
アミド化生成物の調製に有用な酵素を発現させるための細胞株
【課題】ペプチジルグリシンα−アミド化モノオキシゲナーゼ(PAM)、またはその2つの触媒ドメインの一方を発現させるための細胞株を提供する。
【解決手段】非動物供給源の低タンパク質組織培養培地を用いつつ、高レベルの酵素発現が達成される。ロバストな2工程の下流精製により高い酵素純度がもたらされる。得られるPAMまたはそのPHM触媒ドメインは、X−α−ヒドロキシ−GlyまたはX−NH2(Xは、グリシン基が共有結合し得るカルボニル基を有するペプチドまたは任意の化学的化合物である)へのX−Glyの変換の酵素的変換を触媒するために使用される。また、好ましい細胞株の調製方法も示す。
【解決手段】非動物供給源の低タンパク質組織培養培地を用いつつ、高レベルの酵素発現が達成される。ロバストな2工程の下流精製により高い酵素純度がもたらされる。得られるPAMまたはそのPHM触媒ドメインは、X−α−ヒドロキシ−GlyまたはX−NH2(Xは、グリシン基が共有結合し得るカルボニル基を有するペプチドまたは任意の化学的化合物である)へのX−Glyの変換の酵素的変換を触媒するために使用される。また、好ましい細胞株の調製方法も示す。
【発明の詳細な説明】
【技術分野】
【0001】
関連出願の相互参照
本出願は、2005年6月24日に出願された米国特許仮出願第60/693,612号(その開示は引用により本明細書に組み込まれる)の優先権を主張するものである。
【0002】
発明の分野
本発明は、ペプチジルグリシンα−アミド化モノオキシゲナーゼ(PAMもしくはα−AE)、またはその2つの触媒ドメインの一方の発現のための組換え発現ベクターおよび細胞株に関する。本発明はまた、X−α−ヒドロキシ−GlyまたはX−NH2(Xは、グリシン基が共有結合し得るカルボニル基を有するペプチドまたは任意の化学的化合物である)へのX−Glyの酵素的変換を触媒するための、かかるPAM(またはその触媒ドメインの一方)の使用に関する。さらに、本発明は、好ましい細胞株の調製に関する。一部のある実施形態では、CHO K1宿主が用いられる。一部のある実施形態では、発現ベクターは、ヒトメタロチオニンIIAプロモーターおよび/またはSV40エンハンサーを含む。
【背景技術】
【0003】
関連技術の説明
数多くのヒトホルモン、増殖因子、サイトカイン、神経伝達物質、誘導体化脂肪酸および他の重要な生物学的化合物は、その分子構造の実質的な部分としてアミノ酸またはペプチドを有する。多くの疾患は、患者において、これらの生物学的化合物のレベルの上昇に対して陽性に応答する。治療有効量のかかる生物学的に関連性のある化合物は患者に、さまざまな様式で投与され得る。したがって、効率的で費用効果のあるかかる化合物の製造方法は非常に重要である。これは、生物学的化合物が、経口送達用の投薬形態(これは、他の投与様式と比べてバイオアベイラビリティが低いにもかかわらず、通常好ましい投与様式である)に調製される場合、特に言える。
【0004】
哺乳動物細胞および他の真核生物では、ある種の翻訳後プロセシング手順が行なわれ得るが、原核生物では行なわれ得ない。ある種の原核生物、例えば、大腸菌は、バッチ発酵手順で容易に培養され得るため、および遺伝子工学的に充分特性化されているため、組換えDNA(rDNA)手法による哺乳動物タンパク質の産生のための宿主として広く使用されている。しかしながら、多くの哺乳動物タンパク質には、なんらかの型の翻訳後プロセシングが必要とされる。このようなタンパク質が、例えば大腸菌の遺伝子操作によって産生される場合、翻訳後プロセシングは、多くの場合、複雑なインビトロ化学的手順を用いて行なわれなければならないが、該手順は、大規模生産の適用のためには、たいへんな費用がかかる。ペプチドを、哺乳動物宿主を用いて、組換えにより産生させる場合であっても、多くの場合、前駆体を効率的に産生させ、これを、その後でのみさらに修飾させることが望ましい。
【0005】
かかるさらなるプロセシング活性の一例は、ペプチドまたはタンパク質のカルボキシ末端アミノ酸の特異的アミド化を含む。多くの天然に存在するホルモンおよびペプチドはかかる修飾を含み、これは、多くの場合、該タンパク質が生物学的に活性であるべきならば不可欠である。一例はカルシトニンであり、この場合、天然形態のアミド化プロリンを非アミド化プロリン残基で置換すると、生物学的活性の非常に有意な低下がもたらされる。充分な活性に翻訳後アミド化を必要とする他の生物学的ペプチドとしては、限定されないが、成長ホルモン放出因子、他のカルシトニン、カルシトニン遺伝子関連ペプチド、セクレチン、ペプチドYYなどが挙げられる。
【0006】
最大限の有効性のためにアミド化が必要とされる多くの重要な生物学的タンパク質のポリペプチド配列が、例えば、遺伝子操作手法によって製造され得る。しかしながら、重要であり、時には不可欠であるカルボキシ末端のアミド化は、多くの場合、インビトロで行なわなければならない。この点において、高価で煩雑な化学的アミド化手法は回避することが望ましく、したがって、特異的アミド化を行なうためにはアミド化酵素を用いることが望ましい。タンパク質のカルボキシ末端アミノ酸の特異的アミド化は、高頻度で、α−アミド化酵素によって触媒される。
【0007】
ペプチジルグリシンα−アミド化モノオキシゲナーゼ(PAM)は、アミド化ペプチド生成物へのペプチド基質の変換を触媒する。該変換は2工程反応である。PAMは2つの触媒ドメインを有する:ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼ(PHM)は、工程1(中間体への基質の変換)を触媒し、ペプチジルグリシンα−ヒドロキシグリシンα−アミド化リアーゼ(PAL)は、工程2(生成物への中間体の変換)を触媒する。完全長PAMは2つの機能を有し、両方の工程を触媒する。工程2はまた、中間体をルイス塩基と接触させる場合、非酵素的に、効率的になされ得る。
【0008】
自然界では、ほぼ50%のペプチドホルモンおよび神経伝達物質が、PAMによって前述の様式でアミド化される。PAM活性は、数多くの多様な種で認められており、PAMは、ラット、ウシおよびカエルなど多様な種間で有意な構造的相同性を有する傾向にある。また、PAMの機能、基質および補因子は、種間で類似する(高頻度で、同一である)ことが知られている。基質は化合物であり、多くの場合、遊離カルボキシル基を有するグリシン残基を有するペプチドである。PAM触媒アミド化反応は当該技術分野でよく知られている。例えば、ペプチジルグリシンα−アミド化モノオキシゲナーゼを用い、グリシン伸長サケカルシトニン前駆体の、そのC末端がアミド化された(すなわち、前駆体のC末端グリシンの代わりにアミノ基を有する)真のサケカルシトニンへの変換を触媒するペプチドは、米国特許第6,103,495号に詳細に記載されている。
【0009】
PAMおよびPAMを発現する細胞株の供給源は、当該技術分野で知られている。アミド化ペプチドの大規模なPAM触媒産生では、最良のコスト有効性のため、PAMの安定な高収率供給源が必要とされる。さらに、PAMは、潜在的ヒト用医薬品の作製に高頻度に用いられるため、PAM産生(PAM発現細胞株およびこれを培養する培養培地の両方)のための系では、PAM発現中、できるだけ不純物の産生を少なくすることが重要である。特に重要なことは、感染性海綿状脳症(TSE)のリスクを回避するために、哺乳動物タンパク質の使用を最小限に抑えることである。それにもかかわらず、培養培地中の哺乳動物タンパク質増殖因子(TSEの観点からは望ましくない)は、PAM発現細胞株の生存および産物発現の補助において有用であり得る。したがって、当該技術分野において、本発明の場合以外では培地に必要とされ得る哺乳動物タンパク質の非存在下であっても強い発現をもたらし、良好な安定性および生存を示すPAM発現(またはPHM発現)細胞が必要である。
【発明の概要】
【発明が解決しようとする課題】
【0010】
発明の概要
したがって、本発明の目的は、酵素が後に製品の製造(例えば、アミド化医薬品の製造)に使用される場合に問題となり得る哺乳動物型のタンパク質および他の不純物が実質的にない培地中で良好な生存割合および良好な発現収率を有するのに充分強いPAM発現細胞またはPHM発現細胞を提供することである。
【0011】
本発明のさらなる目的は、それ自体は有意な望ましくない不純物を共発現しないPAM発現細胞またはPHM発現細胞を提供することである。
【0012】
さらなる目的は、良好な発現収率をもたらすPAM発現細胞またはPHM発現細胞を提供することである。
【0013】
さらなる目的は、かかる細胞に有用な発現ベクターを提供することである。
さらなる目的は、かかる細胞をトランスフェクトおよび選択するための良好な手法を提供することである。
【0014】
さらなる目的は、酵素反応用の高活性および高純度のPAMまたはPHMを提供し、それにより、良好な酵素反応および結果としてのアミド化生成物を提供することである。
【課題を解決するための手段】
【0015】
これらおよび他の目的は、本明細書に開示する本発明によって提供される。
一実施形態において、本発明は、ペプチジルグリシンα−アミド化モノオキシゲナーゼを発現させるための発現ベクターでトランスフェクトされたCHO K1細胞を提供する。
【0016】
別の実施形態において、本発明は、リボソーム結合部位、プロモーターおよび該プロモーターの上流のSV40エンハンサーを含む制御領域に操作可能に連結されたペプチジルグリシンα−アミド化モノオキシゲナーゼをコードする核酸を有するコード領域を有する組換え発現ベクターを提供する。
【0017】
別の実施形態において、本発明は、リボソーム結合部位およびヒトメタロチオニンIIAプロモーターを含む制御領域に操作可能に連結されたペプチジルグリシンα−アミド化モノオキシゲナーゼをコードする核酸を有するコード領域を有する組換え発現ベクターを提供する。
【0018】
別の実施形態において、本発明は、
(A)第1、第2および第3の発現ベクターの存在下で、潜在的宿主細胞をトランスフェクトする工程であって、該第1のベクターが、第1の選択可能なマーカーをコードするコード領域を含み、該第2のベクターが、第2の選択可能なマーカーをコードするコード領域を含み、該第3のベクターが、ペプチジルグリシンα−アミド化モノオキシゲナーゼをコードするコード領域を含み、該第1のベクターに対する該第3のベクターの濃度比が少なくとも3:1であり、該第2のベクターに対する該第3のベクターの濃度比が少なくとも3:1である工程;
(B)工程(A)で得られた細胞に選択圧をかけ、該第1のベクターでトランスフェクトされた細胞を選択する工程;
(C)工程(B)で得られた細胞に選択圧をかけ、該第2のベクターでトランスフェクトされた細胞を選択する工程;
(D)工程(C)で得られた細胞を限界希釈し、ペプチジルグリシンα−アミド化モノオキシゲナーゼを発現する細胞を選択する工程
を含む、ペプチジルグリシンα−アミド化モノオキシゲナーゼの発現のための細胞株の調製方法を提供する。
【0019】
別の実施形態において、本発明は細胞株UGL 73−26/Mを提供する。
別の実施形態において、本発明は、細胞株UGL 73−26/Mによって発現されるペプチジルグリシンα−アミド化モノオキシゲナーゼを提供する。
【0020】
別の実施形態において、本発明は、
(A)ペプチジルグリシンα−アミド化モノオキシゲナーゼの不純試料を陰イオン交換クロマトグラフィーにかける工程であって、溶出が均一濃度である工程;
(B)工程(A)の溶離液を疎水性相互作用クロマトグラフィーにかける工程であって、硫酸アンモニウムが使用されず、溶出が均一濃度である工程
を含む、ペプチジルグリシンα−アミド化モノオキシゲナーゼを発現および培養培地中への分泌後に精製する方法を提供する。
【0021】
別の実施形態において、本発明は、ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼを発現させるための発現ベクターでトランスフェクトされたCHO K1細胞を提供する。
【0022】
別の実施形態において、本発明は、リボソーム結合部位、プロモーターおよび該プロモーターの上流のSV40エンハンサーを含む制御領域に操作可能に連結されたペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼをコードする核酸を有するコード領域を有する組換え発現ベクターを提供する。
【0023】
別の実施形態において、本発明は、リボソーム結合部位およびヒトメタロチオニンIIAプロモーターを含む制御領域に操作可能に連結されたペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼをコードする核酸を有するコード領域を有する組換え発現ベクターを提供する。
【0024】
別の実施形態において、本発明は、
(A)第1、第2および第3の発現ベクターの存在下で、潜在的宿主細胞をトランスフェクトする工程であって、該第1のベクターが、第1の選択可能なマーカーをコードするコード領域を含み、該第2のベクターが、第2の選択可能なマーカーをコードするコード領域を含み、該第3のベクターが、ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼをコードするコード領域を含み、該第1のベクターに対する該第3のベクターの濃度比が少なくとも3:1であり、該第2のベクターに対する該第3のベクターの濃度比が少なくとも3:1である工程;
(B)工程(A)で得られた細胞に選択圧をかけ、該第1のベクターでトランスフェクトされた細胞を選択する工程;
(C)工程(B)で得られた細胞に選択圧をかけ、該第2のベクターでトランスフェクトされた細胞を選択する工程;
(D)工程(C)で得られた細胞を限界希釈し、ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼを発現する細胞を選択する工程
を含む、ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼの発現のための細胞株の調製方法を提供する。
【0025】
別の実施形態において、本発明は、
(A)ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼの不純試料を陰イオン交換クロマトグラフィーにかける工程であって、溶出が均一濃度である工程;
(B)工程(A)の溶離液を疎水性相互作用クロマトグラフィーにかける工程であって、硫酸アンモニウムが使用されず、溶出が均一濃度である工程
を含む、ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼを発現および培養培地中への分泌後に精製する方法を提供する。
【0026】
所望の酵素は、本発明の細胞および細胞株によって発現され、好ましい実施形態では、本発明の精製手法に従って精製される。次いで、該酵素の存在下で、グリシン残基を有し、遊離酸の形態であり、カルボニル基に結合された前駆体から始まる反応が行なわれる。反応条件および補因子は当該技術分野で知られている。本明細書に記載の実施例1および2に、アミド化生成物の典型的なアミド化反応および精製を示す。好ましいアミド化生成物が得られる。ペプチジルα−ヒドロキシル化モノオキシゲナーゼを用いた場合、中間体が得られ、これは、アミド化生成物を形成するために、ルイス塩基またはペプチジルα−ヒドロキシグリシンα−アミド化リアーゼのいずれかとのさらなる反応を必要とする。
【0027】
酵素産生細胞株、例えば本明細書に記載のものが、酵素を発現して培養培地中に分泌する場合、特定のある実施形態では、続いて、該酵素を精製するための一連の工程が行なわれ得る。収集工程では、ならし培地が細胞から分離される。第1のタンジェンシャルフロー濾過(濃縮、ダイアフィルトレーション)により、低分子量成分が除去される。陰イオン交換クロマトグラフィー工程および疎水性相互作用クロマトグラフィー工程は、主に、タンパク質不純物および培地成分からの精製のために用いられる。ウイルス濾過前の最後のタンジェンシャルフロー濾過、濃縮およびバッファー交換は、望ましくは、保存前に実施される。改善された陰イオン交換クロマトグラフィーおよび疎水性相互作用クロマトグラフィーを、本明細書の特許請求の範囲に示す。
【発明の効果】
【0028】
驚くべきことに、望ましくは安定なCHO K1細胞が、内在ジヒドロ葉酸還元酵素遺伝子の存在にもかかわらず、本発明の状況において使用され得ることがわかった。該内在遺伝子は、ジヒドロ葉酸還元酵素遺伝子を有する選択可能なマーカーに基づくメトトレキサート選択を有意に妨げなかった。
【0029】
PAM遺伝子と、独立したベクター上の2つの選択可能なマーカーとの共トランスフェクションは、本明細書に記載のように、ゲノムのより望ましい部分で起こる共増幅の機会を有意に増大させると考えられる。
【0030】
SV40エンハンサーが使用される場合、エンハンサーは、任意の転写プロモーター、例えば限定されないが、SV40転写プロモーター、本明細書に記載の好ましいメタロチオニンIIAプロモーターなどとともに使用され得ると理解されている。
【0031】
本発明に従って発現および精製されるペプチジルグリシンα−アミド化モノオキシゲナーゼは、望ましくないプロテアーゼ活性を実質的に含まず、本明細書に記載のような基質の効率的なα−アミド化に好適である。
【0032】
本明細書に記載の好ましい細胞株であるUGL 73−26/Mは、良好な長寿命プロフィールを示したため、安定性が非常に重要である規模の拡大に特に有用であると考えられる。
【0033】
同様に、本明細書に記載の好ましい精製手法では、拡張可能な特性および有意な生成物純度が提供される。具体的には、画分収集が有意に単純化される均一濃度の溶出が使用される。先行技術のような疎水性相互作用クロマトグラフィーと共に硫酸アンモニウムを使用することは、硫酸アンモニウムが酵素の沈殿および不活化を引き起こし得るため、望ましくは回避する。
【0034】
類推により、本明細書に記載の発明は、ペプチジルグリシンα−アミド化モノオキシゲナーゼに関して有益であったため、ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼに関して有益であることが予測される。
【0035】
本明細書に記載の好ましい態様の1つ以上を組み合わせて使用され得る。例えば、限定を意図しないが、好ましいプロモーターが、好ましいエンハンサーおよび/または好ましい宿主細胞と一緒に使用され得る。
【0036】
(A)PHMおよび(B)PALまたはルイス塩基のいずれかがPAMの代わりにアミド化に使用される場合、PHMおよびPAL(使用される場合)は、いくつかの様式で得られ得る。一部を以下に示す。
【0037】
PHM
発現されたときPHM活性のみを含むPAM遺伝子の天然に存在する形態の発現
カエルに由来する酵素の一例は、天然に存在するPHM酵素である。Mizunoら(1986),「Peptide C−Terminal a−Amidating Enzyme Purified to Homogeneity From Xenopus laevis Skin」Biochem Biophys.Res.Commun.,137(3)984−991を参照のこと。これは、後に、完全長PAMではなくPHMであることがわかった。Suzukiら(1990),EMBO 9(13)4259−4265を参照のこと。
【0038】
完全長PAMの発現および切断
PAM酵素をPHM活性とPAL活性の間の部位、例えば2塩基性切断部位で切断するため、特異的プロテアーゼが使用され得る。次いで、PHM触媒ドメインが精製によって得られ得る。例えば、Ouafikら,(1992)「The Multifunctional Peptidylglycine a−Amidating Monooxygenase Gene:Exon/Intron Organization of Catalytic,Processing,and Routing Domains」Molecular Endocrinology 6(10)1571−1584には、ラット由来PAM内の2つの触媒ドメインの位置が記載されており、「対の塩基部位での内部タンパク質分解性切断により、2つの触媒ドメインが分離され得る」と記載されている。また、Eipperら(1993),「Peptidylglycine a−Amidating Monooxygenase:A Multifunctional Protein With Catalytic,Processing,and Routing Domains」Protein Science 2,489−497も参照のこと。
【0039】
PAM内への停止コドンまたはリーディングフレームを改変する点変異の導入
代替法として、翻訳停止コドン(TAA、TAG、TGA)が、任意の種由来の任意のPAM cDNA内のPAMの2つの機能ドメイン(PHMおよびPAL)間に導入され得るか、またはリーディングフレームを改変するために、かかる位置内に点変異が導入され得る。
【0040】
PHM cDNAのみを有する発現ベクターの再設計
PCRを用い、PHMドメインのみをコードする切断型PAM遺伝子を合成し、プロモーターまたはエンハンサー/プロモーター配列の下流の発現ベクターに配置し得る。
【0041】
以下に、異なる型のPAM酵素およびその触媒ドメインに関するさらなる参考文献を示す。
【0042】
Mizuno,K.ら,(1987)「Cloning and sequence of cDNA encoding a peptide C−terminal α−amidating enzyme」、Xenopus Laevis Biochem Biophys.Res.Commun.,148(2)546−552
Ohsuye,K.ら,(1988)「Cloning of a cDNA encoding a new C−terminal α−amidating enzyme having a putative membrane−spanning domain」、Xenopus Laevis Biochem.Biophys.Res.Commun.,150(3)1275−1281
Koljekar,A.S.ら,(1997)「Peptidylglycine α−amidating hydroxylating monooxygenase:active site residues,disulfide linkages and tow−domain model of the catalytic core」,Biochemistry 36:13901−13909
Koljekar,A.S.ら,(2002)「Essential features of the catalytic core of Peptidylglycine α−hydroxyglycine α−amidating lyase」,Biochemistry,41:12384−12394
Bertelsen,A.H.ら,(1990)「Cloning and characterization of two alternatively spliced rat α−amidating enzyme cDNAs from rat medullary thyroid carcinoma」,Arch.Biochem.Biophys.,279(1)87−96
Jimenez N.ら,(2003)「Androgen−independent expression of adrenomedullin and peptidylglycine α−amidating monooxygenase in human prostatic carcinoma」,Molecular Carcinogenesis 35:14−24
PAL
天然に存在する形態のPALの発現
PAMの2つの触媒ドメインは、ショウジョウバエと刺胞動物(イソギンチャク)の別々の遺伝子にコードされている。したがって、これらの種に由来するPALをコードする遺伝子は、プロモーターまたはエンハンサー(enchancer)/プロモーター配列の下流の発現ベクターに配置すると、発現され得る。Kolhekar,A.S.ら.(1997)「Neuropeptide Amidation in Drosophila:Separate Genes Encode the Two Enzymes
Catalyzing Amidation」J.Neuroscience 17(4):1363−1376を参照のこと。
【0043】
完全長PAMの発現および切断
完全長の2つの機能を有するPAMを発現させ得、PHMを不活化させるために、PHMドメインとPALドメイン間および/またはPHMドメイン内の特異的プロテアーゼ部位で切断を行ない得る。任意の適当に特異的なプロテアーゼを用い、例えば、2塩基性切断部位でPHM活性をPAL活性から分離し得る。PAL活性/タンパク質は、さらに精製され得る。同様の手順および2塩基性切断部位の位置は、PHMの取得に関して上記に記載している。
【0044】
PAL cDNAのみを有する発現ベクターの再設計
PCRを用い、PALドメインのみをコードする切断型PAM遺伝子を合成し、プロモーターまたはエンハンサー/プロモーター配列の下流の発現ベクターに配置し得る。
【0045】
本発明の他の特徴および利点は、添付の図面、グラフ、表などに言及する本発明の以下の記載から明らかとなろう。
【図面の簡単な説明】
【0046】
【図1】図1は、pHS1のプラスミドマップである。
【図2】図2は、pUC8のプラスミドマップである。
【図3】図3は、pSV402MTのプラスミドマップである。
【図4】図4は、α−AE cDNA遺伝子配列の誘導(derivtion)である。
【図5】図5は、pAE73のプラスミドマップである。
【図6】図6は、73−26浮遊適合スキームである。
【図7】図7は、スピナーフラスコにおける溶存酸素濃度およびα−AE産生性である。
【図8】図8は、DOおよびα−AE産生性に対するスピナーフラスコの容積の効果である。
【図9】図9は、UGL 73−26/Mの攪拌培養pHである。
【図10】図10は、pH制御なしでの攪拌槽バイオリアクターおよびスピナーフラスコのpHプロフィールである。
【図11】図11は、攪拌槽バイオリアクターにおけるUGL 73−26/Mα−AE産生性に対するpHの効果。
【図12】図12は、精製PAM酵素のSDS PAGEである。
【図13】図13は、SDS PAGEゲルのデンシトメトリーのスキャンおよびピーク面積パーセントの計算である。
【図14】図14は、α−AEの産生のプロセスフローダイアグラムである。
【図15】図15は、UGL 73−26/Mの種菌スキームである。
【発明を実施するための形態】
【0047】
発明の実施形態の詳細な説明
本発明によるPAM発現細胞株(内部的にはUGL 73−26/M MWCB 00と示す)は、American Type Culture Collection(ATCC),10801 University Boulevard,Manassas Virginia,20110−2209,U.S.A.に、2005年6月10日あたりに、特許手続きのための微生物寄託の国際認識に関するブダペスト条約に従って寄託された。ATCC受託番号はPTA 6784である。この寄託細胞株は、この条約の下に公布された規定に従い、試料は、その時点で、および該条約によって求められる条件下で、ならびに特許法および該条約の調印国の規定に準じて入手可能となる。例えば、本出願またはこれを優先権主張するか、もしくはこれを参照する任意の他の米国特許出願の米国特許が発行された場合、該寄託物質の入手可能性に関するあらゆる制限は、ブダペスト条約または35 U.S.C.§112に求められる程度まで変更不可的に解除される。
【0048】
本発明のPAM発現ベクターの構築を、以下に詳細に記載する。本明細書に示すプラスミドマップにおいて、「αアミド化酵素」(例えば、図5、pAE73のプラスミドマップを参照)と示す領域は、添付の配列番号1に示されるヌクレオチド配列を有し、添付の配列番号2に示される715アミノ酸の一次翻訳産物をコードする。該翻訳産物は、シグナルペプチドをアミノ酸1〜25に、プロ領域を26〜41に、および成熟ペプチジルグリシンα−アミド化モノオキシゲナーを42〜715に含む。本発明に従って発現および精製される酵素のほとんどは、該プロ領域を含み続け、非常に有効であることが示された。
