
アミロイド−β(1−42)−オリゴマー又はその誘導体を含有する調製物、その製法及びその使用
【課題】Aβ(1−42)の神経変性及び殊にニューロン傷害作用の原因となり、かつ老人性痴呆症、例えば、アルツハイマー病及びダウン−症候群において増大に出現する分子形を提供する。
【解決手段】アミロイド−β(1−42)タンパク質−オリゴマーが、SDS−ゲル電気泳動における見かけの分子量約38kDa及び/又は48kDaを有し、その際、アミロイド−β(1−42)タンパク質−オリゴマーの割合が、調製物中においてアミロイド−β(1−42)タンパク質の全量の少なくとも60質量%である、アミロイド−β(1−42)タンパク質−オリゴマー又は標的化、不動化及び/又は架橋結合アミロイド−β(1−42)タンパク質−オリゴマーの誘導体を含む調製物により解決された。
【解決手段】アミロイド−β(1−42)タンパク質−オリゴマーが、SDS−ゲル電気泳動における見かけの分子量約38kDa及び/又は48kDaを有し、その際、アミロイド−β(1−42)タンパク質−オリゴマーの割合が、調製物中においてアミロイド−β(1−42)タンパク質の全量の少なくとも60質量%である、アミロイド−β(1−42)タンパク質−オリゴマー又は標的化、不動化及び/又は架橋結合アミロイド−β(1−42)タンパク質−オリゴマーの誘導体を含む調製物により解決された。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、アミロイド−β(1−42)−タンパク質の神経変性オリゴマー、このオリゴマーが再現可能な方法及び高収率で得られる特殊製法、及びオリゴマー特異的抗体の製造のための、及びオリゴマー及びその生成と相互作用をし得る物質の発見のための診断剤及び治療剤としてのオリゴマーの使用に関する。
【0002】
本発明は、同様にして、オリゴマーの誘導体、殊に、架橋結合したオリゴマー及びそのアミロイド−β(1−42)−タンパク質の切端形をベースとするオリゴマー、その製造及びその使用にも関する。
【0003】
抗体の製造のための、又は物質の発見のための相応する方法は、抗体自体及び診断剤及び治療剤としての抗体又は物質の使用と全く同様に記載される。
【背景技術】
【0004】
アミロイド−β(1−42)−タンパク質(Aβ(1−42)としても略称される)は、アルツハイマー−及びダウン症候群−患者の脳におけるタンパク質、脂質、炭水化物及び塩から組成される不溶性の細胞外沈着物(老人性又は神経炎性斑)の中心成分である(C.L.Masters et al. PNAS 82, 4245-4249,1985)。水性環境中で重合傾向を有するこのタンパク質は極めて様々な分子形で存在し得る。
【0005】
不溶性タンパク質沈着物と老人性痴呆症、例えば、アルツハイマーの出現又は進展との簡単な相関性は、確実には実証されなかった(R.D.Terry et al Ann. Neurol. 30. 572-580(1991), D.W. Dickson et al. Nurobiol. Aging 16, 285-298(1995))。それに反して、シナップス及び認知感覚の喪失は、より良好にAβ(1−42)の可溶性形と相関すると思われる(L.F. Lue et al. Am. J. Pathol. 155, 853-862,(1999), C.A. McLean et al. Ann. Neurol. 46, 860-866 (1999))。
【0006】
Aβ(1−42)の可溶性形には、老人性痴呆症、例えば、アルツハイマーの原因であるといわれる分子形に関して、実際に2種の異なる仮説がある。
【0007】
1つには、Aβ(1−42)−プロトフィブリルの細胞毒作用が仮定される。更に、これは、150〜250kDaの範囲の分子量を有する可溶性で原線維性の比較的高度に集合したAβ(1−42)−形であり(Arispe et al. PNAS 90. 567(1993), Lashuel et al., Nature 418, 291(2002)) 、これは孔を形成する特性に基づき、カルシウムがニューロン細胞膜を自由に通過するように作用するといわれる。
【0008】
他方では、15〜30kDaの範囲の分子量を有するオリゴマーAβ(1−42)−誘導体(M.P. Lambert et al. PNAS 95, 6448-6453 (1998)) が記載されている。このアミロイドから誘導されるび慢性で老人性痴呆に関与するリガンド(ADDL's Fuer Amyloid Derived Dementing Ligands, US-A 6218506 及びWO 01/10900参照又はfuer Amyloid Derived Diffusible Ligands参照, Lambert et al. supra)としても特徴付けられる非原線維性オリゴマーは、海馬切片中のニューロンの長時間相乗作用率への阻害的影響を示す。
【0009】
勿論、従来のオリゴマー研究の状況は、本来関連のある種についての大きな不透明性を特徴とする。即ち、US−A6218506に、3〜12個のサブユニットを有するADDLsが記載され、これに対して、WO01/10900に記載されたADDLsは24個までのサブユニットを有する。
【0010】
殊に、非変性条件下でのゲル電気泳動分析で、27〜28kDa及び23〜24kDa(US−A6218506)又は約26kDa〜28kDa(WO01/10900)又は17kDa及び27kDa(M.P. Lambert et al. PNAS 95, 6448-6453(1998))の範囲の分子量、及び非変性条件下でのSDS−ゲル電気泳動分析で、17kDa及び22kDa(M.P. Lambert et al. PNAS 95, 6448-6453(1998))又は約22kDa〜約24kDa及び約18kDa〜約19kDa(WO01/10900)の分子量を有する少なくとも2つの形が論じられる。8kDa及び12kDaの範囲の分子量を有するSDS−安定のAβ(1−42)−オリゴマーも、アルツハイマー−患者の脳で、ウエスタン−ブロット(Western Blot)−法によって検証された(C.A. McLean et al. Ann. Neurol. 46, 860-866 (1999))。
【0011】
記載された製法の欠点は、それが不均一なオリゴマー−標本を生じさせることである。
【0012】
即ち、神経変性の原因であるAβ(1−42)の分子形は、再現可能な方法では未だ得られず、まして明白な方法で同定もされ得ていない。
【0013】
しかし、均一な標本の重要性は、例えば、アルツハイマー−患者の能動免疫化のための最初の臨床研究経過から判断され得る。Aβ(1−42)の前集合形のワクチン注射によって、患者の一部で著しい副作用(メンゴ脳炎、出血)が生じ、それというのも、生成した抗体が、細胞の被覆に必要と思われるAβ(1−42)−形も認識し、これによって炎症反応が生じるからである(D. Schenk.; Nat. Rev. Neurosci. 3, 824-828 (2002))。
【0014】
従って、本来的に傷害性のタンパク質形の中和を特に目指す適切な治療には、その同定及び限定の製造が絶対に必要な前提である。
【0015】
更に、Aβ(1−42)−タンパク質のN−末端切断形の出現が、アルツハイマー病と関連して言及されてきた。既に1985年に、死亡したアルツハイマー患者の脳の沈着物中に、Aβ(1−42)の他に、N−末端切断形も検出された(C. Masters et al., PNAS 82, 4245-4249 (1985))。即ち、脳中に存在する一定のプロテアーゼ、例えば、ネプリリシン(NEP 24.11)又はIDE("インシュリン分解酵素(Insulin Degrading Enzyme)"の略語)がAβ(1−42)を分解し得ることも知られている(D.J. Selkoe, Neuron 32, 177-180, (2001))。
【0016】
しかし、アルツハイマー病の病因におけるN−末端切断形の重要性は不明である(E.B. Lee et al. JBS 278, 4458-4466 (2003))。重要なことに、突発性又は家族性アルツハイマー病又はダウン−症候群の若干の患者には、優先的に、この切端形が蓄積されている(J. Naeslund et al. PNAS 91, 8378-8382, (1994), C. Russo et al., Nature 405, 531-532, (2000), T.C. Saido et al., Neuron 14, 457-466, (1995))。新しい研究(N. Sergeant et al. J. of Neurochemistry 85, 1581-1591, (2003)) で、死亡したアルツハイマー−患者の脳中の全不溶性Aβ−ペプチドの60%が、N−末端切断形に基づいていることが示された。
【0017】
モノマーAβ(1−42)−タンパク質及びその一定の断片に照準している抗体は既に記載されている。
【0018】
即ち、WO03/016466及びWO03/016467は、CDR2で一定のグリコシリル化部位が欠けているモノクローナル抗体266及びその類似体に関する。そのヒト化型(Hu−266)も知られている。この印刷物では、他のモノクローナル抗体も記載されていて、それは、抗体226と同様に、Aβ(1−42)のアミノ酸13〜28の範囲でエピトープを認識する。これには、抗体4G8(Lue et al. (1999), supra にも記載されている)及び1C2が属する。更に、McLean et al. (1999), supra に、モノクローナル抗体1E8が記載され、これは、Aβ(1−42)のアミノ酸18〜22の範囲でエピトープを認識すると言われている。
【0019】
更に、Aβ(1−42)のN−末端配列のエピトープを認識する、他の一連の抗体が公知である。それには、市販で得られる、モノクローナル抗体6E10(WO01/10900;Naeslund et al. (1994), supra; Sergeant et al. (2003), supra にも記載されている)及びモノクローナル抗体3D6及び10D5(WO03/016467参照)及びBan50及びNAB228(Lee et al. (2003), supra)が属する。
【0020】
更に、モノクローナル抗体WO2、21F12及び3D6及びポリクローナル血清ADA42が挙げられる(Sergeant et al. (2003), supra) 。
【0021】
前臨床的検査にある抗−Aβ(1−42)−抗体に関する概要は、Schenk et al. (2002), supra にある。
【先行技術文献】
【特許文献】
【0022】
【特許文献1】US-A 6218506
【特許文献2】WO 01/10900
【特許文献3】US−A6218506
【特許文献4】WO03/016466
【特許文献5】WO03/016467
【非特許文献】
【0023】
【非特許文献1】C.L.Masters et al. PNAS 82, 4245-4249,1985
【非特許文献2】R.D.Terry et al Ann. Neurol. 30. 572-580(1991)
【非特許文献3】D.W. Dickson et al. Nurobiol. Aging 16, 285-298(1995)
【非特許文献4】L.F. Lue et al. Am. J. Pathol. 155, 853-862,(1999)
【非特許文献5】C.A. McLean et al. Ann. Neurol. 46, 860-866 (1999)
【非特許文献6】Arispe et al. PNAS 90. 567(1993),
【非特許文献7】Lashuel et al., Nature 418, 291(2002)
【非特許文献8】M.P. Lambert et al. PNAS 95, 6448-6453 (1998)
【非特許文献9】ADDL's Fuer Amyloid Derived Dementing Ligands,
【非特許文献10】fuer Amyloid Derived Diffusible Ligands
【非特許文献11】M.P. Lambert et al. PNAS 95, 6448-6453(1998)
【非特許文献12】C.A. McLean et al. Ann. Neurol. 46, 860-866 (1999)
【非特許文献13】D. Schenk.; Nat. Rev. Neurosci. 3, 824-828 (2002)
【非特許文献14】C. Masters et al., PNAS 82, 4245-4249 (1985)
【非特許文献15】D.J. Selkoe, Neuron 32, 177-180, (2001)
【非特許文献16】E.B. Lee et al. JBS 278, 4458-4466 (2003)
【非特許文献17】J. Naeslund et al. PNAS 91, 8378-8382,(1994)
【非特許文献18】C. Russo et al., Nature 405, 531-532, (2000)
【非特許文献19】T.C. Saido et al., Neuron 14, 457-466, (1995)
【非特許文献20】N. Sergeant et al. J. of Neurochemistry 85, 1581-1591, (2003)
【発明の概要】
【発明が解決しようとする課題】
【0024】
本発明は、Aβ(1−42)の神経変性及び殊にニューロン傷害作用の原因となり、かつ老人性痴呆症、例えば、アルツハイマー病及びダウン−症候群において増大に出現する分子形を得るという本発明の基礎にある課題を、モノマーAβ(1−42)−タンパク質から特異的方法で均一な標本の形で得られる一定のAβ(1−42)−オリゴマーによって、及びこのオリゴマーの一定の誘導体、殊に、架橋結合したオリゴマー及び切端Aβ(1−42)−形をベースとするオリゴマーによって解明する。この製法は、水性媒体中で難溶性のペプチド合成Aβ(1−42)−タンパク質を、高収率で、限定された可溶性オリゴマーに変換させることを可能にする。
【課題を解決するための手段】
【0025】
従って、本発明の目的は、特許請求に定義された目的である。
【0026】
即ち、本発明は、アミロイド−β(1−42)−タンパク質のオリゴマーに関し、この際、このオリゴマーは、SDS−ゲル電気泳動で、約15kDa、20kDa、38kDa又は48kDaの見かけの分子量を示し、又は場合により誘導体化に相応して変化した分子量を有するオリゴマーの誘導体に関する。
【0027】
アミロイド−β(1−42)−タンパク質は、42個のアミノ酸を有するポリペプチドであり、これは、アミロイド−前駆タンパク質(アミロイド前駆タンパク質(Amyloid Precursor Protein)のAPP)から、タンパク質加水分解過程によって誘導される。ヒト変異型のほかに、アミロイド−β(1−42)−タンパク質のイソ型も属し、これは人間以外の生物、ことに他の哺乳類、特にラットに存在する。
【0028】
本発明は、特別な実施態様により、ヒトアミロイド−β(1−42)−タンパク質のオリゴマーに関する。ヒトアミロイド−β(1−42)−タンパク質には、殊に、アミノ酸配列SEQ ID NO:1を有するタンパク質、及び前記の配列から、殊に、アミノ酸交換によって誘導される、その突然変異タンパク質及び全変異型が属する。これに関連して、特に、次のアミノ酸置換が挙げられる:A21G、E22K,E22Q、E22G及びD23N。即ち、アミロイド−β(1−42)−タンパク質の突然変異タンパク質又は全変異型には、本発明により、殊に、アミノ酸配列SEQ ID NO:1を有するタンパク質が属し、この中で、アラニン21、グルタミン酸22及びアスパラギン酸23から選択される1種以上のアミノ酸が、他の、有利にグリシン、リジン、グルタミン及びアスパラギンから選択されるアミノ酸と置換されている。本発明により、殊に、グルタミン又はグリシンによる部位22の置換が特に重要である。
【0029】
本発明は、もう1つの特別な実施態様により、ラットアミロイド−β(1−42)−タンパク質のオリゴマーに関する。ラットアミロイド−β(1−42)−タンパク質には、殊に、アミノ酸配列SEQ ID NO:2を有するタンパク質、及び前記の配列から、殊に、アミノ酸交換によって誘導される、その突然変異タンパク質及び全変異型が属する。これに関連して、特に、ヒトの配列で説明されたアミノ酸置換に相応するアミノ酸配列が挙げられる。
【0030】
アミロイド−β(1−42)−タンパク質は、公知のペプチド合成法によって又は組換えで製造することができる。更に、一連のこのタンパク質は市販で得られる。突然変異タンパク質及び全変異型にも同じことが当てはまる。
【0031】
本発明によるオリゴマーは、アミロイド−β(1−42)−タンパク質のオリゴマー化によって得られる。オリゴマー化は、モノマーアミロイド−タンパク質の非共有集合を含み、従って、このことから、本発明によるオリゴマーは、数種のアミロイド−β(1−42)−タンパク質モノ標識ら組成されていることが判る。
【0032】
オリゴマー化度に応じて、本発明によるオリゴマーは様々な分子量を有する。即ち、オリゴマーに、変性ゲル電気泳動法により、見かけの分子量を配属させることができる。これは、変性標準条件下でのゲル電気泳動法(トリス−グリシン−ゲル、4〜20%、Laemmli UK, Nature 227, 680-685 (1970) 参照)を実施する場合に、オリゴマーA1については約15kDa、オリゴマーA2については約20kDa、オリゴマーB1については約38kDa、オリゴマーB2については約48kDaであり、この際、次の標準タンパク質が同じ条件で次の見かけの分子量を有する:ミオシン250kDa、ウシ−血清アルブミン98kDa、グルタミンヒドロゲナーゼ64kDa、炭酸脱水酵素36kDa、ミオグロビン30kDa、リゾチーム16kDa、アプロチニン6kDa、インシュリンB−鎖4kDa(前−青着色標準(Blue Pre-stained Standard )参照)。もう1つの観点に依り、自然標準条件下でのゲル電気泳動法(トリス−グリシン−ゲル、4〜20%)を実施する場合に、オリゴマーBの分子量は、約64〜90kDaであり、この際、次の標準タンパク質が同じ条件で次の見かけの分子量を有する:ミオシン250kDa、ウシ−血清アルブミン98kDa、グルタミンヒドロゲナーゼ64kDa、カルボアンヒドレーゼ36kDa、ミオグロビン30kDa、リゾチーム16kDa、アプロチニン6kDa、インシュリンB−鎖4kDa(青色前着色標準(Blue Pre-stained Standard )参照)。
【0033】
もう1つの観点により、本発明によるオリゴマーは、ニューロン細胞への親和性を特徴とする。そのことから、オリゴマーは一定の細胞表面タンパク質、殊に、受容体に結合することがわかる。
【0034】
従って、本発明は、予めの細胞構造への本発明によるオリゴマーの結合の測定法にも関し、この際、
i)本発明によるオリゴマーを細胞構造に作用させ、かつ
ii)本発明によるオリゴマーが細胞構造に結合するかどうかを測定する。
【0035】
もう1つの観点により、本発明によるオリゴマーは、神経変性作用を特徴とする。この神経変性作用は、少なくとも1種の本発明によるオリゴマーを、殊に、ニューロン細胞、例えば、神経芽細胞の培養に作用させる場合には、これらの細胞の生存可能性を減少させることで顕示される。この際、細胞の生存可能性は、自体公知の方法で、例えば、本発明によるオリゴマーの作用で生じるアポトーシスの程度を測定することによって評価され得る。このために、好適な試験法、例えば、3−[4,5−ジメチルチアゾル−2−イル]−2,5−ジフェニルテトラゾリウムブロミド(MTT)をベースとする比色法が使用される。生体内では、この神経変性作用は、殊に、ニューロンの発射率(Feuerungsrate)の変性で顕示される。
【0036】
従って、本発明は、本発明によるオリゴマーの活性、殊に、神経毒性の測定法にも関し、この際、
i)本発明によるオリゴマーを細胞に作用させ、かつ
ii)細胞の状態が変化するかどうかを測定する。
【0037】
前記の方法について、本発明によるオリゴマーを前記の方法で、例えば、前記の組成物の形で製造することができる。細胞又は細胞構造物を、有利に、試験管内で、殊に、細胞培養物として又は細胞構造物ではホモジネートとしても製造される。この際、ニューロン細胞及び殊に神経芽細胞を神経毒性の測定に用いる。選択的に細胞を生体内で、殊に、生体、例えば実験動物の一部として、又は生体外で準備することができる。
【0038】
細胞の状態を、通例、オリゴマーの作用前に少なくとも1回及び作用後に少なくとも1回測定する。作用前の状態と作用後の状態との間の比較で差異が生じる場合には、試験したオリゴマーは活性を有する。
【0039】
測定すべき状態の種類は、測定すべき活性の種類に依る。例えば、生存可能性を測定することによって、神経毒性が測定される。それには、オリゴマーの作用前及び作用後の全細胞数に対する生存細胞の数を測定し、相互に比較することができる。
【0040】
前記の結合又は活性の測定方法に基づき、一定の実施態様により、物質が本発明によるオリゴマーの結合を阻害する及び/又はその活性を変性させる、要するに、殊に、減少させる、又は実際に完全に阻止する(阻害する)、又は強化させるかどうかを試験することができる。
【0041】
原則的に、そのために方法を少なくとも2回、1回は試験物質が存在して、もう1回は試験物質が不在で実施する。そのために、試験物質をオリゴマーに、通例、その製造後に添加する。
【0042】
勿論、試験すべき物質がオリゴマーの生成に影響するか否かを測定すべき場合には、試験物質を、有利にオリゴマーの生成前に既に、要するに、その製造前に既に、例えば、オリゴマー生成に使用される1種以上の出発物質に添加する。即ち、本発明による製法を、試験物質の添加下に実施し、引き続き、オリゴマーが生成するかどうか、及びどの程度生成するかを測定する。そのために、この方法で得られる方法生成物が本発明によるオリゴマーの特性、要するに、例えば、その分子量、結合可能性及び活性を有するかどうかを測定することができる。
【0043】
極めて著しい神経変性作用の意において、本発明によるオリゴマーBが有利である。このオリゴマーの誘導体も、この見地において有利であり、この際、殊に、N−末端切断のAβ(1−42)−タンパク質をベースとするオリゴマーが有利である。
【0044】
方法技術的理由から、オリゴマーA又はオリゴマーBは、混合物として、オリゴマーの他に更に少量の他のポリペプチド、殊に、モノマーアミロイド−β(1−42)−タンパク質及び場合により集合アミロイド−β(1−42)−タンパク質の高分子形も含有する組成物の形で生じてもよい。この種類の組成物は、同様に本発明の目的であり、殊に、1種以上の本発明によるオリゴマーの割合が、アミロイド−β(1−42)−タンパク質から誘導されるタンパク質の全量に対して、少なくとも70質量%、有利に少なくとも90質量%、殊に少なくとも95質量%であることを特徴とする。オリゴマーAの標本では、オリゴマーA2(SDS−ゲルで20kDa−帯)の割合が少なくとも50質量%、有利に少なくとも70質量%、殊に少なくとも85質量%である。オリゴマーBの標本では、オリゴマーB1及びB2(SDS−ゲルで38kDa−及び48kDa−帯)の割合は、少なくとも60質量%、有利に少なくとも75質量%、殊に少なくとも90質量%である。
【0045】
本発明によるオリゴマーは誘導体化されていてもよい。この種類の誘導体化の目的は、例えば、殊に、生物学的有効性に関するオリゴマーの物理化学的特性を変性させること、オリゴマーに検出可能な標識を与えること又は不動化させることであってよく、例えば、担体に結合させることができることである。標識化及び不動化は、殊に、診断的使用に重要である。
【0046】
タンパク質生化学的範囲で慣用の好適な標識化は、当業者に公知である。それには、蛍光−標識化、例えば、一定のフルオレセイン−及びテトラメチルローダミン−誘導体、発光標識化、比色標識化、放射性標識化及び磁気標識化及び相補的結合相手、例えば、ビオチン−及びストレプタビジン−誘導体への親和性での標識化が属する。
【0047】
見かけの分子量に関して、オリゴマー誘導体は、非誘導体化オリゴマーに比べて相応に高められていてよい分子量を有することに注意すべきであり、しかしこの際、集合数は同じである。即ち、例えば、SDS−ゲルで38kDaの分子量を有するAβ(1−42)−オリゴマーB1をベースとするビオチン−誘導体は、42kDaの分子量を有する。
【0048】
本発明の特別な実施態様により、オリゴマーは架橋結合している。好適な架橋結合剤は当業者に公知であり、通例、二官能性試剤、例えば、ホルムアルデヒド、グルタルジアルデヒド、ジスクシンイミジルスベレート、ジチオビス(スクシンイミジルプロピオネート)、ジスクシンイミジルタルトレート、ジスルホスクシンイミジルタルトレート、ジメチルアジピミデート、ジメチルピメリデート、ジメチルスベリミデート、ジメチル−3,3’−ジチオビスプロピオニミデート、N−γ−マレイミニドブチルオキシスクシニミドエステル、スクシニミジル−4(N−マレイニミドメチル)−シクロヘキサン−1−カルボキシレート、N−スクシニミジル−(4−イオドアセチル)アミノベンゾエート及びN−スクシンイミジル−3−(2−ピリジルジチオ)プロピオネートである。
【0049】
この種類の架橋結合オリゴマーは、それが安定性であり、そのオリゴマー化度は、通例、もはや不可逆であるという利点を有する。従って、これは特に診断的試験系での使用に、又はオリゴマー特異的抗体の製造のための免疫原として好適である。
【0050】
本発明によるオリゴマーの誘導体には、アミロイド−β(1−42)−タンパク質の断片のオリゴマーも属する。天然に産するプロテアーゼの作用によって得られる断片が有利である。殊に、タンパク質加水分解によって非変性条件下に生理学的緩衝液中で得られる断片は、アミロイド−β(1−42)−タンパク質に比べて、高められたタンパク質加水分解安定性を有する。エンドペプチダーゼの作用によって得られる断片が有利である。本発明の特別な実施態様により、断片は、トリプシン、キモトリプシン、サーモリシン、エラスターゼ、パパイン又はエンドプロテイナーゼGluCの作用によって得られる。もう1つの観点により、その本発明によるオリゴマーが神経変性作用を特徴とする断片が有利である。この観点を更に説明するために、本発明によるアミロイド−β(1−42)−タンパク質のオリゴマーの神経変性作用についての相応する実施が参照される。
【0051】
有利な断片は、構造的に、殊に、それがアミロイド−β(1−42)−タンパク質からN−末端配列の分離によって誘導されることを特徴とする。1観点により、このN−末端配列とは、アミロイド−β(1−42)−タンパク質のN−末端配列の23個まで、有利に21個まで、殊に19個までのアミノ酸を有する断片が重要である。従って、その配列がアミノ酸24〜42個、有利に22〜42個、殊に20〜42個を連続して包含する、本発明によるAβ(1−42)−タンパク質の断片が有利である。もう1つの観点により、分離すべきN−末端配列とは、アミロイド−β(1−42)−タンパク質のN−末端配列の少なくとも7、有利に少なくとも9、殊に少なくとも11個のアミノ酸を有する配列が重要である。従って、本発明により殊に、N−末端で約7〜23、有利に9〜21、殊に11〜19のアミノ酸が切断されているアミロイド−β(1−42)−タンパク質の断片が有利である。この断片は、式 Aβ(x−42)に相応し、ここで、xは8〜24、有利に10〜22、殊に12〜20である。従って、本発明により、Aβ(x−42)−断片は、有利に、次の断片から選択される:Aβ(8−42)、Aβ(9−42)、Aβ(10−42)、Aβ(11−42)、Aβ(12−42)、Aβ(13−42)、Aβ(14−42)、Aβ(15−42)、Aβ(16−42)、Aβ(17−42)、Aβ(18−42)、Aβ(19−42)、Aβ(20−42)、Aβ(21−42)、Aβ(22−42)、Aβ(23−42)及びAβ(24−42)。特殊な断片は、サーモリシンの作用によって得られるアミロイド−β(20−42)−断片及びエンドプロテイナーゼGluCの作用によって得られるアミロイド−β(12−42)−断片である。
【0052】
本発明による誘導体には、C−末端1又は2で他のアミノ酸を有するアミロイド−β(1−42)−タンパク質から誘導される形も属する。即ち、それには、例えば、Aβ(1−43)−タンパク質のオリゴマー又は前記の実施に相応するその誘導体が属する。
【0053】
本発明によるオリゴマーの製法は、実際には、3段階からなり、その第1段階は任意であるが有利であり、第2段階はオリゴマーA及びBの製造のために強制的であり、第3段階は本発明によるオリゴマーBの製造に用いられる。
【0054】
段階1は、タンパク質の変性に関する。そのために、水素橋切断剤、例えば、ヘキサフルオルイソプロパノール(HFIP)をタンパク質に作用させることができる。作用温度が約20〜50℃、殊に約35〜40℃である場合には、数分、例えば、約10〜60分間の作用時間で十分である。蒸発乾燥させた残渣を、引続いて、有利に濃縮形で、水性緩衝液と混合可能な好適な有機溶剤、例えば、ジメチルスルホキシド(DMSO)に溶かし、本発明による方法の第2段階で使用可能な、少なくとも部分的に変性されたタンパク質の懸濁液を得る。必要に応じて、基本懸濁液を暫時、より低温で、例えば、約−20℃で保存することができる。
【0055】
前記の段階1について選択的に、タンパク質を弱酸性の、有利に水溶液中に、例えば、約10ミリモルのHCl−水溶液中に入れることができる。通例、数分間に渡る恒温保持後に、不溶成分を遠心分離する。10000gで数分間が有利である。この方法段階は有利に室温で、即ち、20〜30℃の範囲の温度で実施される。遠心分離後に得られる上澄液は、アミロイド−β(1−42)−タンパク質を含有し、暫時、より低温で、例えば、約−20℃で保存することができる。
【0056】
段階2は、タンパク質からオリゴマーAへのオリゴマー化に関する。そのために、界面活性剤を場合により少なくとも部分的に変性されたタンパク質に、十分なオリゴマーAが生じるまで作用させる。
【0057】
イオン性界面活性剤、殊に、陰イオン性界面活性剤が有利に使用される。
【0058】
有利な実施態様により、式(I)の界面活性剤を使用する:
R−X
[式中、
基Rは、6〜20、有利に10〜14個の炭素原子を有する場合により分枝鎖のアルキル又は6〜20、有利に10〜14個の炭素原子を有する場合により分枝鎖のアルケニルを表わし、
基Xは、酸性基又はその塩を表わし、この際、Xは、有利に、−COO−M+、−SO3−M+及び特に−OSO3−M+から選択され、M+は、水素陽イオン又は有利にアルカリ金属−及びアルカリ土類金属陽イオン及びアンモニウム陽イオンから選択される無機又は有機陽イオンを表わす]。
【0059】
式中のRが非分枝鎖のアルキル基を表わし、この際、この内で殊にアルク−1−イル−基が挙げられる式(I)の界面活性剤が有利である。ドデシル硫酸ナトリウム(SDS)が特に有利である。ラウリル酸及び油酸も有利に使用することができる。界面活性剤ラウロイルサルコシン(Sarkosyl NL-30 又はGardol (登録商標)としても公知である)のNa−塩も特に有利である。
【0060】
界面活性剤の作用時間は、殊に、オリゴマー化されるタンパク質が変性されているかどうか(変性されていれば、どの程度)に依存する。タンパク質が段階1により前以て水素架橋切断剤、要するに、殊に、ヘキサフルオルイソプロパノールで処理されている場合には、作用温度が約20〜50℃、殊に約35〜40℃である場合には、作用時間は数時間の範囲、有利に約1〜20時間、殊に約2〜10時間で十分である。少し変性されたタンパク質又は実際に変性されないタンパク質が生じる場合には、相応により長い作用時間が有利である。アミロイド−β(1−42)−タンパク質が、例えば、段階1について選択的に前記された方法によって予備処理される又は段階2に直接装入される場合には、作用温度が約20〜50℃、殊に約35〜40℃である場合には、作用時間は約5〜30時間、殊に約10〜20時間の範囲で十分である。恒温保持後に、有利に不溶成分を遠心分離する。10000gで数分が有利である。
【0061】
選択すべき界面活性剤−濃度は、使用される界面活性剤に依る。SDSを使用する場合には、0.01〜1質量%、有利に0.05〜0.5質量%の範囲、例えば、約0.2質量%の濃度が有利である。ラウリル酸又は油酸を使用する場合には、少し高い濃度、約0.05〜2質量%、有利に0.1〜0.5質量%の範囲、例えば、約0.5質量%が有利である。
【0062】
界面活性剤の作用は、塩濃度では、ほぼ生理学的範囲の行なわれるべきである。即ち、殊に、NaCl−濃度は、50〜500ミリモル、有利に100〜200ミロモル、特に約140ミリモルの範囲で有利である。
【0063】
得られるオリゴマーA、即ち、殊にオリゴマーA1及び/又はA2を含有する溶液は、暫時、より低い温度、例えば、約−20℃で保存され得る。これをそのものとして本発明による使用に、又はオリゴマーBへの更なる変換に添加することができ、又は先ず更なる処理−又は精製段階に接続させ、それによって殊に、少なくとも1種の本発明によるオリゴマーAを更に富化させることができる。殊に、クロマトグラフィー法により、本発明によるオリゴ標識ら、溶液中に含有される他のタンパク質成分及び殊にアミロイド−β(1−42)−タンパク質から誘導されるものを分離させることができる。殊に、この目的のために、親和クロマトグラフィー法が用いられ、この場合には、例えば、特異的抗体、要するに、殊に本発明によるオリゴマー特異的抗体を使用することができる。
【0064】
段階3は、オリゴマーBへのオリゴマー化に関する。オリゴマーAを含有する組成物がこの段階の出発物質として有利に選択される。その点では、オリゴマーAは、本発明により、オリゴマーBの製造のための中間生成物として重要である。この組成物が段階2から由来する場合には、これは規則的に界面活性剤及び生理学的範囲の塩濃度を含有する。次いで、界面活性剤作用又は塩濃度を減少させることが有利である。これは、界面活性剤−又は塩濃度の減少によって、例えば、有利に水に又は低塩濃度の緩衝液、例えば、トリス(Tris)−HCl、pH7.3での希釈によって行なうことができる。約2〜10の範囲、有利に約3〜8の範囲、殊に約4の希釈ファクターが好適であると判明した。界面活性剤作用の減少は、界面活性剤作用を中和させ得る物質の添加によって達成することもできる。それには、例えば、界面活性剤を複合化させ得る物質、例えば、清浄−及び抽出手段の実施で細胞を安定化させ得る物質、例えば、一定のEO/PO−ブロックコポリマー、殊に、商品名Pluronic(登録商標)F 68 で販売されるブロックコポリマーが属する。アルコキシル化、殊にエトキシル化アルキルフェノール、例えば、Triton(登録商標)X- 系のエトキシル化t−オクチルフェノール、殊にTriton(登録商標)X100、3−(3−コルアミドプロピル−ジメチルアンモニオ)−1−プロパンスルホネート(CHAPS(登録商標))又はアルコキシル化、殊にエトキシル化ソルビタン脂肪酸エステル、例えば、Tween(登録商標)−系、殊にTween(登録商標)20 が、各々臨界ミセル濃度の範囲又はそれ以上の濃度範囲で好適である。
【0065】
引き続き、十分にオリゴマーBが生じるまで溶液を恒温保持する。作用温度が約20〜50℃、殊に約35〜40℃である場合には、作用時間は数時間の範囲、有利に約10〜30時間の範囲、殊に約15〜25時間の範囲で十分である。引き続き、溶液を濃縮させ、場合により生じる残渣を遠心分離する。この場合にも、10000gで数分間が有利であることが判る。遠心分離後に得られる上澄液はオリゴマーBを含有する。
【0066】
得られるオリゴマーB、即ち、殊にオリゴマーB1及び/又はB2を含有する溶液は、暫時、より低い温度で、例えば、約−80℃で保存され得る。これをそのものとして本発明による使用に添加され、又は先ず、更なる処理又は精製段階に接続させることができる。これに関して、オリゴマーAに関する相応の手段についての前記の説明が参照される。
【0067】
更なる処理−又は精製段階は、オリゴマーの製造のために使用される界面活性剤を実際に完全に除去する目的のためにも実施され得る。例えば、オリゴマーBを先ず界面活性剤含有溶液から沈殿させ、取得し、新たに界面活性剤を含まない媒体中に溶かす。タンパク質の沈殿法は当業者によく知られている。本発明により、水性−メタノール性酢酸の添加が有利であると判明した。一定の機械装置に結合することなく、界面活性剤又は界面活性剤−及び塩濃度の変化がタンパク質を限定された溶解形にさせると思われ、このタンパク質は、水性の生理学的緩衝液中に溶解されたタンパク質の出発形とは明らかに異なっており、このことは、例えば、変性又は自然ゲル電気泳動法又はゲル透過クロマトグラフィーにより確認することができる。このことは驚くべきことであり、それというのも、界面活性剤は、通例、タンパク質集合体を解離させる、即ち、下位単位に分解させることができるが、本発明により、集合傾向を有するモノ標識ら出発して限定されたオリゴマーを得ることができるからである。
【0068】
本発明によるオリゴマーの誘導体を製造するために、有利に、前記の本発明による方法を、既に相応に誘導体化されたアミロイド−β(1−42)−タンパク質で実施するようにして行なう。選択的に、オリゴマーの誘導体化を行なうこともでき、それによって、オリゴマーの構造は当然変化されるべきではない。好適なタンパク質化学的手段は、当業者に公知である。
【0069】
本発明によるオリゴマー又はその誘導体の架橋結合は、自体公知の方法で行なうことができる。架橋結合剤として、例えば、グルタルジアルデヒドを使用する場合には、本発明による方法段階2又は3から得られる溶液を、グルタルジアルデヒド−溶液で処理することができる。オリゴマーはグルタルジアルデヒドと室温で数時間後に反応した。次いで、反応は、自体公知の方法で、過剰量のグルタルジアルデヒドと、この目的のために一般に公知の試剤、例えば、エタノールアミンとの反応によって止められ得る。本発明によるオリゴマーAを架橋結合させるか又はBを架橋結合させるかにより、A−CL又はB−CLで表示される本発明による架橋結合オリゴマーの溶液が得られる。
【0070】
殊に、場合により架橋結合したオリゴマーB又はその誘導体について、その合成に続けて、オリゴマーの安定性に影響することなく塩濃度を再び高めることが可能である。このことは、オリゴマーの使用に関して、例えば、使用の際に生理学的条件が有利である場合に重要である(細胞の使用、生体内使用)。
【0071】
アミロイド−β(1−42)−タンパク質の断片のオリゴマーは、原則的に、相応するモノマー断片から出発してオリゴマー化によって、又はアミロイド−β(1−42)−タンパク質のオリゴ標識ら出発してタンパク質加水分解によって製造され得る。即ち、第2変法に従って、次に、本発明による方法段階2又は3から得られる溶液をプロテアーゼで処理することができる。所望のタンパク質加水分解度が達成されたら、プロテアーゼを一般に公知の方法で不活化させる。ここで、生成したオリゴマーを既に前記した方法により取得し、必要に応じて更なる処理−又は精製段階によって更に処理することができる。
【0072】
本発明によるオリゴマー及びこれを含有する組成物は、その均質性及び安定性を特徴とする。これらは、生理学的媒体、例えば、生理学的食塩溶液中に可溶性であり、むしろ球状のその外観によって、原線維形とは異なっている。これらは、他のAβ(1−42)−形、殊に、モノマー、非毒性オリゴマー、Aβ(1−42)を包含する前駆分子APP、プロトフィブリル及び原線維とは明らかに異なっている空間構造を有するという利点を有する。従って、これらは、生体内でも試験管内でも、特異的抗体の生成に特に好適であり、例えば、特異的な能動免疫化を可能にする。この際、免疫化には、病気を起こすオリゴマーだけを使用することが決定的であり得る。他のAβ(1−42)形、例えば、モノマー分子又は低分子量のオリゴマーは、生体中の重要なシグナル機能には不可欠であり得る。同様に、細胞の被覆に重要であると思われる原線維様沈着物の除去は生体を損傷させ得る。
【0073】
同様の方法で、本発明による均質のオリゴマー及びこれを含有する組成物を用いて、治療的可能性、例えば、受動免疫、オリゴマー形の不安定化剤の使用及び受容体(部分)作働剤又は−拮抗剤の使用を実現することができる。即ち、均質オリゴマー標本は、受動免疫化のためのポリクローナル又はモノクローナル抗体の合目的製造も可能にする。このオリゴマー形によって影響される、病気関連の受容体分子又はシグナル分子の発見も、本発明によるオリゴマー及びこれを含有する組成物で可能である。
【0074】
本発明によるオリゴマーは、アミロイド−β−タンパク質関連の生理学的過程へのその関与に基づき、診断的及び治療的価値を有する。即ち、診断的試験管内及び生体内検出法における、本発明によるオリゴマー及びその誘導体の、場合により相応する組成物の形での使用は、本発明の目的である。
【0075】
アミロイド−β−タンパク質関連の生理学的過程には、アミロイドタンパク質(アミロイドーゼ(Amyloidosen))の沈着と関係の過程それが属する。それには、神経組織の構造的変化を生じさせ、例えば、アルツハイマー病及びダウン症候群において重大である過程も、他の組織を襲う過程、例えば、アミロイド−細管異常、例えば、コンゴフィルアミロイド血管病(Congophile Amyloidangiopathie)(CAA)も挙げられる。
【0076】
本発明のもう1つの目的は、オリゴマー特異的抗体の生成のための本発明によるオリゴマー及びその誘導体の、場合により相応する組成物の形での使用である。
【0077】
この際、Aβ(1−42)−タンパク質の切端形をベースとする本発明によるオリゴマーが特に重要である:欠損したN−末端の故に、それを用いて、Aβ(1−42)−タンパク質よりも明らかに選択性の高い免疫応答を生成させることができる。典型的なAβ(1−42)−タンパク質中の強い免疫原のN−末端は、特に、この分子範囲に特異的である抗体、例えば、抗体6E10(F. Signet)及びBAM−10(Fa. Sigma, St. Louis)(これらは、製造者の記載に依れば、N−末端(6E10: Aβ(1-17) 及びBAM-10 Aβ(1-12) に照準している)を生じさせるが、切端Aβ(1−42)−形をベースとする本発明によるオリゴマーを用いて得られる抗体は、オリゴマー特異的範囲を認識し、それによって、他のAβ(1−42)−形に相対する選別性が達成される。この利点は、殊に、能動的ワクチン接種に利用され、それというのも、接種後に生じるより選別性の高い免疫応答(対APP、モノマー形、プロトフィブリル、原線維、斑)が、より少ない副作用(例えば、脳出血、その場でのモノマー−形の生理学的神経栄養活性の侵害)も招くからである。受動免疫化、即ち、抗体治療のために、殊に、モノクローナル抗体をより良好に製造し、選択することができる。
【0078】
この使用の1観点は、治療範囲におけるオリゴマー特異的抗体の生成である。
【0079】
従って、本発明のもう1つの目的は、治療範囲における本発明によるオリゴマー又はその誘導体の、殊にワクチンとしての使用である。
【0080】
そのようなワクチンは、通例、少なくとも1種の本発明によるオリゴマー及び/又は少なくとも1種の本発明によるその誘導体を含有する製薬学的組成物である。そのために、殊に、本発明による、2種以上のオリゴマーを含有する1種の組成物、又は様々な組成物を有する組合せを使用することができる。従って、殊に、オリゴマーB、即ち、B1及びB2又はその誘導体を接種することができる。更に、組成物は、生理学的に認容性の賦形剤及び場合により他の助剤、例えば、免疫刺激剤を含有することができる。
【0081】
好適な賦形剤は、原則的に任意に選択され得るが、賦形剤の種類は、通例、投与方法に依る。即ち、本発明によるワクチンは、殊に、腸管外、例えば、静脈内、筋肉内及び皮下投与に好適な形で組成され得る。この場合には、賦形剤は、有利に水、食塩溶液、アルコール、脂肪、蝋及び/又は緩衝液を包含する。
【0082】
任意の多数の免疫刺激剤が本発明によるワクチン中で使用されてよい。例えば、アジュバントが一緒に入れられてよい。殆どのアジュバントは、急速な異化作用の前に抗原を保護すべき物質、例えば、水酸化アルミニウム又は鉱油、及び脂質A、ボルタデラペルツスシス(Bortadella pertussis) 又はマイコバクテリウム結核(Mycobacterium tuberculosis)から誘導されるタンパク質を含有する。好適なアジュバント、例えば、完全又は不完全フロインドアジュバント;AS−2;アルミニウム塩、例えば、水酸化アルミニウム(場合により、ゲルとして)又は燐酸アルミニウム;カルシウム−、鉄−又は亜鉛塩;アシル化チロシンの不溶性懸濁液;アシル化糖;陽イオン又は陰イオン誘導体化多糖類;ポリホスファゼン(Polyphosphazene) ;生物学的に分解可能な微小球;モノホスホリル−脂質Aが、通例、市販で得られる。サイトカイン、例えば、GM−CSF又はインターロイキン−2、−7又は−12が、アジュバントとして同様に使用される。
【0083】
更に、本発明は抗体の製法に関し、この際、
i)宿主を少なくとも1種の本発明によるオリゴマー、その誘導体又は組成物で免疫化させ、かつ
ii)免疫化への応答として生成した宿主の抗体−含有血清を取得する。
【0084】
抗体の製法の特別な実施態様により、免疫化のために、様々な本発明によるオリゴマー又はオリゴマー−誘導体の混合物を含有する免疫化カクテルを投与する。殊に、方法の経過中に、そのオリゴマー−組成が異なっている数種の免疫化カクテルを投与することが有利である。
【0085】
使用すべきオリゴマー又はオリゴマー−誘導体が非免疫原又は弱免疫原である場合には、それを、担体、有利に担体タンパク質、例えば、ケイホールリンペットヘモシアニン(Keyhole Limpet Hemocyanin)(KLH)、リムルスポリフェヌスヘモシアニン(Limulus Polyphenus Hemocyanin)(LPH)、ウシ血清アルブミン(BSA)又はオバルブミン(OVA)に結合させることによって、その免疫原性を高めることができる。それについて、当業者は、一般に公知である一連の結合可能性が得られる。例えば、グルタルジアルデヒドとの反応が有利であり、例えば、オリゴマー又はオリゴマー−混合物を、好適なペプチド又はペプチド−混合物と、水又は水性溶剤中で恒温保持させることによって可能である。この反応は、有利に環境温度で、要するに、通例、室温で実施され得る。勿論、冷却又はやや加温することも有利であり得る。反応は、通例、数時間内で所望の結果と成り、例えば、2時間の反応時間が通例の範囲である。グルタルジアルデヒド−濃度は、通例、ppm−〜%−範囲、好適には10ppm〜1%、有利に100ppm〜0.5%である。反応パラメーターの最適化は、当業者の知識の範囲にあり、オリゴマーA及びB又はオリゴマー−誘導体が選択された反応条件下に安定性であることを考慮すべきである。
【0086】
免疫化カクテルの製造のために、使用すべき成分を先ず一緒に装入する。得られる成分−混合物を先ず恒温保持することが有利である。これは、有利に環境温度で、通例、室温で行われる。勿論、混合物を冷却又はやや加温することも有利であり得る。恒温保持時間は、通例、数分間から数時間であり、1時間の恒温保持時間が有利であることが実証された。
【0087】
免疫化カクテルは、抗原に付加的に、通例、他の助剤、殊に、免疫化に慣用のアジュバント、例えば、フロインド−アジュバントを含有する。殊に、最初の免疫化には、完全フロインド−アジュバントを使用するが、更なる全ての免疫化を不完全フロインド−アジュバントで実施する。免疫化カクテルの製造のために、有利に前記の成分−混合物としての抗原(免疫原)を1種以上の助剤に加える。この際、抗原は、通例、乳化される。
【0088】
宿主として、殊に、齧歯動物(Nager)又はウサギが好適である。これら又は他の好適な宿主に、免疫化カクテルを有利に皮下注射する。抗体力価は、免疫検定、例えば、宿主−IgGに照準されたヒツジ−抗血清及び標識化オリゴマーで競合的に測定され得る。即ち、測定される宿主が抗体取得に好適であるかどうかを、免疫化の終り頃に決定することができる。例えば、4回の免疫化を実施する場合には、3番目の免疫化後に抗体力価を測定し、次いで、十分な抗体力価を有する動物から抗体を取得することができる。
【0089】
生成した抗体の取得のために、宿主から数週間又は数ヶ月に渡り血液を採取することが有利である。継続して宿主から採血することができる。採血から自体公知の方法で、所望の抗体を含有する血清を取得する。そうして得られる全血清を、必要に応じて、その中に含まれる抗体画分及び殊にオリゴマー−認識抗体を富化させるために、当業者に公知の方法で更に精製することができる。
【0090】
