
アリピプラゾールおよびハロペリドールのパモ酸塩
本発明は、ハロペリドールおよびアリピプラゾールのパモ酸塩が、良好ないし優れた持続性および/または長時間放出性プロフィールをもたらすという知見に関する。したがって、本発明の一局面において、本発明は、ハロペリドールまたはアリピプラゾールのパモ酸塩を含む。好ましくは、パモ酸塩は、ハロペリドールのパモ酸塩に対する比が1:1または2:1であることを特徴とする。パモ酸塩は、針状物または緻密な結晶(例えば、図面に示したもの)などの結晶性であり得る。本発明は、さらに、ハロペリドールおよびアリピプラゾールのパモ酸塩を含有する医薬組成物を投与することを含む、処置を必要とする個体の処置方法に関する。
【発明の詳細な説明】
【発明の詳細な説明】
【0001】
発明の背景
種々の抗精神病薬のポリマー系徐放性デバイスが報告されている。しかしながら、かかる徐放性デバイスは、製造するのに費用がかかり、作製するのが困難な傾向にある。したがって、活性剤の薬理学的プロフィールを最大にし、より費用効率よく作製し得るような薬剤を送達するための改善された方法の必要性がなお存在する。
【0002】
発明の要旨
本発明は、ハロペリドール、オランザピンおよびアリピプラゾールのパモ酸塩が、良好ないし優れた持続性および/または長時間放出性プロフィールをもたらすという知見に関する。したがって、本発明の一局面において、本発明は、ハロペリドール、オランザピンまたはアリピプラゾールのパモ酸塩を含む。好ましくは、パモ酸塩は、薬物、例えばハロペリドールのパモ酸塩に対する比が1:1または2:1であることを特徴とする。パモ酸塩は、針状物または緻密な(dense)結晶(例えば、図面に示したもの)などの結晶性であり得る。
【0003】
本発明は、さらに、ハロペリドールおよびアリピプラゾールのパモ酸塩を含有する医薬組成物を投与することを含む、処置を必要とする個体の処置方法に関する。
【0004】
発明の詳細な説明
本発明は、ハロペリドール、オランザピンおよびアリピプラゾールのパモ酸塩が、良好ないし優れた持続性および/または長時間放出性プロフィールをもたらすという知見に関する。したがって、本発明の一局面において、本発明は、ハロペリドール、オランザピンまたはアリピプラゾールのパモ酸塩を含む。好ましくは、パモ酸塩は、薬物、例えばハロペリドールのパモ酸塩に対する比が1:1または2:1であることを特徴とする。パモ酸塩は、針状物または緻密な結晶(例えば、図面に示したもの)などの結晶性であり得る。
【0005】
本発明は、さらに、ハロペリドール、オランザピンおよびアリピプラゾールのパモ酸塩を含有する医薬組成物を投与することを含む、処置を必要とする個体の処置方法に関する。
【0006】
あるいはまた、本発明のパモ酸塩を製造するために、米国特許第5,006,528号または同第4,734,416号(参照により本明細書に援用される)に記載されたカルボスチリル化合物などの類似の活性剤を使用することができる。
【0007】
有機塩基の電離平衡を以下に示す。
【0008】
【化1】

【0009】
示した化学プロセスは、酸解離定数(Ka)、遊離塩基の溶解度および塩の溶解度積(Ksp)から構成される。アルカリ性条件下では、遊離塩基形態は、安定な形態である。溶液のpHが低下するにしたがって、イオン化した薬物の割合および水溶性が増大する。イオン化した薬物(プロトン化アミン)が高濃度のとき、塩の溶解度積(Ksp)が超過し、その塩形態が析出する。薬物および対イオンの性質がその塩のKspおよび会合した固体状態の特性を決定する。
【0010】
塩基性薬物であるハロペリドールの化学構造および電離平衡は、
【化2】
である。
【0011】
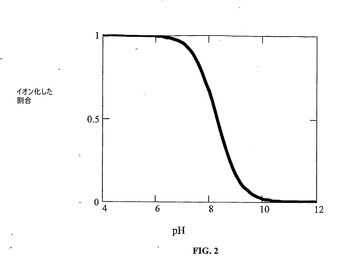
図2は、pHの関数としての薬物のイオン化プロフィールを示す。9より大きいpH値では、薬物は、主に非イオン化状態であるが、pKa未満のpH値では、薬物は正に帯電する。適切な対イオンと結合したプロトン化形態の薬物は、イオン塩の形成を可能にする。
【0012】
塩基の塩を調製するのに使用されている対イオンはさまざまなものがある。塩基性薬物の塩を形成するのに最も頻繁に使用されるアニオンは、塩酸塩の形態である。医薬用塩に使用されるカルボン酸対イオンの2つの例は、
【化3】
である。
【0013】
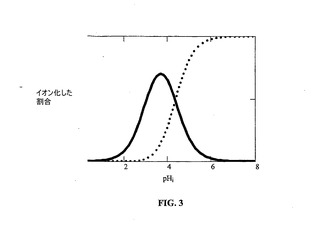
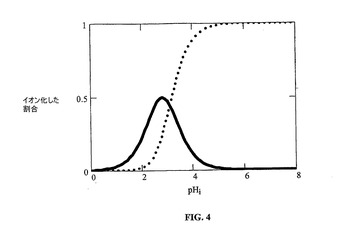
イオン平衡を図3および4に示す。2つの対イオンは、酒石酸塩およびパモ酸塩に溶解特性において広い範囲をもたらす。これらの対イオンの各々は2価であることに注目されたい。したがって、1:1および2:1(プロトン化塩基:対イオン)塩が可能である。塩が形成される溶液状態により、どちらの塩形態(1:1または2:1)が析出するかが決まる。化学構造は、これらの2つの対イオンが非常に異なる構造および特性を有することを示す。酒石酸は、水溶性の高い塩をもたらすが、パモ酸は、溶解性が制限された塩を形成し、したがって、溶解を遅延させるために使用され得る。水溶性形態および水不溶性形態の両方が、薬物送達製剤の開発における使用目的を有する。
【0014】
特許請求の範囲に記載の発明の組成物は、投与目的のための種々の医薬形態を含み得る。本発明の医薬組成物を調製するため、活性成分としての主題の化合物の有効量を、薬学的に許容され得る担体(該担体は、投与に所望される調製物の形態に応じて多種多様な形態をとり得る)と充分に混合された状態で合わせる。これらの医薬組成物は、望ましくは、好ましくは投与に適した単一の投薬形態である。投与としては、経口を含む経腸、または肺系、経皮、皮下もしくは筋肉内注射もしくは埋込みを含む非経口が挙げられる。後者の投与経路では、主題の化合物は、好ましくは水性溶媒中に懸濁され、ソルビタンエステルのポリオキシエチレン誘導体、例えば、ポリソルベート80 (Tween 80 (登録商標))およびポリソルベート20 (Tween 20 (登録商標))、レシチン、ポリオキシエチレンエーテルおよびポリオキシプロピレンエーテル、デオキシコール酸ナトリウムなどの湿潤剤;セルロース誘導体、例えばメチルセルロース、ナトリウムカルボキシメチルセルロースおよびヒドロキシプロピルメチルセルロース、ポリビニルピロリドン、アルギン酸塩、キトサン、デキストラン、ゼラチン、ポリエチレングリコール、ポリオキシエチレンエーテルおよびポリオキシプロピレンエーテルなどの懸濁剤;例えば塩酸などの酸;例えば水酸化ナトリウムなどの塩基;適量の酸(リン酸、コハク酸、酒石酸、乳酸、酢酸、マレイン酸またはクエン酸など)および塩基、特に水酸化ナトリウムまたはリン酸一水素二ナトリウムの混合物を含む緩衝溶液;保存剤、例えば、安息香酸、ベンジルアルコール、ブチル化ヒドロキシアニソール、ブチル化ヒドロキシトルエン、クロルブトール、没食子酸塩、ヒドロキシ安息香酸塩、EDTA、フェノール、クロロクレゾール、メタクレゾール、塩化ベンゾトニウム、ミリスティールγ塩化ピコリニウム、酢酸フェニル水銀、チメロサールなど;張性調整剤、例えば、塩化ナトリウム、デキストロース、マンニトール、ソルビトール、ラクトース、硫酸ナトリウムなどをさらに含み得る。あるいはまた、主題の化合物は、油中に製剤化され得る。この目的に適切な油類は、不揮発油、例えば、ピーナツ油、ゴマ油、綿実油、コーン油、サフラワー油、ヒマシ油、エチルオレエート、大豆油、長鎖脂肪酸または中鎖脂肪酸の合成グリセロールエステルおよびこれらの混合物ならびに他の油類である。また、増粘剤、例えば、例えば、モノステアリン酸アルミニウム、エチルセルロース、トリグリセリド、硬化ヒマシ油などを組成物に添加してもよい。
【0015】
精神病性障害の処置における主題の化合物の有用性を鑑みると、本発明が、精神病性障害を患う温血動物、特にヒトの処置方法を提供することは明白であり、該方法は、薬学的有効量の主題の化合物を医薬用担体と混合して投与することを含む。さらなる局面において、本発明は、主題の化合物の薬としての、特に抗精神病薬としての使用に関する。一般に、有効量は、0.05 mg/kg〜50 mg/kg体重、より好ましくは0.5 mg/kg〜10 mg/kg体重であり得ることが想定される。
【0016】
