
アテローム性動脈硬化症、脂質代謝異常及び関連症状の治療方法
患者にニコチン酸又は別のニコチン酸受容体アゴニストをDP受容体アンタゴニストと併用投与するアテローム性動脈硬化症の治療方法を開示する。DP受容体アンタゴニストはその非投与時に発生する可能性のあるフラッシングを緩和、予防又は解消するために投与する。

【発明の詳細な説明】
【技術分野】
【0001】
ナイアシンないしニコチン酸(ピリジン−3−カルボン酸)は血清高密度リポ蛋白(HDL)値を上昇させるのに有効であることがよく知られている薬剤である。しかし、ニコチン酸は皮膚血管拡張(フラッシングとも言う)を併発することが多い。この副作用はニコチン酸によりプロスタグランジンD2が皮膚内に放出されることに起因し、非常に重度であるため、ニコチン酸治療を中断する患者が多い。本発明は実質的フラッシングを併発せずに治療を進めることができるように、その非投与時に発生する可能性のある皮膚血管拡張を緩和又は解消する化合物とニコチン酸又は別のニコチン酸受容体アゴニストを併用投与することによりアテローム性動脈硬化症、脂質代謝異常、糖尿病及び関連症状を治療する方法に関する。これはヒトではニコチン酸又はニコチン酸受容体アゴニストとDP受容体を阻害する化合物を投与することにより達成される。
【背景技術】
【0002】
各種サブタイプの受容体がプロスタグランジンD2と相互作用する。1種のプロスタグランジンD2受容体は「DP」と呼ばれ、別のプロスタグランジンD2受容体は「CRTH2」として知られる。本発明はその非投与時に発生する可能性のあるフラッシングを予防するか、最小限にするか又は緩和するためにDP受容体のアンタゴニスムを利用する。
【発明の開示】
【発明が解決しようとする課題】
【0003】
従って、本発明の1つの目的はニコチン酸又は別のニコチン酸受容体アゴニストを使用してアテローム性動脈硬化症、脂質代謝異常、糖尿病及び関連症状についてヒトを治療する間に副作用としての実質的フラッシング(頻度及び/又は重度)を解消、抑制又は緩和することである。
【0004】
本発明の別の目的は副作用一般を最小限にするアテローム性動脈硬化症の併用治療を提供することである。
【0005】
本発明の更に別の目的は経口用規定併用医薬組成物を提供することである。
【0006】
以上及び他の目的は本明細書の記載から自明である。
【課題を解決するための手段】
【0007】
治療を必要とするヒト患者におけるアテローム性動脈硬化症の治療方法として、実質的フラッシングを併発せずにアテローム性動脈硬化症を治療するために有効な量であるニコチン酸約1000mgと、約5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg、50mg、75mg、100mg及び150mgから選択される量のDP受容体アンタゴニストを患者に投与することを含む方法を開示する。
【発明を実施するための最良の形態】
【0008】
以下、添付図面に関して本発明を例証する。
【0009】
ナイアシンないしニコチン酸(ピリジン−3−カルボン酸)は高密度リポ蛋白(HDL)値の上昇と、脂質プロフィルの他の有益な変化(超低密度(VLDL)、低密度リポ蛋白(LDL)、トリグリセリド、遊離脂肪酸(FFA)及びリポ蛋白(a)[Lp(a)]の低下)に有効であることがよく知られている薬剤である。ニコチン酸は約50mgから約8g/日等の治療有効用量をヒトに投与した場合にHDL値を上昇させる。しかし、ニコチン酸は皮膚血管拡張(フラッシングとも言う)を併発することが多い。フラッシングは一般に皮膚発赤とほてり、痒み又は炎症を伴う。フラッシングは非常に不快であり、非常に重度であるため、ニコチン酸治療を中断する患者が多い。本発明は実質的フラッシングを併発せずにニコチン酸又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストによりアテローム性動脈硬化症及び本明細書に記載する他の疾患及び症状の治療、予防又は逆行させる方法に関する。これはヒトではニコチン酸又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストと、DP受容体を阻害する化合物を投与し、頻度及び/又は重度においてフラッシング効果を予防するか、緩和するか又は最小限にすることにより達成される。
【0010】
プロスタグランジンD2と相互作用する受容体は「DP」及び「CRTH2」と呼ぶ少なくとも2種類がある。本発明は主にDP受容体のアンタゴニストと併用するニコチン酸又はニコチン酸受容体アゴニストに関する。
【0011】
本発明の有利な1側面は治療を必要とするヒト患者におけるアテローム性動脈硬化症の治療方法として、実質的フラッシングを併発せずにアテローム性動脈硬化症を治療するために有効な量でニコチン酸又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストと、DP受容体アンタゴニストを患者に投与することを含む方法である。
【0012】
本発明の別の有利な側面は治療を必要とするヒト患者における血清HDL値の上昇方法として、ニコチン酸又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストとDP受容体アンタゴニストを患者に投与することを含み、前記併用が実質的フラッシングを併発せずに患者の血清HDL値を上昇させるために有効である方法に関する。
【0013】
本発明の別の有利な側面は治療を必要とするヒト患者における脂質代謝異常の治療方法として、実質的フラッシングを併発せずに脂質代謝異常を治療するために有効な量でニコチン酸又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストと、DP受容体アンタゴニストを患者に投与することを含む方法に関する。
【0014】
本発明の別の有利な側面は治療を必要とするヒト患者における血清VLDL又はLDL値の低下方法として、実質的フラッシングを併発せずに患者の血清VLDL又はLDL値を低下させるために有効な量でニコチン酸又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストと、DP受容体アンタゴニストを患者に投与することを含む方法に関する。
【0015】
本発明の別の有利な側面は治療を必要とするヒト患者における血清トリグリセリド値の低下方法として、実質的フラッシングを併発せずに患者の血清トリグリセリド値を低下させるために有効な量でニコチン酸又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストと、DP受容体アンタゴニストを患者に投与することを含む方法に関する。
【0016】
本発明の別の有利な側面は治療を必要とするヒト患者における血清Lp(a)値の低下方法として、実質的フラッシングを併発せずに患者の血清Lp(a)値を低下させるために有効な量でニコチン酸又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストと、DP受容体アンタゴニストを患者に投与することを含む方法に関する。本明細書で使用するLp(a)とはリポ蛋白質(a)を意味する。
【0017】
本発明の特に有利な側面はニコチン酸又はその塩もしくは溶媒和物を利用する上記各方法に関する。ニコチン酸を使用すると更に有利である。有利な更に別の側面では、DP受容体アンタゴニストは患者におけるフラッシング効果を抑制、緩和又は予防するために有効な量のDP受容体を選択的に調節する。
【0018】
本発明の特に有利な別の側面はニコチン酸を利用し、DP受容体アンタゴニストがDP受容体を選択的に調節し、CRTH2受容体を実質的に調節しない上記各方法に関する。
【0019】
本発明の特に有利な別の側面は治療を必要とするヒト患者におけるアテローム性動脈硬化症、脂質代謝異常、糖尿病又は関連症状の治療方法として、実質的フラッシングを併発せずにアテローム性動脈硬化症、脂質代謝異常、糖尿病又は関連症状を治療するために有効な量で、ニコチン酸又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストと、DP受容体アンタゴニストを患者に併用投与することを含む方法に関する。
【0020】
本発明の特に有利な別の側面は治療を必要とするヒト患者におけるアテローム性動脈硬化症、脂質代謝異常、糖尿病又は関連症状の治療方法として、実質的フラッシングを併発せずにアテローム性動脈硬化症、脂質代謝異常、糖尿病又は関連症状を治療するために有効な量で、a)ナイアシンにより誘発されるフラッシング応答を抑制するために有効な量のアスピリンと、b)ニコチン酸又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストと、c)DP受容体アンタゴニストを患者に併用投与することを含む方法に関する。
【0021】
本発明の別の有利な側面は治療を必要とするヒト患者におけるアテローム性動脈硬化症、脂質代謝異常、糖尿病又は関連症状の治療方法として、ニコチン酸により誘発されるフラッシング応答を抑制又は緩和するために有効な量のアスピリンで患者を予備治療又は治療する段階と、実質的フラッシングを併発せずにアテローム性動脈硬化症、脂質代謝異常、糖尿病又は関連症状を治療するために有効な量のニコチン酸又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストとDP受容体アンタゴニストを併用投与する段階を含む方法に関する。
【0022】
本発明の1側面は実質的フラッシングを併発せずにヒトにおけるアテローム性動脈硬化症を治療するための、ニコチン酸又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストと、DP受容体アンタゴニスト化合物の併用である。
【0023】
本発明の特に有利な別の側面はDP受容体アンタゴニストが化合物AからAJと医薬的に許容可能なその塩及び溶媒和物から構成される群から選択される上記方法に関する。
【0024】
DP受容体を選択的に阻害し、フラッシング効果を抑制するために特に有用な化合物の例としては以下の化合物と、医薬的に許容可能なその塩及び溶媒和物が挙げられる。
【0025】
【化1】
【0026】
本明細書で使用するアテローム性動脈硬化症とは大・中動脈壁の最下層にコレステロールと脂質を含むアテローム斑が沈着することを特徴とする血管疾患の1形態を意味する。アテローム性動脈硬化症は該当医学分野に携わる医師により認識及び理解される血管疾患及び症状を含む。血管再建術後の再狭窄を含むアテローム硬化性心血管疾患、冠状動脈性心臓病(冠動脈疾患又は虚血性心疾患とも言う)、多発梗塞性痴呆を含む脳血管疾患、及び勃起障害を含む末梢血管疾患はいずれもアテローム性動脈硬化症の臨床徴候であるため、「アテローム性動脈硬化症」及び「アテローム硬化性疾患」に含まれる。
【0027】
「脂質代謝異常」はHDL(低)、LDL(高)、VLDL(高)、トリグリセリド(高)、リポ蛋白質(a)(高)、FFA(高)及び他の血清脂質又はその組み合わせ等の異常血漿脂質値を意味するために従来通りの意味で使用する。併発症を伴わない症状でもよいし、糖尿病(糖尿病性脂質代謝異常)、メタボリックシンドローム等の特定関連疾患又は症状の一部でもよい。従って、本発明には併発症を伴わない脂質代謝異常と、基礎症状に関連する脂質代謝異常が含まれる。
【0028】
「患者」なる用語は症状の予防又は治療のために本発明の活性剤を使用する哺乳動物、特にヒトを意味する。患者への薬剤投与は自己投与と他者による投与の両者を含む。患者は既存疾患又は症状の治療を必要とする場合もあるし、アテローム性動脈硬化症の発症の危険を予防又は低減するために予防処置を必要とする場合もある。
【0029】
「治療有効量」なる用語は所望生物学的又は医学的応答を誘発する薬剤の量を意味する。1例として、ニコチン酸は多くの場合には約50mgから約8g/日、好ましくは約0.5gから約3.0g/日の用量を投与する。ニコチン酸の好適用量は約1から2g/日である。
【0030】
「予防有効量」及び「予防するために有効な量」なる用語は予防する必要がある生物学的又は医学的イベントの発生の危険を予防又は低減する薬剤の量を意味する。多くの場合には、予防有効量は治療有効量と同一である。
【0031】
本明細書に記載する本発明は冠状動脈性心臓病イベント、脳血管イベント、及び/又は間欠性跛行の発生、又は潜在性がある場合にはその再発の危険を予防又は低減するために本明細書に記載する化合物及び組成物を投与することを含む。冠状動脈性心臓病イベントはCHD死、心筋梗塞(即ち、心臓発作)、及び冠状血管再建術を含むものとする。脳血管イベントは虚血性又は出血性脳卒中(脳血管障害とも言う)及び一過性脳虚血発作を含むものとする。間欠性跛行は末梢血管疾患の臨床徴候である。本明細書で使用する「アテローム硬化性疾患イベント」なる用語は、冠状動脈性心臓病イベント、脳血管イベント及び間欠性跛行を含むものとする。非致死性アテローム硬化性疾患イベントを過去に1回以上経験した人はこのようなイベントの再発の可能性があるとみなす。
【0032】
従って、本発明はアテローム硬化性疾患イベントの初回又は2回目以降の発生の危険を予防又は低減するための方法として、実質的フラッシングを予防又は最小限にしながらこのようなイベントの危険のある患者に予防有効量の本明細書に記載する化合物を投与することを含む方法も提供する。患者は投与時に既にアテローム硬化性疾患をもつものでもよいし、発症する危険があるものでもよい。
【0033】
本方法は更に実質的フラッシングを予防又は最小限にしながら新規アテローム病変又は斑形成を予防又は遅延させ、既存病変又は斑の進行を予防又は遅延させ、更に既存病変又は斑を退行させる方法に関する。
【0034】
従って、本発明の1側面はアテローム斑進行の停止又は遅延を含むアテローム性動脈硬化症の停止又は遅延方法として、治療を必要とする患者にニコチン酸又は別のニコチン酸受容体アゴニストと共に治療有効量の本明細書に記載のDPアンタゴニストのいずれかを投与することを含む方法に関する。本方法も治療開始時に存在するアテローム斑(即ち、「既存アテローム斑」)の進行の停止又は遅延と、アテローム性動脈硬化症をもつ患者における新規アテローム斑の形成の停止又は遅延を含む。
【0035】
本発明の別の側面はアテローム斑破裂の危険の予防又は低減方法として、治療を必要とする患者にニコチン酸又は別のニコチン酸受容体アゴニストと共に予防有効量の本明細書に記載の化合物のいずれかを投与することを含む方法に関する。本明細書で使用する破裂とは斑が遊離し、場合によっては血管に詰まることを意味する。本発明の別の側面はアテローム性動脈硬化症の発症の危険の予防又は低減方法として、このような治療を必要とする患者に予防有効量の本明細書に記載の化合物を投与することを含む方法に関する。
【0036】
本発明の別の側面はアテローム性動脈硬化症、脂質代謝異常又は関連症状の治療又は予防方法として、フラッシングを抑制又は緩和するのに有効な量のDP受容体アンタゴニストでこのような治療を必要とするヒト患者を予備治療する段階と、その後、実質的フラッシングを併発せずに前記アテローム性動脈硬化症、脂質代謝異常又は関連症状を治療又は予防するために有効な量のニコチン酸、又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストで前記患者を治療する段階を含む方法に関する。
【0037】
本発明の別の側面はHMG Co−Aレダクターゼ阻害剤で患者を予備治療又は治療する段階を更に含む上記方法に関する。
【0038】
本発明の別の側面はHMG Co−Aレダクターゼ阻害剤がシンバスタチンである上記症状の治療又は予防方法に関する。
【0039】
本明細書に記載する方法の1側面は、本明細書に記載する結果を達成するために有効な量のニコチン酸又は別のニコチン酸受容体アゴニスト化合物と、DP受容体を選択的に調節し、CRTH2受容体を実質的に調節しないDP受容体アンタゴニストの併用に関する。従って、DP受容体アンタゴニストはDP受容体に対する親和性(即ち、Ki)がCRTH2受容体に対する親和性の少なくとも約10倍(Ki値の数値が小さい)である。これらの基準に従ってDPと選択的に相互作用する任意化合物を「DP選択的」であるとみなす。
【0040】
「実質的フラッシングを併発せずに」なる用語は治療量のニコチン酸を投与する場合に頻発する副作用を意味する。ニコチン酸のフラッシング効果は一般に患者が治療用量の薬剤に対する耐性を生じるにつれて頻度と重度が低下するが、それでもある程度まではフラッシング効果が生じる。従って、「実質的フラッシングを併発せずに」とはフラッシングが生じる場合にはその重度が低下することを意味するか、又は非投与時よりもフラッシングイベントの発生頻度が低下することを意味する。フラッシング発生率は少なくとも約3分の1低下することが好ましく、発生率は2分の1低下することがより好ましく、フラッシング発生率は約3分の2以上低下することが最も好ましい。同様に、重度は少なくとも約3分の1低下することが好ましく、少なくとも2分の1低下することがより好ましく、少なくとも約3分の2低下することが最も好ましい。当然のことながら、フラッシング発生率及び重度は100%低下することが最も好ましいが、その必要はない。
【0041】
残留フラッシングを抑制又は緩和するためにアスピリンも投与することができる。ニコチン酸により誘発される残留フラッシング応答を抑制又は緩和するために有用なアスピリンの量は一般に治療用量を越えず、典型的には治療用量よりも少なく、約20から25mgから約650mgまでである。特に、本明細書に記載する本発明に有用なアスピリンの量は投与するDP受容体アンタゴニストにより抑制又は緩和されない残留フラッシングを抑制又は緩和するために必要又は有用な量とする。アスピリンは一般にニコチン酸よりも先に投与し、例えばニコチン酸とDP受容体アンタゴニストを投与する約30分から1時間前とするが、ニコチン酸及びDP受容体アンタゴニストと同時に投与してもよい。例えばニコチン酸とDP受容体アンタゴニストを投与する約30分前に単回投与することによりニコチン酸とDP受容体アンタゴニストの前に投与してもよいし、ニコチン酸及びDP受容体アンタゴニストと同時に投与し、必要に応じてDP受容体アンタゴニストにより抑制されない残留フラッシング応答を抑制又は緩和するために十分又は有効な量を約4時間までの間隔で反復投与してもよい。
【0042】
任意特定患者の特定投与レジメン及びレベルは年齢、体重、一般健康状態、性別、食事、投与時間、投与経路、排泄率、薬剤併用及び患者の症状の重篤度等の種々の因子により異なる。症状を予防、逆行、又は進行阻止するために必要な治療有効用量又は予防有効用量を決定する目的でこれらの因子を考慮することは通常の知識をもつ臨床医が可能な範囲内である。本明細書に記載する化合物は患者に該当する症状を治療又は予防するために適切な期間(例えば数カ月、数年又は患者の終生の治療期間)毎日投与することが予想される。
【0043】
特に有利な治療方法の1側面は治療を必要とするヒト患者におけるアテローム性動脈硬化症の治療方法として、実質的フラッシングを併発せずにアテローム性動脈硬化症を治療するために有効な量であるニコチン酸約1000mgと、約5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg、50mg、75mg、100mg及び150mgから選択される量のDP受容体アンタゴニストを患者に投与することを含む方法に関する。
【0044】
特に有利な治療方法の別の側面は治療を必要とするヒト患者におけるアテローム性動脈硬化症の治療方法として、実質的フラッシングを併発せずにアテローム性動脈硬化症を治療するために有効な量であるニコチン酸約2000mgと、約10mg、20mg、30mg、37.5mg、40mg、50mg、75mg、100mg、150mg、200mg及び300mgから選択される量のDP受容体アンタゴニストを患者に投与することを含む方法に関する。用量を調節又は記載するために使用する場合の「約」なる用語は妥当な近似が適用可能であることを表すためにその一般的な意味で使用する。例えば、「約10mg」とは10mgよりも多少多いか又は少ない用量を含み、例えば9mg前後から11mg前後である。「約18.75mg」の用量は17mg前後から20mg前後である。「約20mg」の用量は「約18.75mg」とオーバーラップし、19mg前後から21mg前後の範囲を含む。
【0045】
1種以上の付加活性剤を本明細書に記載する化合物と併用投与することができる。付加活性剤は脂質調節化合物又は他の医薬活性をもつ物質とすることができ、脂質調節効果と他の医薬活性を併有する物質でもよい。利用可能な付加活性剤の例としては限定されないが、そのラクトン化又はジヒドロキシ開環酸体のスタチンとその医薬的に許容可能な塩及びエステル(限定されないが、例えばロバスタチン(米国特許第4,342,767号参照)、シンバスタチン(米国特許第4,444,784号参照)、ジヒドロキシ開環酸シンバスタチン、特にそのアンモニウム又はカルシウム塩、プラバスタチン、特にそのナトリウム塩(米国特許第4,346,227号参照)、フルバスタチン、特にそのナトリウム塩(米国特許第5,354,772号参照)、アトルバスタチン、特にそのカルシウム塩(米国特許第5,273,995号参照)、ピタバスタチン(別称NK−104)(PCT国際出願公開WO97/23200参照)及びロスバスタチン(別称ZD−4522)(CRESTOR(登録商標);米国特許第5,260,440号参照))等のHMG−CoAレダクターゼ阻害剤;HMG−CoAシンターゼ阻害剤;スクアレンエポキシダーゼ阻害剤;スクアレンシンテターゼ阻害剤(別称スクアレンシンターゼ阻害剤)、ACAT−1又はACAT−2の選択的阻害剤とACAT−1及び−2のデュアル阻害剤を含むアシル−補酵素A:コレステロールアシルトランスフェラーゼ(ACAT)阻害剤;ミクロソームトリグリセリド転送蛋白質(MTP)阻害剤;内皮リパーゼ阻害剤;胆汁酸抑制剤;LDL受容体インデューサー;血小板凝集阻害剤(例えば糖蛋白質IIb/IIIaフィブリノーゲン受容体アンタゴニスト及びアスピリン);ヒトペルオキシソーム増殖因子活性化受容体γ(PPARγ)アゴニスト、例えば一般にグリタゾンと呼ばれる化合物(例えばピオグリタゾン及びロシグリタゾン)及びチアゾリジンジオンとして知られる構造クラスに含まれる化合物と、チアゾリジンジオン構造クラスに含まれないPPARγアゴニスト;PPARαアゴニスト(例えばクロフィブレート、微粉状フェノフィブレートを含むフェノフィブレート、及びゲムフィブロジル);PPARデュアルα/γアゴニスト;ビタミンB6(別称ピリドキシン)とその医薬的に許容可能な塩(例えばHCl塩);ビタミンB12(別称シアノコバラミン);葉酸又はその医薬的に許容可能な塩もしくはエステル(例えばナトリウム塩及びメチルグルカミン塩);酸化防止ビタミン類(例えばビタミンC及びE及びβカロチン);β遮断薬;アンギオテンシンIIアンタゴニスト(例えばロサルタン);アンギオテンシン変換酵素阻害剤(例えばエナラプリル及びカプトプリル);レニン阻害剤、カルシウムチャネル遮断薬(例えばニフェジピン及びジルチアゼム);エンドテリンアンタゴニスト;ABC1遺伝子発現を増強する物質;コレステリルエステル転送蛋白質(CETP)阻害化合物、5−リポキシゲナーゼ活性化蛋白質(FLAP)阻害化合物、5−リポキシゲナーゼ(5−LO)阻害化合物、アンタゴニストとアゴニストの両者を含むファルネソイドX受容体(FXR)リガンド;肝臓X受容体(LXR)−αリガンド、LXR−βリガンド、ビスホスホネート化合物(例えばアレンドロン酸ナトリウム);シクロオキシゲナーゼ−2阻害剤(例えばロフェコキシブ及びセレコキシブ)、並びに血管炎症を緩和する化合物が挙げられる。
【0046】
コレステロール吸収抑制剤も本発明で使用することができる。このような化合物はコレステロールが腸内腔から小腸壁の腸細胞内に移動するのを阻止し、血清コレステロール値を低下させる。コレステロール吸収抑制剤の例は米国特許第5,846,966号、5,631,365号、5,767,115号、6,133,001号、5,886,171号、5,856,473号、5,756,470号、5,739,321号、5,919,672号、並びにPCT出願WO00/63703、WO00/60107、WO00/38725、WO00/34240、WO00/20623、WO97/45406、WO97/16424、WO97/16455、及びWO95/08532に記載されている。最も注目に値するコレステロール吸収抑制剤は米国特許第5,767,115号及び5,846,966号に記載されているエゼチミブ、別称1−(4−フルオロフェニル)−3(R)−[3(S)−(4−フルオロフェニル)−3−ヒドロキシプロピル)]−4(S)−(4−ヒドロキシフェニル)−2−アゼチジノンである。
【0047】
コレステロール吸収抑制剤の治療有効量としては約0.01mg/kgから約30mg/kg体重/日、好ましくは約0.1mg/kgから約15mg/kgの用量が挙げられる。
【0048】
糖尿病患者には、本発明で使用する化合物を従来の糖尿病治療薬と併用投与することができる。例えば、本明細書に記載するような治療を受けている糖尿病患者にインスリン又は経口糖尿病治療薬を併用投与することができる。本発明で有用な経口糖尿病治療薬の1例はメトフォルミンである。
【0049】
塩
本明細書で使用するニコチン酸とはピリジン−3−カルボン酸を意味する。しかし、ニコチン酸の塩及び溶媒和物も本発明で使用され、ニコチン酸の多数の医薬的に許容可能な塩及び溶媒和物が本発明で有用である。アルカリ金属塩、特にナトリウム塩とカリウム塩が本明細書に記載する有用な塩を形成する。同様に、アルカリ土類金属、特にカルシウムとマグネシウムも本明細書に記載する有用な塩を形成する。アンモニウム及び置換アンモニウム化合物等の各種アミン塩も本明細書に記載する有用な塩を形成する。同様に、ニコチン酸の溶媒和形態も本発明で有用である。例としては、半水和物、1水和物、2水和物、3水和物及びセスキ水和物が挙げられる。本発明で使用するには遊離酸、ピリジン−3−カルボン酸が特に有利である。
【0050】
本明細書に記載する有用なニコチン酸の用量は約50mg/日から約8g/日を1日1回又は数回に分けて投与する。最初は低用量を使用し、用量を増加してフラッシング効果を更に最小限にすることができる。
【0051】
ニコチン酸以外のニコチン酸受容体アゴニストの用量は広い範囲を取る。一般に、アテローム性動脈硬化症を治療するために有用なニコチン酸受容体アゴニストは約0.01mg/kg/日から約100mg/kg/日を1日1回又は数回に分けて投与する。代表的な用量は約0.1mg/日から約2g/日である。
【0052】
本明細書に記載するDPアンタゴニストは約0.01mg/kg/日から約100mg/kg/日を1日1回又は数回に分けて哺乳動物患者、特にヒトに投与すると、フラッシング効果を緩和又は予防するために有用である。用量は約0.1mg/日から約1.0g/日を1日1回又は数回に分けて投与することが好ましい。
【0053】
本明細書に記載する化合物及び製剤は従来の任意投与経路で投与することができる。好ましい投与経路は経口である。
【0054】
ニコチン酸、又はその塩もしくは溶媒和物、又は他のニコチン酸受容体アゴニストとDPアンタゴニストは本発明の範囲内で1日1回又は複数回、例えば1日2回(bid)、1日3回(tid)又は1日4回(qid)に分けて同時又は順次投与することができる。24時間を越える放出プロフィルを示す徐放製剤等の特に長時間徐放が所望される場合には、1日おきに投与してもよい。しかし、1日1回投与することが好ましい。また、朝晩どちらに投与してもよいが、晩に投与するほうが好ましい。
【0055】
医薬組成物
本明細書に記載する医薬組成物は一般にニコチン酸又は別のニコチン酸受容体アゴニストと、DP受容体アンタゴニストと、医薬的に許容可能なキャリヤーから構成される。
【0056】
適切な経口組成物の例としては錠剤、カプセル剤、トローチ、ロゼンジ、懸濁液、散剤又は顆粒剤、エマルション、シロップ及びエリキシル剤が挙げられる。キャリヤー成分の例としては希釈剤、結合剤、崩壊剤、滑沢剤、甘味剤、フレーバー、着色剤、防腐剤等が挙げられる。希釈剤の例としては、例えば炭酸カルシウム、炭酸ナトリウム、ラクトース、リン酸カルシウム及びリン酸ナトリウムが挙げられる。顆粒化剤及び崩壊剤の例としてはコーンスターチとアルギン酸が挙げられる。結合剤の例としては澱粉、ゼラチン及びアラビアガムがが挙げられる。滑沢剤の例としてはステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸及びタルクが挙げられる。錠剤はコーティングしなくてもよいし、公知技術によりコーティングしてもよい。このようなコーティングは胃腸管での崩壊、従って吸収を遅らせることにより長時間持続作用することができる。
【0057】
本発明の1態様では、ニコチン酸、又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストをDP受容体アンタゴニスト及びキャリヤーと配合し、規定併用製剤を形成する。この規定併用製剤は経口用錠剤又はカプセル剤とすることができる。
【0058】
より特定的には、本発明の別の態様では、ニコチン酸、又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニスト(約1から約1000mg)とDPアンタゴニスト(約1から約500mg)を医薬的に許容可能なキャリヤーと配合し、経口用錠剤又はカプセル剤を提供する。
【0059】
ニコチン酸医薬組成物の製剤では長時間徐放が特に重要であると思われる。徐放錠が特に好ましい。例えば、モノステアリン酸グリセリルやジステアリン酸グリセリル等の時間遅延剤を利用することができる。米国特許第4,256,108号、4,166,452号及び4,265,874号に記載されている技術により製剤をコーティングし、制御放出用浸透圧治療錠を形成してもよい。
【0060】
他の制御放出技術も利用可能であり、本発明に含まれる。徐放錠中のニコチン酸の放出を遅らせるために有用な典型的成分としては、メチルセルロース、エチルセルロース、プロピルセルロース、ヒドロキシプロピルセルロース、ヒドロキシエチルセルロース、ヒドロキシプロピルメチルセルロース、微結晶セルロース、澱粉等の各種セルロース化合物が挙げられる。各種天然及び合成材料も徐放製剤で使用することができる。例としてはアルギン酸と各種アルギン酸塩、ポリビニルピロリドン、トラガカントガム、ローカストビーンガム、グアーガム、ゼラチン、各種長鎖アルコール(例えばセチルアルコール)及び蜜蝋が挙げられる。
【0061】
特に有利な徐放錠は1種以上の上記セルロース化合物とニコチン酸を徐放錠に圧縮してポリマーマトリックスを形成する。DPアンタゴニスト化合物は圧縮前にブレンドに加えてもよいし、マトリックスの外側表面にコーティングしてもよい。
【0062】
より有利な1態様では、ニコチン酸とマトリックス形成材料を配合及び圧縮して徐放コアを形成し、DPアンタゴニスト化合物を1種以上のコーティング剤とブレンドし、コアの外側表面にコーティングする。
【0063】
場合により、上記のような錠剤にHMG Co−Aレダクターゼ阻害剤(例えばシンバスタチン)をコーティングすると更に有利である。従って、この特定態様は3種の活性成分を含み、そのうち、HMG Co−Aレダクターゼ阻害剤とDPアンタゴニストは実質的に摂取直後に放出可能であり、ニコチン酸は上記のように長時間にわたって放出可能である。
【0064】
本発明の徐放錠の典型的放出時間は約1から約48時間、好ましくは約4から約24時間、より好ましくは約8から約16時間である。
【0065】
ハードゼラチンカプセルは別の経口用剤形を構成する。このようなカプセル剤も同様に上記のようなキャリヤー材料と混合した活性成分を含む。ソフトゼラチンカプセルは水混和性溶媒(例えばプロピレングリコール、PEG及びエタノール)、又は油(例えばピーナツ油、流動パラフィン又はオリーブ油)と混合した活性成分を含む。
【0066】
水性懸濁液の製造に適した賦形剤と混合した活性剤を含むものとして水性懸濁液も想定される。このような賦形剤としては懸濁剤(例えばナトリウムカルボキシメチルセルロース、メチルセルロース、ヒドロキシプロピルメチルセルロース、アルギン酸ナトリウム、ポリビニルピロリドン、トラガカントガム及びアラビアガム);分散剤又は湿潤剤(例えばレシチン);防腐剤(例えばパラヒドロキシ安息香酸エチル又はn−プロピル)、着色剤、フレーバー、甘味剤等が挙げられる。
【0067】
水の添加により水性懸濁液を調製するのに適した散剤及び顆粒剤は分散剤又は湿潤剤、懸濁剤及び1種以上の防腐剤と混合した活性成分を提供する。適切な分散剤又は湿潤剤と懸濁剤は上記に挙げたものである。
【0068】
シロップ及びエリキシル剤に製剤化することもできる。
【0069】
特に有利な医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸又はその塩もしくは溶媒和物と、DP受容体アンタゴニストから構成される徐放錠である。
【0070】
特に有利な別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸又はその塩もしくは溶媒和物と、DP受容体アンタゴニストと、HMG Co−Aレダクターゼ阻害剤から構成される徐放錠である。
【0071】
更に特に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、DP受容体アンタゴニストと、シンバスタチンから構成される徐放錠である。
【0072】
特に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、化合物AからAJから構成される群から選択されるDP受容体アンタゴニストから構成される徐放錠である。
【0073】
更に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、化合物A、B、D、E、X、AA、AF、AG、AH、AI及びAJから構成される群から選択されるDP受容体アンタゴニストから構成される。
【0074】
特に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸とDPアンタゴニスト化合物Aから構成される。
【0075】
