
アモキシシリン三水和物
本発明は、平衡相対湿度30%および温度25℃で測定した自由水含有量が0.1重量%未満、好ましくは0.07重量%未満、より好ましくは0.05重量%未満のアモキシシリン三水和物の製品に関する。この製品は、有利には、クラブラン酸との混合物で使用される。本発明はまた、この新製品の調製方法にも関する。
【発明の詳細な説明】
【発明の詳細な説明】
【0001】
本発明は、アモキシシリン三水和物の製品に関する。
【0002】
固体状態のβ−ラクタム系抗生物質内の水は様々な形で存在することができる。水は、例えば結晶水として存在することができる。結晶水とは、β−ラクタム系抗生物質の分子構造に一体化された水のことである。アモキシシリン1分子当り3分子の結晶水を含むアモキシシリン三水和物内の結晶水の量は、約12.9%の結晶水に相当する。自由水とは、大気と交換できる水のことである。自由水の量には、結晶水として存在する水の量は含まれていない。
【0003】
β−ラクタム系抗生物質の試料中の自由水含有量と、この試料に接触している空気の相対湿度とは、相互に影響を及ぼし合っている。β−ラクタム系抗生物質の試料を空気に接触させると、一般に平衡状態が確立されるまで前記試料と空気の間で水の交換が行われる。平衡状態においては、試料とこの試料に接触している空気の間の水の正味の交換はゼロである。一般に、所定温度での上記の平衡状態における所定の相対湿度の値について、β−ラクタム系抗生物質の試料の自由水含有量を決める。この相対湿度の値を平衡相対湿度とも呼ぶ。
【0004】
試料の自由水含有量は、動的蒸気収着によって測定することができる。根底にある原理は、事前に相対湿度を調整した空気に試料を接触させながら、その重量を監視することである。水を吸収または放出する結果として、平衡状態が確立されるまで、試料重量が変化する。この平衡状態における試料重量が、平衡相対湿度に対応する試料重量である。この平衡相対湿度は事前に調整された空気の相対湿度であり、この空気に試料を接触させる。様々な値の平衡相対湿度についてこの手段を繰返すことによって、平衡相対湿度の関数として試料重量を測定することができる。温度Tにおける試料の自由水含有量は、((WERH−Wref)/Wref)×100%によって与えられる。但し、WERH=温度Tにおける平衡相対湿度ERHに対応する試料重量、およびWref=温度Tにおける平衡相対湿度の基準値に対応する試料重量である。この基準値は、前記平衡相対湿度における自由水含有量がゼロに近くなるように選択される。
【0005】
β−ラクタム系抗生物質を利用する際は、自由水の存在が関心事になる可能性がある。これは、例えばアモキシシリン三水和物を第2の薬学的に活性な薬剤、例えばクラブラン酸と混合する場合である。したがって、従来技術は、アモキシシリン三水和物を所定の程度まで乾燥することを提案している。
【0006】
水分活性は平衡相対湿度を100%で除したものとして定義され、β−ラクタム系抗生物質を乾燥するべき限度を規定する方法である。水分活性は、比較的小さい容積を有する密閉されたチャンバーに所定量の試料を入れ、相対湿度が一定になるまで時間の関数として相対湿度を測定することによって測定することができる。この相対湿度がその試料の平衡相対湿度である。水に関連した問題が一定の役割を演ずる用途については、β−ラクタム系抗生物質の水分活性に対して概ね低い値が規定される。
【0007】
乾燥の程度が不十分であると、水に関連した問題が残る。乾燥を過度に行うと、色や安定性などの物性が損なわれる恐れがある。これは、例えば、乾燥を過度に行うと結晶水が追い出されることによるものである可能性がある。
【0008】
本発明者等は、驚くべきことに、平衡相対湿度30%および温度25℃で測定した自由水含有量が0.10重量%未満のアモキシシリン三水和物の製品、ならびにこれを調製する方法を見出した。
【0009】
したがって、本発明は、従来技術による同じ水分活性を有するアモキシシリン三水和物と比べて、水に関する問題が無い、または少ない、アモキシシリン三水和物の製品を提供する。さらに、行われる乾燥の程度を下げることができるので、色や安定性などの特性は損なわれないか、または損なわれる程度が少ない。その自由水含有量が低くなっているので、このアモキシシリン三水和物は、水分の影響を非常に受けやすいクラブラン酸またはその塩と有利に混合することができる。
【0010】
本明細書で用いる、平衡相対湿度30%および温度25℃で測定した自由水含有量は、具体的には、((W30−W10)/W10)×100%として定義される。但し、
W30=温度25℃における平衡相対湿度30%に対応する試料重量、
W10=温度25℃における平衡相対湿度10%に対応する試料重量である。
【0011】
W30およびW10に対する値を含めて、自由水含有量は、動的蒸気収着を用いて、例えばVTI−SGA 100蒸気収着分析装置を用いて測定することが好ましい。この技法を用いて、試料チャンバー内の空気を相対湿度10%で90分間コンディショニングし、その後10%刻みで相対湿度を上昇させ、各値の相対湿度に試料を90分間保持し、ある値の相対湿度で90分後に測定した試料重量を平衡相対湿度に対応した試料重量として用いることによって、重量200mgの試料について吸着項を測定することが好ましい。
【0012】
本発明によるアモキシシリン三水和物の製品は、平衡相対湿度30%および温度25℃で測定した自由水含有量が0.10重量%未満である。好ましくは、本発明によるアモキシシリン三水和物の製品は、平衡相対湿度30%および温度25℃で測定した自由水含有量が0.07重量%未満であり、より好ましくは0.05重量%未満である。自由水含有量に特定の上限はない。平衡相対湿度30%および温度25℃で測定した自由水含有量は、例えば0.01重量%より高くてもよい。
【0013】
本発明によるアモキシシリン三水和物の製品は、任意の適切な形、例えば結晶性粉末または顆粒または結晶性粉末と顆粒の混合物の形のアモキシシリン三水和物とすることができる。好ましい実施形態においては、本発明による製品は、平衡相対湿度30%および温度25℃で測定した自由水含有量が0.1重量%未満、好ましくは0.07重量%未満、より好ましくは0.05重量%未満のアモキシシリン三水和物結晶性粉末である。
【0014】
アモキシシリン三水和物の製品は依然として何らかの不純物を含有している可能性があることは理解されよう。好ましくは、アモキシシリン三水和物の製品は、少なくとも90重量%、好ましくは少なくとも95重量%、より好ましくは少なくとも98重量%のアモキシシリン三水和物を含有する。これらの重量百分率は製品重量に対するものである。好ましくは、本発明によるアモキシシリン三水和物の製品には助剤は含まれていない。
【0015】
本明細書で用いるアモキシシリン三水和物結晶性粉末とは、具体的には、主としてアモキシシリン三水和物の結晶からなる製品のことである。結晶とは、例えば、水またはデンプンのりなどの結合剤、あるいはローラー圧縮成形または押出成形などの機械的な力の助けを借りて、結晶を付着させることによって形成した凝集体のことではないことは理解されよう。通常の取扱い中、例えば乾燥中に、意図せずに凝集体が生成される可能性もある。凝集体は、倍率140倍で光学顕微鏡を用いて観察することができる。本明細書で用いる、主としてアモキシシリン三水和物の結晶からなる製品とは、具体的には、アモキシシリン三水和物の結晶を少なくとも70重量%、好ましくは少なくとも80重量%、より好ましくは少なくとも90重量%、より好ましくは少なくとも95重量%、より好ましくは少なくとも98重量%含む製品のことである。これらの百分率は、風力篩と光学顕微鏡を組み合わせて用いて測定することができる。風力篩い別けは、風力篩Alpine Air Jet 200LS−Nを用いて、重量10gの試料について1200Paで1分間行うのが有利である。光学顕微鏡検査は、前記部分の試料5mgを採取し、この試料を表面積22×40mmの表面上の4滴のパラフィンオイルに懸濁し、倍率140倍を用いて行うのが有利である。
【0016】
アモキシシリン三水和物粉末は依然として何らかの不純物を含有している可能性があることは理解されよう。好ましくは、アモキシシリン三水和物の製品は、少なくとも90重量%、好ましくは少なくとも95重量%、より好ましくは少なくとも98重量%のアモキシシリン三水和物を含有する。これらの重量百分率は結晶性粉末の重量に対するものである。好ましくは、本発明によるアモキシシリン三水和物結晶性粉末には助剤は含まれていない。
【0017】
好ましい実施形態においては、本発明によるアモキシシリン三水和物の製品の水分活性は、0.05より高く、好ましくは0.07より高く、好ましくは0.10より高く、好ましくは0.15より高く、好ましくは0.20より高く、好ましくは0.25より高く、好ましくは0.30より高い。自由水の量が依然として低いままで、アモキシシリン三水和物の特性が低下しない、またはその低下の程度が小さいので、水分含有量が高いことは有利である。水分活性についての特定の下限はない。実際、水分活性は、一般に0.7未満、例えば0.6未満、例えば0.5未満である。但し、これは必要ではない。本明細書で用いるこれらの値は、25℃における水分活性のことである。
【0018】
試料の水分活性を測定する好ましい方法は、比較的小さい容積を有する密閉されたチャンバーに所定量の試料を入れ、相対湿度が一定になる(例えば30分後)まで時間の関数として相対湿度を測定することである。この相対湿度がその試料の平衡相対湿度である。好ましくは、Novasina TH200 Thermoconstanterを用いる。その試料ホルダーの容積は12mlであり、3gの試料を充填される。
【0019】
d10およびd50は、粒度分布を示すための公知の方法である。d50は、結晶の50容積%がその値より小さい粒径を有するような粒径の値を意味する。d50はまた、容積基準の平均粒径のことでもある。同様に、d10は、結晶の10容積%がその値より小さい粒径を有するような粒径の値を意味する。d10およびd50を測定するための好ましい方法は、レーザー回折であり、好ましくはMalvern装置を用いる。d10およびd50のための好適な装置は、粒度分布を示すための公知の方法である。d50は、粒子の50容積%がその値より小さい粒径を有するような粒径の値を意味する。d50はまた、容積基準の平均粒径のことでもある。同様に、d10は、粒子の10容積%がその値より小さい粒径を有するような粒径の値を意味する。d10およびd50を測定するための好ましい方法は、レーザー回折であり、好ましくはMalvern装置を用いる。d10およびd50を測定するための好適な装置は、Malvern Instruments Ltd.、Malvern Ukから入手可能なMalvern粒径測定器2600Cであり、f=300mmおよびビーム長14.30mmの対物レンズを用いる。多分散分析モデルを用いることが有利である。
【0020】
本発明者等は、本発明によるアモキシシリン三水和物結晶性粉末は、d50が増大していることを見出した。したがって、本発明はまた、d50が、10μmより大きい、好ましくは20μmより大きい、より好ましくは30μmより大きい、より好ましくは35μmより大きい、より好ましくは40μmより大きいアモキシシリン三水和物結晶性粉末も提供する。d50についての特定の上限はない。本発明による結晶性粉末のd50は、150μm未満、例えば100μm未満とすることができる。本発明による結晶性粉末は、好ましくはd10が増大しており、好ましくは3μmより大きく、好ましくは5μmより大きく、より好ましくは8μmより大きく、より好ましくは10μmより大きい。本発明による結晶性粉末のd10についての特定の上限はない。本発明による結晶性粉末のd10は、50μm未満とすることができる。
【0021】
アモキシシリン三水和物結晶性粉末は、溶解したアモキシシリンを含む溶液を調製し、前記溶液からアモキシシリンを結晶化させて結晶を形成し、前記溶液からこの結晶を分離し、分離された結晶を乾燥することによって得ることができる。本明細書において用いる結晶性粉末という用語には、それだけに限らないが、この方法によって得られたおよび/または得ることができる乾燥製品が含まれる。