活性剤は、好ましくは、持続性製剤にて投与される。一態様において、活性剤は製剤から少なくとも約24時間、好ましくは約48時間にわたって放出される。
【0017】
また、活性剤は長時間放出製剤にて投与され得る。一態様において、長時間放出製剤は、活性剤を、少なくとも約7日間、好ましくは少なくとも約14日間、あるいはまた、少なくとも2、3、4、6または8週間にわたって放出する。組成物は、好ましくは、筋肉内または皮下などの注射により投与される。
【0018】
一態様において、製剤は、単一または単独投薬量として投与され得る。しかしながら、本発明は、数週間もしくは数ヶ月またはそれより長くにわたって反復投薬を受けている個体などの、不断または長期治療を必要とする個体に特に有用である。かかる投薬計画において、該方法は、第1長時間放出製剤の第1投薬および第2長時間放出製剤の第2投薬を含み得る。第2製剤は、第1のものと同一であってもよく、実質的同一であってもよく、異なっていてもよく、同じ活性剤または異なる活性剤を含み得る。例えば、第2製剤は、第1投薬後、約7日またはそれ以降(例えば、少なくとも約14日または少なくとも約17日)で投与され得、このとき、第1投薬は、1、2、3、4、5、6、7、8、9、10、11、12、13、14日間またはそれより長い期間の薬剤の放出をもたらす。
【0019】
用語「治療有効量」は、任意のパラメータまたは臨床症状の改善をもたらす量を定義することがさらに意図される。実際の投薬量は、各患者により変わり得、必ずしも、すべての疾患症状の消失を意味するものではない。
【0020】
本明細書で使用する場合、用語「個体」、「被験体」または「患者」は、限定されないが、特定の疾患状態に苦しむ、哺乳動物などの温血動物(ヒトを含むが、これに限定されない)をいう。
【0021】
本明細書に記載する処置において使用される化合物の治療有効量は、慣用技術の使用により、および同様の状況下で得られる結果を観察することにより、当業者としての担当診断医によって容易に決定され得る。治療有効投薬量を決定する際には、限定されないが、哺乳動物の種;その体格、年齢および一般健康状態;関与している特定の疾患;疾患の程度または困難さ(involvement)もしくは重篤度;個々の患者の応答;投与する具体的な化合物;投与経路;投与する調製物に特有の生物学的利用能;選択した投与計画;併用薬の使用;および他の関連する状況を含むいくつかの因子が担当診断医によって考慮される。
【0022】
投与の好ましい量および形態は、当業者が決定することができる。製剤調製の当業者は、例えばRemington's Pharmaceutical Sciences, 最新版, Mack Publishing Co.に記載された当該技術分野で公知の製剤技術を用い、選択した化合物の具体的な特徴、処置対象の疾患状態、疾患の病期および他の関連する状況に応じて、投与の適切な形態および様式を容易に選択し得る。
【0023】
医薬組成物は、当該技術分野で公知の技術を用いて製造することができる。典型的には、治療有効量の化合物を、薬学的に許容され得る担体と混合する。
【0024】
本発明の組成物は、注射などにより非経口投与され得る。好ましい投与の方法としては、例えば、筋肉内注射および皮下注射が挙げられる。
【0025】
非経口投与では、該化合物を、生理学的に許容され得る医薬用担体に溶解し、溶液または懸濁液として投与し得る。例えば20℃で少なくとも20 cpの粘度を有する粘性の注射可能な担体が好ましい。他の態様において、懸濁液の液相は、20℃で少なくとも約30 cp、40 cp、50 cpおよび60 cpの粘度を有する。組成物はまた、粘度向上剤、密度向上剤、張性向上剤および/または湿潤剤を含有し得る。好適な医薬用担体の具体例としては、水、生理食塩水、デキストロース溶液、フルクトース溶液、エタノール、または動物油、植物油もしくは合成による油が挙げられる。医薬用担体はまた、当該技術分野で公知の保存剤および緩衝溶液を含んでいてもよい。
【0026】
別の態様において、製剤は、外科的に埋め込み得る。かかる製剤としては、ポリラクチドおよびポリラクチド-コ-グリコリドおよびコラーゲン製剤などの任意の周知の生分解性および生腐食性(bioerodible)担体が挙げられる。かかる物質は、固形の埋没物、スポンジなどの形態であり得る。いずれにせよ、該物質の局所使用では、活性成分は、通常、担体または賦形剤中に約1:1000〜1:20,000の重量比で存在するが、この範囲の比に限定されない。
【0027】
好ましくは、該化合物は長時間放出製剤中に存在する。長時間(徐放性または制御性ともいう)放出調製物は、本明細書に記載する活性剤を取込むか、または封入するポリマー(好ましくはポリラクチドまたはポリラクチド-コ-グリコリドポリマー)の使用により得られ得る。長時間放出製剤は、ポリマー-薬物混合物の噴霧乾燥、エマルジョンを用いる(-based)技術、コアセルベーションを用いる技術、フィルムキャスティング、押出し成形を用いる技術、および長時間放出プロフィールを有するポリマー-薬物微粒子を製造するための他のプロセスにより作製し得る。本発明の薬剤を含めるために使用され得る好適な長時間放出技術としては、限定されないが、例えばWrightに対する米国特許第6,264,987号、同第5,654,008号および/または同第5,792,477号などに記載されたMEDISORB(登録商標)技術;例えばHerbertに対する米国特許第6,358,443号に記載されたPROLEASE(登録商標)技術;Southern Research Instituteにより、例えば米国特許第6,306,425号に記載された技術;および「徐放性微粒子の調製方法(Method of Preparing Sustained Release Microparticles)」という米国特許出願第60/441,946号(2003年1月23日出願)、ならびにALZAMER(登録商標) Depot注射技術を含むAlza Corp.により記載された技術が挙げられる。これらの特許の内容は、参照によりその全体が本明細書に援用される。
【0028】
好ましい一態様において、薬剤は、長時間放出デバイスまたは製剤中に、該デバイスまたは製剤の全重量の少なくとも約5重量%、好ましくは少なくとも約10重量%、より好ましくは少なくとも約30重量%の量で存在する。
【0029】
例えば、コアセルベーション技術により、または界面重合により調製される微粒子(例えば、それぞれ、ヒドロキシメチルセルロースまたはゼラチン-マイクロカプセルおよびポリ-(メチルメタクリレート)マイクロカプセル)、コロイド状薬物送達システム(例えば、リポソーム、アルブミン、微粒子、マイクロエマルジョン、ナノ粒子およびナノカプセル)またはマクロエマルジョン内に活性剤を取り込むこともまた想定される。
【0030】
組成物が注射可能な物質(無針(needle-less)注射を含むが、これに限定されない)として使用される場合、従来の注射可能な担体内に製剤化し得る。好適な担体としては、生体適合性溶液および薬学的に許容され得る溶液が挙げられる。
【0031】
以下の実施例は、例示を目的とするものであって、本発明の範囲を限定するものではない。
【実施例】
【0032】
実施例1:
ハロペリドールのパモ酸塩は、溶媒中でのパモ酸またはパモ酸塩によるハロペリドールの処理により調製することができる。ハロペリドールパモ酸塩は、適切な溶媒、例えばエタノール中にハロペリドールを酢酸とともに含む溶液を、パモ酸二ナトリウム、パモ酸または他のパモ酸塩の溶液に添加し、沈殿するまで1〜3日以上、静置することにより調製することができる。あるいはまた、塩を沈殿させるために、溶液の蒸発、低速もしくは高速冷却または攪拌などの他の方法を使用することもできる。
【0033】
具体的には、酸性化エタノール(5%酢酸)中0.1Mハロペリドール溶液2.5 mlを、エタノール/水(50/50)中パモ酸二ナトリウム塩(2.5 ml)0.1M溶液2.5 mlに添加した。混合物を、室温で1〜3日間、放置した。得られた沈殿物を吸引により濾別し、エタノールで洗浄し、60℃の真空炉内で乾燥し、1:1ハロペリドールパモ酸塩240mgを得た。
【0034】
実施例2:
