
アルキル化半合成グリコサミノグリカンエーテルならびにその製造および使用方法
本明細書においては、アルキル化およびフルオロアルキル化半合成グリコサミノグリカンエーテル(本明細書においては「SAGE」と称する)の合成について記載をする。硫酸化アルキル化およびフルオロアルキル化SAGEの合成についても記載する。本明細書に記載する化合物は、創傷治癒、薬物送達、ならびに多くの炎症性疾患および皮膚障害の治療を含む多くの用途に有用である。
【発明の詳細な説明】
【技術分野】
【0001】
関連出願の相互参照
本出願は、2008年4月4日に出願された米国特許仮出願第61/042,310号に基づく優先権を主張するものである。この出願の教示全体が本明細書に参考として組み込まれる。
【背景技術】
【0002】
背景
乾癬、皮膚炎、ざ瘡、酒さ、皮膚の光老化等の炎症性疾患およびRAGEが媒介するシグナル伝達が関与する数々の疾患によって世界中の人々が苦しめられている。これらの疾患を大局的に見ると、乾癬に悩んでいる人だけでも世界人口の2〜3%すなわち約1億2500万人に上ることがNational Psoriasis Foundationによって報告されている。これらの炎症状態は見た目が悪くなる場合もあるし、治療せずに放置すると深刻な健康上の問題が生じる可能性もある。
【0003】
従来から承認されているこれらの状態の治療は、UV光線療法、コルチコステロイドおよびグルココルチコイド、アシトレチン、シクロスポリン、ならびにメトトレキサートが関与するものであろう。しかしながら、これらの治療はそれぞれ、免疫抑制や肝疾患から、皮膚の菲薄化や先天性欠損症の誘発に至るまで、深刻な副作用を引き起こす可能性がある。これらの治療はある程度または全く無効であるため、その結果に関し患者に不満が残ることが多い。
【0004】
上述した治療に加えてヘパリン治療も試験的に検討されている。ヘパリンすなわち硫酸化された多糖類の用途は、従来、抗凝血剤にほぼ限られていたが、その抗炎症性についてはよく知られている。ヘパリンおよびその誘導体はこれらの炎症性疾患の治療にある程度の可能性を示している。特にヘパリンおよびその誘導体は、炎症カスケードにおける少なくとも3つの重要な現象を阻害する。まず第1に、ヘパリンは、白血球インテグリン、P−およびL−セレクチンに結合してこれらを阻害する。第2に、ヘパリンおよびその誘導体は、陽性荷電PMNプロテアーゼ、ヒト白血球エラスターゼ、およびカテプシンGに結合してこれらを阻害することで炎症カスケードを抑制するため、セレクチンを阻害する最初のヘパリン障壁から逃れたPMNがタンパク質を分解することによる組織損傷が低減される。第3に、ヘパリンおよびその誘導体は、終末糖化産物受容体(RAGE)とそのリガンドとの相互作用を強力に阻害する。
【発明の概要】
【発明が解決しようとする課題】
【0005】
ヘパリンおよびその誘導体がこれらの炎症性疾患を治療する可能性が示されているものの、ヘパリンおよびその誘導体を用いた治療には幾つかの主要な欠点がある。まず第1に、ヘパリンおよびその誘導体は豚から取り出したものであり、したがって、ウイルスの異種間移動の問題が起こる。第2に、ヘパリンの抗凝固性により、この化合物で治療された糖尿病患者が出血過多になる危険性がある。第3に、ヘパリンは、ヘパリンと陽性荷電タンパク質である血小板第4因子(PF−4)との複合体に対する抗体を産生する特定の個体に血小板減少症を誘発する可能性があり、その結果として、破局的な血小板凝集および全身性の逆説的な動静脈塞栓が起こる。したがって、炎症性疾患の治療に用いることが可能であると同時に、他の治療に見られる数々の副作用が回避される化合物を開発するという重要な要求が満たされていない。
【課題を解決するための手段】
【0006】
概要
本明細書においては、アルキル化およびフルオロアルキル化された半合成グリコサミノグリカンエーテル(本明細書においては「SAGE」と称する)の合成について記載する。硫酸化されたアルキル化およびフルオロアルキル化SAGEの合成についても記載する。本明細書に記載する化合物は、多くの治療および化粧用途ならびに多くの炎症性疾患および皮膚障害の治療に有用である。本発明の利点については、以下の記載においてある程度説明するので、その記載からある程度明らかになるか、または以下に記載する態様を実施することによって理解されるであろう。以下に記載する利点は、添付の特許請求の範囲に具体的に指摘する構成要素および組合せによって実現および達成されるであろう。前述した一般的な説明および以下の詳細な説明はいずれも例示および説明のみを目的とするものであって限定を意図するものではないことを理解されたい。
【0007】
添付の図面は以下に説明する幾つかの態様を例示するものであり、これらは本明細書に組み込まれ、その一部を構成するものとする。
【図面の簡単な説明】
【0008】
【図1】アルキル化およびフルオロアルキル化ヒアルロナンならびにその硫酸化誘導体を製造するための合成スキームを示すものである。
【図2】幾つかの例示的なSAGEの構造を示すものである。
【図3】部分O−置換およびメチル化されたHA(P−OSMEHAまたはGM−131201)によるP−セレクチンの阻害を示すものである。
【図4】アルキル化およびフルオロアルキル化ヒアルロナン(硫酸化もされている)を含む硫酸化ヒアルロナン誘導体によるヒト白血球エラスターゼの阻害を示すものである。
【図5】ハイモビリティー・グループ・ボックス・プロテイン−1(HMGB−1)としても周知のアンホテリンと固定化RAGEとの結合に対するP−OSMEHAによる阻害を示すものである。
【図6】S100bカルグラニュリンと固定化RAGEとの結合に対するP−OSMEHA(GM−131201)による阻害を示すものである。
【図7】カルボキシメチルリジン−BSA(CML−BSA)と固定化RAGEとの結合に対するP−OSMEHA(GM−131201)による阻害を示すものである。
【図8】高分子量ヒアルロナン誘導体(硫酸化もされているアルキル化およびフルオロアルキル化ヒアルロナンを含む)のケラチン生成細胞増殖に対する阻害を示すものである。
【図9】低分子量ヒアルロナン誘導体(硫酸化もされているアルキル化およびフルオロアルキル化ヒアルロナンを含む)のケラチン生成細胞増殖に対する阻害を示すものである。
【図10】SAGE(GM−111101)をLL−37と同時に注射した酒さモデルを示すものである。(a)LL−37を注射した皮膚部位ならびに(b)LL−37およびSAGEを同時に注射したモデルの肉眼解剖写真である。(c)LL−37を注射した皮膚試料をH&E染色した断面図である。(d)SAGEと混合したLL−37を注射した皮膚部位をH&E染色した断面図である。(e)LL−37のみ、SAGEを加えたLL−37、またはSAGEのみを注射した群のマウスの皮膚生検により、多形核白血球(PMN)の皮膚浸潤を、PMN酵素であるミエロペルオキシダーゼ(MPO)の活性によって測定したものである。LL−37のみまたはSAGEを加えたLL−37を注射したマウスの(f)紅斑面積および(g)紅斑評点である。
【図11】LL−37酒さモデルのSAGE(GM−111101)局所治療を示すものである。(a)LL−37を注射、(b)LL−37注射直後にSAGE治療、および(c)LL−37注射から12時間後にSAGE治療した皮膚部位の肉眼解剖写真を示すものである。(d)LL−37を注射した皮膚試料をH&E染色した断面図である。(e)LL−37を注射した直後にSAGE治療した皮膚部位をH&E染色した断面図である。(f)LL−37を注射した12時間後にSAGE治療した皮膚部位をH&E染色した断面図である。(g)SAGE治療計画の異なるLL−37注射モデルのMPO活性測定である。SAGEの局所塗布によって治療したLL−37酒さモデルの(h)紅斑面積および(g)紅斑評点を示すものである。
【図12】自然光下の外皮(SAGE1mg/mLで治療)(パネルa)および外皮の蛍光画像(パネルb);自然光下の内皮(パネルc)および蛍光条件下の内皮(パネルd)を示すものである。
【図13】HA誘導体のnHDF細胞増殖に与える影響(a)およびnHEKに与える影響(b)を示すものである。異なる濃度のGM−111101およびGM−212101で治療したマウスの肉眼解剖写真である。GM−111101を0.1mg/mL(e)、GM−111101を1mg/mL(f)、GM−111101を10mg/mL(g)、GM−212101を0.1mg/mL(h)、GM−212101を1mg/mL(i)、およびGM−212101を10mg/mL(j)を、無傷の範囲(c)およびギ酸で刺激した範囲(d)で比較した。
【図14】(a)傷を付けた範囲のSAGEの紅斑評点、(b)傷を付けた範囲のSAGEの浮腫評点、(c)無傷の範囲のSAGEの紅斑評点、(d)無傷の範囲のSAGEの浮腫評点を示すものである。
【図15】クロトン油炎症モデルを用いたSAGE治療を示すものである。クロトン油処理から4時間後の対照(CTL)群であり、CTL群の同じマウスの右(未処理)(パネルa)および左(クロトン油塗布)(パネルb)耳介を比較したものである。PBSを塗布した陰性対照(パネルc)、クロトン油の陽性対照(パネルd)、およびクロトン油の後にSAGE治療を行ったもの(パネルe)についてH&E染色を行ったものである。クロトン油陽性対照群の白血球浸潤および浮腫を確認した。ミエロペルオキシダーゼ(Myeloperoxidase)活性(パネルf)は、多形核白血球活性化の指標であり、SAGE治療後の耳介パンチについて測定したものである。パネルgおよびパネルhは、SAGE治療後の耳介の厚み(浮腫による)および耳介の発赤(炎症による)の変化である(p<0.05)。
【図16】(a)LL−37を注射した皮膚試料をH&E染色した断面図、(b)HAで治療したLL−37注射皮膚部位をH&E染色した断面図、(f)SAGE(GM−111101)で治療したLL−37注射皮膚部位をH&E染色した断面図、(g)HAおよびSAGEで治療したLL−37注射モデルのMPO活性測定、(h)紅斑面積の例、ならびに(g)HAおよびSAGE治療したLL−37酒さモデルの紅斑評点を示すものである。
【図17】ARPE−19細胞内におけるAGEに誘発されるRAGE発現が、AGE産物であるカルボキシメチルリジン−ウシ血清アルブミン(CML−BSA)上で増殖させることによって増大し、修飾ヘパリンによって阻害されることを示すものである。
【図18】対照(a)と比較すると、AGE産物であるCML−BSAがARPE−19細胞内の細胞死を誘導し(b)、これがODSHによって阻害され(c)、SAGE処理によってほぼ解消される(d)ことがわかる。
【図19】ヒト治療用抗凝固血漿ヘパリン濃度に近い濃度である0.4μg/mLにおいても、第XII因子を活性化するヘパリンとは異なり、LMW SAGEが第XII因子を活性化しないことを示している。
【発明を実施するための形態】
【0009】
詳細な説明
本化合物、組成物、および/または方法を開示および説明する前に、以下に説明する態様は、特定の化合物、合成方法、または使用そのものに限定されるものではなく、変形ももちろん可能であることを理解されたい。本明細書において用いられる専門用語も同様に、特定の態様を説明することのみを目的とするものであって、限定を意図するものではないことを理解されたい。
【0010】
本明細書およびそれに続く特許請求の範囲において言及される多くの用語は以下の意味を有するものと定義する。
【0011】
本明細書および添付の特許請求の範囲において用いられる単数形「1つの(a)」、「1つの(an)」、および「その(the)」には、文脈上、そうでないことが明確に指示されていない限り、複数の指示対象も包含されることに留意しなければならない。したがって、例えば、「製剤用担体(a pharmaceutical carrier)」に言及した場合は、この種の担体の2種以上の混合物等も包含される。
【0012】
「任意的な(optional)」または「場合により(optionally)」は、それに続いて記載される事象または状況が起こっても起こらなくてもよいことと、その記載には、当該事象または状況が起こる具体例も起こらない具体例も包含されることとを意味する。例えば、「場合により置換された低級アルキル」という語句は、この低級アルキル基が置換されていてもされていなくてもよいことと、この記載には非置換の低級アルキルおよび置換された低級アルキルの両方が包含されることとを意味している。
【0013】
本明細書および末尾の特許請求の範囲において、組成物または物品の特定の構成要素または成分の重量部に言及する場合は、重量部で表される当該組成物または物品中の当該構成要素または成分と他の任意の構成要素または成分との重量関係を表している。したがって、成分Xを2重量部および成分Yを5重量部含有する化合物中に、XおよびYは2:5の重量比で存在し、当該化合物中に含有されるさらなる成分とは無関係にこのような比で存在する。
【0014】
成分の重量パーセントは、そうでないことが明確に述べられていない限り、当該成分が含まれる配合物または組成物の総重量を基準とする。
【0015】
本明細書および末尾の特許請求の範囲において用いられる、化学種の残基(residue)とは、当該化学種から特定の反応スキームもしくはそれに続く配合により生成するかまたは化学的に生成する部分を指し、当該部分が実際にその化学種から得られたかどうかは関係ない。例えば、少なくとも1個の−OH基を含むヒアルロナンは式Y−OHで表すことができ、式中、Yは、当該ヒアルロナン分子の残り(すなわち残基)である。
【0016】
本明細書において用いられる「治療する(treat)」という用語は、既往症の症状を維持または軽減することとして定義される。本明細書において用いられる「予防(prevent)」という用語は、疾患または機能障害の1種またはそれ以上の症状が発生する可能性をなくすかまたは低下させることとして定義される。本明細書において用いられる「阻害(inhibit)」という用語は、本明細書に記載される化合物が、当該化合物の非存在下における活性と比較した場合に、同活性を完全に消失させるかまたは同活性を低下させる能力である。
【0017】
本明細書においては、アルキル化およびフルオロアルキル化されたヒアルロナンまたはその誘導体について記載する。一態様においては、ヒアルロナンのN−アセチル−グルコサミン残基の少なくとも1個の第1級C−6ヒドロキシルプロトンがアルキル基で置換されている。本明細書において用いられる「アルキル基」という用語は、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、t−ブチル、ペンチル、ヘキシル、ヘプチル、オクチル、デシル、テトラデシル、ヘキサデシル、エイコシル、テトラコシル等の、1〜24個の炭素原子を有する分岐または非分岐の飽和炭化水素基である。一態様においては、アルキル基は、C1〜C10分岐または直鎖アルキル基である。さらなる態様においては、アルキル基はメチルである。アルキル基は、置換されていても非置換であってもよい。アルキル基が置換されている場合、アルキル基上に存在する1個またはそれ以上の水素原子が、これらに限定されるわけではないが、アルキニル、アルケニル、アリール、ハライド、ニトロ、アミノ、エステル、ケトン、アルデヒド、ヒドロキシ、カルボン酸、アラルキル、またはアルコキシ等の1個またはそれ以上の基で置き換えられていてもよい。
【0018】
他の態様においては、ヒアルロナンのN−アセチル−グルコサミン残基の少なくとも1個の第1級C−6ヒドロキシルプロトンがフルオロアルキル基で置換されている。本明細書において用いられる「フルオロアルキル基」という用語は、1〜24個の炭素原子を有する分岐または非分岐の飽和炭化水素基であって、少なくとも1個の水素原子がフッ素で置換されているものである。特定の態様においては、フルオロアルキル基は、少なくとも1個のトリフルオロメチル基を含む。他の態様においては、フルオロアルキル基は、式−CH2(CF2)nCF3(式中、nは、1、2、3、4、5、6、7、8、9、または10の整数である)を有する。一態様においては、フルオロアルキル基は、−CH2CF2CF3または−CH2CF2CF2CF3である。
【0019】
本明細書においては、SAGEをアルキル化またはフルオロアルキル化する方法について記載する。一態様においては、SAGEは、(a)ヒアルロナンまたはその誘導体を、N−アセチル−グルコサミン残基の少なくとも1個の第1級C−6ヒドロキシルプロトンを脱プロトン化するのに十分な量の塩基と反応させ、そして(b)脱プロトン化されたヒアルロナンまたはその誘導体を、少なくとも1個の脱プロトン化された第1級C−6ヒドロキシル基をアルキル化またはフルオロアルキル化するのに十分な時間および濃度でアルキル化剤またはフルオロアルキル化剤と反応させることによって製造される。塩基性条件下ではグリコシド結合も開裂し、修飾過程においてより低分子量のヒアルロナン誘導体が生成する可能性もあることが当業者に理解されるであろう。この塩基性条件下では、酸が脱プロトン化されてカルボキシレートになることおよび第2級ヒドロキシル基が脱プロトン化されることと、これらの求核性部分は、それぞれ、平衡状態における相対的な存在量および陰イオン種の求核性に応じてその後に続くアルキル化に関与する場合があることも理解されるであろう。例えば、2−Oおよび/または3−Oヒドロキシルプロトンは、脱プロトン化およびアルキル化またはフルオロアルキル化され得る。その例を図1に示す。ここでRは、水素、アルキル基、またはアルキル基であってもよい。
【0020】
ヒアルロナン出発物質は、遊離酸またはその塩として存在してもよい。本明細書においては、ヒアルロナン出発物質の誘導体も使用してもよい。誘導体としては、アルキル化またはフルオロアルキル化ステップの前にヒアルロナンに任意の修飾を施したものが含まれる。本明細書においては、幅広い分子量のヒアルロナンを使用してもよい。一態様においては、アルキル化またはフルオロアルキル化前のヒアルロナンの分子量は10kDaを超える。他の態様においては、アルキル化またはフルオロアルキル化前のヒアルロナンの分子量は、25kDa〜1,000kDa、100kDa〜1,000kDa、25kDa〜500kDa、25kDa〜250kDa、または25kDa〜100kDaである。特定の態様においては、ヒアルロナン出発物質またはその誘導体は、動物起源に由来するものではない。これらの態様においては、ヒアルロナンは、細菌等の他の起源に由来するものであってもよい。例えば、組換えバシラス・サブティリス(B.subtilis)発現システムを用いてヒアルロナン出発物質を産生してもよい。
【0021】
まず最初にヒアルロナン出発物質またはその誘導体を、N−アセチル−グルコサミン残基の少なくとも1個の第1級C−6ヒドロキシルプロトンを脱プロトン化するのに十分な量の塩基と反応させる。選択される塩基は変化してもよい。例えば、本明細書においては、水酸化ナトリウムや水酸化カリウム等のアルカリ金属水酸化物を使用してもよい。塩基の濃度または量は、所望のアルキル化またはフルオロアルキル化度に応じて変化させてもよい。一態様においては、塩基の量は、ヒアルロナン出発物質またはその誘導体のN−アセチル−グルコサミン残基の第1級C−6ヒドロキシルプロトンの少なくとも0.001%を脱プロトン化するのに十分なものである。他の態様においては、塩基の量は、ヒアルロナン出発物質またはその誘導体のN−アセチル−グルコサミン残基の第1級C−6ヒドロキシルプロトンの0.001%〜50%、1%〜50%、5%〜45%、5%〜40%、5%〜30%、5%〜20%、10%〜50%、20%〜50%、または30%〜50%を脱プロトン化するのに十分なものである。溶液の塩基性が高くなるほど、鎖開裂反応が起こりやすくなるとともに、達成することができるアルキル化/フルオロアルキル化度が高くなることが理解される。例えば、ヒアルロナンには他のヒドロキシル基が存在する(例えば、2−OHおよび/または3−OHをアルキル化またはフルオロアルキル化される可能性もある)。一態様においては、ヒアルロナン上に存在するすべてのヒドロキシル基をアルキル化またはフルオロアルキル化してもよい。他の態様においては、ヒアルロナン上に存在するヒドロキシルプロトンの0.001%、0.01%、0.1%、1%、5%、10%、20%、30%、40%、50%、60%、70%、80%、90%、95%、100%、またはその任意の範囲を脱プロトン化し、続いてアルキル化またはフルオロアルキル化してもよい。
【0022】
ヒアルロナン出発物質またはその誘導体を塩基で処理した後、脱プロトン化されたヒアルロナンをアルキル化剤またはフルオロアルキル化剤と反応させることによってSAGEが生成する。アルキル化剤の例としては、これらに限定されるものではないが、ハロゲン化アルキルが挙げられる。臭化およびヨウ化アルキルが特に有用である。同様に、フルオロアルキル化剤としては、ハロゲン化フルオロアルキルを挙げることができる。本明細書においては、有機合成に慣用されているアルキル化剤およびフルオロアルキル化剤を使用してもよい。
【0023】
アルキル化およびフルオロアルキル化SAGEを作製するための例示的な合成手順を図1に示す。図1を参照すると、ヒアルロナン(HA)が塩基(例えば、NaOH)およびアルキル化剤(例えば、CH3I)で処理され、ヒアルロナンのN−アセチル−グルコサミン残基の第1級C−6ヒドロキシルプロトンがメチル化され、メチル化ヒアルロナン(MHA)が生成する。図1に、フルオロアルキル化剤(例えば、CF3(CF2)nCH2Br)を用いたフルオロアルキル化ヒアルロナン(FHA)を作製するための例示的な合成手順も示す。
【0024】
特定の態様においては、上述したアルキル化またはフルオロアルキル化SAGEを硫酸化することが望ましい。一態様においては、アルキル化またはフルオロアルキル化SAGEは、アルキル化またはフルオロアルキル化SAGEを硫酸化剤と反応させることによって硫酸化生成物を生成させることにより硫酸化される。硫酸化度は、部分硫酸化から完全硫酸化まで変化してもよい。一般に、アルキル化またはフルオロアルキル化ヒアルロナンまたはその誘導体上に存在する遊離ヒドロキシル基を硫酸化してもよい。一態様においては、少なくとも1個のC−2ヒドロキシルプロトンおよび/またはC−3ヒドロキシルプロトンが硫酸基で置換される。他の態様においては、硫酸化度は、アルキル化またはフルオロアルキル化SAGEの二糖単位当たり0.5、1.0、1.5、2.0、2.5、3.0、3.5、またはその任意の範囲である。一態様においては、アルキル化またはフルオロアルキル化SAGEを、1種またはそれ以上のヒドロキシルプロトンを脱プロトン化するために塩基で処理した後に、硫酸化剤を添加してもよい。硫酸化剤は、ヒドロキシル基または脱プロトン化されたヒドロキシル基と反応させることにより硫酸基を生成する任意の化合物である。SAGEの分子量は、反応条件に応じて変化してもよい。一態様においては、SAGEの分子量は、2kDa〜500kDa、2kDa〜250kDa、2kDa〜100kDa、2kDa〜50kDa、2kDa〜25kDa、または2kDa〜10kDaである。図1に、硫酸化されたアルキル化またはフルオロアルキル化SAGE(それぞれSMHAおよびSFHA)の例示的な合成を示す。
【0025】
図2に、幾つかの例示的なSAGEの構造を示す。各SAGEは、コードGM−XYSTZZで識別される:
Xは、アルキル基の種類であり(1=メチル、2=ペンタフルオロプロピル、3=ヘプタフルオロブチル、4=ベンジルグリシジルエーテル)、
Yは、HAのサイズであり(1=低、2=中、3=高)、
Sは、硫酸化度であり(1=部分、2=完全)、
Tは、アルキル化度であり(1=低、2=高)、
ZZ=ロット番号の連番01または02であり(02は01のバッチと同様に製造され、性状も同じである)。
【0026】
表1に、上で定義したコード体系による数種のSAGEの一覧を示す。
【0027】
【表1】

【0028】
一態様においては、SAGEのアルキル基はメチルであり、ヒアルロナンの少なくとも1個のC−2ヒドロキシルプロトンおよび/またはC−3ヒドロキシルプロトンが硫酸基で置換されている。他の態様においては、SAGEのアルキル基はメチルであり、ヒアルロナンの少なくとも1個のC−2ヒドロキシルプロトンおよび/またはC−3ヒドロキシルプロトンが硫酸基で置換されており、この化合物をアルキル化した後の分子量は2kDa〜200kDaである。この種の化合物の例が、図2に示すGM−111101である。
【0029】
本明細書に記載されたアルキル化およびフルオロアルキル化SAGEはいずれもその薬学的に許容される塩またはエステルであってもよい。薬学的に許容される塩は、遊離酸を適量の薬学的に許容される塩基で処理することによって調製される。代表的な薬学的に許容される塩基は、水酸化アンモニウム、水酸化ナトリウム、水酸化カリウム、水酸化リチウム、水酸化カルシウム、水酸化マグネシウム、水酸化第1鉄、水酸化亜鉛、水酸化銅、水酸化アルミニウム、水酸化第2鉄、イソプロピルアミン、トリメチルアミン、ジエチルアミン、トリエチルアミン、トリフルオロアミン、エタノールアミン、2−ジメチルアミノエタノール、2−ジエチルアミノエタノール、リジン、アルギニン、ヒスチジン等である。一態様においては、反応は、水を単独または水と混合可能な不活性有機溶媒との混合物中において約0℃〜約100℃の温度(室温等)で実施される。構造式Iの化合物対使用される塩基のモル比は、任意の特定の塩に所望される比が得られるように選択される。例えば、遊離酸出発物質のアンモニウム塩を調製する場合は、出発物質を約1当量の薬学的に許容される塩基で処理することによって中性塩を得てもよい。
【0030】
エステル誘導体は、典型的には、以下の実施例に例示するように、化合物の酸形態の前駆体として調製され、したがって、プロドラッグとすることができる。一般に、これらの誘導体は、メチルやエチル等の低級アルキルエステルとなるであろう。アミド誘導体−(CO)NH2、−(CO)NHR、および−(CO)NR2(式中、Rは、上に定義したアルキル基である)は、カルボン酸含有化合物をアンモニアまたは置換アミンと反応させることによって調製してもよい。エステルはまた、脂肪酸エステルであってもよい。例えば、パルミチン酸エステルが調製されており、エステラーゼで活性化されるプロドラッグとして代替的に使用してもよい。
【0031】
本明細書において記載されるSAGEを、生物システムまたは実体が許容できる任意の賦形剤と配合することによって医薬組成物を製造してもよい。このような賦形剤の例としては、これらに限定されるものではないが、水、ヒアルロン酸水溶液、生理食塩水、リンゲル液、デキストロース液、ハンクス液、および他の生理学的バランスのとれた塩の水溶液が挙げられる。不揮発性油、植物油(オリーブ油、ゴマ油等)、トリグリセリド、プロピレングリコール、ポリエチレングリコール、および注射可能な有機エステル(オレイン酸エチル等)等の非水性ビヒクルも使用してもよい。他の有用な配合物としては、カルボキシメチルセルロースナトリウム、ソルビトール、デキストラン等の粘度を高める薬剤を含む懸濁液が挙げられる。賦形剤はまた、等張性および化学的安定性を高めることができる物質等の添加剤も少量含んでいてもよい。緩衝剤の例としては、リン酸緩衝液、重炭酸緩衝液、およびトリス緩衝液が挙げられ、一方、防腐剤の例としては、チメロゾール、クレゾール、ホルマリン、およびベンジルアルコールが挙げられる。特定の態様においては、投与形態に応じてpHを変更してもよい。例えば、組成物のpHは約5〜約6であり、これは局所使用に好適である。さらに、医薬組成物は、本明細書に記載した化合物に加えて、担体、増粘剤、希釈剤、防腐剤、界面活性剤等を含んでいてもよい。
【0032】
医薬組成物はまた、本明細書に記載した化合物と組み合わせて使用される1種またはそれ以上の有効成分も含んでいてもよい。結果として得られる医薬組成物は、薬物および他の生物活性薬剤を、適用部位に接しているかまたは離れている組織に持続的かつ連続的に送達する系を提供してもよい。生物活性薬剤は、それが適用される生体系に局所または全身的な生物学的、生理学的、または治療効果を与えることができるものである。例えば、この薬剤は、数ある機能の中でも特に、感染または炎症の抑制および/または予防、細胞増殖および組織再生の亢進、腫瘍増殖の抑制、鎮痛剤としての作用、抗細胞接着の促進、歯槽骨および歯の喪失の低減、軟骨および体重を支える関節の変性の阻止、ならびに骨成長の促進作用を有するものであってもよい。さらに、本明細書に記載した化合物はいずれも薬学的に許容される化合物の2種以上の組合せを含んでいてもよい。この種の化合物の例としては、これらに限定されるものではないが、抗菌剤、抗炎症剤、麻酔剤等が挙げられる。これらの組成物を薬剤送達素子(drug delivery device)として使用するための方法を以下に詳述する。
【0033】
この医薬組成物は、当該技術分野において周知の技法を用いて調製してもよい。一態様においては、この組成物は、本明細書に記載したSAGEを薬学的に許容される化合物および/または担体と混和(admixing)させることによって調製される。「混和」という用語は、化学反応または物理的相互作用が起きないように2種類の成分を一緒に混合する(mix)こととして定義される。「混和」という用語には、当該化合物と薬学的に許容される化合物とを化学反応または物理的相互作用させることも含まれる。反応性を有する治療用薬物、例えば、求核性基を有するものに当該化合物を共有結合させてもよい。第2に、架橋した多糖中に、薬理活性を有する薬剤を非共有結合的に捕捉してもよい。第3に、静電または疎水性相互作用を用いて、本明細書中に記載した化合物内における薬学的に許容される化合物の維持を促進してもよい。
【0034】
特定の事例におけるSAGEの実際の好ましい量は、利用される特定の化合物、配合される具体的な組成、適用方式、ならびに具体的な位置および治療対象に応じて異なることになることが理解されるであろう。所与の宿主の投与量は、従来の検討を行い、例えば、適切な従来の薬理学的試験手順を用いて、例えば、対象化合物と周知の薬剤との活性の差を通例的に比較することによって決定してもよい。医師および製剤技術者(formulator)、医薬化合物の用量を決定する当業者であれば、推奨基準(Physicians Desk Reference,Barnhart Publishing(1999)に従い問題なく用量を決定するであろう。
【0035】
本明細書に記載する医薬組成物は、局所または全身療法のどちらが所望されるのか、および治療部位に応じて多くの方法で投与することができる。投与は、局所的に(点眼、膣内、直腸内、鼻腔内、経口、または皮膚に直接等)行ってもよい。局所投与製剤としては、軟膏剤、ローション剤、クリーム剤、ゲル剤、滴下剤、坐剤、噴霧剤、液剤、および散剤を挙げることができる。従来の医薬担体、水性、粉末、または油性基剤、増粘剤等が必要であるかまたは望ましい場合もある。投与はまた、エアゾールまたは乾燥微粉末を肺に直接吸入することによって行ってもよい。投与は、炎症または変性した関節の隙間に直接注射することによって行ってもよい。
【0036】
投与用製剤としては、無菌の水性または非水性の液剤、懸濁剤、および乳剤が挙げられる。非水担体の例としては、水、アルコール性/水性の溶液、乳濁液、または懸濁液(生理食塩水および緩衝化した媒質を含む)が挙げられる。開示した組成物および方法を副次的に使用する際に非経口投与用ビヒクルが必要な場合は、塩化ナトリウム溶液、リンゲルブドウ糖液、ブドウ糖および塩化ナトリウム、乳酸リンゲル液、または不揮発性油が挙げられる。開示した組成物および方法を副次的に使用する際に静脈投与用ビヒクルが必要な場合は、液体および栄養補給剤、電解質補給液(リンゲルブドウ糖液をベースとするものなど)等が挙げられる。防腐剤および他の添加剤、例えば、抗菌剤、抗酸化剤、キレート剤、不活性ガス等も存在してもよい。
【0037】
投薬は、治療すべき状態の重症度および応答性に依存するが、通常は、1日1回またはそれ以上となり、当業者が送達を中止すべきであると判断するまで、数日間から数ヶ月間治療過程を継続することとなる。当業者であれば、最適な用量、投薬方法、および反復速度を容易に決定することができる。
【0038】
本明細書に記載されるSAGEおよび医薬組成物は、薬物送達、小分子送達、創傷治癒、炎症性皮膚障害の治療、炎症性歯科疾患(inflammatory dental disorder)の治療、炎症性呼吸器疾患の治療、炎症性眼疾患の治療、熱傷治癒、および組織再生/工学に関連する様々な用途に使用してもよい。一態様においては、本明細書に記載するSAGEおよび組成物は、創傷治癒の改善を必要とする対象者に対しこのような改善を可能にする。本明細書に記載するSAGEおよび医薬組成物は生体適合性を有する材料から構成されているので、精製することなく任意の生体系の内部または表面に直接配置してもよい。SAGEを配置してもよい部位の例として、これらに限定されるものではないが、筋肉や脂肪等の軟組織;骨や軟骨等の硬組織;組織再生部位;歯周ポケット等の隙間;外科的切開またはそれ以外で形成されたポケットまたは空洞;口腔、膣腔、直腸、鼻腔、関節の隙間、眼の盲嚢等の本来の腔所;腹腔およびその中に収容されている臓器;ならびに内部または表面に化合物を配置してもよい他の部位(切り傷、掻き傷、熱傷部位等の皮膚表面の損傷を含む)が挙げられる。組織は、外傷または変性状態によって損傷する可能性があり、その替わりに、本明細書に記載したSAGEおよび組成物を、組織の外傷を防ぐために未損傷の組織に適用できることが見込まれている。SAGEは生分解性であってもよく、天然の酵素が除々に分解するように作用するであろう。SAGEの成分は、SAGEの成分が分解されて、生体系、例えば、細胞や組織等に吸収されることになるという意味で「生分解性」であってもよい。さらに、SAGE、特に、再水和していないものを生体系に適用して、対象部位から体液を吸収してもよい。
【0039】
創傷治癒の場合は、本明細書に記載したSAGEを注射により投与してもよい。SAGEがハイドロゲル薄膜の形態にある場合、多くの臨床使用には注射用ハイドロゲルが好ましく、その理由は主として3つある。まず第1に、注射用ハイドロゲルは、外傷部位において所望される任意の形状に成形することができるであろう。それは、初期のハイドロゲルはゾルまたは成形可能なパテとすることが可能であり、この系を複雑な形状で配置した後に架橋させることによって所望の寸法に適合させることができるためである。第2に、ハイドロゲルは、ゲル形成中に組織に接着することになり、結果として得られる表面の微小な凹凸によって生じる機械的な絡み合いによって組織−ハイドロゲル界面が強化されることになる。第3に、その場で架橋可能なハイドロゲルの導入が、針を用いるかまたは腹腔鏡法によって達成することができ、それによって、手術手技における侵襲が最小限になるであろう。
【0040】
乾癬、ざ瘡、アトピー性皮膚炎、酒さ、またはUV光に影響される光老化等の炎症性皮膚障害の場合、対象とする状態を予防または治療するために、SAGEをエモリエント剤の一部として局所適用してもよい。喘息、慢性閉塞性肺疾患、急性肺傷害、嚢胞性線維症等の呼吸器障害の場合は、気道被覆液に適合する水溶性等張性ビヒクル中にSAGEを溶解し、吸入用エアゾールとして肺または鼻腔に送達させてもよい。別法として、SAGEを微粉末に配合し、乾燥散剤として肺吸入してもよい。眼疾患の場合は、水性ビヒクルにSAGEを入れて点眼薬として局所適用するかまたは針によるかもしくは薬剤定速送達素子を植え込むかのいずれかによって眼に直接注入してもよい。歯周疾患等の歯科疾患の場合は、SAGEを洗口液の成分として添加するかまたは歯肉溝に直接適用するためのクリーム剤もしくは歯肉パック材に配合してもよい。
【0041】
SAGEはまた、糖尿病性血管もしくは腎臓障害または炎症性胃腸疾患等の全身性炎症性疾患を治療または予防するために、非経口的に静脈、筋肉、または皮下のいずれかに注射してもよい。同様に、炎症性および変形性関節症を治療するために関節内にSAGEを注射してもよい。SAGEはまた、炎症性腸疾患を治療するためにカプセル剤で経口投与するかまたは浣腸剤に配合して直腸内投与することにより送達してもよい。
【0042】
本明細書に記載したSAGEおよび組成物は、少なくとも1種の薬学的に許容される化合物の送達を必要とする患者に送達させてもよく、この送達には、この薬学的に許容される化合物を受容することができる少なくとも1つの組織に、本明細書に記載した1種またはそれ以上の組成物を用いて接触させることが含まれる。SAGEを、ヒトまたは非ヒト動物に対する治癒または治療的価値を有する多種多様な放出可能な生物活性物質の担体として使用してもよい。SAGEが担持することができるこのような物質の多くは上述したものである。本発明のゲルに混入させるのに好適な生物活性材料の中には、治療薬、例えば、抗炎症剤、解熱剤、ステロイド性および非ステロイド性抗炎症剤、ホルモン、成長因子、避妊薬、抗ウイルス剤、抗菌剤、抗真菌剤、鎮痛剤、催眠剤、鎮静剤、精神安定剤、抗痙攣薬、筋弛緩剤、局所麻酔剤、鎮痙薬、抗潰瘍薬、ペプチド性アゴニスト、交感神経作用薬、心血管作動薬、抗腫瘍剤、オリゴヌクレオチドおよびその類縁体等が含まれる。生物活性物質は、医薬有効量で添加される。
【0043】
