎水性ビニルモノマーは親水性基を持たない。好ましい疎水性ビニルモノマーとしてはC1−20−ヒドロカルビル(メタ)アクリレート、ブタジエン、スチレン及びビニルナフタレンが挙げられ、C1−20−ヒドロカルビル(メタ)アクリレート(例、メチル(メタ)アクリレート、ブチル(メタ)アクリレート、オクチル(メタ)アクリレート、2−エチルヘキシル(メタ)アクリレート、イソボルニルアクリレート、ラウリルアクリレート、ステアリルアクリレート、ベンジル(メタ)アクリレート、フェノキシエチル(メタ)アクリレート)が好ましく、メチルメタクリレート、ベンジルメタクリレート、2−エチルヘキシルメタクリレート、フェノキシエチルメタクリレートが特に好ましい。これらのヒドロカルビル基は分岐でもよいが、好ましくは直鎖である。
【0117】
少なくとも1つのカルボキシル基を持つポリエステルはジオールモノマーと過剰量のジカルボン酸モノマーとの反応によっても生成される。カルボキシル基はカルボキシル基を持つジオールとジカルボン酸モノマーとの共重合によっても導入できる。
ポリエステルはジカルボン酸とジオールとのエステル化で製造されるのが典型的なものである。
【0118】
カルボキシル基を有するポリエステルは、例えば、カルボキシル基含有化合物と水酸基含有化合物とを、カルボキシル基が残存するように、溶融法、溶剤法などの公知の方法によって脱水縮合反応を行うことにより製造することができる。
【0119】
ポリエステルは、一塩基酸、多塩基酸の如きカルボキシル基を有する化合物と、ジオール、ポリオールの如き水酸基を有する化合物とを適宜選択して脱水縮合させて得られるもの等が挙げられ、さらに、油脂類または脂肪酸類を使用したものがアルキッド樹脂となる。
【0120】
本発明で使用するポリエステルが有するカルボキシル基は、主に、ポリエステルを構成する二塩基酸以上の多塩基酸に由来する未反応のカルボキシル基である。
【0121】
多塩基酸としては、例えば、アジピン酸、(無水)コハク酸、セバシン酸、ダイマー酸、(無水)マレイン酸、(無水)フタル酸、イソフタル酸、テレフタル酸、テトラヒドロ(無水)フタル酸、ヘキサヒドロ(無水)フタル酸、ヘキサヒドロテレフタル酸、2,6−ナフタレンジカルボン酸、(無水)トリメリット酸、(無水)ピロメリット酸などが挙げられる。
【0122】
多塩基酸以外に使用可能なカルボキシル基を有する化合物としては、例えば、テレフタル酸ジメチルの如き酸の低級アルキルエステル類;安息香酸、p−ターシャリブチル安息香酸、ロジン、水添ロジンの如き一塩基酸類;脂肪酸および油脂類;分子末端に1個又は2個のカルボキシル基を有するマクロモノマー類;5−ソジウムスルフォイソフタル酸およびそのジメチルエステル類などが挙げられる。
【0123】
水酸基を有する化合物としては、例えば、エチレングリコール、ネオペンチルグリコール、プロピレングリコール、ジエチレングリコール、ジプロピレングリコール、2−メチル−1,3−プロパンジオール、2,2−ジエチル−1,3−プロパンジオール、1,4−ブタンジオール、1,3−プロパンジオール、1,6−ヘキサンジオール、1,4−シクロヘキサンジメタノール、1,5−ペンタンジオール、ビスフェノールAのアルキレンオキサイド付加物、水添ビスフェノールA、水添ビスフェノールAのアルキレンオキサイド付加物、ポリエチレングリコール、ポリプロピレングリコール、ポリテトラメチレングリコールの如きジオール類;グリセリン、トリメチロールプロパン、トリメチロールエタン、ジグリセリン、ペンタエリスリトール、トリスヒドロキシエチルイソシアヌレートの如きポリオール類;「カージュラE−10」(シェル化学工業株式会社製の合成脂肪酸のグリシジルエステル)などのモノグリシジル化合物類、分子片末端に水酸基を2個有するマクロモノマー類などが挙げられる。
【0124】
また、ポリエステルを合成する際に、ひまし油、12−ヒドロキシステアリン酸などの水酸基含有脂肪酸または油脂類;ジメチロールプロピオン酸、p−ヒドロキシ安息香酸、ε−カプロラクトンの如きカルボキシル基と水酸基とを有する化合物なども使用できる。
【0125】
さらに、二塩基酸の一部をジイソシアネート化合物に代えることもできる。
【0126】
また、カルボキシル基を有するポリエステルは、水酸基を有するポリエステルに、無水マレイン酸、無水フタル酸、テトラヒドロ無水フタル酸、ヘキサヒドロ無水フタル酸、無水トリメリット酸などの無水酸を付加反応せしめる方法によっても製造することができる。
【0127】
水酸基とカルボキシル基とを有するポリエステルは、例えば、ポリエステル樹脂の脱水縮合反応において、公知の方法に従って、水酸基とカルボキシル基とが残存するように反応させることによって容易に製造することができる。
【0128】
第3級アミノ基とカルボキシル基とを有するポリエステルは、例えば、トリエタノールアミン、N−メチルジエタノールアミン、N,N−ジメチルエタノールアミン等の第3級アミノ基と水酸基とを有する化合物を、ポリエステル樹脂を製造する際のアルコール成分として使用することによって容易に製造することができる。
【0129】
ラジカル重合性不飽和基とカルボキシル基を有するポリエステルは、例えば、[1]水酸基とカルボキシル基とを有するポリエステルに、2−メタクリロイルオキシエチルイソシアネートなどのイソシアネート基を有するラジカル重合性不飽和基含有モノマー類、あるいは、無水マレイン酸などのラジカル重合性不飽和基を有する無水酸を付加反応せしめる方法、[2]カルボキシル基を有するポリエステル樹脂に、エポキシ基を有する重合性モノマー類を付加反応せしめる方法、[3]酸成分として無水マレイン酸などのラジカル重合性不飽和基含有モノマーを使用してポリエステル樹脂を合成する方法、等によって容易に製造することができる。
【0130】
ポリウレタンはポリオール成分(例えば、ジ−イソシアネート)とポリオール成分(例えば、ジオール)との縮合反応で好ましく製造される。
カルボキシル基を有するポリウレタンは、例えば、カルボキシル基を導入する成分としてのジメチロールプロピオン酸の如きカルボキシル基と水酸基とを有する化合物を含有するポリオール成分と、ポリイソシアネート成分とを反応させることによって、容易に製造することができる。
【0131】
ポリオール成分としては、ポリエステルの製造方法において掲げたジオール成分のほか、必要に応じて、3官能以上のポリオール化合物を使用することもできる。
【0132】
ポリイソシアネート成分としては、例えば、2,4−トリレンジイソシアネート、2,6−トリレンジイソシアネート、4,4’−ジフェニルメタンジイソシアネート、ヘキサメチレンジイソシアネート、フェニレンジイソシアネート、1,5−ナフタレンジイソシアネート、メタキシリレンジイソシアネート、イソホロンジイソシアネート、水添トリレンジイソシアネート、水添4,4’−ジフェニルメタンジイソシアネート、水添メタキシリレンジイソシアネート、粗製4,4’−ジフェニルメタンジイソシアネートの如きジイソシアネート化合物のほか、ポリメチレンポリフェニルイソシアネートの如きポリイソシアネート化合物も使用できる。
【0133】
ポリウレタンの製造は、常法に従えばよい。例えば、イソシアネート基と反応しない不活性な有機溶剤溶液中で、室温又は40〜100℃程度の温度で付加反応を行うのが好ましい。その際、ジブチル錫ジラウレート等の公知の触媒を使用しても良い。
【0134】
ポリウレタンを製造する際の反応系には、ジアミン、ポリアミン、N−メチルジエタノールアミンの如きN−アルキルジアルカノールアミン;ジヒドラジド化合物などの公知の鎖伸長剤も使用できる。
【0135】
水酸基とカルボキシル基とを有するポリウレタンは、例えば、ポリウレタンを製造する際に、イソシアネート基よりも水酸基が多くなる割合で反応させることにより容易に製造することができる。あるいは、カルボキシル基と末端イソシアネート基とを有するポリイソシアネートに、1分子中に水酸基を2個以上有する化合物を付加反応させることによっても容易に製造することができる。
【0136】
第3級アミノ基とカルボキシル基とを有するポリウレタンは、例えば、ポリオール成分の一部としてN−メチルジエタノールアミンなどのN−アルキルジアルカノールアミンを使用することにより容易に製造することができる。
【0137】
ブロック化イソシアネート基とカルボキシル基とを有するポリウレタンは、例えば、カルボキシル基と末端イソシアネート基とを有するポリイソシアネートに、公知のブロック剤を付加反応させることによって容易に製造することができる。
【0138】
エポキシ基とカルボキシル基とを有するポリウレタンは、例えば、カルボキシル基と末端イソシアネート基とを有するポリイソシアネートに、水酸基とエポキシ基とを有する化合物を付加反応させることによって容易に製造することができる。
【0139】
水酸基とエポキシ基とを有する化合物としては、例えば、グリシドール、グリセリンジグリシジルエーテル、トリメチロールプロパンジグリシジルエーテル、ビスフェノールAのジグリシジルエーテル等が挙げられる。
【0140】
ラジカル重合性不飽和基を、酸性基としてカルボキシル基を有するポリウレタンは、例えば、末端イソシアネート基を有するポリイソシアネートに、前述した如き水酸基を有する重合性モノマー類、及びグリセロールモノ(メタ)アクリレート、トリメチロールプロパンジ(メタ)アクリレート、ペンタエリスリトールトリアクリレートなどの水酸基とラジカル重合性不飽和基とを有する化合物を付加反応せしめる方法等によって容易に製造することができる。
【0141】
加水分解性アルコキシシラン基を、酸性基としてカルボキシル基を有するポリウレタンは、例えば、末端イソシアネート基を有するポリイソシアネートに、γ−メルカプトプロピルトリメトキシシラン、γ−メルカプトプロピルメチルジメトキシシラン、γ−アミノプロピルトリメトキシシラン、γ−アミノプロピルトリエトキシシランの如きイソシアネート基と反応しうる活性水素を有するシランカップリング剤を付加反応させる方法等により容易に製造することができる。
【0142】
ポリマーは微粒子分散物を製造する過程で用いる液体媒体に合うようにまた微粒子分散物に用いられる最終組成物(例えば、インク)中の液体展色剤(ベヒクル)に合うように選ばれる。例えば、微粒子分散物が水性のインクジェット記録用インクに用いられる場合には、好ましくはポリマーは親水性である。
〔分子量〕
