
アデノ随伴ウイルスファージ粒子に関する方法および組成物
本発明の態様は概して、発現の増強を達成するために、1つまたは複数のトランスジーンを腫瘍細胞などの標的細胞に、部位特異的な様式で送達する組成物および方法、ならびにそのような適用において有用なコンストラクトおよび組成物に向けられている。ある種の局面では、対象への処置の適用または対象に対する診断手順の前に、治療的核酸からの発現を評価してもよい。
【発明の詳細な説明】
【技術分野】
【0001】
本出願は、その全体が参照により本明細書に組み入れられる、2006年4月7日に申請された米国特許仮出願第60/744,492号に対する優先権を主張する。
【0002】
米国政府は、国立衛生研究所(NIH)からの助成金に応じた本発明における権利を所有する。
【0003】
I、発明の分野
本発明の態様は一般に、生物学および医学に向けられている。特に、本発明は、対象に治療を提供するためのイメージングと組み合わせてAAVPを用いる遺伝子治療の分野に向けられている。
【背景技術】
【0004】
発明の背景
II、背景
多くの生物学に基づく治療の限界は、生体活性分子の腫瘍細胞またはそれらの周囲のマトリックスへの制御されたおよび効果的な送達を達成することができないことである。遺伝子に基づく治療を利用する目的は、遺伝子発現の結果としての、生体産物の細胞内のそれらの作用の部位への効果的な送達を達成することである。遺伝子に基づく治療はまた、転移される遺伝子材料に特異的なプロモーター/アクチベーターエレメントを含めることによって、これらの生体活性産物の作用のレベル、時期、および持続時間に対する制御を提供し、より効果的な治療的介入を結果的にもたらすことができる。新規のDNAの製剤を用いた様々な体細胞組織および腫瘍への制御された遺伝子送達のための、ならびに細胞特異的で、複製により活性化され、かつ薬物により制御される発現系を用いて遺伝子発現を制御するための方法が開発中である。
【0005】
1つのアプローチにおいて、遺伝子治療は特異的な様式で細胞を標的化しようと試みる。したがって、標的細胞または組織の中に遺伝子を送達するために、治療的遺伝子は何らかの方法で標的化分子に連結される。現在の方法は典型的には、内在化受容体を認識するリガンドまたは抗体などの標的化分子を所望の遺伝子を含む裸のDNAかまたは哺乳動物細胞ウイルスのいずれかに連結する工程を伴う。裸のDNAを用いる場合、それをインビトロで凝縮させて、細胞内への侵入用の小型形状にしなければならない。DNA上の電荷を中和しかつDNAを凝縮させて環状体構造にするためにポリリジンなどのポリカチオンが一般に用いられる。しかしながら、この凝縮過程は十分に理解されているわけではなくかつ制御するのが難しく、したがって、均質な遺伝子治療薬物の製造を極めて厄介なものにしている。
【0006】
ラムダおよび繊維状ファージなどの、バクテリオファージ(ファージ)は、哺乳動物細胞内にDNAを転移しようとする努力において時折用いられる。一般に、ラムダの形質導入は、比較的稀な事象であることが分かっておりかつレポーター遺伝子の発現は弱かった。形質導入効率を増強しようとする努力において、(細胞表面受容体を特異的に標的化するわけではない)リン酸カルシウムまたはリポソームを活用する方法がラムダと併せて用いられた。遺伝子転移は、リン酸カルシウム共沈殿を用いたラムダファージによって、またはDEAE-デキストランもしくはリポポリアミンを用いた繊維状ファージによって観察されている。しかしながら、トランスフェクション効率が低く、非特異的である傾向があり、かつトランスフェクションが面倒であるばかりでなく、細胞タイプに関して無差別でもあるので、哺乳動物細胞内にDNAを導入するこれらの方法は、遺伝子治療適用には実用的でない。
【0007】
現在、真核生物ウイルスは、優れたトランスジーン送達および形質導入を紛れもなく提供するが(Kootstra and Verma, 2003;Machida, 2003)、そのようなベクターのリガンド指向性の標的化は通常、哺乳動物細胞膜受容体に対するそれらの天然の向性の除去を必要とする(Miller et al., 2003;Mizuguchi and Hayakawa, 2004;White et al., 2004)。対照的に、バクテリオファージ(ファージ)などの原核生物ウイルスは通常、哺乳動物細胞形質導入用の不十分なビヒクルと考えられている。しかしながら、「真核生物」ウイルスとしてのそれらの固有の欠点にも関わらず、ファージ粒子は、哺乳動物細胞に対する向性を有さず(Zacher et al., 1980;Barrow and Soothill, 1997;Barbas et al., 2001)かつ低い効率でではあるが、そのような細胞に形質導入するように適合させられてすらいる(Ivanenkov et al., 1999;Larocca et al., 1999;Poul and Marks, 1999;Piersanti et al., 2004)。
【0008】
したがって、治療的適用において有用なベクターを達成したい場合、ベクターを特異的な細胞(または受容体)に標的化しかつ治療的に有用な程度の遺伝子送達および発現を保証するより信頼できる手段がこのように必要とされる。
【発明の開示】
【0009】
発明の概要
本発明の態様は一般に、増強された発現を達成するために、1つまたは複数のトランスジーンを腫瘍細胞などの的細胞に、部位特異的な様式で送達する組成物および方法、ならびにそのような適用において有用なコンストラクトおよび組成物に向けられている。ある種の局面において、対象への処置の実施または対象に対する診断手順の前に、治療的核酸からの発現を検討評価してもよい。さらなる局面において、治療的利益に必要とされる領域または場所における発現の決定または評価を検討評価し、かつ任意の不必要なまたは必要最小限の有益な処置を、代替処置の代わりに是認することができる。
【0010】
任意の特定の理論またはメカニズムに束縛されるわけではないが、本開示は、トランスジーンが1より大きい多重度でゲノムに組み込まれる時にトランスジーン発現が増大し得るという観察に基づいている。特に興味深いのは、ある種のキメラAAVP粒子が、多くの場合コンカテマーとして、1コピーより多くのトランスジーンを細胞に形質導入する能力である。キメラAAVP粒子によって運ばれるレポーター遺伝子の発現によって、形質導入された細胞をモニタリングしてもよい。任意のトランスジーンを本開示のAAVP粒子の中に含め、かつ本開示のAAVP粒子から発現させてもよい。
【0011】
本発明のある種の態様には、対象の標的組織への遺伝子転移および/または対象の標的組織での遺伝子発現を検出するための方法および組成物が含まれ、これらは、以下の工程の1つまたは複数を含む:
(a)本発明で使用し得る1つの工程には、宿主対象中に天然に存在してもまたは存在しなくてもよいレポーター遺伝子を含むAAVPベクターを、対象の標的組織に送達する工程が含まれる。典型的には、レポーター遺伝子は、画像化および/または処置される場所または領域で発現していないと考えられる。ある種の局面において、レポーター遺伝子は、野生型の、突然変異体の、または遺伝子改変されたキナーゼである。さらなる局面において、キナーゼはチミジンキナーゼである。なおさらなる局面において、キナーゼは単純ヘルペスウイルス-チミジンキナーゼ遺伝子またはヒトチミジンキナーゼ2型である。典型的には、転移ベクターまたはAAVPが標的組織の細胞に導入され、レポーター遺伝子が標的組織の細胞で発現し、それによってAAVPベクターによって効率的にトランスフェクトされた細胞でのみ蓄積するレポーター遺伝子産物(タンパク質)が生成される。
(b)使用し得る別の工程は、宿主対象に標識されたレポーター基質を投与する工程であり、ここでレポーター遺伝子産物を発現する細胞は、放射性標識されたヌクレオシド類似体を含む標識されたレポーター基質を代謝して、標識されたレポーター代謝物を産生する。
(c)本方法で使用し得るさらに別の工程には、レポーター基質の標識された代謝物を含む標的組織または細胞を非侵襲的に画像化する工程が含まれる。ある種の局面において、レポーター遺伝子産物によって代謝されない残存レポーター基質を宿主対象から排除した後、対象をイメージングに供し、それによって標的組織への遺伝子転移および標的組織内での発現を検出する。さらなる局面において、代謝されないレポーター基質の排除の代謝に応じて、1、2、3、4、5、6、7、8、9、10、またはそれより多くの分、時間、日、または週の後に、1つまたは複数の対象組織をイメージングに供する。
【0012】
本方法は、レポーター遺伝子産物によって代謝されないもの(発現産物によって代謝されないレポーター基質)の全ての値およびその間の範囲を含む、約、少なくとも、または多くても、60、65、67、70、75、77、80、85、87、90、95、97%もしくはそれより多くを、対象から排除するのに十分な工程(b)後の一定の期間、待機する工程をさらに含むことができる。代謝されない基質には、代謝されない残存レポーター基質に由来する非特異的標識が含まれてもよい。インビトロまたはインビボのトランスフェクション(または形質導入)によって、AAVPベクターを標的組織の細胞に導入することができる。ある種の局面において、静脈内に、腫瘍内に、動脈内に、胸腔内に、気管支内に、および/または経口でAAVPを投与する。
【0013】
ある種の局面において、陽電子放出断層撮影、ガンマカメラ、または単一光子放出コンピュータ断層撮影によるイメージングに適した放射性同位体で、レポーター基質を標識する。レポーター基質および/またはレポーター基質の代謝物は、2H、13C、15N、および19Fを含むがこれらに限定されない安定同位体核種を含む化合物である。さらなる局面において、標識されたレポーター代謝物を陽電子放出断層撮影によって画像化する。なおさらなる局面において、標識されたレポーター代謝物をガンマカメラまたは単一光子放出コンピュータ断層撮影によって画像化する。さらに、なおさらなる局面において、標識されたレポーター基質代謝物を、磁気共鳴イメージングによって画像化する。
【0014】
AAVPベクターは、レポーター遺伝子ならびに適した転写プロモーターおよびエンハンサーエレメントを組み入れ、レポーターおよび治療的遺伝子共発現の組織特異的、組織選択的、または転写因子特異的、またはシグナル伝達特異的な転写活性化を確実にしてもよい。ある種の局面において、対象への複数の細胞または1つの細胞の投与の前に、器官、組織、複数の細胞、または1つの細胞に、プロモーターおよび/またはエンハンサーエレメントなどの転写調節エレメントが機能的に連結されたレポーターを、エクスビボ(インビトロ)でトランスフェクトする。標識された2'-フルオロ-ヌクレオシド類似体には、5-[123I]- 2'-フルオロ-5-ヨード-1β-D-アラビノフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨード-1β-D-アラビノフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨード-1β-D-アラビノフラノシル-ウラシル、5-[18F]-2'-フルオロ-5-フルオロ-1-β-D-アラビノフラノシル-ウラシル;2-[131I]-2'-フルオロ-5-メチル-1-β-D-アラビノフラノシル-ウラシル;5-([11C]-メチル)-2'-フルオロ-5-メチル-1-β-D-アラビノフラノシル-ウラシル;2-[11C]-2'-フルオロ-5-エチル-1-β-D-アラビノフラノシル-ウラシル;5-([11C]-エチル)-2'-フルオロ-5-エチル-1-β-D-アラビノフラノシル-ウラシル;5-(2-[18F]-エチル)-2'-フルオロ-5-(2-フルオロ-エチル)-1-β-D-アラビノフラノシル-ウラシル、5-[123I]- 2'-フルオロ-5-ヨードビニル-1-β-D-アラビノフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨードビニル-1-β-D-アラビノフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨードビニル-1-β-D-アラビノフラノシル-ウラシル;5-[123I]- 2'-フルオロ-5-ヨード-1-β-D-リボフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨード-1-β-D-リボフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨード-1-β-D-リボフラノシル-ウラシル;5-[123I]-2'-フルオロ-5-ヨードビニル-1-β-D-リボフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨードビニル-1-β-D-リボフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨードビニル-1-β-D-リボフラノシル-ウラシル;または9-4-[18F]フルオロ-3-(ヒドロキシメチル)ブチル]グアニンが含まれるが、これらに限定されない。
【0015】
イメージングデータは、磁気共鳴イメージング(MRI)、核医学、陽電子放出断層撮影(PET)、コンピュータ断層撮影(CT)、超音波検査(US)、光学イメージング、赤外線イメージング、インビボ顕微鏡検査、およびX線ラジオグラフィーによって得られたイメージングを具象化することができるが、これらに限定されない。医療機器、薬物、もしくは化合物、造影剤、またはイメージングから追加の情報を引き出すのに使用し得るその他の薬剤もしくは刺激とイメージングとを組み合わせることができる。画像は、病変、組織、標本、系、生命体、対象、または患者に関するこれらの撮画様式を用いて得られ、時間および/または空間の両方において静的または動的な画像であることができる。
【0016】
大規模な生物学的データが得られる組織、標本、系、生命体、または患者にイメージングを適合させることができる。イメージング情報は、各画像、1つもしくは複数のイメージング検討、またはイメージング調査から抽出され、および病変、組織、標本、系、生命体、または患者の形態、組成、構造、生理、遺伝子発現、または機能の違いを具象化し得るが、これらに限定されない、定量的または定性的なイメージング特色からなることができる。イメージング情報の例として、多相造影増強ダイナミックCT、機能的イメージング、磁気共鳴分光法、拡散テンソルイメージング、拡散または灌流に基づくイメージングだけでなく、核医学またはPETによってカプセル化された標的化イメージングからも抽出し得るイメージング特色が含まれるが、これに限定されない。例えば、その全体が参照により本明細書に組み入れられる、米国特許出願公開第20030033616号および第20060223141号を参照されたい。
【0017】
ある種の態様において、本発明は、以下の工程の1つまたは複数を含む対象を処置する方法を含む:
(a)病理学的状態もしくは疾患状態を有する、有することが疑われる、または発症するリスクがある対象に、レポーターをコードする治療的AAVPを投与する工程;および
(b)コードされたレポーターまたはレポーター活性を検出することによって、処置のために標的化された組織または細胞内での治療的AAVPのインサイチュー発現を評価する工程。
【0018】
ある種の局面において、本発明は、標的器官、組織、または細胞でAAVP核酸によって発現される治療的に十分なレベルの治療的遺伝子の発現に基づき、対象に癌処置を施す工程をさらに含むことができる。さらなる局面において、第一の治療的AAVPの発現が治療的に有効なレベルで発現されない場合に、第二の治療的AAVPを投与することができる。第二の治療的AAVPは、第二の標的化リガンドまたはリガンドの組み合わせを含んでもよい。第二のAAVPもまた、標的器官、組織、複数の細胞、または1つの細胞での発現のための第二の制御エレメントを含むことができる。AAVP発現は、レポーターまたはレポーターの活性の非侵襲的検出(例えば、レポータータンパク質によって代謝される標識された基質の検出)によって評価することができる。ある種の局面において、レポーターは治療的タンパク質である。さらなる局面において、治療的タンパク質はプロドラッグ変換酵素である。またさらなる局面において、レポーターは酵素、および特にキナーゼである。ある種の態様において、キナーゼはチミジンキナーゼ、例えば、HSV-tkまたはヒトtk2である。典型的にキナーゼは、検出可能に標識された化合物または標識された基質を修飾または代謝する。ある種の局面において、基質または化合物は、蛍光、化学発光、表面増強ラマン分光法(SERS)、磁気共鳴イメージング(MRI)、コンピュータ断層撮影(CT)、または陽電子放出断層撮影(PET)イメージングによって検出できる検出可能標識を含む。ある種の局面において、検出可能に標識された化合物は、ヌクレオシド類似体である。検出可能に標識された化合物には、フルオロデオキシグルコース(FDG);2'-フルオロ-2'デオキシ-1ベータ-D-アラビオノフラノシル-5-エチル-ウラシル(FEAU);5-[123I]- 2'-フルオロ-5-ヨード-1β-D-アラビノフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨード-1β-D-アラビノフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨード-1β-D-アラビノフラノシル-ウラシル、5-[18F]-2'-フルオロ-5-フルオロ-1-β-D-アラビノフラノシル-ウラシル;2-[11I]-および5-([11C]-メチル)-2'-フルオロ-5-メチル-1-β-D-アラビノフラノシル-ウラシル;2-[11C]-2'-フルオロ-5-エチル-1-β-D-アラビノフラノシル-ウラシル;5-([11C]-エチル)-2'-フルオロ-5-エチル-1-β-D-アラビノフラノシル-ウラシル;5-(2-[18F]-エチル)-2'-フルオロ-5-(2-フルオロ-エチル)-1-β-D-アラビノフラノシル-ウラシル、5-[123I]- 2'-フルオロ-5-ヨードビニル-1-β-D-アラビノフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨードビニル-1-β-D-アラビノフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨードビニル-1-β-D-アラビノフラノシル-ウラシル;5-[123I]- 2'-フルオロ-5-ヨード-1-β-D-リボフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨード-1-β-D-リボフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨード-1-β-D-リボフラノシル-ウラシル;5-[123I]-2'-フルオロ-5-ヨードビニル-1-β-D-リボフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨードビニル-1-β-D-リボフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨードビニル-1-β-D-リボフラノシル-ウラシル;または9-4-[18F]フルオロ-3-(ヒドロキシメチル)ブチル]グアニンが含まれるが、これらに限定されない。
【0019】
さらなる局面において、標識された基質または化合物を

で標識することができる。特定の局面において、検出可能な標識は、131I、125I、123I、111I、99mTc、90Y、186Re、188Re、32P、153Sm、67Ga、201Tl、77Br、または18F標識である。
【0020】
本発明のAAVPは、処置のために標的化される組織または細胞を選択的に標的化する部分を含んでもよい。ある種の局面において、部分は、AAVPのカプシドタンパク質および/もしくは組換えカプシドタンパク質によってコードされるか、またはAAVPのカプシドタンパク質および/もしくは組換えカプシドタンパク質に連結されている。ある種の局面において、カプシドタンパク質は、標的化ペプチドを含む。標的化ペプチドは、環状ペプチド、二環状、および/または直線状ペプチドであることができる。標的化ペプチドは、細胞表面にインテグリンを発現する細胞に選択的に結合する。インテグリンは、αvβ3またはαvβ5インテグリンであることができる。さらなる局面において、ペプチドはRGDモチーフを含む。またさらなる局面において、ペプチドはトランスフェリン受容体を発現する細胞に選択的に結合することができ、そのようなペプチドは、CRTIGPSVCを含むアミノ酸配列を含むことができる。
【0021】
ある種の局面において、対象は、過剰増殖性疾患を有する、有することが疑われる、または発症するリスクがある。過剰増殖性疾患には、線維肉腫、筋肉腫、脂肪肉腫、軟骨肉腫、骨原性肉腫、軟骨腫、血管肉腫、内皮肉腫、リンパ管肉腫、リンパ管内皮肉腫、滑膜腫、中皮腫、ユーイング腫瘍、平滑筋肉腫、横紋筋肉腫、胃癌、食道癌、直腸癌、膵臓癌、卵巣癌、前立腺癌、子宮癌、頭部および頸部の癌、皮膚癌、脳癌、扁平上皮細胞癌腫、脂腺癌腫、乳頭癌腫、乳頭腺癌、嚢胞腺癌、髄様癌、気管支原性癌腫、腎細胞癌腫、肝細胞癌、胆管癌腫、絨毛癌腫、精上皮腫、胎児性癌腫、ウィルムス腫瘍、子宮頸癌、精巣癌、小細胞肺癌腫、非小細胞肺癌腫、膀胱癌腫、上皮性癌腫、神経膠腫、星状細胞腫、髄芽細胞腫、頭蓋咽頭管腫、上衣細胞腫、松果体腫、血管芽細胞腫、聴神経腫、希突起神経膠腫、髄膜腫、メラノーマ、神経芽細胞腫、網膜芽細胞腫、白血病、リンパ腫、またはカポジ肉腫が含まれるが、これらに限定されない。特定の局面において、過剰増殖性疾患は神経膠腫である。
【0022】
さらなる態様において、レポーターおよび/または治療的遺伝子は、組織もしくは細胞選択的プロモーター、または組織もしくは細胞特異的プロモーターに機能的に連結されている。ある種の局面において、発現を評価する工程は、標識された化合物または基質(AAVP核酸を発現する細胞によって代謝され、典型的には非標的組織によって著しい程度までは代謝されない)を投与する工程を含む。
【0023】
治療的AAVPはまた、第二の治療的遺伝子をコードしてもよい。第二の治療的遺伝子は、腫瘍抑制因子、阻害RNA、阻害DNA、またはプロドラッグ変換酵素であることができるが、これらに限定されない。
【0024】
なおさらなる態様において、本発明の組成物は、阻害RNAまたは阻害DNAを含む核酸セグメントを含む治療的AAVP核酸を含むことができる。阻害RNAは、siRNA、miRNA、またはアンチセンスRNAもしくはDNAであることができる。ある種の局面にでは、AAVP核酸はファージ粒子中に含まれる。さらなる局面では、粒子は本明細書に記載するような標的化リガンドを含む。
【0025】
ある種の態様には、AAVP核酸またはそのようなものを含む粒子を投与する工程を含む遺伝子の発現を調整するための組成物および方法が含まれる。
【0026】
本発明に従って、選択された遺伝子またはポリペプチドは、任意のタンパク質、ポリペプチド、またはペプチドを指してもよい。治療的遺伝子またはポリペプチドは、疾患を処置または防止する目的で対象に投与することができる遺伝子またはポリペプチドである。例えば、治療的遺伝子は、癌の処置または防止のために対象に投与される遺伝子であることができる。治療的遺伝子の例として、Rb、CFTR、p16、p21、p27、p57、p73、C-CAM、APC、CTS-1、zac1、scFV ras、DCC、NF-1、NF-2、WT-1、MEN-I、MEN-II、BRCA1、VHL、MMAC1、FCC、MCC、BRCA2、IL-1、IL-2、IL-3、IL-4、IL-5、IL-6、IL-7、IL-8、IL-9、IL-10、IL-11、IL-12、GM-CSF、G-CSF、チミジンキナーゼ、Bax、Bak、Bik、Bim、Bid、Bad、Harakiri、Fas-L、mda-7、fus、インターフェロンα、インターフェロンβ、インターフェロンγ、p53、ABLI、BLC1、BLC6、CBFA1、CBL、CSFIR、ERBA、ERBB、EBRB2、ETS1、ETS2、ETV6、FGR、FOX、FYN、HCR、HRAS、JUN、KRAS、LCK、LYN、MDM2、MLL、MYB、MYC、MYCL1、MYCN、NRAS、PIM1、PML、RET、SRC、TAL1、TCL3、YES、MADH4、RB1、TP53、WT1、TNF、BDNF、CNTF、NGF、IGF、GMF、aFGF、bFGF、NT3、NT5、ApoAI、ApoAIV、ApoE、Rap1A、シトシンデアミナーゼ、Fab、ScFv、BRCA2、zac1、ATM、HIC-1、DPC-4、FHIT、PTEN、ING1、NOEY1、NOEY2、OVCA1、MADR2、53BP2、IRF-1、zac1、DBCCR-1、rks-3、COX-1、TFPI、PGS、Dp、E2F、ras、myc、neu、raf、erb、fms、trk、ret、gsp、hst、abl、E1A、p300、VEGF、FGF、トロンボスポンジン、BAI-1、GDAIF、またはMCCが含まれるが、これらに限定されない。
【0027】
治療的遺伝子のその他の例として、酵素をコードする遺伝子が含まれる。例として、ACPデサチュラーゼ、ACPヒドロキシラーゼ、ADP-グルコースピロホリラーゼ(pyrophorylase)、ATPアーゼ、アルコールデヒドロゲナーゼ、アミラーゼ、アミログルコシダーゼ、カタラーゼ、セルラーゼ、シクロオキシゲナーゼ、デカルボキシラーゼ、デキストリナーゼ、エステラーゼ、DNAポリメラーゼ、RNAポリメラーゼ、ヒアルロンシンターゼ、ガラクトシダーゼ、グルカナーゼ、グルコースオキシダーゼ、GTPアーゼ、ヘリカーゼ、ヘミセルラーゼ、ヒアルロニダーゼ、インテグラーゼ、インベルターゼ、イソメラーゼ、キナーゼ、ラクターゼ、リパーゼ、リポオキシゲナーゼ、リアーゼ、リゾチーム、ペクチンエステラーゼ、ペルオキシダーゼ、ホスファターゼ、ホスホリパーゼ、ホスホリラーゼ、ポリガラクツロナーゼ、プロテイナーゼ、ペプチダーゼ、プラナーゼ、リコンビナーゼ、リバーストランスクリプターゼ、トポイソメラーゼ、キシラナーゼ、レポーター遺伝子、インターロイキン、またはサイトカインが含まれるが、これらに限定されない。
【0028】
治療的遺伝子のさらなる例として、カルバモイルシンセターゼI、オルニチントランスカルバミラーゼ、アルギノスクシネートシンセターゼ、アルギノスクシネートリアーゼ、アルギナーゼ、フマリルアセトアセテートヒドロラーゼ、フェニルアラニンヒドロキシラーゼ、アルファ-1アンチトリプシン、グルコース-6-ホスファターゼ、低密度リポタンパク質受容体、ポルホビリノーゲンデアミナーゼ、第VIII因子、第IX因子、シスタチオンベータ-シンターゼ、分岐鎖ケト酸デカルボキシラーゼ、アルブミン、イソバレリル-CoAデヒドロゲナーゼ、プロピオニルCoAカルボキシラーゼ、メチルマロニルCoAムターゼ、グルタリルCoAデヒドロゲナーゼ、インスリン、-グルコシダーゼ、ピルビン酸カルボキシラーゼ、肝ホスホリラーゼ、ホスホリラーゼキナーゼ、グリシンデカルボキシラーゼ、H-タンパク質、T-タンパク質、メンケス病銅輸送ATPアーゼ、ウィルソン病銅輸送ATPアーゼ、シトシンデアミナーゼ、ヒポキサンチン-グアニンホスホリボシルトランスフェラーゼ、ガラクトース-1-リン酸ウリジルトランスフェラーゼ、フェニルアラニンヒドロキシラーゼ、グルコセルブロシダーゼ、スフィンゴミエリナーゼ、-L-イズロニダーゼ、グルコース-6-リン酸デヒドロゲナーゼ、HSVチミジンキナーゼ、またはヒトチミジンキナーゼをコードする遺伝子が含まれる。
【0029】
治療的遺伝子には、ホルモンをコードする遺伝子も含まれる。例として、成長モルモン、 プロラクチン、胎盤ラクトゲン、黄体形成ホルモン、濾胞刺激ホルモン、絨毛性ゴナドトロピン、甲状腺刺激ホルモン、レプチン、副腎コルチコトロピン、アンギオテンシンI、アンギオテンシンII、β-エンドルフィン、β-メラニン細胞刺激ホルモン、コレシストキニン、エンドセリンI、ガラニン、胃阻害ペプチド、グルカゴン、インスリン、リポトロピン、ニューロフィジン、ソマトスタチン、カルシトニン、カルシトニン遺伝子関連ペプチド、β-カルシトニン遺伝子関連ペプチド、悪性の高カルシウム血症因子、副甲状腺ホルモン関連タンパク質、副甲状腺ホルモン関連タンパク質、グルカゴン様ペプチド、パンクレアスタチン、膵ペプチド、ペプチドYY、PHM、セクレチン、血管作用性小腸ペプチド、オキシトシン、バソプレッシン、バソトシン、エンケファリンアミド、メトルフィンアミド、アルファメラニン細胞刺激ホルモン、心房性ナトリウム利尿因子、アミリン、アミロイドP成分、コルチコトロピン放出ホルモン、成長モルモン放出因子、黄体形成ホルモン放出ホルモン、神経ペプチドY、サブスタンスK、サブスタンスP、またはチロトロピン放出ホルモンをコードする遺伝子が含まれるが、これらに限定されない。
【0030】
本発明のその他の態様は本出願の全体を通して考察される。本発明のある局面に関して考察された任意の態様は、本発明のその他の局面に同様に当てはまり、かつ逆もまた同様である。実施例の項における態様は、本発明の全ての局面に適用可能である本発明の態様であると理解される。
【0031】
本特許請求の範囲および/または本明細書で用いる場合、「阻害すること」、「低下させること」、もしくは「防止」という用語、またはこれらの用語の任意の変形には、所望の結果を達成するための任意の測定可能な減少または完全な阻害が含まれる。
【0032】
本特許請求の範囲および/または本明細書における「含む」という用語と併せて用いられる場合の「1つの(a)」または「1つの(an)」という語の使用は、「1つの(one)」を意味し得るが、それは「1つまたは複数の」、「少なくとも1つの」、および「1つまたは1つよりも多くの」という意味とも一致する。
【0033】
本明細書で考察する任意の態様を本発明の任意の方法および組成物に関して実行することができ、ならびに逆もまた同様であることが企図される。さらに、本発明の組成物およびキットを用いて本発明の方法を達成することができる。
【0034】
本明細書の全体を通じて、「約」という用語を用いて、ある値が、値を決定するために利用する機器または方法についての誤差の標準偏差を含むことを示す。
【0035】
本特許請求の範囲における「または」という用語の使用は、代替物だけを指すことが明瞭に示され、または代替物が相互排他的であるのでなければ、「および/または」を意味するために用いられるが、本開示は、唯一の代替物および「および/または」を指す定義を支持する。
【0036】
本明細書および本特許請求の範囲において用いられる場合、「含む(comprising)」という語(ならびに「含む(comprise)」および「含む(comprises)」などの、含む(comprising)の任意の形態)、「有する(having)」という語(ならびに「有する(have)」および「有する(has)」などの、有する(having)の任意の形態)、「含む(including)」という語(ならびに「含む(includes)」および「含む(include)」などの、含む(including)の任意の形態)、「含む(containing)」という語(ならびに「含む(contains)」および「含む(contain)」などの、含む(containing)の任意の形態)は、包括的または開放型であり、かつ追加の列挙されない要素または方法工程を除外しない。
【0037】
本発明の他の目的、特色、および利点は、以下の詳細な説明から明らかになると考えられる。しかしながら、本発明の精神および範囲の範囲内の様々な変更および修正がこの詳細な説明から当業者に明らかになると考えられるので、詳細な説明および具体的な実施例は、本発明の具体的な態様を示す一方で、例証のみのために与えられることが理解されるべきである。
【0038】
本開示は様々な修正および代替の形態を受け入れることができるが、具体的な実施例態様を、図に示しかつ本明細書でより詳細に記載する。しかしながら、具体的な実施例態様の説明は、開示する特定の形態に本発明を限定するよう意図せず、対照的に本開示は、一部添付の特許請求の範囲によって例証するような全ての修正および同等物を包含するためのものであることが理解されるべきである。
【0039】
発明の詳細な説明
ある種の態様による本開示は、増強された発現を達成するために、腫瘍細胞などの標的細胞に、1つまたは複数のトランスジーンを部位特異的な様式で送達する方法、ならびにそのような適用において有用なコンストラクトおよび組成物に概ね向けられている。ある種の局面において、対象への処置の投与または対象に対する診断手順の前に、治療的核酸からの発現を検討評価してもよい。さらなる局面において、治療的利益が必要とされる領域または場所における発現の決定または評価を検討評価し、任意の不必要なまたは必要最低限の有益な処置を代替処置の代わりに認めることができる。
【0040】
理論またはメカニズムに束縛されることを望まないが、本開示は、トランスジーンが1より大きい多重度でゲノムに組込まれる場合、トランスジーン発現は増加し得るという観察に基づく。特に興味深いのは、ある種のキメラAAVP粒子が、多くの場合コンカテマーとして、1コピーより多くのトランスジーンを細胞に形質導入する能力である。キメラAAVP粒子によって運ばれるレポーター遺伝子の発現によって形質導入された細胞をモニタリングしてもよい。任意のトランスジーンを本開示のAAVP粒子中に含め、本開示のAAVP粒子から発現させてもよい。
【0041】
I、アデノ随伴ウイルス/ファージ(AAVP)粒子
アデノ随伴ウイルス(AAV)はパルボウイルスファミリーの欠損性のメンバーである。AAVゲノムは、プラスまたはマイナス極性の1本鎖DNA分子としてカプセル化されている。両方の極性の鎖がパッケージングされているが、別々のウイルス粒子中にあり、かつ両方の鎖が感染性である。ヒトアデノ随伴ウイルス2型(AAV2)の1本鎖DNAゲノムは、長さが4681塩基対であり、かつ各々145塩基対の逆向き末端反復配列(ITR)が両隣にある。さらに、ウイルスのrepタンパク質は、ITRによる非相同組換えを媒介するように見える。したがって、組換えで役割を果たしかつパルボウイルスの複製およびパッケージングに通常必要とされるITRを、パルボウイルスゲノムが各末端に有する時、本開示のAAVPは通常、ITRまたはその機能的同等物の少なくとも1つの全てまたは一部分を含む。
【0042】
AAVは容易に得られ、遺伝子送達用のベクターとしてのそれらの使用は例えば、Muzyczka, 1992;米国特許第4,797,368号、およびPCT公開公報WO 91/18088に記載されている。AAVベクターの構築は、Lebkowski et al., 1988;Tratschin et al., 1985;Hermonat and Muzyczka, 1984を含む、多数の刊行物に記載されている。
【0043】
本開示は、細菌中で産生される(AAV-M13ベクターなどの)アデノ随伴ウイルス(AAV)バクテリオファージベクター(AAVP)および細胞にAAVPを形質導入することによって、腫瘍細胞などの標的細胞でトランスジーンを発現させるための方法を提供する。一旦精製し、バクテリオファージおよびトランスジーンカセットを持つAAV配列を含む標的化されたバクテリオファージ粒子を用いて、哺乳動物細胞にトランスフェクトする。哺乳動物細胞内へのベクターの内在化の後、トランスジーンは標的細胞のゲノムに組込まれる。本明細書で用いる場合の「ベクター」という用語は、関心対象の核酸の細胞内への送達のための核酸ビヒクルとして定義される。ベクターは、直線状分子または環状分子であってもよい。
【0044】
AAVPは、ファージおよびAAVベクター系の両方の選択されたエレメントを組み合わせ、それによりパッケージング制限のない細菌中で産生し易く、その一方で宿主染色体への組み込みと組み合わせた哺乳動物細胞の感染を可能にするベクターを提供する。適当なエレメントの多くを含むベクターは市販されており、かつ必要な配列を含むよう標準的な方法論によってさらに改変することができる。最低限、本開示の方法を用いた使用のために、ベクターは、プロモーターおよびトランスジーンを含むカセットを受容しなければならない。本開示のAAVPベクターは、標的細胞ゲノムへの組み入れ時に増強されたトランスジーン発現を可能にする。ある種の態様において、トランスジーンはコンカテマーとして標的細胞のゲノムに組み込まれてもよい。
【0045】
とりわけ、AAVPはヘルパーウイルスまたはトランス作用因子を必要としない。さらに、AAVカプシド形成がないので、哺乳動物細胞に対するAAVの天然の向性は消去される。
【0046】
A、AAVP標的化
ファージ粒子の表面上のリガンドの発現によって、本開示のAAVPを特異的な受容体に標的化することができる。ある種の態様において、エンドソームからのベクターの脱出を可能にしまたは促進するペプチドまたはその他の部分(およびそこに付着するかまたはその中に封入された任意の分子)を組み入れ、かつファージの表面に発現させてもよい。そのような「その他の部分」には、それら自体ペプチドではないが、エンドソーム膜を乱す能力を有し、それによってベクターの脱出を容易にする分子、およびさもなければ内部に記載されたペプチド配列のエンドソーム脱出特性を模倣する分子が含まれる(例えば、公開されたPCT公開公報WO 96/10038およびWagner et al., 1992参照)。
【0047】
本開示のAAVPは通常、リガンドが付着している様式に関係なく、1つまたは複数の予め選択されたリガンドを粒子表面に発現する線維状ファージ粒子から構成される。それゆえ、リガンドへの付着の手段が共有結合であるかまたはカプシドタンパク質を介するものかのいずれでも、本開示のAAVPは、受容体へのリガンド結合、それに続くベクターの内在化によって、1つまたは複数のトランスジーンを標的細胞に送達することができる。例えば、粒子表面に発現したリガンド粒子は、αvβ3およびαvβ5インテグリンに選択的に結合する二環状のCDCRGDCFC(RGD-4C)(SEQ ID NO:2)ペプチドであってもよい。これらのインテグリンは、侵入性の腫瘍内皮細胞上に高度に過剰発現している。本明細書で用いる場合、「繊維状ファージ粒子」は、ファージゲノムかまたはファージミドゲノムのいずれかを含む粒子を指す。繊維状カプシドタンパク質に加えて、粒子はその他の分子を含んでもよい。本明細書で用いる場合、「リガンド」は、細胞表面分子に結合しかつ内在化することができる、任意のペプチド、ポリペプチド、タンパク質、またはペプチドミメティックなどの非タンパク質を指す。本明細書で用いる場合、「受容体への結合」は、標準的なインビトロまたはインビボアッセイによってアッセイされた場合の、受容体を特異的に認識し、かつ受容体に検出可能に結合するリガンドの能力を指す。
【0048】
典型的には、本開示のAAVPは、ベクターのリガンド-受容体標的化特性を可能にする標的化ペプチドをコードするオリゴヌクレオチド挿入物を、ファージプラスミドゲノム中に含む。
【0049】
カプシドタンパク質ポリヌクレオチドの全てもしくは一部を標的化リガンドに連結するか、または該ポリヌクレオチドによってコードされるタンパク質の全てまたは一部を標的化リガンドに融合させることによって、ファージカプシドタンパク質またはカプシドタンパク質を改変してもよい。標的化リガンドは、本発明のAAVPの特異的な細胞、組織、および/または器官への結合を導き、導き直し、標的化し、または増強してもよい。
【0050】
標的化されるウイルスは元々、遺伝子治療ベクターの天然の宿主細胞向性により直面する問題を克服するために創り出された。近年、多くの遺伝子治療特許が出されており、キメラ融合糖タンパク質を含むベクター(参照により本明細書に組み入れられる、Kaymanら、米国特許第5,643,756号)およびウイルスカプシドタンパク質に対する抗体を含むベクター(Cottenら、米国特許第5,693,509号)などのベクターは、特異的な細胞にベクターを標的化するのに用いられる異種ポリペプチドを含む。核酸ゲノムが標的非分裂細胞、標的分裂細胞、または増殖性もしくはその他の障害を有する標的細胞に送達されるように、特定の細胞タイプ(例えば、平滑筋細胞、肝細胞、腎細胞、線維芽細胞、ケラチン生成細胞、幹細胞、間葉系幹細胞、骨髄細胞、軟骨細胞、上皮細胞、小腸細胞、新生物性または癌性細胞、および当技術分野において公知のその他の細胞)に粒子が標的化されるように本発明のAAVPを遺伝子改変してもよい。ウイルスを標的化する1つの方法は、細胞の外部表面に分子を有する細胞に優先的に結合させることによって、ウイルスを標的細胞に導くことである。ウイルスを標的化するこの方法は、ウイルスの表面上の標的化リガンドと相互作用する受容体または結合分子を有する細胞または組織に、ウイルスを標的化するのを手助けするために、ウイルスのカプシド上での標的化リガンドの発現またはウイルスのカプシドへの標的化リガンドの組み入れを活用している。ウイルスによる細胞の感染後、遺伝子材料は宿主細胞で処理および発現されることができる。遺伝子材料は宿主細胞のゲノムに組み込まれまたは宿主細胞内でエピソームとして維持されてもよい。
【0051】
本発明のある種の態様において、AAVPを特異的な細胞タイプまたは組織に送達し得るように、カプシドタンパク質を改変して標的化部分を含めてもよい。リガンドに基づく送達系の標的化特異性は、異なる細胞タイプ上のリガンド受容体の分布に基づく。標的化リガンドは、非共有結合または共有結合のいずれかでカプシドタンパク質に会合していてもよい。
【0052】
ある種の態様において、関心対象の異種核酸配列を本発明のウイルスベクターに挿入してもよい。例えば、カプシドタンパク質は、特異的な標的細胞上の受容体に対するリガンドに機能的に連結されてもよい。
【0053】
標的化リガンドは、標的化される領域の特徴的構成要素に特異的な任意のリガンドである。好ましい標的化リガンドには、ポリクローナルもしくはモノクローナル抗体、抗体断片、もしくはキメラ抗体、酵素、ペプチド、もしくはホルモンなどのタンパク質、または単糖類、オリゴ糖類、もしくは多糖類などの糖が含まれる。本発明のある種の態様において、企図される標的化リガンドは、インテグリン、プロテオグリカン、糖タンパク質、受容体、または輸送体と相互作用する。適したリガンドには、標的器官の細胞に、または腫瘍など局部的な病理の結果として循環に曝露される標的器官の構造に、特異的または選択的である任意のものが含まれる。
【0054】
本発明のある種の態様において、耐性の細胞の形質導入を増強させ、標的細胞の形質導入を増加させ、または非所望の細胞の形質導入を限定するために、抗体または環状ペプチド標的化部分(リガンド)をAAVPと会合させてもよい。特定の例における抗体標的化部分は、モノクローナル抗EGF受容体抗体である。EGF受容体は、様々な器官の細胞表面に分布し、かつ火傷、創傷、真皮、および腫瘍に存在する。ペプチド標的化部分はまた、その配列内にRGDインテグリン結合モチーフを含む環状ペプチドであってもよい。細胞表面上のインテグリンに結合するRGDペプチドなどのリガンドは内在化を媒介し、したがって標的化される複合体の送達の効率性を増加させることができる。RGD配列を含み、ペプチドが3〜30アミノ酸長である標的化配列は、RGDFV(SEQ ID NO:3)配列を含んでもよい。その他の態様において、RGDインテグリン結合モチーフは、3〜20アミノ酸長または4〜10アミノ酸長である。本発明の特定の態様において、RGDインテグリン結合モチーフは、5アミノ酸長のペプチドである。その配列内にRGDインテグリン結合モチーフを含む環状ペプチドが好ましいが、直線状ペプチドも本発明において活用してもよい。ヒト癌の診断および治療用の標的化ペプチドの使用に関する第20060239968号の組成物および方法。米国特許出願公開第20060094672号、第20050191294号、第20050187161号、第20050074812号、第20050074747号、第20050037417号、第20050003466号、第20040170955号、第20040131623号、第20040096441号、第20040071689号、第20040048243号、第20030152578号、第20030113320号、および第20010046498号だけでなく、PCT公開公報WO 2006/060171、WO 2006/010070、WO 2005/065418、WO 2005/026195、WO 2004/020999、WO 2003/022991、WO 2002/020822、WO 2002/020769、WO 2002/020723、およびWO 2002/020722も、標的化リガンドを同定する様々な組成物および方法を記載しており、その各々はその全体が参照により本明細書に組み入れられる。
【0055】
本明細書で用いる場合、「リガンド」は、細胞表面分子に結合しかつ内在化することができる、任意のペプチド、ポリペプチド、タンパク質、またはペプチドミメティックなどの非タンパク質を指す。本明細書で用いる場合、「受容体に結合する」とは、標準的なインビトロまたはインビボアッセイによってアッセイされた場合の、受容体を特異的に認識し、受容体に検出可能に結合するリガンドの能力を指す。
【0056】
本発明の文脈中で、リガンドは、融合タンパク質としてまたは化学的コンジュゲーションを通じてのいずれかで、ファージのタンパク質(例えば、カプシドタンパク質)に連結され、細胞に核酸を送達するのに用いられる。断片が適当な細胞表面分子に結合する能力を保持する限り、リガンドの断片を本発明の範囲内で用いてもよい。同様に、置換またはその他の変更はあるが、結合能力を保持するリガンドも用いてよい。なおその上、特定のリガンドは、天然のリガンドのアミノ酸配列を有するポリペプチドだけでなく、リガンドが内皮細胞上のその受容体に結合し、細胞への核酸の送達を結果的にもたらす能力を保持する限り、改変された配列(例えば、天然のタンパク質(ムテイン(mutein))と比較してアミノ酸置換、欠失、挿入、または付加を有する)も指す。
【0057】
リガンドは、その受容体を発現する細胞に結合しかつ内在化される能力を保有するムテインまたは突然変異体タンパク質も包含する。そのようなムテインには、システインの1つまたは複数をセリンと置き換えることによって産生されたものが含まれるが、これに限定されない。典型的には、そのようなムテインは、保存的アミノ酸変化を有すると考えられる。そのようなムテインをコードするDNAは、縮重コドンの置換によって改変されない限り、少なくとも低ストリンジェンシーの条件下で野生型リガンドをコードする天然のDNA配列にハイブリダイズすると考えられる。
【0058】
リガンドをコードするDNAを、公知のアミノ酸もしくはDNA配列に基づいて合成により調製し、当業者に公知の方法(例えば、PCR増幅)を用いて単離し、または市販もしくはその他の源から得てもよい。リガンドをコードするDNAは、縮重コドンの置換によって、または異なるアミノ酸をコードすることによって、上記の配列と異なってもよい。異なる種の相同なリガンド間だけでなく、個々の生命体または種間でも生じる違いなどアミノ酸配列の違いは、リガンドがその受容体に結合する限り許容される。リガンドを天然源から単離するか、または合成(組換え手段もしくは化学合成など)により作製してもよい。
【0059】
本発明の文脈で用いられるリガンドが細胞上の受容体に結合しかつ内在化されること以外に、そのインビボの生物学的活性のいずれかを保持することは必要ではない。リガンドが1つまたは複数の生物学的活性を欠くように改変されていても、例えば、以下の検査または当技術分野において公知のその他の検査によって、結合および内在化をなお容易にアッセイし得る。通常、これらの検査は、リガンドを標識する工程、それを標的細胞とインキュベートする工程、および細胞内の標識を可視化または測定する工程を伴う。例えば、簡潔に述べると、リガンドをFITCで蛍光標識または125Iで放射性標識し、細胞とインキュベートし、内在化について蛍光顕微鏡または共焦点顕微鏡で顕微鏡的に調べてもよい。
【0060】
ファージカプシドタンパク質への付着に備えて、リガンドを組換えまたはその他の手段によって産生してもよい。DNA配列およびこれらのリガンドの配列を得るための方法は周知である。DNA配列に基づいて、遺伝子を(小さいタンパク質について)合成的に合成する、細胞ゲノムDNAまたはcDNAから増幅する、ゲノムDNAまたはcDNAライブラリーなどから単離してもよい。ファージまたはファージミドベクターへのクローニングを容易にするための制限部位を、増幅用のプライマー中などに組み入れてもよい。
【0061】
そのような分子には、癌細胞、内皮細胞、ストローマ細胞、およびそれらと同様の細胞に結合するタンパク質が含まれるが、これらに限定されない。そのようなリガンドには、成長因子およびサイトカインが含まれる。多くの成長因子および成長因子のファミリーは、構造的および機能的特色を共有し、本発明において使用され得る。成長因子のファミリーには、線維芽細胞成長因子FGF-1からFGF-15、および血管内皮成長因子(VEGF)が含まれる。PDGF(血小板由来成長因子)、TGF-α(形質転換成長因子)、TGF-β、HB-EGF、アンギオテンシン、ボンベシス、エリスロポイエチン、幹細胞因子、M-CSF、G-CSF、GM-CSF、およびエンドグリンなどその他の成長因子も、細胞表面上の特異的な同定された受容体に結合し、本発明において使用され得る。インターロイキン、CSF(コロニー刺激因子)、およびインターフェロンを含むサイトカインは、特異的な受容体を有し、本明細書で記載するように使用され得る。
【0062】
例えば、リガンドおよびリガンド/受容体対には、ウロキナーゼ/ウロキナーゼ受容体(GenBankアクセッション番号X02760/X74309);α-1,3フコシルトランスフェラーゼ、α1-アンチトリプシン/E-セレクチン(GenBankアクセッション番号M98825、D38257/M87862);P-セレクチン糖タンパク質リガンド、P-セレクチンリガンド/P-セレクチン(GenBankアクセッション番号U25955、U02297/L23088)、VCAM1/VLA-4(GenBankアクセッション番号X53051/X16983);E9抗原(Blann et al., Atherosclerosis 120:221, 1996)/TGFβ受容体;フィブロネクチン(GenBankアクセッション番号X02761);I型α1-コラーゲン(GenBankアクセッション番号Z74615)、I型β2-コラーゲン(GenBankアクセッション番号Z74616)、ヒアルロン酸/CD44(GenBankアクセッション番号M59040);CD40リガンド(GenBankアクセッション番号L07414)/CD40(GenBankアクセッション番号M83312); elk-1(GenBankアクセッション番号M25269)に対するEFL-3、LERTK-2リガンド(GenBankアクセッション番号L37361、U09304);VE-カドヘリン(GenBankアクセッション番号X79981);カテニンに対するリガンド;LFA-1に対するICAM-3(GenBankアクセッション番号X69819)リガンド、ならびにαvβ3インテグリン(GenBankアクセッション番号U07375、L28832)に対するフォンウェルブランド因子(GenBankアクセッション番号X04385)、フィブリノゲン、およびフィブロネクチン(GenBankアクセッション番号X92461)リガンドが含まれる。
【0063】
