
アミロイドβ線維化阻害ペプチド
【課題】アミロイドβペプチドのミミックペプチドとして機能し、アミロイドβペプチドの線維化を阻害しうるペプチドを提供する。
【解決手段】次式(III):
Z1-X13-Gly-X14-X15-Pro-Trp-Met-Z2 (III)
(式中、X13、X14及びX15は同一、又は異なってα−アミノ酸を表し、Z1及びZ2は同一、又は異なってシステイン又はセリンを表し、Z1及びZ2がシステインを表す場合、これらの間で架橋されていてもよい。)
で示されるアミノ酸配列を含む、アミノ酸残基数9〜30のペプチド;並びに前記ペプチドを含有する医薬組成物及びアミロイドβ線維化阻害剤。
【解決手段】次式(III):
Z1-X13-Gly-X14-X15-Pro-Trp-Met-Z2 (III)
(式中、X13、X14及びX15は同一、又は異なってα−アミノ酸を表し、Z1及びZ2は同一、又は異なってシステイン又はセリンを表し、Z1及びZ2がシステインを表す場合、これらの間で架橋されていてもよい。)
で示されるアミノ酸配列を含む、アミノ酸残基数9〜30のペプチド;並びに前記ペプチドを含有する医薬組成物及びアミロイドβ線維化阻害剤。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、アルツハイマー病の予防・治療剤として有用なアミロイドβ線維化阻害ペプチドに関する。
【背景技術】
【0002】
近年、老人人口の増加に伴い、老人性痴呆症の治療に有効な医薬品の開発が強く望まれている。老人性痴呆症の代表的疾患であるアルツハイマー病は、脳の萎縮、老人斑の沈着及び神経原線維変化を特徴とする神経変性疾患で、アミロイドβペプチドの形状変化と、それに伴う線維化による不溶化分子が神経細胞に沈着し、その毒性により神経細胞の死が誘導され、発症する。
【0003】
アミロイドβペプチド(Aβ)はニューロンアミロイド前駆体蛋白質からβセクレターゼなどによる切断で生じる分解物で、Aβ1-40とAβ1-42の2種が生成するが、Aβ1-42の方がより凝集しやすく、疾患及び神経毒性と相関することが報告されている。
【0004】
アミロイドβペプチドの形状変化及び線維化を阻害できれば、アルツハイマー病の発症を阻止することができる。
【0005】
特許文献1(国際公開第2005/105998号パンフレット)には、Aβ1-42と特異的に結合し、その線維化反応を阻害する活性を有する一本鎖抗体がアルツハイマー病の予防・治療剤として有用であることが記載されている。
【0006】
しかしながら、抗体は高分子の蛋白質であるため高価であり、生産、精製工程の煩雑化、あるいは安定性の欠如といった問題を有する。
【0007】
低分子のアルツハイマー病の予防・治療剤としては、アミロイドβペプチドの生成を抑制する作用を有する薬剤、例えば、ロダニン誘導体(特許文献2:特開平6−192091号公報)、ベンズイミダゾール誘導体(特許文献3:米国特許第5552426号明細書)、ビンポセチン誘導体(特許文献4:国際公開第96/25161号パンフレット)、芳香族アミド誘導体(特許文献5:国際公開第2004/014843号パンフレット)が知られている。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】国際公開第2005/105998号パンフレット
【特許文献2】特開平6−192091号公報
【特許文献3】米国特許第5552426号明細書
【特許文献4】国際公開第96/25161号パンフレット
【特許文献5】国際公開第2004/014843号パンフレット
【発明の概要】
【発明が解決しようとする課題】
【0009】
本発明は、アミロイドβペプチドのミミックペプチドとして機能し、アミロイドβペプチドの線維化を阻害しうるペプチドを提供することを目的とする。
【課題を解決するための手段】
【0010】
本発明の要旨は以下のとおりである。
(1)次式(I):
X1-Asp-X2-X3-X4-Pro-X5-X6 (I)
(式中、X1は分枝アミノ酸を表し、X2、X3、X4、X5及びX6は同一、又は異なってα−アミノ酸を表す。)
で示されるアミノ酸配列を含む、アミノ酸残基数8〜30のペプチド。
(2)前記式(I)において、X2が分枝アミノ酸、ヒドロキシアミノ酸、芳香族アミノ酸又は脂肪族アミノ酸である前記(1)に記載のペプチド。
(3)前記式(I)において、X3が芳香族アミノ酸又は分枝アミノ酸である前記(1)又は(2)に記載のペプチド。
(4)前記式(I)において、X4が脂肪族アミノ酸、ヒドロキシアミノ酸又は分枝アミノ酸である前記(1)〜(3)のいずれかに記載のペプチド。
(5)前記式(I)において、X5が分枝アミノ酸、イミノ酸、脂肪族アミノ酸、塩基性アミノ酸又はヒドロキシアミノ酸である前記(1)〜(4)のいずれかに記載のペプチド。
【0011】
(6)前記式(I)において、X6が塩基性アミノ酸、脂肪族アミノ酸又は分枝アミノ酸である前記(1)〜(5)のいずれかに記載のペプチド。
(7)次式(II):
X7-X8-X9-X10-X1-Asp-X2-X3-X4-Pro-X5-X6-X11-X12-Y1 (II)
(式中、X1、X2、X3、X4、X5及びX6は前記(1)に記載の式(I)中の定義と同一であり、X7、X8、X9、X10、X11及びX12は同一、又は異なってα−アミノ酸又は単結合を表し、Y1はOH又はNH2を表す。)
で示されるペプチド。
(8)前記式(II)において、X7がヒドロキシアミノ酸又は脂肪族アミノ酸である前記(7)に記載のペプチド。
(9)前記式(II)において、X8がヒドロキシアミノ酸又はアミドアミノ酸である前記(7)又は(8)に記載のペプチド。
(10)前記式(II)において、X9が脂肪族アミノ酸、イミノ酸、ヒドロキシアミノ酸又はアミドアミノ酸である前記(7)〜(9)のいずれかに記載のペプチド。
【0012】
(11)前記式(II)において、X10が含硫アミノ酸、分枝アミノ酸、芳香族アミノ酸又はイミノ酸である前記(7)〜(10)のいずれかに記載のペプチド。
(12)前記式(II)において、X11が複素環式アミノ酸、芳香族アミノ酸、脂肪族アミノ酸又は分枝アミノ酸である前記(7)〜(11)のいずれかに記載のペプチド。
(13)前記式(II)において、X12が分枝アミノ酸、酸性アミノ酸又はイミノ酸である前記(7)〜(12)のいずれかに記載のペプチド。
(14)前記式(I)で示されるアミノ酸配列と、次式:
Tyr-Gly-Arg-Lys-Lys-Arg-Arg-Gln-Arg-Arg-Arg
で示されるTAT配列とを含む、アミノ酸残基数19〜30のペプチドである前記(1)〜(13)のいずれかに記載のペプチド。
(15)次式(III):
Z1-X13-Gly-X14-X15-Pro-Trp-Met-Z2 (III)
(式中、X13、X14及びX15は同一、又は異なってα−アミノ酸を表し、Z1及びZ2は同一、又は異なってシステイン又はセリンを表し、Z1及びZ2がシステインを表す場合、これらの間で架橋されていてもよい。)
で示されるアミノ酸配列を含む、アミノ酸残基数9〜30のペプチド。
【0013】
(16)前記式(III)において、X13が芳香族アミノ酸である前記(15)に記載のペプチド。
(17)前記式(III)において、X14がヒドロキシアミノ酸である前記(15)又は(16)に記載のペプチド。
(18)前記式(III)において、X15が塩基性アミノ酸である前記(15)〜(17)のいずれかに記載のペプチド。
(19)前記式(III)で示されるアミノ酸配列と、次式:
Tyr-Gly-Arg-Lys-Lys-Arg-Arg-Gln-Arg-Arg-Arg
で示されるTAT配列とを含む、アミノ酸残基数20〜30のペプチドである前記(15)〜(18)のいずれかに記載のペプチド。
(20)前記(1)〜(19)のいずれかに記載のペプチドを含有する医薬組成物。
【0014】
(21)前記(1)〜(19)のいずれかに記載のペプチドを含有するアミロイドβ線維化阻害剤。
(22)アルツハイマー病の予防・治療剤である前記(21)に記載のアミロイドβ線維化阻害剤。
(23)アミロイドβ線維化阻害活性を有することが確認されていないペプチドから、アミロイドβペプチド1-42と特異的に結合してその線維化反応を阻害する活性を有する一本鎖抗体に結合し、かつアミロイドβ線維化阻害活性を有するペプチドを探索することを特徴とするアミロイドβ線維化阻害ペプチドのスクリーニング方法。
(24)前記(23)に記載のスクリーニング方法により取得されるアミロイドβ線維化阻害ペプチド。
【発明の効果】
【0015】
本発明のペプチドは、アルツハイマー病の予防・治療剤として有用である。
【図面の簡単な説明】
【0016】
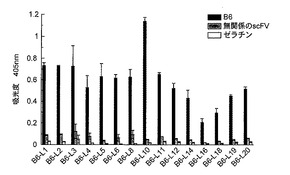
【図1】図1は、B6 scFvに結合したペプチドファージクローンのELISAの結果を示す図である。
【図2】図2は、D1 scFvに結合した12merペプチドファージクローンのELISAの結果を示す図である。
【図3】図3は、D1 scFvに結合したC7CペプチドファージクローンのELISAの結果を示す図である。
【図4】図4は、ペプチドファージによるAβ1-42ペプチドの線維化阻害実験の結果を示す図である。
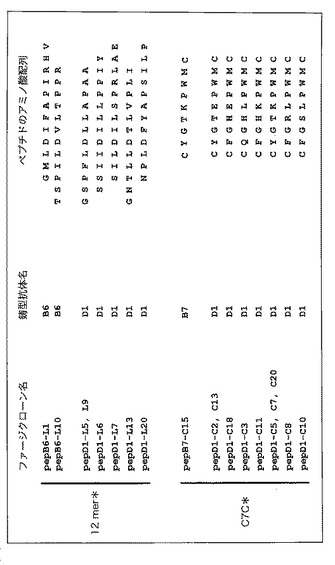
【図5】図5は、Aβ1-42の線維化を阻害するB6 scFv、B7 scFv又はD1 scFvを鋳型として単離したファージクローンの提示するペプチド配列一覧を示す図である。
【図6】図6は、ビオチン化したB6又はB7 scFv結合性配列とTAT配列との融合分子を示す図である。
【図7】図7は、TATとB6-L1の融合ペプチドがB6 scFvに認識されることを示す図である。
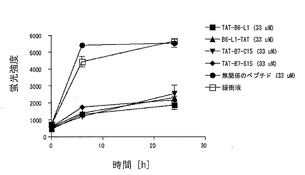
【図8】図8は、TATとB6-L1、B7-C15及びB7-S15それぞれとの融合ペプチドのAβ1-42線維化阻害活性の測定結果を示す図である。
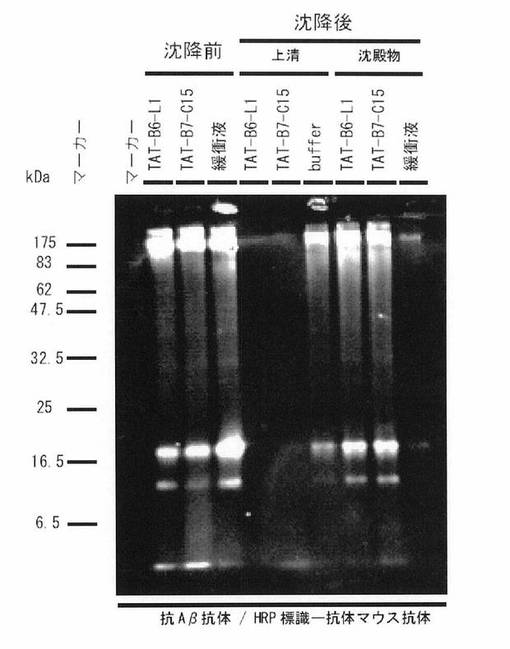
【図9】図9は、TATとB6-L1及びB7-C15それぞれとの融合ペプチドによるAβコンホーマー(Aβオリゴマー及びAβ線維)の沈降実験の結果を示す図である。
【発明を実施するための形態】
【0017】
本発明のペプチドは、前記式(I)又は(III)で示されるアミノ酸配列を含む、アミノ酸残基数30以下のペプチドであり、好ましくは前記式(I)で示されるアミノ酸配列を含む、アミノ酸残基数8〜20のペプチド及び前記式(III)で示されるアミノ酸配列を含む、アミノ酸残基数9〜20のペプチドである。
【0018】
前記式(I)において、X1で表される分枝アミノ酸としては、バリン、ロイシン、イソロイシン等が挙げられる。
【0019】