【実施例】
【0049】
以下、好ましいPAM発現細胞株、その発酵、方法の開発、精製および使用の例を詳述する。
【0050】
以下により詳細に記載するように、PAM遺伝子を、pSV402MT発現ベクター内にクローニングし、PAM発現プラスミドpAE73を創製した。CHO K1細胞をpAE73 DNAでトランスフェクトした。二重選択プロセスおよび限界希釈クローニング後、メトトレキサートの濃度を増加させ、CHO細胞株をさらに増幅した。接着細胞株73−26を、選択なし血清無含有の浮遊培養物UGL 73−26/Mに変換した。UGL 73−26/M MCBおよびMWCBは、BioReliance Corp.において創製した。MCBおよびMWCBの両方を充分に特性化したところ、GMP施設においてのPAMの産生に対し、認可され得ると考えられる。UGL 73−26/M細胞株を適合させ、攪拌槽バイオリアクター内での培養した。発酵の開発により、スピナーフラスコで14日間行なわれる種菌プロセスの定義付けがもたらされた。発酵/細胞培養相は17日間のプロセスである。発酵プロセスの重要なパラメータは以下の通り:第5、10および14日にグルコース添加(2g/L)、溶存酸素濃度≧70%、発酵がその自然な設定点に達するpH、およびタンパク質無含有、非動物由来組織培養培地、Sigma C5467(グルタミンを終濃度2mMまで補充)である。簡便でロバストな下流精製プロセスを、C5467 CHOならし培地からのPAMのために開発した。精製後、定常的に純度の4倍増加および活性酵素の40%の総回収が得られる。該プロセスでは、製造レベルに規模を拡大し易い慣用的な濾過系およびバイオプロセスクロマトグラフィー樹脂が用いられる。細胞培養発酵と下流精製のプロセス全体を、10Lパイロットプラント/製造レベルの規模にした。記載のプロセスは、大量のPAMの産生について確認可能な製造プロセスに従いやすいものであるのがよい。
【0051】
大量のPAMを発現するCHO細胞株UGL 73−26/Mを開発した。UGL 73−26/M用に開発した細胞培養プロセスは、非動物タンパク質由来の培地中のものとした。単純なバッチ細胞培養プロセスが好ましかった。精製スキームの目的は、触媒的に活性な酵素の良好な収率を伴う、ロバストで拡張可能なプロセスを開発することであった。
【0052】
ベクターの構築
該細胞株を作製するために使用される発現ベクターpAE73の創製は、Friedmanら.Bio/Technology 7:359−362(1989)の研究結果を基にした。この論文に記載されたプロセスに従い、本発明者らは、試験用のpAE73発現ベクターの創製を進めた。各ベクターの主要成分は、ヒトメタロチオネインIIA(hMTIIA)プロモーター(pHS1内に位置する、M.Karin氏より受領)、SV40エンハンサー(pSV40由来、ATCC)および本明細書に記載のα−アミド化酵素遺伝子である。
【0053】
pAE73の誘導化
pHS1
pAE73の創製に使用する出発プラスミドは、1990年2月にUCSDのM.Karinから贈与プラスミドpHS1(図1)であった。pHS1は、Dr.Karinの研究室において、pUC8(図2)内にヒトメタロチオネインIIAプロモーターをスプライシングすることにより誘導されたものである。846bpのHind III/Bam H1断片を、pUC8マルチクローニング部位(MCS)のHind III−Bam H1部位内にクローニングした。得られたプラスミドpHS1は、該マルチクローニング部位内に挿入されたヒトメタロチオネインHAプロモーターを有する。
【0054】
pSV401MTおよびpSV402MT
SV40エンハンサーをヒトメタロチオネインプロモーターの上流に挿入することにより、pHS1を発現ベクターpSV401MTまたはpSV402MT(図3)に変換した。SV40 DNA断片は、pSV40プラスミドDNAをHind IIIで消化することにより調製した。1167bpのHind III断片を、pHS1のHind III部位内にクローニングした。SV40エンハンサーは、Hind III DNA断片内に非対称に存在し、したがって、該エンハンサーは、hMTIIAプロモーターに近接して、または遠く離れてのいずれかで位置する。元のプラスミド内のlac Z遺伝子内のものに対するSV40エンハンサー内のBgl I部位の配向により、該プラスミドの配置(designation)が決まる。pSV401MTは、hMTIIAプロモーターから遠く離れたSV40エンハンサーを有し、pSV402MTは、hMTIIAプロモーターに近接したSV40エンハンサーを有する。pSV402MTを、さらなるベクター構築に選択した。
【0055】
pAE73
pAE73は、α−アミド化酵素遺伝子(配列番号1)を、pSV402MTのBam H1部位内にクローニングすることにより調製された。2870bpのα−AE遺伝子断片を、Bgl 1およびBam H1 DNA制限エンドヌクレアーゼでのpAE64の消化後に単離した。pAE64プラスミドは、SV40プロモーター/エンハンサーの下流に、可溶性75kDaのPAMタンパク質を発現するように修飾されたPAM遺伝子を含有する。このPAM DNA配列を別のCHO発現細胞株UGL B3/A1−7に使用し、75kDaのPAMタンパク質を発現させた。PAM遺伝子配列の誘導を図4に示す。
【0056】
2870bpのDNA断片を精製し、該断片のBgl 1末端を、2つの相補DNAオリゴヌクレオチド断片(AE96/AE97)で修飾し、これにより、該断片のBgl 1末端が新たなBam H1末端に変換された。
【0057】
AE96(+)23 5’GATCCATCGATCGCACTAGTGCC 3’
AE97(−)16 5’ACTAGTGCGATCGATG 3’
該修飾PAM DNA断片を、発現ベクターpSV402MTのBam H1部位内にクローニングした。2つのBam H1末端を有するPAM DNA断片は、発現ベクター内にセンスまたはアンチセンスのいずれかの配向に連結され得るため、PAM遺伝子の配向を、該プラスミドのEco R1による制限消化マッピングによって調べた。Eco R1部位と該発現ベクター両方におけるPAM遺伝子により、プラスミドDNAの単純な解析が可能になる。正しい配向のPAMを有するプラスミドをpAE73と表示し、そのプラスミドマップを図5に示す。
【0058】
α−アミド化酵素発現細胞株の創製
トランスフェクションおよびクローニング
3種類のプラスミド:pAE73、pSV2neoおよびpSY2dhfrをCHO K1細胞の形質転換に使用した。トランスフェクト細胞によるプラスミドの組込みにより形質転換細胞株の容易な選択が可能となるため、プラスミドpSV2neoおよびpSV2dhfr(ともにATCCから入手)を使用した。また、プラスミドpSV2dhfrは、特に、CHOゲノム内へのSV40プロモーター/dhfr DNAの組込みにより該遺伝子および他の近位遺伝子の選択的増幅が可能となるため選択された。共増幅される遺伝子は、プラスミドpAE73内に担持されたα−アミド化酵素遺伝子であった。CHO K1細胞は、プラスミドDNAのリン酸カルシウム沈殿により形質転換した。プラスミドは、該細胞内にpAE73:pSV2neo:pSV2dhfrが10:1:1の比でトランスフェクトした。100mmdish1つあたり20μgのpAE73を添加した。トランスフェクションの2日後、形質転換細胞のみが生存可能となるように、細胞を、選択圧下、250mg/LのG418を含有する培地中で培養した。この培地中でのCHO細胞の培養では、pSV2neoプラスミドの安定な組込みが必要とされ得る。トランスフェクションの27日後、G418選択培地中でいったん安定な培養が確立されたら、メトトレキサートを該培養培地に添加した。形質転換細胞のG418プールを、100nM、500nM、1μMまたは5μMのメトトレキサートおよび250mg/LのG418を含有する培地中で培養した。2週間後(トランスフェクションの6週間後)、単離した細胞フォーカスは、クローニングシリンダーにより5μMメトトレキサート+G418培地中で培養した細胞から確立され得る。この方法によって、25のフォーカスから細胞株を確立する試みを行なったが、移した後、2つのフォーカスからしか細胞が培養されず、この手法を却下した。限界希釈クローニングを0.5細胞/ウェルで、1μMメトトレキサート+G418を含有する培地中で培養した細胞から同時に開始した。低濃度のメトトレキサートで培養した形質転換細胞は除外した。限界希釈クローニング開始から3週間後、単離物を24ウェルプレートに移し、次いで、2〜3日間以内に増殖のため100mmdishに移した。CHO K1細胞のトランスフェクション開始から10週間後、73−26と表示する単離物のうちの1つを確立した。該細胞株を低温保存し、α−アミド化酵素発現を評価した。73−26は、この時点の6953U/106細胞/日を産生する最良の産生細胞株の1つであった。
【0059】
細胞株73−26の増幅
73−26の初期クローンを継代し、1μMメトトレキサート+G418を含有する培地中で維持される確立された細胞株を得た。pSV2dhfrプラスミドをトランスフェクションのために選択する目的は、形質転換細胞株を同定するための第2の選択方法が提供されるためだけでなく、細胞を高濃度のメトトレキサートで培養すると、dhfrミニ遺伝子が増幅されることが広く確立されているためであった。Unigene Laboratoriesは、以前に、このプラスミドで形質転換された細胞株を用い、α−アミド化酵素の最大の産生をもたらすのに20〜50μMの濃度のメトトレキサートが必要とされ得ることが示されたことを経験している。α−アミド化酵素細胞株73−26を直接、1μM、20μMまたは50μMのメトトレキサートを含有する培地に分割した。また、すべての培地に、第2の選択方法としてG418も含めた。この細胞株のα−アミド化酵素発現の進行を、種々の時間間隔で表1に示す。
【0060】
【表1】

【0061】
表1のデータは、以前に開発されたB3/A1−7細胞株のものと比べ、この細胞株におけるα−アミド化酵素の発現レベルの増大を示す。α−アミド化酵素遺伝子とdhfr遺伝子の共増幅は、メトトレキサートの濃度を増大させて継代培養を継続するより多くの酵素を発現する該細胞株能力により明白になる。20μMまたは50μMメトトレキサートの存在下で培養した細胞は、選択的増幅の3〜4ヶ月後、最良の発現レベルを有した。継続的に数ヶ月間、継代培養すると、α−アミド化酵素の発現レベルは低下した。したがって、以前に凍結させたこの細胞株のストックを、浮遊適合細胞株の調製に使用した。
【0062】
浮遊適合α−アミド化酵素発現細胞株73−26の特性化
この細胞株の開発の最終目的は、メトトレキサートおよびG418の選択圧なしで安定な方法を開発することである。該細胞株の所望される別の属性は、これが、浮遊培養において比較的高い細胞密度で培養されることであった。該目的を達成するために用いた方法を以下に記載する。
【0063】
浮遊適合無選択細胞株の調製
無選択浮遊適合細胞の調製では、UGL 73−26接着細胞を、血清、メトトレキサートおよびG418から離すことが必要とされた。この課題を達成するため、multi prong attack(図5)を開発した。除去のマトリックス(matrix of removal)は、可能な最終の細胞株に対して最後の数の細胞倍加を付加するという最終目的で確立された。将来的な研究開発のため、以前に凍結させた(1991年12月16日)73−26の20μMメトトレキサート細胞株を解凍し、浮遊適合血清無含有無選択細胞株を確立した。50μMメトトレキサートでのPAM活性はいくぶん高かったが、73−26 50μM細胞株の活性培養物を再確立する試みは成功裏でなかった。該細胞株のα−アミド化酵素産生性を、浮遊適合プロトコルを開始する前に評価した(表2)。
【0064】
【表2】
【0065】
浮遊培養に適合させる前のこの細胞株のPAM活性は、細胞1つあたりの基準で、B3/A1−7対照よりも20倍大きい活性を有した。対照細胞株および73−26両方での酵素活性は、該細胞株を凍結する(1991年12月16日)直前の最初の試験で観察されたものの5〜10倍であった(表1)。
【0066】
4種類の新たな浮遊適合血清無含有細胞株を、図6に示すように作製した。73−26/K−Nと表示する該細胞株を、37日間のプロセス後、低温保存した。
【0067】
浮遊適合73−26のα−アミド化酵素産生
新たな73−26細胞株(73−26/K、73−26/L、73−26/M、73−26/N)のバンク登録細胞を解凍し、スピナーフラスコ内で培養した。該細胞株を、週に3回Ex−Cell301培地中で継代することにより維持した。定期的に培地を完全に交換し、酵素産生性の24時間評価をα−AEアッセイによって評価した。細胞株は、80日間まで培養状態で維持した。以前の細胞株B3/A1−7の半連続的バッチプロセスは40日間プロセスであり、該細胞株の産生安定性を評価するため、この手順の長さの2倍を選択した。この研究のデータを表3に示す。この研究過程において、細胞株73−26/Lは第22日〜第40日の間で終わったが、他のすべての細胞株は、80日の期間の最後まで活発に培養された。
【0068】
【表3】
【0069】
すべての細胞株が、初期の期間ではB3/A1−7よりも有意に多くのα−アミド化酵素を発現したが(<40日間)、73−26/Mおよび73−26/Nのみ、70日後もなお、大量の酵素を産生していた。80日の期間にわたって一貫してより多くのα−アミド化酵素を産生したことから、さらなる開発のためにUGL 73−26/Mを選択した。その他の3種類の細胞株は、80日の試験期間の終了前に培養が終了したか、またはその産生性/細胞/日が最後の20日間で減衰したかのいずれかであった。
【0070】
CHO K1、UGL 73−26、UGL 73−26/M MCBおよびUGL 73−26/M MWCB00のバンク登録前での細胞バンク登録の特性化研究
特性化研究を、前駆細胞株(73−26)である宿主細胞株(CHO K1)およびUGL 73−26/Mシードバンクにおいて行なった。40個のバイアルのUGL 73−26/Mシードバンクを1998年2月4日に調製した。シードバンクの各バイアルには、90%Ex−Cell 301/10%DMSO中4×106細胞/mLが含まれる。実施した研究はすべて、CHO細胞株がMCBおよびMWCBになる必要があるという結果と整合すると評価された(表4参照)。
【0071】
マスター細胞バンクUGL73−26/M細胞株は、BioReliance Corp.において作製された。118MCBのバイアルのいくつかを用い、該細胞株を充分に特性化した。UGL 73−26/M MCBのすべての研究の結果は、CHO起源の細胞株と整合し、感染性因子について陰性である。UGL 73−26/M MCBのバイアルを用い、製造業者の研究用の238個のバイアルの細胞バンクUGL 73−26/M MWCB00が、BioReliance Corp.において創製された。UGL 73−26/M MWCB00の研究のすべての結果は、CHO起源の細胞株と整合し、感染性因子について陰性である。
【0072】
【表4−1】
【0073】
【表4−2】
【0074】
【表4−3】
【0075】
スピナーフラスコおよび攪拌槽バイオリアクターにおけるUGL 73−26/Mのプロセスの開発研究
予備実験では、該細胞株の産生性が多数の生成では一貫して維持され得ないことが示されたため、バッチ発酵プロトコルを検討した。また、バッチプロトコルは、計画の容易さ、易拡張可能性および劇的なバッチ不成功の結果が少ないという利点を提供する。UGL 73−26/Mの発酵プロセスの開発には、該細胞株の細胞内産生性に影響し得るいくつかのパラメータを検討する必要がある。以下に詳述する発酵開発は、溶存酸素(DO)、pHおよび培地補充を検討する主要な研究を示す。研究されなかった発酵パラメータは、インペラーのRPMおよび発酵温度であった。
【0076】
α−アミド化酵素発現に対する溶存酸素の効果
スピナーフラスコ内のCHO培養培地の溶存酸素濃度の効果を調べるための実験を行なった。スピナーフラスコには0.1×106細胞/mLを播種した。2つのスピナーフラスコは、250mLTechneスピナーフラスコ内に150mLの培地を入れ、一方、第3のスピナーフラスコには250mLの培地を入れた。すべてのスピナーフラスコにおいて溶存酸素を毎日測定した。α−AEアッセイによるα−AE産生性の評価のため、清澄ならし培地のアリコートを毎日採取した。この研究では、低タンパク質CHO培地(C1707、Sigma−Aldrich)およびタンパク質無含有CHO培地(C5467、Sigma−Aldrich)の2種類の培養培地を使用した。この2種類の培地中で培養したUGL 73−26/Mの直接比較を図7に示す。2つの培養物中の溶存酸素レベルは著しく異なった。150mLのC1707中で培養した細胞は、等容量のC5467培養培地中で培養した細胞よりも、産生された全酵素/mLが少なかった。両培地の初期の溶存酸素濃度は約80%DOで同じであったが、C5467培地の培養物の溶存酸素濃度は、絶対に50%未満にならなかった。C1707培養物のDO含量は、第9日までに50%未満となり、該培養物のピーク活性は2日後であった。これらのデータは、DO含量/細胞バイアビリティ/α−AE産生性間に相関性が存在し得ることを示す。
【0077】
また、スピナーフラスコの容積も潜在的臨界パラメータとして検討した。スピナーフラスコは一定溶存酸素濃度に維持されないため、培養物が溶存酸素を維持する能力は、培養物の表面:容量比に対し相対的である。スピナーフラスコの培養容積を増大させると、明らかに表面:容積比は変化する。一方のスピナーフラスコの培養容積を150mL(5467s)とし、他方を250mL(5467sa)とした以外は、同一の培養物で開始した。%DOおよびα−AE産生性に対する表面:容積比の変化の効果を図8に示す。
【0078】
図8に示すデータは、培養物の産生性が溶存O2によって大きく影響されることを示す。溶存O2濃度の減少が、培養物による利用のためであれ、表面:容積比の差による効果のためであれ、DO濃度が50%未満の場合、培養物は48〜72時間以内に、より多くのα−アミド化酵素を発現するのを停止する。細胞が最も良好に産生性である培養期に溶存酸素濃度を反映させるためには、バイオリアクター内の溶存O2を70%に維持するのがよい。
【0079】
α−アミド化酵素発現に対する培地の効果
UGL 73−26/Mの培養培地の最も適切な選択肢の選択を容易にするために設計された研究、またはバイオリアクターの培養条件を規定する際の研究のいずれかの一部として、2種類のCHO培養培地C1707およびC5467(Sigma−Aldrich)を用いて数多くの攪拌培養を開始した。両培養培地は規定されたものであり、C1707は、トランスフェリンが添加された低タンパク質培地であり、C5467は非動物タンパク質培地である。培地はともに、購入時に、終濃度2mMまでL−グルタミンを添加する必要があった。以下の表5に、2つの培地のいずれかを用いたスピナーフラスコの多数のサンプリングのデータを示す。
【0080】
【表5】
【0081】
表5のデータでは、いずれかの培地におけるUGL 73−26/M細胞株の酵素産生性の実質的な差は示されていない。Sigma CHO培養培地であるC5467は、動物由来の培地成分を含有しないため、米国および欧州連合の両方において、将来的な規制的ガイドラインに関して、より規制に準拠する培地の選択肢であり得る。Sigma CHO培養培地であるC5467は、将来的な研究のための培地の選択肢である。
【0082】
α−アミド化酵素発現に対するグルコースの効果
UGL73−26/Mのバッチ培養物の栄養状態を検討する研究により、培養開始の4〜5日後、培地中のグルコース濃度は、未使用培地のほぼ50%(2g/L)であることが明らかになった。酵素産生性に対するグルコース補充の効果の徹底的な研究を、さらなるグルコースを培養物に添加する一連のスピナーフラスコ実験として行なった。スピナーフラスコには、2g/Lのグルコースを1〜3回、特定の時間間隔で補充した。α−AE産生性に対するグルコース添加の効果を表6に示す。
【0083】
【表6】
【0084】
【表7】
【0085】
UGL 73−26/Mのバッチ培養物へのグルコースの添加により、培養物の最大産生性は、グルコース添加計画に応じて50〜100%増大した。第5、10および15日目にグルコースを補充したスピナーフラスコでは、非補充対照と比べ、最も多くの酵素が発現されるようである。これらの培養物の平均相対産生性は対照の195%であった。他の補充培養物では、3回補充のものよりほんのわずか産生性が低かった(143〜164%)。さらなるグルコースを補充した培養物はすべて、培地中でのピーク酵素濃度の遅延を示した。ピーク酵素濃度は、グルコース補充培養物では第17/18日目に観察されたのに対し、対照の非補充培養物では第13日目であった(表7)。
【0086】
α−アミド化酵素発現に対するpHの効果
ならし培地のpHは攪拌槽バイオリアクターでは制御され得るが、スピナーフラスコにおいては対処され得ないプロセス変量の1つである。スピナーフラスコでは、培地成分が消費されるにつれて、および細胞の副生成物が産生されるにつれて、培養物のpHは低下する。150mL(5467s)または250mL(5467sa)のいずれかの容積のUGL 73−26/Mの2つの250mLスピナーフラスコのpHプロフィールを、以下に図9に示す。培養物のpHは、バッチの前半では低下し、次いで培養の後半では上昇した。pHの低下は、おそらく、培養前半での培養物中の乳酸濃度の増大によるものである。培養の最後でのpHの上昇は、細胞が該乳酸を炭素供給源として異化することによるものである。培養物の産生性はpHの低下に影響されない(図8)。
【0087】
いくつかの(n=5)6〜10L攪拌槽バイオリアクターは、スピナーフラスコ内の条件を模倣し、pH制御なしで実行した。攪拌槽バイオリアクター内のCHO細胞をC5467培地中で、70%DOおよび37℃にて培養した。バイオリアクター内のならし培地の平均pHプロフィールは、2つのスピナーフラスコで観察されたものと同様である(図10)。バイオリアクター内で溶存酸素濃度は70%に、温度は37℃に維持した。
【0088】
バイオリアクターを上記のようにpH制御なしで実行させたことに加え、一連のバイオリアクターは、該バイオリアクターのpHが設定pH点(7.0〜7.4)に自由になるように実行した。所望のpHに達した時点で、これをそのpHに発酵の持続期間、維持した。攪拌槽バイオリアクター内のCHO細胞をC5467培地中で、70%DOおよび37℃にて培養した。攪拌槽バイオリアクター内でのα−AE産生性に対するpH制御の維持の効果を図11に示す。
【0089】
図11は、pH設定点を開始しなかった場合、それらの実行に対する実行の間、各日のバイオリアクターにおける平均α−AE活性を示す。pHが設定点の0.5pH単位以内になった後でのみpH設定点を設定した場合の個々の実行のデータもまた図11に示す。培養pHと酵素活性と間に明白な相関性がある。pH設定点なしで実行させたバイオリアクターの活性プロフィールは、培養の最初の16日間のすべての実行について優れている。唯一の例外は、pH設定点が7.1である場合の実行に対するプロフィールであった。この差は、おそらく、培養を、その他の実行よりも長く持続させたためであった。平均pH設定点なしの研究でのピークα−AE活性は、第16日目で280,707単位/mLであった。
【0090】
生細胞密度に対するpHの効果はなかった(データ示さず)。培養物は、1.0〜1.5×106細胞/mLのピーク生細胞密度まで培養させた。最大細胞密度は、バイオリアクターにイノキュレートしてから8〜10日後に達成された。pH7.1で実行させた1つの培養物以外は、細胞バイアビリティに対するpHの影響はなかった(データ示さず)。細胞培養物のバイアビリティは、15日以上培養した場合で50%生細胞より大きかった。7.1のpH設定点で実行させた1つのバイオリアクターでは、20日間の培養で>50%生細胞が維持された。
【0091】
研究開発したUGL 73−26/Mの発酵パラメータの概要
α−AE発現細胞株UGL 73−26/Mの確立後、α−アミド化酵素の至適発現のための臨界パラメータを規定するために一連の実験を行なった。動物タンパク質無含有培地(Sigma,C5467)では、該酵素の高レベルの発現が支持されることが測定された。CHO細胞は、L−グルタミンを終濃度2mMで補充したC5467培地中で、バッチ培養物として培養され得る。しかしながら、該培養物は、その産生性をさらに促進するためには、D−グルコース補充を必要とする。至適グルコース補充は、第5、10および15日目で2g/Lであった。バイオリアクターのDO濃度は70%に維持するのがよく、バイオリアクターのpHは、培地の緩衝能とCHO細胞による培地成分の代謝の関数となるpHに調整されるようにするのがよい。
【0092】
下流精製の開発UGL 73−26/M
以下の節に、本発明者らが開発した、UGL 73−26/Mクローンを用いて産生されたPAMの下流精製についてまとめる。代表的な実験のみ(該プロセスの開発作業の論理的な進行を示す)をこの概要に含める。各工程の簡単な説明を示すが、これは、精製プロセスにおけるその工程の機能の記載である。精製実行は、SDS−PAGE(一定タンパク質および一定単位)、PAM活性アッセイならびにBradfordタンパク質アッセイを用いて解析した。実験はすべて、Boonton,NJ試験施設に技術移転する前または直後に完了した。本明細書に記載の方法を用い、PAMバッチ1330−D−1003、1330−D−1004、1330−D−1005、1330−D−1006、1330−1009および1330−1010を製造した。
【0093】
第1のタンジェンシャルフロー濾過(TFF1)
工程の説明:TFF1工程を用い、クロマトグラフィーの前にCHO細胞ならし培地の濃縮およびダイアフィルトレーションを行なう。ならし培地を濃縮し、ダイアフィルトレーションによって導電率を低下させ、陰イオン交換カラムへの結合を助長する。TFF1工程では、再生セルロースPLCTK 30kDa膜(Millipore)を取り付けたPellicon 2 Moduleを使用する。
【0094】
研究開発:この工程の最初の検討では、TFFがクロマトグラフィーの前に必要とされ得るか否かに着目した。水での希釈によるCHOならし培地の導電率の低下を、TFFの代替法として検討した。クロマトグラフィーの前にならし培地を希釈しないと、充填後、PAM活性の大部分が通過画分中に存在した。ならし培地の導電率をほぼ5mSまで低下させると、通過画分中に検出されるPAM活性の量は最小限になった。しかしながら、ならし培地を陰イオン交換カラムに直接充填すると、2つの問題が確認された。一部の培地成分が不可逆的にカラムに結合し、樹脂がひどく変色した。勾配溶出後、大きなUV吸光度特性を示す別の培地成分が、PAM活性とともに共溶出された。
【0095】
