
アルツハイマー病の治療のための安全性に優れた鼻腔内投与可能遺伝子ワクチン
本発明の目的は、アルツハイマー病に対する安全性および有効性の高いワクチン療法を提供することである。アミロイド遺伝子を保持するマイナス鎖RNAウイルスベクターを構築し、24〜25ヶ月齢APPトランスジェニックマウスに鼻腔内投与した。血清抗Aβ42抗体のレベルを測定したところ、コントロールよりも顕著に高かった。組織学的に検討した結果は、前頭葉、頭頂葉、および海馬のいずれにおいても、本発明のベクター投与による老人斑の顕著な減少を示した。脳内のAβレベルは顕著に低下した。さらに、本発明のベクター投与は、中枢神経系におけるリンパ球の浸潤はもたらさなかった。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、マイナス鎖RNAウイルスベクターに基づくアルツハイマー病治療用ワクチンに関する。
【背景技術】
【0002】
急速な高齢化社会を迎えつつある日本では、老年性痴呆は、介護問題も含め社会的に大きな問題となっている。実に、日本の65歳以上の老人のうち、約10%が老年性痴呆であると報告されている。アルツハイマー病は、痴呆性疾患の二大原因の一つであり、罹患人口の約50%を占めるにも関わらず、有効な治療法は現在のところ提供されていない。
【0003】
アルツハイマー病の病理学的所見は、以下の3つの特徴を有する:神経細胞の萎縮/脱落;アミロイドβ(以下、時に「Aβ」とも略す)タンパク質の凝集および沈着による老人斑の形成;ならびに異常タウタンパク質からなる神経原線維変化。アルツハイマー病患者の脳で生じる主要なアミロイドβタンパク質は、アミロイド前駆体タンパク質がβおよびγセクレターゼによる切断からもたらされるアミノ酸40〜43個からなる。老人斑は、中心のアミロイドβおよび取り囲むミクログリア、線維型アストログリア、ならびに異栄養神経突起で構成される凝集体である。現在、アルツハイマー病の病態仮説として、アミロイドβの凝集および沈着による老人斑の形成が原因であるとする「アミロイドカスケード仮説」が有力である。
【0004】
この仮説をもとに、アルツハイマー病の新しい治療法としてのワクチン(免疫)療法が注目されている。アルツハイマー病のワクチン療法は、脳のアミロイドβを免疫学的手法により除去する方法である。Elan社のSchenkらは、前凝集Aβ42をアジュバントと共にPDAPP-トランスジェニックマウスに筋肉内投与し、脳内のアミロイド沈着の減少を確認した(特許文献1および非特許文献1)。Elan社およびWyeth社によって行われた臨床試験では、合成Aβ42ペプチド(AN-1792)がアジュバント(QS21)と共に筋肉内投与され、被験者の血清中にアミロイドのβシート構造を認識する抗Aβ抗体が検出された(非特許文献2)。高次脳機能の改善も報告された(非特許文献3)。残念ながら、フェーズIIにおいて6%(298名中18名)の患者に髄膜脳炎の副作用が生じ、1名の死亡例も報告され、上記臨床試験は中断された。死亡患者の脳組織を病理学的に検討した結果から、新皮質で老人斑の不在が確認され、ワクチンの効果が示唆された。老人斑が消失している領域では、Aβ分解産物のミクログリアによる貪食作用を示す像が確認された。この所見は、Aβに結合した抗体をFcレセプターを介してミクログリアが貪食していることを示している。
【0005】
上記抗Aβ抗体は、ニューロン、グリア細胞、アミロイド前駆体タンパク質(APP)、および細胞内Aβとは反応しないことが確認されている。従って上記副作用は、抗体による脳の炎症とは考えにくい。AN-1792ワクチンを投与するために、アジュバントを必要とする。アジュバントは強い免疫活性化作用を有し、Tリンパ球による細胞性免疫も誘導する。上記副作用は、アジュバント誘導性細胞性免疫により、AβまたはAPPに反応するTh1タイプCD4+ T細胞が脳内に浸潤することで引き起こされる、実験的アレルギー性脳脊髄炎様の髄膜脳炎である可能性がある(非特許文献4)。
【0006】
AN-1792ワクチンにより、Aβペプチドを利用したワクチン療法の病理学的効果が確認された。しかしワクチンの臨床適用のためには、髄膜脳炎などの副作用を軽減することが必須である。このような観点から、N末端断片にT細胞によって認識されるキャリアタンパク質が結合したAβペプチドと、Th2アジュバントとの併用が関与する免疫化方法(特許文献2)、ならびにAβペプチドおよびコレラ毒素Bサブユニットからなる融合タンパク質をコードする核酸を含むアデノウイルスベクター(特許文献3)を含む、改良したワクチン接種法が発案された。さらに本発明者らは、安全性の高い経口ワクチンとして、液性免疫を誘導するAβペプチドのペプチド断片をコードするDNAを含むアデノ随伴ウイルスベクターを開発した(特許文献4)。上記アデノ随伴ウイルスベクターワクチンは、Th2 T細胞が誘導されやすい腸管粘膜免疫系を利用することから、いかなるアジュバントも必要としない。さらに、APPトランスジェニックマウスを用いた実験により、1回の投与で6ヶ月の比較的長期にわたり腸管において抗原提示が可能であること;脳のアミロイド沈着および老人斑形成の減少効果を有すること;他の臓器におけるいかなる観察可能な炎症も認められないことといった、優れた特徴が確認されている。しかしながら、アルツハイマー病のワクチン療法の実用化のため、安全性および有効性のさらに優れたワクチンが必要である。
【0007】
【特許文献1】WO 99/27944
【特許文献2】WO 02/096350
【特許文献3】WO 2004/050876
【特許文献4】WO 2004/111250
【特許文献5】WO 00/70070
【特許文献6】特開2000-253876
【特許文献7】WO 2001/072340
【非特許文献1】Schenk D. et al, Nature 400:173-177, 1999
【非特許文献2】Hock C. et al., Nat. Med. 8:1270-1275, 2002
【非特許文献3】Hock C. et al., Neuron 38, 547-554, 2003
【非特許文献4】臨牀と研究 82(3):439-444, 2005
【発明の開示】
【0008】
本発明は、上記状況に鑑みて達成された。本発明の目的は、アルツハイマー病に対する安全性および有効性の高いワクチン療法の提供である。
【0009】
本発明者らは、上記目的を達成するため、鋭意努力を重ね、アルツハイマー病に対する新規ワクチン療法の開発を試みた。本発明者らは、センダイウイルス(SeV)を用いて遺伝子導入用および遺伝子治療にも使用できるベクターを開発してきた経験を有する。センダイウイルスは、マイナス(−)鎖RNAウイルスである。ウイルスは宿主細胞の細胞質でのみ増殖し、それらの生活環においてDNAフェーズを有さないため、染色体へ組み込まれない。ウイルスは従って、遺伝子導入用ベクターとして利用した場合、高い遺伝的安全性を確実にする。本発明者らは、センダイウイルスに基づく遺伝子導入用ベクターを開発するにあたり、さらに安全性の向上および標的疾患に適したベクターとするために、センダイウイルスゲノムから遺伝子を欠失させた。宿主細胞へのウイルス侵入に関わる膜融合タンパク質(Fタンパク質)の遺伝子をゲノムから欠失させることにより、安全性が高く、二次感染性を引き起こさないベクターへの改良に成功すると共に、ウイルスベクターの効率的な再構成系も確立している(WO 00/70070)。さらにセンダイウイルスベクターを利用したインフルエンザワクチン(特開2000-253876)およびAIDSワクチン(WO 2001/072340)の開発にも成功している。
【0010】
IL-10などのTh2サイトカインは、生体内の免疫応答と密接に関連するTh1/Th2バランスにおいて、重要な役割を果たす。Th1細胞は、IFN-γなどを産生し、細胞性免疫を亢進する。Th1細胞によって産生されたIFN-γは、Th2細胞のIL-10産生を抑制する。一方、Th2細胞は、IL-10などを産生し、液性免疫を亢進させる。Th2細胞によって産生されたIL-10は、Th1細胞におけるIFN-γなどの産生を抑制する。本発明者らは、上記センダイウイルスベクターに、Aβと共にTh2サイトカインをコードする遺伝子を導入することを考え、細胞性免疫による脳髄膜炎などの副作用を抑制するアルツハイマー病に対するワクチンの産生を試みた。次いで本発明者らは、Aβ1-43およびIL-10をコードするセンダイウイルスベクターを構築し、その効果を検討した。まず、該ベクターを24〜25ヶ月齢APPトランスジェニックマウスに鼻腔内投与した。処置後4週間後および8週間後の血清中の抗Aβ42抗体レベルを測定した。コントロール群のマウスでは、抗Aβ抗体は低レベルでしか検出されず、時間と共にレベルが増加した。対照的に、本発明のベクターを投与したマウスの群では、血中に高レベルの抗Aβ抗体が検出され、抗体レベルは4週間後よりも8週間後にわずかに低下することが見出された。組織学的検討は、前頭葉、頭頂葉、および海馬のいずれにおいても、本発明のベクター投与による老人斑の顕著な減少を示した。さらに脳組織中のAβ量をELISA法により測定した。結果は脳組織中Aβの有意な減少を示した。さらに、このベクター投与が中枢神経系におけるリンパ球の浸潤を引き起こすかどうか検討するために、CD3およびIba-1を指標として用いた。結果は、リンパ球の浸潤もミクログリアの病的な(極端な)活性も生じないことを示した。
【0011】
上記検討から、Aβペプチドを発現するマイナス鎖RNAウイルスベクターの極めて優れた特徴が明らかにされた。第一に、このベクターは、極めて高齢のアルツハイマー病モデルマウスにおいて、顕著なAβ減少効果を示した。上記の実験で使用したTg2576マウスは24〜25ヶ月齢であり、ほぼ寿命に近かった。これまでのアルツハイマー病治療方法の研究は、最大18ヶ月までのマウスを用いて実施されてきた。このような高齢マウスにおいて治療効果が確認されたのは、今回が初めてである。そのような高齢のマウスでは、脳に沈着しているAβ量は膨大である。従って本発明のベクターは、よりアルツハイマー病の進行した患者に対しても高い有効性を発揮できる可能性がある。第二に、本発明のベクターは従来のワクチン療法と比較して、低い投与量および少ない投与回数で有効であった。例えば、Aβペプチドとアジュバントを併用して用いた従来の方法を適用した場合、一般的に、免疫確立のために複数回投与が必要である。実際に、AN-1792臨床試験において数回〜十数回の投与が行われた。対照的に、本発明のベクターは単回投与後、顕著な有効性を示した。さらに、アデノ随伴ウイルスベクターによるワクチン療法(WO 2004/111250)において、ワクチン接種は、本明細書に記載の実施例で行ったのと同様の単回投与によって実施し、その投与量は5×1011ゲノム/個体であった。対照的に、本明細書に記載の実施例に適用した投与量は、5×106 CIU(Cell Infectious Unit)/個体(ゲノムコピー数に換算した場合約5×107ゲノム/個体)であった。これら2つのベクターの投与量には約104もの開きがある。第三に、実施例におけるベクターの投与は、マウスモデルにおいて髄膜脳炎を引き起こさなかった。AN-1792臨床試験で観察された髄膜脳炎は、マウスモデルでも確認されているとの報告がある(M. Shoji, et al., 44th Annual Meeting of Japanese Society of Neurology, May 15-17, 2003)。Aβ ペプチドおよびアジュバントを免疫した正常非モデルマウス(C57B6マウス)において、髄膜脳炎の発生も報告された(Furlan R, et al., Vaccination with amyloid-beta peptide induces autoimmune encephalomyelitis in C57/BL6 mice, Brain 126:285-291, 2003)。本明細書に記載の結果は、本発明のベクターがヒトへ投与した場合においても、安全であることを示唆する。
【0012】
上述のとおり、本発明のベクターは、低用量および少投与回数で高いAβ消失効果を示した。センダイウイルスベクターに基づくAIDSワクチンの効果は、一部細胞性免疫によることが確認されている(WO 2001/072340)。実施例で使用したベクターが、副作用を防止するため細胞性免疫よりも液性免疫に依存するよう意図的に設計したにも関わらず、本明細書に示した優れたワクチン効果は、アルツハイマー病治療ワクチンにおいて、液性免疫の重要性を示唆する。Th2サイトカインまたはTh2アジュバントを使用して液性免疫を優位にすることにより、副作用を低減することができる。すなわち、本発明は、Aβをコードするマイナス鎖RNAウイルスベクターおよびその利用、特に下記の発明を提供するものである。
【0013】
[1]アミロイドβをコードする核酸を含む、マイナス鎖RNAウイルスベクター。
[2]Th2サイトカインまたはその部分ペプチドをコードする核酸をさらに含む、[1]のマイナス鎖RNAウイルスベクター。
[3]Th2サイトカインまたはその部分ペプチドが、IL-10またはその部分ペプチドである、[2]のマイナス鎖RNAウイルスベクター。
[4]マイナス鎖RNAウイルスベクターがパラミクソウイルスベクターである、[1]〜[3]のいずれかのベクター。
[5]パラミクソウイルスベクターがセンダイウイルスベクターである、[4]のベクター。
[6]アミロイドβがAβ43またはその部分ペプチドである、[1]〜[5]のいずれかのベクター。
[7]アミロイドβがヒトに由来する、[1]〜[6]のいずれかのベクター。
[8]IL-10が、マウスまたはヒトに由来する、[3]〜[7]のいずれかのベクター。
[9]アミロイドβが、配列番号:1のヌクレオチド配列がコードするポリペプチドまたはその部分である、[1]〜[8]のいずれかのベクター。
[10]IL-10が、配列番号:2または3のヌクレオチド配列がコードするポリペプチドである、[3]〜[9]のいずれかのベクター。
[11]配列番号:5の核酸を含む、[1]〜[10]のいずれかのマイナス鎖RNAウイルスベクター。
[12][1]〜[11]のいずれかのベクターおよび薬学的に許容される担体を含む組成物。
[13](i)Th2サイトカインもしくはその部分ペプチドを発現可能様式でコードする核酸、(ii)Th2サイトカインもしくはその部分ペプチド、または(iii)Th2アジュバントをさらに含む、[12]の組成物。
[14]Th2サイトカイン、その部分ペプチド、またはTh2アジュバントを含む、[13]の組成物。
[15]Th2サイトカインまたはその部分ペプチドを、発現可能様式でコードする核酸を含む、[13]の組成物。
[16]Th2サイトカインが、アミロイドβをコードする核酸を含むマイナス鎖RNAウイルスベクターによってコードされる、[15]の組成物。
[17]Th2サイトカインが、アミロイドβをコードする核酸を含むマイナス鎖RNAウイルスベクター以外のベクターによってコードされる、[15]の組成物。
[18]薬学的組成物である、[12]〜[17]のいずれかの組成物。
[19][1]〜[18]のいずれかのベクターを含む、アルツハイマー病を治療または予防するために用いられる薬学的組成物。
[20](i)Th2サイトカインもしくはその部分ペプチドを発現可能様式でコードする核酸、(ii)Th2サイトカインもしくはその部分ペプチド、または(iii)Th2アジュバントをさらに含む、[19]の組成物。
[21]鼻腔内投与に用いるための、[18]または[20]の薬学的組成物。
[22][1]〜[11]のいずれかのベクター、または該ベクターを含む組成物を投与する段階を含む、アミロイドβの沈着を減少させる方法。
[23][1]〜[11]のいずれかのベクターまたは該ベクターを含む組成物を投与する段階を含む、アルツハイマー病を治療または予防する方法。
[24]投与が鼻腔内投与である、[22]または[23]の方法。
[25]投与が筋肉内投与である、[22]または[23]の方法。
[26]Th2サイトカイン、その部分ペプチド、Th2アジュバント、またはTh2サイトカインもしくはその部分ペプチドをコードするベクターを投与する段階をさらに含む、[22]〜[25]のいずれかの方法。
[27]Th2サイトカイン、その部分ペプチド、またはTh2アジュバントを投与する、[26]の方法。
[28]Th2サイトカインまたはその部分ペプチドをコードするベクターを投与する、[26]の組成物。
【0014】
本発明によって、低用量および少投与回数でアミロイドβ沈着を有効に減少させることのできる、新規マイナス鎖RNAウイルスベクターが提供された。本実施例に記載のベクターは、中枢神経系における炎症を誘導しないことが確認されており、本発明のベクターの投与は、従来のワクチンを投与した場合発生したような脳髄膜炎を引き起こす可能性は低い。本発明のベクターは、アルツハイマー病の極めて有効な治療手段として用いることができる。
【0015】
本発明は、アミロイドβをコードする核酸を含む、マイナス鎖RNAウイルスベクターに関する。
【0016】
本発明において遺伝子とは、遺伝物質または転写単位をコードする核酸を指す。遺伝子はRNAまたはDNAであってよい。本発明においてタンパク質をコードする核酸は、該タンパク質の遺伝子と呼ぶ。遺伝子はタンパク質をコードしていなくてもよく、例えば遺伝子はリボザイムまたはアンチセンスRNAなどの機能的RNAをコードするものでもよい。遺伝子は天然由来または人為的に設計された配列であり得る。本明細書において「DNA」とは、一本鎖DNAおよび二本鎖DNAを含む。「タンパク質をコードする」という語句は、ポリヌクレオチドがタンパク質を適当な条件下で発現できるように、タンパク質のアミノ酸配列をコードするORFをセンスまたはアンチセンス方向で含むことを意味する。
【0017】
マイナス鎖RNAウイルスとは、マイナス鎖RNA(ウイルスタンパク質をコードするセンス鎖と相補的なアンチセンス鎖)をゲノムとして含むウイルスを指す。マイナス鎖RNAウイルスはネガティブ鎖RNAウイルスとも呼ばれる。本発明において用いられるマイナス鎖RNAウイルスとしては、特に一本鎖マイナス鎖RNAウイルス(非分節型(non-segmented)マイナス鎖RNAウイルスとも言う)が好ましい。「一本鎖マイナス鎖RNAウイルス」とは、一本鎖のマイナス鎖RNAをゲノムとして有するウイルスを指す。このようなウイルスとしては、パラミクソウイルス(Paramyxoviridae)(パラミクソウイルス(Paramyxovirus)、モービリウイルス(Morbillivirus)、ルブラウイルス(Rubulavirus)、およびニューモウイルス(Pneumovirus)属などを含む)、ラブドウイルス(Rhabdoviridae)(ベシクロウイルス(Vesiculovirus)、リッサウイルス(Lyssavirus)、およびエフェメロウイルス(Ephemerovirus)属などを含む)、フィロウイルス(Filoviridae)、オルトミクソウイルス(Orthomyxoviridae)(インフルエンザウイルス(Influenza virus)A、B、およびC、ならびにトゴト様ウイルス(Thogoto-like virus)などを含む)、ブニヤウイルス(Bunyaviridae)(ブニヤウイルス(Bunyavirus)、ハンタウイルス(Hantavirus)、ナイロウイルス(Nairovirus)、およびフレボウイルス(Phlebovirus)属などを含む)、ならびにアレナウイルス(Arenaviridae)などの科に属するウイルスが含まれる。
【0018】
本発明において特に好適に用いられるマイナス鎖RNAウイルスを具体例は、例えばパラミクソウイルス科に属するセンダイウイルス(Sendai virus)、ニューカッスル病ウイルス(Newcastle disease virus)、おたふくかぜウイルス(Mumps virus)、麻疹ウイルス(Measles virus)、RSウイルス(Respiratory syncytial virus)、牛疫ウイルス(rinderpest virus)、ジステンパーウイルス(distemper virus)、サルパラインフルエンザウイルス(SV5)、ヒトパラインフルエンザウイルス1、2、および3型;オルトミクソウイルス科に属するインフルエンザウイルス;ラブドウイルス科に属する水疱性口内炎ウイルス(Vesicular stomatitis virus)および狂犬病ウイルス(Rabies virus)を含む。
【0019】
本発明において、好ましくはパラミクソウイルスが用いられる。パラミクソウイルスとはパラミクソウイルス科に属するウイルスまたはその誘導体を指す。パラミクソウイルスは、非分節型ネガティブ鎖RNAをそれらのゲノムとして持つウイルスのグループであり、パラミクソウイルス亜科(Paramyxovirinae)(パラミクソウイルス属とも言うレスピロウイルス属(Respirovirus)、ルブラウイルス属、およびモービリウイルス属を含む)、およびニューモウイルス亜科(Pneumovirinae)(ニューモウイルス属およびメタニューモウイルス(Metapneumovirus)属を含む)を含む。本発明に適用可能なパラミクソウイルスの具体例は、センダイウイルス、ニューカッスル病ウイルス、おたふくかぜウイルス、麻疹ウイルス、RSウイルス、牛疫ウイルス、ジステンパーウイルス、サルパラインフルエンザウイルス(SV5)、ならびにヒトパラインフルエンザウイルス1、2、および3型である。より具体的には、そのような例は、センダイウイルス(SeV)、ヒトパラインフルエンザウイルス-1(HPIV-1)、ヒトパラインフルエンザウイルス-3(HPIV-3)、アザラシジステンパーウイルス(PDV)、犬ジステンパーウイルス(CDV)、イルカモービリウイルス(DMV)、小反芻動物病ウイルス(Peste-des-petits-ruminants virus)(PDPR)、麻疹ウイルス(MV)、牛疫ウイルス(RPV)、ヘンドラウイルス(Hendra virus)(ヘンドラ)、ニパーウイルス(Nipah virus)(ニパー)、ヒトパラインフルエンザウイルス-2(HPIV-2)、サルパラインフルエンザウイルス5(SV5)、ヒトパラインフルエンザウイルス-4a(HPIV-4a)、ヒトパラインフルエンザウイルス-4b(HPIV-4b)、おたふくかぜウイルス(おたふくかぜ)、およびニューカッスル病ウイルス(NDV)を含む。より好ましい例は、センダイウイルス(SeV)、ヒトパラインフルエンザウイルス-1(HPIV-1)、ヒトパラインフルエンザウイルス-3(HPIV-3)、アザラシジステンパーウイルス(PDV)、犬ジステンパーウイルス(CDV)、イルカモービリウイルス(DMV)、小反芻動物病ウイルス(PDPR)、麻疹ウイルス(MV)、牛疫ウイルス(RPV)、ヘンドラウイルス(ヘンドラ)、およびニパーウイルス(ニパー)からなる群より選択されるウイルスである。本発明のウイルスは、好ましくはパラミクソウイルス亜科(レスピロウイルス属、ルブラウイルス属、およびモービリウイルス属を含む)に属するウイルスまたはその誘導体であり、より好ましくはレスピロウイルス属(パラミクソウイルス属とも言う)に属するウイルスまたはその誘導体である。本発明に適用可能なレスピロウイルス属ウイルスの例は、ヒトパラインフルエンザウイルス-1(HPIV-1)、ヒトパラインフルエンザウイルス-3(HPIV-3)、ウシパラインフルエンザウイルス-3(BPIV-3)、センダイウイルス(マウスパラインフルエンザウイルス-1とも呼ばれる)、およびサルパラインフルエンザウイルス-10(SPIV-10)である。本発明において、最も好ましくはセンダイウイルスが用いられる。これらのウイルスは、天然株、野生株、変異株、ラボ継代株、および人為的に構築された株などに由来してもよい。
【0020】
本発明において「ベクター」は、核酸を細胞に導入する担体である。マイナス鎖RNAウイルスベクターとは、マイナス鎖RNAウイルスに由来する、核酸を細胞に導入する担体である。SeVなどのマイナス鎖RNAウイルスは遺伝子導入ベクターとして優れている。マイナス鎖RNAウイルスは、DNAフェーズを持たず、宿主細胞の細胞質でのみ転写および複製を行うため、染色体への組み込みは起こらない。このため染色体異常による癌化または不死化などの安全面における問題が生じない。マイナス鎖RNAウイルスのこの特徴は、ベクターとして用いた場合、安全性に大きく寄与している。異種遺伝子発現に用いた場合、SeVを連続多代継代した場合でさえ、ほとんどヌクレオチド変異が認められず、そのゲノムの高い安定性、および挿入異種遺伝子を長期間にわたって安定に発現することを示す(Yu, D. et al., Genes Cells 2:457-466, 1997)。さらに、SeVにおけるカプシド構造タンパク質の欠失は、挿入遺伝子のサイズおよびそのパッケージングの柔軟性など、性質上のメリットをもたらす。伝播性SeVベクターは、少なくとも5.5kbの外来遺伝子を導入することができ、転写ユニットを付加することによって2種類以上の遺伝子を同時に発現することができる。
【0021】
センダイウイルスはげっ歯類にとっては病原性で肺炎を生じることが知られているが、ヒトに対しては病原性の報告がない。これは、非ヒト霊長類およびヒトへの野生型センダイウイルスの鼻腔内投与が重篤な副作用を引き起こさないというこれまでの報告によって支持された(Hurwitz, J. L. et al., Vaccine 15:533-540, 1997、Slobod, K. S. et al., Vaccine 22:3182-3186, 2004)。さらに利点の例として以下の2点、「高感染性」および「高発現量」を挙げることができる。SeVベクターは細胞膜の糖脂質および糖タンパク質のシアル酸に結合することによって感染する。このシアル酸はほとんどの哺乳動物および鳥類細胞で発現しており、このことが感染スペクトルを広くする、すなわち高感染性に繋がっている。SeVのレプリコンに基づく伝播性ベクターがウイルスを放出する場合、それらは周囲の細胞にも再感染する。多数のRNPが、感染細胞の細胞質において複製され、細胞の分裂を介して娘細胞に分配されるため、持続発現が期待される。さらに、SeVベクターは非常に広い範囲の組織に適用することができる。さらに、細胞質のみで転写および複製が生じるというそれらの特徴的な発現機構は、挿入遺伝子を非常に高いレベルで発現することを示す(Moriya, C. et al., FEBS Lett. 425(1):105-111, 1998;WO 00/70070)。さらに、エンべロープ遺伝子を欠失して非伝播性にしたSeVベクターの回収にも成功している(WO 00/70070;Li, HO. et al., J. Virol. 74(14):6564-6569, 2000)。「高感染性」および「高発現量」を維持して、「安全性」をさらに高めるため、SeVベクターの改良が行われている。
【0022】
本発明のマイナス鎖RNAウイルスベクターは、マイナス鎖RNAウイルスのゲノムRNAを含む。ゲノムRNAとは、マイナス鎖RNAウイルスのあるウイルスタンパク質を含むRNPを形成し、それがゲノム中の遺伝子を発現させる機能を有するRNAを言う。従って核酸は複製され、娘RNPが形成される。マイナス鎖RNAウイルスのRNAは、遺伝子をアンチセンス配列としてコードしている。一般にマイナス鎖RNAウイルスのゲノムにおいて、3'リーダー領域と5'トレイラー領域の間に、ウイルス遺伝子がアンチセンス配列として整列している。各遺伝子のORFの間には、転写終結配列(E配列)-介在配列(I配列)-転写開始配列(S配列)が存在し、これにより各遺伝子のORFをコードするRNAが別々のシストロンとして転写される。本発明のベクターにおけるゲノムRNAは、該RNAによってコードされる遺伝子群の発現、およびRNA自身の自律的な複製に必要なウイルスタンパク質であるN(ヌクレオキャプシド、ヌクレオプロテイン(NP)とも言う)、P(ホスホ)、およびL(ラージ)タンパク質をコードするアンチセンスRNA配列を含む。該RNAはまた、ウイルス粒子の形成に必要なM(マトリックス)タンパク質をコードしていてもよい。さらに該RNAは、ウイルス粒子の感染に必要なエンベロープタンパク質をコードしていてもよい。マイナス鎖RNAウイルスのエンベロープタンパク質には、細胞膜融合を起こすF(融合)タンパク質、および細胞へのウイルス接着に必要なHN(ヘマグルチニン-ノイラミニダーゼ)またはH(ヘマグルチニン)タンパク質が含まれる。しかしながら、ある種の細胞では感染にHNタンパク質は必要なく(Markwell, M.A. et al., Proc. Natl. Acad. Sci. USA 82(4):978-982, 1985)、Fタンパク質のみで感染が成立する。RNAは、Fタンパク質および/またはHNタンパク質以外のエンベロープタンパク質をコードしてもよい。
【0023】
本発明のマイナス鎖RNAウイルスベクターは、例えばマイナス鎖RNAウイルスのゲノムRNAとウイルスタンパク質からなる複合体、すなわちリボヌクレオタンパク質(RNP)であってよい。RNPは、例えば所望のトランスフェクション試薬と組み合わせて細胞に導入することができる。このようなRNPは、具体的にはマイナス鎖RNAウイルスのゲノムRNA、Nタンパク質、Pタンパク質、およびLタンパク質を含む複合体である。RNPは細胞内に導入されると、ウイルスタンパク質の作用によりゲノムRNAからウイルスタンパク質をコードするシストロンが転写されると共に、ゲノム自身が複製され娘RNPが形成される。ゲノムRNAの複製は、該RNAのコピー数の増加をRT-PCR、ノーザンブロットハイブリダイゼーションなどによって検出することにより確認することができる。
【0024】
さらに本発明のマイナス鎖RNAウイルスベクターは、好ましくはマイナス鎖RNAウイルスのウイルス粒子である。「ウイルス粒子」とは、核酸を含む微小粒子を指し、ウイルスタンパク質の作用により細胞から放出される。マイナス鎖RNAウイルスのウイルス粒子は、ゲノムRNAとウイルスタンパク質を含む上記RNPが細胞膜由来の脂質膜(エンベロープという)に含まれた構造を有する。ウイルス粒子は、感染性を有してもよい。感染性とは、マイナス鎖RNAウイルスベクターの細胞接着能および膜融合能に基づき、接着した細胞内に保持した核酸を導入することのできるウイルス粒子の能力を言う。本発明のマイナス鎖RNAウイルスベクターは、伝播性、または非伝播性ベクターであってもよい。「伝播性」とは、ウイルスベクターが宿主細胞に感染した場合、該細胞においてウイルスが自身を複製し、感染性ウイルス粒子を産生することを指す。
【0025】
