
アミロイド沈着に付随する炎症および活性化されたマイクログリアにかかわる脳の炎症の処置法
抗体または非線維状バクテリオファージ抗原をその表面で提示しない線維状バクテリオファージを、アミロイド沈着に付随するおよび/または活性化されたマイクログリアにかかわる脳の炎症を抑制または治療し、アミロイド沈着形成を阻止し、および既に形成されたアミロイド沈着物を脱凝集するために使用する。
【発明の詳細な説明】
【技術分野】
【0001】
関連出願の相互参照
本出願は2005年2月1日出願の米国予備出願第60/648,383の優先権を主張するものであり、そのすべての内容は参照により本願明細書に援用したものとする。
【0002】
背景技術
本発明は、アミロイド沈着形成の抑制および既に形成されたアミロイド沈着の溶解に関するものであり、ならびに脳の炎症ならびに脳内および体内の他の部分のアミロイドまたはアミロイド様の沈着に付随する炎症を治療するための方法および医薬組成物に関する。
【0003】
関連技術の説明
斑形成疾患は、脳におけるアミロイド斑の沈着および神経変性の存在を特徴とする。アミロイド沈着は、ペプチドが凝集して不溶性の塊となることによって形成される。ペプチドの性質は、疾患が異なれば違ってくるが、殆どの場合、凝集体はβプリーツシート構造を有し、コンゴーレッド染料で染色される。早発性アルツハイマー病、遅発性アルツハイマー病および前駆症状アルツハイマー病を含むアルツハイマー病(AD)に加えて、アミロイド沈着を特徴とする他の疾患としては、例えばSAAアミロイド症、遺伝的アイスランド症候群、多発性骨髄種およびプリオン病が挙げられる。動物における最も一般的なプリオン病はヒツジおよびヤギのスクレイピーならびにウシの海綿状脳症(BSE)である(Wilesmith and Wells, 1991)。ヒトにおいて4種のプリオン病が同定されている:(i)クールー、(ii)クロイツフェルトヤコブ病(CJD)、(iii)Gerstmann-Streussler-Sheinker病(GSS)および(iv)致死性家族性不眠症(FFI)である(Gajdusek, 1977; およびTritschler ら. 1992)。
【0004】
プリオン病は、正常細胞プリオンタンパク(PrPC)がそれと対応するスクレイピー・イソ型(PrPSc)へ変換することと関係している。分光学的測定は、PrPCのスクレイピー・イソ型への変換に、立体構造の大きな変換が関与していることを示したが、これはプリオン病が他のアミロイド原性の疾患のように、タンパクの立体構造の障害であることを示唆している。PrPCからPrPScへの変換はα−ヘリックス二次構造の減少(42%から30%へ)とβシート含量の著しい増加(3%から43%)とを伴っている(Caugheyら、1991およびPan等、1993)。この再構成は、非変性の界面活性剤に対する不溶性およびタンパク分解に対する部分的な抵抗性を含む、異常な物理化学的な性質を伴う。先の研究は、ヒトPrPの106−126残基(PrP106−126)と相同性を有する合成ペプチドが、PrPScの病理学的および物理化学的性質の幾つかを有することを示した(Selvagginiら、1993; Tagliaviniら、1993; Forloniら、1993)。このペプチドは、種々の環境において異なった二次構造を獲得し、著明な構造的多形を示す(DeGioiaら、1994)。このペプチドは、緩衝液中においてはベータシート構造をとる傾向にあり、凝集してプロテーアーゼによる分解に部分的に抵抗性を有するアミロイド原線維になる。最近、抗体3F4とそのペプチドエピトープ(PrP104−113)との複合体のX線結晶解析研究が、プリオン病の発症に必須な構造再編成の成分であると考えられるこの可動性の領域の構造図を提供した(Kanyoら、1999)。フォールディング・アンフォールディングおよび/あるいは可溶化−凝集過程に関与している配列のクラスを同定することは、凝集の予防および/あるいは脱凝集の誘導に基づく斑形成疾患の治療に新しい方向を開くかも知れない(Silen and Agard, 1989; Frenkel等、1998; Horiuchi and Caughey, 1999)。
【0005】
アルツハイマー病(AD)は、老年認知症にいたる進行性の疾患である。広義において、この疾患は二つの範疇:老年(典型的には約65歳を超える)に起こる遅発性のもの、および老年よりずっと前に起こる、例えば35と60歳の間に起こる早発性のものに分けられる。この二つの型の疾患では病状は類似しているが、若年において発症する症例においては異常の程度はより重度であり、広範囲に亘っている。この疾患は脳における病変の二つのタイプ、老人斑および神経原線維濃縮体、を特徴とする。老人斑とは、脳組織切片の顕微鏡的解析によって見える、中央における細胞外アミロイド沈着を伴う、径150mmまでの不整の好中球の領域である。神経原線維濃縮体とは、一組になって互いに絡み合っている二本の線維よりなるタウタンパクの細胞内沈着物である。
【0006】
老人斑の主要な構成体はアミロイドベータ(Aβ)あるいはベータ−アミロイドペプチド(βAPあるいはβA)と呼ばれるペプチドである。アミロイドベータペプチドはアミロイド前駆タンパク(APP)と呼ばれる前駆タンパクの39−43アミノ酸の内部断片である。APPタンパク内の数個の突然変異がアルツハイマー病の存在と関連付けられている(Goateら、(1991), バリン717からイソロイシン;Chartier Harlanら、(1991), バリン717からグリシン;Murrellら、(1991), バリン717からフェニルアラニン;Mullanら、(1992)、二重変異、リジン595−メチオニン596からアスパラギン595−ロイシン596)。
【0007】
そのような突然変異は、APPのベータ−アミロイドへのプロセシング、具体的にはAPPをベータ−アミロイドの長型(すなわちAβ1−42およびAβ−43)の増量に導くプロセシングを増加あるいは変化させることにより、アルツハイマー病を引き起こすと考えられている。プレセニリン遺伝子、PS1およびPS2のような他の遺伝子における突然変異は、間接的にAPPのプロセシングに影響をおよぼして、長型のベータ−アミロイドの増量を引き起こすと考えられている(Hardy, TINS 20, 154, 1997参照)。これらの観察はベータ−アミロイド、具体的にはその長型がアルツハイマー病の原因要素であることを示している。
【0008】
自己凝集の証拠のある他のペプチドあるいはタンパクも知られており、これらに限られないが、例えばアミリン(amylin)(Youngら, 1994);ボンベシン(bombesin)、セルレイン(cerulean)、コレシストキニンオクタペプチド(cholecystokinin octapeptide)、エレドイシン(eledoisin)、ガストリン(gastrin)関連ペンタペプチド、ガストリンテトラペプチド、ソマトスタチン(somatostatin)(還元型)、サブスタンスP;およびペプチド、黄体形成ホルモン放出ホルモン、ソマトスタチンN−Tyr(Banks and Kastin, 1992)が挙げられる。
【0009】
アミロイド線維に関する論文は、円筒型のβ−シートが、X線および電子顕微鏡データのあるものと一致する唯一の構造であることを示し、アルツハイマーAβ断片および変異体の線維は、多分二つあるいは三つの同心の円筒型β−シートより成る事を示している(Perutz等, 2002)。完全なAβペプチドは42の残基を含んでいるが、これは円筒型の殻の核を構成する丁度よい数である:この発見およびプロリンが存在しないAβペプチドから成るβ−シートにおける多様で強い静電的相互作用があり得ることが、Aβペプチドのアルツハイマー病患者において見出される細胞外アミロイドプラクを形成する性質を説明する。もしこの解釈が正しければ、アミロイドは中央に水が充填した腔を有する細い管(ナノチューブ)から成っている。インビトロにおけるアミロイドプラークの成長の可逆性は、プラーク内と溶液中におけるβAの定常的平衡を示唆する(Maggio and Mantyh, 1996)。βAのポリメリゼーションは、ペプチド−ペプチド相互作用に依存してβ−プリーツシート原線維を形成すること、およびその反応に他のタンパクがおよぼす促進作用によりアミロイド形成が調節をうけうるかも知れないことが示唆されている。アミロイド形成に干渉することの出来る物質を発見しようとする多くの試みが行われた。最も深く研究された化合物としては、抗体、プロリンのようなベータ−破壊性のアミノ酸から構成されるペプチド、認識モチーフへの荷電基の添加、および構築ブロックとしてのN−メチル化アミノ酸の利用が挙げられる(Gazitによる総説, 2002)。
【0010】
DとLのアミノ酸残基が交互に存在する環状ペプチドは、それ自身を膜に挿入し、膜を脱分極させることにより細菌を殺すナノチューブを形成する(Perutzら、2002)。あるアミロイド線維は導体であって同じ機構で細胞を殺すかも知れないとする示唆もある。
【0011】
ヘリックスのターンの間の間隙にそれ自身を挿入することができるコンゴーレッドのような芳香族化合物は、円筒の殻を不安定にし、このプロセスを開始するかも知れないが、阻止する方が、より有効であり、多分達成しやすいであろう(Perutzら、2002)。
【0012】
本明細書におけるいかなる文書の引用も、そのような文書が関連する先行技術であること、あるいは本出願の請求項の特許性に対して重要であると考えられると、認めることを意図するものではない。いかなる文書の内容あるいは日付に関するいかなる言明も、出願人が出願時に利用できる情報に基づいており、そのような言明の正しさを認めるものではない。
【0013】
発明の概要
本発明は、脳内あるいは体内の他の場所におけるアミロイド沈着に付随する炎症ならびに活性化されたミクログリアにかかわる脳の炎症を抑制または治療するための方法を提供する。その方法は、それを必要とする患者に、野生型の線維状バクテリオファージ、または抗体もしくは非線維状バクテリオファージ抗原をその表面に提示しない線維状バクテリオファージの有効量を投与することを含む。
【0014】
本発明は、また活性成分として線維状バクテリオファージの有効量を含む医薬組成物を提供する。
【0015】
本発明は、更に線維状バクテリオファージを斑形成ペプチドまたは既に形成されたアミロイド沈着物と接触させることにより、アミロイド沈着の形成を抑制し、または既に形成されたアミロイド沈着を脱凝集する方法を提供する。
【0016】
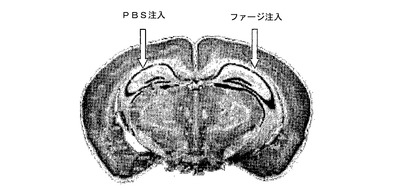
図1は、hAPPトランスジェニックマウスにファージ/PBSの頭蓋内投与を示す脳の断面の模式図である。ファージを5匹のトランスジェニックマウスに脳内注入した。対側半球にPBSを注入した。マウスは、1時間、2日、3日後にと殺した。それらの脳を5μmの切片に切り、斑の量を評価するためにチオフラビン−Sで染色した。
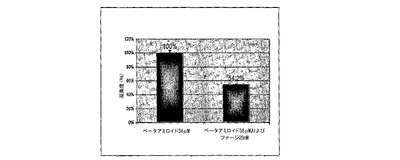
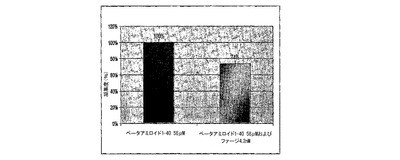
図2Aおよび図2Bは、ThTアッセイを用いて線維状ファージによるβ−アミロイド1−40凝集の脱凝集(図2A)および阻止(図2B)を示すグラフである。図2Aにおいて(脱凝集試験)、β−アミロイド(58μM)を単独で21日間インキュベートした。21日目に異なった濃度のファージを加え一晩インキュベートした。図2Bにおいて(阻止試験)、β−アミロイド(58μM)を単独かあるいは線維状ファージと共に7日間インキュベートし、サンプルをThT溶液に加え、分光光度計によりアミロイド量を測定した。アミロイド含量は、アミロイド原線維と結合するチフラビン−Tを用いて測定した。その蛍光発光は485nm波長で検出した。
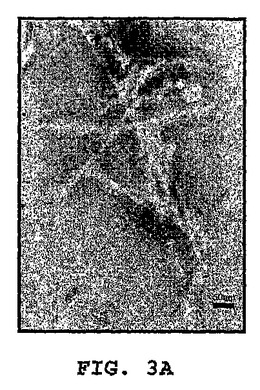
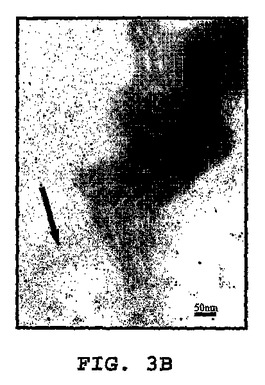
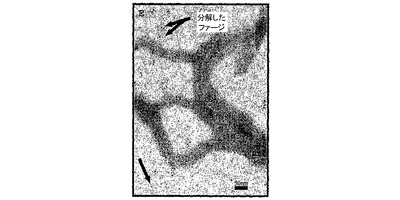
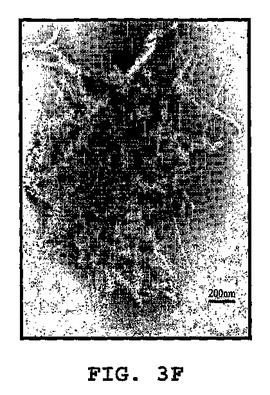
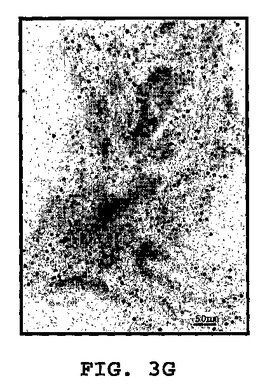
図3A〜3Kは、線維状ファージの存在または非存在下でインキュベートしたβ−アミロイドの電子顕微鏡写真である(倍率 x100K)。図3A:β−アミロイド97μM、PBS中ファージの非存在下。β−アミロイドはマウスモノクローン抗体10D5で染色し、次いでヤギ抗マウス12nm金結合抗体で染色した。図3B〜3C:0.5nM(1x1010)ファージとインキュベートした、β−アミロイド97μM。β−アミロイドはマウスモノクローン抗体10D5および12mn金粒子と結合した二次抗体で染色した。ファージはポリクローナルウサギ抗ファージ血清、次いでヤギ抗ウサギ6nm金結合抗体で標識した。これら二次抗体のシグナルは、ファージまたは一次抗体を含まないサンプルにおいては観察されなかった。図3D:線維状ファージ(fd)(1x1010ファージ)はウサギポリクローナル血清で染色した。二次抗体は6nm金粒子と結合させた。図3E:β−アミロイド近傍の線維状ファージ。ペプチドは、抗ファージ抗体の散在する標識が示すように(矢印)、多分ファージの分解を増大する。この標識は、ファージを単独でインキュベートした時には存在しない(サンプルD)。図3F〜3G:β−アミロイドのみを37℃で10日インキュベートした後(倍率:F−x30K,G−x100K)。図3H〜3I:β−アミロイドを単独で10日間インキュベートし、10日目に線維状ファージを加えて更に16時間インキュベートした(H−X30K,I−Hのx100Kの拡大)。図3J〜3K:アミロイド原線維と線維状ファージの界面。ファージは原線維の軸に沿って並んでいる(×100K)。
図4A〜4Cは、球形ファージを既に凝集したβ−アミロイドに加え、一晩インキュベートしたものの電子顕微鏡写真である。β−アミロイドと球形ファージ(図4Aおよび図4B)。球形ファージ単独(図4C)。(倍率 x100K)。
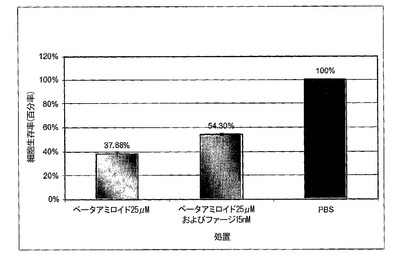
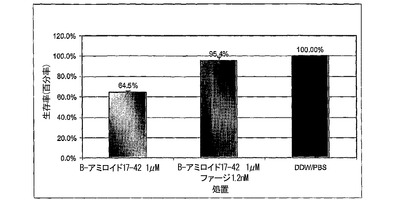
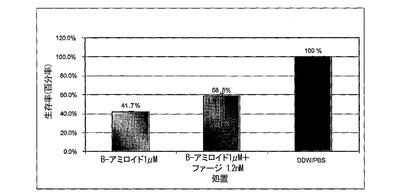
図5A〜5Cは、MTTアッセイを用いた、線維状ファージによるLAN−1細胞に対するβ−アミロイド毒性の阻害を示すグラフである。図5における阻止アッセイに関しては、ファージは25μMのβ−アミロイドに対して1760:1βA/ファージのモル比で加えた。混合物を細胞に加えた。生存100分率は、β−アミロイドの非存在下における細胞の生存率と関連しており(処理−PBS)、100%と考えられる。図5Bおよび5Cにおいて、βA毒性に対して保護するファージの能力を二つの既に凝集させたペプチド:βA1−40およびβA17−42を用いて測定した。両ペプチド(βA1−40およびβA17−42)を37℃で5日間インキュベートした。6日目に線維状ファージを1.2nMの濃度で既に凝集したペプチドに加えて12時間インキュベートし、前日96穴プレートに蒔いた細胞株に加えた。細胞は更に3日間増殖させ、MTTを加えた。3時間後抽出緩衝液を加え、プレートを37℃で一晩インキュベートした。翌日プレートを570nm波長で測定した。
図6Aおよび6Bは、hAPPトランスジェニックマウス(SWE2576, Taconic)の一つの脳半球に注入した線維状ファージ(図6B)と他の半球にPBSのみを注入(図6A)したものを示す。処置3日後にと殺したマウスは、PBS処置半球に比べて、斑量の40%減少を示した。
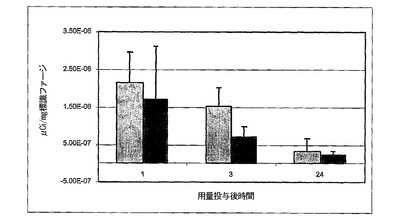
図7は、経鼻投与後のBALB/cマウスにおけるファージの分布を示すグラフである。BALB/cマウスへの経鼻投与後1,3および24時間における脳と嗅球におけるファージ分布の比較は、投与後1時間であってもファージの存在(標識ファージ、μCi/mg)を示していた。それらの濃度はこの1時間の点以後、減少した。
【0017】
発明の詳細な説明
β−アミロイドペプチド(βA)はアルツハイマー病の二つの特徴の内の一つである。このぺプチドは、強い変性剤によってのみ可溶化される脳組織内の線維状の毒性の凝集体を形成する。これらの神経毒性はペプチド凝集型に関連しているので、脳内のアミロイド原線維沈着の程度を、減少または除去する方向への治療法の開発に多くの努力が注がれてきた。
【0018】
生理的な条件下では、合成βAは凝集型を採り、また神経突起からの変化を示し、海馬ニューロンに対する神経毒性を促進する。βAの凝集は、pH、ペプチド濃度、温度およびインキュベーション時間に依存することが示されてきた。
【0019】
本発明者等は、線維状ファージそれ自体が、驚くべきことにインビトロにおいてβAの凝集を阻害し、および既に形成された凝集体を溶解する能力を有することを発見した。
【0020】
本発明者等の研究室においては、構造的および遺伝的レベルの両方でよく理解されている線維状ファージ、M13、f1、およびfd(Greenwoodら、1991)を用いた。この研究室は、線維状バクテリオファージが、ベクターのその不活性な性質と外来分子を輸送する能力の両方を保持しながら、中枢神経系に浸透する性質を示すことを最初に示した(FrenkelとSolomon, 2002)。
【0021】