CHO細胞ならし培地(C5467)を水で希釈し、導電率をほぼ5mSまで低下させ、50mM TRIS、120mM NaCl、0.001%TX−100(pH8.0)で平衡化させたQ−セファロースFFカラム(Pharmacia)に直接充填した。カラムを、100%A(50mM TRIS、120mM NaCl、0.001%TX−100(pH8.0))から68%B(50mM TRIS、475mM NaCl、0.001%TX−100(pH8.0))までの60分かけた直線勾配にかけた。充填直後、大きな暗色のバンドがカラム上部に出現し、クロマトグラフィー実行中、残存した。カラムを2M NaClで清浄にし、1.0M NaOHで殺菌した。清浄手順および殺菌手順を行なっても、暗色バンドはカラムから除去されなかった。続いて、カラムは、より厳しい清浄手順を行なった。カラムを、以下の一連の清浄試薬:0.5M NaOH 1M NaCl、0.1M酢酸、0.1M酢酸 1M NaClおよび70%エタノールにさらした。試験したいずれの条件でも暗色バンドはカラム(NEG:001:225−245)から除去されなかった。着色バンドは、未使用C5467培地中に存在する一成分のアウリントリカルボン酸であると同定された。TRISでpH8.0まで緩衝化したアウリントリカルボン酸の6mg/Lの溶液(培地濃度)を、50mM TRIS、120mM NaCl、0.001%TX−100(pH8.0)で平衡化させたQ−セファロースFFカラムに直接充填した。充填直後、着色バンドがカラム上部に出現した。このカラムは、上記と同じ勾配溶出および清浄手順を行なった。アウリントリカルボン酸は、実行中、陰イオン交換カラムに結合したまま残存した(NEG:001:276−290)。アウリントリカルボン酸は、pH8.0において、水溶液中で容易に重合し、高電荷密度で巨大分子をもたらす。負の電荷を有する巨大分子は、明らかに、DNAとそっくりに陰イオン交換カラムに不可逆的に結合する。この物質をカラムに反復的に適用すると、おそらく充填容量が減少し、したがって、カラム全体の寿命に負の影響を及ぼす。
【0096】
第2の培地成分は、PAMとともに共溶出することがわかった。非ならし培地または未使用培地を、50mM TRIS、120mM NaCl、0.001%TX−100(pH8.0)で平衡化させたQ−セファロースFFカラムに直接充填した。このカラムを再度、上記と同じ勾配溶出条件に供した。該酵素が典型的に溶出される領域に大きなUVピークが観察された(NEG:001:248−257)。ならし培地から精製したピークのSDS−PAGE解析により、該物質は、低分子量タンパク質または非タンパク質性培地成分のいずれかであることが明らかとなった(NEG:001:225−245)。C5467培地はタンパク質水解生成物を含有し、これは、該酵素のピークと一致する最大UVピークの説明となり得る。両方の培地成分であるアウリントリカルボン酸および非同定UVピークは、クロマトグラフィーの前にTFFによって有効に除去される。該酵素は30kDa膜によって保持され(貯留物)、低分子量培地成分はフィルターを通過する(透過物)。
【0097】
再生セルロースPLCTK 30kDa膜(Millipore)を取り付けたPellicon XL装置を用い、ならし培地を10倍に濃縮してダイアフィルトレーションを行なった。該物質は、4〜5容量の25mM TRIS、0.0005%TX−100(pH7.0)を用いて、最終導電率ほぼ3mSまでダイアフィルトレーションを行なった。該物質を、50mM TRIS、120mM NaCl、0.001%TX−100(pH8.0)で平衡化させたQ−セファロースFFカラムに充填した。このカラムは上記のような勾配溶出が行なわれた。樹脂の変色は観察されず、該酵素と共溶出される最大UVピークは存在しなかった。得られた酵素ピークの回収%および比活性を、それぞれ47%および2.1×106U/mgとなるまで測定した(NEG:004:263−283)。
【0098】
TFF工程の種々の段階における酵素安定性を検討した。ならし培地は、最初に、50mM TRIS、0.001%TX−100(pH7.0)でダイアフィルトレーションを行なった後、5倍濃縮した。ダイアフィルトレーションおよび濃縮で得られた試料を、活性の低下および分解について解析した。SDS−PAGEでは分解は観察されず、酵素活性のほぼ95%が回収された(NEG:008:245−251)。
【0099】
陰イオン交換クロマトグラフィー(Q−セファロースFF)
工程の説明:陰イオン交換(AEX)クロマトグラフィー工程により、CHO細胞ならし培地からのPAMの全体精製が提供される。該工程では、主に、高分子量タンパク質が除去される。しかしながら、例えば、該酵素の切断型形態などの一部の低分子量タンパク質もまた除去される。該クロマトグラフィー工程では、定常的に、該酵素2〜3倍の精製がもたらされる。活性酵素の回収%は、典型的には50〜75%である。CHO細胞の発酵後、供給原料流中に存在し得るDNAは、該プロセスのこの段階で有効に除去される。
【0100】
研究開発:Q−セファロースFF工程の開発前に、本発明者らは、陽イオン交換(CEX)クロマトグラフィーを用いてPAMの精製を簡単に検討した。CEXクロマトグラフィーの使用は、最初にTFF工程を伴って、またはなしで検討した。精製は、SP550C(TosoHaas)カラムにおいて行ない、pH5.0〜6.0の移動相を使用した。各場合において、分離および回収の結果は不充分であり、充填後、酵素活性の大部分は通過画分中に確認された(NEG:004:006−029およびNEG:004:102−119)。結合を助長するための移動相のpHの低下は、低pHでの該酵素の安定性の欠如の可能性のため、考慮しなかった。この点で、CEXクロマトグラフィーは却下した。
【0101】
勾配溶出を用いた一連のAEXクロマトグラフィー実行をQ−セファロースFFカラムにおいて検討した。Q−セファロースFF精製はすべて、(1.1×13.7cm)カラムにおいて行ない、180cm/時で実行した。精製開発の初期において、新鮮ならし培地の入手可能性は限定的であり、したがって、これらの実験の多くは、凍結/解凍材料で行なった。ならし培地をTFFによって濃縮/ダイアフィルトレーションし、50mM TRIS、120mM NaCl、0.001%TX−100(pH8.0)で平衡化させたカラムに充填した。このカラムを、100%A(50mM TRIS、120mM NaCl、0.001%TX−100(pH8.0))から68%B(50mM TRIS、475mM NaCl、0.001%TX−100(pH8.0))までの60分かけた塩直線勾配にかけた。pH7.0またはpH8.5のいずれかのHEPESバッファーを用いた同様の勾配実行もまた検討した。概して、この勾配実行では、ほとんど精製はもたらされず、酵素比活性は不充分な結果となった(NEG:004−139−156、NEG:005:119−132、NEG:009:009−022およびNEG:009:024−038)。多くの場合で、酵素活性は、おそらく差示的なグリコシル化の結果として、この勾配全体において散在的であった。
【0102】
開発を継続している際、逐次塩工程を用いた段階的溶出方法を、Q−セファロースFFカラムにおいて検討した。濃縮/ダイアフィルトレーションを行なったならし培地を、50mM TRIS、120mM NaCl、0.001%TX−100(pH8.0)で平衡化させたQ−セファロースカラムに充填した。このカラムは、175mM NaCl、225mM NaClおよび275mM NaClでの逐次段階的溶出が行なわれた。酵素活性は、175mMと225mMの間の塩工程で等しく分布し、全体回収%oはほぼ41%であった。SDS−PAGEによる解析により、175mMおよび225mM両方のNaCl画分において75kDaの酵素の存在が明らかとなった(NEG:005:198−211)。両方の塩工程での酵素活性は、グリコシル化の差またはおそらく活性を保持している該酵素の切断型形態によるものであり得る。
【0103】
スピナーフラスコ実行または10Lバイオリアクター実行のいずれかの新鮮なならし培地を、残りの陰イオン交換実験に使用した。スピナーフラスコからのCHOならし培地00020S3をTFFにさらし、次いで、50mM TRIS、120mM NaCl、0.001%TX−100(pH8.0)で平衡化させたQ−セファロースFF上に充填した。酵素をカラムから、1回の塩工程、50mM TRIS、225mM NaCl、0.001%TX−100(pH8.0)で溶出させた。回収されたピークの回収%および比活性の値は、それぞれ34%および1.9×106U/mgであった(NEG:008:021−033)。同様に、10LバイオリアクターからのCHOならし培地(00021BR、第14日)をTFFにさらし、1回の225mM NaCl工程を用いてQ−Sepharose上で精製した。この実行での回収%および比活性の値は、それぞれ37%および2.3×106U/mgであった(NEG:008:065−075)。
【0104】
また、HEPES移動相を用いた段階的溶出を、活性酵素の回収を改善する取り組みにおいて検討した。2連の精製実行を、50mM HEPES、225mM NaCl、0.001%TX−100(pH8.0)を溶出バッファーとして用いて行なった。10Lバイオリアクター実行(Boontonパイロット施設、実行番号1、第15日)からのCHOならし培地を、これらの精製実行のための投入物質として使用した。両方の精製実行での回収%および比活性は、それぞれ、ほぼ60%および3.0×106U/mgであった(NEG:008:263−288)。HEPES移動相の使用により、酵素の回収%および純度が明らかに改善された。
【0105】
Q−セファロースFF精製に対する細胞バイアビリティの効果を検討した。10Lバイオリアクター実行から、第14日および第21日に収集したCHOならし培地(BOOlOl)の精製を比較した。第14日および第21日における細胞バイアビリティを測定し、それぞれ95%および48%であると決定された。第14日および第21日のCHOならし培地は、TFF、および50mM HEPES、225mM NaCl、0.001%TX−100(pH8.0)を段階的溶出バッファーとして用いたQ−セファロースFF精製が行なわれた。第14日の物質の精製では、49%の回収および2.15×106U/mgの比活性がもたらされたが、第21日での実行では、40%の回収および2.1×106U/mgがもたらされた(NEG:009:114−125およびNEG:009:149−166)。また、10Lバイオリアクター実行からの第12日および第15日のCHOならし培地(00107BR)を用いた精製も行なった。第12日および第15日での細胞バイアビリティを測定し、それぞれ86%および80%であると決定された。各場合において、酵素はQ−セファロースFFカラムから、50mM TRIS、225mM NaCl、0.001%TX−100(pH8.0)を用いて溶出した。第12日の物質では、62%の回収および1.9×106U/mgの比活性がもたらされたが、第15日の物質では、54%の回収および4.3×106U/mgの比活性が得られた(NEG:009:195−203およびNEG:009:227−236)。細胞バイアビリティは、AEXクロマトグラフィー後の全体的な回収%または酵素比活性に対して有意な影響を及ぼさないようであった。
【0106】
実用範囲を確立するため、Q−セファロースFFクロマトグラフィーの臨界パラメータを特定し、検討した。該精製手順に取り組むために設計した計画を規定し、酵素の回収および純度に対する影響を調べるために検討した。2通りのQ−セファロースFF精製実行を、異なる段階的溶出バッファー、50mM TRIS、250mM NaCl、0.001%TX−100(pH7.8)または50mM TRIS、200mM NaCl、0.001%TX−100(pH8.2)(低pH/高塩濃度および高pH/低塩濃度)のいずれかを用いて比較した。これらの実験の論理的説明は、実用範囲を規定するためにクロマトグラフィーを極端な溶出条件に供することであった。10Lバイオリアクター実行からのTFF処理したCHOならし培地(2132−D−1004)を、精製のための投入物質として使用した。低pH/高塩濃度の計画では、ならし培地を、50mM TRIS、120mM NaCl、0.001%TX−100(pH7.8)で平衡化させたカラムに充填した。酵素をカラムから、50mM TRIS、250mM NaCl、0.001%TX−100(pH7.8)で溶出した。酵素の回収%および比活性の値は、それぞれ66%および2.0×106U/mgであった(NEG:010:151−161)。高pH/低塩濃度の計画でのならし培地は、50mM TRIS、120mM NaCl、0.001%TX−100(pH8.2)で平衡化させたカラムに充填し、50mM TRIS、200mM NaCl、0.001%TX−100(pH8.2)で溶出した。この場合の回収%および酵素比活性は、それぞれ66%および2.4×106U/mgであった(NEG:010:140−150)。これらのデータに基づき、Q−セファロースFFカラムは[7.8〜8.2]のpH範囲で実行され得、溶出バッファー中の塩化ナトリウム濃度は[200mM〜250mM NaCl]の間で種々であり得る。
【0107】
30Lバイオリアクター実行に精製規模拡大するのを補助するため、Q−セファロースFFカラムの実用容量を確立した。カラムの実用容量は、2つの様式:[酵素単位/mL樹脂]および[mgタンパク質/mL樹脂]で規定した。この実験の大きな進歩は、通過画分および洗浄工程で確認される酵素活性が合計で≧10%である規定された。TFFにさらされたCHOならし培地(100mL)(2132−D−1002)を、50mM TRIS、120mM NaCl、0.001%TX−100(pH8.0)で平衡化させたQ−セファロースFFカラムに充填した。ならし培地の酵素活性およびタンパク質濃度は、それぞれ1.97×106U/mLおよび2.41mg/mLであると決定された。AEXカラム(Millipore Vantage−L、1.1×13.7cm)を、線速度180cm/時で実行した。50mM TRIS、225mM NaCl、0.001%TX−100(pH8.0)で、段階的溶出を行なった。1%未満の総酵素活性が、カラム通過画分および洗浄工程において確認された。ほぼ51%の酵素単位がメインピークで回収された。13.0mLのカラム容積に対し、実用容量は、[1.51×107U/mL樹脂]または[18.5mgのタンパク質/mL樹脂]のいずれかであると算出された(NEG:010:081−092)。Q−セファロースFF工程の真の充填容量を決定するため、さらなる実験を計画している。
【0108】
疎水性相互作用クロマトグラフィー(フェニル−セファロースFF、低置換)
工程の説明:疎水性相互作用クロマトグラフィー(HIC)工程により、AEX工程後に残留するタンパク質不純物の大部分が除去され、さらに2倍の精製がもたらされる。HIC後のSDS−PAGE解析により、少量の低分子量不純物を伴うのメジャーバンドが明らかになった。典型的には、HIC後の活性酵素の回収%は50〜75%である。
【0109】
研究開発:B3/A1−7クローンから産生されるPAMのために開発した精製プロセスでもHICを用いた。リガンド−タンパク質相互作用促進するため、硫酸アンモニウムを含有する移動相を使用した。硫酸アンモニウムを含有するバッファーへの酵素の長期間の曝露により、予測不可能な沈殿および酵素変性がもたらされた。これらの問題を最小限に抑える試みにおいて、Hofmeisterシリーズで他の塩、例えば、塩化ナトリウムおよびクエン酸ナトリウムを検討した。
【0110】
塩化ナトリウムを含有する移動相を用いた逆直線勾配を、最初に検討した。これらのクロマトグラフィー研究の投入物質は、TFF1で得られたものまたはQ−セファロースFFで得られたもののいずれかとした。最初のTFF工程後の直接HICを、AEX工程を排除する試みにおいて検討した。典型的には、酵素を等容量の水で、その後、等容量の20mM TRIS、4M NaCl(pH7.0)で希釈した。塩添加前の水での希釈により、タンパク質濃度が減少し、「塩析」によって引き起こされる沈殿の可能性が最小限になる。希釈した物質を、10mM TRIS、2.0M NaCl(pH7.0)で平衡化させたフェニル−セファロースFFカラムに充填した。酵素をカラムから、100%A(10mM TRIS、2.0M NaCl(pH7.0))から100%B(10mM TRIS(pH7.0))までの逆直線勾配で、68分間にわたって溶出した。概して、活性酵素の回収%はほぼ50%であった(NEG:005:170−185およびNEG:005:212−226)。
【0111】
また、塩化ナトリウム移動相を用いた段階的溶出方法も検討した。TFF1またはQ−セファロースFFクロマトグラフィーのいずれかにかけられた酵素を、等容量の水で、その後、等容量の20mM TRIS54M NaCl(pH7.0)で希釈した。希釈した物質を、10mM TRIS、2M NaCl(pH7.0)で平衡化させたHICカラムに充填した。このカラムをさらなる平衡バッファーで洗浄した後、10mM TRIS、1.0M NaCl(pH7.0)で洗浄した。酵素をカラムから、10mM TRIS、400mM NaCl(pH7.0)で溶出した。活性酵素の収率は相当であったが、なされた精製はほんのわずかであった(NEG:008:034−046およびNEG:008:135−148)。塩化ナトリウムの使用は、硫酸アンモニウムの場合のように「塩析」を引き起こさないようであったが、高濃度の塩化ナトリウムにおいて、酵素が経時的に変性した。
【0112】
クエン酸ナトリウムは、比較的低濃度でHICカラムへの酵素の結合を促進することがわかった。TFFにさらされたCHOならし培地を等容量の水で、その後、等容量の20mM TRIS、1.0Mのクエン酸ナトリウム(pH7.0)で希釈し、10mM TRIS、0.5M クエン酸ナトリウム(pH7.0)で平衡化させたフェニル−セファロースFFカラムに充填した。このカラムをさらなる平衡バッファーで洗浄し、10mM
TRIS(pH7.0)でストリッピングした。酵素活性は、カラム通過画分または洗浄画分のいずれにおいても確認されなかった。ほぼ50%の酵素活性が10mM TRIS(pH7.0)画分において確認された(NEG:005:064−076)。この実験を、より低い終濃度のクエン酸ナトリウムを用いて繰り返した。この低塩濃度では、酵素を等容量の水で、その後、等容量の20mM TRIS、0.6Mのクエン酸ナトリウム(pH7.0)で希釈した後、10mM TRIS、300mMのクエン酸ナトリウム(pH7.0)で平衡化させた HICカラムに充填した。このカラムをさらなる平衡バッファーで洗浄し、10mM TRIS(pH7.0)でストリッピングした。結果は、本質的に先の実行と同一であり、活性は、通過画分または洗浄画分のいずれにおいても確認されなかった(NEG:008:203−213)。
【0113】
段階的溶出方法の開発を補助するため、クエン酸ナトリウムバッファー系を用いた勾配溶出を検討した。TFFで得られたものを等容量の水で、その後、等容量の20mM TRIS、0.6Mのクエン酸ナトリウム(pH7.0)で希釈した後、10mM TRIS、300mMのクエン酸ナトリウム(pH7.0)中で平衡化させたカラムに充填した。このカラムを100%A(10mM TRIS、300mMのクエン酸ナトリウム(pH7.0))から100%B(10mM TRIS(pH7.0))までの70分間かけて逆直線勾配にかけた。酵素活性のピークは該勾配の最後付近で確認された。他のタンパク質不純物がこの勾配領域で溶出されたとともに、10mM TRIS(pH7.0)カラムストリップ中にまで拡張された(NEG:008:252−262)。10mM TRIS(pH7.0)ストリップ中に溶出される酵素とタンパク質のより良好な分離を達成するため、段階的溶出を検討した。
【0114】
Q−セファロースFF精製(10Lバイオリアクター実行、00107BR、第12日)からのピーク画分(225mM NaCl)を、等容量の水で、その後、等容量の50mM TRIS、600mMのクエン酸ナトリウム(pH7.0)で希釈した。希釈した物質を、25mM TRIS、300mMのクエン酸ナトリウム(pH7.0)で平衡化させたフェニル−セファロースカラムに充填した。このカラムをさらなる平衡バッファーで洗浄した後、25mM TRIS、75mMのクエン酸ナトリウム(pH7.0)での第2の洗浄を行ない、25mM TRIS(pH7.0)でストリッピングした。酵素活性はすべて、75mMのクエン酸ナトリウム画分において確認された。回収%および比活性は、それぞれ42%および2.0×106U/mgであると算出された(NEG:009:204−218)。酵素と他のタンパク質不純物との良好な分離は、この方法を用いて達成された。
【0115】
TFF直後のHIC(AEX工程なし)は、この工程では粗製供給物質流からDNAを排除する効果がないため、却下した。4回のHIC精製を、上記に詳述した塩化ナトリウム段階的溶出方法を用いて行なった。これらの実行の投入物質は、クロマトグラフィーの前にTFFにさらされたB3/A1−7 CHOならし培地とした。このときの本発明者らのDNA内部基準は<10ng/mgタンパク質であった。各場合において、HIC後のメイン酵素ピーク中に存在する残留DNAレベルは該基準より6〜10倍高く、それにより1回の工程でのHIC精製が許容され得ないことがわかった(NEG:006:137−159、NEG:006:176−187およびNEG:006:192−200)。
【0116】
30L容規模拡大の取り組みを補助するため、HIC精製工程の実用容量を検討した。大きな進歩は、カラム通過画分または洗浄画分のいずれかにおいて≧10%の酵素活性の存在と規定された。産生実行のQ−セファロースで得られたもの(125mL、0.368mg/mL、710,563U/mL)1330−D−1003を等容量の水で、その後、等容量の50mM TRIS、0.6Mのクエン酸ナトリウム(pH7.0)で希釈した。希釈した物質を、25mM TRIS、300mMのクエン酸ナトリウム(pH7.0)で平衡化させたフェニル−セファロースFFカラム(Millipore Vantage−L、1.1×15.8cm)上に充填した。このカラムを180cm/時で実行した。このカラムをさらなる平衡バッファーで洗浄し、酵素を25mM TRIS、75mMのクエン酸ナトリウム(pH7.0)で溶出した。酵素活性は、通過画分または洗浄画分のいずれにおいても確認されなかった。メインピークはほぼ75%の酵素活性単位を含んでおり、比活性は2.6×106U/mgであると算出された。15.0mLのカラム容積に対し、実用容量は5.92×106U/mL樹脂であると算出された(NEG:010:111−124)。実用容量は、AEX後にカラムに充填されたタンパク質が極めて少なかったため、[mgタンパク質/mL樹脂]では報告されなかった。HIC工程の真の充填容量を決定するため、さらなる実験を計画している。
【0117】
HIC単位実行のための全体の時間は水希釈を排除し、実行流速を180cm/時から240cm/時に上げることにより劇的に短縮された。クエン酸ナトリウムへの曝露時間および樹脂上で費やされる時間量を最小限に抑えることにより、最終的に、活性酵素の減少の低減が補助され得る。133O−D−1003 からQ−セファロースFFで得られたものを等容量の50mM TRIS、0.6Mのクエン酸ナトリウム(pH7.0)で希釈し、先のバラグラフで記載したHIC段階的溶出方法を用いて精製した。メインピーク(25mM TRIS、75mMのクエン酸ナトリウム(pH7.0))の回収%および比活性は、それぞれ77%および2.4×106U/mgであると算出された(NEG:010:125−139)。クエン酸塩を用いた希釈中またはその後でタンパク質沈殿は観察されなかったが、「塩析」現象を回避するため、該物質はゆっくりと希釈しなければならない。
【0118】
HIC方法は、実用範囲を確立するために移動相の塩濃度を変更することにより取り組んだ。回収%および酵素比活性に対する効果を、2通りの異なる精製手順を行なうことにより検討した。第1の実行では、充填および溶出中のクエン酸塩濃度を減少させ、一方、第2の実行では、充填中のクエン酸塩濃度は減少させたが、溶出バッファーのクエン酸塩濃度は増大させた。10Lバイオリアクター実行からQ−セファロースFFで得られたもの(133O−D−1005)を、等容量の50mM TRIS、0.5M クエン酸ナトリウム(pH7.0)で希釈し、25mM TRIS、250mMのクエン酸ナトリウム(pH7.0)で平衡化させたHICカラムに充填した。このカラムを25mM TRIS、50mMのクエン酸ナトリウム(pH7.0)で洗浄した。ほぼ51%の酵素活性が50mMのクエン酸ナトリウム画分において確認され、1%未満が通過画分および洗浄画分中に確認された。メインピークにおける酵素比活性は1.35×106U/mgであると算出された(NEG:010:175−186)。第2のHIC実行では、Q−セファロースFFで得られたものを、上記のようにして希釈および充填したが、25mM TRIS、100mMのクエン酸ナトリウム(pH7.0)を溶出バッファーとして使用した。この場合、50%の酵素活性が100mMクエン酸塩画分において確認され、2%未満が通過画分および洗浄画分中に確認された。100mMクエン酸塩画分における酵素比活性は2.5×106U/mgであった(NEG:010:163−174)。第1のHIC実行で観察された比活性のほうが低く、これは、溶出バッファー中のクエン酸塩濃度の減少により、通常該酵素と共溶出するストリップ中に見られるさらなるタンパク質がもたらされ、したがって最終純度が低下することを示した。対照的に、溶出バッファー中のクエン酸塩濃度の増大は純度を低下させないようであった。これらのデータに基づき、溶出バッファー中のクエン酸ナトリウムの濃度は、最終純度を損なうことなく75〜100mMの間で変更され得る。
【0119】
第2のタンジェンシャルフロー濾過(TFF2)
工程の説明:TFF2を用いて、ウイルス濾過の前に、フェニル−セファロースFFで得られたものの濃縮およびダイアフィルトレーションを行なう。HICで得られたものを直接TFFにさらし、溶出バッファー(25mM TRIS、75mMのクエン酸ナトリウム(pH7.0))中での長期保存による酵素の不活化を最小限に抑える。フェニル−セファロースFFで得られたものをほぼ3倍に濃縮し、ダイアフィルトレーションを行ない、ウイルス濾過に適したバッファー中に酵素を入れ、その後保存する。TFF2工程では、再生セルロースPLCTK 30kDa膜(Millipore)を取り付けた Pellicon 2 Moduleを使用する。
【0120】
研究開発:TFF2手順は、最初は、B3/A1−7クローンを用いて産生されるPAMのために開発された。該酵素は、種々の条件下の50mM TRIS、25mM NaCl、0.0005%TX−100(pH8.0)中で安定であることが示され、この理由のため、さらなる研究開発は試みなかった。
【0121】
C5467 CHOならし培地由来のPAMの代表的な小規模精製
10Lバイオリアクター実行からのCHO細胞ならし培地(500mL)の試料(00107BR、第15日)を8倍に濃縮し、50mM TRIS、0.001%TX−100(pH8.0)に対してダイアフィルトレーションを行なった。TFF1で得られたもののほぼ3分の1を、上記の精製プロセスの規模縮小形態を用いて精製した(NEG:009:219−249)。精製データを表8にまとめる。この精製実行のこの工程収率は、比較的不充分であり、実際、HIC工程後の酵素比活性は低下した。しかしながら、SDS−PAGE解析により、おおむねα−アミド化酵素は、ほぼ75kDaの単一のメジャーバンドに精製されることが示された(図12、レーン10)。ゲルおよびデンシトメトリースキャン(図13)により、酵素の純度は高く(ほぼ純度67%)、低分子量不純物はごく微量しか存在しないことが明白に示された。該酵素は多くの精製バッファー中で経時的に不活化され、したがって、該プロセスの実施は各工程間で遅滞なく進行させなければならない。精製プロセスにおけるばらつきは、全体収率および比活性を低下させる不活性酵素の存在に起因した。より重要なことには、精製および発酵の開発はともに同時進行させ、これにより、観察されたばらつきがある程度説明され得る。該産生規模における工程収率および比活性は、その後、おそらく該プロセスにおいて該物質をより高速で移動させ、画分を速やかにアッセイした結果、ずっと良好になることがわかった。
【0122】
【表8】
【0123】
PAM下流精製プロセスの概要
