
アマンタジン組成物および使用方法
アマンタジンでの処置を受ける患者における睡眠障害を軽減するためのアマンタジンの夜間投与方法を記載し、夜間投与に適切な持続放出アマンタジンの組成物も記載する。本発明の1つの態様は、アマンタジンを必要とする患者に投与する方法であって、アマンタジンまたはその薬学的に許容され得る塩を含む持続放出(ER)組成物を就寝時刻より前の3時間未満(すなわち、被験体が夜に就寝を望む時間)に経口投与する工程を含む、方法である。この態様はまた、下記の医薬の製造のためのかかる組成物の使用およびアマンタジンの使用を含む。あるいは、組成物を、就寝時間より前の約4時間未満に投与する。
【発明の詳細な説明】
【技術分野】
【0001】
関連出願
本出願は、2009年12月2日に出願された米国仮特許出願第61/266,053号の利益を主張し、この出願は本明細書において参照として援用される。
【背景技術】
【0002】
発明の背景
本発明の分野は、アマンタジンの持続放出組成物(extended release composition)およびその使用である。
【0003】
アマンタジンは、NMDA受容体アンタゴニストによって処置することができる種々の容態(一酸化炭素中毒による神経系の損傷後に生じ得る特発性パーキンソン病(振戦麻痺(Parlysis Agitans))、脳炎後パーキンソニズム、および症候性パーキンソニズムの処置が含まれる)に必要である。アマンタジンはまた、ウイルスM2チャネルインヒビターとしての活性を有し、ウイルス疾患(特に、インフルエンザAウイルス)の感染の予防および治療のために使用される。
【0004】
現在市販されているアマンタジンの形態は、典型的には1日に2回以上投与される即時放出処方物である。アマンタジンの使用は、用量に関連するCNS副作用(眩暈、錯乱、幻覚、不眠症、および悪夢が含まれる)によって制限され(非特許文献1)、この副作用は、アマンタジンを夜間に投与した場合に特に悪化し得る。
【0005】
即時放出アマンタジンは、不眠症および睡眠障害を生じる刺激薬として作用し得ることが公知である。したがって、これらの副作用を最小にするために、最後の用量を、典型的には、午後4時までに投与する。かかるアマンタジンの投与により、夕方または夜に血漿アマンタジン濃度がピークに達し、朝の血漿濃度が非常に低くなる。
【0006】
アマンタジンの持続放出形態は当該分野で説明されている。Guittard et al.の特許文献1およびEdgren et al.の特許文献2は、それぞれ、それぞれ抗ウイルス薬または抗パーキンソン病薬を含む経口浸透圧投薬形態を開示しており、いずれの場合にもアマンタジンは投薬形態中で利用可能な薬物として列挙されている。Smith et al.の特許文献3は、活性薬剤としてNMDA受容体アンタゴニスト(アマンタジンなど)を使用した鎮痛性の即時放出および制御放出の薬学的組成物を開示している。特許文献4、特許文献5、特許文献6、および特許文献7(全てWent et al.に付与)は、それぞれ、任意選択的に制御放出形態のNMDA受容体アンタゴニスト(アマンタジンなど)の投与を開示している。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】米国特許第5,358,721号明細書
【特許文献2】米国特許第6,217,905号明細書
【特許文献3】米国特許第6,194,000号明細書
【特許文献4】米国特許出願公開第2006/0252788号明細書
【特許文献5】米国特許出願公開第2006/0189694号明細書
【特許文献6】米国特許出願公開第2006/0142398号明細書
【特許文献7】米国特許出願公開第2008/0227743号明細書
【非特許文献】
【0008】
【非特許文献1】Gracies JM,Olanow CW;Current and Experimental Therapeutics of Parkinson’s Disease;Neuropsychopharmacology:the Fifth Generation of Progress pp1802;American College of Neuropsychopharmacology 2002
【発明の概要】
【課題を解決するための手段】
【0009】
発明の概要
本発明者らは、睡眠に悪影響を及ぼすことなく朝に起床した際に患者のアマンタジン血漿濃度がより高くなる改良されたアマンタジン処方物が当該分野で必要であることを確認している。さらに、本発明者らは、不眠症および睡眠障害の副作用が軽減され、起床時に有効な血漿濃度のアマンタジンが得られる午後遅くまたは夕方(例えば、午後4時以降)のアマンタジンの投与方法が当該分野で必要であることを確認している。
【0010】
したがって、不眠症や睡眠障害を生じることなく患者が就寝を望む直前に(例えば、就寝時に)患者に投与することができる改良されたアマンタジンの治療方法が当該分野で必要である。さらに、患者が就寝前に摂取することができ、且つ起床時(例えば、朝、ぐっすり眠った後)に適切なアマンタジン血漿濃度が得られるアマンタジン治療が必要である。
【0011】
さらに、多数のパーキンソン病患者は、嚥下が困難であり、複数の投薬を受けている。それ故、治療有効量の薬物を送達し、1日1回投与することができ、小さなサイズの経口投薬形態であり、且つ丸薬の負荷を過度に増加しないアマンタジン治療が必要である。
【0012】
本発明の1つの態様は、アマンタジンを必要とする患者に投与する方法であって、アマンタジンまたはその薬学的に許容され得る塩を含む持続放出(ER)組成物を就寝時刻より前の3時間未満(すなわち、被験体が夜に就寝を望む時間)に経口投与する工程を含む、方法である。この態様はまた、下記の医薬の製造のためのかかる組成物の使用およびアマンタジンの使用を含む。あるいは、組成物を、就寝時間より前の約4時間未満に投与する。
【0013】
第2の態様では、本発明は、アマンタジンでの処置を受けたヒト被験体における睡眠障害の軽減方法であって、アマンタジンまたはその薬学的に許容され得る塩を含む持続放出(ER)組成物を就寝時刻より前の約3時間未満(すなわち、被験体が夜に就寝を望む時間)に投与する工程を含む、方法を提供する。この態様はまた、下記の医薬の製造のためのかかる組成物の使用およびアマンタジンの使用を含む。あるいは、組成物を、就寝時間より前の約4時間未満に投与する。
【0014】
第3の態様では、本発明は、レボドパ誘発性ジスキネジア、疲労、認知症、またはパーキンソン病の任意の他の症状の処置方法であって、アマンタジンまたはその薬学的に許容され得る塩を含む持続放出(ER)組成物を就寝時刻より前の3時間未満(すなわち、被験体が夜に就寝を望む時間)に投与する工程を含む、方法を提供する。この態様はまた、下記の医薬の製造のためのかかる組成物の使用およびアマンタジンの使用を含む。
【0015】
第4の態様では、本発明は、脳損傷、脳外傷、認知症、アルツハイマー病、卒中、ハンチントン病、ALS、多発性硬化症、神経変性疾患、認知症、脳血管性容態、運動障害、脳神経障害、神経精神障害の処置方法であって、アマンタジンまたはその薬学的に許容され得る塩を含む持続放出(ER)組成物を就寝時刻より前の3時間未満(すなわち、被験体が夜に就寝を望む時間)に投与する工程を含む、方法を提供する。この態様はまた、下記の医薬の製造のためのかかる組成物の使用およびアマンタジンの使用を含む。
【0016】
任意の上記態様の1つの実施形態では、就寝時間より前の2.5時間未満、2時間未満、1.5時間未満、1時間未満、または30分未満(すなわち、被験体が夜に就寝を望む時間)に投与する。
【0017】
任意の上記態様の1つの実施形態では、患者はパーキンソン病と診断されている。
【0018】
任意の上記態様の1つの実施形態では、組成物を1日1回投与する。別の態様では、1日用量は200mgを超え、サイズ0、1、または2の1、2、または3つのカプセルで投与する。
【0019】
任意の上記態様の1つの実施形態では、組成物のパーキンソン病患者への投与により、レボドパ誘発性ジスキネジア(LID)が有意に軽減される。特定の実施形態では、組成物の投与により、レボドパ誘発性ジスキネジアが約5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、または80%軽減される。さらなる実施形態では、レボドパ誘発性ジスキネジアの軽減を、LIDの軽減を示す薬物の有効性を評価するためのFDAによって使用される数値的尺度によって測定する。さらなる特定の実施形態では、LIDの軽減の測定で使用される尺度は、UDysRS、UPDRSパートIV(サブスコア32、33)、ジスキネジア評点尺度(DRS)、異常不随意運動尺度(AIMS)、またはこの目的のために開発された他の尺度であり得る。
【0020】
任意の上記態様の1つの実施形態では、組成物のパーキンソン病患者への投与により、パーキンソン病性疲労が有意に軽減される。特定の実施形態では、組成物の投与により、パーキンソン病性疲労が約5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、または60%軽減される。さらなる特定の実施形態では、疲労の軽減を、疲労の軽減を示す薬物の有効性を評価するためのFDAによって使用される数値的尺度によって測定する。さらなる特定の実施形態では、疲労軽減の測定に使用される尺度は、疲労重症度尺度(FSS)であり得る。
【0021】
任意の上記態様の1つの実施形態では、組成物のパーキンソン病患者への投与により、パーキンソン病の症状が有意に軽減される。特定の実施形態では、組成物の投与により、パーキンソンの症状が約5%、10%、15%、20%、25%、30%、35%、または40%軽減される。さらなる特定の実施形態では、パーキンソンの症状の軽減を、パーキンソンの症状の軽減を示す薬物の有効性を評価するためのFDAによって使用される数値的尺度によって測定する。さらなる特定の実施形態では、パーキンソンの症状軽減の測定に使用される尺度は、統合パーキンソン病評点尺度(UPDRS)であり得る。
【0022】
任意の上記態様の1つの実施形態では、組成物を、投与前に、食品、より特定の実施形態では、少量の軟らかい食品(例えば、アップルソースまたはチョコレートプディング)に添加する。食品への添加は、カプセルを開け、内容物を患者の食品上に散りばめることを含み得る。これは、患者が組成物を嚥下できないか嫌がる場合に有利である。
【0023】
任意の上記態様の1つの実施形態では、定常状態の血漿濃度での投与後少なくとも1時間アマンタジンの血漿濃度は増加しない。
【0024】
任意の上記態様の1つの実施形態では、定常状態の血漿濃度での投与後少なくとも2時間アマンタジンの血漿濃度は増加しない。
【0025】
任意の上記態様の1つの実施形態では、定常状態のアマンタジン血漿濃度でのヒト被験体への組成物の投与により、かかる投与の1、2、2.5、または3時間後にアマンタジン血漿濃度が5%、10%、15%、20%、または25%未満増加する。例えば、定常状態のアマンタジン血漿濃度でのヒト被験体への組成物の投与により、アマンタジン血漿濃度が、かかる投与の1、2、2.5、または3時間後に5%未満、かかる投与の1、2、2.5、または3時間後に10%未満、かかる投与の1、2、2.5、または3時間後に15%未満、かかる投与の1、2、2.5、または3時間後に20%未満、またはかかる投与の1、2、2.5、または3時間後に25%未満増加する。
【0026】
任意の上記態様の1つの実施形態では、アマンタジンの単回用量のTmaxは9〜15時間である。さらなる特定の実施形態では、アマンタジンの単回用量のTmaxは、投与10〜14時間後である。別のさらなる特定の実施形態では、アマンタジンの単回用量のTmaxは、投与11〜13時間後である。
【0027】
任意の上記態様の1つの実施形態では、アマンタジンの定常状態のTmaxは7〜13時間である。さらなる特定の実施形態では、アマンタジンの定常状態のTmaxは、投与8〜12時間後である。別のさらなる特定の実施形態では、アマンタジンの定常状態のTmaxは、投与9〜11時間後である。
【0028】
任意の上記態様の1つの実施形態では、アマンタジンのピーク血漿濃度は、単回用量の組成物の投与の6時間後と16時間後との間に達成される。さらなる特定の実施形態では、ピークアマンタジン血漿濃度は、単回用量の組成物の投与の8〜14時間後に達成される。別のさらなる特定の実施形態では、ピークアマンタジン血漿濃度は、単回用量の組成物の投与の10〜12時間後に達成される。さらなる特定の実施形態では、ピークアマンタジン血漿濃度は、単回用量の組成物の投与の6、7、8、9、10、11、または12時間後から約9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、または24時間後に達成される。
【0029】
任意の上記態様の1つの実施形態では、ヒト被験体への組成物の1日1回の経口投与により、投与3時間後の25%未満のアマンタジン濃度の増加によって特徴付けられる定常状態血漿濃度プロフィールが得られる。さらなる特定の実施形態では、定常状態の血漿濃度プロフィールは、投与4時間後の25%未満のアマンタジン濃度の増加によって特徴付けられる。
【0030】
任意の上記態様の1つの実施形態では、組成物を1日1回投与し、定常状態でのCmax/Cmin比は、1.5〜2.0、より具体的には1.7〜1.9、より具体的には約1.8である。
【0031】
任意の上記態様の1つの実施形態では、就寝時刻の組成物のヒト被験体への複数回の投与後の定常状態の血漿濃度プロフィールは、日中の平均血漿濃度(「C−ave−日中」、ヒトPK研究で測定した場合の日中平均アマンタジン血漿濃度として定義)が夜間の平均血漿濃度(「C−ave−夜」、ヒトPK研究で測定した場合の夜間平均アマンタジン血漿濃度として定義)の1.1〜2.0倍であることによって特徴づけられる。より特定の実施形態では、C−ave−日中は、午前5時、午前6時、午前7時、午前8時、または午前9時から午後4時、午後5時、午後6時、午後7時、または午後8時の間(例えば、午前6時と午後4時との間、午前7時と午後6時との間、または午前7時と午後5時との間)で測定した平均アマンタジン血漿濃度である。C−ave−夜は、午後4時、午後5時、午後6時、午後7時、午後8時、午後9時、午後10時、または午後11時から午前5時、午前6時、午前7時、午前8時、または午前9時の間(例えば、午後10時と午前6時との間、午後7時と午前6時との間、または午後8時と午前6時との間)で測定した平均アマンタジン血漿濃度である。
【0032】
任意の上記態様の1つの実施形態では、就寝時刻の組成物のヒト被験体への複数回の投与後の定常状態の血漿濃度プロフィールは、午前中の平均血漿濃度(「C−ave−朝」、午前中にヒトPK研究で測定した場合の平均アマンタジン血漿濃度として定義)が夜間の平均血漿濃度の1.1〜2.0倍であることを特徴とする。1つの実施形態では、C−ave−朝は、午前5時、午前6時、午前7時、午前8時、または午前9時から午前11時、午前11:30時、午後12時、午後12:30時、または午後1:00時の間(例えば、午前5時と午前11時との間または午前7時と午後12時との間)に測定した平均アマンタジン血漿濃度である。より好ましくは、定常状態でのC−ave−朝/C−ave−夜比は1.2〜1.6である。
【0033】
任意の上記態様の1つの実施形態では、組成物の連日投与後の定常状態の血漿濃度プロフィールは、投与8〜12時間後の平均血漿濃度(「C−ave−8−12時間」)が投与から最初の8時間後の平均血漿濃度(「C−ave−0−8時間」)の1.1〜2.0倍であることによって特徴づけられる。より好ましくは、定常状態でのC−ave−8−12時間/C−ave−0−8時間比は、1.2〜1.6である。
【0034】
任意の上記態様の1つの実施形態では、単回用量の組成物のヒト被験体への投与により、以下によって特徴づけられる血漿濃度プロフィールが得られる:AUC0−infの5%未満、より好ましくは3%未満である0〜4時間の断片的AUC、AUC0−infの約5〜15%、好ましくは約8〜12%である0〜8時間の断片的AUC、AUC0−infの約10〜40%、好ましくは約15〜30%である0〜12時間の断片的AUC、AUC0−infの約25〜60%、好ましくは約30〜50%である0〜18時間の断片的AUC、およびAUC0−infの約40〜75%、好ましくは約50〜70%である0〜24時間の断片的AUC。
【0035】
任意の上記態様の1つの実施形態では、組成物のヒト被験体への1日1回の経口投与により、以下によって特徴づけられる定常状態の血漿濃度プロフィールが得られる:AUC24の約2〜25%、好ましくは約5〜20%である0〜4時間の断片的AUC、AUC24の約15〜50%、好ましくは約20〜40%である0〜8時間の断片的AUC、AUC24の約30〜70%、好ましくは約40〜60%である0〜12時間の断片的AUC、およびAUC24の約60〜95%、好ましくは約75〜90%である0〜18時間の断片的AUC。
【0036】
任意の上記態様の1つの実施形態では、組成物のヒト被験体への1日1回の経口投与により、以下によって特徴づけられる定常状態の血漿濃度プロフィールが得られる:AUC24の約15〜40%、好ましくは約20〜32%である0〜8時間の断片的AUC、AUC24の約30〜50%、好ましくは約35〜45%である8〜16時間の断片的AUC、およびAUC24の約20〜35%、好ましくは約25〜33%である16〜24時間の断片的AUC。
【0037】
任意の上記態様の1つの実施形態では、アマンタジンを薬学的に許容され得る塩として投与する。さらなる特定の実施形態では、アマンタジンを塩酸塩または硫酸アマンタジンとして投与する。
【0038】
任意の上記態様の1つの実施形態では、総1日用量50mg〜600mgのアマンタジンまたはその薬学的に許容され得る塩を患者に投与する。より具体的には、アマンタジンまたはその薬学的に許容され得る塩の1日用量は、100〜440mgの範囲であり得る。別の特定の実施形態では、アマンタジンまたはその薬学的に許容され得る塩の1日用量は、260〜420mgの範囲であり得る。別の実施形態では、アマンタジンまたはその薬学的に許容され得る塩の1日用量は、300mg/日を超える。種々の特定の実施形態では、アマンタジンまたはその薬学的に許容され得る塩の1日用量は、50〜75mg、70〜95mg、90〜115mg、110〜135mg、130〜155mg、150〜175mg、170〜195mg、190〜215mg、210〜235mg、230〜255mg、250〜275mg、270〜295mg、290〜305mg、300〜315mg、310〜325mg、320〜335mg、330〜345mg、340〜355mg、350〜365mg、360〜375mg、370〜385mg、380〜395mg、390〜405mg、400〜415mg、410〜425mg、420〜435mg、430〜445mg、または440〜455mgであり得る。
【0039】
任意の上記態様の1つの実施形態では、組成物は、50mg〜600mgのアマンタジンまたはその薬学的に許容され得る塩を含む。より具体的には、組成物は、100mg〜450mgのアマンタジンまたはその薬学的に許容され得る塩を含むことができる。さらにより具体的には、組成物は、130〜210mgのアマンタジンまたはその薬学的に許容され得る塩を含むことができる。種々の特定の実施形態では、組成物を含む投薬形態は、50〜75mg、70〜95mg、90〜115mg、110〜135mg、130〜155mg、150〜175mg、170〜195mg、190〜215mg、210〜235mg、230〜255mg、250〜275mg、270〜295mg、290〜305mg、300〜315mg、310〜325mg、320〜335mg、330〜345mg、340〜355mg、350〜365mg、360〜375mg、370〜385mg、380〜395mg、390〜405mg、400〜415mg、410〜425mg、420〜435mg、430〜445mg、または440〜455mgのアマンタジンまたはその薬学的に許容され得る塩を含む。さらなる特定の実施形態では、組成物は、約110、120、130、140、150、160 170、180、190、210、または220mgのアマンタジンまたはその薬学的に許容され得る塩を含む。別のさらなる特定の実施形態では、組成物は110mgの塩酸アマンタジンを含む。別のさらなる特定の実施形態では、組成物は130mgの塩酸アマンタジンを含む。別のさらなる特定の実施形態では、組成物は170mgの塩酸アマンタジンを含む。別のさらなる特定の実施形態では、組成物は210mgの塩酸アマンタジンを含む。
【0040】
任意の上記態様の1つの実施形態では、組成物を、それぞれ100〜175mgのアマンタジンまたはその薬学的に許容され得る塩を含む1、2、3、または4つの単位投薬形態として投与する。さらなる特定の実施形態では、組成物を、それぞれ100〜175mgのアマンタジンまたはその薬学的に許容され得る塩を含む2つの単位投薬形態として投与する。
【0041】
任意の上記態様の1つの実施形態では、組成物を、それぞれ50〜250mgのアマンタジンまたはその薬学的に許容され得る塩を含む1、2、または3つの単位投薬形態として投与する。さらなる特定の実施形態では、組成物を、それぞれ65〜220mgのアマンタジンまたはその薬学的に許容され得る塩を含む1つまたは2つの単位投薬形態として投与する。
【0042】
任意の上記態様の1つの実施形態では、単回用量の組成物の絶食状態のヒト被験体への経口投与により、1mgのアマンタジンあたり1.0〜2.8ng/mlの最大血漿濃度(Cmax)が得られる。さらなる特定の実施形態では、単回用量の組成物の絶食状態のヒト被験体への経口投与により、1mgのアマンタジンあたり1.6〜2.4ng/mlの最大血漿濃度(Cmax)および1mgのアマンタジンあたり40〜75ng*h/mLのAUC0−inf(t=0からt=無限大までの濃度曲線下面積)が得られる。
【0043】
任意の上記態様の1つの実施形態では、ある用量の組成物のヒト被験体への連日経口投与により、以下のうちの少なくとも1つによって特徴づけられる定常状態血漿濃度プロフィールが得られる:(i)1mgのアマンタジンあたり2.4〜4.2ng/mlのCmax、(ii)1mgのアマンタジンあたり1.1〜2.6ng/mlのCmin、および(iii)1mgのアマンタジンあたり44〜83ng*h/mLのAUC0−24さらなる具体例では、(i)、(ii)、および(iii)の3つ全ての基準を満たす。
【0044】
さらなる特定の実施形態では、定常状態血漿濃度プロフィールは、以下によってさらに特徴づけられる:(iv)投与後少なくとも1時間アマンタジン濃度が増加しないこと、および(v)Cmax/Cmin比が1.5〜2.0であること。さらなる特定の実施形態では、(iv)および(v)の両方の基準を満たす。
【0045】
別のさらなる特定の実施形態では、定常状態の血漿濃度プロフィールは、以下のうちの少なくとも1つによってさらに特徴づけられる:(iv)投与後少なくとも2時間アマンタジン血漿濃度が増加しないこと、および(v)Cmax/Cmin比1.7〜1.9。さらなる特定の実施形態では、(iv)および(v)の両方の基準を満たす。
【0046】
任意の上記態様の1つの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールが、以下のうちの少なくとも1つを示す:(i)2時間で25%以下の溶解、(ii)6時間で40〜80%までの溶解、および(iii)12時間で少なくとも80%の溶解。より特定の実施形態では、基準(i)、(ii)、および(iii)のうちの2つを満たす。さらなる特定の実施形態では、基準(i)、(ii)、および(iii)の3つ全てを満たす。
【0047】
任意の上記態様の1つの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールが、以下のうちの少なくとも1つを示す:(i)2時間で25%以下の溶解、(ii)6時間で25〜55%以下の溶解、および(iii)12時間で少なくとも80%の溶解。より特定の実施形態では、基準(i)、(ii)、および(iii)のうちの2つを満たす。さらなる特定の実施形態では、基準(i)、(ii)、および(iii)の3つ全てを満たす。
【0048】
任意の上記態様の1つの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールが、以下のうちの少なくとも1つを示す:(i)1時間で20%以下の溶解、(ii)2時間で約25〜45%の溶解、(iii)4時間で50〜80%以下の溶解、および(iv)8時間で少なくとも80%の溶解。より特定の実施形態では、基準(i)、(ii)、(iii)、および(iv)のうちの2つを満たす。さらなる特定の実施形態では、基準(i)、(ii)、(iii)、および(iv)の4つ全てを満たす。
【0049】
任意の上記態様の1つの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、アマンタジンのin vitro溶解プロフィールは、以下のアマンタジン放出によってさらに特徴づけられる:(i)1時間で10%以下、(ii)4時間で30〜50%、または(iii)12時間で少なくとも90%。より特定の実施形態では、基準(i)、(ii)、および(iii)のうちの2つを満たす。さらなる特定の実施形態では、(i)、(ii)、および(iii)の3つ全ての基準を満たす。
【0050】
別の態様では、本発明は、ペレット・イン・カプセルを含むかペレット・イン・カプセルからなる薬学的組成物であって、ペレットが、コアシード上にコーティングされたアマンタジンと結合剤との混合物を有するコアシードを含むコア、エチルセルロースを含むコアを取り囲む持続放出コーティング、ポア形成剤(ヒドロキシプロピルメチルセルロースまたはポビドンなど)、および可塑剤を含む、薬学的組成物を提供する。
【0051】
別の態様では、本発明は、上記態様の方法で用いる薬学的組成物であって、組成物が経口投与用であり、経口投与用カプセルを含み、カプセルが複数のペレットを含み、各ペレットは、以下:(a)アマンタジンまたはその薬学的に許容され得る塩を含むペレットコア、および(b)ペレットコアを取り囲む持続放出コーティングを含む、薬学的組成物を提供する。
【0052】
1つの実施形態では、持続放出コーティングは、エチルセルロース、ならびにポビドンおよびヒドロキシプロピルメチルセルロースのうちの少なくとも1つ、ならびに可塑剤を含む。さらなる特定の実施形態では、持続放出コーティングは、エチルセルロース、ポビドン、および可塑剤を含む。
【0053】
1つの実施形態では、ペレットコアは、コアシード上にコーティングされたアマンタジンおよび結合剤を含む。1つの実施形態では、コアシードは、糖スフェア(ノンパレイユ)または微結晶性セルロースシード(例えば、セルフィア(登録商標))である。さらなる特定の実施形態では、コアシードは微結晶性セルロースコアである。別の特定の実施形態では、コアシードの直径は、100ミクロン〜1,000ミクロンの範囲である。さらなる特定の実施形態では、コアシードの直径は、100、200、300、400、500、600、または700ミクロンである。好ましい特定の実施形態では、コアシードの直径は、500ミクロン未満である。
【0054】
1つの実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、アマンタジンまたはその薬学的に許容され得る塩は、20〜80重量%の量、嵩密度0.3〜1.2g/cm3で存在する。
【0055】
1つの実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、アマンタジンまたはその薬学的に許容され得る塩は、40〜60重量%の量、嵩密度0.5〜1.2g/cm3で存在する。
【0056】
1つの実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、アマンタジンまたはその薬学的に許容され得る塩は、60〜80重量%の量、嵩密度0.5〜1.2g/cm3で存在する。
【0057】
1つの実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、結合剤は、8〜25重量%の量で存在する。
【0058】
1つの実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、コアシードは、8〜25重量%の量で存在する。
【0059】
1つの実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、エチルセルロースは、10〜20重量%の量で存在する。
【0060】
1つの実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、ポビドンは、1〜4重量%の量で存在する。
【0061】
1つの実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、可塑剤は、1〜4重量%の量で存在する。
【0062】
1つの実施形態では、コーティングされたペレットの直径は、200ミクロン〜1700ミクロン(micros)の範囲である。さらなる特定の実施形態では、コーティングされたペレットの直径は、200、300、400、500、600、700、800、900、1000、1100、1200、1300、または1500ミクロンである。一定の特定の実施形態では、コーティングされたペレットの直径は、1000ミクロン未満(例えば、500〜1000ミクロン)である。
【0063】
1つの実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、結合剤は、5〜25重量%の量で存在する。
【0064】
1つの実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、コアシードは、1〜15重量%の量で存在する。
【0065】
1つの実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、エチルセルロースは、5〜20重量%の量で存在する。
【0066】
1つの実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、ポビドンは、0.25〜4重量%の量で存在する。
【0067】
1つの実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、可塑剤は、0.25〜4重量%の量で存在する。
【0068】
1つの実施形態では、ペレットは、ペレットコアと持続放出コーティングとの間にシールコーティングをさらに含む。いくつかの実施形態では、不活性コーティングを、薬物コーティング前の不活性コア、薬物コーティングしたペレット上、または制御放出コーティングしたペレット上に適用することができる。別の実施形態では、腸溶コーティングを、薬物コーティングしたペレットまたは制御放出ペレットに適用することができる。
【0069】
1つの実施形態では、ペレットコアは、ヒドロキシプロピルメチルセルロース、コポビドン、およびその混合物からなる群から選択される結合剤を含む。
【0070】
1つの実施形態では、上記組成物を、サイズ3、サイズ2、サイズ1、サイズ0、またはサイズ00カプセルで提供する。
【0071】
1つの実施形態では、治療有効1日用量の上記組成物を、わずか2カプセルで投与する。別の実施形態では、治療有効1日用量の組成物を、わずか3つのサイズ1カプセルで投与する。別の実施形態では、治療有効1日用量の組成物を、わずか2つのサイズ0カプセルで投与する。さらにより好ましい実施形態では、治療有効1日用量の組成物を、わずか2つのサイズ1カプセルで投与する。別の実施形態では、治療有効1日用量の組成物を、わずか3つのサイズ2カプセルで投与する。
【0072】
好ましい実施形態では、上記組成物は、サイズ2カプセル中に50〜110mgのアマンタジンまたはその薬学的に許容され得る塩の量、およびサイズ1カプセル中に110mg〜210mgのアマンタジンまたはその薬学的に許容され得る塩の量で提供される。さらなる実施形態では、上記組成物は、直径300〜1000ミクロンであり、アマンタジンまたはその薬学的に許容され得る塩の含有量が40〜80重量%であり、嵩密度が0.5〜1.2g/cm3であるコーティングされたペレットを含む。さらに好ましい実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、上記組成物のアマンタジンのin vitro溶解プロフィールは、以下のうちの少なくとも1つを示す:(i)2時間で25%以下の溶解、(ii)6時間で40〜80%以下の溶解、および(iii)12時間で少なくとも80%の溶解。より特定の実施形態では、基準(i)、(ii)、および(iii)のうちの2つを満たす。さらなる特定の実施形態では、基準(i)、(ii)、および(iii)の3つ全てを満たす。
【0073】
1つの実施形態では、可塑剤は、中鎖トリグリセリド、フタル酸ジエチル、クエン酸エステル、ポリエチレングリコール、グリセロール、アセチル化グリセリド、およびヒマシ油からなる群から選択される。さらなる特定の実施形態では、可塑剤は中鎖トリグリセリド(例えば、ミグリオール812N)である。
【0074】
別の態様では、本発明は、アマンタジンまたはその薬学的に許容され得る塩を必要とするヒト被験体に投与する方法であって、任意の上記態様の組成物を経口投与する工程を含む、方法を提供する。
【0075】
別の態様では、本発明は、必要とするヒト被験体におけるパーキンソン病の処置方法であって、任意の上記態様の組成物を経口投与する工程を含む、方法を提供する。好ましい態様では、本発明は、必要とするヒト被験体の疾患を処置する方法であって、任意の上記態様の組成物を1日1回夜間に1、2、または3つのカプセルで経口投与する工程を含む、方法を提供する。
【0076】
アマンタジンの必要とする被験体への投与という言及には、NMDAアンタゴニストによって処置、防止、または治癒することができる疾患または容態を有する患者の処置が含まれる。より具体的には、アマンタジンの必要とする患者への投与には、パーキンソン病、脳損傷、脳外傷、認知症、アルツハイマー病、卒中、ハンチントン病、ALS、多発性硬化症、神経変性疾患、認知症、脳血管性容態、運動障害、脳神経障害、神経精神障害を有する患者の処置が含まれる。
【図面の簡単な説明】
【0077】
【図1】図1は、実施例3で言及した3つのアマンタジンER処方物A、B、Cの溶解プロフィールを示す。
【図2A】図2Aおよび2Bは、1日目および9日目の1日2回のアマンタジンIR(A)および1日1回のアマンタジンER(B)の健康な成人の男性および女性の被験体への絶食条件下での投与後の平均血漿濃度時間曲線を示す。
【図2B】図2Aおよび2Bは、1日目および9日目の1日2回のアマンタジンIR(A)および1日1回のアマンタジンER(B)の健康な成人の男性および女性の被験体への絶食条件下での投与後の平均血漿濃度時間曲線を示す。
【図3】図3は、9日目の1日2回のアマンタジンIRおよび1日1回のアマンタジンERの健康な成人の男性および女性の被験体への絶食条件下での投与後のアマンタジンの平均血漿濃度対時間曲線のプロットを示す。
【図4】図4は、1日2回または3回の種々の強度の即時放出アマンタジンの複数回投与後および1日1回の種々の強度のアマンタジンERの投与後のアマンタジンの平均血漿濃度対時間曲線をシミュレーションしている。
【図5】図5は、4つのアマンタジン処置についての平均(SD)血漿アマンタジン濃度対指定時間のプロットを示す。
【図6】図6は、4つのアマンタジン処置についての片対数の平均(SD)血漿アマンタジン濃度対指定時間を示す。
【図7】図7は、実施例12に記載のERアマンタジン処方物についての定常状態血漿濃度の時間プロフィールのシミュレーションを示す。夜間に1日1回投与したERアマンタジン処方物2により、処方物1と比較して定常状態でのピーク血漿濃度の到達時間が約4時間遅延する。
【発明を実施するための形態】
【0078】
発明の詳細な説明
本発明は、アマンタジン処置を受けた患者における睡眠障害の軽減方法を提供する。本方法は、アマンタジンが睡眠を妨害せず、さらに、アマンタジンを服用する多数の患者がしばしば最も必要とする朝の時間に最高に有益となり、さらに、必要に応じてパーキンソン病の症状に対して夜間に適用されるように、アマンタジンを必要とする被験体に投与する工程を含む。夜間適用は、患者が目覚めて再度就寝することを望む場合に有利である。
【0079】
本発明の方法は、夜間投与のためにデザインされた持続放出(ER)アマンタジン組成物を患者に経口投与する工程を含む。組成物を、就寝時刻より前の3時間未満、好ましくは2.5時間未満、2時間未満、1.5時間未満、または1時間未満に服用する。より好ましくは、ERアマンタジン組成物を、就寝時刻より前(すなわち、被験体が夜に就寝を望む時間)の30分未満に服用する。本明細書中で使用する場合、アマンタジンという言及は、その薬学的に許容され得る塩(例えば、塩酸アマンタジン、硫酸アマンタジンなど)が含まれることを意図する。あるいは、組成物を、就寝時間より前の約4時間未満に投与する。
【0080】
本明細書中で使用する場合、「持続放出」には、「制御放出」、「放出調節」、「徐放」、「時効性」、「遅延放出」が含まれ、遅延放出、即時放出、腸溶性コーティングなどと上記のそれぞれとの混合物も含まれる。
【0081】
患者は、アマンタジンが処方される任意の疾患または障害(パーキンソン病、多発性硬化症、薬物誘発性の錐体外路系の反応、レボドパ誘発性ジスキネジア、およびウイルス性疾患(例えば、インフルエンザ、HBV、およびHCV)など)と診断され得る。特定の実施形態では、患者は、パーキンソン病を有する。ここで、本明細書中で使用する場合、パーキンソン病は、パーキンソニズムの診断も含む。1つの実施形態では、患者は早期パーキンソン病を有し、アマンタジンを、単剤療法としてかレボドパを併用せずにモノアミンオキシダーゼB型(MAO−B)インヒビターと組み合わせて使用する。別の実施形態では、患者は後期パーキンソン病を有し、患者は、アマンタジンに加えてレボドパを服用する。別の実施形態では、患者は多発性硬化症を有し、アマンタジンを疲労処置のために使用する。他の実施形態では、患者は、脳損傷、脳損傷、脳外傷、認知症、アルツハイマー病、卒中、ハンチントン病、ALS、多発性硬化症、神経変性疾患、認知症、脳血管性容態、運動障害、脳神経障害、神経精神障害を有する。
【0082】
本発明で用いるERアマンタジン組成物は、被験体の睡眠を妨害しない血漿濃度プロフィールが得られる夜間投与に適している。本発明の組成物により、ヒト被験体への投与の際に、定常状態条件で、ある用量の組成物の投与によってその用量の投与3時間後にアマンタジンの血漿濃度が25%未満増加するようにアマンタジンの血漿濃度の初期漸増が認められるであろう。例えば、ある用量の組成物の投与時の被験体のアマンタジンの定常状態血漿濃度が500ng/mlである場合、3時間後の被験体のアマンタジンの血漿濃度は、625ng/ml未満であろう。好ましくは、アマンタジンの血漿濃度の増加は15%未満、最も好ましくは10%未満である。特に好ましい組成物は、投与後少なくとも1時間、好ましい実施形態では、2時間アマンタジン血漿濃度が増加しないことか、さらに減少すること(定常状態条件で)によってさらに特徴づけられる血漿濃度プロフィールを有する。本発明で用いる組成物は、朝の時間にアマンタジンの最高濃度(Cmax)を提供することによる就寝時刻(すなわち、被験体が夜に就寝を望む時間)の投与にさらに適している。絶食状態での単回用量投与後に測定したCmax到達時間(Tmax)は、少なくとも8時間且つ13、14、15、または16時間まで、少なくとも9時間且つ13、14、15、または16時間まで、または少なくとも10時間且つ13、14、15、または16時間までである。特定の実施形態は、Tmaxは9〜15時間、好ましくは10〜14時間、最も好ましくは11〜13時間である。定常状態では、組成物の1日1回の投与を使用して、Tmaxは7〜13時間、好ましくは8〜12時間、最も好ましくは9〜11時間である。適切なERアマンタジン組成物を、定常状態Cmax/Cmin比1.5〜2.0、好ましくは1.7〜1.9を有し、それにより、最適に変動する組成物が得られることによってさらに特徴づけることができる。
【0083】
より特定の好ましい実施形態では、血漿濃度プロフィールは、単回用量の組成物の投与後に以下のAUCプロフィールを有することによってさらに特徴づけられる:AUC0−infの5%未満、好ましくは3%未満である0〜4時間の断片的AUC、AUC0−infの約5〜15%、好ましくは約8〜12%である0〜8時間の断片的AUC、AUC0−infの約10〜40%、好ましくは約15〜30%である0〜12時間の断片的AUC、AUC0−infの約25〜60%、好ましくは約30〜50%である0〜18時間の断片的AUC、およびAUC0−infの約40〜75%、好ましくは約50〜70%である0〜24時間の断片的AUC。
【0084】
さらに好ましい実施形態では、血漿濃度プロフィールは、以下によって特徴づけられる定常状態条件での組成物の1日1回の投与後のAUCプロフィールを有することによってさらに特徴づけられる:AUC24の約2〜25%、好ましくは約5〜20%である0〜4時間の断片的AUC、AUC24の約15〜50%、好ましくは約20〜40%である0〜8時間の断片的AUC、AUC24の約30〜70%、好ましくは約40〜60%である0〜12時間の断片的AUC、およびAUC24の約60〜95%、好ましくは約75〜90%である0〜18時間の断片的AUC。
【0085】
任意の上記態様のいくつかの実施形態では、就寝時刻の組成物のヒト被験体への複数回の投与後の定常状態の血漿濃度プロフィールは、日中の平均血漿濃度(「C−ave−日中」、ヒトPK研究で測定した場合の日中平均アマンタジン血漿濃度として定義)が夜間の平均血漿濃度(「C−ave−夜」、ヒトPK研究で測定した場合の夜間平均アマンタジン血漿濃度として定義)の1.1〜2.0倍であることによって特徴づけられる。いくつかの実施形態では、定常状態でのC−ave−日中/C−ave−夜比は、以下の範囲の1つである:1.1〜1.9、1.1〜1.8、1.1〜1.7、1.1〜1.6、1.1〜1.5、1.1〜1.4、1.2〜1.9、1.2〜1.7、1.2〜1.6、1.2〜1.5、1.3〜1.9、1.3〜1.8、1.3〜1.7、1.3〜1.6、1.4〜1.9、1.4〜1.8、1.4〜1.7、1.5〜1.9、1.5〜1.8、1.5〜1.7、1.6〜1.9、1.6〜1.8、または1.7〜1.9。いくつかの実施形態では、定常状態でのC−ave−日中/C−ave−夜比は、1.1、1.15、1.2、1.25、1.3、1.35、1.4、1.45、1.5、1.55、1.6、1.65、1.7、1.75、1.8、1.85、1.9、1.95、または2.0である。いくつかの実施形態では、C−ave−日中は、午前5時、午前6時、午前7時、午前8時、または午前9時から午後4時、午後5時、午後6時、午後7時、または午後8時の間に測定した平均アマンタジン血漿濃度であり、C−ave−夜は、午後4時、午後5時、午後6時、午後7時、午後8時、午後9時、午後10時、または午後11時から午前5時、午前6時、午前7時、午前8時、または午前9時までに測定した平均アマンタジン血漿濃度である。いくつかの実施形態では、C−ave−日中は、午前5時と午後8時との間の任意の4〜12時間内に測定した平均アマンタジン血漿濃度であり、C−ave−夜は、午後8時と午前5時との間の任意の4〜12時間に測定した平均アマンタジン血漿濃度である。いくつかの実施形態では、C−ave−日中は、午前5時と午後8時との間の任意の4、5、6、7、8、9、10、11、または12時間内に測定した平均アマンタジン血漿濃度であり、C−ave−夜は、午後8時と午前5時との間の任意の4、5、6、7、8、9、10、11、または12時間内に測定した平均アマンタジン血漿濃度である。
【0086】
本明細書中に記載のいくつかの実施形態では、アマンタジン組成物を、就寝時刻の0〜4時間前に患者に投与する。いくつかの実施形態では、アマンタジン組成物を、就寝時刻の0〜3、0〜2、または0〜1時間前に患者に投与する。いくつかの実施形態では、アマンタジン組成物を、就寝時刻の0〜240分前、0〜180分前(例えば、0〜120分前、0〜60分前、0〜45分前、0〜30分前、0〜15分前、または0〜10分前)に患者に投与する。いくつかの実施形態では、アマンタジン組成物を、就寝時刻の60〜240分前、60〜180分前、60〜120分前、または60〜90分前に患者に投与する。
【0087】
患者への投与には、医療専門家による投与および患者による自己投与が含まれると理解すべきである。
【0088】
本明細書中に別記されない限り、用語「就寝時刻」は、24時間のうちの主な睡眠時間という通常の意味を有する。一般大衆では就寝時刻は夜であるが、就寝時刻が日中である患者(夜に働く患者)が存在する。したがって、いくつかの実施形態では、就寝時刻は、日中または夜間の任意の時間であり得る。
【0089】
本明細書中で使用する場合、他で示さない限り、ヒト被験体における血漿濃度プロフィールまたは特定の薬物動態学的性質(例えば、Cmax、Cmin、AUC、Tmaxなど)という言及は、薬物の薬物動態学的性質を測定するためにデザインされた典型的な第I相臨床試験で決定された健康な成人から得た平均値をいう(例えば、以下の実施例5、6、および7を参照のこと)。本明細書中のTmaxという言及は、他で示さない限り、絶食状態での単回用量の投与後に得た値をいう。
【0090】
本発明のいくつかの実施形態では、本発明のアマンタジンの投与量は、通常はアマンタジンの即時放出組成物のために処方された範囲内または範囲を超える。他の実施形態では、本発明のアマンタジンの投与量は、通常はアマンタジンの即時放出組成物のために処方された範囲を超える。例えば、パーキンソン病処置のためのアマンタジンの推奨用量は、100mg(1日2回投与)である。前述の用量で十分な利点が得られず、且つ患者がかかるより高い用量を許容することができる一部の患者の場合、用量を300mgまたは400mg(分割量)に増加させることができる。最も一般的なアマンタジンの処方用量は100mg〜200mg/日であり、後者は、分割量で投与する。200mg超(例えば、300mg)を、常に分割量で投与する。本発明のために、50〜600mg、より好ましくは200〜450mgの用量をパーキンソン病処置のために投与し、本発明の方法および組成物は、これらの範囲のいずれかによって定義される用量の投与を含むことができる。特定の実施形態では、かかるより高い用量を1日1回投与することができる。さらなる実施形態では、かかるより高い用量を夜間に投与することができる。さらなる実施形態では、かかるより高い用量をサイズ0、1、または2の1、2、または3カプセルの形態で1日1回投与することができる。
【0091】
任意の上記態様の1つの実施形態では、アマンタジンを薬学的に許容され得る塩として投与する。さらなる特定の実施形態では、アマンタジンを塩酸塩または硫酸アマンタジンとして投与する。
【0092】