例えばパラミクソウイルス亜科に属するウイルスにおける各遺伝子は、一般に次のように表記される。一般に、N遺伝子は「NP」とも表記される。
レスピロウイルス属 NP P/C/V M F HN - L
ルブラウイルス属 NP P/V M F HN (SH) L
モービリウイルス属 NP P/C/V M F H - L
【0026】
例えばセンダイウイルスの各遺伝子のヌクレオチド配列のデータベースのアクセッション番号は以下である:N遺伝子について、M29343、M30202、M30203、M30204、M51331、M55565、M69046、およびX17218;P遺伝子について、M30202、M30203、M30204、M55565、M69046、X00583、X17007、およびX17008;M遺伝子について、D11446、K02742、M30202、M30203、M30204、M69046、U31956、X00584、およびX53056;F遺伝子について、D00152、D11446、D17334、D17335、M30202、M30203、M30204、M69046、X00152、およびX02131;HN遺伝子について、D26475、M12397、M30202、M30203、M30204、M69046、X00586、X02808、およびX56131;L遺伝子について、D00053、M30202、M30203、M30204、M69040、X00587、X58886。他のウイルスがコードするウイルス遺伝子の例は以下である:N遺伝子について、CDV、AF014953;DMV、X75961;HPIV-1、D01070;HPIV-2、M55320;HPIV-3、D10025;Mapuera、X85128;Mumps、D86172;MV、K01711;NDV、AF064091;PDPR、X74443;PDV、X75717;RPV、X68311;SeV、X00087;SV5、M81442;およびTupaia、AF079780;P遺伝子については、CDV、X51869;DMV、Z47758;HPIV-l、M74081;HPIV-3、X04721;HPIV-4a、M55975;HPIV-4b、M55976;Mumps、D86173;MV、M89920;NDV、M20302;PDV、X75960;RPV、X68311;SeV、M30202;SV5、AF052755;およびTupaia、AF079780;C遺伝子について、CDV、AF014953;DMV、Z47758;HPIV-1、M74081;HPIV-3、D00047;MV、ABO16162;RPV、X68311;SeV、AB005796;およびTupaia、AF079780;M遺伝子について、CDV、M12669;DMV、Z30087;HPIV-1、S38067;HPIV-2、M62734;HPIV-3、D00130;HPIV-4a、D10241;HPIV-4b、D10242;Mumps、D86171;MV、AB012948;NDV、AF089819;PDPR、Z47977;PDV、X75717;RPV、M34018;SeV、U31956;およびSV5、M32248;F遺伝子について、CDV、M21849;DMV、AJ224704;HPN-1、M22347;HPIV-2、M60182;HPIV-3、X05303、HPIV-4a、D49821;HPIV-4b、D49822;Mumps、D86169;MV、AB003178;NDV、AF048763;PDPR、Z37017;PDV、AJ224706;RPV、M21514;SeV、D17334;およびSV5、AB021962;HN(HまたはG)遺伝子について、CDV、AF112189;DMV、AJ224705;HPIV-1、U709498;HPIV-2、D000865;HPIV-3、AB012132;HPIV-4A、M34033;HPIV-4B、AB006954;Mumps、X99040;MV、K01711;NDV、AF204872;PDPR、Z81358;PDV、Z36979;RPV、AF132934;SeV、U06433;およびSV-5、S76876。しかしながら、各ウイルスについて複数の株が知られており、株の違いにより上記に例示した以外の配列を含む遺伝子も存在する。
【0027】
これらのウイルスタンパク質のORFは、ゲノムRNAにおいて上記のE-I-S配列を用いてアンチセンス配列として整列される。ゲノムRNAの最も3'端に近いORFは、3'リーダー領域と該ORFとの間にS配列のみを必要とし、EおよびI配列は必要ない。さらにゲノムRNAの最も5'端に近いORFは、5'トレイラー領域と該ORFとの間にE配列のみを必要とし、IおよびS配列は必要ない。さらに2つのORFは、例えばIRES配列を用いて単一シストロンとして転写させることもできる。このような場合、これら2つのORFの間にはE-I-S配列は必要ない。例えば野生型のパラミクソウイルスの場合、典型的なRNAゲノムは、3'リーダー領域、N、P、M、F、HN、およびLタンパク質をコードする6つのORF、ならびに5'トレイラー領域をアンチセンスの順で含む。本発明のゲノムRNAにおいて、遺伝子の配置はこれに限定されるものではないが、好ましくは、野生型ウイルスと同様に、3'リーダー領域に続き、この順で整列するN、P、M、F、HN、およびLタンパク質をコードするORF、続く5'トレイラー領域を有する。ある種のマイナス鎖RNAウイルスは、6つのウイルス遺伝子全てを含まないが、そのような場合でも上記の野生型と同様にそれらのウイルス遺伝子を配置することが好ましい。一般に N、P、およびL遺伝子を保持しているベクターは、細胞内で自立的にRNAゲノムから遺伝子を発現することができ、それによってゲノムRNAが複製される。さらにFおよびHN(またはH)遺伝子およびM遺伝子などのエンベロープ構成タンパク質をコードする遺伝子の作用により、感染性のウイルス粒子が形成され、細胞外に放出される。従って、このようなベクターは伝播性ウイルスベクターとなる。AβまたはIL-10を含むポリペプチドをコードする遺伝子は、後述するように、このゲノム中の非タンパク質コード領域に挿入してもよい。
【0028】
さらに、本発明のマイナス鎖RNAウイルスベクターは、野生型マイナス鎖RNAウイルスの遺伝子のいずれかを欠損したものであってよい。ウイルスゲノムRNAは、RNPの再構成に必要なウイルスタンパク質(N、L、およびP)をコードしていれば、エンベロープ構成タンパク質をコードしていなくても細胞内で複製し、遺伝子を発現することができる。例えば、ウイルスの種類に依存して、F、H、HN、G、M、およびM1などのエンベロープ構成タンパク質をコードする遺伝子の少なくとも1つを欠損するベクターが例示できる(WO 00/70055およびWO 00/70070;Li, H.-O. et al., J. Virol. 74(14):6564-6569, 2000)。例えば、M、F、またはHN遺伝子、またはそれらの任意の組み合わせを含まないウイルスベクターも、本発明のベクターとして好適に用いることができる。このようなウイルスベクターの再構成は、例えば、欠損している遺伝子産物を外来的に供給することにより行うことができる。このように調製されたウイルスベクターは、野生型ウイルスと同様に宿主細胞に接着して細胞融合を引き起こすが、細胞に導入されたベクターゲノムは本来のウイルス遺伝子のいずれかを欠損するため、本来のウイルスと同様の感染力を持つ娘ウイルス粒子は形成されない。このため、このようなウイルスは、一回限りの遺伝子導入のための安全で有用なウイルスベクターである。ゲノムから欠損させる遺伝子としては、例えばF遺伝子および/またはHN遺伝子が挙げられる。例えば、F遺伝子が欠損した組換えマイナス鎖RNAウイルスゲノムを発現するプラスミドベクターを、Fタンパク質の発現ベクターならびにNP、P、およびLタンパク質の発現ベクターと共に、宿主細胞にトランスフェクションすることにより、ウイルスベクターの再構成を行うことができる(WO 00/70055およびWO 00/70070;Li, H.-O. et al., J. Virol. 74(14):6564-6569, 2000))。例えば、F遺伝子が染色体に組み込まれた宿主細胞を用いてウイルスを製造することもできる。これらのタンパク質群を外から供給する場合、それらアミノ酸配列は本来のウイルス配列と同じである必要はなく、核酸の導入における活性が天然型のそれと同等かそれ以上ならば、変異または他のウイルス由来の相同遺伝子で代用してもよい。
【0029】
さらに、本発明のウイルスベクターとして、ベクターゲノムが由来するウイルスのエンベロープタンパク質とは異なるタンパク質を含むベクターを作製することもできる。例えば、ウイルス再構成の際に、ベクターの本来のウイルスのエンベロープタンパク質以外のエンベロープタンパク質をパッケージング細胞で発現させることにより、所望のエンベロープタンパク質を含むウイルスベクターを製造することができる。このようなタンパク質に特に制限はなく、例えば、他のウイルスのエンベロープタンパク質、例えば水疱性口内炎ウイルスのGタンパク質(VSV-G)(J. Virology 39:519-528, 1981)を含む。本発明のウイルスベクターには、VSV-Gタンパク質などのように、ゲノムが由来するウイルス以外のウイルスに由来するエンベロープタンパク質を含むシュードタイプウイルスベクターが含まれる。ウイルスRNAゲノムにおいてエンベロープタンパク質がコードされないように設計することにより、ウイルス粒子が細胞に感染した後は、このタンパク質が発現されることはない。
【0030】
さらに、本発明のウイルスベクターは、例えばエンベロープ表面に、接着因子、リガンド、受容体などの特定の細胞に接着し得るタンパク質、抗体もしくはその断片;またはこれらのタンパク質を細胞外ドメインに有し、ウイルスエンベロープ由来のポリペプチドを細胞内ドメインに有するキメラタンパク質を含むベクターであってもよい。これにより、特定の組織を標的とするベクターを産生することもできる。このようなタンパク質は、ウイルスゲノム内にコードされてもよく、またはウイルスベクターの再構成時に、ウイルスゲノム以外の遺伝子(例えば別の発現ベクターまたは宿主細胞染色体上にある遺伝子)の発現により供給されてもよい。
【0031】
さらに本発明のベクターにおいて、例えばウイルスタンパク質による免疫原性を低下させるために、またはRNAの転写効率または複製効率を高めるために、ベクターに含まれる任意のウイルス遺伝子が野生型遺伝子から改変されてもよい。具体的には、例えばマイナス鎖RNAウイルスベクターにおいて、複製因子であるN、P、およびL遺伝子の少なくとも1つを改変し、転写または複製効率を高めることが考えられる。さらに、エンベロープタンパク質の1つであるHNタンパク質は、赤血球凝集活性とノイラミニダーゼ活性の両方を有する。例えば前者の活性を弱める場合、血液中でのウイルスの安定性を向上させることが可能であり、後者の活性を改変する場合、感染能を調節することが可能である。さらに、Fタンパク質を改変することにより膜融合能を調節することもできる。例えば、どちらも細胞表面の抗原分子となり得るFタンパク質またはHNタンパク質の抗原提示エピトープを解析し、これらのタンパク質の抗原性を弱めたウイルスベクターを作製することもできる。さらに、二次放出粒子またはVLP(ウイルス様粒子)の放出を抑制するため、ウイルス遺伝子に温度感受性変異を導入してもよい(WO 2003/025570)。例えば、M遺伝子には、G69E、T116A、およびA183Sを;HN遺伝子には、A262T、G264、およびK461Gを;P遺伝子には、L511Fを;L遺伝子には、N1197SおよびK1795Eのような変異を導入することができる。導入可能な温度感受性変異は上記の変異に限定されない(WO 2003/025570を参照されたい)。
【0032】
さらに本発明のベクターは、アクセサリー遺伝子が欠損したものであってよい。例えばSeVアクセサリー遺伝子の1つであるV遺伝子をノックアウトすることにより、培養細胞における遺伝子発現および複製は障害されることなく、マウスなどの宿主に対するSeVの病原性が顕著に減少する(Kato, A. et al., J. Virol. 71:7266-7272, 1997;Kato, A. et al., EMBO J. 16:578-587, 1997;Curran, J. et al., WO 01/04272, EP1067179)。このような弱毒化ベクターは、インビボまたはエクスビボにおける遺伝子導入用の無毒性ウイルスベクターとして特に有用である。
【0033】
本発明のベクターは、上記のマイナス鎖RNAウイルスベクターのゲノムに、外来遺伝子としてAβをコードする核酸を含む。Aβをコードするアミノ酸数は制限されない。例えば、Aβ1-40、Aβ1-42、およびAβ1-43は全て許容される。Aβとして、天然の全長Aβの抗原性を有する所望の断片または該断片を含むポリペプチドを用いることもできる。このようなポリペプチドは、Aβに対する抗原性を提示するために、天然のAβ(例えばAβ43)のアミノ酸配列由来の連続した6アミノ酸以上、好ましくは連続した7アミノ酸以上、または連続した8アミノ酸以上を含む(Harlow, Antibodies:A laboratory Manual, 1998;Chapter 5 page 76)。例えば、Aβ42(配列番号:27の1〜42)に対するB細胞のエピトープの大半は、N末端の1番目〜15番目のアミノ酸の領域に集中している(Cribbs, D.H. et al., Int. Immunol. 15(4):505-14, 2003)。従って、ベクターから発現させるAβペプチドとして、Aβ42の1番目〜15番目のアミノ酸を含むポリペプチドを好適に用いることができる。または、Aβ42の1番目〜21番目のアミノ酸または4番目〜10番目のアミノ酸を含むポリペプチドを発現させてもよい(特開2005-21149)。さらに、アミロイド繊維形成を阻害し神経を保護する能力について公知の抗Aβモノクローナル抗体10D5および6C6は、Aβ42の3番目〜6番目のアミノ酸に相当する4アミノ酸エピトープ(EFRH;配列番号:27の3〜6)を認識することが報告されている(Frenkel, D. et al., J. Neuroimmunol. 88:85-90, 1998;Frenkel, D. et al., J. Neuroimmunol. 95:136-142, 1999)。同じエピトープを認識するモノクローナル抗体508Fも、Aβによる神経毒性を抑制する(Frenkel, D. et al., J. Neuroimmunol. 106:23-31, 2000)。従って、この配列を含むAβ配列における6〜8アミノ酸以上のポリペプチドを好適に用いることができる。細胞性免疫のエピトープはAβのC末端領域に集中する。従ってAβ1-21などのN末端断片を含み、Aβ22〜43としてC末端付近の配列(配列番号:27の22番目〜43番目)を含まない断片を発現させることで、相対的に細胞性免疫よりも液性免疫を優位にすることができる。本発明のベクターとして、天然に存在するAβペプチド(配列番号:27の1〜39、1〜40、1〜41、1〜42、および1〜43にそれぞれ相当する、Aβ39、Aβ40、Aβ41、Aβ42、およびAβ43)の配列を含むポリペプチドをコードするベクターも好適に用いることができる。短いAβペプチド断片は、適切なキャリアタンパク質と融合させたポリペプチドを作製し、免疫原性を高めるために用いてもよい。キャリアタンパク質には、キーホールリンペットヘモシアニン(KLH)、ウシ血清アルブミン(BSA)、ウサギ血清アルブミン(RSA)、ヒト血清アルブミン(HSA)、オボアルブミン(OVA)、 チログロブリン(TG)などが含まれる。短いペプチドは、約1〜5アミノ酸のスペーサーを介してキャリアタンパク質と融合することができる。抗原ポリペプチド(分泌型の場合、成熟ポリペプチド)の長さ(キャリアタンパク質部分を除くアミノ酸長)は、好ましくは10〜200アミノ酸、より好ましくは15〜100アミノ酸、なおより好ましくは20〜80アミノ酸の範囲である。
【0034】
さらに、ベクターにコードされるAβポリペプチドは好ましくは、細胞からのその分泌を確実にするように、分泌シグナルを含む。分泌シグナル配列として、インターロイキン(IL)-2および組織プラスミノーゲンアクチベーター(tPA)などの所望の分泌タンパク質のシグナル配列を用いることができ、好ましくはアミロイド前駆体タンパク質(APP)のN末端シグナル配列が用いられる。具体的には、アクセッション番号NP_958817の分泌シグナル配列(ヌクレオチド配列は、例えばアクセッション番号NM_201414を参照)を含む。すなわち適した本発明のベクターは、分泌シグナルが付加されたAβペプチドをコードする。
【0035】
さらに本発明のベクターは、外来遺伝子としてTh2サイトカインおよび/または抗炎症性サイトカインをコードする核酸を含んでもよい。Th2サイトカインとは、1型のヘルパーT細胞(Th1細胞)よりも2型のヘルパーT細胞(Th2細胞)によって優位に産生されるサイトカインを指す。具体的にはIL-4、IL-5、IL-6、IL-9、IL-10、およびIL-13が含まれる。本発明において、ベクターにコードされるサイトカインは、好ましくはIL-4、IL-10、およびIL-13からなる群より選択され、最も好ましくはIL-10がコードされる。各サイトカイン遺伝子のヌクレオチド配列およびそのアミノ酸配列は公知である(IL-4:NM_000589、NP_000580、AAH66277、AAH67515、NP_758858、NP_067258、NP_958427;IL-5:NM_000879、NP_000870、CAA31210、NP_034688、NP_068606;IL-6:NM_000600、NP_000591、XP_518992、AAB30962、NP_112445;IL-9:NM_000590、NP_000581、AAH66284、AAH66287、NP_032399、XP_341488;IL-10:NM_000572、NP_000563、CAG46825、NP_034678、NP_036986;IL-13:NM_002188、NP_002179、AAB01681、NP_032381、NP_446280)。
【0036】
ベクターにコードされるIL-10などのTh2サイトカインは、全長(すなわち野生型)でもよく、活性が維持される限り部分ペプチド(すなわち活性断片)でもよい。例えば、N末端のシグナル配列は、適切に他のシグナル配列によって置換してもよい。または、サイトカインは、他のペプチドとの融合タンパク質として発現させてもよい。
【0037】
ベクターにコードされるAβおよびIL-10などのサイトカインの由来に制限はなく、マウス、ラット、モルモット、ウサギ、ブタ、ウシ、ウマ、ロバ、ヤギ、イヌ、チンパンジー、サル、およびヒトを含む任意の哺乳動物も用いることができる。Aβおよびサイトカインの由来は、同じ由来であってもよく、異なる由来であってもよい。投与対象と同種由来のAβおよびサイトカインを用いるのが適当である。これらのサイトカインをコードする核酸のヌクレオチド配列は、上記のデータベースから入手可能である。さらに例を挙げれば、マウスIL-10について、Genbankアクセッション番号AY410237およびNM_010548、ヒトIL-10について、Genbankアクセッション番号AY029171およびNM_000572が利用可能である。本発明のベクターとして好適に用いることのできる「Aβをコードする核酸」は、ヒトAβcDNA(配列番号:1)である。好適に用いることのできる「IL-10をコードする核酸」は、マウスIL-10cDNA配列(配列番号:2)およびヒトIL-10cDNA配列(配列番号:3)である。上記配列番号:1〜3のヌクレオチド配列と異なる配列を有する核酸は、コドンの縮重の結果として、これらが上記ヌクレオチド配列と同じアミノ酸配列を有するのであれば、配列番号:1〜3の配列と同様に、本発明のベクターとして好適に用いることができる。上記外来遺伝子の挿入領域は、例えばゲノムのタンパク質非コード領域内の所望の部位において選択することができる。例えば3'リーダー領域と3'末端に最も近いウイルスタンパク質ORFとの間;各ウイルスタンパク質ORFの間;および/または5'末端に最も近いウイルスタンパク質ORFと5'トレイラー領域の間に外来遺伝子を挿入することができる。さらに、FまたはHN遺伝子などを欠失するゲノムでは、その欠失領域に挿入することができる。パラミクソウイルスに外来遺伝子を導入する場合、ゲノムに挿入されるポリヌクレオチドの鎖長は、好ましくは6の倍数である(J. Virology, 67(8):4822-4830, 1993)。挿入した外来遺伝子とウイルスORFとの間に、E-I-S配列が1セットずつ配置されるようにする。E-I-S配列を用いて2またはそれ以上の外来遺伝子をタンデムに並べて挿入することができる。または、IRESを用いて所望の外来遺伝子を挿入してもよい。
【0038】
ベクターが保持する外来遺伝子の発現レベルは、その遺伝子の上流(マイナス鎖の3'側)に付加する転写開始配列の種類により調節することができる(WO 01/18223)。発現レベルは、ゲノム内の外来遺伝子の挿入位置によって制御することができ;マイナス鎖の3'末端の近くに挿入するほど発現レベルが高く;5'末端の近くに挿入するほど発現レベルが低くなる。このように、外来遺伝子の挿入位置は、所望の遺伝子発現量を得るために、または外来遺伝子の近傍のウイルスタンパク質をコードする遺伝子との組み合わせが最適となるように適切に調節することができる。一般に、Aβの高い発現レベルを得ることが有利であると考えられるため、Aβをコードする外来遺伝子を、効率の高い転写開始配列に連結し、それをマイナス鎖ゲノムの3'末端近くに挿入することが好ましい。具体的には、外来遺伝子は、3'リーダー領域と3'末端に最も近いウイルスタンパク質ORFとの間に挿入される。または、外来遺伝子は、3'末端に一番近いウイルス遺伝子と2番目の遺伝子のORFの間に挿入してもよい。野生型パラミクソウイルスにおいて、ゲノムの3'末端に最も近いウイルスタンパク質遺伝子はN遺伝子であり、2番目に近い遺伝子はP遺伝子である。または、導入遺伝子の高発現レベルが望ましくない場合、適切な効果が得るために、例えばベクターにおいてマイナス鎖ゲノムのなるべく5'側の位置で外来遺伝子を挿入することによって、または効率の低い転写開始配列を選択することによって、ウイルスベクターからの遺伝子発現レベルを低く抑えることができる。
【0039】
Aβを含むポリペプチドおよびTh2サイトカインを含むポリペプチドの2つのポリペプチドをベクターから発現させるため、それぞれのポリペプチドをコードする核酸をベクターのゲノムに挿入する。2つの核酸は別々の部位に挿入してもよく、またはE-I-S配列を介してタンデムに並べて1つの部位に挿入してもよい。S配列として転写開始効率の高い配列を用いることが好ましい。例えば3'-UCCCACUUU-5'(マイナス鎖RNA;配列番号:6)、3'-UCCCAGUUU-5'(マイナス鎖RNA;配列番号:7)、または3'-UCCCACUUA-5'(マイナス鎖RNA;配列番号:8)を、S配列として好適に用いることができる。
【0040】
本発明のベクターは、AβおよびTh2サイトカインをコードする遺伝子を挿入した以外の位置に他の外来遺伝子を保持してもよい。このような外来遺伝子は制限されない。例えばベクターの感染をモニターするためのマーカー遺伝子、またはサイトカインおよびホルモンのような免疫系調節遺伝子であってもよい。本発明のベクターは、シグナル伝達調節因子、サイトカイン、ホルモン、受容体、抗体、およびそれらの断片をコードする1つまたは複数の遺伝子を保持してもよい。本発明のベクターは、生体の標的部位への直接(インビボ)投与、または患者由来細胞もしくはそれ以外の細胞にベクターを感染させ、その細胞を標的部位へ注入する間接(エクスビボ)投与のいずれかにより、遺伝子を導入することができる。
【0041】
本発明のベクターは、アルツハイマー病の治療、進行防止または阻害のための極めて優れた手段として用いることができる。本発明のベクターは、通常大量のAβが蓄積している24〜25ヶ月齢のAPP Tgマウスにおいて、著しいAβ消失効果を示した。この効果は、1回のみの鼻腔内投与によって確認され、本発明のベクターは、患者に対する侵襲性の低い治療法であり得る。中枢神経系における炎症が認められなかったという所見は、使用するベクターの安全性の高さも実証する。
【0042】
本発明のベクターを製造するために、哺乳動物細胞においてマイナス鎖RNAウイルスのゲノムRNA(またはその相補鎖)を含むRNPの再構成に必要なウイルスタンパク質(すなわちN、P、およびLタンパク質)の存在下で、本発明のマイナス鎖RNAウイルスのゲノムRNAをコードするcDNAを転写させる。転写によりマイナス鎖ゲノム(すなわちウイルスゲノムアンチセンス鎖と同様)またはプラス鎖(ウイルスタンパク質をコードするセンス鎖)のいずれかを生成させることによって、ウイルスRNPを再構成することができる。ベクターの再構成効率を高めるために、好ましくはプラス鎖を生成させる。RNA末端は、天然のウイルスゲノムと同じくらい正確に、3'リーダー配列と5'トレイラー配列の末端を反映させることが好ましい。転写産物の5'末端を正確に制御するために、例えば転写開始部位としてT7 RNAポリメラーゼ認識配列を利用し、該RNAポリメラーゼを細胞内で発現させてもよい。転写産物の3'末端を制御するため、例えば転写産物の3'末端に自己切断型リボザイムをコードさせ、このリボザイムにより正確に3'末端が切り出されるようにすることができる(Hasan, M. K. et al., J. Gen. Virol. 78:2813-2820, 1997;Kato, A. et al., EMBO J. 16:578-587, 1997;およびYu, D. et al., Genes Cells 2:457-466, 1997)。
【0043】
例えば外来遺伝子を有する組換えセンダイウイルスベクターは、Hasan, M. K. et al., J. Gen. Virol. 78:2813-2820, 1997;Kato, A. et al., EMBO J. 16:578-587, 1997;およびYu, D. et al., Genes Cells 2:457-466, 1997の記載に基づき、下記のように構築することができる。
【0044】
まず、目的の外来遺伝子のcDNAヌクレオチド配列を含むDNA試料を調製する。DNA試料は、25ng/μl以上の濃度で電気泳動的により単一のプラスミドとして確認できることが好ましい。以下、NotI部位を利用してウイルスゲノムRNAをコードするDNAに外来遺伝子を挿入する場合を例にとって説明する。目的とするcDNAヌクレオチド配列中にNotI認識部位が含まれる場合、部位特異的変異導入法などを用いて、コードするアミノ酸配列を変化させないようにヌクレオチド配列を改変し、NotI部位をあらかじめ除去しておくことが好ましい。この試料から目的の遺伝子断片をPCRにより増幅し回収する。2つのプライマーの5'領域にNotI部位を付加することにより、増幅された断片の両端をNotI部位とする。外来遺伝子をウイルスゲノム上に挿入した後、外来遺伝子のORFとその両側のウイルス遺伝子のORFの間にE-I-S配列が1つずつ配置されるように、プライマー中にE-I-S配列またはその部分を含める。
【0045】
例えば、フォワード合成DNA配列は、NotIによる切断を保証するため、5'側に2つ以上のヌクレオチドの任意の配列(好ましくはGCGおよびGCCなどのNotI認識部位由来の配列が含まれない4ヌクレオチド、およびより好ましくはACTT)を選択し、その3'側にNotI認識部位gcggccgcを付加した形態を有する。スペーサー配列として、任意の9ヌクレオチド、または9に6の倍数を加えた数のヌクレオチドを、その3'側にさらに付加する。さらにその3'側に、開始コドンATGを含む所望のcDNAの約25ヌクレオチドのORFに相当する配列を付加する。フォワード合成オリゴDNAの3'の末端で、最後のヌクレオチドがGまたはCとなるように、所望のcDNAから約25ヌクレオチドを選択することが好ましい。
【0046】
リバース合成DNA配列のために、5'側で任意の2つ以上のヌクレオチド(好ましくはGCGおよびGCCなどのNotI認識部位由来の配列が含まれない4ヌクレオチド、およびより好ましくはACTT)を選択し、その3'側にNotI認識部位「gcggccgc」を付加し、さらにその3'側に長さを調節するためのオリゴDNA断片を付加する。このオリゴDNAの長さは、付加したE-I-S配列を含む最終的なPCR増幅産物であるNotI断片の鎖長が、6の倍数のヌクレオチド数になるように設計する(いわゆる「6のルール(rule of six)」:Kolakofski, D., et al., J. Virol. 72:891-899, 1998;Calain, P. and Roux, L., J. Virol. 67:4822-4830, 1993;Calain, P. and Roux, L., J. Virol. 67:4822-4830, 1993)。このプライマーの挿入されたオリゴDNA断片の3'側にE-I-S配列を付加する場合、センダイウイルスのS配列の相補鎖配列、好ましくは5'-TTTCACCCT-3'(配列番号:9)、5'-TTTGACCCT-3'(配列番号:10)、または5'-ATTCACCCT-3'(配列番号:11)、I配列の相補鎖配列、好ましくは5'-AAG-3'、E配列の相補鎖配列、好ましくは5'-TTTTTCTTACTACGG-3'(配列番号:12)を付加する。さらにその3'側に、最後のヌクレオチドとしてGまたはCを有する相補鎖配列を付加し、長さが所望のcDNA配列の終始コドンから逆に数えて約25ヌクレオチドになるように選択した。このようにして、リバース合成DNAの3'末端とする。
【0047】
PCRは、Taqポリメラーゼまたはその他のDNAポリメラーゼを用いる通常の方法によって実施することができる。増幅した目的断片はNotIで消化した後、pBluescript(商標)(Stratagene)などのプラスミドベクターのNotI部位に挿入する。得られたPCR産物のヌクレオチド配列をシークエンサーで確認し、正しい配列を含むプラスミドを選択する。これらのプラスミドから挿入断片をNotIを用いて切り出し、ゲノムcDNAを含むプラスミドのNotI部位にクローニングする。またプラスミドベクターを用いずにNotI部位に断片を直接挿入することによって、組換えセンダイウイルスcDNAを得ることも可能である。
【0048】