一態様においては、本明細書に記載した硫酸化されたアルキル化およびフルオロアルキル化SAGEは、終末糖化産物受容体(RAGE)、P−セレクチン、またはヒト白血球エラスターゼの活性を阻害することができる。RAGEはヒトの皮膚で高度に発現し、皮膚線維芽細胞、樹状細胞、ケラチン生成細胞、内皮細胞、および単球上に存在している。皮膚が日光に曝されると、RAGEは、終末糖化産物(AGE)およびサイトカインである腫瘍壊死因子−αによって上方制御される。RAGEは、UVに誘起される光老化において顕著な役割を果たし、その場合、UVに誘導されるカルボキシメチルリジン(CML)等のAGE産物にRAGEが連結することによって、皮膚線維芽細胞による細胞外マトリックスの産生が刺激され、それによって皮膚の老化が促進される。乾癬の場合は、炎症の惹起が活性化Tリンパ球に決定的に依存するため、この疾患におけるRAGEの役割は一層顕著になる傾向にある。Tリンパ球はまた、ざ瘡およびアトピー性皮膚炎の機構においても重要な可能性がある。ざ瘡の場合、皮膚のCD3+およびCD4+Tリンパ球ならびにマクロファージが増加することによって毛包の導管内におけるケラチン生成細胞の過剰増殖が刺激され、毛包の導管が閉塞することによってざ瘡面皰が形成される。アトピー性皮膚炎の場合は、皮膚の抗原がTH2リンパ球を活性化することにより、インターロイキン−4(IL−4)やインターロイキン−13(IL−13)等のサイトカインが分泌され、その結果として、好酸球が皮膚に動員される。今度は、好酸球が主要塩基性タンパク質等の陽性荷電した毒素を放出し、アレルギー性皮膚疾患を生じさせる。したがって、本明細書に記載した化合物の強力なRAGE阻害活性は、これらに限定されるものではないが、ざ瘡、湿疹、アトピー性皮膚炎、乾癬、皮膚の光老化等の様々な皮膚疾患の治療に有用となる。
【0044】
成人期においては、RAGEが生体に必ずしも完全に有益なわけではない。悪性腫瘍はオートクライン因子としてアンホテリン(またはハイモビリティー・ボックス・グループ・プロテイン−1、HMGB−1)を分泌し、アンホテリンとRAGEとの相互作用を利用して原発腫瘍の増殖および転移を促進する。組換え型デコイ(可溶型RAGEまたはs−RAGE)を用いてRAGEを阻害すると、腫瘍増殖が減少するとともに転移が阻害される。敗血症の場合は、単球およびマクロファージがアンホテリンを分泌し、これが血管および他の炎症細胞上のRAGEと相互作用することにより、細菌性ショックの重症度が増大する。この相互作用をRAGEの抗体によって阻害することにより、重症敗血症における臓器障害が阻止される。成人期においては、RAGEは、PMN、単球、およびリンパ球の炎症部位への動員を促進する血管内皮細胞接着分子(vascular adhesion receptor)としても機能する。RAGEを阻害することにより、炎症細胞の流入が緩やかになる。このことは多発性硬化症の動物モデルで既に実証されており、血管内皮RAGEとs−RAGEとの競合的阻害により、活性化された脳炎惹起性Tリンパ球の中枢神経系への流入が阻止され、神経の炎症および変性の発症および進行が遅延される。
【0045】
RAGEはまた、PMN、単球、およびリンパ球から分泌される強力な炎症促進因子であるS100カルグラニュリンと呼ばれるカルシウム結合タンパク質ファミリーとも相互作用する。S100カルグラニュリンの上昇は、急性肺傷害および嚢胞性線維症患者の気道分泌物におけるPMN炎症の鋭敏なマーカーである。眼の場合は、加齢黄斑変性においてS100カルグラニュリンとRAGEとの相互作用が顕著な役割を果たし、結果として失明を招く。RAGEはまた、アルツハイマーにおけるβアミロイドペプチドおよびアミロイドタンパクのβシートとも結合する。RAGE−βシート線維の相互作用は、RAGEが関与する神経細胞死および炎症誘発を介して、アルツハイマー型認知症および全身アミロイドーシスにおける臓器障害を媒介する。
【0046】
RAGEは糖尿病においても顕著である。血中グルコースが上昇すると、グルコースのアルデヒド基が細胞内タンパク質のアミンにランダムに結合して共有結合付加体となる。PMNが産生する酸化剤である次亜塩素酸(HOCl)等の酸化剤の存在下においては、今度はこのグルコース部分が酸化される。この酸化されたグリコシル化タンパク質は終末糖化産物(AGE)として周知である。AGEもRAGE受容体に積極的に結合し、RAGEを介するシグナル伝達を惹起する。AGE−RAGEシグナル伝達は、血管内皮機能障害、創傷治癒遅延、およびコントロール状況の悪い糖尿病に特徴的なアテローム性動脈硬化症を加速させる原因となる。眼の場合は、AGE−RAGEシグナル伝達によって網膜微小血管が増殖し、糖尿病性網膜症および失明の原因となる。腎臓においては、AGE−RAGEシグナル伝達が、初期腎肥大、その後に、糖尿病性腎不全(糖尿病性腎症)を招く線維症の原因となる。同様に、AGE−RAGEシグナル伝達は内皮のアポトーシスを起こさせ、血管新生を阻害し、糖尿病皮膚潰瘍の治癒を遅らせる。
【0047】
RAGEを阻害する能力を有するSAGEは、炎症に特に有用な治療薬となる。RAGEは、子宮内において、成長を促進する核タンパクであるアンホテリンすなわちハイモビリティー・ボックス・グループ・プロテイン−1(HMGB−1)に結合する受容体として機能する。ここで、アンホテリン−RAGE相互作用は、神経系の発達に重要な増殖シグナル伝達を惹起する。成人期においては、RAGEは、血管壁細胞、神経組織、心筋細胞、単球およびマクロファージ、Tリンパ球、腎メサンギウム細胞、ならびに皮膚線維芽細胞、樹状細胞、およびケラチン生成細胞に発現する。したがって、一態様においては、本明細書に記載したSAGEおよび組成物は、これらに限定されるものではないが、癌、多発性硬化症、変形性関節症、嚢胞性線維症、鎌状赤血球貧血、炎症性心血管疾患、または炎症性心血管疾患、糖尿病性合併症等のRAGE関連疾患に帰属される様々な異なる病気によって生じる対象者の炎症を安全に軽減または予防するために使用することができる。
【0048】
一態様においては、SAGEおよび組成物を陰性荷電物質として投与することによってカテリシジン由来の陽性荷電した皮膚ペプチドに結合させて阻害し、それによって皮膚障害を治療または予防してもよい。例えば、活性カテリシジンペプチドが皮膚で過剰発現することによって発症することが周知の酒さ性ざ瘡として知られる皮膚状態を、本明細書に記載したSAGEを用いて治療または予防してもよい。SAGEを用いて治療または予防することができる皮膚疾患の例としては、これらに限定されるものではないが、酒さ、アトピー性皮膚炎(湿疹)、アレルギー性接触皮膚炎、乾癬、ヘルペス状皮膚炎、ざ瘡、糖尿病性皮膚潰瘍および他の糖尿病性創傷、熱傷(温熱熱傷の痛みの緩和を含む)、日焼け(日焼けの痛みの緩和を含む)、形成外科手術後の瘢痕予防、光線角化症、虫刺され、ツタウルシによるかぶれ、放射線皮膚炎/熱傷、皮膚治癒の亢進、ケロイド瘢痕の予防および治療、または脂漏性皮膚炎の治療が挙げられる。
【0049】
SAGEがRAGE活性および他の生物学的メカニズムを阻害する能力を有することから、SAGEには皮膚障害の治療または予防以外にも多くの治療用途がある。一態様においては、SAGEを、歯肉炎(歯周疾患)およびアフタ性潰瘍を治療するために歯科および口腔外科で使用してもよい。他の態様においては、SAGEを眼科用途、例えば、加齢性黄斑変性症、糖尿病性網膜症、眼球乾燥症候群、および他の炎症性結膜炎(inflammatory conjunctivitis)、虹彩炎、ブドウ膜炎、アレルギー性結膜炎の治療、白内障手術における消炎補助(anti-inflammatory aid)、または角膜炎症および瘢痕の予防等に使用してもよい。
【0050】
さらなる態様においては、尿生殖器用途(例えば、尿路感染症予防、膀胱および尿路上皮系の移行上皮癌の治療、間質性膀胱炎の治療、および性感染症予防のための膣潤滑/保護薬としての使用)にSAGEを使用してもよい。
【0051】
他の態様においては、嚢胞性線維症、気管支拡張症、鼻炎(アレルギー性および通年性の両方)、副鼻腔炎、肺気腫および慢性気管支炎(COPD)、急性肺傷害/成人型呼吸窮迫症候群、間質性肺線維症、SARS、喘息、ならびに呼吸器合胞体ウイルス等の多くの呼吸器疾患の治療にSAGEを使用してもよい。他の態様においては、SAGEは、いびきおよび閉塞型睡眠時無呼吸の予防および治療、HIV陽性または造血器腫瘍を有する患者等の免疫不全宿主の一般的な呼吸器病原体による感染(ストレプトコッカス・ニューモニエ(Streptococcus pneumoniae)、ヘモフィルス・インフルエンザ、ブドウ球菌属(Staphylococcus)、肺炎マイコプラズマ・ニューモニエ(Mycoplasma pneumoniae)、肺炎クラミジア(Chlamydial pneumonia)、グラム陰性腸内細菌による感染)の予防、または中耳炎の予防および処置に用いることができる。
【0052】
SAGEは、心血管用途(例えば、急性冠症候群またはアテローム性動脈硬化症の治療または予防);血液学/腫瘍学的用途(例えば、鎌状赤血球貧血の予防および治療;転移性疾患の予防および治療;ならびに悪性腫瘍による凝固亢進状態の予防(トルソー症候群));感染症の治療(例えば、脳血管閉塞症候群(cerebral vascular occlusive syndrome)、および熱帯性マラリアにおける腎炎、黄熱病、デング熱、全身性敗血症(systemic sepsis)、ならびに標的細胞のウイルス融合および感染を防ぐためのHIVの補助療法);胃腸疾患の治療(例えば、潰瘍性大腸炎、腸のクローン病、痔核、ならびに胃および食道のストレス潰瘍の予防);リウマチおよび免疫疾患の治療(例えば、変形性関節症、関節リウマチ、全身性エリテマトーデスの予防および治療、血管神経性浮腫、シェーグレン症候群、全身性硬化症、全身性アミロイドーシス、および全身性肥満細胞症の予防および治療);腎疾患(例えば、糖尿病性腎症および糸球体腎炎の予防および治療);ならびに神経系疾患(例えば、多発性硬化症およびアルツハイマー型認知症)に使用してもよい。
【0053】
本明細書に記載したSAGEおよび組成物は、他の関連する療法よりも安全である。例えば、ヘパリンおよび他の硫酸化多糖は動物および臨床研究の両方において糖尿病性合併症を減少させることができ、糖尿病性腎症に特に有効である。しかしながら、抗凝固性を有するヘパリンは、出血の危険性が過度に高くなることから、一般臨床場面で糖尿病性合併症の予防に使用することはできない。本明細書に記載したSAGEおよび組成物は、以下の実施例に示すが、長期治療に関する重要な考慮事項である抗凝固活性が低い。さらに、このことも実施例で示すが、SAGEには毒性がほとんどまたは全くない。
【実施例】
【0054】
以下の実施例は、本明細書に記載および特許請求した化合物、組成物、および方法を作製および評価する方法を当業者に十分に開示および説明するために述べるものであるが、これは単なる例示を目的とするものであって、本発明者らが本発明者らの発明と見なすものの範囲を限定することを意図するものではない。数値(例えば、量や温度等)が確実に正確になるように努めたが、ある程度の誤差およびずれがあることは考慮されたい。特段の指定がない限り、部は重量部、温度は℃単位であるかまたは周囲温度であり、圧力は大気圧付近である。反応条件、例えば、成分の濃度、所望の溶媒、溶媒混合物、温度、圧力、ならびに記載した処理により得られる生成物の純度および収率を最適化するのに用いることができる他の反応範囲および条件には多くの変形および組合せがある。このような工程条件を最適化するためには妥当かつ日常的な実験のみが必要であろう。以下に記載する化合物は、上述および図2に示すコード番号で識別される。
【0055】
I.アルキル化HA誘導体の合成
A.メチルHA(DS−2)の調製
ヒアルロン酸(HA、Novozymes Biopolymers、950kDa)(2.0g)を100mLビーカー内でNaOH(40%w/v)20mLに溶解し、混合物を室温(rt)で2時間撹拌した。結果として得られた粘性液体を、イソプロパノール100mLを含む400mLビーカーに移して混合を継続した。撹拌した混合物にヨードメタン10mLを加え、混合物をrtで24時間撹拌した。結果として得られた懸濁液を濾過して粗生成メチル化HA生成物を回収した。この粗生成MeHAを蒸留水250mLに溶解し、溶液のpHを約7.0に調節し、溶液を蒸留水に対し24時間透析した。この間、外側の水浴を4回交換した。透析したMeHA生成物を凍結乾燥することによりメチルHA1.25gを綿状の網目状物として得た。1H NMR(D2O,δ):1.85(s,3H,NCH3),3.20−3.80(m,10H,OCH+OCH3)。置換度(SD)を1H NMRで測定したところ、SD=2すなわち二糖単位当たりのメチル基が平均2個と見積もられた。SD=[(メチルHAのδ3.20−3.8の積分値)−(HAのδ3.20−3.8の積分値)]/(NCH3の1.85の積分値)。これは、第1級ヒドロキシルおよび少なくとも1個の第2級ヒドロキシル基の両方がこの工程により修飾されたことを示唆している。
【0056】
B.メチルHA(DS−1)の調製
ヒアルロン酸(HA、Novozymes Biopolymers、950kDa)(2.0g)を100mLビーカー内でNaOH(40%w/v)20mLに溶解し、混合物を室温(rt)で2時間撹拌した。結果として得られた粘性液体を、イソプロパノール100mLを含む400mLビーカーに移して混合を継続した。撹拌した混合物にヨードメタン4mLを加え、混合物をrtで24時間撹拌した。結果として得られた懸濁液を濾過して粗生成メチル化HA生成物を回収した。この粗生成MeHAを蒸留水250mLに溶解し、溶液のpHを約7.0に調節し、溶液を蒸留水に対し24時間透析した。この間、外側の水浴を4回交換した。透析したMeHA生成物を凍結乾燥することによりメチルHA1.25gを綿状の網目状物として得た。1H NMR(D2O,δ):1.85(s,3H,NCH3),3.20−3.80(m,10H,OCH+OCH3)。置換度(SD)を1H NMRで測定したところ、SD=1すなわち二糖単位当たりメチル基が平均1個と見積もられた。SD=[(メチルHAのδ3.20−3.8の積分値)−(HAのδ3.20−3.8の積分値)]/(NCH3の1.85の積分値)。このことは、第1級ヒドロキシルおよび少なくとも1個の第2級ヒドロキシル基がこの工程により修飾されたことを示唆している。
【0057】
C.FHA−2(DS−1)の調製
2,2,3,3,4,4−ヘプタフルオロ−1−ブタノール1.0g(5mmol)を含む25mLフラスコに三臭化リン1.3mL(7.5mmol)をゆっくりと加えた。この混合物を60℃で30分間撹拌した後、飽和炭酸水素ナトリウム溶液(15mL)をゆっくりと加えて反応を停止した。この水溶液をジクロロメタン15mLずつで3回抽出し、臭化ヘプタフルオロブチルを含む有機層を濃縮し、残渣を次のステップでさらなる生成に用いた。
【0058】
HA(2.0g)を100mLビーカー内でNaOH(40%w/v)20mLに溶解し、混合物を室温(rt)で2時間撹拌した。結果として得られた粘性液体を、イソプロパノール100mLを含む400mLビーカーに移して混合を継続した。撹拌した混合物にイソプロパノール10mL中の粗生成臭化ヘプタフルオロブチルを加え、混合物をrtで24時間撹拌した。結果として得られた懸濁液を濾過して粗生成FHA−2生成物を回収した。この粗生成FHA−2を蒸留水250mLに溶解し、溶液のpHを約7.0に調節し、溶液を蒸留水に対し24時間透析した。この間、外側の水浴を4回交換した。透析したFHA−2生成物を凍結乾燥することにより、FHA−2として設計された1.5gのFHA−2を綿状の網目状物として得た。1H NMR(D2O,δ):1.82(s,3H,NCH3),3.15−3.80(m,8H,OCH+OCH2)。19F NMR(D2O,δ):−115.3,−120.8。1H NMRで測定した置換度(SD)は1.0であった。SD=[(FHA−2のδ3.15−3.80の積分値)−(HAのδ3.20−3.8の積分値)]/[(NCH3の1.82の積分値)×(2/3)]。
【0059】
D.FHA−2(DS−2)の調製
2,2,3,3,4,4−ヘプタフルオロ−1−ブタノール3.0g(15mmol)を含む25mLフラスコに三臭化リン2.5.mL(16mmol)をゆっくりと加えた。混合物を60℃で30分間撹拌した後、飽和炭酸水素ナトリウム溶液(15mL)をゆっくりと加えて反応を停止させた。この水溶液をジクロロメタン15mLずつで3回抽出し、臭化ヘプタフルオロブチルを含む有機層を濃縮し、残渣を精製することなく次のステップに使用した。
【0060】
HA(2.0g)を100mLビーカー内でNaOH(40%w/v)20mLに溶解し、混合物を室温(rt)で2時間撹拌した。結果として得られた粘性液体を、イソプロパノール100mLを含む400mLビーカーに移して混合を継続した。撹拌した混合物にイソプロパノール10mL中の粗生成臭化ヘプタフルオロブチルを加え、混合物をrtで24時間撹拌した。結果として得られた懸濁液を濾過して粗生成FHA−2生成物を回収した。この粗生成FHA−2を蒸留水250mLに溶解し、溶液のpHを約7.0に調節し、溶液を蒸留水に対し24時間透析した。この間、外側の水浴を4回交換した。透析したFHA−2生成物を凍結乾燥することにより、FHA−2として設計された1.5gのFHA−2を綿状の網目状物として得た。1H NMR(D2O,δ):1.82(s,3H,NCH3),3.15−3.80(m,8H,OCH+OCH2)。19F NMR(D2O,δ):−115.3,−120.8。1H NMRで測定した置換度(SD)は2.0であった。SD=[(FHA−2のδ3.15−3.80の積分値)−(HAのδ3.20−3.8の積分値)]/[(NCH3の1.82の積分値)×(2/3)]。
【0061】
E.FHA−1(DS−1)の調製
2,2,3,3,−ペンタフルオロ−1−プロパノール1.0g(5mmol)を含む25mLフラスコに三臭化リン1.3mL(7.5mmol)をゆっくりと加えた。混合物を60℃で30分間撹拌した後、飽和炭酸水素ナトリウム溶液(15mL)をゆっくりと加えて反応を停止した。この水溶液をジクロロメタン15mLずつで3回抽出し、臭化ヘプタフルオロブチルを含む有機層を濃縮し、残渣を精製することなく次のステップで使用した。
【0062】
HA(2.0g)を100mLビーカー内でNaOH(40%w/v)20mLに溶解し、混合物を室温(rt)で2時間撹拌した。結果として得られた粘性液体を、イソプロパノール100mLを含む400mLビーカーに移して混合を継続した。撹拌した混合物にイソプロパノール10mL中の粗生成臭化ヘプタフルオロブチルを加え、混合物をrtで24時間撹拌した。結果として得られた懸濁液を濾過して粗生成FHA−1生成物を回収した。この粗生成FHA−1を蒸留水250mLに溶解し、溶液のpHを約7.0に調節し、溶液を蒸留水に対し24時間透析した。この間、外側の水浴を4回交換した。透析したFHA−1生成物を凍結乾燥することにより、FHA−1として設計された1.5gのFHA−1を綿状の網目状物として得た。1H NMR(D2O,δ):1.80(s,3H,NCH3),3.15−3.80(m,8H,OCH+OCH2)。19F NMR(D2O,δ):−113.6,−118.0。1H NMRで測定した置換度(SD)は1.0であった。SD=[(FHA−1のδ3.15−3.80の積分値)−(HAのδ3.20−3.8の積分値)]/[(NCH3の1.80の積分値)×(2/3)]。
【0063】
F.FHA−1(DS−2)の調製
2,2,3,3,−ペンタフルオロ−1−プロパノール3.0g(15mmol)を含む25mLフラスコに三臭化リン3mL(18mmol)をゆっくりと加えた。混合物を60℃で30分間撹拌した後、飽和炭酸水素ナトリウム溶液(15mL)をゆっくりと加えて反応を停止した。この水溶液をジクロロメタン15mLずつで3回抽出し、臭化ヘプタフルオロブチルを含む有機層を濃縮し、残渣を精製することなく次のステップで使用した。
【0064】
HA(2.0g)を100mLビーカー内でNaOH(40%w/v)20mLに溶解し、混合物を室温(rt)で2時間撹拌した。結果として得られた粘性液体を、イソプロパノール100mLを含む400mLビーカーに移して混合を継続した。撹拌した混合物にイソプロパノール10mL中の粗生成臭化ヘプタフルオロブチルを加え、混合物をrtで24時間撹拌した。結果として得られた懸濁液を濾過して粗生成FHA−1生成物を回収した。この粗生成FHA−1を蒸留水250mLに溶解し、溶液のpHを約7.0に調節し、溶液を蒸留水に対し24時間透析した。この間、外側の水浴を4回交換した。透析したFHA−1生成物を凍結乾燥することにより、FHA−1として設計された1.5gのFHA−1を綿状の網目状物として得た。1H NMR(D2O,δ):1.80(s,3H,NCH3),3.15−3.80(m,8H,OCH+OCH2)。19F NMR(D2O,δ):−113.6,−118.0。1H NMRで測定した置換度(SD)は2.0であった。SD=[(FHA−1のδ3.15−3.80の積分値)−(HAのδ3.20−3.8の積分値)]/[(NCH3の1.80の積分値)×(2/3)]。
【0065】
G.BGHAの調製
ヒアルロン酸(HA、Novozymes Biopolymers、950kDa)(2.0g)を100mLビーカー内でNaOH(40%w/v)20mLに溶解し、混合物を室温(rt)で2時間撹拌した。結果として得られた粘性液体を、イソプロパノール100mLを含む400mLビーカーに移して混合を継続した。撹拌した混合物にベンジルグリシジルエーテル10mLを加え、混合物をrtで24時間撹拌した。結果として得られた懸濁液を濾過して粗生成BGHA生成物を回収した。この粗生成BGHAを蒸留水250mLに溶解し、溶液のpHを約7.0に調節し、溶液を蒸留水に対し24時間透析した。この間、外側の水浴を4回交換した。透析したBGHA生成物を凍結乾燥することにより、BGHA1.25gを綿状の網目状物として得た。1H NMR(D2O,δ):1.85(s,3H,NCH3),3.20−3.80(m,10H,OCH+OCH3)。置換度(SD)を1H NMRで測定したところ、SDは1未満であった。
【0066】
H.低分子量メチルHA(DS−2)の調製
ヒアルロン酸(HA、Novozymes Biopolymers、53kDa)(2.0g)を100mLビーカー内でNaOH(40%w/v)20mLに溶解し、混合物を室温(rt)で2時間撹拌した。結果として得られた粘性液体を、イソプロパノール100mLを含む400mLビーカーに移して混合を継続した。撹拌した混合物にヨードメタン10mLを加え、混合物をrtで24時間撹拌した。結果として得られた懸濁液を濾過して粗生成低分子メチル化HA生成物を回収した。この粗生成LMW MeHAを蒸留水250mLに溶解し、溶液のpHを約7.0に調節し、溶液を蒸留水に対し24時間透析した。この間、外側の水浴を4回交換した。透析したLMW MeHA生成物を凍結乾燥することにより、メチルHA1.2gを綿状の網目状物として得た。1H NMR(D2O,δ):1.85(s,3H,NCH3),3.20−3.80(m,10H,OCH+OCH3)。1H NMRで測定した置換度(SD)は2であった。
【0067】
I.低分子量メチルHA(DS−1)の調製
ヒアルロン酸(HA、Novozymes Biopolymers、53kDa)(2.0g)を100mLビーカー内でNaOH(40%w/v)20mLに溶解し、混合物を室温(rt)で2時間撹拌した。結果として得られた粘性液体を、イソプロパノール100mLを含む400mLビーカーに移して混合を継続した。撹拌した混合物にヨードメタン4mLを加え、混合物をrtで24時間撹拌した。結果として得られた懸濁液を濾過して、粗生成低分子メチル化HA生成物を回収した。この粗生成LMW MeHAを蒸留水250mLに溶解し、溶液のpHを約7に調節し、溶液を蒸留水に対し24時間透析した。この間、外側の水浴を4回交換した。透析したLMW MeHA生成物を凍結乾燥することにより、LMW MeHA1.2gを綿状の網目状物として得た。1H NMR(D2O,δ):1.85(s,3H,NCH3),3.20−3.80(m,10H,OCH+OCH3)。1H NMRで測定した置換度(SD)は1であった。
【0068】
J.低分子量FHA−2(DS−1)の調製
2,2,3,3,4,4−ヘプタフルオロ−1−ブタノール1.0g(5mmol)を含む25mLフラスコに三臭化リン1.3mL(7.5mmol)をゆっくりと加えた。混合物を60℃で30分間撹拌した後、飽和炭酸水素ナトリウム溶液(15mL)をゆっくりと加えて反応を停止した。この水溶液をジクロロメタン15mLずつで3回抽出し、臭化ヘプタフルオロブチルを含む有機層を濃縮し、残渣を精製することなく次のステップで使用した。
【0069】
HA(2.0g、53kDaを100mLビーカー内でNaOH(40%w/v)20mLに溶解し、混合物を室温(rt)で2時間撹拌した。結果として得られた粘性液体を、イソプロパノール100mLを含む400mLビーカーに移して混合を継続した。撹拌した混合物にイソプロパノール10mL中の粗生成臭化ヘプタフルオロブチルを加え、混合物をrtで24時間撹拌した。結果として得られた懸濁液を濾過してLMW FHA−2生成物を回収した。この粗生成LMW FHA−2を250mLの蒸留水に溶解し、溶液のpHを約7に調節し、溶液を蒸留水に対し24時間透析した。この間、外側の水浴を4回交換した。透析したLMW FHA−2生成物を凍結乾燥することにより、LMW FHA−2として設計された1.5gのLMW FHA−2を綿状の網目状物として得た。1H NMR(D2O,δ):1.82(s,3H,NCH3),3.15−3.80(m,8H,OCH+OCH2)。19F NMR(D2O,δ):−115.3,−120.8。1H NMRで測定した置換度(SD)は1.0であった。
【0070】
K.低分子量FHA−2(DS−2)の調製
2,2,3,3,4,4−ヘプタフルオロ−1−ブタノール3.0g(15mmol)を含む25mLフラスコに三臭化リン2.5mL(16mmol)をゆっくりと加えた。混合物を60℃で30分間撹拌した後、飽和炭酸水素ナトリウム溶液(15mL)をゆっくりと加えて反応を停止した。この水溶液をジクロロメタン15mLずつで3回抽出し、臭化ヘプタフルオロブチルを含む有機層を濃縮し、残渣を精製することなく次のステップで使用した。
【0071】
HA(2.0g、53kDa)を100mLビーカー内でNaOH(40%w/v)20mLに溶解し、混合物を室温(rt)で2時間撹拌した。結果として得られた粘性液体を、イソプロパノール100mLを含む400mLビーカーに移して混合を継続した。撹拌した混合物にイソプロパノール10mL中の粗生成臭化ヘプタフルオロブチルを加え、混合物をrtで24時間撹拌した。結果として得られた懸濁液を濾過してLMW FHA−2生成物を回収した。この粗生成LMW FHA−2を250mLの蒸留水に溶解し、溶液のpHを約7に調節し、溶液を蒸留水に対し24時間透析した。この間、外側の水浴を4回交換した。透析したLMW FHA−2生成物を凍結乾燥することにより、LMW FHA−2として設計された1.5gのLMW FHA−2を綿状の網目状物として得た。1H NMR(D2O,δ):1.82(s,3H,NCH3),3.15−3.80(m,8H,OCH+OCH2)。19F NMR(D2O,δ):−115.3,−120.8。1H NMRで測定した置換度(SD)は2.0であった。
【0072】
L.低分子量FHA−1(DS−1)の調製
2,2,3,3,−ペンタフルオロ−1−プロパノール1.0g(5mmol)を含む25mLフラスコに三臭化リン1.3mL(7.5mmol)をゆっくりと加えた。混合物を60℃で30分間撹拌した後、飽和炭酸水素ナトリウム溶液(15mL)をゆっくりと加えて反応を停止した。この水溶液をジクロロメタン15mLずつで3回抽出し、臭化ヘプタフルオロブチルを含む有機層を濃縮し、残渣を精製することなく次のステップで使用した。
【0073】
HA(2.0g、53kDa)を100mLビーカー内でNaOH(40%w/v)20mLに溶解し、混合物を室温(rt)で2時間撹拌した。結果として得られた粘性液体を、イソプロパノール100mLを含む400mLビーカーに移して混合を継続した。撹拌した混合物に、イソプロパノール10mL中の粗生成臭化ヘプタフルオロブチルを加え、混合物をrtで24時間撹拌した。結果として得られた懸濁液を濾過してLMW FHA−1生成物を回収した。この粗生成LMW FHA−1を250mLの蒸留水に溶解し、溶液のpHを約7に調節し、溶液を蒸留水に対し24時間透析した。この間、外側の水浴を4回交換した。透析したLMW FHA−1生成物を凍結乾燥することにより、LMW FHA−1として設計されたLMW FHA−11.5gを綿状の網目状物として得た。1H NMR(D2O,δ):1.80(s,3H,NCH3),3.15−3.80(m,8H,OCH+OCH2)。19F NMR(D2O,δ):−113.6,−118.0。1H NMRで測定した置換度(SD)は1.0であった。
【0074】
M.LMW FHA−1(DS−2)の調製
2,2,3,3,−ペンタフルオロ−1−プロパノール3.0g(15mmol)を含む25mLフラスコに三臭化リン3mL(18mmol)をゆっくりと加えた。混合物を60℃で30分間撹拌した後、飽和炭酸水素ナトリウム溶液(15mL)をゆっくりと加えて反応を停止した。この水溶液をジクロロメタン15mLずつで3回抽出し、臭化ヘプタフルオロブチルを含む有機層を濃縮し、残渣を精製することなく次のステップで使用した。
【0075】
HA(2.0g、53kDa)を100mLビーカー内でNaOH(40%w/v)20mLに溶解し、混合物を室温(rt)で2時間撹拌した。結果として得られた粘性液体を、イソプロパノール100mLを含む400mLビーカーに移して混合を継続した。撹拌した混合物にイソプロパノール10mL中の粗生成臭化ヘプタフルオロブチルを加え、混合物をrtで24時間撹拌した。結果として得られた懸濁液を濾過してLMW FHA−1生成物を回収した。この粗生成LMW FHA−1を250mLの蒸留水に溶解し、溶液のpHを約7に調節し、溶液を蒸留水に対し24時間透析した。この間、外側の水浴を4回交換した。透析したLMW FHA−1生成物を凍結乾燥することにより、LMW FHA−1として設計された1.5gのLMW FHA−1を綿状の網目状物として得た。1H NMR(D2O,δ):1.80(s,3H,NCH3),3.15−3.80(m,8H,OCH+OCH2)。19F NMR(D2O,δ):−113.6,−118.0。1H NMRで測定した置換度(SD)は2.0であった。
【0076】
N.低分子量BGHAの調製
ヒアルロン酸(HA、Novozymes Biopolymers、53kDa)(2.0g)を100mLビーカー内でNaOH(40%w/v)20mLに溶解し、混合物を室温(rt)で2時間撹拌した。結果として得られた粘性液体を、イソプロパノール100mLを含む400mLビーカーに移して混合を継続した。撹拌した混合物にベンジルグリシジルエーテル10mLを加え、混合物をrtで24時間撹拌した。結果として得られた懸濁液を濾過してLMW BGHA生成物を回収した。この粗生成LMW BGHAを250mLの蒸留水に溶解し、溶液のpHを約7に調節し、溶液を蒸留水に対し24時間透析した。この間、外側の水浴を4回交換した。透析したLMW BGHA生成物を凍結乾燥することにより、LMW BGHA1.25gを綿状の網目状物として得た。1H NMR(D2O,δ):1.85(s,3H,NCH3),3.20−3.80(m,10H,OCH+OCH3)。1H NMRで測定した置換度(SD)は1未満であった。
【0077】
II.アルキル化HA誘導体の硫酸化
1.LMW−P−OSFHA−1(DS−1)(GM−211101)の調製
最初に、脱イオン水100mL中のLMW FHA−1(1.0g)のpHを1NのHClで3.0に調節し、トリブチルアミン1mLを加えることによって、LMW FHA−1(DS−1)のトリブチルアンモニウム(TBA)塩を調製した。この混合物を激しく混合し、凍結乾燥した。結果として得られた塩(FHA−1−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり6mol)のピリジン−三酸化硫黄錯体(0.4g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLで粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化HAを蒸留水に溶解し、100mMのNaCl溶液に対し2日間透析(溶液を1日4回交換)した後、凍結乾燥することにより、生成物(330mg)を収率75%で得た。1H NMRで評価した硫酸化SD=1.0であった。置換度は、文献(Carbohydrate Research、1998、306、35-43)のOCHのNMRシフトと比較することにより決定した。
【0078】
2.LMW−P−OSFHA−2(DS−1)(GM−311101)の調製
蒸留水50mL中のFHA−2(MW53kDaのHA由来のFHA−2)(0.25g)にトリブチルアミン0.5mLを加え、上述した1と同様に処理することによってLMW FHA−2のTBA塩を調製した。結果として得られた塩(LMW FHA−2−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり6mol)のピリジン−三酸化硫黄錯体(0.4g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化LMW FHA−2を水に溶解し、1と同様に透析および凍結乾燥することによって生成物(300mg)を収率68%で得た。1H NMRで評価した硫酸化SD=1.0であった。
【0079】
3.LMW−P−OSMeHA(DS−1)(GM−111101)の調製
1と同様にして、TBA0.5mLおよび蒸留水50mL中のLMW MeHA(DS−1)(MW53kDaのHA由来)(0.5g)からLMW MeHAのTBA塩を調製した。結果として得られた塩(LMW MeHA−TBA)をDMF50mLに溶解し、所要の過剰(MeHA中の利用可能なヒドロキシ基1当量当たり6mol)のピリジン−三酸化硫黄錯体(0.8g)を加えた。40℃で3時間撹拌した後、水100mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール150mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化MeHAを水に溶解し、1と同様に透析および凍結乾燥することによって生成物(540mg)を62%の収率で得た。1H NMRにより硫酸化SD=1.0〜1.5であることが示された。
【0080】
4.LMW−P−OSFHA−1(DS−2)(GM−211201)の調製
蒸留水50mL中のFHA−1(MW53kDaのHA由来のFHA−1)(0.25g)にトリブチルアミン0.5mLを加え、上述した1と同様に処理することによってLMW FHA−1のTBA塩を調製した。結果として得られた塩(FHA−1−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり6mol)のピリジン−三酸化硫黄錯体(0.4g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化HAを蒸留水に溶解し、100mMのNaCl溶液に対し2日間透析(溶液を1日4回交換)した後、凍結乾燥することによって生成物(300mg)を70%の収率で得た。1H NMRで評価した硫酸化SD=1.0であった。
【0081】
5.LMW−P−OSFHA−2(DS−2)(GM−311201)の調製
蒸留水50mL中のFHA−2(MW53kDaのHA由来のFHA−2)(0.25g)にトリブチルアミン0.5mLを加え、上述した1と同様に処理することによってLMW FHA−2のTBA塩を調製した。結果として得られた塩(LMW FHA−2−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり6mol)のピリジン−三酸化硫黄錯体(0.4g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化LMW FHA−2を水に溶解し、1と同様に透析および凍結乾燥することによって生成物(310mg)を収率70%で得た。1H NMRで評価した硫酸化SD=1.0であった。
【0082】
6.LMW−P−OSMeHA(DS−2)(GM−111201)の調製
1と同様にして、TBAを0.5mLおよび蒸留水50mL中のLMW MeHA(DS−1)(MW53kDaのHA由来)(0.5g)からLMW MeHAのTBA塩を調製した。結果として得られた塩(LMW MeHA−TBA)をDMF50mLに溶解し、所要の過剰(MeHA中の利用可能なヒドロキシ基1当量当たり6mol)のピリジン−三酸化硫黄錯体(0.8g)を加えた。40℃で3時間撹拌した後、水100mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール150mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化MeHAを水に溶解し、1と同様に透析および凍結乾燥することによって生成物(560mg)を64%の収率で得た。1H NMRにより、硫酸化SD=1.0〜1.5であることが示された。
【0083】
7.P−OSFHA−1(DS−1)(GM−231101)の調製
蒸留水50mL中のFHA−1(MW950kDaのHA由来のFHA−1)(0.25g)にトリブチルアミン0.5mLを加えて、上述した1と同様に処理することによってFHA−1のTBA塩を調製した。結果として得られた塩(FHA−1−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり6mol)のピリジン−三酸化硫黄錯体(0.4g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化HAを蒸留水に溶解し、100mMのNaCl溶液に対し2日間透析(溶液を1日4回交換)した後、凍結乾燥することによって生成物(300mg)を68%の収率で得た。1H NMRで評価した硫酸化SD=1.0〜1.5であった。
【0084】