その他のリガンドには、CSF-1(GenBankアクセッション番号M11038、M37435);GM-CSF(GenBankアクセッション番号X03021);IFN-α(インターフェロン)(GenBankアクセッション番号A02076;WO 8502862-A);IFN-γ(GenBankアクセッション番号A02137;WO 8502624-A);IL-1-α(インターロイキン1アルファ)(GenBankアクセッション番号X02531、M15329);IL-1-β(インターロイキン1ベータ)(GenBankアクセッション番号X02532、M15330、M15840);IL-1(GenBankアクセッション番号K02770、M54933、M38756);IL-2(GenBankアクセッション番号A14844、A21785、X00695、X00200、X00201、X00202);IL-3(GenBankアクセッション番号M14743、M20137);IL-4(GenBankアクセッション番号M13982);IL-5(GenBankアクセッション番号X04688、J03478);IL-6(GenBankアクセッション番号Y00081、X04602、M54894、M38669、M14584);IL-7(GenBankアクセッション番号J04156);IL-8(GenBankアクセッション番号Z11686);IL-10(GenBankアクセッション番号X78437、M57627);IL-11(GenBankアクセッション番号M57765 M37006);IL-13(GenBankアクセッション番号X69079、U10307);TNF-α(腫瘍壊死因子)(GenBankアクセッション番号A21522);TNF-β(GenBankアクセッション番号D12614);erbB2に対するGP30リガンド(S68256);およびトランスフェリン受容体に対するトランスフェリン(GenBankアクセッション番号DQ923758)が含まれる。
【0064】
またその他のリガンドには、PDGF(GenBankアクセッション番号X03795、X02811)、アンギオテンシン(GenBankアクセッション番号K02215)、ならびにインテグリン受容体に結合するICAM-1(GenBankアクセッション番号X06990)およびVCAM-1(GenBankアクセッション番号X53051) など、全てのRGD含有ペプチドおよびタンパク質が含まれる。その他のリガンドには、TNFα(GenBankアクセッション番号A21522、X01394)、IFN-γ(GenBankアクセッション番号A11033、A11034)、IGF-I(GenBankアクセッション番号A29117、X56773、S61841、X56774、S61860)、IGF-II(GenBankアクセッション番号A00738、X06159、Y00693)、心房性ナトリウム利尿ペプチド(GenBankアクセッション番号X54669)、エンドセリン-1(GenBankアクセッション番号Y00749)、凝固因子Xa(GenBankアクセッション番号L00395、L00396、L29433、N00045、M14327)、TGF-β1(GenBankアクセッション番号A23751)、IL-1β(GenBankアクセッション番号X03833)、IL-1β(GenBankアクセッション番号M15330)、およびエンドグリン(GenBankアクセッション番号X72012)が含まれる。
【0065】
FGFタンパク質のファミリーには現在、FGF-1(酸性FGFまたはaFGF)、FGF-2(塩基性FGFまたはbFGF)、FGF-3(int-2)、FGF-4(hst-1/K-FGF)、FGF-5、FGF-6(hst-2)、FGF-7(ケラチン生成細胞成長因子またはKGF)、FGF-8、FGF-9、FGF-11(WO 96/39507)、FGF-13(WO 96/39508)、FGF-14(WO 96/39506)、および FGF-15(WO 96/39509)が含まれる。FGF受容体に反応性であるその他のポリペプチド、すなわちFGF受容体、好ましくは高親和性FGF受容体と特異的に相互作用し、かつFGF受容体との相互作用によって細胞内にエンドソーム経由で輸送される任意のポリペプチドが、本発明の範囲内で適している。リガンドには、本明細書で同定したリガンドの4、5、6、7、8、9、10、15、20、25、30、またはそれより多くのアミノ酸断片も含まれる。
【0066】
抗体およびその断片も標的化部分として企図される。モノクローナル抗体断片を用いて、脳、心臓、肺、または肝臓を含む動物の異的な器官への送達を標的化してもよい。単一の細胞に特異的なマーカーを欠く細胞へウイルス粒子を標的化するための例示的な方法が記載されている(米国特許第5,849,718号)。例えば、抗体Aが腫瘍に対する特異性だけでなく、正常な心臓および肺組織に対する特異性も有する可能性がある一方、抗体Bは腫瘍だけでなく正常な肝臓に対する特異性も有する。明らかに、腫瘍に抗増殖性核酸を送達するための抗体Aまたは抗体B単独の使用は、事によると心臓および肺または肝臓細胞に対する望まない傷害を結果的にもたらすと考えられる。しかしながら、抗体Aおよび抗体Bを、改良された細胞標的化のために一緒に用いることができる。したがって、抗体Aは、抗増殖性核酸をコードする遺伝子に連結され、かつ受容体を介する摂取系によって腫瘍だけでなく心臓および肺組織に送達される。しかしながら、遺伝子は、これらの細胞で転写されずまたは活性がない。抗体Bは、アクチベーターまたは抗増殖性核酸の転写もしくは活性化に必要な因子をコードする普遍的に活性のある遺伝子に連結することができ、腫瘍および肝臓細胞に送達される。それゆえ、心臓および肺細胞では不活性な抗増殖性核酸のみが送達され、そこではそれが転写されず、副作用を引き起こさない。肝臓細胞では、活性化因子をコードする遺伝子が送達されるが、抗増殖性核酸遺伝子が存在しないので効果を有さない。しかしながら、腫瘍細胞では、両方の遺伝子が送達され、かつ活性化因子が抗増殖性核酸を活性化し、腫瘍特異的な毒性効果をもたらすことができる。
【0067】
本発明の文脈中で、細胞の表面に発現する分子に対する抗体は有用である。そのような抗体には、FGF受容体、VEGF受容体、ウロキナーゼ受容体、E-およびP-セレクチン、VCAM-1、PDGF受容体、TGF受容体、エンドシアリン、αvβ3インテグリン、LFA-1、E9抗原、CD40、カドヘリン、ならびにelk-1に対する抗体が含まれるが、これらに限定されない。細胞によって発現される細胞表面分子に特異的である抗体は、モノクローナルまたはポリクローナル抗血清として容易に作製される。多くのそのような抗体が(例えば、American Type Culture Collection, Rockville, Md.)から入手可能である。あるいは、結合/内在化するリガンドに対する抗体も用いてもよい。そのような戦略において、ファージ粒子はそれらの表面に抗体を有すると考えられ、それはその後リガンドと複合体化すると考えられる。
【0068】
AAVP送達のために標的化される部位に応じて、多くのその他のリガンドをAAVP調製物の標的化工程に利用してもよい。ある種の態様において、受容体を介したエンドサイトーシスによって、AAVPを特異的な細胞タイプに標的化することが企図される。例えば、ラクトシルセラミド、およびアポリポタンパク質E3(「Apo E」)などの、LDL受容体関連タンパク質を標的化するペプチドは、肝臓にリポソームを標的化する際に有用である(Spanjer and Scherphof, 1983;WO 98/0748)。アシアロ糖タンパク質である、アシアロフェツイン(末端ガラクトシル残基を含む)もまた、肝臓にリポソームを標的化することが示されている(Spanjer and Scherphof, 1983;Hara et al., 1995)。マンノシル、フコシル、またはN-アセチルグルコサミンという糖は、ポリペプチドの骨格に連結された場合、高親和性マンノース受容体に結合する(その全体が参照により本明細書に具体的に組み入れられる、米国特許第5,432,260号)。したがって、これらの糖タンパク質を、本発明のAAVPにコンジュゲートすることができ、かつ特異的な細胞(例えば、マクロファージ)を標的化するのに有用であることが企図される。
【0069】
葉酸塩および葉酸受容体もまた、細胞標的化に有用であると記載されている(米国特許第5,871,727号)。この例では、ビタミン葉酸塩がAAVPカプシドタンパク質に連結されている。葉酸受容体は、そのリガンドに対する高い親和性を有し、かつ肺、胸部、および脳腫瘍を含む幾つかの悪性細胞株の表面に過剰発現している。トランスフェリンを介した送達系は、トランスフェリン受容体を発現する幅広い範囲の複製細胞を標的化する(Gilliland et al., 1980)。
【0070】
過剰増殖性疾患の処置のための遺伝子送達用の標的化リガンドの付加は、遺伝子産物が標的化されていない系が可能にするよりも毒性のある遺伝子の送達を可能にする。送達することができるより毒性のある遺伝子の例として、BaxおよびBakなどのプロアポトーシス遺伝子に加えて、アデノウイルスE4orf4およびプロドラッグ6-メチルプリンデオキシリボシドを毒性のあるプリン6-メチルプリンに変換するいわゆる「自殺遺伝子」である、大腸菌(E. coli)プリンヌクレオシドホスホリラーゼなどのウイルスおよびその他の病原菌に由来する遺伝子が含まれる。プロドラッグ療法で用いる自殺遺伝子の他の例には、大腸菌シトシン脱アミノ酵素遺伝子およびHSVチミジンキナーゼ遺伝子がある。
【0071】
ある種の局面において、AAVPを用いて腫瘍血管系を標的化することができる。腫瘍内皮は癌治療のための重要な標的である。その他の組織における最小限の毒性を伴って関心対象の治療的遺伝子を腫瘍内皮に標的化することは、依然として抗血管遺伝子治療の第一義的な目標である。近年、腫瘍内皮を標的化するAAVPが記載されている。本発明者らは、このベクターが、強力な抗血管剤であるヒト腫瘍壊死因子-α(TNF-α)をヒトメラノーマに送達する能力を検討した。AAVPによって内皮単球活性化ポリペプチド-II(EMAP-II)を送達することにより、TNF-α耐性のメラノーマがTNF-α処置に対して感受性になった。
【0072】
2つの遺伝子、TNF-αおよびEMAP-IIを運ぶAAVPベクターをインビトロおよびインビボで評価した。ヒトメラノーマ細胞(M21)をAAVP内在化およびTNF-α遺伝子発現についてインビトロで検討した。ヌードマウス内の皮下で成長した腫瘍であるM21/Pmelを、尾静脈注射によって全身的に処置した。免疫蛍光染色、TaqMan RT-PCR、および免疫組織化学的解析を用いて、標的化されたAAVPの腫瘍血管系への局在化、TNF-α遺伝子発現、およびアポトーシスを調べた。
【0073】
標的化されたAAVPの内在化がM21細胞で観察され、培養上清中の高レベルの機能的に活性のあるTNF-αが結果的にもたらされた。標的化されていないベクターの内在化はこれらの細胞で観察されなかった。AAVPの全身注射は、正常器官への最小限のウイルス局在化を伴う腫瘍に標的化されたウイルス送達を示した。AAVP送達は、TNF-α遺伝子産物の発現を結果的にもたらした。TNF-αの発現は、血管および周囲の腫瘍細胞におけるアポトーシスを誘導し、顕著な腫瘍退化を結果的にもたらした。さらに、EMAP-IIを発現するAAVPの標的化された送達は、TNF-α耐性ヒトメラノーマを、TNF-α処置に対して増感させた。標的化されたAAVPベクターを用いて、腫瘍血管系に特異的に抗血管薬剤を送達し、したがって全身毒性を低下させることができる。
【0074】
特異的な標的化リガンドの付着によって、本発明のAAVPを身体の特異的な領域に標的化し、指定された組織におけるAAVPの速やかな蓄積および濃縮、ならびにそれに応じて、核酸分子の速やかな蓄積および濃縮を提供することができる。本発明での使用が企図されるリガンドを、種々の方法でAAVPにコンジュゲートすることができる。標的化リガンドをカプシドタンパク質に連結するための様々な組成物および方法は、当技術分野において公知である。
【0075】
II、核酸
本開示のAAVPは、1つまたは複数のトランスジーンを標的細胞の核に送達し、それによって標的細胞におけるトランスジーンの発現を増強させる能力を有する。AAVPはまた、トランスジーンカセットのコンカテマーの形成を通じたトランスジーンカセットの変更された運命によって遺伝子発現における利点を授与し、それによって増強された遺伝子発現をもたらす可能性がある。
【0076】
本明細書で用いる場合の、「トランスジーン」という用語は、ある生命体から別の生命体へ転移されている遺伝子または遺伝子材料を指す。トランスジーンは、1つもしくは複数の遺伝子および/または1つもしくは複数のオリゴヌクレオチドを含んでもよい。例えば、トランスジーンは、レポーター遺伝子、自殺遺伝子、プロドラッグ変換酵素、および/または1つもしくは複数の治療的遺伝子を含んでもよい。本明細書で用いる場合、「オリゴヌクレオチド」は、20またはそれより少ない塩基対を持つ短い核酸配列を指す。本明細書で用いる場合の、「治療的遺伝子」という用語は、生命体の疾患、医学的状態、器官、組織、細胞、または生理学的特徴に対する治療的効果を提供する、核酸領域と定義される。本明細書で用いる場合、本明細書で用いる場合の「カセット」という用語は、関心対象のタンパク質、ポリペプチド、またはRNAを発現することができる核酸である。
【0077】
リガンドに加えて、AAVPは、その産物を選択または検出することができるレポーター遺伝子を含むトランスジーンを含んでもよい。本明細書において言及される場合、「レポーター遺伝子」は、蛍光、検出可能に標識された化合物もしくは発色基質、もしくは蛍光基質に対する酵素活性などによって、検出することができ;または成長条件によって選択することができる産物をコードする核酸領域である。そのようなレポーター遺伝子には、緑色蛍光タンパク質(GFP)、β-ガラクトシダーゼ、クロラムフェニコールアセチルトランスフェラーゼ(CAT)、ルシフェラーゼ、ネオマイシンホスホトランスフェラーゼ、分泌アルカリホスファターゼ(SEAP)、ヒト成長ホルモン(HGH)チミジンキナーゼなどが含まれるが、これらに限定されない。選択可能マーカーには、ネオマイシン(G418)、ハイグロマイシンなどなどの、薬物耐性物が含まれる。ある種の態様において、血液、その他の体液、または組織中で検出することができる分泌タンパク質をコードするレポーター遺伝子を用いてレポーター遺伝子の発現レベルを測定してもよい。
【0078】
レポーター遺伝子に加えて、トランスジーンはまた、治療的遺伝子を含んでもよい。そのようなトランスジーンを運ぶAAVPは、とりわけ、インビトロかまたはインビボのいずれかにおいて、対象(例えば、ヒト)を画像化することを可能にしてもよい。インビトロイメージングは、対象全体のまたは対象の標的部の非侵襲的イメージングを可能にしてもよい。これらのトランスジーンの導入後、当技術分野において公知のイメージング技術(例えば、BLIイメージング、PETイメージング、蛍光イメージングなど)を用いて発現を画像化してもよい。本明細書で用いる場合の、「対象」は、任意の哺乳動物実体を指し、例えば、対象は、遺伝子治療またはその他の処置を必要とするヒトであってもよい。
【0079】
その他の態様において、AAVPは自殺遺伝子を含んでもよい。本明細書で用いる場合の「自殺遺伝子」という用語は、プロドラッグの投与時に、遺伝子産物のその宿主細胞を殺傷する化合物への移行をもたらす核酸と定義される。使用してもよい自殺遺伝子/プロドラッグ組み合わせの例は、単純ヘルペスウイルス-チミジンキナーゼ(HSV-tk)およびガンシクロビル、アシクロビル、またはFIAU;オキシドレダクターゼおよびシクロヘキシミド;シトシンデアミナーゼおよび5-フルオロシトシン;チミジンキナーゼチミジレートキナーゼ(Tdk::Tmk)およびAZT;ならびにデオキシシチジンキナーゼおよびシトシンアラビノシドである。ある種の態様において、自殺遺伝子は、その宿主細胞の殺傷の結果として疾患または医学的状態に対する治療的効果を提供することにより、治療的遺伝子の様式で作用する可能性がある。
【0080】
「核酸」という用語は当技術分野において周知である。本明細書で用いる場合の「核酸」は通常、核酸塩基を含む、DNA、RNA、またはその誘導体もしくは類似体の分子(すなわち鎖)を指す。核酸塩基には、例えば、DNA中に見出される天然のプリンもしくはピリミジン塩基(例えば、アデニン「A」、グアニン「G」、チミン「T」、もしくはシトシン「C」)またはRNA(例えば、A、G、ウラシル「U」、もしくはC)が含まれる。「核酸」という用語は、各々「核酸」という用語の亜属として「オリゴヌクレオチド」および「ポリヌクレオチド」という用語を包含する。「オリゴヌクレオチド」という用語は、長さが約8から約100核酸塩基の分子を指す。「ポリヌクレオチド」という用語は、長さが約100核酸塩基よりも大きい少なくとも1つの分子を指す。
【0081】
本明細書中のある種の態様では、「遺伝子」は、転写される核酸を指す。ある種の局面において、遺伝子には、転写、もしくはメッセージ生成に関与する調節性領域、または組成物が含まれる。当業者によって理解されるように、「遺伝子」というこの機能的用語には、遺伝子の転写されない部分の核酸セグメント(遺伝子の転写されないプロモーターもしくはエンハンサー領域を含むが、これらに限定されない)を含む、ゲノム配列、RNAもしくはcDNA配列、またはより小さい人為的に改変された核酸セグメントの両方が含まれる。より小さい人為的に改変された遺伝子核酸セグメントは、タンパク質、ポリペプチド、ドメイン、ペプチド、融合タンパク質、突然変異体、および/もしくはそのような同様のものを発現してもよく、または核酸操作技術を用いて発現するように適合させてもよい。
【0082】
本発明のポリヌクレオチドは、「発現カセット」を形成してもよい。「発現カセット」は、特定の転写単位の発現を提供するポリヌクレオチドである。すなわち、それには遺伝子または転写単位の転写において機能するプロモーターエレメントおよび様々なその他のエレメントが含まれる。発現カセットはまた、より大きい複製ポリヌクレオチドまたは発現ベクターもしくはコンストラクトの一部であってもよい。
【0083】
これらの定義は通常、1本鎖分子を指すが、特異的な態様では、1本鎖分子に部分的に、実質的に、または完全に相補的である追加の鎖も包含すると考えられる。したがって、核酸は、分子を含む特定の配列の1つもしくは複数の相補鎖または「相補物」を含む2本鎖分子を包含してもよい。
【0084】
「他のコード配列から実質的に単離された」とは、関心対象の遺伝子が核酸のコード領域のかなりの部分を形成していること、または核酸が大きい染色体断片、その他の機能的遺伝子、RNA、もしくはcDNAコード領域など、天然のコード核酸の大部分を含まないことを意味する。勿論、これは元々単離されたような核酸を指し、および人間の手によって後で核酸に付加された遺伝子またはコード領域を除外しない。
【0085】
本明細書で用いる場合、「核酸塩基」は、例えば、少なくとも1つの天然の核酸(すなわち、DNAおよびRNA)中に見出される天然の核酸塩基(すなわち、A、T、G、C、またはU)、ならびにそのような核酸塩基の天然または非天然の誘導体および類似体などのヘテロ環状塩基を指す。核酸塩基は通常、天然の核酸塩基対合(例えば、AとT、GとC、およびAとUの間の水素結合)の代わりになり得る様式で、少なくとも1つの天然の核酸塩基との1つまたは複数の水素結合を形成する(「アニールする」または「ハイブリダイズする」)ことができる。
【0086】
本明細書で用いる場合、「ヌクレオチド」は、「骨格部分」をさらに含むヌクレオシドを指す。骨格部分は通常、ヌクレオチドを含む別の分子に、または別のヌクレオチドにヌクレオチドを共有結合で付着させて核酸を形成する。天然のヌクレオチド中の「骨格部分」は典型的には、5炭素糖に共有結合で付着している、リン部分を含む。骨格部分の付着は典型的には、5炭素糖の3'位かまたは5'位のいずれかで生じる。しかしながら、特にヌクレオチドが天然の5炭素糖またはリン部分の誘導体または類似体を含む場合に、その他の種類の付着が当技術分野において公知である。
【0087】
A、発現コンストラクト
本発明の発現コンストラクトには、治療的核酸および/またはイメージングタンパク質をコードする核酸が含まれてもよい。その他の局面において、発現コンストラクトは、本発明の治療的組成物および方法で用いることができる治療的発現コンストラクトであってもよい。ある種の態様では、遺伝子材料を操作して、イメージングタンパク質、標的化タンパク質、および/または治療的遺伝子をコードする発現カセットおよび/または発現コンストラクトを生成してもよい。
【0088】
本発明の態様には、2つの別々の種類の発現カセットまたは発現カセットを含む発現コンストラクトが含まれてもよい。1つのカセットは、イメージングタンパク質、すなわち、直接的に検出可能でありまたは間接的に検出可能である特性の活性を有するタンパク質の発現に用いられる。別の発現カセットは、治療的遺伝子をコードしてもよい。治療的ベクターの文脈では、治療的遺伝子は、疾患状態の予防的または治療的処置において有用な本明細書で考察する治療的遺伝子であってもよい。遺伝子治療の文脈では、遺伝子は、ベクターの骨格を提供するウイルスゲノム以外の源に由来するDNAを含むことを意味する、異種DNAであってもよい。遺伝子は、細菌、ウイルス、酵母、寄生虫、植物、または動物などの原核生物または真核生物源に由来してもよい。異種DNAはまた、複数の源、すなわち、多遺伝子コンストラクトまたは融合タンパク質に由来してもよい。異種DNAにはまた、1つの源に由来し得る調節性領域および異なる源由来の遺伝子が含まれてもよい。
【0089】
B、制御領域
それらがイメージングタンパク質をコードするのであれまたは治療的遺伝子をコードするのであれ、本発明の発現カセットおよび/またはコンストラクトには、典型的に、様々な制御領域が含まれると考えられる。これらの制御領域は、典型的には、関心対象の遺伝子の発現を調整する。
【0090】
1、プロモーター
本出願の全体を通じて、「発現コンストラクト」という用語は、核酸コード配列の一部または全てが転写されることができる遺伝子産物、例えば、イメージングタンパク質または治療的タンパク質の一部または全てをコードする核酸を含む任意の種類の遺伝子コンストラクトを含むことが意味される。転写物はタンパク質に翻訳されてもよいが、それが翻訳される必要はない。ある種の態様において、発現には、遺伝子の転写およびmRNAの遺伝子産物への翻訳の両方が含まれる。その他の態様において、発現には阻害RNAまたはDNAなどの治療的核酸の転写のみが含まれる。
【0091】
遺伝子産物をコードする核酸は、プロモーターの転写制御下にある。「プロモーター」は、遺伝子の特異的な転写を開始するのに必要とされる、細胞の機構、または導入された機構によって認識されるDNA配列を指す。特定の局面において、転写は、構成的、誘導可能、および/または抑制可能であってもよい。「転写制御下」という語句は、RNAポリメラーゼの開始および遺伝子の発現を制御するための、プロモーターが核酸に関して正しい場所および方向にあることを意味する。
【0092】
プロモーターという用語は、ここではRNAポリメラーゼIIのための開始部位の周辺にクラスターを成す転写制御モジュールの群を指すよう用いる。プロモーターがどのように編成されているかに関する考えの大部分は、様々なレトロウイルスプロモーター、HSVチミジンキナーゼ(tk)、およびSV40初期転写単位についての解析を含む、幾つかのウイルスプロモーターの解析に由来している。
【0093】
追加のプロモーターエレメントは、転写の開始の頻度を調節する。最近、多くのプロモーターが同じく出発部位の下流の機能的エレメントを含むことが示されているが、典型的には、これらは出発部位の30〜110 bp上流の領域に位置する。プロモーターエレメント間の間隔は融通が利き、そのためエレメントが互いに対して逆にされたりまたは動かされたりしてもプロモーター機能は保存される。プロモーター次第で、転写を活性化するために個々のエレメントが協調的にかまたは独立にかのいずれかで機能することができるように見える。
【0094】
それが標的化された細胞における核酸の発現を導くことができる限り、関心対象の核酸配列の発現を制御するのに利用される特定のプロモーターが重要であると考えられない。したがって、ヒト細胞が標的化される場合、標的化されるヒト細胞で発現されることができるプロモーターの近辺および該プロモーターの制御下に核酸コード領域を位置付けることが好ましい。一般的に言えば、そのようなプロモーターには、ヒトプロモーターか、ウイルスプロモーターか、またはその組み合わせのいずれかが含まれ得る。
【0095】
様々な態様において、ヒトサイトメガロウイルス即初期遺伝子プロモーター(CMVIE)、SV40初期プロモーター、ラウス肉腫ウイルス長末端反復、β-アクチン、ラットインスリンプロモーター、およびグリセルアルデヒド-3-リン酸デヒドロゲナーゼを用いて、関心対象のコード配列の高レベル発現を得ることができる。発現のレベルが所与の目的のために十分であるならば、関心対象のコード配列の発現を達成することが当技術分野において周知である、その他のウイルス、レトロウイルスまたは哺乳動物細胞または細菌ファージのプロモーターの使用が同じく企図される。周知の特性を持つプロモーターを利用することによって、トランスフェクションまたは形質転換後の関心対象のタンパク質の発現のレベルおよびパターンを最適化することができる。
【0096】
特異的な生理的または合成的シグナルに応答して調節されるプロモーターの選択は、非誘導条件下の細胞と比較した場合の遺伝子産物の誘導可能な発現を可能にすることができる。例えば、1つのトランスジーン、または多シストロン性のベクターを活用した時の複数のトランスジーンの発現が、ベクターを産生する細胞にとって毒性がある場合、トランスジーンのうちの1つまたは複数の発現を禁止しまたは低下させることが望ましい場合がある。プロデューサー細胞株にとって毒性があり得るトランスジーンの例は、プロアポトーシスおよびサイトカイン遺伝子である。トランスジーン産物が毒性であり得る場合に、幾つかの誘導可能なプロモーター系がウイルスベクターの産生に利用可能である。
【0097】
エクジソン系(Invitrogen, Carlsbad, CA)は1つのそのような系である。この系は、哺乳動物細胞における関心対象の遺伝子の調節された発現を可能にするように設計されている。それは、トランスジーンの基礎レベル発現はほとんど可能にしないが、200倍を越える誘導能を可能にする厳しく調節された発現メカニズムからなる。有用であると考えられる別の誘導可能な系は、もとはGossenおよびBujardによって開発された(Gossen and Bujard, 1992;Gossen et al., 1995)Tet-Off(商標)またはTet-On(商標)系(Clontech, Palo Alto, CA)である。この系はまた、高レベルの遺伝子発現がテトラサイクリンまたはドキシサイクリンなどのテトラサイクリン誘導体に応答して調節されるのを可能にする。
【0098】
幾つかの状況において、治療的発現ベクター中のトランスジーンの発現を調節することが望ましい場合がある。例えば、様々に異なる強さの活性を持つ異なるウイルスプロモーターを望ましい発現のレベルに応じて活用してもよい。哺乳動物細胞では、強い転写活性化を提供するために、多くの場合CMV即初期プロモーターが用いられる。低下したレベルのトランスジーンの発現が望ましい場合、あまり強力でないCMVプロモーターの改変版も用いられている。造血細胞におけるトランスジーンの発現が望ましい場合、MLVまたはMMTV由来のLTRなどのレトロウイルスプロモーターが多くの場合用いられる。所望の効果に応じて使用し得るその他のウイルスプロモーターには、SV40、RSV LTR、HIV-1およびHIV-2 LTR、E1A、E2A、またはMLP領域からなどのアデノウイルスプロモーター、AAV LTR、カリフラワーモザイクウイルス、HSV-TK、ならびにトリ肉腫ウイルスが含まれる。
【0099】
同様に、標的化されていない組織に対する潜在的な毒性または望まない効果を低下させるために、組織特異的または選択的プロモーターを用いて特異的な組織または細胞における転写をもたらしてもよい。例えば、PSA、プロバシン、前立腺酸性ホスファターゼ、または前立腺特異的腺カリクレイン(hK2)などのプロモーターを用いて前立腺における遺伝子発現を標的化してもよい。同様に、以下のプロモーターを用いて、その他の組織における遺伝子発現を標的化してもよい(表1)。
【0100】
ある種の徴候において、遺伝子治療ベクターの投与後の特異的な時間に転写を活性化することが望ましい場合がある。これは、ホルモンまたはサイトカイン調節可能であるプロモーターのようなプロモーターを用いて行なわれてもよい。例えば、徴候が特異的なステロイドが産生されまたは送られる生殖腺組織である治療的適用において、アンドロゲンまたはエストロゲンによって調節されるプロモーターの使用が有利である場合がある。ホルモン調節可能であるようなそのようなプロモーターには、MMTV、MT-1、エクジソン、およびルビスコが含まれる。甲状腺、下垂体、および副腎ホルモンに応答性のプロモーターなどのその他のホルモンによって調節されるプロモーターは、本発明において有用であることが期待される。使用し得るサイトカインおよび炎症性タンパク質応答性のプロモーターには、KおよびTキニノーゲン(Kageyama et al., 1987)、c-fos、TNF-アルファ、C反応性タンパク質(Arcone et al., 1988)、ハプトグロビン(Oliviero et al., 1987)、血清アミロイドA2、C/EBPアルファ、IL-1、IL-6(Poli and Cortese, 1989)、補体C3(Wilson et al., 1990)、IL-8、アルファ-1酸性糖タンパク質(Prowse and Baumann, 1988)、 アルファ-1アンチトリプシン、リポタンパク質リパーゼ(Zechner et al., 1988)、アンギオテンシノーゲン(Ron et al., 1990)、フィブリノゲン、c-jun(ホルボールエステル、TNF-アルファ、UV放射、レチノイン酸、および過酸化水素によって誘導可能)、コラゲナーゼ(ホルボールエステルおよびレチノイン酸によって誘導される)、メタロチオネイン(重金属およびグルココルチコイドで誘導可能)、ストロメリシン(ホルボールエステル、インターロイキン-1、およびEGFによって誘導可能)、アルファ-2マクログロブリン、ならびにアルファ-1アンチキモトリプシンが含まれる。また、オステオカルシン、低酸素応答性配列(HRE)、MAGE-4、CEA、アルファ-フェトプロテイン、GRP78/BiP、およびチロシナーゼなどの腫瘍特異的なプロモーターを用いて、腫瘍細胞における遺伝子発現を調節してもよい。
【0101】
単独のまたは別のプロモーターと組み合わせた上記のプロモーターのいずれかが、望ましい作用に応じて本発明に従って有用であり得ることが予想される。さらに、このプロモーターのリストは網羅的または限定的であると解されるべきではなく、当業者は本明細書において開示されたプロモーターおよび方法と併せて使用し得るその他のプロモーターがあるのを知っていると考えられる。
【0102】
(表1)組織特異的プロモーター
【0103】
2、エンハンサー
エンハンサーは、DNAの同じ分子上の遠い位置に位置するプロモーターからの転写を増加させる遺伝子エレメントである。エンハンサーはプロモーターにとてもよく似たように編成されている。すなわち、それらは多くの個々のエレメントから構成され、その各々が1つまたは複数の転写タンパク質に結合する。エンハンサーとプロモーターの間の基本的な差異は操作上のものである。全体としてのエンハンサー領域は、遠隔地での転写を刺激することができなければならず;これはプロモーター領域またはその構成要素エレメントについて当てはまる必要はない。他方、プロモーターは、特定の部位および特定の方向でのRNA合成の開始を導く1つまたは複数のエレメントを有さなければならない一方、エンハンサーはこれらの特異性を欠く。プロモーターおよびエンハンサーは、多くの場合重複および連続し、多くの場合非常によく似たモジュール編成を有するように思われる。
【0104】
以下は、上で列挙された組織特異的プロモーターに追加するプロモーターである、発現コンストラクト中の関心対象の遺伝子をコードする核酸と組み合わせて使用し得る細胞性プロモーター/エンハンサーおよび誘導可能プロモーター/エンハンサーのリストである(表2および表3)。さらに、遺伝子の発現を駆動するために、(真核生物プロモーターデータベース EPDBによる)任意のプロモーター/エンハンサーの組み合わせも使用し得る。適当な細菌ポリメラーゼが送達複合体の一部としてまたは追加の遺伝子発現コンストラクトとしてのいずれかで提供されるならば、真核生物細胞は、ある種の細菌プロモーターからの細胞質転写を支持することができる。
【0105】
本発明の好ましい態様において、治療的発現コンストラクトは、ウイルスまたはウイルスゲノムに由来する人為的に改変されたコンストラクトを含む。ある種のウイルスが受容体を介するエンドサイトーシスによって細胞に侵入する能力および宿主細胞ゲノムに入り込みかつウイルス遺伝子を安定的かつ効率的に発現させる能力によって、それらは哺乳動物細胞への外来遺伝子の転移のための魅力的な候補になっている(Ridgeway, 1988;Nicolas and Rubenstein, 1988;Baichwal and Sugden, 1986;Temin, 1986)。
【0106】
(表2)
【0107】
(表3)
【0108】
C、ポリアデニル化シグナル
ポリアデニル化シグナルが治療的および/イメージングベクターで用いられてもよい。cDNA挿入物が利用される場合、典型的には、遺伝子転写物の適切なポリアデニル化をもたらすためにポリアデニル化シグナルを含めることが望まれる。ポリアデニル化シグナルの性質は本発明の首尾よい実施に必須であるとは考えられておらず、ならびにヒトまたはウシ成長因子ホルモンおよびSV40ポリアデニル化シグナルなどの任意のそのような配列を利用してもよい。ターミネーターも発現カセットのエレメントとして企図される。これらのエレメントは、メッセージレベルを増強させるのにおよびカセットを読み終えてその他の配列に移るのを最小限にするのに役立つことができる。
【0109】
D、治療的遺伝子
本発明のAAVP粒子を用いて、治療的発現ベクターを含む、種々の治療的またはイメージング薬剤を送達してもよい。本発明は、種々の異なる治療的遺伝子の使用を企図する。例えば、酵素、ホルモン、サイトカイン、オンコジーン、受容体、イオンチャネル、腫瘍抑制因子、転写因子、薬物選択可能マーカー、毒素、および様々な抗原をコードする遺伝子が、本発明による使用のための適した遺伝子として企図される。さらに、オンコジーンに由来するアンチセンスおよび阻害RNAコンストラクトは、本発明によるその他の関心対象の「遺伝子」である。
【0110】
本発明に従って、選択された遺伝子またはポリペプチドは、任意のタンパク質、ポリペプチド、またはペプチドを指してもよい。治療的遺伝子またはポリペプチドは、疾患を処置または防止する目的のために対象に投与することができる遺伝子またはポリペプチドである。例えば、治療的遺伝子は、癌の処置または防止のために対象に投与される遺伝子であることができる。治療的遺伝子の例として、Rb、CFTR、p16、p21、p27、p57、p73、C-CAM、APC、CTS-1、zac1、scFV ras、DCC、NF-1、NF-2、WT-1、MEN-I、MEN-II、BRCA1、VHL、MMAC1、FCC、MCC、BRCA2、IL-1、IL-2、IL-3、IL-4、IL-5、IL-6、IL-7、IL-8、IL-9、IL-10、IL-11、IL-12、GM-CSF、G-CSF、チミジンキナーゼ、Bax、Bak、Bik、Bim、Bid、Bad、Harakiri、Fas-L、mda-7、fus、インターフェロンα、インターフェロンβ、インターフェロンγ、ADP、p53、ABLI、BLC1、BLC6、CBFA1、CBL、CSFIR、ERBA、ERBB、EBRB2、ETS1、ETS2、ETV6、FGR、FOX、FYN、HCR、HRAS、JUN、KRAS、LCK、LYN、MDM2、MLL、MYB、MYC、MYCL1、MYCN、NRAS、PIM1、PML、RET、SRC、TAL1、TCL3、YES、MADH4、RB1、TP53、WT1、TNF、BDNF、CNTF、NGF、IGF、GMF、aFGF、bFGF、NT3、NT5、ApoAI、ApoAIV、ApoE、Rap1A、シトシンデアミナーゼ、Fab、ScFv、BRCA2、zac1、ATM、HIC-1、DPC-4、FHIT、PTEN、ING1、NOEY1、NOEY2、OVCA1、MADR2、53BP2、IRF-1、zac1、DBCCR-1、rks-3、COX-1、TFPI、PGS、Dp、E2F、ras、myc、neu、raf、erb、fms、trk、ret、gsp、hst、abl、E1A、p300、VEGF、FGF、トロンボスポンジン、BAI-1、GDAIF、またはMCCが含まれるが、これらに限定されない。
【0111】
治療的遺伝子のその他の例として、酵素をコードする遺伝子が含まれる。例として、ACPデサチュラーゼ、ACPヒドロキシラーゼ、ADP-グルコースピロホリラーゼ、ATPアーゼ、アルコールデヒドロゲナーゼ、アミラーゼ、アミログルコシダーゼ、カタラーゼ、セルラーゼ、シクロオキシゲナーゼ、デカルボキシラーゼ、デキストリナーゼ、エステラーゼ、DNAポリメラーゼ、RNAポリメラーゼ、ヒアルロンシンターゼ、ガラクトシダーゼ、グルカナーゼ、グルコースオキシダーゼ、GTPアーゼ、ヘリカーゼ、ヘミセルラーゼ、ヒアルロニダーゼ、インテグラーゼ、インベルターゼ、イソメラーゼ、キナーゼ、ラクターゼ、リパーゼ、リポオキシゲナーゼ、リアーゼ、リゾチーム、ペクチンエステラーゼ、ペルオキシダーゼ、ホスファターゼ、ホスホリパーゼ、ホスホリラーゼ、ポリガラクツロナーゼ、プロテイナーゼ、ペプチダーゼ、プラナーゼ、リコンビナーゼ、リバーストランスクリプターゼ、トポイソメラーゼ、キシラナーゼ、レポーター遺伝子、インターロイキン、またはサイトカインが含まれるが、これらに限定されない。
【0112】
治療的遺伝子のさらなる例として、カルバモイルシンセターゼI、オルニチントランスカルバミラーゼ、アルギノスクシネートシンセターゼ、アルギノスクシネートリアーゼ、アルギナーゼ、フマリルアセトアセテートヒドロラーゼ、フェニルアラニンヒドロキシラーゼ、アルファ-1アンチトリプシン、グルコース-6-ホスファターゼ、低密度リポタンパク質受容体、ポルホビリノーゲンデアミナーゼ、第VIII因子、第IX因子、シスタチオンベータ-シンターゼ、分岐鎖ケト酸デカルボキシラーゼ、アルブミン、イソバレリル-CoAデヒドロゲナーゼ、プロピオニルCoAカルボキシラーゼ、メチルマロニルCoAムターゼ、グルタリルCoAデヒドロゲナーゼ、インスリン、-グルコシダーゼ、ピルビン酸カルボキシラーゼ、肝ホスホリラーゼ、ホスホリラーゼキナーゼ、グリシンデカルボキシラーゼ、H-タンパク質、T-タンパク質、メンケス病銅輸送ATPアーゼ、ウィルソン病銅輸送ATPアーゼ、シトシンデアミナーゼ、ヒポキサンチン-グアニンホスホリボシルトランスフェラーゼ、ガラクトース-1-リン酸ウリジルトランスフェラーゼ、フェニルアラニンヒドロキシラーゼ、グルコセルブロシダーゼ、スフィンゴミエリナーゼ、-L-イズロニダーゼ、グルコース-6-リン酸デヒドロゲナーゼ、HSVチミジンキナーゼ、またはヒトチミジンキナーゼをコードする遺伝子が含まれる。
【0113】
治療的遺伝子には、ホルモンをコードする遺伝子も含まれる。例として、成長ホルモン、プロラクチン、胎盤ラクトゲン、黄体形成ホルモン、濾胞刺激ホルモン、絨毛性ゴナドトロピン、甲状腺刺激ホルモン、レプチン、副腎コルチコトロピン、アンギオテンシンI、アンギオテンシンII、β-エンドルフィン、β-メラニン細胞刺激ホルモン、コレシストキニン、エンドセリンI、ガラニン、胃阻害ペプチド、グルカゴン、インスリン、リポトロピン、ニューロフィジン、ソマトスタチン、カルシトニン、カルシトニン遺伝子関連ペプチド、β-カルシトニン遺伝子関連ペプチド、悪性の高カルシウム血症因子、副甲状腺ホルモン関連タンパク質、副甲状腺ホルモン関連タンパク質、グルカゴン様ペプチド、パンクレアスタチン、膵ペプチド、ペプチドYY、PHM、セクレチン、血管作用性小腸ペプチド、オキシトシン、バソプレッシン、バソトシン、エンケファリンアミド、メトルフィンアミド、アルファメラニン細胞刺激ホルモン、心房性ナトリウム利尿因子、アミリン、アミロイドP構成要素、コルチコトロピン放出ホルモン、成長ホルモン放出因子、黄体形成ホルモン放出ホルモン、神経ペプチドY、サブスタンスK、サブスタンスP、またはチロトロピン放出ホルモンをコードする遺伝子が含まれるが、これらに限定されない。
【0114】
さらに別の態様において、異種遺伝子には、単鎖抗体が含まれてもよい。単鎖抗体の産生のための方法は当業者に周知である。当業者は、そのような方法のために(参照により本明細書に組み入れられる)米国特許第5,359,046号に差し向けられる。単鎖抗体は、短いペプチドリンカーを用いて重鎖および軽鎖の可変ドメインを一緒に融合し、それによって単一分子上に抗原結合部位を再構成することによって創り出される。
【0115】
E、多遺伝子コンストラクトおよびIRES
本発明のある種の態様において、多遺伝子ポリシストロン性メッセージを創り出すために内部リボソーム結合部位(IRES)エレメントの使用が用いられる(Pelletier and Sonenberg, 1988)。哺乳動物のメッセージ由来のIRES(Macejak and Sarnow, 1991)と同様に、ピカノウイルス(picanovirus)ファミリー(ポリオおよび脳心筋炎)の2つのメンバー由来のIRESエレメントが記載されている(Pelletier and Sonenberg, 1988)。IRESエレメントは、異種オープンリーディングフレームにつなぐことができる。多数のオープンリーディングフレームを一緒に転写し、各々をIRESによって分離し、ポリシストロン性メッセージを創り出すことができる。IRESエレメントのおかげで、各オープンリーディングフレームは、効率的な翻訳のためにリボソームに接近し易い。単一のプロモーター/エンハンサーを用いて多数の遺伝子を効率的に発現させ、単一のメッセージを転写させことができる。任意の異種オープンリーディングフレームをIRESエレメントにつなぐことができる。これには、分泌タンパク質、独立の遺伝子によってコードされる、多サブユニットタンパク質、細胞内または膜結合タンパク質、および選択可能マーカーのための遺伝子が含まれる。このように、幾つかのタンパク質の発現を同時に人為的に改変して単一のコンストラクトおよび単一の選択可能マーカーを持つ細胞にすることができる。
【0116】
F、核酸の調製
例えば、化学合成、酵素的産生、または生物学的産生などの、当業者に公知の任意の技術によって、核酸を作製してもよい。酵素的に産生される核酸の非限定的な例として、PCR(商標)(例えば、各々が参照により本明細書に組み入れられる、米国特許第4,683,202号および米国特許第4,682,195号参照)などの増幅反応、または参照により本明細書に組み入れられる、米国特許第5,645,897号に記載されたオリゴヌクレオチドの合成において酵素によって産生される核酸が含まれる。生物学的に産生される核酸の非限定的な例として、細菌の中で複製される組換えDNAベクター(例えば、参照により本明細書に組み入れられる、Sambrook et al. 2001参照)などの、生細胞の中で産生される(すなわち、複製される)組換え核酸が含まれる。
【0117】
III、薬学的組成物および投与の経路
臨床的適用が企図される場合、意図する適用のために適当な形態でAAVP組成物(治療的組成物)の薬学的組成物を調製する必要があると考えられる。通常、これは、パイロジェンだけでなく、ヒトまたは動物にとって有害であり得るその他の不純物も本質的に含まない組成物を調製する工程を伴っていると考えられる。
【0118】
通常、組成物を患者への導入に好適であるようにするために、適当な塩および緩衝剤を利用することが望まれる。本発明の水性組成物は、薬学的に許容される担体もしくは水性媒体に溶けたまたは分散した有効量のAAVPまたはその他の薬剤を含む。「薬学的にまたは薬理学的に許容される」という語句は、動物またはヒトに投与された時に、不都合な、アレルギー性の、またはその他の厄介な反応を産生しない分子実体および組成物を指す。
【0119】
本明細書で用いる場合、「薬学的に許容される担体」には、任意および全ての溶媒、分散媒体、コーティング剤、抗菌および抗真菌薬剤、等張および吸収遅延薬剤などが含まれる。薬学的に活性な物質のためのそのような媒体および薬剤の使用は、当技術分野において周知である。任意の従来の媒体または薬剤が本発明のAAVP組成物と合わない限りを除き、イメージング試薬としてのまたは治療的組成物におけるその使用が企図される。その他の抗癌薬剤などの、補足の活性成分を組成物中に組み入れることもできる。保管および使用の普通の条件下で、これらの調製物は微生物の成長を防止するための防腐剤を含む。静脈内ビヒクルには、流体および栄養補液が含まれる。防腐剤には、抗微生物薬剤、酸化防止剤、キレート薬剤、および不活性ガスが含まれる。調合薬中の様々な構成要素のpHおよび正確な濃度を周知のパラメーターに従って合わせる。
【0120】
癌などの状態または疾患を画像化することおよび/または改善することなどの、意図された目標に基づいて、組成物の有効量を決定する。「単位用量」という用語は、対象における使用に適した物理的に別個の単位を指し、各単位は、その投与、すなわち、適当な経路および治療レジメンと関連した所望の応答を産生するように計算された所定の数量の治療的組成物を含む。処置の数および単位用量の両方に従って、投与されるべき数量は、処置されるべき対象、対象の状態、および望ましい保護による。治療的組成物の正確な量はまた、施術者の判断によりおよび各々の個人に特有である。
【0121】
2つの活性成分を含む組み合わせ組成物も企図される。特に、本発明は、AAVP組成物および少なくとも1つの第二の治療薬、例えば、抗新生物薬物を含む組成物を提供する。
【0122】
A、非経口投与
本発明の活性組成物を非経口投与用に製剤化し、例えば、静脈内、筋肉内、皮下、または腹腔内経路さえも経由する注射用に製剤化してもよい。第二の薬剤を活性成分として含む水性組成物の調製は、本開示に照らして当業者に公知であると考えられる。典型的には、そのような組成物を、液体溶液かまたは懸濁かのいずれかとして、注射可能なものとして調製することができ;注射前の液体の添加時に溶液または懸濁を調製するために用いるのに適した固形形態を調製することもでき;および調製物を乳化することもできる。
【0123】
注射可能な使用に適した薬学的形態には、滅菌水性溶液または分散;ゴマ油、ピーナッツ油、または水性プロピレングリコールを含む製剤;および滅菌注射可能溶液または分散の即時調製用の滅菌粉末が含まれる。全ての場合において、形態は滅菌されていなければならずおよび容易な注入可能性(syringability)が存在する程度まで流動性でなければならない。
【0124】
担体は、例えば、水、エタノール、ポリオール(例えば、グリセロール、プロピレングリコール、および液体ポリエチレングリコールなど)、その適した混合物、ならびに植物油を含む溶媒または分散媒体であることもできる。例えば、レシチンなどの、コーティング剤の使用によって、分散の場合に必要とされる粒子サイズの維持によって、および界面活性剤の使用によって、適切な流動性を維持することができる。様々な抗菌および抗真菌薬剤、例えば、パラベン、クロロブタノール、フェノール、ソルビン酸、チメロサールなどによって、微生物の作用の防止をもたらすことができる。多くの場合、等張薬剤、例えば、糖または塩化ナトリウムを含めることが好ましいと考えられる。吸収を遅延させる薬剤、例えば、モノステアリン酸アルミニウムおよびゼラチンの組成物での使用によって、注射可能組成物の長期の吸収をもたらすことができる。
【0125】
滅菌注射可能溶液は、必要とされる量の活性化合物を上で列挙された様々なその他の成分と共に適当な溶媒中に組み入れ、必要な場合、それに続いて濾過滅菌することによって調製される。通常、分散は、様々な滅菌された活性成分を塩基性分散媒体および必要とされる上で列挙された成分からの必要とされるその他の成分を含む滅菌ビヒクル中に組み入れることによって調製される。滅菌注射可能溶液の調製用の滅菌粉末の場合、調製の特定の方法は、先に滅菌濾過されたその溶液由来の任意のさらなる所望の成分を加えた活性成分の粉末を産出する真空乾燥およびフリーズドライ技術である。
【0126】
水性溶液での非経口投与のために、例えば、溶液は必要ならば好適に緩衝化されるべきであり、かつ液体希釈剤は最初に十分な生理食塩水またはグルコースで等張にされるべきである。これらの特定の水性溶液はとりわけ、静脈内、筋肉内、皮下、および腹腔内投与に好適である。これに関連して、利用することができる滅菌水性媒体は、本開示に照らして当業者に公知であると考えられる。例えば、1投薬量を1 mLの等張NaCl溶液に溶かし、かつ1000 mlの皮下投与液体に添加するかまたは提案された注入の部位で注射するかのいずれかであり得る(例えば、「Remington's Pharmaceutical Sciences」第15版、1035〜1038および1570〜1580ページ参照)。投薬量の若干の変動は、処置されている対象の状態によって必ず生じると考えられる。投与に責任を負う人は、いずれにしても、個々の対象のための適当な用量を決定すると考えられる。
【0127】
本発明のより良い理解を容易にするために、具体的な態様に関する以下の実施例を挙げる。以下の実施例は、決して、本発明の範囲全体を限定または定義するように読まれるべきではない。
【0128】
IV、実施例
以下の実施例は、本発明の様々な態様を例証するという目的のために与えられ、およびいかなる形であっても本発明を限定するよう意味されない。当業者は、本発明が目的を実行しかつ述べられた目標および利点を得るだけでなく、本明細書において本来備わっている目的を実行し、目標および利点を得るのにも十分適合していることを容易に正しく認識すると考えられる。本実施例は、本明細書に記載する方法と共に、好ましい態様を目下代表するものであり、例示するものであり、および本発明の範囲に対する限定であることが意図されない。特許請求の範囲の範囲によって定義されるような本発明の精神の範囲内に包含されるその中での変化およびその他の使用が当業者の心に浮かぶと考えられる。
【0129】
実施例1
AAVP標的化
A、実験手順
標的化されたAAVP粒子の設計、構築、および作製