線維状バクテリオファージは、環状の一重鎖DNAゲノムを含む構造的に関連したウイルスの一群に属する。これらは宿主を増殖性感染中に殺すことはない。Fプラスミドを含む大腸菌(Escherichia coli)に感染するファージは、集合的にFfバクテリオファージと呼ばれる。これらは哺乳動物細胞には感染しない。
【0022】
線維状バクテリオファージは、約1〜2ミクロン長および直径6nmのフレキシブルな桿体であり、DNAコアを囲むタンパクサブユニットのらせん状の殻を持っている。主要な二つのコートタンパク、タンパクpIIIおよび主要コートタンパクpVIII、は提示されたタンパクのコピー数において異なっている。pIIIが4〜5コピー存在するのに対し、pVIIIは約3000コピー見つかっている。約50残基を有する主要コートタンパクpVIIIサブユニットは主としてアルファヘリックスであり、そのアルファヘリックスの軸は、ウイルス粒子の軸と小角をなしている。タンパク殻は3つの部分から構成されると考えられている:周囲の溶媒と相互作用し、ウイルス粒子の等電点を低くする酸性残基に富んだ、サブユニットのN末端領域によって占められている外表面;タンパクサブユニットが主として相互に作用する、非極性側鎖の19残基のストレッチを含む殻の内側;およびDNAコアと相互作用をする塩基性残基に富んだ、サブユニットのC末端領域によって占められる内側の表面である。実際、全てのタンパク側鎖の相互作用は、サブユニット内で起こるのではなく、コートタンパクアレイにおける異なったサブユニット間で起こるという事実は、これを高分子の集合におけるアルファ−ヘリックスサブユニット間の相互作用の研究のための有用なモデルシステムにすることができる。線維状バクテリオファージの特異な構造が、約16.3MDの大きさを有しているにもかかわらず脳内への侵入を可能にし、ファージの構造がアミロイド原線維自身に似ていることから、βA線維化に干渉する能力に寄与していると考えられる。
【0023】
上述の事を勘案して、本発明者等は、β−アミロイドペプチドの凝集過程に干渉する線維状ファージの能力を調べ、野性型の線維状ファージをβ−アミロイドペプチドとインビトロにおいて、異なる時間間隔と異なる比でインキュベーションすることが、β−アミロイドの阻止および/あるいは脱凝集を引き起こすことを発見した。更に線維状ファージは細胞の生存に対して保護効果を示す。
【0024】
最も興奮するようなデータが、線維状ファージ−β−アミロイド原線維を、スライドグラス上で増殖させたマイクログリア細胞とインキュベートした後に得られた。もしβ−アミロイドがマイクログリアを活性化させるなら、ファージは、マイクログリアを活性化させずにそれを溶解する。ファージ技術は、β−アミロイドの抗凝集剤の新しい事実上無限の原料を提供し、Fc受容体を介してマイクログリアを過剰に活性化させうる抗体の有害な効果を阻止する。
【0025】
バクテリオファージは、遺伝子および/または送達媒体として動物ウイルスよりも明らかに有利である。それらは、動物ウイルスベクターよりも大規模生産および精製を非常に効率よく安価に行うことができる単純なシステムだからである。更に、DNAの大きな部分をファージミドベクターへ効率よくパッケージすることができる。原核生物の感染、構築および複製のために進化してきたことから、バクテリオファージは哺乳動物細胞内で複製もできないし、天然の指向性も有していない。このことで、非特異的な遺伝子送達の可能性は極めて低い。ファージベクターは、動物細胞において複製可能な構成要素をつくる可能性は低いことから、ウイルスよりも潜在的にずっと安全である(Monaciら, 2001)。
【0026】
本発明はアミロイド沈着に付随する、または活性化されたマイクログリアにかかわる脳の炎症を抑制または治療する方法を提供する。更に本方法は、体内の脳以外の場所におけるアミロイド沈着に関連した炎症、例えば多発性骨髄腫や腎アミロイドーシスを抑制または治療する。
【0027】
本方法は、必要とする患者に、野生型の線維状バクテリオファージ、または抗体もしくは非線維状バクテリオファージ抗原をその表面に提示しない線維状バクテリオファージの有効量を導入/投与することを含む。線維状バクテリオファージは、M13、f1またはfdのような線維状バクテリオファージのいずれであってもよい。以下の実施例においてはM13が用いられたが、いかなる他の線維状バクテリオファージであっても、同様な構造と95%を越えるゲノム同一性があることから、同じように行動し、機能することが期待される。
【0028】
本方法を脳の炎症を抑制または治療のために用いる場合、線維状バクテリオファージは、好ましくは活性成分を受容者の嗅覚系を通して受容者の体内に導入するために、経鼻的に投与することが望ましい。
【0029】
本方法は、斑形成疾患において、タンパクが凝集してアミロイド斑または沈着物となるのを抑制するのみならず、また既に形成されたアミロイド沈着、例えばβA原線維を脱凝集させるのに有効である。
【0030】
βA原線維形成または脱凝集に関して、線維状バクテリオファージの抗凝集または脱凝集性は、よく知られたチオフラビンT(ThT)結合アッセイによって測定することができる。βA原線維構造の形成の崩壊、および既に形成されたβA原線維の脱凝集は、ThTの蛍光の実質的に減少することによって示される。
【0031】
本明細書とそれに付随する請求項の目的上、「患者」、「対象」および「受容者」という用語は同義で用いられる。それらの用語には、予防的、実験的または治療的処置の対象である、ヒトおよび他の動物が含まれている。
【0032】
本明細書とそれに伴う請求項の目的上、「ベータアミロイドペプチド」は「β−アミロイドペプチド」、「βAP」、「βA」および「Aβ」と同義である。これらの用語の全ては、アミロイド前駆タンパクから由来した斑形成ペプチドを言う。
【0033】
本明細書においては、「PrPタンパク」、「PrP」、「プリオン」は、適切な条件下で斑形成疾患の原因である凝集体の形成を導くことのできるポリペプチドを言う。例えば、正常細胞プリオンタンパク(PrPC)はそのような条件下で、対応するスクレイピーイソ型に変換するが、それはウシ海綿状脳症(BSE)または狂牛病、ネコ海綿状脳症、クールー、クロイツフェルトヤコブ病(CJD)、Gerstmann-Streussler-Sheinker病(GSS)および致死性家族性不眠症(FFI)のような、これに限らないが、斑形成疾患の原因である。
【0034】
本明細書では「脱凝集」とは、凝集タンパク、典型的には非共有結合によって凝集しているタンパクの可溶化を言う。
【0035】
「治療する」という用語は、疾患の進行を実質的に抑制し、遅らせあるいは逆行させること、疾患の臨床症状を実質的に改善すること、あるいは疾患の臨床症状の出現を実質的に阻止することを意味するよう意図されている。
【0036】
また、本明細書においては、「斑形成疾患」は、これらに限らないが、早発性アルツハイマー病、遅発性アルツハイマー病、発症前アルツハイマー病、SAAアミロイド症、遺伝的アイスランド症候群、老化、多発性骨髄腫、およびヒトに影響することが知られているプリオン病、例えばクールー、クロイツフェルトヤコブ病(CJD)、Gerstmann-Streussler-Sheinker病(GSS)および致死性家族性不眠症(FFI)、および動物に影響することが知られているプリオン病、例えばスクレイピーおよびウシ海綿状脳症(BSE)であって、ベータアミロイド、血清アミロイドA、シスタチンC、IgGカッパ軽鎖またはプリオンタンパクのような凝集タンパク(斑形成ペプチド)による斑形成を特徴とする疾患を言う。
【0037】
上に述べた疾患にかかわるアミロイド斑(アミロイド沈着としても知られている)のほとんどは、脳内に存在するので、いかなる提案される治療様式も、血液脳関門(BBB)を越える能力およびアミロイド斑を溶解する能力を示さなければならない。通常BBBを透過できる分子の平均サイズは約2kDaである。
【0038】
嗅覚の欠陥と中央嗅覚経路における退行的変化がADの臨床経過の初期に影響を受けていることを示す証拠が増えつつある。更にADに関与している解剖学的パターンは、嗅覚経路がADの進展における初期段階でありうることを示唆している。
【0039】
嗅覚受容体ニューロンは、鼻腔の上皮層に存在する双極性の細胞である。それらの軸索は篩板を横切り、脳の嗅球における嗅覚経路の最初のシナプスに突出する。この立体構造により、ウイルスや他の輸送される物質がBBSを通ってCNSに到達することができる通路となっている。
【0040】
以前に示したように、鼻腔内投与(Mathisonら、1998; Chouら, 1997; Draghiaら, 1995)によって、ウイルスおよび高分子が脳脊髄液(CFS)あるいはCNSに直接入ることが可能である。
【0041】
嗅覚受容体ニューロンをアデノウイルスベクターの脳への送達点として使用した例が、文献に報告されている。この方法により、見掛けの毒性なしに、脳内で12日間レポーター遺伝子が発現したことが報告されている(Draghiaら、1995)。
【0042】
従って、本発明の好ましい実施例によれば、斑形成疾患に付随するポリペプチドの凝集体の形成を脱凝集または阻止することが可能な、またはマイクログリアの活性化を抑制することが可能な線維状バクテリオファージは、この経路を介して脳へ送達される。
【0043】
Aβは、末梢組織の細胞によって絶え間なく生産され、血管脳関門(BBB)を越えて、特定のニューロン集団において局在する毒性効果を発揮するので、そのような媒体の経鼻投与は、斑を形成できる末梢Aβの量を最小限にすることにより疾患の進行をも阻止することができる。
【0044】
本発明の医薬調製品には、活性成分として、野性型の線維状バクテリオファージ、または抗体もしくは非線維状バクテリオファージ抗原をその表面に提示しない線維状バクテリオファージが含まれる。該医薬調製品はまた、異なった線維状バクテリオファージの混合物であってもよい。
【0045】
本発明の調製品は、それ自体を、または適切な担体または賦形剤と混合した医薬組成物として、生命体に投与することができる。
【0046】
本明細書においては、「医薬組成物」とは、本明細書に記載された一つまたは複数の活性成分と生理的に適切な担体および賦形剤のような他の化学成分との調製品を言う。医薬組成物の目的は、生命体への化合物の投与を容易にすることである。
【0047】
本明細書において「活性成分」という用語は、生物学的効果を発揮する調製品を言う。
【0048】
今後、「生理的に許容される担体」および「医薬的に許容できる担体」の句は、互いに同じ意味で用いられるが、生命体に有意な刺激を与えず、投与される化合物の生物学的活性および性質を損なわない担体または希釈剤を言う。補助剤はこれらの句の中に含まれる。
【0049】
本明細書において、「賦形剤」という用語は、活性成分の投与を容易にするために医薬組成物に加えられる不活性物質を言う。賦形剤の例としては、炭酸カルシウム、リン酸カルシウム、種々の糖類およびデンプン類、セルロース誘導体、ゼラチン、植物油およびポリエチレングリコールが挙げられるがこれに限定されない。
【0050】
薬剤の処方および投与に関する技術は「Remington's Pharmaceutical Sciences, レミントンの薬剤化学」Mack Publishing Co., Easton, PA, 最新版に捜し出すことができ、参照することにより本明細書に盛り込まれる。
【0051】
適切な投与経路としては、例えば、経口、経直腸、経粘膜、特に経鼻、経腸、または筋肉内、皮下および髄内注射を含む非経口送達、ならびに髄腔内、直接脳室内、静脈内、腹腔内、鼻腔内あるいは眼内注射が挙げられる。
【0052】
あるいは、調製品を全身ではなく、局所的に投与してもよく、例えば、患者の脳に直接調製品を注射する方法が挙げられる。
【0053】
本発明の医薬組成物は、当技術においてよく知られた方法によって製造してもよく、例えば、従来の混合、溶解、造粒、糖衣錠製造、すり潰し、乳化、カプセル化、封入または凍結乾燥法が挙げられる。
【0054】
本発明に従って使用するための医薬組成物は、活性成分を加工して医薬的に使用できる調製品とすることを容易にする賦形剤および補助剤を含む一つあるいは複数の生理的に受容できる担体を用いて、従来の方法で製剤化することができる。適切な剤形は選択される投与経路に依存している。
【0055】
注射剤としては、本発明の活性成分は、水溶液中で、好ましくはHank's溶液、リンゲル液または生理的塩類緩衝液のような生理的に適合する緩衝液中で、製剤されてもよい。経粘膜投与のためには、透過すべき関門に適合した浸透剤がその剤形に用いられる。そのような浸透剤は当技術分野において一般的に周知である。
【0056】
経口投与のためには、化合物は、活性成分と当技術分野において周知の、医薬的に許容できる担体を組み合わせることにより、容易に製剤することができる。そのような担体により、本発明の化合物は、患者が経口的に摂取するための錠剤、丸剤、糖衣剤、カプセル剤、液剤、ゲル剤、シロップ剤、スラリー剤、懸濁剤等として製剤されうる。経口使用される薬理学的調製品は、固体賦形剤を用いて製造可能であり、場合によっては得られた混合物を粉砕し、必要であれば適切な補助剤を加えた後、顆粒の混合物を加工して錠剤または糖衣錠のコアを得る。適切な賦形剤は、具体的には、乳糖、ショ糖、マンニトールまたはソルビトールを含む、糖などの充填剤;トウモロコシデンプン、小麦デンプン、米デンプン、ジャガイモデンプン、ゼラチン、トラガカントゴム、メチルセルロース、ヒドロキシプロピルメチル−セルロースおよびカルボキシメチルセルロースナトリウムのようなセルロース調製品;および/または生理的に許容できる、ポリビニルピロリドン(PVP)のような高分子が挙げられる。必要に応じて、架橋ポリビニルピロリドン、寒天、またはアルギン酸もしくはアルギン酸ナトリウムなどのその塩の崩壊剤を加えてもよい。
【0057】
糖衣錠のコアには、適切なコーティングを施す。このために、濃縮糖溶液を用いてもよいし、場合によっては、アラビアゴム、タルク、ポリビニルピロリドン、カルボポールゲル、ポリエチレングリコール、二酸化チタン、ラッカー溶液、および適切な有機溶媒または溶媒混合物を含んでもよい。活性化合物用量の異なった組み合わせを同定、識別するために、染料または顔料を錠剤や糖衣錠のコーティングに加えてもよい。
【0058】
経口的に用いられる医薬組成物としては、ゼラチンから作られたプッシュフィットカプセル剤やゼラチンおよびグリセリンまたはソルビトールのような可塑剤から作られた、軟、密閉カプセル剤が挙げられる。プッシュフィットカプセル剤は、活性成分を、乳糖のような充填剤、デンプンのような結合剤、タルクやステアリン酸マグネシウムのような潤滑剤、そして場合によっては安定剤と混合して含んでもよい。軟カプセル剤においては、活性成分は油脂、流動パラフィン、または液体ポリエチレングリコールのような適切な液体に溶解あるいは懸濁してもよい。更に安定剤を加えてもよい。経口投与用のすべての製剤は、選択した投与経路に適した剤形でなければならない。
【0059】
頬側投与のためには、組成物は従来の方法で製剤化された錠剤またはロゼンジの剤形を取ってもよい。
【0060】
鼻腔吸入による投与のためには、本発明に従って使用する活性成分は、加圧されたパックからのアエロゾル噴霧の形で、または例えばジクロロジフロロメタン、トリクロロフロロメタン、ジクロローテトラフロロエタンまたは二酸化炭素のような適切な噴霧剤を用いた噴霧器の形でするのが好都合である。加圧アエロゾルの場合は、単位用量は、計量した量を送達する弁を備えることにより決定してもよい。ディスペンサーで使用される、例えばゼラチンのカプセルおよびカートリッジは、化合物と、乳糖またはデンプンのような適当な粉末基剤の混合粉末を含む剤形であってもよい。
【0061】
本明細書に記載される調製品は、非経口投与、例えばボーラス注射または連続注入による剤形であってもよい。注射用の剤形は、例えばアンプルでまたは多回数投与用の容器で場合によっては保存剤を加えて、単位用量で提供してもよい。組成物は、油性または水性の媒体中の懸濁剤、溶液剤または乳剤であってよく、懸濁剤、安定剤および/または分散剤のような製剤用の物質を含んでもよい。
【0062】
非経口投与のための医薬組成品としては、水溶性の活性調製品の水溶液が挙げられる。更に、活性成分の懸濁剤を、適当な油性または水性基剤懸濁注射剤として調製してもよい。適当な脂溶性の溶媒または媒体としては、ごま油のような油脂、またはオレイン酸エチルのような合成脂肪酸エステル、トリグリセリドまたはリポソームが挙げられる。水性の注射懸濁剤は、ナトリウムカルボキシメチルセルロース、ソルビトールまたはデキストランのような懸濁剤の粘度を上げるような物質を含んでもよい。場合によっては懸濁剤は、適当な安定剤、または高濃度の溶液の調製を可能にする活性成分の溶解度を増加させる物質を含んでもよい。
【0063】
あるいは、活性成分は、例えば無菌の発熱成分を含有しない水溶液のような適当な媒体により使用前に構成する粉末剤形でもよい。
【0064】
本発明の調製品は、ココアバターまたは他のグリセリドのような通常の坐剤基剤を用いた坐剤、または保留浣腸のような直腸用の組成物に製剤化してもよい。
【0065】
本発明の範囲で適切に使用される医薬組成物としては、意図する目的を達成するために有効な量の活性成分を含んでいる組成物が挙げられる。より具体的に言えば、治療有効量とは、疾患の症状を阻止、軽減または改善する、あるいは治療を受けている対象の生存期間を延長するのに有効な活性成分の量を意味する。