8.P−OSFHA−2(DS−1)(GM−331101)の調製
蒸留水50mL中のFHA−2(MW950kDaのHA由来のFHA−2)(0.25g)にトリブチルアミン0.5mLを加えて、上述した1と同様に処理することによってFHA−2のTBA塩を調製した。結果として得られた塩(FHA−2−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり6mol)のピリジン−三酸化硫黄錯体(0.4g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化FHA−2を水に溶解し、1と同様に透析および凍結乾燥することによって生成物(320mg)を70%の収率で得た。1H NMRで評価した硫酸化SD=1.0〜1.5であった。
【0085】
9.P−OSMeHA(DS−1)(GM−131101)の調製
1と同様にして、TBAを0.5mLおよび蒸留水50mL中のMeHA(DS−1)(MW950kDaのHA由来)(0.5g)からMeHAのTBA塩を調製した。結果として得られた塩(MeHA−TBA)を、DMF50mLに溶解し、所要の過剰(MeHA中の利用可能なヒドロキシ基1当量当たり6mol)のピリジン−三酸化硫黄錯体(0.8g)を加えた。40℃で3時間撹拌した後、水100mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール150mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化MeHAを水に溶解し、1と同様に透析および凍結乾燥することによって生成物(510mg)を収率60%で得た。1H NMRにより硫酸化SD=1.0〜1.5であることが示された。
【0086】
10.P−OSFHA−1(DS−2)(GM−231201)の調製
蒸留水50mL中のFHA−1(MW950kDaのHA由来のFHA−1)(0.25g)にトリブチルアミン0.5mLを加えて、上述した1と同様に処理することによってFHA−1のTBA塩を調製した。結果として得られた塩(FHA−1−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり6mol)のピリジン−三酸化硫黄錯体(0.4g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化HAを蒸留水に溶解し、100mMのNaCl溶液に対し2日間透析(溶液を1日4回交換)した後、凍結乾燥することによって生成物(280mg)を収率68%で得た。1H NMRで評価した硫酸化SD=1.0〜1.5であった。
【0087】
11.P−OSFHA−2(DS−2)(GM−331201)の調製
蒸留水50mL中のFHA−2(MW950kDaのHA由来のFHA−2)(0.25g)にトリブチルアミン0.5mLを加えて、上述した1と同様に処理することによってFHA−2のTBA塩を調製した。結果として得られた塩(FHA−2−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり6mol)のピリジン−三酸化硫黄錯体(0.4g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化FHA−2を水に溶解し、1と同様に透析および凍結乾燥することによって生成物(300mg)を収率69%で得た。1H NMRで評価した硫酸化SD=1.0〜1.5であった。
【0088】
12.P−OSMeHA(DS−2)(GM−131201)の調製
1と同様にして、TBAを0.5mLおよび蒸留水50mL中のMeHA(DS−1)(MW950kDaのHA由来)(0.5g)から、MeHAのTBA塩を調製した。結果として得られた塩(MeHA−TBA)を、DMF50mLに溶解し、所要の過剰(MeHA中の利用可能なヒドロキシ基1当量当たり6mol)のピリジン−三酸化硫黄錯体(0.8g)を加えた。40℃で3時間撹拌した後、水100mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール150mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化MeHAを水に溶解し、1と同様に透析および凍結乾燥することによって生成物(560mg)を収率64%で得た。1H NMRによりSD=1.0〜1.5であることが示された。
【0089】
13.LMW−F−OSFHA−1(DS−1)(GM−212101)の調製
蒸留水50mL中のFHA−1(MW53kDaのHA由来のFHA−1)(0.25g)にトリブチルアミン0.5mLを加えて、上述した1と同様に処理することによってLMW FHA−1のTBA塩を調製した。結果として得られた塩(FHA−1−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり12mol)のピリジン−三酸化硫黄錯体(0.8g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化HAを蒸留水に溶解し、100mMのNaCl溶液に対し2日間透析(溶液を1日に4回交換)した後、凍結乾燥することによって生成物(300mg)を収率71%で得た。1H NMRで評価したSD=1.5〜2.0であった。
【0090】
14.LMW−F−OSFHA−2(DS−1)(GM−312101)の調製
蒸留水50mL中のFHA−2(MW53kDaのHA由来のFHA−2)(0.25g)にトリブチルアミン0.5mLを加えて、上述した1と同様に処理することによってLMW FHA−2のTBA塩を調製した。結果として得られた塩(LMW FHA−2−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり12mol)のピリジン−三酸化硫黄錯体(0.8g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化LMW FHA−2を水に溶解し、1と同様に透析および凍結乾燥することによって生成物(260mg)を収率65%で得た。1H NMRで評価した硫酸化SD=1.5〜2.0であった。
【0091】
15.LMW−F−OSMeHA(DS−1)(GM−112101)
1と同様にして、TBAを0.5mLおよび蒸留水50mL中のLMW MeHA(DS−1)(MW53kDaのHA由来)(0.5g)からLMW MeHAのTBA塩を調製した。結果として得られた塩(LMW MeHA−TBA)をDMF50mLに溶解し、所要の過剰(MeHA中の利用可能なヒドロキシ基1当量当たり12mol)のピリジン−三酸化硫黄錯体(1.8g)を加えた。40℃で3時間撹拌した後、水100mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール150mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化MeHAを水に溶解し、1と同様に透析および凍結乾燥することによって生成物(480mg)を収率60%で得た。1H NMRにより硫酸化SD=1.5〜2.0であることが示された。
【0092】
16.LMW−F−OSFHA−1(DS−2)(GM−212201)の調製
蒸留水50mL中のFHA−1(MW53kDaのHA由来のFHA−1)(0.25g)にトリブチルアミン0.5mLを加えて、上述した1と同様に処理することによってLMW FHA−1のTBA塩を調製した。結果として得られた塩(FHA−1−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり12mol)のピリジン−三酸化硫黄錯体(0.8g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化HAを蒸留水に溶解し、100mMのNaCl溶液に対し2日間透析(溶液を1日4回交換)した後、凍結乾燥することによって生成物(300mg)を収率70%で得た。1H NMRで評価した硫酸化SD=1.5〜2.0であった。
【0093】
17.LMW−F−OSFHA−2(DS−2)(GM−312201)の調製
蒸留水50mL中のFHA−2(MW53kDaのHA由来のFHA−2)(0.25g)にトリブチルアミン0.5mLを加え、上述した1と同様に処理することによってLMW FHA−2のTBA塩を調製した。結果として得られた塩(LMW FHA−2−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり12mol)のピリジン−三酸化硫黄錯体(0.8g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化LMW FHA−2を水に溶解し、1と同様に透析および凍結乾燥することによって生成物(300mg)を収率68%で得た。1H NMRで評価した硫酸化SD=1.5〜2.0であった。
【0094】
18.LMW−F−OSMeHA(DS−2)(GM−112201)の調製
1と同様にして、TBAを0.5mLおよび蒸留水50mL中のLMW MeHA(DS−1)(MW53kDaのHA由来)(0.5g)からLMW MeHAのTBA塩を調製した。結果として得られた塩(LMW MeHA−TBA)をDMF50mLに溶解し、所要の過剰(MeHA中の利用可能なヒドロキシ基1当量当たり12mol)のピリジン−三酸化硫黄錯体(1.6g)を加えた。40℃で3時間撹拌した後、水100mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール150mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化MeHAを水に溶解し、1と同様に透析および凍結乾燥することによって生成物(550mg)を収率63%で得た。1H NMRにより硫酸化SD=1.5であることが示された。
【0095】
19.F−OSFHA−1(DS−1)(GM−232101)の調製
蒸留水50mL中のFHA−1(MW950kDaのHA由来のFHA−1)(0.25g)にトリブチルアミン0.5mLを加え、上述した1と同様に処理することによってFHA−1のTBA塩を調製した。結果として得られた塩(FHA−1−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり12mol)のピリジン−三酸化硫黄錯体(0.8g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化HAを蒸留水に溶解し、100mMのNaCl溶液に対し2日間透析(溶液を1日4回交換)した後、凍結乾燥することによって生成物(300mg)を収率68%で得た。1H NMRで評価した硫酸化SD=1.5であった。
【0096】
20.F−OSFHA−2(DS−1)(GM−332101)の調製
蒸留水50mL中のFHA−2(MW950kDaHA由来のFHA−2)(0.25g)にトリブチルアミン0.5mLを加えて、上述した1と同様に処理することによってFHA−2のTBA塩を調製した。結果として得られた塩(FHA−2−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり12mol)のピリジン−三酸化硫黄錯体(0.8g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化FHA−2を水に溶解し、1と同様に透析および凍結乾燥することによって生成物(310mg)を収率70%で得た。1H NMRで評価した硫酸化SD=1.5〜2.0であった。
【0097】
21.F−OSMeHA(DS−1)(GM−132101)の調製
1と同様にして、TBAを0.5mLおよび蒸留水50mL中のMeHA(MW950kDaのHA由来)(DS−1)(0.5g)からMeHAのTBA塩を調製した。結果として得られた塩(MeHA−TBA)をDMF50mLに溶解し、所要の過剰(MeHA中の利用可能なヒドロキシ基1当量当たり12mol)のピリジン−三酸化硫黄錯体(1.6g)を加えた。40℃で3時間撹拌した後、水100mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール150mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化MeHAを水に溶解し、1と同様に透析および凍結乾燥することによって生成物(500mg)を収率60%で得た。1H NMRにより硫酸化SD=1.5〜2.0であることが示された。
【0098】
22.F−OSFHA−1(DS−2)(GM−232201)の調製
蒸留水50mL中のFHA−1(MW950kDaのHA由来のFHA−1)(0.25g)にトリブチルアミン0.5mLを加えて、上述した1と同様に処理することによってFHA−1のTBA塩を調製した。結果として得られた塩(FHA−1−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり12mol)のピリジン−三酸化硫黄錯体(0.8g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化HAを蒸留水に溶解し、100mMのNaCl溶液に対し2日間透析(溶液を1日4回交換)した後、凍結乾燥することによって生成物(300mg)を収率69%で得た。1H NMRで評価した硫酸化SD=1.5〜2.0であった。
【0099】
23.F−OSFHA−2(DS−2)(GM−332201)の調製
蒸留水50mL中のFHA−2(MW950kDaのHA由来のFHA−2)(0.25g)にトリブチルアミン0.5mLを加えて、上述した1と同様に処理することによってFHA−2のTBA塩を調製した。結果として得られた塩(FHA−2−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり12mol)のピリジン−三酸化硫黄錯体(0.8g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化FHA−2を水に溶解し、1と同様に透析および凍結乾燥することによって生成物(300mg)を収率69%で得た。1H NMRで評価した硫酸化SD=1.5〜2.0であった。
【0100】
24.F−OSMeHA(DS−2)(GM−132201)の調製
1と同様にして、TBAを0.5mLおよび蒸留水50mL中のMeHA(DS−1)(MW950kDaのHA由来)(0.5g)からMeHAのTBA塩を調製した。結果として得られた塩(MeHA−TBA)をDMF50mLに溶解し、所要の過剰(MeHA中の利用可能なヒドロキシ基1当量当たり12mol)のピリジン−三酸化硫黄錯体(1.6g)を加えた。40℃で3時間撹拌した後、水100mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール150mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化MeHAを水に溶解し、1と同様に透析および凍結乾燥することによって生成物(550mg)を収率63%で得た。1H NMRにより硫酸化SD=1.5〜2.0であることが示された。
【0101】
25.P−OSBGHA(GM−431101)の調製
1と同様にして、TBAを0.5mLおよび蒸留水50mL中のBGHA(MW950kDaのHA由来)(0.5g)からBGHAのTBA塩を調製した。結果として得られた塩(BGHA−TBA)をDMF50mLに溶解し、所要の過剰(BGHA中の利用可能なヒドロキシル基1当量当たり6mol)のピリジン−三酸化硫黄錯体(0.8g)を加えた。40℃で3時間撹拌した後、水100mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール150mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成O−硫酸化BGHAを水に溶解し、1と同様に透析および凍結乾燥することによって生成物(500mg)を収率60%で得た。1H NMRにより硫酸化SD<1であることが示された。
【0102】
26.F−OSBGHA(GM−432101)の調製
1と同様にして、TBAを0.5mLおよび蒸留水50mL中のBGHA(MW950kDaのHA由来)(0.5g)からBGHAのTBA塩を調製した。結果として得られた塩(BGHA−TBA)をDMF50mLに溶解し、所要の過剰(BGHA中の利用可能なヒドロキシル基1当量当たり12mol)のピリジン−三酸化硫黄錯体(1.6g)を加えた。40℃で3時間撹拌した後、水100mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール150mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成O−硫酸化BGHAを水に溶解し、1と同様に透析および凍結乾燥することによって生成物(520mg)を収率61%で得た。1H NMRにより硫酸化SD<1であることが示された。
【0103】
27.P−OSBGHA(GM−411101)の調製
1と同様にして、TBAを0.5mLおよび蒸留水50mL中のBGHA(MW53kDaのHA由来)(0.5g)からBGHAのTBA塩を調製した。結果として得られた塩(BGHA−TBA)をDMF50mLに溶解し、所要の過剰(BGHA中の利用可能なヒドロキシル基1当量当たり6mol)のピリジン−三酸化硫黄錯体(0.8g)を加えた。40℃で3時間撹拌した後、水100mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール150mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成O−硫酸化BGHAを水に溶解し、1と同様に透析および凍結乾燥することによって生成物(500mg)を収率60%で得た。1H NMRにより硫酸化SD<1であることが示された。
【0104】
28.P−OSBGHA(GM−412101)の調製
1と同様にして、TBAを0.5mLおよび蒸留水50mL中のBGHA(MW53kDaのHA由来)(0.5g)からBGHAのTBA塩を調製した。結果として得られた塩(BGHA−TBA)をDMF50mLに溶解し、所要の過剰(BGHA中の利用可能なヒドロキシル基1当量当たり12mol)のピリジン−三酸化硫黄錯体(1.6g)を加えた。40℃で3時間撹拌した後、水100mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール150mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成O−硫酸化BGHAを水に溶解し、1と同様に透析および凍結乾燥することによって生成物(500mg)を収率60%で得た。1H NMRにより硫酸化SD<1であることが示された。
【0105】
III.蛍光SAGEおよびパルミトイル化SAGEの調製
a.LMW−F−OSFHA−1(DS−2)蛍光体複合体の調製
LMW−F−OSFHA−1(50mg)およびNHS(40mg)を水10mLに溶解し、次いで、DMF4mL中のAlaxa fluo@488(1mg)を加えた。次いで、pHを4.75に調節した後、固体形態のEDCI100mgを加えた。NaOH溶液を加えてpHを4.75に維持した。溶液をアルミニウム箔で覆って室温で一夜撹拌した。次いで、生成物を蒸留水、続いて、メタノール/水溶液(50/50、v/v)で透析(カットオフMW3500)することにより精製し、さらにゲル濾過カラム(Sephadex G−25)で精製した。さらにPD−10カラムで精製し、凍結乾燥によって40mgを得た。
【0106】
b.LMW−P−OSMeHA(DS−1)蛍光体複合体の調製
LMW−P−OSMeHA(50mg)およびNHS(40mg)を水10mLに溶解した後、DMF4mL中のAlaxa fluo@488(1mg)を加えた。次いで、PHを4.75に調節した後、固体形態のEDCI100mgを加えた。NaOH溶液を加えてpHを4.75に維持した。溶液をアルミニウム箔で覆って室温で一夜撹拌した。次いで、生成物を蒸留水、続いてメタノール/水溶液(50/50、v/v)で透析(カットオフMW3500)することにより精製し、さらにゲル濾過カラム(Sephadex G-25)で精製した。さらにPD−10カラムで精製し、凍結乾燥によって43mgを得た。
【0107】
c.パルミトイル化SAGEの調製
LMW−P−OSMeHA(50mg)をDMF50mLに溶解し、このDMF溶液を撹拌しながらトリエチルアミン(0.3mL)を加えた。5分後に塩化パルミトイル(0.5mL)を加えた。結果として得られた混合物を一夜撹拌し続けた。溶液を蒸発させ、残渣を蒸留水に溶解し、1日間透析し(水を4回交換した)、凍結乾燥することにより35mgを得た。
【0108】
IV.SAGEは、P−セレクチン、ヒト白血球エラスターゼ、およびRAGEとそのあらゆるリガンドとの相互作用の強力な阻害物質である。
材料:ポリクローナルヤギ抗ヒトRAGE、組換え型ヒトハイモビリティー・ボックス・プロテイン−1(HMGB−1)、組換え型ヒトP−セレクチン/Fcキメラ、組換え型ヒトRAGE/Fcキメラ、ヒトアズロシジン、およびポリクローナルヤギ抗ヒトアズロシジンを、R&D Systems(Minneapolis,MN)より購入した。ヒトS100bカルグラニュリンはCalbiochem(San Diego,CA)より入手した。終末糖化産物カルボキシメチルリジン−ウシ血清アルブミン(CML−BSA)は、MBL International(Woburn,MA)より入手した。U937ヒト単球細胞は、American Type Culture Collection(Manassas,VA)より入手した。Aタンパク、西洋ワサビペルオキシダーゼ結合ウサギ抗ヤギIgG、炭酸−重炭酸緩衝液、およびウシ血清アルブミンブロッカー(10×)は、Piercenet(Rockford,IL)より入手した。カルセインAM、ダルベッコ変法イーグル培地(DMEM)、エチレンジアミン四酢酸(EDTA)、ウシ胎児血清(FBS)、HEPES、非必須アミノ酸、ペニシリン/ストレプトマイシン/L−グルタミン溶液、RPMI−1640(L−グルタミン非含有)、および炭酸水素ナトリウムはInvitrogen(Carlsbad,CA)より入手した。高結合能96穴マイクロプレートは、Corning Life Sciences(Corning,NY)より入手した。他の明記されていない薬品はすべてSigma-Aldrich(St.Louis,MO)より購入した。
【0109】
細胞培養:U937単球を、10%加熱不活性化FBS、2mM L−グルタミン、1mM ピルビン酸ナトリウム、0.1mM MEM非必須アミノ酸、100単位/mL ペニシリン、および100mg/mL ストレプトマイシンを添加したRPMI−1640を用いて、加湿CO2(5%)−空気(95%)中、懸濁培養により37℃で培養した。1〜5代の細胞を用いて実験を行った。
【0110】
細胞結合アッセイ:U937単球とP−セレクチンまたはRAGEとの結合に対するSAGEの影響を、Aタンパク8μg/mL(50μg/ウェル)を含む0.2M炭酸−重炭酸緩衝液(pH9.4)でコーティングした高結合能マイクロプレートを用いて調査した。プレートを1%BSAを含むリン酸緩衝生理食塩水(PBS−BSA)で洗浄し、P−セレクチン−FcまたはRAGE−Fcキメラ(50μL、1μg含有)を各ウェルに分注して、それぞれ室温で2時間または4℃で一夜インキュベートした。インキュベート後、ウェルをPBS−BSAで洗浄した。20mMのHEPES緩衝液(125mM NaCl、2mMカルシウム、および2mMマグネシウム含有)で段階希釈(0〜1000μg/mL)したSAGE50μLを各ウェルに加え、室温で15分間インキュベートした。陰性対照として、カルシウムを捕捉することによって細胞との結合を阻止するように、選択したウェルに10mM EDTA50μLを加えた。インキュベーション完了後、U937細胞(105細胞/ウェル、製造業者の指示に従いカルセイン標識)50μLを各ウェルに加えて、プレートをさらに室温で30分間インキュベートした。次いで、ウェルをPBSで3回洗浄した後、トリス−TritonX−100含有緩衝液100μLを加えて、結合した細胞を溶解した。マイクロプレートリーダーを用いて494nmで励起、517nmで発光させて蛍光を測定した。
【0111】
固相結合分析:固相結合分析を用いて、RAGEとそのリガンドとの結合をSAGEが阻害する能力を調査した。RAGEとリガンドとの結合に及ぼすSAGEの影響を調査するために、ポリビニル96穴プレートを特定のリガンド(CML−BSA、HMGB−1、またはS100bカルグラニュリン)5μg/ウェルでコーティングした。プレートを4℃で一夜インキュベートした後、PBS−0.05%Tween−20(PBST)で3回洗浄した。これとは別に、RAGE−Fcキメラ(PBST−0.1%BSA中、0.5μg/mL含有)100μLを等量の段階希釈したSAGE(PBST−BSA中、0.001から1000μg/mL)と一緒に4℃でインキュベートした。翌日、RAGE−SAGE混合物各50μLを、リガンドをコーティングした各ウェルに移し、37℃で2時間インキュベートした。次いで、ウェルをPBSTで4回洗浄した。結合したRAGEを検出するために、抗RAGE抗体(0.5μg/mL)50μLを各ウェルに加え、混合物を室温で1時間インキュベートし、ウェルを再度PBSTで4回洗浄した。西洋ワサビペルオキシダーゼ結合二次抗体(抗体をPBSTで1:10000に希釈;50μL/ウェル)を加え、ウェルを室温で1時間インキュベートした後、PBSTで1回洗浄した。テトラメチルベンジジン色素原(TMB single solution色素原)50μLを加えて呈色反応を開始し、15分後、1NのHClを50μL加えることによって停止した。自動マイクロプレートリーダーを用いて450nmの吸光度を測定した。
【0112】
酵素的試験:陽性荷電PMNプロテアーゼHLEのSAGEによる阻害を評価するために、活性検定(Fryer A、Huang Y-C、Rao G、Jacoby D、Mancilla E、Whorton R、Piantadosi CA、Kennedy T、Hoidal J、選択的O−脱硫酸化により肺で薬理活性を維持する非抗凝固性ヘパリンが生成する(Selective O-desulfation produces nonanticoagulant heparin that retains pharmacologic activity in the lung.)、J Pharmacol Exp Ther 282:208-219、1997)を用いて、精製HLEが発色基質を切断する能力を測定した。HLE(100nM)をSAGE(1〜100nM)と一緒に0.5M HEPES緩衝液中で15分間インキュベートした。インキュベート後、エラスターゼ基質であるSuc−Ala−Ala−Val−p−ニトロアニリン(p−NA)を反応混合物に加えて最終濃度を0.3mMとした。405nmの吸光度を測定することにより、加水分解によるp−NAの遊離を15分間追跡した。SAGEが第XII因子または補体を活性化する能力を評価するために、汚染された市販のヘパリンの毒性をスクリーニングするために最近用いられているものと類似の活性検定を用いた(Kishimoto TK、Viswanathan K、Ganguly T、Elankumaran S、Smith S、Pelzer K、Lansing JC、Sriranganathan N、Zhao G、Galcheva-Gargova Z、Al-Hakim A、Bailey GS、Fraser B、Roy S、Rogers-Cotrone T、Buhse L、Whary M、Fox J、Nasr M、Dal Pan GJ、Shriver Z、Langer RS、Venkataranam G、Austen KF、Woodcock J、Sasisekharan R、臨床有害事象と接触系の活性化に関連する汚染ヘハ゜リン(Contaminated hepalin associated with adverse clinical events and activation of the contact system.)、N Engl J Med 358:2457-2467、2008;Guerrini M、Beccati D、Shriver Z、Naggi A、Viswanathan K、Bisio A、Capila I、Lansing JC、Guglieri S、Fraser B、Al-Hakim A、Gunay NS、Zhang Z、Robinson L、Buhse L、Nasr M、Woodcock J、Langer R、Venkataraman G、Linhardt RJ、Casu B、Torri G、Sasisekharan R、過硫酸化されたコンドロイチン硫酸が臨床有害事象に関連するヘパリンの汚染源である(Oversulfated chondroitin sulfate is a contaminant in hepalin associated with adverse clinical events.)、Nat Biotech 26:669-675、2008)。プールしたヒト血漿(5μL)をSAHA(0.1〜1000μg/mL)100μLと一緒に、Triton X−100含有0.05M HEPES中、5℃で5分間インキュベートした。D−シクロヒドロチロシル−Glyc−L−Arg−p−NAを0.5mMを加えて405nmの吸光度変化を追跡することにより、ハーゲマン因子に特異的なアミダーゼ活性を測定した(Silverberg M、Dunn JT、Garen L、Kaplan AP、ヒトハーゲマン因子の自己活性化。合成基質を用いた実証(Autoactivation of human Hageman factor. Demonstration using a synthetic substrate.)、J Biol Chem 255:7281-7286、1980)。D−Pro−Phe−Arg−p−NAを加えて、450nmの吸光度変化を追跡することにより、活性カリクレインに特異的なアミダーゼ活性を測定した。
【0113】
結果:検定の結果を表2に示す。SAGEはP−セレクチンの強力な阻害剤である。P−セレクチン糖タンパクリガンド−1(PSGL−1)を介してP−セレクチンに強力に接着するU937ヒト単球のコンペティターを介した置換を、蛍光標識細胞を用いて調査した。図3から、SAGEがU937とP−セレクチンとの結合を0.5μg/mLの50%阻止濃度(IC50)で阻害することがわかる。
【0114】
【表2】
【0115】
第2に、高度に硫酸化されたポリアニオンと同様、アルキル化および硫酸化されたSAGEは多形核白血球プロテアーゼの強力な阻害剤である。図4は、アルキル化および/またはフルオロアルキル化ならびに硫酸化されたSAGEがヒト白血球エラスターゼ(HLE)を驚くほど強力なIC50値で阻害することを示すものである。具体的には、アルキル化されていない、完全にO−硫酸化されたHA(F−OSHA)は、HLEに対するIC50が0.66nMである。修飾および硫酸化されたHA誘導体のIC50値は、部分O−硫酸化カルボキシメチル化HA(P−OSCHMHA)が1.89nM、部分O−硫酸化HA(P−OSHA)が1.97nM、部分O−硫酸化メチル化HA(P−OSMEHA;GM−131101)が3.46nMであった。
【0116】
第3に、SAGEは、RAGEの極めて強力な阻害剤である。SAGEは、RAGEおよびアンホテリン(HMGB−1)の相互作用をIC50が1.1μg/mLで阻害し(図5)、RAGEおよびS100カルグラニュリンの相互作用をIC50が60ng/mL(図6)で阻害し、RAGEとAGE産物であるカルボキシメチル−リジンBSAとの結合をIC50が44ng/mLで阻害する(図7)。これらの値は、ヘパリンおよび2−O,3−O脱硫酸化ヘパリンを用いて本発明者らが測定したRAGE−リガンド阻害に関する対応するレベルよりも5〜10倍強力である。
【0117】
第4に、SAGEは、ヒトケラチン生成細胞増殖の強力な阻害剤である。これらの検定においては、ヒト新生児表皮ケラチン生成細胞をSAGEまたは他の阻害剤の存在下および非存在下に培養し、染色液を加えることによって増殖を測定(存在する生細胞数に正比例して低くなる)した。図に示した吸光度値は、対照(相対吸光度を1.0とした)に対して標準化したものである。図8から、高分子量部分O−硫酸化HA(P−OSHA)および完全O−硫酸化HA(F−OSHA)を100μg/mLの濃度で添加した場合、ヘパリン誘導体である2−O,3−O脱硫酸化ヘパリン(ODSH)よりもケラチン生成細胞の増殖抑制に関し有効であったことがわかる。部分硫酸化カルボキシメチル化HA(P−OSCMHA)およびメチル化HA(P−OSMEHA;GM−131101)はケラチン生成細胞増殖の阻害活性がより低いことから、硫酸化度がより高い方が有利であることがわかる。図9から、本検定においては、全体的により高分子量の誘導体よりもより低分子量の(LMW)誘導体がケラチン生成細胞増殖の低下に関し有効であることがわかる。特に、LMW部分O−硫酸化MeHA(LMW−P−OMEHA;GM−111201)、部分O−硫酸化CMHA(LMW−P−OSCMHA)、および部分O−硫酸化53kDa HAそのもの(LMW−P−OSHA)はいずれも1μg/mLという低濃度においても対照よりも増殖を低下させている。
【0118】
V.酒さおよび炎症の治療に関するIn Vivo試験
材料および方法
薬品:GM−111101、GM−131101、GM−312101、およびGM−212101について評価した。培地はAmerican Type Culture Collection(ATCC)より購入した。EpiLife培地はInvitrogen(Madison,WI)より購入した。
【0119】
細胞:ヒト皮膚線維芽細胞(nHDF)はATCCより購入した。ヒト新生児皮膚ケラチン生成細胞(HEKn)はInvitrogen(Madison,WI)より入手した。
【0120】
細胞毒性:96穴平底マイクロプレートの培地100μLを含む各ウェルに4000個のnHDF細胞を播種し、5%CO2中、37℃で12時間インキュベートした。すべての培地を、各種硫酸化ヒアルロナン(HA)誘導体を含む完全培地で、各段の最終濃度が10、100、1000、10000、100000、1000000ng/mLとなるように交換した。48時間後、各ウェルにMTS(Promega,Madison,WI)20μLをピペットで分注し、細胞をさらに2時間インキュベートした。試料の490nmにおける吸光度を96穴プレートリーダーで測定した。
【0121】
マウスの皮膚刺激性試験:マウスの皮膚に対するGM−111101およびGM−212101の皮膚刺激性を判定するためにin vivo試験を実施した。それぞれ0.1mg/mLおよび1mg/mLの2種の異なる濃度で2種の被験試薬を調製した。10%ギ酸およびPBSをそれぞれ陽性および陰性対照として用いた(n=6)。過去に実験に使用したことがなく、試験開始よりも前に皮膚炎、外傷、または臨床に不利な徴候を示していないことが認められているBalb/cマウスを計画された試験条件に無作為に群分けした。動物の背部毛を、試料を塗布する少なくとも4時間前に電気バリカンですべて刈毛しておいた。被験物質を塗布する直前に、各マウスの被験部位の下側の区域に滅菌針を用いて皮膚に4本の平行な傷を付け、被験部位の上側の区域は無傷のままとした。麻酔下で2種類の被験溶液の試料0.5mLを被験部位全体に塗布し、皮膚の約2.5cm角の範囲にガーゼを二重にして載せた。この貼布にプラスチックを裏張りし、非反応性テープで覆い、被験部位全体を包帯で巻いた。動物をそれぞれのケージに戻した。試剤を施用してから24時間後、包帯および濡れた試験用ガーゼを取り除いた。被験部位を水道水で拭き、残留している被験化合物を除去した。化合物を塗布してから24および72時間後に被験部位の皮膚反応をFHSAが推奨するドレイズ評価基準に従い調査した。試験終了後、試験品の一次刺激指数(P.I.I.)を求めることになる。P.I.I.評点が5.00以上の物質が陽性とみなされることになり、この物質が皮膚の一次刺激物とみなされることになる。
【0122】
動物:市販のBalb/cマウスを、Univ.of Utah veterinary medicine department and vivarium承認の供給業者から購入した。受領書に従い規定の期間検疫を行った後、すぐに試験に用いることができる。
【0123】