本方法の特別な実施態様により、免疫原として使用されるオリゴマー、その誘導体又は免疫原として使用される組成物中に含有される少なくとも1種のオリゴマー又はその誘導体を特異的に認識する少なくとも1種の血清抗体を選択する。これに関する特異性は、特に、モノマーアミロイド−β(1−42)−タンパク質及び本発明によるオリゴマーよりも高い分子量を有するオリゴマー又はマルチマーアミロイド−β(1−42)−タンパク質−集合体に比べて、他の、殊に類似タンパク質に対するよりも、免疫原に対する抗体のより高い結合親和性を意味する。モノクローナルオリゴマー特異的抗体も、この方法で取得することができる。当然、そのために、宿主から脾臓組織を取り出し、そうして得られる脾臓リンパ球から出発して、モノクローナル抗体を生産するハイブリドーマを常法で確立することが有利である。
【0091】
抗体製造の詳細な説明
哺乳動物の免疫系の一部は、全体として無数の様々な抗体特異性から構成される抗体−レパートリーを有するB−リンパ球である。一定の抗原に対する通常の免疫応答は、このレパートリーからの抗原を特異的に結合する1種以上の抗体の選別を意味し、免疫応答の結果は、刺激抗原を特異的に認識し(かつ最終的には排除する)かつ、この際、抗体の周囲から他の分子を無視するというこの抗体の能力に、少なくとも部分的に基づいている。
【0092】
一定の目的抗原を特異的に認識する抗体の有用性は、モノクローナル抗体技術の発展を導いた。今や、標準されたハイブリドーマ技術は、関与する抗原のための単一の特異性を有する抗体の製造を可能にする。最近では、組換え抗体技術、例えば、抗体バンクの試験管内−スクリーニングが開発されている。この技術を用いて、関与する抗原のための単一の特異性を有する抗体を同様に製造することができる。
【0093】
本発明による方法で、関与する抗原を生体内又は試験管内で抗体−レパートリーに作用させることができる。
【0094】
1実施態様により、動物を生体内において抗原で免疫化させることによって、抗原をレパートリーに作用させることができる。この生体内−法は、更に、動物のリンパ球から一連のハイブリドーマを確立し、抗原を特異的に結合する抗体を分泌するハイブリドーマを選択することを包含する。免疫化すべき動物とは、例えば、マウス、ラット、ウサギ、ニワトリ、ラクダ(Kamelid)又はヒツジ、又は前記した動物の1種の形質転換型、例えば、抗原刺激によってヒト抗体を生じさせるヒト免疫グロブリン遺伝子を有する形質転換マウスである。免疫化され得る動物のその他の型には、ヒト末梢単核血球で再構成されている重症複合免疫不全(SCID)マウス(キメラhu-PBMC-SCID-マウス)又はリンパ様細胞又はその前駆体で再構成されている重症複合免疫不全マウス、同様に、致死的全身放射線照射で処理され、引き続き、重症複合免疫不全(SCID)マウスからの骨髄細胞で放射線照射に対して保護され、引き続き、機能的ヒトリンパ球を移植されているマウス(いわゆる、トリメラ−系(Trimera-System))が属する。免疫化すべき動物のもう1種の型は、そのゲノムにおいて、関与する抗原をコードする内在性遺伝子が、例えば、相同的組換えによって遮断された("ノックアウト(knocked out )")動物(例えば、マウス)であり、即ち、この動物は、抗原での免疫化後に、抗原を異種として認識する。ELISA−技術がそれに属する公知のスクリーニング−法を使用することによって、この方法で製造されたポリクローナル又はモノクローナル抗体を特徴づけ、選択することは当業者には明白であるが、これに限定されるものではない。
【0095】
もう1つの実施態様により、組換え抗体バンクを抗原でスクリーニングすることによって、抗原を抗体−レパートリー上へ試験管内で作用させることができる。組換え抗体バンクは、例えば、バクテリオファージの表面上又は酵母細胞の表面上又は細菌細胞の表面上で発現されてよい。多様な実施態様において、組換え抗体バンクは、例えば、scFv−バンク又はFab−バンクである。もう1つの実施態様により、抗体バンクはRNA−タンパク質−融合として発現される。
【0096】
本発明による抗体の製造のためのもう1つの方法は、生体内−及び試験管内−法からの組合せを包含する。例えば、動物を抗原で生体内免疫化させ、引き続き、動物のリンパ様細胞から製造される組換え抗体バンク又は単一ドメイン−抗体バンク(例えば、重鎖及び/又は軽鎖を有する)を抗原で試験管内スクリーニングすることによって、抗原を抗体−レパートリー上に作用させることができる。もう1つの方法により、動物を抗原で生体内免疫化させ、引き続き、動物のリンパ様細胞から製造した組換え抗体バンク又は単一ドメイン−バンクを親和性成熟させることによって、抗原を抗体−レパートリー上に作用させることができる。もう1つの方法により、動物を抗原で生体内免疫化させ、引き続き、関与する抗体を分泌する単一の抗体−生産細胞を選択し、この選択された細胞から重鎖及び軽鎖の可変領域のcDNAを取得し(例えば、PCRにより)、重鎖及び軽鎖の可変領域を試験管内で哺乳動物宿主細胞中で発現させる(これは、リンパ球−抗体−選択法、"又は選択リンパ球抗体法(Selected Lymphocyte Antibody Method)"のSLAMとして表示される)(それによって選択された抗体−遺伝子配列を更に選択し、操作することができる)ことによって、抗原を抗体−レパートリー上に作用させることができる。更に、重鎖及び軽鎖の抗体遺伝子を哺乳動物細胞中に発現させ、所望の結合親和性を有する抗体を分泌する哺乳動物細胞を選択することによって、モノクローナル抗体を発現クローニングによって選択することができる。
【0097】
従って、本発明の観点は、限定された抗原をスクリーニング及び逆スクリーニングに使用することである。即ち、本発明によるオリゴマー又はその誘導体を結合しないが、他の型のA(1−42)−タンパク質、APP、アミロイド原線維又はアミロイド斑及び一連のその他の非類似抗原及び組織を結合するポリクローナル及びモノクローナル抗体を本発明により選別することができる。
【0098】
抗体選別は明確な抗原に基づくということは、当業者に十分知られている。これに反して、余り明確ではない抗原を使用する場合には、それは十分には選別されない。要するに、試験管内での作用及び選別は、親和性クロマトグラフィーに似ていて、この際、所望の抗原の"リガンド"は、抗原を十分な親和性では結合しないそれから分離される。従って、他の抗体の巨大プールからの所望抗体の富化度は、抗原品質の直接的結果である。意外にも、本発明によるオリゴマー及びその誘導体は、好適で適切な選別的抗体を富化させ、A(1−42)−タンパク質と関連する他の型及び他の非類似抗原を認識する抗体から効果的に分離させる抗原である。
【0099】
本発明による抗体の製法を用いて、様々な型の抗体を製造することができる。それには、実際に、ヒト抗体、キメラ抗体、ヒト化抗体及びCDR−グラフト−抗体及びその抗原−結合部分が属する。
【0100】
次に、本発明による抗体の製法を記載する。この際、生体内−法、試験管内−法、又は2法を含む組合せを区別する。
【0101】
生体内−法
生体内で生じた抗体を生産する細胞から、モノクローナル抗体を、標準された技術、例えば、Kohler 及びMilstein (1975, Nature 256: 495-497 )(同様に、Brown et al. (1981) J. Immunol 127: 539-46; Brown et al. (1980) J Biol Chem 255: 4980-83; Yeh et al. (1976) PNAS 76: 2927-31; 及びYeh et al. (1982) Int. J. Cancer 29: 269-75 参照)によって本来記載されたハイブリドーマ技術によって製造することができる。モノクローナル抗体ハイブリドーマの生産技術は十分に公知である(一般に、R. H. Kenneth, in Monoclonal Antibodies: A New Dimension In Biological Analyses, Plenum Publishing Corp., New York, New York (1980); E. A. Lerner (1981) Yale J. Biol. Med. 54: 387-402; M. L. Gefter et al. (1977) Somatic Cell Genet., 3: 231-36 参照)。要するに、不死化細胞系(典型的には、骨髄腫)を、本発明によるオリゴマー又はその誘導体で免疫化した哺乳動物のリンパ球(典型的には、脾細胞又はリンパ節細胞又は末梢血液リンパ球)と融合させ、生じるハイブリドーマ細胞の培養上澄液をスクリーニングし、本発明によるオリゴマー又はその誘導体の特異性を有するモノクローナル抗体を生産するハイブリドーマを同定する。そのために、多数の周知されているプロトコルの任意の1つを、リンパ球及び不死化細胞系の融合のために使用することができる(同様に、G. Galfre et al. (1977) Nature 266: 550-52; Gefter et al. Somatic Cell Genet., cited supra; Lerner, Yale J. Biol. Med. , cited supra; Kenneth, Monoclonal Antibodies, cited supra参照)。更に、同様に使用可能である方法の様々な変法が当業者に公知である。典型的には、不死化細胞系(例えば、骨髄腫細胞系)は、同一哺乳動物種、例えば、リンパ球から由来する。例えば、本発明による免疫原調製物で免疫化されたマウスからのリンパ球を不死化マウス細胞系と融合させることによって、マウスハイブリドーマを確立することができる。有利な不死化細胞系は、ハイポキサンチン、アミノプテリン及びチミジンを含有する培地(HAT−培地)に感受性のあるマウス−骨髄腫細胞系である。多数の骨髄腫細胞系の任意の1つ、例えば、P3−NS1/1−Ag4−1−、P3−x63−Ag8.653−又はSp2/O−Ag14−骨髄腫系(Myelomlinie)を融合相手として標準的に使用することができる。この骨髄腫細胞系は、アメリカンタイプカルチャーコレクション(American Type Culture Collection) (ATCC), ロックビル(Rockville, MD) から得られる。典型的には、HAT−感受性マウス−骨髄腫細胞を、マウス−脾細胞と、ポリエチレングリコール(PEG)の使用下に融合させる。融合から得られるハイブリドーマ細胞を、次いで、HAT−培地の使用下に選別し、それによって、非融合化及び非生産的融合化骨髄腫細胞を死滅させる(非融合化脾細胞は、形質転換されないので、数日間後に死滅する)。本発明によるオリゴマー又はその誘導体を特異的に認識するモノクローナル抗体−生産性ハイブリドーマ細胞は、ハイブリドーマ培養上澄液をそのような抗体についてスクリーニングすることによって、例えば、標準−ELISA−検定を使用することによって同定され、本発明によるオリゴマー又はその誘導体を特異的に結合することができる抗体が選別される。
【0102】
所望抗体の種類に応じて、様々な宿主動物を生体内−免疫化に使用することができる。関与する抗原の内在型自体を発現する宿主を使用することができる。選択的に、関与抗原の内在Versionのために不全にされた宿主を使用することができる。例えば、相応する内在遺伝子での相同的組換えを経て一定の内在タンパク質のために不全にされたマウス(即ち、ノックアウト−マウス)は、マウスがそれで免疫化されるタンパク質に対して体液性応答を生じさせ、従って、タンパク質に対する高親和性のモノクローナル抗体の製造に使用され得ることが示された(例えば、Roes, J. et al. (1995) J. Immunol. Methods 183: 231-237; Lunn, M. P. et al. (2000) J. Neurochem. 75: 404-412 参照)。
【0103】
本発明によるオリゴマー又はその誘導体に対する非ヒト抗体の製造のために、多数の非−人間の哺乳動物が抗体製造のための宿主として好適である。それには、マウス、ラット、ニワトリ、ラクダ、ウサギ及びヤギ(及びそのノックアウト−型)が属するが、ハイブリドーマ製造にはマウスが有利である。更に、実際に、二重の特異性を有するヒト抗原に対するヒト抗体の製造のために、ヒト抗体−レパートリーを発現する非−ヒト宿主動物を使用することができる。そのような非−ヒト動物には、ヒト−免疫グロブリン−導入遺伝子を有する導入遺伝子動物(例えば、マウス)(キメラhu-PBMC-SCID-マウス)及びヒト/マウス−放射線照射キメラが属し、これらは次に詳記されている。
【0104】
1実施態様により、本発明によるオリゴマー又はその誘導体で免疫化される動物は、非−ヒト哺乳動物、有利に、ヒト免疫グロブリン遺伝子による形質転換のマウスであり、従って、非−ヒト哺乳動物は抗原刺激によりヒト抗体を作る。典型的には、そのような動物中に、ヒト生殖系列立体配置を有する重鎖及び軽鎖のための免疫グロブリン−導入遺伝子が導入され、この際、動物は、重鎖及び軽鎖のためのその内在座が不活性であるように変えられた。そのような動物を抗原(例えば、ヒト抗原)で刺激する場合には、ヒト免疫グロブリン配列から由来する抗体(即ち、ヒト抗体)が生産される。そのような動物のリンパ球から、標準ハイブリドーマ技術により、ヒトモノクローナル抗体を作ることができる。ヒト免疫グロブリンの形質転換マウス及びヒト抗体の製造の際のその使用の更なる説明について、例えば、US−特許第5939598、WO96/33735、WO96/34096、WO98/24893及びWO99/53049(Abgenix Inc.)及びUS−特許第5545806、第5569825、第5625126、第5633425、第5661016、第5770429、第5814318、第5877397及びWO99/45962(Genpharm Inc.)が参照される;同様に、MacQuitty, J. J. 及びKay, R. M. (1992) Science 257: 1188; Taylor, L. D. et al. (1992) Nucleic Acids Res. 20: 6287-6295; Lonberg, N. et al. (1994) Nature 368: 856-859; Lonberg, N. 及びHuszar, D. (1995) Int. Rev. Immunol. 13: 65-93; Harding, F. A. 及びLonberg, N. (1995) Ann. N. Y. Acad. Sci. 764: 536-546; Fishwild, D. M. et al. (1996) Nature Biotechnology 14: 845-851; Mendez, M. J. et al. (1997) Nature Genetics 15: 146-156; Green, L. L. 及びJakobovits, A. (1998) J. Exp. Med. 188: 483-495; Green, L. L. (1999) J. Immunol. Methods 231: 11-23; Yang, X. D. et al. (1999) J. Leukoc. Biol. 66: 401-410; Gallo, M. L. et al. (2000) Eur. J. Immunol. 30: 534-540が参照される。
【0105】
もう1つの実施態様により、本発明によるオリゴマー又はその誘導体で免疫化される動物は、ヒト末梢単核血球又はリンパ様細胞又はその前駆体で再構成された重症複合免疫不全(SCID)のマウスであってよい。キメラhu-PBMC-SCID-マウスとして特徴付けられるそのようなマウスは、明らかに、抗原刺激によるヒト免疫グロブリン応答を生産する。これらのマウス及び抗体の生成のためのその使用の更なる説明について、例えば、Leader, K. A. et al. (1992) Immunology 76: 229-234; Bombil, F. et al. (1996) Immunobiol. 195: 360-375; Murpy, W. J. et al. (1996) Semin. Immunol. 8: 233-241; Herz, U. et al. (1997) Int. Arch. Allergy Immunol. 113: 150-152; Albert, S. E. et al. (1997) J. Immunol. 159: 1393-1403; Nguyen, H. et al. (1997) Microbiol. Immunol. 41: 901-907; Arai, K. et al. (1998) J. Immunol. Methods 217: 79-85; Yoshinari, K. 及びArai, K. (1998) Hybridoma 17: 41-45; Hutchins, W. A. et al. (1999) Hybridoma 18: 121-129; Murphy, W. J. et al. (1999) Clin. Immunol. 90: 22-27; Smithson, S. L. et al. (1999) Mol. Immunol. 36: 113-124; Chamat, S. et al. (1999) J. Infect. Diseases 180: 268-277; 及びHeard, C. et al. (1999) Molec. Med. 5: 35-45が参照される。
【0106】
もう1つの実施態様により、本発明によるオリゴマー又はその誘導体で免疫化される動物は、致死的全身放射線照射で処理され、引き続き、重症併合免疫不全(SCID)のマウスからの骨髄細胞で照射に対して保護され、かつ引き続き、機能的ヒトリンパ球を移植されているマウスである。トリメラ−系として特徴付けられるこのキメラ型は、ヒトモノクローナル抗体を製造するために、マウスを関与抗原で免疫化させ、引き続き、モノクローナル抗体を標準ハイブリドーマ技術の使用下に製造することによって使用される。これらのマウス及び抗体の製造のためのその使用の更なる説明について、例えば、Eren, R. et al. (1998) Immunology 93: 154-161; Reisner, Y及びDagan, S. (1998) Trends Biotechnol. 16: 242-246; Ilan, E. et al. (1999) Hepatology 29: 553-562; 及びBocher, W. O. et al. (1999) Immunology 96: 634-641が参照される。
【0107】
試験管内−法
本発明による抗体を免疫化及び選別によって製造するために、選択的に、オリゴマー又はその誘導体に特異的に結合する免疫グロブリン−バンクの構成員を分離するために、組換え集合免疫グロブリン−バンク(ライブラリー)を本発明によるオリゴマー又はその誘導体でスクリーニングすることによって、本発明による抗体を同定及び分離することができる。ディスプレー−バンクの製造及びスクリーニングのためのキットは市販で得られる(例えば、ファルマシア(Pharmacia)製の組換えファージ抗体システム(Recombinant Phage Antibody System)カタログ−番号27−9400−01;及びストラタゲン(Stratagene)製のSurfZAP(登録商標)Ohage Display Kit、カタログ−番号240612)。多数の実施態様で、ディスプレー−バンクは、scFv−バンク又はFab−バンクである。組換え抗体バンクのスクリーニングのためのファージディスプレー−技術は十分に記載されている。抗体−ディスプレー−バンクの製造及びスクリーニングの際に有利に使用され得る方法及び化合物の例は、例えば、McCafferty et al. WO92/01047、US−特許第5969108及びEP589877(殊に、scFvのディスプレーを記載)、Ladner et al. US−特許第5223409、第5403484、第5571698、第5837500及びEP436597(例えば、pIII−融合を記載);Dower et al. WO91/17271、US−特許第5427908、US−特許第5580717及びEP527839(殊に、Fabのディスプレーを記載);Winter et al. International Publication WO92/20791及びEP368684(殊に、可変免疫グロブリンドメインの配列のクローニングを記載);Griffiths et al. US−特許第5885793及びEP589877(組換えバンクの使用下にヒト抗原に対するヒト抗体の単離を記載);Garrard et al. WO92/09690(殊に、ファージ発現技術を記載);Knappik et al. WO97/08320(ヒト組換え抗体バンクHuCalを記載);Salfeld et al. WO97/29131(ヒト抗原に対する組換えヒト抗体(ヒト−腫瘍−壊死−因子アルファ)の製造及び組換え抗体の試験管内−親和性成熟を記載)及びSalfeld et al. U. S. Provisional Application 第60/126603及びこれに基づく特許出願(同様に、ヒト抗原に対する組換えヒト抗体(ヒトインターロイキン−12)の製造及び組換え抗体の試験管内−親和性成熟を記載)にある。
【0108】
組換え抗体バンクのスクリーニングの更なる記載は科学的刊行物、例えば、Fuchs et al. (1991) Bio/Technology 9:1370-1372;Hay et al. (1992) Hum Antibod Hybridomas 3:81-85; Huse et al. (1989) Science 246:1275-1281; Griffiths et al. (1993) EMBO J 12:725-735; Hawkins et al. (1992) J Mol Biol 226:889-896; Clarkson et al. (1991) Nature 352:624-628; Gram et al. (1992) PNAS 89:3576-3580); Garrad et al. (1991) Bio/Technology 9:1373-1377; Hoogenboom et al. (1991) Nuc Acid Res 19:4133-4137; Barbas et al. (1991) PNAS 88:7978-7982; McCafferty et al. Nature (1990) 348:552-554; 及びKnappik et al. (2000) J. Mol. Biol. 296:57-86 にある。
【0109】
バクテリオファージ−ディスプレー−系の使用のために、選択的に、組換え抗体バンクを、酵母細胞又は細菌細胞の表面上に発現させることができる。酵母細胞の表面上に発現するバンクの製造及びスクリーニングの方法は、WO99/36569に記載されている。細菌細胞の表面上に発現されるバンクの製造及びスクリーニングの方法は、WO98/49286に比較的正確に記載されている。
【0110】
関与する抗体が組合せバンクから同定されると直ちに、抗体の軽鎖及び重鎖をコードするDNAが、標準分子生物学的技術により、例えば、バンクのスクリーニングの間に分離したディスプレー−パッケージ(例えば、ファージ)からのDNAのPCR−増幅によって単離される。PCRを製造し得る抗体軽鎖及び重鎖のための遺伝子のヌクレオチド配列は、当業者に公知である。多数のそのような配列は、例えば、Kabat, E. A., et al. (1991) Sequences of Proteins of Immunological Interest, Fifth Edition, U. S. Department of Health und Human Services, NIH Publication Nr. 91-3242 及びヒト生殖系列(Humankeimbahn) VBASE の配列のためのデータバンク(Datenbank fuer Sequenzen)に記載されている。
【0111】
本発明による抗体又は抗体部分は、宿主細胞中で免疫グロブリン軽鎖及び重鎖のための遺伝子を組換え発現させることによって製造され得る。抗体を組換え発現させるために、抗体の免疫グロブリン軽鎖及び重鎖をコードするDNA−断片を有する1つ以上の組換え発現ベクターで宿主細胞をトランスフェクションさせ、従って、宿主細胞中で軽鎖及び重鎖が発現し、有利に宿主細胞を培養する培地中に分泌される。この培地から、抗体を取得することができる。抗体重鎖及び軽鎖のための遺伝子を得て、この遺伝子を組換え発現ベクターに組込み、ベクターを宿主細胞に導入させるために、標準組換えDNA−法を使用する。この種類の方法は、例えば、Sambrook, Fritsch 及び Maniatis (Hrsg.), Molecular Cloninng; A Laboratory Manual, Second Edition, Cold Spring Harber, N. Y., (1989), Ausubel, F. M. et al. (Hrsg.) Current Protocols in Molecular Biology, Greene Publishing Associates, (1989) 及びBoss et al. のUS−特許第4816397に記載されている。
【0112】
関与する抗体のVH−及びVL−分節をコードするDNA−断片が得られると直ちに、このDNA−断片は、標準組換えDNA−技術により、例えば、可変領域の遺伝子を全長の抗体鎖の遺伝子、Fab−断片の遺伝子又はscFv−遺伝子に変えるために更に操作され得る。この操作では、VL−又はVH−コードDNA−断片は、更にもう1種のタンパク質、例えば、定常抗体領域又は可変リンカーをコードするDNA−断片と操作的に結合される。ここで、"操作的結合"とは、2つのDNA−断片によってコードされるアミノ酸配列が読み枠(フレーム)中に留まるように、2つのDNA−断片が相互に結合されていることを意味すべきである。
【0113】
VH−領域をコードする単離DNAは、VH−領域をコードするDNAをもう1つの重鎖の定常領域(CH1、CH2及びCH3)をコードするDNA−配列と操作的に結合させることによって、全長の重鎖の遺伝子に変えられ得る。ヒト重鎖の定常領域の遺伝子の配列はよく知られていて(例えば、Kabat, E. A., et al. (1991) Sequences of Proteins of Immunological Interest, Fifth Edition, U. S. Department of Health und Human Services, NIH Publication Nr. 91-3242参照)、この領域にかかるDNA−断片は標準PCR−増幅によって得られ得る。重鎖の定常領域は、IgG1、IgG2、IgG3、IgG4、IgGA、IgGE、IgGM又はIgGDからの定常領域であってよく、この際、IgG1又はIgG4からの定常領域が有利である。重鎖のFab−断片の遺伝子を得るために、VH−コードDNAを、重鎖の定常領域CH1だけをコードするもう1つのDNA−配列と操作的に結合させることができる。
【0114】
VL−コードDNAを、軽鎖の定常領域CLをコードするもう1つのDNA−配列と操作的に結合させることによって、VL−領域をコードする単離DNAを、全長の軽鎖の遺伝子(及びFab−軽鎖の遺伝子)に変換することができる。ヒト軽鎖の定常領域の遺伝子配列はよく知られていて(例えば、Kabat, E. A., et al. (1991) Sequences of Proteins of Immunological Interest, Fifth Edition, U. S. Department of Health und Human Services, NIH Publication Nr. 91-3242参照)、この領域にかかるDNA−断片は標準PCR−増幅によって得られ得る。軽鎖の定常領域は、定常のカッパ−又はラムダ−領域であってよく、この際、定常のカッパ−領域が有利である。
【0115】
scFv−遺伝子を生産するために、VH−及びVL−コードDNA−断片を、可変リンカー、例えば、アミノ酸配列(Gly4−Ser)3をコードするもう1つの断片と操作的に結合させることができ、従って、VH−及びVL−配列は連続単鎖タンパク質として発現され、この際、VL−及びVH−領域は可変リンカーを経て相互に結合している(Bird et al. (1988) Science 242:423-426; Huston et al. (1988) Proc. Natl. Acad. Sci. USA 85:5879-5883; McCafferty et al., Nature (1990) 348:552-554 参照)。
【0116】
本発明によるオリゴマー又はその誘導体の特異性を有するVH−及びVL−単一ドメインは、前記の方法で、単一ドメイン−バンクから単離され得る。所望の特異性を有する2本のVH−単一ドメイン−鎖(CH1有り又は無し)又は2本のVL−鎖又はVH−及びVL−鎖からの一組を、本発明によるオリゴマー又はその誘導体を体から除去するために使用することができる。
【0117】
本発明による組換え抗体又は抗体部分を発現するために、部分的長さ又は全長の軽鎖及び重鎖をコードするDNAを発現ベクターに挿入させることができ、そうして遺伝子は転写及び翻訳調節配列と操作的に結合している。この関連において、"操作的結合"とは、転写及び翻訳調節配列が、ベクター内で、抗体遺伝子の転写及び翻訳の調節のためにその意図する機能を果たすように、抗体遺伝子がベクター中で連結していることを意味するべきである。
【0118】
発現ベクター及び発現調節配列は、それらが、使用される発現宿主細胞と適合性であるように選択される。抗体軽鎖の遺伝子及び抗体重鎖の遺伝子は、別々のベクターに挿入されてよく、又は2つの遺伝子は同じ発現ベクターに挿入され、これは常例である。抗体遺伝子は発現ベクター中で標準法により挿入される(例えば、抗体遺伝子断片及びベクターでの相補的制限切断部位のライゲーション、又は制限切断部位が存在していない場合には、平滑末端のライゲーション)。軽鎖及び重鎖の配列の挿入前に、既に発現ベクターは定常抗体領域の配列を有する。例えば、重鎖又は軽鎖のための定常領域を既にコードする発現ベクターにVH−及びVL−配列を挿入させることによって、VH−及びVL−配列を全長の抗体遺伝子に変換させる方法があり、従って、VH−分節は1つ以上のCH−分節とベクター内で操作的に結合していて、かつVL−分節もCL−分節とベクター内で操作的に結合している。付加的に又は選択的に、組換え発現ベクターは、宿主細胞からの抗体鎖の分泌を容易にするシグナルペプチドをコードすることができる。抗体鎖の遺伝子は、ベクター中へクローン化されることができ、従って、シグナルペプチドは読み枠で抗体鎖の遺伝子のN−末端と結合している。シグナルペプチドは、免疫グロブリン−シグナルペプチド又は異種シグナルペプチドであってよい(即ち、非−免疫グロブリン−タンパク質からのシグナルペプチド)。抗体鎖の遺伝子に付加的に、本発明による発現ベクターは、宿主細胞中で抗体鎖の遺伝子の発現を調節する調節配列を有することができる。"調節配列"とは、抗体鎖の遺伝子の転写又は翻訳を調節するプロモーター、エンハンサー及び他の発現調節要素(例えば、ポリアデニル化シグナル)を含むべきである。そのような調節配列は、例えば、Goeddel; Gene Expression Technology: Methods in Enzymology 185, Academic Press, San Diego, CA (1990)に記載されている。調節配列の選別が属する発現ベクターの設計は、因子、例えば形質転換宿主細胞の選別、タンパク質の所望される発現強度等に依存してよい。哺乳動物宿主細胞における発現の有利な調節配列には、哺乳動物細胞中に強いタンパク質発現を引き起こすウイルス要素、例えば、サイトメガロウイルス(CMV)(例えば、CMV−プロモーター/エンハンサー)、サル−ウイルス40(SV40)(例えば、SV40−プロモーター/エンハンサー)、アデノウイルス(例えば、後期アデノウイルス−主要プロモーター(アデノウイルス主要後期プロモーターのAdMLP)及びポリオー標識ら由来するプロモーター及び/又はエンハンサーが属する。ウイルス調節要素及びその配列の更なる記載について、例えば、StinskiによるUS−特許第5168062、Bell et al.によるUS−特許第4510245及びSchaffner et al.によるUS−特許第4968615が参照される。
【0119】
抗体鎖及び調節配列の遺伝子に付加的に、本発明による組換え発現ベクターは、付加的な配列、例えば、宿主細胞へのベクターの複製を調節する配列(例えば、複製出発点)及び選別可能な標識遺伝子を有することができる。選別可能な標識遺伝子は、ベクターがその中に導入された宿主細胞の選別を容易にする(例えば、全てAxel et al. によるUS−特許第4399216、4634665及び5179017参照)。例えば、選別可能な標識遺伝子は、ベクターが挿入された宿主細胞を、作用物質、例えばG418、ハイグロマイシン又はメトトレキセートに対して耐性にさせることは通常である。有利な選別可能な標識遺伝子には、ジヒドロ葉酸レダクターゼ(DHFR)の遺伝子(メトトレキセート−選別/増幅を伴うdhfr−宿主細胞における使用のために)及び新生−遺伝子(G418−選別のための)が属する。
【0120】
軽鎖及び重鎖の発現のために、軽鎖及び重鎖をコードする1種以上の発現ベクターを、標準技術によって、宿主細胞にトランスフェクションさせる。"トランスフェクション"の様々な形は、原核又は真核宿主細胞への外来DNAの導入のために通例使用される多数の技術、例えば、電気穿孔、燐酸カルシウム−沈降、DEAE−デキストラン−トランスフェクション等を理解すべきである。本発明による抗体を原核宿主細胞又は真核宿主細胞で発現することは理論的には可能であるが、真核細胞、殊に、哺乳動物宿主細胞での抗体の発現が有利であり、それというのも、正確に折りたたまれ、免疫学的に能動的な抗体が集合して分泌される確率が、そのような真核細胞、殊に哺乳動物細胞において、原核細胞におけるよりも高いからである。抗体遺伝子の原核発現について、それが能動抗体の高収率の生産には無効であるということが報告されている(Boss, M. A.及びWood, C. R. (1985) Immunology Today 6:12-13)。
【0121】
本発明による組換え抗体の発現に有利である哺乳動物宿主細胞には、CHO−細胞(Urlaub 及び Chasin, (1980) Proc. Natl. Acad. Sci. USA 77:4216-4220 に記載され、例えば、R. J. Kaufman 及び P. A. Sharp (1982) Mol. Biol. 159:601-621に記載されているDHFR−選別可能な標識と併用されるdhfr−CHO−細胞を含む)、NSO−骨髄腫細胞、COS−細胞及びSP2−細胞が属する。抗体遺伝子をコードする組換え発現ベクターが、哺乳動物宿主細胞に導入される場合には、抗体が宿主細胞中に発現され又は有利に宿主細胞が増殖する培地に抗体が分泌されるまでの間、宿主細胞を培養することによって抗体は製造される。タンパク質の精製のための標準法を使用して、抗体を培地から取得することができる。
【0122】
完全抗体の一部、例えば、Fab−断片又はscFv−分子を製造するために、同様に宿主細胞を使用することができる。当然、前記の方法の変法は発明に属する。例えば、本発明による抗体の軽鎖又は重鎖(しかし、両方ではない)をコードするDNAで宿主細胞をトランスフェクションすることは所望に値し得る。関与抗原の結合のためには必要ではない軽鎖又は重鎖が存在する場合には、そのような軽鎖又はそのような重鎖又は両方をコードするDNAを、組換えDNA−技術によって、部分的に又は完全に除去することができる。そのような切端DNA−分子によって発現される配列も、本発明による抗体に属する。更に、1本の重鎖及び1本の軽鎖が本発明による抗体であり、かつ他の重鎖及び軽鎖が関与抗原以外の抗原の特異性を有する二官能抗体は、本発明による抗体を第二の抗体と標準化学的方法で架橋結合させることによって製造され得る。
【0123】
本発明による抗体又はその抗原−結合部分の組換え発現のための有利な系において、抗体重鎖も抗体軽鎖もコードする組換え発現ベクターを、燐酸カルシウム−促進トランスフェクションによって、dhfr−CHO−細胞に導入させる。遺伝子の強い転写を引き起こすために、組換え発現ベクター内で、抗体重鎖及び抗体軽鎖の遺伝子を、各々、調節CMV−エンハンサー/AdMLP−プロモーター要素と操作的に結合させる。組換え発現ベクターは、メトトレキセート−選別/増幅を使用することによって、ベクターとトランスフェクションするCHO−細胞を選別し得るDHFR−遺伝子も有する。選別された形質転換宿主細胞を培養し、そうして抗体重鎖及び抗体軽鎖を発現させ、完全抗体を培地から取得する。標準分子生物学的技術を使用して、組換え発現ベクターを製造し、宿主細胞をトランスフェクションし、形質転換体を選別し、宿主細胞を培養しかつ抗体を培地から取得することができる。即ち、本発明は、本発明による宿主細胞を本発明による組換え抗体が合成されるまで好適な培地中で培養することによる、本発明による組換え抗体の合成法に関する。この方法は、更に、組換え抗体を培地から単離することも包含し得る。
【0124】
ファージ−ディスプレーによる組換え抗体バンクのスクリーニングに選択的に、本発明による抗体を同定するために、大きな組合せバンクのスクリーニング用の当業者に公知の他の方法を使用することができる。選択的発現系の1方法では、例えば、Szostak 及びRobertsによるWO−98/31700及びRoberts, R. W. 及びSzostak, J. W. (1997) Proc. Natl. Acad. Sci. USA 94:12297-12302に記載されているように、組換え抗体バンクをRNA−タンパク質−融合の形で発現させる。この系では、その3’−末端にピューロマイシン、ペプチジル受容体−抗生物質を有する合成mRNAの試験管内−翻訳によって、mRNA及びそれがコードするペプチド又はタンパク質の間に、共有融合を生成させる。即ち、mRNAの複合混合物(例えば、組合せバンク)をからの特異的mRNAは、コードされるペプチド又はタンパク質(例えば、抗体又はその一部分の)の特性により、例えば、本発明によるオリゴマー又はその誘導体への抗体又はその一部分の結合により富化される。そのようなバンクのスクリーニングから取得される、抗体又はその一部分をコードする核酸配列を、組換え手段によって前記の方法で発現させ(例えば、哺乳動物宿主細胞中で)かつ、更なる経過でmRNA−ペプチド−融合をスクリーニングし、ここで当初の1種以上の選択された配列に突然変異を導入させるか、又は前記の方法で組換え抗体の試験管内−親和性成熟のための他の方法を使用することによって、更に親和性成熟をさせることができる。
【0125】
生体内−及び試験管内−法の組合せ
生体内−及び試験管内法の組合せ、例えば、オリゴマー−又は誘導体結合抗体の生産を刺激するために、先ず本発明によるオリゴマー又はその誘導体を宿主動物における生体内で抗体−レパートリー上に作用させ、引続いて、1種以上の試験管内−技術による更なる抗体選別及び/又は抗体成熟(即ち、最適化)を実施する方法を使用することによって、本発明による抗体を同様に製造することができる。1実施態様により、先ず、抗原に対する抗体−応答を刺激するために、非−ヒト動物(例えば、マウス、ラット、ウサギ、ニワトリ、ラクダ、ヤギ又はその形質転換型又はキメラマウス)を本発明によるオリゴマー又はその誘導体で免疫化させ、引き続き、オリゴマー又は誘導体の作用によって生体内で刺激されたリンパ球からの免疫グロブリン配列の使用下にファージ−ディスプレー−抗体バンクを製造し、かつスクリーニングさせる組合せ方法が包含される。この組合せ方法の第1段階は、生体内−方法に関連する前記の方法で実施され得るが、この方法の第2段階は、試験管内−方法に関連する前記の方法で実施され得る。刺激されたリンパ球から製造されたファージ−ディスプレー−バンクを、引き続き、試験管内−スクリーニングすることを伴う、非−ヒト動物の超免疫の有利な方法には、BioSite Inc.,により記載されている方法(例えば、WO98/47343、WO91/17271、US−特許第5427908及びUS−特許第5580717参照)が属する。
【0126】
もう1つの実施態様により、先ず、オリゴマー又はその誘導体に対する抗体応答を刺激するために、非−ヒト動物(例えば、マウス、ラット、ウサギ、ニワトリ、ラクダ、ヤギ又はノックアウト−及び/又はその形質転換型又はキメラマウス)を本発明によるオリゴマー又はその誘導体で免疫化させ、ハイブリドーマ(例えば、免疫化動物から製造される)をスクリーニングすることによって、所望の特異性を有する抗体を生産するリンパ球を選別する組合せ法が包含される。抗体又は単一ドメイン抗体の遺伝子を、選別されたクローンから分離させ(標準法、例えば、逆転写酵素−ポリメラーゼ連鎖反応により)、試験管内−親和性成熟させ、それによって、1種以上の選別された抗体の結合特性を改善することができる。この方法の第1段階は生体内−法に関連する前記の方法で実施され得るが、この方法の第2段階は試験管内−法に関連する前記の方法で、殊に、試験管内−親和性成熟法、例えば、WO−97/29131及びWO−00/56772に記載されている方法によって実施され得る。
【0127】
もう1つの組合せ方法では、リンパ球−抗体−選別法(SLAM)として当業者に公知であり、US−特許第5627052、WO−92/02551及びBabcock, J. S. et al. (1996) Proc. Natl. Acad. Sci. USA 93:7843-7848に記載されている方法を使用することによって、単離された単一リンパ球から組換え抗体を生成させる。この方法では、オリゴマー又はその誘導体に対する抗体応答を刺激するために、先ず、非−ヒト動物(例えば、マウス、ラット、ウサギ、ニワトリ、ラクダ、ヤギ又はその形質転換型又はキメラマウス)を、生体内で、本発明によるオリゴマー又はその誘導体で免疫化させ、次いで、抗原特異的溶血斑検定を使用して、関与する抗体を分泌する単一細胞を選別する。そのために、オリゴマー又はその誘導体、又は関与の構造的類似配列をヒツジ−赤血球に結合させることができ、この際、リンカー、例えば、ビオチンを使用し、それによって好適な特異性を有する抗体を分泌する単一細胞が、溶血斑検定の使用下に同定され得る。関与抗体を分泌する細胞の検定に続いて、細胞から軽鎖及び重鎖の可変領域のcDNAを逆転写酵素−PCRによって取得し、この可変領域を、好適な定常免疫グロブリン領域(例えば、ヒト定常領域)と関連して、哺乳動物宿主細胞、例えば、COS−又はCHO−細胞に発現させることができる。生体内で選別されたリンパ球から由来する増幅された免疫グロブリン配列でトランスフェクションされた宿主細胞を、次いで、所望の特異性を有する抗体を発現させる細胞を分離するために、例えば、トランスフェクションさせた細胞を拡散させることによって、もう1つの試験管内−分析及び−選別させることができる。増幅された免疫グロブリン配列を、更に、試験管内で扱うことができる。
【0128】
前記の方法により、本発明によるオリゴマー又はその誘導体のための、及び構造的に類似の合成配列のための特異性を有する抗体を製造することもできる。そのために、
i)本発明によるオリゴマー又はその誘導体及び構造的に類似の合成配列に共通である構造的特徴を包含する抗原を製造する;
ii)抗原を抗体−レパートリー上に作用させる;かつ
iii)2種の構造的に類似の配列に結合する抗体をレパートリーから選別し、それによって所望の特異性を有する抗体を得る。
【0129】
前記の方法により得られるオリゴマー特異的抗体は、アミロイド−β関連の痴呆症の治療剤を製造するための、又はアミロイド−β関連の痴呆症の診断用組成物を製造するためのその使用と全く同様に本発明の目的である。
【0130】
本発明により得られる抗体には、殊に、前記の方法で得ることができる抗血清が属する。これは全血清、即ち、宿主から得られる凝固可能な細胞成分の分離後の血液、又は、殊に免疫グロブリン−画分及び有利にオリゴマー認識免疫グロブリン−画分が富化されているこの血清の画分であってよい。この種類の画分は、抗体−精製と関連して前記された方法で得られ得る。
【0131】
本発明による抗血清はポリクローナルであり、即ち、これは様々な特異性の、通例様々な部類及び下位部類の抗体を含有し、通例、全てのL−連鎖−イソ型が存在していて、数種のタンパク質エピトープが知られている。
【0132】
免疫原として本発明による異なるオリゴマーを使用する場合には、本発明による抗血清は、通例、架橋結合性である。
【0133】
もう1つの観点により、本発明により得られる抗体には、モノクローナル抗体、殊に、キメラ及びヒト化抗体、及びそのオリゴマー結合断片が属する。
【0134】
本発明の目的は、本発明によるオリゴマー又はその誘導体に特異的に結合するタンパク質及び殊に抗体、即ち、本発明によるオリゴマー又はその誘導体の特異性を有する抗体である。本発明の目的は、同様に、このタンパク質又は抗体の一部、殊に、その抗原結合部分、即ち、本発明によるオリゴマー又はその誘導体を結合するタンパク質−又は抗体部分である。
【0135】
本発明による抗体を有利に選別する場合には、それは本発明によるオリゴマー又はその誘導体への特異的結合のための一定の結合速度(例えば、高い親和性、低い解離、低いオフ−速度、強い中和活性)を有する。
【0136】
即ち、KD=10−6〜10−12Mの範囲の本発明によるオリゴマー又はその誘導体の親和性を有するタンパク質及び殊に抗体が有利である。Kd=10−8Mよりも高い親和性、Kd=10−9Mよりも高い親和性、Kd=10−10Mよりも高い親和性又はKd=10−11Mよりも高い親和性で結合する高親和性タンパク質及び殊に抗体が特に有利である。
【0137】
もう1つの観点により、他のAβ(1−42)−形、殊に、モノマーAβ(1−42)−タンパク質及び/又はモノマーAβ(1−40)−タンパク質を、比較的低い親和性、殊にKd=10−8Mよりも低い親和性で結合するタンパク質及び殊に抗体が有利である。
【0138】
従って、本発明により特に、モノマーAβ(1−42)−タンパク質及び/又はモノマーAβ(1−40)−タンパク質よりも高い親和性でオリゴマー又はその誘導体を結合するタンパク質及び殊に抗体が有利である。10、100又は1000の親和性比率が特に有利である。
【0139】
もう1つの観点により、本発明による抗体を、それがオリゴマー又はその誘導体をkoff−速度定数0.1s−1以下で結合するように選択することができる。速度定数koff1x10−2s−1以下、1x10−3s−1以下、1x10−4s−1以下、1x10−5s−1以下が記載順に有利になる。
【0140】
更に、本発明による抗体を、それが本発明によるオリゴマー又はその誘導体の活性、殊に神経毒活性を、1x10−6M以下のIC50で阻害するように選択することができる。1x10−7M以下、1x10−8M以下、1x10−9M以下、1x10−10M以下、又は1x10−11M以下の阻害定数IC50が記載順に有利になる。
【0141】
抗体とは、有利に、単離された抗体のことである。もう1つの観点により、抗体は中和抗体である。本発明による抗体には、モノクローナル抗体及び組換え抗体が属する。多数の実施態様により、抗体は、完全に単一種から由来するアミノ酸配列、例えば、ヒト抗体又はマウス抗体を包含することができる。他の実施態様により、抗体は、キメラ抗体又はCDR−グラフト−抗体又は他の型のヒト化抗体であってよい。
【0142】
"抗体"とは、4本のポリペプチド鎖、2本の重(H)鎖及び2本の軽(L)鎖から形成されている免疫グロブリン分子に関する。この連鎖は、通例、ジスルフィド−結合によって相互に結合されている。各重鎖は、重鎖の可変領域(ここでは、HCVR又はVHとして略される)及び重鎖の定常領域から組成している。重鎖の定常領域は、3つのドメインCH1、CH2及びCH3から形成される。各軽鎖は、軽鎖の可変領域(ここでは、LCVR又はVLとして略される)及び軽鎖の定常領域から組成している。軽鎖の定常領域はドメインCLから形成される。VH−及びVL−領域は、更に、相補性決定領域(相補性決定領域(Complementarity Determining Region)のCDR)として表示される超可変領域に細分され、かつ骨格領域(骨格領域(Frame Work Region)のFR)として表示される保存領域と混在していてよい。各VH−及びVL−領域は、N−末端からC−末端へ次の順序で配置されている3つのCDRs及び4つのFRsから形成される:FR1、CDR1、FR2、CDR2、FR3、CDR3、FR4。
【0143】