前記式(I)、(II)又は(III)において、X2、X3、X4、X5、X6、X7、X8、X9、X10、X11、X12、X13、X14又はX15で表されるα−アミノ酸としては、例えば、グリシン、アラニン等の脂肪族アミノ酸;バリン、ロイシン、イソロイシン等の分枝アミノ酸;セリン、トレオニン等のヒドロキシアミノ酸;アスパラギン酸、グルタミン酸等の酸性アミノ酸;アスパラギン、グルタミン等のアミドアミノ酸;リシン、ヒドロキシリシン、アルギニン等の塩基性アミノ酸;システイン、シスチン、メチオニン等の含硫アミノ酸;フェニルアラニン、チロシン等の芳香族アミノ酸;トリプトファン、ヒスチジン等の複素環式アミノ酸;プロリン、4−ヒドロキシプロリン等のイミノ酸が挙げられる。
【0020】
前記式(I)及び(II)において、X2で表されるα−アミノ酸としては、好ましくは分枝アミノ酸、ヒドロキシアミノ酸、芳香族アミノ酸、脂肪族アミノ酸、更に好ましくはバリン、ロイシン、イソロイシン、トレオニン、フェニルアラニンが挙げられ;X3で表されるα−アミノ酸としては、好ましくは芳香族アミノ酸、分枝アミノ酸、更に好ましくはフェニルアラニン、ロイシン、チロシンが挙げられ;X4で表されるα−アミノ酸としては、好ましくは脂肪族アミノ酸、ヒドロキシアミノ酸、分枝アミノ酸、更に好ましくはアラニン、トレオニン、ロイシン、セリン、バリンが挙げられ;X5で表されるα−アミノ酸としては、好ましくは分枝アミノ酸、イミノ酸、脂肪族アミノ酸、塩基性アミノ酸、ヒドロキシアミノ酸、更に好ましくはロイシン、イソロイシン、プロリン、アラニン、アルギニン、セリンが挙げられ;X6で表されるα−アミノ酸としては、好ましくは塩基性アミノ酸、脂肪族アミノ酸、分枝アミノ酸、更に好ましくはアルギニン、アラニン、ロイシン、イソロイシンが挙げられる。
【0021】
前記式(II)において、X7で表されるα−アミノ酸としては、好ましくはヒドロキシアミノ酸、脂肪族アミノ酸、更に好ましくはトレオニン、グリシンが挙げられ;X8で表されるα−アミノ酸としては、好ましくはヒドロキシアミノ酸、アミドアミノ酸、更に好ましくはセリン、アスパラギンが挙げられ;X9で表されるα−アミノ酸としては、好ましくは脂肪族アミノ酸、イミノ酸、ヒドロキシアミノ酸、アミドアミノ酸、更に好ましくはグリシン、プロリン、セリン、トレオニン、アスパラギンが挙げられ;X10で表されるα−アミノ酸としては、好ましくは含硫アミノ酸、分枝アミノ酸、芳香族アミノ酸、イミノ酸、更に好ましくはメチオニン、ロイシン、イソロイシン、フェニルアラニン、プロリンが挙げられ;X11で表されるα−アミノ酸としては、好ましくは複素環式アミノ酸、芳香族アミノ酸、脂肪族アミノ酸又は分枝アミノ酸、更に好ましくはヒスチジン、チロシン、アラニン、ロイシンが挙げられ;X12で表されるα−アミノ酸としては、好ましくは分枝アミノ酸、酸性アミノ酸、イミノ酸、更に好ましくはバリン、グルタミン酸、プロリンが挙げられる。
【0022】
前記式(III)において、X13で表されるα−アミノ酸としては、好ましくは芳香族アミノ酸、アミドアミノ酸、更に好ましくはチロシン、フェニルアラニン、グルタミン、最も好ましくはチロシンが挙げられ;X14で表されるα−アミノ酸としては、好ましくはヒドロキシアミノ酸、複素環式アミノ酸、塩基性アミノ酸、更に好ましくはセリン、トレオニン、ヒスチジン、アルギニン、最も好ましくはトレオニンが挙げられ;X15で表されるα−アミノ酸としては、好ましくは塩基性アミノ酸、酸性アミノ酸、分枝アミノ酸、更に好ましくはリシン、グルタミン酸、ロイシン、最も好ましくはリシンが挙げられる。
【0023】
本発明のペプチドは、公知の方法によって化学合成することもでき、例えばペプチド自動合成装置によって合成することができる。この基本的な合成過程はR.B. Merifield, Advance in Enzymology 32: 221-296(1969)に記載の方法を適用できる。この方法は、カルボキシル末端のアミノ酸を樹脂担体に共有結合させておき、α−アミノ基の保護基の除去、保護アミノ酸の縮合を順次繰返して、アミノ末端に向けてペプチド鎖を延長させ目的のアミノ酸配列を有するペプチド樹脂を得ることを原理とするものである。
【0024】
各アミノ酸の縮合やα−アミノ基の保護基の除去等は、Boc, Fmoc法を利用してほぼ同一の条件でなされ、中間体の精製も行わないため、合成に際しては一般に高度な熟練は要求されない。しかもこの方法は迅速であり、種々のペプチドを合成するに際し、非常に便利な方法である。こうして得られた保護ペプチド樹脂を、例えば無水フッ化水素、トリフルオロメタンスルホン酸もしくはトリフルオロ酢酸と種々の添加物の共存下に反応させることにより、ペプチドを樹脂から脱離させることと全保護基の除去を一段階で行うことができる。
【0025】
樹脂担体としてカルボキシル末端カルボン酸型ペプチド合成用樹脂を用いれば、カルボキシル末端がカルボキシル基であるペプチド、例えば前記式(II)において、Y1がOHであるペプチドを得ることができ、カルボキシル末端アミド型ペプチド合成用樹脂を用いれば、カルボキシル末端カルボン酸がアミド化されたペプチド、例えば前記式(II)において、Y1がNH2であるペプチドを得ることができる。
【0026】
得られたペプチド粗製物は、ペプチドを精製する公知の手段で精製することができる。例えば、ゲル濾過、陽イオン交換樹脂もしくは陰イオン交換樹脂を用いるイオン交換クロマトグラフィー、更には疎水クロマトグラフィー、分配吸着クロマトグラフィーなど、種々の原理によるカラムクロマトグラフィーや高速液体クロマトグラフィーが挙げられる。
【0027】
本発明のペプチドは、複数のシステイン残基を含む場合には、それらのシステイン残基の間で架橋されていてもよい。例えば、次式(IIIa):
Cys1-X13-Gly-X14-X15-Pro-Trp-Met-Cys2 (IIIa)
(式中、X13、X14及びX15は同一、又は異なってα−アミノ酸を表し、Cys1とCys2の間で架橋されていてもよい。)
で示されるアミノ酸配列を含む、アミノ酸残基数9〜30、好ましくは9〜20のペプチドは、Cys1とCys2の間で架橋されていてもよい。
【0028】
これらの架橋としては、システイン残基間を直接ジスルフィド架橋してもよく、またスペーサーとしてジスルフィド化合物を用いて、スペーサーを介してジスルフィド架橋してもよい。ジスルフィド架橋は、例えばペプチドの希釈水溶液をK3[Fe(CN)6]で酸化、又は酸性条件下のヨウ素で酸化することにより形成することができる。
【0029】
本発明のペプチドは種々の塩の形で得ることできる。その塩としては、例えば無機酸や、蟻酸、酢酸、酒石酸、クエン酸などの有機酸との塩、もしくはナトリウムやアンモニアなどの無機塩基や、トリエチルアミン、エチルアミン、メチルアミンなどの有機塩基との塩が挙げられる。
【0030】
ペプチドのアミノ基のビオチン化はアミノ酸誘導体を樹脂上で縮合する通常のHOBt−DCC, HBTu−HOBt方法などで可能である。
【0031】
本発明のスクリーニング方法は、アミロイドβ線維化阻害活性を有することが確認されていないペプチドから、Aβ1-42と特異的に結合してその線維化反応を阻害する活性を有する一本鎖抗体に結合し、かつアミロイドβ線維化阻害活性を有するペプチドを探索することを特徴とするものである。
【0032】
Aβ1-42と特異的に結合し、その線維化反応を阻害する活性を有する一本鎖抗体(scFv)としては、好ましくは、WO2005/105998に記載されているヒト一本鎖可変領域フラグメントscFv(B6、B7、D1、F10)が挙げられる。本明細書において、これらの抗体を鋳型と称する。
【0033】
B6scFv(WO2005/105998のクローンB6)のVH鎖のアミノ酸配列を配列番号1に、VL鎖のアミノ酸配列を配列番号2に示す。
【0034】
B7scFv(WO2005/105998のクローンB7)のVH鎖のアミノ酸配列を配列番号3に、VL鎖のアミノ酸配列を配列番号4に示す。
【0035】
D1scFv(WO2005/105998のクローンD1)のVH鎖のアミノ酸配列を配列番号5に、VL鎖のアミノ酸配列を配列番号6に示す。
【0036】
F10scFv(WO2005/105998のクローンF10)のVH鎖のアミノ酸配列を配列番号7に、VL鎖のアミノ酸配列を配列番号8に示す。
【0037】
本発明のペプチドを得るための方法としては、例えば以下に述べるペプチドライブラリー法が挙げられる。更に、ペプチドライブラリー法は、バクテリオファージを用いる方法と、化学合成でライブラリーを作製する方法に分けられる。
【0038】
ファージランダムペプチドライブラリーを構築する方法は、例えばM13系ファージのコートプロテイン(例えばgeneIII蛋白やVIII蛋白)の遺伝子にランダムな配列を有する合成遺伝子を連結し、作製すればよい。その方法としては、Science, 249, 386 (1990), Proc. Natl. Acad. Sci. USA, 87, 6378 (1990) 等に記載された方法を用いることができる。挿入する遺伝子の大きさは、発現されるペプチドが安定であれば特に制限はないが、作製したライブラリーがより多くのランダムな配列を網羅し、更に標的分子に結合能を有するためには6から15アミノ酸が好ましい。鋳型とするscFvに結合するペプチドファージを選択するためには、カラムやマイクロタイタープレート上に精製した前記scFvを直接あるいは抗IgG抗体等を介して固定化し、前記ライブラリーを接触させる。その後、非結合ファージは洗浄操作で洗い流す。洗浄後、結合しているファージを酸で溶出し、中和した後、大腸菌に感染させ増幅させる。この操作(パニング)を3回か4回繰り返すと、前記scFvに親和性のあるファージが濃縮される。ここで単一なクローンを得るためには、再度大腸菌にファージを感染させ、抗生物質を含んだ寒天培地上でシングルコロニーを形成させる。個々のコロニーを液体培地で培養した後、上清中のファージをポリエチレングリコール等で沈殿濃縮し、その塩基配列を決定すれば、ペプチドの構造を知ることができる。
【0039】
ランダムなアミノ酸配列を有するペプチドライブラリーは、前記のファージを用いる方法のほか、化学合成で作製することも可能である。その方法としては、ビーズを用いる方法(Nature, 354, 82 (1991))、液相フォーカシング法(Nature, 354, 84 (1991))、マイクロプレート法(Science, 251, 767 (1991))等が挙げられる。
【0040】
ライブラリーより得られた配列を有するペプチドを大量に調製するためには、人工的にペプチドを合成する方法や、遺伝子組換え技術を利用して大腸菌、酵母、昆虫細胞、動物細胞等で発現させる方法が挙げられる。
【0041】