LL37ペプチドおよびSAGEを注射した酒さモデル:皮膚の慢性疾患、特に、酒さが顔面に現れた場合は、患者の人生に忘れ難い傷跡を残す。我々の種は他人の外見を受け容れたり拒否したりし、普通とは違う外見を持つ、自分とは異なる者がいると思わず尻込みしてしまう。皮膚疾患は多くの場合生命に関わることはないが、普通の人には推し量れない形で生活を一変させてしまう。酒さは、このように生活を一変させてしまう病気の1つである。米国の人口の3%すなわち約1400万人の米国人が酒さを患っている、顔面の見た目が悪くなる一般的な皮膚疾患であり、普通は30〜50歳で発症する。主としてケルト系の白人が冒され、男性よりも女性の方が問題を抱えているようである。患者の3分の1以上は家族歴があり、それが遺伝病であることを強く示唆している。顔面紅潮や凸凹した鼻を伴う酒さは、飲酒のし過ぎであるというありがちな誤解を受けることから、特に汚名を着せられやすい病気である。酒さは幾つかの臨床表現型を示す。最も一般に知られている症状は、頬や鼻の毛細血管が拡張することによる一過性または持続性の顔面中心部の紅潮および紅斑を特徴とするものである。この表現型は、「赤ら顔」程度から、容易に認められる持続的な血管拡張に及ぶ。この亜型には有効な局所治療がない。その次に広く知られている症状は、顔の中心の凸状部に発生する丘疹および膿疱であり、紅斑の上に重なることが多い。生検によると、その皮膚には多くのPMNが浸潤している。酒さ性ざ瘡は、抗生物質の局所または全身投与によって経験的に治療が行われているが、皮膚感染症例との関連が明確ではない。マクロライドは、局所または全身のいずれの治療にも最も多く用いられており、その抗菌活性よりもむしろPMNの炎症部位への遊走能を遅延させる能力によって改善が得られる可能性がある。第3に、そしてより稀な表現型であることは幸いであるが、酒さにより、鼻の皮脂腺および結合組織の両方が過形成することによって鼻瘤が生じ、昔で言うところの「W.C.Fields」の鼻または「酔っぱらいの鼻(whiskey nose)」になる。この症状の治療には、肥厚化した組織を除去するために外科的に組織を切除するかまたはレーザー治療が必要となる。第4の表現型においては、酒さによって眼瞼の炎症を伴う眼の乾燥および痒み(眼瞼炎)、光過敏、かすみ目、および結膜炎が生じる場合がある。疾患が長引くと、角膜炎や、それどころか角膜瘢痕が生じる可能性がある。眼瞼および下眼瞼表面にあるマイボーム腺の炎症によって起こるこの表現型は一般的なものであり、臨床的には眼科医がよく診察する「眼球乾燥」症候群と重複する。肌の色が白く眼の色が明るい人の日光が当たる顔面領域のみに酒さが発生することは、この状態を引き起こす太陽放射の病原的意義を示している。
【0124】
最近になって酒さの病因が解明された(Yamasaki K、Di Nardo A、Bardan A、Murakami M、Ohtake T、Coda A、Dorschner RA、Bonnart C、Descargues P、Hovnanian A、Morhenn VB、Gallop RL、セリンプロテアーゼ活性の亢進とカテリシジンの増加が酒さによる皮膚の炎症を促進する(Increased serine protease activity and cathelicidin promotes skin inflammation in rosacea.)、Nat Med 13:975-980、2007;Bevins CL、Liu F-T、酒さ:皮膚本来の免疫がうまく機能していない?(Rosacea:skin innate immunity gone awry?)、Nat Med 13:904−906、2007)。Yamasakiらの見事な研究により、酒さを伴う皮膚には、カテリシジンおよびそのプロセシングプロテアーゼであるSCTEが高濃度で存在することが実証された。哺乳類の抗菌ペプチドの主要なファミリーであるカテリシジンは、多くの臓器の白血球および上皮細胞で発現し、ここで細菌に対する自然免疫応答を媒介する(Nizet V、Ohtake T、Lauth X、Trowbridge Jk、Rudisill J、Dorschner RA、Prestonjamasp V、Piraino J、Huttner K、Gallop RL、生得的な抗菌ペプチドが侵襲性の細菌感染から皮膚を防御する(Innate antimicrobial peptide protects the skin from invasive bacterial infection.)、Nature 414:454-457、2001)。カテリシジンはまた、血管増殖、PMN遊走、および創傷治癒のシグナルも伝達する(Bevins、ibid)。ヒトカテリシジンは、ケラチン生成細胞から18kDaのhCAP18プロペプチドとして分泌され、SCTEによって切断されて、C末端に活性を持つ抗菌ペプチドLL−37(37個のアミノ酸長および2個のN末端ロイシンより命名)となる。酒さを伴う皮膚には、不適切な高濃度のカテリシジンおよびSCTEの両方が確認されているが、刺激となる菌の侵入ははっきりしていない。初代ヒトケリチノサイテスを覆う培地にLL−37を添加すると、IL−8等の走化性サイトカインの産生が大幅に増強される。酒さを伴う皮膚に認められる濃度と類似の濃度でLL−37をマウスに皮内注射すると紅斑が生じ、PMNの真皮内浸潤が強力に刺激される。SCTEを皮内注射した場合も野生型マウスに皮膚の紅斑およびPMNの真皮内浸潤が認められるが、カテリシジン−/−マウスには認められない。したがって、酒さの特徴である炎症、過度の血管新生、および皮脂腺過形成を生じさせる炎症促進性の陽性荷電皮膚ペプチドが局所的に過剰発現することがこの疾患を媒介しているという考えを支持する一連の証拠が増えつつある。酒さ患者のほぼ全員から、両親に一定のパターンの反応性顔面紅潮や発赤があると聞き取ることができることから、酒さに遺伝的要因があることは臨床医には明らかである。
【0125】
酒さのモデルを作製するために、LL−37を48時間の間、12時間毎に皮内注射した。Yamasakiらが報告したように、このモデルの皮膚には、紅斑および多形核白血球(PMN)の顕著な皮内浸潤が生じた。Balb/cマウスを調査前に剃毛し、背部の皮膚を剥き出しにした。24時間後、剃毛した皮膚に、ビヒクル40μuL(リン酸緩衝生理食塩水PBS)、陽性荷電ペプチド(PBS中、濃度320μM)、SAGE(PBS中320〜1280μM)、または陽性荷電ペプチド+SAGE混合物(ペプチド+1〜4倍モル濃度のSAGE)を、31ゲージの針を用いて、無傷の表皮に水疱が隆起するように計画された方法で皮内注射し、それによって表皮下または真皮レベルに投与していることが認識されるようにした。注射用に選択するSAGEは、新たに合成した12種類のSAGEから選択した。これらは、生化学的試験において、ヒト白血球エラスターゼ(他の陽性荷電タンパク質として)の阻害剤としても、終末糖化産物受容体(RAGE)の4種類の一般的なリガンドによる活性化の拮抗剤としても、活性が極めて高いという試験結果が得られたものである。
【0126】
注射前にSAGEおよびペプチドをPBS中で一緒に混合し、注射前に室温で15分間インキュベートした。その後、12時間毎に反復注射を行った。最初の注射から48時間後(合計4回注射)、動物にペントバルビタール25mg/kgを腹腔内投与して軽麻酔を行った。マウスが睡眠状態になったら、皮膚の注射した部位を写真撮影して、紅斑および浮腫の重症度を視認できるように記録した。紅斑の強さを発赤評点(1〜5)で評価し、紅斑の範囲をノギスで測定した。次いで、6mm穴のパンチを用いて皮膚の注射した範囲を切除生検してヘマトキシリン−エオシン染色によって組織病理学的変化を検査するとともにミエロペルオキシダーゼ(MPO)活性を測定することによってPMN浸潤を評価した。皮膚表面の代表的な画像および各皮膚の組織像を高倍率のマイクロスコープで観察した。
【0127】
SAGEで局所治療した酒さモデル:Balb/cマウス背部のLL37を作用させる皮膚の範囲を予め剃毛した。次いで、SAGEを5%(有効エモリエント成分)を含むヒアルロナン−ベースのエモリエント剤またはヒアルロン酸ベースのエモリエント剤を単独でこの皮膚の範囲に12時間毎に局所塗布することを開始した。24時間後、ビヒクル40μL(PBS)または陽性荷電ペプチド(濃度320μM)を剃毛した皮膚の皮下に上述した方法で注射した。注射およびエモリエント剤の局所塗布をその後12時間毎に繰り返した。最初の注射から48時間後(合計4回注射)、動物を上述したように軽麻酔した。皮膚の注射した範囲の写真を撮影することによって紅斑および浮腫の重症度を視認できるように記録した。紅斑の強さを発赤評点(1〜5)として評価し、紅斑の範囲をノギスで測定した。次いで、注射した皮膚の部位を6mm穴のパンチを用いて切除生検し、H&E染色によって組織病理学的変化を検査するとともにミエロペルオキシダーゼ(MPO)活性を測定することによってPMN浸潤を評価した。
【0128】
SAGEの真皮への侵入:調査を行う前に、Balb/cマウス背部の皮膚の範囲を剃毛して剥き出しにした。12時間毎にSAGEを皮膚に局所塗布した。48時間後、動物を安楽死させて皮膚を生検した。次いで、皮膚の一部を蛍光顕微鏡で調査することによって、SAGEが皮膚に侵入した深さを測定した。
【0129】
クロトン油炎症モデル:PMNが媒介する皮膚炎症の他のモデルとしてクロトン油を用いた。クロトン油は、皮膚細胞のプロテインキナーゼCを活性化するフォルボールエステルを含有している。その結果として、皮膚細胞が大量のケモカインおよびケモタキシンを産生し、そのシグナル伝達によって血液中のPMNが流入する。活性化されたPMNは皮膚組織に紅斑および浮腫を生じさせる。クロトン油に誘発される炎症は、皮膚科で使用される抗炎症化合物のスクリーニングにおいて、PMNが媒介する皮膚炎症モデルとしてよく用いられている。
【0130】
このモデルを作製するために、クロトン油(Sigma-Aldrich,St.Louis,MO)をアセトンと混合して0.8%溶液とした。ピペットを用いてマウスの片方の耳介の各面に10μLを塗布し、他方の耳介は対照としてそのままにした。4、8、および24時間後、軟骨隆起より遠位の頂部付近の耳介の厚みを測定した。そして、対照の耳介の厚みからの変化を浮腫の指標とした。紅斑の強さを発赤評点(1〜5)として評価し、紅斑の範囲をノギスで測定した。24時間後に測定した後、マウスを安楽死させてすぐに耳介のパンチ生検(6mm穴パンチ)を実施し、重量を測定し、凍結させて−80℃で保管し、H&E染色によって組織病理学的変化を検査するとともにミエロペルオキシダーゼ(MPO)活性を測定することによってPMN浸潤を評価した。手順を標準化し、誤差を減らすために、すべての耳介に関する測定および生検を1人の検査員が行った。残りの耳介を取り除き、免疫組織化学用に包埋して凍結させた。
【0131】
ミエロペルオキシダーゼ(MPO)測定。各マウスに対し組織生検(直径6mm穴のパンチ)を即座に実施し、重量を測定し、−80℃で凍結させて保管した。組織のMPO活性を、Suzukiらの方法(Suzuki K、Ota H、Sasagawa S、Sakatani T、Fujikura T、ヒト多形核白血球中のミエロペルオキシダーゼ測定方法(Assay method for myeloperoxidase in human polymorphonuclear leukocytes.)、Anal Biochem 132:345-352、1983)をYoungら(Young JM、Spires DA、Bedord CJ、Wagner B、Ballaron SJ、De oung LM、局所的なアラキドン酸に対するマウス耳介の炎症応答(The mouse ear inflammatory response to tpical arachidonic acid.)、J Invest Dermatol 82:367-371、1984)によって改変されたものを用いて測定した。各マウスの生検組織を、ヘキサデシルトリメチル−アンモニウムブロミド(HTAB)0.5%を含む80mMのリン酸緩衝生理食塩水(PBS)(pH5.4)0.75mLに入れた。各試料を小型の実験室用Tissue Tearor Homogenizer Model 985-370(Biospec Products,Bartlesville,OK)で4℃で45秒間ホモジナイズした。ホモジネートを定量的に微小遠心管に移し、PBS中のHTABを0.75mLを加えた。試料1.5mLを4℃に維持しながら12000×gで15分間遠心分離した。結果として得られた上清の試料30uLをトリプリケートで96穴マイクロタイタープレートのウェルに加えた。MPO測定を行うため、80mMのPBS(pH5.4)100uL、0.22MのPBS(pH5.4)85uL、および0.017%の過酸化水素15uLを含む混合物200uLを各ウェルに加えた。8%ジメチルホルムアミド水溶液中の18.4mMのテトラメチルベンジジンHClを20uLを加えて反応を開始した。マイクロタイタープレートを37℃で3分間インキュベートした後、氷に載せた。1.46Mの酢酸ナトリウム30uLを加えて反応を停止した。MPO酵素活性を波長630nmの吸光度で評価した。MPO活性を光学濃度(OD)/生検として表した。
【0132】
統計解析。in vitro試験に関する実験はすべてトリプリケートで実施した。Aspin−Welch検定を用いて平均値を比較することにより試料の有意差を計算した。p<0.05を有意差ありと宣言した。
【0133】
結果
LL37ペプチドおよびSAGEを注射した酒さモデル:陽性荷電カテリシジンを直接中和すると皮膚内の炎症活性が阻害されるかどうかを確認するために、LL37単独、SAGE(GM−111101)単独、ビヒクル(PBS)単独、またはLL37およびSAGEの混合物をマウス背部の剃毛した範囲に、その後12時間毎に皮下注射した。48時間後にマウスを屠殺し、異なる処理群の肉眼解剖写真を撮影した(図10aおよび10b)。ヘマトキシリン−エオシン染色を用いた組織学的研究から、白血球の浸潤の増大および顕著な皮膚の浮腫が認められる一方、誘発直後にSAGEを投与すると皮膚の腫脹反応が阻止されることが示された(図10cおよび10d)。
【0134】
皮膚の評点の個々の結果を紅斑面積(図10f)および紅斑の発赤評点(図10g)で表した。48時間後、SAGE処理群は、紅斑面積が劇的に減少するとともに発赤評点が大幅に低下することが示された。注射の48時間後に組織のパンチ生検を行い、PMN浸潤の指標としてミエロペルオキシダーゼ活性を測定した。SAGEをLL−37ペプチドと同時投与するとMPO活性が大幅に(50%)低下した(図10e)。したがって、SAGEをLL−37ペプチドと同時に注射することによりLL−37カテリシジンペプチドの炎症活性が実質的に誘導された。このことは、SAGEがLL−37が媒介する炎症を阻害し、酒さの治療に有用であることを示唆している。
【0135】
SAGEによる酒さの局所治療モデル:SAGE(GM−111101)の局所治療を用いることにより、LL−37を作用させてから時間が経過した後に治療を行ってもペプチドに誘発される皮膚炎症を阻止できるのかどうかを試験した。これに従い、マウス背部の皮膚の範囲にLL−37を注射した直後にSAGEを塗布した。肉眼解剖写真は、LL−37を適用してから48時間後に強度の浮腫および紅斑が現れたことを示している(図11a、11h、および11i)。一方、SAGEを用いた局所治療を行うと、直後の治療(図11b)および12時間遅れてからの治療(図11c)の両方において、発赤および患部が大幅に減少した。H&E染色から、2つのSAGE治療群と比較して、白血球浸潤および皮膚の浮腫がはるかに高いことが示され、これは、SAGEが皮膚腫脹反応およびMPO活性の阻害剤であるという結果と一致していた(図11g)。これらの結果は、酒さによるカテリシジンを介した炎症を治療するために、SAGEを従来の薬学的に許容されるエモリエント剤中で局所的に適用してもよいことを示唆している。
【0136】
SAGEの真皮侵入:SAGEが真皮に侵入するレベルを確認する。SAGE化合物である蛍光体−GM−212101および蛍光体−GM−111101を被験品として使用し、Balb/cマウスの背部の皮膚の一部分に傷を付けて触れないようにして、蛍光化合物を0.1mg/mL、1mg/mL、および10mg/mLを塗布した。24時間後にマウスを屠殺し、皮膚の被験部位全体を切除し、自然光および長波長UV光条件下の両方で撮影した(図12)。自然光およびUV光条件下の両方で、皮膚の治療部位の内側および外側の両方に蛍光物質の層が認められた。SAGEの顕著な侵入による分布はマイクロメータースケールにまで及んでいた。
【0137】
細胞毒性および皮膚刺激性のin Vivo試験:SAGE誘導体(SAGEs)であるGM−131101、GM−312101、およびGM−212101の細胞毒性をnHDF細胞内で評価した。結果を図13aに示す。どの化合物も濃度10mg/mLまではnHEKにも無毒であることがわかった(図13b)。in vivo皮膚刺激性試験に関する異なる治療群のマウスの肉眼解剖写真を図13c〜13jに示す。被験物質の一次刺激指数を求めたところ、GM−111101およびGM−212101は両方とも0.00であった。マウスの皮膚に炎症は認められなかった(図14)。GM−111101およびGM−212101は両方とも細胞毒性が認められなかった。この試験から、すべてのSAGEの濃度閾値を求めることができた。16 CFR 1500のFHSA Regulationsの指針において、実験による評点が5.00未満の物質は皮膚の一次刺激物ではないと定めされているように、この試験条件下におけるこの被験試薬は一次皮膚刺激物ではないとみなされるであろう。これらの結果は、SAGE自体には皮膚刺激性がなく、炎症性皮膚疾患の治療に安全に用いることができることを示している。
【0138】
クロトン油炎症モデル:簡便で非常に再現性の高いPMN媒介皮膚炎症モデルとして、マウス皮膚へのクロトン油塗布を用いた。このモデルを用いてSAGEの抗炎症活性を試験した。異なる治療群のマウスの肉眼解剖写真を図15aおよび15bに示すとともに、治療および未治療の耳介の両方の耳介厚を測定して、5つの群すべてで比較した。個々の皮膚評点の結果を、傷付けた範囲および無傷の範囲の紅斑で表した。この結果から、SAGE(GM−111101)治療群の発赤および厚みが未治療群と比較して大幅に低下したことが示された(図15gおよび15h)。組織病理学検査により、クロトン油を塗布した耳介においては、浸潤白血球数の増加および顕著な真皮の浮腫が見られた一方で、誘発直後にSAGEを投与すると耳介腫脹反応が阻害される(これはビヒクルで処理したマウスに匹敵していた)ことがわかった。これらの組織学的発見を、測定値によってさらに確認した。クロトン油塗布後の耳介パンチ生検によってMPO活性も測定した。クロトン油塗布直後からSAGE治療を開始して4時間毎に実施したところ、MPO活性が有意に低下した(図15f)。これらの結果は、SAGEを酒さ以外の炎症性皮膚疾患の局所治療に用いることができることを示唆している。
【0139】
酒さモデルのヒアルロン酸(HA)による局所治療:前述のLL−37酒さモデルの局所治療を用いてSAGE(GM−111101)対HAの比較を行った。その結果から、HAの局所投与では炎症(図16b、16e、および7f)およびMPO活性(図16d)が軽減されないことがはっきりと示された。これとは逆に、SAGEは抗炎症性が非常に増強されており(図16c〜f)、炎症および酒さの阻害剤とみなすことができるであろう。これらのデータは、SAGEの薬理活性はヒアルロン酸には備わっておらず、ヒアルロナンに新規な修飾を施さなければならないことを示唆している。
【0140】
ラットの静脈内投与によるIn Vivo急性毒性試験:本試験の目的は、SAGEであるGM−111101およびGM−212101を静脈内投与によりラットに単回投与した場合の急性毒性の評価に加えて、GM−111101を7日間の間に1日1回、単回投与量を投与することによる毒性を評価することにあった。
【0141】
スプラーグドーリー(Sprague-Dawley)雌雄各3匹のラットから構成される1つの投与群に、GM−111101を3mg/kgを単回投与し、次いで、1週間後にGM−111101を10mg/kgを単回投与し、最後に、最後の単回投与から1週間後に、GM−111101を10mg/kgの1日1回投与を開始して合計7日間実施した。スプラーグドーリー雌雄各3匹のラットの2つの群には、GM−111101を30または100mg/kgを投与した。さらに、スプラーグドーリー雌雄各3匹のラットの2つの群には、GM−212101を30または100mg/kgを投与した。スプラーグドーリー雌雄各3匹のラットの2つの群には、塩化ナトリウム0.9%を注射して陰性対照群とした。すべての用量は、投与量を1mL/kgとし、尾骨静脈から静脈注射することによって投与した。直前に記録された体重に基づき投与量を計算した。
【0142】
急激に(単回)投与した動物にはいずれも、注射直後ならびに0日目の用量投与の2時間および4時間後にも同様に、臨床的毒性の明らかな徴候が認められた。さらに、すべての生存動物について、臨床的毒性の目に見える徴候について1日1回、1〜14日目まで観察した。1日1回、7日間投与したすべての動物について、臨床的毒性の目に見える徴候について1日1回、1〜14日目まで観察した。0日目(急激投与)または1日目(反復投与)の用量投与前、6または7日目、および14日目の終了前に体重を記録した。試験から14日目にそれぞれの生存動物について肉眼的剖検による評価を行った。試験途中に死亡した動物は、死亡率を観察した後、すぐに肉眼的剖検により検査した。
【0143】
GM−212101を30mg/kg投与した群の1匹の雌動物に、用量投与後の最初の2時間以内に、中等度の歩行異常、中等度の運動失調、および尿が赤みがかった橙色に変色するという臨床徴候が認められた。これらの所見は、軽い運動失調を除いて、4時間後の観察時間の時点では既に存在せず、残りの試験の間はずっとそのままであった。運動失調以外の所見は、用量投与の翌日にはもはや存在しなかった。さらに、GM−212101を100mg/kg投与した群の1匹の雄のラットが、用量投与後の最初の4分以内に死亡していた。すべての動物は試験期間中に体重が増加した。試験中に死亡したGM−212101を100mg/kg投与した群の1匹の動物の胸腺全体に直径約1〜2mmの暗赤色の病巣が観察されたことを除いて、視認できる病変は剖検では観察されなかった。
【0144】
この試験結果に基づくと、GM−111101は、3、10、30、および100mg/kgの急激な単回投与ならびに7日間の10mg/kgの反復投与を含む、評価を行ったどの用量においても毒性の徴候が見られなかった。したがって、ラットにGM−111101を静脈内投与した場合の最大無作用量(NOEL)は少なくとも100mg/kgとみなされる。GM−212101は、30および100mg/kgの用量で毒性または致死性の徴候が見られた。したがって、ラットにGM−212101を静脈内投与した場合のNOELは10mg/kgとみなされる。GM−111101のすべての用量において死亡が認められず、GM−212101を100mg/kg投与した動物の17%にしか死亡が認められなかったことから、ラットにGM−111101およびGM−212101を静脈投与した場合のLD50は100mg/kgを超えるとみなされる。この結果は、SAGEが疾患の全身および注射治療に安全に用いられることを示唆している。
【0145】
VI.加齢黄斑変性の治療に関するSAGEの調査。
活性化された補体およびRAGEが培養RPE細胞における血管形成および炎症促進シグナル伝達を誘発する:培養RPE細胞におけるRAGEの生物学を調査する実験を、ARPE−19ヒトRPE細胞株を用いて実施した。免疫ブロットに示すように(図17)、ARPE−19細胞は、細胞可溶化物中で45〜50kDaの範囲の少なくとも4種のRAGEのアイソフォームを発現し、これらのアイソフォームを条件培地に分泌する。AGE産物であるCML−BSAでコーティングされたプレートで細胞を増殖させた場合、4種類すべてのRAGEアイソフォームの発現が大幅に上方制御された(図17の免疫ブロットの右側を左側と比較)。RAGEが連結することによって転写因子NF−κBが活性化されるため、RAGEの発現が増加することによって炎症促進性のシグナル伝達が大幅に促進される。この系に非抗凝固性(nonanticoagulant)ヘパリンである2−O,3−O脱硫酸化ヘパリン(ODSH)を加えると、ARPE−19細胞上でのCML−BSAとRAGEとの相互作用が阻害されることにより、RAGE発現の上方制御が阻止された。こうすることにより、RAGEが連結することによって炎症促進性のRAGE自体の発現が「フィードフォワード」的に増大することが阻止されるであろう。
【0146】
AGE産物がRPE細胞死を誘導する能力を調査した。ARPE−19細胞を丸型カバーガラスでコンフルエントになるまで増殖させた後、新しい皿に移し、25μMのAGE−BSAに40時間曝露した。次いで、Molecular Probes Fixable Live/Dead Cell Stain Kit(L11101,Eugene,OR)を用いて細胞死の検査を行った。各パネルにカバーガラス全体および生/死細胞界面の拡大図を示す。核がDAPIによって赤色に染色され、死細胞が緑色に染色される。A.対照BSA。カバーガラスの縁部に死細胞が若干認められたが、界面に生および死細胞が散在している。B.AGE−BSA(25μM)。顕著な細胞死が縁部周囲に認められ、生および死細胞の境界がはっきりとしている。C.AGE−BSA(25μM)+ODSH(200μM)。ODSHによってある程度保護されるようであるが、顕著な細胞死が依然として起こっている。D.AGE−BSA(25μM)+P−OSMeHA(200μM)(GM−111101)。
【0147】
図18に示すように、Live/Dead Cell Stain kit(Molecular Probes)を用いた緑色染色により測定すると、培養ARPE−19細胞をAGEで処理することによって顕著な細胞死が誘導されている(図18B)。細胞をODSHと共にインキュベートすることにより細胞死が低減されるが(図18C)、対応する濃度のSAGA P−OSMeHA(GM−111101)ではほぼ完全に阻止された(図18D)。細胞死は、丸型カバーガラス上で培養した細胞の縁部から内側に向かって進行しているようであった。RAGEはRPE細胞内で顕著に発現し、他のヒト上皮細胞と同様に、基底膜上で選択的に発現するであろう。したがって、培地中においては、AGEは最初は縁部の単層の基底に位置するRAGEにのみ到達して溶解することができ、内側に進行すると予想される細胞死を相次いで起こさせるであろう。これとは対照的に、AGEが網膜色素上皮およびブルッフ膜の間に蓄積されるドルーゼン内にある場合は、基底に位置するRAGEにすぐに到達することとなり、加齢黄斑変性においていわゆる「地図状萎縮」を形成する、RPEの限局的な細胞死を媒介するAGE/RAGEシグナル伝達が最適化される。したがって、SAGEであるGM−111101は、AGEに誘導されるRPE細胞死をほぼ完全に阻止する。これらの結果は、失明を招く加齢黄斑変性等の重要な眼疾患の治療にSAGEが有効となり得ることを示唆している。
【0148】
SAGEは無毒性であり非抗凝固性である:O−硫酸化およびメチル化されたHA(P−OSMeHA)、完全O−硫酸化およびペンタフルオロプロピル化されたHA(F−OSFHA−1)ならびに完全O−硫酸化およびメチル化されたHA(F−OSMeHA)を、培養したヒト皮膚上皮細胞または線維芽細胞に適用し、細胞毒性アッセイ(CellTiter96(登録商標)Aqueous One assay,Promega)により調査すると、濃度が1mg/mLでさえもSAGEは増殖を阻害せず、細胞毒性も示さない。SAGEはまた、非抗凝固性でもない。低分子量硫酸化フルオロアルキル化HAは抗−Xaを示さず、抗IIa抗凝固活性が<0.2U/mgであり、これと比較すると、未分画ヘパリンの場合はそれぞれ150U/mgである。
【0149】
ヘパリンとは異なり、高度に帯電した多価陰イオン性高分子は、ハーゲマン因子(第XIIa因子)を活性化し、二次的にキニンを活性化する、内因性または接触凝固カスケードの強力な誘発物質である。SAGEの内因性凝固を刺激する能力(ハーゲマン因子の活性化)についてスクリーニングを行った。プールしたヒト血漿をヘパリンまたは低分子量(Lmw)硫酸化フルオロアルキル化HA(Lmw−OSFHA−1、Lmw−OSFHA−2)とインキュベートし、D−シクロヒドロチロシル−Gly−Arg−p−NAを基質として用いてアミド分解活性を測定した。第XII因子の活性化能力について試験すると、図19に示すように、低分子量(50kDa)SAGEは、炎症を薬理学的に阻害する濃度よりも10〜100倍高いSAGE濃度でさえも、市販の医療用ヘパリンよりもはるかに安全性が高いようである。
【0150】
本出願を通して様々な刊行物が参照される。本明細書に記載した化合物、組成物、および方法をより十分に説明するために、これらの刊行物の開示内容全体を本出願に参考として組み込むものとする。
【0151】
本明細書に記載した化合物、組成物、および方法の様々な修正および変形が可能である。本明細書に記載した化合物、組成物、および方法の他の態様は、本明細書に開示した化合物、組成物、および方法の詳細および実施を考察することによって明らかになるであろう。この詳細および実施例は例示的なものとみなされることを意図している。
【技術分野】
【0001】
関連出願の相互参照
本出願は、2008年4月4日に出願された米国特許仮出願第61/042,310号に基づく優先権を主張するものである。この出願の教示全体が本明細書に参考として組み込まれる。
【背景技術】
【0002】
背景
乾癬、皮膚炎、ざ瘡、酒さ、皮膚の光老化等の炎症性疾患およびRAGEが媒介するシグナル伝達が関与する数々の疾患によって世界中の人々が苦しめられている。これらの疾患を大局的に見ると、乾癬に悩んでいる人だけでも世界人口の2〜3%すなわち約1億2500万人に上ることがNational Psoriasis Foundationによって報告されている。これらの炎症状態は見た目が悪くなる場合もあるし、治療せずに放置すると深刻な健康上の問題が生じる可能性もある。
【0003】
従来から承認されているこれらの状態の治療は、UV光線療法、コルチコステロイドおよびグルココルチコイド、アシトレチン、シクロスポリン、ならびにメトトレキサートが関与するものであろう。しかしながら、これらの治療はそれぞれ、免疫抑制や肝疾患から、皮膚の菲薄化や先天性欠損症の誘発に至るまで、深刻な副作用を引き起こす可能性がある。これらの治療はある程度または全く無効であるため、その結果に関し患者に不満が残ることが多い。
【0004】
上述した治療に加えてヘパリン治療も試験的に検討されている。ヘパリンすなわち硫酸化された多糖類の用途は、従来、抗凝血剤にほぼ限られていたが、その抗炎症性についてはよく知られている。ヘパリンおよびその誘導体はこれらの炎症性疾患の治療にある程度の可能性を示している。特にヘパリンおよびその誘導体は、炎症カスケードにおける少なくとも3つの重要な現象を阻害する。まず第1に、ヘパリンは、白血球インテグリン、P−およびL−セレクチンに結合してこれらを阻害する。第2に、ヘパリンおよびその誘導体は、陽性荷電PMNプロテアーゼ、ヒト白血球エラスターゼ、およびカテプシンGに結合してこれらを阻害することで炎症カスケードを抑制するため、セレクチンを阻害する最初のヘパリン障壁から逃れたPMNがタンパク質を分解することによる組織損傷が低減される。第3に、ヘパリンおよびその誘導体は、終末糖化産物受容体(RAGE)とそのリガンドとの相互作用を強力に阻害する。
【発明の概要】
【発明が解決しようとする課題】
【0005】
ヘパリンおよびその誘導体がこれらの炎症性疾患を治療する可能性が示されているものの、ヘパリンおよびその誘導体を用いた治療には幾つかの主要な欠点がある。まず第1に、ヘパリンおよびその誘導体は豚から取り出したものであり、したがって、ウイルスの異種間移動の問題が起こる。第2に、ヘパリンの抗凝固性により、この化合物で治療された糖尿病患者が出血過多になる危険性がある。第3に、ヘパリンは、ヘパリンと陽性荷電タンパク質である血小板第4因子(PF−4)との複合体に対する抗体を産生する特定の個体に血小板減少症を誘発する可能性があり、その結果として、破局的な血小板凝集および全身性の逆説的な動静脈塞栓が起こる。したがって、炎症性疾患の治療に用いることが可能であると同時に、他の治療に見られる数々の副作用が回避される化合物を開発するという重要な要求が満たされていない。
【課題を解決するための手段】
【0006】
概要
本明細書においては、アルキル化およびフルオロアルキル化された半合成グリコサミノグリカンエーテル(本明細書においては「SAGE」と称する)の合成について記載する。硫酸化されたアルキル化およびフルオロアルキル化SAGEの合成についても記載する。本明細書に記載する化合物は、多くの治療および化粧用途ならびに多くの炎症性疾患および皮膚障害の治療に有用である。本発明の利点については、以下の記載においてある程度説明するので、その記載からある程度明らかになるか、または以下に記載する態様を実施することによって理解されるであろう。以下に記載する利点は、添付の特許請求の範囲に具体的に指摘する構成要素および組合せによって実現および達成されるであろう。前述した一般的な説明および以下の詳細な説明はいずれも例示および説明のみを目的とするものであって限定を意図するものではないことを理解されたい。
【0007】
添付の図面は以下に説明する幾つかの態様を例示するものであり、これらは本明細書に組み込まれ、その一部を構成するものとする。
【図面の簡単な説明】
【0008】
【図1】アルキル化およびフルオロアルキル化ヒアルロナンならびにその硫酸化誘導体を製造するための合成スキームを示すものである。
【図2】幾つかの例示的なSAGEの構造を示すものである。
【図3】部分O−置換およびメチル化されたHA(P−OSMEHAまたはGM−131201)によるP−セレクチンの阻害を示すものである。
【図4】アルキル化およびフルオロアルキル化ヒアルロナン(硫酸化もされている)を含む硫酸化ヒアルロナン誘導体によるヒト白血球エラスターゼの阻害を示すものである。
【図5】ハイモビリティー・グループ・ボックス・プロテイン−1(HMGB−1)としても周知のアンホテリンと固定化RAGEとの結合に対するP−OSMEHAによる阻害を示すものである。
【図6】S100bカルグラニュリンと固定化RAGEとの結合に対するP−OSMEHA(GM−131201)による阻害を示すものである。
【図7】カルボキシメチルリジン−BSA(CML−BSA)と固定化RAGEとの結合に対するP−OSMEHA(GM−131201)による阻害を示すものである。
【図8】高分子量ヒアルロナン誘導体(硫酸化もされているアルキル化およびフルオロアルキル化ヒアルロナンを含む)のケラチン生成細胞増殖に対する阻害を示すものである。
【図9】低分子量ヒアルロナン誘導体(硫酸化もされているアルキル化およびフルオロアルキル化ヒアルロナンを含む)のケラチン生成細胞増殖に対する阻害を示すものである。
【図10】SAGE(GM−111101)をLL−37と同時に注射した酒さモデルを示すものである。(a)LL−37を注射した皮膚部位ならびに(b)LL−37およびSAGEを同時に注射したモデルの肉眼解剖写真である。(c)LL−37を注射した皮膚試料をH&E染色した断面図である。(d)SAGEと混合したLL−37を注射した皮膚部位をH&E染色した断面図である。(e)LL−37のみ、SAGEを加えたLL−37、またはSAGEのみを注射した群のマウスの皮膚生検により、多形核白血球(PMN)の皮膚浸潤を、PMN酵素であるミエロペルオキシダーゼ(MPO)の活性によって測定したものである。LL−37のみまたはSAGEを加えたLL−37を注射したマウスの(f)紅斑面積および(g)紅斑評点である。
【図11】LL−37酒さモデルのSAGE(GM−111101)局所治療を示すものである。(a)LL−37を注射、(b)LL−37注射直後にSAGE治療、および(c)LL−37注射から12時間後にSAGE治療した皮膚部位の肉眼解剖写真を示すものである。(d)LL−37を注射した皮膚試料をH&E染色した断面図である。(e)LL−37を注射した直後にSAGE治療した皮膚部位をH&E染色した断面図である。(f)LL−37を注射した12時間後にSAGE治療した皮膚部位をH&E染色した断面図である。(g)SAGE治療計画の異なるLL−37注射モデルのMPO活性測定である。SAGEの局所塗布によって治療したLL−37酒さモデルの(h)紅斑面積および(g)紅斑評点を示すものである。
【図12】自然光下の外皮(SAGE1mg/mLで治療)(パネルa)および外皮の蛍光画像(パネルb);自然光下の内皮(パネルc)および蛍光条件下の内皮(パネルd)を示すものである。
【図13】HA誘導体のnHDF細胞増殖に与える影響(a)およびnHEKに与える影響(b)を示すものである。異なる濃度のGM−111101およびGM−212101で治療したマウスの肉眼解剖写真である。GM−111101を0.1mg/mL(e)、GM−111101を1mg/mL(f)、GM−111101を10mg/mL(g)、GM−212101を0.1mg/mL(h)、GM−212101を1mg/mL(i)、およびGM−212101を10mg/mL(j)を、無傷の範囲(c)およびギ酸で刺激した範囲(d)で比較した。
【図14】(a)傷を付けた範囲のSAGEの紅斑評点、(b)傷を付けた範囲のSAGEの浮腫評点、(c)無傷の範囲のSAGEの紅斑評点、(d)無傷の範囲のSAGEの浮腫評点を示すものである。
【図15】クロトン油炎症モデルを用いたSAGE治療を示すものである。クロトン油処理から4時間後の対照(CTL)群であり、CTL群の同じマウスの右(未処理)(パネルa)および左(クロトン油塗布)(パネルb)耳介を比較したものである。PBSを塗布した陰性対照(パネルc)、クロトン油の陽性対照(パネルd)、およびクロトン油の後にSAGE治療を行ったもの(パネルe)についてH&E染色を行ったものである。クロトン油陽性対照群の白血球浸潤および浮腫を確認した。ミエロペルオキシダーゼ(Myeloperoxidase)活性(パネルf)は、多形核白血球活性化の指標であり、SAGE治療後の耳介パンチについて測定したものである。パネルgおよびパネルhは、SAGE治療後の耳介の厚み(浮腫による)および耳介の発赤(炎症による)の変化である(p<0.05)。
【図16】(a)LL−37を注射した皮膚試料をH&E染色した断面図、(b)HAで治療したLL−37注射皮膚部位をH&E染色した断面図、(f)SAGE(GM−111101)で治療したLL−37注射皮膚部位をH&E染色した断面図、(g)HAおよびSAGEで治療したLL−37注射モデルのMPO活性測定、(h)紅斑面積の例、ならびに(g)HAおよびSAGE治療したLL−37酒さモデルの紅斑評点を示すものである。