例えば、組換えセンダイウイルスゲノムcDNAは、文献記載の方法に従って構築することができる(Yu, D. et al., Genes Cells 2:457-466, 1997;Hasan, M. K. et al., J. Gen. Virol. 78:2813-2820, 1997)。例えば、NotI制限部位を含む18bpのスペーサー配列(5'-(G)-CGGCCGCAGATCTTCACG-3')(配列番号:13)を、クローニングされたセンダイウイルスゲノムcDNA(pSeV(+))内のリーダー配列とNタンパク質のORFとの間に挿入し、デルタ肝炎ウイルスのアンチゲノム鎖由来の自己切断リボザイム部位を含むプラスミドpSeV18+b(+)を得る(Hasan, M. K. et al., J. General Virology 78:2813-2820, 1997)。pSeV18+b(+)のNotI部位に外来遺伝子断片を挿入することにより、所望の外来遺伝子を含む組換えセンダイウイルスcDNAを得ることができる。
【0049】
このように調製した組換えウイルスのゲノムRNAをコードするDNAを、上記のウイルスタンパク質(L、P、およびN)存在下で細胞内で転写させることにより、本発明のベクターを再構成することができる。本発明は、本発明のベクターの作製のための、本発明のベクターのウイルスゲノムRNAをコードするDNAを提供する。本発明はまた、本発明のベクターの作製に適用するための、ベクターのゲノムRNAをコードするDNAの使用に関する。組換えウイルスは、文献における公知の方法により再構成することができる(WO 97/16539; WO 97/16538; WO 00/70055; WO 00/70070; WO 01/18223; WO 03/025570; Durbin, A. P. et al., Virology 235: 323-332, 1997; Whelan, S. P. et al., Proc. Natl. Acad. Sci. USA 92: 8388-8392, 1995; Schnell. M. J. et al., EMBO J. 13: 4195-4203, 1994; Radecke, F. et al., EMBO J. 14: 5773-5784, 1995; Lawson, N. D. et al., Proc. Natl. Acad. Sci. USA 92: 4477-4481, 1995; Garcin, D. et al., EMBO J. 14: 6087-6094, 1995; Kato, A. et al., Genes Cells 1: 569-579, 1996; Baron, M. D. and Barrett, T., J. Virol. 71: 1265-1271, 1997; Bridgen, A. and Elliott, R. M., Proc. Natl. Acad. Sci. USA 93: 15400-15404, 1996; Hasan, M. K. et al., J. Gen. Virol. 78: 2813-2820, 1997; Kato, A. et al., EMBO J. 16: 578-587, 1997; Yu, D. et al., Genes Cells 2: 457-466, 1997; Tokusumi, T. et al., Virus Res. 86: 33-38, 2002; Li, H.-O. et al., J. Virol. 74: 6564-6569, 2000)。これらの方法により、パラインフルエンザウイルス、水疱性口内炎ウイルス、狂犬病ウイルス、麻疹ウイルス、リンダーペストウイルス、およびセンダイウイルスを含むマイナス鎖RNAウイルスを、DNAから再構成させることができる。これらの方法に従って、本発明のベクターを再構成させることができる。F遺伝子、HN遺伝子、および/またはM遺伝子などを欠失させたウイルスベクターDNAは、感染性のウイルス粒子を形成しない。しかしながら欠失した遺伝子および/または他のウイルスのエンベロープタンパク質をコードする遺伝子を宿主細胞に導入し発現させることにより、感染性のウイルス粒子を形成させることが可能である。
【0050】
具体的には、ウイルスは以下の工程に従って作製することができる:(a)マイナス鎖RNAウイルスのゲノムRNA(マイナス鎖RNA)またはその相補鎖(プラス鎖)をコードするcDNAを、N、P、およびLタンパク質を発現する細胞内で転写させる工程、ならびに(b)それらの細胞またはその培養上清から該ゲノムRNAを含むRNA複合体を回収する工程。転写のために、ゲノムRNAをコードするDNAは適当なプロモーターの下流に連結される。転写されたゲノムRNAはN、L、およびPタンパク質の存在下で複製されRNP複合体を形成する。次いでM、HN、およびFタンパク質の存在下でエンベロープに包まれたウイルス粒子が形成される。ゲノムRNAをコードするDNAは、例えばT7プロモーターの下流に連結し、T7 RNAポリメラーゼによりRNAに転写させることができる。T7ポリメラーゼ認識配列を含むもの以外にも、所望のプロモーターを利用することができる。または、インビトロで転写させたRNAを細胞にトランスフェクトしてもよい。N、L、およびPタンパク質は、プラスミドなどの適当な発現ベクターを用いて細胞内で発現させることができる。プロモーターの例には、CMVプロモーターおよびCAGプロモーターが含まれる(Niwa, H. et al.Gene. 108:193-199, 1991;特開平3-168087)。
【0051】
DNAからのゲノムRNAの最初の転写に必要なT7 RNAポリメラーゼなどの酵素は、これらを発現するプラスミドベクターまたはウイルスベクターの導入によって、または、例えば細胞の染色体にこれらの酵素の遺伝子を、発現を誘導できるように組み込み、ウイルス再構成時に発現を誘導することによって供給することができる。さらにゲノムRNA、およびベクター再構成に必要なウイルスタンパク質は、例えばこれらを発現するプラスミドの導入によって供給する。これらのウイルスタンパク質の供給において、野生型またはある種の変異マイナス鎖RNAウイルスのヘルパーウイルスを用いることもできるが、これらのウイルスの混入が発生し得るため好ましくない。
【0052】
ゲノムRNAを発現するDNAを細胞内に導入する方法には、例えば以下を含む:(i)目的の細胞が取り込めるようなDNA沈殿物を作製する方法;(ii)低い細胞毒性を有し、かつ目的の細胞による取り込みに適したDNAを含む陽性に荷電した複合体を作製する方法;および(iii)目的の細胞へDNA分子が通り抜けるのに十分なサイズの穴を、細胞膜上に電気パルスによって瞬間的に開ける方法。
【0053】
方法(ii)において、様々なトランスフェクション試薬、例えば、DOTMA(Roche)、Superfect(QIAGEN #301305)、DOTAP、DOPE、DOSPER(Roche #1811169)などが利用できる。(i)の例は、リン酸カルシウムを用いたトランスフェクション法であり、この方法によって細胞内に移入したDNAは貧食小胞に取り込まれるが、核内にも十分な量のDNAが入ることが知られている(Graham, F. L. and Van Der Eb, J., Virology 52:456, 1973;Wigler, M. and Silverstein, S., Cell 11:223, 1977)。ChenおよびOkayamaは移入技術の最適化を検討し、(1)細胞と共沈殿物のインキュベーション条件が、2〜4%CO2、35℃、15〜24時間であり、(2)直鎖状DNAより環状DNAが高い活性を有し、かつ(3)沈殿混合物が20〜30μg/mlのDNA濃度を有する場合、最適な沈殿が得られることを報告している(Chen, C. and Okayama, H., Mol. Cell. Biol. 7:2745, 1987)。(ii)の方法は、一過的なトランスフェクションに適している。以前はDEAE-デキストラン(Sigma #D-9885 M.W. 5×105)混合物を所望のDNA濃度比に基づいて調製するトランスフェクション方法が知られていた。複合体の多くはエンドソームの中で分解されてしまうため、効果を高めるためにクロロキンを加えることもできる(Calos, M. P., Proc. Natl. Acad. Sci. USA 80:3015, 1983)。(iii)の方法は電気穿孔法と呼ばれ、細胞選択性がないため、(i)および(ii)の方法がより汎用性が高い。方法の効率は、パルス電流の持続時間、パルスの形、電界(電極間のギャップ、電圧)の強さ、緩衝液の導電率、DNA濃度、および細胞密度の最適条件下で良好とされる。
【0054】
以上3つのカテゴリーの中で(ii)の方法は、操作が簡便で多量の細胞を用いて多数の試料を検討することができるため、ベクター再構成においてDNAの細胞への導入に、トランスフェクション試薬が適している。好ましくは、Superfect Transfection Reagent(QIAGEN, Cat No.301305)、DOSPER Liposomal Transfection Reagent(Roche, Cat No.1811169)、およびTransIT(Mirus, Cat No.MIR2300)のようなトランスフェクション試薬が用いられる。しかしこれらに制限されない。
【0055】
具体的に、cDNAからのウイルスの再構成は、例えば以下のように行うことができる。
【0056】
約6〜24穴のプラスチックプレートまたは100mmペトリ皿などで、10%ウシ胎児血清(FCS)および抗生物質(100ユニット/mlペニシリンGおよび100μg/mlストレプトマイシン)を含む最少必須培地(MEM)を用いて、サル腎臓由来LLC-MK2をほぼ100%コンフルエントまで培養し、例えばT7 RNAポリメラーゼを発現し、かつ1μg/mlソラレン存在下で20分のUV照射により不活化した組換えワクシニアウイルスvTF7-3を、2 PFU/細胞で感染させる(Fuerst, T. R. et al., Proc. Natl. Acad. Sci. USA 83:8122-8126,1986、Kato, A. et al., Genes Cells 1:569-579, 1996)。ソラレンの添加量およびUV照射時間は適切に調整することができる。感染の1時間後、2〜60μg、より好ましくは3〜20μgの組換えセンダイウイルスのゲノムRNAをコードするDNAを、ウイルスRNPの生成に必須なトランスに作用するウイルスタンパク質を発現するプラスミド(0.5〜24μgのpGEM-N、0.25〜12μgのpGEM-P、および0.5〜24μgのpGEM-L)(Kato, A. et al., Genes Cells 1:569-579, 1996)と共に、Superfect(QIAGEN社)を用いたリポフェクション法などによりトランスフェクションする。N、P、およびLタンパク質をコードする発現ベクターの量比は、好ましくは2:1:2であり、プラスミド量は、例えば1〜4μgのpGEM-N、0.5〜2μgのpGEM-P、および1〜4μgのpGEM-Lの範囲内で適切に調整した。
【0057】
トランスフェクションを行った細胞は、所望により100μg/mlのリファンピシン(Sigma)およびシトシンアラビノシド(AraC)、より好ましくは40μg/mlのシトシンアラビノシド(AraC)(Sigma)のみを含む無血清MEMで培養する。ワクシニアウイルスによる細胞毒性を最少にとどめ、ウイルスの回収率を最大にするように薬剤の最適濃度を設定する(Kato, A. et al., Genes Cells 1:569-579, 1996)。トランスフェクションから48〜72時間培養後、細胞を回収し、凍結融解を3回繰り返して細胞を破砕する。RNPを含む破砕物をLLC-MK2細胞に感染させ、さらに培養する。または、培養上清を回収し、LLC-MK2細胞の培養液に添加して感染させ培養する。トランスフェクションは、例えばリポフェクトアミンまたはポリカチオニックリポソームなどと共に複合体を形成させ、それを細胞に導入することによって実行することができる。具体的には、様々なトランスフェクション試薬、例えば、DOTMA(Roche)、Superfect(QIAGEN #301305)、DOTAP、DOPE、DOSPER(Roche #1811169)が利用できる。エンドソーム中での分解を防ぐため、クロロキンを加えることもできる(Calos, M. P., Proc. Natl. Acad. Sci. USA 80:3015, 1983)。RNPが導入された細胞において、RNPからのウイルス遺伝子の発現およびRNPの複製の結果、ベクターが増幅する。得られたウイルス溶液(上清)を希釈(例えば106倍)し、新しい細胞を用いて増幅を繰り返すことにより、ワクシニアウイルスvTF7-3は完全に除去することができる。そのような増幅を、例えば3回以上繰り返す。得られたベクターは-80℃で保存することができる。伝播能を持たず、エンベロープタンパク質をコードする遺伝子を欠損したウイルスベクターを再構成させるために、エンベロープタンパク質を発現するLLC-MK2細胞をトランスフェクションに使用してもよく、またはエンベロープ発現プラスミドを共にトランスフェクションしてもよい。または、トランスフェクションを行った細胞にエンベロープタンパク質を発現するLLK-MK2細胞をさらに重層して培養することによって、欠損型ウイルスベクターを増幅することもできる(WO 00/70055およびWO 00/70070を参照されたい)。
【0058】
回収されたウイルスの力価は、例えばCIU(Cell-Infectious Unit)または赤血球凝集活性(HA)を測定することにより決定することができる(WO 00/70070;Kato, A. et al., Genes Cells 1:569-579, 1996;Yonemitsu, Y. & Kaneda, Y., Hemaggulutinating virus of Japan-liposome-mediated gene delivery to vascular cells. Ed. by Baker AH. Molecular Biology of Vascular Diseases, Method in Molecular Medicine:Humana Press:pp. 295-306, 1999)。GFP(緑色蛍光タンパク質)マーカー遺伝子などを保持したベクターの力価は、マーカーを指標に直接感染細胞をカウントすることにより、定量することができる(例えばGFP-CIUとして)。このように測定した力価は、CIUと同等に扱うことができる(WO 00/70070)。
【0059】
ウイルスベクターを再構成することができる限り、再構成に用いる宿主細胞は特に制限されない。例えば、センダイウイルスベクターなどの再構成において、サル腎臓由来のLLC-MK2細胞(ATCC CCL-7)およびCV-1細胞(例えばATCC CCL-70)、ハムスター腎臓由来のBHK細胞(例えばATCC CCL-10)などの培養細胞、ヒト由来細胞を用いることができる。これらの細胞に適当なエンベロープタンパク質を発現させることで、そのタンパク質をエンベロープに含む感染性ウイルス粒子を得ることもできる。さらに、大量にセンダイウイルスベクターを得るために、上記の宿主から得られたウイルスベクターを発育鶏卵に感染させ、該ベクターを増幅することができる。鶏卵を使ったウイルスベクターの作製方法は既に開発されている(Nakanishi,M. et al. ed.「State-of-the-Art Technology Protocol in Neuroscience Research III (Shinkei Kagaku Kenkyu-no Sentan Gijutu Protocol III); Molecular Neuron Physiology (Bunshi Sinkeisaibou Seirigaku)」, Koseisha, Osaka, pp. 153-172, 1993)。具体的には、例えば、受精卵を培養器に入れ9〜12日間、37〜38℃で培養し、胚を成長させる。ウイルスベクターを尿膜腔へ接種し、数日間(例えば3日間)卵を培養してウイルスベクターを増殖させる。培養期間などの条件は、増幅する組換えセンダイウイルスにより変わり得る。その後、ウイルスを含んだ尿膜腔液を回収する。通常の方法に従って、センダイウイルスベクターを尿膜腔液から分離および精製することができる(Tashiro, M.,「Virus Experiment Protocol」, Nagai, Ishihama, ed., Medical View Co., Ltd., pp. 68-73, 1995)。
【0060】
例えば、F遺伝子を欠失したセンダイウイルスベクターの再構成と産生は、以下のように行うことができる(WO 00/70055およびWO 00/70070を参照されたい)。
【0061】
<1>F遺伝子欠失型センダイウイルスゲノムcDNAおよびF発現プラスミドの構築
センダイウイルス(SeV)全長ゲノムcDNA、pSeV18+b(+)(Hasan, M. K. et al., J. General Virology 78:2813-2820, 1997)(「pSeV18+b(+)」は「pSeV18+」とも言う)のcDNAをSphI/KpnIで消化して断片(14673bp)を生成する。断片をpUC18にクローニングしてプラスミドpUC18/KSを調製し、pUC18/KS上でF遺伝子欠損部位を構築する。F遺伝子の欠失を、PCRおよびライゲーション方法の組み合わせで作製する。結果としてF遺伝子のORF(ATG-TGA=1698bp)が除かれる。例えばatgcatgccggcagatga(配列番号:14)で連結し、F遺伝子欠損型SeVゲノムcDNA(pSeV18+/ΔF)を構築する。Fの上流領域についてプライマー[フォワード:5'-gttgagtactgcaagagc/配列番号:15、リバース:5'-tttgccggcatgcatgtttcccaaggggagagttttgcaacc/配列番号:16]を用いて形成されたPCR産物、およびFの下流領域についてプライマー[フォワード:5'-atgcatgccggcagatga/配列番号:17、リバース:5'-tgggtgaatgagagaatcagc/配列番号:18]を用いて形成されたPCR産物を、EcoT22I制限酵素部位を介してライゲーションした。このように得られたプラスミドをSacIとSalIで消化し、F遺伝子欠損部位を含む領域をカバーする断片(4931bp)を回収してpUC18にクローニングし、pUC18/dFSSを形成する。このpUC18/dFSSをDraIIIで消化して、ライゲーションを介して、回収した断片をpSeV18+のF遺伝子を含む領域をカバーするDraIII断片と置き換え、プラスミドpSeV18+/ΔFを得る。
【0062】
外来遺伝子は、例えばpUC18/dFSSのF遺伝子欠失部位にあるNsiIおよびNgoMIVの制限酵素部位に挿入する。このために、例えば外来遺伝子断片を、NsiI-tailedプライマーおよびNgoMIV-tailedプライマーを用いて増幅してもよい。
【0063】
<2>SeV-Fタンパク質を発現するヘルパー細胞の作製
センダイウイルスのF遺伝子(SeV-F)を発現するCre/loxP誘導型プラスミドの構築のため、SeV-F遺伝子をPCRで増幅し、Cre DNAリコンビナーゼにより遺伝子産物を発現するように設計したプラスミドpCALNdlw(Arai, T. et al., J. Virology 72:1115-1121, 1998)のユニークSwaI部位に挿入し、pCALNdLw/Fプラスミドを構築する。
【0064】
F遺伝子欠損ゲノムから感染ウイルス粒子を回収するため、SeV-Fタンパク質を発現するヘルパー細胞株を樹立する。例えばSeVの増殖に一般に用いられるサル腎臓由来LLC-MK2細胞を用いることができる。LLC-MK2細胞は、10%の熱不活化ウシ胎児血清(FBS)、ペニシリンGナトリウム(50ユニット/ml)、およびストレプトマイシン50μg/mlを添加したMEMで37℃、5%CO2で培養する。SeV-F遺伝子産物は細胞傷害性であるため、Cre DNAリコンビナーゼによりF遺伝子産物を発現するように設計した上記pCALNdLw/Fプラスミドを、mammalian transfection kit(Stratagene)を用いてリン酸カルシウム法により、周知のプロトコールに従って、LLC-MK2細胞にトランスフェクションする。
【0065】
10cmプレートで40%コンフルエントまで生育したLLC-MK2細胞に、pCALNdLw/Fプラスミド(10μg)を導入し、細胞を10%FBS/MEM(10ml)にて、37℃の5%CO2インキュベーター中で24時間培養する。次いで細胞をはがし、培地(10ml)に懸濁する。懸濁液を10cmシャーレ5枚に播種する:5ml 1枚、2ml 各2枚、および0.2ml 各2枚。G418(GIBCO-BRL)(1200μg/ml)および10%FBSを含むMEM(10ml)にて、細胞を14日間培養し、2日毎に培地交換する。遺伝子が安定して導入された細胞株を選択する。上記培地で生育した細胞はG418に耐性であり、次いでクローニングリングを用いて回収する。回収した各細胞クローンは、10cmプレートでコンフルエントになるまで培養を続ける。
【0066】
Fタンパク質の発現は、細胞を6cmシャーレにてコンフルエントまで生育させた後、Saitoらの方法(Saito et al., Nucl. Acids Res. 23:3816-3821, 1995;Arai, T.et al., J. Virol 72:1115-1121, 1998)により、例えばMOI=3でアデノウイルスAxCANCreを細胞に感染させることによって誘導する。
【0067】
<3>F遺伝子欠失センダイウイルス(SeV/ΔF)の再構成および増幅
外来遺伝子が挿入された上記pSeV18+/ΔFプラスミドを、以下のようにLLC-MK2細胞にトランスフェクションする。LLC-MK2細胞を5×106細胞/シャーレで100mmのシャーレに播種する。T7 RNAポリメラーゼによりゲノムRNAの転写を実施する場合、細胞を24時間培養し、次いでソラレンと長波長紫外線(365nm)で20分間処理したT7 RNAポリメラーゼを発現する組換えワクシニアウイルス(PLWUV-VacT7:Fuerst, T.R. et al., Proc. Natl. Acad. Sci. USA 83:8122-8126, 1986)を、MOI=2程度で室温で1時間感染させる。ワクシニアウイルスへの紫外線照射には、例えば15ワットバルブ5本が装備されたUV Stratalinker(商標)2400(カタログ番号400676(100V), Stratagene, La Jolla, CA, USA)を用いることができる。細胞を無血清MEMで洗浄した後、適切なリポフェクション試薬を用いて、ゲノムRNAを発現するプラスミド、ならびにパラミクソウイルスのN、P、L、およびFプラスHNタンパク質を発現する発現プラスミドをトランスフェクションする。これらのプラスミドの量は、これに限定されないが、好ましくは順に 6:2:1:2:2とすることができる。例えば、ゲノムRNA発現プラスミド、ならびにN、P、L、および FプラスHNタンパク質を発現する発現プラスミド(pGEM/NP、pGEM/P、pGEM/L、およびpGEM/F-HN;WO 00/70070, Kato, A. et al., Genes Cells 1:569-579, 1996)を、それぞれ12μg、4μg、2μg、4μg、および4μg/シャーレの量でトランスフェクションする。数時間培養後、無血清MEMで細胞を2回洗浄し、40μg/mlのシトシンβ-D-アラビノフラノシド(AraC:Sigma, St.Louis, MO)および7.5μg/mlのトリプシン(Gibco-BRL, Rockville, MD)を含むMEMで培養する。これらの細胞を回収し、ペレットをOpti-MEM に懸濁する(107細胞/ml)。凍結融解を3回繰り返してリポフェクション試薬DOSPER(Boehringer Mannheim)と混合し(106細胞/25μl DOSPER)室温で15分放置した後、上記でクローニングしたF発現ヘルパー細胞にトランスフェクション(12-ウェル-プレート中106細胞/ウェル)し、無血清MEM(40μg/ml AraCおよび7.5μg/mlトリプシンを含む)で培養し、上清を回収する。F以外の遺伝子、例えばHNおよび/またはM遺伝子を欠損したウイルスはいずれも、これと同様の方法で調製することができる。
【0068】
ウイルス遺伝子欠損型ベクターの作製において、例えば、異なるウイルス遺伝子がウイルスゲノムから欠損している2種またはそれ以上のベクターを、同じ細胞に導入する場合、各ベクターの欠損したウイルスタンパク質が他のベクターからの発現により供給され、そのような相互補完が、感染力のあるウイルス粒子の構築、および複製サイクルを介したウイルスベクターの増幅を可能にする。結果として、個々のウイルス遺伝子欠損型ウイルスベクターの混合物を低コストで大量に産生することができる。このようなウイルスは、ウイルス遺伝子が欠損しているため、無傷のウイルスよりゲノムサイズが小さい。このようなウイルスは、サイズの大きい外来遺伝子を保持することができる。さらに、ウイルス遺伝子の欠損により非伝播性であるこのようなウイルスは、一旦細胞外に放出された後、単に希釈されると、感染力の維持が困難であることから、不稔化するため、環境へのそれらの放出が管理できるという利点がある。例えばAβをコードするベクターとTh2サイトカインをコードするベクターとを互いに相補できるように別々に構築し、共感染に用いてもよい。本発明は、Aβを含むポリペプチドをコードするマイナス鎖RNAウイルスベクター、およびTh2サイトカインをコードするマイナス鎖RNAウイルスベクターを含む組成物を提供する。本発明はまた、Aβを含むポリペプチドをコードするマイナス鎖RNAウイルスベクター、およびTh2サイトカインをコードするマイナス鎖RNAウイルスベクターを含むキットを提供する。これらの組成物およびキットは、アミロイドβ沈着を減少させるために使用できる。
【0069】
伝播性のマイナス鎖RNAウイルスベクターを個体または細胞に投与後、例えば治療の完了時にウイルスベクターの増殖を抑止する必要が生じた場合、RNA依存性RNAポリメラーゼ阻害剤を投与することによって、宿主に障害を与えずにウイルスベクターの増殖だけを特異的に抑止することもできる。
【0070】
本発明の方法によれば、本発明のウイルスベクターは、例えば1×105 CIU/ml以上、好ましくは1×106 CIU/ml以上、より好ましくは5×106 CIU/ml以上、より好ましくは1×107 CIU/ml以上、より好ましくは5×107 CIU/ml以上、より好ましくは1×108 CIU/ml以上、より好ましくは5×108 CIU/ml以上の力価でウイルス産生細胞の培養培地中に放出させることが可能である。ウイルスの力価は、本明細書および他に記載の方法により測定することができる(Kiyotani, K. et al., Virology 177(1):65-74, 1990;WO 00/70070)。
【0071】
回収したウイルスベクターは実質的に純粋になるよう精製することができる。ベクターの精製は、濾過、遠心分離、およびカラム精製を含む文献において公知の精製および分離方法、またはその組み合わせにより行うことができる。「実質的に純粋」とは、ウイルスベクターが、それが存在する試料中の成分として主要な割合を占めることを言う。典型的には、実質的に純粋なウイルスベクターは、ウイルスベクター由来のタンパク質の割合が、試料中の全タンパク質(しかしキャリアおよび安定剤として加えたタンパク質は除く)の10%以上、好ましくは20%以上、より好ましくは50%以上、好ましくは70%以上、より好ましくは80%以上、およびさらに好ましくは90%以上であることを確認することによって同定できる。ウイルスの具体的な精製方法の例として、セルロース硫酸エステルまたは架橋ポリサッカライド硫酸エステルを用いる方法(特公昭62-30752号公報、特公昭62-33879号公報、および特公昭62-30753号公報)、ならびにフコース硫酸を含む多糖および/またはその分解物にウイルスを吸着させる方法(WO 97/32010)がある。
【0072】
ベクターを含む組成物の調製において、ベクターは必要に応じて薬学的に許容される所望の担体または媒体と組み合わせることができる。「薬学的に許容される担体または媒体」とは、ベクターと共に投与することが可能であり、ベクターによる遺伝子導入を有意に阻害しない材料である。例えばベクターを生理食塩水またはリン酸緩衝生理食塩水(PBS)で適切に希釈して組成物を形成することができる。ベクターを鶏卵などにおいて増殖させた場合、「薬学的に許容される担体または媒体」は尿膜腔液を含んでよい。さらにベクターを含む組成物は、脱イオン水および5%デキストロース水溶液などの担体または媒体を含んでもよい。さらに、上記以外にも、植物油、懸濁剤、界面活性剤、安定剤、殺生物剤などが含まれてもよい。保存剤またはその他の添加剤を含むこともできる。本発明のベクターを含む組成物は試薬および薬剤として有用である。本発明は、Aβペプチドをコードするマイナス鎖RNAウイルスベクターまたは該ベクターが導入された細胞、および薬学的に許容される担体または媒体を含む組成物を製造する工程を含む、抗アミロイド沈着薬の製造方法に関する。本発明は、抗アミロイド沈着薬の製造における、マイナス鎖RNAウイルスベクター、該ベクターを産生する細胞、または該ベクターが導入された細胞の使用にも関する。
【0073】
本発明のベクターを含む組成物は、バイオポリマーなどの有機物、ハイドロキシアパタイトなどの無機物、具体的にはコラーゲンマトリックス、ポリ乳酸ポリマーまたはコポリマー、ポリエチレングリコールポリマーまたはコポリマーおよびそれらの化学的誘導体を、担体として組み合わせて含み得る。
【0074】