更に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、DPアンタゴニスト化合物Bから構成される。
【0076】
更に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、DPアンタゴニスト化合物Dから構成される。
【0077】
更に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、DPアンタゴニスト化合物Eから構成される。
【0078】
更に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、DPアンタゴニスト化合物Xから構成される。
【0079】
更に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、DPアンタゴニスト化合物AAから構成される。
【0080】
更に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、DPアンタゴニスト化合物AFから構成される。
【0081】
更に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、DPアンタゴニスト化合物AGから構成される。
【0082】
更に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、DPアンタゴニスト化合物AHから構成される。
【0083】
更に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、DPアンタゴニスト化合物AIから構成される。
【0084】
更に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、DPアンタゴニスト化合物AJから構成される。
【0085】
更に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、上記DPアンタゴニスト化合物の1種と、シンバスタチンから構成される。
【0086】
更に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、化合物AからAJから選択されるDP受容体アンタゴニストと、シンバスタチンから構成される徐放錠である。
【0087】
特に有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸約500mg、750mg又は1000mgと、約5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg、50mg、75mg、100mg及び150mgから選択される量のA、B、D、E、X、AA、AF、AG、AH、AI及びAJから構成される群から選択されるDPアンタゴニスト、又は医薬的に許容可能なその塩もしくは溶媒和物を含有する。
【0088】
より特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg及び50mgから選択される量のA、B、D、E、X、AA、AF、AG、AH、AI及びAJから構成される群から選択されるDPアンタゴニスト、又は医薬的に許容可能なその塩もしくは溶媒和物を含有する。
【0089】
より特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、A、B、D、E、X、AA、AF、AG、AH、AI及びAJから構成される群から選択されるDPアンタゴニスト、又は医薬的に許容可能なその塩もしくは溶媒和物5mg、10mg、15mg、18.75mg、20mg、25mg又は37.5mgを含有する。
【0090】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、A、B、D、E、X、AA、AF、AG、AH、AI及びAJから構成される群から選択されるDPアンタゴニスト、又は医薬的に許容可能なその塩もしくは溶媒和物5mg、10mg、15mg、20mg、25mg、又は37.5mgを含有する。
【0091】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物A又は医薬的に許容可能なその塩もしくは溶媒和物5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgを含有する。
【0092】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物B又は医薬的に許容可能なその塩もしくは溶媒和物5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgを含有する。
【0093】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物D又は医薬的に許容可能なその塩もしくは溶媒和物5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgを含有する。
【0094】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物E又は医薬的に許容可能なその塩もしくは溶媒和物5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgを含有する。
【0095】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物X又は医薬的に許容可能なその塩もしくは溶媒和物5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgを含有する。
【0096】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物AA又は医薬的に許容可能なその塩もしくは溶媒和物5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgを含有する。
【0097】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物AF又は医薬的に許容可能なその塩もしくは溶媒和物5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgを含有する。
【0098】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物AG又は医薬的に許容可能なその塩もしくは溶媒和物5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgを含有する。
【0099】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物AH又は医薬的に許容可能なその塩もしくは溶媒和物5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgを含有する。
【0100】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物AI又は医薬的に許容可能なその塩もしくは溶媒和物5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgを含有する。
【0101】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物AJ又は医薬的に許容可能なその塩もしくは溶媒和物5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgを含有する。
【0102】
更に特に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、化合物A、B、D、E、X、AA、AF、AG、AH、AI及びAJから構成される群から選択されるDP受容体アンタゴニストと、シンバスタチンから構成される徐放錠に関する。
【0103】
特に有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸約500mg、750mg又は1000mgと、約5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg、50mg、75mg、100mg及び150mgの量のA、B、D、E、X、AA、AF、AG、AH、AI及びAJから構成される群から選択されるDPアンタゴニスト、又は医薬的に許容可能なその塩もしくは溶媒和物と、シンバスタチン約10mg、20mg又は40mgを含有する。
【0104】
より特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg及び50mgから選択される量のA、B、D、E、X、AA、AF、AG、AH、AI及びAJから構成される群から選択されるDPアンタゴニスト、又は医薬的に許容可能なその塩もしくは溶媒和物と、シンバスタチン約10mg、20mg又は40mgを含有する。
【0105】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、A、B、D、E、X、AA、AF、AG、AH、AI及びAJから構成される群から選択されるDPアンタゴニスト、又は医薬的に許容可能なその塩もしくは溶媒和物5mg、10mg、15mg、18.75mg、20mg、25mg又は37.5mgと、シンバスタチン約10mg、20mg又は40mgを含有する。
【0106】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、A、B、D、E、X、AA、AF、AG、AH、AI及びAJから構成される群から選択されるDPアンタゴニスト、又は医薬的に許容可能なその塩もしくは溶媒和物10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgと、シンバスタチン約10mg、20mg又は40mgを含有する。
【0107】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物A又は医薬的に許容可能なその塩もしくは溶媒和物10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgと、シンバスタチン約10mg、20mg又は40mgを含有する。
【0108】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物B又は医薬的に許容可能なその塩もしくは溶媒和物10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgと、シンバスタチン約10mg、20mg又は40mgを含有する。
【0109】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物D又は医薬的に許容可能なその塩もしくは溶媒和物10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgと、シンバスタチン約10mg、20mg又は40mgを含有する。
【0110】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物E又は医薬的に許容可能なその塩もしくは溶媒和物10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgと、シンバスタチン約10mg、20mg又は40mgを含有する。
【0111】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物X又は医薬的に許容可能なその塩もしくは溶媒和物10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgと、シンバスタチン約10mg、20mg又は40mgを含有する。
【0112】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物AA又は医薬的に許容可能なその塩もしくは溶媒和物10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgと、シンバスタチン約10mg、20mg又は40mgを含有する。
【0113】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物AF又は医薬的に許容可能なその塩もしくは溶媒和物10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgと、シンバスタチン約10mg、20mg又は40mgを含有する。
【0114】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物AG又は医薬的に許容可能なその塩もしくは溶媒和物10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgと、シンバスタチン約10mg、20mg又は40mgを含有する。
【0115】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物AH又は医薬的に許容可能なその塩もしくは溶媒和物10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgと、シンバスタチン約10mg、20mg又は40mgを含有する。
【0116】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物AI又は医薬的に許容可能なその塩もしくは溶媒和物10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgと、シンバスタチン約10mg、20mg又は40mgを含有する。
【0117】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物AJ又は医薬的に許容可能なその塩もしくは溶媒和物10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgと、シンバスタチン約10mg、20mg又は40mgを含有する。
【0118】
「組成物」なる用語は上記医薬組成物に加え、活性成分又は賦形剤成分の任意2種以上の配合、錯化もしくは凝集、成分の1種以上の解離、又は成分の1種以上の他の型の反応もしくは相互作用により直接又は間接的に得られる任意製剤も含む。従って、本発明の医薬組成物は本発明の化合物と、任意付加活性成分と、医薬的に許容可能な賦形剤を混合又は他の方法で配合することにより製造される任意組成物を含む。
【0119】
本発明の別の側面は医薬の製造におけるニコチン酸又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストとDPアンタゴニストの使用に関する。この医薬は本明細書に記載する用途をもつ。
【0120】
より特定的には、本発明の別の側面は医薬の製造におけるニコチン酸又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストと、DPアンタゴニストと、HMG Co−Aレダクターゼ阻害剤(例えばシンバスタチン)の使用に関する。この医薬は本明細書に記載する用途をもつ。
【0121】
医薬組成物の例を以下に挙げる。
【0122】
【表1】
【0123】
【表2】
【0124】
【表3】
【0125】
製剤例1から5の手順
1.DPアンタゴニスト含有層
化合物D、微結晶セルロース、ラクトース一水和物、クロスカルメロースナトリウム及び黄酸化鉄をDiosna P10高剪断ミキサーグラニュレーターで乾式混合する。別に、ヒドロキシプロピルセルロースを含有する水溶液を調製し、高剪断ミキサーグラニュレーターに加える。ブレンドを湿式造粒し、Strea−1流動層乾燥機を使用して乾燥する。乾燥した顆粒を粉砕する。粉砕した顆粒をApex 17Lドラムブレンダーでステアリン酸マグネシウムとブレンドし、DPアンタゴニスト顆粒を形成する。
【0126】
2.ニコチン酸含有層
ニコチン酸の一部をコロイド状二酸化ケイ素とプレブレンドする。ヒドロキシプロピルメチルセルロース、ニコチン酸含有混合物、コロイド状二酸化ケイ素、微結晶セルロース及びニコチン酸の残量を適切なブレンダーに加え、乾式混合する。ブレンドをAlexanderwerk WP 120ローラーコンパクターで乾式造粒し、粉砕する。粉砕した顆粒をHarbruc 65Lブレンダーでステアリルフマル酸ナトリウムとブレンドし、ニコチン酸顆粒を形成する。
【0127】
3.錠剤化手順
粉砕し、滑沢剤を添加した上記顆粒をRiva二層錠剤プレスで二層錠剤に圧縮する。
【0128】
製剤例6の手順
1.DPアンタゴニスト含有層1
上記製剤例1から5に記載した手順に従って成分をブレンドし、造粒する。
【0129】
2.ニコチン酸含有層
ヒドロキシプロピルメチルセルロース、ニコチン酸及びポビドンをApex 17Lブレンダーに加え、乾式混合する。混合物をApex 17Lドラムブレンダーでステアリン酸とブレンドし、ニコチン酸ブレンドを形成する。
【0130】
3.錠剤化手順
粉砕し、滑沢剤を添加したニコチン酸及びDPアンタゴニストブレンド顆粒をRiva二層錠剤プレスで二層錠剤に圧縮する。
【0131】
【表4】
【0132】
【表5】
【0133】
【表6】
【0134】
製剤例7から11の手順
1.DPアンタゴニスト/シンバスタチン層
化合物D、シンバスタチン、微結晶セルロース、ラクトース一水和物、クロスカルメロースナトリウム、黄酸化鉄及びヒドロキシプロピルメチルセルロースをDiosna P−6高剪断ミキサーグラニュレーターに加え、乾式混合する。BHAとクエン酸を含有する水性アルコール溶液を調製し、高剪断ミキサーに加え、ブレンドを湿式造粒する。顆粒をStrea−1流動層乾燥機で乾燥し、粉砕し、粉砕した顆粒をApex 17Lブレンダーで(予め凝塊を分解しておいた)ステアリン酸マグネシウムとブレンドする。
【0135】
2.ニコチン酸層
ニコチン酸層は上記製剤例1から5に記載したように作製する。
【0136】
3.錠剤化手順
粉砕し、滑沢剤を添加したニコチン酸とDPアンタゴニスト/シンバスタチン顆粒をRiva二層錠剤プレスで二層錠剤に圧縮する。
【0137】
製剤例12の手順
1.DPアンタゴニスト層
上記製剤例1から5に記載した手順に従って成分をブレンドし、造粒する。
【0138】
2.ニコチン酸層
ヒドロキシプロピルメチルセルロース、ニコチン酸及びポビドンをApex 17Lブレンダーに加え、乾式混合する。混合物をApex 17Lドラムブレンダーでステアリン酸とブレンドし、ニコチン酸ブレンドを形成する。
【0139】
3.錠剤化手順
粉砕し、滑沢剤を添加したニコチン酸及びDPアンタゴニストブレンド顆粒をRiva二層錠剤プレスで二層錠剤に圧縮する。
【0140】
【表7】
【0141】
【表8】
【0142】
製剤例13の手順
1.DPアンタゴニスト層
化合物E、微結晶セルロース、ラクトース一水和物及びクロスカルメロースナトリウムをDiosna P10高剪断ミキサーグラニュレーターで乾式混合する。別に、ヒドロキシプロピルセルロースを含有する水溶液を調製し、高剪断ミキサーグラニュレーターに加える。ブレンドを湿式造粒し、Strea−1流動層乾燥機を使用して乾燥する。乾燥した顆粒を粉砕する。粉砕した顆粒をApex 17Lドラムブレンダーでステアリン酸マグネシウム及びステアリルフマル酸ナトリウムとブレンドし、DPアンタゴニスト顆粒を形成する。
【0143】
2.ニコチン酸層
ニコチン酸層は上記製剤例1から5に記載したように作製する。
【0144】
3.錠剤化手順
粉砕し、滑沢剤を添加したニコチン酸及びDPアンタゴニストブレンド顆粒をRiva二層錠剤プレスで二層錠剤に圧縮する。
【0145】
製剤例14Aから14Cの手順
1.DPアンタゴニスト/シンバスタチン層
化合物E、シンバスタチン、微結晶セルロース、ラクトース一水和物、クロスカルメロースナトリウム、黄酸化鉄、FDC Blue No.2及びヒドロキシプロピルメチルセルロースをDiosna P−6高剪断ミキサーグラニュレーターに加え、乾式混合する。BHAとクエン酸を含有する水性アルコール溶液を調製し、高剪断ミキサーに加え、ブレンドを湿式造粒する。顆粒をStrea−1流動層乾燥機で乾燥し、粉砕し、粉砕した顆粒をApex 17Lブレンダーで(予め凝塊を分解しておいた)ステアリン酸マグネシウム及びステアリルフマル酸ナトリウムとブレンドする。
【0146】
2.ニコチン酸層
ニコチン酸層は上記製剤例1から5に記載したように作製する。
【0147】
3.錠剤化手順
粉砕し、滑沢剤を添加したニコチン酸とDPアンタゴニスト/シンバスタチンの顆粒をRiva二層錠剤プレスで二層錠剤に圧縮する。
【0148】
標準ニコチン酸受容体アゴニストであるニコチン酸に加え、多数のニコチン酸受容体アゴニストが記載されている。以下の刊行物がニコチン酸受容体アゴニストである化合物を開示している:
Lorenzen,A.ら,Molecular Pharmacology 59:349−357(2001)、
Lorenzen,A.ら,Biochemical Pharmacology 64:645−648(2002)、
Soga,T.ら,Biochemical and Biophysical Research Comm.303:364−369(2003)、
Tunaru,S.ら,Nature Medicine 9:352−355(2003)、
Wise,A.ら,Journal of Biological Chemistry 278:9869−9874(2003)、及び
Van Herk,T.ら Journal of Medicinal Chemistry 46:3945−3951(2003)。
【0149】
なお、van Herkらに開示されているもの等のニコチン酸受容体の部分アゴニストも本発明の組成物及び治療方法に含まれる。
【0150】
更に、ニコチン酸受容体はWO02/084298A2(公開日2002年10月24日)と、Soga,T.ら、Tunaru,S.ら及びWise,A.ら(上記引用)に記載され、特性決定されている。
【0151】
多数のDP受容体アンタゴニスト化合物が発表されており、本発明の方法で有用であり、本発明の方法に含まれる。例えば、DP受容体アンタゴニストはWO01/79169(公開日2001年10月25日)、EP 1305286(公開日2003年5月2日)、WO02/094830(公開日2002年11月28日)及びWO03/062200(公開日2003年7月31日)に従って得ることができる。化合物ABはWO01/66520A1(公開日2001年9月13日)の記載に従って合成することができ;化合物ACはWO03/022814A1(公開日2003年3月20日)の記載に従って合成することができ、化合物AD及びAEはWO03/078409(公開日2003年9月25日)の記載に従って合成することができる。本発明で使用される他の代表的なDPアンタゴニスト化合物は以下の実施例に従って合成することができる。
【0152】
(実施例1)
[5−[(4−クロロフェニル)チオ]−4−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸(化合物G)
【0153】
【化2】
【0154】
ステップ1:4−クロロニコチンアルデヒド
標記化合物はF.Marsaisら,J.Heterocyclic Chem.,25,81(1988)により記載されているように製造した。
【0155】
ステップ2:4−(メチルチオ)ニコチンアルデヒド
NaSMe(9.5g,135mmol)のMeOH(250mL)溶液にMeOH(250mL)中のステップ1の4−クロロニコチンアルデヒド(13.5g,94.4mmol)を加えた。反応混合物を60℃に15分間維持した。反応混合物をNH4ClとEtOAcに注いだ。有機相を分離し、H2Oで洗浄し、Na2SO4で乾燥した。次に、ヘキサン中50%EtOAcを使用して化合物をシリカゲルで精製すると、標記化合物が得られた。
【0156】
ステップ3:(2Z)−2−アジド−3−[4−(メチルチオ)ピリジン−3−イル]プロプ−2−エン酸メチル
4−(メチルチオ)ニコチンアルデヒド(4.8g,31mmol)とアジド酢酸メチル(9.0g,78mmol)のMeOH(50mL)溶液を−12℃のNaOMeの25%MeOH溶液(16.9mL,78mmol)に加えた。内部温度をモニターし、30分間の添加中、−10℃から−12℃に維持した。得られた混合物を次に氷浴中で数時間撹拌した後、冷所にて氷浴中で一晩撹拌した。次に懸濁液を氷とNH4Clの混合物に注ぎ、10分間撹拌後にスラリーを濾過した。生成物を冷H2Oで洗浄した後に減圧乾燥すると、標記化合物が多少の塩を含有するベージュ色固体(7.4g)として得られた。次にEtOAcを使用して化合物をシリカゲルで精製した。
【0157】
ステップ4:4−(メチルチオ)−1H−ピロロ[2,3−b]ピリジン−2−カルボン酸メチル
ステップ3の化合物(0.40g,1.6mmol)のキシレン(16mL)懸濁液を140℃までゆっくりと加熱した。140℃で15分後に、黄色溶液を室温まで冷却した。窒素形成により発熱する可能性があるので、注意が必要である。その後、懸濁液を0℃まで冷却し、濾過し、キシレンで洗浄すると、標記化合物が得られた。
【0158】
ステップ5:4−(メチルチオ)−6−オキソ−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−7−カルボン酸エチル
ステップ4の化合物(0.35g,1.6mmol)の0℃のDMF(20mL)溶液にNaH(1.2当量)を加えた。5分後にnBu4NI(0.10g)と4−ブロモ酪酸エチル(0.40mL)を加えた。室温で1時間後に反応混合物を飽和NH4ClとEtOAcに注いだ。有機相を分離し、H2Oで洗浄し、Na2SO4で乾燥した。蒸発後、粗生成物をフラッシュクロマトグラフィーにより精製した。このビスエステルを次にTHF(7.0mL)に溶かし、カリウムtert−ブトキシドの1.06M THF溶液(2.2mL)を0℃で加えた。室温で1時間後に反応混合物を飽和NH4ClとEtOAcに注いだ。有機相を分離し、Na2SO4で乾燥し、減圧乾燥すると、標記化合物がエチルエステルとメチルエステルの混合物として得られた。
【0159】
ステップ6:4−(メチルチオ)−8,9−ジヒドロピリド[3,2−b]インドリジン−6(7H)−オン
ステップ5の化合物(0.32g)にEtOH(8.0mL)と濃Cl(2.0mL)を加えた。得られた懸濁液を5時間還流した。反応混合物をEtOAcとNa2CO3に分配した。有機相を分離し、蒸発させると、標記化合物が得られた。
【0160】
ステップ7:(2E,2Z)−[4−(メチルチオ)−8,9−ジヒドロピリド[3,2−b]インドリジン−6(7H)−イリデン]エタン酸エチル
ホスホノ酢酸トリエチル(0.45g,2.17mmol)のDMF溶液(12mL)に80%NaH(0.06g,2.00mmol)とステップ6の化合物(0.22g,1.00mmol)を加えた。55℃で4時間後に反応混合物を飽和NH4ClとEtOAcに注いだ。有機相を分離し、減圧蒸発させた。粗生成物をフラッシュクロマトグラフィーにより精製すると、標記化合物が得られた。
【0161】
ステップ8:[4−(メチルチオ)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸エチル
熱溶解を使用してステップ7の化合物をMeOH−THFに溶かした。前記溶液を冷却し、室温でPtO2を加え、得られた混合物を大気圧水素下に18時間維持した。CH2Cl2を使用して反応混合物をセライトで注意深く濾過した。濾液を減圧蒸発させると、標記化合物が得られた。あるいは、ステップ7の化合物を40PSI H2下にEtOAc中Pd(OH)2で18時間水素化してもよい。
【0162】
ステップ9:[4−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸エチル
ステップ8の化合物(0.08g,0.27mmol)のMeOH(3.0mL)溶液にNa2WO4(0.10g)と30%H2O2(600μL)を加えた。1時間後に反応混合物をH2OとEtOAcに分配した。有機相をH2Oで洗浄し、分離し、蒸発させた。標記化合物をフラッシュクロマトグラフィーにより精製した。
【0163】
ステップ10:[5−[(4−クロロフェニル)チオ]−4−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸エチル
4,4’−ジクロロジフェニルジスルフィド(0.24g)の1,2−ジクロロエタン溶液(2.0mL)にSO2Cl2(50μL)を加えた。ステップ9の化合物(0.05g)のDMF(2.0mL)溶液に前記混合物(から180μL)を加えた。反応後に1H NMRを測定し、出発材料がなくなるまで室温に維持した。反応混合物を飽和NaHCO3とEtOAcに注いだ。有機相を分離し、蒸発させ、標記化合物をフラッシュクロマトグラフィーにより精製した。
【0164】
ステップ11:[5−[(4−クロロフェニル)チオ]−4−(メチルスルホニル)−67,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸
THF−MeOHの1/1混合物に溶かしたステップ10の化合物を1N NaOHを加えた。室温で18時間後に反応混合物を飽和NH4ClとEtOAcに分配した。有機相を分離し、Na2SO4で乾燥し、蒸発させると、標記化合物が得られた。
1H NMR(500MHz,アセトン−d6)δ 11.00(bs,1H),8.60(d,1H),7.80(d,1H),7.20(d,2H),7.00(d,2H),4.65(m,1H),4.20(m,1H),3.75(m,1H),3.35(s,3H),2.80−2.10(m,6H)。
【0165】
(実施例2)
[5−[(4−クロロフェニル)チオ]−4−(メチルチオ)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸(化合物H)
【0166】
【化3】
【0167】
標記化合物は実施例1,ステップ8の化合物から実施例1,ステップ10及び11に記載したと同様に製造することができる。m/z418。
【0168】
(実施例3)
[5−[(3,4−ジクロロフェニル)チオ]−4−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸(化合物I)
【0169】
【化4】
【0170】
標記化合物はステップ10でビス(3,4−ジクロロフェニル)ジスルフィドを使用した以外は実施例1に記載したように製造した。
1H NMR(500MHz,アセトン−d6)δ 8.55(d,1H),7.85(d,1H),7.35(d,1H),7.15(s,1H),6.95(d,1H),4.60(m,1H),4.15(m,1H),3.80(m,1H),3.40(s,3H),2.80−2.10(m,6H)。
【0171】
ヘキサン中30%イソプロパノール 17%エタノール 0.2%酢酸を流速8ml/分で使用してChiralecel ODカラム25cm×20mmでエナンチオマーを分離した。ヘキサン中35%イソプロパノール 0.2%酢酸を流速1.0ml/分で使用してChiralecel ODカラム25cm×4.6mmでその純度を確認した。高移動度エナンチオマーTr=9.7分、低移動度エナンチオマーTr=11.1分。
【0172】
(実施例4)
[5−(4−クロロベンゾイル)−4−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸(化合物J)
【0173】
【化5】
【0174】
ステップ1:[5−(4−クロロベンゾイル)−4−(メチルチオ)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸エチル
4−クロロベンゾイルクロライド(0.30g,1.7mmol)の1,2−ジクロロエタン(6.0mL)溶液にAlCl3(0.24g,1.8mmole)を加えた。5分後に実施例1ステップ8からの[4−(メチルチオ)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸エチル(0.15g,0.47mmole)の1,2−ジクロロエタン(6.0mL)溶液を前記混合物に加えた。80℃で4時間後に、反応混合物をEtOAcとNaHCO3に分配した。有機相を分離し、Na2SO4で乾燥し、蒸発させた。標記化合物をフラッシュクロマトグラフィーにより精製した。
【0175】
ステップ2:[5−(4−クロロベンゾイル)−4−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸エチル
[5−(4−クロロベンゾイル)−4−(メチルチオ)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸エチル(0.12g,0.27mmole)のMeOH(5.0mL)溶液にNa2WO4(0.1g)と30%H2O2(300μL)を加えた。反応混合物を55℃で1時間撹拌した。次に反応混合物をH2OとEtOAcに分配した。有機相をH2Oで洗浄し、Na2SO4で乾燥し、蒸発させた。標記化合物をフラッシュクロマトグラフィーにより精製した。
【0176】
ステップ3:[5−(4−クロロベンゾイル)−4−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸
[5−(4−クロロベンゾイル)−4−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸エチルを実施例1ステップ11に記載したように処理すると、標記化合物が得られた。
1H NMR(500MHz,アセトン−d6)δ 8.55(d,1H),7.90(d,2H),7.65(d,1H),7.45(d,2H),4.55(m,1H),4.25(m,1H),3.45(m,1H),3.20(s,3H),2.05−3.00(m,6H)。m/z446。
【0177】
(実施例5)