【図17】ARPE−19細胞内におけるAGEに誘発されるRAGE発現が、AGE産物であるカルボキシメチルリジン−ウシ血清アルブミン(CML−BSA)上で増殖させることによって増大し、修飾ヘパリンによって阻害されることを示すものである。
【図18】対照(a)と比較すると、AGE産物であるCML−BSAがARPE−19細胞内の細胞死を誘導し(b)、これがODSHによって阻害され(c)、SAGE処理によってほぼ解消される(d)ことがわかる。
【図19】ヒト治療用抗凝固血漿ヘパリン濃度に近い濃度である0.4μg/mLにおいても、第XII因子を活性化するヘパリンとは異なり、LMW SAGEが第XII因子を活性化しないことを示している。
【発明を実施するための形態】
【0009】
詳細な説明
本化合物、組成物、および/または方法を開示および説明する前に、以下に説明する態様は、特定の化合物、合成方法、または使用そのものに限定されるものではなく、変形ももちろん可能であることを理解されたい。本明細書において用いられる専門用語も同様に、特定の態様を説明することのみを目的とするものであって、限定を意図するものではないことを理解されたい。
【0010】
本明細書およびそれに続く特許請求の範囲において言及される多くの用語は以下の意味を有するものと定義する。
【0011】
本明細書および添付の特許請求の範囲において用いられる単数形「1つの(a)」、「1つの(an)」、および「その(the)」には、文脈上、そうでないことが明確に指示されていない限り、複数の指示対象も包含されることに留意しなければならない。したがって、例えば、「製剤用担体(a pharmaceutical carrier)」に言及した場合は、この種の担体の2種以上の混合物等も包含される。
【0012】
「任意的な(optional)」または「場合により(optionally)」は、それに続いて記載される事象または状況が起こっても起こらなくてもよいことと、その記載には、当該事象または状況が起こる具体例も起こらない具体例も包含されることとを意味する。例えば、「場合により置換された低級アルキル」という語句は、この低級アルキル基が置換されていてもされていなくてもよいことと、この記載には非置換の低級アルキルおよび置換された低級アルキルの両方が包含されることとを意味している。
【0013】
本明細書および末尾の特許請求の範囲において、組成物または物品の特定の構成要素または成分の重量部に言及する場合は、重量部で表される当該組成物または物品中の当該構成要素または成分と他の任意の構成要素または成分との重量関係を表している。したがって、成分Xを2重量部および成分Yを5重量部含有する化合物中に、XおよびYは2:5の重量比で存在し、当該化合物中に含有されるさらなる成分とは無関係にこのような比で存在する。
【0014】
成分の重量パーセントは、そうでないことが明確に述べられていない限り、当該成分が含まれる配合物または組成物の総重量を基準とする。
【0015】
本明細書および末尾の特許請求の範囲において用いられる、化学種の残基(residue)とは、当該化学種から特定の反応スキームもしくはそれに続く配合により生成するかまたは化学的に生成する部分を指し、当該部分が実際にその化学種から得られたかどうかは関係ない。例えば、少なくとも1個の−OH基を含むヒアルロナンは式Y−OHで表すことができ、式中、Yは、当該ヒアルロナン分子の残り(すなわち残基)である。
【0016】
本明細書において用いられる「治療する(treat)」という用語は、既往症の症状を維持または軽減することとして定義される。本明細書において用いられる「予防(prevent)」という用語は、疾患または機能障害の1種またはそれ以上の症状が発生する可能性をなくすかまたは低下させることとして定義される。本明細書において用いられる「阻害(inhibit)」という用語は、本明細書に記載される化合物が、当該化合物の非存在下における活性と比較した場合に、同活性を完全に消失させるかまたは同活性を低下させる能力である。
【0017】
本明細書においては、アルキル化およびフルオロアルキル化されたヒアルロナンまたはその誘導体について記載する。一態様においては、ヒアルロナンのN−アセチル−グルコサミン残基の少なくとも1個の第1級C−6ヒドロキシルプロトンがアルキル基で置換されている。本明細書において用いられる「アルキル基」という用語は、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、t−ブチル、ペンチル、ヘキシル、ヘプチル、オクチル、デシル、テトラデシル、ヘキサデシル、エイコシル、テトラコシル等の、1〜24個の炭素原子を有する分岐または非分岐の飽和炭化水素基である。一態様においては、アルキル基は、C1〜C10分岐または直鎖アルキル基である。さらなる態様においては、アルキル基はメチルである。アルキル基は、置換されていても非置換であってもよい。アルキル基が置換されている場合、アルキル基上に存在する1個またはそれ以上の水素原子が、これらに限定されるわけではないが、アルキニル、アルケニル、アリール、ハライド、ニトロ、アミノ、エステル、ケトン、アルデヒド、ヒドロキシ、カルボン酸、アラルキル、またはアルコキシ等の1個またはそれ以上の基で置き換えられていてもよい。
【0018】
他の態様においては、ヒアルロナンのN−アセチル−グルコサミン残基の少なくとも1個の第1級C−6ヒドロキシルプロトンがフルオロアルキル基で置換されている。本明細書において用いられる「フルオロアルキル基」という用語は、1〜24個の炭素原子を有する分岐または非分岐の飽和炭化水素基であって、少なくとも1個の水素原子がフッ素で置換されているものである。特定の態様においては、フルオロアルキル基は、少なくとも1個のトリフルオロメチル基を含む。他の態様においては、フルオロアルキル基は、式−CH2(CF2)nCF3(式中、nは、1、2、3、4、5、6、7、8、9、または10の整数である)を有する。一態様においては、フルオロアルキル基は、−CH2CF2CF3または−CH2CF2CF2CF3である。
【0019】
本明細書においては、SAGEをアルキル化またはフルオロアルキル化する方法について記載する。一態様においては、SAGEは、(a)ヒアルロナンまたはその誘導体を、N−アセチル−グルコサミン残基の少なくとも1個の第1級C−6ヒドロキシルプロトンを脱プロトン化するのに十分な量の塩基と反応させ、そして(b)脱プロトン化されたヒアルロナンまたはその誘導体を、少なくとも1個の脱プロトン化された第1級C−6ヒドロキシル基をアルキル化またはフルオロアルキル化するのに十分な時間および濃度でアルキル化剤またはフルオロアルキル化剤と反応させることによって製造される。塩基性条件下ではグリコシド結合も開裂し、修飾過程においてより低分子量のヒアルロナン誘導体が生成する可能性もあることが当業者に理解されるであろう。この塩基性条件下では、酸が脱プロトン化されてカルボキシレートになることおよび第2級ヒドロキシル基が脱プロトン化されることと、これらの求核性部分は、それぞれ、平衡状態における相対的な存在量および陰イオン種の求核性に応じてその後に続くアルキル化に関与する場合があることも理解されるであろう。例えば、2−Oおよび/または3−Oヒドロキシルプロトンは、脱プロトン化およびアルキル化またはフルオロアルキル化され得る。その例を図1に示す。ここでRは、水素、アルキル基、またはアルキル基であってもよい。
【0020】
ヒアルロナン出発物質は、遊離酸またはその塩として存在してもよい。本明細書においては、ヒアルロナン出発物質の誘導体も使用してもよい。誘導体としては、アルキル化またはフルオロアルキル化ステップの前にヒアルロナンに任意の修飾を施したものが含まれる。本明細書においては、幅広い分子量のヒアルロナンを使用してもよい。一態様においては、アルキル化またはフルオロアルキル化前のヒアルロナンの分子量は10kDaを超える。他の態様においては、アルキル化またはフルオロアルキル化前のヒアルロナンの分子量は、25kDa〜1,000kDa、100kDa〜1,000kDa、25kDa〜500kDa、25kDa〜250kDa、または25kDa〜100kDaである。特定の態様においては、ヒアルロナン出発物質またはその誘導体は、動物起源に由来するものではない。これらの態様においては、ヒアルロナンは、細菌等の他の起源に由来するものであってもよい。例えば、組換えバシラス・サブティリス(B.subtilis)発現システムを用いてヒアルロナン出発物質を産生してもよい。
【0021】
まず最初にヒアルロナン出発物質またはその誘導体を、N−アセチル−グルコサミン残基の少なくとも1個の第1級C−6ヒドロキシルプロトンを脱プロトン化するのに十分な量の塩基と反応させる。選択される塩基は変化してもよい。例えば、本明細書においては、水酸化ナトリウムや水酸化カリウム等のアルカリ金属水酸化物を使用してもよい。塩基の濃度または量は、所望のアルキル化またはフルオロアルキル化度に応じて変化させてもよい。一態様においては、塩基の量は、ヒアルロナン出発物質またはその誘導体のN−アセチル−グルコサミン残基の第1級C−6ヒドロキシルプロトンの少なくとも0.001%を脱プロトン化するのに十分なものである。他の態様においては、塩基の量は、ヒアルロナン出発物質またはその誘導体のN−アセチル−グルコサミン残基の第1級C−6ヒドロキシルプロトンの0.001%〜50%、1%〜50%、5%〜45%、5%〜40%、5%〜30%、5%〜20%、10%〜50%、20%〜50%、または30%〜50%を脱プロトン化するのに十分なものである。溶液の塩基性が高くなるほど、鎖開裂反応が起こりやすくなるとともに、達成することができるアルキル化/フルオロアルキル化度が高くなることが理解される。例えば、ヒアルロナンには他のヒドロキシル基が存在する(例えば、2−OHおよび/または3−OHをアルキル化またはフルオロアルキル化される可能性もある)。一態様においては、ヒアルロナン上に存在するすべてのヒドロキシル基をアルキル化またはフルオロアルキル化してもよい。他の態様においては、ヒアルロナン上に存在するヒドロキシルプロトンの0.001%、0.01%、0.1%、1%、5%、10%、20%、30%、40%、50%、60%、70%、80%、90%、95%、100%、またはその任意の範囲を脱プロトン化し、続いてアルキル化またはフルオロアルキル化してもよい。
【0022】
ヒアルロナン出発物質またはその誘導体を塩基で処理した後、脱プロトン化されたヒアルロナンをアルキル化剤またはフルオロアルキル化剤と反応させることによってSAGEが生成する。アルキル化剤の例としては、これらに限定されるものではないが、ハロゲン化アルキルが挙げられる。臭化およびヨウ化アルキルが特に有用である。同様に、フルオロアルキル化剤としては、ハロゲン化フルオロアルキルを挙げることができる。本明細書においては、有機合成に慣用されているアルキル化剤およびフルオロアルキル化剤を使用してもよい。
【0023】
アルキル化およびフルオロアルキル化SAGEを作製するための例示的な合成手順を図1に示す。図1を参照すると、ヒアルロナン(HA)が塩基(例えば、NaOH)およびアルキル化剤(例えば、CH3I)で処理され、ヒアルロナンのN−アセチル−グルコサミン残基の第1級C−6ヒドロキシルプロトンがメチル化され、メチル化ヒアルロナン(MHA)が生成する。図1に、フルオロアルキル化剤(例えば、CF3(CF2)nCH2Br)を用いたフルオロアルキル化ヒアルロナン(FHA)を作製するための例示的な合成手順も示す。
【0024】
特定の態様においては、上述したアルキル化またはフルオロアルキル化SAGEを硫酸化することが望ましい。一態様においては、アルキル化またはフルオロアルキル化SAGEは、アルキル化またはフルオロアルキル化SAGEを硫酸化剤と反応させることによって硫酸化生成物を生成させることにより硫酸化される。硫酸化度は、部分硫酸化から完全硫酸化まで変化してもよい。一般に、アルキル化またはフルオロアルキル化ヒアルロナンまたはその誘導体上に存在する遊離ヒドロキシル基を硫酸化してもよい。一態様においては、少なくとも1個のC−2ヒドロキシルプロトンおよび/またはC−3ヒドロキシルプロトンが硫酸基で置換される。他の態様においては、硫酸化度は、アルキル化またはフルオロアルキル化SAGEの二糖単位当たり0.5、1.0、1.5、2.0、2.5、3.0、3.5、またはその任意の範囲である。一態様においては、アルキル化またはフルオロアルキル化SAGEを、1種またはそれ以上のヒドロキシルプロトンを脱プロトン化するために塩基で処理した後に、硫酸化剤を添加してもよい。硫酸化剤は、ヒドロキシル基または脱プロトン化されたヒドロキシル基と反応させることにより硫酸基を生成する任意の化合物である。SAGEの分子量は、反応条件に応じて変化してもよい。一態様においては、SAGEの分子量は、2kDa〜500kDa、2kDa〜250kDa、2kDa〜100kDa、2kDa〜50kDa、2kDa〜25kDa、または2kDa〜10kDaである。図1に、硫酸化されたアルキル化またはフルオロアルキル化SAGE(それぞれSMHAおよびSFHA)の例示的な合成を示す。
【0025】
図2に、幾つかの例示的なSAGEの構造を示す。各SAGEは、コードGM−XYSTZZで識別される:
Xは、アルキル基の種類であり(1=メチル、2=ペンタフルオロプロピル、3=ヘプタフルオロブチル、4=ベンジルグリシジルエーテル)、
Yは、HAのサイズであり(1=低、2=中、3=高)、
Sは、硫酸化度であり(1=部分、2=完全)、
Tは、アルキル化度であり(1=低、2=高)、
ZZ=ロット番号の連番01または02であり(02は01のバッチと同様に製造され、性状も同じである)。
【0026】
表1に、上で定義したコード体系による数種のSAGEの一覧を示す。
【0027】
【表1】
【0028】
一態様においては、SAGEのアルキル基はメチルであり、ヒアルロナンの少なくとも1個のC−2ヒドロキシルプロトンおよび/またはC−3ヒドロキシルプロトンが硫酸基で置換されている。他の態様においては、SAGEのアルキル基はメチルであり、ヒアルロナンの少なくとも1個のC−2ヒドロキシルプロトンおよび/またはC−3ヒドロキシルプロトンが硫酸基で置換されており、この化合物をアルキル化した後の分子量は2kDa〜200kDaである。この種の化合物の例が、図2に示すGM−111101である。
【0029】
本明細書に記載されたアルキル化およびフルオロアルキル化SAGEはいずれもその薬学的に許容される塩またはエステルであってもよい。薬学的に許容される塩は、遊離酸を適量の薬学的に許容される塩基で処理することによって調製される。代表的な薬学的に許容される塩基は、水酸化アンモニウム、水酸化ナトリウム、水酸化カリウム、水酸化リチウム、水酸化カルシウム、水酸化マグネシウム、水酸化第1鉄、水酸化亜鉛、水酸化銅、水酸化アルミニウム、水酸化第2鉄、イソプロピルアミン、トリメチルアミン、ジエチルアミン、トリエチルアミン、トリフルオロアミン、エタノールアミン、2−ジメチルアミノエタノール、2−ジエチルアミノエタノール、リジン、アルギニン、ヒスチジン等である。一態様においては、反応は、水を単独または水と混合可能な不活性有機溶媒との混合物中において約0℃〜約100℃の温度(室温等)で実施される。構造式Iの化合物対使用される塩基のモル比は、任意の特定の塩に所望される比が得られるように選択される。例えば、遊離酸出発物質のアンモニウム塩を調製する場合は、出発物質を約1当量の薬学的に許容される塩基で処理することによって中性塩を得てもよい。
【0030】
エステル誘導体は、典型的には、以下の実施例に例示するように、化合物の酸形態の前駆体として調製され、したがって、プロドラッグとすることができる。一般に、これらの誘導体は、メチルやエチル等の低級アルキルエステルとなるであろう。アミド誘導体−(CO)NH2、−(CO)NHR、および−(CO)NR2(式中、Rは、上に定義したアルキル基である)は、カルボン酸含有化合物をアンモニアまたは置換アミンと反応させることによって調製してもよい。エステルはまた、脂肪酸エステルであってもよい。例えば、パルミチン酸エステルが調製されており、エステラーゼで活性化されるプロドラッグとして代替的に使用してもよい。
【0031】
本明細書において記載されるSAGEを、生物システムまたは実体が許容できる任意の賦形剤と配合することによって医薬組成物を製造してもよい。このような賦形剤の例としては、これらに限定されるものではないが、水、ヒアルロン酸水溶液、生理食塩水、リンゲル液、デキストロース液、ハンクス液、および他の生理学的バランスのとれた塩の水溶液が挙げられる。不揮発性油、植物油(オリーブ油、ゴマ油等)、トリグリセリド、プロピレングリコール、ポリエチレングリコール、および注射可能な有機エステル(オレイン酸エチル等)等の非水性ビヒクルも使用してもよい。他の有用な配合物としては、カルボキシメチルセルロースナトリウム、ソルビトール、デキストラン等の粘度を高める薬剤を含む懸濁液が挙げられる。賦形剤はまた、等張性および化学的安定性を高めることができる物質等の添加剤も少量含んでいてもよい。緩衝剤の例としては、リン酸緩衝液、重炭酸緩衝液、およびトリス緩衝液が挙げられ、一方、防腐剤の例としては、チメロゾール、クレゾール、ホルマリン、およびベンジルアルコールが挙げられる。特定の態様においては、投与形態に応じてpHを変更してもよい。例えば、組成物のpHは約5〜約6であり、これは局所使用に好適である。さらに、医薬組成物は、本明細書に記載した化合物に加えて、担体、増粘剤、希釈剤、防腐剤、界面活性剤等を含んでいてもよい。
【0032】
医薬組成物はまた、本明細書に記載した化合物と組み合わせて使用される1種またはそれ以上の有効成分も含んでいてもよい。結果として得られる医薬組成物は、薬物および他の生物活性薬剤を、適用部位に接しているかまたは離れている組織に持続的かつ連続的に送達する系を提供してもよい。生物活性薬剤は、それが適用される生体系に局所または全身的な生物学的、生理学的、または治療効果を与えることができるものである。例えば、この薬剤は、数ある機能の中でも特に、感染または炎症の抑制および/または予防、細胞増殖および組織再生の亢進、腫瘍増殖の抑制、鎮痛剤としての作用、抗細胞接着の促進、歯槽骨および歯の喪失の低減、軟骨および体重を支える関節の変性の阻止、ならびに骨成長の促進作用を有するものであってもよい。さらに、本明細書に記載した化合物はいずれも薬学的に許容される化合物の2種以上の組合せを含んでいてもよい。この種の化合物の例としては、これらに限定されるものではないが、抗菌剤、抗炎症剤、麻酔剤等が挙げられる。これらの組成物を薬剤送達素子(drug delivery device)として使用するための方法を以下に詳述する。
【0033】
この医薬組成物は、当該技術分野において周知の技法を用いて調製してもよい。一態様においては、この組成物は、本明細書に記載したSAGEを薬学的に許容される化合物および/または担体と混和(admixing)させることによって調製される。「混和」という用語は、化学反応または物理的相互作用が起きないように2種類の成分を一緒に混合する(mix)こととして定義される。「混和」という用語には、当該化合物と薬学的に許容される化合物とを化学反応または物理的相互作用させることも含まれる。反応性を有する治療用薬物、例えば、求核性基を有するものに当該化合物を共有結合させてもよい。第2に、架橋した多糖中に、薬理活性を有する薬剤を非共有結合的に捕捉してもよい。第3に、静電または疎水性相互作用を用いて、本明細書中に記載した化合物内における薬学的に許容される化合物の維持を促進してもよい。
【0034】
特定の事例におけるSAGEの実際の好ましい量は、利用される特定の化合物、配合される具体的な組成、適用方式、ならびに具体的な位置および治療対象に応じて異なることになることが理解されるであろう。所与の宿主の投与量は、従来の検討を行い、例えば、適切な従来の薬理学的試験手順を用いて、例えば、対象化合物と周知の薬剤との活性の差を通例的に比較することによって決定してもよい。医師および製剤技術者(formulator)、医薬化合物の用量を決定する当業者であれば、推奨基準(Physicians Desk Reference,Barnhart Publishing(1999)に従い問題なく用量を決定するであろう。
【0035】
本明細書に記載する医薬組成物は、局所または全身療法のどちらが所望されるのか、および治療部位に応じて多くの方法で投与することができる。投与は、局所的に(点眼、膣内、直腸内、鼻腔内、経口、または皮膚に直接等)行ってもよい。局所投与製剤としては、軟膏剤、ローション剤、クリーム剤、ゲル剤、滴下剤、坐剤、噴霧剤、液剤、および散剤を挙げることができる。従来の医薬担体、水性、粉末、または油性基剤、増粘剤等が必要であるかまたは望ましい場合もある。投与はまた、エアゾールまたは乾燥微粉末を肺に直接吸入することによって行ってもよい。投与は、炎症または変性した関節の隙間に直接注射することによって行ってもよい。
【0036】
投与用製剤としては、無菌の水性または非水性の液剤、懸濁剤、および乳剤が挙げられる。非水担体の例としては、水、アルコール性/水性の溶液、乳濁液、または懸濁液(生理食塩水および緩衝化した媒質を含む)が挙げられる。開示した組成物および方法を副次的に使用する際に非経口投与用ビヒクルが必要な場合は、塩化ナトリウム溶液、リンゲルブドウ糖液、ブドウ糖および塩化ナトリウム、乳酸リンゲル液、または不揮発性油が挙げられる。開示した組成物および方法を副次的に使用する際に静脈投与用ビヒクルが必要な場合は、液体および栄養補給剤、電解質補給液(リンゲルブドウ糖液をベースとするものなど)等が挙げられる。防腐剤および他の添加剤、例えば、抗菌剤、抗酸化剤、キレート剤、不活性ガス等も存在してもよい。
【0037】
投薬は、治療すべき状態の重症度および応答性に依存するが、通常は、1日1回またはそれ以上となり、当業者が送達を中止すべきであると判断するまで、数日間から数ヶ月間治療過程を継続することとなる。当業者であれば、最適な用量、投薬方法、および反復速度を容易に決定することができる。
【0038】
本明細書に記載されるSAGEおよび医薬組成物は、薬物送達、小分子送達、創傷治癒、炎症性皮膚障害の治療、炎症性歯科疾患(inflammatory dental disorder)の治療、炎症性呼吸器疾患の治療、炎症性眼疾患の治療、熱傷治癒、および組織再生/工学に関連する様々な用途に使用してもよい。一態様においては、本明細書に記載するSAGEおよび組成物は、創傷治癒の改善を必要とする対象者に対しこのような改善を可能にする。本明細書に記載するSAGEおよび医薬組成物は生体適合性を有する材料から構成されているので、精製することなく任意の生体系の内部または表面に直接配置してもよい。SAGEを配置してもよい部位の例として、これらに限定されるものではないが、筋肉や脂肪等の軟組織;骨や軟骨等の硬組織;組織再生部位;歯周ポケット等の隙間;外科的切開またはそれ以外で形成されたポケットまたは空洞;口腔、膣腔、直腸、鼻腔、関節の隙間、眼の盲嚢等の本来の腔所;腹腔およびその中に収容されている臓器;ならびに内部または表面に化合物を配置してもよい他の部位(切り傷、掻き傷、熱傷部位等の皮膚表面の損傷を含む)が挙げられる。組織は、外傷または変性状態によって損傷する可能性があり、その替わりに、本明細書に記載したSAGEおよび組成物を、組織の外傷を防ぐために未損傷の組織に適用できることが見込まれている。SAGEは生分解性であってもよく、天然の酵素が除々に分解するように作用するであろう。SAGEの成分は、SAGEの成分が分解されて、生体系、例えば、細胞や組織等に吸収されることになるという意味で「生分解性」であってもよい。さらに、SAGE、特に、再水和していないものを生体系に適用して、対象部位から体液を吸収してもよい。
【0039】
創傷治癒の場合は、本明細書に記載したSAGEを注射により投与してもよい。SAGEがハイドロゲル薄膜の形態にある場合、多くの臨床使用には注射用ハイドロゲルが好ましく、その理由は主として3つある。まず第1に、注射用ハイドロゲルは、外傷部位において所望される任意の形状に成形することができるであろう。それは、初期のハイドロゲルはゾルまたは成形可能なパテとすることが可能であり、この系を複雑な形状で配置した後に架橋させることによって所望の寸法に適合させることができるためである。第2に、ハイドロゲルは、ゲル形成中に組織に接着することになり、結果として得られる表面の微小な凹凸によって生じる機械的な絡み合いによって組織−ハイドロゲル界面が強化されることになる。第3に、その場で架橋可能なハイドロゲルの導入が、針を用いるかまたは腹腔鏡法によって達成することができ、それによって、手術手技における侵襲が最小限になるであろう。
【0040】
乾癬、ざ瘡、アトピー性皮膚炎、酒さ、またはUV光に影響される光老化等の炎症性皮膚障害の場合、対象とする状態を予防または治療するために、SAGEをエモリエント剤の一部として局所適用してもよい。喘息、慢性閉塞性肺疾患、急性肺傷害、嚢胞性線維症等の呼吸器障害の場合は、気道被覆液に適合する水溶性等張性ビヒクル中にSAGEを溶解し、吸入用エアゾールとして肺または鼻腔に送達させてもよい。別法として、SAGEを微粉末に配合し、乾燥散剤として肺吸入してもよい。眼疾患の場合は、水性ビヒクルにSAGEを入れて点眼薬として局所適用するかまたは針によるかもしくは薬剤定速送達素子を植え込むかのいずれかによって眼に直接注入してもよい。歯周疾患等の歯科疾患の場合は、SAGEを洗口液の成分として添加するかまたは歯肉溝に直接適用するためのクリーム剤もしくは歯肉パック材に配合してもよい。
【0041】
SAGEはまた、糖尿病性血管もしくは腎臓障害または炎症性胃腸疾患等の全身性炎症性疾患を治療または予防するために、非経口的に静脈、筋肉、または皮下のいずれかに注射してもよい。同様に、炎症性および変形性関節症を治療するために関節内にSAGEを注射してもよい。SAGEはまた、炎症性腸疾患を治療するためにカプセル剤で経口投与するかまたは浣腸剤に配合して直腸内投与することにより送達してもよい。
【0042】
本明細書に記載したSAGEおよび組成物は、少なくとも1種の薬学的に許容される化合物の送達を必要とする患者に送達させてもよく、この送達には、この薬学的に許容される化合物を受容することができる少なくとも1つの組織に、本明細書に記載した1種またはそれ以上の組成物を用いて接触させることが含まれる。SAGEを、ヒトまたは非ヒト動物に対する治癒または治療的価値を有する多種多様な放出可能な生物活性物質の担体として使用してもよい。SAGEが担持することができるこのような物質の多くは上述したものである。本発明のゲルに混入させるのに好適な生物活性材料の中には、治療薬、例えば、抗炎症剤、解熱剤、ステロイド性および非ステロイド性抗炎症剤、ホルモン、成長因子、避妊薬、抗ウイルス剤、抗菌剤、抗真菌剤、鎮痛剤、催眠剤、鎮静剤、精神安定剤、抗痙攣薬、筋弛緩剤、局所麻酔剤、鎮痙薬、抗潰瘍薬、ペプチド性アゴニスト、交感神経作用薬、心血管作動薬、抗腫瘍剤、オリゴヌクレオチドおよびその類縁体等が含まれる。生物活性物質は、医薬有効量で添加される。
【0043】
一態様においては、本明細書に記載した硫酸化されたアルキル化およびフルオロアルキル化SAGEは、終末糖化産物受容体(RAGE)、P−セレクチン、またはヒト白血球エラスターゼの活性を阻害することができる。RAGEはヒトの皮膚で高度に発現し、皮膚線維芽細胞、樹状細胞、ケラチン生成細胞、内皮細胞、および単球上に存在している。皮膚が日光に曝されると、RAGEは、終末糖化産物(AGE)およびサイトカインである腫瘍壊死因子−αによって上方制御される。RAGEは、UVに誘起される光老化において顕著な役割を果たし、その場合、UVに誘導されるカルボキシメチルリジン(CML)等のAGE産物にRAGEが連結することによって、皮膚線維芽細胞による細胞外マトリックスの産生が刺激され、それによって皮膚の老化が促進される。乾癬の場合は、炎症の惹起が活性化Tリンパ球に決定的に依存するため、この疾患におけるRAGEの役割は一層顕著になる傾向にある。Tリンパ球はまた、ざ瘡およびアトピー性皮膚炎の機構においても重要な可能性がある。ざ瘡の場合、皮膚のCD3+およびCD4+Tリンパ球ならびにマクロファージが増加することによって毛包の導管内におけるケラチン生成細胞の過剰増殖が刺激され、毛包の導管が閉塞することによってざ瘡面皰が形成される。アトピー性皮膚炎の場合は、皮膚の抗原がTH2リンパ球を活性化することにより、インターロイキン−4(IL−4)やインターロイキン−13(IL−13)等のサイトカインが分泌され、その結果として、好酸球が皮膚に動員される。今度は、好酸球が主要塩基性タンパク質等の陽性荷電した毒素を放出し、アレルギー性皮膚疾患を生じさせる。したがって、本明細書に記載した化合物の強力なRAGE阻害活性は、これらに限定されるものではないが、ざ瘡、湿疹、アトピー性皮膚炎、乾癬、皮膚の光老化等の様々な皮膚疾患の治療に有用となる。
【0044】
成人期においては、RAGEが生体に必ずしも完全に有益なわけではない。悪性腫瘍はオートクライン因子としてアンホテリン(またはハイモビリティー・ボックス・グループ・プロテイン−1、HMGB−1)を分泌し、アンホテリンとRAGEとの相互作用を利用して原発腫瘍の増殖および転移を促進する。組換え型デコイ(可溶型RAGEまたはs−RAGE)を用いてRAGEを阻害すると、腫瘍増殖が減少するとともに転移が阻害される。敗血症の場合は、単球およびマクロファージがアンホテリンを分泌し、これが血管および他の炎症細胞上のRAGEと相互作用することにより、細菌性ショックの重症度が増大する。この相互作用をRAGEの抗体によって阻害することにより、重症敗血症における臓器障害が阻止される。成人期においては、RAGEは、PMN、単球、およびリンパ球の炎症部位への動員を促進する血管内皮細胞接着分子(vascular adhesion receptor)としても機能する。RAGEを阻害することにより、炎症細胞の流入が緩やかになる。このことは多発性硬化症の動物モデルで既に実証されており、血管内皮RAGEとs−RAGEとの競合的阻害により、活性化された脳炎惹起性Tリンパ球の中枢神経系への流入が阻止され、神経の炎症および変性の発症および進行が遅延される。
【0045】
RAGEはまた、PMN、単球、およびリンパ球から分泌される強力な炎症促進因子であるS100カルグラニュリンと呼ばれるカルシウム結合タンパク質ファミリーとも相互作用する。S100カルグラニュリンの上昇は、急性肺傷害および嚢胞性線維症患者の気道分泌物におけるPMN炎症の鋭敏なマーカーである。眼の場合は、加齢黄斑変性においてS100カルグラニュリンとRAGEとの相互作用が顕著な役割を果たし、結果として失明を招く。RAGEはまた、アルツハイマーにおけるβアミロイドペプチドおよびアミロイドタンパクのβシートとも結合する。RAGE−βシート線維の相互作用は、RAGEが関与する神経細胞死および炎症誘発を介して、アルツハイマー型認知症および全身アミロイドーシスにおける臓器障害を媒介する。
【0046】
RAGEは糖尿病においても顕著である。血中グルコースが上昇すると、グルコースのアルデヒド基が細胞内タンパク質のアミンにランダムに結合して共有結合付加体となる。PMNが産生する酸化剤である次亜塩素酸(HOCl)等の酸化剤の存在下においては、今度はこのグルコース部分が酸化される。この酸化されたグリコシル化タンパク質は終末糖化産物(AGE)として周知である。AGEもRAGE受容体に積極的に結合し、RAGEを介するシグナル伝達を惹起する。AGE−RAGEシグナル伝達は、血管内皮機能障害、創傷治癒遅延、およびコントロール状況の悪い糖尿病に特徴的なアテローム性動脈硬化症を加速させる原因となる。眼の場合は、AGE−RAGEシグナル伝達によって網膜微小血管が増殖し、糖尿病性網膜症および失明の原因となる。腎臓においては、AGE−RAGEシグナル伝達が、初期腎肥大、その後に、糖尿病性腎不全(糖尿病性腎症)を招く線維症の原因となる。同様に、AGE−RAGEシグナル伝達は内皮のアポトーシスを起こさせ、血管新生を阻害し、糖尿病皮膚潰瘍の治癒を遅らせる。
【0047】
RAGEを阻害する能力を有するSAGEは、炎症に特に有用な治療薬となる。RAGEは、子宮内において、成長を促進する核タンパクであるアンホテリンすなわちハイモビリティー・ボックス・グループ・プロテイン−1(HMGB−1)に結合する受容体として機能する。ここで、アンホテリン−RAGE相互作用は、神経系の発達に重要な増殖シグナル伝達を惹起する。成人期においては、RAGEは、血管壁細胞、神経組織、心筋細胞、単球およびマクロファージ、Tリンパ球、腎メサンギウム細胞、ならびに皮膚線維芽細胞、樹状細胞、およびケラチン生成細胞に発現する。したがって、一態様においては、本明細書に記載したSAGEおよび組成物は、これらに限定されるものではないが、癌、多発性硬化症、変形性関節症、嚢胞性線維症、鎌状赤血球貧血、炎症性心血管疾患、または炎症性心血管疾患、糖尿病性合併症等のRAGE関連疾患に帰属される様々な異なる病気によって生じる対象者の炎症を安全に軽減または予防するために使用することができる。
【0048】
一態様においては、SAGEおよび組成物を陰性荷電物質として投与することによってカテリシジン由来の陽性荷電した皮膚ペプチドに結合させて阻害し、それによって皮膚障害を治療または予防してもよい。例えば、活性カテリシジンペプチドが皮膚で過剰発現することによって発症することが周知の酒さ性ざ瘡として知られる皮膚状態を、本明細書に記載したSAGEを用いて治療または予防してもよい。SAGEを用いて治療または予防することができる皮膚疾患の例としては、これらに限定されるものではないが、酒さ、アトピー性皮膚炎(湿疹)、アレルギー性接触皮膚炎、乾癬、ヘルペス状皮膚炎、ざ瘡、糖尿病性皮膚潰瘍および他の糖尿病性創傷、熱傷(温熱熱傷の痛みの緩和を含む)、日焼け(日焼けの痛みの緩和を含む)、形成外科手術後の瘢痕予防、光線角化症、虫刺され、ツタウルシによるかぶれ、放射線皮膚炎/熱傷、皮膚治癒の亢進、ケロイド瘢痕の予防および治療、または脂漏性皮膚炎の治療が挙げられる。
【0049】
SAGEがRAGE活性および他の生物学的メカニズムを阻害する能力を有することから、SAGEには皮膚障害の治療または予防以外にも多くの治療用途がある。一態様においては、SAGEを、歯肉炎(歯周疾患)およびアフタ性潰瘍を治療するために歯科および口腔外科で使用してもよい。他の態様においては、SAGEを眼科用途、例えば、加齢性黄斑変性症、糖尿病性網膜症、眼球乾燥症候群、および他の炎症性結膜炎(inflammatory conjunctivitis)、虹彩炎、ブドウ膜炎、アレルギー性結膜炎の治療、白内障手術における消炎補助(anti-inflammatory aid)、または角膜炎症および瘢痕の予防等に使用してもよい。
【0050】
さらなる態様においては、尿生殖器用途(例えば、尿路感染症予防、膀胱および尿路上皮系の移行上皮癌の治療、間質性膀胱炎の治療、および性感染症予防のための膣潤滑/保護薬としての使用)にSAGEを使用してもよい。
【0051】
他の態様においては、嚢胞性線維症、気管支拡張症、鼻炎(アレルギー性および通年性の両方)、副鼻腔炎、肺気腫および慢性気管支炎(COPD)、急性肺傷害/成人型呼吸窮迫症候群、間質性肺線維症、SARS、喘息、ならびに呼吸器合胞体ウイルス等の多くの呼吸器疾患の治療にSAGEを使用してもよい。他の態様においては、SAGEは、いびきおよび閉塞型睡眠時無呼吸の予防および治療、HIV陽性または造血器腫瘍を有する患者等の免疫不全宿主の一般的な呼吸器病原体による感染(ストレプトコッカス・ニューモニエ(Streptococcus pneumoniae)、ヘモフィルス・インフルエンザ、ブドウ球菌属(Staphylococcus)、肺炎マイコプラズマ・ニューモニエ(Mycoplasma pneumoniae)、肺炎クラミジア(Chlamydial pneumonia)、グラム陰性腸内細菌による感染)の予防、または中耳炎の予防および処置に用いることができる。
【0052】
SAGEは、心血管用途(例えば、急性冠症候群またはアテローム性動脈硬化症の治療または予防);血液学/腫瘍学的用途(例えば、鎌状赤血球貧血の予防および治療;転移性疾患の予防および治療;ならびに悪性腫瘍による凝固亢進状態の予防(トルソー症候群));感染症の治療(例えば、脳血管閉塞症候群(cerebral vascular occlusive syndrome)、および熱帯性マラリアにおける腎炎、黄熱病、デング熱、全身性敗血症(systemic sepsis)、ならびに標的細胞のウイルス融合および感染を防ぐためのHIVの補助療法);胃腸疾患の治療(例えば、潰瘍性大腸炎、腸のクローン病、痔核、ならびに胃および食道のストレス潰瘍の予防);リウマチおよび免疫疾患の治療(例えば、変形性関節症、関節リウマチ、全身性エリテマトーデスの予防および治療、血管神経性浮腫、シェーグレン症候群、全身性硬化症、全身性アミロイドーシス、および全身性肥満細胞症の予防および治療);腎疾患(例えば、糖尿病性腎症および糸球体腎炎の予防および治療);ならびに神経系疾患(例えば、多発性硬化症およびアルツハイマー型認知症)に使用してもよい。
【0053】
本明細書に記載したSAGEおよび組成物は、他の関連する療法よりも安全である。例えば、ヘパリンおよび他の硫酸化多糖は動物および臨床研究の両方において糖尿病性合併症を減少させることができ、糖尿病性腎症に特に有効である。しかしながら、抗凝固性を有するヘパリンは、出血の危険性が過度に高くなることから、一般臨床場面で糖尿病性合併症の予防に使用することはできない。本明細書に記載したSAGEおよび組成物は、以下の実施例に示すが、長期治療に関する重要な考慮事項である抗凝固活性が低い。さらに、このことも実施例で示すが、SAGEには毒性がほとんどまたは全くない。