酸性化エタノール(5%酢酸)中0.25Mハロペリドール溶液2.5 mlを、エタノール/水(75/25)中0.05Mパモ酸二ナトリウム塩溶液12.5 mlに添加した。混合物を、室温で1〜3日間、放置した。得られた沈殿物を吸引により濾別し、エタノールで洗浄し、60℃の真空炉内で乾燥し、2:1ハロペリドールパモ酸塩206mgを得た。
【0035】
実施例3:
酸性化エタノール(5%酢酸)中0.25Mハロペリドール溶液2.5 mlを、エタノール/水(50/50)中0.1Mパモ酸二ナトリウム塩溶液6.25 mlに添加した。混合物を、室温で1〜3日間、放置した。得られた沈殿物を吸引により濾別し、エタノールで洗浄し、60℃の真空炉内で乾燥し、2:1ハロペリドールパモ酸塩264mgを得た。
【0036】
実施例4:
酸性化エタノール(5%酢酸)中0.05Mハロぺリドール溶液5mlを、エタノール/水(50/50)中0.25Mパモ酸二ナトリウム塩溶液1mlに添加した。混合物を、室温で1〜3日間放置した。生じた沈殿物を吸引で濾過し、エタノールで洗浄し60℃の真空炉内で乾燥して、1:1パモ酸ハロぺリドール塩107mgを得た。
【0037】
実施例5:
酸性化エタノール(5%酢酸)中0.05Mハロぺリドール溶液5mlを、エタノール/水(50/50)中0.1Mパモ酸二ナトリウム塩溶液2.5mlに添加した。混合物を、室温で1〜3日間放置した。生じた沈殿物を吸引で濾過し、エタノールで洗浄し60℃の真空炉内で乾燥して、1:1パモ酸ハロぺリドール塩119mgを得た。
【0038】
実施例6:
酸性化エタノール中アリピプラゾール溶液(0.05〜0.5M)を、水/エタノール(100/0〜0/100)の混合物中パモ酸二ナトリウム塩溶液(0.05〜0.5M)に添加した。混合物を、室温で1〜3日間放置した。生じた沈殿物を吸引で濾過し、溶媒で洗浄し60℃の真空炉内で乾燥した。
【0039】
実施例7:
ハロペリドールおよびオランザピン調剤の単回皮下用量の投与に続いてのラット中のハロペリドールおよびオランザピンの薬物動態の評価
【0040】
種および系統: Sprague-Dawley ラット雄;450+/−50グラム
【0041】
試験群:4群、12被験体
A群:20mgのハロペリドールバルク(bulk)薬物を1回SC注
射した3匹のラット
B群:40mgのパモ酸ハロペリドールを1回SC注射した3匹の
ラット
C群:20mgのオランザピンバルク薬物を1回SC注射した3匹
のラット
D群:50mgのオランザピンバルク薬物を1回SC注射した3匹
のラット
【0042】
注射の経路: 肩甲間部への皮下(SC)注射
【0043】
注射賦形剤: 水性希釈剤:水、3%低粘度CMC、0.1%Tween 20、0.9%NaCl中
【0044】
投薬容量: 投薬量懸濁液は以下のようにして製剤化した。
A群:0.75mL希釈剤中の20mg粉末
B群:0.75mL希釈剤中の40mg微粒子
C群:0.75mL希釈剤中の20mg粉末
D群:0.75mL希釈剤中の50mg粉末
【0045】
血液採取:
ハロタンによる麻酔の後に血液試料を尾静脈より採取した。抗凝固剤を含まないシリンジを、血液採取に使用し、次いで全血をK2 EDTAおよび混合ビーズ(Microtainer(登録商標);MFG♯ BD365974)を含むチューブに移した。血液試料を処理して(チューブを15〜20回上下にして、>14000gで2分間遠心分離する)血漿を分離した。この方法で準備した血漿試料を標識されたプレインチューブ(Microtainer(登録商標);MFG♯ BD5962)に移し、<−70℃で凍らせて保存した。
【0046】
血液容量: 初めの24時間のそれぞれの時間点では少なくとも血液250μLであり、その後のそれぞれの時間点では400μL
【0047】
血漿を得るための時間点:
2h 24h 3d 10d*
4h 32h 4d 14d*
8h 2d 7d
*注:B群だけは7日目以降の時間点を有した。また、いずれの群についても、血漿濃度が定量限界より低くなった時、その群は終了とした。
【0048】
観察された結果は、パモ酸塩が良から優の長時間放出プロフィールを表したことを示す。
【0049】
実施例8:
ポリマーおよびパモ酸塩を含む注射可能な微粒子は、効率が良く簡易な1つの溶媒処理を使用して調製され得る。PLGポリマーおよび塩は1つの溶媒中で共溶解され得、(2)溶媒を真空乾燥または昇華によって取り除き、ポリマー/薬物マトリックスを形成し、(3)該マトリックスを粉砕し、粉末を作製し、(4)該生じた粉末を圧縮し、圧縮されたマトリックスを形成し、(5)該圧縮されたマトリックスを粉砕すると、緻密で注射可能な微粒子製剤が形成され得る。好ましくは、パモ酸塩充填は、微粒子組成物の最終重量の約10%または約30%以上(w/w)であり得る。溶媒、例えば塩化メチレン、アセトン、ジメチルスルホキシド(DMSO)、アセトニトリル、および酢酸エチルは使用に好適である。
【0050】
適切なポリマーとしては、
ラクチド:グリコリド比;固有粘度;末端基;溶媒
A30%(w/w)50:50、0.75dL/g;酸末端基;CH2Cl2
B30%(w/w)75:25;0.60dL/g;ラウリルエステル末端基;CH2Cl2
C25 30%(w/w)50:50;0.61dL/g;ラウリルエステル末端基;CH2Cl2
が挙げられる。
【0051】
パモ酸塩/ポリマー/溶媒混合物は、ポリテトラフルオロエチレン平面型(およそ1インチ×1インチ×1/2インチ奥行き)または3インチ直径ジャーのいずれか中に注ぎ込まれ得、例えばフィルムを形成する。該フィルムはFTS Dura-Dry Lyophilizer (Kinetic Systems, Inc., Santa Clara, CA)または真空炉中のいずれかで乾燥され得る。最大真空、雰囲気圧力、高温、周囲温度、および乾燥時間の変化を含む様々な環境下で乾燥されたフィルムが作られ得る。
【0052】
該フィルムは、14,000rpmで操作する24枚歯の Retsch Ultra Centrifugal Mill (Retsch, Inc., Newtown, PA)を使用して粉砕され得る。粉砕する前に、採集皿を、液体窒素で満たす。採集皿から集められた、生じた粉末は、続く圧縮工程を助ける流動性生成物である。フィルムを粉砕して作製された粉末の一部は、この時点で分析するために、保持され得る。これらの粉末は、以下に記載されるフィルム粉末の続く圧縮および再粉砕によって作られた粉末との比較のために保持され得る。
【0053】
粉砕された粉末の一部は、Carver Model C Press (Carver, Inc., Wabash, IN)および約1/4インチまたは約1/2インチ円筒ダイのいずれかを使用して、圧縮され得る。約50〜約300ミリグラムの粉砕された粉末を、ダイ中に充填し、室温で約30秒間、約5000ポンドのマシンセッティングで粉砕し、ペレットを形成する。
【0054】
圧縮されたマトリックスを、14000rpmで操作する24枚歯の Retsch Ultra Centrifugal Mill(Retsch, Inc., Newtown, PA)を使用して引き続き粉砕する。粉砕する前に、採集皿を液体窒素で満たす。最終粉末を採集皿から集め、分析のためにバイアル中に入れた。
【0055】
本明細書に記載された、持続放出組成物は、エマルジョン、コアセルベーション、および低温マイクロカプセル化技術のいずれかによって調製され得る。それぞれの技術に関連する一般的な処理を、以下に記載する。
【0056】
コアセルベーション-S/O/O処理
コアセルベーション処理は、本明細書中では溶媒-油-油(S/O/O)処理ともいうが、薬物と有機ポリマー溶液を用いた油中溶媒型エマルジョンの形成を要する。次いで、相分離を誘導させるためおよびポリマーを沈殿させるために、油(典型的にシリコーン油)を油中水型エマルジョンに加える。次いで、初期微粒子を、油とポリマー溶媒を取り除く溶媒中で失活させる。パモ酸塩を溶媒-油-油(S/O/O)エマルジョン系を使用したPLGポリマー中にカプセル化する。初期初期微粒子を、S/O/O中で内部エマルジョン工程において形成して、その後コアセルベーション化と硬化工程に供する。微粒子を集めて乾燥し、バイアルに充填する。さらに、完全処理のそれぞれの工程のさらなる詳細は、以下に記す。
【0057】
内部エマルジョン形成