人工的にペプチドを合成する方法は、一般的なペプチド合成法により容易に行うことができ、例えば固相合成法で行うことが簡便であり、目的の配列に、欠失、置換、挿入又は付加を施した変異体を作製することも容易である(細胞工学別冊、抗ペプチド抗体実験プロトコール、p.26-46、秀潤社)。また、非天然型アミノ酸の導入、各アミノ酸残基の化学修飾やシステイン残基を導入することにより分子内を環化させて構造を安定化させる等の修飾を施してもよい。
【0042】
遺伝子組換え技術を利用する場合、得られたアミノ酸配列から、codon usageに従ってDNA配列を設定し(Molecular Cloning, Appendix D1参照, マニアティスら;Cold Spring Habor Laboratory社、1989)、宿主の細胞に導入することは、技術的に確立されている。更に、塩基配列に変異を導入することにより、アミノ酸を他の残基に変換することも可能である。例えば大腸菌で発現させる場合は、得られたDNA配列をプロモーター配列、例えばトリプトファン合成酵素オペロン(Trp)、ラクトースオペロン(lac)プロモーターに結合し、リボゾーム結合配列、例えばシャインダルガルノ(SD)配列や転写終結因子認識部位を付加することが望ましい。作製した発現ベクターを大腸菌に導入する方法等はMolecular Cloning (マニアティスら;Cold Spring Habor Laboratory社、1989)記載の方法が使用できる。発現産物を精製する方法は、例えば各種クロマトグラフィーを用いることができる。
【0043】
得られたペプチドがアミロイドβ線維化阻害活性を有するか否かを調べる方法としては、例えば、チオフラビンT(ThT)法(H. Levine, III, Protein Science 21: 404-410 (1993))が挙げられる。
【0044】
本発明のスクリーニング方法に従ってアミロイドβ線維化阻害活性が確認されたペプチドは、該ペプチドをリード化合物とし、これに、1もしくは2以上のアミノ酸残基の欠失、置換、挿入もしくは付加、又は修飾を施し、アミロイドβ線維化阻害活性を有するペプチドを得ることにより、類縁のアミロイドβ線維化阻害ペプチドを製造することができる。このペプチドがアミロイドβ線維化阻害活性を有するか否かは、前記の方法と同様にして調べることができる。
【0045】
本発明は、また、前記スクリーニング方法により取得されるアミロイドβ線維化阻害ペプチドを提供する。ここで、「前記スクリーニング方法により取得される」とは、前記スクリーニング方法により実際にアミロイドβ線維化阻害活性が確認されたペプチドをいう。前記ペプチドのアミノ酸残基数は、通常6〜40、好ましくは8〜20、更に好ましくは8〜15である。
【0046】
本発明のペプチドは、アミロイドβ線維化阻害活性を有し、アルツハイマー病の予防・治療剤として有用である。
【0047】
本発明のアミロイドβ線維化阻害ペプチドは、そのままもしくは自体公知の薬学的に許容される担体、賦形剤等と混合した医薬組成物として経口又は非経口的に投与することができる。
【0048】
経口投与のための剤形としては、具体的には錠剤、丸剤、カプセル剤、顆粒剤、細粒剤、散剤、シロップ剤、乳剤、懸濁剤等が挙げられる。かかる剤形は、自体公知の方法によって製造され、製剤分野において通常用いられる担体もしくは賦形剤を含有するものである。例えば錠剤用の担体、賦形剤としては、ラクトース、マルトース、ショ糖、澱粉、ステアリン酸マグネシウム等が挙げられる。
【0049】
非経口投与のための剤形としては、例えば、点眼剤、軟膏剤、注射剤、湿布剤、坐薬、経鼻吸収剤、経肺吸収剤、経皮吸収剤、局所徐放剤等が挙げられる。溶液製剤は自体公知の方法、例えば、アミロイドβ線維化阻害ペプチドを通常、注射剤に用いられる無菌の水溶液に溶解、更には乳化して、リポソームに包埋させた状態で調製されうる。ペプチドは生体内でペプチダーゼ等による分解を受け易く、また目的の場所への到達性の問題等があることから、ここに述べたリポソームを含む適切なデリバリー法の活用は好ましい使用形態の一つである。ペプチドのデリバリー法の活用については前記リポソームを用いる方法はその一つであるが、これに限られたものではない。固体製剤は、自体公知の方法、例えば、アミロイドβ線維化阻害ペプチドにマンニトール、トレハロース、ソルビトール、ラクトース、グルコース等を賦形剤として加え、そのまま凍結乾燥することにより調製されうる。更にこれを粉体化して用いることもできる。また、これら粉体をポリ乳酸やグリコール酸等と混合し固体化して用いることもできる。ゲル化剤は、自体公知の方法、例えば、アミロイドβ線維化阻害ペプチドをグリセリン、ポリエチレングリコール、メチルセルロース、カルボキシメチルセルロース、ヒアルロン酸、コンドロイチン硫酸等の増粘剤や多糖に溶解した状態で調製されうる。
【0050】
いずれの製剤においても、安定化剤としてヒト血清アルブミン、ヒト免疫グロブリン、α2マクログロブリン、アミノ酸等を添加することができ、また分散剤あるいは吸収促進剤としてアミロイドβ線維化阻害ペプチドの活性を損なわない範囲でアルコール、糖アルコール、イオン性界面活性剤、非イオン性界面活性剤等を添加することができる。また、微量金属や有機酸塩も必要に応じて加えることができる。
【0051】
本発明の医薬組成物において、有効成分であるペプチドとしての投与量は、当該ペプチドの活性、患者の年令、体重、疾患の種類又は程度により異なるが、一般には、経口投与では、通常1日0.001〜1000mg/kg体重であり、静脈内、筋肉内又は皮下投与では、通常1日0.001〜1000mg/kg体重である。投与回数は、通常経口投与では1日1〜3回、注射剤では1日1〜2回である。
【0052】
本明細書は、本願の優先権の基礎である特願2006−125769の明細書及び/又は図面に記載される内容を包含する。
【実施例】
【0053】
以下、製造例及び実施例を挙げて本発明を更に詳細に説明するが、本発明はこれらに限定されるものではない。
【0054】
(実施例1)
Kaji et al., J. Biochem. 129: 577-583 (2001)及びS. Hashiguchi et al., J. Biochem. 133: 43-49 (2003)に従って以下の実験を行った。
【0055】
1.材料及び方法
(1)E-tagカラムによるscFvの精製
Aβに結合するscFvを発現させるために、Hashiguchiらの方法(J. Biochem. 133: 43-49 (2003))によりscFvファージクローンを大腸菌株HB2151に感染させた。得られた培地上清からE-tagカラムAmersham Biosciences)を用いてアミロイド特異的scFv(B6、B7、D1、F10)を精製した。
【0056】
(2)ペプチドファージライブラリー
ファージのgeneIII蛋白のアミノ末端に12個のランダムなアミノ酸配列を提示したPh.D.-12ライブラリー(New England Biolabs、MA)及びファージのgeneIII蛋白のアミノ末端にシステインとシステインの間に7つのアミノ酸配列を含むペプチドを提示するPh.D.-C7Cライブラリー(New England Biolabs、MA)を用いた。
【0057】
(3)バイオパニング
鋳型となるヒト一本鎖抗体(scFv:B6、B7、D1又はF10)又はコントロールscFv 1μg/100μl/well (0.1 M NaHCO3 pH 8.6)をMaxisorp plateに4℃で6時間コートした。ブロッキング溶液としてそれぞれのウェルを1stパニングでは0.5%ゼラチン,2ndパニングでは0.25% BSA,3rdパニングでは0.5%ゼラチンを用いて4℃で14時間ブロックした。12mer又はC7Cペプチドファージライブラリーをブロッキング溶液で希釈後(1.5 x 1012 pfu/100 μl) 30分静置後、コントロールscFvで吸収操作を行った。すなわち、コントロールscFvをコートしたウェルを0.2% Tween 20/PBS (PBST)で3回洗浄しファージ溶液を加え室温で1時間反応させ、コントロールscFvと結合しなかったファージ溶液を回収した。このファージ溶液を鋳型とするscFvをコートしたウェルに加え室温で1時間反応させた。0.2% PBSTを用いて10回洗浄後1 mg/ml BSA / 0.1 M グリシン塩酸 pH 2.2を100 μl加え室温で5分置くことにより結合したファージを回収し、ただちに1 M トリス塩酸pH 9.1を15 μl加え中和した。回収したファージを大腸菌ER2738に感染させ増幅させ、2, 3ラウンドのパニングに用いた(1.5 x 1012 pfu/100 μl)。
【0058】
(4)ファージクローンの単離
パニングを2回行った後、回収したファージをER2738に感染させLB/Tet/X-gal plateで培養した。溶菌してできたプラークを再度ER2738に感染させ、ファージクローンを単離した。
【0059】
(5)ELISA
パニングで鋳型としたscFv又はコントロールscFv 50 ng/40 μl/well (0.1 M NaHCO3 pH 8.6)をELISAプレートに4℃で6時間コートした。0.5%ゼラチンを用いて4℃で13時間ブロックした。0.2% PBSTを用いてウェルを3回洗浄後、単離したペプチドファージクローン溶液(約1.6 x 1011 virions/40 μl)を加え室温で1時間反応させた。結合したファージは40μlのビオチン化抗M13モノクローナル抗体(1000倍希釈)、ストレプトアビジンアルカリフォスファターゼ(1000倍希釈)を反応させた後、基質パラニトロフェニルリン酸を用いて検出した。
【0060】
(6)DNA塩基配列解析
特異性の認められたファージクローンをフェノール/クロロホルム処理して除蛋白した後、エタノール沈殿でDNAを精製し、塩基配列決定の鋳型とした。primer-96gIII(1 pg/μl) 5’-CCC TCA TAG TTA GCG TAA CG-3’(New England Biolabs, MA)を用いて、ランダムなペプチド配列が挿入されている領域の遺伝子増幅を行い、DNA塩基配列決定を行った。
【0061】
(7)ペプチド合成
本発明のペプチドは、一般的な固相ペプチド合成法、Fmoc/HBTu+HOBt法により合成した。本発明のペプチドB6-L1, B7-C15, B7-S15のそれぞれにTAT配列を連結したペプチドを合成した。なお、B7-C15については、カルボキシル末端側にグリシンを付加したものにTAT配列を連結した。ちなみにTAT配列は脳血管関門を通過する機能をもつペプチド配列である(P. Jarver and Ulo Langel, The use of cell-penetrating peptides as a tool for gene regulation, DDT, 9: 395-401, 2004)。