【0022】
所与の平衡相対湿度での自由水含有量が低下した結晶性粉末を調製するステップは、好ましくは、この方法、具体的には結晶化、分離および乾燥の各ステップを、乾燥結晶の粒経、具体的にはd50および/またはd10が増大するような条件下で実施するステップを含むことが見出された。
【0023】
好ましい結晶化条件としては、溶液から結晶化するアモキシシリン三水和物の粒径が増大しているような結晶化条件が挙げられる。これは、例えば比較的長い滞在時間を適用すること、水溶液において比較的低いアモキシシリン濃度を適用すること、または純度の高い水溶液を使用することによって実現することができる。その他の好ましい条件については以下に説明する。
【0024】
例えば分離および/または乾燥時の機械的な衝撃の程度が粒径に影響を及ぼすことがある。分離中の機械的な衝撃は、例えば遠心分離中に与えることができる。乾燥中の機械的な衝撃は、例えば接触式乾燥機、例えばVrieco−Nauta接触式乾燥機、またはフラッシュドライヤーを用いて与えることができる。機械的な衝撃はまた、空気輸送、例えば分離工程から乾燥工程へのアモキシシリン三水和物の空気輸送を利用して与えることもできる。過大な機械的衝撃を加えると、粒径の望ましくない低下をもたらす恐れがある。本発明によって提供されるこの洞察を用い、かつ機械的な力を変化させることによって、当業者は、望ましくない粒径の低下を回避することができる条件を見出すことができる。
【0025】
上記を考慮して、本発明はまた、溶液からアモキシシリン三水和物を結晶化させるステップと、前記溶液から結晶を分離するステップと、分離された結晶を乾燥するステップとを含むアモキシシリン三水和物結晶性粉末の調製方法であって、この方法、好ましくは結晶化、分離および/または乾燥の各ステップが、得られる結晶性粉末のd50が10μmより大きく、好ましくは20μmより大きく、より好ましくは30μmより大きく、特に35μmより大きく、より好ましくは40μmより大きくなるような条件下で行われる方法を提供する。d50には特定の上限はない。この方法、好ましくは結晶化、分離および/または乾燥の各ステップは、例えば、得られる結晶性粉末のd50が150μm未満、例えば100μm未満になるような条件下で行うことができる。この方法、好ましくは結晶化、分離および/または乾燥の各ステップは、乾燥した結晶のd10が3μmより大きく、好ましくは5μmより大きく、より好ましくは8μmより大きく、より好ましくは10μmより大きくなるような条件下で行われることが好ましい。d10には特定の上限はない。この方法、好ましくは結晶化、分離および/または乾燥の各ステップは、例えば、得られる結晶性粉末のd10が50μm未満になるような条件下で行うことができる。
【0026】
本発明者等は、自由水含有量が低下したアモキシシリン三水和物の結晶は、好ましくは、以下に記載の好ましいプロセス条件を適用することによって得られることを見出した。
【0027】
好ましくは、本発明によるアモキシシリン三水和物結晶性粉末の調製方法は、6−アミノ−ペニシラン酸またはその塩を、担体上に固定化された酵素の存在下、活性状態のp−ヒドロキシフェニルグリシンと反応させることによってアモキシシリンを調製するステップと、塩酸を含有する、アモキシシリン含有水溶液を形成するステップと、前記水溶液からアモキシシリン三水和物を結晶化させるステップとを含む。
【0028】
好ましくは、アモキシシリン三水和物を結晶化させる溶液は水溶液である。任意の適切な水溶液を使用することができる。適切な水溶液としては、水:有機溶媒の重量比が、100:0から70:30の範囲、好ましくは100:0から80:20の範囲、好ましくは100:0から90:10の範囲、好ましくは100:0から95:5の範囲、好ましくは100:0から99:1の範囲の溶液が挙げられる。
【0029】
好ましくは、アモキシシリン三水和物を結晶化させる溶液は、1,000,000重量部のアモキシシリン(溶解した形であるか否かにかかわらず、アモキシシリンの総濃度)当り、200重量部未満のタンパク質、好ましくは100重量部未満のタンパク質、より好ましくは50重量部未満のタンパク質、より好ましくは35重量部未満のタンパク質を含有する。
【0030】
好ましくは、アモキシシリン三水和物を結晶化させる溶液は、アモキシシリン濃度(溶解した形であるか否かにかかわらず、アモキシシリンの総濃度)が、0.6モル/l未満、好ましくは0.5モル/l未満、より好ましくは0.4モル/l未満、より好ましくは0.3モル/l未満の水溶液である。
【0031】
アモキシシリン三水和物を結晶化させる水溶液は、塩酸または塩化物を含有する溶液であることが好ましい。アモキシシリン三水和物を結晶化させる水溶液は、アモキシシリン(溶解した形であるか否かにかかわらず、アモキシシリンの総濃度)1モル当り0.9から5モルの塩酸または塩化物、好ましくはアモキシシリン1モル当り0.9から3モルの塩酸または塩化物、より好ましくはアモキシシリン1モル当り0.9から1.5モルの塩酸または塩化物を含有することが好ましい。アモキシシリンを結晶化させる水溶液は、アモキシシリン1モル当り1.0モルを超える塩酸または塩化物を含有することが好ましい。
【0032】
アモキシシリン三水和物は、pH2から7の範囲、好ましくは3から6の範囲で水溶液から結晶化させることが好ましい。この方法は、好ましくはpH2から5の範囲、好ましくは3から4の範囲の第1のステップと、第1のステップより高いpH、好ましくはpH4から7の範囲、好ましくは4.5から6の範囲の第2のステップとで、アモキシシリン三水和物を水溶液から結晶化させるステップを含むことが好ましい。
【0033】
5℃から40℃の範囲、好ましくは10から30℃の範囲、より好ましくは15から25℃の範囲の温度で、水溶液からアモキシシリン三水和物を結晶化させることが好ましい。
【0034】
アモキシシリン三水和物を結晶化させる溶液は、任意の適切な方法で調製することができる。溶解したアモキシシリンを含有する水溶液は、アモキシシリン三水和物を溶解することによって調製することができる。アモキシシリン三水和物を溶液に添加し、添加したアモキシシリン三水和物を溶解させることが可能である。また、溶液中にin situでアモキシシリン三水和物の結晶を形成することによって水性懸濁液を調製し、前記懸濁液中のアモキシシリン三水和物の結晶を溶解させることも可能である。アモキシシリンの調製方法においては、溶解したアモキシシリンを含有する水溶液であって、アモキシシリン濃度が0.6モル/l未満、好ましくは0.5モル/l未満、より好ましくは0.4モル/l未満、より好ましくは0.3モル/l未満の水溶液を調製するステップを含むことが好ましい。この方法は、溶解したアモキシシリンを含有する水溶液であって、pHが0から1.5の範囲、好ましくは0.5から1.2の範囲の水溶液を調製するステップを含むことが好ましい。アモキシシリンの溶解は、任意の適切な方法、例えばアモキシシリン三水和物の結晶を含有する水性懸濁液に酸を添加することによって、好ましくは塩酸を添加することによって行うことができる。酸好ましくは塩酸の添加量は、アモキシシリン1モル当り塩酸0.9から5モル、好ましくはアモキシシリン1モル当り塩酸0.9から3モル、より好ましくはアモキシシリン1モル当り塩酸0.9から1.5モルとすることができる。アモキシシリン1モル当り1.0モルを超える塩酸を添加することが好ましい。好ましい実施形態においては、この方法は、(水)溶液または(水性)懸濁液を、pH1.5未満、好ましくは1.2未満に、60分未満、好ましくは30分未満、より好ましくは15分未満、より好ましくは10分未満、より好ましくは8分未満の時間保持するステップを含む。これにより、アモキシシリンの純度を改良することができる。この方法は、水溶液または水性懸濁液を、急速混合ミキサー、例えばスタティックミキサーを用いて、酸と混合するステップを含むことが好ましい。これにより、水溶液または水性懸濁液を低pHに保持する時間を短縮することができる。酸と水性懸濁液との混合は、例えば−5℃より高い、例えば5℃より高い、例えば10℃より高い、例えば15℃より高い、例えば50℃より低い、例えば40℃より低い任意の適切な温度で行うことができる。この方法は、前記結晶化ステップの前に溶液を濾過するステップを含むことが好ましい。この方法は、溶解したアモキシシリンを含有する水溶液であって、そのpHが好ましくは0から1.5の範囲、好ましくは0.5から1.2の範囲の水溶液を濾過するステップを含むことが好ましい。溶液は、任意の適切なフィルターを通すことができる。孔径が40μm未満、好ましくは20μm未満、好ましくは10μm未満、より好ましくは5μm未満のフィルターを使用することが好ましい。
【0035】
アモキシシリン三水和物は、有利には、例えばNaOHなどの塩基を添加することによってpHを上昇させて前記水溶液から結晶化させることができる。
【0036】
結晶化は、バッチ式でまたは連続的に行うことができる。バッチ式で行う場合、種晶を水溶液に添加することが好ましい。好ましくは、結晶化は連続的に行われる。
【0037】
アモキシシリンは、6−アミノ−ペニシラン酸またはその誘導体、例えば6−アミノ−ペニシラン酸の塩を、水性反応媒体中で酵素の存在下、活性状態のp−ヒドロキシフェニルグリシンから選択されるアシル化剤と反応させることによって調製することが好ましい。活性状態のp−ヒドロキシフェニルグリシンは、好ましくは、p−ヒドロキシフェニルグリシンのエステルまたはアミドである。好適なエステルとしては、例えば、炭素原子1から4個のアルキルエステル、例えばメチルエステル、エチルエステル、n−プロピルまたはイソプロピルエステルが挙げられる。グリコールエステル、例えばエチレングリコールエステルも使用することができる。−CONH2基が置換されていないアミドも使用することができる。
【0038】
酵素は、加水分解活性を有する任意の酵素(加水分解酵素)とすることができる。酵素は、例えばアシラーゼ、とりわけペニシリンGアシラーゼ、アミダーゼまたはエステラーゼとすることができる。酵素は、様々な天然微生物、例えば真菌および細菌から分離することができる。ペニシリンアシラーゼを生成することが見出されている微生物は、例えば、アセトバクター属、エーロモナス属、アルカリゲネス属、アファノクラジウム属(Aphanocladium)、桿菌属(Bacillus sp.)、セファロスポリウム属、エシェリヒア属、フラボバクテリウム属、クライベラ属(Kluyvera)、マイコプラナ属(Mycoplana)、プロタミノバクター属(Protaminobacter)、シュードモナス属またはキサントモナス属の種が挙げられる。
【0039】
酵素の存在下においてアモキシシリンを調製する方法は、国際公開第9201061号パンフレット、国際公開第9417800号パンフレット、国際公開第9704086号パンフレット、国際公開第9820120号パンフレット、欧州特許出願公開第771357号明細書に記載されており、これらの内容を参照により本明細書に組み込む。
【0040】
反応は、任意の適切なpH、好ましくは5から9の範囲、好ましくは5.5から8の範囲、より好ましくは6から7.5の範囲のpHで行うことができる。反応は、任意の適切な温度、例えば、0から40℃の範囲、好ましくは0から30℃の範囲、より好ましくは0から15℃の範囲の温度で行うことができる。
【0041】
生成したアモキシシリンは、反応を行った条件で結晶化することができる。アモキシシリンの結晶化は、例えば、5から8の範囲、好ましくは5.5から7.5の範囲のpHで行うことが好ましい。
【0042】
好ましくは、酵素は担体上に固定化された酵素である。任意の適切な担体を使用することができる。好ましくは、担体はゲル化剤および遊離アミノ基を含むポリマーを含む。好ましくは、ポリマーはアルギン酸アンモニウム、キトサン、ペクチン、またはポリエチレンイミンから選択される。好ましくは、ゲル化剤はゼラチンである。この担体およびその調製は、欧州特許出願公開第222462号明細書および国際公開第9704086号パンフレットに記載されている。固定化の前に、イオン交換クロマトグラフィーを用いて、分離された酵素を精製することが好ましい。
