
アミロイド斑結合及び造影のためのピペラジン誘導体及びそれらの使用
本発明は、式(I)の化合物、それらの合成、及び特に患者におけるアミロイド沈着物の検出のためのそれらの使用に関する。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、アミロイド沈着物に結合しかつ造影するのに有用な新規化合物、並びにアルツハイマー病及びアミロイドーシスの検出又は治療におけるそれらの使用に関する。
【背景技術】
【0002】
アルツハイマー病(AD)は、記憶、認知、及び行動の安定性の喪失により特徴付けられる進行性神経変性障害である。ADは、β−アミロイドペプチド(Aβ)の線維状沈着物により構成される細胞外老人斑及び過剰リン酸化されたタウの対らせん状細線維(PHF)により構成された神経原線維変化(FT)により、病理学的に定義される。Aβペプチドを構成している39〜43個のアミノ酸は、より大きいアミロイド前駆体タンパク質(APP)に由来している。アミロイド形成経路において、Aβペプチドは、APPからβ−及びγ−セクレターゼの逐次タンパク質分解により、切断される。Aβペプチドは、可溶性タンパク質として放出され、かつ正常な加齢脳内の脳脊髄液(CSF)中で低レベルで検出されることができる。ADの進行時に、Aβペプチドは凝集し、かつ脳の実質及び脈管構造内にアミロイド沈着物を形成し、これは組織学的試験において、びまん性老人斑及び血管アミロイドとして、剖検時に検出され得る(最新の総説については、Blennowら、Lancet., 368(9533): 387-403 (2006年7月29日)を参照のこと)。
【0003】
アルツハイマー病は、世界中で、大きい健康上及び社会経済学上の問題となり始めている。本疾患の早期検出及び有効治療に関する技術及び方法を開発するために、膨大な努力が成されている。現在、記憶障害クリニックの専門的状況(academic setting)においてADの診断は、およそ85〜90%の精度である(Petrella JRら、Radiology, 226:315-36 (2003))。これは、同様の症状を引き起こす様々な疾患の排除、並びに慎重な神経学的及び精神医学的検査、更には神経心理学的試験を基にしている。しかし依然、脳の剖検時組織検査がこの疾患の唯一の確定診断である。従って本疾患のひとつの病理学的特徴―脳内のアミロイド凝集物の沈着―のインビボ検出は、ADの早期検出及び他の認知症からの識別に大きい影響を有すると考えられる。加えて開発中のほとんどの疾患修飾療法は、脳内のアミロイド負荷の軽減を目的としている。従って脳内のアミロイド負荷の造影は、患者階層化及び治療モニタリングにとって必須のツールを提供することができる。
【0004】
加えてアミロイド沈着物は、アミロイドーシスにおいて役割を果たすこともわかっており、ここではアミロイドタンパク質が、様々な臓器及び/又は組織内に異常に沈着され、疾患を引き起こす。最近の総説については、Chitiらの論文、Annu Rev Biochem., 75:333-66 (2006)を参照のこと。
【0005】
脳内のアミロイド凝集物の可視化のための可能性のあるリガンドは、アミロイドへの高い結合親和性を示さなければならず、かつ血液脳関門を超えなければならない。AD患者の脳内でのトレーサーの結合パターンに関してヒトにおいて既に研究されているPETトレーサーは、[F−18]FDDNP(Shoghi-Jadidら、Am J Geriatr Psychiatry, 10:24-35 (2002))、[C−11]PIB(Klunkら、Ann Neurol. 55:306-319 (2004))、[C−11]SB−13(Verhoeffら、Am J Geriatr Psychiatry, 12:584-595 (2004);[F−18]Bay94−9172(Roweら、Lancet Neurol, 7:129-135 (2008));[C−11]BF227(Kudoら、J Nucl. Med, 49:554-561 (2007));及び、[F−18]PIB(Farrarら、Turku PET Symposium, Abstract 49(2007))がある。最近の総説については、Lockhardtの論文、Drug Discov Today, 11:1093-1099 (2006)、Henriksenらの論文、Eur. J. Nucl. Med. Mol. Imaging, (2007)、Cohenの論文、Mol. Imaging Biol. 9:204-2162007、Nordbergの論文、Curr. Opin Biol., 20:398-402 (2007)、Smallらの論文、Neurology 7:161-172 (2008)、Nordbergの論文、Eur. J. Nucl. Med. Mol. Imaging, 35, S46-S50(2008)を参照されたい。
【0006】
現在最も有望なPETトレーサーは、脳内のアミロイド沈着物へのそれらの特異的結合に加え、特にAD患者の白質脳領域内に加え海馬(HC)において、不都合な非特異的蓄積を示す。概して非特異的バックグラウンド結合は、画質に干渉し、かつ例えばアミロイドの定量及び本疾患の極初期の診断を損なう場合がある。
【発明の概要】
【発明が解決しようとする課題】
【0007】
すなわち、本発明の前提となる課題は、本疾患患者におけるアミロイド沈着物を、高い特異性で、アミロイド関連疾患の初期の段階で検出するのに適した化合物を提供することであった。
【0008】
本発明は、アミロイドβへの親和性が高く、脳から非特異的シグナルが迅速に排除される、新規トレーサーを提供することにより、本課題を解決する。
【課題を解決するための手段】
【0009】
本発明は、アミロイド沈着物に結合し、かつ血液脳関門を通過することができ、従って患者のアルツハイマー病及びアミロイドーシスを、好ましくは該疾患の初期において、診断するのに有用な化合物に関する。
【0010】
一態様によれば、本発明は、式Iの化合物、及び医薬として許容し得るその塩又はプロドラッグに関する。
【化1】
(式中、
−Yは:
F、Cl、Br、I、H、
18F、19F、76Br、123I、125I、11C、3H、13N、15O等の検出可能な標識;
トシル、ブロシル、ノシル、トリフラート、スルホネート、置換スルホネート、メシラート、及びノナフレートなどの脱離基;並びに
芳香族C原子に直接結合される場合は、ヨードニウム−アリールI+−アリール、トリアルキルアンモニウム、好ましくはトリメチルアンモニウム、及びNO2からなる群から選択され;
−Arは、1又は2個のアルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基により任意に置換されていてもよい、単環、二環又は三環式の芳香環又は芳香族複素環系からなる群から選択され;
ここで、該アルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基は、任意に置換されていてもよく、ここで該置換基は、好ましくはオキソ又はヒドロキシルから選択され、
該アルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基は、1〜5個の酸素原子、−SO−又は−SO2−基により隔てられていてもよく、好ましくは、該置換基はポリエチレングリコール部分であり、
該アルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基は、C3−C6シクロアルキル部分を含んでいてもよく、
該単環、二環又は三環式の芳香環又は芳香族複素環系は、芳香族C原子に直接結合されている電子求引基により更に置換されていてもよく、好ましい電子求引基は、−CN又は−CF3であり;
好ましくは、Arは、フェニル、2−、3−、又は4−ピリジル、ピリミジル、ピラジル、プロピルピリミジン−2−イル、エトキシフェニル、(CH2CH2O)3フェニル、アルキルフェニル、アルコキシフェニル、N−アルキルインドリル、及びアルキルピリジルからなる群から選択され、
更に好ましい実施形態によれば、
【化2】
であり;
−Bは、直接結合、1〜10個のC原子からなる分枝又は非分枝のアルキル又はアルキレン鎖からなる群から選択され、ここで該アルキレン鎖は、1又は2個の不飽和結合を含んでいてもよく、
該アルキル又はアルキレン鎖は、N、S、SO、SO2又はOにより任意に隔てられていてもよく、
該アルキル又はアルキレン鎖は、オキソ又は−OHにより任意に置換され;
好ましくは、Bは、直接結合、CONH−CH2CO、CO−(CH2)2CO、(CH2)nCO、O(CH2)nCO(但しn=1〜10)、及び(CH=CH)COからなる群から選択され、
さらに好ましくは、
【化3】
であり;
−Aは、直接結合、及びCO−NH、CS−NHからなる群から選択され;
−Ar’は、1又は2個のアルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基により任意に置換されていてもよい、単環又は二環式芳香環又は芳香族複素環系からなる群から選択され;
該アルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基は、任意に置換されていてもよく、該置換基は、好ましくはオキソ又はヒドロキシルから選択され、
該アルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基は、1〜5個の酸素原子により隔てられていてもよく、好ましくは、該置換基は、ポリエチレングリコール部分であり、
該アルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基は、C3−C6シクロアルキル部分を含んでいてもよく;
好ましくは、Ar’は、フェニル、2−、3−、又は4−ピリジル、ピリミジル、ピラジル、プロピルピリミジン−2−イル、エトキシフェニル、(CH2CH2O)3フェニル、アルキルフェニル、アルコキシフェニル、N−アルキルインドリル、フェニル、ベンゾフラニル、インドリル及びアルキルピリジルからなる群から選択され、
より好ましくは、Ar’は、フェニル、ベンゾフラニル、及びインドリルからなる群から選択され、
さらに好ましくは、
【化4】
であり;
最も好ましくは、Ar’は、フェニルであり;
−Xは、直接結合、又は好ましくは非限定的にオキソ若しくはチオである1若しくは2個の置換基により任意に置換されていてもよい、C1−C3アルキル鎖からなる群から選択され;
該アルキル鎖は、1〜2個のO、N、S、SO又はSO2基により隔てられていてもよく;
好ましくは、Xは、直接結合、OCH2、NHCO、CH2O、CONH、NHCS、又はCSNHからなる群から選択され;
−Ar”は、1又は2個のアルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基により任意に置換されていてもよい、単環又は二環式芳香環又は芳香族複素環系からなる群から選択され、
該アルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基は、任意に置換されていてもよく、該置換基は、好ましくはオキソ又はヒドロキシルから選択され、
該アルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基は、1〜5個の酸素原子により隔てられていてもよく、好ましくは、該置換基は、ポリエチレングリコール部分であり、
該アルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基は、C3−C6シクロアルキル部分を含んでいてもよく、
より好ましくはAr”は、フェニル、1−フェニル、1−ナフチル、2−ナフチル、及びそれらに対応する複素環全てからなる群から選択され、
更に好ましくは、
【化5】
である。
【0011】
Ar’並びにAr”は:
F、Cl、Br、I、H、
18F、19F、76Br、123I、125I、11C、3H、13N、15O等の検出可能な標識;
トシル、ブロシル、ノシル、トリフラート、スルホネート、置換スルホネート、メシラート、及びノナフレートなどの脱離基;並びに
芳香族C原子に直接結合される場合は、ヨードニウム−アリールI+−アリール、トリアルキルアンモニウム、好ましくはトリメチルアンモニウム、及びNO2
により、任意に置換されていてもよい。
【図面の簡単な説明】
【0012】
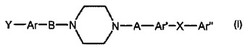
【図1】図1は、アルツハイマー病患者(AD)及びAβプラークを伴わない対照(HC/FTD)(健常対照/前頭側頭型認知症)の皮質由来の凍結切片への3gの結合のオートラジオグラフィー分析を示す。AD試料の斑豊富領域における特異的結合を矢印で示す。
【図2】図2は、アルツハイマー病患者(AD)及びAβプラークを伴わない対照(HC/FTD)(健常対照/前頭側頭型認知症)の皮質由来の凍結切片への実施例10eの結合のオートラジオグラフィー分析を示す。AD試料の斑豊富領域における特異的結合を矢印で示す。
【図3】図3は、アルツハイマー病患者(AD)及びAβプラークを伴わない対照(HC/FTD)(健常対照/前頭側頭型認知症)の皮質由来の凍結切片への実施例11cの結合のオートラジオグラフィー分析を示す。AD試料の斑豊富領域における特異的結合を矢印で示す。
【図4】図4は、アルツハイマー病患者(AD)及びAβプラークを伴わない対照(HC/FTD)(健常対照/前頭側頭型認知症)の皮質由来の凍結切片への実施例12cの結合のオートラジオグラフィー分析を示す。AD試料の斑豊富領域における特異的結合を矢印で示す。
【図5】図5は、AD患者由来の脳ホモジネートを使用する競合アッセイにおいて測定された、選択された化合物の[nM]でのIC50値を示す。
【図6】図6A及び図6Bは、実施例14の化合物のHPLC分析を示す。
【図7】図7A及び図7Bは、実施例15の化合物のHPLC分析を示す。
【図8】図8は、アルツハイマー病患者(AD)及び健常対照(HC)の皮質由来の凍結切片への14fの結合のオートラジオグラフィー分析を示す。AD試料の斑豊富領域における特異的結合を矢印で示す。
【図9】図9は、AD患者由来の脳ホモジネートを使用する競合アッセイにおいて測定された、選択された化合物の[nM]でのIC50値を示す。
【発明を実施するための形態】
【0013】
インビトロ及びインビボ双方の診断目的には、放射性核種又は蛍光標識等の検出可能な標識を含んでなり、又はそれらを有する式Iの化合物が好ましい。インビトロ使用の場合、新鮮凍結試料又はパラフィン試料等の組織切片が分析され得る。
【0014】
式Iの化合物の好ましい実施形態は以下に示すとおりであるが、これらを化合物1d/e、2d/e、3g/h、4f/g、5b/c、6f、7d、8g、9d/e、10、11、12、13、14、15、16、17、18、19、20、21及び22と称する。これらの好ましい実施形態は、例えば式Iの記号Y、Ar、B、A、Ar’、X、及びAr”により表される様々な基の例でもある。
【0015】
「アルキル」とは、炭素及び水素のみからなり、1〜8個の炭素原子を有し、不飽和部位を含まない、直鎖又は分枝鎖の基をいう。例えば、メチル、エチル、n−プロピル、1−メチルエチル(イソ−プロピル)、n−ブチル、n−ペンチル、1,1−ジメチルエチル(t−ブチル)、n−ヘプチル等が挙げられる。「アルコキシ」とは、−Oアルキル基という式で表される基である。ここでアルキルは先に定義されるとおりである。
【0016】
本発明との関連において好ましい塩は、医薬として許容し得る本発明の化合物の塩である。本発明は、それ自体は医薬適用に適していないが、例えば本発明の化合物の単離又は精製等に使用できる塩も含む。
【0017】
本発明の化合物の医薬として許容し得る塩は、無機酸、カルボン酸及びスルホン酸の酸付加塩、例えば、塩酸、臭化水素酸、硫酸、リン酸、メタンスルホン酸、エタンスルホン酸、トルエンスルホン酸、ベンゼンスルホン酸、ナフタレンジスルホン酸、酢酸、トリフルオロ酢酸、プロピオン酸、乳酸、酒石酸、リンゴ酸、クエン酸、フマル酸、マレイン酸及び安息香酸の塩を含む。
【0018】
本発明の化合物の医薬として許容し得る塩の例として、好ましくは、アルカリ金属塩(例えば、ナトリウム塩及びカリウム塩)、アルカリ土類金属塩(例えば、カルシウム塩及びマグネシウム塩)、並びにアンモニア、又は炭素原子1〜16個を有する有機アミン、例えば、好ましくは、エチルアミン、ジエチルアミン、トリエチルアミン、エチルジイソプロピルアミン、モノエタノールアミン、ジエタノールアミン、トリエタノールアミン、ジシクロヘキシルアミン、ジメチルアミノエタノール、プロカイン、ジベンジルアミン、N−メチル−モルホリン、アルギニン、リシン、エチレンジアミン及びN−メチルピペリジンなどに由来したアンモニウム塩などの、慣例的塩基の塩も含む。
【0019】
更に本発明は、本発明の化合物のプロドラッグも含む。用語「プロドラッグ」は、それらの化合物の一部に関してそれらは生物学的に活性又は不活性であるが、それらが体内で消費される間に本発明の化合物に転換される(例えば、代謝又は加水分解により)化合物を含む。
【0020】
特に本発明は、式(I)のカルボン酸の加水分解可能なエステル誘導体も含む。これらは、生理的媒体において、特にインビボにおいて、酵素的又は化学的手段により加水分解され、遊離カルボン酸を生じることができるエステルであると理解される。このようなエステルは好ましくは、アルキル基がヒドロキシル、(C1−C4)−アルコキシ、アミノ、モノ−(C1−C4)−アルキルアミノ及び/又はジ−(C1−C4)−アルキル−アミノにより置換されてよい、直鎖又は分枝された(C1−C6)−アルキルエステルである。特に好ましいのは、式(I)の化合物のメチルエステル又はエチルエステルである。
【0021】
本発明の文脈において、他に特定されない限りは、置換基は下記の意味を有する。
本発明の文脈において、アルキルは、各場合に言及された炭素原子数を有する、直鎖又は分枝鎖のアルキルラジカルを表す。下記のラジカルは、一例として及び好ましいものとして言及されている:メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、1−メチルプロピル、tert−ブチル、n−ペンチル、イソペンチル、1−エチルプロピル、1−メチルブチル、2−メチルブチル、3−メチルブチル、n−ヘキシル、1−メチルペンチル、2−メチルペンチル、3−メチルペンチル、4−メチルペンチル、3,3−ジメチルブチル、1−エチルブチル、2−エチルブチル、1,4−ジメチルペンチル、4,4−ジメチルペンチル及び1,4,4−トリメチルペンチル。
【0022】
本発明の文脈において、シクロアルキルは、炭素原子3〜7個を有する、単環の飽和されたアルキルラジカルを表す。下記のラジカルは、一例として及び好ましいものとして言及されている:シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル及びシクロヘプチル。
【0023】
本発明の文脈において、アルコキシは、炭素原子1〜4個を有する、直鎖又は分枝されたアルコキシラジカルを表す。下記のラジカルは、一例として及び好ましいものとして言及されている:メトキシ、エトキシ、n−プロポキシ、イソプロポキシ、1−メチルプロパオキシ、n−ブトキシ、イソブトキシ及びtert−ブトキシ。
本発明の文脈において、ハロゲンは、フッ素、塩素、臭素及びヨウ素を含む。好ましいのはフッ素である。
【0024】
本発明の化合物のラジカルが置換されている場合、これらのラジカルは、他に特定されない限りは、単−又は多置換されることができる。本発明の文脈において、2回以上出現する全てのラジカルの意味は、互いに独立している。1、2又は3個の同一又は異なる置換基による置換が、好ましい。とりわけ好ましいのは、1個の置換基による置換である。
【0025】
本発明の化合物又は医薬として許容し得るその塩は、それらの構造内に不斉炭素原子を有することができる。従って本発明の化合物及び医薬として許容し得るその塩は、単独のエナンチオマー、ジアステレオ異性体、ラセミ体、及びエナンチオマーとジアステレオマーの混合物として存在してよい。そのような単独のエナンチオマー、ジアステレオ異性体、ラセミ体、及びそれらの混合物は全て、本発明の範囲内である。
【0026】
先に及び本明細書において説明された化合物は、本発明の好ましい実施形態において、Aβペプチドへ結合される。
【0027】
本発明の別の態様は、特にヒトなどの哺乳動物における、患者のアルツハイマー病及び/又はアミロイドーシスを診断及び/又は治療するための先に及び本明細書において説明された式Iの化合物の使用である。
【0028】
アルツハイマー病及び/又はアミロイドーシスの患者の治療は、好ましくは、放射性標識を有さないが、Yが例えば水素である、式Iの本発明の化合物により実施されることができる。
【0029】
好ましくは、この診断における本発明の化合物の使用は、ポジトロン放出断層撮影(PET)、単光子放出型コンピュータ断層撮影(SPECT)、磁気共鳴(MR)スペクトル分析法又は断層撮影法を使用し実行される。
【0030】
本発明の別の態様は、アミロイド沈着物の造影方法に関する。そのような方法は、a)検出可能な標識を含む先に及び本明細書において説明された化合物を、哺乳動物へ投与すること、及びb)アミロイド沈着物に特異的に結合されている本化合物から発生する(steming)シグナルを検出することを含む。この特異的結合は、本発明の化合物のアミロイド沈着物への高い結合親和性の結果である。
【0031】
更なる態様において、本発明は、アルツハイマー病又はアミロイドーシスの患者を診断する方法に関する。この方法は、a)先に及び本明細書に説明されたヒトにおける本化合物の検出のための検出可能な標識を伴う本発明の化合物を、そのような診断が必要なヒトへ投与すること、及びb)本化合物のヒトへの投与から生じる、検出可能な標識からのシグナルを、好ましくはγ線カメラの使用、ポジトロン放出断層撮影(PET)、又は単光子放出型コンピュータ断層撮影(SPECT)により、測定することを含む。
【0032】
本発明の更なる実施形態は、本発明の化合物を患者へ投与すること、及び本発明の造影方法を適用することを含む、患者におけるアルツハイマー病の除外を含む、アルツハイマー病のような他の神経学的障害の診断方法を含む。
【0033】
本発明の更なる態様は、放射標識された式Iの化合物を含有する、アミロイド沈着物の造影のための診断用組成物に関する。
【0034】
本発明の診断方法は、剖検時診断方法においても使用することができる。
【0035】
更に本発明の診断方法は、アルツハイマー病、神経変性障害又はアミロイドーシスの療法のモニタリングに使用することもできる。
【0036】
更に本発明の診断方法は、アルツハイマー病を除外することにより、アルツハイマー病以外の神経学的障害を診断するためにも使用することができる。
【0037】
本発明の更なる態様において、本発明は、本明細書に説明されたような式Iの化合物を、そのような治療を必要とするヒトに投与することを含む、アミロイドーシス又はアルツハイマー病を治療又は予防する方法を含む。
【0038】
本発明の更なる態様は、本明細書に説明されたような本発明の化合物を、任意に好適な担体及び/又は添加剤と一緒に含有する医薬組成物に関する。
【0039】
更に、本発明の化合物は、例えば、ハイスループットスクリーニング法及びインビトロアッセイなどの、スクリーニングにおけるツールとして使用されることもできる。
【0040】
本発明は、本明細書に説明されたような式Iの本発明の化合物を合成する方法にも関する。本発明の化合物の全般的合成方法は、以下である。
【0041】
F−18放射標識
本発明の更なる態様は、式Iの化合物をフッ素化剤と反応させる工程を含む、放射標識された式Iの化合物を製造するための、式Iの化合物の放射性フッ素化の方法に関する。有用な放射性フッ素化法は、当業者に周知である。
【0042】
好ましい実施形態において、前記フッ素化剤は、4,7,13,16,21,24−ヘキサオキサ−1,10ジアザビシクロ[8.8.8]−ヘキサコサンK18F(クラウンエーテル塩クリプトフィックスK18F)、K18F、H18F、KH18F2又は18Fのテトラアルキルアンモニウム塩である。より好ましいフッ素化剤は、K18F、H18F、又はKH18F2である。
【0043】
使用される溶媒は、ジメチルホルムアミド、DMF、ジメチルスルホキシド、DMSO、アセトニトリル、MeCN、ジメチルアセトアミド、DMA、DMAAなどであり、好ましくはDMSO、MeCN又はDMFである。これらの溶媒は、先に記した溶媒の混合物であることもできる。
【0044】
[F−18]放射標識手順は、当業者に周知である。例えば、放射標識は、以下に説明されたように実行されることができる。
【0045】
[F−18]フッ化物は、18O(p,n)18F反応のために[O−18]水が充填された銀標的(1mL)を使用するサイクロ(登録商標)トロン内における陽子衝撃により生成されることができる。この水性[F−18]フッ化物は、カートリッジ(例えば、QMA樹脂カートリッジ、Waters社、Sep Pak Light QMA部品番号:WAT023525)を通過させることができる。次に捕獲された[F−18]フッ化物は、例えばクリプトフィックスK2.2.2/K2CO3溶液の添加により、カートリッジから溶離されることができる(クリプトフィックスは4,7,13,16,21,24−ヘキサオキサ−1,10−ジアザビシクロ[8.8.8]−ヘキサコサンである)。この前駆体の求核置換は、NBu4OH、(NBu4)2CO3、K2CO3などの塩基の存在下、高温で働くことが好ましい。クリプトフィックス(K2.2.2)などのクラウンエーテルの添加は、特に塩基としてのK2CO3の存在下で、この反応に正に影響を及ぼすことができる。
【0046】
フッ化カリウムクリプトフィックス錯体は、アセトニトリルの逐次添加による、繰り返しの共沸蒸留により好ましくは乾燥される。アセトニトリル、DMF、DMSOなどの溶媒は、反応溶媒として使用されることができる。この標識生成物は、カートリッジを使用する固相抽出により精製されることができる。好ましいカートリッジは、Sep−Pak Plus C18カートリッジ(Waters社、WAT020515)である。このカートリッジは、水によりすすぐことができ、かつこの化合物は、アセトニトリルにより溶離されることができる。溶離された化合物は、水により希釈されることができ、かつ次に分取HPLC精製に供されることができる。好ましいHPLCカラムは、Gemini 5μ C18 110Å、250×10mm(Phenomenex社、00G-4435-N0)などの、逆相カラムである。緩衝溶液、酸、水などの、アセトニトリル、メタノール、エタノールなどの有機溶媒との混合液は、移動相として使用されることができる。
【0047】
その後この溶液は、例えば濃縮及び溶媒交換のために、カートリッジを通過される水により希釈されることができる。
【0048】
F−18化合物:アルキル−F及び(ヘテロ)アリール−Fの全般的合成
一般式Iのアルキル−F−18化合物(式IでY=18F)の前駆体は、例えばトシラート、ブロシラート、ノシラート、メシラート、トリフラート、ノナフレートなど(式IでY=脱離基)であり、これらは当該技術分野において公知の方法に従い各ヒドロキシ化合物から合成されることができる(J. March、Advanced Organic Chemistry、第4版、1992、John Wiley & Sons社、352ff頁)。追加の方法は、実施例3f、4e及び5aに説明されており、かつ例えばTsO−(CH2)n−OTsのような、好適なビス(トシラート)などによる合成を含む。
【0049】
一般式Iのアルキル−F−18化合物(式IでY=18F)のその他の前駆体は、例えばヨウ化物及び臭化物などであり、かつそれらの各フッ化物への転化も当該技術分野において公知である(J. March、前記参照)。
【0050】
一般式Iのアリール−F−18化合物の前駆体は、例えば、当該技術分野において公知の方法により本発明の各F−18化合物へ転換されることができる、アリール又はヘテロアリールブロミド、ニトロ化合物、トリアルキルアンモニウム、アリールヨードニウムである(L. Cai、S. Lu、V. Pike、Eur. J. Org. Chem, 2853-2873 (2008))。これらの前駆体のための出発材料は市販されているか、又は当該技術分野において公知の方法により合成されることができる(R.C. Larock、Comprehensive Organic Transformations、VCH Publishers社、1989)。
【0051】
トシラート、ブロシラート、ノシラート、メシラート、トリフラート、ノナフレートなどのための出発材料としてのヒドロキシ化合物の合成は、以下を含む。
【0052】
・OH−保護基の脱保護。非常に多用途な保護基のひとつとして、アセチル保護基が挙げられる。多くのその他のものが、当該技術分野において公知であり、例えばT.W. Greene及びP.G.M. Wuts、Protective Groups in Organic Synthesis、第3版、1999年、John Wiley & Sons社、17ff頁を参照し、同じく例えば実施例3d及び4cも参照されたい。
【0053】
・実施例1b及び2bに示すような、Hal=Br、Iである、
【化6】
の、Pd触媒された置換による、ヒドロキシアルキル基の導入。
【0054】
・実施例6dに示すような、B’が−CO−(CH2)n−COOHであるヒドロキシ化合物HO−Ar−B’は、フリーデル・クラフツアシル化により合成することができる。
【0055】
Y=Br、I、HO、保護基−O−である、一般式Iの化合物は、下記により合成されることができる。
【化7】
【0056】
・任意の位置及び配列順での−CO−NH−基を確立するための、アミドカップリング。
又は
【0057】
・実施例1a及び2aに説明されたように、任意に、尿素結合(−NH−CO−NH−)を構築するための、ホスゲン又はホスゲン同等物の使用による、ピペラジン誘導体の、好適なアミノ成分との反応による。
【0058】
アリール−ピペラジンは、市販されているか、又は文献に従い、例えばKlaparsらの文献(Journal of the American Chemical Society, 124, 7421-28 (2002))及びそこに引用された文献に従い、合成されることができる。
【0059】
本発明の更なる態様は、本発明の放射標識されない化合物を備えるキットに言及しており、この化合物は任意に、乾燥状態であるか、又は不活性の医薬として許容し得る担体及び/若しくは溶媒及び/若しくは補助物質が添加されている。好ましい実施形態において、本キットは、該キットの化合物を含む、1個又は複数の密閉容器を備えている。好ましい実施形態において、本キットは、Yが、トシル、ブロシル、ノシル、トリフラート、スルホネート、置換スルホネート、メシラート、ノナフレート、ヨードニウム−アリールI+−アリール、トリアルキルアンモニウム、トリメチルアンモニウム、又はNO2などの脱離基である、本明細書に説明された式Iの化合物を備えている。
【0060】
更に別の本発明の態様は、哺乳動物において、アミロイドの形成を阻害するか、又はアミロイドの病原性を調節する方法に関する。この方法は、式Iの化合物が、アミロイドの形成を阻害するか、又はアミロイドの病原性を調節する上で有効である量で投与されることを含む。
【0061】
発明の好ましい化合物
本発明の種々の態様によれば、特定の式Iの化合物が好ましい。斯かる好ましい化合物を以下に示す。化合物に付した記号は、対応する化合物の合成を示す実施例を表す。
【0062】
【化8】
【0063】
【化9】
【0064】
【化10】
【0065】
より一層好ましい本発明の実施形態は、各F−18化合物である。
【0066】
本発明の化合物は、アミロイドβに高親和性を持ち、かつ非特異的シグナルが脳から迅速に排泄される、新規トレーサーを表している。
【0067】
特に本発明は、以下に関する:
【0068】
(1)式Iの化合物、又は医薬として許容し得るその塩若しくはプロドラッグ:
【化11】
(式中、
−Yは、F、Cl、Br、I、H、18F、19F、76Br、123I、125I、11C、3H、13N、15O;
脱離基、トシル、ブロシル、ノシル、トリフラート、スルホネート、置換スルホネート、メシラート、及びノナフレート;並びに、芳香族C原子に直接結合される場合は、ヨードニウム−アリールI+−アリール、トリアルキルアンモニウム、好ましくはトリメチルアンモニウム、及びNO2からなる群から選択され;
−Arは、置換又は非置換の、単環、二環又は三環式の、芳香環又は芳香族複素環系からなる群から選択され;
−Bは、直接結合、1〜10個のC原子からなる分枝又は非分枝のアルキル又はアルキレン鎖からなる群から選択され;
−Aは、直接結合、及びCO−NH、CS−NHからなる群から選択され;
−Ar’は、置換又は非置換の、単環又は二環式の、芳香環又は芳香族複素環系からなる群から選択され;
−Xは、直接結合、又は、置換若しくは非置換のC1−C3アルキル鎖からなる群から選択され;
−Ar”は、置換又は非置換の、単環又は二環式の、芳香環又は芳香族複素環系からなる群から選択される)。
【0069】
(2)1番の化合物、式中:
−Arの芳香族複素環又は芳香環系は、1又は2個のアルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基により任意に置換されており;
−単環、二環又は三環式の芳香環又は芳香族複素環系は、芳香族C原子に直接結合された電子求引基により更に置換されてよく;
−Bのアルキレン鎖は、1又は2個の不飽和結合を含んでいてもよく、ここでアルキル又はアルキレン鎖は、任意にN、S、SO、SO2又はOにより隔てられ、ここでアルキル又はアルキレン鎖は、オキソ又は−OHにより任意に置換されており;
−Ar’の芳香族複素環又は芳香環系は、1又は2個のアルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基により任意に置換されていてもよく、ここで単環、二環又は三環式の芳香環又は芳香族複素環系は、芳香族C原子に直接結合された電子求引基により更に置換されてよく;
−XのC1−C3アルキル鎖は、オキソ又はチオであってよい1又は2個の置換基により任意に置換され、ここでアルキル鎖は、1〜2個のO、N、S、SO又はSO2基により隔てられていてもよく;
−Ar”の芳香族複素環又は芳香環系は、1又は2個のアルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基により任意に置換され、ここで単環、二環又は三環式の芳香環又は芳香族複素環系は、芳香族C原子に直接結合された電子求引基により更に置換されてよい。
【0070】
(3)1番又は2番の化合物、式中:
−Ar、Ar’及びAr”の任意のアルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基は、オキソ又はヒドロキシルからなる群から選択され;
Ar、Ar’及びAr”のアルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基は、1〜5個の酸素原子により隔てられていてもよく、好ましくはこの置換基はポリエチレングリコール部分であり;
更にAr、Ar’及びAr”のアルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基は、C3−C6シクロアルキル部分を含んでいてもよく、ここでAr、Ar’及びAr”の任意の電子求引基は、−CN又はCF3からなる群から選択され;
−Bは、直接結合、CONH−CH2CO、CO−(CH2)2CO、(CH2)nCO、O(CH2)nCO(但しn=1〜10)、(CH=CH)CO、
【化12】
からなる群から任意に選択され;
−Xは、直接結合、OCH2、NHCO、CH2O、CONH、NHCS、又はCSNHからなる群から任意に選択される。
【0071】
(4)1番−3番の化合物、式中:
−Arは、プロピルピリミジン−2−イル、エトキシフェニル、(CH2CH2O)3フェニル、アルキルフェニル、アルコキシフェニル、N−アルキルインドリル、及びアルキルピリジル、
【化13】
からなる群から任意に選択され;
−Ar’は、プロピルピリミジン−2−イル、エトキシフェニル、(CH2CH2O)3フェニル、アルキルフェニル、アルコキシフェニル、N−アルキルインドリル、フェニル、ベンゾフラニル、インドリル及びアルキルピリジルからなる群から任意に選択され;
−Ar”は、フェニル、1−フェニル、1−ナフチル、2−ナフチル、及びそれらに対応する複素環全てからなる群から任意に選択される。
【0072】
(5)下記からなる群から選択される化合物:
【化14】
【化15】
【化16】
【0073】
(6)Fが、18Fの意味を有する、5番の化合物。
【0074】
(7)放射性核種又は蛍光標識などの、検出可能な標識を含む、1番、2番、3番又は4番の化合物。
【0075】
(8)検出可能な標識が、18Fである、7番の化合物。
【0076】
(9)診断用化合物としての、1番−8番の化合物。
【0077】
(10)医薬品としての、1番−5番又は7番の化合物。
【0078】
(11)アルツハイマー病、神経変性障害、又はアミロイドーシスからなる群から選択される疾患のための診断用化合物としての、8番の化合物。