【0066】
治療有効量の決定は、特に本明細書で提供された詳細な開示の観点から、当業者の能力の範囲内に十分ある。
【0067】
本発明の方法において使用されるいかなる調製品に対しても、治療有効量または投与量は、最初はインビトロおよび細胞培養アッセイから推測することができる。例えば投与量は、循環中の所望の抗体濃度または力価を達成するために動物モデルにおいて式化できる。そのような情報は、ヒトでの有用な投与量をより正確に決めるために使用される。
【0068】
本明細書に記載される活性成分の毒性と治療効果は、インビトロにて、細胞培養または実験動物において標準的な医薬手法で決定することができる。これらインビトロでの、および細胞培養アッセイおよび動物実験から得られるデータは、ヒトにおいて用いられる投与量の範囲を計算するために使用される。投与量は使用された剤形および投与経路に依存して変わりうる。正しい剤形、投与経路および投与量は患者の状態を考慮して個々の医師によって選択されることができる(例えば、Finglら「治療の薬理学的基礎」、第1章p.1(1975)参照)。
【0069】
投与量と投与間隔は、凝集を阻止し、既に存在する凝集体を脱凝集させるために十分な線維状バクテリオファージの血漿中または脳内レベル(最小有効濃度MEC)を与えるために個人ごとに調整できる。MECはそれぞれの調製品によって異なるであろうが、そのインビトロデータから推定することができる。MECを達成するために必要な投与量は、個々の特徴と投与経路に依存するであろう。結合アッセイを、血漿中濃度を決定するために用いることができる。
【0070】
投与間隔もまた、MEC値を用いて決定することができる。調製品は、10〜90%、好ましくは30〜90%、最も好ましくは50〜90%の時間、血漿中レベルをMECの上に維持するレジメンを用いて投与されるべきである。
【0071】
治療する状態の重度と反応性によつて、投与形態は、単回投与または複数回投与であることができ、治療コースは、数日から数週間、または治癒する、もしくは疾患状態の減少が達成されるまで継続される。
【0072】
投与されるべき組成物の量は、もちろん、治療対象、疾患の重度、投与方式、処方する医師の判断等に依存する。
【0073】
本発明の組成物は、必要に応じて、活性成分を含む1つ以上の単位投与量剤形を入れたFDA認可キットのようなパックまたはディスペンサー器具で提供されてもよい。該パックは、ブリスターパックのような金属またはプラスチックのフォイルを含んでいてもよい。パックまたはディスペンサー器具は、投与法についての指示書を添付していてもよい。パックまたはディスペンサー器具はまた、容器に付随した通知を伴ってもよく、その通知は医薬品の製造、使用および販売を規制する政府機関によって指定された形式で、組成の形態もしくはヒトまたは動物投与用にその機関から受けた認可を反映している。そのような通知は、例えば、米国食品医薬品局によって認可された処方医薬品のラベルであってもよいし、また認可製品の添付文書であってもよい。適合する医薬用担体中に製剤された本発明の調製品を含む組成物もまた調製して、適切な容器中に入れ、上に述べたように表示条件の治療のためのラベルを付けてもよい。
【0074】
本発明を一般的に説明したが、同じことを次の実施例を参照することでより容易に理解
できるであろうが、実施例は例示するために示しているものであり、本発明を限定することを意図しているものではない。
【0075】
実施例
材料と方法
ファージの増殖および精製
線維状ファージ(M13)は50μg/mlカナマイシンを含む2YTブロス中の形質転換TG1培養物から調製した。細菌細胞を遠心により沈殿させ、澄明な上清をデカンテーションにより得た。2.5M NaCl中の20%ポリエチレングリコール(分子量8000Da)溶液の当初量の20%容を加えて、4℃で3時間静置してファージを上清から沈殿させた。ファージを遠心分離して(9000rpm、1時間)ペレットとし、その後上清の3%容のPBSに再懸濁した。細菌の残渣は更に7000rpmで遠心分離して除き、ファージを、ポリエチレングリコールを加えて沈殿させて再濃縮した。ペレットを最終的に増殖培地の当初量の0.001容のPBSに再懸濁した(Hartら, 1994)。
【0076】
β−アミロイド凝集へのファージの干渉:
βA凝集を阻止し、既に形成された凝集体を脱凝集するファージの能力を、三つの方法で分析した:
【0077】
1.チオフラビン−T蛍光アッセイ
βA凝集を、アミロイド原線維との結合により発光スペクトルが変化するチオフラビン−T(ThT)色素を用いて測定した(LeVine, 1993)。凝集体形成を阻止するファージの能力を評価するために、βA1−40 577μM(Bachem)8μlを37℃で14日間、単独で(PBSで80μlに希釈し、最終濃度58μMとした)または線維状ファージと共に(3x1012ファージ/ml、72μl)インキュベートした。それぞれの反応混合液(20μl)の蛍光を、50mMグリシン緩衝液、pH9、中のThT(2μM)980μlを加えた後測定した。蛍光はPerkin-Elmer Model LS-50蛍光光度計を用いて、励起波長435nmおよび発光波長485nmで測定した。
【0078】
ファージの脱凝集活性は以下のようにして検査した:βA(58μM)を37℃で21日間インキュベートして凝集を促進した。ファージを加え(ファージ濃度1014ファージ/mlの溶液と等量)、凝集したβAと共に更に17時間インキュベートした。凝集の程度を上記のごとくThT蛍光を用いて評価した。
【0079】
2.電子顕微鏡(EM):
β−アミロイドと線維状ファージの相互作用。βA凝集のレベルをEMを用いて可視化した。βA形成の阻止については、ペプチド(289μM)10μlを、単独で(PBS20μlを加えた)または各種ファージ濃度(以下の濃度のファージ20μl:1x1014ファージ/ml、1x1012ファージ/mlおよび1x1010ファージ/ml)で、37℃で9日間インキュベートした。
【0080】
ファージの脱凝集活性は、各種濃度のファージを、既に凝集したβAに加えることによって証明した。PBSに溶解したβ−アミロイド1−40(193μM)の20μlを37℃で10日間インキュベートした。10日目にPBSまたは各種濃度のファージ(1x1014ファージ/ml、1x1012ファージ/ml)10μlをサンプルに加え、更に16時間インキュベートした。
【0081】
阻止および脱凝集アッセイの両方において、ファージとβAをカーボン蒸着被膜をほどこしたフォルムバー(formvar)200#グリッドに付着させた。アミロイドおよびファージの免疫標識を、種々のサイズの金結合抗体(Linら、1999)を用いて行ったが、これはβ−アミロイド原線維と、アミロイド線維と似ている可能性のあるファージを容易に識別するためである。β−アミロイド原線維はモノクローナル抗体(mAb)105Dおよび二次抗体としてヤギ抗マウス−12nm金粒子結合抗体(Electron Microscopy Sciences, Washington, USA)で染色した。ファージを標識するために、ファージに対するウサギポリクローナル血清を用い、二次抗体を6nm金粒子と結合させた(Electron Microscopy Sciences)。サンプルは酢酸ウラン(Sigma)水溶液(2%重量/体積)でネガティブ染色を行った。グリッドはJEOL1200EX電子顕微鏡を用いて、拡大率30Kおよび100Kで可視化した。
【0082】
β−アミロイドとクロロホルム処理ファージとの相互作用
ファージ(1014ファージ/ml)200μlを200μlのクロロホルムに加えた。溶液を室温で3分間に6回ボルテックス処理を行った(それぞれ10秒)(Griffithら、1981)。遠心管を13,200rpmで遠心分離し、水相を別の管に移し、ドラフト中に置き、残っているクロロホルムを蒸発させた。β−アミロイドと球形ファージの間の相互作用を電子顕微鏡によって分析した。
【0083】
β−アミロイド(97μM)を37℃で13日インキュベートした。13日目にPBSまたは各種濃度のS−ファージ(クロロホルム処理)(5x1013ファージ/ml、5x1011ファージ/ml)をサンプルに加え、更に16時間インキュベートした。β−アミロイドの凝集の程度はJEOL1200EX電子顕微鏡で先に述べたように可視化した。
【0084】
3.MTTアッセイ:
ヒト神経細胞株の生存率に対するβAの毒性からファージによる保護を評価するために、MTTアッセイを行った。アッセイは生存している細胞内の、3−(4,5−ジメチルチアゾールー2−イル)−2,5−ジフェニルテトラゾリウムブロマイド(MTT)のMTT−ホルマザンへの還元に基づいている。MTT−ホルマザンの濃度は、分光光度計により570nmで測定したが、細胞の生存率と直接相関していた。
【0085】
LAN−1ヒト神経細胞株は、10%ウシ胎児血清と100ユニット/mlペニシリン/ストレプトマイシンを補充したRPMI中で培養し、37℃、5%CO2下でインキュベートした。神経毒性アッセイのために、培養したLAN−1細胞を96穴プレートに104細胞/100μl/ウエルの密度で播種した。投与量依存性の神経毒性を、β−アミロイド(289μM)サンプル単独で、またはβ−アミロイド(289μM)とファージ濃度を変えて測定した。
【0086】
βAの神経毒性に対するファージ保護効果を試験するために、ペプチド2μlを8μlの各種濃度のファージ(5x1013/ml、5x1012/ml、5x1011/mlファージ)と37℃で4日間インキュベートした。細胞を播種してプレートに接着してから24時間後にサンプルを加えた。プレートを37℃で2日間インキュベートした後、細胞の生存率を、先に記述したように3−(4,5−ジメチルチアゾール−2−イル)−2,5−ジフェニルテトラゾリウムブロマイド(MTT)を用いて細胞の酸化還元活性を測定して評価した(Solomonら, 1997)。MTT(Sigma)を最終濃度1mg/mlでウエルに加え、細胞と共に更に37℃で3時間インキュベートした。細胞溶解緩衝液(20%重量/体積SDSを含む50%ジメチルホルムアミド溶液、pH7.4)を加え、プレートを一晩37℃でインキュベートした。MTTの還元は、自動マイクロプレート分光光度計を用いて570nmのODの変化により比色法で測定した。
【0087】
既に形成されたβA原線維の脱凝集アッセイにおいて、神経毒因子として二つのペプチド(β−アミロイド17−42およびβ−アミロイド1−40)を用いた。凝集したペプチドから細胞生存を守るファージの能力は以下に示すように試験した。第一の実験において、細胞を既に凝集したβA1−40と5日間インキュベートしたが、一方第二の実験においてはβ−アミロイド17−42を神経毒性ペプチドとして用いた。先に述べたように、細胞を96穴プレートに104細胞/100μl/ウエルの密度で播種し、細胞を24時間付着させた後にサンプルを加えた。凝集ペプチド(288μM−保存溶液)2μlに8μlのファージ(1014ファージ/ml、1013ファージ/ml、1012ファージ/ml)を加えて一晩インキュベートした。サンプルを細胞に加えて、更に2日間インキュベートした。MTTを最終濃度1mg/mlで加え、MTTホルマザン含量の測定のために、3時間後に溶解緩衝液を加えた。細胞の生存率は570nmにおけるOD変化を定量化して評価した。
【0088】
線維状ファージのインビボ投与
ADモデル、野生型およびトランスジェニックマウスを線維状ファージで攻撃した:
【0089】
抗凝集性の評価
抗凝集剤としてのファージ活性の最大能力を評価するために、ファージをトランスジェニックマウス(Taconic, Appswe(2576),10ヶ月齢)の頭蓋内に以下のようにして注射した:
【0090】
線維状ファージ溶液(1014ファージ/ml)2.5μlを10分かけて一方の半球へ(Bregma−2.8mm、側方2.5mm、腹側2.5mm)注射し、一方対側にはリン酸緩衝食塩水(PBS)をコントロールとして注射した(図1)。ファージがアミロイド沈着を脱凝集させるに要する時間は知られていないので、処置されたマウスは異なった時間点でと殺した。第一の群は、注射後1時間に4%パラホルムアルデヒドを用いて心臓内灌流を行った。第二の群は投与後2日目に、そして最後の群は3日後に灌流を行った。それらの脳は4%パラホルムアルデヒド中で一晩固定後、マイクロトームを用いて切片とした。チオフラビンーS(ThS)染色を行い、斑量を評価した。この目的のために、切片はMayer'sヘマトキシリンで染色し、核の自己蛍光を抑え、そして洗浄後ThS溶液(1%)を3分間適用した。1%酢酸を20分間用いて差別化を行い、スライドを洗浄後乾燥し、抗退色マウント剤を用いてマウントした。斑量はLEICA Qwinプログラムを用いて計算した。
【0091】
放射能標識線維状ファージの生体内分布
線維状ファージをI125で放射能標識した。ファージを、結合していないヨウ素からG25セファデックスカラムを用いて精製し、先に記載したように、溶出液をポリエチレングリコール(PEG)を用いて更に沈殿させた。9匹のBALB/cマウスを3群に分けた。それぞれのマウスに経鼻的に9マイクロキュリーI125で標識されたファージ100μl(1.25x1012ファージ)を1時間かけて与えた。第一のマウス群は4%パラホルムアルデヒドを用いた心臓内灌流1時間後にと殺した。第二の群は3時間後そして最後の群は24時間後にと殺した。灌流後、脳および末梢器官を取り出し、それらのガンマ線を測定した。
【0092】
線維状ファージの経鼻投与
線維状ファージの効果を十分に評価するために、ファージをSWE/APP2576トランスジェニックマウス(Taconic、10ヶ月齢)に経鼻的に投与した。ファージ溶液(5x1012/ml)20μlを2週間毎に4ヶ月間投与し、認識機能を評価した。9回目の接種後、記憶改善におよぼすファージの影響を研究するために、対象認識試験を行った。第一日目に、マウスを二つの新しい対象に20分間触れさせた。翌日一つの対象を取り替えて、新しい物を探るマウスの好奇心を試験した。認識指標を、各マウスが新しい対象の近くで過ごす時間を、各マウスが二つの対象の近くで過ごす全時間で割った値として計算した。従って、値が0.5を越えることは、古い物を認識し、新しい対象の周りを探査するためにより多くの時間を過ごしていることを示唆している。
【0093】
結果
線維状ファージのインビトロにおける抗凝集性を、アミロイド形成の減少および阻止について評価した。チオフラビンーTアッセイにおいて、ファージはβA原繊維の形成を阻止するよりはβA原繊維の脱凝集においてより有効であった。ペプチドを線維状ファージとモル比1:10,000(ファージ対βA)でインキュベートした時、βA凝集の26%減少が観察された(図2B)。既に凝集したβAにファージ(28nM)を加えた結果、アミロイド原線維の45%減少に至った(図2A)。
【0094】
ファージとβ−アミロイドペプチド間の物理的な相互作用の電子顕微鏡による可視化は、免疫金染色β−アミロイドとファージとのその複合体を用いて行った。ファージがβ−アミロイドペプチドと相互作用をする時は、ファージが存在しない時に比べて集合体はより小さく、より分散していた。
興味あることには、ファージをβ−アミロイドペプチドとインキュベーションすると、ファージが分解した。これはファージ粒子上にないファージの主要コートタンパクに対する抗体の強い標識化から知ることができる(図3C)。この現象は、ファージをβ−アミロイドペプチドと一緒に一晩インキュベートした時(脱凝集アッセイにおいて)、またはファージを単独で同じ期間(図3D)インキュベートした時には観察されなかった。図3(F−K)は、形成されたアミロイド凝集体におよぼすファージの効果を示す。図3Fは、12nm金粒子によって標識された集合体におけるアミロイド原線維を示す。高倍率(図3G)は、異なる大きさの凝集した針状原線維を示す。図3H−Kは、束状になっているファージを示す(矢頭印)が、それはアミロイド針に接着すると分散する(矢)。アミロイドの集合体は小さくそして小さい細いアミロイド原線維を含んでいる。特に興味があるのは、ファージとアミロイド原線維との並行した組織構造である(白い矢)。
【0095】
pVIII分子は3つの立体配置で存在することが示されている。完全なファージにおいては、該タンパクは、少なくとも90%α−ヘリックスであるが、クロロホルム/水の界面に曝されると、形に大きな変化が起こる。これは温度に依存する過程である。2℃では桿形(I−型)であるが、25℃では球形構造(S−型)を取る。S−型への変換はコートタンパクのヘリックス含量の実質的な減少を伴って起こるが、トリプトファン26の環境における有意な変化を伴う(Robert and Dunker, 1993)。
【0096】
クロロホルムによるファージの3分間の処理により、球形のファージはβA凝集の場所に存在しているが、それらは通常原線維の端末に存在しており、可溶化には寄与していないことを示す(図4A−B)。
【0097】
MTT:
線維状ファージがβA神経毒からLAN−1細胞を防御することは、MTTアッセイを用いて示された。ファージをβAとインキュベートした場合、神経細胞の生存率は、βAのみの存在下で増殖した細胞と比較して増加した。細胞培養に加えられたファージの最高量が、最も有意な効果(細胞生存率17%の増加)をもたらした(図5A)。
【0098】