任意の上記態様の1つの実施形態では、総1日用量50mg〜600mgのアマンタジンまたはその薬学的に許容され得る塩を患者に投与する。より具体的には、アマンタジンまたはその薬学的に許容され得る塩の1日用量は、100mg〜440mgの範囲であり得る。別の特定の実施形態では、アマンタジンまたはその薬学的に許容され得る塩の1日用量は、260mg〜420mgの範囲であり得る。別の実施形態では、アマンタジンまたはその薬学的に許容され得る塩の1日用量は、300mg/日を超える。種々の特定の実施形態では、アマンタジンまたはその薬学的に許容され得る塩の1日用量は、50〜75mg、70〜95mg、90〜115mg、110〜135mg、130〜155mg、150〜175mg、170〜195mg、190〜215mg、210〜235mg、230〜255mg、250〜275mg、270〜295mg、290〜305mg、300〜315mg、310〜325mg、320〜335mg、330〜345mg、340〜355mg、350〜365mg、360〜375mg、370〜385mg、380〜395mg、390〜405mg、400〜415mg、410〜425mg、420〜435mg、430〜445mg、または440〜455mgであり得る。
【0093】
任意の上記態様の1つの実施形態では、組成物は、50〜600mgのアマンタジンまたはその薬学的に許容され得る塩を含む。より具体的には、組成物は、100〜450mgのアマンタジンまたはその薬学的に許容され得る塩を含むことができる。さらにより具体的には、組成物は、130〜210mgのアマンタジンまたはその薬学的に許容され得る塩を含むことができる。種々の特定の実施形態では、投薬形態は、50〜75mg、70〜95mg、90〜115mg、110〜135mg、130〜155mg、150〜175mg、170〜195mg、190〜215mg、210〜235mg、230〜255mg、250〜275mg、270〜295mg、290〜305mg、300〜315mg、310〜325mg、320〜335mg、330〜345mg、340〜355mg、350〜365mg、360〜375mg、370〜385mg、380〜395mg、390〜405mg、400〜415mg、410〜425mg、420〜435mg、430〜445mg、または440〜455mgのアマンタジンまたはその薬学的に許容され得る塩を含む。さらなる特定の実施形態では、組成物は、約110、120、130、140、150、160 170、180、190、210、または220mgのアマンタジンまたはその薬学的に許容され得る塩を含む。別のさらなる特定の実施形態では、組成物は110mgの塩酸アマンタジンを含む。別のさらなる特定の実施形態では、組成物は130mgの塩酸アマンタジンを含む。別のさらなる特定の実施形態では、組成物は170mgの塩酸アマンタジンを含む。別のさらなる特定の実施形態では、組成物は210mgの塩酸アマンタジンを含む。
【0094】
任意の上記態様の1つの実施形態では、組成物は、約50mg、60mg、70mg、80mg、90mg、100mg、110mg、120mg、130mg、140mg、150mg、160mg、170mg、180mg、190mg、200mg、210mg、220mg、230mg、240mg、250mg、260mgのアマンタジンまたはその薬学的に許容され得る塩から約75mg、85mg、95mg、105mg、115mg、125mg、135mg、145mg、155mg、165mg、175mg、185mg、195mg、205mg、215mg、225mg、235mg、245mg、255mg、265mg、275mg、285mg、295mg、305mg、315mg、325mg、335mg、345mg、355mg、365mg、375mg、385mg、395mg、405mg、415mg、425mg、435mg、445mgまでのアマンタジンまたはその薬学的に許容され得る塩を含む。
【0095】
本発明の特定の実施形態では、被験体のアマンタジンの全1日用量を、就寝時刻より前(すなわち、被験体が夜に就寝を望む時間)の約3、2、または1時間未満に1回投与する。他の実施形態では、アマンタジンの1日用量の少なくとも半分を、前述の就寝時刻前に服用する。好ましくは、アマンタジン用量の少なくとも2/3を、前述の就寝時刻前に服用し、残りを午前中または午後に服用する。アマンタジンの午前中または昼の用量を、従来の即時放出投薬形態または持続放出形態で提供することができる。
【0096】
任意の上記態様の1つの実施形態では、組成物のパーキンソン病患者への投与により、レボドパ誘発性ジスキネジアが有意に軽減される。特定の実施形態では、組成物の投与により、レボドパ誘発性ジスキネジアが約5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、または80%軽減される。さらなる実施形態では、レボドパ誘発性ジスキネジアの軽減を、LID処置の有効性を評価し、認可薬を認可するためのFDAまたは他の規制当局によって使用または承認される数値的尺度によって測定する。さらなる特定の実施形態では、LID軽減の測定で使用される尺度は、UDysRS、UPDRSパートIV(サブスコア32、33)、ジスキネジア評点尺度(DRS)、異常不随意運動尺度(AIMS)、Rushジスキネジア評点尺度、パーキンソン病ジスキネジア尺度(PDYS−26)、Obesoジスキネジア評点尺度(CAPIT)、臨床ジスキネジア評点尺度(CDRS)、日常生活ジスキネジアのLang−Fahn活動、またはこの目的のために開発された他の尺度であり得る。
【0097】
任意の上記態様の1つの実施形態では、組成物のパーキンソン病患者への投与により、パーキンソン病性疲労が有意に軽減される。特定の実施形態では、組成物の投与により、パーキンソン病性疲労が約5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、または60%軽減される。さらなる特定の実施形態では、疲労の軽減を、疲労処置の有効性を評価し、認可薬を認可するためのFDAまたは他の規制当局によって使用または承認される数値的尺度によって測定する。さらなる特定の実施形態では、疲労軽減の測定に使用される尺度は、疲労重症度尺度(FSS)、疲労評価目録、慢性疾患治療の機能的評価−疲労(FACIT疲労)、多次元疲労目録(MFI−20)、パーキンソン疲労尺度(PFS−16)、および疲労重症度目録であり得る。他の特定の実施形態では、疲労の軽減を、比較臨床試験においてプラセボと比較して測定する。他の実施形態では、疲労の軽減を、比較臨床試験においてベースラインと比較して測定する。
【0098】
任意の上記態様の1つの実施形態では、組成物のパーキンソン病患者への投与により、パーキンソン病の症状が有意に軽減される。特定の実施形態では、組成物の投与により、パーキンソンの症状が約5%、10%、15%、20%、25%、30%、35%、または40%軽減される。さらなる特定の実施形態では、パーキンソンの症状の軽減を、パーキンソンの症状処置の有効性を評価し、認可薬を認可するためのFDAまたは他の規制当局によって使用または承認される数値的尺度によって測定する。さらなる特定の実施形態では、パーキンソンの症状軽減の測定に使用される尺度は、統合パーキンソン病評点尺度(UPDRS)であり得る。統合パーキンソン病評点尺度(UPDRS、MDS改訂)−パートI:日常生活で経験する非運動性の状況(13項目)、パートII:日常生活で経験する運動性の状況(13項目)−パートIII:運動性の検査(33スコア化項目)−パートI: 精神状態、行動、および気分−パートII:日常生活における活動−パートIII:運動性の検査(27スコア化項目)Hoehn and Yahrの病期分類尺度(オリジナルまたは改訂版)。
【0099】
任意の上記態様の1つの実施形態では、組成物のパーキンソン病患者への投与により、レボドパ誘発性ジスキネジアが有意に軽減される。特定の実施形態では、組成物の投与により、レボドパ誘発性ジスキネジアが約5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、または80%軽減される。さらなる実施形態では、レボドパ誘発性ジスキネジアの軽減を、LIDの軽減を示す薬物の有効性を評価するためのFDAによって使用される数値的尺度によって測定する。さらなる特定の実施形態では、LID軽減の測定に使用される尺度は、UDysRS、UPDRSパートIV(サブスコア32、33)、ジスキネジア評点尺度(DRS)、異常不随意運動尺度(AIMS)、またはこの目的のために開発された他の尺度であり得る。他の特定の実施形態では、LIDの軽減を、比較臨床試験においてプラセボと比較して測定する。他の実施形態では、LIDの軽減を、比較臨床試験においてベースラインと比較して測定する。
【0100】
任意の上記態様の1つの実施形態では、組成物のパーキンソン病患者への投与により、パーキンソン病性疲労が有意に軽減される。特定の実施形態では、組成物の投与により、パーキンソン病性疲労が約5%、10%、15%、20%、25%、30%、35%、または40%軽減される。さらなる特定の実施形態では、疲労の軽減を、疲労の軽減を示す薬物の有効性を評価するためのFDAによって使用される数値的尺度によって測定する。さらなる特定の実施形態では、疲労軽減の測定に使用される尺度は、疲労重症度尺度(FSS)であり得る。
【0101】
任意の上記態様の1つの実施形態では、組成物のパーキンソン病患者への投与により、パーキンソン病の症状が有意に軽減される。特定の実施形態では、組成物の投与により、パーキンソンの症状が約5%、10%、15%、20%、25%、30%、35%、または40%軽減される。さらなる特定の実施形態では、パーキンソンの症状の軽減を、パーキンソンの症状の軽減を示す薬物の有効性を評価するためのFDAによって使用される数値的尺度によって測定する。さらなる特定の実施形態では、パーキンソンの症状軽減の測定に使用される尺度は、統合パーキンソン病評点尺度(UPDRS)であり得る。他の特定の実施形態では、パーキンソン病の症状の軽減を、比較臨床試験においてプラセボと比較して測定する。他の実施形態では、パーキンソン病の症状の軽減を、比較臨床試験においてベースラインと比較して測定する。
【0102】
持続放出処方物
本発明の方法での使用に適切な持続放出アマンタジン組成物を、種々の持続放出テクノロジー(上記の背景の項で参照した特許公報(公報はその全体が本明細書中で参考として援用される)に記載のテクノロジーなど)を使用して作製することができる。いくつかの実施形態では、本発明は、カプセル投薬形態のペレットである。いくつかの実施形態では、ペレットはペレットコアを含み、ペレットコアは、少なくとも1つの薬物層および少なくとも1つの持続放出コーティング層でコーティングされている。いくつかの実施形態では、ペレットを、少なくとも1つの薬物層、中間層(シールコートなど)、および持続放出コーティング層でコーティングする。いくつかの実施形態では、ペレット、薬物層、またはその両方は、1つ以上の結合剤を含む。
【0103】
いくつかの実施形態では、投薬単位は、複数のコーティングしたペレットを含む。いくつかの実施形態では、ペレットの直径は、例えば、300〜1700ミクロン、いくつかの場合、500〜1200ミクロンである。ペレットは、例えば、不活性物質(糖スフェア、微結晶性セルロース(MCC)スフェア、デンプンペレットなど)を含むであろう。いくつかの実施形態では、ペレットを、他の過程(ペレット化、押し出し、球形化など)またはその組み合わせによって調製することができる。コアペレットは、塩酸アマンタジンおよび薬学的に許容され得る賦形剤から構成されるであろう。
【0104】
コーティングしたペレット
ペレットコアを、有効成分(例えば、アマンタジンまたはその薬学的に許容され得る塩および/もしくは多形)でコーティングする。いくつかの実施形態では、有効成分に加えて、ペレットはまた、1つ以上の結合剤(例えば、ヒドロキシプロピルメチルセルロース、コポビドン、ポビドン、ヒドロキシプロピルセルロース、ヒドロキシエチルセルロース、メチルセルロース、カルボキシメチルセルロースなど)を含む。いくつかの実施形態では、ペレットはまた、1つ以上のさらなる賦形剤(粘着防止剤(例えば、タルク、ステアリン酸マグネシウムなど)など)を含む。
【0105】
いくつかの実施形態では、ペレットコアを、従来のコーティング技術(流動化層コーティング、パンコーティングなど)によって、有効成分、および任意選択的な1つ以上の結合剤、粘着防止剤、および/または溶媒を含む薬物層でコーティングする。
【0106】
中間層コーティング
いくつかの実施形態では、ペレットを、中間層(シールコートなど)でコーティングする。いくつかの実施形態では、シールコートは、持続放出コーティング中の成分によるペレットコア中の成分の妨害の防止、ペレットコア中の成分のペレットコアから持続放出層への拡散の防止などに適応する。本明細書中に記載のように、本発明のシールコートは、1つ以上のフィルム形成ポリマー(ヒドロキシプロピルメチルセルロース(HPMC)、コポビドン、ポビドン、ポリビニルピロリドン、ヒドロキシプロピルセルロース、ヒドロキシエチルセルロース、メチルセルロース、カルボキシメチルセルロース、またはその任意の組み合わせなどが含まれるが、これらに限定されない)を含むことができる。
【0107】
シールコートは、可塑剤(プロピレングリコール、トリアセチン、ポリエチレングリコール、クエン酸トリブチルなど)および任意選択的な粘着防止剤(ステアリン酸マグネシウム、ケイ酸カルシウム、ケイ酸マグネシウム、およびコロイド状二酸化ケイ素、またはタルクなど)のような他の添加物をさらに含むことができる。
【0108】
上記の可塑剤および粘着防止剤は別として、シールコートは、任意選択的に、当業者に公知の緩衝液、着色剤、乳白剤、界面活性剤、または基剤を含むことができる。
【0109】
シールコーティングを、従来のコーティング技術(流動化層コーティング、パンコーティングなど)を使用してコアに適用することができる。いくつかの実施形態では、薬物コーティングしたペレットコアを、流動化層コーティングまたはパンコーティングによって1つ以上の結合剤、粘着防止剤、および/または溶媒を任意選択的に含むシールコート層でコーティングする。
【0110】
結合剤
いくつかの実施形態では、ペレットコア、中間コーティング層、またはその両方のいずれかは、1つ以上の結合剤(例えば、フィルム形成ポリマー)を含むことができる。本明細書中で使用する適切な結合剤には、例えば、アルギン酸およびその塩、セルロース誘導体(カルボキシメチルセルロース、メチルセルロース(例えば、メトセル(登録商標))、ヒドロキシプロピルメチルセルロース、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース(例えば、クルセル(登録商標))、エチルセルロース(例えば、エトセル(登録商標))、および微結晶性セルロース(例えば、アビセル(登録商標))など);微結晶性デキストロース、アミロース、マグネシウムケイ酸アルミニウム、多糖酸、ベントナイト、ゼラチン、ポリビニルピロリドン/酢酸ビニルコポリマー、クロスポビドン、ポビドン、デンプン、アルファ化デンプン、トラガカント、デキストリン、糖(スクロース(例えば、ダイパック(登録商標))、グルコース、デキストロース、糖蜜、マンニトール、ソルビトール、キシリトール(例えば、キシリタブ(登録商標))、およびラクトースなど)、天然ゴムまたは合成ゴム(アカシア、トラガカント、ガッチゴムなど)、イサポール外皮の粘液、ポリビニルピロリドン(例えば、ポリビドン(登録商標)CL、コリドン(登録商標)CL、ポリプラスドン(登録商標)XL−10)、カラマツのアラビノガラクタン(larch arabogalactan)、ビーガム(登録商標)、ポリエチレングリコール、ワックス、およびアルギン酸ナトリウムなどが含まれる。
【0111】
持続放出コーティング
ペレットを、持続放出コーティングでコーティングする。持続放出コーティングは、投薬形態の使用環境への導入後の一定期間コーティングされた薬物コアからの薬物の放出を遅延させるように構成される。いくつかの実施形態では、持続放出コーティングは、1つ以上のpH依存性またはpH非依存性の持続放出賦形剤を含む。pH非依存性持続放出ポリマーの例には、エチルセルロース、ヒドロキシプロピルメチルセルロース、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース、カルボキシメチルセルロース、エチルアクリラートのコポリマー、メチルメタクリラート(例えば、オイドラギットRS)などが含まれる。pH依存性持続放出賦形剤の例には、メタクリル酸(methacrylic acic)コポリマー、ヒドロキシプロピルメチル酢酸セルローススクシナート、ヒドロキシプロピルメチルセルロースフタラート、およびセルロースアセタートフタラートなどが含まれる。持続放出コーティングには、ポア形成物質(ポビドン、ポリエチレングリコール、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロースなど)、糖(スクロース、マンニトール、ラクトースなど)、および塩(塩化ナトリウム、クエン酸ナトリウムなど)、可塑剤(アセチル化クエン酸化エステル、アセチル化グリセリド、ヒマシ油、クエン酸エステル、セバジン酸ジブチル、モノステアリン酸グリセリル、フタル酸ジエチル、グリセロール、中鎖トリグリセリド、プロピレングリコール、ポリエチレングリコールなど)も含まれ得る。持続放出コーティングには、1つ以上のさらなる賦形剤(潤滑剤(例えば、ステアリン酸マグネシウム、タルクなど)など)も含まれ得る。
【0112】
持続放出コーティングを、従来のコーティング技術(流動化層コーティング、パンコーティングなど)を使用して適用することができる。任意選択的にシールコートを含む薬物コーティングしたペレットコアを、流動化層コーティングによって持続放出コーティングでコーティングする。
【0113】
持続放出賦形剤(コーティングポリマー)
本明細書中に記載のように、例示的な持続放出賦形剤には、不溶性プラスチック、親水性ポリマー、および脂肪族化合物が含まれるが、これらに限定されない。プラスチック母材には、メチルアクリラート−メチルメタクリラート、ポリ塩化ビニル、およびポリエチレンが含まれるが、これらに限定されない。親水性ポリマーには、セルロースポリマー(メチルセルロースおよびエチルセルロースなど)、ヒドロキシアルキルセルロース(ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、カルボキシメチルセルロースナトリウムなど)、および架橋アクリル酸ポリマー(カーボポール(登録商標)934、ポリエチレンオキシドなど)、およびその混合物が含まれるが、これらに限定されない。脂肪族化合物には、種々のワックス(カルナウバ蝋およびトリステアリン酸グリセリルなど)およびワックス型物質(硬化ヒマシ油または硬化植物油が含まれる)、またはその混合物が含まれるが、これらに限定されない。
【0114】
一定の実施形態では、可塑性物質は、薬学的に許容され得るアクリルポリマー(アクリル酸およびメタクリル酸コポリマー、メチルメタクリラート、メチルメタクリラートコポリマー、エトキシエチルメタクリラート、シアノエチルメタクリラート、アミノアルキルメタクリラートコポリマー、ポリ(アクリル酸)、ポリ(メタクリル酸)、メタクリル酸 アルキルアミンコポリマーポリ(メチルメタクリラート)、ポリ(メタクリル酸)(無水物)、ポリメタクリラート、ポリアクリルアミド、ポリ(メタクリル酸無水物)、およびグリシジルメタクリラートコポリマーが含まれるが、これらに限定されない)であり得る。
【0115】
一定の他の実施形態では、アクリルポリマーは、1つ以上のアンモニオメタクリラートコポリマーから構成される。アンモニオメタクリラートコポリマーは当該分野で周知であり、アクリル酸エステルおよびメタクリル酸エステルの低含有量の第四級アンモニウム基との完全重合したコポリマーとしてNF XVIIに記載されている。
【0116】
さらなる他の実施形態では、アクリルポリマーは、アクリル樹脂ラッカー(Rohm Pharmaから商標名オイドラギット(登録商標)で市販されているものなど)である。さらなる実施形態では、アクリルポリマーは、Rohm Pharmaからそれぞれ商標名オイドラギット(登録商標)RL30Dおよびオイドラギット(登録商標)RS30Dで市販されている2つのアクリル樹脂ラッカーの混合物を含む。オイドラギット(登録商標)RL30Dおよびオイドラギット(登録商標)RS30Dは、アクリル酸エステルおよびメタクリル酸エステルの低含有量の第四級アンモニウム基とのコポリマーであり、アンモニウム基と中性(メタクリル酸)アクリル酸エステルとのモル比がオイドラギットRL30Dでは1:20であり、オイドラギット(登録商標)RS30Dでは1:40である。平均分子量は約150,000である。オイドラギット(Edragit)(登録商標)S−100およびオイドラギット(登録商標)L−100も本明細書中の使用に適切である。コード名RL(高透過性)およびRS(低透過性)は、これらの薬剤の透過性をいう。オイドラギット(登録商標)RL/RS混合物は、水および消化液に不溶性を示す。しかし、これを含むように形成された多粒子系は、水溶液および消化液中で膨潤性および透過性を示す。
【0117】
上記のポリマー(オイドラギット(登録商標)RL/RSなど)を、最終的に所望の溶解プロフィールを有する持続放出処方物を得るために、任意の所望の比で混合することができる。当業者は、他のアクリルポリマー(例えば、オイドラギット(登録商標)Lなど)も使用することができると認識するであろう。
【0118】
ポア形成物質
いくつかの実施形態では、持続放出コーティングは、ポア形成物質を含む。持続放出コーティングでの使用に適切なポア形成物質は、有機または無機の薬剤であり得、使用環境下でコーティングから溶解、抽出、または浸出することができる材料が含まれる。ポア形成物質の例には、有機化合物(モノサッカリド、オリゴサッカリド、およびポリサッカリド(スクロース、グルコース、フルクトース、マンニトール、マンノース、ガラクトース、ラクトース、ソルビトール、プルラン、デキストランなど);水溶性親水性ポリマーなどの使用環境下で溶解性を示すポリマー(ポビドン、クロスポビドン、ポリエチレングリコール、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ヒドロキシアルキルセルロース、カルボキシアルキルセルロース、セルロースエーテル、アクリル樹脂、ポリビニルピロリドン、架橋ポリビニルピロリドン、ポリエチレンオキシド、カーボワックス、およびカーボポール(登録商標)など)、ジオール、ポリオール、多価アルコール、ポリアルキレングリコール、ポリエチレングリコール、ポリプロピレングリコール、またはそのブロック重合体、ポリグリコール、ポリ(α−Ω)アルキレンジオールなど);無機化合物(アルカリ金属塩、炭酸リチウム、塩化ナトリウム、臭化ナトリウム、塩化カリウム、硫酸カリウム、リン酸カリウム、酢酸ナトリウム、クエン酸ナトリウム、適切なカルシウム塩など)など)が含まれるが、これらに限定されない。一定の実施形態では、可塑剤を、ポア形成物質として使用することもできる。
【0119】
カプセル
持続放出ペレットを、ペレット投薬チャンバーを備えたエンカプスレータの使用によって適切なカプセルに導入する。カプセルサイズは、00、0、0EL、1、1EL、2、2EL、3、4、または5であり得る。理想的な薬物動態学的性質および血漿濃度プロフィールを提供する特に好ましい組成物は、典型的には直径が約500μm〜1.2mm、好ましくは約700μm〜1000μmの複数のペレットを含み、各ペレットがアマンタジンおよび結合剤、ならびに上記の所望の薬物動態学的性質およびアマンタジン血漿濃度プロフィールを得るためにアマンタジン放出を持続させるコアを囲む持続放出コーティングを含む、ペレット・イン・カプセル組成物である。
【0120】
いくつかの実施形態では、ペレット・イン・カプセル中のペレットは、サイズ0以下、好ましくはサイズ1以下のカプセルである。平均ペレット直径は、いくつかの実施形態では、500μm〜1200μmの範囲(例えば、500μm〜1100μm、500μm〜1000μm、500μm〜900μm、500μm〜800μm、500μm〜700μm、600μm〜1100μm、600μm〜1000μm、600μm〜900μm、600μm〜800μm、600μm〜700μm、700μm〜1100μm、700μm〜1000μm、700μm〜900μm、または700μm〜800μm)であり得る。いくつかの実施形態では、平均粒子直径は、±10%(例えば、500μm、550μm、600μm、650μm、700μm、750μm、800μm、850μm、900μm、950μm、1000μm、1050μm、1100μm、1150μm、または1200μm)である。
【0121】
本発明の1つの好ましい組成物は、ペレット・イン・カプセル組成物であって、各ペレットがコアシード上にコーティングしたアマンタジンと結合剤との混合物を有するコアシードを含むコア、ならびにエチルセルロース、ポア形成剤(ヒドロキシプロピルメチルセルロースまたはポビドンなど)、および可塑剤を含むコアを囲む持続放出コーティングを含む、ペレット・イン・カプセル組成物である。いくつかの実施形態では、ペレットは、ペレットコアと持続放出コーティングとの間にシールコーティングをさらに含むことができる。ペレットを、当該分野で公知の方法(以下の実施例1に記載の方法など)を使用して処方する。特定の実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、アマンタジンは、20〜80重量%、45〜70重量%、40〜50重量%、45〜55重量%、50〜60重量%、55〜65重量%、60〜70重量%、65〜75重量%、70〜80重量%、または40〜60重量%の量で存在し、結合剤(好ましくはヒドロキシプロピルメチルセルロース、コポビドン、またはその混合物である)は、8〜25重量%の量で存在し、コアシード(好ましくは、糖スフェア(ノンパレイユ)または微結晶性セルロースシード(例えば、セルフィア(登録商標)))は、1〜25重量%の量で存在し、エチルセルロースは、10〜20重量%の量で存在し、ポア形成剤(好ましくは、ポビドン)は、1〜4重量%の量で存在し、可塑剤は、1〜4重量%の量で存在する。別の特定の実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、アマンタジンは、50〜70重量%の量で存在し、結合剤(好ましくは、ヒドロキシプロピルメチルセルロース、コポビドン、またはその混合物である)は、5〜25重量%の量で存在し、コアシード(好ましくは、糖スフェア(ノンパレイユ)または微結晶性セルロースシード(例えば、セルフィア(登録商標)))は、5〜15重量%の量で存在し、エチルセルロースは、1〜15重量%の量で存在し、ポア形成剤(好ましくは、ポビドン)は、0.25〜4重量%の量で存在し、可塑剤は、0.25〜4重量%の量で存在する。
【0122】
本発明のさらなる実施形態を、以下の表に「種々のアマンタジンERカプセルサイズ1処方物」というタイトルで示す。本明細書中に記載の方法および組成物を用いて、本明細書中に記載の所望の溶解性および標的薬物動態学的プロフィールを達成する処方物を作製することができる。より具体的には、これらの結果を得るために本明細書中に記載している製造方法および組成物を使用して、治療有効用量のアマンタジンをわずか2つのサイズ1(またはそれ未満(例えば、サイズ2または3))カプセルにて1日1回投与することができる。特に、本明細書中に記載の組成物および製造方法を使用して、より高い薬物負荷を達成することができる。いくつかの実施形態では、より小さなコアペレットサイズを使用して必要な溶解プロフィールを有し、同時により小さなコア上の薬物層を増加させるが、持続放出コートが変化しないというより高い薬物負荷を達成することができる。いくつかの実施形態では、本明細書中に記載の別の製造アプローチ(例えば、押し出しおよび球形化)を使用して、所望の溶解プロフィールを実現するためのさらにより高い薬物負荷を達成することができ、それにより、適切な薬物動態学的プロフィールにてより高いアマンタジン薬負荷が可能であり、その結果、治療的により有効であり、少なくとも同様に許容され、比較的小さなサイズのカプセル(例えば、サイズ1、2、または3)に充填することができ、患者への投与を容易にすることができる組成物が得られる。
【0123】
【表1A】

いくつかの実施形態では、アマンタジンまたはその薬学的に許容され得る塩は、20〜80重量%の量(ペレットコアおよび持続放出コーティングを合わせた重量に基づく)、嵩密度0.3〜1.2g/cm3で存在する。いくつかの実施形態では、アマンタジンまたはその薬学的に許容され得る塩は、20〜77.5重量%、20〜75重量%、20〜72.5重量%、20〜70重量%、20〜67.5重量%、20〜65重量%、20〜62.5重量%、20〜60重量%、20〜57.5重量%、20〜55重量%、20〜52.5重量%、20〜50重量%、20〜47.5重量%、20〜45重量%、20〜42.5重量%、20〜40重量%、20〜37.5重量%、20〜35重量%、20〜32.5重量%、20〜30重量%、30〜80重量%、30〜77.5重量%、30〜75重量%、30〜72.5重量%、30〜70重量%、30〜67.5重量%、30〜65重量%、30〜62.5重量%、30〜60重量%、30〜57.5重量%、30〜55重量%、30〜52.5重量%、30〜50重量%、30〜47.5重量%、30〜45重量%、30〜42.5重量%、30〜40重量%、40〜80重量%、40〜77.5重量%、40〜75重量%、40〜72.5重量%、40〜70重量%、40〜67.5重量%、40〜65重量%、40〜62.5重量%、40〜60重量%、40〜57.5重量%、40〜55重量%、40〜52.5重量%、40〜50重量%、40〜47.5重量%、40〜45重量%、50〜80重量%、50〜77.5重量%、50〜75重量%、50〜72.5重量%、50〜70重量%、50〜67.5重量%、50〜65重量%、50〜62.5重量%、50〜60重量%、50〜57.5重量%、50〜55重量%、60〜80重量%、60〜77.5重量%、60〜75重量%、60〜72.5重量%、60〜70重量%、60〜67.5重量%、60〜65重量%の量で存在する。いくつかの実施形態では、嵩密度は、0.3〜1.2g/cm3、0.3〜1.15g/cm3、0.3〜1.1g/cm3、0.3〜1.05g/cm3、0.3〜1.0g/cm3、0.3〜0.9g/cm3、0.3〜0.8g/cm3、0.3〜0.7g/cm3、0.3〜0.6g/cm3、0.3〜0.5g/cm3、0.3〜0.4g/cm3、0.4〜1.2g/cm3、0.4〜1.15g/cm3、0.4〜1.1g/cm3、0.4〜1.05g/cm3、0.4〜1.0g/cm3、0.4〜0.9g/cm3、0.4〜0.8g/cm3、0.4〜0.7g/cm3、0.4〜0.6g/cm3、0.4〜0.5g/cm3、0.5〜1.2g/cm3、0.5〜1.15g/cm3、0.5〜1.1g/cm3、0.5〜1.05g/cm3、0.5〜1.0g/cm3、0.5〜0.9g/cm3、0.5〜0.8g/cm3、0.5〜0.7g/cm3、0.5〜0.6g/cm3、0.6〜1.2g/cm3、0.6〜1.15g/cm3、0.6〜1.1g/cm3、0.6〜1.05g/cm3、0.6〜1.0g/cm3、0.6〜0.9g/cm3、0.6〜0.8g/cm3、0.6〜0.7g/cm3、0.7〜1.2g/cm3、0.7〜1.15g/cm3、0.7〜1.1g/cm3、0.7〜1.05g/cm3、0.7〜1.0g/cm3、0.7〜0.9g/cm3、0.7〜0.8g/cm3、0.5〜1.2g/cm3、0.8〜1.15g/cm3、0.8〜1.1g/cm3、0.8〜1.05g/cm3、0.8〜1.0g/cm3、0.8〜0.9g/cm3、0.9〜1.2g/cm3、0.9〜1.15g/cm3、0.9〜1.1g/cm3、0.9〜1.05g/cm3、または0.9〜1.0g/cm3である。いくつかの実施形態では、組成物は、カプセル処方物中にペレットを含む投薬単位で存在し、カプセルサイズは、サイズ00、サイズ0、サイズ1、サイズ2、またはサイズ3である。いくつかの好ましい実施形態では、投薬単位は、サイズ0、1、2、または3カプセル中に50〜250mgのアマンタジンを含むペレットを含む。いくつかの実施形態では、投薬単位は、サイズ0、1、2、または3カプセル、好ましくはサイズ1、2、または3カプセル中に100〜250mg(例えば、100〜200mg)のアマンタジンを含むペレットを含む。さらなる特定の実施形態では、投薬単位は、約110、120、130、140、150、160 170、180、190、210、または220mgのアマンタジンまたはその薬学的に許容され得る塩を含む。別のさらなる特定の実施形態では、投薬単位は、110mg塩酸アマンタジンを含む。別のさらなる特定の実施形態では、投薬単位は、130mg塩酸アマンタジンを含む。別のさらなる特定の実施形態では、投薬単位は、170mg塩酸アマンタジンを含む。別のさらなる特定の実施形態では、投薬単位は、210mg塩酸アマンタジンを含む。
【0124】
適切な可塑剤には、中鎖トリグリセリド、フタル酸ジエチル、クエン酸エステル、ポリエチレングリコール、グリセロール、アセチル化グリセリド、およびヒマシ油などが含まれる。ペレットを、所望のアマンタジン強度が得られるようにカプセルに充填する。この組成物の利点は、就寝時刻前の投与に適切な組成物を作製する所望の放出特性が得られることである。さらなる利点は、持続放出コーティングが、組成物の放出特性に悪影響を及ぼすことなく丸薬の嚥下が困難な患者への投与のためにカプセルを開け、ペレットを食品上に振りかけることができるような十分な耐久性があることである。組成物を食品上に振りかけることによって投与する場合、30分以内、好ましくは15分以内に消費される軟らかい食品(アップルソースまたはチョコレートプディングなど)を使用することが好ましい。上記組成物のなおさらなる利点は、バッチ毎の再現性および貯蔵安定性が非常に良好であることである。
【0125】
いくつかの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての測定した場合、本発明の組成物は、2時間で25%以下、6時間で40〜80%、12時間で少なくとも80%のアマンタジンのin vitro溶解プロフィールを有する。より好ましくは、in vitro溶解は、1時間で10%以下、4時間で30〜50%、および12時間で少なくとも90%のアマンタジン放出によってさらに特徴づけられる。
【0126】
さらなる実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての測定した場合、本発明の組成物のサイズ1カプセル中の110mg〜210mgのERアマンタジンは、2時間で25%以下、6時間で40〜80%、12時間で少なくとも80%のアマンタジンのin vitro溶解プロフィールを有する。より好ましくは、in vitro溶解は、1時間で10%以下、4時間で30〜50%、および12時間で少なくとも90%のアマンタジン放出によってさらに特徴づけられる。
【0127】
任意の上記態様の1つの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールが、以下のうちの少なくとも1つを示す:(i)2時間で25%以下の溶解、(ii)6時間で25〜55%以下の溶解、および(iii)12時間で少なくとも80%の溶解。より特定の実施形態では、基準(i)、(ii)、および(iii)のうちの2つを満たす。さらなる特定の実施形態では、基準(i)、(ii)、および(iii)の3つ全てを満たす。
【0128】
任意の上記態様の1つの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールが、以下のうちの少なくとも1つを示す:(i)1時間で20%以下の溶解、(ii)2時間で約25〜45%の溶解、(iii)4時間で50〜80%以下の溶解、および(iii)8時間で少なくとも80%の溶解。より特定の実施形態では、基準(i)、(ii)、および(iii)のうちの2つを満たす。さらなる特定の実施形態では、基準(i)、(ii)、および(iii)の3つ全てを満たす。
【0129】
本発明の好ましいペレット・イン・カプセル組成物は、上記のin vitro溶解性を有することに加えて、前述の就寝時刻前の投与に適切な組成物を作製する上記の薬物動態学的性質(例えば、in vivo放出プロフィール、Tmax、Cmax/Cmin比など)のいずれかを有する。組成物は、単回用量のカプセルの絶食状態のヒト被験体への経口投与後に1mgのアマンタジンあたり1.6〜2.4ng/mlのCmaxおよび1mgのアマンタジンあたり40〜75ng*h/mLのAUC0−infが得られることによってさらに特徴づけられる。好ましいペレット・イン・カプセル組成物は、カプセルのヒト被験体への1日1回の経口投与によって1mgのアマンタジンあたり2.4〜4.2ng/mlのCmax、1mgのアマンタジンあたり1.1〜2.6ng/mlのCmin、および1mgのアマンタジンあたり48〜73ng*h/mLのAUC0−24が得られるという定常状態の血漿濃度によってさらに特徴づけられる。
【0130】
上記ペレット・イン・カプセル組成物を、アマンタジン治療に適切な強度で提供することができる。典型的な強度は、少なくとも約50mg〜約250mgの範囲である。特定の実施形態では、カプセル強度は、70mg、80mg、90mg、110mg、120mg、125mg、130mg、140mg、150mg、160mg、160mg、170mg、180mg、190mg、210mg、および220mgであり、これらにより、アマンタジンHClの即時放出処方物の100mg錠剤(例えば、シンメトレル(登録商標)または他のFDAオレンジブックリファレンス掲載薬物)に等しい単回用量AUC0−inf/mgが得られる。1、2、または3つのかかるカプセルを、就寝時刻前に被験体に投与することができる。好ましい実施形態では、220mgと650mgとの間のアマンタジンを、2カプセルの適切なER処方物を使用して1日1回投与する。
【0131】
本発明を、以下の番号をつけた実施形態に関して記載することもできる。
1.アマンタジンを必要とする被験体に投与する方法で用いるアマンタジンまたはその薬学的に許容され得る塩を含む持続放出(ER)組成物であって、方法が就寝時刻より前(すなわち、被験体が夜に就寝を望む時間)の3時間未満に組成物を経口投与する工程を含む、組成物。
2.必要とする被験体に対するNMDA受容体によって媒介される疾患の処置のための医薬の製造におけるアマンタジンまたはその薬学的に許容され得る塩の使用であって、医薬が持続放出(ER)組成物であり、処置が就寝時刻より前(すなわち、被験体が夜に就寝を望む時間)の3時間未満に組成物を経口投与する工程を含む、使用。
3.アマンタジンでの処置を受けるヒト被験体における睡眠障害の軽減方法で用いるアマンタジンまたはその薬学的に許容され得る塩を含む持続放出(ER)組成物であって、方法が就寝時刻より前(すなわち、被験体が夜に就寝を望む時間)の3時間未満に組成物を投与する工程を含む、組成物。
4.アマンタジンでの処置を受けるヒト被験体における睡眠障害の軽減のための医薬の製造におけるアマンタジンまたはその薬学的に許容され得る塩の使用であって、医薬が持続放出(ER)組成物であり、就寝時刻より前(すなわち、被験体が夜に就寝を望む時間)の3時間未満の投与に適合している、使用。
5.就寝時刻より前の1時間未満に投与する、実施形態1から4のいずれか1つに記載の使用または組成物。
6.患者がパーキンソン病と診断されている、実施形態1から5のいずれか1つに記載の使用または組成物。
7.組成物を1日1回投与する、実施形態1から6のいずれか1つに記載の使用または組成物。
8.組成物を投与前に食品に添加する、実施形態1から7のいずれか1つに記載の使用または組成物。
9.アマンタジンの血漿濃度が、定常状態での投与後少なくとも1時間増加しない、実施形態1から8のいずれか1つに記載の使用または組成物。
10.アマンタジンの血漿濃度が、定常状態での投与後少なくとも2時間増加しない、実施形態1から9のいずれか1つに記載の使用または組成物。
11.投与後のアマンタジンの単回用量のTmaxが9〜15時間であり、そして/または定常状態のTmaxが7〜13時間である、実施形態1から10のいずれか1つに記載の組成物の使用。
12.アマンタジンの単回用量のTmaxが投与後10〜14時間であり、そして/または定常状態のTmaxが投与後8〜12時間である、実施形態1から11のいずれか1つに記載の使用または組成物。
13.アマンタジンの単回用量のTmaxが投与後9〜15時間であり、そして/または定常状態のTmaxが7〜13時間である、実施形態1から10のいずれか1つに記載の組成物の使用。
14.アマンタジンの単回用量のTmaxが投与後10〜14時間であり、そして/または定常状態のTmaxが投与後8〜12時間である、実施形態1から11のいずれか1つに記載の使用または組成物。
15.アマンタジンの単回用量のTmaxが投与後9〜15時間であり、そして/または定常状態のTmaxが7〜13時間である、実施形態1から10のいずれか1つに記載の組成物の使用。
16.アマンタジンの単回用量のTmaxが投与後10〜14時間であり、そして/または定常状態のTmaxが投与後8〜12時間である、実施形態1から11のいずれか1つに記載の使用または組成物。
17.アマンタジンの単回用量のTmaxが投与後11〜13時間であり、そして/または定常状態のTmaxが投与後9〜11時間である、実施形態1から12のいずれか1つに記載の使用または組成物。
18.ヒト被験体への組成物の1日1回の経口投与により、投与3時間後の25%未満のアマンタジン濃度の増加によって特徴付けられる定常状態血漿濃度プロフィールが得られる、実施形態1から13のいずれか1つに記載の使用または組成物。
19.Cmax/Cmin比が1.5〜2.0である、実施形態1から14のいずれか1つに記載の使用または組成物。
20.Cmax/Cmin比が1.7〜1.9である、実施形態1から15のいずれか1つに記載の使用または組成物。
21.アマンタジンが塩酸アマンタジンまたは硫酸アマンタジンである、実施形態1から16のいずれか1つに記載の使用または組成物。
22.組成物が50〜600mgのアマンタジンまたはその薬学的に許容され得る塩を含む、実施形態1から17のいずれか1つに記載の使用または組成物。
23.組成物を、それぞれ100〜175mgのアマンタジンまたはその薬学的に許容され得る塩を含む1つ、2つ、3つ、または4つの単位投薬形態として投与する、実施形態18に記載の使用または組成物。
24.組成物が200〜420mgのアマンタジンまたはその薬学的に許容され得る塩を含む、実施形態1から19のいずれか1つに記載の使用または組成物。
25.組成物を、110〜175mgのアマンタジンまたはその薬学的に許容され得る塩をそれぞれ含む2つの単位投薬形態として投与する、実施形態20に記載の使用または組成物。
26.組成物が50〜200mgのアマンタジンまたはその薬学的に許容され得る塩を含む、実施形態1から17のいずれか1つに記載の使用または組成物。
27.組成物が100〜125mgのアマンタジンまたはその薬学的に許容され得る塩を含む、実施形態22に記載の使用または組成物。
28.組成物が110mgの塩酸アマンタジンを含む、実施形態23に記載の使用または組成物。
29.単回用量の組成物の絶食状態のヒト被験体への経口投与により、1mgのアマンタジンあたり1.6〜2.4ng/mlのアマンタジンの最大血漿濃度(Cmax)および1mgのアマンタジンあたり40〜75ng*h/mLのAUC0−infが得られる、実施形態1から24のいずれか1つに記載の使用または組成物。
30.ヒト被験体へのある用量の組成物の1日1回の経口投与により、以下:
(i)1mgのアマンタジンあたり2.4〜4.2ng/mlのCmax、
(ii)1mgのアマンタジンあたり1.1〜2.6ng/mlのCmin、および
(iii)1mgのアマンタジンあたり44〜83ng*h/mLのAUC0−24
によって特徴付けられる定常状態血漿アマンタジン濃度プロフィールが得られる、実施形態1から25のいずれか1つに記載の使用または組成物。
31.定常状態血漿濃度プロフィールが、以下:
(iv)投与後少なくとも1時間アマンタジンの血漿濃度が増加しないこと、および
(v)Cmax/Cmin比1.5〜2.0
によってさらに特徴づけられる、実施形態26に記載の使用または組成物。
32.定常状態血漿濃度プロフィールが、以下:
(iv)投与後少なくとも2時間アマンタジン濃度が増加しないこと、および
(v)Cmax/Cmin比1.7〜1.9
によってさらに特徴づけられる、実施形態27に記載の使用または組成物。
33.溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールが、2時間で25%以下、6時間で55〜85%、および12時間で少なくとも80%である、実施形態1から28のいずれか1つに記載の使用または組成物。
34.アマンタジンのin vitro溶解プロフィールが、1時間で10%以下、4時間で30〜50%、および12時間で少なくとも90%のアマンタジンの放出によってさらに特徴づけられる、実施形態29に記載の使用または組成物。
35.単回用量の組成物の投与後の組成物のAUCプロフィールが、以下:AUC0−infの5%未満である0〜4時間の断片的AUC、AUC0−infの約5〜15%である0〜8時間の断片的AUC、AUC0−infの約10〜40%である0〜12時間の断片的AUC、AUC0−infの約25〜60%である0〜18時間の断片的AUC、およびAUC0−infの約40〜75%である0〜24時間の断片的AUC
によって特徴付けられる、実施形態1から30のいずれか1つに記載の使用または組成物。
36.定常状態条件での組成物の1日1回の投与後の組成物のAUCプロフィールが、以下:AUC24の約2〜25%である0〜4時間の断片的AUC、AUC24の約15〜50%である0〜8時間の断片的AUC、AUC24の約30〜70%である0〜12時間の断片的AUC、およびAUC24の約60〜95%である0〜18時間の断片的AUCによって特徴づけられる、実施形態1から31のいずれか1つに記載の使用または組成物。
37.実施形態1、3、または5から32のいずれか1つに具体化した薬学的組成物または実施形態2、4、または5から32のいずれか1つの使用であって、組成物が経口投与用であり、経口投与用カプセルを含み、カプセルが複数のペレットを含み、各ペレットが、以下:
(a)アマンタジンまたはその薬学的に許容され得る塩を含むペレットコア、および
(b)ペレットコアを取り囲む持続放出コーティング
を含む、薬学的組成物または使用。
38.持続放出コーティングが、エチルセルロース、ならびにポビドンおよびヒドロキシプロピルメチルセルロースのうちの少なくとも1つ、ならびに可塑剤を含む、実施形態32に記載の使用または組成物。
39.ペレットコアが、コアシード上にコーティングしたアマンタジンまたはその薬学的に許容され得る塩および結合剤を含む、実施形態33または34のいずれか1つに記載の使用または組成物。
40.ペレットコアと持続放出コーティングとを合わせた重量に基づいて、アマンタジンが40〜60重量%の量で存在し、結合剤が8〜25重量%の量で存在し、コアシードが8〜25重量%の量で存在し、エチルセルロースが10〜20重量%の量で存在し、ポビドンが1〜4重量%の量で存在し、可塑剤が1〜4重量%の量で存在する、実施形態35に記載の使用または組成物。
41.ペレットコアと持続放出コーティングとの間にシールコーティングをさらに含む、実施形態33から36のいずれか1つに記載の使用または組成物。
42.ペレットコアが、ヒドロキシプロピルメチルセルロース、コポビドン、およびその混合物からなる群から選択される結合剤を含む、実施形態35から37のいずれか1つに記載の使用または組成物。