[5−(4−ブロモフェニル)チオ]−4−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸(化合物K)
【0178】
【化6】
【0179】
標記化合物は4,4’−ジブロモジフェニルジスルフィドを使用した以外は実施例1に記載したように製造した。
1H NMR(500MHz,アセトン−d6)δ 8.60(d,1H),7.80(d,1H),7.35(d,2H),7.00(d,2H),4.65(m,1H),4.20(m,1H),3.80(m,1H),3.35(s,3H),2.80−2.10(m,6H)。
【0180】
(実施例6)
方法1
[9−[(3,4−ジクロロフェニル)チオ]−1−(メチルスルホニル)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル]酢酸(化合物L)
【0181】
【化7】
【0182】
ステップ1:2−(メチルチオ)ニコチンアルデヒド
標記化合物は溶液を55℃に2時間加熱した以外は実施例1ステップ2に記載したように2−ブロモニコチンアルデヒド(A.Numata Synthesis 1999 p.306)から製造した。
【0183】
ステップ2:(2Z)−2−アジド−3−[2−(メチルチオ)ピリジン−3−イル]プロプ−2−エン酸メチル
標記化合物は実施例1ステップ3に記載したように製造した。
【0184】
ステップ3:4−(メチルチオ)−1H−ピロロ[3,2−c]ピリジン−2−カルボン酸メチル
(2Z)−2−アジド−3−[2−(メチルチオ)ピリジン−3−イル]プロプ−2−エン酸メチル(1.00g,4.00mmol)のメシチレン(50mL)溶液を160℃に1時間加熱した。反応混合物を室温まで冷却した後、0℃まで冷却し、沈殿を濾過し、冷メシチレンで洗浄すると、標記化合物が得られた。
【0185】
ステップ4:1−(メチルチオ)−8−オキソ−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−7−カルボン酸メチル
4−(メチルチオ)−1H−ピロロ[3,2−c]ピリジン−2−カルボン酸メチル(0.30g,1.35mmol)のTHF(3mL)−トルエン(12.0mL)懸濁液にカリウムtert−ブトキシド(1.42mL/1.41mmol)の1.06M THF溶液とアクリル酸メチル(300μL)を加えた。得られた混合物を80℃に18時間加熱した。混合物をEtOAcとNH4Clに分配し、セライトで濾過した。有機相を分離し、Na2SO4で乾燥し、濾過すると、標記化合物が得られた。
【0186】
ステップ5:1−(メチルチオ)−6,7−ジヒドロ−8H−ピリド[3,4−b]ピロリジン−8−オン
実施例1ステップ6に記載したように1−(メチルチオ)−8−オキソ−7,8−ジヒドロ−6H−ピリド[3,4−b]pピロリジン−7−カルボン酸メチルを標記化合物に変換した。
【0187】
ステップ6:[8−ヒドロキシ−1−(メチルチオ)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル]酢酸メチル
THF(3.0mL)中の1−(メチルチオ)−6,7−ジヒドロ−8H−ピリド[3,4−b]ピロリジン−8−オン(0.15g,0.68mmol)、ブロモ酢酸メチル(0.34mL)、Zn−Cu(0.226g)の混合物を2時間音波処理した。次に、反応が完了するまで混合物を60℃に5分間加熱した。反応混合物をEtOAcとNH4Clに分配した。有機相を分離し、Na2SO4で乾燥し、濾過し、減圧蒸発させると、標記化合物が得られた。化合物をフラッシュクロマトグラフィーにより精製した。
【0188】
ステップ7:[1−(メチルチオ)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル]酢酸メチル
CH3CN(3.2mL)中のNaI(0.300g)にTMSCl(0.266mL)を加えた。この混合物を水浴中で[8−ヒドロキシ−1−(メチルチオ)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル]酢酸メチル(0.15g,0.515mmol)のCH3CN(1.5mL)懸濁液に加えた。30分後に反応混合物をEtOAcとNaHCO3に分配した。有機相を分離し、チオ硫酸ナトリウムで洗浄し、MgSO4で乾燥し、蒸発させた。標記化合物をフラッシュクロマトグラフィーにより精製した。
【0189】
ステップ8:[1−(メチルスルホニル)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル]酢酸メチル
実施例1ステップ9に記載したように[1−(メチルチオ)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル]酢酸メチルを標記化合物に変換した。
【0190】
ステップ9:[9−[(3,4−ジクロロフェニル)チオ]−1−(メチルスルホニル)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル]酢酸
ステップ10でビス(3,4−ジクロロフェニル)ジスルフィドを使用した以外は実施例1,ステップ10及び11に記載したように[1−(メチルスルホニル)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル]酢酸メチルを標記化合物に変換した。
1H NMR(500MHz,アセトン−d6)δ 8.35(d,1H)7.80(d,1H),7.35(d,1H),7.15(s,1H),6.95(d,1H),4.55(m,1H),4.35(m,1H)1 3.90(m,1H),3.30(s,3H),3.15(m,1H),3.05(m,1H),2.80(m,1H),2.50(m,1H)。
【0191】
(実施例6)
方法2
[9−[(3,4−ジクロロフェニル)チオ]−1−(メチルスルホニル)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル]酢酸
ステップ1:1−(メチルチオ)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−オール
実施例6,方法1ステップ5からの1−(メチルチオ)−6,7−ジヒドロ−8H−ピリド[3,4−b]ピロリジン−8−オン(0.55g,2.2mmol)のEtOH(10mL)−THF(1mL)懸濁液にNaBH4(0.10g,2.6mmol)を0℃で加えた。室温で30分後にアセトンを加えることにより反応をクエンチした。溶媒を減圧蒸発させ、EtOACとH2Oを残渣に加えた。有機相を分離し、MgSO4で乾燥し、蒸発させた。標記化合物をEtOAc/ヘキサンで洗浄し、濾過した。
【0192】
ステップ2:2−[1−(メチルチオ)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル]マロン酸ジメチル
1−(メチルチオ)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−オール(0.54g,2.1mmol)の−78℃のTHF(10mL)懸濁液にTHF中1M NaHMDS(2.35mL,2.4mmol)とクロロリン酸ジフェニル(0.53mL,2.6mmol)を加えた。30分後にマロン酸ジメチル(0.73mL,6.4mmol)とTHF中1M NaHMDS(6.8mL,6.8mmol)を加えた。反応混合物を0℃まで昇温させた後、室温まで昇温させた。次に混合物をETOAcとNH4Clに分配した。有機相をMgSO4で乾燥し、濾過し、蒸発させた。標記化合物をフラッシュクロマトグラフィーにより精製した。
【0193】
ステップ3:[1−(メチルチオ)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル]−酢酸メチル
2−[1−(メチルチオ)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル]マロン酸ジメチル(0.59g,2.17mmol)とDMSO(4mL)の混合物にH2O(0.45mL)中のNaCl(0.45g)を加えた。150℃で18時間後に反応混合物をETOAcとH2Oに分配した。有機相を分離し、Na2SO4で乾燥し、蒸発させた。次に標記化合物をフラッシュクロマトグラフィーにより精製した。
【0194】
ステップ4:[9−[(3,4−ジクロロフェニル)チオ]−1−(メチルスルホニル)−7,8−ジヒドロ−6H−ピリド[3.4−b]ピロリジン−8−イル]酢酸
標記化合物は[1−(メチルチオ)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル]酢酸メチルから実施例6,方法1,ステップ8から9に記載したように得られた。
【0195】
(実施例7)
[10−[(3,4−ジクロロフェニル)スルファニル]−1−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,4−b]インドリジン−9−イル]酢酸(化合物M)
【0196】
【化8】
【0197】
ステップ1:[1−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,4−b]インドリジン−9−イル]酢酸エチル
標記化合物は実施例6,ステップ3の生成物から実施例1,ステップ5から9に記載したと同様に製造した。
【0198】
ステップ2:[10−[(3,4−ジクロロフェニル)スルファニル]−1−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,4−b]インドリジン−9−イル]酢酸
ステップ10でビス(3,4−ジクロロフェニル)ジスルフィドを使用した以外は実施例1,ステップ10から11と同様にステップ1の生成物を標記化合物に変換した。
MS M+1=485。
【0199】
(実施例8)
(4−(メチルスルホニル)−5−{[4−(トリフルオロメチル)フェニル]チオ}−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル)酢酸(化合物N)
【0200】
【化9】
【0201】
標記化合物はビス[4−トリフルオロメチル)フェニル]ジスルフィドを使用した以外は実施例1に記載したように製造した。
1H NMR(500MHz,アセトン−d6)δ 8.55(d,1H),7.75(d,1H),7.45(d,2H),7.15(d,2H),4.55(m,1H),4.15(m,1H),3.80(m,1H),3.30(s,3H),2.80−2.10(m,6H)。m/z513(M+1)。
【0202】
(実施例9)
[5−[(2−クロロ−4−フルオロフェニル)チオ]−4−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸(化合物O)
【0203】
【化10】
【0204】
標記化合物はビス(2−クロロ−4−フルオロフェニル)ジスルフィドを使用した以外は実施例1に記載したように製造した。
m/z469(M+1)。
【0205】
(実施例10)
[4−(メチルスルホニル)−5−(2−ナフチルチオ)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸(化合物P)
【0206】
【化11】
【0207】
標記化合物はジ(2−ナフチル)ジスルフィドを使用した以外は実施例1に記載したように製造した。
M/z467(M+1)。
【0208】
(実施例11)
[5−[(2,3−ジクロロフェニル)チオ]−4−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸(化合物Q)
【0209】
【化12】
【0210】
標記化合物はビス(2,3−ジクロロフェニル)ジスルフィドを使用した以外は実施例1に記載したように製造した。
1H NMR(500MHz,アセトン−d6)δ 8.85(d,1H),7.80(ds 1H),7.30(d,1H),7.00(t,1H),6.60(d,1H),4.60(m,1H),4.20(m,1H),3.80(m,1H),3.40(s,3H),2.80−2.10(m,6H)。
【0211】
(実施例12)
[5−[(4−メチルフェニル)チオ]−4−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸(化合物R)
【0212】
【化13】
【0213】
標記化合物はp−トリルジスルフィドを使用した以外は実施例1に記載したように製造した。
1H NMR(500MHz,アセトン−d6)δ 8.55(d,1H),7.80(d,1H),6.95(m,4H),4.60(m,1H),4.15(m,1H),3.80(m,1H),3.35(s,3H),2.80−2.10(m,6H)。
【0214】
(実施例13)
[4−(メチルスルホニル)−5−(フェニルチオ)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸(化合物S)
【0215】
【化14】
【0216】
標記化合物はジフェニルジスルフィドを使用した以外は実施例1に記載したように製造した。
1H NMR(500MHz,アセトン−d6)δ 8.55(d,1H),7.80(d,1H),7.15−6.90(m,5H),4.60(m,1H),4.15(m,1H),3.75(m,1H),3.30(s,3H),2.80−2.10(m,6H)。
【0217】
(実施例14)
[5−[(2,4−ジクロロフェニル)チオ]−4−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸(化合物T)
【0218】
【化15】
【0219】
標記化合物はビス(2,4−ジクロロフェニル)ジスルフィドを使用した以外は実施例1に記載したように製造した。ジスルフィドはエーテル中Br2を使用して2,4−ジクロロチオフェニルから製造した。
1H NMR(500MHz,アセトン−d6)δ 8.55(d,1H),7.85(d,1H),7.35(s,1H),7.00(d,1H),6.65(d,1H),4.55(m,1H),4.15(m,1H),3.80(m,1H),3.35(s,3H),2.80−2.10(m,6H)。
【0220】
(実施例15)
[5−[(4−クロロフェニル)チオ]−4−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[4,3−b]インドリジン−6−イル]酢酸(化合物U)
【0221】
【化16】
【0222】
標記化合物は還流下にアジドをデカリンに加えることにより末端環化を実施した以外は実施例1に記載したように3−クロロニコチンアルデヒド(Heterocycles p.151,1993)から製造した。
1H NMR(500MHz,アセトン−d6)δ 9.20(s,1H),8.85(s,1H),7.20(d,2H),7.00(d,2H),4.70(m,1H),4.30(m,1H),3.75(m,1H),3.35(s,3H),2.80−2.10(m,6H)。
【0223】
(実施例16)
[9−[(4−クロロフェニル)チオ]−1−(メチルスルホニル)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル]酢酸(化合物V)
【0224】
【化17】
【0225】
標記化合物はステップ10でビス(4−クロロフェニル)ジスルフィドを使用した以外は実施例1ステップ10及び11に要約した手順に記載したように実施例6方法1ステップ8の生成物から製造した。
1H NMR(500MHz,アセトン−d6)δ 8.25−8.3(m,1H),7.71−7.75(m,1H),7.12−7.17(m,2H),6.97−7.04(m,2H),4.45−4.51(m,1H),4.32−4.39(m,1H),3.73−3.80(m,1H),3.29(s,3H),3.15−3.21(m,1H),2.99−3.08(m,1H),2.66−2.73(m,1H),2.46−2.54(m,1H)。
【0226】
(実施例17)
(−)−[(4−クロロベンジル)−7−フルオロ−5−メタンスルホニル)−1,2,3,4−テトラヒドロシクロペンタ[b]インドール−3−イル]酢酸(化合物E)
【0227】
【化18】
【0228】
ステップ1:(+/−)−(7−フルオロ−1,2,3,4−テトラヒドロシクロペンタ[b]インドール−3−イル)酢酸エチルエステル
【0229】
【化19】
【0230】
4−フルオロ−2−ヨードアニリン10.00g、2−(2−オキソシクロペンチル)酢酸エチル6.57g及びp−トルエンスルホン酸121mgのベンゼン(100ml)溶液をDean−StarkトラップでN2雰囲気下に24時間還流した。その後、ベンゼンを留去した。次に、DMF60mlを加え、溶液を脱気した後、ヒューニッヒ塩基19mlとPd(OAc)2 405mgを順次加えた。溶液を115℃まで3時間加熱した後、室温まで冷却した、反応をクエンチするために、1N HCl 300mlと酢酸エチル200mlを加え、混合物をセライトで濾過した。相分離し、酸相を酢酸エチル200mlで2回抽出した。有機相を合わせてブラインで洗浄し、無水Na2SO4で乾燥し、セライトで濾過し、濃縮した。100%トルエンを溶離液として粗材料をフラッシュクロマトグラフィーにより更に精製すると、標記化合物が得られた。
1H NMR(アセトン−d6)δ 9.76(br s,1H),7.34(dd,1H),7.03(d,1H),6.78(td,1H),4.14(q,2H),3.57(m,1H),2.85−2.55(m,5H),2.15(m,1H),1.22(t,3H)。
【0231】
【化20】
【0232】
ステップ2:(+/−)−(7−フルオロ−1,2,3,4−テトラヒドロシクロペンタ[b]インドール−3−イル)酢酸
ステップ1からのエステル1.24gの室温のテトラヒドロフラン(THF)(14mL)溶液にMeOH 7mLと2N NaOH 7mLを順次加えた。2.5時間後に、酢酸エチル(EtOAc)/1N HClを入れた分液漏斗に反応混合物を注入した。相分離し、酸相をEtOAcで2回抽出した。有機層を合わせてブラインで洗浄し、無水Na2SO4で乾燥し、蒸発乾涸すると、粗油状物が得られ、次段階でそのまま使用した(純度>90%)。
1H NMR(アセトン−d6)δ 10.90(br s,1H),9.77(br s,1H),7.34(dd,1H),7.04(dd,1H),6.79(td,1H),3.56(m,1H),2.90−2.50(m,5H),2.16(m,1H)。MS(−APCI)m/z232.2(M−H)−。
【0233】
ステップ3:(+/−)−(5−ブロモ−7−フルオロ−1,2,3,4−テトラヒドロシクロペンタ[b]インドール−3−イル)酢酸
【0234】
【化21】
【0235】
ステップ2からの酸(純度>90%)2.20gのピリジン(30mL)溶液に三臭化ピリジニウム(純度90%)6.85gを−40℃で加えた。懸濁液を10分間0℃で撹拌し、30分間かけて室温まで昇温させた。次に、高真空下で加熱せずに溶媒を除去した。粗材料をAcOH 40mLに溶かし、Zn微粉2.88gを0℃の冷溶液に少量ずつ加えた。懸濁液を15分間15℃で撹拌し、室温まで更に15分間昇温させた。この時点で1N HClを加えることにより反応混合物をクエンチし、ブライン/EtOAcを入れた分液漏斗にこの混合物を注入した。層分離し、有機層を水、ブラインで洗浄し、無水Na2SO4で乾燥し、濃縮した。この材料をそれ以上精製せずに次段階で使用した。
1H NMR(アセトン−d6)δ 10.77(br s,1H),9.84(br s,1H),7.09(m,2H),3.60(m,1H),2.95−2.65(m,4H),2.56(dd,1H),2.19(m,1H)。
【0236】
ステップ4:(+/−)−[5−ブロモ−4−(4−クロロベンジル)−7−フルオロ−1,2,3,4−テトラヒドロシクロペンタ[b]インドール−3−イル]酢酸
【0237】
【化22】
【0238】
TLCでモニターしながら酸が完全に消費されるまでステップ3からの酸2.13gのTHF(10mL)溶液に過剰のジアゾメタンのエーテル溶液を加えた。次に、溶媒を減圧除去した。こうして形成された粗メチルエステルのDMF(20mL)溶液にNaH懸濁液(油中60%)539mgを−78℃で加えた。懸濁液を10分間0℃で撹拌し、再び−78℃まで冷却し、4−クロロベンジルブロマイド1.70gで処理した。5分後に温度を0℃まで上げ、混合物を20分間撹拌した。この時点でAcOH 2mLを加えることにより反応をクエンチし、1N HCl/EtOAcを入れた分液漏斗にこの混合物を注入した。層分離し、有機層をブラインで洗浄し、無水Na2SO4で乾燥し、濃縮した。ステップ2に記載した手順を使用してアルキル化物を水素化した。EtOAc/ヘキサンの存在下にトリチュレーションにより粗材料を更に精製すると、標記化合物が得られた。
1H NMR(アセトン−d6)δ 10.70(br s,1H),7.31(d,2H),7.18(d,1H),7.06(d,1H),6.92(d,2H),5.90(d,1H),5.74(d,1H),3.61(m,1H),3.00−2.70(m,3H),2.65(dd,1H),2.39(dd,1H),2.26(m,1H)。MS(−APCI)m/z436.3,434.5(M−H)−。
【0239】
ステップ5:(+)−[5−ブロモ−4−(4−クロロベンジル)−7−フルオロ−1,2,3,4−テトラヒドロシクロペンタ[b]インドール−3−イル]酢酸
【0240】
【化23】
【0241】
ステップ4の酸2.35gの80℃のEtOH(130mL)溶液に(S)−(−)−1−(1−ナフチル)エチルアミン780μLを加えた。溶液を室温まで冷却し、一晩撹拌した。回収した塩(1.7g)をEtOH 200mLで再び再結晶した。濾過後、得られた白色固体塩を1N HClで中和し、生成物をEtOAcで抽出した。有機層をブラインで洗浄し、無水Na2SO4で乾燥し、濃縮した。EtOAcを溶離液として材料をSiO2パッドで濾過すると、標記エナンチオマーが得られた。2種のエナンチオマーの保持時間は夫々7.5分と9.4分であった[ChiralPak ADカラム,ヘキサン/2−プロパノール/酢酸(95:5:0.1)]。高極性エナンチオマーは98%eeであった。
ee=98%;保持時間=9.4分[ChiralPak ADカラム:250×4.6mm,ヘキサン/2−プロパノール/酢酸(75:25:0.1)];[α]D21=+39.2°(c 1.0,MeOH)。
【0242】
ステップ6:(−)−[4−(4−クロロベンジル)−7−フルオロ−5−(メタンスルホニル)−1,2,3,4−テトラヒドロシクロペンタ[b]インドール−3−イル]酢酸及びナトリウム塩
まず、ステップ5からの酸(15.4g)をジアゾメタンでエステル化した。こうして形成されたエステルをN−メチルピロリジノン中メタンスルフィン酸ナトリウム塩16.3g及びCuI(I)30.2gと混合することによりスルホニル化を実施した。懸濁液をN2流下に脱気し、150℃まで加熱し、3時間撹拌した後、室温まで冷却した。反応をクエンチするために、酢酸エチル500mlとヘキサン500mlを加え、EtOAcを溶離液として混合物をSiO2パッドで濾過した。有機相を濃縮した。粗油状物をEtOAcに溶かし、水で3回、ブラインで1回洗浄し、無水Na2SO4で乾燥し、濾過し、濃縮した。EtOAc中100%トルエン→50%トルエンの勾配を溶離液として粗材料をフラッシュクロマトグラフィーにより更に精製すると、スルホン酸エステル14gが得られ、ステップ2に記載した手順を使用して加水分解した。酢酸イソプロピル/ヘプタンとCH2Cl2/ヘキサンで順次2回再結晶後に標記化合物が得られた。
1H NMR(500MHz アセトン−d6)δ 10.73(br s,1H),7.57(d,2H,J=8.8Hz),7.31(m,1H),7.29(m,1H),6.84(d,2H,J=8.8Hz),6.29(d,1H,JAB=17.8Hz),5.79(d,1H,JAB=17.8Hz),3.43(m,1H),2.98(s,3H),2.94(m,1H),2.85−2.65(m,3H),2.42(dd,1H,J1=16.1Hz,J2=10.3Hz),2.27(m,1H)。13C NMR(125MHz アセトン−d6)δ 173.0,156.5(d,JCF=237Hz),153.9,139.2,133.7,133.3,130.0(d,JCF=8.9Hz),129.6,128.2,127.5(d,JCF=7.6Hz),122.2(d,JCF=4.2Hz),112.3(d,JCF=29.4Hz),111.0(d,JCF=22.6Hz)5 50.8,44.7,38.6,36.6,36.5,23.3。MS(−APCI)m/z436.1,434.1(M−H)−。
ee=97%;保持時間=15.3分[ChiralCel ODカラム:250×4.6mm,ヘキサン/2−プロパノール/エタノール/酢酸(90:5:5:0.2)];[α]D21=−29.3°(c1.0,MeOH)。Mp175.0℃。
【0243】
EtOH(100mL)中の上記酸化合物6.45g(14.80mmol)を1N NaOH水溶液14.80mLで処理することによりナトリウム塩を製造した。有機溶媒を減圧除去し、粗固体を還流下にイソプロピルアルコール1.2Lに溶かした。溶媒の蒸留により最終容量を500mLまで減らした。室温まで冷却することによりナトリウム塩を結晶化させた。結晶ナトリウム塩をH2Oに懸濁し、ドライアイス浴で凍結させ、高真空下に凍結乾燥すると、標記化合物がナトリウム塩として得られた。
1H NMR(500MHz DMSO−d6)δ 7.63(dd,1H,J1=8.5Hz,J2=2.6Hz),7.47(dd,1H,J1=9.7Hz,J2=2.6Hz),7.33(d,2H,J=8.4Hz),6.70(d,2H,J=8.4Hz),6.06(d,1H,J=17.9Hz),5.76(d,1H,J=17.9Hz),3.29(m,1H),3.08(s,3H),2.80(m,1H),2.69(m,1H),2.55(m,1H),2.18(m,2H),1.93(dd,1H,J1=14.4Hz,J2=9.7Hz)。
【0244】
(実施例17A)
(+/−)−[5−ブロモ−4−(4−クロロベンジル)−7−フルオロ−1,2,3,4−テトラヒドロシクロペンタ[b]インドール−3−イル)酢酸(実施例17,ステップ4)の代替手順
ステップ1:(+/−)−7−フルオロ−1,2,3,4−テトラヒドロシクロペンタ[b]インドール−3−イル)酢酸ジシクロヘキシルアミン(DCSA)塩
2−ブロモ−4−フルオロアニリンの0.526Mキシレン溶液を(2−オキソシクロペンチル)酢酸エチル(1.5当量)及び硫酸(0.02当量)と共に20時間加熱還流した。Dean−Stark装置で水を共沸除去した。反応後にNMRを測定し、20時間後に、一般に所望イミン中間体への80から85%変換が認められた。反応混合物を1M重炭酸ナトリウム(0.2容量)で15分間洗浄し、有機フラクションを蒸発させた。残留シロップを減圧(0.5mm Hg)蒸留した。残留キシレンを30℃で蒸留した後、過剰のケトンと未反応アニリンを50から110℃で回収し、イミンを110から180℃フラクションで薄茶色透明液体として純度83%で回収した。
【0245】
次にイミン中間体を酢酸カリウム(3当量)、テトラ−n−ブチルアンモニウムクロライド1水和物(1当量)、酢酸パラジウム(0.03当量)及びN,N−ジメチルアセトアミド(イミン最終濃度=0.365M)の脱気混合物に加えた。反応混合物を115℃まで5時間加熱し、室温まで冷却した。次に3N KOH(3当量)を加え、混合物を室温で1時間撹拌した。反応混合物を水(1.0容量)で希釈し、トルエン(3×0.75容量)で洗浄した。水相を3N HClでpH1まで酸性化し、tertブチルメチルエーテル(2×0.75容量)で抽出した。有機フラクションを合わせて水洗した(0.75容量)。薄茶色透明溶液にジシクロヘキシルアミン(1当量)を加え、溶液を室温で16時間撹拌した。塩を濾過し、酢酸エチル、tertブチルメチルエーテルで洗浄し、乾燥すると、標記化合物が得られた。アッセイ:94 A%。
1H NMR(500mHz,CDCl3):δ 9.24(s,1H),7.16−7.08(m,2H),6.82(t,1H),6.2(br,2H),3.6−3.5(m,1H),3.04−2.97(m,2H),2.88−2.70(m,3H),2.66(dd,1H),2.45−2.37(m,1H),2.13−2.05(m,2.05),1.83(d,4H),1.67(d,2H),1.55−1.43(m,4H),1.33−1.11(m,6H)。
【0246】
ステップ2:(+/−)−(5−ブロモ−7−フルオロ−1,2,3,4−テトラヒドロシクロペンタ[b]インドール−3−イル)酢酸
上記ステップ1からのDCHA塩のジクロロメタン(0.241M溶液)中スラリーを−20から−15℃まで冷却した。ピリジン(2当量)を一度に加え、温度を−20℃から−15℃に維持しながら30から45分間かけてスラリーに臭素(2.5当量)を滴下した。(臭素を約1/3添加した時点では、反応混合物は高粘性であり、効率的な撹拌が必要であった。その後、臭素を約1/2添加した時点で混合物は再び「低粘性」になった。)添加の完了後、反応混合物を更に1時間−15℃でエージングさせた。次に酢酸(3.04当量)を5分間かけて加え、亜鉛微粉(3.04当量)を少量ずつ加えた。(亜鉛の一部は−15℃で加え、混合物を約5分間エージングさせて発熱を進行させた(約−15℃から−10℃))。約30分間かけて亜鉛を約5回加えることによりこの操作を繰返した。発熱が観察されなくなったら、残余の亜鉛をより迅速に加えた。全操作は約30から45分間を要した。
【0247】
添加の完了後、バッチを室温まで昇温させ、1時間エージングさせ、濃縮した。反応混合物をメチルt−ブチルエーテル(MTBE,0.8容量)に交換し、10%酢酸水溶液(0.8容量)を加えた。混合物(塩の結晶化、例えばピリジウム)を室温で1時間エージングさせ、ソルカフロックで濾過した。ソルカフロックパッドをMTBE(約0.2容量)でリンスし、濾液(2相,MTBE/水)を抽出器に移した。有機相を水洗した(0.8容量)。MTBE抽出相を濃縮し、イソプロピルアルコール(IPA,0.25容量)に交換し、化合物を結晶化させた。水(0.25容量)を加え、バッチを1時間エージングさせた。更に水(0.33容量)を1時間加えた。水添加の完了後、バッチを更に1時間エージングさせ、濾過し、30/70 IPA/水(0.15容量)でリンスした。結晶化したブロモ酸を+45℃のオーブンで乾燥した。
【0248】
ステップ3:(+/−)−[5−ブロモ−4−(4−クロロベンジル)−7−フルオロ−1,2,3,4−テトラヒドロシクロペンタ[b]インドール−3−イル)−酢酸
ステップ2のブロモ酸をジメチルアセトアミド(0.416M溶液)に溶かし、炭酸セシウム(2.5当量)を一度に加えた。スラリーに4−クロロベンジルクロライド(2.5当量)を一度に加え、バッチを50℃まで20時間加熱した。バッチを室温まで冷却し、5N水酸化ナトリウム(4.00当量)を5分間かけて加えた(温度は+40℃まで上昇した)。反応混合物を50℃で約3時間エージングさせ、室温まで冷却し、L抽出器に移した。溶液を酢酸イソプロピル(IPAc,2容量)で希釈し、+15℃まで冷却した。溶液を5N HClでpHから2まで酸性化した。層分離し、有機層を水洗した(2×2容量)。IPAc溶液を濃縮し、IPA(0.8容量)に交換し、生成物を結晶化させた。水(8L)を2時間加え、バッチを濾過すると、標記化合物が得られた。バッチを+40℃のオーブンで24時間乾燥してもよい。
【0249】
(実施例18)
(+/−)−{4−[1−(4−クロロフェニル)エチル]−7−フルオロ−5−メタンスルホニル−1,2,3,4−テトラヒドロシクロペンタ[b]インドール−3−イル]酢酸(化合物X)
【0250】
【化24】
【0251】
標記化合物はPCT WO03/062200(公開日2003年7月30日)の記載に従って合成した。
【0252】
(実施例19)
(+/−)−[9−(4−クロロベンジル)−6−フルオロ−メタンスルホニル−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸(化合物Y)
【0253】
【化25】
【0254】
標記化合物はPCT WO03/062200(公開日2003年7月30日)の記載に従って合成した。
【0255】
(実施例20)
[4−(4−クロロベンジル)−7−フルオロ−5−メタンスルホニル−1−オキソ−1,2,3,4−テトラヒドロシクロペンタ[b]インドール−3−イル)酢酸(化合物Z)
【0256】
【化26】
【0257】
標記化合物はPCT WO03/062200(公開日2003年7月30日)の記載に従って合成した。
【0258】
(実施例21)
{9−[(3,4−ジクロロフェニル)チオ]−1−イソプロピル−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル}酢酸(エナンチオマーA及びエナンチオマーB)(化合物AA)
【0259】
【化27】
【0260】
ステップ1:2−クロロニコチンアルデヒド
ジイソプロピルアミン(110mL,780mmol)のTHF(500mL)溶液に−40℃のn−BuLi(300mL,750mmol)の2.5Mヘキサン溶液を加えた。5分後に、反応混合物を−95℃まで冷却した後に、DMPU(15mL)と2−クロロピリジン(50mL,532mmol)を順次加えた。得られた混合物を次に昇温させ、−78℃で4時間撹拌した。その後、黄色い懸濁液を再び−95℃まで冷却した後、DMF(70mL)を加えた。最終反応混合物を−78℃まで昇温させ、この温度で1.5時間撹拌した。反応混合物を冷HCl水溶液(3N,800mL)に注ぎ、5分間撹拌した。濃NH4OH水溶液を加えてpHを7.5に調整した。水層をEtOAcで3回抽出した。有機層を合わせてNH4Cl水溶液とブラインで洗浄し、無水Na2SO4で乾燥し、濾過し、濃縮した。100%ヘキサン→100%EtOAcの勾配を溶離液として粗材料をシリカゲルパッドにより更に精製し、生成物を冷ヘキサン中で結晶化させると、標記化合物が薄黄色固体として得られた。