分散剤の重量平均分子量は10000以上200000以下が好ましく、さらに15000以上150000以下であることが好ましく、中でも20000以上100000以下であることがより好ましい。10000以上では印画物の画質が優れ好ましい一方、200000以下では、粘度が高くなるのを抑制でき、さらに貯蔵安定性がの低下を防ぎ、好ましい。
【0143】
〔D/P値〕
分散剤の含有量は、顔料100質量部に対して20〜100質量部の範囲であることが好ましく、より好ましくは25〜90質量部の範囲であり、さらに好ましくは30〜70質量部の範囲である。また、分散剤は、単独で用いても、複数のものを組み合わせて用いてもよい。
分散剤の含有量が20質量部未満の場合、分散剤の量が顔料に対して不十分になり、貯蔵安定性が不十分になる。一方、100質量部超過の場合、粘度が高くなり、さらに貯蔵安定性が低下するため不適である。
前記顔料分散物中の着色剤の含有量をP、分散剤の含有量をDとし、含有量Dと含有量Pとの比をD/P値としたときに、D/P値が0.15以上1.0以下であることが好ましく、0.16以上0.8以下であることがより好ましく、0.17以上0.7以下であることが更に好ましい。。
【0144】
〔酸価〕
分散剤は架橋剤と架橋するために十分な酸価をもつ必要があり、少なくとも50mgKOH/g以上の酸価をもつものが好ましい。
全ての態様において、上記の酸価は好ましくは70〜200mgKOH/gであり、より好ましくは70〜160mgKOH/gである。係る酸価をもつ分散剤は改良された保存安定性を与える。
また、50mgKOH/gより低いと、水系溶媒への溶解性が低いため不適である。
【0145】
〔溶解性〕
分散剤は水不溶性、水溶性のどちらでも良いが、水への溶解性として、1g/100mL以上であることが好ましく、さらに好ましくは、3g/100mL以上であり、特に好ましくは5g/100mL以上である。
1g/(100m)L未満では、水への溶解性が低いために、顔料粒子に吸着しにくくなり、分散性が低下する場合がある。
【0146】
〔架橋〕
前記水系分散物が、架橋剤により架橋されていることが好ましい。
本発明のより好ましい形態は、分散剤は架橋する前に顔料表面に吸着し、相対的に安定な分散物が形成され、そしてこの分散工程に引き続き、架橋剤を用いて架橋する工程を実施することによりより高度な保存安定性を有し、印画物の画質に優れる分散物が得られる。
少なくとも50mg/KOH以上の酸価をもつ分散剤を用いる場合には、架橋剤はオリゴマー分散基を持っていても、持たなくてもよい。「オリゴマー」という言葉は分子量に上限はないし、また繰り返し単位の上限もない意味で用いる。1以上のオリゴマー分散基を持つ架橋剤は生じた微粒子分散物の安定性を増加させる。この増加された安定性はインクジェット記録に用いる液体展色剤(ビヒクル)において特に有用である。それは50mg/KOH未満の酸価をもつ分散剤では分散が困難であるからである。
【0147】
オリゴマー分散基は好ましくはポリアルキレンオキシドであり、より好ましくはポリC2−4−アルキレンオキシドであり、特に好ましくはポリエチレンオキシドである。ポリアルキレンオキシドは生じた微粒子分散物の安定性を改良する。ポリアルキレンオキシドは好ましくは3〜200、より好ましくは5〜50、特に好ましくは5〜20のアルキレンオキシド繰り返し単位を有する。
【0148】
架橋剤は2つ以上のエポキシ基を持つことが好ましい。少なくとも2つのエポキシ基を持つ好ましい架橋剤はエピクロロヒドリン誘導体である。2つ以上のエポキシ基を持ち、オリゴマー分散基を持たない架橋剤はエチレングリコールジグリシジルエーテル、レゾルシノールジグリシジルエーテル、ネオペンチルグリコールジグリシジルエーテル、1,6−ヘキサンジオールジグリシジルエーテル、ハロゲン化されたビスフェノールAジグリシジルエーテル、トリメチロールプロパンポリグリシジルエーテル、ポリグリセロールポリグリシジルエーテル、グリセロールポリグリシジルエーテル、ペンタエリスリトールポリグリシジルエーテル、ジグリセロールポリグリシジルエーテル、ソルビトールポリグリシジルエーテル、及びポリブタジエンジグリシジルエーテルである。2つのエポキシ基を持ち、かつ1以上のオリゴマー分散基をもつ好ましい架橋剤はジエチレングリコールジグリシジルエーテル、ポリエチレングリコールジグリシジルエーテル、またはジプロピレングリコールジグリシジルエーテルである。
また、無水フタル酸、無水コハク酸等の酸無水物も架橋剤として用いることができる。
【0149】
〔温度、pH〕
本発明では架橋反応は100℃以下、pH6以上で行うことが好ましい。更に好ましい架橋反応は30℃〜90℃、より好ましくは40℃〜85℃である。
架橋反応の好ましいpHは7〜10であり、より好ましくは8〜9.5である。
架橋剤がさらにカルボキシ基を含み、カルボキシ基とエポキシ基の間の架橋反応を100℃以下、pH6以上で行うことが好ましい。
【0150】
架橋反応は水系で行うため、100℃以下が好ましい。逆に、低温では架橋反応の進行が遅くなるため、好ましくなく、30℃以上が好ましく、40℃以上がさらに好ましい。
【0151】
pHが10超過では、架橋反応で熱を加えるとポリマーが加水分解してしまう可能性がある。一方、pHが6未満では、顔料分散物が凝集を起こしやすくなり不安定になってしまうので、好ましくない。
【0152】
〔膜精製〕
膜精製には逆浸透膜(NF膜)、限外ろ過膜(UF膜)を使用することができ、加圧してもしなくても良いが、加圧する場合の方が、精製に要する時間が短くなり、効率的である。UF膜としては、分画分子量10000以上150000以下が好ましく、20000以上100000以下がより好ましい。10000未満では精製するための時間が長くなってしまうため、非効率である。一方、150000超過では、分散剤が流出してしまう可能性があるため、好ましくない。
【0153】
本発明のアゾ顔料の用途としては、画像、特にカラー画像を形成するための画像記録材料が挙げられ、具体的には、以下に詳述するインクジェット方式記録材料を始めとして、感熱記録材料、感圧記録材料、電子写真方式を用いる記録材料、転写式ハロゲン化銀感光材料、印刷インク、記録ペン等があり、好ましくはインクジェット方式記録材料、感熱記録材料、電子写真方式を用いる記録材料であり、更に好ましくはインクジェット方式記録材料である。
【0154】
また、CCDなどの固体撮像素子やLCD、PDP等のディスプレーで用いられるカラー画像を記録・再現するためのカラーフィルター、各種繊維の染色の為の染色液にも適用できる。
【0155】
以上に説明した本発明の製造方法によれば、非晶質なアゾ化合物から結晶変換することで、結晶成長をコントロールし、易分散なアゾ顔料を製造することができる。
【0156】
工程(b)において式(1)で表されるアゾ化合物が反応液中の少なくとも一部が溶解しているため、特に、アゾ化合物溶解液の調整方法が上記形態(ii)である場合は、カップリング反応がよりスムーズに進行してより高純度のアゾ化合物を得ることができる。このことは、最終的に得られるアゾ顔料の高効率な製造に寄与する。
【0157】
ところで、上記したように、カップリング反応後の反応液中で析出した特許文献6の顔料(一度、粒子として得られた顔料)を有機溶媒に溶解させることは困難である。すなわち、顔料の粒子径をより微細なものにするべく、特許文献6の顔料を有機溶剤に溶解させようとする場合、大量の有機溶媒が使用する必要があり、製造コストが増大する上、大量の有機溶媒を使用したとしても溶解させられない場合も多く、ましてや完全に溶解させることは非常に難しい。
【0158】
一方、本発明によれば、工程(b)の終了後には、大量の有機溶媒を使用することなく(高濃度で)、目的とするアゾ化合物の少なくとも一部が溶解した溶解液を得ることができ、このことは、最終的に得られるアゾ顔料の高効率な製造に寄与する。また、工程(b)で得られたアゾ化合物溶解液と、このアゾ化合物に対する貧溶媒とを混合することにより、アゾ顔料を微粒子として、析出させることができる。
【0159】
以上のように、本発明のアゾ顔料の製造方法によれば、易分散なアゾ顔料微粒子を高効率かつ低コストで製造することができる。
【0160】
[着色組成物]
本発明の着色組成物は、上記した本発明のアゾ顔料、その塩、水和物又は溶媒和物を少なくとも1種含有する。本発明の着色組成物は、媒体を含有させることができるが、媒体として溶媒を用いた場合は特にインクジェット記録用インクとして好適である。本発明の着色組成物は、媒体として、親油性媒体や水性媒体を用いて、それらの中に、本発明の顔料を分散させることによって作製することができる。好ましくは、水性媒体を用いる場合である。本発明の着色組成物には、媒体を除いたインク用組成物も含まれる。本発明の着色組成物は、必要に応じてその他の添加剤を、本発明の効果を害しない範囲内において含有しうる。その他の添加剤としては、例えば、乾燥防止剤(湿潤剤)、褪色防止剤、乳化安定剤、浸透促進剤、紫外線吸収剤、防腐剤、防黴剤、pH調整剤、表面張力調整剤、消泡剤、粘度調整剤、分散剤、分散安定剤、防錆剤、キレート剤等の公知の添加剤(特開2003−306623号公報に記載)が挙げられる。これらの各種添加剤は、水溶性インクの場合にはインク液に直接添加する。油溶性インクの場合には、アゾ顔料分散物の調製後分散物に添加するのが一般的であるが、調製時に油相又は水相に添加してもよい。
【実施例】
【0161】
以下、本発明を実施例に基づきさらに詳細に説明するが、本発明はこれらの実施例に何ら限定されるものではない。なお、実施例中、「部」とは質量部を表す。
【0162】
<アゾ化合物、アゾ顔料の製造>
【0163】
以下の実施例にて得られたアゾ化合物の1次粒子径については、透過型顕微鏡(日本電子(株)製:JEM−1010電子顕微鏡)を用いて目視にて観察した。
【0164】
アゾ化合物、アゾ顔料のX線回折の測定は、日本工業規格JISK0131(X線回析分析通則)に準じて、粉末X線回折測定装置RINT2500(株式会社リガク製)にてCuKα線を用い、次の条件で行ったものである。
【0165】
使用測定器:Rigaku社製 自動X線回折装置RINT2500
X線管球:Cu
管電圧:55KV
管電流:280mA
スキャン方法:2θ/θスキャン
スキャン速度:6deg./min
サンプリング間隔:0.100deg.
スタート角度(2θ):5deg.
ストップ角度(2θ):55deg.
ダイバージェンススリット:2deg.
スキャッタリングスリット:2deg.