43.可塑剤が、中鎖トリグリセリド、フタル酸ジエチル、クエン酸エステル、ポリエチレングリコール、グリセロール、アセチル化グリセリド、およびヒマシ油からなる群から選択される、実施形態42から38のいずれか1つに記載の使用または組成物。
44.組成物を経口投与する工程を含む、必要とするヒト被験体におけるパーキンソン病の処置方法で用いる実施形態33から39のいずれか1つに記載の組成物。
【0132】
本明細書中のいくつかの実施形態は、アマンタジンを必要とする被験体に投与する方法であって、アマンタジンまたはその薬学的に許容され得る塩を含む持続放出(ER)組成物を就寝時刻より前の3時間未満に経口投与する工程を含む、方法を提供する。いくつかの実施形態では、就寝時刻より前の1時間未満に投与する。いくつかの実施形態では、患者はパーキンソン病と診断されている。いくつかの実施形態では、組成物を1日1回投与する。いくつかの実施形態では、組成物を投与前に食品に添加する。いくつかの実施形態では、アマンタジンの血漿濃度は、投与後少なくとも1時間増加しない。いくつかの実施形態では、アマンタジンの血漿濃度は、投与後少なくとも2時間増加しない。いくつかの実施形態では、アマンタジンの単回用量のTmaxは9〜15時間であり、そして/または定常状態のTmaxは7〜13時間である。いくつかの実施形態では、アマンタジンの単回用量のTmaxは投与後10〜14時間であり、そして/または定常状態のTmaxは8〜12時間である。いくつかの実施形態では、アマンタジンの単回用量のTmaxは投与後11〜13時間であり、そして/または定常状態のTmaxは9〜11時間である。いくつかの実施形態では、ヒト被験体への組成物の1日1回の経口投与により、投与3時間後の25%未満のアマンタジン濃度の増加によって特徴付けられる定常状態血漿濃度プロフィールが得られる。いくつかの実施形態では、PK曲線のCmax/Cmin比は1.5〜2.0である。いくつかの実施形態では、PK曲線のCmax/Cmin比は1.7〜1.9である。いくつかの実施形態では、定常状態でのC−ave−日中/C−ave−夜比は1.2〜1.6である。いくつかの実施形態では、定常状態でのC−ave−朝/C−ave−夜比は1.3〜1.5である。いくつかの実施形態では、定常状態での日中の平均アマンタジン血漿濃度(C−ave−日中)は500〜2000ng/mlである。いくつかの実施形態では、定常状態での朝の平均アマンタジン血漿濃度(C−ave−朝)は500〜2000ng/mlである。いくつかの実施形態では、アマンタジンは塩酸アマンタジンまたは硫酸アマンタジンである。いくつかの実施形態では、組成物は50〜600mgのアマンタジンまたはその薬学的に許容され得る塩を含む。いくつかの実施形態では、組成物を、それぞれ100〜175mgのアマンタジンまたはその薬学的に許容され得る塩を含む1つ、2つ、3つ、または4つの単位投薬形態として投与する。いくつかの実施形態では、組成物を、それぞれ130〜210mgの持続放出アマンタジンまたはその薬学的に許容され得る塩を含む1つまたは2つの単位投薬形態として投与する。いくつかの実施形態では、組成物は、カプセルサイズ番号1のカプセル内に存在する。いくつかの実施形態では、組成物は、200〜350mgのアマンタジンまたはその薬学的に許容され得る塩を含む。いくつかの実施形態では、組成物を、それぞれ100〜175mgのアマンタジンまたはその薬学的に許容され得る塩を含む2つの単位投薬形態として投与する。いくつかの実施形態では、組成物は、50〜200mgのアマンタジンまたはその薬学的に許容され得る塩を含む。いくつかの実施形態では、組成物は、100〜125mgのアマンタジンまたはその薬学的に許容され得る塩を含む。いくつかの実施形態では、組成物は110mgの塩酸アマンタジンを含む。いくつかの実施形態では、絶食状態のヒト被験体への単回用量の組成物の経口投与により、1mgのアマンタジンあたり1.6〜2.4ng/mlの最大血漿濃度(Cmax)、および1mgのアマンタジンあたり40〜75ng*h/mLのAUC0−infが得られる。いくつかの実施形態では、ヒト被験体へのある用量の組成物の1日1回の経口投与により、以下によって特徴付けられる定常状態血漿濃度プロフィールが得られる:(a)1mgのアマンタジンあたり2.4〜4.2ng/mlのCmax;(b)1mgのアマンタジンあたり1.1〜2.6ng/mlのCmin、および(c)1mgのアマンタジンあたり44〜83ng*h/mLのAUC0−24。いくつかの実施形態では、定常状態血漿濃度プロフィールは、以下によってさらに特徴づけられる:(d)投与後少なくとも1時間アマンタジンの血漿濃度が増加しないこと、および(e)Cmax/Cmin比1.5〜2.0。いくつかの実施形態では、定常状態血漿濃度プロフィールは、以下によってさらに特徴づけられる:(f)投与後少なくとも2時間アマンタジン濃度が増加しないこと、および(g)Cmax/Cmin比1.7〜1.9。いくつかの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールは、2時間で25%以下、6時間で55〜85%、および12時間で少なくとも80%である。いくつかの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールは、2時間で25%以下、6時間で25〜55%、および12時間で少なくとも80%である。いくつかの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールは、1時間で20%以下、2時間で25〜45%、4時間で50〜80%、および8時間で少なくとも80%である。いくつかの実施形態では、アマンタジンのin vitro溶解プロフィールは、1時間で10%以下、4時間で30〜50%、および12時間で少なくとも90%のアマンタジン放出によってさらに特徴付けられる。いくつかの実施形態では、単回用量の組成物の投与後の組成物のAUCプロフィールは、以下によって特徴付けられる:AUC0−infの5%未満である0〜4時間の断片的AUC、AUC0−infの約5〜15%である0〜8時間の断片的AUC、AUC0−infの約10〜40%である0〜12時間の断片的AUC、AUC0−infの約25〜60%である0〜18時間の断片的AUC、およびAUC0−infの約40〜75%である0〜24時間の断片的AUC。いくつかの実施形態では、定常状態条件での組成物の1日1回の投与後の組成物のAUCプロフィールは、以下によって特徴づけられる:AUC24の約2〜25%である0〜4時間の断片的AUC、AUC24の約15〜50%である0〜8時間の断片的AUC、AUC24の約30〜70%である0〜12時間の断片的AUC、およびAUC24の約60〜95%である0〜18時間の断片的AUC。
【0133】
本明細書中のいくつかの実施形態は、アマンタジンでの処置を受けるヒト被験体における睡眠障害の軽減方法であって、アマンタジンまたはその薬学的に許容され得る塩を含む持続放出(ER)組成物を就寝時刻より前の3時間未満に投与する工程を含む、方法を提供する。いくつかの実施形態では、就寝時刻より前の1時間未満に投与する。いくつかの実施形態では、患者はパーキンソン病と診断されている。いくつかの実施形態では、組成物を1日1回投与する。いくつかの実施形態では、組成物を投与前に食品に添加する。いくつかの実施形態では、アマンタジンの血漿濃度は、投与後少なくとも1時間増加しない。いくつかの実施形態では、アマンタジンの血漿濃度は、投与後少なくとも2時間増加しない。いくつかの実施形態では、アマンタジンの単回用量のTmaxは9〜15時間であり、そして/または定常状態のTmaxは7〜13時間である。いくつかの実施形態では、アマンタジンの単回用量のTmaxは投与後10〜14時間であり、そして/または定常状態のTmaxは8〜12時間である。いくつかの実施形態では、アマンタジンの単回用量のTmaxは投与後11〜13時間であり、そして/または定常状態のTmaxは9〜11時間である。いくつかの実施形態では、ヒト被験体への組成物の1日1回の経口投与により、投与3時間後の25%未満のアマンタジン濃度の増加によって特徴付けられる定常状態血漿濃度プロフィールが得られる。いくつかの実施形態では、PK曲線のCmax/Cmin比は1.5〜2.0である。いくつかの実施形態では、PK曲線のCmax/Cmin比は1.7〜1.9である。いくつかの実施形態では、定常状態でのC−ave−日中/C−ave−夜比は1.2〜1.6である。いくつかの実施形態では、定常状態でのC−ave−朝/C−ave−夜比は1.3〜1.5である。いくつかの実施形態では、定常状態での日中の平均アマンタジン血漿濃度(C−ave−日中)は500〜2000ng/mlである。いくつかの実施形態では、定常状態での朝の平均アマンタジン血漿濃度(C−ave−朝)は500〜2000ng/mlである。いくつかの実施形態では、アマンタジンは塩酸アマンタジンまたは硫酸アマンタジンである。いくつかの実施形態では、組成物は50〜600mgのアマンタジンまたはその薬学的に許容され得る塩を含む。いくつかの実施形態では、組成物を、それぞれ100〜175mgのアマンタジンまたはその薬学的に許容され得る塩を含む1つ、2つ、3つ、または4つの単位投薬形態として投与する。いくつかの実施形態では、組成物を、それぞれ130〜210mgの持続放出アマンタジンまたはその薬学的に許容され得る塩を含む1つまたは2つの単位投薬形態として投与する。いくつかの実施形態では、組成物は、カプセルサイズ番号1のカプセル内に存在する。いくつかの実施形態では、組成物は、200〜350mgのアマンタジンまたはその薬学的に許容され得る塩を含む。いくつかの実施形態では、組成物を、それぞれ100〜175mgのアマンタジンまたはその薬学的に許容され得る塩を含む2つの単位投薬形態として投与する。いくつかの実施形態では、組成物は、50〜200mgのアマンタジンまたはその薬学的に許容され得る塩を含む。いくつかの実施形態では、組成物は、100〜125mgのアマンタジンまたはその薬学的に許容され得る塩を含む。いくつかの実施形態では、組成物は110mgの塩酸アマンタジンを含む。いくつかの実施形態では、絶食状態のヒト被験体への単回用量の組成物の経口投与により、1mgのアマンタジンあたり1.6〜2.4ng/mlの最大血漿濃度(Cmax)、および1mgのアマンタジンあたり40〜75ng*h/mLのAUC0−infが得られる。いくつかの実施形態では、ヒト被験体へのある用量の組成物の1日1回の経口投与により、以下によって特徴付けられる定常状態血漿濃度プロフィールが得られる:(a)1mgのアマンタジンあたり2.4〜4.2ng/mlのCmax;(b)1mgのアマンタジンあたり1.1〜2.6ng/mlのCmin、および(c)1mgのアマンタジンあたり44〜83ng*h/mLのAUC0−24。いくつかの実施形態では、定常状態血漿濃度プロフィールは、以下によってさらに特徴づけられる:(d)投与後少なくとも1時間アマンタジンの血漿濃度が増加しないこと、および(e)Cmax/Cmin比1.5〜2.0。いくつかの実施形態では、定常状態血漿濃度プロフィールは、以下によってさらに特徴づけられる:(f)投与後少なくとも2時間アマンタジン濃度が増加しないこと、および(g)Cmax/Cmin比1.7〜1.9。いくつかの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールは、2時間で25%以下、6時間で55〜85%、および12時間で少なくとも80%である。いくつかの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールは、2時間で25%以下、6時間で25〜55%、および12時間で少なくとも80%である。いくつかの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールは、1時間で20%以下、2時間で25〜45%、4時間で50〜80%、および8時間で少なくとも80%である。いくつかの実施形態では、アマンタジンのin vitro溶解プロフィールは、1時間で10%以下、4時間で30〜50%、および12時間で少なくとも90%のアマンタジン放出によってさらに特徴付けられる。いくつかの実施形態では、単回用量の組成物の投与後の組成物のAUCプロフィールは、以下によって特徴付けられる:AUC0−infの5%未満である0〜4時間の断片的AUC、AUC0−infの約5〜15%である0〜8時間の断片的AUC、AUC0−infの約10〜40%である0〜12時間の断片的AUC、AUC0−infの約25〜60%である0〜18時間の断片的AUC、およびAUC0−infの約40〜75%である0〜24時間の断片的AUC。いくつかの実施形態では、定常状態条件での組成物の1日1回の投与後の組成物のAUCプロフィールは、以下によって特徴づけられる:AUC24の約2〜25%である0〜4時間の断片的AUC、AUC24の約15〜50%である0〜8時間の断片的AUC、AUC24の約30〜70%である0〜12時間の断片的AUC、およびAUC24の約60〜95%である0〜18時間の断片的AUC。
【0134】
本明細書中のいくつかの実施形態は、パーキンソン病患者におけるレボドパ誘発性ジスキネジアの処置方法であって、アマンタジンまたはその薬学的に許容され得る塩を含む持続放出(ER)組成物を就寝時刻より前の3時間未満に1日1回経口投与する工程を含む、方法を提供する。いくつかの実施形態では、就寝時刻より前の1時間未満に投与する。いくつかの実施形態では、患者はパーキンソン病と診断されている。いくつかの実施形態では、組成物を1日1回投与する。いくつかの実施形態では、組成物を投与前に食品に添加する。いくつかの実施形態では、アマンタジンの血漿濃度は、投与後少なくとも1時間増加しない。いくつかの実施形態では、アマンタジンの血漿濃度は、投与後少なくとも2時間増加しない。いくつかの実施形態では、アマンタジンの単回用量のTmaxは9〜15時間であり、そして/または定常状態のTmaxは7〜13時間である。いくつかの実施形態では、アマンタジンの単回用量のTmaxは投与後10〜14時間であり、そして/または定常状態のTmaxは8〜12時間である。いくつかの実施形態では、アマンタジンの単回用量のTmaxは投与後11〜13時間であり、そして/または定常状態のTmaxは9〜11時間である。いくつかの実施形態では、ヒト被験体への組成物の1日1回の経口投与により、投与3時間後の25%未満のアマンタジン濃度の増加によって特徴付けられる定常状態血漿濃度プロフィールが得られる。いくつかの実施形態では、PK曲線のCmax/Cmin比は1.5〜2.0である。いくつかの実施形態では、PK曲線のCmax/Cmin比は1.7〜1.9である。いくつかの実施形態では、定常状態でのC−ave−日中/C−ave−夜比は1.2〜1.6である。いくつかの実施形態では、定常状態でのC−ave−朝/C−ave−夜比は1.3〜1.5である。いくつかの実施形態では、定常状態での日中の平均アマンタジン血漿濃度(C−ave−日中)は500〜2000ng/mlである。いくつかの実施形態では、定常状態での朝の平均アマンタジン血漿濃度(C−ave−朝)は500〜2000ng/mlである。いくつかの実施形態では、アマンタジンは塩酸アマンタジンまたは硫酸アマンタジンである。いくつかの実施形態では、組成物は50〜600mgのアマンタジンまたはその薬学的に許容され得る塩を含む。いくつかの実施形態では、組成物を、それぞれ100〜175mgのアマンタジンまたはその薬学的に許容され得る塩を含む1つ、2つ、3つ、または4つの単位投薬形態として投与する。いくつかの実施形態では、組成物を、それぞれ130〜210mgの持続放出アマンタジンまたはその薬学的に許容され得る塩を含む1つまたは2つの単位投薬形態として投与する。いくつかの実施形態では、組成物は、カプセルサイズ番号1のカプセル内に存在する。いくつかの実施形態では、組成物は、200〜350mgのアマンタジンまたはその薬学的に許容され得る塩を含む。いくつかの実施形態では、組成物を、それぞれ100〜175mgのアマンタジンまたはその薬学的に許容され得る塩を含む2つの単位投薬形態として投与する。いくつかの実施形態では、組成物は、50〜200mgのアマンタジンまたはその薬学的に許容され得る塩を含む。いくつかの実施形態では、組成物は、100〜125mgのアマンタジンまたはその薬学的に許容され得る塩を含む。いくつかの実施形態では、組成物は110mgの塩酸アマンタジンを含む。いくつかの実施形態では、絶食状態のヒト被験体への単回用量の組成物の経口投与により、1mgのアマンタジンあたり1.6〜2.4ng/mlの最大血漿濃度(Cmax)、および1mgのアマンタジンあたり40〜75ng*h/mLのAUC0−infが得られる。いくつかの実施形態では、ヒト被験体へのある用量の組成物の1日1回の経口投与により、以下によって特徴付けられる定常状態血漿濃度プロフィールが得られる:(a)1mgのアマンタジンあたり2.4〜4.2ng/mlのCmax;(b)1mgのアマンタジンあたり1.1〜2.6ng/mlのCmin、および(c)1mgのアマンタジンあたり44〜83ng*h/mLのAUC0−24。いくつかの実施形態では、定常状態血漿濃度プロフィールは、以下によってさらに特徴づけられる:(d)投与後少なくとも1時間アマンタジンの血漿濃度が増加しないこと、および(e)Cmax/Cmin比1.5〜2.0。いくつかの実施形態では、定常状態血漿濃度プロフィールは、以下によってさらに特徴づけられる:(f)投与後少なくとも2時間アマンタジン濃度が増加しないこと、および(g)Cmax/Cmin比1.7〜1.9。いくつかの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールは、2時間で25%以下、6時間で55〜85%、および12時間で少なくとも80%である。いくつかの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールは、2時間で25%以下、6時間で25〜55%、および12時間で少なくとも80%である。いくつかの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールは、1時間で20%以下、2時間で25〜45%、4時間で50〜80%、および8時間で少なくとも80%である。いくつかの実施形態では、アマンタジンのin vitro溶解プロフィールは、1時間で10%以下、4時間で30〜50%、および12時間で少なくとも90%のアマンタジン放出によってさらに特徴付けられる。いくつかの実施形態では、単回用量の組成物の投与後の組成物のAUCプロフィールは、以下によって特徴付けられる:AUC0−infの5%未満である0〜4時間の断片的AUC、AUC0−infの約5〜15%である0〜8時間の断片的AUC、AUC0−infの約10〜40%である0〜12時間の断片的AUC、AUC0−infの約25〜60%である0〜18時間の断片的AUC、およびAUC0−infの約40〜75%である0〜24時間の断片的AUC。いくつかの実施形態では、定常状態条件での組成物の1日1回の投与後の組成物のAUCプロフィールは、以下によって特徴づけられる:AUC24の約2〜25%である0〜4時間の断片的AUC、AUC24の約15〜50%である0〜8時間の断片的AUC、AUC24の約30〜70%である0〜12時間の断片的AUC、およびAUC24の約60〜95%である0〜18時間の断片的AUC。
【0135】
本明細書中のいくつかの実施形態は、本明細書中に記載の方法のいずれかのための薬学的組成物であって、組成物が経口投与用であり、経口投与用カプセルを含み、カプセルが複数のペレットを含み、各ペレットは、以下:(a)アマンタジンまたはその薬学的に許容され得る塩を含むペレットコア、および(b)ペレットコアを取り囲む持続放出コーティングを含む、薬学的組成物を提供する。いくつかの実施形態では、持続放出コーティングは、エチルセルロース、ならびにポビドンおよびヒドロキシプロピルメチルセルロースのうちの少なくとも1つ、ならびに可塑剤を含む。いくつかの実施形態では、ペレットコアは、コアシード上にコーティングされたアマンタジンまたはその薬学的に許容され得る塩および結合剤を含む。いくつかの実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、アマンタジンは40〜60重量%の量で存在し、結合剤は1〜25重量%の量で存在し、コアシードは8〜25重量%の量で存在し、エチルセルロースは10〜20重量%の量で存在し、ポビドンは1〜4重量%の量で存在し、可塑剤は1〜4重量%の量で存在する。いくつかの実施形態では、組成物は、さらに、ペレットコアと持続放出コーティングとの間にシールコーティングを含む。いくつかの実施形態では、ペレットコアは、ヒドロキシプロピルメチルセルロース、コポビドン、およびその混合物からなる群から選択される結合剤を含む。いくつかの実施形態では、可塑剤は、中鎖トリグリセリド、フタル酸ジエチル、クエン酸エステル、ポリエチレングリコール、グリセロール、アセチル化グリセリド、およびヒマシ油からなる群から選択される。
【0136】
本明細書中のいくつかの実施形態は、アマンタジンまたはその薬学的に許容され得る塩を必要とするヒト被験体に投与する方法であって、経口投与用のカプセル中にアマンタジンを含む薬学的組成物を経口投与する工程を含み、カプセルが複数のペレットを含み、各ペレットは、以下:(a)アマンタジンまたはその薬学的に許容され得る塩を含むペレットコア、および(b)ペレットコアを取り囲む持続放出コーティングを含む、方法を提供する。いくつかの実施形態では、持続放出コーティングは、エチルセルロース、ならびにポビドンおよびヒドロキシプロピルメチルセルロースのうちの少なくとも1つ、ならびに可塑剤を含む。いくつかの実施形態では、ペレットコアは、コアシード上にコーティングされたアマンタジンまたはその薬学的に許容され得る塩および結合剤を含む。いくつかの実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、アマンタジンは40〜60重量%の量で存在し、結合剤は1〜25重量%の量で存在し、コアシードは8〜25重量%の量で存在し、エチルセルロースは10〜20重量%の量で存在し、ポビドンは1〜4重量%の量で存在し、可塑剤は1〜4重量%の量で存在する。いくつかの実施形態では、組成物は、さらに、ペレットコアと持続放出コーティングとの間にシールコーティングを含む。いくつかの実施形態では、ペレットコアは、ヒドロキシプロピルメチルセルロース、コポビドン、およびその混合物からなる群から選択される結合剤を含む。いくつかの実施形態では、可塑剤は、中鎖トリグリセリド、フタル酸ジエチル、クエン酸エステル、ポリエチレングリコール、グリセロール、アセチル化グリセリド、およびヒマシ油からなる群から選択される。いくつかの実施形態は、必要とするヒト被験体のパーキンソン病の処置を含む。
【0137】
本明細書中のいくつかの実施形態は、必要とする患者への1日1回の経口投与に適切な薬学的組成物であって、組成物は、治療有効量のアマンタジンまたはその薬学的に許容され得る塩を1日1回の投与において2つ以下のサイズ0以下のカプセルとして投与することができる持続放出形態で含む、薬学的組成物を提供する。いくつかの実施形態では、組成物は、110〜220mgのアマンタジンまたはその薬学的に許容され得る塩を含む。いくつかの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールが、2時間で25%以下、6時間で40〜80%、および12時間で少なくとも80%である。いくつかの実施形態では、組成物は複数のペレットを含み、各ペレットは、以下:(a)アマンタジンまたはその薬学的に許容され得る塩を含むペレットコア、および(b)ペレットコアを取り囲む持続放出コーティングを含む。いくつかの実施形態では、持続放出コーティングは、エチルセルロース、ならびにポビドンおよびヒドロキシプロピルメチルセルロースのうちの少なくとも1つ、ならびに可塑剤を含む。いくつかの実施形態では、ペレットコアは、コアシード上にコーティングされたアマンタジンまたはその薬学的に許容され得る塩および結合剤を含む。いくつかの実施形態では、組成物はアマンタジンを含み、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、アマンタジンは40〜70重量%の量で存在する。いくつかの実施形態では、ペレットコアは、直径が100ミクロンと500ミクロンとの間の糖または微結晶性セルロースを含むコアシードを含む。いくつかの実施形態では、嵩密度は0.5gm/cm3と1gm/cm3との間である。いくつかの実施形態では、組成物は、ペレットコアと持続放出コーティングとの間にシールコーティングを含む。いくつかの実施形態では、ペレットコアは、ヒドロキシプロピルメチルセルロース、コポビドン、およびその混合物からなる群から選択される結合剤を含む。いくつかの実施形態では、可塑剤は、中鎖トリグリセリド、フタル酸ジエチル、クエン酸エステル、ポリエチレングリコール、グリセロール、アセチル化グリセリド、およびヒマシ油からなる群から選択される。
【0138】
本明細書中のいくつかの実施形態は、ヒト被験体におけるパーキンソン病の処置方法であって、治療有効量のアマンタジンまたはその薬学的に許容され得る塩を1日1回の投与において2つ以下のサイズ0以下のカプセルとして投与することができる持続放出形態で含む組成物を経口投与する工程を含む、方法を提供する。いくつかの実施形態では、組成物は、110〜220mgのアマンタジンまたはその薬学的に許容され得る塩を含む。いくつかの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールが、2時間で25%以下、6時間で40〜80%、および12時間で少なくとも80%である。いくつかの実施形態では、組成物は複数のペレットを含み、各ペレットは、以下:(a)アマンタジンまたはその薬学的に許容され得る塩を含むペレットコア、および(b)ペレットコアを取り囲む持続放出コーティングを含む。いくつかの実施形態では、持続放出コーティングは、エチルセルロース、ならびにポビドンおよびヒドロキシプロピルメチルセルロースのうちの少なくとも1つ、ならびに可塑剤を含む。いくつかの実施形態では、ペレットコアは、コアシード上にコーティングされたアマンタジンまたはその薬学的に許容され得る塩および結合剤を含む。いくつかの実施形態では、組成物はアマンタジンを含み、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、アマンタジンは40〜70重量%の量で存在する。いくつかの実施形態では、ペレットコアは、直径が100ミクロンと500ミクロンとの間の糖または微結晶性セルロースを含むコアシードを含む。いくつかの実施形態では、嵩密度は0.5gm/cm3と1gm/cm3との間である。いくつかの実施形態では、組成物は、ペレットコアと持続放出コーティングとの間にシールコーティングを含む。いくつかの実施形態では、ペレットコアは、ヒドロキシプロピルメチルセルロース、コポビドン、およびその混合物からなる群から選択される結合剤を含む。いくつかの実施形態では、可塑剤は、中鎖トリグリセリド、フタル酸ジエチル、クエン酸エステル、ポリエチレングリコール、グリセロール、アセチル化グリセリド、およびヒマシ油からなる群から選択される。
【0139】
本明細書中のいくつかの実施形態は、ヒト被験体におけるレボドパ誘発性ジスキネジアの処置方法であって、治療有効量のアマンタジンまたはその薬学的に許容され得る塩を1日1回の投与において2つ以下のサイズ0以下のカプセルとして投与することができる持続放出形態で含む組成物を経口投与する工程を含む、方法を提供する。本明細書中のいくつかの実施形態は、ヒト被験体における外傷性脳損傷の処置方法であって、治療有効量のアマンタジンまたはその薬学的に許容され得る塩を1日1回の投与において2つ以下のサイズ0以下のカプセルとして投与することができる持続放出形態で含む組成物を経口投与する工程を含む、方法を提供する。いくつかの実施形態は、ヒト被験体における外傷性脳損傷の処置方法であって、治療有効量のアマンタジンまたはその薬学的に許容され得る塩を1日1回の投与において2つ以下のサイズ0以下のカプセルとして投与することができる持続放出形態で含む組成物を経口投与する工程を含む、方法を提供する。いくつかの実施形態は、ヒト被験体における疲労の処置方法であって、治療有効量のアマンタジンまたはその薬学的に許容され得る塩を1日1回の投与において2つ以下のサイズ0以下のカプセルとして投与することができる持続放出形態で含む組成物を経口投与する工程を含む、方法を提供する。いくつかの実施形態では、組成物は、110〜220mgのアマンタジンまたはその薬学的に許容され得る塩を含む。いくつかの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールが、2時間で25%以下、6時間で40〜80%、および12時間で少なくとも80%である。いくつかの実施形態では、組成物は複数のペレットを含み、各ペレットは、以下:(a)アマンタジンまたはその薬学的に許容され得る塩を含むペレットコア、および(b)ペレットコアを取り囲む持続放出コーティングを含む。いくつかの実施形態では、持続放出コーティングは、エチルセルロース、ならびにポビドンおよびヒドロキシプロピルメチルセルロースのうちの少なくとも1つ、ならびに可塑剤を含む。いくつかの実施形態では、ペレットコアは、コアシード上にコーティングされたアマンタジンまたはその薬学的に許容され得る塩および結合剤を含む。いくつかの実施形態では、組成物はアマンタジンを含み、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、アマンタジンは40〜70重量%の量で存在する。いくつかの実施形態では、ペレットコアは、直径が100ミクロンと500ミクロンとの間の糖または微結晶性セルロースを含むコアシードを含む。いくつかの実施形態では、嵩密度は0.5gm/cm3と1gm/cm3との間である。いくつかの実施形態では、組成物は、ペレットコアと持続放出コーティングとの間にシールコーティングを含む。いくつかの実施形態では、ペレットコアは、ヒドロキシプロピルメチルセルロース、コポビドン、およびその混合物からなる群から選択される結合剤を含む。いくつかの実施形態では、可塑剤は、中鎖トリグリセリド、フタル酸ジエチル、クエン酸エステル、ポリエチレングリコール、グリセロール、アセチル化グリセリド、およびヒマシ油からなる群から選択される。いくつかの実施形態では、本方法は、就寝時刻より前の3時間未満に患者に組成物を投与する工程を含む。
【0140】
本発明は、以下の実施例を参照してより深く理解することができ、実施例は、特許請求の範囲の範囲を制限することを意図しない。
【実施例】
【0141】
実施例1: アマンタジンを持続放出コーティングしたペレット処方物
夜間投与のためにデザインしたアマンタジンHClを持続放出コーティングしたペレット組成物を、以下の表1に示す成分および相対量を使用して調製した。各組成物について、薬物コーティング液を、撹拌しながらHPMC 5cpsおよびコポビドンをイソプロピルアルコールに添加することによって調製した。精製水をこの分散液に添加し、透明な溶液が形成されるまで撹拌し続けた。次いで、薬物(アマンタジンHCl)をこの結合剤溶液に添加し、薬物が完全に溶解するまで撹拌し続けた。最後に、タルクを添加し、撹拌によって均一に分散させた。
【0142】
セルフィアビーズ(篩サイズ♯35〜♯50(すなわち、300〜500ミクロン))を、Wursterコーティングユニットにロードした。薬物コーティング分散液を、ビーズ上に噴霧し、その後に一定期間乾燥させた。得られた薬物コーティングしたペレットを篩にかけて、篩♯18と♯24との間の画分(直径およそ700μm〜1mm)を保持した。
【0143】
シールコーティング液を、撹拌しながらHPMC 5cpsをイソプロピルアルコールに添加することによって調製した。精製水をこの分散液に添加し、透明な溶液が形成されるまで撹拌し続けた。タルクを添加し、撹拌によって均一に分散させた。篩にかけた薬物コーティングしたペレットを、Wursterコーティングユニット中にロードした。シールコーティング分散液を薬物コーティングしたペレット上に噴霧し、その後に一定期間乾燥させてペレット中に残存する溶媒および水を除去した。得られたシールコーティングしたペレットを篩にかけて、篩♯18と♯24との間の画分を保持した。
【0144】
ERコーティング液を、イソプロピルアルコールおよび精製水中にエチルセルロース(粘度7cps)を溶解し、透明な溶液が形成されるまで撹拌することによって調製した。次いで、ポビドンK−90をこの透明な溶液に溶解し、その後に撹拌しながら可塑剤ミグリオール812Nを添加して透明な溶液を形成させた。篩にかけたシールコーティングしたペレットを、Wursterコーティングユニット中にロードした。ERコーティング液をシールコーティングしたペレット上に噴霧し、その後に一定期間乾燥させてERコートに影響を与え、ペレット中に残存する溶媒および水を除去した。乾燥後、ステアリン酸マグネシウムを、アニュラス領域中のコーティングしたペレットのベッド上部に噴霧し、その後にWursterユニット中でペレットを再循環させてステアリン酸マグネシウムをコーティングしたペレットとブレンドした。得られたERコーティングされたペレットを篩にかけて、篩♯18と♯24との間の画分を保持した。
【0145】
所望の重量の単位用量を含むERコーティングしたペレットを、ペレット投薬チャンバーを備えたエンカプスレータを使用して空の1硬ゼラチンカプセルシェル(100〜140mg強度についてはサイズ1)に充填した。
【0146】
【表1】
溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、上記で調製したカプセルのin vitro溶解を試験した。所望の溶解規格を満たすカプセルは、2時間で25%以下、6時間で40〜80%、および12時間で少なくとも80%の薬物を放出した。例示的な溶解プロフィールでは、1時間で薬物放出0%、2時間で12%放出、4時間で43%放出、6時間で68%放出、8時間で83%放出、10時間で92%放出、および12時間で97%放出であった。上記方法にしたがって調製したカプセルは、良好な貯蔵安定性およびスケールアップの際のバッチ毎の再現性を示した。
【0147】
実施例2:より大量の薬物を負荷したアマンタジンを持続放出コーティングしたペレット処方物
夜間投与のためにデザインしたアマンタジンHClを持続放出コーティングしたペレット組成物を、以下の表2に示す成分および相対量ならびに実施例1に記載の製造過程を使用して調製した。
【0148】
不活性コアの直径は、200〜300ミクロンである。コーティングしたペレットの直径は、600〜1200ミクロンである。コーティングしたペレットの嵩密度は、0.7〜1.2g/cm3である。
【0149】
所望の重量の単位用量を含むERコーティングしたペレットを、ペレット投薬チャンバーを備えたエンカプスレータを使用して空の硬ゼラチンカプセルシェル(170mg強度についてはサイズ1)に充填する。
【0150】
【表2】
溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、上記で調製したカプセルのin vitro溶解を試験し、このカプセルは2時間で25%以下、6時間で40〜80%、および12時間で少なくとも80%の薬物を放出する。
【0151】
実施例3: アマンタジン持続放出コーティングしたペレット処方物
夜間投与に適切なアマンタジンHCl持続放出コーティングしたペレット組成物を、以下の表3に示した成分および相対量ならびに実施例1に記載の製造過程を使用して調製した。
【0152】
単位用量を含む所望の重量のERコーティングしたペレットを、ペレット投薬チャンバーを備えたエンカプスレータを使用して空の番号1硬ゼラチンカプセルシェル(強度100mg)に充填した。
【0153】
【表3】
溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、上記で調製したカプセルのin vitro溶解を試験した。結果を図1に示す。
【0154】
実施例4:押出球形化によって作製したアマンタジン持続放出処方物
夜間投与のためにデザインしたアマンタジンHCl持続放出組成物を、以下の表4に示す成分および相対量ならびに下記の製造過程を使用して調製する。
【0155】
アマンタジンHCl、微結晶性セルロース、およびラクトース一水和物のブレンドを調製し、湿塊を、ポビドン水溶液を使用して高剪断造粒機中で調製する。1mm篩を使用して湿塊を押し出し、押し出された塊をスフェロナイザーを使用して球形化する。ペレットを箱形乾燥機で乾燥させてコアペレットを得る。コアペレットを、パンコーター中にて持続放出コーティング液でコーティングする。所望の重量の単位用量を含むERコーティングしたペレットを、ペレット投薬チャンバーを備えたエンカプスレータを使用して空の1硬ゼラチンカプセルシェル(170mg強度)に充填する。
【0156】
【表4】
溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、上記で調製したカプセルのin vitro溶解を試験し、このカプセルは2時間で25%以下、6時間で40〜80%、および12時間で少なくとも80%の薬物を放出する。
【0157】
実施例5:IRアマンタジンと比較したアマンタジンERの処方物の薬物動態学的測定
目的:本研究の主な目的は、実施例3の持続放出処方物のPK特性を確認し、絶食条件下で健康な成人被験体に単回用量として投与した100mgのフィルムコーティングしたIRアマンタジンHCl錠(シンメトレル(登録商標))と比較して異なる放出特性を有する実施例3に記載のアマンタジンHClのERカプセルの3つのプロトタイプ処方物の薬物動態学的プロフィール、安全性、および耐容性を決定することであった。
【0158】
研究デザイン:これは第1相、無作為抽出、単回用量、非盲検、4期、交差、絶食状態の薬物動態学的研究であり、単回用量100mgの異なる放出特性を有するアマンタジンERカプセルの3つの処方物を単回用量100mgの市販のアマンタジンIR錠(シンメトレル(登録商標))と比較した。図1に示すように、3つのER処方物は、in vitroでのアマンタジン放出速度が異なっていた。
【0159】
方法:被験体を、研究スクリーニングの21日以内の第1期に入院させた。入院翌日に被験体に投与し、投与24時間後に退院させた。被験体に、退院後、投与56時間後および152時間後に追跡調査のために再度来院するように依頼した。各投与期間を、休薬のために少なくとも7日間の間隔をあけた。
【0160】
一晩の絶食後、240mLの水を使用して座りながら、処方物を被験体に投与した。血液サンプルを、各投与から0(投与前)、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、18、24(退院)、および56時間後に採取した。血漿サンプルを、有効な液体クロマトグラフィ/タンデム質量分析(LC/MS/MS)法によってアマンタジンについてアッセイした。薬物動態学的パラメーターを、WinNonlinソフトウェア(バージョン4.1以降;Pharsight Corporation)を使用した非コンパートメント解析を使用して計算した。
【0161】
分散分析(ANOVA)を、線形混合効果モデルを使用した治験薬の単回投与後のデータから決定されたCmaxおよびAUC0−∞の自然対数に対して行った。モデルは、被験体、順序、期間、およびレジメンの効果を含んでいた。被験体に対する効果が無作為であった一方で、順序、期間、およびレジメンの効果を固定した。AUC(ER処方物についての相対的生物学的利用能)およびCmaxの両方についてのER対IR比を計算した。研究を通して有害事象をモニタリングした。生命兆候(脈拍数、血圧、および体温)、臨床検査測定値(生化学、血液学、および尿検査)、およびECGを、研究中の種々の時点で収集した。
【0162】
結果: 全部で20人の被験体が研究に参加した。平均年齢は25.5歳(20〜38歳の範囲)であった。研究は、8人の男性被験体(40%)および12人の女性被験体(60%)から構成され、平均体型指数(BMI)は23.6kg/m2±2.85であった。人種構成は100%コーカサス人であった。15人の被験体に、全部で4つの処置を施した。
【0163】
本研究のPKの結果は、シンメトレル(登録商標)と比較したCmax値の減少およびTmaxの増加に基づいて、3つ全てのアマンタジンER処方物の吸収速度が減少したことを示していた(表5、図5、6)。IR処方物は、最高の平均Cmax(277±73.9ng/mL)および最短のTmaxの中央値(4時間)を有していた。処方物A、B、およびCは、徐々にCmax値が低下し、Tmax値が延長された。処方物A、B、およびCについて、それぞれ、Cmaxは204±61.4から166±34.8、149±34.4ng/mLへと減少し、Tmaxの中央値は7.0時間から11.0時間、14.0時間へと増加した。AUC0−∞によって測定した場合の総アマンタジン曝露は、シンメトレル(登録商標)よりも3つ全てのアマンタジンER処方物でわずかに低いが、3つ全ての処方物は、許容され得る生物学的利用能(85〜95%)を有していた。
【0164】
【表5】
【0165】
【表6】
実施例6:アマンタジンERの食品−効果評価
目的:主な目的は、夜間投与に適切なアマンタジンER処方物が食品と共に投与した場合に優れた生物学的利用能を示すことを証明することであった。本発明者らは、高脂肪食および絶食状態の両方で投与した場合の100mgカプセルのアマンタジンER処方物(実施例3、処方物B)の薬物動態を決定した。
【0166】
研究デザイン:これは、満腹状態および絶食状態の健康な成人(18〜45歳)の男性被験体および女性被験体における100mg処方物Iの単回投与と比較するための第1相、無作為抽出、単回用量、非盲検、2期、交差の食品−効果研究であった。研究は、21日目から−2日目のスクリーニング期(計画した投与日前)および2つの処置期間(期間1および期間2)(処置期間の間に8日間の休薬期間を含む)からなっていた。
【0167】
方法:一晩の絶食後、絶食条件については周囲温度の240mLの水を使用して座りながら、処方物を被験体に投与した。摂食条件については、一晩の絶食後、被験体に高脂肪および高カロリーの試験食(Guidance for Industry Food−Effect Bioavailability and Fed Bioequivalence Studies,December 2002)を朝食として与え、試験薬投与の30分前以内に完全に消費するように要求した。被験体を2つの順序のうちの1つに無作為抽出し、順序は、それぞれ、8日間の休薬期間をおいた摂食条件および絶食条件下での処置的投与から構成された。
【0168】
各期間について、薬物動態学的血液サンプルを、投与前ならびに各期間における投与から1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、18、24、28、48、72、96、および144時間後に採取した。被験体を、研究用製品投与の少なくとも15時間前に臨床施設に収容し、各期間において研究用製品投与の少なくとも28時間臨床施設に滞在させた。各期間における28時間後のサンプルを、外来を基本として採取した。血漿中のアマンタジンを、有効なLC/MS/MS法によって定量した。アマンタジンについてWinNonlin Professionalソフトウェア−バージョン5.0.1(Pharsight Corporation,USA)を使用して、薬物動態学的パラメーターを、非コンパートメントモデルによる薬物濃度−時間プロフィールから計算した。母平均のln変換したCmax、AUClast、およびAUC∞摂食/絶食比についての点推定および90%信頼区間(CI)が完全に80%〜125%の標準的許容範囲内にある場合、食品効果なしと定義した。アマンタジンの全統計解析を、SAS(登録商標)リリース9.1.3(SAS Institute Inc.,USA)のPROC MIXEDを使用して行った。
【0169】
日常的な安全性のモニタリングを、全被験体において投与中および投与後に行った。
【0170】
結果:全部で26人の被験体が研究に参加し、19人(73%)が男性であり、7人(27%)が女性であった。平均年齢は26歳(19〜44の範囲)であり、平均BMIは22.4kg/m2(18.1〜29.8の範囲)であった。人種構成は100%アジア人であった。全被験体に少なくとも1用量の治験薬を投与し、安全性解析に参加させた。24人(92.3%)の被験体が研究を完了し、薬物動態解析に参加させた。2人の被験体(7.7%)が、プロトコール逸脱のために研究完了前に処置を中止した。
【0171】
本研究の結果(表7)は、処方物Bの単回用量の薬物動態は食品の影響を受けないことを示す。食品を使用するか使用しないで投与したアマンタジン吸収のCmaxによって測定される速度ならびにAUC0−lastおよびAUC0−∞によって測定される範囲は等価であった(表8)。
【0172】
【表7】
【0173】
【表8】
結論:本研究の結果は、アマンタジンERの単回用量の薬物動態は食品の影響を受けないことを示す。食品を使用するか使用しないで投与したアマンタジン吸収のCmaxによって測定される速度ならびにAUC0−lastおよびAUC0−∞によって測定される範囲は等価であった。
【0174】
実施例7:絶食条件下での健康な成人におけるアマンタジンHCl ERカプセルの1日1回投与とアマンタジンHCl IR錠剤の1日2回投与とを比較した薬物動態研究。
【0175】
目的:本研究の主な目的は、反復投与または慢性投与下での定常状態で、夜間投与に適切なERアマンタジン処方物の薬物動態学を測定することであった。これにより、夜間に投与したERアマンタジン処方物のさらなる安全性および有効性の研究のための重要なPKパラメーター(すなわち、C−ave−朝、C−ave−日中、C−ave−夜)の計算が可能である。本発明者らは、市販の即時放出(IR)処方物として1日2回投与したアマンタジンHClと1日1回のアマンタジン持続放出(ER)処方物(実施例3、処方物B)との単回用量および反復用量の薬物動態を比較した。
【0176】
研究デザイン: これは、2期、複数回用量の交差研究であった。スクリーニング21日後、26人の健康な男性および女性の被験体を無作為抽出して、第I期に2つの処置(アマンタジンER 200mgを1日1回またはアマンタジンIR 100mgを1日2回)のうちの1つを施し、次いで、第II期に交差させて他方の処置を施した。
【0177】
方法:1日目に治験薬投与を開始した。2日目に治験薬を投与しなかった。3日目に複数回投与を開始し、7日間継続させた(9日目まで)。8日間の休薬期間により、用量投与の間隔をあけた。治験薬を、240mLの飲料水と共に投与した。投与1時間以内は他の流動物を許可しなかった。各期間中、薬物動態解析用の血液サンプルを、投与前ならびに最初の投与の1、2、3、4、5、6、8、10、11、12、13、14、15、16、17、18、20、24、28、36、および48時間後に採取した。朝のトラフ値(投与前)用の血液サンプルを、7日目および8日目に採取した。9日目の朝の投与の直前ならびにその1、2、3、4、5、6、8、10、11、12、13、14、15、16、17、18、20、24、28、48、72、および96時間後に血液サンプルを再度採取した。9日目の朝の投与から28時間後のサンプルを、各期間に外来を基本として採取した。血漿アマンタジンを、有効なLC/MS/MS法によって定量した。薬物動態学的パラメーターを、アマンタジンについてWinNonlin Professionalソフトウェアバージョン5.0.1(Pharsight Corporation,USA)を使用した非コンパートメントモデルによる薬物濃度−時間プロフィールから計算した。