【0261】
ステップ2:(2Z)−2−アジド−3−(2−クロロピリジン−3−イル)プロプ−2−エン酸メチル
2−クロロニコチンアルデヒド(20.0g,139.9mmol)とアジド酢酸メチル(32.2mL,349.7mmol)のMeOH(168mL)溶液を−20℃のNaOMeの25%MeOH溶液(80mL,349mmol)に加えた。内部温度をモニターし、30分間の添加中、から−20℃に維持した。得られた混合物を次に氷浴中で数時間撹拌した後、冷所にて氷浴中で一晩撹拌した。次に懸濁液を氷とNH4Clの混合物に注ぎ、スラリーを10分間撹拌後に濾過した。生成物を冷H2Oで洗浄した後、減圧乾燥した。粗材料をCH2Cl2に溶かし、MgSO4を加えた。懸濁液をシリカゲルパッドで濾過し、CH2Cl2て洗浄した。濾液を減圧濃縮すると、標記生成物のベージュ色沈殿(20g)が得られた。
【0262】
ステップ3:4−クロロ−1H−ピロロ[3,2−c]ピリジン−2−カルボン酸メチル
(2Z)−2−アジド−3−[2−クロロピリジン−3−イル]プロプ−2−エン酸メチル(21g,88mmol)のメシチレン(880mL)溶液を1時間加熱還流した。反応混合物を室温まで冷却した後、0℃まで冷却し、沈殿を濾過し、冷ヘキサンで洗浄した。材料を1:20 EtOAc/ヘキサン中で一晩撹拌すると、濾過後に標記生成物が薄黄色固体(13.2g)として得られた。
【0263】
ステップ4:1−クロロ−8−オキソ−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−7−カルボン酸メチル
4−クロロ−1H−ピロロ[3,2−c]ピリジン−2−カルボン酸メチル(12.5g,59mmol)のTHF(116mL)−トルエン(460mL)懸濁液にカリウムtert−ブトキシド(64mL,64mmol)の1.0MTHF溶液とアクリル酸メチル(55mL,611mmol)を加えた。得られた混合物を100℃に18時間加熱した。その後、懸濁液を室温まで冷却し、飽和NH4Cl水溶液(400mL)とヘキサン(400mL)の混合物に注いだ。固形分をデカントし、濾過し、H2Oとヘキサンで洗浄すると、標記化合物が得られた。
【0264】
ステップ5:1−クロロ−6,7−ジヒドロ−8H−ピリド[3,4−b]ピロリジン−8−オン
前段階の化合物に100℃で1時間撹拌下にイソプロパノール(8.0mL)と濃HCl(2.0mL)を加えた。反応混合物をEtOAcとNa2CO3に分配した。有機相を分離し、蒸発させると、標記化合物が得られた。
【0265】
ステップ6:1−イソプロペニル−6,7−ジヒドロ−8H−ピリド[3,4−b]ピロリジン−8−オン
DMF(100mL)中の1−クロロ−6,7−ジヒドロ−8H−ピリド[3,4−b]ピロリジン−8−オン(5.0g,24.3mmol)、トリス(ジベンジリデンアセトン)ジパラジウム(0)(1.0g,1.09mmol)及びトリフェニルアルシン(2.70g,8.82mmol)の混合物にトリブチルイソプロペニルスタナン(9.60g,29.00mmol)を加えた。得られた混合物を脱気し、78℃に18時間加熱した。溶媒を減圧蒸発させた。得られた混合物にCH2Cl2とセライトを加えた後、セライトで濾過した。標記化合物をフラッシュクロマトグラフィーにより精製した(ヘキサン中50%→100%EtOAc)。
【0266】
ステップ7:(2E)−(1−イソプロペニル−6,7−ジヒドロ−8H−ピリド[3,4−b]ピロリジン−8−イリデン)エタン酸エチル
1−イソプロペニル−6,7−ジヒドロ−8H−ピリド[3,4−b]ピロリジン−8−オン(0.60g,2.8mmol)とホスホノ酢酸トリエチル(1.00g,4.46mmol)の−78℃のTHF(24mL)溶液に80%NaH(0.12g,4.00mmol)を加え、反応混合物を0℃まで昇温させた後、室温まで昇温させた。反応混合物を飽和NH4ClとEtOAcに注いだ。有機相を分離し、Na2SO4で乾燥し、蒸発させた。標記化合物をフラッシュクロマトグラフィーにより精製した(ヘキサン中40%EtOAc)。
【0267】
ステップ8:(1−イソプロピル−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル)酢酸エチル
(2E)−(1−イソプロペニル−6,7−ジヒドロ−8H−ピリド[3,4−b]ピロリジン−8−イリデン)エタン酸エチル(0.40g,1.4mmol)のMeOH(20mL)溶液にPd(OH)2(0.20g)を加えた。混合物を1気圧H2下に3時間撹拌した。混合物をセライトで濾過し、蒸発させると、標記化合物が得られた。
【0268】
ステップ9:{9−[(3,4−ジクロロフェニル)チオ]−イソプロピル−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル}酢酸エチル
ビス(3,4−ジクロロフェニル)ジスルフィド(0.24g,0.67mmol)のCH2Cl2(5.6mL)溶液にSO2Cl2(0.036mL)を加えた。得られた黄色い混合物を室温で1時間撹拌した。この溶液を(1−イソプロピル−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル)酢酸エチル(0.15g,0.52mmol)の0℃のDMF(5.6mL)溶液に加えた。0℃で1.5時間後に、反応混合物を飽和NaHCO3とEtOAcに注いだ。有機相を分離し、Na2SO4で乾燥し、濾過し、蒸発させた。標記化合物をフラッシュクロマトグラフィーにより精製した(ヘキサン中30%→40%EtOAc)。
【0269】
ステップ10:{9−[(3,4−ジクロロフェニル)チオ]−イソプロピル−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル}酢酸
{9−[(3,4−ジクロロフェニル)チオ]−イソプロピル−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル}酢酸エチル(0.23g,0.50mmol)のTHF(5mL)及びMeOH(2.5mL)溶液に1.0M NaOH(1.5mL,1.5mmol)を加えた。室温で18時間後にHOAc(0.25mL)を加え、溶媒を蒸発させた。残渣をEtOAc/H2Oに取り、有機層をH2Oとブラインで洗浄した。乾燥(Na2SO4)後、溶液を濾過し、蒸発させた。残渣を1:1 EtOAc:ヘキサンの存在下で撹拌すると、濾過後に標記化合物が白色固体として得られた。
1H NMR(MeOH−d4)δ 1.14−1.26(m,6H),2.47−2.56(m,1H),2.56−2.64(m,1H),2.94−3.05(m,2H),3.81−3.89(m,1H),4.22−4.30(m,1H),4.33−4.44(m,2H),6.93−6.99(m,1H),7.14−7.19(m,1H),7.33−7.39(m,1H),7.54−7.59(m,1H),8.16−8.21(m,1H)。
【0270】
CH2N2を使用してステップ10の生成物をそのメチルエステルに変換し、ヘキサン中12%2−プロパノールを流速6mL/分で溶離液として使用してエステルをキラル固定相(chiralcel ODカラム2×25cm)でHPLC分離した。エナンチオマーA(低極性)は保持時間31.9分であり、エナンチオマーB(高極性)は保持時間35.5分である。A及びBの両者を実施例17ステップ10と同様に加水分解すると、標記化合物のエナンチオマーA及びBが得られた。
【0271】
(実施例22)
((1R)−6−フルオロ−8−(メチルスルホニル)−9−{(1S)−1−[4−(トリフルオロメチル)フェニル]エチル}−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸(化合物AJ)
【0272】
【化28】
【0273】
ステップ1:塩化2−(2−ブロモ−4−フルオロフェニル)ヒドラジニウム
2−ブロモ−4−フルオロアニリンの−10℃の濃HCl(1.5M)懸濁液にNaNO2(1.1当量)の10.0M水溶液をゆっくりと加えた。混合物を0℃で2.5時間撹拌した。次に内部温度を10℃未満に維持しながらSnCl2(3.8M)の冷(−30℃)濃HCl溶液をゆっくりと加えた。得られた混合物を10℃で20分間、次いで室温で1時間機械的に撹拌した。高粘性スラリーを濾過し、固体を一晩風乾した。固体を冷HClに再懸濁し、再び濾過した。乾燥した材料をEt2Oに懸濁し、10分間撹拌し、濾過し、一晩風乾すると、標記化合物がベージュ色固体として得られた。
【0274】
ステップ2:(+/−)−(8−ブロモ−6−フルオロ−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル)酢酸エチル
ステップ1の化合物(1当量)のAcOH(0.5M)懸濁液に(2−オキソシクロヘキシル)酢酸エチル(1当量)を加えた。混合物を還流下に16時間撹拌し、冷却し、AcOHを減圧蒸発により除去した。残渣をEtOAcで希釈し、水と飽和NaHCO3水溶液で洗浄した。有機層をNa2SO4で乾燥し、濃縮した。次にトルエンを溶離液として残渣をシリカゲルパッドで濾過した。濾液を濃縮し、ヘキサン中で撹拌すると、濾過後に標記化合物が白色固体として得られた。MS(+APCI)m/z354.2(M+H)+。
【0275】
ステップ3:(+/−)−[6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸エチル
ステップ2の化合物(1当量)の無水DMSO(0.28M)溶液にメタンスルフィン酸ナトリウム(3当量)とヨウ化銅(3当量)を加えた。N2を混合物中に5分間バブリングした後、反応混合物を100℃でN2雰囲気下に撹拌した。12時間後に更にメタンスルフィン酸ナトリウム(2当量)とヨウ化銅(2当量)を加えた。混合物を更に12時間100℃で撹拌し、冷却し、EtOAcで希釈し、1N HClを加えて混合物を酸性化した。懸濁液を30分間撹拌し、セライトで濾過した。濾液を水洗し、Na2SO4で乾燥し、濃縮した。まず非極性不純物を除去するために溶離液としてトルエンを使用し、次いで所望生成物を溶出させるためにヘキサン/EtOAcの2:1混合物を使用して残渣をシリカゲルパッドで濾液した。ヘキサン/EtOAc混合物による溶離後の濾液を濃縮すると、標記化合物が薄黄色固体として得られた。MS(−APCI)m/z352.1(M−H)−。
【0276】
ステップ4:[(1R)−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸エチル
ヘキサン中15%iPrOHの混合物を溶離液としてステップ3からのラセミ混合物をchiralpak AD分取カラムで分取HPLCにより分離した。最終生成物の活性に基づき、高極性エナンチオマー(保持時間が長い)が標記化合物であることが確認された。
【0277】
ステップ5:[(1R)−9−[(1S)−1−(4−クロロフェニル)エチル]−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸エチル
ステップ4の化合物(1当量)、トリフェニルホスフィン(1.5当量)及び(1R)−1−(4−クロロフェニル)エタノール(1.5当量,参考例1に記載の一般手順に従って製造)のTHF(0.175M)溶液にアゾジカルボン酸ジ−tert−ブチル(THF中2.1M,1.5当量)を10分間かけて加えた。混合物を室温で2時間撹拌し、濃縮した。トルエン中7%EtOAcを溶離液として残渣をシリカゲルフラッシュクロマトグラフィーにより精製すると、所望生成物(純度から90%)が得られ、次段階でそのまま使用した。
【0278】
ステップ6:[(1R)−9−[(1S)−1−(4−クロロフェニル)エチル]−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸及び[(1S)−9−[(1S)−1−(4−クロロフェニル)エチル]−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸
ステップ5の化合物のTHFとメタノール(0.1M)の2:1混合物溶液に1N LiOH水溶液(3当量)を加えた。混合物を室温で2時間撹拌し、AcOHを加え、溶媒を蒸発により除去した。残渣をEtOAc/H2Oに取り、有機層をブラインで洗浄し、Na2SO4で乾燥し、濾過し、濃縮した。残渣をヘキサン中30%EtOAcでスウィッシュし、生成物をジエチルエーテルに懸濁し、45分間音波処理し、濾過し、高真空下に50℃で24時間乾燥すると、標記化合物が白色固体として得られた。MS(−APCI)m/z462.1(M−H)。
【0279】
別法として、(+/−)−[6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸エチルをステップ5のアルキル化反応に使用し、[(1R)−9−[(1S)−1−(4−クロロフェニル)エチル]−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸エチルと[(1S)−9−[(1S)−1−(4−クロロフェニル)エチル]−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸エチルの2種のジアステレオマーの混合物を得た。以下の手順を使用してジアステレオマー混合物を選択的加水分解により分解すると、所望[(1R)−9−[(1S)−1−(4−クロロフェニル)エチル]−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸が得られた。
【0280】
分解:
[(1R)−9−[(1S)−1−(4−クロロフェニル)エチル]−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸エチルと[(1S)−9−[(1S)−1−(4−クロロフェニル)エチル]−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸エチルのジアステレオマー混合物(1当量)をTHF/MeOHの3.5/1混合物(0.25M)に溶かし、0℃に冷却した。1N LiOH水溶液(1当量)をゆっくりと加え、12時間又は[(1R)−9−[(1S)−1−(4−クロロフェニル)エチル]−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸エチルがほぼ完全に加水分解されるまで混合物を0℃で撹拌し、他方のジアステレオマーはこれらの条件下で僅かしか加水分解されなかった。AcOHを加え、溶媒を蒸発により除去した。残渣をEtOAc/H2Oに取り、有機層をブラインで洗浄し、Na2SO4で乾燥し、濾過し、濃縮した。1%AcOHを加えたヘキサン中40%EtOAcを溶離液として[(1S)−9−[(1S)−1−(4−クロロフェニル)エチル]−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸エチルと[(1R)−9−[(1S)−1−(4−クロロフェニル)エチル]−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸をフラッシュクロマトグラフィーにより分離すると、所望[(1R)−9−[(1S)−1−(4−クロロフェニル)エチル]−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸がde>90%で得られ、ヘキサン中30%EtOAcでスウィッシュすると、所望化合物が白色固体としてde>95%で得られた。
【0281】
ステップ7:[(1R)−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸メチル
[(1R)−9−[(1S)−1−(4−クロロフェニル)エチル]−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸(MeOH中[α]D=−226°)のMeOH(0.1M)溶液に炭素担持10%パラジウム(10%wt/wt)を加えた。N2流を混合物に5分間バブリングした。反応混合物を室温でH2雰囲気(バルーン)下に24時間撹拌し、CH2Cl2を溶離液としてセライトパッドで濾過した。溶媒を減圧蒸発により除去し、残渣をMeOHでスウィッシュすると、化合物[(1R)−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸メチルが得られた。
【0282】
【化29】
【0283】
ステップ8:((1R)−6−フルオロ−8−(メチルスルホニル)−9−{(1S)−1−[4−(トリフルオロメチル)フェニル]エチル}−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル)酢酸(化合物AJ)
ステップ7の化合物(1当量)、トリフェニルホスフィン(1.5当量)及び(1R)−1−[4−(トリフルオロメチル)フェニル]エタノール(1.5当量)のTHF(0.2M)溶液にアゾジカルボン酸ジ−tert−ブチル(THF中1M,1.5当量)を20分間かけて加えた。混合物を室温で2時間撹拌し、濃縮した。トルエン中10%EtOAcを溶離液として残渣をシリカゲルフラッシュクロマトグラフィーにより精製すると、((1R)−6−フルオロ−8−(メチルスルホニル)−9−{(1S)−1−[4−(トリフルオロメチル)フェニル]エチル}−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル)酢酸メチル(純度から90%)が得られ、次段階でそのまま使用した。
【0284】
上記エステル(1当量)の0℃のTHF/MeOHの3.5/1混合物(0.25M)溶液に1N LiOH水溶液(1当量)をゆっくりと加え、16時間又はエステルがほぼ完全に加水分解されるまで混合物を0℃で撹拌し、これらの条件下で他方の副ジアステレオマーは加水分解速度が著しく遅い。AcOHを加え、溶媒を減圧除去した。残渣をEtOAc/H2Oに取り、有機層をブラインで洗浄し、Na2SO4で乾燥し、濾過し、濃縮した。未反応メチルエステルを除去するために、まず10%EtOAc/トルエン、次いで1%AcOHを加えた60%EtOAc/トルエンを溶離液として残渣をシリカゲルパッドで濾過した。残渣を30%EtOAc/ヘキサンでスウィッシュし、高真空下に50℃で16時間乾燥すると、標記化合物がde及びee>95%(キラルHPLCにより確認)で白色固体として得られた。MS(−APCI)m/z496.0(M−H)−。MeOH中[α]D=−181°。
【0285】
生物学
選択的DPアンタゴニストとして機能する本発明で使用する化合物は一般にDPに対する親和性(Ki)がCRTH2受容体に対する親和性(Ki)の少なくとも約10倍(Ki値の数値が小さい)である。本発明で使用する典型的DPアンタゴニストはDP受容体に対する選択性がCRTH2受容体の少なくとも約10倍である。より特定的には、選択的DP受容体アンタゴニストはDP受容体に対する選択性がCRTH2受容体の少なくとも約100倍である。更に特定的には、DP選択的アンタゴニスト化合物はDP受容体に対する選択性がCRTH2受容体の少なくとも約800から1000倍であり、即ちDP受容体に対する親和性(Ki)がCRTH2受容体に対する親和性(Ki)の800から1000倍である。
【0286】
本明細書で使用する場合には、ある化合物が「DP受容体を選択的に調節する」とき、この化合物は治療用量で達成可能な濃度でDP受容体と結合し、これを阻害するが、このような治療達成可能な濃度でCRTH2を実質的に調節しない。
【0287】
一般に、本発明で使用するDPアンタゴニストはCRTH2受容体に対する親和性(Ki)が約0.5マイクロモル以上である。CRTH2に対する結合親和性が約0.5マイクロモル以上であり、DP受容体に対する選択性がCRTH2の少なくとも約10倍である化合物がこのような選択的DPアンタゴニストの不在下でニコチン酸を投与する場合に認められるフラッシング効果を抑制するのに有用である。
【0288】
組換えヒトDP及びCRTH2受容体に対する化合物の親和性及び選択性の測定
Abramovitz Mら,Biochem.Biophys.Acta(2000)1483:285−293、及びSawyer Nら,Br.J.Pharmacol.(2002);137:1163−1172に記載されているような放射性リガンド結合アッセイを使用してDP及びCRTH2に対する化合物の受容体親和性及び選択性を測定した。要約すると、ヒト胚性腎(HEK)293EBNA(Epstein Barr virus Nuclear Antigen:エプスタイン・ハケールウイルス核抗原)細胞(HEK293E細胞株と呼ぶ)を使用してヒトDP及びCRTH2受容体を各々発現する安定な細胞株を樹立した。これらの組換え細胞株から調製した膜フラクションを平衡競合放射性リガンド結合アッセイで利用し、DP及びCRTH2受容体に対する化合物の親和性と選択性を測定した。
【0289】
全長コーディング配列に対応するDP及びCRTH2 cDNAを哺乳動物発現ベクターpCEP4(Invitrogen)の適当な部位にサブクローニングし、HEK293E細胞で発現させた。分画遠心(いずれも4℃にて1000×gで10分後、160,000×gで30分)後にプロテアーゼ阻害剤(2mM AEBSF,10μM E−64,100μMロイペプチン及び0.05mg/mLペプスタチン)の存在下に氷上で30分間800psi窒素キャビテーションにより細胞を溶解させることにより膜を調製した。1mM EDTAを加えた10mM HEPES/KOH(pH7.4)にDounceホモジナイゼーション(Dounce A;10ストローク)により160,000×gペレットを約5から10mg/mL蛋白質で再懸濁し、液体窒素で凍結させ、−80℃で保存した。1mM EDTA,10mM MnCl2及び0.7nM [3H]PGD2(200Ci/mmol)を加えた10mM HEPES/KOH(pH7.4)中、最終インキュベーション容量0.2mLで受容体結合アッセイを実施した。160,000×gフラクションからの膜蛋白質(DPでは約30μg、CRTH2では10μg)を加えることにより反応を開始した。リガンドをジメチルスルホキシド(DMSO)に加え、ジメチルスルホキシドは全インキュベーションで1%(v/v)の一定値に維持した。10μM非放射性PGD2の存在下で非特異的結合を測定した。インキュベーションはミニオービタルシェーカーで室温にて60分間実施した。Tomtec Mach III 96ウェル半自動セルハーベスターを使用してEDTAを添加しないアッセイインキュベーションバッファー(4℃)で予め湿潤させておいた96ウェルUnifilter GF/C(Canberra Packard)で迅速濾過により結合アッセイを終了した。フィルターを同一バッファー3から4mLで洗浄し、90分間55℃で乾燥し、1450 MicroBeta(Wallac)カウンターを使用してUltima Gold F(Canberra Packard)50μLを添加するシンチレーション計数により個々のフィルターに結合した残留放射能を測定した。
【0290】
競合剤の不在下で全結合から非特異的結合を引いた値として最大特異的結合を定義した。各化合物濃度で特異的結合を測定し、最大特異的結合の百分率として表した。試験化合物濃度の関数として最大特異的結合百分率を表すことによりS字状平衡競合曲線を作成し、4パラメーター式に基づくシンプレックス法による非線形最小二乗曲線フィットルーチンを利用してカスタム設計ソフトウェアパッケージにより分析し、変曲点(InPt)を決定した。式Ki=InPt/1+([放射性リガンド]/Kd)(式中、Kdは放射性リガンド−受容体相互作用の平衡解離定数である)から平衡阻害定数(Ki)を計算することにより試験化合物の結合親和性を決定した。InPtを決定できない場合には、IC50を使用した(即ち最大特異的結合の50%を阻害するために必要な試験化合物の濃度)。
【0291】
一般に、本発明で使用する化合物はDP受容体に対するKiが約0.4nMから約16.3nMである。同様に、本発明で使用する化合物は一般にCRTH2受容体に対するKiが約180nMから約22,000nM又はそれ以上である。
【0292】
ニコチン酸により誘発されるマウスの血管拡張に対する化合物の効果
本明細書に記載する選択的DPアンタゴニストの効力はヒトニコチン酸により誘発されるフラッシングのマウスモデルを使用してフラッシング抑制効果を測定することにより実証することができる。(対照として)ビヒクル又はDPアンタゴニストで予め処置しておいたマウスにニコチン酸を投与後にマウス耳の血流(ヒトにおけるフラッシングの顕著な要素である血管拡張の尺度)を測定する。具体的には、雄C57BL/6マウス(から25g)を試験で使用した。各試験群でマウス5匹を評価した。ネンブタールを最終濃度5mg/mlまで水で希釈し、0.3ml/匹を腹腔内注射した。DPアンタゴニストを5%ヒドロキシプロピルβ−シクロデキストリンに最終濃度5mg/mlで溶かし、化合物を容量0.2ml/匹(から40mpk)で腹腔内投与した。ニコチン酸を5%ヒドロキシプロピルβ−シクロデキストリンに最終濃度12.5mg/mlで溶かした。ニコチン酸ストック溶液を2N NaOHでpH7.4に調整し、0.2ml/匹(から100mpk)を皮下注射した。
【0293】
ニコチン酸投与の5分前から開始して30秒おきに15分間レーザードップラー血流イメージャー(PeriScan PIM II,Perimed,Sweden)でマウス耳皮膚の血流をモニターした。ビヒクル又はニコチン酸投与後10分間の平均血流変化百分率を計算し、時間に対する平均血流変化百分率のグラフを1匹毎に作成した。次に、各グラフから平均血流の曲線下面積(AUC)(%Δ×分)を計算し、結果を各群の平均AUC±SEMで表した。
【0294】
化合物DはPGD−2により誘発されるマウスの血管拡張を抑制した(図1)。試験したDPアンタゴニストはニコチン酸により誘発されるマウスの血管拡張を抑制し、その他、選択化合物のデータを図2及び3に示す。
【0295】
ニコチン酸、又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストのフラッシング効果のアスピリン抑制に関して、ヒトフラッシングの相関としてDP受容体(DP1)を遺伝的に欠失するマウスを使用し、ニコチン酸により誘発される血管拡張のマウスモデルで試験した処、ニコチン酸により誘発される血管拡張が多少生じる場合もあるが、血管拡張の程度はDP受容体をもつマウスよりも低いことが判明した。従って、ニコチン酸により誘発される血管拡張の一部はDP受容体に依存しないと思われる。このDP非依存性血管拡張はCOX−1/COX−II阻害剤であるアスピリンをマウスに投与又は予備投与することにより抑制することができる。ヒト用量は約20から25mgから約650mgまでであり、ニコチン酸投与の約1時間から約30分間前から同時投与までとし、必要に応じて約4時間までの間隔で反復する。従って、DP受容体アンタゴニストとアスピリンの併用はヒトにおけるニコチン酸により誘発されるフラッシングを抑制し、DP受容体又はアスピリン単独では抑制されないフラッシングの一部又は全部を緩和するために特に有効である。
【0296】
本明細書に引用する全特許、特許出願及び刊行物は参考資料としてその開示内容全体を本明細書に組込む。本明細書には所定の好ましい態様を詳細に記載したが、多数の代替態様が本発明の範囲に含まれるとみなされる。
【図面の簡単な説明】
【0297】
【図1】化合物DがプロスタグランジンD2により誘発されるマウスの血管拡張を抑制することを示すグラフである。
【図2】化合物Dがニコチン酸により誘発されるマウスの血管拡張を抑制することを示すグラフである。
【図3】他の選択化合物がニコチン酸により誘発されるマウスの血管拡張を抑制することを示すグラフである。
【技術分野】
【0001】
ナイアシンないしニコチン酸(ピリジン−3−カルボン酸)は血清高密度リポ蛋白(HDL)値を上昇させるのに有効であることがよく知られている薬剤である。しかし、ニコチン酸は皮膚血管拡張(フラッシングとも言う)を併発することが多い。この副作用はニコチン酸によりプロスタグランジンD2が皮膚内に放出されることに起因し、非常に重度であるため、ニコチン酸治療を中断する患者が多い。本発明は実質的フラッシングを併発せずに治療を進めることができるように、その非投与時に発生する可能性のある皮膚血管拡張を緩和又は解消する化合物とニコチン酸又は別のニコチン酸受容体アゴニストを併用投与することによりアテローム性動脈硬化症、脂質代謝異常、糖尿病及び関連症状を治療する方法に関する。これはヒトではニコチン酸又はニコチン酸受容体アゴニストとDP受容体を阻害する化合物を投与することにより達成される。
【背景技術】
【0002】
各種サブタイプの受容体がプロスタグランジンD2と相互作用する。1種のプロスタグランジンD2受容体は「DP」と呼ばれ、別のプロスタグランジンD2受容体は「CRTH2」として知られる。本発明はその非投与時に発生する可能性のあるフラッシングを予防するか、最小限にするか又は緩和するためにDP受容体のアンタゴニスムを利用する。
【発明の開示】
【発明が解決しようとする課題】
【0003】
従って、本発明の1つの目的はニコチン酸又は別のニコチン酸受容体アゴニストを使用してアテローム性動脈硬化症、脂質代謝異常、糖尿病及び関連症状についてヒトを治療する間に副作用としての実質的フラッシング(頻度及び/又は重度)を解消、抑制又は緩和することである。
【0004】
本発明の別の目的は副作用一般を最小限にするアテローム性動脈硬化症の併用治療を提供することである。
【0005】
本発明の更に別の目的は経口用規定併用医薬組成物を提供することである。
【0006】
以上及び他の目的は本明細書の記載から自明である。
【課題を解決するための手段】
【0007】
治療を必要とするヒト患者におけるアテローム性動脈硬化症の治療方法として、実質的フラッシングを併発せずにアテローム性動脈硬化症を治療するために有効な量であるニコチン酸約1000mgと、約5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg、50mg、75mg、100mg及び150mgから選択される量のDP受容体アンタゴニストを患者に投与することを含む方法を開示する。
【発明を実施するための最良の形態】
【0008】
以下、添付図面に関して本発明を例証する。
【0009】
ナイアシンないしニコチン酸(ピリジン−3−カルボン酸)は高密度リポ蛋白(HDL)値の上昇と、脂質プロフィルの他の有益な変化(超低密度(VLDL)、低密度リポ蛋白(LDL)、トリグリセリド、遊離脂肪酸(FFA)及びリポ蛋白(a)[Lp(a)]の低下)に有効であることがよく知られている薬剤である。ニコチン酸は約50mgから約8g/日等の治療有効用量をヒトに投与した場合にHDL値を上昇させる。しかし、ニコチン酸は皮膚血管拡張(フラッシングとも言う)を併発することが多い。フラッシングは一般に皮膚発赤とほてり、痒み又は炎症を伴う。フラッシングは非常に不快であり、非常に重度であるため、ニコチン酸治療を中断する患者が多い。本発明は実質的フラッシングを併発せずにニコチン酸又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストによりアテローム性動脈硬化症及び本明細書に記載する他の疾患及び症状の治療、予防又は逆行させる方法に関する。これはヒトではニコチン酸又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストと、DP受容体を阻害する化合物を投与し、頻度及び/又は重度においてフラッシング効果を予防するか、緩和するか又は最小限にすることにより達成される。
【0010】
プロスタグランジンD2と相互作用する受容体は「DP」及び「CRTH2」と呼ぶ少なくとも2種類がある。本発明は主にDP受容体のアンタゴニストと併用するニコチン酸又はニコチン酸受容体アゴニストに関する。
【0011】
本発明の有利な1側面は治療を必要とするヒト患者におけるアテローム性動脈硬化症の治療方法として、実質的フラッシングを併発せずにアテローム性動脈硬化症を治療するために有効な量でニコチン酸又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストと、DP受容体アンタゴニストを患者に投与することを含む方法である。
【0012】
本発明の別の有利な側面は治療を必要とするヒト患者における血清HDL値の上昇方法として、ニコチン酸又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストとDP受容体アンタゴニストを患者に投与することを含み、前記併用が実質的フラッシングを併発せずに患者の血清HDL値を上昇させるために有効である方法に関する。
【0013】
本発明の別の有利な側面は治療を必要とするヒト患者における脂質代謝異常の治療方法として、実質的フラッシングを併発せずに脂質代謝異常を治療するために有効な量でニコチン酸又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストと、DP受容体アンタゴニストを患者に投与することを含む方法に関する。
【0014】
本発明の別の有利な側面は治療を必要とするヒト患者における血清VLDL又はLDL値の低下方法として、実質的フラッシングを併発せずに患者の血清VLDL又はLDL値を低下させるために有効な量でニコチン酸又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストと、DP受容体アンタゴニストを患者に投与することを含む方法に関する。
【0015】
本発明の別の有利な側面は治療を必要とするヒト患者における血清トリグリセリド値の低下方法として、実質的フラッシングを併発せずに患者の血清トリグリセリド値を低下させるために有効な量でニコチン酸又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストと、DP受容体アンタゴニストを患者に投与することを含む方法に関する。
【0016】