【0043】
好ましくは、酵素は担体上に固定された酵素であり、この方法は、固定化酵素から生成したアモキシシリンを含む製品を分離するステップを含むことが好ましい。固定化酵素から製品を分離する前記ステップは、任意の適切な方法、例えば重力または固定化酵素の主要部分を透過させないスクリーンを用いて行うことができる。好ましくは、固定化酵素から分離された製品は、アモキシシリン1,000,000重量部当り200重量部未満のタンパク質、好ましくは100重量部未満のタンパク質、より好ましくは50重量部未満のタンパク質、より好ましくはアモキシシリン1,000,000重量部当り35重量部未満のタンパク質を含有する。これは、担体上に十分に固定化された酵素を使用して、少量のタンパク質がアモキシシリン三水和物から分離することを避けることによって実現することが好ましい。この実施形態は、得られた最終のアモキシシリン三水和物が、アモキシシリン1,000,000重量部当り200重量部未満のタンパク質、好ましくは100重量部未満のタンパク質、より好ましくは50重量部未満のタンパク質、より好ましくはアモキシシリン1,000,000重量部当り35重量部未満のタンパク質を含有するという利点を有する。固定化酵素から分離された製品は、溶解した状態でアモキシシリンを含有する水溶液とすることができる。固定化酵素から分離された製品は、ウェットケーキとすることもできる。分離された製品は、好ましくは、アモキシシリン三水和物を含む水性懸濁液である。好ましくは、この方法は、前記アモキシシリン三水和物の結晶を溶解して、溶解したアモキシシリンを含有する水溶液を形成するステップを含む。
【0044】
本発明はまた、本発明による方法によって得ることができるアモキシシリン三水和物結晶性粉末にも関する。
【0045】
本発明による製品は、医薬組成物の調製に有利に使用することができる。
【0046】
本発明によるアモキシシリン三水和物の製品は、有利には、薬学的に許容される助剤および/または第2の薬学的に活性な薬剤と混合することができる。本発明によるアモキシシリン三水和物の製品は、結晶性粉末と助剤の合計重量に対して、例えば0から50重量%の範囲、好ましくは0から40重量%の範囲、好ましくは0から30重量%の範囲、より好ましくは0から20重量%の範囲、好ましくは1重量%超の助剤と混合することができる。アモキシシリン三水和物の製品は、例えば、クラブラン酸塩、好ましくはクラブラン酸カリウムと混合することができる。アモキシシリン:クラブラン酸の重量比は、好ましくは1:1から15:1の範囲であり、好ましくは2:1から10:1の範囲であり、好ましくは4:1から8:1の範囲である。これらの重量比は、無水アモキシシリンと酸の形のクラブラン酸について計算する。したがって、本発明はまた、本発明によるアモキシシリン三水和物の製品を助剤および/または第2の薬学的に活性な薬剤と混合するステップを含む方法によって得られる混合物にも関する。本発明はまた、(i)本発明によるアモキシシリン三水和物粉末の製品、ならびに(ii)第2の薬学的に活性な薬剤と助剤、または第2の薬学的に活性な薬剤のみ、を含む混合物も提供する。
【0047】
第2の薬学的に活性な薬剤としては、クラブラン酸塩、好ましくはクラブラン酸カリウムを用いることが好ましい。
【0048】
助剤としては、例えば、フィラー、乾式結合剤、分解剤、湿潤剤、湿式結合剤、潤滑剤、流動化剤などを使用することができる。助剤の例としては、ラクトース、デンプン、ベントナイト、炭酸カルシウム、マンニトール、微結晶性セルロース、ポリソルベート、ラウリル硫酸ナトリウム、カルボキシメチルセルロースNa、アルギン酸ナトリウム、ステアリン酸マグネシウム、二酸化ケイ素、滑石が挙げられる。
【0049】
一実施形態においては、混合物は、0から50重量%の範囲、好ましくは0から40重量%の範囲、好ましくは0から30重量%の範囲、より好ましくは0から20重量%の範囲、好ましくは1重量%超の助剤を含有する。これらの重量百分率は、アモキシシリン三水和物と助剤の合計重量に対するものである。
【0050】
好ましくは、アモキシシリン:クラブラン酸の重量比は、1:1から15:1の範囲であり、好ましくは2:1から10:1の範囲であり、好ましくは4:1から8:1の範囲である。これらの重量比は、無水アモキシシリンと酸の形のクラブラン酸について計算する。
【0051】
本発明による製品または混合物は、有利には、医薬用のカプセル、例えばゼラチンカプセルの充填用に用いることができる。したがって、本発明はまた、本発明による製品を含むカプセル、または本発明による混合物を含むカプセルにも関する。本発明による製品または本発明による混合物は、任意の適切な方法でカプセルに供給することができる。本発明はまた、本発明による製品または本発明による混合物の、カプセルを充填するための使用にも関する。
【0052】
本発明はまた、本発明による製品を圧縮して、または本発明による混合物を圧縮して、圧縮製品を製造するステップを含む方法も提供する。圧縮製品は、例えば、顆粒または錠剤とすることができる。本発明はまた、圧縮された形で本発明による製品を含む、または圧縮された形で本発明による混合物を含む、顆粒または錠剤にも関する。
【0053】
本発明はまた、本発明による結晶性粉末または本発明による混合物を、任意選択で助剤および/または第2の薬学的に活性な薬剤と組み合わせて、ローラー圧縮機に供給してコンパクトを製造するステップと、このコンパクトを粉砕して顆粒を製造するステップとを含む、顆粒調製方法にも関する。製造された顆粒は、有利には、篩いにかけて所望の粒度分布を得ることができる。本発明はまた、この方法によって得られる顆粒にも関する。
【0054】
本発明はまた、本発明による結晶または本発明による混合物を、例えば湿潤液に溶解した結合剤と混合するステップと、結晶を湿式または乾式で圧縮するステップと、得られたコンパクトを篩いにかけて顆粒化するステップとを含む、顆粒調製方法にも関する。本発明はまた、この方法によって得られる顆粒にも関する。
【0055】
本発明はまた、本発明による結晶性粉末または本発明による混合物からペーストを形成するステップと、このペーストを10℃から80℃の温度で混練するステップと、このペーストを2軸押出機で押出成形するステップと、所望により、得られた顆粒を乾燥するステップとを含む方法にも関する。本発明はまた、この方法によって得られる顆粒にも関する。
【0056】
本発明はまた、任意選択で助剤および/または薬学的に活性な薬剤と混ぜて、本発明による顆粒を圧縮して錠剤を調製するステップを含む方法にも関する。本発明はまた、この方法によって得られる錠剤にも関する。
【0057】
本発明者等はまた、流れ特性に関してアモキシシリン三水和物結晶性粉末の物性を改良することができることも見出した。
【0058】
好ましい実施形態においては、本発明によるアモキシシリン三水和物粉末の嵩密度は0.45g/mlより高い。本発明のこの態様による結晶性粉末は、造粒、成形、アグロメレーションまたは凝集などのプロセスにかけることなく流れ特性が改良され、色特性が優れ、安定性が優れ、且つ溶解速度が高い。この結晶性粉末を、さらに造粒、成形、アグロメレーションまたは凝集などのプロセスにかけることが望ましい場合にも、本発明のこの態様による結晶性粉末は流れ特性が改良されているので、これらのプロセスは容易に適用できる。さらに、所与のサイズのカプセルに入れることができる結晶性粉末の量を増やすことができる。嵩密度は、米国薬局方24、I法(1913頁)に従って測定することが好ましい。好ましくは、この結晶性粉末は、嵩密度が、0.46g/mlより高く、好ましくは0.5g/mlより高く、より好ましくは0.55g/mlより高い。これにより流れ特性がさらに改良される。さらに、結晶性粉末を所定の容積、例えばカプセルに入れることができるので、嵩密度が高いと有利である。嵩密度には特定の上限はない。嵩密度は、0.8g/mlより低く、例えば0.7g/mlより低くすることができる。
【0059】
好ましい実施形態においては、本発明による結晶性粉末は、タップ密度が、0.6g/mlより高く、好ましくは0.7より高く、より好ましくは0.8g/mlより高い。タップ密度が高いと流れ特性が良くなる。さらに、より多くの製品を所定の容積、例えばカプセルに入れることができるので、タップ密度が高いと有利である。タップ密度には特定の上限はない。タップ密度は、1.2g/mlより低く、例えば1.1g/mlより低く、例えば1.0g/mlより低くすることができる。タップ密度は、米国薬局方24、II法(1914頁)に従って測定することが好ましい。
【0060】
好ましい実施形態においては、本発明による結晶性粉末の嵩密度とタップ密度は、dt/dbの比が、1.7未満、好ましくは1.6未満、好ましくは1.5未満、好ましくは1.45未満である(但し、dt=タップ密度、およびdb=嵩密度)。これにより流れ特性が改良される。dt/dbの比に特定の下限はない。dt/dbの比は、1.05より高く、例えば1.1より高くてもよい。
【0061】
好ましい実施形態においては、本発明による結晶性粉末の嵩密度とタップ密度は、dt/dbの比が、1.7未満、好ましくは1.6未満、好ましくは1.5未満、好ましくは1.45未満である。この結晶性粉末は、既知の粉末に比べて流れ特性が改良されている。dt/dbの比には特定の上限はない。dt/dbの比は、1.05より高く、例えば1.1より高くすることができる。
【0062】
好ましい実施形態においては、本発明による結晶性粉末の嵩密度とタップ密度は、((dt−db)/dt)×100%で定義される圧縮率が、40%未満、好ましくは35%未満、より好ましくは30%未満である。これにより流れ特性が改良される。圧縮率には特定の下限はない。圧縮率は、例えば10%より高くてもよい。
【0063】
本発明者等は、好ましくは、流れ特性、嵩密度および/またはタップ密度が改良された結晶性粉末は、d50が増大していることを見出した。
【0064】
驚くべきことに、結晶化、分離、および/または乾燥条件を選ぶことによって、流れ特性が改良された、具体的には嵩密度が高い、および/またはタップ密度が高い結晶性粉末が得られることが見出された。
【0065】
流れ特性が改良された、具体的には嵩密度が高い、および/またはタップ密度が高い結晶性粉末を調製するステップは、好ましくは、この方法、具体的には結晶化、分離および乾燥の各ステップを、乾燥結晶の粒経、具体的にはd50および/またはd10が増大するような条件下で実施するステップを含むことが見出された。
【0066】
さらに、特にサイズが増大した結晶について、例えば結晶化、分離および/または乾燥時の機械的な衝撃の程度が、嵩密度およびタップ密度に影響を及ぼすことが見出された。例えば結晶の乾燥中および/または分離中あるいは輸送中に結晶に機械的な力を加えると、驚くべきことに、機械的な衝撃がない状態と比べて嵩密度およびタップ密度が増大することが見出された。しかし、機械的な力が高すぎると、嵩密度およびタップ密度は低下することが見出されている。分離中の機械的な衝撃は、例えば遠心分離中に与えることができる。乾燥中の機械的な衝撃は、例えば接触式乾燥機、例えばVrieco−Nauta接触式乾燥機、またはフラッシュドライヤーを用いて与えることができる。機械的な衝撃はまた、空気輸送、例えば分離工程から乾燥工程へのアモキシシリン三水和物の空気輸送を利用して与えることもできる。どんな科学理論にも拘束されたくはないが、限られた大きさの機械的な衝撃には、比較的大きな針状の結晶を破壊する効果があるために、嵩密度および/またはタップ密度が上昇すると考えられる。しかし、過大な機械的な力を用いると細かすぎる結晶が生成するため、嵩密度および/またはタップ密度が低下することになると思われる。本発明によって提供されるこの洞察を用い、かつ機械的な力を変化させることによって、当業者は、最適な嵩密度および/またはタップ密度を得る条件を見出すことができる。