【実施例】
【0054】
以下の実施例は、本明細書に記載および特許請求した化合物、組成物、および方法を作製および評価する方法を当業者に十分に開示および説明するために述べるものであるが、これは単なる例示を目的とするものであって、本発明者らが本発明者らの発明と見なすものの範囲を限定することを意図するものではない。数値(例えば、量や温度等)が確実に正確になるように努めたが、ある程度の誤差およびずれがあることは考慮されたい。特段の指定がない限り、部は重量部、温度は℃単位であるかまたは周囲温度であり、圧力は大気圧付近である。反応条件、例えば、成分の濃度、所望の溶媒、溶媒混合物、温度、圧力、ならびに記載した処理により得られる生成物の純度および収率を最適化するのに用いることができる他の反応範囲および条件には多くの変形および組合せがある。このような工程条件を最適化するためには妥当かつ日常的な実験のみが必要であろう。以下に記載する化合物は、上述および図2に示すコード番号で識別される。
【0055】
I.アルキル化HA誘導体の合成
A.メチルHA(DS−2)の調製
ヒアルロン酸(HA、Novozymes Biopolymers、950kDa)(2.0g)を100mLビーカー内でNaOH(40%w/v)20mLに溶解し、混合物を室温(rt)で2時間撹拌した。結果として得られた粘性液体を、イソプロパノール100mLを含む400mLビーカーに移して混合を継続した。撹拌した混合物にヨードメタン10mLを加え、混合物をrtで24時間撹拌した。結果として得られた懸濁液を濾過して粗生成メチル化HA生成物を回収した。この粗生成MeHAを蒸留水250mLに溶解し、溶液のpHを約7.0に調節し、溶液を蒸留水に対し24時間透析した。この間、外側の水浴を4回交換した。透析したMeHA生成物を凍結乾燥することによりメチルHA1.25gを綿状の網目状物として得た。1H NMR(D2O,δ):1.85(s,3H,NCH3),3.20−3.80(m,10H,OCH+OCH3)。置換度(SD)を1H NMRで測定したところ、SD=2すなわち二糖単位当たりのメチル基が平均2個と見積もられた。SD=[(メチルHAのδ3.20−3.8の積分値)−(HAのδ3.20−3.8の積分値)]/(NCH3の1.85の積分値)。これは、第1級ヒドロキシルおよび少なくとも1個の第2級ヒドロキシル基の両方がこの工程により修飾されたことを示唆している。
【0056】
B.メチルHA(DS−1)の調製
ヒアルロン酸(HA、Novozymes Biopolymers、950kDa)(2.0g)を100mLビーカー内でNaOH(40%w/v)20mLに溶解し、混合物を室温(rt)で2時間撹拌した。結果として得られた粘性液体を、イソプロパノール100mLを含む400mLビーカーに移して混合を継続した。撹拌した混合物にヨードメタン4mLを加え、混合物をrtで24時間撹拌した。結果として得られた懸濁液を濾過して粗生成メチル化HA生成物を回収した。この粗生成MeHAを蒸留水250mLに溶解し、溶液のpHを約7.0に調節し、溶液を蒸留水に対し24時間透析した。この間、外側の水浴を4回交換した。透析したMeHA生成物を凍結乾燥することによりメチルHA1.25gを綿状の網目状物として得た。1H NMR(D2O,δ):1.85(s,3H,NCH3),3.20−3.80(m,10H,OCH+OCH3)。置換度(SD)を1H NMRで測定したところ、SD=1すなわち二糖単位当たりメチル基が平均1個と見積もられた。SD=[(メチルHAのδ3.20−3.8の積分値)−(HAのδ3.20−3.8の積分値)]/(NCH3の1.85の積分値)。このことは、第1級ヒドロキシルおよび少なくとも1個の第2級ヒドロキシル基がこの工程により修飾されたことを示唆している。
【0057】
C.FHA−2(DS−1)の調製
2,2,3,3,4,4−ヘプタフルオロ−1−ブタノール1.0g(5mmol)を含む25mLフラスコに三臭化リン1.3mL(7.5mmol)をゆっくりと加えた。この混合物を60℃で30分間撹拌した後、飽和炭酸水素ナトリウム溶液(15mL)をゆっくりと加えて反応を停止した。この水溶液をジクロロメタン15mLずつで3回抽出し、臭化ヘプタフルオロブチルを含む有機層を濃縮し、残渣を次のステップでさらなる生成に用いた。
【0058】
HA(2.0g)を100mLビーカー内でNaOH(40%w/v)20mLに溶解し、混合物を室温(rt)で2時間撹拌した。結果として得られた粘性液体を、イソプロパノール100mLを含む400mLビーカーに移して混合を継続した。撹拌した混合物にイソプロパノール10mL中の粗生成臭化ヘプタフルオロブチルを加え、混合物をrtで24時間撹拌した。結果として得られた懸濁液を濾過して粗生成FHA−2生成物を回収した。この粗生成FHA−2を蒸留水250mLに溶解し、溶液のpHを約7.0に調節し、溶液を蒸留水に対し24時間透析した。この間、外側の水浴を4回交換した。透析したFHA−2生成物を凍結乾燥することにより、FHA−2として設計された1.5gのFHA−2を綿状の網目状物として得た。1H NMR(D2O,δ):1.82(s,3H,NCH3),3.15−3.80(m,8H,OCH+OCH2)。19F NMR(D2O,δ):−115.3,−120.8。1H NMRで測定した置換度(SD)は1.0であった。SD=[(FHA−2のδ3.15−3.80の積分値)−(HAのδ3.20−3.8の積分値)]/[(NCH3の1.82の積分値)×(2/3)]。
【0059】
D.FHA−2(DS−2)の調製
2,2,3,3,4,4−ヘプタフルオロ−1−ブタノール3.0g(15mmol)を含む25mLフラスコに三臭化リン2.5.mL(16mmol)をゆっくりと加えた。混合物を60℃で30分間撹拌した後、飽和炭酸水素ナトリウム溶液(15mL)をゆっくりと加えて反応を停止させた。この水溶液をジクロロメタン15mLずつで3回抽出し、臭化ヘプタフルオロブチルを含む有機層を濃縮し、残渣を精製することなく次のステップに使用した。
【0060】
HA(2.0g)を100mLビーカー内でNaOH(40%w/v)20mLに溶解し、混合物を室温(rt)で2時間撹拌した。結果として得られた粘性液体を、イソプロパノール100mLを含む400mLビーカーに移して混合を継続した。撹拌した混合物にイソプロパノール10mL中の粗生成臭化ヘプタフルオロブチルを加え、混合物をrtで24時間撹拌した。結果として得られた懸濁液を濾過して粗生成FHA−2生成物を回収した。この粗生成FHA−2を蒸留水250mLに溶解し、溶液のpHを約7.0に調節し、溶液を蒸留水に対し24時間透析した。この間、外側の水浴を4回交換した。透析したFHA−2生成物を凍結乾燥することにより、FHA−2として設計された1.5gのFHA−2を綿状の網目状物として得た。1H NMR(D2O,δ):1.82(s,3H,NCH3),3.15−3.80(m,8H,OCH+OCH2)。19F NMR(D2O,δ):−115.3,−120.8。1H NMRで測定した置換度(SD)は2.0であった。SD=[(FHA−2のδ3.15−3.80の積分値)−(HAのδ3.20−3.8の積分値)]/[(NCH3の1.82の積分値)×(2/3)]。
【0061】
E.FHA−1(DS−1)の調製
2,2,3,3,−ペンタフルオロ−1−プロパノール1.0g(5mmol)を含む25mLフラスコに三臭化リン1.3mL(7.5mmol)をゆっくりと加えた。混合物を60℃で30分間撹拌した後、飽和炭酸水素ナトリウム溶液(15mL)をゆっくりと加えて反応を停止した。この水溶液をジクロロメタン15mLずつで3回抽出し、臭化ヘプタフルオロブチルを含む有機層を濃縮し、残渣を精製することなく次のステップで使用した。
【0062】
HA(2.0g)を100mLビーカー内でNaOH(40%w/v)20mLに溶解し、混合物を室温(rt)で2時間撹拌した。結果として得られた粘性液体を、イソプロパノール100mLを含む400mLビーカーに移して混合を継続した。撹拌した混合物にイソプロパノール10mL中の粗生成臭化ヘプタフルオロブチルを加え、混合物をrtで24時間撹拌した。結果として得られた懸濁液を濾過して粗生成FHA−1生成物を回収した。この粗生成FHA−1を蒸留水250mLに溶解し、溶液のpHを約7.0に調節し、溶液を蒸留水に対し24時間透析した。この間、外側の水浴を4回交換した。透析したFHA−1生成物を凍結乾燥することにより、FHA−1として設計された1.5gのFHA−1を綿状の網目状物として得た。1H NMR(D2O,δ):1.80(s,3H,NCH3),3.15−3.80(m,8H,OCH+OCH2)。19F NMR(D2O,δ):−113.6,−118.0。1H NMRで測定した置換度(SD)は1.0であった。SD=[(FHA−1のδ3.15−3.80の積分値)−(HAのδ3.20−3.8の積分値)]/[(NCH3の1.80の積分値)×(2/3)]。
【0063】
F.FHA−1(DS−2)の調製
2,2,3,3,−ペンタフルオロ−1−プロパノール3.0g(15mmol)を含む25mLフラスコに三臭化リン3mL(18mmol)をゆっくりと加えた。混合物を60℃で30分間撹拌した後、飽和炭酸水素ナトリウム溶液(15mL)をゆっくりと加えて反応を停止した。この水溶液をジクロロメタン15mLずつで3回抽出し、臭化ヘプタフルオロブチルを含む有機層を濃縮し、残渣を精製することなく次のステップで使用した。
【0064】
HA(2.0g)を100mLビーカー内でNaOH(40%w/v)20mLに溶解し、混合物を室温(rt)で2時間撹拌した。結果として得られた粘性液体を、イソプロパノール100mLを含む400mLビーカーに移して混合を継続した。撹拌した混合物にイソプロパノール10mL中の粗生成臭化ヘプタフルオロブチルを加え、混合物をrtで24時間撹拌した。結果として得られた懸濁液を濾過して粗生成FHA−1生成物を回収した。この粗生成FHA−1を蒸留水250mLに溶解し、溶液のpHを約7.0に調節し、溶液を蒸留水に対し24時間透析した。この間、外側の水浴を4回交換した。透析したFHA−1生成物を凍結乾燥することにより、FHA−1として設計された1.5gのFHA−1を綿状の網目状物として得た。1H NMR(D2O,δ):1.80(s,3H,NCH3),3.15−3.80(m,8H,OCH+OCH2)。19F NMR(D2O,δ):−113.6,−118.0。1H NMRで測定した置換度(SD)は2.0であった。SD=[(FHA−1のδ3.15−3.80の積分値)−(HAのδ3.20−3.8の積分値)]/[(NCH3の1.80の積分値)×(2/3)]。
【0065】
G.BGHAの調製
ヒアルロン酸(HA、Novozymes Biopolymers、950kDa)(2.0g)を100mLビーカー内でNaOH(40%w/v)20mLに溶解し、混合物を室温(rt)で2時間撹拌した。結果として得られた粘性液体を、イソプロパノール100mLを含む400mLビーカーに移して混合を継続した。撹拌した混合物にベンジルグリシジルエーテル10mLを加え、混合物をrtで24時間撹拌した。結果として得られた懸濁液を濾過して粗生成BGHA生成物を回収した。この粗生成BGHAを蒸留水250mLに溶解し、溶液のpHを約7.0に調節し、溶液を蒸留水に対し24時間透析した。この間、外側の水浴を4回交換した。透析したBGHA生成物を凍結乾燥することにより、BGHA1.25gを綿状の網目状物として得た。1H NMR(D2O,δ):1.85(s,3H,NCH3),3.20−3.80(m,10H,OCH+OCH3)。置換度(SD)を1H NMRで測定したところ、SDは1未満であった。
【0066】
H.低分子量メチルHA(DS−2)の調製
ヒアルロン酸(HA、Novozymes Biopolymers、53kDa)(2.0g)を100mLビーカー内でNaOH(40%w/v)20mLに溶解し、混合物を室温(rt)で2時間撹拌した。結果として得られた粘性液体を、イソプロパノール100mLを含む400mLビーカーに移して混合を継続した。撹拌した混合物にヨードメタン10mLを加え、混合物をrtで24時間撹拌した。結果として得られた懸濁液を濾過して粗生成低分子メチル化HA生成物を回収した。この粗生成LMW MeHAを蒸留水250mLに溶解し、溶液のpHを約7.0に調節し、溶液を蒸留水に対し24時間透析した。この間、外側の水浴を4回交換した。透析したLMW MeHA生成物を凍結乾燥することにより、メチルHA1.2gを綿状の網目状物として得た。1H NMR(D2O,δ):1.85(s,3H,NCH3),3.20−3.80(m,10H,OCH+OCH3)。1H NMRで測定した置換度(SD)は2であった。
【0067】
I.低分子量メチルHA(DS−1)の調製
ヒアルロン酸(HA、Novozymes Biopolymers、53kDa)(2.0g)を100mLビーカー内でNaOH(40%w/v)20mLに溶解し、混合物を室温(rt)で2時間撹拌した。結果として得られた粘性液体を、イソプロパノール100mLを含む400mLビーカーに移して混合を継続した。撹拌した混合物にヨードメタン4mLを加え、混合物をrtで24時間撹拌した。結果として得られた懸濁液を濾過して、粗生成低分子メチル化HA生成物を回収した。この粗生成LMW MeHAを蒸留水250mLに溶解し、溶液のpHを約7に調節し、溶液を蒸留水に対し24時間透析した。この間、外側の水浴を4回交換した。透析したLMW MeHA生成物を凍結乾燥することにより、LMW MeHA1.2gを綿状の網目状物として得た。1H NMR(D2O,δ):1.85(s,3H,NCH3),3.20−3.80(m,10H,OCH+OCH3)。1H NMRで測定した置換度(SD)は1であった。
【0068】
J.低分子量FHA−2(DS−1)の調製
2,2,3,3,4,4−ヘプタフルオロ−1−ブタノール1.0g(5mmol)を含む25mLフラスコに三臭化リン1.3mL(7.5mmol)をゆっくりと加えた。混合物を60℃で30分間撹拌した後、飽和炭酸水素ナトリウム溶液(15mL)をゆっくりと加えて反応を停止した。この水溶液をジクロロメタン15mLずつで3回抽出し、臭化ヘプタフルオロブチルを含む有機層を濃縮し、残渣を精製することなく次のステップで使用した。
【0069】
HA(2.0g、53kDaを100mLビーカー内でNaOH(40%w/v)20mLに溶解し、混合物を室温(rt)で2時間撹拌した。結果として得られた粘性液体を、イソプロパノール100mLを含む400mLビーカーに移して混合を継続した。撹拌した混合物にイソプロパノール10mL中の粗生成臭化ヘプタフルオロブチルを加え、混合物をrtで24時間撹拌した。結果として得られた懸濁液を濾過してLMW FHA−2生成物を回収した。この粗生成LMW FHA−2を250mLの蒸留水に溶解し、溶液のpHを約7に調節し、溶液を蒸留水に対し24時間透析した。この間、外側の水浴を4回交換した。透析したLMW FHA−2生成物を凍結乾燥することにより、LMW FHA−2として設計された1.5gのLMW FHA−2を綿状の網目状物として得た。1H NMR(D2O,δ):1.82(s,3H,NCH3),3.15−3.80(m,8H,OCH+OCH2)。19F NMR(D2O,δ):−115.3,−120.8。1H NMRで測定した置換度(SD)は1.0であった。
【0070】
K.低分子量FHA−2(DS−2)の調製
2,2,3,3,4,4−ヘプタフルオロ−1−ブタノール3.0g(15mmol)を含む25mLフラスコに三臭化リン2.5mL(16mmol)をゆっくりと加えた。混合物を60℃で30分間撹拌した後、飽和炭酸水素ナトリウム溶液(15mL)をゆっくりと加えて反応を停止した。この水溶液をジクロロメタン15mLずつで3回抽出し、臭化ヘプタフルオロブチルを含む有機層を濃縮し、残渣を精製することなく次のステップで使用した。
【0071】
HA(2.0g、53kDa)を100mLビーカー内でNaOH(40%w/v)20mLに溶解し、混合物を室温(rt)で2時間撹拌した。結果として得られた粘性液体を、イソプロパノール100mLを含む400mLビーカーに移して混合を継続した。撹拌した混合物にイソプロパノール10mL中の粗生成臭化ヘプタフルオロブチルを加え、混合物をrtで24時間撹拌した。結果として得られた懸濁液を濾過してLMW FHA−2生成物を回収した。この粗生成LMW FHA−2を250mLの蒸留水に溶解し、溶液のpHを約7に調節し、溶液を蒸留水に対し24時間透析した。この間、外側の水浴を4回交換した。透析したLMW FHA−2生成物を凍結乾燥することにより、LMW FHA−2として設計された1.5gのLMW FHA−2を綿状の網目状物として得た。1H NMR(D2O,δ):1.82(s,3H,NCH3),3.15−3.80(m,8H,OCH+OCH2)。19F NMR(D2O,δ):−115.3,−120.8。1H NMRで測定した置換度(SD)は2.0であった。
【0072】
L.低分子量FHA−1(DS−1)の調製
2,2,3,3,−ペンタフルオロ−1−プロパノール1.0g(5mmol)を含む25mLフラスコに三臭化リン1.3mL(7.5mmol)をゆっくりと加えた。混合物を60℃で30分間撹拌した後、飽和炭酸水素ナトリウム溶液(15mL)をゆっくりと加えて反応を停止した。この水溶液をジクロロメタン15mLずつで3回抽出し、臭化ヘプタフルオロブチルを含む有機層を濃縮し、残渣を精製することなく次のステップで使用した。
【0073】
HA(2.0g、53kDa)を100mLビーカー内でNaOH(40%w/v)20mLに溶解し、混合物を室温(rt)で2時間撹拌した。結果として得られた粘性液体を、イソプロパノール100mLを含む400mLビーカーに移して混合を継続した。撹拌した混合物に、イソプロパノール10mL中の粗生成臭化ヘプタフルオロブチルを加え、混合物をrtで24時間撹拌した。結果として得られた懸濁液を濾過してLMW FHA−1生成物を回収した。この粗生成LMW FHA−1を250mLの蒸留水に溶解し、溶液のpHを約7に調節し、溶液を蒸留水に対し24時間透析した。この間、外側の水浴を4回交換した。透析したLMW FHA−1生成物を凍結乾燥することにより、LMW FHA−1として設計されたLMW FHA−11.5gを綿状の網目状物として得た。1H NMR(D2O,δ):1.80(s,3H,NCH3),3.15−3.80(m,8H,OCH+OCH2)。19F NMR(D2O,δ):−113.6,−118.0。1H NMRで測定した置換度(SD)は1.0であった。
【0074】
M.LMW FHA−1(DS−2)の調製
2,2,3,3,−ペンタフルオロ−1−プロパノール3.0g(15mmol)を含む25mLフラスコに三臭化リン3mL(18mmol)をゆっくりと加えた。混合物を60℃で30分間撹拌した後、飽和炭酸水素ナトリウム溶液(15mL)をゆっくりと加えて反応を停止した。この水溶液をジクロロメタン15mLずつで3回抽出し、臭化ヘプタフルオロブチルを含む有機層を濃縮し、残渣を精製することなく次のステップで使用した。
【0075】
HA(2.0g、53kDa)を100mLビーカー内でNaOH(40%w/v)20mLに溶解し、混合物を室温(rt)で2時間撹拌した。結果として得られた粘性液体を、イソプロパノール100mLを含む400mLビーカーに移して混合を継続した。撹拌した混合物にイソプロパノール10mL中の粗生成臭化ヘプタフルオロブチルを加え、混合物をrtで24時間撹拌した。結果として得られた懸濁液を濾過してLMW FHA−1生成物を回収した。この粗生成LMW FHA−1を250mLの蒸留水に溶解し、溶液のpHを約7に調節し、溶液を蒸留水に対し24時間透析した。この間、外側の水浴を4回交換した。透析したLMW FHA−1生成物を凍結乾燥することにより、LMW FHA−1として設計された1.5gのLMW FHA−1を綿状の網目状物として得た。1H NMR(D2O,δ):1.80(s,3H,NCH3),3.15−3.80(m,8H,OCH+OCH2)。19F NMR(D2O,δ):−113.6,−118.0。1H NMRで測定した置換度(SD)は2.0であった。
【0076】
N.低分子量BGHAの調製
ヒアルロン酸(HA、Novozymes Biopolymers、53kDa)(2.0g)を100mLビーカー内でNaOH(40%w/v)20mLに溶解し、混合物を室温(rt)で2時間撹拌した。結果として得られた粘性液体を、イソプロパノール100mLを含む400mLビーカーに移して混合を継続した。撹拌した混合物にベンジルグリシジルエーテル10mLを加え、混合物をrtで24時間撹拌した。結果として得られた懸濁液を濾過してLMW BGHA生成物を回収した。この粗生成LMW BGHAを250mLの蒸留水に溶解し、溶液のpHを約7に調節し、溶液を蒸留水に対し24時間透析した。この間、外側の水浴を4回交換した。透析したLMW BGHA生成物を凍結乾燥することにより、LMW BGHA1.25gを綿状の網目状物として得た。1H NMR(D2O,δ):1.85(s,3H,NCH3),3.20−3.80(m,10H,OCH+OCH3)。1H NMRで測定した置換度(SD)は1未満であった。
【0077】
II.アルキル化HA誘導体の硫酸化
1.LMW−P−OSFHA−1(DS−1)(GM−211101)の調製
最初に、脱イオン水100mL中のLMW FHA−1(1.0g)のpHを1NのHClで3.0に調節し、トリブチルアミン1mLを加えることによって、LMW FHA−1(DS−1)のトリブチルアンモニウム(TBA)塩を調製した。この混合物を激しく混合し、凍結乾燥した。結果として得られた塩(FHA−1−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり6mol)のピリジン−三酸化硫黄錯体(0.4g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLで粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化HAを蒸留水に溶解し、100mMのNaCl溶液に対し2日間透析(溶液を1日4回交換)した後、凍結乾燥することにより、生成物(330mg)を収率75%で得た。1H NMRで評価した硫酸化SD=1.0であった。置換度は、文献(Carbohydrate Research、1998、306、35-43)のOCHのNMRシフトと比較することにより決定した。
【0078】
2.LMW−P−OSFHA−2(DS−1)(GM−311101)の調製
蒸留水50mL中のFHA−2(MW53kDaのHA由来のFHA−2)(0.25g)にトリブチルアミン0.5mLを加え、上述した1と同様に処理することによってLMW FHA−2のTBA塩を調製した。結果として得られた塩(LMW FHA−2−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり6mol)のピリジン−三酸化硫黄錯体(0.4g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化LMW FHA−2を水に溶解し、1と同様に透析および凍結乾燥することによって生成物(300mg)を収率68%で得た。1H NMRで評価した硫酸化SD=1.0であった。
【0079】
3.LMW−P−OSMeHA(DS−1)(GM−111101)の調製
1と同様にして、TBA0.5mLおよび蒸留水50mL中のLMW MeHA(DS−1)(MW53kDaのHA由来)(0.5g)からLMW MeHAのTBA塩を調製した。結果として得られた塩(LMW MeHA−TBA)をDMF50mLに溶解し、所要の過剰(MeHA中の利用可能なヒドロキシ基1当量当たり6mol)のピリジン−三酸化硫黄錯体(0.8g)を加えた。40℃で3時間撹拌した後、水100mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール150mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化MeHAを水に溶解し、1と同様に透析および凍結乾燥することによって生成物(540mg)を62%の収率で得た。1H NMRにより硫酸化SD=1.0〜1.5であることが示された。
【0080】
4.LMW−P−OSFHA−1(DS−2)(GM−211201)の調製
蒸留水50mL中のFHA−1(MW53kDaのHA由来のFHA−1)(0.25g)にトリブチルアミン0.5mLを加え、上述した1と同様に処理することによってLMW FHA−1のTBA塩を調製した。結果として得られた塩(FHA−1−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり6mol)のピリジン−三酸化硫黄錯体(0.4g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化HAを蒸留水に溶解し、100mMのNaCl溶液に対し2日間透析(溶液を1日4回交換)した後、凍結乾燥することによって生成物(300mg)を70%の収率で得た。1H NMRで評価した硫酸化SD=1.0であった。
【0081】
5.LMW−P−OSFHA−2(DS−2)(GM−311201)の調製
蒸留水50mL中のFHA−2(MW53kDaのHA由来のFHA−2)(0.25g)にトリブチルアミン0.5mLを加え、上述した1と同様に処理することによってLMW FHA−2のTBA塩を調製した。結果として得られた塩(LMW FHA−2−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり6mol)のピリジン−三酸化硫黄錯体(0.4g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化LMW FHA−2を水に溶解し、1と同様に透析および凍結乾燥することによって生成物(310mg)を収率70%で得た。1H NMRで評価した硫酸化SD=1.0であった。
【0082】
6.LMW−P−OSMeHA(DS−2)(GM−111201)の調製
1と同様にして、TBAを0.5mLおよび蒸留水50mL中のLMW MeHA(DS−1)(MW53kDaのHA由来)(0.5g)からLMW MeHAのTBA塩を調製した。結果として得られた塩(LMW MeHA−TBA)をDMF50mLに溶解し、所要の過剰(MeHA中の利用可能なヒドロキシ基1当量当たり6mol)のピリジン−三酸化硫黄錯体(0.8g)を加えた。40℃で3時間撹拌した後、水100mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール150mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化MeHAを水に溶解し、1と同様に透析および凍結乾燥することによって生成物(560mg)を64%の収率で得た。1H NMRにより、硫酸化SD=1.0〜1.5であることが示された。
【0083】
7.P−OSFHA−1(DS−1)(GM−231101)の調製
蒸留水50mL中のFHA−1(MW950kDaのHA由来のFHA−1)(0.25g)にトリブチルアミン0.5mLを加えて、上述した1と同様に処理することによってFHA−1のTBA塩を調製した。結果として得られた塩(FHA−1−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり6mol)のピリジン−三酸化硫黄錯体(0.4g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化HAを蒸留水に溶解し、100mMのNaCl溶液に対し2日間透析(溶液を1日4回交換)した後、凍結乾燥することによって生成物(300mg)を68%の収率で得た。1H NMRで評価した硫酸化SD=1.0〜1.5であった。
【0084】
8.P−OSFHA−2(DS−1)(GM−331101)の調製
蒸留水50mL中のFHA−2(MW950kDaのHA由来のFHA−2)(0.25g)にトリブチルアミン0.5mLを加えて、上述した1と同様に処理することによってFHA−2のTBA塩を調製した。結果として得られた塩(FHA−2−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり6mol)のピリジン−三酸化硫黄錯体(0.4g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化FHA−2を水に溶解し、1と同様に透析および凍結乾燥することによって生成物(320mg)を70%の収率で得た。1H NMRで評価した硫酸化SD=1.0〜1.5であった。
【0085】
9.P−OSMeHA(DS−1)(GM−131101)の調製
1と同様にして、TBAを0.5mLおよび蒸留水50mL中のMeHA(DS−1)(MW950kDaのHA由来)(0.5g)からMeHAのTBA塩を調製した。結果として得られた塩(MeHA−TBA)を、DMF50mLに溶解し、所要の過剰(MeHA中の利用可能なヒドロキシ基1当量当たり6mol)のピリジン−三酸化硫黄錯体(0.8g)を加えた。40℃で3時間撹拌した後、水100mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール150mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化MeHAを水に溶解し、1と同様に透析および凍結乾燥することによって生成物(510mg)を収率60%で得た。1H NMRにより硫酸化SD=1.0〜1.5であることが示された。
【0086】
10.P−OSFHA−1(DS−2)(GM−231201)の調製
蒸留水50mL中のFHA−1(MW950kDaのHA由来のFHA−1)(0.25g)にトリブチルアミン0.5mLを加えて、上述した1と同様に処理することによってFHA−1のTBA塩を調製した。結果として得られた塩(FHA−1−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり6mol)のピリジン−三酸化硫黄錯体(0.4g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化HAを蒸留水に溶解し、100mMのNaCl溶液に対し2日間透析(溶液を1日4回交換)した後、凍結乾燥することによって生成物(280mg)を収率68%で得た。1H NMRで評価した硫酸化SD=1.0〜1.5であった。
【0087】
11.P−OSFHA−2(DS−2)(GM−331201)の調製
蒸留水50mL中のFHA−2(MW950kDaのHA由来のFHA−2)(0.25g)にトリブチルアミン0.5mLを加えて、上述した1と同様に処理することによってFHA−2のTBA塩を調製した。結果として得られた塩(FHA−2−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり6mol)のピリジン−三酸化硫黄錯体(0.4g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化FHA−2を水に溶解し、1と同様に透析および凍結乾燥することによって生成物(300mg)を収率69%で得た。1H NMRで評価した硫酸化SD=1.0〜1.5であった。
【0088】
12.P−OSMeHA(DS−2)(GM−131201)の調製
1と同様にして、TBAを0.5mLおよび蒸留水50mL中のMeHA(DS−1)(MW950kDaのHA由来)(0.5g)から、MeHAのTBA塩を調製した。結果として得られた塩(MeHA−TBA)を、DMF50mLに溶解し、所要の過剰(MeHA中の利用可能なヒドロキシ基1当量当たり6mol)のピリジン−三酸化硫黄錯体(0.8g)を加えた。40℃で3時間撹拌した後、水100mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール150mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化MeHAを水に溶解し、1と同様に透析および凍結乾燥することによって生成物(560mg)を収率64%で得た。1H NMRによりSD=1.0〜1.5であることが示された。
【0089】
13.LMW−F−OSFHA−1(DS−1)(GM−212101)の調製
蒸留水50mL中のFHA−1(MW53kDaのHA由来のFHA−1)(0.25g)にトリブチルアミン0.5mLを加えて、上述した1と同様に処理することによってLMW FHA−1のTBA塩を調製した。結果として得られた塩(FHA−1−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり12mol)のピリジン−三酸化硫黄錯体(0.8g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化HAを蒸留水に溶解し、100mMのNaCl溶液に対し2日間透析(溶液を1日に4回交換)した後、凍結乾燥することによって生成物(300mg)を収率71%で得た。1H NMRで評価したSD=1.5〜2.0であった。
【0090】
14.LMW−F−OSFHA−2(DS−1)(GM−312101)の調製
蒸留水50mL中のFHA−2(MW53kDaのHA由来のFHA−2)(0.25g)にトリブチルアミン0.5mLを加えて、上述した1と同様に処理することによってLMW FHA−2のTBA塩を調製した。結果として得られた塩(LMW FHA−2−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり12mol)のピリジン−三酸化硫黄錯体(0.8g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化LMW FHA−2を水に溶解し、1と同様に透析および凍結乾燥することによって生成物(260mg)を収率65%で得た。1H NMRで評価した硫酸化SD=1.5〜2.0であった。
【0091】
15.LMW−F−OSMeHA(DS−1)(GM−112101)
1と同様にして、TBAを0.5mLおよび蒸留水50mL中のLMW MeHA(DS−1)(MW53kDaのHA由来)(0.5g)からLMW MeHAのTBA塩を調製した。結果として得られた塩(LMW MeHA−TBA)をDMF50mLに溶解し、所要の過剰(MeHA中の利用可能なヒドロキシ基1当量当たり12mol)のピリジン−三酸化硫黄錯体(1.8g)を加えた。40℃で3時間撹拌した後、水100mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール150mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化MeHAを水に溶解し、1と同様に透析および凍結乾燥することによって生成物(480mg)を収率60%で得た。1H NMRにより硫酸化SD=1.5〜2.0であることが示された。
【0092】
16.LMW−F−OSFHA−1(DS−2)(GM−212201)の調製
蒸留水50mL中のFHA−1(MW53kDaのHA由来のFHA−1)(0.25g)にトリブチルアミン0.5mLを加えて、上述した1と同様に処理することによってLMW FHA−1のTBA塩を調製した。結果として得られた塩(FHA−1−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり12mol)のピリジン−三酸化硫黄錯体(0.8g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化HAを蒸留水に溶解し、100mMのNaCl溶液に対し2日間透析(溶液を1日4回交換)した後、凍結乾燥することによって生成物(300mg)を収率70%で得た。1H NMRで評価した硫酸化SD=1.5〜2.0であった。
【0093】
17.LMW−F−OSFHA−2(DS−2)(GM−312201)の調製
蒸留水50mL中のFHA−2(MW53kDaのHA由来のFHA−2)(0.25g)にトリブチルアミン0.5mLを加え、上述した1と同様に処理することによってLMW FHA−2のTBA塩を調製した。結果として得られた塩(LMW FHA−2−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり12mol)のピリジン−三酸化硫黄錯体(0.8g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化LMW FHA−2を水に溶解し、1と同様に透析および凍結乾燥することによって生成物(300mg)を収率68%で得た。1H NMRで評価した硫酸化SD=1.5〜2.0であった。
【0094】
18.LMW−F−OSMeHA(DS−2)(GM−112201)の調製
1と同様にして、TBAを0.5mLおよび蒸留水50mL中のLMW MeHA(DS−1)(MW53kDaのHA由来)(0.5g)からLMW MeHAのTBA塩を調製した。結果として得られた塩(LMW MeHA−TBA)をDMF50mLに溶解し、所要の過剰(MeHA中の利用可能なヒドロキシ基1当量当たり12mol)のピリジン−三酸化硫黄錯体(1.6g)を加えた。40℃で3時間撹拌した後、水100mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール150mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化MeHAを水に溶解し、1と同様に透析および凍結乾燥することによって生成物(550mg)を収率63%で得た。1H NMRにより硫酸化SD=1.5であることが示された。