本発明の別の有利な側面は治療を必要とするヒト患者における血清Lp(a)値の低下方法として、実質的フラッシングを併発せずに患者の血清Lp(a)値を低下させるために有効な量でニコチン酸又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストと、DP受容体アンタゴニストを患者に投与することを含む方法に関する。本明細書で使用するLp(a)とはリポ蛋白質(a)を意味する。
【0017】
本発明の特に有利な側面はニコチン酸又はその塩もしくは溶媒和物を利用する上記各方法に関する。ニコチン酸を使用すると更に有利である。有利な更に別の側面では、DP受容体アンタゴニストは患者におけるフラッシング効果を抑制、緩和又は予防するために有効な量のDP受容体を選択的に調節する。
【0018】
本発明の特に有利な別の側面はニコチン酸を利用し、DP受容体アンタゴニストがDP受容体を選択的に調節し、CRTH2受容体を実質的に調節しない上記各方法に関する。
【0019】
本発明の特に有利な別の側面は治療を必要とするヒト患者におけるアテローム性動脈硬化症、脂質代謝異常、糖尿病又は関連症状の治療方法として、実質的フラッシングを併発せずにアテローム性動脈硬化症、脂質代謝異常、糖尿病又は関連症状を治療するために有効な量で、ニコチン酸又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストと、DP受容体アンタゴニストを患者に併用投与することを含む方法に関する。
【0020】
本発明の特に有利な別の側面は治療を必要とするヒト患者におけるアテローム性動脈硬化症、脂質代謝異常、糖尿病又は関連症状の治療方法として、実質的フラッシングを併発せずにアテローム性動脈硬化症、脂質代謝異常、糖尿病又は関連症状を治療するために有効な量で、a)ナイアシンにより誘発されるフラッシング応答を抑制するために有効な量のアスピリンと、b)ニコチン酸又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストと、c)DP受容体アンタゴニストを患者に併用投与することを含む方法に関する。
【0021】
本発明の別の有利な側面は治療を必要とするヒト患者におけるアテローム性動脈硬化症、脂質代謝異常、糖尿病又は関連症状の治療方法として、ニコチン酸により誘発されるフラッシング応答を抑制又は緩和するために有効な量のアスピリンで患者を予備治療又は治療する段階と、実質的フラッシングを併発せずにアテローム性動脈硬化症、脂質代謝異常、糖尿病又は関連症状を治療するために有効な量のニコチン酸又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストとDP受容体アンタゴニストを併用投与する段階を含む方法に関する。
【0022】
本発明の1側面は実質的フラッシングを併発せずにヒトにおけるアテローム性動脈硬化症を治療するための、ニコチン酸又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストと、DP受容体アンタゴニスト化合物の併用である。
【0023】
本発明の特に有利な別の側面はDP受容体アンタゴニストが化合物AからAJと医薬的に許容可能なその塩及び溶媒和物から構成される群から選択される上記方法に関する。
【0024】
DP受容体を選択的に阻害し、フラッシング効果を抑制するために特に有用な化合物の例としては以下の化合物と、医薬的に許容可能なその塩及び溶媒和物が挙げられる。
【0025】
【化1】
【0026】
本明細書で使用するアテローム性動脈硬化症とは大・中動脈壁の最下層にコレステロールと脂質を含むアテローム斑が沈着することを特徴とする血管疾患の1形態を意味する。アテローム性動脈硬化症は該当医学分野に携わる医師により認識及び理解される血管疾患及び症状を含む。血管再建術後の再狭窄を含むアテローム硬化性心血管疾患、冠状動脈性心臓病(冠動脈疾患又は虚血性心疾患とも言う)、多発梗塞性痴呆を含む脳血管疾患、及び勃起障害を含む末梢血管疾患はいずれもアテローム性動脈硬化症の臨床徴候であるため、「アテローム性動脈硬化症」及び「アテローム硬化性疾患」に含まれる。
【0027】
「脂質代謝異常」はHDL(低)、LDL(高)、VLDL(高)、トリグリセリド(高)、リポ蛋白質(a)(高)、FFA(高)及び他の血清脂質又はその組み合わせ等の異常血漿脂質値を意味するために従来通りの意味で使用する。併発症を伴わない症状でもよいし、糖尿病(糖尿病性脂質代謝異常)、メタボリックシンドローム等の特定関連疾患又は症状の一部でもよい。従って、本発明には併発症を伴わない脂質代謝異常と、基礎症状に関連する脂質代謝異常が含まれる。
【0028】
「患者」なる用語は症状の予防又は治療のために本発明の活性剤を使用する哺乳動物、特にヒトを意味する。患者への薬剤投与は自己投与と他者による投与の両者を含む。患者は既存疾患又は症状の治療を必要とする場合もあるし、アテローム性動脈硬化症の発症の危険を予防又は低減するために予防処置を必要とする場合もある。
【0029】
「治療有効量」なる用語は所望生物学的又は医学的応答を誘発する薬剤の量を意味する。1例として、ニコチン酸は多くの場合には約50mgから約8g/日、好ましくは約0.5gから約3.0g/日の用量を投与する。ニコチン酸の好適用量は約1から2g/日である。
【0030】
「予防有効量」及び「予防するために有効な量」なる用語は予防する必要がある生物学的又は医学的イベントの発生の危険を予防又は低減する薬剤の量を意味する。多くの場合には、予防有効量は治療有効量と同一である。
【0031】
本明細書に記載する本発明は冠状動脈性心臓病イベント、脳血管イベント、及び/又は間欠性跛行の発生、又は潜在性がある場合にはその再発の危険を予防又は低減するために本明細書に記載する化合物及び組成物を投与することを含む。冠状動脈性心臓病イベントはCHD死、心筋梗塞(即ち、心臓発作)、及び冠状血管再建術を含むものとする。脳血管イベントは虚血性又は出血性脳卒中(脳血管障害とも言う)及び一過性脳虚血発作を含むものとする。間欠性跛行は末梢血管疾患の臨床徴候である。本明細書で使用する「アテローム硬化性疾患イベント」なる用語は、冠状動脈性心臓病イベント、脳血管イベント及び間欠性跛行を含むものとする。非致死性アテローム硬化性疾患イベントを過去に1回以上経験した人はこのようなイベントの再発の可能性があるとみなす。
【0032】
従って、本発明はアテローム硬化性疾患イベントの初回又は2回目以降の発生の危険を予防又は低減するための方法として、実質的フラッシングを予防又は最小限にしながらこのようなイベントの危険のある患者に予防有効量の本明細書に記載する化合物を投与することを含む方法も提供する。患者は投与時に既にアテローム硬化性疾患をもつものでもよいし、発症する危険があるものでもよい。
【0033】
本方法は更に実質的フラッシングを予防又は最小限にしながら新規アテローム病変又は斑形成を予防又は遅延させ、既存病変又は斑の進行を予防又は遅延させ、更に既存病変又は斑を退行させる方法に関する。
【0034】
従って、本発明の1側面はアテローム斑進行の停止又は遅延を含むアテローム性動脈硬化症の停止又は遅延方法として、治療を必要とする患者にニコチン酸又は別のニコチン酸受容体アゴニストと共に治療有効量の本明細書に記載のDPアンタゴニストのいずれかを投与することを含む方法に関する。本方法も治療開始時に存在するアテローム斑(即ち、「既存アテローム斑」)の進行の停止又は遅延と、アテローム性動脈硬化症をもつ患者における新規アテローム斑の形成の停止又は遅延を含む。
【0035】
本発明の別の側面はアテローム斑破裂の危険の予防又は低減方法として、治療を必要とする患者にニコチン酸又は別のニコチン酸受容体アゴニストと共に予防有効量の本明細書に記載の化合物のいずれかを投与することを含む方法に関する。本明細書で使用する破裂とは斑が遊離し、場合によっては血管に詰まることを意味する。本発明の別の側面はアテローム性動脈硬化症の発症の危険の予防又は低減方法として、このような治療を必要とする患者に予防有効量の本明細書に記載の化合物を投与することを含む方法に関する。
【0036】
本発明の別の側面はアテローム性動脈硬化症、脂質代謝異常又は関連症状の治療又は予防方法として、フラッシングを抑制又は緩和するのに有効な量のDP受容体アンタゴニストでこのような治療を必要とするヒト患者を予備治療する段階と、その後、実質的フラッシングを併発せずに前記アテローム性動脈硬化症、脂質代謝異常又は関連症状を治療又は予防するために有効な量のニコチン酸、又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストで前記患者を治療する段階を含む方法に関する。
【0037】
本発明の別の側面はHMG Co−Aレダクターゼ阻害剤で患者を予備治療又は治療する段階を更に含む上記方法に関する。
【0038】
本発明の別の側面はHMG Co−Aレダクターゼ阻害剤がシンバスタチンである上記症状の治療又は予防方法に関する。
【0039】
本明細書に記載する方法の1側面は、本明細書に記載する結果を達成するために有効な量のニコチン酸又は別のニコチン酸受容体アゴニスト化合物と、DP受容体を選択的に調節し、CRTH2受容体を実質的に調節しないDP受容体アンタゴニストの併用に関する。従って、DP受容体アンタゴニストはDP受容体に対する親和性(即ち、Ki)がCRTH2受容体に対する親和性の少なくとも約10倍(Ki値の数値が小さい)である。これらの基準に従ってDPと選択的に相互作用する任意化合物を「DP選択的」であるとみなす。
【0040】
「実質的フラッシングを併発せずに」なる用語は治療量のニコチン酸を投与する場合に頻発する副作用を意味する。ニコチン酸のフラッシング効果は一般に患者が治療用量の薬剤に対する耐性を生じるにつれて頻度と重度が低下するが、それでもある程度まではフラッシング効果が生じる。従って、「実質的フラッシングを併発せずに」とはフラッシングが生じる場合にはその重度が低下することを意味するか、又は非投与時よりもフラッシングイベントの発生頻度が低下することを意味する。フラッシング発生率は少なくとも約3分の1低下することが好ましく、発生率は2分の1低下することがより好ましく、フラッシング発生率は約3分の2以上低下することが最も好ましい。同様に、重度は少なくとも約3分の1低下することが好ましく、少なくとも2分の1低下することがより好ましく、少なくとも約3分の2低下することが最も好ましい。当然のことながら、フラッシング発生率及び重度は100%低下することが最も好ましいが、その必要はない。
【0041】
残留フラッシングを抑制又は緩和するためにアスピリンも投与することができる。ニコチン酸により誘発される残留フラッシング応答を抑制又は緩和するために有用なアスピリンの量は一般に治療用量を越えず、典型的には治療用量よりも少なく、約20から25mgから約650mgまでである。特に、本明細書に記載する本発明に有用なアスピリンの量は投与するDP受容体アンタゴニストにより抑制又は緩和されない残留フラッシングを抑制又は緩和するために必要又は有用な量とする。アスピリンは一般にニコチン酸よりも先に投与し、例えばニコチン酸とDP受容体アンタゴニストを投与する約30分から1時間前とするが、ニコチン酸及びDP受容体アンタゴニストと同時に投与してもよい。例えばニコチン酸とDP受容体アンタゴニストを投与する約30分前に単回投与することによりニコチン酸とDP受容体アンタゴニストの前に投与してもよいし、ニコチン酸及びDP受容体アンタゴニストと同時に投与し、必要に応じてDP受容体アンタゴニストにより抑制されない残留フラッシング応答を抑制又は緩和するために十分又は有効な量を約4時間までの間隔で反復投与してもよい。
【0042】
任意特定患者の特定投与レジメン及びレベルは年齢、体重、一般健康状態、性別、食事、投与時間、投与経路、排泄率、薬剤併用及び患者の症状の重篤度等の種々の因子により異なる。症状を予防、逆行、又は進行阻止するために必要な治療有効用量又は予防有効用量を決定する目的でこれらの因子を考慮することは通常の知識をもつ臨床医が可能な範囲内である。本明細書に記載する化合物は患者に該当する症状を治療又は予防するために適切な期間(例えば数カ月、数年又は患者の終生の治療期間)毎日投与することが予想される。
【0043】
特に有利な治療方法の1側面は治療を必要とするヒト患者におけるアテローム性動脈硬化症の治療方法として、実質的フラッシングを併発せずにアテローム性動脈硬化症を治療するために有効な量であるニコチン酸約1000mgと、約5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg、50mg、75mg、100mg及び150mgから選択される量のDP受容体アンタゴニストを患者に投与することを含む方法に関する。
【0044】
特に有利な治療方法の別の側面は治療を必要とするヒト患者におけるアテローム性動脈硬化症の治療方法として、実質的フラッシングを併発せずにアテローム性動脈硬化症を治療するために有効な量であるニコチン酸約2000mgと、約10mg、20mg、30mg、37.5mg、40mg、50mg、75mg、100mg、150mg、200mg及び300mgから選択される量のDP受容体アンタゴニストを患者に投与することを含む方法に関する。用量を調節又は記載するために使用する場合の「約」なる用語は妥当な近似が適用可能であることを表すためにその一般的な意味で使用する。例えば、「約10mg」とは10mgよりも多少多いか又は少ない用量を含み、例えば9mg前後から11mg前後である。「約18.75mg」の用量は17mg前後から20mg前後である。「約20mg」の用量は「約18.75mg」とオーバーラップし、19mg前後から21mg前後の範囲を含む。
【0045】
1種以上の付加活性剤を本明細書に記載する化合物と併用投与することができる。付加活性剤は脂質調節化合物又は他の医薬活性をもつ物質とすることができ、脂質調節効果と他の医薬活性を併有する物質でもよい。利用可能な付加活性剤の例としては限定されないが、そのラクトン化又はジヒドロキシ開環酸体のスタチンとその医薬的に許容可能な塩及びエステル(限定されないが、例えばロバスタチン(米国特許第4,342,767号参照)、シンバスタチン(米国特許第4,444,784号参照)、ジヒドロキシ開環酸シンバスタチン、特にそのアンモニウム又はカルシウム塩、プラバスタチン、特にそのナトリウム塩(米国特許第4,346,227号参照)、フルバスタチン、特にそのナトリウム塩(米国特許第5,354,772号参照)、アトルバスタチン、特にそのカルシウム塩(米国特許第5,273,995号参照)、ピタバスタチン(別称NK−104)(PCT国際出願公開WO97/23200参照)及びロスバスタチン(別称ZD−4522)(CRESTOR(登録商標);米国特許第5,260,440号参照))等のHMG−CoAレダクターゼ阻害剤;HMG−CoAシンターゼ阻害剤;スクアレンエポキシダーゼ阻害剤;スクアレンシンテターゼ阻害剤(別称スクアレンシンターゼ阻害剤)、ACAT−1又はACAT−2の選択的阻害剤とACAT−1及び−2のデュアル阻害剤を含むアシル−補酵素A:コレステロールアシルトランスフェラーゼ(ACAT)阻害剤;ミクロソームトリグリセリド転送蛋白質(MTP)阻害剤;内皮リパーゼ阻害剤;胆汁酸抑制剤;LDL受容体インデューサー;血小板凝集阻害剤(例えば糖蛋白質IIb/IIIaフィブリノーゲン受容体アンタゴニスト及びアスピリン);ヒトペルオキシソーム増殖因子活性化受容体γ(PPARγ)アゴニスト、例えば一般にグリタゾンと呼ばれる化合物(例えばピオグリタゾン及びロシグリタゾン)及びチアゾリジンジオンとして知られる構造クラスに含まれる化合物と、チアゾリジンジオン構造クラスに含まれないPPARγアゴニスト;PPARαアゴニスト(例えばクロフィブレート、微粉状フェノフィブレートを含むフェノフィブレート、及びゲムフィブロジル);PPARデュアルα/γアゴニスト;ビタミンB6(別称ピリドキシン)とその医薬的に許容可能な塩(例えばHCl塩);ビタミンB12(別称シアノコバラミン);葉酸又はその医薬的に許容可能な塩もしくはエステル(例えばナトリウム塩及びメチルグルカミン塩);酸化防止ビタミン類(例えばビタミンC及びE及びβカロチン);β遮断薬;アンギオテンシンIIアンタゴニスト(例えばロサルタン);アンギオテンシン変換酵素阻害剤(例えばエナラプリル及びカプトプリル);レニン阻害剤、カルシウムチャネル遮断薬(例えばニフェジピン及びジルチアゼム);エンドテリンアンタゴニスト;ABC1遺伝子発現を増強する物質;コレステリルエステル転送蛋白質(CETP)阻害化合物、5−リポキシゲナーゼ活性化蛋白質(FLAP)阻害化合物、5−リポキシゲナーゼ(5−LO)阻害化合物、アンタゴニストとアゴニストの両者を含むファルネソイドX受容体(FXR)リガンド;肝臓X受容体(LXR)−αリガンド、LXR−βリガンド、ビスホスホネート化合物(例えばアレンドロン酸ナトリウム);シクロオキシゲナーゼ−2阻害剤(例えばロフェコキシブ及びセレコキシブ)、並びに血管炎症を緩和する化合物が挙げられる。
【0046】
コレステロール吸収抑制剤も本発明で使用することができる。このような化合物はコレステロールが腸内腔から小腸壁の腸細胞内に移動するのを阻止し、血清コレステロール値を低下させる。コレステロール吸収抑制剤の例は米国特許第5,846,966号、5,631,365号、5,767,115号、6,133,001号、5,886,171号、5,856,473号、5,756,470号、5,739,321号、5,919,672号、並びにPCT出願WO00/63703、WO00/60107、WO00/38725、WO00/34240、WO00/20623、WO97/45406、WO97/16424、WO97/16455、及びWO95/08532に記載されている。最も注目に値するコレステロール吸収抑制剤は米国特許第5,767,115号及び5,846,966号に記載されているエゼチミブ、別称1−(4−フルオロフェニル)−3(R)−[3(S)−(4−フルオロフェニル)−3−ヒドロキシプロピル)]−4(S)−(4−ヒドロキシフェニル)−2−アゼチジノンである。
【0047】
コレステロール吸収抑制剤の治療有効量としては約0.01mg/kgから約30mg/kg体重/日、好ましくは約0.1mg/kgから約15mg/kgの用量が挙げられる。
【0048】
糖尿病患者には、本発明で使用する化合物を従来の糖尿病治療薬と併用投与することができる。例えば、本明細書に記載するような治療を受けている糖尿病患者にインスリン又は経口糖尿病治療薬を併用投与することができる。本発明で有用な経口糖尿病治療薬の1例はメトフォルミンである。
【0049】
塩
本明細書で使用するニコチン酸とはピリジン−3−カルボン酸を意味する。しかし、ニコチン酸の塩及び溶媒和物も本発明で使用され、ニコチン酸の多数の医薬的に許容可能な塩及び溶媒和物が本発明で有用である。アルカリ金属塩、特にナトリウム塩とカリウム塩が本明細書に記載する有用な塩を形成する。同様に、アルカリ土類金属、特にカルシウムとマグネシウムも本明細書に記載する有用な塩を形成する。アンモニウム及び置換アンモニウム化合物等の各種アミン塩も本明細書に記載する有用な塩を形成する。同様に、ニコチン酸の溶媒和形態も本発明で有用である。例としては、半水和物、1水和物、2水和物、3水和物及びセスキ水和物が挙げられる。本発明で使用するには遊離酸、ピリジン−3−カルボン酸が特に有利である。
【0050】
本明細書に記載する有用なニコチン酸の用量は約50mg/日から約8g/日を1日1回又は数回に分けて投与する。最初は低用量を使用し、用量を増加してフラッシング効果を更に最小限にすることができる。
【0051】
ニコチン酸以外のニコチン酸受容体アゴニストの用量は広い範囲を取る。一般に、アテローム性動脈硬化症を治療するために有用なニコチン酸受容体アゴニストは約0.01mg/kg/日から約100mg/kg/日を1日1回又は数回に分けて投与する。代表的な用量は約0.1mg/日から約2g/日である。
【0052】
本明細書に記載するDPアンタゴニストは約0.01mg/kg/日から約100mg/kg/日を1日1回又は数回に分けて哺乳動物患者、特にヒトに投与すると、フラッシング効果を緩和又は予防するために有用である。用量は約0.1mg/日から約1.0g/日を1日1回又は数回に分けて投与することが好ましい。
【0053】
本明細書に記載する化合物及び製剤は従来の任意投与経路で投与することができる。好ましい投与経路は経口である。
【0054】
ニコチン酸、又はその塩もしくは溶媒和物、又は他のニコチン酸受容体アゴニストとDPアンタゴニストは本発明の範囲内で1日1回又は複数回、例えば1日2回(bid)、1日3回(tid)又は1日4回(qid)に分けて同時又は順次投与することができる。24時間を越える放出プロフィルを示す徐放製剤等の特に長時間徐放が所望される場合には、1日おきに投与してもよい。しかし、1日1回投与することが好ましい。また、朝晩どちらに投与してもよいが、晩に投与するほうが好ましい。
【0055】
医薬組成物
本明細書に記載する医薬組成物は一般にニコチン酸又は別のニコチン酸受容体アゴニストと、DP受容体アンタゴニストと、医薬的に許容可能なキャリヤーから構成される。
【0056】
適切な経口組成物の例としては錠剤、カプセル剤、トローチ、ロゼンジ、懸濁液、散剤又は顆粒剤、エマルション、シロップ及びエリキシル剤が挙げられる。キャリヤー成分の例としては希釈剤、結合剤、崩壊剤、滑沢剤、甘味剤、フレーバー、着色剤、防腐剤等が挙げられる。希釈剤の例としては、例えば炭酸カルシウム、炭酸ナトリウム、ラクトース、リン酸カルシウム及びリン酸ナトリウムが挙げられる。顆粒化剤及び崩壊剤の例としてはコーンスターチとアルギン酸が挙げられる。結合剤の例としては澱粉、ゼラチン及びアラビアガムがが挙げられる。滑沢剤の例としてはステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸及びタルクが挙げられる。錠剤はコーティングしなくてもよいし、公知技術によりコーティングしてもよい。このようなコーティングは胃腸管での崩壊、従って吸収を遅らせることにより長時間持続作用することができる。
【0057】
本発明の1態様では、ニコチン酸、又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストをDP受容体アンタゴニスト及びキャリヤーと配合し、規定併用製剤を形成する。この規定併用製剤は経口用錠剤又はカプセル剤とすることができる。
【0058】
より特定的には、本発明の別の態様では、ニコチン酸、又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニスト(約1から約1000mg)とDPアンタゴニスト(約1から約500mg)を医薬的に許容可能なキャリヤーと配合し、経口用錠剤又はカプセル剤を提供する。
【0059】
ニコチン酸医薬組成物の製剤では長時間徐放が特に重要であると思われる。徐放錠が特に好ましい。例えば、モノステアリン酸グリセリルやジステアリン酸グリセリル等の時間遅延剤を利用することができる。米国特許第4,256,108号、4,166,452号及び4,265,874号に記載されている技術により製剤をコーティングし、制御放出用浸透圧治療錠を形成してもよい。
【0060】
他の制御放出技術も利用可能であり、本発明に含まれる。徐放錠中のニコチン酸の放出を遅らせるために有用な典型的成分としては、メチルセルロース、エチルセルロース、プロピルセルロース、ヒドロキシプロピルセルロース、ヒドロキシエチルセルロース、ヒドロキシプロピルメチルセルロース、微結晶セルロース、澱粉等の各種セルロース化合物が挙げられる。各種天然及び合成材料も徐放製剤で使用することができる。例としてはアルギン酸と各種アルギン酸塩、ポリビニルピロリドン、トラガカントガム、ローカストビーンガム、グアーガム、ゼラチン、各種長鎖アルコール(例えばセチルアルコール)及び蜜蝋が挙げられる。
【0061】
特に有利な徐放錠は1種以上の上記セルロース化合物とニコチン酸を徐放錠に圧縮してポリマーマトリックスを形成する。DPアンタゴニスト化合物は圧縮前にブレンドに加えてもよいし、マトリックスの外側表面にコーティングしてもよい。
【0062】
より有利な1態様では、ニコチン酸とマトリックス形成材料を配合及び圧縮して徐放コアを形成し、DPアンタゴニスト化合物を1種以上のコーティング剤とブレンドし、コアの外側表面にコーティングする。
【0063】
場合により、上記のような錠剤にHMG Co−Aレダクターゼ阻害剤(例えばシンバスタチン)をコーティングすると更に有利である。従って、この特定態様は3種の活性成分を含み、そのうち、HMG Co−Aレダクターゼ阻害剤とDPアンタゴニストは実質的に摂取直後に放出可能であり、ニコチン酸は上記のように長時間にわたって放出可能である。
【0064】
本発明の徐放錠の典型的放出時間は約1から約48時間、好ましくは約4から約24時間、より好ましくは約8から約16時間である。
【0065】
ハードゼラチンカプセルは別の経口用剤形を構成する。このようなカプセル剤も同様に上記のようなキャリヤー材料と混合した活性成分を含む。ソフトゼラチンカプセルは水混和性溶媒(例えばプロピレングリコール、PEG及びエタノール)、又は油(例えばピーナツ油、流動パラフィン又はオリーブ油)と混合した活性成分を含む。
【0066】
水性懸濁液の製造に適した賦形剤と混合した活性剤を含むものとして水性懸濁液も想定される。このような賦形剤としては懸濁剤(例えばナトリウムカルボキシメチルセルロース、メチルセルロース、ヒドロキシプロピルメチルセルロース、アルギン酸ナトリウム、ポリビニルピロリドン、トラガカントガム及びアラビアガム);分散剤又は湿潤剤(例えばレシチン);防腐剤(例えばパラヒドロキシ安息香酸エチル又はn−プロピル)、着色剤、フレーバー、甘味剤等が挙げられる。
【0067】
水の添加により水性懸濁液を調製するのに適した散剤及び顆粒剤は分散剤又は湿潤剤、懸濁剤及び1種以上の防腐剤と混合した活性成分を提供する。適切な分散剤又は湿潤剤と懸濁剤は上記に挙げたものである。
【0068】
シロップ及びエリキシル剤に製剤化することもできる。
【0069】
特に有利な医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸又はその塩もしくは溶媒和物と、DP受容体アンタゴニストから構成される徐放錠である。
【0070】
特に有利な別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸又はその塩もしくは溶媒和物と、DP受容体アンタゴニストと、HMG Co−Aレダクターゼ阻害剤から構成される徐放錠である。
【0071】
更に特に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、DP受容体アンタゴニストと、シンバスタチンから構成される徐放錠である。
【0072】
特に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、化合物AからAJから構成される群から選択されるDP受容体アンタゴニストから構成される徐放錠である。
【0073】
更に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、化合物A、B、D、E、X、AA、AF、AG、AH、AI及びAJから構成される群から選択されるDP受容体アンタゴニストから構成される。
【0074】
特に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸とDPアンタゴニスト化合物Aから構成される。
【0075】
更に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、DPアンタゴニスト化合物Bから構成される。
【0076】
更に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、DPアンタゴニスト化合物Dから構成される。
【0077】
更に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、DPアンタゴニスト化合物Eから構成される。
【0078】
更に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、DPアンタゴニスト化合物Xから構成される。
【0079】
更に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、DPアンタゴニスト化合物AAから構成される。
【0080】
更に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、DPアンタゴニスト化合物AFから構成される。
【0081】
更に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、DPアンタゴニスト化合物AGから構成される。
【0082】
更に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、DPアンタゴニスト化合物AHから構成される。
【0083】
更に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、DPアンタゴニスト化合物AIから構成される。
【0084】
更に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、DPアンタゴニスト化合物AJから構成される。
【0085】
更に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、上記DPアンタゴニスト化合物の1種と、シンバスタチンから構成される。
【0086】
更に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、化合物AからAJから選択されるDP受容体アンタゴニストと、シンバスタチンから構成される徐放錠である。
【0087】
特に有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸約500mg、750mg又は1000mgと、約5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg、50mg、75mg、100mg及び150mgから選択される量のA、B、D、E、X、AA、AF、AG、AH、AI及びAJから構成される群から選択されるDPアンタゴニスト、又は医薬的に許容可能なその塩もしくは溶媒和物を含有する。
【0088】
より特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg及び50mgから選択される量のA、B、D、E、X、AA、AF、AG、AH、AI及びAJから構成される群から選択されるDPアンタゴニスト、又は医薬的に許容可能なその塩もしくは溶媒和物を含有する。
【0089】
より特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、A、B、D、E、X、AA、AF、AG、AH、AI及びAJから構成される群から選択されるDPアンタゴニスト、又は医薬的に許容可能なその塩もしくは溶媒和物5mg、10mg、15mg、18.75mg、20mg、25mg又は37.5mgを含有する。
【0090】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、A、B、D、E、X、AA、AF、AG、AH、AI及びAJから構成される群から選択されるDPアンタゴニスト、又は医薬的に許容可能なその塩もしくは溶媒和物5mg、10mg、15mg、20mg、25mg、又は37.5mgを含有する。
【0091】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物A又は医薬的に許容可能なその塩もしくは溶媒和物5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgを含有する。
【0092】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物B又は医薬的に許容可能なその塩もしくは溶媒和物5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgを含有する。
【0093】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物D又は医薬的に許容可能なその塩もしくは溶媒和物5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgを含有する。
【0094】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物E又は医薬的に許容可能なその塩もしくは溶媒和物5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgを含有する。
【0095】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物X又は医薬的に許容可能なその塩もしくは溶媒和物5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgを含有する。
【0096】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物AA又は医薬的に許容可能なその塩もしくは溶媒和物5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgを含有する。
【0097】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物AF又は医薬的に許容可能なその塩もしくは溶媒和物5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgを含有する。
【0098】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物AG又は医薬的に許容可能なその塩もしくは溶媒和物5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgを含有する。