RGD-4CファージおよびRGD-4C AAVPを中間体(RGD-4C fUSE5-MCS)の作製ならびにその後のRGD-4CファージコンストラクトおよびRGD-4C AAVPの作製:という2工程過程で人為的に改変した。RGD-4C fUSES-MCSは、特異的な標的化ペプチドRGD-4Cをコードするオリゴヌクレオチド挿入物および真核生物発現カセットの挿入用のマルチクローニング部位(MCS)を有するfMCSプラスミドの断片を含んだ。RGD-4Cファージ由来fUSE5 DNAおよびファージ由来fMCS DNAを宿主大腸菌(MC1061)のライセートから精製した。本発明者らは、RGD-4C FUSE5プラスミドの5.4 kb BamHIISacII断片をfMCSプラスミドの4.1 kb BamHI/SacII断片にライゲートすることによって、中間体RGD-4C fUSE5-MCS を得た。次に、本発明者らは、pAAV-eGFP プラスミド(増強されたGFP;Stratagene)のITRからITRまでのPacI断片(2.8 kb)をRGD 4C fUSE5-MSCのPstI部位にクローニングすることによって、標的化されたAAVP-GFPを創り出した。簡潔に述べると、pAAVをPacIで消化して2.8 kb断片を放出し、それをDNAポリメラーゼで平滑化し、かつRGD-4C fUSE5-MSCの平滑化されたPstI部位にクローニングした。ITRがない標的化されたファージコンストラクトを作製するために、pAAV-eGFPのITRの間に位置しかつpCMV-GFPおよびSV40ポリAを含む2.3 kb断片をEcoRI消化で放出し、DNAポリメラーゼで平滑化し、その後RGD-4C fUSE5-MSCのMCSにクローニングした。GFPを発現する細胞を選択するために、pQBIホスホグリセリン酸キナーゼ-1(PGK;QBIOgene)プロモーターのおよびGFPneo融合配列を含むBamHI-Sac1断片をAAVPまたは対照ファージコンストラクトのNotI部位にクローニングし、GFPを発現する細胞がG418耐性であることを確実にした。pQBI PGKのGFPneo断片をBamHIおよびSac1消化で放出し、かつDNAポリメラーゼで平滑化し;その後NotIへのリン酸化リンカーを付加した。NotI消化後、1.57 kb GFPneo断片をAAVPまたは非キメラファージコンストラクトのNotI部位にクローニングした。最後に、HSVtkまたはLucの遺伝子を運ぶ標的化されたAAVP粒子を作製するために、HSVtkまたはLucを含むBamHI-Not1断片をpAAVプラスミドのBamHI-Not1部位にサブクローニングし、GFPを取り替えた。ITR-HSVtk-ITRまたはITR-Luc-ITR断片をpAAV-HSVtkおよびpAAV-Lucから除去し、その後RGD-4C fUSE5-MCSに挿入した。コンストラクトをDNAシークエンシングおよび制限解析で確認し、宿主大腸菌(MC1061)の培養上清から精製し、PBSに再懸濁し、および再遠心分離した。結果として得られた上清を大腸菌(k91Kan)で滴定した。連続希釈をテトラサイクリンおよびカナマイシンを含むルリア-ベルターニ(LB)寒天プレート上にプレーティングしならびにコロニー計数によって形質導入単位(TU)を決定した。
【0130】
哺乳動物細胞表面結合および内在化アッセイ
本発明者らは、(BRASILと呼ばれる)選択的相互作用リガンド法(Giordano et al., 2001)のバイオパニングおよび迅速解析を用いて、インタクトな細胞へのファージ結合を評価した。簡潔に述べると、KS1767細胞をエチレンジアミン四酢酸(EDTA)で剥がし、かつ1% BSAを含むダルベッコの改変イーグル培地(DMEM)にml当たり4 x 106細胞で再懸濁した。細胞懸濁(50 μl)を109 TUのRGD-4C AAVPかまたはスクランブルバージョンのRGD-4C(CDCFGDCRC(SEQ ID NO:2)、CDCGFDCRC(SEQ ID NO:3)、CRCDGFCDC(SEQ ID NO:4))、突然変異体RGE-4Cペプチド、もしくは標的化されていない対照を提示するAAVPクローンのいずれかとインキュベートした。2時間後、AAVP/細胞混合物(水性相)をフタル酸ジブチル:シクロヘキサン(9:1 [v:v]、D = 1.03 g/ml)からなる非混和性の有機相(400 μlエッペンドルフチューブ中200 μl溶液)の上に移し、かつ10,000 gで10分間4℃で遠心分離した。その後、チューブを液体窒素中で瞬間凍結し、チーブの底を切り取り、および細胞-AAVPペレットを単離しならびに膜に結合したAAVPを回収した(Giordano et al., 2001)。
【0131】
細胞内在化のために、KS1767細胞を組織チャンバースライド(Lab-Tek II Chamber Slide System; Nalge Nunc International Corp.)で増殖させ、PBSで2回洗浄し、109 TUのRGD-4C AAVPまたはスクランブルバージョンのRGD-4CもしくはRGE-4Cを提示する対照AAVPと1% BSAを含むDMEM中で37℃でインキュベートし、および4時間のインキュベーション後にPBSで洗浄して、結合していないAAVPを除去した。細胞を20 mMグリシン(pH 2.3)で濯ぐことにより、細胞膜に結合したクローンを化学的に溶出した。次に、細胞をPBSで3回洗浄し、4%パラホルムアルデヒド(PFA)を含むPBSでRTで15分間固定し、PBSで洗浄し、0.2% Triton X-100で透過処理し、PBSで洗浄し、および1% BSAを含むPBSでブロッキングした。その後、細胞を1:200希釈の一次抗M13バクテリオファージ抗体(Amersham)と1% BSAを含むPBS中でRTで2時間インキュベートし、PBSで洗浄し、および1:200希釈のCy3-コンジュゲートされた抗ウサギ二次抗体と1% BSAを含むPBS中で1時間RTでインキュベートした。最後に、細胞をPBSで洗浄し、4% PFAを含むPBSで固定し、マウントし、光学蛍光顕微鏡で可視化した。
【0132】
AAVP粒子によって形質導入された細胞からの組換えAAVのレスキュー
ヒト293細胞にRGD-4C AAVP-GFPneoまたはRGD-4Cファージ-GFPneoを感染させた。感染4日後、細胞にAAV rep-およびcap-発現プラスミド(pXX2)(Xiao et al., 1998)をトランスフェクトしならびに野生型アデノウイルス5型(Ad)を重感染させた。このように、AAV repおよびcap遺伝子をトランスフェクションによって供給し、かつアデノウイルスヘルパー機能を重感染によって提供した。アデノウイルス感染72時間後に細胞を採取し、その後上清を用いて新しい293細胞に感染させた。48時間後にFACSを用いることによってGFP発現を解析した。このアッセイにおいて、機能的な組換えAAVは、RGD 4C AAVP-GFPキメラを形質導入した細胞からのみ作製されたが、非キメラファージ-GFPまたは幾つかの対照を形質導入した細胞からは作製されなかった。同様の結果はまた、全てのRGD-4C AAVPクローンを用いて得られたが、ファージクローンのいずれを用いても得られなかった。
【0133】
クローン性の哺乳動物細胞株の作製
ヒト293細胞にRGD-4C AAVP-GFPneoまたはRGD-4Cファージ-GFPneoを(各々の場合に細胞当たり106 TUで)感染させた。単一クローン(群当たりn=9)をG418選択下で単離し、かつ選択12週後にFACSでGFP発現について解析した。安定クローンをファージ-GFPneoについてはファージクローン#1-9およびAAVP-GFPneoについてはAAVP #1-9クローンと名付けた。
【0134】
腫瘍モデル
動物実験法は、施設の動物研究委員会によって審査および承認された。腫瘍を有するマウスを記載されたように樹立した(Pasqualini et al., 1997;Arap et al., 1998; Ellerby et al., 1999;Hajitou et al., 2001;Arap et al., 2004;Marchib et al., 2004)。アベルチンBの腹腔内投与によってまたはガス(2%イソフルランおよび98%酸素)吸入によってマウスに麻酔をかけた。標的化されたコンストラクトまたは対照を静脈内投与した。腫瘍細胞をトリプシン処理し、計数し、遠心分離し、および血清を含まない培地に再懸濁した。カポジ肉腫(KS 1767)、膀胱癌腫(UC3)、または前立腺癌腫(DU145)株由来の合計106細胞を6週齢免疫不全ヌードマウスに皮下に埋め込んだ。EF43-FGF4マウス乳腫瘍細胞(5 x 104)を6週齢メスBALBIc免疫適格マウスに皮下に埋め込んだ。腫瘍体積を記載されたように計算し(Pasqualini et al., 1997;Arap et al., 1998;Ellerby et al., 1999;Hajitou et al., 2001;Arap et al., 2004;Marchib et al., 2004)および平均腫瘍体積 * 標準偏差(SD)として表した。腫瘍が-50 mm2(小さいとみなされる)または-150 mm2(大きいとみなされる)のDU145由来異種移植片の体積に達した時(0日目)、腫瘍を有するマウスは単一静脈内用量のRGD-4C AAVP-HSVtk、または対照を受けた。サイズが一致した腫瘍を有するマウスのコホートにおいて2日後にGVC処置(1日当たり80 mg/kg、腹腔内)を始めた。
【0135】
腫瘍を有するマウスにおける分子-遺伝子イメージング
非侵襲的な分子イメージングのために、本発明者らはヒト細胞株DU145に基づいた前立腺癌のモデルを用い、その中で右肩の皮下部に腫瘍異種移植片を持つオスのヌードマウスを用いた。ホタルLuc遺伝子発現を画像化するために、腫瘍を有するマウスは、単一用量(150 mg/kg)の基質D-ルシフェリン(Xenogen)を腹腔内投与により受けた。Luc遺伝子を運ぶ標的化されたRGD-4C AAVPまたは対照(標的化されていないAAVP-Luc、もしくはスクランブルのRGD-4C AAVPLuc)の尾静脈投与後にIn Vivo Imaging System 200(IVIS200;Xenogen, CA)を用いることによって、光子の放出を画像化した。イメージングパラメーター:画像獲得時間、1分;ビニング、2;フィルターなし;フルストップ(flstop)、1;視野、10 cm。光子/秒/cm2/srと表される、シグナル強度を測定するための腫瘍に対して関心対象の領域(ROI)を手作業で定義した。
【0136】
BLIは実験系におけるトランスジーン発現細胞の生存率を検討評価することができるが、それは臨床的に適用可能ではない。したがって、樹立された腫瘍異種移植片の生存率を検討評価するために、[18F]-FDG 100 μCi/マウスの静脈内投与2時間後にmicroPETスキャナー(Concorde Microsystems, TN)を用いてマウスを画像化した。[18F]-FDGは商業的に(PETNet, Houston, TX)得られた。HSVtk遺伝子発現を画像化するために、放射性標識ヌクレオシド類似体[18F]-FEAUの静脈内投与1〜2時間後にPETイメージングを行なった。コンピュータ制御されたポジショニングベッドが装着され、10.8-cmの横断方向の視野および8-cmの軸方向の視野(FOV)を有し、それがセプタムを有さずおよび3次元リストモードでもっぱら操作される、microPET R4(Concorde Microsystems, Inc.)でPETイメージングを行なった。350〜750 keVのエネルギーウィンドウおよび6 nsの時間ウィンドウを用いて完全3次元リストモードデータを収集した。全ての生データをまず3次元サイノグラムの中にソートし、次いでASIPRO VMソフトウェア(Concord Microsystems, TN)を用いたフーリエリビニングおよびOSEM画像再構成を行なった。画像ピクセルサイズは、横断方向に1.2 mmスライス厚でおよそ1 mmであった。
【0137】
もとは Alauddinら(2003)によって記載されたように、l-ブロモ-2-デオキシ-2-[18F]フルオロ-3,5-ジ-O-ベンゾイル-α-D-アラビノフラノースによる凝縮のためのピリミジン塩基として5-エチルウラシル-2,5-ビス-トリメチルシリルエーテルを用いることによって、放射性標識[l8F]-FEAUを99%を越える放射化学的純度まで合成した。腫瘍ならびにその他の器官および組織における[18F]-FEAUまたは[18F]-FDG由来の放射能濃度を定量するために、関心対象の領域を画像上に描きかつ測定された値を nCi/mm2から%グラム当たりの注射された用量(%ID/g;Tjuvajev et al., 1998)に変換した。上で記載されたように、HSVtk遺伝子を運ぶ標的化されたAAVPまたは対照の標的化されていないAAVPの投与後3、5、10、および16日目に繰り返しの[18F]-FEAU PETイメージングを行ない;11日目から19日目にGCV処置を投与した。注目すべきことに、そうでない場合にはHSVtk酵素によるリン酸化をFEAUと競合すると考えられる、GCVの十分な消去を可能にするために、16日目のPETイメージングをGCV投薬の24時間後に行なった。[18F]-FDGを用いたPETイメージングをAAVP投与後17日目に繰り返し、もしあれば、残存する腫瘍の生存率を検討評価した。
【0138】
免疫組織化学
麻酔をかけたマウスを殺し、4% PFAを含むPBSを灌流させた。ラット抗マウスCD31抗体(BD Biosciences)を用いることにより凍結切片で腫瘍血管新生を検討評価した。TUNELキット(Promega)を用いてパラフィン包埋切片に対してアポトーシス解析を行なった。組織でのファージ免疫検出のために、パラフィン切片をウサギ抗ファージ一次抗体(Sigma)、次いでペルオキシダーゼがコンジュゲートされた抗ウサギ二次抗体(Dako)とインキュベートした。基質-色原体3,3'-ジアミノベンジジンでスライドを現像し、ヘマトキシリンで対比染色した。GFP免疫染色のために、器官および腫瘍を2% PFAを含むPBS中で2時間固定し、15%スクロースを含むPBS中で48時間平衡化した。凍結切片を4% PFAを含むPBS中で20分間後固定しならびに1% BSAおよび0.1% Triton X-100を含むPBS(PBS-T)中の5%ヤギ血清でブロッキングした。次に、組織切片を2%ヤギ血清および1% BSA 中のウサギ親和性精製GFP抗体(Molecular Probes)とインキュベートした。その後、切片をPBS-Tおよび1% BSA中の二次抗体であるAlexaFluor 488コンジュゲートされたヤギ抗ウサギ(Molecular Probes)で染色した。PBS灌流動物から除去された腫瘍のアセトン固定凍結切片に対してαvインテグリン免疫染色を行なった。切片を一次ラット抗インテグリンαvモノクローナル抗体(Chemicon)と1時間、次いで二次Cy3コンジュゲートされたヤギ抗ラット抗体(Jackson ImmunoResearch)とインキュベートした。
【0139】
哺乳動物細胞において機能的であるリガンド指向性粒子
組換えAAVおよび(RGD-4Cファージ(Pasqualini et al., 1997;Arap et al., 1998)と名付けられた)二重環状ペプチドCDCRGDCFC(SEQ ID NO:2)を提示するfd-tetファージクローンから構成される標的化されたキメラウイルスを構築した。RGD-4Cペプチドは、腫瘍細胞および腫瘍血管の新生血管形成内皮の両方で過剰発現される細胞表面受容体である、αvインテグリンに結合する(Brooks et al., 1994;Pasqualini et al., 1997;Arap et al., 1998;Sipkins et al., 1998;Ellerby et al., 1999;Hood et al., 2002)。(AAV/ファージ;AAVPとさらに称される)キメラウイルスを得るために、本発明者らは、組換えAAV由来の真核生物遺伝子カセットをRGD-4Cファージ(RGD-4C AAVP)、挿入物のないファージ(標的化されていないAAVP)、またはスクランブルのRGD-4C AAVPもしくは(RGE-4Cと名付けられた)DからEへの突然変異体AAVPなどの、対照ペプチドを提示するファージのゲノム間領域に挿入し、およびそれをファージDNAと共にファージカプシド中にパッケージングした(図7)。結果として得られるαvインテグリンに標的化されたキメラウイルスのシスエレメントが依然として機能的であることを示すために、本発明者らは、RGD-4Cペプチドのリガンド特性およびAAVPの文脈における逆向き末端反復(ITR)のレスキュー特性を評価した。まず、ペプチド特異性を評価するために、哺乳動物細胞に結合も感染もしない、標的化されていないAAVPまたはRGE-4Cもしくは様々なスクランブルバージョンのRGD-4C配列(図1A)などの陰性対照ペプチドを提示するAAVPとは対照的に、RGD-4C AAVPがαvインテグリンを発現する哺乳動物細胞に結合することが示された。レポーター遺伝子を運ぶRGD-4C AAVPは、対照と比べて、リガンド指向性の内在化(図1B)および哺乳動物細胞の形質導入(図1C)を媒介することができることも示された。細胞内在化実験については、陰性対照として標的化されていないAAVP、様々なRGD-4CスクランブルのAAVP、またはRGE-4C AAVPが含まれ(図1B);細胞形質導入実験については、標的化されていないAAVP(図lC)、スクランブルのRGD-4C AAVP、またはRGE-4C AAVPが陰性対照の役割を果たした。これらの結果と一致して、本発明者らは、合成RGD-4Cペプチドが様々な標的化されたRGD-4Cファージに基づくコンストラクトの細胞結合および内在化を特異的に阻害することを以前に示している(Giordano et al., 2001;Chen et al., 2004、および未公表の結果)。最後に、哺乳動物細胞形質導入は、標的化されていないAAVP(図1D)、スクランブルのRGD-4C、またはRGE-4Cなどの陰性対照と比べて、合成RGD-4Cペプチドによって特異的に競合させることもできることが示された。これらの結果(図1Aおよび1B)のうちの幾つかが、細胞膜からAAVPを除去するためのグリシン(低pH)洗浄工程の選択的失敗から結果的に生じるアーティファクトを表し得るという可能性を除外するために、温度(氷冷)対照実験を行ない(Giordano et al., 2001)、そこでは細胞結合は観察されたが、RGD-4C AAVPによって媒介される内在化は観察されなかった。
【0140】
次に、ITRがAAVP粒子中で依然として機能的であるかどうかを評価するために、本発明者らはレスキュー実験を行なった。本発明者らは、機能的な組換えAAV粒子がRGD 4C AAVPを形質導入した哺乳動物細胞のみから作製されるが、陰性対照コンストラクトを形質導入した細胞からは作製されないことを示している(図1E)。これらのデータは、RGD-4C AAVP粒子を結果的にもたらす遺伝子のキメラ化が、(i)リガンド指向性RGD-4Cファージのペプチド標的化特性または(ii)RGD-4C AAVP により形質導入された哺乳動物細胞から組換えAAV粒子をレスキューする能力を本来変化させないことを立証している。レポータートランスジーン発現の並列時間経過は、RGD-4C AAVP形質導入が、RGD-4Cファージの形質導入と比較してずっとより長い間検出可能であることを明らかにした(表4)。
【0141】
(表4)インビトロでのトランスジーン発現
2人独立の観察者が、時点当たり三つ組みウェルの293細胞で、半定量的にGFP発現を得点化した。
【0142】
トランスジーン発現の分子メカニズム
AAVP粒子によって媒介されるトランスジーン発現の分子メカニズムについての見識を得るために、本発明者らは、哺乳動物細胞における形質導入されたゲノムの運命を詳しく調べた。まず、GFPneo発現AAVPを用いて個々の形質導入された細胞クローンの選択を可能にすることにより、安定に形質導入された細胞株を作製した。RGD-4C AAVP-GFPneoかまたはAAV ITRを欠くRGD-4Cファージ-GFPneoのいずれかを用いて、αvインテグリンを発現するヒト293細胞(Nakamura et al., 2002)に形質導入し、およびG418選択下でクローンを単離した。レスキュー実験により、機能的な組換えAAV-GFPneoは全てのRGD-4C AAVP-GFPneo 293細胞クローンから作製されたが、RGD-4Cファージ-GFPneoクローンからは何も作製されなかったことが示され、したがってキメラの個々のクローンは機能的なITRを含むことが確認された。非キメラRGD-4Cファージ-GFPneoを形質導入した細胞はG418耐性を保持したが、GFP発現はRGD-4C AAVP-GFPneoクローンのGFP発現よりも一般に弱かった(図2A)。その後、本発明者らは、ゲノムDNAの広範囲にわたる制限酵素消化、それに続くサザンブロッティングおよびポリメラーゼ連鎖反応(PCR)に基づく解析によって、安定に形質導入されたクローンにおけるGFPneoトランスジーンカセットの運命を明らかにしようと試みた(図2、図8、および表5)。トランスジーンカセットの持続性を証明するために、解析前にゲノムDNAをAflIIおよびXhoIで消化して全長トランスジーンカセットの放出を検出した。そのような放出は、非キメラRGD-4C ファージコンストラクトによって形質導入されたクローンの33%(9クローンのうちn=3)でのみと比較して、RGD-4C AAVPによって形質導入されたクローンの100%(9クローンのうちn=9)で観察された(図2B)。ゲノムDNAの別の制限消化を設計して、トランスジーンカセットの潜在的なコンカテマー形態を検出した(Lieber et al., 1999; Hsiao et al., 2001)。トランスジーンカセットの頭-尾コンカテマーの存在はRGD-4C AAVPによって形質導入されたクローンの67%(9クローンのうちn=6)で検出されたが、そのようなコンカテマーは非キメラRGD-4C ファージコンストラクトによって形質導入されたクローンでは検出されなかった(図2C)。トランスジーンカセットのあり得る追加のコンカテマー形態を同定するために、コンストラクトの5'および3'末端の両隣にあるプライマーを用いることによって、マルチプレックスPCRを行なった。この場合もやはり、コンカテマーは非キメラRGD-4C ファージコンストラクトによって形質導入されたクローンでは見出されなかったが、その一方でRGD-4C AAVPによって形質導入されたクローンの100%がコンカテマー形態を含み(9クローンのうちn=9)、それらは全て頭-尾方向で見出された(図8B)。その上、より小さいPCR産物のtopoクローニングにより、ITR欠失を持つトランスジーンカセットの頭-尾方向が明らかにされた(図8C)(Yang et al., 1997)。最後に、大きいPCR産物は配列決定することができなかったが、それらのサイズから、接合部位でのインタクトなITRを持つコンカテマーの存在が示唆された。各々の単一クローンについてのDNAの個々の解析も詳述している(表5)。理論またはメカニズムに束縛されることを望まないが、これらのデータから、哺乳動物トランスジーンカセット全体の維持、エピソームDNAのより良好な持続、トランスジーンカセットのコンカテマーの形成を通じた、またはおそらくはこれらの相互排他的でないメカニズムの組み合わせによるトランスジーンカセットの変更された運命によって、AAVPが遺伝子発現における利点を授与し得ることが示唆される。これらの観察は、AAVの理解における最近の進展と一致する(McCarty et al., 2004)。
【0143】
(表5)RGD-4C AAVP-GFneoまたはRGD-4Cファージ-GFneoによって安定に形質導入された293細胞クローンにおけるベクターDNAの運命
【0144】
インビボでの腫瘍標的化および分子-遺伝子イメージング
標的化されたキメラウイルス粒子の中心的エレメント(すなわち、RGD-4CペプチドおよびAAV ITR)がインタクトかつ機能的であることを示した後でならびに哺乳動物細胞におけるAAVPを介する遺伝子発現の分子メカニズムを解明した後で、AAVPの全身投与後の腫瘍への遺伝子送達の特異性および有効性を評価した。初めの前臨床的モデルとして、ヒトカポジ肉腫KS 1767細胞に由来する皮下腫瘍異種移植片を持つヌードマウスを用いた(Arap et al., 1998;Ellerby et al., 1999)。まず、ウイルスコンストラクトがマウスにおけるKS1767由来異種移植片を標的化することを確認するために、RGD-4C AAVPかまたは幾つかの陰性対照(標的化されていないAAVP、スクランブルのRGD-4C AAVP、もしくはRGE-4C AAVP)のうちの1つのいずれかを静脈内投与した。3〜5分の循環時間の後、腫瘍血管系における強い抗AAVP染色がRGD-4C AAVPを受けたマウスで観察されたが、対照マウスでは観察されなかった(図3A)。緑色蛍光タンパク質(GFP)をコードするRGD-4C AAVP変異体をレポーターとして用いて、インサイチュー免疫蛍光顕微鏡イメージングを用いることによって、このベクター(RGD-4C AAVP-GFP)がKS1767由来異種移植片に形質導入することができるかどうかを明らかにした。腫瘍を有するマウスへのRGD-4C AAVP-GFPかまたは陰性対照コンストラクトのいずれかの全身投与の7日後に、腫瘍におけるおよび異なる器官におけるGFPに対する免疫染色を行なった。免疫蛍光により、主にRGD4C AAVP-GFPを受けたマウスにおける腫瘍血管および周囲の腫瘍細胞でのGFP発現が明らかになった。対照的に、標的化されていないAAVP-GFP、スクランブルのAAVP-GFP、または突然変異体AAVP-GFPを受けた対照マウス由来の腫瘍ではGFP染色は検出されなかった(図3B)。この染色パターンから、リガンド指向性の形質導入は腫瘍の血管内皮におけるαvインテグリンの標的化によって媒介されることが示唆される(Brooks et al., 1994;Pasqualini et al., 1997;Arap et al., 1998;Sipkins et al., 1998;Ellerby et al., 1999;Giordano et al., 2001;Hood et al., 2002;Chen et al., 2004)。矛盾のないことには、幾つかの非標的対照器官(脳、肝臓、膵臓、および腎臓)はGFPの組織発現を欠いていた(図9)。まとめると、RGD-4C AAVP粒子はリガンド指向性のメカニズムによって腫瘍異種移植片を特異的に標的化し、かつインビボでの全身投与後にそれらに形質導入できることをこれらのデータは示している。
【0145】
次に、本発明者らは、RGD-4C AAVPの全身投与後に生きている腫瘍を有するマウスにおける、それぞれホタルルシフェラーゼ(Luc)およびHSVtkレポーター遺伝子発現の時間的力学および空間的不均一性の非侵襲的なモニタリングのための前臨床的な生体発光イメージング(BLI)および[18F]-FEAUを用いた臨床適用可能な分子-遺伝子PETイメージングの有効性を検討評価した。この特に普及している腫瘍は依然として、患者の中で適切に画像化するための課題であるので、これらの分子-遺伝子イメージング検討をヒト前立腺癌の前臨床モデルで実行した。本発明者らはまず、腫瘍を有するマウスにおけるホタルルシフェラーゼ(Luc)トランスジーンレポーターのインビボイメージング用の標準的な実験の仕組みを用いた(図4A)。それがマウスにおけるレポーター遺伝子イメージングのための非常に感度の良い方法でありおよび画像中の非特異的バックグラウンド活性をほとんど有さないので(Gross and Piwnica-Worms, 2005a;Gelovani and Blasberg, 2003;Uhrbom et al., 2004;Walensky et al., 2004)、本発明者らは、Luc発現のBLIを選択した。非常に腫瘍特異的なLucの発現がRGD-4C AAVP-Lucを受けているマウスにおけるDU145腫瘍で観察された。対照的に、腫瘍に関連した生体発光シグナルは、標的化されていないAAVP-LucまたはスクランブルのRGD-4C AAVP-Lucを受けている対照マウスで観察することができなかった。全ての種類のAAVPベクター(標的化されていないAAVP-Luc、スクランブルのRGD-4C AAVP-Luc、RGE-4C AAVP-Luc、およびRGD-4C AAVP-Luc)について、生体発光は肝臓、脾臓、または腎臓などの正常器官では観察されなかった。これらのデータによって、RGD-4C AAVP-GFPを用いた免疫蛍光顕微鏡イメージング検討で観察されたRGD-4C AAVPを介する標的化およびトランスジーン発現の腫瘍特異性が確認されている。ここで示された先の結果と一致して(図9)、非特異的な肝細胞によるファージ粒子のクリアランス(Geier et al., 1973;Pasqualini et al., 1997;Arap et al., 1998;Barbas et al., 2001)にもかかわらず、そのような現象は望ましくない肝臓の形質導入を結果的にもたらさないことが分布の動態から示唆される。これらの観察は、哺乳動物のウイルス遺伝子送達ベクターによる(肝臓などの)正常器官の十分に立証された非特異的な形質導入(Shayakhmetov et al., 2005)とは際立って対照的である。インビボでBLIを用いることによって、腫瘍内のLucレポータートランスジーン発現はAAVP投与3日後に明瞭に検出可能でありおよび徐々に増加して10日目までに最高レベルに達した。Lucレポーター遺伝子発現の繰り返しの2次元BLIが1日おきに行なわれならびにAAVPによって媒介されるレポータートランスジーン発現の特異性、時間的力学、および空間的不均一性を検討するための初期の費用効果的な戦略を提供した。しかしながら、Lucレポーター遺伝子発現のBLIは臨床適用可能ではないので、本発明者らは次に、自殺遺伝子(ガンシクロビル;GCVと組み合わせた場合)としてならびにHSVtk特異的な放射性標識ヌクレオシド類似体(例えば、[124I]-FAIU、[18F]-FHBG、および[18F]-FEAU)を用いる臨床適用可能なPETイメージング用のレポータートランスジーンとしての両方の役割を果たすことができる、HSVtk遺伝子をAAVPベクターに導入した。
【0146】
以前の検討によって、HSVtk発現のPETイメージングは、トランスジーン発現の場所、大きさ、および持続時間を定義する能力を提供することが立証された(Tjuvajev et al., 1998;Tjuvajev et al., 1999;Ray et al., 2001;Massoud and Gambhir, 2003)。HSVtk形質導入細胞株およびインビボの腫瘍における蓄積した放射性標識トレーサーの大きさはHSVtk発現のレベルと相関することも以前に明らかにされている(Blasberg and Tjuvajev, 2003;Gross and Piwnica-Worms, 2005a;Tai and Laforest, 2005)。ここで示された検討において、本発明者らは、とりわけ薬物動態的考察(全ての正常器官および組織における非常に低いバックグラウンド活性)(Kang et al., 2005)から、その他のヌクレオシド類似体よりも良いHSVtk酵素用の放射性標識基質である放射性標識ヌクレオシド類似体2'-[18F]-フルオロ-2'-デオキシ-l-β-D-アラビノ-フラノシル-5-エチル-ウラシル([18F]-FEAU)を選択し、合成し、および使用した。(0、3、5、10、および16日目に)[18F]-FEAUを用いた繰り返しのPETイメージングを用いることにより、本発明者らは、ヌードマウスにおけるDU145由来腫瘍異種移植片ならびにその他の器官および組織中のRGD-4C AAVP-HSVtkまたは標的化されていないAAVP-HSVtkの単一全身投与後のHSVtk遺伝子発現の時間的力学および空間的不均一性を可視化および定量した(図4B)。RGD4C AAVP-HSVtkかまたは標的化されていないAAVP-HSVtkのいずれかの投与前の腫瘍異種移植片サイズ(およそ150 mm2)だけでなく、RGD4C AAVP-HSVtkかまたは標的化されていないAAVP-HSVtkのいずれかの投与後の腫瘍成長速度もマウスの両方のコホートでよく似ていた(図4C)。[18F]-FEAUを用いたPETイメージングにより、RGD-4C AAVP-HSVtk の投与後の最初の5日間の腫瘍におけるHSVtkトランスジーン発現のレベルの漸進的な増加(% グラム当たり投与された静脈内用量の増加)、それに続くベクター投与後10日目に向けてのHSVtk発現レベルの漸進的な安定化が明らかにされた。対照的に、標的化されていないAAVP-HSVtkを受けている対照の腫瘍を有するマウスでは、ほんのわずかな[18F]-FEAUの腫瘍蓄積の増加が3日目に観察され、それは速やかにバックグラウンドレベルまで減少した(図4D)。前述のBLI実験と一致して、[18F]-FEAU PET検出可能なHSVtk発現は、非標的器官または組織では観察されなかった(図4B)。実際、PET画像における低レベルで不均一な活性は、正常なバックグラウンド活性を表し、それは非標的組織に対する腫瘍でのHSVtk発現の特異性を人為的に「改善する」ために低レベルの放射能の切り捨てがなされていないことを示すために画像の中で意図的に強調された。腫瘍が確実に触診可能なサイズ(およそ350〜400 mm2)まで成長し、かつHSVtk発現のプラトーが腫瘍において達成された時、GCVによる処置を動物の全てのコホートで開始した(図4C)。[18F]-フルオロデオキシグルコース([18F]-FDG)を用いたPETイメージングは、グルコース代謝およびGCVによって誘導される腫瘍生存率の変化をモニタリングする役目を果たした。GCV治療の開始2日前(ベクター投与後9日目)に、両群のマウスにおけるDU145腫瘍は生存可能でかつ [18F]-FDGを活発に蓄積した(図4E)。GCV治療後、RGD-4C AAVP-HSVtkを受けたマウスにおける腫瘍の体積は、標的化されていないAAVP-HSVtkを受けたマウスにおいてよりも有意に小さかった(p<0.05;図4C)。その上、[18F]-FDGの蓄積の減少によって証明されているように腫瘍異種移植片もまた代謝が抑制された(図4E)。(FEAUとの競合を避けるために)最後のGCV用量の24時間後に得られる、PET画像における[18F]-FEAU蓄積の際立った減少によって証明されているように、RGD-4C AAVP-HSVtkを投与されたマウスの腫瘍におけるHSVtk発現のレベルもまたGCV治療後に有意に減少した(図4D)。これらの検討により、RGD-4C AAVPによる腫瘍標的化の特異性が確認され、かつHSVtkトランスジーン発現のレベルがGCVの効果的なプロドラッグ活性化にとって十分に高いことが示されている。
【0147】
その他の前臨床モデルで有効性を水平に評価するために、本発明者らは、異なる種および組織学的起源由来の腫瘍細胞株のパネルを集めならびに免疫抑制または免疫適格マウスで腫瘍を発生させた。カポジ肉腫(KS 1767)に由来する腫瘍を有するマウスのコホートは、単一静脈内用量のRGD-4C AAVP-HSVtkかまたは標的化されていないAAVP-HSVtk(対照)のいずれか、それに続く全ての群におけるGCV処置を全身的に受けた。ビヒクルで処置されたマウスまたは標的化されていないAAVPを受けたマウスと比較した場合、RGD-4C AAVP-HSVtkを受けた腫瘍を有するマウスで顕著な腫瘍成長抑制が観察された(図5A)。同様の腫瘍成長抑制効果は、より大きい腫瘍異種移植片を処置した場合(図5D)でも、ヌードマウスにおけるUC3由来膀胱癌腫(図5B)およびDU145由来前立腺癌腫(図5C)で観察され、かつ示されたイメージングデータ(図4)と一致した。観察された抗腫瘍効果が種特異的かまたは異種移植片特異的かのいずれかである可能性を除外するために、本発明者らは、標準的なマウス腫瘍モデルに対するRGD-4C AAVP-HSVtk の有効性を解析しようと試みた。本発明者らは、免疫適格マウスにおいて高度に血管が発達した腫瘍の速やかな成長を誘導するためにEF43-FGF4マウス乳細胞が皮下投与されている(Hajitou et al., 2001)同種同系腫瘍を選んだ。まず、本発明者らは、EF43 FGF4由来腫瘍へのRGD-4C AAVPのリガンド指向性ホーミングを抗ファージ免疫染色により示している(図10)。RGD-4C AAVP-GFPかまたは標的化されていないAAVP-GFPのいずれかを同種同系乳腫瘍を持つマウスに3〜5分の循環時間の間静脈内投与した。標的化されていないAAVP-GFPと違って、RGD-4C AAVP GFPは腫瘍における強い抗ファージ染色を産生した(図10A)。次に、本発明者らは、静脈内投与7日後に抗GFP抗体を用いた免疫蛍光を行ない、RGD-4C AAVP-GFPを受けたマウスにおける腫瘍での強いGFP発現を明らかにし;対照的に、標的化されていないAAVPGFPを受けたマウス由来の腫瘍ではGFP染色が検出されず;矛盾のないことには、抗avインテグリン抗体はEF43-FGF4腫瘍での強い発現を検出した(図10B)。この場合もやはり、単一全身用量のRGD-4C AAVP-HSVtk、それに続くGCVによって、EF43-FGF-4腫瘍の成長が顕著に阻害された(図5E〜Gおよび図6)。その上、治療の終結後に腫瘍が成長して元に戻った場合、RGD-4C AAVP-HSVtkの繰り返された投与によって、EF43-FGF4腫瘍成長が再び阻害され、かつ腫瘍を有するマウスの生存が改善された(図5F)。ファージに基づく粒子は免疫原性であることが公知であるが、この特色はそれ自身を標的化することを通じて調整することができる(Trepel et al., 2001)。事実、RGD-4C AAVP-HSVtkプラスGCVは、非常に高い力価の循環抗ファージIgGにもかかわらず、ファージワクチンを接種した免疫適格マウスに対して驚くべき程に有効であり続けた(図5G)。選択的な実験において、ビヒクル単独、ビヒクルプラスGCV、標的化されていないAAVP、標的化されていないAAVPプラスGCV、標的化されたRGD-4C AAVP、標的化されたRGD-4C AAVP-GFP、および標的化されたRGD-4C AAVP-GFPプラスGCV(模擬形質導入)を含む陰性実験対照の広範囲にわたるパネルを用いた(図6A)。
【0148】
処置後効果について調査するために、本発明者らは、治療7日後に回収されたEF43 FGF4腫瘍の詳細な組織病理学的解析を得た。単一全身用量のRGD-4C AAVP-HSVtkプラスGCVによって引き起こされた徹底的な腫瘍破壊が顕著であった。具体的には、ヘマトキシリンおよびエオシン(H&E)染色によって、腫瘍の中心部の一様な破壊およびほんのわずかの生存している外部縁が明らかになり;対照的に、標的化されていないAAVP-HSVtkはそのような効果を有さなかった(図6B)。抗CD31抗体を用いた染色によって、腫瘍中心領域内の乱された腫瘍血管および外部縁に向かう保存された血管系の両方が確認された一方、標的化されていないAAVP-HSVtkで処置された腫瘍では損傷が観察されなかった(図6B)。HSVtk/GCV戦略は細胞のアポトーシス死と関連するので(Hamel et al., 1996)、本発明者らはまた、アポトーシス細胞に印をする、ターミナルデオキシヌクレオチジルトランスフェラーゼを介するdUTPビオチンニックエンド標識化(TUNEL)染色について腫瘍を評価した。RGD-4C AAVP-HSVtkおよびGCVで処置された腫瘍では、TUNEL染色によって、腫瘍中心領域におけるアポトーシスは検出されたが、外部縁内では検出されず、一方標的化されていないAAVP-HSVtkキメラを受けたマウス由来の腫瘍ではアポトーシスが検出されなかった(図6B)。同じ実験プロトコルで処置された腫瘍を有するマウスから除去された対照器官からは、組織病理学的異常が明らかにならなかった(図11)。まとめると、これらの結果は、単一全身用量のRGD-4C AAVP-HSVtkプラスGCV維持が、腫瘍成長を抑制できることを示している。
【0149】
各々の腫瘍区画(すなわち、腫瘍細胞対腫瘍血管内皮および/またはストローマ)を標的化することの相対的寄与は、リガンド-受容体系および使用されるモデルに依存すると考えられる。例えば、腫瘍細胞における膜標的(すなわち、αvインテグリン)の発現は、低い(EF43 FGF4)から高い(KS1767)まで様々に異なると考えられる。その上、使用される各モデルの形質導入(および適用)のための具体的な至適用量を除いて、トランスジーン発現を調べるための至適時期もある。換言すれば、レポーター遺伝子発現の化学量論は、個々の細胞におけるレポーター発現のレベルおよびパターンだけでなく、増殖しているトランスジーン発現細胞対瀕死のトランスジーン発現細胞の相対的な数にも依存する。したがって、本発明者らは、これらの実施例のための固定されたパラメーターを用いたが、ケースバイケースの基準での標的化されたAAVP至適用量および時間枠のさらなる決定が依然として適用される。
【0150】
本発明者らは、とりわけ異なる前臨床的および臨床的な分子-遺伝子イメージング設定と組み合わせて標的化されたAAVPを用いて、分子腫瘍学を遥かに越える広範囲の現在手に負えない生物学的問題を扱うことができることを企図する。例えば、(CMVプロモーターの代わりに)組織および/または疾患特異的プロモーターを用いた標的部位へのコンストラクトの全身的リガンド指向性送達によって、それらの対応する天然の遺伝子の発現をインビボでモニタリングすることが可能になると考えられ;そのようなプロモーターに駆動されるレポーター活性の転写によって、細胞トラフィッキングおよび生着の検討が可能になると考えられる。レポータートランスアクチベーション、相補、または再構成戦略による基質特異的分解、タンパク質-タンパク質相互作用、およびにその他の分子事象の実験的モニタリング(Luker et al., 2004;De and Gambhir, 2005;Gross and Piwnica-Worms 2005b)などの幾つかの非侵襲的イメージング適用を細胞でおよび動物丸ごとで利用することができる。本発明者らは最近、生体センサーおよび細胞標的化薬剤としての金ナノ粒子およびバクテリオファージのネットワークを記載しており(Souza et al., 2006)、そのような技術をリガンド指向性AAVPと組み合わせて分子-遺伝子イメージングをさらに改良することができる。話は変わるが、(AAVPベクターと対照的に)ITRのないファージに基づくコンストラクトは陰性実験対照としての役割を果たすことができるので、AAVPそれ自体がトランスジーン持続性および染色体組み込みにおけるITR構造の機械的役割を検討するための適した試薬を提供する可能性がある。したがって、協力する多くのその他の全身的な標的化および分子イメージング適用が、この新規のプラットフォームおよびその派生ツールによって比較的短い時間枠で可能である。
【0151】
実施例2
腫瘍血管系にTNF-αを標的化するためのAAVPの使用
A.方法
細胞培養
ヒト臍帯静脈内皮細胞(HUVEC)をCambrex(Walkersville, Maryland)から入手し、以前に記載されたように内皮細胞成長培地-2中で培養した(Tandle et al., 2005)。全ての実験を3〜5代継代のHUVECを用いて実施した。10%血清、2 mM グルタミン、100 u/ml ペニシリン、100 μg/ml ストレプトマイシン、100 μg/mlゲンタマイシン、およびファンギゾンを含むRPMI 1640培地中でM21ヒトメラノーマ細胞を成長させた。 10% 血清、2 mM グルタミン、100 u/ml ペニシリン、100 μg/ml ストレプトマイシン、100 μg/mlゲンタマイシン、およびファンギゾンを含むDMEM培地中でPmel細胞を成長させた。
【0152】
TNF-α/EMAP-IIを発現する標的化されたAAVPの構築
AAVP骨格の一般的な設計および構築は、Hajitouら(2006)に記載されている。TNF-αを発現するAAVPコンストラクトを2工程で創り出した。まず、pG1SiTNF由来の880 bpのNotI/HindIII断片を消化し、pAAV-eGFP/NotI/HindIIIベクターにライゲートしてGFP遺伝子配列を取り替えた(Hwu et al., 1993)。第二の工程において、fMCS/RGDMCSおよびAAV-TNFをPvuIIで消化した。その後、逆向き末端反復(ITR)を持つAAV-TNF-αをfMCS-/RGDMCS PvuII部位に再びライゲートし、AAVPベクターを得た。したがって、pfdTNF-αが標的化されていないベクターである一方、pRGDTNFは細胞表面αvインテグリン受容体に対する結合親和性のある標的化されたベクターである。
【0153】
EMAP-IIを発現するAAVPコンストラクトを3工程で創り出した。成熟EMAP-IIを発現する、pET-20bプラスミドは、The Scripps Research Institute, La Jolla, CAのPaul Schimmelから快く寄贈された。第一の工程において、PCR産物中に制限酵素部位および分泌可能シグナル配列を組み入れるための3つのプライマーを用いて、pET-20bEMAP-IIからEMAP-II配列を増幅させた。プライマー1は、5'末端にNotI制限酵素部位、それに続いて遺伝子産物の細胞外分泌のための(pSecTag2ベクター, Invitrogen, Carlsbad, Californiaから設計された)シグナル配列およびEMAP-II配列がある132 bpのフォワードプライマーである。プライマー2は、PCR産物の増幅を容易にするためのプライマー1のより短いバージョンである。プライマー3は、3'末端にHindIII制限酵素部位があるリバースプライマーである。PCR増幅により、NotIおよびHindIII酵素部位ならびにシグナル配列がある667 bpの産物が作製された。第二の工程において、EMAP-II PCR産物をpCRII-TOPOクローニングベクター(Invitrogen, Carlsbad, California)にクローニングした。結果として得られたクローンを配列決定し、その後667 bpのNotI/HindIII断片を先に説明されたようなpAAV-eGFP/NotI/HindIIIベクターにライゲートした。第三の工程において、fMCS/RGDMCSおよびAAV-EMAP-IIをPvuIIで消化しならびにライゲートしてAAVPベクターを得た。したがって、pfdEMAP-IIが標的化されていないベクターである一方、pRGDEMAP-IIは細胞表面αvインテグリン受容体に対する結合親和性のある標的化されたベクターである。プライマー1:
;プライマー2:
;プライマー3:
【0154】
AAVP粒子精製
標的化されていないおよび標的化されたAAVP粒子を得るために、DNAをエレクトロポレートしてMC1061大腸菌に入れた。細胞およびウイルス粒子を培養上清から精製した。大規模なAAVP粒子を許容性のk91Kan細胞から精製した。細菌形質導入単位(TU)の数を決定するために、k91細胞をファージ粒子の連続希釈に感染させおよびテトラサイクリンおよびカナマイシンを含むルリア-ベルターニ寒天プレート上にプレーティングした。その後、細菌コロニーの数を計数することによってTUを決定した。
【0155】
インビトロファージ内在化アッセイ
M21細胞を6ウェル組織培養プレート中で終夜成長させた。ベクター内在化を検討するために、細胞を培地で洗浄しその後37℃で3時間ウイルス粒子を感染させた。インキュベーション後、ウイルス内在化を停止するためにプレートを氷上に5分間置いた。ハンクスの平衡塩溶液(HBSS)中での細胞の徹底的洗浄によって結合していない粒子を除去した。1時間氷上でのサブチリシン(カルシウムおよびマグネシウムを含まないHBSS中の3 mg/ml サブチリシン、20 mM Tris pH 7.5、2 mM EDTA pH 8.0)による処置によって、細胞外ウイルス粒子を不活化した(Ivanenkov et al., 1999)。その後、細胞を穏やかなピペッティングで剥がし、サブチリンを2 mM EDTAを用いて氷上15分で不活化した。溶解緩衝剤(10 mM Tris pH 7.5、2 mM EDTA pH 8.0、および2%オルトバナジン酸ナトリウム)を用いることによって細胞を溶解させ、内在化したAAVP粒子を放出させた。その後、上で記載されたようなTUとして内在化したAAVP濃度を決定した。
【0156】
免疫蛍光(IF)アッセイ
IFを用いてM21における内在化したウイルス粒子を観察した。簡潔に述べると、8ウェル Lab-Tekチャンバーガラススライド(Nunc, Rochester, New York)上で成長した細胞を10%血清を含むDMEMを用いることによって37℃で16時間AAVP粒子に感染させた。感染後、細胞をPBSで洗浄し、チャンバーを除去し、細胞を3.7%ホルムアルデヒドで10分間固定した。細胞をPBS中の0.1%サポニン(Sigma, St. Louis, Missouri)で透過処理し、ならびにブロッキング緩衝剤(1% BSA, 0.025%アジ化ナトリウム、および0.1%サポニンを含むPBS)で15分間ブロッキングした。透過緩衝剤で洗浄した後、細胞をマウス抗バクテリオファージ抗体で1時間、その後FITCコンジュゲートされた抗マウスIgG抗体で1時間インキュベートした。ガスケットを剥がしならびに細胞をAntifade(MP Biomedicals, Solon, Ohio)を用いてマウントし、Zeiss Axiovert蛍光顕微鏡下で調べた。
【0157】
ファージによる遺伝子発現
内在化アッセイと同様に、M21細胞をAAVP粒子に感染させた。培地を48時間で取り替えた。4日目および12日目に、培養上清を収集し、ELISA(Invitrogen, Carlsbad, California)で分泌可能なサイトカインレベルを測定した。
【0158】
組織因子(TF)アッセイ
分泌されたTNF-α/EMAP-IIが機能的であるかどうかを調べるために、本発明者らは、内皮細胞ECでTF合成を誘導するその能力を調べた。簡潔に述べると、ウェル当たり2 x 105 HUVECを6ウェル組織培養プレート上にプレーティングした。翌日、細胞をM21培養上清で6時間血清を含まないRPMI培地中で処置した。細胞をPBSで洗浄し、25 mM Tris pH 7.5と10分間室温でインキュベートした。その後、培養プレートを-80℃で2時間インキュベートした。総細胞ライセートを組織因子アッセイ緩衝剤(20 mM Tris pH 7.5、150 mM NaCl、および0.1% BSA)中で調製した。ライセートを13,000 rpmで10分間の遠心分離できれいにした。Amelung KC 4A Micro Coagulation Analyzer(Sigma, St. Louis, Missouri)の中でCaCl2(Sigma, St. Louis, Missouri)の存在下で第VIII因子欠損血漿(Geroge King Biomedical Inc, Overland Park, Kansas)の凝集に必要とされる時間を測定することによって、100 μl ライセートを組織因子の存在について解析した。公知の組織因子濃度でプロットされた標準校正曲線を用いることによって、第VIII因子欠損血漿を凝集させるのに必要な時間を組織因子単位に変換した。
【0159】
免疫蛍光染色によるインビボAAVP送達および検出
NIH Animal Care and Use Committeeによって承認されたプロトコルに従って全ての動物実験を実施した。Jackson Laboratoriesからメスの無胸腺ヌードマウスを入手し、National Cancer Institutesの動物施設で収容した。ヒトメラノーマ細胞(4 x 106)をヌードマウスの右脇腹に皮下に播種した。腫瘍体積(mm3)を3次元で測定し、長さ x 幅 x 高さ x 0.52として算出した。腫瘍体積がおよそ100〜150 mm3に達した時、1 x 1011 AAVP粒子を(尾静脈を通して)静脈内注射した。様々な時間間隔で動物に麻酔をかけた。切除された腫瘍組織ならびに対照組織(腎臓および肝臓)をさらなる解析のために凍結した。
【0160】