抗体の"抗原結合部分"(又は単に"抗体部分")とは、本発明によるオリゴマー又はその誘導体への特異性を有する抗体の1種以上の断片に関し、この際、1種以上の断片は、依然として、オリゴマー又はその誘導体を特異的に結合する能力を有する。抗体の抗原結合機能は、完全抗体の断片によって認められ得ることが示されている。結合断片の例には、抗体の概念"抗原結合部分"の意において、(i)Fab−断片、即ち、VL−、VH−、CL−及びCH1−ドメインから組成される一価の断片;(ii)F(ab')2−断片、即ち、ジスルフィド−橋を経て蝶番−領域で相互に結合した2つのFab−断片を包含する二価の断片;(iii)VH−及びCH1−ドメインから組成するFd−断片;(iv)抗体の単一アームのFL−及びVH−ドメインから組成するFv−断片;(v)VH−ドメイン又はVH、CH1、CH2、DH3又はVH、CH2、CH3から成るdAb−断片(Ward, et al. (1989) Nature 341:544-546);及び(vi)分離された相補性決定領域(CDR)が属する。Fv−断片の2つのドメイン、つまりVL及びVHが、別々の遺伝子によってコードされるとしても、これらは更に合成リンカーと組換え法の使用下に相互に結合され、それによって、これらを単一のタンパク質鎖として製造することができ、その中で、VL−及びVH−領域は一価の分子を形成するために集合することができる(単鎖−Fv(ScFv)として知られる;例えば、Bird et al. (1988) Science 242:423-426; 及びHuston et al. (1998) Proc. Natl. Acad. Sci. USA 85:5879-5883参照)。この種類の単鎖−抗体は、抗体の概念"抗原結合部分"によっても把握されるべきである。他の型の単鎖−抗体、例えば"ジアボデーズ(Diabodies)"も同様にこれに属する。ジアボデーズは、VH−及びVL−ドメインが単一のポリペプチド鎖上で発現される二価の二重特異的抗体であり、その際、2つのドメインが同一鎖上で集合し得るには短すぎる1つのリンカーが使用されるが、それによって、そのドメインが強制的に他の鎖の相補性ドメインと対にされ、2つの抗原結合部位が形成される(例えば、Holliger, P., et al. (1993) Proc. Natl. Acad. Sci. USA 90:6444-6448; Poljak, R. J. et al. (1994) Structure 2:1121-1123参照)。
【0144】
更に、抗体又はその抗原結合部分は、抗体又は抗体部分と1種以上の他のタンパク質又はペプチドとの共有又は非共有会合によって形成される、より大きな免疫接着分子の一部であってよい。そのような免疫接着分子には、テトラマーscFv−分子を製造するためのストレプトアビジン−コア領域の使用(Kipriyanov, S. M., et al. (1995) Human Antibodies und Hybridomas 6:93-101)及び二価のビオチニル化scFv−分子を作製するためのシステイン基、標識ペプチド及びC−末端ポリヒスチジン−タグの使用が属する((Kipriyanov, S. M., et al. (1994) Mol. Immunol. 31:1047-1058)。抗体部分、例えば、Fab及びF(ab')2−断片は、慣用の技術、例えば、パパイン又はペプシンでの消化を使用することによって、全抗体から製造され得る。更に、抗体、抗体部分及び免疫接着分子は、標準組換えDNA−技術を使用することによって得ることができる。"本発明によるオリゴマー又はその誘導体への特異性を有する分離抗体"は、様々な抗原特異性を有する他の抗体を実際に含まない、本発明によるオリゴマー又はその誘導体への特異性を有する抗体、要するに、殊に、前記のようなAβ(1−42)−タンパク質の他の型に特異的に結合する抗体を含まない抗体を意味する。
【0145】
"中和抗体"とは、一定の抗原へのその結合が抗原の生物学的活性の阻害を引き起こす抗体を意味する。この抗原の生物学活性の阻害は、抗原の生物学的活性のための1種以上の指標を測定することによって評価することができ、この際、好適な試験管内−又は生体内−検定を使用する。
【0146】
"モノクローナル抗体"とは、ハイブリドー標識ら由来する抗体を意味する(例えば、ハイブリドーマ技術、例えば、Kohler 及び Milsteinによる標準ハイブリドーマ法により製造されるハイブリドー標識ら分泌される抗体)。従って、ハイブリドー標識ら由来し、本発明によるオリゴマー又はその誘導体への特異性を有する抗体は、モノクローナル抗体として表示される。
【0147】
"組換え抗体"とは、組換え手段で製造され、発現され、生成され又は分離される抗体、例えば、宿主細胞にトランスフェクションされた組換え発現ベクターの使用下に発現される抗体;組換え組合せ抗体バンクから分離される抗体;ヒト免疫グロブリン遺伝子によって形質転換された動物(例えば、マウス)から分離される抗体(例えば、Taylor, L. D., et al. (1992) Nucl. Acids. Res. 20:6287-6295参照);又は一定の免疫グロブリン−遺伝子配列(ヒト免疫グロブリン−遺伝子配列)を、他のDNA−配列と集合させる何か別の方法で、製造され、発現され、生成され又は分離される抗体に関する。組換え抗体には、例えば、キメラ、CDR−グラフト−及びヒト化抗体が属する。
【0148】
"ヒト抗体"とは、その可変及び定常領域が、例えば、Kabat et al. (Kabat, et al. (1991) Sequences of Proteins of Immunological Interest, Fifth Edition, U. S. Department of Health und Human Services, NIH Publication Nr. 91-3242参照)により記載されているヒト生殖系列の免疫グロブリン配列に相応し、又はそれから由来する抗体に関する。当然、本発明によるヒト抗体は、例えば、CDRs及び殊にCDR3に、ヒト生殖系列−免疫グロブリン配列によってコードされない(例えば、試験管内でランダム又は部位特異的な突然変異誘発によって又は生体内で体細胞突然変異によって導入される突然変異)アミノ酸基を包含し得る。本発明による組換えヒト抗体は可変領域を有し、ヒト生殖系列の免疫グロブリン配列から由来する定常領域も包含する(Kabat, et al. (1991) Sequences of Proteins of Immunological Interest, Fifth, Edition, U.S. Department of Health und Human Services, NIH Publication Nr. 91-3242参照)。一定の実施態様により、この種類の組換えヒト抗体を、当然、試験管内−突然変異誘発(又は、ヒトIg−配列による形質転換性動物を使用する場合には、体細胞生体内突然変異誘発)させ、従って、組換え抗体のVH−及びVL−領域のアミノ酸配列は、それがヒト生殖系列のVH−及びVL−配列と関連している又はそれから由来するとはいえ、生体内ヒト抗体−生殖系列レパートリー内に自然には存在しない配列である。一定の実施態様により、この種類の組換え抗体は、選別的突然変異誘発又は復帰突然変異、又は両方の結果である。
【0149】
"復帰突然変異"とは、ヒト抗体の体細胞突然変異アミノ酸の若干又は全部を、相同生殖系列抗体配列の相応する生殖系列基と交代させる方法に関する。本発明によるヒト抗体の重鎖及び軽鎖の配列を、別々に、最大の相同性を有する配列を同定するために、VBASE−データバンク中の生殖系列配列と比較する。そのような偏向アミノ酸をコードする限定ヌクレオチド位置で突然変異させることによって、本発明によるヒト抗体中の刷込みを生殖系列配列上に復帰させる。この方法で復帰突然変異の候補として同定される各アミノ酸の抗原結合に対する直接的又は間接的重要性が調査されるべきであり、突然変異後に所望のヒト抗体特性を侵害するアミノ酸は、完成のヒト抗体中に一緒に包含されるべきではない。復帰突然変異のためのアミノ酸の数をできるだけ少なく保つために、隣接の生殖系列配列とは偏向するが、第二の生殖系列配列の相応するアミノ酸配列と同一であるアミノ酸位置は不変のままであってよいが、第二の生殖系列配列は、本発明によるヒト抗体配列と、当該アミノ酸の両側上の少なくとも10個及び有利に12個のアミノ酸で同一である又は共−直線性であることが前提である。復帰突然変異は、抗体最適化の任意段階で行なうことができる。
【0150】
"キメラ抗体"とは、種からの重鎖及び軽鎖の可変領域の配列を包含するが、その中でVH及び/又はVLからの1つ以上のCDR−領域の配列が他の種のCDR−配列と交代している抗体、例えば、マウスからの重鎖及び軽鎖の可変領域を有し、その中で1つ以上のマウス−CDRs(例えば、CDR3)がヒトCDR−配列と交代している抗体に関する。
【0151】
"ヒト化抗体"とは、非ヒト種(例えば、マウス、ラット、ウサギ、ニワトリ、ラクダ、ヤギ)からの重鎖及び軽鎖の可変領域の配列を包含するが、更にもっと"ヒト類似性"にするために、即ち、ヒト生殖系列の可変配列を類似性にするために、少なくとも一部のVH−及び/又はVL−配列が変更されている抗体に関する。ヒト化抗体の種類は、相応する非ヒトCDR−配列を交代させるために、ヒトCDR−配列が非ヒトVH−及びVL−配列中に導入されているCDR−グラフト抗体である。
【0152】
抗体の結合反応速度を測定するための方法は、表面プラスモン共鳴による。"表面プラスモン共鳴"とは、タンパク質濃度の変化をバイオセンサーマトリックスで検出することによって、生体特異的相互作用を分析することができる光学的現象に関し、この際、例えば、BlAcore−Systemを使用する(Pharmacia Biosensor AB, Uppsala, Sweden und Piscataway, NJ)。他の記載については、Joensson, U., et al. (1993) Ann. Biol. Clin. 51:19-26; Joensson, U., et al. (1991) Biotechniques 11:620-627; Johnsson, B., et al. (1995) J. Mol. Recognit. 8:125-131; 及びJohnnson, B., et al. (1991) Anal. Biochem. 198:268-277が参照される。
【0153】
"Koff "とは、抗体/抗原−複合体からの抗体の解離についてのオフ−速度定数に関する。
【0154】
"Kd"とは、一定の抗体−抗原−相互作用の解離定数に関する。
【0155】
本発明による抗体の結合親和性は、標準試験管内−免疫検定、例えば、ELISA−又はBlAcore−分析を使用することによって評価され得る。
【0156】
抗体の他に、タンパク質は、T−細胞受容体から由来する分子又はT−細胞受容体から由来する受容体ドメイン、又は受容体ドメインと免疫グロブリンのFc−部分との融合タンパク質であってよい。
【0157】
本発明の目的は、本発明によるタンパク質及び殊に本発明による抗体及び場合により製薬学的に認容性の賦形剤を含有する製薬学的製剤(組成物)でもある。本発明による製薬学的組成物は、更に少なくとも1種の付加的治療剤、例えば、本発明による抗体がその軽減のために使用可能である病気の治療のための1種以上の付加的治療剤を含有することができる。本発明による抗体を、例えば、本発明によるオリゴマーに結合させる場合には、製薬学的組成物は、更に、オリゴマーの活性が重要である病気の治療のために使用可能である1種以上の付加的な治療剤を含有することができる。
【0158】
製薬学的に認容性の賦形剤には、生理学的に適合性である限り、全ての溶剤、分散媒体、被覆剤、抗菌剤及び抗菌類剤、等張剤及び吸収遅延剤等が属する。製薬学的に認容性の賦形剤には、例えば、水、食塩溶液、燐酸塩−緩衝化食塩溶液、デキストロース、グリセリン、エタノール等、及びその組合せが属する。多くの場合には、等張剤、例えば、糖、ポリアルコール、例えば、マンニトール又はソルビトール、又は塩化ナトリウムを併用することが有利である。製薬学的認容性賦形剤は、更に、抗体の品質保持又は有効性を高める少量の補助物質、例えば、湿潤剤又は乳化剤、保存剤又は緩衝液を含有することができる。
【0159】
製薬学的組成物は、例えば、腸管外投与に好適であってよい。この際、抗体は抗体含量0.1〜250mg/mlを有する注射可能な溶液として調製されることが有利である。注射可能な溶液は、投与形として、液体又は凍結乾燥形で、フリントガラス又はバイアル、アンプル又は充填注射器中に調製されていてよい。緩衝液はL−ヒスチジン(1〜50ミリモル、有利に5〜10ミリモル)を含有し、pH−値5.0〜7.0、有利に6.0を有することができる。他の好適な緩衝液には、コハク酸ナトリウム−、クエン酸ナトリウム−、燐酸ナトリウム−又は燐酸カリウム−緩衝液が属するが、これに限定されるものではない。溶液の張度を濃度0〜300ミリモル(液状投与形については、有利に150ミリモル)に調整するために、塩化ナトリウムを使用することができる。凍結乾燥形のために、凍結保護剤、例えば、スクロース(例えば、0〜10%、有利に0.5〜1.0%)を一緒に含有することができる。他の好適な凍結保護剤には、トレハロース及びラクトースが属する。凍結乾燥投与形のために、充填剤、例えば、マンニトール(例えば、1〜10%、有利に2〜4%)を一緒に含有することができる。安定剤、例えばL−メチオニン(例えば、51〜50ミリモル、有利に5〜10ミリモル)を液状でも、凍結乾燥投与形でも使用することができる。他の好適な充填剤には、グリシン及びアルギニンが属する。同様に、界面活性剤、例えば、ポリソルベート−80(例えば、0〜0.05%、有利に0.005〜0.01%)を使用することができる。他の界面活性剤には、ポリソルベート−20及びBRIJ−界面活性剤が属する。
【0160】
本発明による組成物は、多数の形を取ることができる。これには、液状、半固形及び固形の投与形、例えば、液状溶液(例えば、注射可能な及び注入可能な溶液)、分散液又は懸濁液、錠剤、丸剤、粉末、リポソーム及び座薬が属する。有利な形は、意図される投与方法及び治療的使用に依存する。典型的には、注射可能な又は注入可能な溶液の形の組成物、例えば、ヒトの受動免疫のために他の抗体との類似性を有する組成物が有利である。有利な投与方法は、腸管外(例えば、静脈内、皮下、腹膜内、筋肉内)である。有利な実施態様により、抗体は静脈内注入又は注射によって投与される。もう1つの有利な実施態様により、抗体は、筋肉内又は皮下注射によって投与される。
【0161】
治療的組成物は、典型的には無菌で、製造ー及び貯蔵条件下に安定性でなければならない。組成物は溶液、微細エマルジョン、分散液、リポソーム又は作用物質高濃度に好適な他の次数化構造として組成され得る。注射可能な無菌溶液は、活性化合物(即ち、抗体)を必要量で好適な溶剤中に、場合により必要に応じて1種以上の前記内容物の組合せと一緒に装入し、引き続き、無菌濾過することによって製造され得る。分散液は、通例、基礎分散媒体及び場合により他の必要な内容物を含有する無菌ビヒクル中に活性化合物を入れることによって製造される。注射可能な無菌溶液を製造するための凍結乾燥無菌粉末の場合には、真空乾燥及び噴霧乾燥が有利な製法であり、その方法で、前以て無菌濾過した溶液から活性内容物及び場合により他の所望される内容物の粉末が得られる。溶液の適切な流動性は、例えば、被覆剤、例えば、分散液の場合には、必要な粒度を維持するレシチンを使用し、又は界面活性剤を使用することによって維持され得る。注射可能な組成物の延滞吸収は、吸収を遅延させる薬剤、例えば、モノステアレート塩及びゼラチンを組成物中に一緒に入れることによって達成され得る。
【0162】
本発明による抗体は、当業者に公知である多数の方法で投与され得るが、多数の治療的使用には、皮下注射、静脈内注射又は注入が有利な投与方法である。当業者は、投与の方法及び/又は種類は所望の結果に依ることを周知である。一定の実施態様により、活性化合物を、化合物を急速な放出に対して保護する賦形剤と一緒に、即ち、例えば、調節放出の組成物を製造することができ、それには、インプラント、経皮プラスター及び微細カプセル投与系が属する。生物学的に分解可能な生物適合性ポリマー、例えば、エチレンビニルアセテート、ポリ酸無水物、ポリグリコール酸、コラーゲン、ポリオルトエステル及びポリ乳酸を使用することができる。そのような組成物の製法は、当業者に一般に公知であり、例えば、Sustained und Controlled Release Drug Delivery Systems, J.R.Robinson, ed., Marcel Dekker, Inc., New York, 1978が参照される。
【0163】
一定の実施態様により、例えば、不活性希釈剤又は同化可能な食可能賦形剤中の本発明による抗体を経口投与することができる。抗体(及び所望の場合には、他の内容物)を硬質−又は軟質ゼラチンカプセルに包入させ、打錠して錠剤にし又は食物に直接添加することもできる。治療的経口投与のために、抗体を賦形剤と混合させ、嚥下可能な錠剤、バッカル錠剤、カプセル剤、エリキシル、懸濁液、シロップ等の形で使用することができる。本発明による抗体を、腸管外投与とは別の方法により投与すべき場合には、その不活性化を阻害する材料から被覆を選択することが必要であり得る。
【0164】
本発明による抗体は、それが結合する本発明によるオリゴマー又はその誘導体の活性を試験管内でも生体内でも中和することができる。従って、抗体は、本発明によるオリゴマー又はその誘導体の活性を阻害するために、例えば、オリゴマー又はその誘導体を含有する細胞培養液中又はオリゴマー又はその誘導体が存在しているヒト個体又は哺乳動物中で使用される。1実施態様により、本発明の目的は、本発明によるオリゴマー又はその誘導体の活性の阻害法であり、この際、本発明による抗体をオリゴマー又はその誘導体に、オリゴマー又はその誘導体の活性が阻害されるように作用させることができる。この際、活性は、例えば試験管内で阻害され得る。例えば、本発明による抗体は、本発明によるオリゴマー又はその誘導体を含有する又は含有すると考えられる細胞培養液に添加して、培養液中のオリゴマー又はその誘導体を阻害することができる。選択的に、オリゴマー又はその誘導体の活性を、1個体において生体内で阻害することができる。
【0165】
即ち、本発明のもう1つの目的は、アミロイド−β−タンパク質が関与していて、殊に本発明によるオリゴマー又はその誘導体の活性が関与する病気にかかっている個体における本発明によるオリゴマー又はその誘導体の活性の阻害法に関する。この方法は、抗体が結合するオリゴマー又はその誘導体の活性を阻害する目的で、少なくとも1種の本発明による抗体を個体に投与することを包含する。個体は有利に人間である。本発明による抗体はヒト個体に治療目的のために投与することができる。更に、本発明による抗体を非ヒト哺乳動物に獣医的目的のために、又は一定の病気の動物モデルの範囲で投与することができる。その種類の動物モデルは、本発明による抗体の治療的有効性を評価するために有益である(例えば、投与量及び投与の時間経過を試験するために)。
【0166】
本発明によるオリゴマー又はその誘導体が関与する病気には、殊に、その発生及び/又は経過に本発明によるオリゴマー又はその誘導体が関与している病気が属する。従って、殊に、本発明によるオリゴマー又はその誘導体が、証明的に又は推測的に病気の病態生理学に起因する、又は病気の発生及び/又は経過に寄与する因子である病気が考えられる。従って、本発明によるオリゴマー又はその誘導体の活性の阻害が、病気の症状及び/又は進行を緩和することができる病気がそれに属する。そのような病気は、例えば、一定の病気にかかっている個体の体液中の本発明によるオリゴマー又はその誘導体の高められた濃度によって証明される(例えば、血清、血漿、CSF、ウリン等中の高められた濃度)。このことは、例えば、本発明による抗体を使用することによって検証することができる。本発明によるオリゴマー又はその誘導体は、神経変性要素、認識欠如、神経毒性要素及び炎症性要素が関与している多数の病気と関連している病変において決定的な役割を果たしている。
【0167】
本発明による抗体は、前記の病気の治療に使用可能である1種以上の付加的な治療剤と一緒に投与され得る。
【0168】
本発明の製薬学的組成物は、通例、少なくとも1種の本発明による抗体の治療的有効量又は予防的有効量を含有する。所望の処置によって、例えば、治療的処置が所望されるか又は予防的処置が所望されるかどうかによって、投与計画を選択し、適合させる。例えば、単一用量、時間配分で数回の分別用量又は治療状況の要求により増加又は減少用量を投与することができる。投与を容易にさせ、投与の一様性を保証するために、殊に、腸管外用組成物を単一用量形で組成させることが有利である。
【0169】
本発明による抗体の治療的又は予防的有効量は、0.1〜20mg/kg及び有利に1〜10mg/kgの範囲であってよいが、これに限定されるものではない。この量は、当然、緩和すべき状態の種類及び重大性によって変化され得る。
【0170】
抗体の診断的使用の範囲で、定性又は定量的な特異的オリゴマー−測定が、殊に、病気に関連するアミロイド−β(1−42)−型の診断に用いられる。この関係における特異性は、充分な感受性を有する一定のオリゴマー又はオリゴマー−混合物を検出し得る可能性を意味する。本発明による抗体は、10ng/試料mlよりも少ない、有利に1ng/試料mlよりも少ない、特に有利に100pg/試料mlよりも少ない感受性を有利に有する。従って、少なくとも、試料1mlに対する各々前記のオリゴマー濃度を、有利により少ない濃度も、本発明による抗体で検出できることが考えられる。
【0171】
測定は免疫学的に行なわれる。これは、原則的には、抗体を使用する各分析的又は診断的試験法で行なわれ得る。この方法には、凝集−及び沈殿−法、免疫検定法、免疫組織化学法及び免疫ブロット−法、例えば、ウエスタン−ブロット法又はドットブロット−法が属する。生体内−法には、例えば、画像法も属する。
【0172】
免疫検定法における使用が有利である。競合的免疫検定、即ち、抗体結合について抗原及び標識化抗原(トレーサー)を競合させることも、サンドイッチ−免疫検定法、即ち、抗原への特異的抗体の結合を第二の、大抵は標識化された抗体で確認することも好適である。これらの検定法は、均一であっても(即ち、固相及び液相へ分離しない)、不均一であってもよい(即ち、例えば、固相結合抗体を介して結合標識体を非結合体から分離することができる)。様々な不均一及び均一免疫検定−形は、標識化及び測定法により決められる部類、例えば、RIAs(放射線免疫検定法)、ELISA(固相酵素免疫検定法)、FIA(蛍光−免疫検定法)、LIA(発光−免疫検定法)、TEFIA(時間的分解FIA)、IMAC(免疫活性化)、EMIT(酵素多重化免疫試験法)、TIA(ターボジメトリック免疫検定法)、I−PCR(免疫−PCR)に配属される。
【0173】
本発明によるオリゴマー−測定には、競合的免疫検定法が有利である。この際、標識化オリゴマー(トレーサー)は、使用抗体への結合について、試料の測量すべきオリゴマーと競合する。置換されたトレーサーの量から、標準曲線によって、試料中の抗原量、即ち、オリゴマー量を測定することができる。
【0174】
この目的に使用される標識は、酵素が有利であることが判明した。例えば、ペルオキシダーゼ、殊に西洋わさび−ペルオキシダーゼ、アルカリ性ホスファターゼ及びβ−D−ガラクトシダーゼをベースとする系を使用することができる。その変換を、例えば、測光的に追跡し得る特異的基質がこの酵素のために使用される。好適な基質系は、アルカリ性ホスファターゼについては、p−ニトロフェニルホスフェート(p−NPP)、5−ブロム−4−クロル−3−インドリルホスフェート/ニトロブルー−テトラゾリウム(BCIP/NPT)、ファースト−レッド/ネフトール−AS−TS−ホスフェート;ペルオキシダーゼについては、2,2−アジノ−ビス−(3−エチルベンズチアゾリン−6−スルホン酸)(ABTS)、o−フェニレンジアミン(OPT)、3,3’,5,5’−テトラメチルベンジジン(TMB)、o−ジアニシジン、5−アミノサリチル酸、3−ジメチルアミノ安息香酸(DMAB)及び3−メチル−2−ベンゾチアゾリンヒドラゾン(MBTH);β−D−ガラクトシダーゼについては、o−ニトロフェニル−β−D−ガラクトシド(o−NPG)、p−ニトロフェニル−β−D−ガラクトシド及び4−メチルウムベリ(umbelli)フェニル−β−D−ガラクトシド(MUG)をベースとする。多くの場合において、これらの基質系は既製形で、例えば、他の試薬、例えば、有利に緩衝液等も含有し得る錠剤の形で、市販で得られる。
【0175】
トレーサーとして、標識化オリゴマーが使用される。この意において、一定のオリゴマーの測定には、測定すべきオリゴマーを標識化し、トレーサーとして使用することができる。
【0176】
トレーサーの製造のためのオリゴマーへの標識の結合は、自体公知の方法で行なうことができる。本発明によるオリゴマーの誘導体化のための前記の実施が同様に引用される。更に、タンパク質の複合のために有利に変性された一連の標識、例えば、ビオチン−、アビジン−、エキストラビジン−又はストレプトアビジン−複合酵素、マレイミド−活性化酵素等が得られる。これらの標識は、オリゴマー又は、必要に応じて、相応に誘導体化されたオリゴマーと直接反応してトレーサーを得ることができる。例えば、ストレプトアビジン−ペルオキシダーゼ−複合体を使用する場合には、これは先ずオリゴマーのビオチニル化を必要とする。相応することは逆の順序にも当てはまる。この目的についても、好適な方法は当業者に公知である。
【0177】
不均一免疫検定−型を選択する場合には、抗原−抗体−複合体を、分離のために、例えば、担体に結合した抗−イディオタイプ抗体、例えば、ウサギ−IgGに照準した抗体を介して担体に結合させることができる。相応する抗体を担持している担体、殊に、微小滴定プレートは公知であり、部分的に市販で得られる。
【0178】
本発明のもう1つの目的は、少なくとも1種の前記の抗体及び他の成分を有する免疫検定−セットである。この際、本発明によるオリゴマー−測定の実施のための手段の、通例、包装ユニットとしての集成装置が重要である。できるだけ簡単な取扱いのために、この手段は、有利に実際に使用完成されている。有利な装置は免疫検定をキット−型で提供する。キットは、通例、成分の別々の装置用の数個の容器を包含する。全成分は、使用完成された希釈液で、希釈用濃縮物として又は溶解又は懸濁用乾燥物質又は凍結乾燥物として準備されていてよい;単一成分又は全成分は凍結されている又は環境温度で使用するまで貯蔵されていてよい。血清は、例えば、−20℃でショック凍結され、従って、この場合には、免疫検定を使用前に有利に凍結温度で保持しなければならない。
【0179】
免疫検定に添加される他の成分は、免疫検定の種類に依る。通例、抗血清標準タンパク質、場合により必要なトレーサー及び対照血清と一緒に添加される。更に、有利に抗体を担持させた微小滴定プレート、例えば、基質の試験、洗浄又は反応のための緩衝液、及び酵素基質自体を添加することができる。
【0180】
実験室及び臨床における免疫検定の一般的原則及び助剤としての抗体の製造及び使用は、例えば、Antibodies, A Laboratory Manual (Harlow, E., and Lane, D., Ed., Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, 1988)にある。
【0181】
更に、本発明によるオリゴマーの集合を阻害し又はその解離を促進させる物質も重要である。この種類の物質は、前記のアミロイド−β−関連病、例えば、アルツハイマー病の治療に殊に可能な治療剤である。
【0182】
従って、本発明の目的は、物質又は物質混合物の特性付けのための方法でもあり、この際、
i)物質又は物質混合物を好適な方法で準備する;
ii)物質又は物質混合物を、少なくとも1種の本発明によるオリゴマー、その誘導体又は組成物に作用させる;及び
iii)物質又は物質混合物の一定部分が、オリゴマー、その誘導体又は組成物中に含有される少なくとも1種のオリゴマー又はその誘導体に結合しているかどうかを測定する。
【0183】
この方法は、本発明によるオリゴマーへの親和性を有する天然の結合相手、例えば、細胞表面抗原、殊に、受容体及び可溶性リガンド、例えば、一定のタンパク質及びメディエーターを同定するために、生物学的起源の混合物、例えば、細胞標本及び−抽出物に実施することもできる。
【0184】
物質の単なる結合に加えて、本発明によるオリゴマーとの相互作用及び本発明によるオリゴマーへの影響も本方法の目的であってよい。即ち、殊に、
物質が本発明によるオリゴマーに対するアミロイド−β−タンパク質の集合を変性させ、殊に、阻害させることができるかどうか;
物質が、本発明によるオリゴマーの解離を変性させ、殊に、促進させることができるかどうか;
本発明によるオリゴマーが、結合相手の機能変化を呼び起こし、例えば、受容体で共働的、部分的共働的、拮抗的又は反共働的作用を有するかどうかを測定することができる。
【0185】
この方法では、通例、試験管内−スクリーニング−法が重要であり、この方法で、多数の様々な物質から、将来の使用を展望して最も有望であると思われる物質を選別することができる。例えば、組合せ化学によって、多数のポテンシャル作用物質を包含する広範囲の物質バンクを設定することができる。所望の活性を有する物質による組合せ物質ライブラリーの精査は自動可能である。スクリーニング−ロボットは、有利に微小滴定プレート上に配置された単一検定の効率的な評価に用いられる。即ち、本発明は、スクリーニング−法、即ち、一次スクリーニング−法にも、二次スクリーニング−法にも関し、その際、有利に、次に記載した方法の少なくとも1法が使用される。数種の方法を使用する場合には、方法を時間的にずらして又は同時に、検査すべき物質の1試料及び同一試料又は異なる試料で行なうことができる。
【0186】
この種類の方法の特に効果的な実施技術は、作用物質スクリーニングの範囲で公知のシンチレーション近接検定(略称SPA)である。この検定法の実施のためのキット及び成分は、例えば、Amersham Pharmacia Biotech.で市販されて入手することができる。原則的に、溶解化又は膜結合受容体は、シンチレーション物質を含有する蛍光微小球上で固定化される。例えば、放射性リガンドが固定化受容体に結合する場合には、シンチレーション物質と放射性リガンドとの間に空間的近接が生じるので、シンチレーション物質は励起して発光する。
【0187】
この種類の方法のもう1つの特に効果的な実施技術は、作用物質スクリーニングの範囲で公知のフラッシュプレートR−技術である。この検定法の実施のためのキット及び成分は、例えば、NENR Life Science Productsで市販されて得られる。この原則は、同様にシンチレーション物質で被覆される微小滴定プレート(96又は384個の)に基づく。
【0188】
この方法により、オリゴマー、その誘導体又は相応する組成物中に含有される少なくとも1種のオリゴマー又はその誘導体に結合するリガンドとして同定可能な物質又は物質混合物からの一部は、アミロイド−β−関連病、殊に、痴呆症の治療剤を製造するための、又はアミロイド−β−関連病、殊に、痴呆症の診断用組成物を製造するためのその使用と全く同様に本発明の目的である。
【図面の簡単な説明】
【0189】
【図1】図1は、Aβ(1−42)−オリゴマーA−標本(線A);Aβ(1−42)−オリゴマーB−標本(線B);標準タンパク質(分子標識タンパク質、線C)のSDS−PAGEを示す
【図2】図2は、Aβ(1−42)−オリゴマーA−標本(線A);Aβ(1−42)−オリゴマーA−CL−標本(線A’);Aβ(1−42)−オリゴマーB−標本(線B);Aβ(1−42)−オリゴマーB−CL−標本(線B’);標準タンパク質(分子標識タンパク質、線C)のSDS−PAGEを示す
【図3】図3は、ビオチン−Aβ(1−42)−オリゴマーB−標本(線A);標準タンパク質(分子標識タンパク質、線B)のSDS−PAGEを示す
【図4】図4は、フルオレスセイン−Aβ(1−42)−オリゴマーB−標本(線A);標準タンパク質(分子標識タンパク質、線B)のSDS−PAGEを示す
【図5】図5は、Aβ(1−42)−凍結乾燥物を含有する溶液の、Aβ(1−42)−オリゴマーBを含有する標本と比較したゲル透過クロマトグラフィーを示す
【図6】図6は、Aβ(1−42)−オリゴマーB−標本(線A);標準タンパク質(分子標識タンパク質、線B)のNATIVE−PAGEを示す
【図7】図7は、(A)モノマーAβ(1−42)−タンパク質及び(B)Aβ(1−42)−オリゴマーA及び(C)Aβ(1−42)−オリゴマーの、ヒト神経芽細胞腫−細胞系IMR−32の表面への結合を示す
【図8】図8は、マウスの皮質ニューロンをAβ(1−42)−オリゴマーBで処理した後の神経毒作用(%)(この際、誤差バーは信頼区間95%に相応する)を示す
【図9】図9は、トリプシン(線2)、キモトリプシン(線3)、サーモリシン(線4)、エラスターゼ(線5)、パパイン(線6)で処理した又は未処理(線7)であったAβ(1−42)−標本;標準タンパク質(分子標識タンパク質、線1)のSDS−PAGEを示す
【図10】図10は、例6bからのAβ(1−42)−オリゴマーB−標本(欄1);例1aからのHFIP−処理したAβ(1−42)−モノマー(欄2);例15aからのサーモリシンで分解したAβ(1−42)−オリゴマーB−標本(欄3);例14aからのグルタルアルデヒドと架橋結合したAβ(1−42)−オリゴマーB−標本(欄4);M. P. Lambert et al., J. Neurochem. 79,595-605(2001))による4℃又は室温又は37℃で製造したADDL(欄5,6又は7);0.1%NH4OH中に溶かしたAβ(1−42)(欄8);例27からのAβ(1−42)−原線維−標本(欄9);及びPBS中で希釈したFa. SigmaのAPP(欄10)100ピコモル(pmol)(行A);10ピコモル(行B);1ピコモル(行C);0.1ピコモル(行D)又は0.01ピコモル(行E)と、a)モノクローナル抗体6E10;b)例25dからのポリクローナル抗血清(d1);c)例25cからのポリクローナル抗血清(c1);d)例25aからのポリクローナル抗血清(a1);及びe)例25aからのポリクローナル抗血清(a2)との反応性のドットブロットを示す
【図11】図11は、例15aからのAβ(20−42)−オリゴマーB−標本(線B);例15bからのAβ(12−42)−オリゴマーB−標本(線C);例6bからのAβ(1−42)−オリゴマーB−標本(線A);及び標準タンパク質(分子標識タンパク質、線D)のSDS−PAGEを示す
【図12】図12は、例6bからのAβ(1−42)−オリゴマーB−標本の、例14aからのAβ(1−42)−オリゴマーB−CL−標本に比較したゲル透過クロマトグラフィーを示す
【図13】図13は、モノマーAβ(1−42)−タンパク質(左側);Aβ(1−42)−オリゴマー(12−マー、A);及びタンパク質加水分解によって得られるAβ(12−42)−オリゴマー(12−マー、C)及びAβ(20−42)−オリゴマー(12−マー、B)の図解表示を示す
【図14】図14は、海馬断片へのAβ(1−42)−オリゴマーBの作用下に、時間に依存する興奮性シナプス後電位(fEPSP)を示す
【図15】図15は、海馬ラットニューロンへのAβ(1−42)−オリゴマーB(a)の、非特異的結合蛍光(b)に比較した結合を示す免疫蛍光画像を示す
【実施例】
【0190】
次に実施例につき本発明を詳説するが、この範囲に限定されるものではない。
【0191】
他の記載のない限り、本発明によるオリゴマーの濃度を、モノマーのAβ(1−42)−ポリペプチドのモルで表わす。概念β−アミロイド(1−42)−タンパク質は、概念アミロイド−β(1−42)−タンパク質に相応する。他の記載のない限り、使用したタンパク質(ポリペプチド)は、ヒト起源である。
【0192】
例1
a)ヒトAβ(1−42)の基本懸濁液の製造
ヒトβ−アミロイド(1−42)−タンパク質(略称:Aβ(1−42);ペプチド合成原料、凍結乾燥物、Fa. Bachem, Deutschland)2mgを、1,1,1,3,3,3−ヘキサフルオル−2−プロパノール800μl中に溶かし、エッペンドルフ容器中37℃で30分間恒温保持する。その後に、真空濃縮機(Speed Vac)中で蒸発乾固させる。残渣をDMSO88μlに入れ、この際、Aβ(1−42)−基本懸濁液5ミリモルが生じる。これを−20℃で保存することができる。
【0193】
b)ラット−Aβ(1−42)の基本懸濁液の製造
ラット−β−アミロイド(1−42)−タンパク質(略称:ラット−Aβ(1−42);ペプチド合成原料、凍結乾燥物、Fa. Bachem, Deutschland)2mgを、1,1,1,3,3,3−ヘキサフルオル−2−プロパノール800μl中に溶かし、エッペンドルフ容器中37℃で30分間恒温保持する。その後に、真空濃縮機(Speed Vac)中で蒸発乾固させる。残渣をDMSO88μlに入れ、この際、ラット−Aβ(1−42)−基本懸濁液5ミリモルが生じる。これを−20℃で保存することができる。
【0194】
例2
a)分子量15kDa及び20kDaを有するAβ(1−42)−オリゴマー[Aβ(1−42)−オリゴマーA]の製造;SDSの使用
例1aからの基本溶液60μlにPBS−緩衝液(燐酸ナトリウム20ミリモル、NaCl140ミリモル、pH7.4)690μlを加え、2%のドデシル硫酸ナトリウム(SDS)−溶液75μlで0.2%SDSの含量に調整する。その後に、37℃で5時間恒温保持し、10000gで10分間遠心分離する。このAβ(1−42)−オリゴマーA−標本(Aβ(1−42)約400マイクロモル)を−20℃で保存することができる。
【0195】
b)分子量15kDa及び20kDaを有するラット−Aβ(1−42)−オリゴマー[ラット−Aβ(1−42)−オリゴマーA]の製造;SDSの使用
例1bからの基本溶液60μlにPBS−緩衝液(燐酸ナトリウム20ミリモル、NaCl140ミリモル、pH7.4)690μlを加え、2%のドデシル硫酸ナトリウム(SDS)−溶液75μlで0.2%SDSの含量に調整する。その後に、37℃で5時間恒温保持し、10000gで10分間遠心分離する。このラット−Aβ(1−42)−オリゴマーA−標本(ラット−Aβ(1−42)約400マイクロモル)を−20℃で保存することができる。
【0196】
c)分子量15kDa及び20kDaを有するAβ(1−42)−オリゴマー[Aβ(1−42)−オリゴマーA]の製造;ラウリル酸の使用
例1aからの基本溶液60μlにPBS−緩衝液(燐酸ナトリウム20ミリモル、NaCl140ミリモル、pH7.4)690μlを加え、5%のラウリル酸−溶液75μlで0.5%ラウリル酸の含量に調整する。その後に、37℃で5時間恒温保持し、10000gで10分間遠心分離する。このAβ(1−42)−オリゴマーA−標本(Aβ(1−42)約400マイクロモル)を−20℃で保存することができる。
【0197】
d)分子量15kDa及び20kDaを有するAβ(1−42)−オリゴマー[Aβ(1−42)−オリゴマーA]の製造;油酸の使用
例1aからの基本溶液60μlにPBS−緩衝液(燐酸ナトリウム20ミリモル、NaCl140ミリモル、pH7.4)690μlを加え、5%の油酸−溶液75μlで0.5%油酸の含量に調整する。その後に、37℃で5時間恒温保持し、10000gで10分間遠心分離する。このAβ(1−42)−オリゴマーA−標本(Aβ(1−42)約400マイクロモル)を−20℃で保存することができる。
【0198】
e)分子量15kDa及び20kDaを有するAβ(1−42)−オリゴマー[Aβ(1−42)−オリゴマーA]の製造;ラウロイルサルコシンの使用
例1aからの基本溶液60μlにPBS−緩衝液(燐酸ナトリウム20ミリモル、NaCl140ミリモル、pH7.4)690μlを加え、5%のラウロイルサルコシン−溶液75μlで0.5%ラウロイルサルコシンの含量に調整する。その後に、37℃で5時間恒温保持し、10000gで10分間遠心分離する。このAβ(1−42)−オリゴマーA−標本(Aβ(1−42)約400マイクロモル)を−20℃で保存することができる。
【0199】
例3
a)分子量15kDa及び20kDaを有するAβ(1−42)−オリゴマー[Aβ(1−42)−オリゴマーA]の製造のための選択的製法
ヒトβ−アミロイド(1−42)−タンパク質(略称:Aβ(1−42);ペプチド合成原料、凍結乾燥物、Fa. Bachem, Deutschland)1mgを、HCl−水溶液10ミリモル220μl中に入れ、室温で10分間恒温保持する。不溶成分を10000gで5分間遠心分離する。上澄液(Aβ(1−42) 1ミリモル)は、Aβ(1−42)−タンパク質を含有し、次のように更に処理される:
上澄液1μlにPBS−緩衝液9μl及び2%のSDS−溶液1μlを加え、37℃で16時間恒温保持する。そうして得られるhAβ(1−42)−オリゴマーA−標本(100マイクロモル)を−20℃で保存することができる。
【0200】
b)分子量15kDa及び20kDaを有するラット−Aβ(1−42)−オリゴマー[ラット−Aβ(1−42)−オリゴマーA]の製造のための選択的製法
ラット−β−アミロイド(1−42)−タンパク質(略称:ラット−Aβ(1−42);ペプチド合成原料、凍結乾燥物、Fa. Bachem, Deutschland)1mgを、HCl−水溶液10ミリモル220μl中に入れ、室温で10分間恒温保持する。不溶成分を10000gで5分間遠心分離する。上澄液(ラット−Aβ(1−42) 1ミリモル)は、ラット−Aβ(1−42)−タンパク質を含有し、次のように更に処理される:
上澄液1μlにPBS−緩衝液9μl及び2%のSDS−溶液1μlを加え、37℃で16時間恒温保持する。そうして得られるラット−Aβ(1−42)−オリゴマーA−標本(100マイクロモル)を−20℃で保存することができる。
【0201】
例4
a)分子量15kDa及び20kDaを有するAβ(1−42)−オリゴマー[Aβ(1−42)−オリゴマーA]の製造のための選択的製法
ヒトβ−アミロイド(1−42)−タンパク質(略称:Aβ(1−42);ペプチド合成原料、凍結乾燥物、Fa. Bachem, Deutschland)1mgを、1%SDS/H2O44μl中に溶かす(Aβ(1−42) 5ミリモル)。この溶液5μlにPBS40μl及び2%SDS5μlを加え、37℃で16時間恒温保持する。不溶成分を10000gで5分間遠心分離する。そうして得られるAβ(1−42)−オリゴマーA−標本(Aβ(1−42) 500マイクロモル)を−20℃で保存することができる。
【0202】
b)分子量15kDa及び20kDaを有するラット−Aβ(1−42)−オリゴマー[ラット−Aβ(1−42)−オリゴマーA]の製造のための選択的製法
ラット−β−アミロイド(1−42)−タンパク質(略称:ラット−Aβ(1−42);ペプチド合成原料、凍結乾燥物、Fa. Bachem, Deutschland)1mgを、1%SDS/H2O44μl中に溶かす(ラット−Aβ(1−42) 5ミリモル)。この溶液5μlにPBS40μl及び2%SDS5μlを加え、37℃で16時間恒温保持する。不溶成分を10000gで5分間遠心分離する。そうして得られるラット−Aβ(1−42)−オリゴマーA−標本(Aβ(1−42) 500マイクロモル)を−20℃で保存することができる。
【0203】
例5
a)分子量38kDa及び48kDaを有するAβ(1−42)−オリゴマー[Aβ(1−42)−オリゴマーB]の製造
例2aにより得られるAβ(1−42)−オリゴマーA−溶液を、水2475mlで希釈し(0.05%SDS−含量、Aβ(1−42) 0.1ミリモル)、37℃で20時間恒温保持する。このAβ(1−42)−オリゴマーB−標本の部分標本を−80℃で凍結させ、更なる研究のために保存する。
b)分子量38kDa及び48kDaを有するラット−Aβ(1−42)−オリゴマー[ラット−Aβ(1−42)−オリゴマーB]の製造
例2bにより得られるラット−Aβ(1−42)−オリゴマーA−溶液を、水2475mlで希釈し(0.05%SDS−含量、ラット−Aβ(1−42) 0.1ミリモル)、37℃で20時間恒温保持する。このラット−Aβ(1−42)−オリゴマーB−標本の部分標本を−80℃で凍結させ、更なる研究のために保存する。
【0204】
c)分子量38kDa及び48kDaを有するAβ(1−42)−オリゴマー[Aβ(1−42)−オリゴマーB]の選択的製造
例2cにより得られるAβ(1−42)−オリゴマーA−溶液を、水2475mlで希釈し(0.125%ラウリル酸−含量、Aβ(1−42) 0.1ミリモル)、37℃で20時間恒温保持する。このAβ(1−42)−オリゴマーB−標本の部分標本を−80℃で凍結させ、更なる研究のために保存する。
【0205】
d)分子量38kDa及び48kDaを有するAβ(1−42)−オリゴマー[Aβ(1−42)−オリゴマーB]の選択的製造
例2dにより得られるAβ(1−42)−オリゴマーA−溶液を、水2475mlで希釈し(0.125%油酸−含量、Aβ(1−42) 0.1ミリモル)、37℃で20時間恒温保持する。このAβ(1−42)−オリゴマーB−標本の部分標本を−80℃で凍結させ、更なる研究のために保存する。
e)分子量38kDa及び48kDaを有するAβ(1−42)−オリゴマーB[Aβ(1−42)−オリゴマーB]の選択的製造
例2eにより得られるAβ(1−42)−オリゴマーA−溶液を、水2475mlで希釈し(0.125%ラウロイルサルコシン−含量、Aβ(1−42) 0.1ミリモル)、37℃で20時間恒温保持する。このAβ(1−42)−オリゴマーB−標本の部分標本を−80℃で凍結させ、更なる研究のために保存する。
【0206】
f)SDSを含まない、分子量38kDa及び48kDaを有するAβ(1−42)−オリゴマーB[Aβ(1−42)−オリゴマーB]の製造
例6bにより製造したAβ(1−42)−オリゴマーB−標本10μlに、比率4%/33%/63%の酢酸/メタノール/水−混合物250μlを加え、氷上0℃で30分間恒温保持する。遠心分離(10000gで10分間)後に、上澄液を取り出し、沈殿したタンパク質残渣を緩衝液(燐酸−Na20ミリモル、NaCl140ミリモル、pH7.4)200μl中に入れる。そうして得られる標本は、SDSを含まない形の溶解したAβ(1−42)−オリゴマーBを含有し、−20℃で保存され得る。
【0207】
例6
a)分子量38kDa及び48kDaを有するAβ(1−42)−オリゴマー[Aβ(1−42)−オリゴマーB]の透析及び濃縮
例5aにより製造したAβ(1−42)−オリゴマーB−標本に、0.1%プルロニック(Pluronic)(登録商標)F68(Fa. BASF)を含有するPBS−緩衝液30mlを加え、Fa. Amicon のCentriprep YM, 30 KD中で3mlに濃縮させる。場合により存在する残渣を遠心分離(10000gで5分間)によって除去する。上澄液を取り出す。このAβ(1−42)−オリゴマーB−標本の部分標本を−80℃で凍結させ、更なる研究のために保存することができる。
【0208】
b)分子量38kDa及び48kDaを有するAβ(1−42)−オリゴマーB[Aβ(1−42)−オリゴマーB]の濃縮物の製造
例5aにより得られるAβ(1−42)−オリゴマーB−標本72.6mlを、30 KD−Centriprep−細管YM(Amicon)上で2mlに濃縮させる。濃縮物を10000gで10分間遠心分離する。上澄液を取り出し、透析管中で、緩衝液(燐酸ナトリウム5ミリモル、塩化ナトリウム35ミリモル、pH7.4)1lに対して6℃で16時間透析する。透析物を10000gで10分間遠心分離する。上澄液を取り出し、更なる研究のために−80℃で保存することができる。
【0209】
例7
ビオチン−Aβ(1−42)−基本懸濁液の製造
ビオチン−β−アミロイド(1−42)−タンパク質(略称:ビオチン−Aβ(1−42);ペプチド合成原料、凍結乾燥物、AnaSpec)0.5mgを、1,1,1,3,3,3−ヘキサフルオル−2−プロパノール200μl中に溶かし、エッペンドルフ容器中37℃で30分間恒温保持する。その後に、真空濃縮機(Speed Vac)中で蒸発乾固させる。残渣をDMSO20.5μlに入れ、この際、ビオチン−Aβ(1−42)−基本懸濁液5ミリモルが生じる。これを−20℃で保存することができる。
【0210】
例8
分子量17kDa及び22kDaを有するビオチン−Aβ(1−42)−オリゴマー[ビオチン−Aβ(1−42)−オリゴマーA]の製造
例7からの基本懸濁液2μlに、PBS−緩衝液(燐酸ナトリウム20ミリモル、NaCl140ミリモル、pH7.4)23μlを加え、2%のSDS−溶液2.4μlで0.2%SDSの含量に調整する。その後に、37℃で6時間恒温保持し、不溶成分を10000gで5分間遠心分離する。そうして得られるビオチン−Aβ(1−42)−オリゴマーA−標本を−20℃で保存することができる。
【0211】
例9
分子量42kDa及び52kDaを有するビオチン−Aβ(1−42)−オリゴマー[ビオチン−Aβ(1−42)−オリゴマーB]の製造
例8により得られるビオチン−Aβ(1−42)−オリゴマーA−溶液を、水82μlで希釈し(0.05%SDS−含量、Aβ0.1ミリモル)、37℃で16時間恒温保持する。不溶成分を10000gで5分間遠心分離する。このビオチン−Aβ(1−42)−オリゴマーB−標本を−20℃で凍結させ、更なる研究のために保存することができる(図3)。