レシービングスリット:0.6mm
縦型ゴニオメータ使用
【0166】
[実施例1]
亜硝酸ナトリウム2.2gを水50mLに溶解させた。別に式(2)で表されるアミノ化合物5.8gを濃塩酸50mLに溶解させた後、内温−10℃まで冷却した。この中に内温が0℃以下になるように先述の亜硝酸ナトリウム水溶液を滴下した。内温−10℃〜0℃にて1時間攪拌した後、内温0℃以下にて尿素1.8gを加えた。添加終了後15分間同温度にて攪拌し、ジアゾニウム塩溶液を得た。別に式(3)の化合物5gをメタノール175mLに加えた後昇温し、還流下溶解させた。この溶液を内温0℃まで冷却し、先述のジアゾニウム塩溶液を内温が10℃以下になるように添加した。内温10℃にて1時間攪拌した後、析出している固体を濾別した。メタノール、水で十分にかけ洗いを行った後、水300mLに懸濁させ、28%アンモニア水溶液を添加してpHを6.0に調整した。析出している固体を濾別して水で十分にかけ洗いを行い、60℃にて乾燥後、非晶質なアゾ化合物(1)−1を9.8g得た。
得られた非晶質なアゾ化合物(1)−1の1次粒子の長軸方向の長さは、約0.5μmであった。
非晶質なアゾ化合物(1)−1のX線回折の測定を上記の条件により行ったところ、特徴的なX線回折ピークが見られなかった。
【0167】
得られた非晶質なアゾ化合物(1)−1 5gをエチレングリコール50mLに懸濁させた。内温100℃まで昇温した後、同温度にて2時間攪拌した。内温30℃まで冷却した後、析出している固体を濾別し、δ型結晶形態のアゾ顔料(1)−2を4.5g得た。
得られたδ型結晶形態のアゾ顔料(1)−2の1次粒子の長軸方向の長さは、約0.6μmであった。
得られたδ型結晶形態のアゾ顔料(1)−2のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図1に示す。
【0168】
[実施例2]
亜硝酸ナトリウム2.2gを水50mLに溶解させた。別に式(2)で表されるアミノ化合物5.8gを濃塩酸50mLに溶解させた後、内温−10℃まで冷却した。この中に内温が0℃以下になるように先述の亜硝酸ナトリウム水溶液を滴下した。内温−10℃〜0℃にて1時間攪拌した後、内温0℃以下にて尿素1.8gを加えた。添加終了後15分間同温度にて攪拌し、ジアゾニウム塩溶液を得た。このジアゾニウム塩溶液に式(3)の化合物5gを内温が5℃以下になるように少しずつ加えた。添加終了後、内温10℃まで昇温し、同温度にて3時間攪拌した後、析出している固体を濾別した。水で十分にかけ洗いを行った後、水200mLに懸濁させ、28%アンモニア水溶液を添加してpHを6.0に調整した。固体を濾別して水で十分にかけ洗いを行い、60℃にて乾燥後、非晶質なアゾ化合物(1)−3を9.9g得た。
得られたアゾ化合物(1)−3の1次粒子の長軸方向の長さは、約0.3μmであった。
アゾ化合物(1)−3のX線回折の測定を上記の条件により行ったところ、特徴的なX線回折ピークが見られなかった。
【0169】
得られた非晶質なアゾ化合物(1)−3 5gをエチレングリコール50mLに懸濁させた。内温120℃まで昇温した後、同温度にて2時間攪拌した。内温30℃まで冷却した後、固体を濾別し、δ型結晶形態のアゾ顔料(1)−4を4.5g得た。
得られたアゾ顔料(1)−4の1次粒子の長軸方向の長さは、約0.5μmであった。
得られたアゾ顔料(1)−4のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°及び9.7°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図2に示す。
【0170】
[実施例3]
式(2)の化合物34.6gを酢酸150gに懸濁し、硫酸24gを内温が20℃〜30℃になるように滴下した。さらに内温が20℃〜30℃になるように43%ニトロシル硫酸の硫酸溶液48.6gを滴下し、内温20℃にて1時間攪拌後、尿素0.28gを添加してジアゾニウム塩溶液を得た。このジアゾニウム塩溶液に式(3)の化合物30gを内温が20℃〜30℃になるように分割して添加し、内温25℃にて1時間攪拌し、アゾ化合物の均一反応液を得た。別に360gのメタノールを内温25℃にて用意し、上述のアゾ化合物の均一反応液を内温が30℃以下になるように添加し、10分間攪拌した後、析出している固体を濾別した。300mLのメタノールでかけ洗いした後、水900mLに懸濁させ、28%アンモニウム水溶液を添加してpHを6.0に調整した。固体を濾別し、ζ型結晶形態のアゾ顔料(1)−5を得た。
得られたアゾ顔料(1)−5の1次粒子の長軸方向の長さは、約2μmであった。
得られたアゾ顔料(1)−5のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が6.5°、6.7°、9.1°および21.3°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図3に示す。
【0171】
得られたアゾ顔料(1)−5 5gを硫酸50mLに溶解させ、水300mLに内温が15℃以下になるように添加した。析出している固体を濾別し、十分に水でかけ洗いした後、水300mLに懸濁させ、28%アンモニア水溶液を添加してpHを6.1に調整した。固体を濾別して、水で十分にかけ洗いを行い、60℃にて乾燥後、非晶質なアゾ化合物(1)−6を3.9g得た。
得られたアゾ化合物(1)−6の1次粒子の長軸方向の長さは、約0.2μmであった。
アゾ化合物(1)−6のX線回折の測定を上記の条件により行ったところ、特徴的なX線回折ピークが見られなかった。
【0172】
得られた非晶質なアゾ化合物(1)−6 3gをエチレングリコール30mLに懸濁させた。内温120℃まで昇温した後、同温度にて2時間攪拌した。内温30℃まで冷却した後、固体を濾別し、δ型結晶形態のアゾ顔料(1)−7を2.4g得た。
得られたアゾ顔料(1)−7の1次粒子の長軸方向の長さは、約0.3μmであった。
得られたアゾ顔料(1)−7のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図4に示す。
【0173】
[実施例4]
実施例3で得られたアゾ顔料(1)−5 5gをリン酸50mLに溶解させ、水300mLに内温が15℃以下になるように添加した。析出している固体を濾別し、十分に水でかけ洗いした後、水300mLに懸濁させ、28%アンモニア水溶液を添加してpHを7.2に調整した。固体を濾別して、水で十分にかけ洗いを行い、60℃にて乾燥後、非晶質なアゾ化合物(1)−8を4.2g得た。
得られたアゾ化合物(1)−8の1次粒子の長軸方向の長さは、約0.2μmであった。
アゾ化合物(1)−8のX線回折の測定を上記の条件により行ったところ、特徴的なX線回折ピークが見られなかった。
【0174】
得られた非晶質なアゾ化合物(1)−8 3gをエチレングリコール30mLに懸濁させた。内温120℃まで昇温した後、同温度にて2時間攪拌した。内温30℃まで冷却した後、固体を濾別し、δ型結晶形態のアゾ顔料(1)−9を2.4g得た。
得られたアゾ顔料(1)−9の1次粒子の長軸方向の長さは、約0.2μmであった。
得られたアゾ顔料(1)−9のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図5に示す。
【0175】
[実施例5]
式(2)の化合物11.5gを酢酸50gに懸濁させ、内温が20℃〜30℃になるようにニトロシル硫酸の43%硫酸溶液16.2gを滴下した。内温20℃にて1時間攪拌した後、尿素0.1gを添加してジアゾニウム塩溶液を得た。別に式(3)の化合物10gを酢酸100mLに溶解させ、上述のジアゾニウム塩溶液に内温が20℃〜25℃になるように滴下した。内温20℃にて1時間攪拌し、アゾ化合物(1)の均一反応液を得た。別に水150gを用意し、内温20℃〜25℃にて上述のアゾ化合物(1)の均一反応液を滴下した。析出している固体を濾別した後、水で十分にかけ洗いを行い、水200mLに懸濁させ、28%アンモニア水溶液を添加してpHを6.2に調整した。固体を濾別して、水で十分にかけ洗い、非晶質なアゾ化合物(1)−10を得た。
得られたアゾ化合物(1)−10の1次粒子の長軸方向の長さは、約0.2μmであった。
水分測定を行ったところ、水の含率が68%だった。
アゾ化合物(1)−10の一部を乾燥し、X線回折の測定を上記の条件により行ったところ、特徴的なX線回折ピークが見られなかった。
【0176】
得られた非晶質な含水のアゾ化合物(1)−10 10gをエチレングリコール30mLに懸濁させた。内温95℃まで昇温した後、同温度にて2時間攪拌した。内温30℃まで冷却した後、固体を濾別し、δ型結晶形態のアゾ顔料(1)−11を2.9g得た。
得られたアゾ顔料(1)−11の1次粒子の長軸方向の長さは、約0.15μmであった。
得られたアゾ顔料(1)−11のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図6に示す。
【0177】
[実施例6]
実施例5で得られた非晶質な含水のアゾ化合物(1)−10を乾燥し、アゾ化合物(1)−12を得た。アゾ化合物(1)−12、10gをエチレングリコール100mLに懸濁させ、内温120℃まで昇温した後、同温度にて2時間攪拌した。内温30℃まで冷却した後、固体を濾別し、δ型結晶形態のアゾ顔料(1)−13を9.1g得た。
得られたアゾ顔料(1)−13の1次粒子の長軸方向の長さは、約0.2μmであった。
得られたアゾ顔料(1)−13のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図7に示す。
【0178】
[実施例7]
アゾ化合物(1)−12、10gをエチレングリコール50mL、水50mLの混合溶媒に懸濁させ、内温95℃まで昇温した後、同温度にて2時間攪拌した。内温30℃まで冷却した後、固体を濾別し、δ型結晶形態のアゾ顔料(1)−14を9.3g得た。
得られたアゾ顔料(1)−14の1次粒子の長軸方向の長さは、約0.2μmであった。