【0067】
本発明はまた、本発明による結晶性粉末を篩い別けするステップを含む方法も提供する。これにより、結晶性粉末の物性がさらに改良される。好ましくは、風力篩が用いられる。
【0068】
本発明を、以下の実施例を用いてさらに説明するが、これらに限定されるものではない。
【0069】
実施例および比較実験
固定化酵素の調製
国際公開第9212782号パンフレットに記載されたようにして大腸菌ペニシリンアシラーゼを分離し、イオン交換クロマトグラフィーを用いて精製し、欧州特許出願公開第222462号明細書および国際公開第9704086号パンフレットに記載されたようにして固定化した。
【0070】
ペニシリンGアシラーゼ活性の定義としては、以下を用いる:1単位(U)は、標準条件(ペニシリンGカリウム塩100g/l、0.05Mリン酸カリウム緩衝液、pH8.0、28℃)下、ペニシリンG1μモル当り1分間に加水分解する酵素の量に相当する。
【0071】
アモキシシリンの製造
162.2gの6−APA(6−アミノ−ペニシラン酸)および184.8gのHPGM(D(−)−p−ヒドロキシフェニルグリシンメチルエステル)を、450mlの水に懸濁させた。懸濁液を、10℃の温度まで冷却した。この反応混合物に、32850単位の固定化ペニシリンアシラーゼを加え、水を加えて最終容積を1500mlにした。この混合物を6時間かき混ぜた。反応中pHは6.9に上昇し、反応の終わりにはpHは6.2に下がった。この混合物に750mlの水を加え、(メッシュが100マイクロメートルの)篩いで懸濁液を2時間かけて濾過し、固定化酵素を分離した。アモキシシリン三水和物結晶を含有する、得られた懸濁液を0℃に冷却した。懸濁液は、アモキシシリン三水和物に対して50ppm未満のタンパク質(1,000,000重量部のアモキシシリン三水和物当り50重量部未満のタンパク質)を含有していた。
【0072】
上記のようにして得られた、アモキシシリンを水に含有する水性懸濁液(懸濁液1リットル当りアモキシシリン三水和物100g)を、pH1の溶液が得られるように、スタティックミキサーを用いて32重量%HCl溶液と(温度25℃で)混合した。スタティックミキサー中の滞在時間は1.5分であった。得られた酸性溶液を、2つのフィルターを通してポンプで送った。第1のフィルターの孔径は40□mであり、第2のフィルターの孔径は4.5μmであった。これらのフィルターにおける滞在時間は約3分であった。濾過した酸性溶液を、第1の撹拌槽に送った。この槽においては、8MNaOH溶液を添加することによってpH3.7が保持される。第1の槽における温度は17から23℃の範囲である。第1の槽における滞在時間は45分である。第1の槽の内容物を、8MNaOH溶液を添加することによってpH5.0が保持される第2の撹拌槽に送る。第2の撹拌槽における温度は17から23℃の範囲である。第2の撹拌槽における滞在時間は15分である。槽2の内容物を、温度が1から5℃に保持された第3の撹拌槽に送る。第3の槽における滞在時間は4時間より長い。第3の撹拌槽の内容物を、逆フィルター遠心分離機に送ってアモキシシリンの結晶を分離し、固体物質86重量%を含有するウェットケーキを得る。このウェットケーキを水で洗い、円錐形の真空接触式乾燥機(Vrieco−Nauta)に空気輸送し、この中で30から40℃の温度および30ミリバールの圧力で7時間乾燥した。
【0073】
粒度分布の測定
粒度分布(d10およびd50を含めて)を、対物レンズf=300mm、malvern試料測定ユニットPS1、およびMalvern乾燥粉末フィーダーPS64を有する、Malvern粒径測定器2600Cを用いて測定した。ビーム長は14.30mmであった。多分散分析モデルを用いた。
【0074】
吸着等温線の測定
VTI−SGA 100蒸気収着分析装置を用いて、動的蒸気収着を用いて吸着等温線を求めた。重量200mgの試料を用いた。試料チャンバー内の空気を相対湿度10%で90分間コンディショニングした。その後、10%刻みで相対湿度を上昇させ、各値の相対湿度に試料を90分間保持した。90分後の試料重量を平衡相対湿度に対応した試料重量として用いた。温度は25℃とした。
【0075】
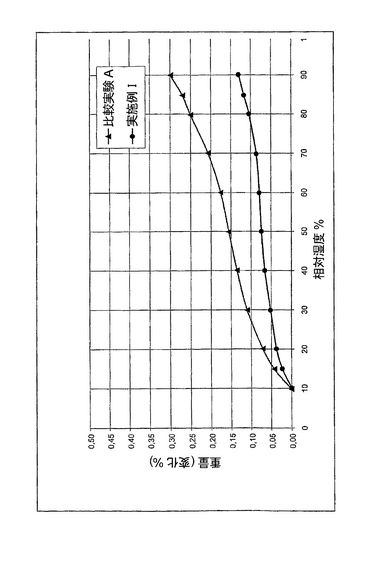
実施例I
上記の方法を用いて1バッチのアモキシシリン三水和物結晶性粉末を調製した。吸着等温線、ならびに粒度分布を測定した。図1に吸着等温線を示す。d50およびd10を表に示す。
【0076】
平衡相対湿度30%、25℃で測定した自由水含有量は0.05重量%であった。
【0077】
比較実験A
アモキシシリンを調製する化学的方法で、希HClおよびイソプロパノール中にアモキシシリンを含有する溶液を得た。この溶液を撹拌槽に送った。温度20℃でpHを3.7に保持した。その後、NaOHを加えてpHを5.0に上げた。得られた混合物を、1から5℃で3から12時間容器内に保持する。このアモキシシリンを、遠心分離機を用いて分離し、流動床乾燥機を用いて乾燥した。
【0078】
吸着等温線、ならびに粒度分布を測定した。図1に吸着等温線を示す。d50およびd10を表に示す。平衡相対湿度30%、25℃で測定した自由水含有量は0.11重量%であった。
【0079】
【表1】

【0080】
実施例1と比較実験Aを比較すると、実施例Iのアモキシシリン粉末は比較実験Aのアモキシシリン粉末より少ない自由水を含有していることが分かる。
【0081】
実施例II
実施例Iの方法で得られたアモキシシリン三水和物粉末を、水分活性0.15まで乾燥する。この粉末を、4対1の重量比でクラブラン酸カリウムと混合する(無水アモキシシリンとクラブラン酸として計算した)。この混合物は安定である。
【0082】
実施例III
実施例Iの方法で得られたアモキシシリン粉末を、水分活性0.2まで乾燥する。この粉末を、4対1の比でクラブラン酸カリウムと混合する。この混合物は安定である。
【0083】
実施例IV
実施例Iの方法で得られたアモキシシリン粉末を、水分活性0.15まで乾燥する。この粉末を、4対1の比でクラブラン酸カリウムと混合する。この混合物は安定である。
【0084】
実施例V
乾燥を、円錐形の真空接触式乾燥機(Vrieco−Nauta)を用いず、材料に機械的衝撃を与えない乾燥機(Ventilation stove)を用いて行った点を変えて実施例Iを繰返した。乾燥は、35℃の温度で16時間行った。d50およびd10は、それぞれ66.3μmおよび17.4μmである。嵩密度およびタップ密度は、それぞれ0.25g/mlおよび0.47g/mlである。
【0085】
実施例VI
実施例Iに従ってアモキシシリン三水和物粉末の別のバッチを調製した(d50およびd10はそれぞれ61μmおよび19μmであり、嵩密度およびタップ密度はそれぞれ0.58g/mlおよび0.79g/mlである)。このバッチを風力篩い別け(Hosakawa Alpine製の風力篩200LS−N)にかけた。篩い別けは、75μmのスクリーンを用いて10分間行った。前記篩い別け中に生成した少量の凝集物を、振動篩(425μm)を用いて篩上分(篩いを通過しなかった部分)から取除き、その後得られた篩上分の結晶のタップ密度、嵩密度、d50、d10を測定した。d50およびd10は、それぞれ88μmおよび36μmである。嵩密度およびタップ密度は、それぞれ0.59g/mlおよび0.74g/mlである。
【図面の簡単な説明】
【0086】
【図1】図1は実施例Iおよび比較実験Aの吸着等温線を示す。
【発明の詳細な説明】
【0001】
本発明は、アモキシシリン三水和物の製品に関する。
【0002】
固体状態のβ−ラクタム系抗生物質内の水は様々な形で存在することができる。水は、例えば結晶水として存在することができる。結晶水とは、β−ラクタム系抗生物質の分子構造に一体化された水のことである。アモキシシリン1分子当り3分子の結晶水を含むアモキシシリン三水和物内の結晶水の量は、約12.9%の結晶水に相当する。自由水とは、大気と交換できる水のことである。自由水の量には、結晶水として存在する水の量は含まれていない。
【0003】
β−ラクタム系抗生物質の試料中の自由水含有量と、この試料に接触している空気の相対湿度とは、相互に影響を及ぼし合っている。β−ラクタム系抗生物質の試料を空気に接触させると、一般に平衡状態が確立されるまで前記試料と空気の間で水の交換が行われる。平衡状態においては、試料とこの試料に接触している空気の間の水の正味の交換はゼロである。一般に、所定温度での上記の平衡状態における所定の相対湿度の値について、β−ラクタム系抗生物質の試料の自由水含有量を決める。この相対湿度の値を平衡相対湿度とも呼ぶ。
【0004】
試料の自由水含有量は、動的蒸気収着によって測定することができる。根底にある原理は、事前に相対湿度を調整した空気に試料を接触させながら、その重量を監視することである。水を吸収または放出する結果として、平衡状態が確立されるまで、試料重量が変化する。この平衡状態における試料重量が、平衡相対湿度に対応する試料重量である。この平衡相対湿度は事前に調整された空気の相対湿度であり、この空気に試料を接触させる。様々な値の平衡相対湿度についてこの手段を繰返すことによって、平衡相対湿度の関数として試料重量を測定することができる。温度Tにおける試料の自由水含有量は、((WERH−Wref)/Wref)×100%によって与えられる。但し、WERH=温度Tにおける平衡相対湿度ERHに対応する試料重量、およびWref=温度Tにおける平衡相対湿度の基準値に対応する試料重量である。この基準値は、前記平衡相対湿度における自由水含有量がゼロに近くなるように選択される。
【0005】
β−ラクタム系抗生物質を利用する際は、自由水の存在が関心事になる可能性がある。これは、例えばアモキシシリン三水和物を第2の薬学的に活性な薬剤、例えばクラブラン酸と混合する場合である。したがって、従来技術は、アモキシシリン三水和物を所定の程度まで乾燥することを提案している。
【0006】
水分活性は平衡相対湿度を100%で除したものとして定義され、β−ラクタム系抗生物質を乾燥するべき限度を規定する方法である。水分活性は、比較的小さい容積を有する密閉されたチャンバーに所定量の試料を入れ、相対湿度が一定になるまで時間の関数として相対湿度を測定することによって測定することができる。この相対湿度がその試料の平衡相対湿度である。水に関連した問題が一定の役割を演ずる用途については、β−ラクタム系抗生物質の水分活性に対して概ね低い値が規定される。
【0007】
乾燥の程度が不十分であると、水に関連した問題が残る。乾燥を過度に行うと、色や安定性などの物性が損なわれる恐れがある。これは、例えば、乾燥を過度に行うと結晶水が追い出されることによるものである可能性がある。
【0008】
本発明者等は、驚くべきことに、平衡相対湿度30%および温度25℃で測定した自由水含有量が0.10重量%未満のアモキシシリン三水和物の製品、ならびにこれを調製する方法を見出した。
【0009】
したがって、本発明は、従来技術による同じ水分活性を有するアモキシシリン三水和物と比べて、水に関する問題が無い、または少ない、アモキシシリン三水和物の製品を提供する。さらに、行われる乾燥の程度を下げることができるので、色や安定性などの特性は損なわれないか、または損なわれる程度が少ない。その自由水含有量が低くなっているので、このアモキシシリン三水和物は、水分の影響を非常に受けやすいクラブラン酸またはその塩と有利に混合することができる。
【0010】
本明細書で用いる、平衡相対湿度30%および温度25℃で測定した自由水含有量は、具体的には、((W30−W10)/W10)×100%として定義される。但し、
W30=温度25℃における平衡相対湿度30%に対応する試料重量、
W10=温度25℃における平衡相対湿度10%に対応する試料重量である。
【0011】