【0212】
例10
フルオレスセイン−Aβ(1−42)−基本懸濁液の製造
フルオレスセイン−β−アミロイド(1−42)−タンパク質(略称:フルオレスセイン−Aβ(1−42);ペプチド合成原料、凍結乾燥物、AnaSpec)0.5mgを、1,1,1,3,3,3−ヘキサフルオル−2−プロパノール200μl中に溶かし、エッペンドルフ容器中37℃で30分間恒温保持する。その後に、真空濃縮機(Speed Vac)中で蒸発乾固させる。残渣をDMSO20.5μlに入れ、この際、フルオレスセイン−Aβ(1−42)−基本懸濁液5ミリモルが生じる。これを−20℃で保存することができる。
【0213】
例11
分子量17kDa及び22kDaを有するフルオレスセイン−Aβ(1−42)−オリゴマー[フルオレスセイン−Aβ(1−42)−オリゴマーA]の製造
例10からの基本懸濁液2μlにPBS−緩衝液(燐酸ナトリウム20ミリモル、NaCl140ミリモル、pH7.4)23μlを加え、2%のSDS−溶液2.4μlで0.2%SDSの含量に調整する。その後に、37℃で6時間恒温保持する。不溶成分を10000gで10分間遠心分離する。そうして得られるフルオレスセイン−Aβ(1−42)−オリゴマーA−標本を−20℃で保存することができる。
【0214】
例12
分子量42kDa及び52kDaを有するフルオレスセイン−Aβ(1−42)−オリゴマー[フルオレスセイン−Aβ(1−42)−オリゴマーB]の製造
例11により得られるフルオレスセイン−Aβ(1−42)−オリゴマーA−溶液を、水82μlで希釈し(0.05%SDS−含量、Aβ(1−42) 0.1ミリモル)、37℃で16時間恒温保持する。フルオレスセイン−Aβ(1−42)−オリゴマーB−標本を−80℃で凍結させ、更なる研究のために保存することができる(図4)。
【0215】
例13
例2aからのAβ(1−42)−オリゴマーA[Aβ(1−42)−オリゴマーA−CL]の架橋結合
例2aにより製造したAβ(1−42)−オリゴマーA−溶液10μlをPBS7.5μl、0.2%SDSで、Aβ(1−42)−含量100マイクロモルに希釈する。この溶液に新規に製造したグルタルジアルデヒド−水溶液10ミリモル1μlを加え、これを室温で3時間攪拌する。過剰のグルタルジアルデヒドを飽和させるために、試料に、エタノールアミン−水溶液100ミリモル1μl、pH7.4を加え、1時間攪拌する。そうして得られる標本は、架橋結合したAβ(1−42)−オリゴマーAを含有し、Aβ(1−42)−オリゴマーA−CL−標本と表示される。
【0216】
例14
a)例5aからのAβ(1−42)−オリゴマーB[Aβ(1−42)−オリゴマーB−CL]の架橋結合
例5aにより製造したAβ(1−42)−オリゴマーB−溶液10μlに、新規に製造したグルタルジアルデヒド−水溶液10ミリモル1μlを加え、室温で3時間攪拌する。過剰のグルタルジアルデヒドを飽和させるために、試料に、エタノールアミン−水溶液100ミリモル1μl、pH7.4を加え、1時間攪拌する。そうして得られる標本は、架橋結合したAβ(1−42)−オリゴマーBを含有し、Aβ(1−42)−オリゴマーB−CL−標本と表示される。
【0217】
b)Aβ(1−42)−オリゴマー[Aβ(1−42)−オリゴマーB−CL]の架橋結合のための選択的方法
例5aにより製造したAβ(1−42)−オリゴマーB−溶液72.6mlに、新規に製造したグルタルジアルデヒド−水溶液10ミリモル7,26mlを加え、室温で2時間攪拌する。過剰のグルタルジアルデヒドを飽和させるために、試料に、緩衝液(燐酸−Na20ミリモル、NaCl140ミリモル、エタノールアミン500ミリモル、pH7.4)726μlを加え、室温で30分間攪拌する。成分を30kDa−セントリプレプ(Centriprep)−細管15ml上で3mlに濃縮させる。濃縮物を10000gで10分間遠心分離する。上澄液を取り出し、透析管中で、燐酸−Na115ミリモル、NaCl35ミリモル、pH7.4に対して、6℃で16時間透析する。その後に、透析物を10000gで10分間遠心分離し、上澄液を取り出す。上澄液を更なる研究のために−80℃で保存することができる。そうして得られる標本は、架橋結合したAβ(1−42)−オリゴマーBを含有し、Aβ(1−42)−オリゴマーB−CL−標本と表示される。
【0218】
例15
a)サーモリシンでの分解による、Aβ(1−42)−オリゴマーBから出発する切端されたAβ(20−42)−オリゴマーの製造:
例6bにより製造したAβ(1−42)−オリゴマーB−標本1.59mlに、緩衝液(MES/NaOH50ミリモル、pH7.4)38ml及びサーモリシン−水溶液(Fa. Roche)1mg/ml 200μlを加える。成分を室温で20時間攪拌する。その後に、EDTA−水溶液100ミリモル(pH7.4)80μlを加え、更に、1%のSDS−溶液400μlでSDS−含量0.01%に調整する。成分を、30kDa−セントリプレプ−細管15mlを介して約1mlに濃縮させる。濃縮物に、緩衝液(MES/NaOH50ミリモル、SDS0.02%、pH7.4)9mlを加え、再び1mlに濃縮させる。濃縮物を、透析管中で、緩衝液(燐酸−Na5ミリモル、NaCl35ミリモル)1lに対して、6℃で16時間透析する。透析物を2%のSDS−水溶液で、SDS−含量0.1%に調整する。試料を10000gで10分間遠心分離し、上澄液を取り出す。
【0219】
そうして得られる物質を更に分析する(SDS−ポリアクリルアミドゲル−電気泳動;図11参照);生成した切端オリゴマーの質量分析により、オリゴマーは切端Aβ(20−42)から成ることが明らかである。
【0220】
b)エンドプロテイナーゼGluCでの分解による、Aβ(1−42)−オリゴマーBから出発する切端Aβ(12−42)−オリゴマーの製造:
例6bにより製造したAβ(1−42)−オリゴマーB−標本2mlに、緩衝液(燐酸ナトリウム5ミリモル、塩化ナトリウム35ミリモル、pH7.4)38ml及びエンドプロテイナーゼGluC(Fa. Roche)1mg/ml(水中)150μlを加える。成分を室温で6時間攪拌する。引き続き、もう1度、水中のエンドプロテイナーゼGluC(Fa. Roche)1mg/ml 150μlを加える。成分を室温で更に16時間攪拌する。その後に、DIFP−溶液5モル8μlを加える。成分を、30kDa−セントリプレプ−細管15ml上で約1mlに濃縮させる。濃縮物に、緩衝液(燐酸ナトリウム5ミリモル、塩化ナトリウム35ミリモル、pH7.4)9mlを加え、再び1mlに濃縮させる。濃縮物を、透析管中で、緩衝液(燐酸−Na5ミリモル、NaCl35ミリモル)1lに対して、6℃で16時間透析する。透析物を1%のSDS−水溶液で、SDS−含量0.1%に調整する。試料を10000gで10分間遠心分離し、上澄液を取り出す。
【0221】
そうして得られる物質を更に分析する(SDS−ポリアクリルアミドゲル−電気泳動;図11参照);生成した切端オリゴマーの質量分析により、オリゴマーは切端Aβ(12−42)から成ることが判明する。
【0222】
Aβ(1−42)−オリゴマーの特徴付け
例16
SDS−ポリアクリルアミドゲル電気泳動(SDS−PAGE)
変性条件下での分子量の特徴付けのために、例2a、5a、9、12、13及び14aからのオリゴマーA及びBを含有する標本を、変性条件下に、標準条件により、4〜20%のトリス−グリシン−SDS−PAGEで分析する。
【0223】
SDS−PAGEの評価(図1)により、オリゴマーA−標本中になお存在する出発タンパク質Aβ(1−42)が約4kDaでの帯として現われるが、約15kDa(より弱い帯)及び約20kDa(主帯)で、例2aからのオリゴマーA1又はA2(図1において、1又は2で表示される)が見られることが明らかである。
【0224】
オリゴマーB−標本では、比較的に少ない出発タンパク質Aβ(1−42)が検出される(約4kDaでの比較的弱い帯)。これに対して、約38kDa及び約48kDaで、例5aからのオリゴマーB1又はB2(図1において、3及び4で表示される(矢印参照))が現われる。例9及び12からのビオチン及びフルオレスセインで誘導体化されたオリゴマーBについて、約42kDa及び約52kDaと一致するより高い分子量が明らかである(図3、4)。
【0225】
各々架橋結合生成物A−CL及びB−CLを含有する標本の分析(例13及び14a;図2)は、2つの試料がそのオリゴマー化度(線AとA’及び線BとB’を比較)を実際に保持していることを示す。オリゴマーA及びBについて僅かに異なる泳動は、僅かに変化したSDS−結合力によって及びN−末端のアミノ基又はリジン基の変性によって説明することができる。
【0226】
切端Aβ(20−42)−オリゴマー(例15aから)の分析は、38/48kDa二重帯(図11、線A)が、サーモリシンでのN−末端ペプチドのタンパク質加水分解によって、28/38kDa二重帯(図11;線B)に移動することを示す。同様に、切端Aβ(12−42)−オリゴマー(例15bから)の分析は、38/48kDa二重帯(図11、線A)が、エンドプロテアーゼGlu-CでのN−末端ペプチドのタンパク質加水分解によって、33/40kDa二重帯(図11;線C)に移動することを示す。
【0227】
例17
ゲル透過クロマトグラフィー
非変性条件下での分子量特性を詳細に調べるために、ゲル透過クロマトグラフィー(GPC)を、FPLC−法で、スパロース(Superose) 12 HR 10/30 カラムを用いて実施する。GPCは4℃で行なわれる。
【0228】
カラムを5カラム容量のPBS−緩衝液で平衡させ(流量0.5m/分、検出UV、214nm)、先ず、標準タンパク質で検定する。引き続き、例5aからのAβ(1−42)−オリゴマーB−標本(図5、下方)及び、比較のために同一濃度で、新規に秤量してPBS中に溶かしたAβ(1−42)−凍結乾燥物(図5、上方)を、10000gで5−分間不溶成分の遠心分離後に分析する。
【0229】
その評価により、Aβ(1−42)−オリゴマーB−標本は、分子量範囲約50kDaの主ピークを特徴とするタンパク質画分を示すことが明らかである。SDS−PAGEにおける変性条件下と同様に、このタンパク質画分は、分子量範囲約16kDaの主ピークを特徴とするモノマーAβ(1−42)−タンパク質とは著しく異なっている。
【0230】
例6bからのAβ(1−42)−オリゴマーB及び例14aからのAβ(1−42)−オリゴマーB−CLの非変性条件下での分子量特性を調べるために、ゲル透過クロマトグラフィー(GPC)をFPLC−法でスパロース12 HR 10/30−カラムを用いて室温で実施する。カラムを5カラム容量のPBS−緩衝液で平衡させ(流量0.5m/分、検出UV、214nm)、先ず、標準タンパク質で検定する。引き続き、例6bからのAβ(1−42)−オリゴマーBをPBS緩衝液で1mg/mlに希釈し、かつ例14aからのAβ(1−42)−オリゴマーB−CLをPBS−緩衝液で1mg/mlに希釈し、分析する(図12)。
【0231】
その評価により、減少されたSDS−含量を有するAβ(1−42)−オリゴマーB−標本は、分子量範囲約100kDaの主ピークを特徴とするタンパク質画分を示すことが明らかである。それに比較して、減少されたSDS−含量を有するAβ(1−42)−オリゴマーB−CL−標本は、分子量範囲約60kDaの主ピークを特徴とするタンパク質画分を示す。
【0232】
例18
例5aからのAβ(1−42)−オリゴマーBの自然ポリアクリルアミドゲル電気泳動(NATIVE−PAGE)
自然条件化での分子量の特徴付けのために、例5aからのオリゴマーBを含有する標本を、非変性条件下に、4〜20%トリス−グリシン−ゲル中で分析する。
【0233】
標本中に含有される界面活性剤を、非イオン性界面活性剤トリトン(Triton)X−100で中和する。そのために、(4%トリトンX−100)1μlを例5aからの標本10μlにピペットで入れ、室温で5分間高温保持する。その後に、10μlに、同一容量の自然試料緩衝液(トリス1モル4ml、pH6.8、グリシン8ml、H2O50ml中のブロムフェノールブルー1ml)を加え、電気泳動(流動緩衝液:トリス7.5g、グリシン36g、H2O2.5lへ)を実施する。
【0234】
NATIVE−PAGEの評価により、オリゴマーB−標本中になお存在する出発ペプチドAβ(1−42)が約28kDaでの帯として(線A、1で表示)現われ、例5aからの標本の主ピークが、見かけの分子量64〜90kDa(線A、2で表示)で認められることが明らかである(図6)。
【0235】
ゲル透過クロマトグラフィー(例17参照)又は自然ゲル電気泳動(例18参照)と同様に、自然分子量−分析法でも、本発明によるオリゴマー、殊に、オリゴマーBに分子量を割り当て得ることは重要である。
【0236】
SDS−PAGEにおいて分子量約38及び48kDaを割り当てられ得るオリゴマーBは、SDSの複合化後に、自然ゲル電気泳動法で、選別された標準タンパク質に対して、約64〜90kDaの分子量範囲の帯として検出される。この方法により、正確な分子量を読み取ることは期待されず、それというのも、オリゴマーの移動特性は分子量の大きさに加えてその固有荷電によって実際に測定されるからである。当然、この結果から、限定されたオリゴマー種が存在することは推論され得る。
【0237】
例19
a)生理学的緩衝液中の異なるタンパク質濃度におけるAβ(1−42)−オリゴマーBにおける安定性
例6bにより得られるAβ(1−42)−オリゴマーBを、次の条件下に、PBS−緩衝液中の安定性について試験する。
【0238】
5mg/mlを、PBSを用いて2回の希釈段階で0.08mg/mlに希釈する。全ての得られる溶液を室温で24時間恒温保持する。続いて、帯パターンを、凍結された対照標本に比較して、SDS−PAGEで分析する。帯パターンは全試料で同一である。
【0239】
b)生理学的緩衝液における異なる温度で及び異なる時間後のAβ(1−42)−オリゴマーBの安定性
例6bにより得られるAβ(1−42)−オリゴマーBを、PBS−緩衝液で0.5mg/mlに希釈し、24時間又は96時間室温又は370℃で高温保持する。続いて、帯パターンをSDS−PAGEで分析する。帯パターンは全試料で同一である。
【0240】
例20
異なるプロテアーゼ(トリプシン、キモトリプシン、サーモリシン、エラスターゼ、パパイン)によるAβ(1−42)−オリゴマーBのタンパク質加水分解
例6により得られるAβ(1−42)−オリゴマーB−標本の部分標本を、緩衝液(燐酸−Na20ミリモル、NaCl140ミリモル、pH7.4)で0.5mg/mlに希釈し、次の条件下に、各々、図9に挙げたプロテアーゼ溶液の質量の1/50と共に、20時間37℃及びpH7.4で恒温保持する。その後に、成分の部分標本1μgをSDS−PAGEで分析する(図9)。
【0241】
SDS−PAGEは、Aβ(1−42)−オリゴマーB−標本から出発して、全プロテアーゼが、選択された限定のタンパク質加水分解条件下に、38/48kDaでの二重帯を約32/28kDaでのkDa二重帯へ戻すことを示す。
【0242】
例21
ラット血漿中のAβ(1−42)−オリゴマーBの安定性
例6bにより製造したAβ(1−42)−オリゴマーB−標本4μlを、ラット血漿76μlと共に、0時間、1時間、2時間、4時間及び8時間室温で恒温保持する。恒温保持をドライアイス中での凍結によって止める。
【0243】
引き続き、全試料をSDS−試料緩衝液−添加後に、SDS−PAGEで分析する。その後に、安定性の評価を、ウエスタンブロット法で、Aβ(1−42)−オリゴマーBの変色を介して行なう。検出のために、Aβ(1−42)照準の抗体6E10(Fa. Signet)を使用する。帯は、アルカリ性ホスファターゼに結合される抗−マウス−IgG−抗体及び基質NBT/BCIPの添加によって可視される。観察される帯パターンの分子量も強度も、2時間、4時間及び8時間に渡って殆ど無変化のままである。
【0244】
この結果から、Aβ(1−42)−オリゴマーBが高い血漿安定性を有することが推論できる。従って、血漿中の生物学的半減期は、8時間以上の範囲である。
【0245】
例22
分子量17kDa及び22kDaを有するビオチン−Aβ(1−42)−オリゴマー[ビオチン−Aβ(1−42)−オリゴマーA]又は分子量42kDa及び52kDaを有するビオチン−Aβ(1−42)−オリゴマー[ビオチン−Aβ(1−42)−オリゴマーB]のヒト神経細胞の表面への結合
ヒトβ−アミロイド(1−42)−タンパク質及び2つのAβ(1−42)−オリゴマーA−及びAβ(1−42)−オリゴマーB−標本の、ヒト神経芽細胞系IMR−32(ATCC番号:CCL−127)への結合を、FACScan(Beckton Dickinson)によって調べる。IMR−32細胞の懸濁液(細胞1.5x106/PBS0.1ml)を、ビオチン−標識化ヒトβ−アミロイド(1−42)−タンパク質(ペプチド合成原料、凍結乾燥物、AnaSpec)及びビオチン−標識化オリゴマーA又はBを含有する標本(例8又は9)と共に、20分間37℃で恒温保持する。引き続き、細胞を緩衝液(PBSプラス1%BSA)で洗浄し、ストレプトアビジンイソチオシアネート結合のフルオレセイン(Sigma)と共に20分間室温で恒温保持する。緩衝液での清浄段階後に、IMR−32細胞の表面への結合をFACScanで分析する。断続線は、ビオチン−標識化成分の不在で、基底値蛍光を示す。個々の標本の添加は細胞−会合蛍光の強力な増加を導き、濃い線によって描かれている。細胞表面へのオリゴマーA(図7B)及びB(図7C)の結合は、モノマーβ−アミロイド(1−42)−タンパク質(図7A)の結合とは明らかに異なっている。データは、ヒト細胞表面上のオリゴマーの特異的結合部位を示す。
【0246】
例23
icv−投与後のラット脳ホモジネート中のAβ(1−42)−オリゴマーBの検出
例6bより得られるAβ(1−42)−オリゴマーB−標本10ナノモルを、ラットにicv丸剤として投与する。脳を15分間後又は120分間後に解剖する。未処理対照動物の脳を同様に解剖する。そのために、50ml入りファルコン(Falcon)試験管中で、ラット脳各1gに、破砕緩衝液A(燐酸ナトリウム5ミリモル200ml、NaCl35ミリモル、蔗糖300ミリモル、pH7.4に調整、プロテアーゼ阻害剤−カクテルComplete(登録商標)、Fa. Rocheの4錠を加える)9mlを加え、氷上の超音波(Ultra Turrax)中で2分間破砕する。溶液を20分間放置し、素早く振動させ、部分標本8x1mlに分ける(=ホモジネート)。
【0247】
検出を定量的に行なうために、先ず、PBS中のAβ(1−42)−オリゴマーB−標本の一連の標準を、濃度範囲1.58ng/μl〜0.005ng/μlで調製する。
【0248】
更に、正対照として、未処理ラットの対照脳からのホモジネートを一緒に経過させ、同様の方法で、この脳ホモジネート中のAβ(1−42)−オリゴマーB−標本の一連の標準を調製する。その後に、ホモジネートを超遠心分離機において100000gで1時間遠心分離させ、上澄液を次の分析に使用する。
【0249】
2つの標準列で測定した値(PBS対対照脳−ホモジネート−上澄液)の比較から、先ず、正対照中のAβ(1−42)−オリゴマーBの含量を定量的に測定し、次いで、それに比較して、処理ラットの脳試料においても同様に測定する。
【0250】
1)ドットブロット−検出法
標準列−試料及び処理動物からの試料抽出物1μlを、ニトロセルロース−紙上に滴下し、Aβ(1−42)を抗体6E10(抗−Aβ(1−42);Fa. Signet)で検出する。抗−マウスIgGに結合したアルカリ性ホスファターゼを用いて、呈色剤NBT/BCIPの添加によって呈色させる。
【0251】
未処理ラットの脳抽出物中ではAβ(1−42)(0.01ナノモル/g)は検出不可能であるが、処理の15分間後に殺したラットにおいて、正対照の相応する濃度との呈色強度の比較によって、Aβ(1−42)約0.4ナノモル/gが検出可能であり、かつ処理の120分間後に殺したラットにおいて、同様の方法により、約0.2ナノモル/gがなお検出可能である。このことから、外部から投与されたAβ(1−42)−オリゴマーBについて、約105分間の平均的な生物学的半減期が判明する。
【0252】
2)ウエスタンブロット−検出法
ドットブロット法で分析した全試料を、同様にウエスタンブロット法で分析する。ウエスタンブロット法は、同様に、mMAb6E10(抗−Aβ(1−42);Fa. Signet)及び更に抗−マウス−IgGに結合したアルカリ性ホスファターゼを用いて、呈色剤NBT/BCIPの添加によって展開させる。
【0253】
抗Aβ(1−42)−反応帯は38/48でのみ出現し、これはAβ(1−42)−オリゴマーBの見かけの分子量に相応し、即ち、オリゴマー構造は、生体内適用でも2時間後もなお保持されたままである。
【0254】
更に、ドットブロット−法の場合と同様に、15分間−ラット(0.4ナノモル/g)及び120分間−ラット(0.2ナノモル/g)の脳において、正対照におけるAβ(1−42)−オリゴマーBの相応に強く呈色した帯との呈色強度の比較によって、同じ濃度が評価され得る。
【0255】
例24
分子量38kDa及び48kDaを有するAβ(1−42)−オリゴマー[Aβ(1−42)−オリゴマーB]の、マウスの大脳皮質ニューロンへの神経毒作用
ネズミの大脳皮質ニューロンの標本及び培養は、文献(Choi et al. (1987) J. Neurosci. 7, 357-368)による神経膠細胞との混合培養として行なわれる。胚の場合には、展開14〜15日目に、大脳皮膚及び深部脳範囲の皮部を機械的に除去する。0.05%のトリプシン溶液中37℃で5〜7分間の恒温保持し、及び引続いて、孔を縮小させたパスツールピペットを数回通過させることよって細胞を細かく分離させる。細胞数の測定後に、ポリ−L−オルニチン及びラミニンで被覆された細胞培養原料上で2cm2当たり、保存培地(グルタミン0.8ミリモル、ブドウ糖18ミリモル、NaHCO3 23ミリモル及び10%馬血清を有する最少必須培地)0.5ml中で430000個の細胞を接種する。細胞の培養及びその後の恒温保持は、空気湿潤の細胞培養箱中で37℃、5%CO2で行なわれる。培養3〜5日間後に、神経膠−細胞の増加を、(+)−5−フルオル−2’−デスオキシウリジン/ウリジン−混合物(各10マイクロモル)との1日間の恒温保持によって止める。培養14日間後に、Aβ(1−42)−オリゴマーBの毒性作用を調べる。そのために、細胞を15分間脳細胞緩衝液(NaCl120ミリモル、KCl5.4ミリモル、CaCl21.8ミリモル、ブドウ糖15ミリモル、HEPES25ミリモル、pH7.2)中で恒温保持する。対照細胞1を脳細胞緩衝液中でL−グルタメート300マイクロモルと共に同じ時間恒温保持する。Aβ−オリゴマーの基本溶液を、血清を含まない培地(グルタミン0.8ミリモル、ブドウ糖20ミリモル、NaHCO3 26ミリモルを有する最少必須培地)で、様々な最終濃度に希釈し、24時間恒温保持する。L−グルタメートで処理した対照細胞1を平行して血清を含まない培地中で恒温保持する。もう1つの群の細胞(対照細胞2)を、脳細胞緩衝液だけで、血清を含まない培地で処理する。24時間後に細胞培養上澄液を除去し、残留細胞を蒸留水中で20分間の恒温保持によって破砕し、2つの溶液中で酵素、乳酸デヒドロゲナーゼ(LDH)の活性を酵素的に調べる。評価のために、細胞培養上澄液のLDH−活性対上澄液及び残留細胞からのLDH−活性の合計の比率を測定し、4回の測定の平均値を出す。3回の実験を実施し、この際、各実験条件は4重で存在している(n=12)。グルタメートで処理した細胞(対照細胞1)における平均値はニューロン死100%とみなし、L−グルタメートでも、Aβ−オリゴマーでも処理していない細胞(対照細胞2)の平均値はニューロン死0%とみなし、Aβ−オリゴマーで処理した細胞値は相応に換算される。各使用濃度における全測定の神経毒性の平均値は、分子量38kDa及び48kDaを有するAβ(1−42)−オリゴマー[Aβ(1−42)−オリゴマーB]の明らかな毒性作用を示す。図8参照。
【0256】
例25
抗体の製造
免疫化に使用されるカクテルは、全例で、実際に次の成分を有するアジュバント(Fa. Biogenes)を含有する:
パラフィン油95%
ツィーン(Tween)40 2.4%
コレステリン0.1%
リポ多糖類0.1%
アジュバントは、安定したエマルジョンが得られるまで、抗原の溶液と比率2:1で混合される。エマルジョンを注射し、抗原がそれから連続的に遊離されるデポーを形成する。
【0257】
a)例15aからの切端Aβ(20−42)−オリゴマーBでのウサギの免疫化によるポリクローナル抗血清の製造
2匹のウサギを、標準プロトコルにより、例15aからの非複合化Aβ(20−42)−オリゴマーB−標本で免疫化させる。
【0258】
1日目 前免疫血清の第1免疫化及び採取
7日目 第1免疫増強
14日目 第2免疫増強
28日目 第3免疫増強及び出血
35日目 採血
抗血清を、ウサギ血液から、室温での静置及び引き続き室温での遠心分離によって得る。そうして得られる血清を血清(a1)と表示する。
【0259】
例15aからのAβ(20−42)−オリゴマーB−標本2mgを、LPHにグルタルジアルデヒドで標準条件下に結合させ、他の2匹のウサギを、標準プロトコルにより、この複合Aβ(20−42)−オリゴマーBで免疫化させる。
【0260】
1日目 前免疫血清の第1免疫化及び採取
7日目 第1免疫増強
14日目 第2免疫増強
28日目 第3免疫増強及び出血
35日目 採血
抗血清をウサギ血液から、室温での静置及び引き続き室温での遠心分離によって得る。そうして得られる血清を血清(a2)と表示する。
【0261】
b)例15bからの切端Aβ(12−42)−オリゴマーBでのウサギの免疫化によるポリクローナル抗血清の製造
2匹のウサギを、標準プロトコルにより、例15bからの非複合化Aβ(12−42)−オリゴマーB−標本で免疫化させる。
【0262】
1日目 前免疫血清の第1免疫化及び採取
7日目 第1免疫増強
14日目 第2免疫増強
28日目 第3免疫増強及び出血
35日目 採血
抗血清を、ウサギ血液から、室温での静置及び引き続き室温での遠心分離によって得る。そうして得られる血清を血清(b1)と表示する。
【0263】
例15bからのAβ(12−42)−オリゴマーB−標本2mgを、LPHにグルタルジアルデヒドで標準条件下に結合させ、他の2匹のウサギを、標準プロトコルにより、この複合Aβ(12−42)−オリゴマーBで免疫化させる。
【0264】
1日目 前免疫血清の第1免疫化及び採取
7日目 第1免疫増強
14日目 第2免疫増強
28日目 第3免疫増強及び出血
35日目 採血
抗血清をウサギ血液から、室温での静置及び引き続き室温での遠心分離によって得る。そうして得られる血清を血清(b2)と表示する。
【0265】
c)例14aからのAβ(1−42)−オリゴマーB−CLでのウサギの免疫化によるポリクローナル抗血清の製造
2匹のウサギを、標準プロトコルにより、例14aからの非複合化Aβ(1−42)−オリゴマーB−CLー標本で免疫化させる。
【0266】
1日目 前免疫血清の第1免疫化及び採取
7日目 第1免疫増強
14日目 第2免疫増強
28日目 第3免疫増強及び出血
35日目 採血
抗血清を、ウサギ血液から、室温での静置及び引き続き室温での遠心分離によって得る。そうして得られる血清を血清(c1)と表示する。
【0267】
d)例6bからの切端Aβ(1−42)−オリゴマーBー標本でのウサギの免疫化によるポリクローナル抗血清の製造
2匹のウサギを、標準プロトコルにより、例6bからの非複合化Aβ(1−42)−オリゴマーB−標本で免疫化させる。
【0268】
1日目 前免疫血清の第1免疫化及び採取
7日目 第1免疫増強
14日目 第2免疫増強
28日目 第3免疫増強及び出血
35日目 採血
抗血清を、ウサギ血液から、室温での静置及び引き続き室温での遠心分離によって得る。そうして得られる血清を血清(d1)と表示する。
【0269】
例6bからのAβ(1−42)−オリゴマーB−標本2mgを、LPHにグルタルジアルデヒドで標準条件下に結合させ、他の2匹のウサギを、標準プロトコルにより、この複合Aβ(1−42)−オリゴマーBー標本で免疫化させる。
【0270】
1日目 前免疫血清の第1免疫化及び採取
7日目 第1免疫増強
14日目 第2免疫増強
28日目 第3免疫増強及び出血
35日目 採血
抗血清をウサギ血液から、室温での静置及び引き続き室温での遠心分離によって得る。そうして得られる血清を血清(d2)と表示する。
【0271】
例26
例25の免疫化からのポリクローナル抗血清の、様々なAβ(1−42)−型とのその架橋結合についての特徴づけ
ポリクローナル抗血清の特徴付けのために、PBS中100ピコモル(pmol)/μl〜0.01ピコモル/μlの範囲の希釈列で様々なAβ(1−42)−型を製造した。試料各1μlをニトロセルロース−膜上に載せた:検出は例25からの相応するウサギ血清で行なった。比較として、mMAb 6E10(Fa. Signet)での検出を実施した。抗−ウサギ−IgGに結合したアルカリ性ホスファターゼ(比較用:抗−マウス−IgGに結合したアルカリ性ホスファターゼ)及び呈色剤NBT/BCIPの添加で呈色させた。図10はそうして得られるドットブロットを示し、これは数値的に次のように評価される:
【表1】

【0272】
表中の数字は、最低可視量の抗原を[ピコモル;APPの場合を除いて、各々モノマーに対する]で示す(検出限度)。
【0273】
例15aからの非複合Aβ(20−42)−オリゴマーでの免疫化によって得られたウサギ血清(a1)との免疫学的反応の比較は、このオリゴマーに照準している抗体が、他の形、例えば、原線維、APP、及びモノマーと僅かしか架橋結合しないことを明らかに示す。これに対して、Aβ(1−42)−オリゴマーBとの明らかにより強い架橋結合(列1参照)及び更に安定化CL−抗原との架橋結合が認められる。このデータは、APP、モノマー、及び原線維構造に比較して、本オリゴマー形の明らかに異なる構造を示す。抗体はオリゴマー−特異的構造に結合する。
【0274】
例15aからの複合Aβ(20−42)−オリゴマーでの免疫化によって得られたウサギ血清(a2)との免疫学的反応の比較は、このオリゴマーに照準している抗体が、他の形、例えば、原線維及びモノマーと僅かしか架橋結合しないことを明らかに示す。これに対して、Aβ(1−42)−オリゴマーBとの弱い架橋結合(列1参照)及び更に安定化CL−抗原との、及びAPPとの架橋結合が認められる。このデータは、モノマー及び原線維構造に比較して、本オリゴマー形の同様に明らかに異なる構造を示す。
【0275】
これに比較して、例6bからの非複合Aβ(1−42)−オリゴマーでの免疫化によって得られたウサギ血清(d1)及び例14aからのAβ(1−42)−オリゴマーB−CLでの免疫化によって得られたウサギ血清(c1)との免疫学的反応は、このオリゴマーに照準している抗体が、他の形、例えば、原線維、APP及びモノマーと、Aβ(1−42)−オリゴマーBと同様に(列1参照)比較可能に強く架橋結合することを示す。この抗体は、mMab 6E10(Fa. Signet)に比較可能な免疫反応を示し、オリゴマー−特異的構造には結合しない。
【0276】
相応する方法で、マウスを例6aからのAβ(1−42)−オリゴマーで免疫化させ、自体公知の方法で、モノクローナル抗体を確立した。この際、10個のハイブリドーマの内2個がモノクローナル抗体を分泌し、その結合プロフィールは、殊に、前記で試験した反応性に関して、抗血清のそれと(a1)類似している。
【0277】
例27
Aβ(1−42)−原線維の製造
0.1%NH4OH中のAβ(1−42)−溶液(2mg/ml)100μlを、緩衝液(燐酸−Na20ミリモル、NaCl140ミリモル、pH7.4)300μlで、0.5mg/mlに希釈し、HCl0.1モルでpH7.4に調製する(Aβ(1−42) 100マイクロモル)。試料を24時間37℃で恒温保持し、その後に、10分間10000gで遠心分離する。得られるタンパク質残渣を、緩衝液(燐酸−Na20ミリモル、NaCl140ミリモル、pH7.4)400μlで再懸濁させる。そうして得られるAβ(1−42)−原線維−標本を−20℃で保存し、更なる研究に使用することができる。
【0278】
例28
Aβ(1−42)−オリゴマーB−CL(例14bから)の、海馬切片への作用
厚さ400μMの海馬横断切片を、切片浸漬室中34℃で少なくとも1時間、ガス処理したリンガー液(NaCl124ミリモル、KCl4.9ミリモル、MgSO4 1.3ミリモル、CaCl2 2.5ミリモル、KH2PO4 1.2ミリモル、NaHCO3 25.6ミリモル、ブドウ糖10ミリモル)の灌流下に適応させた。その後に、単極の刺激電極により、シェ−ファー副軸索を刺激し、放線状層中の興奮性シナプス後電位((fEPSP)を導出させた。最高のfEPSPを発生させる刺激強度の30%で試験パルスを与えた。長時間−相乗作用の誘発のために、単一パルスを200μsの幅で100パルスを3回、いずれも10分間施した(強い強縮)。得られるfEPSPの相乗作用を少なくとも240分間記録した。例14bからのオリゴマーB−CLの洗浄を、最初の強縮の20分間前に開始し、最後の強縮の10分間後に終了した。fEPSPsの上昇(勾配)を調べ、時間に対して作図した。
【0279】
図14は、Aβ(1−42)−オリゴマーの洗浄が、海馬における長時間相乗作用を、特に維持相("維持相(maintenance phase)")で阻害することを示す。従って、Aβ(1−42)−オリゴマーB−CLは、神経細胞の情報蓄積(細胞記憶)への影響を有する。
【0280】
例29
Aβ(1−42)−オリゴマーB(例5bから)の、一次海馬ラット−ニューロンへの結合
例5bからのラット−Aβ(1−42)−オリゴマーBの、一次海馬ラットニューロンへの結合を調べた。海馬−ニューロンを、ポリ−L−リジン−被覆のデッキガラス上でB27−サプリメントを有する神経基礎培地中で培養し、培養の14日目に使用した。ニューロン細胞膜へのオリゴマーの結合は、新鮮培地中で、オリゴマー200ナノモル(全モノマーAβ−濃度)の添加及び37℃で15分間の恒温保持によって行なわれた。Aβ−含有培地の除去後に、培地で2回洗浄し、細胞を引続いて3.7%ホルムアルデヒド中に固定させた。細胞を更に洗浄した後に、非特異的結合部位をPBS−緩衝液中10%標準ロバ−血清で室温で90分間遮断した。第1抗体として、6E10(マウスから)を1:2000の希釈で、2時間室温で適用した。細胞を再び洗浄し、マウスに照準していて蛍光色素Cy3がそれに結合している第2抗体(ロバから)と共に2時間室温で恒温保持した。細胞を再び洗浄した後に、ニューロンを有するデッキガラスを、封埋培地を有するスライド上に固定した。結合Aβ−オリゴマーを有する海馬ニューロンは、蛍光顕微鏡で表示された。対照として、第1抗体6E10が省略されている成分を使用した。従って、この対照は、Aβに基づいていない非特異的蛍光を示す。
【0281】
図15aで見られるように、オリゴマーはニューロンの細胞表面にドット法で結合する。それに対して、第1抗体6E 10を含まない対照は、ニューロンへの第2抗体の結合による僅かな非特異的蛍光だけを示す(図15b)。
【技術分野】
【0001】
本発明は、アミロイド−β(1−42)−タンパク質の神経変性オリゴマー、このオリゴマーが再現可能な方法及び高収率で得られる特殊製法、及びオリゴマー特異的抗体の製造のための、及びオリゴマー及びその生成と相互作用をし得る物質の発見のための診断剤及び治療剤としてのオリゴマーの使用に関する。
【0002】
本発明は、同様にして、オリゴマーの誘導体、殊に、架橋結合したオリゴマー及びそのアミロイド−β(1−42)−タンパク質の切端形をベースとするオリゴマー、その製造及びその使用にも関する。
【0003】
抗体の製造のための、又は物質の発見のための相応する方法は、抗体自体及び診断剤及び治療剤としての抗体又は物質の使用と全く同様に記載される。
【背景技術】
【0004】
アミロイド−β(1−42)−タンパク質(Aβ(1−42)としても略称される)は、アルツハイマー−及びダウン症候群−患者の脳におけるタンパク質、脂質、炭水化物及び塩から組成される不溶性の細胞外沈着物(老人性又は神経炎性斑)の中心成分である(C.L.Masters et al. PNAS 82, 4245-4249,1985)。水性環境中で重合傾向を有するこのタンパク質は極めて様々な分子形で存在し得る。
【0005】
不溶性タンパク質沈着物と老人性痴呆症、例えば、アルツハイマーの出現又は進展との簡単な相関性は、確実には実証されなかった(R.D.Terry et al Ann. Neurol. 30. 572-580(1991), D.W. Dickson et al. Nurobiol. Aging 16, 285-298(1995))。それに反して、シナップス及び認知感覚の喪失は、より良好にAβ(1−42)の可溶性形と相関すると思われる(L.F. Lue et al. Am. J. Pathol. 155, 853-862,(1999), C.A. McLean et al. Ann. Neurol. 46, 860-866 (1999))。
【0006】
Aβ(1−42)の可溶性形には、老人性痴呆症、例えば、アルツハイマーの原因であるといわれる分子形に関して、実際に2種の異なる仮説がある。
【0007】
1つには、Aβ(1−42)−プロトフィブリルの細胞毒作用が仮定される。更に、これは、150〜250kDaの範囲の分子量を有する可溶性で原線維性の比較的高度に集合したAβ(1−42)−形であり(Arispe et al. PNAS 90. 567(1993), Lashuel et al., Nature 418, 291(2002)) 、これは孔を形成する特性に基づき、カルシウムがニューロン細胞膜を自由に通過するように作用するといわれる。
【0008】
他方では、15〜30kDaの範囲の分子量を有するオリゴマーAβ(1−42)−誘導体(M.P. Lambert et al. PNAS 95, 6448-6453 (1998)) が記載されている。このアミロイドから誘導されるび慢性で老人性痴呆に関与するリガンド(ADDL's Fuer Amyloid Derived Dementing Ligands, US-A 6218506 及びWO 01/10900参照又はfuer Amyloid Derived Diffusible Ligands参照, Lambert et al. supra)としても特徴付けられる非原線維性オリゴマーは、海馬切片中のニューロンの長時間相乗作用率への阻害的影響を示す。
【0009】
勿論、従来のオリゴマー研究の状況は、本来関連のある種についての大きな不透明性を特徴とする。即ち、US−A6218506に、3〜12個のサブユニットを有するADDLsが記載され、これに対して、WO01/10900に記載されたADDLsは24個までのサブユニットを有する。
【0010】
殊に、非変性条件下でのゲル電気泳動分析で、27〜28kDa及び23〜24kDa(US−A6218506)又は約26kDa〜28kDa(WO01/10900)又は17kDa及び27kDa(M.P. Lambert et al. PNAS 95, 6448-6453(1998))の範囲の分子量、及び非変性条件下でのSDS−ゲル電気泳動分析で、17kDa及び22kDa(M.P. Lambert et al. PNAS 95, 6448-6453(1998))又は約22kDa〜約24kDa及び約18kDa〜約19kDa(WO01/10900)の分子量を有する少なくとも2つの形が論じられる。8kDa及び12kDaの範囲の分子量を有するSDS−安定のAβ(1−42)−オリゴマーも、アルツハイマー−患者の脳で、ウエスタン−ブロット(Western Blot)−法によって検証された(C.A. McLean et al. Ann. Neurol. 46, 860-866 (1999))。
【0011】
記載された製法の欠点は、それが不均一なオリゴマー−標本を生じさせることである。
【0012】
即ち、神経変性の原因であるAβ(1−42)の分子形は、再現可能な方法では未だ得られず、まして明白な方法で同定もされ得ていない。
【0013】
しかし、均一な標本の重要性は、例えば、アルツハイマー−患者の能動免疫化のための最初の臨床研究経過から判断され得る。Aβ(1−42)の前集合形のワクチン注射によって、患者の一部で著しい副作用(メンゴ脳炎、出血)が生じ、それというのも、生成した抗体が、細胞の被覆に必要と思われるAβ(1−42)−形も認識し、これによって炎症反応が生じるからである(D. Schenk.; Nat. Rev. Neurosci. 3, 824-828 (2002))。
【0014】
従って、本来的に傷害性のタンパク質形の中和を特に目指す適切な治療には、その同定及び限定の製造が絶対に必要な前提である。
【0015】
更に、Aβ(1−42)−タンパク質のN−末端切断形の出現が、アルツハイマー病と関連して言及されてきた。既に1985年に、死亡したアルツハイマー患者の脳の沈着物中に、Aβ(1−42)の他に、N−末端切断形も検出された(C. Masters et al., PNAS 82, 4245-4249 (1985))。即ち、脳中に存在する一定のプロテアーゼ、例えば、ネプリリシン(NEP 24.11)又はIDE("インシュリン分解酵素(Insulin Degrading Enzyme)"の略語)がAβ(1−42)を分解し得ることも知られている(D.J. Selkoe, Neuron 32, 177-180, (2001))。
【0016】
しかし、アルツハイマー病の病因におけるN−末端切断形の重要性は不明である(E.B. Lee et al. JBS 278, 4458-4466 (2003))。重要なことに、突発性又は家族性アルツハイマー病又はダウン−症候群の若干の患者には、優先的に、この切端形が蓄積されている(J. Naeslund et al. PNAS 91, 8378-8382, (1994), C. Russo et al., Nature 405, 531-532, (2000), T.C. Saido et al., Neuron 14, 457-466, (1995))。新しい研究(N. Sergeant et al. J. of Neurochemistry 85, 1581-1591, (2003)) で、死亡したアルツハイマー−患者の脳中の全不溶性Aβ−ペプチドの60%が、N−末端切断形に基づいていることが示された。
【0017】
モノマーAβ(1−42)−タンパク質及びその一定の断片に照準している抗体は既に記載されている。
【0018】
即ち、WO03/016466及びWO03/016467は、CDR2で一定のグリコシリル化部位が欠けているモノクローナル抗体266及びその類似体に関する。そのヒト化型(Hu−266)も知られている。この印刷物では、他のモノクローナル抗体も記載されていて、それは、抗体226と同様に、Aβ(1−42)のアミノ酸13〜28の範囲でエピトープを認識する。これには、抗体4G8(Lue et al. (1999), supra にも記載されている)及び1C2が属する。更に、McLean et al. (1999), supra に、モノクローナル抗体1E8が記載され、これは、Aβ(1−42)のアミノ酸18〜22の範囲でエピトープを認識すると言われている。
【0019】
更に、Aβ(1−42)のN−末端配列のエピトープを認識する、他の一連の抗体が公知である。それには、市販で得られる、モノクローナル抗体6E10(WO01/10900;Naeslund et al. (1994), supra; Sergeant et al. (2003), supra にも記載されている)及びモノクローナル抗体3D6及び10D5(WO03/016467参照)及びBan50及びNAB228(Lee et al. (2003), supra)が属する。
【0020】
更に、モノクローナル抗体WO2、21F12及び3D6及びポリクローナル血清ADA42が挙げられる(Sergeant et al. (2003), supra) 。
【0021】
前臨床的検査にある抗−Aβ(1−42)−抗体に関する概要は、Schenk et al. (2002), supra にある。
【先行技術文献】
【特許文献】
【0022】
【特許文献1】US-A 6218506
【特許文献2】WO 01/10900
【特許文献3】US−A6218506
【特許文献4】WO03/016466
【特許文献5】WO03/016467
【非特許文献】
【0023】
【非特許文献1】C.L.Masters et al. PNAS 82, 4245-4249,1985
【非特許文献2】R.D.Terry et al Ann. Neurol. 30. 572-580(1991)
【非特許文献3】D.W. Dickson et al. Nurobiol. Aging 16, 285-298(1995)
【非特許文献4】L.F. Lue et al. Am. J. Pathol. 155, 853-862,(1999)
【非特許文献5】C.A. McLean et al. Ann. Neurol. 46, 860-866 (1999)
【非特許文献6】Arispe et al. PNAS 90. 567(1993),
【非特許文献7】Lashuel et al., Nature 418, 291(2002)
【非特許文献8】M.P. Lambert et al. PNAS 95, 6448-6453 (1998)
【非特許文献9】ADDL's Fuer Amyloid Derived Dementing Ligands,
【非特許文献10】fuer Amyloid Derived Diffusible Ligands
【非特許文献11】M.P. Lambert et al. PNAS 95, 6448-6453(1998)
【非特許文献12】C.A. McLean et al. Ann. Neurol. 46, 860-866 (1999)
【非特許文献13】D. Schenk.; Nat. Rev. Neurosci. 3, 824-828 (2002)
【非特許文献14】C. Masters et al., PNAS 82, 4245-4249 (1985)
【非特許文献15】D.J. Selkoe, Neuron 32, 177-180, (2001)
【非特許文献16】E.B. Lee et al. JBS 278, 4458-4466 (2003)
【非特許文献17】J. Naeslund et al. PNAS 91, 8378-8382,(1994)
【非特許文献18】C. Russo et al., Nature 405, 531-532, (2000)
【非特許文献19】T.C. Saido et al., Neuron 14, 457-466, (1995)
【非特許文献20】N. Sergeant et al. J. of Neurochemistry 85, 1581-1591, (2003)
【発明の概要】
【発明が解決しようとする課題】
【0024】
本発明は、Aβ(1−42)の神経変性及び殊にニューロン傷害作用の原因となり、かつ老人性痴呆症、例えば、アルツハイマー病及びダウン−症候群において増大に出現する分子形を得るという本発明の基礎にある課題を、モノマーAβ(1−42)−タンパク質から特異的方法で均一な標本の形で得られる一定のAβ(1−42)−オリゴマーによって、及びこのオリゴマーの一定の誘導体、殊に、架橋結合したオリゴマー及び切端Aβ(1−42)−形をベースとするオリゴマーによって解明する。この製法は、水性媒体中で難溶性のペプチド合成Aβ(1−42)−タンパク質を、高収率で、限定された可溶性オリゴマーに変換させることを可能にする。
【課題を解決するための手段】
【0025】
従って、本発明の目的は、特許請求に定義された目的である。
【0026】
即ち、本発明は、アミロイド−β(1−42)−タンパク質のオリゴマーに関し、この際、このオリゴマーは、SDS−ゲル電気泳動で、約15kDa、20kDa、38kDa又は48kDaの見かけの分子量を示し、又は場合により誘導体化に相応して変化した分子量を有するオリゴマーの誘導体に関する。
【0027】
アミロイド−β(1−42)−タンパク質は、42個のアミノ酸を有するポリペプチドであり、これは、アミロイド−前駆タンパク質(アミロイド前駆タンパク質(Amyloid Precursor Protein)のAPP)から、タンパク質加水分解過程によって誘導される。ヒト変異型のほかに、アミロイド−β(1−42)−タンパク質のイソ型も属し、これは人間以外の生物、ことに他の哺乳類、特にラットに存在する。
【0028】