得られたアゾ顔料(1)−14のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図8に示す。
【0179】
[実施例8]
アゾ化合物(1)−12、10gをエチレングリコール5mL、水95mLの混合溶媒に懸濁させ、内温85℃まで昇温した後、同温度にて2時間攪拌した。内温30℃まで冷却した後、固体を濾別し、δ型結晶形態のアゾ顔料(1)−15を9.5g得た。
得られたアゾ顔料(1)−15の1次粒子の長軸方向の長さは、約0.15μmであった。
得られたアゾ顔料(1)−15のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図9に示す。
【0180】
[実施例9]
アゾ化合物(1)−12、10gをイソプロパノール40mL、水60mLの混合溶媒に懸濁させ、内温80℃まで昇温した後、同温度にて2時間攪拌した。内温30℃まで冷却した後、固体を濾別し、δ型結晶形態のアゾ顔料(1)−16を8.2g得た。
得られたアゾ顔料(1)−16の1次粒子の長軸方向の長さは、約5μmであった。
得られたアゾ顔料(1)−16のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図10に示す。
【0181】
[実施例10]
アゾ化合物(1)−12、10gをイソブチルアルコール100mL、水10mLに懸濁させ、内温80℃まで昇温した後、同温度にて2時間攪拌した。内温30℃まで冷却した後、固体を濾別し、δ型結晶形態のアゾ顔料(1)−17を7.9g得た。
得られたアゾ顔料(1)−17の1次粒子の長軸方向の長さは、約15μmであった。
得られたアゾ顔料(1)−17のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図11に示す。
【0182】
[実施例11]
アゾ化合物(1)−12、10gを酢酸ブチル100mLに懸濁させ、内温90℃まで昇温した後、同温度にて2時間攪拌した。内温30℃まで冷却した後、固体を濾別し、δ型結晶形態のアゾ顔料(1)−18を8.5g得た。
得られたアゾ顔料(1)−18の1次粒子の長軸方向の長さは、約20μmであった。
得られたアゾ顔料(1)−18のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図12に示す。
【0183】
[実施例11−2]
アゾ化合物(1)−12、10gをメタノール100mLに懸濁させた。室温にて2時間攪拌した後に固体を濾別しζ型結晶形態のアゾ顔料(1)−101を9.4g得た。
得られたアゾ顔料(1)−101の1次粒子の長軸方向の長さは約10μmであった。
得られたアゾ顔料(1)−101のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が6.5°、6.7°、9.1°、及び21.3°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図13に示す。
【0184】
[実施例12]
式(2)の化合物11.5gを酢酸50gに懸濁させ、内温が20℃〜30℃になるようにニトロシル硫酸の43%硫酸溶液16.2gを滴下した。内温20℃にて1時間攪拌した後、尿素0.1gを添加してジアゾニウム塩溶液を得た。別に式(3)の化合物10gを酢酸100mLに溶解させ、上述のジアゾニウム塩溶液に内温が20℃〜25℃になるように滴下した。内温20℃にて1時間攪拌し、アゾ化合物(1)の均一反応液を得た。別に水150gを用意し、内温20℃〜25℃にて上述のアゾ化合物(1)の均一反応液を滴下した。析出している固体を濾別した後、水で十分にかけ洗いを行い、非晶質なアゾ化合物(1)−19を得た。
得られたアゾ化合物(1)−19の1次粒子の長軸方向の長さは、約0.2μmであった。
CuKα特性X線回折図を図14に示す。
アゾ化合物(1)−19のX線回折の測定を上記の条件により行ったところ、特徴的なX線回折ピークが見られなかった。
【0185】
得られた非晶質なアゾ化合物(1)−19を水120mL、エチレングリコール180mLの混合溶媒に懸濁させた。28%アンモニア水溶液でpHを6.28に調整した後、内温85℃まで昇温し、同温度にて2時間攪拌した。内温30℃まで冷却した後、結晶を濾別し、水で十分にかけ洗いを行い、δ型結晶形態のアゾ顔料(1)−20を19.5g得た。
得られたアゾ顔料(1)−20の1次粒子の長軸方向の長さは、約0.3μmであった。
得られたアゾ顔料(1)−20のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図15に示す。
【0186】
[実施例12−2]
実施例12において、アゾ化合物(1)の均一反応液をエチレングリコール150gに滴下すること以外は実施例12と同様にして、非晶質なアゾ化合物(1)−102を得た。得られたアゾ化合物(1)−102の1次粒子の長軸方向の長さは約0.7μmであった。
アゾ化合物(1)−102のX線回折の測定を上記の条件により行ったところ、特徴的なX線回折ピークが見られなかった。
続いて、実施例12と同様の操作で結晶変換を行い、δ型結晶形態のアゾ顔料(1)−103を19.1g得た。
得られたアゾ顔料(1)−103の1次粒子の長軸方向の長さは、約0.7μmであった。
得られたアゾ顔料(1)−103のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図16に示す。
【0187】
[実施例13]
式(2)の化合物11.5gを酢酸50gに懸濁させ、内温が20℃〜30℃になるようにニトロシル硫酸の43%硫酸溶液16.2gを滴下した。内温20℃にて1時間攪拌した後、尿素0.1gを添加してジアゾニウム塩溶液を得た。別に式(3)の化合物10gを酢酸100mLに溶解させ、上述のジアゾニウム塩溶液に内温が20℃〜25℃になるように滴下した。内温20℃にて1時間攪拌し、アゾ化合物(1)の均一反応液を得た。別に水150gを用意し、内温20℃〜25℃にて上述のアゾ化合物(1)の均一反応液を滴下した。固体(非晶質なアゾ化合物)が析出した懸濁液を同温度にて30分間攪拌した後、エチレングリコール20mLを添加し、内温85℃まで昇温し、同温度にて2時間攪拌した。内温30℃まで冷却した後、析出している結晶を濾別し、水で十分にかけ洗いを行い、δ型結晶形態のアゾ顔料(1)−21を19.9g得た。
得られたアゾ顔料(1)−21の1次粒子の長軸方向の長さは、約0.2μmであった。
得られたアゾ顔料(1)−21のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図17に示す。
【0188】
[実施例14]
式(2)の化合物11.5gを酢酸50gに懸濁させ、内温が20℃〜30℃になるようにニトロシル硫酸の43%硫酸溶液16.2gを滴下した。内温20℃にて1時間攪拌した後、尿素0.1gを添加してジアゾニウム塩溶液を得た。別に式(3)の化合物10gを酢酸100mLに溶解させ、上述のジアゾニウム塩溶液に内温が20℃〜25℃になるように滴下した。内温20℃にて1時間攪拌し、アゾ化合物(1)の均一反応液を得た。別に水150gを用意し、内温20℃〜25℃にて上述のアゾ化合物(1)の均一反応液を滴下した。固体(非晶質なアゾ化合物)が析出した懸濁液を同温度にて30分間攪拌した後、エチレングリコール20mLを添加した。内温30℃以下になるように28%アンモニア水溶液を添加してpHを4.01に調整した後、内温85℃まで昇温し、同温度にて2時間攪拌した。内温30℃まで冷却した後、析出している結晶を濾別し、水で十分にかけ洗いを行い、δ型結晶形態のアゾ顔料(1)−22を19.9g得た。
得られたアゾ顔料(1)−22の1次粒子の長軸方向の長さは、約0.5μmであった。
得られたアゾ顔料(1)−22のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図18に示す。
【0189】
[実施例15]
式(2)の化合物11.5gを酢酸50gに懸濁させ、内温が20℃〜30℃になるようにニトロシル硫酸の43%硫酸溶液16.2gを滴下した。内温20℃にて1時間攪拌した後、尿素0.1gを添加してジアゾニウム塩溶液を得た。別に式(3)の化合物10gを酢酸100mLに溶解させ、上述のジアゾニウム塩溶液に内温が20℃〜25℃になるように滴下した。内温20℃にて1時間攪拌し、アゾ化合物(1)の均一反応液を得た。別に水150gを用意し、内温20℃〜25℃にて上述のアゾ化合物(1)の均一反応液を滴下した。固体(非晶質なアゾ化合物)が析出した懸濁液を同温度にて30分間攪拌した後、エチレングリコール20mLを添加し、内温85℃まで昇温し、同温度にて2時間攪拌した。内温30℃まで冷却した後、内温が30℃以下になるように28%アンモニア水溶液を添加してpHを6.50に調整した。析出している結晶を濾別し、水で十分にかけ洗いを行い、δ型結晶形態のアゾ顔料(1)−23を19.9g得た。
得られたアゾ顔料(1)−23の1次粒子の長軸方向の長さは、約0.4μmであった。
得られたアゾ顔料(1)−23のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図19に示す。
【0190】
[実施例16]
式(2)の化合物11.5gを酢酸50gに懸濁させ、内温が20℃〜30℃になるようにニトロシル硫酸の43%硫酸溶液16.2gを滴下した。内温20℃にて1時間攪拌した後、尿素0.1gを添加してジアゾニウム塩溶液を得た。別に式(3)の化合物10gを酢酸100mLに溶解させ、上述のジアゾニウム塩溶液に内温が20℃〜25℃になるように滴下した。内温20℃にて1時間攪拌し、アゾ化合物(1)の均一反応液を得た。別に水150gを用意し、内温20℃〜25℃にて上述のアゾ化合物(1)の均一反応液を滴下した。固体(非晶質なアゾ化合物)が析出した懸濁液を同温度にて30分間攪拌した後、エチレングリコール20mLを添加し、内温85℃まで昇温し、同温度にて2時間攪拌した。内温30℃まで冷却した後、析出している結晶を濾別し、水で十分にかけ洗いを行い、1%炭酸水素ナトリウム水溶液100mLでかけ洗いを行った。さらに水で十分にかけ洗いを行い、δ型結晶形態のアゾ顔料(1)−24を19.8g得た。