【0062】
なお、B6-L1, B7-C15, B7-S15のそれぞれの配列のアミノ末端側にTAT配列を融合したものがTAT-B6-L1, TAT-B7-C15, TAT-B7-S15であり、B6-L1の配列のカルボキシル末端側にTAT配列を融合したものがB6-L1-TATである。このペプチドの結合を検出するためにペプチドのアミノ末端にビオチンが結合しているペプチド配列として合成した。
【0063】
B6-L1, B7-C15, B7-S15、及びこれらとTAT配列との融合分子のアミノ酸配列と配列番号との関係は以下のとおりである。
B6-L1:配列番号9;
B7-C15:配列番号16
B7-S15:配列番号23
TAT-B6-L1:配列番号24
B6-L1-TAT:配列番号25
TAT-B7-C15:配列番号26
TAT-B7-S15:配列番号27
【0064】
(8)アミロイドβ線維化阻害実験
20 mMリン酸バッファー pH 7.0を用いて40 μMに希釈したAβ1-42ペプチドに合成したペプチド(TAT-B6-L1, B6-L1-TAT, TAT-B7-C15, TAT-B7-S15)を様々な濃度(1.65 nM, 16.5 nM, 33 nM)で加えた。0, 6, 24時間に10 μlのサンプルを回収し11 μMのチオフラビンT(Sigma)/リン酸バッファー90 μlを加えマルチラベルプレートカウンター、Wallac 1420 ARVOsx (Perkin-Elmer; Wellesley, MA)を用い、450nmの励起光により発生する482nmの蛍光強度を測定した。
【0065】
(9)Aβ線維化阻害ペプチドによるAβコンホーマー(Aβオリゴマー及びAβ線維)の沈降実験
Aβ1-42を20 mMリン酸バッファーpH 7.0に40 μMの濃度で溶解し、その1.5時間目に、40 μMに調製したTAT-B6-L1又はTAT-B7-C15ペプチドを液量比(1:1)で加え37℃で1時間反応させた(最終濃度:Aβ[20 μM]/合成ペプチド[20 μM])。反応後、免疫沈降前のサンプルを「沈降前」として一部サンプリングし、残りのサンプルを20 μg/μlに調製したM-280ストレプロアビジン-マグネットビーズ(Dynal Biotech, Oslo, Norway)と液量比(1:1)で氷上で1時間反応させた(最終濃度:Aβ[10 μM]/合成ペプチド[10 μM]/ビーズ[10 μg/μl])。反応後、マグネットによりビーズを沈降させ上清を「上清」とした。沈降したビーズに回収した上清と等液量のPBSを加え「沈殿物」とした。得られた各サンプルを電気泳動し、抗Aβ抗体によるWestern blot解析を行った。
【0066】
2.結果
(1)Aβ特異的scFvに結合するペプチドファージクローン
(a)B6 scFvに結合したペプチドファージクローンのELISAの結果を図1に示す。無関係のscFv(コントロールscFv)としてFv1E1というAβの線維化を阻害しない抗体を用いた。ファージライブラリーは12merのアミノ酸配列を提示するPh.D.-12ライブラリー(New England Biolabs、MA)を用いた。実験は前記(5)ELISAで記したとおりに行った。
(b)D1 scFvに結合した12merペプチドファージクローンのELISAの結果を図2に示す。無関係のscFv(コントロールscFv)としてMY5RというAβの線維化を阻害しない抗体を用いた。ファージライブラリーは12merのアミノ酸配列を提示するPh.D.-12ライブラリー(New England Biolabs、MA)を用いた。実験は前記(5)ELISAで記したとおりに行った。
(c)D1 scFvに結合したC7CペプチドファージクローンのELISAの結果を図3に示す。無関係のscFv(コントロールscFv)としてMY5RというAβの線維化を阻害しない抗体を用いた。ファージライブラリーはシステインとシステインの間に7つのアミノ酸配列を含むペプチドを提示するPh.D.-C7Cライブラリー(New England Biolabs、MA)を用いた。実験は前記(5)ELISAで記したとおりに行った。
【0067】
(2)B6結合性ペプチドファージのAβ1-42線維化阻害活性
B6結合性ペプチドファージによるAβ1-42ペプチドの線維化阻害実験の結果を図4に示す。Aβ1-42線維化阻害実験は前記(8)線維化阻害実験で記したとおりに行った。pepB6-L1及びpepB6-L10は、Aβ1-42の線維化を阻害するB6 scFvを鋳型として、それに結合するファージクローンであり、pep1E1-L1, 1-L2, 1-L4, 1-L12は、Aβ1-42の線維化を阻害しないFv1E1 scFv(WO2005−105998段落0074〜0075に記載)を鋳型として単離したファージクローンである。ここで用いたファージクローンの濃度は3.0 x 1012 virions/mlである。
【0068】
(3)Aβ特異的scFvのエピトープ(結合性配列)
Aβ1-42の線維化を阻害するB6 scFv、B7 scFv又はD1 scFvを鋳型として単離したファージクローンの提示するペプチドのアミノ酸配列一覧を図5に示す。ファージライブラリーとしてPh.D.-12ライブラリー(図5中12mer*と略)又はPh.D.-C7Cライブラリー(図5中C7C*と略)を使用した。pepB7-C15とpepD1-C5,C7,C20は同一の配列であった。番号の前についているLは12merのライブラリーから、CはC7Cのライブラリーから単離されたクローンであることを示している。
【0069】
各ファージクローンの提示するペプチドのアミノ酸配列と配列番号との関係は以下のとおりである。
pepB6-L1:配列番号9;
pepB6-L10:配列番号10;
pepD1-L5,L9:配列番号11;
pepD1-L6:配列番号12;
pepD1-L7:配列番号13;
pepD1-L13:配列番号14;
pepD1-L20:配列番号15;
pepB7-C15 (pepD1-C5,C7,C20):配列番号16;
pepD1-C2,C13:配列番号17;
pepD1-C18:配列番号18;
pepD1-C3:配列番号19;
pepD1-C11:配列番号20;
pepD1-C8:配列番号21;
pepD1-C10:配列番号22
【0070】
(4)ビオチン化したB6又はB7 scFv結合性配列とTAT配列との融合分子
ビオチン化したB6 scFv結合性配列とTAT配列との融合分子を図6に示す。B6-L1, B7-C15, B7-S15のそれぞれの配列のアミノ末端側にTAT配列を融合したものがTAT-B6-L1, TAT-B7-C15, TAT-B7-S15であり、B6-L1の配列のカルボキシル末端側にTAT配列を融合したものがB6-L1-TATである。TAT配列は脳血管関門を通過する機能をもつペプチド配列である(P. Jarver and Ulo Langel, The use of cell-penetrating peptides as a tool for gene regulation, DDT, 9: 395-402, 2004)。これらのペプチドのアミノ末端をビオチンで標識し、このペプチドの結合を酵素標識したアビジンを用いて定量を行った。
【0071】
(5)TATとB6-L1の融合ペプチドはB6 scFvに認識された(図7)。合成ペプチド、TAT-B6-L1あるいはB6-L1-TATを200 ng/ wellの条件でELISAプレートにコートし、種々の濃度のB6 scFvを添加しその結合活性を検討した。コントロールペプチドとして無関係のペプチド(GSGGGSCGYWRSEWGLCG)を用いた。B6 scFvの量依存的な結合反応が確認された。
【0072】
(6)TATとB6-L1, B7-C15, B7-S15それぞれとの融合ペプチドのAβ1-42線維化阻害活性
TATとB6-L1, B7-C15, B7-S15それぞれとの融合ペプチドのAβ1-42線維化阻害活性の測定結果を図8に示す。線維化阻害実験は前記線維化阻害実験で記したとおりに行った。TAT-B6-L1, B6-L1-TAT, TAT-B7-C15, TAT-B7-S15は線維化阻害活性があることが明らかとなった。前記のコントロールペプチドは全く阻害活性を示さなかった。
【0073】
(7)TAT-B6-L1, TAT-B7-C15ペプチドによるAβコンホーマー(Aβオリゴマー及びAβ線維)の沈降実験
TAT-B6-L1, TAT-B7-C15ペプチドの沈降実験の結果を図9に示す。Aβ1-42を20 mMリン酸バッファー pH 7.0に40 μMの濃度で溶解し、その1.5時間目にビオチン化TAT-B6-L1又はビオチン化TAT-B7-C15ペプチドを加え結合物をストレプトアビジン-マグネットビーズで沈降させた。沈降前はAβオリゴマーやAβ線維などのバンドが確認できるが、TAT-B6-L1, TAT-B7-C15ペプチドで沈降を行った上清にはAβオリゴマーやAβ線維は確認されず、沈殿物の方で確認できた。この結果からAβ1-42溶解後1.5時間目に形成されるAβオリゴマーやAβ線維とTAT-B6-L1, TAT-B7-C15ペプチドが結合することが明らかとなった。無関係なコントロールペプチドでは沈降反応は認められなかった(図9には示されていない)。
【0074】
本明細書中で引用した全ての刊行物、特許及び特許出願をそのまま参考として本明細書中にとり入れるものとする。
【技術分野】
【0001】
本発明は、アルツハイマー病の予防・治療剤として有用なアミロイドβ線維化阻害ペプチドに関する。
【背景技術】
【0002】
近年、老人人口の増加に伴い、老人性痴呆症の治療に有効な医薬品の開発が強く望まれている。老人性痴呆症の代表的疾患であるアルツハイマー病は、脳の萎縮、老人斑の沈着及び神経原線維変化を特徴とする神経変性疾患で、アミロイドβペプチドの形状変化と、それに伴う線維化による不溶化分子が神経細胞に沈着し、その毒性により神経細胞の死が誘導され、発症する。
【0003】
アミロイドβペプチド(Aβ)はニューロンアミロイド前駆体蛋白質からβセクレターゼなどによる切断で生じる分解物で、Aβ1-40とAβ1-42の2種が生成するが、Aβ1-42の方がより凝集しやすく、疾患及び神経毒性と相関することが報告されている。
【0004】
アミロイドβペプチドの形状変化及び線維化を阻害できれば、アルツハイマー病の発症を阻止することができる。
【0005】
特許文献1(国際公開第2005/105998号パンフレット)には、Aβ1-42と特異的に結合し、その線維化反応を阻害する活性を有する一本鎖抗体がアルツハイマー病の予防・治療剤として有用であることが記載されている。
【0006】
しかしながら、抗体は高分子の蛋白質であるため高価であり、生産、精製工程の煩雑化、あるいは安定性の欠如といった問題を有する。