β−アミロイド17−42の毒性からのファージによる保護は、β−アミロイド1−40の毒性を阻止する能力と比較して、より高かった。ファージを既に凝集したβA17−42に加えた場合、細胞の生存率は、βAのみで増殖した細胞と比較して30%増加した(図5B)。同じファージ濃度をβA1−40に加えた場合、細胞生存率は17%増加したのみであった(図5C)。より高い濃度のファージを加えた場合、βA処理を受けた細胞に比較して、細胞生存率を25%に増加させることに成功した。
【0099】
この差に関する可能性のある理由としては、β−アミロイドのN末端がファージ活性を妨害するかも知れない、またはβ−アミロイド17−42はβ−アミロイド1−40よりも早く凝集するという事実に拠るのかも知れない(Pikeら、1995)。β−アミロイド1−40に対するファージの親和性は、ペプチド凝集に伴って増加する。ELISAは、可溶性β−アミロイド、1時間凝集β−アミロイドまたは3週間凝集β−アミロイドと結合するファージについて調べ、βA1−40に結合するファージにおいて有意な差を示した。それ故、高度に凝集したペプチドに対するファージのより高い親和性が、β−アミロイドβ−アミロイド17−42とのより強い相互作用を説明する可能性がある。
【0100】
線維状ファージの脳内注射によるβ−アミロイド斑の脱凝集のインビボでの研究において、注射後1時間でと殺したでマウスにおいては、斑量の減少は観察されなかった。処置後2日目にと殺したマウスにおいては、線維状ファージで処置された半球において、PBS注射半球に比較して、斑量の25%減少が観察された。処置後3日目にと殺したマウスにおいては、ファージ注射を受けた半球において、斑量の40%減少が示された(図6Aおよび6B)。
【0101】
放射能標識線維状ファージの脳内分布:
経鼻投与後1時間で早くも、線維状ファージは嗅球と脳内で検出された(図7)。ファージの消失速度を決定するためには群は小さすぎるが(n=3)、ファージの濃度は1時間後にピークに達し、24時間以内に殆ど完全に除去されると考えられる。マウス間のバラつきが大きいのは、異なった量のファージを飲み込み、吸入する結果であろう。脳実質細胞への侵入量は、与えられた量の0.0009%から0.018%とさまざまであった。血清抗体濃度の0.1%が脳に到達し斑量を減少させることは言及する価値がある。これらの予備的なデータは、BALB/cマウスにおいてファージが短時間で脳に侵入することができ、1時間後に消失が始まることを示している。それ故、線維状ファージの経鼻投与によって、アミロイド沈着を有するトランスジェニックマウス治療の効果を試験することができる。
【0102】
トランスジェニックマウスへの線維状ファージの繰り返し投与:
認識機能における有意な改善が、ファージ処置を受けない群に比較して、ファージ処置を受けたマウスにおいて認められた(データは示されない)。研究の終わりに、治療効果の総合的な推測を得るために、マウスの斑量、マイクログリア活性化、シナプス密度および有害事象の可能性を評価するための組織化学的な分析が行われる。
【0103】
考察
本発明者の研究室は、βAのN末端に部位特異的なモノクローナル抗体(mAbs)が、既に形成されたβA原線維に結合し、アミロイドを脱凝縮させて、無定型状態にし、それらの神経毒性を阻害することを、先に報告した(Solomonら, 1996, 1997)。この活性はまた、ADのためのトランスジェニックマウスにおいて、インビボでも示された(Bardら、2000)。経鼻的に抗−凝集抗体断片を脳斑に送達するために、βAに対するscFvを提示するファージを用いた(Frenkelら, 2002)。トランスジェニックマウスにおいてscFvを脳へ侵入させ、アミロイド沈着と結合させることを可能にする媒体としてファージを用いた。
【0104】
投与回数と、海馬および臭球において検出されたファージの量の間に直接、関連性があることが示された。ファージの線形構造が、種々の膜を通っての侵入性を与えることが示唆されている。ファージ投与後のマウスの脳において、いかなる病理的な兆候も、観察されなかったことは言及する価値がある(Frenkelら、2002)。
【0105】
βA原線維をThTおよび蛍光標識抗ファージ抗体との両方で可視化し、脳から線維状ファージが毒性効果を引き起こさずに消失することが組織学的研究において示された。先に報告された実験において、線維状ファージはマウスに静脈内注射され、その後、異なった器官から回収され、体内における循環の間、その完全性は影響を受けながったことが示された。
【0106】
アミロイドの繊維化は、静電的な相互作用よりは、疎水性の相互作用によって駆り立てられていると見なされている。βAは二つの疎水性部を含んでいるが、その中央部は、17〜21残基と、30〜40残基を含むC末端領域から構成される。固体NMRから得られた実験的な制約に基づいたモデルによれば、ペプチドの立体構造は、残基25〜29によって形成された180°曲げによって分離された二つのβ鎖を含んでいる。該β鎖は、側鎖−側鎖接触を通して相互作用する二つの既知の平行なβ−シートを形成する。疎水性の側鎖は外側の表面に露出しており、疎水性の面を形成する一方、荷電した極性の他の側鎖は反対側の面に、曲がりの凸面および原線維が成長するに連れて溶媒和されるN末端部に分布している(Petkovaら, 2002)。
【0107】
一方、主要ファージコートタンパクは3つの部分より構成されている:外表面(サブユニットのN末端領域によって占められており、周囲の溶媒と相互作用しウイルス粒子に低い等電点を与える酸性残基に富んでいる);殻の内部(タンパクサブユニットが主として互いに相互作用する、非極性側鎖の19残基ストレッチを含む);内部表面(サブユニットのC末端領域によって占められており、DNAコアと相互作用する塩基性残基に富んでいる)。ほぼ全てのタンパク側鎖の相互作用は、サブユニット内ではなく、コートタンパクアレイにおける異なったサブユニット間であるという事実のため、これは、高分子集合体におけるα−ヘリックスサブユニット間の相互作用の研究のための有用なモデルシステムになっている。
【0108】
本研究において、ファージは、脳にβAに対する抗体を送達する媒体としての、報告されている機能に加えて、抗凝集性を有していることが示された。実施例において提出されたデータから、線維状ファージはβ−アミロイドと相互作用し、その凝集過程に干渉し、その可溶化を引き起こすことさえできる。現在、診断は、β−アミロイド斑がすでに形成された疾患の後期になされることから、脱凝集性は大きな価値を有する。アミロイド斑を有するTgマウスにファージを頭蓋内注射した後、最大効果は3日後のみに観察されることから、この過程は時間依存性である。この効果は、電子顕微鏡写真において見ることができるように、ファージの長く薄い線維としての特異な構造がアミロイド原線維にそって組織化するのを可能とする結果であろう。この理論は、線形の構造を失った球状ファージはアミロイドの形成を阻害することができなかった事実によって明らかである。ファージ活性に寄与する主要な別の因子は、ベータシート構造に干渉するかも知れないアルファヘリックス(プロテイン8)の含量が高いことである。pVIIIサブユニットはヘリックスのアレイに封入されて、ssDNAゲノムを囲む管状構造を形成している。pVIIIのC末端は、環状構造の内側に露出しており、負に荷電したssDNAゲノムと相互作用する正荷電の残基を含んでいる。中間部は、サブユニット間の相互作用の原因である疎水性のアミノ酸に富んでいる。最後に、屈曲しやすい負荷電のN末端部は粒子の外側に露出している(Marvin, 1998)。
【0109】
線維状バクテリオファージは、他のベクターよりも明らかな利点を有する。線維状ファージ、M13、f1およびfdは構造的にも遺伝子レベルでもよく理解されている(Wilson and Finley 1998; Rodi and Makowski, 1999)。それらは抗原および/または抗体を提示するために、遺伝子操作されており、その表面に外来タンパクを提示する、異なった生物システムに用いられた(Scott等、1990; McCaffertyら、 1990)。原核生物の感染、集合および複製に関して進化したことから、バクテリオファージは、哺乳動物細胞で増殖することもできず、それに対する天然指向性も示さない。このことは、非特異的な遺伝子送達の機会を最小とする。ファージベクターは、動物細胞において複製可能な実態を作ることは有りそうもないことから、ウイルスよりもより安全である可能性が高い。
【0110】
脱凝集剤としてバクテリオファージを用いる別の利点は、細菌培養の増殖により、より多くの材料を得る事ができることから、製造が容易であることである。放射能標識アッセイによれば、ファージは脳実質細胞に1時間未満で侵入する。それらの消失は多分その後すぐ始まるであろう。投与された用量から侵入したファージの百分率は0.0009%から0.018%までまちまちである。マウス間のバラつきを最小にするような、更なる研究が行われるべきであろう。用量を最小にし、点滴のそれぞれの滴下の間隔を長くすることは、良いことであろう。対照的に、抗体の血清濃度から0.1%が脳に達している。ファージの分子量は抗体のそれよりも二桁大きいが、経鼻投与はファージ侵入を引き起こすことになりうる。
【0111】
本発明を完全に説明したので、本発明の精神と範囲から逸脱することなしに、また必要以上の実験を行うことなしに、相当するパラメータ、濃度および条件の広い範囲内で、当業者は、当然のことながら同様のことを行うことができる。
【0112】
本発明を、その具体的な実施態様と関連して説明したが、更に変更することができることは理解される。本出願は、本発明の原理に一般的に従って、いかなる変異、使用または適応を包含することを意図しているが、本開示からの以下のような逸脱をも含める:すなわち本発明に関する技術内で、既知のまた慣習的な慣行内にあるもの、および添付の請求の範囲において以下のように記述された、上記の本質的な特徴にも適用してもよい。
【0113】
本明細書において引用されている専門誌の記事、抄録、公表されたあるいは対応する米国あるいは外国の特許出願(米国または外国で発行された特許)、または他の全ての参考文献を含む、全ての参考資料は、引用において提示されたデータすべて、表、図およびテクスト含めて、参照により本明細書に援用される。更に、引用文献内の全内容も、参照により本明細書に援用される。
【0114】
既知の段階法、従来の段階法、既知の方法または従来の方法の引用は、本発明のいかなる態様、説明または実施例も、関連技術において開示され、教示されあるいは示唆されることを認めるものではない。
【0115】
具体的な実施態様についての前述の説明により、本発明の一般的な性質が十分に明らかにされるので、他者は、当業の技術内での知識(本明細書で引用された文献の内容を含めて)を適用することにより、過度の実験をしないで、また本発明の一般的な概念から逸脱しないで、具体的な実施態様のような種々の応用のために容易に変更および/あるいは適応することができる。それ故、そのような適応および変更は、本明細書に提示される教示と指導に基づく、開示された実施態様と同等の意味と範囲内にあるように意図している。本明細書における言い回しまたは述語は、ここに提示される教示や指導に照らして、当技術分野における通常技術の一つと組み合わせて当業者に解釈されるように説明するためのものであり、限定するためのものではないことを理解されたい。
【0116】
【表1】

【図面の簡単な説明】
【0117】
【図1】脳の断面の模式図である。
【図2A】β−アミロイド1−40凝集の脱凝集および阻止を示すグラフである。
【図2B】β−アミロイド1−40凝集の脱凝集および阻止を示すグラフである。
【図3A】β−アミロイドの電子顕微鏡写真である。
【図3B】β−アミロイドの電子顕微鏡写真である。
【図3C】β−アミロイドの電子顕微鏡写真である。
【図3D】β−アミロイドの電子顕微鏡写真である。
【図3E】β−アミロイドの電子顕微鏡写真である。
【図3F】β−アミロイドの電子顕微鏡写真である。
【図3G】β−アミロイドの電子顕微鏡写真である。
【図3H】β−アミロイドの電子顕微鏡写真である。
【図3I】β−アミロイドの電子顕微鏡写真である。
【図3J】β−アミロイドの電子顕微鏡写真である。
【図3K】β−アミロイドの電子顕微鏡写真である。
【図4A】電子顕微鏡写真である。
【図4B】電子顕微鏡写真である。
【図4C】電子顕微鏡写真である。
【図5A】β−アミロイド毒性の阻害を示すグラフである。
【図5B】β−アミロイド毒性の阻害を示すグラフである。
【図5C】β−アミロイド毒性の阻害を示すグラフである。
【図6A】線維状ファージを注入した図である。
【図6B】PBSのみを注入した図である。
【図7】マウスにおけるファージの分布を示すグラフである。
【技術分野】
【0001】
関連出願の相互参照
本出願は2005年2月1日出願の米国予備出願第60/648,383の優先権を主張するものであり、そのすべての内容は参照により本願明細書に援用したものとする。
【0002】
背景技術
本発明は、アミロイド沈着形成の抑制および既に形成されたアミロイド沈着の溶解に関するものであり、ならびに脳の炎症ならびに脳内および体内の他の部分のアミロイドまたはアミロイド様の沈着に付随する炎症を治療するための方法および医薬組成物に関する。
【0003】
関連技術の説明
斑形成疾患は、脳におけるアミロイド斑の沈着および神経変性の存在を特徴とする。アミロイド沈着は、ペプチドが凝集して不溶性の塊となることによって形成される。ペプチドの性質は、疾患が異なれば違ってくるが、殆どの場合、凝集体はβプリーツシート構造を有し、コンゴーレッド染料で染色される。早発性アルツハイマー病、遅発性アルツハイマー病および前駆症状アルツハイマー病を含むアルツハイマー病(AD)に加えて、アミロイド沈着を特徴とする他の疾患としては、例えばSAAアミロイド症、遺伝的アイスランド症候群、多発性骨髄種およびプリオン病が挙げられる。動物における最も一般的なプリオン病はヒツジおよびヤギのスクレイピーならびにウシの海綿状脳症(BSE)である(Wilesmith and Wells, 1991)。ヒトにおいて4種のプリオン病が同定されている:(i)クールー、(ii)クロイツフェルトヤコブ病(CJD)、(iii)Gerstmann-Streussler-Sheinker病(GSS)および(iv)致死性家族性不眠症(FFI)である(Gajdusek, 1977; およびTritschler ら. 1992)。
【0004】
プリオン病は、正常細胞プリオンタンパク(PrPC)がそれと対応するスクレイピー・イソ型(PrPSc)へ変換することと関係している。分光学的測定は、PrPCのスクレイピー・イソ型への変換に、立体構造の大きな変換が関与していることを示したが、これはプリオン病が他のアミロイド原性の疾患のように、タンパクの立体構造の障害であることを示唆している。PrPCからPrPScへの変換はα−ヘリックス二次構造の減少(42%から30%へ)とβシート含量の著しい増加(3%から43%)とを伴っている(Caugheyら、1991およびPan等、1993)。この再構成は、非変性の界面活性剤に対する不溶性およびタンパク分解に対する部分的な抵抗性を含む、異常な物理化学的な性質を伴う。先の研究は、ヒトPrPの106−126残基(PrP106−126)と相同性を有する合成ペプチドが、PrPScの病理学的および物理化学的性質の幾つかを有することを示した(Selvagginiら、1993; Tagliaviniら、1993; Forloniら、1993)。このペプチドは、種々の環境において異なった二次構造を獲得し、著明な構造的多形を示す(DeGioiaら、1994)。このペプチドは、緩衝液中においてはベータシート構造をとる傾向にあり、凝集してプロテーアーゼによる分解に部分的に抵抗性を有するアミロイド原線維になる。最近、抗体3F4とそのペプチドエピトープ(PrP104−113)との複合体のX線結晶解析研究が、プリオン病の発症に必須な構造再編成の成分であると考えられるこの可動性の領域の構造図を提供した(Kanyoら、1999)。フォールディング・アンフォールディングおよび/あるいは可溶化−凝集過程に関与している配列のクラスを同定することは、凝集の予防および/あるいは脱凝集の誘導に基づく斑形成疾患の治療に新しい方向を開くかも知れない(Silen and Agard, 1989; Frenkel等、1998; Horiuchi and Caughey, 1999)。
【0005】
アルツハイマー病(AD)は、老年認知症にいたる進行性の疾患である。広義において、この疾患は二つの範疇:老年(典型的には約65歳を超える)に起こる遅発性のもの、および老年よりずっと前に起こる、例えば35と60歳の間に起こる早発性のものに分けられる。この二つの型の疾患では病状は類似しているが、若年において発症する症例においては異常の程度はより重度であり、広範囲に亘っている。この疾患は脳における病変の二つのタイプ、老人斑および神経原線維濃縮体、を特徴とする。老人斑とは、脳組織切片の顕微鏡的解析によって見える、中央における細胞外アミロイド沈着を伴う、径150mmまでの不整の好中球の領域である。神経原線維濃縮体とは、一組になって互いに絡み合っている二本の線維よりなるタウタンパクの細胞内沈着物である。
【0006】