油中溶媒型エマルジョンは超音波処理を使用して作製する。エマルジョンの溶媒相は溶解した薬物および様々な賦形剤を水中に含んだ。PLG相は、塩化メチレンに溶解したポリマーを含んだ。
【0058】
コアセルベーション形成
コアセルベーションは、攪拌しながらシリコーン油を制御された割合で内部エマルジョンに加えることによって誘導され得、初期微粒子を形成する。形成された初期微粒子は比較的軟らかく、硬化を要する。
【0059】
微粒子の硬化
初期微粒子を穏やかに攪拌しながら、ヘプタン/エタノール混合溶媒に加える。混合溶媒は初期微粒子を硬化する。約3℃で約1時間硬化させた後、溶媒混合物をデカントし、純ヘプタンを3℃で加え、約1時間混ぜた。
【0060】
微粒子乾燥と収集
硬化段階の後、微粒子を乾燥チャンバの中の微細メッシュ細孔板に移して集める。硬化用の容器の、最終ヘプタン洗浄が行われる。温度を約3℃から約38℃に上げていきつつ、4日間、微粒子を窒素ガスにより乾燥させる。
【0061】
一般的に、PLGを塩化メチレンに溶解する。溶媒中に薬物およびいくらかの賦形剤を溶解させて内部相を調製する。次いで、プローブ音波処理をしながら、水溶液をポリマー溶液中に注入する。次いで、得られたエマルジョンをエマルジョン反応器に加える。シリコーン油(350センチストークス)を約1000rpmで攪拌させながら、ぜん動ポンプによってゆっくりと反応器に加える。次いで、混合物をn-ヘプタンに加える。約2時間攪拌させた後、微粒子を濾過により単離し、一晩中真空乾燥する。
【0062】
エマルジョン処理-S/O/W処理
エマルジョン処理は、溶媒-油-水(S/O/W)処理とも言う。簡単には、薬物の溶液はポリマー溶液中に分散され、外側の水相(例えば、PVA)中に乳化される。次いで、微粒子を水性クエンチ(quench)中で硬化する。
【0063】
典型的な実験では、PLG(1.96g)を塩化メチレン(22.5g)中に溶解し、薬物を溶解する(1.75g溶媒中に20mgの薬物)。次いで、薬物溶液をシリンジ内に吸引し、プローブ音波処理をしながらポリマー溶液中に注入する。次いで、得られたエマルジョンを、5%ポリビニルアルコール(PVA)水溶液125gを含んだエマルジョン反応器中に素早く加える。反応器中の攪拌速度を約800RPMに設定した。混合物を約1.5分間攪拌し、水クエンチ(10℃で2.8L)中に加える。クエンチ中で約2時間後、硬化した微粒子を濾過により単離し、一晩中真空乾燥する。
【0064】
低温処理
低温処理では、噴霧化を用いて薬物を含んだポリマー溶液の小滴を形成した。次いで、初期微粒子を液体窒素中で凍結して、該ポリマー溶媒を、次のエタノール抽出技術によって取り除く。
【0065】
微粒子を作製するための低温処理は、(1)乾燥凍結物または乾燥薬物物質の作製、および(2)低温、無水技術を使用したマイクロカプセル化の2つの工程を含む。乾燥凍結物は、2−流動ノズルを用いた薬物および賦形剤の混合物の噴霧化、噴霧化された小滴の凍結、並びに乾燥凍結を用いた凍結小滴の乾燥によって調剤する。当該技術分野で公知のいずれの好適な乾燥する方法が使用され得ることを、理解されるべきである。特に、凍結小滴を、−26℃の棚(shelf)および96mTorrチャンバ圧力の第一乾燥条件で約7日間、続いて約20℃および0mTorrでの更なる3日間の二次乾燥によって、乾燥する。
【0066】
パモ酸塩を含んだ微粒子は、すべて10%薬物以上の名目上の標的負荷で、低温、無水処理で生産され得る。薬物を、塩化メチレン中に溶解した4A PLG3〜20%により成り立つ有機溶媒中で懸濁する。この懸濁液を氷上で約4分間音波処理し、次いで該懸濁液を、超音波ノズルを使用して噴霧化し、凍結エタノールの吸着床上に層をなす液体窒素に接触させることによって凍結する。該試料を、微粒子の硬化および溶媒の抽出を可能にするために、−80℃まで温める。次いで、該微粒子を濾過し乾燥する。
【0067】
固体/油/水(S/O/W)および固体/油/油(S/O/O)処理
固体薬物をまた、上記エマルジョンおよびコアセルベーション処理の修正されたバージョンを使用してカプセル化し得る。これらの修正された処理は、固体/油/水(S/O/W)および固体/油/油(S/O/O)を言う。例えば、固体薬物を、3〜20%PLGを含む塩化メチレン中で懸濁し、氷上で約4分間音波処理する。次の処理はW/O/OまたはW/O/Wの方法と類似した方法によって行われる。
【0068】
ポリマー:
使用するのに好適な、特定のPLGポリマーの例を以下に列挙する。以下の例で使用される全てのポリマーは、リスト中の前方に配置し、全ての列挙されたポリマーは、Cincinnati, OHのAlkermes, Inc.から購入され、以下のように表示され得る。
ポリマー2A:ポリ(ラクチド−コ−グリコリド)、50:50のラクチド:グリコリド比、12.3kD 分子量;IV=0.15(dL/g)、
ポリマー2A−1:ポリ(ラクチド−コ−グリコリド)、65:35のラクチド:グリコリド比、16kD 分子量;IV=0.19(dL/g)、
ポリマー2.5A:ポリ(ラクチド−コ−グリコリド)、50:50のラクチド:グリコリド比、25kD 分子量;IV=0.24(dL/g)、
ポリマー3A:ポリ(ラクチド−コ−グリコリド)、50:50のラクチド:グリコリド比、47kD 分子量;IV=0.38(dL/g)、
ポリマー3.5A:ポリ(ラクチド−コ−グリコリド)、50:50のラクチド:グリコリド比、分子量 決定されない.;IV=0.42(dL/g)、
ポリマー4A:ポリ(ラクチド−コ−グリコリド)、50:50のラクチド:グリコリド比、45-64kD 分子量;IV=0.45-0.47(dL/g)、
ポリマー4A-1:ポリ(ラクチド−コ−グリコリド)、65:35のラクチド:グリコリド比、53kD 分子量;IV=0.43(dL/g)、
【0069】
本発明の変更および変形は、本発明の前述の詳細な説明から、当業者に明らかである。このような変更および変形は、請求の範囲内に包含されることを意図する。
【0070】
本明細書で引用された特許、特許出願出版物、および記事は、参照として全体に援用される。
【図面の簡単な説明】
【0071】
【図1】図1は、ハロペリドールパモ酸塩の2つの塩形態を示す。
【図2】図2は、pHの関数としての薬物のイオン化プロフィールを示す。9より大きいpH値では、薬物は主に非イオン化状態であるが、pKa未満のpH値では、薬物は正に帯電する。
【図3】図3は、酒石酸塩およびパモ酸塩のイオン平衡を示す。モノアニオンを実線で示し、ジアニオンを点線で示す。
【図4】図4は、酒石酸塩およびパモ酸塩のイオン平衡を示す。モノアニオンを実線で示し、ジアニオンを点線で示す。
【発明の詳細な説明】
【0001】
発明の背景
種々の抗精神病薬のポリマー系徐放性デバイスが報告されている。しかしながら、かかる徐放性デバイスは、製造するのに費用がかかり、作製するのが困難な傾向にある。したがって、活性剤の薬理学的プロフィールを最大にし、より費用効率よく作製し得るような薬剤を送達するための改善された方法の必要性がなお存在する。
【0002】
発明の要旨
本発明は、ハロペリドール、オランザピンおよびアリピプラゾールのパモ酸塩が、良好ないし優れた持続性および/または長時間放出性プロフィールをもたらすという知見に関する。したがって、本発明の一局面において、本発明は、ハロペリドール、オランザピンまたはアリピプラゾールのパモ酸塩を含む。好ましくは、パモ酸塩は、薬物、例えばハロペリドールのパモ酸塩に対する比が1:1または2:1であることを特徴とする。パモ酸塩は、針状物または緻密な(dense)結晶(例えば、図面に示したもの)などの結晶性であり得る。
【0003】
本発明は、さらに、ハロペリドールおよびアリピプラゾールのパモ酸塩を含有する医薬組成物を投与することを含む、処置を必要とする個体の処置方法に関する。
【0004】
発明の詳細な説明
本発明は、ハロペリドール、オランザピンおよびアリピプラゾールのパモ酸塩が、良好ないし優れた持続性および/または長時間放出性プロフィールをもたらすという知見に関する。したがって、本発明の一局面において、本発明は、ハロペリドール、オランザピンまたはアリピプラゾールのパモ酸塩を含む。好ましくは、パモ酸塩は、薬物、例えばハロペリドールのパモ酸塩に対する比が1:1または2:1であることを特徴とする。パモ酸塩は、針状物または緻密な結晶(例えば、図面に示したもの)などの結晶性であり得る。