技術移転のパイロット施設に渡す情報を反映する各プロセス工程の精製手順を以下に詳述する。SDS−PAGEおよびデンシトメトリースキャンを含む代表的な小規模精製実行のデータを、表8および図11および12に含める。
【0124】
TFF1手順:清澄CHO細胞ならし培地を5倍に濃縮し、50mM TRIS、0.001%TX−100(pH8.0)で、最終導電率4〜6mSが達成されるまでダイアフィルトレーションを行なう。濾過中、膜貫通圧力を≦10psiに維持する。
【0125】
AEX手順:TFFによって濃縮/ダイアフィルトレーションした清澄CHO細胞ならし培地を、50mM TRIS、120mM NaCl、0.001%TX−100(pH8.0)で平衡化させたQ−セファロースFF(Amersham Biosciences)カラムに充填する。このカラムを180cm/時で実行し、UV吸光度を280nmにおいてモニターする。カラムをさらなる平衡バッファーで洗浄し、PAMをカラムから、50mM TRIS、225mM NaCl(pH8.0)で溶出する。カラムを2.0M NaClで清浄にし、1.0M NaOHで殺菌する。カラムは、各実行間では10mM NaOH中に保存する。
【0126】
HIC手順:Q−セファロースで得られたものを、等容量の50mM TRIS、600mMクエン酸塩(pH7.0)で希釈し、25mM TRIS、300mMクエン酸塩(pH7.0)で平衡化させたフェニル−セファロースFF(Amersham Biosciences)上に充填する。カラムを240cm/時で実行し、UV吸光度を280nmにおいてモニターする。カラムを25mM TRIS、300mMクエン酸塩(pH7.0)で洗浄し、PAMをカラムから、25mM TRIS、75mMクエン酸塩(pH7.0)で溶出する。カラムを、それぞれ25mM TRIS(pH7.0)および1.0M NaOHで清浄し、殺菌する。カラムは、各実行間では10mM NaOH中に保存する。
【0127】
TFF2手順:フェニル−セファロースFFで得られたものを最初にほぼ3倍に濃縮し、50mM TRIS、25mM NaCl、0.0005%TX−100(pH8.0)を用いてダイアフィルトレーションを行ない、ウイルス濾過に適した容量までさらに濃縮し、保存する。
【0128】
10L攪拌槽バイオリアクタープロセス実行
α−アミド化酵素の10L発酵および精製プロセスの概要
いくつかの10L攪拌槽バイオリアクター実行に従って精製α−AEを調製した。α−アミド化酵素を誘導するためのプロセス工程は、上記および図14に示すように、14日間の種菌相、17日間の発酵相および2日間の精製スキームであった。これらの実験で使用した培養培地は、タンパク質無含有CHO培地C5467(Sigma−Aldrich)であった。これらの実行のプロセスの詳細はすべて、適切なバッチの記録を見るとよい。
【0129】
プロセスフローダイアグラムである図14は、UGL 73−26/Mクローンを用いて産生されるα−AE/PAMのために開発した最終プロセス工程を示す。
【0130】
第1相−種菌
10L攪拌槽バイオリアクター用の種菌を調製するため、UGL 73−26/M MWCB00のバイアルの1つを解凍した。このクリオバイアルの細胞を取り出し、新鮮培地中に入れた。細胞ペレットを新たな培地中に再懸濁し、C5467 CHO培地を入れたスピナーフラスコに添加した。次の14日間、培養物を、400mLの培地+細胞を入れた4つのフラスコ内に増殖した(種菌スキーム図15を参照)。
【0131】
培養物を、種菌相l2の最後でのα−AE発現について試験した。第12日目、平均酵素活性濃度は>13,000U/mLであった(表9パネルC参照)、培養物および細胞バイアビリティの生細胞計数を各時点で行ない、種菌培養物を修正した。培養物のバイアビリティは、概ね、この相の間で95%よりも大きかった(表9パネルA参照)。種菌培養物の平均総生細胞計数は、第14日目で2.03×109であった(表9パネルB参照)。
【0132】
【表9−1】
【0133】
【表9−2】
【0134】
第2相−発酵
10Lのバイオリアクターは、各種菌相の終了時に開始した。バイオリアクターに、未使用タンパク質無含有培地中1×105細胞/mLでL−グルタミン(Sigma,C5467)を播種した。バイオリアクターパラメータを、下記の設定点;温度=37℃、RPM=60、pH=維持するpH設定点なしに設定し、pHは流動的にし(開始時、pHはpH7.5より大きくならなりようにする)、DO設定点は70%DOにする。未使用培地の溶存酸素濃度は、70%より大きくし、バイオリアクターの溶存酸素濃度は70%まで流動的にさせ、次いで、該設定点に維持した。バイオリアクターには、第5、10および14日目に2g/Lのグルコースを補充した。第17日目にならし培地を収集した。収集物質をMillipore Opticap Filterに通して清澄にした。後述する最初の2つの発酵では、第17日目ではなく第18日目に2131−D−1003および2131−D−1004を収集した。培養物の最大細胞密度は第10〜17/18日目の間で達成され、維持された。これらの発酵の平均最大細胞密度は1.5−1.6×106細胞/mLであった(表10パネルB)。培養物のバイアビリティは、培養の最初の14日間で>80%であり、収集した日の平均バイアビリティは76.0%であった(表10パネルA)。培養物の産生性を、収集時、第17/18日目に評価した。平均清澄収集物は378,567単位のα−AE/mLを含んでいた(表10パネルC)。
【0135】
【表10】
【0136】
第3相−精製
該精製プロセスは、上記のような5つの異なる工程を有する。清澄収集物を濃縮し、50mM TRIS、0.001%TX−100(pH8.0)に対してダイアフィルトレーションした。酵素物質を平均してほぼ4倍に濃縮した(データ示さず。バッチの記録参照)。このプロセス物質をQ−セファロースカラムに適用し、225mM NaClを含有する50mM TRIS、0.001%Tritonバッファー中でのNaCl段階的勾配に従って溶出した。酵素溶液を50mM TRIS、600mMクエン酸(pH7.0)で希釈し、フェニルセファロースカラムに適用した。α−アミド化酵素をカラムから、クエン酸塩の工程勾配に従って溶出した。酵素物質を最終容量ほぼ1Lまで濃縮した。6つの酵素バッチのうち4つをウイルス除去フィルターに通して処理した。2つのバッチはMillipore NFPフィルターに通して処理し(1330−D−1005および1330−D−1006)、2つのバッチは、Pall Trincor DV50フィルターに通した(1330−1009および1330−1010)。各々のプロセス工程のデータを以下の表11に示す。
【0137】
最初の工程TFF1は、α−アミド化酵素活性単位の保持に関してほぼ定量的な工程であった。10Lバイオリアクター実行からのTFF濃縮物は、1.60×106単位/mgタンパク質の平均比活性を有する(表11)。Q−セファロースクロマトグラフィーカラムに適用したTFF1濃縮物は、カラムからほぼ3Lで溶出された。酵素比活性は、このプロセス工程で、3.33×106単位/mgまでほぼ2倍に増大した。Q−セファロース溶出液のタンパク質濃度および酵素活性濃度は非常に一貫していた(表11参照)。フェニル−セファロースクロマトグラフィー後、平均酵素比活性は、3.33×106から4.96×106単位/mgタンパク質まで増大した。フェニル−セファロース溶出液を、第2のTFF工程を用いて1Lまで濃縮した。この濃度工程後、比活性に変化はなかった(表11)。一部の酵素バッチからのTFF2濃縮物を、ウイルス除去フィルターに適用した。4つのうち3つの酵素バッチの比活性が低下した(表11)。最終精製酵素の平均比活性は、350,000単位/mgから4.61×106単位/mgタンパク質低下した。この比活性の低下は、おそらく一部の該酵素の不活化によるものであった。
【0138】
【表11−1】
【0139】
【表11−2】
【0140】
精製プロセスの各工程の一貫性を示すこのデータは重要である。しかしながら、本当に重要なのは、該プロセスの各工程での所望の生成物の回収パーセントである。表12は、α−アミド化酵素精製プロセスの各工程での回収パーセント、および4回または5回のプロセス工程後のプロセス全体収率を示す。表12は、酵素が両方のTFF工程から定量的に回収されたことを示す。クロマトグラフィー工程、Q−セファロースおよびフェニルセファロースでの平均回収パーセントは、それぞれ65.3%および72.0%である。試験した両方のウイルス除去フィルターで約80%のα−AE活性が回収された。TFF2またはウイルス濾過を通した清澄回収物質の酵素精製プロセスの全体収率は、それぞれ40.8%または38.8%であった。
【0141】
【表12】
【0142】
結論
高レベルのα−AE活性を発現する安定な充分特性化されたCHO細胞株UGL 73−26/Mが開発された。高レベルの酵素発現は、非動物供給源の低タンパク質含有組織培養培地C5467(Sigma)を用いる17日間のバッチ発酵プロセスにおいて達成される。臨界発酵パラメータ(例えば、pH、DOおよびグルコース濃度など)を検討し、最適化した。また、酵素がほぼ均一に精製され得るロバストな2工程の下流精製プロセスも開発した。この発酵および精製プロセスの一貫性は、製造レベルへの規模拡大に充分適している。
【0143】
以下に、本発明の細胞によって発現されるPAMを用いたアミド化生成物の産生を示す実施例を示す。
【0144】
実施例1:ペプチジルグリシンα−アミド化モノオキシゲナーゼを用いたグリシン増殖副甲状腺ホルモン断片のアミド化対応物への変換
ピルビン酸塩を用いたrhPTH(l−34)Gly35−OHのアミド化
rhPTH(l−34)Gly35−OHのアミド化に用いた成分および終濃度を表12に示す。アミド化の簡単な説明は以下の通りである。
【0145】
【表13】
【0146】
・ほぼ12.4gのrhPTH(l−34)Gly35−OHを含む1,900mLの25mM MES、200mM NaCl(pH 6.0)を、攪拌器およびガススパージャーを取り付けたガラス槽内に供給した。
【0147】
・この溶液に、以下の成分:3025mLの水、741mLの250mM MES pH 6.3、1.03mLの3mM 硫酸銅、124mLのヨウ化カリウム、62mLの190プルーフエタノール、124mLの400mMピルビン酸ナトリウムおよび124mLの100mMのアスコルビン酸ナトリウムを記載順に添加した。
【0148】
・反応槽を水浴中に入れ、反応混合物を25〜27℃まで攪拌しながら昇温させた。
・反応混合物のpHを21mLの2M HClによって5.8に調整した。酸素のスパージングを開始したが、スパージング速度は、反応混合物の過剰な起泡が回避されるように調整した。
【0149】
・47mLのPAMを添加し、反応混合物を25〜27℃で4時間35分間インキュベートした(インキュベーション期間中、酸素スパージングを行なった)。
【0150】
・反応混合物を74mLの2MのHClでpH2.4に酸性化した。
この実施例で用いたPAM酵素は、上記のようにして構築し、本明細書においてUGL 73−26/Mと表示する本発明の好ましいCHO K1細胞から発現された。また、この細胞株を用いて上記のATCC寄託物を提供した。
【0151】
グリシン増殖前駆体は、既知の供給源から得られるものであってもよく、既知の様式で生成されたものであってもよい。例えば、これは、米国特許第6,103,495号(その実施例1〜2)に記載のものと同様の様式で発酵によって生成され、アミド化の前に米国特許第6,103,495号(その実施例3)に記載のようにして精製され得る。上記の本発明のアミド化に用いた具体的な前駆体は、係属中の米国特許出願第11/076,260号(2005年3月9日出願および2005年10月6日に公開番号US 2005/0221442で公開)の細胞株によって発現させた。
【0152】
アミド化に使用する酵素がペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼ(PHM)である場合、PHMをPAMに変えて、上記のものと同じ反応混合物が使用され得る。また、4〜6時間のインキュベーション期間の最後に、反応混合物のpHを塩基の添加によって8〜9に上昇させる。反応混合物は、反応終了前にさらに4〜8時間攪拌する。PHMは、PAMを本明細書に教示したとおりに発現させた後、PHM触媒ドメインをPAM分子の残部から分離することにより得られ得る。あるいはまた、本明細書において使用されるベクターは、PHMの後でPALの前の触媒ドメインで翻訳が停止されるように修飾されていてもよい。あるいはまた、該ベクターは、第1の例では、上記のPAMコード領域の代わりにPHMコード領域を用いて構築され得る。PHMは、PAM のN末端部分のみ(最初の約40kDa)を発現させることにより得られ得る。PAMおよびその触媒ドメインの位置は文献に報告されている。かかる任意のPAMまたはPHMは、本発明に従って有用であると考えられる。例えば、Mizunoら,BBRC Vol.148,No.2,pp.546−52(1987)(その開示は引用により本明細書に組み込まれる)を参照のこと。特にMizunoの「AE1」を参照のこと。カエルの皮膚は、天然状態でPHMを発現することが知られている。
【0153】
実施例2:アミド化後精製
陽イオン交換(CEX)クロマトグラフィー
残留rhPTH(l−34)Gly35−OHからのrhPTH(l−34)−NH2の精製を、CEXクロマトグラフィーを用いて行なった。CEXクロマトグラフィー方法の簡単な説明は以下の通りである。アミド化で得られたものを酸性化して25mM MES(pH 6.5)で平衡化させたToyopearl SP650M(Tosoh Bioscience LLC)カラム(9cm×19cm)上に充填した。カラムを180cm/時で実行し、カラム流出液のUV吸光度を280nmでモニターした。このカラムを25mM MES(pH6.5)で、カラム流出液pHのpHが6.5に戻るまで洗浄した。このカラムを25mM MES,80mM NaCl(pH 6.5)で、洗浄ピークが完全に溶出され、安定なUVベースラインが得られるまで洗浄した。生成物rhPTH(l−34)−NH2をカラムから、25mM MES,200mM NaCl(pH 6.5)で溶出した。UVピークをすべて回収したが、プール基準を決定するため、画分をRP−HPLCによってスクリーニングした。
【0154】
逆相(RP)クロマトグラフィー
RPクロマトグラフィーを用い、塩形態のペプチドを塩化物から酢酸塩に交換した;RPクロマトグラフィーでは、ほんのわずかのペプチド精製しかもたらされない。CEXクロマトグラフィーで得られたものを3容量の333mM酢酸ナトリウムで希釈し、充分混合した。混合物は、充填前に室温で75分間放置した。酢酸塩希釈試料を、250mM酢酸ナトリウム(pH7.5)で平衡化させた Amberchrom CG300 M(Tosoh Bioscience LLC)カラム(6cm×17cm)上に充填した。カラムを180cm/時で実行し、カラム流出液のUV吸光度を280nmでモニターした。カラムを250mM酢酸ナトリウム(pH7.5)で60分間洗浄した。カラムを0.1%酢酸中で平衡化させた。生成物rhPTH(l−34)−NH2をカラムから、0.1%酢酸、40%エタノールで溶出した。UVピークをすべて回収した。
【0155】
rhPTH(l−34)−NH2の特性化
RPクロマトグラフィーで得られたものを濃縮し、凍結乾燥によって白色の綿毛様粉末にし、11.8g(アミド化から95%の全体収率)のrhPTH(l−34)−NH2を得た。rhPTH(l−34)−NH2の分子質量は、エレクトロスプレーイオン化質量分析(ESI−MS)によって4,116.9Daであると決定され、これは、平均分子質量の計算値4,116.8Daと整合した。
【0156】
本発明は、特別の実施形態に関して説明されており、多くの他の変形および修飾、ならびに他の使用は、当業者に明らかである。したがって、本発明は、本明細書における特異的開示、または付け加えられた特許請求の範囲に限定されない。
【技術分野】
【0001】
関連出願の相互参照
本出願は、2005年6月24日に出願された米国特許仮出願第60/693,612号(その開示は引用により本明細書に組み込まれる)の優先権を主張するものである。
【0002】
発明の分野
本発明は、ペプチジルグリシンα−アミド化モノオキシゲナーゼ(PAMもしくはα−AE)、またはその2つの触媒ドメインの一方の発現のための組換え発現ベクターおよび細胞株に関する。本発明はまた、X−α−ヒドロキシ−GlyまたはX−NH2(Xは、グリシン基が共有結合し得るカルボニル基を有するペプチドまたは任意の化学的化合物である)へのX−Glyの酵素的変換を触媒するための、かかるPAM(またはその触媒ドメインの一方)の使用に関する。さらに、本発明は、好ましい細胞株の調製に関する。一部のある実施形態では、CHO K1宿主が用いられる。一部のある実施形態では、発現ベクターは、ヒトメタロチオニンIIAプロモーターおよび/またはSV40エンハンサーを含む。
【背景技術】
【0003】
関連技術の説明
数多くのヒトホルモン、増殖因子、サイトカイン、神経伝達物質、誘導体化脂肪酸および他の重要な生物学的化合物は、その分子構造の実質的な部分としてアミノ酸またはペプチドを有する。多くの疾患は、患者において、これらの生物学的化合物のレベルの上昇に対して陽性に応答する。治療有効量のかかる生物学的に関連性のある化合物は患者に、さまざまな様式で投与され得る。したがって、効率的で費用効果のあるかかる化合物の製造方法は非常に重要である。これは、生物学的化合物が、経口送達用の投薬形態(これは、他の投与様式と比べてバイオアベイラビリティが低いにもかかわらず、通常好ましい投与様式である)に調製される場合、特に言える。
【0004】
哺乳動物細胞および他の真核生物では、ある種の翻訳後プロセシング手順が行なわれ得るが、原核生物では行なわれ得ない。ある種の原核生物、例えば、大腸菌は、バッチ発酵手順で容易に培養され得るため、および遺伝子工学的に充分特性化されているため、組換えDNA(rDNA)手法による哺乳動物タンパク質の産生のための宿主として広く使用されている。しかしながら、多くの哺乳動物タンパク質には、なんらかの型の翻訳後プロセシングが必要とされる。このようなタンパク質が、例えば大腸菌の遺伝子操作によって産生される場合、翻訳後プロセシングは、多くの場合、複雑なインビトロ化学的手順を用いて行なわれなければならないが、該手順は、大規模生産の適用のためには、たいへんな費用がかかる。ペプチドを、哺乳動物宿主を用いて、組換えにより産生させる場合であっても、多くの場合、前駆体を効率的に産生させ、これを、その後でのみさらに修飾させることが望ましい。
【0005】
かかるさらなるプロセシング活性の一例は、ペプチドまたはタンパク質のカルボキシ末端アミノ酸の特異的アミド化を含む。多くの天然に存在するホルモンおよびペプチドはかかる修飾を含み、これは、多くの場合、該タンパク質が生物学的に活性であるべきならば不可欠である。一例はカルシトニンであり、この場合、天然形態のアミド化プロリンを非アミド化プロリン残基で置換すると、生物学的活性の非常に有意な低下がもたらされる。充分な活性に翻訳後アミド化を必要とする他の生物学的ペプチドとしては、限定されないが、成長ホルモン放出因子、他のカルシトニン、カルシトニン遺伝子関連ペプチド、セクレチン、ペプチドYYなどが挙げられる。
【0006】
最大限の有効性のためにアミド化が必要とされる多くの重要な生物学的タンパク質のポリペプチド配列が、例えば、遺伝子操作手法によって製造され得る。しかしながら、重要であり、時には不可欠であるカルボキシ末端のアミド化は、多くの場合、インビトロで行なわなければならない。この点において、高価で煩雑な化学的アミド化手法は回避することが望ましく、したがって、特異的アミド化を行なうためにはアミド化酵素を用いることが望ましい。タンパク質のカルボキシ末端アミノ酸の特異的アミド化は、高頻度で、α−アミド化酵素によって触媒される。
【0007】
ペプチジルグリシンα−アミド化モノオキシゲナーゼ(PAM)は、アミド化ペプチド生成物へのペプチド基質の変換を触媒する。該変換は2工程反応である。PAMは2つの触媒ドメインを有する:ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼ(PHM)は、工程1(中間体への基質の変換)を触媒し、ペプチジルグリシンα−ヒドロキシグリシンα−アミド化リアーゼ(PAL)は、工程2(生成物への中間体の変換)を触媒する。完全長PAMは2つの機能を有し、両方の工程を触媒する。工程2はまた、中間体をルイス塩基と接触させる場合、非酵素的に、効率的になされ得る。
【0008】
自然界では、ほぼ50%のペプチドホルモンおよび神経伝達物質が、PAMによって前述の様式でアミド化される。PAM活性は、数多くの多様な種で認められており、PAMは、ラット、ウシおよびカエルなど多様な種間で有意な構造的相同性を有する傾向にある。また、PAMの機能、基質および補因子は、種間で類似する(高頻度で、同一である)ことが知られている。基質は化合物であり、多くの場合、遊離カルボキシル基を有するグリシン残基を有するペプチドである。PAM触媒アミド化反応は当該技術分野でよく知られている。例えば、ペプチジルグリシンα−アミド化モノオキシゲナーゼを用い、グリシン伸長サケカルシトニン前駆体の、そのC末端がアミド化された(すなわち、前駆体のC末端グリシンの代わりにアミノ基を有する)真のサケカルシトニンへの変換を触媒するペプチドは、米国特許第6,103,495号に詳細に記載されている。
【0009】
PAMおよびPAMを発現する細胞株の供給源は、当該技術分野で知られている。アミド化ペプチドの大規模なPAM触媒産生では、最良のコスト有効性のため、PAMの安定な高収率供給源が必要とされる。さらに、PAMは、潜在的ヒト用医薬品の作製に高頻度に用いられるため、PAM産生(PAM発現細胞株およびこれを培養する培養培地の両方)のための系では、PAM発現中、できるだけ不純物の産生を少なくすることが重要である。特に重要なことは、感染性海綿状脳症(TSE)のリスクを回避するために、哺乳動物タンパク質の使用を最小限に抑えることである。それにもかかわらず、培養培地中の哺乳動物タンパク質増殖因子(TSEの観点からは望ましくない)は、PAM発現細胞株の生存および産物発現の補助において有用であり得る。したがって、当該技術分野において、本発明の場合以外では培地に必要とされ得る哺乳動物タンパク質の非存在下であっても強い発現をもたらし、良好な安定性および生存を示すPAM発現(またはPHM発現)細胞が必要である。
【発明の概要】
【発明が解決しようとする課題】
【0010】
発明の概要
したがって、本発明の目的は、酵素が後に製品の製造(例えば、アミド化医薬品の製造)に使用される場合に問題となり得る哺乳動物型のタンパク質および他の不純物が実質的にない培地中で良好な生存割合および良好な発現収率を有するのに充分強いPAM発現細胞またはPHM発現細胞を提供することである。
【0011】
本発明のさらなる目的は、それ自体は有意な望ましくない不純物を共発現しないPAM発現細胞またはPHM発現細胞を提供することである。
【0012】
さらなる目的は、良好な発現収率をもたらすPAM発現細胞またはPHM発現細胞を提供することである。
【0013】
さらなる目的は、かかる細胞に有用な発現ベクターを提供することである。
さらなる目的は、かかる細胞をトランスフェクトおよび選択するための良好な手法を提供することである。
【0014】
さらなる目的は、酵素反応用の高活性および高純度のPAMまたはPHMを提供し、それにより、良好な酵素反応および結果としてのアミド化生成物を提供することである。
【課題を解決するための手段】
【0015】
これらおよび他の目的は、本明細書に開示する本発明によって提供される。
一実施形態において、本発明は、ペプチジルグリシンα−アミド化モノオキシゲナーゼを発現させるための発現ベクターでトランスフェクトされたCHO K1細胞を提供する。
【0016】
別の実施形態において、本発明は、リボソーム結合部位、プロモーターおよび該プロモーターの上流のSV40エンハンサーを含む制御領域に操作可能に連結されたペプチジルグリシンα−アミド化モノオキシゲナーゼをコードする核酸を有するコード領域を有する組換え発現ベクターを提供する。
【0017】
別の実施形態において、本発明は、リボソーム結合部位およびヒトメタロチオニンIIAプロモーターを含む制御領域に操作可能に連結されたペプチジルグリシンα−アミド化モノオキシゲナーゼをコードする核酸を有するコード領域を有する組換え発現ベクターを提供する。
【0018】
別の実施形態において、本発明は、
(A)第1、第2および第3の発現ベクターの存在下で、潜在的宿主細胞をトランスフェクトする工程であって、該第1のベクターが、第1の選択可能なマーカーをコードするコード領域を含み、該第2のベクターが、第2の選択可能なマーカーをコードするコード領域を含み、該第3のベクターが、ペプチジルグリシンα−アミド化モノオキシゲナーゼをコードするコード領域を含み、該第1のベクターに対する該第3のベクターの濃度比が少なくとも3:1であり、該第2のベクターに対する該第3のベクターの濃度比が少なくとも3:1である工程;
(B)工程(A)で得られた細胞に選択圧をかけ、該第1のベクターでトランスフェクトされた細胞を選択する工程;
(C)工程(B)で得られた細胞に選択圧をかけ、該第2のベクターでトランスフェクトされた細胞を選択する工程;
(D)工程(C)で得られた細胞を限界希釈し、ペプチジルグリシンα−アミド化モノオキシゲナーゼを発現する細胞を選択する工程
を含む、ペプチジルグリシンα−アミド化モノオキシゲナーゼの発現のための細胞株の調製方法を提供する。
【0019】
別の実施形態において、本発明は細胞株UGL 73−26/Mを提供する。
別の実施形態において、本発明は、細胞株UGL 73−26/Mによって発現されるペプチジルグリシンα−アミド化モノオキシゲナーゼを提供する。
【0020】
別の実施形態において、本発明は、
(A)ペプチジルグリシンα−アミド化モノオキシゲナーゼの不純試料を陰イオン交換クロマトグラフィーにかける工程であって、溶出が均一濃度である工程;
(B)工程(A)の溶離液を疎水性相互作用クロマトグラフィーにかける工程であって、硫酸アンモニウムが使用されず、溶出が均一濃度である工程
を含む、ペプチジルグリシンα−アミド化モノオキシゲナーゼを発現および培養培地中への分泌後に精製する方法を提供する。
【0021】
別の実施形態において、本発明は、ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼを発現させるための発現ベクターでトランスフェクトされたCHO K1細胞を提供する。
【0022】
別の実施形態において、本発明は、リボソーム結合部位、プロモーターおよび該プロモーターの上流のSV40エンハンサーを含む制御領域に操作可能に連結されたペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼをコードする核酸を有するコード領域を有する組換え発現ベクターを提供する。
【0023】
別の実施形態において、本発明は、リボソーム結合部位およびヒトメタロチオニンIIAプロモーターを含む制御領域に操作可能に連結されたペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼをコードする核酸を有するコード領域を有する組換え発現ベクターを提供する。
【0024】
別の実施形態において、本発明は、
(A)第1、第2および第3の発現ベクターの存在下で、潜在的宿主細胞をトランスフェクトする工程であって、該第1のベクターが、第1の選択可能なマーカーをコードするコード領域を含み、該第2のベクターが、第2の選択可能なマーカーをコードするコード領域を含み、該第3のベクターが、ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼをコードするコード領域を含み、該第1のベクターに対する該第3のベクターの濃度比が少なくとも3:1であり、該第2のベクターに対する該第3のベクターの濃度比が少なくとも3:1である工程;