老人斑の主要な構成体はアミロイドベータ(Aβ)あるいはベータ−アミロイドペプチド(βAPあるいはβA)と呼ばれるペプチドである。アミロイドベータペプチドはアミロイド前駆タンパク(APP)と呼ばれる前駆タンパクの39−43アミノ酸の内部断片である。APPタンパク内の数個の突然変異がアルツハイマー病の存在と関連付けられている(Goateら、(1991), バリン717からイソロイシン;Chartier Harlanら、(1991), バリン717からグリシン;Murrellら、(1991), バリン717からフェニルアラニン;Mullanら、(1992)、二重変異、リジン595−メチオニン596からアスパラギン595−ロイシン596)。
【0007】
そのような突然変異は、APPのベータ−アミロイドへのプロセシング、具体的にはAPPをベータ−アミロイドの長型(すなわちAβ1−42およびAβ−43)の増量に導くプロセシングを増加あるいは変化させることにより、アルツハイマー病を引き起こすと考えられている。プレセニリン遺伝子、PS1およびPS2のような他の遺伝子における突然変異は、間接的にAPPのプロセシングに影響をおよぼして、長型のベータ−アミロイドの増量を引き起こすと考えられている(Hardy, TINS 20, 154, 1997参照)。これらの観察はベータ−アミロイド、具体的にはその長型がアルツハイマー病の原因要素であることを示している。
【0008】
自己凝集の証拠のある他のペプチドあるいはタンパクも知られており、これらに限られないが、例えばアミリン(amylin)(Youngら, 1994);ボンベシン(bombesin)、セルレイン(cerulean)、コレシストキニンオクタペプチド(cholecystokinin octapeptide)、エレドイシン(eledoisin)、ガストリン(gastrin)関連ペンタペプチド、ガストリンテトラペプチド、ソマトスタチン(somatostatin)(還元型)、サブスタンスP;およびペプチド、黄体形成ホルモン放出ホルモン、ソマトスタチンN−Tyr(Banks and Kastin, 1992)が挙げられる。
【0009】
アミロイド線維に関する論文は、円筒型のβ−シートが、X線および電子顕微鏡データのあるものと一致する唯一の構造であることを示し、アルツハイマーAβ断片および変異体の線維は、多分二つあるいは三つの同心の円筒型β−シートより成る事を示している(Perutz等, 2002)。完全なAβペプチドは42の残基を含んでいるが、これは円筒型の殻の核を構成する丁度よい数である:この発見およびプロリンが存在しないAβペプチドから成るβ−シートにおける多様で強い静電的相互作用があり得ることが、Aβペプチドのアルツハイマー病患者において見出される細胞外アミロイドプラクを形成する性質を説明する。もしこの解釈が正しければ、アミロイドは中央に水が充填した腔を有する細い管(ナノチューブ)から成っている。インビトロにおけるアミロイドプラークの成長の可逆性は、プラーク内と溶液中におけるβAの定常的平衡を示唆する(Maggio and Mantyh, 1996)。βAのポリメリゼーションは、ペプチド−ペプチド相互作用に依存してβ−プリーツシート原線維を形成すること、およびその反応に他のタンパクがおよぼす促進作用によりアミロイド形成が調節をうけうるかも知れないことが示唆されている。アミロイド形成に干渉することの出来る物質を発見しようとする多くの試みが行われた。最も深く研究された化合物としては、抗体、プロリンのようなベータ−破壊性のアミノ酸から構成されるペプチド、認識モチーフへの荷電基の添加、および構築ブロックとしてのN−メチル化アミノ酸の利用が挙げられる(Gazitによる総説, 2002)。
【0010】
DとLのアミノ酸残基が交互に存在する環状ペプチドは、それ自身を膜に挿入し、膜を脱分極させることにより細菌を殺すナノチューブを形成する(Perutzら、2002)。あるアミロイド線維は導体であって同じ機構で細胞を殺すかも知れないとする示唆もある。
【0011】
ヘリックスのターンの間の間隙にそれ自身を挿入することができるコンゴーレッドのような芳香族化合物は、円筒の殻を不安定にし、このプロセスを開始するかも知れないが、阻止する方が、より有効であり、多分達成しやすいであろう(Perutzら、2002)。
【0012】
本明細書におけるいかなる文書の引用も、そのような文書が関連する先行技術であること、あるいは本出願の請求項の特許性に対して重要であると考えられると、認めることを意図するものではない。いかなる文書の内容あるいは日付に関するいかなる言明も、出願人が出願時に利用できる情報に基づいており、そのような言明の正しさを認めるものではない。
【0013】
発明の概要
本発明は、脳内あるいは体内の他の場所におけるアミロイド沈着に付随する炎症ならびに活性化されたミクログリアにかかわる脳の炎症を抑制または治療するための方法を提供する。その方法は、それを必要とする患者に、野生型の線維状バクテリオファージ、または抗体もしくは非線維状バクテリオファージ抗原をその表面に提示しない線維状バクテリオファージの有効量を投与することを含む。
【0014】
本発明は、また活性成分として線維状バクテリオファージの有効量を含む医薬組成物を提供する。
【0015】
本発明は、更に線維状バクテリオファージを斑形成ペプチドまたは既に形成されたアミロイド沈着物と接触させることにより、アミロイド沈着の形成を抑制し、または既に形成されたアミロイド沈着を脱凝集する方法を提供する。
【0016】
図1は、hAPPトランスジェニックマウスにファージ/PBSの頭蓋内投与を示す脳の断面の模式図である。ファージを5匹のトランスジェニックマウスに脳内注入した。対側半球にPBSを注入した。マウスは、1時間、2日、3日後にと殺した。それらの脳を5μmの切片に切り、斑の量を評価するためにチオフラビン−Sで染色した。
図2Aおよび図2Bは、ThTアッセイを用いて線維状ファージによるβ−アミロイド1−40凝集の脱凝集(図2A)および阻止(図2B)を示すグラフである。図2Aにおいて(脱凝集試験)、β−アミロイド(58μM)を単独で21日間インキュベートした。21日目に異なった濃度のファージを加え一晩インキュベートした。図2Bにおいて(阻止試験)、β−アミロイド(58μM)を単独かあるいは線維状ファージと共に7日間インキュベートし、サンプルをThT溶液に加え、分光光度計によりアミロイド量を測定した。アミロイド含量は、アミロイド原線維と結合するチフラビン−Tを用いて測定した。その蛍光発光は485nm波長で検出した。
図3A〜3Kは、線維状ファージの存在または非存在下でインキュベートしたβ−アミロイドの電子顕微鏡写真である(倍率 x100K)。図3A:β−アミロイド97μM、PBS中ファージの非存在下。β−アミロイドはマウスモノクローン抗体10D5で染色し、次いでヤギ抗マウス12nm金結合抗体で染色した。図3B〜3C:0.5nM(1x1010)ファージとインキュベートした、β−アミロイド97μM。β−アミロイドはマウスモノクローン抗体10D5および12mn金粒子と結合した二次抗体で染色した。ファージはポリクローナルウサギ抗ファージ血清、次いでヤギ抗ウサギ6nm金結合抗体で標識した。これら二次抗体のシグナルは、ファージまたは一次抗体を含まないサンプルにおいては観察されなかった。図3D:線維状ファージ(fd)(1x1010ファージ)はウサギポリクローナル血清で染色した。二次抗体は6nm金粒子と結合させた。図3E:β−アミロイド近傍の線維状ファージ。ペプチドは、抗ファージ抗体の散在する標識が示すように(矢印)、多分ファージの分解を増大する。この標識は、ファージを単独でインキュベートした時には存在しない(サンプルD)。図3F〜3G:β−アミロイドのみを37℃で10日インキュベートした後(倍率:F−x30K,G−x100K)。図3H〜3I:β−アミロイドを単独で10日間インキュベートし、10日目に線維状ファージを加えて更に16時間インキュベートした(H−X30K,I−Hのx100Kの拡大)。図3J〜3K:アミロイド原線維と線維状ファージの界面。ファージは原線維の軸に沿って並んでいる(×100K)。
図4A〜4Cは、球形ファージを既に凝集したβ−アミロイドに加え、一晩インキュベートしたものの電子顕微鏡写真である。β−アミロイドと球形ファージ(図4Aおよび図4B)。球形ファージ単独(図4C)。(倍率 x100K)。
図5A〜5Cは、MTTアッセイを用いた、線維状ファージによるLAN−1細胞に対するβ−アミロイド毒性の阻害を示すグラフである。図5における阻止アッセイに関しては、ファージは25μMのβ−アミロイドに対して1760:1βA/ファージのモル比で加えた。混合物を細胞に加えた。生存100分率は、β−アミロイドの非存在下における細胞の生存率と関連しており(処理−PBS)、100%と考えられる。図5Bおよび5Cにおいて、βA毒性に対して保護するファージの能力を二つの既に凝集させたペプチド:βA1−40およびβA17−42を用いて測定した。両ペプチド(βA1−40およびβA17−42)を37℃で5日間インキュベートした。6日目に線維状ファージを1.2nMの濃度で既に凝集したペプチドに加えて12時間インキュベートし、前日96穴プレートに蒔いた細胞株に加えた。細胞は更に3日間増殖させ、MTTを加えた。3時間後抽出緩衝液を加え、プレートを37℃で一晩インキュベートした。翌日プレートを570nm波長で測定した。
図6Aおよび6Bは、hAPPトランスジェニックマウス(SWE2576, Taconic)の一つの脳半球に注入した線維状ファージ(図6B)と他の半球にPBSのみを注入(図6A)したものを示す。処置3日後にと殺したマウスは、PBS処置半球に比べて、斑量の40%減少を示した。
図7は、経鼻投与後のBALB/cマウスにおけるファージの分布を示すグラフである。BALB/cマウスへの経鼻投与後1,3および24時間における脳と嗅球におけるファージ分布の比較は、投与後1時間であってもファージの存在(標識ファージ、μCi/mg)を示していた。それらの濃度はこの1時間の点以後、減少した。
【0017】
発明の詳細な説明
β−アミロイドペプチド(βA)はアルツハイマー病の二つの特徴の内の一つである。このぺプチドは、強い変性剤によってのみ可溶化される脳組織内の線維状の毒性の凝集体を形成する。これらの神経毒性はペプチド凝集型に関連しているので、脳内のアミロイド原線維沈着の程度を、減少または除去する方向への治療法の開発に多くの努力が注がれてきた。
【0018】
生理的な条件下では、合成βAは凝集型を採り、また神経突起からの変化を示し、海馬ニューロンに対する神経毒性を促進する。βAの凝集は、pH、ペプチド濃度、温度およびインキュベーション時間に依存することが示されてきた。
【0019】
本発明者等は、線維状ファージそれ自体が、驚くべきことにインビトロにおいてβAの凝集を阻害し、および既に形成された凝集体を溶解する能力を有することを発見した。
【0020】
本発明者等の研究室においては、構造的および遺伝的レベルの両方でよく理解されている線維状ファージ、M13、f1、およびfd(Greenwoodら、1991)を用いた。この研究室は、線維状バクテリオファージが、ベクターのその不活性な性質と外来分子を輸送する能力の両方を保持しながら、中枢神経系に浸透する性質を示すことを最初に示した(FrenkelとSolomon, 2002)。
【0021】
線維状バクテリオファージは、環状の一重鎖DNAゲノムを含む構造的に関連したウイルスの一群に属する。これらは宿主を増殖性感染中に殺すことはない。Fプラスミドを含む大腸菌(Escherichia coli)に感染するファージは、集合的にFfバクテリオファージと呼ばれる。これらは哺乳動物細胞には感染しない。
【0022】
線維状バクテリオファージは、約1〜2ミクロン長および直径6nmのフレキシブルな桿体であり、DNAコアを囲むタンパクサブユニットのらせん状の殻を持っている。主要な二つのコートタンパク、タンパクpIIIおよび主要コートタンパクpVIII、は提示されたタンパクのコピー数において異なっている。pIIIが4〜5コピー存在するのに対し、pVIIIは約3000コピー見つかっている。約50残基を有する主要コートタンパクpVIIIサブユニットは主としてアルファヘリックスであり、そのアルファヘリックスの軸は、ウイルス粒子の軸と小角をなしている。タンパク殻は3つの部分から構成されると考えられている:周囲の溶媒と相互作用し、ウイルス粒子の等電点を低くする酸性残基に富んだ、サブユニットのN末端領域によって占められている外表面;タンパクサブユニットが主として相互に作用する、非極性側鎖の19残基のストレッチを含む殻の内側;およびDNAコアと相互作用をする塩基性残基に富んだ、サブユニットのC末端領域によって占められる内側の表面である。実際、全てのタンパク側鎖の相互作用は、サブユニット内で起こるのではなく、コートタンパクアレイにおける異なったサブユニット間で起こるという事実は、これを高分子の集合におけるアルファ−ヘリックスサブユニット間の相互作用の研究のための有用なモデルシステムにすることができる。線維状バクテリオファージの特異な構造が、約16.3MDの大きさを有しているにもかかわらず脳内への侵入を可能にし、ファージの構造がアミロイド原線維自身に似ていることから、βA線維化に干渉する能力に寄与していると考えられる。
【0023】
上述の事を勘案して、本発明者等は、β−アミロイドペプチドの凝集過程に干渉する線維状ファージの能力を調べ、野性型の線維状ファージをβ−アミロイドペプチドとインビトロにおいて、異なる時間間隔と異なる比でインキュベーションすることが、β−アミロイドの阻止および/あるいは脱凝集を引き起こすことを発見した。更に線維状ファージは細胞の生存に対して保護効果を示す。
【0024】
最も興奮するようなデータが、線維状ファージ−β−アミロイド原線維を、スライドグラス上で増殖させたマイクログリア細胞とインキュベートした後に得られた。もしβ−アミロイドがマイクログリアを活性化させるなら、ファージは、マイクログリアを活性化させずにそれを溶解する。ファージ技術は、β−アミロイドの抗凝集剤の新しい事実上無限の原料を提供し、Fc受容体を介してマイクログリアを過剰に活性化させうる抗体の有害な効果を阻止する。
【0025】
バクテリオファージは、遺伝子および/または送達媒体として動物ウイルスよりも明らかに有利である。それらは、動物ウイルスベクターよりも大規模生産および精製を非常に効率よく安価に行うことができる単純なシステムだからである。更に、DNAの大きな部分をファージミドベクターへ効率よくパッケージすることができる。原核生物の感染、構築および複製のために進化してきたことから、バクテリオファージは哺乳動物細胞内で複製もできないし、天然の指向性も有していない。このことで、非特異的な遺伝子送達の可能性は極めて低い。ファージベクターは、動物細胞において複製可能な構成要素をつくる可能性は低いことから、ウイルスよりも潜在的にずっと安全である(Monaciら, 2001)。
【0026】
本発明はアミロイド沈着に付随する、または活性化されたマイクログリアにかかわる脳の炎症を抑制または治療する方法を提供する。更に本方法は、体内の脳以外の場所におけるアミロイド沈着に関連した炎症、例えば多発性骨髄腫や腎アミロイドーシスを抑制または治療する。
【0027】
本方法は、必要とする患者に、野生型の線維状バクテリオファージ、または抗体もしくは非線維状バクテリオファージ抗原をその表面に提示しない線維状バクテリオファージの有効量を導入/投与することを含む。線維状バクテリオファージは、M13、f1またはfdのような線維状バクテリオファージのいずれであってもよい。以下の実施例においてはM13が用いられたが、いかなる他の線維状バクテリオファージであっても、同様な構造と95%を越えるゲノム同一性があることから、同じように行動し、機能することが期待される。
【0028】
本方法を脳の炎症を抑制または治療のために用いる場合、線維状バクテリオファージは、好ましくは活性成分を受容者の嗅覚系を通して受容者の体内に導入するために、経鼻的に投与することが望ましい。
【0029】
本方法は、斑形成疾患において、タンパクが凝集してアミロイド斑または沈着物となるのを抑制するのみならず、また既に形成されたアミロイド沈着、例えばβA原線維を脱凝集させるのに有効である。
【0030】
βA原線維形成または脱凝集に関して、線維状バクテリオファージの抗凝集または脱凝集性は、よく知られたチオフラビンT(ThT)結合アッセイによって測定することができる。βA原線維構造の形成の崩壊、および既に形成されたβA原線維の脱凝集は、ThTの蛍光の実質的に減少することによって示される。
【0031】
本明細書とそれに付随する請求項の目的上、「患者」、「対象」および「受容者」という用語は同義で用いられる。それらの用語には、予防的、実験的または治療的処置の対象である、ヒトおよび他の動物が含まれている。
【0032】
本明細書とそれに伴う請求項の目的上、「ベータアミロイドペプチド」は「β−アミロイドペプチド」、「βAP」、「βA」および「Aβ」と同義である。これらの用語の全ては、アミロイド前駆タンパクから由来した斑形成ペプチドを言う。
【0033】
本明細書においては、「PrPタンパク」、「PrP」、「プリオン」は、適切な条件下で斑形成疾患の原因である凝集体の形成を導くことのできるポリペプチドを言う。例えば、正常細胞プリオンタンパク(PrPC)はそのような条件下で、対応するスクレイピーイソ型に変換するが、それはウシ海綿状脳症(BSE)または狂牛病、ネコ海綿状脳症、クールー、クロイツフェルトヤコブ病(CJD)、Gerstmann-Streussler-Sheinker病(GSS)および致死性家族性不眠症(FFI)のような、これに限らないが、斑形成疾患の原因である。
【0034】