得られたアゾ顔料(1)−24の1次粒子の長軸方向の長さは、約0.15μmであった。
得られたアゾ顔料(1)−24のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図20に示す。
【0191】
[実施例17]
実施例6で得られたアゾ化合物(1)−12、10gをエチレングリコール100mLに懸濁させ、室温にて24時間攪拌した。固体を濾別し、δ型結晶形態のアゾ顔料(1)−25を9.5g得た。
得られたアゾ顔料(1)−25の1次粒子の長軸方向の長さは、約0.2μmであった。
得られたアゾ顔料(1)−25のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図21に示す。
【0192】
[実施例18]
式(2)の化合物11.4gを90%酢酸50gに懸濁させ、内温が10〜20℃となるまで冷却した。この温度範囲で、硫酸4g、続いてニトロシル硫酸の43%硫酸溶液16.2gを滴下した。内温10〜20℃にて1時間攪拌した後、尿素0.1gを添加してジアゾニウム塩溶液を得た。このジアゾニウム塩溶液の中に、式(3)の化合物10gを内温が10〜20℃になるように分割添加した。内温10〜20℃にて1時間攪拌し、アゾ化合物(1)の均一反応液を得た。別に水150gを用意し、内温20℃〜25℃にて上述のアゾ化合物(1)の均一反応液を滴下した。固体(非晶質なアゾ化合物)が析出した懸濁液を同温度にて15分間攪拌した後、エチレングリコール20gを添加した。続いて、内温70℃まで昇温し、同温度にて1時間攪拌して結晶変換を行った。内温30℃まで冷却した後、析出している結晶を濾別し、水およびメタノールで十分にかけ洗いを行い、δ型結晶形態のアゾ顔料(1)−27を19.9g得た。
得られたアゾ顔料(1)−27の1次粒子の長軸方向の長さは、約0.5μmであった。
得られたアゾ顔料(1)−27のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図22に示す。
[実施例18−2]
式(2)の化合物11.4gを90%酢酸50gに懸濁させ、内温が10〜20℃となるまで冷却した。この温度範囲で、硫酸4g、続いてニトロシル硫酸の43%硫酸溶液16.2gを滴下した。内温10〜20℃にて1時間攪拌した後、尿素0.1gを添加してジアゾニウム塩溶液を得た。このジアゾニウム塩溶液の中に、式(3)の化合物10gを内温が10〜20℃になるように分割添加した。内温10〜20℃にて1時間攪拌し、アゾ化合物(1)の均一反応液を得た。別に水150gを用意し、内温40℃〜45℃にて上述のアゾ化合物(1)の均一反応液を滴下した。固体(非晶質なアゾ化合物)が析出した懸濁液を同温度にて15分間攪拌した後、エチレングリコール20gを添加した。同温度にて16時間攪拌して結晶変換を行った。内温30℃まで冷却した後、析出している結晶を濾別し、水およびメタノールで十分にかけ洗いを行い、δ型結晶形態のアゾ顔料(1)−104を19.9g得た。
得られたアゾ顔料(1)−104の1次粒子の長軸方向の長さは、約0.4μmであった。
得られたアゾ顔料(1)−104のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図23に示す。
【0193】
[比較例1]
実施例3で得られたアゾ顔料(1)−5 5gをエチレングリコール50mLに懸濁させ、内温120℃まで昇温した後、同温度にて2時間攪拌した。内温30℃まで冷却した後、析出している固体を濾別し、δ型結晶形態のアゾ顔料(1)−26を3.9g得た。
得られたアゾ顔料(1)−26を光学顕微鏡(ニコン(株)製:ECLIPSE LV150)で目視にて観察したところ、1次粒子の長軸方向の長さは、約80μmであった。
得られたアゾ顔料(1)−26のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図24に示す。
【0194】
【化12】
【0195】
【化13】
【0196】
【化14】
【0197】
<顔料分散物の作製>
【0198】
上記実施例1で合成したδ型結晶形態アゾ顔料(1)−2を2.5部、オレイン酸ナトリウム0.5部、グリセリン5部、水42部を混合し、直径0.1mmのジルコニアビーズ100部とともに遊星型ボールミルを用いて毎分300回転、3時間分散を行った。分散終了後、ジルコニアビーズを分離し、黄色の顔料分散物1を得た。
上記実施例2〜17で合成したδ型結晶形態アゾ顔料についてもそれぞれ同様の操作を行い、黄色の顔料分散物2〜17を得た。
【0199】
比較のために、上記比較例1で合成したδ型結晶形態アゾ顔料(1)−26を2.5部、オレイン酸ナトリウム0.5部、グリセリン5部、水42部を混合し、直径0.1mmのジルコニアビーズ100部とともに遊星型ボールミルを用いて毎分300回転、8時間分散を行った。分散終了後、ジルコニアビーズを分離し、黄色の比較顔料分散物1を得た。
さらに上記実施例1にて合成した非晶質なアゾ化合物(1)−1を2.5部、オレイン酸ナトリウム0.5部、グリセリン5部、水42部を混合し、直径0.1mmのジルコニアビーズ100部とともに遊星型ボールミルを用いて毎分300回転、7時間分散を行った。分散終了後、ジルコニアビーズを分離し、黄色の比較顔料分散物2を得た。
【0200】
顔料分散物中の顔料の体積平均粒子径を、日機装(株)製Nanotrac150(UPA−EX150)を用いて測定した。測定結果を表1に示す。
【0201】
【表1】
【0202】
以上の結果より、本発明の製造方法により得られる特定の結晶構造を有するアゾ顔料は、より短時間で体積平均粒子径(nm)の小さな分散物を調製することが可能であることが分かった。
【0203】
〔比表面積が50m2/g以上の顔料を含有する顔料分散物の製造方法と性能評価〕
<非晶質なアゾ顔料の結晶変換工程を含まないアゾ顔料(1)の製造>
【0204】
[合成例1]α型結晶形態アゾ顔料(1)−29の製造
前記式(2)で表される化合物67.5gをリン酸530mLに溶解させ、氷冷して内温を3℃まで冷却した。内温が4℃以下になるように15分間かけて亜硝酸ナトリウム26.9gを分割して添加した。添加終了後、同温度にて50分間攪拌し、尿素18.6gを分割して添加し、ジアゾニウム塩溶液を得た。別に前記式(3)で表される化合物47.9gをメタノール1680mLに加え、還流下完溶させた。氷冷して内温を0℃まで冷却し、ここに上述のジアゾニウム塩溶液を内温が10℃以下になるように30分かけて添加した。内温5℃にて1時間30分攪拌した後、水1.6Lに添加し、30分間攪拌した。析出している結晶を濾別し、水1Lでかけ洗った。得られた結晶を水2.5Lに懸濁させ、28%アンモニア水を加えてpHが6.1になるように調製した。結晶を濾別し、水で十分にかけ洗いを行い、γ型結晶形態アゾ顔料を得た。得られた結晶をアセトン1.5Lに懸濁させ、昇温して還流下2時間攪拌した。結晶を熱時にて濾別して、アセトンで十分かけ洗いを行い、β型結晶形態アゾ顔料(1)−28を103.5g得た。
得られたβ型結晶形態アゾ顔料(1)−28を60℃24時間乾燥させ、α型結晶形態アゾ顔料(1)−29を92.8g(収率88.8%)得た。
【0205】
[合成例2]ε型結晶形態アゾ顔料(1)−31の製造
50gの酢酸、8gの硫酸からなる混合溶媒にニトロシル硫酸の43%硫酸溶液16.2gを20分かけて加えた。この溶液を3℃まで冷却し、前記式(2)で表される化合物11.55gを粉末で添加しジアゾ化反応を行った。同温度で1時間攪拌後、余分のニトロシル硫酸を尿素0.094gで失活させ、ジアゾニウム化合物調製液を得た。
この上述のジアゾニウム化合物調製液の中に前記式(3)で表される化合物10gを粉体にて5℃以下で分割添加した。添加終了後、20℃へ昇温し2時間反応させてアゾ化合物溶解液を得た。なお、カップリング反応中、顔料の析出は見られず、アゾ化合物溶解液は、得られたアゾ化合物を完全に溶解している状態であった。
メタノール150mLからなる貧溶媒を用意し5℃、200rpmで攪拌した。この貧溶媒中に上述のアゾ化合物溶解液を滴下した。
そのまま15分攪拌後、生成した結晶を濾別し、前記式(1)で表されるζ型結晶形態のアゾ顔料(1)−30を得た。
【0206】
得られたζ型結晶形態のアゾ顔料(1)−30の結晶を水200mLに懸濁させ、28%アンモニア水を加えてpHを6.0に調製した。析出している結晶(ζ型)を濾別し、水で十分にかけ洗いを行い、60℃で24時間乾燥させた。得られた結晶(ζ型)をアセトン200mLに懸濁させ、昇温して還流下2時間攪拌した。室温まで冷却し、結晶を濾別し、前記式(1)で表されるη型結晶形態のアゾ顔料を得た。得られた結晶をアセトンで十分にかけ洗いを行い、60℃にて24時間乾燥させて前記式(1)で表されるε型結晶形態アゾ顔料(1)−31を18.9g得た。
【0207】
[合成例3]δ型結晶形態のアゾ顔料(1)−32の合成
前記式(2)の化合物34.6gを酢酸150gに懸濁し、硫酸24gを内温が20℃〜30℃になるように滴下した。さらに内温が20℃〜30℃になるように43%ニトロシル硫酸の硫酸溶液48.6gを滴下し、内温20℃にて1時間攪拌後、尿素0.28gを添加してジアゾニウム塩溶液を得た。このジアゾニウム塩溶液に前記式(3)の化合物30gを内温が20℃〜30℃になるように分割して添加し、内温25℃にて1時間攪拌し、アゾ化合物の均一反応液を得た。別に360gのメタノールを内温25℃にて用意し、上述のアゾ化合物の均一反応液を内温が30℃以下になるように添加し、10分間攪拌した後、析出している固体を濾別した。300mLのメタノールでかけ洗いした後、水900mLに懸濁させ、28%アンモニウム水溶液を添加してpHを6.0に調整した。析出している固体を濾別し、前記式(1)で表されるアゾ顔料を得た。
得られたアゾ顔料の1次粒子の長軸方向の長さは、約2μmであった。