【0095】
19.F−OSFHA−1(DS−1)(GM−232101)の調製
蒸留水50mL中のFHA−1(MW950kDaのHA由来のFHA−1)(0.25g)にトリブチルアミン0.5mLを加え、上述した1と同様に処理することによってFHA−1のTBA塩を調製した。結果として得られた塩(FHA−1−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり12mol)のピリジン−三酸化硫黄錯体(0.8g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化HAを蒸留水に溶解し、100mMのNaCl溶液に対し2日間透析(溶液を1日4回交換)した後、凍結乾燥することによって生成物(300mg)を収率68%で得た。1H NMRで評価した硫酸化SD=1.5であった。
【0096】
20.F−OSFHA−2(DS−1)(GM−332101)の調製
蒸留水50mL中のFHA−2(MW950kDaHA由来のFHA−2)(0.25g)にトリブチルアミン0.5mLを加えて、上述した1と同様に処理することによってFHA−2のTBA塩を調製した。結果として得られた塩(FHA−2−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり12mol)のピリジン−三酸化硫黄錯体(0.8g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化FHA−2を水に溶解し、1と同様に透析および凍結乾燥することによって生成物(310mg)を収率70%で得た。1H NMRで評価した硫酸化SD=1.5〜2.0であった。
【0097】
21.F−OSMeHA(DS−1)(GM−132101)の調製
1と同様にして、TBAを0.5mLおよび蒸留水50mL中のMeHA(MW950kDaのHA由来)(DS−1)(0.5g)からMeHAのTBA塩を調製した。結果として得られた塩(MeHA−TBA)をDMF50mLに溶解し、所要の過剰(MeHA中の利用可能なヒドロキシ基1当量当たり12mol)のピリジン−三酸化硫黄錯体(1.6g)を加えた。40℃で3時間撹拌した後、水100mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール150mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化MeHAを水に溶解し、1と同様に透析および凍結乾燥することによって生成物(500mg)を収率60%で得た。1H NMRにより硫酸化SD=1.5〜2.0であることが示された。
【0098】
22.F−OSFHA−1(DS−2)(GM−232201)の調製
蒸留水50mL中のFHA−1(MW950kDaのHA由来のFHA−1)(0.25g)にトリブチルアミン0.5mLを加えて、上述した1と同様に処理することによってFHA−1のTBA塩を調製した。結果として得られた塩(FHA−1−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり12mol)のピリジン−三酸化硫黄錯体(0.8g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化HAを蒸留水に溶解し、100mMのNaCl溶液に対し2日間透析(溶液を1日4回交換)した後、凍結乾燥することによって生成物(300mg)を収率69%で得た。1H NMRで評価した硫酸化SD=1.5〜2.0であった。
【0099】
23.F−OSFHA−2(DS−2)(GM−332201)の調製
蒸留水50mL中のFHA−2(MW950kDaのHA由来のFHA−2)(0.25g)にトリブチルアミン0.5mLを加えて、上述した1と同様に処理することによってFHA−2のTBA塩を調製した。結果として得られた塩(FHA−2−TBA)をDMF25mLに溶解し、所要の過剰(HA中の利用可能なヒドロキシ基1当量当たり12mol)のピリジン−三酸化硫黄錯体(0.8g)を加えた。40℃で3時間撹拌した後、水50mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール75mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化FHA−2を水に溶解し、1と同様に透析および凍結乾燥することによって生成物(300mg)を収率69%で得た。1H NMRで評価した硫酸化SD=1.5〜2.0であった。
【0100】
24.F−OSMeHA(DS−2)(GM−132201)の調製
1と同様にして、TBAを0.5mLおよび蒸留水50mL中のMeHA(DS−1)(MW950kDaのHA由来)(0.5g)からMeHAのTBA塩を調製した。結果として得られた塩(MeHA−TBA)をDMF50mLに溶解し、所要の過剰(MeHA中の利用可能なヒドロキシ基1当量当たり12mol)のピリジン−三酸化硫黄錯体(1.6g)を加えた。40℃で3時間撹拌した後、水100mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール150mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成部分O−硫酸化MeHAを水に溶解し、1と同様に透析および凍結乾燥することによって生成物(550mg)を収率63%で得た。1H NMRにより硫酸化SD=1.5〜2.0であることが示された。
【0101】
25.P−OSBGHA(GM−431101)の調製
1と同様にして、TBAを0.5mLおよび蒸留水50mL中のBGHA(MW950kDaのHA由来)(0.5g)からBGHAのTBA塩を調製した。結果として得られた塩(BGHA−TBA)をDMF50mLに溶解し、所要の過剰(BGHA中の利用可能なヒドロキシル基1当量当たり6mol)のピリジン−三酸化硫黄錯体(0.8g)を加えた。40℃で3時間撹拌した後、水100mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール150mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成O−硫酸化BGHAを水に溶解し、1と同様に透析および凍結乾燥することによって生成物(500mg)を収率60%で得た。1H NMRにより硫酸化SD<1であることが示された。
【0102】
26.F−OSBGHA(GM−432101)の調製
1と同様にして、TBAを0.5mLおよび蒸留水50mL中のBGHA(MW950kDaのHA由来)(0.5g)からBGHAのTBA塩を調製した。結果として得られた塩(BGHA−TBA)をDMF50mLに溶解し、所要の過剰(BGHA中の利用可能なヒドロキシル基1当量当たり12mol)のピリジン−三酸化硫黄錯体(1.6g)を加えた。40℃で3時間撹拌した後、水100mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール150mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成O−硫酸化BGHAを水に溶解し、1と同様に透析および凍結乾燥することによって生成物(520mg)を収率61%で得た。1H NMRにより硫酸化SD<1であることが示された。
【0103】
27.P−OSBGHA(GM−411101)の調製
1と同様にして、TBAを0.5mLおよび蒸留水50mL中のBGHA(MW53kDaのHA由来)(0.5g)からBGHAのTBA塩を調製した。結果として得られた塩(BGHA−TBA)をDMF50mLに溶解し、所要の過剰(BGHA中の利用可能なヒドロキシル基1当量当たり6mol)のピリジン−三酸化硫黄錯体(0.8g)を加えた。40℃で3時間撹拌した後、水100mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール150mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成O−硫酸化BGHAを水に溶解し、1と同様に透析および凍結乾燥することによって生成物(500mg)を収率60%で得た。1H NMRにより硫酸化SD<1であることが示された。
【0104】
28.P−OSBGHA(GM−412101)の調製
1と同様にして、TBAを0.5mLおよび蒸留水50mL中のBGHA(MW53kDaのHA由来)(0.5g)からBGHAのTBA塩を調製した。結果として得られた塩(BGHA−TBA)をDMF50mLに溶解し、所要の過剰(BGHA中の利用可能なヒドロキシル基1当量当たり12mol)のピリジン−三酸化硫黄錯体(1.6g)を加えた。40℃で3時間撹拌した後、水100mLを加えて反応を停止し、無水酢酸ナトリウムで飽和した冷エタノール150mLを加えて粗生成物を析出させた後、濾過して回収した。結果として得られた粗生成O−硫酸化BGHAを水に溶解し、1と同様に透析および凍結乾燥することによって生成物(500mg)を収率60%で得た。1H NMRにより硫酸化SD<1であることが示された。
【0105】
III.蛍光SAGEおよびパルミトイル化SAGEの調製
a.LMW−F−OSFHA−1(DS−2)蛍光体複合体の調製
LMW−F−OSFHA−1(50mg)およびNHS(40mg)を水10mLに溶解し、次いで、DMF4mL中のAlaxa fluo@488(1mg)を加えた。次いで、pHを4.75に調節した後、固体形態のEDCI100mgを加えた。NaOH溶液を加えてpHを4.75に維持した。溶液をアルミニウム箔で覆って室温で一夜撹拌した。次いで、生成物を蒸留水、続いて、メタノール/水溶液(50/50、v/v)で透析(カットオフMW3500)することにより精製し、さらにゲル濾過カラム(Sephadex G−25)で精製した。さらにPD−10カラムで精製し、凍結乾燥によって40mgを得た。
【0106】
b.LMW−P−OSMeHA(DS−1)蛍光体複合体の調製
LMW−P−OSMeHA(50mg)およびNHS(40mg)を水10mLに溶解した後、DMF4mL中のAlaxa fluo@488(1mg)を加えた。次いで、PHを4.75に調節した後、固体形態のEDCI100mgを加えた。NaOH溶液を加えてpHを4.75に維持した。溶液をアルミニウム箔で覆って室温で一夜撹拌した。次いで、生成物を蒸留水、続いてメタノール/水溶液(50/50、v/v)で透析(カットオフMW3500)することにより精製し、さらにゲル濾過カラム(Sephadex G-25)で精製した。さらにPD−10カラムで精製し、凍結乾燥によって43mgを得た。
【0107】
c.パルミトイル化SAGEの調製
LMW−P−OSMeHA(50mg)をDMF50mLに溶解し、このDMF溶液を撹拌しながらトリエチルアミン(0.3mL)を加えた。5分後に塩化パルミトイル(0.5mL)を加えた。結果として得られた混合物を一夜撹拌し続けた。溶液を蒸発させ、残渣を蒸留水に溶解し、1日間透析し(水を4回交換した)、凍結乾燥することにより35mgを得た。
【0108】
IV.SAGEは、P−セレクチン、ヒト白血球エラスターゼ、およびRAGEとそのあらゆるリガンドとの相互作用の強力な阻害物質である。
材料:ポリクローナルヤギ抗ヒトRAGE、組換え型ヒトハイモビリティー・ボックス・プロテイン−1(HMGB−1)、組換え型ヒトP−セレクチン/Fcキメラ、組換え型ヒトRAGE/Fcキメラ、ヒトアズロシジン、およびポリクローナルヤギ抗ヒトアズロシジンを、R&D Systems(Minneapolis,MN)より購入した。ヒトS100bカルグラニュリンはCalbiochem(San Diego,CA)より入手した。終末糖化産物カルボキシメチルリジン−ウシ血清アルブミン(CML−BSA)は、MBL International(Woburn,MA)より入手した。U937ヒト単球細胞は、American Type Culture Collection(Manassas,VA)より入手した。Aタンパク、西洋ワサビペルオキシダーゼ結合ウサギ抗ヤギIgG、炭酸−重炭酸緩衝液、およびウシ血清アルブミンブロッカー(10×)は、Piercenet(Rockford,IL)より入手した。カルセインAM、ダルベッコ変法イーグル培地(DMEM)、エチレンジアミン四酢酸(EDTA)、ウシ胎児血清(FBS)、HEPES、非必須アミノ酸、ペニシリン/ストレプトマイシン/L−グルタミン溶液、RPMI−1640(L−グルタミン非含有)、および炭酸水素ナトリウムはInvitrogen(Carlsbad,CA)より入手した。高結合能96穴マイクロプレートは、Corning Life Sciences(Corning,NY)より入手した。他の明記されていない薬品はすべてSigma-Aldrich(St.Louis,MO)より購入した。
【0109】
細胞培養:U937単球を、10%加熱不活性化FBS、2mM L−グルタミン、1mM ピルビン酸ナトリウム、0.1mM MEM非必須アミノ酸、100単位/mL ペニシリン、および100mg/mL ストレプトマイシンを添加したRPMI−1640を用いて、加湿CO2(5%)−空気(95%)中、懸濁培養により37℃で培養した。1〜5代の細胞を用いて実験を行った。
【0110】
細胞結合アッセイ:U937単球とP−セレクチンまたはRAGEとの結合に対するSAGEの影響を、Aタンパク8μg/mL(50μg/ウェル)を含む0.2M炭酸−重炭酸緩衝液(pH9.4)でコーティングした高結合能マイクロプレートを用いて調査した。プレートを1%BSAを含むリン酸緩衝生理食塩水(PBS−BSA)で洗浄し、P−セレクチン−FcまたはRAGE−Fcキメラ(50μL、1μg含有)を各ウェルに分注して、それぞれ室温で2時間または4℃で一夜インキュベートした。インキュベート後、ウェルをPBS−BSAで洗浄した。20mMのHEPES緩衝液(125mM NaCl、2mMカルシウム、および2mMマグネシウム含有)で段階希釈(0〜1000μg/mL)したSAGE50μLを各ウェルに加え、室温で15分間インキュベートした。陰性対照として、カルシウムを捕捉することによって細胞との結合を阻止するように、選択したウェルに10mM EDTA50μLを加えた。インキュベーション完了後、U937細胞(105細胞/ウェル、製造業者の指示に従いカルセイン標識)50μLを各ウェルに加えて、プレートをさらに室温で30分間インキュベートした。次いで、ウェルをPBSで3回洗浄した後、トリス−TritonX−100含有緩衝液100μLを加えて、結合した細胞を溶解した。マイクロプレートリーダーを用いて494nmで励起、517nmで発光させて蛍光を測定した。
【0111】
固相結合分析:固相結合分析を用いて、RAGEとそのリガンドとの結合をSAGEが阻害する能力を調査した。RAGEとリガンドとの結合に及ぼすSAGEの影響を調査するために、ポリビニル96穴プレートを特定のリガンド(CML−BSA、HMGB−1、またはS100bカルグラニュリン)5μg/ウェルでコーティングした。プレートを4℃で一夜インキュベートした後、PBS−0.05%Tween−20(PBST)で3回洗浄した。これとは別に、RAGE−Fcキメラ(PBST−0.1%BSA中、0.5μg/mL含有)100μLを等量の段階希釈したSAGE(PBST−BSA中、0.001から1000μg/mL)と一緒に4℃でインキュベートした。翌日、RAGE−SAGE混合物各50μLを、リガンドをコーティングした各ウェルに移し、37℃で2時間インキュベートした。次いで、ウェルをPBSTで4回洗浄した。結合したRAGEを検出するために、抗RAGE抗体(0.5μg/mL)50μLを各ウェルに加え、混合物を室温で1時間インキュベートし、ウェルを再度PBSTで4回洗浄した。西洋ワサビペルオキシダーゼ結合二次抗体(抗体をPBSTで1:10000に希釈;50μL/ウェル)を加え、ウェルを室温で1時間インキュベートした後、PBSTで1回洗浄した。テトラメチルベンジジン色素原(TMB single solution色素原)50μLを加えて呈色反応を開始し、15分後、1NのHClを50μL加えることによって停止した。自動マイクロプレートリーダーを用いて450nmの吸光度を測定した。
【0112】
酵素的試験:陽性荷電PMNプロテアーゼHLEのSAGEによる阻害を評価するために、活性検定(Fryer A、Huang Y-C、Rao G、Jacoby D、Mancilla E、Whorton R、Piantadosi CA、Kennedy T、Hoidal J、選択的O−脱硫酸化により肺で薬理活性を維持する非抗凝固性ヘパリンが生成する(Selective O-desulfation produces nonanticoagulant heparin that retains pharmacologic activity in the lung.)、J Pharmacol Exp Ther 282:208-219、1997)を用いて、精製HLEが発色基質を切断する能力を測定した。HLE(100nM)をSAGE(1〜100nM)と一緒に0.5M HEPES緩衝液中で15分間インキュベートした。インキュベート後、エラスターゼ基質であるSuc−Ala−Ala−Val−p−ニトロアニリン(p−NA)を反応混合物に加えて最終濃度を0.3mMとした。405nmの吸光度を測定することにより、加水分解によるp−NAの遊離を15分間追跡した。SAGEが第XII因子または補体を活性化する能力を評価するために、汚染された市販のヘパリンの毒性をスクリーニングするために最近用いられているものと類似の活性検定を用いた(Kishimoto TK、Viswanathan K、Ganguly T、Elankumaran S、Smith S、Pelzer K、Lansing JC、Sriranganathan N、Zhao G、Galcheva-Gargova Z、Al-Hakim A、Bailey GS、Fraser B、Roy S、Rogers-Cotrone T、Buhse L、Whary M、Fox J、Nasr M、Dal Pan GJ、Shriver Z、Langer RS、Venkataranam G、Austen KF、Woodcock J、Sasisekharan R、臨床有害事象と接触系の活性化に関連する汚染ヘハ゜リン(Contaminated hepalin associated with adverse clinical events and activation of the contact system.)、N Engl J Med 358:2457-2467、2008;Guerrini M、Beccati D、Shriver Z、Naggi A、Viswanathan K、Bisio A、Capila I、Lansing JC、Guglieri S、Fraser B、Al-Hakim A、Gunay NS、Zhang Z、Robinson L、Buhse L、Nasr M、Woodcock J、Langer R、Venkataraman G、Linhardt RJ、Casu B、Torri G、Sasisekharan R、過硫酸化されたコンドロイチン硫酸が臨床有害事象に関連するヘパリンの汚染源である(Oversulfated chondroitin sulfate is a contaminant in hepalin associated with adverse clinical events.)、Nat Biotech 26:669-675、2008)。プールしたヒト血漿(5μL)をSAHA(0.1〜1000μg/mL)100μLと一緒に、Triton X−100含有0.05M HEPES中、5℃で5分間インキュベートした。D−シクロヒドロチロシル−Glyc−L−Arg−p−NAを0.5mMを加えて405nmの吸光度変化を追跡することにより、ハーゲマン因子に特異的なアミダーゼ活性を測定した(Silverberg M、Dunn JT、Garen L、Kaplan AP、ヒトハーゲマン因子の自己活性化。合成基質を用いた実証(Autoactivation of human Hageman factor. Demonstration using a synthetic substrate.)、J Biol Chem 255:7281-7286、1980)。D−Pro−Phe−Arg−p−NAを加えて、450nmの吸光度変化を追跡することにより、活性カリクレインに特異的なアミダーゼ活性を測定した。
【0113】
結果:検定の結果を表2に示す。SAGEはP−セレクチンの強力な阻害剤である。P−セレクチン糖タンパクリガンド−1(PSGL−1)を介してP−セレクチンに強力に接着するU937ヒト単球のコンペティターを介した置換を、蛍光標識細胞を用いて調査した。図3から、SAGEがU937とP−セレクチンとの結合を0.5μg/mLの50%阻止濃度(IC50)で阻害することがわかる。
【0114】
【表2】
【0115】
第2に、高度に硫酸化されたポリアニオンと同様、アルキル化および硫酸化されたSAGEは多形核白血球プロテアーゼの強力な阻害剤である。図4は、アルキル化および/またはフルオロアルキル化ならびに硫酸化されたSAGEがヒト白血球エラスターゼ(HLE)を驚くほど強力なIC50値で阻害することを示すものである。具体的には、アルキル化されていない、完全にO−硫酸化されたHA(F−OSHA)は、HLEに対するIC50が0.66nMである。修飾および硫酸化されたHA誘導体のIC50値は、部分O−硫酸化カルボキシメチル化HA(P−OSCHMHA)が1.89nM、部分O−硫酸化HA(P−OSHA)が1.97nM、部分O−硫酸化メチル化HA(P−OSMEHA;GM−131101)が3.46nMであった。
【0116】
第3に、SAGEは、RAGEの極めて強力な阻害剤である。SAGEは、RAGEおよびアンホテリン(HMGB−1)の相互作用をIC50が1.1μg/mLで阻害し(図5)、RAGEおよびS100カルグラニュリンの相互作用をIC50が60ng/mL(図6)で阻害し、RAGEとAGE産物であるカルボキシメチル−リジンBSAとの結合をIC50が44ng/mLで阻害する(図7)。これらの値は、ヘパリンおよび2−O,3−O脱硫酸化ヘパリンを用いて本発明者らが測定したRAGE−リガンド阻害に関する対応するレベルよりも5〜10倍強力である。
【0117】
第4に、SAGEは、ヒトケラチン生成細胞増殖の強力な阻害剤である。これらの検定においては、ヒト新生児表皮ケラチン生成細胞をSAGEまたは他の阻害剤の存在下および非存在下に培養し、染色液を加えることによって増殖を測定(存在する生細胞数に正比例して低くなる)した。図に示した吸光度値は、対照(相対吸光度を1.0とした)に対して標準化したものである。図8から、高分子量部分O−硫酸化HA(P−OSHA)および完全O−硫酸化HA(F−OSHA)を100μg/mLの濃度で添加した場合、ヘパリン誘導体である2−O,3−O脱硫酸化ヘパリン(ODSH)よりもケラチン生成細胞の増殖抑制に関し有効であったことがわかる。部分硫酸化カルボキシメチル化HA(P−OSCMHA)およびメチル化HA(P−OSMEHA;GM−131101)はケラチン生成細胞増殖の阻害活性がより低いことから、硫酸化度がより高い方が有利であることがわかる。図9から、本検定においては、全体的により高分子量の誘導体よりもより低分子量の(LMW)誘導体がケラチン生成細胞増殖の低下に関し有効であることがわかる。特に、LMW部分O−硫酸化MeHA(LMW−P−OMEHA;GM−111201)、部分O−硫酸化CMHA(LMW−P−OSCMHA)、および部分O−硫酸化53kDa HAそのもの(LMW−P−OSHA)はいずれも1μg/mLという低濃度においても対照よりも増殖を低下させている。
【0118】
V.酒さおよび炎症の治療に関するIn Vivo試験
材料および方法
薬品:GM−111101、GM−131101、GM−312101、およびGM−212101について評価した。培地はAmerican Type Culture Collection(ATCC)より購入した。EpiLife培地はInvitrogen(Madison,WI)より購入した。
【0119】
細胞:ヒト皮膚線維芽細胞(nHDF)はATCCより購入した。ヒト新生児皮膚ケラチン生成細胞(HEKn)はInvitrogen(Madison,WI)より入手した。
【0120】
細胞毒性:96穴平底マイクロプレートの培地100μLを含む各ウェルに4000個のnHDF細胞を播種し、5%CO2中、37℃で12時間インキュベートした。すべての培地を、各種硫酸化ヒアルロナン(HA)誘導体を含む完全培地で、各段の最終濃度が10、100、1000、10000、100000、1000000ng/mLとなるように交換した。48時間後、各ウェルにMTS(Promega,Madison,WI)20μLをピペットで分注し、細胞をさらに2時間インキュベートした。試料の490nmにおける吸光度を96穴プレートリーダーで測定した。
【0121】
マウスの皮膚刺激性試験:マウスの皮膚に対するGM−111101およびGM−212101の皮膚刺激性を判定するためにin vivo試験を実施した。それぞれ0.1mg/mLおよび1mg/mLの2種の異なる濃度で2種の被験試薬を調製した。10%ギ酸およびPBSをそれぞれ陽性および陰性対照として用いた(n=6)。過去に実験に使用したことがなく、試験開始よりも前に皮膚炎、外傷、または臨床に不利な徴候を示していないことが認められているBalb/cマウスを計画された試験条件に無作為に群分けした。動物の背部毛を、試料を塗布する少なくとも4時間前に電気バリカンですべて刈毛しておいた。被験物質を塗布する直前に、各マウスの被験部位の下側の区域に滅菌針を用いて皮膚に4本の平行な傷を付け、被験部位の上側の区域は無傷のままとした。麻酔下で2種類の被験溶液の試料0.5mLを被験部位全体に塗布し、皮膚の約2.5cm角の範囲にガーゼを二重にして載せた。この貼布にプラスチックを裏張りし、非反応性テープで覆い、被験部位全体を包帯で巻いた。動物をそれぞれのケージに戻した。試剤を施用してから24時間後、包帯および濡れた試験用ガーゼを取り除いた。被験部位を水道水で拭き、残留している被験化合物を除去した。化合物を塗布してから24および72時間後に被験部位の皮膚反応をFHSAが推奨するドレイズ評価基準に従い調査した。試験終了後、試験品の一次刺激指数(P.I.I.)を求めることになる。P.I.I.評点が5.00以上の物質が陽性とみなされることになり、この物質が皮膚の一次刺激物とみなされることになる。
【0122】
動物:市販のBalb/cマウスを、Univ.of Utah veterinary medicine department and vivarium承認の供給業者から購入した。受領書に従い規定の期間検疫を行った後、すぐに試験に用いることができる。
【0123】
LL37ペプチドおよびSAGEを注射した酒さモデル:皮膚の慢性疾患、特に、酒さが顔面に現れた場合は、患者の人生に忘れ難い傷跡を残す。我々の種は他人の外見を受け容れたり拒否したりし、普通とは違う外見を持つ、自分とは異なる者がいると思わず尻込みしてしまう。皮膚疾患は多くの場合生命に関わることはないが、普通の人には推し量れない形で生活を一変させてしまう。酒さは、このように生活を一変させてしまう病気の1つである。米国の人口の3%すなわち約1400万人の米国人が酒さを患っている、顔面の見た目が悪くなる一般的な皮膚疾患であり、普通は30〜50歳で発症する。主としてケルト系の白人が冒され、男性よりも女性の方が問題を抱えているようである。患者の3分の1以上は家族歴があり、それが遺伝病であることを強く示唆している。顔面紅潮や凸凹した鼻を伴う酒さは、飲酒のし過ぎであるというありがちな誤解を受けることから、特に汚名を着せられやすい病気である。酒さは幾つかの臨床表現型を示す。最も一般に知られている症状は、頬や鼻の毛細血管が拡張することによる一過性または持続性の顔面中心部の紅潮および紅斑を特徴とするものである。この表現型は、「赤ら顔」程度から、容易に認められる持続的な血管拡張に及ぶ。この亜型には有効な局所治療がない。その次に広く知られている症状は、顔の中心の凸状部に発生する丘疹および膿疱であり、紅斑の上に重なることが多い。生検によると、その皮膚には多くのPMNが浸潤している。酒さ性ざ瘡は、抗生物質の局所または全身投与によって経験的に治療が行われているが、皮膚感染症例との関連が明確ではない。マクロライドは、局所または全身のいずれの治療にも最も多く用いられており、その抗菌活性よりもむしろPMNの炎症部位への遊走能を遅延させる能力によって改善が得られる可能性がある。第3に、そしてより稀な表現型であることは幸いであるが、酒さにより、鼻の皮脂腺および結合組織の両方が過形成することによって鼻瘤が生じ、昔で言うところの「W.C.Fields」の鼻または「酔っぱらいの鼻(whiskey nose)」になる。この症状の治療には、肥厚化した組織を除去するために外科的に組織を切除するかまたはレーザー治療が必要となる。第4の表現型においては、酒さによって眼瞼の炎症を伴う眼の乾燥および痒み(眼瞼炎)、光過敏、かすみ目、および結膜炎が生じる場合がある。疾患が長引くと、角膜炎や、それどころか角膜瘢痕が生じる可能性がある。眼瞼および下眼瞼表面にあるマイボーム腺の炎症によって起こるこの表現型は一般的なものであり、臨床的には眼科医がよく診察する「眼球乾燥」症候群と重複する。肌の色が白く眼の色が明るい人の日光が当たる顔面領域のみに酒さが発生することは、この状態を引き起こす太陽放射の病原的意義を示している。
【0124】
最近になって酒さの病因が解明された(Yamasaki K、Di Nardo A、Bardan A、Murakami M、Ohtake T、Coda A、Dorschner RA、Bonnart C、Descargues P、Hovnanian A、Morhenn VB、Gallop RL、セリンプロテアーゼ活性の亢進とカテリシジンの増加が酒さによる皮膚の炎症を促進する(Increased serine protease activity and cathelicidin promotes skin inflammation in rosacea.)、Nat Med 13:975-980、2007;Bevins CL、Liu F-T、酒さ:皮膚本来の免疫がうまく機能していない?(Rosacea:skin innate immunity gone awry?)、Nat Med 13:904−906、2007)。Yamasakiらの見事な研究により、酒さを伴う皮膚には、カテリシジンおよびそのプロセシングプロテアーゼであるSCTEが高濃度で存在することが実証された。哺乳類の抗菌ペプチドの主要なファミリーであるカテリシジンは、多くの臓器の白血球および上皮細胞で発現し、ここで細菌に対する自然免疫応答を媒介する(Nizet V、Ohtake T、Lauth X、Trowbridge Jk、Rudisill J、Dorschner RA、Prestonjamasp V、Piraino J、Huttner K、Gallop RL、生得的な抗菌ペプチドが侵襲性の細菌感染から皮膚を防御する(Innate antimicrobial peptide protects the skin from invasive bacterial infection.)、Nature 414:454-457、2001)。カテリシジンはまた、血管増殖、PMN遊走、および創傷治癒のシグナルも伝達する(Bevins、ibid)。ヒトカテリシジンは、ケラチン生成細胞から18kDaのhCAP18プロペプチドとして分泌され、SCTEによって切断されて、C末端に活性を持つ抗菌ペプチドLL−37(37個のアミノ酸長および2個のN末端ロイシンより命名)となる。酒さを伴う皮膚には、不適切な高濃度のカテリシジンおよびSCTEの両方が確認されているが、刺激となる菌の侵入ははっきりしていない。初代ヒトケリチノサイテスを覆う培地にLL−37を添加すると、IL−8等の走化性サイトカインの産生が大幅に増強される。酒さを伴う皮膚に認められる濃度と類似の濃度でLL−37をマウスに皮内注射すると紅斑が生じ、PMNの真皮内浸潤が強力に刺激される。SCTEを皮内注射した場合も野生型マウスに皮膚の紅斑およびPMNの真皮内浸潤が認められるが、カテリシジン−/−マウスには認められない。したがって、酒さの特徴である炎症、過度の血管新生、および皮脂腺過形成を生じさせる炎症促進性の陽性荷電皮膚ペプチドが局所的に過剰発現することがこの疾患を媒介しているという考えを支持する一連の証拠が増えつつある。酒さ患者のほぼ全員から、両親に一定のパターンの反応性顔面紅潮や発赤があると聞き取ることができることから、酒さに遺伝的要因があることは臨床医には明らかである。
【0125】
酒さのモデルを作製するために、LL−37を48時間の間、12時間毎に皮内注射した。Yamasakiらが報告したように、このモデルの皮膚には、紅斑および多形核白血球(PMN)の顕著な皮内浸潤が生じた。Balb/cマウスを調査前に剃毛し、背部の皮膚を剥き出しにした。24時間後、剃毛した皮膚に、ビヒクル40μuL(リン酸緩衝生理食塩水PBS)、陽性荷電ペプチド(PBS中、濃度320μM)、SAGE(PBS中320〜1280μM)、または陽性荷電ペプチド+SAGE混合物(ペプチド+1〜4倍モル濃度のSAGE)を、31ゲージの針を用いて、無傷の表皮に水疱が隆起するように計画された方法で皮内注射し、それによって表皮下または真皮レベルに投与していることが認識されるようにした。注射用に選択するSAGEは、新たに合成した12種類のSAGEから選択した。これらは、生化学的試験において、ヒト白血球エラスターゼ(他の陽性荷電タンパク質として)の阻害剤としても、終末糖化産物受容体(RAGE)の4種類の一般的なリガンドによる活性化の拮抗剤としても、活性が極めて高いという試験結果が得られたものである。
【0126】
注射前にSAGEおよびペプチドをPBS中で一緒に混合し、注射前に室温で15分間インキュベートした。その後、12時間毎に反復注射を行った。最初の注射から48時間後(合計4回注射)、動物にペントバルビタール25mg/kgを腹腔内投与して軽麻酔を行った。マウスが睡眠状態になったら、皮膚の注射した部位を写真撮影して、紅斑および浮腫の重症度を視認できるように記録した。紅斑の強さを発赤評点(1〜5)で評価し、紅斑の範囲をノギスで測定した。次いで、6mm穴のパンチを用いて皮膚の注射した範囲を切除生検してヘマトキシリン−エオシン染色によって組織病理学的変化を検査するとともにミエロペルオキシダーゼ(MPO)活性を測定することによってPMN浸潤を評価した。皮膚表面の代表的な画像および各皮膚の組織像を高倍率のマイクロスコープで観察した。
【0127】
SAGEで局所治療した酒さモデル:Balb/cマウス背部のLL37を作用させる皮膚の範囲を予め剃毛した。次いで、SAGEを5%(有効エモリエント成分)を含むヒアルロナン−ベースのエモリエント剤またはヒアルロン酸ベースのエモリエント剤を単独でこの皮膚の範囲に12時間毎に局所塗布することを開始した。24時間後、ビヒクル40μL(PBS)または陽性荷電ペプチド(濃度320μM)を剃毛した皮膚の皮下に上述した方法で注射した。注射およびエモリエント剤の局所塗布をその後12時間毎に繰り返した。最初の注射から48時間後(合計4回注射)、動物を上述したように軽麻酔した。皮膚の注射した範囲の写真を撮影することによって紅斑および浮腫の重症度を視認できるように記録した。紅斑の強さを発赤評点(1〜5)として評価し、紅斑の範囲をノギスで測定した。次いで、注射した皮膚の部位を6mm穴のパンチを用いて切除生検し、H&E染色によって組織病理学的変化を検査するとともにミエロペルオキシダーゼ(MPO)活性を測定することによってPMN浸潤を評価した。
【0128】
SAGEの真皮への侵入:調査を行う前に、Balb/cマウス背部の皮膚の範囲を剃毛して剥き出しにした。12時間毎にSAGEを皮膚に局所塗布した。48時間後、動物を安楽死させて皮膚を生検した。次いで、皮膚の一部を蛍光顕微鏡で調査することによって、SAGEが皮膚に侵入した深さを測定した。
【0129】
クロトン油炎症モデル:PMNが媒介する皮膚炎症の他のモデルとしてクロトン油を用いた。クロトン油は、皮膚細胞のプロテインキナーゼCを活性化するフォルボールエステルを含有している。その結果として、皮膚細胞が大量のケモカインおよびケモタキシンを産生し、そのシグナル伝達によって血液中のPMNが流入する。活性化されたPMNは皮膚組織に紅斑および浮腫を生じさせる。クロトン油に誘発される炎症は、皮膚科で使用される抗炎症化合物のスクリーニングにおいて、PMNが媒介する皮膚炎症モデルとしてよく用いられている。
【0130】
このモデルを作製するために、クロトン油(Sigma-Aldrich,St.Louis,MO)をアセトンと混合して0.8%溶液とした。ピペットを用いてマウスの片方の耳介の各面に10μLを塗布し、他方の耳介は対照としてそのままにした。4、8、および24時間後、軟骨隆起より遠位の頂部付近の耳介の厚みを測定した。そして、対照の耳介の厚みからの変化を浮腫の指標とした。紅斑の強さを発赤評点(1〜5)として評価し、紅斑の範囲をノギスで測定した。24時間後に測定した後、マウスを安楽死させてすぐに耳介のパンチ生検(6mm穴パンチ)を実施し、重量を測定し、凍結させて−80℃で保管し、H&E染色によって組織病理学的変化を検査するとともにミエロペルオキシダーゼ(MPO)活性を測定することによってPMN浸潤を評価した。手順を標準化し、誤差を減らすために、すべての耳介に関する測定および生検を1人の検査員が行った。残りの耳介を取り除き、免疫組織化学用に包埋して凍結させた。
【0131】