本明細書では「脱凝集」とは、凝集タンパク、典型的には非共有結合によって凝集しているタンパクの可溶化を言う。
【0035】
「治療する」という用語は、疾患の進行を実質的に抑制し、遅らせあるいは逆行させること、疾患の臨床症状を実質的に改善すること、あるいは疾患の臨床症状の出現を実質的に阻止することを意味するよう意図されている。
【0036】
また、本明細書においては、「斑形成疾患」は、これらに限らないが、早発性アルツハイマー病、遅発性アルツハイマー病、発症前アルツハイマー病、SAAアミロイド症、遺伝的アイスランド症候群、老化、多発性骨髄腫、およびヒトに影響することが知られているプリオン病、例えばクールー、クロイツフェルトヤコブ病(CJD)、Gerstmann-Streussler-Sheinker病(GSS)および致死性家族性不眠症(FFI)、および動物に影響することが知られているプリオン病、例えばスクレイピーおよびウシ海綿状脳症(BSE)であって、ベータアミロイド、血清アミロイドA、シスタチンC、IgGカッパ軽鎖またはプリオンタンパクのような凝集タンパク(斑形成ペプチド)による斑形成を特徴とする疾患を言う。
【0037】
上に述べた疾患にかかわるアミロイド斑(アミロイド沈着としても知られている)のほとんどは、脳内に存在するので、いかなる提案される治療様式も、血液脳関門(BBB)を越える能力およびアミロイド斑を溶解する能力を示さなければならない。通常BBBを透過できる分子の平均サイズは約2kDaである。
【0038】
嗅覚の欠陥と中央嗅覚経路における退行的変化がADの臨床経過の初期に影響を受けていることを示す証拠が増えつつある。更にADに関与している解剖学的パターンは、嗅覚経路がADの進展における初期段階でありうることを示唆している。
【0039】
嗅覚受容体ニューロンは、鼻腔の上皮層に存在する双極性の細胞である。それらの軸索は篩板を横切り、脳の嗅球における嗅覚経路の最初のシナプスに突出する。この立体構造により、ウイルスや他の輸送される物質がBBSを通ってCNSに到達することができる通路となっている。
【0040】
以前に示したように、鼻腔内投与(Mathisonら、1998; Chouら, 1997; Draghiaら, 1995)によって、ウイルスおよび高分子が脳脊髄液(CFS)あるいはCNSに直接入ることが可能である。
【0041】
嗅覚受容体ニューロンをアデノウイルスベクターの脳への送達点として使用した例が、文献に報告されている。この方法により、見掛けの毒性なしに、脳内で12日間レポーター遺伝子が発現したことが報告されている(Draghiaら、1995)。
【0042】
従って、本発明の好ましい実施例によれば、斑形成疾患に付随するポリペプチドの凝集体の形成を脱凝集または阻止することが可能な、またはマイクログリアの活性化を抑制することが可能な線維状バクテリオファージは、この経路を介して脳へ送達される。
【0043】
Aβは、末梢組織の細胞によって絶え間なく生産され、血管脳関門(BBB)を越えて、特定のニューロン集団において局在する毒性効果を発揮するので、そのような媒体の経鼻投与は、斑を形成できる末梢Aβの量を最小限にすることにより疾患の進行をも阻止することができる。
【0044】
本発明の医薬調製品には、活性成分として、野性型の線維状バクテリオファージ、または抗体もしくは非線維状バクテリオファージ抗原をその表面に提示しない線維状バクテリオファージが含まれる。該医薬調製品はまた、異なった線維状バクテリオファージの混合物であってもよい。
【0045】
本発明の調製品は、それ自体を、または適切な担体または賦形剤と混合した医薬組成物として、生命体に投与することができる。
【0046】
本明細書においては、「医薬組成物」とは、本明細書に記載された一つまたは複数の活性成分と生理的に適切な担体および賦形剤のような他の化学成分との調製品を言う。医薬組成物の目的は、生命体への化合物の投与を容易にすることである。
【0047】
本明細書において「活性成分」という用語は、生物学的効果を発揮する調製品を言う。
【0048】
今後、「生理的に許容される担体」および「医薬的に許容できる担体」の句は、互いに同じ意味で用いられるが、生命体に有意な刺激を与えず、投与される化合物の生物学的活性および性質を損なわない担体または希釈剤を言う。補助剤はこれらの句の中に含まれる。
【0049】
本明細書において、「賦形剤」という用語は、活性成分の投与を容易にするために医薬組成物に加えられる不活性物質を言う。賦形剤の例としては、炭酸カルシウム、リン酸カルシウム、種々の糖類およびデンプン類、セルロース誘導体、ゼラチン、植物油およびポリエチレングリコールが挙げられるがこれに限定されない。
【0050】
薬剤の処方および投与に関する技術は「Remington's Pharmaceutical Sciences, レミントンの薬剤化学」Mack Publishing Co., Easton, PA, 最新版に捜し出すことができ、参照することにより本明細書に盛り込まれる。
【0051】
適切な投与経路としては、例えば、経口、経直腸、経粘膜、特に経鼻、経腸、または筋肉内、皮下および髄内注射を含む非経口送達、ならびに髄腔内、直接脳室内、静脈内、腹腔内、鼻腔内あるいは眼内注射が挙げられる。
【0052】
あるいは、調製品を全身ではなく、局所的に投与してもよく、例えば、患者の脳に直接調製品を注射する方法が挙げられる。
【0053】
本発明の医薬組成物は、当技術においてよく知られた方法によって製造してもよく、例えば、従来の混合、溶解、造粒、糖衣錠製造、すり潰し、乳化、カプセル化、封入または凍結乾燥法が挙げられる。
【0054】
本発明に従って使用するための医薬組成物は、活性成分を加工して医薬的に使用できる調製品とすることを容易にする賦形剤および補助剤を含む一つあるいは複数の生理的に受容できる担体を用いて、従来の方法で製剤化することができる。適切な剤形は選択される投与経路に依存している。
【0055】
注射剤としては、本発明の活性成分は、水溶液中で、好ましくはHank's溶液、リンゲル液または生理的塩類緩衝液のような生理的に適合する緩衝液中で、製剤されてもよい。経粘膜投与のためには、透過すべき関門に適合した浸透剤がその剤形に用いられる。そのような浸透剤は当技術分野において一般的に周知である。
【0056】
経口投与のためには、化合物は、活性成分と当技術分野において周知の、医薬的に許容できる担体を組み合わせることにより、容易に製剤することができる。そのような担体により、本発明の化合物は、患者が経口的に摂取するための錠剤、丸剤、糖衣剤、カプセル剤、液剤、ゲル剤、シロップ剤、スラリー剤、懸濁剤等として製剤されうる。経口使用される薬理学的調製品は、固体賦形剤を用いて製造可能であり、場合によっては得られた混合物を粉砕し、必要であれば適切な補助剤を加えた後、顆粒の混合物を加工して錠剤または糖衣錠のコアを得る。適切な賦形剤は、具体的には、乳糖、ショ糖、マンニトールまたはソルビトールを含む、糖などの充填剤;トウモロコシデンプン、小麦デンプン、米デンプン、ジャガイモデンプン、ゼラチン、トラガカントゴム、メチルセルロース、ヒドロキシプロピルメチル−セルロースおよびカルボキシメチルセルロースナトリウムのようなセルロース調製品;および/または生理的に許容できる、ポリビニルピロリドン(PVP)のような高分子が挙げられる。必要に応じて、架橋ポリビニルピロリドン、寒天、またはアルギン酸もしくはアルギン酸ナトリウムなどのその塩の崩壊剤を加えてもよい。
【0057】
糖衣錠のコアには、適切なコーティングを施す。このために、濃縮糖溶液を用いてもよいし、場合によっては、アラビアゴム、タルク、ポリビニルピロリドン、カルボポールゲル、ポリエチレングリコール、二酸化チタン、ラッカー溶液、および適切な有機溶媒または溶媒混合物を含んでもよい。活性化合物用量の異なった組み合わせを同定、識別するために、染料または顔料を錠剤や糖衣錠のコーティングに加えてもよい。
【0058】
経口的に用いられる医薬組成物としては、ゼラチンから作られたプッシュフィットカプセル剤やゼラチンおよびグリセリンまたはソルビトールのような可塑剤から作られた、軟、密閉カプセル剤が挙げられる。プッシュフィットカプセル剤は、活性成分を、乳糖のような充填剤、デンプンのような結合剤、タルクやステアリン酸マグネシウムのような潤滑剤、そして場合によっては安定剤と混合して含んでもよい。軟カプセル剤においては、活性成分は油脂、流動パラフィン、または液体ポリエチレングリコールのような適切な液体に溶解あるいは懸濁してもよい。更に安定剤を加えてもよい。経口投与用のすべての製剤は、選択した投与経路に適した剤形でなければならない。
【0059】
頬側投与のためには、組成物は従来の方法で製剤化された錠剤またはロゼンジの剤形を取ってもよい。
【0060】
鼻腔吸入による投与のためには、本発明に従って使用する活性成分は、加圧されたパックからのアエロゾル噴霧の形で、または例えばジクロロジフロロメタン、トリクロロフロロメタン、ジクロローテトラフロロエタンまたは二酸化炭素のような適切な噴霧剤を用いた噴霧器の形でするのが好都合である。加圧アエロゾルの場合は、単位用量は、計量した量を送達する弁を備えることにより決定してもよい。ディスペンサーで使用される、例えばゼラチンのカプセルおよびカートリッジは、化合物と、乳糖またはデンプンのような適当な粉末基剤の混合粉末を含む剤形であってもよい。
【0061】
本明細書に記載される調製品は、非経口投与、例えばボーラス注射または連続注入による剤形であってもよい。注射用の剤形は、例えばアンプルでまたは多回数投与用の容器で場合によっては保存剤を加えて、単位用量で提供してもよい。組成物は、油性または水性の媒体中の懸濁剤、溶液剤または乳剤であってよく、懸濁剤、安定剤および/または分散剤のような製剤用の物質を含んでもよい。
【0062】
非経口投与のための医薬組成品としては、水溶性の活性調製品の水溶液が挙げられる。更に、活性成分の懸濁剤を、適当な油性または水性基剤懸濁注射剤として調製してもよい。適当な脂溶性の溶媒または媒体としては、ごま油のような油脂、またはオレイン酸エチルのような合成脂肪酸エステル、トリグリセリドまたはリポソームが挙げられる。水性の注射懸濁剤は、ナトリウムカルボキシメチルセルロース、ソルビトールまたはデキストランのような懸濁剤の粘度を上げるような物質を含んでもよい。場合によっては懸濁剤は、適当な安定剤、または高濃度の溶液の調製を可能にする活性成分の溶解度を増加させる物質を含んでもよい。
【0063】
あるいは、活性成分は、例えば無菌の発熱成分を含有しない水溶液のような適当な媒体により使用前に構成する粉末剤形でもよい。
【0064】
本発明の調製品は、ココアバターまたは他のグリセリドのような通常の坐剤基剤を用いた坐剤、または保留浣腸のような直腸用の組成物に製剤化してもよい。
【0065】
本発明の範囲で適切に使用される医薬組成物としては、意図する目的を達成するために有効な量の活性成分を含んでいる組成物が挙げられる。より具体的に言えば、治療有効量とは、疾患の症状を阻止、軽減または改善する、あるいは治療を受けている対象の生存期間を延長するのに有効な活性成分の量を意味する。
【0066】
治療有効量の決定は、特に本明細書で提供された詳細な開示の観点から、当業者の能力の範囲内に十分ある。
【0067】
本発明の方法において使用されるいかなる調製品に対しても、治療有効量または投与量は、最初はインビトロおよび細胞培養アッセイから推測することができる。例えば投与量は、循環中の所望の抗体濃度または力価を達成するために動物モデルにおいて式化できる。そのような情報は、ヒトでの有用な投与量をより正確に決めるために使用される。
【0068】
本明細書に記載される活性成分の毒性と治療効果は、インビトロにて、細胞培養または実験動物において標準的な医薬手法で決定することができる。これらインビトロでの、および細胞培養アッセイおよび動物実験から得られるデータは、ヒトにおいて用いられる投与量の範囲を計算するために使用される。投与量は使用された剤形および投与経路に依存して変わりうる。正しい剤形、投与経路および投与量は患者の状態を考慮して個々の医師によって選択されることができる(例えば、Finglら「治療の薬理学的基礎」、第1章p.1(1975)参照)。
【0069】
投与量と投与間隔は、凝集を阻止し、既に存在する凝集体を脱凝集させるために十分な線維状バクテリオファージの血漿中または脳内レベル(最小有効濃度MEC)を与えるために個人ごとに調整できる。MECはそれぞれの調製品によって異なるであろうが、そのインビトロデータから推定することができる。MECを達成するために必要な投与量は、個々の特徴と投与経路に依存するであろう。結合アッセイを、血漿中濃度を決定するために用いることができる。
【0070】
投与間隔もまた、MEC値を用いて決定することができる。調製品は、10〜90%、好ましくは30〜90%、最も好ましくは50〜90%の時間、血漿中レベルをMECの上に維持するレジメンを用いて投与されるべきである。
【0071】
治療する状態の重度と反応性によつて、投与形態は、単回投与または複数回投与であることができ、治療コースは、数日から数週間、または治癒する、もしくは疾患状態の減少が達成されるまで継続される。
【0072】
投与されるべき組成物の量は、もちろん、治療対象、疾患の重度、投与方式、処方する医師の判断等に依存する。
【0073】
本発明の組成物は、必要に応じて、活性成分を含む1つ以上の単位投与量剤形を入れたFDA認可キットのようなパックまたはディスペンサー器具で提供されてもよい。該パックは、ブリスターパックのような金属またはプラスチックのフォイルを含んでいてもよい。パックまたはディスペンサー器具は、投与法についての指示書を添付していてもよい。パックまたはディスペンサー器具はまた、容器に付随した通知を伴ってもよく、その通知は医薬品の製造、使用および販売を規制する政府機関によって指定された形式で、組成の形態もしくはヒトまたは動物投与用にその機関から受けた認可を反映している。そのような通知は、例えば、米国食品医薬品局によって認可された処方医薬品のラベルであってもよいし、また認可製品の添付文書であってもよい。適合する医薬用担体中に製剤された本発明の調製品を含む組成物もまた調製して、適切な容器中に入れ、上に述べたように表示条件の治療のためのラベルを付けてもよい。
【0074】
本発明を一般的に説明したが、同じことを次の実施例を参照することでより容易に理解
できるであろうが、実施例は例示するために示しているものであり、本発明を限定することを意図しているものではない。
【0075】
実施例
材料と方法
ファージの増殖および精製
線維状ファージ(M13)は50μg/mlカナマイシンを含む2YTブロス中の形質転換TG1培養物から調製した。細菌細胞を遠心により沈殿させ、澄明な上清をデカンテーションにより得た。2.5M NaCl中の20%ポリエチレングリコール(分子量8000Da)溶液の当初量の20%容を加えて、4℃で3時間静置してファージを上清から沈殿させた。ファージを遠心分離して(9000rpm、1時間)ペレットとし、その後上清の3%容のPBSに再懸濁した。細菌の残渣は更に7000rpmで遠心分離して除き、ファージを、ポリエチレングリコールを加えて沈殿させて再濃縮した。ペレットを最終的に増殖培地の当初量の0.001容のPBSに再懸濁した(Hartら, 1994)。
【0076】
β−アミロイド凝集へのファージの干渉:
βA凝集を阻止し、既に形成された凝集体を脱凝集するファージの能力を、三つの方法で分析した:
【0077】
1.チオフラビン−T蛍光アッセイ
βA凝集を、アミロイド原線維との結合により発光スペクトルが変化するチオフラビン−T(ThT)色素を用いて測定した(LeVine, 1993)。凝集体形成を阻止するファージの能力を評価するために、βA1−40 577μM(Bachem)8μlを37℃で14日間、単独で(PBSで80μlに希釈し、最終濃度58μMとした)または線維状ファージと共に(3x1012ファージ/ml、72μl)インキュベートした。それぞれの反応混合液(20μl)の蛍光を、50mMグリシン緩衝液、pH9、中のThT(2μM)980μlを加えた後測定した。蛍光はPerkin-Elmer Model LS-50蛍光光度計を用いて、励起波長435nmおよび発光波長485nmで測定した。
【0078】
ファージの脱凝集活性は以下のようにして検査した:βA(58μM)を37℃で21日間インキュベートして凝集を促進した。ファージを加え(ファージ濃度1014ファージ/mlの溶液と等量)、凝集したβAと共に更に17時間インキュベートした。凝集の程度を上記のごとくThT蛍光を用いて評価した。
【0079】
2.電子顕微鏡(EM):
β−アミロイドと線維状ファージの相互作用。βA凝集のレベルをEMを用いて可視化した。βA形成の阻止については、ペプチド(289μM)10μlを、単独で(PBS20μlを加えた)または各種ファージ濃度(以下の濃度のファージ20μl:1x1014ファージ/ml、1x1012ファージ/mlおよび1x1010ファージ/ml)で、37℃で9日間インキュベートした。
【0080】
ファージの脱凝集活性は、各種濃度のファージを、既に凝集したβAに加えることによって証明した。PBSに溶解したβ−アミロイド1−40(193μM)の20μlを37℃で10日間インキュベートした。10日目にPBSまたは各種濃度のファージ(1x1014ファージ/ml、1x1012ファージ/ml)10μlをサンプルに加え、更に16時間インキュベートした。