本発明は、特別な実施態様により、ヒトアミロイド−β(1−42)−タンパク質のオリゴマーに関する。ヒトアミロイド−β(1−42)−タンパク質には、殊に、アミノ酸配列SEQ ID NO:1を有するタンパク質、及び前記の配列から、殊に、アミノ酸交換によって誘導される、その突然変異タンパク質及び全変異型が属する。これに関連して、特に、次のアミノ酸置換が挙げられる:A21G、E22K,E22Q、E22G及びD23N。即ち、アミロイド−β(1−42)−タンパク質の突然変異タンパク質又は全変異型には、本発明により、殊に、アミノ酸配列SEQ ID NO:1を有するタンパク質が属し、この中で、アラニン21、グルタミン酸22及びアスパラギン酸23から選択される1種以上のアミノ酸が、他の、有利にグリシン、リジン、グルタミン及びアスパラギンから選択されるアミノ酸と置換されている。本発明により、殊に、グルタミン又はグリシンによる部位22の置換が特に重要である。
【0029】
本発明は、もう1つの特別な実施態様により、ラットアミロイド−β(1−42)−タンパク質のオリゴマーに関する。ラットアミロイド−β(1−42)−タンパク質には、殊に、アミノ酸配列SEQ ID NO:2を有するタンパク質、及び前記の配列から、殊に、アミノ酸交換によって誘導される、その突然変異タンパク質及び全変異型が属する。これに関連して、特に、ヒトの配列で説明されたアミノ酸置換に相応するアミノ酸配列が挙げられる。
【0030】
アミロイド−β(1−42)−タンパク質は、公知のペプチド合成法によって又は組換えで製造することができる。更に、一連のこのタンパク質は市販で得られる。突然変異タンパク質及び全変異型にも同じことが当てはまる。
【0031】
本発明によるオリゴマーは、アミロイド−β(1−42)−タンパク質のオリゴマー化によって得られる。オリゴマー化は、モノマーアミロイド−タンパク質の非共有集合を含み、従って、このことから、本発明によるオリゴマーは、数種のアミロイド−β(1−42)−タンパク質モノ標識ら組成されていることが判る。
【0032】
オリゴマー化度に応じて、本発明によるオリゴマーは様々な分子量を有する。即ち、オリゴマーに、変性ゲル電気泳動法により、見かけの分子量を配属させることができる。これは、変性標準条件下でのゲル電気泳動法(トリス−グリシン−ゲル、4〜20%、Laemmli UK, Nature 227, 680-685 (1970) 参照)を実施する場合に、オリゴマーA1については約15kDa、オリゴマーA2については約20kDa、オリゴマーB1については約38kDa、オリゴマーB2については約48kDaであり、この際、次の標準タンパク質が同じ条件で次の見かけの分子量を有する:ミオシン250kDa、ウシ−血清アルブミン98kDa、グルタミンヒドロゲナーゼ64kDa、炭酸脱水酵素36kDa、ミオグロビン30kDa、リゾチーム16kDa、アプロチニン6kDa、インシュリンB−鎖4kDa(前−青着色標準(Blue Pre-stained Standard )参照)。もう1つの観点に依り、自然標準条件下でのゲル電気泳動法(トリス−グリシン−ゲル、4〜20%)を実施する場合に、オリゴマーBの分子量は、約64〜90kDaであり、この際、次の標準タンパク質が同じ条件で次の見かけの分子量を有する:ミオシン250kDa、ウシ−血清アルブミン98kDa、グルタミンヒドロゲナーゼ64kDa、カルボアンヒドレーゼ36kDa、ミオグロビン30kDa、リゾチーム16kDa、アプロチニン6kDa、インシュリンB−鎖4kDa(青色前着色標準(Blue Pre-stained Standard )参照)。
【0033】
もう1つの観点により、本発明によるオリゴマーは、ニューロン細胞への親和性を特徴とする。そのことから、オリゴマーは一定の細胞表面タンパク質、殊に、受容体に結合することがわかる。
【0034】
従って、本発明は、予めの細胞構造への本発明によるオリゴマーの結合の測定法にも関し、この際、
i)本発明によるオリゴマーを細胞構造に作用させ、かつ
ii)本発明によるオリゴマーが細胞構造に結合するかどうかを測定する。
【0035】
もう1つの観点により、本発明によるオリゴマーは、神経変性作用を特徴とする。この神経変性作用は、少なくとも1種の本発明によるオリゴマーを、殊に、ニューロン細胞、例えば、神経芽細胞の培養に作用させる場合には、これらの細胞の生存可能性を減少させることで顕示される。この際、細胞の生存可能性は、自体公知の方法で、例えば、本発明によるオリゴマーの作用で生じるアポトーシスの程度を測定することによって評価され得る。このために、好適な試験法、例えば、3−[4,5−ジメチルチアゾル−2−イル]−2,5−ジフェニルテトラゾリウムブロミド(MTT)をベースとする比色法が使用される。生体内では、この神経変性作用は、殊に、ニューロンの発射率(Feuerungsrate)の変性で顕示される。
【0036】
従って、本発明は、本発明によるオリゴマーの活性、殊に、神経毒性の測定法にも関し、この際、
i)本発明によるオリゴマーを細胞に作用させ、かつ
ii)細胞の状態が変化するかどうかを測定する。
【0037】
前記の方法について、本発明によるオリゴマーを前記の方法で、例えば、前記の組成物の形で製造することができる。細胞又は細胞構造物を、有利に、試験管内で、殊に、細胞培養物として又は細胞構造物ではホモジネートとしても製造される。この際、ニューロン細胞及び殊に神経芽細胞を神経毒性の測定に用いる。選択的に細胞を生体内で、殊に、生体、例えば実験動物の一部として、又は生体外で準備することができる。
【0038】
細胞の状態を、通例、オリゴマーの作用前に少なくとも1回及び作用後に少なくとも1回測定する。作用前の状態と作用後の状態との間の比較で差異が生じる場合には、試験したオリゴマーは活性を有する。
【0039】
測定すべき状態の種類は、測定すべき活性の種類に依る。例えば、生存可能性を測定することによって、神経毒性が測定される。それには、オリゴマーの作用前及び作用後の全細胞数に対する生存細胞の数を測定し、相互に比較することができる。
【0040】
前記の結合又は活性の測定方法に基づき、一定の実施態様により、物質が本発明によるオリゴマーの結合を阻害する及び/又はその活性を変性させる、要するに、殊に、減少させる、又は実際に完全に阻止する(阻害する)、又は強化させるかどうかを試験することができる。
【0041】
原則的に、そのために方法を少なくとも2回、1回は試験物質が存在して、もう1回は試験物質が不在で実施する。そのために、試験物質をオリゴマーに、通例、その製造後に添加する。
【0042】
勿論、試験すべき物質がオリゴマーの生成に影響するか否かを測定すべき場合には、試験物質を、有利にオリゴマーの生成前に既に、要するに、その製造前に既に、例えば、オリゴマー生成に使用される1種以上の出発物質に添加する。即ち、本発明による製法を、試験物質の添加下に実施し、引き続き、オリゴマーが生成するかどうか、及びどの程度生成するかを測定する。そのために、この方法で得られる方法生成物が本発明によるオリゴマーの特性、要するに、例えば、その分子量、結合可能性及び活性を有するかどうかを測定することができる。
【0043】
極めて著しい神経変性作用の意において、本発明によるオリゴマーBが有利である。このオリゴマーの誘導体も、この見地において有利であり、この際、殊に、N−末端切断のAβ(1−42)−タンパク質をベースとするオリゴマーが有利である。
【0044】
方法技術的理由から、オリゴマーA又はオリゴマーBは、混合物として、オリゴマーの他に更に少量の他のポリペプチド、殊に、モノマーアミロイド−β(1−42)−タンパク質及び場合により集合アミロイド−β(1−42)−タンパク質の高分子形も含有する組成物の形で生じてもよい。この種類の組成物は、同様に本発明の目的であり、殊に、1種以上の本発明によるオリゴマーの割合が、アミロイド−β(1−42)−タンパク質から誘導されるタンパク質の全量に対して、少なくとも70質量%、有利に少なくとも90質量%、殊に少なくとも95質量%であることを特徴とする。オリゴマーAの標本では、オリゴマーA2(SDS−ゲルで20kDa−帯)の割合が少なくとも50質量%、有利に少なくとも70質量%、殊に少なくとも85質量%である。オリゴマーBの標本では、オリゴマーB1及びB2(SDS−ゲルで38kDa−及び48kDa−帯)の割合は、少なくとも60質量%、有利に少なくとも75質量%、殊に少なくとも90質量%である。
【0045】
本発明によるオリゴマーは誘導体化されていてもよい。この種類の誘導体化の目的は、例えば、殊に、生物学的有効性に関するオリゴマーの物理化学的特性を変性させること、オリゴマーに検出可能な標識を与えること又は不動化させることであってよく、例えば、担体に結合させることができることである。標識化及び不動化は、殊に、診断的使用に重要である。
【0046】
タンパク質生化学的範囲で慣用の好適な標識化は、当業者に公知である。それには、蛍光−標識化、例えば、一定のフルオレセイン−及びテトラメチルローダミン−誘導体、発光標識化、比色標識化、放射性標識化及び磁気標識化及び相補的結合相手、例えば、ビオチン−及びストレプタビジン−誘導体への親和性での標識化が属する。
【0047】
見かけの分子量に関して、オリゴマー誘導体は、非誘導体化オリゴマーに比べて相応に高められていてよい分子量を有することに注意すべきであり、しかしこの際、集合数は同じである。即ち、例えば、SDS−ゲルで38kDaの分子量を有するAβ(1−42)−オリゴマーB1をベースとするビオチン−誘導体は、42kDaの分子量を有する。
【0048】
本発明の特別な実施態様により、オリゴマーは架橋結合している。好適な架橋結合剤は当業者に公知であり、通例、二官能性試剤、例えば、ホルムアルデヒド、グルタルジアルデヒド、ジスクシンイミジルスベレート、ジチオビス(スクシンイミジルプロピオネート)、ジスクシンイミジルタルトレート、ジスルホスクシンイミジルタルトレート、ジメチルアジピミデート、ジメチルピメリデート、ジメチルスベリミデート、ジメチル−3,3’−ジチオビスプロピオニミデート、N−γ−マレイミニドブチルオキシスクシニミドエステル、スクシニミジル−4(N−マレイニミドメチル)−シクロヘキサン−1−カルボキシレート、N−スクシニミジル−(4−イオドアセチル)アミノベンゾエート及びN−スクシンイミジル−3−(2−ピリジルジチオ)プロピオネートである。
【0049】
この種類の架橋結合オリゴマーは、それが安定性であり、そのオリゴマー化度は、通例、もはや不可逆であるという利点を有する。従って、これは特に診断的試験系での使用に、又はオリゴマー特異的抗体の製造のための免疫原として好適である。
【0050】
本発明によるオリゴマーの誘導体には、アミロイド−β(1−42)−タンパク質の断片のオリゴマーも属する。天然に産するプロテアーゼの作用によって得られる断片が有利である。殊に、タンパク質加水分解によって非変性条件下に生理学的緩衝液中で得られる断片は、アミロイド−β(1−42)−タンパク質に比べて、高められたタンパク質加水分解安定性を有する。エンドペプチダーゼの作用によって得られる断片が有利である。本発明の特別な実施態様により、断片は、トリプシン、キモトリプシン、サーモリシン、エラスターゼ、パパイン又はエンドプロテイナーゼGluCの作用によって得られる。もう1つの観点により、その本発明によるオリゴマーが神経変性作用を特徴とする断片が有利である。この観点を更に説明するために、本発明によるアミロイド−β(1−42)−タンパク質のオリゴマーの神経変性作用についての相応する実施が参照される。
【0051】
有利な断片は、構造的に、殊に、それがアミロイド−β(1−42)−タンパク質からN−末端配列の分離によって誘導されることを特徴とする。1観点により、このN−末端配列とは、アミロイド−β(1−42)−タンパク質のN−末端配列の23個まで、有利に21個まで、殊に19個までのアミノ酸を有する断片が重要である。従って、その配列がアミノ酸24〜42個、有利に22〜42個、殊に20〜42個を連続して包含する、本発明によるAβ(1−42)−タンパク質の断片が有利である。もう1つの観点により、分離すべきN−末端配列とは、アミロイド−β(1−42)−タンパク質のN−末端配列の少なくとも7、有利に少なくとも9、殊に少なくとも11個のアミノ酸を有する配列が重要である。従って、本発明により殊に、N−末端で約7〜23、有利に9〜21、殊に11〜19のアミノ酸が切断されているアミロイド−β(1−42)−タンパク質の断片が有利である。この断片は、式 Aβ(x−42)に相応し、ここで、xは8〜24、有利に10〜22、殊に12〜20である。従って、本発明により、Aβ(x−42)−断片は、有利に、次の断片から選択される:Aβ(8−42)、Aβ(9−42)、Aβ(10−42)、Aβ(11−42)、Aβ(12−42)、Aβ(13−42)、Aβ(14−42)、Aβ(15−42)、Aβ(16−42)、Aβ(17−42)、Aβ(18−42)、Aβ(19−42)、Aβ(20−42)、Aβ(21−42)、Aβ(22−42)、Aβ(23−42)及びAβ(24−42)。特殊な断片は、サーモリシンの作用によって得られるアミロイド−β(20−42)−断片及びエンドプロテイナーゼGluCの作用によって得られるアミロイド−β(12−42)−断片である。
【0052】
本発明による誘導体には、C−末端1又は2で他のアミノ酸を有するアミロイド−β(1−42)−タンパク質から誘導される形も属する。即ち、それには、例えば、Aβ(1−43)−タンパク質のオリゴマー又は前記の実施に相応するその誘導体が属する。
【0053】
本発明によるオリゴマーの製法は、実際には、3段階からなり、その第1段階は任意であるが有利であり、第2段階はオリゴマーA及びBの製造のために強制的であり、第3段階は本発明によるオリゴマーBの製造に用いられる。
【0054】
段階1は、タンパク質の変性に関する。そのために、水素橋切断剤、例えば、ヘキサフルオルイソプロパノール(HFIP)をタンパク質に作用させることができる。作用温度が約20〜50℃、殊に約35〜40℃である場合には、数分、例えば、約10〜60分間の作用時間で十分である。蒸発乾燥させた残渣を、引続いて、有利に濃縮形で、水性緩衝液と混合可能な好適な有機溶剤、例えば、ジメチルスルホキシド(DMSO)に溶かし、本発明による方法の第2段階で使用可能な、少なくとも部分的に変性されたタンパク質の懸濁液を得る。必要に応じて、基本懸濁液を暫時、より低温で、例えば、約−20℃で保存することができる。
【0055】
前記の段階1について選択的に、タンパク質を弱酸性の、有利に水溶液中に、例えば、約10ミリモルのHCl−水溶液中に入れることができる。通例、数分間に渡る恒温保持後に、不溶成分を遠心分離する。10000gで数分間が有利である。この方法段階は有利に室温で、即ち、20〜30℃の範囲の温度で実施される。遠心分離後に得られる上澄液は、アミロイド−β(1−42)−タンパク質を含有し、暫時、より低温で、例えば、約−20℃で保存することができる。
【0056】
段階2は、タンパク質からオリゴマーAへのオリゴマー化に関する。そのために、界面活性剤を場合により少なくとも部分的に変性されたタンパク質に、十分なオリゴマーAが生じるまで作用させる。
【0057】
イオン性界面活性剤、殊に、陰イオン性界面活性剤が有利に使用される。
【0058】
有利な実施態様により、式(I)の界面活性剤を使用する:
R−X
[式中、
基Rは、6〜20、有利に10〜14個の炭素原子を有する場合により分枝鎖のアルキル又は6〜20、有利に10〜14個の炭素原子を有する場合により分枝鎖のアルケニルを表わし、
基Xは、酸性基又はその塩を表わし、この際、Xは、有利に、−COO−M+、−SO3−M+及び特に−OSO3−M+から選択され、M+は、水素陽イオン又は有利にアルカリ金属−及びアルカリ土類金属陽イオン及びアンモニウム陽イオンから選択される無機又は有機陽イオンを表わす]。
【0059】
式中のRが非分枝鎖のアルキル基を表わし、この際、この内で殊にアルク−1−イル−基が挙げられる式(I)の界面活性剤が有利である。ドデシル硫酸ナトリウム(SDS)が特に有利である。ラウリル酸及び油酸も有利に使用することができる。界面活性剤ラウロイルサルコシン(Sarkosyl NL-30 又はGardol (登録商標)としても公知である)のNa−塩も特に有利である。
【0060】
界面活性剤の作用時間は、殊に、オリゴマー化されるタンパク質が変性されているかどうか(変性されていれば、どの程度)に依存する。タンパク質が段階1により前以て水素架橋切断剤、要するに、殊に、ヘキサフルオルイソプロパノールで処理されている場合には、作用温度が約20〜50℃、殊に約35〜40℃である場合には、作用時間は数時間の範囲、有利に約1〜20時間、殊に約2〜10時間で十分である。少し変性されたタンパク質又は実際に変性されないタンパク質が生じる場合には、相応により長い作用時間が有利である。アミロイド−β(1−42)−タンパク質が、例えば、段階1について選択的に前記された方法によって予備処理される又は段階2に直接装入される場合には、作用温度が約20〜50℃、殊に約35〜40℃である場合には、作用時間は約5〜30時間、殊に約10〜20時間の範囲で十分である。恒温保持後に、有利に不溶成分を遠心分離する。10000gで数分が有利である。
【0061】
選択すべき界面活性剤−濃度は、使用される界面活性剤に依る。SDSを使用する場合には、0.01〜1質量%、有利に0.05〜0.5質量%の範囲、例えば、約0.2質量%の濃度が有利である。ラウリル酸又は油酸を使用する場合には、少し高い濃度、約0.05〜2質量%、有利に0.1〜0.5質量%の範囲、例えば、約0.5質量%が有利である。
【0062】
界面活性剤の作用は、塩濃度では、ほぼ生理学的範囲の行なわれるべきである。即ち、殊に、NaCl−濃度は、50〜500ミリモル、有利に100〜200ミロモル、特に約140ミリモルの範囲で有利である。
【0063】
得られるオリゴマーA、即ち、殊にオリゴマーA1及び/又はA2を含有する溶液は、暫時、より低い温度、例えば、約−20℃で保存され得る。これをそのものとして本発明による使用に、又はオリゴマーBへの更なる変換に添加することができ、又は先ず更なる処理−又は精製段階に接続させ、それによって殊に、少なくとも1種の本発明によるオリゴマーAを更に富化させることができる。殊に、クロマトグラフィー法により、本発明によるオリゴ標識ら、溶液中に含有される他のタンパク質成分及び殊にアミロイド−β(1−42)−タンパク質から誘導されるものを分離させることができる。殊に、この目的のために、親和クロマトグラフィー法が用いられ、この場合には、例えば、特異的抗体、要するに、殊に本発明によるオリゴマー特異的抗体を使用することができる。
【0064】
段階3は、オリゴマーBへのオリゴマー化に関する。オリゴマーAを含有する組成物がこの段階の出発物質として有利に選択される。その点では、オリゴマーAは、本発明により、オリゴマーBの製造のための中間生成物として重要である。この組成物が段階2から由来する場合には、これは規則的に界面活性剤及び生理学的範囲の塩濃度を含有する。次いで、界面活性剤作用又は塩濃度を減少させることが有利である。これは、界面活性剤−又は塩濃度の減少によって、例えば、有利に水に又は低塩濃度の緩衝液、例えば、トリス(Tris)−HCl、pH7.3での希釈によって行なうことができる。約2〜10の範囲、有利に約3〜8の範囲、殊に約4の希釈ファクターが好適であると判明した。界面活性剤作用の減少は、界面活性剤作用を中和させ得る物質の添加によって達成することもできる。それには、例えば、界面活性剤を複合化させ得る物質、例えば、清浄−及び抽出手段の実施で細胞を安定化させ得る物質、例えば、一定のEO/PO−ブロックコポリマー、殊に、商品名Pluronic(登録商標)F 68 で販売されるブロックコポリマーが属する。アルコキシル化、殊にエトキシル化アルキルフェノール、例えば、Triton(登録商標)X- 系のエトキシル化t−オクチルフェノール、殊にTriton(登録商標)X100、3−(3−コルアミドプロピル−ジメチルアンモニオ)−1−プロパンスルホネート(CHAPS(登録商標))又はアルコキシル化、殊にエトキシル化ソルビタン脂肪酸エステル、例えば、Tween(登録商標)−系、殊にTween(登録商標)20 が、各々臨界ミセル濃度の範囲又はそれ以上の濃度範囲で好適である。
【0065】
引き続き、十分にオリゴマーBが生じるまで溶液を恒温保持する。作用温度が約20〜50℃、殊に約35〜40℃である場合には、作用時間は数時間の範囲、有利に約10〜30時間の範囲、殊に約15〜25時間の範囲で十分である。引き続き、溶液を濃縮させ、場合により生じる残渣を遠心分離する。この場合にも、10000gで数分間が有利であることが判る。遠心分離後に得られる上澄液はオリゴマーBを含有する。
【0066】
得られるオリゴマーB、即ち、殊にオリゴマーB1及び/又はB2を含有する溶液は、暫時、より低い温度で、例えば、約−80℃で保存され得る。これをそのものとして本発明による使用に添加され、又は先ず、更なる処理又は精製段階に接続させることができる。これに関して、オリゴマーAに関する相応の手段についての前記の説明が参照される。
【0067】
更なる処理−又は精製段階は、オリゴマーの製造のために使用される界面活性剤を実際に完全に除去する目的のためにも実施され得る。例えば、オリゴマーBを先ず界面活性剤含有溶液から沈殿させ、取得し、新たに界面活性剤を含まない媒体中に溶かす。タンパク質の沈殿法は当業者によく知られている。本発明により、水性−メタノール性酢酸の添加が有利であると判明した。一定の機械装置に結合することなく、界面活性剤又は界面活性剤−及び塩濃度の変化がタンパク質を限定された溶解形にさせると思われ、このタンパク質は、水性の生理学的緩衝液中に溶解されたタンパク質の出発形とは明らかに異なっており、このことは、例えば、変性又は自然ゲル電気泳動法又はゲル透過クロマトグラフィーにより確認することができる。このことは驚くべきことであり、それというのも、界面活性剤は、通例、タンパク質集合体を解離させる、即ち、下位単位に分解させることができるが、本発明により、集合傾向を有するモノ標識ら出発して限定されたオリゴマーを得ることができるからである。
【0068】
本発明によるオリゴマーの誘導体を製造するために、有利に、前記の本発明による方法を、既に相応に誘導体化されたアミロイド−β(1−42)−タンパク質で実施するようにして行なう。選択的に、オリゴマーの誘導体化を行なうこともでき、それによって、オリゴマーの構造は当然変化されるべきではない。好適なタンパク質化学的手段は、当業者に公知である。
【0069】
本発明によるオリゴマー又はその誘導体の架橋結合は、自体公知の方法で行なうことができる。架橋結合剤として、例えば、グルタルジアルデヒドを使用する場合には、本発明による方法段階2又は3から得られる溶液を、グルタルジアルデヒド−溶液で処理することができる。オリゴマーはグルタルジアルデヒドと室温で数時間後に反応した。次いで、反応は、自体公知の方法で、過剰量のグルタルジアルデヒドと、この目的のために一般に公知の試剤、例えば、エタノールアミンとの反応によって止められ得る。本発明によるオリゴマーAを架橋結合させるか又はBを架橋結合させるかにより、A−CL又はB−CLで表示される本発明による架橋結合オリゴマーの溶液が得られる。
【0070】
殊に、場合により架橋結合したオリゴマーB又はその誘導体について、その合成に続けて、オリゴマーの安定性に影響することなく塩濃度を再び高めることが可能である。このことは、オリゴマーの使用に関して、例えば、使用の際に生理学的条件が有利である場合に重要である(細胞の使用、生体内使用)。
【0071】
アミロイド−β(1−42)−タンパク質の断片のオリゴマーは、原則的に、相応するモノマー断片から出発してオリゴマー化によって、又はアミロイド−β(1−42)−タンパク質のオリゴ標識ら出発してタンパク質加水分解によって製造され得る。即ち、第2変法に従って、次に、本発明による方法段階2又は3から得られる溶液をプロテアーゼで処理することができる。所望のタンパク質加水分解度が達成されたら、プロテアーゼを一般に公知の方法で不活化させる。ここで、生成したオリゴマーを既に前記した方法により取得し、必要に応じて更なる処理−又は精製段階によって更に処理することができる。
【0072】
本発明によるオリゴマー及びこれを含有する組成物は、その均質性及び安定性を特徴とする。これらは、生理学的媒体、例えば、生理学的食塩溶液中に可溶性であり、むしろ球状のその外観によって、原線維形とは異なっている。これらは、他のAβ(1−42)−形、殊に、モノマー、非毒性オリゴマー、Aβ(1−42)を包含する前駆分子APP、プロトフィブリル及び原線維とは明らかに異なっている空間構造を有するという利点を有する。従って、これらは、生体内でも試験管内でも、特異的抗体の生成に特に好適であり、例えば、特異的な能動免疫化を可能にする。この際、免疫化には、病気を起こすオリゴマーだけを使用することが決定的であり得る。他のAβ(1−42)形、例えば、モノマー分子又は低分子量のオリゴマーは、生体中の重要なシグナル機能には不可欠であり得る。同様に、細胞の被覆に重要であると思われる原線維様沈着物の除去は生体を損傷させ得る。
【0073】
同様の方法で、本発明による均質のオリゴマー及びこれを含有する組成物を用いて、治療的可能性、例えば、受動免疫、オリゴマー形の不安定化剤の使用及び受容体(部分)作働剤又は−拮抗剤の使用を実現することができる。即ち、均質オリゴマー標本は、受動免疫化のためのポリクローナル又はモノクローナル抗体の合目的製造も可能にする。このオリゴマー形によって影響される、病気関連の受容体分子又はシグナル分子の発見も、本発明によるオリゴマー及びこれを含有する組成物で可能である。
【0074】
本発明によるオリゴマーは、アミロイド−β−タンパク質関連の生理学的過程へのその関与に基づき、診断的及び治療的価値を有する。即ち、診断的試験管内及び生体内検出法における、本発明によるオリゴマー及びその誘導体の、場合により相応する組成物の形での使用は、本発明の目的である。
【0075】
アミロイド−β−タンパク質関連の生理学的過程には、アミロイドタンパク質(アミロイドーゼ(Amyloidosen))の沈着と関係の過程それが属する。それには、神経組織の構造的変化を生じさせ、例えば、アルツハイマー病及びダウン症候群において重大である過程も、他の組織を襲う過程、例えば、アミロイド−細管異常、例えば、コンゴフィルアミロイド血管病(Congophile Amyloidangiopathie)(CAA)も挙げられる。
【0076】
本発明のもう1つの目的は、オリゴマー特異的抗体の生成のための本発明によるオリゴマー及びその誘導体の、場合により相応する組成物の形での使用である。
【0077】
この際、Aβ(1−42)−タンパク質の切端形をベースとする本発明によるオリゴマーが特に重要である:欠損したN−末端の故に、それを用いて、Aβ(1−42)−タンパク質よりも明らかに選択性の高い免疫応答を生成させることができる。典型的なAβ(1−42)−タンパク質中の強い免疫原のN−末端は、特に、この分子範囲に特異的である抗体、例えば、抗体6E10(F. Signet)及びBAM−10(Fa. Sigma, St. Louis)(これらは、製造者の記載に依れば、N−末端(6E10: Aβ(1-17) 及びBAM-10 Aβ(1-12) に照準している)を生じさせるが、切端Aβ(1−42)−形をベースとする本発明によるオリゴマーを用いて得られる抗体は、オリゴマー特異的範囲を認識し、それによって、他のAβ(1−42)−形に相対する選別性が達成される。この利点は、殊に、能動的ワクチン接種に利用され、それというのも、接種後に生じるより選別性の高い免疫応答(対APP、モノマー形、プロトフィブリル、原線維、斑)が、より少ない副作用(例えば、脳出血、その場でのモノマー−形の生理学的神経栄養活性の侵害)も招くからである。受動免疫化、即ち、抗体治療のために、殊に、モノクローナル抗体をより良好に製造し、選択することができる。
【0078】
この使用の1観点は、治療範囲におけるオリゴマー特異的抗体の生成である。
【0079】
従って、本発明のもう1つの目的は、治療範囲における本発明によるオリゴマー又はその誘導体の、殊にワクチンとしての使用である。
【0080】
そのようなワクチンは、通例、少なくとも1種の本発明によるオリゴマー及び/又は少なくとも1種の本発明によるその誘導体を含有する製薬学的組成物である。そのために、殊に、本発明による、2種以上のオリゴマーを含有する1種の組成物、又は様々な組成物を有する組合せを使用することができる。従って、殊に、オリゴマーB、即ち、B1及びB2又はその誘導体を接種することができる。更に、組成物は、生理学的に認容性の賦形剤及び場合により他の助剤、例えば、免疫刺激剤を含有することができる。
【0081】
好適な賦形剤は、原則的に任意に選択され得るが、賦形剤の種類は、通例、投与方法に依る。即ち、本発明によるワクチンは、殊に、腸管外、例えば、静脈内、筋肉内及び皮下投与に好適な形で組成され得る。この場合には、賦形剤は、有利に水、食塩溶液、アルコール、脂肪、蝋及び/又は緩衝液を包含する。
【0082】
任意の多数の免疫刺激剤が本発明によるワクチン中で使用されてよい。例えば、アジュバントが一緒に入れられてよい。殆どのアジュバントは、急速な異化作用の前に抗原を保護すべき物質、例えば、水酸化アルミニウム又は鉱油、及び脂質A、ボルタデラペルツスシス(Bortadella pertussis) 又はマイコバクテリウム結核(Mycobacterium tuberculosis)から誘導されるタンパク質を含有する。好適なアジュバント、例えば、完全又は不完全フロインドアジュバント;AS−2;アルミニウム塩、例えば、水酸化アルミニウム(場合により、ゲルとして)又は燐酸アルミニウム;カルシウム−、鉄−又は亜鉛塩;アシル化チロシンの不溶性懸濁液;アシル化糖;陽イオン又は陰イオン誘導体化多糖類;ポリホスファゼン(Polyphosphazene) ;生物学的に分解可能な微小球;モノホスホリル−脂質Aが、通例、市販で得られる。サイトカイン、例えば、GM−CSF又はインターロイキン−2、−7又は−12が、アジュバントとして同様に使用される。
【0083】
更に、本発明は抗体の製法に関し、この際、
i)宿主を少なくとも1種の本発明によるオリゴマー、その誘導体又は組成物で免疫化させ、かつ
ii)免疫化への応答として生成した宿主の抗体−含有血清を取得する。
【0084】
抗体の製法の特別な実施態様により、免疫化のために、様々な本発明によるオリゴマー又はオリゴマー−誘導体の混合物を含有する免疫化カクテルを投与する。殊に、方法の経過中に、そのオリゴマー−組成が異なっている数種の免疫化カクテルを投与することが有利である。
【0085】
使用すべきオリゴマー又はオリゴマー−誘導体が非免疫原又は弱免疫原である場合には、それを、担体、有利に担体タンパク質、例えば、ケイホールリンペットヘモシアニン(Keyhole Limpet Hemocyanin)(KLH)、リムルスポリフェヌスヘモシアニン(Limulus Polyphenus Hemocyanin)(LPH)、ウシ血清アルブミン(BSA)又はオバルブミン(OVA)に結合させることによって、その免疫原性を高めることができる。それについて、当業者は、一般に公知である一連の結合可能性が得られる。例えば、グルタルジアルデヒドとの反応が有利であり、例えば、オリゴマー又はオリゴマー−混合物を、好適なペプチド又はペプチド−混合物と、水又は水性溶剤中で恒温保持させることによって可能である。この反応は、有利に環境温度で、要するに、通例、室温で実施され得る。勿論、冷却又はやや加温することも有利であり得る。反応は、通例、数時間内で所望の結果と成り、例えば、2時間の反応時間が通例の範囲である。グルタルジアルデヒド−濃度は、通例、ppm−〜%−範囲、好適には10ppm〜1%、有利に100ppm〜0.5%である。反応パラメーターの最適化は、当業者の知識の範囲にあり、オリゴマーA及びB又はオリゴマー−誘導体が選択された反応条件下に安定性であることを考慮すべきである。
【0086】
免疫化カクテルの製造のために、使用すべき成分を先ず一緒に装入する。得られる成分−混合物を先ず恒温保持することが有利である。これは、有利に環境温度で、通例、室温で行われる。勿論、混合物を冷却又はやや加温することも有利であり得る。恒温保持時間は、通例、数分間から数時間であり、1時間の恒温保持時間が有利であることが実証された。
【0087】
免疫化カクテルは、抗原に付加的に、通例、他の助剤、殊に、免疫化に慣用のアジュバント、例えば、フロインド−アジュバントを含有する。殊に、最初の免疫化には、完全フロインド−アジュバントを使用するが、更なる全ての免疫化を不完全フロインド−アジュバントで実施する。免疫化カクテルの製造のために、有利に前記の成分−混合物としての抗原(免疫原)を1種以上の助剤に加える。この際、抗原は、通例、乳化される。
【0088】
宿主として、殊に、齧歯動物(Nager)又はウサギが好適である。これら又は他の好適な宿主に、免疫化カクテルを有利に皮下注射する。抗体力価は、免疫検定、例えば、宿主−IgGに照準されたヒツジ−抗血清及び標識化オリゴマーで競合的に測定され得る。即ち、測定される宿主が抗体取得に好適であるかどうかを、免疫化の終り頃に決定することができる。例えば、4回の免疫化を実施する場合には、3番目の免疫化後に抗体力価を測定し、次いで、十分な抗体力価を有する動物から抗体を取得することができる。
【0089】
生成した抗体の取得のために、宿主から数週間又は数ヶ月に渡り血液を採取することが有利である。継続して宿主から採血することができる。採血から自体公知の方法で、所望の抗体を含有する血清を取得する。そうして得られる全血清を、必要に応じて、その中に含まれる抗体画分及び殊にオリゴマー−認識抗体を富化させるために、当業者に公知の方法で更に精製することができる。
【0090】
本方法の特別な実施態様により、免疫原として使用されるオリゴマー、その誘導体又は免疫原として使用される組成物中に含有される少なくとも1種のオリゴマー又はその誘導体を特異的に認識する少なくとも1種の血清抗体を選択する。これに関する特異性は、特に、モノマーアミロイド−β(1−42)−タンパク質及び本発明によるオリゴマーよりも高い分子量を有するオリゴマー又はマルチマーアミロイド−β(1−42)−タンパク質−集合体に比べて、他の、殊に類似タンパク質に対するよりも、免疫原に対する抗体のより高い結合親和性を意味する。モノクローナルオリゴマー特異的抗体も、この方法で取得することができる。当然、そのために、宿主から脾臓組織を取り出し、そうして得られる脾臓リンパ球から出発して、モノクローナル抗体を生産するハイブリドーマを常法で確立することが有利である。
【0091】
抗体製造の詳細な説明
哺乳動物の免疫系の一部は、全体として無数の様々な抗体特異性から構成される抗体−レパートリーを有するB−リンパ球である。一定の抗原に対する通常の免疫応答は、このレパートリーからの抗原を特異的に結合する1種以上の抗体の選別を意味し、免疫応答の結果は、刺激抗原を特異的に認識し(かつ最終的には排除する)かつ、この際、抗体の周囲から他の分子を無視するというこの抗体の能力に、少なくとも部分的に基づいている。
【0092】
一定の目的抗原を特異的に認識する抗体の有用性は、モノクローナル抗体技術の発展を導いた。今や、標準されたハイブリドーマ技術は、関与する抗原のための単一の特異性を有する抗体の製造を可能にする。最近では、組換え抗体技術、例えば、抗体バンクの試験管内−スクリーニングが開発されている。この技術を用いて、関与する抗原のための単一の特異性を有する抗体を同様に製造することができる。
【0093】
本発明による方法で、関与する抗原を生体内又は試験管内で抗体−レパートリーに作用させることができる。
【0094】
1実施態様により、動物を生体内において抗原で免疫化させることによって、抗原をレパートリーに作用させることができる。この生体内−法は、更に、動物のリンパ球から一連のハイブリドーマを確立し、抗原を特異的に結合する抗体を分泌するハイブリドーマを選択することを包含する。免疫化すべき動物とは、例えば、マウス、ラット、ウサギ、ニワトリ、ラクダ(Kamelid)又はヒツジ、又は前記した動物の1種の形質転換型、例えば、抗原刺激によってヒト抗体を生じさせるヒト免疫グロブリン遺伝子を有する形質転換マウスである。免疫化され得る動物のその他の型には、ヒト末梢単核血球で再構成されている重症複合免疫不全(SCID)マウス(キメラhu-PBMC-SCID-マウス)又はリンパ様細胞又はその前駆体で再構成されている重症複合免疫不全マウス、同様に、致死的全身放射線照射で処理され、引き続き、重症複合免疫不全(SCID)マウスからの骨髄細胞で放射線照射に対して保護され、引き続き、機能的ヒトリンパ球を移植されているマウス(いわゆる、トリメラ−系(Trimera-System))が属する。免疫化すべき動物のもう1種の型は、そのゲノムにおいて、関与する抗原をコードする内在性遺伝子が、例えば、相同的組換えによって遮断された("ノックアウト(knocked out )")動物(例えば、マウス)であり、即ち、この動物は、抗原での免疫化後に、抗原を異種として認識する。ELISA−技術がそれに属する公知のスクリーニング−法を使用することによって、この方法で製造されたポリクローナル又はモノクローナル抗体を特徴づけ、選択することは当業者には明白であるが、これに限定されるものではない。
【0095】
もう1つの実施態様により、組換え抗体バンクを抗原でスクリーニングすることによって、抗原を抗体−レパートリー上へ試験管内で作用させることができる。組換え抗体バンクは、例えば、バクテリオファージの表面上又は酵母細胞の表面上又は細菌細胞の表面上で発現されてよい。多様な実施態様において、組換え抗体バンクは、例えば、scFv−バンク又はFab−バンクである。もう1つの実施態様により、抗体バンクはRNA−タンパク質−融合として発現される。
【0096】
本発明による抗体の製造のためのもう1つの方法は、生体内−及び試験管内−法からの組合せを包含する。例えば、動物を抗原で生体内免疫化させ、引き続き、動物のリンパ様細胞から製造される組換え抗体バンク又は単一ドメイン−抗体バンク(例えば、重鎖及び/又は軽鎖を有する)を抗原で試験管内スクリーニングすることによって、抗原を抗体−レパートリー上に作用させることができる。もう1つの方法により、動物を抗原で生体内免疫化させ、引き続き、動物のリンパ様細胞から製造した組換え抗体バンク又は単一ドメイン−バンクを親和性成熟させることによって、抗原を抗体−レパートリー上に作用させることができる。もう1つの方法により、動物を抗原で生体内免疫化させ、引き続き、関与する抗体を分泌する単一の抗体−生産細胞を選択し、この選択された細胞から重鎖及び軽鎖の可変領域のcDNAを取得し(例えば、PCRにより)、重鎖及び軽鎖の可変領域を試験管内で哺乳動物宿主細胞中で発現させる(これは、リンパ球−抗体−選択法、"又は選択リンパ球抗体法(Selected Lymphocyte Antibody Method)"のSLAMとして表示される)(それによって選択された抗体−遺伝子配列を更に選択し、操作することができる)ことによって、抗原を抗体−レパートリー上に作用させることができる。更に、重鎖及び軽鎖の抗体遺伝子を哺乳動物細胞中に発現させ、所望の結合親和性を有する抗体を分泌する哺乳動物細胞を選択することによって、モノクローナル抗体を発現クローニングによって選択することができる。
【0097】
従って、本発明の観点は、限定された抗原をスクリーニング及び逆スクリーニングに使用することである。即ち、本発明によるオリゴマー又はその誘導体を結合しないが、他の型のA(1−42)−タンパク質、APP、アミロイド原線維又はアミロイド斑及び一連のその他の非類似抗原及び組織を結合するポリクローナル及びモノクローナル抗体を本発明により選別することができる。
【0098】
抗体選別は明確な抗原に基づくということは、当業者に十分知られている。これに反して、余り明確ではない抗原を使用する場合には、それは十分には選別されない。要するに、試験管内での作用及び選別は、親和性クロマトグラフィーに似ていて、この際、所望の抗原の"リガンド"は、抗原を十分な親和性では結合しないそれから分離される。従って、他の抗体の巨大プールからの所望抗体の富化度は、抗原品質の直接的結果である。意外にも、本発明によるオリゴマー及びその誘導体は、好適で適切な選別的抗体を富化させ、A(1−42)−タンパク質と関連する他の型及び他の非類似抗原を認識する抗体から効果的に分離させる抗原である。
【0099】
本発明による抗体の製法を用いて、様々な型の抗体を製造することができる。それには、実際に、ヒト抗体、キメラ抗体、ヒト化抗体及びCDR−グラフト−抗体及びその抗原−結合部分が属する。
【0100】
次に、本発明による抗体の製法を記載する。この際、生体内−法、試験管内−法、又は2法を含む組合せを区別する。
【0101】
生体内−法
生体内で生じた抗体を生産する細胞から、モノクローナル抗体を、標準された技術、例えば、Kohler 及びMilstein (1975, Nature 256: 495-497 )(同様に、Brown et al. (1981) J. Immunol 127: 539-46; Brown et al. (1980) J Biol Chem 255: 4980-83; Yeh et al. (1976) PNAS 76: 2927-31; 及びYeh et al. (1982) Int. J. Cancer 29: 269-75 参照)によって本来記載されたハイブリドーマ技術によって製造することができる。モノクローナル抗体ハイブリドーマの生産技術は十分に公知である(一般に、R. H. Kenneth, in Monoclonal Antibodies: A New Dimension In Biological Analyses, Plenum Publishing Corp., New York, New York (1980); E. A. Lerner (1981) Yale J. Biol. Med. 54: 387-402; M. L. Gefter et al. (1977) Somatic Cell Genet., 3: 231-36 参照)。要するに、不死化細胞系(典型的には、骨髄腫)を、本発明によるオリゴマー又はその誘導体で免疫化した哺乳動物のリンパ球(典型的には、脾細胞又はリンパ節細胞又は末梢血液リンパ球)と融合させ、生じるハイブリドーマ細胞の培養上澄液をスクリーニングし、本発明によるオリゴマー又はその誘導体の特異性を有するモノクローナル抗体を生産するハイブリドーマを同定する。そのために、多数の周知されているプロトコルの任意の1つを、リンパ球及び不死化細胞系の融合のために使用することができる(同様に、G. Galfre et al. (1977) Nature 266: 550-52; Gefter et al. Somatic Cell Genet., cited supra; Lerner, Yale J. Biol. Med. , cited supra; Kenneth, Monoclonal Antibodies, cited supra参照)。更に、同様に使用可能である方法の様々な変法が当業者に公知である。典型的には、不死化細胞系(例えば、骨髄腫細胞系)は、同一哺乳動物種、例えば、リンパ球から由来する。例えば、本発明による免疫原調製物で免疫化されたマウスからのリンパ球を不死化マウス細胞系と融合させることによって、マウスハイブリドーマを確立することができる。有利な不死化細胞系は、ハイポキサンチン、アミノプテリン及びチミジンを含有する培地(HAT−培地)に感受性のあるマウス−骨髄腫細胞系である。多数の骨髄腫細胞系の任意の1つ、例えば、P3−NS1/1−Ag4−1−、P3−x63−Ag8.653−又はSp2/O−Ag14−骨髄腫系(Myelomlinie)を融合相手として標準的に使用することができる。この骨髄腫細胞系は、アメリカンタイプカルチャーコレクション(American Type Culture Collection) (ATCC), ロックビル(Rockville, MD) から得られる。典型的には、HAT−感受性マウス−骨髄腫細胞を、マウス−脾細胞と、ポリエチレングリコール(PEG)の使用下に融合させる。融合から得られるハイブリドーマ細胞を、次いで、HAT−培地の使用下に選別し、それによって、非融合化及び非生産的融合化骨髄腫細胞を死滅させる(非融合化脾細胞は、形質転換されないので、数日間後に死滅する)。本発明によるオリゴマー又はその誘導体を特異的に認識するモノクローナル抗体−生産性ハイブリドーマ細胞は、ハイブリドーマ培養上澄液をそのような抗体についてスクリーニングすることによって、例えば、標準−ELISA−検定を使用することによって同定され、本発明によるオリゴマー又はその誘導体を特異的に結合することができる抗体が選別される。
【0102】
所望抗体の種類に応じて、様々な宿主動物を生体内−免疫化に使用することができる。関与する抗原の内在型自体を発現する宿主を使用することができる。選択的に、関与抗原の内在Versionのために不全にされた宿主を使用することができる。例えば、相応する内在遺伝子での相同的組換えを経て一定の内在タンパク質のために不全にされたマウス(即ち、ノックアウト−マウス)は、マウスがそれで免疫化されるタンパク質に対して体液性応答を生じさせ、従って、タンパク質に対する高親和性のモノクローナル抗体の製造に使用され得ることが示された(例えば、Roes, J. et al. (1995) J. Immunol. Methods 183: 231-237; Lunn, M. P. et al. (2000) J. Neurochem. 75: 404-412 参照)。
【0103】
本発明によるオリゴマー又はその誘導体に対する非ヒト抗体の製造のために、多数の非−人間の哺乳動物が抗体製造のための宿主として好適である。それには、マウス、ラット、ニワトリ、ラクダ、ウサギ及びヤギ(及びそのノックアウト−型)が属するが、ハイブリドーマ製造にはマウスが有利である。更に、実際に、二重の特異性を有するヒト抗原に対するヒト抗体の製造のために、ヒト抗体−レパートリーを発現する非−ヒト宿主動物を使用することができる。そのような非−ヒト動物には、ヒト−免疫グロブリン−導入遺伝子を有する導入遺伝子動物(例えば、マウス)(キメラhu-PBMC-SCID-マウス)及びヒト/マウス−放射線照射キメラが属し、これらは次に詳記されている。
【0104】
1実施態様により、本発明によるオリゴマー又はその誘導体で免疫化される動物は、非−ヒト哺乳動物、有利に、ヒト免疫グロブリン遺伝子による形質転換のマウスであり、従って、非−ヒト哺乳動物は抗原刺激によりヒト抗体を作る。典型的には、そのような動物中に、ヒト生殖系列立体配置を有する重鎖及び軽鎖のための免疫グロブリン−導入遺伝子が導入され、この際、動物は、重鎖及び軽鎖のためのその内在座が不活性であるように変えられた。そのような動物を抗原(例えば、ヒト抗原)で刺激する場合には、ヒト免疫グロブリン配列から由来する抗体(即ち、ヒト抗体)が生産される。そのような動物のリンパ球から、標準ハイブリドーマ技術により、ヒトモノクローナル抗体を作ることができる。ヒト免疫グロブリンの形質転換マウス及びヒト抗体の製造の際のその使用の更なる説明について、例えば、US−特許第5939598、WO96/33735、WO96/34096、WO98/24893及びWO99/53049(Abgenix Inc.)及びUS−特許第5545806、第5569825、第5625126、第5633425、第5661016、第5770429、第5814318、第5877397及びWO99/45962(Genpharm Inc.)が参照される;同様に、MacQuitty, J. J. 及びKay, R. M. (1992) Science 257: 1188; Taylor, L. D. et al. (1992) Nucleic Acids Res. 20: 6287-6295; Lonberg, N. et al. (1994) Nature 368: 856-859; Lonberg, N. 及びHuszar, D. (1995) Int. Rev. Immunol. 13: 65-93; Harding, F. A. 及びLonberg, N. (1995) Ann. N. Y. Acad. Sci. 764: 536-546; Fishwild, D. M. et al. (1996) Nature Biotechnology 14: 845-851; Mendez, M. J. et al. (1997) Nature Genetics 15: 146-156; Green, L. L. 及びJakobovits, A. (1998) J. Exp. Med. 188: 483-495; Green, L. L. (1999) J. Immunol. Methods 231: 11-23; Yang, X. D. et al. (1999) J. Leukoc. Biol. 66: 401-410; Gallo, M. L. et al. (2000) Eur. J. Immunol. 30: 534-540が参照される。
【0105】