【0178】
統計解析を行って、市販の即時放出(IR)処方物として1日2回投与したアマンタジンHClと1日1回の持続放出(ER)処方物(処方物B)とを比較して単回用量および反復用量の薬物動態プロフィールを評価した。分散分析(ANOVA)を、線形混合効果モデルを使用した研究9日目の治験薬投与後のデータから決定したCmax、Cmin、およびAUC24の自然対数に対して行った。モデルは、順序、期間、レジメンについての固定効果および変量効果を含んでいた。信頼区間を使用して、等価な評価のための2つの片側検定手順を行った。信頼区間を、ANOVAモデルのフレームワーク内で得た平均対数の相違についての信頼区間のエンドポイントの累乗によって得た。自然対数変換データ由来の信頼区間の上限および下限を、逆数で累乗して幾何平均の比についての90%信頼区間を得た。累乗した90%信頼区間が区間(80.00%、125.00%)内に完全に含まれる場合、同等性が確率された。
【0179】
反復測定ANOVAを、未変換データおよびln変換データの両方に対する5%有意水準での7、8、および9日目についてのCminの比較のために行った。反復測定ANOVA検定によって有意でないことが見い出された場合、定常状態が証明された。アマンタジンの統計解析を、SAS(登録商標)リリース9.1.3(SAS Institute Inc.、USA)のPROC MIXEDを使用して行った。
【0180】
日常的な安全性モニタリングを、全被験体における投与中および投与後ならびに研究の終了時に行った。
【0181】
結果:全部で26人の被験体が研究に参加し、22人(84.6%)が男性であり、4人(15.4%)が女性であった。平均年齢は26歳(19〜42の範囲)であり、平均BMIは22.9kg/m2(18.1〜28.8の範囲)であった。人種構成は100%アジア人であった。全被験体に少なくとも1用量の治験薬を投与し、安全性解析に参加させた。24人(92.3%)の被験体が研究を完了し、薬物動態解析に参加させた。2人の被験体(7.7%)が、投与12時間以内の嘔吐(薬物動態の排除基準)のために研究完了前にPK解析を中止した。
【0182】
半減期から予想されるように、アマンタジンERの1日1回の投与およびアマンタジンIRの1日2回の投与により、1日目と比較して9日目に、より高いCmaxおよびAUCによって測定されたように蓄積が認められた(表9および図2)。7、8、および9日目の類似のトラフレベルによって証明されるように、両処方物について9日目までに定常状態が達成された(データ示さず)。定常状態で(9日目)、血漿濃度(図2、表9)および薬物動態学的パラメーター(表9)は、両処方物で類似していた。さらに、処方物は、定常状態のCmax、Cmin、およびAUC0−24によって測定した場合にアマンタジンの吸収範囲および吸収速度に関して同等であり(表9)、この同等性は、80%〜125%の範囲内のアマンタジンIRに対するアマンタジンERの試験の最小2乗平均対定常状態のCmax、Cmin、およびAUC0−24の基準の比の90%CIによって定義される。
【0183】
【表9】
1日1回の夜間投与についてのERアマンタジン処方物の安定性の決定で重要な一定のさらなるPKパラメーターも表10中に報告する。
【0184】
【表10】
結論:ERアマンタジン処方物は、所望の定常状態PK特性を示し、この特性は、夜間投与ならびに所望の有効性および耐容性という利点の達成に同様に適切であろう。具体的には、夜間に1日1回投与したERアマンタジン処方物により、投与0〜8時間後と比較して投与8〜12時間後のアマンタジン血漿濃度の初期増加は比較的ゆっくりであり、平均アマンタジン血漿濃度が高い。したがって、夜間に投与した場合、IRアマンタジンと比較して、夜間アマンタジン血漿濃度に対する平均期間の比がより高い。したがって、この処方物は、現行の実務よりも高い用量での投与に十分に適しており、それにより、LID、疲労、およびパーキンソン病の処置において比較的十分に許容され、潜在的に優れた有効性を提供すると期待される。
【0185】
実施例8:健康なボランティアにおけるアマンタジンHCl ERカプセルの夜間1回投与とアマンタジンHCl IR錠の1日2回の投与との比較研究。
【0186】
目的:主な目的は、健康なボランティアにおいて就寝時刻に1日1回投与したアマンタジン持続放出(ER)カプセル(処方物B)と1日2回投与したアマンタジン即時放出(IR)錠とを睡眠に及ぼす影響について比較することである。このER処方物は、Cave、日中/Cave、夜=1.30を示す。
【0187】
研究デザイン:これは、終夜睡眠ポリグラフ(PSG)および標準化された質問表(スタンフォード眠気尺度(SSS);改訂エプワース眠気尺度(m−ESS)/カロリンスカ眠気尺度(KSS);トロント病院俊敏試験(THAT)/ZOGIM俊敏試験(ZOGIM−A);眠気/俊敏性の視覚的アナログ尺度(VAS))によって評価した場合に30人の健康なボランティアにおけるアマンタジンERカプセル、QHS、アマンタジンIR錠BID、およびカフェインカプレット(実薬対照)の睡眠に及ぼす影響を比較するための単一施設、二重盲検、3つの偽薬、無作為抽出の交差研究である。
【0188】
治験薬を、3つの投与期間に投与する。1投与期間あたり1日投薬量の1つの薬物を投与する。各投与日を、1週間の休薬期間によって間隔をあける。アマンタジンER(処方物B)の1日投薬量は、1つの220mg錠(または2×110mgカプセル)からなり、これを就寝時刻(QHS;本研究目的については23時と定義する)に投与する。アマンタジンIRの1日投薬量は、1つの100mgカプセルからなり、これを1日2回(BID;本研究目的については8時および16時と定義する)投与する。カフェインの1日投薬量は、1つの100mgカプセルからなり、これを1日3回(TID;本研究目的については8時、16時、および23時と定義する)投与する。
【0189】
全被験体に、1日3回(8時、16時、および23時)投与する。各投与時間に、各被験体に活性薬物または適合するプラセボのいずれかを3つの各処置のために投与する。特定の投与時間に投与したカプセル、錠剤、またはカプレットが活性治験薬を含むかそれら自体であるかどうかと無関係に、被験体を割り当てられる投与の順序および期間にしたがってプラセボダミーを決定する。
【0190】
適格規準を満たす同意した被験体を3つの処置順序(群)のうちの1つに等しく無作為抽出し、各順序は、上記のように1週間の休薬期間によって間隔をあけた3つの1日処置期間を含む。さらに、各二重盲検投与日前に1日の単盲検のプラセボ導入を行う。これにより、被験体はPSG記録条件下にて臨床研究病棟(CRU)での睡眠に順応し、個別のベースライン(BL)PSG特性を確立することが可能である。
【0191】
各投与期間について、被験体を、活性治験薬投与1日目の前日に睡眠検査室を備えたCRUに入院させる。被験体をCRU内で一晩滞在させ、活性薬物投与日は終日滞在させる。被験体を再度一晩滞在させ、次いで、翌日の朝にCRUを退院させる。第1の投与期間のために、CRUへの入院日(−1日目)はスクリーニング期の最終日を構成し、CRUからの退院日は第1の休薬期間の初日(2日目)を構成する。第2の投与期間のために、CRUへの再入院日(7日目)は第1の休薬期間の最終日を構成し、退院日(9日目)は第2の休薬期間の初日を構成するであろう。第3の投与期間のために、CRUへの再入院日(14日目)は第2の休薬期間の最終日を構成し、退院日(16日目)は追跡期の初日を構成する。
【0192】
CRUへの入院日(再入院日)に、被験体に日常検査および生命兆候試験を行う。被験体に、単盲検様式で16時および23時に各プラセボダミー(アマンタジンER、アマンタジンIR、およびカフェインについて)を投与する。被験体に有害事象(AE)について質問し、各投与の直前に生命兆候をチェックした。16時の投与前に、日常的な臨床検査および薬物中毒のスクリーニング検査のために採血する。被験体を、PSG記録条件下にて睡眠検査室内で一晩過ごさせる。
【0193】
活性治験薬の投与日に、被験体は7時に起床し、一連の睡眠および俊敏性の質問表に記入する。被験体に、8時、16時、および23時に治験薬(活性薬またはプラセボ)を投与する。被験体にAEについて質問し、各投与の直前に生命兆候をチェックした。23時の投与前に血漿アマンタジン濃度を測定するために採血する。
【0194】
活性治験薬の投与の翌日に、被験体は7時に起床し、一連の睡眠および俊敏性の質問表に記入する。8時前(すなわち、最後の投与時間から9時間後)に、被験体にAEについて質問し、生命兆候をチェックした。また、血漿アマンタジン濃度の測定のために採血する。任意のAEを報告するためのサイトへの連絡に関する指示を、CRUからの退院前の被験体に概説する。次の投与期間のためのPSUへの再訪(これを、2期および3期のための再訪に適用する)または電話による接触(これを、第3の投与期間後の追跡に適用する)のためのスケジュールを概説する。
【0195】
全被験体に対してCRUから退院3日後(19日目)に追跡電話調査を行う。
【0196】
AEおよび併用薬を、研究を通してモニタリングする。血漿濃度の測定のために、血液サンプルを1、8、および15日目の23時の投与直前および2、9、および16日目のおよそ8時に採取する。
【0197】
被験体による各時点での睡眠パラメーターならびに眠気および俊敏性の測定を列挙する。PSGおよび質問表の各要素由来の合成スコアおよびスコアの両方について作表し、分析する。測定した各パラメーターについて、記述要約統計量を、順序および処置によって計算する(平均(または必要に応じて中央値)、範囲、および標準偏差(SD)が含まれる)。
【0198】
選択した結果について推測統計学を行う。これは、分散と比較した処置群にわたる平均の間の相違の規模が処置結果の相違の可能性を示唆している。連続変数データを、パラメトリック統計学(適切な追加の事後解析および/または対応のあるT検定を使用した反復測定分散分析)によって分析する。分類データおよび正規分布に適合しないデータを、ノンパラメトリック統計(ウィルコクソン符合付き順位検定)によって分析する。PSGデータを、多変量解析および/またはスペクトル分析によって評価することもできる。
【0199】
結果:PSGおよび標準化された睡眠質問表(例えば、SSS、m−ESS、KSS、THAT、ZOGIM−A、またはVAS)によって測定した場合のアマンタジンIRのBID投与と比較して220mgのアマンタジンERのQD投与が睡眠障害を増加させないことまたは減少は、就寝時刻での1日1回の投与にアマンタジンERが適切であることを証明している。
【0200】
実施例9:パーキンソン病患者における夜間の1日1回のアマンタジンHCl ERカプセル投与と1日2回のアマンタジンHCl IRカプセル投与との睡眠に及ぼす影響および有効性の比較研究。
【0201】
目的:アマンタジン持続放出(ER)カプセルの睡眠に及ぼす影響および有効性を比較すること。
【0202】
研究デザイン:これは、120人のパーキンソン病患者におけるアマンタジン持続放出(ER)カプセルの睡眠に及ぼす影響および有効性を比較するための多施設、二重盲検の無作為抽出研究であり、この研究は、UPDRS(統合パーキンソン病評点尺度)、UPDRS−IV(統合パーキンソン病評点尺度 パートIV)、AIMS(異常不随意運動尺度)、終夜睡眠ポリグラフ(PSG)、および標準化された質問表(スタンフォード眠気尺度(SSS)、改訂エプワース眠気尺度(m−ESS)/カロリンスカ眠気尺度(KSS)、トロント病院俊敏試験(THAT)/ZOGIM俊敏試験(ZOGIM−A)、眠気/俊敏性の視覚的アナログ尺度(VAS))によって評価する。
【0203】
全治験薬を経口投与する。処置Aは、午前中に投与したプラセボカプセルおよび就寝時刻に投与した2つの110mgのアマンタジン(ER)カプセルおよびプラセボカプセルからなる。処置Bは、午前中に投与したプラセボカプセルおよび就寝時刻に投与した3つの110mgのアマンタジン(ER)カプセルからなる。処置Cは、午前中に投与した100mgのアマンタジンIRカプセルならびに就寝時刻に投与した100mgのアマンタジンIRカプセルおよび2つのプラセボカプセルからなる。処置Dは、午前中に投与したプラセボカプセルおよび就寝時刻に投与した3つのプラセボカプセルからなる。
【0204】
適格規準を満たす同意した被験体を3つの処置群のうちの1つに等しく無作為抽出し、各処置期間は14日間である。さらに、各二重盲検投与日前に1日の単盲検のプラセボ導入を行う。これにより、被験体はPSG記録条件下にて臨床研究病棟(CRU)での睡眠に順応し、個別のベースライン(BL)PSG特性を確立することが可能である。
【0205】
各投与期間について、被験体を、活性治験薬投与1日目の前日に睡眠検査室を備えたCRUに入院させる。被験体をCRU内で一晩滞在させ、活性薬物投与日は終日滞在させる。被験体を再度一晩滞在させ、次いで、翌日の朝にCRUを退院させる。
【0206】
標準的なスコアリング方法(UPDRSおよびAIMが含まれる)を使用して、1、7、および14日目の午前中にパーキンソンスコアを記録する。
【0207】
AEおよび併用薬を、研究を通してモニタリングする。
【0208】
被験体による各時点での睡眠パラメーターならびに眠気および俊敏性の測定を列挙する。PSGおよび質問表の各要素由来の合成スコアおよびスコアの両方について作表し、分析する。測定した各パラメーターについて、記述要約統計量を、順序および処置によって計算する(平均(または必要に応じて中央値)、範囲、および標準偏差(SD)が含まれる)。
【0209】
選択した結果について推測統計学を行う。これは、分散と比較した処置群にわたる平均の間の相違の規模が処置結果の相違の可能性を示唆している。連続変数データを、パラメトリック統計学(適切な追加の事後解析および/または対応のあるt検定を使用した反復測定分散分析)によって分析する。分類データおよび正規分布に適合しないデータを、ノンパラメトリック統計(ウィルコクソン符合付き順位検定)によって分析する。PSGデータを、多変量解析および/またはスペクトル分析によって評価することもできる。
【0210】
結果:PSGおよび標準化された睡眠質問表(例えば、SSS、m−ESS、KSS、THAT、ZOGIM−A、またはVAS)によって測定した場合のUPDRS、UPDRS−IV、AIMの改善、睡眠障害が増加しないことまたは減少は、就寝時刻での1日1回の投与にアマンタジンERが適切であることを証明している。
【0211】
実施例10.夜間に投与したより高い強度のアマンタジンER処方物の薬物動態学的特徴のシミュレーション
目的:目的は、実施例7に記載の臨床研究で作成したデータを使用して異なる用量レベルでの種々のIRおよびERアマンタジンレジメンの定常状態の血漿濃度−時間プロフィールを予想し、夜間に投与したより高い強度のアマンタジンER処方物の利点を示すことである。
【0212】
方法論:実施例7に記載の研究手順(ADS−5101−MD−104)あたり複数の用量のアマンタジンのER処方物およびIR処方物を投与された健康なボランティア由来の血漿濃度−時間プロフィールを使用して、2つの処方物を各々記載した薬物動態モデルを作製した。本研究は、26人の健康な成人の男性および女性のボランティアにおける1日1回のアマンタジンERカプセルおよび1日2回のアマンタジンIR錠を比較する非盲検、無作為抽出、2処置、2期の二元交差研究であった。24人の固体由来の完全なデータを本演習で使用した。薬物動態評価のための血液サンプルを、1日目の単回投与後および9日目の定常状態で採取した。分析の第1段階では、WinNonlin 5.2.1(Pharsight Corp.,Mountain View,CA)を使用して、1次インプットおよび1次アウトプット、加重1/y(yはアマンタジン血漿濃度である)を用いた一区画モデルを単回用量(1日目)および反復用量(9日目)のアマンタジンIRおよびER投与後に得た各個体の血漿濃度−時間データにフィッティングした。フィッティングを、両処方物について個別であるが、両日について同時に行った。使用したモデル仮定は、用量比例性および時間関数としての一定クリアランスを含む。
【0213】
モデルを以下の式によって示す:
【0214】
【数1】
(式中、Cは血漿濃度であり、Fは絶対生物学的利用能であり、Dは用量であり、Vは分布容積であり、kaは吸収速度定数であり、kは消失速度定数であり、tは時間であり、tlagは吸収時間のずれである)。適合度を、予想される各モデルと研究ADS−5101−MD−104から得た濃度−時間データとの比較によって検証した。式1を各個体の血漿濃度―時間データにフィッティングした後、24人の各被験体についてのV/F、ka、k、およびtlagのモデルパラメーター推定値を得た。定常状態での予測度を、ER処方物およびIR処方物としての200mgの連日投薬後に得られたアマンタジンの実測データと推定定常状態濃度との比較によって確認した(9日目)。
【0215】
第2の分析工程では、以下の投与レジメンにしたがって、各モデルパラメーター推定値を使用して、各パラメーター推定値の式1への再挿入および連続した7日間の投与の寄与の加算によって両処方物についての各個体の定常状態濃度−時間プロフィールをシミュレーションした。
1.260、340、および420mgのER処方物の定常状態への1日1回(QD)の投与
2.100mgのIR処方物の定常状態への1日3回(TID)の投与
3.100mgのIR処方物の定常状態への1日2回(BID)の投与
結果: 図4は、種々のERアマンタジン用量についての定常状態血漿濃度時間プロフィールのシミュレーションを、IRアマンタジンの種々のレジメンと共に示す。表11は、夜間に投与した場合にERアマンタジンの有効性および耐容性に影響を及ぼす薬物動態学的パラメーター値をまとめている。
【0216】
【表11】
表11および図中に示すように、夜間に1日1回投与したERアマンタジン処方物により、IRアマンタジンと比較して平均日中/夜間アマンタジン血漿濃度比が高く、比較的十分な耐容性を示すと予想される。またER処方物により、平均日中アマンタジン血漿濃度が100mgを1日2回投与したIRアマンタジンの1.3〜2.2倍となり、以下の実施例11に記載の臨床研究で患者に投与した場合に有効性が有意に増強されると予想される。
【0217】
実施例11:パーキンソン病におけるレボドパ誘発性ジスキネジア処置のためのアマンタジン持続放出経口カプセルの有効性および安全性についての無作為抽出、二重盲検、プラセボ−対照研究
研究目的:本研究を、パーキンソン病(PD)を有する被験体におけるレボドパ誘発性ジスキネジア(LID)の処置のための夜間に1日1回投与したアマンタジン持続放出(ER)経口カプセルの用量範囲を確認するためにデザインする。さらに、研究を、PDを有する被験体のLIDの処置のための1日1回投与したアマンタジンER経口カプセルの安全性および耐容性を証明するためにデザインする。最後に、研究を、パーキンソン病患者におけるアマンタジンER投与レジメンの定常状態薬物動態を確認し、C−ave−日中、C−ave−朝、C−ave−朝/C−ave−夜、およびC−ave−日中/C−ave−夜をアマンタジンの有効性および耐容性と相関させるためにデザインする。
【0218】
研究デザイン:これは、PDおよびLIDを有する被験体におけるアマンタジンERの多施設、無作為抽出、二重盲検、プラセボ−対照の4−アーム並行群研究であろう。適格規準を満たす同意した被験体を、以下の4つの処置(それぞれ、1日1回、夜間に8週間投与する)のうちの1つを施すために1:1:1:1に無作為抽出するであろう。
・処置A: プラセボ、
・処置B: 260mgアマンタジンER(ADS−5102)、
・処置C: 340mgアマンタジンER(ADS−5102)
・処置D: 420mgアマンタジンER(ADS−5102)
処置CまたはD(高用量アマンタジン群)に無作為抽出される被験体に、二重盲検様式で、260mgアマンタジンERを1週目に1日1回投与し、2週目の最初に340mgまたは420mgのいずれかの1日1回投与に増加させるであろう。8週目まで投与を継続するであろう。
【0219】
ベースライン来院および無作為抽出の完了後、被験体は、1、2、4、6、および8週間の投与後にクリニックに再度来院し、治験薬の最後の投与から14日後に追跡調査のために来院するであろう。研究のための訪問および評価を、可能な場合、朝の時間(午前9時から午後1時)に計画するであろう。2つの24時間のダイアリー一式を、無作為抽出の48時間前および選択した研究訪問の48時間前に完了するであろう。ダイアリーを使用して、以下の5つの条件を30分間隔でスコアリングするであろう:睡眠、オフ、ジスキネジアなしのオン、厄介でないジスキネジアを伴うオン、厄介なジスキネジアを伴うオン。
【0220】
選択した研究訪問で、血液サンプルを、アマンタジン血漿濃度の決定および定常状態母集団薬物動態学の評価のために採取するであろう。研究中の被験体の参加期間は、12週間までであり、2週間(最大)のスクリーニング期間、8週間(最大)の処置期間、および2週間の追跡期間を含むであろう。その割り当てた治験薬に耐容性を示すことができない被験体は、治験薬を恒久的に中断し、治験薬の最後の投与後に2週間にわたって安全性についての追跡を継続するであろう。
【0221】
患者の適格規準:被験体が選択基準を満たし、且つ排除基準を満たさない場合、被験体は研究に参加する資格がある。選択した鍵となる基準は以下である。
【0222】
選択基準:
・共同体で暮らす(すなわち、施設で暮らしていない)男性または女性の成人、
・30歳と75歳との間(両端を含む)、
・歩行能力または補助器具(例えば、歩行器またはステッキ)の使用によって歩行能力があること(被験体が必要な研究訪問のために来院することができること)、
・必要に応じて研究訪問のために被験体に付き添うための博識で信頼できる介護者/研究パートナー、
・現行のIRB/IEC承認されたインフォームドコンセント用紙に署名していること、
・訓練後、被験体が協力的であり、24時間のホームダイアリーを理解し、完了することができること(介護者が補助してよい)、
・ジスキネジアを伴う特発性パーキンソン病(スクリーニング中にMDS−UPDRSスコアを決定するであろうが、最小スコアは必要ない)、
・スクリーニング前の少なくとも30日間に抗パーキンソン病薬(レボドパが含まれる)のレジメンを安定に受けており、研究参加中はこのレジメンの継続に協力的であること、
・最小UDysRSスコアとして定義したジスキネジアの存在。
【0223】
排除基準:
・認知に影響を及ぼし得る他の神経疾患(アルツハイマー型認知症、ハンチントン病、レヴィ小体痴呆、前頭側頭型痴呆、大脳皮質基底核変性、または卒中もしくは脳外傷に続発する運動障害もしくは感覚障害が含まれるが、これらに限定されない)の存在、
・スクリーニング中の24未満のミニメンタルステート試験(MMSE)スコアによって証明される認知障害の存在、
・DSM−IV−TRによる急性の主要な精神障害(例えば、大鬱病性障害)または被験体の研究評価を完了する能力に影響を及ぼし得る症状(例えば、幻覚、興奮、偏執症)の存在、
・被験体の研究評価を完了する能力を損なうであろう感覚障害(例えば、聴覚、視覚)の存在、
・スクリーニング前の2年以内のDSM−IV基準によるアルコールまたは薬物の依存症および乱用歴、
・痙攣(小児期の熱性痙攣を除く)の病歴、
・スクリーニング前の2年以内の卒中またはTIAの病歴、
・スクリーニング前の2年以内の心筋梗塞、NYHA鬱血性心不全クラス3または4、または心房細動の病歴、
・スクリーニング前の5年以内の癌の病歴(以下を例外とする:適切に処置された非黒色腫性皮膚癌、局在性膀胱癌、非転移性前立腺癌、または上皮子宮頸癌(これらの例外は、研究登録前の医学的モニタリングを使用して考察し、承認されなければならない))、
・任意の以下の検査所見の異常:ヘモグロビン10g/dL未満、WBC数3.0×109/L未満、好中球数1.5×109/L未満、リンパ球数0.5×109/L未満、血小板数100×109/L未満、ヘモグロビンA1Cが9%超、またはアスパラギン酸アミノトランスフェラーゼ(AST)および/またはアラニンアミノトランスフェラーゼ(ALT)が正常上限の2倍超、
・腎臓病食の修正(MDRD)またはコッククロフト・ゴールト式によってGFRが50mL/分/1.73m2未満と評価されたこと、
・任意の臨床的に有意なECG異常、
・経口カプセルを嚥下できないか、経口投薬の使用を妨げる胃腸管吸収不良の病歴、
研究エンドポイント:主な有効性エンドポイントは、ベースラインから8週目までの統合ジスキネジア評点尺度(UDysRS)スコアの変化であろう。鍵となる副次エンドポイントには、以下が含まれるであろう:
・標準化されたPDホームダイアリーに基づいた、厄介なジスキネジアのないオンタイム(ジスキネジアなしのオン+厄介でないジスキネジアを伴うオン)、
・統合パーキンソン病評点尺度(MDS−UPDRS)(総合評点)、
・疲労重症度尺度(FSS)によって測定された疲労。この尺度は、評点尺度1(全く異なる)から7(非常にそう思う)を使用して患者によって完了される9つの質問を含む。この疲労尺度は、スクリーニングおよび重症度の評定の両方についてMDSで推奨されている(2010)
・安全性(有害事象、安全性に関連する治験薬の中断、生命兆候、および臨床検査が含まれる)。
【0224】
以下の伝統的尺度と新規の尺度との組み合わせを、この第2相研究のために選択した:
・統合ジスキネジア評点尺度(UDysRS)を、主要結果項目のために使用するであろう。この尺度は以下の4つのパートを有し、可能なトータルスコアは104である:
I:ジスキネジアの影響がある歴史的能力障害(患者が認識している)
II:ジスキネジアの影響がない歴史的能力障害(患者が認識している)
III:客観的機能障害(4つの認められた活動に基づくジスキネジア重症度、解剖学的分布、および型)
IV:パートIIIの活動に基づく客観的能力障害
・標準化されたパーキンソン病ホームダイアリー(試験ダイアリーII、〔33〕を提案する)に基づいた厄介なジスキネジアのないオンタイムは、副次結果項目であろう。この尺度は多数の研究で使用されており、種々の成功が得られている〔34〕。しかし、大部分のKOLは、被験体が報告したダイアリーのデータを収集しなければならず、主要結果項目を補助する必要があると考えられる。
・統合パーキンソン病評点尺度(UPDRS)、パートIV、項目32(ジスキネジアの持続時間: 0=なし、4=歩行日の76〜100%)および33(ジスキネジアの能力障害: 0=障害なし、4=完全な障害)は副次結果項目であろう。この尺度はPDで長年使用されている伝統的な尺度であり、これらの項目は大部分のLID研究で使用されている。
・認知尺度: 包括的な介護者の印象、鬱病、および他の尺度を使用して、ERアマンタジンの精神状態に対する利点を測定するであろう。
【0225】
統計学的方法
有効性解析:有効性解析集団は、少なくとも1つのベースライン後有効性評価を与える全ての無作為抽出および投与された被験体を含むであろう。UDysRS尺度の有効性エンドポイントのために、ベースラインから8週目までの変化を、要因としての処置群および共変量としてのUDysRSベースライン値を用いた共分散分析(ANCOVA)モデルを使用して分析するであろう。主要解析では、5%有意水準での両側検定を使用して、260mgのADS−5102群をプラセボ群と比較するであろう。主な比較が統計的に有意である場合(P<0.05)、5%有意水準での両側検定も使用して、340mgおよび420mgのADS−5102群をプラセボと比較するであろう。
【0226】
副次エンドポイントを、主要評価項目について記載したANCOVAモデルと同型を使用して分析するであろう。5%有意水準での両側検定を使用して、処置群間の全ての二次比較を行うであろう。最終観測値延長(LOCF)アプローチを、欠測データのために使用するであろう。主要有効性解析を、プロトコール準拠集団(8週目の有効性評価を行う有効性解析集団の部分集団)のために繰り返すであろう。
【0227】
安全性解析:安全性解析集団は、少なくとも1用量の治験薬を投与される全ての無作為抽出された被験体を含むであろう。全ての安全性エンドポイントを、最初の用量から追跡完了まで(最後の用量の治験薬の投与から2週間後)分析するであろう。また、ADS−5102アマンタジンERでの最初の投与の耐容性を評価するために、安全性解析を治験薬での処置の最初の2週間に報告された安全性に関して行うであろう。
【0228】
結果:本研究から以下の改善が予想され、以下の表に示す。さらなるエンドポイントを以下に記載する。
・許容され得る主要評価項目(例えば、UDysRS)によって測定したジスキネジアスコアの有意な(20〜60%)減少
・厄介なジスキネジアのないオンタイムの増加(20〜60%)
・5%から20%へのUPDRSの改善。
・5%から60%へのパーキンソン病性疲労(FSS)の改善。
・5%から20%へのPGIによる気分の改善。
【0229】
【表12】
実施例12.夜間投与に適切な遅延放出コートを使用したアマンタジンER処方物の薬物動態学的特徴のシミュレーション。
【0230】
目的:目的は、夜間投与に適切なアマンタジンの2つの別のER処方物(処方物1(実施例7で試験した処方物である)および処方物2(実施例7で試験したが、持続放出コート上部に遅延放出保護膜を有する処方物である))の薬物動態プロフィールを評価することである。
【0231】
実施例7に記載の研究手順(ADS−5101−MD−104)あたり複数の用量のアマンタジンのER処方物およびIR処方物を投与された健康なボランティア由来の血漿濃度−時間プロフィールを使用して、2つの処方物を各々記載した薬物動態モデルを作製した。本研究は、26人の健康な成人の男性および女性のボランティアにおける1日1回のアマンタジンERカプセルおよび1日2回のアマンタジンIR錠を比較する非盲検、無作為抽出、2処置、2期の二元交差研究であった。24人の固体由来の完全なデータを本演習で使用した。薬物動態評価のための血液サンプルを、1日目の単回投与後および9日目の定常状態で採取した。分析の第1段階では、WinNonlin 5.2.1(Pharsight Corp.,Mountain View,CA)を使用して、1次インプットおよび1次アウトプット、加重1/y(yはアマンタジン血漿濃度である)を用いた一区画モデルを単回用量(1日目)および反復用量(9日目)のアマンタジンIRおよびER投与後に得た各個体の血漿濃度−時間データにフィッティングした。フィッティングを、両処方物について個別であるが、両日について同時に行った。使用したモデル仮定は、用量比例性および時間関数としての一定クリアランスを含む。
【0232】
モデルを以下の式によって示す:
【0233】
【数2】
(式中、Cは血漿濃度であり、Fは絶対生物学的利用能であり、Dは用量であり、Vは分布容積であり、kaは吸収速度定数であり、kは消失速度定数であり、tは時間であり、tlagは吸収時間のずれである)。適合度を、予想される各モデルと研究ADS−5101−MD−104から得た濃度−時間データとの比較によって検証した。式1を各個体の血漿濃度―時間データにフィッティングした後、24人の各被験体についてのV/F、ka、k、およびtlagのモデルパラメーター推定値を得た。定常状態での予測度を、ER処方物およびIR処方物としての200mgの連日投薬後に得られたアマンタジンの実測データと推定定常状態濃度との比較によって確認した(9日目)。
【0234】
第2の分析工程では、以下の投与レジメンにしたがって、各モデルパラメーター推定値を使用して、各パラメーター推定値の式1への再挿入および連続した7日間の投与の寄与の加算によって両処方物についての各個体の定常状態濃度−時間プロフィールをシミュレーションした。
1.200mgのER処方物1の定常状態への1日1回(QD)の投与
2.200mgのER処方物2の定常状態への1日1回(QD)の投与
結果: 図7は、2つのERアマンタジン処方物についての定常状態血漿濃度時間プロフィールのシミュレーションを示す。(アマンタジン血漿濃度をy上に示し、日数をx軸に示す)。図7に示すように、夜間に1日1回投与したERアマンタジン処方物2により、処方物1と比較して定常状態でのピーク血漿濃度の達成が約4時間遅延する。したがって、持続放出コート上部に遅延放出コートを含む処方物は、アマンタジンへの日中血漿曝露を最大にする一方で、定常状態での夜間の血漿曝露を最小するという点で非常に好ましい薬物動態プロフィールを有する。
【0235】
本発明の好ましい実施形態を本明細書中に示し、且つ説明しているが、かかる実施形態は、例示のみを目的として提供している。当業者は、本発明を逸脱することなく多数の変形形態、変更形態、および置換形態をすぐに思いつくであろう。本明細書中に記載の本発明の実施形態の種々の代替形態を本発明の実施で使用することができると理解すべきである。本明細書中に引用した全てのリファレンスは、その全体が本明細書中で参考として援用される。
【技術分野】
【0001】
関連出願
本出願は、2009年12月2日に出願された米国仮特許出願第61/266,053号の利益を主張し、この出願は本明細書において参照として援用される。
【背景技術】
【0002】
発明の背景
本発明の分野は、アマンタジンの持続放出組成物(extended release composition)およびその使用である。
【0003】
アマンタジンは、NMDA受容体アンタゴニストによって処置することができる種々の容態(一酸化炭素中毒による神経系の損傷後に生じ得る特発性パーキンソン病(振戦麻痺(Parlysis Agitans))、脳炎後パーキンソニズム、および症候性パーキンソニズムの処置が含まれる)に必要である。アマンタジンはまた、ウイルスM2チャネルインヒビターとしての活性を有し、ウイルス疾患(特に、インフルエンザAウイルス)の感染の予防および治療のために使用される。
【0004】
現在市販されているアマンタジンの形態は、典型的には1日に2回以上投与される即時放出処方物である。アマンタジンの使用は、用量に関連するCNS副作用(眩暈、錯乱、幻覚、不眠症、および悪夢が含まれる)によって制限され(非特許文献1)、この副作用は、アマンタジンを夜間に投与した場合に特に悪化し得る。
【0005】
即時放出アマンタジンは、不眠症および睡眠障害を生じる刺激薬として作用し得ることが公知である。したがって、これらの副作用を最小にするために、最後の用量を、典型的には、午後4時までに投与する。かかるアマンタジンの投与により、夕方または夜に血漿アマンタジン濃度がピークに達し、朝の血漿濃度が非常に低くなる。
【0006】
アマンタジンの持続放出形態は当該分野で説明されている。Guittard et al.の特許文献1およびEdgren et al.の特許文献2は、それぞれ、それぞれ抗ウイルス薬または抗パーキンソン病薬を含む経口浸透圧投薬形態を開示しており、いずれの場合にもアマンタジンは投薬形態中で利用可能な薬物として列挙されている。Smith et al.の特許文献3は、活性薬剤としてNMDA受容体アンタゴニスト(アマンタジンなど)を使用した鎮痛性の即時放出および制御放出の薬学的組成物を開示している。特許文献4、特許文献5、特許文献6、および特許文献7(全てWent et al.に付与)は、それぞれ、任意選択的に制御放出形態のNMDA受容体アンタゴニスト(アマンタジンなど)の投与を開示している。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】米国特許第5,358,721号明細書
【特許文献2】米国特許第6,217,905号明細書
【特許文献3】米国特許第6,194,000号明細書
【特許文献4】米国特許出願公開第2006/0252788号明細書
【特許文献5】米国特許出願公開第2006/0189694号明細書
【特許文献6】米国特許出願公開第2006/0142398号明細書
【特許文献7】米国特許出願公開第2008/0227743号明細書
【非特許文献】
【0008】
【非特許文献1】Gracies JM,Olanow CW;Current and Experimental Therapeutics of Parkinson’s Disease;Neuropsychopharmacology:the Fifth Generation of Progress pp1802;American College of Neuropsychopharmacology 2002
【発明の概要】
【課題を解決するための手段】
【0009】
発明の概要
本発明者らは、睡眠に悪影響を及ぼすことなく朝に起床した際に患者のアマンタジン血漿濃度がより高くなる改良されたアマンタジン処方物が当該分野で必要であることを確認している。さらに、本発明者らは、不眠症および睡眠障害の副作用が軽減され、起床時に有効な血漿濃度のアマンタジンが得られる午後遅くまたは夕方(例えば、午後4時以降)のアマンタジンの投与方法が当該分野で必要であることを確認している。
【0010】
したがって、不眠症や睡眠障害を生じることなく患者が就寝を望む直前に(例えば、就寝時に)患者に投与することができる改良されたアマンタジンの治療方法が当該分野で必要である。さらに、患者が就寝前に摂取することができ、且つ起床時(例えば、朝、ぐっすり眠った後)に適切なアマンタジン血漿濃度が得られるアマンタジン治療が必要である。
【0011】
さらに、多数のパーキンソン病患者は、嚥下が困難であり、複数の投薬を受けている。それ故、治療有効量の薬物を送達し、1日1回投与することができ、小さなサイズの経口投薬形態であり、且つ丸薬の負荷を過度に増加しないアマンタジン治療が必要である。
【0012】
本発明の1つの態様は、アマンタジンを必要とする患者に投与する方法であって、アマンタジンまたはその薬学的に許容され得る塩を含む持続放出(ER)組成物を就寝時刻より前の3時間未満(すなわち、被験体が夜に就寝を望む時間)に経口投与する工程を含む、方法である。この態様はまた、下記の医薬の製造のためのかかる組成物の使用およびアマンタジンの使用を含む。あるいは、組成物を、就寝時間より前の約4時間未満に投与する。
【0013】
第2の態様では、本発明は、アマンタジンでの処置を受けたヒト被験体における睡眠障害の軽減方法であって、アマンタジンまたはその薬学的に許容され得る塩を含む持続放出(ER)組成物を就寝時刻より前の約3時間未満(すなわち、被験体が夜に就寝を望む時間)に投与する工程を含む、方法を提供する。この態様はまた、下記の医薬の製造のためのかかる組成物の使用およびアマンタジンの使用を含む。あるいは、組成物を、就寝時間より前の約4時間未満に投与する。
【0014】
第3の態様では、本発明は、レボドパ誘発性ジスキネジア、疲労、認知症、またはパーキンソン病の任意の他の症状の処置方法であって、アマンタジンまたはその薬学的に許容され得る塩を含む持続放出(ER)組成物を就寝時刻より前の3時間未満(すなわち、被験体が夜に就寝を望む時間)に投与する工程を含む、方法を提供する。この態様はまた、下記の医薬の製造のためのかかる組成物の使用およびアマンタジンの使用を含む。
【0015】
第4の態様では、本発明は、脳損傷、脳外傷、認知症、アルツハイマー病、卒中、ハンチントン病、ALS、多発性硬化症、神経変性疾患、認知症、脳血管性容態、運動障害、脳神経障害、神経精神障害の処置方法であって、アマンタジンまたはその薬学的に許容され得る塩を含む持続放出(ER)組成物を就寝時刻より前の3時間未満(すなわち、被験体が夜に就寝を望む時間)に投与する工程を含む、方法を提供する。この態様はまた、下記の医薬の製造のためのかかる組成物の使用およびアマンタジンの使用を含む。
【0016】
任意の上記態様の1つの実施形態では、就寝時間より前の2.5時間未満、2時間未満、1.5時間未満、1時間未満、または30分未満(すなわち、被験体が夜に就寝を望む時間)に投与する。
【0017】
任意の上記態様の1つの実施形態では、患者はパーキンソン病と診断されている。
【0018】
任意の上記態様の1つの実施形態では、組成物を1日1回投与する。別の態様では、1日用量は200mgを超え、サイズ0、1、または2の1、2、または3つのカプセルで投与する。
【0019】
任意の上記態様の1つの実施形態では、組成物のパーキンソン病患者への投与により、レボドパ誘発性ジスキネジア(LID)が有意に軽減される。特定の実施形態では、組成物の投与により、レボドパ誘発性ジスキネジアが約5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、または80%軽減される。さらなる実施形態では、レボドパ誘発性ジスキネジアの軽減を、LIDの軽減を示す薬物の有効性を評価するためのFDAによって使用される数値的尺度によって測定する。さらなる特定の実施形態では、LIDの軽減の測定で使用される尺度は、UDysRS、UPDRSパートIV(サブスコア32、33)、ジスキネジア評点尺度(DRS)、異常不随意運動尺度(AIMS)、またはこの目的のために開発された他の尺度であり得る。
【0020】
任意の上記態様の1つの実施形態では、組成物のパーキンソン病患者への投与により、パーキンソン病性疲労が有意に軽減される。特定の実施形態では、組成物の投与により、パーキンソン病性疲労が約5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、または60%軽減される。さらなる特定の実施形態では、疲労の軽減を、疲労の軽減を示す薬物の有効性を評価するためのFDAによって使用される数値的尺度によって測定する。さらなる特定の実施形態では、疲労軽減の測定に使用される尺度は、疲労重症度尺度(FSS)であり得る。
【0021】
任意の上記態様の1つの実施形態では、組成物のパーキンソン病患者への投与により、パーキンソン病の症状が有意に軽減される。特定の実施形態では、組成物の投与により、パーキンソンの症状が約5%、10%、15%、20%、25%、30%、35%、または40%軽減される。さらなる特定の実施形態では、パーキンソンの症状の軽減を、パーキンソンの症状の軽減を示す薬物の有効性を評価するためのFDAによって使用される数値的尺度によって測定する。さらなる特定の実施形態では、パーキンソンの症状軽減の測定に使用される尺度は、統合パーキンソン病評点尺度(UPDRS)であり得る。
【0022】
任意の上記態様の1つの実施形態では、組成物を、投与前に、食品、より特定の実施形態では、少量の軟らかい食品(例えば、アップルソースまたはチョコレートプディング)に添加する。食品への添加は、カプセルを開け、内容物を患者の食品上に散りばめることを含み得る。これは、患者が組成物を嚥下できないか嫌がる場合に有利である。
【0023】
任意の上記態様の1つの実施形態では、定常状態の血漿濃度での投与後少なくとも1時間アマンタジンの血漿濃度は増加しない。
【0024】
任意の上記態様の1つの実施形態では、定常状態の血漿濃度での投与後少なくとも2時間アマンタジンの血漿濃度は増加しない。
【0025】
任意の上記態様の1つの実施形態では、定常状態のアマンタジン血漿濃度でのヒト被験体への組成物の投与により、かかる投与の1、2、2.5、または3時間後にアマンタジン血漿濃度が5%、10%、15%、20%、または25%未満増加する。例えば、定常状態のアマンタジン血漿濃度でのヒト被験体への組成物の投与により、アマンタジン血漿濃度が、かかる投与の1、2、2.5、または3時間後に5%未満、かかる投与の1、2、2.5、または3時間後に10%未満、かかる投与の1、2、2.5、または3時間後に15%未満、かかる投与の1、2、2.5、または3時間後に20%未満、またはかかる投与の1、2、2.5、または3時間後に25%未満増加する。
【0026】
任意の上記態様の1つの実施形態では、アマンタジンの単回用量のTmaxは9〜15時間である。さらなる特定の実施形態では、アマンタジンの単回用量のTmaxは、投与10〜14時間後である。別のさらなる特定の実施形態では、アマンタジンの単回用量のTmaxは、投与11〜13時間後である。
【0027】
任意の上記態様の1つの実施形態では、アマンタジンの定常状態のTmaxは7〜13時間である。さらなる特定の実施形態では、アマンタジンの定常状態のTmaxは、投与8〜12時間後である。別のさらなる特定の実施形態では、アマンタジンの定常状態のTmaxは、投与9〜11時間後である。
【0028】
任意の上記態様の1つの実施形態では、アマンタジンのピーク血漿濃度は、単回用量の組成物の投与の6時間後と16時間後との間に達成される。さらなる特定の実施形態では、ピークアマンタジン血漿濃度は、単回用量の組成物の投与の8〜14時間後に達成される。別のさらなる特定の実施形態では、ピークアマンタジン血漿濃度は、単回用量の組成物の投与の10〜12時間後に達成される。さらなる特定の実施形態では、ピークアマンタジン血漿濃度は、単回用量の組成物の投与の6、7、8、9、10、11、または12時間後から約9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、または24時間後に達成される。
【0029】
任意の上記態様の1つの実施形態では、ヒト被験体への組成物の1日1回の経口投与により、投与3時間後の25%未満のアマンタジン濃度の増加によって特徴付けられる定常状態血漿濃度プロフィールが得られる。さらなる特定の実施形態では、定常状態の血漿濃度プロフィールは、投与4時間後の25%未満のアマンタジン濃度の増加によって特徴付けられる。
【0030】
任意の上記態様の1つの実施形態では、組成物を1日1回投与し、定常状態でのCmax/Cmin比は、1.5〜2.0、より具体的には1.7〜1.9、より具体的には約1.8である。
【0031】
任意の上記態様の1つの実施形態では、就寝時刻の組成物のヒト被験体への複数回の投与後の定常状態の血漿濃度プロフィールは、日中の平均血漿濃度(「C−ave−日中」、ヒトPK研究で測定した場合の日中平均アマンタジン血漿濃度として定義)が夜間の平均血漿濃度(「C−ave−夜」、ヒトPK研究で測定した場合の夜間平均アマンタジン血漿濃度として定義)の1.1〜2.0倍であることによって特徴づけられる。より特定の実施形態では、C−ave−日中は、午前5時、午前6時、午前7時、午前8時、または午前9時から午後4時、午後5時、午後6時、午後7時、または午後8時の間(例えば、午前6時と午後4時との間、午前7時と午後6時との間、または午前7時と午後5時との間)で測定した平均アマンタジン血漿濃度である。C−ave−夜は、午後4時、午後5時、午後6時、午後7時、午後8時、午後9時、午後10時、または午後11時から午前5時、午前6時、午前7時、午前8時、または午前9時の間(例えば、午後10時と午前6時との間、午後7時と午前6時との間、または午後8時と午前6時との間)で測定した平均アマンタジン血漿濃度である。
【0032】
任意の上記態様の1つの実施形態では、就寝時刻の組成物のヒト被験体への複数回の投与後の定常状態の血漿濃度プロフィールは、午前中の平均血漿濃度(「C−ave−朝」、午前中にヒトPK研究で測定した場合の平均アマンタジン血漿濃度として定義)が夜間の平均血漿濃度の1.1〜2.0倍であることを特徴とする。1つの実施形態では、C−ave−朝は、午前5時、午前6時、午前7時、午前8時、または午前9時から午前11時、午前11:30時、午後12時、午後12:30時、または午後1:00時の間(例えば、午前5時と午前11時との間または午前7時と午後12時との間)に測定した平均アマンタジン血漿濃度である。より好ましくは、定常状態でのC−ave−朝/C−ave−夜比は1.2〜1.6である。
【0033】