AAVPの存在を検出するために、5 μM厚の凍結切片を二重IF染色を用いて染色した。簡潔に述べると、切片を4%パラホルムアルデヒド中で5分間固定し、その後PBS中で10分間2回洗浄した。透過処理を1% Triton-X-100を含むPBS中で10分間行なった。切片をImage-iT FXシグナル増強剤と30分間室温(RT)でインキュベートし、その後1% Triton-X-100を含むPBS中で3回洗浄した。非特異的結合を5%ヤギ血清を用いて30分間RTでブロッキングした。一次抗体:1:2000希釈の抗-fdバクテリオファージ抗体(Sigma, St. Louis, Missouri)および1:50希釈の抗マウスCD31(BD Biosciences, San Jose, California)を終夜4℃で適用した。切片を1% Triton-X-100を含むPBS中で10分間3回洗浄し、その後二次抗体(Invitrogen, Carlsbad, California):各1:400希釈のヤギ抗ウサギAlexa Fluor 594およびヤギ抗ラットAlexa Fluor 488と30分間暗中でインキュベートした。切片を1% Triton-X-100を含むPBS中で10分間3回洗浄し、もう一度PBSで洗浄し、その後DAPI(Vector Labs, Burlingame, California)入りのVectashieldマウンティング媒体中でマウントした。
【0161】
インビボでのTNF-α発現
TNF-αタンパク質発現を検出するために、総細胞ライセートをプロテアーゼ阻害剤カクテル(Roche, Branchburg, New Jersey)を含む溶解緩衝剤(50 mM Tris pH 7.4、140 mM NaCl、0.1% SDS、1% NP40、および0.5%デオキシコール酸ナトリウム)を用いて5 μM 凍結組織切片から調製した。ライセートを13,000 rpmで10分間の遠心分離によってきれいにした。BioRADからのタンパク質アッセイ試薬を用いてタンパク質の量を定量した。50 μgの総タンパク質と等しいライセートの量をヒトTNF-αについてELISA(Invitrogen, Carlsbad, California)でアッセイした。
【0162】
TNF-α発現の局在化を調べるために、5 μM 凍結組織を以下のように染色した。簡潔に述べると、切片を4%パラホルムアルデヒドを含むPBS中で20分間固定し、PBSで3回各5分間洗浄し、ならびに非特異的結合を5%ヤギ血清を用いて20分間ブロッキングした。切片を1:200希釈された抗fd抗体(Sigma, St. Louis, Missouri)かまたは1:100希釈されたTNF-α抗体(Novus Biologicals, Littleton, Colorado)のいずれか、またはラット抗マウスCD31 (BD Biosciences, San Diego, California)と1時間インキュベートし、その後洗浄緩衝剤(50 mM Tris pH 7.6および0.02% Tween-20を含むPBS)中で各々5分の3回の洗浄を行なった。内在性ペルオキシダーゼを3%過酸化水素水で5分間ブロッキングし、その後洗浄し、1:200希釈の二次抗マウス抗体-HRPまたはビオチン化ロバ抗ラット抗体と30分間インキュベートした。切片をジアミノベンジジン四塩酸塩基質(Dako, Carpinteria, California)で5分間現像し、ヘマトキシリンで30秒間対比染色し、水道水で濯ぎ、脱水し、きれいにし、ならびにマウントした。
【0163】
アポトーシスアッセイ
製造元の勧める行為に従って、インサイチューアポトーシス検出キット、TACS TdT(R&D Systems, Minneapolis, Minnesota)を用いて、アポトーシスを検出した。
【0164】
腫瘍成長解析
ヒトメラノーマ細胞(3 x 106)をヌードマウスの右脇腹に皮下に播種した。腫瘍体積がおよそ100 mm3に達した時、尾静脈を通して1 x 1011 AAVP粒子を投与した。AAVP粒子投与を7日後にもう一度繰り返した。3日毎に盲式で腫瘍体積を測定することによって腫瘍を有するマウスを追跡調査した。
【0165】
統計解析
分散の解析(ANOVA)およびテューキー比較事後検定(GraphPad Instat Software, Inc., San Diego, California)を用いることによって群を比較した。P値<0.05を統計的有意とみなした。
【0166】
B.結果
哺乳動物細胞によって内在化されるAAVP粒子
以前の検討により、ファージ粒子はインテグリンを介する受容体内在化によって内在化され得ることが示されている(Hajitou et al., 2006)。M21細胞はそれらの細胞表面にαvβ3受容体を発現する(データは示さない)。M21細胞がAAVP粒子を内在化することができるかどうかを調べるために、本発明者らは、細胞にAAVP粒子を感染させ、その後内在化されたファージを計数した。感染後、細胞外ウイルス全体をサブチリシン処置によって不活化し、細胞を溶解し、内在化されたファージをライセート中で回収した。内在化されたAAVP濃度をTUとして測定した(表6)。標的化されたヌル(RGD)または標的化されたTNF-α発現AAVP(RGDTNF-α)に感染した細胞とは対照的に、標的化されていないヌル(fd)かまたはTNF-αを発現する標的化されていないウイルス(fdTNF-α)のいずれかに感染したM21細胞による最低限のAAVP内在化があった。
【0167】
(表6)ファージ内在化アッセイ
【0168】
IFを用いて、M21細胞内部での内在化されたAAVP粒子の局在化を可視化した。標的化されていないTNF-α発現AAVP(fdTNF-α)に感染した細胞は、ファージ局在化を示さなかった(図12A、上のパネル)。対照的に、TNF-αを発現する標的化されたAAVP(RGDTNF-α)に感染した細胞は、M21細胞内部での増強された局在化を示した(図12A、下のパネル)。
【0169】
AAVPを介する遺伝子発現
標的化されたAAVPが、哺乳動物細胞に感染しかつ内部に局在化できることを明らかにした後で、本発明者らは、ウイルス感染が遺伝子産物の発現をもたらすことができるかどうかを調べた。M21細胞にTNF-αを発現するAAVPを、2通りで感染させ、およびTNF-α遺伝子産物の産生をELISAで測定した。遺伝子産物は分泌可能でありかつ培養上清中で検出することができた(図12B)。感染の5日後に検査された上清は、800 pg/mlのTNF-αレベルを示した。12日目に検査された遺伝子産物は、4日目よりも高かった。希釈剤対照(PBS)、標的化されていない空(fd)、TNF-αを発現する標的化されていないウイルス(fdTNF-α)、および標的化されたヌルAAVP(RGD)は、検出可能なレベルのTNF-α分泌を有さなかった(図12B)。
【0170】
AAVP感染M21細胞によって分泌されたTNF-αが機能的であるかどうかを調べるために;本発明者らは、ECで組織因子(TF)合成を誘導するその能力を検査した。分泌されたTNF-αは、ECでTF発現を誘導することができた(図12C)。組換えTNF-αを陽性対照として用いた。TF誘導は、培養上清を TNF-αモノクローナル抗体とインキュベートすることによって阻止することができた。感染後23日で解析された培養上清も機能的TNF-α分泌を示した(図12C)。したがって、AAVPによる単一感染により、感染後23日までの機能的遺伝子産物の産生が結果的にもたらされた。
【0171】
AAVPによるインビボ腫瘍標的化
本発明者らは、哺乳動物細胞のAAVP感染およびインビトロでのTNF-α遺伝子産物の機能的発現を観察した。AAVPのインビボ標的化を評価するために、腫瘍を有するヌードマウスに尾静脈を通してウイルスを全身注射した。
【0172】
希釈剤(PBS)かまたはRGDTNF-α AAVPのいずれかを注射されたマウスに注射後15分、1日、2日、3日、4日、8日、および10日で麻酔をかけた。腫瘍組織由来の凍結切片を二重IF染色によってウイルス粒子の存在について解析した。図13で示されるように、AAVP粒子は赤く染まり(Alexa Flour 594)、血管は緑色に染まり(Alexa Flour 488)、およびDAPIは核染色を示している。PBSを注射された動物はどれもいかなる時点でもAAVPの存在を示さなかった。PBSを15分間注射された動物由来の代表的な腫瘍切片を示す(図13A)。RGDTNF -αを発現するAAVPを注射された動物は、血管におけるウイルス粒子の共局在化を示した(図13B〜H)。ウイルス粒子の最大の蓄積は15分後に注射された動物で検出された(図13B)。AAVPの存在は、検査された全ての時点、1日目(図13C)、2日目(図13D)、3日目(図13E)、4日目(図13F)、8日目(図13G)、および10日目(図13H)で検出された。しかしながら、本発明者らは、時間の経過に伴う検出可能なウイルス粒子の漸進的な減少に注目した。
【0173】
インビボで正常組織を標的化しないAAVP
インビボでの腫瘍血管へのAAVP標的化の特異性を調べるために、本発明者らは、PBSかまたは異なる時点でRGDTNF-αを発現するAAVPのいずれかを注射したヌードマウス 由来の2つの対照組織を調べた(図14および図15)。先に記載されたような二重IF染色を用いてウイルスの存在を検出した。
【0174】
図14で示されたように、本発明者らは、1日目に肝臓組織でウイルス粒子の若干の染色を観察した(図14A)。しかしながら、ウイルス染色は2日目までに最低限のレベルまで低下し(図14B)、3日目(図14C)、8日目(図14D)、および10日目(図14E)にはウイルス染色は観察されなかった。図15は、ウイルス粒子の存在について染色された腎臓切片を示す。AAVP粒子は、検査された時点、1日目(図15A)、2日目(図15B)、3日目(図15C)、8日目(図15D)、および10日目(図15E)のいずれにおいても腎臓で検出されなかった。それにもかかわらず、異なる時点での腎臓組織は全て、良好な血管染色を示した。
【0175】
AAVPによる機能的遺伝子産物のインビボ発現
腫瘍血管系に標的化されたAAVP粒子が遺伝子産物を発現することができるかどうかを調べるために、本発明者らは、2通りで、3日目、4日目、8日目、および10日目の腫瘍組織をTNF-αタンパク質レベルについてELISAで解析した(図16)。本発明者らは、検査した組織全てにおいて基礎レベルの内在性TNF-α発現を観察した。PBSを注射された動物は、任意の検査された時点で内在性レベルを超えるいかなる増加も示さなかった。RGDTNF-α AAVPを注射されたマウスは、4日目から始まりかつ10日目まで徐々に増加するTNF-α発現を示した(図16)。
【0176】
遺伝子産物が産生されている細胞タイプを決定するために、本発明者らは、TNF-αに特異的な抗体を用いて組織切片を染色した。本発明者らは、血管周辺のTNF-α染色の存在を観察した(図17A)。TNF-α発現の効果を見るために、本発明者らは、アポトーシスについて、腫瘍切片を染色した。TACSブルー標識を用いてDNA断片を検出した。アポトーシス細胞は青く染まっている(図17B、左のパネル)。アポトーシス細胞をネクローシス細胞と区別するために、試料を核ファストレッドで対比染色し、アポトーシスの形態学的検証を手助けした。本発明者らは、血管およびアポトーシスを経ている周囲の腫瘍細胞を観察した。右のパネルは、CD31特異的抗体で染色された血管を示す(図17B、右のパネル)。
【0177】
腫瘍成長解析
本発明者らは、2つの異なる腫瘍モデル、TNF-α感受性(M21)およびTNF-α耐性(Pmel)において、ヌードマウス中で成長した腫瘍異種移植片に対するAAVPの効果を解析し、TNF-αを発現するAAVPの処置有効性を調べた。TNF-αに感受性である、ヒトメラノーマM21腫瘍をヌードマウス中で皮下に成長させた。腫瘍進展後、マウスを様々なAAVPコンストラクトまたはPBSで尾静脈注射を通じて全身的に処置した。動物を27日間追跡調査した。TNF-αを発現する標的化されたAAVP(RGDTNF-α)を用いたM21腫瘍の処置は、特徴的な中心腫瘍ネクローシスおよび腫瘍縮小を示した。異なるコホートについて最後に測定された時点である、27日目に、PBS処置群は743±383(±SD)mm3の平均腫瘍体積を有し、標的化されていないfdTNF群は613±155(±SD)mm3の平均腫瘍体積を有し、ヌルの標的化されたRGDファージは、622±141(±SD)mm3の平均腫瘍体積を有し、および標的化されたTNFα発現群は358±98(±SD)mm3の平均腫瘍体積を有した(p<0.048)(図18A)。RGDTNF-α群における腫瘍体積の低下は、20日目から統計的に有意であった。
【0178】
TNF-α耐性腫瘍モデルでは、TNF-αを投与する前にEMAP-IIを発現するAAVPを用いてそれらを前処置することによって、ヒトメラノーマPmel腫瘍をTNF-α効果に対して増感させた。以前の検討において、本発明者らは、EMAP-IIのウイルスベクター送達が、全身送達されたTNF-αの効果に対して耐性腫瘍を増感させることができることを示した。ヌードマウス中で皮下に成長した腫瘍をEMAP-IIを発現するAAVPを用いて全身的に処置し、その後組換えTNF-α(rTNF-α)を用いた全身処置を行なった。処置後2週間、腫瘍を追跡調査した。図18Bで示されたように、EMAP-IIを発現する標的化されていないAAVP(fdEMAP)で処置されたマウスは、PBS単独で処置されたマウスと比較して同様の腫瘍成長を示した。rTNF-αかまたはEMAP-IIを発現する標的化されたAAVP(RGDEMAP-II)単独のいずれかで処置されたマウスは非常にわずかの効果を示し、かつPBSまたはfdEMAP-II群と有意には異ならなかった。しかしながら、RGD-EMAP-IIウイルスおよびrTNF-αの組み合わせで処置されたマウスは、腫瘍体積の有意な低下を示した(p = 0.007)。
【0179】
実施例3
トランスフェリン/トランスフェリン受容体系を標的化するヒトトランスフェリンペプチド
培養中のヒト神経膠腫細胞のCRTIGPSVC(SEQ ID NO:1)AAVP-Lucによる形質導入。U87ヒト由来神経膠腫細胞を40,000細胞/ウェルの濃度で24ウェルプレート上に播きおよび終夜37℃で培養した。次の日、Nature Methodのプロトコルに従って、細胞をAAVPとインキュベートした。RGD-4C AAVP GFPおよびRGD-4C AAVP Lucを形質導入効率についての陽性対照として用いた。画像は7日目に撮った。ファージ摂取は低いが、細胞を鉄AAVPヒドロゲル中で培養した場合に劇的に増加する(プレートおよびグラフの最後の行)(図19A〜19B)。本発明者らは、U87ヒト由来神経膠芽腫細胞を持つ動物への全身投与後に、CRTIGPSVC(SEQ ID NO:1)AAVP-Lucの特異性および有効性を評価した。ファージ注射の7日後にルシフェラーゼ活性を測定した。
【0180】
参照
以下の参照は、それらが本明細書において示された詳細を補足する例示的で手続き的なまたはその他の詳細を提供する程度まで、参照により本明細書に具体的に組み入れられる。
【図面の簡単な説明】
【0181】
以下の図面は、本明細書の一部を形成しかつ本発明のある種の局面をさらに示すために含まれる。本発明は、本明細書において提示された具体的な態様の詳細な説明と組み合わせたこれらの図面の1つまたは複数への参照によってより良く理解され得る。本開示の幾つかの具体的な実施例態様は、以下の説明および付随する図面を一部参照することによって理解され得る。
【0182】
【図1】図1Aは、標的化されていないAAVPまたはRGE-4Cもしくは様々なスクランブルバージョンのRGD-4C配列などの陰性対照ペプチドを提示するAAVPと対照的に、αvインテグリンを発現する哺乳動物細胞へのRGD-4C AAVPの結合を示すグラフである。図1Bは、レポーター遺伝子を運びリガンド指向性の内在化を示すRGD-4C AAVPを示す画像である。図1Cは、RGD-4C AAVP-β-ガラクトシダーゼによって媒介されたKS1767細胞への標的化された遺伝子転移を示す。図1Dは、合成RGD-4Cペプチドによるが、関連のない対照ペプチドにはよらない形質導入の阻害を示し;非特異的な形質導入レベルを標的化されていないAAVPを用いることによって決定した。抗β-gal抗体を染色に用いおよび遺伝子発現を免疫蛍光によって検出した。図1Eは、RGD-4C AAVPを感染させた細胞からの組換えAAVのレスキューを示す。ヒト293細胞を標的化されたRGD-4C AAVP-GFP(106形質導入単位/細胞)または陰性対照(標的化された非キメラRGD-4Cファージ-GFP、標的化されていないAAVP-GFP)とインキュベートした。感染4日後、細胞にAAV rep-およびcap-発現プラスミドをトランスフェクトしならびに野生型アデノウイルス5型(Ad)を重感染させた。アデノウイルス感染72時間後に細胞を採取し、その後上清を用いて新しい293細胞に感染させた。48時間後フローサイトメトリーによってGFP発現を解析した。各コンストラクトからのレスキュー後にバックグラウンドを超えて産生された組換えAAV-GFPの平均増加を示す。
【図2】図2Aは、AAVP-GFPneo(n=9)またはファージ-GFPneo(n=9)のいずれかを安定に形質導入された293クローンのフローサイトメトリー解析の概要を示すヒストグラムである。白抜きの三角は緑色蛍光タンパク質(GFP)陽性細胞のパーセンテージを示し、黒い棒はGFP発現レベル(平均蛍光強度;MFI)を表す。図2Bは、RGD-4C AAVP-GFPneoまたはRGD-4Cファージ-GFPneoを形質導入されたクローン性の細胞株におけるトランスジーンカセットの持続のサザンブロット解析を示す。形質導入されていない293親細胞または形質導入された細胞クローンの各々(各群について#1〜9)由来の総細胞DNAをAflII-XhoIで二重消化した(トランスジーンカセットの両隣にあるコンストラクトDNA内部に各酵素当たり1つの制限消化部位)。図2Cは、サザンブロットによるトランスジーンカセットの潜在的な頭尾コンカテマーの解析を示す。サザンブロッティングの前に総細胞DNAをXhoIで消化した(3'ITRの隣のトランスジーンカセット内部に単一の制限消化部位)。
【図3】図3Aは、KS1767由来腫瘍異種移植片を持つ深く麻酔されたヌードマウスへのRGD-4C AAVP(5 x 1010 TU)または陰性対照(標的化されていないAAVP、スクランブルのRGD-4C AAVP、もしくはRGE-4C AAVP)の(尾静脈を通じた静脈内への)全身投与後のKS1767由来異種移植片におけるファージに対する免疫組織化学染色を示す。AAVPコンストラクトを5分間循環させ、それに続いて灌流および腫瘍の外科手術除去を行なった。ファージに対するポリクローナル抗体をパラフィン包埋腫瘍切片に対する染色に用いた。矢印は、腫瘍血管におけるファージ染色を示す。図3Bは、RGD 4C AAVP-GFPまたは示されたような陰性対照(標的化されていない、スクランブルの、もしくは突然変異体)のいずれかの全身投与7日後のKS1767由来異種移植片におけるGFP発現の免疫蛍光解析を示す。
【図4】図4Aは、標的化されたAAVPの全身送達後のホタルルシフェラーゼ発現のインビボ生体発光イメージングを示す。DU145由来腫瘍異種移植片を持つヌードマウスは、全身単一用量のRGD-4C AAVP-Luc(5 x 1011 TU、静脈内)または対照(標的化されていないAAVP-Luc、もしくはスクランブルのRGD-4C AAVP-Luc)のいずれかを受けた。10日後、腫瘍を有するマウスの生物発光イメージング(BLI)を行ない、トランスジーン発現を検討評価した。校正目盛をパネル中に提供する。図4Bは、標的化されたRGD-4C AAVP-HSVtkの全身送達後の腫瘍を有するマウスにおけるマルチトレーサーPETイメージングを示す。DU145由来腫瘍異種移植片を持つヌードマウス(各コホート当たりn=9の腫瘍を有するマウス)は、全身単一用量(5 x 1011 TU、静脈内)のRGD-4C AAVP-HSVtkまたは標的化されていないAAVP-HSVtkを受けた。GCV処置の前および後に得られた[18F]-FDGおよび[18F]-FEAUを用いたPET画像を示す。T、腫瘍;H、心臓;BR、脳;BL、膀胱。校正目盛をパネル内に提供する。[18F]-FDGおよび[18F]-FEAU生体分布の解釈を簡単にするために、代表的な腫瘍を有するマウスのPET画像および写真画像の重ね合わせ面付け(overimposition)を行なった。図4Cは、AAVP投与後の個々の腫瘍異種移植片の成長曲線を示す。図4Dは、AAVP投与後の異なる日における[18F]-FEAUを用いた繰り返しのPETイメージングによって検討評価された場合のHSVtk遺伝子発現の時間的力学を示す。図4Eは、[18F]-FDG PETを用いて検討評価された場合のGCV治療の前および後の腫瘍生存能力の変化を示す。
【図5】図5Aは、時間に対するプロットされた平均腫瘍体積±標準偏差(SD)を示すグラフである。KS1767細胞に由来する(およそ50 mm2でサイズが一致した)樹立されたヒト異種移植片を持つ免疫不全ヌードマウスのコホートを用いた。マウスは、RGD-4C AAVP-HSVtkかまたは対照(標的化されていないAAVP-HSVtk、RGD-4C AAVP-GFP、もしくはビヒクル単独)のいずれかの単一全身投与(5 x 1010 TU、静脈内)を受けた。処置後2日目から実験の終わりまで、ガンシクロビル(GCV)をマウスに投与した。RGD-4C AAVP-HSVtkで処置されたが後にGCVで処置されていない追加の対照群の他は、全てのマウスがGCVを受けた。図5Bは、時間に対するプロットされた平均腫瘍体積±標準偏差(SD)を示すグラフである。膀胱UC3癌腫細胞に由来する(およそ50 mm2でサイズが一致した)樹立されたヒト異種移植片を持つ免疫不全ヌードマウスのコホート。マウスは、RGD-4C AAVP-HSVtkかまたは対照(標的化されていないAAVP-HSVtk、RGD-4C AAVP-GFP、もしくはビヒクル単独)のいずれかの単一全身投与(5 x 1010 TU、静脈内)を受けた。処置後2日目から実験の終わりまで、ガンシクロビル(GCV)をマウスに投与した。RGD-4C AAVP-HSVtkで処置されたが後にGCVで処置されていない追加の対照群の他は、全てのマウスがGCVを受けた。図5Cは、時間に対するプロットされた平均腫瘍体積±標準偏差(SD)を示すグラフである。前立腺DU145癌腫細胞に由来する(およそ50 mm2でサイズが一致した)樹立されたヒト異種移植片を持つ免疫不全ヌードマウスのコホート。マウスは、RGD-4C AAVP-HSVtkかまたは対照(標的化されていないAAVP-HSVtk、RGD-4C AAVP-GFP、もしくはビヒクル単独)のいずれかの単一全身投与(5 x 1010 TU、静脈内)を受けた。処置後2日目から実験の終わりまで、ガンシクロビル(GCV)をマウスに投与した。RGD-4C AAVP-HSVtkで処置されたが後にGCVで処置されていない追加の対照群の他は、全てのマウスがGCVを受けた。示されているのは、時間に対するプロットされた平均腫瘍体積±標準偏差(SD)である。図5Dは、単一全身用量(5 x 1010 TU、静脈内)のRGD-4C AAVP-HSVtkによる(およそ150 mm2の)大きいDU145由来異種移植片の成長阻害を示すグラフである。図5Eは、単一静脈内用量(5 x 1010 TU)のRGD-4C AAVP-HSVtkによる免疫適格BALB/cマウスにおける(およそ50 mm2でサイズが一致した)EF43-FGF4マウス乳癌腫の腫瘍成長の阻害を示すグラフである。図5Fは、同種同系のEF43-FGF4腫瘍を持つ免疫適格BALB/cマウスへの繰り返された用量のAAVP(各々5 x 1010 TU、静脈内)によるRGD-4C AAVP-HSVtkおよびGCVの長期にわたる有効性を示すグラフである。腫瘍を有するマウスコホート(処置群当たりn=10マウス)を用いた独立の実験で治療結果を一貫して観察した。矢印は、AAVP投与の時間を示す。図5Gは、ファージに対する液性免疫応答のRGD-4C AAVP-HSVtkを用いた治療に対する効果を示すグラフである。BALB/cマウス免疫適格マウス(群当たりn=7マウス)はまず、3全身用量のRGD-4C AAVP-GFP(週当たり1010 TUで3週間、静脈内)を受けることにより、「ワクチン接種」された。その後、マウスにEF43-FGF4細胞を埋め込んだ。腫瘍が樹立された(およそ50 mm2でサイズが一致した)場合、腫瘍を有するマウスは、1全身単一用量のRGD-4C AAVP-HSVtkまたは標的化されていないAAVP-HSVtk、それに続いて(AAVPの投与後2日目に始められた)GCV維持を受けた。ELISAで高力価(およそ1:10,000まで)の循環抗ファージIgGの存在を確認するために、ワクチン接種を開始する前および後にならびにAAVP投与前のワクチン接種計画の最後に再度、マウスから血清試料を収集した。ワクチン接種は、抗ファージ抗体の存在にもかかわらず、抗腫瘍効果に影響を及ぼすようには見えなかった。
【図6】図6Aは、治療の全ての実験群由来の腫瘍を有するマウス(上のパネル)および対応する外科手術で除去された腫瘍(下のパネル)の画像である(BALB/c免疫適格マウスにおけるEF43-FGF4乳腫瘍)。図6Bは、EF43-FGF4で処置された腫瘍の組織病理学的解析である。EF43-FGF4腫瘍を回収し、切片を作製し、および染色した。標的化されていないAAVP-HSVtkで処置された腫瘍(左のパネル)、外部縁と中心の腫瘍部の間の境界(真ん中のパネル)、およびRGD-4C AAVP-HSVtkで処置された腫瘍の中心の腫瘍部(右のパネル)を連続腫瘍切片の低倍率挿入図からの高倍率図として示す。腫瘍切片のヘマトキシリンおよびエオシン(H&E)染色、抗CD31免疫染色、およびTUNEL染色を示す。矢印は、腫瘍血管およびアポトーシス細胞を示す。
【図7】RGD-4C AAVPの構築の組み立てを示す図解であり、クローニング戦略および結果として得られるベクターの構造が描かれている。
【図8】図8Aは、293形質導入クローン性細胞株中のRGD-4C AAVP-GFPneoまたはRGD-4Cファージ-GFPneo由来のDNAのサザンブロット解析を示す。形質導入されていない293親細胞または形質導入された個々の安定細胞クローン(各ベクターについて#1〜9)由来の総細胞DNAをStuI(ベクターDNA内に制限消化部位なし)、Xba-I(ベクターDNA内に単一の制限消化部位)、またはSacI-MluIとインキュベートした。結果として得られるDNA断片を示されたように0.8%アガロースゲル上で分離し、ナイロン膜に移し、および標識されたneoプローブとハイブリダイズさせた。消化されたベクタープラスミドも対照として用いた。図8Bは、RGD-4Cファージ-GFPneoまたはRGD-4C AAVPGFPneoを安定に形質導入した293クローン性細胞株中のトランスジーンカセットのコンカテマーのPCR解析を示す。形質導入されていない293親細胞は陰性対照としての役割を果たした;示されたように追加の陰性対照および100-bp分子マーカー(Invitrogen)も示す。RGD-4C AAVPおよびRGD-4Cファージに対応するDNAにおけるコンカテマー形態を同定するために、トランスジーンカセットの5'および3'末端近くにアニールするプライマーを用いたネスティッドPCRを行なった。矢印はプライマーを示す(H、頭;T、尾)。PCR増幅産物は2%アガロースゲルで検出され、もっぱらAAVPを形質導入された細胞クローンにおけるコンカテマーのベクターDNAを明らかにした。図8Cは、AAVP DNAにおける異なるコンカテマー形態のシークエンシング結果を示し(大文字はトランスジーンカセットの3'末端を意味し、イタリック文字はトランスジーンカセットの5'末端を意味する)、欠失したITRを持つ頭-尾コンカテマーを明らかにした。
【図9】RGD-4C AAVP-GFPかまたは標的化されていないAAVP-GFPのいずれかのマウスへの静脈内用量(5 x 1010 TU)7日後の対照器官である脳、肝臓、膵臓、および腎臓におけるレポーター遺伝子発現GFPの免疫染色を示す画像である。
【図10】図10Aは、EF43-FGF4腫瘍を持つ免疫適格BALB/cマウスへのRGD-4C AAVP(右のパネル)または標的化されていないAAVP(真ん中のパネル)の静脈内投与後のAAVPに対する免疫組織化学的染色の画像である。コンストラクトを5分間循環させ、それに続いて記載されたように灌流および組織回収を行なった。ファージに対するポリクローナル抗体を染色に用いた。左のパネルは、抗CD31抗体を用いることによる腫瘍血管染色を示す。矢印は腫瘍血管を示す。図10Bは、EF43-FGF4腫瘍を持つマウスへの標的化されていないAAVP-GFP(左のパネル)またはRGD-4C AAVP-GFP(真ん中のパネル)の静脈内投与1週間後のEF43-FGF4腫瘍におけるGFP発現の免疫蛍光解析の画像である。EF43-FGF4腫瘍におけるαv-インテグリンに対する免疫染色も示す(右のパネル)。
【図11】標的化されていないAAVP-HSVtk、RGD-4C AAVP-HSVtkベクター、またはビヒクル単独に加えてGCV維持で処置されたマウス由来の対照器官である肝臓、心臓、および腎臓の組織学的解析の画像が示されていることを示す。毒性の組織学的兆候は、これらの器官でH&E染色によって検出されなかった。
【図12】RGD標的化されたAAVPを感染させた哺乳動物細胞は機能的な遺伝子産物を発現する。(図12A)ヒトメラノーマ細胞、M21にAAVP;標的化されていないTNF-α発現ファージfdTNF-α(上のパネル)および標的化されたTNF-α発現ウイルスRGDTNF-α(下のパネル)を感染させならびにfd特異的一次抗体、次いでFITC標識二次抗体を用いて検出した。(図12B)PBS、空の標的化されていないウイルス(fd)、空の標的化されたウイルス(RGD)、標的化されていないTNF-α発現ウイルス(fdTNF-α)、および標的化されたTNF-α発現ウイルスRGDTNF-αを感染させたM21細胞。感染5日後、培養上清を解析し、ELISAでTNF-αを測定した。感染12日後の培養上清も示す。(図12C)ヒト臍帯静脈内皮細胞(HUVEC)を様々な群;PBS、fd(標的化されていないヌルウイルス)、RGD(標的化されたヌルウイルス)、fdTNF(標的化されていないTNF-α発現ウイルス)、およびRGDTNF(標的化されたTNF-α発現ウイルス)で感染させたM21細胞由来の5日目の上清で処置しならびに組織因子(TF)産生について解析した。組換えTNF-αを陽性対照として用いた。特異性を調べるために、HUVECに適用する前に、RGDTNF 感染細胞由来のM21上清をTNF-α特異的抗体とインキュベートした。感染後23日目の上清もTF分泌について検査した。
【図13】AAVPは腫瘍血管系に特異的に標的化される。バクテリオファージ特異的抗体およびCD31血管抗体で染色されたPBSかまたはRGDTNF-αのいずれかを注射されたヒトメラノーマ異種移植片。Alexa Fluor 488、Alexa Fluor 594、およびDAPIを用いて検出を行ない、それぞれ血管、AAVP、および細胞核を可視化した(250X)。PBSを注射された動物はどれも、いずれの時点でもAAVPの存在を示さなかった。PBSを15分間注射された動物由来の代表的な腫瘍切片を示す(図13A)。RGDTNF-αを発現するAAVPを注射された動物は、早ければ15分(図13B)ならびにその後の時点:1日目(図13C)、2日目(図13D)、3日目(図13E)、4日目(図13F)、8日目(図13G)、および10日目(図13H)での血管におけるウイルス粒子の共局在化を示した。
【図14】AAVP粒子は正常肝臓組織では検出されなかった。バクテリオファージ特異的抗体およびCD31血管抗体で染色されたRGDTNF-αを注射されたヒトメラノーマ異種移植片を持つ動物由来の肝臓。Alexa Fluor 488、Alexa Fluor 594、およびDAPIを用いて検出を行ない、それぞれ血管、AAVP、および細胞核を可視化した(250X)。RGDTNF-αを発現するAAVPを注射された動物は、肝臓組織における1日目(図14A)でのウイルス粒子の若干の染色を示した。しかしながら、ウイルス染色は、2日目(図14B)までに最低限のレベルまで低下し、3日目(図14C)、8日目(図14D)、および10日目(図14E)時点でウイルス染色は観察されなかった。異なる時点での肝臓組織は全て、良好な血管染色を示した。
【図15】AAVP粒子は正常腎臓組織では検出されなかった。RGDTNF-αを注射されたヒトメラノーマ異種移植片を持つ動物由来の腎臓をバクテリオファージ特異的抗体およびCD31血管抗体で染色した。Alexa Fluor 488、Alexa Fluor 594、およびDAPIを用いて検出を行ない、それぞれ血管、AAVP、および細胞核を可視化した(250X)。本発明者らは、検査した時点、1日目(図15A)、2日目(図15B)、3日目(図15C)、8日目(図15D)、および10日目(図15E)のいずれにおいても腎臓におけるAAVPの存在を観察しなかった。それにもかかわらず、異なる時点での腎臓組織は全て、良好な血管染色を示した。
【図16】インビボにおいて、AAVPは特異的な遺伝子産物を発現する。PBSかまたはRGDTNF-αのいずれかを注射されたヒトメラノーマ異種移植片を持つ動物由来の凍結切片を用いて総タンパク質を抽出した。50 μgの総タンパク質を用いて、ELISAにより2通りでTNF-αタンパク質レベルを測定した。内在性TNF-α発現の基礎レベルを検査した全ての組織で観察した。PBSを注射された動物は、検査した任意の時点で内在性レベルを超えるいかなる増加も示さなかった。RGDTNF-α AAVPを注射されたマウスは、4日目に始まりおよび10日目まで徐々に増加するTNF-α発現を示した。
【図17】AAVPは血管系でTNF-αを発現しかつインビボでアポトーシスを誘導する。(図17A)免疫組織化学的解析を用いてTNF-α特異的抗体で染色されたRGDTNF-α AAVPを注射されたヒトメラノーマ異種移植片を持つ動物由来の凍結切片。TNF-α染色は血管の周辺に茶色の染色として見られる(左のパネル、100X;右のパネル、400X)。(図17B)アポトーシス細胞を検出するために染色されたRGDTNF-α AAVPを注射されたヒトメラノーマ異種移植片を持つ動物由来の凍結切片。血管および周囲の腫瘍細胞は、TACSブルー標識で染色された青く色付いた細胞に見られるアポトーシスを示した(左のパネル、200X)。試料は核ファストレッドで対比染色されている。右のパネルは、CD31特異的抗体で染色された血管を示す(200X)。
【図18】AAVPを用いたTNF-α感受性ヒトメラノーマ、M21(図18A)およびTNF-α耐性ヒトメラノーマPmel(図18B)の処置。皮下に埋め込まれたM21/Pmel腫瘍を持つヌードマウスをAAVPで尾静脈注射を通じて全身的に処置し、腫瘍体積を異なる時点で測定した。処置後の異なる日に対してプロットされた腫瘍体積。(図18A)RGDTNF-αで処置された動物は、20日目から統計的に有意な(p<0.05)腫瘍体積の低下を示した。(図18B)AAVPによるEMAP-IIの送達、それに続く組換えTNF-αを用いた処置によって、TNF-α耐性ヒトメラノーマをTNF-α治療に対して感受性にした。rTNF-αかまたはEMAP-IIを発現する標的化されたAAVP(RGDEMAP-II)単独のいずれかで処置されたマウスは、非常にわずかの効果を示し、PBSまたはfdEMAP-II群と有意には異ならなかった。RGD-EMAP-IIウイルスおよびrTNF-αの組み合わせで処置されたマウスは腫瘍体積の有意な低下を示した(p=0.007)。
【図19】図19Aおよび19Bは、培養中のヒト神経膠腫細胞のCRTIGPSVC(SEQ ID NO:1)AAVP-Lucによる形質導入を示す。U87ヒト由来神経膠腫細胞を40,000細胞/ウェルの濃度で24ウェルプレート上に播きおよび終夜37℃で培養した。次の日、 Nature Methodのプロトコルに従って、細胞をAAVPとインキュベートした。RGD-4C AAVP GFPおよびRGD-4C AAVP Lucを形質導入効率についての陽性対照として用いた。画像は7日目に撮った。ファージの取り込みは低いが、細胞を鉄AAVPヒドロゲル中で培養した場合に劇的に増加する(プレートおよびグラフの最後の行)。
【図1A】
【図1B】
【図1C】
【図1D】
【図1E】
【図2A】
【図2B】
【図2C】
【技術分野】
【0001】
本出願は、その全体が参照により本明細書に組み入れられる、2006年4月7日に申請された米国特許仮出願第60/744,492号に対する優先権を主張する。
【0002】
米国政府は、国立衛生研究所(NIH)からの助成金に応じた本発明における権利を所有する。
【0003】
I、発明の分野
本発明の態様は一般に、生物学および医学に向けられている。特に、本発明は、対象に治療を提供するためのイメージングと組み合わせてAAVPを用いる遺伝子治療の分野に向けられている。
【背景技術】
【0004】
発明の背景
II、背景
多くの生物学に基づく治療の限界は、生体活性分子の腫瘍細胞またはそれらの周囲のマトリックスへの制御されたおよび効果的な送達を達成することができないことである。遺伝子に基づく治療を利用する目的は、遺伝子発現の結果としての、生体産物の細胞内のそれらの作用の部位への効果的な送達を達成することである。遺伝子に基づく治療はまた、転移される遺伝子材料に特異的なプロモーター/アクチベーターエレメントを含めることによって、これらの生体活性産物の作用のレベル、時期、および持続時間に対する制御を提供し、より効果的な治療的介入を結果的にもたらすことができる。新規のDNAの製剤を用いた様々な体細胞組織および腫瘍への制御された遺伝子送達のための、ならびに細胞特異的で、複製により活性化され、かつ薬物により制御される発現系を用いて遺伝子発現を制御するための方法が開発中である。
【0005】
1つのアプローチにおいて、遺伝子治療は特異的な様式で細胞を標的化しようと試みる。したがって、標的細胞または組織の中に遺伝子を送達するために、治療的遺伝子は何らかの方法で標的化分子に連結される。現在の方法は典型的には、内在化受容体を認識するリガンドまたは抗体などの標的化分子を所望の遺伝子を含む裸のDNAかまたは哺乳動物細胞ウイルスのいずれかに連結する工程を伴う。裸のDNAを用いる場合、それをインビトロで凝縮させて、細胞内への侵入用の小型形状にしなければならない。DNA上の電荷を中和しかつDNAを凝縮させて環状体構造にするためにポリリジンなどのポリカチオンが一般に用いられる。しかしながら、この凝縮過程は十分に理解されているわけではなくかつ制御するのが難しく、したがって、均質な遺伝子治療薬物の製造を極めて厄介なものにしている。
【0006】
ラムダおよび繊維状ファージなどの、バクテリオファージ(ファージ)は、哺乳動物細胞内にDNAを転移しようとする努力において時折用いられる。一般に、ラムダの形質導入は、比較的稀な事象であることが分かっておりかつレポーター遺伝子の発現は弱かった。形質導入効率を増強しようとする努力において、(細胞表面受容体を特異的に標的化するわけではない)リン酸カルシウムまたはリポソームを活用する方法がラムダと併せて用いられた。遺伝子転移は、リン酸カルシウム共沈殿を用いたラムダファージによって、またはDEAE-デキストランもしくはリポポリアミンを用いた繊維状ファージによって観察されている。しかしながら、トランスフェクション効率が低く、非特異的である傾向があり、かつトランスフェクションが面倒であるばかりでなく、細胞タイプに関して無差別でもあるので、哺乳動物細胞内にDNAを導入するこれらの方法は、遺伝子治療適用には実用的でない。
【0007】
現在、真核生物ウイルスは、優れたトランスジーン送達および形質導入を紛れもなく提供するが(Kootstra and Verma, 2003;Machida, 2003)、そのようなベクターのリガンド指向性の標的化は通常、哺乳動物細胞膜受容体に対するそれらの天然の向性の除去を必要とする(Miller et al., 2003;Mizuguchi and Hayakawa, 2004;White et al., 2004)。対照的に、バクテリオファージ(ファージ)などの原核生物ウイルスは通常、哺乳動物細胞形質導入用の不十分なビヒクルと考えられている。しかしながら、「真核生物」ウイルスとしてのそれらの固有の欠点にも関わらず、ファージ粒子は、哺乳動物細胞に対する向性を有さず(Zacher et al., 1980;Barrow and Soothill, 1997;Barbas et al., 2001)かつ低い効率でではあるが、そのような細胞に形質導入するように適合させられてすらいる(Ivanenkov et al., 1999;Larocca et al., 1999;Poul and Marks, 1999;Piersanti et al., 2004)。
【0008】
したがって、治療的適用において有用なベクターを達成したい場合、ベクターを特異的な細胞(または受容体)に標的化しかつ治療的に有用な程度の遺伝子送達および発現を保証するより信頼できる手段がこのように必要とされる。
【発明の開示】
【0009】
発明の概要
本発明の態様は一般に、増強された発現を達成するために、1つまたは複数のトランスジーンを腫瘍細胞などの的細胞に、部位特異的な様式で送達する組成物および方法、ならびにそのような適用において有用なコンストラクトおよび組成物に向けられている。ある種の局面において、対象への処置の実施または対象に対する診断手順の前に、治療的核酸からの発現を検討評価してもよい。さらなる局面において、治療的利益に必要とされる領域または場所における発現の決定または評価を検討評価し、かつ任意の不必要なまたは必要最小限の有益な処置を、代替処置の代わりに是認することができる。
【0010】
任意の特定の理論またはメカニズムに束縛されるわけではないが、本開示は、トランスジーンが1より大きい多重度でゲノムに組み込まれる時にトランスジーン発現が増大し得るという観察に基づいている。特に興味深いのは、ある種のキメラAAVP粒子が、多くの場合コンカテマーとして、1コピーより多くのトランスジーンを細胞に形質導入する能力である。キメラAAVP粒子によって運ばれるレポーター遺伝子の発現によって、形質導入された細胞をモニタリングしてもよい。任意のトランスジーンを本開示のAAVP粒子の中に含め、かつ本開示のAAVP粒子から発現させてもよい。
【0011】
本発明のある種の態様には、対象の標的組織への遺伝子転移および/または対象の標的組織での遺伝子発現を検出するための方法および組成物が含まれ、これらは、以下の工程の1つまたは複数を含む:
(a)本発明で使用し得る1つの工程には、宿主対象中に天然に存在してもまたは存在しなくてもよいレポーター遺伝子を含むAAVPベクターを、対象の標的組織に送達する工程が含まれる。典型的には、レポーター遺伝子は、画像化および/または処置される場所または領域で発現していないと考えられる。ある種の局面において、レポーター遺伝子は、野生型の、突然変異体の、または遺伝子改変されたキナーゼである。さらなる局面において、キナーゼはチミジンキナーゼである。なおさらなる局面において、キナーゼは単純ヘルペスウイルス-チミジンキナーゼ遺伝子またはヒトチミジンキナーゼ2型である。典型的には、転移ベクターまたはAAVPが標的組織の細胞に導入され、レポーター遺伝子が標的組織の細胞で発現し、それによってAAVPベクターによって効率的にトランスフェクトされた細胞でのみ蓄積するレポーター遺伝子産物(タンパク質)が生成される。
(b)使用し得る別の工程は、宿主対象に標識されたレポーター基質を投与する工程であり、ここでレポーター遺伝子産物を発現する細胞は、放射性標識されたヌクレオシド類似体を含む標識されたレポーター基質を代謝して、標識されたレポーター代謝物を産生する。
(c)本方法で使用し得るさらに別の工程には、レポーター基質の標識された代謝物を含む標的組織または細胞を非侵襲的に画像化する工程が含まれる。ある種の局面において、レポーター遺伝子産物によって代謝されない残存レポーター基質を宿主対象から排除した後、対象をイメージングに供し、それによって標的組織への遺伝子転移および標的組織内での発現を検出する。さらなる局面において、代謝されないレポーター基質の排除の代謝に応じて、1、2、3、4、5、6、7、8、9、10、またはそれより多くの分、時間、日、または週の後に、1つまたは複数の対象組織をイメージングに供する。
【0012】
本方法は、レポーター遺伝子産物によって代謝されないもの(発現産物によって代謝されないレポーター基質)の全ての値およびその間の範囲を含む、約、少なくとも、または多くても、60、65、67、70、75、77、80、85、87、90、95、97%もしくはそれより多くを、対象から排除するのに十分な工程(b)後の一定の期間、待機する工程をさらに含むことができる。代謝されない基質には、代謝されない残存レポーター基質に由来する非特異的標識が含まれてもよい。インビトロまたはインビボのトランスフェクション(または形質導入)によって、AAVPベクターを標的組織の細胞に導入することができる。ある種の局面において、静脈内に、腫瘍内に、動脈内に、胸腔内に、気管支内に、および/または経口でAAVPを投与する。
【0013】
ある種の局面において、陽電子放出断層撮影、ガンマカメラ、または単一光子放出コンピュータ断層撮影によるイメージングに適した放射性同位体で、レポーター基質を標識する。レポーター基質および/またはレポーター基質の代謝物は、2H、13C、15N、および19Fを含むがこれらに限定されない安定同位体核種を含む化合物である。さらなる局面において、標識されたレポーター代謝物を陽電子放出断層撮影によって画像化する。なおさらなる局面において、標識されたレポーター代謝物をガンマカメラまたは単一光子放出コンピュータ断層撮影によって画像化する。さらに、なおさらなる局面において、標識されたレポーター基質代謝物を、磁気共鳴イメージングによって画像化する。
【0014】
AAVPベクターは、レポーター遺伝子ならびに適した転写プロモーターおよびエンハンサーエレメントを組み入れ、レポーターおよび治療的遺伝子共発現の組織特異的、組織選択的、または転写因子特異的、またはシグナル伝達特異的な転写活性化を確実にしてもよい。ある種の局面において、対象への複数の細胞または1つの細胞の投与の前に、器官、組織、複数の細胞、または1つの細胞に、プロモーターおよび/またはエンハンサーエレメントなどの転写調節エレメントが機能的に連結されたレポーターを、エクスビボ(インビトロ)でトランスフェクトする。標識された2'-フルオロ-ヌクレオシド類似体には、5-[123I]- 2'-フルオロ-5-ヨード-1β-D-アラビノフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨード-1β-D-アラビノフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨード-1β-D-アラビノフラノシル-ウラシル、5-[18F]-2'-フルオロ-5-フルオロ-1-β-D-アラビノフラノシル-ウラシル;2-[131I]-2'-フルオロ-5-メチル-1-β-D-アラビノフラノシル-ウラシル;5-([11C]-メチル)-2'-フルオロ-5-メチル-1-β-D-アラビノフラノシル-ウラシル;2-[11C]-2'-フルオロ-5-エチル-1-β-D-アラビノフラノシル-ウラシル;5-([11C]-エチル)-2'-フルオロ-5-エチル-1-β-D-アラビノフラノシル-ウラシル;5-(2-[18F]-エチル)-2'-フルオロ-5-(2-フルオロ-エチル)-1-β-D-アラビノフラノシル-ウラシル、5-[123I]- 2'-フルオロ-5-ヨードビニル-1-β-D-アラビノフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨードビニル-1-β-D-アラビノフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨードビニル-1-β-D-アラビノフラノシル-ウラシル;5-[123I]- 2'-フルオロ-5-ヨード-1-β-D-リボフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨード-1-β-D-リボフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨード-1-β-D-リボフラノシル-ウラシル;5-[123I]-2'-フルオロ-5-ヨードビニル-1-β-D-リボフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨードビニル-1-β-D-リボフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨードビニル-1-β-D-リボフラノシル-ウラシル;または9-4-[18F]フルオロ-3-(ヒドロキシメチル)ブチル]グアニンが含まれるが、これらに限定されない。
【0015】
イメージングデータは、磁気共鳴イメージング(MRI)、核医学、陽電子放出断層撮影(PET)、コンピュータ断層撮影(CT)、超音波検査(US)、光学イメージング、赤外線イメージング、インビボ顕微鏡検査、およびX線ラジオグラフィーによって得られたイメージングを具象化することができるが、これらに限定されない。医療機器、薬物、もしくは化合物、造影剤、またはイメージングから追加の情報を引き出すのに使用し得るその他の薬剤もしくは刺激とイメージングとを組み合わせることができる。画像は、病変、組織、標本、系、生命体、対象、または患者に関するこれらの撮画様式を用いて得られ、時間および/または空間の両方において静的または動的な画像であることができる。
【0016】
大規模な生物学的データが得られる組織、標本、系、生命体、または患者にイメージングを適合させることができる。イメージング情報は、各画像、1つもしくは複数のイメージング検討、またはイメージング調査から抽出され、および病変、組織、標本、系、生命体、または患者の形態、組成、構造、生理、遺伝子発現、または機能の違いを具象化し得るが、これらに限定されない、定量的または定性的なイメージング特色からなることができる。イメージング情報の例として、多相造影増強ダイナミックCT、機能的イメージング、磁気共鳴分光法、拡散テンソルイメージング、拡散または灌流に基づくイメージングだけでなく、核医学またはPETによってカプセル化された標的化イメージングからも抽出し得るイメージング特色が含まれるが、これに限定されない。例えば、その全体が参照により本明細書に組み入れられる、米国特許出願公開第20030033616号および第20060223141号を参照されたい。
【0017】
ある種の態様において、本発明は、以下の工程の1つまたは複数を含む対象を処置する方法を含む:
(a)病理学的状態もしくは疾患状態を有する、有することが疑われる、または発症するリスクがある対象に、レポーターをコードする治療的AAVPを投与する工程;および
(b)コードされたレポーターまたはレポーター活性を検出することによって、処置のために標的化された組織または細胞内での治療的AAVPのインサイチュー発現を評価する工程。
【0018】