もう1つの実施態様により、本発明によるオリゴマー又はその誘導体で免疫化される動物は、ヒト末梢単核血球又はリンパ様細胞又はその前駆体で再構成された重症複合免疫不全(SCID)のマウスであってよい。キメラhu-PBMC-SCID-マウスとして特徴付けられるそのようなマウスは、明らかに、抗原刺激によるヒト免疫グロブリン応答を生産する。これらのマウス及び抗体の生成のためのその使用の更なる説明について、例えば、Leader, K. A. et al. (1992) Immunology 76: 229-234; Bombil, F. et al. (1996) Immunobiol. 195: 360-375; Murpy, W. J. et al. (1996) Semin. Immunol. 8: 233-241; Herz, U. et al. (1997) Int. Arch. Allergy Immunol. 113: 150-152; Albert, S. E. et al. (1997) J. Immunol. 159: 1393-1403; Nguyen, H. et al. (1997) Microbiol. Immunol. 41: 901-907; Arai, K. et al. (1998) J. Immunol. Methods 217: 79-85; Yoshinari, K. 及びArai, K. (1998) Hybridoma 17: 41-45; Hutchins, W. A. et al. (1999) Hybridoma 18: 121-129; Murphy, W. J. et al. (1999) Clin. Immunol. 90: 22-27; Smithson, S. L. et al. (1999) Mol. Immunol. 36: 113-124; Chamat, S. et al. (1999) J. Infect. Diseases 180: 268-277; 及びHeard, C. et al. (1999) Molec. Med. 5: 35-45が参照される。
【0106】
もう1つの実施態様により、本発明によるオリゴマー又はその誘導体で免疫化される動物は、致死的全身放射線照射で処理され、引き続き、重症併合免疫不全(SCID)のマウスからの骨髄細胞で照射に対して保護され、かつ引き続き、機能的ヒトリンパ球を移植されているマウスである。トリメラ−系として特徴付けられるこのキメラ型は、ヒトモノクローナル抗体を製造するために、マウスを関与抗原で免疫化させ、引き続き、モノクローナル抗体を標準ハイブリドーマ技術の使用下に製造することによって使用される。これらのマウス及び抗体の製造のためのその使用の更なる説明について、例えば、Eren, R. et al. (1998) Immunology 93: 154-161; Reisner, Y及びDagan, S. (1998) Trends Biotechnol. 16: 242-246; Ilan, E. et al. (1999) Hepatology 29: 553-562; 及びBocher, W. O. et al. (1999) Immunology 96: 634-641が参照される。
【0107】
試験管内−法
本発明による抗体を免疫化及び選別によって製造するために、選択的に、オリゴマー又はその誘導体に特異的に結合する免疫グロブリン−バンクの構成員を分離するために、組換え集合免疫グロブリン−バンク(ライブラリー)を本発明によるオリゴマー又はその誘導体でスクリーニングすることによって、本発明による抗体を同定及び分離することができる。ディスプレー−バンクの製造及びスクリーニングのためのキットは市販で得られる(例えば、ファルマシア(Pharmacia)製の組換えファージ抗体システム(Recombinant Phage Antibody System)カタログ−番号27−9400−01;及びストラタゲン(Stratagene)製のSurfZAP(登録商標)Ohage Display Kit、カタログ−番号240612)。多数の実施態様で、ディスプレー−バンクは、scFv−バンク又はFab−バンクである。組換え抗体バンクのスクリーニングのためのファージディスプレー−技術は十分に記載されている。抗体−ディスプレー−バンクの製造及びスクリーニングの際に有利に使用され得る方法及び化合物の例は、例えば、McCafferty et al. WO92/01047、US−特許第5969108及びEP589877(殊に、scFvのディスプレーを記載)、Ladner et al. US−特許第5223409、第5403484、第5571698、第5837500及びEP436597(例えば、pIII−融合を記載);Dower et al. WO91/17271、US−特許第5427908、US−特許第5580717及びEP527839(殊に、Fabのディスプレーを記載);Winter et al. International Publication WO92/20791及びEP368684(殊に、可変免疫グロブリンドメインの配列のクローニングを記載);Griffiths et al. US−特許第5885793及びEP589877(組換えバンクの使用下にヒト抗原に対するヒト抗体の単離を記載);Garrard et al. WO92/09690(殊に、ファージ発現技術を記載);Knappik et al. WO97/08320(ヒト組換え抗体バンクHuCalを記載);Salfeld et al. WO97/29131(ヒト抗原に対する組換えヒト抗体(ヒト−腫瘍−壊死−因子アルファ)の製造及び組換え抗体の試験管内−親和性成熟を記載)及びSalfeld et al. U. S. Provisional Application 第60/126603及びこれに基づく特許出願(同様に、ヒト抗原に対する組換えヒト抗体(ヒトインターロイキン−12)の製造及び組換え抗体の試験管内−親和性成熟を記載)にある。
【0108】
組換え抗体バンクのスクリーニングの更なる記載は科学的刊行物、例えば、Fuchs et al. (1991) Bio/Technology 9:1370-1372;Hay et al. (1992) Hum Antibod Hybridomas 3:81-85; Huse et al. (1989) Science 246:1275-1281; Griffiths et al. (1993) EMBO J 12:725-735; Hawkins et al. (1992) J Mol Biol 226:889-896; Clarkson et al. (1991) Nature 352:624-628; Gram et al. (1992) PNAS 89:3576-3580); Garrad et al. (1991) Bio/Technology 9:1373-1377; Hoogenboom et al. (1991) Nuc Acid Res 19:4133-4137; Barbas et al. (1991) PNAS 88:7978-7982; McCafferty et al. Nature (1990) 348:552-554; 及びKnappik et al. (2000) J. Mol. Biol. 296:57-86 にある。
【0109】
バクテリオファージ−ディスプレー−系の使用のために、選択的に、組換え抗体バンクを、酵母細胞又は細菌細胞の表面上に発現させることができる。酵母細胞の表面上に発現するバンクの製造及びスクリーニングの方法は、WO99/36569に記載されている。細菌細胞の表面上に発現されるバンクの製造及びスクリーニングの方法は、WO98/49286に比較的正確に記載されている。
【0110】
関与する抗体が組合せバンクから同定されると直ちに、抗体の軽鎖及び重鎖をコードするDNAが、標準分子生物学的技術により、例えば、バンクのスクリーニングの間に分離したディスプレー−パッケージ(例えば、ファージ)からのDNAのPCR−増幅によって単離される。PCRを製造し得る抗体軽鎖及び重鎖のための遺伝子のヌクレオチド配列は、当業者に公知である。多数のそのような配列は、例えば、Kabat, E. A., et al. (1991) Sequences of Proteins of Immunological Interest, Fifth Edition, U. S. Department of Health und Human Services, NIH Publication Nr. 91-3242 及びヒト生殖系列(Humankeimbahn) VBASE の配列のためのデータバンク(Datenbank fuer Sequenzen)に記載されている。
【0111】
本発明による抗体又は抗体部分は、宿主細胞中で免疫グロブリン軽鎖及び重鎖のための遺伝子を組換え発現させることによって製造され得る。抗体を組換え発現させるために、抗体の免疫グロブリン軽鎖及び重鎖をコードするDNA−断片を有する1つ以上の組換え発現ベクターで宿主細胞をトランスフェクションさせ、従って、宿主細胞中で軽鎖及び重鎖が発現し、有利に宿主細胞を培養する培地中に分泌される。この培地から、抗体を取得することができる。抗体重鎖及び軽鎖のための遺伝子を得て、この遺伝子を組換え発現ベクターに組込み、ベクターを宿主細胞に導入させるために、標準組換えDNA−法を使用する。この種類の方法は、例えば、Sambrook, Fritsch 及び Maniatis (Hrsg.), Molecular Cloninng; A Laboratory Manual, Second Edition, Cold Spring Harber, N. Y., (1989), Ausubel, F. M. et al. (Hrsg.) Current Protocols in Molecular Biology, Greene Publishing Associates, (1989) 及びBoss et al. のUS−特許第4816397に記載されている。
【0112】
関与する抗体のVH−及びVL−分節をコードするDNA−断片が得られると直ちに、このDNA−断片は、標準組換えDNA−技術により、例えば、可変領域の遺伝子を全長の抗体鎖の遺伝子、Fab−断片の遺伝子又はscFv−遺伝子に変えるために更に操作され得る。この操作では、VL−又はVH−コードDNA−断片は、更にもう1種のタンパク質、例えば、定常抗体領域又は可変リンカーをコードするDNA−断片と操作的に結合される。ここで、"操作的結合"とは、2つのDNA−断片によってコードされるアミノ酸配列が読み枠(フレーム)中に留まるように、2つのDNA−断片が相互に結合されていることを意味すべきである。
【0113】
VH−領域をコードする単離DNAは、VH−領域をコードするDNAをもう1つの重鎖の定常領域(CH1、CH2及びCH3)をコードするDNA−配列と操作的に結合させることによって、全長の重鎖の遺伝子に変えられ得る。ヒト重鎖の定常領域の遺伝子の配列はよく知られていて(例えば、Kabat, E. A., et al. (1991) Sequences of Proteins of Immunological Interest, Fifth Edition, U. S. Department of Health und Human Services, NIH Publication Nr. 91-3242参照)、この領域にかかるDNA−断片は標準PCR−増幅によって得られ得る。重鎖の定常領域は、IgG1、IgG2、IgG3、IgG4、IgGA、IgGE、IgGM又はIgGDからの定常領域であってよく、この際、IgG1又はIgG4からの定常領域が有利である。重鎖のFab−断片の遺伝子を得るために、VH−コードDNAを、重鎖の定常領域CH1だけをコードするもう1つのDNA−配列と操作的に結合させることができる。
【0114】
VL−コードDNAを、軽鎖の定常領域CLをコードするもう1つのDNA−配列と操作的に結合させることによって、VL−領域をコードする単離DNAを、全長の軽鎖の遺伝子(及びFab−軽鎖の遺伝子)に変換することができる。ヒト軽鎖の定常領域の遺伝子配列はよく知られていて(例えば、Kabat, E. A., et al. (1991) Sequences of Proteins of Immunological Interest, Fifth Edition, U. S. Department of Health und Human Services, NIH Publication Nr. 91-3242参照)、この領域にかかるDNA−断片は標準PCR−増幅によって得られ得る。軽鎖の定常領域は、定常のカッパ−又はラムダ−領域であってよく、この際、定常のカッパ−領域が有利である。
【0115】
scFv−遺伝子を生産するために、VH−及びVL−コードDNA−断片を、可変リンカー、例えば、アミノ酸配列(Gly4−Ser)3をコードするもう1つの断片と操作的に結合させることができ、従って、VH−及びVL−配列は連続単鎖タンパク質として発現され、この際、VL−及びVH−領域は可変リンカーを経て相互に結合している(Bird et al. (1988) Science 242:423-426; Huston et al. (1988) Proc. Natl. Acad. Sci. USA 85:5879-5883; McCafferty et al., Nature (1990) 348:552-554 参照)。
【0116】
本発明によるオリゴマー又はその誘導体の特異性を有するVH−及びVL−単一ドメインは、前記の方法で、単一ドメイン−バンクから単離され得る。所望の特異性を有する2本のVH−単一ドメイン−鎖(CH1有り又は無し)又は2本のVL−鎖又はVH−及びVL−鎖からの一組を、本発明によるオリゴマー又はその誘導体を体から除去するために使用することができる。
【0117】
本発明による組換え抗体又は抗体部分を発現するために、部分的長さ又は全長の軽鎖及び重鎖をコードするDNAを発現ベクターに挿入させることができ、そうして遺伝子は転写及び翻訳調節配列と操作的に結合している。この関連において、"操作的結合"とは、転写及び翻訳調節配列が、ベクター内で、抗体遺伝子の転写及び翻訳の調節のためにその意図する機能を果たすように、抗体遺伝子がベクター中で連結していることを意味するべきである。
【0118】
発現ベクター及び発現調節配列は、それらが、使用される発現宿主細胞と適合性であるように選択される。抗体軽鎖の遺伝子及び抗体重鎖の遺伝子は、別々のベクターに挿入されてよく、又は2つの遺伝子は同じ発現ベクターに挿入され、これは常例である。抗体遺伝子は発現ベクター中で標準法により挿入される(例えば、抗体遺伝子断片及びベクターでの相補的制限切断部位のライゲーション、又は制限切断部位が存在していない場合には、平滑末端のライゲーション)。軽鎖及び重鎖の配列の挿入前に、既に発現ベクターは定常抗体領域の配列を有する。例えば、重鎖又は軽鎖のための定常領域を既にコードする発現ベクターにVH−及びVL−配列を挿入させることによって、VH−及びVL−配列を全長の抗体遺伝子に変換させる方法があり、従って、VH−分節は1つ以上のCH−分節とベクター内で操作的に結合していて、かつVL−分節もCL−分節とベクター内で操作的に結合している。付加的に又は選択的に、組換え発現ベクターは、宿主細胞からの抗体鎖の分泌を容易にするシグナルペプチドをコードすることができる。抗体鎖の遺伝子は、ベクター中へクローン化されることができ、従って、シグナルペプチドは読み枠で抗体鎖の遺伝子のN−末端と結合している。シグナルペプチドは、免疫グロブリン−シグナルペプチド又は異種シグナルペプチドであってよい(即ち、非−免疫グロブリン−タンパク質からのシグナルペプチド)。抗体鎖の遺伝子に付加的に、本発明による発現ベクターは、宿主細胞中で抗体鎖の遺伝子の発現を調節する調節配列を有することができる。"調節配列"とは、抗体鎖の遺伝子の転写又は翻訳を調節するプロモーター、エンハンサー及び他の発現調節要素(例えば、ポリアデニル化シグナル)を含むべきである。そのような調節配列は、例えば、Goeddel; Gene Expression Technology: Methods in Enzymology 185, Academic Press, San Diego, CA (1990)に記載されている。調節配列の選別が属する発現ベクターの設計は、因子、例えば形質転換宿主細胞の選別、タンパク質の所望される発現強度等に依存してよい。哺乳動物宿主細胞における発現の有利な調節配列には、哺乳動物細胞中に強いタンパク質発現を引き起こすウイルス要素、例えば、サイトメガロウイルス(CMV)(例えば、CMV−プロモーター/エンハンサー)、サル−ウイルス40(SV40)(例えば、SV40−プロモーター/エンハンサー)、アデノウイルス(例えば、後期アデノウイルス−主要プロモーター(アデノウイルス主要後期プロモーターのAdMLP)及びポリオー標識ら由来するプロモーター及び/又はエンハンサーが属する。ウイルス調節要素及びその配列の更なる記載について、例えば、StinskiによるUS−特許第5168062、Bell et al.によるUS−特許第4510245及びSchaffner et al.によるUS−特許第4968615が参照される。
【0119】
抗体鎖及び調節配列の遺伝子に付加的に、本発明による組換え発現ベクターは、付加的な配列、例えば、宿主細胞へのベクターの複製を調節する配列(例えば、複製出発点)及び選別可能な標識遺伝子を有することができる。選別可能な標識遺伝子は、ベクターがその中に導入された宿主細胞の選別を容易にする(例えば、全てAxel et al. によるUS−特許第4399216、4634665及び5179017参照)。例えば、選別可能な標識遺伝子は、ベクターが挿入された宿主細胞を、作用物質、例えばG418、ハイグロマイシン又はメトトレキセートに対して耐性にさせることは通常である。有利な選別可能な標識遺伝子には、ジヒドロ葉酸レダクターゼ(DHFR)の遺伝子(メトトレキセート−選別/増幅を伴うdhfr−宿主細胞における使用のために)及び新生−遺伝子(G418−選別のための)が属する。
【0120】
軽鎖及び重鎖の発現のために、軽鎖及び重鎖をコードする1種以上の発現ベクターを、標準技術によって、宿主細胞にトランスフェクションさせる。"トランスフェクション"の様々な形は、原核又は真核宿主細胞への外来DNAの導入のために通例使用される多数の技術、例えば、電気穿孔、燐酸カルシウム−沈降、DEAE−デキストラン−トランスフェクション等を理解すべきである。本発明による抗体を原核宿主細胞又は真核宿主細胞で発現することは理論的には可能であるが、真核細胞、殊に、哺乳動物宿主細胞での抗体の発現が有利であり、それというのも、正確に折りたたまれ、免疫学的に能動的な抗体が集合して分泌される確率が、そのような真核細胞、殊に哺乳動物細胞において、原核細胞におけるよりも高いからである。抗体遺伝子の原核発現について、それが能動抗体の高収率の生産には無効であるということが報告されている(Boss, M. A.及びWood, C. R. (1985) Immunology Today 6:12-13)。
【0121】
本発明による組換え抗体の発現に有利である哺乳動物宿主細胞には、CHO−細胞(Urlaub 及び Chasin, (1980) Proc. Natl. Acad. Sci. USA 77:4216-4220 に記載され、例えば、R. J. Kaufman 及び P. A. Sharp (1982) Mol. Biol. 159:601-621に記載されているDHFR−選別可能な標識と併用されるdhfr−CHO−細胞を含む)、NSO−骨髄腫細胞、COS−細胞及びSP2−細胞が属する。抗体遺伝子をコードする組換え発現ベクターが、哺乳動物宿主細胞に導入される場合には、抗体が宿主細胞中に発現され又は有利に宿主細胞が増殖する培地に抗体が分泌されるまでの間、宿主細胞を培養することによって抗体は製造される。タンパク質の精製のための標準法を使用して、抗体を培地から取得することができる。
【0122】
完全抗体の一部、例えば、Fab−断片又はscFv−分子を製造するために、同様に宿主細胞を使用することができる。当然、前記の方法の変法は発明に属する。例えば、本発明による抗体の軽鎖又は重鎖(しかし、両方ではない)をコードするDNAで宿主細胞をトランスフェクションすることは所望に値し得る。関与抗原の結合のためには必要ではない軽鎖又は重鎖が存在する場合には、そのような軽鎖又はそのような重鎖又は両方をコードするDNAを、組換えDNA−技術によって、部分的に又は完全に除去することができる。そのような切端DNA−分子によって発現される配列も、本発明による抗体に属する。更に、1本の重鎖及び1本の軽鎖が本発明による抗体であり、かつ他の重鎖及び軽鎖が関与抗原以外の抗原の特異性を有する二官能抗体は、本発明による抗体を第二の抗体と標準化学的方法で架橋結合させることによって製造され得る。
【0123】
本発明による抗体又はその抗原−結合部分の組換え発現のための有利な系において、抗体重鎖も抗体軽鎖もコードする組換え発現ベクターを、燐酸カルシウム−促進トランスフェクションによって、dhfr−CHO−細胞に導入させる。遺伝子の強い転写を引き起こすために、組換え発現ベクター内で、抗体重鎖及び抗体軽鎖の遺伝子を、各々、調節CMV−エンハンサー/AdMLP−プロモーター要素と操作的に結合させる。組換え発現ベクターは、メトトレキセート−選別/増幅を使用することによって、ベクターとトランスフェクションするCHO−細胞を選別し得るDHFR−遺伝子も有する。選別された形質転換宿主細胞を培養し、そうして抗体重鎖及び抗体軽鎖を発現させ、完全抗体を培地から取得する。標準分子生物学的技術を使用して、組換え発現ベクターを製造し、宿主細胞をトランスフェクションし、形質転換体を選別し、宿主細胞を培養しかつ抗体を培地から取得することができる。即ち、本発明は、本発明による宿主細胞を本発明による組換え抗体が合成されるまで好適な培地中で培養することによる、本発明による組換え抗体の合成法に関する。この方法は、更に、組換え抗体を培地から単離することも包含し得る。
【0124】
ファージ−ディスプレーによる組換え抗体バンクのスクリーニングに選択的に、本発明による抗体を同定するために、大きな組合せバンクのスクリーニング用の当業者に公知の他の方法を使用することができる。選択的発現系の1方法では、例えば、Szostak 及びRobertsによるWO−98/31700及びRoberts, R. W. 及びSzostak, J. W. (1997) Proc. Natl. Acad. Sci. USA 94:12297-12302に記載されているように、組換え抗体バンクをRNA−タンパク質−融合の形で発現させる。この系では、その3’−末端にピューロマイシン、ペプチジル受容体−抗生物質を有する合成mRNAの試験管内−翻訳によって、mRNA及びそれがコードするペプチド又はタンパク質の間に、共有融合を生成させる。即ち、mRNAの複合混合物(例えば、組合せバンク)をからの特異的mRNAは、コードされるペプチド又はタンパク質(例えば、抗体又はその一部分の)の特性により、例えば、本発明によるオリゴマー又はその誘導体への抗体又はその一部分の結合により富化される。そのようなバンクのスクリーニングから取得される、抗体又はその一部分をコードする核酸配列を、組換え手段によって前記の方法で発現させ(例えば、哺乳動物宿主細胞中で)かつ、更なる経過でmRNA−ペプチド−融合をスクリーニングし、ここで当初の1種以上の選択された配列に突然変異を導入させるか、又は前記の方法で組換え抗体の試験管内−親和性成熟のための他の方法を使用することによって、更に親和性成熟をさせることができる。
【0125】
生体内−及び試験管内−法の組合せ
生体内−及び試験管内法の組合せ、例えば、オリゴマー−又は誘導体結合抗体の生産を刺激するために、先ず本発明によるオリゴマー又はその誘導体を宿主動物における生体内で抗体−レパートリー上に作用させ、引続いて、1種以上の試験管内−技術による更なる抗体選別及び/又は抗体成熟(即ち、最適化)を実施する方法を使用することによって、本発明による抗体を同様に製造することができる。1実施態様により、先ず、抗原に対する抗体−応答を刺激するために、非−ヒト動物(例えば、マウス、ラット、ウサギ、ニワトリ、ラクダ、ヤギ又はその形質転換型又はキメラマウス)を本発明によるオリゴマー又はその誘導体で免疫化させ、引き続き、オリゴマー又は誘導体の作用によって生体内で刺激されたリンパ球からの免疫グロブリン配列の使用下にファージ−ディスプレー−抗体バンクを製造し、かつスクリーニングさせる組合せ方法が包含される。この組合せ方法の第1段階は、生体内−方法に関連する前記の方法で実施され得るが、この方法の第2段階は、試験管内−方法に関連する前記の方法で実施され得る。刺激されたリンパ球から製造されたファージ−ディスプレー−バンクを、引き続き、試験管内−スクリーニングすることを伴う、非−ヒト動物の超免疫の有利な方法には、BioSite Inc.,により記載されている方法(例えば、WO98/47343、WO91/17271、US−特許第5427908及びUS−特許第5580717参照)が属する。
【0126】
もう1つの実施態様により、先ず、オリゴマー又はその誘導体に対する抗体応答を刺激するために、非−ヒト動物(例えば、マウス、ラット、ウサギ、ニワトリ、ラクダ、ヤギ又はノックアウト−及び/又はその形質転換型又はキメラマウス)を本発明によるオリゴマー又はその誘導体で免疫化させ、ハイブリドーマ(例えば、免疫化動物から製造される)をスクリーニングすることによって、所望の特異性を有する抗体を生産するリンパ球を選別する組合せ法が包含される。抗体又は単一ドメイン抗体の遺伝子を、選別されたクローンから分離させ(標準法、例えば、逆転写酵素−ポリメラーゼ連鎖反応により)、試験管内−親和性成熟させ、それによって、1種以上の選別された抗体の結合特性を改善することができる。この方法の第1段階は生体内−法に関連する前記の方法で実施され得るが、この方法の第2段階は試験管内−法に関連する前記の方法で、殊に、試験管内−親和性成熟法、例えば、WO−97/29131及びWO−00/56772に記載されている方法によって実施され得る。
【0127】
もう1つの組合せ方法では、リンパ球−抗体−選別法(SLAM)として当業者に公知であり、US−特許第5627052、WO−92/02551及びBabcock, J. S. et al. (1996) Proc. Natl. Acad. Sci. USA 93:7843-7848に記載されている方法を使用することによって、単離された単一リンパ球から組換え抗体を生成させる。この方法では、オリゴマー又はその誘導体に対する抗体応答を刺激するために、先ず、非−ヒト動物(例えば、マウス、ラット、ウサギ、ニワトリ、ラクダ、ヤギ又はその形質転換型又はキメラマウス)を、生体内で、本発明によるオリゴマー又はその誘導体で免疫化させ、次いで、抗原特異的溶血斑検定を使用して、関与する抗体を分泌する単一細胞を選別する。そのために、オリゴマー又はその誘導体、又は関与の構造的類似配列をヒツジ−赤血球に結合させることができ、この際、リンカー、例えば、ビオチンを使用し、それによって好適な特異性を有する抗体を分泌する単一細胞が、溶血斑検定の使用下に同定され得る。関与抗体を分泌する細胞の検定に続いて、細胞から軽鎖及び重鎖の可変領域のcDNAを逆転写酵素−PCRによって取得し、この可変領域を、好適な定常免疫グロブリン領域(例えば、ヒト定常領域)と関連して、哺乳動物宿主細胞、例えば、COS−又はCHO−細胞に発現させることができる。生体内で選別されたリンパ球から由来する増幅された免疫グロブリン配列でトランスフェクションされた宿主細胞を、次いで、所望の特異性を有する抗体を発現させる細胞を分離するために、例えば、トランスフェクションさせた細胞を拡散させることによって、もう1つの試験管内−分析及び−選別させることができる。増幅された免疫グロブリン配列を、更に、試験管内で扱うことができる。
【0128】
前記の方法により、本発明によるオリゴマー又はその誘導体のための、及び構造的に類似の合成配列のための特異性を有する抗体を製造することもできる。そのために、
i)本発明によるオリゴマー又はその誘導体及び構造的に類似の合成配列に共通である構造的特徴を包含する抗原を製造する;
ii)抗原を抗体−レパートリー上に作用させる;かつ
iii)2種の構造的に類似の配列に結合する抗体をレパートリーから選別し、それによって所望の特異性を有する抗体を得る。
【0129】
前記の方法により得られるオリゴマー特異的抗体は、アミロイド−β関連の痴呆症の治療剤を製造するための、又はアミロイド−β関連の痴呆症の診断用組成物を製造するためのその使用と全く同様に本発明の目的である。
【0130】
本発明により得られる抗体には、殊に、前記の方法で得ることができる抗血清が属する。これは全血清、即ち、宿主から得られる凝固可能な細胞成分の分離後の血液、又は、殊に免疫グロブリン−画分及び有利にオリゴマー認識免疫グロブリン−画分が富化されているこの血清の画分であってよい。この種類の画分は、抗体−精製と関連して前記された方法で得られ得る。
【0131】
本発明による抗血清はポリクローナルであり、即ち、これは様々な特異性の、通例様々な部類及び下位部類の抗体を含有し、通例、全てのL−連鎖−イソ型が存在していて、数種のタンパク質エピトープが知られている。
【0132】
免疫原として本発明による異なるオリゴマーを使用する場合には、本発明による抗血清は、通例、架橋結合性である。
【0133】
もう1つの観点により、本発明により得られる抗体には、モノクローナル抗体、殊に、キメラ及びヒト化抗体、及びそのオリゴマー結合断片が属する。
【0134】
本発明の目的は、本発明によるオリゴマー又はその誘導体に特異的に結合するタンパク質及び殊に抗体、即ち、本発明によるオリゴマー又はその誘導体の特異性を有する抗体である。本発明の目的は、同様に、このタンパク質又は抗体の一部、殊に、その抗原結合部分、即ち、本発明によるオリゴマー又はその誘導体を結合するタンパク質−又は抗体部分である。
【0135】
本発明による抗体を有利に選別する場合には、それは本発明によるオリゴマー又はその誘導体への特異的結合のための一定の結合速度(例えば、高い親和性、低い解離、低いオフ−速度、強い中和活性)を有する。
【0136】
即ち、KD=10−6〜10−12Mの範囲の本発明によるオリゴマー又はその誘導体の親和性を有するタンパク質及び殊に抗体が有利である。Kd=10−8Mよりも高い親和性、Kd=10−9Mよりも高い親和性、Kd=10−10Mよりも高い親和性又はKd=10−11Mよりも高い親和性で結合する高親和性タンパク質及び殊に抗体が特に有利である。
【0137】
もう1つの観点により、他のAβ(1−42)−形、殊に、モノマーAβ(1−42)−タンパク質及び/又はモノマーAβ(1−40)−タンパク質を、比較的低い親和性、殊にKd=10−8Mよりも低い親和性で結合するタンパク質及び殊に抗体が有利である。
【0138】
従って、本発明により特に、モノマーAβ(1−42)−タンパク質及び/又はモノマーAβ(1−40)−タンパク質よりも高い親和性でオリゴマー又はその誘導体を結合するタンパク質及び殊に抗体が有利である。10、100又は1000の親和性比率が特に有利である。
【0139】
もう1つの観点により、本発明による抗体を、それがオリゴマー又はその誘導体をkoff−速度定数0.1s−1以下で結合するように選択することができる。速度定数koff1x10−2s−1以下、1x10−3s−1以下、1x10−4s−1以下、1x10−5s−1以下が記載順に有利になる。
【0140】
更に、本発明による抗体を、それが本発明によるオリゴマー又はその誘導体の活性、殊に神経毒活性を、1x10−6M以下のIC50で阻害するように選択することができる。1x10−7M以下、1x10−8M以下、1x10−9M以下、1x10−10M以下、又は1x10−11M以下の阻害定数IC50が記載順に有利になる。
【0141】
抗体とは、有利に、単離された抗体のことである。もう1つの観点により、抗体は中和抗体である。本発明による抗体には、モノクローナル抗体及び組換え抗体が属する。多数の実施態様により、抗体は、完全に単一種から由来するアミノ酸配列、例えば、ヒト抗体又はマウス抗体を包含することができる。他の実施態様により、抗体は、キメラ抗体又はCDR−グラフト−抗体又は他の型のヒト化抗体であってよい。
【0142】
"抗体"とは、4本のポリペプチド鎖、2本の重(H)鎖及び2本の軽(L)鎖から形成されている免疫グロブリン分子に関する。この連鎖は、通例、ジスルフィド−結合によって相互に結合されている。各重鎖は、重鎖の可変領域(ここでは、HCVR又はVHとして略される)及び重鎖の定常領域から組成している。重鎖の定常領域は、3つのドメインCH1、CH2及びCH3から形成される。各軽鎖は、軽鎖の可変領域(ここでは、LCVR又はVLとして略される)及び軽鎖の定常領域から組成している。軽鎖の定常領域はドメインCLから形成される。VH−及びVL−領域は、更に、相補性決定領域(相補性決定領域(Complementarity Determining Region)のCDR)として表示される超可変領域に細分され、かつ骨格領域(骨格領域(Frame Work Region)のFR)として表示される保存領域と混在していてよい。各VH−及びVL−領域は、N−末端からC−末端へ次の順序で配置されている3つのCDRs及び4つのFRsから形成される:FR1、CDR1、FR2、CDR2、FR3、CDR3、FR4。
【0143】
抗体の"抗原結合部分"(又は単に"抗体部分")とは、本発明によるオリゴマー又はその誘導体への特異性を有する抗体の1種以上の断片に関し、この際、1種以上の断片は、依然として、オリゴマー又はその誘導体を特異的に結合する能力を有する。抗体の抗原結合機能は、完全抗体の断片によって認められ得ることが示されている。結合断片の例には、抗体の概念"抗原結合部分"の意において、(i)Fab−断片、即ち、VL−、VH−、CL−及びCH1−ドメインから組成される一価の断片;(ii)F(ab')2−断片、即ち、ジスルフィド−橋を経て蝶番−領域で相互に結合した2つのFab−断片を包含する二価の断片;(iii)VH−及びCH1−ドメインから組成するFd−断片;(iv)抗体の単一アームのFL−及びVH−ドメインから組成するFv−断片;(v)VH−ドメイン又はVH、CH1、CH2、DH3又はVH、CH2、CH3から成るdAb−断片(Ward, et al. (1989) Nature 341:544-546);及び(vi)分離された相補性決定領域(CDR)が属する。Fv−断片の2つのドメイン、つまりVL及びVHが、別々の遺伝子によってコードされるとしても、これらは更に合成リンカーと組換え法の使用下に相互に結合され、それによって、これらを単一のタンパク質鎖として製造することができ、その中で、VL−及びVH−領域は一価の分子を形成するために集合することができる(単鎖−Fv(ScFv)として知られる;例えば、Bird et al. (1988) Science 242:423-426; 及びHuston et al. (1998) Proc. Natl. Acad. Sci. USA 85:5879-5883参照)。この種類の単鎖−抗体は、抗体の概念"抗原結合部分"によっても把握されるべきである。他の型の単鎖−抗体、例えば"ジアボデーズ(Diabodies)"も同様にこれに属する。ジアボデーズは、VH−及びVL−ドメインが単一のポリペプチド鎖上で発現される二価の二重特異的抗体であり、その際、2つのドメインが同一鎖上で集合し得るには短すぎる1つのリンカーが使用されるが、それによって、そのドメインが強制的に他の鎖の相補性ドメインと対にされ、2つの抗原結合部位が形成される(例えば、Holliger, P., et al. (1993) Proc. Natl. Acad. Sci. USA 90:6444-6448; Poljak, R. J. et al. (1994) Structure 2:1121-1123参照)。
【0144】
更に、抗体又はその抗原結合部分は、抗体又は抗体部分と1種以上の他のタンパク質又はペプチドとの共有又は非共有会合によって形成される、より大きな免疫接着分子の一部であってよい。そのような免疫接着分子には、テトラマーscFv−分子を製造するためのストレプトアビジン−コア領域の使用(Kipriyanov, S. M., et al. (1995) Human Antibodies und Hybridomas 6:93-101)及び二価のビオチニル化scFv−分子を作製するためのシステイン基、標識ペプチド及びC−末端ポリヒスチジン−タグの使用が属する((Kipriyanov, S. M., et al. (1994) Mol. Immunol. 31:1047-1058)。抗体部分、例えば、Fab及びF(ab')2−断片は、慣用の技術、例えば、パパイン又はペプシンでの消化を使用することによって、全抗体から製造され得る。更に、抗体、抗体部分及び免疫接着分子は、標準組換えDNA−技術を使用することによって得ることができる。"本発明によるオリゴマー又はその誘導体への特異性を有する分離抗体"は、様々な抗原特異性を有する他の抗体を実際に含まない、本発明によるオリゴマー又はその誘導体への特異性を有する抗体、要するに、殊に、前記のようなAβ(1−42)−タンパク質の他の型に特異的に結合する抗体を含まない抗体を意味する。
【0145】
"中和抗体"とは、一定の抗原へのその結合が抗原の生物学的活性の阻害を引き起こす抗体を意味する。この抗原の生物学活性の阻害は、抗原の生物学的活性のための1種以上の指標を測定することによって評価することができ、この際、好適な試験管内−又は生体内−検定を使用する。
【0146】
"モノクローナル抗体"とは、ハイブリドー標識ら由来する抗体を意味する(例えば、ハイブリドーマ技術、例えば、Kohler 及び Milsteinによる標準ハイブリドーマ法により製造されるハイブリドー標識ら分泌される抗体)。従って、ハイブリドー標識ら由来し、本発明によるオリゴマー又はその誘導体への特異性を有する抗体は、モノクローナル抗体として表示される。
【0147】
"組換え抗体"とは、組換え手段で製造され、発現され、生成され又は分離される抗体、例えば、宿主細胞にトランスフェクションされた組換え発現ベクターの使用下に発現される抗体;組換え組合せ抗体バンクから分離される抗体;ヒト免疫グロブリン遺伝子によって形質転換された動物(例えば、マウス)から分離される抗体(例えば、Taylor, L. D., et al. (1992) Nucl. Acids. Res. 20:6287-6295参照);又は一定の免疫グロブリン−遺伝子配列(ヒト免疫グロブリン−遺伝子配列)を、他のDNA−配列と集合させる何か別の方法で、製造され、発現され、生成され又は分離される抗体に関する。組換え抗体には、例えば、キメラ、CDR−グラフト−及びヒト化抗体が属する。
【0148】
"ヒト抗体"とは、その可変及び定常領域が、例えば、Kabat et al. (Kabat, et al. (1991) Sequences of Proteins of Immunological Interest, Fifth Edition, U. S. Department of Health und Human Services, NIH Publication Nr. 91-3242参照)により記載されているヒト生殖系列の免疫グロブリン配列に相応し、又はそれから由来する抗体に関する。当然、本発明によるヒト抗体は、例えば、CDRs及び殊にCDR3に、ヒト生殖系列−免疫グロブリン配列によってコードされない(例えば、試験管内でランダム又は部位特異的な突然変異誘発によって又は生体内で体細胞突然変異によって導入される突然変異)アミノ酸基を包含し得る。本発明による組換えヒト抗体は可変領域を有し、ヒト生殖系列の免疫グロブリン配列から由来する定常領域も包含する(Kabat, et al. (1991) Sequences of Proteins of Immunological Interest, Fifth, Edition, U.S. Department of Health und Human Services, NIH Publication Nr. 91-3242参照)。一定の実施態様により、この種類の組換えヒト抗体を、当然、試験管内−突然変異誘発(又は、ヒトIg−配列による形質転換性動物を使用する場合には、体細胞生体内突然変異誘発)させ、従って、組換え抗体のVH−及びVL−領域のアミノ酸配列は、それがヒト生殖系列のVH−及びVL−配列と関連している又はそれから由来するとはいえ、生体内ヒト抗体−生殖系列レパートリー内に自然には存在しない配列である。一定の実施態様により、この種類の組換え抗体は、選別的突然変異誘発又は復帰突然変異、又は両方の結果である。
【0149】
"復帰突然変異"とは、ヒト抗体の体細胞突然変異アミノ酸の若干又は全部を、相同生殖系列抗体配列の相応する生殖系列基と交代させる方法に関する。本発明によるヒト抗体の重鎖及び軽鎖の配列を、別々に、最大の相同性を有する配列を同定するために、VBASE−データバンク中の生殖系列配列と比較する。そのような偏向アミノ酸をコードする限定ヌクレオチド位置で突然変異させることによって、本発明によるヒト抗体中の刷込みを生殖系列配列上に復帰させる。この方法で復帰突然変異の候補として同定される各アミノ酸の抗原結合に対する直接的又は間接的重要性が調査されるべきであり、突然変異後に所望のヒト抗体特性を侵害するアミノ酸は、完成のヒト抗体中に一緒に包含されるべきではない。復帰突然変異のためのアミノ酸の数をできるだけ少なく保つために、隣接の生殖系列配列とは偏向するが、第二の生殖系列配列の相応するアミノ酸配列と同一であるアミノ酸位置は不変のままであってよいが、第二の生殖系列配列は、本発明によるヒト抗体配列と、当該アミノ酸の両側上の少なくとも10個及び有利に12個のアミノ酸で同一である又は共−直線性であることが前提である。復帰突然変異は、抗体最適化の任意段階で行なうことができる。
【0150】
"キメラ抗体"とは、種からの重鎖及び軽鎖の可変領域の配列を包含するが、その中でVH及び/又はVLからの1つ以上のCDR−領域の配列が他の種のCDR−配列と交代している抗体、例えば、マウスからの重鎖及び軽鎖の可変領域を有し、その中で1つ以上のマウス−CDRs(例えば、CDR3)がヒトCDR−配列と交代している抗体に関する。
【0151】
"ヒト化抗体"とは、非ヒト種(例えば、マウス、ラット、ウサギ、ニワトリ、ラクダ、ヤギ)からの重鎖及び軽鎖の可変領域の配列を包含するが、更にもっと"ヒト類似性"にするために、即ち、ヒト生殖系列の可変配列を類似性にするために、少なくとも一部のVH−及び/又はVL−配列が変更されている抗体に関する。ヒト化抗体の種類は、相応する非ヒトCDR−配列を交代させるために、ヒトCDR−配列が非ヒトVH−及びVL−配列中に導入されているCDR−グラフト抗体である。
【0152】
抗体の結合反応速度を測定するための方法は、表面プラスモン共鳴による。"表面プラスモン共鳴"とは、タンパク質濃度の変化をバイオセンサーマトリックスで検出することによって、生体特異的相互作用を分析することができる光学的現象に関し、この際、例えば、BlAcore−Systemを使用する(Pharmacia Biosensor AB, Uppsala, Sweden und Piscataway, NJ)。他の記載については、Joensson, U., et al. (1993) Ann. Biol. Clin. 51:19-26; Joensson, U., et al. (1991) Biotechniques 11:620-627; Johnsson, B., et al. (1995) J. Mol. Recognit. 8:125-131; 及びJohnnson, B., et al. (1991) Anal. Biochem. 198:268-277が参照される。
【0153】
"Koff "とは、抗体/抗原−複合体からの抗体の解離についてのオフ−速度定数に関する。
【0154】
"Kd"とは、一定の抗体−抗原−相互作用の解離定数に関する。
【0155】
本発明による抗体の結合親和性は、標準試験管内−免疫検定、例えば、ELISA−又はBlAcore−分析を使用することによって評価され得る。
【0156】
抗体の他に、タンパク質は、T−細胞受容体から由来する分子又はT−細胞受容体から由来する受容体ドメイン、又は受容体ドメインと免疫グロブリンのFc−部分との融合タンパク質であってよい。
【0157】
本発明の目的は、本発明によるタンパク質及び殊に本発明による抗体及び場合により製薬学的に認容性の賦形剤を含有する製薬学的製剤(組成物)でもある。本発明による製薬学的組成物は、更に少なくとも1種の付加的治療剤、例えば、本発明による抗体がその軽減のために使用可能である病気の治療のための1種以上の付加的治療剤を含有することができる。本発明による抗体を、例えば、本発明によるオリゴマーに結合させる場合には、製薬学的組成物は、更に、オリゴマーの活性が重要である病気の治療のために使用可能である1種以上の付加的な治療剤を含有することができる。
【0158】
製薬学的に認容性の賦形剤には、生理学的に適合性である限り、全ての溶剤、分散媒体、被覆剤、抗菌剤及び抗菌類剤、等張剤及び吸収遅延剤等が属する。製薬学的に認容性の賦形剤には、例えば、水、食塩溶液、燐酸塩−緩衝化食塩溶液、デキストロース、グリセリン、エタノール等、及びその組合せが属する。多くの場合には、等張剤、例えば、糖、ポリアルコール、例えば、マンニトール又はソルビトール、又は塩化ナトリウムを併用することが有利である。製薬学的認容性賦形剤は、更に、抗体の品質保持又は有効性を高める少量の補助物質、例えば、湿潤剤又は乳化剤、保存剤又は緩衝液を含有することができる。
【0159】
製薬学的組成物は、例えば、腸管外投与に好適であってよい。この際、抗体は抗体含量0.1〜250mg/mlを有する注射可能な溶液として調製されることが有利である。注射可能な溶液は、投与形として、液体又は凍結乾燥形で、フリントガラス又はバイアル、アンプル又は充填注射器中に調製されていてよい。緩衝液はL−ヒスチジン(1〜50ミリモル、有利に5〜10ミリモル)を含有し、pH−値5.0〜7.0、有利に6.0を有することができる。他の好適な緩衝液には、コハク酸ナトリウム−、クエン酸ナトリウム−、燐酸ナトリウム−又は燐酸カリウム−緩衝液が属するが、これに限定されるものではない。溶液の張度を濃度0〜300ミリモル(液状投与形については、有利に150ミリモル)に調整するために、塩化ナトリウムを使用することができる。凍結乾燥形のために、凍結保護剤、例えば、スクロース(例えば、0〜10%、有利に0.5〜1.0%)を一緒に含有することができる。他の好適な凍結保護剤には、トレハロース及びラクトースが属する。凍結乾燥投与形のために、充填剤、例えば、マンニトール(例えば、1〜10%、有利に2〜4%)を一緒に含有することができる。安定剤、例えばL−メチオニン(例えば、51〜50ミリモル、有利に5〜10ミリモル)を液状でも、凍結乾燥投与形でも使用することができる。他の好適な充填剤には、グリシン及びアルギニンが属する。同様に、界面活性剤、例えば、ポリソルベート−80(例えば、0〜0.05%、有利に0.005〜0.01%)を使用することができる。他の界面活性剤には、ポリソルベート−20及びBRIJ−界面活性剤が属する。
【0160】
本発明による組成物は、多数の形を取ることができる。これには、液状、半固形及び固形の投与形、例えば、液状溶液(例えば、注射可能な及び注入可能な溶液)、分散液又は懸濁液、錠剤、丸剤、粉末、リポソーム及び座薬が属する。有利な形は、意図される投与方法及び治療的使用に依存する。典型的には、注射可能な又は注入可能な溶液の形の組成物、例えば、ヒトの受動免疫のために他の抗体との類似性を有する組成物が有利である。有利な投与方法は、腸管外(例えば、静脈内、皮下、腹膜内、筋肉内)である。有利な実施態様により、抗体は静脈内注入又は注射によって投与される。もう1つの有利な実施態様により、抗体は、筋肉内又は皮下注射によって投与される。
【0161】
治療的組成物は、典型的には無菌で、製造ー及び貯蔵条件下に安定性でなければならない。組成物は溶液、微細エマルジョン、分散液、リポソーム又は作用物質高濃度に好適な他の次数化構造として組成され得る。注射可能な無菌溶液は、活性化合物(即ち、抗体)を必要量で好適な溶剤中に、場合により必要に応じて1種以上の前記内容物の組合せと一緒に装入し、引き続き、無菌濾過することによって製造され得る。分散液は、通例、基礎分散媒体及び場合により他の必要な内容物を含有する無菌ビヒクル中に活性化合物を入れることによって製造される。注射可能な無菌溶液を製造するための凍結乾燥無菌粉末の場合には、真空乾燥及び噴霧乾燥が有利な製法であり、その方法で、前以て無菌濾過した溶液から活性内容物及び場合により他の所望される内容物の粉末が得られる。溶液の適切な流動性は、例えば、被覆剤、例えば、分散液の場合には、必要な粒度を維持するレシチンを使用し、又は界面活性剤を使用することによって維持され得る。注射可能な組成物の延滞吸収は、吸収を遅延させる薬剤、例えば、モノステアレート塩及びゼラチンを組成物中に一緒に入れることによって達成され得る。