ミエロペルオキシダーゼ(MPO)測定。各マウスに対し組織生検(直径6mm穴のパンチ)を即座に実施し、重量を測定し、−80℃で凍結させて保管した。組織のMPO活性を、Suzukiらの方法(Suzuki K、Ota H、Sasagawa S、Sakatani T、Fujikura T、ヒト多形核白血球中のミエロペルオキシダーゼ測定方法(Assay method for myeloperoxidase in human polymorphonuclear leukocytes.)、Anal Biochem 132:345-352、1983)をYoungら(Young JM、Spires DA、Bedord CJ、Wagner B、Ballaron SJ、De oung LM、局所的なアラキドン酸に対するマウス耳介の炎症応答(The mouse ear inflammatory response to tpical arachidonic acid.)、J Invest Dermatol 82:367-371、1984)によって改変されたものを用いて測定した。各マウスの生検組織を、ヘキサデシルトリメチル−アンモニウムブロミド(HTAB)0.5%を含む80mMのリン酸緩衝生理食塩水(PBS)(pH5.4)0.75mLに入れた。各試料を小型の実験室用Tissue Tearor Homogenizer Model 985-370(Biospec Products,Bartlesville,OK)で4℃で45秒間ホモジナイズした。ホモジネートを定量的に微小遠心管に移し、PBS中のHTABを0.75mLを加えた。試料1.5mLを4℃に維持しながら12000×gで15分間遠心分離した。結果として得られた上清の試料30uLをトリプリケートで96穴マイクロタイタープレートのウェルに加えた。MPO測定を行うため、80mMのPBS(pH5.4)100uL、0.22MのPBS(pH5.4)85uL、および0.017%の過酸化水素15uLを含む混合物200uLを各ウェルに加えた。8%ジメチルホルムアミド水溶液中の18.4mMのテトラメチルベンジジンHClを20uLを加えて反応を開始した。マイクロタイタープレートを37℃で3分間インキュベートした後、氷に載せた。1.46Mの酢酸ナトリウム30uLを加えて反応を停止した。MPO酵素活性を波長630nmの吸光度で評価した。MPO活性を光学濃度(OD)/生検として表した。
【0132】
統計解析。in vitro試験に関する実験はすべてトリプリケートで実施した。Aspin−Welch検定を用いて平均値を比較することにより試料の有意差を計算した。p<0.05を有意差ありと宣言した。
【0133】
結果
LL37ペプチドおよびSAGEを注射した酒さモデル:陽性荷電カテリシジンを直接中和すると皮膚内の炎症活性が阻害されるかどうかを確認するために、LL37単独、SAGE(GM−111101)単独、ビヒクル(PBS)単独、またはLL37およびSAGEの混合物をマウス背部の剃毛した範囲に、その後12時間毎に皮下注射した。48時間後にマウスを屠殺し、異なる処理群の肉眼解剖写真を撮影した(図10aおよび10b)。ヘマトキシリン−エオシン染色を用いた組織学的研究から、白血球の浸潤の増大および顕著な皮膚の浮腫が認められる一方、誘発直後にSAGEを投与すると皮膚の腫脹反応が阻止されることが示された(図10cおよび10d)。
【0134】
皮膚の評点の個々の結果を紅斑面積(図10f)および紅斑の発赤評点(図10g)で表した。48時間後、SAGE処理群は、紅斑面積が劇的に減少するとともに発赤評点が大幅に低下することが示された。注射の48時間後に組織のパンチ生検を行い、PMN浸潤の指標としてミエロペルオキシダーゼ活性を測定した。SAGEをLL−37ペプチドと同時投与するとMPO活性が大幅に(50%)低下した(図10e)。したがって、SAGEをLL−37ペプチドと同時に注射することによりLL−37カテリシジンペプチドの炎症活性が実質的に誘導された。このことは、SAGEがLL−37が媒介する炎症を阻害し、酒さの治療に有用であることを示唆している。
【0135】
SAGEによる酒さの局所治療モデル:SAGE(GM−111101)の局所治療を用いることにより、LL−37を作用させてから時間が経過した後に治療を行ってもペプチドに誘発される皮膚炎症を阻止できるのかどうかを試験した。これに従い、マウス背部の皮膚の範囲にLL−37を注射した直後にSAGEを塗布した。肉眼解剖写真は、LL−37を適用してから48時間後に強度の浮腫および紅斑が現れたことを示している(図11a、11h、および11i)。一方、SAGEを用いた局所治療を行うと、直後の治療(図11b)および12時間遅れてからの治療(図11c)の両方において、発赤および患部が大幅に減少した。H&E染色から、2つのSAGE治療群と比較して、白血球浸潤および皮膚の浮腫がはるかに高いことが示され、これは、SAGEが皮膚腫脹反応およびMPO活性の阻害剤であるという結果と一致していた(図11g)。これらの結果は、酒さによるカテリシジンを介した炎症を治療するために、SAGEを従来の薬学的に許容されるエモリエント剤中で局所的に適用してもよいことを示唆している。
【0136】
SAGEの真皮侵入:SAGEが真皮に侵入するレベルを確認する。SAGE化合物である蛍光体−GM−212101および蛍光体−GM−111101を被験品として使用し、Balb/cマウスの背部の皮膚の一部分に傷を付けて触れないようにして、蛍光化合物を0.1mg/mL、1mg/mL、および10mg/mLを塗布した。24時間後にマウスを屠殺し、皮膚の被験部位全体を切除し、自然光および長波長UV光条件下の両方で撮影した(図12)。自然光およびUV光条件下の両方で、皮膚の治療部位の内側および外側の両方に蛍光物質の層が認められた。SAGEの顕著な侵入による分布はマイクロメータースケールにまで及んでいた。
【0137】
細胞毒性および皮膚刺激性のin Vivo試験:SAGE誘導体(SAGEs)であるGM−131101、GM−312101、およびGM−212101の細胞毒性をnHDF細胞内で評価した。結果を図13aに示す。どの化合物も濃度10mg/mLまではnHEKにも無毒であることがわかった(図13b)。in vivo皮膚刺激性試験に関する異なる治療群のマウスの肉眼解剖写真を図13c〜13jに示す。被験物質の一次刺激指数を求めたところ、GM−111101およびGM−212101は両方とも0.00であった。マウスの皮膚に炎症は認められなかった(図14)。GM−111101およびGM−212101は両方とも細胞毒性が認められなかった。この試験から、すべてのSAGEの濃度閾値を求めることができた。16 CFR 1500のFHSA Regulationsの指針において、実験による評点が5.00未満の物質は皮膚の一次刺激物ではないと定めされているように、この試験条件下におけるこの被験試薬は一次皮膚刺激物ではないとみなされるであろう。これらの結果は、SAGE自体には皮膚刺激性がなく、炎症性皮膚疾患の治療に安全に用いることができることを示している。
【0138】
クロトン油炎症モデル:簡便で非常に再現性の高いPMN媒介皮膚炎症モデルとして、マウス皮膚へのクロトン油塗布を用いた。このモデルを用いてSAGEの抗炎症活性を試験した。異なる治療群のマウスの肉眼解剖写真を図15aおよび15bに示すとともに、治療および未治療の耳介の両方の耳介厚を測定して、5つの群すべてで比較した。個々の皮膚評点の結果を、傷付けた範囲および無傷の範囲の紅斑で表した。この結果から、SAGE(GM−111101)治療群の発赤および厚みが未治療群と比較して大幅に低下したことが示された(図15gおよび15h)。組織病理学検査により、クロトン油を塗布した耳介においては、浸潤白血球数の増加および顕著な真皮の浮腫が見られた一方で、誘発直後にSAGEを投与すると耳介腫脹反応が阻害される(これはビヒクルで処理したマウスに匹敵していた)ことがわかった。これらの組織学的発見を、測定値によってさらに確認した。クロトン油塗布後の耳介パンチ生検によってMPO活性も測定した。クロトン油塗布直後からSAGE治療を開始して4時間毎に実施したところ、MPO活性が有意に低下した(図15f)。これらの結果は、SAGEを酒さ以外の炎症性皮膚疾患の局所治療に用いることができることを示唆している。
【0139】
酒さモデルのヒアルロン酸(HA)による局所治療:前述のLL−37酒さモデルの局所治療を用いてSAGE(GM−111101)対HAの比較を行った。その結果から、HAの局所投与では炎症(図16b、16e、および7f)およびMPO活性(図16d)が軽減されないことがはっきりと示された。これとは逆に、SAGEは抗炎症性が非常に増強されており(図16c〜f)、炎症および酒さの阻害剤とみなすことができるであろう。これらのデータは、SAGEの薬理活性はヒアルロン酸には備わっておらず、ヒアルロナンに新規な修飾を施さなければならないことを示唆している。
【0140】
ラットの静脈内投与によるIn Vivo急性毒性試験:本試験の目的は、SAGEであるGM−111101およびGM−212101を静脈内投与によりラットに単回投与した場合の急性毒性の評価に加えて、GM−111101を7日間の間に1日1回、単回投与量を投与することによる毒性を評価することにあった。
【0141】
スプラーグドーリー(Sprague-Dawley)雌雄各3匹のラットから構成される1つの投与群に、GM−111101を3mg/kgを単回投与し、次いで、1週間後にGM−111101を10mg/kgを単回投与し、最後に、最後の単回投与から1週間後に、GM−111101を10mg/kgの1日1回投与を開始して合計7日間実施した。スプラーグドーリー雌雄各3匹のラットの2つの群には、GM−111101を30または100mg/kgを投与した。さらに、スプラーグドーリー雌雄各3匹のラットの2つの群には、GM−212101を30または100mg/kgを投与した。スプラーグドーリー雌雄各3匹のラットの2つの群には、塩化ナトリウム0.9%を注射して陰性対照群とした。すべての用量は、投与量を1mL/kgとし、尾骨静脈から静脈注射することによって投与した。直前に記録された体重に基づき投与量を計算した。
【0142】
急激に(単回)投与した動物にはいずれも、注射直後ならびに0日目の用量投与の2時間および4時間後にも同様に、臨床的毒性の明らかな徴候が認められた。さらに、すべての生存動物について、臨床的毒性の目に見える徴候について1日1回、1〜14日目まで観察した。1日1回、7日間投与したすべての動物について、臨床的毒性の目に見える徴候について1日1回、1〜14日目まで観察した。0日目(急激投与)または1日目(反復投与)の用量投与前、6または7日目、および14日目の終了前に体重を記録した。試験から14日目にそれぞれの生存動物について肉眼的剖検による評価を行った。試験途中に死亡した動物は、死亡率を観察した後、すぐに肉眼的剖検により検査した。
【0143】
GM−212101を30mg/kg投与した群の1匹の雌動物に、用量投与後の最初の2時間以内に、中等度の歩行異常、中等度の運動失調、および尿が赤みがかった橙色に変色するという臨床徴候が認められた。これらの所見は、軽い運動失調を除いて、4時間後の観察時間の時点では既に存在せず、残りの試験の間はずっとそのままであった。運動失調以外の所見は、用量投与の翌日にはもはや存在しなかった。さらに、GM−212101を100mg/kg投与した群の1匹の雄のラットが、用量投与後の最初の4分以内に死亡していた。すべての動物は試験期間中に体重が増加した。試験中に死亡したGM−212101を100mg/kg投与した群の1匹の動物の胸腺全体に直径約1〜2mmの暗赤色の病巣が観察されたことを除いて、視認できる病変は剖検では観察されなかった。
【0144】
この試験結果に基づくと、GM−111101は、3、10、30、および100mg/kgの急激な単回投与ならびに7日間の10mg/kgの反復投与を含む、評価を行ったどの用量においても毒性の徴候が見られなかった。したがって、ラットにGM−111101を静脈内投与した場合の最大無作用量(NOEL)は少なくとも100mg/kgとみなされる。GM−212101は、30および100mg/kgの用量で毒性または致死性の徴候が見られた。したがって、ラットにGM−212101を静脈内投与した場合のNOELは10mg/kgとみなされる。GM−111101のすべての用量において死亡が認められず、GM−212101を100mg/kg投与した動物の17%にしか死亡が認められなかったことから、ラットにGM−111101およびGM−212101を静脈投与した場合のLD50は100mg/kgを超えるとみなされる。この結果は、SAGEが疾患の全身および注射治療に安全に用いられることを示唆している。
【0145】
VI.加齢黄斑変性の治療に関するSAGEの調査。
活性化された補体およびRAGEが培養RPE細胞における血管形成および炎症促進シグナル伝達を誘発する:培養RPE細胞におけるRAGEの生物学を調査する実験を、ARPE−19ヒトRPE細胞株を用いて実施した。免疫ブロットに示すように(図17)、ARPE−19細胞は、細胞可溶化物中で45〜50kDaの範囲の少なくとも4種のRAGEのアイソフォームを発現し、これらのアイソフォームを条件培地に分泌する。AGE産物であるCML−BSAでコーティングされたプレートで細胞を増殖させた場合、4種類すべてのRAGEアイソフォームの発現が大幅に上方制御された(図17の免疫ブロットの右側を左側と比較)。RAGEが連結することによって転写因子NF−κBが活性化されるため、RAGEの発現が増加することによって炎症促進性のシグナル伝達が大幅に促進される。この系に非抗凝固性(nonanticoagulant)ヘパリンである2−O,3−O脱硫酸化ヘパリン(ODSH)を加えると、ARPE−19細胞上でのCML−BSAとRAGEとの相互作用が阻害されることにより、RAGE発現の上方制御が阻止された。こうすることにより、RAGEが連結することによって炎症促進性のRAGE自体の発現が「フィードフォワード」的に増大することが阻止されるであろう。
【0146】
AGE産物がRPE細胞死を誘導する能力を調査した。ARPE−19細胞を丸型カバーガラスでコンフルエントになるまで増殖させた後、新しい皿に移し、25μMのAGE−BSAに40時間曝露した。次いで、Molecular Probes Fixable Live/Dead Cell Stain Kit(L11101,Eugene,OR)を用いて細胞死の検査を行った。各パネルにカバーガラス全体および生/死細胞界面の拡大図を示す。核がDAPIによって赤色に染色され、死細胞が緑色に染色される。A.対照BSA。カバーガラスの縁部に死細胞が若干認められたが、界面に生および死細胞が散在している。B.AGE−BSA(25μM)。顕著な細胞死が縁部周囲に認められ、生および死細胞の境界がはっきりとしている。C.AGE−BSA(25μM)+ODSH(200μM)。ODSHによってある程度保護されるようであるが、顕著な細胞死が依然として起こっている。D.AGE−BSA(25μM)+P−OSMeHA(200μM)(GM−111101)。
【0147】
図18に示すように、Live/Dead Cell Stain kit(Molecular Probes)を用いた緑色染色により測定すると、培養ARPE−19細胞をAGEで処理することによって顕著な細胞死が誘導されている(図18B)。細胞をODSHと共にインキュベートすることにより細胞死が低減されるが(図18C)、対応する濃度のSAGA P−OSMeHA(GM−111101)ではほぼ完全に阻止された(図18D)。細胞死は、丸型カバーガラス上で培養した細胞の縁部から内側に向かって進行しているようであった。RAGEはRPE細胞内で顕著に発現し、他のヒト上皮細胞と同様に、基底膜上で選択的に発現するであろう。したがって、培地中においては、AGEは最初は縁部の単層の基底に位置するRAGEにのみ到達して溶解することができ、内側に進行すると予想される細胞死を相次いで起こさせるであろう。これとは対照的に、AGEが網膜色素上皮およびブルッフ膜の間に蓄積されるドルーゼン内にある場合は、基底に位置するRAGEにすぐに到達することとなり、加齢黄斑変性においていわゆる「地図状萎縮」を形成する、RPEの限局的な細胞死を媒介するAGE/RAGEシグナル伝達が最適化される。したがって、SAGEであるGM−111101は、AGEに誘導されるRPE細胞死をほぼ完全に阻止する。これらの結果は、失明を招く加齢黄斑変性等の重要な眼疾患の治療にSAGEが有効となり得ることを示唆している。
【0148】
SAGEは無毒性であり非抗凝固性である:O−硫酸化およびメチル化されたHA(P−OSMeHA)、完全O−硫酸化およびペンタフルオロプロピル化されたHA(F−OSFHA−1)ならびに完全O−硫酸化およびメチル化されたHA(F−OSMeHA)を、培養したヒト皮膚上皮細胞または線維芽細胞に適用し、細胞毒性アッセイ(CellTiter96(登録商標)Aqueous One assay,Promega)により調査すると、濃度が1mg/mLでさえもSAGEは増殖を阻害せず、細胞毒性も示さない。SAGEはまた、非抗凝固性でもない。低分子量硫酸化フルオロアルキル化HAは抗−Xaを示さず、抗IIa抗凝固活性が<0.2U/mgであり、これと比較すると、未分画ヘパリンの場合はそれぞれ150U/mgである。
【0149】
ヘパリンとは異なり、高度に帯電した多価陰イオン性高分子は、ハーゲマン因子(第XIIa因子)を活性化し、二次的にキニンを活性化する、内因性または接触凝固カスケードの強力な誘発物質である。SAGEの内因性凝固を刺激する能力(ハーゲマン因子の活性化)についてスクリーニングを行った。プールしたヒト血漿をヘパリンまたは低分子量(Lmw)硫酸化フルオロアルキル化HA(Lmw−OSFHA−1、Lmw−OSFHA−2)とインキュベートし、D−シクロヒドロチロシル−Gly−Arg−p−NAを基質として用いてアミド分解活性を測定した。第XII因子の活性化能力について試験すると、図19に示すように、低分子量(50kDa)SAGEは、炎症を薬理学的に阻害する濃度よりも10〜100倍高いSAGE濃度でさえも、市販の医療用ヘパリンよりもはるかに安全性が高いようである。
【0150】
本出願を通して様々な刊行物が参照される。本明細書に記載した化合物、組成物、および方法をより十分に説明するために、これらの刊行物の開示内容全体を本出願に参考として組み込むものとする。
【0151】
本明細書に記載した化合物、組成物、および方法の様々な修正および変形が可能である。本明細書に記載した化合物、組成物、および方法の他の態様は、本明細書に開示した化合物、組成物、および方法の詳細および実施を考察することによって明らかになるであろう。この詳細および実施例は例示的なものとみなされることを意図している。
【特許請求の範囲】
【請求項1】
ヒアルロナンもしくはその誘導体を含む化合物またはその薬学的に許容される塩もしくはエステルであって、N−アセチル−グルコサミン残基の少なくとも1個の第1級C−6ヒドロキシルプロトンがアルキル基またはフルオロアルキル基で置換されている、化合物。
【請求項2】
前記アルキル基が、C1〜C10分岐または直鎖アルキル基を含む、請求項1に記載の化合物。
【請求項3】
前記アルキル基が、メチル、エチル、プロピル、イソプロピル、ブチル、ペンチル、またはヘキシルを含む、請求項1に記載の化合物。
【請求項4】
前記アルキル基が、メチルである、請求項1に記載の化合物。
【請求項5】
前記フルオロアルキル基が、少なくとも1個のトリフルオロメチル基を含む、請求項1に記載の化合物。
【請求項6】
前記フルオロアルキル基が、式−CH2(CF2)nCF3(式中、nは、0〜10の整数である)を含む、請求項1に記載の化合物。
【請求項7】
nが、1、2、3、4、または5である、請求項6に記載の化合物。
【請求項8】
前記N−アセチル−グルコサミン残基の前記第1級C−6ヒドロキシルプロトンの少なくとも1%が、アルキル基またはフルオロアルキル基で置換されている、請求項1〜7のいずれか一項に記載の化合物。
【請求項9】
前記N−アセチル−グルコサミン残基の前記第1級C−6ヒドロキシルプロトンの1%〜50%が、アルキル基またはフルオロアルキル基で置換されている、請求項1〜7のいずれか一項に記載の化合物。
【請求項10】
少なくとも1個のC−2ヒドロキシルプロトンおよびC−3ヒドロキシルプロトンがアルキル基またはフルオロアルキル基で置換されている、請求項1〜7のいずれか一項に記載の化合物。
【請求項11】
前記ヒアルロナンのアルキル化またはフルオロアルキル化前の分子量が10kDaを超える、請求項1〜10のいずれか一項に記載の化合物。
【請求項12】
前記ヒアルロナンのアルキル化またはフルオロアルキル化前の分子量が40kDa〜2000kDaである、請求項1〜10のいずれか一項に記載の化合物。
【請求項13】
少なくとも1個のC−2ヒドロキシルプロトンまたはC−3ヒドロキシルプロトンが硫酸基で置換されている、請求項1〜12のいずれか一項に記載の化合物。
【請求項14】
少なくとも1個のC−2ヒドロキシルプロトンおよびC−3ヒドロキシルプロトンが硫酸基で置換されている、請求項1〜12のいずれか一項に記載の化合物。
【請求項15】
前記化合物の硫酸化度が、二糖単位当たり0.5〜3.5である、請求項13または14に記載の化合物。
【請求項16】
前記アルキル基がメチルであり、かつ少なくとも1個のC−2ヒドロキシルプロトンおよび/またはC−3ヒドロキシルプロトンが硫酸基で置換されている、請求項1に記載の化合物。
【請求項17】
前記アルキル基がメチルであり、少なくとも1個のC−2ヒドロキシルプロトンおよび/またはC−3ヒドロキシルプロトンが硫酸基で置換されており、かつ前記化合物の分子量が2kDa〜10kDaである、請求項1に記載の化合物。
【請求項18】
前記フルオロアルキル基が−CH2CF2CF3であり、かつ少なくとも1個のC−2ヒドロキシルプロトンおよび/またはC−3ヒドロキシルプロトンが硫酸基で置換されている、請求項1に記載の化合物。
【請求項19】
前記ヒアルロナンまたはその誘導体が、動物起源に由来するものではない、請求項1〜18のいずれか一項に記載の化合物。
【請求項20】
薬学的に許容される化合物および請求項1〜19のいずれか一項に記載の化合物を含む、医薬組成物。
【請求項21】
請求項1〜18のいずれか一項に記載の化合物を含む、医薬組成物。
【請求項22】
前記組成物のpHが5〜6である、請求項20または21に記載の組成物。
【請求項23】
創傷治癒の改善を必要とする対象者にこのような改善を施すための方法であって、前記患者の前記創傷を、請求項1〜19のいずれか一項に記載の化合物と接触させることを含む、方法。
【請求項24】
少なくとも1種の薬学的に許容される化合物の送達を必要とする患者にこのような送達を行う方法であって、前記薬学的に許容される化合物を受容することができる少なくとも1つの組織に請求項1〜19のいずれか一項に記載の化合物を接触させることを含み、前記化合物が、前記薬学的に許容される化合物を含む、方法。
【請求項25】
眼科学的状態、尿生殖器状態、呼吸器状態、心血管状態、胃腸疾患、リウマチおよび免疫疾患、腎疾患、変性関節疾患、歯科もしくは口腔疾患、または神経疾患を治療または予防するための、請求項1〜19のいずれか一項に記載の化合物の使用。
【請求項26】
歯科および口腔外科における、請求項1〜19のいずれか一項に記載の化合物の使用。
【請求項27】
皮膚障害を治療または予防するための方法であって、請求項1〜19のいずれか一項に記載の化合物を前記皮膚の表面に適用することを含む、方法。
【請求項28】
前記皮膚障害が、酒さ、アトピー性皮膚炎(湿疹)、アレルギー性接触皮膚炎、乾癬、ヘルペス状皮膚炎、ざ瘡、糖尿病性皮膚潰瘍および他の糖尿病性創傷、熱傷、日光皮膚炎、瘢痕予防、光線角化症、虫刺されによる炎症、ツタウルシによるかぶれ、放射線皮膚炎/熱傷、または脂漏性皮膚炎を含む、請求項27に記載の方法。
【請求項29】
対象者の炎症を軽減または予防するための方法であって、前記対象者に、有効量の請求項1〜19のいずれか一項に記載の化合物を投与することを含む、方法。
【請求項30】
前記炎症が、癌、多発性硬化症、変形性関節症、関節リウマチ、アルツハイマーのベータアミロイド、歯周疾患、炎症性腸疾患、喘息、鼻炎、副鼻腔炎、慢性閉塞性肺疾患、急性肺傷害、嚢胞性線維症、鎌状赤血球貧血、炎症性心血管疾患、炎症性肺疾患、炎症性眼疾患、炎症性脳疾患、または炎症性腸管疾患により生じる、請求項29に記載の方法。
【請求項31】
眼疾患を治療または予防するための方法であって、前記眼に請求項1〜19のいずれか一項に記載の化合物を投与することを含む、方法。
【請求項32】
前記眼疾患が、加齢黄斑変性、糖尿病性網膜症、眼球乾燥症候群、結膜炎、虹彩炎、ブドウ膜炎、アレルギー性結膜炎、または角膜炎および瘢痕を含む、請求項31に記載の方法。
【請求項33】
前記化合物が眼内投与される、請求項31に記載の方法。
【請求項34】
P−セレクチンの活性を低下させるかまたは阻害するための、請求項1〜19のいずれか一項に記載の化合物の使用。
【請求項35】
RAGEの活性を低下させるかまたは阻害するための、請求項1〜19のいずれか一項に記載の化合物の使用。
【請求項36】
ヒト白血球エラスターゼの活性を低下させるかまたは阻害するための、請求項1〜19のいずれか一項に記載の化合物の使用。
【請求項37】
ヒアルロナンまたはその誘導体をアルキル化またはフルオロアルキル化するための方法であって、(a)前記ヒアルロナンまたはその誘導体を、N−アセチル−グルコサミン残基の少なくとも1個の第1級C−6ヒドロキシルプロトンを脱プロトン化するのに十分な量の塩基と反応させることと、(b)前記脱プロトン化されたヒアルロナンまたはその誘導体を、アルキル化剤またはフルオロアルキル化剤と、少なくとも1個の脱プロトン化された第1級C−6ヒドロキシル基をアルキル化またはフルオロアルキル化するのに十分な時間および濃度で反応させることとを含む、方法。
【請求項38】
前記塩基が、アルカリ金属水酸化物を含む、請求項37に記載の方法。
【請求項39】
前記アルキル化剤が、ハロゲン化アルキルを含む、請求項37に記載の方法。
【請求項40】
前記フルオロアルキル化剤が、ハロゲン化フルオロアルキルを含む、請求項37に記載の方法。
【請求項41】
ステップ(b)の後に、前記アルキル化もしくはフルオロアルキル化ヒアルロナンまたはその誘導体を硫酸化剤と反応させることによって硫酸化物を生成させることを含む、前記アルキル化もしくはフルオロアルキル化ヒアルロナンまたはその誘導体を硫酸化することをさらに含む、請求項37に記載の方法。
【請求項42】
前記硫酸化物が、部分硫酸化されている、請求項41に記載の方法。
【請求項43】
前記硫酸化物が、完全硫酸化されている、請求項41に記載の方法。
【請求項1】
ヒアルロナンもしくはその誘導体を含む化合物またはその薬学的に許容される塩もしくはエステルであって、N−アセチル−グルコサミン残基の少なくとも1個の第1級C−6ヒドロキシルプロトンがアルキル基またはフルオロアルキル基で置換されている、化合物。
【請求項2】
前記アルキル基が、C1〜C10分岐または直鎖アルキル基を含む、請求項1に記載の化合物。
【請求項3】
前記アルキル基が、メチル、エチル、プロピル、イソプロピル、ブチル、ペンチル、またはヘキシルを含む、請求項1に記載の化合物。
【請求項4】
前記アルキル基が、メチルである、請求項1に記載の化合物。
【請求項5】
前記フルオロアルキル基が、少なくとも1個のトリフルオロメチル基を含む、請求項1に記載の化合物。
【請求項6】
前記フルオロアルキル基が、式−CH2(CF2)nCF3(式中、nは、0〜10の整数である)を含む、請求項1に記載の化合物。
【請求項7】
nが、1、2、3、4、または5である、請求項6に記載の化合物。
【請求項8】
前記N−アセチル−グルコサミン残基の前記第1級C−6ヒドロキシルプロトンの少なくとも1%が、アルキル基またはフルオロアルキル基で置換されている、請求項1〜7のいずれか一項に記載の化合物。
【請求項9】
前記N−アセチル−グルコサミン残基の前記第1級C−6ヒドロキシルプロトンの1%〜50%が、アルキル基またはフルオロアルキル基で置換されている、請求項1〜7のいずれか一項に記載の化合物。
【請求項10】
少なくとも1個のC−2ヒドロキシルプロトンおよびC−3ヒドロキシルプロトンがアルキル基またはフルオロアルキル基で置換されている、請求項1〜7のいずれか一項に記載の化合物。
【請求項11】
前記ヒアルロナンのアルキル化またはフルオロアルキル化前の分子量が10kDaを超える、請求項1〜10のいずれか一項に記載の化合物。
【請求項12】
前記ヒアルロナンのアルキル化またはフルオロアルキル化前の分子量が40kDa〜2000kDaである、請求項1〜10のいずれか一項に記載の化合物。
【請求項13】
少なくとも1個のC−2ヒドロキシルプロトンまたはC−3ヒドロキシルプロトンが硫酸基で置換されている、請求項1〜12のいずれか一項に記載の化合物。
【請求項14】
少なくとも1個のC−2ヒドロキシルプロトンおよびC−3ヒドロキシルプロトンが硫酸基で置換されている、請求項1〜12のいずれか一項に記載の化合物。
【請求項15】
前記化合物の硫酸化度が、二糖単位当たり0.5〜3.5である、請求項13または14に記載の化合物。
【請求項16】
前記アルキル基がメチルであり、かつ少なくとも1個のC−2ヒドロキシルプロトンおよび/またはC−3ヒドロキシルプロトンが硫酸基で置換されている、請求項1に記載の化合物。
【請求項17】
前記アルキル基がメチルであり、少なくとも1個のC−2ヒドロキシルプロトンおよび/またはC−3ヒドロキシルプロトンが硫酸基で置換されており、かつ前記化合物の分子量が2kDa〜10kDaである、請求項1に記載の化合物。
【請求項18】
前記フルオロアルキル基が−CH2CF2CF3であり、かつ少なくとも1個のC−2ヒドロキシルプロトンおよび/またはC−3ヒドロキシルプロトンが硫酸基で置換されている、請求項1に記載の化合物。
【請求項19】
前記ヒアルロナンまたはその誘導体が、動物起源に由来するものではない、請求項1〜18のいずれか一項に記載の化合物。
【請求項20】
薬学的に許容される化合物および請求項1〜19のいずれか一項に記載の化合物を含む、医薬組成物。
【請求項21】
請求項1〜18のいずれか一項に記載の化合物を含む、医薬組成物。
【請求項22】
前記組成物のpHが5〜6である、請求項20または21に記載の組成物。
【請求項23】
創傷治癒の改善を必要とする対象者にこのような改善を施すための方法であって、前記患者の前記創傷を、請求項1〜19のいずれか一項に記載の化合物と接触させることを含む、方法。
【請求項24】
少なくとも1種の薬学的に許容される化合物の送達を必要とする患者にこのような送達を行う方法であって、前記薬学的に許容される化合物を受容することができる少なくとも1つの組織に請求項1〜19のいずれか一項に記載の化合物を接触させることを含み、前記化合物が、前記薬学的に許容される化合物を含む、方法。
【請求項25】
眼科学的状態、尿生殖器状態、呼吸器状態、心血管状態、胃腸疾患、リウマチおよび免疫疾患、腎疾患、変性関節疾患、歯科もしくは口腔疾患、または神経疾患を治療または予防するための、請求項1〜19のいずれか一項に記載の化合物の使用。
【請求項26】
歯科および口腔外科における、請求項1〜19のいずれか一項に記載の化合物の使用。
【請求項27】
皮膚障害を治療または予防するための方法であって、請求項1〜19のいずれか一項に記載の化合物を前記皮膚の表面に適用することを含む、方法。
【請求項28】
前記皮膚障害が、酒さ、アトピー性皮膚炎(湿疹)、アレルギー性接触皮膚炎、乾癬、ヘルペス状皮膚炎、ざ瘡、糖尿病性皮膚潰瘍および他の糖尿病性創傷、熱傷、日光皮膚炎、瘢痕予防、光線角化症、虫刺されによる炎症、ツタウルシによるかぶれ、放射線皮膚炎/熱傷、または脂漏性皮膚炎を含む、請求項27に記載の方法。
【請求項29】
対象者の炎症を軽減または予防するための方法であって、前記対象者に、有効量の請求項1〜19のいずれか一項に記載の化合物を投与することを含む、方法。
【請求項30】
前記炎症が、癌、多発性硬化症、変形性関節症、関節リウマチ、アルツハイマーのベータアミロイド、歯周疾患、炎症性腸疾患、喘息、鼻炎、副鼻腔炎、慢性閉塞性肺疾患、急性肺傷害、嚢胞性線維症、鎌状赤血球貧血、炎症性心血管疾患、炎症性肺疾患、炎症性眼疾患、炎症性脳疾患、または炎症性腸管疾患により生じる、請求項29に記載の方法。
【請求項31】
眼疾患を治療または予防するための方法であって、前記眼に請求項1〜19のいずれか一項に記載の化合物を投与することを含む、方法。
【請求項32】
前記眼疾患が、加齢黄斑変性、糖尿病性網膜症、眼球乾燥症候群、結膜炎、虹彩炎、ブドウ膜炎、アレルギー性結膜炎、または角膜炎および瘢痕を含む、請求項31に記載の方法。
【請求項33】
前記化合物が眼内投与される、請求項31に記載の方法。
【請求項34】
P−セレクチンの活性を低下させるかまたは阻害するための、請求項1〜19のいずれか一項に記載の化合物の使用。
【請求項35】
RAGEの活性を低下させるかまたは阻害するための、請求項1〜19のいずれか一項に記載の化合物の使用。
【請求項36】
ヒト白血球エラスターゼの活性を低下させるかまたは阻害するための、請求項1〜19のいずれか一項に記載の化合物の使用。
【請求項37】
ヒアルロナンまたはその誘導体をアルキル化またはフルオロアルキル化するための方法であって、(a)前記ヒアルロナンまたはその誘導体を、N−アセチル−グルコサミン残基の少なくとも1個の第1級C−6ヒドロキシルプロトンを脱プロトン化するのに十分な量の塩基と反応させることと、(b)前記脱プロトン化されたヒアルロナンまたはその誘導体を、アルキル化剤またはフルオロアルキル化剤と、少なくとも1個の脱プロトン化された第1級C−6ヒドロキシル基をアルキル化またはフルオロアルキル化するのに十分な時間および濃度で反応させることとを含む、方法。
【請求項38】
前記塩基が、アルカリ金属水酸化物を含む、請求項37に記載の方法。
【請求項39】
前記アルキル化剤が、ハロゲン化アルキルを含む、請求項37に記載の方法。
【請求項40】
前記フルオロアルキル化剤が、ハロゲン化フルオロアルキルを含む、請求項37に記載の方法。
【請求項41】
ステップ(b)の後に、前記アルキル化もしくはフルオロアルキル化ヒアルロナンまたはその誘導体を硫酸化剤と反応させることによって硫酸化物を生成させることを含む、前記アルキル化もしくはフルオロアルキル化ヒアルロナンまたはその誘導体を硫酸化することをさらに含む、請求項37に記載の方法。
【請求項42】
前記硫酸化物が、部分硫酸化されている、請求項41に記載の方法。
【請求項43】
前記硫酸化物が、完全硫酸化されている、請求項41に記載の方法。
【図1】

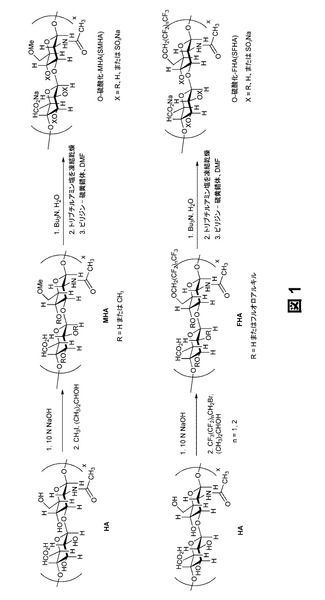
【図2】

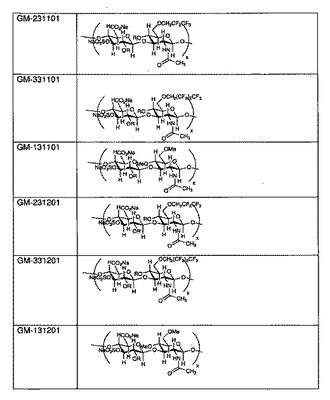
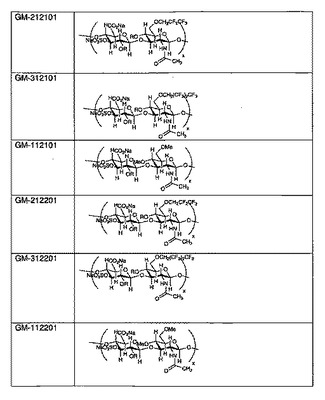
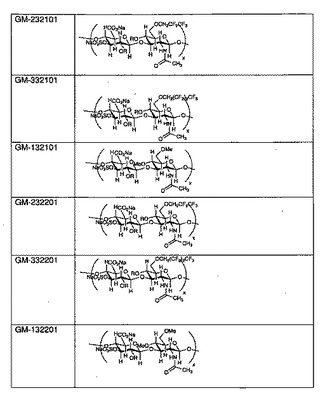
【図3】
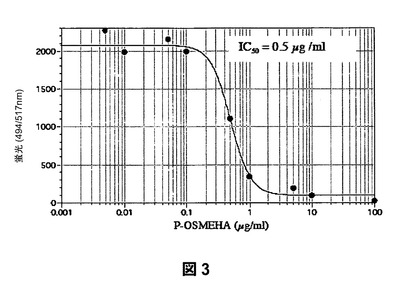
【図4】
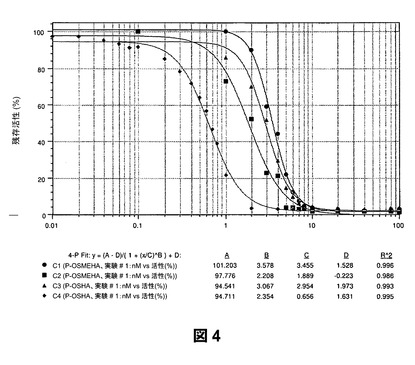
【図5】
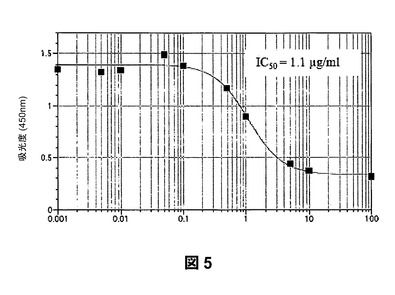
【図6】
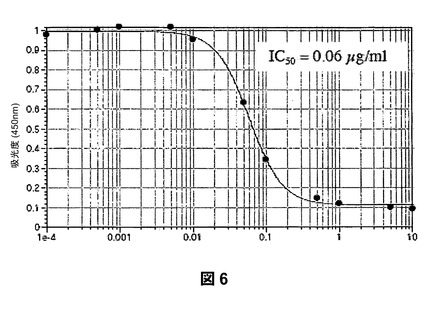
【図7】
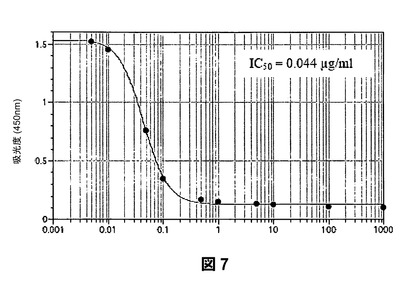
【図8】
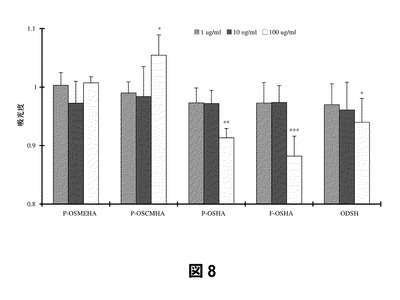
【図9】
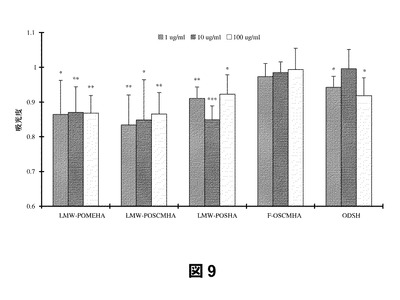
【図10】
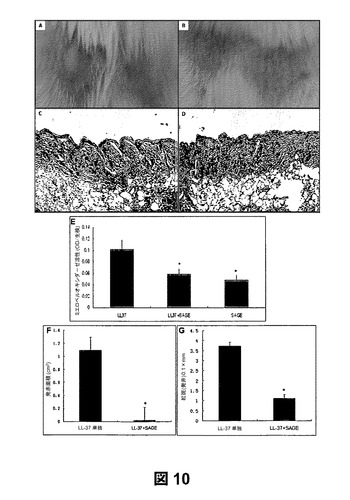
【図11】
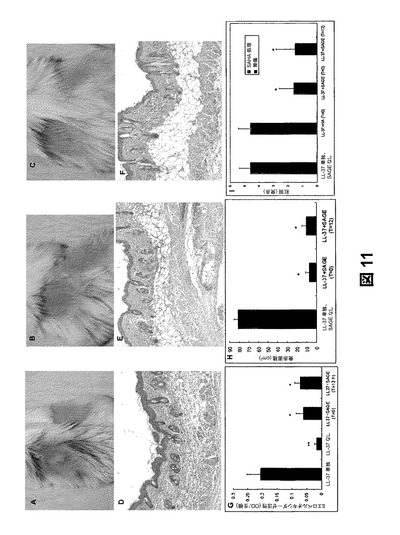
【図12】
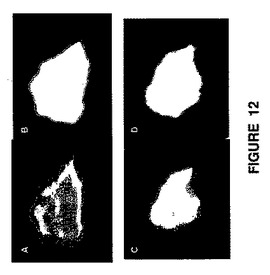
【図13】
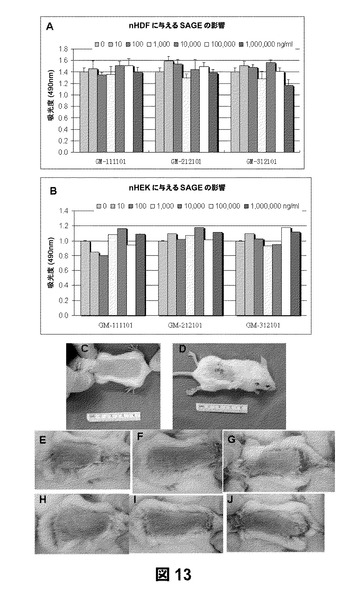
【図14】
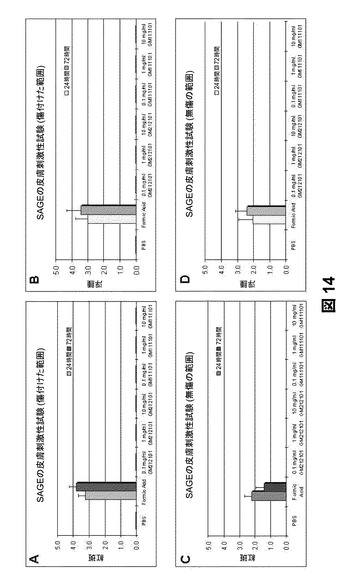
【図15】
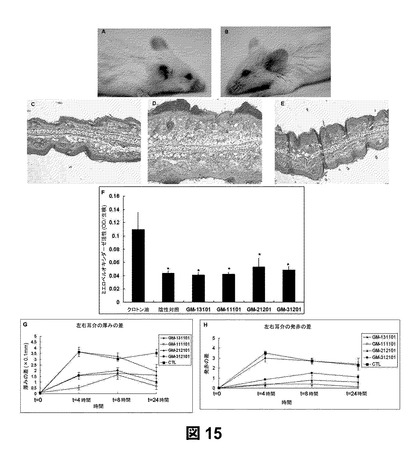
【図16】

【図17】
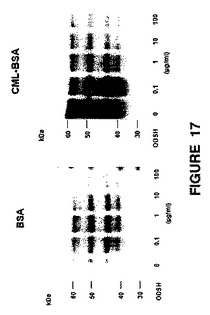
【図18】

【図19】
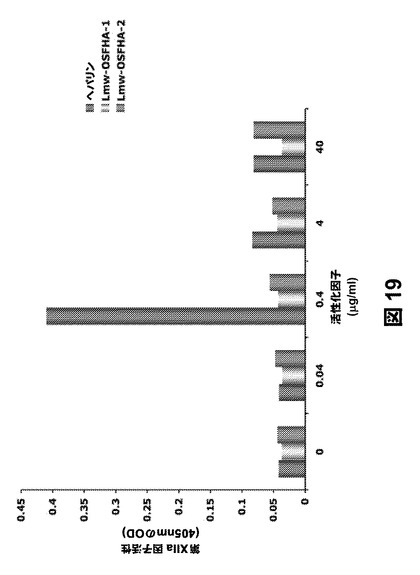
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【公表番号】特表2011−516672(P2011−516672A)
【公表日】平成23年5月26日(2011.5.26)
【国際特許分類】
【出願番号】特願2011−503216(P2011−503216)
【出願日】平成21年4月3日(2009.4.3)
【国際出願番号】PCT/US2009/039498
【国際公開番号】WO2009/124266
【国際公開日】平成21年10月8日(2009.10.8)
【出願人】(506051429)ユニバーシティ オブ ユタ リサーチ ファウンデーション (25)
【Fターム(参考)】
【公表日】平成23年5月26日(2011.5.26)
【国際特許分類】
【出願日】平成21年4月3日(2009.4.3)
【国際出願番号】PCT/US2009/039498
【国際公開番号】WO2009/124266
【国際公開日】平成21年10月8日(2009.10.8)
【出願人】(506051429)ユニバーシティ オブ ユタ リサーチ ファウンデーション (25)
【Fターム(参考)】
[ Back to top ]