本発明の組成物は、Th2サイトカインおよび/またはTh2サイトカインをコードする核酸を、発現可能な様式で含んでもよい。このようなTh2サイトカインは、全長(野生型)ポリペプチド、または活性を保持する限り部分ペプチドであってよい。例えば、N末端および/またはC末端アミノ酸(例えば1〜30アミノ酸、具体的には1、2、3、4、5、10、15、20、または25アミノ酸)の欠失は、サイトカイン活性に影響しないだろう。さらに、N末端シグナル配列は、他のシグナル配列によって適切に置換することができる。または、サイトカインは他のペプチドとの融合タンパク質として発現させてもよい。Th2サイトカインをコードする核酸は、発現ベクターに挿入するか、またはアクチンプロモーター、EF1プロモーター、CMVプロモーター、CAGプロモーターなどの適切なプロモーターに接続してもよい。核酸は、マイナス鎖RNAウイルスベクターまたは任意の他の発現ベクター内に含まれてもよい。Th2サイトカインがマイナス鎖RNAウイルスベクターにコードされる場合、Th2サイトカインが同じベクター内にAβとしてコードされてもよく、またはTh2サイトカインが別のマイナス鎖RNAウイルスベクター内にコードされてもよい。具体的に、Aβをコードする本発明のマイナス鎖RNAウイルスベクターがTh2サイトカインをコードしない場合、Th1/Th2バランスは、Th2サイトカインまたはTh2サイトカインベクターを、Aβをコードする本発明のベクターと組み合わせて同時投与することにより、Th2にシフトさせることができる。例えば、用いることができるTh2サイトカインは、IL-4、IL-5、IL-6、IL-9、IL-10、およびIL-13、またはそれらの組み合わせである。Th2サイトカインは好ましくは、IL-4、IL-10、およびIL-13からなる群より選択される。
【0075】
本発明の組成物はまた、アジュバントを含んでもよい。具体的に、本発明のベクターがTh2サイトカインをコードしない場合であっても、Th2アジュバントを含む組成物を用いて、Th1/Th2バランスをTh2にシフトさせることができる。Th2アジュバントとは、1型のヘルパーT細胞(Th1細胞)よりも2型のヘルパーT細胞(Th2細胞)を優位に活性化するアジュバントを指す。具体的には、アジュバントは、水酸化アルミニウム(alum)、コレラ毒素(Bサブユニット)、マンソン住血吸虫卵抽出タンパク質(例えばLacto-N-fucopentaose III)などを含む(Grun, J. L. and P. H. Maurer, Cellular Immunol. 121: 134-145, 1989; Holmgren, J. et al., Vaccine 11:1179-1184, 1993; Wilson, A.D. et al., Vaccine 11: 113-118, 1993; Lindsay, D.S. et al, Int. Arch. Allergy Immunol. 105: 281-288, 1994; Xu-Amano, J. et al., J. Exp. Med. 178: 1309-1320, 1993; Okano, M. et al., J. Immunol. 167(1):442-50, 2001)。本発明の組成物は、任意の他の成分を含んでもよい。例えば、それらは投与をモニターするためのマーカー、または免疫系調節因子、シグナル伝達調節因子、サイトカイン、ホルモン、受容体、抗体、およびそれらの断片のような1つもしくは複数の化合物を含んでもよい。本発明の組成物は、シグナル伝達調節因子、シグナル伝達調節因子、サイトカイン、ホルモン、受容体、抗体、およびそれらの断片などの1つもしくは複数のポリペプチドをコードする1つまたは複数のベクターを含んでもよい。
【0076】
製造されたマイナス鎖RNAウイルスまたは該ウイルスを含む組成物を個体に投与することにより、Aβに対する免疫反応が誘導され、それによってAβ沈着を低下させる。ベクターは、Th2サイトカイン、Th2サイトカイン発現ベクター、またはTh2アジュバントと組み合わせて投与してもよい。ベクターの投与は、インビボ投与でも、細胞を介したエクスビボ投与でもよい。接種するマイナス鎖RNAウイルスベクターは、保持する抗原ポリペプチド遺伝子をゲノムRNAによって発現する能力を有する限り、感染性ウイルス粒子である必要はなく、非感染性ウイルス粒子およびウイルスのコア(ゲノムおよびゲノム結合ウイルスタンパク質を含むRNP複合体)であってもよい。本明細書においてマイナス鎖RNAウイルスベクターとは、マイナス鎖RNAウイルス由来のゲノムRNA、ならびにゲノムRNAの複製およびウイルスが保持する遺伝子の発現に必要なウイルスタンパク質を含むリボヌクレオプロテイン(RNP)複合体を含み、感染した細胞において該ゲノムRNAを複製し、保持する遺伝子を発現する複合体を言う。RNPは、例えばマイナス鎖RNAウイルスのゲノムRNA、ならびにN、L、およびPタンパク質を含む複合体である。具体的には、本発明のマイナス鎖RNAウイルスベクターには、感染性ウイルス粒子、非感染性粒子(ウイルス様粒子(VLP)とも言う)、およびマイナス鎖RNAウイルスのヌクレオカプシドなどの、ゲノムRNAおよびゲノムRNAに結合するウイルスタンパク質を含むRNPが含まれる。ウイルス粒子からエンベロープを除去したRNP(ウイルスコア)であっても、細胞に導入されれば、細胞内においてウイルスゲノムRNAを複製することができる(WO 97/16538;WO 00/70055)。RNPまたはVLPを望ましくは、例えばトランスフェクション試薬などと組み合わせて接種してもよい(WO 00/70055;WO 00/70070)。
【0077】
細胞を介してベクターを接種する場合、適当な培養細胞または接種対象動物から採取した細胞などに、マイナス鎖RNAウイルスベクターを導入する。マイナス鎖RNAウイルスをインビトロ(例えば試験管またはシャーレ内)で細胞に感染させる場合、感染は、例えば培養液または生理食塩水など所望の生理的水溶液中で、インビトロ(またはエクスビボ)で実施する。MOI(多重感染度;すなわち細胞1つあたりの感染ウイルス数)は、好ましくは1〜1000の間の範囲、より好ましくは2〜500、さらに好ましくは3〜300、さらに好ましくは5〜100である。マイナス鎖RNAウイルスと細胞との接触は短い時間でも十分である。例えば1分以上、好ましくは3分以上、5分以上、10分以上、または20分以上接触させればよく、例えば1〜60分程度、より特定すれば5分〜30分程度であってよい。または、それ以上の時間接触させてもよく、例えば数日間またはそれ以上接触させてもよい。
【0078】
ウイルスゲノムRNAを含むRNPおよび非感染性ウイルス粒子(ウイルス様粒子(VLP))は、周知のトランスフェクション方法により細胞に導入することができる。具体的には、リン酸カルシウム法(Chen, C. & Okayama, H., BioTechniques 6:632-638, 1988; Chen, C. and Okayama, H., Mol. Cell. Biol. 7: 2745, 1987)、DEAE-デキストラン法(Rosenthal, N., Methods Enzymol. 152:704-709, 1987)、様々なリポソームベースのトランスフェクション試薬(Sambrook, J. et al., Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, 1989)、およびエレクトロポレーション法(Ausubel, F. et al., In Current Protocols in Molecular Biology, John Wiley and Sons, NY, Vol. 1, Ch. 5 and 9, 1994)など、当業者に公知の様々な技術で細胞にトランスフェクションすることが可能である。エンドソームでの分解を抑制するため、トランスフェクションにおいてクロロキンを加えることもできる(Calos, M. P., Proc. Natl. Acad. Sci. USA 80: 3015, 1983)。トランスフェクション試薬には、DOTMA(Roche)、Superfect Transfection Reagent(QIAGEN, Cat No.301305)、DOTAP、DOPE、DOSPER(Roche #1811169)、TransIT-LT1(Mirus, Product No. MIR 2300)、CalPhos(商標)Mammalian Transfection Kit(Clontech #K2051-1)、CLONfectin(商標)(Clontech #8020-1)などが含まれる。エンベロープウイルスはウイルス粒子形成の際に宿主細胞由来のタンパク質を取り込むことが知られている。このようなタンパク質は、細胞に導入する場合、ある抗原性を有し、細胞傷害性の原因となり得る(J. Biol. Chem., 272: 16578-16584, 1997)。従って、エンベロープが除去されたRNPを用いることには利点がある(WO 00/70055)。
【0079】
ウイルスゲノムRNAの発現ベクター、およびそのゲノムRNAの複製に必要なウイルスタンパク質(N、PおよびLタンパク質)をコードする発現ベクターを細胞に導入することにより、細胞内でウイルスRNPを直接形成させることもできる。ウイルスベクターが導入された細胞をこのように調製してもよい。
【0080】
マイナス鎖RNAウイルスベクターが導入された細胞を調製したら、これを約12時間〜5日間(好ましくは1〜3日間)培養し、Aβペプチドを発現させてもよい。得られた細胞は、直接、または細胞ライセートとして動物に接種することができるが、好ましくはAβペプチドを発現する細胞が接種に用いられる。シグナルペプチドを有するAβを、ベクターから発現させ、細胞外に分泌させてもよい。ベクターを導入した細胞のライセートは、界面活性剤で細胞膜を溶解する手順または、凍結-融解サイクルの繰り返しを含む手順によって調製することができる。Triton X-100およびNonidet P-40などの非イオン性界面活性剤が、0.1〜1%の濃度範囲で適用され得る。例えばPBSで洗浄した後、遠心によって細胞塊を回収し、TNE緩衝液[25mM Tris-HCl(pH7.5)、150mM NaCl、1mM EDTA、1% Nonidet P-40]に再懸濁し、氷上で10〜30分間、懸濁液をインキュベートすることで得られる。抗原として使用するタンパク質が細胞質に可溶性である場合、調製したライセートを遠心(10,000×g、10分)し、不要な不溶性画分を沈殿として除去し、得られた上清を免疫に用いることができる。界面活性剤が望ましくない部位に、ライセートを投与する場合、洗浄後、細胞をPBSに再懸濁し、5〜6回の凍結-融解サイクルを繰り返して細胞を破壊することによって、ライセートを調製してもよい。ライセートは、トランスフェクション試薬と共に投与してもよい。
【0081】
または、Aβペプチドをコードするマイナス鎖RNAウイルスベクターを生成する核酸を動物に投与し、動物の体内でマイナス鎖RNAウイルスベクターによって発現されるAβペプチドを生成させることによって、免疫を誘導することができる。マイナス鎖RNAウイルスベクターを生成する核酸とは、組換えマイナス鎖RNAウイルスベクターの生成に必要なRNAおよびタンパク質群を発現する核酸のセットである。具体的には、Aβをコードするマイナス鎖ウイルスのゲノムRNAまたはその相補鎖(アンチゲノムRNA;すなわちプラス鎖)をコードする核酸、およびマイナス鎖RNAウイルスのゲノムRNAを含むRNPを構成するウイルスタンパク質群をコードする核酸を含む核酸のセットを言う。これらの核酸は、コードするRNAおよびタンパク質を発現できるように適当なプロモーターを含む。そのようなプロモーターは、例えば、CMVプロモーター(Foecking, M.K, and Hofstetter H., Gene 45: 101-105, 1986)、レトロウイルスLTR(Shinnik, T. M. et al., Nature, 293: 543-548, 1981)、EF1プロモーター、およびCAGプロモーター(Niwa, H. et al., Gene. 108: 193-199, 1991、特開平3-168087)を含む。核酸として、プラスミドベクターを好適に用いることができる。マイナス鎖RNAウイルスのゲノムは、ゲノムRNAを含むRNPを構成するウイルスタンパク質群(典型的にはN、L、およびP)をコードする遺伝子を保持し、かつ親の野生型ウイルスが持つ全てのエンベロープ構成タンパク質遺伝子を保持するものが好ましい。マイナス鎖RNAウイルスゲノムが、エンベロープ構成タンパク質の任意の遺伝子を欠損する場合、感染性ウイルス粒子の形成を相補するエンベロープタンパク質(例えばゲノムで欠損するエンベロープタンパク質またはVSV-Gなどの他のエンベロープタンパク質)をコードする核酸を、上記の核酸のセットに含めてもよい。これらの核酸を動物に投与することにより、動物内で組換えマイナス鎖RNAウイルスが生成される。マイナス鎖RNAウイルスのゲノムRNAを含むRNPを構成するウイルスタンパク質群とは、ウイルスゲノムRNAと共にRNPを形成し、ヌクレオカプシドを構成するタンパク質を言う。これらはゲノムの複製および遺伝子発現に必要なタンパク質群であり、典型的には、N、P、およびLタンパク質を含む。具体的に本発明は、(i)Aβをコードするマイナス鎖RNAウイルスのゲノムRNAまたはその相補鎖をコードする核酸、および(ii)N、P、およびLタンパク質をコードする核酸を、動物に接種する工程を含む方法にも関する。この動物においてゲノムRNAを含むRNPが形成され、Aβペプチドが発現する。ゲノムには、Th2サイトカインがさらにコードされてもよい。
【0082】
または、あらかじめゲノムRNAまたはその相補鎖をコードする核酸、および上記RNPを構成するウイルスタンパク質群が導入された細胞、またはその細胞のライセートを接種してもよい。マイナス鎖RNAウイルスベクターを生成する核酸の詳細については、以下の文献を参照することができる:WO 97/16539; WO 97/16538; WO 00/70055; WO 00/70070; WO 01/18223; WO 03/025570; Durbin, A. P. et al., Virology 235: 323-332, 1997; Whelan, S. P. et al., Proc. Natl. Acad. Sci. USA 92: 8388-8392, 1995; Schnell. M. J. et al., EMBO J. 13: 4195-4203, 1994; Radecke, F. et al., EMBO J. 14: 5773-5784, 1995; Lawson, N. D. et al., Proc. Natl. Acad. Sci. USA 92: 4477-4481, 1995; Garcin, D. et al., EMBO J. 14: 6087-6094, 1995; Kato, A. et al., Genes Cells 1: 569-579, 1996; Baron, M. D. and Barrett, T., J. Virol. 71: 1265-1271, 1997; Bridgen, A. and Elliott, R. M., Proc. Natl. Acad. Sci. USA 93: 15400-15404, 1996; Hasan, M. K. et al., J. Gen. Virol. 78: 2813-2820, 1997; Kato, A. et al., EMBO J. 16: 578-587, 1997; Yu, D. et al., Genes Cells 2: 457-466, 1997; Tokusumi, T. et al., Virus Res. 2002: 86; 33-38, 1997; Li, H.-O. et al., J. Virol., 74: 6564-6569, 2000。
【0083】
マイナス鎖RNAウイルスベクターを生成する核酸を動物に接種する場合、Aβをコードするマイナス鎖RNAウイルスのゲノムRNAまたはその相補鎖をコードする核酸、ならびにN、P、およびLタンパク質をコードする核酸を、5:0.5:0.5:2の重量比で動物に導入してもよい。核酸の比は適切に調整してよい。例えば発現プラスミドを用いる場合、AβをコードするウイルスゲノムRNA(プラス鎖またはマイナス鎖)をコードするプラスミド5μg〜1000μg、ならびにN発現プラスミド0.5μg〜200μg、P発現プラスミド0.5μg〜200μg、およびL発現プラスミド2μg〜500μgを用いて投与を実施することができる。核酸の投与は、例えば裸DNAを直接、またはトランスフェクション試薬と混合した後、注入することによって実施することができる。トランスフェクション試薬には、例えばリポフェクトアミンおよびポリカチオニックリポソームが含まれる。具体的には、DOTMA(Roche)、Superfect(QIAGEN #301305)、DOTAP、DOPE、DOSPER(Roche #1811169)、TransIT-LT1(Mirus, Product No. MIR 2300)などが利用できる。
【0084】
本発明のベクターの投与量は、疾患の種類、患者の体重、年齢、性別、および症状、投与目的、導入する組成物の投与形態、投与方法、導入する遺伝子のタイプなどにより異なる。当業者は適切な投与量を決定することが可能である。投与経路は適切に選択することができ、例えば経皮、鼻腔内、経気管支、筋内、腹腔内、静脈内、および皮下の経路を含むがそれらに限定されない。特に筋肉内投与(例えば腓腹筋)、皮下投与、点鼻投与またはスプレー、手掌または足蹠皮内投与、脾臓直接投与、腹腔内投与などが好ましい。接種部位は、一ヵ所または複数ヵ所、例えば2〜15ヶ所であってよい。接種量は、対象動物、接種部位、および接種回数などに応じて適切に調整してよい。ベクターは、約105〜約1011 CIU/ml、より好ましくは約107〜約109 CIU/ml、最も好ましくは約1×108〜約5×108 CIU/mlの範囲内の量で、薬学上許容される担体と組み合わせて投与することが好ましい。ヒト対する1回あたりの投与量は、ウイルス力価に換算して、1×104 CIU〜5×1011 CIU(細胞感染単位)、および好ましくは2×105 CIU〜2×1010 CIUである。投与回数は、1回、または副作用が臨床上許容される範囲内である限り複数回可能である。1日の投与回数についても同様に決定してもよい。単回投与だけでも有意な効果を発揮できるが、ベクターを2回以上導入することにより、より強い効果が得られる。
【0085】
複数回投与する場合、初回投与から例えば約1年後に再度投与してもよい。好ましい投与経路は、例えば鼻腔内投与または筋肉内投与である。より強い効果のために、投与を繰り返してよい。複数回投与には、好ましくは、Aβをコードする核酸を保持する上記のマイナス鎖RNAウイルスベクター、ウイルスベクターを生成する核酸、ウイルスベクターまたはベクターを生成する核酸が導入された細胞、または細胞のライセートの接種を用いてもよい。
【0086】
細胞を介して接種(エクスビボ投与)する場合、例えばヒト細胞、好ましくは自己の細胞にマイナス鎖RNAウイルスベクターを感染させ、104〜109細胞、および好ましくは105〜108細胞、またはそのライセートを用いることができる。このベクターは非ヒト動物に接種するのに用いることもでき、投与量は、例えば目的の動物とヒトとの体重または投与標的部位の容積比(例えば平均値)に基づき、上記の投与量を換算することができる。本発明のベクターを含む組成物の投与対象として、免疫系を有する所望の脊椎動物(ヒトおよび非ヒト脊椎動物)が含まれ、好ましくは鳥類および哺乳動物であり、より好ましくは哺乳動物(ヒトおよび非ヒト哺乳動物を含む)である。具体的には、ヒト、サルなどの非ヒト霊長類、マウスおよびラットなどのげっ歯類、ウサギ、ヤギ、ヒツジ、ブタ、ウシ、およびイヌなどその他の全ての哺乳動物が含まれる。投与対象となる動物は、例えばアルツハイマー病に罹患した個体、Aβレベルが亢進している個体、Aβ沈着が亢進している個体、アルツハイマー型変異遺伝子を持つ個体などである。
【0087】
本発明の方法により、Aβに対する免疫反応が誘導される。Aβに対する免疫反応は、抗Aβ抗体の産生、脳組織内のAβ量の減少、およびAβ沈着の減少などにより確認することができる。抗Aβ抗体の産生は、血中の抗Aβ抗体の検出によって調べてもよい。抗体レベルは、ELISA(enzyme-linked immunosorbent assay)およびオークタロニー法により測定することができる。ELISA法は、例えばマイクロプレートに抗原を吸着させ、抗血清を調製し、調製した抗血清を2倍段階希釈(開始溶液1:1000)し、希釈した抗血清プレートに加え抗原抗体反応を行わせることによって実施する。次いで発色のために、免疫動物の抗体をペルオキシダーゼ酵素標識した異種抗体と反応させ、二次抗体とする。吸光度が最大発色吸光度の1/2である場合、抗体の希釈倍率に基づいて、抗体価を算出することができる。または、オークタロニー法において、寒天ゲル内に抗原および抗体が拡散し、免疫沈降反応の結果として白い沈降線が形成される。沈降線は、免疫沈降反応が生じた場合の抗血清の希釈倍率である抗体価を測定するのに用いることができる。脳組織におけるAβレベルは、例えば脳組織の抽出物およびBiosource ELISA kitなどを用いて測定することができる。
【0088】
Aβ沈着(老人斑)の減少効果は、例えば以下の手順によって測定することができる:脳組織切片を70%ギ酸で処理し、5%H2O2で内因性のペルオキシダーゼを失活させた後、抗Aβ抗体と切片を反応させ、ペルオキシダーゼ標識二次抗体を用いてDAB染色を行う。染色後、顕微鏡での観察によりAβ蓄積部分の面積を測定することができる。本発明のベクターを投与しない場合に比べ、蓄積部分の面積の割合が減少すれば、Aβ沈着レベルが減少したと判断できる。または、1-フルオロ-2,5-ビス-(3-ヒドロキシカルボニル-4-ヒドロキシ)スチリルベンゼン(FSB)などのアミロイドに親和性の化合物を静注投与した後、MRIを用いて、生きた対象において老人斑を観察することができる(Higuchi M et al., Nat. Neurosci. 8(4):527-33, 2005; Sato, K. et al., Eur. J. Med. Chem. 39: 573, 2004; Klunk, W. E. et al., Ann. Neurol. 55(3):306-19, 2004)。このような非侵襲的なアミロイドイメージング技術により、本発明のベクターの効果を確認することができる。
【0089】
本発明はまた、以下の工程を含む、アミロイドβに対する免疫反応を測定する方法に関する:本発明のベクターまたはベクターを含む組成物を、アミロイドβ沈着を有する対象に導入する工程、および対象における抗アミロイドβ抗体を検出する工程。本発明はまた、以下の工程を含む、アミロイドβ沈着を測定する方法に関する:本発明のベクターまたはベクターを含む組成物を、アミロイドβ沈着を有する対象に投与する工程、および対象におけるアミロイドβ沈着のレベルを検出する工程。これらの方法により、アミロイドβに対する免疫反応および/またはアミロイドβ沈着の減少効果のモニタリングが可能である。
【0090】
本発明を実施するための最良の形態
以下実施例により本発明を具体的に説明するが、これらに限定されるとは解釈されない。本明細書中に引用された文献は、参照により組み込まれる。
【0091】
[実施例1]Aβ遺伝子を保持したマイナス鎖RNAウイルスゲノムcDNAの構築
(1−1)NotI断片の構築(マイナス鎖RNAウイルスの転写シグナルの付加)
鋳型として、ヒトAβペプチド1〜43(配列番号:27)にアミロイド前駆体タンパク質(APP:アクセッション番号AF282245)の分泌シグナル(1〜18アミノ酸)を付加することによって分泌型としたAβペプチド(配列番号:28)をコードする遺伝子(特開2005-21149;配列番号:1)、ならびにphAbeta-NnotI(配列番号:19)およびphAbeta-CnotI(配列番号:20)の2種のプライマーを用いてPCRを行った。得られたPCR産物を、NotIで消化し、次いでpBluescript(商標)II KS(Stratagene)にサブクローニングし、センダイウイルスの転写シグナルを含むAβペプチド遺伝子のNotI断片(配列番号:21)を構築した。マウスIL-10(mIL10)についても同様に、鋳型としてmIL10 cDNA(アクセッション番号NM_010548;配列番号:2)、ならびにpmIL10-N(配列番号:22)およびpmIL10-C(配列番号:23)の2種のプライマーを用いてPCRを行った。得られたPCR産物を、NotIで消化し、次いでpBluescript(商標)II KS(Stratagene)にサブクローニングし、センダイウイルスの転写シグナルを含むmIL10遺伝子のNotI断片(配列番号:24)を構築した。
【0092】
配列番号:1
ATGCTGCCCGGTTTGGCACTGCTCCTGCTGGCCGCCTGGACGGCTCGGGCGCTTGATGCAGAATTCCGACATGACTCAGGATATGAAGTTCATCATCAAAAATTGGTGTTCTTTGCAGAAGATGTGGGTTCAAACAAAGGTGCAATCATTGGACTCATGGTGGGCGGTGTTGTCATAGCGACTTAA
配列番号:19
ATTGCGGCCGCCAAGGTTCACTTATGCTGCCCGGTTTGGCACTGCTCCTG
配列番号:20
ATTGCGGCCGCGATGAACTTTCACCCTAAGTTTTTCTTACTACGGTTAAGTCGCTATGACAACACCGCCCACCATGAGTCC
配列番号:21
gcggccgccaaggttcacttATGCTGCCCGGTTTGGCACTGCTCCTGCTGGCCGCCTGGACGGCTCGGGCGCTTGATGCAGAATTCCGACATGACTCAGGATATGAAGTTCATCATCAAAAATTGGTGTTCTTTGCAGAAGATGTGGGTTCAAACAAAGGTGCAATCATTGGACTCATGGTGGGCGGTGTTGTCATAGCGACTTAAccgtagtaagaaaaacttagggtgaaagttcatcgcggccgc
配列番号:2
ATGCCTGGCTCAGCACTGCTATGCTGCCTGCTCTTACTGACTGGCATGAGGATCAGCAGGGGCCAGTACAGCCGGGAAGACAATAACTGCACCCACTTCCCAGTCGGCCAGAGCCACATGCTCCTAGAGCTGCGGACTGCCTTCAGCCAGGTGAAGACTTTCTTTCAAACAAAGGACCAGCTGGACAACATACTGCTAACCGACTCCTTAATGCAGGACTTTAAGGGTTACTTGGGTTGCCAAGCCTTATCGGAAATGATCCAGTTTTACCTGGTAGAAGTGATGCCCCAGGCAGAGAAGCATGGCCCAGAAATCAAGGAGCATTTGAATTCCCTGGGTGAGAAGCTGAAGACCCTCAGGATGCGGCTGAGGCGCTGTCATCGATTTCTCCCCTGTGAAAATAAGAGCAAGGCAGTGGAGCAGGTGAAGAGTGATTTTAATAAGCTCCAAGACCAAGGTGTCTACAAGGCCATGAATGAATTTGACATCTTCATCAACTGCATAGAAGCATACATGATGATCAAAATGAAAAGCTAA
配列番号:22
ACTTGCGGCCGCCAAAGTTCAATGCCTGGCTCAGCACTGCTATGCTGCCTG
配列番号:23
ATCCGCGGCCGCGATGAACTTTCACCCTAAGTTTTTCTTACTACGGTTAGCTTTTCATTTTGATCATCATGTATGCTTC
配列番号:24
gcggccgccaaagttcaATGCCTGGCTCAGCACTGCTATGCTGCCTGCTCTTACTGACTGGCATGAGGATCAGCAGGGGCCAGTACAGCCGGGAAGACAATAACTGCACCCACTTCCCAGTCGGCCAGAGCCACATGCTCCTAGAGCTGCGGACTGCCTTCAGCCAGGTGAAGACTTTCTTTCAAACAAAGGACCAGCTGGACAACATACTGCTAACCGACTCCTTAATGCAGGACTTTAAGGGTTACTTGGGTTGCCAAGCCTTATCGGAAATGATCCAGTTTTACCTGGTAGAAGTGATGCCCCAGGCAGAGAAGCATGGCCCAGAAATCAAGGAGCATTTGAATTCCCTGGGTGAGAAGCTGAAGACCCTCAGGATGCGGCTGAGGCGCTGTCATCGATTTCTCCCCTGTGAAAATAAGAGCAAGGCAGTGGAGCAGGTGAAGAGTGATTTTAATAAGCTCCAAGACCAAGGTGTCTACAAGGCCATGAATGAATTTGACATCTTCATCAACTGCATAGAAGCATACATGATGATCAAAATGAAAAGCTAAccgtagtaagaaaaacttagggtgaaagttcatcgcggccgc
【0093】
(1−2)F遺伝子欠失部位へのmIL10 NotI断片の挿入
9つの点でアミノ酸変異(Mタンパク質、G69E、T116A、およびA183S、HNタンパク質;A262T、G264R、およびK461G、Pタンパク質;L511F、Lタンパク質;N1197SおよびK1795E)を導入したF遺伝子欠失型SeVベクター(WO 2003/025570)のcDNA(pSeV18+NotI/TSΔF)を、SalIおよびNheIで消化し、Litmus38(New England Biolabs)へサブクローニングした。次いで、MおよびHN遺伝子を含む、得られたLitmus TSΔFプラスミドのF遺伝子欠失部位に、NotI切断部位を挿入するための変異を導入した。鋳型としてLitmus TSΔF、ならびにF_lit3008_NotI(配列番号:25)およびR_lit3020_NotI(配列番号:26)の2種のプライマーを用いたPCRにより、NotI切断部位を挿入した(図1上)。このLitmus TSΔF NotIをNotIで消化後、mIL10遺伝子NotI断片を挿入した(図1下)。
配列番号25:AAGCGGCCGCCTCAAACAAGCACAGATCATGGATG
配列番号26:AGGCGGCCGCTTAATTAACCAAGCACTCACAAGG
【0094】
(1−3)Aβ遺伝子を保持したF遺伝子欠失型SeV cDNAの構築
NotIで消化したpSeV18+NotI/TSΔF上のNotI部位に、Aβペプチド遺伝子NotI断片を挿入し、Aβ遺伝子を保持したF遺伝子欠失型SeV cDNA(pSeV18+Aβ/TSΔF)を構築した(図2)。
【0095】
(1−4)Aβ遺伝子およびIL-10を両方保持したF遺伝子欠失型SeVゲノムcDNAの構築