【0099】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物AH又は医薬的に許容可能なその塩もしくは溶媒和物5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgを含有する。
【0100】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物AI又は医薬的に許容可能なその塩もしくは溶媒和物5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgを含有する。
【0101】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物AJ又は医薬的に許容可能なその塩もしくは溶媒和物5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgを含有する。
【0102】
更に特に有利な更に別の医薬組成物は医薬的に許容可能なキャリヤーと共に、ニコチン酸と、化合物A、B、D、E、X、AA、AF、AG、AH、AI及びAJから構成される群から選択されるDP受容体アンタゴニストと、シンバスタチンから構成される徐放錠に関する。
【0103】
特に有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸約500mg、750mg又は1000mgと、約5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg、50mg、75mg、100mg及び150mgの量のA、B、D、E、X、AA、AF、AG、AH、AI及びAJから構成される群から選択されるDPアンタゴニスト、又は医薬的に許容可能なその塩もしくは溶媒和物と、シンバスタチン約10mg、20mg又は40mgを含有する。
【0104】
より特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、5mg、10mg、15mg、18.75mg、20mg、25mg、37.5mg及び50mgから選択される量のA、B、D、E、X、AA、AF、AG、AH、AI及びAJから構成される群から選択されるDPアンタゴニスト、又は医薬的に許容可能なその塩もしくは溶媒和物と、シンバスタチン約10mg、20mg又は40mgを含有する。
【0105】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、A、B、D、E、X、AA、AF、AG、AH、AI及びAJから構成される群から選択されるDPアンタゴニスト、又は医薬的に許容可能なその塩もしくは溶媒和物5mg、10mg、15mg、18.75mg、20mg、25mg又は37.5mgと、シンバスタチン約10mg、20mg又は40mgを含有する。
【0106】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、A、B、D、E、X、AA、AF、AG、AH、AI及びAJから構成される群から選択されるDPアンタゴニスト、又は医薬的に許容可能なその塩もしくは溶媒和物10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgと、シンバスタチン約10mg、20mg又は40mgを含有する。
【0107】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物A又は医薬的に許容可能なその塩もしくは溶媒和物10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgと、シンバスタチン約10mg、20mg又は40mgを含有する。
【0108】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物B又は医薬的に許容可能なその塩もしくは溶媒和物10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgと、シンバスタチン約10mg、20mg又は40mgを含有する。
【0109】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物D又は医薬的に許容可能なその塩もしくは溶媒和物10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgと、シンバスタチン約10mg、20mg又は40mgを含有する。
【0110】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物E又は医薬的に許容可能なその塩もしくは溶媒和物10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgと、シンバスタチン約10mg、20mg又は40mgを含有する。
【0111】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物X又は医薬的に許容可能なその塩もしくは溶媒和物10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgと、シンバスタチン約10mg、20mg又は40mgを含有する。
【0112】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物AA又は医薬的に許容可能なその塩もしくは溶媒和物10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgと、シンバスタチン約10mg、20mg又は40mgを含有する。
【0113】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物AF又は医薬的に許容可能なその塩もしくは溶媒和物10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgと、シンバスタチン約10mg、20mg又は40mgを含有する。
【0114】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物AG又は医薬的に許容可能なその塩もしくは溶媒和物10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgと、シンバスタチン約10mg、20mg又は40mgを含有する。
【0115】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物AH又は医薬的に許容可能なその塩もしくは溶媒和物10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgと、シンバスタチン約10mg、20mg又は40mgを含有する。
【0116】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物AI又は医薬的に許容可能なその塩もしくは溶媒和物10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgと、シンバスタチン約10mg、20mg又は40mgを含有する。
【0117】
更に特定的には、有利な製剤は医薬的に許容可能なキャリヤーと共に、ニコチン酸1000mgと、化合物AJ又は医薬的に許容可能なその塩もしくは溶媒和物10mg、15mg、18.75mg、20mg、25mg、37.5mg又は50mgと、シンバスタチン約10mg、20mg又は40mgを含有する。
【0118】
「組成物」なる用語は上記医薬組成物に加え、活性成分又は賦形剤成分の任意2種以上の配合、錯化もしくは凝集、成分の1種以上の解離、又は成分の1種以上の他の型の反応もしくは相互作用により直接又は間接的に得られる任意製剤も含む。従って、本発明の医薬組成物は本発明の化合物と、任意付加活性成分と、医薬的に許容可能な賦形剤を混合又は他の方法で配合することにより製造される任意組成物を含む。
【0119】
本発明の別の側面は医薬の製造におけるニコチン酸又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストとDPアンタゴニストの使用に関する。この医薬は本明細書に記載する用途をもつ。
【0120】
より特定的には、本発明の別の側面は医薬の製造におけるニコチン酸又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストと、DPアンタゴニストと、HMG Co−Aレダクターゼ阻害剤(例えばシンバスタチン)の使用に関する。この医薬は本明細書に記載する用途をもつ。
【0121】
医薬組成物の例を以下に挙げる。
【0122】
【表1】
【0123】
【表2】
【0124】
【表3】
【0125】
製剤例1から5の手順
1.DPアンタゴニスト含有層
化合物D、微結晶セルロース、ラクトース一水和物、クロスカルメロースナトリウム及び黄酸化鉄をDiosna P10高剪断ミキサーグラニュレーターで乾式混合する。別に、ヒドロキシプロピルセルロースを含有する水溶液を調製し、高剪断ミキサーグラニュレーターに加える。ブレンドを湿式造粒し、Strea−1流動層乾燥機を使用して乾燥する。乾燥した顆粒を粉砕する。粉砕した顆粒をApex 17Lドラムブレンダーでステアリン酸マグネシウムとブレンドし、DPアンタゴニスト顆粒を形成する。
【0126】
2.ニコチン酸含有層
ニコチン酸の一部をコロイド状二酸化ケイ素とプレブレンドする。ヒドロキシプロピルメチルセルロース、ニコチン酸含有混合物、コロイド状二酸化ケイ素、微結晶セルロース及びニコチン酸の残量を適切なブレンダーに加え、乾式混合する。ブレンドをAlexanderwerk WP 120ローラーコンパクターで乾式造粒し、粉砕する。粉砕した顆粒をHarbruc 65Lブレンダーでステアリルフマル酸ナトリウムとブレンドし、ニコチン酸顆粒を形成する。
【0127】
3.錠剤化手順
粉砕し、滑沢剤を添加した上記顆粒をRiva二層錠剤プレスで二層錠剤に圧縮する。
【0128】
製剤例6の手順
1.DPアンタゴニスト含有層1
上記製剤例1から5に記載した手順に従って成分をブレンドし、造粒する。
【0129】
2.ニコチン酸含有層
ヒドロキシプロピルメチルセルロース、ニコチン酸及びポビドンをApex 17Lブレンダーに加え、乾式混合する。混合物をApex 17Lドラムブレンダーでステアリン酸とブレンドし、ニコチン酸ブレンドを形成する。
【0130】
3.錠剤化手順
粉砕し、滑沢剤を添加したニコチン酸及びDPアンタゴニストブレンド顆粒をRiva二層錠剤プレスで二層錠剤に圧縮する。
【0131】
【表4】
【0132】
【表5】
【0133】
【表6】
【0134】
製剤例7から11の手順
1.DPアンタゴニスト/シンバスタチン層
化合物D、シンバスタチン、微結晶セルロース、ラクトース一水和物、クロスカルメロースナトリウム、黄酸化鉄及びヒドロキシプロピルメチルセルロースをDiosna P−6高剪断ミキサーグラニュレーターに加え、乾式混合する。BHAとクエン酸を含有する水性アルコール溶液を調製し、高剪断ミキサーに加え、ブレンドを湿式造粒する。顆粒をStrea−1流動層乾燥機で乾燥し、粉砕し、粉砕した顆粒をApex 17Lブレンダーで(予め凝塊を分解しておいた)ステアリン酸マグネシウムとブレンドする。
【0135】
2.ニコチン酸層
ニコチン酸層は上記製剤例1から5に記載したように作製する。
【0136】
3.錠剤化手順
粉砕し、滑沢剤を添加したニコチン酸とDPアンタゴニスト/シンバスタチン顆粒をRiva二層錠剤プレスで二層錠剤に圧縮する。
【0137】
製剤例12の手順
1.DPアンタゴニスト層
上記製剤例1から5に記載した手順に従って成分をブレンドし、造粒する。
【0138】
2.ニコチン酸層
ヒドロキシプロピルメチルセルロース、ニコチン酸及びポビドンをApex 17Lブレンダーに加え、乾式混合する。混合物をApex 17Lドラムブレンダーでステアリン酸とブレンドし、ニコチン酸ブレンドを形成する。
【0139】
3.錠剤化手順
粉砕し、滑沢剤を添加したニコチン酸及びDPアンタゴニストブレンド顆粒をRiva二層錠剤プレスで二層錠剤に圧縮する。
【0140】
【表7】
【0141】
【表8】
【0142】
製剤例13の手順
1.DPアンタゴニスト層
化合物E、微結晶セルロース、ラクトース一水和物及びクロスカルメロースナトリウムをDiosna P10高剪断ミキサーグラニュレーターで乾式混合する。別に、ヒドロキシプロピルセルロースを含有する水溶液を調製し、高剪断ミキサーグラニュレーターに加える。ブレンドを湿式造粒し、Strea−1流動層乾燥機を使用して乾燥する。乾燥した顆粒を粉砕する。粉砕した顆粒をApex 17Lドラムブレンダーでステアリン酸マグネシウム及びステアリルフマル酸ナトリウムとブレンドし、DPアンタゴニスト顆粒を形成する。
【0143】
2.ニコチン酸層
ニコチン酸層は上記製剤例1から5に記載したように作製する。
【0144】
3.錠剤化手順
粉砕し、滑沢剤を添加したニコチン酸及びDPアンタゴニストブレンド顆粒をRiva二層錠剤プレスで二層錠剤に圧縮する。
【0145】
製剤例14Aから14Cの手順
1.DPアンタゴニスト/シンバスタチン層
化合物E、シンバスタチン、微結晶セルロース、ラクトース一水和物、クロスカルメロースナトリウム、黄酸化鉄、FDC Blue No.2及びヒドロキシプロピルメチルセルロースをDiosna P−6高剪断ミキサーグラニュレーターに加え、乾式混合する。BHAとクエン酸を含有する水性アルコール溶液を調製し、高剪断ミキサーに加え、ブレンドを湿式造粒する。顆粒をStrea−1流動層乾燥機で乾燥し、粉砕し、粉砕した顆粒をApex 17Lブレンダーで(予め凝塊を分解しておいた)ステアリン酸マグネシウム及びステアリルフマル酸ナトリウムとブレンドする。
【0146】
2.ニコチン酸層
ニコチン酸層は上記製剤例1から5に記載したように作製する。
【0147】
3.錠剤化手順
粉砕し、滑沢剤を添加したニコチン酸とDPアンタゴニスト/シンバスタチンの顆粒をRiva二層錠剤プレスで二層錠剤に圧縮する。
【0148】
標準ニコチン酸受容体アゴニストであるニコチン酸に加え、多数のニコチン酸受容体アゴニストが記載されている。以下の刊行物がニコチン酸受容体アゴニストである化合物を開示している:
Lorenzen,A.ら,Molecular Pharmacology 59:349−357(2001)、
Lorenzen,A.ら,Biochemical Pharmacology 64:645−648(2002)、
Soga,T.ら,Biochemical and Biophysical Research Comm.303:364−369(2003)、
Tunaru,S.ら,Nature Medicine 9:352−355(2003)、
Wise,A.ら,Journal of Biological Chemistry 278:9869−9874(2003)、及び
Van Herk,T.ら Journal of Medicinal Chemistry 46:3945−3951(2003)。
【0149】
なお、van Herkらに開示されているもの等のニコチン酸受容体の部分アゴニストも本発明の組成物及び治療方法に含まれる。
【0150】
更に、ニコチン酸受容体はWO02/084298A2(公開日2002年10月24日)と、Soga,T.ら、Tunaru,S.ら及びWise,A.ら(上記引用)に記載され、特性決定されている。
【0151】
多数のDP受容体アンタゴニスト化合物が発表されており、本発明の方法で有用であり、本発明の方法に含まれる。例えば、DP受容体アンタゴニストはWO01/79169(公開日2001年10月25日)、EP 1305286(公開日2003年5月2日)、WO02/094830(公開日2002年11月28日)及びWO03/062200(公開日2003年7月31日)に従って得ることができる。化合物ABはWO01/66520A1(公開日2001年9月13日)の記載に従って合成することができ;化合物ACはWO03/022814A1(公開日2003年3月20日)の記載に従って合成することができ、化合物AD及びAEはWO03/078409(公開日2003年9月25日)の記載に従って合成することができる。本発明で使用される他の代表的なDPアンタゴニスト化合物は以下の実施例に従って合成することができる。
【0152】
(実施例1)
[5−[(4−クロロフェニル)チオ]−4−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸(化合物G)
【0153】
【化2】
【0154】
ステップ1:4−クロロニコチンアルデヒド
標記化合物はF.Marsaisら,J.Heterocyclic Chem.,25,81(1988)により記載されているように製造した。
【0155】
ステップ2:4−(メチルチオ)ニコチンアルデヒド
NaSMe(9.5g,135mmol)のMeOH(250mL)溶液にMeOH(250mL)中のステップ1の4−クロロニコチンアルデヒド(13.5g,94.4mmol)を加えた。反応混合物を60℃に15分間維持した。反応混合物をNH4ClとEtOAcに注いだ。有機相を分離し、H2Oで洗浄し、Na2SO4で乾燥した。次に、ヘキサン中50%EtOAcを使用して化合物をシリカゲルで精製すると、標記化合物が得られた。
【0156】
ステップ3:(2Z)−2−アジド−3−[4−(メチルチオ)ピリジン−3−イル]プロプ−2−エン酸メチル
4−(メチルチオ)ニコチンアルデヒド(4.8g,31mmol)とアジド酢酸メチル(9.0g,78mmol)のMeOH(50mL)溶液を−12℃のNaOMeの25%MeOH溶液(16.9mL,78mmol)に加えた。内部温度をモニターし、30分間の添加中、−10℃から−12℃に維持した。得られた混合物を次に氷浴中で数時間撹拌した後、冷所にて氷浴中で一晩撹拌した。次に懸濁液を氷とNH4Clの混合物に注ぎ、10分間撹拌後にスラリーを濾過した。生成物を冷H2Oで洗浄した後に減圧乾燥すると、標記化合物が多少の塩を含有するベージュ色固体(7.4g)として得られた。次にEtOAcを使用して化合物をシリカゲルで精製した。
【0157】
ステップ4:4−(メチルチオ)−1H−ピロロ[2,3−b]ピリジン−2−カルボン酸メチル
ステップ3の化合物(0.40g,1.6mmol)のキシレン(16mL)懸濁液を140℃までゆっくりと加熱した。140℃で15分後に、黄色溶液を室温まで冷却した。窒素形成により発熱する可能性があるので、注意が必要である。その後、懸濁液を0℃まで冷却し、濾過し、キシレンで洗浄すると、標記化合物が得られた。
【0158】
ステップ5:4−(メチルチオ)−6−オキソ−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−7−カルボン酸エチル
ステップ4の化合物(0.35g,1.6mmol)の0℃のDMF(20mL)溶液にNaH(1.2当量)を加えた。5分後にnBu4NI(0.10g)と4−ブロモ酪酸エチル(0.40mL)を加えた。室温で1時間後に反応混合物を飽和NH4ClとEtOAcに注いだ。有機相を分離し、H2Oで洗浄し、Na2SO4で乾燥した。蒸発後、粗生成物をフラッシュクロマトグラフィーにより精製した。このビスエステルを次にTHF(7.0mL)に溶かし、カリウムtert−ブトキシドの1.06M THF溶液(2.2mL)を0℃で加えた。室温で1時間後に反応混合物を飽和NH4ClとEtOAcに注いだ。有機相を分離し、Na2SO4で乾燥し、減圧乾燥すると、標記化合物がエチルエステルとメチルエステルの混合物として得られた。
【0159】
ステップ6:4−(メチルチオ)−8,9−ジヒドロピリド[3,2−b]インドリジン−6(7H)−オン
ステップ5の化合物(0.32g)にEtOH(8.0mL)と濃Cl(2.0mL)を加えた。得られた懸濁液を5時間還流した。反応混合物をEtOAcとNa2CO3に分配した。有機相を分離し、蒸発させると、標記化合物が得られた。
【0160】
ステップ7:(2E,2Z)−[4−(メチルチオ)−8,9−ジヒドロピリド[3,2−b]インドリジン−6(7H)−イリデン]エタン酸エチル
ホスホノ酢酸トリエチル(0.45g,2.17mmol)のDMF溶液(12mL)に80%NaH(0.06g,2.00mmol)とステップ6の化合物(0.22g,1.00mmol)を加えた。55℃で4時間後に反応混合物を飽和NH4ClとEtOAcに注いだ。有機相を分離し、減圧蒸発させた。粗生成物をフラッシュクロマトグラフィーにより精製すると、標記化合物が得られた。
【0161】
ステップ8:[4−(メチルチオ)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸エチル
熱溶解を使用してステップ7の化合物をMeOH−THFに溶かした。前記溶液を冷却し、室温でPtO2を加え、得られた混合物を大気圧水素下に18時間維持した。CH2Cl2を使用して反応混合物をセライトで注意深く濾過した。濾液を減圧蒸発させると、標記化合物が得られた。あるいは、ステップ7の化合物を40PSI H2下にEtOAc中Pd(OH)2で18時間水素化してもよい。
【0162】
ステップ9:[4−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸エチル
ステップ8の化合物(0.08g,0.27mmol)のMeOH(3.0mL)溶液にNa2WO4(0.10g)と30%H2O2(600μL)を加えた。1時間後に反応混合物をH2OとEtOAcに分配した。有機相をH2Oで洗浄し、分離し、蒸発させた。標記化合物をフラッシュクロマトグラフィーにより精製した。
【0163】
ステップ10:[5−[(4−クロロフェニル)チオ]−4−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸エチル
4,4’−ジクロロジフェニルジスルフィド(0.24g)の1,2−ジクロロエタン溶液(2.0mL)にSO2Cl2(50μL)を加えた。ステップ9の化合物(0.05g)のDMF(2.0mL)溶液に前記混合物(から180μL)を加えた。反応後に1H NMRを測定し、出発材料がなくなるまで室温に維持した。反応混合物を飽和NaHCO3とEtOAcに注いだ。有機相を分離し、蒸発させ、標記化合物をフラッシュクロマトグラフィーにより精製した。
【0164】
ステップ11:[5−[(4−クロロフェニル)チオ]−4−(メチルスルホニル)−67,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸
THF−MeOHの1/1混合物に溶かしたステップ10の化合物を1N NaOHを加えた。室温で18時間後に反応混合物を飽和NH4ClとEtOAcに分配した。有機相を分離し、Na2SO4で乾燥し、蒸発させると、標記化合物が得られた。
1H NMR(500MHz,アセトン−d6)δ 11.00(bs,1H),8.60(d,1H),7.80(d,1H),7.20(d,2H),7.00(d,2H),4.65(m,1H),4.20(m,1H),3.75(m,1H),3.35(s,3H),2.80−2.10(m,6H)。
【0165】
(実施例2)
[5−[(4−クロロフェニル)チオ]−4−(メチルチオ)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸(化合物H)
【0166】
【化3】
【0167】
標記化合物は実施例1,ステップ8の化合物から実施例1,ステップ10及び11に記載したと同様に製造することができる。m/z418。
【0168】
(実施例3)
[5−[(3,4−ジクロロフェニル)チオ]−4−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸(化合物I)
【0169】
【化4】
【0170】
標記化合物はステップ10でビス(3,4−ジクロロフェニル)ジスルフィドを使用した以外は実施例1に記載したように製造した。
1H NMR(500MHz,アセトン−d6)δ 8.55(d,1H),7.85(d,1H),7.35(d,1H),7.15(s,1H),6.95(d,1H),4.60(m,1H),4.15(m,1H),3.80(m,1H),3.40(s,3H),2.80−2.10(m,6H)。
【0171】
ヘキサン中30%イソプロパノール 17%エタノール 0.2%酢酸を流速8ml/分で使用してChiralecel ODカラム25cm×20mmでエナンチオマーを分離した。ヘキサン中35%イソプロパノール 0.2%酢酸を流速1.0ml/分で使用してChiralecel ODカラム25cm×4.6mmでその純度を確認した。高移動度エナンチオマーTr=9.7分、低移動度エナンチオマーTr=11.1分。
【0172】
(実施例4)
[5−(4−クロロベンゾイル)−4−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸(化合物J)
【0173】
【化5】
【0174】
ステップ1:[5−(4−クロロベンゾイル)−4−(メチルチオ)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸エチル
4−クロロベンゾイルクロライド(0.30g,1.7mmol)の1,2−ジクロロエタン(6.0mL)溶液にAlCl3(0.24g,1.8mmole)を加えた。5分後に実施例1ステップ8からの[4−(メチルチオ)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸エチル(0.15g,0.47mmole)の1,2−ジクロロエタン(6.0mL)溶液を前記混合物に加えた。80℃で4時間後に、反応混合物をEtOAcとNaHCO3に分配した。有機相を分離し、Na2SO4で乾燥し、蒸発させた。標記化合物をフラッシュクロマトグラフィーにより精製した。
【0175】
ステップ2:[5−(4−クロロベンゾイル)−4−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸エチル
[5−(4−クロロベンゾイル)−4−(メチルチオ)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸エチル(0.12g,0.27mmole)のMeOH(5.0mL)溶液にNa2WO4(0.1g)と30%H2O2(300μL)を加えた。反応混合物を55℃で1時間撹拌した。次に反応混合物をH2OとEtOAcに分配した。有機相をH2Oで洗浄し、Na2SO4で乾燥し、蒸発させた。標記化合物をフラッシュクロマトグラフィーにより精製した。
【0176】
ステップ3:[5−(4−クロロベンゾイル)−4−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸
[5−(4−クロロベンゾイル)−4−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸エチルを実施例1ステップ11に記載したように処理すると、標記化合物が得られた。
1H NMR(500MHz,アセトン−d6)δ 8.55(d,1H),7.90(d,2H),7.65(d,1H),7.45(d,2H),4.55(m,1H),4.25(m,1H),3.45(m,1H),3.20(s,3H),2.05−3.00(m,6H)。m/z446。
【0177】
(実施例5)
[5−(4−ブロモフェニル)チオ]−4−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸(化合物K)
【0178】
【化6】
【0179】
標記化合物は4,4’−ジブロモジフェニルジスルフィドを使用した以外は実施例1に記載したように製造した。
1H NMR(500MHz,アセトン−d6)δ 8.60(d,1H),7.80(d,1H),7.35(d,2H),7.00(d,2H),4.65(m,1H),4.20(m,1H),3.80(m,1H),3.35(s,3H),2.80−2.10(m,6H)。
【0180】
(実施例6)
方法1
[9−[(3,4−ジクロロフェニル)チオ]−1−(メチルスルホニル)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル]酢酸(化合物L)
【0181】
【化7】
【0182】
ステップ1:2−(メチルチオ)ニコチンアルデヒド
標記化合物は溶液を55℃に2時間加熱した以外は実施例1ステップ2に記載したように2−ブロモニコチンアルデヒド(A.Numata Synthesis 1999 p.306)から製造した。
【0183】
ステップ2:(2Z)−2−アジド−3−[2−(メチルチオ)ピリジン−3−イル]プロプ−2−エン酸メチル
標記化合物は実施例1ステップ3に記載したように製造した。
【0184】
ステップ3:4−(メチルチオ)−1H−ピロロ[3,2−c]ピリジン−2−カルボン酸メチル
(2Z)−2−アジド−3−[2−(メチルチオ)ピリジン−3−イル]プロプ−2−エン酸メチル(1.00g,4.00mmol)のメシチレン(50mL)溶液を160℃に1時間加熱した。反応混合物を室温まで冷却した後、0℃まで冷却し、沈殿を濾過し、冷メシチレンで洗浄すると、標記化合物が得られた。
【0185】
ステップ4:1−(メチルチオ)−8−オキソ−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−7−カルボン酸メチル
4−(メチルチオ)−1H−ピロロ[3,2−c]ピリジン−2−カルボン酸メチル(0.30g,1.35mmol)のTHF(3mL)−トルエン(12.0mL)懸濁液にカリウムtert−ブトキシド(1.42mL/1.41mmol)の1.06M THF溶液とアクリル酸メチル(300μL)を加えた。得られた混合物を80℃に18時間加熱した。混合物をEtOAcとNH4Clに分配し、セライトで濾過した。有機相を分離し、Na2SO4で乾燥し、濾過すると、標記化合物が得られた。
【0186】
ステップ5:1−(メチルチオ)−6,7−ジヒドロ−8H−ピリド[3,4−b]ピロリジン−8−オン
実施例1ステップ6に記載したように1−(メチルチオ)−8−オキソ−7,8−ジヒドロ−6H−ピリド[3,4−b]pピロリジン−7−カルボン酸メチルを標記化合物に変換した。
【0187】
ステップ6:[8−ヒドロキシ−1−(メチルチオ)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル]酢酸メチル
THF(3.0mL)中の1−(メチルチオ)−6,7−ジヒドロ−8H−ピリド[3,4−b]ピロリジン−8−オン(0.15g,0.68mmol)、ブロモ酢酸メチル(0.34mL)、Zn−Cu(0.226g)の混合物を2時間音波処理した。次に、反応が完了するまで混合物を60℃に5分間加熱した。反応混合物をEtOAcとNH4Clに分配した。有機相を分離し、Na2SO4で乾燥し、濾過し、減圧蒸発させると、標記化合物が得られた。化合物をフラッシュクロマトグラフィーにより精製した。
【0188】
ステップ7:[1−(メチルチオ)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル]酢酸メチル
CH3CN(3.2mL)中のNaI(0.300g)にTMSCl(0.266mL)を加えた。この混合物を水浴中で[8−ヒドロキシ−1−(メチルチオ)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル]酢酸メチル(0.15g,0.515mmol)のCH3CN(1.5mL)懸濁液に加えた。30分後に反応混合物をEtOAcとNaHCO3に分配した。有機相を分離し、チオ硫酸ナトリウムで洗浄し、MgSO4で乾燥し、蒸発させた。標記化合物をフラッシュクロマトグラフィーにより精製した。
【0189】
ステップ8:[1−(メチルスルホニル)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル]酢酸メチル
実施例1ステップ9に記載したように[1−(メチルチオ)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル]酢酸メチルを標記化合物に変換した。
【0190】
ステップ9:[9−[(3,4−ジクロロフェニル)チオ]−1−(メチルスルホニル)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル]酢酸
ステップ10でビス(3,4−ジクロロフェニル)ジスルフィドを使用した以外は実施例1,ステップ10及び11に記載したように[1−(メチルスルホニル)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル]酢酸メチルを標記化合物に変換した。
1H NMR(500MHz,アセトン−d6)δ 8.35(d,1H)7.80(d,1H),7.35(d,1H),7.15(s,1H),6.95(d,1H),4.55(m,1H),4.35(m,1H)1 3.90(m,1H),3.30(s,3H),3.15(m,1H),3.05(m,1H),2.80(m,1H),2.50(m,1H)。
【0191】
(実施例6)
方法2
[9−[(3,4−ジクロロフェニル)チオ]−1−(メチルスルホニル)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル]酢酸
ステップ1:1−(メチルチオ)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−オール
実施例6,方法1ステップ5からの1−(メチルチオ)−6,7−ジヒドロ−8H−ピリド[3,4−b]ピロリジン−8−オン(0.55g,2.2mmol)のEtOH(10mL)−THF(1mL)懸濁液にNaBH4(0.10g,2.6mmol)を0℃で加えた。室温で30分後にアセトンを加えることにより反応をクエンチした。溶媒を減圧蒸発させ、EtOACとH2Oを残渣に加えた。有機相を分離し、MgSO4で乾燥し、蒸発させた。標記化合物をEtOAc/ヘキサンで洗浄し、濾過した。
【0192】
ステップ2:2−[1−(メチルチオ)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル]マロン酸ジメチル