(B)工程(A)で得られた細胞に選択圧をかけ、該第1のベクターでトランスフェクトされた細胞を選択する工程;
(C)工程(B)で得られた細胞に選択圧をかけ、該第2のベクターでトランスフェクトされた細胞を選択する工程;
(D)工程(C)で得られた細胞を限界希釈し、ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼを発現する細胞を選択する工程
を含む、ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼの発現のための細胞株の調製方法を提供する。
【0025】
別の実施形態において、本発明は、
(A)ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼの不純試料を陰イオン交換クロマトグラフィーにかける工程であって、溶出が均一濃度である工程;
(B)工程(A)の溶離液を疎水性相互作用クロマトグラフィーにかける工程であって、硫酸アンモニウムが使用されず、溶出が均一濃度である工程
を含む、ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼを発現および培養培地中への分泌後に精製する方法を提供する。
【0026】
所望の酵素は、本発明の細胞および細胞株によって発現され、好ましい実施形態では、本発明の精製手法に従って精製される。次いで、該酵素の存在下で、グリシン残基を有し、遊離酸の形態であり、カルボニル基に結合された前駆体から始まる反応が行なわれる。反応条件および補因子は当該技術分野で知られている。本明細書に記載の実施例1および2に、アミド化生成物の典型的なアミド化反応および精製を示す。好ましいアミド化生成物が得られる。ペプチジルα−ヒドロキシル化モノオキシゲナーゼを用いた場合、中間体が得られ、これは、アミド化生成物を形成するために、ルイス塩基またはペプチジルα−ヒドロキシグリシンα−アミド化リアーゼのいずれかとのさらなる反応を必要とする。
【0027】
酵素産生細胞株、例えば本明細書に記載のものが、酵素を発現して培養培地中に分泌する場合、特定のある実施形態では、続いて、該酵素を精製するための一連の工程が行なわれ得る。収集工程では、ならし培地が細胞から分離される。第1のタンジェンシャルフロー濾過(濃縮、ダイアフィルトレーション)により、低分子量成分が除去される。陰イオン交換クロマトグラフィー工程および疎水性相互作用クロマトグラフィー工程は、主に、タンパク質不純物および培地成分からの精製のために用いられる。ウイルス濾過前の最後のタンジェンシャルフロー濾過、濃縮およびバッファー交換は、望ましくは、保存前に実施される。改善された陰イオン交換クロマトグラフィーおよび疎水性相互作用クロマトグラフィーを、本明細書の特許請求の範囲に示す。
【発明の効果】
【0028】
驚くべきことに、望ましくは安定なCHO K1細胞が、内在ジヒドロ葉酸還元酵素遺伝子の存在にもかかわらず、本発明の状況において使用され得ることがわかった。該内在遺伝子は、ジヒドロ葉酸還元酵素遺伝子を有する選択可能なマーカーに基づくメトトレキサート選択を有意に妨げなかった。
【0029】
PAM遺伝子と、独立したベクター上の2つの選択可能なマーカーとの共トランスフェクションは、本明細書に記載のように、ゲノムのより望ましい部分で起こる共増幅の機会を有意に増大させると考えられる。
【0030】
SV40エンハンサーが使用される場合、エンハンサーは、任意の転写プロモーター、例えば限定されないが、SV40転写プロモーター、本明細書に記載の好ましいメタロチオニンIIAプロモーターなどとともに使用され得ると理解されている。
【0031】
本発明に従って発現および精製されるペプチジルグリシンα−アミド化モノオキシゲナーゼは、望ましくないプロテアーゼ活性を実質的に含まず、本明細書に記載のような基質の効率的なα−アミド化に好適である。
【0032】
本明細書に記載の好ましい細胞株であるUGL 73−26/Mは、良好な長寿命プロフィールを示したため、安定性が非常に重要である規模の拡大に特に有用であると考えられる。
【0033】
同様に、本明細書に記載の好ましい精製手法では、拡張可能な特性および有意な生成物純度が提供される。具体的には、画分収集が有意に単純化される均一濃度の溶出が使用される。先行技術のような疎水性相互作用クロマトグラフィーと共に硫酸アンモニウムを使用することは、硫酸アンモニウムが酵素の沈殿および不活化を引き起こし得るため、望ましくは回避する。
【0034】
類推により、本明細書に記載の発明は、ペプチジルグリシンα−アミド化モノオキシゲナーゼに関して有益であったため、ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼに関して有益であることが予測される。
【0035】
本明細書に記載の好ましい態様の1つ以上を組み合わせて使用され得る。例えば、限定を意図しないが、好ましいプロモーターが、好ましいエンハンサーおよび/または好ましい宿主細胞と一緒に使用され得る。
【0036】
(A)PHMおよび(B)PALまたはルイス塩基のいずれかがPAMの代わりにアミド化に使用される場合、PHMおよびPAL(使用される場合)は、いくつかの様式で得られ得る。一部を以下に示す。
【0037】
PHM
発現されたときPHM活性のみを含むPAM遺伝子の天然に存在する形態の発現
カエルに由来する酵素の一例は、天然に存在するPHM酵素である。Mizunoら(1986),「Peptide C−Terminal a−Amidating Enzyme Purified to Homogeneity From Xenopus laevis Skin」Biochem Biophys.Res.Commun.,137(3)984−991を参照のこと。これは、後に、完全長PAMではなくPHMであることがわかった。Suzukiら(1990),EMBO 9(13)4259−4265を参照のこと。
【0038】
完全長PAMの発現および切断
PAM酵素をPHM活性とPAL活性の間の部位、例えば2塩基性切断部位で切断するため、特異的プロテアーゼが使用され得る。次いで、PHM触媒ドメインが精製によって得られ得る。例えば、Ouafikら,(1992)「The Multifunctional Peptidylglycine a−Amidating Monooxygenase Gene:Exon/Intron Organization of Catalytic,Processing,and Routing Domains」Molecular Endocrinology 6(10)1571−1584には、ラット由来PAM内の2つの触媒ドメインの位置が記載されており、「対の塩基部位での内部タンパク質分解性切断により、2つの触媒ドメインが分離され得る」と記載されている。また、Eipperら(1993),「Peptidylglycine a−Amidating Monooxygenase:A Multifunctional Protein With Catalytic,Processing,and Routing Domains」Protein Science 2,489−497も参照のこと。
【0039】
PAM内への停止コドンまたはリーディングフレームを改変する点変異の導入
代替法として、翻訳停止コドン(TAA、TAG、TGA)が、任意の種由来の任意のPAM cDNA内のPAMの2つの機能ドメイン(PHMおよびPAL)間に導入され得るか、またはリーディングフレームを改変するために、かかる位置内に点変異が導入され得る。
【0040】
PHM cDNAのみを有する発現ベクターの再設計
PCRを用い、PHMドメインのみをコードする切断型PAM遺伝子を合成し、プロモーターまたはエンハンサー/プロモーター配列の下流の発現ベクターに配置し得る。
【0041】
以下に、異なる型のPAM酵素およびその触媒ドメインに関するさらなる参考文献を示す。
【0042】
Mizuno,K.ら,(1987)「Cloning and sequence of cDNA encoding a peptide C−terminal α−amidating enzyme」、Xenopus Laevis Biochem Biophys.Res.Commun.,148(2)546−552
Ohsuye,K.ら,(1988)「Cloning of a cDNA encoding a new C−terminal α−amidating enzyme having a putative membrane−spanning domain」、Xenopus Laevis Biochem.Biophys.Res.Commun.,150(3)1275−1281
Koljekar,A.S.ら,(1997)「Peptidylglycine α−amidating hydroxylating monooxygenase:active site residues,disulfide linkages and tow−domain model of the catalytic core」,Biochemistry 36:13901−13909
Koljekar,A.S.ら,(2002)「Essential features of the catalytic core of Peptidylglycine α−hydroxyglycine α−amidating lyase」,Biochemistry,41:12384−12394
Bertelsen,A.H.ら,(1990)「Cloning and characterization of two alternatively spliced rat α−amidating enzyme cDNAs from rat medullary thyroid carcinoma」,Arch.Biochem.Biophys.,279(1)87−96
Jimenez N.ら,(2003)「Androgen−independent expression of adrenomedullin and peptidylglycine α−amidating monooxygenase in human prostatic carcinoma」,Molecular Carcinogenesis 35:14−24
PAL
天然に存在する形態のPALの発現
PAMの2つの触媒ドメインは、ショウジョウバエと刺胞動物(イソギンチャク)の別々の遺伝子にコードされている。したがって、これらの種に由来するPALをコードする遺伝子は、プロモーターまたはエンハンサー(enchancer)/プロモーター配列の下流の発現ベクターに配置すると、発現され得る。Kolhekar,A.S.ら.(1997)「Neuropeptide Amidation in Drosophila:Separate Genes Encode the Two Enzymes
Catalyzing Amidation」J.Neuroscience 17(4):1363−1376を参照のこと。
【0043】
完全長PAMの発現および切断
完全長の2つの機能を有するPAMを発現させ得、PHMを不活化させるために、PHMドメインとPALドメイン間および/またはPHMドメイン内の特異的プロテアーゼ部位で切断を行ない得る。任意の適当に特異的なプロテアーゼを用い、例えば、2塩基性切断部位でPHM活性をPAL活性から分離し得る。PAL活性/タンパク質は、さらに精製され得る。同様の手順および2塩基性切断部位の位置は、PHMの取得に関して上記に記載している。
【0044】
PAL cDNAのみを有する発現ベクターの再設計
PCRを用い、PALドメインのみをコードする切断型PAM遺伝子を合成し、プロモーターまたはエンハンサー/プロモーター配列の下流の発現ベクターに配置し得る。
【0045】
本発明の他の特徴および利点は、添付の図面、グラフ、表などに言及する本発明の以下の記載から明らかとなろう。
【図面の簡単な説明】
【0046】
【図1】図1は、pHS1のプラスミドマップである。
【図2】図2は、pUC8のプラスミドマップである。
【図3】図3は、pSV402MTのプラスミドマップである。
【図4】図4は、α−AE cDNA遺伝子配列の誘導(derivtion)である。
【図5】図5は、pAE73のプラスミドマップである。
【図6】図6は、73−26浮遊適合スキームである。
【図7】図7は、スピナーフラスコにおける溶存酸素濃度およびα−AE産生性である。
【図8】図8は、DOおよびα−AE産生性に対するスピナーフラスコの容積の効果である。
【図9】図9は、UGL 73−26/Mの攪拌培養pHである。
【図10】図10は、pH制御なしでの攪拌槽バイオリアクターおよびスピナーフラスコのpHプロフィールである。
【図11】図11は、攪拌槽バイオリアクターにおけるUGL 73−26/Mα−AE産生性に対するpHの効果。
【図12】図12は、精製PAM酵素のSDS PAGEである。
【図13】図13は、SDS PAGEゲルのデンシトメトリーのスキャンおよびピーク面積パーセントの計算である。
【図14】図14は、α−AEの産生のプロセスフローダイアグラムである。
【図15】図15は、UGL 73−26/Mの種菌スキームである。
【発明を実施するための形態】
【0047】
発明の実施形態の詳細な説明
本発明によるPAM発現細胞株(内部的にはUGL 73−26/M MWCB 00と示す)は、American Type Culture Collection(ATCC),10801 University Boulevard,Manassas Virginia,20110−2209,U.S.A.に、2005年6月10日あたりに、特許手続きのための微生物寄託の国際認識に関するブダペスト条約に従って寄託された。ATCC受託番号はPTA 6784である。この寄託細胞株は、この条約の下に公布された規定に従い、試料は、その時点で、および該条約によって求められる条件下で、ならびに特許法および該条約の調印国の規定に準じて入手可能となる。例えば、本出願またはこれを優先権主張するか、もしくはこれを参照する任意の他の米国特許出願の米国特許が発行された場合、該寄託物質の入手可能性に関するあらゆる制限は、ブダペスト条約または35 U.S.C.§112に求められる程度まで変更不可的に解除される。
【0048】
本発明のPAM発現ベクターの構築を、以下に詳細に記載する。本明細書に示すプラスミドマップにおいて、「αアミド化酵素」(例えば、図5、pAE73のプラスミドマップを参照)と示す領域は、添付の配列番号1に示されるヌクレオチド配列を有し、添付の配列番号2に示される715アミノ酸の一次翻訳産物をコードする。該翻訳産物は、シグナルペプチドをアミノ酸1〜25に、プロ領域を26〜41に、および成熟ペプチジルグリシンα−アミド化モノオキシゲナーを42〜715に含む。本発明に従って発現および精製される酵素のほとんどは、該プロ領域を含み続け、非常に有効であることが示された。
【実施例】
【0049】
以下、好ましいPAM発現細胞株、その発酵、方法の開発、精製および使用の例を詳述する。
【0050】
以下により詳細に記載するように、PAM遺伝子を、pSV402MT発現ベクター内にクローニングし、PAM発現プラスミドpAE73を創製した。CHO K1細胞をpAE73 DNAでトランスフェクトした。二重選択プロセスおよび限界希釈クローニング後、メトトレキサートの濃度を増加させ、CHO細胞株をさらに増幅した。接着細胞株73−26を、選択なし血清無含有の浮遊培養物UGL 73−26/Mに変換した。UGL 73−26/M MCBおよびMWCBは、BioReliance Corp.において創製した。MCBおよびMWCBの両方を充分に特性化したところ、GMP施設においてのPAMの産生に対し、認可され得ると考えられる。UGL 73−26/M細胞株を適合させ、攪拌槽バイオリアクター内での培養した。発酵の開発により、スピナーフラスコで14日間行なわれる種菌プロセスの定義付けがもたらされた。発酵/細胞培養相は17日間のプロセスである。発酵プロセスの重要なパラメータは以下の通り:第5、10および14日にグルコース添加(2g/L)、溶存酸素濃度≧70%、発酵がその自然な設定点に達するpH、およびタンパク質無含有、非動物由来組織培養培地、Sigma C5467(グルタミンを終濃度2mMまで補充)である。簡便でロバストな下流精製プロセスを、C5467 CHOならし培地からのPAMのために開発した。精製後、定常的に純度の4倍増加および活性酵素の40%の総回収が得られる。該プロセスでは、製造レベルに規模を拡大し易い慣用的な濾過系およびバイオプロセスクロマトグラフィー樹脂が用いられる。細胞培養発酵と下流精製のプロセス全体を、10Lパイロットプラント/製造レベルの規模にした。記載のプロセスは、大量のPAMの産生について確認可能な製造プロセスに従いやすいものであるのがよい。
【0051】
大量のPAMを発現するCHO細胞株UGL 73−26/Mを開発した。UGL 73−26/M用に開発した細胞培養プロセスは、非動物タンパク質由来の培地中のものとした。単純なバッチ細胞培養プロセスが好ましかった。精製スキームの目的は、触媒的に活性な酵素の良好な収率を伴う、ロバストで拡張可能なプロセスを開発することであった。
【0052】
ベクターの構築
該細胞株を作製するために使用される発現ベクターpAE73の創製は、Friedmanら.Bio/Technology 7:359−362(1989)の研究結果を基にした。この論文に記載されたプロセスに従い、本発明者らは、試験用のpAE73発現ベクターの創製を進めた。各ベクターの主要成分は、ヒトメタロチオネインIIA(hMTIIA)プロモーター(pHS1内に位置する、M.Karin氏より受領)、SV40エンハンサー(pSV40由来、ATCC)および本明細書に記載のα−アミド化酵素遺伝子である。
【0053】
pAE73の誘導化
pHS1
pAE73の創製に使用する出発プラスミドは、1990年2月にUCSDのM.Karinから贈与プラスミドpHS1(図1)であった。pHS1は、Dr.Karinの研究室において、pUC8(図2)内にヒトメタロチオネインIIAプロモーターをスプライシングすることにより誘導されたものである。846bpのHind III/Bam H1断片を、pUC8マルチクローニング部位(MCS)のHind III−Bam H1部位内にクローニングした。得られたプラスミドpHS1は、該マルチクローニング部位内に挿入されたヒトメタロチオネインHAプロモーターを有する。
【0054】
pSV401MTおよびpSV402MT
SV40エンハンサーをヒトメタロチオネインプロモーターの上流に挿入することにより、pHS1を発現ベクターpSV401MTまたはpSV402MT(図3)に変換した。SV40 DNA断片は、pSV40プラスミドDNAをHind IIIで消化することにより調製した。1167bpのHind III断片を、pHS1のHind III部位内にクローニングした。SV40エンハンサーは、Hind III DNA断片内に非対称に存在し、したがって、該エンハンサーは、hMTIIAプロモーターに近接して、または遠く離れてのいずれかで位置する。元のプラスミド内のlac Z遺伝子内のものに対するSV40エンハンサー内のBgl I部位の配向により、該プラスミドの配置(designation)が決まる。pSV401MTは、hMTIIAプロモーターから遠く離れたSV40エンハンサーを有し、pSV402MTは、hMTIIAプロモーターに近接したSV40エンハンサーを有する。pSV402MTを、さらなるベクター構築に選択した。
【0055】
pAE73
pAE73は、α−アミド化酵素遺伝子(配列番号1)を、pSV402MTのBam H1部位内にクローニングすることにより調製された。2870bpのα−AE遺伝子断片を、Bgl 1およびBam H1 DNA制限エンドヌクレアーゼでのpAE64の消化後に単離した。pAE64プラスミドは、SV40プロモーター/エンハンサーの下流に、可溶性75kDaのPAMタンパク質を発現するように修飾されたPAM遺伝子を含有する。このPAM DNA配列を別のCHO発現細胞株UGL B3/A1−7に使用し、75kDaのPAMタンパク質を発現させた。PAM遺伝子配列の誘導を図4に示す。
【0056】
2870bpのDNA断片を精製し、該断片のBgl 1末端を、2つの相補DNAオリゴヌクレオチド断片(AE96/AE97)で修飾し、これにより、該断片のBgl 1末端が新たなBam H1末端に変換された。
【0057】
AE96(+)23 5’GATCCATCGATCGCACTAGTGCC 3’
AE97(−)16 5’ACTAGTGCGATCGATG 3’
該修飾PAM DNA断片を、発現ベクターpSV402MTのBam H1部位内にクローニングした。2つのBam H1末端を有するPAM DNA断片は、発現ベクター内にセンスまたはアンチセンスのいずれかの配向に連結され得るため、PAM遺伝子の配向を、該プラスミドのEco R1による制限消化マッピングによって調べた。Eco R1部位と該発現ベクター両方におけるPAM遺伝子により、プラスミドDNAの単純な解析が可能になる。正しい配向のPAMを有するプラスミドをpAE73と表示し、そのプラスミドマップを図5に示す。
【0058】
α−アミド化酵素発現細胞株の創製
トランスフェクションおよびクローニング
3種類のプラスミド:pAE73、pSV2neoおよびpSY2dhfrをCHO K1細胞の形質転換に使用した。トランスフェクト細胞によるプラスミドの組込みにより形質転換細胞株の容易な選択が可能となるため、プラスミドpSV2neoおよびpSV2dhfr(ともにATCCから入手)を使用した。また、プラスミドpSV2dhfrは、特に、CHOゲノム内へのSV40プロモーター/dhfr DNAの組込みにより該遺伝子および他の近位遺伝子の選択的増幅が可能となるため選択された。共増幅される遺伝子は、プラスミドpAE73内に担持されたα−アミド化酵素遺伝子であった。CHO K1細胞は、プラスミドDNAのリン酸カルシウム沈殿により形質転換した。プラスミドは、該細胞内にpAE73:pSV2neo:pSV2dhfrが10:1:1の比でトランスフェクトした。100mmdish1つあたり20μgのpAE73を添加した。トランスフェクションの2日後、形質転換細胞のみが生存可能となるように、細胞を、選択圧下、250mg/LのG418を含有する培地中で培養した。この培地中でのCHO細胞の培養では、pSV2neoプラスミドの安定な組込みが必要とされ得る。トランスフェクションの27日後、G418選択培地中でいったん安定な培養が確立されたら、メトトレキサートを該培養培地に添加した。形質転換細胞のG418プールを、100nM、500nM、1μMまたは5μMのメトトレキサートおよび250mg/LのG418を含有する培地中で培養した。2週間後(トランスフェクションの6週間後)、単離した細胞フォーカスは、クローニングシリンダーにより5μMメトトレキサート+G418培地中で培養した細胞から確立され得る。この方法によって、25のフォーカスから細胞株を確立する試みを行なったが、移した後、2つのフォーカスからしか細胞が培養されず、この手法を却下した。限界希釈クローニングを0.5細胞/ウェルで、1μMメトトレキサート+G418を含有する培地中で培養した細胞から同時に開始した。低濃度のメトトレキサートで培養した形質転換細胞は除外した。限界希釈クローニング開始から3週間後、単離物を24ウェルプレートに移し、次いで、2〜3日間以内に増殖のため100mmdishに移した。CHO K1細胞のトランスフェクション開始から10週間後、73−26と表示する単離物のうちの1つを確立した。該細胞株を低温保存し、α−アミド化酵素発現を評価した。73−26は、この時点の6953U/106細胞/日を産生する最良の産生細胞株の1つであった。
【0059】
細胞株73−26の増幅
73−26の初期クローンを継代し、1μMメトトレキサート+G418を含有する培地中で維持される確立された細胞株を得た。pSV2dhfrプラスミドをトランスフェクションのために選択する目的は、形質転換細胞株を同定するための第2の選択方法が提供されるためだけでなく、細胞を高濃度のメトトレキサートで培養すると、dhfrミニ遺伝子が増幅されることが広く確立されているためであった。Unigene Laboratoriesは、以前に、このプラスミドで形質転換された細胞株を用い、α−アミド化酵素の最大の産生をもたらすのに20〜50μMの濃度のメトトレキサートが必要とされ得ることが示されたことを経験している。α−アミド化酵素細胞株73−26を直接、1μM、20μMまたは50μMのメトトレキサートを含有する培地に分割した。また、すべての培地に、第2の選択方法としてG418も含めた。この細胞株のα−アミド化酵素発現の進行を、種々の時間間隔で表1に示す。
【0060】
【表1】
【0061】
表1のデータは、以前に開発されたB3/A1−7細胞株のものと比べ、この細胞株におけるα−アミド化酵素の発現レベルの増大を示す。α−アミド化酵素遺伝子とdhfr遺伝子の共増幅は、メトトレキサートの濃度を増大させて継代培養を継続するより多くの酵素を発現する該細胞株能力により明白になる。20μMまたは50μMメトトレキサートの存在下で培養した細胞は、選択的増幅の3〜4ヶ月後、最良の発現レベルを有した。継続的に数ヶ月間、継代培養すると、α−アミド化酵素の発現レベルは低下した。したがって、以前に凍結させたこの細胞株のストックを、浮遊適合細胞株の調製に使用した。
【0062】
浮遊適合α−アミド化酵素発現細胞株73−26の特性化
この細胞株の開発の最終目的は、メトトレキサートおよびG418の選択圧なしで安定な方法を開発することである。該細胞株の所望される別の属性は、これが、浮遊培養において比較的高い細胞密度で培養されることであった。該目的を達成するために用いた方法を以下に記載する。
【0063】
浮遊適合無選択細胞株の調製
無選択浮遊適合細胞の調製では、UGL 73−26接着細胞を、血清、メトトレキサートおよびG418から離すことが必要とされた。この課題を達成するため、multi prong attack(図5)を開発した。除去のマトリックス(matrix of removal)は、可能な最終の細胞株に対して最後の数の細胞倍加を付加するという最終目的で確立された。将来的な研究開発のため、以前に凍結させた(1991年12月16日)73−26の20μMメトトレキサート細胞株を解凍し、浮遊適合血清無含有無選択細胞株を確立した。50μMメトトレキサートでのPAM活性はいくぶん高かったが、73−26 50μM細胞株の活性培養物を再確立する試みは成功裏でなかった。該細胞株のα−アミド化酵素産生性を、浮遊適合プロトコルを開始する前に評価した(表2)。
【0064】
【表2】
【0065】
浮遊培養に適合させる前のこの細胞株のPAM活性は、細胞1つあたりの基準で、B3/A1−7対照よりも20倍大きい活性を有した。対照細胞株および73−26両方での酵素活性は、該細胞株を凍結する(1991年12月16日)直前の最初の試験で観察されたものの5〜10倍であった(表1)。
【0066】
4種類の新たな浮遊適合血清無含有細胞株を、図6に示すように作製した。73−26/K−Nと表示する該細胞株を、37日間のプロセス後、低温保存した。
【0067】
浮遊適合73−26のα−アミド化酵素産生
新たな73−26細胞株(73−26/K、73−26/L、73−26/M、73−26/N)のバンク登録細胞を解凍し、スピナーフラスコ内で培養した。該細胞株を、週に3回Ex−Cell301培地中で継代することにより維持した。定期的に培地を完全に交換し、酵素産生性の24時間評価をα−AEアッセイによって評価した。細胞株は、80日間まで培養状態で維持した。以前の細胞株B3/A1−7の半連続的バッチプロセスは40日間プロセスであり、該細胞株の産生安定性を評価するため、この手順の長さの2倍を選択した。この研究のデータを表3に示す。この研究過程において、細胞株73−26/Lは第22日〜第40日の間で終わったが、他のすべての細胞株は、80日の期間の最後まで活発に培養された。