【0079】
(12)アルツハイマー病、神経変性障害、又はアミロイドーシスからなる群から選択される疾患の治療のための医薬品としての、10番の化合物。
【0080】
(13)好適な前駆分子をフッ素化剤と反応させることを含む、1番−8番に従う、フッ素化された化合物の調製方法。
【0081】
(14)1番−5番又は7番の化合物の治療的有効量を、哺乳動物へ投与することを含む、該哺乳動物におけるアルツハイマー病、神経変性障害又はアミロイドーシスからなる群から選択される障害の治療又は予防の方法。
【0082】
(15)1番−5番又は7番の化合物の治療的有効量が、哺乳動物へ投与される、該哺乳動物におけるアルツハイマー病、神経変性障害又はアミロイドーシスからなる群から選択される疾患の治療における1番−5番又は7番の化合物の使用。
【0083】
(16)6番又は8番の化合物を、哺乳動物へ投与することを含む、アルツハイマー病、神経変性障害又はアミロイドーシスからなる群から選択される該哺乳動物における疾患を診断する方法。
【0084】
(17)前記哺乳動物を造影し、かつ造影シグナルを検出することを含む、16番の方法。
【0085】
(18)前記造影が、PET、SPECT、MRスペクトル分析法、及びMR−断層撮影法からなる群から選択される造影方法を用いて実行される、17番の方法。
【0086】
(19)療法の作用がモニタリングされる、16番−18番の方法。
【0087】
(20)前記方法が、哺乳動物の試料をインビトロにおいて分析することを含み、ここで該哺乳動物又は試料が、6番又は8番の化合物により処理される、該哺乳動物におけるアルツハイマー病、神経変性障害又はアミロイドーシスからなる群から選択される疾患を診断又は療法をモニタリングする方法。
【0088】
(21)試料が脳脊髄液である、20番の方法。
【0089】
(22)1番−8番の化合物を備えるキット。
【0090】
本キットは、1番−8番の化合物を含む、1個又は複数の密封バイアルを備えることができる。
【0091】
本発明は更に、1番−8番の化合物を含有する医薬組成物又は診断用組成物、好ましくは放射標識された化合物を含有するそのような組成物、より一層好ましくはF−18により標識された化合物を含有するそのような組成物に関する。
【0092】
本発明は更に、A及びBが直接結合である場合、Ar又はAr’は、6員の芳香環系であることを条件とする、1番の化合物に関する。
【0093】
本発明は更に、A又はBが直接結合である場合、Ar又はAr’は、6員の芳香環系であることを条件とする、1番の化合物に関する。
【0094】
本発明は更に、A又はBが直接結合である場合、この直接結合した残基Ar又はAr’は、6員の芳香環系であることを条件とする、1番の化合物に関する。
【0095】
本発明の放射標識された化合物及び診断用組成物は、βアミロイド造影に有用である。
【実施例】
【0096】
これらの組成物を合成及び標識する方法は、下記実施例においてより十分に例示されている。これらの実施例は、前述の方法のある態様及び有利な結果を例示しており、かつこれらは例証として示されているが限定として示されているものではない。
【0097】
実施例1
a)4−(5−ブロモ−ピリミジン−2−イル)−ピペラジン−1−カルボン酸(4−ベンジルオキシ−フェニル)−アミド
【化17】
ジクロロメタン10mL中の4−ベンジルオキシ−フェニルアミン塩酸塩(Aldrich社)707mg(3mmol)及びN,N−エチルジイソプロピルアミン1.13mL(6.6mmol)の溶液を、ジクロロメタン10mL中のトリホスゲン297mg(1mmol)の溶液に、0℃で滴下した。この反応混合液を、0℃で15分間攪拌し、その後ジクロロメタン10mL中の5−ブロモ−2−ピペラジン−1−イル−ピリミジン0.73g(3mmol)及びN,N−エチルジイソプロピルアミン1.13mL(6.6mmol)の混合液を添加した。0℃で30分間及び室温で一晩経過後、溶媒を蒸発させ、残渣を、ジクロロメタン/酢酸エチル勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:0.93g(66%)
元素分析:
理論値:C 56.42、H 4.73、Br 17.06、N 14.95
実測値:C 56.11、H 4.83、Br 16.94、N 14.77
【0098】
b)4−[5−(3−ヒドロキシ−プロピル)−ピリミジン−2−イル]−ピペラジン−1−カルボン酸(4−ベンジルオキシフェニル)−アミド
【化18】
無水テトラヒドロフラン(THF)9mL中のアリルアルコール0.21mL(3mmol)の溶液へ、THF中の9−ボラビシクロ−(3.3.1)−ノナン0.5M溶液18mL(9mmol)を0℃で添加した。この混合液を、0℃で更に15分間、室温で5時間攪拌した(=溶液A)。
ブロモ誘導体化合物1aの0.70g(1.5mmol)を、無水DMF 10mL中に懸濁し、かつテトラキス(トリフェニルホスフィン)パラジウム(0)0.35g(0.3mmol)及び3M炭酸カリウム水溶液4mL(12mmol)を添加した(=懸濁液B)。
溶液Aを、懸濁液Bに添加し、65℃で一晩攪拌した。溶媒の蒸発後、残渣を、ジクロロメタン/メタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:110mg(16%)
元素分析:
理論値:C 67.10、H 6.53、N 15.65
実測値:C 66.92、H 6.41、N 15.88
【0099】
c)トルエン−4−スルホン酸3−{2−[4−(4−ベンジルオキシ−フェニルカルバモイル)−ピペラジン−1−イル]−ピリミジン−5−イル}−プロピルエステル
【化19】
無水ピリジン3mL中のヒドロキシ誘導体1bの0.45g(1mmol)の懸濁液へ、無水ピリジン2mL中のp−トルエンスルホニルクロリド0.25g(1.3mmol)の溶液を、0℃で滴下した。30分間攪拌した後、この混合液を、蒸発乾固させ、かつその残渣を、ジクロロメタン/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:379mg(63%)
元素分析:
理論値:C 63.88、H 5.86、N 11.64、S 5.33
実測値: C 63.67、H 5.97、N 11.48、S 5.02
【0100】
d)4−[5−(3−[F−18]フルオロ−プロピル)−ピリミジン−2−イル]−ピペラジン−1−カルボン酸(4−ベンジルオキシ−フェニル)−アミド
【化20】
[F−18]フッ化物を、18O(p,n)18F反応のために[O−18]水が充填された低容積、定圧、銀標的(1mL)を使用するサイクロ(登録商標)トロン内における陽子衝撃により生成した。この水性[F−18]フッ化物(約1mL)は、QMA樹脂カートリッジ(Waters社、Sep Pak Light QMA部品番号:WAT023525)を通過させた。捕獲された[F−18]フッ化物を、クリプトフィックスK2.2.2/K2CO3溶液(K2.2.2 5mg/アセトニトリル1.5mL+K2CO3 1mg/水0.5mL)の添加により、カートリッジから溶離させた。この混合物を、アセトニトリル1mLを逐次添加しながら、繰り返しの(2×)共沸蒸留により乾燥させた。このトシラート前駆体1cを、無水ジメチルホルムアミド(100μL)及びアセトニトリル(400μL)中に溶解し、乾燥カリウム/クリプトフィックス/フッ化物錯体に添加した。この溶液を、110℃で6分間加熱し、その後室温で5分間冷却した。この溶液を、水で希釈し、最終容積10mLとし、かつSep−Pak Plus C18カートリッジ(Waters社、WAT020515)を通過させた。このカートリッジを、水(20ml)ですすぎ、本化合物をアセトニトリル(3mL)により溶離させた。溶離された化合物を水で希釈し、5mLを得、これを分取HPLC精製に供した。Gemini 5μ C18 110Å、250×10mm(Phenomenex社、00G-4435-N0)を、固定相として使用した。水(A)/アセトニトリル(B)の3/7混合液を、流量3mL/分の移動相として、使用した。
この溶液を、水で希釈し、最終容積20mLとし、Sep−Pak light C18カートリッジ(Waters社、WAT023501)を通過させた。このカートリッジを、水(10ml)ですすぎ、本化合物をエタノール(0.5mL)により溶離させた。最終生成物を、Gemini 5μ C18 110Å、250×4.6mm(Phenomenex社、00G-4435-E0)HPLCカラム上で、アイソクラティックな水/アセトニトリル(2/8;v/v)混合液を、流量1mL/分で使用するHPLCにより特徴決定した。保持時間(tR):4.8分。衝撃の最後に減衰補正(d.c.)した放射化学的収率(RCY):44%。放射化学的純度(RCP):98%。
【0101】
e)4−[5−(3−フルオロ−プロピル)−ピリミジン−2−イル]−ピペラジン−1−カルボン酸(4−ベンジルオキシ−フェニル)−アミド(HPLC標準)
【化21】
ジクロロメタン10mL中のヒドロキシ誘導体化合物1bの67mg(0.15mmol)へ、ジエチルアミノ硫黄トリフルオリド(DAST)48mg(0.3mmol)を0℃で添加した。0℃で30分後、水を添加し、有機相を硫酸ナトリウム上で乾燥し、その有機溶媒を蒸発させた後、残渣を、ジクロロメタン/酢酸エチル勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:40mg(60%)
元素分析:
理論値:C 66.80、H 6.28、F 4.23、N 15.58
実測値:C 66.54、H 6.49、F 4.06、N 15.77
【0102】
実施例2
a)4−(5−ブロモ−ピリミジン−2−イル)−ピペラジン−1−カルボン酸(2−フェニル−ベンゾフラン−5−イル)−アミド
【化22】
ジクロロメタン20mL中の2−フェニル−ベンゾフラン−5−イルアミン塩酸塩(ChemBridge社、サンディエゴ、米国)628mg(3mmol)及びN,N−エチルジイソプロピルアミン1.13mL(6.6mmol)の溶液を、ジクロロメタン10mL中のトリホスゲン297mg(1mmol)の溶液に、0℃で滴下した。この反応混合液を、0℃で30分間攪拌し、その後ジクロロメタン20mL中の5−ブロモ−2−ピペラジン−1−イル−ピリミジン0.73g(3mmol)及びN,N−エチルジイソプロピルアミン0.56mL(3.3mmol)の混合液を添加した。0℃で1時間及び室温で一晩経過後、溶媒を蒸発させ、残渣を、ジクロロメタン/酢酸エチル勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:0.81g(56%)
元素分析:
理論値:C 57.75、H 4.21、Br 16.70、N 14.64
実測値:C 57.43、H 4.37、Br 16.11、N 14.45
【0103】
b)4−[5−(3−ヒドロキシ−プロピル)−ピリミジン−2−イル]−ピペラジン−1−カルボン酸(2−フェニル−ベンゾフラン−5−イル)−アミド
【化23】
無水テトラヒドロフラン(THF)9mL中のアリルアルコール0.21mL(3mmol)の溶液へ、THF中の9−ボラビシクロ−(3.3.1)−ノナン0.5M溶液18mL(9mmol)を0℃で添加した。この混合液を、0℃で更に15分間、室温で5時間攪拌した(=溶液A)。
化合物2aの0.72g(1.5mmol)を、無水DMF 10mL中に懸濁し、かつテトラキス(トリフェニルホスフィン)パラジウム(0)0.35g(0.3mmol)及び3M炭酸カリウム水溶液4mL(12mmol)を添加した(=懸濁液B)。
溶液Aを、懸濁液Bに添加し、65℃で一晩攪拌した。溶媒の蒸発後、残渣を、ジクロロメタン/メタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:85mg(12%)
元素分析:
理論値:C 68.25、H 5.95、N 15.31
実測値:C 68.05、H 6.12、N 15.20
【0104】
c)トルエン−4−スルホン酸3−{2−[4−(2−フェニル−ベンゾフラン−5−イルカルバモイル)−ピペラジン−1−イル]−ピリミジン−5−イル}−プロピルエステル
【化24】
無水ピリジン3mL中のヒドロキシ誘導体2bの0.46g(1mmol)の懸濁液へ、無水ピリジン2mL中のp−トルエンスルホニルクロリド0.25g(1.3mmol)の溶液を、0℃で滴下した。30分間攪拌した後、この混合液を、蒸発乾固させ、かつその残渣を、ジクロロメタン/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:434mg(71%)
元素分析:
理論値:C 64.80、H 5.44、N 11.45、S 5.24
実測値:C 64.66、H 5.23、N 11.74、S 4.97
【0105】
d)4−[5−(3−[F−18]フルオロ−プロピル)−ピリミジン−2−イル]−ピペラジン−1−カルボン酸(2−フェニル−ベンゾフラン−5−イル)−アミド
【化25】
トシラート前駆体2cを、無水ジメチルホルムアミド(100μL)及びアセトニトリル(400μL)中に溶解し、乾燥カリウム/クリプトフィックス/フッ化物錯体に添加した(上記参照)。この溶液を、110℃で6分間加熱し、その後室温で5分間冷却した。この溶液を、水で希釈し、最終容積10mLとし、かつSep−Pak Plus C18カートリッジ(Waters社、WAT020515)を通過させた。このカートリッジを、水(20ml)ですすぎ、本化合物をアセトニトリル(3mL)により溶離させた。溶離された化合物を水で希釈し、5mLを得、これを分取HPLC精製に供した。Gemini 5μ C18 110Å、250×10mm(Phenomenex社、00G-4435-N0)を、固定相として使用した。水(A)/アセトニトリル(B)の3/7混合液を、流量3mL/分の移動相として、使用した。
【0106】
この溶液を、水で希釈し、最終容積20mLとし、Sep−Pak light C18カートリッジ(Waters社、WAT023501)を通過させた。このカートリッジを、水(10ml)ですすぎ、本化合物をエタノール(0.5mL)により溶離させた。最終生成物を、Gemini 5μ C18 110Å、250×4.6mm(Phenomenex社、00G-4435-E0)HPLCカラム上で、アイソクラティックな水/アセトニトリル(2/8;v/v)混合液を、流量1mL/分で使用するHPLCにより特徴決定した。
保持時間(tR):4.1分。RCY(d.c.):38%。RCP:97%。
【0107】
e)4−[5−(3−フルオロ−プロピル)−ピリミジン−2−イル]−ピペラジン−1−カルボン酸(2−フェニル−ベンゾフラン−5−イル)−アミド(HPLC標準)
【化26】
ジクロロメタン10mL中のヒドロキシ誘導体化合物2bの69mg(0.15mmol)へ、ジエチルアミノ硫黄トリフルオリド(DAST)48mg(0.3mmol)を0℃で添加した。0℃で30分後、水を添加し、有機相を硫酸ナトリウム上で乾燥し、その有機溶媒を蒸発させた後、残渣を、ジクロロメタン/酢酸エチル勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:15mg(22%)
元素分析:
理論値:C 67.96、H 5.70、F 4.13、N 15.24
実測値:C 67.67、H 5.96、F 3.96、N 15.22
【0108】
実施例3
a)(3−アセトキシ−ベンゾイルアミノ)−酢酸ベンジルエステル
【化27】
グリシンベンジルエステルp−トルエンスルホン酸30.4g(90mmol)を、二相システムのジクロロメタン及び飽和炭酸水素ナトリウム水溶液中に溶解した。有機相を、硫酸マグネシウム上で乾燥し、次に蒸発させた。
遊離アミン13.09g(79.25mmol)を得、これを更に精製することなく、引き続きのカップリング反応において使用した。
【0109】
THF 150mL及びトリエチルアミン11mL(79.25mmol)中の3−アセトキシ−安息香酸14.28g(79.25mmol)の−15℃の溶液に、クロロギ酸イソブチル11.39mL(87.2mmol)を滴下し、この溶液を、この温度で更に15分間維持した。その後この冷溶液へ、THF 50mL及びジクロロメタン50mL中のグリシンベンジルエステル13.09g及びトリエチルアミン11mL(79.25mmol)をゆっくり添加し、更に15分間その温度を10℃未満に維持し、その後室温とした。一晩攪拌した後、溶媒を蒸発させ、残渣を、酢酸エチル/エタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:24.6g(95%)。
元素分析:
理論値:C 66.05、H 5.23、N 4.28
実測値:C 65.84、H 5.43、N 4.16
【0110】
b)(3−アセトキシ−ベンゾイルアミノ)−酢酸
【化28】
メタノール300mL中のベンジルエステル3aの19.64g(60mmol)の溶液に、チャコール上に担持されたPd(10%)3gを添加し、この懸濁液を水素下、室温で一晩攪拌した。触媒を濾過除去し、溶媒を蒸発させた。
収量:14.2g(定量的)。
元素分析:
理論値:C 55.70、H 4.67、N 5.90
実測値:C 55.66、H 4.49、N 5.77
【0111】
c)1−(4−ベンジルオキシ−フェニル)−ピペラジン
【化29】
ガラス器具は全て、100℃で乾燥した。トルエン60mL中のピペラジン4.32g(50.16mmol)の溶液へ、トリス(ジベンジリデンアセトン)ジパラジウム(0)459mg(0.5mmol)及びBINAP(2,2’−ビス(ジフェニルホスフィノ)−1,1’−ビナフチル)423mg(0.68mmol)を添加した。その後THF 40mL中の4−ベンジルオキシ−ブロモベンゼン12g(45.6mmol)の溶液を添加し、引き続きTHF中のナトリウムt−ブチラート6.56g(68.27mmol)の懸濁液を添加した。
【0112】
この反応混合物を、3時間還流し、室温で一晩攪拌した。溶媒を蒸発させた後、残渣を、ジクロロメタン/メタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:12.2g(45.7%)。
元素分析:
理論値:C 76.09、H 7.51、N 10.44
実測値:C 75.91、H 7.76、N 10.20
【0113】
d)酢酸3−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチルカルバモイル}−フェニルエステル
【化30】
THF 70mL及びトリエチルアミン0.40mL(2.87mmol)中の(3−アセトキシ−ベンゾイルアミノ)−酢酸3bの654mg(2.76mmol)の−15℃の溶液へ、クロロギ酸イソブチル0.396mL(3.03mmol)を滴下し、この溶液を、この温度で更に15分間維持した。その後この冷溶液へ、THF 30mL及びジクロロメタン30mL中の1−(4−ベンジルオキシ−フェニル)−ピペラジン3cの740mg及びトリエチルアミン1.7mL(12.25mmol)をゆっくり添加し、更に15分間その温度を10℃未満に維持し、その後室温とした。一晩攪拌した後、溶媒を蒸発させ、残渣を、ヘキサン/酢酸エチル勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:390mg(30%)。
元素分析:
理論値:C 68.98、H 6.00、N 8.62
実測値:C 69.09、H 5.81、N 8.42
【0114】
e)N−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−3−ヒドロキシ−ベンズアミド
【化31】
酢酸塩3dの230mg(0.47mmol)を、エタノール30mLに溶解し、0℃に冷却した。3N NaOH 1.5mLを添加した後、この溶液を1時間攪拌し、そのpHがpH7を下回るまで、氷酢酸を添加し、溶媒を蒸発させた。粗生成物を、エタノールから結晶化した。
収量:200mg(95%)。
元素分析:
理論値:C 70.10、H 6.11、N 9.43
実測値:C 69.88、H 6.27、N 9.20
【0115】
f)トルエン−4−スルホン酸2−(3−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチルカルバモイル}−フェノキシ)−エチルエステル
【化32】
DMF 10mL中のN−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−3−ヒドロキシ−ベンズアミド(3e)80mg(0.18mmol)の溶液に、炭酸カリウム62mg(0.45mmol)及び1,2ビス(トシルオキシ)エタン100mg(0.27mmol)(Aldrich社)を添加した。この混合物を60℃で3時間加熱した。室温で一晩攪拌した後、溶媒を蒸発させ、残渣を、酢酸エチル/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:56mg(48%)。
この化合物は、HPLCにより純度>95%を有し、F−18標識化の前駆体として適している。
【0116】
g)N−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−3−(2−[F−18]フルオロ−エトキシ)−ベンズアミド
【化33】
トシラート前駆体3fを、無水ジメチルホルムアミド(100μL)及びアセトニトリル(400μL)に溶解し、乾燥カリウム/クリプトフィックス/フッ化物錯体(前記参照)に添加した。この溶液を110℃で6分間加熱し、次に室温で5分間冷却した。この溶液を、水で希釈し、最終容積10mLとし、Sep−Pak Plus C18カートリッジ(Waters社、WAT020501)を通過させた。このカートリッジを、水(20ml)ですすぎ、本化合物をアセトニトリル(3mL)により溶離させた。溶離された化合物を水で希釈し、5mLを得、これを分取HPLC精製に供した。Gemini 5μ C18 110Å、250×10mm(Phenomenex社、00G-4435-N0)を、固定相として使用した。水(A)/アセトニトリル(B)の3/7混合液を、流量3mL/分で移動相として使用した。
【0117】
この溶液を水で希釈し、最終容積20mLとし、Sep−Pak light C18カートリッジ(Waters社、WAT023501)を通過させた。このカートリッジを、水(10ml)ですすぎ、本化合物をエタノール(0.5mL)により溶離させた。最終生成物を、Gemini 5μ C18 110Å、250×4.6mm(Phenomenex社、00G-4435-E0)HPLCカラム上で、アイソクラティックな水/アセトニトリル(2/8;v/v)混合液を、流量1mL/分で使用するHPLCにより特徴決定した。保持時間(tR):4.3分。RCY(d.c.):38%。RCP:99%。
【0118】
h)N−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−3−(2−フルオロ−エトキシ)−ベンズアミド(HPLC標準)
【化34】
DMF 5mL中のN−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−3−ヒドロキシ−ベンズアミド(3e)50mg(0.11mmol)の溶液に、炭酸カリウム34mg(0.25mmol)及び1−フルオロ−2−ブロモエタン(ABCR社、独国)10μL(0.13mmol)を添加した。この混合物を、60℃で3時間加熱した。溶媒を蒸発させ、残渣を、酢酸エチル/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:30mg(54%)。
元素分析:
理論値:C 68.42、H 6.15、F 3.86、N 8.55
実測値:C 68.17、H 6.28、F 3.64、N 8.71
【0119】
実施例4
a)酢酸3−{2−[4−(4−ニトロフェニル)−ピペラジン−1−イル]−2−オキソ−エチルカルバモイル}−フェニルエステル
【化35】
DMF 100mL中の(3−アセトキシ−ベンゾイルアミノ)−酢酸(3b)3.43g(14.5mmol)及び1−(4−ニトロフェニル)ピペラジン(Aldrich社)2.0g(9.65mmol)の溶液へ、PyBOP(ヘキサフルオロリン酸(ベンゾトリアゾール−1−イルオキシ)トリピロリジノホスホニウム)5.0g(9.65mmol)及びN−エチル−N,N−ジイソプロピルアミン5mLを添加し、かつこの反応混合液を、室温で一晩攪拌した。溶媒を蒸発させた後、残渣を、酢酸エチル/エタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:1.2g(29.2%)。
元素分析:
理論値:C 59.15、H 5.20、N 13.14
実測値:C 59.38、H 5.01、N 13.25
【0120】
b)酢酸3−{2−[4−(4−アミノ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチルカルバモイル}−フェニルエステル
【化36】
メタノール/ジクロロメタン(1:1)50mL中のニトロ化合物4a 853mg(2mmol)の溶液へ、触媒量のチャコール上に担持されたPd(10%)を添加し、この懸濁液を、水素下、室温で一晩攪拌した。触媒を濾過により除去し、溶媒を蒸発させた。
収量:795mg(定量的)。この生成物は、更に精製することなく次工程において使用した。
【0121】
c)酢酸3−{2−[4−(4−ベンゾイルアミノ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチルカルバモイル}−フェニルエステル
【化37】
DMF 20mL中の酢酸3−{2−[4−(4−アミノ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチルカルバモイル}−フェニルエステル(4b) 396mg(1mmol)及び安息香酸134mg(1.1mmol)の溶液へ、PyBOP 590mg(1.14mmol)及びN−エチル−N,N−ジイソプロピルアミン0.35mL(2mmol)を添加し、反応混合物を室温で攪拌した。4時間後、溶媒を蒸発させ、残渣を、ジクロロメタン/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:0.31g(62.1%)。
元素分析:
理論値:C 67.19、H 5.64、N 11.19
実測値:C 66.94、H 5.88、N 11.02
【0122】
d)N−{2−[4−(4−ベンゾイルアミノ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−3−ヒドロキシ−ベンズアミド
【化38】
酢酸塩4cの235mg(0.47mmol)を、エタノール30mL中に溶解し、0℃に冷却した。3N NaOH 1.5mLの添加後、この溶液を1時間攪拌し、そのpHがpH7を下回るまで氷酢酸を添加し、溶媒を蒸発させた。粗生成物をエタノールから結晶化した。
収量:177mg(82%)。
元素分析:
理論値:C 68.11、H 5.72、N 12.22
実測値:C 67.91、H 5.60、N 12.00
【0123】
e)トルエン−4−スルホン酸2−(3−{2−[4−(4−ベンゾイルアミノ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチルカルバモイル}−フェノキシ)−エチルエステル
【化39】
DMF 10mL中のN−{2−[4−(4−ベンゾイルアミノ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−3−ヒドロキシ−ベンズアミド(4d)83mg(0.18mmol)の溶液へ、炭酸カリウム62mg(0.45mmol)及び1,2ビス(トシルオキシ)エタン(Aldrich社)100mg(0.27mmol)を添加した。この混合物を60℃で3時間加熱した。室温で一晩攪拌後、溶媒を蒸発させ、残渣を、酢酸エチル/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:60mg(51%)。
この化合物は、HPLCにより純度>95%を有し、かつF−18標識化の前駆体として適している。
【0124】
f)N−{2−[4−(4−ベンゾイルアミノ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−3−(2−[F−18]フルオロ−エトキシ)−ベンズアミド
【化40】
トシラート前駆体4eを、無水ジメチルホルムアミド(100μL)及びアセトニトリル(400μL)中に溶解し、乾燥カリウム/クリプトフィックス/フッ化物錯体(前記参照)に添加した。この溶液を、110℃で6分間加熱し、その後室温で5分間冷却した。この溶液を、水で希釈し、最終容積10mLとし、かつSep−Pak Plus C18カートリッジ(Waters社、WAT020515)を通過させた。このカートリッジを、水(20ml)ですすぎ、本化合物をアセトニトリル(3mL)により溶離させた。溶離された化合物を水で希釈し、5mLを得、これを分取HPLC精製に供した。Gemini 5μ C18 110Å、250×10mm(Phenomenex社、00G-4435-N0)を、固定相として使用した。水(A)/アセトニトリル(B)の3/7混合液を、流量3mL/分の移動相として、使用した。
【0125】
この溶液を、水で希釈し、最終容積20mLとし、Sep−Pak light C18カートリッジ(Waters社、WAT023501)を通過させた。このカートリッジを、水(10ml)ですすぎ、本化合物をエタノール(0.5mL)により溶離させた。最終生成物を、Gemini 5μ C18 110Å、250×4.6mm(Phenomenex社、00G-4435-E0)HPLCカラム上で、アイソクラティックな水/アセトニトリル(2/8;v/v)混合液を、流量1mL/分で使用するHPLCにより特徴決定した。保持時間(tR):4.1分。RCY (d.c.):32%。RCP:99%。
【0126】
g)N−{2−[4−(4−ベンゾイルアミノ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−3−(2−フルオロ−エトキシ)−ベンズアミド(HPLC標準)
【化41】
DMF 5mL中のN−{2−[4−(4−ベンゾイルアミノ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−3−ヒドロキシ−ベンズアミド(4d)50mg(0.11mmol)の溶液へ、炭酸カリウム34mg(0.25mmol)及び1−フルオロ−2−ブロモエタン(ABCR社、独国)10μL(0.13mmol)を添加した。この混合物を60℃で3時間加熱した。溶媒を蒸発させ、残渣を、酢酸エチル/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:34mg(62%)。
元素分析:
理論値:C 66.65、H 5.79、F 3.77、N 11.10
実測値:C 66.43、H 5.89、F 3.41、N 11.31
【0127】
実施例5
a)トルエン−4−スルホン酸2−{2−[2−(3−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチルカルバモイル}−フェノキシ)−エトキシ]−エトキシ}−エチルエステル
【化42】
DMF 10mL中のN−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−3−ヒドロキシ−ベンズアミド(3e)80mg(0.18mmol)の溶液へ、炭酸カリウム62mg(0.45mmol)及びジ−p−トルエンスルホン酸トリ(エチレングリコール)(Aldrich社)124mg(0.27mmol)を添加した。この混合物を、60℃で3時間加熱した。室温で一晩攪拌した後、溶媒を蒸発させ、残渣を、酢酸エチル/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:54mg(41%)。
この化合物は、HPLCにより純度>95%を有し、かつF−18標識化の前駆体として適している。
【0128】
b)N−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−3−{2−[2−(2−[F−18]フルオロ−エトキシ)−エトキシ]−エトキシ}−ベンズアミド
【化43】
トシラート前駆体5aを、無水ジメチルホルムアミド(100μL)及びアセトニトリル(400μL)中に溶解し、乾燥カリウム/クリプトフィックス/フッ化物錯体(前記参照)に添加した。この溶液を、110℃で6分間加熱し、その後室温で5分間冷却した。この溶液を、水で希釈し、最終容積10mLとし、かつSep−Pak Plus C18カートリッジ(Waters社、WAT020515)を通過させた。このカートリッジを、水(20ml)ですすぎ、本化合物をアセトニトリル(3mL)により溶離させた。溶離された化合物を水で希釈し、5mLを得、これを分取HPLC精製に供した。Gemini 5μ C18 110Å、250×10mm(Phenomenex社、00G-4435-N0)を、固定相として使用した。水(A)/アセトニトリル(B)の3/7混合液を、流量3mL/分の移動相として、使用した。
【0129】
この溶液を、水で希釈し、最終容積20mLとし、Sep−Pak light C18カートリッジ(Waters社、WAT023501)を通過させた。このカートリッジを、水(10ml)ですすぎ、本化合物をエタノール(0.5mL)により溶離させた。最終生成物を、Gemini 5μ C18 110Å、250×4.6mm(Phenomenex社、00G-4435-E0)HPLCカラム上で、アイソクラティックな水/アセトニトリル(2/8;v/v)混合液を、流量1mL/分で使用するHPLCにより特徴決定した。保持時間(tR):4.2分。RCY (d.c.):42%。RCP:99%。
【0130】
c)N−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−3−{2−[2−(2−フルオロ−エトキシ)−エトキシ]−エトキシ}−ベンズアミド(HPLC標準)
【化44】
トシラート前駆体5aの73mg(0.10mmol)を、アセトニトリル2mL中に溶解した。アセトニトリル1mL中のフッ化カリウム6.8mg(0.12mmol)及びクリプトフィックス44.4mgを添加し、100℃で10分間インキュベーションした。この反応を、分析用HPLCによりチェックした。この反応が完了した後、混合物を蒸発させ、かつ残渣を、酢酸エチル/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:28mg(49%)。
元素分析:
理論値:C 66.31、H 6.61、F 3.28、N 7.25
実測値:C 66.07、H 6.73、F 3.11、N 7.38
【0131】
実施例6
a)酢酸4−{4−[4−(4−ニトロフェニル)−ピペラジン−1−イル]−4−オキソ−ブチリル}−フェニルエステル
【化45】
DMF 20mL中の4−(4−アセトキシフェニル)−4−オキソ酪酸(Arch. Pharm.社(ワインハイム、独国) 284;292(1951))1.18g(5mmol)及び1−(4−ニトロフェニル)ピペラジン(Aldrich社)1.04g(5mmol)の溶液に、N−エチル−N,N−ジイソプロピルアミン1.71mL(10mmol)及びヘキサフルオロリン酸2−(1H−ベンゾトリアゾール−1−イル)−テトラメチルウロニウム(HBTU)1.9g(5mmol)を添加した。この反応混合液を、室温で4時間攪拌し、その後溶媒を蒸発させ、残渣を、酢酸エチル/エタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:1.53g(72%)。
元素分析:
理論値:C 62.11、H 5.45、N 9.88
実測値:C 61.89、H 5.54、N 9.61
【0132】
b)酢酸4−{4−[4−(4−アミノ−フェニル)−ピペラジン−1−イル]−4−オキソ−ブチリル}−フェニルエステル
【化46】
メタノール/DMF(3:1)50mL中のニトロ化合物6aの851mg(2mmol)の溶液へ、触媒量のチャコール上に担持されたPd(10%)を添加し、この懸濁液を、水素下、室温で一晩攪拌した。触媒を濾過により除去し、溶媒を蒸発させた。
収量:791mg(定量的)。この生成物は、更に精製することなく次工程において使用した。
【0133】
c)酢酸4−[4−(4−{4−[(ナフタレン−2−カルボニル)−アミノ]−フェニル}−ピペラジン−1−イル)−4−オキソ−ブチリル]−フェニルエステル
【化47】
DMF 20mL中の酢酸 4−{4−[4−(4−アミノ−フェニル)−ピペラジン−1−イル]−4−オキソ−ブチリル}−フェニルエステル(6b)395mg(1mmol)及び2−ナフタレンカルボン酸(Aldrich社)190mg(1.1mmol)の溶液へ、PyBOP 590mg(1.14mmol)及びN−エチル−N,N−ジイソプロピルアミン0.35mL(2mmol)を添加し、反応混合物を室温で攪拌した。4時間後、溶媒を蒸発させ、残渣を、ジクロロメタン/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:374mg(68%)。