SalIおよびNheIでpSeV18+Aβ/TSΔFを消化した後、MおよびHN遺伝子を含まない断片を回収した。一方、Litmus TSΔF-mIL10をSalIおよびNheIで消化し、MおよびHN遺伝子を含む断片を調製した。この2つの断片をライゲーションし、Aβ遺伝子およびIL-10遺伝子を両方保持したF遺伝子欠失型SeVゲノムcDNA(pSeV18+Aβ/TSΔF-mIL10)を構築した(図3)。
【0096】
[実施例2]センダイウイルスベクターの再構成と増幅
ウイルスの再構成および増幅は、Liらの報告した方法(Li, H.-O. et al., J. Virology 74: 6564-6569, 2000; WO 00/70070)、およびその改良法(PCT/JP2005/00705)に従って行った。ウイルスベクターがF遺伝子欠失型であるため、Fタンパク質を供給したヘルパー細胞を利用した。このヘルパー細胞は、Cre/loxP誘導発現システムを用いて調製した。このシステムは、Cre DNA リコンビナーゼにより遺伝子産物を誘導発現するように設計されたプラスミドpCALNdLw(Arai, T. et al., J. Virol. 72: 1115-1121, 1988)を利用して、pCALNdLwプラスミドで形質転換したトランスフォーマントに、Cre DNAリコンビナーゼを発現する組換えアデノウイルス(AxCANCre)を、Saitoらの方法(Saito, I. et al., Nucl. Acid Res. 23: 3816-3821, 1995; Arai, T. et al., J. Virol. 72: 1115-1121, 1998)に従って感染させた。
【0097】
上記の方法によって、Aβ遺伝子を保持したF遺伝子欠失型SeVベクター(SeV18+Aβ/TSΔF)、ならびにAβ遺伝子およびIL-10遺伝子を両方保持したF遺伝子欠失型SeVベクター(SeV18+Aβ/TSΔF-mIL10)を調製した。SeV18+Aβ/TSΔF-mIL10のプラス鎖をコードするDNA配列を配列番号:4に、ゲノムRNA(マイナス鎖)のヌクレオチド配列を配列番号:5に示す。コントロールとして、Aβ遺伝子およびIL-10遺伝子の代わりに、F遺伝子欠失部位にLacZ遺伝子を保持するSeVベクター(SeV18+ LacZ /TSΔF;「SeV LacZ」とも称す)を調製した。
【0098】
[実施例3]アルツハイマー病動物モデルにおけるSeV-Aβ1-43/mIL10の鼻腔内投与効果
(3−1)動物および投与方法
24〜25ヶ月齢のAPPトランスジェニックマウス(Tg2576)(Hsiao, K., et al., Science 274:99-102, 1996)を用い、本発明のSeV18+Aβ/TSΔF-mIL10(以下、「SeV-Aβ1-43/mIL-10」とも記載)の効果を検討した。マウスを4匹ずつ2群に分け、一方を治療群とし、もう一方をコントロール群とした。治療群には、SeV-Aβ1-43/mIL-10(1匹あたり5×106 CIU)を、コントロール群には、SeV LacZ(1匹あたり5×106 CIU)を鼻腔内投与した。
【0099】
(3−2)血清中の抗Aβ抗体の測定
上記処置4週間後および8週間後、マウスから血液を採取し、血清中の抗Aβ42抗体量を測定した。Aβ1-42ペプチド(5μg/mL)を、96ウェルプレート(Nunc, MaxiSorp)の各ウェルに吸着させた。5%脱脂乳/TBS-T緩衝液でブロックした後、マウス血清を加え(500倍希釈)、ペルオキシダーゼ標識抗マウスIgG抗体で検出した。抗体価の評価は、ELISAリーダーでの吸光度測定(O.D.450)により行った。
【0100】
結果を図4に示す。コントロール群と比較し、治療群の全個体において抗体価の顕著な増加が観察された。
【0101】
(3−3)SeV-Aβ1-43/mIL-10の老人斑消失効果
上記センダイウイルスベクターを鼻腔内投与したマウスを、投与後8週間(26または27ヶ月齢)で解剖し、前頭葉皮質、頭頂葉、および海馬の皮質の脳組織切片を調製した。上記組織の凍結切片を、以下のように処理した。Aβタンパク質または老人斑を検出するために、組織切片を70%ギ酸で処理し、5% H2O2で内因性のペルオキシダーゼを失活させた。次いで組織切片をラビット抗pan-Aβ抗体(1000倍希釈)と反応させ、ペルオキシダーゼ標識二次抗体を加え、DAB染色を行った。染色した切片を、顕微鏡に連結させた3CCDカメラを用いて観察し、各領域におけるAβ蓄積部分の面積を測定し、面積率を計算した。
【0102】
切片の染色結果を図5(前頭葉)、図6(頭頂葉)、および図7(海馬)に示す。Aβ沈着面積率を図8に示す。前頭葉、頭頂葉、および海馬のいずれにおいても、SeV-Aβ1-43/mIL-10投与により、老人斑がコントロールの10〜20%に顕著に減少したことが確認された。
【0103】
(3−4)SeV-Aβ1-43/mIL投与の安全性の検討
上記(3-3)と同様の手順で、投与後8週間(26〜27ヶ月齢)の治療群およびコントロール群から、前頭葉および海馬組織の凍結切片を調製した。抗CD3抗体(T細胞)、抗Iba-1抗体(ミクログリア)を用いて、凍結切片をABC法にて染色し、中枢神経系におけるリンパ球の浸潤、およびミクログリアの変化が生じているか確認した。図9に示すように、Tリンパ球(pan T細胞マーカーであるCD3陽性の細胞)の浸潤は、治療群またはコントロール群のいずれにおいても、前頭葉および海馬の両方で観察されなかった。従って、本ベクターを治療に用いることにより、中枢神経系の炎症を起こす可能性が非常に低いことが示された。さらに、ミクログリアマーカーであるIBA-1陽性細胞の量は、コントロール群と治療群の間で同程度であり、治療群における極端な活性化(蓄積)は生じないことが確認された。
【0104】
(3−5)脳組織におけるAβの測定
マウス大脳および小脳を正中裂にそって切断し、半球を-80℃に急速冷凍して保存した。脳半球を1mlのTBS溶液中でホモジェナイズし、卓上超遠心機で100,000 g、1時間遠心した。可溶性画分(TBS画分)を保存した。不溶画分を2%SDSに溶解しホモジェナイズ後、さらに100,000 g、1時間遠心した。得られた可溶性画分(2%SDS画分)を保存した。不溶画分を70%ギ酸に溶解しホモジェナイズ後、再び100,000 g、1時間遠心した。得られた可溶性画分(ギ酸画分)を保存した。脳組織中のAβ40および42は、Biosource ELISA kitを用いて測定した。TBS画分を4倍希釈し;2%SDS画分は400〜2000倍希釈し;ギ酸画分は1M Tris溶液で1000倍希釈した。次いで希釈した画分は、測定のため、ELISA希釈液でさらに希釈(2〜10倍)した。
【0105】
図10に示すように、SDS画分、ギ酸画分、およびTBS画分の全てにおいて、SeV-Aβ1-43/mIL-10投与による脳組織中のAβレベルの顕著な減少が確認された。
【0106】
[実施例4]アルツハイマー病動物モデルにおけるSeV-Aβ1-43/mIL-10筋肉内投与効果
(4−1)動物および投与方法
投与経路以外、上記鼻腔内投与の場合と全く同様に行った。5×106 CIUの投与量で、SeV-Aβ1-43/mIL-10ベクターを後肢大腿筋に投与した。
【0107】
(4−2)血清中の抗Aβ抗体の測定
上記鼻腔内投与の場合と同様の手法によって、SeV-Aβ1-43/mIL-10ベクターを筋肉内投与したマウスの血清中の抗Aβ抗体量を測定した。結果を図11に示す。
【0108】
(4−3)SeV-Aβ1-43/mILの老人斑消失効果
上記鼻腔内投与の場合と同様の方法によって、SeV-Aβ1-43/mIL-10ベクターの筋肉内投与後の前頭葉、頭頂葉、および海馬における老人斑消失効果を検討した。結果を、図12(前頭葉)、図13(頭頂葉)、および図14(海馬)に示す。Aβ沈着面積率を図15に示す。各領域は、一定のAβ沈着減少を示した。
【0109】
(4−4)SeV-Aβ1-43/mIL-10投与の安全性の検討
上記鼻腔内投与の場合と同様の方法によって、SeV-Aβ1-43/mIL-10ベクターの筋肉内投与後の中枢神経系におけるリンパ球の浸潤の存在を確認した。図16に示すように、コントロール群または治療群のいずれも、前頭葉におけるTリンパ球の浸潤は観察されなかった。
【0110】
(4−5)脳組織におけるAβの測定
上記鼻腔内投与の場合と同様の方法によって、SeV-Aβ1-43/mIL-10ベクターの筋肉内投与後の脳組織中のAβ40およびAβ42を測定した。結果を図17に示す。いくつかの可溶性画分においてAβレベルの減少が確認された。
【0111】
[実施例5]正常マウスにおけるSeV-Aβ1-43/hIL-10の鼻腔内投与による抗体価の上昇
(5−1)動物および投与方法
本発明のSeV18+Aβ1-43/TSΔF-hIL10(以下「SeV-Aβ1-43/hIL-10」とも称する)の効果を、6週齢C57BL/6Nマウスを用いて試験した。マウスを、各6匹のマウスからなる3つの群に分けた。SeV-Aβ1-43/hIL-10(群1:5×105 CIU/匹、群2:5×106 CIU/匹、群3:5×107 CIU/匹)を、各マウスに鼻腔内投与した。
【0112】
(5−2)血漿中の抗Aβ抗体の測定
投与前ならびに投与の2、4、および8週間後、マウスから血液を採取し、血漿中に存在する抗Aβ42抗体量を測定した。Aβ1-42ペプチド(4μg/mL)を、96ウェルプレート(Nunc, MaxiSorp)の各ウェルに吸着させた。1%BSA/2%ヤギ血清/TBS-T緩衝液でブロックした後、マウス血漿(300倍希釈)を加え、ペルオキシダーゼ標識抗マウスIgG抗体で検出した。抗体価の評価は、ELISAリーダーでの吸光度測定(O.D.450)により行った。
【0113】
結果を図18に示す。抗Aβ抗体量は、3つの群全てで経時的に増加した。ベクター投与によって、投与量依存性の様式で、抗体量は増加したが、群2(5×106 CIU/匹)および群3(5×107 CIU/匹)の間で抗体量の有意な差は観察されなかった。結果は、本発明の試薬(SeV-Aβ1-43/hIL-10)の投与による抗Aβ抗体の誘導は、アルツハイマー病モデルマウス(APP Tgマウス)に特異的ではないが、正常マウスにおいても十分達成されることを示す。
【0114】
[実施例6]正常マウスへの鼻腔内投与におけるアルツハイマー病に対する治療的ベクターをコードするサイトカインの効果
(6−1)動物および投与方法
いくつかのサイトカインをコードする遺伝子を、上記のアルツハイマー病に対する治療用ベクターに挿入した。これらのセンダイウイルスベクターの効果は、6週齢C57BL/6Nマウスを用いて試験した。マウスを、各6匹のマウスからなる3つの群に分けた。(Aβペプチドをコードする遺伝子を保持する、ならびにマウスIL-4、IL-5、またはIL-6をコードする遺伝子を保持する;それぞれSeV-Aβ1-42/mIL-4、SeV-Aβ1-42/mIL-5、またはSeV-Aβ1-42/mIL-6と称される)センダイウイルスベクターを、各マウスに鼻腔内投与した(5×106 CIU/10μl)。
【0115】
(6−2)血漿中の抗Aβ抗体の測定
投与前ならびに投与の1および2週間後、マウスから血液を採取し、血漿中に存在する抗Aβ抗体量を測定した。Aβ1-42ペプチド(4μg/mL)を、96ウェルプレート(Nunc, MaxiSorp)の各ウェルに吸着させた。1%BSA/2%ヤギ血清/TBS-T緩衝液でブロックした後、マウス血漿(300倍希釈)を加え、ペルオキシダーゼ標識抗マウスIgG抗体で検出した。抗体価の評価は、ELISAリーダーでの吸光度測定(O.D.450)により行った。
【0116】
結果を図19に示す。抗Aβ抗体量は、3つの群全てで経時的に増加した。結果は、IL-4、IL-5、およびIL-6が抗Aβ抗体を誘導するのに効果的で、Aβ抗原をコードする核酸、ならびにIL-4、IL-5、およびIL-6より選択される1つまたは複数のサイトカインの両方を保持するセンダイウイルスベクターを含む製剤が、アルツハイマー病に対する治療において、抗Aβ抗体を誘導するのに好ましいことを示す。
【0117】
産業上の利用可能性
本発明によって、アミロイドβ沈着レベルを有効に減少させることのできる新規マイナス鎖RNAウイルスベクターが提供された。本発明のベクターは、アルツハイマー病の治療に有用である。
【図面の簡単な説明】
【0118】
【図1】Litmus SalI/NheIfrg-MtsHNtsPLmut-dF mIL10(以下、Litmus TS-dF mIL10と略す)の構築過程を示す図である。Litmus TS-dFのF遺伝子欠失部位に変異を導入してNotI部位を作製し、そこにNotIを用いて切り出したmIL-10遺伝子を挿入した。
【図2】pSeV18+Aβ/MtsHNtsP511Lmut-dF(以下、pSeV18+Aβ/TS-dFと略す)の構築過程を示す図である。pBluescript(商標)のNotI部位に挿入されているAβ遺伝子を切り出し、pSeV18+NotI/TS-dFのNotI部位に挿入した。
【図3】pSeV18+Aβ/MtsHNtsP511Lmut-dF mIL10 (以下、pSeV18+Aβ/TS-dF mIL10と略す)の構築過程を示す図である。pSeV18+Aβ/TS-dFとLitmus TS-dF mIL10から、SalIおよびNheIを用いて、必要な断片を切り出し、交換した。
【図4】SeV-Aβ1-43/mIL-10またはコントロールベクターの鼻腔内投与の4および8週間後のAPPトランスジェニックマウスの血清中の抗Aβ抗体レベルを示す図である。
【図5】SeV-Aβ1-43/mIL-10 またはコントロールベクターの鼻腔内投与8週間後のAPPトランスジェニックマウスの前頭葉における老人斑を示す図である。
【図6】SeV-Aβ1-43/mIL-10またはコントロールベクターの鼻腔内投与8週間後のAPPトランスジェニックマウスの頭頂葉における老人斑を示す図である。
【図7】SeV-Aβ1-43/mIL-10またはコントロールベクターの鼻腔内投与8週間後のAPPトランスジェニックマウスの海馬における老人斑を示す図である。
【図8】SeV-Aβ1-43/mIL-10またはコントロールベクターの鼻腔内投与8週間後のAPPトランスジェニックマウスにおける老人斑(前頭葉、頭頂葉、および海馬)を定量化した結果を示す図である。
【図9】SeV-Aβ1-43/mIL-10またはコントロールベクターの鼻腔内投与8週間後のAPPトランスジェニックマウスの中枢神経系におけるリンパ球浸潤を検討した結果を示す。前頭葉におけるT細胞の浸潤を、CD3を指標として検出した。海馬におけるミクログリアの変化を、Iba-1を指標として検出した。
【図10】SeV-Aβ1-43/mIL-10またはコントロールベクターの鼻腔内投与8週間後のAPPトランスジェニックマウスの脳組織におけるAβ量を示す図のセットである。
【図11】SeV-Aβ1-43/mIL-10またはコントロールベクターの筋肉内投与4および8週間後のAPPトランスジェニックマウスの血清中のAβ抗体レベルを示す図である。
【図12】SeV-Aβ1-43/mIL-10またはコントロールベクターの筋肉内投与8週間後のAPPトランスジェニックマウスの前頭葉における老人斑を示す写真である。
【図13】SeV-Aβ1-43/mIL-10またはコントロールベクターの筋肉内投与8週間後のAPPトランスジェニックマウスの頭頂葉における老人斑を示す写真である。
【図14】SeV-Aβ1-43/mIL-10またはコントロールベクターの筋肉内投与8週間後のAPPトランスジェニックマウスの海馬における老人斑を示す写真である。
【図15】SeV-Aβ1-43/mIL-10またはコントロールベクターの筋肉内投与8週間後のAPPトランスジェニックマウスにおける老人斑(前頭葉、頭頂葉、および海馬)を定量化した結果を示す図である。
【図16】SeV-Aβ1-43/mIL-10またはコントロールベクターの筋肉内投与8週間後の前頭葉におけるリンパ球浸潤を検討した結果を示す写真である。
【図17】SeV-Aβ1-43/mIL-10またはコントロールベクターの筋肉内投与8週間後の脳組織におけるAβ量を示す図のセットである。
【図18】SeV-Aβ1-43/mIL-10の鼻腔内投与後の正常マウスの血清抗Aβ抗体レベルを示す図である。
【図19】サイトカインをコードするセンダイウイルス治療用ベクターの鼻腔内投与後の正常マウスの血清抗Aβ抗体レベルを示す図である。
【技術分野】
【0001】
本発明は、マイナス鎖RNAウイルスベクターに基づくアルツハイマー病治療用ワクチンに関する。
【背景技術】
【0002】
急速な高齢化社会を迎えつつある日本では、老年性痴呆は、介護問題も含め社会的に大きな問題となっている。実に、日本の65歳以上の老人のうち、約10%が老年性痴呆であると報告されている。アルツハイマー病は、痴呆性疾患の二大原因の一つであり、罹患人口の約50%を占めるにも関わらず、有効な治療法は現在のところ提供されていない。
【0003】
アルツハイマー病の病理学的所見は、以下の3つの特徴を有する:神経細胞の萎縮/脱落;アミロイドβ(以下、時に「Aβ」とも略す)タンパク質の凝集および沈着による老人斑の形成;ならびに異常タウタンパク質からなる神経原線維変化。アルツハイマー病患者の脳で生じる主要なアミロイドβタンパク質は、アミロイド前駆体タンパク質がβおよびγセクレターゼによる切断からもたらされるアミノ酸40〜43個からなる。老人斑は、中心のアミロイドβおよび取り囲むミクログリア、線維型アストログリア、ならびに異栄養神経突起で構成される凝集体である。現在、アルツハイマー病の病態仮説として、アミロイドβの凝集および沈着による老人斑の形成が原因であるとする「アミロイドカスケード仮説」が有力である。
【0004】
この仮説をもとに、アルツハイマー病の新しい治療法としてのワクチン(免疫)療法が注目されている。アルツハイマー病のワクチン療法は、脳のアミロイドβを免疫学的手法により除去する方法である。Elan社のSchenkらは、前凝集Aβ42をアジュバントと共にPDAPP-トランスジェニックマウスに筋肉内投与し、脳内のアミロイド沈着の減少を確認した(特許文献1および非特許文献1)。Elan社およびWyeth社によって行われた臨床試験では、合成Aβ42ペプチド(AN-1792)がアジュバント(QS21)と共に筋肉内投与され、被験者の血清中にアミロイドのβシート構造を認識する抗Aβ抗体が検出された(非特許文献2)。高次脳機能の改善も報告された(非特許文献3)。残念ながら、フェーズIIにおいて6%(298名中18名)の患者に髄膜脳炎の副作用が生じ、1名の死亡例も報告され、上記臨床試験は中断された。死亡患者の脳組織を病理学的に検討した結果から、新皮質で老人斑の不在が確認され、ワクチンの効果が示唆された。老人斑が消失している領域では、Aβ分解産物のミクログリアによる貪食作用を示す像が確認された。この所見は、Aβに結合した抗体をFcレセプターを介してミクログリアが貪食していることを示している。
【0005】
上記抗Aβ抗体は、ニューロン、グリア細胞、アミロイド前駆体タンパク質(APP)、および細胞内Aβとは反応しないことが確認されている。従って上記副作用は、抗体による脳の炎症とは考えにくい。AN-1792ワクチンを投与するために、アジュバントを必要とする。アジュバントは強い免疫活性化作用を有し、Tリンパ球による細胞性免疫も誘導する。上記副作用は、アジュバント誘導性細胞性免疫により、AβまたはAPPに反応するTh1タイプCD4+ T細胞が脳内に浸潤することで引き起こされる、実験的アレルギー性脳脊髄炎様の髄膜脳炎である可能性がある(非特許文献4)。
【0006】
AN-1792ワクチンにより、Aβペプチドを利用したワクチン療法の病理学的効果が確認された。しかしワクチンの臨床適用のためには、髄膜脳炎などの副作用を軽減することが必須である。このような観点から、N末端断片にT細胞によって認識されるキャリアタンパク質が結合したAβペプチドと、Th2アジュバントとの併用が関与する免疫化方法(特許文献2)、ならびにAβペプチドおよびコレラ毒素Bサブユニットからなる融合タンパク質をコードする核酸を含むアデノウイルスベクター(特許文献3)を含む、改良したワクチン接種法が発案された。さらに本発明者らは、安全性の高い経口ワクチンとして、液性免疫を誘導するAβペプチドのペプチド断片をコードするDNAを含むアデノ随伴ウイルスベクターを開発した(特許文献4)。上記アデノ随伴ウイルスベクターワクチンは、Th2 T細胞が誘導されやすい腸管粘膜免疫系を利用することから、いかなるアジュバントも必要としない。さらに、APPトランスジェニックマウスを用いた実験により、1回の投与で6ヶ月の比較的長期にわたり腸管において抗原提示が可能であること;脳のアミロイド沈着および老人斑形成の減少効果を有すること;他の臓器におけるいかなる観察可能な炎症も認められないことといった、優れた特徴が確認されている。しかしながら、アルツハイマー病のワクチン療法の実用化のため、安全性および有効性のさらに優れたワクチンが必要である。
【0007】
【特許文献1】WO 99/27944
【特許文献2】WO 02/096350
【特許文献3】WO 2004/050876
【特許文献4】WO 2004/111250
【特許文献5】WO 00/70070
【特許文献6】特開2000-253876
【特許文献7】WO 2001/072340
【非特許文献1】Schenk D. et al, Nature 400:173-177, 1999
【非特許文献2】Hock C. et al., Nat. Med. 8:1270-1275, 2002
【非特許文献3】Hock C. et al., Neuron 38, 547-554, 2003
【非特許文献4】臨牀と研究 82(3):439-444, 2005
【発明の開示】
【0008】
本発明は、上記状況に鑑みて達成された。本発明の目的は、アルツハイマー病に対する安全性および有効性の高いワクチン療法の提供である。
【0009】
本発明者らは、上記目的を達成するため、鋭意努力を重ね、アルツハイマー病に対する新規ワクチン療法の開発を試みた。本発明者らは、センダイウイルス(SeV)を用いて遺伝子導入用および遺伝子治療にも使用できるベクターを開発してきた経験を有する。センダイウイルスは、マイナス(−)鎖RNAウイルスである。ウイルスは宿主細胞の細胞質でのみ増殖し、それらの生活環においてDNAフェーズを有さないため、染色体へ組み込まれない。ウイルスは従って、遺伝子導入用ベクターとして利用した場合、高い遺伝的安全性を確実にする。本発明者らは、センダイウイルスに基づく遺伝子導入用ベクターを開発するにあたり、さらに安全性の向上および標的疾患に適したベクターとするために、センダイウイルスゲノムから遺伝子を欠失させた。宿主細胞へのウイルス侵入に関わる膜融合タンパク質(Fタンパク質)の遺伝子をゲノムから欠失させることにより、安全性が高く、二次感染性を引き起こさないベクターへの改良に成功すると共に、ウイルスベクターの効率的な再構成系も確立している(WO 00/70070)。さらにセンダイウイルスベクターを利用したインフルエンザワクチン(特開2000-253876)およびAIDSワクチン(WO 2001/072340)の開発にも成功している。
【0010】
IL-10などのTh2サイトカインは、生体内の免疫応答と密接に関連するTh1/Th2バランスにおいて、重要な役割を果たす。Th1細胞は、IFN-γなどを産生し、細胞性免疫を亢進する。Th1細胞によって産生されたIFN-γは、Th2細胞のIL-10産生を抑制する。一方、Th2細胞は、IL-10などを産生し、液性免疫を亢進させる。Th2細胞によって産生されたIL-10は、Th1細胞におけるIFN-γなどの産生を抑制する。本発明者らは、上記センダイウイルスベクターに、Aβと共にTh2サイトカインをコードする遺伝子を導入することを考え、細胞性免疫による脳髄膜炎などの副作用を抑制するアルツハイマー病に対するワクチンの産生を試みた。次いで本発明者らは、Aβ1-43およびIL-10をコードするセンダイウイルスベクターを構築し、その効果を検討した。まず、該ベクターを24〜25ヶ月齢APPトランスジェニックマウスに鼻腔内投与した。処置後4週間後および8週間後の血清中の抗Aβ42抗体レベルを測定した。コントロール群のマウスでは、抗Aβ抗体は低レベルでしか検出されず、時間と共にレベルが増加した。対照的に、本発明のベクターを投与したマウスの群では、血中に高レベルの抗Aβ抗体が検出され、抗体レベルは4週間後よりも8週間後にわずかに低下することが見出された。組織学的検討は、前頭葉、頭頂葉、および海馬のいずれにおいても、本発明のベクター投与による老人斑の顕著な減少を示した。さらに脳組織中のAβ量をELISA法により測定した。結果は脳組織中Aβの有意な減少を示した。さらに、このベクター投与が中枢神経系におけるリンパ球の浸潤を引き起こすかどうか検討するために、CD3およびIba-1を指標として用いた。結果は、リンパ球の浸潤もミクログリアの病的な(極端な)活性も生じないことを示した。
【0011】
上記検討から、Aβペプチドを発現するマイナス鎖RNAウイルスベクターの極めて優れた特徴が明らかにされた。第一に、このベクターは、極めて高齢のアルツハイマー病モデルマウスにおいて、顕著なAβ減少効果を示した。上記の実験で使用したTg2576マウスは24〜25ヶ月齢であり、ほぼ寿命に近かった。これまでのアルツハイマー病治療方法の研究は、最大18ヶ月までのマウスを用いて実施されてきた。このような高齢マウスにおいて治療効果が確認されたのは、今回が初めてである。そのような高齢のマウスでは、脳に沈着しているAβ量は膨大である。従って本発明のベクターは、よりアルツハイマー病の進行した患者に対しても高い有効性を発揮できる可能性がある。第二に、本発明のベクターは従来のワクチン療法と比較して、低い投与量および少ない投与回数で有効であった。例えば、Aβペプチドとアジュバントを併用して用いた従来の方法を適用した場合、一般的に、免疫確立のために複数回投与が必要である。実際に、AN-1792臨床試験において数回〜十数回の投与が行われた。対照的に、本発明のベクターは単回投与後、顕著な有効性を示した。さらに、アデノ随伴ウイルスベクターによるワクチン療法(WO 2004/111250)において、ワクチン接種は、本明細書に記載の実施例で行ったのと同様の単回投与によって実施し、その投与量は5×1011ゲノム/個体であった。対照的に、本明細書に記載の実施例に適用した投与量は、5×106 CIU(Cell Infectious Unit)/個体(ゲノムコピー数に換算した場合約5×107ゲノム/個体)であった。これら2つのベクターの投与量には約104もの開きがある。第三に、実施例におけるベクターの投与は、マウスモデルにおいて髄膜脳炎を引き起こさなかった。AN-1792臨床試験で観察された髄膜脳炎は、マウスモデルでも確認されているとの報告がある(M. Shoji, et al., 44th Annual Meeting of Japanese Society of Neurology, May 15-17, 2003)。Aβ ペプチドおよびアジュバントを免疫した正常非モデルマウス(C57B6マウス)において、髄膜脳炎の発生も報告された(Furlan R, et al., Vaccination with amyloid-beta peptide induces autoimmune encephalomyelitis in C57/BL6 mice, Brain 126:285-291, 2003)。本明細書に記載の結果は、本発明のベクターがヒトへ投与した場合においても、安全であることを示唆する。
【0012】
上述のとおり、本発明のベクターは、低用量および少投与回数で高いAβ消失効果を示した。センダイウイルスベクターに基づくAIDSワクチンの効果は、一部細胞性免疫によることが確認されている(WO 2001/072340)。実施例で使用したベクターが、副作用を防止するため細胞性免疫よりも液性免疫に依存するよう意図的に設計したにも関わらず、本明細書に示した優れたワクチン効果は、アルツハイマー病治療ワクチンにおいて、液性免疫の重要性を示唆する。Th2サイトカインまたはTh2アジュバントを使用して液性免疫を優位にすることにより、副作用を低減することができる。すなわち、本発明は、Aβをコードするマイナス鎖RNAウイルスベクターおよびその利用、特に下記の発明を提供するものである。
【0013】
[1]アミロイドβをコードする核酸を含む、マイナス鎖RNAウイルスベクター。
[2]Th2サイトカインまたはその部分ペプチドをコードする核酸をさらに含む、[1]のマイナス鎖RNAウイルスベクター。
[3]Th2サイトカインまたはその部分ペプチドが、IL-10またはその部分ペプチドである、[2]のマイナス鎖RNAウイルスベクター。
[4]マイナス鎖RNAウイルスベクターがパラミクソウイルスベクターである、[1]〜[3]のいずれかのベクター。
[5]パラミクソウイルスベクターがセンダイウイルスベクターである、[4]のベクター。
[6]アミロイドβがAβ43またはその部分ペプチドである、[1]〜[5]のいずれかのベクター。
[7]アミロイドβがヒトに由来する、[1]〜[6]のいずれかのベクター。
[8]IL-10が、マウスまたはヒトに由来する、[3]〜[7]のいずれかのベクター。
[9]アミロイドβが、配列番号:1のヌクレオチド配列がコードするポリペプチドまたはその部分である、[1]〜[8]のいずれかのベクター。
[10]IL-10が、配列番号:2または3のヌクレオチド配列がコードするポリペプチドである、[3]〜[9]のいずれかのベクター。
[11]配列番号:5の核酸を含む、[1]〜[10]のいずれかのマイナス鎖RNAウイルスベクター。
[12][1]〜[11]のいずれかのベクターおよび薬学的に許容される担体を含む組成物。
[13](i)Th2サイトカインもしくはその部分ペプチドを発現可能様式でコードする核酸、(ii)Th2サイトカインもしくはその部分ペプチド、または(iii)Th2アジュバントをさらに含む、[12]の組成物。
[14]Th2サイトカイン、その部分ペプチド、またはTh2アジュバントを含む、[13]の組成物。
[15]Th2サイトカインまたはその部分ペプチドを、発現可能様式でコードする核酸を含む、[13]の組成物。
[16]Th2サイトカインが、アミロイドβをコードする核酸を含むマイナス鎖RNAウイルスベクターによってコードされる、[15]の組成物。
[17]Th2サイトカインが、アミロイドβをコードする核酸を含むマイナス鎖RNAウイルスベクター以外のベクターによってコードされる、[15]の組成物。