【0162】
本発明による抗体は、当業者に公知である多数の方法で投与され得るが、多数の治療的使用には、皮下注射、静脈内注射又は注入が有利な投与方法である。当業者は、投与の方法及び/又は種類は所望の結果に依ることを周知である。一定の実施態様により、活性化合物を、化合物を急速な放出に対して保護する賦形剤と一緒に、即ち、例えば、調節放出の組成物を製造することができ、それには、インプラント、経皮プラスター及び微細カプセル投与系が属する。生物学的に分解可能な生物適合性ポリマー、例えば、エチレンビニルアセテート、ポリ酸無水物、ポリグリコール酸、コラーゲン、ポリオルトエステル及びポリ乳酸を使用することができる。そのような組成物の製法は、当業者に一般に公知であり、例えば、Sustained und Controlled Release Drug Delivery Systems, J.R.Robinson, ed., Marcel Dekker, Inc., New York, 1978が参照される。
【0163】
一定の実施態様により、例えば、不活性希釈剤又は同化可能な食可能賦形剤中の本発明による抗体を経口投与することができる。抗体(及び所望の場合には、他の内容物)を硬質−又は軟質ゼラチンカプセルに包入させ、打錠して錠剤にし又は食物に直接添加することもできる。治療的経口投与のために、抗体を賦形剤と混合させ、嚥下可能な錠剤、バッカル錠剤、カプセル剤、エリキシル、懸濁液、シロップ等の形で使用することができる。本発明による抗体を、腸管外投与とは別の方法により投与すべき場合には、その不活性化を阻害する材料から被覆を選択することが必要であり得る。
【0164】
本発明による抗体は、それが結合する本発明によるオリゴマー又はその誘導体の活性を試験管内でも生体内でも中和することができる。従って、抗体は、本発明によるオリゴマー又はその誘導体の活性を阻害するために、例えば、オリゴマー又はその誘導体を含有する細胞培養液中又はオリゴマー又はその誘導体が存在しているヒト個体又は哺乳動物中で使用される。1実施態様により、本発明の目的は、本発明によるオリゴマー又はその誘導体の活性の阻害法であり、この際、本発明による抗体をオリゴマー又はその誘導体に、オリゴマー又はその誘導体の活性が阻害されるように作用させることができる。この際、活性は、例えば試験管内で阻害され得る。例えば、本発明による抗体は、本発明によるオリゴマー又はその誘導体を含有する又は含有すると考えられる細胞培養液に添加して、培養液中のオリゴマー又はその誘導体を阻害することができる。選択的に、オリゴマー又はその誘導体の活性を、1個体において生体内で阻害することができる。
【0165】
即ち、本発明のもう1つの目的は、アミロイド−β−タンパク質が関与していて、殊に本発明によるオリゴマー又はその誘導体の活性が関与する病気にかかっている個体における本発明によるオリゴマー又はその誘導体の活性の阻害法に関する。この方法は、抗体が結合するオリゴマー又はその誘導体の活性を阻害する目的で、少なくとも1種の本発明による抗体を個体に投与することを包含する。個体は有利に人間である。本発明による抗体はヒト個体に治療目的のために投与することができる。更に、本発明による抗体を非ヒト哺乳動物に獣医的目的のために、又は一定の病気の動物モデルの範囲で投与することができる。その種類の動物モデルは、本発明による抗体の治療的有効性を評価するために有益である(例えば、投与量及び投与の時間経過を試験するために)。
【0166】
本発明によるオリゴマー又はその誘導体が関与する病気には、殊に、その発生及び/又は経過に本発明によるオリゴマー又はその誘導体が関与している病気が属する。従って、殊に、本発明によるオリゴマー又はその誘導体が、証明的に又は推測的に病気の病態生理学に起因する、又は病気の発生及び/又は経過に寄与する因子である病気が考えられる。従って、本発明によるオリゴマー又はその誘導体の活性の阻害が、病気の症状及び/又は進行を緩和することができる病気がそれに属する。そのような病気は、例えば、一定の病気にかかっている個体の体液中の本発明によるオリゴマー又はその誘導体の高められた濃度によって証明される(例えば、血清、血漿、CSF、ウリン等中の高められた濃度)。このことは、例えば、本発明による抗体を使用することによって検証することができる。本発明によるオリゴマー又はその誘導体は、神経変性要素、認識欠如、神経毒性要素及び炎症性要素が関与している多数の病気と関連している病変において決定的な役割を果たしている。
【0167】
本発明による抗体は、前記の病気の治療に使用可能である1種以上の付加的な治療剤と一緒に投与され得る。
【0168】
本発明の製薬学的組成物は、通例、少なくとも1種の本発明による抗体の治療的有効量又は予防的有効量を含有する。所望の処置によって、例えば、治療的処置が所望されるか又は予防的処置が所望されるかどうかによって、投与計画を選択し、適合させる。例えば、単一用量、時間配分で数回の分別用量又は治療状況の要求により増加又は減少用量を投与することができる。投与を容易にさせ、投与の一様性を保証するために、殊に、腸管外用組成物を単一用量形で組成させることが有利である。
【0169】
本発明による抗体の治療的又は予防的有効量は、0.1〜20mg/kg及び有利に1〜10mg/kgの範囲であってよいが、これに限定されるものではない。この量は、当然、緩和すべき状態の種類及び重大性によって変化され得る。
【0170】
抗体の診断的使用の範囲で、定性又は定量的な特異的オリゴマー−測定が、殊に、病気に関連するアミロイド−β(1−42)−型の診断に用いられる。この関係における特異性は、充分な感受性を有する一定のオリゴマー又はオリゴマー−混合物を検出し得る可能性を意味する。本発明による抗体は、10ng/試料mlよりも少ない、有利に1ng/試料mlよりも少ない、特に有利に100pg/試料mlよりも少ない感受性を有利に有する。従って、少なくとも、試料1mlに対する各々前記のオリゴマー濃度を、有利により少ない濃度も、本発明による抗体で検出できることが考えられる。
【0171】
測定は免疫学的に行なわれる。これは、原則的には、抗体を使用する各分析的又は診断的試験法で行なわれ得る。この方法には、凝集−及び沈殿−法、免疫検定法、免疫組織化学法及び免疫ブロット−法、例えば、ウエスタン−ブロット法又はドットブロット−法が属する。生体内−法には、例えば、画像法も属する。
【0172】
免疫検定法における使用が有利である。競合的免疫検定、即ち、抗体結合について抗原及び標識化抗原(トレーサー)を競合させることも、サンドイッチ−免疫検定法、即ち、抗原への特異的抗体の結合を第二の、大抵は標識化された抗体で確認することも好適である。これらの検定法は、均一であっても(即ち、固相及び液相へ分離しない)、不均一であってもよい(即ち、例えば、固相結合抗体を介して結合標識体を非結合体から分離することができる)。様々な不均一及び均一免疫検定−形は、標識化及び測定法により決められる部類、例えば、RIAs(放射線免疫検定法)、ELISA(固相酵素免疫検定法)、FIA(蛍光−免疫検定法)、LIA(発光−免疫検定法)、TEFIA(時間的分解FIA)、IMAC(免疫活性化)、EMIT(酵素多重化免疫試験法)、TIA(ターボジメトリック免疫検定法)、I−PCR(免疫−PCR)に配属される。
【0173】
本発明によるオリゴマー−測定には、競合的免疫検定法が有利である。この際、標識化オリゴマー(トレーサー)は、使用抗体への結合について、試料の測量すべきオリゴマーと競合する。置換されたトレーサーの量から、標準曲線によって、試料中の抗原量、即ち、オリゴマー量を測定することができる。
【0174】
この目的に使用される標識は、酵素が有利であることが判明した。例えば、ペルオキシダーゼ、殊に西洋わさび−ペルオキシダーゼ、アルカリ性ホスファターゼ及びβ−D−ガラクトシダーゼをベースとする系を使用することができる。その変換を、例えば、測光的に追跡し得る特異的基質がこの酵素のために使用される。好適な基質系は、アルカリ性ホスファターゼについては、p−ニトロフェニルホスフェート(p−NPP)、5−ブロム−4−クロル−3−インドリルホスフェート/ニトロブルー−テトラゾリウム(BCIP/NPT)、ファースト−レッド/ネフトール−AS−TS−ホスフェート;ペルオキシダーゼについては、2,2−アジノ−ビス−(3−エチルベンズチアゾリン−6−スルホン酸)(ABTS)、o−フェニレンジアミン(OPT)、3,3’,5,5’−テトラメチルベンジジン(TMB)、o−ジアニシジン、5−アミノサリチル酸、3−ジメチルアミノ安息香酸(DMAB)及び3−メチル−2−ベンゾチアゾリンヒドラゾン(MBTH);β−D−ガラクトシダーゼについては、o−ニトロフェニル−β−D−ガラクトシド(o−NPG)、p−ニトロフェニル−β−D−ガラクトシド及び4−メチルウムベリ(umbelli)フェニル−β−D−ガラクトシド(MUG)をベースとする。多くの場合において、これらの基質系は既製形で、例えば、他の試薬、例えば、有利に緩衝液等も含有し得る錠剤の形で、市販で得られる。
【0175】
トレーサーとして、標識化オリゴマーが使用される。この意において、一定のオリゴマーの測定には、測定すべきオリゴマーを標識化し、トレーサーとして使用することができる。
【0176】
トレーサーの製造のためのオリゴマーへの標識の結合は、自体公知の方法で行なうことができる。本発明によるオリゴマーの誘導体化のための前記の実施が同様に引用される。更に、タンパク質の複合のために有利に変性された一連の標識、例えば、ビオチン−、アビジン−、エキストラビジン−又はストレプトアビジン−複合酵素、マレイミド−活性化酵素等が得られる。これらの標識は、オリゴマー又は、必要に応じて、相応に誘導体化されたオリゴマーと直接反応してトレーサーを得ることができる。例えば、ストレプトアビジン−ペルオキシダーゼ−複合体を使用する場合には、これは先ずオリゴマーのビオチニル化を必要とする。相応することは逆の順序にも当てはまる。この目的についても、好適な方法は当業者に公知である。
【0177】
不均一免疫検定−型を選択する場合には、抗原−抗体−複合体を、分離のために、例えば、担体に結合した抗−イディオタイプ抗体、例えば、ウサギ−IgGに照準した抗体を介して担体に結合させることができる。相応する抗体を担持している担体、殊に、微小滴定プレートは公知であり、部分的に市販で得られる。
【0178】
本発明のもう1つの目的は、少なくとも1種の前記の抗体及び他の成分を有する免疫検定−セットである。この際、本発明によるオリゴマー−測定の実施のための手段の、通例、包装ユニットとしての集成装置が重要である。できるだけ簡単な取扱いのために、この手段は、有利に実際に使用完成されている。有利な装置は免疫検定をキット−型で提供する。キットは、通例、成分の別々の装置用の数個の容器を包含する。全成分は、使用完成された希釈液で、希釈用濃縮物として又は溶解又は懸濁用乾燥物質又は凍結乾燥物として準備されていてよい;単一成分又は全成分は凍結されている又は環境温度で使用するまで貯蔵されていてよい。血清は、例えば、−20℃でショック凍結され、従って、この場合には、免疫検定を使用前に有利に凍結温度で保持しなければならない。
【0179】
免疫検定に添加される他の成分は、免疫検定の種類に依る。通例、抗血清標準タンパク質、場合により必要なトレーサー及び対照血清と一緒に添加される。更に、有利に抗体を担持させた微小滴定プレート、例えば、基質の試験、洗浄又は反応のための緩衝液、及び酵素基質自体を添加することができる。
【0180】
実験室及び臨床における免疫検定の一般的原則及び助剤としての抗体の製造及び使用は、例えば、Antibodies, A Laboratory Manual (Harlow, E., and Lane, D., Ed., Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, 1988)にある。
【0181】
更に、本発明によるオリゴマーの集合を阻害し又はその解離を促進させる物質も重要である。この種類の物質は、前記のアミロイド−β−関連病、例えば、アルツハイマー病の治療に殊に可能な治療剤である。
【0182】
従って、本発明の目的は、物質又は物質混合物の特性付けのための方法でもあり、この際、
i)物質又は物質混合物を好適な方法で準備する;
ii)物質又は物質混合物を、少なくとも1種の本発明によるオリゴマー、その誘導体又は組成物に作用させる;及び
iii)物質又は物質混合物の一定部分が、オリゴマー、その誘導体又は組成物中に含有される少なくとも1種のオリゴマー又はその誘導体に結合しているかどうかを測定する。
【0183】
この方法は、本発明によるオリゴマーへの親和性を有する天然の結合相手、例えば、細胞表面抗原、殊に、受容体及び可溶性リガンド、例えば、一定のタンパク質及びメディエーターを同定するために、生物学的起源の混合物、例えば、細胞標本及び−抽出物に実施することもできる。
【0184】
物質の単なる結合に加えて、本発明によるオリゴマーとの相互作用及び本発明によるオリゴマーへの影響も本方法の目的であってよい。即ち、殊に、
物質が本発明によるオリゴマーに対するアミロイド−β−タンパク質の集合を変性させ、殊に、阻害させることができるかどうか;
物質が、本発明によるオリゴマーの解離を変性させ、殊に、促進させることができるかどうか;
本発明によるオリゴマーが、結合相手の機能変化を呼び起こし、例えば、受容体で共働的、部分的共働的、拮抗的又は反共働的作用を有するかどうかを測定することができる。
【0185】
この方法では、通例、試験管内−スクリーニング−法が重要であり、この方法で、多数の様々な物質から、将来の使用を展望して最も有望であると思われる物質を選別することができる。例えば、組合せ化学によって、多数のポテンシャル作用物質を包含する広範囲の物質バンクを設定することができる。所望の活性を有する物質による組合せ物質ライブラリーの精査は自動可能である。スクリーニング−ロボットは、有利に微小滴定プレート上に配置された単一検定の効率的な評価に用いられる。即ち、本発明は、スクリーニング−法、即ち、一次スクリーニング−法にも、二次スクリーニング−法にも関し、その際、有利に、次に記載した方法の少なくとも1法が使用される。数種の方法を使用する場合には、方法を時間的にずらして又は同時に、検査すべき物質の1試料及び同一試料又は異なる試料で行なうことができる。
【0186】
この種類の方法の特に効果的な実施技術は、作用物質スクリーニングの範囲で公知のシンチレーション近接検定(略称SPA)である。この検定法の実施のためのキット及び成分は、例えば、Amersham Pharmacia Biotech.で市販されて入手することができる。原則的に、溶解化又は膜結合受容体は、シンチレーション物質を含有する蛍光微小球上で固定化される。例えば、放射性リガンドが固定化受容体に結合する場合には、シンチレーション物質と放射性リガンドとの間に空間的近接が生じるので、シンチレーション物質は励起して発光する。
【0187】
この種類の方法のもう1つの特に効果的な実施技術は、作用物質スクリーニングの範囲で公知のフラッシュプレートR−技術である。この検定法の実施のためのキット及び成分は、例えば、NENR Life Science Productsで市販されて得られる。この原則は、同様にシンチレーション物質で被覆される微小滴定プレート(96又は384個の)に基づく。
【0188】
この方法により、オリゴマー、その誘導体又は相応する組成物中に含有される少なくとも1種のオリゴマー又はその誘導体に結合するリガンドとして同定可能な物質又は物質混合物からの一部は、アミロイド−β−関連病、殊に、痴呆症の治療剤を製造するための、又はアミロイド−β−関連病、殊に、痴呆症の診断用組成物を製造するためのその使用と全く同様に本発明の目的である。
【図面の簡単な説明】
【0189】
【図1】図1は、Aβ(1−42)−オリゴマーA−標本(線A);Aβ(1−42)−オリゴマーB−標本(線B);標準タンパク質(分子標識タンパク質、線C)のSDS−PAGEを示す
【図2】図2は、Aβ(1−42)−オリゴマーA−標本(線A);Aβ(1−42)−オリゴマーA−CL−標本(線A’);Aβ(1−42)−オリゴマーB−標本(線B);Aβ(1−42)−オリゴマーB−CL−標本(線B’);標準タンパク質(分子標識タンパク質、線C)のSDS−PAGEを示す
【図3】図3は、ビオチン−Aβ(1−42)−オリゴマーB−標本(線A);標準タンパク質(分子標識タンパク質、線B)のSDS−PAGEを示す
【図4】図4は、フルオレスセイン−Aβ(1−42)−オリゴマーB−標本(線A);標準タンパク質(分子標識タンパク質、線B)のSDS−PAGEを示す
【図5】図5は、Aβ(1−42)−凍結乾燥物を含有する溶液の、Aβ(1−42)−オリゴマーBを含有する標本と比較したゲル透過クロマトグラフィーを示す
【図6】図6は、Aβ(1−42)−オリゴマーB−標本(線A);標準タンパク質(分子標識タンパク質、線B)のNATIVE−PAGEを示す
【図7】図7は、(A)モノマーAβ(1−42)−タンパク質及び(B)Aβ(1−42)−オリゴマーA及び(C)Aβ(1−42)−オリゴマーの、ヒト神経芽細胞腫−細胞系IMR−32の表面への結合を示す
【図8】図8は、マウスの皮質ニューロンをAβ(1−42)−オリゴマーBで処理した後の神経毒作用(%)(この際、誤差バーは信頼区間95%に相応する)を示す
【図9】図9は、トリプシン(線2)、キモトリプシン(線3)、サーモリシン(線4)、エラスターゼ(線5)、パパイン(線6)で処理した又は未処理(線7)であったAβ(1−42)−標本;標準タンパク質(分子標識タンパク質、線1)のSDS−PAGEを示す
【図10】図10は、例6bからのAβ(1−42)−オリゴマーB−標本(欄1);例1aからのHFIP−処理したAβ(1−42)−モノマー(欄2);例15aからのサーモリシンで分解したAβ(1−42)−オリゴマーB−標本(欄3);例14aからのグルタルアルデヒドと架橋結合したAβ(1−42)−オリゴマーB−標本(欄4);M. P. Lambert et al., J. Neurochem. 79,595-605(2001))による4℃又は室温又は37℃で製造したADDL(欄5,6又は7);0.1%NH4OH中に溶かしたAβ(1−42)(欄8);例27からのAβ(1−42)−原線維−標本(欄9);及びPBS中で希釈したFa. SigmaのAPP(欄10)100ピコモル(pmol)(行A);10ピコモル(行B);1ピコモル(行C);0.1ピコモル(行D)又は0.01ピコモル(行E)と、a)モノクローナル抗体6E10;b)例25dからのポリクローナル抗血清(d1);c)例25cからのポリクローナル抗血清(c1);d)例25aからのポリクローナル抗血清(a1);及びe)例25aからのポリクローナル抗血清(a2)との反応性のドットブロットを示す
【図11】図11は、例15aからのAβ(20−42)−オリゴマーB−標本(線B);例15bからのAβ(12−42)−オリゴマーB−標本(線C);例6bからのAβ(1−42)−オリゴマーB−標本(線A);及び標準タンパク質(分子標識タンパク質、線D)のSDS−PAGEを示す
【図12】図12は、例6bからのAβ(1−42)−オリゴマーB−標本の、例14aからのAβ(1−42)−オリゴマーB−CL−標本に比較したゲル透過クロマトグラフィーを示す
【図13】図13は、モノマーAβ(1−42)−タンパク質(左側);Aβ(1−42)−オリゴマー(12−マー、A);及びタンパク質加水分解によって得られるAβ(12−42)−オリゴマー(12−マー、C)及びAβ(20−42)−オリゴマー(12−マー、B)の図解表示を示す
【図14】図14は、海馬断片へのAβ(1−42)−オリゴマーBの作用下に、時間に依存する興奮性シナプス後電位(fEPSP)を示す
【図15】図15は、海馬ラットニューロンへのAβ(1−42)−オリゴマーB(a)の、非特異的結合蛍光(b)に比較した結合を示す免疫蛍光画像を示す
【実施例】
【0190】
次に実施例につき本発明を詳説するが、この範囲に限定されるものではない。
【0191】
他の記載のない限り、本発明によるオリゴマーの濃度を、モノマーのAβ(1−42)−ポリペプチドのモルで表わす。概念β−アミロイド(1−42)−タンパク質は、概念アミロイド−β(1−42)−タンパク質に相応する。他の記載のない限り、使用したタンパク質(ポリペプチド)は、ヒト起源である。
【0192】
例1
a)ヒトAβ(1−42)の基本懸濁液の製造
ヒトβ−アミロイド(1−42)−タンパク質(略称:Aβ(1−42);ペプチド合成原料、凍結乾燥物、Fa. Bachem, Deutschland)2mgを、1,1,1,3,3,3−ヘキサフルオル−2−プロパノール800μl中に溶かし、エッペンドルフ容器中37℃で30分間恒温保持する。その後に、真空濃縮機(Speed Vac)中で蒸発乾固させる。残渣をDMSO88μlに入れ、この際、Aβ(1−42)−基本懸濁液5ミリモルが生じる。これを−20℃で保存することができる。
【0193】
b)ラット−Aβ(1−42)の基本懸濁液の製造
ラット−β−アミロイド(1−42)−タンパク質(略称:ラット−Aβ(1−42);ペプチド合成原料、凍結乾燥物、Fa. Bachem, Deutschland)2mgを、1,1,1,3,3,3−ヘキサフルオル−2−プロパノール800μl中に溶かし、エッペンドルフ容器中37℃で30分間恒温保持する。その後に、真空濃縮機(Speed Vac)中で蒸発乾固させる。残渣をDMSO88μlに入れ、この際、ラット−Aβ(1−42)−基本懸濁液5ミリモルが生じる。これを−20℃で保存することができる。
【0194】
例2
a)分子量15kDa及び20kDaを有するAβ(1−42)−オリゴマー[Aβ(1−42)−オリゴマーA]の製造;SDSの使用
例1aからの基本溶液60μlにPBS−緩衝液(燐酸ナトリウム20ミリモル、NaCl140ミリモル、pH7.4)690μlを加え、2%のドデシル硫酸ナトリウム(SDS)−溶液75μlで0.2%SDSの含量に調整する。その後に、37℃で5時間恒温保持し、10000gで10分間遠心分離する。このAβ(1−42)−オリゴマーA−標本(Aβ(1−42)約400マイクロモル)を−20℃で保存することができる。
【0195】
b)分子量15kDa及び20kDaを有するラット−Aβ(1−42)−オリゴマー[ラット−Aβ(1−42)−オリゴマーA]の製造;SDSの使用
例1bからの基本溶液60μlにPBS−緩衝液(燐酸ナトリウム20ミリモル、NaCl140ミリモル、pH7.4)690μlを加え、2%のドデシル硫酸ナトリウム(SDS)−溶液75μlで0.2%SDSの含量に調整する。その後に、37℃で5時間恒温保持し、10000gで10分間遠心分離する。このラット−Aβ(1−42)−オリゴマーA−標本(ラット−Aβ(1−42)約400マイクロモル)を−20℃で保存することができる。
【0196】
c)分子量15kDa及び20kDaを有するAβ(1−42)−オリゴマー[Aβ(1−42)−オリゴマーA]の製造;ラウリル酸の使用
例1aからの基本溶液60μlにPBS−緩衝液(燐酸ナトリウム20ミリモル、NaCl140ミリモル、pH7.4)690μlを加え、5%のラウリル酸−溶液75μlで0.5%ラウリル酸の含量に調整する。その後に、37℃で5時間恒温保持し、10000gで10分間遠心分離する。このAβ(1−42)−オリゴマーA−標本(Aβ(1−42)約400マイクロモル)を−20℃で保存することができる。
【0197】
d)分子量15kDa及び20kDaを有するAβ(1−42)−オリゴマー[Aβ(1−42)−オリゴマーA]の製造;油酸の使用
例1aからの基本溶液60μlにPBS−緩衝液(燐酸ナトリウム20ミリモル、NaCl140ミリモル、pH7.4)690μlを加え、5%の油酸−溶液75μlで0.5%油酸の含量に調整する。その後に、37℃で5時間恒温保持し、10000gで10分間遠心分離する。このAβ(1−42)−オリゴマーA−標本(Aβ(1−42)約400マイクロモル)を−20℃で保存することができる。
【0198】
e)分子量15kDa及び20kDaを有するAβ(1−42)−オリゴマー[Aβ(1−42)−オリゴマーA]の製造;ラウロイルサルコシンの使用
例1aからの基本溶液60μlにPBS−緩衝液(燐酸ナトリウム20ミリモル、NaCl140ミリモル、pH7.4)690μlを加え、5%のラウロイルサルコシン−溶液75μlで0.5%ラウロイルサルコシンの含量に調整する。その後に、37℃で5時間恒温保持し、10000gで10分間遠心分離する。このAβ(1−42)−オリゴマーA−標本(Aβ(1−42)約400マイクロモル)を−20℃で保存することができる。
【0199】
例3
a)分子量15kDa及び20kDaを有するAβ(1−42)−オリゴマー[Aβ(1−42)−オリゴマーA]の製造のための選択的製法
ヒトβ−アミロイド(1−42)−タンパク質(略称:Aβ(1−42);ペプチド合成原料、凍結乾燥物、Fa. Bachem, Deutschland)1mgを、HCl−水溶液10ミリモル220μl中に入れ、室温で10分間恒温保持する。不溶成分を10000gで5分間遠心分離する。上澄液(Aβ(1−42) 1ミリモル)は、Aβ(1−42)−タンパク質を含有し、次のように更に処理される:
上澄液1μlにPBS−緩衝液9μl及び2%のSDS−溶液1μlを加え、37℃で16時間恒温保持する。そうして得られるhAβ(1−42)−オリゴマーA−標本(100マイクロモル)を−20℃で保存することができる。
【0200】
b)分子量15kDa及び20kDaを有するラット−Aβ(1−42)−オリゴマー[ラット−Aβ(1−42)−オリゴマーA]の製造のための選択的製法
ラット−β−アミロイド(1−42)−タンパク質(略称:ラット−Aβ(1−42);ペプチド合成原料、凍結乾燥物、Fa. Bachem, Deutschland)1mgを、HCl−水溶液10ミリモル220μl中に入れ、室温で10分間恒温保持する。不溶成分を10000gで5分間遠心分離する。上澄液(ラット−Aβ(1−42) 1ミリモル)は、ラット−Aβ(1−42)−タンパク質を含有し、次のように更に処理される:
上澄液1μlにPBS−緩衝液9μl及び2%のSDS−溶液1μlを加え、37℃で16時間恒温保持する。そうして得られるラット−Aβ(1−42)−オリゴマーA−標本(100マイクロモル)を−20℃で保存することができる。
【0201】
例4
a)分子量15kDa及び20kDaを有するAβ(1−42)−オリゴマー[Aβ(1−42)−オリゴマーA]の製造のための選択的製法
ヒトβ−アミロイド(1−42)−タンパク質(略称:Aβ(1−42);ペプチド合成原料、凍結乾燥物、Fa. Bachem, Deutschland)1mgを、1%SDS/H2O44μl中に溶かす(Aβ(1−42) 5ミリモル)。この溶液5μlにPBS40μl及び2%SDS5μlを加え、37℃で16時間恒温保持する。不溶成分を10000gで5分間遠心分離する。そうして得られるAβ(1−42)−オリゴマーA−標本(Aβ(1−42) 500マイクロモル)を−20℃で保存することができる。
【0202】
b)分子量15kDa及び20kDaを有するラット−Aβ(1−42)−オリゴマー[ラット−Aβ(1−42)−オリゴマーA]の製造のための選択的製法
ラット−β−アミロイド(1−42)−タンパク質(略称:ラット−Aβ(1−42);ペプチド合成原料、凍結乾燥物、Fa. Bachem, Deutschland)1mgを、1%SDS/H2O44μl中に溶かす(ラット−Aβ(1−42) 5ミリモル)。この溶液5μlにPBS40μl及び2%SDS5μlを加え、37℃で16時間恒温保持する。不溶成分を10000gで5分間遠心分離する。そうして得られるラット−Aβ(1−42)−オリゴマーA−標本(Aβ(1−42) 500マイクロモル)を−20℃で保存することができる。
【0203】
例5
a)分子量38kDa及び48kDaを有するAβ(1−42)−オリゴマー[Aβ(1−42)−オリゴマーB]の製造
例2aにより得られるAβ(1−42)−オリゴマーA−溶液を、水2475mlで希釈し(0.05%SDS−含量、Aβ(1−42) 0.1ミリモル)、37℃で20時間恒温保持する。このAβ(1−42)−オリゴマーB−標本の部分標本を−80℃で凍結させ、更なる研究のために保存する。
b)分子量38kDa及び48kDaを有するラット−Aβ(1−42)−オリゴマー[ラット−Aβ(1−42)−オリゴマーB]の製造
例2bにより得られるラット−Aβ(1−42)−オリゴマーA−溶液を、水2475mlで希釈し(0.05%SDS−含量、ラット−Aβ(1−42) 0.1ミリモル)、37℃で20時間恒温保持する。このラット−Aβ(1−42)−オリゴマーB−標本の部分標本を−80℃で凍結させ、更なる研究のために保存する。
【0204】
c)分子量38kDa及び48kDaを有するAβ(1−42)−オリゴマー[Aβ(1−42)−オリゴマーB]の選択的製造
例2cにより得られるAβ(1−42)−オリゴマーA−溶液を、水2475mlで希釈し(0.125%ラウリル酸−含量、Aβ(1−42) 0.1ミリモル)、37℃で20時間恒温保持する。このAβ(1−42)−オリゴマーB−標本の部分標本を−80℃で凍結させ、更なる研究のために保存する。
【0205】
d)分子量38kDa及び48kDaを有するAβ(1−42)−オリゴマー[Aβ(1−42)−オリゴマーB]の選択的製造
例2dにより得られるAβ(1−42)−オリゴマーA−溶液を、水2475mlで希釈し(0.125%油酸−含量、Aβ(1−42) 0.1ミリモル)、37℃で20時間恒温保持する。このAβ(1−42)−オリゴマーB−標本の部分標本を−80℃で凍結させ、更なる研究のために保存する。
e)分子量38kDa及び48kDaを有するAβ(1−42)−オリゴマーB[Aβ(1−42)−オリゴマーB]の選択的製造
例2eにより得られるAβ(1−42)−オリゴマーA−溶液を、水2475mlで希釈し(0.125%ラウロイルサルコシン−含量、Aβ(1−42) 0.1ミリモル)、37℃で20時間恒温保持する。このAβ(1−42)−オリゴマーB−標本の部分標本を−80℃で凍結させ、更なる研究のために保存する。
【0206】
f)SDSを含まない、分子量38kDa及び48kDaを有するAβ(1−42)−オリゴマーB[Aβ(1−42)−オリゴマーB]の製造
例6bにより製造したAβ(1−42)−オリゴマーB−標本10μlに、比率4%/33%/63%の酢酸/メタノール/水−混合物250μlを加え、氷上0℃で30分間恒温保持する。遠心分離(10000gで10分間)後に、上澄液を取り出し、沈殿したタンパク質残渣を緩衝液(燐酸−Na20ミリモル、NaCl140ミリモル、pH7.4)200μl中に入れる。そうして得られる標本は、SDSを含まない形の溶解したAβ(1−42)−オリゴマーBを含有し、−20℃で保存され得る。
【0207】
例6
a)分子量38kDa及び48kDaを有するAβ(1−42)−オリゴマー[Aβ(1−42)−オリゴマーB]の透析及び濃縮
例5aにより製造したAβ(1−42)−オリゴマーB−標本に、0.1%プルロニック(Pluronic)(登録商標)F68(Fa. BASF)を含有するPBS−緩衝液30mlを加え、Fa. Amicon のCentriprep YM, 30 KD中で3mlに濃縮させる。場合により存在する残渣を遠心分離(10000gで5分間)によって除去する。上澄液を取り出す。このAβ(1−42)−オリゴマーB−標本の部分標本を−80℃で凍結させ、更なる研究のために保存することができる。
【0208】
b)分子量38kDa及び48kDaを有するAβ(1−42)−オリゴマーB[Aβ(1−42)−オリゴマーB]の濃縮物の製造
例5aにより得られるAβ(1−42)−オリゴマーB−標本72.6mlを、30 KD−Centriprep−細管YM(Amicon)上で2mlに濃縮させる。濃縮物を10000gで10分間遠心分離する。上澄液を取り出し、透析管中で、緩衝液(燐酸ナトリウム5ミリモル、塩化ナトリウム35ミリモル、pH7.4)1lに対して6℃で16時間透析する。透析物を10000gで10分間遠心分離する。上澄液を取り出し、更なる研究のために−80℃で保存することができる。
【0209】
例7
ビオチン−Aβ(1−42)−基本懸濁液の製造
ビオチン−β−アミロイド(1−42)−タンパク質(略称:ビオチン−Aβ(1−42);ペプチド合成原料、凍結乾燥物、AnaSpec)0.5mgを、1,1,1,3,3,3−ヘキサフルオル−2−プロパノール200μl中に溶かし、エッペンドルフ容器中37℃で30分間恒温保持する。その後に、真空濃縮機(Speed Vac)中で蒸発乾固させる。残渣をDMSO20.5μlに入れ、この際、ビオチン−Aβ(1−42)−基本懸濁液5ミリモルが生じる。これを−20℃で保存することができる。
【0210】
例8
分子量17kDa及び22kDaを有するビオチン−Aβ(1−42)−オリゴマー[ビオチン−Aβ(1−42)−オリゴマーA]の製造
例7からの基本懸濁液2μlに、PBS−緩衝液(燐酸ナトリウム20ミリモル、NaCl140ミリモル、pH7.4)23μlを加え、2%のSDS−溶液2.4μlで0.2%SDSの含量に調整する。その後に、37℃で6時間恒温保持し、不溶成分を10000gで5分間遠心分離する。そうして得られるビオチン−Aβ(1−42)−オリゴマーA−標本を−20℃で保存することができる。
【0211】
例9
分子量42kDa及び52kDaを有するビオチン−Aβ(1−42)−オリゴマー[ビオチン−Aβ(1−42)−オリゴマーB]の製造
例8により得られるビオチン−Aβ(1−42)−オリゴマーA−溶液を、水82μlで希釈し(0.05%SDS−含量、Aβ0.1ミリモル)、37℃で16時間恒温保持する。不溶成分を10000gで5分間遠心分離する。このビオチン−Aβ(1−42)−オリゴマーB−標本を−20℃で凍結させ、更なる研究のために保存することができる(図3)。
【0212】
例10
フルオレスセイン−Aβ(1−42)−基本懸濁液の製造
フルオレスセイン−β−アミロイド(1−42)−タンパク質(略称:フルオレスセイン−Aβ(1−42);ペプチド合成原料、凍結乾燥物、AnaSpec)0.5mgを、1,1,1,3,3,3−ヘキサフルオル−2−プロパノール200μl中に溶かし、エッペンドルフ容器中37℃で30分間恒温保持する。その後に、真空濃縮機(Speed Vac)中で蒸発乾固させる。残渣をDMSO20.5μlに入れ、この際、フルオレスセイン−Aβ(1−42)−基本懸濁液5ミリモルが生じる。これを−20℃で保存することができる。
【0213】
例11
分子量17kDa及び22kDaを有するフルオレスセイン−Aβ(1−42)−オリゴマー[フルオレスセイン−Aβ(1−42)−オリゴマーA]の製造
例10からの基本懸濁液2μlにPBS−緩衝液(燐酸ナトリウム20ミリモル、NaCl140ミリモル、pH7.4)23μlを加え、2%のSDS−溶液2.4μlで0.2%SDSの含量に調整する。その後に、37℃で6時間恒温保持する。不溶成分を10000gで10分間遠心分離する。そうして得られるフルオレスセイン−Aβ(1−42)−オリゴマーA−標本を−20℃で保存することができる。
【0214】
例12
分子量42kDa及び52kDaを有するフルオレスセイン−Aβ(1−42)−オリゴマー[フルオレスセイン−Aβ(1−42)−オリゴマーB]の製造
例11により得られるフルオレスセイン−Aβ(1−42)−オリゴマーA−溶液を、水82μlで希釈し(0.05%SDS−含量、Aβ(1−42) 0.1ミリモル)、37℃で16時間恒温保持する。フルオレスセイン−Aβ(1−42)−オリゴマーB−標本を−80℃で凍結させ、更なる研究のために保存することができる(図4)。
【0215】
例13
例2aからのAβ(1−42)−オリゴマーA[Aβ(1−42)−オリゴマーA−CL]の架橋結合
例2aにより製造したAβ(1−42)−オリゴマーA−溶液10μlをPBS7.5μl、0.2%SDSで、Aβ(1−42)−含量100マイクロモルに希釈する。この溶液に新規に製造したグルタルジアルデヒド−水溶液10ミリモル1μlを加え、これを室温で3時間攪拌する。過剰のグルタルジアルデヒドを飽和させるために、試料に、エタノールアミン−水溶液100ミリモル1μl、pH7.4を加え、1時間攪拌する。そうして得られる標本は、架橋結合したAβ(1−42)−オリゴマーAを含有し、Aβ(1−42)−オリゴマーA−CL−標本と表示される。
【0216】
例14
a)例5aからのAβ(1−42)−オリゴマーB[Aβ(1−42)−オリゴマーB−CL]の架橋結合
例5aにより製造したAβ(1−42)−オリゴマーB−溶液10μlに、新規に製造したグルタルジアルデヒド−水溶液10ミリモル1μlを加え、室温で3時間攪拌する。過剰のグルタルジアルデヒドを飽和させるために、試料に、エタノールアミン−水溶液100ミリモル1μl、pH7.4を加え、1時間攪拌する。そうして得られる標本は、架橋結合したAβ(1−42)−オリゴマーBを含有し、Aβ(1−42)−オリゴマーB−CL−標本と表示される。
【0217】
b)Aβ(1−42)−オリゴマー[Aβ(1−42)−オリゴマーB−CL]の架橋結合のための選択的方法
例5aにより製造したAβ(1−42)−オリゴマーB−溶液72.6mlに、新規に製造したグルタルジアルデヒド−水溶液10ミリモル7,26mlを加え、室温で2時間攪拌する。過剰のグルタルジアルデヒドを飽和させるために、試料に、緩衝液(燐酸−Na20ミリモル、NaCl140ミリモル、エタノールアミン500ミリモル、pH7.4)726μlを加え、室温で30分間攪拌する。成分を30kDa−セントリプレプ(Centriprep)−細管15ml上で3mlに濃縮させる。濃縮物を10000gで10分間遠心分離する。上澄液を取り出し、透析管中で、燐酸−Na115ミリモル、NaCl35ミリモル、pH7.4に対して、6℃で16時間透析する。その後に、透析物を10000gで10分間遠心分離し、上澄液を取り出す。上澄液を更なる研究のために−80℃で保存することができる。そうして得られる標本は、架橋結合したAβ(1−42)−オリゴマーBを含有し、Aβ(1−42)−オリゴマーB−CL−標本と表示される。
【0218】
例15
a)サーモリシンでの分解による、Aβ(1−42)−オリゴマーBから出発する切端されたAβ(20−42)−オリゴマーの製造:
例6bにより製造したAβ(1−42)−オリゴマーB−標本1.59mlに、緩衝液(MES/NaOH50ミリモル、pH7.4)38ml及びサーモリシン−水溶液(Fa. Roche)1mg/ml 200μlを加える。成分を室温で20時間攪拌する。その後に、EDTA−水溶液100ミリモル(pH7.4)80μlを加え、更に、1%のSDS−溶液400μlでSDS−含量0.01%に調整する。成分を、30kDa−セントリプレプ−細管15mlを介して約1mlに濃縮させる。濃縮物に、緩衝液(MES/NaOH50ミリモル、SDS0.02%、pH7.4)9mlを加え、再び1mlに濃縮させる。濃縮物を、透析管中で、緩衝液(燐酸−Na5ミリモル、NaCl35ミリモル)1lに対して、6℃で16時間透析する。透析物を2%のSDS−水溶液で、SDS−含量0.1%に調整する。試料を10000gで10分間遠心分離し、上澄液を取り出す。
【0219】
そうして得られる物質を更に分析する(SDS−ポリアクリルアミドゲル−電気泳動;図11参照);生成した切端オリゴマーの質量分析により、オリゴマーは切端Aβ(20−42)から成ることが明らかである。
【0220】
b)エンドプロテイナーゼGluCでの分解による、Aβ(1−42)−オリゴマーBから出発する切端Aβ(12−42)−オリゴマーの製造:
例6bにより製造したAβ(1−42)−オリゴマーB−標本2mlに、緩衝液(燐酸ナトリウム5ミリモル、塩化ナトリウム35ミリモル、pH7.4)38ml及びエンドプロテイナーゼGluC(Fa. Roche)1mg/ml(水中)150μlを加える。成分を室温で6時間攪拌する。引き続き、もう1度、水中のエンドプロテイナーゼGluC(Fa. Roche)1mg/ml 150μlを加える。成分を室温で更に16時間攪拌する。その後に、DIFP−溶液5モル8μlを加える。成分を、30kDa−セントリプレプ−細管15ml上で約1mlに濃縮させる。濃縮物に、緩衝液(燐酸ナトリウム5ミリモル、塩化ナトリウム35ミリモル、pH7.4)9mlを加え、再び1mlに濃縮させる。濃縮物を、透析管中で、緩衝液(燐酸−Na5ミリモル、NaCl35ミリモル)1lに対して、6℃で16時間透析する。透析物を1%のSDS−水溶液で、SDS−含量0.1%に調整する。試料を10000gで10分間遠心分離し、上澄液を取り出す。
【0221】
そうして得られる物質を更に分析する(SDS−ポリアクリルアミドゲル−電気泳動;図11参照);生成した切端オリゴマーの質量分析により、オリゴマーは切端Aβ(12−42)から成ることが判明する。
【0222】
Aβ(1−42)−オリゴマーの特徴付け
例16
SDS−ポリアクリルアミドゲル電気泳動(SDS−PAGE)
変性条件下での分子量の特徴付けのために、例2a、5a、9、12、13及び14aからのオリゴマーA及びBを含有する標本を、変性条件下に、標準条件により、4〜20%のトリス−グリシン−SDS−PAGEで分析する。
【0223】
SDS−PAGEの評価(図1)により、オリゴマーA−標本中になお存在する出発タンパク質Aβ(1−42)が約4kDaでの帯として現われるが、約15kDa(より弱い帯)及び約20kDa(主帯)で、例2aからのオリゴマーA1又はA2(図1において、1又は2で表示される)が見られることが明らかである。
【0224】
オリゴマーB−標本では、比較的に少ない出発タンパク質Aβ(1−42)が検出される(約4kDaでの比較的弱い帯)。これに対して、約38kDa及び約48kDaで、例5aからのオリゴマーB1又はB2(図1において、3及び4で表示される(矢印参照))が現われる。例9及び12からのビオチン及びフルオレスセインで誘導体化されたオリゴマーBについて、約42kDa及び約52kDaと一致するより高い分子量が明らかである(図3、4)。
【0225】
各々架橋結合生成物A−CL及びB−CLを含有する標本の分析(例13及び14a;図2)は、2つの試料がそのオリゴマー化度(線AとA’及び線BとB’を比較)を実際に保持していることを示す。オリゴマーA及びBについて僅かに異なる泳動は、僅かに変化したSDS−結合力によって及びN−末端のアミノ基又はリジン基の変性によって説明することができる。
【0226】
切端Aβ(20−42)−オリゴマー(例15aから)の分析は、38/48kDa二重帯(図11、線A)が、サーモリシンでのN−末端ペプチドのタンパク質加水分解によって、28/38kDa二重帯(図11;線B)に移動することを示す。同様に、切端Aβ(12−42)−オリゴマー(例15bから)の分析は、38/48kDa二重帯(図11、線A)が、エンドプロテアーゼGlu-CでのN−末端ペプチドのタンパク質加水分解によって、33/40kDa二重帯(図11;線C)に移動することを示す。
【0227】
例17
ゲル透過クロマトグラフィー
非変性条件下での分子量特性を詳細に調べるために、ゲル透過クロマトグラフィー(GPC)を、FPLC−法で、スパロース(Superose) 12 HR 10/30 カラムを用いて実施する。GPCは4℃で行なわれる。
【0228】
カラムを5カラム容量のPBS−緩衝液で平衡させ(流量0.5m/分、検出UV、214nm)、先ず、標準タンパク質で検定する。引き続き、例5aからのAβ(1−42)−オリゴマーB−標本(図5、下方)及び、比較のために同一濃度で、新規に秤量してPBS中に溶かしたAβ(1−42)−凍結乾燥物(図5、上方)を、10000gで5−分間不溶成分の遠心分離後に分析する。
【0229】
その評価により、Aβ(1−42)−オリゴマーB−標本は、分子量範囲約50kDaの主ピークを特徴とするタンパク質画分を示すことが明らかである。SDS−PAGEにおける変性条件下と同様に、このタンパク質画分は、分子量範囲約16kDaの主ピークを特徴とするモノマーAβ(1−42)−タンパク質とは著しく異なっている。
【0230】
例6bからのAβ(1−42)−オリゴマーB及び例14aからのAβ(1−42)−オリゴマーB−CLの非変性条件下での分子量特性を調べるために、ゲル透過クロマトグラフィー(GPC)をFPLC−法でスパロース12 HR 10/30−カラムを用いて室温で実施する。カラムを5カラム容量のPBS−緩衝液で平衡させ(流量0.5m/分、検出UV、214nm)、先ず、標準タンパク質で検定する。引き続き、例6bからのAβ(1−42)−オリゴマーBをPBS緩衝液で1mg/mlに希釈し、かつ例14aからのAβ(1−42)−オリゴマーB−CLをPBS−緩衝液で1mg/mlに希釈し、分析する(図12)。
【0231】
その評価により、減少されたSDS−含量を有するAβ(1−42)−オリゴマーB−標本は、分子量範囲約100kDaの主ピークを特徴とするタンパク質画分を示すことが明らかである。それに比較して、減少されたSDS−含量を有するAβ(1−42)−オリゴマーB−CL−標本は、分子量範囲約60kDaの主ピークを特徴とするタンパク質画分を示す。
【0232】
例18
例5aからのAβ(1−42)−オリゴマーBの自然ポリアクリルアミドゲル電気泳動(NATIVE−PAGE)
自然条件化での分子量の特徴付けのために、例5aからのオリゴマーBを含有する標本を、非変性条件下に、4〜20%トリス−グリシン−ゲル中で分析する。
【0233】
標本中に含有される界面活性剤を、非イオン性界面活性剤トリトン(Triton)X−100で中和する。そのために、(4%トリトンX−100)1μlを例5aからの標本10μlにピペットで入れ、室温で5分間高温保持する。その後に、10μlに、同一容量の自然試料緩衝液(トリス1モル4ml、pH6.8、グリシン8ml、H2O50ml中のブロムフェノールブルー1ml)を加え、電気泳動(流動緩衝液:トリス7.5g、グリシン36g、H2O2.5lへ)を実施する。
【0234】
NATIVE−PAGEの評価により、オリゴマーB−標本中になお存在する出発ペプチドAβ(1−42)が約28kDaでの帯として(線A、1で表示)現われ、例5aからの標本の主ピークが、見かけの分子量64〜90kDa(線A、2で表示)で認められることが明らかである(図6)。
【0235】
ゲル透過クロマトグラフィー(例17参照)又は自然ゲル電気泳動(例18参照)と同様に、自然分子量−分析法でも、本発明によるオリゴマー、殊に、オリゴマーBに分子量を割り当て得ることは重要である。
【0236】
SDS−PAGEにおいて分子量約38及び48kDaを割り当てられ得るオリゴマーBは、SDSの複合化後に、自然ゲル電気泳動法で、選別された標準タンパク質に対して、約64〜90kDaの分子量範囲の帯として検出される。この方法により、正確な分子量を読み取ることは期待されず、それというのも、オリゴマーの移動特性は分子量の大きさに加えてその固有荷電によって実際に測定されるからである。当然、この結果から、限定されたオリゴマー種が存在することは推論され得る。
【0237】
例19
a)生理学的緩衝液中の異なるタンパク質濃度におけるAβ(1−42)−オリゴマーBにおける安定性
例6bにより得られるAβ(1−42)−オリゴマーBを、次の条件下に、PBS−緩衝液中の安定性について試験する。
【0238】
5mg/mlを、PBSを用いて2回の希釈段階で0.08mg/mlに希釈する。全ての得られる溶液を室温で24時間恒温保持する。続いて、帯パターンを、凍結された対照標本に比較して、SDS−PAGEで分析する。帯パターンは全試料で同一である。
【0239】
b)生理学的緩衝液における異なる温度で及び異なる時間後のAβ(1−42)−オリゴマーBの安定性
例6bにより得られるAβ(1−42)−オリゴマーBを、PBS−緩衝液で0.5mg/mlに希釈し、24時間又は96時間室温又は370℃で高温保持する。続いて、帯パターンをSDS−PAGEで分析する。帯パターンは全試料で同一である。
【0240】
例20
異なるプロテアーゼ(トリプシン、キモトリプシン、サーモリシン、エラスターゼ、パパイン)によるAβ(1−42)−オリゴマーBのタンパク質加水分解
例6により得られるAβ(1−42)−オリゴマーB−標本の部分標本を、緩衝液(燐酸−Na20ミリモル、NaCl140ミリモル、pH7.4)で0.5mg/mlに希釈し、次の条件下に、各々、図9に挙げたプロテアーゼ溶液の質量の1/50と共に、20時間37℃及びpH7.4で恒温保持する。その後に、成分の部分標本1μgをSDS−PAGEで分析する(図9)。
【0241】
SDS−PAGEは、Aβ(1−42)−オリゴマーB−標本から出発して、全プロテアーゼが、選択された限定のタンパク質加水分解条件下に、38/48kDaでの二重帯を約32/28kDaでのkDa二重帯へ戻すことを示す。
【0242】
例21
ラット血漿中のAβ(1−42)−オリゴマーBの安定性