W30およびW10に対する値を含めて、自由水含有量は、動的蒸気収着を用いて、例えばVTI−SGA 100蒸気収着分析装置を用いて測定することが好ましい。この技法を用いて、試料チャンバー内の空気を相対湿度10%で90分間コンディショニングし、その後10%刻みで相対湿度を上昇させ、各値の相対湿度に試料を90分間保持し、ある値の相対湿度で90分後に測定した試料重量を平衡相対湿度に対応した試料重量として用いることによって、重量200mgの試料について吸着項を測定することが好ましい。
【0012】
本発明によるアモキシシリン三水和物の製品は、平衡相対湿度30%および温度25℃で測定した自由水含有量が0.10重量%未満である。好ましくは、本発明によるアモキシシリン三水和物の製品は、平衡相対湿度30%および温度25℃で測定した自由水含有量が0.07重量%未満であり、より好ましくは0.05重量%未満である。自由水含有量に特定の上限はない。平衡相対湿度30%および温度25℃で測定した自由水含有量は、例えば0.01重量%より高くてもよい。
【0013】
本発明によるアモキシシリン三水和物の製品は、任意の適切な形、例えば結晶性粉末または顆粒または結晶性粉末と顆粒の混合物の形のアモキシシリン三水和物とすることができる。好ましい実施形態においては、本発明による製品は、平衡相対湿度30%および温度25℃で測定した自由水含有量が0.1重量%未満、好ましくは0.07重量%未満、より好ましくは0.05重量%未満のアモキシシリン三水和物結晶性粉末である。
【0014】
アモキシシリン三水和物の製品は依然として何らかの不純物を含有している可能性があることは理解されよう。好ましくは、アモキシシリン三水和物の製品は、少なくとも90重量%、好ましくは少なくとも95重量%、より好ましくは少なくとも98重量%のアモキシシリン三水和物を含有する。これらの重量百分率は製品重量に対するものである。好ましくは、本発明によるアモキシシリン三水和物の製品には助剤は含まれていない。
【0015】
本明細書で用いるアモキシシリン三水和物結晶性粉末とは、具体的には、主としてアモキシシリン三水和物の結晶からなる製品のことである。結晶とは、例えば、水またはデンプンのりなどの結合剤、あるいはローラー圧縮成形または押出成形などの機械的な力の助けを借りて、結晶を付着させることによって形成した凝集体のことではないことは理解されよう。通常の取扱い中、例えば乾燥中に、意図せずに凝集体が生成される可能性もある。凝集体は、倍率140倍で光学顕微鏡を用いて観察することができる。本明細書で用いる、主としてアモキシシリン三水和物の結晶からなる製品とは、具体的には、アモキシシリン三水和物の結晶を少なくとも70重量%、好ましくは少なくとも80重量%、より好ましくは少なくとも90重量%、より好ましくは少なくとも95重量%、より好ましくは少なくとも98重量%含む製品のことである。これらの百分率は、風力篩と光学顕微鏡を組み合わせて用いて測定することができる。風力篩い別けは、風力篩Alpine Air Jet 200LS−Nを用いて、重量10gの試料について1200Paで1分間行うのが有利である。光学顕微鏡検査は、前記部分の試料5mgを採取し、この試料を表面積22×40mmの表面上の4滴のパラフィンオイルに懸濁し、倍率140倍を用いて行うのが有利である。
【0016】
アモキシシリン三水和物粉末は依然として何らかの不純物を含有している可能性があることは理解されよう。好ましくは、アモキシシリン三水和物の製品は、少なくとも90重量%、好ましくは少なくとも95重量%、より好ましくは少なくとも98重量%のアモキシシリン三水和物を含有する。これらの重量百分率は結晶性粉末の重量に対するものである。好ましくは、本発明によるアモキシシリン三水和物結晶性粉末には助剤は含まれていない。
【0017】
好ましい実施形態においては、本発明によるアモキシシリン三水和物の製品の水分活性は、0.05より高く、好ましくは0.07より高く、好ましくは0.10より高く、好ましくは0.15より高く、好ましくは0.20より高く、好ましくは0.25より高く、好ましくは0.30より高い。自由水の量が依然として低いままで、アモキシシリン三水和物の特性が低下しない、またはその低下の程度が小さいので、水分含有量が高いことは有利である。水分活性についての特定の下限はない。実際、水分活性は、一般に0.7未満、例えば0.6未満、例えば0.5未満である。但し、これは必要ではない。本明細書で用いるこれらの値は、25℃における水分活性のことである。
【0018】
試料の水分活性を測定する好ましい方法は、比較的小さい容積を有する密閉されたチャンバーに所定量の試料を入れ、相対湿度が一定になる(例えば30分後)まで時間の関数として相対湿度を測定することである。この相対湿度がその試料の平衡相対湿度である。好ましくは、Novasina TH200 Thermoconstanterを用いる。その試料ホルダーの容積は12mlであり、3gの試料を充填される。
【0019】
d10およびd50は、粒度分布を示すための公知の方法である。d50は、結晶の50容積%がその値より小さい粒径を有するような粒径の値を意味する。d50はまた、容積基準の平均粒径のことでもある。同様に、d10は、結晶の10容積%がその値より小さい粒径を有するような粒径の値を意味する。d10およびd50を測定するための好ましい方法は、レーザー回折であり、好ましくはMalvern装置を用いる。d10およびd50のための好適な装置は、粒度分布を示すための公知の方法である。d50は、粒子の50容積%がその値より小さい粒径を有するような粒径の値を意味する。d50はまた、容積基準の平均粒径のことでもある。同様に、d10は、粒子の10容積%がその値より小さい粒径を有するような粒径の値を意味する。d10およびd50を測定するための好ましい方法は、レーザー回折であり、好ましくはMalvern装置を用いる。d10およびd50を測定するための好適な装置は、Malvern Instruments Ltd.、Malvern Ukから入手可能なMalvern粒径測定器2600Cであり、f=300mmおよびビーム長14.30mmの対物レンズを用いる。多分散分析モデルを用いることが有利である。
【0020】
本発明者等は、本発明によるアモキシシリン三水和物結晶性粉末は、d50が増大していることを見出した。したがって、本発明はまた、d50が、10μmより大きい、好ましくは20μmより大きい、より好ましくは30μmより大きい、より好ましくは35μmより大きい、より好ましくは40μmより大きいアモキシシリン三水和物結晶性粉末も提供する。d50についての特定の上限はない。本発明による結晶性粉末のd50は、150μm未満、例えば100μm未満とすることができる。本発明による結晶性粉末は、好ましくはd10が増大しており、好ましくは3μmより大きく、好ましくは5μmより大きく、より好ましくは8μmより大きく、より好ましくは10μmより大きい。本発明による結晶性粉末のd10についての特定の上限はない。本発明による結晶性粉末のd10は、50μm未満とすることができる。
【0021】
アモキシシリン三水和物結晶性粉末は、溶解したアモキシシリンを含む溶液を調製し、前記溶液からアモキシシリンを結晶化させて結晶を形成し、前記溶液からこの結晶を分離し、分離された結晶を乾燥することによって得ることができる。本明細書において用いる結晶性粉末という用語には、それだけに限らないが、この方法によって得られたおよび/または得ることができる乾燥製品が含まれる。
【0022】
所与の平衡相対湿度での自由水含有量が低下した結晶性粉末を調製するステップは、好ましくは、この方法、具体的には結晶化、分離および乾燥の各ステップを、乾燥結晶の粒経、具体的にはd50および/またはd10が増大するような条件下で実施するステップを含むことが見出された。
【0023】
好ましい結晶化条件としては、溶液から結晶化するアモキシシリン三水和物の粒径が増大しているような結晶化条件が挙げられる。これは、例えば比較的長い滞在時間を適用すること、水溶液において比較的低いアモキシシリン濃度を適用すること、または純度の高い水溶液を使用することによって実現することができる。その他の好ましい条件については以下に説明する。
【0024】
例えば分離および/または乾燥時の機械的な衝撃の程度が粒径に影響を及ぼすことがある。分離中の機械的な衝撃は、例えば遠心分離中に与えることができる。乾燥中の機械的な衝撃は、例えば接触式乾燥機、例えばVrieco−Nauta接触式乾燥機、またはフラッシュドライヤーを用いて与えることができる。機械的な衝撃はまた、空気輸送、例えば分離工程から乾燥工程へのアモキシシリン三水和物の空気輸送を利用して与えることもできる。過大な機械的衝撃を加えると、粒径の望ましくない低下をもたらす恐れがある。本発明によって提供されるこの洞察を用い、かつ機械的な力を変化させることによって、当業者は、望ましくない粒径の低下を回避することができる条件を見出すことができる。
【0025】
上記を考慮して、本発明はまた、溶液からアモキシシリン三水和物を結晶化させるステップと、前記溶液から結晶を分離するステップと、分離された結晶を乾燥するステップとを含むアモキシシリン三水和物結晶性粉末の調製方法であって、この方法、好ましくは結晶化、分離および/または乾燥の各ステップが、得られる結晶性粉末のd50が10μmより大きく、好ましくは20μmより大きく、より好ましくは30μmより大きく、特に35μmより大きく、より好ましくは40μmより大きくなるような条件下で行われる方法を提供する。d50には特定の上限はない。この方法、好ましくは結晶化、分離および/または乾燥の各ステップは、例えば、得られる結晶性粉末のd50が150μm未満、例えば100μm未満になるような条件下で行うことができる。この方法、好ましくは結晶化、分離および/または乾燥の各ステップは、乾燥した結晶のd10が3μmより大きく、好ましくは5μmより大きく、より好ましくは8μmより大きく、より好ましくは10μmより大きくなるような条件下で行われることが好ましい。d10には特定の上限はない。この方法、好ましくは結晶化、分離および/または乾燥の各ステップは、例えば、得られる結晶性粉末のd10が50μm未満になるような条件下で行うことができる。
【0026】
本発明者等は、自由水含有量が低下したアモキシシリン三水和物の結晶は、好ましくは、以下に記載の好ましいプロセス条件を適用することによって得られることを見出した。
【0027】
好ましくは、本発明によるアモキシシリン三水和物結晶性粉末の調製方法は、6−アミノ−ペニシラン酸またはその塩を、担体上に固定化された酵素の存在下、活性状態のp−ヒドロキシフェニルグリシンと反応させることによってアモキシシリンを調製するステップと、塩酸を含有する、アモキシシリン含有水溶液を形成するステップと、前記水溶液からアモキシシリン三水和物を結晶化させるステップとを含む。
【0028】
好ましくは、アモキシシリン三水和物を結晶化させる溶液は水溶液である。任意の適切な水溶液を使用することができる。適切な水溶液としては、水:有機溶媒の重量比が、100:0から70:30の範囲、好ましくは100:0から80:20の範囲、好ましくは100:0から90:10の範囲、好ましくは100:0から95:5の範囲、好ましくは100:0から99:1の範囲の溶液が挙げられる。
【0029】
好ましくは、アモキシシリン三水和物を結晶化させる溶液は、1,000,000重量部のアモキシシリン(溶解した形であるか否かにかかわらず、アモキシシリンの総濃度)当り、200重量部未満のタンパク質、好ましくは100重量部未満のタンパク質、より好ましくは50重量部未満のタンパク質、より好ましくは35重量部未満のタンパク質を含有する。
【0030】
好ましくは、アモキシシリン三水和物を結晶化させる溶液は、アモキシシリン濃度(溶解した形であるか否かにかかわらず、アモキシシリンの総濃度)が、0.6モル/l未満、好ましくは0.5モル/l未満、より好ましくは0.4モル/l未満、より好ましくは0.3モル/l未満の水溶液である。
【0031】