【0007】
低分子のアルツハイマー病の予防・治療剤としては、アミロイドβペプチドの生成を抑制する作用を有する薬剤、例えば、ロダニン誘導体(特許文献2:特開平6−192091号公報)、ベンズイミダゾール誘導体(特許文献3:米国特許第5552426号明細書)、ビンポセチン誘導体(特許文献4:国際公開第96/25161号パンフレット)、芳香族アミド誘導体(特許文献5:国際公開第2004/014843号パンフレット)が知られている。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】国際公開第2005/105998号パンフレット
【特許文献2】特開平6−192091号公報
【特許文献3】米国特許第5552426号明細書
【特許文献4】国際公開第96/25161号パンフレット
【特許文献5】国際公開第2004/014843号パンフレット
【発明の概要】
【発明が解決しようとする課題】
【0009】
本発明は、アミロイドβペプチドのミミックペプチドとして機能し、アミロイドβペプチドの線維化を阻害しうるペプチドを提供することを目的とする。
【課題を解決するための手段】
【0010】
本発明の要旨は以下のとおりである。
(1)次式(I):
X1-Asp-X2-X3-X4-Pro-X5-X6 (I)
(式中、X1は分枝アミノ酸を表し、X2、X3、X4、X5及びX6は同一、又は異なってα−アミノ酸を表す。)
で示されるアミノ酸配列を含む、アミノ酸残基数8〜30のペプチド。
(2)前記式(I)において、X2が分枝アミノ酸、ヒドロキシアミノ酸、芳香族アミノ酸又は脂肪族アミノ酸である前記(1)に記載のペプチド。
(3)前記式(I)において、X3が芳香族アミノ酸又は分枝アミノ酸である前記(1)又は(2)に記載のペプチド。
(4)前記式(I)において、X4が脂肪族アミノ酸、ヒドロキシアミノ酸又は分枝アミノ酸である前記(1)〜(3)のいずれかに記載のペプチド。
(5)前記式(I)において、X5が分枝アミノ酸、イミノ酸、脂肪族アミノ酸、塩基性アミノ酸又はヒドロキシアミノ酸である前記(1)〜(4)のいずれかに記載のペプチド。
【0011】
(6)前記式(I)において、X6が塩基性アミノ酸、脂肪族アミノ酸又は分枝アミノ酸である前記(1)〜(5)のいずれかに記載のペプチド。
(7)次式(II):
X7-X8-X9-X10-X1-Asp-X2-X3-X4-Pro-X5-X6-X11-X12-Y1 (II)
(式中、X1、X2、X3、X4、X5及びX6は前記(1)に記載の式(I)中の定義と同一であり、X7、X8、X9、X10、X11及びX12は同一、又は異なってα−アミノ酸又は単結合を表し、Y1はOH又はNH2を表す。)
で示されるペプチド。
(8)前記式(II)において、X7がヒドロキシアミノ酸又は脂肪族アミノ酸である前記(7)に記載のペプチド。
(9)前記式(II)において、X8がヒドロキシアミノ酸又はアミドアミノ酸である前記(7)又は(8)に記載のペプチド。
(10)前記式(II)において、X9が脂肪族アミノ酸、イミノ酸、ヒドロキシアミノ酸又はアミドアミノ酸である前記(7)〜(9)のいずれかに記載のペプチド。
【0012】
(11)前記式(II)において、X10が含硫アミノ酸、分枝アミノ酸、芳香族アミノ酸又はイミノ酸である前記(7)〜(10)のいずれかに記載のペプチド。
(12)前記式(II)において、X11が複素環式アミノ酸、芳香族アミノ酸、脂肪族アミノ酸又は分枝アミノ酸である前記(7)〜(11)のいずれかに記載のペプチド。
(13)前記式(II)において、X12が分枝アミノ酸、酸性アミノ酸又はイミノ酸である前記(7)〜(12)のいずれかに記載のペプチド。
(14)前記式(I)で示されるアミノ酸配列と、次式:
Tyr-Gly-Arg-Lys-Lys-Arg-Arg-Gln-Arg-Arg-Arg
で示されるTAT配列とを含む、アミノ酸残基数19〜30のペプチドである前記(1)〜(13)のいずれかに記載のペプチド。
(15)次式(III):
Z1-X13-Gly-X14-X15-Pro-Trp-Met-Z2 (III)
(式中、X13、X14及びX15は同一、又は異なってα−アミノ酸を表し、Z1及びZ2は同一、又は異なってシステイン又はセリンを表し、Z1及びZ2がシステインを表す場合、これらの間で架橋されていてもよい。)
で示されるアミノ酸配列を含む、アミノ酸残基数9〜30のペプチド。
【0013】
(16)前記式(III)において、X13が芳香族アミノ酸である前記(15)に記載のペプチド。
(17)前記式(III)において、X14がヒドロキシアミノ酸である前記(15)又は(16)に記載のペプチド。
(18)前記式(III)において、X15が塩基性アミノ酸である前記(15)〜(17)のいずれかに記載のペプチド。
(19)前記式(III)で示されるアミノ酸配列と、次式:
Tyr-Gly-Arg-Lys-Lys-Arg-Arg-Gln-Arg-Arg-Arg
で示されるTAT配列とを含む、アミノ酸残基数20〜30のペプチドである前記(15)〜(18)のいずれかに記載のペプチド。
(20)前記(1)〜(19)のいずれかに記載のペプチドを含有する医薬組成物。
【0014】
(21)前記(1)〜(19)のいずれかに記載のペプチドを含有するアミロイドβ線維化阻害剤。
(22)アルツハイマー病の予防・治療剤である前記(21)に記載のアミロイドβ線維化阻害剤。
(23)アミロイドβ線維化阻害活性を有することが確認されていないペプチドから、アミロイドβペプチド1-42と特異的に結合してその線維化反応を阻害する活性を有する一本鎖抗体に結合し、かつアミロイドβ線維化阻害活性を有するペプチドを探索することを特徴とするアミロイドβ線維化阻害ペプチドのスクリーニング方法。
(24)前記(23)に記載のスクリーニング方法により取得されるアミロイドβ線維化阻害ペプチド。
【発明の効果】
【0015】
本発明のペプチドは、アルツハイマー病の予防・治療剤として有用である。
【図面の簡単な説明】
【0016】
【図1】図1は、B6 scFvに結合したペプチドファージクローンのELISAの結果を示す図である。
【図2】図2は、D1 scFvに結合した12merペプチドファージクローンのELISAの結果を示す図である。
【図3】図3は、D1 scFvに結合したC7CペプチドファージクローンのELISAの結果を示す図である。
【図4】図4は、ペプチドファージによるAβ1-42ペプチドの線維化阻害実験の結果を示す図である。
【図5】図5は、Aβ1-42の線維化を阻害するB6 scFv、B7 scFv又はD1 scFvを鋳型として単離したファージクローンの提示するペプチド配列一覧を示す図である。
【図6】図6は、ビオチン化したB6又はB7 scFv結合性配列とTAT配列との融合分子を示す図である。
【図7】図7は、TATとB6-L1の融合ペプチドがB6 scFvに認識されることを示す図である。
【図8】図8は、TATとB6-L1、B7-C15及びB7-S15それぞれとの融合ペプチドのAβ1-42線維化阻害活性の測定結果を示す図である。
【図9】図9は、TATとB6-L1及びB7-C15それぞれとの融合ペプチドによるAβコンホーマー(Aβオリゴマー及びAβ線維)の沈降実験の結果を示す図である。
【発明を実施するための形態】
【0017】
本発明のペプチドは、前記式(I)又は(III)で示されるアミノ酸配列を含む、アミノ酸残基数30以下のペプチドであり、好ましくは前記式(I)で示されるアミノ酸配列を含む、アミノ酸残基数8〜20のペプチド及び前記式(III)で示されるアミノ酸配列を含む、アミノ酸残基数9〜20のペプチドである。
【0018】
前記式(I)において、X1で表される分枝アミノ酸としては、バリン、ロイシン、イソロイシン等が挙げられる。
【0019】
前記式(I)、(II)又は(III)において、X2、X3、X4、X5、X6、X7、X8、X9、X10、X11、X12、X13、X14又はX15で表されるα−アミノ酸としては、例えば、グリシン、アラニン等の脂肪族アミノ酸;バリン、ロイシン、イソロイシン等の分枝アミノ酸;セリン、トレオニン等のヒドロキシアミノ酸;アスパラギン酸、グルタミン酸等の酸性アミノ酸;アスパラギン、グルタミン等のアミドアミノ酸;リシン、ヒドロキシリシン、アルギニン等の塩基性アミノ酸;システイン、シスチン、メチオニン等の含硫アミノ酸;フェニルアラニン、チロシン等の芳香族アミノ酸;トリプトファン、ヒスチジン等の複素環式アミノ酸;プロリン、4−ヒドロキシプロリン等のイミノ酸が挙げられる。
【0020】
前記式(I)及び(II)において、X2で表されるα−アミノ酸としては、好ましくは分枝アミノ酸、ヒドロキシアミノ酸、芳香族アミノ酸、脂肪族アミノ酸、更に好ましくはバリン、ロイシン、イソロイシン、トレオニン、フェニルアラニンが挙げられ;X3で表されるα−アミノ酸としては、好ましくは芳香族アミノ酸、分枝アミノ酸、更に好ましくはフェニルアラニン、ロイシン、チロシンが挙げられ;X4で表されるα−アミノ酸としては、好ましくは脂肪族アミノ酸、ヒドロキシアミノ酸、分枝アミノ酸、更に好ましくはアラニン、トレオニン、ロイシン、セリン、バリンが挙げられ;X5で表されるα−アミノ酸としては、好ましくは分枝アミノ酸、イミノ酸、脂肪族アミノ酸、塩基性アミノ酸、ヒドロキシアミノ酸、更に好ましくはロイシン、イソロイシン、プロリン、アラニン、アルギニン、セリンが挙げられ;X6で表されるα−アミノ酸としては、好ましくは塩基性アミノ酸、脂肪族アミノ酸、分枝アミノ酸、更に好ましくはアルギニン、アラニン、ロイシン、イソロイシンが挙げられる。
【0021】
前記式(II)において、X7で表されるα−アミノ酸としては、好ましくはヒドロキシアミノ酸、脂肪族アミノ酸、更に好ましくはトレオニン、グリシンが挙げられ;X8で表されるα−アミノ酸としては、好ましくはヒドロキシアミノ酸、アミドアミノ酸、更に好ましくはセリン、アスパラギンが挙げられ;X9で表されるα−アミノ酸としては、好ましくは脂肪族アミノ酸、イミノ酸、ヒドロキシアミノ酸、アミドアミノ酸、更に好ましくはグリシン、プロリン、セリン、トレオニン、アスパラギンが挙げられ;X10で表されるα−アミノ酸としては、好ましくは含硫アミノ酸、分枝アミノ酸、芳香族アミノ酸、イミノ酸、更に好ましくはメチオニン、ロイシン、イソロイシン、フェニルアラニン、プロリンが挙げられ;X11で表されるα−アミノ酸としては、好ましくは複素環式アミノ酸、芳香族アミノ酸、脂肪族アミノ酸又は分枝アミノ酸、更に好ましくはヒスチジン、チロシン、アラニン、ロイシンが挙げられ;X12で表されるα−アミノ酸としては、好ましくは分枝アミノ酸、酸性アミノ酸、イミノ酸、更に好ましくはバリン、グルタミン酸、プロリンが挙げられる。
【0022】