【0081】
阻止および脱凝集アッセイの両方において、ファージとβAをカーボン蒸着被膜をほどこしたフォルムバー(formvar)200#グリッドに付着させた。アミロイドおよびファージの免疫標識を、種々のサイズの金結合抗体(Linら、1999)を用いて行ったが、これはβ−アミロイド原線維と、アミロイド線維と似ている可能性のあるファージを容易に識別するためである。β−アミロイド原線維はモノクローナル抗体(mAb)105Dおよび二次抗体としてヤギ抗マウス−12nm金粒子結合抗体(Electron Microscopy Sciences, Washington, USA)で染色した。ファージを標識するために、ファージに対するウサギポリクローナル血清を用い、二次抗体を6nm金粒子と結合させた(Electron Microscopy Sciences)。サンプルは酢酸ウラン(Sigma)水溶液(2%重量/体積)でネガティブ染色を行った。グリッドはJEOL1200EX電子顕微鏡を用いて、拡大率30Kおよび100Kで可視化した。
【0082】
β−アミロイドとクロロホルム処理ファージとの相互作用
ファージ(1014ファージ/ml)200μlを200μlのクロロホルムに加えた。溶液を室温で3分間に6回ボルテックス処理を行った(それぞれ10秒)(Griffithら、1981)。遠心管を13,200rpmで遠心分離し、水相を別の管に移し、ドラフト中に置き、残っているクロロホルムを蒸発させた。β−アミロイドと球形ファージの間の相互作用を電子顕微鏡によって分析した。
【0083】
β−アミロイド(97μM)を37℃で13日インキュベートした。13日目にPBSまたは各種濃度のS−ファージ(クロロホルム処理)(5x1013ファージ/ml、5x1011ファージ/ml)をサンプルに加え、更に16時間インキュベートした。β−アミロイドの凝集の程度はJEOL1200EX電子顕微鏡で先に述べたように可視化した。
【0084】
3.MTTアッセイ:
ヒト神経細胞株の生存率に対するβAの毒性からファージによる保護を評価するために、MTTアッセイを行った。アッセイは生存している細胞内の、3−(4,5−ジメチルチアゾールー2−イル)−2,5−ジフェニルテトラゾリウムブロマイド(MTT)のMTT−ホルマザンへの還元に基づいている。MTT−ホルマザンの濃度は、分光光度計により570nmで測定したが、細胞の生存率と直接相関していた。
【0085】
LAN−1ヒト神経細胞株は、10%ウシ胎児血清と100ユニット/mlペニシリン/ストレプトマイシンを補充したRPMI中で培養し、37℃、5%CO2下でインキュベートした。神経毒性アッセイのために、培養したLAN−1細胞を96穴プレートに104細胞/100μl/ウエルの密度で播種した。投与量依存性の神経毒性を、β−アミロイド(289μM)サンプル単独で、またはβ−アミロイド(289μM)とファージ濃度を変えて測定した。
【0086】
βAの神経毒性に対するファージ保護効果を試験するために、ペプチド2μlを8μlの各種濃度のファージ(5x1013/ml、5x1012/ml、5x1011/mlファージ)と37℃で4日間インキュベートした。細胞を播種してプレートに接着してから24時間後にサンプルを加えた。プレートを37℃で2日間インキュベートした後、細胞の生存率を、先に記述したように3−(4,5−ジメチルチアゾール−2−イル)−2,5−ジフェニルテトラゾリウムブロマイド(MTT)を用いて細胞の酸化還元活性を測定して評価した(Solomonら, 1997)。MTT(Sigma)を最終濃度1mg/mlでウエルに加え、細胞と共に更に37℃で3時間インキュベートした。細胞溶解緩衝液(20%重量/体積SDSを含む50%ジメチルホルムアミド溶液、pH7.4)を加え、プレートを一晩37℃でインキュベートした。MTTの還元は、自動マイクロプレート分光光度計を用いて570nmのODの変化により比色法で測定した。
【0087】
既に形成されたβA原線維の脱凝集アッセイにおいて、神経毒因子として二つのペプチド(β−アミロイド17−42およびβ−アミロイド1−40)を用いた。凝集したペプチドから細胞生存を守るファージの能力は以下に示すように試験した。第一の実験において、細胞を既に凝集したβA1−40と5日間インキュベートしたが、一方第二の実験においてはβ−アミロイド17−42を神経毒性ペプチドとして用いた。先に述べたように、細胞を96穴プレートに104細胞/100μl/ウエルの密度で播種し、細胞を24時間付着させた後にサンプルを加えた。凝集ペプチド(288μM−保存溶液)2μlに8μlのファージ(1014ファージ/ml、1013ファージ/ml、1012ファージ/ml)を加えて一晩インキュベートした。サンプルを細胞に加えて、更に2日間インキュベートした。MTTを最終濃度1mg/mlで加え、MTTホルマザン含量の測定のために、3時間後に溶解緩衝液を加えた。細胞の生存率は570nmにおけるOD変化を定量化して評価した。
【0088】
線維状ファージのインビボ投与
ADモデル、野生型およびトランスジェニックマウスを線維状ファージで攻撃した:
【0089】
抗凝集性の評価
抗凝集剤としてのファージ活性の最大能力を評価するために、ファージをトランスジェニックマウス(Taconic, Appswe(2576),10ヶ月齢)の頭蓋内に以下のようにして注射した:
【0090】
線維状ファージ溶液(1014ファージ/ml)2.5μlを10分かけて一方の半球へ(Bregma−2.8mm、側方2.5mm、腹側2.5mm)注射し、一方対側にはリン酸緩衝食塩水(PBS)をコントロールとして注射した(図1)。ファージがアミロイド沈着を脱凝集させるに要する時間は知られていないので、処置されたマウスは異なった時間点でと殺した。第一の群は、注射後1時間に4%パラホルムアルデヒドを用いて心臓内灌流を行った。第二の群は投与後2日目に、そして最後の群は3日後に灌流を行った。それらの脳は4%パラホルムアルデヒド中で一晩固定後、マイクロトームを用いて切片とした。チオフラビンーS(ThS)染色を行い、斑量を評価した。この目的のために、切片はMayer'sヘマトキシリンで染色し、核の自己蛍光を抑え、そして洗浄後ThS溶液(1%)を3分間適用した。1%酢酸を20分間用いて差別化を行い、スライドを洗浄後乾燥し、抗退色マウント剤を用いてマウントした。斑量はLEICA Qwinプログラムを用いて計算した。
【0091】
放射能標識線維状ファージの生体内分布
線維状ファージをI125で放射能標識した。ファージを、結合していないヨウ素からG25セファデックスカラムを用いて精製し、先に記載したように、溶出液をポリエチレングリコール(PEG)を用いて更に沈殿させた。9匹のBALB/cマウスを3群に分けた。それぞれのマウスに経鼻的に9マイクロキュリーI125で標識されたファージ100μl(1.25x1012ファージ)を1時間かけて与えた。第一のマウス群は4%パラホルムアルデヒドを用いた心臓内灌流1時間後にと殺した。第二の群は3時間後そして最後の群は24時間後にと殺した。灌流後、脳および末梢器官を取り出し、それらのガンマ線を測定した。
【0092】
線維状ファージの経鼻投与
線維状ファージの効果を十分に評価するために、ファージをSWE/APP2576トランスジェニックマウス(Taconic、10ヶ月齢)に経鼻的に投与した。ファージ溶液(5x1012/ml)20μlを2週間毎に4ヶ月間投与し、認識機能を評価した。9回目の接種後、記憶改善におよぼすファージの影響を研究するために、対象認識試験を行った。第一日目に、マウスを二つの新しい対象に20分間触れさせた。翌日一つの対象を取り替えて、新しい物を探るマウスの好奇心を試験した。認識指標を、各マウスが新しい対象の近くで過ごす時間を、各マウスが二つの対象の近くで過ごす全時間で割った値として計算した。従って、値が0.5を越えることは、古い物を認識し、新しい対象の周りを探査するためにより多くの時間を過ごしていることを示唆している。
【0093】
結果
線維状ファージのインビトロにおける抗凝集性を、アミロイド形成の減少および阻止について評価した。チオフラビンーTアッセイにおいて、ファージはβA原繊維の形成を阻止するよりはβA原繊維の脱凝集においてより有効であった。ペプチドを線維状ファージとモル比1:10,000(ファージ対βA)でインキュベートした時、βA凝集の26%減少が観察された(図2B)。既に凝集したβAにファージ(28nM)を加えた結果、アミロイド原線維の45%減少に至った(図2A)。
【0094】
ファージとβ−アミロイドペプチド間の物理的な相互作用の電子顕微鏡による可視化は、免疫金染色β−アミロイドとファージとのその複合体を用いて行った。ファージがβ−アミロイドペプチドと相互作用をする時は、ファージが存在しない時に比べて集合体はより小さく、より分散していた。
興味あることには、ファージをβ−アミロイドペプチドとインキュベーションすると、ファージが分解した。これはファージ粒子上にないファージの主要コートタンパクに対する抗体の強い標識化から知ることができる(図3C)。この現象は、ファージをβ−アミロイドペプチドと一緒に一晩インキュベートした時(脱凝集アッセイにおいて)、またはファージを単独で同じ期間(図3D)インキュベートした時には観察されなかった。図3(F−K)は、形成されたアミロイド凝集体におよぼすファージの効果を示す。図3Fは、12nm金粒子によって標識された集合体におけるアミロイド原線維を示す。高倍率(図3G)は、異なる大きさの凝集した針状原線維を示す。図3H−Kは、束状になっているファージを示す(矢頭印)が、それはアミロイド針に接着すると分散する(矢)。アミロイドの集合体は小さくそして小さい細いアミロイド原線維を含んでいる。特に興味があるのは、ファージとアミロイド原線維との並行した組織構造である(白い矢)。
【0095】
pVIII分子は3つの立体配置で存在することが示されている。完全なファージにおいては、該タンパクは、少なくとも90%α−ヘリックスであるが、クロロホルム/水の界面に曝されると、形に大きな変化が起こる。これは温度に依存する過程である。2℃では桿形(I−型)であるが、25℃では球形構造(S−型)を取る。S−型への変換はコートタンパクのヘリックス含量の実質的な減少を伴って起こるが、トリプトファン26の環境における有意な変化を伴う(Robert and Dunker, 1993)。
【0096】
クロロホルムによるファージの3分間の処理により、球形のファージはβA凝集の場所に存在しているが、それらは通常原線維の端末に存在しており、可溶化には寄与していないことを示す(図4A−B)。
【0097】
MTT:
線維状ファージがβA神経毒からLAN−1細胞を防御することは、MTTアッセイを用いて示された。ファージをβAとインキュベートした場合、神経細胞の生存率は、βAのみの存在下で増殖した細胞と比較して増加した。細胞培養に加えられたファージの最高量が、最も有意な効果(細胞生存率17%の増加)をもたらした(図5A)。
【0098】
β−アミロイド17−42の毒性からのファージによる保護は、β−アミロイド1−40の毒性を阻止する能力と比較して、より高かった。ファージを既に凝集したβA17−42に加えた場合、細胞の生存率は、βAのみで増殖した細胞と比較して30%増加した(図5B)。同じファージ濃度をβA1−40に加えた場合、細胞生存率は17%増加したのみであった(図5C)。より高い濃度のファージを加えた場合、βA処理を受けた細胞に比較して、細胞生存率を25%に増加させることに成功した。
【0099】
この差に関する可能性のある理由としては、β−アミロイドのN末端がファージ活性を妨害するかも知れない、またはβ−アミロイド17−42はβ−アミロイド1−40よりも早く凝集するという事実に拠るのかも知れない(Pikeら、1995)。β−アミロイド1−40に対するファージの親和性は、ペプチド凝集に伴って増加する。ELISAは、可溶性β−アミロイド、1時間凝集β−アミロイドまたは3週間凝集β−アミロイドと結合するファージについて調べ、βA1−40に結合するファージにおいて有意な差を示した。それ故、高度に凝集したペプチドに対するファージのより高い親和性が、β−アミロイドβ−アミロイド17−42とのより強い相互作用を説明する可能性がある。
【0100】
線維状ファージの脳内注射によるβ−アミロイド斑の脱凝集のインビボでの研究において、注射後1時間でと殺したでマウスにおいては、斑量の減少は観察されなかった。処置後2日目にと殺したマウスにおいては、線維状ファージで処置された半球において、PBS注射半球に比較して、斑量の25%減少が観察された。処置後3日目にと殺したマウスにおいては、ファージ注射を受けた半球において、斑量の40%減少が示された(図6Aおよび6B)。
【0101】
放射能標識線維状ファージの脳内分布:
経鼻投与後1時間で早くも、線維状ファージは嗅球と脳内で検出された(図7)。ファージの消失速度を決定するためには群は小さすぎるが(n=3)、ファージの濃度は1時間後にピークに達し、24時間以内に殆ど完全に除去されると考えられる。マウス間のバラつきが大きいのは、異なった量のファージを飲み込み、吸入する結果であろう。脳実質細胞への侵入量は、与えられた量の0.0009%から0.018%とさまざまであった。血清抗体濃度の0.1%が脳に到達し斑量を減少させることは言及する価値がある。これらの予備的なデータは、BALB/cマウスにおいてファージが短時間で脳に侵入することができ、1時間後に消失が始まることを示している。それ故、線維状ファージの経鼻投与によって、アミロイド沈着を有するトランスジェニックマウス治療の効果を試験することができる。
【0102】
トランスジェニックマウスへの線維状ファージの繰り返し投与:
認識機能における有意な改善が、ファージ処置を受けない群に比較して、ファージ処置を受けたマウスにおいて認められた(データは示されない)。研究の終わりに、治療効果の総合的な推測を得るために、マウスの斑量、マイクログリア活性化、シナプス密度および有害事象の可能性を評価するための組織化学的な分析が行われる。
【0103】
考察
本発明者の研究室は、βAのN末端に部位特異的なモノクローナル抗体(mAbs)が、既に形成されたβA原線維に結合し、アミロイドを脱凝縮させて、無定型状態にし、それらの神経毒性を阻害することを、先に報告した(Solomonら, 1996, 1997)。この活性はまた、ADのためのトランスジェニックマウスにおいて、インビボでも示された(Bardら、2000)。経鼻的に抗−凝集抗体断片を脳斑に送達するために、βAに対するscFvを提示するファージを用いた(Frenkelら, 2002)。トランスジェニックマウスにおいてscFvを脳へ侵入させ、アミロイド沈着と結合させることを可能にする媒体としてファージを用いた。
【0104】
投与回数と、海馬および臭球において検出されたファージの量の間に直接、関連性があることが示された。ファージの線形構造が、種々の膜を通っての侵入性を与えることが示唆されている。ファージ投与後のマウスの脳において、いかなる病理的な兆候も、観察されなかったことは言及する価値がある(Frenkelら、2002)。
【0105】
βA原線維をThTおよび蛍光標識抗ファージ抗体との両方で可視化し、脳から線維状ファージが毒性効果を引き起こさずに消失することが組織学的研究において示された。先に報告された実験において、線維状ファージはマウスに静脈内注射され、その後、異なった器官から回収され、体内における循環の間、その完全性は影響を受けながったことが示された。
【0106】
アミロイドの繊維化は、静電的な相互作用よりは、疎水性の相互作用によって駆り立てられていると見なされている。βAは二つの疎水性部を含んでいるが、その中央部は、17〜21残基と、30〜40残基を含むC末端領域から構成される。固体NMRから得られた実験的な制約に基づいたモデルによれば、ペプチドの立体構造は、残基25〜29によって形成された180°曲げによって分離された二つのβ鎖を含んでいる。該β鎖は、側鎖−側鎖接触を通して相互作用する二つの既知の平行なβ−シートを形成する。疎水性の側鎖は外側の表面に露出しており、疎水性の面を形成する一方、荷電した極性の他の側鎖は反対側の面に、曲がりの凸面および原線維が成長するに連れて溶媒和されるN末端部に分布している(Petkovaら, 2002)。
【0107】
一方、主要ファージコートタンパクは3つの部分より構成されている:外表面(サブユニットのN末端領域によって占められており、周囲の溶媒と相互作用しウイルス粒子に低い等電点を与える酸性残基に富んでいる);殻の内部(タンパクサブユニットが主として互いに相互作用する、非極性側鎖の19残基ストレッチを含む);内部表面(サブユニットのC末端領域によって占められており、DNAコアと相互作用する塩基性残基に富んでいる)。ほぼ全てのタンパク側鎖の相互作用は、サブユニット内ではなく、コートタンパクアレイにおける異なったサブユニット間であるという事実のため、これは、高分子集合体におけるα−ヘリックスサブユニット間の相互作用の研究のための有用なモデルシステムになっている。
【0108】