ある種の局面において、本発明は、標的器官、組織、または細胞でAAVP核酸によって発現される治療的に十分なレベルの治療的遺伝子の発現に基づき、対象に癌処置を施す工程をさらに含むことができる。さらなる局面において、第一の治療的AAVPの発現が治療的に有効なレベルで発現されない場合に、第二の治療的AAVPを投与することができる。第二の治療的AAVPは、第二の標的化リガンドまたはリガンドの組み合わせを含んでもよい。第二のAAVPもまた、標的器官、組織、複数の細胞、または1つの細胞での発現のための第二の制御エレメントを含むことができる。AAVP発現は、レポーターまたはレポーターの活性の非侵襲的検出(例えば、レポータータンパク質によって代謝される標識された基質の検出)によって評価することができる。ある種の局面において、レポーターは治療的タンパク質である。さらなる局面において、治療的タンパク質はプロドラッグ変換酵素である。またさらなる局面において、レポーターは酵素、および特にキナーゼである。ある種の態様において、キナーゼはチミジンキナーゼ、例えば、HSV-tkまたはヒトtk2である。典型的にキナーゼは、検出可能に標識された化合物または標識された基質を修飾または代謝する。ある種の局面において、基質または化合物は、蛍光、化学発光、表面増強ラマン分光法(SERS)、磁気共鳴イメージング(MRI)、コンピュータ断層撮影(CT)、または陽電子放出断層撮影(PET)イメージングによって検出できる検出可能標識を含む。ある種の局面において、検出可能に標識された化合物は、ヌクレオシド類似体である。検出可能に標識された化合物には、フルオロデオキシグルコース(FDG);2'-フルオロ-2'デオキシ-1ベータ-D-アラビオノフラノシル-5-エチル-ウラシル(FEAU);5-[123I]- 2'-フルオロ-5-ヨード-1β-D-アラビノフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨード-1β-D-アラビノフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨード-1β-D-アラビノフラノシル-ウラシル、5-[18F]-2'-フルオロ-5-フルオロ-1-β-D-アラビノフラノシル-ウラシル;2-[11I]-および5-([11C]-メチル)-2'-フルオロ-5-メチル-1-β-D-アラビノフラノシル-ウラシル;2-[11C]-2'-フルオロ-5-エチル-1-β-D-アラビノフラノシル-ウラシル;5-([11C]-エチル)-2'-フルオロ-5-エチル-1-β-D-アラビノフラノシル-ウラシル;5-(2-[18F]-エチル)-2'-フルオロ-5-(2-フルオロ-エチル)-1-β-D-アラビノフラノシル-ウラシル、5-[123I]- 2'-フルオロ-5-ヨードビニル-1-β-D-アラビノフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨードビニル-1-β-D-アラビノフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨードビニル-1-β-D-アラビノフラノシル-ウラシル;5-[123I]- 2'-フルオロ-5-ヨード-1-β-D-リボフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨード-1-β-D-リボフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨード-1-β-D-リボフラノシル-ウラシル;5-[123I]-2'-フルオロ-5-ヨードビニル-1-β-D-リボフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨードビニル-1-β-D-リボフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨードビニル-1-β-D-リボフラノシル-ウラシル;または9-4-[18F]フルオロ-3-(ヒドロキシメチル)ブチル]グアニンが含まれるが、これらに限定されない。
【0019】
さらなる局面において、標識された基質または化合物を
で標識することができる。特定の局面において、検出可能な標識は、131I、125I、123I、111I、99mTc、90Y、186Re、188Re、32P、153Sm、67Ga、201Tl、77Br、または18F標識である。
【0020】
本発明のAAVPは、処置のために標的化される組織または細胞を選択的に標的化する部分を含んでもよい。ある種の局面において、部分は、AAVPのカプシドタンパク質および/もしくは組換えカプシドタンパク質によってコードされるか、またはAAVPのカプシドタンパク質および/もしくは組換えカプシドタンパク質に連結されている。ある種の局面において、カプシドタンパク質は、標的化ペプチドを含む。標的化ペプチドは、環状ペプチド、二環状、および/または直線状ペプチドであることができる。標的化ペプチドは、細胞表面にインテグリンを発現する細胞に選択的に結合する。インテグリンは、αvβ3またはαvβ5インテグリンであることができる。さらなる局面において、ペプチドはRGDモチーフを含む。またさらなる局面において、ペプチドはトランスフェリン受容体を発現する細胞に選択的に結合することができ、そのようなペプチドは、CRTIGPSVCを含むアミノ酸配列を含むことができる。
【0021】
ある種の局面において、対象は、過剰増殖性疾患を有する、有することが疑われる、または発症するリスクがある。過剰増殖性疾患には、線維肉腫、筋肉腫、脂肪肉腫、軟骨肉腫、骨原性肉腫、軟骨腫、血管肉腫、内皮肉腫、リンパ管肉腫、リンパ管内皮肉腫、滑膜腫、中皮腫、ユーイング腫瘍、平滑筋肉腫、横紋筋肉腫、胃癌、食道癌、直腸癌、膵臓癌、卵巣癌、前立腺癌、子宮癌、頭部および頸部の癌、皮膚癌、脳癌、扁平上皮細胞癌腫、脂腺癌腫、乳頭癌腫、乳頭腺癌、嚢胞腺癌、髄様癌、気管支原性癌腫、腎細胞癌腫、肝細胞癌、胆管癌腫、絨毛癌腫、精上皮腫、胎児性癌腫、ウィルムス腫瘍、子宮頸癌、精巣癌、小細胞肺癌腫、非小細胞肺癌腫、膀胱癌腫、上皮性癌腫、神経膠腫、星状細胞腫、髄芽細胞腫、頭蓋咽頭管腫、上衣細胞腫、松果体腫、血管芽細胞腫、聴神経腫、希突起神経膠腫、髄膜腫、メラノーマ、神経芽細胞腫、網膜芽細胞腫、白血病、リンパ腫、またはカポジ肉腫が含まれるが、これらに限定されない。特定の局面において、過剰増殖性疾患は神経膠腫である。
【0022】
さらなる態様において、レポーターおよび/または治療的遺伝子は、組織もしくは細胞選択的プロモーター、または組織もしくは細胞特異的プロモーターに機能的に連結されている。ある種の局面において、発現を評価する工程は、標識された化合物または基質(AAVP核酸を発現する細胞によって代謝され、典型的には非標的組織によって著しい程度までは代謝されない)を投与する工程を含む。
【0023】
治療的AAVPはまた、第二の治療的遺伝子をコードしてもよい。第二の治療的遺伝子は、腫瘍抑制因子、阻害RNA、阻害DNA、またはプロドラッグ変換酵素であることができるが、これらに限定されない。
【0024】
なおさらなる態様において、本発明の組成物は、阻害RNAまたは阻害DNAを含む核酸セグメントを含む治療的AAVP核酸を含むことができる。阻害RNAは、siRNA、miRNA、またはアンチセンスRNAもしくはDNAであることができる。ある種の局面にでは、AAVP核酸はファージ粒子中に含まれる。さらなる局面では、粒子は本明細書に記載するような標的化リガンドを含む。
【0025】
ある種の態様には、AAVP核酸またはそのようなものを含む粒子を投与する工程を含む遺伝子の発現を調整するための組成物および方法が含まれる。
【0026】
本発明に従って、選択された遺伝子またはポリペプチドは、任意のタンパク質、ポリペプチド、またはペプチドを指してもよい。治療的遺伝子またはポリペプチドは、疾患を処置または防止する目的で対象に投与することができる遺伝子またはポリペプチドである。例えば、治療的遺伝子は、癌の処置または防止のために対象に投与される遺伝子であることができる。治療的遺伝子の例として、Rb、CFTR、p16、p21、p27、p57、p73、C-CAM、APC、CTS-1、zac1、scFV ras、DCC、NF-1、NF-2、WT-1、MEN-I、MEN-II、BRCA1、VHL、MMAC1、FCC、MCC、BRCA2、IL-1、IL-2、IL-3、IL-4、IL-5、IL-6、IL-7、IL-8、IL-9、IL-10、IL-11、IL-12、GM-CSF、G-CSF、チミジンキナーゼ、Bax、Bak、Bik、Bim、Bid、Bad、Harakiri、Fas-L、mda-7、fus、インターフェロンα、インターフェロンβ、インターフェロンγ、p53、ABLI、BLC1、BLC6、CBFA1、CBL、CSFIR、ERBA、ERBB、EBRB2、ETS1、ETS2、ETV6、FGR、FOX、FYN、HCR、HRAS、JUN、KRAS、LCK、LYN、MDM2、MLL、MYB、MYC、MYCL1、MYCN、NRAS、PIM1、PML、RET、SRC、TAL1、TCL3、YES、MADH4、RB1、TP53、WT1、TNF、BDNF、CNTF、NGF、IGF、GMF、aFGF、bFGF、NT3、NT5、ApoAI、ApoAIV、ApoE、Rap1A、シトシンデアミナーゼ、Fab、ScFv、BRCA2、zac1、ATM、HIC-1、DPC-4、FHIT、PTEN、ING1、NOEY1、NOEY2、OVCA1、MADR2、53BP2、IRF-1、zac1、DBCCR-1、rks-3、COX-1、TFPI、PGS、Dp、E2F、ras、myc、neu、raf、erb、fms、trk、ret、gsp、hst、abl、E1A、p300、VEGF、FGF、トロンボスポンジン、BAI-1、GDAIF、またはMCCが含まれるが、これらに限定されない。
【0027】
治療的遺伝子のその他の例として、酵素をコードする遺伝子が含まれる。例として、ACPデサチュラーゼ、ACPヒドロキシラーゼ、ADP-グルコースピロホリラーゼ(pyrophorylase)、ATPアーゼ、アルコールデヒドロゲナーゼ、アミラーゼ、アミログルコシダーゼ、カタラーゼ、セルラーゼ、シクロオキシゲナーゼ、デカルボキシラーゼ、デキストリナーゼ、エステラーゼ、DNAポリメラーゼ、RNAポリメラーゼ、ヒアルロンシンターゼ、ガラクトシダーゼ、グルカナーゼ、グルコースオキシダーゼ、GTPアーゼ、ヘリカーゼ、ヘミセルラーゼ、ヒアルロニダーゼ、インテグラーゼ、インベルターゼ、イソメラーゼ、キナーゼ、ラクターゼ、リパーゼ、リポオキシゲナーゼ、リアーゼ、リゾチーム、ペクチンエステラーゼ、ペルオキシダーゼ、ホスファターゼ、ホスホリパーゼ、ホスホリラーゼ、ポリガラクツロナーゼ、プロテイナーゼ、ペプチダーゼ、プラナーゼ、リコンビナーゼ、リバーストランスクリプターゼ、トポイソメラーゼ、キシラナーゼ、レポーター遺伝子、インターロイキン、またはサイトカインが含まれるが、これらに限定されない。
【0028】
治療的遺伝子のさらなる例として、カルバモイルシンセターゼI、オルニチントランスカルバミラーゼ、アルギノスクシネートシンセターゼ、アルギノスクシネートリアーゼ、アルギナーゼ、フマリルアセトアセテートヒドロラーゼ、フェニルアラニンヒドロキシラーゼ、アルファ-1アンチトリプシン、グルコース-6-ホスファターゼ、低密度リポタンパク質受容体、ポルホビリノーゲンデアミナーゼ、第VIII因子、第IX因子、シスタチオンベータ-シンターゼ、分岐鎖ケト酸デカルボキシラーゼ、アルブミン、イソバレリル-CoAデヒドロゲナーゼ、プロピオニルCoAカルボキシラーゼ、メチルマロニルCoAムターゼ、グルタリルCoAデヒドロゲナーゼ、インスリン、-グルコシダーゼ、ピルビン酸カルボキシラーゼ、肝ホスホリラーゼ、ホスホリラーゼキナーゼ、グリシンデカルボキシラーゼ、H-タンパク質、T-タンパク質、メンケス病銅輸送ATPアーゼ、ウィルソン病銅輸送ATPアーゼ、シトシンデアミナーゼ、ヒポキサンチン-グアニンホスホリボシルトランスフェラーゼ、ガラクトース-1-リン酸ウリジルトランスフェラーゼ、フェニルアラニンヒドロキシラーゼ、グルコセルブロシダーゼ、スフィンゴミエリナーゼ、-L-イズロニダーゼ、グルコース-6-リン酸デヒドロゲナーゼ、HSVチミジンキナーゼ、またはヒトチミジンキナーゼをコードする遺伝子が含まれる。
【0029】
治療的遺伝子には、ホルモンをコードする遺伝子も含まれる。例として、成長モルモン、 プロラクチン、胎盤ラクトゲン、黄体形成ホルモン、濾胞刺激ホルモン、絨毛性ゴナドトロピン、甲状腺刺激ホルモン、レプチン、副腎コルチコトロピン、アンギオテンシンI、アンギオテンシンII、β-エンドルフィン、β-メラニン細胞刺激ホルモン、コレシストキニン、エンドセリンI、ガラニン、胃阻害ペプチド、グルカゴン、インスリン、リポトロピン、ニューロフィジン、ソマトスタチン、カルシトニン、カルシトニン遺伝子関連ペプチド、β-カルシトニン遺伝子関連ペプチド、悪性の高カルシウム血症因子、副甲状腺ホルモン関連タンパク質、副甲状腺ホルモン関連タンパク質、グルカゴン様ペプチド、パンクレアスタチン、膵ペプチド、ペプチドYY、PHM、セクレチン、血管作用性小腸ペプチド、オキシトシン、バソプレッシン、バソトシン、エンケファリンアミド、メトルフィンアミド、アルファメラニン細胞刺激ホルモン、心房性ナトリウム利尿因子、アミリン、アミロイドP成分、コルチコトロピン放出ホルモン、成長モルモン放出因子、黄体形成ホルモン放出ホルモン、神経ペプチドY、サブスタンスK、サブスタンスP、またはチロトロピン放出ホルモンをコードする遺伝子が含まれるが、これらに限定されない。
【0030】
本発明のその他の態様は本出願の全体を通して考察される。本発明のある局面に関して考察された任意の態様は、本発明のその他の局面に同様に当てはまり、かつ逆もまた同様である。実施例の項における態様は、本発明の全ての局面に適用可能である本発明の態様であると理解される。
【0031】
本特許請求の範囲および/または本明細書で用いる場合、「阻害すること」、「低下させること」、もしくは「防止」という用語、またはこれらの用語の任意の変形には、所望の結果を達成するための任意の測定可能な減少または完全な阻害が含まれる。
【0032】
本特許請求の範囲および/または本明細書における「含む」という用語と併せて用いられる場合の「1つの(a)」または「1つの(an)」という語の使用は、「1つの(one)」を意味し得るが、それは「1つまたは複数の」、「少なくとも1つの」、および「1つまたは1つよりも多くの」という意味とも一致する。
【0033】
本明細書で考察する任意の態様を本発明の任意の方法および組成物に関して実行することができ、ならびに逆もまた同様であることが企図される。さらに、本発明の組成物およびキットを用いて本発明の方法を達成することができる。
【0034】
本明細書の全体を通じて、「約」という用語を用いて、ある値が、値を決定するために利用する機器または方法についての誤差の標準偏差を含むことを示す。
【0035】
本特許請求の範囲における「または」という用語の使用は、代替物だけを指すことが明瞭に示され、または代替物が相互排他的であるのでなければ、「および/または」を意味するために用いられるが、本開示は、唯一の代替物および「および/または」を指す定義を支持する。
【0036】
本明細書および本特許請求の範囲において用いられる場合、「含む(comprising)」という語(ならびに「含む(comprise)」および「含む(comprises)」などの、含む(comprising)の任意の形態)、「有する(having)」という語(ならびに「有する(have)」および「有する(has)」などの、有する(having)の任意の形態)、「含む(including)」という語(ならびに「含む(includes)」および「含む(include)」などの、含む(including)の任意の形態)、「含む(containing)」という語(ならびに「含む(contains)」および「含む(contain)」などの、含む(containing)の任意の形態)は、包括的または開放型であり、かつ追加の列挙されない要素または方法工程を除外しない。
【0037】
本発明の他の目的、特色、および利点は、以下の詳細な説明から明らかになると考えられる。しかしながら、本発明の精神および範囲の範囲内の様々な変更および修正がこの詳細な説明から当業者に明らかになると考えられるので、詳細な説明および具体的な実施例は、本発明の具体的な態様を示す一方で、例証のみのために与えられることが理解されるべきである。
【0038】
本開示は様々な修正および代替の形態を受け入れることができるが、具体的な実施例態様を、図に示しかつ本明細書でより詳細に記載する。しかしながら、具体的な実施例態様の説明は、開示する特定の形態に本発明を限定するよう意図せず、対照的に本開示は、一部添付の特許請求の範囲によって例証するような全ての修正および同等物を包含するためのものであることが理解されるべきである。
【0039】
発明の詳細な説明
ある種の態様による本開示は、増強された発現を達成するために、腫瘍細胞などの標的細胞に、1つまたは複数のトランスジーンを部位特異的な様式で送達する方法、ならびにそのような適用において有用なコンストラクトおよび組成物に概ね向けられている。ある種の局面において、対象への処置の投与または対象に対する診断手順の前に、治療的核酸からの発現を検討評価してもよい。さらなる局面において、治療的利益が必要とされる領域または場所における発現の決定または評価を検討評価し、任意の不必要なまたは必要最低限の有益な処置を代替処置の代わりに認めることができる。
【0040】
理論またはメカニズムに束縛されることを望まないが、本開示は、トランスジーンが1より大きい多重度でゲノムに組込まれる場合、トランスジーン発現は増加し得るという観察に基づく。特に興味深いのは、ある種のキメラAAVP粒子が、多くの場合コンカテマーとして、1コピーより多くのトランスジーンを細胞に形質導入する能力である。キメラAAVP粒子によって運ばれるレポーター遺伝子の発現によって形質導入された細胞をモニタリングしてもよい。任意のトランスジーンを本開示のAAVP粒子中に含め、本開示のAAVP粒子から発現させてもよい。
【0041】
I、アデノ随伴ウイルス/ファージ(AAVP)粒子
アデノ随伴ウイルス(AAV)はパルボウイルスファミリーの欠損性のメンバーである。AAVゲノムは、プラスまたはマイナス極性の1本鎖DNA分子としてカプセル化されている。両方の極性の鎖がパッケージングされているが、別々のウイルス粒子中にあり、かつ両方の鎖が感染性である。ヒトアデノ随伴ウイルス2型(AAV2)の1本鎖DNAゲノムは、長さが4681塩基対であり、かつ各々145塩基対の逆向き末端反復配列(ITR)が両隣にある。さらに、ウイルスのrepタンパク質は、ITRによる非相同組換えを媒介するように見える。したがって、組換えで役割を果たしかつパルボウイルスの複製およびパッケージングに通常必要とされるITRを、パルボウイルスゲノムが各末端に有する時、本開示のAAVPは通常、ITRまたはその機能的同等物の少なくとも1つの全てまたは一部分を含む。
【0042】
AAVは容易に得られ、遺伝子送達用のベクターとしてのそれらの使用は例えば、Muzyczka, 1992;米国特許第4,797,368号、およびPCT公開公報WO 91/18088に記載されている。AAVベクターの構築は、Lebkowski et al., 1988;Tratschin et al., 1985;Hermonat and Muzyczka, 1984を含む、多数の刊行物に記載されている。
【0043】
本開示は、細菌中で産生される(AAV-M13ベクターなどの)アデノ随伴ウイルス(AAV)バクテリオファージベクター(AAVP)および細胞にAAVPを形質導入することによって、腫瘍細胞などの標的細胞でトランスジーンを発現させるための方法を提供する。一旦精製し、バクテリオファージおよびトランスジーンカセットを持つAAV配列を含む標的化されたバクテリオファージ粒子を用いて、哺乳動物細胞にトランスフェクトする。哺乳動物細胞内へのベクターの内在化の後、トランスジーンは標的細胞のゲノムに組込まれる。本明細書で用いる場合の「ベクター」という用語は、関心対象の核酸の細胞内への送達のための核酸ビヒクルとして定義される。ベクターは、直線状分子または環状分子であってもよい。
【0044】
AAVPは、ファージおよびAAVベクター系の両方の選択されたエレメントを組み合わせ、それによりパッケージング制限のない細菌中で産生し易く、その一方で宿主染色体への組み込みと組み合わせた哺乳動物細胞の感染を可能にするベクターを提供する。適当なエレメントの多くを含むベクターは市販されており、かつ必要な配列を含むよう標準的な方法論によってさらに改変することができる。最低限、本開示の方法を用いた使用のために、ベクターは、プロモーターおよびトランスジーンを含むカセットを受容しなければならない。本開示のAAVPベクターは、標的細胞ゲノムへの組み入れ時に増強されたトランスジーン発現を可能にする。ある種の態様において、トランスジーンはコンカテマーとして標的細胞のゲノムに組み込まれてもよい。
【0045】
とりわけ、AAVPはヘルパーウイルスまたはトランス作用因子を必要としない。さらに、AAVカプシド形成がないので、哺乳動物細胞に対するAAVの天然の向性は消去される。
【0046】
A、AAVP標的化
ファージ粒子の表面上のリガンドの発現によって、本開示のAAVPを特異的な受容体に標的化することができる。ある種の態様において、エンドソームからのベクターの脱出を可能にしまたは促進するペプチドまたはその他の部分(およびそこに付着するかまたはその中に封入された任意の分子)を組み入れ、かつファージの表面に発現させてもよい。そのような「その他の部分」には、それら自体ペプチドではないが、エンドソーム膜を乱す能力を有し、それによってベクターの脱出を容易にする分子、およびさもなければ内部に記載されたペプチド配列のエンドソーム脱出特性を模倣する分子が含まれる(例えば、公開されたPCT公開公報WO 96/10038およびWagner et al., 1992参照)。
【0047】
本開示のAAVPは通常、リガンドが付着している様式に関係なく、1つまたは複数の予め選択されたリガンドを粒子表面に発現する線維状ファージ粒子から構成される。それゆえ、リガンドへの付着の手段が共有結合であるかまたはカプシドタンパク質を介するものかのいずれでも、本開示のAAVPは、受容体へのリガンド結合、それに続くベクターの内在化によって、1つまたは複数のトランスジーンを標的細胞に送達することができる。例えば、粒子表面に発現したリガンド粒子は、αvβ3およびαvβ5インテグリンに選択的に結合する二環状のCDCRGDCFC(RGD-4C)(SEQ ID NO:2)ペプチドであってもよい。これらのインテグリンは、侵入性の腫瘍内皮細胞上に高度に過剰発現している。本明細書で用いる場合、「繊維状ファージ粒子」は、ファージゲノムかまたはファージミドゲノムのいずれかを含む粒子を指す。繊維状カプシドタンパク質に加えて、粒子はその他の分子を含んでもよい。本明細書で用いる場合、「リガンド」は、細胞表面分子に結合しかつ内在化することができる、任意のペプチド、ポリペプチド、タンパク質、またはペプチドミメティックなどの非タンパク質を指す。本明細書で用いる場合、「受容体への結合」は、標準的なインビトロまたはインビボアッセイによってアッセイされた場合の、受容体を特異的に認識し、かつ受容体に検出可能に結合するリガンドの能力を指す。
【0048】
典型的には、本開示のAAVPは、ベクターのリガンド-受容体標的化特性を可能にする標的化ペプチドをコードするオリゴヌクレオチド挿入物を、ファージプラスミドゲノム中に含む。
【0049】
カプシドタンパク質ポリヌクレオチドの全てもしくは一部を標的化リガンドに連結するか、または該ポリヌクレオチドによってコードされるタンパク質の全てまたは一部を標的化リガンドに融合させることによって、ファージカプシドタンパク質またはカプシドタンパク質を改変してもよい。標的化リガンドは、本発明のAAVPの特異的な細胞、組織、および/または器官への結合を導き、導き直し、標的化し、または増強してもよい。
【0050】
標的化されるウイルスは元々、遺伝子治療ベクターの天然の宿主細胞向性により直面する問題を克服するために創り出された。近年、多くの遺伝子治療特許が出されており、キメラ融合糖タンパク質を含むベクター(参照により本明細書に組み入れられる、Kaymanら、米国特許第5,643,756号)およびウイルスカプシドタンパク質に対する抗体を含むベクター(Cottenら、米国特許第5,693,509号)などのベクターは、特異的な細胞にベクターを標的化するのに用いられる異種ポリペプチドを含む。核酸ゲノムが標的非分裂細胞、標的分裂細胞、または増殖性もしくはその他の障害を有する標的細胞に送達されるように、特定の細胞タイプ(例えば、平滑筋細胞、肝細胞、腎細胞、線維芽細胞、ケラチン生成細胞、幹細胞、間葉系幹細胞、骨髄細胞、軟骨細胞、上皮細胞、小腸細胞、新生物性または癌性細胞、および当技術分野において公知のその他の細胞)に粒子が標的化されるように本発明のAAVPを遺伝子改変してもよい。ウイルスを標的化する1つの方法は、細胞の外部表面に分子を有する細胞に優先的に結合させることによって、ウイルスを標的細胞に導くことである。ウイルスを標的化するこの方法は、ウイルスの表面上の標的化リガンドと相互作用する受容体または結合分子を有する細胞または組織に、ウイルスを標的化するのを手助けするために、ウイルスのカプシド上での標的化リガンドの発現またはウイルスのカプシドへの標的化リガンドの組み入れを活用している。ウイルスによる細胞の感染後、遺伝子材料は宿主細胞で処理および発現されることができる。遺伝子材料は宿主細胞のゲノムに組み込まれまたは宿主細胞内でエピソームとして維持されてもよい。
【0051】
本発明のある種の態様において、AAVPを特異的な細胞タイプまたは組織に送達し得るように、カプシドタンパク質を改変して標的化部分を含めてもよい。リガンドに基づく送達系の標的化特異性は、異なる細胞タイプ上のリガンド受容体の分布に基づく。標的化リガンドは、非共有結合または共有結合のいずれかでカプシドタンパク質に会合していてもよい。
【0052】
ある種の態様において、関心対象の異種核酸配列を本発明のウイルスベクターに挿入してもよい。例えば、カプシドタンパク質は、特異的な標的細胞上の受容体に対するリガンドに機能的に連結されてもよい。
【0053】
標的化リガンドは、標的化される領域の特徴的構成要素に特異的な任意のリガンドである。好ましい標的化リガンドには、ポリクローナルもしくはモノクローナル抗体、抗体断片、もしくはキメラ抗体、酵素、ペプチド、もしくはホルモンなどのタンパク質、または単糖類、オリゴ糖類、もしくは多糖類などの糖が含まれる。本発明のある種の態様において、企図される標的化リガンドは、インテグリン、プロテオグリカン、糖タンパク質、受容体、または輸送体と相互作用する。適したリガンドには、標的器官の細胞に、または腫瘍など局部的な病理の結果として循環に曝露される標的器官の構造に、特異的または選択的である任意のものが含まれる。
【0054】
本発明のある種の態様において、耐性の細胞の形質導入を増強させ、標的細胞の形質導入を増加させ、または非所望の細胞の形質導入を限定するために、抗体または環状ペプチド標的化部分(リガンド)をAAVPと会合させてもよい。特定の例における抗体標的化部分は、モノクローナル抗EGF受容体抗体である。EGF受容体は、様々な器官の細胞表面に分布し、かつ火傷、創傷、真皮、および腫瘍に存在する。ペプチド標的化部分はまた、その配列内にRGDインテグリン結合モチーフを含む環状ペプチドであってもよい。細胞表面上のインテグリンに結合するRGDペプチドなどのリガンドは内在化を媒介し、したがって標的化される複合体の送達の効率性を増加させることができる。RGD配列を含み、ペプチドが3〜30アミノ酸長である標的化配列は、RGDFV(SEQ ID NO:3)配列を含んでもよい。その他の態様において、RGDインテグリン結合モチーフは、3〜20アミノ酸長または4〜10アミノ酸長である。本発明の特定の態様において、RGDインテグリン結合モチーフは、5アミノ酸長のペプチドである。その配列内にRGDインテグリン結合モチーフを含む環状ペプチドが好ましいが、直線状ペプチドも本発明において活用してもよい。ヒト癌の診断および治療用の標的化ペプチドの使用に関する第20060239968号の組成物および方法。米国特許出願公開第20060094672号、第20050191294号、第20050187161号、第20050074812号、第20050074747号、第20050037417号、第20050003466号、第20040170955号、第20040131623号、第20040096441号、第20040071689号、第20040048243号、第20030152578号、第20030113320号、および第20010046498号だけでなく、PCT公開公報WO 2006/060171、WO 2006/010070、WO 2005/065418、WO 2005/026195、WO 2004/020999、WO 2003/022991、WO 2002/020822、WO 2002/020769、WO 2002/020723、およびWO 2002/020722も、標的化リガンドを同定する様々な組成物および方法を記載しており、その各々はその全体が参照により本明細書に組み入れられる。
【0055】
本明細書で用いる場合、「リガンド」は、細胞表面分子に結合しかつ内在化することができる、任意のペプチド、ポリペプチド、タンパク質、またはペプチドミメティックなどの非タンパク質を指す。本明細書で用いる場合、「受容体に結合する」とは、標準的なインビトロまたはインビボアッセイによってアッセイされた場合の、受容体を特異的に認識し、受容体に検出可能に結合するリガンドの能力を指す。
【0056】
本発明の文脈中で、リガンドは、融合タンパク質としてまたは化学的コンジュゲーションを通じてのいずれかで、ファージのタンパク質(例えば、カプシドタンパク質)に連結され、細胞に核酸を送達するのに用いられる。断片が適当な細胞表面分子に結合する能力を保持する限り、リガンドの断片を本発明の範囲内で用いてもよい。同様に、置換またはその他の変更はあるが、結合能力を保持するリガンドも用いてよい。なおその上、特定のリガンドは、天然のリガンドのアミノ酸配列を有するポリペプチドだけでなく、リガンドが内皮細胞上のその受容体に結合し、細胞への核酸の送達を結果的にもたらす能力を保持する限り、改変された配列(例えば、天然のタンパク質(ムテイン(mutein))と比較してアミノ酸置換、欠失、挿入、または付加を有する)も指す。
【0057】
リガンドは、その受容体を発現する細胞に結合しかつ内在化される能力を保有するムテインまたは突然変異体タンパク質も包含する。そのようなムテインには、システインの1つまたは複数をセリンと置き換えることによって産生されたものが含まれるが、これに限定されない。典型的には、そのようなムテインは、保存的アミノ酸変化を有すると考えられる。そのようなムテインをコードするDNAは、縮重コドンの置換によって改変されない限り、少なくとも低ストリンジェンシーの条件下で野生型リガンドをコードする天然のDNA配列にハイブリダイズすると考えられる。
【0058】
リガンドをコードするDNAを、公知のアミノ酸もしくはDNA配列に基づいて合成により調製し、当業者に公知の方法(例えば、PCR増幅)を用いて単離し、または市販もしくはその他の源から得てもよい。リガンドをコードするDNAは、縮重コドンの置換によって、または異なるアミノ酸をコードすることによって、上記の配列と異なってもよい。異なる種の相同なリガンド間だけでなく、個々の生命体または種間でも生じる違いなどアミノ酸配列の違いは、リガンドがその受容体に結合する限り許容される。リガンドを天然源から単離するか、または合成(組換え手段もしくは化学合成など)により作製してもよい。
【0059】
本発明の文脈で用いられるリガンドが細胞上の受容体に結合しかつ内在化されること以外に、そのインビボの生物学的活性のいずれかを保持することは必要ではない。リガンドが1つまたは複数の生物学的活性を欠くように改変されていても、例えば、以下の検査または当技術分野において公知のその他の検査によって、結合および内在化をなお容易にアッセイし得る。通常、これらの検査は、リガンドを標識する工程、それを標的細胞とインキュベートする工程、および細胞内の標識を可視化または測定する工程を伴う。例えば、簡潔に述べると、リガンドをFITCで蛍光標識または125Iで放射性標識し、細胞とインキュベートし、内在化について蛍光顕微鏡または共焦点顕微鏡で顕微鏡的に調べてもよい。
【0060】
ファージカプシドタンパク質への付着に備えて、リガンドを組換えまたはその他の手段によって産生してもよい。DNA配列およびこれらのリガンドの配列を得るための方法は周知である。DNA配列に基づいて、遺伝子を(小さいタンパク質について)合成的に合成する、細胞ゲノムDNAまたはcDNAから増幅する、ゲノムDNAまたはcDNAライブラリーなどから単離してもよい。ファージまたはファージミドベクターへのクローニングを容易にするための制限部位を、増幅用のプライマー中などに組み入れてもよい。
【0061】
そのような分子には、癌細胞、内皮細胞、ストローマ細胞、およびそれらと同様の細胞に結合するタンパク質が含まれるが、これらに限定されない。そのようなリガンドには、成長因子およびサイトカインが含まれる。多くの成長因子および成長因子のファミリーは、構造的および機能的特色を共有し、本発明において使用され得る。成長因子のファミリーには、線維芽細胞成長因子FGF-1からFGF-15、および血管内皮成長因子(VEGF)が含まれる。PDGF(血小板由来成長因子)、TGF-α(形質転換成長因子)、TGF-β、HB-EGF、アンギオテンシン、ボンベシス、エリスロポイエチン、幹細胞因子、M-CSF、G-CSF、GM-CSF、およびエンドグリンなどその他の成長因子も、細胞表面上の特異的な同定された受容体に結合し、本発明において使用され得る。インターロイキン、CSF(コロニー刺激因子)、およびインターフェロンを含むサイトカインは、特異的な受容体を有し、本明細書で記載するように使用され得る。
【0062】
例えば、リガンドおよびリガンド/受容体対には、ウロキナーゼ/ウロキナーゼ受容体(GenBankアクセッション番号X02760/X74309);α-1,3フコシルトランスフェラーゼ、α1-アンチトリプシン/E-セレクチン(GenBankアクセッション番号M98825、D38257/M87862);P-セレクチン糖タンパク質リガンド、P-セレクチンリガンド/P-セレクチン(GenBankアクセッション番号U25955、U02297/L23088)、VCAM1/VLA-4(GenBankアクセッション番号X53051/X16983);E9抗原(Blann et al., Atherosclerosis 120:221, 1996)/TGFβ受容体;フィブロネクチン(GenBankアクセッション番号X02761);I型α1-コラーゲン(GenBankアクセッション番号Z74615)、I型β2-コラーゲン(GenBankアクセッション番号Z74616)、ヒアルロン酸/CD44(GenBankアクセッション番号M59040);CD40リガンド(GenBankアクセッション番号L07414)/CD40(GenBankアクセッション番号M83312); elk-1(GenBankアクセッション番号M25269)に対するEFL-3、LERTK-2リガンド(GenBankアクセッション番号L37361、U09304);VE-カドヘリン(GenBankアクセッション番号X79981);カテニンに対するリガンド;LFA-1に対するICAM-3(GenBankアクセッション番号X69819)リガンド、ならびにαvβ3インテグリン(GenBankアクセッション番号U07375、L28832)に対するフォンウェルブランド因子(GenBankアクセッション番号X04385)、フィブリノゲン、およびフィブロネクチン(GenBankアクセッション番号X92461)リガンドが含まれる。
【0063】
その他のリガンドには、CSF-1(GenBankアクセッション番号M11038、M37435);GM-CSF(GenBankアクセッション番号X03021);IFN-α(インターフェロン)(GenBankアクセッション番号A02076;WO 8502862-A);IFN-γ(GenBankアクセッション番号A02137;WO 8502624-A);IL-1-α(インターロイキン1アルファ)(GenBankアクセッション番号X02531、M15329);IL-1-β(インターロイキン1ベータ)(GenBankアクセッション番号X02532、M15330、M15840);IL-1(GenBankアクセッション番号K02770、M54933、M38756);IL-2(GenBankアクセッション番号A14844、A21785、X00695、X00200、X00201、X00202);IL-3(GenBankアクセッション番号M14743、M20137);IL-4(GenBankアクセッション番号M13982);IL-5(GenBankアクセッション番号X04688、J03478);IL-6(GenBankアクセッション番号Y00081、X04602、M54894、M38669、M14584);IL-7(GenBankアクセッション番号J04156);IL-8(GenBankアクセッション番号Z11686);IL-10(GenBankアクセッション番号X78437、M57627);IL-11(GenBankアクセッション番号M57765 M37006);IL-13(GenBankアクセッション番号X69079、U10307);TNF-α(腫瘍壊死因子)(GenBankアクセッション番号A21522);TNF-β(GenBankアクセッション番号D12614);erbB2に対するGP30リガンド(S68256);およびトランスフェリン受容体に対するトランスフェリン(GenBankアクセッション番号DQ923758)が含まれる。
【0064】
またその他のリガンドには、PDGF(GenBankアクセッション番号X03795、X02811)、アンギオテンシン(GenBankアクセッション番号K02215)、ならびにインテグリン受容体に結合するICAM-1(GenBankアクセッション番号X06990)およびVCAM-1(GenBankアクセッション番号X53051) など、全てのRGD含有ペプチドおよびタンパク質が含まれる。その他のリガンドには、TNFα(GenBankアクセッション番号A21522、X01394)、IFN-γ(GenBankアクセッション番号A11033、A11034)、IGF-I(GenBankアクセッション番号A29117、X56773、S61841、X56774、S61860)、IGF-II(GenBankアクセッション番号A00738、X06159、Y00693)、心房性ナトリウム利尿ペプチド(GenBankアクセッション番号X54669)、エンドセリン-1(GenBankアクセッション番号Y00749)、凝固因子Xa(GenBankアクセッション番号L00395、L00396、L29433、N00045、M14327)、TGF-β1(GenBankアクセッション番号A23751)、IL-1β(GenBankアクセッション番号X03833)、IL-1β(GenBankアクセッション番号M15330)、およびエンドグリン(GenBankアクセッション番号X72012)が含まれる。
【0065】
FGFタンパク質のファミリーには現在、FGF-1(酸性FGFまたはaFGF)、FGF-2(塩基性FGFまたはbFGF)、FGF-3(int-2)、FGF-4(hst-1/K-FGF)、FGF-5、FGF-6(hst-2)、FGF-7(ケラチン生成細胞成長因子またはKGF)、FGF-8、FGF-9、FGF-11(WO 96/39507)、FGF-13(WO 96/39508)、FGF-14(WO 96/39506)、および FGF-15(WO 96/39509)が含まれる。FGF受容体に反応性であるその他のポリペプチド、すなわちFGF受容体、好ましくは高親和性FGF受容体と特異的に相互作用し、かつFGF受容体との相互作用によって細胞内にエンドソーム経由で輸送される任意のポリペプチドが、本発明の範囲内で適している。リガンドには、本明細書で同定したリガンドの4、5、6、7、8、9、10、15、20、25、30、またはそれより多くのアミノ酸断片も含まれる。
【0066】
抗体およびその断片も標的化部分として企図される。モノクローナル抗体断片を用いて、脳、心臓、肺、または肝臓を含む動物の異的な器官への送達を標的化してもよい。単一の細胞に特異的なマーカーを欠く細胞へウイルス粒子を標的化するための例示的な方法が記載されている(米国特許第5,849,718号)。例えば、抗体Aが腫瘍に対する特異性だけでなく、正常な心臓および肺組織に対する特異性も有する可能性がある一方、抗体Bは腫瘍だけでなく正常な肝臓に対する特異性も有する。明らかに、腫瘍に抗増殖性核酸を送達するための抗体Aまたは抗体B単独の使用は、事によると心臓および肺または肝臓細胞に対する望まない傷害を結果的にもたらすと考えられる。しかしながら、抗体Aおよび抗体Bを、改良された細胞標的化のために一緒に用いることができる。したがって、抗体Aは、抗増殖性核酸をコードする遺伝子に連結され、かつ受容体を介する摂取系によって腫瘍だけでなく心臓および肺組織に送達される。しかしながら、遺伝子は、これらの細胞で転写されずまたは活性がない。抗体Bは、アクチベーターまたは抗増殖性核酸の転写もしくは活性化に必要な因子をコードする普遍的に活性のある遺伝子に連結することができ、腫瘍および肝臓細胞に送達される。それゆえ、心臓および肺細胞では不活性な抗増殖性核酸のみが送達され、そこではそれが転写されず、副作用を引き起こさない。肝臓細胞では、活性化因子をコードする遺伝子が送達されるが、抗増殖性核酸遺伝子が存在しないので効果を有さない。しかしながら、腫瘍細胞では、両方の遺伝子が送達され、かつ活性化因子が抗増殖性核酸を活性化し、腫瘍特異的な毒性効果をもたらすことができる。
【0067】
本発明の文脈中で、細胞の表面に発現する分子に対する抗体は有用である。そのような抗体には、FGF受容体、VEGF受容体、ウロキナーゼ受容体、E-およびP-セレクチン、VCAM-1、PDGF受容体、TGF受容体、エンドシアリン、αvβ3インテグリン、LFA-1、E9抗原、CD40、カドヘリン、ならびにelk-1に対する抗体が含まれるが、これらに限定されない。細胞によって発現される細胞表面分子に特異的である抗体は、モノクローナルまたはポリクローナル抗血清として容易に作製される。多くのそのような抗体が(例えば、American Type Culture Collection, Rockville, Md.)から入手可能である。あるいは、結合/内在化するリガンドに対する抗体も用いてもよい。そのような戦略において、ファージ粒子はそれらの表面に抗体を有すると考えられ、それはその後リガンドと複合体化すると考えられる。
【0068】
AAVP送達のために標的化される部位に応じて、多くのその他のリガンドをAAVP調製物の標的化工程に利用してもよい。ある種の態様において、受容体を介したエンドサイトーシスによって、AAVPを特異的な細胞タイプに標的化することが企図される。例えば、ラクトシルセラミド、およびアポリポタンパク質E3(「Apo E」)などの、LDL受容体関連タンパク質を標的化するペプチドは、肝臓にリポソームを標的化する際に有用である(Spanjer and Scherphof, 1983;WO 98/0748)。アシアロ糖タンパク質である、アシアロフェツイン(末端ガラクトシル残基を含む)もまた、肝臓にリポソームを標的化することが示されている(Spanjer and Scherphof, 1983;Hara et al., 1995)。マンノシル、フコシル、またはN-アセチルグルコサミンという糖は、ポリペプチドの骨格に連結された場合、高親和性マンノース受容体に結合する(その全体が参照により本明細書に具体的に組み入れられる、米国特許第5,432,260号)。したがって、これらの糖タンパク質を、本発明のAAVPにコンジュゲートすることができ、かつ特異的な細胞(例えば、マクロファージ)を標的化するのに有用であることが企図される。
【0069】
葉酸塩および葉酸受容体もまた、細胞標的化に有用であると記載されている(米国特許第5,871,727号)。この例では、ビタミン葉酸塩がAAVPカプシドタンパク質に連結されている。葉酸受容体は、そのリガンドに対する高い親和性を有し、かつ肺、胸部、および脳腫瘍を含む幾つかの悪性細胞株の表面に過剰発現している。トランスフェリンを介した送達系は、トランスフェリン受容体を発現する幅広い範囲の複製細胞を標的化する(Gilliland et al., 1980)。
【0070】
過剰増殖性疾患の処置のための遺伝子送達用の標的化リガンドの付加は、遺伝子産物が標的化されていない系が可能にするよりも毒性のある遺伝子の送達を可能にする。送達することができるより毒性のある遺伝子の例として、BaxおよびBakなどのプロアポトーシス遺伝子に加えて、アデノウイルスE4orf4およびプロドラッグ6-メチルプリンデオキシリボシドを毒性のあるプリン6-メチルプリンに変換するいわゆる「自殺遺伝子」である、大腸菌(E. coli)プリンヌクレオシドホスホリラーゼなどのウイルスおよびその他の病原菌に由来する遺伝子が含まれる。プロドラッグ療法で用いる自殺遺伝子の他の例には、大腸菌シトシン脱アミノ酵素遺伝子およびHSVチミジンキナーゼ遺伝子がある。
【0071】
ある種の局面において、AAVPを用いて腫瘍血管系を標的化することができる。腫瘍内皮は癌治療のための重要な標的である。その他の組織における最小限の毒性を伴って関心対象の治療的遺伝子を腫瘍内皮に標的化することは、依然として抗血管遺伝子治療の第一義的な目標である。近年、腫瘍内皮を標的化するAAVPが記載されている。本発明者らは、このベクターが、強力な抗血管剤であるヒト腫瘍壊死因子-α(TNF-α)をヒトメラノーマに送達する能力を検討した。AAVPによって内皮単球活性化ポリペプチド-II(EMAP-II)を送達することにより、TNF-α耐性のメラノーマがTNF-α処置に対して感受性になった。
【0072】
2つの遺伝子、TNF-αおよびEMAP-IIを運ぶAAVPベクターをインビトロおよびインビボで評価した。ヒトメラノーマ細胞(M21)をAAVP内在化およびTNF-α遺伝子発現についてインビトロで検討した。ヌードマウス内の皮下で成長した腫瘍であるM21/Pmelを、尾静脈注射によって全身的に処置した。免疫蛍光染色、TaqMan RT-PCR、および免疫組織化学的解析を用いて、標的化されたAAVPの腫瘍血管系への局在化、TNF-α遺伝子発現、およびアポトーシスを調べた。
【0073】
標的化されたAAVPの内在化がM21細胞で観察され、培養上清中の高レベルの機能的に活性のあるTNF-αが結果的にもたらされた。標的化されていないベクターの内在化はこれらの細胞で観察されなかった。AAVPの全身注射は、正常器官への最小限のウイルス局在化を伴う腫瘍に標的化されたウイルス送達を示した。AAVP送達は、TNF-α遺伝子産物の発現を結果的にもたらした。TNF-αの発現は、血管および周囲の腫瘍細胞におけるアポトーシスを誘導し、顕著な腫瘍退化を結果的にもたらした。さらに、EMAP-IIを発現するAAVPの標的化された送達は、TNF-α耐性ヒトメラノーマを、TNF-α処置に対して増感させた。標的化されたAAVPベクターを用いて、腫瘍血管系に特異的に抗血管薬剤を送達し、したがって全身毒性を低下させることができる。
【0074】
特異的な標的化リガンドの付着によって、本発明のAAVPを身体の特異的な領域に標的化し、指定された組織におけるAAVPの速やかな蓄積および濃縮、ならびにそれに応じて、核酸分子の速やかな蓄積および濃縮を提供することができる。本発明での使用が企図されるリガンドを、種々の方法でAAVPにコンジュゲートすることができる。標的化リガンドをカプシドタンパク質に連結するための様々な組成物および方法は、当技術分野において公知である。
【0075】
II、核酸
本開示のAAVPは、1つまたは複数のトランスジーンを標的細胞の核に送達し、それによって標的細胞におけるトランスジーンの発現を増強させる能力を有する。AAVPはまた、トランスジーンカセットのコンカテマーの形成を通じたトランスジーンカセットの変更された運命によって遺伝子発現における利点を授与し、それによって増強された遺伝子発現をもたらす可能性がある。
【0076】
本明細書で用いる場合の、「トランスジーン」という用語は、ある生命体から別の生命体へ転移されている遺伝子または遺伝子材料を指す。トランスジーンは、1つもしくは複数の遺伝子および/または1つもしくは複数のオリゴヌクレオチドを含んでもよい。例えば、トランスジーンは、レポーター遺伝子、自殺遺伝子、プロドラッグ変換酵素、および/または1つもしくは複数の治療的遺伝子を含んでもよい。本明細書で用いる場合、「オリゴヌクレオチド」は、20またはそれより少ない塩基対を持つ短い核酸配列を指す。本明細書で用いる場合の、「治療的遺伝子」という用語は、生命体の疾患、医学的状態、器官、組織、細胞、または生理学的特徴に対する治療的効果を提供する、核酸領域と定義される。本明細書で用いる場合、本明細書で用いる場合の「カセット」という用語は、関心対象のタンパク質、ポリペプチド、またはRNAを発現することができる核酸である。
【0077】
リガンドに加えて、AAVPは、その産物を選択または検出することができるレポーター遺伝子を含むトランスジーンを含んでもよい。本明細書において言及される場合、「レポーター遺伝子」は、蛍光、検出可能に標識された化合物もしくは発色基質、もしくは蛍光基質に対する酵素活性などによって、検出することができ;または成長条件によって選択することができる産物をコードする核酸領域である。そのようなレポーター遺伝子には、緑色蛍光タンパク質(GFP)、β-ガラクトシダーゼ、クロラムフェニコールアセチルトランスフェラーゼ(CAT)、ルシフェラーゼ、ネオマイシンホスホトランスフェラーゼ、分泌アルカリホスファターゼ(SEAP)、ヒト成長ホルモン(HGH)チミジンキナーゼなどが含まれるが、これらに限定されない。選択可能マーカーには、ネオマイシン(G418)、ハイグロマイシンなどなどの、薬物耐性物が含まれる。ある種の態様において、血液、その他の体液、または組織中で検出することができる分泌タンパク質をコードするレポーター遺伝子を用いてレポーター遺伝子の発現レベルを測定してもよい。
【0078】
レポーター遺伝子に加えて、トランスジーンはまた、治療的遺伝子を含んでもよい。そのようなトランスジーンを運ぶAAVPは、とりわけ、インビトロかまたはインビボのいずれかにおいて、対象(例えば、ヒト)を画像化することを可能にしてもよい。インビトロイメージングは、対象全体のまたは対象の標的部の非侵襲的イメージングを可能にしてもよい。これらのトランスジーンの導入後、当技術分野において公知のイメージング技術(例えば、BLIイメージング、PETイメージング、蛍光イメージングなど)を用いて発現を画像化してもよい。本明細書で用いる場合の、「対象」は、任意の哺乳動物実体を指し、例えば、対象は、遺伝子治療またはその他の処置を必要とするヒトであってもよい。
【0079】
その他の態様において、AAVPは自殺遺伝子を含んでもよい。本明細書で用いる場合の「自殺遺伝子」という用語は、プロドラッグの投与時に、遺伝子産物のその宿主細胞を殺傷する化合物への移行をもたらす核酸と定義される。使用してもよい自殺遺伝子/プロドラッグ組み合わせの例は、単純ヘルペスウイルス-チミジンキナーゼ(HSV-tk)およびガンシクロビル、アシクロビル、またはFIAU;オキシドレダクターゼおよびシクロヘキシミド;シトシンデアミナーゼおよび5-フルオロシトシン;チミジンキナーゼチミジレートキナーゼ(Tdk::Tmk)およびAZT;ならびにデオキシシチジンキナーゼおよびシトシンアラビノシドである。ある種の態様において、自殺遺伝子は、その宿主細胞の殺傷の結果として疾患または医学的状態に対する治療的効果を提供することにより、治療的遺伝子の様式で作用する可能性がある。
【0080】