1−(メチルチオ)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−オール(0.54g,2.1mmol)の−78℃のTHF(10mL)懸濁液にTHF中1M NaHMDS(2.35mL,2.4mmol)とクロロリン酸ジフェニル(0.53mL,2.6mmol)を加えた。30分後にマロン酸ジメチル(0.73mL,6.4mmol)とTHF中1M NaHMDS(6.8mL,6.8mmol)を加えた。反応混合物を0℃まで昇温させた後、室温まで昇温させた。次に混合物をETOAcとNH4Clに分配した。有機相をMgSO4で乾燥し、濾過し、蒸発させた。標記化合物をフラッシュクロマトグラフィーにより精製した。
【0193】
ステップ3:[1−(メチルチオ)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル]−酢酸メチル
2−[1−(メチルチオ)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル]マロン酸ジメチル(0.59g,2.17mmol)とDMSO(4mL)の混合物にH2O(0.45mL)中のNaCl(0.45g)を加えた。150℃で18時間後に反応混合物をETOAcとH2Oに分配した。有機相を分離し、Na2SO4で乾燥し、蒸発させた。次に標記化合物をフラッシュクロマトグラフィーにより精製した。
【0194】
ステップ4:[9−[(3,4−ジクロロフェニル)チオ]−1−(メチルスルホニル)−7,8−ジヒドロ−6H−ピリド[3.4−b]ピロリジン−8−イル]酢酸
標記化合物は[1−(メチルチオ)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル]酢酸メチルから実施例6,方法1,ステップ8から9に記載したように得られた。
【0195】
(実施例7)
[10−[(3,4−ジクロロフェニル)スルファニル]−1−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,4−b]インドリジン−9−イル]酢酸(化合物M)
【0196】
【化8】
【0197】
ステップ1:[1−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,4−b]インドリジン−9−イル]酢酸エチル
標記化合物は実施例6,ステップ3の生成物から実施例1,ステップ5から9に記載したと同様に製造した。
【0198】
ステップ2:[10−[(3,4−ジクロロフェニル)スルファニル]−1−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,4−b]インドリジン−9−イル]酢酸
ステップ10でビス(3,4−ジクロロフェニル)ジスルフィドを使用した以外は実施例1,ステップ10から11と同様にステップ1の生成物を標記化合物に変換した。
MS M+1=485。
【0199】
(実施例8)
(4−(メチルスルホニル)−5−{[4−(トリフルオロメチル)フェニル]チオ}−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル)酢酸(化合物N)
【0200】
【化9】
【0201】
標記化合物はビス[4−トリフルオロメチル)フェニル]ジスルフィドを使用した以外は実施例1に記載したように製造した。
1H NMR(500MHz,アセトン−d6)δ 8.55(d,1H),7.75(d,1H),7.45(d,2H),7.15(d,2H),4.55(m,1H),4.15(m,1H),3.80(m,1H),3.30(s,3H),2.80−2.10(m,6H)。m/z513(M+1)。
【0202】
(実施例9)
[5−[(2−クロロ−4−フルオロフェニル)チオ]−4−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸(化合物O)
【0203】
【化10】
【0204】
標記化合物はビス(2−クロロ−4−フルオロフェニル)ジスルフィドを使用した以外は実施例1に記載したように製造した。
m/z469(M+1)。
【0205】
(実施例10)
[4−(メチルスルホニル)−5−(2−ナフチルチオ)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸(化合物P)
【0206】
【化11】
【0207】
標記化合物はジ(2−ナフチル)ジスルフィドを使用した以外は実施例1に記載したように製造した。
M/z467(M+1)。
【0208】
(実施例11)
[5−[(2,3−ジクロロフェニル)チオ]−4−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸(化合物Q)
【0209】
【化12】
【0210】
標記化合物はビス(2,3−ジクロロフェニル)ジスルフィドを使用した以外は実施例1に記載したように製造した。
1H NMR(500MHz,アセトン−d6)δ 8.85(d,1H),7.80(ds 1H),7.30(d,1H),7.00(t,1H),6.60(d,1H),4.60(m,1H),4.20(m,1H),3.80(m,1H),3.40(s,3H),2.80−2.10(m,6H)。
【0211】
(実施例12)
[5−[(4−メチルフェニル)チオ]−4−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸(化合物R)
【0212】
【化13】
【0213】
標記化合物はp−トリルジスルフィドを使用した以外は実施例1に記載したように製造した。
1H NMR(500MHz,アセトン−d6)δ 8.55(d,1H),7.80(d,1H),6.95(m,4H),4.60(m,1H),4.15(m,1H),3.80(m,1H),3.35(s,3H),2.80−2.10(m,6H)。
【0214】
(実施例13)
[4−(メチルスルホニル)−5−(フェニルチオ)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸(化合物S)
【0215】
【化14】
【0216】
標記化合物はジフェニルジスルフィドを使用した以外は実施例1に記載したように製造した。
1H NMR(500MHz,アセトン−d6)δ 8.55(d,1H),7.80(d,1H),7.15−6.90(m,5H),4.60(m,1H),4.15(m,1H),3.75(m,1H),3.30(s,3H),2.80−2.10(m,6H)。
【0217】
(実施例14)
[5−[(2,4−ジクロロフェニル)チオ]−4−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[3,2−b]インドリジン−6−イル]酢酸(化合物T)
【0218】
【化15】
【0219】
標記化合物はビス(2,4−ジクロロフェニル)ジスルフィドを使用した以外は実施例1に記載したように製造した。ジスルフィドはエーテル中Br2を使用して2,4−ジクロロチオフェニルから製造した。
1H NMR(500MHz,アセトン−d6)δ 8.55(d,1H),7.85(d,1H),7.35(s,1H),7.00(d,1H),6.65(d,1H),4.55(m,1H),4.15(m,1H),3.80(m,1H),3.35(s,3H),2.80−2.10(m,6H)。
【0220】
(実施例15)
[5−[(4−クロロフェニル)チオ]−4−(メチルスルホニル)−6,7,8,9−テトラヒドロピリド[4,3−b]インドリジン−6−イル]酢酸(化合物U)
【0221】
【化16】
【0222】
標記化合物は還流下にアジドをデカリンに加えることにより末端環化を実施した以外は実施例1に記載したように3−クロロニコチンアルデヒド(Heterocycles p.151,1993)から製造した。
1H NMR(500MHz,アセトン−d6)δ 9.20(s,1H),8.85(s,1H),7.20(d,2H),7.00(d,2H),4.70(m,1H),4.30(m,1H),3.75(m,1H),3.35(s,3H),2.80−2.10(m,6H)。
【0223】
(実施例16)
[9−[(4−クロロフェニル)チオ]−1−(メチルスルホニル)−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル]酢酸(化合物V)
【0224】
【化17】
【0225】
標記化合物はステップ10でビス(4−クロロフェニル)ジスルフィドを使用した以外は実施例1ステップ10及び11に要約した手順に記載したように実施例6方法1ステップ8の生成物から製造した。
1H NMR(500MHz,アセトン−d6)δ 8.25−8.3(m,1H),7.71−7.75(m,1H),7.12−7.17(m,2H),6.97−7.04(m,2H),4.45−4.51(m,1H),4.32−4.39(m,1H),3.73−3.80(m,1H),3.29(s,3H),3.15−3.21(m,1H),2.99−3.08(m,1H),2.66−2.73(m,1H),2.46−2.54(m,1H)。
【0226】
(実施例17)
(−)−[(4−クロロベンジル)−7−フルオロ−5−メタンスルホニル)−1,2,3,4−テトラヒドロシクロペンタ[b]インドール−3−イル]酢酸(化合物E)
【0227】
【化18】
【0228】
ステップ1:(+/−)−(7−フルオロ−1,2,3,4−テトラヒドロシクロペンタ[b]インドール−3−イル)酢酸エチルエステル
【0229】
【化19】
【0230】
4−フルオロ−2−ヨードアニリン10.00g、2−(2−オキソシクロペンチル)酢酸エチル6.57g及びp−トルエンスルホン酸121mgのベンゼン(100ml)溶液をDean−StarkトラップでN2雰囲気下に24時間還流した。その後、ベンゼンを留去した。次に、DMF60mlを加え、溶液を脱気した後、ヒューニッヒ塩基19mlとPd(OAc)2 405mgを順次加えた。溶液を115℃まで3時間加熱した後、室温まで冷却した、反応をクエンチするために、1N HCl 300mlと酢酸エチル200mlを加え、混合物をセライトで濾過した。相分離し、酸相を酢酸エチル200mlで2回抽出した。有機相を合わせてブラインで洗浄し、無水Na2SO4で乾燥し、セライトで濾過し、濃縮した。100%トルエンを溶離液として粗材料をフラッシュクロマトグラフィーにより更に精製すると、標記化合物が得られた。
1H NMR(アセトン−d6)δ 9.76(br s,1H),7.34(dd,1H),7.03(d,1H),6.78(td,1H),4.14(q,2H),3.57(m,1H),2.85−2.55(m,5H),2.15(m,1H),1.22(t,3H)。
【0231】
【化20】
【0232】
ステップ2:(+/−)−(7−フルオロ−1,2,3,4−テトラヒドロシクロペンタ[b]インドール−3−イル)酢酸
ステップ1からのエステル1.24gの室温のテトラヒドロフラン(THF)(14mL)溶液にMeOH 7mLと2N NaOH 7mLを順次加えた。2.5時間後に、酢酸エチル(EtOAc)/1N HClを入れた分液漏斗に反応混合物を注入した。相分離し、酸相をEtOAcで2回抽出した。有機層を合わせてブラインで洗浄し、無水Na2SO4で乾燥し、蒸発乾涸すると、粗油状物が得られ、次段階でそのまま使用した(純度>90%)。
1H NMR(アセトン−d6)δ 10.90(br s,1H),9.77(br s,1H),7.34(dd,1H),7.04(dd,1H),6.79(td,1H),3.56(m,1H),2.90−2.50(m,5H),2.16(m,1H)。MS(−APCI)m/z232.2(M−H)−。
【0233】
ステップ3:(+/−)−(5−ブロモ−7−フルオロ−1,2,3,4−テトラヒドロシクロペンタ[b]インドール−3−イル)酢酸
【0234】
【化21】
【0235】
ステップ2からの酸(純度>90%)2.20gのピリジン(30mL)溶液に三臭化ピリジニウム(純度90%)6.85gを−40℃で加えた。懸濁液を10分間0℃で撹拌し、30分間かけて室温まで昇温させた。次に、高真空下で加熱せずに溶媒を除去した。粗材料をAcOH 40mLに溶かし、Zn微粉2.88gを0℃の冷溶液に少量ずつ加えた。懸濁液を15分間15℃で撹拌し、室温まで更に15分間昇温させた。この時点で1N HClを加えることにより反応混合物をクエンチし、ブライン/EtOAcを入れた分液漏斗にこの混合物を注入した。層分離し、有機層を水、ブラインで洗浄し、無水Na2SO4で乾燥し、濃縮した。この材料をそれ以上精製せずに次段階で使用した。
1H NMR(アセトン−d6)δ 10.77(br s,1H),9.84(br s,1H),7.09(m,2H),3.60(m,1H),2.95−2.65(m,4H),2.56(dd,1H),2.19(m,1H)。
【0236】
ステップ4:(+/−)−[5−ブロモ−4−(4−クロロベンジル)−7−フルオロ−1,2,3,4−テトラヒドロシクロペンタ[b]インドール−3−イル]酢酸
【0237】
【化22】
【0238】
TLCでモニターしながら酸が完全に消費されるまでステップ3からの酸2.13gのTHF(10mL)溶液に過剰のジアゾメタンのエーテル溶液を加えた。次に、溶媒を減圧除去した。こうして形成された粗メチルエステルのDMF(20mL)溶液にNaH懸濁液(油中60%)539mgを−78℃で加えた。懸濁液を10分間0℃で撹拌し、再び−78℃まで冷却し、4−クロロベンジルブロマイド1.70gで処理した。5分後に温度を0℃まで上げ、混合物を20分間撹拌した。この時点でAcOH 2mLを加えることにより反応をクエンチし、1N HCl/EtOAcを入れた分液漏斗にこの混合物を注入した。層分離し、有機層をブラインで洗浄し、無水Na2SO4で乾燥し、濃縮した。ステップ2に記載した手順を使用してアルキル化物を水素化した。EtOAc/ヘキサンの存在下にトリチュレーションにより粗材料を更に精製すると、標記化合物が得られた。
1H NMR(アセトン−d6)δ 10.70(br s,1H),7.31(d,2H),7.18(d,1H),7.06(d,1H),6.92(d,2H),5.90(d,1H),5.74(d,1H),3.61(m,1H),3.00−2.70(m,3H),2.65(dd,1H),2.39(dd,1H),2.26(m,1H)。MS(−APCI)m/z436.3,434.5(M−H)−。
【0239】
ステップ5:(+)−[5−ブロモ−4−(4−クロロベンジル)−7−フルオロ−1,2,3,4−テトラヒドロシクロペンタ[b]インドール−3−イル]酢酸
【0240】
【化23】
【0241】
ステップ4の酸2.35gの80℃のEtOH(130mL)溶液に(S)−(−)−1−(1−ナフチル)エチルアミン780μLを加えた。溶液を室温まで冷却し、一晩撹拌した。回収した塩(1.7g)をEtOH 200mLで再び再結晶した。濾過後、得られた白色固体塩を1N HClで中和し、生成物をEtOAcで抽出した。有機層をブラインで洗浄し、無水Na2SO4で乾燥し、濃縮した。EtOAcを溶離液として材料をSiO2パッドで濾過すると、標記エナンチオマーが得られた。2種のエナンチオマーの保持時間は夫々7.5分と9.4分であった[ChiralPak ADカラム,ヘキサン/2−プロパノール/酢酸(95:5:0.1)]。高極性エナンチオマーは98%eeであった。
ee=98%;保持時間=9.4分[ChiralPak ADカラム:250×4.6mm,ヘキサン/2−プロパノール/酢酸(75:25:0.1)];[α]D21=+39.2°(c 1.0,MeOH)。
【0242】
ステップ6:(−)−[4−(4−クロロベンジル)−7−フルオロ−5−(メタンスルホニル)−1,2,3,4−テトラヒドロシクロペンタ[b]インドール−3−イル]酢酸及びナトリウム塩
まず、ステップ5からの酸(15.4g)をジアゾメタンでエステル化した。こうして形成されたエステルをN−メチルピロリジノン中メタンスルフィン酸ナトリウム塩16.3g及びCuI(I)30.2gと混合することによりスルホニル化を実施した。懸濁液をN2流下に脱気し、150℃まで加熱し、3時間撹拌した後、室温まで冷却した。反応をクエンチするために、酢酸エチル500mlとヘキサン500mlを加え、EtOAcを溶離液として混合物をSiO2パッドで濾過した。有機相を濃縮した。粗油状物をEtOAcに溶かし、水で3回、ブラインで1回洗浄し、無水Na2SO4で乾燥し、濾過し、濃縮した。EtOAc中100%トルエン→50%トルエンの勾配を溶離液として粗材料をフラッシュクロマトグラフィーにより更に精製すると、スルホン酸エステル14gが得られ、ステップ2に記載した手順を使用して加水分解した。酢酸イソプロピル/ヘプタンとCH2Cl2/ヘキサンで順次2回再結晶後に標記化合物が得られた。
1H NMR(500MHz アセトン−d6)δ 10.73(br s,1H),7.57(d,2H,J=8.8Hz),7.31(m,1H),7.29(m,1H),6.84(d,2H,J=8.8Hz),6.29(d,1H,JAB=17.8Hz),5.79(d,1H,JAB=17.8Hz),3.43(m,1H),2.98(s,3H),2.94(m,1H),2.85−2.65(m,3H),2.42(dd,1H,J1=16.1Hz,J2=10.3Hz),2.27(m,1H)。13C NMR(125MHz アセトン−d6)δ 173.0,156.5(d,JCF=237Hz),153.9,139.2,133.7,133.3,130.0(d,JCF=8.9Hz),129.6,128.2,127.5(d,JCF=7.6Hz),122.2(d,JCF=4.2Hz),112.3(d,JCF=29.4Hz),111.0(d,JCF=22.6Hz)5 50.8,44.7,38.6,36.6,36.5,23.3。MS(−APCI)m/z436.1,434.1(M−H)−。
ee=97%;保持時間=15.3分[ChiralCel ODカラム:250×4.6mm,ヘキサン/2−プロパノール/エタノール/酢酸(90:5:5:0.2)];[α]D21=−29.3°(c1.0,MeOH)。Mp175.0℃。
【0243】
EtOH(100mL)中の上記酸化合物6.45g(14.80mmol)を1N NaOH水溶液14.80mLで処理することによりナトリウム塩を製造した。有機溶媒を減圧除去し、粗固体を還流下にイソプロピルアルコール1.2Lに溶かした。溶媒の蒸留により最終容量を500mLまで減らした。室温まで冷却することによりナトリウム塩を結晶化させた。結晶ナトリウム塩をH2Oに懸濁し、ドライアイス浴で凍結させ、高真空下に凍結乾燥すると、標記化合物がナトリウム塩として得られた。
1H NMR(500MHz DMSO−d6)δ 7.63(dd,1H,J1=8.5Hz,J2=2.6Hz),7.47(dd,1H,J1=9.7Hz,J2=2.6Hz),7.33(d,2H,J=8.4Hz),6.70(d,2H,J=8.4Hz),6.06(d,1H,J=17.9Hz),5.76(d,1H,J=17.9Hz),3.29(m,1H),3.08(s,3H),2.80(m,1H),2.69(m,1H),2.55(m,1H),2.18(m,2H),1.93(dd,1H,J1=14.4Hz,J2=9.7Hz)。
【0244】
(実施例17A)
(+/−)−[5−ブロモ−4−(4−クロロベンジル)−7−フルオロ−1,2,3,4−テトラヒドロシクロペンタ[b]インドール−3−イル)酢酸(実施例17,ステップ4)の代替手順
ステップ1:(+/−)−7−フルオロ−1,2,3,4−テトラヒドロシクロペンタ[b]インドール−3−イル)酢酸ジシクロヘキシルアミン(DCSA)塩
2−ブロモ−4−フルオロアニリンの0.526Mキシレン溶液を(2−オキソシクロペンチル)酢酸エチル(1.5当量)及び硫酸(0.02当量)と共に20時間加熱還流した。Dean−Stark装置で水を共沸除去した。反応後にNMRを測定し、20時間後に、一般に所望イミン中間体への80から85%変換が認められた。反応混合物を1M重炭酸ナトリウム(0.2容量)で15分間洗浄し、有機フラクションを蒸発させた。残留シロップを減圧(0.5mm Hg)蒸留した。残留キシレンを30℃で蒸留した後、過剰のケトンと未反応アニリンを50から110℃で回収し、イミンを110から180℃フラクションで薄茶色透明液体として純度83%で回収した。
【0245】
次にイミン中間体を酢酸カリウム(3当量)、テトラ−n−ブチルアンモニウムクロライド1水和物(1当量)、酢酸パラジウム(0.03当量)及びN,N−ジメチルアセトアミド(イミン最終濃度=0.365M)の脱気混合物に加えた。反応混合物を115℃まで5時間加熱し、室温まで冷却した。次に3N KOH(3当量)を加え、混合物を室温で1時間撹拌した。反応混合物を水(1.0容量)で希釈し、トルエン(3×0.75容量)で洗浄した。水相を3N HClでpH1まで酸性化し、tertブチルメチルエーテル(2×0.75容量)で抽出した。有機フラクションを合わせて水洗した(0.75容量)。薄茶色透明溶液にジシクロヘキシルアミン(1当量)を加え、溶液を室温で16時間撹拌した。塩を濾過し、酢酸エチル、tertブチルメチルエーテルで洗浄し、乾燥すると、標記化合物が得られた。アッセイ:94 A%。
1H NMR(500mHz,CDCl3):δ 9.24(s,1H),7.16−7.08(m,2H),6.82(t,1H),6.2(br,2H),3.6−3.5(m,1H),3.04−2.97(m,2H),2.88−2.70(m,3H),2.66(dd,1H),2.45−2.37(m,1H),2.13−2.05(m,2.05),1.83(d,4H),1.67(d,2H),1.55−1.43(m,4H),1.33−1.11(m,6H)。
【0246】
ステップ2:(+/−)−(5−ブロモ−7−フルオロ−1,2,3,4−テトラヒドロシクロペンタ[b]インドール−3−イル)酢酸
上記ステップ1からのDCHA塩のジクロロメタン(0.241M溶液)中スラリーを−20から−15℃まで冷却した。ピリジン(2当量)を一度に加え、温度を−20℃から−15℃に維持しながら30から45分間かけてスラリーに臭素(2.5当量)を滴下した。(臭素を約1/3添加した時点では、反応混合物は高粘性であり、効率的な撹拌が必要であった。その後、臭素を約1/2添加した時点で混合物は再び「低粘性」になった。)添加の完了後、反応混合物を更に1時間−15℃でエージングさせた。次に酢酸(3.04当量)を5分間かけて加え、亜鉛微粉(3.04当量)を少量ずつ加えた。(亜鉛の一部は−15℃で加え、混合物を約5分間エージングさせて発熱を進行させた(約−15℃から−10℃))。約30分間かけて亜鉛を約5回加えることによりこの操作を繰返した。発熱が観察されなくなったら、残余の亜鉛をより迅速に加えた。全操作は約30から45分間を要した。
【0247】
添加の完了後、バッチを室温まで昇温させ、1時間エージングさせ、濃縮した。反応混合物をメチルt−ブチルエーテル(MTBE,0.8容量)に交換し、10%酢酸水溶液(0.8容量)を加えた。混合物(塩の結晶化、例えばピリジウム)を室温で1時間エージングさせ、ソルカフロックで濾過した。ソルカフロックパッドをMTBE(約0.2容量)でリンスし、濾液(2相,MTBE/水)を抽出器に移した。有機相を水洗した(0.8容量)。MTBE抽出相を濃縮し、イソプロピルアルコール(IPA,0.25容量)に交換し、化合物を結晶化させた。水(0.25容量)を加え、バッチを1時間エージングさせた。更に水(0.33容量)を1時間加えた。水添加の完了後、バッチを更に1時間エージングさせ、濾過し、30/70 IPA/水(0.15容量)でリンスした。結晶化したブロモ酸を+45℃のオーブンで乾燥した。
【0248】
ステップ3:(+/−)−[5−ブロモ−4−(4−クロロベンジル)−7−フルオロ−1,2,3,4−テトラヒドロシクロペンタ[b]インドール−3−イル)−酢酸
ステップ2のブロモ酸をジメチルアセトアミド(0.416M溶液)に溶かし、炭酸セシウム(2.5当量)を一度に加えた。スラリーに4−クロロベンジルクロライド(2.5当量)を一度に加え、バッチを50℃まで20時間加熱した。バッチを室温まで冷却し、5N水酸化ナトリウム(4.00当量)を5分間かけて加えた(温度は+40℃まで上昇した)。反応混合物を50℃で約3時間エージングさせ、室温まで冷却し、L抽出器に移した。溶液を酢酸イソプロピル(IPAc,2容量)で希釈し、+15℃まで冷却した。溶液を5N HClでpHから2まで酸性化した。層分離し、有機層を水洗した(2×2容量)。IPAc溶液を濃縮し、IPA(0.8容量)に交換し、生成物を結晶化させた。水(8L)を2時間加え、バッチを濾過すると、標記化合物が得られた。バッチを+40℃のオーブンで24時間乾燥してもよい。
【0249】
(実施例18)
(+/−)−{4−[1−(4−クロロフェニル)エチル]−7−フルオロ−5−メタンスルホニル−1,2,3,4−テトラヒドロシクロペンタ[b]インドール−3−イル]酢酸(化合物X)
【0250】
【化24】
【0251】
標記化合物はPCT WO03/062200(公開日2003年7月30日)の記載に従って合成した。
【0252】
(実施例19)
(+/−)−[9−(4−クロロベンジル)−6−フルオロ−メタンスルホニル−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸(化合物Y)
【0253】
【化25】
【0254】
標記化合物はPCT WO03/062200(公開日2003年7月30日)の記載に従って合成した。
【0255】
(実施例20)
[4−(4−クロロベンジル)−7−フルオロ−5−メタンスルホニル−1−オキソ−1,2,3,4−テトラヒドロシクロペンタ[b]インドール−3−イル)酢酸(化合物Z)
【0256】
【化26】
【0257】
標記化合物はPCT WO03/062200(公開日2003年7月30日)の記載に従って合成した。
【0258】
(実施例21)
{9−[(3,4−ジクロロフェニル)チオ]−1−イソプロピル−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル}酢酸(エナンチオマーA及びエナンチオマーB)(化合物AA)
【0259】
【化27】
【0260】
ステップ1:2−クロロニコチンアルデヒド
ジイソプロピルアミン(110mL,780mmol)のTHF(500mL)溶液に−40℃のn−BuLi(300mL,750mmol)の2.5Mヘキサン溶液を加えた。5分後に、反応混合物を−95℃まで冷却した後に、DMPU(15mL)と2−クロロピリジン(50mL,532mmol)を順次加えた。得られた混合物を次に昇温させ、−78℃で4時間撹拌した。その後、黄色い懸濁液を再び−95℃まで冷却した後、DMF(70mL)を加えた。最終反応混合物を−78℃まで昇温させ、この温度で1.5時間撹拌した。反応混合物を冷HCl水溶液(3N,800mL)に注ぎ、5分間撹拌した。濃NH4OH水溶液を加えてpHを7.5に調整した。水層をEtOAcで3回抽出した。有機層を合わせてNH4Cl水溶液とブラインで洗浄し、無水Na2SO4で乾燥し、濾過し、濃縮した。100%ヘキサン→100%EtOAcの勾配を溶離液として粗材料をシリカゲルパッドにより更に精製し、生成物を冷ヘキサン中で結晶化させると、標記化合物が薄黄色固体として得られた。
【0261】
ステップ2:(2Z)−2−アジド−3−(2−クロロピリジン−3−イル)プロプ−2−エン酸メチル
2−クロロニコチンアルデヒド(20.0g,139.9mmol)とアジド酢酸メチル(32.2mL,349.7mmol)のMeOH(168mL)溶液を−20℃のNaOMeの25%MeOH溶液(80mL,349mmol)に加えた。内部温度をモニターし、30分間の添加中、から−20℃に維持した。得られた混合物を次に氷浴中で数時間撹拌した後、冷所にて氷浴中で一晩撹拌した。次に懸濁液を氷とNH4Clの混合物に注ぎ、スラリーを10分間撹拌後に濾過した。生成物を冷H2Oで洗浄した後、減圧乾燥した。粗材料をCH2Cl2に溶かし、MgSO4を加えた。懸濁液をシリカゲルパッドで濾過し、CH2Cl2て洗浄した。濾液を減圧濃縮すると、標記生成物のベージュ色沈殿(20g)が得られた。
【0262】
ステップ3:4−クロロ−1H−ピロロ[3,2−c]ピリジン−2−カルボン酸メチル
(2Z)−2−アジド−3−[2−クロロピリジン−3−イル]プロプ−2−エン酸メチル(21g,88mmol)のメシチレン(880mL)溶液を1時間加熱還流した。反応混合物を室温まで冷却した後、0℃まで冷却し、沈殿を濾過し、冷ヘキサンで洗浄した。材料を1:20 EtOAc/ヘキサン中で一晩撹拌すると、濾過後に標記生成物が薄黄色固体(13.2g)として得られた。
【0263】
ステップ4:1−クロロ−8−オキソ−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−7−カルボン酸メチル
4−クロロ−1H−ピロロ[3,2−c]ピリジン−2−カルボン酸メチル(12.5g,59mmol)のTHF(116mL)−トルエン(460mL)懸濁液にカリウムtert−ブトキシド(64mL,64mmol)の1.0MTHF溶液とアクリル酸メチル(55mL,611mmol)を加えた。得られた混合物を100℃に18時間加熱した。その後、懸濁液を室温まで冷却し、飽和NH4Cl水溶液(400mL)とヘキサン(400mL)の混合物に注いだ。固形分をデカントし、濾過し、H2Oとヘキサンで洗浄すると、標記化合物が得られた。
【0264】
ステップ5:1−クロロ−6,7−ジヒドロ−8H−ピリド[3,4−b]ピロリジン−8−オン
前段階の化合物に100℃で1時間撹拌下にイソプロパノール(8.0mL)と濃HCl(2.0mL)を加えた。反応混合物をEtOAcとNa2CO3に分配した。有機相を分離し、蒸発させると、標記化合物が得られた。
【0265】
ステップ6:1−イソプロペニル−6,7−ジヒドロ−8H−ピリド[3,4−b]ピロリジン−8−オン
DMF(100mL)中の1−クロロ−6,7−ジヒドロ−8H−ピリド[3,4−b]ピロリジン−8−オン(5.0g,24.3mmol)、トリス(ジベンジリデンアセトン)ジパラジウム(0)(1.0g,1.09mmol)及びトリフェニルアルシン(2.70g,8.82mmol)の混合物にトリブチルイソプロペニルスタナン(9.60g,29.00mmol)を加えた。得られた混合物を脱気し、78℃に18時間加熱した。溶媒を減圧蒸発させた。得られた混合物にCH2Cl2とセライトを加えた後、セライトで濾過した。標記化合物をフラッシュクロマトグラフィーにより精製した(ヘキサン中50%→100%EtOAc)。
【0266】
ステップ7:(2E)−(1−イソプロペニル−6,7−ジヒドロ−8H−ピリド[3,4−b]ピロリジン−8−イリデン)エタン酸エチル
1−イソプロペニル−6,7−ジヒドロ−8H−ピリド[3,4−b]ピロリジン−8−オン(0.60g,2.8mmol)とホスホノ酢酸トリエチル(1.00g,4.46mmol)の−78℃のTHF(24mL)溶液に80%NaH(0.12g,4.00mmol)を加え、反応混合物を0℃まで昇温させた後、室温まで昇温させた。反応混合物を飽和NH4ClとEtOAcに注いだ。有機相を分離し、Na2SO4で乾燥し、蒸発させた。標記化合物をフラッシュクロマトグラフィーにより精製した(ヘキサン中40%EtOAc)。
【0267】
ステップ8:(1−イソプロピル−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル)酢酸エチル
(2E)−(1−イソプロペニル−6,7−ジヒドロ−8H−ピリド[3,4−b]ピロリジン−8−イリデン)エタン酸エチル(0.40g,1.4mmol)のMeOH(20mL)溶液にPd(OH)2(0.20g)を加えた。混合物を1気圧H2下に3時間撹拌した。混合物をセライトで濾過し、蒸発させると、標記化合物が得られた。
【0268】
ステップ9:{9−[(3,4−ジクロロフェニル)チオ]−イソプロピル−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル}酢酸エチル
ビス(3,4−ジクロロフェニル)ジスルフィド(0.24g,0.67mmol)のCH2Cl2(5.6mL)溶液にSO2Cl2(0.036mL)を加えた。得られた黄色い混合物を室温で1時間撹拌した。この溶液を(1−イソプロピル−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル)酢酸エチル(0.15g,0.52mmol)の0℃のDMF(5.6mL)溶液に加えた。0℃で1.5時間後に、反応混合物を飽和NaHCO3とEtOAcに注いだ。有機相を分離し、Na2SO4で乾燥し、濾過し、蒸発させた。標記化合物をフラッシュクロマトグラフィーにより精製した(ヘキサン中30%→40%EtOAc)。
【0269】
ステップ10:{9−[(3,4−ジクロロフェニル)チオ]−イソプロピル−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル}酢酸
{9−[(3,4−ジクロロフェニル)チオ]−イソプロピル−7,8−ジヒドロ−6H−ピリド[3,4−b]ピロリジン−8−イル}酢酸エチル(0.23g,0.50mmol)のTHF(5mL)及びMeOH(2.5mL)溶液に1.0M NaOH(1.5mL,1.5mmol)を加えた。室温で18時間後にHOAc(0.25mL)を加え、溶媒を蒸発させた。残渣をEtOAc/H2Oに取り、有機層をH2Oとブラインで洗浄した。乾燥(Na2SO4)後、溶液を濾過し、蒸発させた。残渣を1:1 EtOAc:ヘキサンの存在下で撹拌すると、濾過後に標記化合物が白色固体として得られた。
1H NMR(MeOH−d4)δ 1.14−1.26(m,6H),2.47−2.56(m,1H),2.56−2.64(m,1H),2.94−3.05(m,2H),3.81−3.89(m,1H),4.22−4.30(m,1H),4.33−4.44(m,2H),6.93−6.99(m,1H),7.14−7.19(m,1H),7.33−7.39(m,1H),7.54−7.59(m,1H),8.16−8.21(m,1H)。
【0270】