【0068】
【表3】
【0069】
すべての細胞株が、初期の期間ではB3/A1−7よりも有意に多くのα−アミド化酵素を発現したが(<40日間)、73−26/Mおよび73−26/Nのみ、70日後もなお、大量の酵素を産生していた。80日の期間にわたって一貫してより多くのα−アミド化酵素を産生したことから、さらなる開発のためにUGL 73−26/Mを選択した。その他の3種類の細胞株は、80日の試験期間の終了前に培養が終了したか、またはその産生性/細胞/日が最後の20日間で減衰したかのいずれかであった。
【0070】
CHO K1、UGL 73−26、UGL 73−26/M MCBおよびUGL 73−26/M MWCB00のバンク登録前での細胞バンク登録の特性化研究
特性化研究を、前駆細胞株(73−26)である宿主細胞株(CHO K1)およびUGL 73−26/Mシードバンクにおいて行なった。40個のバイアルのUGL 73−26/Mシードバンクを1998年2月4日に調製した。シードバンクの各バイアルには、90%Ex−Cell 301/10%DMSO中4×106細胞/mLが含まれる。実施した研究はすべて、CHO細胞株がMCBおよびMWCBになる必要があるという結果と整合すると評価された(表4参照)。
【0071】
マスター細胞バンクUGL73−26/M細胞株は、BioReliance Corp.において作製された。118MCBのバイアルのいくつかを用い、該細胞株を充分に特性化した。UGL 73−26/M MCBのすべての研究の結果は、CHO起源の細胞株と整合し、感染性因子について陰性である。UGL 73−26/M MCBのバイアルを用い、製造業者の研究用の238個のバイアルの細胞バンクUGL 73−26/M MWCB00が、BioReliance Corp.において創製された。UGL 73−26/M MWCB00の研究のすべての結果は、CHO起源の細胞株と整合し、感染性因子について陰性である。
【0072】
【表4−1】
【0073】
【表4−2】
【0074】
【表4−3】
【0075】
スピナーフラスコおよび攪拌槽バイオリアクターにおけるUGL 73−26/Mのプロセスの開発研究
予備実験では、該細胞株の産生性が多数の生成では一貫して維持され得ないことが示されたため、バッチ発酵プロトコルを検討した。また、バッチプロトコルは、計画の容易さ、易拡張可能性および劇的なバッチ不成功の結果が少ないという利点を提供する。UGL 73−26/Mの発酵プロセスの開発には、該細胞株の細胞内産生性に影響し得るいくつかのパラメータを検討する必要がある。以下に詳述する発酵開発は、溶存酸素(DO)、pHおよび培地補充を検討する主要な研究を示す。研究されなかった発酵パラメータは、インペラーのRPMおよび発酵温度であった。
【0076】
α−アミド化酵素発現に対する溶存酸素の効果
スピナーフラスコ内のCHO培養培地の溶存酸素濃度の効果を調べるための実験を行なった。スピナーフラスコには0.1×106細胞/mLを播種した。2つのスピナーフラスコは、250mLTechneスピナーフラスコ内に150mLの培地を入れ、一方、第3のスピナーフラスコには250mLの培地を入れた。すべてのスピナーフラスコにおいて溶存酸素を毎日測定した。α−AEアッセイによるα−AE産生性の評価のため、清澄ならし培地のアリコートを毎日採取した。この研究では、低タンパク質CHO培地(C1707、Sigma−Aldrich)およびタンパク質無含有CHO培地(C5467、Sigma−Aldrich)の2種類の培養培地を使用した。この2種類の培地中で培養したUGL 73−26/Mの直接比較を図7に示す。2つの培養物中の溶存酸素レベルは著しく異なった。150mLのC1707中で培養した細胞は、等容量のC5467培養培地中で培養した細胞よりも、産生された全酵素/mLが少なかった。両培地の初期の溶存酸素濃度は約80%DOで同じであったが、C5467培地の培養物の溶存酸素濃度は、絶対に50%未満にならなかった。C1707培養物のDO含量は、第9日までに50%未満となり、該培養物のピーク活性は2日後であった。これらのデータは、DO含量/細胞バイアビリティ/α−AE産生性間に相関性が存在し得ることを示す。
【0077】
また、スピナーフラスコの容積も潜在的臨界パラメータとして検討した。スピナーフラスコは一定溶存酸素濃度に維持されないため、培養物が溶存酸素を維持する能力は、培養物の表面:容量比に対し相対的である。スピナーフラスコの培養容積を増大させると、明らかに表面:容積比は変化する。一方のスピナーフラスコの培養容積を150mL(5467s)とし、他方を250mL(5467sa)とした以外は、同一の培養物で開始した。%DOおよびα−AE産生性に対する表面:容積比の変化の効果を図8に示す。
【0078】
図8に示すデータは、培養物の産生性が溶存O2によって大きく影響されることを示す。溶存O2濃度の減少が、培養物による利用のためであれ、表面:容積比の差による効果のためであれ、DO濃度が50%未満の場合、培養物は48〜72時間以内に、より多くのα−アミド化酵素を発現するのを停止する。細胞が最も良好に産生性である培養期に溶存酸素濃度を反映させるためには、バイオリアクター内の溶存O2を70%に維持するのがよい。
【0079】
α−アミド化酵素発現に対する培地の効果
UGL 73−26/Mの培養培地の最も適切な選択肢の選択を容易にするために設計された研究、またはバイオリアクターの培養条件を規定する際の研究のいずれかの一部として、2種類のCHO培養培地C1707およびC5467(Sigma−Aldrich)を用いて数多くの攪拌培養を開始した。両培養培地は規定されたものであり、C1707は、トランスフェリンが添加された低タンパク質培地であり、C5467は非動物タンパク質培地である。培地はともに、購入時に、終濃度2mMまでL−グルタミンを添加する必要があった。以下の表5に、2つの培地のいずれかを用いたスピナーフラスコの多数のサンプリングのデータを示す。
【0080】
【表5】
【0081】
表5のデータでは、いずれかの培地におけるUGL 73−26/M細胞株の酵素産生性の実質的な差は示されていない。Sigma CHO培養培地であるC5467は、動物由来の培地成分を含有しないため、米国および欧州連合の両方において、将来的な規制的ガイドラインに関して、より規制に準拠する培地の選択肢であり得る。Sigma CHO培養培地であるC5467は、将来的な研究のための培地の選択肢である。
【0082】
α−アミド化酵素発現に対するグルコースの効果
UGL73−26/Mのバッチ培養物の栄養状態を検討する研究により、培養開始の4〜5日後、培地中のグルコース濃度は、未使用培地のほぼ50%(2g/L)であることが明らかになった。酵素産生性に対するグルコース補充の効果の徹底的な研究を、さらなるグルコースを培養物に添加する一連のスピナーフラスコ実験として行なった。スピナーフラスコには、2g/Lのグルコースを1〜3回、特定の時間間隔で補充した。α−AE産生性に対するグルコース添加の効果を表6に示す。
【0083】
【表6】
【0084】
【表7】
【0085】
UGL 73−26/Mのバッチ培養物へのグルコースの添加により、培養物の最大産生性は、グルコース添加計画に応じて50〜100%増大した。第5、10および15日目にグルコースを補充したスピナーフラスコでは、非補充対照と比べ、最も多くの酵素が発現されるようである。これらの培養物の平均相対産生性は対照の195%であった。他の補充培養物では、3回補充のものよりほんのわずか産生性が低かった(143〜164%)。さらなるグルコースを補充した培養物はすべて、培地中でのピーク酵素濃度の遅延を示した。ピーク酵素濃度は、グルコース補充培養物では第17/18日目に観察されたのに対し、対照の非補充培養物では第13日目であった(表7)。
【0086】
α−アミド化酵素発現に対するpHの効果
ならし培地のpHは攪拌槽バイオリアクターでは制御され得るが、スピナーフラスコにおいては対処され得ないプロセス変量の1つである。スピナーフラスコでは、培地成分が消費されるにつれて、および細胞の副生成物が産生されるにつれて、培養物のpHは低下する。150mL(5467s)または250mL(5467sa)のいずれかの容積のUGL 73−26/Mの2つの250mLスピナーフラスコのpHプロフィールを、以下に図9に示す。培養物のpHは、バッチの前半では低下し、次いで培養の後半では上昇した。pHの低下は、おそらく、培養前半での培養物中の乳酸濃度の増大によるものである。培養の最後でのpHの上昇は、細胞が該乳酸を炭素供給源として異化することによるものである。培養物の産生性はpHの低下に影響されない(図8)。
【0087】
いくつかの(n=5)6〜10L攪拌槽バイオリアクターは、スピナーフラスコ内の条件を模倣し、pH制御なしで実行した。攪拌槽バイオリアクター内のCHO細胞をC5467培地中で、70%DOおよび37℃にて培養した。バイオリアクター内のならし培地の平均pHプロフィールは、2つのスピナーフラスコで観察されたものと同様である(図10)。バイオリアクター内で溶存酸素濃度は70%に、温度は37℃に維持した。
【0088】
バイオリアクターを上記のようにpH制御なしで実行させたことに加え、一連のバイオリアクターは、該バイオリアクターのpHが設定pH点(7.0〜7.4)に自由になるように実行した。所望のpHに達した時点で、これをそのpHに発酵の持続期間、維持した。攪拌槽バイオリアクター内のCHO細胞をC5467培地中で、70%DOおよび37℃にて培養した。攪拌槽バイオリアクター内でのα−AE産生性に対するpH制御の維持の効果を図11に示す。
【0089】
図11は、pH設定点を開始しなかった場合、それらの実行に対する実行の間、各日のバイオリアクターにおける平均α−AE活性を示す。pHが設定点の0.5pH単位以内になった後でのみpH設定点を設定した場合の個々の実行のデータもまた図11に示す。培養pHと酵素活性と間に明白な相関性がある。pH設定点なしで実行させたバイオリアクターの活性プロフィールは、培養の最初の16日間のすべての実行について優れている。唯一の例外は、pH設定点が7.1である場合の実行に対するプロフィールであった。この差は、おそらく、培養を、その他の実行よりも長く持続させたためであった。平均pH設定点なしの研究でのピークα−AE活性は、第16日目で280,707単位/mLであった。
【0090】
生細胞密度に対するpHの効果はなかった(データ示さず)。培養物は、1.0〜1.5×106細胞/mLのピーク生細胞密度まで培養させた。最大細胞密度は、バイオリアクターにイノキュレートしてから8〜10日後に達成された。pH7.1で実行させた1つの培養物以外は、細胞バイアビリティに対するpHの影響はなかった(データ示さず)。細胞培養物のバイアビリティは、15日以上培養した場合で50%生細胞より大きかった。7.1のpH設定点で実行させた1つのバイオリアクターでは、20日間の培養で>50%生細胞が維持された。
【0091】
研究開発したUGL 73−26/Mの発酵パラメータの概要
α−AE発現細胞株UGL 73−26/Mの確立後、α−アミド化酵素の至適発現のための臨界パラメータを規定するために一連の実験を行なった。動物タンパク質無含有培地(Sigma,C5467)では、該酵素の高レベルの発現が支持されることが測定された。CHO細胞は、L−グルタミンを終濃度2mMで補充したC5467培地中で、バッチ培養物として培養され得る。しかしながら、該培養物は、その産生性をさらに促進するためには、D−グルコース補充を必要とする。至適グルコース補充は、第5、10および15日目で2g/Lであった。バイオリアクターのDO濃度は70%に維持するのがよく、バイオリアクターのpHは、培地の緩衝能とCHO細胞による培地成分の代謝の関数となるpHに調整されるようにするのがよい。
【0092】
下流精製の開発UGL 73−26/M
以下の節に、本発明者らが開発した、UGL 73−26/Mクローンを用いて産生されたPAMの下流精製についてまとめる。代表的な実験のみ(該プロセスの開発作業の論理的な進行を示す)をこの概要に含める。各工程の簡単な説明を示すが、これは、精製プロセスにおけるその工程の機能の記載である。精製実行は、SDS−PAGE(一定タンパク質および一定単位)、PAM活性アッセイならびにBradfordタンパク質アッセイを用いて解析した。実験はすべて、Boonton,NJ試験施設に技術移転する前または直後に完了した。本明細書に記載の方法を用い、PAMバッチ1330−D−1003、1330−D−1004、1330−D−1005、1330−D−1006、1330−1009および1330−1010を製造した。
【0093】
第1のタンジェンシャルフロー濾過(TFF1)
工程の説明:TFF1工程を用い、クロマトグラフィーの前にCHO細胞ならし培地の濃縮およびダイアフィルトレーションを行なう。ならし培地を濃縮し、ダイアフィルトレーションによって導電率を低下させ、陰イオン交換カラムへの結合を助長する。TFF1工程では、再生セルロースPLCTK 30kDa膜(Millipore)を取り付けたPellicon 2 Moduleを使用する。
【0094】
研究開発:この工程の最初の検討では、TFFがクロマトグラフィーの前に必要とされ得るか否かに着目した。水での希釈によるCHOならし培地の導電率の低下を、TFFの代替法として検討した。クロマトグラフィーの前にならし培地を希釈しないと、充填後、PAM活性の大部分が通過画分中に存在した。ならし培地の導電率をほぼ5mSまで低下させると、通過画分中に検出されるPAM活性の量は最小限になった。しかしながら、ならし培地を陰イオン交換カラムに直接充填すると、2つの問題が確認された。一部の培地成分が不可逆的にカラムに結合し、樹脂がひどく変色した。勾配溶出後、大きなUV吸光度特性を示す別の培地成分が、PAM活性とともに共溶出された。
【0095】
CHO細胞ならし培地(C5467)を水で希釈し、導電率をほぼ5mSまで低下させ、50mM TRIS、120mM NaCl、0.001%TX−100(pH8.0)で平衡化させたQ−セファロースFFカラム(Pharmacia)に直接充填した。カラムを、100%A(50mM TRIS、120mM NaCl、0.001%TX−100(pH8.0))から68%B(50mM TRIS、475mM NaCl、0.001%TX−100(pH8.0))までの60分かけた直線勾配にかけた。充填直後、大きな暗色のバンドがカラム上部に出現し、クロマトグラフィー実行中、残存した。カラムを2M NaClで清浄にし、1.0M NaOHで殺菌した。清浄手順および殺菌手順を行なっても、暗色バンドはカラムから除去されなかった。続いて、カラムは、より厳しい清浄手順を行なった。カラムを、以下の一連の清浄試薬:0.5M NaOH 1M NaCl、0.1M酢酸、0.1M酢酸 1M NaClおよび70%エタノールにさらした。試験したいずれの条件でも暗色バンドはカラム(NEG:001:225−245)から除去されなかった。着色バンドは、未使用C5467培地中に存在する一成分のアウリントリカルボン酸であると同定された。TRISでpH8.0まで緩衝化したアウリントリカルボン酸の6mg/Lの溶液(培地濃度)を、50mM TRIS、120mM NaCl、0.001%TX−100(pH8.0)で平衡化させたQ−セファロースFFカラムに直接充填した。充填直後、着色バンドがカラム上部に出現した。このカラムは、上記と同じ勾配溶出および清浄手順を行なった。アウリントリカルボン酸は、実行中、陰イオン交換カラムに結合したまま残存した(NEG:001:276−290)。アウリントリカルボン酸は、pH8.0において、水溶液中で容易に重合し、高電荷密度で巨大分子をもたらす。負の電荷を有する巨大分子は、明らかに、DNAとそっくりに陰イオン交換カラムに不可逆的に結合する。この物質をカラムに反復的に適用すると、おそらく充填容量が減少し、したがって、カラム全体の寿命に負の影響を及ぼす。
【0096】
第2の培地成分は、PAMとともに共溶出することがわかった。非ならし培地または未使用培地を、50mM TRIS、120mM NaCl、0.001%TX−100(pH8.0)で平衡化させたQ−セファロースFFカラムに直接充填した。このカラムを再度、上記と同じ勾配溶出条件に供した。該酵素が典型的に溶出される領域に大きなUVピークが観察された(NEG:001:248−257)。ならし培地から精製したピークのSDS−PAGE解析により、該物質は、低分子量タンパク質または非タンパク質性培地成分のいずれかであることが明らかとなった(NEG:001:225−245)。C5467培地はタンパク質水解生成物を含有し、これは、該酵素のピークと一致する最大UVピークの説明となり得る。両方の培地成分であるアウリントリカルボン酸および非同定UVピークは、クロマトグラフィーの前にTFFによって有効に除去される。該酵素は30kDa膜によって保持され(貯留物)、低分子量培地成分はフィルターを通過する(透過物)。
【0097】
再生セルロースPLCTK 30kDa膜(Millipore)を取り付けたPellicon XL装置を用い、ならし培地を10倍に濃縮してダイアフィルトレーションを行なった。該物質は、4〜5容量の25mM TRIS、0.0005%TX−100(pH7.0)を用いて、最終導電率ほぼ3mSまでダイアフィルトレーションを行なった。該物質を、50mM TRIS、120mM NaCl、0.001%TX−100(pH8.0)で平衡化させたQ−セファロースFFカラムに充填した。このカラムは上記のような勾配溶出が行なわれた。樹脂の変色は観察されず、該酵素と共溶出される最大UVピークは存在しなかった。得られた酵素ピークの回収%および比活性を、それぞれ47%および2.1×106U/mgとなるまで測定した(NEG:004:263−283)。
【0098】
TFF工程の種々の段階における酵素安定性を検討した。ならし培地は、最初に、50mM TRIS、0.001%TX−100(pH7.0)でダイアフィルトレーションを行なった後、5倍濃縮した。ダイアフィルトレーションおよび濃縮で得られた試料を、活性の低下および分解について解析した。SDS−PAGEでは分解は観察されず、酵素活性のほぼ95%が回収された(NEG:008:245−251)。
【0099】
陰イオン交換クロマトグラフィー(Q−セファロースFF)
工程の説明:陰イオン交換(AEX)クロマトグラフィー工程により、CHO細胞ならし培地からのPAMの全体精製が提供される。該工程では、主に、高分子量タンパク質が除去される。しかしながら、例えば、該酵素の切断型形態などの一部の低分子量タンパク質もまた除去される。該クロマトグラフィー工程では、定常的に、該酵素2〜3倍の精製がもたらされる。活性酵素の回収%は、典型的には50〜75%である。CHO細胞の発酵後、供給原料流中に存在し得るDNAは、該プロセスのこの段階で有効に除去される。
【0100】
研究開発:Q−セファロースFF工程の開発前に、本発明者らは、陽イオン交換(CEX)クロマトグラフィーを用いてPAMの精製を簡単に検討した。CEXクロマトグラフィーの使用は、最初にTFF工程を伴って、またはなしで検討した。精製は、SP550C(TosoHaas)カラムにおいて行ない、pH5.0〜6.0の移動相を使用した。各場合において、分離および回収の結果は不充分であり、充填後、酵素活性の大部分は通過画分中に確認された(NEG:004:006−029およびNEG:004:102−119)。結合を助長するための移動相のpHの低下は、低pHでの該酵素の安定性の欠如の可能性のため、考慮しなかった。この点で、CEXクロマトグラフィーは却下した。
【0101】
勾配溶出を用いた一連のAEXクロマトグラフィー実行をQ−セファロースFFカラムにおいて検討した。Q−セファロースFF精製はすべて、(1.1×13.7cm)カラムにおいて行ない、180cm/時で実行した。精製開発の初期において、新鮮ならし培地の入手可能性は限定的であり、したがって、これらの実験の多くは、凍結/解凍材料で行なった。ならし培地をTFFによって濃縮/ダイアフィルトレーションし、50mM TRIS、120mM NaCl、0.001%TX−100(pH8.0)で平衡化させたカラムに充填した。このカラムを、100%A(50mM TRIS、120mM NaCl、0.001%TX−100(pH8.0))から68%B(50mM TRIS、475mM NaCl、0.001%TX−100(pH8.0))までの60分かけた塩直線勾配にかけた。pH7.0またはpH8.5のいずれかのHEPESバッファーを用いた同様の勾配実行もまた検討した。概して、この勾配実行では、ほとんど精製はもたらされず、酵素比活性は不充分な結果となった(NEG:004−139−156、NEG:005:119−132、NEG:009:009−022およびNEG:009:024−038)。多くの場合で、酵素活性は、おそらく差示的なグリコシル化の結果として、この勾配全体において散在的であった。
【0102】
開発を継続している際、逐次塩工程を用いた段階的溶出方法を、Q−セファロースFFカラムにおいて検討した。濃縮/ダイアフィルトレーションを行なったならし培地を、50mM TRIS、120mM NaCl、0.001%TX−100(pH8.0)で平衡化させたQ−セファロースカラムに充填した。このカラムは、175mM NaCl、225mM NaClおよび275mM NaClでの逐次段階的溶出が行なわれた。酵素活性は、175mMと225mMの間の塩工程で等しく分布し、全体回収%oはほぼ41%であった。SDS−PAGEによる解析により、175mMおよび225mM両方のNaCl画分において75kDaの酵素の存在が明らかとなった(NEG:005:198−211)。両方の塩工程での酵素活性は、グリコシル化の差またはおそらく活性を保持している該酵素の切断型形態によるものであり得る。
【0103】
スピナーフラスコ実行または10Lバイオリアクター実行のいずれかの新鮮なならし培地を、残りの陰イオン交換実験に使用した。スピナーフラスコからのCHOならし培地00020S3をTFFにさらし、次いで、50mM TRIS、120mM NaCl、0.001%TX−100(pH8.0)で平衡化させたQ−セファロースFF上に充填した。酵素をカラムから、1回の塩工程、50mM TRIS、225mM NaCl、0.001%TX−100(pH8.0)で溶出させた。回収されたピークの回収%および比活性の値は、それぞれ34%および1.9×106U/mgであった(NEG:008:021−033)。同様に、10LバイオリアクターからのCHOならし培地(00021BR、第14日)をTFFにさらし、1回の225mM NaCl工程を用いてQ−Sepharose上で精製した。この実行での回収%および比活性の値は、それぞれ37%および2.3×106U/mgであった(NEG:008:065−075)。
【0104】
また、HEPES移動相を用いた段階的溶出を、活性酵素の回収を改善する取り組みにおいて検討した。2連の精製実行を、50mM HEPES、225mM NaCl、0.001%TX−100(pH8.0)を溶出バッファーとして用いて行なった。10Lバイオリアクター実行(Boontonパイロット施設、実行番号1、第15日)からのCHOならし培地を、これらの精製実行のための投入物質として使用した。両方の精製実行での回収%および比活性は、それぞれ、ほぼ60%および3.0×106U/mgであった(NEG:008:263−288)。HEPES移動相の使用により、酵素の回収%および純度が明らかに改善された。
【0105】
Q−セファロースFF精製に対する細胞バイアビリティの効果を検討した。10Lバイオリアクター実行から、第14日および第21日に収集したCHOならし培地(BOOlOl)の精製を比較した。第14日および第21日における細胞バイアビリティを測定し、それぞれ95%および48%であると決定された。第14日および第21日のCHOならし培地は、TFF、および50mM HEPES、225mM NaCl、0.001%TX−100(pH8.0)を段階的溶出バッファーとして用いたQ−セファロースFF精製が行なわれた。第14日の物質の精製では、49%の回収および2.15×106U/mgの比活性がもたらされたが、第21日での実行では、40%の回収および2.1×106U/mgがもたらされた(NEG:009:114−125およびNEG:009:149−166)。また、10Lバイオリアクター実行からの第12日および第15日のCHOならし培地(00107BR)を用いた精製も行なった。第12日および第15日での細胞バイアビリティを測定し、それぞれ86%および80%であると決定された。各場合において、酵素はQ−セファロースFFカラムから、50mM TRIS、225mM NaCl、0.001%TX−100(pH8.0)を用いて溶出した。第12日の物質では、62%の回収および1.9×106U/mgの比活性がもたらされたが、第15日の物質では、54%の回収および4.3×106U/mgの比活性が得られた(NEG:009:195−203およびNEG:009:227−236)。細胞バイアビリティは、AEXクロマトグラフィー後の全体的な回収%または酵素比活性に対して有意な影響を及ぼさないようであった。
【0106】
実用範囲を確立するため、Q−セファロースFFクロマトグラフィーの臨界パラメータを特定し、検討した。該精製手順に取り組むために設計した計画を規定し、酵素の回収および純度に対する影響を調べるために検討した。2通りのQ−セファロースFF精製実行を、異なる段階的溶出バッファー、50mM TRIS、250mM NaCl、0.001%TX−100(pH7.8)または50mM TRIS、200mM NaCl、0.001%TX−100(pH8.2)(低pH/高塩濃度および高pH/低塩濃度)のいずれかを用いて比較した。これらの実験の論理的説明は、実用範囲を規定するためにクロマトグラフィーを極端な溶出条件に供することであった。10Lバイオリアクター実行からのTFF処理したCHOならし培地(2132−D−1004)を、精製のための投入物質として使用した。低pH/高塩濃度の計画では、ならし培地を、50mM TRIS、120mM NaCl、0.001%TX−100(pH7.8)で平衡化させたカラムに充填した。酵素をカラムから、50mM TRIS、250mM NaCl、0.001%TX−100(pH7.8)で溶出した。酵素の回収%および比活性の値は、それぞれ66%および2.0×106U/mgであった(NEG:010:151−161)。高pH/低塩濃度の計画でのならし培地は、50mM TRIS、120mM NaCl、0.001%TX−100(pH8.2)で平衡化させたカラムに充填し、50mM TRIS、200mM NaCl、0.001%TX−100(pH8.2)で溶出した。この場合の回収%および酵素比活性は、それぞれ66%および2.4×106U/mgであった(NEG:010:140−150)。これらのデータに基づき、Q−セファロースFFカラムは[7.8〜8.2]のpH範囲で実行され得、溶出バッファー中の塩化ナトリウム濃度は[200mM〜250mM NaCl]の間で種々であり得る。
【0107】