前記式(III)において、X13で表されるα−アミノ酸としては、好ましくは芳香族アミノ酸、アミドアミノ酸、更に好ましくはチロシン、フェニルアラニン、グルタミン、最も好ましくはチロシンが挙げられ;X14で表されるα−アミノ酸としては、好ましくはヒドロキシアミノ酸、複素環式アミノ酸、塩基性アミノ酸、更に好ましくはセリン、トレオニン、ヒスチジン、アルギニン、最も好ましくはトレオニンが挙げられ;X15で表されるα−アミノ酸としては、好ましくは塩基性アミノ酸、酸性アミノ酸、分枝アミノ酸、更に好ましくはリシン、グルタミン酸、ロイシン、最も好ましくはリシンが挙げられる。
【0023】
本発明のペプチドは、公知の方法によって化学合成することもでき、例えばペプチド自動合成装置によって合成することができる。この基本的な合成過程はR.B. Merifield, Advance in Enzymology 32: 221-296(1969)に記載の方法を適用できる。この方法は、カルボキシル末端のアミノ酸を樹脂担体に共有結合させておき、α−アミノ基の保護基の除去、保護アミノ酸の縮合を順次繰返して、アミノ末端に向けてペプチド鎖を延長させ目的のアミノ酸配列を有するペプチド樹脂を得ることを原理とするものである。
【0024】
各アミノ酸の縮合やα−アミノ基の保護基の除去等は、Boc, Fmoc法を利用してほぼ同一の条件でなされ、中間体の精製も行わないため、合成に際しては一般に高度な熟練は要求されない。しかもこの方法は迅速であり、種々のペプチドを合成するに際し、非常に便利な方法である。こうして得られた保護ペプチド樹脂を、例えば無水フッ化水素、トリフルオロメタンスルホン酸もしくはトリフルオロ酢酸と種々の添加物の共存下に反応させることにより、ペプチドを樹脂から脱離させることと全保護基の除去を一段階で行うことができる。
【0025】
樹脂担体としてカルボキシル末端カルボン酸型ペプチド合成用樹脂を用いれば、カルボキシル末端がカルボキシル基であるペプチド、例えば前記式(II)において、Y1がOHであるペプチドを得ることができ、カルボキシル末端アミド型ペプチド合成用樹脂を用いれば、カルボキシル末端カルボン酸がアミド化されたペプチド、例えば前記式(II)において、Y1がNH2であるペプチドを得ることができる。
【0026】
得られたペプチド粗製物は、ペプチドを精製する公知の手段で精製することができる。例えば、ゲル濾過、陽イオン交換樹脂もしくは陰イオン交換樹脂を用いるイオン交換クロマトグラフィー、更には疎水クロマトグラフィー、分配吸着クロマトグラフィーなど、種々の原理によるカラムクロマトグラフィーや高速液体クロマトグラフィーが挙げられる。
【0027】
本発明のペプチドは、複数のシステイン残基を含む場合には、それらのシステイン残基の間で架橋されていてもよい。例えば、次式(IIIa):
Cys1-X13-Gly-X14-X15-Pro-Trp-Met-Cys2 (IIIa)
(式中、X13、X14及びX15は同一、又は異なってα−アミノ酸を表し、Cys1とCys2の間で架橋されていてもよい。)
で示されるアミノ酸配列を含む、アミノ酸残基数9〜30、好ましくは9〜20のペプチドは、Cys1とCys2の間で架橋されていてもよい。
【0028】
これらの架橋としては、システイン残基間を直接ジスルフィド架橋してもよく、またスペーサーとしてジスルフィド化合物を用いて、スペーサーを介してジスルフィド架橋してもよい。ジスルフィド架橋は、例えばペプチドの希釈水溶液をK3[Fe(CN)6]で酸化、又は酸性条件下のヨウ素で酸化することにより形成することができる。
【0029】
本発明のペプチドは種々の塩の形で得ることできる。その塩としては、例えば無機酸や、蟻酸、酢酸、酒石酸、クエン酸などの有機酸との塩、もしくはナトリウムやアンモニアなどの無機塩基や、トリエチルアミン、エチルアミン、メチルアミンなどの有機塩基との塩が挙げられる。
【0030】
ペプチドのアミノ基のビオチン化はアミノ酸誘導体を樹脂上で縮合する通常のHOBt−DCC, HBTu−HOBt方法などで可能である。
【0031】
本発明のスクリーニング方法は、アミロイドβ線維化阻害活性を有することが確認されていないペプチドから、Aβ1-42と特異的に結合してその線維化反応を阻害する活性を有する一本鎖抗体に結合し、かつアミロイドβ線維化阻害活性を有するペプチドを探索することを特徴とするものである。
【0032】
Aβ1-42と特異的に結合し、その線維化反応を阻害する活性を有する一本鎖抗体(scFv)としては、好ましくは、WO2005/105998に記載されているヒト一本鎖可変領域フラグメントscFv(B6、B7、D1、F10)が挙げられる。本明細書において、これらの抗体を鋳型と称する。
【0033】
B6scFv(WO2005/105998のクローンB6)のVH鎖のアミノ酸配列を配列番号1に、VL鎖のアミノ酸配列を配列番号2に示す。
【0034】
B7scFv(WO2005/105998のクローンB7)のVH鎖のアミノ酸配列を配列番号3に、VL鎖のアミノ酸配列を配列番号4に示す。
【0035】
D1scFv(WO2005/105998のクローンD1)のVH鎖のアミノ酸配列を配列番号5に、VL鎖のアミノ酸配列を配列番号6に示す。
【0036】
F10scFv(WO2005/105998のクローンF10)のVH鎖のアミノ酸配列を配列番号7に、VL鎖のアミノ酸配列を配列番号8に示す。
【0037】
本発明のペプチドを得るための方法としては、例えば以下に述べるペプチドライブラリー法が挙げられる。更に、ペプチドライブラリー法は、バクテリオファージを用いる方法と、化学合成でライブラリーを作製する方法に分けられる。
【0038】
ファージランダムペプチドライブラリーを構築する方法は、例えばM13系ファージのコートプロテイン(例えばgeneIII蛋白やVIII蛋白)の遺伝子にランダムな配列を有する合成遺伝子を連結し、作製すればよい。その方法としては、Science, 249, 386 (1990), Proc. Natl. Acad. Sci. USA, 87, 6378 (1990) 等に記載された方法を用いることができる。挿入する遺伝子の大きさは、発現されるペプチドが安定であれば特に制限はないが、作製したライブラリーがより多くのランダムな配列を網羅し、更に標的分子に結合能を有するためには6から15アミノ酸が好ましい。鋳型とするscFvに結合するペプチドファージを選択するためには、カラムやマイクロタイタープレート上に精製した前記scFvを直接あるいは抗IgG抗体等を介して固定化し、前記ライブラリーを接触させる。その後、非結合ファージは洗浄操作で洗い流す。洗浄後、結合しているファージを酸で溶出し、中和した後、大腸菌に感染させ増幅させる。この操作(パニング)を3回か4回繰り返すと、前記scFvに親和性のあるファージが濃縮される。ここで単一なクローンを得るためには、再度大腸菌にファージを感染させ、抗生物質を含んだ寒天培地上でシングルコロニーを形成させる。個々のコロニーを液体培地で培養した後、上清中のファージをポリエチレングリコール等で沈殿濃縮し、その塩基配列を決定すれば、ペプチドの構造を知ることができる。
【0039】
ランダムなアミノ酸配列を有するペプチドライブラリーは、前記のファージを用いる方法のほか、化学合成で作製することも可能である。その方法としては、ビーズを用いる方法(Nature, 354, 82 (1991))、液相フォーカシング法(Nature, 354, 84 (1991))、マイクロプレート法(Science, 251, 767 (1991))等が挙げられる。
【0040】
ライブラリーより得られた配列を有するペプチドを大量に調製するためには、人工的にペプチドを合成する方法や、遺伝子組換え技術を利用して大腸菌、酵母、昆虫細胞、動物細胞等で発現させる方法が挙げられる。
【0041】
人工的にペプチドを合成する方法は、一般的なペプチド合成法により容易に行うことができ、例えば固相合成法で行うことが簡便であり、目的の配列に、欠失、置換、挿入又は付加を施した変異体を作製することも容易である(細胞工学別冊、抗ペプチド抗体実験プロトコール、p.26-46、秀潤社)。また、非天然型アミノ酸の導入、各アミノ酸残基の化学修飾やシステイン残基を導入することにより分子内を環化させて構造を安定化させる等の修飾を施してもよい。
【0042】
遺伝子組換え技術を利用する場合、得られたアミノ酸配列から、codon usageに従ってDNA配列を設定し(Molecular Cloning, Appendix D1参照, マニアティスら;Cold Spring Habor Laboratory社、1989)、宿主の細胞に導入することは、技術的に確立されている。更に、塩基配列に変異を導入することにより、アミノ酸を他の残基に変換することも可能である。例えば大腸菌で発現させる場合は、得られたDNA配列をプロモーター配列、例えばトリプトファン合成酵素オペロン(Trp)、ラクトースオペロン(lac)プロモーターに結合し、リボゾーム結合配列、例えばシャインダルガルノ(SD)配列や転写終結因子認識部位を付加することが望ましい。作製した発現ベクターを大腸菌に導入する方法等はMolecular Cloning (マニアティスら;Cold Spring Habor Laboratory社、1989)記載の方法が使用できる。発現産物を精製する方法は、例えば各種クロマトグラフィーを用いることができる。
【0043】
得られたペプチドがアミロイドβ線維化阻害活性を有するか否かを調べる方法としては、例えば、チオフラビンT(ThT)法(H. Levine, III, Protein Science 21: 404-410 (1993))が挙げられる。
【0044】
本発明のスクリーニング方法に従ってアミロイドβ線維化阻害活性が確認されたペプチドは、該ペプチドをリード化合物とし、これに、1もしくは2以上のアミノ酸残基の欠失、置換、挿入もしくは付加、又は修飾を施し、アミロイドβ線維化阻害活性を有するペプチドを得ることにより、類縁のアミロイドβ線維化阻害ペプチドを製造することができる。このペプチドがアミロイドβ線維化阻害活性を有するか否かは、前記の方法と同様にして調べることができる。
【0045】
本発明は、また、前記スクリーニング方法により取得されるアミロイドβ線維化阻害ペプチドを提供する。ここで、「前記スクリーニング方法により取得される」とは、前記スクリーニング方法により実際にアミロイドβ線維化阻害活性が確認されたペプチドをいう。前記ペプチドのアミノ酸残基数は、通常6〜40、好ましくは8〜20、更に好ましくは8〜15である。
【0046】
本発明のペプチドは、アミロイドβ線維化阻害活性を有し、アルツハイマー病の予防・治療剤として有用である。
【0047】
本発明のアミロイドβ線維化阻害ペプチドは、そのままもしくは自体公知の薬学的に許容される担体、賦形剤等と混合した医薬組成物として経口又は非経口的に投与することができる。