【0005】
本発明は、さらに、ハロペリドール、オランザピンおよびアリピプラゾールのパモ酸塩を含有する医薬組成物を投与することを含む、処置を必要とする個体の処置方法に関する。
【0006】
あるいはまた、本発明のパモ酸塩を製造するために、米国特許第5,006,528号または同第4,734,416号(参照により本明細書に援用される)に記載されたカルボスチリル化合物などの類似の活性剤を使用することができる。
【0007】
有機塩基の電離平衡を以下に示す。
【0008】
【化1】
【0009】
示した化学プロセスは、酸解離定数(Ka)、遊離塩基の溶解度および塩の溶解度積(Ksp)から構成される。アルカリ性条件下では、遊離塩基形態は、安定な形態である。溶液のpHが低下するにしたがって、イオン化した薬物の割合および水溶性が増大する。イオン化した薬物(プロトン化アミン)が高濃度のとき、塩の溶解度積(Ksp)が超過し、その塩形態が析出する。薬物および対イオンの性質がその塩のKspおよび会合した固体状態の特性を決定する。
【0010】
塩基性薬物であるハロペリドールの化学構造および電離平衡は、
【化2】
である。
【0011】
図2は、pHの関数としての薬物のイオン化プロフィールを示す。9より大きいpH値では、薬物は、主に非イオン化状態であるが、pKa未満のpH値では、薬物は正に帯電する。適切な対イオンと結合したプロトン化形態の薬物は、イオン塩の形成を可能にする。
【0012】
塩基の塩を調製するのに使用されている対イオンはさまざまなものがある。塩基性薬物の塩を形成するのに最も頻繁に使用されるアニオンは、塩酸塩の形態である。医薬用塩に使用されるカルボン酸対イオンの2つの例は、
【化3】
である。
【0013】
イオン平衡を図3および4に示す。2つの対イオンは、酒石酸塩およびパモ酸塩に溶解特性において広い範囲をもたらす。これらの対イオンの各々は2価であることに注目されたい。したがって、1:1および2:1(プロトン化塩基:対イオン)塩が可能である。塩が形成される溶液状態により、どちらの塩形態(1:1または2:1)が析出するかが決まる。化学構造は、これらの2つの対イオンが非常に異なる構造および特性を有することを示す。酒石酸は、水溶性の高い塩をもたらすが、パモ酸は、溶解性が制限された塩を形成し、したがって、溶解を遅延させるために使用され得る。水溶性形態および水不溶性形態の両方が、薬物送達製剤の開発における使用目的を有する。
【0014】
特許請求の範囲に記載の発明の組成物は、投与目的のための種々の医薬形態を含み得る。本発明の医薬組成物を調製するため、活性成分としての主題の化合物の有効量を、薬学的に許容され得る担体(該担体は、投与に所望される調製物の形態に応じて多種多様な形態をとり得る)と充分に混合された状態で合わせる。これらの医薬組成物は、望ましくは、好ましくは投与に適した単一の投薬形態である。投与としては、経口を含む経腸、または肺系、経皮、皮下もしくは筋肉内注射もしくは埋込みを含む非経口が挙げられる。後者の投与経路では、主題の化合物は、好ましくは水性溶媒中に懸濁され、ソルビタンエステルのポリオキシエチレン誘導体、例えば、ポリソルベート80 (Tween 80 (登録商標))およびポリソルベート20 (Tween 20 (登録商標))、レシチン、ポリオキシエチレンエーテルおよびポリオキシプロピレンエーテル、デオキシコール酸ナトリウムなどの湿潤剤;セルロース誘導体、例えばメチルセルロース、ナトリウムカルボキシメチルセルロースおよびヒドロキシプロピルメチルセルロース、ポリビニルピロリドン、アルギン酸塩、キトサン、デキストラン、ゼラチン、ポリエチレングリコール、ポリオキシエチレンエーテルおよびポリオキシプロピレンエーテルなどの懸濁剤;例えば塩酸などの酸;例えば水酸化ナトリウムなどの塩基;適量の酸(リン酸、コハク酸、酒石酸、乳酸、酢酸、マレイン酸またはクエン酸など)および塩基、特に水酸化ナトリウムまたはリン酸一水素二ナトリウムの混合物を含む緩衝溶液;保存剤、例えば、安息香酸、ベンジルアルコール、ブチル化ヒドロキシアニソール、ブチル化ヒドロキシトルエン、クロルブトール、没食子酸塩、ヒドロキシ安息香酸塩、EDTA、フェノール、クロロクレゾール、メタクレゾール、塩化ベンゾトニウム、ミリスティールγ塩化ピコリニウム、酢酸フェニル水銀、チメロサールなど;張性調整剤、例えば、塩化ナトリウム、デキストロース、マンニトール、ソルビトール、ラクトース、硫酸ナトリウムなどをさらに含み得る。あるいはまた、主題の化合物は、油中に製剤化され得る。この目的に適切な油類は、不揮発油、例えば、ピーナツ油、ゴマ油、綿実油、コーン油、サフラワー油、ヒマシ油、エチルオレエート、大豆油、長鎖脂肪酸または中鎖脂肪酸の合成グリセロールエステルおよびこれらの混合物ならびに他の油類である。また、増粘剤、例えば、例えば、モノステアリン酸アルミニウム、エチルセルロース、トリグリセリド、硬化ヒマシ油などを組成物に添加してもよい。
【0015】
精神病性障害の処置における主題の化合物の有用性を鑑みると、本発明が、精神病性障害を患う温血動物、特にヒトの処置方法を提供することは明白であり、該方法は、薬学的有効量の主題の化合物を医薬用担体と混合して投与することを含む。さらなる局面において、本発明は、主題の化合物の薬としての、特に抗精神病薬としての使用に関する。一般に、有効量は、0.05 mg/kg〜50 mg/kg体重、より好ましくは0.5 mg/kg〜10 mg/kg体重であり得ることが想定される。
【0016】
活性剤は、好ましくは、持続性製剤にて投与される。一態様において、活性剤は製剤から少なくとも約24時間、好ましくは約48時間にわたって放出される。
【0017】
また、活性剤は長時間放出製剤にて投与され得る。一態様において、長時間放出製剤は、活性剤を、少なくとも約7日間、好ましくは少なくとも約14日間、あるいはまた、少なくとも2、3、4、6または8週間にわたって放出する。組成物は、好ましくは、筋肉内または皮下などの注射により投与される。
【0018】
一態様において、製剤は、単一または単独投薬量として投与され得る。しかしながら、本発明は、数週間もしくは数ヶ月またはそれより長くにわたって反復投薬を受けている個体などの、不断または長期治療を必要とする個体に特に有用である。かかる投薬計画において、該方法は、第1長時間放出製剤の第1投薬および第2長時間放出製剤の第2投薬を含み得る。第2製剤は、第1のものと同一であってもよく、実質的同一であってもよく、異なっていてもよく、同じ活性剤または異なる活性剤を含み得る。例えば、第2製剤は、第1投薬後、約7日またはそれ以降(例えば、少なくとも約14日または少なくとも約17日)で投与され得、このとき、第1投薬は、1、2、3、4、5、6、7、8、9、10、11、12、13、14日間またはそれより長い期間の薬剤の放出をもたらす。
【0019】
用語「治療有効量」は、任意のパラメータまたは臨床症状の改善をもたらす量を定義することがさらに意図される。実際の投薬量は、各患者により変わり得、必ずしも、すべての疾患症状の消失を意味するものではない。
【0020】
本明細書で使用する場合、用語「個体」、「被験体」または「患者」は、限定されないが、特定の疾患状態に苦しむ、哺乳動物などの温血動物(ヒトを含むが、これに限定されない)をいう。
【0021】
本明細書に記載する処置において使用される化合物の治療有効量は、慣用技術の使用により、および同様の状況下で得られる結果を観察することにより、当業者としての担当診断医によって容易に決定され得る。治療有効投薬量を決定する際には、限定されないが、哺乳動物の種;その体格、年齢および一般健康状態;関与している特定の疾患;疾患の程度または困難さ(involvement)もしくは重篤度;個々の患者の応答;投与する具体的な化合物;投与経路;投与する調製物に特有の生物学的利用能;選択した投与計画;併用薬の使用;および他の関連する状況を含むいくつかの因子が担当診断医によって考慮される。
【0022】
投与の好ましい量および形態は、当業者が決定することができる。製剤調製の当業者は、例えばRemington's Pharmaceutical Sciences, 最新版, Mack Publishing Co.に記載された当該技術分野で公知の製剤技術を用い、選択した化合物の具体的な特徴、処置対象の疾患状態、疾患の病期および他の関連する状況に応じて、投与の適切な形態および様式を容易に選択し得る。