任意の上記態様の1つの実施形態では、組成物の連日投与後の定常状態の血漿濃度プロフィールは、投与8〜12時間後の平均血漿濃度(「C−ave−8−12時間」)が投与から最初の8時間後の平均血漿濃度(「C−ave−0−8時間」)の1.1〜2.0倍であることによって特徴づけられる。より好ましくは、定常状態でのC−ave−8−12時間/C−ave−0−8時間比は、1.2〜1.6である。
【0034】
任意の上記態様の1つの実施形態では、単回用量の組成物のヒト被験体への投与により、以下によって特徴づけられる血漿濃度プロフィールが得られる:AUC0−infの5%未満、より好ましくは3%未満である0〜4時間の断片的AUC、AUC0−infの約5〜15%、好ましくは約8〜12%である0〜8時間の断片的AUC、AUC0−infの約10〜40%、好ましくは約15〜30%である0〜12時間の断片的AUC、AUC0−infの約25〜60%、好ましくは約30〜50%である0〜18時間の断片的AUC、およびAUC0−infの約40〜75%、好ましくは約50〜70%である0〜24時間の断片的AUC。
【0035】
任意の上記態様の1つの実施形態では、組成物のヒト被験体への1日1回の経口投与により、以下によって特徴づけられる定常状態の血漿濃度プロフィールが得られる:AUC24の約2〜25%、好ましくは約5〜20%である0〜4時間の断片的AUC、AUC24の約15〜50%、好ましくは約20〜40%である0〜8時間の断片的AUC、AUC24の約30〜70%、好ましくは約40〜60%である0〜12時間の断片的AUC、およびAUC24の約60〜95%、好ましくは約75〜90%である0〜18時間の断片的AUC。
【0036】
任意の上記態様の1つの実施形態では、組成物のヒト被験体への1日1回の経口投与により、以下によって特徴づけられる定常状態の血漿濃度プロフィールが得られる:AUC24の約15〜40%、好ましくは約20〜32%である0〜8時間の断片的AUC、AUC24の約30〜50%、好ましくは約35〜45%である8〜16時間の断片的AUC、およびAUC24の約20〜35%、好ましくは約25〜33%である16〜24時間の断片的AUC。
【0037】
任意の上記態様の1つの実施形態では、アマンタジンを薬学的に許容され得る塩として投与する。さらなる特定の実施形態では、アマンタジンを塩酸塩または硫酸アマンタジンとして投与する。
【0038】
任意の上記態様の1つの実施形態では、総1日用量50mg〜600mgのアマンタジンまたはその薬学的に許容され得る塩を患者に投与する。より具体的には、アマンタジンまたはその薬学的に許容され得る塩の1日用量は、100〜440mgの範囲であり得る。別の特定の実施形態では、アマンタジンまたはその薬学的に許容され得る塩の1日用量は、260〜420mgの範囲であり得る。別の実施形態では、アマンタジンまたはその薬学的に許容され得る塩の1日用量は、300mg/日を超える。種々の特定の実施形態では、アマンタジンまたはその薬学的に許容され得る塩の1日用量は、50〜75mg、70〜95mg、90〜115mg、110〜135mg、130〜155mg、150〜175mg、170〜195mg、190〜215mg、210〜235mg、230〜255mg、250〜275mg、270〜295mg、290〜305mg、300〜315mg、310〜325mg、320〜335mg、330〜345mg、340〜355mg、350〜365mg、360〜375mg、370〜385mg、380〜395mg、390〜405mg、400〜415mg、410〜425mg、420〜435mg、430〜445mg、または440〜455mgであり得る。
【0039】
任意の上記態様の1つの実施形態では、組成物は、50mg〜600mgのアマンタジンまたはその薬学的に許容され得る塩を含む。より具体的には、組成物は、100mg〜450mgのアマンタジンまたはその薬学的に許容され得る塩を含むことができる。さらにより具体的には、組成物は、130〜210mgのアマンタジンまたはその薬学的に許容され得る塩を含むことができる。種々の特定の実施形態では、組成物を含む投薬形態は、50〜75mg、70〜95mg、90〜115mg、110〜135mg、130〜155mg、150〜175mg、170〜195mg、190〜215mg、210〜235mg、230〜255mg、250〜275mg、270〜295mg、290〜305mg、300〜315mg、310〜325mg、320〜335mg、330〜345mg、340〜355mg、350〜365mg、360〜375mg、370〜385mg、380〜395mg、390〜405mg、400〜415mg、410〜425mg、420〜435mg、430〜445mg、または440〜455mgのアマンタジンまたはその薬学的に許容され得る塩を含む。さらなる特定の実施形態では、組成物は、約110、120、130、140、150、160 170、180、190、210、または220mgのアマンタジンまたはその薬学的に許容され得る塩を含む。別のさらなる特定の実施形態では、組成物は110mgの塩酸アマンタジンを含む。別のさらなる特定の実施形態では、組成物は130mgの塩酸アマンタジンを含む。別のさらなる特定の実施形態では、組成物は170mgの塩酸アマンタジンを含む。別のさらなる特定の実施形態では、組成物は210mgの塩酸アマンタジンを含む。
【0040】
任意の上記態様の1つの実施形態では、組成物を、それぞれ100〜175mgのアマンタジンまたはその薬学的に許容され得る塩を含む1、2、3、または4つの単位投薬形態として投与する。さらなる特定の実施形態では、組成物を、それぞれ100〜175mgのアマンタジンまたはその薬学的に許容され得る塩を含む2つの単位投薬形態として投与する。
【0041】
任意の上記態様の1つの実施形態では、組成物を、それぞれ50〜250mgのアマンタジンまたはその薬学的に許容され得る塩を含む1、2、または3つの単位投薬形態として投与する。さらなる特定の実施形態では、組成物を、それぞれ65〜220mgのアマンタジンまたはその薬学的に許容され得る塩を含む1つまたは2つの単位投薬形態として投与する。
【0042】
任意の上記態様の1つの実施形態では、単回用量の組成物の絶食状態のヒト被験体への経口投与により、1mgのアマンタジンあたり1.0〜2.8ng/mlの最大血漿濃度(Cmax)が得られる。さらなる特定の実施形態では、単回用量の組成物の絶食状態のヒト被験体への経口投与により、1mgのアマンタジンあたり1.6〜2.4ng/mlの最大血漿濃度(Cmax)および1mgのアマンタジンあたり40〜75ng*h/mLのAUC0−inf(t=0からt=無限大までの濃度曲線下面積)が得られる。
【0043】
任意の上記態様の1つの実施形態では、ある用量の組成物のヒト被験体への連日経口投与により、以下のうちの少なくとも1つによって特徴づけられる定常状態血漿濃度プロフィールが得られる:(i)1mgのアマンタジンあたり2.4〜4.2ng/mlのCmax、(ii)1mgのアマンタジンあたり1.1〜2.6ng/mlのCmin、および(iii)1mgのアマンタジンあたり44〜83ng*h/mLのAUC0−24さらなる具体例では、(i)、(ii)、および(iii)の3つ全ての基準を満たす。
【0044】
さらなる特定の実施形態では、定常状態血漿濃度プロフィールは、以下によってさらに特徴づけられる:(iv)投与後少なくとも1時間アマンタジン濃度が増加しないこと、および(v)Cmax/Cmin比が1.5〜2.0であること。さらなる特定の実施形態では、(iv)および(v)の両方の基準を満たす。
【0045】
別のさらなる特定の実施形態では、定常状態の血漿濃度プロフィールは、以下のうちの少なくとも1つによってさらに特徴づけられる:(iv)投与後少なくとも2時間アマンタジン血漿濃度が増加しないこと、および(v)Cmax/Cmin比1.7〜1.9。さらなる特定の実施形態では、(iv)および(v)の両方の基準を満たす。
【0046】
任意の上記態様の1つの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールが、以下のうちの少なくとも1つを示す:(i)2時間で25%以下の溶解、(ii)6時間で40〜80%までの溶解、および(iii)12時間で少なくとも80%の溶解。より特定の実施形態では、基準(i)、(ii)、および(iii)のうちの2つを満たす。さらなる特定の実施形態では、基準(i)、(ii)、および(iii)の3つ全てを満たす。
【0047】
任意の上記態様の1つの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールが、以下のうちの少なくとも1つを示す:(i)2時間で25%以下の溶解、(ii)6時間で25〜55%以下の溶解、および(iii)12時間で少なくとも80%の溶解。より特定の実施形態では、基準(i)、(ii)、および(iii)のうちの2つを満たす。さらなる特定の実施形態では、基準(i)、(ii)、および(iii)の3つ全てを満たす。
【0048】
任意の上記態様の1つの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールが、以下のうちの少なくとも1つを示す:(i)1時間で20%以下の溶解、(ii)2時間で約25〜45%の溶解、(iii)4時間で50〜80%以下の溶解、および(iv)8時間で少なくとも80%の溶解。より特定の実施形態では、基準(i)、(ii)、(iii)、および(iv)のうちの2つを満たす。さらなる特定の実施形態では、基準(i)、(ii)、(iii)、および(iv)の4つ全てを満たす。
【0049】
任意の上記態様の1つの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、アマンタジンのin vitro溶解プロフィールは、以下のアマンタジン放出によってさらに特徴づけられる:(i)1時間で10%以下、(ii)4時間で30〜50%、または(iii)12時間で少なくとも90%。より特定の実施形態では、基準(i)、(ii)、および(iii)のうちの2つを満たす。さらなる特定の実施形態では、(i)、(ii)、および(iii)の3つ全ての基準を満たす。
【0050】
別の態様では、本発明は、ペレット・イン・カプセルを含むかペレット・イン・カプセルからなる薬学的組成物であって、ペレットが、コアシード上にコーティングされたアマンタジンと結合剤との混合物を有するコアシードを含むコア、エチルセルロースを含むコアを取り囲む持続放出コーティング、ポア形成剤(ヒドロキシプロピルメチルセルロースまたはポビドンなど)、および可塑剤を含む、薬学的組成物を提供する。
【0051】
別の態様では、本発明は、上記態様の方法で用いる薬学的組成物であって、組成物が経口投与用であり、経口投与用カプセルを含み、カプセルが複数のペレットを含み、各ペレットは、以下:(a)アマンタジンまたはその薬学的に許容され得る塩を含むペレットコア、および(b)ペレットコアを取り囲む持続放出コーティングを含む、薬学的組成物を提供する。
【0052】
1つの実施形態では、持続放出コーティングは、エチルセルロース、ならびにポビドンおよびヒドロキシプロピルメチルセルロースのうちの少なくとも1つ、ならびに可塑剤を含む。さらなる特定の実施形態では、持続放出コーティングは、エチルセルロース、ポビドン、および可塑剤を含む。
【0053】
1つの実施形態では、ペレットコアは、コアシード上にコーティングされたアマンタジンおよび結合剤を含む。1つの実施形態では、コアシードは、糖スフェア(ノンパレイユ)または微結晶性セルロースシード(例えば、セルフィア(登録商標))である。さらなる特定の実施形態では、コアシードは微結晶性セルロースコアである。別の特定の実施形態では、コアシードの直径は、100ミクロン〜1,000ミクロンの範囲である。さらなる特定の実施形態では、コアシードの直径は、100、200、300、400、500、600、または700ミクロンである。好ましい特定の実施形態では、コアシードの直径は、500ミクロン未満である。
【0054】
1つの実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、アマンタジンまたはその薬学的に許容され得る塩は、20〜80重量%の量、嵩密度0.3〜1.2g/cm3で存在する。
【0055】
1つの実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、アマンタジンまたはその薬学的に許容され得る塩は、40〜60重量%の量、嵩密度0.5〜1.2g/cm3で存在する。
【0056】
1つの実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、アマンタジンまたはその薬学的に許容され得る塩は、60〜80重量%の量、嵩密度0.5〜1.2g/cm3で存在する。
【0057】
1つの実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、結合剤は、8〜25重量%の量で存在する。
【0058】
1つの実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、コアシードは、8〜25重量%の量で存在する。
【0059】
1つの実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、エチルセルロースは、10〜20重量%の量で存在する。
【0060】
1つの実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、ポビドンは、1〜4重量%の量で存在する。
【0061】
1つの実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、可塑剤は、1〜4重量%の量で存在する。
【0062】
1つの実施形態では、コーティングされたペレットの直径は、200ミクロン〜1700ミクロン(micros)の範囲である。さらなる特定の実施形態では、コーティングされたペレットの直径は、200、300、400、500、600、700、800、900、1000、1100、1200、1300、または1500ミクロンである。一定の特定の実施形態では、コーティングされたペレットの直径は、1000ミクロン未満(例えば、500〜1000ミクロン)である。
【0063】
1つの実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、結合剤は、5〜25重量%の量で存在する。
【0064】
1つの実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、コアシードは、1〜15重量%の量で存在する。
【0065】
1つの実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、エチルセルロースは、5〜20重量%の量で存在する。
【0066】
1つの実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、ポビドンは、0.25〜4重量%の量で存在する。
【0067】
1つの実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、可塑剤は、0.25〜4重量%の量で存在する。
【0068】
1つの実施形態では、ペレットは、ペレットコアと持続放出コーティングとの間にシールコーティングをさらに含む。いくつかの実施形態では、不活性コーティングを、薬物コーティング前の不活性コア、薬物コーティングしたペレット上、または制御放出コーティングしたペレット上に適用することができる。別の実施形態では、腸溶コーティングを、薬物コーティングしたペレットまたは制御放出ペレットに適用することができる。
【0069】
1つの実施形態では、ペレットコアは、ヒドロキシプロピルメチルセルロース、コポビドン、およびその混合物からなる群から選択される結合剤を含む。
【0070】
1つの実施形態では、上記組成物を、サイズ3、サイズ2、サイズ1、サイズ0、またはサイズ00カプセルで提供する。
【0071】
1つの実施形態では、治療有効1日用量の上記組成物を、わずか2カプセルで投与する。別の実施形態では、治療有効1日用量の組成物を、わずか3つのサイズ1カプセルで投与する。別の実施形態では、治療有効1日用量の組成物を、わずか2つのサイズ0カプセルで投与する。さらにより好ましい実施形態では、治療有効1日用量の組成物を、わずか2つのサイズ1カプセルで投与する。別の実施形態では、治療有効1日用量の組成物を、わずか3つのサイズ2カプセルで投与する。
【0072】
好ましい実施形態では、上記組成物は、サイズ2カプセル中に50〜110mgのアマンタジンまたはその薬学的に許容され得る塩の量、およびサイズ1カプセル中に110mg〜210mgのアマンタジンまたはその薬学的に許容され得る塩の量で提供される。さらなる実施形態では、上記組成物は、直径300〜1000ミクロンであり、アマンタジンまたはその薬学的に許容され得る塩の含有量が40〜80重量%であり、嵩密度が0.5〜1.2g/cm3であるコーティングされたペレットを含む。さらに好ましい実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、上記組成物のアマンタジンのin vitro溶解プロフィールは、以下のうちの少なくとも1つを示す:(i)2時間で25%以下の溶解、(ii)6時間で40〜80%以下の溶解、および(iii)12時間で少なくとも80%の溶解。より特定の実施形態では、基準(i)、(ii)、および(iii)のうちの2つを満たす。さらなる特定の実施形態では、基準(i)、(ii)、および(iii)の3つ全てを満たす。
【0073】
1つの実施形態では、可塑剤は、中鎖トリグリセリド、フタル酸ジエチル、クエン酸エステル、ポリエチレングリコール、グリセロール、アセチル化グリセリド、およびヒマシ油からなる群から選択される。さらなる特定の実施形態では、可塑剤は中鎖トリグリセリド(例えば、ミグリオール812N)である。
【0074】
別の態様では、本発明は、アマンタジンまたはその薬学的に許容され得る塩を必要とするヒト被験体に投与する方法であって、任意の上記態様の組成物を経口投与する工程を含む、方法を提供する。
【0075】
別の態様では、本発明は、必要とするヒト被験体におけるパーキンソン病の処置方法であって、任意の上記態様の組成物を経口投与する工程を含む、方法を提供する。好ましい態様では、本発明は、必要とするヒト被験体の疾患を処置する方法であって、任意の上記態様の組成物を1日1回夜間に1、2、または3つのカプセルで経口投与する工程を含む、方法を提供する。
【0076】
アマンタジンの必要とする被験体への投与という言及には、NMDAアンタゴニストによって処置、防止、または治癒することができる疾患または容態を有する患者の処置が含まれる。より具体的には、アマンタジンの必要とする患者への投与には、パーキンソン病、脳損傷、脳外傷、認知症、アルツハイマー病、卒中、ハンチントン病、ALS、多発性硬化症、神経変性疾患、認知症、脳血管性容態、運動障害、脳神経障害、神経精神障害を有する患者の処置が含まれる。
【図面の簡単な説明】
【0077】
【図1】図1は、実施例3で言及した3つのアマンタジンER処方物A、B、Cの溶解プロフィールを示す。
【図2A】図2Aおよび2Bは、1日目および9日目の1日2回のアマンタジンIR(A)および1日1回のアマンタジンER(B)の健康な成人の男性および女性の被験体への絶食条件下での投与後の平均血漿濃度時間曲線を示す。
【図2B】図2Aおよび2Bは、1日目および9日目の1日2回のアマンタジンIR(A)および1日1回のアマンタジンER(B)の健康な成人の男性および女性の被験体への絶食条件下での投与後の平均血漿濃度時間曲線を示す。
【図3】図3は、9日目の1日2回のアマンタジンIRおよび1日1回のアマンタジンERの健康な成人の男性および女性の被験体への絶食条件下での投与後のアマンタジンの平均血漿濃度対時間曲線のプロットを示す。
【図4】図4は、1日2回または3回の種々の強度の即時放出アマンタジンの複数回投与後および1日1回の種々の強度のアマンタジンERの投与後のアマンタジンの平均血漿濃度対時間曲線をシミュレーションしている。
【図5】図5は、4つのアマンタジン処置についての平均(SD)血漿アマンタジン濃度対指定時間のプロットを示す。
【図6】図6は、4つのアマンタジン処置についての片対数の平均(SD)血漿アマンタジン濃度対指定時間を示す。
【図7】図7は、実施例12に記載のERアマンタジン処方物についての定常状態血漿濃度の時間プロフィールのシミュレーションを示す。夜間に1日1回投与したERアマンタジン処方物2により、処方物1と比較して定常状態でのピーク血漿濃度の到達時間が約4時間遅延する。
【発明を実施するための形態】
【0078】
発明の詳細な説明
本発明は、アマンタジン処置を受けた患者における睡眠障害の軽減方法を提供する。本方法は、アマンタジンが睡眠を妨害せず、さらに、アマンタジンを服用する多数の患者がしばしば最も必要とする朝の時間に最高に有益となり、さらに、必要に応じてパーキンソン病の症状に対して夜間に適用されるように、アマンタジンを必要とする被験体に投与する工程を含む。夜間適用は、患者が目覚めて再度就寝することを望む場合に有利である。
【0079】
本発明の方法は、夜間投与のためにデザインされた持続放出(ER)アマンタジン組成物を患者に経口投与する工程を含む。組成物を、就寝時刻より前の3時間未満、好ましくは2.5時間未満、2時間未満、1.5時間未満、または1時間未満に服用する。より好ましくは、ERアマンタジン組成物を、就寝時刻より前(すなわち、被験体が夜に就寝を望む時間)の30分未満に服用する。本明細書中で使用する場合、アマンタジンという言及は、その薬学的に許容され得る塩(例えば、塩酸アマンタジン、硫酸アマンタジンなど)が含まれることを意図する。あるいは、組成物を、就寝時間より前の約4時間未満に投与する。
【0080】
本明細書中で使用する場合、「持続放出」には、「制御放出」、「放出調節」、「徐放」、「時効性」、「遅延放出」が含まれ、遅延放出、即時放出、腸溶性コーティングなどと上記のそれぞれとの混合物も含まれる。
【0081】
患者は、アマンタジンが処方される任意の疾患または障害(パーキンソン病、多発性硬化症、薬物誘発性の錐体外路系の反応、レボドパ誘発性ジスキネジア、およびウイルス性疾患(例えば、インフルエンザ、HBV、およびHCV)など)と診断され得る。特定の実施形態では、患者は、パーキンソン病を有する。ここで、本明細書中で使用する場合、パーキンソン病は、パーキンソニズムの診断も含む。1つの実施形態では、患者は早期パーキンソン病を有し、アマンタジンを、単剤療法としてかレボドパを併用せずにモノアミンオキシダーゼB型(MAO−B)インヒビターと組み合わせて使用する。別の実施形態では、患者は後期パーキンソン病を有し、患者は、アマンタジンに加えてレボドパを服用する。別の実施形態では、患者は多発性硬化症を有し、アマンタジンを疲労処置のために使用する。他の実施形態では、患者は、脳損傷、脳損傷、脳外傷、認知症、アルツハイマー病、卒中、ハンチントン病、ALS、多発性硬化症、神経変性疾患、認知症、脳血管性容態、運動障害、脳神経障害、神経精神障害を有する。
【0082】
本発明で用いるERアマンタジン組成物は、被験体の睡眠を妨害しない血漿濃度プロフィールが得られる夜間投与に適している。本発明の組成物により、ヒト被験体への投与の際に、定常状態条件で、ある用量の組成物の投与によってその用量の投与3時間後にアマンタジンの血漿濃度が25%未満増加するようにアマンタジンの血漿濃度の初期漸増が認められるであろう。例えば、ある用量の組成物の投与時の被験体のアマンタジンの定常状態血漿濃度が500ng/mlである場合、3時間後の被験体のアマンタジンの血漿濃度は、625ng/ml未満であろう。好ましくは、アマンタジンの血漿濃度の増加は15%未満、最も好ましくは10%未満である。特に好ましい組成物は、投与後少なくとも1時間、好ましい実施形態では、2時間アマンタジン血漿濃度が増加しないことか、さらに減少すること(定常状態条件で)によってさらに特徴づけられる血漿濃度プロフィールを有する。本発明で用いる組成物は、朝の時間にアマンタジンの最高濃度(Cmax)を提供することによる就寝時刻(すなわち、被験体が夜に就寝を望む時間)の投与にさらに適している。絶食状態での単回用量投与後に測定したCmax到達時間(Tmax)は、少なくとも8時間且つ13、14、15、または16時間まで、少なくとも9時間且つ13、14、15、または16時間まで、または少なくとも10時間且つ13、14、15、または16時間までである。特定の実施形態は、Tmaxは9〜15時間、好ましくは10〜14時間、最も好ましくは11〜13時間である。定常状態では、組成物の1日1回の投与を使用して、Tmaxは7〜13時間、好ましくは8〜12時間、最も好ましくは9〜11時間である。適切なERアマンタジン組成物を、定常状態Cmax/Cmin比1.5〜2.0、好ましくは1.7〜1.9を有し、それにより、最適に変動する組成物が得られることによってさらに特徴づけることができる。
【0083】
より特定の好ましい実施形態では、血漿濃度プロフィールは、単回用量の組成物の投与後に以下のAUCプロフィールを有することによってさらに特徴づけられる:AUC0−infの5%未満、好ましくは3%未満である0〜4時間の断片的AUC、AUC0−infの約5〜15%、好ましくは約8〜12%である0〜8時間の断片的AUC、AUC0−infの約10〜40%、好ましくは約15〜30%である0〜12時間の断片的AUC、AUC0−infの約25〜60%、好ましくは約30〜50%である0〜18時間の断片的AUC、およびAUC0−infの約40〜75%、好ましくは約50〜70%である0〜24時間の断片的AUC。
【0084】
さらに好ましい実施形態では、血漿濃度プロフィールは、以下によって特徴づけられる定常状態条件での組成物の1日1回の投与後のAUCプロフィールを有することによってさらに特徴づけられる:AUC24の約2〜25%、好ましくは約5〜20%である0〜4時間の断片的AUC、AUC24の約15〜50%、好ましくは約20〜40%である0〜8時間の断片的AUC、AUC24の約30〜70%、好ましくは約40〜60%である0〜12時間の断片的AUC、およびAUC24の約60〜95%、好ましくは約75〜90%である0〜18時間の断片的AUC。
【0085】
任意の上記態様のいくつかの実施形態では、就寝時刻の組成物のヒト被験体への複数回の投与後の定常状態の血漿濃度プロフィールは、日中の平均血漿濃度(「C−ave−日中」、ヒトPK研究で測定した場合の日中平均アマンタジン血漿濃度として定義)が夜間の平均血漿濃度(「C−ave−夜」、ヒトPK研究で測定した場合の夜間平均アマンタジン血漿濃度として定義)の1.1〜2.0倍であることによって特徴づけられる。いくつかの実施形態では、定常状態でのC−ave−日中/C−ave−夜比は、以下の範囲の1つである:1.1〜1.9、1.1〜1.8、1.1〜1.7、1.1〜1.6、1.1〜1.5、1.1〜1.4、1.2〜1.9、1.2〜1.7、1.2〜1.6、1.2〜1.5、1.3〜1.9、1.3〜1.8、1.3〜1.7、1.3〜1.6、1.4〜1.9、1.4〜1.8、1.4〜1.7、1.5〜1.9、1.5〜1.8、1.5〜1.7、1.6〜1.9、1.6〜1.8、または1.7〜1.9。いくつかの実施形態では、定常状態でのC−ave−日中/C−ave−夜比は、1.1、1.15、1.2、1.25、1.3、1.35、1.4、1.45、1.5、1.55、1.6、1.65、1.7、1.75、1.8、1.85、1.9、1.95、または2.0である。いくつかの実施形態では、C−ave−日中は、午前5時、午前6時、午前7時、午前8時、または午前9時から午後4時、午後5時、午後6時、午後7時、または午後8時の間に測定した平均アマンタジン血漿濃度であり、C−ave−夜は、午後4時、午後5時、午後6時、午後7時、午後8時、午後9時、午後10時、または午後11時から午前5時、午前6時、午前7時、午前8時、または午前9時までに測定した平均アマンタジン血漿濃度である。いくつかの実施形態では、C−ave−日中は、午前5時と午後8時との間の任意の4〜12時間内に測定した平均アマンタジン血漿濃度であり、C−ave−夜は、午後8時と午前5時との間の任意の4〜12時間に測定した平均アマンタジン血漿濃度である。いくつかの実施形態では、C−ave−日中は、午前5時と午後8時との間の任意の4、5、6、7、8、9、10、11、または12時間内に測定した平均アマンタジン血漿濃度であり、C−ave−夜は、午後8時と午前5時との間の任意の4、5、6、7、8、9、10、11、または12時間内に測定した平均アマンタジン血漿濃度である。
【0086】
本明細書中に記載のいくつかの実施形態では、アマンタジン組成物を、就寝時刻の0〜4時間前に患者に投与する。いくつかの実施形態では、アマンタジン組成物を、就寝時刻の0〜3、0〜2、または0〜1時間前に患者に投与する。いくつかの実施形態では、アマンタジン組成物を、就寝時刻の0〜240分前、0〜180分前(例えば、0〜120分前、0〜60分前、0〜45分前、0〜30分前、0〜15分前、または0〜10分前)に患者に投与する。いくつかの実施形態では、アマンタジン組成物を、就寝時刻の60〜240分前、60〜180分前、60〜120分前、または60〜90分前に患者に投与する。
【0087】
患者への投与には、医療専門家による投与および患者による自己投与が含まれると理解すべきである。
【0088】
本明細書中に別記されない限り、用語「就寝時刻」は、24時間のうちの主な睡眠時間という通常の意味を有する。一般大衆では就寝時刻は夜であるが、就寝時刻が日中である患者(夜に働く患者)が存在する。したがって、いくつかの実施形態では、就寝時刻は、日中または夜間の任意の時間であり得る。
【0089】
本明細書中で使用する場合、他で示さない限り、ヒト被験体における血漿濃度プロフィールまたは特定の薬物動態学的性質(例えば、Cmax、Cmin、AUC、Tmaxなど)という言及は、薬物の薬物動態学的性質を測定するためにデザインされた典型的な第I相臨床試験で決定された健康な成人から得た平均値をいう(例えば、以下の実施例5、6、および7を参照のこと)。本明細書中のTmaxという言及は、他で示さない限り、絶食状態での単回用量の投与後に得た値をいう。
【0090】
本発明のいくつかの実施形態では、本発明のアマンタジンの投与量は、通常はアマンタジンの即時放出組成物のために処方された範囲内または範囲を超える。他の実施形態では、本発明のアマンタジンの投与量は、通常はアマンタジンの即時放出組成物のために処方された範囲を超える。例えば、パーキンソン病処置のためのアマンタジンの推奨用量は、100mg(1日2回投与)である。前述の用量で十分な利点が得られず、且つ患者がかかるより高い用量を許容することができる一部の患者の場合、用量を300mgまたは400mg(分割量)に増加させることができる。最も一般的なアマンタジンの処方用量は100mg〜200mg/日であり、後者は、分割量で投与する。200mg超(例えば、300mg)を、常に分割量で投与する。本発明のために、50〜600mg、より好ましくは200〜450mgの用量をパーキンソン病処置のために投与し、本発明の方法および組成物は、これらの範囲のいずれかによって定義される用量の投与を含むことができる。特定の実施形態では、かかるより高い用量を1日1回投与することができる。さらなる実施形態では、かかるより高い用量を夜間に投与することができる。さらなる実施形態では、かかるより高い用量をサイズ0、1、または2の1、2、または3カプセルの形態で1日1回投与することができる。
【0091】
任意の上記態様の1つの実施形態では、アマンタジンを薬学的に許容され得る塩として投与する。さらなる特定の実施形態では、アマンタジンを塩酸塩または硫酸アマンタジンとして投与する。
【0092】
任意の上記態様の1つの実施形態では、総1日用量50mg〜600mgのアマンタジンまたはその薬学的に許容され得る塩を患者に投与する。より具体的には、アマンタジンまたはその薬学的に許容され得る塩の1日用量は、100mg〜440mgの範囲であり得る。別の特定の実施形態では、アマンタジンまたはその薬学的に許容され得る塩の1日用量は、260mg〜420mgの範囲であり得る。別の実施形態では、アマンタジンまたはその薬学的に許容され得る塩の1日用量は、300mg/日を超える。種々の特定の実施形態では、アマンタジンまたはその薬学的に許容され得る塩の1日用量は、50〜75mg、70〜95mg、90〜115mg、110〜135mg、130〜155mg、150〜175mg、170〜195mg、190〜215mg、210〜235mg、230〜255mg、250〜275mg、270〜295mg、290〜305mg、300〜315mg、310〜325mg、320〜335mg、330〜345mg、340〜355mg、350〜365mg、360〜375mg、370〜385mg、380〜395mg、390〜405mg、400〜415mg、410〜425mg、420〜435mg、430〜445mg、または440〜455mgであり得る。
【0093】
任意の上記態様の1つの実施形態では、組成物は、50〜600mgのアマンタジンまたはその薬学的に許容され得る塩を含む。より具体的には、組成物は、100〜450mgのアマンタジンまたはその薬学的に許容され得る塩を含むことができる。さらにより具体的には、組成物は、130〜210mgのアマンタジンまたはその薬学的に許容され得る塩を含むことができる。種々の特定の実施形態では、投薬形態は、50〜75mg、70〜95mg、90〜115mg、110〜135mg、130〜155mg、150〜175mg、170〜195mg、190〜215mg、210〜235mg、230〜255mg、250〜275mg、270〜295mg、290〜305mg、300〜315mg、310〜325mg、320〜335mg、330〜345mg、340〜355mg、350〜365mg、360〜375mg、370〜385mg、380〜395mg、390〜405mg、400〜415mg、410〜425mg、420〜435mg、430〜445mg、または440〜455mgのアマンタジンまたはその薬学的に許容され得る塩を含む。さらなる特定の実施形態では、組成物は、約110、120、130、140、150、160 170、180、190、210、または220mgのアマンタジンまたはその薬学的に許容され得る塩を含む。別のさらなる特定の実施形態では、組成物は110mgの塩酸アマンタジンを含む。別のさらなる特定の実施形態では、組成物は130mgの塩酸アマンタジンを含む。別のさらなる特定の実施形態では、組成物は170mgの塩酸アマンタジンを含む。別のさらなる特定の実施形態では、組成物は210mgの塩酸アマンタジンを含む。
【0094】
任意の上記態様の1つの実施形態では、組成物は、約50mg、60mg、70mg、80mg、90mg、100mg、110mg、120mg、130mg、140mg、150mg、160mg、170mg、180mg、190mg、200mg、210mg、220mg、230mg、240mg、250mg、260mgのアマンタジンまたはその薬学的に許容され得る塩から約75mg、85mg、95mg、105mg、115mg、125mg、135mg、145mg、155mg、165mg、175mg、185mg、195mg、205mg、215mg、225mg、235mg、245mg、255mg、265mg、275mg、285mg、295mg、305mg、315mg、325mg、335mg、345mg、355mg、365mg、375mg、385mg、395mg、405mg、415mg、425mg、435mg、445mgまでのアマンタジンまたはその薬学的に許容され得る塩を含む。
【0095】
本発明の特定の実施形態では、被験体のアマンタジンの全1日用量を、就寝時刻より前(すなわち、被験体が夜に就寝を望む時間)の約3、2、または1時間未満に1回投与する。他の実施形態では、アマンタジンの1日用量の少なくとも半分を、前述の就寝時刻前に服用する。好ましくは、アマンタジン用量の少なくとも2/3を、前述の就寝時刻前に服用し、残りを午前中または午後に服用する。アマンタジンの午前中または昼の用量を、従来の即時放出投薬形態または持続放出形態で提供することができる。
【0096】
任意の上記態様の1つの実施形態では、組成物のパーキンソン病患者への投与により、レボドパ誘発性ジスキネジアが有意に軽減される。特定の実施形態では、組成物の投与により、レボドパ誘発性ジスキネジアが約5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、または80%軽減される。さらなる実施形態では、レボドパ誘発性ジスキネジアの軽減を、LID処置の有効性を評価し、認可薬を認可するためのFDAまたは他の規制当局によって使用または承認される数値的尺度によって測定する。さらなる特定の実施形態では、LID軽減の測定で使用される尺度は、UDysRS、UPDRSパートIV(サブスコア32、33)、ジスキネジア評点尺度(DRS)、異常不随意運動尺度(AIMS)、Rushジスキネジア評点尺度、パーキンソン病ジスキネジア尺度(PDYS−26)、Obesoジスキネジア評点尺度(CAPIT)、臨床ジスキネジア評点尺度(CDRS)、日常生活ジスキネジアのLang−Fahn活動、またはこの目的のために開発された他の尺度であり得る。
【0097】
任意の上記態様の1つの実施形態では、組成物のパーキンソン病患者への投与により、パーキンソン病性疲労が有意に軽減される。特定の実施形態では、組成物の投与により、パーキンソン病性疲労が約5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、または60%軽減される。さらなる特定の実施形態では、疲労の軽減を、疲労処置の有効性を評価し、認可薬を認可するためのFDAまたは他の規制当局によって使用または承認される数値的尺度によって測定する。さらなる特定の実施形態では、疲労軽減の測定に使用される尺度は、疲労重症度尺度(FSS)、疲労評価目録、慢性疾患治療の機能的評価−疲労(FACIT疲労)、多次元疲労目録(MFI−20)、パーキンソン疲労尺度(PFS−16)、および疲労重症度目録であり得る。他の特定の実施形態では、疲労の軽減を、比較臨床試験においてプラセボと比較して測定する。他の実施形態では、疲労の軽減を、比較臨床試験においてベースラインと比較して測定する。
【0098】
任意の上記態様の1つの実施形態では、組成物のパーキンソン病患者への投与により、パーキンソン病の症状が有意に軽減される。特定の実施形態では、組成物の投与により、パーキンソンの症状が約5%、10%、15%、20%、25%、30%、35%、または40%軽減される。さらなる特定の実施形態では、パーキンソンの症状の軽減を、パーキンソンの症状処置の有効性を評価し、認可薬を認可するためのFDAまたは他の規制当局によって使用または承認される数値的尺度によって測定する。さらなる特定の実施形態では、パーキンソンの症状軽減の測定に使用される尺度は、統合パーキンソン病評点尺度(UPDRS)であり得る。統合パーキンソン病評点尺度(UPDRS、MDS改訂)−パートI:日常生活で経験する非運動性の状況(13項目)、パートII:日常生活で経験する運動性の状況(13項目)−パートIII:運動性の検査(33スコア化項目)−パートI: 精神状態、行動、および気分−パートII:日常生活における活動−パートIII:運動性の検査(27スコア化項目)Hoehn and Yahrの病期分類尺度(オリジナルまたは改訂版)。
【0099】
任意の上記態様の1つの実施形態では、組成物のパーキンソン病患者への投与により、レボドパ誘発性ジスキネジアが有意に軽減される。特定の実施形態では、組成物の投与により、レボドパ誘発性ジスキネジアが約5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、または80%軽減される。さらなる実施形態では、レボドパ誘発性ジスキネジアの軽減を、LIDの軽減を示す薬物の有効性を評価するためのFDAによって使用される数値的尺度によって測定する。さらなる特定の実施形態では、LID軽減の測定に使用される尺度は、UDysRS、UPDRSパートIV(サブスコア32、33)、ジスキネジア評点尺度(DRS)、異常不随意運動尺度(AIMS)、またはこの目的のために開発された他の尺度であり得る。他の特定の実施形態では、LIDの軽減を、比較臨床試験においてプラセボと比較して測定する。他の実施形態では、LIDの軽減を、比較臨床試験においてベースラインと比較して測定する。
【0100】
任意の上記態様の1つの実施形態では、組成物のパーキンソン病患者への投与により、パーキンソン病性疲労が有意に軽減される。特定の実施形態では、組成物の投与により、パーキンソン病性疲労が約5%、10%、15%、20%、25%、30%、35%、または40%軽減される。さらなる特定の実施形態では、疲労の軽減を、疲労の軽減を示す薬物の有効性を評価するためのFDAによって使用される数値的尺度によって測定する。さらなる特定の実施形態では、疲労軽減の測定に使用される尺度は、疲労重症度尺度(FSS)であり得る。
【0101】
任意の上記態様の1つの実施形態では、組成物のパーキンソン病患者への投与により、パーキンソン病の症状が有意に軽減される。特定の実施形態では、組成物の投与により、パーキンソンの症状が約5%、10%、15%、20%、25%、30%、35%、または40%軽減される。さらなる特定の実施形態では、パーキンソンの症状の軽減を、パーキンソンの症状の軽減を示す薬物の有効性を評価するためのFDAによって使用される数値的尺度によって測定する。さらなる特定の実施形態では、パーキンソンの症状軽減の測定に使用される尺度は、統合パーキンソン病評点尺度(UPDRS)であり得る。他の特定の実施形態では、パーキンソン病の症状の軽減を、比較臨床試験においてプラセボと比較して測定する。他の実施形態では、パーキンソン病の症状の軽減を、比較臨床試験においてベースラインと比較して測定する。
【0102】
持続放出処方物
本発明の方法での使用に適切な持続放出アマンタジン組成物を、種々の持続放出テクノロジー(上記の背景の項で参照した特許公報(公報はその全体が本明細書中で参考として援用される)に記載のテクノロジーなど)を使用して作製することができる。いくつかの実施形態では、本発明は、カプセル投薬形態のペレットである。いくつかの実施形態では、ペレットはペレットコアを含み、ペレットコアは、少なくとも1つの薬物層および少なくとも1つの持続放出コーティング層でコーティングされている。いくつかの実施形態では、ペレットを、少なくとも1つの薬物層、中間層(シールコートなど)、および持続放出コーティング層でコーティングする。いくつかの実施形態では、ペレット、薬物層、またはその両方は、1つ以上の結合剤を含む。
【0103】
いくつかの実施形態では、投薬単位は、複数のコーティングしたペレットを含む。いくつかの実施形態では、ペレットの直径は、例えば、300〜1700ミクロン、いくつかの場合、500〜1200ミクロンである。ペレットは、例えば、不活性物質(糖スフェア、微結晶性セルロース(MCC)スフェア、デンプンペレットなど)を含むであろう。いくつかの実施形態では、ペレットを、他の過程(ペレット化、押し出し、球形化など)またはその組み合わせによって調製することができる。コアペレットは、塩酸アマンタジンおよび薬学的に許容され得る賦形剤から構成されるであろう。
【0104】
コーティングしたペレット
ペレットコアを、有効成分(例えば、アマンタジンまたはその薬学的に許容され得る塩および/もしくは多形)でコーティングする。いくつかの実施形態では、有効成分に加えて、ペレットはまた、1つ以上の結合剤(例えば、ヒドロキシプロピルメチルセルロース、コポビドン、ポビドン、ヒドロキシプロピルセルロース、ヒドロキシエチルセルロース、メチルセルロース、カルボキシメチルセルロースなど)を含む。いくつかの実施形態では、ペレットはまた、1つ以上のさらなる賦形剤(粘着防止剤(例えば、タルク、ステアリン酸マグネシウムなど)など)を含む。
【0105】
いくつかの実施形態では、ペレットコアを、従来のコーティング技術(流動化層コーティング、パンコーティングなど)によって、有効成分、および任意選択的な1つ以上の結合剤、粘着防止剤、および/または溶媒を含む薬物層でコーティングする。
【0106】
中間層コーティング
いくつかの実施形態では、ペレットを、中間層(シールコートなど)でコーティングする。いくつかの実施形態では、シールコートは、持続放出コーティング中の成分によるペレットコア中の成分の妨害の防止、ペレットコア中の成分のペレットコアから持続放出層への拡散の防止などに適応する。本明細書中に記載のように、本発明のシールコートは、1つ以上のフィルム形成ポリマー(ヒドロキシプロピルメチルセルロース(HPMC)、コポビドン、ポビドン、ポリビニルピロリドン、ヒドロキシプロピルセルロース、ヒドロキシエチルセルロース、メチルセルロース、カルボキシメチルセルロース、またはその任意の組み合わせなどが含まれるが、これらに限定されない)を含むことができる。
【0107】
シールコートは、可塑剤(プロピレングリコール、トリアセチン、ポリエチレングリコール、クエン酸トリブチルなど)および任意選択的な粘着防止剤(ステアリン酸マグネシウム、ケイ酸カルシウム、ケイ酸マグネシウム、およびコロイド状二酸化ケイ素、またはタルクなど)のような他の添加物をさらに含むことができる。
【0108】
上記の可塑剤および粘着防止剤は別として、シールコートは、任意選択的に、当業者に公知の緩衝液、着色剤、乳白剤、界面活性剤、または基剤を含むことができる。
【0109】
シールコーティングを、従来のコーティング技術(流動化層コーティング、パンコーティングなど)を使用してコアに適用することができる。いくつかの実施形態では、薬物コーティングしたペレットコアを、流動化層コーティングまたはパンコーティングによって1つ以上の結合剤、粘着防止剤、および/または溶媒を任意選択的に含むシールコート層でコーティングする。
【0110】
結合剤
いくつかの実施形態では、ペレットコア、中間コーティング層、またはその両方のいずれかは、1つ以上の結合剤(例えば、フィルム形成ポリマー)を含むことができる。本明細書中で使用する適切な結合剤には、例えば、アルギン酸およびその塩、セルロース誘導体(カルボキシメチルセルロース、メチルセルロース(例えば、メトセル(登録商標))、ヒドロキシプロピルメチルセルロース、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース(例えば、クルセル(登録商標))、エチルセルロース(例えば、エトセル(登録商標))、および微結晶性セルロース(例えば、アビセル(登録商標))など);微結晶性デキストロース、アミロース、マグネシウムケイ酸アルミニウム、多糖酸、ベントナイト、ゼラチン、ポリビニルピロリドン/酢酸ビニルコポリマー、クロスポビドン、ポビドン、デンプン、アルファ化デンプン、トラガカント、デキストリン、糖(スクロース(例えば、ダイパック(登録商標))、グルコース、デキストロース、糖蜜、マンニトール、ソルビトール、キシリトール(例えば、キシリタブ(登録商標))、およびラクトースなど)、天然ゴムまたは合成ゴム(アカシア、トラガカント、ガッチゴムなど)、イサポール外皮の粘液、ポリビニルピロリドン(例えば、ポリビドン(登録商標)CL、コリドン(登録商標)CL、ポリプラスドン(登録商標)XL−10)、カラマツのアラビノガラクタン(larch arabogalactan)、ビーガム(登録商標)、ポリエチレングリコール、ワックス、およびアルギン酸ナトリウムなどが含まれる。
【0111】
持続放出コーティング