元素分析:
理論値:C 72.12、H 5.69、N 7.65
実測値:C 72.30、H 5.61、N 7.42
【0134】
d)ナフタレン−2−カルボン酸(4−{4−[4−(4−ヒドロキシ−フェニル)−4−オキソ−ブチリル]−ピペラジン−1−イル}−フェニル)−アミド
【化48】
酢酸塩6cの242mg(0.44mmol)を、エタノール/DMF(1:1)30mL中に溶解し、0℃に冷却した。2N NaOH 2.2mLの添加後、この溶液を30分間攪拌し、次にそのpHがpH7を下回るまで氷酢酸を添加し、溶媒を蒸発させた。粗生成物を水から結晶化し、真空下40℃で乾燥した。
収量:223mg(定量的)。
元素分析:
理論値:C 73.35、H 5.76、N 8.28
実測値:C 73.10、H 5.91、N 8.25
【0135】
e)トルエン−4−スルホン酸2−{4−[4−(4−{4−[(ナフタレン−2−カルボニル)−アミノ]−フェニル}−ピペラジン−1−イル)−4−オキソ−ブチリル]−フェノキシ}−エチルエステル
【化49】
DMF 10mL中のナフタレン−2−カルボン酸(4−{4−[4−(4−ヒドロキシ−フェニル)−4−オキソ−ブチリル]−ピペラジン−1−イル}−フェニル)−アミド(6d)91mg(0.18mmol)の溶液へ、炭酸カリウム62mg(0.45mmol)及び1,2ビス(トシルオキシ)エタン(Aldrich社)100mg(0.27mmol)を添加した。この混合物を60℃で3時間加熱した。室温で一晩攪拌した後、溶媒を蒸発させ、残渣を、酢酸エチル/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:57mg(46%)。
この化合物は、HPLCにより純度>95%を有し、かつF−18標識化の前駆体として適している。
【0136】
f)ナフタレン−2−カルボン酸[4−(4−{4−[4−(2−[F−18]フルオロ−エトキシ)−フェニル]−4−オキソ−ブチリル}−ピペラジン−1−イル)−フェニル]−アミド
【化50】
トシラート前駆体6eを、無水ジメチルホルムアミド(100μL)及びアセトニトリル(400μL)中に溶解し、乾燥カリウム/クリプトフィックス/フッ化物錯体(前記参照)に添加した。この溶液を、110℃で6分間加熱し、その後室温で5分間冷却した。この溶液を、水で希釈し、最終容積10mLとし、かつSep−Pak Plus C18カートリッジ(Waters社、WAT020515)を通過させた。このカートリッジを、水(20ml)ですすぎ、本化合物をアセトニトリル(3mL)により溶離させた。溶離された化合物を水で希釈し、5mLを得、これを分取HPLC精製に供した。Gemini 5μ C18 110Å、250×10mm(Phenomenex社、00G-4435-N0)を、固定相として使用した。水(A)/アセトニトリル(B)の3/7混合液を、流量3mL/分の移動相として、使用した。
【0137】
この溶液を、水で希釈し、最終容積20mLとし、Sep−Pak light C18カートリッジ(Waters社、WAT023501)を通過させた。このカートリッジを、水(10ml)ですすぎ、本化合物をエタノール(0.5mL)により溶離させた。最終生成物を、Gemini 5μ C18 110Å、250×4.6mm(Phenomenex社、00G-4435-E0)HPLCカラム上で、アイソクラティックな水/アセトニトリル(2/8;v/v)混合液を、流量1mL/分で使用するHPLCにより特徴決定した。保持時間(tR):5.6分。RCY (d.c.):35%。RCP:98%。
【0138】
実施例7
a)酢酸4−[4−(4−{4−[(ナフタレン−1−カルボニル)−アミノ]−フェニル}−ピペラジン−1−イル)−4−オキソ−ブチリル]−フェニルエステル
【化51】
DMF 20mL中の酢酸4−{4−[4−(4−アミノ−フェニル)−ピペラジン−1−イル]−4−オキソ−ブチリル}−フェニルエステル(6b)395mg(1mmol)及び1−ナフタレンカルボン酸(Aldrich社)190mg(1.1mmol)の溶液に、PyBOP 590mg(1.14mmol)及びN−エチル−N,N−ジイソプロピルアミン0.35mL(2mmol)を添加し、かつこの反応混合液を、室温で攪拌した。4時間後、溶媒を蒸発させ、残渣を、ジクロロメタン/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:336mg(61%)。
元素分析:
理論値:C 72.12、H 5.69、N 7.65
実測値:C 71.94、H 5.77、N 7.59
【0139】
b)ナフタレン−1−カルボン酸(4−{4−[4−(4−ヒドロキシ−フェニル)−4−オキソ−ブチリル]−ピペラジン−1−イル}−フェニル)−アミド
【化52】
酢酸塩7aの242mg(0.44mmol)を、エタノール/DMF(1:1)30mLに溶解し、0℃に冷却した。2N NaOH 2.2mLを添加した後、この溶液を30分間攪拌し、次にそのpHがpH7を下回るまで、氷酢酸を添加し、溶媒を蒸発させた。粗生成物を水から結晶化し、真空下40℃で乾燥した。
収量:223mg(定量的)。
元素分析:
理論値:C 73.35、H 5.76、N 8.28
実測値:C 73.21、H 5.62、N 8.11
【0140】
c)トルエン−4−スルホン酸2−{4−[4−(4−{4−[(ナフタレン−1−カルボニル)−アミノ]−フェニル}−ピペラジン−1−イル)−4−オキソ−ブチリル]−フェノキシ}−エチルエステル
【化53】
DMF 10mL中のナフタレン−1−カルボン酸(4−{4−[4−(4−ヒドロキシ−フェニル)−4−オキソ−ブチリル]−ピペラジン−1−イル}−フェニル)−アミド(7b) 91mg(0.18mmol)の溶液へ、炭酸カリウム62mg(0.45mmol)及び1,2ビス(トシルオキシ)エタン(Aldrich社)100mg(0.27mmol)を添加した。この混合物を60℃で3時間加熱した。室温で一晩攪拌した後、溶媒を蒸発させ、残渣を、酢酸エチル/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:63mg(51%)。
この化合物は、HPLCにより純度>95%を有し、かつF−18標識化の前駆体として適している。
【0141】
d)ナフタレン−1−カルボン酸[4−(4−{4−[4−(2−[F−18]フルオロ−エトキシ)−フェニル]−4−オキソ−ブチリル}−ピペラジン−1−イル)−フェニル]−アミド
【化54】
トシラート前駆体7cを、無水ジメチルホルムアミド(100μL)及びアセトニトリル(400μL)中に溶解し、乾燥カリウム/クリプトフィックス/フッ化物錯体(前記参照)に添加した。この溶液を、110℃で6分間加熱し、その後室温で5分間冷却した。この溶液を、水で希釈し、最終容積10mLとし、かつSep−Pak Plus C18カートリッジ(Waters社、WAT020515)を通過させた。このカートリッジを、水(20ml)ですすぎ、本化合物をアセトニトリル(3mL)により溶離させた。溶離された化合物を水で希釈し、5mLを得、これを分取HPLC精製に供した。Gemini 5μ C18 110Å、250×10mm(Phenomenex社、00G-4435-N0)を、固定相として使用した。水(A)/アセトニトリル(B)の3/7混合液を、流量3mL/分の移動相として、使用した。
【0142】
この溶液を、水で希釈し、最終容積20mLとし、Sep−Pak light C18カートリッジ(Waters社、WAT023501)を通過させた。このカートリッジを、水(10ml)ですすぎ、本化合物をエタノール(0.5mL)により溶離させた。最終生成物を、Gemini 5μ C18 110Å、250×4.6mm(Phenomenex社、00G-4435-E0)HPLCカラム上で、アイソクラティックな水/アセトニトリル(2/8;v/v)混合液を、流量1mL/分で使用するHPLCにより特徴決定した。保持時間(tR):6.0分。RCY (d.c.):32%。RCP:97%。
【0143】
実施例8
a)酢酸4−{4−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−4−オキソブチリル}−フェニルエステル
【化55】
THF 70mL及びトリエチルアミン0.40mL(2.87mmol)中の4−(4−アセトキシフェニル)−4−オキソ酪酸(Arch. Pharm.社(ワインハイム、独国) 284; 292 (1951))652mg(2.76mmol)の−15℃の溶液へ、クロロギ酸イソブチル0.396mL(3.03mmol)を滴下し、この溶液を更に15分間この温度で維持した。次にこの冷溶液へ、THF 30mL及びジクロロメタン30mL中の1−(4−ベンジルオキシ−フェニル)−ピペラジン3c 740mg及びトリエチルアミン1.7mL(12.25mmol)を添加し、その温度を10℃未満に更に15分間維持し、その後室温とした。一晩攪拌した後、溶媒を蒸発させ、かつ残渣を、ヘキサン/酢酸エチル勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:219mg(45%)。
元素分析:
理論値:C 71.59、H 6.21、N 5.76
実測値:C 71.33、H 6.46、N 5.60
【0144】
b)1−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−4−(4−ヒドロキシフェニル)−ブタン−1,4−ジオン
【化56】
酢酸塩8aの229mg(0.47mmol)を、エタノール30mLに溶解し、0℃に冷却した。3N NaOH 1.5mLの添加後、この溶液を1時間攪拌し、そのpHがpH7を下回るまで、氷酢酸を添加し、溶媒を蒸発させた。粗生成物をエタノールから結晶化した。
収量:186mg(89%)。
元素分析:
理論値:C 72.95、H 6.35、N 6.30
実測値:C 72.78、H 6.27、N 6.41
【0145】
c)トルエン−4−スルホン酸2−(4−{4−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−4−オキソブチリル}−フェノキシ)−エチルエステル
【化57】
DMF 10mL中の1−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−4−(4−ヒドロキシフェニル)−ブタン−1,4−ジオン(8b)80mg(0.18mmol)の溶液へ、炭酸カリウム62mg(0.45mmol)及び1,2ビス(トシルオキシ)エタン(Aldrich社)100mg(0.27mmol)を添加した。この混合物を60℃で3時間加熱した。室温で一晩攪拌した後、溶媒を蒸発させ、残渣を、酢酸エチル/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:60mg(52%)。
この化合物は、HPLCにより純度>95%を有し、かつF−18標識化の前駆体として適している。
【0146】
d)1−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−4−[4−(2−[F−18]フルオロ−エトキシ)−フェニル]−ブタン−1,4−ジオン
【化58】
トシラート前駆体8cを、無水ジメチルホルムアミド(100μL)及びアセトニトリル(400μL)中に溶解し、乾燥カリウム/クリプトフィックス/フッ化物錯体(前記参照)に添加した。この溶液を、110℃で6分間加熱し、その後室温で5分間冷却した。この溶液を、水で希釈し、最終容積10mLとし、かつSep−Pak Plus C18カートリッジ(Waters社、WAT020515)を通過させた。このカートリッジを、水(20ml)ですすぎ、本化合物をアセトニトリル(3mL)により溶離させた。溶離された化合物を水で希釈し、5mLを得、これを分取HPLC精製に供した。Gemini 5μ C18 110Å、250×10mm(Phenomenex社、00G-4435-N0)を、固定相として使用した。水(A)/アセトニトリル(B)の3/7混合液を、流量3mL/分の移動相として、使用した。
【0147】
この溶液を、水で希釈し、最終容積20mLとし、Sep−Pak light C18カートリッジ(Waters社、WAT023501)を通過させた。このカートリッジを、水(10ml)ですすぎ、本化合物をエタノール(0.5mL)により溶離させた。最終生成物を、Gemini 5μ C18 110Å、250×4.6mm(Phenomenex社、00G-4435-E0)HPLCカラム上で、アイソクラティックな水/アセトニトリル(2/8;v/v)混合液を、流量1mL/分で使用するHPLCにより特徴決定した。保持時間(tR):4.7分。RCY (d.c.):45%。RCP:99%。
【0148】
実施例9
a)酢酸3−[2−(4−{4−[(ナフタレン−2−カルボニル)−アミノ]−フェニル}−ピペラジン−1−イル)−2−オキソ−エチルカルバモイル]−フェニルエステル
【化59】
DMF 20mL中の酢酸3−{2−[4−(4−アミノ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチルカルバモイル}−フェニルエステル(4b)396mg(1mmol)及び2−ナフタレンカルボン酸(Aldrich社)190mg(1.1mmol)の溶液へ、PyBOP 590mg(1.14mmol)及びN−エチル−N,N−ジイソプロピルアミン0.35mL(2mmol)を添加し、かつこの反応混合液を、室温で攪拌した。4時間後、溶媒を蒸発させ、残渣を、ジクロロメタン/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:0.25g(43.9%)。
元素分析:
理論値:C 69.80、H 5.49、N 10.18
実測値:C 69.55、H 5.61、N 10.03
【0149】
b)ナフタレン−2−カルボン酸(4−{4−[2−(3−ヒドロキシ−ベンゾイルアミノ)−アセチル]−ピペラジン−1−イル}−フェニル)−アミド
【化60】
酢酸塩9aの240mg(0.44mmol)を、エタノール/DMF(1:1)30mL中に溶解し、0℃に冷却した。2N NaOH 2.2mLの添加後、この溶液を30分間攪拌し、次にそのpHがpH7を下回るまで、氷酢酸を添加し、溶媒を蒸発させた。粗生成物を水から結晶化し、真空中で40℃で乾燥した。
収量:220mg(定量的)。
元素分析:
理論値:C 70.85、H 5.55、N 11.02
実測値:C 70.53、H 5.77、N 10.85
【0150】
c)トルエン−4−スルホン酸2−{3−[2−(4−{4−[(ナフタレン−2−カルボニル)−アミノ]−フェニル}−ピペラジン−1−イル)−2−オキソ−エチルカルバモイル]−フェノキシ}−エチルエステル
【化61】
DMF 10mL中のナフタレン−2−カルボン酸(4−{4−[2−(3−ヒドロキシ−ベンゾイルアミノ)−アセチル]−ピペラジン−1−イル}−フェニル)−アミド(9b)92mg(0.18mmol)の溶液へ、炭酸カリウム62mg(0.45mmol)及び1,2ビス(トシルオキシ)エタン(Aldrich社)100mg(0.27mmol)を添加した。この混合物を60℃で3時間加熱した。室温で一晩攪拌した後、溶媒を蒸発させ、残渣を、酢酸エチル/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:58mg(46%)。
この化合物は、HPLCにより純度>95%を有し、かつF−18標識化の前駆体として適している。
【0151】
d)ナフタレン−2−カルボン酸[4−(4−{2−[3−(2−[F−18]フルオロ−エトキシ)−ベンゾイルアミノ]−アセチル}−ピペラジン−1−イル)−フェニル]−アミド
【化62】
トシラート前駆体9cを、無水ジメチルホルムアミド(100μL)及びアセトニトリル(400μL)中に溶解し、乾燥カリウム/クリプトフィックス/フッ化物錯体(前記参照)に添加した。この溶液を、110℃で6分間加熱し、その後室温で5分間冷却した。この溶液を、水で希釈し、最終容積10mLとし、かつSep−Pak Plus C18カートリッジ(Waters社、WAT020515)を通過させた。このカートリッジを、水(20ml)ですすぎ、本化合物をアセトニトリル(3mL)により溶離させた。溶離された化合物を水で希釈し、5mLを得、これを分取HPLC精製に供した。Gemini 5μ C18 110Å、250×10mm(Phenomenex社、00G-4435-N0)を、固定相として使用した。水(A)/アセトニトリル(B)の3/7混合液を、流量3mL/分の移動相として、使用した。
【0152】
この溶液を、水で希釈し、最終容積20mLとし、Sep−Pak light C18カートリッジ(Waters社、WAT023501)を通過させた。このカートリッジを、水(10ml)ですすぎ、本化合物をエタノール(0.5mL)により溶離させた。最終生成物を、Gemini 5μ C18 110Å、250×4.6mm(Phenomenex社、00G-4435-E0)HPLCカラム上で、アイソクラティックな水/アセトニトリル(2/8;v/v)混合液を、流量1mL/分で使用するHPLCにより特徴決定した。保持時間(tR):4.6分。RCY (d.c.):39%。RCP:99%。
【0153】
e)ナフタレン−2−カルボン酸[4−(4−{2−[3−(2−フルオロ−エトキシ)−ベンゾイルアミノ]−アセチル}−ピペラジン−1−イル)−フェニル]−アミド(HPLC−標準)
【化63】
DMF 5mL中のナフタレン−2−カルボン酸(4−{4−[2−(3−ヒドロキシ−ベンゾイルアミノ)−アセチル]−ピペラジン−1−イル}−フェニル)−アミド(9b)56mg(0.11mmol)の溶液へ、炭酸カリウム34mg(0.25mmol)及び1−フルオロ−2−ブロモエタン(ABCR社、独国)10μL(0.13mmol)を添加した。この混合物を60℃で3時間加熱した。溶媒を蒸発させ、残渣を、ジメチルスルホキシドに溶かし、水/アセトニトリル勾配を使用し、RP−18上で2回クロマトグラフィーにかけた。
収量:24mg(40%)。
元素分析:
理論値:C 69.30、H 5.63、F 3.43、N 10.10
実測値:C 69.15、H 5.81、F 3.22、N 10.28
【0154】
実施例10
a){2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−カルバミン酸tert−ブチルエステル
【化64】
THF 800mL及びトリエチルアミン8mL(57.71mmol)中のt−ブトキシカルボニル−グリシン(Aldrich社)7.7g(43.97mmol)の−15℃の溶液に、クロロギ酸イソブチル5.75mL(43.97mmol)を滴下し、この溶液を、この温度で更に15分間維持した。次にこの冷溶液へ、THF/ジクロロメタン(1:1)300mL中の1−(4−ベンジルオキシフェニル)−ピペラジン(3c)11.8g及びトリエチルアミン29mL(210mmol)をゆっくり添加し、その温度を、10℃未満に更に15分間維持し、次に室温とした。一晩攪拌した後、溶媒を蒸発させ、かつ残渣を酢酸エチルに溶かした。この溶液を、水性炭酸ナトリウム、水、1M HCl水溶液、飽和塩化ナトリウム水溶液により連続して洗浄し、最後に硫酸マグネシウム上で乾燥し、その後蒸発させた。この残渣を、ヘキサン/酢酸エチル勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:15.51g(82.9%)。
元素分析:
理論値:C 67.74、H 7.34、N 9.87
実測値:C 67.33、H 7.47、N 9.62
【0155】
b)2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル−アンモニウムクロリド
【化65】
{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−カルバミン酸tert−ブチルエステル(10a)17.0g(39.95mmol)を、ジエチルエーテル中の2N HCl 300mL中で懸濁し、かつ室温で一晩攪拌した。沈殿物を濾過し、エーテルで洗浄し、真空中40℃で乾燥した。
収量:14.5g(定量的)。この生成物は、更に精製することなく次工程において使用した。
【0156】
c)N−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−6−ブロモ−ニコチンアミド
【化66】
DMF 20mL中の2−ブロモ−5−ピリジンカルボン酸(Aldrich社)202mg(1mmol)及び2−[4−(4−ベンジルオキシフェニル)−ピペラジン−1−イル]−2−オキソエチル−アンモニウムクロリド(10b)398mg(1.1mmol)の溶液へ、PyBOP 624mg(1.2mmol)及びN−エチル−N,N−ジイソプロピルアミン0.61mLを添加し、この反応混合液を、室温で一晩攪拌した。溶媒を蒸発させた後、残渣を、酢酸エチル/エタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:183mg(35.9%)。
元素分析:
理論値:C 58.95、H 4.95、Br 15.69、N 11.00
実測値:C 58.66、H 5.18、Br 15.20、N 11.36
この化合物は、HPLCにより純度>95%を有し、かつF−18標識化の前駆体として適している。
【0157】
d)N−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−6−フルオロ−ニコチンアミド
【化67】
DMF 20mL中の2−フルオロ−5−ピリジンカルボン酸(Aldrich社)141mg(1mmol)及び2−[4−(4−ベンジルオキシフェニル)−ピペラジン−1−イル]−2−オキソエチル−アンモニウムクロリド(10b)398mg(1.1mmol)の溶液へ、PyBOP 624mg(1.2mmol)及びN−エチル−N,N−ジイソプロピルアミン0.61mLを添加し、この反応混合液を、室温で一晩攪拌した。溶媒を蒸発させた後、残渣を、酢酸エチル/エタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:302mg(67.3%)。
元素分析:
理論値:C 66.95、H 5.62、F 4.24、N 12.49
実測値:C 66.84、H 5.97、F 3.95、N 12.52
【0158】
e)N−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−6−[F−18]−フルオロ−ニコチンアミド
【化68】
水性[18F]フッ化物(5.1GBq)を、QMAカートリッジ(Waters社、Sep Pak Light QMA、部品番号:WAT023525)上に捕獲し、Wheatonバイアル(5ml)中へ、MeCN 0.95ml中K2.2.2 5mg+水50μl中K2CO3 1mgにより溶離させた。この溶媒を、120℃で10分間、窒素流れ下で加熱することにより、除去した。無水MeCN(1ml)を添加し、前述のように蒸発させた。無水DMSO 700μl中の前駆体10c(5mg)の溶液を添加した。180℃で30分間加熱した後、粗反応混合物を総容積まで水で希釈し、分取HPLCにより精製した:ACE 5−C18−HL 250mm×10mm、Advanced Chromatography Technologies社;カタログ番号:ACE 321-2510;アイソクラチック、0.1%トリフルオロ酢酸中35%アセトニトリル、流量:4ml/分;tR=18分。収集されたHPLC画分を、水40mlで希釈し、Sep−Pak light C18カートリッジ(Waters社、WAT023501)上に固定し、これを水5mlで洗浄し、エタノール1mlで溶離し、生成物1015MBqを送り出した(36%、減衰補正;放射化学的純度>99%)。所望の生成物を、分析用HPLC:Agilent社 ZORBAX 300SB-C18 250×4.6mm;5μm Agilent社;PN 880995-902上での、非−放射性F−19フルオロ標準との同時注入により、特徴付けた;A):水+0.1%TFA、B):MeCN+0.1%TFA、0分〜10分、35%B;10分〜10分30秒、35%Bから100%B;1mL/分(tR=7.6分)、RCP:99%(HPLC)。
【0159】
実施例11
a)N−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−2−ブロモ−イソニコチンアミド
【化69】
DMF 20mL中の2−ブロモ−4−ピリジンカルボン酸(Alfa社)202mg(1mmol)及び2−[4−(4−ベンジルオキシフェニル)−ピペラジン−1−イル]−2−オキソエチル−アンモニウムクロリド(10b)398mg(1.1mmol)の溶液へ、PyBOP 624mg(1.2mmol)及びN−エチル−N,N−ジイソプロピルアミン0.61mLを添加し、この反応混合物を、室温で一晩攪拌した。溶媒を蒸発させた後、残渣を、酢酸エチル/エタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。生成物含有画分を収集し、酢酸エチルから再結晶化した。
収量:165mg(32.4%)。
元素分析:
理論値:C 58.95、H 4.95、Br 15.69、N 11.00
実測値:C 58.32、H 5.09、Br 15.11、N 10.73
この化合物は、HPLCにより純度>95%を有し、かつF−18標識化の前駆体として適している。
【0160】
b)N−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−2−フルオロ−イソニコチンアミド
【化70】
DMF 20mL中の2−フルオロ−4−ピリジンカルボン酸(Aldrich社)141mg(1mmol)及び2−[4−(4−ベンジルオキシフェニル)−ピペラジン−1−イル]−2−オキソエチル−アンモニウムクロリド(10b)398mg(1.1mmol)の溶液へ、PyBOP 624mg(1.2mmol)及びN−エチル−N,N−ジイソプロピルアミン0.61mLを添加し、この反応混合物を、室温で一晩攪拌した。溶媒を蒸発させた後、残渣を、酢酸エチル/エタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:326mg(72.7%)。
元素分析:
理論値:C 66.95、H 5.62、F 4.24、N 12.49
実測値:C 66.58、H 5.81、F 4.03、N 12.68
【0161】
c)N−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−2−[F−18]−フルオロ−イソニコチンアミド
【化71】
水性[18F]フッ化物(4.9GBq)を、QMAカートリッジ(Waters社、Sep Pak Light QMA、部品番号:WAT023525)上に捕獲し、Wheatonバイアル(5ml)中へ、MeCN 0.95ml中K2.2.2 5mg+水50μl中K2CO3 1mgにより溶離させた。この溶媒を、120℃で10分間、窒素流れ下で加熱することにより、除去した。無水MeCN(1ml)を添加し、前述のように蒸発させた。無水DMSO 700μl中の前駆体11a(5mg)の溶液を添加した。180℃で30分間加熱した後、粗反応混合物を総容積5mLまで水で希釈し、分取HPLCにより精製した:ACE 5−C18−HL 250mm×10mm、Advanced Chromatography Technologies社;カタログ番号:ACE 321-2510;アイソクラチック、0.1%トリフルオロ酢酸中35%アセトニトリル、流量:4ml/分;tR=18分。収集されたHPLC画分を、水40mlで希釈し、Sep−Pak light C18カートリッジ(Waters社、WAT023501)上に固定し、これを水5mlで洗浄し、エタノール1mlで溶離し、生成物1293MBqを送り出し(44%、減衰補正)、これは分析用HPLC:Agilent社 ZORBAX 300SB-C18 50×4.6mm;5μm Agilent社;PN 880995-902を使用する、非−放射性F−19フルオロ標準との同時注入により、特徴付け、かつ再確認した;A):水+0.1%TFA、B):MeCN+0.1%TFA、0分〜10分、35%B;10分〜10分30秒、35%Bから100%B;1mL/分(tR=7.5分)、RCP:>99%(HPLC)。
【0162】
実施例12
a)N−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−2−ブロモ−ニコチンアミド
【化72】
DMF 20mL中の2−ブロモ−ニコチン酸(Aldrich社)202mg(1mmol)及び2−[4−(4−ベンジルオキシフェニル)−ピペラジン−1−イル]−2−オキソエチル−アンモニウムクロリド(10b)398mg(1.1mmol)の溶液へ、PyBOP 624mg(1.2mmol)及びN−エチル−N,N−ジイソプロピルアミン0.61mLを添加し、この反応混合物を、室温で一晩攪拌した。溶媒を蒸発させた後、残渣を、酢酸エチル/エタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:210mg(41.2%)。
元素分析:
理論値:C 58.95、H 4.95、Br 15.69、N 11.00
実測値:C 59.11、H 4.75、Br 15.39、N 11.03
この化合物は、HPLCにより純度>95%を有し、かつF−18標識化の前駆体として適している。
【0163】
b)N−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−2−フルオロ−ニコチンアミド
【化73】
DMF 20mL中の2−フルオロ−ニコチン酸(Aldrich社)141mg(1mmol)及び2−[4−(4−ベンジルオキシフェニル)−ピペラジン−1−イル]−2−オキソエチル−アンモニウムクロリド(10b)398mg(1.1mmol)の溶液へ、PyBOP 624mg(1.2mmol)及びN−エチル−N,N−ジイソプロピルアミン0.61mLを添加し、この反応混合物を、室温で一晩攪拌した。溶媒を蒸発させた後、残渣を、酢酸エチル/エタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:326mg(72.7%)。
元素分析:
理論値:C 66.95、H 5.62、F 4.24、N 12.49
実測値:C 66.71、H 5.79、F 3.88、N 12.30
【0164】
c)N−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−2−[F−18]−フルオロ−ニコチンアミド
【化74】
水性[18F]フッ化物(7GBq)を、QMAカートリッジ(Waters社、Sep Pak Light QMA、部品番号:WAT023525)上に捕獲し、Wheatonバイアル(5ml)中へ、MeCN 0.95ml中K2.2.2 5mg+水50μl中K2CO3 1mgにより溶離させた。その溶媒を、120℃で10分間、窒素流れ下で加熱することにより、除去した。無水MeCN(1ml)を添加し、前述のように蒸発させた。無水DMSO 700μl中の前駆体12a(5mg)の溶液を添加した。180℃で30分間加熱した後、粗反応混合物を、総容積5mLまで水で希釈し、分取HPLCにより精製した:ACE 5−C18−HL 250mm×10mm、Advanced Chromatography Technologies社;カタログ番号:ACE 321-2510;アイソクラチック、0.1%トリフルオロ酢酸中35%アセトニトリル、流量:4ml/分;tR=19分。収集されたHPLC画分を、水40mlで希釈し、Sep−Pak light C18カートリッジ(Waters社、WAT023501)上に固定し、これを水5mlで洗浄し、エタノール1mlで溶離し、生成物2015MBqを送り出し(49%、減衰補正)、これは分析用HPLC:Agilent ZORBAX 300SB-C18 50×4.6mm;5μm Agilent;PN 880995-902を使用する、非−放射性F−19フルオロ標準との同時注入により、特徴付け、かつ再確認した;A):水+0.1%TFA、B):MeCN+0.1%TFA、0分〜10分、35%B;10分〜10分30秒、35%Bから100%B;1mL/分(tR=7.8分)、RCP:>99%(HPLC)。
【0165】
実施例13:生物学的データ
a)方法
ヒト脳ホモジネートを使用する結合試験
トリチウム標識されたアミロイドリガンドとの競合アッセイを、96−ウェルプレート(Greiner bio-one社;カタログ番号651201;ロット番号06260130)において、AD患者由来の脳ホモジネートを使用し行った。ホモジネートは、AD患者の灰白質及び白質を含む解離した前頭皮質を、リン酸緩衝食塩水(PBS、pH7.4)中でホモジナイズすることにより(Ultra-Turrax、設定2、30秒間、24000rpm)調製した。このホモジネートは、濃度100mg湿組織/mlとし、300μlアリコートに分割し、−80℃で貯蔵した。
【0166】
様々な濃度の標識されない被験物質を、PBS、0.1%BSA中で、100μg/mlホモジネート及び10nMトリチウム標識されたリガンドと共に(最終容積200μl)、室温で3時間インキュベーションした。引き続き、この結合混合物を、Filtermate 196収集器(Packard社)を使用し、Whatman GF/Bフィルター(PBS、0.1%BSAで湿らせたもの)を通して濾過した。その後フィルターを、PBS、0.1%BSAで2回洗浄し、かつシンチレーター40μlを各ウェルに添加し、その後結合した放射活性を、TopCount装置(Perkin Elmer社)を用いて測定した。非特異的結合は、この反応混合物へ、トリチウム標識されたリガンドの1000倍のアクセス(access of 1000x)を添加することにより、評価した。最後にIC50値を、好適な解析ソフトウェアの助けを借りて算出した。
【0167】
オートラジオグラフィー解析
アルツハイマー型認知症患者、前頭側頭型認知症患者及び年齢が合致した対照からの前頭葉の新鮮凍結切片に加え、パラフィン包埋切片を、本試験に使用した。
クライオスタット(Leica社、独国)上で厚さ18μmにスライスした凍結切片、及び滑走式ミクロトーム(Leica社)上で厚さ6μmにスライスしたパラフィン切片を、スライドガラス(Superfrost Plus, Fa.Menzel社, ブラウンシュバイク、独国)上に搭載した。凍結切片は、−20℃で数晩にわたりスライドに接着させた。パラフィン切片は、慣習的組織学的方法を用いて、脱パラフィンした。結合試験のために、切片を、25mM Hepes緩衝液、pH7.4、0.1%(BSA)中に希釈された、10Bq/μlのF−18標識された被験化合物(200〜300μl/スライド)と一緒に、室温で1.5時間、加湿チャンバー内でインキュベーションした。ブロック試験のために、このインキュベーション混合物に、非標識の被験物質の1000倍アクセスを添加した。ハイブリダイゼーション後、切片を、Hepes緩衝液、0.1%BSAにより4回(又は代わりに40%エタノールで2回)洗浄し、最後に蒸留水に10秒間2回浸した。風乾した切片を、造影プレートに曝し、シグナルを放射線画像解析装置(Fuji BAS5000)により検出した。
【0168】
生体内分布
生体内分布試験及び排泄試験は、雄のNMRIマウス(体重約30g;1時点につき動物3匹)で行った。これらの動物は、通常の実験室条件下、温度22±2℃及び12時間の暗/明リズムで飼育した。飼料及び水は、自由に摂取させた。試験開始前少なくとも3日間の馴化期間の間に、動物は、臨床的に試験し、異常な臨床徴候が存在しないことを確認した。本被験化合物の100μl中約150kBqの尾静脈からの静脈内注射の2、5、30、60、240分後に、尿及び糞便を定量的に収集した。同時点で、動物を、イソフルラン麻酔下で断頭により屠殺し、以下の臓器及び組織を、γ線カウンターを使用する放射活性測定のために摘出した:脾臓、肝臓、腎臓、肺、大腿、心臓、脳、脂肪、甲状腺、筋肉、皮膚、血液、尾部、胃(内容物なし)、精巣、腸(内容物なし)、膵臓、副腎、及び生体残存部。解析のために、組織重量当たりの注射された投与量の減衰補正率(%ID/g±標準偏差)を算出した。
【0169】
b)結果
【表1】
【0170】
オートラジオグラフィー
図1は、アルツハイマー病患者(AD)及びAβプラークを伴わない対照(HC/FTD)(健常対照/前頭側頭型認知症)の皮質由来の凍結切片への3gの結合のオートラジオグラフィー分析を示す。AD試料の斑豊富領域における特異的結合を矢印で示す。
【0171】
【表2】
【0172】
オートラジオグラフィー
図2は、アルツハイマー病患者(AD)及びAβプラークを伴わない対照(HC/FTD)(健常対照/前頭側頭型認知症)の皮質由来の凍結切片への実施例10eの結合のオートラジオグラフィー分析を示す。AD試料の斑豊富領域における特異的結合を矢印で示す。