【0023】
医薬組成物は、当該技術分野で公知の技術を用いて製造することができる。典型的には、治療有効量の化合物を、薬学的に許容され得る担体と混合する。
【0024】
本発明の組成物は、注射などにより非経口投与され得る。好ましい投与の方法としては、例えば、筋肉内注射および皮下注射が挙げられる。
【0025】
非経口投与では、該化合物を、生理学的に許容され得る医薬用担体に溶解し、溶液または懸濁液として投与し得る。例えば20℃で少なくとも20 cpの粘度を有する粘性の注射可能な担体が好ましい。他の態様において、懸濁液の液相は、20℃で少なくとも約30 cp、40 cp、50 cpおよび60 cpの粘度を有する。組成物はまた、粘度向上剤、密度向上剤、張性向上剤および/または湿潤剤を含有し得る。好適な医薬用担体の具体例としては、水、生理食塩水、デキストロース溶液、フルクトース溶液、エタノール、または動物油、植物油もしくは合成による油が挙げられる。医薬用担体はまた、当該技術分野で公知の保存剤および緩衝溶液を含んでいてもよい。
【0026】
別の態様において、製剤は、外科的に埋め込み得る。かかる製剤としては、ポリラクチドおよびポリラクチド-コ-グリコリドおよびコラーゲン製剤などの任意の周知の生分解性および生腐食性(bioerodible)担体が挙げられる。かかる物質は、固形の埋没物、スポンジなどの形態であり得る。いずれにせよ、該物質の局所使用では、活性成分は、通常、担体または賦形剤中に約1:1000〜1:20,000の重量比で存在するが、この範囲の比に限定されない。
【0027】
好ましくは、該化合物は長時間放出製剤中に存在する。長時間(徐放性または制御性ともいう)放出調製物は、本明細書に記載する活性剤を取込むか、または封入するポリマー(好ましくはポリラクチドまたはポリラクチド-コ-グリコリドポリマー)の使用により得られ得る。長時間放出製剤は、ポリマー-薬物混合物の噴霧乾燥、エマルジョンを用いる(-based)技術、コアセルベーションを用いる技術、フィルムキャスティング、押出し成形を用いる技術、および長時間放出プロフィールを有するポリマー-薬物微粒子を製造するための他のプロセスにより作製し得る。本発明の薬剤を含めるために使用され得る好適な長時間放出技術としては、限定されないが、例えばWrightに対する米国特許第6,264,987号、同第5,654,008号および/または同第5,792,477号などに記載されたMEDISORB(登録商標)技術;例えばHerbertに対する米国特許第6,358,443号に記載されたPROLEASE(登録商標)技術;Southern Research Instituteにより、例えば米国特許第6,306,425号に記載された技術;および「徐放性微粒子の調製方法(Method of Preparing Sustained Release Microparticles)」という米国特許出願第60/441,946号(2003年1月23日出願)、ならびにALZAMER(登録商標) Depot注射技術を含むAlza Corp.により記載された技術が挙げられる。これらの特許の内容は、参照によりその全体が本明細書に援用される。
【0028】
好ましい一態様において、薬剤は、長時間放出デバイスまたは製剤中に、該デバイスまたは製剤の全重量の少なくとも約5重量%、好ましくは少なくとも約10重量%、より好ましくは少なくとも約30重量%の量で存在する。
【0029】
例えば、コアセルベーション技術により、または界面重合により調製される微粒子(例えば、それぞれ、ヒドロキシメチルセルロースまたはゼラチン-マイクロカプセルおよびポリ-(メチルメタクリレート)マイクロカプセル)、コロイド状薬物送達システム(例えば、リポソーム、アルブミン、微粒子、マイクロエマルジョン、ナノ粒子およびナノカプセル)またはマクロエマルジョン内に活性剤を取り込むこともまた想定される。
【0030】
組成物が注射可能な物質(無針(needle-less)注射を含むが、これに限定されない)として使用される場合、従来の注射可能な担体内に製剤化し得る。好適な担体としては、生体適合性溶液および薬学的に許容され得る溶液が挙げられる。
【0031】
以下の実施例は、例示を目的とするものであって、本発明の範囲を限定するものではない。
【実施例】
【0032】
実施例1:
ハロペリドールのパモ酸塩は、溶媒中でのパモ酸またはパモ酸塩によるハロペリドールの処理により調製することができる。ハロペリドールパモ酸塩は、適切な溶媒、例えばエタノール中にハロペリドールを酢酸とともに含む溶液を、パモ酸二ナトリウム、パモ酸または他のパモ酸塩の溶液に添加し、沈殿するまで1〜3日以上、静置することにより調製することができる。あるいはまた、塩を沈殿させるために、溶液の蒸発、低速もしくは高速冷却または攪拌などの他の方法を使用することもできる。
【0033】
具体的には、酸性化エタノール(5%酢酸)中0.1Mハロペリドール溶液2.5 mlを、エタノール/水(50/50)中パモ酸二ナトリウム塩(2.5 ml)0.1M溶液2.5 mlに添加した。混合物を、室温で1〜3日間、放置した。得られた沈殿物を吸引により濾別し、エタノールで洗浄し、60℃の真空炉内で乾燥し、1:1ハロペリドールパモ酸塩240mgを得た。
【0034】
実施例2:
酸性化エタノール(5%酢酸)中0.25Mハロペリドール溶液2.5 mlを、エタノール/水(75/25)中0.05Mパモ酸二ナトリウム塩溶液12.5 mlに添加した。混合物を、室温で1〜3日間、放置した。得られた沈殿物を吸引により濾別し、エタノールで洗浄し、60℃の真空炉内で乾燥し、2:1ハロペリドールパモ酸塩206mgを得た。
【0035】
実施例3:
酸性化エタノール(5%酢酸)中0.25Mハロペリドール溶液2.5 mlを、エタノール/水(50/50)中0.1Mパモ酸二ナトリウム塩溶液6.25 mlに添加した。混合物を、室温で1〜3日間、放置した。得られた沈殿物を吸引により濾別し、エタノールで洗浄し、60℃の真空炉内で乾燥し、2:1ハロペリドールパモ酸塩264mgを得た。
【0036】
実施例4:
酸性化エタノール(5%酢酸)中0.05Mハロぺリドール溶液5mlを、エタノール/水(50/50)中0.25Mパモ酸二ナトリウム塩溶液1mlに添加した。混合物を、室温で1〜3日間放置した。生じた沈殿物を吸引で濾過し、エタノールで洗浄し60℃の真空炉内で乾燥して、1:1パモ酸ハロぺリドール塩107mgを得た。
【0037】
実施例5:
酸性化エタノール(5%酢酸)中0.05Mハロぺリドール溶液5mlを、エタノール/水(50/50)中0.1Mパモ酸二ナトリウム塩溶液2.5mlに添加した。混合物を、室温で1〜3日間放置した。生じた沈殿物を吸引で濾過し、エタノールで洗浄し60℃の真空炉内で乾燥して、1:1パモ酸ハロぺリドール塩119mgを得た。
【0038】
実施例6:
酸性化エタノール中アリピプラゾール溶液(0.05〜0.5M)を、水/エタノール(100/0〜0/100)の混合物中パモ酸二ナトリウム塩溶液(0.05〜0.5M)に添加した。混合物を、室温で1〜3日間放置した。生じた沈殿物を吸引で濾過し、溶媒で洗浄し60℃の真空炉内で乾燥した。
【0039】
実施例7:
ハロペリドールおよびオランザピン調剤の単回皮下用量の投与に続いてのラット中のハロペリドールおよびオランザピンの薬物動態の評価
【0040】
種および系統: Sprague-Dawley ラット雄;450+/−50グラム
【0041】
試験群:4群、12被験体
A群:20mgのハロペリドールバルク(bulk)薬物を1回SC注
射した3匹のラット
B群:40mgのパモ酸ハロペリドールを1回SC注射した3匹の
ラット
C群:20mgのオランザピンバルク薬物を1回SC注射した3匹
のラット
D群:50mgのオランザピンバルク薬物を1回SC注射した3匹
のラット
【0042】
注射の経路: 肩甲間部への皮下(SC)注射
【0043】
注射賦形剤: 水性希釈剤:水、3%低粘度CMC、0.1%Tween 20、0.9%NaCl中
【0044】
投薬容量: 投薬量懸濁液は以下のようにして製剤化した。
A群:0.75mL希釈剤中の20mg粉末