30Lバイオリアクター実行に精製規模拡大するのを補助するため、Q−セファロースFFカラムの実用容量を確立した。カラムの実用容量は、2つの様式:[酵素単位/mL樹脂]および[mgタンパク質/mL樹脂]で規定した。この実験の大きな進歩は、通過画分および洗浄工程で確認される酵素活性が合計で≧10%である規定された。TFFにさらされたCHOならし培地(100mL)(2132−D−1002)を、50mM TRIS、120mM NaCl、0.001%TX−100(pH8.0)で平衡化させたQ−セファロースFFカラムに充填した。ならし培地の酵素活性およびタンパク質濃度は、それぞれ1.97×106U/mLおよび2.41mg/mLであると決定された。AEXカラム(Millipore Vantage−L、1.1×13.7cm)を、線速度180cm/時で実行した。50mM TRIS、225mM NaCl、0.001%TX−100(pH8.0)で、段階的溶出を行なった。1%未満の総酵素活性が、カラム通過画分および洗浄工程において確認された。ほぼ51%の酵素単位がメインピークで回収された。13.0mLのカラム容積に対し、実用容量は、[1.51×107U/mL樹脂]または[18.5mgのタンパク質/mL樹脂]のいずれかであると算出された(NEG:010:081−092)。Q−セファロースFF工程の真の充填容量を決定するため、さらなる実験を計画している。
【0108】
疎水性相互作用クロマトグラフィー(フェニル−セファロースFF、低置換)
工程の説明:疎水性相互作用クロマトグラフィー(HIC)工程により、AEX工程後に残留するタンパク質不純物の大部分が除去され、さらに2倍の精製がもたらされる。HIC後のSDS−PAGE解析により、少量の低分子量不純物を伴うのメジャーバンドが明らかになった。典型的には、HIC後の活性酵素の回収%は50〜75%である。
【0109】
研究開発:B3/A1−7クローンから産生されるPAMのために開発した精製プロセスでもHICを用いた。リガンド−タンパク質相互作用促進するため、硫酸アンモニウムを含有する移動相を使用した。硫酸アンモニウムを含有するバッファーへの酵素の長期間の曝露により、予測不可能な沈殿および酵素変性がもたらされた。これらの問題を最小限に抑える試みにおいて、Hofmeisterシリーズで他の塩、例えば、塩化ナトリウムおよびクエン酸ナトリウムを検討した。
【0110】
塩化ナトリウムを含有する移動相を用いた逆直線勾配を、最初に検討した。これらのクロマトグラフィー研究の投入物質は、TFF1で得られたものまたはQ−セファロースFFで得られたもののいずれかとした。最初のTFF工程後の直接HICを、AEX工程を排除する試みにおいて検討した。典型的には、酵素を等容量の水で、その後、等容量の20mM TRIS、4M NaCl(pH7.0)で希釈した。塩添加前の水での希釈により、タンパク質濃度が減少し、「塩析」によって引き起こされる沈殿の可能性が最小限になる。希釈した物質を、10mM TRIS、2.0M NaCl(pH7.0)で平衡化させたフェニル−セファロースFFカラムに充填した。酵素をカラムから、100%A(10mM TRIS、2.0M NaCl(pH7.0))から100%B(10mM TRIS(pH7.0))までの逆直線勾配で、68分間にわたって溶出した。概して、活性酵素の回収%はほぼ50%であった(NEG:005:170−185およびNEG:005:212−226)。
【0111】
また、塩化ナトリウム移動相を用いた段階的溶出方法も検討した。TFF1またはQ−セファロースFFクロマトグラフィーのいずれかにかけられた酵素を、等容量の水で、その後、等容量の20mM TRIS54M NaCl(pH7.0)で希釈した。希釈した物質を、10mM TRIS、2M NaCl(pH7.0)で平衡化させたHICカラムに充填した。このカラムをさらなる平衡バッファーで洗浄した後、10mM TRIS、1.0M NaCl(pH7.0)で洗浄した。酵素をカラムから、10mM TRIS、400mM NaCl(pH7.0)で溶出した。活性酵素の収率は相当であったが、なされた精製はほんのわずかであった(NEG:008:034−046およびNEG:008:135−148)。塩化ナトリウムの使用は、硫酸アンモニウムの場合のように「塩析」を引き起こさないようであったが、高濃度の塩化ナトリウムにおいて、酵素が経時的に変性した。
【0112】
クエン酸ナトリウムは、比較的低濃度でHICカラムへの酵素の結合を促進することがわかった。TFFにさらされたCHOならし培地を等容量の水で、その後、等容量の20mM TRIS、1.0Mのクエン酸ナトリウム(pH7.0)で希釈し、10mM TRIS、0.5M クエン酸ナトリウム(pH7.0)で平衡化させたフェニル−セファロースFFカラムに充填した。このカラムをさらなる平衡バッファーで洗浄し、10mM
TRIS(pH7.0)でストリッピングした。酵素活性は、カラム通過画分または洗浄画分のいずれにおいても確認されなかった。ほぼ50%の酵素活性が10mM TRIS(pH7.0)画分において確認された(NEG:005:064−076)。この実験を、より低い終濃度のクエン酸ナトリウムを用いて繰り返した。この低塩濃度では、酵素を等容量の水で、その後、等容量の20mM TRIS、0.6Mのクエン酸ナトリウム(pH7.0)で希釈した後、10mM TRIS、300mMのクエン酸ナトリウム(pH7.0)で平衡化させた HICカラムに充填した。このカラムをさらなる平衡バッファーで洗浄し、10mM TRIS(pH7.0)でストリッピングした。結果は、本質的に先の実行と同一であり、活性は、通過画分または洗浄画分のいずれにおいても確認されなかった(NEG:008:203−213)。
【0113】
段階的溶出方法の開発を補助するため、クエン酸ナトリウムバッファー系を用いた勾配溶出を検討した。TFFで得られたものを等容量の水で、その後、等容量の20mM TRIS、0.6Mのクエン酸ナトリウム(pH7.0)で希釈した後、10mM TRIS、300mMのクエン酸ナトリウム(pH7.0)中で平衡化させたカラムに充填した。このカラムを100%A(10mM TRIS、300mMのクエン酸ナトリウム(pH7.0))から100%B(10mM TRIS(pH7.0))までの70分間かけて逆直線勾配にかけた。酵素活性のピークは該勾配の最後付近で確認された。他のタンパク質不純物がこの勾配領域で溶出されたとともに、10mM TRIS(pH7.0)カラムストリップ中にまで拡張された(NEG:008:252−262)。10mM TRIS(pH7.0)ストリップ中に溶出される酵素とタンパク質のより良好な分離を達成するため、段階的溶出を検討した。
【0114】
Q−セファロースFF精製(10Lバイオリアクター実行、00107BR、第12日)からのピーク画分(225mM NaCl)を、等容量の水で、その後、等容量の50mM TRIS、600mMのクエン酸ナトリウム(pH7.0)で希釈した。希釈した物質を、25mM TRIS、300mMのクエン酸ナトリウム(pH7.0)で平衡化させたフェニル−セファロースカラムに充填した。このカラムをさらなる平衡バッファーで洗浄した後、25mM TRIS、75mMのクエン酸ナトリウム(pH7.0)での第2の洗浄を行ない、25mM TRIS(pH7.0)でストリッピングした。酵素活性はすべて、75mMのクエン酸ナトリウム画分において確認された。回収%および比活性は、それぞれ42%および2.0×106U/mgであると算出された(NEG:009:204−218)。酵素と他のタンパク質不純物との良好な分離は、この方法を用いて達成された。
【0115】
TFF直後のHIC(AEX工程なし)は、この工程では粗製供給物質流からDNAを排除する効果がないため、却下した。4回のHIC精製を、上記に詳述した塩化ナトリウム段階的溶出方法を用いて行なった。これらの実行の投入物質は、クロマトグラフィーの前にTFFにさらされたB3/A1−7 CHOならし培地とした。このときの本発明者らのDNA内部基準は<10ng/mgタンパク質であった。各場合において、HIC後のメイン酵素ピーク中に存在する残留DNAレベルは該基準より6〜10倍高く、それにより1回の工程でのHIC精製が許容され得ないことがわかった(NEG:006:137−159、NEG:006:176−187およびNEG:006:192−200)。
【0116】
30L容規模拡大の取り組みを補助するため、HIC精製工程の実用容量を検討した。大きな進歩は、カラム通過画分または洗浄画分のいずれかにおいて≧10%の酵素活性の存在と規定された。産生実行のQ−セファロースで得られたもの(125mL、0.368mg/mL、710,563U/mL)1330−D−1003を等容量の水で、その後、等容量の50mM TRIS、0.6Mのクエン酸ナトリウム(pH7.0)で希釈した。希釈した物質を、25mM TRIS、300mMのクエン酸ナトリウム(pH7.0)で平衡化させたフェニル−セファロースFFカラム(Millipore Vantage−L、1.1×15.8cm)上に充填した。このカラムを180cm/時で実行した。このカラムをさらなる平衡バッファーで洗浄し、酵素を25mM TRIS、75mMのクエン酸ナトリウム(pH7.0)で溶出した。酵素活性は、通過画分または洗浄画分のいずれにおいても確認されなかった。メインピークはほぼ75%の酵素活性単位を含んでおり、比活性は2.6×106U/mgであると算出された。15.0mLのカラム容積に対し、実用容量は5.92×106U/mL樹脂であると算出された(NEG:010:111−124)。実用容量は、AEX後にカラムに充填されたタンパク質が極めて少なかったため、[mgタンパク質/mL樹脂]では報告されなかった。HIC工程の真の充填容量を決定するため、さらなる実験を計画している。
【0117】
HIC単位実行のための全体の時間は水希釈を排除し、実行流速を180cm/時から240cm/時に上げることにより劇的に短縮された。クエン酸ナトリウムへの曝露時間および樹脂上で費やされる時間量を最小限に抑えることにより、最終的に、活性酵素の減少の低減が補助され得る。133O−D−1003 からQ−セファロースFFで得られたものを等容量の50mM TRIS、0.6Mのクエン酸ナトリウム(pH7.0)で希釈し、先のバラグラフで記載したHIC段階的溶出方法を用いて精製した。メインピーク(25mM TRIS、75mMのクエン酸ナトリウム(pH7.0))の回収%および比活性は、それぞれ77%および2.4×106U/mgであると算出された(NEG:010:125−139)。クエン酸塩を用いた希釈中またはその後でタンパク質沈殿は観察されなかったが、「塩析」現象を回避するため、該物質はゆっくりと希釈しなければならない。
【0118】
HIC方法は、実用範囲を確立するために移動相の塩濃度を変更することにより取り組んだ。回収%および酵素比活性に対する効果を、2通りの異なる精製手順を行なうことにより検討した。第1の実行では、充填および溶出中のクエン酸塩濃度を減少させ、一方、第2の実行では、充填中のクエン酸塩濃度は減少させたが、溶出バッファーのクエン酸塩濃度は増大させた。10Lバイオリアクター実行からQ−セファロースFFで得られたもの(133O−D−1005)を、等容量の50mM TRIS、0.5M クエン酸ナトリウム(pH7.0)で希釈し、25mM TRIS、250mMのクエン酸ナトリウム(pH7.0)で平衡化させたHICカラムに充填した。このカラムを25mM TRIS、50mMのクエン酸ナトリウム(pH7.0)で洗浄した。ほぼ51%の酵素活性が50mMのクエン酸ナトリウム画分において確認され、1%未満が通過画分および洗浄画分中に確認された。メインピークにおける酵素比活性は1.35×106U/mgであると算出された(NEG:010:175−186)。第2のHIC実行では、Q−セファロースFFで得られたものを、上記のようにして希釈および充填したが、25mM TRIS、100mMのクエン酸ナトリウム(pH7.0)を溶出バッファーとして使用した。この場合、50%の酵素活性が100mMクエン酸塩画分において確認され、2%未満が通過画分および洗浄画分中に確認された。100mMクエン酸塩画分における酵素比活性は2.5×106U/mgであった(NEG:010:163−174)。第1のHIC実行で観察された比活性のほうが低く、これは、溶出バッファー中のクエン酸塩濃度の減少により、通常該酵素と共溶出するストリップ中に見られるさらなるタンパク質がもたらされ、したがって最終純度が低下することを示した。対照的に、溶出バッファー中のクエン酸塩濃度の増大は純度を低下させないようであった。これらのデータに基づき、溶出バッファー中のクエン酸ナトリウムの濃度は、最終純度を損なうことなく75〜100mMの間で変更され得る。
【0119】
第2のタンジェンシャルフロー濾過(TFF2)
工程の説明:TFF2を用いて、ウイルス濾過の前に、フェニル−セファロースFFで得られたものの濃縮およびダイアフィルトレーションを行なう。HICで得られたものを直接TFFにさらし、溶出バッファー(25mM TRIS、75mMのクエン酸ナトリウム(pH7.0))中での長期保存による酵素の不活化を最小限に抑える。フェニル−セファロースFFで得られたものをほぼ3倍に濃縮し、ダイアフィルトレーションを行ない、ウイルス濾過に適したバッファー中に酵素を入れ、その後保存する。TFF2工程では、再生セルロースPLCTK 30kDa膜(Millipore)を取り付けた Pellicon 2 Moduleを使用する。
【0120】
研究開発:TFF2手順は、最初は、B3/A1−7クローンを用いて産生されるPAMのために開発された。該酵素は、種々の条件下の50mM TRIS、25mM NaCl、0.0005%TX−100(pH8.0)中で安定であることが示され、この理由のため、さらなる研究開発は試みなかった。
【0121】
C5467 CHOならし培地由来のPAMの代表的な小規模精製
10Lバイオリアクター実行からのCHO細胞ならし培地(500mL)の試料(00107BR、第15日)を8倍に濃縮し、50mM TRIS、0.001%TX−100(pH8.0)に対してダイアフィルトレーションを行なった。TFF1で得られたもののほぼ3分の1を、上記の精製プロセスの規模縮小形態を用いて精製した(NEG:009:219−249)。精製データを表8にまとめる。この精製実行のこの工程収率は、比較的不充分であり、実際、HIC工程後の酵素比活性は低下した。しかしながら、SDS−PAGE解析により、おおむねα−アミド化酵素は、ほぼ75kDaの単一のメジャーバンドに精製されることが示された(図12、レーン10)。ゲルおよびデンシトメトリースキャン(図13)により、酵素の純度は高く(ほぼ純度67%)、低分子量不純物はごく微量しか存在しないことが明白に示された。該酵素は多くの精製バッファー中で経時的に不活化され、したがって、該プロセスの実施は各工程間で遅滞なく進行させなければならない。精製プロセスにおけるばらつきは、全体収率および比活性を低下させる不活性酵素の存在に起因した。より重要なことには、精製および発酵の開発はともに同時進行させ、これにより、観察されたばらつきがある程度説明され得る。該産生規模における工程収率および比活性は、その後、おそらく該プロセスにおいて該物質をより高速で移動させ、画分を速やかにアッセイした結果、ずっと良好になることがわかった。
【0122】
【表8】
【0123】
PAM下流精製プロセスの概要
技術移転のパイロット施設に渡す情報を反映する各プロセス工程の精製手順を以下に詳述する。SDS−PAGEおよびデンシトメトリースキャンを含む代表的な小規模精製実行のデータを、表8および図11および12に含める。
【0124】
TFF1手順:清澄CHO細胞ならし培地を5倍に濃縮し、50mM TRIS、0.001%TX−100(pH8.0)で、最終導電率4〜6mSが達成されるまでダイアフィルトレーションを行なう。濾過中、膜貫通圧力を≦10psiに維持する。
【0125】
AEX手順:TFFによって濃縮/ダイアフィルトレーションした清澄CHO細胞ならし培地を、50mM TRIS、120mM NaCl、0.001%TX−100(pH8.0)で平衡化させたQ−セファロースFF(Amersham Biosciences)カラムに充填する。このカラムを180cm/時で実行し、UV吸光度を280nmにおいてモニターする。カラムをさらなる平衡バッファーで洗浄し、PAMをカラムから、50mM TRIS、225mM NaCl(pH8.0)で溶出する。カラムを2.0M NaClで清浄にし、1.0M NaOHで殺菌する。カラムは、各実行間では10mM NaOH中に保存する。
【0126】
HIC手順:Q−セファロースで得られたものを、等容量の50mM TRIS、600mMクエン酸塩(pH7.0)で希釈し、25mM TRIS、300mMクエン酸塩(pH7.0)で平衡化させたフェニル−セファロースFF(Amersham Biosciences)上に充填する。カラムを240cm/時で実行し、UV吸光度を280nmにおいてモニターする。カラムを25mM TRIS、300mMクエン酸塩(pH7.0)で洗浄し、PAMをカラムから、25mM TRIS、75mMクエン酸塩(pH7.0)で溶出する。カラムを、それぞれ25mM TRIS(pH7.0)および1.0M NaOHで清浄し、殺菌する。カラムは、各実行間では10mM NaOH中に保存する。
【0127】
TFF2手順:フェニル−セファロースFFで得られたものを最初にほぼ3倍に濃縮し、50mM TRIS、25mM NaCl、0.0005%TX−100(pH8.0)を用いてダイアフィルトレーションを行ない、ウイルス濾過に適した容量までさらに濃縮し、保存する。
【0128】
10L攪拌槽バイオリアクタープロセス実行
α−アミド化酵素の10L発酵および精製プロセスの概要
いくつかの10L攪拌槽バイオリアクター実行に従って精製α−AEを調製した。α−アミド化酵素を誘導するためのプロセス工程は、上記および図14に示すように、14日間の種菌相、17日間の発酵相および2日間の精製スキームであった。これらの実験で使用した培養培地は、タンパク質無含有CHO培地C5467(Sigma−Aldrich)であった。これらの実行のプロセスの詳細はすべて、適切なバッチの記録を見るとよい。
【0129】
プロセスフローダイアグラムである図14は、UGL 73−26/Mクローンを用いて産生されるα−AE/PAMのために開発した最終プロセス工程を示す。
【0130】
第1相−種菌
10L攪拌槽バイオリアクター用の種菌を調製するため、UGL 73−26/M MWCB00のバイアルの1つを解凍した。このクリオバイアルの細胞を取り出し、新鮮培地中に入れた。細胞ペレットを新たな培地中に再懸濁し、C5467 CHO培地を入れたスピナーフラスコに添加した。次の14日間、培養物を、400mLの培地+細胞を入れた4つのフラスコ内に増殖した(種菌スキーム図15を参照)。
【0131】
培養物を、種菌相l2の最後でのα−AE発現について試験した。第12日目、平均酵素活性濃度は>13,000U/mLであった(表9パネルC参照)、培養物および細胞バイアビリティの生細胞計数を各時点で行ない、種菌培養物を修正した。培養物のバイアビリティは、概ね、この相の間で95%よりも大きかった(表9パネルA参照)。種菌培養物の平均総生細胞計数は、第14日目で2.03×109であった(表9パネルB参照)。
【0132】
【表9−1】
【0133】
【表9−2】
【0134】
第2相−発酵
10Lのバイオリアクターは、各種菌相の終了時に開始した。バイオリアクターに、未使用タンパク質無含有培地中1×105細胞/mLでL−グルタミン(Sigma,C5467)を播種した。バイオリアクターパラメータを、下記の設定点;温度=37℃、RPM=60、pH=維持するpH設定点なしに設定し、pHは流動的にし(開始時、pHはpH7.5より大きくならなりようにする)、DO設定点は70%DOにする。未使用培地の溶存酸素濃度は、70%より大きくし、バイオリアクターの溶存酸素濃度は70%まで流動的にさせ、次いで、該設定点に維持した。バイオリアクターには、第5、10および14日目に2g/Lのグルコースを補充した。第17日目にならし培地を収集した。収集物質をMillipore Opticap Filterに通して清澄にした。後述する最初の2つの発酵では、第17日目ではなく第18日目に2131−D−1003および2131−D−1004を収集した。培養物の最大細胞密度は第10〜17/18日目の間で達成され、維持された。これらの発酵の平均最大細胞密度は1.5−1.6×106細胞/mLであった(表10パネルB)。培養物のバイアビリティは、培養の最初の14日間で>80%であり、収集した日の平均バイアビリティは76.0%であった(表10パネルA)。培養物の産生性を、収集時、第17/18日目に評価した。平均清澄収集物は378,567単位のα−AE/mLを含んでいた(表10パネルC)。
【0135】
【表10】
【0136】
第3相−精製
該精製プロセスは、上記のような5つの異なる工程を有する。清澄収集物を濃縮し、50mM TRIS、0.001%TX−100(pH8.0)に対してダイアフィルトレーションした。酵素物質を平均してほぼ4倍に濃縮した(データ示さず。バッチの記録参照)。このプロセス物質をQ−セファロースカラムに適用し、225mM NaClを含有する50mM TRIS、0.001%Tritonバッファー中でのNaCl段階的勾配に従って溶出した。酵素溶液を50mM TRIS、600mMクエン酸(pH7.0)で希釈し、フェニルセファロースカラムに適用した。α−アミド化酵素をカラムから、クエン酸塩の工程勾配に従って溶出した。酵素物質を最終容量ほぼ1Lまで濃縮した。6つの酵素バッチのうち4つをウイルス除去フィルターに通して処理した。2つのバッチはMillipore NFPフィルターに通して処理し(1330−D−1005および1330−D−1006)、2つのバッチは、Pall Trincor DV50フィルターに通した(1330−1009および1330−1010)。各々のプロセス工程のデータを以下の表11に示す。
【0137】
最初の工程TFF1は、α−アミド化酵素活性単位の保持に関してほぼ定量的な工程であった。10Lバイオリアクター実行からのTFF濃縮物は、1.60×106単位/mgタンパク質の平均比活性を有する(表11)。Q−セファロースクロマトグラフィーカラムに適用したTFF1濃縮物は、カラムからほぼ3Lで溶出された。酵素比活性は、このプロセス工程で、3.33×106単位/mgまでほぼ2倍に増大した。Q−セファロース溶出液のタンパク質濃度および酵素活性濃度は非常に一貫していた(表11参照)。フェニル−セファロースクロマトグラフィー後、平均酵素比活性は、3.33×106から4.96×106単位/mgタンパク質まで増大した。フェニル−セファロース溶出液を、第2のTFF工程を用いて1Lまで濃縮した。この濃度工程後、比活性に変化はなかった(表11)。一部の酵素バッチからのTFF2濃縮物を、ウイルス除去フィルターに適用した。4つのうち3つの酵素バッチの比活性が低下した(表11)。最終精製酵素の平均比活性は、350,000単位/mgから4.61×106単位/mgタンパク質低下した。この比活性の低下は、おそらく一部の該酵素の不活化によるものであった。
【0138】
【表11−1】
【0139】
【表11−2】
【0140】
精製プロセスの各工程の一貫性を示すこのデータは重要である。しかしながら、本当に重要なのは、該プロセスの各工程での所望の生成物の回収パーセントである。表12は、α−アミド化酵素精製プロセスの各工程での回収パーセント、および4回または5回のプロセス工程後のプロセス全体収率を示す。表12は、酵素が両方のTFF工程から定量的に回収されたことを示す。クロマトグラフィー工程、Q−セファロースおよびフェニルセファロースでの平均回収パーセントは、それぞれ65.3%および72.0%である。試験した両方のウイルス除去フィルターで約80%のα−AE活性が回収された。TFF2またはウイルス濾過を通した清澄回収物質の酵素精製プロセスの全体収率は、それぞれ40.8%または38.8%であった。
【0141】
【表12】
【0142】
結論
高レベルのα−AE活性を発現する安定な充分特性化されたCHO細胞株UGL 73−26/Mが開発された。高レベルの酵素発現は、非動物供給源の低タンパク質含有組織培養培地C5467(Sigma)を用いる17日間のバッチ発酵プロセスにおいて達成される。臨界発酵パラメータ(例えば、pH、DOおよびグルコース濃度など)を検討し、最適化した。また、酵素がほぼ均一に精製され得るロバストな2工程の下流精製プロセスも開発した。この発酵および精製プロセスの一貫性は、製造レベルへの規模拡大に充分適している。
【0143】
以下に、本発明の細胞によって発現されるPAMを用いたアミド化生成物の産生を示す実施例を示す。
【0144】
実施例1:ペプチジルグリシンα−アミド化モノオキシゲナーゼを用いたグリシン増殖副甲状腺ホルモン断片のアミド化対応物への変換
ピルビン酸塩を用いたrhPTH(l−34)Gly35−OHのアミド化
rhPTH(l−34)Gly35−OHのアミド化に用いた成分および終濃度を表12に示す。アミド化の簡単な説明は以下の通りである。
【0145】
【表13】
【0146】
・ほぼ12.4gのrhPTH(l−34)Gly35−OHを含む1,900mLの25mM MES、200mM NaCl(pH 6.0)を、攪拌器およびガススパージャーを取り付けたガラス槽内に供給した。
【0147】
・この溶液に、以下の成分:3025mLの水、741mLの250mM MES pH 6.3、1.03mLの3mM 硫酸銅、124mLのヨウ化カリウム、62mLの190プルーフエタノール、124mLの400mMピルビン酸ナトリウムおよび124mLの100mMのアスコルビン酸ナトリウムを記載順に添加した。
【0148】
・反応槽を水浴中に入れ、反応混合物を25〜27℃まで攪拌しながら昇温させた。
・反応混合物のpHを21mLの2M HClによって5.8に調整した。酸素のスパージングを開始したが、スパージング速度は、反応混合物の過剰な起泡が回避されるように調整した。
【0149】
・47mLのPAMを添加し、反応混合物を25〜27℃で4時間35分間インキュベートした(インキュベーション期間中、酸素スパージングを行なった)。
【0150】
・反応混合物を74mLの2MのHClでpH2.4に酸性化した。
この実施例で用いたPAM酵素は、上記のようにして構築し、本明細書においてUGL 73−26/Mと表示する本発明の好ましいCHO K1細胞から発現された。また、この細胞株を用いて上記のATCC寄託物を提供した。
【0151】
グリシン増殖前駆体は、既知の供給源から得られるものであってもよく、既知の様式で生成されたものであってもよい。例えば、これは、米国特許第6,103,495号(その実施例1〜2)に記載のものと同様の様式で発酵によって生成され、アミド化の前に米国特許第6,103,495号(その実施例3)に記載のようにして精製され得る。上記の本発明のアミド化に用いた具体的な前駆体は、係属中の米国特許出願第11/076,260号(2005年3月9日出願および2005年10月6日に公開番号US 2005/0221442で公開)の細胞株によって発現させた。
【0152】
アミド化に使用する酵素がペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼ(PHM)である場合、PHMをPAMに変えて、上記のものと同じ反応混合物が使用され得る。また、4〜6時間のインキュベーション期間の最後に、反応混合物のpHを塩基の添加によって8〜9に上昇させる。反応混合物は、反応終了前にさらに4〜8時間攪拌する。PHMは、PAMを本明細書に教示したとおりに発現させた後、PHM触媒ドメインをPAM分子の残部から分離することにより得られ得る。あるいはまた、本明細書において使用されるベクターは、PHMの後でPALの前の触媒ドメインで翻訳が停止されるように修飾されていてもよい。あるいはまた、該ベクターは、第1の例では、上記のPAMコード領域の代わりにPHMコード領域を用いて構築され得る。PHMは、PAM のN末端部分のみ(最初の約40kDa)を発現させることにより得られ得る。PAMおよびその触媒ドメインの位置は文献に報告されている。かかる任意のPAMまたはPHMは、本発明に従って有用であると考えられる。例えば、Mizunoら,BBRC Vol.148,No.2,pp.546−52(1987)(その開示は引用により本明細書に組み込まれる)を参照のこと。特にMizunoの「AE1」を参照のこと。カエルの皮膚は、天然状態でPHMを発現することが知られている。