アモキシシリン三水和物を結晶化させる水溶液は、塩酸または塩化物を含有する溶液であることが好ましい。アモキシシリン三水和物を結晶化させる水溶液は、アモキシシリン(溶解した形であるか否かにかかわらず、アモキシシリンの総濃度)1モル当り0.9から5モルの塩酸または塩化物、好ましくはアモキシシリン1モル当り0.9から3モルの塩酸または塩化物、より好ましくはアモキシシリン1モル当り0.9から1.5モルの塩酸または塩化物を含有することが好ましい。アモキシシリンを結晶化させる水溶液は、アモキシシリン1モル当り1.0モルを超える塩酸または塩化物を含有することが好ましい。
【0032】
アモキシシリン三水和物は、pH2から7の範囲、好ましくは3から6の範囲で水溶液から結晶化させることが好ましい。この方法は、好ましくはpH2から5の範囲、好ましくは3から4の範囲の第1のステップと、第1のステップより高いpH、好ましくはpH4から7の範囲、好ましくは4.5から6の範囲の第2のステップとで、アモキシシリン三水和物を水溶液から結晶化させるステップを含むことが好ましい。
【0033】
5℃から40℃の範囲、好ましくは10から30℃の範囲、より好ましくは15から25℃の範囲の温度で、水溶液からアモキシシリン三水和物を結晶化させることが好ましい。
【0034】
アモキシシリン三水和物を結晶化させる溶液は、任意の適切な方法で調製することができる。溶解したアモキシシリンを含有する水溶液は、アモキシシリン三水和物を溶解することによって調製することができる。アモキシシリン三水和物を溶液に添加し、添加したアモキシシリン三水和物を溶解させることが可能である。また、溶液中にin situでアモキシシリン三水和物の結晶を形成することによって水性懸濁液を調製し、前記懸濁液中のアモキシシリン三水和物の結晶を溶解させることも可能である。アモキシシリンの調製方法においては、溶解したアモキシシリンを含有する水溶液であって、アモキシシリン濃度が0.6モル/l未満、好ましくは0.5モル/l未満、より好ましくは0.4モル/l未満、より好ましくは0.3モル/l未満の水溶液を調製するステップを含むことが好ましい。この方法は、溶解したアモキシシリンを含有する水溶液であって、pHが0から1.5の範囲、好ましくは0.5から1.2の範囲の水溶液を調製するステップを含むことが好ましい。アモキシシリンの溶解は、任意の適切な方法、例えばアモキシシリン三水和物の結晶を含有する水性懸濁液に酸を添加することによって、好ましくは塩酸を添加することによって行うことができる。酸好ましくは塩酸の添加量は、アモキシシリン1モル当り塩酸0.9から5モル、好ましくはアモキシシリン1モル当り塩酸0.9から3モル、より好ましくはアモキシシリン1モル当り塩酸0.9から1.5モルとすることができる。アモキシシリン1モル当り1.0モルを超える塩酸を添加することが好ましい。好ましい実施形態においては、この方法は、(水)溶液または(水性)懸濁液を、pH1.5未満、好ましくは1.2未満に、60分未満、好ましくは30分未満、より好ましくは15分未満、より好ましくは10分未満、より好ましくは8分未満の時間保持するステップを含む。これにより、アモキシシリンの純度を改良することができる。この方法は、水溶液または水性懸濁液を、急速混合ミキサー、例えばスタティックミキサーを用いて、酸と混合するステップを含むことが好ましい。これにより、水溶液または水性懸濁液を低pHに保持する時間を短縮することができる。酸と水性懸濁液との混合は、例えば−5℃より高い、例えば5℃より高い、例えば10℃より高い、例えば15℃より高い、例えば50℃より低い、例えば40℃より低い任意の適切な温度で行うことができる。この方法は、前記結晶化ステップの前に溶液を濾過するステップを含むことが好ましい。この方法は、溶解したアモキシシリンを含有する水溶液であって、そのpHが好ましくは0から1.5の範囲、好ましくは0.5から1.2の範囲の水溶液を濾過するステップを含むことが好ましい。溶液は、任意の適切なフィルターを通すことができる。孔径が40μm未満、好ましくは20μm未満、好ましくは10μm未満、より好ましくは5μm未満のフィルターを使用することが好ましい。
【0035】
アモキシシリン三水和物は、有利には、例えばNaOHなどの塩基を添加することによってpHを上昇させて前記水溶液から結晶化させることができる。
【0036】
結晶化は、バッチ式でまたは連続的に行うことができる。バッチ式で行う場合、種晶を水溶液に添加することが好ましい。好ましくは、結晶化は連続的に行われる。
【0037】
アモキシシリンは、6−アミノ−ペニシラン酸またはその誘導体、例えば6−アミノ−ペニシラン酸の塩を、水性反応媒体中で酵素の存在下、活性状態のp−ヒドロキシフェニルグリシンから選択されるアシル化剤と反応させることによって調製することが好ましい。活性状態のp−ヒドロキシフェニルグリシンは、好ましくは、p−ヒドロキシフェニルグリシンのエステルまたはアミドである。好適なエステルとしては、例えば、炭素原子1から4個のアルキルエステル、例えばメチルエステル、エチルエステル、n−プロピルまたはイソプロピルエステルが挙げられる。グリコールエステル、例えばエチレングリコールエステルも使用することができる。−CONH2基が置換されていないアミドも使用することができる。
【0038】
酵素は、加水分解活性を有する任意の酵素(加水分解酵素)とすることができる。酵素は、例えばアシラーゼ、とりわけペニシリンGアシラーゼ、アミダーゼまたはエステラーゼとすることができる。酵素は、様々な天然微生物、例えば真菌および細菌から分離することができる。ペニシリンアシラーゼを生成することが見出されている微生物は、例えば、アセトバクター属、エーロモナス属、アルカリゲネス属、アファノクラジウム属(Aphanocladium)、桿菌属(Bacillus sp.)、セファロスポリウム属、エシェリヒア属、フラボバクテリウム属、クライベラ属(Kluyvera)、マイコプラナ属(Mycoplana)、プロタミノバクター属(Protaminobacter)、シュードモナス属またはキサントモナス属の種が挙げられる。
【0039】
酵素の存在下においてアモキシシリンを調製する方法は、国際公開第9201061号パンフレット、国際公開第9417800号パンフレット、国際公開第9704086号パンフレット、国際公開第9820120号パンフレット、欧州特許出願公開第771357号明細書に記載されており、これらの内容を参照により本明細書に組み込む。
【0040】
反応は、任意の適切なpH、好ましくは5から9の範囲、好ましくは5.5から8の範囲、より好ましくは6から7.5の範囲のpHで行うことができる。反応は、任意の適切な温度、例えば、0から40℃の範囲、好ましくは0から30℃の範囲、より好ましくは0から15℃の範囲の温度で行うことができる。
【0041】
生成したアモキシシリンは、反応を行った条件で結晶化することができる。アモキシシリンの結晶化は、例えば、5から8の範囲、好ましくは5.5から7.5の範囲のpHで行うことが好ましい。
【0042】
好ましくは、酵素は担体上に固定化された酵素である。任意の適切な担体を使用することができる。好ましくは、担体はゲル化剤および遊離アミノ基を含むポリマーを含む。好ましくは、ポリマーはアルギン酸アンモニウム、キトサン、ペクチン、またはポリエチレンイミンから選択される。好ましくは、ゲル化剤はゼラチンである。この担体およびその調製は、欧州特許出願公開第222462号明細書および国際公開第9704086号パンフレットに記載されている。固定化の前に、イオン交換クロマトグラフィーを用いて、分離された酵素を精製することが好ましい。
【0043】
好ましくは、酵素は担体上に固定された酵素であり、この方法は、固定化酵素から生成したアモキシシリンを含む製品を分離するステップを含むことが好ましい。固定化酵素から製品を分離する前記ステップは、任意の適切な方法、例えば重力または固定化酵素の主要部分を透過させないスクリーンを用いて行うことができる。好ましくは、固定化酵素から分離された製品は、アモキシシリン1,000,000重量部当り200重量部未満のタンパク質、好ましくは100重量部未満のタンパク質、より好ましくは50重量部未満のタンパク質、より好ましくはアモキシシリン1,000,000重量部当り35重量部未満のタンパク質を含有する。これは、担体上に十分に固定化された酵素を使用して、少量のタンパク質がアモキシシリン三水和物から分離することを避けることによって実現することが好ましい。この実施形態は、得られた最終のアモキシシリン三水和物が、アモキシシリン1,000,000重量部当り200重量部未満のタンパク質、好ましくは100重量部未満のタンパク質、より好ましくは50重量部未満のタンパク質、より好ましくはアモキシシリン1,000,000重量部当り35重量部未満のタンパク質を含有するという利点を有する。固定化酵素から分離された製品は、溶解した状態でアモキシシリンを含有する水溶液とすることができる。固定化酵素から分離された製品は、ウェットケーキとすることもできる。分離された製品は、好ましくは、アモキシシリン三水和物を含む水性懸濁液である。好ましくは、この方法は、前記アモキシシリン三水和物の結晶を溶解して、溶解したアモキシシリンを含有する水溶液を形成するステップを含む。
【0044】
本発明はまた、本発明による方法によって得ることができるアモキシシリン三水和物結晶性粉末にも関する。
【0045】
本発明による製品は、医薬組成物の調製に有利に使用することができる。
【0046】
本発明によるアモキシシリン三水和物の製品は、有利には、薬学的に許容される助剤および/または第2の薬学的に活性な薬剤と混合することができる。本発明によるアモキシシリン三水和物の製品は、結晶性粉末と助剤の合計重量に対して、例えば0から50重量%の範囲、好ましくは0から40重量%の範囲、好ましくは0から30重量%の範囲、より好ましくは0から20重量%の範囲、好ましくは1重量%超の助剤と混合することができる。アモキシシリン三水和物の製品は、例えば、クラブラン酸塩、好ましくはクラブラン酸カリウムと混合することができる。アモキシシリン:クラブラン酸の重量比は、好ましくは1:1から15:1の範囲であり、好ましくは2:1から10:1の範囲であり、好ましくは4:1から8:1の範囲である。これらの重量比は、無水アモキシシリンと酸の形のクラブラン酸について計算する。したがって、本発明はまた、本発明によるアモキシシリン三水和物の製品を助剤および/または第2の薬学的に活性な薬剤と混合するステップを含む方法によって得られる混合物にも関する。本発明はまた、(i)本発明によるアモキシシリン三水和物粉末の製品、ならびに(ii)第2の薬学的に活性な薬剤と助剤、または第2の薬学的に活性な薬剤のみ、を含む混合物も提供する。
【0047】
第2の薬学的に活性な薬剤としては、クラブラン酸塩、好ましくはクラブラン酸カリウムを用いることが好ましい。
【0048】
助剤としては、例えば、フィラー、乾式結合剤、分解剤、湿潤剤、湿式結合剤、潤滑剤、流動化剤などを使用することができる。助剤の例としては、ラクトース、デンプン、ベントナイト、炭酸カルシウム、マンニトール、微結晶性セルロース、ポリソルベート、ラウリル硫酸ナトリウム、カルボキシメチルセルロースNa、アルギン酸ナトリウム、ステアリン酸マグネシウム、二酸化ケイ素、滑石が挙げられる。
【0049】
一実施形態においては、混合物は、0から50重量%の範囲、好ましくは0から40重量%の範囲、好ましくは0から30重量%の範囲、より好ましくは0から20重量%の範囲、好ましくは1重量%超の助剤を含有する。これらの重量百分率は、アモキシシリン三水和物と助剤の合計重量に対するものである。
【0050】
好ましくは、アモキシシリン:クラブラン酸の重量比は、1:1から15:1の範囲であり、好ましくは2:1から10:1の範囲であり、好ましくは4:1から8:1の範囲である。これらの重量比は、無水アモキシシリンと酸の形のクラブラン酸について計算する。
【0051】
本発明による製品または混合物は、有利には、医薬用のカプセル、例えばゼラチンカプセルの充填用に用いることができる。したがって、本発明はまた、本発明による製品を含むカプセル、または本発明による混合物を含むカプセルにも関する。本発明による製品または本発明による混合物は、任意の適切な方法でカプセルに供給することができる。本発明はまた、本発明による製品または本発明による混合物の、カプセルを充填するための使用にも関する。