「核酸」という用語は当技術分野において周知である。本明細書で用いる場合の「核酸」は通常、核酸塩基を含む、DNA、RNA、またはその誘導体もしくは類似体の分子(すなわち鎖)を指す。核酸塩基には、例えば、DNA中に見出される天然のプリンもしくはピリミジン塩基(例えば、アデニン「A」、グアニン「G」、チミン「T」、もしくはシトシン「C」)またはRNA(例えば、A、G、ウラシル「U」、もしくはC)が含まれる。「核酸」という用語は、各々「核酸」という用語の亜属として「オリゴヌクレオチド」および「ポリヌクレオチド」という用語を包含する。「オリゴヌクレオチド」という用語は、長さが約8から約100核酸塩基の分子を指す。「ポリヌクレオチド」という用語は、長さが約100核酸塩基よりも大きい少なくとも1つの分子を指す。
【0081】
本明細書中のある種の態様では、「遺伝子」は、転写される核酸を指す。ある種の局面において、遺伝子には、転写、もしくはメッセージ生成に関与する調節性領域、または組成物が含まれる。当業者によって理解されるように、「遺伝子」というこの機能的用語には、遺伝子の転写されない部分の核酸セグメント(遺伝子の転写されないプロモーターもしくはエンハンサー領域を含むが、これらに限定されない)を含む、ゲノム配列、RNAもしくはcDNA配列、またはより小さい人為的に改変された核酸セグメントの両方が含まれる。より小さい人為的に改変された遺伝子核酸セグメントは、タンパク質、ポリペプチド、ドメイン、ペプチド、融合タンパク質、突然変異体、および/もしくはそのような同様のものを発現してもよく、または核酸操作技術を用いて発現するように適合させてもよい。
【0082】
本発明のポリヌクレオチドは、「発現カセット」を形成してもよい。「発現カセット」は、特定の転写単位の発現を提供するポリヌクレオチドである。すなわち、それには遺伝子または転写単位の転写において機能するプロモーターエレメントおよび様々なその他のエレメントが含まれる。発現カセットはまた、より大きい複製ポリヌクレオチドまたは発現ベクターもしくはコンストラクトの一部であってもよい。
【0083】
これらの定義は通常、1本鎖分子を指すが、特異的な態様では、1本鎖分子に部分的に、実質的に、または完全に相補的である追加の鎖も包含すると考えられる。したがって、核酸は、分子を含む特定の配列の1つもしくは複数の相補鎖または「相補物」を含む2本鎖分子を包含してもよい。
【0084】
「他のコード配列から実質的に単離された」とは、関心対象の遺伝子が核酸のコード領域のかなりの部分を形成していること、または核酸が大きい染色体断片、その他の機能的遺伝子、RNA、もしくはcDNAコード領域など、天然のコード核酸の大部分を含まないことを意味する。勿論、これは元々単離されたような核酸を指し、および人間の手によって後で核酸に付加された遺伝子またはコード領域を除外しない。
【0085】
本明細書で用いる場合、「核酸塩基」は、例えば、少なくとも1つの天然の核酸(すなわち、DNAおよびRNA)中に見出される天然の核酸塩基(すなわち、A、T、G、C、またはU)、ならびにそのような核酸塩基の天然または非天然の誘導体および類似体などのヘテロ環状塩基を指す。核酸塩基は通常、天然の核酸塩基対合(例えば、AとT、GとC、およびAとUの間の水素結合)の代わりになり得る様式で、少なくとも1つの天然の核酸塩基との1つまたは複数の水素結合を形成する(「アニールする」または「ハイブリダイズする」)ことができる。
【0086】
本明細書で用いる場合、「ヌクレオチド」は、「骨格部分」をさらに含むヌクレオシドを指す。骨格部分は通常、ヌクレオチドを含む別の分子に、または別のヌクレオチドにヌクレオチドを共有結合で付着させて核酸を形成する。天然のヌクレオチド中の「骨格部分」は典型的には、5炭素糖に共有結合で付着している、リン部分を含む。骨格部分の付着は典型的には、5炭素糖の3'位かまたは5'位のいずれかで生じる。しかしながら、特にヌクレオチドが天然の5炭素糖またはリン部分の誘導体または類似体を含む場合に、その他の種類の付着が当技術分野において公知である。
【0087】
A、発現コンストラクト
本発明の発現コンストラクトには、治療的核酸および/またはイメージングタンパク質をコードする核酸が含まれてもよい。その他の局面において、発現コンストラクトは、本発明の治療的組成物および方法で用いることができる治療的発現コンストラクトであってもよい。ある種の態様では、遺伝子材料を操作して、イメージングタンパク質、標的化タンパク質、および/または治療的遺伝子をコードする発現カセットおよび/または発現コンストラクトを生成してもよい。
【0088】
本発明の態様には、2つの別々の種類の発現カセットまたは発現カセットを含む発現コンストラクトが含まれてもよい。1つのカセットは、イメージングタンパク質、すなわち、直接的に検出可能でありまたは間接的に検出可能である特性の活性を有するタンパク質の発現に用いられる。別の発現カセットは、治療的遺伝子をコードしてもよい。治療的ベクターの文脈では、治療的遺伝子は、疾患状態の予防的または治療的処置において有用な本明細書で考察する治療的遺伝子であってもよい。遺伝子治療の文脈では、遺伝子は、ベクターの骨格を提供するウイルスゲノム以外の源に由来するDNAを含むことを意味する、異種DNAであってもよい。遺伝子は、細菌、ウイルス、酵母、寄生虫、植物、または動物などの原核生物または真核生物源に由来してもよい。異種DNAはまた、複数の源、すなわち、多遺伝子コンストラクトまたは融合タンパク質に由来してもよい。異種DNAにはまた、1つの源に由来し得る調節性領域および異なる源由来の遺伝子が含まれてもよい。
【0089】
B、制御領域
それらがイメージングタンパク質をコードするのであれまたは治療的遺伝子をコードするのであれ、本発明の発現カセットおよび/またはコンストラクトには、典型的に、様々な制御領域が含まれると考えられる。これらの制御領域は、典型的には、関心対象の遺伝子の発現を調整する。
【0090】
1、プロモーター
本出願の全体を通じて、「発現コンストラクト」という用語は、核酸コード配列の一部または全てが転写されることができる遺伝子産物、例えば、イメージングタンパク質または治療的タンパク質の一部または全てをコードする核酸を含む任意の種類の遺伝子コンストラクトを含むことが意味される。転写物はタンパク質に翻訳されてもよいが、それが翻訳される必要はない。ある種の態様において、発現には、遺伝子の転写およびmRNAの遺伝子産物への翻訳の両方が含まれる。その他の態様において、発現には阻害RNAまたはDNAなどの治療的核酸の転写のみが含まれる。
【0091】
遺伝子産物をコードする核酸は、プロモーターの転写制御下にある。「プロモーター」は、遺伝子の特異的な転写を開始するのに必要とされる、細胞の機構、または導入された機構によって認識されるDNA配列を指す。特定の局面において、転写は、構成的、誘導可能、および/または抑制可能であってもよい。「転写制御下」という語句は、RNAポリメラーゼの開始および遺伝子の発現を制御するための、プロモーターが核酸に関して正しい場所および方向にあることを意味する。
【0092】
プロモーターという用語は、ここではRNAポリメラーゼIIのための開始部位の周辺にクラスターを成す転写制御モジュールの群を指すよう用いる。プロモーターがどのように編成されているかに関する考えの大部分は、様々なレトロウイルスプロモーター、HSVチミジンキナーゼ(tk)、およびSV40初期転写単位についての解析を含む、幾つかのウイルスプロモーターの解析に由来している。
【0093】
追加のプロモーターエレメントは、転写の開始の頻度を調節する。最近、多くのプロモーターが同じく出発部位の下流の機能的エレメントを含むことが示されているが、典型的には、これらは出発部位の30〜110 bp上流の領域に位置する。プロモーターエレメント間の間隔は融通が利き、そのためエレメントが互いに対して逆にされたりまたは動かされたりしてもプロモーター機能は保存される。プロモーター次第で、転写を活性化するために個々のエレメントが協調的にかまたは独立にかのいずれかで機能することができるように見える。
【0094】
それが標的化された細胞における核酸の発現を導くことができる限り、関心対象の核酸配列の発現を制御するのに利用される特定のプロモーターが重要であると考えられない。したがって、ヒト細胞が標的化される場合、標的化されるヒト細胞で発現されることができるプロモーターの近辺および該プロモーターの制御下に核酸コード領域を位置付けることが好ましい。一般的に言えば、そのようなプロモーターには、ヒトプロモーターか、ウイルスプロモーターか、またはその組み合わせのいずれかが含まれ得る。
【0095】
様々な態様において、ヒトサイトメガロウイルス即初期遺伝子プロモーター(CMVIE)、SV40初期プロモーター、ラウス肉腫ウイルス長末端反復、β-アクチン、ラットインスリンプロモーター、およびグリセルアルデヒド-3-リン酸デヒドロゲナーゼを用いて、関心対象のコード配列の高レベル発現を得ることができる。発現のレベルが所与の目的のために十分であるならば、関心対象のコード配列の発現を達成することが当技術分野において周知である、その他のウイルス、レトロウイルスまたは哺乳動物細胞または細菌ファージのプロモーターの使用が同じく企図される。周知の特性を持つプロモーターを利用することによって、トランスフェクションまたは形質転換後の関心対象のタンパク質の発現のレベルおよびパターンを最適化することができる。
【0096】
特異的な生理的または合成的シグナルに応答して調節されるプロモーターの選択は、非誘導条件下の細胞と比較した場合の遺伝子産物の誘導可能な発現を可能にすることができる。例えば、1つのトランスジーン、または多シストロン性のベクターを活用した時の複数のトランスジーンの発現が、ベクターを産生する細胞にとって毒性がある場合、トランスジーンのうちの1つまたは複数の発現を禁止しまたは低下させることが望ましい場合がある。プロデューサー細胞株にとって毒性があり得るトランスジーンの例は、プロアポトーシスおよびサイトカイン遺伝子である。トランスジーン産物が毒性であり得る場合に、幾つかの誘導可能なプロモーター系がウイルスベクターの産生に利用可能である。
【0097】
エクジソン系(Invitrogen, Carlsbad, CA)は1つのそのような系である。この系は、哺乳動物細胞における関心対象の遺伝子の調節された発現を可能にするように設計されている。それは、トランスジーンの基礎レベル発現はほとんど可能にしないが、200倍を越える誘導能を可能にする厳しく調節された発現メカニズムからなる。有用であると考えられる別の誘導可能な系は、もとはGossenおよびBujardによって開発された(Gossen and Bujard, 1992;Gossen et al., 1995)Tet-Off(商標)またはTet-On(商標)系(Clontech, Palo Alto, CA)である。この系はまた、高レベルの遺伝子発現がテトラサイクリンまたはドキシサイクリンなどのテトラサイクリン誘導体に応答して調節されるのを可能にする。
【0098】
幾つかの状況において、治療的発現ベクター中のトランスジーンの発現を調節することが望ましい場合がある。例えば、様々に異なる強さの活性を持つ異なるウイルスプロモーターを望ましい発現のレベルに応じて活用してもよい。哺乳動物細胞では、強い転写活性化を提供するために、多くの場合CMV即初期プロモーターが用いられる。低下したレベルのトランスジーンの発現が望ましい場合、あまり強力でないCMVプロモーターの改変版も用いられている。造血細胞におけるトランスジーンの発現が望ましい場合、MLVまたはMMTV由来のLTRなどのレトロウイルスプロモーターが多くの場合用いられる。所望の効果に応じて使用し得るその他のウイルスプロモーターには、SV40、RSV LTR、HIV-1およびHIV-2 LTR、E1A、E2A、またはMLP領域からなどのアデノウイルスプロモーター、AAV LTR、カリフラワーモザイクウイルス、HSV-TK、ならびにトリ肉腫ウイルスが含まれる。
【0099】
同様に、標的化されていない組織に対する潜在的な毒性または望まない効果を低下させるために、組織特異的または選択的プロモーターを用いて特異的な組織または細胞における転写をもたらしてもよい。例えば、PSA、プロバシン、前立腺酸性ホスファターゼ、または前立腺特異的腺カリクレイン(hK2)などのプロモーターを用いて前立腺における遺伝子発現を標的化してもよい。同様に、以下のプロモーターを用いて、その他の組織における遺伝子発現を標的化してもよい(表1)。
【0100】
ある種の徴候において、遺伝子治療ベクターの投与後の特異的な時間に転写を活性化することが望ましい場合がある。これは、ホルモンまたはサイトカイン調節可能であるプロモーターのようなプロモーターを用いて行なわれてもよい。例えば、徴候が特異的なステロイドが産生されまたは送られる生殖腺組織である治療的適用において、アンドロゲンまたはエストロゲンによって調節されるプロモーターの使用が有利である場合がある。ホルモン調節可能であるようなそのようなプロモーターには、MMTV、MT-1、エクジソン、およびルビスコが含まれる。甲状腺、下垂体、および副腎ホルモンに応答性のプロモーターなどのその他のホルモンによって調節されるプロモーターは、本発明において有用であることが期待される。使用し得るサイトカインおよび炎症性タンパク質応答性のプロモーターには、KおよびTキニノーゲン(Kageyama et al., 1987)、c-fos、TNF-アルファ、C反応性タンパク質(Arcone et al., 1988)、ハプトグロビン(Oliviero et al., 1987)、血清アミロイドA2、C/EBPアルファ、IL-1、IL-6(Poli and Cortese, 1989)、補体C3(Wilson et al., 1990)、IL-8、アルファ-1酸性糖タンパク質(Prowse and Baumann, 1988)、 アルファ-1アンチトリプシン、リポタンパク質リパーゼ(Zechner et al., 1988)、アンギオテンシノーゲン(Ron et al., 1990)、フィブリノゲン、c-jun(ホルボールエステル、TNF-アルファ、UV放射、レチノイン酸、および過酸化水素によって誘導可能)、コラゲナーゼ(ホルボールエステルおよびレチノイン酸によって誘導される)、メタロチオネイン(重金属およびグルココルチコイドで誘導可能)、ストロメリシン(ホルボールエステル、インターロイキン-1、およびEGFによって誘導可能)、アルファ-2マクログロブリン、ならびにアルファ-1アンチキモトリプシンが含まれる。また、オステオカルシン、低酸素応答性配列(HRE)、MAGE-4、CEA、アルファ-フェトプロテイン、GRP78/BiP、およびチロシナーゼなどの腫瘍特異的なプロモーターを用いて、腫瘍細胞における遺伝子発現を調節してもよい。
【0101】
単独のまたは別のプロモーターと組み合わせた上記のプロモーターのいずれかが、望ましい作用に応じて本発明に従って有用であり得ることが予想される。さらに、このプロモーターのリストは網羅的または限定的であると解されるべきではなく、当業者は本明細書において開示されたプロモーターおよび方法と併せて使用し得るその他のプロモーターがあるのを知っていると考えられる。
【0102】
(表1)組織特異的プロモーター
【0103】
2、エンハンサー
エンハンサーは、DNAの同じ分子上の遠い位置に位置するプロモーターからの転写を増加させる遺伝子エレメントである。エンハンサーはプロモーターにとてもよく似たように編成されている。すなわち、それらは多くの個々のエレメントから構成され、その各々が1つまたは複数の転写タンパク質に結合する。エンハンサーとプロモーターの間の基本的な差異は操作上のものである。全体としてのエンハンサー領域は、遠隔地での転写を刺激することができなければならず;これはプロモーター領域またはその構成要素エレメントについて当てはまる必要はない。他方、プロモーターは、特定の部位および特定の方向でのRNA合成の開始を導く1つまたは複数のエレメントを有さなければならない一方、エンハンサーはこれらの特異性を欠く。プロモーターおよびエンハンサーは、多くの場合重複および連続し、多くの場合非常によく似たモジュール編成を有するように思われる。
【0104】
以下は、上で列挙された組織特異的プロモーターに追加するプロモーターである、発現コンストラクト中の関心対象の遺伝子をコードする核酸と組み合わせて使用し得る細胞性プロモーター/エンハンサーおよび誘導可能プロモーター/エンハンサーのリストである(表2および表3)。さらに、遺伝子の発現を駆動するために、(真核生物プロモーターデータベース EPDBによる)任意のプロモーター/エンハンサーの組み合わせも使用し得る。適当な細菌ポリメラーゼが送達複合体の一部としてまたは追加の遺伝子発現コンストラクトとしてのいずれかで提供されるならば、真核生物細胞は、ある種の細菌プロモーターからの細胞質転写を支持することができる。
【0105】
本発明の好ましい態様において、治療的発現コンストラクトは、ウイルスまたはウイルスゲノムに由来する人為的に改変されたコンストラクトを含む。ある種のウイルスが受容体を介するエンドサイトーシスによって細胞に侵入する能力および宿主細胞ゲノムに入り込みかつウイルス遺伝子を安定的かつ効率的に発現させる能力によって、それらは哺乳動物細胞への外来遺伝子の転移のための魅力的な候補になっている(Ridgeway, 1988;Nicolas and Rubenstein, 1988;Baichwal and Sugden, 1986;Temin, 1986)。
【0106】
(表2)
【0107】
(表3)
【0108】
C、ポリアデニル化シグナル
ポリアデニル化シグナルが治療的および/イメージングベクターで用いられてもよい。cDNA挿入物が利用される場合、典型的には、遺伝子転写物の適切なポリアデニル化をもたらすためにポリアデニル化シグナルを含めることが望まれる。ポリアデニル化シグナルの性質は本発明の首尾よい実施に必須であるとは考えられておらず、ならびにヒトまたはウシ成長因子ホルモンおよびSV40ポリアデニル化シグナルなどの任意のそのような配列を利用してもよい。ターミネーターも発現カセットのエレメントとして企図される。これらのエレメントは、メッセージレベルを増強させるのにおよびカセットを読み終えてその他の配列に移るのを最小限にするのに役立つことができる。
【0109】
D、治療的遺伝子
本発明のAAVP粒子を用いて、治療的発現ベクターを含む、種々の治療的またはイメージング薬剤を送達してもよい。本発明は、種々の異なる治療的遺伝子の使用を企図する。例えば、酵素、ホルモン、サイトカイン、オンコジーン、受容体、イオンチャネル、腫瘍抑制因子、転写因子、薬物選択可能マーカー、毒素、および様々な抗原をコードする遺伝子が、本発明による使用のための適した遺伝子として企図される。さらに、オンコジーンに由来するアンチセンスおよび阻害RNAコンストラクトは、本発明によるその他の関心対象の「遺伝子」である。
【0110】
本発明に従って、選択された遺伝子またはポリペプチドは、任意のタンパク質、ポリペプチド、またはペプチドを指してもよい。治療的遺伝子またはポリペプチドは、疾患を処置または防止する目的のために対象に投与することができる遺伝子またはポリペプチドである。例えば、治療的遺伝子は、癌の処置または防止のために対象に投与される遺伝子であることができる。治療的遺伝子の例として、Rb、CFTR、p16、p21、p27、p57、p73、C-CAM、APC、CTS-1、zac1、scFV ras、DCC、NF-1、NF-2、WT-1、MEN-I、MEN-II、BRCA1、VHL、MMAC1、FCC、MCC、BRCA2、IL-1、IL-2、IL-3、IL-4、IL-5、IL-6、IL-7、IL-8、IL-9、IL-10、IL-11、IL-12、GM-CSF、G-CSF、チミジンキナーゼ、Bax、Bak、Bik、Bim、Bid、Bad、Harakiri、Fas-L、mda-7、fus、インターフェロンα、インターフェロンβ、インターフェロンγ、ADP、p53、ABLI、BLC1、BLC6、CBFA1、CBL、CSFIR、ERBA、ERBB、EBRB2、ETS1、ETS2、ETV6、FGR、FOX、FYN、HCR、HRAS、JUN、KRAS、LCK、LYN、MDM2、MLL、MYB、MYC、MYCL1、MYCN、NRAS、PIM1、PML、RET、SRC、TAL1、TCL3、YES、MADH4、RB1、TP53、WT1、TNF、BDNF、CNTF、NGF、IGF、GMF、aFGF、bFGF、NT3、NT5、ApoAI、ApoAIV、ApoE、Rap1A、シトシンデアミナーゼ、Fab、ScFv、BRCA2、zac1、ATM、HIC-1、DPC-4、FHIT、PTEN、ING1、NOEY1、NOEY2、OVCA1、MADR2、53BP2、IRF-1、zac1、DBCCR-1、rks-3、COX-1、TFPI、PGS、Dp、E2F、ras、myc、neu、raf、erb、fms、trk、ret、gsp、hst、abl、E1A、p300、VEGF、FGF、トロンボスポンジン、BAI-1、GDAIF、またはMCCが含まれるが、これらに限定されない。
【0111】
治療的遺伝子のその他の例として、酵素をコードする遺伝子が含まれる。例として、ACPデサチュラーゼ、ACPヒドロキシラーゼ、ADP-グルコースピロホリラーゼ、ATPアーゼ、アルコールデヒドロゲナーゼ、アミラーゼ、アミログルコシダーゼ、カタラーゼ、セルラーゼ、シクロオキシゲナーゼ、デカルボキシラーゼ、デキストリナーゼ、エステラーゼ、DNAポリメラーゼ、RNAポリメラーゼ、ヒアルロンシンターゼ、ガラクトシダーゼ、グルカナーゼ、グルコースオキシダーゼ、GTPアーゼ、ヘリカーゼ、ヘミセルラーゼ、ヒアルロニダーゼ、インテグラーゼ、インベルターゼ、イソメラーゼ、キナーゼ、ラクターゼ、リパーゼ、リポオキシゲナーゼ、リアーゼ、リゾチーム、ペクチンエステラーゼ、ペルオキシダーゼ、ホスファターゼ、ホスホリパーゼ、ホスホリラーゼ、ポリガラクツロナーゼ、プロテイナーゼ、ペプチダーゼ、プラナーゼ、リコンビナーゼ、リバーストランスクリプターゼ、トポイソメラーゼ、キシラナーゼ、レポーター遺伝子、インターロイキン、またはサイトカインが含まれるが、これらに限定されない。
【0112】
治療的遺伝子のさらなる例として、カルバモイルシンセターゼI、オルニチントランスカルバミラーゼ、アルギノスクシネートシンセターゼ、アルギノスクシネートリアーゼ、アルギナーゼ、フマリルアセトアセテートヒドロラーゼ、フェニルアラニンヒドロキシラーゼ、アルファ-1アンチトリプシン、グルコース-6-ホスファターゼ、低密度リポタンパク質受容体、ポルホビリノーゲンデアミナーゼ、第VIII因子、第IX因子、シスタチオンベータ-シンターゼ、分岐鎖ケト酸デカルボキシラーゼ、アルブミン、イソバレリル-CoAデヒドロゲナーゼ、プロピオニルCoAカルボキシラーゼ、メチルマロニルCoAムターゼ、グルタリルCoAデヒドロゲナーゼ、インスリン、-グルコシダーゼ、ピルビン酸カルボキシラーゼ、肝ホスホリラーゼ、ホスホリラーゼキナーゼ、グリシンデカルボキシラーゼ、H-タンパク質、T-タンパク質、メンケス病銅輸送ATPアーゼ、ウィルソン病銅輸送ATPアーゼ、シトシンデアミナーゼ、ヒポキサンチン-グアニンホスホリボシルトランスフェラーゼ、ガラクトース-1-リン酸ウリジルトランスフェラーゼ、フェニルアラニンヒドロキシラーゼ、グルコセルブロシダーゼ、スフィンゴミエリナーゼ、-L-イズロニダーゼ、グルコース-6-リン酸デヒドロゲナーゼ、HSVチミジンキナーゼ、またはヒトチミジンキナーゼをコードする遺伝子が含まれる。
【0113】
治療的遺伝子には、ホルモンをコードする遺伝子も含まれる。例として、成長ホルモン、プロラクチン、胎盤ラクトゲン、黄体形成ホルモン、濾胞刺激ホルモン、絨毛性ゴナドトロピン、甲状腺刺激ホルモン、レプチン、副腎コルチコトロピン、アンギオテンシンI、アンギオテンシンII、β-エンドルフィン、β-メラニン細胞刺激ホルモン、コレシストキニン、エンドセリンI、ガラニン、胃阻害ペプチド、グルカゴン、インスリン、リポトロピン、ニューロフィジン、ソマトスタチン、カルシトニン、カルシトニン遺伝子関連ペプチド、β-カルシトニン遺伝子関連ペプチド、悪性の高カルシウム血症因子、副甲状腺ホルモン関連タンパク質、副甲状腺ホルモン関連タンパク質、グルカゴン様ペプチド、パンクレアスタチン、膵ペプチド、ペプチドYY、PHM、セクレチン、血管作用性小腸ペプチド、オキシトシン、バソプレッシン、バソトシン、エンケファリンアミド、メトルフィンアミド、アルファメラニン細胞刺激ホルモン、心房性ナトリウム利尿因子、アミリン、アミロイドP構成要素、コルチコトロピン放出ホルモン、成長ホルモン放出因子、黄体形成ホルモン放出ホルモン、神経ペプチドY、サブスタンスK、サブスタンスP、またはチロトロピン放出ホルモンをコードする遺伝子が含まれるが、これらに限定されない。
【0114】
さらに別の態様において、異種遺伝子には、単鎖抗体が含まれてもよい。単鎖抗体の産生のための方法は当業者に周知である。当業者は、そのような方法のために(参照により本明細書に組み入れられる)米国特許第5,359,046号に差し向けられる。単鎖抗体は、短いペプチドリンカーを用いて重鎖および軽鎖の可変ドメインを一緒に融合し、それによって単一分子上に抗原結合部位を再構成することによって創り出される。
【0115】
E、多遺伝子コンストラクトおよびIRES
本発明のある種の態様において、多遺伝子ポリシストロン性メッセージを創り出すために内部リボソーム結合部位(IRES)エレメントの使用が用いられる(Pelletier and Sonenberg, 1988)。哺乳動物のメッセージ由来のIRES(Macejak and Sarnow, 1991)と同様に、ピカノウイルス(picanovirus)ファミリー(ポリオおよび脳心筋炎)の2つのメンバー由来のIRESエレメントが記載されている(Pelletier and Sonenberg, 1988)。IRESエレメントは、異種オープンリーディングフレームにつなぐことができる。多数のオープンリーディングフレームを一緒に転写し、各々をIRESによって分離し、ポリシストロン性メッセージを創り出すことができる。IRESエレメントのおかげで、各オープンリーディングフレームは、効率的な翻訳のためにリボソームに接近し易い。単一のプロモーター/エンハンサーを用いて多数の遺伝子を効率的に発現させ、単一のメッセージを転写させことができる。任意の異種オープンリーディングフレームをIRESエレメントにつなぐことができる。これには、分泌タンパク質、独立の遺伝子によってコードされる、多サブユニットタンパク質、細胞内または膜結合タンパク質、および選択可能マーカーのための遺伝子が含まれる。このように、幾つかのタンパク質の発現を同時に人為的に改変して単一のコンストラクトおよび単一の選択可能マーカーを持つ細胞にすることができる。
【0116】
F、核酸の調製
例えば、化学合成、酵素的産生、または生物学的産生などの、当業者に公知の任意の技術によって、核酸を作製してもよい。酵素的に産生される核酸の非限定的な例として、PCR(商標)(例えば、各々が参照により本明細書に組み入れられる、米国特許第4,683,202号および米国特許第4,682,195号参照)などの増幅反応、または参照により本明細書に組み入れられる、米国特許第5,645,897号に記載されたオリゴヌクレオチドの合成において酵素によって産生される核酸が含まれる。生物学的に産生される核酸の非限定的な例として、細菌の中で複製される組換えDNAベクター(例えば、参照により本明細書に組み入れられる、Sambrook et al. 2001参照)などの、生細胞の中で産生される(すなわち、複製される)組換え核酸が含まれる。
【0117】
III、薬学的組成物および投与の経路
臨床的適用が企図される場合、意図する適用のために適当な形態でAAVP組成物(治療的組成物)の薬学的組成物を調製する必要があると考えられる。通常、これは、パイロジェンだけでなく、ヒトまたは動物にとって有害であり得るその他の不純物も本質的に含まない組成物を調製する工程を伴っていると考えられる。
【0118】
通常、組成物を患者への導入に好適であるようにするために、適当な塩および緩衝剤を利用することが望まれる。本発明の水性組成物は、薬学的に許容される担体もしくは水性媒体に溶けたまたは分散した有効量のAAVPまたはその他の薬剤を含む。「薬学的にまたは薬理学的に許容される」という語句は、動物またはヒトに投与された時に、不都合な、アレルギー性の、またはその他の厄介な反応を産生しない分子実体および組成物を指す。
【0119】
本明細書で用いる場合、「薬学的に許容される担体」には、任意および全ての溶媒、分散媒体、コーティング剤、抗菌および抗真菌薬剤、等張および吸収遅延薬剤などが含まれる。薬学的に活性な物質のためのそのような媒体および薬剤の使用は、当技術分野において周知である。任意の従来の媒体または薬剤が本発明のAAVP組成物と合わない限りを除き、イメージング試薬としてのまたは治療的組成物におけるその使用が企図される。その他の抗癌薬剤などの、補足の活性成分を組成物中に組み入れることもできる。保管および使用の普通の条件下で、これらの調製物は微生物の成長を防止するための防腐剤を含む。静脈内ビヒクルには、流体および栄養補液が含まれる。防腐剤には、抗微生物薬剤、酸化防止剤、キレート薬剤、および不活性ガスが含まれる。調合薬中の様々な構成要素のpHおよび正確な濃度を周知のパラメーターに従って合わせる。
【0120】
癌などの状態または疾患を画像化することおよび/または改善することなどの、意図された目標に基づいて、組成物の有効量を決定する。「単位用量」という用語は、対象における使用に適した物理的に別個の単位を指し、各単位は、その投与、すなわち、適当な経路および治療レジメンと関連した所望の応答を産生するように計算された所定の数量の治療的組成物を含む。処置の数および単位用量の両方に従って、投与されるべき数量は、処置されるべき対象、対象の状態、および望ましい保護による。治療的組成物の正確な量はまた、施術者の判断によりおよび各々の個人に特有である。
【0121】
2つの活性成分を含む組み合わせ組成物も企図される。特に、本発明は、AAVP組成物および少なくとも1つの第二の治療薬、例えば、抗新生物薬物を含む組成物を提供する。
【0122】
A、非経口投与
本発明の活性組成物を非経口投与用に製剤化し、例えば、静脈内、筋肉内、皮下、または腹腔内経路さえも経由する注射用に製剤化してもよい。第二の薬剤を活性成分として含む水性組成物の調製は、本開示に照らして当業者に公知であると考えられる。典型的には、そのような組成物を、液体溶液かまたは懸濁かのいずれかとして、注射可能なものとして調製することができ;注射前の液体の添加時に溶液または懸濁を調製するために用いるのに適した固形形態を調製することもでき;および調製物を乳化することもできる。
【0123】
注射可能な使用に適した薬学的形態には、滅菌水性溶液または分散;ゴマ油、ピーナッツ油、または水性プロピレングリコールを含む製剤;および滅菌注射可能溶液または分散の即時調製用の滅菌粉末が含まれる。全ての場合において、形態は滅菌されていなければならずおよび容易な注入可能性(syringability)が存在する程度まで流動性でなければならない。
【0124】
担体は、例えば、水、エタノール、ポリオール(例えば、グリセロール、プロピレングリコール、および液体ポリエチレングリコールなど)、その適した混合物、ならびに植物油を含む溶媒または分散媒体であることもできる。例えば、レシチンなどの、コーティング剤の使用によって、分散の場合に必要とされる粒子サイズの維持によって、および界面活性剤の使用によって、適切な流動性を維持することができる。様々な抗菌および抗真菌薬剤、例えば、パラベン、クロロブタノール、フェノール、ソルビン酸、チメロサールなどによって、微生物の作用の防止をもたらすことができる。多くの場合、等張薬剤、例えば、糖または塩化ナトリウムを含めることが好ましいと考えられる。吸収を遅延させる薬剤、例えば、モノステアリン酸アルミニウムおよびゼラチンの組成物での使用によって、注射可能組成物の長期の吸収をもたらすことができる。
【0125】
滅菌注射可能溶液は、必要とされる量の活性化合物を上で列挙された様々なその他の成分と共に適当な溶媒中に組み入れ、必要な場合、それに続いて濾過滅菌することによって調製される。通常、分散は、様々な滅菌された活性成分を塩基性分散媒体および必要とされる上で列挙された成分からの必要とされるその他の成分を含む滅菌ビヒクル中に組み入れることによって調製される。滅菌注射可能溶液の調製用の滅菌粉末の場合、調製の特定の方法は、先に滅菌濾過されたその溶液由来の任意のさらなる所望の成分を加えた活性成分の粉末を産出する真空乾燥およびフリーズドライ技術である。
【0126】
水性溶液での非経口投与のために、例えば、溶液は必要ならば好適に緩衝化されるべきであり、かつ液体希釈剤は最初に十分な生理食塩水またはグルコースで等張にされるべきである。これらの特定の水性溶液はとりわけ、静脈内、筋肉内、皮下、および腹腔内投与に好適である。これに関連して、利用することができる滅菌水性媒体は、本開示に照らして当業者に公知であると考えられる。例えば、1投薬量を1 mLの等張NaCl溶液に溶かし、かつ1000 mlの皮下投与液体に添加するかまたは提案された注入の部位で注射するかのいずれかであり得る(例えば、「Remington's Pharmaceutical Sciences」第15版、1035〜1038および1570〜1580ページ参照)。投薬量の若干の変動は、処置されている対象の状態によって必ず生じると考えられる。投与に責任を負う人は、いずれにしても、個々の対象のための適当な用量を決定すると考えられる。
【0127】
本発明のより良い理解を容易にするために、具体的な態様に関する以下の実施例を挙げる。以下の実施例は、決して、本発明の範囲全体を限定または定義するように読まれるべきではない。
【0128】
IV、実施例
以下の実施例は、本発明の様々な態様を例証するという目的のために与えられ、およびいかなる形であっても本発明を限定するよう意味されない。当業者は、本発明が目的を実行しかつ述べられた目標および利点を得るだけでなく、本明細書において本来備わっている目的を実行し、目標および利点を得るのにも十分適合していることを容易に正しく認識すると考えられる。本実施例は、本明細書に記載する方法と共に、好ましい態様を目下代表するものであり、例示するものであり、および本発明の範囲に対する限定であることが意図されない。特許請求の範囲の範囲によって定義されるような本発明の精神の範囲内に包含されるその中での変化およびその他の使用が当業者の心に浮かぶと考えられる。
【0129】
実施例1
AAVP標的化
A、実験手順
標的化されたAAVP粒子の設計、構築、および作製
RGD-4CファージおよびRGD-4C AAVPを中間体(RGD-4C fUSE5-MCS)の作製ならびにその後のRGD-4CファージコンストラクトおよびRGD-4C AAVPの作製:という2工程過程で人為的に改変した。RGD-4C fUSES-MCSは、特異的な標的化ペプチドRGD-4Cをコードするオリゴヌクレオチド挿入物および真核生物発現カセットの挿入用のマルチクローニング部位(MCS)を有するfMCSプラスミドの断片を含んだ。RGD-4Cファージ由来fUSE5 DNAおよびファージ由来fMCS DNAを宿主大腸菌(MC1061)のライセートから精製した。本発明者らは、RGD-4C FUSE5プラスミドの5.4 kb BamHIISacII断片をfMCSプラスミドの4.1 kb BamHI/SacII断片にライゲートすることによって、中間体RGD-4C fUSE5-MCS を得た。次に、本発明者らは、pAAV-eGFP プラスミド(増強されたGFP;Stratagene)のITRからITRまでのPacI断片(2.8 kb)をRGD 4C fUSE5-MSCのPstI部位にクローニングすることによって、標的化されたAAVP-GFPを創り出した。簡潔に述べると、pAAVをPacIで消化して2.8 kb断片を放出し、それをDNAポリメラーゼで平滑化し、かつRGD-4C fUSE5-MSCの平滑化されたPstI部位にクローニングした。ITRがない標的化されたファージコンストラクトを作製するために、pAAV-eGFPのITRの間に位置しかつpCMV-GFPおよびSV40ポリAを含む2.3 kb断片をEcoRI消化で放出し、DNAポリメラーゼで平滑化し、その後RGD-4C fUSE5-MSCのMCSにクローニングした。GFPを発現する細胞を選択するために、pQBIホスホグリセリン酸キナーゼ-1(PGK;QBIOgene)プロモーターのおよびGFPneo融合配列を含むBamHI-Sac1断片をAAVPまたは対照ファージコンストラクトのNotI部位にクローニングし、GFPを発現する細胞がG418耐性であることを確実にした。pQBI PGKのGFPneo断片をBamHIおよびSac1消化で放出し、かつDNAポリメラーゼで平滑化し;その後NotIへのリン酸化リンカーを付加した。NotI消化後、1.57 kb GFPneo断片をAAVPまたは非キメラファージコンストラクトのNotI部位にクローニングした。最後に、HSVtkまたはLucの遺伝子を運ぶ標的化されたAAVP粒子を作製するために、HSVtkまたはLucを含むBamHI-Not1断片をpAAVプラスミドのBamHI-Not1部位にサブクローニングし、GFPを取り替えた。ITR-HSVtk-ITRまたはITR-Luc-ITR断片をpAAV-HSVtkおよびpAAV-Lucから除去し、その後RGD-4C fUSE5-MCSに挿入した。コンストラクトをDNAシークエンシングおよび制限解析で確認し、宿主大腸菌(MC1061)の培養上清から精製し、PBSに再懸濁し、および再遠心分離した。結果として得られた上清を大腸菌(k91Kan)で滴定した。連続希釈をテトラサイクリンおよびカナマイシンを含むルリア-ベルターニ(LB)寒天プレート上にプレーティングしならびにコロニー計数によって形質導入単位(TU)を決定した。
【0130】
哺乳動物細胞表面結合および内在化アッセイ
本発明者らは、(BRASILと呼ばれる)選択的相互作用リガンド法(Giordano et al., 2001)のバイオパニングおよび迅速解析を用いて、インタクトな細胞へのファージ結合を評価した。簡潔に述べると、KS1767細胞をエチレンジアミン四酢酸(EDTA)で剥がし、かつ1% BSAを含むダルベッコの改変イーグル培地(DMEM)にml当たり4 x 106細胞で再懸濁した。細胞懸濁(50 μl)を109 TUのRGD-4C AAVPかまたはスクランブルバージョンのRGD-4C(CDCFGDCRC(SEQ ID NO:2)、CDCGFDCRC(SEQ ID NO:3)、CRCDGFCDC(SEQ ID NO:4))、突然変異体RGE-4Cペプチド、もしくは標的化されていない対照を提示するAAVPクローンのいずれかとインキュベートした。2時間後、AAVP/細胞混合物(水性相)をフタル酸ジブチル:シクロヘキサン(9:1 [v:v]、D = 1.03 g/ml)からなる非混和性の有機相(400 μlエッペンドルフチューブ中200 μl溶液)の上に移し、かつ10,000 gで10分間4℃で遠心分離した。その後、チューブを液体窒素中で瞬間凍結し、チーブの底を切り取り、および細胞-AAVPペレットを単離しならびに膜に結合したAAVPを回収した(Giordano et al., 2001)。
【0131】
細胞内在化のために、KS1767細胞を組織チャンバースライド(Lab-Tek II Chamber Slide System; Nalge Nunc International Corp.)で増殖させ、PBSで2回洗浄し、109 TUのRGD-4C AAVPまたはスクランブルバージョンのRGD-4CもしくはRGE-4Cを提示する対照AAVPと1% BSAを含むDMEM中で37℃でインキュベートし、および4時間のインキュベーション後にPBSで洗浄して、結合していないAAVPを除去した。細胞を20 mMグリシン(pH 2.3)で濯ぐことにより、細胞膜に結合したクローンを化学的に溶出した。次に、細胞をPBSで3回洗浄し、4%パラホルムアルデヒド(PFA)を含むPBSでRTで15分間固定し、PBSで洗浄し、0.2% Triton X-100で透過処理し、PBSで洗浄し、および1% BSAを含むPBSでブロッキングした。その後、細胞を1:200希釈の一次抗M13バクテリオファージ抗体(Amersham)と1% BSAを含むPBS中でRTで2時間インキュベートし、PBSで洗浄し、および1:200希釈のCy3-コンジュゲートされた抗ウサギ二次抗体と1% BSAを含むPBS中で1時間RTでインキュベートした。最後に、細胞をPBSで洗浄し、4% PFAを含むPBSで固定し、マウントし、光学蛍光顕微鏡で可視化した。
【0132】
AAVP粒子によって形質導入された細胞からの組換えAAVのレスキュー
ヒト293細胞にRGD-4C AAVP-GFPneoまたはRGD-4Cファージ-GFPneoを感染させた。感染4日後、細胞にAAV rep-およびcap-発現プラスミド(pXX2)(Xiao et al., 1998)をトランスフェクトしならびに野生型アデノウイルス5型(Ad)を重感染させた。このように、AAV repおよびcap遺伝子をトランスフェクションによって供給し、かつアデノウイルスヘルパー機能を重感染によって提供した。アデノウイルス感染72時間後に細胞を採取し、その後上清を用いて新しい293細胞に感染させた。48時間後にFACSを用いることによってGFP発現を解析した。このアッセイにおいて、機能的な組換えAAVは、RGD 4C AAVP-GFPキメラを形質導入した細胞からのみ作製されたが、非キメラファージ-GFPまたは幾つかの対照を形質導入した細胞からは作製されなかった。同様の結果はまた、全てのRGD-4C AAVPクローンを用いて得られたが、ファージクローンのいずれを用いても得られなかった。
【0133】
クローン性の哺乳動物細胞株の作製
ヒト293細胞にRGD-4C AAVP-GFPneoまたはRGD-4Cファージ-GFPneoを(各々の場合に細胞当たり106 TUで)感染させた。単一クローン(群当たりn=9)をG418選択下で単離し、かつ選択12週後にFACSでGFP発現について解析した。安定クローンをファージ-GFPneoについてはファージクローン#1-9およびAAVP-GFPneoについてはAAVP #1-9クローンと名付けた。
【0134】
腫瘍モデル
動物実験法は、施設の動物研究委員会によって審査および承認された。腫瘍を有するマウスを記載されたように樹立した(Pasqualini et al., 1997;Arap et al., 1998; Ellerby et al., 1999;Hajitou et al., 2001;Arap et al., 2004;Marchib et al., 2004)。アベルチンBの腹腔内投与によってまたはガス(2%イソフルランおよび98%酸素)吸入によってマウスに麻酔をかけた。標的化されたコンストラクトまたは対照を静脈内投与した。腫瘍細胞をトリプシン処理し、計数し、遠心分離し、および血清を含まない培地に再懸濁した。カポジ肉腫(KS 1767)、膀胱癌腫(UC3)、または前立腺癌腫(DU145)株由来の合計106細胞を6週齢免疫不全ヌードマウスに皮下に埋め込んだ。EF43-FGF4マウス乳腫瘍細胞(5 x 104)を6週齢メスBALBIc免疫適格マウスに皮下に埋め込んだ。腫瘍体積を記載されたように計算し(Pasqualini et al., 1997;Arap et al., 1998;Ellerby et al., 1999;Hajitou et al., 2001;Arap et al., 2004;Marchib et al., 2004)および平均腫瘍体積 * 標準偏差(SD)として表した。腫瘍が-50 mm2(小さいとみなされる)または-150 mm2(大きいとみなされる)のDU145由来異種移植片の体積に達した時(0日目)、腫瘍を有するマウスは単一静脈内用量のRGD-4C AAVP-HSVtk、または対照を受けた。サイズが一致した腫瘍を有するマウスのコホートにおいて2日後にGVC処置(1日当たり80 mg/kg、腹腔内)を始めた。
【0135】
腫瘍を有するマウスにおける分子-遺伝子イメージング
非侵襲的な分子イメージングのために、本発明者らはヒト細胞株DU145に基づいた前立腺癌のモデルを用い、その中で右肩の皮下部に腫瘍異種移植片を持つオスのヌードマウスを用いた。ホタルLuc遺伝子発現を画像化するために、腫瘍を有するマウスは、単一用量(150 mg/kg)の基質D-ルシフェリン(Xenogen)を腹腔内投与により受けた。Luc遺伝子を運ぶ標的化されたRGD-4C AAVPまたは対照(標的化されていないAAVP-Luc、もしくはスクランブルのRGD-4C AAVPLuc)の尾静脈投与後にIn Vivo Imaging System 200(IVIS200;Xenogen, CA)を用いることによって、光子の放出を画像化した。イメージングパラメーター:画像獲得時間、1分;ビニング、2;フィルターなし;フルストップ(flstop)、1;視野、10 cm。光子/秒/cm2/srと表される、シグナル強度を測定するための腫瘍に対して関心対象の領域(ROI)を手作業で定義した。
【0136】
BLIは実験系におけるトランスジーン発現細胞の生存率を検討評価することができるが、それは臨床的に適用可能ではない。したがって、樹立された腫瘍異種移植片の生存率を検討評価するために、[18F]-FDG 100 μCi/マウスの静脈内投与2時間後にmicroPETスキャナー(Concorde Microsystems, TN)を用いてマウスを画像化した。[18F]-FDGは商業的に(PETNet, Houston, TX)得られた。HSVtk遺伝子発現を画像化するために、放射性標識ヌクレオシド類似体[18F]-FEAUの静脈内投与1〜2時間後にPETイメージングを行なった。コンピュータ制御されたポジショニングベッドが装着され、10.8-cmの横断方向の視野および8-cmの軸方向の視野(FOV)を有し、それがセプタムを有さずおよび3次元リストモードでもっぱら操作される、microPET R4(Concorde Microsystems, Inc.)でPETイメージングを行なった。350〜750 keVのエネルギーウィンドウおよび6 nsの時間ウィンドウを用いて完全3次元リストモードデータを収集した。全ての生データをまず3次元サイノグラムの中にソートし、次いでASIPRO VMソフトウェア(Concord Microsystems, TN)を用いたフーリエリビニングおよびOSEM画像再構成を行なった。画像ピクセルサイズは、横断方向に1.2 mmスライス厚でおよそ1 mmであった。
【0137】
もとは Alauddinら(2003)によって記載されたように、l-ブロモ-2-デオキシ-2-[18F]フルオロ-3,5-ジ-O-ベンゾイル-α-D-アラビノフラノースによる凝縮のためのピリミジン塩基として5-エチルウラシル-2,5-ビス-トリメチルシリルエーテルを用いることによって、放射性標識[l8F]-FEAUを99%を越える放射化学的純度まで合成した。腫瘍ならびにその他の器官および組織における[18F]-FEAUまたは[18F]-FDG由来の放射能濃度を定量するために、関心対象の領域を画像上に描きかつ測定された値を nCi/mm2から%グラム当たりの注射された用量(%ID/g;Tjuvajev et al., 1998)に変換した。上で記載されたように、HSVtk遺伝子を運ぶ標的化されたAAVPまたは対照の標的化されていないAAVPの投与後3、5、10、および16日目に繰り返しの[18F]-FEAU PETイメージングを行ない;11日目から19日目にGCV処置を投与した。注目すべきことに、そうでない場合にはHSVtk酵素によるリン酸化をFEAUと競合すると考えられる、GCVの十分な消去を可能にするために、16日目のPETイメージングをGCV投薬の24時間後に行なった。[18F]-FDGを用いたPETイメージングをAAVP投与後17日目に繰り返し、もしあれば、残存する腫瘍の生存率を検討評価した。
【0138】
免疫組織化学
麻酔をかけたマウスを殺し、4% PFAを含むPBSを灌流させた。ラット抗マウスCD31抗体(BD Biosciences)を用いることにより凍結切片で腫瘍血管新生を検討評価した。TUNELキット(Promega)を用いてパラフィン包埋切片に対してアポトーシス解析を行なった。組織でのファージ免疫検出のために、パラフィン切片をウサギ抗ファージ一次抗体(Sigma)、次いでペルオキシダーゼがコンジュゲートされた抗ウサギ二次抗体(Dako)とインキュベートした。基質-色原体3,3'-ジアミノベンジジンでスライドを現像し、ヘマトキシリンで対比染色した。GFP免疫染色のために、器官および腫瘍を2% PFAを含むPBS中で2時間固定し、15%スクロースを含むPBS中で48時間平衡化した。凍結切片を4% PFAを含むPBS中で20分間後固定しならびに1% BSAおよび0.1% Triton X-100を含むPBS(PBS-T)中の5%ヤギ血清でブロッキングした。次に、組織切片を2%ヤギ血清および1% BSA 中のウサギ親和性精製GFP抗体(Molecular Probes)とインキュベートした。その後、切片をPBS-Tおよび1% BSA中の二次抗体であるAlexaFluor 488コンジュゲートされたヤギ抗ウサギ(Molecular Probes)で染色した。PBS灌流動物から除去された腫瘍のアセトン固定凍結切片に対してαvインテグリン免疫染色を行なった。切片を一次ラット抗インテグリンαvモノクローナル抗体(Chemicon)と1時間、次いで二次Cy3コンジュゲートされたヤギ抗ラット抗体(Jackson ImmunoResearch)とインキュベートした。
【0139】
哺乳動物細胞において機能的であるリガンド指向性粒子
組換えAAVおよび(RGD-4Cファージ(Pasqualini et al., 1997;Arap et al., 1998)と名付けられた)二重環状ペプチドCDCRGDCFC(SEQ ID NO:2)を提示するfd-tetファージクローンから構成される標的化されたキメラウイルスを構築した。RGD-4Cペプチドは、腫瘍細胞および腫瘍血管の新生血管形成内皮の両方で過剰発現される細胞表面受容体である、αvインテグリンに結合する(Brooks et al., 1994;Pasqualini et al., 1997;Arap et al., 1998;Sipkins et al., 1998;Ellerby et al., 1999;Hood et al., 2002)。(AAV/ファージ;AAVPとさらに称される)キメラウイルスを得るために、本発明者らは、組換えAAV由来の真核生物遺伝子カセットをRGD-4Cファージ(RGD-4C AAVP)、挿入物のないファージ(標的化されていないAAVP)、またはスクランブルのRGD-4C AAVPもしくは(RGE-4Cと名付けられた)DからEへの突然変異体AAVPなどの、対照ペプチドを提示するファージのゲノム間領域に挿入し、およびそれをファージDNAと共にファージカプシド中にパッケージングした(図7)。結果として得られるαvインテグリンに標的化されたキメラウイルスのシスエレメントが依然として機能的であることを示すために、本発明者らは、RGD-4Cペプチドのリガンド特性およびAAVPの文脈における逆向き末端反復(ITR)のレスキュー特性を評価した。まず、ペプチド特異性を評価するために、哺乳動物細胞に結合も感染もしない、標的化されていないAAVPまたはRGE-4Cもしくは様々なスクランブルバージョンのRGD-4C配列(図1A)などの陰性対照ペプチドを提示するAAVPとは対照的に、RGD-4C AAVPがαvインテグリンを発現する哺乳動物細胞に結合することが示された。レポーター遺伝子を運ぶRGD-4C AAVPは、対照と比べて、リガンド指向性の内在化(図1B)および哺乳動物細胞の形質導入(図1C)を媒介することができることも示された。細胞内在化実験については、陰性対照として標的化されていないAAVP、様々なRGD-4CスクランブルのAAVP、またはRGE-4C AAVPが含まれ(図1B);細胞形質導入実験については、標的化されていないAAVP(図lC)、スクランブルのRGD-4C AAVP、またはRGE-4C AAVPが陰性対照の役割を果たした。これらの結果と一致して、本発明者らは、合成RGD-4Cペプチドが様々な標的化されたRGD-4Cファージに基づくコンストラクトの細胞結合および内在化を特異的に阻害することを以前に示している(Giordano et al., 2001;Chen et al., 2004、および未公表の結果)。最後に、哺乳動物細胞形質導入は、標的化されていないAAVP(図1D)、スクランブルのRGD-4C、またはRGE-4Cなどの陰性対照と比べて、合成RGD-4Cペプチドによって特異的に競合させることもできることが示された。これらの結果(図1Aおよび1B)のうちの幾つかが、細胞膜からAAVPを除去するためのグリシン(低pH)洗浄工程の選択的失敗から結果的に生じるアーティファクトを表し得るという可能性を除外するために、温度(氷冷)対照実験を行ない(Giordano et al., 2001)、そこでは細胞結合は観察されたが、RGD-4C AAVPによって媒介される内在化は観察されなかった。