CH2N2を使用してステップ10の生成物をそのメチルエステルに変換し、ヘキサン中12%2−プロパノールを流速6mL/分で溶離液として使用してエステルをキラル固定相(chiralcel ODカラム2×25cm)でHPLC分離した。エナンチオマーA(低極性)は保持時間31.9分であり、エナンチオマーB(高極性)は保持時間35.5分である。A及びBの両者を実施例17ステップ10と同様に加水分解すると、標記化合物のエナンチオマーA及びBが得られた。
【0271】
(実施例22)
((1R)−6−フルオロ−8−(メチルスルホニル)−9−{(1S)−1−[4−(トリフルオロメチル)フェニル]エチル}−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸(化合物AJ)
【0272】
【化28】
【0273】
ステップ1:塩化2−(2−ブロモ−4−フルオロフェニル)ヒドラジニウム
2−ブロモ−4−フルオロアニリンの−10℃の濃HCl(1.5M)懸濁液にNaNO2(1.1当量)の10.0M水溶液をゆっくりと加えた。混合物を0℃で2.5時間撹拌した。次に内部温度を10℃未満に維持しながらSnCl2(3.8M)の冷(−30℃)濃HCl溶液をゆっくりと加えた。得られた混合物を10℃で20分間、次いで室温で1時間機械的に撹拌した。高粘性スラリーを濾過し、固体を一晩風乾した。固体を冷HClに再懸濁し、再び濾過した。乾燥した材料をEt2Oに懸濁し、10分間撹拌し、濾過し、一晩風乾すると、標記化合物がベージュ色固体として得られた。
【0274】
ステップ2:(+/−)−(8−ブロモ−6−フルオロ−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル)酢酸エチル
ステップ1の化合物(1当量)のAcOH(0.5M)懸濁液に(2−オキソシクロヘキシル)酢酸エチル(1当量)を加えた。混合物を還流下に16時間撹拌し、冷却し、AcOHを減圧蒸発により除去した。残渣をEtOAcで希釈し、水と飽和NaHCO3水溶液で洗浄した。有機層をNa2SO4で乾燥し、濃縮した。次にトルエンを溶離液として残渣をシリカゲルパッドで濾過した。濾液を濃縮し、ヘキサン中で撹拌すると、濾過後に標記化合物が白色固体として得られた。MS(+APCI)m/z354.2(M+H)+。
【0275】
ステップ3:(+/−)−[6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸エチル
ステップ2の化合物(1当量)の無水DMSO(0.28M)溶液にメタンスルフィン酸ナトリウム(3当量)とヨウ化銅(3当量)を加えた。N2を混合物中に5分間バブリングした後、反応混合物を100℃でN2雰囲気下に撹拌した。12時間後に更にメタンスルフィン酸ナトリウム(2当量)とヨウ化銅(2当量)を加えた。混合物を更に12時間100℃で撹拌し、冷却し、EtOAcで希釈し、1N HClを加えて混合物を酸性化した。懸濁液を30分間撹拌し、セライトで濾過した。濾液を水洗し、Na2SO4で乾燥し、濃縮した。まず非極性不純物を除去するために溶離液としてトルエンを使用し、次いで所望生成物を溶出させるためにヘキサン/EtOAcの2:1混合物を使用して残渣をシリカゲルパッドで濾液した。ヘキサン/EtOAc混合物による溶離後の濾液を濃縮すると、標記化合物が薄黄色固体として得られた。MS(−APCI)m/z352.1(M−H)−。
【0276】
ステップ4:[(1R)−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸エチル
ヘキサン中15%iPrOHの混合物を溶離液としてステップ3からのラセミ混合物をchiralpak AD分取カラムで分取HPLCにより分離した。最終生成物の活性に基づき、高極性エナンチオマー(保持時間が長い)が標記化合物であることが確認された。
【0277】
ステップ5:[(1R)−9−[(1S)−1−(4−クロロフェニル)エチル]−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸エチル
ステップ4の化合物(1当量)、トリフェニルホスフィン(1.5当量)及び(1R)−1−(4−クロロフェニル)エタノール(1.5当量,参考例1に記載の一般手順に従って製造)のTHF(0.175M)溶液にアゾジカルボン酸ジ−tert−ブチル(THF中2.1M,1.5当量)を10分間かけて加えた。混合物を室温で2時間撹拌し、濃縮した。トルエン中7%EtOAcを溶離液として残渣をシリカゲルフラッシュクロマトグラフィーにより精製すると、所望生成物(純度から90%)が得られ、次段階でそのまま使用した。
【0278】
ステップ6:[(1R)−9−[(1S)−1−(4−クロロフェニル)エチル]−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸及び[(1S)−9−[(1S)−1−(4−クロロフェニル)エチル]−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸
ステップ5の化合物のTHFとメタノール(0.1M)の2:1混合物溶液に1N LiOH水溶液(3当量)を加えた。混合物を室温で2時間撹拌し、AcOHを加え、溶媒を蒸発により除去した。残渣をEtOAc/H2Oに取り、有機層をブラインで洗浄し、Na2SO4で乾燥し、濾過し、濃縮した。残渣をヘキサン中30%EtOAcでスウィッシュし、生成物をジエチルエーテルに懸濁し、45分間音波処理し、濾過し、高真空下に50℃で24時間乾燥すると、標記化合物が白色固体として得られた。MS(−APCI)m/z462.1(M−H)。
【0279】
別法として、(+/−)−[6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸エチルをステップ5のアルキル化反応に使用し、[(1R)−9−[(1S)−1−(4−クロロフェニル)エチル]−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸エチルと[(1S)−9−[(1S)−1−(4−クロロフェニル)エチル]−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸エチルの2種のジアステレオマーの混合物を得た。以下の手順を使用してジアステレオマー混合物を選択的加水分解により分解すると、所望[(1R)−9−[(1S)−1−(4−クロロフェニル)エチル]−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸が得られた。
【0280】
分解:
[(1R)−9−[(1S)−1−(4−クロロフェニル)エチル]−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸エチルと[(1S)−9−[(1S)−1−(4−クロロフェニル)エチル]−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸エチルのジアステレオマー混合物(1当量)をTHF/MeOHの3.5/1混合物(0.25M)に溶かし、0℃に冷却した。1N LiOH水溶液(1当量)をゆっくりと加え、12時間又は[(1R)−9−[(1S)−1−(4−クロロフェニル)エチル]−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸エチルがほぼ完全に加水分解されるまで混合物を0℃で撹拌し、他方のジアステレオマーはこれらの条件下で僅かしか加水分解されなかった。AcOHを加え、溶媒を蒸発により除去した。残渣をEtOAc/H2Oに取り、有機層をブラインで洗浄し、Na2SO4で乾燥し、濾過し、濃縮した。1%AcOHを加えたヘキサン中40%EtOAcを溶離液として[(1S)−9−[(1S)−1−(4−クロロフェニル)エチル]−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸エチルと[(1R)−9−[(1S)−1−(4−クロロフェニル)エチル]−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸をフラッシュクロマトグラフィーにより分離すると、所望[(1R)−9−[(1S)−1−(4−クロロフェニル)エチル]−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸がde>90%で得られ、ヘキサン中30%EtOAcでスウィッシュすると、所望化合物が白色固体としてde>95%で得られた。
【0281】
ステップ7:[(1R)−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸メチル
[(1R)−9−[(1S)−1−(4−クロロフェニル)エチル]−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸(MeOH中[α]D=−226°)のMeOH(0.1M)溶液に炭素担持10%パラジウム(10%wt/wt)を加えた。N2流を混合物に5分間バブリングした。反応混合物を室温でH2雰囲気(バルーン)下に24時間撹拌し、CH2Cl2を溶離液としてセライトパッドで濾過した。溶媒を減圧蒸発により除去し、残渣をMeOHでスウィッシュすると、化合物[(1R)−6−フルオロ−8−(メチルスルホニル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル]酢酸メチルが得られた。
【0282】
【化29】
【0283】
ステップ8:((1R)−6−フルオロ−8−(メチルスルホニル)−9−{(1S)−1−[4−(トリフルオロメチル)フェニル]エチル}−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル)酢酸(化合物AJ)
ステップ7の化合物(1当量)、トリフェニルホスフィン(1.5当量)及び(1R)−1−[4−(トリフルオロメチル)フェニル]エタノール(1.5当量)のTHF(0.2M)溶液にアゾジカルボン酸ジ−tert−ブチル(THF中1M,1.5当量)を20分間かけて加えた。混合物を室温で2時間撹拌し、濃縮した。トルエン中10%EtOAcを溶離液として残渣をシリカゲルフラッシュクロマトグラフィーにより精製すると、((1R)−6−フルオロ−8−(メチルスルホニル)−9−{(1S)−1−[4−(トリフルオロメチル)フェニル]エチル}−2,3,4,9−テトラヒドロ−1H−カルバゾール−1−イル)酢酸メチル(純度から90%)が得られ、次段階でそのまま使用した。
【0284】
上記エステル(1当量)の0℃のTHF/MeOHの3.5/1混合物(0.25M)溶液に1N LiOH水溶液(1当量)をゆっくりと加え、16時間又はエステルがほぼ完全に加水分解されるまで混合物を0℃で撹拌し、これらの条件下で他方の副ジアステレオマーは加水分解速度が著しく遅い。AcOHを加え、溶媒を減圧除去した。残渣をEtOAc/H2Oに取り、有機層をブラインで洗浄し、Na2SO4で乾燥し、濾過し、濃縮した。未反応メチルエステルを除去するために、まず10%EtOAc/トルエン、次いで1%AcOHを加えた60%EtOAc/トルエンを溶離液として残渣をシリカゲルパッドで濾過した。残渣を30%EtOAc/ヘキサンでスウィッシュし、高真空下に50℃で16時間乾燥すると、標記化合物がde及びee>95%(キラルHPLCにより確認)で白色固体として得られた。MS(−APCI)m/z496.0(M−H)−。MeOH中[α]D=−181°。
【0285】
生物学
選択的DPアンタゴニストとして機能する本発明で使用する化合物は一般にDPに対する親和性(Ki)がCRTH2受容体に対する親和性(Ki)の少なくとも約10倍(Ki値の数値が小さい)である。本発明で使用する典型的DPアンタゴニストはDP受容体に対する選択性がCRTH2受容体の少なくとも約10倍である。より特定的には、選択的DP受容体アンタゴニストはDP受容体に対する選択性がCRTH2受容体の少なくとも約100倍である。更に特定的には、DP選択的アンタゴニスト化合物はDP受容体に対する選択性がCRTH2受容体の少なくとも約800から1000倍であり、即ちDP受容体に対する親和性(Ki)がCRTH2受容体に対する親和性(Ki)の800から1000倍である。
【0286】
本明細書で使用する場合には、ある化合物が「DP受容体を選択的に調節する」とき、この化合物は治療用量で達成可能な濃度でDP受容体と結合し、これを阻害するが、このような治療達成可能な濃度でCRTH2を実質的に調節しない。
【0287】
一般に、本発明で使用するDPアンタゴニストはCRTH2受容体に対する親和性(Ki)が約0.5マイクロモル以上である。CRTH2に対する結合親和性が約0.5マイクロモル以上であり、DP受容体に対する選択性がCRTH2の少なくとも約10倍である化合物がこのような選択的DPアンタゴニストの不在下でニコチン酸を投与する場合に認められるフラッシング効果を抑制するのに有用である。
【0288】
組換えヒトDP及びCRTH2受容体に対する化合物の親和性及び選択性の測定
Abramovitz Mら,Biochem.Biophys.Acta(2000)1483:285−293、及びSawyer Nら,Br.J.Pharmacol.(2002);137:1163−1172に記載されているような放射性リガンド結合アッセイを使用してDP及びCRTH2に対する化合物の受容体親和性及び選択性を測定した。要約すると、ヒト胚性腎(HEK)293EBNA(Epstein Barr virus Nuclear Antigen:エプスタイン・ハケールウイルス核抗原)細胞(HEK293E細胞株と呼ぶ)を使用してヒトDP及びCRTH2受容体を各々発現する安定な細胞株を樹立した。これらの組換え細胞株から調製した膜フラクションを平衡競合放射性リガンド結合アッセイで利用し、DP及びCRTH2受容体に対する化合物の親和性と選択性を測定した。
【0289】
全長コーディング配列に対応するDP及びCRTH2 cDNAを哺乳動物発現ベクターpCEP4(Invitrogen)の適当な部位にサブクローニングし、HEK293E細胞で発現させた。分画遠心(いずれも4℃にて1000×gで10分後、160,000×gで30分)後にプロテアーゼ阻害剤(2mM AEBSF,10μM E−64,100μMロイペプチン及び0.05mg/mLペプスタチン)の存在下に氷上で30分間800psi窒素キャビテーションにより細胞を溶解させることにより膜を調製した。1mM EDTAを加えた10mM HEPES/KOH(pH7.4)にDounceホモジナイゼーション(Dounce A;10ストローク)により160,000×gペレットを約5から10mg/mL蛋白質で再懸濁し、液体窒素で凍結させ、−80℃で保存した。1mM EDTA,10mM MnCl2及び0.7nM [3H]PGD2(200Ci/mmol)を加えた10mM HEPES/KOH(pH7.4)中、最終インキュベーション容量0.2mLで受容体結合アッセイを実施した。160,000×gフラクションからの膜蛋白質(DPでは約30μg、CRTH2では10μg)を加えることにより反応を開始した。リガンドをジメチルスルホキシド(DMSO)に加え、ジメチルスルホキシドは全インキュベーションで1%(v/v)の一定値に維持した。10μM非放射性PGD2の存在下で非特異的結合を測定した。インキュベーションはミニオービタルシェーカーで室温にて60分間実施した。Tomtec Mach III 96ウェル半自動セルハーベスターを使用してEDTAを添加しないアッセイインキュベーションバッファー(4℃)で予め湿潤させておいた96ウェルUnifilter GF/C(Canberra Packard)で迅速濾過により結合アッセイを終了した。フィルターを同一バッファー3から4mLで洗浄し、90分間55℃で乾燥し、1450 MicroBeta(Wallac)カウンターを使用してUltima Gold F(Canberra Packard)50μLを添加するシンチレーション計数により個々のフィルターに結合した残留放射能を測定した。
【0290】
競合剤の不在下で全結合から非特異的結合を引いた値として最大特異的結合を定義した。各化合物濃度で特異的結合を測定し、最大特異的結合の百分率として表した。試験化合物濃度の関数として最大特異的結合百分率を表すことによりS字状平衡競合曲線を作成し、4パラメーター式に基づくシンプレックス法による非線形最小二乗曲線フィットルーチンを利用してカスタム設計ソフトウェアパッケージにより分析し、変曲点(InPt)を決定した。式Ki=InPt/1+([放射性リガンド]/Kd)(式中、Kdは放射性リガンド−受容体相互作用の平衡解離定数である)から平衡阻害定数(Ki)を計算することにより試験化合物の結合親和性を決定した。InPtを決定できない場合には、IC50を使用した(即ち最大特異的結合の50%を阻害するために必要な試験化合物の濃度)。
【0291】
一般に、本発明で使用する化合物はDP受容体に対するKiが約0.4nMから約16.3nMである。同様に、本発明で使用する化合物は一般にCRTH2受容体に対するKiが約180nMから約22,000nM又はそれ以上である。
【0292】
ニコチン酸により誘発されるマウスの血管拡張に対する化合物の効果
本明細書に記載する選択的DPアンタゴニストの効力はヒトニコチン酸により誘発されるフラッシングのマウスモデルを使用してフラッシング抑制効果を測定することにより実証することができる。(対照として)ビヒクル又はDPアンタゴニストで予め処置しておいたマウスにニコチン酸を投与後にマウス耳の血流(ヒトにおけるフラッシングの顕著な要素である血管拡張の尺度)を測定する。具体的には、雄C57BL/6マウス(から25g)を試験で使用した。各試験群でマウス5匹を評価した。ネンブタールを最終濃度5mg/mlまで水で希釈し、0.3ml/匹を腹腔内注射した。DPアンタゴニストを5%ヒドロキシプロピルβ−シクロデキストリンに最終濃度5mg/mlで溶かし、化合物を容量0.2ml/匹(から40mpk)で腹腔内投与した。ニコチン酸を5%ヒドロキシプロピルβ−シクロデキストリンに最終濃度12.5mg/mlで溶かした。ニコチン酸ストック溶液を2N NaOHでpH7.4に調整し、0.2ml/匹(から100mpk)を皮下注射した。
【0293】
ニコチン酸投与の5分前から開始して30秒おきに15分間レーザードップラー血流イメージャー(PeriScan PIM II,Perimed,Sweden)でマウス耳皮膚の血流をモニターした。ビヒクル又はニコチン酸投与後10分間の平均血流変化百分率を計算し、時間に対する平均血流変化百分率のグラフを1匹毎に作成した。次に、各グラフから平均血流の曲線下面積(AUC)(%Δ×分)を計算し、結果を各群の平均AUC±SEMで表した。
【0294】
化合物DはPGD−2により誘発されるマウスの血管拡張を抑制した(図1)。試験したDPアンタゴニストはニコチン酸により誘発されるマウスの血管拡張を抑制し、その他、選択化合物のデータを図2及び3に示す。
【0295】
ニコチン酸、又はその塩もしくは溶媒和物、又は別のニコチン酸受容体アゴニストのフラッシング効果のアスピリン抑制に関して、ヒトフラッシングの相関としてDP受容体(DP1)を遺伝的に欠失するマウスを使用し、ニコチン酸により誘発される血管拡張のマウスモデルで試験した処、ニコチン酸により誘発される血管拡張が多少生じる場合もあるが、血管拡張の程度はDP受容体をもつマウスよりも低いことが判明した。従って、ニコチン酸により誘発される血管拡張の一部はDP受容体に依存しないと思われる。このDP非依存性血管拡張はCOX−1/COX−II阻害剤であるアスピリンをマウスに投与又は予備投与することにより抑制することができる。ヒト用量は約20から25mgから約650mgまでであり、ニコチン酸投与の約1時間から約30分間前から同時投与までとし、必要に応じて約4時間までの間隔で反復する。従って、DP受容体アンタゴニストとアスピリンの併用はヒトにおけるニコチン酸により誘発されるフラッシングを抑制し、DP受容体又はアスピリン単独では抑制されないフラッシングの一部又は全部を緩和するために特に有効である。
【0296】
本明細書に引用する全特許、特許出願及び刊行物は参考資料としてその開示内容全体を本明細書に組込む。本明細書には所定の好ましい態様を詳細に記載したが、多数の代替態様が本発明の範囲に含まれるとみなされる。
【図面の簡単な説明】
【0297】
【図1】化合物DがプロスタグランジンD2により誘発されるマウスの血管拡張を抑制することを示すグラフである。
【図2】化合物Dがニコチン酸により誘発されるマウスの血管拡張を抑制することを示すグラフである。
【図3】他の選択化合物がニコチン酸により誘発されるマウスの血管拡張を抑制することを示すグラフである。
【特許請求の範囲】
【請求項1】
治療を必要とするヒト患者におけるアテローム性動脈硬化症の治療方法であって、実質的フラッシングを併発せずにアテローム性動脈硬化症を治療するために有効な量であるニコチン酸約1000mgと、約18.75mg、20mg、37.5mg、50mg、75mg、100mg及び150mgから選択される量のDP受容体アンタゴニストを患者に投与することを含む前記方法。
【請求項2】
治療を必要とするヒト患者におけるアテローム性動脈硬化症の治療方法であって、実質的フラッシングを併発せずにアテローム性動脈硬化症を治療するために有効な量であるニコチン酸約2000mgと、約37.5mg、75mg、100mg、150mg、200mg及び300mgから選択される量のDP受容体アンタゴニストを患者に投与することを含む前記方法。
【請求項3】
ニコチン酸約1000mgと、18.75mg、20mg、37.5mg、50mg、75mg、100mg及び150mgから選択される量のA、B、D、E、X、AA、AF、AG、AH、AI及びAJから構成される群から選択されるDPアンタゴニスト、又は医薬的に許容可能なその塩もしくは溶媒和物を医薬的に許容可能なキャリヤーと共に含有する医薬組成物。
【請求項4】
ニコチン酸1000mgと、A、B、D、E、X、AA、AF、AG、AH、AI及びAJから構成される群から選択されるDPアンタゴニスト、又は医薬的に許容可能なその塩もしくは溶媒和物20mg、75mg、100mg又は150mgを医薬的に許容可能なキャリヤーと共に含有する請求項3に記載の医薬組成物。
【請求項5】
ニコチン酸1000mgと、A、B、D、E、X、AA、AF、AG、AH、AI及びAJから構成される群から選択されるDPアンタゴニスト、又は医薬的に許容可能なその塩もしくは溶媒和物20mgを医薬的に許容可能なキャリヤーと共に含有する請求項4に記載の医薬組成物。
【請求項6】
ニコチン酸1000mgと、化合物A又は医薬的に許容可能なその塩もしくは溶媒和物20mgを医薬的に許容可能なキャリヤーと共に含有する請求項5に記載の医薬組成物。
【請求項7】
ニコチン酸1000mgと、化合物B又は医薬的に許容可能なその塩もしくは溶媒和物20mgを医薬的に許容可能なキャリヤーと共に含有する請求項5に記載の医薬組成物。
【請求項8】
ニコチン酸1000mgと、化合物D又は医薬的に許容可能なその塩もしくは溶媒和物20mgを医薬的に許容可能なキャリヤーと共に含有する請求項5に記載の医薬組成物。
【請求項9】
ニコチン酸1000mgと、化合物E又は医薬的に許容可能なその塩もしくは溶媒和物20mgを医薬的に許容可能なキャリヤーと共に含有する請求項5に記載の医薬組成物。
【請求項10】
ニコチン酸1000mgと、化合物X又は医薬的に許容可能なその塩もしくは溶媒和物20mgを医薬的に許容可能なキャリヤーと共に含有する請求項5に記載の医薬組成物。
【請求項11】
ニコチン酸1000mgと、化合物AA又は医薬的に許容可能なその塩もしくは溶媒和物20mgを医薬的に許容可能なキャリヤーと共に含有する請求項5に記載の医薬組成物。
【請求項12】
ニコチン酸1000mgと、化合物AF又は医薬的に許容可能なその塩もしくは溶媒和物20mgを医薬的に許容可能なキャリヤーと共に含有する請求項5に記載の医薬組成物。
【請求項13】
ニコチン酸1000mgと、化合物AG又は医薬的に許容可能なその塩もしくは溶媒和物20mgを医薬的に許容可能なキャリヤーと共に含有する請求項5に記載の医薬組成物。
【請求項14】
ニコチン酸1000mgと、化合物AH又は医薬的に許容可能なその塩もしくは溶媒和物20mgを医薬的に許容可能なキャリヤーと共に含有する請求項5に記載の医薬組成物。
【請求項15】
ニコチン酸1000mgと、化合物AI又は医薬的に許容可能なその塩もしくは溶媒和物20mgを医薬的に許容可能なキャリヤーと共に含有する請求項5に記載の医薬組成物。
【請求項16】
ニコチン酸1000mgと、化合物AJ又は医薬的に許容可能なその塩もしくは溶媒和物20mgを医薬的に許容可能なキャリヤーと共に含有する請求項5に記載の医薬組成物。
【請求項17】
シンバスタチン約10mg、20mg又は40mgを更に含有する請求項3に記載の医薬組成物。
【請求項18】
シンバスタチン約10mg、20mg又は40mgを更に含有する請求項4に記載の医薬組成物。
【請求項19】
シンバスタチン約10mg、20mg又は40mgを更に含有する請求項5に記載の医薬組成物。
【請求項20】
シンバスタチン約10mg、20mg又は40mgを更に含有する請求項6に記載の医薬組成物。
【請求項21】
シンバスタチン約10mg、20mg又は40mgを更に含有する請求項7に記載の医薬組成物。
【請求項22】
シンバスタチン約10mg、20mg又は40mgを更に含有する請求項8に記載の医薬組成物。
【請求項23】
シンバスタチン約10mg、20mg又は40mgを更に含有する請求項9に記載の医薬組成物。
【請求項24】
シンバスタチン約10mg、20mg又は40mgを更に含有する請求項10に記載の医薬組成物。
【請求項25】
シンバスタチン約10mg、20mg又は40mgを更に含有する請求項11に記載の医薬組成物。
【請求項26】
シンバスタチン約10mg、20mg又は40mgを更に含有する請求項12に記載の医薬組成物。
【請求項27】
シンバスタチン約10mg、20mg又は40mgを更に含有する請求項13に記載の医薬組成物。
【請求項28】
シンバスタチン約10mg、20mg又は40mgを更に含有する請求項14に記載の医薬組成物。
【請求項29】
シンバスタチン約10mg、20mg又は40mgを更に含有する請求項15に記載の医薬組成物。
【請求項30】
シンバスタチン約10mg、20mg又は40mgを更に含有する請求項16に記載の医薬組成物。
【請求項31】
シンバスタチン約10mg、20mg又は40mgを更に含有する請求項17に記載の医薬組成物。
【請求項1】
治療を必要とするヒト患者におけるアテローム性動脈硬化症の治療方法であって、実質的フラッシングを併発せずにアテローム性動脈硬化症を治療するために有効な量であるニコチン酸約1000mgと、約18.75mg、20mg、37.5mg、50mg、75mg、100mg及び150mgから選択される量のDP受容体アンタゴニストを患者に投与することを含む前記方法。
【請求項2】
治療を必要とするヒト患者におけるアテローム性動脈硬化症の治療方法であって、実質的フラッシングを併発せずにアテローム性動脈硬化症を治療するために有効な量であるニコチン酸約2000mgと、約37.5mg、75mg、100mg、150mg、200mg及び300mgから選択される量のDP受容体アンタゴニストを患者に投与することを含む前記方法。
【請求項3】
ニコチン酸約1000mgと、18.75mg、20mg、37.5mg、50mg、75mg、100mg及び150mgから選択される量のA、B、D、E、X、AA、AF、AG、AH、AI及びAJから構成される群から選択されるDPアンタゴニスト、又は医薬的に許容可能なその塩もしくは溶媒和物を医薬的に許容可能なキャリヤーと共に含有する医薬組成物。
【請求項4】
ニコチン酸1000mgと、A、B、D、E、X、AA、AF、AG、AH、AI及びAJから構成される群から選択されるDPアンタゴニスト、又は医薬的に許容可能なその塩もしくは溶媒和物20mg、75mg、100mg又は150mgを医薬的に許容可能なキャリヤーと共に含有する請求項3に記載の医薬組成物。
【請求項5】
ニコチン酸1000mgと、A、B、D、E、X、AA、AF、AG、AH、AI及びAJから構成される群から選択されるDPアンタゴニスト、又は医薬的に許容可能なその塩もしくは溶媒和物20mgを医薬的に許容可能なキャリヤーと共に含有する請求項4に記載の医薬組成物。
【請求項6】
ニコチン酸1000mgと、化合物A又は医薬的に許容可能なその塩もしくは溶媒和物20mgを医薬的に許容可能なキャリヤーと共に含有する請求項5に記載の医薬組成物。
【請求項7】
ニコチン酸1000mgと、化合物B又は医薬的に許容可能なその塩もしくは溶媒和物20mgを医薬的に許容可能なキャリヤーと共に含有する請求項5に記載の医薬組成物。
【請求項8】
ニコチン酸1000mgと、化合物D又は医薬的に許容可能なその塩もしくは溶媒和物20mgを医薬的に許容可能なキャリヤーと共に含有する請求項5に記載の医薬組成物。
【請求項9】
ニコチン酸1000mgと、化合物E又は医薬的に許容可能なその塩もしくは溶媒和物20mgを医薬的に許容可能なキャリヤーと共に含有する請求項5に記載の医薬組成物。
【請求項10】
ニコチン酸1000mgと、化合物X又は医薬的に許容可能なその塩もしくは溶媒和物20mgを医薬的に許容可能なキャリヤーと共に含有する請求項5に記載の医薬組成物。
【請求項11】
ニコチン酸1000mgと、化合物AA又は医薬的に許容可能なその塩もしくは溶媒和物20mgを医薬的に許容可能なキャリヤーと共に含有する請求項5に記載の医薬組成物。
【請求項12】
ニコチン酸1000mgと、化合物AF又は医薬的に許容可能なその塩もしくは溶媒和物20mgを医薬的に許容可能なキャリヤーと共に含有する請求項5に記載の医薬組成物。
【請求項13】
ニコチン酸1000mgと、化合物AG又は医薬的に許容可能なその塩もしくは溶媒和物20mgを医薬的に許容可能なキャリヤーと共に含有する請求項5に記載の医薬組成物。
【請求項14】
ニコチン酸1000mgと、化合物AH又は医薬的に許容可能なその塩もしくは溶媒和物20mgを医薬的に許容可能なキャリヤーと共に含有する請求項5に記載の医薬組成物。
【請求項15】
ニコチン酸1000mgと、化合物AI又は医薬的に許容可能なその塩もしくは溶媒和物20mgを医薬的に許容可能なキャリヤーと共に含有する請求項5に記載の医薬組成物。
【請求項16】
ニコチン酸1000mgと、化合物AJ又は医薬的に許容可能なその塩もしくは溶媒和物20mgを医薬的に許容可能なキャリヤーと共に含有する請求項5に記載の医薬組成物。
【請求項17】
シンバスタチン約10mg、20mg又は40mgを更に含有する請求項3に記載の医薬組成物。
【請求項18】
シンバスタチン約10mg、20mg又は40mgを更に含有する請求項4に記載の医薬組成物。
【請求項19】
シンバスタチン約10mg、20mg又は40mgを更に含有する請求項5に記載の医薬組成物。
【請求項20】
シンバスタチン約10mg、20mg又は40mgを更に含有する請求項6に記載の医薬組成物。
【請求項21】
シンバスタチン約10mg、20mg又は40mgを更に含有する請求項7に記載の医薬組成物。
【請求項22】
シンバスタチン約10mg、20mg又は40mgを更に含有する請求項8に記載の医薬組成物。
【請求項23】
シンバスタチン約10mg、20mg又は40mgを更に含有する請求項9に記載の医薬組成物。
【請求項24】
シンバスタチン約10mg、20mg又は40mgを更に含有する請求項10に記載の医薬組成物。
【請求項25】
シンバスタチン約10mg、20mg又は40mgを更に含有する請求項11に記載の医薬組成物。
【請求項26】
シンバスタチン約10mg、20mg又は40mgを更に含有する請求項12に記載の医薬組成物。
【請求項27】
シンバスタチン約10mg、20mg又は40mgを更に含有する請求項13に記載の医薬組成物。
【請求項28】
シンバスタチン約10mg、20mg又は40mgを更に含有する請求項14に記載の医薬組成物。
【請求項29】
シンバスタチン約10mg、20mg又は40mgを更に含有する請求項15に記載の医薬組成物。
【請求項30】
シンバスタチン約10mg、20mg又は40mgを更に含有する請求項16に記載の医薬組成物。
【請求項31】
シンバスタチン約10mg、20mg又は40mgを更に含有する請求項17に記載の医薬組成物。
【図1】

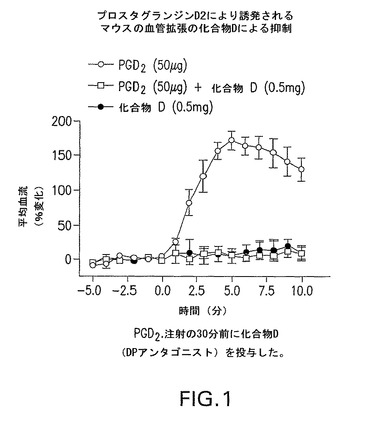
【図2】
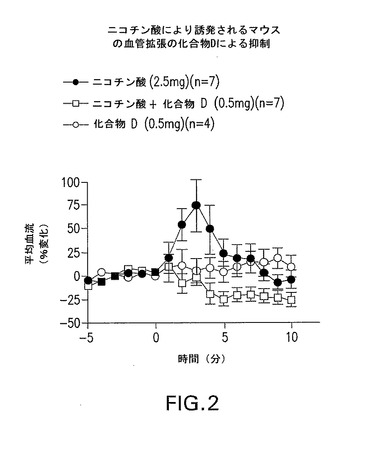
【図3】
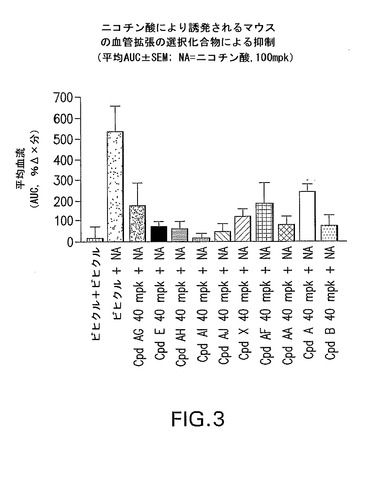
【図2】
【図3】
【公表番号】特表2008−530250(P2008−530250A)
【公表日】平成20年8月7日(2008.8.7)
【国際特許分類】
【出願番号】特願2007−556436(P2007−556436)
【出願日】平成18年2月15日(2006.2.15)
【国際出願番号】PCT/US2006/006951
【国際公開番号】WO2006/089309
【国際公開日】平成18年8月24日(2006.8.24)
【出願人】(390023526)メルク エンド カムパニー インコーポレーテッド (924)
【氏名又は名称原語表記】MERCK & COMPANY INCOPORATED
【出願人】(390035482)メルク シャープ エンド ドーム リミテッド (81)
【Fターム(参考)】
【公表日】平成20年8月7日(2008.8.7)
【国際特許分類】
【出願日】平成18年2月15日(2006.2.15)
【国際出願番号】PCT/US2006/006951
【国際公開番号】WO2006/089309
【国際公開日】平成18年8月24日(2006.8.24)
【出願人】(390023526)メルク エンド カムパニー インコーポレーテッド (924)
【氏名又は名称原語表記】MERCK & COMPANY INCOPORATED
【出願人】(390035482)メルク シャープ エンド ドーム リミテッド (81)
【Fターム(参考)】
[ Back to top ]