ペレットを、持続放出コーティングでコーティングする。持続放出コーティングは、投薬形態の使用環境への導入後の一定期間コーティングされた薬物コアからの薬物の放出を遅延させるように構成される。いくつかの実施形態では、持続放出コーティングは、1つ以上のpH依存性またはpH非依存性の持続放出賦形剤を含む。pH非依存性持続放出ポリマーの例には、エチルセルロース、ヒドロキシプロピルメチルセルロース、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース、カルボキシメチルセルロース、エチルアクリラートのコポリマー、メチルメタクリラート(例えば、オイドラギットRS)などが含まれる。pH依存性持続放出賦形剤の例には、メタクリル酸(methacrylic acic)コポリマー、ヒドロキシプロピルメチル酢酸セルローススクシナート、ヒドロキシプロピルメチルセルロースフタラート、およびセルロースアセタートフタラートなどが含まれる。持続放出コーティングには、ポア形成物質(ポビドン、ポリエチレングリコール、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロースなど)、糖(スクロース、マンニトール、ラクトースなど)、および塩(塩化ナトリウム、クエン酸ナトリウムなど)、可塑剤(アセチル化クエン酸化エステル、アセチル化グリセリド、ヒマシ油、クエン酸エステル、セバジン酸ジブチル、モノステアリン酸グリセリル、フタル酸ジエチル、グリセロール、中鎖トリグリセリド、プロピレングリコール、ポリエチレングリコールなど)も含まれ得る。持続放出コーティングには、1つ以上のさらなる賦形剤(潤滑剤(例えば、ステアリン酸マグネシウム、タルクなど)など)も含まれ得る。
【0112】
持続放出コーティングを、従来のコーティング技術(流動化層コーティング、パンコーティングなど)を使用して適用することができる。任意選択的にシールコートを含む薬物コーティングしたペレットコアを、流動化層コーティングによって持続放出コーティングでコーティングする。
【0113】
持続放出賦形剤(コーティングポリマー)
本明細書中に記載のように、例示的な持続放出賦形剤には、不溶性プラスチック、親水性ポリマー、および脂肪族化合物が含まれるが、これらに限定されない。プラスチック母材には、メチルアクリラート−メチルメタクリラート、ポリ塩化ビニル、およびポリエチレンが含まれるが、これらに限定されない。親水性ポリマーには、セルロースポリマー(メチルセルロースおよびエチルセルロースなど)、ヒドロキシアルキルセルロース(ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、カルボキシメチルセルロースナトリウムなど)、および架橋アクリル酸ポリマー(カーボポール(登録商標)934、ポリエチレンオキシドなど)、およびその混合物が含まれるが、これらに限定されない。脂肪族化合物には、種々のワックス(カルナウバ蝋およびトリステアリン酸グリセリルなど)およびワックス型物質(硬化ヒマシ油または硬化植物油が含まれる)、またはその混合物が含まれるが、これらに限定されない。
【0114】
一定の実施形態では、可塑性物質は、薬学的に許容され得るアクリルポリマー(アクリル酸およびメタクリル酸コポリマー、メチルメタクリラート、メチルメタクリラートコポリマー、エトキシエチルメタクリラート、シアノエチルメタクリラート、アミノアルキルメタクリラートコポリマー、ポリ(アクリル酸)、ポリ(メタクリル酸)、メタクリル酸 アルキルアミンコポリマーポリ(メチルメタクリラート)、ポリ(メタクリル酸)(無水物)、ポリメタクリラート、ポリアクリルアミド、ポリ(メタクリル酸無水物)、およびグリシジルメタクリラートコポリマーが含まれるが、これらに限定されない)であり得る。
【0115】
一定の他の実施形態では、アクリルポリマーは、1つ以上のアンモニオメタクリラートコポリマーから構成される。アンモニオメタクリラートコポリマーは当該分野で周知であり、アクリル酸エステルおよびメタクリル酸エステルの低含有量の第四級アンモニウム基との完全重合したコポリマーとしてNF XVIIに記載されている。
【0116】
さらなる他の実施形態では、アクリルポリマーは、アクリル樹脂ラッカー(Rohm Pharmaから商標名オイドラギット(登録商標)で市販されているものなど)である。さらなる実施形態では、アクリルポリマーは、Rohm Pharmaからそれぞれ商標名オイドラギット(登録商標)RL30Dおよびオイドラギット(登録商標)RS30Dで市販されている2つのアクリル樹脂ラッカーの混合物を含む。オイドラギット(登録商標)RL30Dおよびオイドラギット(登録商標)RS30Dは、アクリル酸エステルおよびメタクリル酸エステルの低含有量の第四級アンモニウム基とのコポリマーであり、アンモニウム基と中性(メタクリル酸)アクリル酸エステルとのモル比がオイドラギットRL30Dでは1:20であり、オイドラギット(登録商標)RS30Dでは1:40である。平均分子量は約150,000である。オイドラギット(Edragit)(登録商標)S−100およびオイドラギット(登録商標)L−100も本明細書中の使用に適切である。コード名RL(高透過性)およびRS(低透過性)は、これらの薬剤の透過性をいう。オイドラギット(登録商標)RL/RS混合物は、水および消化液に不溶性を示す。しかし、これを含むように形成された多粒子系は、水溶液および消化液中で膨潤性および透過性を示す。
【0117】
上記のポリマー(オイドラギット(登録商標)RL/RSなど)を、最終的に所望の溶解プロフィールを有する持続放出処方物を得るために、任意の所望の比で混合することができる。当業者は、他のアクリルポリマー(例えば、オイドラギット(登録商標)Lなど)も使用することができると認識するであろう。
【0118】
ポア形成物質
いくつかの実施形態では、持続放出コーティングは、ポア形成物質を含む。持続放出コーティングでの使用に適切なポア形成物質は、有機または無機の薬剤であり得、使用環境下でコーティングから溶解、抽出、または浸出することができる材料が含まれる。ポア形成物質の例には、有機化合物(モノサッカリド、オリゴサッカリド、およびポリサッカリド(スクロース、グルコース、フルクトース、マンニトール、マンノース、ガラクトース、ラクトース、ソルビトール、プルラン、デキストランなど);水溶性親水性ポリマーなどの使用環境下で溶解性を示すポリマー(ポビドン、クロスポビドン、ポリエチレングリコール、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ヒドロキシアルキルセルロース、カルボキシアルキルセルロース、セルロースエーテル、アクリル樹脂、ポリビニルピロリドン、架橋ポリビニルピロリドン、ポリエチレンオキシド、カーボワックス、およびカーボポール(登録商標)など)、ジオール、ポリオール、多価アルコール、ポリアルキレングリコール、ポリエチレングリコール、ポリプロピレングリコール、またはそのブロック重合体、ポリグリコール、ポリ(α−Ω)アルキレンジオールなど);無機化合物(アルカリ金属塩、炭酸リチウム、塩化ナトリウム、臭化ナトリウム、塩化カリウム、硫酸カリウム、リン酸カリウム、酢酸ナトリウム、クエン酸ナトリウム、適切なカルシウム塩など)など)が含まれるが、これらに限定されない。一定の実施形態では、可塑剤を、ポア形成物質として使用することもできる。
【0119】
カプセル
持続放出ペレットを、ペレット投薬チャンバーを備えたエンカプスレータの使用によって適切なカプセルに導入する。カプセルサイズは、00、0、0EL、1、1EL、2、2EL、3、4、または5であり得る。理想的な薬物動態学的性質および血漿濃度プロフィールを提供する特に好ましい組成物は、典型的には直径が約500μm〜1.2mm、好ましくは約700μm〜1000μmの複数のペレットを含み、各ペレットがアマンタジンおよび結合剤、ならびに上記の所望の薬物動態学的性質およびアマンタジン血漿濃度プロフィールを得るためにアマンタジン放出を持続させるコアを囲む持続放出コーティングを含む、ペレット・イン・カプセル組成物である。
【0120】
いくつかの実施形態では、ペレット・イン・カプセル中のペレットは、サイズ0以下、好ましくはサイズ1以下のカプセルである。平均ペレット直径は、いくつかの実施形態では、500μm〜1200μmの範囲(例えば、500μm〜1100μm、500μm〜1000μm、500μm〜900μm、500μm〜800μm、500μm〜700μm、600μm〜1100μm、600μm〜1000μm、600μm〜900μm、600μm〜800μm、600μm〜700μm、700μm〜1100μm、700μm〜1000μm、700μm〜900μm、または700μm〜800μm)であり得る。いくつかの実施形態では、平均粒子直径は、±10%(例えば、500μm、550μm、600μm、650μm、700μm、750μm、800μm、850μm、900μm、950μm、1000μm、1050μm、1100μm、1150μm、または1200μm)である。
【0121】
本発明の1つの好ましい組成物は、ペレット・イン・カプセル組成物であって、各ペレットがコアシード上にコーティングしたアマンタジンと結合剤との混合物を有するコアシードを含むコア、ならびにエチルセルロース、ポア形成剤(ヒドロキシプロピルメチルセルロースまたはポビドンなど)、および可塑剤を含むコアを囲む持続放出コーティングを含む、ペレット・イン・カプセル組成物である。いくつかの実施形態では、ペレットは、ペレットコアと持続放出コーティングとの間にシールコーティングをさらに含むことができる。ペレットを、当該分野で公知の方法(以下の実施例1に記載の方法など)を使用して処方する。特定の実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、アマンタジンは、20〜80重量%、45〜70重量%、40〜50重量%、45〜55重量%、50〜60重量%、55〜65重量%、60〜70重量%、65〜75重量%、70〜80重量%、または40〜60重量%の量で存在し、結合剤(好ましくはヒドロキシプロピルメチルセルロース、コポビドン、またはその混合物である)は、8〜25重量%の量で存在し、コアシード(好ましくは、糖スフェア(ノンパレイユ)または微結晶性セルロースシード(例えば、セルフィア(登録商標)))は、1〜25重量%の量で存在し、エチルセルロースは、10〜20重量%の量で存在し、ポア形成剤(好ましくは、ポビドン)は、1〜4重量%の量で存在し、可塑剤は、1〜4重量%の量で存在する。別の特定の実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、アマンタジンは、50〜70重量%の量で存在し、結合剤(好ましくは、ヒドロキシプロピルメチルセルロース、コポビドン、またはその混合物である)は、5〜25重量%の量で存在し、コアシード(好ましくは、糖スフェア(ノンパレイユ)または微結晶性セルロースシード(例えば、セルフィア(登録商標)))は、5〜15重量%の量で存在し、エチルセルロースは、1〜15重量%の量で存在し、ポア形成剤(好ましくは、ポビドン)は、0.25〜4重量%の量で存在し、可塑剤は、0.25〜4重量%の量で存在する。
【0122】
本発明のさらなる実施形態を、以下の表に「種々のアマンタジンERカプセルサイズ1処方物」というタイトルで示す。本明細書中に記載の方法および組成物を用いて、本明細書中に記載の所望の溶解性および標的薬物動態学的プロフィールを達成する処方物を作製することができる。より具体的には、これらの結果を得るために本明細書中に記載している製造方法および組成物を使用して、治療有効用量のアマンタジンをわずか2つのサイズ1(またはそれ未満(例えば、サイズ2または3))カプセルにて1日1回投与することができる。特に、本明細書中に記載の組成物および製造方法を使用して、より高い薬物負荷を達成することができる。いくつかの実施形態では、より小さなコアペレットサイズを使用して必要な溶解プロフィールを有し、同時により小さなコア上の薬物層を増加させるが、持続放出コートが変化しないというより高い薬物負荷を達成することができる。いくつかの実施形態では、本明細書中に記載の別の製造アプローチ(例えば、押し出しおよび球形化)を使用して、所望の溶解プロフィールを実現するためのさらにより高い薬物負荷を達成することができ、それにより、適切な薬物動態学的プロフィールにてより高いアマンタジン薬負荷が可能であり、その結果、治療的により有効であり、少なくとも同様に許容され、比較的小さなサイズのカプセル(例えば、サイズ1、2、または3)に充填することができ、患者への投与を容易にすることができる組成物が得られる。
【0123】
【表1A】
いくつかの実施形態では、アマンタジンまたはその薬学的に許容され得る塩は、20〜80重量%の量(ペレットコアおよび持続放出コーティングを合わせた重量に基づく)、嵩密度0.3〜1.2g/cm3で存在する。いくつかの実施形態では、アマンタジンまたはその薬学的に許容され得る塩は、20〜77.5重量%、20〜75重量%、20〜72.5重量%、20〜70重量%、20〜67.5重量%、20〜65重量%、20〜62.5重量%、20〜60重量%、20〜57.5重量%、20〜55重量%、20〜52.5重量%、20〜50重量%、20〜47.5重量%、20〜45重量%、20〜42.5重量%、20〜40重量%、20〜37.5重量%、20〜35重量%、20〜32.5重量%、20〜30重量%、30〜80重量%、30〜77.5重量%、30〜75重量%、30〜72.5重量%、30〜70重量%、30〜67.5重量%、30〜65重量%、30〜62.5重量%、30〜60重量%、30〜57.5重量%、30〜55重量%、30〜52.5重量%、30〜50重量%、30〜47.5重量%、30〜45重量%、30〜42.5重量%、30〜40重量%、40〜80重量%、40〜77.5重量%、40〜75重量%、40〜72.5重量%、40〜70重量%、40〜67.5重量%、40〜65重量%、40〜62.5重量%、40〜60重量%、40〜57.5重量%、40〜55重量%、40〜52.5重量%、40〜50重量%、40〜47.5重量%、40〜45重量%、50〜80重量%、50〜77.5重量%、50〜75重量%、50〜72.5重量%、50〜70重量%、50〜67.5重量%、50〜65重量%、50〜62.5重量%、50〜60重量%、50〜57.5重量%、50〜55重量%、60〜80重量%、60〜77.5重量%、60〜75重量%、60〜72.5重量%、60〜70重量%、60〜67.5重量%、60〜65重量%の量で存在する。いくつかの実施形態では、嵩密度は、0.3〜1.2g/cm3、0.3〜1.15g/cm3、0.3〜1.1g/cm3、0.3〜1.05g/cm3、0.3〜1.0g/cm3、0.3〜0.9g/cm3、0.3〜0.8g/cm3、0.3〜0.7g/cm3、0.3〜0.6g/cm3、0.3〜0.5g/cm3、0.3〜0.4g/cm3、0.4〜1.2g/cm3、0.4〜1.15g/cm3、0.4〜1.1g/cm3、0.4〜1.05g/cm3、0.4〜1.0g/cm3、0.4〜0.9g/cm3、0.4〜0.8g/cm3、0.4〜0.7g/cm3、0.4〜0.6g/cm3、0.4〜0.5g/cm3、0.5〜1.2g/cm3、0.5〜1.15g/cm3、0.5〜1.1g/cm3、0.5〜1.05g/cm3、0.5〜1.0g/cm3、0.5〜0.9g/cm3、0.5〜0.8g/cm3、0.5〜0.7g/cm3、0.5〜0.6g/cm3、0.6〜1.2g/cm3、0.6〜1.15g/cm3、0.6〜1.1g/cm3、0.6〜1.05g/cm3、0.6〜1.0g/cm3、0.6〜0.9g/cm3、0.6〜0.8g/cm3、0.6〜0.7g/cm3、0.7〜1.2g/cm3、0.7〜1.15g/cm3、0.7〜1.1g/cm3、0.7〜1.05g/cm3、0.7〜1.0g/cm3、0.7〜0.9g/cm3、0.7〜0.8g/cm3、0.5〜1.2g/cm3、0.8〜1.15g/cm3、0.8〜1.1g/cm3、0.8〜1.05g/cm3、0.8〜1.0g/cm3、0.8〜0.9g/cm3、0.9〜1.2g/cm3、0.9〜1.15g/cm3、0.9〜1.1g/cm3、0.9〜1.05g/cm3、または0.9〜1.0g/cm3である。いくつかの実施形態では、組成物は、カプセル処方物中にペレットを含む投薬単位で存在し、カプセルサイズは、サイズ00、サイズ0、サイズ1、サイズ2、またはサイズ3である。いくつかの好ましい実施形態では、投薬単位は、サイズ0、1、2、または3カプセル中に50〜250mgのアマンタジンを含むペレットを含む。いくつかの実施形態では、投薬単位は、サイズ0、1、2、または3カプセル、好ましくはサイズ1、2、または3カプセル中に100〜250mg(例えば、100〜200mg)のアマンタジンを含むペレットを含む。さらなる特定の実施形態では、投薬単位は、約110、120、130、140、150、160 170、180、190、210、または220mgのアマンタジンまたはその薬学的に許容され得る塩を含む。別のさらなる特定の実施形態では、投薬単位は、110mg塩酸アマンタジンを含む。別のさらなる特定の実施形態では、投薬単位は、130mg塩酸アマンタジンを含む。別のさらなる特定の実施形態では、投薬単位は、170mg塩酸アマンタジンを含む。別のさらなる特定の実施形態では、投薬単位は、210mg塩酸アマンタジンを含む。
【0124】
適切な可塑剤には、中鎖トリグリセリド、フタル酸ジエチル、クエン酸エステル、ポリエチレングリコール、グリセロール、アセチル化グリセリド、およびヒマシ油などが含まれる。ペレットを、所望のアマンタジン強度が得られるようにカプセルに充填する。この組成物の利点は、就寝時刻前の投与に適切な組成物を作製する所望の放出特性が得られることである。さらなる利点は、持続放出コーティングが、組成物の放出特性に悪影響を及ぼすことなく丸薬の嚥下が困難な患者への投与のためにカプセルを開け、ペレットを食品上に振りかけることができるような十分な耐久性があることである。組成物を食品上に振りかけることによって投与する場合、30分以内、好ましくは15分以内に消費される軟らかい食品(アップルソースまたはチョコレートプディングなど)を使用することが好ましい。上記組成物のなおさらなる利点は、バッチ毎の再現性および貯蔵安定性が非常に良好であることである。
【0125】
いくつかの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての測定した場合、本発明の組成物は、2時間で25%以下、6時間で40〜80%、12時間で少なくとも80%のアマンタジンのin vitro溶解プロフィールを有する。より好ましくは、in vitro溶解は、1時間で10%以下、4時間で30〜50%、および12時間で少なくとも90%のアマンタジン放出によってさらに特徴づけられる。
【0126】
さらなる実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての測定した場合、本発明の組成物のサイズ1カプセル中の110mg〜210mgのERアマンタジンは、2時間で25%以下、6時間で40〜80%、12時間で少なくとも80%のアマンタジンのin vitro溶解プロフィールを有する。より好ましくは、in vitro溶解は、1時間で10%以下、4時間で30〜50%、および12時間で少なくとも90%のアマンタジン放出によってさらに特徴づけられる。
【0127】
任意の上記態様の1つの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールが、以下のうちの少なくとも1つを示す:(i)2時間で25%以下の溶解、(ii)6時間で25〜55%以下の溶解、および(iii)12時間で少なくとも80%の溶解。より特定の実施形態では、基準(i)、(ii)、および(iii)のうちの2つを満たす。さらなる特定の実施形態では、基準(i)、(ii)、および(iii)の3つ全てを満たす。
【0128】
任意の上記態様の1つの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールが、以下のうちの少なくとも1つを示す:(i)1時間で20%以下の溶解、(ii)2時間で約25〜45%の溶解、(iii)4時間で50〜80%以下の溶解、および(iii)8時間で少なくとも80%の溶解。より特定の実施形態では、基準(i)、(ii)、および(iii)のうちの2つを満たす。さらなる特定の実施形態では、基準(i)、(ii)、および(iii)の3つ全てを満たす。
【0129】
本発明の好ましいペレット・イン・カプセル組成物は、上記のin vitro溶解性を有することに加えて、前述の就寝時刻前の投与に適切な組成物を作製する上記の薬物動態学的性質(例えば、in vivo放出プロフィール、Tmax、Cmax/Cmin比など)のいずれかを有する。組成物は、単回用量のカプセルの絶食状態のヒト被験体への経口投与後に1mgのアマンタジンあたり1.6〜2.4ng/mlのCmaxおよび1mgのアマンタジンあたり40〜75ng*h/mLのAUC0−infが得られることによってさらに特徴づけられる。好ましいペレット・イン・カプセル組成物は、カプセルのヒト被験体への1日1回の経口投与によって1mgのアマンタジンあたり2.4〜4.2ng/mlのCmax、1mgのアマンタジンあたり1.1〜2.6ng/mlのCmin、および1mgのアマンタジンあたり48〜73ng*h/mLのAUC0−24が得られるという定常状態の血漿濃度によってさらに特徴づけられる。
【0130】
上記ペレット・イン・カプセル組成物を、アマンタジン治療に適切な強度で提供することができる。典型的な強度は、少なくとも約50mg〜約250mgの範囲である。特定の実施形態では、カプセル強度は、70mg、80mg、90mg、110mg、120mg、125mg、130mg、140mg、150mg、160mg、160mg、170mg、180mg、190mg、210mg、および220mgであり、これらにより、アマンタジンHClの即時放出処方物の100mg錠剤(例えば、シンメトレル(登録商標)または他のFDAオレンジブックリファレンス掲載薬物)に等しい単回用量AUC0−inf/mgが得られる。1、2、または3つのかかるカプセルを、就寝時刻前に被験体に投与することができる。好ましい実施形態では、220mgと650mgとの間のアマンタジンを、2カプセルの適切なER処方物を使用して1日1回投与する。
【0131】
本発明を、以下の番号をつけた実施形態に関して記載することもできる。
1.アマンタジンを必要とする被験体に投与する方法で用いるアマンタジンまたはその薬学的に許容され得る塩を含む持続放出(ER)組成物であって、方法が就寝時刻より前(すなわち、被験体が夜に就寝を望む時間)の3時間未満に組成物を経口投与する工程を含む、組成物。
2.必要とする被験体に対するNMDA受容体によって媒介される疾患の処置のための医薬の製造におけるアマンタジンまたはその薬学的に許容され得る塩の使用であって、医薬が持続放出(ER)組成物であり、処置が就寝時刻より前(すなわち、被験体が夜に就寝を望む時間)の3時間未満に組成物を経口投与する工程を含む、使用。
3.アマンタジンでの処置を受けるヒト被験体における睡眠障害の軽減方法で用いるアマンタジンまたはその薬学的に許容され得る塩を含む持続放出(ER)組成物であって、方法が就寝時刻より前(すなわち、被験体が夜に就寝を望む時間)の3時間未満に組成物を投与する工程を含む、組成物。
4.アマンタジンでの処置を受けるヒト被験体における睡眠障害の軽減のための医薬の製造におけるアマンタジンまたはその薬学的に許容され得る塩の使用であって、医薬が持続放出(ER)組成物であり、就寝時刻より前(すなわち、被験体が夜に就寝を望む時間)の3時間未満の投与に適合している、使用。
5.就寝時刻より前の1時間未満に投与する、実施形態1から4のいずれか1つに記載の使用または組成物。
6.患者がパーキンソン病と診断されている、実施形態1から5のいずれか1つに記載の使用または組成物。
7.組成物を1日1回投与する、実施形態1から6のいずれか1つに記載の使用または組成物。
8.組成物を投与前に食品に添加する、実施形態1から7のいずれか1つに記載の使用または組成物。
9.アマンタジンの血漿濃度が、定常状態での投与後少なくとも1時間増加しない、実施形態1から8のいずれか1つに記載の使用または組成物。
10.アマンタジンの血漿濃度が、定常状態での投与後少なくとも2時間増加しない、実施形態1から9のいずれか1つに記載の使用または組成物。
11.投与後のアマンタジンの単回用量のTmaxが9〜15時間であり、そして/または定常状態のTmaxが7〜13時間である、実施形態1から10のいずれか1つに記載の組成物の使用。
12.アマンタジンの単回用量のTmaxが投与後10〜14時間であり、そして/または定常状態のTmaxが投与後8〜12時間である、実施形態1から11のいずれか1つに記載の使用または組成物。
13.アマンタジンの単回用量のTmaxが投与後9〜15時間であり、そして/または定常状態のTmaxが7〜13時間である、実施形態1から10のいずれか1つに記載の組成物の使用。
14.アマンタジンの単回用量のTmaxが投与後10〜14時間であり、そして/または定常状態のTmaxが投与後8〜12時間である、実施形態1から11のいずれか1つに記載の使用または組成物。
15.アマンタジンの単回用量のTmaxが投与後9〜15時間であり、そして/または定常状態のTmaxが7〜13時間である、実施形態1から10のいずれか1つに記載の組成物の使用。
16.アマンタジンの単回用量のTmaxが投与後10〜14時間であり、そして/または定常状態のTmaxが投与後8〜12時間である、実施形態1から11のいずれか1つに記載の使用または組成物。
17.アマンタジンの単回用量のTmaxが投与後11〜13時間であり、そして/または定常状態のTmaxが投与後9〜11時間である、実施形態1から12のいずれか1つに記載の使用または組成物。
18.ヒト被験体への組成物の1日1回の経口投与により、投与3時間後の25%未満のアマンタジン濃度の増加によって特徴付けられる定常状態血漿濃度プロフィールが得られる、実施形態1から13のいずれか1つに記載の使用または組成物。
19.Cmax/Cmin比が1.5〜2.0である、実施形態1から14のいずれか1つに記載の使用または組成物。
20.Cmax/Cmin比が1.7〜1.9である、実施形態1から15のいずれか1つに記載の使用または組成物。
21.アマンタジンが塩酸アマンタジンまたは硫酸アマンタジンである、実施形態1から16のいずれか1つに記載の使用または組成物。
22.組成物が50〜600mgのアマンタジンまたはその薬学的に許容され得る塩を含む、実施形態1から17のいずれか1つに記載の使用または組成物。
23.組成物を、それぞれ100〜175mgのアマンタジンまたはその薬学的に許容され得る塩を含む1つ、2つ、3つ、または4つの単位投薬形態として投与する、実施形態18に記載の使用または組成物。
24.組成物が200〜420mgのアマンタジンまたはその薬学的に許容され得る塩を含む、実施形態1から19のいずれか1つに記載の使用または組成物。
25.組成物を、110〜175mgのアマンタジンまたはその薬学的に許容され得る塩をそれぞれ含む2つの単位投薬形態として投与する、実施形態20に記載の使用または組成物。
26.組成物が50〜200mgのアマンタジンまたはその薬学的に許容され得る塩を含む、実施形態1から17のいずれか1つに記載の使用または組成物。
27.組成物が100〜125mgのアマンタジンまたはその薬学的に許容され得る塩を含む、実施形態22に記載の使用または組成物。
28.組成物が110mgの塩酸アマンタジンを含む、実施形態23に記載の使用または組成物。
29.単回用量の組成物の絶食状態のヒト被験体への経口投与により、1mgのアマンタジンあたり1.6〜2.4ng/mlのアマンタジンの最大血漿濃度(Cmax)および1mgのアマンタジンあたり40〜75ng*h/mLのAUC0−infが得られる、実施形態1から24のいずれか1つに記載の使用または組成物。
30.ヒト被験体へのある用量の組成物の1日1回の経口投与により、以下:
(i)1mgのアマンタジンあたり2.4〜4.2ng/mlのCmax、
(ii)1mgのアマンタジンあたり1.1〜2.6ng/mlのCmin、および
(iii)1mgのアマンタジンあたり44〜83ng*h/mLのAUC0−24
によって特徴付けられる定常状態血漿アマンタジン濃度プロフィールが得られる、実施形態1から25のいずれか1つに記載の使用または組成物。
31.定常状態血漿濃度プロフィールが、以下:
(iv)投与後少なくとも1時間アマンタジンの血漿濃度が増加しないこと、および
(v)Cmax/Cmin比1.5〜2.0
によってさらに特徴づけられる、実施形態26に記載の使用または組成物。
32.定常状態血漿濃度プロフィールが、以下:
(iv)投与後少なくとも2時間アマンタジン濃度が増加しないこと、および
(v)Cmax/Cmin比1.7〜1.9
によってさらに特徴づけられる、実施形態27に記載の使用または組成物。
33.溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールが、2時間で25%以下、6時間で55〜85%、および12時間で少なくとも80%である、実施形態1から28のいずれか1つに記載の使用または組成物。
34.アマンタジンのin vitro溶解プロフィールが、1時間で10%以下、4時間で30〜50%、および12時間で少なくとも90%のアマンタジンの放出によってさらに特徴づけられる、実施形態29に記載の使用または組成物。
35.単回用量の組成物の投与後の組成物のAUCプロフィールが、以下:AUC0−infの5%未満である0〜4時間の断片的AUC、AUC0−infの約5〜15%である0〜8時間の断片的AUC、AUC0−infの約10〜40%である0〜12時間の断片的AUC、AUC0−infの約25〜60%である0〜18時間の断片的AUC、およびAUC0−infの約40〜75%である0〜24時間の断片的AUC
によって特徴付けられる、実施形態1から30のいずれか1つに記載の使用または組成物。
36.定常状態条件での組成物の1日1回の投与後の組成物のAUCプロフィールが、以下:AUC24の約2〜25%である0〜4時間の断片的AUC、AUC24の約15〜50%である0〜8時間の断片的AUC、AUC24の約30〜70%である0〜12時間の断片的AUC、およびAUC24の約60〜95%である0〜18時間の断片的AUCによって特徴づけられる、実施形態1から31のいずれか1つに記載の使用または組成物。
37.実施形態1、3、または5から32のいずれか1つに具体化した薬学的組成物または実施形態2、4、または5から32のいずれか1つの使用であって、組成物が経口投与用であり、経口投与用カプセルを含み、カプセルが複数のペレットを含み、各ペレットが、以下:
(a)アマンタジンまたはその薬学的に許容され得る塩を含むペレットコア、および
(b)ペレットコアを取り囲む持続放出コーティング
を含む、薬学的組成物または使用。
38.持続放出コーティングが、エチルセルロース、ならびにポビドンおよびヒドロキシプロピルメチルセルロースのうちの少なくとも1つ、ならびに可塑剤を含む、実施形態32に記載の使用または組成物。
39.ペレットコアが、コアシード上にコーティングしたアマンタジンまたはその薬学的に許容され得る塩および結合剤を含む、実施形態33または34のいずれか1つに記載の使用または組成物。
40.ペレットコアと持続放出コーティングとを合わせた重量に基づいて、アマンタジンが40〜60重量%の量で存在し、結合剤が8〜25重量%の量で存在し、コアシードが8〜25重量%の量で存在し、エチルセルロースが10〜20重量%の量で存在し、ポビドンが1〜4重量%の量で存在し、可塑剤が1〜4重量%の量で存在する、実施形態35に記載の使用または組成物。
41.ペレットコアと持続放出コーティングとの間にシールコーティングをさらに含む、実施形態33から36のいずれか1つに記載の使用または組成物。
42.ペレットコアが、ヒドロキシプロピルメチルセルロース、コポビドン、およびその混合物からなる群から選択される結合剤を含む、実施形態35から37のいずれか1つに記載の使用または組成物。
43.可塑剤が、中鎖トリグリセリド、フタル酸ジエチル、クエン酸エステル、ポリエチレングリコール、グリセロール、アセチル化グリセリド、およびヒマシ油からなる群から選択される、実施形態42から38のいずれか1つに記載の使用または組成物。
44.組成物を経口投与する工程を含む、必要とするヒト被験体におけるパーキンソン病の処置方法で用いる実施形態33から39のいずれか1つに記載の組成物。
【0132】
本明細書中のいくつかの実施形態は、アマンタジンを必要とする被験体に投与する方法であって、アマンタジンまたはその薬学的に許容され得る塩を含む持続放出(ER)組成物を就寝時刻より前の3時間未満に経口投与する工程を含む、方法を提供する。いくつかの実施形態では、就寝時刻より前の1時間未満に投与する。いくつかの実施形態では、患者はパーキンソン病と診断されている。いくつかの実施形態では、組成物を1日1回投与する。いくつかの実施形態では、組成物を投与前に食品に添加する。いくつかの実施形態では、アマンタジンの血漿濃度は、投与後少なくとも1時間増加しない。いくつかの実施形態では、アマンタジンの血漿濃度は、投与後少なくとも2時間増加しない。いくつかの実施形態では、アマンタジンの単回用量のTmaxは9〜15時間であり、そして/または定常状態のTmaxは7〜13時間である。いくつかの実施形態では、アマンタジンの単回用量のTmaxは投与後10〜14時間であり、そして/または定常状態のTmaxは8〜12時間である。いくつかの実施形態では、アマンタジンの単回用量のTmaxは投与後11〜13時間であり、そして/または定常状態のTmaxは9〜11時間である。いくつかの実施形態では、ヒト被験体への組成物の1日1回の経口投与により、投与3時間後の25%未満のアマンタジン濃度の増加によって特徴付けられる定常状態血漿濃度プロフィールが得られる。いくつかの実施形態では、PK曲線のCmax/Cmin比は1.5〜2.0である。いくつかの実施形態では、PK曲線のCmax/Cmin比は1.7〜1.9である。いくつかの実施形態では、定常状態でのC−ave−日中/C−ave−夜比は1.2〜1.6である。いくつかの実施形態では、定常状態でのC−ave−朝/C−ave−夜比は1.3〜1.5である。いくつかの実施形態では、定常状態での日中の平均アマンタジン血漿濃度(C−ave−日中)は500〜2000ng/mlである。いくつかの実施形態では、定常状態での朝の平均アマンタジン血漿濃度(C−ave−朝)は500〜2000ng/mlである。いくつかの実施形態では、アマンタジンは塩酸アマンタジンまたは硫酸アマンタジンである。いくつかの実施形態では、組成物は50〜600mgのアマンタジンまたはその薬学的に許容され得る塩を含む。いくつかの実施形態では、組成物を、それぞれ100〜175mgのアマンタジンまたはその薬学的に許容され得る塩を含む1つ、2つ、3つ、または4つの単位投薬形態として投与する。いくつかの実施形態では、組成物を、それぞれ130〜210mgの持続放出アマンタジンまたはその薬学的に許容され得る塩を含む1つまたは2つの単位投薬形態として投与する。いくつかの実施形態では、組成物は、カプセルサイズ番号1のカプセル内に存在する。いくつかの実施形態では、組成物は、200〜350mgのアマンタジンまたはその薬学的に許容され得る塩を含む。いくつかの実施形態では、組成物を、それぞれ100〜175mgのアマンタジンまたはその薬学的に許容され得る塩を含む2つの単位投薬形態として投与する。いくつかの実施形態では、組成物は、50〜200mgのアマンタジンまたはその薬学的に許容され得る塩を含む。いくつかの実施形態では、組成物は、100〜125mgのアマンタジンまたはその薬学的に許容され得る塩を含む。いくつかの実施形態では、組成物は110mgの塩酸アマンタジンを含む。いくつかの実施形態では、絶食状態のヒト被験体への単回用量の組成物の経口投与により、1mgのアマンタジンあたり1.6〜2.4ng/mlの最大血漿濃度(Cmax)、および1mgのアマンタジンあたり40〜75ng*h/mLのAUC0−infが得られる。いくつかの実施形態では、ヒト被験体へのある用量の組成物の1日1回の経口投与により、以下によって特徴付けられる定常状態血漿濃度プロフィールが得られる:(a)1mgのアマンタジンあたり2.4〜4.2ng/mlのCmax;(b)1mgのアマンタジンあたり1.1〜2.6ng/mlのCmin、および(c)1mgのアマンタジンあたり44〜83ng*h/mLのAUC0−24。いくつかの実施形態では、定常状態血漿濃度プロフィールは、以下によってさらに特徴づけられる:(d)投与後少なくとも1時間アマンタジンの血漿濃度が増加しないこと、および(e)Cmax/Cmin比1.5〜2.0。いくつかの実施形態では、定常状態血漿濃度プロフィールは、以下によってさらに特徴づけられる:(f)投与後少なくとも2時間アマンタジン濃度が増加しないこと、および(g)Cmax/Cmin比1.7〜1.9。いくつかの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールは、2時間で25%以下、6時間で55〜85%、および12時間で少なくとも80%である。いくつかの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールは、2時間で25%以下、6時間で25〜55%、および12時間で少なくとも80%である。いくつかの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールは、1時間で20%以下、2時間で25〜45%、4時間で50〜80%、および8時間で少なくとも80%である。いくつかの実施形態では、アマンタジンのin vitro溶解プロフィールは、1時間で10%以下、4時間で30〜50%、および12時間で少なくとも90%のアマンタジン放出によってさらに特徴付けられる。いくつかの実施形態では、単回用量の組成物の投与後の組成物のAUCプロフィールは、以下によって特徴付けられる:AUC0−infの5%未満である0〜4時間の断片的AUC、AUC0−infの約5〜15%である0〜8時間の断片的AUC、AUC0−infの約10〜40%である0〜12時間の断片的AUC、AUC0−infの約25〜60%である0〜18時間の断片的AUC、およびAUC0−infの約40〜75%である0〜24時間の断片的AUC。いくつかの実施形態では、定常状態条件での組成物の1日1回の投与後の組成物のAUCプロフィールは、以下によって特徴づけられる:AUC24の約2〜25%である0〜4時間の断片的AUC、AUC24の約15〜50%である0〜8時間の断片的AUC、AUC24の約30〜70%である0〜12時間の断片的AUC、およびAUC24の約60〜95%である0〜18時間の断片的AUC。
【0133】
本明細書中のいくつかの実施形態は、アマンタジンでの処置を受けるヒト被験体における睡眠障害の軽減方法であって、アマンタジンまたはその薬学的に許容され得る塩を含む持続放出(ER)組成物を就寝時刻より前の3時間未満に投与する工程を含む、方法を提供する。いくつかの実施形態では、就寝時刻より前の1時間未満に投与する。いくつかの実施形態では、患者はパーキンソン病と診断されている。いくつかの実施形態では、組成物を1日1回投与する。いくつかの実施形態では、組成物を投与前に食品に添加する。いくつかの実施形態では、アマンタジンの血漿濃度は、投与後少なくとも1時間増加しない。いくつかの実施形態では、アマンタジンの血漿濃度は、投与後少なくとも2時間増加しない。いくつかの実施形態では、アマンタジンの単回用量のTmaxは9〜15時間であり、そして/または定常状態のTmaxは7〜13時間である。いくつかの実施形態では、アマンタジンの単回用量のTmaxは投与後10〜14時間であり、そして/または定常状態のTmaxは8〜12時間である。いくつかの実施形態では、アマンタジンの単回用量のTmaxは投与後11〜13時間であり、そして/または定常状態のTmaxは9〜11時間である。いくつかの実施形態では、ヒト被験体への組成物の1日1回の経口投与により、投与3時間後の25%未満のアマンタジン濃度の増加によって特徴付けられる定常状態血漿濃度プロフィールが得られる。いくつかの実施形態では、PK曲線のCmax/Cmin比は1.5〜2.0である。いくつかの実施形態では、PK曲線のCmax/Cmin比は1.7〜1.9である。いくつかの実施形態では、定常状態でのC−ave−日中/C−ave−夜比は1.2〜1.6である。いくつかの実施形態では、定常状態でのC−ave−朝/C−ave−夜比は1.3〜1.5である。いくつかの実施形態では、定常状態での日中の平均アマンタジン血漿濃度(C−ave−日中)は500〜2000ng/mlである。いくつかの実施形態では、定常状態での朝の平均アマンタジン血漿濃度(C−ave−朝)は500〜2000ng/mlである。いくつかの実施形態では、アマンタジンは塩酸アマンタジンまたは硫酸アマンタジンである。いくつかの実施形態では、組成物は50〜600mgのアマンタジンまたはその薬学的に許容され得る塩を含む。いくつかの実施形態では、組成物を、それぞれ100〜175mgのアマンタジンまたはその薬学的に許容され得る塩を含む1つ、2つ、3つ、または4つの単位投薬形態として投与する。いくつかの実施形態では、組成物を、それぞれ130〜210mgの持続放出アマンタジンまたはその薬学的に許容され得る塩を含む1つまたは2つの単位投薬形態として投与する。いくつかの実施形態では、組成物は、カプセルサイズ番号1のカプセル内に存在する。いくつかの実施形態では、組成物は、200〜350mgのアマンタジンまたはその薬学的に許容され得る塩を含む。いくつかの実施形態では、組成物を、それぞれ100〜175mgのアマンタジンまたはその薬学的に許容され得る塩を含む2つの単位投薬形態として投与する。いくつかの実施形態では、組成物は、50〜200mgのアマンタジンまたはその薬学的に許容され得る塩を含む。いくつかの実施形態では、組成物は、100〜125mgのアマンタジンまたはその薬学的に許容され得る塩を含む。いくつかの実施形態では、組成物は110mgの塩酸アマンタジンを含む。いくつかの実施形態では、絶食状態のヒト被験体への単回用量の組成物の経口投与により、1mgのアマンタジンあたり1.6〜2.4ng/mlの最大血漿濃度(Cmax)、および1mgのアマンタジンあたり40〜75ng*h/mLのAUC0−infが得られる。いくつかの実施形態では、ヒト被験体へのある用量の組成物の1日1回の経口投与により、以下によって特徴付けられる定常状態血漿濃度プロフィールが得られる:(a)1mgのアマンタジンあたり2.4〜4.2ng/mlのCmax;(b)1mgのアマンタジンあたり1.1〜2.6ng/mlのCmin、および(c)1mgのアマンタジンあたり44〜83ng*h/mLのAUC0−24。いくつかの実施形態では、定常状態血漿濃度プロフィールは、以下によってさらに特徴づけられる:(d)投与後少なくとも1時間アマンタジンの血漿濃度が増加しないこと、および(e)Cmax/Cmin比1.5〜2.0。いくつかの実施形態では、定常状態血漿濃度プロフィールは、以下によってさらに特徴づけられる:(f)投与後少なくとも2時間アマンタジン濃度が増加しないこと、および(g)Cmax/Cmin比1.7〜1.9。いくつかの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールは、2時間で25%以下、6時間で55〜85%、および12時間で少なくとも80%である。いくつかの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールは、2時間で25%以下、6時間で25〜55%、および12時間で少なくとも80%である。いくつかの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールは、1時間で20%以下、2時間で25〜45%、4時間で50〜80%、および8時間で少なくとも80%である。いくつかの実施形態では、アマンタジンのin vitro溶解プロフィールは、1時間で10%以下、4時間で30〜50%、および12時間で少なくとも90%のアマンタジン放出によってさらに特徴付けられる。いくつかの実施形態では、単回用量の組成物の投与後の組成物のAUCプロフィールは、以下によって特徴付けられる:AUC0−infの5%未満である0〜4時間の断片的AUC、AUC0−infの約5〜15%である0〜8時間の断片的AUC、AUC0−infの約10〜40%である0〜12時間の断片的AUC、AUC0−infの約25〜60%である0〜18時間の断片的AUC、およびAUC0−infの約40〜75%である0〜24時間の断片的AUC。いくつかの実施形態では、定常状態条件での組成物の1日1回の投与後の組成物のAUCプロフィールは、以下によって特徴づけられる:AUC24の約2〜25%である0〜4時間の断片的AUC、AUC24の約15〜50%である0〜8時間の断片的AUC、AUC24の約30〜70%である0〜12時間の断片的AUC、およびAUC24の約60〜95%である0〜18時間の断片的AUC。
【0134】