【0153】
実施例2:アミド化後精製
陽イオン交換(CEX)クロマトグラフィー
残留rhPTH(l−34)Gly35−OHからのrhPTH(l−34)−NH2の精製を、CEXクロマトグラフィーを用いて行なった。CEXクロマトグラフィー方法の簡単な説明は以下の通りである。アミド化で得られたものを酸性化して25mM MES(pH 6.5)で平衡化させたToyopearl SP650M(Tosoh Bioscience LLC)カラム(9cm×19cm)上に充填した。カラムを180cm/時で実行し、カラム流出液のUV吸光度を280nmでモニターした。このカラムを25mM MES(pH6.5)で、カラム流出液pHのpHが6.5に戻るまで洗浄した。このカラムを25mM MES,80mM NaCl(pH 6.5)で、洗浄ピークが完全に溶出され、安定なUVベースラインが得られるまで洗浄した。生成物rhPTH(l−34)−NH2をカラムから、25mM MES,200mM NaCl(pH 6.5)で溶出した。UVピークをすべて回収したが、プール基準を決定するため、画分をRP−HPLCによってスクリーニングした。
【0154】
逆相(RP)クロマトグラフィー
RPクロマトグラフィーを用い、塩形態のペプチドを塩化物から酢酸塩に交換した;RPクロマトグラフィーでは、ほんのわずかのペプチド精製しかもたらされない。CEXクロマトグラフィーで得られたものを3容量の333mM酢酸ナトリウムで希釈し、充分混合した。混合物は、充填前に室温で75分間放置した。酢酸塩希釈試料を、250mM酢酸ナトリウム(pH7.5)で平衡化させた Amberchrom CG300 M(Tosoh Bioscience LLC)カラム(6cm×17cm)上に充填した。カラムを180cm/時で実行し、カラム流出液のUV吸光度を280nmでモニターした。カラムを250mM酢酸ナトリウム(pH7.5)で60分間洗浄した。カラムを0.1%酢酸中で平衡化させた。生成物rhPTH(l−34)−NH2をカラムから、0.1%酢酸、40%エタノールで溶出した。UVピークをすべて回収した。
【0155】
rhPTH(l−34)−NH2の特性化
RPクロマトグラフィーで得られたものを濃縮し、凍結乾燥によって白色の綿毛様粉末にし、11.8g(アミド化から95%の全体収率)のrhPTH(l−34)−NH2を得た。rhPTH(l−34)−NH2の分子質量は、エレクトロスプレーイオン化質量分析(ESI−MS)によって4,116.9Daであると決定され、これは、平均分子質量の計算値4,116.8Daと整合した。
【0156】
本発明は、特別の実施形態に関して説明されており、多くの他の変形および修飾、ならびに他の使用は、当業者に明らかである。したがって、本発明は、本明細書における特異的開示、または付け加えられた特許請求の範囲に限定されない。
【特許請求の範囲】
【請求項1】
(A)第1、第2および第3の発現ベクターの存在下で、潜在的宿主細胞をトランスフェクトする工程であって、前記第1のベクターが、第1の選択可能なマーカーをコードするコード領域を含み、前記第2のベクターが、第2の選択可能なマーカーをコードするコード領域を含み、前記第3のベクターが、ペプチジルグリシンα−アミド化モノオキシゲナーゼをコードするコード領域を含み、前記第1のベクターに対する前記第3のベクターの濃度比が少なくとも3:1であり、前記第2のベクターに対する前記第3のベクターの濃度比が少なくとも3:1である工程;
(B)工程(A)で得られた細胞に選択圧をかけ、前記第1のベクターでトランスフェクトされた細胞を選択する工程;
(C)工程(B)で得られた細胞に選択圧をかけ、前記第2のベクターでトランスフェクトされた細胞を選択する工程;
(D)工程(C)で得られた細胞を限界希釈し、ペプチジルグリシンα−アミド化モノオキシゲナーゼ(monoxygenase)を発現する細胞を選択する工程
を含む、ペプチジルグリシンα−アミド化モノオキシゲナーゼの発現のための細胞株の調製方法。
【請求項2】
工程(D)で得られた細胞に増幅した選択圧をかけることにより、ペプチジルグリシンα−アミド化モノオキシゲナーゼを増幅する工程をさらに含む、請求項1に記載の方法。
【請求項3】
前記第1の選択可能なマーカーが、ネオマイシン耐性をコードする遺伝子である、請求項1に記載の方法。
【請求項4】
前記第2の選択可能なマーカーが、ジヒドロ葉酸還元酵素をコードする遺伝子である、請求項1に記載の方法。
【請求項5】
前記選択可能なマーカーの少なくとも一方がジヒドロ葉酸還元酵素をコードする遺伝子であり、前記増幅がメトトレキサートの存在下で行なわれる、請求項2に記載の方法。
【請求項6】
前記第1のベクターに対する前記第3のベクターの濃度比が少なくとも10:1であり、前記第2のベクターに対する前記第3のベクターの濃度比が少なくとも10:1である、請求項1に記載の方法。
【請求項7】
前記第3のベクターが、リボソーム結合部位、プロモーターおよび前記プロモーターの上流のSV40エンハンサーを含む制御領域に操作可能に連結されたペプチジルグリシンα−アミド化モノオキシゲナーゼをコードする核酸を有するコード領域を含む、請求項1に記載の方法。
【請求項8】
前記第3のベクターが、リボソーム結合部位、ヒトメタロチオニンを含む制御領域、プロモーターおよび前記プロモーターの上流のエンハンサーに操作可能に連結されたペプチジルグリシンα−アミド化モノオキシゲナーゼをコードする核酸を有するコード領域を含む、請求項1に記載の方法。
【請求項9】
前記エンハンサーがSV40エンハンサーである、請求項8に記載の方法。
【請求項10】
前記潜在的宿主細胞がCHO K1細胞を含む、請求項1に記載の方法。
【請求項11】
請求項1に記載の方法で得られる細胞株。
【請求項12】
(A)ペプチジルグリシンα−アミド化モノオキシゲナーゼの不純試料を陰イオン交換クロマトグラフィーにかける工程であって、溶出が均一濃度である工程;
(B)工程(A)の溶離液を疎水性相互作用クロマトグラフィーにかける工程であって、硫酸アンモニウムが使用されず、溶出が均一濃度である工程
を含む、ペプチジルグリシンα−アミド化モノオキシゲナーゼを発現および培養培地中への分泌後に精製する方法。
【請求項13】
工程(B)においてカラム結合を助長するために、クエン酸ナトリウムが用いられる、請求項12に記載の方法。
【請求項14】
ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼを発現させるための発現ベクターでトランスフェクトされたCHO K1細胞。
【請求項15】
前記発現ベクターが、リボソーム結合部位、プロモーターおよび前記プロモーターの上流のSV40エンハンサーを含む制御領域に操作可能に連結されたペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼをコードする核酸を有するコード領域を有する、請求項14に記載の形質転換細胞。
【請求項16】
前記発現ベクターが、リボソーム結合部位およびヒトメタロチオニンIIAプロモーターを有する制御領域に操作可能に連結されたペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼをコードする核酸を有するコード領域を有する、請求項14に記載の形質転換細胞。
【請求項17】
前記ベクターが、前記プロモーターの上流にエンハンサーをさらに含む、請求項16に記載の形質転換細胞。
【請求項18】
前記エンハンサーがSV40エンハンサーである、請求項17に記載の形質転換細胞。
【請求項19】
リボソーム結合部位、プロモーターおよび前記プロモーターの上流のSV40エンハンサーを含む制御領域に操作可能に連結されたペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼをコードする核酸を有するコード領域を有する組換え発現ベクター。
【請求項20】
リボソーム結合部位およびヒトメタロチオニンIIAプロモーターを含む制御領域に操作可能に連結されたペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼをコードする核酸を有するコード領域を有する組換え発現ベクター。
【請求項21】
前記ベクターが前記プロモーターの上流にエンハンサーをさらに含む、請求項40に記載のベクター。
【請求項22】
前記エンハンサーがSV40エンハンサーである、請求項21に記載のベクター。
【請求項23】
(A)第1、第2および第3の発現ベクターの存在下で、潜在的宿主細胞をトランスフェクトする工程であって、前記第1のベクターが、第1の選択可能なマーカーをコードするコード領域を含み、前記第2のベクターが、第2の選択可能なマーカーをコードするコード領域を含み、前記第3のベクターが、ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼをコードするコード領域を含み、前記第1のベクターに対する前記第3のベクターの濃度比が少なくとも3:1であり、前記第2のベクターに対する前記第3のベクターの濃度比が少なくとも3:1である工程;
(B)工程(A)で得られた細胞に選択圧をかけ、前記第1のベクターでトランスフェクトされた細胞を選択する工程;
(C)工程(B)で得られた細胞に選択圧をかけ、前記第2のベクターでトランスフェクトされた細胞を選択する工程;
(D)工程(C)で得られた細胞を限界希釈し、ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼを発現する細胞を選択する工程
を含む、ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼの発現のための細胞株の調製方法。
【請求項24】
工程(D)で得られた細胞に増幅した選択圧をかけることにより、ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼを増幅する工程をさらに含む、請求項23記載の方法。
【請求項25】
前記第1の選択可能なマーカーが、ネオマイシン耐性をコードする遺伝子である、請求項23記載の方法。
【請求項26】
前記第2の選択可能なマーカーが、ジヒドロ葉酸還元酵素をコードする遺伝子である、請求項23記載の方法。
【請求項27】
前記選択可能なマーカーの少なくとも一方がジヒドロ葉酸還元酵素をコードする遺伝子であり、前記増幅がメトトレキサートの存在下で行なわれる、請求項24記載の方法。
【請求項28】
前記第1のベクターに対する前記第3のベクターの濃度比が少なくとも10:1であり、前記第2のベクターに対する前記第3のベクターの濃度比が少なくとも10:1である、請求項23記載の方法。
【請求項29】
前記第3のベクターが、リボソーム結合部位、プロモーターおよび前記プロモーターの上流のSV40エンハンサーを含む制御領域に操作可能に連結されたペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼをコードする核酸を有するコード領域を含む、請求項23記載の方法。
【請求項30】
前記第3のベクターが、リボソーム結合部位、ヒトメタロチオニンを含む制御領域、プロモーターおよび前記プロモーターの上流のエンハンサーに操作可能に連結されたペプチジルグリシンα−アミド化モノオキシゲナーゼをコードする核酸を有するコード領域を含む、請求項23記載の方法。
【請求項31】
前記エンハンサーがSV40エンハンサーである、請求項30記載の方法。
【請求項32】
前記潜在的宿主細胞がCHO K1細胞を含む、請求項23記載の方法。
【請求項33】
請求項23〜32のいずれか1項に記載の方法で得られる細胞株。
【請求項34】
請求項19〜22のいずれか1項に記載のベクターでトランスフェクトされた宿主細胞を含む細胞株。
【請求項35】
請求項14〜18のいずれか1項に記載の細胞または細胞株によって発現されるペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼ。
【請求項36】
グリシン残基を有し、遊離酸の形態であり、カルボニル基に結合された前駆体を、請求項35に記載のペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼの存在下で反応させた後、ルイス塩基またはペプチジルα−ヒドロキシグリシンα−アミド化リアーゼの存在下で、さらに反応させることを含む、アミド化生成物のインビトロ産生方法。
【請求項37】
前記前駆体が、C末端グリシンを有するペプチドである、請求項36記載の方法。
【請求項38】
請求項36記載の方法で得られるアミド化生成物。
【請求項39】
(A)ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼの不純試料を陰イオン交換クロマトグラフィーにかける工程であって、溶出が均一濃度である工程;
(B)工程(A)の溶離液を疎水性相互作用クロマトグラフィーにかける工程であって、硫酸アンモニウムが使用されず、溶出が均一濃度である工程
を含む、ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼを発現および培養培地中への分泌後に精製する方法。
【請求項40】
工程(B)においてカラム結合を助長するために、クエン酸ナトリウムが用いられる、請求項39記載の方法。
【請求項41】
(A)グリシン残基を有し、遊離酸の形態であり、カルボニル基に結合された前駆体を、ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼの存在下で反応させることにより、ヒドロキシル化中間体を形成すること;および
(B)前記中間体を、ルイス塩基またはペプチジルα−ヒドロキシグリシンα−アミド化リアーゼのいずれかと同時またはその後反応させること
を含む、アミド化生成物のインビトロ産生方法であって
前記ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼが、請求項14〜18のいずれか1項に記載の細胞または細胞株によって発現された、産生方法。
【請求項42】
工程(B)でルイス塩基が用いられる、請求項41記載の方法。
【請求項1】
(A)第1、第2および第3の発現ベクターの存在下で、潜在的宿主細胞をトランスフェクトする工程であって、前記第1のベクターが、第1の選択可能なマーカーをコードするコード領域を含み、前記第2のベクターが、第2の選択可能なマーカーをコードするコード領域を含み、前記第3のベクターが、ペプチジルグリシンα−アミド化モノオキシゲナーゼをコードするコード領域を含み、前記第1のベクターに対する前記第3のベクターの濃度比が少なくとも3:1であり、前記第2のベクターに対する前記第3のベクターの濃度比が少なくとも3:1である工程;
(B)工程(A)で得られた細胞に選択圧をかけ、前記第1のベクターでトランスフェクトされた細胞を選択する工程;
(C)工程(B)で得られた細胞に選択圧をかけ、前記第2のベクターでトランスフェクトされた細胞を選択する工程;
(D)工程(C)で得られた細胞を限界希釈し、ペプチジルグリシンα−アミド化モノオキシゲナーゼ(monoxygenase)を発現する細胞を選択する工程
を含む、ペプチジルグリシンα−アミド化モノオキシゲナーゼの発現のための細胞株の調製方法。
【請求項2】
工程(D)で得られた細胞に増幅した選択圧をかけることにより、ペプチジルグリシンα−アミド化モノオキシゲナーゼを増幅する工程をさらに含む、請求項1に記載の方法。
【請求項3】
前記第1の選択可能なマーカーが、ネオマイシン耐性をコードする遺伝子である、請求項1に記載の方法。
【請求項4】
前記第2の選択可能なマーカーが、ジヒドロ葉酸還元酵素をコードする遺伝子である、請求項1に記載の方法。
【請求項5】
前記選択可能なマーカーの少なくとも一方がジヒドロ葉酸還元酵素をコードする遺伝子であり、前記増幅がメトトレキサートの存在下で行なわれる、請求項2に記載の方法。
【請求項6】
前記第1のベクターに対する前記第3のベクターの濃度比が少なくとも10:1であり、前記第2のベクターに対する前記第3のベクターの濃度比が少なくとも10:1である、請求項1に記載の方法。
【請求項7】
前記第3のベクターが、リボソーム結合部位、プロモーターおよび前記プロモーターの上流のSV40エンハンサーを含む制御領域に操作可能に連結されたペプチジルグリシンα−アミド化モノオキシゲナーゼをコードする核酸を有するコード領域を含む、請求項1に記載の方法。
【請求項8】
前記第3のベクターが、リボソーム結合部位、ヒトメタロチオニンを含む制御領域、プロモーターおよび前記プロモーターの上流のエンハンサーに操作可能に連結されたペプチジルグリシンα−アミド化モノオキシゲナーゼをコードする核酸を有するコード領域を含む、請求項1に記載の方法。
【請求項9】
前記エンハンサーがSV40エンハンサーである、請求項8に記載の方法。
【請求項10】
前記潜在的宿主細胞がCHO K1細胞を含む、請求項1に記載の方法。
【請求項11】
請求項1に記載の方法で得られる細胞株。
【請求項12】
(A)ペプチジルグリシンα−アミド化モノオキシゲナーゼの不純試料を陰イオン交換クロマトグラフィーにかける工程であって、溶出が均一濃度である工程;
(B)工程(A)の溶離液を疎水性相互作用クロマトグラフィーにかける工程であって、硫酸アンモニウムが使用されず、溶出が均一濃度である工程
を含む、ペプチジルグリシンα−アミド化モノオキシゲナーゼを発現および培養培地中への分泌後に精製する方法。
【請求項13】
工程(B)においてカラム結合を助長するために、クエン酸ナトリウムが用いられる、請求項12に記載の方法。
【請求項14】
ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼを発現させるための発現ベクターでトランスフェクトされたCHO K1細胞。
【請求項15】
前記発現ベクターが、リボソーム結合部位、プロモーターおよび前記プロモーターの上流のSV40エンハンサーを含む制御領域に操作可能に連結されたペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼをコードする核酸を有するコード領域を有する、請求項14に記載の形質転換細胞。
【請求項16】
前記発現ベクターが、リボソーム結合部位およびヒトメタロチオニンIIAプロモーターを有する制御領域に操作可能に連結されたペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼをコードする核酸を有するコード領域を有する、請求項14に記載の形質転換細胞。
【請求項17】
前記ベクターが、前記プロモーターの上流にエンハンサーをさらに含む、請求項16に記載の形質転換細胞。
【請求項18】
前記エンハンサーがSV40エンハンサーである、請求項17に記載の形質転換細胞。
【請求項19】
リボソーム結合部位、プロモーターおよび前記プロモーターの上流のSV40エンハンサーを含む制御領域に操作可能に連結されたペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼをコードする核酸を有するコード領域を有する組換え発現ベクター。
【請求項20】
リボソーム結合部位およびヒトメタロチオニンIIAプロモーターを含む制御領域に操作可能に連結されたペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼをコードする核酸を有するコード領域を有する組換え発現ベクター。
【請求項21】
前記ベクターが前記プロモーターの上流にエンハンサーをさらに含む、請求項40に記載のベクター。
【請求項22】
前記エンハンサーがSV40エンハンサーである、請求項21に記載のベクター。
【請求項23】
(A)第1、第2および第3の発現ベクターの存在下で、潜在的宿主細胞をトランスフェクトする工程であって、前記第1のベクターが、第1の選択可能なマーカーをコードするコード領域を含み、前記第2のベクターが、第2の選択可能なマーカーをコードするコード領域を含み、前記第3のベクターが、ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼをコードするコード領域を含み、前記第1のベクターに対する前記第3のベクターの濃度比が少なくとも3:1であり、前記第2のベクターに対する前記第3のベクターの濃度比が少なくとも3:1である工程;
(B)工程(A)で得られた細胞に選択圧をかけ、前記第1のベクターでトランスフェクトされた細胞を選択する工程;
(C)工程(B)で得られた細胞に選択圧をかけ、前記第2のベクターでトランスフェクトされた細胞を選択する工程;
(D)工程(C)で得られた細胞を限界希釈し、ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼを発現する細胞を選択する工程
を含む、ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼの発現のための細胞株の調製方法。
【請求項24】
工程(D)で得られた細胞に増幅した選択圧をかけることにより、ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼを増幅する工程をさらに含む、請求項23記載の方法。
【請求項25】
前記第1の選択可能なマーカーが、ネオマイシン耐性をコードする遺伝子である、請求項23記載の方法。
【請求項26】
前記第2の選択可能なマーカーが、ジヒドロ葉酸還元酵素をコードする遺伝子である、請求項23記載の方法。
【請求項27】
前記選択可能なマーカーの少なくとも一方がジヒドロ葉酸還元酵素をコードする遺伝子であり、前記増幅がメトトレキサートの存在下で行なわれる、請求項24記載の方法。
【請求項28】
前記第1のベクターに対する前記第3のベクターの濃度比が少なくとも10:1であり、前記第2のベクターに対する前記第3のベクターの濃度比が少なくとも10:1である、請求項23記載の方法。
【請求項29】
前記第3のベクターが、リボソーム結合部位、プロモーターおよび前記プロモーターの上流のSV40エンハンサーを含む制御領域に操作可能に連結されたペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼをコードする核酸を有するコード領域を含む、請求項23記載の方法。
【請求項30】
前記第3のベクターが、リボソーム結合部位、ヒトメタロチオニンを含む制御領域、プロモーターおよび前記プロモーターの上流のエンハンサーに操作可能に連結されたペプチジルグリシンα−アミド化モノオキシゲナーゼをコードする核酸を有するコード領域を含む、請求項23記載の方法。
【請求項31】
前記エンハンサーがSV40エンハンサーである、請求項30記載の方法。
【請求項32】
前記潜在的宿主細胞がCHO K1細胞を含む、請求項23記載の方法。
【請求項33】
請求項23〜32のいずれか1項に記載の方法で得られる細胞株。
【請求項34】
請求項19〜22のいずれか1項に記載のベクターでトランスフェクトされた宿主細胞を含む細胞株。
【請求項35】
請求項14〜18のいずれか1項に記載の細胞または細胞株によって発現されるペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼ。
【請求項36】
グリシン残基を有し、遊離酸の形態であり、カルボニル基に結合された前駆体を、請求項35に記載のペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼの存在下で反応させた後、ルイス塩基またはペプチジルα−ヒドロキシグリシンα−アミド化リアーゼの存在下で、さらに反応させることを含む、アミド化生成物のインビトロ産生方法。
【請求項37】
前記前駆体が、C末端グリシンを有するペプチドである、請求項36記載の方法。
【請求項38】
請求項36記載の方法で得られるアミド化生成物。
【請求項39】
(A)ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼの不純試料を陰イオン交換クロマトグラフィーにかける工程であって、溶出が均一濃度である工程;
(B)工程(A)の溶離液を疎水性相互作用クロマトグラフィーにかける工程であって、硫酸アンモニウムが使用されず、溶出が均一濃度である工程
を含む、ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼを発現および培養培地中への分泌後に精製する方法。
【請求項40】
工程(B)においてカラム結合を助長するために、クエン酸ナトリウムが用いられる、請求項39記載の方法。
【請求項41】
(A)グリシン残基を有し、遊離酸の形態であり、カルボニル基に結合された前駆体を、ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼの存在下で反応させることにより、ヒドロキシル化中間体を形成すること;および
(B)前記中間体を、ルイス塩基またはペプチジルα−ヒドロキシグリシンα−アミド化リアーゼのいずれかと同時またはその後反応させること
を含む、アミド化生成物のインビトロ産生方法であって
前記ペプチジルグリシンα−ヒドロキシル化モノオキシゲナーゼが、請求項14〜18のいずれか1項に記載の細胞または細胞株によって発現された、産生方法。
【請求項42】
工程(B)でルイス塩基が用いられる、請求項41記載の方法。
【図4】

【図6】
【図7】
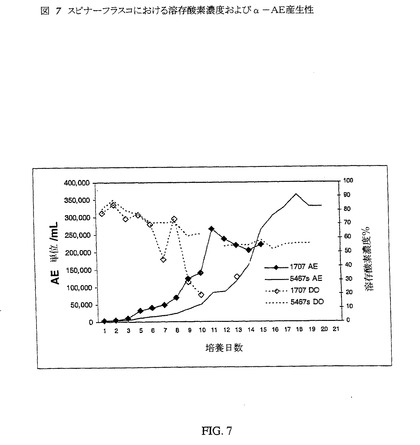
【図8】
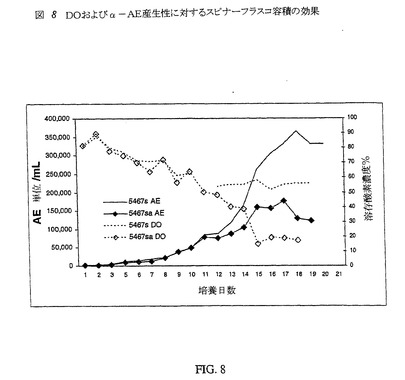
【図9】
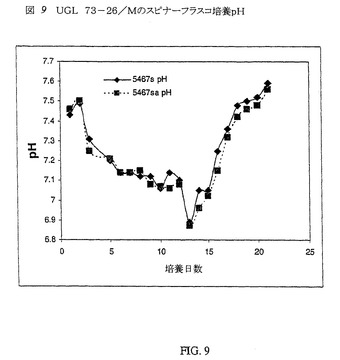
【図10】
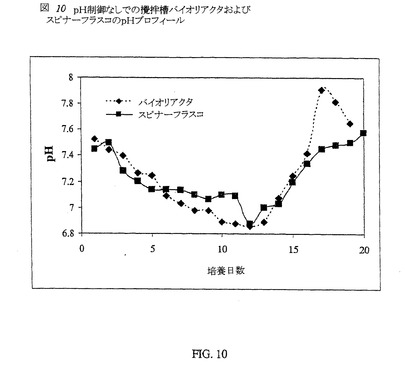
【図11】
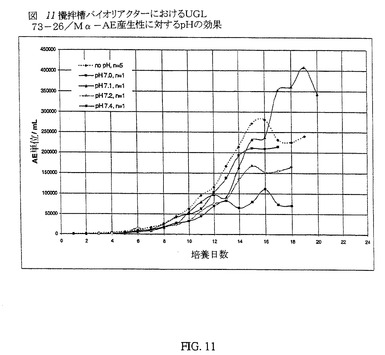
【図14】
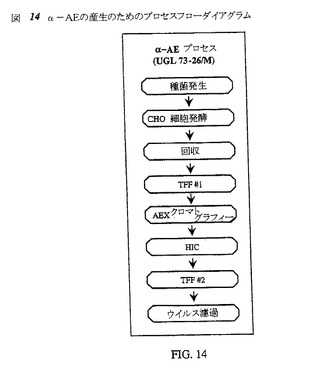
【図15】
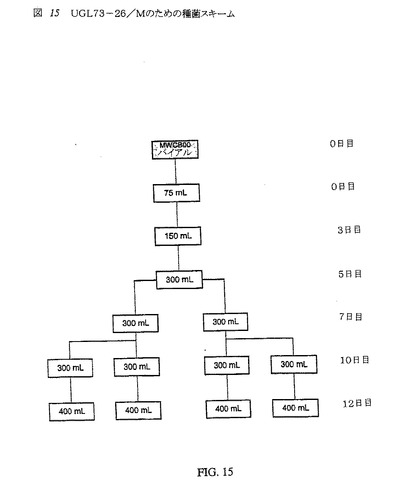
【図1】
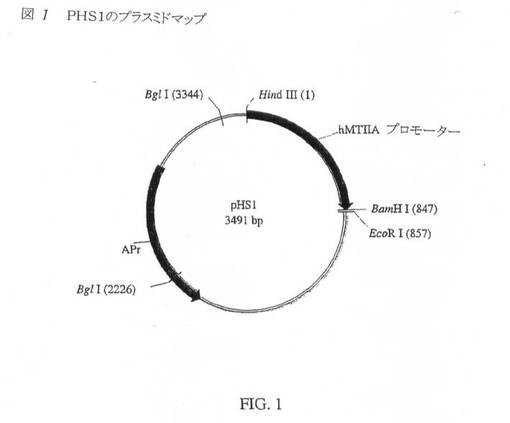
【図2】
【図3】
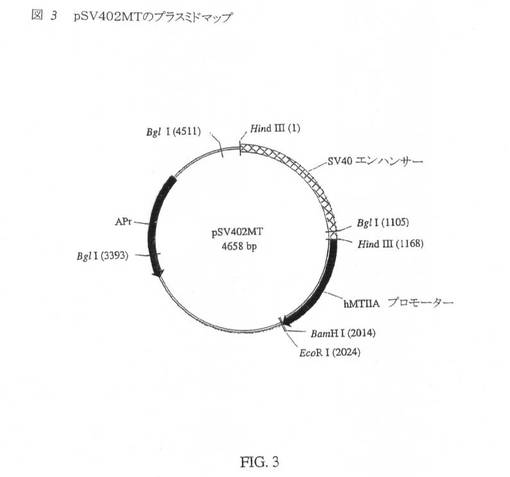
【図5】
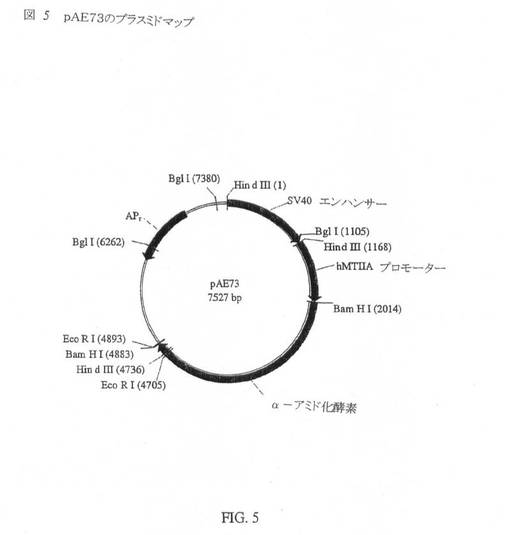
【図12】
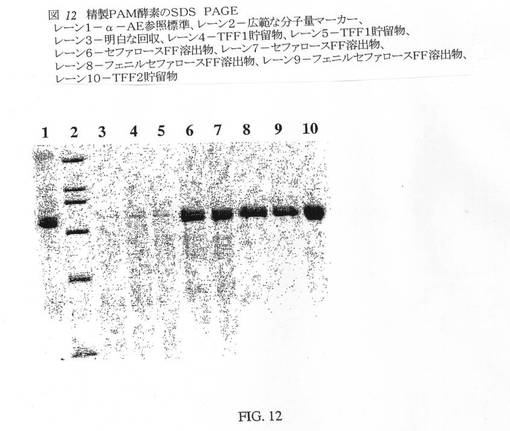
【図13】
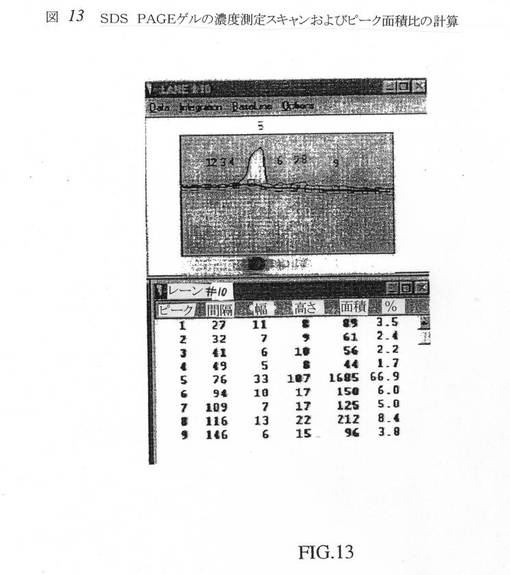
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図14】
【図15】
【図1】
【図2】
【図3】
【図5】
【図12】
【図13】
【公開番号】特開2011−139714(P2011−139714A)
【公開日】平成23年7月21日(2011.7.21)
【国際特許分類】
【出願番号】特願2011−94071(P2011−94071)
【出願日】平成23年4月20日(2011.4.20)
【分割の表示】特願2008−518476(P2008−518476)の分割
【原出願日】平成18年6月23日(2006.6.23)
【出願人】(503195300)ユニジーン・ラボラトリーズ・インコーポレーテッド (17)
【Fターム(参考)】
【公開日】平成23年7月21日(2011.7.21)
【国際特許分類】
【出願日】平成23年4月20日(2011.4.20)
【分割の表示】特願2008−518476(P2008−518476)の分割
【原出願日】平成18年6月23日(2006.6.23)
【出願人】(503195300)ユニジーン・ラボラトリーズ・インコーポレーテッド (17)
【Fターム(参考)】
[ Back to top ]