[18]薬学的組成物である、[12]〜[17]のいずれかの組成物。
[19][1]〜[18]のいずれかのベクターを含む、アルツハイマー病を治療または予防するために用いられる薬学的組成物。
[20](i)Th2サイトカインもしくはその部分ペプチドを発現可能様式でコードする核酸、(ii)Th2サイトカインもしくはその部分ペプチド、または(iii)Th2アジュバントをさらに含む、[19]の組成物。
[21]鼻腔内投与に用いるための、[18]または[20]の薬学的組成物。
[22][1]〜[11]のいずれかのベクター、または該ベクターを含む組成物を投与する段階を含む、アミロイドβの沈着を減少させる方法。
[23][1]〜[11]のいずれかのベクターまたは該ベクターを含む組成物を投与する段階を含む、アルツハイマー病を治療または予防する方法。
[24]投与が鼻腔内投与である、[22]または[23]の方法。
[25]投与が筋肉内投与である、[22]または[23]の方法。
[26]Th2サイトカイン、その部分ペプチド、Th2アジュバント、またはTh2サイトカインもしくはその部分ペプチドをコードするベクターを投与する段階をさらに含む、[22]〜[25]のいずれかの方法。
[27]Th2サイトカイン、その部分ペプチド、またはTh2アジュバントを投与する、[26]の方法。
[28]Th2サイトカインまたはその部分ペプチドをコードするベクターを投与する、[26]の組成物。
【0014】
本発明によって、低用量および少投与回数でアミロイドβ沈着を有効に減少させることのできる、新規マイナス鎖RNAウイルスベクターが提供された。本実施例に記載のベクターは、中枢神経系における炎症を誘導しないことが確認されており、本発明のベクターの投与は、従来のワクチンを投与した場合発生したような脳髄膜炎を引き起こす可能性は低い。本発明のベクターは、アルツハイマー病の極めて有効な治療手段として用いることができる。
【0015】
本発明は、アミロイドβをコードする核酸を含む、マイナス鎖RNAウイルスベクターに関する。
【0016】
本発明において遺伝子とは、遺伝物質または転写単位をコードする核酸を指す。遺伝子はRNAまたはDNAであってよい。本発明においてタンパク質をコードする核酸は、該タンパク質の遺伝子と呼ぶ。遺伝子はタンパク質をコードしていなくてもよく、例えば遺伝子はリボザイムまたはアンチセンスRNAなどの機能的RNAをコードするものでもよい。遺伝子は天然由来または人為的に設計された配列であり得る。本明細書において「DNA」とは、一本鎖DNAおよび二本鎖DNAを含む。「タンパク質をコードする」という語句は、ポリヌクレオチドがタンパク質を適当な条件下で発現できるように、タンパク質のアミノ酸配列をコードするORFをセンスまたはアンチセンス方向で含むことを意味する。
【0017】
マイナス鎖RNAウイルスとは、マイナス鎖RNA(ウイルスタンパク質をコードするセンス鎖と相補的なアンチセンス鎖)をゲノムとして含むウイルスを指す。マイナス鎖RNAウイルスはネガティブ鎖RNAウイルスとも呼ばれる。本発明において用いられるマイナス鎖RNAウイルスとしては、特に一本鎖マイナス鎖RNAウイルス(非分節型(non-segmented)マイナス鎖RNAウイルスとも言う)が好ましい。「一本鎖マイナス鎖RNAウイルス」とは、一本鎖のマイナス鎖RNAをゲノムとして有するウイルスを指す。このようなウイルスとしては、パラミクソウイルス(Paramyxoviridae)(パラミクソウイルス(Paramyxovirus)、モービリウイルス(Morbillivirus)、ルブラウイルス(Rubulavirus)、およびニューモウイルス(Pneumovirus)属などを含む)、ラブドウイルス(Rhabdoviridae)(ベシクロウイルス(Vesiculovirus)、リッサウイルス(Lyssavirus)、およびエフェメロウイルス(Ephemerovirus)属などを含む)、フィロウイルス(Filoviridae)、オルトミクソウイルス(Orthomyxoviridae)(インフルエンザウイルス(Influenza virus)A、B、およびC、ならびにトゴト様ウイルス(Thogoto-like virus)などを含む)、ブニヤウイルス(Bunyaviridae)(ブニヤウイルス(Bunyavirus)、ハンタウイルス(Hantavirus)、ナイロウイルス(Nairovirus)、およびフレボウイルス(Phlebovirus)属などを含む)、ならびにアレナウイルス(Arenaviridae)などの科に属するウイルスが含まれる。
【0018】
本発明において特に好適に用いられるマイナス鎖RNAウイルスを具体例は、例えばパラミクソウイルス科に属するセンダイウイルス(Sendai virus)、ニューカッスル病ウイルス(Newcastle disease virus)、おたふくかぜウイルス(Mumps virus)、麻疹ウイルス(Measles virus)、RSウイルス(Respiratory syncytial virus)、牛疫ウイルス(rinderpest virus)、ジステンパーウイルス(distemper virus)、サルパラインフルエンザウイルス(SV5)、ヒトパラインフルエンザウイルス1、2、および3型;オルトミクソウイルス科に属するインフルエンザウイルス;ラブドウイルス科に属する水疱性口内炎ウイルス(Vesicular stomatitis virus)および狂犬病ウイルス(Rabies virus)を含む。
【0019】
本発明において、好ましくはパラミクソウイルスが用いられる。パラミクソウイルスとはパラミクソウイルス科に属するウイルスまたはその誘導体を指す。パラミクソウイルスは、非分節型ネガティブ鎖RNAをそれらのゲノムとして持つウイルスのグループであり、パラミクソウイルス亜科(Paramyxovirinae)(パラミクソウイルス属とも言うレスピロウイルス属(Respirovirus)、ルブラウイルス属、およびモービリウイルス属を含む)、およびニューモウイルス亜科(Pneumovirinae)(ニューモウイルス属およびメタニューモウイルス(Metapneumovirus)属を含む)を含む。本発明に適用可能なパラミクソウイルスの具体例は、センダイウイルス、ニューカッスル病ウイルス、おたふくかぜウイルス、麻疹ウイルス、RSウイルス、牛疫ウイルス、ジステンパーウイルス、サルパラインフルエンザウイルス(SV5)、ならびにヒトパラインフルエンザウイルス1、2、および3型である。より具体的には、そのような例は、センダイウイルス(SeV)、ヒトパラインフルエンザウイルス-1(HPIV-1)、ヒトパラインフルエンザウイルス-3(HPIV-3)、アザラシジステンパーウイルス(PDV)、犬ジステンパーウイルス(CDV)、イルカモービリウイルス(DMV)、小反芻動物病ウイルス(Peste-des-petits-ruminants virus)(PDPR)、麻疹ウイルス(MV)、牛疫ウイルス(RPV)、ヘンドラウイルス(Hendra virus)(ヘンドラ)、ニパーウイルス(Nipah virus)(ニパー)、ヒトパラインフルエンザウイルス-2(HPIV-2)、サルパラインフルエンザウイルス5(SV5)、ヒトパラインフルエンザウイルス-4a(HPIV-4a)、ヒトパラインフルエンザウイルス-4b(HPIV-4b)、おたふくかぜウイルス(おたふくかぜ)、およびニューカッスル病ウイルス(NDV)を含む。より好ましい例は、センダイウイルス(SeV)、ヒトパラインフルエンザウイルス-1(HPIV-1)、ヒトパラインフルエンザウイルス-3(HPIV-3)、アザラシジステンパーウイルス(PDV)、犬ジステンパーウイルス(CDV)、イルカモービリウイルス(DMV)、小反芻動物病ウイルス(PDPR)、麻疹ウイルス(MV)、牛疫ウイルス(RPV)、ヘンドラウイルス(ヘンドラ)、およびニパーウイルス(ニパー)からなる群より選択されるウイルスである。本発明のウイルスは、好ましくはパラミクソウイルス亜科(レスピロウイルス属、ルブラウイルス属、およびモービリウイルス属を含む)に属するウイルスまたはその誘導体であり、より好ましくはレスピロウイルス属(パラミクソウイルス属とも言う)に属するウイルスまたはその誘導体である。本発明に適用可能なレスピロウイルス属ウイルスの例は、ヒトパラインフルエンザウイルス-1(HPIV-1)、ヒトパラインフルエンザウイルス-3(HPIV-3)、ウシパラインフルエンザウイルス-3(BPIV-3)、センダイウイルス(マウスパラインフルエンザウイルス-1とも呼ばれる)、およびサルパラインフルエンザウイルス-10(SPIV-10)である。本発明において、最も好ましくはセンダイウイルスが用いられる。これらのウイルスは、天然株、野生株、変異株、ラボ継代株、および人為的に構築された株などに由来してもよい。
【0020】
本発明において「ベクター」は、核酸を細胞に導入する担体である。マイナス鎖RNAウイルスベクターとは、マイナス鎖RNAウイルスに由来する、核酸を細胞に導入する担体である。SeVなどのマイナス鎖RNAウイルスは遺伝子導入ベクターとして優れている。マイナス鎖RNAウイルスは、DNAフェーズを持たず、宿主細胞の細胞質でのみ転写および複製を行うため、染色体への組み込みは起こらない。このため染色体異常による癌化または不死化などの安全面における問題が生じない。マイナス鎖RNAウイルスのこの特徴は、ベクターとして用いた場合、安全性に大きく寄与している。異種遺伝子発現に用いた場合、SeVを連続多代継代した場合でさえ、ほとんどヌクレオチド変異が認められず、そのゲノムの高い安定性、および挿入異種遺伝子を長期間にわたって安定に発現することを示す(Yu, D. et al., Genes Cells 2:457-466, 1997)。さらに、SeVにおけるカプシド構造タンパク質の欠失は、挿入遺伝子のサイズおよびそのパッケージングの柔軟性など、性質上のメリットをもたらす。伝播性SeVベクターは、少なくとも5.5kbの外来遺伝子を導入することができ、転写ユニットを付加することによって2種類以上の遺伝子を同時に発現することができる。
【0021】
センダイウイルスはげっ歯類にとっては病原性で肺炎を生じることが知られているが、ヒトに対しては病原性の報告がない。これは、非ヒト霊長類およびヒトへの野生型センダイウイルスの鼻腔内投与が重篤な副作用を引き起こさないというこれまでの報告によって支持された(Hurwitz, J. L. et al., Vaccine 15:533-540, 1997、Slobod, K. S. et al., Vaccine 22:3182-3186, 2004)。さらに利点の例として以下の2点、「高感染性」および「高発現量」を挙げることができる。SeVベクターは細胞膜の糖脂質および糖タンパク質のシアル酸に結合することによって感染する。このシアル酸はほとんどの哺乳動物および鳥類細胞で発現しており、このことが感染スペクトルを広くする、すなわち高感染性に繋がっている。SeVのレプリコンに基づく伝播性ベクターがウイルスを放出する場合、それらは周囲の細胞にも再感染する。多数のRNPが、感染細胞の細胞質において複製され、細胞の分裂を介して娘細胞に分配されるため、持続発現が期待される。さらに、SeVベクターは非常に広い範囲の組織に適用することができる。さらに、細胞質のみで転写および複製が生じるというそれらの特徴的な発現機構は、挿入遺伝子を非常に高いレベルで発現することを示す(Moriya, C. et al., FEBS Lett. 425(1):105-111, 1998;WO 00/70070)。さらに、エンべロープ遺伝子を欠失して非伝播性にしたSeVベクターの回収にも成功している(WO 00/70070;Li, HO. et al., J. Virol. 74(14):6564-6569, 2000)。「高感染性」および「高発現量」を維持して、「安全性」をさらに高めるため、SeVベクターの改良が行われている。
【0022】
本発明のマイナス鎖RNAウイルスベクターは、マイナス鎖RNAウイルスのゲノムRNAを含む。ゲノムRNAとは、マイナス鎖RNAウイルスのあるウイルスタンパク質を含むRNPを形成し、それがゲノム中の遺伝子を発現させる機能を有するRNAを言う。従って核酸は複製され、娘RNPが形成される。マイナス鎖RNAウイルスのRNAは、遺伝子をアンチセンス配列としてコードしている。一般にマイナス鎖RNAウイルスのゲノムにおいて、3'リーダー領域と5'トレイラー領域の間に、ウイルス遺伝子がアンチセンス配列として整列している。各遺伝子のORFの間には、転写終結配列(E配列)-介在配列(I配列)-転写開始配列(S配列)が存在し、これにより各遺伝子のORFをコードするRNAが別々のシストロンとして転写される。本発明のベクターにおけるゲノムRNAは、該RNAによってコードされる遺伝子群の発現、およびRNA自身の自律的な複製に必要なウイルスタンパク質であるN(ヌクレオキャプシド、ヌクレオプロテイン(NP)とも言う)、P(ホスホ)、およびL(ラージ)タンパク質をコードするアンチセンスRNA配列を含む。該RNAはまた、ウイルス粒子の形成に必要なM(マトリックス)タンパク質をコードしていてもよい。さらに該RNAは、ウイルス粒子の感染に必要なエンベロープタンパク質をコードしていてもよい。マイナス鎖RNAウイルスのエンベロープタンパク質には、細胞膜融合を起こすF(融合)タンパク質、および細胞へのウイルス接着に必要なHN(ヘマグルチニン-ノイラミニダーゼ)またはH(ヘマグルチニン)タンパク質が含まれる。しかしながら、ある種の細胞では感染にHNタンパク質は必要なく(Markwell, M.A. et al., Proc. Natl. Acad. Sci. USA 82(4):978-982, 1985)、Fタンパク質のみで感染が成立する。RNAは、Fタンパク質および/またはHNタンパク質以外のエンベロープタンパク質をコードしてもよい。
【0023】
本発明のマイナス鎖RNAウイルスベクターは、例えばマイナス鎖RNAウイルスのゲノムRNAとウイルスタンパク質からなる複合体、すなわちリボヌクレオタンパク質(RNP)であってよい。RNPは、例えば所望のトランスフェクション試薬と組み合わせて細胞に導入することができる。このようなRNPは、具体的にはマイナス鎖RNAウイルスのゲノムRNA、Nタンパク質、Pタンパク質、およびLタンパク質を含む複合体である。RNPは細胞内に導入されると、ウイルスタンパク質の作用によりゲノムRNAからウイルスタンパク質をコードするシストロンが転写されると共に、ゲノム自身が複製され娘RNPが形成される。ゲノムRNAの複製は、該RNAのコピー数の増加をRT-PCR、ノーザンブロットハイブリダイゼーションなどによって検出することにより確認することができる。
【0024】
さらに本発明のマイナス鎖RNAウイルスベクターは、好ましくはマイナス鎖RNAウイルスのウイルス粒子である。「ウイルス粒子」とは、核酸を含む微小粒子を指し、ウイルスタンパク質の作用により細胞から放出される。マイナス鎖RNAウイルスのウイルス粒子は、ゲノムRNAとウイルスタンパク質を含む上記RNPが細胞膜由来の脂質膜(エンベロープという)に含まれた構造を有する。ウイルス粒子は、感染性を有してもよい。感染性とは、マイナス鎖RNAウイルスベクターの細胞接着能および膜融合能に基づき、接着した細胞内に保持した核酸を導入することのできるウイルス粒子の能力を言う。本発明のマイナス鎖RNAウイルスベクターは、伝播性、または非伝播性ベクターであってもよい。「伝播性」とは、ウイルスベクターが宿主細胞に感染した場合、該細胞においてウイルスが自身を複製し、感染性ウイルス粒子を産生することを指す。
【0025】
例えばパラミクソウイルス亜科に属するウイルスにおける各遺伝子は、一般に次のように表記される。一般に、N遺伝子は「NP」とも表記される。
レスピロウイルス属 NP P/C/V M F HN - L
ルブラウイルス属 NP P/V M F HN (SH) L
モービリウイルス属 NP P/C/V M F H - L
【0026】
例えばセンダイウイルスの各遺伝子のヌクレオチド配列のデータベースのアクセッション番号は以下である:N遺伝子について、M29343、M30202、M30203、M30204、M51331、M55565、M69046、およびX17218;P遺伝子について、M30202、M30203、M30204、M55565、M69046、X00583、X17007、およびX17008;M遺伝子について、D11446、K02742、M30202、M30203、M30204、M69046、U31956、X00584、およびX53056;F遺伝子について、D00152、D11446、D17334、D17335、M30202、M30203、M30204、M69046、X00152、およびX02131;HN遺伝子について、D26475、M12397、M30202、M30203、M30204、M69046、X00586、X02808、およびX56131;L遺伝子について、D00053、M30202、M30203、M30204、M69040、X00587、X58886。他のウイルスがコードするウイルス遺伝子の例は以下である:N遺伝子について、CDV、AF014953;DMV、X75961;HPIV-1、D01070;HPIV-2、M55320;HPIV-3、D10025;Mapuera、X85128;Mumps、D86172;MV、K01711;NDV、AF064091;PDPR、X74443;PDV、X75717;RPV、X68311;SeV、X00087;SV5、M81442;およびTupaia、AF079780;P遺伝子については、CDV、X51869;DMV、Z47758;HPIV-l、M74081;HPIV-3、X04721;HPIV-4a、M55975;HPIV-4b、M55976;Mumps、D86173;MV、M89920;NDV、M20302;PDV、X75960;RPV、X68311;SeV、M30202;SV5、AF052755;およびTupaia、AF079780;C遺伝子について、CDV、AF014953;DMV、Z47758;HPIV-1、M74081;HPIV-3、D00047;MV、ABO16162;RPV、X68311;SeV、AB005796;およびTupaia、AF079780;M遺伝子について、CDV、M12669;DMV、Z30087;HPIV-1、S38067;HPIV-2、M62734;HPIV-3、D00130;HPIV-4a、D10241;HPIV-4b、D10242;Mumps、D86171;MV、AB012948;NDV、AF089819;PDPR、Z47977;PDV、X75717;RPV、M34018;SeV、U31956;およびSV5、M32248;F遺伝子について、CDV、M21849;DMV、AJ224704;HPN-1、M22347;HPIV-2、M60182;HPIV-3、X05303、HPIV-4a、D49821;HPIV-4b、D49822;Mumps、D86169;MV、AB003178;NDV、AF048763;PDPR、Z37017;PDV、AJ224706;RPV、M21514;SeV、D17334;およびSV5、AB021962;HN(HまたはG)遺伝子について、CDV、AF112189;DMV、AJ224705;HPIV-1、U709498;HPIV-2、D000865;HPIV-3、AB012132;HPIV-4A、M34033;HPIV-4B、AB006954;Mumps、X99040;MV、K01711;NDV、AF204872;PDPR、Z81358;PDV、Z36979;RPV、AF132934;SeV、U06433;およびSV-5、S76876。しかしながら、各ウイルスについて複数の株が知られており、株の違いにより上記に例示した以外の配列を含む遺伝子も存在する。
【0027】
これらのウイルスタンパク質のORFは、ゲノムRNAにおいて上記のE-I-S配列を用いてアンチセンス配列として整列される。ゲノムRNAの最も3'端に近いORFは、3'リーダー領域と該ORFとの間にS配列のみを必要とし、EおよびI配列は必要ない。さらにゲノムRNAの最も5'端に近いORFは、5'トレイラー領域と該ORFとの間にE配列のみを必要とし、IおよびS配列は必要ない。さらに2つのORFは、例えばIRES配列を用いて単一シストロンとして転写させることもできる。このような場合、これら2つのORFの間にはE-I-S配列は必要ない。例えば野生型のパラミクソウイルスの場合、典型的なRNAゲノムは、3'リーダー領域、N、P、M、F、HN、およびLタンパク質をコードする6つのORF、ならびに5'トレイラー領域をアンチセンスの順で含む。本発明のゲノムRNAにおいて、遺伝子の配置はこれに限定されるものではないが、好ましくは、野生型ウイルスと同様に、3'リーダー領域に続き、この順で整列するN、P、M、F、HN、およびLタンパク質をコードするORF、続く5'トレイラー領域を有する。ある種のマイナス鎖RNAウイルスは、6つのウイルス遺伝子全てを含まないが、そのような場合でも上記の野生型と同様にそれらのウイルス遺伝子を配置することが好ましい。一般に N、P、およびL遺伝子を保持しているベクターは、細胞内で自立的にRNAゲノムから遺伝子を発現することができ、それによってゲノムRNAが複製される。さらにFおよびHN(またはH)遺伝子およびM遺伝子などのエンベロープ構成タンパク質をコードする遺伝子の作用により、感染性のウイルス粒子が形成され、細胞外に放出される。従って、このようなベクターは伝播性ウイルスベクターとなる。AβまたはIL-10を含むポリペプチドをコードする遺伝子は、後述するように、このゲノム中の非タンパク質コード領域に挿入してもよい。
【0028】
さらに、本発明のマイナス鎖RNAウイルスベクターは、野生型マイナス鎖RNAウイルスの遺伝子のいずれかを欠損したものであってよい。ウイルスゲノムRNAは、RNPの再構成に必要なウイルスタンパク質(N、L、およびP)をコードしていれば、エンベロープ構成タンパク質をコードしていなくても細胞内で複製し、遺伝子を発現することができる。例えば、ウイルスの種類に依存して、F、H、HN、G、M、およびM1などのエンベロープ構成タンパク質をコードする遺伝子の少なくとも1つを欠損するベクターが例示できる(WO 00/70055およびWO 00/70070;Li, H.-O. et al., J. Virol. 74(14):6564-6569, 2000)。例えば、M、F、またはHN遺伝子、またはそれらの任意の組み合わせを含まないウイルスベクターも、本発明のベクターとして好適に用いることができる。このようなウイルスベクターの再構成は、例えば、欠損している遺伝子産物を外来的に供給することにより行うことができる。このように調製されたウイルスベクターは、野生型ウイルスと同様に宿主細胞に接着して細胞融合を引き起こすが、細胞に導入されたベクターゲノムは本来のウイルス遺伝子のいずれかを欠損するため、本来のウイルスと同様の感染力を持つ娘ウイルス粒子は形成されない。このため、このようなウイルスは、一回限りの遺伝子導入のための安全で有用なウイルスベクターである。ゲノムから欠損させる遺伝子としては、例えばF遺伝子および/またはHN遺伝子が挙げられる。例えば、F遺伝子が欠損した組換えマイナス鎖RNAウイルスゲノムを発現するプラスミドベクターを、Fタンパク質の発現ベクターならびにNP、P、およびLタンパク質の発現ベクターと共に、宿主細胞にトランスフェクションすることにより、ウイルスベクターの再構成を行うことができる(WO 00/70055およびWO 00/70070;Li, H.-O. et al., J. Virol. 74(14):6564-6569, 2000))。例えば、F遺伝子が染色体に組み込まれた宿主細胞を用いてウイルスを製造することもできる。これらのタンパク質群を外から供給する場合、それらアミノ酸配列は本来のウイルス配列と同じである必要はなく、核酸の導入における活性が天然型のそれと同等かそれ以上ならば、変異または他のウイルス由来の相同遺伝子で代用してもよい。
【0029】
さらに、本発明のウイルスベクターとして、ベクターゲノムが由来するウイルスのエンベロープタンパク質とは異なるタンパク質を含むベクターを作製することもできる。例えば、ウイルス再構成の際に、ベクターの本来のウイルスのエンベロープタンパク質以外のエンベロープタンパク質をパッケージング細胞で発現させることにより、所望のエンベロープタンパク質を含むウイルスベクターを製造することができる。このようなタンパク質に特に制限はなく、例えば、他のウイルスのエンベロープタンパク質、例えば水疱性口内炎ウイルスのGタンパク質(VSV-G)(J. Virology 39:519-528, 1981)を含む。本発明のウイルスベクターには、VSV-Gタンパク質などのように、ゲノムが由来するウイルス以外のウイルスに由来するエンベロープタンパク質を含むシュードタイプウイルスベクターが含まれる。ウイルスRNAゲノムにおいてエンベロープタンパク質がコードされないように設計することにより、ウイルス粒子が細胞に感染した後は、このタンパク質が発現されることはない。
【0030】
さらに、本発明のウイルスベクターは、例えばエンベロープ表面に、接着因子、リガンド、受容体などの特定の細胞に接着し得るタンパク質、抗体もしくはその断片;またはこれらのタンパク質を細胞外ドメインに有し、ウイルスエンベロープ由来のポリペプチドを細胞内ドメインに有するキメラタンパク質を含むベクターであってもよい。これにより、特定の組織を標的とするベクターを産生することもできる。このようなタンパク質は、ウイルスゲノム内にコードされてもよく、またはウイルスベクターの再構成時に、ウイルスゲノム以外の遺伝子(例えば別の発現ベクターまたは宿主細胞染色体上にある遺伝子)の発現により供給されてもよい。
【0031】
さらに本発明のベクターにおいて、例えばウイルスタンパク質による免疫原性を低下させるために、またはRNAの転写効率または複製効率を高めるために、ベクターに含まれる任意のウイルス遺伝子が野生型遺伝子から改変されてもよい。具体的には、例えばマイナス鎖RNAウイルスベクターにおいて、複製因子であるN、P、およびL遺伝子の少なくとも1つを改変し、転写または複製効率を高めることが考えられる。さらに、エンベロープタンパク質の1つであるHNタンパク質は、赤血球凝集活性とノイラミニダーゼ活性の両方を有する。例えば前者の活性を弱める場合、血液中でのウイルスの安定性を向上させることが可能であり、後者の活性を改変する場合、感染能を調節することが可能である。さらに、Fタンパク質を改変することにより膜融合能を調節することもできる。例えば、どちらも細胞表面の抗原分子となり得るFタンパク質またはHNタンパク質の抗原提示エピトープを解析し、これらのタンパク質の抗原性を弱めたウイルスベクターを作製することもできる。さらに、二次放出粒子またはVLP(ウイルス様粒子)の放出を抑制するため、ウイルス遺伝子に温度感受性変異を導入してもよい(WO 2003/025570)。例えば、M遺伝子には、G69E、T116A、およびA183Sを;HN遺伝子には、A262T、G264、およびK461Gを;P遺伝子には、L511Fを;L遺伝子には、N1197SおよびK1795Eのような変異を導入することができる。導入可能な温度感受性変異は上記の変異に限定されない(WO 2003/025570を参照されたい)。
【0032】
さらに本発明のベクターは、アクセサリー遺伝子が欠損したものであってよい。例えばSeVアクセサリー遺伝子の1つであるV遺伝子をノックアウトすることにより、培養細胞における遺伝子発現および複製は障害されることなく、マウスなどの宿主に対するSeVの病原性が顕著に減少する(Kato, A. et al., J. Virol. 71:7266-7272, 1997;Kato, A. et al., EMBO J. 16:578-587, 1997;Curran, J. et al., WO 01/04272, EP1067179)。このような弱毒化ベクターは、インビボまたはエクスビボにおける遺伝子導入用の無毒性ウイルスベクターとして特に有用である。
【0033】
本発明のベクターは、上記のマイナス鎖RNAウイルスベクターのゲノムに、外来遺伝子としてAβをコードする核酸を含む。Aβをコードするアミノ酸数は制限されない。例えば、Aβ1-40、Aβ1-42、およびAβ1-43は全て許容される。Aβとして、天然の全長Aβの抗原性を有する所望の断片または該断片を含むポリペプチドを用いることもできる。このようなポリペプチドは、Aβに対する抗原性を提示するために、天然のAβ(例えばAβ43)のアミノ酸配列由来の連続した6アミノ酸以上、好ましくは連続した7アミノ酸以上、または連続した8アミノ酸以上を含む(Harlow, Antibodies:A laboratory Manual, 1998;Chapter 5 page 76)。例えば、Aβ42(配列番号:27の1〜42)に対するB細胞のエピトープの大半は、N末端の1番目〜15番目のアミノ酸の領域に集中している(Cribbs, D.H. et al., Int. Immunol. 15(4):505-14, 2003)。従って、ベクターから発現させるAβペプチドとして、Aβ42の1番目〜15番目のアミノ酸を含むポリペプチドを好適に用いることができる。または、Aβ42の1番目〜21番目のアミノ酸または4番目〜10番目のアミノ酸を含むポリペプチドを発現させてもよい(特開2005-21149)。さらに、アミロイド繊維形成を阻害し神経を保護する能力について公知の抗Aβモノクローナル抗体10D5および6C6は、Aβ42の3番目〜6番目のアミノ酸に相当する4アミノ酸エピトープ(EFRH;配列番号:27の3〜6)を認識することが報告されている(Frenkel, D. et al., J. Neuroimmunol. 88:85-90, 1998;Frenkel, D. et al., J. Neuroimmunol. 95:136-142, 1999)。同じエピトープを認識するモノクローナル抗体508Fも、Aβによる神経毒性を抑制する(Frenkel, D. et al., J. Neuroimmunol. 106:23-31, 2000)。従って、この配列を含むAβ配列における6〜8アミノ酸以上のポリペプチドを好適に用いることができる。細胞性免疫のエピトープはAβのC末端領域に集中する。従ってAβ1-21などのN末端断片を含み、Aβ22〜43としてC末端付近の配列(配列番号:27の22番目〜43番目)を含まない断片を発現させることで、相対的に細胞性免疫よりも液性免疫を優位にすることができる。本発明のベクターとして、天然に存在するAβペプチド(配列番号:27の1〜39、1〜40、1〜41、1〜42、および1〜43にそれぞれ相当する、Aβ39、Aβ40、Aβ41、Aβ42、およびAβ43)の配列を含むポリペプチドをコードするベクターも好適に用いることができる。短いAβペプチド断片は、適切なキャリアタンパク質と融合させたポリペプチドを作製し、免疫原性を高めるために用いてもよい。キャリアタンパク質には、キーホールリンペットヘモシアニン(KLH)、ウシ血清アルブミン(BSA)、ウサギ血清アルブミン(RSA)、ヒト血清アルブミン(HSA)、オボアルブミン(OVA)、 チログロブリン(TG)などが含まれる。短いペプチドは、約1〜5アミノ酸のスペーサーを介してキャリアタンパク質と融合することができる。抗原ポリペプチド(分泌型の場合、成熟ポリペプチド)の長さ(キャリアタンパク質部分を除くアミノ酸長)は、好ましくは10〜200アミノ酸、より好ましくは15〜100アミノ酸、なおより好ましくは20〜80アミノ酸の範囲である。