【0173】
【表3】
【0174】
オートラジオグラフィー
図3は、アルツハイマー病患者(AD)及びAβプラークを伴わない対照(HC/FTD)(健常対照/前頭側頭型認知症)の皮質由来の凍結切片への実施例11cの結合のオートラジオグラフィー分析を示す。AD試料の斑豊富領域における特異的結合を矢印で示す。
【0175】
【表4】
【0176】
オートラジオグラフィー
図4は、アルツハイマー病患者(AD)及びAβプラークを伴わない対照(HC/FTD)(健常対照/前頭側頭型認知症)の皮質由来の凍結切片への実施例12cの結合のオートラジオグラフィー分析を示す。AD試料の斑豊富領域における特異的結合を矢印で示す。
【0177】
選択された化合物のIC50値
図5は、AD患者由来の脳ホモジネートを使用する競合アッセイにおいて測定された、選択された化合物の[nM]でのIC50値を示す。
【0178】
実施例14
a)5−ベンジルオキシ−2−ブロモ−ピリジン
【化75】
DMF 400mL中の2−ブロモ−5−ヒドロキシピリジン10.0g(57.47mmol)の溶液へ、臭化ベンジル14.75g(86.21mmol)及び炭酸カリウム23.82g(172.4mmol)を添加した。この混合物を、60℃で6時間及び室温で一晩攪拌した。この懸濁液を濾過し、溶媒を蒸発させた後、残渣を、ジクロロメタン/メタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:14.82g(96.7%)。
MS(ESIpos):m/z=264, 266 [M+H]+
1H-NMR (300MHz, CHCl3-d): δ [ppm]=5.10 (s, 2H), 7.16 (dd, 1H), 7.32-7.47 (m, 6H), 8.14 (d, 1H)。
【0179】
b)1−(5−ベンジルオキシ−ピリジン−2−イル)−ピペラジン
【化76】
ガラス製品は全て、100℃で乾燥した。トルエン180mL中のピペラジン5.27g(61.22mmol)の溶液へ、トリス(ジベンジリデンアセトン)ジパラジウム(0) 561mg(0.61mmol)及びBINAP(2,2’−ビス(ジフェニルホスフィノ)−1,1’−ビナフチル)520mg(0.83mmol)を添加した。その後THF中の5−ベンジルオキシ−2−ブロモ−ピリジン(実施例14a)14.7g(55.66mmol)の溶液、引き続きTHF中のナトリウムt−ブチラート8.02g(83.48mmol)の懸濁液を添加した。この反応混合物を、6時間還流し、室温で一晩攪拌した。溶媒を蒸発させた後、残渣を、ジクロロメタン/メタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:7.12g(47.0%)。
MS (ESIpos):m/z=270 [M+H]+
1H-NMR (300MHz, CHCl3-d): δ [ppm]=2.97-3.07 (m, 4H), 3.36-3.46 (m, 4H), 5.04 (s, 2H), 6.63 (d, 1H), 7.21 (dd, 1H), 7.29-7.48 (m, 5H), 8.00 (d, 1H)。
【0180】
c)(2−{4−[5−(ベンジルオキシ)ピリジン−2−イル]ピペラジン−1−イル}−2−オキソエチル)カルバミン酸tert−ブチル
【化77】
THF 500mL及びトリエチルアミン(35.87mmol)5mL中のt−ブトキシカルボニル−グリシン(Aldrich社)4.63g(26.43mmol)の−15℃の溶液へ、クロロギ酸イソブチル3.43mL(26.43mmol)を滴下し、この溶液を、この温度で更に15分間維持した。その後、この冷溶液へ、THF/ジクロロメタン(1:1)200mL中の1−(5−ベンジルオキシ−ピリジン−2−イル)−ピペラジン(14b)7.12g及びトリエチルアミン18mL(129mmol)をゆっくり添加し、その温度を−10℃以下に更に15分間維持し、その後室温とした。一晩攪拌した後、溶媒を蒸発させ、残渣を酢酸エチルに溶かした。この溶液を、水性炭酸ナトリウム、水、1M HCl水溶液、飽和塩化ナトリウム水溶液で連続して洗浄し、最後に硫酸マグネシウム上で乾燥し、その後蒸発させた。この残渣を、ヘキサン/酢酸エチル勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:8.04g(70.6%)。
MS (ESIpos):m/z=427 [M+H]+
1H-NMR (300MHz, CHCl3-d): δ [ppm]=1.46 (s, 9H), 3.36-3.45 (m, 2H), 3.51 (br. s., 4H), 3.70-3.81 (m, 2H), 4.02 (d, 2H), 5.05 (s, 2H), 5.53 (br. s., 1H), 6.65 (d, 1H), 7.23 (dd, 1H), 7.30-7.48 (m, 5H), 8.00 (d, 1H)。
【0181】
d)N−(2−{4−[5−(ベンジルオキシ)ピリジン−2−イル]ピペラジン−1−イル}−2−オキソエチル)−2−フルオロピリジン−4−カルボキサミド
【化78】
(2−{4−[5−(ベンジルオキシ)ピリジン−2−イル]ピペラジン−1−イル}−2−オキソエチル)カルバミン酸tert−ブチル(14c)8.0g(18.76mmol)を、ジエチルエーテル中2N HClの160mL中に懸濁し、室温で一晩攪拌した。沈殿物を濾過し、エーテルで洗浄し、真空中で40℃で乾燥した。
収量:7.4g(定量的)。この生成物は、更に精製することなく次工程において使用した。
MS (ESIpos):m/z=327 [M+H]+
【0182】
DMF 40mL中の2−フルオロピリジン−4−カルボン酸(Aldrich社)177mg(1.25mmol)及び前述のように調製した塩酸塩501mg(1.38mmol)の溶液へ、PyBOP 784mg(1.5mmol)及びN−エチル−N,N−ジイソプロピルアミン0.80mLを添加し、この反応混合物を室温で一晩攪拌した。溶媒を蒸発させた後、残渣を、酢酸エチル/エタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:315mg(50.2%)。
MS (ESIpos):m/z=449 [M+H]+
1H-NMR (400MHz, DMSO-d6): δ [ppm]=3.37 (br. s., 2H), 3.44 (br. s., 2H), 3.52-3.66 (m, 4H), 4.22 (d, 2H), 5.07 (s, 2H), 6.83 (d, 1H), 7.27-7.47 (m, 6H), 7.53 (s, 1H), 7.70-7.81 (m, 1H), 7.95 (d, 1H), 8.39 (d, 1H), 9.01 (t, 1H)。
【0183】
e)N−(2−{4−[5−(ベンジルオキシ)ピリジン−2−イル]ピペラジン−1−イル}−2−オキソエチル)−2−ブロモピリジン−4−カルボキサミド
【化79】
(2−{4−[5−(ベンジルオキシ)ピリジン−2−イル]ピペラジン−1−イル}−2−オキソエチル)カルバミン酸tert−ブチル(14c)8.0g(18.76mmol)を、ジエチルエーテル中2N HClの160mL中で懸濁し、室温で一晩攪拌した。沈殿物を濾過し、エーテルで洗浄し、真空中で40℃で乾燥した。
収量:7.4g(定量的)。この生成物は、更に精製することなく次工程において使用した。
MS (ESIpos):m/z=327 [M+H]+
【0184】
DMF 160mL中の2−ブロモピリジン−4−カルボン酸(Aldrich社)1.01g(5.01mmol)及び前述のように調製した塩酸塩2.0g(5.51mmol)の溶液へ、PyBOP 3.13g(6.0mmol)及びN−エチル−N,N−ジイソプロピルアミン2.75mLを添加し、この反応混合物を室温で一晩攪拌した。溶媒を蒸発させた後、残渣を、酢酸エチル/エタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:739mg(27.7%)。
MS (ESIpos):m/z=510, 512 [M+H]+
1H-NMR (400MHz, DMSO-d6): δ [ppm]=2.72 (s, 1H), 2.88 (s, 1H), 3.40-3.48 (m, 2H), 3.51-3.64 (m, 4H), 4.21 (d, 2H), 5.07 (s, 2H), 6.86 (d, 1H), 7.25-7.48 (m, 6H), 7.81 (dd, 1H), 7.95 (d, 1H), 8.02 (s, 1H), 8.56 (d, 1H), 9.06 (s, 1H)。
【0185】
f)[18F]−N−(2−{4−[5−(ベンジルオキシ)ピリジン−2−イル]ピペラジン−1−イル}−2−オキソエチル)−2−フルオロピリジン−4−カルボキサミド
【化80】
水性[18F]フッ化物38.7GBqを、QMAカートリッジ(Waters社)上に捕獲し、反応器中へ、TBAOH溶液2mL(MeCN 1.5mL、H2O 0.3mL+TBAOH溶液(40%)8μL)により溶離させた。その溶媒を、120℃で10分間、窒素流れ下で加熱することにより、除去した。無水MeCN(1ml)を添加し、前述のように蒸発させた。無水DMSO 500μl中の前駆体14e(5mg)の溶液を添加した。180℃で20分間加熱した後、粗反応混合物を水/MeCN(50:50)4mLで希釈し、分取HPLCにより精製した:ACE 5−C18−HL 250mm×10mm;アイソクラチック、0.1%トリフルオロ酢酸を含有する水中25%アセトニトリル、流量:4ml/分;tR=〜22分。収集されたHPLC画分を、水40mlで希釈し、Sep−Pak plus short tC18カートリッジ(Waters社)上に固定し、これを水5mlで洗浄し、エタノール1mlで溶離し、EtOH 1000μl中、F−18標識された生成物3.5GBqを送り出し(再結晶収率15.5%、減衰補正;>96% HPLC)、全体の合成時間は80分間未満であった。望ましいF−18標識された生成物14f(tR=3.2分)を、分析用HPLCを用い分析した:ACE3−C18 50mm×4.6mm;溶媒勾配:出発時5%アセトニトリルから、7分間で0.1%トリフルオロ酢酸中95%アセトニトリル、流量:2mL/分、及びこの分析用HPLC上での、対応する非−放射性F−19フルオロ標準14d(tR=3.1分)との同時注入により、確認した。
HPLC分析は、図6A及び図6Bに示されている。
【0186】
実施例15
a)2−{4−[4−(ベンジルオキシ)フェニル]ピペラジン−1−イル}−2−オキソエチルアセテート
【化81】
THF 300mL及びトリエチルアミン7.5mL(54.3mmol)中のアセトキシ酢酸1.42g (12mmol)(Aldrich社)の−15℃溶液へ、クロロギ酸イソブチル1.73mL(13.23mmol)を滴下し、かつこの溶液を、この温度で更に15分間維持した。その後、この冷溶液へ、THF/ジクロロメタン(1:1)240mL中の1−(4−ベンジルオキシフェニル)−ピペラジン(3c)3.23g(12mmol)及びトリエチルアミン1.74mL(12.5mmol)をゆっくり添加し、その温度を−10℃以下に更に15分間維持し、その後室温とした。一晩攪拌した後、溶媒を蒸発させ、残渣を酢酸エチルに溶かした。この溶液を、水性炭酸ナトリウム、水、1M HCl水溶液、飽和塩化ナトリウム水溶液で連続して洗浄し、最後に硫酸マグネシウム上で乾燥し、その後蒸発させた。この残渣を、ヘキサン/酢酸エチル勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:1.12g(25.2%)。
MS (ESIpos):m/z=369 [M+H]+
1H-NMR (300MHz, CHCl3-d): δ [ppm]=2.21 (s, 3H), 3.02-3.14 (m, 4H), 3.55 (br. s., 2H), 3.78 (br. s., 2H), 4.78 (s, 2H), 5.03 (s, 2H), 6.83-7.00 (m, 4H), 7.29-7.48 (m, 5H)。
【0187】
b)1−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−ヒドロキシ−エタノン
【化82】
酢酸エステル15aの246mg(0.67mmol)を、エタノール40mL中に溶解し、0℃に冷却した。3N NaOH 2.1mLの添加後、この溶液を1時間攪拌し、そのpHがpH7を下回るまで、氷酢酸を添加し、溶媒を蒸発させた。粗生成物をエタノールから結晶化した。
収量:116mg(52.7%)。
MS (ESIpos):m/z=327 [M+H]+
1H-NMR (300MHz, CHCl3-d): δ [ppm]=3.01-3.14 (m, 4H), 3.37-3.49 (m, 2H), 3.63 (t, 1H), 3.78-3.88 (m, 2H), 4.22 (d, 2H), 5.04 (s, 2H), 6.83-7.00 (m, 4H), 7.29-7.49 (m, 5H)。
【0188】
c)2−{4−[4−(ベンジルオキシ)フェニル]ピペラジン−1−イル}−2−オキソエチル 2−フルオロピリジン−4−カルボキシラート
【化83】
THF 5mL及びトリエチルアミン29μL(0.2mmol)中の2−フルオロピリジン−4−カルボン酸(Aldrich社) 28.2mg(0.2mmol)の−15℃溶液へ、クロロギ酸イソブチル31.6μL(0.22mmol)を滴下し、この溶液を、この温度で更に15分間維持した。その後、この冷溶液へ、THF/ジクロロメタン(1:1) 5mL中の1−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−ヒドロキシ−エタノン(15b)65.28mg(0.2mmol)及びトリエチルアミン125μL(0.9mmol)をゆっくり添加し、その温度を−10℃以下に更に15分間維持し、その後室温とした。一晩攪拌した後、溶媒を蒸発させ、残渣を酢酸エチルに溶かした。この溶液を、水性炭酸ナトリウム、水、1M HCl水溶液、飽和塩化ナトリウム水溶液で連続して洗浄し、最後に硫酸マグネシウム上で乾燥し、その後蒸発させた。この残渣を、ヘキサン/酢酸エチル勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:30mg(33.4%)。
MS (ESIpos):m/z=450 [M+H]+
1H-NMR (400MHz, CHCl3-d): δ [ppm]=3.03-3.18 (m, 4H), 3.61 (br. s., 2H), 3.81 (br. s., 2H), 5.06 (d, 4H), 6.87-6.99 (m, 4H), 7.29-7.47 (m, 5H), 7.55-7.62 (m, 1H), 7.83 (dt, 1H), 8.40 (d, 1H)。
【0189】
d)2−{4−[4−(ベンジルオキシ)フェニル]ピペラジン−1−イル}−2−オキソエチル2−ブロモピリジン−4−カルボキシラート
【化84】
THF 25mL及びトリエチルアミン0.2mL(1.44mmol)中の2−ブロモピリジン−4−カルボン酸(Aldrich社)185.7mg(0.92mmol)の−15℃溶液へ、クロロギ酸イソブチル132.3μL(1.01mmol)を滴下し、この溶液を、この温度で更に15分間維持した。その後、この冷溶液へ、THF/ジクロロメタン(1:1)24mL中の1−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−ヒドロキシ−エタノン(15b)300mg(0.92mmol)及びトリエチルアミン0.6mL(4.3mmol)をゆっくり添加し、その温度を−10℃以下に更に15分間維持し、その後室温とした。一晩攪拌した後、溶媒を蒸発させ、残渣を酢酸エチルに溶かした。この溶液を、水性炭酸ナトリウム、水、1M HCl水溶液、飽和塩化ナトリウム水溶液で連続して洗浄し、最後に硫酸マグネシウム上で乾燥し、その後蒸発させた。この残渣を、ヘキサン/酢酸エチル勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:145mg(30.6%)。
MS (ESIpos):m/z=510, 512 [M+H]+
1H-NMR (400MHz, CHCl3-d): δ [ppm]=3.11 (d, 4H), 3.60 (br. s., 2H), 3.80 (br. s., 2H), 5.05 (d, 4H), 6.86-6.99 (m, 4H), 7.29-7.48 (m, 5H), 7.89 (dd, 1H), 8.13 (s, 1H), 8.56 (d, 1H)。
【0190】
e)[18F]−2−{4−[4−(ベンジルオキシ)フェニル]ピペラジン−1−イル}−2−オキソエチル2−フルオロピリジン−4−カルボキシラート
【化85】
水性[18F]フッ化物19GBqを、QMAカートリッジ(Waters社)上に捕獲し、反応器中へ、TBAOH溶液2mL(MeCN 1.5mL、H2O 0.3mL+TBAOH溶液(40%)8μL)により溶離させた。その溶媒を、120℃で10分間、窒素流れ下で加熱することにより、除去した。無水MeCN(1ml)を添加し、前述のように蒸発させた。無水DMSO 500μl中の前駆体15d(5mg)の溶液を添加した。180℃で20分間加熱した後、粗反応混合物を水/MeCN(50:50)4mLで希釈し、分取HPLCにより精製した:ACE 5−C18−HL 250mm×10mm;アイソクラチック、0.1%トリフルオロ酢酸を含む水中40%アセトニトリル、流量:4ml/分;tR=〜24分。収集されたHPLC画分を、水40mlで希釈し、Sep−Pak plus short tC18カートリッジ(Waters社)上に固定し、これを水5mlで洗浄し、エタノール1mlで溶離し、EtOH 1000μl中、F−18標識された生成物15e 0.2GBqを送り出し(再結晶収率2%、減衰補正;>98% HPLC)、全体の合成時間は80分未満であった。望ましいF−18標識された生成物15e(tR=4.3分)を、分析用HPLCを用い分析し:ACE3−C18 50mm×4.6mm;溶媒勾配:出発時5%アセトニトリルから、7分間で0.1%トリフルオロ酢酸中95%アセトニトリル、流量:2mL/分、及びこの分析用HPLC上での、対応する非−放射性F−19フルオロ標準15c(tR=4.2分)との同時注入により、確認した。
HPLC分析は、図7A及び図7Bに示されている。
【0191】
実施例16
a)2−フルオロ−N−{2−[4−(4−ヒドロキシフェニル)ピペラジン−1−イル]−2−オキソエチル}ピリジン−4−カルボキサミド
【化86】
メタノール70mL中のN−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−2−フルオロ−イソニコチンアミド(11b)280mg(0.62mmol)の溶液に、活性炭上に担持されたパラジウム(10%)100mgを添加し、この懸濁液を水素大気下で一晩室温で攪拌した。その後これを、触媒から濾過除去し、その溶液を真空中で蒸発させた。生成物は、更に精製することなく、次工程において使用した。
収量:175mg(54.8%)。
MS (ESIpos):m/z=359 [M+H]+。
【0192】
b)4−(4−{N−[(2−フルオロピリジン−4−イル)カルボニル]グリシル}ピペラジン−1−イル)フェニルベンゾエート
【化87】
THF 10mL及びトリエチルアミン90μL(0.65mmol)中の安息香酸57.9mg(0.47mmol)の−15℃溶液へ、クロロギ酸イソブチル62.06μL(0.47mmol)を滴下し、この溶液を、この温度で更に15分間維持した。その後この冷溶液へ、THF/ジクロロメタン(1:1)20mL中の先に調製した実施例16a 170mg(0.47mmol)、及びトリエチルアミン0.4mL(2.87mmol)をゆっくり添加し、更に15分間その温度を−10℃未満に維持し、その後室温とした。一晩攪拌した後、溶媒を蒸発させ、残渣を、酢酸エチルに溶かした。この溶液を、水性炭酸ナトリウム、水、1M HCl水溶液、飽和塩化ナトリウム水溶液で連続して洗浄し、最後に硫酸マグネシウム上で乾燥し、その後蒸発させた。この残渣を、ヘキサン/酢酸エチル勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:23mg(9.5%)。
MS (ESIpos): m/z=463 [M+H]+
1H-NMR (400MHz, DMSO-d6): δ [ppm]=3.15 (br. s., 2H), 3.22 (br. s., 2H), 3.66 (br. s., 4H), 4.24 (d, 2H), 7.00-7.11 (m, 2H), 7.11-7.21 (m, 2H), 7.50-7.66 (m, 3H), 7.68-7.82 (m, 2H), 8.12 (d, 2H), 8.41 (d, 1H), 9.03 (t, 1H)。
【0193】
c){2−[4−(4−ヒドロキシフェニル)ピペラジン−1−イル]−2−オキソエチル}カルバミン酸tert−ブチル
【化88】
メタノール100mL中の{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−カルバミン酸tert−ブチルエステル(10a)2.5g(5.88mmol)の溶液に、活性炭上に担持されたパラジウム(10%)1gを添加し、この懸濁液を水素大気下で一晩室温で攪拌した。その後これを、触媒から濾過除去し、その溶液を真空中で蒸発させた。生成物は、更に精製することなく、次工程において使用した。
収量:1.92g(95.9%)。
MS (ESIpos):m/z=336 [M+H]+。
【0194】
d)2−ブロモ−N−{2−[4−(4−ヒドロキシフェニル)ピペラジン−1−イル]−2−オキソエチル}ピリジン−4−カルボキサミド
【化89】
{2−[4−(4−ヒドロキシフェニル)ピペラジン−1−イル]−2−オキソエチル}カルバミン酸tert−ブチル(16c)1.8g(5.37mmol)を、ジエチルエーテル中の2N HCl 60mL中に懸濁し、室温で一晩攪拌した。沈殿物を濾過し、エーテルで洗浄し、真空中40℃で乾燥した。
収量:1.8g(定量的)。生成物は、更に精製することなく、次工程において使用した。
【0195】
DMF 60mL中の2−ブロモ−4−ピリジンカルボン酸(Alfa社)338mg(1.67mmol)及び先に調製した塩酸塩500mg(1.84mmol)の溶液へ、PyBOP 1.045g(2mmol)及びN−エチル−N,N−ジイソプロピルアミン2.86mL(16.73mmol)を添加し、この反応混合物を、室温で一晩攪拌した。溶媒を蒸発させた後、残渣を、酢酸エチル/エタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。生成物含有画分を収集し、かつ酢酸エチルから再結晶化した。
収量:284mg(40.5%)。
MS (ESIpos):m/z=419, 421 [M+H]+
1H-NMR (300MHz, CH3OH-d4): δ [ppm]=1.49 (dt, 4H), 2.17 (dt, 4H), 2.76 (s, 2H), 5.11-5.22 (m, 2H), 5.29-5.40 (m, 2H), 6.23 (dd, 1H), 6.43-6.50 (m, 1H), 6.89-6.98 (m, 1H)。
【0196】
e)4−(4−{N−[(2−ブロモピリジン−4−イル)カルボニル]グリシル}ピペラジン−1−イル)フェニルベンゾエート
【化90】
DMF 45mL中の2−ブロモ−N−{2−[4−(4−ヒドロキシフェニル)ピペラジン−1−イル]−2−オキソエチル}ピリジン−4−カルボキサミド(16d)218mg(0.52mmol)の溶液へ、1,3−ジシクロヘキシルカルボジイミド214.56mg(1.04mmol)及び4−ジメチルアミノピリジン(DMAP)20mgを添加し、室温で40分間攪拌した。その後安息香酸127mg(1.04mmol)を添加し、この反応混合物を、この温度で3日間攪拌した。この溶媒を蒸発させ、残渣を、酢酸エチル/エタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:222mg(81.6%)。
MS (ESIpos):m/z=523, 525 [M+H]+
1H-NMR (400MHz, DMSO-d6): δ [ppm]=3.25 (br. s., 4H), 3.65 (br. s., 4H), 4.23 (d, 2H), 6.99-7.10 (m, 2H), 7.10-7.21 (m, 2H), 7.53-7.65 (m, 2H), 7.67-7.77 (m, 1H), 7.83 (dd, 1H), 7.99-8.06 (m, 1H), 8.06-8.17 (m, 2H), 8.55 (d, 1H), 9.05 (t, 1H)。
【0197】
f)[18F]−4−(4−{N−[(2−フルオロピリジン−4−イル)カルボニル]グリシル}ピペラジン−1−イル)フェニルベンゾエート
【化91】
水性[18F]フッ化物1GBqを、QMAカートリッジ(Waters社)上に捕獲し、反応器中へ、TBAOH溶液2mL(MeCN 1.5mL、H2O 0.3mL+TBAOH溶液(40%)8μL)により溶離させた。その溶媒を、120℃で10分間、窒素流れ下で加熱することにより、除去した。無水MeCN(1ml)を添加し、前述のように蒸発させた。無水DMSO 500μl中の前駆体16e(5mg)の溶液を添加した。180℃で20分間加熱した後、粗反応混合物を分析用HPLCを用い分析し:ACE3−C18 50mm×4.6mm;溶媒勾配:出発5%アセトニトリルから、7分間で0.1%トリフルオロ酢酸中の95%アセトニトリル、流量:2mL/分、かつこの分析用HPLC上での、対応する非−放射性F−19フルオロ標準15cとの同時注入により、確認した(tR=3.7分)。粗生成物は、分取HPLCにより精製することができる:ACE 5−C18−HL 250mm×10mm;アイソクラチック、0.1%トリフルオロ酢酸を含有する水中35%アセトニトリル、流量:4ml/分。
【0198】
実施例17:生物学的データ
実施例14及び15の化合物の生物学的データを、実施例13に説明したように得た。
【0199】
図8は、アルツハイマー病患者(AD)及び健常対照(HC)の皮質由来の凍結切片への14fの結合のオートラジオグラフィー分析を示す。AD試料の斑豊富領域における特異的結合を矢印で示す。
【0200】
図9は、AD患者由来の脳ホモジネートを使用する競合アッセイにおいて測定された、選択された化合物の[nM]でのIC50値を示す。
【0201】
【表5】
【技術分野】
【0001】
本発明は、アミロイド沈着物に結合しかつ造影するのに有用な新規化合物、並びにアルツハイマー病及びアミロイドーシスの検出又は治療におけるそれらの使用に関する。
【背景技術】
【0002】
アルツハイマー病(AD)は、記憶、認知、及び行動の安定性の喪失により特徴付けられる進行性神経変性障害である。ADは、β−アミロイドペプチド(Aβ)の線維状沈着物により構成される細胞外老人斑及び過剰リン酸化されたタウの対らせん状細線維(PHF)により構成された神経原線維変化(FT)により、病理学的に定義される。Aβペプチドを構成している39〜43個のアミノ酸は、より大きいアミロイド前駆体タンパク質(APP)に由来している。アミロイド形成経路において、Aβペプチドは、APPからβ−及びγ−セクレターゼの逐次タンパク質分解により、切断される。Aβペプチドは、可溶性タンパク質として放出され、かつ正常な加齢脳内の脳脊髄液(CSF)中で低レベルで検出されることができる。ADの進行時に、Aβペプチドは凝集し、かつ脳の実質及び脈管構造内にアミロイド沈着物を形成し、これは組織学的試験において、びまん性老人斑及び血管アミロイドとして、剖検時に検出され得る(最新の総説については、Blennowら、Lancet., 368(9533): 387-403 (2006年7月29日)を参照のこと)。
【0003】
アルツハイマー病は、世界中で、大きい健康上及び社会経済学上の問題となり始めている。本疾患の早期検出及び有効治療に関する技術及び方法を開発するために、膨大な努力が成されている。現在、記憶障害クリニックの専門的状況(academic setting)においてADの診断は、およそ85〜90%の精度である(Petrella JRら、Radiology, 226:315-36 (2003))。これは、同様の症状を引き起こす様々な疾患の排除、並びに慎重な神経学的及び精神医学的検査、更には神経心理学的試験を基にしている。しかし依然、脳の剖検時組織検査がこの疾患の唯一の確定診断である。従って本疾患のひとつの病理学的特徴―脳内のアミロイド凝集物の沈着―のインビボ検出は、ADの早期検出及び他の認知症からの識別に大きい影響を有すると考えられる。加えて開発中のほとんどの疾患修飾療法は、脳内のアミロイド負荷の軽減を目的としている。従って脳内のアミロイド負荷の造影は、患者階層化及び治療モニタリングにとって必須のツールを提供することができる。
【0004】
加えてアミロイド沈着物は、アミロイドーシスにおいて役割を果たすこともわかっており、ここではアミロイドタンパク質が、様々な臓器及び/又は組織内に異常に沈着され、疾患を引き起こす。最近の総説については、Chitiらの論文、Annu Rev Biochem., 75:333-66 (2006)を参照のこと。
【0005】
脳内のアミロイド凝集物の可視化のための可能性のあるリガンドは、アミロイドへの高い結合親和性を示さなければならず、かつ血液脳関門を超えなければならない。AD患者の脳内でのトレーサーの結合パターンに関してヒトにおいて既に研究されているPETトレーサーは、[F−18]FDDNP(Shoghi-Jadidら、Am J Geriatr Psychiatry, 10:24-35 (2002))、[C−11]PIB(Klunkら、Ann Neurol. 55:306-319 (2004))、[C−11]SB−13(Verhoeffら、Am J Geriatr Psychiatry, 12:584-595 (2004);[F−18]Bay94−9172(Roweら、Lancet Neurol, 7:129-135 (2008));[C−11]BF227(Kudoら、J Nucl. Med, 49:554-561 (2007));及び、[F−18]PIB(Farrarら、Turku PET Symposium, Abstract 49(2007))がある。最近の総説については、Lockhardtの論文、Drug Discov Today, 11:1093-1099 (2006)、Henriksenらの論文、Eur. J. Nucl. Med. Mol. Imaging, (2007)、Cohenの論文、Mol. Imaging Biol. 9:204-2162007、Nordbergの論文、Curr. Opin Biol., 20:398-402 (2007)、Smallらの論文、Neurology 7:161-172 (2008)、Nordbergの論文、Eur. J. Nucl. Med. Mol. Imaging, 35, S46-S50(2008)を参照されたい。
【0006】
現在最も有望なPETトレーサーは、脳内のアミロイド沈着物へのそれらの特異的結合に加え、特にAD患者の白質脳領域内に加え海馬(HC)において、不都合な非特異的蓄積を示す。概して非特異的バックグラウンド結合は、画質に干渉し、かつ例えばアミロイドの定量及び本疾患の極初期の診断を損なう場合がある。
【発明の概要】
【発明が解決しようとする課題】
【0007】
すなわち、本発明の前提となる課題は、本疾患患者におけるアミロイド沈着物を、高い特異性で、アミロイド関連疾患の初期の段階で検出するのに適した化合物を提供することであった。
【0008】
本発明は、アミロイドβへの親和性が高く、脳から非特異的シグナルが迅速に排除される、新規トレーサーを提供することにより、本課題を解決する。
【課題を解決するための手段】
【0009】
本発明は、アミロイド沈着物に結合し、かつ血液脳関門を通過することができ、従って患者のアルツハイマー病及びアミロイドーシスを、好ましくは該疾患の初期において、診断するのに有用な化合物に関する。
【0010】
一態様によれば、本発明は、式Iの化合物、及び医薬として許容し得るその塩又はプロドラッグに関する。
【化1】
(式中、
−Yは:
F、Cl、Br、I、H、
18F、19F、76Br、123I、125I、11C、3H、13N、15O等の検出可能な標識;
トシル、ブロシル、ノシル、トリフラート、スルホネート、置換スルホネート、メシラート、及びノナフレートなどの脱離基;並びに
芳香族C原子に直接結合される場合は、ヨードニウム−アリールI+−アリール、トリアルキルアンモニウム、好ましくはトリメチルアンモニウム、及びNO2からなる群から選択され;
−Arは、1又は2個のアルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基により任意に置換されていてもよい、単環、二環又は三環式の芳香環又は芳香族複素環系からなる群から選択され;
ここで、該アルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基は、任意に置換されていてもよく、ここで該置換基は、好ましくはオキソ又はヒドロキシルから選択され、
該アルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基は、1〜5個の酸素原子、−SO−又は−SO2−基により隔てられていてもよく、好ましくは、該置換基はポリエチレングリコール部分であり、
該アルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基は、C3−C6シクロアルキル部分を含んでいてもよく、
該単環、二環又は三環式の芳香環又は芳香族複素環系は、芳香族C原子に直接結合されている電子求引基により更に置換されていてもよく、好ましい電子求引基は、−CN又は−CF3であり;
好ましくは、Arは、フェニル、2−、3−、又は4−ピリジル、ピリミジル、ピラジル、プロピルピリミジン−2−イル、エトキシフェニル、(CH2CH2O)3フェニル、アルキルフェニル、アルコキシフェニル、N−アルキルインドリル、及びアルキルピリジルからなる群から選択され、
更に好ましい実施形態によれば、
【化2】
であり;
−Bは、直接結合、1〜10個のC原子からなる分枝又は非分枝のアルキル又はアルキレン鎖からなる群から選択され、ここで該アルキレン鎖は、1又は2個の不飽和結合を含んでいてもよく、
該アルキル又はアルキレン鎖は、N、S、SO、SO2又はOにより任意に隔てられていてもよく、
該アルキル又はアルキレン鎖は、オキソ又は−OHにより任意に置換され;
好ましくは、Bは、直接結合、CONH−CH2CO、CO−(CH2)2CO、(CH2)nCO、O(CH2)nCO(但しn=1〜10)、及び(CH=CH)COからなる群から選択され、
さらに好ましくは、
【化3】
であり;
−Aは、直接結合、及びCO−NH、CS−NHからなる群から選択され;
−Ar’は、1又は2個のアルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基により任意に置換されていてもよい、単環又は二環式芳香環又は芳香族複素環系からなる群から選択され;
該アルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基は、任意に置換されていてもよく、該置換基は、好ましくはオキソ又はヒドロキシルから選択され、
該アルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基は、1〜5個の酸素原子により隔てられていてもよく、好ましくは、該置換基は、ポリエチレングリコール部分であり、
該アルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基は、C3−C6シクロアルキル部分を含んでいてもよく;
好ましくは、Ar’は、フェニル、2−、3−、又は4−ピリジル、ピリミジル、ピラジル、プロピルピリミジン−2−イル、エトキシフェニル、(CH2CH2O)3フェニル、アルキルフェニル、アルコキシフェニル、N−アルキルインドリル、フェニル、ベンゾフラニル、インドリル及びアルキルピリジルからなる群から選択され、
より好ましくは、Ar’は、フェニル、ベンゾフラニル、及びインドリルからなる群から選択され、
さらに好ましくは、
【化4】
であり;
最も好ましくは、Ar’は、フェニルであり;
−Xは、直接結合、又は好ましくは非限定的にオキソ若しくはチオである1若しくは2個の置換基により任意に置換されていてもよい、C1−C3アルキル鎖からなる群から選択され;
該アルキル鎖は、1〜2個のO、N、S、SO又はSO2基により隔てられていてもよく;
好ましくは、Xは、直接結合、OCH2、NHCO、CH2O、CONH、NHCS、又はCSNHからなる群から選択され;
−Ar”は、1又は2個のアルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基により任意に置換されていてもよい、単環又は二環式芳香環又は芳香族複素環系からなる群から選択され、
該アルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基は、任意に置換されていてもよく、該置換基は、好ましくはオキソ又はヒドロキシルから選択され、
該アルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基は、1〜5個の酸素原子により隔てられていてもよく、好ましくは、該置換基は、ポリエチレングリコール部分であり、
該アルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基は、C3−C6シクロアルキル部分を含んでいてもよく、
より好ましくはAr”は、フェニル、1−フェニル、1−ナフチル、2−ナフチル、及びそれらに対応する複素環全てからなる群から選択され、
更に好ましくは、
【化5】
である。
【0011】
Ar’並びにAr”は:
F、Cl、Br、I、H、
18F、19F、76Br、123I、125I、11C、3H、13N、15O等の検出可能な標識;
トシル、ブロシル、ノシル、トリフラート、スルホネート、置換スルホネート、メシラート、及びノナフレートなどの脱離基;並びに
芳香族C原子に直接結合される場合は、ヨードニウム−アリールI+−アリール、トリアルキルアンモニウム、好ましくはトリメチルアンモニウム、及びNO2
により、任意に置換されていてもよい。
【図面の簡単な説明】
【0012】
【図1】図1は、アルツハイマー病患者(AD)及びAβプラークを伴わない対照(HC/FTD)(健常対照/前頭側頭型認知症)の皮質由来の凍結切片への3gの結合のオートラジオグラフィー分析を示す。AD試料の斑豊富領域における特異的結合を矢印で示す。
【図2】図2は、アルツハイマー病患者(AD)及びAβプラークを伴わない対照(HC/FTD)(健常対照/前頭側頭型認知症)の皮質由来の凍結切片への実施例10eの結合のオートラジオグラフィー分析を示す。AD試料の斑豊富領域における特異的結合を矢印で示す。
【図3】図3は、アルツハイマー病患者(AD)及びAβプラークを伴わない対照(HC/FTD)(健常対照/前頭側頭型認知症)の皮質由来の凍結切片への実施例11cの結合のオートラジオグラフィー分析を示す。AD試料の斑豊富領域における特異的結合を矢印で示す。
【図4】図4は、アルツハイマー病患者(AD)及びAβプラークを伴わない対照(HC/FTD)(健常対照/前頭側頭型認知症)の皮質由来の凍結切片への実施例12cの結合のオートラジオグラフィー分析を示す。AD試料の斑豊富領域における特異的結合を矢印で示す。
【図5】図5は、AD患者由来の脳ホモジネートを使用する競合アッセイにおいて測定された、選択された化合物の[nM]でのIC50値を示す。
【図6】図6A及び図6Bは、実施例14の化合物のHPLC分析を示す。
【図7】図7A及び図7Bは、実施例15の化合物のHPLC分析を示す。
【図8】図8は、アルツハイマー病患者(AD)及び健常対照(HC)の皮質由来の凍結切片への14fの結合のオートラジオグラフィー分析を示す。AD試料の斑豊富領域における特異的結合を矢印で示す。
【図9】図9は、AD患者由来の脳ホモジネートを使用する競合アッセイにおいて測定された、選択された化合物の[nM]でのIC50値を示す。
【発明を実施するための形態】
【0013】
インビトロ及びインビボ双方の診断目的には、放射性核種又は蛍光標識等の検出可能な標識を含んでなり、又はそれらを有する式Iの化合物が好ましい。インビトロ使用の場合、新鮮凍結試料又はパラフィン試料等の組織切片が分析され得る。
【0014】
式Iの化合物の好ましい実施形態は以下に示すとおりであるが、これらを化合物1d/e、2d/e、3g/h、4f/g、5b/c、6f、7d、8g、9d/e、10、11、12、13、14、15、16、17、18、19、20、21及び22と称する。これらの好ましい実施形態は、例えば式Iの記号Y、Ar、B、A、Ar’、X、及びAr”により表される様々な基の例でもある。
【0015】
「アルキル」とは、炭素及び水素のみからなり、1〜8個の炭素原子を有し、不飽和部位を含まない、直鎖又は分枝鎖の基をいう。例えば、メチル、エチル、n−プロピル、1−メチルエチル(イソ−プロピル)、n−ブチル、n−ペンチル、1,1−ジメチルエチル(t−ブチル)、n−ヘプチル等が挙げられる。「アルコキシ」とは、−Oアルキル基という式で表される基である。ここでアルキルは先に定義されるとおりである。
【0016】
本発明との関連において好ましい塩は、医薬として許容し得る本発明の化合物の塩である。本発明は、それ自体は医薬適用に適していないが、例えば本発明の化合物の単離又は精製等に使用できる塩も含む。
【0017】
本発明の化合物の医薬として許容し得る塩は、無機酸、カルボン酸及びスルホン酸の酸付加塩、例えば、塩酸、臭化水素酸、硫酸、リン酸、メタンスルホン酸、エタンスルホン酸、トルエンスルホン酸、ベンゼンスルホン酸、ナフタレンジスルホン酸、酢酸、トリフルオロ酢酸、プロピオン酸、乳酸、酒石酸、リンゴ酸、クエン酸、フマル酸、マレイン酸及び安息香酸の塩を含む。
【0018】
本発明の化合物の医薬として許容し得る塩の例として、好ましくは、アルカリ金属塩(例えば、ナトリウム塩及びカリウム塩)、アルカリ土類金属塩(例えば、カルシウム塩及びマグネシウム塩)、並びにアンモニア、又は炭素原子1〜16個を有する有機アミン、例えば、好ましくは、エチルアミン、ジエチルアミン、トリエチルアミン、エチルジイソプロピルアミン、モノエタノールアミン、ジエタノールアミン、トリエタノールアミン、ジシクロヘキシルアミン、ジメチルアミノエタノール、プロカイン、ジベンジルアミン、N−メチル−モルホリン、アルギニン、リシン、エチレンジアミン及びN−メチルピペリジンなどに由来したアンモニウム塩などの、慣例的塩基の塩も含む。
【0019】
更に本発明は、本発明の化合物のプロドラッグも含む。用語「プロドラッグ」は、それらの化合物の一部に関してそれらは生物学的に活性又は不活性であるが、それらが体内で消費される間に本発明の化合物に転換される(例えば、代謝又は加水分解により)化合物を含む。
【0020】
特に本発明は、式(I)のカルボン酸の加水分解可能なエステル誘導体も含む。これらは、生理的媒体において、特にインビボにおいて、酵素的又は化学的手段により加水分解され、遊離カルボン酸を生じることができるエステルであると理解される。このようなエステルは好ましくは、アルキル基がヒドロキシル、(C1−C4)−アルコキシ、アミノ、モノ−(C1−C4)−アルキルアミノ及び/又はジ−(C1−C4)−アルキル−アミノにより置換されてよい、直鎖又は分枝された(C1−C6)−アルキルエステルである。特に好ましいのは、式(I)の化合物のメチルエステル又はエチルエステルである。
【0021】
本発明の文脈において、他に特定されない限りは、置換基は下記の意味を有する。
本発明の文脈において、アルキルは、各場合に言及された炭素原子数を有する、直鎖又は分枝鎖のアルキルラジカルを表す。下記のラジカルは、一例として及び好ましいものとして言及されている:メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、1−メチルプロピル、tert−ブチル、n−ペンチル、イソペンチル、1−エチルプロピル、1−メチルブチル、2−メチルブチル、3−メチルブチル、n−ヘキシル、1−メチルペンチル、2−メチルペンチル、3−メチルペンチル、4−メチルペンチル、3,3−ジメチルブチル、1−エチルブチル、2−エチルブチル、1,4−ジメチルペンチル、4,4−ジメチルペンチル及び1,4,4−トリメチルペンチル。
【0022】
本発明の文脈において、シクロアルキルは、炭素原子3〜7個を有する、単環の飽和されたアルキルラジカルを表す。下記のラジカルは、一例として及び好ましいものとして言及されている:シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル及びシクロヘプチル。
【0023】
本発明の文脈において、アルコキシは、炭素原子1〜4個を有する、直鎖又は分枝されたアルコキシラジカルを表す。下記のラジカルは、一例として及び好ましいものとして言及されている:メトキシ、エトキシ、n−プロポキシ、イソプロポキシ、1−メチルプロパオキシ、n−ブトキシ、イソブトキシ及びtert−ブトキシ。
本発明の文脈において、ハロゲンは、フッ素、塩素、臭素及びヨウ素を含む。好ましいのはフッ素である。
【0024】
本発明の化合物のラジカルが置換されている場合、これらのラジカルは、他に特定されない限りは、単−又は多置換されることができる。本発明の文脈において、2回以上出現する全てのラジカルの意味は、互いに独立している。1、2又は3個の同一又は異なる置換基による置換が、好ましい。とりわけ好ましいのは、1個の置換基による置換である。
【0025】
本発明の化合物又は医薬として許容し得るその塩は、それらの構造内に不斉炭素原子を有することができる。従って本発明の化合物及び医薬として許容し得るその塩は、単独のエナンチオマー、ジアステレオ異性体、ラセミ体、及びエナンチオマーとジアステレオマーの混合物として存在してよい。そのような単独のエナンチオマー、ジアステレオ異性体、ラセミ体、及びそれらの混合物は全て、本発明の範囲内である。
【0026】
先に及び本明細書において説明された化合物は、本発明の好ましい実施形態において、Aβペプチドへ結合される。
【0027】
本発明の別の態様は、特にヒトなどの哺乳動物における、患者のアルツハイマー病及び/又はアミロイドーシスを診断及び/又は治療するための先に及び本明細書において説明された式Iの化合物の使用である。
【0028】
アルツハイマー病及び/又はアミロイドーシスの患者の治療は、好ましくは、放射性標識を有さないが、Yが例えば水素である、式Iの本発明の化合物により実施されることができる。
【0029】
好ましくは、この診断における本発明の化合物の使用は、ポジトロン放出断層撮影(PET)、単光子放出型コンピュータ断層撮影(SPECT)、磁気共鳴(MR)スペクトル分析法又は断層撮影法を使用し実行される。
【0030】
本発明の別の態様は、アミロイド沈着物の造影方法に関する。そのような方法は、a)検出可能な標識を含む先に及び本明細書において説明された化合物を、哺乳動物へ投与すること、及びb)アミロイド沈着物に特異的に結合されている本化合物から発生する(steming)シグナルを検出することを含む。この特異的結合は、本発明の化合物のアミロイド沈着物への高い結合親和性の結果である。
【0031】
更なる態様において、本発明は、アルツハイマー病又はアミロイドーシスの患者を診断する方法に関する。この方法は、a)先に及び本明細書に説明されたヒトにおける本化合物の検出のための検出可能な標識を伴う本発明の化合物を、そのような診断が必要なヒトへ投与すること、及びb)本化合物のヒトへの投与から生じる、検出可能な標識からのシグナルを、好ましくはγ線カメラの使用、ポジトロン放出断層撮影(PET)、又は単光子放出型コンピュータ断層撮影(SPECT)により、測定することを含む。
【0032】
本発明の更なる実施形態は、本発明の化合物を患者へ投与すること、及び本発明の造影方法を適用することを含む、患者におけるアルツハイマー病の除外を含む、アルツハイマー病のような他の神経学的障害の診断方法を含む。
【0033】
本発明の更なる態様は、放射標識された式Iの化合物を含有する、アミロイド沈着物の造影のための診断用組成物に関する。
【0034】
本発明の診断方法は、剖検時診断方法においても使用することができる。
【0035】
更に本発明の診断方法は、アルツハイマー病、神経変性障害又はアミロイドーシスの療法のモニタリングに使用することもできる。
【0036】
更に本発明の診断方法は、アルツハイマー病を除外することにより、アルツハイマー病以外の神経学的障害を診断するためにも使用することができる。
【0037】
本発明の更なる態様において、本発明は、本明細書に説明されたような式Iの化合物を、そのような治療を必要とするヒトに投与することを含む、アミロイドーシス又はアルツハイマー病を治療又は予防する方法を含む。
【0038】
本発明の更なる態様は、本明細書に説明されたような本発明の化合物を、任意に好適な担体及び/又は添加剤と一緒に含有する医薬組成物に関する。
【0039】
更に、本発明の化合物は、例えば、ハイスループットスクリーニング法及びインビトロアッセイなどの、スクリーニングにおけるツールとして使用されることもできる。
【0040】
本発明は、本明細書に説明されたような式Iの本発明の化合物を合成する方法にも関する。本発明の化合物の全般的合成方法は、以下である。
【0041】
F−18放射標識
本発明の更なる態様は、式Iの化合物をフッ素化剤と反応させる工程を含む、放射標識された式Iの化合物を製造するための、式Iの化合物の放射性フッ素化の方法に関する。有用な放射性フッ素化法は、当業者に周知である。
【0042】
好ましい実施形態において、前記フッ素化剤は、4,7,13,16,21,24−ヘキサオキサ−1,10ジアザビシクロ[8.8.8]−ヘキサコサンK18F(クラウンエーテル塩クリプトフィックスK18F)、K18F、H18F、KH18F2又は18Fのテトラアルキルアンモニウム塩である。より好ましいフッ素化剤は、K18F、H18F、又はKH18F2である。
【0043】
使用される溶媒は、ジメチルホルムアミド、DMF、ジメチルスルホキシド、DMSO、アセトニトリル、MeCN、ジメチルアセトアミド、DMA、DMAAなどであり、好ましくはDMSO、MeCN又はDMFである。これらの溶媒は、先に記した溶媒の混合物であることもできる。
【0044】
[F−18]放射標識手順は、当業者に周知である。例えば、放射標識は、以下に説明されたように実行されることができる。
【0045】
[F−18]フッ化物は、18O(p,n)18F反応のために[O−18]水が充填された銀標的(1mL)を使用するサイクロ(登録商標)トロン内における陽子衝撃により生成されることができる。この水性[F−18]フッ化物は、カートリッジ(例えば、QMA樹脂カートリッジ、Waters社、Sep Pak Light QMA部品番号:WAT023525)を通過させることができる。次に捕獲された[F−18]フッ化物は、例えばクリプトフィックスK2.2.2/K2CO3溶液の添加により、カートリッジから溶離されることができる(クリプトフィックスは4,7,13,16,21,24−ヘキサオキサ−1,10−ジアザビシクロ[8.8.8]−ヘキサコサンである)。この前駆体の求核置換は、NBu4OH、(NBu4)2CO3、K2CO3などの塩基の存在下、高温で働くことが好ましい。クリプトフィックス(K2.2.2)などのクラウンエーテルの添加は、特に塩基としてのK2CO3の存在下で、この反応に正に影響を及ぼすことができる。
【0046】
フッ化カリウムクリプトフィックス錯体は、アセトニトリルの逐次添加による、繰り返しの共沸蒸留により好ましくは乾燥される。アセトニトリル、DMF、DMSOなどの溶媒は、反応溶媒として使用されることができる。この標識生成物は、カートリッジを使用する固相抽出により精製されることができる。好ましいカートリッジは、Sep−Pak Plus C18カートリッジ(Waters社、WAT020515)である。このカートリッジは、水によりすすぐことができ、かつこの化合物は、アセトニトリルにより溶離されることができる。溶離された化合物は、水により希釈されることができ、かつ次に分取HPLC精製に供されることができる。好ましいHPLCカラムは、Gemini 5μ C18 110Å、250×10mm(Phenomenex社、00G-4435-N0)などの、逆相カラムである。緩衝溶液、酸、水などの、アセトニトリル、メタノール、エタノールなどの有機溶媒との混合液は、移動相として使用されることができる。
【0047】
その後この溶液は、例えば濃縮及び溶媒交換のために、カートリッジを通過される水により希釈されることができる。
【0048】
F−18化合物:アルキル−F及び(ヘテロ)アリール−Fの全般的合成
一般式Iのアルキル−F−18化合物(式IでY=18F)の前駆体は、例えばトシラート、ブロシラート、ノシラート、メシラート、トリフラート、ノナフレートなど(式IでY=脱離基)であり、これらは当該技術分野において公知の方法に従い各ヒドロキシ化合物から合成されることができる(J. March、Advanced Organic Chemistry、第4版、1992、John Wiley & Sons社、352ff頁)。追加の方法は、実施例3f、4e及び5aに説明されており、かつ例えばTsO−(CH2)n−OTsのような、好適なビス(トシラート)などによる合成を含む。
【0049】
一般式Iのアルキル−F−18化合物(式IでY=18F)のその他の前駆体は、例えばヨウ化物及び臭化物などであり、かつそれらの各フッ化物への転化も当該技術分野において公知である(J. March、前記参照)。
【0050】
一般式Iのアリール−F−18化合物の前駆体は、例えば、当該技術分野において公知の方法により本発明の各F−18化合物へ転換されることができる、アリール又はヘテロアリールブロミド、ニトロ化合物、トリアルキルアンモニウム、アリールヨードニウムである(L. Cai、S. Lu、V. Pike、Eur. J. Org. Chem, 2853-2873 (2008))。これらの前駆体のための出発材料は市販されているか、又は当該技術分野において公知の方法により合成されることができる(R.C. Larock、Comprehensive Organic Transformations、VCH Publishers社、1989)。
【0051】
トシラート、ブロシラート、ノシラート、メシラート、トリフラート、ノナフレートなどのための出発材料としてのヒドロキシ化合物の合成は、以下を含む。
【0052】
・OH−保護基の脱保護。非常に多用途な保護基のひとつとして、アセチル保護基が挙げられる。多くのその他のものが、当該技術分野において公知であり、例えばT.W. Greene及びP.G.M. Wuts、Protective Groups in Organic Synthesis、第3版、1999年、John Wiley & Sons社、17ff頁を参照し、同じく例えば実施例3d及び4cも参照されたい。
【0053】
・実施例1b及び2bに示すような、Hal=Br、Iである、
【化6】
の、Pd触媒された置換による、ヒドロキシアルキル基の導入。
【0054】
・実施例6dに示すような、B’が−CO−(CH2)n−COOHであるヒドロキシ化合物HO−Ar−B’は、フリーデル・クラフツアシル化により合成することができる。
【0055】
Y=Br、I、HO、保護基−O−である、一般式Iの化合物は、下記により合成されることができる。
【化7】
【0056】
・任意の位置及び配列順での−CO−NH−基を確立するための、アミドカップリング。
又は
【0057】
・実施例1a及び2aに説明されたように、任意に、尿素結合(−NH−CO−NH−)を構築するための、ホスゲン又はホスゲン同等物の使用による、ピペラジン誘導体の、好適なアミノ成分との反応による。
【0058】
アリール−ピペラジンは、市販されているか、又は文献に従い、例えばKlaparsらの文献(Journal of the American Chemical Society, 124, 7421-28 (2002))及びそこに引用された文献に従い、合成されることができる。
【0059】
本発明の更なる態様は、本発明の放射標識されない化合物を備えるキットに言及しており、この化合物は任意に、乾燥状態であるか、又は不活性の医薬として許容し得る担体及び/若しくは溶媒及び/若しくは補助物質が添加されている。好ましい実施形態において、本キットは、該キットの化合物を含む、1個又は複数の密閉容器を備えている。好ましい実施形態において、本キットは、Yが、トシル、ブロシル、ノシル、トリフラート、スルホネート、置換スルホネート、メシラート、ノナフレート、ヨードニウム−アリールI+−アリール、トリアルキルアンモニウム、トリメチルアンモニウム、又はNO2などの脱離基である、本明細書に説明された式Iの化合物を備えている。
【0060】
更に別の本発明の態様は、哺乳動物において、アミロイドの形成を阻害するか、又はアミロイドの病原性を調節する方法に関する。この方法は、式Iの化合物が、アミロイドの形成を阻害するか、又はアミロイドの病原性を調節する上で有効である量で投与されることを含む。
【0061】
発明の好ましい化合物
本発明の種々の態様によれば、特定の式Iの化合物が好ましい。斯かる好ましい化合物を以下に示す。化合物に付した記号は、対応する化合物の合成を示す実施例を表す。
【0062】
【化8】
【0063】
【化9】
【0064】
【化10】
【0065】
より一層好ましい本発明の実施形態は、各F−18化合物である。
【0066】
本発明の化合物は、アミロイドβに高親和性を持ち、かつ非特異的シグナルが脳から迅速に排泄される、新規トレーサーを表している。
【0067】
特に本発明は、以下に関する:
【0068】
(1)式Iの化合物、又は医薬として許容し得るその塩若しくはプロドラッグ:
【化11】
(式中、
−Yは、F、Cl、Br、I、H、18F、19F、76Br、123I、125I、11C、3H、13N、15O;
脱離基、トシル、ブロシル、ノシル、トリフラート、スルホネート、置換スルホネート、メシラート、及びノナフレート;並びに、芳香族C原子に直接結合される場合は、ヨードニウム−アリールI+−アリール、トリアルキルアンモニウム、好ましくはトリメチルアンモニウム、及びNO2からなる群から選択され;
−Arは、置換又は非置換の、単環、二環又は三環式の、芳香環又は芳香族複素環系からなる群から選択され;
−Bは、直接結合、1〜10個のC原子からなる分枝又は非分枝のアルキル又はアルキレン鎖からなる群から選択され;
−Aは、直接結合、及びCO−NH、CS−NHからなる群から選択され;
−Ar’は、置換又は非置換の、単環又は二環式の、芳香環又は芳香族複素環系からなる群から選択され;
−Xは、直接結合、又は、置換若しくは非置換のC1−C3アルキル鎖からなる群から選択され;
−Ar”は、置換又は非置換の、単環又は二環式の、芳香環又は芳香族複素環系からなる群から選択される)。
【0069】
(2)1番の化合物、式中:
−Arの芳香族複素環又は芳香環系は、1又は2個のアルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基により任意に置換されており;
−単環、二環又は三環式の芳香環又は芳香族複素環系は、芳香族C原子に直接結合された電子求引基により更に置換されてよく;
−Bのアルキレン鎖は、1又は2個の不飽和結合を含んでいてもよく、ここでアルキル又はアルキレン鎖は、任意にN、S、SO、SO2又はOにより隔てられ、ここでアルキル又はアルキレン鎖は、オキソ又は−OHにより任意に置換されており;
−Ar’の芳香族複素環又は芳香環系は、1又は2個のアルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基により任意に置換されていてもよく、ここで単環、二環又は三環式の芳香環又は芳香族複素環系は、芳香族C原子に直接結合された電子求引基により更に置換されてよく;
−XのC1−C3アルキル鎖は、オキソ又はチオであってよい1又は2個の置換基により任意に置換され、ここでアルキル鎖は、1〜2個のO、N、S、SO又はSO2基により隔てられていてもよく;
−Ar”の芳香族複素環又は芳香環系は、1又は2個のアルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基により任意に置換され、ここで単環、二環又は三環式の芳香環又は芳香族複素環系は、芳香族C原子に直接結合された電子求引基により更に置換されてよい。
【0070】
(3)1番又は2番の化合物、式中:
−Ar、Ar’及びAr”の任意のアルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基は、オキソ又はヒドロキシルからなる群から選択され;
Ar、Ar’及びAr”のアルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基は、1〜5個の酸素原子により隔てられていてもよく、好ましくはこの置換基はポリエチレングリコール部分であり;
更にAr、Ar’及びAr”のアルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基は、C3−C6シクロアルキル部分を含んでいてもよく、ここでAr、Ar’及びAr”の任意の電子求引基は、−CN又はCF3からなる群から選択され;
−Bは、直接結合、CONH−CH2CO、CO−(CH2)2CO、(CH2)nCO、O(CH2)nCO(但しn=1〜10)、(CH=CH)CO、
【化12】
からなる群から任意に選択され;
−Xは、直接結合、OCH2、NHCO、CH2O、CONH、NHCS、又はCSNHからなる群から任意に選択される。
【0071】
(4)1番−3番の化合物、式中:
−Arは、プロピルピリミジン−2−イル、エトキシフェニル、(CH2CH2O)3フェニル、アルキルフェニル、アルコキシフェニル、N−アルキルインドリル、及びアルキルピリジル、
【化13】
からなる群から任意に選択され;
−Ar’は、プロピルピリミジン−2−イル、エトキシフェニル、(CH2CH2O)3フェニル、アルキルフェニル、アルコキシフェニル、N−アルキルインドリル、フェニル、ベンゾフラニル、インドリル及びアルキルピリジルからなる群から任意に選択され;
−Ar”は、フェニル、1−フェニル、1−ナフチル、2−ナフチル、及びそれらに対応する複素環全てからなる群から任意に選択される。
【0072】
(5)下記からなる群から選択される化合物:
【化14】
【化15】
【化16】
【0073】
(6)Fが、18Fの意味を有する、5番の化合物。
【0074】
(7)放射性核種又は蛍光標識などの、検出可能な標識を含む、1番、2番、3番又は4番の化合物。
【0075】
(8)検出可能な標識が、18Fである、7番の化合物。
【0076】
(9)診断用化合物としての、1番−8番の化合物。
【0077】
(10)医薬品としての、1番−5番又は7番の化合物。
【0078】
(11)アルツハイマー病、神経変性障害、又はアミロイドーシスからなる群から選択される疾患のための診断用化合物としての、8番の化合物。
【0079】
(12)アルツハイマー病、神経変性障害、又はアミロイドーシスからなる群から選択される疾患の治療のための医薬品としての、10番の化合物。
【0080】
(13)好適な前駆分子をフッ素化剤と反応させることを含む、1番−8番に従う、フッ素化された化合物の調製方法。
【0081】
(14)1番−5番又は7番の化合物の治療的有効量を、哺乳動物へ投与することを含む、該哺乳動物におけるアルツハイマー病、神経変性障害又はアミロイドーシスからなる群から選択される障害の治療又は予防の方法。
【0082】
(15)1番−5番又は7番の化合物の治療的有効量が、哺乳動物へ投与される、該哺乳動物におけるアルツハイマー病、神経変性障害又はアミロイドーシスからなる群から選択される疾患の治療における1番−5番又は7番の化合物の使用。
【0083】
(16)6番又は8番の化合物を、哺乳動物へ投与することを含む、アルツハイマー病、神経変性障害又はアミロイドーシスからなる群から選択される該哺乳動物における疾患を診断する方法。
【0084】
(17)前記哺乳動物を造影し、かつ造影シグナルを検出することを含む、16番の方法。
【0085】
(18)前記造影が、PET、SPECT、MRスペクトル分析法、及びMR−断層撮影法からなる群から選択される造影方法を用いて実行される、17番の方法。
【0086】
(19)療法の作用がモニタリングされる、16番−18番の方法。
【0087】
(20)前記方法が、哺乳動物の試料をインビトロにおいて分析することを含み、ここで該哺乳動物又は試料が、6番又は8番の化合物により処理される、該哺乳動物におけるアルツハイマー病、神経変性障害又はアミロイドーシスからなる群から選択される疾患を診断又は療法をモニタリングする方法。
【0088】
(21)試料が脳脊髄液である、20番の方法。
【0089】
(22)1番−8番の化合物を備えるキット。
【0090】
本キットは、1番−8番の化合物を含む、1個又は複数の密封バイアルを備えることができる。
【0091】
本発明は更に、1番−8番の化合物を含有する医薬組成物又は診断用組成物、好ましくは放射標識された化合物を含有するそのような組成物、より一層好ましくはF−18により標識された化合物を含有するそのような組成物に関する。
【0092】
本発明は更に、A及びBが直接結合である場合、Ar又はAr’は、6員の芳香環系であることを条件とする、1番の化合物に関する。
【0093】
本発明は更に、A又はBが直接結合である場合、Ar又はAr’は、6員の芳香環系であることを条件とする、1番の化合物に関する。
【0094】
本発明は更に、A又はBが直接結合である場合、この直接結合した残基Ar又はAr’は、6員の芳香環系であることを条件とする、1番の化合物に関する。
【0095】
本発明の放射標識された化合物及び診断用組成物は、βアミロイド造影に有用である。
【実施例】
【0096】
これらの組成物を合成及び標識する方法は、下記実施例においてより十分に例示されている。これらの実施例は、前述の方法のある態様及び有利な結果を例示しており、かつこれらは例証として示されているが限定として示されているものではない。
【0097】
実施例1
a)4−(5−ブロモ−ピリミジン−2−イル)−ピペラジン−1−カルボン酸(4−ベンジルオキシ−フェニル)−アミド
【化17】
ジクロロメタン10mL中の4−ベンジルオキシ−フェニルアミン塩酸塩(Aldrich社)707mg(3mmol)及びN,N−エチルジイソプロピルアミン1.13mL(6.6mmol)の溶液を、ジクロロメタン10mL中のトリホスゲン297mg(1mmol)の溶液に、0℃で滴下した。この反応混合液を、0℃で15分間攪拌し、その後ジクロロメタン10mL中の5−ブロモ−2−ピペラジン−1−イル−ピリミジン0.73g(3mmol)及びN,N−エチルジイソプロピルアミン1.13mL(6.6mmol)の混合液を添加した。0℃で30分間及び室温で一晩経過後、溶媒を蒸発させ、残渣を、ジクロロメタン/酢酸エチル勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:0.93g(66%)
元素分析:
理論値:C 56.42、H 4.73、Br 17.06、N 14.95
実測値:C 56.11、H 4.83、Br 16.94、N 14.77
【0098】
b)4−[5−(3−ヒドロキシ−プロピル)−ピリミジン−2−イル]−ピペラジン−1−カルボン酸(4−ベンジルオキシフェニル)−アミド
【化18】
無水テトラヒドロフラン(THF)9mL中のアリルアルコール0.21mL(3mmol)の溶液へ、THF中の9−ボラビシクロ−(3.3.1)−ノナン0.5M溶液18mL(9mmol)を0℃で添加した。この混合液を、0℃で更に15分間、室温で5時間攪拌した(=溶液A)。
ブロモ誘導体化合物1aの0.70g(1.5mmol)を、無水DMF 10mL中に懸濁し、かつテトラキス(トリフェニルホスフィン)パラジウム(0)0.35g(0.3mmol)及び3M炭酸カリウム水溶液4mL(12mmol)を添加した(=懸濁液B)。
溶液Aを、懸濁液Bに添加し、65℃で一晩攪拌した。溶媒の蒸発後、残渣を、ジクロロメタン/メタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:110mg(16%)
元素分析:
理論値:C 67.10、H 6.53、N 15.65
実測値:C 66.92、H 6.41、N 15.88
【0099】
c)トルエン−4−スルホン酸3−{2−[4−(4−ベンジルオキシ−フェニルカルバモイル)−ピペラジン−1−イル]−ピリミジン−5−イル}−プロピルエステル
【化19】
無水ピリジン3mL中のヒドロキシ誘導体1bの0.45g(1mmol)の懸濁液へ、無水ピリジン2mL中のp−トルエンスルホニルクロリド0.25g(1.3mmol)の溶液を、0℃で滴下した。30分間攪拌した後、この混合液を、蒸発乾固させ、かつその残渣を、ジクロロメタン/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:379mg(63%)
元素分析:
理論値:C 63.88、H 5.86、N 11.64、S 5.33
実測値: C 63.67、H 5.97、N 11.48、S 5.02
【0100】
d)4−[5−(3−[F−18]フルオロ−プロピル)−ピリミジン−2−イル]−ピペラジン−1−カルボン酸(4−ベンジルオキシ−フェニル)−アミド
【化20】
[F−18]フッ化物を、18O(p,n)18F反応のために[O−18]水が充填された低容積、定圧、銀標的(1mL)を使用するサイクロ(登録商標)トロン内における陽子衝撃により生成した。この水性[F−18]フッ化物(約1mL)は、QMA樹脂カートリッジ(Waters社、Sep Pak Light QMA部品番号:WAT023525)を通過させた。捕獲された[F−18]フッ化物を、クリプトフィックスK2.2.2/K2CO3溶液(K2.2.2 5mg/アセトニトリル1.5mL+K2CO3 1mg/水0.5mL)の添加により、カートリッジから溶離させた。この混合物を、アセトニトリル1mLを逐次添加しながら、繰り返しの(2×)共沸蒸留により乾燥させた。このトシラート前駆体1cを、無水ジメチルホルムアミド(100μL)及びアセトニトリル(400μL)中に溶解し、乾燥カリウム/クリプトフィックス/フッ化物錯体に添加した。この溶液を、110℃で6分間加熱し、その後室温で5分間冷却した。この溶液を、水で希釈し、最終容積10mLとし、かつSep−Pak Plus C18カートリッジ(Waters社、WAT020515)を通過させた。このカートリッジを、水(20ml)ですすぎ、本化合物をアセトニトリル(3mL)により溶離させた。溶離された化合物を水で希釈し、5mLを得、これを分取HPLC精製に供した。Gemini 5μ C18 110Å、250×10mm(Phenomenex社、00G-4435-N0)を、固定相として使用した。水(A)/アセトニトリル(B)の3/7混合液を、流量3mL/分の移動相として、使用した。
この溶液を、水で希釈し、最終容積20mLとし、Sep−Pak light C18カートリッジ(Waters社、WAT023501)を通過させた。このカートリッジを、水(10ml)ですすぎ、本化合物をエタノール(0.5mL)により溶離させた。最終生成物を、Gemini 5μ C18 110Å、250×4.6mm(Phenomenex社、00G-4435-E0)HPLCカラム上で、アイソクラティックな水/アセトニトリル(2/8;v/v)混合液を、流量1mL/分で使用するHPLCにより特徴決定した。保持時間(tR):4.8分。衝撃の最後に減衰補正(d.c.)した放射化学的収率(RCY):44%。放射化学的純度(RCP):98%。
【0101】
e)4−[5−(3−フルオロ−プロピル)−ピリミジン−2−イル]−ピペラジン−1−カルボン酸(4−ベンジルオキシ−フェニル)−アミド(HPLC標準)
【化21】
ジクロロメタン10mL中のヒドロキシ誘導体化合物1bの67mg(0.15mmol)へ、ジエチルアミノ硫黄トリフルオリド(DAST)48mg(0.3mmol)を0℃で添加した。0℃で30分後、水を添加し、有機相を硫酸ナトリウム上で乾燥し、その有機溶媒を蒸発させた後、残渣を、ジクロロメタン/酢酸エチル勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:40mg(60%)
元素分析:
理論値:C 66.80、H 6.28、F 4.23、N 15.58
実測値:C 66.54、H 6.49、F 4.06、N 15.77
【0102】
実施例2
a)4−(5−ブロモ−ピリミジン−2−イル)−ピペラジン−1−カルボン酸(2−フェニル−ベンゾフラン−5−イル)−アミド
【化22】
ジクロロメタン20mL中の2−フェニル−ベンゾフラン−5−イルアミン塩酸塩(ChemBridge社、サンディエゴ、米国)628mg(3mmol)及びN,N−エチルジイソプロピルアミン1.13mL(6.6mmol)の溶液を、ジクロロメタン10mL中のトリホスゲン297mg(1mmol)の溶液に、0℃で滴下した。この反応混合液を、0℃で30分間攪拌し、その後ジクロロメタン20mL中の5−ブロモ−2−ピペラジン−1−イル−ピリミジン0.73g(3mmol)及びN,N−エチルジイソプロピルアミン0.56mL(3.3mmol)の混合液を添加した。0℃で1時間及び室温で一晩経過後、溶媒を蒸発させ、残渣を、ジクロロメタン/酢酸エチル勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:0.81g(56%)
元素分析:
理論値:C 57.75、H 4.21、Br 16.70、N 14.64
実測値:C 57.43、H 4.37、Br 16.11、N 14.45
【0103】