B群:0.75mL希釈剤中の40mg微粒子
C群:0.75mL希釈剤中の20mg粉末
D群:0.75mL希釈剤中の50mg粉末
【0045】
血液採取:
ハロタンによる麻酔の後に血液試料を尾静脈より採取した。抗凝固剤を含まないシリンジを、血液採取に使用し、次いで全血をK2 EDTAおよび混合ビーズ(Microtainer(登録商標);MFG♯ BD365974)を含むチューブに移した。血液試料を処理して(チューブを15〜20回上下にして、>14000gで2分間遠心分離する)血漿を分離した。この方法で準備した血漿試料を標識されたプレインチューブ(Microtainer(登録商標);MFG♯ BD5962)に移し、<−70℃で凍らせて保存した。
【0046】
血液容量: 初めの24時間のそれぞれの時間点では少なくとも血液250μLであり、その後のそれぞれの時間点では400μL
【0047】
血漿を得るための時間点:
2h 24h 3d 10d*
4h 32h 4d 14d*
8h 2d 7d
*注:B群だけは7日目以降の時間点を有した。また、いずれの群についても、血漿濃度が定量限界より低くなった時、その群は終了とした。
【0048】
観察された結果は、パモ酸塩が良から優の長時間放出プロフィールを表したことを示す。
【0049】
実施例8:
ポリマーおよびパモ酸塩を含む注射可能な微粒子は、効率が良く簡易な1つの溶媒処理を使用して調製され得る。PLGポリマーおよび塩は1つの溶媒中で共溶解され得、(2)溶媒を真空乾燥または昇華によって取り除き、ポリマー/薬物マトリックスを形成し、(3)該マトリックスを粉砕し、粉末を作製し、(4)該生じた粉末を圧縮し、圧縮されたマトリックスを形成し、(5)該圧縮されたマトリックスを粉砕すると、緻密で注射可能な微粒子製剤が形成され得る。好ましくは、パモ酸塩充填は、微粒子組成物の最終重量の約10%または約30%以上(w/w)であり得る。溶媒、例えば塩化メチレン、アセトン、ジメチルスルホキシド(DMSO)、アセトニトリル、および酢酸エチルは使用に好適である。
【0050】
適切なポリマーとしては、
ラクチド:グリコリド比;固有粘度;末端基;溶媒
A30%(w/w)50:50、0.75dL/g;酸末端基;CH2Cl2
B30%(w/w)75:25;0.60dL/g;ラウリルエステル末端基;CH2Cl2
C25 30%(w/w)50:50;0.61dL/g;ラウリルエステル末端基;CH2Cl2
が挙げられる。
【0051】
パモ酸塩/ポリマー/溶媒混合物は、ポリテトラフルオロエチレン平面型(およそ1インチ×1インチ×1/2インチ奥行き)または3インチ直径ジャーのいずれか中に注ぎ込まれ得、例えばフィルムを形成する。該フィルムはFTS Dura-Dry Lyophilizer (Kinetic Systems, Inc., Santa Clara, CA)または真空炉中のいずれかで乾燥され得る。最大真空、雰囲気圧力、高温、周囲温度、および乾燥時間の変化を含む様々な環境下で乾燥されたフィルムが作られ得る。
【0052】
該フィルムは、14,000rpmで操作する24枚歯の Retsch Ultra Centrifugal Mill (Retsch, Inc., Newtown, PA)を使用して粉砕され得る。粉砕する前に、採集皿を、液体窒素で満たす。採集皿から集められた、生じた粉末は、続く圧縮工程を助ける流動性生成物である。フィルムを粉砕して作製された粉末の一部は、この時点で分析するために、保持され得る。これらの粉末は、以下に記載されるフィルム粉末の続く圧縮および再粉砕によって作られた粉末との比較のために保持され得る。
【0053】
粉砕された粉末の一部は、Carver Model C Press (Carver, Inc., Wabash, IN)および約1/4インチまたは約1/2インチ円筒ダイのいずれかを使用して、圧縮され得る。約50〜約300ミリグラムの粉砕された粉末を、ダイ中に充填し、室温で約30秒間、約5000ポンドのマシンセッティングで粉砕し、ペレットを形成する。
【0054】
圧縮されたマトリックスを、14000rpmで操作する24枚歯の Retsch Ultra Centrifugal Mill(Retsch, Inc., Newtown, PA)を使用して引き続き粉砕する。粉砕する前に、採集皿を液体窒素で満たす。最終粉末を採集皿から集め、分析のためにバイアル中に入れた。
【0055】
本明細書に記載された、持続放出組成物は、エマルジョン、コアセルベーション、および低温マイクロカプセル化技術のいずれかによって調製され得る。それぞれの技術に関連する一般的な処理を、以下に記載する。
【0056】
コアセルベーション-S/O/O処理
コアセルベーション処理は、本明細書中では溶媒-油-油(S/O/O)処理ともいうが、薬物と有機ポリマー溶液を用いた油中溶媒型エマルジョンの形成を要する。次いで、相分離を誘導させるためおよびポリマーを沈殿させるために、油(典型的にシリコーン油)を油中水型エマルジョンに加える。次いで、初期微粒子を、油とポリマー溶媒を取り除く溶媒中で失活させる。パモ酸塩を溶媒-油-油(S/O/O)エマルジョン系を使用したPLGポリマー中にカプセル化する。初期初期微粒子を、S/O/O中で内部エマルジョン工程において形成して、その後コアセルベーション化と硬化工程に供する。微粒子を集めて乾燥し、バイアルに充填する。さらに、完全処理のそれぞれの工程のさらなる詳細は、以下に記す。
【0057】
内部エマルジョン形成
油中溶媒型エマルジョンは超音波処理を使用して作製する。エマルジョンの溶媒相は溶解した薬物および様々な賦形剤を水中に含んだ。PLG相は、塩化メチレンに溶解したポリマーを含んだ。
【0058】
コアセルベーション形成
コアセルベーションは、攪拌しながらシリコーン油を制御された割合で内部エマルジョンに加えることによって誘導され得、初期微粒子を形成する。形成された初期微粒子は比較的軟らかく、硬化を要する。
【0059】
微粒子の硬化
初期微粒子を穏やかに攪拌しながら、ヘプタン/エタノール混合溶媒に加える。混合溶媒は初期微粒子を硬化する。約3℃で約1時間硬化させた後、溶媒混合物をデカントし、純ヘプタンを3℃で加え、約1時間混ぜた。
【0060】
微粒子乾燥と収集
硬化段階の後、微粒子を乾燥チャンバの中の微細メッシュ細孔板に移して集める。硬化用の容器の、最終ヘプタン洗浄が行われる。温度を約3℃から約38℃に上げていきつつ、4日間、微粒子を窒素ガスにより乾燥させる。
【0061】
一般的に、PLGを塩化メチレンに溶解する。溶媒中に薬物およびいくらかの賦形剤を溶解させて内部相を調製する。次いで、プローブ音波処理をしながら、水溶液をポリマー溶液中に注入する。次いで、得られたエマルジョンをエマルジョン反応器に加える。シリコーン油(350センチストークス)を約1000rpmで攪拌させながら、ぜん動ポンプによってゆっくりと反応器に加える。次いで、混合物をn-ヘプタンに加える。約2時間攪拌させた後、微粒子を濾過により単離し、一晩中真空乾燥する。
【0062】
エマルジョン処理-S/O/W処理
エマルジョン処理は、溶媒-油-水(S/O/W)処理とも言う。簡単には、薬物の溶液はポリマー溶液中に分散され、外側の水相(例えば、PVA)中に乳化される。次いで、微粒子を水性クエンチ(quench)中で硬化する。
【0063】
典型的な実験では、PLG(1.96g)を塩化メチレン(22.5g)中に溶解し、薬物を溶解する(1.75g溶媒中に20mgの薬物)。次いで、薬物溶液をシリンジ内に吸引し、プローブ音波処理をしながらポリマー溶液中に注入する。次いで、得られたエマルジョンを、5%ポリビニルアルコール(PVA)水溶液125gを含んだエマルジョン反応器中に素早く加える。反応器中の攪拌速度を約800RPMに設定した。混合物を約1.5分間攪拌し、水クエンチ(10℃で2.8L)中に加える。クエンチ中で約2時間後、硬化した微粒子を濾過により単離し、一晩中真空乾燥する。
【0064】
低温処理
低温処理では、噴霧化を用いて薬物を含んだポリマー溶液の小滴を形成した。次いで、初期微粒子を液体窒素中で凍結して、該ポリマー溶媒を、次のエタノール抽出技術によって取り除く。
【0065】