【0140】
次に、ITRがAAVP粒子中で依然として機能的であるかどうかを評価するために、本発明者らはレスキュー実験を行なった。本発明者らは、機能的な組換えAAV粒子がRGD 4C AAVPを形質導入した哺乳動物細胞のみから作製されるが、陰性対照コンストラクトを形質導入した細胞からは作製されないことを示している(図1E)。これらのデータは、RGD-4C AAVP粒子を結果的にもたらす遺伝子のキメラ化が、(i)リガンド指向性RGD-4Cファージのペプチド標的化特性または(ii)RGD-4C AAVP により形質導入された哺乳動物細胞から組換えAAV粒子をレスキューする能力を本来変化させないことを立証している。レポータートランスジーン発現の並列時間経過は、RGD-4C AAVP形質導入が、RGD-4Cファージの形質導入と比較してずっとより長い間検出可能であることを明らかにした(表4)。
【0141】
(表4)インビトロでのトランスジーン発現
2人独立の観察者が、時点当たり三つ組みウェルの293細胞で、半定量的にGFP発現を得点化した。
【0142】
トランスジーン発現の分子メカニズム
AAVP粒子によって媒介されるトランスジーン発現の分子メカニズムについての見識を得るために、本発明者らは、哺乳動物細胞における形質導入されたゲノムの運命を詳しく調べた。まず、GFPneo発現AAVPを用いて個々の形質導入された細胞クローンの選択を可能にすることにより、安定に形質導入された細胞株を作製した。RGD-4C AAVP-GFPneoかまたはAAV ITRを欠くRGD-4Cファージ-GFPneoのいずれかを用いて、αvインテグリンを発現するヒト293細胞(Nakamura et al., 2002)に形質導入し、およびG418選択下でクローンを単離した。レスキュー実験により、機能的な組換えAAV-GFPneoは全てのRGD-4C AAVP-GFPneo 293細胞クローンから作製されたが、RGD-4Cファージ-GFPneoクローンからは何も作製されなかったことが示され、したがってキメラの個々のクローンは機能的なITRを含むことが確認された。非キメラRGD-4Cファージ-GFPneoを形質導入した細胞はG418耐性を保持したが、GFP発現はRGD-4C AAVP-GFPneoクローンのGFP発現よりも一般に弱かった(図2A)。その後、本発明者らは、ゲノムDNAの広範囲にわたる制限酵素消化、それに続くサザンブロッティングおよびポリメラーゼ連鎖反応(PCR)に基づく解析によって、安定に形質導入されたクローンにおけるGFPneoトランスジーンカセットの運命を明らかにしようと試みた(図2、図8、および表5)。トランスジーンカセットの持続性を証明するために、解析前にゲノムDNAをAflIIおよびXhoIで消化して全長トランスジーンカセットの放出を検出した。そのような放出は、非キメラRGD-4C ファージコンストラクトによって形質導入されたクローンの33%(9クローンのうちn=3)でのみと比較して、RGD-4C AAVPによって形質導入されたクローンの100%(9クローンのうちn=9)で観察された(図2B)。ゲノムDNAの別の制限消化を設計して、トランスジーンカセットの潜在的なコンカテマー形態を検出した(Lieber et al., 1999; Hsiao et al., 2001)。トランスジーンカセットの頭-尾コンカテマーの存在はRGD-4C AAVPによって形質導入されたクローンの67%(9クローンのうちn=6)で検出されたが、そのようなコンカテマーは非キメラRGD-4C ファージコンストラクトによって形質導入されたクローンでは検出されなかった(図2C)。トランスジーンカセットのあり得る追加のコンカテマー形態を同定するために、コンストラクトの5'および3'末端の両隣にあるプライマーを用いることによって、マルチプレックスPCRを行なった。この場合もやはり、コンカテマーは非キメラRGD-4C ファージコンストラクトによって形質導入されたクローンでは見出されなかったが、その一方でRGD-4C AAVPによって形質導入されたクローンの100%がコンカテマー形態を含み(9クローンのうちn=9)、それらは全て頭-尾方向で見出された(図8B)。その上、より小さいPCR産物のtopoクローニングにより、ITR欠失を持つトランスジーンカセットの頭-尾方向が明らかにされた(図8C)(Yang et al., 1997)。最後に、大きいPCR産物は配列決定することができなかったが、それらのサイズから、接合部位でのインタクトなITRを持つコンカテマーの存在が示唆された。各々の単一クローンについてのDNAの個々の解析も詳述している(表5)。理論またはメカニズムに束縛されることを望まないが、これらのデータから、哺乳動物トランスジーンカセット全体の維持、エピソームDNAのより良好な持続、トランスジーンカセットのコンカテマーの形成を通じた、またはおそらくはこれらの相互排他的でないメカニズムの組み合わせによるトランスジーンカセットの変更された運命によって、AAVPが遺伝子発現における利点を授与し得ることが示唆される。これらの観察は、AAVの理解における最近の進展と一致する(McCarty et al., 2004)。
【0143】
(表5)RGD-4C AAVP-GFneoまたはRGD-4Cファージ-GFneoによって安定に形質導入された293細胞クローンにおけるベクターDNAの運命
【0144】
インビボでの腫瘍標的化および分子-遺伝子イメージング
標的化されたキメラウイルス粒子の中心的エレメント(すなわち、RGD-4CペプチドおよびAAV ITR)がインタクトかつ機能的であることを示した後でならびに哺乳動物細胞におけるAAVPを介する遺伝子発現の分子メカニズムを解明した後で、AAVPの全身投与後の腫瘍への遺伝子送達の特異性および有効性を評価した。初めの前臨床的モデルとして、ヒトカポジ肉腫KS 1767細胞に由来する皮下腫瘍異種移植片を持つヌードマウスを用いた(Arap et al., 1998;Ellerby et al., 1999)。まず、ウイルスコンストラクトがマウスにおけるKS1767由来異種移植片を標的化することを確認するために、RGD-4C AAVPかまたは幾つかの陰性対照(標的化されていないAAVP、スクランブルのRGD-4C AAVP、もしくはRGE-4C AAVP)のうちの1つのいずれかを静脈内投与した。3〜5分の循環時間の後、腫瘍血管系における強い抗AAVP染色がRGD-4C AAVPを受けたマウスで観察されたが、対照マウスでは観察されなかった(図3A)。緑色蛍光タンパク質(GFP)をコードするRGD-4C AAVP変異体をレポーターとして用いて、インサイチュー免疫蛍光顕微鏡イメージングを用いることによって、このベクター(RGD-4C AAVP-GFP)がKS1767由来異種移植片に形質導入することができるかどうかを明らかにした。腫瘍を有するマウスへのRGD-4C AAVP-GFPかまたは陰性対照コンストラクトのいずれかの全身投与の7日後に、腫瘍におけるおよび異なる器官におけるGFPに対する免疫染色を行なった。免疫蛍光により、主にRGD4C AAVP-GFPを受けたマウスにおける腫瘍血管および周囲の腫瘍細胞でのGFP発現が明らかになった。対照的に、標的化されていないAAVP-GFP、スクランブルのAAVP-GFP、または突然変異体AAVP-GFPを受けた対照マウス由来の腫瘍ではGFP染色は検出されなかった(図3B)。この染色パターンから、リガンド指向性の形質導入は腫瘍の血管内皮におけるαvインテグリンの標的化によって媒介されることが示唆される(Brooks et al., 1994;Pasqualini et al., 1997;Arap et al., 1998;Sipkins et al., 1998;Ellerby et al., 1999;Giordano et al., 2001;Hood et al., 2002;Chen et al., 2004)。矛盾のないことには、幾つかの非標的対照器官(脳、肝臓、膵臓、および腎臓)はGFPの組織発現を欠いていた(図9)。まとめると、RGD-4C AAVP粒子はリガンド指向性のメカニズムによって腫瘍異種移植片を特異的に標的化し、かつインビボでの全身投与後にそれらに形質導入できることをこれらのデータは示している。
【0145】
次に、本発明者らは、RGD-4C AAVPの全身投与後に生きている腫瘍を有するマウスにおける、それぞれホタルルシフェラーゼ(Luc)およびHSVtkレポーター遺伝子発現の時間的力学および空間的不均一性の非侵襲的なモニタリングのための前臨床的な生体発光イメージング(BLI)および[18F]-FEAUを用いた臨床適用可能な分子-遺伝子PETイメージングの有効性を検討評価した。この特に普及している腫瘍は依然として、患者の中で適切に画像化するための課題であるので、これらの分子-遺伝子イメージング検討をヒト前立腺癌の前臨床モデルで実行した。本発明者らはまず、腫瘍を有するマウスにおけるホタルルシフェラーゼ(Luc)トランスジーンレポーターのインビボイメージング用の標準的な実験の仕組みを用いた(図4A)。それがマウスにおけるレポーター遺伝子イメージングのための非常に感度の良い方法でありおよび画像中の非特異的バックグラウンド活性をほとんど有さないので(Gross and Piwnica-Worms, 2005a;Gelovani and Blasberg, 2003;Uhrbom et al., 2004;Walensky et al., 2004)、本発明者らは、Luc発現のBLIを選択した。非常に腫瘍特異的なLucの発現がRGD-4C AAVP-Lucを受けているマウスにおけるDU145腫瘍で観察された。対照的に、腫瘍に関連した生体発光シグナルは、標的化されていないAAVP-LucまたはスクランブルのRGD-4C AAVP-Lucを受けている対照マウスで観察することができなかった。全ての種類のAAVPベクター(標的化されていないAAVP-Luc、スクランブルのRGD-4C AAVP-Luc、RGE-4C AAVP-Luc、およびRGD-4C AAVP-Luc)について、生体発光は肝臓、脾臓、または腎臓などの正常器官では観察されなかった。これらのデータによって、RGD-4C AAVP-GFPを用いた免疫蛍光顕微鏡イメージング検討で観察されたRGD-4C AAVPを介する標的化およびトランスジーン発現の腫瘍特異性が確認されている。ここで示された先の結果と一致して(図9)、非特異的な肝細胞によるファージ粒子のクリアランス(Geier et al., 1973;Pasqualini et al., 1997;Arap et al., 1998;Barbas et al., 2001)にもかかわらず、そのような現象は望ましくない肝臓の形質導入を結果的にもたらさないことが分布の動態から示唆される。これらの観察は、哺乳動物のウイルス遺伝子送達ベクターによる(肝臓などの)正常器官の十分に立証された非特異的な形質導入(Shayakhmetov et al., 2005)とは際立って対照的である。インビボでBLIを用いることによって、腫瘍内のLucレポータートランスジーン発現はAAVP投与3日後に明瞭に検出可能でありおよび徐々に増加して10日目までに最高レベルに達した。Lucレポーター遺伝子発現の繰り返しの2次元BLIが1日おきに行なわれならびにAAVPによって媒介されるレポータートランスジーン発現の特異性、時間的力学、および空間的不均一性を検討するための初期の費用効果的な戦略を提供した。しかしながら、Lucレポーター遺伝子発現のBLIは臨床適用可能ではないので、本発明者らは次に、自殺遺伝子(ガンシクロビル;GCVと組み合わせた場合)としてならびにHSVtk特異的な放射性標識ヌクレオシド類似体(例えば、[124I]-FAIU、[18F]-FHBG、および[18F]-FEAU)を用いる臨床適用可能なPETイメージング用のレポータートランスジーンとしての両方の役割を果たすことができる、HSVtk遺伝子をAAVPベクターに導入した。
【0146】
以前の検討によって、HSVtk発現のPETイメージングは、トランスジーン発現の場所、大きさ、および持続時間を定義する能力を提供することが立証された(Tjuvajev et al., 1998;Tjuvajev et al., 1999;Ray et al., 2001;Massoud and Gambhir, 2003)。HSVtk形質導入細胞株およびインビボの腫瘍における蓄積した放射性標識トレーサーの大きさはHSVtk発現のレベルと相関することも以前に明らかにされている(Blasberg and Tjuvajev, 2003;Gross and Piwnica-Worms, 2005a;Tai and Laforest, 2005)。ここで示された検討において、本発明者らは、とりわけ薬物動態的考察(全ての正常器官および組織における非常に低いバックグラウンド活性)(Kang et al., 2005)から、その他のヌクレオシド類似体よりも良いHSVtk酵素用の放射性標識基質である放射性標識ヌクレオシド類似体2'-[18F]-フルオロ-2'-デオキシ-l-β-D-アラビノ-フラノシル-5-エチル-ウラシル([18F]-FEAU)を選択し、合成し、および使用した。(0、3、5、10、および16日目に)[18F]-FEAUを用いた繰り返しのPETイメージングを用いることにより、本発明者らは、ヌードマウスにおけるDU145由来腫瘍異種移植片ならびにその他の器官および組織中のRGD-4C AAVP-HSVtkまたは標的化されていないAAVP-HSVtkの単一全身投与後のHSVtk遺伝子発現の時間的力学および空間的不均一性を可視化および定量した(図4B)。RGD4C AAVP-HSVtkかまたは標的化されていないAAVP-HSVtkのいずれかの投与前の腫瘍異種移植片サイズ(およそ150 mm2)だけでなく、RGD4C AAVP-HSVtkかまたは標的化されていないAAVP-HSVtkのいずれかの投与後の腫瘍成長速度もマウスの両方のコホートでよく似ていた(図4C)。[18F]-FEAUを用いたPETイメージングにより、RGD-4C AAVP-HSVtk の投与後の最初の5日間の腫瘍におけるHSVtkトランスジーン発現のレベルの漸進的な増加(% グラム当たり投与された静脈内用量の増加)、それに続くベクター投与後10日目に向けてのHSVtk発現レベルの漸進的な安定化が明らかにされた。対照的に、標的化されていないAAVP-HSVtkを受けている対照の腫瘍を有するマウスでは、ほんのわずかな[18F]-FEAUの腫瘍蓄積の増加が3日目に観察され、それは速やかにバックグラウンドレベルまで減少した(図4D)。前述のBLI実験と一致して、[18F]-FEAU PET検出可能なHSVtk発現は、非標的器官または組織では観察されなかった(図4B)。実際、PET画像における低レベルで不均一な活性は、正常なバックグラウンド活性を表し、それは非標的組織に対する腫瘍でのHSVtk発現の特異性を人為的に「改善する」ために低レベルの放射能の切り捨てがなされていないことを示すために画像の中で意図的に強調された。腫瘍が確実に触診可能なサイズ(およそ350〜400 mm2)まで成長し、かつHSVtk発現のプラトーが腫瘍において達成された時、GCVによる処置を動物の全てのコホートで開始した(図4C)。[18F]-フルオロデオキシグルコース([18F]-FDG)を用いたPETイメージングは、グルコース代謝およびGCVによって誘導される腫瘍生存率の変化をモニタリングする役目を果たした。GCV治療の開始2日前(ベクター投与後9日目)に、両群のマウスにおけるDU145腫瘍は生存可能でかつ [18F]-FDGを活発に蓄積した(図4E)。GCV治療後、RGD-4C AAVP-HSVtkを受けたマウスにおける腫瘍の体積は、標的化されていないAAVP-HSVtkを受けたマウスにおいてよりも有意に小さかった(p<0.05;図4C)。その上、[18F]-FDGの蓄積の減少によって証明されているように腫瘍異種移植片もまた代謝が抑制された(図4E)。(FEAUとの競合を避けるために)最後のGCV用量の24時間後に得られる、PET画像における[18F]-FEAU蓄積の際立った減少によって証明されているように、RGD-4C AAVP-HSVtkを投与されたマウスの腫瘍におけるHSVtk発現のレベルもまたGCV治療後に有意に減少した(図4D)。これらの検討により、RGD-4C AAVPによる腫瘍標的化の特異性が確認され、かつHSVtkトランスジーン発現のレベルがGCVの効果的なプロドラッグ活性化にとって十分に高いことが示されている。
【0147】
その他の前臨床モデルで有効性を水平に評価するために、本発明者らは、異なる種および組織学的起源由来の腫瘍細胞株のパネルを集めならびに免疫抑制または免疫適格マウスで腫瘍を発生させた。カポジ肉腫(KS 1767)に由来する腫瘍を有するマウスのコホートは、単一静脈内用量のRGD-4C AAVP-HSVtkかまたは標的化されていないAAVP-HSVtk(対照)のいずれか、それに続く全ての群におけるGCV処置を全身的に受けた。ビヒクルで処置されたマウスまたは標的化されていないAAVPを受けたマウスと比較した場合、RGD-4C AAVP-HSVtkを受けた腫瘍を有するマウスで顕著な腫瘍成長抑制が観察された(図5A)。同様の腫瘍成長抑制効果は、より大きい腫瘍異種移植片を処置した場合(図5D)でも、ヌードマウスにおけるUC3由来膀胱癌腫(図5B)およびDU145由来前立腺癌腫(図5C)で観察され、かつ示されたイメージングデータ(図4)と一致した。観察された抗腫瘍効果が種特異的かまたは異種移植片特異的かのいずれかである可能性を除外するために、本発明者らは、標準的なマウス腫瘍モデルに対するRGD-4C AAVP-HSVtk の有効性を解析しようと試みた。本発明者らは、免疫適格マウスにおいて高度に血管が発達した腫瘍の速やかな成長を誘導するためにEF43-FGF4マウス乳細胞が皮下投与されている(Hajitou et al., 2001)同種同系腫瘍を選んだ。まず、本発明者らは、EF43 FGF4由来腫瘍へのRGD-4C AAVPのリガンド指向性ホーミングを抗ファージ免疫染色により示している(図10)。RGD-4C AAVP-GFPかまたは標的化されていないAAVP-GFPのいずれかを同種同系乳腫瘍を持つマウスに3〜5分の循環時間の間静脈内投与した。標的化されていないAAVP-GFPと違って、RGD-4C AAVP GFPは腫瘍における強い抗ファージ染色を産生した(図10A)。次に、本発明者らは、静脈内投与7日後に抗GFP抗体を用いた免疫蛍光を行ない、RGD-4C AAVP-GFPを受けたマウスにおける腫瘍での強いGFP発現を明らかにし;対照的に、標的化されていないAAVPGFPを受けたマウス由来の腫瘍ではGFP染色が検出されず;矛盾のないことには、抗avインテグリン抗体はEF43-FGF4腫瘍での強い発現を検出した(図10B)。この場合もやはり、単一全身用量のRGD-4C AAVP-HSVtk、それに続くGCVによって、EF43-FGF-4腫瘍の成長が顕著に阻害された(図5E〜Gおよび図6)。その上、治療の終結後に腫瘍が成長して元に戻った場合、RGD-4C AAVP-HSVtkの繰り返された投与によって、EF43-FGF4腫瘍成長が再び阻害され、かつ腫瘍を有するマウスの生存が改善された(図5F)。ファージに基づく粒子は免疫原性であることが公知であるが、この特色はそれ自身を標的化することを通じて調整することができる(Trepel et al., 2001)。事実、RGD-4C AAVP-HSVtkプラスGCVは、非常に高い力価の循環抗ファージIgGにもかかわらず、ファージワクチンを接種した免疫適格マウスに対して驚くべき程に有効であり続けた(図5G)。選択的な実験において、ビヒクル単独、ビヒクルプラスGCV、標的化されていないAAVP、標的化されていないAAVPプラスGCV、標的化されたRGD-4C AAVP、標的化されたRGD-4C AAVP-GFP、および標的化されたRGD-4C AAVP-GFPプラスGCV(模擬形質導入)を含む陰性実験対照の広範囲にわたるパネルを用いた(図6A)。
【0148】
処置後効果について調査するために、本発明者らは、治療7日後に回収されたEF43 FGF4腫瘍の詳細な組織病理学的解析を得た。単一全身用量のRGD-4C AAVP-HSVtkプラスGCVによって引き起こされた徹底的な腫瘍破壊が顕著であった。具体的には、ヘマトキシリンおよびエオシン(H&E)染色によって、腫瘍の中心部の一様な破壊およびほんのわずかの生存している外部縁が明らかになり;対照的に、標的化されていないAAVP-HSVtkはそのような効果を有さなかった(図6B)。抗CD31抗体を用いた染色によって、腫瘍中心領域内の乱された腫瘍血管および外部縁に向かう保存された血管系の両方が確認された一方、標的化されていないAAVP-HSVtkで処置された腫瘍では損傷が観察されなかった(図6B)。HSVtk/GCV戦略は細胞のアポトーシス死と関連するので(Hamel et al., 1996)、本発明者らはまた、アポトーシス細胞に印をする、ターミナルデオキシヌクレオチジルトランスフェラーゼを介するdUTPビオチンニックエンド標識化(TUNEL)染色について腫瘍を評価した。RGD-4C AAVP-HSVtkおよびGCVで処置された腫瘍では、TUNEL染色によって、腫瘍中心領域におけるアポトーシスは検出されたが、外部縁内では検出されず、一方標的化されていないAAVP-HSVtkキメラを受けたマウス由来の腫瘍ではアポトーシスが検出されなかった(図6B)。同じ実験プロトコルで処置された腫瘍を有するマウスから除去された対照器官からは、組織病理学的異常が明らかにならなかった(図11)。まとめると、これらの結果は、単一全身用量のRGD-4C AAVP-HSVtkプラスGCV維持が、腫瘍成長を抑制できることを示している。
【0149】
各々の腫瘍区画(すなわち、腫瘍細胞対腫瘍血管内皮および/またはストローマ)を標的化することの相対的寄与は、リガンド-受容体系および使用されるモデルに依存すると考えられる。例えば、腫瘍細胞における膜標的(すなわち、αvインテグリン)の発現は、低い(EF43 FGF4)から高い(KS1767)まで様々に異なると考えられる。その上、使用される各モデルの形質導入(および適用)のための具体的な至適用量を除いて、トランスジーン発現を調べるための至適時期もある。換言すれば、レポーター遺伝子発現の化学量論は、個々の細胞におけるレポーター発現のレベルおよびパターンだけでなく、増殖しているトランスジーン発現細胞対瀕死のトランスジーン発現細胞の相対的な数にも依存する。したがって、本発明者らは、これらの実施例のための固定されたパラメーターを用いたが、ケースバイケースの基準での標的化されたAAVP至適用量および時間枠のさらなる決定が依然として適用される。
【0150】
本発明者らは、とりわけ異なる前臨床的および臨床的な分子-遺伝子イメージング設定と組み合わせて標的化されたAAVPを用いて、分子腫瘍学を遥かに越える広範囲の現在手に負えない生物学的問題を扱うことができることを企図する。例えば、(CMVプロモーターの代わりに)組織および/または疾患特異的プロモーターを用いた標的部位へのコンストラクトの全身的リガンド指向性送達によって、それらの対応する天然の遺伝子の発現をインビボでモニタリングすることが可能になると考えられ;そのようなプロモーターに駆動されるレポーター活性の転写によって、細胞トラフィッキングおよび生着の検討が可能になると考えられる。レポータートランスアクチベーション、相補、または再構成戦略による基質特異的分解、タンパク質-タンパク質相互作用、およびにその他の分子事象の実験的モニタリング(Luker et al., 2004;De and Gambhir, 2005;Gross and Piwnica-Worms 2005b)などの幾つかの非侵襲的イメージング適用を細胞でおよび動物丸ごとで利用することができる。本発明者らは最近、生体センサーおよび細胞標的化薬剤としての金ナノ粒子およびバクテリオファージのネットワークを記載しており(Souza et al., 2006)、そのような技術をリガンド指向性AAVPと組み合わせて分子-遺伝子イメージングをさらに改良することができる。話は変わるが、(AAVPベクターと対照的に)ITRのないファージに基づくコンストラクトは陰性実験対照としての役割を果たすことができるので、AAVPそれ自体がトランスジーン持続性および染色体組み込みにおけるITR構造の機械的役割を検討するための適した試薬を提供する可能性がある。したがって、協力する多くのその他の全身的な標的化および分子イメージング適用が、この新規のプラットフォームおよびその派生ツールによって比較的短い時間枠で可能である。
【0151】
実施例2
腫瘍血管系にTNF-αを標的化するためのAAVPの使用
A.方法
細胞培養
ヒト臍帯静脈内皮細胞(HUVEC)をCambrex(Walkersville, Maryland)から入手し、以前に記載されたように内皮細胞成長培地-2中で培養した(Tandle et al., 2005)。全ての実験を3〜5代継代のHUVECを用いて実施した。10%血清、2 mM グルタミン、100 u/ml ペニシリン、100 μg/ml ストレプトマイシン、100 μg/mlゲンタマイシン、およびファンギゾンを含むRPMI 1640培地中でM21ヒトメラノーマ細胞を成長させた。 10% 血清、2 mM グルタミン、100 u/ml ペニシリン、100 μg/ml ストレプトマイシン、100 μg/mlゲンタマイシン、およびファンギゾンを含むDMEM培地中でPmel細胞を成長させた。
【0152】
TNF-α/EMAP-IIを発現する標的化されたAAVPの構築
AAVP骨格の一般的な設計および構築は、Hajitouら(2006)に記載されている。TNF-αを発現するAAVPコンストラクトを2工程で創り出した。まず、pG1SiTNF由来の880 bpのNotI/HindIII断片を消化し、pAAV-eGFP/NotI/HindIIIベクターにライゲートしてGFP遺伝子配列を取り替えた(Hwu et al., 1993)。第二の工程において、fMCS/RGDMCSおよびAAV-TNFをPvuIIで消化した。その後、逆向き末端反復(ITR)を持つAAV-TNF-αをfMCS-/RGDMCS PvuII部位に再びライゲートし、AAVPベクターを得た。したがって、pfdTNF-αが標的化されていないベクターである一方、pRGDTNFは細胞表面αvインテグリン受容体に対する結合親和性のある標的化されたベクターである。
【0153】
EMAP-IIを発現するAAVPコンストラクトを3工程で創り出した。成熟EMAP-IIを発現する、pET-20bプラスミドは、The Scripps Research Institute, La Jolla, CAのPaul Schimmelから快く寄贈された。第一の工程において、PCR産物中に制限酵素部位および分泌可能シグナル配列を組み入れるための3つのプライマーを用いて、pET-20bEMAP-IIからEMAP-II配列を増幅させた。プライマー1は、5'末端にNotI制限酵素部位、それに続いて遺伝子産物の細胞外分泌のための(pSecTag2ベクター, Invitrogen, Carlsbad, Californiaから設計された)シグナル配列およびEMAP-II配列がある132 bpのフォワードプライマーである。プライマー2は、PCR産物の増幅を容易にするためのプライマー1のより短いバージョンである。プライマー3は、3'末端にHindIII制限酵素部位があるリバースプライマーである。PCR増幅により、NotIおよびHindIII酵素部位ならびにシグナル配列がある667 bpの産物が作製された。第二の工程において、EMAP-II PCR産物をpCRII-TOPOクローニングベクター(Invitrogen, Carlsbad, California)にクローニングした。結果として得られたクローンを配列決定し、その後667 bpのNotI/HindIII断片を先に説明されたようなpAAV-eGFP/NotI/HindIIIベクターにライゲートした。第三の工程において、fMCS/RGDMCSおよびAAV-EMAP-IIをPvuIIで消化しならびにライゲートしてAAVPベクターを得た。したがって、pfdEMAP-IIが標的化されていないベクターである一方、pRGDEMAP-IIは細胞表面αvインテグリン受容体に対する結合親和性のある標的化されたベクターである。プライマー1:
;プライマー2:
;プライマー3:
【0154】
AAVP粒子精製
標的化されていないおよび標的化されたAAVP粒子を得るために、DNAをエレクトロポレートしてMC1061大腸菌に入れた。細胞およびウイルス粒子を培養上清から精製した。大規模なAAVP粒子を許容性のk91Kan細胞から精製した。細菌形質導入単位(TU)の数を決定するために、k91細胞をファージ粒子の連続希釈に感染させおよびテトラサイクリンおよびカナマイシンを含むルリア-ベルターニ寒天プレート上にプレーティングした。その後、細菌コロニーの数を計数することによってTUを決定した。
【0155】
インビトロファージ内在化アッセイ
M21細胞を6ウェル組織培養プレート中で終夜成長させた。ベクター内在化を検討するために、細胞を培地で洗浄しその後37℃で3時間ウイルス粒子を感染させた。インキュベーション後、ウイルス内在化を停止するためにプレートを氷上に5分間置いた。ハンクスの平衡塩溶液(HBSS)中での細胞の徹底的洗浄によって結合していない粒子を除去した。1時間氷上でのサブチリシン(カルシウムおよびマグネシウムを含まないHBSS中の3 mg/ml サブチリシン、20 mM Tris pH 7.5、2 mM EDTA pH 8.0)による処置によって、細胞外ウイルス粒子を不活化した(Ivanenkov et al., 1999)。その後、細胞を穏やかなピペッティングで剥がし、サブチリンを2 mM EDTAを用いて氷上15分で不活化した。溶解緩衝剤(10 mM Tris pH 7.5、2 mM EDTA pH 8.0、および2%オルトバナジン酸ナトリウム)を用いることによって細胞を溶解させ、内在化したAAVP粒子を放出させた。その後、上で記載されたようなTUとして内在化したAAVP濃度を決定した。
【0156】
免疫蛍光(IF)アッセイ
IFを用いてM21における内在化したウイルス粒子を観察した。簡潔に述べると、8ウェル Lab-Tekチャンバーガラススライド(Nunc, Rochester, New York)上で成長した細胞を10%血清を含むDMEMを用いることによって37℃で16時間AAVP粒子に感染させた。感染後、細胞をPBSで洗浄し、チャンバーを除去し、細胞を3.7%ホルムアルデヒドで10分間固定した。細胞をPBS中の0.1%サポニン(Sigma, St. Louis, Missouri)で透過処理し、ならびにブロッキング緩衝剤(1% BSA, 0.025%アジ化ナトリウム、および0.1%サポニンを含むPBS)で15分間ブロッキングした。透過緩衝剤で洗浄した後、細胞をマウス抗バクテリオファージ抗体で1時間、その後FITCコンジュゲートされた抗マウスIgG抗体で1時間インキュベートした。ガスケットを剥がしならびに細胞をAntifade(MP Biomedicals, Solon, Ohio)を用いてマウントし、Zeiss Axiovert蛍光顕微鏡下で調べた。
【0157】
ファージによる遺伝子発現
内在化アッセイと同様に、M21細胞をAAVP粒子に感染させた。培地を48時間で取り替えた。4日目および12日目に、培養上清を収集し、ELISA(Invitrogen, Carlsbad, California)で分泌可能なサイトカインレベルを測定した。
【0158】
組織因子(TF)アッセイ
分泌されたTNF-α/EMAP-IIが機能的であるかどうかを調べるために、本発明者らは、内皮細胞ECでTF合成を誘導するその能力を調べた。簡潔に述べると、ウェル当たり2 x 105 HUVECを6ウェル組織培養プレート上にプレーティングした。翌日、細胞をM21培養上清で6時間血清を含まないRPMI培地中で処置した。細胞をPBSで洗浄し、25 mM Tris pH 7.5と10分間室温でインキュベートした。その後、培養プレートを-80℃で2時間インキュベートした。総細胞ライセートを組織因子アッセイ緩衝剤(20 mM Tris pH 7.5、150 mM NaCl、および0.1% BSA)中で調製した。ライセートを13,000 rpmで10分間の遠心分離できれいにした。Amelung KC 4A Micro Coagulation Analyzer(Sigma, St. Louis, Missouri)の中でCaCl2(Sigma, St. Louis, Missouri)の存在下で第VIII因子欠損血漿(Geroge King Biomedical Inc, Overland Park, Kansas)の凝集に必要とされる時間を測定することによって、100 μl ライセートを組織因子の存在について解析した。公知の組織因子濃度でプロットされた標準校正曲線を用いることによって、第VIII因子欠損血漿を凝集させるのに必要な時間を組織因子単位に変換した。
【0159】
免疫蛍光染色によるインビボAAVP送達および検出
NIH Animal Care and Use Committeeによって承認されたプロトコルに従って全ての動物実験を実施した。Jackson Laboratoriesからメスの無胸腺ヌードマウスを入手し、National Cancer Institutesの動物施設で収容した。ヒトメラノーマ細胞(4 x 106)をヌードマウスの右脇腹に皮下に播種した。腫瘍体積(mm3)を3次元で測定し、長さ x 幅 x 高さ x 0.52として算出した。腫瘍体積がおよそ100〜150 mm3に達した時、1 x 1011 AAVP粒子を(尾静脈を通して)静脈内注射した。様々な時間間隔で動物に麻酔をかけた。切除された腫瘍組織ならびに対照組織(腎臓および肝臓)をさらなる解析のために凍結した。
【0160】
AAVPの存在を検出するために、5 μM厚の凍結切片を二重IF染色を用いて染色した。簡潔に述べると、切片を4%パラホルムアルデヒド中で5分間固定し、その後PBS中で10分間2回洗浄した。透過処理を1% Triton-X-100を含むPBS中で10分間行なった。切片をImage-iT FXシグナル増強剤と30分間室温(RT)でインキュベートし、その後1% Triton-X-100を含むPBS中で3回洗浄した。非特異的結合を5%ヤギ血清を用いて30分間RTでブロッキングした。一次抗体:1:2000希釈の抗-fdバクテリオファージ抗体(Sigma, St. Louis, Missouri)および1:50希釈の抗マウスCD31(BD Biosciences, San Jose, California)を終夜4℃で適用した。切片を1% Triton-X-100を含むPBS中で10分間3回洗浄し、その後二次抗体(Invitrogen, Carlsbad, California):各1:400希釈のヤギ抗ウサギAlexa Fluor 594およびヤギ抗ラットAlexa Fluor 488と30分間暗中でインキュベートした。切片を1% Triton-X-100を含むPBS中で10分間3回洗浄し、もう一度PBSで洗浄し、その後DAPI(Vector Labs, Burlingame, California)入りのVectashieldマウンティング媒体中でマウントした。
【0161】
インビボでのTNF-α発現
TNF-αタンパク質発現を検出するために、総細胞ライセートをプロテアーゼ阻害剤カクテル(Roche, Branchburg, New Jersey)を含む溶解緩衝剤(50 mM Tris pH 7.4、140 mM NaCl、0.1% SDS、1% NP40、および0.5%デオキシコール酸ナトリウム)を用いて5 μM 凍結組織切片から調製した。ライセートを13,000 rpmで10分間の遠心分離によってきれいにした。BioRADからのタンパク質アッセイ試薬を用いてタンパク質の量を定量した。50 μgの総タンパク質と等しいライセートの量をヒトTNF-αについてELISA(Invitrogen, Carlsbad, California)でアッセイした。
【0162】
TNF-α発現の局在化を調べるために、5 μM 凍結組織を以下のように染色した。簡潔に述べると、切片を4%パラホルムアルデヒドを含むPBS中で20分間固定し、PBSで3回各5分間洗浄し、ならびに非特異的結合を5%ヤギ血清を用いて20分間ブロッキングした。切片を1:200希釈された抗fd抗体(Sigma, St. Louis, Missouri)かまたは1:100希釈されたTNF-α抗体(Novus Biologicals, Littleton, Colorado)のいずれか、またはラット抗マウスCD31 (BD Biosciences, San Diego, California)と1時間インキュベートし、その後洗浄緩衝剤(50 mM Tris pH 7.6および0.02% Tween-20を含むPBS)中で各々5分の3回の洗浄を行なった。内在性ペルオキシダーゼを3%過酸化水素水で5分間ブロッキングし、その後洗浄し、1:200希釈の二次抗マウス抗体-HRPまたはビオチン化ロバ抗ラット抗体と30分間インキュベートした。切片をジアミノベンジジン四塩酸塩基質(Dako, Carpinteria, California)で5分間現像し、ヘマトキシリンで30秒間対比染色し、水道水で濯ぎ、脱水し、きれいにし、ならびにマウントした。
【0163】
アポトーシスアッセイ
製造元の勧める行為に従って、インサイチューアポトーシス検出キット、TACS TdT(R&D Systems, Minneapolis, Minnesota)を用いて、アポトーシスを検出した。
【0164】
腫瘍成長解析
ヒトメラノーマ細胞(3 x 106)をヌードマウスの右脇腹に皮下に播種した。腫瘍体積がおよそ100 mm3に達した時、尾静脈を通して1 x 1011 AAVP粒子を投与した。AAVP粒子投与を7日後にもう一度繰り返した。3日毎に盲式で腫瘍体積を測定することによって腫瘍を有するマウスを追跡調査した。
【0165】
統計解析
分散の解析(ANOVA)およびテューキー比較事後検定(GraphPad Instat Software, Inc., San Diego, California)を用いることによって群を比較した。P値<0.05を統計的有意とみなした。
【0166】
B.結果
哺乳動物細胞によって内在化されるAAVP粒子
以前の検討により、ファージ粒子はインテグリンを介する受容体内在化によって内在化され得ることが示されている(Hajitou et al., 2006)。M21細胞はそれらの細胞表面にαvβ3受容体を発現する(データは示さない)。M21細胞がAAVP粒子を内在化することができるかどうかを調べるために、本発明者らは、細胞にAAVP粒子を感染させ、その後内在化されたファージを計数した。感染後、細胞外ウイルス全体をサブチリシン処置によって不活化し、細胞を溶解し、内在化されたファージをライセート中で回収した。内在化されたAAVP濃度をTUとして測定した(表6)。標的化されたヌル(RGD)または標的化されたTNF-α発現AAVP(RGDTNF-α)に感染した細胞とは対照的に、標的化されていないヌル(fd)かまたはTNF-αを発現する標的化されていないウイルス(fdTNF-α)のいずれかに感染したM21細胞による最低限のAAVP内在化があった。
【0167】
(表6)ファージ内在化アッセイ
【0168】
IFを用いて、M21細胞内部での内在化されたAAVP粒子の局在化を可視化した。標的化されていないTNF-α発現AAVP(fdTNF-α)に感染した細胞は、ファージ局在化を示さなかった(図12A、上のパネル)。対照的に、TNF-αを発現する標的化されたAAVP(RGDTNF-α)に感染した細胞は、M21細胞内部での増強された局在化を示した(図12A、下のパネル)。
【0169】
AAVPを介する遺伝子発現
標的化されたAAVPが、哺乳動物細胞に感染しかつ内部に局在化できることを明らかにした後で、本発明者らは、ウイルス感染が遺伝子産物の発現をもたらすことができるかどうかを調べた。M21細胞にTNF-αを発現するAAVPを、2通りで感染させ、およびTNF-α遺伝子産物の産生をELISAで測定した。遺伝子産物は分泌可能でありかつ培養上清中で検出することができた(図12B)。感染の5日後に検査された上清は、800 pg/mlのTNF-αレベルを示した。12日目に検査された遺伝子産物は、4日目よりも高かった。希釈剤対照(PBS)、標的化されていない空(fd)、TNF-αを発現する標的化されていないウイルス(fdTNF-α)、および標的化されたヌルAAVP(RGD)は、検出可能なレベルのTNF-α分泌を有さなかった(図12B)。
【0170】
AAVP感染M21細胞によって分泌されたTNF-αが機能的であるかどうかを調べるために;本発明者らは、ECで組織因子(TF)合成を誘導するその能力を検査した。分泌されたTNF-αは、ECでTF発現を誘導することができた(図12C)。組換えTNF-αを陽性対照として用いた。TF誘導は、培養上清を TNF-αモノクローナル抗体とインキュベートすることによって阻止することができた。感染後23日で解析された培養上清も機能的TNF-α分泌を示した(図12C)。したがって、AAVPによる単一感染により、感染後23日までの機能的遺伝子産物の産生が結果的にもたらされた。
【0171】
AAVPによるインビボ腫瘍標的化
本発明者らは、哺乳動物細胞のAAVP感染およびインビトロでのTNF-α遺伝子産物の機能的発現を観察した。AAVPのインビボ標的化を評価するために、腫瘍を有するヌードマウスに尾静脈を通してウイルスを全身注射した。
【0172】
希釈剤(PBS)かまたはRGDTNF-α AAVPのいずれかを注射されたマウスに注射後15分、1日、2日、3日、4日、8日、および10日で麻酔をかけた。腫瘍組織由来の凍結切片を二重IF染色によってウイルス粒子の存在について解析した。図13で示されるように、AAVP粒子は赤く染まり(Alexa Flour 594)、血管は緑色に染まり(Alexa Flour 488)、およびDAPIは核染色を示している。PBSを注射された動物はどれもいかなる時点でもAAVPの存在を示さなかった。PBSを15分間注射された動物由来の代表的な腫瘍切片を示す(図13A)。RGDTNF -αを発現するAAVPを注射された動物は、血管におけるウイルス粒子の共局在化を示した(図13B〜H)。ウイルス粒子の最大の蓄積は15分後に注射された動物で検出された(図13B)。AAVPの存在は、検査された全ての時点、1日目(図13C)、2日目(図13D)、3日目(図13E)、4日目(図13F)、8日目(図13G)、および10日目(図13H)で検出された。しかしながら、本発明者らは、時間の経過に伴う検出可能なウイルス粒子の漸進的な減少に注目した。
【0173】
インビボで正常組織を標的化しないAAVP
インビボでの腫瘍血管へのAAVP標的化の特異性を調べるために、本発明者らは、PBSかまたは異なる時点でRGDTNF-αを発現するAAVPのいずれかを注射したヌードマウス 由来の2つの対照組織を調べた(図14および図15)。先に記載されたような二重IF染色を用いてウイルスの存在を検出した。
【0174】
図14で示されたように、本発明者らは、1日目に肝臓組織でウイルス粒子の若干の染色を観察した(図14A)。しかしながら、ウイルス染色は2日目までに最低限のレベルまで低下し(図14B)、3日目(図14C)、8日目(図14D)、および10日目(図14E)にはウイルス染色は観察されなかった。図15は、ウイルス粒子の存在について染色された腎臓切片を示す。AAVP粒子は、検査された時点、1日目(図15A)、2日目(図15B)、3日目(図15C)、8日目(図15D)、および10日目(図15E)のいずれにおいても腎臓で検出されなかった。それにもかかわらず、異なる時点での腎臓組織は全て、良好な血管染色を示した。
【0175】
AAVPによる機能的遺伝子産物のインビボ発現
腫瘍血管系に標的化されたAAVP粒子が遺伝子産物を発現することができるかどうかを調べるために、本発明者らは、2通りで、3日目、4日目、8日目、および10日目の腫瘍組織をTNF-αタンパク質レベルについてELISAで解析した(図16)。本発明者らは、検査した組織全てにおいて基礎レベルの内在性TNF-α発現を観察した。PBSを注射された動物は、任意の検査された時点で内在性レベルを超えるいかなる増加も示さなかった。RGDTNF-α AAVPを注射されたマウスは、4日目から始まりかつ10日目まで徐々に増加するTNF-α発現を示した(図16)。
【0176】
遺伝子産物が産生されている細胞タイプを決定するために、本発明者らは、TNF-αに特異的な抗体を用いて組織切片を染色した。本発明者らは、血管周辺のTNF-α染色の存在を観察した(図17A)。TNF-α発現の効果を見るために、本発明者らは、アポトーシスについて、腫瘍切片を染色した。TACSブルー標識を用いてDNA断片を検出した。アポトーシス細胞は青く染まっている(図17B、左のパネル)。アポトーシス細胞をネクローシス細胞と区別するために、試料を核ファストレッドで対比染色し、アポトーシスの形態学的検証を手助けした。本発明者らは、血管およびアポトーシスを経ている周囲の腫瘍細胞を観察した。右のパネルは、CD31特異的抗体で染色された血管を示す(図17B、右のパネル)。
【0177】
腫瘍成長解析
本発明者らは、2つの異なる腫瘍モデル、TNF-α感受性(M21)およびTNF-α耐性(Pmel)において、ヌードマウス中で成長した腫瘍異種移植片に対するAAVPの効果を解析し、TNF-αを発現するAAVPの処置有効性を調べた。TNF-αに感受性である、ヒトメラノーマM21腫瘍をヌードマウス中で皮下に成長させた。腫瘍進展後、マウスを様々なAAVPコンストラクトまたはPBSで尾静脈注射を通じて全身的に処置した。動物を27日間追跡調査した。TNF-αを発現する標的化されたAAVP(RGDTNF-α)を用いたM21腫瘍の処置は、特徴的な中心腫瘍ネクローシスおよび腫瘍縮小を示した。異なるコホートについて最後に測定された時点である、27日目に、PBS処置群は743±383(±SD)mm3の平均腫瘍体積を有し、標的化されていないfdTNF群は613±155(±SD)mm3の平均腫瘍体積を有し、ヌルの標的化されたRGDファージは、622±141(±SD)mm3の平均腫瘍体積を有し、および標的化されたTNFα発現群は358±98(±SD)mm3の平均腫瘍体積を有した(p<0.048)(図18A)。RGDTNF-α群における腫瘍体積の低下は、20日目から統計的に有意であった。
【0178】
TNF-α耐性腫瘍モデルでは、TNF-αを投与する前にEMAP-IIを発現するAAVPを用いてそれらを前処置することによって、ヒトメラノーマPmel腫瘍をTNF-α効果に対して増感させた。以前の検討において、本発明者らは、EMAP-IIのウイルスベクター送達が、全身送達されたTNF-αの効果に対して耐性腫瘍を増感させることができることを示した。ヌードマウス中で皮下に成長した腫瘍をEMAP-IIを発現するAAVPを用いて全身的に処置し、その後組換えTNF-α(rTNF-α)を用いた全身処置を行なった。処置後2週間、腫瘍を追跡調査した。図18Bで示されたように、EMAP-IIを発現する標的化されていないAAVP(fdEMAP)で処置されたマウスは、PBS単独で処置されたマウスと比較して同様の腫瘍成長を示した。rTNF-αかまたはEMAP-IIを発現する標的化されたAAVP(RGDEMAP-II)単独のいずれかで処置されたマウスは非常にわずかの効果を示し、かつPBSまたはfdEMAP-II群と有意には異ならなかった。しかしながら、RGD-EMAP-IIウイルスおよびrTNF-αの組み合わせで処置されたマウスは、腫瘍体積の有意な低下を示した(p = 0.007)。
【0179】
実施例3
トランスフェリン/トランスフェリン受容体系を標的化するヒトトランスフェリンペプチド
培養中のヒト神経膠腫細胞のCRTIGPSVC(SEQ ID NO:1)AAVP-Lucによる形質導入。U87ヒト由来神経膠腫細胞を40,000細胞/ウェルの濃度で24ウェルプレート上に播きおよび終夜37℃で培養した。次の日、Nature Methodのプロトコルに従って、細胞をAAVPとインキュベートした。RGD-4C AAVP GFPおよびRGD-4C AAVP Lucを形質導入効率についての陽性対照として用いた。画像は7日目に撮った。ファージ摂取は低いが、細胞を鉄AAVPヒドロゲル中で培養した場合に劇的に増加する(プレートおよびグラフの最後の行)(図19A〜19B)。本発明者らは、U87ヒト由来神経膠芽腫細胞を持つ動物への全身投与後に、CRTIGPSVC(SEQ ID NO:1)AAVP-Lucの特異性および有効性を評価した。ファージ注射の7日後にルシフェラーゼ活性を測定した。
【0180】
参照
以下の参照は、それらが本明細書において示された詳細を補足する例示的で手続き的なまたはその他の詳細を提供する程度まで、参照により本明細書に具体的に組み入れられる。
【図面の簡単な説明】
【0181】
以下の図面は、本明細書の一部を形成しかつ本発明のある種の局面をさらに示すために含まれる。本発明は、本明細書において提示された具体的な態様の詳細な説明と組み合わせたこれらの図面の1つまたは複数への参照によってより良く理解され得る。本開示の幾つかの具体的な実施例態様は、以下の説明および付随する図面を一部参照することによって理解され得る。
【0182】
【図1】図1Aは、標的化されていないAAVPまたはRGE-4Cもしくは様々なスクランブルバージョンのRGD-4C配列などの陰性対照ペプチドを提示するAAVPと対照的に、αvインテグリンを発現する哺乳動物細胞へのRGD-4C AAVPの結合を示すグラフである。図1Bは、レポーター遺伝子を運びリガンド指向性の内在化を示すRGD-4C AAVPを示す画像である。図1Cは、RGD-4C AAVP-β-ガラクトシダーゼによって媒介されたKS1767細胞への標的化された遺伝子転移を示す。図1Dは、合成RGD-4Cペプチドによるが、関連のない対照ペプチドにはよらない形質導入の阻害を示し;非特異的な形質導入レベルを標的化されていないAAVPを用いることによって決定した。抗β-gal抗体を染色に用いおよび遺伝子発現を免疫蛍光によって検出した。図1Eは、RGD-4C AAVPを感染させた細胞からの組換えAAVのレスキューを示す。ヒト293細胞を標的化されたRGD-4C AAVP-GFP(106形質導入単位/細胞)または陰性対照(標的化された非キメラRGD-4Cファージ-GFP、標的化されていないAAVP-GFP)とインキュベートした。感染4日後、細胞にAAV rep-およびcap-発現プラスミドをトランスフェクトしならびに野生型アデノウイルス5型(Ad)を重感染させた。アデノウイルス感染72時間後に細胞を採取し、その後上清を用いて新しい293細胞に感染させた。48時間後フローサイトメトリーによってGFP発現を解析した。各コンストラクトからのレスキュー後にバックグラウンドを超えて産生された組換えAAV-GFPの平均増加を示す。