【0034】
さらに、ベクターにコードされるAβポリペプチドは好ましくは、細胞からのその分泌を確実にするように、分泌シグナルを含む。分泌シグナル配列として、インターロイキン(IL)-2および組織プラスミノーゲンアクチベーター(tPA)などの所望の分泌タンパク質のシグナル配列を用いることができ、好ましくはアミロイド前駆体タンパク質(APP)のN末端シグナル配列が用いられる。具体的には、アクセッション番号NP_958817の分泌シグナル配列(ヌクレオチド配列は、例えばアクセッション番号NM_201414を参照)を含む。すなわち適した本発明のベクターは、分泌シグナルが付加されたAβペプチドをコードする。
【0035】
さらに本発明のベクターは、外来遺伝子としてTh2サイトカインおよび/または抗炎症性サイトカインをコードする核酸を含んでもよい。Th2サイトカインとは、1型のヘルパーT細胞(Th1細胞)よりも2型のヘルパーT細胞(Th2細胞)によって優位に産生されるサイトカインを指す。具体的にはIL-4、IL-5、IL-6、IL-9、IL-10、およびIL-13が含まれる。本発明において、ベクターにコードされるサイトカインは、好ましくはIL-4、IL-10、およびIL-13からなる群より選択され、最も好ましくはIL-10がコードされる。各サイトカイン遺伝子のヌクレオチド配列およびそのアミノ酸配列は公知である(IL-4:NM_000589、NP_000580、AAH66277、AAH67515、NP_758858、NP_067258、NP_958427;IL-5:NM_000879、NP_000870、CAA31210、NP_034688、NP_068606;IL-6:NM_000600、NP_000591、XP_518992、AAB30962、NP_112445;IL-9:NM_000590、NP_000581、AAH66284、AAH66287、NP_032399、XP_341488;IL-10:NM_000572、NP_000563、CAG46825、NP_034678、NP_036986;IL-13:NM_002188、NP_002179、AAB01681、NP_032381、NP_446280)。
【0036】
ベクターにコードされるIL-10などのTh2サイトカインは、全長(すなわち野生型)でもよく、活性が維持される限り部分ペプチド(すなわち活性断片)でもよい。例えば、N末端のシグナル配列は、適切に他のシグナル配列によって置換してもよい。または、サイトカインは、他のペプチドとの融合タンパク質として発現させてもよい。
【0037】
ベクターにコードされるAβおよびIL-10などのサイトカインの由来に制限はなく、マウス、ラット、モルモット、ウサギ、ブタ、ウシ、ウマ、ロバ、ヤギ、イヌ、チンパンジー、サル、およびヒトを含む任意の哺乳動物も用いることができる。Aβおよびサイトカインの由来は、同じ由来であってもよく、異なる由来であってもよい。投与対象と同種由来のAβおよびサイトカインを用いるのが適当である。これらのサイトカインをコードする核酸のヌクレオチド配列は、上記のデータベースから入手可能である。さらに例を挙げれば、マウスIL-10について、Genbankアクセッション番号AY410237およびNM_010548、ヒトIL-10について、Genbankアクセッション番号AY029171およびNM_000572が利用可能である。本発明のベクターとして好適に用いることのできる「Aβをコードする核酸」は、ヒトAβcDNA(配列番号:1)である。好適に用いることのできる「IL-10をコードする核酸」は、マウスIL-10cDNA配列(配列番号:2)およびヒトIL-10cDNA配列(配列番号:3)である。上記配列番号:1〜3のヌクレオチド配列と異なる配列を有する核酸は、コドンの縮重の結果として、これらが上記ヌクレオチド配列と同じアミノ酸配列を有するのであれば、配列番号:1〜3の配列と同様に、本発明のベクターとして好適に用いることができる。上記外来遺伝子の挿入領域は、例えばゲノムのタンパク質非コード領域内の所望の部位において選択することができる。例えば3'リーダー領域と3'末端に最も近いウイルスタンパク質ORFとの間;各ウイルスタンパク質ORFの間;および/または5'末端に最も近いウイルスタンパク質ORFと5'トレイラー領域の間に外来遺伝子を挿入することができる。さらに、FまたはHN遺伝子などを欠失するゲノムでは、その欠失領域に挿入することができる。パラミクソウイルスに外来遺伝子を導入する場合、ゲノムに挿入されるポリヌクレオチドの鎖長は、好ましくは6の倍数である(J. Virology, 67(8):4822-4830, 1993)。挿入した外来遺伝子とウイルスORFとの間に、E-I-S配列が1セットずつ配置されるようにする。E-I-S配列を用いて2またはそれ以上の外来遺伝子をタンデムに並べて挿入することができる。または、IRESを用いて所望の外来遺伝子を挿入してもよい。
【0038】
ベクターが保持する外来遺伝子の発現レベルは、その遺伝子の上流(マイナス鎖の3'側)に付加する転写開始配列の種類により調節することができる(WO 01/18223)。発現レベルは、ゲノム内の外来遺伝子の挿入位置によって制御することができ;マイナス鎖の3'末端の近くに挿入するほど発現レベルが高く;5'末端の近くに挿入するほど発現レベルが低くなる。このように、外来遺伝子の挿入位置は、所望の遺伝子発現量を得るために、または外来遺伝子の近傍のウイルスタンパク質をコードする遺伝子との組み合わせが最適となるように適切に調節することができる。一般に、Aβの高い発現レベルを得ることが有利であると考えられるため、Aβをコードする外来遺伝子を、効率の高い転写開始配列に連結し、それをマイナス鎖ゲノムの3'末端近くに挿入することが好ましい。具体的には、外来遺伝子は、3'リーダー領域と3'末端に最も近いウイルスタンパク質ORFとの間に挿入される。または、外来遺伝子は、3'末端に一番近いウイルス遺伝子と2番目の遺伝子のORFの間に挿入してもよい。野生型パラミクソウイルスにおいて、ゲノムの3'末端に最も近いウイルスタンパク質遺伝子はN遺伝子であり、2番目に近い遺伝子はP遺伝子である。または、導入遺伝子の高発現レベルが望ましくない場合、適切な効果が得るために、例えばベクターにおいてマイナス鎖ゲノムのなるべく5'側の位置で外来遺伝子を挿入することによって、または効率の低い転写開始配列を選択することによって、ウイルスベクターからの遺伝子発現レベルを低く抑えることができる。
【0039】
Aβを含むポリペプチドおよびTh2サイトカインを含むポリペプチドの2つのポリペプチドをベクターから発現させるため、それぞれのポリペプチドをコードする核酸をベクターのゲノムに挿入する。2つの核酸は別々の部位に挿入してもよく、またはE-I-S配列を介してタンデムに並べて1つの部位に挿入してもよい。S配列として転写開始効率の高い配列を用いることが好ましい。例えば3'-UCCCACUUU-5'(マイナス鎖RNA;配列番号:6)、3'-UCCCAGUUU-5'(マイナス鎖RNA;配列番号:7)、または3'-UCCCACUUA-5'(マイナス鎖RNA;配列番号:8)を、S配列として好適に用いることができる。
【0040】
本発明のベクターは、AβおよびTh2サイトカインをコードする遺伝子を挿入した以外の位置に他の外来遺伝子を保持してもよい。このような外来遺伝子は制限されない。例えばベクターの感染をモニターするためのマーカー遺伝子、またはサイトカインおよびホルモンのような免疫系調節遺伝子であってもよい。本発明のベクターは、シグナル伝達調節因子、サイトカイン、ホルモン、受容体、抗体、およびそれらの断片をコードする1つまたは複数の遺伝子を保持してもよい。本発明のベクターは、生体の標的部位への直接(インビボ)投与、または患者由来細胞もしくはそれ以外の細胞にベクターを感染させ、その細胞を標的部位へ注入する間接(エクスビボ)投与のいずれかにより、遺伝子を導入することができる。
【0041】
本発明のベクターは、アルツハイマー病の治療、進行防止または阻害のための極めて優れた手段として用いることができる。本発明のベクターは、通常大量のAβが蓄積している24〜25ヶ月齢のAPP Tgマウスにおいて、著しいAβ消失効果を示した。この効果は、1回のみの鼻腔内投与によって確認され、本発明のベクターは、患者に対する侵襲性の低い治療法であり得る。中枢神経系における炎症が認められなかったという所見は、使用するベクターの安全性の高さも実証する。
【0042】
本発明のベクターを製造するために、哺乳動物細胞においてマイナス鎖RNAウイルスのゲノムRNA(またはその相補鎖)を含むRNPの再構成に必要なウイルスタンパク質(すなわちN、P、およびLタンパク質)の存在下で、本発明のマイナス鎖RNAウイルスのゲノムRNAをコードするcDNAを転写させる。転写によりマイナス鎖ゲノム(すなわちウイルスゲノムアンチセンス鎖と同様)またはプラス鎖(ウイルスタンパク質をコードするセンス鎖)のいずれかを生成させることによって、ウイルスRNPを再構成することができる。ベクターの再構成効率を高めるために、好ましくはプラス鎖を生成させる。RNA末端は、天然のウイルスゲノムと同じくらい正確に、3'リーダー配列と5'トレイラー配列の末端を反映させることが好ましい。転写産物の5'末端を正確に制御するために、例えば転写開始部位としてT7 RNAポリメラーゼ認識配列を利用し、該RNAポリメラーゼを細胞内で発現させてもよい。転写産物の3'末端を制御するため、例えば転写産物の3'末端に自己切断型リボザイムをコードさせ、このリボザイムにより正確に3'末端が切り出されるようにすることができる(Hasan, M. K. et al., J. Gen. Virol. 78:2813-2820, 1997;Kato, A. et al., EMBO J. 16:578-587, 1997;およびYu, D. et al., Genes Cells 2:457-466, 1997)。
【0043】
例えば外来遺伝子を有する組換えセンダイウイルスベクターは、Hasan, M. K. et al., J. Gen. Virol. 78:2813-2820, 1997;Kato, A. et al., EMBO J. 16:578-587, 1997;およびYu, D. et al., Genes Cells 2:457-466, 1997の記載に基づき、下記のように構築することができる。
【0044】
まず、目的の外来遺伝子のcDNAヌクレオチド配列を含むDNA試料を調製する。DNA試料は、25ng/μl以上の濃度で電気泳動的により単一のプラスミドとして確認できることが好ましい。以下、NotI部位を利用してウイルスゲノムRNAをコードするDNAに外来遺伝子を挿入する場合を例にとって説明する。目的とするcDNAヌクレオチド配列中にNotI認識部位が含まれる場合、部位特異的変異導入法などを用いて、コードするアミノ酸配列を変化させないようにヌクレオチド配列を改変し、NotI部位をあらかじめ除去しておくことが好ましい。この試料から目的の遺伝子断片をPCRにより増幅し回収する。2つのプライマーの5'領域にNotI部位を付加することにより、増幅された断片の両端をNotI部位とする。外来遺伝子をウイルスゲノム上に挿入した後、外来遺伝子のORFとその両側のウイルス遺伝子のORFの間にE-I-S配列が1つずつ配置されるように、プライマー中にE-I-S配列またはその部分を含める。
【0045】
例えば、フォワード合成DNA配列は、NotIによる切断を保証するため、5'側に2つ以上のヌクレオチドの任意の配列(好ましくはGCGおよびGCCなどのNotI認識部位由来の配列が含まれない4ヌクレオチド、およびより好ましくはACTT)を選択し、その3'側にNotI認識部位gcggccgcを付加した形態を有する。スペーサー配列として、任意の9ヌクレオチド、または9に6の倍数を加えた数のヌクレオチドを、その3'側にさらに付加する。さらにその3'側に、開始コドンATGを含む所望のcDNAの約25ヌクレオチドのORFに相当する配列を付加する。フォワード合成オリゴDNAの3'の末端で、最後のヌクレオチドがGまたはCとなるように、所望のcDNAから約25ヌクレオチドを選択することが好ましい。
【0046】
リバース合成DNA配列のために、5'側で任意の2つ以上のヌクレオチド(好ましくはGCGおよびGCCなどのNotI認識部位由来の配列が含まれない4ヌクレオチド、およびより好ましくはACTT)を選択し、その3'側にNotI認識部位「gcggccgc」を付加し、さらにその3'側に長さを調節するためのオリゴDNA断片を付加する。このオリゴDNAの長さは、付加したE-I-S配列を含む最終的なPCR増幅産物であるNotI断片の鎖長が、6の倍数のヌクレオチド数になるように設計する(いわゆる「6のルール(rule of six)」:Kolakofski, D., et al., J. Virol. 72:891-899, 1998;Calain, P. and Roux, L., J. Virol. 67:4822-4830, 1993;Calain, P. and Roux, L., J. Virol. 67:4822-4830, 1993)。このプライマーの挿入されたオリゴDNA断片の3'側にE-I-S配列を付加する場合、センダイウイルスのS配列の相補鎖配列、好ましくは5'-TTTCACCCT-3'(配列番号:9)、5'-TTTGACCCT-3'(配列番号:10)、または5'-ATTCACCCT-3'(配列番号:11)、I配列の相補鎖配列、好ましくは5'-AAG-3'、E配列の相補鎖配列、好ましくは5'-TTTTTCTTACTACGG-3'(配列番号:12)を付加する。さらにその3'側に、最後のヌクレオチドとしてGまたはCを有する相補鎖配列を付加し、長さが所望のcDNA配列の終始コドンから逆に数えて約25ヌクレオチドになるように選択した。このようにして、リバース合成DNAの3'末端とする。
【0047】
PCRは、Taqポリメラーゼまたはその他のDNAポリメラーゼを用いる通常の方法によって実施することができる。増幅した目的断片はNotIで消化した後、pBluescript(商標)(Stratagene)などのプラスミドベクターのNotI部位に挿入する。得られたPCR産物のヌクレオチド配列をシークエンサーで確認し、正しい配列を含むプラスミドを選択する。これらのプラスミドから挿入断片をNotIを用いて切り出し、ゲノムcDNAを含むプラスミドのNotI部位にクローニングする。またプラスミドベクターを用いずにNotI部位に断片を直接挿入することによって、組換えセンダイウイルスcDNAを得ることも可能である。
【0048】
例えば、組換えセンダイウイルスゲノムcDNAは、文献記載の方法に従って構築することができる(Yu, D. et al., Genes Cells 2:457-466, 1997;Hasan, M. K. et al., J. Gen. Virol. 78:2813-2820, 1997)。例えば、NotI制限部位を含む18bpのスペーサー配列(5'-(G)-CGGCCGCAGATCTTCACG-3')(配列番号:13)を、クローニングされたセンダイウイルスゲノムcDNA(pSeV(+))内のリーダー配列とNタンパク質のORFとの間に挿入し、デルタ肝炎ウイルスのアンチゲノム鎖由来の自己切断リボザイム部位を含むプラスミドpSeV18+b(+)を得る(Hasan, M. K. et al., J. General Virology 78:2813-2820, 1997)。pSeV18+b(+)のNotI部位に外来遺伝子断片を挿入することにより、所望の外来遺伝子を含む組換えセンダイウイルスcDNAを得ることができる。
【0049】
このように調製した組換えウイルスのゲノムRNAをコードするDNAを、上記のウイルスタンパク質(L、P、およびN)存在下で細胞内で転写させることにより、本発明のベクターを再構成することができる。本発明は、本発明のベクターの作製のための、本発明のベクターのウイルスゲノムRNAをコードするDNAを提供する。本発明はまた、本発明のベクターの作製に適用するための、ベクターのゲノムRNAをコードするDNAの使用に関する。組換えウイルスは、文献における公知の方法により再構成することができる(WO 97/16539; WO 97/16538; WO 00/70055; WO 00/70070; WO 01/18223; WO 03/025570; Durbin, A. P. et al., Virology 235: 323-332, 1997; Whelan, S. P. et al., Proc. Natl. Acad. Sci. USA 92: 8388-8392, 1995; Schnell. M. J. et al., EMBO J. 13: 4195-4203, 1994; Radecke, F. et al., EMBO J. 14: 5773-5784, 1995; Lawson, N. D. et al., Proc. Natl. Acad. Sci. USA 92: 4477-4481, 1995; Garcin, D. et al., EMBO J. 14: 6087-6094, 1995; Kato, A. et al., Genes Cells 1: 569-579, 1996; Baron, M. D. and Barrett, T., J. Virol. 71: 1265-1271, 1997; Bridgen, A. and Elliott, R. M., Proc. Natl. Acad. Sci. USA 93: 15400-15404, 1996; Hasan, M. K. et al., J. Gen. Virol. 78: 2813-2820, 1997; Kato, A. et al., EMBO J. 16: 578-587, 1997; Yu, D. et al., Genes Cells 2: 457-466, 1997; Tokusumi, T. et al., Virus Res. 86: 33-38, 2002; Li, H.-O. et al., J. Virol. 74: 6564-6569, 2000)。これらの方法により、パラインフルエンザウイルス、水疱性口内炎ウイルス、狂犬病ウイルス、麻疹ウイルス、リンダーペストウイルス、およびセンダイウイルスを含むマイナス鎖RNAウイルスを、DNAから再構成させることができる。これらの方法に従って、本発明のベクターを再構成させることができる。F遺伝子、HN遺伝子、および/またはM遺伝子などを欠失させたウイルスベクターDNAは、感染性のウイルス粒子を形成しない。しかしながら欠失した遺伝子および/または他のウイルスのエンベロープタンパク質をコードする遺伝子を宿主細胞に導入し発現させることにより、感染性のウイルス粒子を形成させることが可能である。
【0050】
具体的には、ウイルスは以下の工程に従って作製することができる:(a)マイナス鎖RNAウイルスのゲノムRNA(マイナス鎖RNA)またはその相補鎖(プラス鎖)をコードするcDNAを、N、P、およびLタンパク質を発現する細胞内で転写させる工程、ならびに(b)それらの細胞またはその培養上清から該ゲノムRNAを含むRNA複合体を回収する工程。転写のために、ゲノムRNAをコードするDNAは適当なプロモーターの下流に連結される。転写されたゲノムRNAはN、L、およびPタンパク質の存在下で複製されRNP複合体を形成する。次いでM、HN、およびFタンパク質の存在下でエンベロープに包まれたウイルス粒子が形成される。ゲノムRNAをコードするDNAは、例えばT7プロモーターの下流に連結し、T7 RNAポリメラーゼによりRNAに転写させることができる。T7ポリメラーゼ認識配列を含むもの以外にも、所望のプロモーターを利用することができる。または、インビトロで転写させたRNAを細胞にトランスフェクトしてもよい。N、L、およびPタンパク質は、プラスミドなどの適当な発現ベクターを用いて細胞内で発現させることができる。プロモーターの例には、CMVプロモーターおよびCAGプロモーターが含まれる(Niwa, H. et al.Gene. 108:193-199, 1991;特開平3-168087)。
【0051】
DNAからのゲノムRNAの最初の転写に必要なT7 RNAポリメラーゼなどの酵素は、これらを発現するプラスミドベクターまたはウイルスベクターの導入によって、または、例えば細胞の染色体にこれらの酵素の遺伝子を、発現を誘導できるように組み込み、ウイルス再構成時に発現を誘導することによって供給することができる。さらにゲノムRNA、およびベクター再構成に必要なウイルスタンパク質は、例えばこれらを発現するプラスミドの導入によって供給する。これらのウイルスタンパク質の供給において、野生型またはある種の変異マイナス鎖RNAウイルスのヘルパーウイルスを用いることもできるが、これらのウイルスの混入が発生し得るため好ましくない。
【0052】
ゲノムRNAを発現するDNAを細胞内に導入する方法には、例えば以下を含む:(i)目的の細胞が取り込めるようなDNA沈殿物を作製する方法;(ii)低い細胞毒性を有し、かつ目的の細胞による取り込みに適したDNAを含む陽性に荷電した複合体を作製する方法;および(iii)目的の細胞へDNA分子が通り抜けるのに十分なサイズの穴を、細胞膜上に電気パルスによって瞬間的に開ける方法。
【0053】
方法(ii)において、様々なトランスフェクション試薬、例えば、DOTMA(Roche)、Superfect(QIAGEN #301305)、DOTAP、DOPE、DOSPER(Roche #1811169)などが利用できる。(i)の例は、リン酸カルシウムを用いたトランスフェクション法であり、この方法によって細胞内に移入したDNAは貧食小胞に取り込まれるが、核内にも十分な量のDNAが入ることが知られている(Graham, F. L. and Van Der Eb, J., Virology 52:456, 1973;Wigler, M. and Silverstein, S., Cell 11:223, 1977)。ChenおよびOkayamaは移入技術の最適化を検討し、(1)細胞と共沈殿物のインキュベーション条件が、2〜4%CO2、35℃、15〜24時間であり、(2)直鎖状DNAより環状DNAが高い活性を有し、かつ(3)沈殿混合物が20〜30μg/mlのDNA濃度を有する場合、最適な沈殿が得られることを報告している(Chen, C. and Okayama, H., Mol. Cell. Biol. 7:2745, 1987)。(ii)の方法は、一過的なトランスフェクションに適している。以前はDEAE-デキストラン(Sigma #D-9885 M.W. 5×105)混合物を所望のDNA濃度比に基づいて調製するトランスフェクション方法が知られていた。複合体の多くはエンドソームの中で分解されてしまうため、効果を高めるためにクロロキンを加えることもできる(Calos, M. P., Proc. Natl. Acad. Sci. USA 80:3015, 1983)。(iii)の方法は電気穿孔法と呼ばれ、細胞選択性がないため、(i)および(ii)の方法がより汎用性が高い。方法の効率は、パルス電流の持続時間、パルスの形、電界(電極間のギャップ、電圧)の強さ、緩衝液の導電率、DNA濃度、および細胞密度の最適条件下で良好とされる。
【0054】
以上3つのカテゴリーの中で(ii)の方法は、操作が簡便で多量の細胞を用いて多数の試料を検討することができるため、ベクター再構成においてDNAの細胞への導入に、トランスフェクション試薬が適している。好ましくは、Superfect Transfection Reagent(QIAGEN, Cat No.301305)、DOSPER Liposomal Transfection Reagent(Roche, Cat No.1811169)、およびTransIT(Mirus, Cat No.MIR2300)のようなトランスフェクション試薬が用いられる。しかしこれらに制限されない。
【0055】
具体的に、cDNAからのウイルスの再構成は、例えば以下のように行うことができる。
【0056】
約6〜24穴のプラスチックプレートまたは100mmペトリ皿などで、10%ウシ胎児血清(FCS)および抗生物質(100ユニット/mlペニシリンGおよび100μg/mlストレプトマイシン)を含む最少必須培地(MEM)を用いて、サル腎臓由来LLC-MK2をほぼ100%コンフルエントまで培養し、例えばT7 RNAポリメラーゼを発現し、かつ1μg/mlソラレン存在下で20分のUV照射により不活化した組換えワクシニアウイルスvTF7-3を、2 PFU/細胞で感染させる(Fuerst, T. R. et al., Proc. Natl. Acad. Sci. USA 83:8122-8126,1986、Kato, A. et al., Genes Cells 1:569-579, 1996)。ソラレンの添加量およびUV照射時間は適切に調整することができる。感染の1時間後、2〜60μg、より好ましくは3〜20μgの組換えセンダイウイルスのゲノムRNAをコードするDNAを、ウイルスRNPの生成に必須なトランスに作用するウイルスタンパク質を発現するプラスミド(0.5〜24μgのpGEM-N、0.25〜12μgのpGEM-P、および0.5〜24μgのpGEM-L)(Kato, A. et al., Genes Cells 1:569-579, 1996)と共に、Superfect(QIAGEN社)を用いたリポフェクション法などによりトランスフェクションする。N、P、およびLタンパク質をコードする発現ベクターの量比は、好ましくは2:1:2であり、プラスミド量は、例えば1〜4μgのpGEM-N、0.5〜2μgのpGEM-P、および1〜4μgのpGEM-Lの範囲内で適切に調整した。
【0057】
トランスフェクションを行った細胞は、所望により100μg/mlのリファンピシン(Sigma)およびシトシンアラビノシド(AraC)、より好ましくは40μg/mlのシトシンアラビノシド(AraC)(Sigma)のみを含む無血清MEMで培養する。ワクシニアウイルスによる細胞毒性を最少にとどめ、ウイルスの回収率を最大にするように薬剤の最適濃度を設定する(Kato, A. et al., Genes Cells 1:569-579, 1996)。トランスフェクションから48〜72時間培養後、細胞を回収し、凍結融解を3回繰り返して細胞を破砕する。RNPを含む破砕物をLLC-MK2細胞に感染させ、さらに培養する。または、培養上清を回収し、LLC-MK2細胞の培養液に添加して感染させ培養する。トランスフェクションは、例えばリポフェクトアミンまたはポリカチオニックリポソームなどと共に複合体を形成させ、それを細胞に導入することによって実行することができる。具体的には、様々なトランスフェクション試薬、例えば、DOTMA(Roche)、Superfect(QIAGEN #301305)、DOTAP、DOPE、DOSPER(Roche #1811169)が利用できる。エンドソーム中での分解を防ぐため、クロロキンを加えることもできる(Calos, M. P., Proc. Natl. Acad. Sci. USA 80:3015, 1983)。RNPが導入された細胞において、RNPからのウイルス遺伝子の発現およびRNPの複製の結果、ベクターが増幅する。得られたウイルス溶液(上清)を希釈(例えば106倍)し、新しい細胞を用いて増幅を繰り返すことにより、ワクシニアウイルスvTF7-3は完全に除去することができる。そのような増幅を、例えば3回以上繰り返す。得られたベクターは-80℃で保存することができる。伝播能を持たず、エンベロープタンパク質をコードする遺伝子を欠損したウイルスベクターを再構成させるために、エンベロープタンパク質を発現するLLC-MK2細胞をトランスフェクションに使用してもよく、またはエンベロープ発現プラスミドを共にトランスフェクションしてもよい。または、トランスフェクションを行った細胞にエンベロープタンパク質を発現するLLK-MK2細胞をさらに重層して培養することによって、欠損型ウイルスベクターを増幅することもできる(WO 00/70055およびWO 00/70070を参照されたい)。
【0058】
回収されたウイルスの力価は、例えばCIU(Cell-Infectious Unit)または赤血球凝集活性(HA)を測定することにより決定することができる(WO 00/70070;Kato, A. et al., Genes Cells 1:569-579, 1996;Yonemitsu, Y. & Kaneda, Y., Hemaggulutinating virus of Japan-liposome-mediated gene delivery to vascular cells. Ed. by Baker AH. Molecular Biology of Vascular Diseases, Method in Molecular Medicine:Humana Press:pp. 295-306, 1999)。GFP(緑色蛍光タンパク質)マーカー遺伝子などを保持したベクターの力価は、マーカーを指標に直接感染細胞をカウントすることにより、定量することができる(例えばGFP-CIUとして)。このように測定した力価は、CIUと同等に扱うことができる(WO 00/70070)。
【0059】