例6bにより製造したAβ(1−42)−オリゴマーB−標本4μlを、ラット血漿76μlと共に、0時間、1時間、2時間、4時間及び8時間室温で恒温保持する。恒温保持をドライアイス中での凍結によって止める。
【0243】
引き続き、全試料をSDS−試料緩衝液−添加後に、SDS−PAGEで分析する。その後に、安定性の評価を、ウエスタンブロット法で、Aβ(1−42)−オリゴマーBの変色を介して行なう。検出のために、Aβ(1−42)照準の抗体6E10(Fa. Signet)を使用する。帯は、アルカリ性ホスファターゼに結合される抗−マウス−IgG−抗体及び基質NBT/BCIPの添加によって可視される。観察される帯パターンの分子量も強度も、2時間、4時間及び8時間に渡って殆ど無変化のままである。
【0244】
この結果から、Aβ(1−42)−オリゴマーBが高い血漿安定性を有することが推論できる。従って、血漿中の生物学的半減期は、8時間以上の範囲である。
【0245】
例22
分子量17kDa及び22kDaを有するビオチン−Aβ(1−42)−オリゴマー[ビオチン−Aβ(1−42)−オリゴマーA]又は分子量42kDa及び52kDaを有するビオチン−Aβ(1−42)−オリゴマー[ビオチン−Aβ(1−42)−オリゴマーB]のヒト神経細胞の表面への結合
ヒトβ−アミロイド(1−42)−タンパク質及び2つのAβ(1−42)−オリゴマーA−及びAβ(1−42)−オリゴマーB−標本の、ヒト神経芽細胞系IMR−32(ATCC番号:CCL−127)への結合を、FACScan(Beckton Dickinson)によって調べる。IMR−32細胞の懸濁液(細胞1.5x106/PBS0.1ml)を、ビオチン−標識化ヒトβ−アミロイド(1−42)−タンパク質(ペプチド合成原料、凍結乾燥物、AnaSpec)及びビオチン−標識化オリゴマーA又はBを含有する標本(例8又は9)と共に、20分間37℃で恒温保持する。引き続き、細胞を緩衝液(PBSプラス1%BSA)で洗浄し、ストレプトアビジンイソチオシアネート結合のフルオレセイン(Sigma)と共に20分間室温で恒温保持する。緩衝液での清浄段階後に、IMR−32細胞の表面への結合をFACScanで分析する。断続線は、ビオチン−標識化成分の不在で、基底値蛍光を示す。個々の標本の添加は細胞−会合蛍光の強力な増加を導き、濃い線によって描かれている。細胞表面へのオリゴマーA(図7B)及びB(図7C)の結合は、モノマーβ−アミロイド(1−42)−タンパク質(図7A)の結合とは明らかに異なっている。データは、ヒト細胞表面上のオリゴマーの特異的結合部位を示す。
【0246】
例23
icv−投与後のラット脳ホモジネート中のAβ(1−42)−オリゴマーBの検出
例6bより得られるAβ(1−42)−オリゴマーB−標本10ナノモルを、ラットにicv丸剤として投与する。脳を15分間後又は120分間後に解剖する。未処理対照動物の脳を同様に解剖する。そのために、50ml入りファルコン(Falcon)試験管中で、ラット脳各1gに、破砕緩衝液A(燐酸ナトリウム5ミリモル200ml、NaCl35ミリモル、蔗糖300ミリモル、pH7.4に調整、プロテアーゼ阻害剤−カクテルComplete(登録商標)、Fa. Rocheの4錠を加える)9mlを加え、氷上の超音波(Ultra Turrax)中で2分間破砕する。溶液を20分間放置し、素早く振動させ、部分標本8x1mlに分ける(=ホモジネート)。
【0247】
検出を定量的に行なうために、先ず、PBS中のAβ(1−42)−オリゴマーB−標本の一連の標準を、濃度範囲1.58ng/μl〜0.005ng/μlで調製する。
【0248】
更に、正対照として、未処理ラットの対照脳からのホモジネートを一緒に経過させ、同様の方法で、この脳ホモジネート中のAβ(1−42)−オリゴマーB−標本の一連の標準を調製する。その後に、ホモジネートを超遠心分離機において100000gで1時間遠心分離させ、上澄液を次の分析に使用する。
【0249】
2つの標準列で測定した値(PBS対対照脳−ホモジネート−上澄液)の比較から、先ず、正対照中のAβ(1−42)−オリゴマーBの含量を定量的に測定し、次いで、それに比較して、処理ラットの脳試料においても同様に測定する。
【0250】
1)ドットブロット−検出法
標準列−試料及び処理動物からの試料抽出物1μlを、ニトロセルロース−紙上に滴下し、Aβ(1−42)を抗体6E10(抗−Aβ(1−42);Fa. Signet)で検出する。抗−マウスIgGに結合したアルカリ性ホスファターゼを用いて、呈色剤NBT/BCIPの添加によって呈色させる。
【0251】
未処理ラットの脳抽出物中ではAβ(1−42)(0.01ナノモル/g)は検出不可能であるが、処理の15分間後に殺したラットにおいて、正対照の相応する濃度との呈色強度の比較によって、Aβ(1−42)約0.4ナノモル/gが検出可能であり、かつ処理の120分間後に殺したラットにおいて、同様の方法により、約0.2ナノモル/gがなお検出可能である。このことから、外部から投与されたAβ(1−42)−オリゴマーBについて、約105分間の平均的な生物学的半減期が判明する。
【0252】
2)ウエスタンブロット−検出法
ドットブロット法で分析した全試料を、同様にウエスタンブロット法で分析する。ウエスタンブロット法は、同様に、mMAb6E10(抗−Aβ(1−42);Fa. Signet)及び更に抗−マウス−IgGに結合したアルカリ性ホスファターゼを用いて、呈色剤NBT/BCIPの添加によって展開させる。
【0253】
抗Aβ(1−42)−反応帯は38/48でのみ出現し、これはAβ(1−42)−オリゴマーBの見かけの分子量に相応し、即ち、オリゴマー構造は、生体内適用でも2時間後もなお保持されたままである。
【0254】
更に、ドットブロット−法の場合と同様に、15分間−ラット(0.4ナノモル/g)及び120分間−ラット(0.2ナノモル/g)の脳において、正対照におけるAβ(1−42)−オリゴマーBの相応に強く呈色した帯との呈色強度の比較によって、同じ濃度が評価され得る。
【0255】
例24
分子量38kDa及び48kDaを有するAβ(1−42)−オリゴマー[Aβ(1−42)−オリゴマーB]の、マウスの大脳皮質ニューロンへの神経毒作用
ネズミの大脳皮質ニューロンの標本及び培養は、文献(Choi et al. (1987) J. Neurosci. 7, 357-368)による神経膠細胞との混合培養として行なわれる。胚の場合には、展開14〜15日目に、大脳皮膚及び深部脳範囲の皮部を機械的に除去する。0.05%のトリプシン溶液中37℃で5〜7分間の恒温保持し、及び引続いて、孔を縮小させたパスツールピペットを数回通過させることよって細胞を細かく分離させる。細胞数の測定後に、ポリ−L−オルニチン及びラミニンで被覆された細胞培養原料上で2cm2当たり、保存培地(グルタミン0.8ミリモル、ブドウ糖18ミリモル、NaHCO3 23ミリモル及び10%馬血清を有する最少必須培地)0.5ml中で430000個の細胞を接種する。細胞の培養及びその後の恒温保持は、空気湿潤の細胞培養箱中で37℃、5%CO2で行なわれる。培養3〜5日間後に、神経膠−細胞の増加を、(+)−5−フルオル−2’−デスオキシウリジン/ウリジン−混合物(各10マイクロモル)との1日間の恒温保持によって止める。培養14日間後に、Aβ(1−42)−オリゴマーBの毒性作用を調べる。そのために、細胞を15分間脳細胞緩衝液(NaCl120ミリモル、KCl5.4ミリモル、CaCl21.8ミリモル、ブドウ糖15ミリモル、HEPES25ミリモル、pH7.2)中で恒温保持する。対照細胞1を脳細胞緩衝液中でL−グルタメート300マイクロモルと共に同じ時間恒温保持する。Aβ−オリゴマーの基本溶液を、血清を含まない培地(グルタミン0.8ミリモル、ブドウ糖20ミリモル、NaHCO3 26ミリモルを有する最少必須培地)で、様々な最終濃度に希釈し、24時間恒温保持する。L−グルタメートで処理した対照細胞1を平行して血清を含まない培地中で恒温保持する。もう1つの群の細胞(対照細胞2)を、脳細胞緩衝液だけで、血清を含まない培地で処理する。24時間後に細胞培養上澄液を除去し、残留細胞を蒸留水中で20分間の恒温保持によって破砕し、2つの溶液中で酵素、乳酸デヒドロゲナーゼ(LDH)の活性を酵素的に調べる。評価のために、細胞培養上澄液のLDH−活性対上澄液及び残留細胞からのLDH−活性の合計の比率を測定し、4回の測定の平均値を出す。3回の実験を実施し、この際、各実験条件は4重で存在している(n=12)。グルタメートで処理した細胞(対照細胞1)における平均値はニューロン死100%とみなし、L−グルタメートでも、Aβ−オリゴマーでも処理していない細胞(対照細胞2)の平均値はニューロン死0%とみなし、Aβ−オリゴマーで処理した細胞値は相応に換算される。各使用濃度における全測定の神経毒性の平均値は、分子量38kDa及び48kDaを有するAβ(1−42)−オリゴマー[Aβ(1−42)−オリゴマーB]の明らかな毒性作用を示す。図8参照。
【0256】
例25
抗体の製造
免疫化に使用されるカクテルは、全例で、実際に次の成分を有するアジュバント(Fa. Biogenes)を含有する:
パラフィン油95%
ツィーン(Tween)40 2.4%
コレステリン0.1%
リポ多糖類0.1%
アジュバントは、安定したエマルジョンが得られるまで、抗原の溶液と比率2:1で混合される。エマルジョンを注射し、抗原がそれから連続的に遊離されるデポーを形成する。
【0257】
a)例15aからの切端Aβ(20−42)−オリゴマーBでのウサギの免疫化によるポリクローナル抗血清の製造
2匹のウサギを、標準プロトコルにより、例15aからの非複合化Aβ(20−42)−オリゴマーB−標本で免疫化させる。
【0258】
1日目 前免疫血清の第1免疫化及び採取
7日目 第1免疫増強
14日目 第2免疫増強
28日目 第3免疫増強及び出血
35日目 採血
抗血清を、ウサギ血液から、室温での静置及び引き続き室温での遠心分離によって得る。そうして得られる血清を血清(a1)と表示する。
【0259】
例15aからのAβ(20−42)−オリゴマーB−標本2mgを、LPHにグルタルジアルデヒドで標準条件下に結合させ、他の2匹のウサギを、標準プロトコルにより、この複合Aβ(20−42)−オリゴマーBで免疫化させる。
【0260】
1日目 前免疫血清の第1免疫化及び採取
7日目 第1免疫増強
14日目 第2免疫増強
28日目 第3免疫増強及び出血
35日目 採血
抗血清をウサギ血液から、室温での静置及び引き続き室温での遠心分離によって得る。そうして得られる血清を血清(a2)と表示する。
【0261】
b)例15bからの切端Aβ(12−42)−オリゴマーBでのウサギの免疫化によるポリクローナル抗血清の製造
2匹のウサギを、標準プロトコルにより、例15bからの非複合化Aβ(12−42)−オリゴマーB−標本で免疫化させる。
【0262】
1日目 前免疫血清の第1免疫化及び採取
7日目 第1免疫増強
14日目 第2免疫増強
28日目 第3免疫増強及び出血
35日目 採血
抗血清を、ウサギ血液から、室温での静置及び引き続き室温での遠心分離によって得る。そうして得られる血清を血清(b1)と表示する。
【0263】
例15bからのAβ(12−42)−オリゴマーB−標本2mgを、LPHにグルタルジアルデヒドで標準条件下に結合させ、他の2匹のウサギを、標準プロトコルにより、この複合Aβ(12−42)−オリゴマーBで免疫化させる。
【0264】
1日目 前免疫血清の第1免疫化及び採取
7日目 第1免疫増強
14日目 第2免疫増強
28日目 第3免疫増強及び出血
35日目 採血
抗血清をウサギ血液から、室温での静置及び引き続き室温での遠心分離によって得る。そうして得られる血清を血清(b2)と表示する。
【0265】
c)例14aからのAβ(1−42)−オリゴマーB−CLでのウサギの免疫化によるポリクローナル抗血清の製造
2匹のウサギを、標準プロトコルにより、例14aからの非複合化Aβ(1−42)−オリゴマーB−CLー標本で免疫化させる。
【0266】
1日目 前免疫血清の第1免疫化及び採取
7日目 第1免疫増強
14日目 第2免疫増強
28日目 第3免疫増強及び出血
35日目 採血
抗血清を、ウサギ血液から、室温での静置及び引き続き室温での遠心分離によって得る。そうして得られる血清を血清(c1)と表示する。
【0267】
d)例6bからの切端Aβ(1−42)−オリゴマーBー標本でのウサギの免疫化によるポリクローナル抗血清の製造
2匹のウサギを、標準プロトコルにより、例6bからの非複合化Aβ(1−42)−オリゴマーB−標本で免疫化させる。
【0268】
1日目 前免疫血清の第1免疫化及び採取
7日目 第1免疫増強
14日目 第2免疫増強
28日目 第3免疫増強及び出血
35日目 採血
抗血清を、ウサギ血液から、室温での静置及び引き続き室温での遠心分離によって得る。そうして得られる血清を血清(d1)と表示する。
【0269】
例6bからのAβ(1−42)−オリゴマーB−標本2mgを、LPHにグルタルジアルデヒドで標準条件下に結合させ、他の2匹のウサギを、標準プロトコルにより、この複合Aβ(1−42)−オリゴマーBー標本で免疫化させる。
【0270】
1日目 前免疫血清の第1免疫化及び採取
7日目 第1免疫増強
14日目 第2免疫増強
28日目 第3免疫増強及び出血
35日目 採血
抗血清をウサギ血液から、室温での静置及び引き続き室温での遠心分離によって得る。そうして得られる血清を血清(d2)と表示する。
【0271】
例26
例25の免疫化からのポリクローナル抗血清の、様々なAβ(1−42)−型とのその架橋結合についての特徴づけ
ポリクローナル抗血清の特徴付けのために、PBS中100ピコモル(pmol)/μl〜0.01ピコモル/μlの範囲の希釈列で様々なAβ(1−42)−型を製造した。試料各1μlをニトロセルロース−膜上に載せた:検出は例25からの相応するウサギ血清で行なった。比較として、mMAb 6E10(Fa. Signet)での検出を実施した。抗−ウサギ−IgGに結合したアルカリ性ホスファターゼ(比較用:抗−マウス−IgGに結合したアルカリ性ホスファターゼ)及び呈色剤NBT/BCIPの添加で呈色させた。図10はそうして得られるドットブロットを示し、これは数値的に次のように評価される:
【表1】
【0272】
表中の数字は、最低可視量の抗原を[ピコモル;APPの場合を除いて、各々モノマーに対する]で示す(検出限度)。
【0273】
例15aからの非複合Aβ(20−42)−オリゴマーでの免疫化によって得られたウサギ血清(a1)との免疫学的反応の比較は、このオリゴマーに照準している抗体が、他の形、例えば、原線維、APP、及びモノマーと僅かしか架橋結合しないことを明らかに示す。これに対して、Aβ(1−42)−オリゴマーBとの明らかにより強い架橋結合(列1参照)及び更に安定化CL−抗原との架橋結合が認められる。このデータは、APP、モノマー、及び原線維構造に比較して、本オリゴマー形の明らかに異なる構造を示す。抗体はオリゴマー−特異的構造に結合する。
【0274】
例15aからの複合Aβ(20−42)−オリゴマーでの免疫化によって得られたウサギ血清(a2)との免疫学的反応の比較は、このオリゴマーに照準している抗体が、他の形、例えば、原線維及びモノマーと僅かしか架橋結合しないことを明らかに示す。これに対して、Aβ(1−42)−オリゴマーBとの弱い架橋結合(列1参照)及び更に安定化CL−抗原との、及びAPPとの架橋結合が認められる。このデータは、モノマー及び原線維構造に比較して、本オリゴマー形の同様に明らかに異なる構造を示す。
【0275】
これに比較して、例6bからの非複合Aβ(1−42)−オリゴマーでの免疫化によって得られたウサギ血清(d1)及び例14aからのAβ(1−42)−オリゴマーB−CLでの免疫化によって得られたウサギ血清(c1)との免疫学的反応は、このオリゴマーに照準している抗体が、他の形、例えば、原線維、APP及びモノマーと、Aβ(1−42)−オリゴマーBと同様に(列1参照)比較可能に強く架橋結合することを示す。この抗体は、mMab 6E10(Fa. Signet)に比較可能な免疫反応を示し、オリゴマー−特異的構造には結合しない。
【0276】
相応する方法で、マウスを例6aからのAβ(1−42)−オリゴマーで免疫化させ、自体公知の方法で、モノクローナル抗体を確立した。この際、10個のハイブリドーマの内2個がモノクローナル抗体を分泌し、その結合プロフィールは、殊に、前記で試験した反応性に関して、抗血清のそれと(a1)類似している。
【0277】
例27
Aβ(1−42)−原線維の製造
0.1%NH4OH中のAβ(1−42)−溶液(2mg/ml)100μlを、緩衝液(燐酸−Na20ミリモル、NaCl140ミリモル、pH7.4)300μlで、0.5mg/mlに希釈し、HCl0.1モルでpH7.4に調製する(Aβ(1−42) 100マイクロモル)。試料を24時間37℃で恒温保持し、その後に、10分間10000gで遠心分離する。得られるタンパク質残渣を、緩衝液(燐酸−Na20ミリモル、NaCl140ミリモル、pH7.4)400μlで再懸濁させる。そうして得られるAβ(1−42)−原線維−標本を−20℃で保存し、更なる研究に使用することができる。
【0278】
例28
Aβ(1−42)−オリゴマーB−CL(例14bから)の、海馬切片への作用
厚さ400μMの海馬横断切片を、切片浸漬室中34℃で少なくとも1時間、ガス処理したリンガー液(NaCl124ミリモル、KCl4.9ミリモル、MgSO4 1.3ミリモル、CaCl2 2.5ミリモル、KH2PO4 1.2ミリモル、NaHCO3 25.6ミリモル、ブドウ糖10ミリモル)の灌流下に適応させた。その後に、単極の刺激電極により、シェ−ファー副軸索を刺激し、放線状層中の興奮性シナプス後電位((fEPSP)を導出させた。最高のfEPSPを発生させる刺激強度の30%で試験パルスを与えた。長時間−相乗作用の誘発のために、単一パルスを200μsの幅で100パルスを3回、いずれも10分間施した(強い強縮)。得られるfEPSPの相乗作用を少なくとも240分間記録した。例14bからのオリゴマーB−CLの洗浄を、最初の強縮の20分間前に開始し、最後の強縮の10分間後に終了した。fEPSPsの上昇(勾配)を調べ、時間に対して作図した。
【0279】
図14は、Aβ(1−42)−オリゴマーの洗浄が、海馬における長時間相乗作用を、特に維持相("維持相(maintenance phase)")で阻害することを示す。従って、Aβ(1−42)−オリゴマーB−CLは、神経細胞の情報蓄積(細胞記憶)への影響を有する。
【0280】
例29
Aβ(1−42)−オリゴマーB(例5bから)の、一次海馬ラット−ニューロンへの結合
例5bからのラット−Aβ(1−42)−オリゴマーBの、一次海馬ラットニューロンへの結合を調べた。海馬−ニューロンを、ポリ−L−リジン−被覆のデッキガラス上でB27−サプリメントを有する神経基礎培地中で培養し、培養の14日目に使用した。ニューロン細胞膜へのオリゴマーの結合は、新鮮培地中で、オリゴマー200ナノモル(全モノマーAβ−濃度)の添加及び37℃で15分間の恒温保持によって行なわれた。Aβ−含有培地の除去後に、培地で2回洗浄し、細胞を引続いて3.7%ホルムアルデヒド中に固定させた。細胞を更に洗浄した後に、非特異的結合部位をPBS−緩衝液中10%標準ロバ−血清で室温で90分間遮断した。第1抗体として、6E10(マウスから)を1:2000の希釈で、2時間室温で適用した。細胞を再び洗浄し、マウスに照準していて蛍光色素Cy3がそれに結合している第2抗体(ロバから)と共に2時間室温で恒温保持した。細胞を再び洗浄した後に、ニューロンを有するデッキガラスを、封埋培地を有するスライド上に固定した。結合Aβ−オリゴマーを有する海馬ニューロンは、蛍光顕微鏡で表示された。対照として、第1抗体6E10が省略されている成分を使用した。従って、この対照は、Aβに基づいていない非特異的蛍光を示す。
【0281】
図15aで見られるように、オリゴマーはニューロンの細胞表面にドット法で結合する。それに対して、第1抗体6E 10を含まない対照は、ニューロンへの第2抗体の結合による僅かな非特異的蛍光だけを示す(図15b)。
【特許請求の範囲】
【請求項1】
アミロイド−β(1−42)タンパク質−オリゴマーが、SDS−ゲル電気泳動における見かけの分子量約38kDa及び/又は48kDaを有し、その際、アミロイド−β(1−42)タンパク質−オリゴマーの割合が、調製物中においてアミロイド−β(1−42)タンパク質の全量の少なくとも60質量%である、アミロイド−β(1−42)タンパク質−オリゴマー又は標的化、不動化及び/又は架橋結合されたアミロイド−β(1−42)タンパク質−オリゴマーの誘導体を含む調製物。
【請求項2】
標識が、検出可能な標識である、請求項1に記載の調製物。
【請求項3】
標識が、蛍光性、発光性、比色性、放射性又は磁気性標識、又は相補性結合相手への親和性を有する標識である、請求項2に記載の調製物。
【請求項4】
調製物中のアミロイド−β(1−42)タンパク質−オリゴマーの割合が、アミロイド−β(1−42)タンパク質の全量の少なくとも75質量%である、請求項1から3までのいずれか1項に記載の調製物。
【請求項5】
調製物中のアミロイド−β(1−42)タンパク質−オリゴマーの割合が、アミロイド−β(1−42)タンパク質の全量の少なくとも90質量%である、請求項1から3までのいずれか1項に記載の調製物。
【請求項6】
アミロイド−β(1−42)タンパク質−オリゴマー又はその誘導体が、モノマーアミロイド−β(1−42)タンパク質又はモノマーアミロイド−β(1−42)タンパク質の標識化された誘導体へ界面活性剤を作用させ、その後に界面活性作用を減少させ、かつ更に恒温保持し、得られたオリゴマー又は誘導体を場合により標識化、不動化及び/又は架橋結合し、かつアミロイド−β(1−42)タンパク質−オリゴマー又はその誘導体を含有する調製物を取得することにより得られる、請求項1から5までのいずれか1項に記載の調製物。
【請求項7】
アミロイド−β(1−42)タンパク質−オリゴマー、アミロイド−β(1−43)タンパク質−オリゴマー又はアミロイド−β(1−44)タンパク質−オリゴマーであるか、あるいは、アミロイド−β(1−42)タンパク質−オリゴマー、アミロイド−β(1−43)タンパク質−オリゴマー又はアミロイド−β(1−44)タンパク質−オリゴマーの標識化、不動化及び/又は架橋結合された誘導体を製造する方法において、この場合、この方法が、モノマーアミロイド−β(1−42)タンパク質、モノマーアミロイド−β(1−43)タンパク質又はモノマーアミロイド−β(1−44)タンパク質であるか、あるいは、モノマーアミロイド−β(1−42)タンパク質、モノマーアミロイド−β(1−43)タンパク質又はモノマーアミロイド−β(1−44)タンパク質の標識化された誘導体へ界面活性剤を作用させ、その後に界面活性作用を減少させ、かつ更に恒温保持し、得られたオリゴマー又は誘導体を場合により標識化、不動化及び/又は架橋結合し、かつ、前記オリゴマー又は誘導体を取得することを含む、アミロイド−β(1−42)タンパク質−オリゴマー、アミロイド−β(1−43)タンパク質−オリゴマー又はアミロイド−β(1−44)タンパク質−オリゴマーであるか、あるいは、アミロイド−β(1−42)タンパク質−オリゴマー、アミロイド−β(1−43)タンパク質−オリゴマー又はアミロイド−β(1−44)タンパク質−オリゴマーの標識化、不動化及び/又は架橋された誘導体を製造する方法。
【請求項8】
界面活性剤がイオン性である、請求項7に記載の方法。
【請求項9】
界面活性剤がドデシル硫酸ナトリウムである、請求項8に記載の方法。
【請求項10】
界面活性剤の作用時間が約1〜20時間である、請求項7から9までのいずれか1項に記載の方法。
【請求項11】
作用温度が約20〜50℃である、請求項10に記載の方法。
【請求項12】
恒温保持時間が約10〜30時間である、請求項7から9までのいずれか1項に記載の方法。
【請求項13】
恒温保持温度が約20〜50℃である、請求項12に記載の方法。
【請求項14】
アミロイド−β(1−42)タンパク質、アミロイド−β(1−43)タンパク質、アミロイド−β(1−44)タンパク質又はそれらの誘導体が、界面活性剤を作用させる前に少なくとも部分的に変性されている、請求項7から13までのいずれか1項に記載の方法。
【請求項15】
タンパク質が、水素橋−切断剤を作用させることにより変性される、請求項14に記載の方法。
【請求項16】
水素橋−切断剤がHFIPである、請求項15に記載の方法。
【請求項17】
請求項7から16までのいずれか1項に記載の方法によって得られたアミロイド−β(1−42)タンパク質−オリゴマー、アミロイド−β(1−43)タンパク質−オリゴマー又はアミロイド−β(1−44)タンパク質−オリゴマーであるか、あるいは、アミロイド−β(1−42)タンパク質−オリゴマー、アミロイド−β(1−43)タンパク質−オリゴマー又はアミロイド−β(1−44)タンパク質−オリゴマーの標識化、不動化及び/又は架橋結合された誘導体。
【請求項18】
請求項1から6までのいずれか1項に記載の調製物又は請求項17に記載のオリゴマー又は誘導体並びに場合により製薬学的に認容性の賦形剤を含む、製薬学的組成物。
【請求項19】
組成物がワクチンである、請求項18に記載の組成物。
【請求項20】
試験管内検出法における、請求項1から6まで又は17のいずれか1項に記載の調製物、オリゴマー又は誘導体の使用。
【請求項21】
生体内検出法において、診断において使用するための組成物を製造するための、請求項1から6まで又は17のいずれか1項に記載の調製物、オリゴマー又は誘導体の使用。
【請求項22】
能動免疫のための製薬学的組成物を製造するための、請求項1から6又は17のいずれか1項に記載の調製物、オリゴマー又は誘導体の使用。
【請求項23】
アミロイド−β関連の病気の治療用ワクチンを製造するための、請求項1から6又は17のいずれか1項に記載の調製物、オリゴマー又は誘導体の使用。
【請求項24】
アミロイド−β関連の病気がアルツハイマー病である、請求項23に記載の使用。
【請求項25】
物質又は物質混合物を特徴付けるための試験管内法において、
i)物質又は物質混合物を得て、
ii)物質又は物質混合物を、請求項1から6又は17のいずれか1項に記載の調製物、オリゴマー又はその誘導体に作用させ、かつ
iii)物質又は物質混合物の一定部分が、調製物、オリゴマー又はその誘導体に結合するかどうかを測定する方法を含む、物質又は物質混合物を特徴付けるための試験管内法。
【請求項26】
請求項1から6又は17のいずれか1項に記載のオリゴマー又は誘導体と特異的に結合する抗体又はその抗原結合部位。
【請求項27】
請求項1から6まで又は17のいずれか1項に記載のオリゴマー又は誘導体と、モノマーアミロイドβ(1−42)タンパク質よりも高い親和性で結合する、抗体又はその抗原結合部位。
【請求項28】
KD=10−6〜10−12Mの範囲の親和性でオリゴマー又はその誘導体と結合する、請求項26又は27に記載の抗体又はその抗原結合部位。
【請求項29】
Kd=10−8よりも高い親和性、Kd=10−9よりも高い親和性、Kd=10−10よりも高い親和性又はKd=10−11よりも高い親和性で、オリゴマー又はその誘導体と結合する、請求項26に記載の抗体又は抗原結合部位。
【請求項30】
Kd=10−8よりも低い親和性で、モノマーアミロイドβ(1−42)タンパク質−モノマー及び/又はモノマーアミロイドβ(1−40)タンパク質と結合する、請求項26から29までのいずれか1項に記載の抗体又はその抗原結合部位。
【請求項31】
オリゴマー又はその誘導体に対する前記抗体の親和性が、モノマーアミロイドβ(1−42)タンパク質及び/又はモノマーアミロイドβ(1−40)タンパク質に対する親和性よりも少なくとも10倍高いか、モノマーアミロイドβ(1−42)タンパク質及び/又はモノマーアミロイドβ(1−40)タンパク質に対する親和性よりも少なくとも100倍高いか、又はモノマーアミロイドβ(1−42)タンパク質及び/又はモノマーアミロイドβ(1−40)タンパク質に対する親和性よりも少なくとも1000倍高い、請求項26から30までのいずれか1項に記載の抗体又はその抗原結合部位。
【請求項32】
抗体が、表面プラスモン共鳴により測定したkoff−速度定数0.1s−1以下でオリゴマー又はその誘導体と結合する、請求項26から31までのいずれか1項に記載の抗体又はその抗原結合部位。
【請求項33】
抗体が、オリゴマー又はその誘導体の活性を、1x10−5以下の阻害定数IC50で阻害する、請求項26から32までのいずれか1項に記載の抗体又はその抗原結合部位。
【請求項34】
抗体がポリクローナル抗体又はモノクローナル抗体である、請求項26から33までのいずれか1項に記載の抗体又はその抗原結合部位。
【請求項35】
抗原が実際にヒト抗体、キメラ抗体又はCDRグラフト抗体である、請求項26から34までのいずれか1項に記載の抗体又はその抗原結合部位。
【請求項36】
抗体を製造する方法において、この場合、この方法が、
i)請求項1から6まで又は17のいずれか1項に記載の少なくとも1種のオリゴマー又は誘導体を用いて宿主を免疫化し;かつ、
ii)前記免疫化に対する応答として生じた抗体含有宿主血清を取得することを含む、抗体を製造する方法、但し、ヒトにおいて実施する方法を除く。
【請求項37】
免疫原として使用されたオリゴマー又は誘導体を認識する、血清の少なくとも1種の抗体が選択される、請求項36に記載の方法。
【請求項38】
請求項1から6まで又は17のいずれか1項に記載のオリゴマー又はその誘導体に特異的な抗体を製造するための方法において、この場合、この方法が、
i)前記オリゴマー又はその誘導体を含む抗原を得て;
ii)前記抗原を抗体レパートリーに作用させ;かつ、
iii)前記レパートリーから前記オリゴマー又は誘導体と特異的に結合する抗原を選択する、請求項1から6まで又は17のいずれか1項に記載のオリゴマー又はその誘導体に特異的な抗体を製造するための方法、但し、ヒトにおいて実施する方法を除く。
【請求項39】
動物を抗原で免疫化させることにより、抗原を生体内で抗体レパートリーに作用させる、請求項38に記載の方法。
【請求項40】
更に、一連のハイブリドーマを動物のリンパ球から確立し、かつオリゴマー又はその誘導体と特異的に結合する抗体を分泌するハイブリドーマを選択する、請求項39に記載の方法。
【請求項41】
動物が、マウス、ラット、ニワトリ、ラクダ、カニクイザル、ウサギ及びヤギから成る群から選択される、請求項39又は40に記載の方法。
【請求項42】
動物が、APP欠損を有するノックアウトマウス、抗原刺激後にヒト抗体を生じさせるヒト免疫グロブリン遺伝子を有するトランスジェニックマウス、ヒト末梢単核血球又はリンパ様細胞又はその前駆体で再構成されている、重症併合免疫不全(SCID)のマウス及び
致死的全身放射線照射で処理され、引き続き、重症併合免疫不全(SCID)のマウスからの骨髄細胞で照射に対して保護され、引き続き、機能的ヒトリンパ球又はその前駆体を移植されているマウスから成る群から選択される、請求項41に記載の方法。
【請求項43】
組換え体の抗体バンクを抗原でスクリーニングすることにより、試験管内で抗原を抗体レパートリーに作用させる、請求項38に記載の方法。
【請求項44】
組換え体の抗体バンクをバクテリオファージの表面で、酵母細胞の表面で、細菌細胞の表面で又はRNA−タンパク質−融合として発現させるか、あるいは、組換え体の抗体バンクが、scFv−バンク又はFab−バンク又は単一ドメイン−バンクである、請求項43に記載の方法。
【請求項45】
動物を抗原で生体内免疫化させ、かつ、動物のリンパ様細胞から製造される組換え体の抗体バンクを抗原でスクリーニングすることによってか、動物を抗原で生体内免疫化させ、かつ動物のリンパ様細胞から製造される組換え体の抗体バンクを試験管内で親和性成熟させることによってか、あるいは、動物を抗原で免疫化させ、抗原結合抗体を分泌する個々の細胞を選択し、かつ単一の細胞から重鎖又は軽鎖の可変領域のcDNAsを取得することによって、抗原を抗体レパートリーに作用させる、請求項38に記載の方法。
【請求項46】
請求項36から45までのいずれか1項に記載の方法によって得られた抗体又はその抗原結合部位。
【請求項47】
請求項26から35又は46のいずれか1項に記載の抗体又はその抗原結合部位並びに場合により製薬学的に認容性の賦形剤を含む、製薬学的組成物。
【請求項48】
アミロイドβ−関連の病気を治療するための医薬品を製造するための、請求項26から35又は46までのいずれか1項に記載の抗体又はその抗原結合部位の使用。
【請求項49】
治療が受動免疫である、請求項48に記載の使用。
【請求項50】
アミロイドβ−関連の病気を診断するための組成物を製造するための、請求項26から35まで又は46のいずれか1項に記載の抗体又はその抗原結合部位の使用。
【請求項51】
アミロイドβ−関連の病気がアルツハイマー病である、請求項48から50までのいずれか1項に記載の使用。
【請求項1】
アミロイド−β(1−42)タンパク質−オリゴマーが、SDS−ゲル電気泳動における見かけの分子量約38kDa及び/又は48kDaを有し、その際、アミロイド−β(1−42)タンパク質−オリゴマーの割合が、調製物中においてアミロイド−β(1−42)タンパク質の全量の少なくとも60質量%である、アミロイド−β(1−42)タンパク質−オリゴマー又は標的化、不動化及び/又は架橋結合されたアミロイド−β(1−42)タンパク質−オリゴマーの誘導体を含む調製物。
【請求項2】
標識が、検出可能な標識である、請求項1に記載の調製物。
【請求項3】
標識が、蛍光性、発光性、比色性、放射性又は磁気性標識、又は相補性結合相手への親和性を有する標識である、請求項2に記載の調製物。
【請求項4】
調製物中のアミロイド−β(1−42)タンパク質−オリゴマーの割合が、アミロイド−β(1−42)タンパク質の全量の少なくとも75質量%である、請求項1から3までのいずれか1項に記載の調製物。
【請求項5】
調製物中のアミロイド−β(1−42)タンパク質−オリゴマーの割合が、アミロイド−β(1−42)タンパク質の全量の少なくとも90質量%である、請求項1から3までのいずれか1項に記載の調製物。
【請求項6】
アミロイド−β(1−42)タンパク質−オリゴマー又はその誘導体が、モノマーアミロイド−β(1−42)タンパク質又はモノマーアミロイド−β(1−42)タンパク質の標識化された誘導体へ界面活性剤を作用させ、その後に界面活性作用を減少させ、かつ更に恒温保持し、得られたオリゴマー又は誘導体を場合により標識化、不動化及び/又は架橋結合し、かつアミロイド−β(1−42)タンパク質−オリゴマー又はその誘導体を含有する調製物を取得することにより得られる、請求項1から5までのいずれか1項に記載の調製物。
【請求項7】
アミロイド−β(1−42)タンパク質−オリゴマー、アミロイド−β(1−43)タンパク質−オリゴマー又はアミロイド−β(1−44)タンパク質−オリゴマーであるか、あるいは、アミロイド−β(1−42)タンパク質−オリゴマー、アミロイド−β(1−43)タンパク質−オリゴマー又はアミロイド−β(1−44)タンパク質−オリゴマーの標識化、不動化及び/又は架橋結合された誘導体を製造する方法において、この場合、この方法が、モノマーアミロイド−β(1−42)タンパク質、モノマーアミロイド−β(1−43)タンパク質又はモノマーアミロイド−β(1−44)タンパク質であるか、あるいは、モノマーアミロイド−β(1−42)タンパク質、モノマーアミロイド−β(1−43)タンパク質又はモノマーアミロイド−β(1−44)タンパク質の標識化された誘導体へ界面活性剤を作用させ、その後に界面活性作用を減少させ、かつ更に恒温保持し、得られたオリゴマー又は誘導体を場合により標識化、不動化及び/又は架橋結合し、かつ、前記オリゴマー又は誘導体を取得することを含む、アミロイド−β(1−42)タンパク質−オリゴマー、アミロイド−β(1−43)タンパク質−オリゴマー又はアミロイド−β(1−44)タンパク質−オリゴマーであるか、あるいは、アミロイド−β(1−42)タンパク質−オリゴマー、アミロイド−β(1−43)タンパク質−オリゴマー又はアミロイド−β(1−44)タンパク質−オリゴマーの標識化、不動化及び/又は架橋された誘導体を製造する方法。
【請求項8】
界面活性剤がイオン性である、請求項7に記載の方法。
【請求項9】
界面活性剤がドデシル硫酸ナトリウムである、請求項8に記載の方法。
【請求項10】
界面活性剤の作用時間が約1〜20時間である、請求項7から9までのいずれか1項に記載の方法。
【請求項11】
作用温度が約20〜50℃である、請求項10に記載の方法。
【請求項12】
恒温保持時間が約10〜30時間である、請求項7から9までのいずれか1項に記載の方法。
【請求項13】
恒温保持温度が約20〜50℃である、請求項12に記載の方法。
【請求項14】
アミロイド−β(1−42)タンパク質、アミロイド−β(1−43)タンパク質、アミロイド−β(1−44)タンパク質又はそれらの誘導体が、界面活性剤を作用させる前に少なくとも部分的に変性されている、請求項7から13までのいずれか1項に記載の方法。
【請求項15】
タンパク質が、水素橋−切断剤を作用させることにより変性される、請求項14に記載の方法。
【請求項16】
水素橋−切断剤がHFIPである、請求項15に記載の方法。
【請求項17】
請求項7から16までのいずれか1項に記載の方法によって得られたアミロイド−β(1−42)タンパク質−オリゴマー、アミロイド−β(1−43)タンパク質−オリゴマー又はアミロイド−β(1−44)タンパク質−オリゴマーであるか、あるいは、アミロイド−β(1−42)タンパク質−オリゴマー、アミロイド−β(1−43)タンパク質−オリゴマー又はアミロイド−β(1−44)タンパク質−オリゴマーの標識化、不動化及び/又は架橋結合された誘導体。
【請求項18】
請求項1から6までのいずれか1項に記載の調製物又は請求項17に記載のオリゴマー又は誘導体並びに場合により製薬学的に認容性の賦形剤を含む、製薬学的組成物。
【請求項19】
組成物がワクチンである、請求項18に記載の組成物。
【請求項20】
試験管内検出法における、請求項1から6まで又は17のいずれか1項に記載の調製物、オリゴマー又は誘導体の使用。
【請求項21】
生体内検出法において、診断において使用するための組成物を製造するための、請求項1から6まで又は17のいずれか1項に記載の調製物、オリゴマー又は誘導体の使用。
【請求項22】
能動免疫のための製薬学的組成物を製造するための、請求項1から6又は17のいずれか1項に記載の調製物、オリゴマー又は誘導体の使用。
【請求項23】
アミロイド−β関連の病気の治療用ワクチンを製造するための、請求項1から6又は17のいずれか1項に記載の調製物、オリゴマー又は誘導体の使用。
【請求項24】
アミロイド−β関連の病気がアルツハイマー病である、請求項23に記載の使用。
【請求項25】
物質又は物質混合物を特徴付けるための試験管内法において、
i)物質又は物質混合物を得て、
ii)物質又は物質混合物を、請求項1から6又は17のいずれか1項に記載の調製物、オリゴマー又はその誘導体に作用させ、かつ
iii)物質又は物質混合物の一定部分が、調製物、オリゴマー又はその誘導体に結合するかどうかを測定する方法を含む、物質又は物質混合物を特徴付けるための試験管内法。
【請求項26】
請求項1から6又は17のいずれか1項に記載のオリゴマー又は誘導体と特異的に結合する抗体又はその抗原結合部位。
【請求項27】
請求項1から6まで又は17のいずれか1項に記載のオリゴマー又は誘導体と、モノマーアミロイドβ(1−42)タンパク質よりも高い親和性で結合する、抗体又はその抗原結合部位。
【請求項28】
KD=10−6〜10−12Mの範囲の親和性でオリゴマー又はその誘導体と結合する、請求項26又は27に記載の抗体又はその抗原結合部位。
【請求項29】
Kd=10−8よりも高い親和性、Kd=10−9よりも高い親和性、Kd=10−10よりも高い親和性又はKd=10−11よりも高い親和性で、オリゴマー又はその誘導体と結合する、請求項26に記載の抗体又は抗原結合部位。
【請求項30】
Kd=10−8よりも低い親和性で、モノマーアミロイドβ(1−42)タンパク質−モノマー及び/又はモノマーアミロイドβ(1−40)タンパク質と結合する、請求項26から29までのいずれか1項に記載の抗体又はその抗原結合部位。
【請求項31】
オリゴマー又はその誘導体に対する前記抗体の親和性が、モノマーアミロイドβ(1−42)タンパク質及び/又はモノマーアミロイドβ(1−40)タンパク質に対する親和性よりも少なくとも10倍高いか、モノマーアミロイドβ(1−42)タンパク質及び/又はモノマーアミロイドβ(1−40)タンパク質に対する親和性よりも少なくとも100倍高いか、又はモノマーアミロイドβ(1−42)タンパク質及び/又はモノマーアミロイドβ(1−40)タンパク質に対する親和性よりも少なくとも1000倍高い、請求項26から30までのいずれか1項に記載の抗体又はその抗原結合部位。
【請求項32】
抗体が、表面プラスモン共鳴により測定したkoff−速度定数0.1s−1以下でオリゴマー又はその誘導体と結合する、請求項26から31までのいずれか1項に記載の抗体又はその抗原結合部位。
【請求項33】
抗体が、オリゴマー又はその誘導体の活性を、1x10−5以下の阻害定数IC50で阻害する、請求項26から32までのいずれか1項に記載の抗体又はその抗原結合部位。
【請求項34】
抗体がポリクローナル抗体又はモノクローナル抗体である、請求項26から33までのいずれか1項に記載の抗体又はその抗原結合部位。
【請求項35】
抗原が実際にヒト抗体、キメラ抗体又はCDRグラフト抗体である、請求項26から34までのいずれか1項に記載の抗体又はその抗原結合部位。
【請求項36】
抗体を製造する方法において、この場合、この方法が、
i)請求項1から6まで又は17のいずれか1項に記載の少なくとも1種のオリゴマー又は誘導体を用いて宿主を免疫化し;かつ、
ii)前記免疫化に対する応答として生じた抗体含有宿主血清を取得することを含む、抗体を製造する方法、但し、ヒトにおいて実施する方法を除く。
【請求項37】
免疫原として使用されたオリゴマー又は誘導体を認識する、血清の少なくとも1種の抗体が選択される、請求項36に記載の方法。
【請求項38】
請求項1から6まで又は17のいずれか1項に記載のオリゴマー又はその誘導体に特異的な抗体を製造するための方法において、この場合、この方法が、
i)前記オリゴマー又はその誘導体を含む抗原を得て;
ii)前記抗原を抗体レパートリーに作用させ;かつ、
iii)前記レパートリーから前記オリゴマー又は誘導体と特異的に結合する抗原を選択する、請求項1から6まで又は17のいずれか1項に記載のオリゴマー又はその誘導体に特異的な抗体を製造するための方法、但し、ヒトにおいて実施する方法を除く。
【請求項39】
動物を抗原で免疫化させることにより、抗原を生体内で抗体レパートリーに作用させる、請求項38に記載の方法。
【請求項40】
更に、一連のハイブリドーマを動物のリンパ球から確立し、かつオリゴマー又はその誘導体と特異的に結合する抗体を分泌するハイブリドーマを選択する、請求項39に記載の方法。
【請求項41】
動物が、マウス、ラット、ニワトリ、ラクダ、カニクイザル、ウサギ及びヤギから成る群から選択される、請求項39又は40に記載の方法。
【請求項42】
動物が、APP欠損を有するノックアウトマウス、抗原刺激後にヒト抗体を生じさせるヒト免疫グロブリン遺伝子を有するトランスジェニックマウス、ヒト末梢単核血球又はリンパ様細胞又はその前駆体で再構成されている、重症併合免疫不全(SCID)のマウス及び
致死的全身放射線照射で処理され、引き続き、重症併合免疫不全(SCID)のマウスからの骨髄細胞で照射に対して保護され、引き続き、機能的ヒトリンパ球又はその前駆体を移植されているマウスから成る群から選択される、請求項41に記載の方法。
【請求項43】
組換え体の抗体バンクを抗原でスクリーニングすることにより、試験管内で抗原を抗体レパートリーに作用させる、請求項38に記載の方法。
【請求項44】
組換え体の抗体バンクをバクテリオファージの表面で、酵母細胞の表面で、細菌細胞の表面で又はRNA−タンパク質−融合として発現させるか、あるいは、組換え体の抗体バンクが、scFv−バンク又はFab−バンク又は単一ドメイン−バンクである、請求項43に記載の方法。
【請求項45】
動物を抗原で生体内免疫化させ、かつ、動物のリンパ様細胞から製造される組換え体の抗体バンクを抗原でスクリーニングすることによってか、動物を抗原で生体内免疫化させ、かつ動物のリンパ様細胞から製造される組換え体の抗体バンクを試験管内で親和性成熟させることによってか、あるいは、動物を抗原で免疫化させ、抗原結合抗体を分泌する個々の細胞を選択し、かつ単一の細胞から重鎖又は軽鎖の可変領域のcDNAsを取得することによって、抗原を抗体レパートリーに作用させる、請求項38に記載の方法。
【請求項46】
請求項36から45までのいずれか1項に記載の方法によって得られた抗体又はその抗原結合部位。
【請求項47】
請求項26から35又は46のいずれか1項に記載の抗体又はその抗原結合部位並びに場合により製薬学的に認容性の賦形剤を含む、製薬学的組成物。
【請求項48】
アミロイドβ−関連の病気を治療するための医薬品を製造するための、請求項26から35又は46までのいずれか1項に記載の抗体又はその抗原結合部位の使用。
【請求項49】
治療が受動免疫である、請求項48に記載の使用。
【請求項50】
アミロイドβ−関連の病気を診断するための組成物を製造するための、請求項26から35まで又は46のいずれか1項に記載の抗体又はその抗原結合部位の使用。
【請求項51】
アミロイドβ−関連の病気がアルツハイマー病である、請求項48から50までのいずれか1項に記載の使用。
【図7】

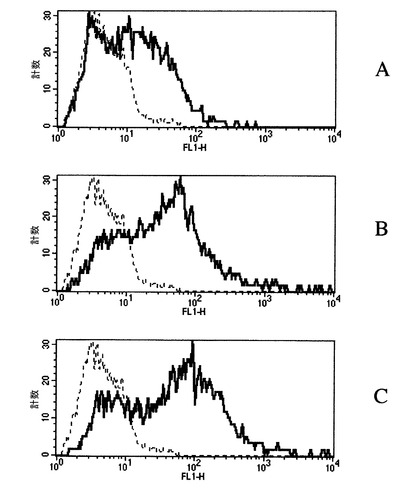
【図1】
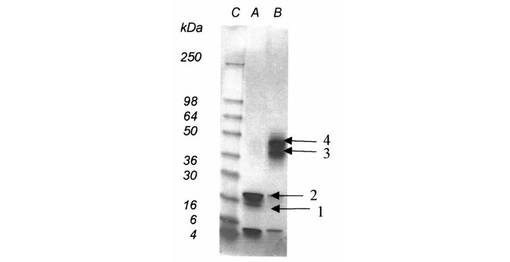
【図2】
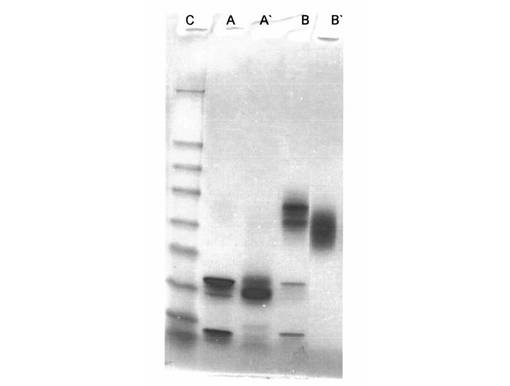
【図3】
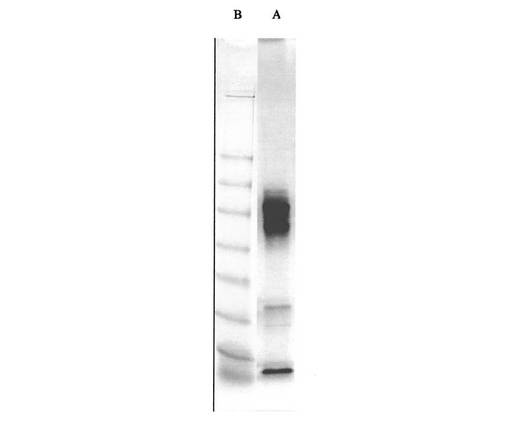
【図4】
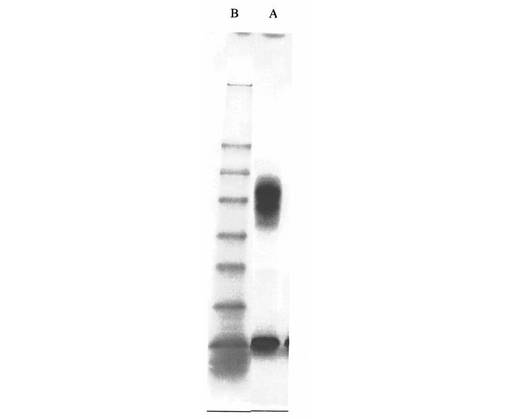
【図5】
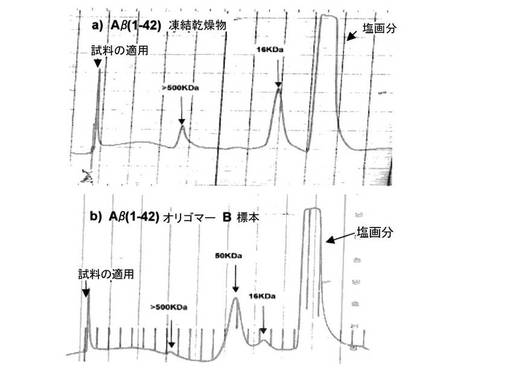
【図6】
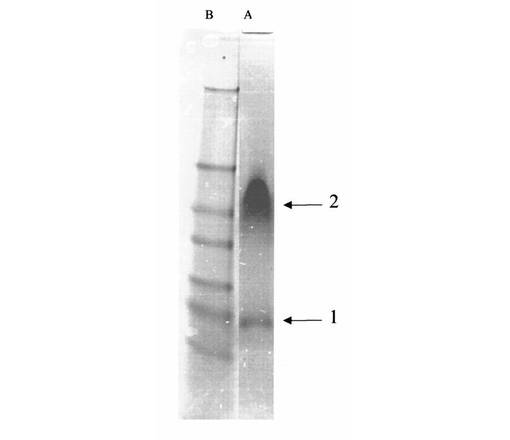
【図8】
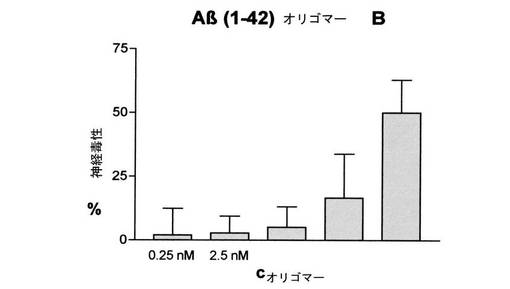
【図9】
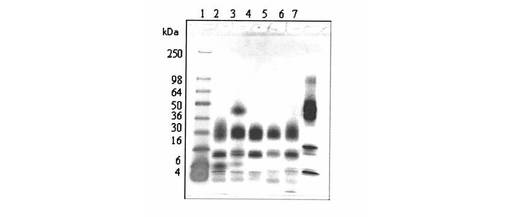
【図10】
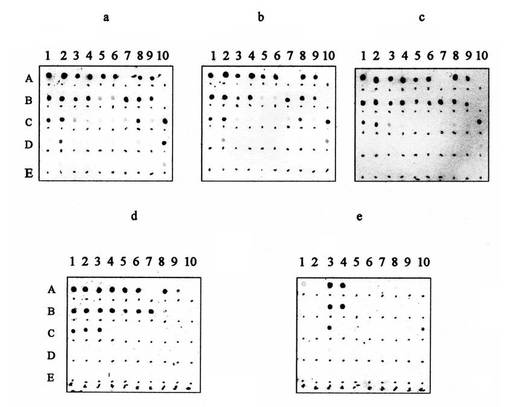
【図11】
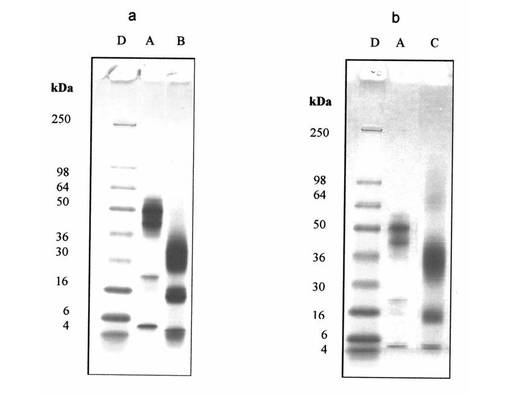
【図12】
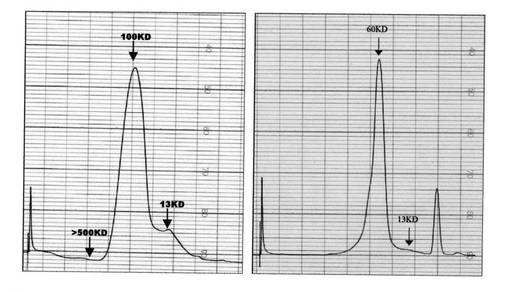
【図13】
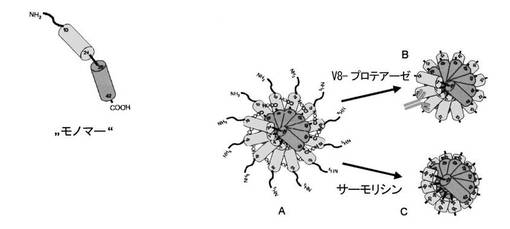
【図14】
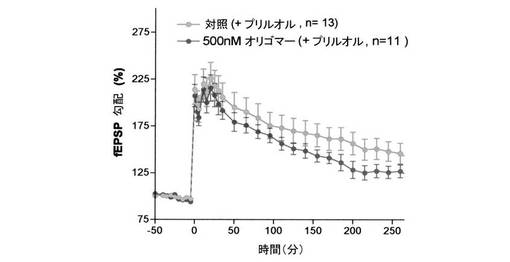
【図15】
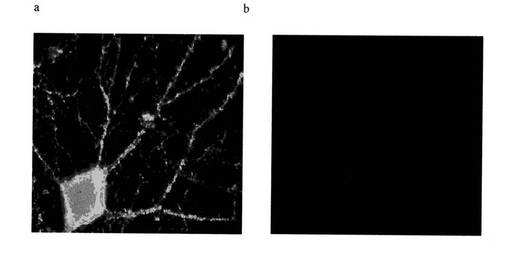
【図1】
【図2】
【図3】
【図4】
【図5】
【図6】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【公開番号】特開2010−168385(P2010−168385A)
【公開日】平成22年8月5日(2010.8.5)
【国際特許分類】
【出願番号】特願2010−21337(P2010−21337)
【出願日】平成22年2月2日(2010.2.2)
【分割の表示】特願2006−501701(P2006−501701)の分割
【原出願日】平成16年2月2日(2004.2.2)
【出願人】(502159343)アボット ゲゼルシャフト ミット ベシュレンクテル ハフツング ウント コンパニー コマンディトゲゼルシャフト (24)
【氏名又は名称原語表記】Abbott GmbH & Co. KG
【住所又は居所原語表記】Max−Planck−Ring 2, D−65205 Wiesbaden, Germany
【Fターム(参考)】
【公開日】平成22年8月5日(2010.8.5)
【国際特許分類】
【出願日】平成22年2月2日(2010.2.2)
【分割の表示】特願2006−501701(P2006−501701)の分割
【原出願日】平成16年2月2日(2004.2.2)
【出願人】(502159343)アボット ゲゼルシャフト ミット ベシュレンクテル ハフツング ウント コンパニー コマンディトゲゼルシャフト (24)
【氏名又は名称原語表記】Abbott GmbH & Co. KG
【住所又は居所原語表記】Max−Planck−Ring 2, D−65205 Wiesbaden, Germany
【Fターム(参考)】
[ Back to top ]