【図2】図2Aは、AAVP-GFPneo(n=9)またはファージ-GFPneo(n=9)のいずれかを安定に形質導入された293クローンのフローサイトメトリー解析の概要を示すヒストグラムである。白抜きの三角は緑色蛍光タンパク質(GFP)陽性細胞のパーセンテージを示し、黒い棒はGFP発現レベル(平均蛍光強度;MFI)を表す。図2Bは、RGD-4C AAVP-GFPneoまたはRGD-4Cファージ-GFPneoを形質導入されたクローン性の細胞株におけるトランスジーンカセットの持続のサザンブロット解析を示す。形質導入されていない293親細胞または形質導入された細胞クローンの各々(各群について#1〜9)由来の総細胞DNAをAflII-XhoIで二重消化した(トランスジーンカセットの両隣にあるコンストラクトDNA内部に各酵素当たり1つの制限消化部位)。図2Cは、サザンブロットによるトランスジーンカセットの潜在的な頭尾コンカテマーの解析を示す。サザンブロッティングの前に総細胞DNAをXhoIで消化した(3'ITRの隣のトランスジーンカセット内部に単一の制限消化部位)。
【図3】図3Aは、KS1767由来腫瘍異種移植片を持つ深く麻酔されたヌードマウスへのRGD-4C AAVP(5 x 1010 TU)または陰性対照(標的化されていないAAVP、スクランブルのRGD-4C AAVP、もしくはRGE-4C AAVP)の(尾静脈を通じた静脈内への)全身投与後のKS1767由来異種移植片におけるファージに対する免疫組織化学染色を示す。AAVPコンストラクトを5分間循環させ、それに続いて灌流および腫瘍の外科手術除去を行なった。ファージに対するポリクローナル抗体をパラフィン包埋腫瘍切片に対する染色に用いた。矢印は、腫瘍血管におけるファージ染色を示す。図3Bは、RGD 4C AAVP-GFPまたは示されたような陰性対照(標的化されていない、スクランブルの、もしくは突然変異体)のいずれかの全身投与7日後のKS1767由来異種移植片におけるGFP発現の免疫蛍光解析を示す。
【図4】図4Aは、標的化されたAAVPの全身送達後のホタルルシフェラーゼ発現のインビボ生体発光イメージングを示す。DU145由来腫瘍異種移植片を持つヌードマウスは、全身単一用量のRGD-4C AAVP-Luc(5 x 1011 TU、静脈内)または対照(標的化されていないAAVP-Luc、もしくはスクランブルのRGD-4C AAVP-Luc)のいずれかを受けた。10日後、腫瘍を有するマウスの生物発光イメージング(BLI)を行ない、トランスジーン発現を検討評価した。校正目盛をパネル中に提供する。図4Bは、標的化されたRGD-4C AAVP-HSVtkの全身送達後の腫瘍を有するマウスにおけるマルチトレーサーPETイメージングを示す。DU145由来腫瘍異種移植片を持つヌードマウス(各コホート当たりn=9の腫瘍を有するマウス)は、全身単一用量(5 x 1011 TU、静脈内)のRGD-4C AAVP-HSVtkまたは標的化されていないAAVP-HSVtkを受けた。GCV処置の前および後に得られた[18F]-FDGおよび[18F]-FEAUを用いたPET画像を示す。T、腫瘍;H、心臓;BR、脳;BL、膀胱。校正目盛をパネル内に提供する。[18F]-FDGおよび[18F]-FEAU生体分布の解釈を簡単にするために、代表的な腫瘍を有するマウスのPET画像および写真画像の重ね合わせ面付け(overimposition)を行なった。図4Cは、AAVP投与後の個々の腫瘍異種移植片の成長曲線を示す。図4Dは、AAVP投与後の異なる日における[18F]-FEAUを用いた繰り返しのPETイメージングによって検討評価された場合のHSVtk遺伝子発現の時間的力学を示す。図4Eは、[18F]-FDG PETを用いて検討評価された場合のGCV治療の前および後の腫瘍生存能力の変化を示す。
【図5】図5Aは、時間に対するプロットされた平均腫瘍体積±標準偏差(SD)を示すグラフである。KS1767細胞に由来する(およそ50 mm2でサイズが一致した)樹立されたヒト異種移植片を持つ免疫不全ヌードマウスのコホートを用いた。マウスは、RGD-4C AAVP-HSVtkかまたは対照(標的化されていないAAVP-HSVtk、RGD-4C AAVP-GFP、もしくはビヒクル単独)のいずれかの単一全身投与(5 x 1010 TU、静脈内)を受けた。処置後2日目から実験の終わりまで、ガンシクロビル(GCV)をマウスに投与した。RGD-4C AAVP-HSVtkで処置されたが後にGCVで処置されていない追加の対照群の他は、全てのマウスがGCVを受けた。図5Bは、時間に対するプロットされた平均腫瘍体積±標準偏差(SD)を示すグラフである。膀胱UC3癌腫細胞に由来する(およそ50 mm2でサイズが一致した)樹立されたヒト異種移植片を持つ免疫不全ヌードマウスのコホート。マウスは、RGD-4C AAVP-HSVtkかまたは対照(標的化されていないAAVP-HSVtk、RGD-4C AAVP-GFP、もしくはビヒクル単独)のいずれかの単一全身投与(5 x 1010 TU、静脈内)を受けた。処置後2日目から実験の終わりまで、ガンシクロビル(GCV)をマウスに投与した。RGD-4C AAVP-HSVtkで処置されたが後にGCVで処置されていない追加の対照群の他は、全てのマウスがGCVを受けた。図5Cは、時間に対するプロットされた平均腫瘍体積±標準偏差(SD)を示すグラフである。前立腺DU145癌腫細胞に由来する(およそ50 mm2でサイズが一致した)樹立されたヒト異種移植片を持つ免疫不全ヌードマウスのコホート。マウスは、RGD-4C AAVP-HSVtkかまたは対照(標的化されていないAAVP-HSVtk、RGD-4C AAVP-GFP、もしくはビヒクル単独)のいずれかの単一全身投与(5 x 1010 TU、静脈内)を受けた。処置後2日目から実験の終わりまで、ガンシクロビル(GCV)をマウスに投与した。RGD-4C AAVP-HSVtkで処置されたが後にGCVで処置されていない追加の対照群の他は、全てのマウスがGCVを受けた。示されているのは、時間に対するプロットされた平均腫瘍体積±標準偏差(SD)である。図5Dは、単一全身用量(5 x 1010 TU、静脈内)のRGD-4C AAVP-HSVtkによる(およそ150 mm2の)大きいDU145由来異種移植片の成長阻害を示すグラフである。図5Eは、単一静脈内用量(5 x 1010 TU)のRGD-4C AAVP-HSVtkによる免疫適格BALB/cマウスにおける(およそ50 mm2でサイズが一致した)EF43-FGF4マウス乳癌腫の腫瘍成長の阻害を示すグラフである。図5Fは、同種同系のEF43-FGF4腫瘍を持つ免疫適格BALB/cマウスへの繰り返された用量のAAVP(各々5 x 1010 TU、静脈内)によるRGD-4C AAVP-HSVtkおよびGCVの長期にわたる有効性を示すグラフである。腫瘍を有するマウスコホート(処置群当たりn=10マウス)を用いた独立の実験で治療結果を一貫して観察した。矢印は、AAVP投与の時間を示す。図5Gは、ファージに対する液性免疫応答のRGD-4C AAVP-HSVtkを用いた治療に対する効果を示すグラフである。BALB/cマウス免疫適格マウス(群当たりn=7マウス)はまず、3全身用量のRGD-4C AAVP-GFP(週当たり1010 TUで3週間、静脈内)を受けることにより、「ワクチン接種」された。その後、マウスにEF43-FGF4細胞を埋め込んだ。腫瘍が樹立された(およそ50 mm2でサイズが一致した)場合、腫瘍を有するマウスは、1全身単一用量のRGD-4C AAVP-HSVtkまたは標的化されていないAAVP-HSVtk、それに続いて(AAVPの投与後2日目に始められた)GCV維持を受けた。ELISAで高力価(およそ1:10,000まで)の循環抗ファージIgGの存在を確認するために、ワクチン接種を開始する前および後にならびにAAVP投与前のワクチン接種計画の最後に再度、マウスから血清試料を収集した。ワクチン接種は、抗ファージ抗体の存在にもかかわらず、抗腫瘍効果に影響を及ぼすようには見えなかった。
【図6】図6Aは、治療の全ての実験群由来の腫瘍を有するマウス(上のパネル)および対応する外科手術で除去された腫瘍(下のパネル)の画像である(BALB/c免疫適格マウスにおけるEF43-FGF4乳腫瘍)。図6Bは、EF43-FGF4で処置された腫瘍の組織病理学的解析である。EF43-FGF4腫瘍を回収し、切片を作製し、および染色した。標的化されていないAAVP-HSVtkで処置された腫瘍(左のパネル)、外部縁と中心の腫瘍部の間の境界(真ん中のパネル)、およびRGD-4C AAVP-HSVtkで処置された腫瘍の中心の腫瘍部(右のパネル)を連続腫瘍切片の低倍率挿入図からの高倍率図として示す。腫瘍切片のヘマトキシリンおよびエオシン(H&E)染色、抗CD31免疫染色、およびTUNEL染色を示す。矢印は、腫瘍血管およびアポトーシス細胞を示す。
【図7】RGD-4C AAVPの構築の組み立てを示す図解であり、クローニング戦略および結果として得られるベクターの構造が描かれている。
【図8】図8Aは、293形質導入クローン性細胞株中のRGD-4C AAVP-GFPneoまたはRGD-4Cファージ-GFPneo由来のDNAのサザンブロット解析を示す。形質導入されていない293親細胞または形質導入された個々の安定細胞クローン(各ベクターについて#1〜9)由来の総細胞DNAをStuI(ベクターDNA内に制限消化部位なし)、Xba-I(ベクターDNA内に単一の制限消化部位)、またはSacI-MluIとインキュベートした。結果として得られるDNA断片を示されたように0.8%アガロースゲル上で分離し、ナイロン膜に移し、および標識されたneoプローブとハイブリダイズさせた。消化されたベクタープラスミドも対照として用いた。図8Bは、RGD-4Cファージ-GFPneoまたはRGD-4C AAVPGFPneoを安定に形質導入した293クローン性細胞株中のトランスジーンカセットのコンカテマーのPCR解析を示す。形質導入されていない293親細胞は陰性対照としての役割を果たした;示されたように追加の陰性対照および100-bp分子マーカー(Invitrogen)も示す。RGD-4C AAVPおよびRGD-4Cファージに対応するDNAにおけるコンカテマー形態を同定するために、トランスジーンカセットの5'および3'末端近くにアニールするプライマーを用いたネスティッドPCRを行なった。矢印はプライマーを示す(H、頭;T、尾)。PCR増幅産物は2%アガロースゲルで検出され、もっぱらAAVPを形質導入された細胞クローンにおけるコンカテマーのベクターDNAを明らかにした。図8Cは、AAVP DNAにおける異なるコンカテマー形態のシークエンシング結果を示し(大文字はトランスジーンカセットの3'末端を意味し、イタリック文字はトランスジーンカセットの5'末端を意味する)、欠失したITRを持つ頭-尾コンカテマーを明らかにした。
【図9】RGD-4C AAVP-GFPかまたは標的化されていないAAVP-GFPのいずれかのマウスへの静脈内用量(5 x 1010 TU)7日後の対照器官である脳、肝臓、膵臓、および腎臓におけるレポーター遺伝子発現GFPの免疫染色を示す画像である。
【図10】図10Aは、EF43-FGF4腫瘍を持つ免疫適格BALB/cマウスへのRGD-4C AAVP(右のパネル)または標的化されていないAAVP(真ん中のパネル)の静脈内投与後のAAVPに対する免疫組織化学的染色の画像である。コンストラクトを5分間循環させ、それに続いて記載されたように灌流および組織回収を行なった。ファージに対するポリクローナル抗体を染色に用いた。左のパネルは、抗CD31抗体を用いることによる腫瘍血管染色を示す。矢印は腫瘍血管を示す。図10Bは、EF43-FGF4腫瘍を持つマウスへの標的化されていないAAVP-GFP(左のパネル)またはRGD-4C AAVP-GFP(真ん中のパネル)の静脈内投与1週間後のEF43-FGF4腫瘍におけるGFP発現の免疫蛍光解析の画像である。EF43-FGF4腫瘍におけるαv-インテグリンに対する免疫染色も示す(右のパネル)。
【図11】標的化されていないAAVP-HSVtk、RGD-4C AAVP-HSVtkベクター、またはビヒクル単独に加えてGCV維持で処置されたマウス由来の対照器官である肝臓、心臓、および腎臓の組織学的解析の画像が示されていることを示す。毒性の組織学的兆候は、これらの器官でH&E染色によって検出されなかった。
【図12】RGD標的化されたAAVPを感染させた哺乳動物細胞は機能的な遺伝子産物を発現する。(図12A)ヒトメラノーマ細胞、M21にAAVP;標的化されていないTNF-α発現ファージfdTNF-α(上のパネル)および標的化されたTNF-α発現ウイルスRGDTNF-α(下のパネル)を感染させならびにfd特異的一次抗体、次いでFITC標識二次抗体を用いて検出した。(図12B)PBS、空の標的化されていないウイルス(fd)、空の標的化されたウイルス(RGD)、標的化されていないTNF-α発現ウイルス(fdTNF-α)、および標的化されたTNF-α発現ウイルスRGDTNF-αを感染させたM21細胞。感染5日後、培養上清を解析し、ELISAでTNF-αを測定した。感染12日後の培養上清も示す。(図12C)ヒト臍帯静脈内皮細胞(HUVEC)を様々な群;PBS、fd(標的化されていないヌルウイルス)、RGD(標的化されたヌルウイルス)、fdTNF(標的化されていないTNF-α発現ウイルス)、およびRGDTNF(標的化されたTNF-α発現ウイルス)で感染させたM21細胞由来の5日目の上清で処置しならびに組織因子(TF)産生について解析した。組換えTNF-αを陽性対照として用いた。特異性を調べるために、HUVECに適用する前に、RGDTNF 感染細胞由来のM21上清をTNF-α特異的抗体とインキュベートした。感染後23日目の上清もTF分泌について検査した。
【図13】AAVPは腫瘍血管系に特異的に標的化される。バクテリオファージ特異的抗体およびCD31血管抗体で染色されたPBSかまたはRGDTNF-αのいずれかを注射されたヒトメラノーマ異種移植片。Alexa Fluor 488、Alexa Fluor 594、およびDAPIを用いて検出を行ない、それぞれ血管、AAVP、および細胞核を可視化した(250X)。PBSを注射された動物はどれも、いずれの時点でもAAVPの存在を示さなかった。PBSを15分間注射された動物由来の代表的な腫瘍切片を示す(図13A)。RGDTNF-αを発現するAAVPを注射された動物は、早ければ15分(図13B)ならびにその後の時点:1日目(図13C)、2日目(図13D)、3日目(図13E)、4日目(図13F)、8日目(図13G)、および10日目(図13H)での血管におけるウイルス粒子の共局在化を示した。
【図14】AAVP粒子は正常肝臓組織では検出されなかった。バクテリオファージ特異的抗体およびCD31血管抗体で染色されたRGDTNF-αを注射されたヒトメラノーマ異種移植片を持つ動物由来の肝臓。Alexa Fluor 488、Alexa Fluor 594、およびDAPIを用いて検出を行ない、それぞれ血管、AAVP、および細胞核を可視化した(250X)。RGDTNF-αを発現するAAVPを注射された動物は、肝臓組織における1日目(図14A)でのウイルス粒子の若干の染色を示した。しかしながら、ウイルス染色は、2日目(図14B)までに最低限のレベルまで低下し、3日目(図14C)、8日目(図14D)、および10日目(図14E)時点でウイルス染色は観察されなかった。異なる時点での肝臓組織は全て、良好な血管染色を示した。
【図15】AAVP粒子は正常腎臓組織では検出されなかった。RGDTNF-αを注射されたヒトメラノーマ異種移植片を持つ動物由来の腎臓をバクテリオファージ特異的抗体およびCD31血管抗体で染色した。Alexa Fluor 488、Alexa Fluor 594、およびDAPIを用いて検出を行ない、それぞれ血管、AAVP、および細胞核を可視化した(250X)。本発明者らは、検査した時点、1日目(図15A)、2日目(図15B)、3日目(図15C)、8日目(図15D)、および10日目(図15E)のいずれにおいても腎臓におけるAAVPの存在を観察しなかった。それにもかかわらず、異なる時点での腎臓組織は全て、良好な血管染色を示した。
【図16】インビボにおいて、AAVPは特異的な遺伝子産物を発現する。PBSかまたはRGDTNF-αのいずれかを注射されたヒトメラノーマ異種移植片を持つ動物由来の凍結切片を用いて総タンパク質を抽出した。50 μgの総タンパク質を用いて、ELISAにより2通りでTNF-αタンパク質レベルを測定した。内在性TNF-α発現の基礎レベルを検査した全ての組織で観察した。PBSを注射された動物は、検査した任意の時点で内在性レベルを超えるいかなる増加も示さなかった。RGDTNF-α AAVPを注射されたマウスは、4日目に始まりおよび10日目まで徐々に増加するTNF-α発現を示した。
【図17】AAVPは血管系でTNF-αを発現しかつインビボでアポトーシスを誘導する。(図17A)免疫組織化学的解析を用いてTNF-α特異的抗体で染色されたRGDTNF-α AAVPを注射されたヒトメラノーマ異種移植片を持つ動物由来の凍結切片。TNF-α染色は血管の周辺に茶色の染色として見られる(左のパネル、100X;右のパネル、400X)。(図17B)アポトーシス細胞を検出するために染色されたRGDTNF-α AAVPを注射されたヒトメラノーマ異種移植片を持つ動物由来の凍結切片。血管および周囲の腫瘍細胞は、TACSブルー標識で染色された青く色付いた細胞に見られるアポトーシスを示した(左のパネル、200X)。試料は核ファストレッドで対比染色されている。右のパネルは、CD31特異的抗体で染色された血管を示す(200X)。
【図18】AAVPを用いたTNF-α感受性ヒトメラノーマ、M21(図18A)およびTNF-α耐性ヒトメラノーマPmel(図18B)の処置。皮下に埋め込まれたM21/Pmel腫瘍を持つヌードマウスをAAVPで尾静脈注射を通じて全身的に処置し、腫瘍体積を異なる時点で測定した。処置後の異なる日に対してプロットされた腫瘍体積。(図18A)RGDTNF-αで処置された動物は、20日目から統計的に有意な(p<0.05)腫瘍体積の低下を示した。(図18B)AAVPによるEMAP-IIの送達、それに続く組換えTNF-αを用いた処置によって、TNF-α耐性ヒトメラノーマをTNF-α治療に対して感受性にした。rTNF-αかまたはEMAP-IIを発現する標的化されたAAVP(RGDEMAP-II)単独のいずれかで処置されたマウスは、非常にわずかの効果を示し、PBSまたはfdEMAP-II群と有意には異ならなかった。RGD-EMAP-IIウイルスおよびrTNF-αの組み合わせで処置されたマウスは腫瘍体積の有意な低下を示した(p=0.007)。
【図19】図19Aおよび19Bは、培養中のヒト神経膠腫細胞のCRTIGPSVC(SEQ ID NO:1)AAVP-Lucによる形質導入を示す。U87ヒト由来神経膠腫細胞を40,000細胞/ウェルの濃度で24ウェルプレート上に播きおよび終夜37℃で培養した。次の日、 Nature Methodのプロトコルに従って、細胞をAAVPとインキュベートした。RGD-4C AAVP GFPおよびRGD-4C AAVP Lucを形質導入効率についての陽性対照として用いた。画像は7日目に撮った。ファージの取り込みは低いが、細胞を鉄AAVPヒドロゲル中で培養した場合に劇的に増加する(プレートおよびグラフの最後の行)。
【図1A】
【図1B】
【図1C】
【図1D】
【図1E】
【図2A】
【図2B】
【図2C】
【特許請求の範囲】
【請求項1】
以下の工程を含む、対象を処置する方法:
(a)病理学的状態もしくは疾患状態を有する、有することが疑われる、または発症するリスクがある対象に、レポーターをコードする治療的AAVPを投与する工程;および
(b)コードされたレポーターまたはレポーター活性を検出することによって、処置のために標的化された組織または細胞内での治療的AAVPのインサイチュー発現を評価する工程。
【請求項2】
標的器官、組織、または細胞内でAAVP核酸によって発現される治療的に十分なレベルの治療的遺伝子の発現に基づいて、対象に癌処置を施す工程をさらに含む、請求項1記載の方法。
【請求項3】
第一の治療的AAVPの発現が治療的に有効なレベルで発現されない場合に、第二の治療的AAVPを投与する、請求項2記載の方法。
【請求項4】
AAVP発現の評価が、レポーターまたはレポーターの活性の非侵襲的検出によるものである、請求項1記載の方法。
【請求項5】
レポーターが治療的タンパク質である、請求項1記載の方法。
【請求項6】
治療的タンパク質がプロドラッグ変換酵素である、請求項5記載の方法。
【請求項7】
レポーターが酵素である、請求項4記載の方法。
【請求項8】
酵素がキナーゼである、請求項7記載の方法。
【請求項9】
キナーゼがチミジンキナーゼである、請求項8記載の方法。
【請求項10】
キナーゼが検出可能に標識された化合物を修飾する、請求項8記載の方法。
【請求項11】
検出可能な標識が、蛍光、化学発光、表面増強ラマン分光法(SERS)、磁気共鳴イメージング(MRI)、コンピュータ断層撮影(CT)、または陽電子放出断層撮影(PET)イメージングによって検出可能である、請求項10記載の方法。
【請求項12】
検出可能に標識された化合物が、ヌクレオシド類似体である、請求項10記載の方法。
【請求項13】
検出可能に標識された化合物が、フルオロデオキシグルコース(FDG);2'-フルオロ-2'デオキシ-1ベータ-D-アラビオノフラノシル-5-エチル-ウラシル(FEAU);5-[123I]-2'-フルオロ-5-ヨード-1β-D-アラビノフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨード-1β-D-アラビノフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨード-1β-D-アラビノフラノシル-ウラシル、5-[18F]-2'-フルオロ-5-フルオロ-1-β-D-アラビノフラノシル-ウラシル;2-[11I]-および5-([11C]-メチル)-2'-フルオロ-5-メチル-1-β-D-アラビノフラノシル-ウラシル;2-[11C]-2'-フルオロ-5-エチル-1-β-D-アラビノフラノシル-ウラシル;5-([11C]-エチル)-2'-フルオロ-5-エチル-1-β-D-アラビノフラノシル-ウラシル;5-(2-[18F]-エチル)-2'-フルオロ-5-(2-フルオロ-エチル)-1-β-D-アラビノフラノシル-ウラシル、5-[123I]-2'-フルオロ-5-ヨードビニル-1-β-D-アラビノフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨードビニル-1-β-D-アラビノフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨードビニル-1-β-D-アラビノフラノシル-ウラシル;5-[123I]-2'-フルオロ-5-ヨード-1-β-D-リボフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨード-1-β-D-リボフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨード-1-β-D-リボフラノシル-ウラシル;5-[123I]-2'-フルオロ-5-ヨードビニル-1-β-D-リボフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨードビニル-1-β-D-リボフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨードビニル-1-β-D-リボフラノシル-ウラシル;または9-4-[18F]フルオロ-3-(ヒドロキシメチル)ブチル]グアニンである、請求項10記載の方法。
【請求項14】
検出可能な標識が
を含む、請求項10記載の方法。
【請求項15】
検出可能な標識が、131I、125I、123I、111I、99mTc、90Y、186Re、188Re、32P、153Sm、67Ga、201Tl、77Br、または18F標識である、請求項14記載の方法。
【請求項16】
AAVPが、処置のために標的化される組織または細胞を選択的に標的化する部分を含む、請求項1記載の方法。
【請求項17】
部分が、AAVPのカプシドタンパク質によってコードされる、請求項16記載の方法。
【請求項18】
表面タンパク質が組換えカプシドタンパク質である、請求項17記載の方法。
【請求項19】
組換えカプシドタンパク質が標的化ペプチドを含む、請求項18記載の方法。
【請求項20】
標的化ペプチドが環状ペプチドである、請求項19記載の方法。
【請求項21】
標的化ペプチドが直線状ペプチドである、請求項19記載の方法。
【請求項22】
標的化ペプチドが、細胞表面にインテグリンを発現する細胞に選択的に結合する、請求項19記載の方法。
【請求項23】
インテグリンがαvβ3またはαvβ5インテグリンである、請求項20記載の方法。
【請求項24】
ペプチドがRGDモチーフを含む、請求項22記載の方法。
【請求項25】
ペプチドがトランスフェリン受容体を発現する細胞に選択的に結合する、請求項19記載の方法。
【請求項26】
ペプチドがCRTIGPSVCを含むアミノ酸配列を含む、請求項25記載の方法。
【請求項27】
対象が、過剰増殖性疾患を有する、有することが疑われる、または発症するリスクがある、請求項1記載の方法。
【請求項28】
過剰増殖性疾患が、線維肉腫、筋肉腫、脂肪肉腫、軟骨肉腫、骨原性肉腫、軟骨腫、血管肉腫、内皮肉腫、リンパ管肉腫、リンパ管内皮肉腫、滑膜腫、中皮腫、ユーイング腫瘍、平滑筋肉腫、横紋筋肉腫、胃癌、食道癌、直腸癌、膵臓癌、卵巣癌、前立腺癌、子宮癌、頭部および頸部の癌、皮膚癌、脳癌、扁平上皮細胞癌腫、脂腺癌腫、乳頭癌腫、乳頭腺癌、嚢胞腺癌、髄様癌、気管支原性癌腫、腎細胞癌腫、肝細胞癌、胆管癌腫、絨毛癌腫、精上皮腫、胎児性癌腫、ウィルムス腫瘍、子宮頸癌、精巣癌、小細胞肺癌腫、非小細胞肺癌腫、膀胱癌腫、上皮性癌腫、神経膠腫、星状細胞腫、髄芽細胞腫、頭蓋咽頭管腫、上衣細胞腫、松果体腫、血管芽細胞腫、聴神経腫、希突起神経膠腫、髄膜腫、メラノーマ、神経芽細胞腫、網膜芽細胞腫、白血病、リンパ腫、またはカポジ肉腫である、請求項27記載の方法。
【請求項29】
レポーター遺伝子が、組織もしくは細胞選択的プロモーター、または組織もしくは細胞特異的プロモーターに機能的に連結される、請求項1記載の方法。
【請求項30】
発現を評価する工程が、AAVP核酸を発現する細胞によって選択的に代謝される標識を投与する工程を含む、請求項1記載の方法。
【請求項31】
治療的AAVPが第二の治療的遺伝子をコードする、請求項1記載の方法。
【請求項32】
第二の治療的遺伝子が、腫瘍抑制因子、阻害RNA、阻害DNA、またはプロドラッグ変換酵素である、請求項31記載の方法。
【請求項33】
阻害RNAまたは阻害DNAを含む核酸セグメントを含む、治療的AAVP核酸。
【請求項34】
阻害RNAが、siRNA、miRNA、またはアンチセンスRNAである、請求項33記載の核酸。
【請求項35】
請求項33記載のAAVP核酸を含むファージ粒子。
【請求項36】
標的化リガンドをさらに含む、請求項35記載のファージ粒子。
【請求項37】
請求項26記載のAAVP核酸を投与する工程を含む、遺伝子の発現を調整する方法。
【請求項38】
以下の工程を含む、宿主対象の標的組織への遺伝子転移および宿主対象の標的組織内での発現を検出する方法:
(a)宿主対象中に天然に存在しないレポーター遺伝子を含むAAVPベクターを宿主対象の標的組織に送達する工程であって、レポーター遺伝子が野生型の、突然変異体の、もしくは遺伝子改変された単純ヘルペスウイルス-チミジンキナーゼ遺伝子、またはヒトチミジンキナーゼ2型からなる群より選択され、かつ転移ベクターが標的組織の細胞に導入され、レポーター遺伝子が標的組織の細胞で発現し、それによってAAVPベクターによって効率的にトランスフェクトされた細胞でのみ蓄積するレポーター遺伝子産物(タンパク質)が生成される、工程;
(b)宿主対象に標識されたレポーター基質を投与する工程であって、工程(a)のレポーター遺伝子産物を発現する細胞が、放射性標識されたヌクレオシド類似体を含む標識されたレポーター基質を代謝して、標識されたレポーター代謝物を産生する、工程;ならびに
(c)レポーター遺伝子産物によって代謝されない残存レポーター基質を宿主対象から排除した後、レポーター基質の標識された代謝物を含む標的組織または細胞を非侵襲的に画像化し、それによって標的組織への遺伝子転移および標的組織内での発現を検出する工程。
【請求項39】
レポーター遺伝子産物によって代謝されない残存レポーター基質に由来する非特異的標識の少なくとも67%を、対象から排除するのに十分な工程(b)後の期間、待機する工程をさらに含む、請求項1記載の方法。
【請求項40】
レポーター遺伝子産物によって代謝されない残存レポーター基質に由来する非特異的標識の少なくとも80%を、対象から排除するのに十分な工程(b)後の期間、待機する工程をさらに含む、請求項1記載の方法。
【請求項41】
レポーター遺伝子産物によって代謝されない残存レポーター基質に由来する非特異的標識の少なくとも90%を、対象から排除するのに十分な工程(b)後の期間、待機する工程をさらに含む、請求項1記載の方法。
【請求項42】
AAVPベクターが、インビトロまたはインビボのトランスフェクション(または形質導入)によって標的組織の細胞に導入される、請求項1記載の方法。
【請求項43】
レポーター基質が、陽電子放出断層撮影、ガンマカメラ、または単一光子放出コンピュータ断層撮影によるイメージングに適した放射性同位元素で標識される、請求項1記載の方法。
【請求項44】
レポーター基質およびレポーター基質の代謝物が、2H、13C、15N、および19Fからなる群より選択される安定同位体核種を含む化合物である、請求項1記載の方法。
【請求項45】
標識されたレポーター基質代謝物が、陽電子放出断層撮影によって画像化される、請求項1記載の方法。
【請求項46】
標識されたレポーター基質代謝物が、ガンマカメラまたは単一光子放出コンピュータ断層撮影によって画像化される、請求項1記載の方法。
【請求項47】
標識されたレポーター基質代謝物が、磁気共鳴イメージングによって画像化される、請求項1記載の方法。
【請求項48】
AAVPベクターが、レポーター遺伝子ならびに適した転写プロモーターおよびエンハンサーエレメントを組み入れ、レポーターと治療的遺伝子との共発現の組織特異的、転写因子特異的、またはシグナル伝達特異的な転写活性化を確実にする、請求項1記載の方法。
【請求項49】
宿主対象への複数の細胞(または1つの細胞)の投与の前に、AAVPベクターを用いて、複数の細胞(または1つの細胞)が、レポーター遺伝子ならびに適した転写プロモーターおよびエンハンサーエレメントをエクスビボ(インビトロ)でトランスフェクトされる、請求項48記載の方法。
【請求項50】
標識された2'-フルオロ-ヌクレオシド類似体が、5-[123I]-2'-フルオロ-5-ヨード-1β-D-アラビノフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨード-1β-D-アラビノフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨード-1β-D-アラビノフラノシル-ウラシル、5-[18F]-2'-フルオロ-5-フルオロ-1-β-D-アラビノフラノシル-ウラシル;2-[11I]-および5-([11C]-メチル)-2'-フルオロ-5-メチル-1-β-D-アラビノフラノシル-ウラシル;2-[11C]-2'-フルオロ-5-エチル-1-β-D-アラビノフラノシル-ウラシル;5-([11C]-エチル)-2'-フルオロ-5-エチル-1-β-D-アラビノフラノシル-ウラシル;5-(2-[18F]-エチル)-2'-フルオロ-5-(2-フルオロ-エチル)-1-β-D-アラビノフラノシル-ウラシル、5-[123I]-2'-フルオロ-5-ヨードビニル-1-β-D-アラビノフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨードビニル-1-β-D-アラビノフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨードビニル-1-β-D-アラビノフラノシル-ウラシル;5-[123I]-2'-フルオロ-5-ヨード-1-β-D-リボフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨード-1-β-D-リボフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨード-1-β-D-リボフラノシル-ウラシル;5-[123I]-2'-フルオロ-5-ヨードビニル-1-β-D-リボフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨードビニル-1-β-D-リボフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨードビニル-1-β-D-リボフラノシル-ウラシル;または9-4-[18F]フルオロ-3-(ヒドロキシメチル)ブチル]グアニンである、請求項38記載の方法。
【請求項1】
以下の工程を含む、対象を処置する方法:
(a)病理学的状態もしくは疾患状態を有する、有することが疑われる、または発症するリスクがある対象に、レポーターをコードする治療的AAVPを投与する工程;および
(b)コードされたレポーターまたはレポーター活性を検出することによって、処置のために標的化された組織または細胞内での治療的AAVPのインサイチュー発現を評価する工程。
【請求項2】
標的器官、組織、または細胞内でAAVP核酸によって発現される治療的に十分なレベルの治療的遺伝子の発現に基づいて、対象に癌処置を施す工程をさらに含む、請求項1記載の方法。
【請求項3】
第一の治療的AAVPの発現が治療的に有効なレベルで発現されない場合に、第二の治療的AAVPを投与する、請求項2記載の方法。
【請求項4】
AAVP発現の評価が、レポーターまたはレポーターの活性の非侵襲的検出によるものである、請求項1記載の方法。
【請求項5】
レポーターが治療的タンパク質である、請求項1記載の方法。
【請求項6】
治療的タンパク質がプロドラッグ変換酵素である、請求項5記載の方法。
【請求項7】
レポーターが酵素である、請求項4記載の方法。
【請求項8】
酵素がキナーゼである、請求項7記載の方法。
【請求項9】
キナーゼがチミジンキナーゼである、請求項8記載の方法。
【請求項10】
キナーゼが検出可能に標識された化合物を修飾する、請求項8記載の方法。
【請求項11】
検出可能な標識が、蛍光、化学発光、表面増強ラマン分光法(SERS)、磁気共鳴イメージング(MRI)、コンピュータ断層撮影(CT)、または陽電子放出断層撮影(PET)イメージングによって検出可能である、請求項10記載の方法。
【請求項12】
検出可能に標識された化合物が、ヌクレオシド類似体である、請求項10記載の方法。
【請求項13】
検出可能に標識された化合物が、フルオロデオキシグルコース(FDG);2'-フルオロ-2'デオキシ-1ベータ-D-アラビオノフラノシル-5-エチル-ウラシル(FEAU);5-[123I]-2'-フルオロ-5-ヨード-1β-D-アラビノフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨード-1β-D-アラビノフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨード-1β-D-アラビノフラノシル-ウラシル、5-[18F]-2'-フルオロ-5-フルオロ-1-β-D-アラビノフラノシル-ウラシル;2-[11I]-および5-([11C]-メチル)-2'-フルオロ-5-メチル-1-β-D-アラビノフラノシル-ウラシル;2-[11C]-2'-フルオロ-5-エチル-1-β-D-アラビノフラノシル-ウラシル;5-([11C]-エチル)-2'-フルオロ-5-エチル-1-β-D-アラビノフラノシル-ウラシル;5-(2-[18F]-エチル)-2'-フルオロ-5-(2-フルオロ-エチル)-1-β-D-アラビノフラノシル-ウラシル、5-[123I]-2'-フルオロ-5-ヨードビニル-1-β-D-アラビノフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨードビニル-1-β-D-アラビノフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨードビニル-1-β-D-アラビノフラノシル-ウラシル;5-[123I]-2'-フルオロ-5-ヨード-1-β-D-リボフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨード-1-β-D-リボフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨード-1-β-D-リボフラノシル-ウラシル;5-[123I]-2'-フルオロ-5-ヨードビニル-1-β-D-リボフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨードビニル-1-β-D-リボフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨードビニル-1-β-D-リボフラノシル-ウラシル;または9-4-[18F]フルオロ-3-(ヒドロキシメチル)ブチル]グアニンである、請求項10記載の方法。
【請求項14】
検出可能な標識が
を含む、請求項10記載の方法。
【請求項15】
検出可能な標識が、131I、125I、123I、111I、99mTc、90Y、186Re、188Re、32P、153Sm、67Ga、201Tl、77Br、または18F標識である、請求項14記載の方法。
【請求項16】
AAVPが、処置のために標的化される組織または細胞を選択的に標的化する部分を含む、請求項1記載の方法。
【請求項17】
部分が、AAVPのカプシドタンパク質によってコードされる、請求項16記載の方法。
【請求項18】
表面タンパク質が組換えカプシドタンパク質である、請求項17記載の方法。
【請求項19】
組換えカプシドタンパク質が標的化ペプチドを含む、請求項18記載の方法。
【請求項20】
標的化ペプチドが環状ペプチドである、請求項19記載の方法。
【請求項21】
標的化ペプチドが直線状ペプチドである、請求項19記載の方法。
【請求項22】
標的化ペプチドが、細胞表面にインテグリンを発現する細胞に選択的に結合する、請求項19記載の方法。
【請求項23】
インテグリンがαvβ3またはαvβ5インテグリンである、請求項20記載の方法。
【請求項24】
ペプチドがRGDモチーフを含む、請求項22記載の方法。
【請求項25】
ペプチドがトランスフェリン受容体を発現する細胞に選択的に結合する、請求項19記載の方法。
【請求項26】
ペプチドがCRTIGPSVCを含むアミノ酸配列を含む、請求項25記載の方法。
【請求項27】
対象が、過剰増殖性疾患を有する、有することが疑われる、または発症するリスクがある、請求項1記載の方法。
【請求項28】
過剰増殖性疾患が、線維肉腫、筋肉腫、脂肪肉腫、軟骨肉腫、骨原性肉腫、軟骨腫、血管肉腫、内皮肉腫、リンパ管肉腫、リンパ管内皮肉腫、滑膜腫、中皮腫、ユーイング腫瘍、平滑筋肉腫、横紋筋肉腫、胃癌、食道癌、直腸癌、膵臓癌、卵巣癌、前立腺癌、子宮癌、頭部および頸部の癌、皮膚癌、脳癌、扁平上皮細胞癌腫、脂腺癌腫、乳頭癌腫、乳頭腺癌、嚢胞腺癌、髄様癌、気管支原性癌腫、腎細胞癌腫、肝細胞癌、胆管癌腫、絨毛癌腫、精上皮腫、胎児性癌腫、ウィルムス腫瘍、子宮頸癌、精巣癌、小細胞肺癌腫、非小細胞肺癌腫、膀胱癌腫、上皮性癌腫、神経膠腫、星状細胞腫、髄芽細胞腫、頭蓋咽頭管腫、上衣細胞腫、松果体腫、血管芽細胞腫、聴神経腫、希突起神経膠腫、髄膜腫、メラノーマ、神経芽細胞腫、網膜芽細胞腫、白血病、リンパ腫、またはカポジ肉腫である、請求項27記載の方法。
【請求項29】
レポーター遺伝子が、組織もしくは細胞選択的プロモーター、または組織もしくは細胞特異的プロモーターに機能的に連結される、請求項1記載の方法。
【請求項30】
発現を評価する工程が、AAVP核酸を発現する細胞によって選択的に代謝される標識を投与する工程を含む、請求項1記載の方法。
【請求項31】
治療的AAVPが第二の治療的遺伝子をコードする、請求項1記載の方法。
【請求項32】
第二の治療的遺伝子が、腫瘍抑制因子、阻害RNA、阻害DNA、またはプロドラッグ変換酵素である、請求項31記載の方法。
【請求項33】
阻害RNAまたは阻害DNAを含む核酸セグメントを含む、治療的AAVP核酸。
【請求項34】
阻害RNAが、siRNA、miRNA、またはアンチセンスRNAである、請求項33記載の核酸。
【請求項35】
請求項33記載のAAVP核酸を含むファージ粒子。
【請求項36】
標的化リガンドをさらに含む、請求項35記載のファージ粒子。
【請求項37】
請求項26記載のAAVP核酸を投与する工程を含む、遺伝子の発現を調整する方法。
【請求項38】
以下の工程を含む、宿主対象の標的組織への遺伝子転移および宿主対象の標的組織内での発現を検出する方法:
(a)宿主対象中に天然に存在しないレポーター遺伝子を含むAAVPベクターを宿主対象の標的組織に送達する工程であって、レポーター遺伝子が野生型の、突然変異体の、もしくは遺伝子改変された単純ヘルペスウイルス-チミジンキナーゼ遺伝子、またはヒトチミジンキナーゼ2型からなる群より選択され、かつ転移ベクターが標的組織の細胞に導入され、レポーター遺伝子が標的組織の細胞で発現し、それによってAAVPベクターによって効率的にトランスフェクトされた細胞でのみ蓄積するレポーター遺伝子産物(タンパク質)が生成される、工程;
(b)宿主対象に標識されたレポーター基質を投与する工程であって、工程(a)のレポーター遺伝子産物を発現する細胞が、放射性標識されたヌクレオシド類似体を含む標識されたレポーター基質を代謝して、標識されたレポーター代謝物を産生する、工程;ならびに
(c)レポーター遺伝子産物によって代謝されない残存レポーター基質を宿主対象から排除した後、レポーター基質の標識された代謝物を含む標的組織または細胞を非侵襲的に画像化し、それによって標的組織への遺伝子転移および標的組織内での発現を検出する工程。
【請求項39】
レポーター遺伝子産物によって代謝されない残存レポーター基質に由来する非特異的標識の少なくとも67%を、対象から排除するのに十分な工程(b)後の期間、待機する工程をさらに含む、請求項1記載の方法。
【請求項40】
レポーター遺伝子産物によって代謝されない残存レポーター基質に由来する非特異的標識の少なくとも80%を、対象から排除するのに十分な工程(b)後の期間、待機する工程をさらに含む、請求項1記載の方法。
【請求項41】
レポーター遺伝子産物によって代謝されない残存レポーター基質に由来する非特異的標識の少なくとも90%を、対象から排除するのに十分な工程(b)後の期間、待機する工程をさらに含む、請求項1記載の方法。
【請求項42】
AAVPベクターが、インビトロまたはインビボのトランスフェクション(または形質導入)によって標的組織の細胞に導入される、請求項1記載の方法。
【請求項43】
レポーター基質が、陽電子放出断層撮影、ガンマカメラ、または単一光子放出コンピュータ断層撮影によるイメージングに適した放射性同位元素で標識される、請求項1記載の方法。
【請求項44】
レポーター基質およびレポーター基質の代謝物が、2H、13C、15N、および19Fからなる群より選択される安定同位体核種を含む化合物である、請求項1記載の方法。
【請求項45】
標識されたレポーター基質代謝物が、陽電子放出断層撮影によって画像化される、請求項1記載の方法。
【請求項46】
標識されたレポーター基質代謝物が、ガンマカメラまたは単一光子放出コンピュータ断層撮影によって画像化される、請求項1記載の方法。
【請求項47】
標識されたレポーター基質代謝物が、磁気共鳴イメージングによって画像化される、請求項1記載の方法。
【請求項48】
AAVPベクターが、レポーター遺伝子ならびに適した転写プロモーターおよびエンハンサーエレメントを組み入れ、レポーターと治療的遺伝子との共発現の組織特異的、転写因子特異的、またはシグナル伝達特異的な転写活性化を確実にする、請求項1記載の方法。
【請求項49】
宿主対象への複数の細胞(または1つの細胞)の投与の前に、AAVPベクターを用いて、複数の細胞(または1つの細胞)が、レポーター遺伝子ならびに適した転写プロモーターおよびエンハンサーエレメントをエクスビボ(インビトロ)でトランスフェクトされる、請求項48記載の方法。
【請求項50】
標識された2'-フルオロ-ヌクレオシド類似体が、5-[123I]-2'-フルオロ-5-ヨード-1β-D-アラビノフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨード-1β-D-アラビノフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨード-1β-D-アラビノフラノシル-ウラシル、5-[18F]-2'-フルオロ-5-フルオロ-1-β-D-アラビノフラノシル-ウラシル;2-[11I]-および5-([11C]-メチル)-2'-フルオロ-5-メチル-1-β-D-アラビノフラノシル-ウラシル;2-[11C]-2'-フルオロ-5-エチル-1-β-D-アラビノフラノシル-ウラシル;5-([11C]-エチル)-2'-フルオロ-5-エチル-1-β-D-アラビノフラノシル-ウラシル;5-(2-[18F]-エチル)-2'-フルオロ-5-(2-フルオロ-エチル)-1-β-D-アラビノフラノシル-ウラシル、5-[123I]-2'-フルオロ-5-ヨードビニル-1-β-D-アラビノフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨードビニル-1-β-D-アラビノフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨードビニル-1-β-D-アラビノフラノシル-ウラシル;5-[123I]-2'-フルオロ-5-ヨード-1-β-D-リボフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨード-1-β-D-リボフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨード-1-β-D-リボフラノシル-ウラシル;5-[123I]-2'-フルオロ-5-ヨードビニル-1-β-D-リボフラノシル-ウラシル;5-[124I]-2'-フルオロ-5-ヨードビニル-1-β-D-リボフラノシル-ウラシル;5-[131I]-2'-フルオロ-5-ヨードビニル-1-β-D-リボフラノシル-ウラシル;または9-4-[18F]フルオロ-3-(ヒドロキシメチル)ブチル]グアニンである、請求項38記載の方法。
【図3】


【図4A】
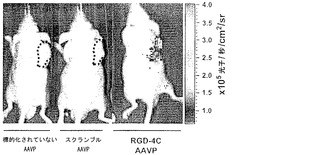
【図4B】
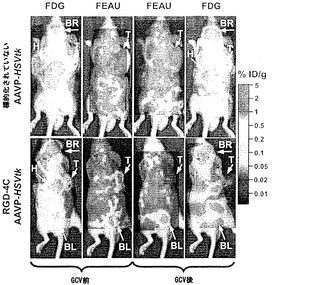
【図4C】
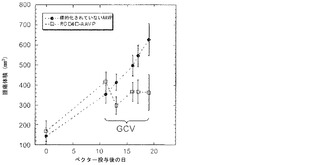
【図4D】
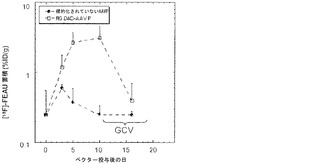
【図4E】

【図5A】
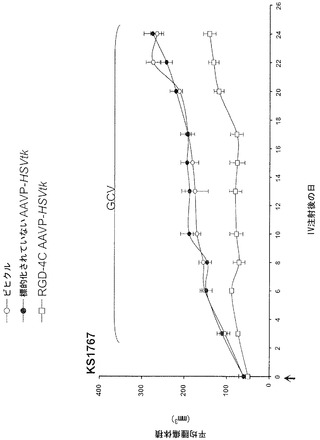
【図5B】
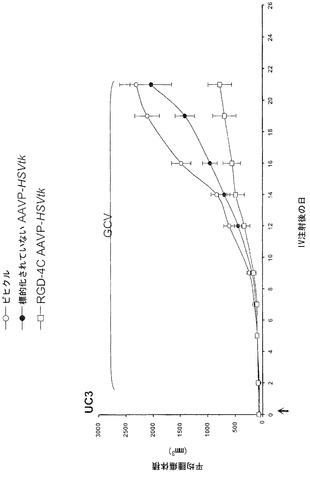
【図5C】
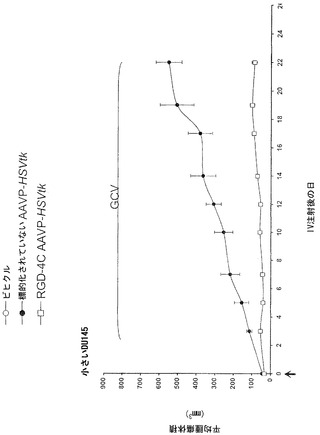
【図5D】
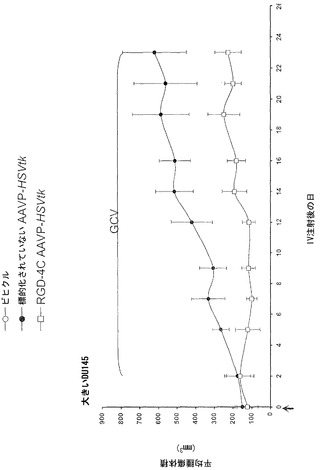
【図5E】
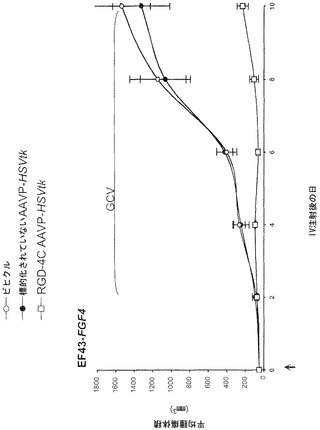
【図5F】

【図5G】
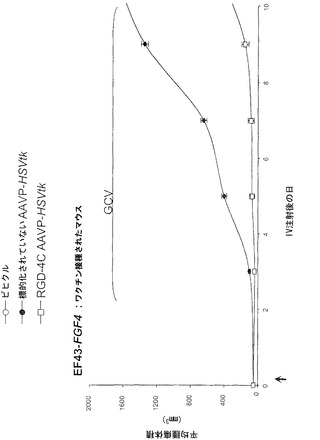
【図6A】
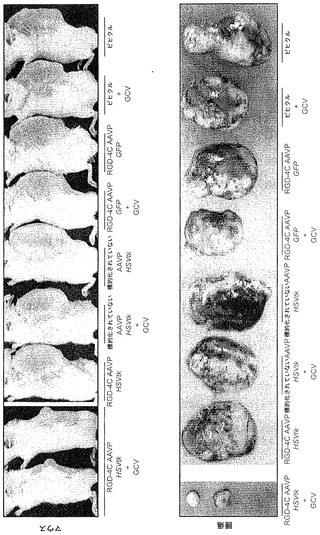
【図6B】
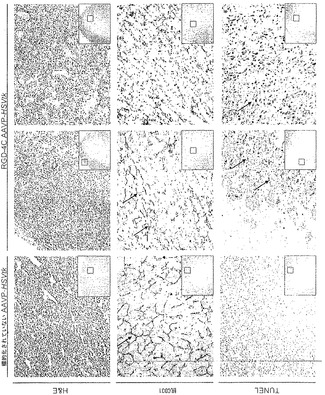
【図7】
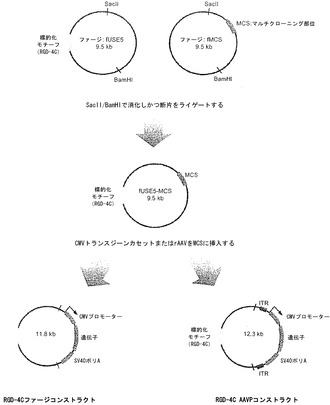
【図8A−1】

【図8A−2】
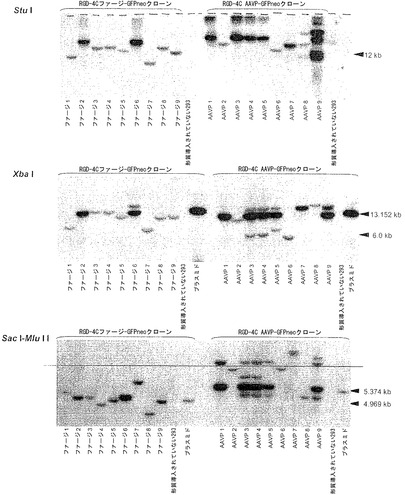
【図8B】
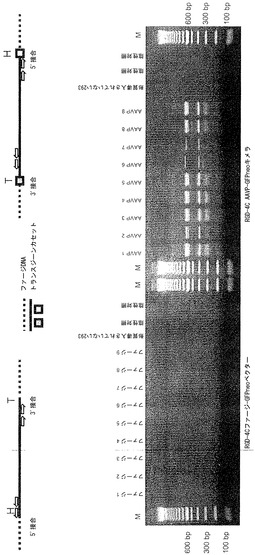
【図8C】

【図9】

【図10】
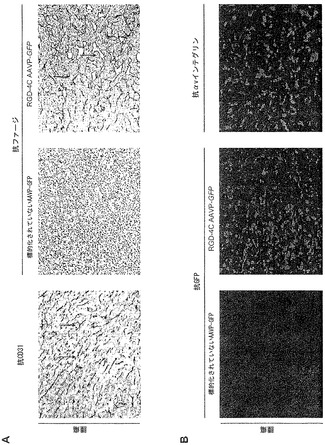
【図11】
【図12】
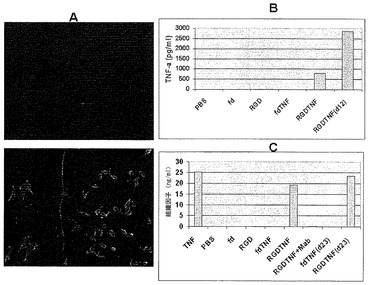
【図13】
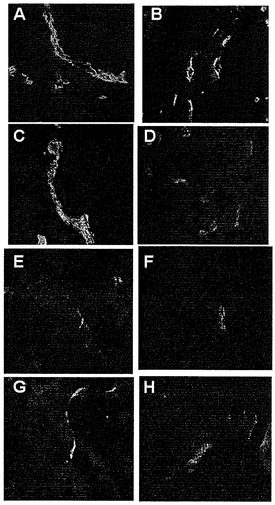
【図14】
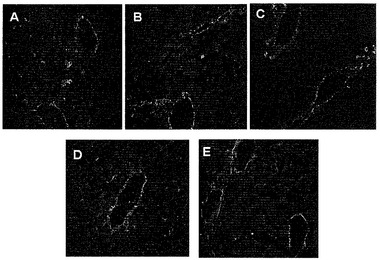
【図15】
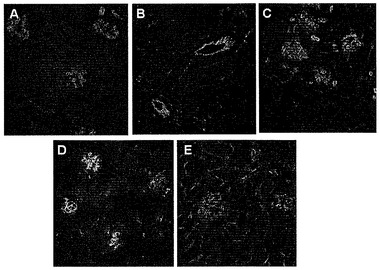
【図16】
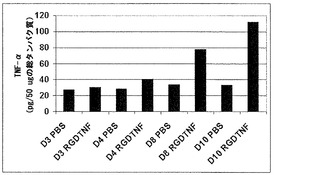
【図17】
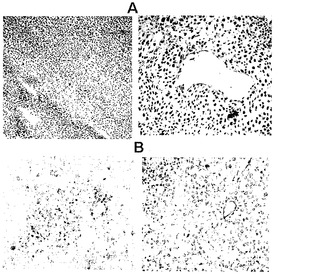
【図18】
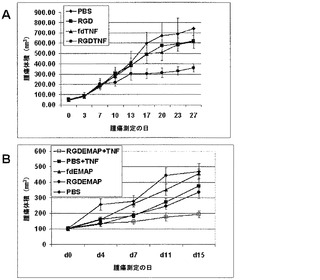
【図19】
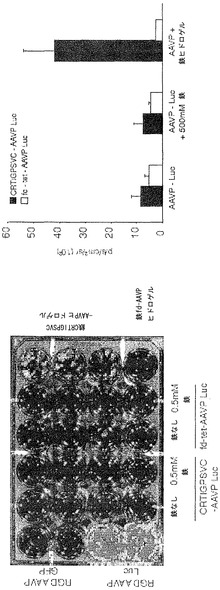
【図4A】
【図4B】
【図4C】
【図4D】
【図4E】
【図5A】
【図5B】
【図5C】
【図5D】
【図5E】
【図5F】
【図5G】
【図6A】
【図6B】
【図7】
【図8A−1】
【図8A−2】
【図8B】
【図8C】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【公表番号】特表2009−534314(P2009−534314A)
【公表日】平成21年9月24日(2009.9.24)
【国際特許分類】
【出願番号】特願2009−504507(P2009−504507)
【出願日】平成19年4月9日(2007.4.9)
【国際出願番号】PCT/US2007/066265
【国際公開番号】WO2007/118245
【国際公開日】平成19年10月18日(2007.10.18)
【出願人】(507410825)ザ ボード オブ リージェンツ オブ ザ ユニバーシティー オブ テキサス システム (4)
【Fターム(参考)】
【公表日】平成21年9月24日(2009.9.24)
【国際特許分類】
【出願日】平成19年4月9日(2007.4.9)
【国際出願番号】PCT/US2007/066265
【国際公開番号】WO2007/118245
【国際公開日】平成19年10月18日(2007.10.18)
【出願人】(507410825)ザ ボード オブ リージェンツ オブ ザ ユニバーシティー オブ テキサス システム (4)
【Fターム(参考)】
[ Back to top ]