本明細書中のいくつかの実施形態は、パーキンソン病患者におけるレボドパ誘発性ジスキネジアの処置方法であって、アマンタジンまたはその薬学的に許容され得る塩を含む持続放出(ER)組成物を就寝時刻より前の3時間未満に1日1回経口投与する工程を含む、方法を提供する。いくつかの実施形態では、就寝時刻より前の1時間未満に投与する。いくつかの実施形態では、患者はパーキンソン病と診断されている。いくつかの実施形態では、組成物を1日1回投与する。いくつかの実施形態では、組成物を投与前に食品に添加する。いくつかの実施形態では、アマンタジンの血漿濃度は、投与後少なくとも1時間増加しない。いくつかの実施形態では、アマンタジンの血漿濃度は、投与後少なくとも2時間増加しない。いくつかの実施形態では、アマンタジンの単回用量のTmaxは9〜15時間であり、そして/または定常状態のTmaxは7〜13時間である。いくつかの実施形態では、アマンタジンの単回用量のTmaxは投与後10〜14時間であり、そして/または定常状態のTmaxは8〜12時間である。いくつかの実施形態では、アマンタジンの単回用量のTmaxは投与後11〜13時間であり、そして/または定常状態のTmaxは9〜11時間である。いくつかの実施形態では、ヒト被験体への組成物の1日1回の経口投与により、投与3時間後の25%未満のアマンタジン濃度の増加によって特徴付けられる定常状態血漿濃度プロフィールが得られる。いくつかの実施形態では、PK曲線のCmax/Cmin比は1.5〜2.0である。いくつかの実施形態では、PK曲線のCmax/Cmin比は1.7〜1.9である。いくつかの実施形態では、定常状態でのC−ave−日中/C−ave−夜比は1.2〜1.6である。いくつかの実施形態では、定常状態でのC−ave−朝/C−ave−夜比は1.3〜1.5である。いくつかの実施形態では、定常状態での日中の平均アマンタジン血漿濃度(C−ave−日中)は500〜2000ng/mlである。いくつかの実施形態では、定常状態での朝の平均アマンタジン血漿濃度(C−ave−朝)は500〜2000ng/mlである。いくつかの実施形態では、アマンタジンは塩酸アマンタジンまたは硫酸アマンタジンである。いくつかの実施形態では、組成物は50〜600mgのアマンタジンまたはその薬学的に許容され得る塩を含む。いくつかの実施形態では、組成物を、それぞれ100〜175mgのアマンタジンまたはその薬学的に許容され得る塩を含む1つ、2つ、3つ、または4つの単位投薬形態として投与する。いくつかの実施形態では、組成物を、それぞれ130〜210mgの持続放出アマンタジンまたはその薬学的に許容され得る塩を含む1つまたは2つの単位投薬形態として投与する。いくつかの実施形態では、組成物は、カプセルサイズ番号1のカプセル内に存在する。いくつかの実施形態では、組成物は、200〜350mgのアマンタジンまたはその薬学的に許容され得る塩を含む。いくつかの実施形態では、組成物を、それぞれ100〜175mgのアマンタジンまたはその薬学的に許容され得る塩を含む2つの単位投薬形態として投与する。いくつかの実施形態では、組成物は、50〜200mgのアマンタジンまたはその薬学的に許容され得る塩を含む。いくつかの実施形態では、組成物は、100〜125mgのアマンタジンまたはその薬学的に許容され得る塩を含む。いくつかの実施形態では、組成物は110mgの塩酸アマンタジンを含む。いくつかの実施形態では、絶食状態のヒト被験体への単回用量の組成物の経口投与により、1mgのアマンタジンあたり1.6〜2.4ng/mlの最大血漿濃度(Cmax)、および1mgのアマンタジンあたり40〜75ng*h/mLのAUC0−infが得られる。いくつかの実施形態では、ヒト被験体へのある用量の組成物の1日1回の経口投与により、以下によって特徴付けられる定常状態血漿濃度プロフィールが得られる:(a)1mgのアマンタジンあたり2.4〜4.2ng/mlのCmax;(b)1mgのアマンタジンあたり1.1〜2.6ng/mlのCmin、および(c)1mgのアマンタジンあたり44〜83ng*h/mLのAUC0−24。いくつかの実施形態では、定常状態血漿濃度プロフィールは、以下によってさらに特徴づけられる:(d)投与後少なくとも1時間アマンタジンの血漿濃度が増加しないこと、および(e)Cmax/Cmin比1.5〜2.0。いくつかの実施形態では、定常状態血漿濃度プロフィールは、以下によってさらに特徴づけられる:(f)投与後少なくとも2時間アマンタジン濃度が増加しないこと、および(g)Cmax/Cmin比1.7〜1.9。いくつかの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールは、2時間で25%以下、6時間で55〜85%、および12時間で少なくとも80%である。いくつかの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールは、2時間で25%以下、6時間で25〜55%、および12時間で少なくとも80%である。いくつかの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールは、1時間で20%以下、2時間で25〜45%、4時間で50〜80%、および8時間で少なくとも80%である。いくつかの実施形態では、アマンタジンのin vitro溶解プロフィールは、1時間で10%以下、4時間で30〜50%、および12時間で少なくとも90%のアマンタジン放出によってさらに特徴付けられる。いくつかの実施形態では、単回用量の組成物の投与後の組成物のAUCプロフィールは、以下によって特徴付けられる:AUC0−infの5%未満である0〜4時間の断片的AUC、AUC0−infの約5〜15%である0〜8時間の断片的AUC、AUC0−infの約10〜40%である0〜12時間の断片的AUC、AUC0−infの約25〜60%である0〜18時間の断片的AUC、およびAUC0−infの約40〜75%である0〜24時間の断片的AUC。いくつかの実施形態では、定常状態条件での組成物の1日1回の投与後の組成物のAUCプロフィールは、以下によって特徴づけられる:AUC24の約2〜25%である0〜4時間の断片的AUC、AUC24の約15〜50%である0〜8時間の断片的AUC、AUC24の約30〜70%である0〜12時間の断片的AUC、およびAUC24の約60〜95%である0〜18時間の断片的AUC。
【0135】
本明細書中のいくつかの実施形態は、本明細書中に記載の方法のいずれかのための薬学的組成物であって、組成物が経口投与用であり、経口投与用カプセルを含み、カプセルが複数のペレットを含み、各ペレットは、以下:(a)アマンタジンまたはその薬学的に許容され得る塩を含むペレットコア、および(b)ペレットコアを取り囲む持続放出コーティングを含む、薬学的組成物を提供する。いくつかの実施形態では、持続放出コーティングは、エチルセルロース、ならびにポビドンおよびヒドロキシプロピルメチルセルロースのうちの少なくとも1つ、ならびに可塑剤を含む。いくつかの実施形態では、ペレットコアは、コアシード上にコーティングされたアマンタジンまたはその薬学的に許容され得る塩および結合剤を含む。いくつかの実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、アマンタジンは40〜60重量%の量で存在し、結合剤は1〜25重量%の量で存在し、コアシードは8〜25重量%の量で存在し、エチルセルロースは10〜20重量%の量で存在し、ポビドンは1〜4重量%の量で存在し、可塑剤は1〜4重量%の量で存在する。いくつかの実施形態では、組成物は、さらに、ペレットコアと持続放出コーティングとの間にシールコーティングを含む。いくつかの実施形態では、ペレットコアは、ヒドロキシプロピルメチルセルロース、コポビドン、およびその混合物からなる群から選択される結合剤を含む。いくつかの実施形態では、可塑剤は、中鎖トリグリセリド、フタル酸ジエチル、クエン酸エステル、ポリエチレングリコール、グリセロール、アセチル化グリセリド、およびヒマシ油からなる群から選択される。
【0136】
本明細書中のいくつかの実施形態は、アマンタジンまたはその薬学的に許容され得る塩を必要とするヒト被験体に投与する方法であって、経口投与用のカプセル中にアマンタジンを含む薬学的組成物を経口投与する工程を含み、カプセルが複数のペレットを含み、各ペレットは、以下:(a)アマンタジンまたはその薬学的に許容され得る塩を含むペレットコア、および(b)ペレットコアを取り囲む持続放出コーティングを含む、方法を提供する。いくつかの実施形態では、持続放出コーティングは、エチルセルロース、ならびにポビドンおよびヒドロキシプロピルメチルセルロースのうちの少なくとも1つ、ならびに可塑剤を含む。いくつかの実施形態では、ペレットコアは、コアシード上にコーティングされたアマンタジンまたはその薬学的に許容され得る塩および結合剤を含む。いくつかの実施形態では、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、アマンタジンは40〜60重量%の量で存在し、結合剤は1〜25重量%の量で存在し、コアシードは8〜25重量%の量で存在し、エチルセルロースは10〜20重量%の量で存在し、ポビドンは1〜4重量%の量で存在し、可塑剤は1〜4重量%の量で存在する。いくつかの実施形態では、組成物は、さらに、ペレットコアと持続放出コーティングとの間にシールコーティングを含む。いくつかの実施形態では、ペレットコアは、ヒドロキシプロピルメチルセルロース、コポビドン、およびその混合物からなる群から選択される結合剤を含む。いくつかの実施形態では、可塑剤は、中鎖トリグリセリド、フタル酸ジエチル、クエン酸エステル、ポリエチレングリコール、グリセロール、アセチル化グリセリド、およびヒマシ油からなる群から選択される。いくつかの実施形態は、必要とするヒト被験体のパーキンソン病の処置を含む。
【0137】
本明細書中のいくつかの実施形態は、必要とする患者への1日1回の経口投与に適切な薬学的組成物であって、組成物は、治療有効量のアマンタジンまたはその薬学的に許容され得る塩を1日1回の投与において2つ以下のサイズ0以下のカプセルとして投与することができる持続放出形態で含む、薬学的組成物を提供する。いくつかの実施形態では、組成物は、110〜220mgのアマンタジンまたはその薬学的に許容され得る塩を含む。いくつかの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールが、2時間で25%以下、6時間で40〜80%、および12時間で少なくとも80%である。いくつかの実施形態では、組成物は複数のペレットを含み、各ペレットは、以下:(a)アマンタジンまたはその薬学的に許容され得る塩を含むペレットコア、および(b)ペレットコアを取り囲む持続放出コーティングを含む。いくつかの実施形態では、持続放出コーティングは、エチルセルロース、ならびにポビドンおよびヒドロキシプロピルメチルセルロースのうちの少なくとも1つ、ならびに可塑剤を含む。いくつかの実施形態では、ペレットコアは、コアシード上にコーティングされたアマンタジンまたはその薬学的に許容され得る塩および結合剤を含む。いくつかの実施形態では、組成物はアマンタジンを含み、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、アマンタジンは40〜70重量%の量で存在する。いくつかの実施形態では、ペレットコアは、直径が100ミクロンと500ミクロンとの間の糖または微結晶性セルロースを含むコアシードを含む。いくつかの実施形態では、嵩密度は0.5gm/cm3と1gm/cm3との間である。いくつかの実施形態では、組成物は、ペレットコアと持続放出コーティングとの間にシールコーティングを含む。いくつかの実施形態では、ペレットコアは、ヒドロキシプロピルメチルセルロース、コポビドン、およびその混合物からなる群から選択される結合剤を含む。いくつかの実施形態では、可塑剤は、中鎖トリグリセリド、フタル酸ジエチル、クエン酸エステル、ポリエチレングリコール、グリセロール、アセチル化グリセリド、およびヒマシ油からなる群から選択される。
【0138】
本明細書中のいくつかの実施形態は、ヒト被験体におけるパーキンソン病の処置方法であって、治療有効量のアマンタジンまたはその薬学的に許容され得る塩を1日1回の投与において2つ以下のサイズ0以下のカプセルとして投与することができる持続放出形態で含む組成物を経口投与する工程を含む、方法を提供する。いくつかの実施形態では、組成物は、110〜220mgのアマンタジンまたはその薬学的に許容され得る塩を含む。いくつかの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールが、2時間で25%以下、6時間で40〜80%、および12時間で少なくとも80%である。いくつかの実施形態では、組成物は複数のペレットを含み、各ペレットは、以下:(a)アマンタジンまたはその薬学的に許容され得る塩を含むペレットコア、および(b)ペレットコアを取り囲む持続放出コーティングを含む。いくつかの実施形態では、持続放出コーティングは、エチルセルロース、ならびにポビドンおよびヒドロキシプロピルメチルセルロースのうちの少なくとも1つ、ならびに可塑剤を含む。いくつかの実施形態では、ペレットコアは、コアシード上にコーティングされたアマンタジンまたはその薬学的に許容され得る塩および結合剤を含む。いくつかの実施形態では、組成物はアマンタジンを含み、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、アマンタジンは40〜70重量%の量で存在する。いくつかの実施形態では、ペレットコアは、直径が100ミクロンと500ミクロンとの間の糖または微結晶性セルロースを含むコアシードを含む。いくつかの実施形態では、嵩密度は0.5gm/cm3と1gm/cm3との間である。いくつかの実施形態では、組成物は、ペレットコアと持続放出コーティングとの間にシールコーティングを含む。いくつかの実施形態では、ペレットコアは、ヒドロキシプロピルメチルセルロース、コポビドン、およびその混合物からなる群から選択される結合剤を含む。いくつかの実施形態では、可塑剤は、中鎖トリグリセリド、フタル酸ジエチル、クエン酸エステル、ポリエチレングリコール、グリセロール、アセチル化グリセリド、およびヒマシ油からなる群から選択される。
【0139】
本明細書中のいくつかの実施形態は、ヒト被験体におけるレボドパ誘発性ジスキネジアの処置方法であって、治療有効量のアマンタジンまたはその薬学的に許容され得る塩を1日1回の投与において2つ以下のサイズ0以下のカプセルとして投与することができる持続放出形態で含む組成物を経口投与する工程を含む、方法を提供する。本明細書中のいくつかの実施形態は、ヒト被験体における外傷性脳損傷の処置方法であって、治療有効量のアマンタジンまたはその薬学的に許容され得る塩を1日1回の投与において2つ以下のサイズ0以下のカプセルとして投与することができる持続放出形態で含む組成物を経口投与する工程を含む、方法を提供する。いくつかの実施形態は、ヒト被験体における外傷性脳損傷の処置方法であって、治療有効量のアマンタジンまたはその薬学的に許容され得る塩を1日1回の投与において2つ以下のサイズ0以下のカプセルとして投与することができる持続放出形態で含む組成物を経口投与する工程を含む、方法を提供する。いくつかの実施形態は、ヒト被験体における疲労の処置方法であって、治療有効量のアマンタジンまたはその薬学的に許容され得る塩を1日1回の投与において2つ以下のサイズ0以下のカプセルとして投与することができる持続放出形態で含む組成物を経口投与する工程を含む、方法を提供する。いくつかの実施形態では、組成物は、110〜220mgのアマンタジンまたはその薬学的に許容され得る塩を含む。いくつかの実施形態では、溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、組成物のアマンタジンのin vitro溶解プロフィールが、2時間で25%以下、6時間で40〜80%、および12時間で少なくとも80%である。いくつかの実施形態では、組成物は複数のペレットを含み、各ペレットは、以下:(a)アマンタジンまたはその薬学的に許容され得る塩を含むペレットコア、および(b)ペレットコアを取り囲む持続放出コーティングを含む。いくつかの実施形態では、持続放出コーティングは、エチルセルロース、ならびにポビドンおよびヒドロキシプロピルメチルセルロースのうちの少なくとも1つ、ならびに可塑剤を含む。いくつかの実施形態では、ペレットコアは、コアシード上にコーティングされたアマンタジンまたはその薬学的に許容され得る塩および結合剤を含む。いくつかの実施形態では、組成物はアマンタジンを含み、ペレットコアと持続放出コーティングとを合わせた重量に基づいて、アマンタジンは40〜70重量%の量で存在する。いくつかの実施形態では、ペレットコアは、直径が100ミクロンと500ミクロンとの間の糖または微結晶性セルロースを含むコアシードを含む。いくつかの実施形態では、嵩密度は0.5gm/cm3と1gm/cm3との間である。いくつかの実施形態では、組成物は、ペレットコアと持続放出コーティングとの間にシールコーティングを含む。いくつかの実施形態では、ペレットコアは、ヒドロキシプロピルメチルセルロース、コポビドン、およびその混合物からなる群から選択される結合剤を含む。いくつかの実施形態では、可塑剤は、中鎖トリグリセリド、フタル酸ジエチル、クエン酸エステル、ポリエチレングリコール、グリセロール、アセチル化グリセリド、およびヒマシ油からなる群から選択される。いくつかの実施形態では、本方法は、就寝時刻より前の3時間未満に患者に組成物を投与する工程を含む。
【0140】
本発明は、以下の実施例を参照してより深く理解することができ、実施例は、特許請求の範囲の範囲を制限することを意図しない。
【実施例】
【0141】
実施例1: アマンタジンを持続放出コーティングしたペレット処方物
夜間投与のためにデザインしたアマンタジンHClを持続放出コーティングしたペレット組成物を、以下の表1に示す成分および相対量を使用して調製した。各組成物について、薬物コーティング液を、撹拌しながらHPMC 5cpsおよびコポビドンをイソプロピルアルコールに添加することによって調製した。精製水をこの分散液に添加し、透明な溶液が形成されるまで撹拌し続けた。次いで、薬物(アマンタジンHCl)をこの結合剤溶液に添加し、薬物が完全に溶解するまで撹拌し続けた。最後に、タルクを添加し、撹拌によって均一に分散させた。
【0142】
セルフィアビーズ(篩サイズ♯35〜♯50(すなわち、300〜500ミクロン))を、Wursterコーティングユニットにロードした。薬物コーティング分散液を、ビーズ上に噴霧し、その後に一定期間乾燥させた。得られた薬物コーティングしたペレットを篩にかけて、篩♯18と♯24との間の画分(直径およそ700μm〜1mm)を保持した。
【0143】
シールコーティング液を、撹拌しながらHPMC 5cpsをイソプロピルアルコールに添加することによって調製した。精製水をこの分散液に添加し、透明な溶液が形成されるまで撹拌し続けた。タルクを添加し、撹拌によって均一に分散させた。篩にかけた薬物コーティングしたペレットを、Wursterコーティングユニット中にロードした。シールコーティング分散液を薬物コーティングしたペレット上に噴霧し、その後に一定期間乾燥させてペレット中に残存する溶媒および水を除去した。得られたシールコーティングしたペレットを篩にかけて、篩♯18と♯24との間の画分を保持した。
【0144】
ERコーティング液を、イソプロピルアルコールおよび精製水中にエチルセルロース(粘度7cps)を溶解し、透明な溶液が形成されるまで撹拌することによって調製した。次いで、ポビドンK−90をこの透明な溶液に溶解し、その後に撹拌しながら可塑剤ミグリオール812Nを添加して透明な溶液を形成させた。篩にかけたシールコーティングしたペレットを、Wursterコーティングユニット中にロードした。ERコーティング液をシールコーティングしたペレット上に噴霧し、その後に一定期間乾燥させてERコートに影響を与え、ペレット中に残存する溶媒および水を除去した。乾燥後、ステアリン酸マグネシウムを、アニュラス領域中のコーティングしたペレットのベッド上部に噴霧し、その後にWursterユニット中でペレットを再循環させてステアリン酸マグネシウムをコーティングしたペレットとブレンドした。得られたERコーティングされたペレットを篩にかけて、篩♯18と♯24との間の画分を保持した。
【0145】
所望の重量の単位用量を含むERコーティングしたペレットを、ペレット投薬チャンバーを備えたエンカプスレータを使用して空の1硬ゼラチンカプセルシェル(100〜140mg強度についてはサイズ1)に充填した。
【0146】
【表1】
溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、上記で調製したカプセルのin vitro溶解を試験した。所望の溶解規格を満たすカプセルは、2時間で25%以下、6時間で40〜80%、および12時間で少なくとも80%の薬物を放出した。例示的な溶解プロフィールでは、1時間で薬物放出0%、2時間で12%放出、4時間で43%放出、6時間で68%放出、8時間で83%放出、10時間で92%放出、および12時間で97%放出であった。上記方法にしたがって調製したカプセルは、良好な貯蔵安定性およびスケールアップの際のバッチ毎の再現性を示した。
【0147】
実施例2:より大量の薬物を負荷したアマンタジンを持続放出コーティングしたペレット処方物
夜間投与のためにデザインしたアマンタジンHClを持続放出コーティングしたペレット組成物を、以下の表2に示す成分および相対量ならびに実施例1に記載の製造過程を使用して調製した。
【0148】
不活性コアの直径は、200〜300ミクロンである。コーティングしたペレットの直径は、600〜1200ミクロンである。コーティングしたペレットの嵩密度は、0.7〜1.2g/cm3である。
【0149】
所望の重量の単位用量を含むERコーティングしたペレットを、ペレット投薬チャンバーを備えたエンカプスレータを使用して空の硬ゼラチンカプセルシェル(170mg強度についてはサイズ1)に充填する。
【0150】
【表2】
溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、上記で調製したカプセルのin vitro溶解を試験し、このカプセルは2時間で25%以下、6時間で40〜80%、および12時間で少なくとも80%の薬物を放出する。
【0151】
実施例3: アマンタジン持続放出コーティングしたペレット処方物
夜間投与に適切なアマンタジンHCl持続放出コーティングしたペレット組成物を、以下の表3に示した成分および相対量ならびに実施例1に記載の製造過程を使用して調製した。
【0152】
単位用量を含む所望の重量のERコーティングしたペレットを、ペレット投薬チャンバーを備えたエンカプスレータを使用して空の番号1硬ゼラチンカプセルシェル(強度100mg)に充填した。
【0153】
【表3】
溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、上記で調製したカプセルのin vitro溶解を試験した。結果を図1に示す。
【0154】
実施例4:押出球形化によって作製したアマンタジン持続放出処方物
夜間投与のためにデザインしたアマンタジンHCl持続放出組成物を、以下の表4に示す成分および相対量ならびに下記の製造過程を使用して調製する。
【0155】
アマンタジンHCl、微結晶性セルロース、およびラクトース一水和物のブレンドを調製し、湿塊を、ポビドン水溶液を使用して高剪断造粒機中で調製する。1mm篩を使用して湿塊を押し出し、押し出された塊をスフェロナイザーを使用して球形化する。ペレットを箱形乾燥機で乾燥させてコアペレットを得る。コアペレットを、パンコーター中にて持続放出コーティング液でコーティングする。所望の重量の単位用量を含むERコーティングしたペレットを、ペレット投薬チャンバーを備えたエンカプスレータを使用して空の1硬ゼラチンカプセルシェル(170mg強度)に充填する。
【0156】
【表4】
溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、上記で調製したカプセルのin vitro溶解を試験し、このカプセルは2時間で25%以下、6時間で40〜80%、および12時間で少なくとも80%の薬物を放出する。
【0157】
実施例5:IRアマンタジンと比較したアマンタジンERの処方物の薬物動態学的測定
目的:本研究の主な目的は、実施例3の持続放出処方物のPK特性を確認し、絶食条件下で健康な成人被験体に単回用量として投与した100mgのフィルムコーティングしたIRアマンタジンHCl錠(シンメトレル(登録商標))と比較して異なる放出特性を有する実施例3に記載のアマンタジンHClのERカプセルの3つのプロトタイプ処方物の薬物動態学的プロフィール、安全性、および耐容性を決定することであった。
【0158】
研究デザイン:これは第1相、無作為抽出、単回用量、非盲検、4期、交差、絶食状態の薬物動態学的研究であり、単回用量100mgの異なる放出特性を有するアマンタジンERカプセルの3つの処方物を単回用量100mgの市販のアマンタジンIR錠(シンメトレル(登録商標))と比較した。図1に示すように、3つのER処方物は、in vitroでのアマンタジン放出速度が異なっていた。
【0159】
方法:被験体を、研究スクリーニングの21日以内の第1期に入院させた。入院翌日に被験体に投与し、投与24時間後に退院させた。被験体に、退院後、投与56時間後および152時間後に追跡調査のために再度来院するように依頼した。各投与期間を、休薬のために少なくとも7日間の間隔をあけた。
【0160】
一晩の絶食後、240mLの水を使用して座りながら、処方物を被験体に投与した。血液サンプルを、各投与から0(投与前)、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、18、24(退院)、および56時間後に採取した。血漿サンプルを、有効な液体クロマトグラフィ/タンデム質量分析(LC/MS/MS)法によってアマンタジンについてアッセイした。薬物動態学的パラメーターを、WinNonlinソフトウェア(バージョン4.1以降;Pharsight Corporation)を使用した非コンパートメント解析を使用して計算した。
【0161】
分散分析(ANOVA)を、線形混合効果モデルを使用した治験薬の単回投与後のデータから決定されたCmaxおよびAUC0−∞の自然対数に対して行った。モデルは、被験体、順序、期間、およびレジメンの効果を含んでいた。被験体に対する効果が無作為であった一方で、順序、期間、およびレジメンの効果を固定した。AUC(ER処方物についての相対的生物学的利用能)およびCmaxの両方についてのER対IR比を計算した。研究を通して有害事象をモニタリングした。生命兆候(脈拍数、血圧、および体温)、臨床検査測定値(生化学、血液学、および尿検査)、およびECGを、研究中の種々の時点で収集した。
【0162】
結果: 全部で20人の被験体が研究に参加した。平均年齢は25.5歳(20〜38歳の範囲)であった。研究は、8人の男性被験体(40%)および12人の女性被験体(60%)から構成され、平均体型指数(BMI)は23.6kg/m2±2.85であった。人種構成は100%コーカサス人であった。15人の被験体に、全部で4つの処置を施した。
【0163】
本研究のPKの結果は、シンメトレル(登録商標)と比較したCmax値の減少およびTmaxの増加に基づいて、3つ全てのアマンタジンER処方物の吸収速度が減少したことを示していた(表5、図5、6)。IR処方物は、最高の平均Cmax(277±73.9ng/mL)および最短のTmaxの中央値(4時間)を有していた。処方物A、B、およびCは、徐々にCmax値が低下し、Tmax値が延長された。処方物A、B、およびCについて、それぞれ、Cmaxは204±61.4から166±34.8、149±34.4ng/mLへと減少し、Tmaxの中央値は7.0時間から11.0時間、14.0時間へと増加した。AUC0−∞によって測定した場合の総アマンタジン曝露は、シンメトレル(登録商標)よりも3つ全てのアマンタジンER処方物でわずかに低いが、3つ全ての処方物は、許容され得る生物学的利用能(85〜95%)を有していた。
【0164】
【表5】
【0165】
【表6】
実施例6:アマンタジンERの食品−効果評価
目的:主な目的は、夜間投与に適切なアマンタジンER処方物が食品と共に投与した場合に優れた生物学的利用能を示すことを証明することであった。本発明者らは、高脂肪食および絶食状態の両方で投与した場合の100mgカプセルのアマンタジンER処方物(実施例3、処方物B)の薬物動態を決定した。
【0166】
研究デザイン:これは、満腹状態および絶食状態の健康な成人(18〜45歳)の男性被験体および女性被験体における100mg処方物Iの単回投与と比較するための第1相、無作為抽出、単回用量、非盲検、2期、交差の食品−効果研究であった。研究は、21日目から−2日目のスクリーニング期(計画した投与日前)および2つの処置期間(期間1および期間2)(処置期間の間に8日間の休薬期間を含む)からなっていた。
【0167】
方法:一晩の絶食後、絶食条件については周囲温度の240mLの水を使用して座りながら、処方物を被験体に投与した。摂食条件については、一晩の絶食後、被験体に高脂肪および高カロリーの試験食(Guidance for Industry Food−Effect Bioavailability and Fed Bioequivalence Studies,December 2002)を朝食として与え、試験薬投与の30分前以内に完全に消費するように要求した。被験体を2つの順序のうちの1つに無作為抽出し、順序は、それぞれ、8日間の休薬期間をおいた摂食条件および絶食条件下での処置的投与から構成された。
【0168】
各期間について、薬物動態学的血液サンプルを、投与前ならびに各期間における投与から1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、18、24、28、48、72、96、および144時間後に採取した。被験体を、研究用製品投与の少なくとも15時間前に臨床施設に収容し、各期間において研究用製品投与の少なくとも28時間臨床施設に滞在させた。各期間における28時間後のサンプルを、外来を基本として採取した。血漿中のアマンタジンを、有効なLC/MS/MS法によって定量した。アマンタジンについてWinNonlin Professionalソフトウェア−バージョン5.0.1(Pharsight Corporation,USA)を使用して、薬物動態学的パラメーターを、非コンパートメントモデルによる薬物濃度−時間プロフィールから計算した。母平均のln変換したCmax、AUClast、およびAUC∞摂食/絶食比についての点推定および90%信頼区間(CI)が完全に80%〜125%の標準的許容範囲内にある場合、食品効果なしと定義した。アマンタジンの全統計解析を、SAS(登録商標)リリース9.1.3(SAS Institute Inc.,USA)のPROC MIXEDを使用して行った。
【0169】
日常的な安全性のモニタリングを、全被験体において投与中および投与後に行った。
【0170】
結果:全部で26人の被験体が研究に参加し、19人(73%)が男性であり、7人(27%)が女性であった。平均年齢は26歳(19〜44の範囲)であり、平均BMIは22.4kg/m2(18.1〜29.8の範囲)であった。人種構成は100%アジア人であった。全被験体に少なくとも1用量の治験薬を投与し、安全性解析に参加させた。24人(92.3%)の被験体が研究を完了し、薬物動態解析に参加させた。2人の被験体(7.7%)が、プロトコール逸脱のために研究完了前に処置を中止した。
【0171】
本研究の結果(表7)は、処方物Bの単回用量の薬物動態は食品の影響を受けないことを示す。食品を使用するか使用しないで投与したアマンタジン吸収のCmaxによって測定される速度ならびにAUC0−lastおよびAUC0−∞によって測定される範囲は等価であった(表8)。
【0172】
【表7】
【0173】
【表8】
結論:本研究の結果は、アマンタジンERの単回用量の薬物動態は食品の影響を受けないことを示す。食品を使用するか使用しないで投与したアマンタジン吸収のCmaxによって測定される速度ならびにAUC0−lastおよびAUC0−∞によって測定される範囲は等価であった。
【0174】
実施例7:絶食条件下での健康な成人におけるアマンタジンHCl ERカプセルの1日1回投与とアマンタジンHCl IR錠剤の1日2回投与とを比較した薬物動態研究。
【0175】
目的:本研究の主な目的は、反復投与または慢性投与下での定常状態で、夜間投与に適切なERアマンタジン処方物の薬物動態学を測定することであった。これにより、夜間に投与したERアマンタジン処方物のさらなる安全性および有効性の研究のための重要なPKパラメーター(すなわち、C−ave−朝、C−ave−日中、C−ave−夜)の計算が可能である。本発明者らは、市販の即時放出(IR)処方物として1日2回投与したアマンタジンHClと1日1回のアマンタジン持続放出(ER)処方物(実施例3、処方物B)との単回用量および反復用量の薬物動態を比較した。
【0176】
研究デザイン: これは、2期、複数回用量の交差研究であった。スクリーニング21日後、26人の健康な男性および女性の被験体を無作為抽出して、第I期に2つの処置(アマンタジンER 200mgを1日1回またはアマンタジンIR 100mgを1日2回)のうちの1つを施し、次いで、第II期に交差させて他方の処置を施した。
【0177】
方法:1日目に治験薬投与を開始した。2日目に治験薬を投与しなかった。3日目に複数回投与を開始し、7日間継続させた(9日目まで)。8日間の休薬期間により、用量投与の間隔をあけた。治験薬を、240mLの飲料水と共に投与した。投与1時間以内は他の流動物を許可しなかった。各期間中、薬物動態解析用の血液サンプルを、投与前ならびに最初の投与の1、2、3、4、5、6、8、10、11、12、13、14、15、16、17、18、20、24、28、36、および48時間後に採取した。朝のトラフ値(投与前)用の血液サンプルを、7日目および8日目に採取した。9日目の朝の投与の直前ならびにその1、2、3、4、5、6、8、10、11、12、13、14、15、16、17、18、20、24、28、48、72、および96時間後に血液サンプルを再度採取した。9日目の朝の投与から28時間後のサンプルを、各期間に外来を基本として採取した。血漿アマンタジンを、有効なLC/MS/MS法によって定量した。薬物動態学的パラメーターを、アマンタジンについてWinNonlin Professionalソフトウェアバージョン5.0.1(Pharsight Corporation,USA)を使用した非コンパートメントモデルによる薬物濃度−時間プロフィールから計算した。
【0178】
統計解析を行って、市販の即時放出(IR)処方物として1日2回投与したアマンタジンHClと1日1回の持続放出(ER)処方物(処方物B)とを比較して単回用量および反復用量の薬物動態プロフィールを評価した。分散分析(ANOVA)を、線形混合効果モデルを使用した研究9日目の治験薬投与後のデータから決定したCmax、Cmin、およびAUC24の自然対数に対して行った。モデルは、順序、期間、レジメンについての固定効果および変量効果を含んでいた。信頼区間を使用して、等価な評価のための2つの片側検定手順を行った。信頼区間を、ANOVAモデルのフレームワーク内で得た平均対数の相違についての信頼区間のエンドポイントの累乗によって得た。自然対数変換データ由来の信頼区間の上限および下限を、逆数で累乗して幾何平均の比についての90%信頼区間を得た。累乗した90%信頼区間が区間(80.00%、125.00%)内に完全に含まれる場合、同等性が確率された。
【0179】
反復測定ANOVAを、未変換データおよびln変換データの両方に対する5%有意水準での7、8、および9日目についてのCminの比較のために行った。反復測定ANOVA検定によって有意でないことが見い出された場合、定常状態が証明された。アマンタジンの統計解析を、SAS(登録商標)リリース9.1.3(SAS Institute Inc.、USA)のPROC MIXEDを使用して行った。
【0180】
日常的な安全性モニタリングを、全被験体における投与中および投与後ならびに研究の終了時に行った。
【0181】
結果:全部で26人の被験体が研究に参加し、22人(84.6%)が男性であり、4人(15.4%)が女性であった。平均年齢は26歳(19〜42の範囲)であり、平均BMIは22.9kg/m2(18.1〜28.8の範囲)であった。人種構成は100%アジア人であった。全被験体に少なくとも1用量の治験薬を投与し、安全性解析に参加させた。24人(92.3%)の被験体が研究を完了し、薬物動態解析に参加させた。2人の被験体(7.7%)が、投与12時間以内の嘔吐(薬物動態の排除基準)のために研究完了前にPK解析を中止した。
【0182】
半減期から予想されるように、アマンタジンERの1日1回の投与およびアマンタジンIRの1日2回の投与により、1日目と比較して9日目に、より高いCmaxおよびAUCによって測定されたように蓄積が認められた(表9および図2)。7、8、および9日目の類似のトラフレベルによって証明されるように、両処方物について9日目までに定常状態が達成された(データ示さず)。定常状態で(9日目)、血漿濃度(図2、表9)および薬物動態学的パラメーター(表9)は、両処方物で類似していた。さらに、処方物は、定常状態のCmax、Cmin、およびAUC0−24によって測定した場合にアマンタジンの吸収範囲および吸収速度に関して同等であり(表9)、この同等性は、80%〜125%の範囲内のアマンタジンIRに対するアマンタジンERの試験の最小2乗平均対定常状態のCmax、Cmin、およびAUC0−24の基準の比の90%CIによって定義される。
【0183】
【表9】
1日1回の夜間投与についてのERアマンタジン処方物の安定性の決定で重要な一定のさらなるPKパラメーターも表10中に報告する。
【0184】
【表10】
結論:ERアマンタジン処方物は、所望の定常状態PK特性を示し、この特性は、夜間投与ならびに所望の有効性および耐容性という利点の達成に同様に適切であろう。具体的には、夜間に1日1回投与したERアマンタジン処方物により、投与0〜8時間後と比較して投与8〜12時間後のアマンタジン血漿濃度の初期増加は比較的ゆっくりであり、平均アマンタジン血漿濃度が高い。したがって、夜間に投与した場合、IRアマンタジンと比較して、夜間アマンタジン血漿濃度に対する平均期間の比がより高い。したがって、この処方物は、現行の実務よりも高い用量での投与に十分に適しており、それにより、LID、疲労、およびパーキンソン病の処置において比較的十分に許容され、潜在的に優れた有効性を提供すると期待される。
【0185】
実施例8:健康なボランティアにおけるアマンタジンHCl ERカプセルの夜間1回投与とアマンタジンHCl IR錠の1日2回の投与との比較研究。
【0186】
目的:主な目的は、健康なボランティアにおいて就寝時刻に1日1回投与したアマンタジン持続放出(ER)カプセル(処方物B)と1日2回投与したアマンタジン即時放出(IR)錠とを睡眠に及ぼす影響について比較することである。このER処方物は、Cave、日中/Cave、夜=1.30を示す。
【0187】
研究デザイン:これは、終夜睡眠ポリグラフ(PSG)および標準化された質問表(スタンフォード眠気尺度(SSS);改訂エプワース眠気尺度(m−ESS)/カロリンスカ眠気尺度(KSS);トロント病院俊敏試験(THAT)/ZOGIM俊敏試験(ZOGIM−A);眠気/俊敏性の視覚的アナログ尺度(VAS))によって評価した場合に30人の健康なボランティアにおけるアマンタジンERカプセル、QHS、アマンタジンIR錠BID、およびカフェインカプレット(実薬対照)の睡眠に及ぼす影響を比較するための単一施設、二重盲検、3つの偽薬、無作為抽出の交差研究である。
【0188】
治験薬を、3つの投与期間に投与する。1投与期間あたり1日投薬量の1つの薬物を投与する。各投与日を、1週間の休薬期間によって間隔をあける。アマンタジンER(処方物B)の1日投薬量は、1つの220mg錠(または2×110mgカプセル)からなり、これを就寝時刻(QHS;本研究目的については23時と定義する)に投与する。アマンタジンIRの1日投薬量は、1つの100mgカプセルからなり、これを1日2回(BID;本研究目的については8時および16時と定義する)投与する。カフェインの1日投薬量は、1つの100mgカプセルからなり、これを1日3回(TID;本研究目的については8時、16時、および23時と定義する)投与する。
【0189】
全被験体に、1日3回(8時、16時、および23時)投与する。各投与時間に、各被験体に活性薬物または適合するプラセボのいずれかを3つの各処置のために投与する。特定の投与時間に投与したカプセル、錠剤、またはカプレットが活性治験薬を含むかそれら自体であるかどうかと無関係に、被験体を割り当てられる投与の順序および期間にしたがってプラセボダミーを決定する。
【0190】
適格規準を満たす同意した被験体を3つの処置順序(群)のうちの1つに等しく無作為抽出し、各順序は、上記のように1週間の休薬期間によって間隔をあけた3つの1日処置期間を含む。さらに、各二重盲検投与日前に1日の単盲検のプラセボ導入を行う。これにより、被験体はPSG記録条件下にて臨床研究病棟(CRU)での睡眠に順応し、個別のベースライン(BL)PSG特性を確立することが可能である。
【0191】
各投与期間について、被験体を、活性治験薬投与1日目の前日に睡眠検査室を備えたCRUに入院させる。被験体をCRU内で一晩滞在させ、活性薬物投与日は終日滞在させる。被験体を再度一晩滞在させ、次いで、翌日の朝にCRUを退院させる。第1の投与期間のために、CRUへの入院日(−1日目)はスクリーニング期の最終日を構成し、CRUからの退院日は第1の休薬期間の初日(2日目)を構成する。第2の投与期間のために、CRUへの再入院日(7日目)は第1の休薬期間の最終日を構成し、退院日(9日目)は第2の休薬期間の初日を構成するであろう。第3の投与期間のために、CRUへの再入院日(14日目)は第2の休薬期間の最終日を構成し、退院日(16日目)は追跡期の初日を構成する。
【0192】
CRUへの入院日(再入院日)に、被験体に日常検査および生命兆候試験を行う。被験体に、単盲検様式で16時および23時に各プラセボダミー(アマンタジンER、アマンタジンIR、およびカフェインについて)を投与する。被験体に有害事象(AE)について質問し、各投与の直前に生命兆候をチェックした。16時の投与前に、日常的な臨床検査および薬物中毒のスクリーニング検査のために採血する。被験体を、PSG記録条件下にて睡眠検査室内で一晩過ごさせる。
【0193】
活性治験薬の投与日に、被験体は7時に起床し、一連の睡眠および俊敏性の質問表に記入する。被験体に、8時、16時、および23時に治験薬(活性薬またはプラセボ)を投与する。被験体にAEについて質問し、各投与の直前に生命兆候をチェックした。23時の投与前に血漿アマンタジン濃度を測定するために採血する。
【0194】
活性治験薬の投与の翌日に、被験体は7時に起床し、一連の睡眠および俊敏性の質問表に記入する。8時前(すなわち、最後の投与時間から9時間後)に、被験体にAEについて質問し、生命兆候をチェックした。また、血漿アマンタジン濃度の測定のために採血する。任意のAEを報告するためのサイトへの連絡に関する指示を、CRUからの退院前の被験体に概説する。次の投与期間のためのPSUへの再訪(これを、2期および3期のための再訪に適用する)または電話による接触(これを、第3の投与期間後の追跡に適用する)のためのスケジュールを概説する。
【0195】
全被験体に対してCRUから退院3日後(19日目)に追跡電話調査を行う。
【0196】
AEおよび併用薬を、研究を通してモニタリングする。血漿濃度の測定のために、血液サンプルを1、8、および15日目の23時の投与直前および2、9、および16日目のおよそ8時に採取する。
【0197】
被験体による各時点での睡眠パラメーターならびに眠気および俊敏性の測定を列挙する。PSGおよび質問表の各要素由来の合成スコアおよびスコアの両方について作表し、分析する。測定した各パラメーターについて、記述要約統計量を、順序および処置によって計算する(平均(または必要に応じて中央値)、範囲、および標準偏差(SD)が含まれる)。
【0198】
選択した結果について推測統計学を行う。これは、分散と比較した処置群にわたる平均の間の相違の規模が処置結果の相違の可能性を示唆している。連続変数データを、パラメトリック統計学(適切な追加の事後解析および/または対応のあるT検定を使用した反復測定分散分析)によって分析する。分類データおよび正規分布に適合しないデータを、ノンパラメトリック統計(ウィルコクソン符合付き順位検定)によって分析する。PSGデータを、多変量解析および/またはスペクトル分析によって評価することもできる。
【0199】
結果:PSGおよび標準化された睡眠質問表(例えば、SSS、m−ESS、KSS、THAT、ZOGIM−A、またはVAS)によって測定した場合のアマンタジンIRのBID投与と比較して220mgのアマンタジンERのQD投与が睡眠障害を増加させないことまたは減少は、就寝時刻での1日1回の投与にアマンタジンERが適切であることを証明している。
【0200】
実施例9:パーキンソン病患者における夜間の1日1回のアマンタジンHCl ERカプセル投与と1日2回のアマンタジンHCl IRカプセル投与との睡眠に及ぼす影響および有効性の比較研究。
【0201】
目的:アマンタジン持続放出(ER)カプセルの睡眠に及ぼす影響および有効性を比較すること。
【0202】
研究デザイン:これは、120人のパーキンソン病患者におけるアマンタジン持続放出(ER)カプセルの睡眠に及ぼす影響および有効性を比較するための多施設、二重盲検の無作為抽出研究であり、この研究は、UPDRS(統合パーキンソン病評点尺度)、UPDRS−IV(統合パーキンソン病評点尺度 パートIV)、AIMS(異常不随意運動尺度)、終夜睡眠ポリグラフ(PSG)、および標準化された質問表(スタンフォード眠気尺度(SSS)、改訂エプワース眠気尺度(m−ESS)/カロリンスカ眠気尺度(KSS)、トロント病院俊敏試験(THAT)/ZOGIM俊敏試験(ZOGIM−A)、眠気/俊敏性の視覚的アナログ尺度(VAS))によって評価する。
【0203】
全治験薬を経口投与する。処置Aは、午前中に投与したプラセボカプセルおよび就寝時刻に投与した2つの110mgのアマンタジン(ER)カプセルおよびプラセボカプセルからなる。処置Bは、午前中に投与したプラセボカプセルおよび就寝時刻に投与した3つの110mgのアマンタジン(ER)カプセルからなる。処置Cは、午前中に投与した100mgのアマンタジンIRカプセルならびに就寝時刻に投与した100mgのアマンタジンIRカプセルおよび2つのプラセボカプセルからなる。処置Dは、午前中に投与したプラセボカプセルおよび就寝時刻に投与した3つのプラセボカプセルからなる。
【0204】
適格規準を満たす同意した被験体を3つの処置群のうちの1つに等しく無作為抽出し、各処置期間は14日間である。さらに、各二重盲検投与日前に1日の単盲検のプラセボ導入を行う。これにより、被験体はPSG記録条件下にて臨床研究病棟(CRU)での睡眠に順応し、個別のベースライン(BL)PSG特性を確立することが可能である。
【0205】
各投与期間について、被験体を、活性治験薬投与1日目の前日に睡眠検査室を備えたCRUに入院させる。被験体をCRU内で一晩滞在させ、活性薬物投与日は終日滞在させる。被験体を再度一晩滞在させ、次いで、翌日の朝にCRUを退院させる。
【0206】
標準的なスコアリング方法(UPDRSおよびAIMが含まれる)を使用して、1、7、および14日目の午前中にパーキンソンスコアを記録する。
【0207】
AEおよび併用薬を、研究を通してモニタリングする。
【0208】
被験体による各時点での睡眠パラメーターならびに眠気および俊敏性の測定を列挙する。PSGおよび質問表の各要素由来の合成スコアおよびスコアの両方について作表し、分析する。測定した各パラメーターについて、記述要約統計量を、順序および処置によって計算する(平均(または必要に応じて中央値)、範囲、および標準偏差(SD)が含まれる)。
【0209】
選択した結果について推測統計学を行う。これは、分散と比較した処置群にわたる平均の間の相違の規模が処置結果の相違の可能性を示唆している。連続変数データを、パラメトリック統計学(適切な追加の事後解析および/または対応のあるt検定を使用した反復測定分散分析)によって分析する。分類データおよび正規分布に適合しないデータを、ノンパラメトリック統計(ウィルコクソン符合付き順位検定)によって分析する。PSGデータを、多変量解析および/またはスペクトル分析によって評価することもできる。
【0210】
結果:PSGおよび標準化された睡眠質問表(例えば、SSS、m−ESS、KSS、THAT、ZOGIM−A、またはVAS)によって測定した場合のUPDRS、UPDRS−IV、AIMの改善、睡眠障害が増加しないことまたは減少は、就寝時刻での1日1回の投与にアマンタジンERが適切であることを証明している。
【0211】
実施例10.夜間に投与したより高い強度のアマンタジンER処方物の薬物動態学的特徴のシミュレーション
目的:目的は、実施例7に記載の臨床研究で作成したデータを使用して異なる用量レベルでの種々のIRおよびERアマンタジンレジメンの定常状態の血漿濃度−時間プロフィールを予想し、夜間に投与したより高い強度のアマンタジンER処方物の利点を示すことである。
【0212】