b)4−[5−(3−ヒドロキシ−プロピル)−ピリミジン−2−イル]−ピペラジン−1−カルボン酸(2−フェニル−ベンゾフラン−5−イル)−アミド
【化23】
無水テトラヒドロフラン(THF)9mL中のアリルアルコール0.21mL(3mmol)の溶液へ、THF中の9−ボラビシクロ−(3.3.1)−ノナン0.5M溶液18mL(9mmol)を0℃で添加した。この混合液を、0℃で更に15分間、室温で5時間攪拌した(=溶液A)。
化合物2aの0.72g(1.5mmol)を、無水DMF 10mL中に懸濁し、かつテトラキス(トリフェニルホスフィン)パラジウム(0)0.35g(0.3mmol)及び3M炭酸カリウム水溶液4mL(12mmol)を添加した(=懸濁液B)。
溶液Aを、懸濁液Bに添加し、65℃で一晩攪拌した。溶媒の蒸発後、残渣を、ジクロロメタン/メタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:85mg(12%)
元素分析:
理論値:C 68.25、H 5.95、N 15.31
実測値:C 68.05、H 6.12、N 15.20
【0104】
c)トルエン−4−スルホン酸3−{2−[4−(2−フェニル−ベンゾフラン−5−イルカルバモイル)−ピペラジン−1−イル]−ピリミジン−5−イル}−プロピルエステル
【化24】
無水ピリジン3mL中のヒドロキシ誘導体2bの0.46g(1mmol)の懸濁液へ、無水ピリジン2mL中のp−トルエンスルホニルクロリド0.25g(1.3mmol)の溶液を、0℃で滴下した。30分間攪拌した後、この混合液を、蒸発乾固させ、かつその残渣を、ジクロロメタン/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:434mg(71%)
元素分析:
理論値:C 64.80、H 5.44、N 11.45、S 5.24
実測値:C 64.66、H 5.23、N 11.74、S 4.97
【0105】
d)4−[5−(3−[F−18]フルオロ−プロピル)−ピリミジン−2−イル]−ピペラジン−1−カルボン酸(2−フェニル−ベンゾフラン−5−イル)−アミド
【化25】
トシラート前駆体2cを、無水ジメチルホルムアミド(100μL)及びアセトニトリル(400μL)中に溶解し、乾燥カリウム/クリプトフィックス/フッ化物錯体に添加した(上記参照)。この溶液を、110℃で6分間加熱し、その後室温で5分間冷却した。この溶液を、水で希釈し、最終容積10mLとし、かつSep−Pak Plus C18カートリッジ(Waters社、WAT020515)を通過させた。このカートリッジを、水(20ml)ですすぎ、本化合物をアセトニトリル(3mL)により溶離させた。溶離された化合物を水で希釈し、5mLを得、これを分取HPLC精製に供した。Gemini 5μ C18 110Å、250×10mm(Phenomenex社、00G-4435-N0)を、固定相として使用した。水(A)/アセトニトリル(B)の3/7混合液を、流量3mL/分の移動相として、使用した。
【0106】
この溶液を、水で希釈し、最終容積20mLとし、Sep−Pak light C18カートリッジ(Waters社、WAT023501)を通過させた。このカートリッジを、水(10ml)ですすぎ、本化合物をエタノール(0.5mL)により溶離させた。最終生成物を、Gemini 5μ C18 110Å、250×4.6mm(Phenomenex社、00G-4435-E0)HPLCカラム上で、アイソクラティックな水/アセトニトリル(2/8;v/v)混合液を、流量1mL/分で使用するHPLCにより特徴決定した。
保持時間(tR):4.1分。RCY(d.c.):38%。RCP:97%。
【0107】
e)4−[5−(3−フルオロ−プロピル)−ピリミジン−2−イル]−ピペラジン−1−カルボン酸(2−フェニル−ベンゾフラン−5−イル)−アミド(HPLC標準)
【化26】
ジクロロメタン10mL中のヒドロキシ誘導体化合物2bの69mg(0.15mmol)へ、ジエチルアミノ硫黄トリフルオリド(DAST)48mg(0.3mmol)を0℃で添加した。0℃で30分後、水を添加し、有機相を硫酸ナトリウム上で乾燥し、その有機溶媒を蒸発させた後、残渣を、ジクロロメタン/酢酸エチル勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:15mg(22%)
元素分析:
理論値:C 67.96、H 5.70、F 4.13、N 15.24
実測値:C 67.67、H 5.96、F 3.96、N 15.22
【0108】
実施例3
a)(3−アセトキシ−ベンゾイルアミノ)−酢酸ベンジルエステル
【化27】
グリシンベンジルエステルp−トルエンスルホン酸30.4g(90mmol)を、二相システムのジクロロメタン及び飽和炭酸水素ナトリウム水溶液中に溶解した。有機相を、硫酸マグネシウム上で乾燥し、次に蒸発させた。
遊離アミン13.09g(79.25mmol)を得、これを更に精製することなく、引き続きのカップリング反応において使用した。
【0109】
THF 150mL及びトリエチルアミン11mL(79.25mmol)中の3−アセトキシ−安息香酸14.28g(79.25mmol)の−15℃の溶液に、クロロギ酸イソブチル11.39mL(87.2mmol)を滴下し、この溶液を、この温度で更に15分間維持した。その後この冷溶液へ、THF 50mL及びジクロロメタン50mL中のグリシンベンジルエステル13.09g及びトリエチルアミン11mL(79.25mmol)をゆっくり添加し、更に15分間その温度を10℃未満に維持し、その後室温とした。一晩攪拌した後、溶媒を蒸発させ、残渣を、酢酸エチル/エタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:24.6g(95%)。
元素分析:
理論値:C 66.05、H 5.23、N 4.28
実測値:C 65.84、H 5.43、N 4.16
【0110】
b)(3−アセトキシ−ベンゾイルアミノ)−酢酸
【化28】
メタノール300mL中のベンジルエステル3aの19.64g(60mmol)の溶液に、チャコール上に担持されたPd(10%)3gを添加し、この懸濁液を水素下、室温で一晩攪拌した。触媒を濾過除去し、溶媒を蒸発させた。
収量:14.2g(定量的)。
元素分析:
理論値:C 55.70、H 4.67、N 5.90
実測値:C 55.66、H 4.49、N 5.77
【0111】
c)1−(4−ベンジルオキシ−フェニル)−ピペラジン
【化29】
ガラス器具は全て、100℃で乾燥した。トルエン60mL中のピペラジン4.32g(50.16mmol)の溶液へ、トリス(ジベンジリデンアセトン)ジパラジウム(0)459mg(0.5mmol)及びBINAP(2,2’−ビス(ジフェニルホスフィノ)−1,1’−ビナフチル)423mg(0.68mmol)を添加した。その後THF 40mL中の4−ベンジルオキシ−ブロモベンゼン12g(45.6mmol)の溶液を添加し、引き続きTHF中のナトリウムt−ブチラート6.56g(68.27mmol)の懸濁液を添加した。
【0112】
この反応混合物を、3時間還流し、室温で一晩攪拌した。溶媒を蒸発させた後、残渣を、ジクロロメタン/メタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:12.2g(45.7%)。
元素分析:
理論値:C 76.09、H 7.51、N 10.44
実測値:C 75.91、H 7.76、N 10.20
【0113】
d)酢酸3−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチルカルバモイル}−フェニルエステル
【化30】
THF 70mL及びトリエチルアミン0.40mL(2.87mmol)中の(3−アセトキシ−ベンゾイルアミノ)−酢酸3bの654mg(2.76mmol)の−15℃の溶液へ、クロロギ酸イソブチル0.396mL(3.03mmol)を滴下し、この溶液を、この温度で更に15分間維持した。その後この冷溶液へ、THF 30mL及びジクロロメタン30mL中の1−(4−ベンジルオキシ−フェニル)−ピペラジン3cの740mg及びトリエチルアミン1.7mL(12.25mmol)をゆっくり添加し、更に15分間その温度を10℃未満に維持し、その後室温とした。一晩攪拌した後、溶媒を蒸発させ、残渣を、ヘキサン/酢酸エチル勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:390mg(30%)。
元素分析:
理論値:C 68.98、H 6.00、N 8.62
実測値:C 69.09、H 5.81、N 8.42
【0114】
e)N−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−3−ヒドロキシ−ベンズアミド
【化31】
酢酸塩3dの230mg(0.47mmol)を、エタノール30mLに溶解し、0℃に冷却した。3N NaOH 1.5mLを添加した後、この溶液を1時間攪拌し、そのpHがpH7を下回るまで、氷酢酸を添加し、溶媒を蒸発させた。粗生成物を、エタノールから結晶化した。
収量:200mg(95%)。
元素分析:
理論値:C 70.10、H 6.11、N 9.43
実測値:C 69.88、H 6.27、N 9.20
【0115】
f)トルエン−4−スルホン酸2−(3−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチルカルバモイル}−フェノキシ)−エチルエステル
【化32】
DMF 10mL中のN−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−3−ヒドロキシ−ベンズアミド(3e)80mg(0.18mmol)の溶液に、炭酸カリウム62mg(0.45mmol)及び1,2ビス(トシルオキシ)エタン100mg(0.27mmol)(Aldrich社)を添加した。この混合物を60℃で3時間加熱した。室温で一晩攪拌した後、溶媒を蒸発させ、残渣を、酢酸エチル/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:56mg(48%)。
この化合物は、HPLCにより純度>95%を有し、F−18標識化の前駆体として適している。
【0116】
g)N−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−3−(2−[F−18]フルオロ−エトキシ)−ベンズアミド
【化33】
トシラート前駆体3fを、無水ジメチルホルムアミド(100μL)及びアセトニトリル(400μL)に溶解し、乾燥カリウム/クリプトフィックス/フッ化物錯体(前記参照)に添加した。この溶液を110℃で6分間加熱し、次に室温で5分間冷却した。この溶液を、水で希釈し、最終容積10mLとし、Sep−Pak Plus C18カートリッジ(Waters社、WAT020501)を通過させた。このカートリッジを、水(20ml)ですすぎ、本化合物をアセトニトリル(3mL)により溶離させた。溶離された化合物を水で希釈し、5mLを得、これを分取HPLC精製に供した。Gemini 5μ C18 110Å、250×10mm(Phenomenex社、00G-4435-N0)を、固定相として使用した。水(A)/アセトニトリル(B)の3/7混合液を、流量3mL/分で移動相として使用した。
【0117】
この溶液を水で希釈し、最終容積20mLとし、Sep−Pak light C18カートリッジ(Waters社、WAT023501)を通過させた。このカートリッジを、水(10ml)ですすぎ、本化合物をエタノール(0.5mL)により溶離させた。最終生成物を、Gemini 5μ C18 110Å、250×4.6mm(Phenomenex社、00G-4435-E0)HPLCカラム上で、アイソクラティックな水/アセトニトリル(2/8;v/v)混合液を、流量1mL/分で使用するHPLCにより特徴決定した。保持時間(tR):4.3分。RCY(d.c.):38%。RCP:99%。
【0118】
h)N−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−3−(2−フルオロ−エトキシ)−ベンズアミド(HPLC標準)
【化34】
DMF 5mL中のN−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−3−ヒドロキシ−ベンズアミド(3e)50mg(0.11mmol)の溶液に、炭酸カリウム34mg(0.25mmol)及び1−フルオロ−2−ブロモエタン(ABCR社、独国)10μL(0.13mmol)を添加した。この混合物を、60℃で3時間加熱した。溶媒を蒸発させ、残渣を、酢酸エチル/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:30mg(54%)。
元素分析:
理論値:C 68.42、H 6.15、F 3.86、N 8.55
実測値:C 68.17、H 6.28、F 3.64、N 8.71
【0119】
実施例4
a)酢酸3−{2−[4−(4−ニトロフェニル)−ピペラジン−1−イル]−2−オキソ−エチルカルバモイル}−フェニルエステル
【化35】
DMF 100mL中の(3−アセトキシ−ベンゾイルアミノ)−酢酸(3b)3.43g(14.5mmol)及び1−(4−ニトロフェニル)ピペラジン(Aldrich社)2.0g(9.65mmol)の溶液へ、PyBOP(ヘキサフルオロリン酸(ベンゾトリアゾール−1−イルオキシ)トリピロリジノホスホニウム)5.0g(9.65mmol)及びN−エチル−N,N−ジイソプロピルアミン5mLを添加し、かつこの反応混合液を、室温で一晩攪拌した。溶媒を蒸発させた後、残渣を、酢酸エチル/エタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:1.2g(29.2%)。
元素分析:
理論値:C 59.15、H 5.20、N 13.14
実測値:C 59.38、H 5.01、N 13.25
【0120】
b)酢酸3−{2−[4−(4−アミノ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチルカルバモイル}−フェニルエステル
【化36】
メタノール/ジクロロメタン(1:1)50mL中のニトロ化合物4a 853mg(2mmol)の溶液へ、触媒量のチャコール上に担持されたPd(10%)を添加し、この懸濁液を、水素下、室温で一晩攪拌した。触媒を濾過により除去し、溶媒を蒸発させた。
収量:795mg(定量的)。この生成物は、更に精製することなく次工程において使用した。
【0121】
c)酢酸3−{2−[4−(4−ベンゾイルアミノ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチルカルバモイル}−フェニルエステル
【化37】
DMF 20mL中の酢酸3−{2−[4−(4−アミノ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチルカルバモイル}−フェニルエステル(4b) 396mg(1mmol)及び安息香酸134mg(1.1mmol)の溶液へ、PyBOP 590mg(1.14mmol)及びN−エチル−N,N−ジイソプロピルアミン0.35mL(2mmol)を添加し、反応混合物を室温で攪拌した。4時間後、溶媒を蒸発させ、残渣を、ジクロロメタン/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:0.31g(62.1%)。
元素分析:
理論値:C 67.19、H 5.64、N 11.19
実測値:C 66.94、H 5.88、N 11.02
【0122】
d)N−{2−[4−(4−ベンゾイルアミノ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−3−ヒドロキシ−ベンズアミド
【化38】
酢酸塩4cの235mg(0.47mmol)を、エタノール30mL中に溶解し、0℃に冷却した。3N NaOH 1.5mLの添加後、この溶液を1時間攪拌し、そのpHがpH7を下回るまで氷酢酸を添加し、溶媒を蒸発させた。粗生成物をエタノールから結晶化した。
収量:177mg(82%)。
元素分析:
理論値:C 68.11、H 5.72、N 12.22
実測値:C 67.91、H 5.60、N 12.00
【0123】
e)トルエン−4−スルホン酸2−(3−{2−[4−(4−ベンゾイルアミノ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチルカルバモイル}−フェノキシ)−エチルエステル
【化39】
DMF 10mL中のN−{2−[4−(4−ベンゾイルアミノ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−3−ヒドロキシ−ベンズアミド(4d)83mg(0.18mmol)の溶液へ、炭酸カリウム62mg(0.45mmol)及び1,2ビス(トシルオキシ)エタン(Aldrich社)100mg(0.27mmol)を添加した。この混合物を60℃で3時間加熱した。室温で一晩攪拌後、溶媒を蒸発させ、残渣を、酢酸エチル/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:60mg(51%)。
この化合物は、HPLCにより純度>95%を有し、かつF−18標識化の前駆体として適している。
【0124】
f)N−{2−[4−(4−ベンゾイルアミノ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−3−(2−[F−18]フルオロ−エトキシ)−ベンズアミド
【化40】
トシラート前駆体4eを、無水ジメチルホルムアミド(100μL)及びアセトニトリル(400μL)中に溶解し、乾燥カリウム/クリプトフィックス/フッ化物錯体(前記参照)に添加した。この溶液を、110℃で6分間加熱し、その後室温で5分間冷却した。この溶液を、水で希釈し、最終容積10mLとし、かつSep−Pak Plus C18カートリッジ(Waters社、WAT020515)を通過させた。このカートリッジを、水(20ml)ですすぎ、本化合物をアセトニトリル(3mL)により溶離させた。溶離された化合物を水で希釈し、5mLを得、これを分取HPLC精製に供した。Gemini 5μ C18 110Å、250×10mm(Phenomenex社、00G-4435-N0)を、固定相として使用した。水(A)/アセトニトリル(B)の3/7混合液を、流量3mL/分の移動相として、使用した。
【0125】
この溶液を、水で希釈し、最終容積20mLとし、Sep−Pak light C18カートリッジ(Waters社、WAT023501)を通過させた。このカートリッジを、水(10ml)ですすぎ、本化合物をエタノール(0.5mL)により溶離させた。最終生成物を、Gemini 5μ C18 110Å、250×4.6mm(Phenomenex社、00G-4435-E0)HPLCカラム上で、アイソクラティックな水/アセトニトリル(2/8;v/v)混合液を、流量1mL/分で使用するHPLCにより特徴決定した。保持時間(tR):4.1分。RCY (d.c.):32%。RCP:99%。
【0126】
g)N−{2−[4−(4−ベンゾイルアミノ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−3−(2−フルオロ−エトキシ)−ベンズアミド(HPLC標準)
【化41】
DMF 5mL中のN−{2−[4−(4−ベンゾイルアミノ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−3−ヒドロキシ−ベンズアミド(4d)50mg(0.11mmol)の溶液へ、炭酸カリウム34mg(0.25mmol)及び1−フルオロ−2−ブロモエタン(ABCR社、独国)10μL(0.13mmol)を添加した。この混合物を60℃で3時間加熱した。溶媒を蒸発させ、残渣を、酢酸エチル/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:34mg(62%)。
元素分析:
理論値:C 66.65、H 5.79、F 3.77、N 11.10
実測値:C 66.43、H 5.89、F 3.41、N 11.31
【0127】
実施例5
a)トルエン−4−スルホン酸2−{2−[2−(3−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチルカルバモイル}−フェノキシ)−エトキシ]−エトキシ}−エチルエステル
【化42】
DMF 10mL中のN−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−3−ヒドロキシ−ベンズアミド(3e)80mg(0.18mmol)の溶液へ、炭酸カリウム62mg(0.45mmol)及びジ−p−トルエンスルホン酸トリ(エチレングリコール)(Aldrich社)124mg(0.27mmol)を添加した。この混合物を、60℃で3時間加熱した。室温で一晩攪拌した後、溶媒を蒸発させ、残渣を、酢酸エチル/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:54mg(41%)。
この化合物は、HPLCにより純度>95%を有し、かつF−18標識化の前駆体として適している。
【0128】
b)N−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−3−{2−[2−(2−[F−18]フルオロ−エトキシ)−エトキシ]−エトキシ}−ベンズアミド
【化43】
トシラート前駆体5aを、無水ジメチルホルムアミド(100μL)及びアセトニトリル(400μL)中に溶解し、乾燥カリウム/クリプトフィックス/フッ化物錯体(前記参照)に添加した。この溶液を、110℃で6分間加熱し、その後室温で5分間冷却した。この溶液を、水で希釈し、最終容積10mLとし、かつSep−Pak Plus C18カートリッジ(Waters社、WAT020515)を通過させた。このカートリッジを、水(20ml)ですすぎ、本化合物をアセトニトリル(3mL)により溶離させた。溶離された化合物を水で希釈し、5mLを得、これを分取HPLC精製に供した。Gemini 5μ C18 110Å、250×10mm(Phenomenex社、00G-4435-N0)を、固定相として使用した。水(A)/アセトニトリル(B)の3/7混合液を、流量3mL/分の移動相として、使用した。
【0129】
この溶液を、水で希釈し、最終容積20mLとし、Sep−Pak light C18カートリッジ(Waters社、WAT023501)を通過させた。このカートリッジを、水(10ml)ですすぎ、本化合物をエタノール(0.5mL)により溶離させた。最終生成物を、Gemini 5μ C18 110Å、250×4.6mm(Phenomenex社、00G-4435-E0)HPLCカラム上で、アイソクラティックな水/アセトニトリル(2/8;v/v)混合液を、流量1mL/分で使用するHPLCにより特徴決定した。保持時間(tR):4.2分。RCY (d.c.):42%。RCP:99%。
【0130】
c)N−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−3−{2−[2−(2−フルオロ−エトキシ)−エトキシ]−エトキシ}−ベンズアミド(HPLC標準)
【化44】
トシラート前駆体5aの73mg(0.10mmol)を、アセトニトリル2mL中に溶解した。アセトニトリル1mL中のフッ化カリウム6.8mg(0.12mmol)及びクリプトフィックス44.4mgを添加し、100℃で10分間インキュベーションした。この反応を、分析用HPLCによりチェックした。この反応が完了した後、混合物を蒸発させ、かつ残渣を、酢酸エチル/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:28mg(49%)。
元素分析:
理論値:C 66.31、H 6.61、F 3.28、N 7.25
実測値:C 66.07、H 6.73、F 3.11、N 7.38
【0131】
実施例6
a)酢酸4−{4−[4−(4−ニトロフェニル)−ピペラジン−1−イル]−4−オキソ−ブチリル}−フェニルエステル
【化45】
DMF 20mL中の4−(4−アセトキシフェニル)−4−オキソ酪酸(Arch. Pharm.社(ワインハイム、独国) 284;292(1951))1.18g(5mmol)及び1−(4−ニトロフェニル)ピペラジン(Aldrich社)1.04g(5mmol)の溶液に、N−エチル−N,N−ジイソプロピルアミン1.71mL(10mmol)及びヘキサフルオロリン酸2−(1H−ベンゾトリアゾール−1−イル)−テトラメチルウロニウム(HBTU)1.9g(5mmol)を添加した。この反応混合液を、室温で4時間攪拌し、その後溶媒を蒸発させ、残渣を、酢酸エチル/エタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:1.53g(72%)。
元素分析:
理論値:C 62.11、H 5.45、N 9.88
実測値:C 61.89、H 5.54、N 9.61
【0132】
b)酢酸4−{4−[4−(4−アミノ−フェニル)−ピペラジン−1−イル]−4−オキソ−ブチリル}−フェニルエステル
【化46】
メタノール/DMF(3:1)50mL中のニトロ化合物6aの851mg(2mmol)の溶液へ、触媒量のチャコール上に担持されたPd(10%)を添加し、この懸濁液を、水素下、室温で一晩攪拌した。触媒を濾過により除去し、溶媒を蒸発させた。
収量:791mg(定量的)。この生成物は、更に精製することなく次工程において使用した。
【0133】
c)酢酸4−[4−(4−{4−[(ナフタレン−2−カルボニル)−アミノ]−フェニル}−ピペラジン−1−イル)−4−オキソ−ブチリル]−フェニルエステル
【化47】
DMF 20mL中の酢酸 4−{4−[4−(4−アミノ−フェニル)−ピペラジン−1−イル]−4−オキソ−ブチリル}−フェニルエステル(6b)395mg(1mmol)及び2−ナフタレンカルボン酸(Aldrich社)190mg(1.1mmol)の溶液へ、PyBOP 590mg(1.14mmol)及びN−エチル−N,N−ジイソプロピルアミン0.35mL(2mmol)を添加し、反応混合物を室温で攪拌した。4時間後、溶媒を蒸発させ、残渣を、ジクロロメタン/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:374mg(68%)。
元素分析:
理論値:C 72.12、H 5.69、N 7.65
実測値:C 72.30、H 5.61、N 7.42
【0134】
d)ナフタレン−2−カルボン酸(4−{4−[4−(4−ヒドロキシ−フェニル)−4−オキソ−ブチリル]−ピペラジン−1−イル}−フェニル)−アミド
【化48】
酢酸塩6cの242mg(0.44mmol)を、エタノール/DMF(1:1)30mL中に溶解し、0℃に冷却した。2N NaOH 2.2mLの添加後、この溶液を30分間攪拌し、次にそのpHがpH7を下回るまで氷酢酸を添加し、溶媒を蒸発させた。粗生成物を水から結晶化し、真空下40℃で乾燥した。
収量:223mg(定量的)。
元素分析:
理論値:C 73.35、H 5.76、N 8.28
実測値:C 73.10、H 5.91、N 8.25
【0135】
e)トルエン−4−スルホン酸2−{4−[4−(4−{4−[(ナフタレン−2−カルボニル)−アミノ]−フェニル}−ピペラジン−1−イル)−4−オキソ−ブチリル]−フェノキシ}−エチルエステル
【化49】
DMF 10mL中のナフタレン−2−カルボン酸(4−{4−[4−(4−ヒドロキシ−フェニル)−4−オキソ−ブチリル]−ピペラジン−1−イル}−フェニル)−アミド(6d)91mg(0.18mmol)の溶液へ、炭酸カリウム62mg(0.45mmol)及び1,2ビス(トシルオキシ)エタン(Aldrich社)100mg(0.27mmol)を添加した。この混合物を60℃で3時間加熱した。室温で一晩攪拌した後、溶媒を蒸発させ、残渣を、酢酸エチル/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:57mg(46%)。
この化合物は、HPLCにより純度>95%を有し、かつF−18標識化の前駆体として適している。
【0136】
f)ナフタレン−2−カルボン酸[4−(4−{4−[4−(2−[F−18]フルオロ−エトキシ)−フェニル]−4−オキソ−ブチリル}−ピペラジン−1−イル)−フェニル]−アミド
【化50】
トシラート前駆体6eを、無水ジメチルホルムアミド(100μL)及びアセトニトリル(400μL)中に溶解し、乾燥カリウム/クリプトフィックス/フッ化物錯体(前記参照)に添加した。この溶液を、110℃で6分間加熱し、その後室温で5分間冷却した。この溶液を、水で希釈し、最終容積10mLとし、かつSep−Pak Plus C18カートリッジ(Waters社、WAT020515)を通過させた。このカートリッジを、水(20ml)ですすぎ、本化合物をアセトニトリル(3mL)により溶離させた。溶離された化合物を水で希釈し、5mLを得、これを分取HPLC精製に供した。Gemini 5μ C18 110Å、250×10mm(Phenomenex社、00G-4435-N0)を、固定相として使用した。水(A)/アセトニトリル(B)の3/7混合液を、流量3mL/分の移動相として、使用した。
【0137】
この溶液を、水で希釈し、最終容積20mLとし、Sep−Pak light C18カートリッジ(Waters社、WAT023501)を通過させた。このカートリッジを、水(10ml)ですすぎ、本化合物をエタノール(0.5mL)により溶離させた。最終生成物を、Gemini 5μ C18 110Å、250×4.6mm(Phenomenex社、00G-4435-E0)HPLCカラム上で、アイソクラティックな水/アセトニトリル(2/8;v/v)混合液を、流量1mL/分で使用するHPLCにより特徴決定した。保持時間(tR):5.6分。RCY (d.c.):35%。RCP:98%。
【0138】
実施例7
a)酢酸4−[4−(4−{4−[(ナフタレン−1−カルボニル)−アミノ]−フェニル}−ピペラジン−1−イル)−4−オキソ−ブチリル]−フェニルエステル
【化51】
DMF 20mL中の酢酸4−{4−[4−(4−アミノ−フェニル)−ピペラジン−1−イル]−4−オキソ−ブチリル}−フェニルエステル(6b)395mg(1mmol)及び1−ナフタレンカルボン酸(Aldrich社)190mg(1.1mmol)の溶液に、PyBOP 590mg(1.14mmol)及びN−エチル−N,N−ジイソプロピルアミン0.35mL(2mmol)を添加し、かつこの反応混合液を、室温で攪拌した。4時間後、溶媒を蒸発させ、残渣を、ジクロロメタン/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:336mg(61%)。
元素分析:
理論値:C 72.12、H 5.69、N 7.65
実測値:C 71.94、H 5.77、N 7.59
【0139】
b)ナフタレン−1−カルボン酸(4−{4−[4−(4−ヒドロキシ−フェニル)−4−オキソ−ブチリル]−ピペラジン−1−イル}−フェニル)−アミド
【化52】
酢酸塩7aの242mg(0.44mmol)を、エタノール/DMF(1:1)30mLに溶解し、0℃に冷却した。2N NaOH 2.2mLを添加した後、この溶液を30分間攪拌し、次にそのpHがpH7を下回るまで、氷酢酸を添加し、溶媒を蒸発させた。粗生成物を水から結晶化し、真空下40℃で乾燥した。
収量:223mg(定量的)。
元素分析:
理論値:C 73.35、H 5.76、N 8.28
実測値:C 73.21、H 5.62、N 8.11
【0140】
c)トルエン−4−スルホン酸2−{4−[4−(4−{4−[(ナフタレン−1−カルボニル)−アミノ]−フェニル}−ピペラジン−1−イル)−4−オキソ−ブチリル]−フェノキシ}−エチルエステル
【化53】
DMF 10mL中のナフタレン−1−カルボン酸(4−{4−[4−(4−ヒドロキシ−フェニル)−4−オキソ−ブチリル]−ピペラジン−1−イル}−フェニル)−アミド(7b) 91mg(0.18mmol)の溶液へ、炭酸カリウム62mg(0.45mmol)及び1,2ビス(トシルオキシ)エタン(Aldrich社)100mg(0.27mmol)を添加した。この混合物を60℃で3時間加熱した。室温で一晩攪拌した後、溶媒を蒸発させ、残渣を、酢酸エチル/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:63mg(51%)。
この化合物は、HPLCにより純度>95%を有し、かつF−18標識化の前駆体として適している。
【0141】
d)ナフタレン−1−カルボン酸[4−(4−{4−[4−(2−[F−18]フルオロ−エトキシ)−フェニル]−4−オキソ−ブチリル}−ピペラジン−1−イル)−フェニル]−アミド
【化54】
トシラート前駆体7cを、無水ジメチルホルムアミド(100μL)及びアセトニトリル(400μL)中に溶解し、乾燥カリウム/クリプトフィックス/フッ化物錯体(前記参照)に添加した。この溶液を、110℃で6分間加熱し、その後室温で5分間冷却した。この溶液を、水で希釈し、最終容積10mLとし、かつSep−Pak Plus C18カートリッジ(Waters社、WAT020515)を通過させた。このカートリッジを、水(20ml)ですすぎ、本化合物をアセトニトリル(3mL)により溶離させた。溶離された化合物を水で希釈し、5mLを得、これを分取HPLC精製に供した。Gemini 5μ C18 110Å、250×10mm(Phenomenex社、00G-4435-N0)を、固定相として使用した。水(A)/アセトニトリル(B)の3/7混合液を、流量3mL/分の移動相として、使用した。
【0142】
この溶液を、水で希釈し、最終容積20mLとし、Sep−Pak light C18カートリッジ(Waters社、WAT023501)を通過させた。このカートリッジを、水(10ml)ですすぎ、本化合物をエタノール(0.5mL)により溶離させた。最終生成物を、Gemini 5μ C18 110Å、250×4.6mm(Phenomenex社、00G-4435-E0)HPLCカラム上で、アイソクラティックな水/アセトニトリル(2/8;v/v)混合液を、流量1mL/分で使用するHPLCにより特徴決定した。保持時間(tR):6.0分。RCY (d.c.):32%。RCP:97%。
【0143】
実施例8
a)酢酸4−{4−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−4−オキソブチリル}−フェニルエステル
【化55】
THF 70mL及びトリエチルアミン0.40mL(2.87mmol)中の4−(4−アセトキシフェニル)−4−オキソ酪酸(Arch. Pharm.社(ワインハイム、独国) 284; 292 (1951))652mg(2.76mmol)の−15℃の溶液へ、クロロギ酸イソブチル0.396mL(3.03mmol)を滴下し、この溶液を更に15分間この温度で維持した。次にこの冷溶液へ、THF 30mL及びジクロロメタン30mL中の1−(4−ベンジルオキシ−フェニル)−ピペラジン3c 740mg及びトリエチルアミン1.7mL(12.25mmol)を添加し、その温度を10℃未満に更に15分間維持し、その後室温とした。一晩攪拌した後、溶媒を蒸発させ、かつ残渣を、ヘキサン/酢酸エチル勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:219mg(45%)。
元素分析:
理論値:C 71.59、H 6.21、N 5.76
実測値:C 71.33、H 6.46、N 5.60
【0144】
b)1−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−4−(4−ヒドロキシフェニル)−ブタン−1,4−ジオン
【化56】
酢酸塩8aの229mg(0.47mmol)を、エタノール30mLに溶解し、0℃に冷却した。3N NaOH 1.5mLの添加後、この溶液を1時間攪拌し、そのpHがpH7を下回るまで、氷酢酸を添加し、溶媒を蒸発させた。粗生成物をエタノールから結晶化した。
収量:186mg(89%)。
元素分析:
理論値:C 72.95、H 6.35、N 6.30
実測値:C 72.78、H 6.27、N 6.41
【0145】
c)トルエン−4−スルホン酸2−(4−{4−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−4−オキソブチリル}−フェノキシ)−エチルエステル
【化57】
DMF 10mL中の1−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−4−(4−ヒドロキシフェニル)−ブタン−1,4−ジオン(8b)80mg(0.18mmol)の溶液へ、炭酸カリウム62mg(0.45mmol)及び1,2ビス(トシルオキシ)エタン(Aldrich社)100mg(0.27mmol)を添加した。この混合物を60℃で3時間加熱した。室温で一晩攪拌した後、溶媒を蒸発させ、残渣を、酢酸エチル/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:60mg(52%)。