得られたアゾ顔料のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が6.5°、6.7°、9.1°および21.3°に特徴的なX線回折ピークを示し、ζ型結晶形態を有することが判明した。
上記で得られたζ型結晶形態アゾ顔料 5gをエチレングリコール50mLに懸濁させ、内温120℃まで昇温した後、同温度にて2時間攪拌した。内温30℃まで冷却した後、析出している固体を濾別し、前記式(1)で表されるδ型結晶形態のアゾ顔料(1)−32を3.9g得た。
得られたδ型結晶形態のアゾ顔料(1)−32を光学顕微鏡(ニコン(株)製:ECLIPSE LV150)で目視にて観察したところ、1次粒子の長軸方向の長さは、約80μmであった。
得られたδ型結晶形態のアゾ顔料(1)−32のX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示した。
【0208】
<ミリング>
〔実施例19及び比較例2〕
実施例19のアゾ顔料は、実施例1で得られたアゾ顔料(1)−2をミリングすることなく用いた。同様に、比較例2のアゾ顔料は、合成例2の途中で生成するアゾ顔料(1)−30をミリングすることなくそのまま用いた。
【0209】
その他の実施例20及び比較例3〜6のアゾ顔料は、下記のとおりミリングしたものを用いた。
〔実施例20〕
以下の組成となるように、スーパーミキサーに粗アゾ顔料及び食塩を投入して混合した。スーパーミキサーを回転させながらジエチレングリコールを少しずつ添加して混合物(以下、「予備混合物」ということがある)を調製した。
・実施例1で得られたアゾ顔料(1)−2 ・・・150g
・食塩(ナイカイ塩業(株)製 ナクルUM−05) ・・・1500g
・ジエチレングリコール ・・・300g
続いて、連続式1軸混練機(浅田鉄工(株)製、ミラクルKCK−L)の磨砕部および押し出し部の5箇所の温度を15〜20℃に、軸回転数50rpmに設定し、上記で得られた予備混合物を投入し、混練物を得た。この時、電流値(負荷)は約4Aで、吐出量は50g/分、吐出物の温度は16℃であった。
こうして得られた混練物を1%希塩酸5000gへ投入して攪拌処理を行った後、濾過および十分に水洗をして食塩およびジエチレングリコールを除去し、乾燥した。得られたδ型結晶形態アゾ顔料(1)−2−AのX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.6°、10.7°、17.3°、18.9°、20.0°、及び26.7°に特徴的なX線回折ピークを示した。
CuKα特性X線回折図を図25に示す。
【0210】
〔比較例3〜6〕
・比較例3:合成例3のアゾ顔料(1)−32
ブラッグ角(2θ±0.2°)が4.8°、7.1°、9.6°、10.7°、17.3°、18.9°、20.0°、及び26.7°に特徴的なX線回折ピークを示すδ型アゾ顔料(1)−32−Aを得た。
CuKα特性X線回折図を図26に示す。
【0211】
・比較例4:合成例1のアゾ顔料(1)−29
ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示すδ型アゾ顔料(1)−29−Aを得た。
CuKα特性X線回折図を図27に示す。
【0212】
・比較例5:合成例2のアゾ顔料(1)−31
ブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的なX線回折ピークを示すδ型アゾ顔料(1)−31−Aを得た。
CuKα特性X線回折図を図28に示す。
【0213】
・比較例6:合成例2の途中で生成するアゾ顔料(1)−30
ブラッグ角(2θ±0.2°)が6.6°、9.2°、10.3°および21.4°に特徴的なX線回折ピークを示すζ型アゾ顔料(1)−30−Aを得た。
CuKα特性X線回折図を図29に示す。
【0214】
〔実施例21〕
実施例20において、食塩の使用量を750gに変えたこと以外は実施例6と同様にして、δ型結晶アゾ顔料(1)−2−Bを得た。δ型結晶形態アゾ顔料(1)−2−BのX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.1°、9.5°、10.7°、17.3°、18.9°、20.0°、及び26.7°に特徴的なX線回折ピークを示した。
【0215】
〔実施例22〕
実施例20において、ジエチレングリコールの使用量を400gに変えたこと以外は実施例6と同様にして、δ型結晶アゾ顔料(1)−2−Cを得た。δ型結晶形態アゾ顔料(1)−2−CのX線回折の測定を上記の条件により行ったところ、ブラッグ角(2θ±0.2°)が4.8°、7.1°、9.5°、10.7°、17.3°、18.9°、20.0°、及び26.7°に特徴的なX線回折ピークを示した。
【0216】
<BET比表面積の測定>
あらかじめ80℃において真空乾燥した顔料0.1gを試料セルに加え、比表面積測定装置「MONOSORB MS−17」(ユアサアイオニクス(株)製)を用いて測定を行った。なお、測定には、He:N2=7:3の混合ガスを用いた。
【0217】
<顔料分散物の製造>
上記実施例20で合成したδ型結晶形態アゾ顔料(1)−2−Aを10g、オレイン酸ナトリウム5g、グリセリン10g、水75gを混合し、直径0.1mmのジルコニアビーズ375gとともにサンドグラインダミルTSG1(アイメックス社製)を用いて毎分1500回転、45℃で分散を行った。体積平均粒子径Mvが100nm以下となるまで分散を行った後、ジルコニアビーズを分離し、顔料分散物を得た。
実施例19及び比較例2のミリングしなかったアゾ顔料、及び、比較例3〜6で合成した各アゾ顔料についても同様の方法により、顔料分散物を得た。
実施例20及び比較例2〜6の顔料分散物の下記評価は、実施例19の顔料分散物との相対的な評価により行った。
【0218】
<分散性評価>
分散性は、上記顔料分散物の製造において、体積平均粒子径Mvが100nm以下となるまでの時間で評価した(日機装(株)製Nanotrac150(UPA−EX150)を用いて測定)。ミリング前のδ型結晶系アゾ顔料より更に優れるものをA、ミリング前のδ型結晶系アゾ顔料と同程度のものをB、ミリング前のδ型結晶系アゾ顔料より劣るものをCとした。結果を表2に示す。
【0219】
<着色力評価>
上記実施例及び比較例で得られた顔料分散物を水で10質量倍に希釈してからNo.3のバーコーターを用いてエプソン社製フォトマット紙に塗布した。得られた塗布物の画像濃度を反射濃度計(X−Rite社製X−Rite938)を用いて測定し、「着色力(OD:Optical Density)」を以下の基準で評価した。ODがミリング前のδ型結晶系アゾ顔料より更に優れるものをA、ミリング前のδ型結晶系アゾ顔料と同程度のものをB、ミリング前のδ型結晶系アゾ顔料より劣るものをCとした。結果を表2に示す。
【0220】
【表2】
【0221】
[合成例4]
窒素雰囲気下、ジプロピレングリコール58.7gを内温70℃に昇温し、ここにメタクリル酸を10.8g、メタクリル酸ベンジルを39.4g、V−601を1.2g、ジプロピレングリコールを58.7gを混合した溶液を3時間かけて滴下した。同温度にてさらに1時間攪拌した後、V−601(重合開始剤:和光純薬社製)を0.6g添加し、同温度にてさらに2時間攪拌した。同温度にて50%水酸化カリウム水溶液を11.3g滴下した後、同温度で1時間攪拌した。室温にまで冷却し、メタクリル酸ベンジル(66.7モル%)、メタクリル酸(33.3モル%)の共重合体(Mw=83,000、酸価140mgKOH)のジプロピレングリコール溶液を得た。
【0222】
[合成例5]
合成例4のV−601の量を1.2gから2.5gに増量し、さらに温度を86℃にし、同様の操作を行うことで、メタクリル酸ベンジル(66.7モル%)、メタクリル酸(33.3モル%)の共重合体(Mw=25,000、酸価128mgKOH)のジプロピレングリコール溶液を得た。
【0223】
[合成例6]
窒素雰囲気下、ジプロピレングリコール41.1gを内温70℃に昇温し、ここにメタクリル酸を9.6g、メタクリル酸メチルを16.8g、メタクリル酸2−エチルヘキシルを8.9g、V-601を2.5g、ジプロピレングリコールを41.1gを混合した溶液を3時間かけて滴下した。他の操作は合成例4と同様に行うことで、メタクリル酸メチル(47.8モル%)、メタクリル酸(31.8モル%)、メタクリル酸2−エチルヘキシル(20.4モル%)の共重合体(Mw=83,000、酸価154mgKOH)のジプロピレングリコール溶液を得た。
【0224】
〔実施例23〕
上記実施例20で合成したδ型結晶形態アゾ顔料(1)−2−Aを20gに合成例4で得られた分散剤(メタクリル酸ベンジル(66.7モル%)、メタクリル酸(33.3モル%)の共重合体のジプロピレングリコール溶液、Mw=83,000、酸価140mgKOH)32.2g(固形分含率30.8%、固形分9.9g)、水58gを混合し、直径0.1mmφのジルコニアビーズ375gとともにサンドグラインダミルTSG1(アイメックス社製)を用いて毎分1500回転、45℃で分散を3時間行った後、ジルコニアビーズを分離し、水で洗浄して、顔料濃度15.4重量%の粗顔料分散液(1)を99g得た(平均体積粒子径Mv=91.7nm)。
得られた粗顔料分散液(1)99gにデナコールEX−321(ナガセケムテックス株式会社製)を0.43g、6.18%のホウ酸水溶液3.02g、水40gを加え、70℃にて5時間攪拌した。反応終了後、室温まで冷却し、孔径1.0μmのフィルターを通して粗大粒子を除去した後、遠心分離機でさらに粗大粒子を沈降させた(7000rpm、10分間)。沈降した固体を除去した後、分画分子量50,000のフィルターを用いて、水で十分に洗浄し、顔料濃度10.8%の顔料分散液(1)を121g得た。
【0225】
〔実施例24〕