【0048】
経口投与のための剤形としては、具体的には錠剤、丸剤、カプセル剤、顆粒剤、細粒剤、散剤、シロップ剤、乳剤、懸濁剤等が挙げられる。かかる剤形は、自体公知の方法によって製造され、製剤分野において通常用いられる担体もしくは賦形剤を含有するものである。例えば錠剤用の担体、賦形剤としては、ラクトース、マルトース、ショ糖、澱粉、ステアリン酸マグネシウム等が挙げられる。
【0049】
非経口投与のための剤形としては、例えば、点眼剤、軟膏剤、注射剤、湿布剤、坐薬、経鼻吸収剤、経肺吸収剤、経皮吸収剤、局所徐放剤等が挙げられる。溶液製剤は自体公知の方法、例えば、アミロイドβ線維化阻害ペプチドを通常、注射剤に用いられる無菌の水溶液に溶解、更には乳化して、リポソームに包埋させた状態で調製されうる。ペプチドは生体内でペプチダーゼ等による分解を受け易く、また目的の場所への到達性の問題等があることから、ここに述べたリポソームを含む適切なデリバリー法の活用は好ましい使用形態の一つである。ペプチドのデリバリー法の活用については前記リポソームを用いる方法はその一つであるが、これに限られたものではない。固体製剤は、自体公知の方法、例えば、アミロイドβ線維化阻害ペプチドにマンニトール、トレハロース、ソルビトール、ラクトース、グルコース等を賦形剤として加え、そのまま凍結乾燥することにより調製されうる。更にこれを粉体化して用いることもできる。また、これら粉体をポリ乳酸やグリコール酸等と混合し固体化して用いることもできる。ゲル化剤は、自体公知の方法、例えば、アミロイドβ線維化阻害ペプチドをグリセリン、ポリエチレングリコール、メチルセルロース、カルボキシメチルセルロース、ヒアルロン酸、コンドロイチン硫酸等の増粘剤や多糖に溶解した状態で調製されうる。
【0050】
いずれの製剤においても、安定化剤としてヒト血清アルブミン、ヒト免疫グロブリン、α2マクログロブリン、アミノ酸等を添加することができ、また分散剤あるいは吸収促進剤としてアミロイドβ線維化阻害ペプチドの活性を損なわない範囲でアルコール、糖アルコール、イオン性界面活性剤、非イオン性界面活性剤等を添加することができる。また、微量金属や有機酸塩も必要に応じて加えることができる。
【0051】
本発明の医薬組成物において、有効成分であるペプチドとしての投与量は、当該ペプチドの活性、患者の年令、体重、疾患の種類又は程度により異なるが、一般には、経口投与では、通常1日0.001〜1000mg/kg体重であり、静脈内、筋肉内又は皮下投与では、通常1日0.001〜1000mg/kg体重である。投与回数は、通常経口投与では1日1〜3回、注射剤では1日1〜2回である。
【0052】
本明細書は、本願の優先権の基礎である特願2006−125769の明細書及び/又は図面に記載される内容を包含する。
【実施例】
【0053】
以下、製造例及び実施例を挙げて本発明を更に詳細に説明するが、本発明はこれらに限定されるものではない。
【0054】
(実施例1)
Kaji et al., J. Biochem. 129: 577-583 (2001)及びS. Hashiguchi et al., J. Biochem. 133: 43-49 (2003)に従って以下の実験を行った。
【0055】
1.材料及び方法
(1)E-tagカラムによるscFvの精製
Aβに結合するscFvを発現させるために、Hashiguchiらの方法(J. Biochem. 133: 43-49 (2003))によりscFvファージクローンを大腸菌株HB2151に感染させた。得られた培地上清からE-tagカラムAmersham Biosciences)を用いてアミロイド特異的scFv(B6、B7、D1、F10)を精製した。
【0056】
(2)ペプチドファージライブラリー
ファージのgeneIII蛋白のアミノ末端に12個のランダムなアミノ酸配列を提示したPh.D.-12ライブラリー(New England Biolabs、MA)及びファージのgeneIII蛋白のアミノ末端にシステインとシステインの間に7つのアミノ酸配列を含むペプチドを提示するPh.D.-C7Cライブラリー(New England Biolabs、MA)を用いた。
【0057】
(3)バイオパニング
鋳型となるヒト一本鎖抗体(scFv:B6、B7、D1又はF10)又はコントロールscFv 1μg/100μl/well (0.1 M NaHCO3 pH 8.6)をMaxisorp plateに4℃で6時間コートした。ブロッキング溶液としてそれぞれのウェルを1stパニングでは0.5%ゼラチン,2ndパニングでは0.25% BSA,3rdパニングでは0.5%ゼラチンを用いて4℃で14時間ブロックした。12mer又はC7Cペプチドファージライブラリーをブロッキング溶液で希釈後(1.5 x 1012 pfu/100 μl) 30分静置後、コントロールscFvで吸収操作を行った。すなわち、コントロールscFvをコートしたウェルを0.2% Tween 20/PBS (PBST)で3回洗浄しファージ溶液を加え室温で1時間反応させ、コントロールscFvと結合しなかったファージ溶液を回収した。このファージ溶液を鋳型とするscFvをコートしたウェルに加え室温で1時間反応させた。0.2% PBSTを用いて10回洗浄後1 mg/ml BSA / 0.1 M グリシン塩酸 pH 2.2を100 μl加え室温で5分置くことにより結合したファージを回収し、ただちに1 M トリス塩酸pH 9.1を15 μl加え中和した。回収したファージを大腸菌ER2738に感染させ増幅させ、2, 3ラウンドのパニングに用いた(1.5 x 1012 pfu/100 μl)。
【0058】
(4)ファージクローンの単離
パニングを2回行った後、回収したファージをER2738に感染させLB/Tet/X-gal plateで培養した。溶菌してできたプラークを再度ER2738に感染させ、ファージクローンを単離した。
【0059】
(5)ELISA
パニングで鋳型としたscFv又はコントロールscFv 50 ng/40 μl/well (0.1 M NaHCO3 pH 8.6)をELISAプレートに4℃で6時間コートした。0.5%ゼラチンを用いて4℃で13時間ブロックした。0.2% PBSTを用いてウェルを3回洗浄後、単離したペプチドファージクローン溶液(約1.6 x 1011 virions/40 μl)を加え室温で1時間反応させた。結合したファージは40μlのビオチン化抗M13モノクローナル抗体(1000倍希釈)、ストレプトアビジンアルカリフォスファターゼ(1000倍希釈)を反応させた後、基質パラニトロフェニルリン酸を用いて検出した。
【0060】
(6)DNA塩基配列解析
特異性の認められたファージクローンをフェノール/クロロホルム処理して除蛋白した後、エタノール沈殿でDNAを精製し、塩基配列決定の鋳型とした。primer-96gIII(1 pg/μl) 5’-CCC TCA TAG TTA GCG TAA CG-3’(New England Biolabs, MA)を用いて、ランダムなペプチド配列が挿入されている領域の遺伝子増幅を行い、DNA塩基配列決定を行った。
【0061】
(7)ペプチド合成
本発明のペプチドは、一般的な固相ペプチド合成法、Fmoc/HBTu+HOBt法により合成した。本発明のペプチドB6-L1, B7-C15, B7-S15のそれぞれにTAT配列を連結したペプチドを合成した。なお、B7-C15については、カルボキシル末端側にグリシンを付加したものにTAT配列を連結した。ちなみにTAT配列は脳血管関門を通過する機能をもつペプチド配列である(P. Jarver and Ulo Langel, The use of cell-penetrating peptides as a tool for gene regulation, DDT, 9: 395-401, 2004)。
【0062】
なお、B6-L1, B7-C15, B7-S15のそれぞれの配列のアミノ末端側にTAT配列を融合したものがTAT-B6-L1, TAT-B7-C15, TAT-B7-S15であり、B6-L1の配列のカルボキシル末端側にTAT配列を融合したものがB6-L1-TATである。このペプチドの結合を検出するためにペプチドのアミノ末端にビオチンが結合しているペプチド配列として合成した。
【0063】
B6-L1, B7-C15, B7-S15、及びこれらとTAT配列との融合分子のアミノ酸配列と配列番号との関係は以下のとおりである。
B6-L1:配列番号9;
B7-C15:配列番号16
B7-S15:配列番号23
TAT-B6-L1:配列番号24
B6-L1-TAT:配列番号25
TAT-B7-C15:配列番号26
TAT-B7-S15:配列番号27
【0064】
(8)アミロイドβ線維化阻害実験
20 mMリン酸バッファー pH 7.0を用いて40 μMに希釈したAβ1-42ペプチドに合成したペプチド(TAT-B6-L1, B6-L1-TAT, TAT-B7-C15, TAT-B7-S15)を様々な濃度(1.65 nM, 16.5 nM, 33 nM)で加えた。0, 6, 24時間に10 μlのサンプルを回収し11 μMのチオフラビンT(Sigma)/リン酸バッファー90 μlを加えマルチラベルプレートカウンター、Wallac 1420 ARVOsx (Perkin-Elmer; Wellesley, MA)を用い、450nmの励起光により発生する482nmの蛍光強度を測定した。
【0065】
(9)Aβ線維化阻害ペプチドによるAβコンホーマー(Aβオリゴマー及びAβ線維)の沈降実験
Aβ1-42を20 mMリン酸バッファーpH 7.0に40 μMの濃度で溶解し、その1.5時間目に、40 μMに調製したTAT-B6-L1又はTAT-B7-C15ペプチドを液量比(1:1)で加え37℃で1時間反応させた(最終濃度:Aβ[20 μM]/合成ペプチド[20 μM])。反応後、免疫沈降前のサンプルを「沈降前」として一部サンプリングし、残りのサンプルを20 μg/μlに調製したM-280ストレプロアビジン-マグネットビーズ(Dynal Biotech, Oslo, Norway)と液量比(1:1)で氷上で1時間反応させた(最終濃度:Aβ[10 μM]/合成ペプチド[10 μM]/ビーズ[10 μg/μl])。反応後、マグネットによりビーズを沈降させ上清を「上清」とした。沈降したビーズに回収した上清と等液量のPBSを加え「沈殿物」とした。得られた各サンプルを電気泳動し、抗Aβ抗体によるWestern blot解析を行った。
【0066】
2.結果
(1)Aβ特異的scFvに結合するペプチドファージクローン