この化合物は、HPLCにより純度>95%を有し、かつF−18標識化の前駆体として適している。
【0146】
d)1−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−4−[4−(2−[F−18]フルオロ−エトキシ)−フェニル]−ブタン−1,4−ジオン
【化58】
トシラート前駆体8cを、無水ジメチルホルムアミド(100μL)及びアセトニトリル(400μL)中に溶解し、乾燥カリウム/クリプトフィックス/フッ化物錯体(前記参照)に添加した。この溶液を、110℃で6分間加熱し、その後室温で5分間冷却した。この溶液を、水で希釈し、最終容積10mLとし、かつSep−Pak Plus C18カートリッジ(Waters社、WAT020515)を通過させた。このカートリッジを、水(20ml)ですすぎ、本化合物をアセトニトリル(3mL)により溶離させた。溶離された化合物を水で希釈し、5mLを得、これを分取HPLC精製に供した。Gemini 5μ C18 110Å、250×10mm(Phenomenex社、00G-4435-N0)を、固定相として使用した。水(A)/アセトニトリル(B)の3/7混合液を、流量3mL/分の移動相として、使用した。
【0147】
この溶液を、水で希釈し、最終容積20mLとし、Sep−Pak light C18カートリッジ(Waters社、WAT023501)を通過させた。このカートリッジを、水(10ml)ですすぎ、本化合物をエタノール(0.5mL)により溶離させた。最終生成物を、Gemini 5μ C18 110Å、250×4.6mm(Phenomenex社、00G-4435-E0)HPLCカラム上で、アイソクラティックな水/アセトニトリル(2/8;v/v)混合液を、流量1mL/分で使用するHPLCにより特徴決定した。保持時間(tR):4.7分。RCY (d.c.):45%。RCP:99%。
【0148】
実施例9
a)酢酸3−[2−(4−{4−[(ナフタレン−2−カルボニル)−アミノ]−フェニル}−ピペラジン−1−イル)−2−オキソ−エチルカルバモイル]−フェニルエステル
【化59】
DMF 20mL中の酢酸3−{2−[4−(4−アミノ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチルカルバモイル}−フェニルエステル(4b)396mg(1mmol)及び2−ナフタレンカルボン酸(Aldrich社)190mg(1.1mmol)の溶液へ、PyBOP 590mg(1.14mmol)及びN−エチル−N,N−ジイソプロピルアミン0.35mL(2mmol)を添加し、かつこの反応混合液を、室温で攪拌した。4時間後、溶媒を蒸発させ、残渣を、ジクロロメタン/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:0.25g(43.9%)。
元素分析:
理論値:C 69.80、H 5.49、N 10.18
実測値:C 69.55、H 5.61、N 10.03
【0149】
b)ナフタレン−2−カルボン酸(4−{4−[2−(3−ヒドロキシ−ベンゾイルアミノ)−アセチル]−ピペラジン−1−イル}−フェニル)−アミド
【化60】
酢酸塩9aの240mg(0.44mmol)を、エタノール/DMF(1:1)30mL中に溶解し、0℃に冷却した。2N NaOH 2.2mLの添加後、この溶液を30分間攪拌し、次にそのpHがpH7を下回るまで、氷酢酸を添加し、溶媒を蒸発させた。粗生成物を水から結晶化し、真空中で40℃で乾燥した。
収量:220mg(定量的)。
元素分析:
理論値:C 70.85、H 5.55、N 11.02
実測値:C 70.53、H 5.77、N 10.85
【0150】
c)トルエン−4−スルホン酸2−{3−[2−(4−{4−[(ナフタレン−2−カルボニル)−アミノ]−フェニル}−ピペラジン−1−イル)−2−オキソ−エチルカルバモイル]−フェノキシ}−エチルエステル
【化61】
DMF 10mL中のナフタレン−2−カルボン酸(4−{4−[2−(3−ヒドロキシ−ベンゾイルアミノ)−アセチル]−ピペラジン−1−イル}−フェニル)−アミド(9b)92mg(0.18mmol)の溶液へ、炭酸カリウム62mg(0.45mmol)及び1,2ビス(トシルオキシ)エタン(Aldrich社)100mg(0.27mmol)を添加した。この混合物を60℃で3時間加熱した。室温で一晩攪拌した後、溶媒を蒸発させ、残渣を、酢酸エチル/ヘキサン勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:58mg(46%)。
この化合物は、HPLCにより純度>95%を有し、かつF−18標識化の前駆体として適している。
【0151】
d)ナフタレン−2−カルボン酸[4−(4−{2−[3−(2−[F−18]フルオロ−エトキシ)−ベンゾイルアミノ]−アセチル}−ピペラジン−1−イル)−フェニル]−アミド
【化62】
トシラート前駆体9cを、無水ジメチルホルムアミド(100μL)及びアセトニトリル(400μL)中に溶解し、乾燥カリウム/クリプトフィックス/フッ化物錯体(前記参照)に添加した。この溶液を、110℃で6分間加熱し、その後室温で5分間冷却した。この溶液を、水で希釈し、最終容積10mLとし、かつSep−Pak Plus C18カートリッジ(Waters社、WAT020515)を通過させた。このカートリッジを、水(20ml)ですすぎ、本化合物をアセトニトリル(3mL)により溶離させた。溶離された化合物を水で希釈し、5mLを得、これを分取HPLC精製に供した。Gemini 5μ C18 110Å、250×10mm(Phenomenex社、00G-4435-N0)を、固定相として使用した。水(A)/アセトニトリル(B)の3/7混合液を、流量3mL/分の移動相として、使用した。
【0152】
この溶液を、水で希釈し、最終容積20mLとし、Sep−Pak light C18カートリッジ(Waters社、WAT023501)を通過させた。このカートリッジを、水(10ml)ですすぎ、本化合物をエタノール(0.5mL)により溶離させた。最終生成物を、Gemini 5μ C18 110Å、250×4.6mm(Phenomenex社、00G-4435-E0)HPLCカラム上で、アイソクラティックな水/アセトニトリル(2/8;v/v)混合液を、流量1mL/分で使用するHPLCにより特徴決定した。保持時間(tR):4.6分。RCY (d.c.):39%。RCP:99%。
【0153】
e)ナフタレン−2−カルボン酸[4−(4−{2−[3−(2−フルオロ−エトキシ)−ベンゾイルアミノ]−アセチル}−ピペラジン−1−イル)−フェニル]−アミド(HPLC−標準)
【化63】
DMF 5mL中のナフタレン−2−カルボン酸(4−{4−[2−(3−ヒドロキシ−ベンゾイルアミノ)−アセチル]−ピペラジン−1−イル}−フェニル)−アミド(9b)56mg(0.11mmol)の溶液へ、炭酸カリウム34mg(0.25mmol)及び1−フルオロ−2−ブロモエタン(ABCR社、独国)10μL(0.13mmol)を添加した。この混合物を60℃で3時間加熱した。溶媒を蒸発させ、残渣を、ジメチルスルホキシドに溶かし、水/アセトニトリル勾配を使用し、RP−18上で2回クロマトグラフィーにかけた。
収量:24mg(40%)。
元素分析:
理論値:C 69.30、H 5.63、F 3.43、N 10.10
実測値:C 69.15、H 5.81、F 3.22、N 10.28
【0154】
実施例10
a){2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−カルバミン酸tert−ブチルエステル
【化64】
THF 800mL及びトリエチルアミン8mL(57.71mmol)中のt−ブトキシカルボニル−グリシン(Aldrich社)7.7g(43.97mmol)の−15℃の溶液に、クロロギ酸イソブチル5.75mL(43.97mmol)を滴下し、この溶液を、この温度で更に15分間維持した。次にこの冷溶液へ、THF/ジクロロメタン(1:1)300mL中の1−(4−ベンジルオキシフェニル)−ピペラジン(3c)11.8g及びトリエチルアミン29mL(210mmol)をゆっくり添加し、その温度を、10℃未満に更に15分間維持し、次に室温とした。一晩攪拌した後、溶媒を蒸発させ、かつ残渣を酢酸エチルに溶かした。この溶液を、水性炭酸ナトリウム、水、1M HCl水溶液、飽和塩化ナトリウム水溶液により連続して洗浄し、最後に硫酸マグネシウム上で乾燥し、その後蒸発させた。この残渣を、ヘキサン/酢酸エチル勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:15.51g(82.9%)。
元素分析:
理論値:C 67.74、H 7.34、N 9.87
実測値:C 67.33、H 7.47、N 9.62
【0155】
b)2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル−アンモニウムクロリド
【化65】
{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−カルバミン酸tert−ブチルエステル(10a)17.0g(39.95mmol)を、ジエチルエーテル中の2N HCl 300mL中で懸濁し、かつ室温で一晩攪拌した。沈殿物を濾過し、エーテルで洗浄し、真空中40℃で乾燥した。
収量:14.5g(定量的)。この生成物は、更に精製することなく次工程において使用した。
【0156】
c)N−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−6−ブロモ−ニコチンアミド
【化66】
DMF 20mL中の2−ブロモ−5−ピリジンカルボン酸(Aldrich社)202mg(1mmol)及び2−[4−(4−ベンジルオキシフェニル)−ピペラジン−1−イル]−2−オキソエチル−アンモニウムクロリド(10b)398mg(1.1mmol)の溶液へ、PyBOP 624mg(1.2mmol)及びN−エチル−N,N−ジイソプロピルアミン0.61mLを添加し、この反応混合液を、室温で一晩攪拌した。溶媒を蒸発させた後、残渣を、酢酸エチル/エタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:183mg(35.9%)。
元素分析:
理論値:C 58.95、H 4.95、Br 15.69、N 11.00
実測値:C 58.66、H 5.18、Br 15.20、N 11.36
この化合物は、HPLCにより純度>95%を有し、かつF−18標識化の前駆体として適している。
【0157】
d)N−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−6−フルオロ−ニコチンアミド
【化67】
DMF 20mL中の2−フルオロ−5−ピリジンカルボン酸(Aldrich社)141mg(1mmol)及び2−[4−(4−ベンジルオキシフェニル)−ピペラジン−1−イル]−2−オキソエチル−アンモニウムクロリド(10b)398mg(1.1mmol)の溶液へ、PyBOP 624mg(1.2mmol)及びN−エチル−N,N−ジイソプロピルアミン0.61mLを添加し、この反応混合液を、室温で一晩攪拌した。溶媒を蒸発させた後、残渣を、酢酸エチル/エタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:302mg(67.3%)。
元素分析:
理論値:C 66.95、H 5.62、F 4.24、N 12.49
実測値:C 66.84、H 5.97、F 3.95、N 12.52
【0158】
e)N−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−6−[F−18]−フルオロ−ニコチンアミド
【化68】
水性[18F]フッ化物(5.1GBq)を、QMAカートリッジ(Waters社、Sep Pak Light QMA、部品番号:WAT023525)上に捕獲し、Wheatonバイアル(5ml)中へ、MeCN 0.95ml中K2.2.2 5mg+水50μl中K2CO3 1mgにより溶離させた。この溶媒を、120℃で10分間、窒素流れ下で加熱することにより、除去した。無水MeCN(1ml)を添加し、前述のように蒸発させた。無水DMSO 700μl中の前駆体10c(5mg)の溶液を添加した。180℃で30分間加熱した後、粗反応混合物を総容積まで水で希釈し、分取HPLCにより精製した:ACE 5−C18−HL 250mm×10mm、Advanced Chromatography Technologies社;カタログ番号:ACE 321-2510;アイソクラチック、0.1%トリフルオロ酢酸中35%アセトニトリル、流量:4ml/分;tR=18分。収集されたHPLC画分を、水40mlで希釈し、Sep−Pak light C18カートリッジ(Waters社、WAT023501)上に固定し、これを水5mlで洗浄し、エタノール1mlで溶離し、生成物1015MBqを送り出した(36%、減衰補正;放射化学的純度>99%)。所望の生成物を、分析用HPLC:Agilent社 ZORBAX 300SB-C18 250×4.6mm;5μm Agilent社;PN 880995-902上での、非−放射性F−19フルオロ標準との同時注入により、特徴付けた;A):水+0.1%TFA、B):MeCN+0.1%TFA、0分〜10分、35%B;10分〜10分30秒、35%Bから100%B;1mL/分(tR=7.6分)、RCP:99%(HPLC)。
【0159】
実施例11
a)N−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−2−ブロモ−イソニコチンアミド
【化69】
DMF 20mL中の2−ブロモ−4−ピリジンカルボン酸(Alfa社)202mg(1mmol)及び2−[4−(4−ベンジルオキシフェニル)−ピペラジン−1−イル]−2−オキソエチル−アンモニウムクロリド(10b)398mg(1.1mmol)の溶液へ、PyBOP 624mg(1.2mmol)及びN−エチル−N,N−ジイソプロピルアミン0.61mLを添加し、この反応混合物を、室温で一晩攪拌した。溶媒を蒸発させた後、残渣を、酢酸エチル/エタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。生成物含有画分を収集し、酢酸エチルから再結晶化した。
収量:165mg(32.4%)。
元素分析:
理論値:C 58.95、H 4.95、Br 15.69、N 11.00
実測値:C 58.32、H 5.09、Br 15.11、N 10.73
この化合物は、HPLCにより純度>95%を有し、かつF−18標識化の前駆体として適している。
【0160】
b)N−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−2−フルオロ−イソニコチンアミド
【化70】
DMF 20mL中の2−フルオロ−4−ピリジンカルボン酸(Aldrich社)141mg(1mmol)及び2−[4−(4−ベンジルオキシフェニル)−ピペラジン−1−イル]−2−オキソエチル−アンモニウムクロリド(10b)398mg(1.1mmol)の溶液へ、PyBOP 624mg(1.2mmol)及びN−エチル−N,N−ジイソプロピルアミン0.61mLを添加し、この反応混合物を、室温で一晩攪拌した。溶媒を蒸発させた後、残渣を、酢酸エチル/エタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:326mg(72.7%)。
元素分析:
理論値:C 66.95、H 5.62、F 4.24、N 12.49
実測値:C 66.58、H 5.81、F 4.03、N 12.68
【0161】
c)N−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−2−[F−18]−フルオロ−イソニコチンアミド
【化71】
水性[18F]フッ化物(4.9GBq)を、QMAカートリッジ(Waters社、Sep Pak Light QMA、部品番号:WAT023525)上に捕獲し、Wheatonバイアル(5ml)中へ、MeCN 0.95ml中K2.2.2 5mg+水50μl中K2CO3 1mgにより溶離させた。この溶媒を、120℃で10分間、窒素流れ下で加熱することにより、除去した。無水MeCN(1ml)を添加し、前述のように蒸発させた。無水DMSO 700μl中の前駆体11a(5mg)の溶液を添加した。180℃で30分間加熱した後、粗反応混合物を総容積5mLまで水で希釈し、分取HPLCにより精製した:ACE 5−C18−HL 250mm×10mm、Advanced Chromatography Technologies社;カタログ番号:ACE 321-2510;アイソクラチック、0.1%トリフルオロ酢酸中35%アセトニトリル、流量:4ml/分;tR=18分。収集されたHPLC画分を、水40mlで希釈し、Sep−Pak light C18カートリッジ(Waters社、WAT023501)上に固定し、これを水5mlで洗浄し、エタノール1mlで溶離し、生成物1293MBqを送り出し(44%、減衰補正)、これは分析用HPLC:Agilent社 ZORBAX 300SB-C18 50×4.6mm;5μm Agilent社;PN 880995-902を使用する、非−放射性F−19フルオロ標準との同時注入により、特徴付け、かつ再確認した;A):水+0.1%TFA、B):MeCN+0.1%TFA、0分〜10分、35%B;10分〜10分30秒、35%Bから100%B;1mL/分(tR=7.5分)、RCP:>99%(HPLC)。
【0162】
実施例12
a)N−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−2−ブロモ−ニコチンアミド
【化72】
DMF 20mL中の2−ブロモ−ニコチン酸(Aldrich社)202mg(1mmol)及び2−[4−(4−ベンジルオキシフェニル)−ピペラジン−1−イル]−2−オキソエチル−アンモニウムクロリド(10b)398mg(1.1mmol)の溶液へ、PyBOP 624mg(1.2mmol)及びN−エチル−N,N−ジイソプロピルアミン0.61mLを添加し、この反応混合物を、室温で一晩攪拌した。溶媒を蒸発させた後、残渣を、酢酸エチル/エタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:210mg(41.2%)。
元素分析:
理論値:C 58.95、H 4.95、Br 15.69、N 11.00
実測値:C 59.11、H 4.75、Br 15.39、N 11.03
この化合物は、HPLCにより純度>95%を有し、かつF−18標識化の前駆体として適している。
【0163】
b)N−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−2−フルオロ−ニコチンアミド
【化73】
DMF 20mL中の2−フルオロ−ニコチン酸(Aldrich社)141mg(1mmol)及び2−[4−(4−ベンジルオキシフェニル)−ピペラジン−1−イル]−2−オキソエチル−アンモニウムクロリド(10b)398mg(1.1mmol)の溶液へ、PyBOP 624mg(1.2mmol)及びN−エチル−N,N−ジイソプロピルアミン0.61mLを添加し、この反応混合物を、室温で一晩攪拌した。溶媒を蒸発させた後、残渣を、酢酸エチル/エタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:326mg(72.7%)。
元素分析:
理論値:C 66.95、H 5.62、F 4.24、N 12.49
実測値:C 66.71、H 5.79、F 3.88、N 12.30
【0164】
c)N−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−2−[F−18]−フルオロ−ニコチンアミド
【化74】
水性[18F]フッ化物(7GBq)を、QMAカートリッジ(Waters社、Sep Pak Light QMA、部品番号:WAT023525)上に捕獲し、Wheatonバイアル(5ml)中へ、MeCN 0.95ml中K2.2.2 5mg+水50μl中K2CO3 1mgにより溶離させた。その溶媒を、120℃で10分間、窒素流れ下で加熱することにより、除去した。無水MeCN(1ml)を添加し、前述のように蒸発させた。無水DMSO 700μl中の前駆体12a(5mg)の溶液を添加した。180℃で30分間加熱した後、粗反応混合物を、総容積5mLまで水で希釈し、分取HPLCにより精製した:ACE 5−C18−HL 250mm×10mm、Advanced Chromatography Technologies社;カタログ番号:ACE 321-2510;アイソクラチック、0.1%トリフルオロ酢酸中35%アセトニトリル、流量:4ml/分;tR=19分。収集されたHPLC画分を、水40mlで希釈し、Sep−Pak light C18カートリッジ(Waters社、WAT023501)上に固定し、これを水5mlで洗浄し、エタノール1mlで溶離し、生成物2015MBqを送り出し(49%、減衰補正)、これは分析用HPLC:Agilent ZORBAX 300SB-C18 50×4.6mm;5μm Agilent;PN 880995-902を使用する、非−放射性F−19フルオロ標準との同時注入により、特徴付け、かつ再確認した;A):水+0.1%TFA、B):MeCN+0.1%TFA、0分〜10分、35%B;10分〜10分30秒、35%Bから100%B;1mL/分(tR=7.8分)、RCP:>99%(HPLC)。
【0165】
実施例13:生物学的データ
a)方法
ヒト脳ホモジネートを使用する結合試験
トリチウム標識されたアミロイドリガンドとの競合アッセイを、96−ウェルプレート(Greiner bio-one社;カタログ番号651201;ロット番号06260130)において、AD患者由来の脳ホモジネートを使用し行った。ホモジネートは、AD患者の灰白質及び白質を含む解離した前頭皮質を、リン酸緩衝食塩水(PBS、pH7.4)中でホモジナイズすることにより(Ultra-Turrax、設定2、30秒間、24000rpm)調製した。このホモジネートは、濃度100mg湿組織/mlとし、300μlアリコートに分割し、−80℃で貯蔵した。
【0166】
様々な濃度の標識されない被験物質を、PBS、0.1%BSA中で、100μg/mlホモジネート及び10nMトリチウム標識されたリガンドと共に(最終容積200μl)、室温で3時間インキュベーションした。引き続き、この結合混合物を、Filtermate 196収集器(Packard社)を使用し、Whatman GF/Bフィルター(PBS、0.1%BSAで湿らせたもの)を通して濾過した。その後フィルターを、PBS、0.1%BSAで2回洗浄し、かつシンチレーター40μlを各ウェルに添加し、その後結合した放射活性を、TopCount装置(Perkin Elmer社)を用いて測定した。非特異的結合は、この反応混合物へ、トリチウム標識されたリガンドの1000倍のアクセス(access of 1000x)を添加することにより、評価した。最後にIC50値を、好適な解析ソフトウェアの助けを借りて算出した。
【0167】
オートラジオグラフィー解析
アルツハイマー型認知症患者、前頭側頭型認知症患者及び年齢が合致した対照からの前頭葉の新鮮凍結切片に加え、パラフィン包埋切片を、本試験に使用した。
クライオスタット(Leica社、独国)上で厚さ18μmにスライスした凍結切片、及び滑走式ミクロトーム(Leica社)上で厚さ6μmにスライスしたパラフィン切片を、スライドガラス(Superfrost Plus, Fa.Menzel社, ブラウンシュバイク、独国)上に搭載した。凍結切片は、−20℃で数晩にわたりスライドに接着させた。パラフィン切片は、慣習的組織学的方法を用いて、脱パラフィンした。結合試験のために、切片を、25mM Hepes緩衝液、pH7.4、0.1%(BSA)中に希釈された、10Bq/μlのF−18標識された被験化合物(200〜300μl/スライド)と一緒に、室温で1.5時間、加湿チャンバー内でインキュベーションした。ブロック試験のために、このインキュベーション混合物に、非標識の被験物質の1000倍アクセスを添加した。ハイブリダイゼーション後、切片を、Hepes緩衝液、0.1%BSAにより4回(又は代わりに40%エタノールで2回)洗浄し、最後に蒸留水に10秒間2回浸した。風乾した切片を、造影プレートに曝し、シグナルを放射線画像解析装置(Fuji BAS5000)により検出した。
【0168】
生体内分布
生体内分布試験及び排泄試験は、雄のNMRIマウス(体重約30g;1時点につき動物3匹)で行った。これらの動物は、通常の実験室条件下、温度22±2℃及び12時間の暗/明リズムで飼育した。飼料及び水は、自由に摂取させた。試験開始前少なくとも3日間の馴化期間の間に、動物は、臨床的に試験し、異常な臨床徴候が存在しないことを確認した。本被験化合物の100μl中約150kBqの尾静脈からの静脈内注射の2、5、30、60、240分後に、尿及び糞便を定量的に収集した。同時点で、動物を、イソフルラン麻酔下で断頭により屠殺し、以下の臓器及び組織を、γ線カウンターを使用する放射活性測定のために摘出した:脾臓、肝臓、腎臓、肺、大腿、心臓、脳、脂肪、甲状腺、筋肉、皮膚、血液、尾部、胃(内容物なし)、精巣、腸(内容物なし)、膵臓、副腎、及び生体残存部。解析のために、組織重量当たりの注射された投与量の減衰補正率(%ID/g±標準偏差)を算出した。
【0169】
b)結果
【表1】
【0170】
オートラジオグラフィー
図1は、アルツハイマー病患者(AD)及びAβプラークを伴わない対照(HC/FTD)(健常対照/前頭側頭型認知症)の皮質由来の凍結切片への3gの結合のオートラジオグラフィー分析を示す。AD試料の斑豊富領域における特異的結合を矢印で示す。
【0171】
【表2】
【0172】
オートラジオグラフィー
図2は、アルツハイマー病患者(AD)及びAβプラークを伴わない対照(HC/FTD)(健常対照/前頭側頭型認知症)の皮質由来の凍結切片への実施例10eの結合のオートラジオグラフィー分析を示す。AD試料の斑豊富領域における特異的結合を矢印で示す。
【0173】
【表3】
【0174】
オートラジオグラフィー
図3は、アルツハイマー病患者(AD)及びAβプラークを伴わない対照(HC/FTD)(健常対照/前頭側頭型認知症)の皮質由来の凍結切片への実施例11cの結合のオートラジオグラフィー分析を示す。AD試料の斑豊富領域における特異的結合を矢印で示す。
【0175】
【表4】
【0176】
オートラジオグラフィー
図4は、アルツハイマー病患者(AD)及びAβプラークを伴わない対照(HC/FTD)(健常対照/前頭側頭型認知症)の皮質由来の凍結切片への実施例12cの結合のオートラジオグラフィー分析を示す。AD試料の斑豊富領域における特異的結合を矢印で示す。
【0177】
選択された化合物のIC50値
図5は、AD患者由来の脳ホモジネートを使用する競合アッセイにおいて測定された、選択された化合物の[nM]でのIC50値を示す。
【0178】
実施例14
a)5−ベンジルオキシ−2−ブロモ−ピリジン
【化75】
DMF 400mL中の2−ブロモ−5−ヒドロキシピリジン10.0g(57.47mmol)の溶液へ、臭化ベンジル14.75g(86.21mmol)及び炭酸カリウム23.82g(172.4mmol)を添加した。この混合物を、60℃で6時間及び室温で一晩攪拌した。この懸濁液を濾過し、溶媒を蒸発させた後、残渣を、ジクロロメタン/メタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:14.82g(96.7%)。
MS(ESIpos):m/z=264, 266 [M+H]+
1H-NMR (300MHz, CHCl3-d): δ [ppm]=5.10 (s, 2H), 7.16 (dd, 1H), 7.32-7.47 (m, 6H), 8.14 (d, 1H)。
【0179】
b)1−(5−ベンジルオキシ−ピリジン−2−イル)−ピペラジン
【化76】
ガラス製品は全て、100℃で乾燥した。トルエン180mL中のピペラジン5.27g(61.22mmol)の溶液へ、トリス(ジベンジリデンアセトン)ジパラジウム(0) 561mg(0.61mmol)及びBINAP(2,2’−ビス(ジフェニルホスフィノ)−1,1’−ビナフチル)520mg(0.83mmol)を添加した。その後THF中の5−ベンジルオキシ−2−ブロモ−ピリジン(実施例14a)14.7g(55.66mmol)の溶液、引き続きTHF中のナトリウムt−ブチラート8.02g(83.48mmol)の懸濁液を添加した。この反応混合物を、6時間還流し、室温で一晩攪拌した。溶媒を蒸発させた後、残渣を、ジクロロメタン/メタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:7.12g(47.0%)。
MS (ESIpos):m/z=270 [M+H]+
1H-NMR (300MHz, CHCl3-d): δ [ppm]=2.97-3.07 (m, 4H), 3.36-3.46 (m, 4H), 5.04 (s, 2H), 6.63 (d, 1H), 7.21 (dd, 1H), 7.29-7.48 (m, 5H), 8.00 (d, 1H)。
【0180】
c)(2−{4−[5−(ベンジルオキシ)ピリジン−2−イル]ピペラジン−1−イル}−2−オキソエチル)カルバミン酸tert−ブチル
【化77】
THF 500mL及びトリエチルアミン(35.87mmol)5mL中のt−ブトキシカルボニル−グリシン(Aldrich社)4.63g(26.43mmol)の−15℃の溶液へ、クロロギ酸イソブチル3.43mL(26.43mmol)を滴下し、この溶液を、この温度で更に15分間維持した。その後、この冷溶液へ、THF/ジクロロメタン(1:1)200mL中の1−(5−ベンジルオキシ−ピリジン−2−イル)−ピペラジン(14b)7.12g及びトリエチルアミン18mL(129mmol)をゆっくり添加し、その温度を−10℃以下に更に15分間維持し、その後室温とした。一晩攪拌した後、溶媒を蒸発させ、残渣を酢酸エチルに溶かした。この溶液を、水性炭酸ナトリウム、水、1M HCl水溶液、飽和塩化ナトリウム水溶液で連続して洗浄し、最後に硫酸マグネシウム上で乾燥し、その後蒸発させた。この残渣を、ヘキサン/酢酸エチル勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:8.04g(70.6%)。
MS (ESIpos):m/z=427 [M+H]+
1H-NMR (300MHz, CHCl3-d): δ [ppm]=1.46 (s, 9H), 3.36-3.45 (m, 2H), 3.51 (br. s., 4H), 3.70-3.81 (m, 2H), 4.02 (d, 2H), 5.05 (s, 2H), 5.53 (br. s., 1H), 6.65 (d, 1H), 7.23 (dd, 1H), 7.30-7.48 (m, 5H), 8.00 (d, 1H)。
【0181】
d)N−(2−{4−[5−(ベンジルオキシ)ピリジン−2−イル]ピペラジン−1−イル}−2−オキソエチル)−2−フルオロピリジン−4−カルボキサミド
【化78】
(2−{4−[5−(ベンジルオキシ)ピリジン−2−イル]ピペラジン−1−イル}−2−オキソエチル)カルバミン酸tert−ブチル(14c)8.0g(18.76mmol)を、ジエチルエーテル中2N HClの160mL中に懸濁し、室温で一晩攪拌した。沈殿物を濾過し、エーテルで洗浄し、真空中で40℃で乾燥した。
収量:7.4g(定量的)。この生成物は、更に精製することなく次工程において使用した。
MS (ESIpos):m/z=327 [M+H]+
【0182】
DMF 40mL中の2−フルオロピリジン−4−カルボン酸(Aldrich社)177mg(1.25mmol)及び前述のように調製した塩酸塩501mg(1.38mmol)の溶液へ、PyBOP 784mg(1.5mmol)及びN−エチル−N,N−ジイソプロピルアミン0.80mLを添加し、この反応混合物を室温で一晩攪拌した。溶媒を蒸発させた後、残渣を、酢酸エチル/エタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:315mg(50.2%)。
MS (ESIpos):m/z=449 [M+H]+
1H-NMR (400MHz, DMSO-d6): δ [ppm]=3.37 (br. s., 2H), 3.44 (br. s., 2H), 3.52-3.66 (m, 4H), 4.22 (d, 2H), 5.07 (s, 2H), 6.83 (d, 1H), 7.27-7.47 (m, 6H), 7.53 (s, 1H), 7.70-7.81 (m, 1H), 7.95 (d, 1H), 8.39 (d, 1H), 9.01 (t, 1H)。
【0183】
e)N−(2−{4−[5−(ベンジルオキシ)ピリジン−2−イル]ピペラジン−1−イル}−2−オキソエチル)−2−ブロモピリジン−4−カルボキサミド
【化79】
(2−{4−[5−(ベンジルオキシ)ピリジン−2−イル]ピペラジン−1−イル}−2−オキソエチル)カルバミン酸tert−ブチル(14c)8.0g(18.76mmol)を、ジエチルエーテル中2N HClの160mL中で懸濁し、室温で一晩攪拌した。沈殿物を濾過し、エーテルで洗浄し、真空中で40℃で乾燥した。
収量:7.4g(定量的)。この生成物は、更に精製することなく次工程において使用した。
MS (ESIpos):m/z=327 [M+H]+
【0184】
DMF 160mL中の2−ブロモピリジン−4−カルボン酸(Aldrich社)1.01g(5.01mmol)及び前述のように調製した塩酸塩2.0g(5.51mmol)の溶液へ、PyBOP 3.13g(6.0mmol)及びN−エチル−N,N−ジイソプロピルアミン2.75mLを添加し、この反応混合物を室温で一晩攪拌した。溶媒を蒸発させた後、残渣を、酢酸エチル/エタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:739mg(27.7%)。
MS (ESIpos):m/z=510, 512 [M+H]+
1H-NMR (400MHz, DMSO-d6): δ [ppm]=2.72 (s, 1H), 2.88 (s, 1H), 3.40-3.48 (m, 2H), 3.51-3.64 (m, 4H), 4.21 (d, 2H), 5.07 (s, 2H), 6.86 (d, 1H), 7.25-7.48 (m, 6H), 7.81 (dd, 1H), 7.95 (d, 1H), 8.02 (s, 1H), 8.56 (d, 1H), 9.06 (s, 1H)。
【0185】
f)[18F]−N−(2−{4−[5−(ベンジルオキシ)ピリジン−2−イル]ピペラジン−1−イル}−2−オキソエチル)−2−フルオロピリジン−4−カルボキサミド
【化80】
水性[18F]フッ化物38.7GBqを、QMAカートリッジ(Waters社)上に捕獲し、反応器中へ、TBAOH溶液2mL(MeCN 1.5mL、H2O 0.3mL+TBAOH溶液(40%)8μL)により溶離させた。その溶媒を、120℃で10分間、窒素流れ下で加熱することにより、除去した。無水MeCN(1ml)を添加し、前述のように蒸発させた。無水DMSO 500μl中の前駆体14e(5mg)の溶液を添加した。180℃で20分間加熱した後、粗反応混合物を水/MeCN(50:50)4mLで希釈し、分取HPLCにより精製した:ACE 5−C18−HL 250mm×10mm;アイソクラチック、0.1%トリフルオロ酢酸を含有する水中25%アセトニトリル、流量:4ml/分;tR=〜22分。収集されたHPLC画分を、水40mlで希釈し、Sep−Pak plus short tC18カートリッジ(Waters社)上に固定し、これを水5mlで洗浄し、エタノール1mlで溶離し、EtOH 1000μl中、F−18標識された生成物3.5GBqを送り出し(再結晶収率15.5%、減衰補正;>96% HPLC)、全体の合成時間は80分間未満であった。望ましいF−18標識された生成物14f(tR=3.2分)を、分析用HPLCを用い分析した:ACE3−C18 50mm×4.6mm;溶媒勾配:出発時5%アセトニトリルから、7分間で0.1%トリフルオロ酢酸中95%アセトニトリル、流量:2mL/分、及びこの分析用HPLC上での、対応する非−放射性F−19フルオロ標準14d(tR=3.1分)との同時注入により、確認した。
HPLC分析は、図6A及び図6Bに示されている。
【0186】
実施例15
a)2−{4−[4−(ベンジルオキシ)フェニル]ピペラジン−1−イル}−2−オキソエチルアセテート
【化81】