微粒子を作製するための低温処理は、(1)乾燥凍結物または乾燥薬物物質の作製、および(2)低温、無水技術を使用したマイクロカプセル化の2つの工程を含む。乾燥凍結物は、2−流動ノズルを用いた薬物および賦形剤の混合物の噴霧化、噴霧化された小滴の凍結、並びに乾燥凍結を用いた凍結小滴の乾燥によって調剤する。当該技術分野で公知のいずれの好適な乾燥する方法が使用され得ることを、理解されるべきである。特に、凍結小滴を、−26℃の棚(shelf)および96mTorrチャンバ圧力の第一乾燥条件で約7日間、続いて約20℃および0mTorrでの更なる3日間の二次乾燥によって、乾燥する。
【0066】
パモ酸塩を含んだ微粒子は、すべて10%薬物以上の名目上の標的負荷で、低温、無水処理で生産され得る。薬物を、塩化メチレン中に溶解した4A PLG3〜20%により成り立つ有機溶媒中で懸濁する。この懸濁液を氷上で約4分間音波処理し、次いで該懸濁液を、超音波ノズルを使用して噴霧化し、凍結エタノールの吸着床上に層をなす液体窒素に接触させることによって凍結する。該試料を、微粒子の硬化および溶媒の抽出を可能にするために、−80℃まで温める。次いで、該微粒子を濾過し乾燥する。
【0067】
固体/油/水(S/O/W)および固体/油/油(S/O/O)処理
固体薬物をまた、上記エマルジョンおよびコアセルベーション処理の修正されたバージョンを使用してカプセル化し得る。これらの修正された処理は、固体/油/水(S/O/W)および固体/油/油(S/O/O)を言う。例えば、固体薬物を、3〜20%PLGを含む塩化メチレン中で懸濁し、氷上で約4分間音波処理する。次の処理はW/O/OまたはW/O/Wの方法と類似した方法によって行われる。
【0068】
ポリマー:
使用するのに好適な、特定のPLGポリマーの例を以下に列挙する。以下の例で使用される全てのポリマーは、リスト中の前方に配置し、全ての列挙されたポリマーは、Cincinnati, OHのAlkermes, Inc.から購入され、以下のように表示され得る。
ポリマー2A:ポリ(ラクチド−コ−グリコリド)、50:50のラクチド:グリコリド比、12.3kD 分子量;IV=0.15(dL/g)、
ポリマー2A−1:ポリ(ラクチド−コ−グリコリド)、65:35のラクチド:グリコリド比、16kD 分子量;IV=0.19(dL/g)、
ポリマー2.5A:ポリ(ラクチド−コ−グリコリド)、50:50のラクチド:グリコリド比、25kD 分子量;IV=0.24(dL/g)、
ポリマー3A:ポリ(ラクチド−コ−グリコリド)、50:50のラクチド:グリコリド比、47kD 分子量;IV=0.38(dL/g)、
ポリマー3.5A:ポリ(ラクチド−コ−グリコリド)、50:50のラクチド:グリコリド比、分子量 決定されない.;IV=0.42(dL/g)、
ポリマー4A:ポリ(ラクチド−コ−グリコリド)、50:50のラクチド:グリコリド比、45-64kD 分子量;IV=0.45-0.47(dL/g)、
ポリマー4A-1:ポリ(ラクチド−コ−グリコリド)、65:35のラクチド:グリコリド比、53kD 分子量;IV=0.43(dL/g)、
【0069】
本発明の変更および変形は、本発明の前述の詳細な説明から、当業者に明らかである。このような変更および変形は、請求の範囲内に包含されることを意図する。
【0070】
本明細書で引用された特許、特許出願出版物、および記事は、参照として全体に援用される。
【図面の簡単な説明】
【0071】
【図1】図1は、ハロペリドールパモ酸塩の2つの塩形態を示す。
【図2】図2は、pHの関数としての薬物のイオン化プロフィールを示す。9より大きいpH値では、薬物は主に非イオン化状態であるが、pKa未満のpH値では、薬物は正に帯電する。
【図3】図3は、酒石酸塩およびパモ酸塩のイオン平衡を示す。モノアニオンを実線で示し、ジアニオンを点線で示す。
【図4】図4は、酒石酸塩およびパモ酸塩のイオン平衡を示す。モノアニオンを実線で示し、ジアニオンを点線で示す。
【特許請求の範囲】
【請求項1】
ハロペリドールのパモ酸塩。
【請求項2】
ハロペリドールのパモ酸塩に対する比が1:1または2:1である、請求項1記載のパモ酸塩。
【請求項3】
該塩が針状物である、請求項1記載のパモ酸塩。
【請求項4】
該塩が結晶性である、請求項1記載のパモ酸塩。
【請求項5】
アリピプラゾールのパモ酸塩。
【請求項6】
アリピプラゾールのパモ酸塩に対する比が1:1または2:1である、請求項4記載のパモ酸塩。
【請求項7】
該塩が結晶性である、請求項5記載のパモ酸塩。
【請求項8】
ハロペリドールおよびアリピプラゾールからなる群より選択される活性剤のパモ酸塩を含有してなる医薬組成物を投与することを含む、処置を必要とする個体の処置方法。
【請求項9】
組成物が注射により投与される、請求項8記載の方法。
【請求項10】
組成物が筋肉内または皮下に投与される、請求項8記載の方法。
【請求項11】
組成物が、有効量の活性剤を少なくとも約24時間にわたって放出する、請求項8記載の方法。
【請求項12】
組成物が、有効量の活性剤を少なくとも約48時間にわたって放出する、請求項8記載の方法。
【請求項13】
該組成物が前記パモ酸塩および粘性の水性担体から本質的になる、請求項8記載の方法。
【請求項14】
該組成物が生分解性ポリマーおよび前記パモ酸塩を含有してなる長時間放出製剤である、請求項8記載の方法。
【請求項15】
長時間放出製剤がポリラクチドおよび前記パモ酸塩を含有してなる、請求項14記載の方法。
【請求項16】
長時間放出製剤がポリラクチド-コ-グリコリドおよび前記パモ酸塩を含有する、請求項14記載の方法。
【請求項1】
ハロペリドールのパモ酸塩。
【請求項2】
ハロペリドールのパモ酸塩に対する比が1:1または2:1である、請求項1記載のパモ酸塩。
【請求項3】
該塩が針状物である、請求項1記載のパモ酸塩。
【請求項4】
該塩が結晶性である、請求項1記載のパモ酸塩。
【請求項5】
アリピプラゾールのパモ酸塩。
【請求項6】
アリピプラゾールのパモ酸塩に対する比が1:1または2:1である、請求項4記載のパモ酸塩。
【請求項7】
該塩が結晶性である、請求項5記載のパモ酸塩。
【請求項8】
ハロペリドールおよびアリピプラゾールからなる群より選択される活性剤のパモ酸塩を含有してなる医薬組成物を投与することを含む、処置を必要とする個体の処置方法。
【請求項9】
組成物が注射により投与される、請求項8記載の方法。
【請求項10】
組成物が筋肉内または皮下に投与される、請求項8記載の方法。
【請求項11】
組成物が、有効量の活性剤を少なくとも約24時間にわたって放出する、請求項8記載の方法。
【請求項12】
組成物が、有効量の活性剤を少なくとも約48時間にわたって放出する、請求項8記載の方法。
【請求項13】
該組成物が前記パモ酸塩および粘性の水性担体から本質的になる、請求項8記載の方法。
【請求項14】
該組成物が生分解性ポリマーおよび前記パモ酸塩を含有してなる長時間放出製剤である、請求項8記載の方法。
【請求項15】
長時間放出製剤がポリラクチドおよび前記パモ酸塩を含有してなる、請求項14記載の方法。
【請求項16】
長時間放出製剤がポリラクチド-コ-グリコリドおよび前記パモ酸塩を含有する、請求項14記載の方法。
【図1】


【図2】
【図3】
【図4】
【図2】
【図3】
【図4】
【公表番号】特表2007−501235(P2007−501235A)
【公表日】平成19年1月25日(2007.1.25)
【国際特許分類】
【出願番号】特願2006−522613(P2006−522613)
【出願日】平成16年7月29日(2004.7.29)
【国際出願番号】PCT/US2004/024344
【国際公開番号】WO2005/016261
【国際公開日】平成17年2月24日(2005.2.24)
【出願人】(505444972)アルカーメス コントロールド セラピューティクス,ツー (4)
【Fターム(参考)】
【公表日】平成19年1月25日(2007.1.25)
【国際特許分類】
【出願日】平成16年7月29日(2004.7.29)
【国際出願番号】PCT/US2004/024344
【国際公開番号】WO2005/016261
【国際公開日】平成17年2月24日(2005.2.24)
【出願人】(505444972)アルカーメス コントロールド セラピューティクス,ツー (4)
【Fターム(参考)】
[ Back to top ]