【0052】
本発明はまた、本発明による製品を圧縮して、または本発明による混合物を圧縮して、圧縮製品を製造するステップを含む方法も提供する。圧縮製品は、例えば、顆粒または錠剤とすることができる。本発明はまた、圧縮された形で本発明による製品を含む、または圧縮された形で本発明による混合物を含む、顆粒または錠剤にも関する。
【0053】
本発明はまた、本発明による結晶性粉末または本発明による混合物を、任意選択で助剤および/または第2の薬学的に活性な薬剤と組み合わせて、ローラー圧縮機に供給してコンパクトを製造するステップと、このコンパクトを粉砕して顆粒を製造するステップとを含む、顆粒調製方法にも関する。製造された顆粒は、有利には、篩いにかけて所望の粒度分布を得ることができる。本発明はまた、この方法によって得られる顆粒にも関する。
【0054】
本発明はまた、本発明による結晶または本発明による混合物を、例えば湿潤液に溶解した結合剤と混合するステップと、結晶を湿式または乾式で圧縮するステップと、得られたコンパクトを篩いにかけて顆粒化するステップとを含む、顆粒調製方法にも関する。本発明はまた、この方法によって得られる顆粒にも関する。
【0055】
本発明はまた、本発明による結晶性粉末または本発明による混合物からペーストを形成するステップと、このペーストを10℃から80℃の温度で混練するステップと、このペーストを2軸押出機で押出成形するステップと、所望により、得られた顆粒を乾燥するステップとを含む方法にも関する。本発明はまた、この方法によって得られる顆粒にも関する。
【0056】
本発明はまた、任意選択で助剤および/または薬学的に活性な薬剤と混ぜて、本発明による顆粒を圧縮して錠剤を調製するステップを含む方法にも関する。本発明はまた、この方法によって得られる錠剤にも関する。
【0057】
本発明者等はまた、流れ特性に関してアモキシシリン三水和物結晶性粉末の物性を改良することができることも見出した。
【0058】
好ましい実施形態においては、本発明によるアモキシシリン三水和物粉末の嵩密度は0.45g/mlより高い。本発明のこの態様による結晶性粉末は、造粒、成形、アグロメレーションまたは凝集などのプロセスにかけることなく流れ特性が改良され、色特性が優れ、安定性が優れ、且つ溶解速度が高い。この結晶性粉末を、さらに造粒、成形、アグロメレーションまたは凝集などのプロセスにかけることが望ましい場合にも、本発明のこの態様による結晶性粉末は流れ特性が改良されているので、これらのプロセスは容易に適用できる。さらに、所与のサイズのカプセルに入れることができる結晶性粉末の量を増やすことができる。嵩密度は、米国薬局方24、I法(1913頁)に従って測定することが好ましい。好ましくは、この結晶性粉末は、嵩密度が、0.46g/mlより高く、好ましくは0.5g/mlより高く、より好ましくは0.55g/mlより高い。これにより流れ特性がさらに改良される。さらに、結晶性粉末を所定の容積、例えばカプセルに入れることができるので、嵩密度が高いと有利である。嵩密度には特定の上限はない。嵩密度は、0.8g/mlより低く、例えば0.7g/mlより低くすることができる。
【0059】
好ましい実施形態においては、本発明による結晶性粉末は、タップ密度が、0.6g/mlより高く、好ましくは0.7より高く、より好ましくは0.8g/mlより高い。タップ密度が高いと流れ特性が良くなる。さらに、より多くの製品を所定の容積、例えばカプセルに入れることができるので、タップ密度が高いと有利である。タップ密度には特定の上限はない。タップ密度は、1.2g/mlより低く、例えば1.1g/mlより低く、例えば1.0g/mlより低くすることができる。タップ密度は、米国薬局方24、II法(1914頁)に従って測定することが好ましい。
【0060】
好ましい実施形態においては、本発明による結晶性粉末の嵩密度とタップ密度は、dt/dbの比が、1.7未満、好ましくは1.6未満、好ましくは1.5未満、好ましくは1.45未満である(但し、dt=タップ密度、およびdb=嵩密度)。これにより流れ特性が改良される。dt/dbの比に特定の下限はない。dt/dbの比は、1.05より高く、例えば1.1より高くてもよい。
【0061】
好ましい実施形態においては、本発明による結晶性粉末の嵩密度とタップ密度は、dt/dbの比が、1.7未満、好ましくは1.6未満、好ましくは1.5未満、好ましくは1.45未満である。この結晶性粉末は、既知の粉末に比べて流れ特性が改良されている。dt/dbの比には特定の上限はない。dt/dbの比は、1.05より高く、例えば1.1より高くすることができる。
【0062】
好ましい実施形態においては、本発明による結晶性粉末の嵩密度とタップ密度は、((dt−db)/dt)×100%で定義される圧縮率が、40%未満、好ましくは35%未満、より好ましくは30%未満である。これにより流れ特性が改良される。圧縮率には特定の下限はない。圧縮率は、例えば10%より高くてもよい。
【0063】
本発明者等は、好ましくは、流れ特性、嵩密度および/またはタップ密度が改良された結晶性粉末は、d50が増大していることを見出した。
【0064】
驚くべきことに、結晶化、分離、および/または乾燥条件を選ぶことによって、流れ特性が改良された、具体的には嵩密度が高い、および/またはタップ密度が高い結晶性粉末が得られることが見出された。
【0065】
流れ特性が改良された、具体的には嵩密度が高い、および/またはタップ密度が高い結晶性粉末を調製するステップは、好ましくは、この方法、具体的には結晶化、分離および乾燥の各ステップを、乾燥結晶の粒経、具体的にはd50および/またはd10が増大するような条件下で実施するステップを含むことが見出された。
【0066】
さらに、特にサイズが増大した結晶について、例えば結晶化、分離および/または乾燥時の機械的な衝撃の程度が、嵩密度およびタップ密度に影響を及ぼすことが見出された。例えば結晶の乾燥中および/または分離中あるいは輸送中に結晶に機械的な力を加えると、驚くべきことに、機械的な衝撃がない状態と比べて嵩密度およびタップ密度が増大することが見出された。しかし、機械的な力が高すぎると、嵩密度およびタップ密度は低下することが見出されている。分離中の機械的な衝撃は、例えば遠心分離中に与えることができる。乾燥中の機械的な衝撃は、例えば接触式乾燥機、例えばVrieco−Nauta接触式乾燥機、またはフラッシュドライヤーを用いて与えることができる。機械的な衝撃はまた、空気輸送、例えば分離工程から乾燥工程へのアモキシシリン三水和物の空気輸送を利用して与えることもできる。どんな科学理論にも拘束されたくはないが、限られた大きさの機械的な衝撃には、比較的大きな針状の結晶を破壊する効果があるために、嵩密度および/またはタップ密度が上昇すると考えられる。しかし、過大な機械的な力を用いると細かすぎる結晶が生成するため、嵩密度および/またはタップ密度が低下することになると思われる。本発明によって提供されるこの洞察を用い、かつ機械的な力を変化させることによって、当業者は、最適な嵩密度および/またはタップ密度を得る条件を見出すことができる。
【0067】
本発明はまた、本発明による結晶性粉末を篩い別けするステップを含む方法も提供する。これにより、結晶性粉末の物性がさらに改良される。好ましくは、風力篩が用いられる。
【0068】
本発明を、以下の実施例を用いてさらに説明するが、これらに限定されるものではない。
【0069】
実施例および比較実験
固定化酵素の調製
国際公開第9212782号パンフレットに記載されたようにして大腸菌ペニシリンアシラーゼを分離し、イオン交換クロマトグラフィーを用いて精製し、欧州特許出願公開第222462号明細書および国際公開第9704086号パンフレットに記載されたようにして固定化した。
【0070】
ペニシリンGアシラーゼ活性の定義としては、以下を用いる:1単位(U)は、標準条件(ペニシリンGカリウム塩100g/l、0.05Mリン酸カリウム緩衝液、pH8.0、28℃)下、ペニシリンG1μモル当り1分間に加水分解する酵素の量に相当する。
【0071】
アモキシシリンの製造
162.2gの6−APA(6−アミノ−ペニシラン酸)および184.8gのHPGM(D(−)−p−ヒドロキシフェニルグリシンメチルエステル)を、450mlの水に懸濁させた。懸濁液を、10℃の温度まで冷却した。この反応混合物に、32850単位の固定化ペニシリンアシラーゼを加え、水を加えて最終容積を1500mlにした。この混合物を6時間かき混ぜた。反応中pHは6.9に上昇し、反応の終わりにはpHは6.2に下がった。この混合物に750mlの水を加え、(メッシュが100マイクロメートルの)篩いで懸濁液を2時間かけて濾過し、固定化酵素を分離した。アモキシシリン三水和物結晶を含有する、得られた懸濁液を0℃に冷却した。懸濁液は、アモキシシリン三水和物に対して50ppm未満のタンパク質(1,000,000重量部のアモキシシリン三水和物当り50重量部未満のタンパク質)を含有していた。
【0072】
上記のようにして得られた、アモキシシリンを水に含有する水性懸濁液(懸濁液1リットル当りアモキシシリン三水和物100g)を、pH1の溶液が得られるように、スタティックミキサーを用いて32重量%HCl溶液と(温度25℃で)混合した。スタティックミキサー中の滞在時間は1.5分であった。得られた酸性溶液を、2つのフィルターを通してポンプで送った。第1のフィルターの孔径は40□mであり、第2のフィルターの孔径は4.5μmであった。これらのフィルターにおける滞在時間は約3分であった。濾過した酸性溶液を、第1の撹拌槽に送った。この槽においては、8MNaOH溶液を添加することによってpH3.7が保持される。第1の槽における温度は17から23℃の範囲である。第1の槽における滞在時間は45分である。第1の槽の内容物を、8MNaOH溶液を添加することによってpH5.0が保持される第2の撹拌槽に送る。第2の撹拌槽における温度は17から23℃の範囲である。第2の撹拌槽における滞在時間は15分である。槽2の内容物を、温度が1から5℃に保持された第3の撹拌槽に送る。第3の槽における滞在時間は4時間より長い。第3の撹拌槽の内容物を、逆フィルター遠心分離機に送ってアモキシシリンの結晶を分離し、固体物質86重量%を含有するウェットケーキを得る。このウェットケーキを水で洗い、円錐形の真空接触式乾燥機(Vrieco−Nauta)に空気輸送し、この中で30から40℃の温度および30ミリバールの圧力で7時間乾燥した。
【0073】
粒度分布の測定
粒度分布(d10およびd50を含めて)を、対物レンズf=300mm、malvern試料測定ユニットPS1、およびMalvern乾燥粉末フィーダーPS64を有する、Malvern粒径測定器2600Cを用いて測定した。ビーム長は14.30mmであった。多分散分析モデルを用いた。
【0074】
吸着等温線の測定
VTI−SGA 100蒸気収着分析装置を用いて、動的蒸気収着を用いて吸着等温線を求めた。重量200mgの試料を用いた。試料チャンバー内の空気を相対湿度10%で90分間コンディショニングした。その後、10%刻みで相対湿度を上昇させ、各値の相対湿度に試料を90分間保持した。90分後の試料重量を平衡相対湿度に対応した試料重量として用いた。温度は25℃とした。
【0075】
実施例I
上記の方法を用いて1バッチのアモキシシリン三水和物結晶性粉末を調製した。吸着等温線、ならびに粒度分布を測定した。図1に吸着等温線を示す。d50およびd10を表に示す。
【0076】
平衡相対湿度30%、25℃で測定した自由水含有量は0.05重量%であった。
【0077】
比較実験A
アモキシシリンを調製する化学的方法で、希HClおよびイソプロパノール中にアモキシシリンを含有する溶液を得た。この溶液を撹拌槽に送った。温度20℃でpHを3.7に保持した。その後、NaOHを加えてpHを5.0に上げた。得られた混合物を、1から5℃で3から12時間容器内に保持する。このアモキシシリンを、遠心分離機を用いて分離し、流動床乾燥機を用いて乾燥した。