THF 300mL及びトリエチルアミン7.5mL(54.3mmol)中のアセトキシ酢酸1.42g (12mmol)(Aldrich社)の−15℃溶液へ、クロロギ酸イソブチル1.73mL(13.23mmol)を滴下し、かつこの溶液を、この温度で更に15分間維持した。その後、この冷溶液へ、THF/ジクロロメタン(1:1)240mL中の1−(4−ベンジルオキシフェニル)−ピペラジン(3c)3.23g(12mmol)及びトリエチルアミン1.74mL(12.5mmol)をゆっくり添加し、その温度を−10℃以下に更に15分間維持し、その後室温とした。一晩攪拌した後、溶媒を蒸発させ、残渣を酢酸エチルに溶かした。この溶液を、水性炭酸ナトリウム、水、1M HCl水溶液、飽和塩化ナトリウム水溶液で連続して洗浄し、最後に硫酸マグネシウム上で乾燥し、その後蒸発させた。この残渣を、ヘキサン/酢酸エチル勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:1.12g(25.2%)。
MS (ESIpos):m/z=369 [M+H]+
1H-NMR (300MHz, CHCl3-d): δ [ppm]=2.21 (s, 3H), 3.02-3.14 (m, 4H), 3.55 (br. s., 2H), 3.78 (br. s., 2H), 4.78 (s, 2H), 5.03 (s, 2H), 6.83-7.00 (m, 4H), 7.29-7.48 (m, 5H)。
【0187】
b)1−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−ヒドロキシ−エタノン
【化82】
酢酸エステル15aの246mg(0.67mmol)を、エタノール40mL中に溶解し、0℃に冷却した。3N NaOH 2.1mLの添加後、この溶液を1時間攪拌し、そのpHがpH7を下回るまで、氷酢酸を添加し、溶媒を蒸発させた。粗生成物をエタノールから結晶化した。
収量:116mg(52.7%)。
MS (ESIpos):m/z=327 [M+H]+
1H-NMR (300MHz, CHCl3-d): δ [ppm]=3.01-3.14 (m, 4H), 3.37-3.49 (m, 2H), 3.63 (t, 1H), 3.78-3.88 (m, 2H), 4.22 (d, 2H), 5.04 (s, 2H), 6.83-7.00 (m, 4H), 7.29-7.49 (m, 5H)。
【0188】
c)2−{4−[4−(ベンジルオキシ)フェニル]ピペラジン−1−イル}−2−オキソエチル 2−フルオロピリジン−4−カルボキシラート
【化83】
THF 5mL及びトリエチルアミン29μL(0.2mmol)中の2−フルオロピリジン−4−カルボン酸(Aldrich社) 28.2mg(0.2mmol)の−15℃溶液へ、クロロギ酸イソブチル31.6μL(0.22mmol)を滴下し、この溶液を、この温度で更に15分間維持した。その後、この冷溶液へ、THF/ジクロロメタン(1:1) 5mL中の1−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−ヒドロキシ−エタノン(15b)65.28mg(0.2mmol)及びトリエチルアミン125μL(0.9mmol)をゆっくり添加し、その温度を−10℃以下に更に15分間維持し、その後室温とした。一晩攪拌した後、溶媒を蒸発させ、残渣を酢酸エチルに溶かした。この溶液を、水性炭酸ナトリウム、水、1M HCl水溶液、飽和塩化ナトリウム水溶液で連続して洗浄し、最後に硫酸マグネシウム上で乾燥し、その後蒸発させた。この残渣を、ヘキサン/酢酸エチル勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:30mg(33.4%)。
MS (ESIpos):m/z=450 [M+H]+
1H-NMR (400MHz, CHCl3-d): δ [ppm]=3.03-3.18 (m, 4H), 3.61 (br. s., 2H), 3.81 (br. s., 2H), 5.06 (d, 4H), 6.87-6.99 (m, 4H), 7.29-7.47 (m, 5H), 7.55-7.62 (m, 1H), 7.83 (dt, 1H), 8.40 (d, 1H)。
【0189】
d)2−{4−[4−(ベンジルオキシ)フェニル]ピペラジン−1−イル}−2−オキソエチル2−ブロモピリジン−4−カルボキシラート
【化84】
THF 25mL及びトリエチルアミン0.2mL(1.44mmol)中の2−ブロモピリジン−4−カルボン酸(Aldrich社)185.7mg(0.92mmol)の−15℃溶液へ、クロロギ酸イソブチル132.3μL(1.01mmol)を滴下し、この溶液を、この温度で更に15分間維持した。その後、この冷溶液へ、THF/ジクロロメタン(1:1)24mL中の1−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−ヒドロキシ−エタノン(15b)300mg(0.92mmol)及びトリエチルアミン0.6mL(4.3mmol)をゆっくり添加し、その温度を−10℃以下に更に15分間維持し、その後室温とした。一晩攪拌した後、溶媒を蒸発させ、残渣を酢酸エチルに溶かした。この溶液を、水性炭酸ナトリウム、水、1M HCl水溶液、飽和塩化ナトリウム水溶液で連続して洗浄し、最後に硫酸マグネシウム上で乾燥し、その後蒸発させた。この残渣を、ヘキサン/酢酸エチル勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:145mg(30.6%)。
MS (ESIpos):m/z=510, 512 [M+H]+
1H-NMR (400MHz, CHCl3-d): δ [ppm]=3.11 (d, 4H), 3.60 (br. s., 2H), 3.80 (br. s., 2H), 5.05 (d, 4H), 6.86-6.99 (m, 4H), 7.29-7.48 (m, 5H), 7.89 (dd, 1H), 8.13 (s, 1H), 8.56 (d, 1H)。
【0190】
e)[18F]−2−{4−[4−(ベンジルオキシ)フェニル]ピペラジン−1−イル}−2−オキソエチル2−フルオロピリジン−4−カルボキシラート
【化85】
水性[18F]フッ化物19GBqを、QMAカートリッジ(Waters社)上に捕獲し、反応器中へ、TBAOH溶液2mL(MeCN 1.5mL、H2O 0.3mL+TBAOH溶液(40%)8μL)により溶離させた。その溶媒を、120℃で10分間、窒素流れ下で加熱することにより、除去した。無水MeCN(1ml)を添加し、前述のように蒸発させた。無水DMSO 500μl中の前駆体15d(5mg)の溶液を添加した。180℃で20分間加熱した後、粗反応混合物を水/MeCN(50:50)4mLで希釈し、分取HPLCにより精製した:ACE 5−C18−HL 250mm×10mm;アイソクラチック、0.1%トリフルオロ酢酸を含む水中40%アセトニトリル、流量:4ml/分;tR=〜24分。収集されたHPLC画分を、水40mlで希釈し、Sep−Pak plus short tC18カートリッジ(Waters社)上に固定し、これを水5mlで洗浄し、エタノール1mlで溶離し、EtOH 1000μl中、F−18標識された生成物15e 0.2GBqを送り出し(再結晶収率2%、減衰補正;>98% HPLC)、全体の合成時間は80分未満であった。望ましいF−18標識された生成物15e(tR=4.3分)を、分析用HPLCを用い分析し:ACE3−C18 50mm×4.6mm;溶媒勾配:出発時5%アセトニトリルから、7分間で0.1%トリフルオロ酢酸中95%アセトニトリル、流量:2mL/分、及びこの分析用HPLC上での、対応する非−放射性F−19フルオロ標準15c(tR=4.2分)との同時注入により、確認した。
HPLC分析は、図7A及び図7Bに示されている。
【0191】
実施例16
a)2−フルオロ−N−{2−[4−(4−ヒドロキシフェニル)ピペラジン−1−イル]−2−オキソエチル}ピリジン−4−カルボキサミド
【化86】
メタノール70mL中のN−{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−2−フルオロ−イソニコチンアミド(11b)280mg(0.62mmol)の溶液に、活性炭上に担持されたパラジウム(10%)100mgを添加し、この懸濁液を水素大気下で一晩室温で攪拌した。その後これを、触媒から濾過除去し、その溶液を真空中で蒸発させた。生成物は、更に精製することなく、次工程において使用した。
収量:175mg(54.8%)。
MS (ESIpos):m/z=359 [M+H]+。
【0192】
b)4−(4−{N−[(2−フルオロピリジン−4−イル)カルボニル]グリシル}ピペラジン−1−イル)フェニルベンゾエート
【化87】
THF 10mL及びトリエチルアミン90μL(0.65mmol)中の安息香酸57.9mg(0.47mmol)の−15℃溶液へ、クロロギ酸イソブチル62.06μL(0.47mmol)を滴下し、この溶液を、この温度で更に15分間維持した。その後この冷溶液へ、THF/ジクロロメタン(1:1)20mL中の先に調製した実施例16a 170mg(0.47mmol)、及びトリエチルアミン0.4mL(2.87mmol)をゆっくり添加し、更に15分間その温度を−10℃未満に維持し、その後室温とした。一晩攪拌した後、溶媒を蒸発させ、残渣を、酢酸エチルに溶かした。この溶液を、水性炭酸ナトリウム、水、1M HCl水溶液、飽和塩化ナトリウム水溶液で連続して洗浄し、最後に硫酸マグネシウム上で乾燥し、その後蒸発させた。この残渣を、ヘキサン/酢酸エチル勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:23mg(9.5%)。
MS (ESIpos): m/z=463 [M+H]+
1H-NMR (400MHz, DMSO-d6): δ [ppm]=3.15 (br. s., 2H), 3.22 (br. s., 2H), 3.66 (br. s., 4H), 4.24 (d, 2H), 7.00-7.11 (m, 2H), 7.11-7.21 (m, 2H), 7.50-7.66 (m, 3H), 7.68-7.82 (m, 2H), 8.12 (d, 2H), 8.41 (d, 1H), 9.03 (t, 1H)。
【0193】
c){2−[4−(4−ヒドロキシフェニル)ピペラジン−1−イル]−2−オキソエチル}カルバミン酸tert−ブチル
【化88】
メタノール100mL中の{2−[4−(4−ベンジルオキシ−フェニル)−ピペラジン−1−イル]−2−オキソ−エチル}−カルバミン酸tert−ブチルエステル(10a)2.5g(5.88mmol)の溶液に、活性炭上に担持されたパラジウム(10%)1gを添加し、この懸濁液を水素大気下で一晩室温で攪拌した。その後これを、触媒から濾過除去し、その溶液を真空中で蒸発させた。生成物は、更に精製することなく、次工程において使用した。
収量:1.92g(95.9%)。
MS (ESIpos):m/z=336 [M+H]+。
【0194】
d)2−ブロモ−N−{2−[4−(4−ヒドロキシフェニル)ピペラジン−1−イル]−2−オキソエチル}ピリジン−4−カルボキサミド
【化89】
{2−[4−(4−ヒドロキシフェニル)ピペラジン−1−イル]−2−オキソエチル}カルバミン酸tert−ブチル(16c)1.8g(5.37mmol)を、ジエチルエーテル中の2N HCl 60mL中に懸濁し、室温で一晩攪拌した。沈殿物を濾過し、エーテルで洗浄し、真空中40℃で乾燥した。
収量:1.8g(定量的)。生成物は、更に精製することなく、次工程において使用した。
【0195】
DMF 60mL中の2−ブロモ−4−ピリジンカルボン酸(Alfa社)338mg(1.67mmol)及び先に調製した塩酸塩500mg(1.84mmol)の溶液へ、PyBOP 1.045g(2mmol)及びN−エチル−N,N−ジイソプロピルアミン2.86mL(16.73mmol)を添加し、この反応混合物を、室温で一晩攪拌した。溶媒を蒸発させた後、残渣を、酢酸エチル/エタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。生成物含有画分を収集し、かつ酢酸エチルから再結晶化した。
収量:284mg(40.5%)。
MS (ESIpos):m/z=419, 421 [M+H]+
1H-NMR (300MHz, CH3OH-d4): δ [ppm]=1.49 (dt, 4H), 2.17 (dt, 4H), 2.76 (s, 2H), 5.11-5.22 (m, 2H), 5.29-5.40 (m, 2H), 6.23 (dd, 1H), 6.43-6.50 (m, 1H), 6.89-6.98 (m, 1H)。
【0196】
e)4−(4−{N−[(2−ブロモピリジン−4−イル)カルボニル]グリシル}ピペラジン−1−イル)フェニルベンゾエート
【化90】
DMF 45mL中の2−ブロモ−N−{2−[4−(4−ヒドロキシフェニル)ピペラジン−1−イル]−2−オキソエチル}ピリジン−4−カルボキサミド(16d)218mg(0.52mmol)の溶液へ、1,3−ジシクロヘキシルカルボジイミド214.56mg(1.04mmol)及び4−ジメチルアミノピリジン(DMAP)20mgを添加し、室温で40分間攪拌した。その後安息香酸127mg(1.04mmol)を添加し、この反応混合物を、この温度で3日間攪拌した。この溶媒を蒸発させ、残渣を、酢酸エチル/エタノール勾配を使用し、シリカゲル上でクロマトグラフィーにかけた。
収量:222mg(81.6%)。
MS (ESIpos):m/z=523, 525 [M+H]+
1H-NMR (400MHz, DMSO-d6): δ [ppm]=3.25 (br. s., 4H), 3.65 (br. s., 4H), 4.23 (d, 2H), 6.99-7.10 (m, 2H), 7.10-7.21 (m, 2H), 7.53-7.65 (m, 2H), 7.67-7.77 (m, 1H), 7.83 (dd, 1H), 7.99-8.06 (m, 1H), 8.06-8.17 (m, 2H), 8.55 (d, 1H), 9.05 (t, 1H)。
【0197】
f)[18F]−4−(4−{N−[(2−フルオロピリジン−4−イル)カルボニル]グリシル}ピペラジン−1−イル)フェニルベンゾエート
【化91】
水性[18F]フッ化物1GBqを、QMAカートリッジ(Waters社)上に捕獲し、反応器中へ、TBAOH溶液2mL(MeCN 1.5mL、H2O 0.3mL+TBAOH溶液(40%)8μL)により溶離させた。その溶媒を、120℃で10分間、窒素流れ下で加熱することにより、除去した。無水MeCN(1ml)を添加し、前述のように蒸発させた。無水DMSO 500μl中の前駆体16e(5mg)の溶液を添加した。180℃で20分間加熱した後、粗反応混合物を分析用HPLCを用い分析し:ACE3−C18 50mm×4.6mm;溶媒勾配:出発5%アセトニトリルから、7分間で0.1%トリフルオロ酢酸中の95%アセトニトリル、流量:2mL/分、かつこの分析用HPLC上での、対応する非−放射性F−19フルオロ標準15cとの同時注入により、確認した(tR=3.7分)。粗生成物は、分取HPLCにより精製することができる:ACE 5−C18−HL 250mm×10mm;アイソクラチック、0.1%トリフルオロ酢酸を含有する水中35%アセトニトリル、流量:4ml/分。
【0198】
実施例17:生物学的データ
実施例14及び15の化合物の生物学的データを、実施例13に説明したように得た。
【0199】
図8は、アルツハイマー病患者(AD)及び健常対照(HC)の皮質由来の凍結切片への14fの結合のオートラジオグラフィー分析を示す。AD試料の斑豊富領域における特異的結合を矢印で示す。
【0200】
図9は、AD患者由来の脳ホモジネートを使用する競合アッセイにおいて測定された、選択された化合物の[nM]でのIC50値を示す。
【0201】
【表5】
【特許請求の範囲】
【請求項1】
式Iの化合物、又は、医薬として許容し得るその塩若しくはプロドラッグを含有する診断用組成物:
【化1】
(式中、
−Yは:
F、Cl、Br、I、H、18F、19F、76Br、123I、125I、11C、3H、13N、15O;
脱離基、トシル、ブロシル、ノシル、トリフラート、スルホネート、置換スルホネート、メシラート、及びノナフレート;並びに
芳香族C原子に直接結合される場合は、ヨードニウム−アリールI+−アリール、トリアルキルアンモニウム、好ましくはトリメチルアンモニウム、及びNO2
からなる群から選択され;
−Arは、置換又は非置換の、単環、二環又は三環式の、芳香環又は芳香族複素環系からなる群から選択され;
−Bは、直接結合、1〜10個のC原子からなる分枝又は非分枝のアルキル又はアルキレン鎖からなる群から選択され;
−Aは、直接結合、及びCO−NH、CS−NHからなる群から選択され;
−Ar’は、置換又は非置換の、単環又は二環式の、芳香環又は芳香族複素環系からなる群から選択され;
−Xは、直接結合、又は、置換若しくは非置換のC1−C3アルキル鎖からなる群から選択され;
−Ar”は、置換又は非置換の、単環又は二環式の、芳香環又は芳香族複素環系からなる群から選択される)。
【請求項2】
Yが、18Fである、請求項1記載の組成物。
【請求項3】
式Iの化合物、又は、医薬として許容し得るその塩若しくはプロドラッグ:
【化2】
(式中、
−Yは:
F、Cl、Br、I、H、18F、19F、76Br、123I、125I、11C、3H、13N、15O;
脱離基、トシル、ブロシル、ノシル、トリフラート、スルホネート、置換スルホネート、メシラート、及びノナフレート;並びに
芳香族C原子に直接結合される場合は、ヨードニウム−アリールI+−アリール、トリアルキルアンモニウム、好ましくはトリメチルアンモニウム、及びNO2
からなる群から選択され;
−Arは、置換又は非置換の、単環、二環又は三環式の、芳香環又は芳香族複素環系からなる群から選択され;
−Bは、直接結合、1〜10個のC原子からなる分枝又は非分枝のアルキル又はアルキレン鎖からなる群から選択され;
−Aは、直接結合、及びCO−NH、CS−NHからなる群から選択され;
−Ar’は、置換又は非置換の、単環又は二環式の、芳香環又は芳香族複素環系からなる群から選択され;
−Xは、直接結合、又は、置換若しくは非置換のC1−C3アルキル鎖からなる群から選択され;
−Ar”は、置換又は非置換の、単環又は二環式の、芳香環又は芳香族複素環系からなる群から選択され、
但し、A及び/又はBが直接結合である場合、直接結合された残基Ar又はAr’は、6員の芳香環系である)。
【請求項4】
式Iの化合物、又は、医薬として許容し得るその塩若しくはプロドラッグ:
【化3】
(式中、
−Yは:
F、Cl、Br、I、H、18F、19F、76Br、123I、125I、11C、3H、13N、15O;
脱離基、トシル、ブロシル、ノシル、トリフラート、スルホネート、置換スルホネート、メシラート、及びノナフレート;並びに
芳香族C原子に直接結合される場合は、ヨードニウム−アリールI+−アリール、トリアルキルアンモニウム、好ましくはトリメチルアンモニウム、及びNO2
からなる群から選択され;
−Arは、1又は2個のアルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基により置換された、単環、二環又は三環式の芳香環又は芳香族複素環系からなる群から選択され;
−Bは、直接結合、1〜10個のC原子からなる分枝又は非分枝のアルキル又はアルキレン鎖からなる群から選択され;
−Aは、直接結合、及びCO−NH、CS−NHからなる群から選択され;
−Ar’は、1又は2個のアルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基により置換された単環又は二環式芳香環又は芳香族複素環系からなる群から選択され、ここで該単環、二環又は三環式の芳香環又は芳香族複素環系は、芳香族C原子に直接結合された電子求引基により更に置換されており;
−Xは、直接結合、又は、置換若しくは非置換のC1−C3アルキル鎖からなる群から選択され;
−Ar”は、置換又は非置換の、単環又は二環式の、芳香環又は芳香族複素環系からなる群から選択される)。
【請求項5】
前記式中、
−Ar、Ar’及びAr”のアルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基が、オキソ又はヒドロキシルからなる群から選択され、
Ar、Ar’及びAr”のアルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基は、1〜5個の酸素原子により隔てられており、好ましくはこれらの置換基はポリエチレングリコール部分であり、
更にAr、Ar’及びAr”のアルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基が、C3−C6シクロアルキル部分を含み、ここでAr、Ar’及びAr”の任意の電子求引基が、−CN又はCF3からなる群から選択され;
−Bが、直接結合、CONH−CH2CO、CO−(CH2)2CO、(CH2)nCO、O(CH2)nCO(但しn=1〜10)、(CH=CH)CO、
【化4】
からなる群から選択され;
−Xが、直接結合、OCH2、NHCO、CH2O、CONH、NHCS、又はCSNHからなる群から選択される、請求項3又は4記載の化合物。
【請求項6】
前記式中、
−Arが、プロピルピリミジン−2−イル、エトキシフェニル、(CH2CH2O)3フェニル、アルキルフェニル、アルコキシフェニル、N−アルキルインドリル、及びアルキルピリジル、
【化5】
からなる群から選択され;
−Ar’が、プロピルピリミジン−2−イル、エトキシフェニル、(CH2CH2O)3フェニル、アルキルフェニル、アルコキシフェニル、N−アルキルインドリル、フェニル、ベンゾフラニル、インドリル及びアルキルピリジルからなる群から選択され;
−Ar”が、フェニル、1−フェニル、1−ナフチル、2−ナフチル、及びそれらに対応する複素環全てからなる群から選択される、請求項3〜5の何れか一項に記載の化合物。
【請求項7】
下記からなる群から選択される、化合物:
【化6】
【化7】
【化8】
【請求項8】
Fが、18Fを意味する、請求項7記載の化合物。
【請求項9】
放射性核種又は蛍光標識等の検出可能な標識を含む、請求項3、4、5又は6記載の化合物。
【請求項10】
検出可能な標識が、18Fである、請求項9記載の化合物。
【請求項11】
診断用化合物としての、請求項3〜10の何れか一項に記載の化合物。
【請求項12】
医薬品としての、請求項3〜7の何れか一項に記載の化合物。
【請求項13】
アルツハイマー病、神経変性障害、又はアミロイドーシスからなる群から選択される疾患のための診断用化合物としての、請求項8、9又は10記載の化合物。
【請求項14】
アルツハイマー病、神経変性障害、又はアミロイドーシスからなる群から選択される疾患の治療のための医薬品としての、請求項12記載の化合物。
【請求項15】
請求項3〜10の何れか一項に記載のフッ素化された化合物、又は、請求項1若しくは2記載の組成物に使用される請求項1若しくは2記載の化合物を調製する方法であって、好適な前駆分子をフッ素化剤と反応させることを含む、方法。
【請求項16】
哺乳動物におけるアルツハイマー病、神経変性障害又はアミロイドーシスからなる群から選択される障害を治療又は予防する方法であって、請求項3〜7及び12の何れか一項に記載の化合物の治療的有効量を、該哺乳動物へ投与することを含む、方法。
【請求項17】
哺乳動物におけるアルツハイマー病、神経変性障害又はアミロイドーシスからなる群から選択される疾患の治療における請求項3〜7及び12の何れか一項に記載の化合物の使用であって、該化合物の治療的有効量が該哺乳動物へ投与される、使用。
【請求項18】
アルツハイマー病、神経変性障害又はアミロイドーシスからなる群から選択される疾患を哺乳動物において診断する方法であって、請求項8〜11の何れか一項に記載の化合物又は請求項1若しくは2記載の組成物を該哺乳動物へ投与することを含む、方法。
【請求項19】
前記方法が、該哺乳動物を造影すること、及び造影シグナルを検出することを含む、請求項18記載の方法。
【請求項20】
前記造影が、PET、SPECT、MRスペクトル分析法、及びMR−断層撮影法からなる群から選択される造影方法を用いて実行される、請求項19記載の方法。
【請求項21】
療法の作用がモニタリングされる、請求項18〜20の何れか一項に記載の方法。
【請求項22】
哺乳動物におけるアルツハイマー病、神経変性障害又はアミロイドーシスからなる群から選択される疾患を診断又は療法をモニタリングする方法であって、該方法が、該哺乳動物の試料をインビトロにおいて分析することを含み、ここで該哺乳動物又は試料が、請求項3〜11の何れか一項に記載の化合物若しくは請求項1又は2記載の組成物により処理されることを含む、方法。
【請求項23】
試料が脳脊髄液である、請求項22記載の方法。
【請求項24】
密封バイアル中に、請求項3〜14の何れか一項に記載の化合物、又は、請求項1若しくは2記載の組成物を備える、キット。
【請求項25】
Fが脱離基により置換されている、請求項7記載の化合物。
【請求項26】
脱離基が、トシル、ブロシル、ノシル、トリフラート、スルホネート、置換スルホネート、メシラート、ノナフレート;並びに、芳香族C原子に直接結合される場合は、ヨードニウム−アリールI+−アリール、トリアルキルアンモニウム、及びNO2からなる群から選択される、請求項25記載の化合物。
【請求項27】
請求項25又は26記載の対応する化合物をフッ素化剤と反応させることを含む、請求項7又は8記載の化合物を調製する方法。
【請求項1】
式Iの化合物、又は、医薬として許容し得るその塩若しくはプロドラッグを含有する診断用組成物:
【化1】
(式中、
−Yは:
F、Cl、Br、I、H、18F、19F、76Br、123I、125I、11C、3H、13N、15O;
脱離基、トシル、ブロシル、ノシル、トリフラート、スルホネート、置換スルホネート、メシラート、及びノナフレート;並びに
芳香族C原子に直接結合される場合は、ヨードニウム−アリールI+−アリール、トリアルキルアンモニウム、好ましくはトリメチルアンモニウム、及びNO2
からなる群から選択され;
−Arは、置換又は非置換の、単環、二環又は三環式の、芳香環又は芳香族複素環系からなる群から選択され;
−Bは、直接結合、1〜10個のC原子からなる分枝又は非分枝のアルキル又はアルキレン鎖からなる群から選択され;
−Aは、直接結合、及びCO−NH、CS−NHからなる群から選択され;
−Ar’は、置換又は非置換の、単環又は二環式の、芳香環又は芳香族複素環系からなる群から選択され;
−Xは、直接結合、又は、置換若しくは非置換のC1−C3アルキル鎖からなる群から選択され;
−Ar”は、置換又は非置換の、単環又は二環式の、芳香環又は芳香族複素環系からなる群から選択される)。
【請求項2】
Yが、18Fである、請求項1記載の組成物。
【請求項3】
式Iの化合物、又は、医薬として許容し得るその塩若しくはプロドラッグ:
【化2】
(式中、
−Yは:
F、Cl、Br、I、H、18F、19F、76Br、123I、125I、11C、3H、13N、15O;
脱離基、トシル、ブロシル、ノシル、トリフラート、スルホネート、置換スルホネート、メシラート、及びノナフレート;並びに
芳香族C原子に直接結合される場合は、ヨードニウム−アリールI+−アリール、トリアルキルアンモニウム、好ましくはトリメチルアンモニウム、及びNO2
からなる群から選択され;
−Arは、置換又は非置換の、単環、二環又は三環式の、芳香環又は芳香族複素環系からなる群から選択され;
−Bは、直接結合、1〜10個のC原子からなる分枝又は非分枝のアルキル又はアルキレン鎖からなる群から選択され;
−Aは、直接結合、及びCO−NH、CS−NHからなる群から選択され;
−Ar’は、置換又は非置換の、単環又は二環式の、芳香環又は芳香族複素環系からなる群から選択され;
−Xは、直接結合、又は、置換若しくは非置換のC1−C3アルキル鎖からなる群から選択され;
−Ar”は、置換又は非置換の、単環又は二環式の、芳香環又は芳香族複素環系からなる群から選択され、
但し、A及び/又はBが直接結合である場合、直接結合された残基Ar又はAr’は、6員の芳香環系である)。
【請求項4】
式Iの化合物、又は、医薬として許容し得るその塩若しくはプロドラッグ:
【化3】
(式中、
−Yは:
F、Cl、Br、I、H、18F、19F、76Br、123I、125I、11C、3H、13N、15O;
脱離基、トシル、ブロシル、ノシル、トリフラート、スルホネート、置換スルホネート、メシラート、及びノナフレート;並びに
芳香族C原子に直接結合される場合は、ヨードニウム−アリールI+−アリール、トリアルキルアンモニウム、好ましくはトリメチルアンモニウム、及びNO2
からなる群から選択され;
−Arは、1又は2個のアルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基により置換された、単環、二環又は三環式の芳香環又は芳香族複素環系からなる群から選択され;
−Bは、直接結合、1〜10個のC原子からなる分枝又は非分枝のアルキル又はアルキレン鎖からなる群から選択され;
−Aは、直接結合、及びCO−NH、CS−NHからなる群から選択され;
−Ar’は、1又は2個のアルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基により置換された単環又は二環式芳香環又は芳香族複素環系からなる群から選択され、ここで該単環、二環又は三環式の芳香環又は芳香族複素環系は、芳香族C原子に直接結合された電子求引基により更に置換されており;
−Xは、直接結合、又は、置換若しくは非置換のC1−C3アルキル鎖からなる群から選択され;
−Ar”は、置換又は非置換の、単環又は二環式の、芳香環又は芳香族複素環系からなる群から選択される)。
【請求項5】
前記式中、
−Ar、Ar’及びAr”のアルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基が、オキソ又はヒドロキシルからなる群から選択され、
Ar、Ar’及びAr”のアルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基は、1〜5個の酸素原子により隔てられており、好ましくはこれらの置換基はポリエチレングリコール部分であり、
更にAr、Ar’及びAr”のアルキル、アルキレン、アルキン置換基及び/又はアルコキシ置換基が、C3−C6シクロアルキル部分を含み、ここでAr、Ar’及びAr”の任意の電子求引基が、−CN又はCF3からなる群から選択され;
−Bが、直接結合、CONH−CH2CO、CO−(CH2)2CO、(CH2)nCO、O(CH2)nCO(但しn=1〜10)、(CH=CH)CO、
【化4】
からなる群から選択され;
−Xが、直接結合、OCH2、NHCO、CH2O、CONH、NHCS、又はCSNHからなる群から選択される、請求項3又は4記載の化合物。
【請求項6】
前記式中、
−Arが、プロピルピリミジン−2−イル、エトキシフェニル、(CH2CH2O)3フェニル、アルキルフェニル、アルコキシフェニル、N−アルキルインドリル、及びアルキルピリジル、
【化5】
からなる群から選択され;
−Ar’が、プロピルピリミジン−2−イル、エトキシフェニル、(CH2CH2O)3フェニル、アルキルフェニル、アルコキシフェニル、N−アルキルインドリル、フェニル、ベンゾフラニル、インドリル及びアルキルピリジルからなる群から選択され;
−Ar”が、フェニル、1−フェニル、1−ナフチル、2−ナフチル、及びそれらに対応する複素環全てからなる群から選択される、請求項3〜5の何れか一項に記載の化合物。
【請求項7】
下記からなる群から選択される、化合物:
【化6】
【化7】
【化8】
【請求項8】
Fが、18Fを意味する、請求項7記載の化合物。
【請求項9】
放射性核種又は蛍光標識等の検出可能な標識を含む、請求項3、4、5又は6記載の化合物。
【請求項10】
検出可能な標識が、18Fである、請求項9記載の化合物。
【請求項11】
診断用化合物としての、請求項3〜10の何れか一項に記載の化合物。
【請求項12】
医薬品としての、請求項3〜7の何れか一項に記載の化合物。
【請求項13】
アルツハイマー病、神経変性障害、又はアミロイドーシスからなる群から選択される疾患のための診断用化合物としての、請求項8、9又は10記載の化合物。
【請求項14】
アルツハイマー病、神経変性障害、又はアミロイドーシスからなる群から選択される疾患の治療のための医薬品としての、請求項12記載の化合物。
【請求項15】
請求項3〜10の何れか一項に記載のフッ素化された化合物、又は、請求項1若しくは2記載の組成物に使用される請求項1若しくは2記載の化合物を調製する方法であって、好適な前駆分子をフッ素化剤と反応させることを含む、方法。
【請求項16】
哺乳動物におけるアルツハイマー病、神経変性障害又はアミロイドーシスからなる群から選択される障害を治療又は予防する方法であって、請求項3〜7及び12の何れか一項に記載の化合物の治療的有効量を、該哺乳動物へ投与することを含む、方法。
【請求項17】
哺乳動物におけるアルツハイマー病、神経変性障害又はアミロイドーシスからなる群から選択される疾患の治療における請求項3〜7及び12の何れか一項に記載の化合物の使用であって、該化合物の治療的有効量が該哺乳動物へ投与される、使用。
【請求項18】
アルツハイマー病、神経変性障害又はアミロイドーシスからなる群から選択される疾患を哺乳動物において診断する方法であって、請求項8〜11の何れか一項に記載の化合物又は請求項1若しくは2記載の組成物を該哺乳動物へ投与することを含む、方法。
【請求項19】
前記方法が、該哺乳動物を造影すること、及び造影シグナルを検出することを含む、請求項18記載の方法。
【請求項20】
前記造影が、PET、SPECT、MRスペクトル分析法、及びMR−断層撮影法からなる群から選択される造影方法を用いて実行される、請求項19記載の方法。
【請求項21】
療法の作用がモニタリングされる、請求項18〜20の何れか一項に記載の方法。
【請求項22】
哺乳動物におけるアルツハイマー病、神経変性障害又はアミロイドーシスからなる群から選択される疾患を診断又は療法をモニタリングする方法であって、該方法が、該哺乳動物の試料をインビトロにおいて分析することを含み、ここで該哺乳動物又は試料が、請求項3〜11の何れか一項に記載の化合物若しくは請求項1又は2記載の組成物により処理されることを含む、方法。
【請求項23】
試料が脳脊髄液である、請求項22記載の方法。
【請求項24】
密封バイアル中に、請求項3〜14の何れか一項に記載の化合物、又は、請求項1若しくは2記載の組成物を備える、キット。
【請求項25】
Fが脱離基により置換されている、請求項7記載の化合物。
【請求項26】
脱離基が、トシル、ブロシル、ノシル、トリフラート、スルホネート、置換スルホネート、メシラート、ノナフレート;並びに、芳香族C原子に直接結合される場合は、ヨードニウム−アリールI+−アリール、トリアルキルアンモニウム、及びNO2からなる群から選択される、請求項25記載の化合物。
【請求項27】
請求項25又は26記載の対応する化合物をフッ素化剤と反応させることを含む、請求項7又は8記載の化合物を調製する方法。
【図1】

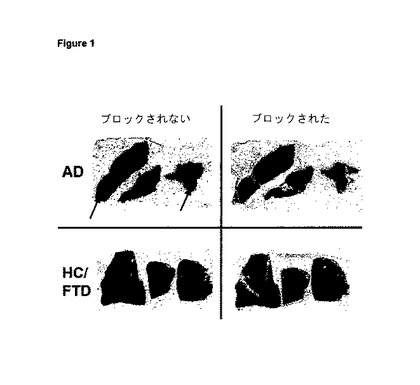
【図2】
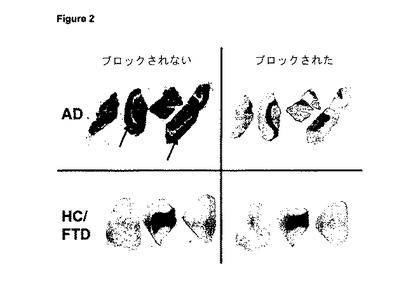
【図3】

【図4】
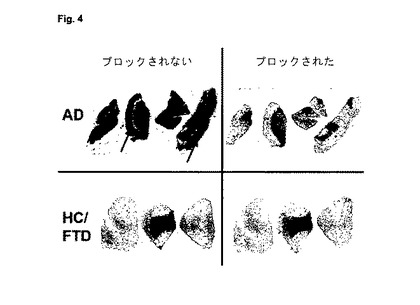
【図5】
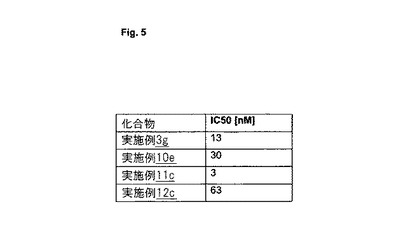
【図6】
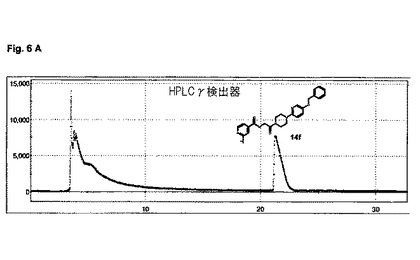
【図6B】
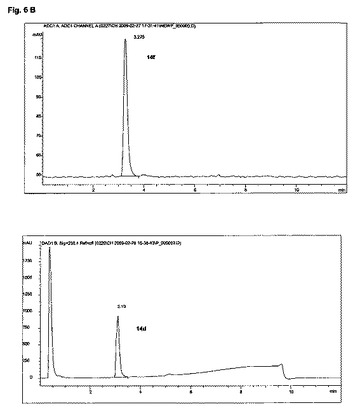
【図7】
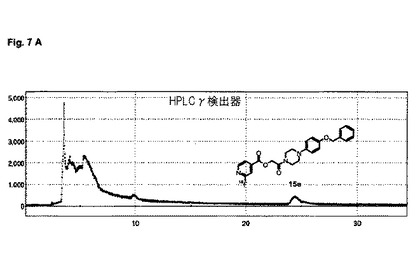
【図7B】
【図8】

【図9】
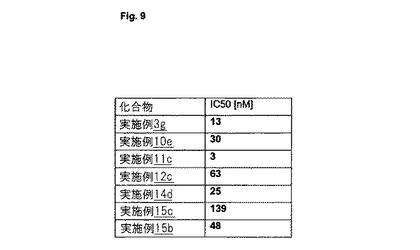
【図2】
【図3】
【図4】
【図5】
【図6】
【図6B】
【図7】
【図7B】
【図8】
【図9】
【公表番号】特表2012−502070(P2012−502070A)
【公表日】平成24年1月26日(2012.1.26)
【国際特許分類】
【出願番号】特願2011−526397(P2011−526397)
【出願日】平成21年9月4日(2009.9.4)
【国際出願番号】PCT/EP2009/006406
【国際公開番号】WO2010/028776
【国際公開日】平成22年3月18日(2010.3.18)
【出願人】(300049958)バイエル ファーマ アクチエンゲゼルシャフト (357)
【Fターム(参考)】
【公表日】平成24年1月26日(2012.1.26)
【国際特許分類】
【出願日】平成21年9月4日(2009.9.4)
【国際出願番号】PCT/EP2009/006406
【国際公開番号】WO2010/028776
【国際公開日】平成22年3月18日(2010.3.18)
【出願人】(300049958)バイエル ファーマ アクチエンゲゼルシャフト (357)
【Fターム(参考)】
[ Back to top ]