上記実施例6で合成したδ型結晶形態アゾ顔料(1)−3−Aを20gに合成例5で得られた分散剤(メタクリル酸ベンジル(66.7モル%)、メタクリル酸(33.3モル%)の共重合体のジプロピレングリコール溶液、Mw=25,000、酸価128mgKOH)28.6g(固形分含率35%、固形分10.0g)、水58gを混合し、直径0.1mmφのジルコニアビーズ375gとともにサンドグラインダミルTSG1(アイメックス社製)を用いて毎分1500回転、45℃で分散を2時間行った後、ジルコニアビーズを分離し、水で洗浄して、顔料濃度14.6重量%の粗顔料分散液(2)を109g得た(平均体積粒子径Mv=89.6nm)。
得られた粗顔料分散液(2)109gにデナコールEX−321(ナガセケムテックス株式会社製)を0.17g、6.18%のホウ酸水溶液1.19g、水50gを加え、70℃にて5時間攪拌した。反応終了後、室温まで冷却し、孔径1.0μmのフィルターを通して粗大粒子を除去した後、遠心分離機でさらに粗大粒子を沈降させた(7000rpm、10分間)。沈降した固体を除去した後、分画分子量50,000のフィルターを用いて、水で十分に洗浄し、顔料濃度9.7%の顔料分散液(2)を149g得た。
【0226】
〔実施例25〕
上記実施例6で合成したδ型結晶形態アゾ顔料(1)−3−Aを20gに合成例6で得られた分散剤(メタクリル酸メチル(47.8モル%)、メタクリル酸(31.8モル%)、メタクリル酸2−エチルヘキシル(20.4モル%)の共重合体のジプロピレングリコール溶液、Mw=83,000、酸価154mgKOH)28.4g(固形分含率35.2%、固形分10.0g)、水62gを混合し、直径0.1mmφのジルコニアビーズ375gとともにサンドグラインダミルTSG1(アイメックス社製)を用いて毎分1500回転、45℃で分散を3時間行った後、ジルコニアビーズを分離し、水で洗浄して、顔料濃度13.9重量%の粗顔料分散液(3)を112g得た(平均体積粒子径Mv=96.7nm)。
得られた粗顔料分散液(3)112gにデナコールEX−321(ナガセケムテックス株式会社製)を0.77g、6.18%のホウ酸水溶液5.4g、水30gを加え、70℃にて5時間攪拌した。反応終了後、室温まで冷却し、孔径1.0μmのフィルターを通して粗大粒子を除去した後、遠心分離機でさらに粗大粒子を沈降させた(7000rpm、10分間)。沈降した固体を除去した後、分画分子量50,000のフィルターを用いて、水で十分に洗浄し、顔料濃度10.4%の顔料分散液(3)を127g得た。
【0227】
〔実施例26〕
上記合成例2で合成したδ型結晶形態アゾ顔料(1)−2を20gに合成例4で得られた分散剤(メタクリル酸ベンジル(66.7モル%)、メタクリル酸(33.3モル%)の共重合体のジプロピレングリコール溶液、Mw=83,000、酸価140mgKOH)32.4g(固形分含率30.8%、固形分10.0g)、水46gを混合し、直径0.1mmφのジルコニアビーズ375gとともにサンドグラインダミルTSG1(アイメックス社製)を用いて毎分1500回転、45℃で分散を9時間行った後、ジルコニアビーズを分離し、水で洗浄して、顔料濃度15.3重量%の粗顔料分散液(4)を98g得た(平均体積粒子径Mv=98.2nm)。
得られた粗顔料分散液(4)98gにデナコールEX−321(ナガセケムテックス株式会社製)を0.75g、6.18%のホウ酸水溶液5.3g、水50gを加え、70℃にて5時間攪拌した。反応終了後、室温まで冷却し、孔径1.0μmのフィルターを通して粗大粒子を除去した後、遠心分離機でさらに粗大粒子を沈降させた(7000rpm、10分間)。沈降した固体を除去した後、分画分子量50,000のフィルターを用いて、水で十分に洗浄し、顔料濃度9.4%の顔料分散液(4)を145g得た。
【0228】
〔比較例7〕
P.Y.128(Cromophtal Yellow 8GN、チバ社製)を20gに合成例5で得られた分散剤(メタクリル酸ベンジル(66.7モル%)、メタクリル酸(33.3モル%)の共重合体のジプロピレングリコール溶液、Mw=83,000、酸価140mgKOH)32.4g(固形分含率30.8%、固形分10.0g)、水46gを混合し、直径0.1mmφのジルコニアビーズ375gとともにサンドグラインダミルTSG1(アイメックス社製)を用いて毎分1500回転、45℃で分散を6時間行った後、ジルコニアビーズを分離し、水で洗浄して、顔料濃度16.3重量%の比較粗顔料分散液(1)を81g得た(平均体積粒子径Mv=93.4nm)。
得られた比較粗顔料分散液(1)81gにデナコールEX−321(ナガセケムテックス株式会社製)を0.66g、6.18%のホウ酸水溶液4.7g、水45gを加え、70℃にて5時間攪拌した。反応終了後、室温まで冷却し、孔径1.0μmのフィルターを通して粗大粒子を除去した後、遠心分離機でさらに粗大粒子を沈降させた(7000rpm、10分間)。沈降した固体を除去した後、分画分子量50,000のフィルターを用いて、水で十分に洗浄し、顔料濃度10.2%の比較顔料分散液(4)を115g得た。
【0229】
<着色力評価>
上記実施例23〜26で得られた顔料分散物を水で10質量倍に希釈してからNo.3のバーコーターを用いてエプソン社製フォトマット紙に塗布した。得られた塗布物の画像濃度を反射濃度計(X−Rite社製X−Rite938)を用いて測定し、「着色力(OD:Optical Density)」を以下の基準で評価した。ODが実施例20より更に優れるものをA、実施例20と同程度のものをB、実施例20より劣るが、実施例19より優れるものをC、実施例19と同程度のものをD、実施例19より劣るものをEとした。結果を表3に示す。
【0230】
【表3】
【0231】
〔実施例27〕
実施例23で得られた顔料分散液(1)を固形分で5質量%、グリセリン10質量%、2−ピロリドン5質量%、1,2−ヘキサンジオール2質量%、トリエチレングリコールモノブチルエーテル2質量%、プロピレングリコール0.5質量%、サーフィノール465を1質量%、イオン交換水74.5質量%になるように各成分を加えて、得られた混合液を1.2μmのフィルター(アセチルセルロース膜、外径:25mm、富士フイルム(株)社製)を取り付けた容量20mLのシリンジで濾過し、粗大粒子を除去することにより顔料インク(1)を得た。
【0232】
〔実施例28、29〕
上記実施例27における顔料分散液(1)を、実施例24で得られた顔料分散液(2)及び実施例25で得られた顔料分散液(3)に代えて、それぞれ顔料インク(2)及び顔料インク(3)を得た。
【0233】
〔比較例8〕
セイコーエプソン(株)社製ICY−42を用いて、下記評価を行った。
【0234】
<評価>
インク液をセイコーエプソン(株)社製インクジェットプリンターPX−V630のイエローインク液のカートリッジに装てんし、受像シートはセイコーエプソン(株)社製写真用紙クリスピア<高光沢>にカラー設定:色補正なし、印刷品質:フォトで、イエローのO.D.が1.0となるように印画したものおよびベタ印画したものを作製し、耐光性および濃度を評価した。
【0235】
<着色力評価>
ベタ印画したものの画像濃度を反射濃度計(X−Rite社製X−Rite938)を用いて測定し、「着色力(OD:Optical Density)」を以下の基準で評価した。ODが比較例8より優れるものをA、比較例8と同程度のものをB、比較例8より劣るものをCとした。結果を表4に示す。
【0236】
<耐光性評価>
O.D.が1.0となるように印画したものを、フェードメーターを用いてキセノン光(99000lux;TACフィルター存在下)を28日間照射し、キセノン照射前後の画像濃度を反射濃度計を用いて測定し、色素残存率[(照射後濃度/照射前濃度)×100%]が90%以上の場合をA、90%未満の場合をBとして評価した。結果を表4に示す。
【0237】
【表4】
【特許請求の範囲】
【請求項1】
非晶質な下記式(1)で表されるアゾ化合物、その塩、水和物又は溶媒和物を結晶変換することを特徴とする下記式(1)で表されるアゾ顔料の製造方法。
【化1】
【請求項2】
非晶質の前記式(1)で表されるアゾ化合物、その塩、水和物又は溶媒和物を溶媒中で加熱攪拌することにより結晶変換することを特徴とする請求項1に記載のアゾ顔料の製造方法。
【請求項3】
非晶質な前記式(1)で表されるアゾ化合物を、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が4.8°、7.2°及び9.7°に特徴的X線回折ピークを有する結晶形に結晶変換することを特徴とする請求項1又は2に記載のアゾ顔料の製造方法。
【請求項4】
非晶質な前記式(1)で表されるアゾ化合物を、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が4.8°、7.2°、9.7°、20.1°、及び26.8°に特徴的X線回折ピークを有する結晶形に結晶変換することを特徴とする請求項1〜3のいずれか一項に記載のアゾ顔料の製造方法。
【請求項5】
非晶質な前記式(1)で表されるアゾ化合物を、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的X線回折ピークを有する結晶形に結晶変換することを特徴とする請求項1〜4のいずれか一項に記載のアゾ顔料の製造方法。
【請求項6】
(i)非晶質な前記式(1)で表されるアゾ化合物を、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が4.8°、7.2°及び9.7°に特徴的X線回折ピークを有する結晶形に結晶変換する工程、及び、(ii)前記工程(i)で得たアゾ顔料と、水溶性無機塩と、水溶性有機溶剤とを含む混合物を混練して、窒素吸着法によるBET比表面積を50m2/g以上の結晶にする工程、を含むことを特徴とする、請求項3〜5のいずれか一項に記載のアゾ顔料、又はその互変異性体の製造方法。
【請求項7】
(a)ジアゾ化剤と下記式(2)で表されるアミノ化合物とを混合させる工程、(b)前記工程(a)で得た反応生成物と下記式(3)で表されるカップリング成分とを混合することにより反応を行い、該反応により生成した下記式(1)で表されるアゾ化合物の少なくとも一部が溶解した溶液を得る工程、(c)前記工程(b)で得た溶液と、前記アゾ化合物に対する貧溶媒とを混合して、下記式(1)で表される非晶質なアゾ化合物を晶析させる工程、を含むことを特徴とする請求項1〜6のいずれか一項に記載のアゾ顔料の製造方法。