本研究において、ファージは、脳にβAに対する抗体を送達する媒体としての、報告されている機能に加えて、抗凝集性を有していることが示された。実施例において提出されたデータから、線維状ファージはβ−アミロイドと相互作用し、その凝集過程に干渉し、その可溶化を引き起こすことさえできる。現在、診断は、β−アミロイド斑がすでに形成された疾患の後期になされることから、脱凝集性は大きな価値を有する。アミロイド斑を有するTgマウスにファージを頭蓋内注射した後、最大効果は3日後のみに観察されることから、この過程は時間依存性である。この効果は、電子顕微鏡写真において見ることができるように、ファージの長く薄い線維としての特異な構造がアミロイド原線維にそって組織化するのを可能とする結果であろう。この理論は、線形の構造を失った球状ファージはアミロイドの形成を阻害することができなかった事実によって明らかである。ファージ活性に寄与する主要な別の因子は、ベータシート構造に干渉するかも知れないアルファヘリックス(プロテイン8)の含量が高いことである。pVIIIサブユニットはヘリックスのアレイに封入されて、ssDNAゲノムを囲む管状構造を形成している。pVIIIのC末端は、環状構造の内側に露出しており、負に荷電したssDNAゲノムと相互作用する正荷電の残基を含んでいる。中間部は、サブユニット間の相互作用の原因である疎水性のアミノ酸に富んでいる。最後に、屈曲しやすい負荷電のN末端部は粒子の外側に露出している(Marvin, 1998)。
【0109】
線維状バクテリオファージは、他のベクターよりも明らかな利点を有する。線維状ファージ、M13、f1およびfdは構造的にも遺伝子レベルでもよく理解されている(Wilson and Finley 1998; Rodi and Makowski, 1999)。それらは抗原および/または抗体を提示するために、遺伝子操作されており、その表面に外来タンパクを提示する、異なった生物システムに用いられた(Scott等、1990; McCaffertyら、 1990)。原核生物の感染、集合および複製に関して進化したことから、バクテリオファージは、哺乳動物細胞で増殖することもできず、それに対する天然指向性も示さない。このことは、非特異的な遺伝子送達の機会を最小とする。ファージベクターは、動物細胞において複製可能な実態を作ることは有りそうもないことから、ウイルスよりもより安全である可能性が高い。
【0110】
脱凝集剤としてバクテリオファージを用いる別の利点は、細菌培養の増殖により、より多くの材料を得る事ができることから、製造が容易であることである。放射能標識アッセイによれば、ファージは脳実質細胞に1時間未満で侵入する。それらの消失は多分その後すぐ始まるであろう。投与された用量から侵入したファージの百分率は0.0009%から0.018%までまちまちである。マウス間のバラつきを最小にするような、更なる研究が行われるべきであろう。用量を最小にし、点滴のそれぞれの滴下の間隔を長くすることは、良いことであろう。対照的に、抗体の血清濃度から0.1%が脳に達している。ファージの分子量は抗体のそれよりも二桁大きいが、経鼻投与はファージ侵入を引き起こすことになりうる。
【0111】
本発明を完全に説明したので、本発明の精神と範囲から逸脱することなしに、また必要以上の実験を行うことなしに、相当するパラメータ、濃度および条件の広い範囲内で、当業者は、当然のことながら同様のことを行うことができる。
【0112】
本発明を、その具体的な実施態様と関連して説明したが、更に変更することができることは理解される。本出願は、本発明の原理に一般的に従って、いかなる変異、使用または適応を包含することを意図しているが、本開示からの以下のような逸脱をも含める:すなわち本発明に関する技術内で、既知のまた慣習的な慣行内にあるもの、および添付の請求の範囲において以下のように記述された、上記の本質的な特徴にも適用してもよい。
【0113】
本明細書において引用されている専門誌の記事、抄録、公表されたあるいは対応する米国あるいは外国の特許出願(米国または外国で発行された特許)、または他の全ての参考文献を含む、全ての参考資料は、引用において提示されたデータすべて、表、図およびテクスト含めて、参照により本明細書に援用される。更に、引用文献内の全内容も、参照により本明細書に援用される。
【0114】
既知の段階法、従来の段階法、既知の方法または従来の方法の引用は、本発明のいかなる態様、説明または実施例も、関連技術において開示され、教示されあるいは示唆されることを認めるものではない。
【0115】
具体的な実施態様についての前述の説明により、本発明の一般的な性質が十分に明らかにされるので、他者は、当業の技術内での知識(本明細書で引用された文献の内容を含めて)を適用することにより、過度の実験をしないで、また本発明の一般的な概念から逸脱しないで、具体的な実施態様のような種々の応用のために容易に変更および/あるいは適応することができる。それ故、そのような適応および変更は、本明細書に提示される教示と指導に基づく、開示された実施態様と同等の意味と範囲内にあるように意図している。本明細書における言い回しまたは述語は、ここに提示される教示や指導に照らして、当技術分野における通常技術の一つと組み合わせて当業者に解釈されるように説明するためのものであり、限定するためのものではないことを理解されたい。
【0116】
【表1】
【図面の簡単な説明】
【0117】
【図1】脳の断面の模式図である。
【図2A】β−アミロイド1−40凝集の脱凝集および阻止を示すグラフである。
【図2B】β−アミロイド1−40凝集の脱凝集および阻止を示すグラフである。
【図3A】β−アミロイドの電子顕微鏡写真である。
【図3B】β−アミロイドの電子顕微鏡写真である。
【図3C】β−アミロイドの電子顕微鏡写真である。
【図3D】β−アミロイドの電子顕微鏡写真である。
【図3E】β−アミロイドの電子顕微鏡写真である。
【図3F】β−アミロイドの電子顕微鏡写真である。
【図3G】β−アミロイドの電子顕微鏡写真である。
【図3H】β−アミロイドの電子顕微鏡写真である。
【図3I】β−アミロイドの電子顕微鏡写真である。
【図3J】β−アミロイドの電子顕微鏡写真である。
【図3K】β−アミロイドの電子顕微鏡写真である。
【図4A】電子顕微鏡写真である。
【図4B】電子顕微鏡写真である。
【図4C】電子顕微鏡写真である。
【図5A】β−アミロイド毒性の阻害を示すグラフである。
【図5B】β−アミロイド毒性の阻害を示すグラフである。
【図5C】β−アミロイド毒性の阻害を示すグラフである。
【図6A】線維状ファージを注入した図である。
【図6B】PBSのみを注入した図である。
【図7】マウスにおけるファージの分布を示すグラフである。
【特許請求の範囲】
【請求項1】
アミロイド沈着に付随する炎症、およびアミロイド沈着に付随する、または活性化されたマイクログリアにかかわる脳の炎症を抑制または治療する方法であって、それを必要とする患者に、アミロイド沈着に付随する炎症、およびアミロイド沈着に付随する、または活性化されたマイクログリアにかかわる脳の炎症を抑制または治療するために、野性型の線維状バクテリオファージ、または抗体もしくは非線維状バクテリオファージ抗原をその表面に提示しない線維状バクテリオファージを有効量投与することからなる方法。
【請求項2】
該線維状バクテリオファージが、M13、f1およびfdバクテリオファージ、ならびにそれらの混合物よりなる群から選択される、請求項1に記載の方法。
【請求項3】
該線維状バクテリオファージがM13である、請求項1に記載の方法。
【請求項4】
該脳の炎症が、線維状バクテリオファージの有効量を経鼻的に投与することにより抑制または治療される、請求項1に記載の方法。
【請求項5】
アミロイド沈着に付随する、または活性化されたマイクログリアにかかわる該脳の炎症が、斑形成疾患である、請求項1に記載の方法。
【請求項6】
該斑形成疾患がアルツハイマー病である、請求項5に記載の方法。
【請求項7】
該斑形成疾患がプリオン病である、請求項5に記載の方法。
【請求項8】
該アミロイド沈着に付随する炎症が多発性骨髄腫である、請求項1に記載の方法。
【請求項9】
該アミロイド沈着に付随する炎症が腎アミロイドーシスである、請求項1に記載の方法。
【請求項10】
医薬的に許容できる担体または賦形剤、および活性成分として、野性型の線維状バクテリオファージ、または抗体もしくは非線維状バクテリオファージ抗原をその表面に提示しない線維状バクテリオファージを含む医薬組成物。
【請求項11】
前記線維状バクテリオファージがM13、f1、fdおよびそれらの混合物よりなる群から選択される、請求項10に記載の医薬組成物。
【請求項12】
線維状バクテリオファージを斑形成ペプチドと接触させてアミロイド沈着を抑制する、アミロイド沈着の形成を抑制する方法であって、該線維状バクテリオファージが、野生型の線維状バクテリオファージ、または抗体もしくは非線維状バクテリオファージ抗原をその表面に提示しない線維状バクテリオファージである方法。
【請求項13】
該線維状バクテリオファージがM13、f1、およびfdバクテリオファージならびにそれらの混合物よりなる群から選択される、請求項12に記載の方法。
【請求項14】
線維状バクテリオファージを既に形成されたアミロイド沈着と接触させて、既に形成されたアミロイド沈着を脱凝集する、既に形成されたアミロイド沈着を脱凝集する方法であって、該線維状バクテリオファージが野生型の線維状バクテリオファージ、または抗体もしくは非線維状バクテリオファージ抗原をその表面に提示しない線維状バクテリオファージである方法。
【請求項15】
該線維状バクテリオファージがM13、f1、およびfdバクテリオファージならびにそれらの混合物よりなる群から選択される、請求項14に記載の方法。
【請求項16】
アミロイド沈着に付随する炎症、およびアミロイド沈着に付随する、または活性化されたマイクログリアにかかわる脳の炎症を抑制または治療する医薬の調製における、野性型の線維状バクテリオファージ、または抗体もしくは非線維状バクテリオファージ抗原をその表面に提示しない線維状バクテリオファージの使用。
【請求項17】
アミロイド沈着に付随する、または活性化されたマイクログリアにかかわる該脳の炎症が、アルツハイマー病およびプリオン病より選択される斑形成疾患である、請求項16に記載の使用。
【請求項18】
アミロイド沈着に付随する該炎症が多発性骨髄腫および腎アミロイドーシスより選択される、請求項16に記載の使用。
【請求項19】
該線維状バクテリオファージがM13、f1、fdバクテリオファージおよびそれらのいずれかの混合物より選択される、請求項16に記載の使用。
【請求項20】
アミロイド沈着の形成を抑制するための医薬の調製における、野生型の線維状バクテリオファージ、または抗体もしくは非線維状バクテリオファージ抗原をその表面に提示しない線維状バクテリオファージの使用。
【請求項21】
該線維状バクテリオファージがM13、f1、fdバクテリオファージおよびそれらのいずれかの混合物より選択される、請求項20に記載の使用。
【請求項22】
既に形成されたアミロイド沈着を脱凝集するための薬剤の調製における、野生型の線維状バクテリオファージ、または抗体もしくは非線維状バクテリオファージ抗原をその表面に提示しない線維状バクテリオファージの使用。
【請求項23】
該線維状バクテリオファージがM13、f1、fdバクテリオファージおよびそれらのいずれかの混合物より選択される、請求項22に記載の使用。
【請求項1】
アミロイド沈着に付随する炎症、およびアミロイド沈着に付随する、または活性化されたマイクログリアにかかわる脳の炎症を抑制または治療する方法であって、それを必要とする患者に、アミロイド沈着に付随する炎症、およびアミロイド沈着に付随する、または活性化されたマイクログリアにかかわる脳の炎症を抑制または治療するために、野性型の線維状バクテリオファージ、または抗体もしくは非線維状バクテリオファージ抗原をその表面に提示しない線維状バクテリオファージを有効量投与することからなる方法。
【請求項2】
該線維状バクテリオファージが、M13、f1およびfdバクテリオファージ、ならびにそれらの混合物よりなる群から選択される、請求項1に記載の方法。
【請求項3】
該線維状バクテリオファージがM13である、請求項1に記載の方法。
【請求項4】
該脳の炎症が、線維状バクテリオファージの有効量を経鼻的に投与することにより抑制または治療される、請求項1に記載の方法。
【請求項5】
アミロイド沈着に付随する、または活性化されたマイクログリアにかかわる該脳の炎症が、斑形成疾患である、請求項1に記載の方法。
【請求項6】
該斑形成疾患がアルツハイマー病である、請求項5に記載の方法。
【請求項7】
該斑形成疾患がプリオン病である、請求項5に記載の方法。
【請求項8】
該アミロイド沈着に付随する炎症が多発性骨髄腫である、請求項1に記載の方法。
【請求項9】
該アミロイド沈着に付随する炎症が腎アミロイドーシスである、請求項1に記載の方法。
【請求項10】
医薬的に許容できる担体または賦形剤、および活性成分として、野性型の線維状バクテリオファージ、または抗体もしくは非線維状バクテリオファージ抗原をその表面に提示しない線維状バクテリオファージを含む医薬組成物。
【請求項11】
前記線維状バクテリオファージがM13、f1、fdおよびそれらの混合物よりなる群から選択される、請求項10に記載の医薬組成物。
【請求項12】
線維状バクテリオファージを斑形成ペプチドと接触させてアミロイド沈着を抑制する、アミロイド沈着の形成を抑制する方法であって、該線維状バクテリオファージが、野生型の線維状バクテリオファージ、または抗体もしくは非線維状バクテリオファージ抗原をその表面に提示しない線維状バクテリオファージである方法。
【請求項13】
該線維状バクテリオファージがM13、f1、およびfdバクテリオファージならびにそれらの混合物よりなる群から選択される、請求項12に記載の方法。
【請求項14】
線維状バクテリオファージを既に形成されたアミロイド沈着と接触させて、既に形成されたアミロイド沈着を脱凝集する、既に形成されたアミロイド沈着を脱凝集する方法であって、該線維状バクテリオファージが野生型の線維状バクテリオファージ、または抗体もしくは非線維状バクテリオファージ抗原をその表面に提示しない線維状バクテリオファージである方法。
【請求項15】
該線維状バクテリオファージがM13、f1、およびfdバクテリオファージならびにそれらの混合物よりなる群から選択される、請求項14に記載の方法。
【請求項16】
アミロイド沈着に付随する炎症、およびアミロイド沈着に付随する、または活性化されたマイクログリアにかかわる脳の炎症を抑制または治療する医薬の調製における、野性型の線維状バクテリオファージ、または抗体もしくは非線維状バクテリオファージ抗原をその表面に提示しない線維状バクテリオファージの使用。
【請求項17】
アミロイド沈着に付随する、または活性化されたマイクログリアにかかわる該脳の炎症が、アルツハイマー病およびプリオン病より選択される斑形成疾患である、請求項16に記載の使用。
【請求項18】
アミロイド沈着に付随する該炎症が多発性骨髄腫および腎アミロイドーシスより選択される、請求項16に記載の使用。
【請求項19】
該線維状バクテリオファージがM13、f1、fdバクテリオファージおよびそれらのいずれかの混合物より選択される、請求項16に記載の使用。
【請求項20】
アミロイド沈着の形成を抑制するための医薬の調製における、野生型の線維状バクテリオファージ、または抗体もしくは非線維状バクテリオファージ抗原をその表面に提示しない線維状バクテリオファージの使用。
【請求項21】
該線維状バクテリオファージがM13、f1、fdバクテリオファージおよびそれらのいずれかの混合物より選択される、請求項20に記載の使用。
【請求項22】
既に形成されたアミロイド沈着を脱凝集するための薬剤の調製における、野生型の線維状バクテリオファージ、または抗体もしくは非線維状バクテリオファージ抗原をその表面に提示しない線維状バクテリオファージの使用。
【請求項23】
該線維状バクテリオファージがM13、f1、fdバクテリオファージおよびそれらのいずれかの混合物より選択される、請求項22に記載の使用。
【図1】

【図2A】
【図2B】
【図3A】
【図3B】
【図3C】

【図3D】

【図3E】
【図3F】
【図3G】
【図3H】

【図3I】

【図3J】

【図3K】

【図4A】

【図4B】

【図4C】

【図5A】
【図5B】
【図5C】
【図6A】

【図6B】

【図7】
【図2A】
【図2B】
【図3A】
【図3B】
【図3C】
【図3D】
【図3E】
【図3F】
【図3G】
【図3H】
【図3I】
【図3J】
【図3K】
【図4A】
【図4B】
【図4C】
【図5A】
【図5B】
【図5C】
【図6A】
【図6B】
【図7】
【公表番号】特表2008−528688(P2008−528688A)
【公表日】平成20年7月31日(2008.7.31)
【国際特許分類】
【出願番号】特願2007−554157(P2007−554157)
【出願日】平成18年1月31日(2006.1.31)
【国際出願番号】PCT/US2006/003291
【国際公開番号】WO2006/083795
【国際公開日】平成18年8月10日(2006.8.10)
【出願人】(502435775)ラモット アット テル−アビブ ユニバーシティ リミティド (3)
【Fターム(参考)】
【公表日】平成20年7月31日(2008.7.31)
【国際特許分類】
【出願日】平成18年1月31日(2006.1.31)
【国際出願番号】PCT/US2006/003291
【国際公開番号】WO2006/083795
【国際公開日】平成18年8月10日(2006.8.10)
【出願人】(502435775)ラモット アット テル−アビブ ユニバーシティ リミティド (3)
【Fターム(参考)】
[ Back to top ]