(a)B6 scFvに結合したペプチドファージクローンのELISAの結果を図1に示す。無関係のscFv(コントロールscFv)としてFv1E1というAβの線維化を阻害しない抗体を用いた。ファージライブラリーは12merのアミノ酸配列を提示するPh.D.-12ライブラリー(New England Biolabs、MA)を用いた。実験は前記(5)ELISAで記したとおりに行った。
(b)D1 scFvに結合した12merペプチドファージクローンのELISAの結果を図2に示す。無関係のscFv(コントロールscFv)としてMY5RというAβの線維化を阻害しない抗体を用いた。ファージライブラリーは12merのアミノ酸配列を提示するPh.D.-12ライブラリー(New England Biolabs、MA)を用いた。実験は前記(5)ELISAで記したとおりに行った。
(c)D1 scFvに結合したC7CペプチドファージクローンのELISAの結果を図3に示す。無関係のscFv(コントロールscFv)としてMY5RというAβの線維化を阻害しない抗体を用いた。ファージライブラリーはシステインとシステインの間に7つのアミノ酸配列を含むペプチドを提示するPh.D.-C7Cライブラリー(New England Biolabs、MA)を用いた。実験は前記(5)ELISAで記したとおりに行った。
【0067】
(2)B6結合性ペプチドファージのAβ1-42線維化阻害活性
B6結合性ペプチドファージによるAβ1-42ペプチドの線維化阻害実験の結果を図4に示す。Aβ1-42線維化阻害実験は前記(8)線維化阻害実験で記したとおりに行った。pepB6-L1及びpepB6-L10は、Aβ1-42の線維化を阻害するB6 scFvを鋳型として、それに結合するファージクローンであり、pep1E1-L1, 1-L2, 1-L4, 1-L12は、Aβ1-42の線維化を阻害しないFv1E1 scFv(WO2005−105998段落0074〜0075に記載)を鋳型として単離したファージクローンである。ここで用いたファージクローンの濃度は3.0 x 1012 virions/mlである。
【0068】
(3)Aβ特異的scFvのエピトープ(結合性配列)
Aβ1-42の線維化を阻害するB6 scFv、B7 scFv又はD1 scFvを鋳型として単離したファージクローンの提示するペプチドのアミノ酸配列一覧を図5に示す。ファージライブラリーとしてPh.D.-12ライブラリー(図5中12mer*と略)又はPh.D.-C7Cライブラリー(図5中C7C*と略)を使用した。pepB7-C15とpepD1-C5,C7,C20は同一の配列であった。番号の前についているLは12merのライブラリーから、CはC7Cのライブラリーから単離されたクローンであることを示している。
【0069】
各ファージクローンの提示するペプチドのアミノ酸配列と配列番号との関係は以下のとおりである。
pepB6-L1:配列番号9;
pepB6-L10:配列番号10;
pepD1-L5,L9:配列番号11;
pepD1-L6:配列番号12;
pepD1-L7:配列番号13;
pepD1-L13:配列番号14;
pepD1-L20:配列番号15;
pepB7-C15 (pepD1-C5,C7,C20):配列番号16;
pepD1-C2,C13:配列番号17;
pepD1-C18:配列番号18;
pepD1-C3:配列番号19;
pepD1-C11:配列番号20;
pepD1-C8:配列番号21;
pepD1-C10:配列番号22
【0070】
(4)ビオチン化したB6又はB7 scFv結合性配列とTAT配列との融合分子
ビオチン化したB6 scFv結合性配列とTAT配列との融合分子を図6に示す。B6-L1, B7-C15, B7-S15のそれぞれの配列のアミノ末端側にTAT配列を融合したものがTAT-B6-L1, TAT-B7-C15, TAT-B7-S15であり、B6-L1の配列のカルボキシル末端側にTAT配列を融合したものがB6-L1-TATである。TAT配列は脳血管関門を通過する機能をもつペプチド配列である(P. Jarver and Ulo Langel, The use of cell-penetrating peptides as a tool for gene regulation, DDT, 9: 395-402, 2004)。これらのペプチドのアミノ末端をビオチンで標識し、このペプチドの結合を酵素標識したアビジンを用いて定量を行った。
【0071】
(5)TATとB6-L1の融合ペプチドはB6 scFvに認識された(図7)。合成ペプチド、TAT-B6-L1あるいはB6-L1-TATを200 ng/ wellの条件でELISAプレートにコートし、種々の濃度のB6 scFvを添加しその結合活性を検討した。コントロールペプチドとして無関係のペプチド(GSGGGSCGYWRSEWGLCG)を用いた。B6 scFvの量依存的な結合反応が確認された。
【0072】
(6)TATとB6-L1, B7-C15, B7-S15それぞれとの融合ペプチドのAβ1-42線維化阻害活性
TATとB6-L1, B7-C15, B7-S15それぞれとの融合ペプチドのAβ1-42線維化阻害活性の測定結果を図8に示す。線維化阻害実験は前記線維化阻害実験で記したとおりに行った。TAT-B6-L1, B6-L1-TAT, TAT-B7-C15, TAT-B7-S15は線維化阻害活性があることが明らかとなった。前記のコントロールペプチドは全く阻害活性を示さなかった。
【0073】
(7)TAT-B6-L1, TAT-B7-C15ペプチドによるAβコンホーマー(Aβオリゴマー及びAβ線維)の沈降実験
TAT-B6-L1, TAT-B7-C15ペプチドの沈降実験の結果を図9に示す。Aβ1-42を20 mMリン酸バッファー pH 7.0に40 μMの濃度で溶解し、その1.5時間目にビオチン化TAT-B6-L1又はビオチン化TAT-B7-C15ペプチドを加え結合物をストレプトアビジン-マグネットビーズで沈降させた。沈降前はAβオリゴマーやAβ線維などのバンドが確認できるが、TAT-B6-L1, TAT-B7-C15ペプチドで沈降を行った上清にはAβオリゴマーやAβ線維は確認されず、沈殿物の方で確認できた。この結果からAβ1-42溶解後1.5時間目に形成されるAβオリゴマーやAβ線維とTAT-B6-L1, TAT-B7-C15ペプチドが結合することが明らかとなった。無関係なコントロールペプチドでは沈降反応は認められなかった(図9には示されていない)。
【0074】
本明細書中で引用した全ての刊行物、特許及び特許出願をそのまま参考として本明細書中にとり入れるものとする。
【特許請求の範囲】
【請求項1】
次式(III):
Z1-X13-Gly-X14-X15-Pro-Trp-Met-Z2 (III)
(式中、X13、X14及びX15は同一、又は異なってα−アミノ酸を表し、Z1及びZ2は同一、又は異なってシステイン又はセリンを表し、Z1及びZ2がシステインを表す場合、これらの間で架橋されていてもよい。)
で示されるアミノ酸配列を含む、アミノ酸残基数9〜30のペプチド。
【請求項2】
前記式(III)において、X13が芳香族アミノ酸である請求項1記載のペプチド。
【請求項3】
前記式(III)において、X14がヒドロキシアミノ酸である請求項1又は2記載のペプチド。
【請求項4】
前記式(III)において、X15が塩基性アミノ酸である請求項1〜3のいずれか1項に記載のペプチド。
【請求項5】
前記式(III)において、X13が芳香族アミノ酸であり、X14がヒドロキシアミノ酸であり、X15が塩基性アミノ酸である請求項1記載のペプチド。
【請求項6】
前記式(III)で示されるアミノ酸配列と、次式:
Tyr-Gly-Arg-Lys-Lys-Arg-Arg-Gln-Arg-Arg-Arg
で示されるTAT配列とを含む、アミノ酸残基数20〜30のペプチドである請求項1〜5のいずれか1項に記載のペプチド。
【請求項7】
請求項1〜6のいずれか1項に記載のペプチドを含有する医薬組成物。
【請求項8】
請求項1〜6のいずれか1項に記載のペプチドを含有するアミロイドβ線維化阻害剤。
【請求項9】
アルツハイマー病の予防・治療剤である請求項8記載のアミロイドβ線維化阻害剤。
【請求項1】
次式(III):
Z1-X13-Gly-X14-X15-Pro-Trp-Met-Z2 (III)
(式中、X13、X14及びX15は同一、又は異なってα−アミノ酸を表し、Z1及びZ2は同一、又は異なってシステイン又はセリンを表し、Z1及びZ2がシステインを表す場合、これらの間で架橋されていてもよい。)
で示されるアミノ酸配列を含む、アミノ酸残基数9〜30のペプチド。
【請求項2】
前記式(III)において、X13が芳香族アミノ酸である請求項1記載のペプチド。
【請求項3】
前記式(III)において、X14がヒドロキシアミノ酸である請求項1又は2記載のペプチド。
【請求項4】
前記式(III)において、X15が塩基性アミノ酸である請求項1〜3のいずれか1項に記載のペプチド。
【請求項5】
前記式(III)において、X13が芳香族アミノ酸であり、X14がヒドロキシアミノ酸であり、X15が塩基性アミノ酸である請求項1記載のペプチド。
【請求項6】
前記式(III)で示されるアミノ酸配列と、次式:
Tyr-Gly-Arg-Lys-Lys-Arg-Arg-Gln-Arg-Arg-Arg
で示されるTAT配列とを含む、アミノ酸残基数20〜30のペプチドである請求項1〜5のいずれか1項に記載のペプチド。
【請求項7】
請求項1〜6のいずれか1項に記載のペプチドを含有する医薬組成物。
【請求項8】
請求項1〜6のいずれか1項に記載のペプチドを含有するアミロイドβ線維化阻害剤。
【請求項9】
アルツハイマー病の予防・治療剤である請求項8記載のアミロイドβ線維化阻害剤。
【図1】


【図2】

【図3】

【図4】

【図5】
【図6】

【図7】

【図8】
【図9】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公開番号】特開2012−116851(P2012−116851A)
【公開日】平成24年6月21日(2012.6.21)
【国際特許分類】
【出願番号】特願2012−15584(P2012−15584)
【出願日】平成24年1月27日(2012.1.27)
【分割の表示】特願2008−513322(P2008−513322)の分割
【原出願日】平成19年4月24日(2007.4.24)
【出願人】(504258527)国立大学法人 鹿児島大学 (284)
【Fターム(参考)】
【公開日】平成24年6月21日(2012.6.21)
【国際特許分類】
【出願日】平成24年1月27日(2012.1.27)
【分割の表示】特願2008−513322(P2008−513322)の分割
【原出願日】平成19年4月24日(2007.4.24)
【出願人】(504258527)国立大学法人 鹿児島大学 (284)
【Fターム(参考)】
[ Back to top ]