【化2】
【化3】
【化4】
【請求項8】
前記工程(b)において、得られた溶液が酸性溶液であることを特徴とする請求項7に記載のアゾ顔料の製造方法。
【請求項9】
前記酸性溶液が、酢酸及び硫酸の少なくとも一方を含むことを特徴とする請求項8に記載のアゾ顔料の製造方法。
【請求項10】
前記工程(b)において、得られた溶液が、カップリング反応により生成した前記式(1)で表されるアゾ化合物が完全に溶解した溶液であることを特徴とする請求項7〜9のいずれか一項に記載のアゾ顔料の製造方法。
【請求項11】
前記(c)工程において、前記工程(b)で得られた前記式(1)で表されるアゾ化合物の溶解度が1g/L以下である貧溶媒と混合することを特徴とする請求項7〜10のいずれか一項に記載のアゾ顔料の製造方法。
【請求項12】
前記貧溶媒が、水及び、炭素数1〜3のアルコール、炭素数1〜6のグリコールからなる群から選ばれる1種以上の溶媒を含むことを特徴とする請求項7〜11のいずれか一項に記載のアゾ顔料の製造方法。
【請求項13】
請求項1〜12のいずれか一項に記載の製造方法により得られるアゾ顔料。
【請求項14】
請求項13に記載のアゾ顔料、分散剤、及び水を含む顔料分散物。
【請求項15】
分散剤が水溶性高分子であることを特徴とする請求項14に記載の水系顔料分散物。
【請求項16】
前記水溶性高分子分散剤が少なくとも1つのカルボキシ基を有し、少なくとも50mgKOH/g以上の酸価を有することを特徴とする請求項15に記載の水系顔料分散物。
【請求項17】
前記水系顔料分散物が、架橋剤により架橋されていることを特徴とする請求項15又は16に記載の水系顔料分散物。
【請求項1】
非晶質な下記式(1)で表されるアゾ化合物、その塩、水和物又は溶媒和物を結晶変換することを特徴とする下記式(1)で表されるアゾ顔料の製造方法。
【化1】
【請求項2】
非晶質の前記式(1)で表されるアゾ化合物、その塩、水和物又は溶媒和物を溶媒中で加熱攪拌することにより結晶変換することを特徴とする請求項1に記載のアゾ顔料の製造方法。
【請求項3】
非晶質な前記式(1)で表されるアゾ化合物を、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が4.8°、7.2°及び9.7°に特徴的X線回折ピークを有する結晶形に結晶変換することを特徴とする請求項1又は2に記載のアゾ顔料の製造方法。
【請求項4】
非晶質な前記式(1)で表されるアゾ化合物を、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が4.8°、7.2°、9.7°、20.1°、及び26.8°に特徴的X線回折ピークを有する結晶形に結晶変換することを特徴とする請求項1〜3のいずれか一項に記載のアゾ顔料の製造方法。
【請求項5】
非晶質な前記式(1)で表されるアゾ化合物を、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が4.8°、7.2°、9.5°、9.7°、10.7°、17.4°、19.0°、20.1°、及び26.8°に特徴的X線回折ピークを有する結晶形に結晶変換することを特徴とする請求項1〜4のいずれか一項に記載のアゾ顔料の製造方法。
【請求項6】
(i)非晶質な前記式(1)で表されるアゾ化合物を、CuKα特性X線回折におけるブラッグ角(2θ±0.2°)が4.8°、7.2°及び9.7°に特徴的X線回折ピークを有する結晶形に結晶変換する工程、及び、(ii)前記工程(i)で得たアゾ顔料と、水溶性無機塩と、水溶性有機溶剤とを含む混合物を混練して、窒素吸着法によるBET比表面積を50m2/g以上の結晶にする工程、を含むことを特徴とする、請求項3〜5のいずれか一項に記載のアゾ顔料、又はその互変異性体の製造方法。
【請求項7】
(a)ジアゾ化剤と下記式(2)で表されるアミノ化合物とを混合させる工程、(b)前記工程(a)で得た反応生成物と下記式(3)で表されるカップリング成分とを混合することにより反応を行い、該反応により生成した下記式(1)で表されるアゾ化合物の少なくとも一部が溶解した溶液を得る工程、(c)前記工程(b)で得た溶液と、前記アゾ化合物に対する貧溶媒とを混合して、下記式(1)で表される非晶質なアゾ化合物を晶析させる工程、を含むことを特徴とする請求項1〜6のいずれか一項に記載のアゾ顔料の製造方法。
【化2】
【化3】
【化4】
【請求項8】
前記工程(b)において、得られた溶液が酸性溶液であることを特徴とする請求項7に記載のアゾ顔料の製造方法。
【請求項9】
前記酸性溶液が、酢酸及び硫酸の少なくとも一方を含むことを特徴とする請求項8に記載のアゾ顔料の製造方法。
【請求項10】
前記工程(b)において、得られた溶液が、カップリング反応により生成した前記式(1)で表されるアゾ化合物が完全に溶解した溶液であることを特徴とする請求項7〜9のいずれか一項に記載のアゾ顔料の製造方法。
【請求項11】
前記(c)工程において、前記工程(b)で得られた前記式(1)で表されるアゾ化合物の溶解度が1g/L以下である貧溶媒と混合することを特徴とする請求項7〜10のいずれか一項に記載のアゾ顔料の製造方法。
【請求項12】
前記貧溶媒が、水及び、炭素数1〜3のアルコール、炭素数1〜6のグリコールからなる群から選ばれる1種以上の溶媒を含むことを特徴とする請求項7〜11のいずれか一項に記載のアゾ顔料の製造方法。
【請求項13】
請求項1〜12のいずれか一項に記載の製造方法により得られるアゾ顔料。
【請求項14】
請求項13に記載のアゾ顔料、分散剤、及び水を含む顔料分散物。
【請求項15】
分散剤が水溶性高分子であることを特徴とする請求項14に記載の水系顔料分散物。
【請求項16】
前記水溶性高分子分散剤が少なくとも1つのカルボキシ基を有し、少なくとも50mgKOH/g以上の酸価を有することを特徴とする請求項15に記載の水系顔料分散物。
【請求項17】
前記水系顔料分散物が、架橋剤により架橋されていることを特徴とする請求項15又は16に記載の水系顔料分散物。
【図1】

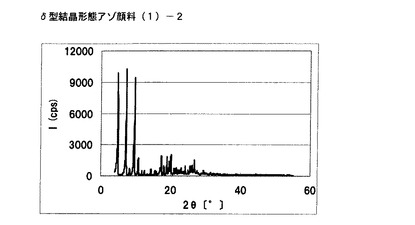
【図2】
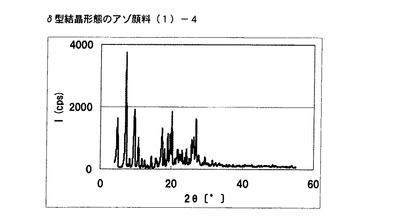
【図3】
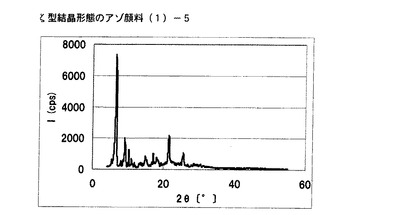
【図4】
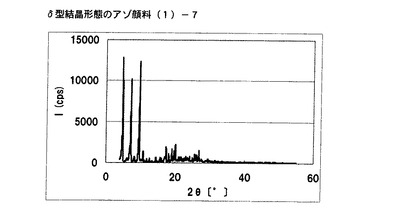
【図5】
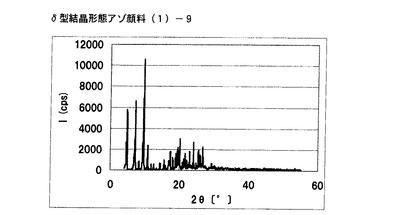
【図6】
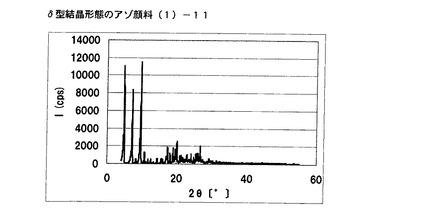
【図7】
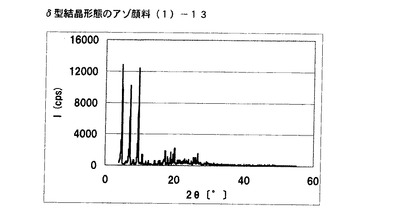
【図8】
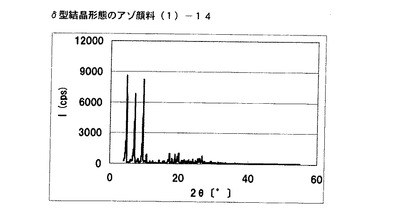
【図9】
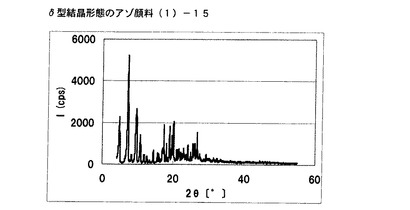
【図10】
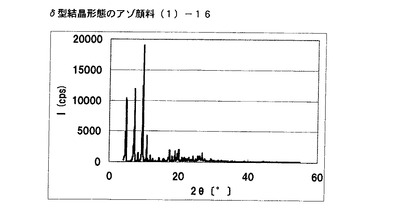
【図11】
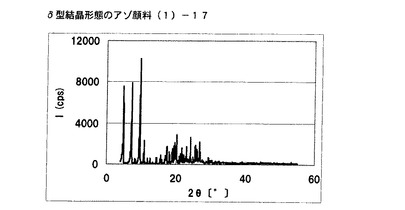
【図12】
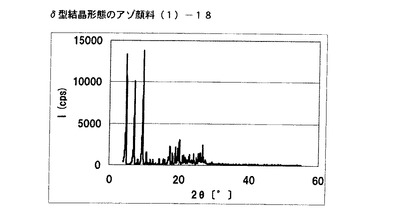
【図13】
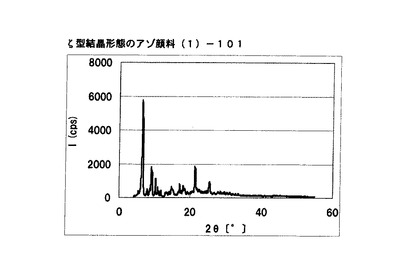
【図14】
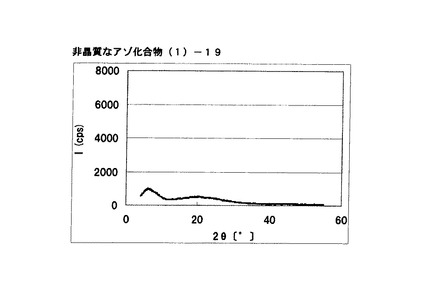
【図15】
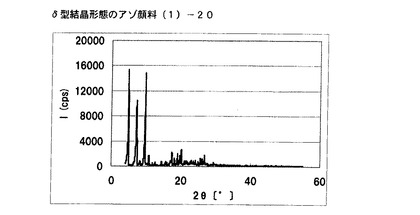
【図16】
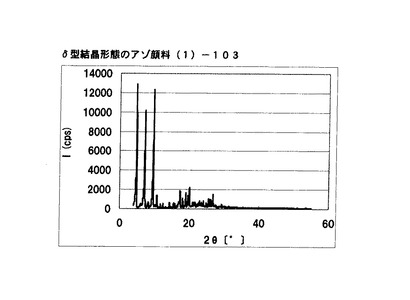
【図17】
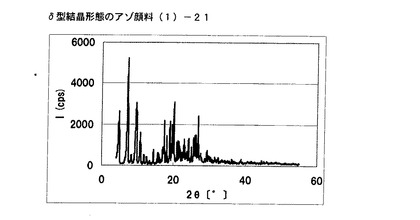
【図18】
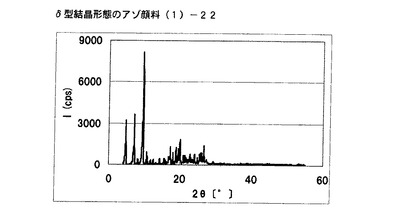
【図19】
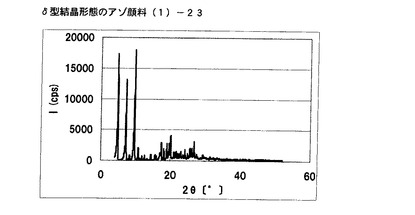
【図20】
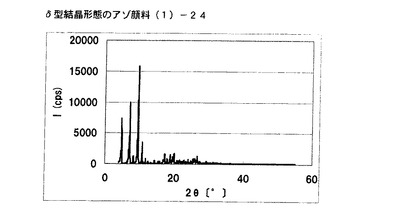
【図21】
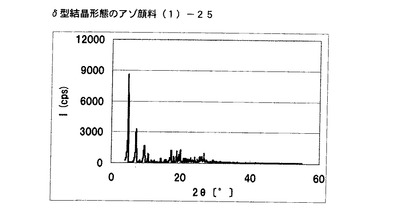
【図22】
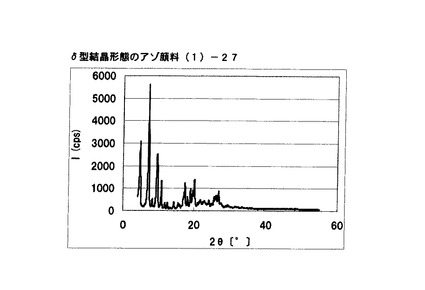
【図23】
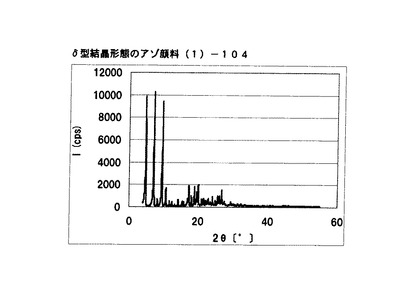
【図24】
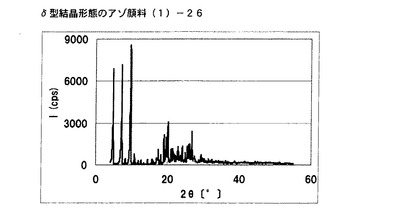
【図25】
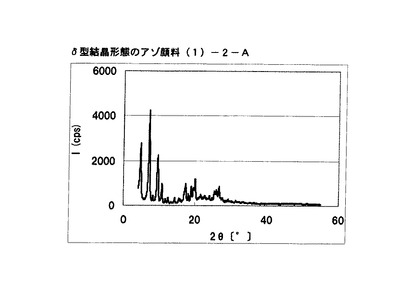
【図26】
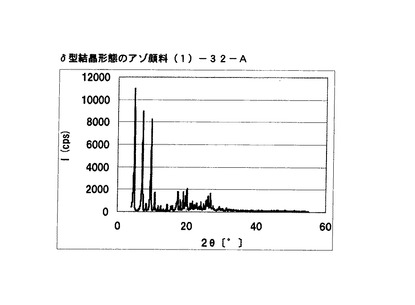
【図27】

【図28】
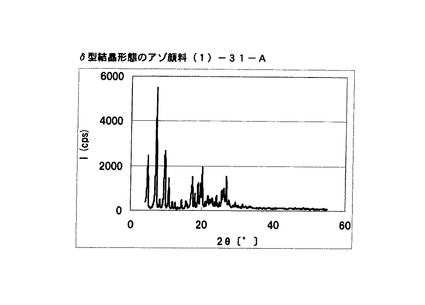
【図29】
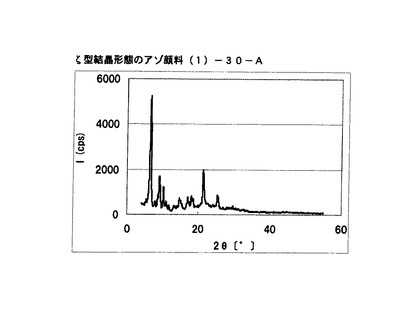
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【公開番号】特開2013−49827(P2013−49827A)
【公開日】平成25年3月14日(2013.3.14)
【国際特許分類】
【出願番号】特願2012−33394(P2012−33394)
【出願日】平成24年2月17日(2012.2.17)
【出願人】(306037311)富士フイルム株式会社 (25,513)
【Fターム(参考)】
【公開日】平成25年3月14日(2013.3.14)
【国際特許分類】
【出願日】平成24年2月17日(2012.2.17)
【出願人】(306037311)富士フイルム株式会社 (25,513)
【Fターム(参考)】
[ Back to top ]