方法論:実施例7に記載の研究手順(ADS−5101−MD−104)あたり複数の用量のアマンタジンのER処方物およびIR処方物を投与された健康なボランティア由来の血漿濃度−時間プロフィールを使用して、2つの処方物を各々記載した薬物動態モデルを作製した。本研究は、26人の健康な成人の男性および女性のボランティアにおける1日1回のアマンタジンERカプセルおよび1日2回のアマンタジンIR錠を比較する非盲検、無作為抽出、2処置、2期の二元交差研究であった。24人の固体由来の完全なデータを本演習で使用した。薬物動態評価のための血液サンプルを、1日目の単回投与後および9日目の定常状態で採取した。分析の第1段階では、WinNonlin 5.2.1(Pharsight Corp.,Mountain View,CA)を使用して、1次インプットおよび1次アウトプット、加重1/y(yはアマンタジン血漿濃度である)を用いた一区画モデルを単回用量(1日目)および反復用量(9日目)のアマンタジンIRおよびER投与後に得た各個体の血漿濃度−時間データにフィッティングした。フィッティングを、両処方物について個別であるが、両日について同時に行った。使用したモデル仮定は、用量比例性および時間関数としての一定クリアランスを含む。
【0213】
モデルを以下の式によって示す:
【0214】
【数1】
(式中、Cは血漿濃度であり、Fは絶対生物学的利用能であり、Dは用量であり、Vは分布容積であり、kaは吸収速度定数であり、kは消失速度定数であり、tは時間であり、tlagは吸収時間のずれである)。適合度を、予想される各モデルと研究ADS−5101−MD−104から得た濃度−時間データとの比較によって検証した。式1を各個体の血漿濃度―時間データにフィッティングした後、24人の各被験体についてのV/F、ka、k、およびtlagのモデルパラメーター推定値を得た。定常状態での予測度を、ER処方物およびIR処方物としての200mgの連日投薬後に得られたアマンタジンの実測データと推定定常状態濃度との比較によって確認した(9日目)。
【0215】
第2の分析工程では、以下の投与レジメンにしたがって、各モデルパラメーター推定値を使用して、各パラメーター推定値の式1への再挿入および連続した7日間の投与の寄与の加算によって両処方物についての各個体の定常状態濃度−時間プロフィールをシミュレーションした。
1.260、340、および420mgのER処方物の定常状態への1日1回(QD)の投与
2.100mgのIR処方物の定常状態への1日3回(TID)の投与
3.100mgのIR処方物の定常状態への1日2回(BID)の投与
結果: 図4は、種々のERアマンタジン用量についての定常状態血漿濃度時間プロフィールのシミュレーションを、IRアマンタジンの種々のレジメンと共に示す。表11は、夜間に投与した場合にERアマンタジンの有効性および耐容性に影響を及ぼす薬物動態学的パラメーター値をまとめている。
【0216】
【表11】
表11および図中に示すように、夜間に1日1回投与したERアマンタジン処方物により、IRアマンタジンと比較して平均日中/夜間アマンタジン血漿濃度比が高く、比較的十分な耐容性を示すと予想される。またER処方物により、平均日中アマンタジン血漿濃度が100mgを1日2回投与したIRアマンタジンの1.3〜2.2倍となり、以下の実施例11に記載の臨床研究で患者に投与した場合に有効性が有意に増強されると予想される。
【0217】
実施例11:パーキンソン病におけるレボドパ誘発性ジスキネジア処置のためのアマンタジン持続放出経口カプセルの有効性および安全性についての無作為抽出、二重盲検、プラセボ−対照研究
研究目的:本研究を、パーキンソン病(PD)を有する被験体におけるレボドパ誘発性ジスキネジア(LID)の処置のための夜間に1日1回投与したアマンタジン持続放出(ER)経口カプセルの用量範囲を確認するためにデザインする。さらに、研究を、PDを有する被験体のLIDの処置のための1日1回投与したアマンタジンER経口カプセルの安全性および耐容性を証明するためにデザインする。最後に、研究を、パーキンソン病患者におけるアマンタジンER投与レジメンの定常状態薬物動態を確認し、C−ave−日中、C−ave−朝、C−ave−朝/C−ave−夜、およびC−ave−日中/C−ave−夜をアマンタジンの有効性および耐容性と相関させるためにデザインする。
【0218】
研究デザイン:これは、PDおよびLIDを有する被験体におけるアマンタジンERの多施設、無作為抽出、二重盲検、プラセボ−対照の4−アーム並行群研究であろう。適格規準を満たす同意した被験体を、以下の4つの処置(それぞれ、1日1回、夜間に8週間投与する)のうちの1つを施すために1:1:1:1に無作為抽出するであろう。
・処置A: プラセボ、
・処置B: 260mgアマンタジンER(ADS−5102)、
・処置C: 340mgアマンタジンER(ADS−5102)
・処置D: 420mgアマンタジンER(ADS−5102)
処置CまたはD(高用量アマンタジン群)に無作為抽出される被験体に、二重盲検様式で、260mgアマンタジンERを1週目に1日1回投与し、2週目の最初に340mgまたは420mgのいずれかの1日1回投与に増加させるであろう。8週目まで投与を継続するであろう。
【0219】
ベースライン来院および無作為抽出の完了後、被験体は、1、2、4、6、および8週間の投与後にクリニックに再度来院し、治験薬の最後の投与から14日後に追跡調査のために来院するであろう。研究のための訪問および評価を、可能な場合、朝の時間(午前9時から午後1時)に計画するであろう。2つの24時間のダイアリー一式を、無作為抽出の48時間前および選択した研究訪問の48時間前に完了するであろう。ダイアリーを使用して、以下の5つの条件を30分間隔でスコアリングするであろう:睡眠、オフ、ジスキネジアなしのオン、厄介でないジスキネジアを伴うオン、厄介なジスキネジアを伴うオン。
【0220】
選択した研究訪問で、血液サンプルを、アマンタジン血漿濃度の決定および定常状態母集団薬物動態学の評価のために採取するであろう。研究中の被験体の参加期間は、12週間までであり、2週間(最大)のスクリーニング期間、8週間(最大)の処置期間、および2週間の追跡期間を含むであろう。その割り当てた治験薬に耐容性を示すことができない被験体は、治験薬を恒久的に中断し、治験薬の最後の投与後に2週間にわたって安全性についての追跡を継続するであろう。
【0221】
患者の適格規準:被験体が選択基準を満たし、且つ排除基準を満たさない場合、被験体は研究に参加する資格がある。選択した鍵となる基準は以下である。
【0222】
選択基準:
・共同体で暮らす(すなわち、施設で暮らしていない)男性または女性の成人、
・30歳と75歳との間(両端を含む)、
・歩行能力または補助器具(例えば、歩行器またはステッキ)の使用によって歩行能力があること(被験体が必要な研究訪問のために来院することができること)、
・必要に応じて研究訪問のために被験体に付き添うための博識で信頼できる介護者/研究パートナー、
・現行のIRB/IEC承認されたインフォームドコンセント用紙に署名していること、
・訓練後、被験体が協力的であり、24時間のホームダイアリーを理解し、完了することができること(介護者が補助してよい)、
・ジスキネジアを伴う特発性パーキンソン病(スクリーニング中にMDS−UPDRSスコアを決定するであろうが、最小スコアは必要ない)、
・スクリーニング前の少なくとも30日間に抗パーキンソン病薬(レボドパが含まれる)のレジメンを安定に受けており、研究参加中はこのレジメンの継続に協力的であること、
・最小UDysRSスコアとして定義したジスキネジアの存在。
【0223】
排除基準:
・認知に影響を及ぼし得る他の神経疾患(アルツハイマー型認知症、ハンチントン病、レヴィ小体痴呆、前頭側頭型痴呆、大脳皮質基底核変性、または卒中もしくは脳外傷に続発する運動障害もしくは感覚障害が含まれるが、これらに限定されない)の存在、
・スクリーニング中の24未満のミニメンタルステート試験(MMSE)スコアによって証明される認知障害の存在、
・DSM−IV−TRによる急性の主要な精神障害(例えば、大鬱病性障害)または被験体の研究評価を完了する能力に影響を及ぼし得る症状(例えば、幻覚、興奮、偏執症)の存在、
・被験体の研究評価を完了する能力を損なうであろう感覚障害(例えば、聴覚、視覚)の存在、
・スクリーニング前の2年以内のDSM−IV基準によるアルコールまたは薬物の依存症および乱用歴、
・痙攣(小児期の熱性痙攣を除く)の病歴、
・スクリーニング前の2年以内の卒中またはTIAの病歴、
・スクリーニング前の2年以内の心筋梗塞、NYHA鬱血性心不全クラス3または4、または心房細動の病歴、
・スクリーニング前の5年以内の癌の病歴(以下を例外とする:適切に処置された非黒色腫性皮膚癌、局在性膀胱癌、非転移性前立腺癌、または上皮子宮頸癌(これらの例外は、研究登録前の医学的モニタリングを使用して考察し、承認されなければならない))、
・任意の以下の検査所見の異常:ヘモグロビン10g/dL未満、WBC数3.0×109/L未満、好中球数1.5×109/L未満、リンパ球数0.5×109/L未満、血小板数100×109/L未満、ヘモグロビンA1Cが9%超、またはアスパラギン酸アミノトランスフェラーゼ(AST)および/またはアラニンアミノトランスフェラーゼ(ALT)が正常上限の2倍超、
・腎臓病食の修正(MDRD)またはコッククロフト・ゴールト式によってGFRが50mL/分/1.73m2未満と評価されたこと、
・任意の臨床的に有意なECG異常、
・経口カプセルを嚥下できないか、経口投薬の使用を妨げる胃腸管吸収不良の病歴、
研究エンドポイント:主な有効性エンドポイントは、ベースラインから8週目までの統合ジスキネジア評点尺度(UDysRS)スコアの変化であろう。鍵となる副次エンドポイントには、以下が含まれるであろう:
・標準化されたPDホームダイアリーに基づいた、厄介なジスキネジアのないオンタイム(ジスキネジアなしのオン+厄介でないジスキネジアを伴うオン)、
・統合パーキンソン病評点尺度(MDS−UPDRS)(総合評点)、
・疲労重症度尺度(FSS)によって測定された疲労。この尺度は、評点尺度1(全く異なる)から7(非常にそう思う)を使用して患者によって完了される9つの質問を含む。この疲労尺度は、スクリーニングおよび重症度の評定の両方についてMDSで推奨されている(2010)
・安全性(有害事象、安全性に関連する治験薬の中断、生命兆候、および臨床検査が含まれる)。
【0224】
以下の伝統的尺度と新規の尺度との組み合わせを、この第2相研究のために選択した:
・統合ジスキネジア評点尺度(UDysRS)を、主要結果項目のために使用するであろう。この尺度は以下の4つのパートを有し、可能なトータルスコアは104である:
I:ジスキネジアの影響がある歴史的能力障害(患者が認識している)
II:ジスキネジアの影響がない歴史的能力障害(患者が認識している)
III:客観的機能障害(4つの認められた活動に基づくジスキネジア重症度、解剖学的分布、および型)
IV:パートIIIの活動に基づく客観的能力障害
・標準化されたパーキンソン病ホームダイアリー(試験ダイアリーII、〔33〕を提案する)に基づいた厄介なジスキネジアのないオンタイムは、副次結果項目であろう。この尺度は多数の研究で使用されており、種々の成功が得られている〔34〕。しかし、大部分のKOLは、被験体が報告したダイアリーのデータを収集しなければならず、主要結果項目を補助する必要があると考えられる。
・統合パーキンソン病評点尺度(UPDRS)、パートIV、項目32(ジスキネジアの持続時間: 0=なし、4=歩行日の76〜100%)および33(ジスキネジアの能力障害: 0=障害なし、4=完全な障害)は副次結果項目であろう。この尺度はPDで長年使用されている伝統的な尺度であり、これらの項目は大部分のLID研究で使用されている。
・認知尺度: 包括的な介護者の印象、鬱病、および他の尺度を使用して、ERアマンタジンの精神状態に対する利点を測定するであろう。
【0225】
統計学的方法
有効性解析:有効性解析集団は、少なくとも1つのベースライン後有効性評価を与える全ての無作為抽出および投与された被験体を含むであろう。UDysRS尺度の有効性エンドポイントのために、ベースラインから8週目までの変化を、要因としての処置群および共変量としてのUDysRSベースライン値を用いた共分散分析(ANCOVA)モデルを使用して分析するであろう。主要解析では、5%有意水準での両側検定を使用して、260mgのADS−5102群をプラセボ群と比較するであろう。主な比較が統計的に有意である場合(P<0.05)、5%有意水準での両側検定も使用して、340mgおよび420mgのADS−5102群をプラセボと比較するであろう。
【0226】
副次エンドポイントを、主要評価項目について記載したANCOVAモデルと同型を使用して分析するであろう。5%有意水準での両側検定を使用して、処置群間の全ての二次比較を行うであろう。最終観測値延長(LOCF)アプローチを、欠測データのために使用するであろう。主要有効性解析を、プロトコール準拠集団(8週目の有効性評価を行う有効性解析集団の部分集団)のために繰り返すであろう。
【0227】
安全性解析:安全性解析集団は、少なくとも1用量の治験薬を投与される全ての無作為抽出された被験体を含むであろう。全ての安全性エンドポイントを、最初の用量から追跡完了まで(最後の用量の治験薬の投与から2週間後)分析するであろう。また、ADS−5102アマンタジンERでの最初の投与の耐容性を評価するために、安全性解析を治験薬での処置の最初の2週間に報告された安全性に関して行うであろう。
【0228】
結果:本研究から以下の改善が予想され、以下の表に示す。さらなるエンドポイントを以下に記載する。
・許容され得る主要評価項目(例えば、UDysRS)によって測定したジスキネジアスコアの有意な(20〜60%)減少
・厄介なジスキネジアのないオンタイムの増加(20〜60%)
・5%から20%へのUPDRSの改善。
・5%から60%へのパーキンソン病性疲労(FSS)の改善。
・5%から20%へのPGIによる気分の改善。
【0229】
【表12】
実施例12.夜間投与に適切な遅延放出コートを使用したアマンタジンER処方物の薬物動態学的特徴のシミュレーション。
【0230】
目的:目的は、夜間投与に適切なアマンタジンの2つの別のER処方物(処方物1(実施例7で試験した処方物である)および処方物2(実施例7で試験したが、持続放出コート上部に遅延放出保護膜を有する処方物である))の薬物動態プロフィールを評価することである。
【0231】
実施例7に記載の研究手順(ADS−5101−MD−104)あたり複数の用量のアマンタジンのER処方物およびIR処方物を投与された健康なボランティア由来の血漿濃度−時間プロフィールを使用して、2つの処方物を各々記載した薬物動態モデルを作製した。本研究は、26人の健康な成人の男性および女性のボランティアにおける1日1回のアマンタジンERカプセルおよび1日2回のアマンタジンIR錠を比較する非盲検、無作為抽出、2処置、2期の二元交差研究であった。24人の固体由来の完全なデータを本演習で使用した。薬物動態評価のための血液サンプルを、1日目の単回投与後および9日目の定常状態で採取した。分析の第1段階では、WinNonlin 5.2.1(Pharsight Corp.,Mountain View,CA)を使用して、1次インプットおよび1次アウトプット、加重1/y(yはアマンタジン血漿濃度である)を用いた一区画モデルを単回用量(1日目)および反復用量(9日目)のアマンタジンIRおよびER投与後に得た各個体の血漿濃度−時間データにフィッティングした。フィッティングを、両処方物について個別であるが、両日について同時に行った。使用したモデル仮定は、用量比例性および時間関数としての一定クリアランスを含む。
【0232】
モデルを以下の式によって示す:
【0233】
【数2】
(式中、Cは血漿濃度であり、Fは絶対生物学的利用能であり、Dは用量であり、Vは分布容積であり、kaは吸収速度定数であり、kは消失速度定数であり、tは時間であり、tlagは吸収時間のずれである)。適合度を、予想される各モデルと研究ADS−5101−MD−104から得た濃度−時間データとの比較によって検証した。式1を各個体の血漿濃度―時間データにフィッティングした後、24人の各被験体についてのV/F、ka、k、およびtlagのモデルパラメーター推定値を得た。定常状態での予測度を、ER処方物およびIR処方物としての200mgの連日投薬後に得られたアマンタジンの実測データと推定定常状態濃度との比較によって確認した(9日目)。
【0234】
第2の分析工程では、以下の投与レジメンにしたがって、各モデルパラメーター推定値を使用して、各パラメーター推定値の式1への再挿入および連続した7日間の投与の寄与の加算によって両処方物についての各個体の定常状態濃度−時間プロフィールをシミュレーションした。
1.200mgのER処方物1の定常状態への1日1回(QD)の投与
2.200mgのER処方物2の定常状態への1日1回(QD)の投与
結果: 図7は、2つのERアマンタジン処方物についての定常状態血漿濃度時間プロフィールのシミュレーションを示す。(アマンタジン血漿濃度をy上に示し、日数をx軸に示す)。図7に示すように、夜間に1日1回投与したERアマンタジン処方物2により、処方物1と比較して定常状態でのピーク血漿濃度の達成が約4時間遅延する。したがって、持続放出コート上部に遅延放出コートを含む処方物は、アマンタジンへの日中血漿曝露を最大にする一方で、定常状態での夜間の血漿曝露を最小するという点で非常に好ましい薬物動態プロフィールを有する。
【0235】
本発明の好ましい実施形態を本明細書中に示し、且つ説明しているが、かかる実施形態は、例示のみを目的として提供している。当業者は、本発明を逸脱することなく多数の変形形態、変更形態、および置換形態をすぐに思いつくであろう。本明細書中に記載の本発明の実施形態の種々の代替形態を本発明の実施で使用することができると理解すべきである。本明細書中に引用した全てのリファレンスは、その全体が本明細書中で参考として援用される。
【特許請求の範囲】
【請求項1】
アマンタジンを必要とする被験体に投与する方法であって、アマンタジンまたはその薬学的に許容され得る塩を含む持続放出(ER)組成物を就寝時刻より前の3時間未満に経口投与する工程を含む、方法。
【請求項2】
アマンタジンでの処置を受けるヒト被験体における睡眠障害を軽減する方法であって、アマンタジンまたはその薬学的に許容され得る塩を含む持続放出(ER)組成物を就寝時刻より前の3時間未満に投与する工程を含む、方法。
【請求項3】
パーキンソン病患者におけるレボドパ誘発性ジスキネジアを処置する方法であって、アマンタジンまたはその薬学的に許容され得る塩を含む持続放出(ER)組成物を就寝時刻より前の3時間未満に1日1回経口投与する工程を含む、方法。
【請求項4】
就寝時刻より前の1時間未満に投与する、請求項1、2、または3に記載の方法。
【請求項5】
前記患者がパーキンソン病と診断されている、前記請求項のいずれか1項に記載の方法。
【請求項6】
前記組成物を1日1回投与する、前記請求項のいずれか1項に記載の方法。
【請求項7】
前記組成物を投与前に食品に添加する、前記請求項のいずれか1項に記載の方法。
【請求項8】
アマンタジンの血漿濃度が、投与後少なくとも1時間増加しない、前記請求項のいずれか1項に記載の方法。
【請求項9】
アマンタジンの血漿濃度が、投与後少なくとも2時間増加しない、前記請求項のいずれか1項に記載の方法。
【請求項10】
前記アマンタジンの単回用量のTmaxが9〜15時間であり、そして/または定常状態のTmaxが7〜13時間である、前記請求項のいずれか1項に記載の方法。
【請求項11】
前記アマンタジンの単回用量のTmaxが投与後10〜14時間であり、そして/または定常状態のTmaxが8〜12時間である、前記請求項のいずれか1項に記載の方法。
【請求項12】
前記アマンタジンの単回用量のTmaxが投与後11〜13時間であり、そして/または定常状態のTmaxが9〜11時間である、前記請求項のいずれか1項に記載の方法。
【請求項13】
ヒト被験体への前記組成物の1日1回の経口投与により、投与3時間後の25%未満のアマンタジン濃度の増加によって特徴付けられる定常状態血漿濃度プロフィールが得られる、前記請求項のいずれか1項に記載の方法。
【請求項14】
Cmax/Cmin比が1.5〜2.0である、前記請求項のいずれか1項に記載の方法。
【請求項15】
Cmax/Cmin比が1.7〜1.9である、前記請求項のいずれか1項に記載の方法。
【請求項16】
定常状態でのC−ave−日中/C−ave−夜比が1.2〜1.6である、前記請求項のいずれかに記載の方法。
【請求項17】
定常状態でのC−ave−朝/C−ave−夜比が1.3〜1.5である、前記請求項のいずれかに記載の方法。
【請求項18】
定常状態での日中の平均アマンタジン血漿濃度(C−ave−日中)が500〜2000ng/mlである、前記請求項のいずれかに記載の方法。
【請求項19】
定常状態での朝の平均アマンタジン血漿濃度(C−ave−朝)が500〜2000ng/mlである、前記請求項のいずれかに記載の方法。
【請求項20】
前記アマンタジンが塩酸アマンタジンまたは硫酸アマンタジンである、前記請求項のいずれか1項に記載の方法。
【請求項21】
前記組成物が50〜600mgのアマンタジンまたはその薬学的に許容され得る塩を含む、前記請求項のいずれかに記載の方法。
【請求項22】
前記組成物を、それぞれ100〜175mgのアマンタジンまたはその薬学的に許容され得る塩を含む1つ、2つ、3つ、または4つの単位投薬形態として投与する、請求項21に記載の方法。
【請求項23】
前記組成物を、それぞれ130〜210mgの持続放出アマンタジンまたはその薬学的に許容され得る塩を含む1つまたは2つの単位投薬形態として投与する、請求項21に記載の方法。
【請求項24】
カプセルサイズが番号1である、請求項22に記載の組成物。
【請求項25】
前記組成物が200〜350mgのアマンタジンまたはその薬学的に許容され得る塩を含む、前記請求項のいずれか1項に記載の方法。
【請求項26】
前記組成物を、それぞれ100〜175mgのアマンタジンまたはその薬学的に許容され得る塩を含む2つの単位投薬形態として投与する、請求項25に記載の方法。
【請求項27】
前記組成物が50〜200mgのアマンタジンまたはその薬学的に許容され得る塩を含む、請求項1から20のいずれか1項に記載の方法。
【請求項28】
前記組成物が100〜125mgのアマンタジンまたはその薬学的に許容され得る塩を含む、請求項27に記載の方法。
【請求項29】
前記組成物が110mgの塩酸アマンタジンを含む、請求項28に記載の方法。
【請求項30】
絶食状態のヒト被験体への単回用量の前記組成物の経口投与により、1mgのアマンタジンあたり1.6〜2.4ng/mlの最大血漿濃度(Cmax)、および1mgのアマンタジンあたり40〜75ng*h/mLのAUC0−infが得られる、前記請求項のいずれかに記載の方法。
【請求項31】
ヒト被験体へのある用量の前記組成物の1日1回の経口投与により、以下:
(i)1mgのアマンタジンあたり2.4〜4.2ng/mlのCmax、
(ii)1mgのアマンタジンあたり1.1〜2.6ng/mlのCmin、および
(iii)1mgのアマンタジンあたり44〜83ng*h/mLのAUC0−24
によって特徴付けられる定常状態血漿濃度プロフィールが得られる、前記請求項のいずれかに記載の方法。
【請求項32】
前記定常状態血漿濃度プロフィールが、以下:
(iv)投与後少なくとも1時間アマンタジンの血漿濃度が増加しないこと、および
(v)Cmax/Cmin比1.5〜2.0
によってさらに特徴づけられる、請求項31に記載の方法。
【請求項33】
前記定常状態血漿濃度プロフィールが、以下:
(iv)投与後少なくとも2時間アマンタジン濃度が増加しないこと、および
(v)Cmax/Cmin比1.7〜1.9
によってさらに特徴づけられる、請求項32に記載の方法。
【請求項34】
溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、前記組成物のアマンタジンのin vitro溶解プロフィールが、2時間で25%以下、6時間で40〜80%、および12時間で少なくとも80%である、前記請求項のいずれかに記載の方法。
【請求項35】
溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、前記組成物のアマンタジンのin vitro溶解プロフィールが、2時間で25%以下、6時間で25〜55%、および12時間で少なくとも80%である、前記請求項のいずれかに記載の方法。
【請求項36】
溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、前記組成物のアマンタジンのin vitro溶解プロフィールが、1時間で20%以下、2時間で25〜45%、4時間で50〜80%、および8時間で少なくとも80%である、前記請求項のいずれかに記載の方法。
【請求項37】
前記アマンタジンのin vitro溶解プロフィールが、1時間で10%以下、4時間で30〜50%、および12時間で少なくとも90%のアマンタジン放出によってさらに特徴付けられる、請求項34に記載の方法。
【請求項38】
単回用量の前記組成物の投与後の該組成物のAUCプロフィールが、以下:AUC0−infの5%未満である0〜4時間の断片的AUC、AUC0−infの約5〜15%である0〜8時間の断片的AUC、AUC0−infの約10〜40%である0〜12時間の断片的AUC、AUC0−infの約25〜60%である0〜18時間の断片的AUC、およびAUC0−infの約40〜75%である0〜24時間の断片的AUCによって特徴付けられる、前記請求項のいずれかに記載の方法。
【請求項39】
定常状態条件での前記組成物の1日1回の投与後の該組成物のAUCプロフィールが、以下:AUC24の約2〜25%である0〜4時間の断片的AUC、AUC24の約15〜50%である0〜8時間の断片的AUC、AUC24の約30〜70%である0〜12時間の断片的AUC、およびAUC24の約60〜95%である0〜18時間の断片的AUCによって特徴づけられる、前記請求項のいずれかに記載の方法。
【請求項40】
請求項8から39のいずれか1項に記載の方法で用いる薬学的組成物であって、該組成物が経口投与用であり、経口投与用カプセルを含み、前記カプセルが複数のペレットを含み、各ペレットが、以下:
(a)アマンタジンまたはその薬学的に許容され得る塩を含むペレットコア、および
(b)該ペレットコアを取り囲む持続放出コーティング
を含む、薬学的組成物。
【請求項41】
前記持続放出コーティングが、エチルセルロース、ならびにポビドンおよびヒドロキシプロピルメチルセルロースのうちの少なくとも1つ、ならびに可塑剤を含む、請求項40に記載の薬学的組成物。
【請求項42】
前記ペレットコアが、コアシード上にコーティングされたアマンタジンまたはその薬学的に許容され得る塩および結合剤を含む、請求項40または41のいずれか1項に記載の薬学的組成物。
【請求項43】
前記ペレットコアと持続放出コーティングとを合わせた重量に基づいて、前記アマンタジンが40〜60重量%の量で存在し、前記結合剤が1〜25重量%の量で存在し、前記コアシードが8〜25重量%の量で存在し、前記エチルセルロースが10〜20重量%の量で存在し、前記ポビドンが1〜4重量%の量で存在し、前記可塑剤が1〜4重量%の量で存在する、請求項42に記載の薬学的組成物。
【請求項44】
前記ペレットコアと前記持続放出コーティングとの間にシールコーティングをさらに含む、請求項40から43のいずれか1項に記載の薬学的組成物。
【請求項45】
前記ペレットコアが、ヒドロキシプロピルメチルセルロース、コポビドン、およびその混合物からなる群から選択される結合剤を含む、請求項42から44のいずれか1項に記載の薬学的組成物。
【請求項46】
前記可塑剤が、中鎖トリグリセリド、フタル酸ジエチル、クエン酸エステル、ポリエチレングリコール、グリセロール、アセチル化グリセリド、およびヒマシ油からなる群から選択される、請求項42から45のいずれか1項に記載の薬学的組成物。
【請求項47】
アマンタジンまたはその薬学的に許容され得る塩を必要とするヒト被験体に投与する方法であって、請求項40から46のいずれか1項に記載の組成物を経口投与する工程を含む、方法。
【請求項48】
必要とするヒト被験体におけるパーキンソン病を処置する方法であって、請求項40から46のいずれか1項に記載の組成物を経口投与する工程を含む、方法。
【請求項49】
必要とする患者への1日1回の経口投与に適切な薬学的組成物であって、該組成物は、治療有効量のアマンタジンまたはその薬学的に許容され得る塩を1日1回の投与において2つ以下のサイズ0以下のカプセルとして投与することができる持続放出形態で含む、薬学的組成物。
【請求項50】
110〜230mgのアマンタジンまたはその薬学的に許容され得る塩を含む、請求項47に記載の組成物。
【請求項51】
溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、アマンタジンのin vitro溶解プロフィールが、2時間で25%以下、6時間で40〜80%、および12時間で少なくとも80%である、請求項47に記載の薬学的組成物。
【請求項52】
複数のペレットを含み、各ペレットが、以下:(a)アマンタジンまたはその薬学的に許容され得る塩を含むペレットコア、および(b)該ペレットコアを取り囲む持続放出コーティングを含む、請求項47に記載の薬学的組成物。
【請求項53】
前記持続放出コーティングが、エチルセルロース、ならびにポビドンおよびヒドロキシプロピルメチルセルロースのうちの少なくとも1つ、ならびに可塑剤を含む、請求項50に記載の薬学的組成物。
【請求項54】
前記ペレットコアが、コアシード上にコーティングされたアマンタジンまたはその薬学的に許容され得る塩および結合剤を含む、請求項51のいずれか1項に記載の薬学的組成物。
【請求項55】
前記ペレットコアと持続放出コーティングとを合わせた重量に基づいて、前記アマンタジンが40〜70重量%の量で存在する、請求項52に記載の薬学的組成物。
【請求項56】
前記ペレットコアが、直径が100ミクロンと500ミクロンとの間の糖または微結晶性セルロースを含むコアシードを含む、請求項52に記載の薬学的組成物。
【請求項57】
嵩密度が0.5gm/cm3と1gm/cm3との間である、請求項47に記載の薬学的組成物。
【請求項58】
前記ペレットコアと前記持続放出コーティングとの間にシールコーティングをさらに含む、請求項50から53のいずれか1項に記載の薬学的組成物。
【請求項59】
前記ペレットコアが、ヒドロキシプロピルメチルセルロース、コポビドン、およびその混合物からなる群から選択される結合剤を含む、請求項50から53のいずれか1項に記載の薬学的組成物。
【請求項60】
前記可塑剤が、中鎖トリグリセリド、フタル酸ジエチル、クエン酸エステル、ポリエチレングリコール、グリセロール、アセチル化グリセリド、およびヒマシ油からなる群から選択される、請求項50から53のいずれか1項に記載の薬学的組成物。
【請求項61】
遅延放出コーティングをさらに含む、請求項50から53のいずれか1項に記載の薬学的組成物。
【請求項62】
ヒト被験体におけるパーキンソン病を処置する方法であって、請求項47から61のいずれか1項に記載の組成物を経口投与する工程を含む、方法。
【請求項63】
ヒト被験体におけるレボドパ誘発性ジスキネジアを処置する方法であって、請求項47から61のいずれか1項に記載の組成物を経口投与する工程を含む、方法。
【請求項64】
ヒト被験体における外傷性脳損傷を処置する方法であって、請求項47から61のいずれか1項に記載の組成物を経口投与する工程を含む、方法。
【請求項65】
ヒト被験体における疲労を処置する方法であって、請求項47から61のいずれか1項に記載の組成物を経口投与する工程を含む、方法。
【請求項66】
請求項47から61のいずれか1項に記載の組成物を就寝時刻より前の3時間未満に投与する工程を含む、請求項62から67のいずれか1項に記載の方法。
【請求項1】
アマンタジンを必要とする被験体に投与する方法であって、アマンタジンまたはその薬学的に許容され得る塩を含む持続放出(ER)組成物を就寝時刻より前の3時間未満に経口投与する工程を含む、方法。
【請求項2】
アマンタジンでの処置を受けるヒト被験体における睡眠障害を軽減する方法であって、アマンタジンまたはその薬学的に許容され得る塩を含む持続放出(ER)組成物を就寝時刻より前の3時間未満に投与する工程を含む、方法。
【請求項3】
パーキンソン病患者におけるレボドパ誘発性ジスキネジアを処置する方法であって、アマンタジンまたはその薬学的に許容され得る塩を含む持続放出(ER)組成物を就寝時刻より前の3時間未満に1日1回経口投与する工程を含む、方法。
【請求項4】
就寝時刻より前の1時間未満に投与する、請求項1、2、または3に記載の方法。
【請求項5】
前記患者がパーキンソン病と診断されている、前記請求項のいずれか1項に記載の方法。
【請求項6】
前記組成物を1日1回投与する、前記請求項のいずれか1項に記載の方法。
【請求項7】
前記組成物を投与前に食品に添加する、前記請求項のいずれか1項に記載の方法。
【請求項8】
アマンタジンの血漿濃度が、投与後少なくとも1時間増加しない、前記請求項のいずれか1項に記載の方法。
【請求項9】
アマンタジンの血漿濃度が、投与後少なくとも2時間増加しない、前記請求項のいずれか1項に記載の方法。
【請求項10】
前記アマンタジンの単回用量のTmaxが9〜15時間であり、そして/または定常状態のTmaxが7〜13時間である、前記請求項のいずれか1項に記載の方法。
【請求項11】
前記アマンタジンの単回用量のTmaxが投与後10〜14時間であり、そして/または定常状態のTmaxが8〜12時間である、前記請求項のいずれか1項に記載の方法。
【請求項12】
前記アマンタジンの単回用量のTmaxが投与後11〜13時間であり、そして/または定常状態のTmaxが9〜11時間である、前記請求項のいずれか1項に記載の方法。
【請求項13】
ヒト被験体への前記組成物の1日1回の経口投与により、投与3時間後の25%未満のアマンタジン濃度の増加によって特徴付けられる定常状態血漿濃度プロフィールが得られる、前記請求項のいずれか1項に記載の方法。
【請求項14】
Cmax/Cmin比が1.5〜2.0である、前記請求項のいずれか1項に記載の方法。
【請求項15】
Cmax/Cmin比が1.7〜1.9である、前記請求項のいずれか1項に記載の方法。
【請求項16】
定常状態でのC−ave−日中/C−ave−夜比が1.2〜1.6である、前記請求項のいずれかに記載の方法。
【請求項17】
定常状態でのC−ave−朝/C−ave−夜比が1.3〜1.5である、前記請求項のいずれかに記載の方法。
【請求項18】
定常状態での日中の平均アマンタジン血漿濃度(C−ave−日中)が500〜2000ng/mlである、前記請求項のいずれかに記載の方法。
【請求項19】
定常状態での朝の平均アマンタジン血漿濃度(C−ave−朝)が500〜2000ng/mlである、前記請求項のいずれかに記載の方法。
【請求項20】
前記アマンタジンが塩酸アマンタジンまたは硫酸アマンタジンである、前記請求項のいずれか1項に記載の方法。
【請求項21】
前記組成物が50〜600mgのアマンタジンまたはその薬学的に許容され得る塩を含む、前記請求項のいずれかに記載の方法。
【請求項22】
前記組成物を、それぞれ100〜175mgのアマンタジンまたはその薬学的に許容され得る塩を含む1つ、2つ、3つ、または4つの単位投薬形態として投与する、請求項21に記載の方法。
【請求項23】
前記組成物を、それぞれ130〜210mgの持続放出アマンタジンまたはその薬学的に許容され得る塩を含む1つまたは2つの単位投薬形態として投与する、請求項21に記載の方法。
【請求項24】
カプセルサイズが番号1である、請求項22に記載の組成物。
【請求項25】
前記組成物が200〜350mgのアマンタジンまたはその薬学的に許容され得る塩を含む、前記請求項のいずれか1項に記載の方法。
【請求項26】
前記組成物を、それぞれ100〜175mgのアマンタジンまたはその薬学的に許容され得る塩を含む2つの単位投薬形態として投与する、請求項25に記載の方法。
【請求項27】
前記組成物が50〜200mgのアマンタジンまたはその薬学的に許容され得る塩を含む、請求項1から20のいずれか1項に記載の方法。
【請求項28】
前記組成物が100〜125mgのアマンタジンまたはその薬学的に許容され得る塩を含む、請求項27に記載の方法。
【請求項29】
前記組成物が110mgの塩酸アマンタジンを含む、請求項28に記載の方法。
【請求項30】
絶食状態のヒト被験体への単回用量の前記組成物の経口投与により、1mgのアマンタジンあたり1.6〜2.4ng/mlの最大血漿濃度(Cmax)、および1mgのアマンタジンあたり40〜75ng*h/mLのAUC0−infが得られる、前記請求項のいずれかに記載の方法。
【請求項31】
ヒト被験体へのある用量の前記組成物の1日1回の経口投与により、以下:
(i)1mgのアマンタジンあたり2.4〜4.2ng/mlのCmax、
(ii)1mgのアマンタジンあたり1.1〜2.6ng/mlのCmin、および
(iii)1mgのアマンタジンあたり44〜83ng*h/mLのAUC0−24
によって特徴付けられる定常状態血漿濃度プロフィールが得られる、前記請求項のいずれかに記載の方法。
【請求項32】
前記定常状態血漿濃度プロフィールが、以下:
(iv)投与後少なくとも1時間アマンタジンの血漿濃度が増加しないこと、および
(v)Cmax/Cmin比1.5〜2.0
によってさらに特徴づけられる、請求項31に記載の方法。
【請求項33】
前記定常状態血漿濃度プロフィールが、以下:
(iv)投与後少なくとも2時間アマンタジン濃度が増加しないこと、および
(v)Cmax/Cmin比1.7〜1.9
によってさらに特徴づけられる、請求項32に記載の方法。
【請求項34】
溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、前記組成物のアマンタジンのin vitro溶解プロフィールが、2時間で25%以下、6時間で40〜80%、および12時間で少なくとも80%である、前記請求項のいずれかに記載の方法。
【請求項35】
溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、前記組成物のアマンタジンのin vitro溶解プロフィールが、2時間で25%以下、6時間で25〜55%、および12時間で少なくとも80%である、前記請求項のいずれかに記載の方法。
【請求項36】
溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、前記組成物のアマンタジンのin vitro溶解プロフィールが、1時間で20%以下、2時間で25〜45%、4時間で50〜80%、および8時間で少なくとも80%である、前記請求項のいずれかに記載の方法。
【請求項37】
前記アマンタジンのin vitro溶解プロフィールが、1時間で10%以下、4時間で30〜50%、および12時間で少なくとも90%のアマンタジン放出によってさらに特徴付けられる、請求項34に記載の方法。
【請求項38】
単回用量の前記組成物の投与後の該組成物のAUCプロフィールが、以下:AUC0−infの5%未満である0〜4時間の断片的AUC、AUC0−infの約5〜15%である0〜8時間の断片的AUC、AUC0−infの約10〜40%である0〜12時間の断片的AUC、AUC0−infの約25〜60%である0〜18時間の断片的AUC、およびAUC0−infの約40〜75%である0〜24時間の断片的AUCによって特徴付けられる、前記請求項のいずれかに記載の方法。
【請求項39】
定常状態条件での前記組成物の1日1回の投与後の該組成物のAUCプロフィールが、以下:AUC24の約2〜25%である0〜4時間の断片的AUC、AUC24の約15〜50%である0〜8時間の断片的AUC、AUC24の約30〜70%である0〜12時間の断片的AUC、およびAUC24の約60〜95%である0〜18時間の断片的AUCによって特徴づけられる、前記請求項のいずれかに記載の方法。
【請求項40】
請求項8から39のいずれか1項に記載の方法で用いる薬学的組成物であって、該組成物が経口投与用であり、経口投与用カプセルを含み、前記カプセルが複数のペレットを含み、各ペレットが、以下:
(a)アマンタジンまたはその薬学的に許容され得る塩を含むペレットコア、および
(b)該ペレットコアを取り囲む持続放出コーティング
を含む、薬学的組成物。
【請求項41】
前記持続放出コーティングが、エチルセルロース、ならびにポビドンおよびヒドロキシプロピルメチルセルロースのうちの少なくとも1つ、ならびに可塑剤を含む、請求項40に記載の薬学的組成物。
【請求項42】
前記ペレットコアが、コアシード上にコーティングされたアマンタジンまたはその薬学的に許容され得る塩および結合剤を含む、請求項40または41のいずれか1項に記載の薬学的組成物。
【請求項43】
前記ペレットコアと持続放出コーティングとを合わせた重量に基づいて、前記アマンタジンが40〜60重量%の量で存在し、前記結合剤が1〜25重量%の量で存在し、前記コアシードが8〜25重量%の量で存在し、前記エチルセルロースが10〜20重量%の量で存在し、前記ポビドンが1〜4重量%の量で存在し、前記可塑剤が1〜4重量%の量で存在する、請求項42に記載の薬学的組成物。
【請求項44】
前記ペレットコアと前記持続放出コーティングとの間にシールコーティングをさらに含む、請求項40から43のいずれか1項に記載の薬学的組成物。
【請求項45】
前記ペレットコアが、ヒドロキシプロピルメチルセルロース、コポビドン、およびその混合物からなる群から選択される結合剤を含む、請求項42から44のいずれか1項に記載の薬学的組成物。
【請求項46】
前記可塑剤が、中鎖トリグリセリド、フタル酸ジエチル、クエン酸エステル、ポリエチレングリコール、グリセロール、アセチル化グリセリド、およびヒマシ油からなる群から選択される、請求項42から45のいずれか1項に記載の薬学的組成物。
【請求項47】
アマンタジンまたはその薬学的に許容され得る塩を必要とするヒト被験体に投与する方法であって、請求項40から46のいずれか1項に記載の組成物を経口投与する工程を含む、方法。
【請求項48】
必要とするヒト被験体におけるパーキンソン病を処置する方法であって、請求項40から46のいずれか1項に記載の組成物を経口投与する工程を含む、方法。
【請求項49】
必要とする患者への1日1回の経口投与に適切な薬学的組成物であって、該組成物は、治療有効量のアマンタジンまたはその薬学的に許容され得る塩を1日1回の投与において2つ以下のサイズ0以下のカプセルとして投与することができる持続放出形態で含む、薬学的組成物。
【請求項50】
110〜230mgのアマンタジンまたはその薬学的に許容され得る塩を含む、請求項47に記載の組成物。
【請求項51】
溶解媒質として37℃の500mlの水を用いた50rpmでのUSP Apparatus II(パドル)を用いての、アマンタジンのin vitro溶解プロフィールが、2時間で25%以下、6時間で40〜80%、および12時間で少なくとも80%である、請求項47に記載の薬学的組成物。
【請求項52】
複数のペレットを含み、各ペレットが、以下:(a)アマンタジンまたはその薬学的に許容され得る塩を含むペレットコア、および(b)該ペレットコアを取り囲む持続放出コーティングを含む、請求項47に記載の薬学的組成物。
【請求項53】
前記持続放出コーティングが、エチルセルロース、ならびにポビドンおよびヒドロキシプロピルメチルセルロースのうちの少なくとも1つ、ならびに可塑剤を含む、請求項50に記載の薬学的組成物。
【請求項54】
前記ペレットコアが、コアシード上にコーティングされたアマンタジンまたはその薬学的に許容され得る塩および結合剤を含む、請求項51のいずれか1項に記載の薬学的組成物。
【請求項55】
前記ペレットコアと持続放出コーティングとを合わせた重量に基づいて、前記アマンタジンが40〜70重量%の量で存在する、請求項52に記載の薬学的組成物。
【請求項56】
前記ペレットコアが、直径が100ミクロンと500ミクロンとの間の糖または微結晶性セルロースを含むコアシードを含む、請求項52に記載の薬学的組成物。
【請求項57】
嵩密度が0.5gm/cm3と1gm/cm3との間である、請求項47に記載の薬学的組成物。
【請求項58】
前記ペレットコアと前記持続放出コーティングとの間にシールコーティングをさらに含む、請求項50から53のいずれか1項に記載の薬学的組成物。
【請求項59】
前記ペレットコアが、ヒドロキシプロピルメチルセルロース、コポビドン、およびその混合物からなる群から選択される結合剤を含む、請求項50から53のいずれか1項に記載の薬学的組成物。
【請求項60】
前記可塑剤が、中鎖トリグリセリド、フタル酸ジエチル、クエン酸エステル、ポリエチレングリコール、グリセロール、アセチル化グリセリド、およびヒマシ油からなる群から選択される、請求項50から53のいずれか1項に記載の薬学的組成物。
【請求項61】
遅延放出コーティングをさらに含む、請求項50から53のいずれか1項に記載の薬学的組成物。
【請求項62】
ヒト被験体におけるパーキンソン病を処置する方法であって、請求項47から61のいずれか1項に記載の組成物を経口投与する工程を含む、方法。
【請求項63】
ヒト被験体におけるレボドパ誘発性ジスキネジアを処置する方法であって、請求項47から61のいずれか1項に記載の組成物を経口投与する工程を含む、方法。
【請求項64】
ヒト被験体における外傷性脳損傷を処置する方法であって、請求項47から61のいずれか1項に記載の組成物を経口投与する工程を含む、方法。
【請求項65】
ヒト被験体における疲労を処置する方法であって、請求項47から61のいずれか1項に記載の組成物を経口投与する工程を含む、方法。
【請求項66】
請求項47から61のいずれか1項に記載の組成物を就寝時刻より前の3時間未満に投与する工程を含む、請求項62から67のいずれか1項に記載の方法。
【図1】

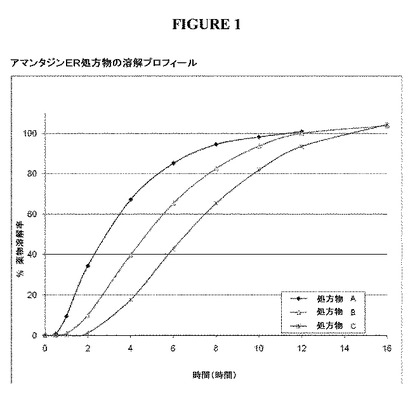
【図2A】
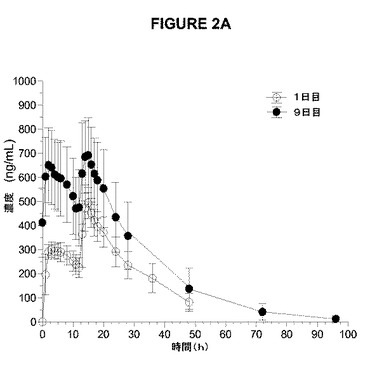
【図2B】
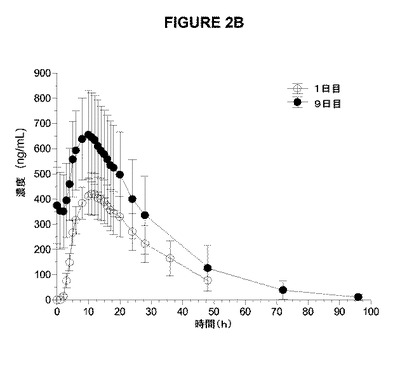
【図3】
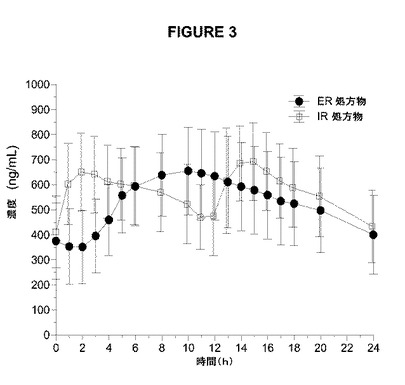
【図5】
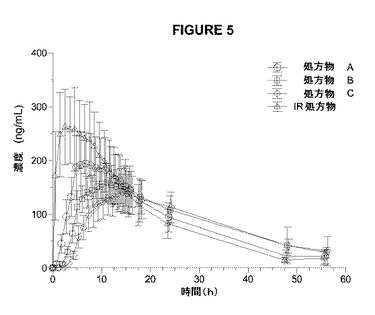
【図6】
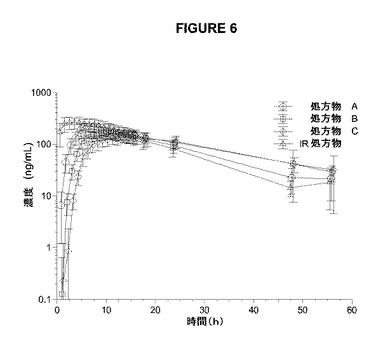
【図4】

【図7】
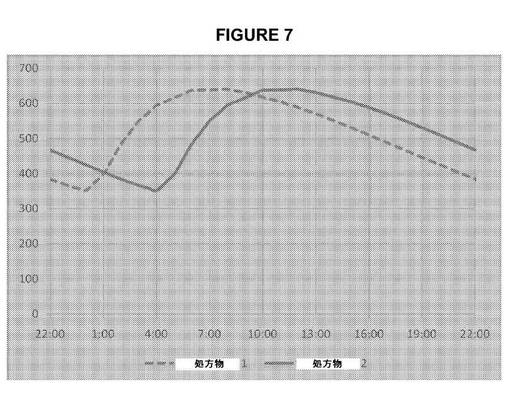
【図2A】
【図2B】
【図3】
【図5】
【図6】
【図4】
【図7】
【公表番号】特表2013−512919(P2013−512919A)
【公表日】平成25年4月18日(2013.4.18)
【国際特許分類】
【出願番号】特願2012−542198(P2012−542198)
【出願日】平成22年12月2日(2010.12.2)
【国際出願番号】PCT/US2010/058789
【国際公開番号】WO2011/069010
【国際公開日】平成23年6月9日(2011.6.9)
【出願人】(503206226)アダマス ファーマシューティカルズ, インコーポレイテッド (1)
【Fターム(参考)】
【公表日】平成25年4月18日(2013.4.18)
【国際特許分類】
【出願日】平成22年12月2日(2010.12.2)
【国際出願番号】PCT/US2010/058789
【国際公開番号】WO2011/069010
【国際公開日】平成23年6月9日(2011.6.9)
【出願人】(503206226)アダマス ファーマシューティカルズ, インコーポレイテッド (1)
【Fターム(参考)】
[ Back to top ]