【0078】
吸着等温線、ならびに粒度分布を測定した。図1に吸着等温線を示す。d50およびd10を表に示す。平衡相対湿度30%、25℃で測定した自由水含有量は0.11重量%であった。
【0079】
【表1】
【0080】
実施例1と比較実験Aを比較すると、実施例Iのアモキシシリン粉末は比較実験Aのアモキシシリン粉末より少ない自由水を含有していることが分かる。
【0081】
実施例II
実施例Iの方法で得られたアモキシシリン三水和物粉末を、水分活性0.15まで乾燥する。この粉末を、4対1の重量比でクラブラン酸カリウムと混合する(無水アモキシシリンとクラブラン酸として計算した)。この混合物は安定である。
【0082】
実施例III
実施例Iの方法で得られたアモキシシリン粉末を、水分活性0.2まで乾燥する。この粉末を、4対1の比でクラブラン酸カリウムと混合する。この混合物は安定である。
【0083】
実施例IV
実施例Iの方法で得られたアモキシシリン粉末を、水分活性0.15まで乾燥する。この粉末を、4対1の比でクラブラン酸カリウムと混合する。この混合物は安定である。
【0084】
実施例V
乾燥を、円錐形の真空接触式乾燥機(Vrieco−Nauta)を用いず、材料に機械的衝撃を与えない乾燥機(Ventilation stove)を用いて行った点を変えて実施例Iを繰返した。乾燥は、35℃の温度で16時間行った。d50およびd10は、それぞれ66.3μmおよび17.4μmである。嵩密度およびタップ密度は、それぞれ0.25g/mlおよび0.47g/mlである。
【0085】
実施例VI
実施例Iに従ってアモキシシリン三水和物粉末の別のバッチを調製した(d50およびd10はそれぞれ61μmおよび19μmであり、嵩密度およびタップ密度はそれぞれ0.58g/mlおよび0.79g/mlである)。このバッチを風力篩い別け(Hosakawa Alpine製の風力篩200LS−N)にかけた。篩い別けは、75μmのスクリーンを用いて10分間行った。前記篩い別け中に生成した少量の凝集物を、振動篩(425μm)を用いて篩上分(篩いを通過しなかった部分)から取除き、その後得られた篩上分の結晶のタップ密度、嵩密度、d50、d10を測定した。d50およびd10は、それぞれ88μmおよび36μmである。嵩密度およびタップ密度は、それぞれ0.59g/mlおよび0.74g/mlである。
【図面の簡単な説明】
【0086】
【図1】図1は実施例Iおよび比較実験Aの吸着等温線を示す。
【特許請求の範囲】
【請求項1】
平衡相対湿度30%および温度25℃で測定した自由水含有量が0.1重量%未満のアモキシシリン三水和物の製品。
【請求項2】
平衡相対湿度30%および温度25℃で測定した自由水含有量が、0.07重量%未満、より好ましくは0.05重量%未満である、請求項1に記載の製品。
【請求項3】
温度25℃で測定した水分活性が、0.05より高い、好ましくは0.07より高い、好ましくは0.10より高い、好ましくは0.15より高い、好ましくは0.20より高い、好ましくは0.25より高い、好ましくは0.30より高い、請求項1に記載の製品。
【請求項4】
前記製品がアモキシシリン三水和物結晶性粉末である、請求項1から3のいずれか一項に記載の製品。
【請求項5】
d50が、10μmより大きい、好ましくは20μmより大きい、より好ましくは30μmより大きい、より好ましくは35μmより大きい、より好ましくは40μmより大きい、請求項4に記載の結晶性粉末。
【請求項6】
d10が、3μmより大きい、好ましくは5μmより大きい、より好ましくは8μmより大きい、より好ましくは10μmより大きい、請求項4または請求項5に記載の結晶性粉末。
【請求項7】
嵩密度が、0.45g/mlより高い、好ましくは0.5g/mlより高い、より好ましくは0.55g/mlより高い、請求項4から6のいずれか一項に記載の結晶性粉末。
【請求項8】
タップ密度が、0.6g/mlより高い、好ましくは0.7より高い、より好ましくは0.8g/mlより高い、請求項4から7のいずれか一項に記載の結晶性粉末。
【請求項9】
請求項1から8のいずれか一項に記載の製品を、第2の薬学的に活性な薬剤および/または助剤と混合するステップを含む方法。
【請求項10】
(i)請求項1から8のいずれか一項に記載の製品と、
(ii)第2の薬学的に活性な薬剤および/または助剤と、
を含む混合物。
【請求項11】
前記第2の薬学的に活性な薬剤が、クラブラン酸塩、好ましくはクラブラン酸カリウムである、請求項9に記載の方法または請求項10に記載の混合物。
【請求項12】
請求項1から8のいずれか一項に記載の製品、または請求項10に記載の混合物を圧縮して圧縮製品を製造するステップを含む方法。
【請求項13】
前記圧縮製品が顆粒または錠剤である、請求項12に記載の方法。
【請求項14】
請求項1から8のいずれか一項に記載の製品の、医薬組成物調製のための使用。
【請求項15】
請求項1から8のいずれか一項に記載の製品、または請求項10に記載の混合物の、錠剤を調製するための、またはカプセルを充填するための使用。
【請求項16】
6−アミノ−ペニシラン酸またはその塩を、担体上に固定化された酵素の存在下、活性状態のp−ヒドロキシフェニルグリシンと反応させることによってアモキシシリンを調製するステップと、
塩酸を含有する、アモキシシリン含有水溶液を形成するステップと、
前記水溶液からアモキシシリン三水和物を結晶化させるステップと、
を含む、アモキシシリン三水和物の調製方法。
【請求項17】
アモキシシリンを結晶化させる水溶液は、アモキシシリン濃度が、0.6モル/l未満、好ましくは0.5モル/l未満、より好ましくは0.4モル/l未満、より好ましくは0.3モル/l未満である、請求項16に記載の方法。
【請求項18】
アモキシシリンを結晶化させる溶液が、アモキシシリン1,000,000重量部当り、200重量部未満のタンパク質、好ましくは100重量部未満のタンパク質、より好ましくは50重量部未満のタンパク質、より好ましくは35重量部未満のタンパク質を前記溶液中に含有する、請求項16または請求項17に記載の方法。
【請求項19】
前記水溶液から結晶を分離するステップと、分離された結晶を乾燥するステップとを含み、d50が、10μmより大きい、好ましくは20μmより大きい、より好ましくは30μmより大きい、より好ましくは35μmより大きい、より好ましくは40μmより大きい結晶性粉末を得る、請求項16から18のいずれか一項に記載の方法。
【請求項20】
d10が、3μmより大きい、好ましくは5μmより大きい、より好ましくは8μmより大きい、より好ましくは10μmより大きい結晶性粉末を得る、請求項16から19のいずれか一項に記載の方法。
【請求項1】
平衡相対湿度30%および温度25℃で測定した自由水含有量が0.1重量%未満のアモキシシリン三水和物の製品。
【請求項2】
平衡相対湿度30%および温度25℃で測定した自由水含有量が、0.07重量%未満、より好ましくは0.05重量%未満である、請求項1に記載の製品。
【請求項3】
温度25℃で測定した水分活性が、0.05より高い、好ましくは0.07より高い、好ましくは0.10より高い、好ましくは0.15より高い、好ましくは0.20より高い、好ましくは0.25より高い、好ましくは0.30より高い、請求項1に記載の製品。
【請求項4】
前記製品がアモキシシリン三水和物結晶性粉末である、請求項1から3のいずれか一項に記載の製品。
【請求項5】
d50が、10μmより大きい、好ましくは20μmより大きい、より好ましくは30μmより大きい、より好ましくは35μmより大きい、より好ましくは40μmより大きい、請求項4に記載の結晶性粉末。
【請求項6】
d10が、3μmより大きい、好ましくは5μmより大きい、より好ましくは8μmより大きい、より好ましくは10μmより大きい、請求項4または請求項5に記載の結晶性粉末。
【請求項7】
嵩密度が、0.45g/mlより高い、好ましくは0.5g/mlより高い、より好ましくは0.55g/mlより高い、請求項4から6のいずれか一項に記載の結晶性粉末。
【請求項8】
タップ密度が、0.6g/mlより高い、好ましくは0.7より高い、より好ましくは0.8g/mlより高い、請求項4から7のいずれか一項に記載の結晶性粉末。
【請求項9】
請求項1から8のいずれか一項に記載の製品を、第2の薬学的に活性な薬剤および/または助剤と混合するステップを含む方法。
【請求項10】
(i)請求項1から8のいずれか一項に記載の製品と、
(ii)第2の薬学的に活性な薬剤および/または助剤と、
を含む混合物。
【請求項11】
前記第2の薬学的に活性な薬剤が、クラブラン酸塩、好ましくはクラブラン酸カリウムである、請求項9に記載の方法または請求項10に記載の混合物。
【請求項12】
請求項1から8のいずれか一項に記載の製品、または請求項10に記載の混合物を圧縮して圧縮製品を製造するステップを含む方法。
【請求項13】
前記圧縮製品が顆粒または錠剤である、請求項12に記載の方法。
【請求項14】
請求項1から8のいずれか一項に記載の製品の、医薬組成物調製のための使用。
【請求項15】
請求項1から8のいずれか一項に記載の製品、または請求項10に記載の混合物の、錠剤を調製するための、またはカプセルを充填するための使用。
【請求項16】
6−アミノ−ペニシラン酸またはその塩を、担体上に固定化された酵素の存在下、活性状態のp−ヒドロキシフェニルグリシンと反応させることによってアモキシシリンを調製するステップと、
塩酸を含有する、アモキシシリン含有水溶液を形成するステップと、
前記水溶液からアモキシシリン三水和物を結晶化させるステップと、
を含む、アモキシシリン三水和物の調製方法。
【請求項17】
アモキシシリンを結晶化させる水溶液は、アモキシシリン濃度が、0.6モル/l未満、好ましくは0.5モル/l未満、より好ましくは0.4モル/l未満、より好ましくは0.3モル/l未満である、請求項16に記載の方法。
【請求項18】
アモキシシリンを結晶化させる溶液が、アモキシシリン1,000,000重量部当り、200重量部未満のタンパク質、好ましくは100重量部未満のタンパク質、より好ましくは50重量部未満のタンパク質、より好ましくは35重量部未満のタンパク質を前記溶液中に含有する、請求項16または請求項17に記載の方法。
【請求項19】
前記水溶液から結晶を分離するステップと、分離された結晶を乾燥するステップとを含み、d50が、10μmより大きい、好ましくは20μmより大きい、より好ましくは30μmより大きい、より好ましくは35μmより大きい、より好ましくは40μmより大きい結晶性粉末を得る、請求項16から18のいずれか一項に記載の方法。
【請求項20】
d10が、3μmより大きい、好ましくは5μmより大きい、より好ましくは8μmより大きい、より好ましくは10μmより大きい結晶性粉末を得る、請求項16から19のいずれか一項に記載の方法。
【図1】

【公表番号】特表2006−520765(P2006−520765A)
【公表日】平成18年9月14日(2006.9.14)
【国際特許分類】
【出願番号】特願2006−504807(P2006−504807)
【出願日】平成16年3月19日(2004.3.19)
【国際出願番号】PCT/EP2004/003031
【国際公開番号】WO2004/082662
【国際公開日】平成16年9月30日(2004.9.30)
【出願人】(505220217)デーエスエム アイピー アセッツ ベー. ヴェー. (29)
【Fターム(参考)】
【公表日】平成18年9月14日(2006.9.14)
【国際特許分類】
【出願日】平成16年3月19日(2004.3.19)
【国際出願番号】PCT/EP2004/003031
【国際公開番号】WO2004/082662
【国際公開日】平成16年9月30日(2004.9.30)
【出願人】(505220217)デーエスエム アイピー アセッツ ベー. ヴェー. (29)
【Fターム(参考)】
[ Back to top ]