ウイルスベクターを再構成することができる限り、再構成に用いる宿主細胞は特に制限されない。例えば、センダイウイルスベクターなどの再構成において、サル腎臓由来のLLC-MK2細胞(ATCC CCL-7)およびCV-1細胞(例えばATCC CCL-70)、ハムスター腎臓由来のBHK細胞(例えばATCC CCL-10)などの培養細胞、ヒト由来細胞を用いることができる。これらの細胞に適当なエンベロープタンパク質を発現させることで、そのタンパク質をエンベロープに含む感染性ウイルス粒子を得ることもできる。さらに、大量にセンダイウイルスベクターを得るために、上記の宿主から得られたウイルスベクターを発育鶏卵に感染させ、該ベクターを増幅することができる。鶏卵を使ったウイルスベクターの作製方法は既に開発されている(Nakanishi,M. et al. ed.「State-of-the-Art Technology Protocol in Neuroscience Research III (Shinkei Kagaku Kenkyu-no Sentan Gijutu Protocol III); Molecular Neuron Physiology (Bunshi Sinkeisaibou Seirigaku)」, Koseisha, Osaka, pp. 153-172, 1993)。具体的には、例えば、受精卵を培養器に入れ9〜12日間、37〜38℃で培養し、胚を成長させる。ウイルスベクターを尿膜腔へ接種し、数日間(例えば3日間)卵を培養してウイルスベクターを増殖させる。培養期間などの条件は、増幅する組換えセンダイウイルスにより変わり得る。その後、ウイルスを含んだ尿膜腔液を回収する。通常の方法に従って、センダイウイルスベクターを尿膜腔液から分離および精製することができる(Tashiro, M.,「Virus Experiment Protocol」, Nagai, Ishihama, ed., Medical View Co., Ltd., pp. 68-73, 1995)。
【0060】
例えば、F遺伝子を欠失したセンダイウイルスベクターの再構成と産生は、以下のように行うことができる(WO 00/70055およびWO 00/70070を参照されたい)。
【0061】
<1>F遺伝子欠失型センダイウイルスゲノムcDNAおよびF発現プラスミドの構築
センダイウイルス(SeV)全長ゲノムcDNA、pSeV18+b(+)(Hasan, M. K. et al., J. General Virology 78:2813-2820, 1997)(「pSeV18+b(+)」は「pSeV18+」とも言う)のcDNAをSphI/KpnIで消化して断片(14673bp)を生成する。断片をpUC18にクローニングしてプラスミドpUC18/KSを調製し、pUC18/KS上でF遺伝子欠損部位を構築する。F遺伝子の欠失を、PCRおよびライゲーション方法の組み合わせで作製する。結果としてF遺伝子のORF(ATG-TGA=1698bp)が除かれる。例えばatgcatgccggcagatga(配列番号:14)で連結し、F遺伝子欠損型SeVゲノムcDNA(pSeV18+/ΔF)を構築する。Fの上流領域についてプライマー[フォワード:5'-gttgagtactgcaagagc/配列番号:15、リバース:5'-tttgccggcatgcatgtttcccaaggggagagttttgcaacc/配列番号:16]を用いて形成されたPCR産物、およびFの下流領域についてプライマー[フォワード:5'-atgcatgccggcagatga/配列番号:17、リバース:5'-tgggtgaatgagagaatcagc/配列番号:18]を用いて形成されたPCR産物を、EcoT22I制限酵素部位を介してライゲーションした。このように得られたプラスミドをSacIとSalIで消化し、F遺伝子欠損部位を含む領域をカバーする断片(4931bp)を回収してpUC18にクローニングし、pUC18/dFSSを形成する。このpUC18/dFSSをDraIIIで消化して、ライゲーションを介して、回収した断片をpSeV18+のF遺伝子を含む領域をカバーするDraIII断片と置き換え、プラスミドpSeV18+/ΔFを得る。
【0062】
外来遺伝子は、例えばpUC18/dFSSのF遺伝子欠失部位にあるNsiIおよびNgoMIVの制限酵素部位に挿入する。このために、例えば外来遺伝子断片を、NsiI-tailedプライマーおよびNgoMIV-tailedプライマーを用いて増幅してもよい。
【0063】
<2>SeV-Fタンパク質を発現するヘルパー細胞の作製
センダイウイルスのF遺伝子(SeV-F)を発現するCre/loxP誘導型プラスミドの構築のため、SeV-F遺伝子をPCRで増幅し、Cre DNAリコンビナーゼにより遺伝子産物を発現するように設計したプラスミドpCALNdlw(Arai, T. et al., J. Virology 72:1115-1121, 1998)のユニークSwaI部位に挿入し、pCALNdLw/Fプラスミドを構築する。
【0064】
F遺伝子欠損ゲノムから感染ウイルス粒子を回収するため、SeV-Fタンパク質を発現するヘルパー細胞株を樹立する。例えばSeVの増殖に一般に用いられるサル腎臓由来LLC-MK2細胞を用いることができる。LLC-MK2細胞は、10%の熱不活化ウシ胎児血清(FBS)、ペニシリンGナトリウム(50ユニット/ml)、およびストレプトマイシン50μg/mlを添加したMEMで37℃、5%CO2で培養する。SeV-F遺伝子産物は細胞傷害性であるため、Cre DNAリコンビナーゼによりF遺伝子産物を発現するように設計した上記pCALNdLw/Fプラスミドを、mammalian transfection kit(Stratagene)を用いてリン酸カルシウム法により、周知のプロトコールに従って、LLC-MK2細胞にトランスフェクションする。
【0065】
10cmプレートで40%コンフルエントまで生育したLLC-MK2細胞に、pCALNdLw/Fプラスミド(10μg)を導入し、細胞を10%FBS/MEM(10ml)にて、37℃の5%CO2インキュベーター中で24時間培養する。次いで細胞をはがし、培地(10ml)に懸濁する。懸濁液を10cmシャーレ5枚に播種する:5ml 1枚、2ml 各2枚、および0.2ml 各2枚。G418(GIBCO-BRL)(1200μg/ml)および10%FBSを含むMEM(10ml)にて、細胞を14日間培養し、2日毎に培地交換する。遺伝子が安定して導入された細胞株を選択する。上記培地で生育した細胞はG418に耐性であり、次いでクローニングリングを用いて回収する。回収した各細胞クローンは、10cmプレートでコンフルエントになるまで培養を続ける。
【0066】
Fタンパク質の発現は、細胞を6cmシャーレにてコンフルエントまで生育させた後、Saitoらの方法(Saito et al., Nucl. Acids Res. 23:3816-3821, 1995;Arai, T.et al., J. Virol 72:1115-1121, 1998)により、例えばMOI=3でアデノウイルスAxCANCreを細胞に感染させることによって誘導する。
【0067】
<3>F遺伝子欠失センダイウイルス(SeV/ΔF)の再構成および増幅
外来遺伝子が挿入された上記pSeV18+/ΔFプラスミドを、以下のようにLLC-MK2細胞にトランスフェクションする。LLC-MK2細胞を5×106細胞/シャーレで100mmのシャーレに播種する。T7 RNAポリメラーゼによりゲノムRNAの転写を実施する場合、細胞を24時間培養し、次いでソラレンと長波長紫外線(365nm)で20分間処理したT7 RNAポリメラーゼを発現する組換えワクシニアウイルス(PLWUV-VacT7:Fuerst, T.R. et al., Proc. Natl. Acad. Sci. USA 83:8122-8126, 1986)を、MOI=2程度で室温で1時間感染させる。ワクシニアウイルスへの紫外線照射には、例えば15ワットバルブ5本が装備されたUV Stratalinker(商標)2400(カタログ番号400676(100V), Stratagene, La Jolla, CA, USA)を用いることができる。細胞を無血清MEMで洗浄した後、適切なリポフェクション試薬を用いて、ゲノムRNAを発現するプラスミド、ならびにパラミクソウイルスのN、P、L、およびFプラスHNタンパク質を発現する発現プラスミドをトランスフェクションする。これらのプラスミドの量は、これに限定されないが、好ましくは順に 6:2:1:2:2とすることができる。例えば、ゲノムRNA発現プラスミド、ならびにN、P、L、および FプラスHNタンパク質を発現する発現プラスミド(pGEM/NP、pGEM/P、pGEM/L、およびpGEM/F-HN;WO 00/70070, Kato, A. et al., Genes Cells 1:569-579, 1996)を、それぞれ12μg、4μg、2μg、4μg、および4μg/シャーレの量でトランスフェクションする。数時間培養後、無血清MEMで細胞を2回洗浄し、40μg/mlのシトシンβ-D-アラビノフラノシド(AraC:Sigma, St.Louis, MO)および7.5μg/mlのトリプシン(Gibco-BRL, Rockville, MD)を含むMEMで培養する。これらの細胞を回収し、ペレットをOpti-MEM に懸濁する(107細胞/ml)。凍結融解を3回繰り返してリポフェクション試薬DOSPER(Boehringer Mannheim)と混合し(106細胞/25μl DOSPER)室温で15分放置した後、上記でクローニングしたF発現ヘルパー細胞にトランスフェクション(12-ウェル-プレート中106細胞/ウェル)し、無血清MEM(40μg/ml AraCおよび7.5μg/mlトリプシンを含む)で培養し、上清を回収する。F以外の遺伝子、例えばHNおよび/またはM遺伝子を欠損したウイルスはいずれも、これと同様の方法で調製することができる。
【0068】
ウイルス遺伝子欠損型ベクターの作製において、例えば、異なるウイルス遺伝子がウイルスゲノムから欠損している2種またはそれ以上のベクターを、同じ細胞に導入する場合、各ベクターの欠損したウイルスタンパク質が他のベクターからの発現により供給され、そのような相互補完が、感染力のあるウイルス粒子の構築、および複製サイクルを介したウイルスベクターの増幅を可能にする。結果として、個々のウイルス遺伝子欠損型ウイルスベクターの混合物を低コストで大量に産生することができる。このようなウイルスは、ウイルス遺伝子が欠損しているため、無傷のウイルスよりゲノムサイズが小さい。このようなウイルスは、サイズの大きい外来遺伝子を保持することができる。さらに、ウイルス遺伝子の欠損により非伝播性であるこのようなウイルスは、一旦細胞外に放出された後、単に希釈されると、感染力の維持が困難であることから、不稔化するため、環境へのそれらの放出が管理できるという利点がある。例えばAβをコードするベクターとTh2サイトカインをコードするベクターとを互いに相補できるように別々に構築し、共感染に用いてもよい。本発明は、Aβを含むポリペプチドをコードするマイナス鎖RNAウイルスベクター、およびTh2サイトカインをコードするマイナス鎖RNAウイルスベクターを含む組成物を提供する。本発明はまた、Aβを含むポリペプチドをコードするマイナス鎖RNAウイルスベクター、およびTh2サイトカインをコードするマイナス鎖RNAウイルスベクターを含むキットを提供する。これらの組成物およびキットは、アミロイドβ沈着を減少させるために使用できる。
【0069】
伝播性のマイナス鎖RNAウイルスベクターを個体または細胞に投与後、例えば治療の完了時にウイルスベクターの増殖を抑止する必要が生じた場合、RNA依存性RNAポリメラーゼ阻害剤を投与することによって、宿主に障害を与えずにウイルスベクターの増殖だけを特異的に抑止することもできる。
【0070】
本発明の方法によれば、本発明のウイルスベクターは、例えば1×105 CIU/ml以上、好ましくは1×106 CIU/ml以上、より好ましくは5×106 CIU/ml以上、より好ましくは1×107 CIU/ml以上、より好ましくは5×107 CIU/ml以上、より好ましくは1×108 CIU/ml以上、より好ましくは5×108 CIU/ml以上の力価でウイルス産生細胞の培養培地中に放出させることが可能である。ウイルスの力価は、本明細書および他に記載の方法により測定することができる(Kiyotani, K. et al., Virology 177(1):65-74, 1990;WO 00/70070)。
【0071】
回収したウイルスベクターは実質的に純粋になるよう精製することができる。ベクターの精製は、濾過、遠心分離、およびカラム精製を含む文献において公知の精製および分離方法、またはその組み合わせにより行うことができる。「実質的に純粋」とは、ウイルスベクターが、それが存在する試料中の成分として主要な割合を占めることを言う。典型的には、実質的に純粋なウイルスベクターは、ウイルスベクター由来のタンパク質の割合が、試料中の全タンパク質(しかしキャリアおよび安定剤として加えたタンパク質は除く)の10%以上、好ましくは20%以上、より好ましくは50%以上、好ましくは70%以上、より好ましくは80%以上、およびさらに好ましくは90%以上であることを確認することによって同定できる。ウイルスの具体的な精製方法の例として、セルロース硫酸エステルまたは架橋ポリサッカライド硫酸エステルを用いる方法(特公昭62-30752号公報、特公昭62-33879号公報、および特公昭62-30753号公報)、ならびにフコース硫酸を含む多糖および/またはその分解物にウイルスを吸着させる方法(WO 97/32010)がある。
【0072】
ベクターを含む組成物の調製において、ベクターは必要に応じて薬学的に許容される所望の担体または媒体と組み合わせることができる。「薬学的に許容される担体または媒体」とは、ベクターと共に投与することが可能であり、ベクターによる遺伝子導入を有意に阻害しない材料である。例えばベクターを生理食塩水またはリン酸緩衝生理食塩水(PBS)で適切に希釈して組成物を形成することができる。ベクターを鶏卵などにおいて増殖させた場合、「薬学的に許容される担体または媒体」は尿膜腔液を含んでよい。さらにベクターを含む組成物は、脱イオン水および5%デキストロース水溶液などの担体または媒体を含んでもよい。さらに、上記以外にも、植物油、懸濁剤、界面活性剤、安定剤、殺生物剤などが含まれてもよい。保存剤またはその他の添加剤を含むこともできる。本発明のベクターを含む組成物は試薬および薬剤として有用である。本発明は、Aβペプチドをコードするマイナス鎖RNAウイルスベクターまたは該ベクターが導入された細胞、および薬学的に許容される担体または媒体を含む組成物を製造する工程を含む、抗アミロイド沈着薬の製造方法に関する。本発明は、抗アミロイド沈着薬の製造における、マイナス鎖RNAウイルスベクター、該ベクターを産生する細胞、または該ベクターが導入された細胞の使用にも関する。
【0073】
本発明のベクターを含む組成物は、バイオポリマーなどの有機物、ハイドロキシアパタイトなどの無機物、具体的にはコラーゲンマトリックス、ポリ乳酸ポリマーまたはコポリマー、ポリエチレングリコールポリマーまたはコポリマーおよびそれらの化学的誘導体を、担体として組み合わせて含み得る。
【0074】
本発明の組成物は、Th2サイトカインおよび/またはTh2サイトカインをコードする核酸を、発現可能な様式で含んでもよい。このようなTh2サイトカインは、全長(野生型)ポリペプチド、または活性を保持する限り部分ペプチドであってよい。例えば、N末端および/またはC末端アミノ酸(例えば1〜30アミノ酸、具体的には1、2、3、4、5、10、15、20、または25アミノ酸)の欠失は、サイトカイン活性に影響しないだろう。さらに、N末端シグナル配列は、他のシグナル配列によって適切に置換することができる。または、サイトカインは他のペプチドとの融合タンパク質として発現させてもよい。Th2サイトカインをコードする核酸は、発現ベクターに挿入するか、またはアクチンプロモーター、EF1プロモーター、CMVプロモーター、CAGプロモーターなどの適切なプロモーターに接続してもよい。核酸は、マイナス鎖RNAウイルスベクターまたは任意の他の発現ベクター内に含まれてもよい。Th2サイトカインがマイナス鎖RNAウイルスベクターにコードされる場合、Th2サイトカインが同じベクター内にAβとしてコードされてもよく、またはTh2サイトカインが別のマイナス鎖RNAウイルスベクター内にコードされてもよい。具体的に、Aβをコードする本発明のマイナス鎖RNAウイルスベクターがTh2サイトカインをコードしない場合、Th1/Th2バランスは、Th2サイトカインまたはTh2サイトカインベクターを、Aβをコードする本発明のベクターと組み合わせて同時投与することにより、Th2にシフトさせることができる。例えば、用いることができるTh2サイトカインは、IL-4、IL-5、IL-6、IL-9、IL-10、およびIL-13、またはそれらの組み合わせである。Th2サイトカインは好ましくは、IL-4、IL-10、およびIL-13からなる群より選択される。
【0075】
本発明の組成物はまた、アジュバントを含んでもよい。具体的に、本発明のベクターがTh2サイトカインをコードしない場合であっても、Th2アジュバントを含む組成物を用いて、Th1/Th2バランスをTh2にシフトさせることができる。Th2アジュバントとは、1型のヘルパーT細胞(Th1細胞)よりも2型のヘルパーT細胞(Th2細胞)を優位に活性化するアジュバントを指す。具体的には、アジュバントは、水酸化アルミニウム(alum)、コレラ毒素(Bサブユニット)、マンソン住血吸虫卵抽出タンパク質(例えばLacto-N-fucopentaose III)などを含む(Grun, J. L. and P. H. Maurer, Cellular Immunol. 121: 134-145, 1989; Holmgren, J. et al., Vaccine 11:1179-1184, 1993; Wilson, A.D. et al., Vaccine 11: 113-118, 1993; Lindsay, D.S. et al, Int. Arch. Allergy Immunol. 105: 281-288, 1994; Xu-Amano, J. et al., J. Exp. Med. 178: 1309-1320, 1993; Okano, M. et al., J. Immunol. 167(1):442-50, 2001)。本発明の組成物は、任意の他の成分を含んでもよい。例えば、それらは投与をモニターするためのマーカー、または免疫系調節因子、シグナル伝達調節因子、サイトカイン、ホルモン、受容体、抗体、およびそれらの断片のような1つもしくは複数の化合物を含んでもよい。本発明の組成物は、シグナル伝達調節因子、シグナル伝達調節因子、サイトカイン、ホルモン、受容体、抗体、およびそれらの断片などの1つもしくは複数のポリペプチドをコードする1つまたは複数のベクターを含んでもよい。
【0076】
製造されたマイナス鎖RNAウイルスまたは該ウイルスを含む組成物を個体に投与することにより、Aβに対する免疫反応が誘導され、それによってAβ沈着を低下させる。ベクターは、Th2サイトカイン、Th2サイトカイン発現ベクター、またはTh2アジュバントと組み合わせて投与してもよい。ベクターの投与は、インビボ投与でも、細胞を介したエクスビボ投与でもよい。接種するマイナス鎖RNAウイルスベクターは、保持する抗原ポリペプチド遺伝子をゲノムRNAによって発現する能力を有する限り、感染性ウイルス粒子である必要はなく、非感染性ウイルス粒子およびウイルスのコア(ゲノムおよびゲノム結合ウイルスタンパク質を含むRNP複合体)であってもよい。本明細書においてマイナス鎖RNAウイルスベクターとは、マイナス鎖RNAウイルス由来のゲノムRNA、ならびにゲノムRNAの複製およびウイルスが保持する遺伝子の発現に必要なウイルスタンパク質を含むリボヌクレオプロテイン(RNP)複合体を含み、感染した細胞において該ゲノムRNAを複製し、保持する遺伝子を発現する複合体を言う。RNPは、例えばマイナス鎖RNAウイルスのゲノムRNA、ならびにN、L、およびPタンパク質を含む複合体である。具体的には、本発明のマイナス鎖RNAウイルスベクターには、感染性ウイルス粒子、非感染性粒子(ウイルス様粒子(VLP)とも言う)、およびマイナス鎖RNAウイルスのヌクレオカプシドなどの、ゲノムRNAおよびゲノムRNAに結合するウイルスタンパク質を含むRNPが含まれる。ウイルス粒子からエンベロープを除去したRNP(ウイルスコア)であっても、細胞に導入されれば、細胞内においてウイルスゲノムRNAを複製することができる(WO 97/16538;WO 00/70055)。RNPまたはVLPを望ましくは、例えばトランスフェクション試薬などと組み合わせて接種してもよい(WO 00/70055;WO 00/70070)。
【0077】
細胞を介してベクターを接種する場合、適当な培養細胞または接種対象動物から採取した細胞などに、マイナス鎖RNAウイルスベクターを導入する。マイナス鎖RNAウイルスをインビトロ(例えば試験管またはシャーレ内)で細胞に感染させる場合、感染は、例えば培養液または生理食塩水など所望の生理的水溶液中で、インビトロ(またはエクスビボ)で実施する。MOI(多重感染度;すなわち細胞1つあたりの感染ウイルス数)は、好ましくは1〜1000の間の範囲、より好ましくは2〜500、さらに好ましくは3〜300、さらに好ましくは5〜100である。マイナス鎖RNAウイルスと細胞との接触は短い時間でも十分である。例えば1分以上、好ましくは3分以上、5分以上、10分以上、または20分以上接触させればよく、例えば1〜60分程度、より特定すれば5分〜30分程度であってよい。または、それ以上の時間接触させてもよく、例えば数日間またはそれ以上接触させてもよい。
【0078】
ウイルスゲノムRNAを含むRNPおよび非感染性ウイルス粒子(ウイルス様粒子(VLP))は、周知のトランスフェクション方法により細胞に導入することができる。具体的には、リン酸カルシウム法(Chen, C. & Okayama, H., BioTechniques 6:632-638, 1988; Chen, C. and Okayama, H., Mol. Cell. Biol. 7: 2745, 1987)、DEAE-デキストラン法(Rosenthal, N., Methods Enzymol. 152:704-709, 1987)、様々なリポソームベースのトランスフェクション試薬(Sambrook, J. et al., Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, 1989)、およびエレクトロポレーション法(Ausubel, F. et al., In Current Protocols in Molecular Biology, John Wiley and Sons, NY, Vol. 1, Ch. 5 and 9, 1994)など、当業者に公知の様々な技術で細胞にトランスフェクションすることが可能である。エンドソームでの分解を抑制するため、トランスフェクションにおいてクロロキンを加えることもできる(Calos, M. P., Proc. Natl. Acad. Sci. USA 80: 3015, 1983)。トランスフェクション試薬には、DOTMA(Roche)、Superfect Transfection Reagent(QIAGEN, Cat No.301305)、DOTAP、DOPE、DOSPER(Roche #1811169)、TransIT-LT1(Mirus, Product No. MIR 2300)、CalPhos(商標)Mammalian Transfection Kit(Clontech #K2051-1)、CLONfectin(商標)(Clontech #8020-1)などが含まれる。エンベロープウイルスはウイルス粒子形成の際に宿主細胞由来のタンパク質を取り込むことが知られている。このようなタンパク質は、細胞に導入する場合、ある抗原性を有し、細胞傷害性の原因となり得る(J. Biol. Chem., 272: 16578-16584, 1997)。従って、エンベロープが除去されたRNPを用いることには利点がある(WO 00/70055)。
【0079】
ウイルスゲノムRNAの発現ベクター、およびそのゲノムRNAの複製に必要なウイルスタンパク質(N、PおよびLタンパク質)をコードする発現ベクターを細胞に導入することにより、細胞内でウイルスRNPを直接形成させることもできる。ウイルスベクターが導入された細胞をこのように調製してもよい。
【0080】
マイナス鎖RNAウイルスベクターが導入された細胞を調製したら、これを約12時間〜5日間(好ましくは1〜3日間)培養し、Aβペプチドを発現させてもよい。得られた細胞は、直接、または細胞ライセートとして動物に接種することができるが、好ましくはAβペプチドを発現する細胞が接種に用いられる。シグナルペプチドを有するAβを、ベクターから発現させ、細胞外に分泌させてもよい。ベクターを導入した細胞のライセートは、界面活性剤で細胞膜を溶解する手順または、凍結-融解サイクルの繰り返しを含む手順によって調製することができる。Triton X-100およびNonidet P-40などの非イオン性界面活性剤が、0.1〜1%の濃度範囲で適用され得る。例えばPBSで洗浄した後、遠心によって細胞塊を回収し、TNE緩衝液[25mM Tris-HCl(pH7.5)、150mM NaCl、1mM EDTA、1% Nonidet P-40]に再懸濁し、氷上で10〜30分間、懸濁液をインキュベートすることで得られる。抗原として使用するタンパク質が細胞質に可溶性である場合、調製したライセートを遠心(10,000×g、10分)し、不要な不溶性画分を沈殿として除去し、得られた上清を免疫に用いることができる。界面活性剤が望ましくない部位に、ライセートを投与する場合、洗浄後、細胞をPBSに再懸濁し、5〜6回の凍結-融解サイクルを繰り返して細胞を破壊することによって、ライセートを調製してもよい。ライセートは、トランスフェクション試薬と共に投与してもよい。
【0081】
または、Aβペプチドをコードするマイナス鎖RNAウイルスベクターを生成する核酸を動物に投与し、動物の体内でマイナス鎖RNAウイルスベクターによって発現されるAβペプチドを生成させることによって、免疫を誘導することができる。マイナス鎖RNAウイルスベクターを生成する核酸とは、組換えマイナス鎖RNAウイルスベクターの生成に必要なRNAおよびタンパク質群を発現する核酸のセットである。具体的には、Aβをコードするマイナス鎖ウイルスのゲノムRNAまたはその相補鎖(アンチゲノムRNA;すなわちプラス鎖)をコードする核酸、およびマイナス鎖RNAウイルスのゲノムRNAを含むRNPを構成するウイルスタンパク質群をコードする核酸を含む核酸のセットを言う。これらの核酸は、コードするRNAおよびタンパク質を発現できるように適当なプロモーターを含む。そのようなプロモーターは、例えば、CMVプロモーター(Foecking, M.K, and Hofstetter H., Gene 45: 101-105, 1986)、レトロウイルスLTR(Shinnik, T. M. et al., Nature, 293: 543-548, 1981)、EF1プロモーター、およびCAGプロモーター(Niwa, H. et al., Gene. 108: 193-199, 1991、特開平3-168087)を含む。核酸として、プラスミドベクターを好適に用いることができる。マイナス鎖RNAウイルスのゲノムは、ゲノムRNAを含むRNPを構成するウイルスタンパク質群(典型的にはN、L、およびP)をコードする遺伝子を保持し、かつ親の野生型ウイルスが持つ全てのエンベロープ構成タンパク質遺伝子を保持するものが好ましい。マイナス鎖RNAウイルスゲノムが、エンベロープ構成タンパク質の任意の遺伝子を欠損する場合、感染性ウイルス粒子の形成を相補するエンベロープタンパク質(例えばゲノムで欠損するエンベロープタンパク質またはVSV-Gなどの他のエンベロープタンパク質)をコードする核酸を、上記の核酸のセットに含めてもよい。これらの核酸を動物に投与することにより、動物内で組換えマイナス鎖RNAウイルスが生成される。マイナス鎖RNAウイルスのゲノムRNAを含むRNPを構成するウイルスタンパク質群とは、ウイルスゲノムRNAと共にRNPを形成し、ヌクレオカプシドを構成するタンパク質を言う。これらはゲノムの複製および遺伝子発現に必要なタンパク質群であり、典型的には、N、P、およびLタンパク質を含む。具体的に本発明は、(i)Aβをコードするマイナス鎖RNAウイルスのゲノムRNAまたはその相補鎖をコードする核酸、および(ii)N、P、およびLタンパク質をコードする核酸を、動物に接種する工程を含む方法にも関する。この動物においてゲノムRNAを含むRNPが形成され、Aβペプチドが発現する。ゲノムには、Th2サイトカインがさらにコードされてもよい。
【0082】
または、あらかじめゲノムRNAまたはその相補鎖をコードする核酸、および上記RNPを構成するウイルスタンパク質群が導入された細胞、またはその細胞のライセートを接種してもよい。マイナス鎖RNAウイルスベクターを生成する核酸の詳細については、以下の文献を参照することができる:WO 97/16539; WO 97/16538; WO 00/70055; WO 00/70070; WO 01/18223; WO 03/025570; Durbin, A. P. et al., Virology 235: 323-332, 1997; Whelan, S. P. et al., Proc. Natl. Acad. Sci. USA 92: 8388-8392, 1995; Schnell. M. J. et al., EMBO J. 13: 4195-4203, 1994; Radecke, F. et al., EMBO J. 14: 5773-5784, 1995; Lawson, N. D. et al., Proc. Natl. Acad. Sci. USA 92: 4477-4481, 1995; Garcin, D. et al., EMBO J. 14: 6087-6094, 1995; Kato, A. et al., Genes Cells 1: 569-579, 1996; Baron, M. D. and Barrett, T., J. Virol. 71: 1265-1271, 1997; Bridgen, A. and Elliott, R. M., Proc. Natl. Acad. Sci. USA 93: 15400-15404, 1996; Hasan, M. K. et al., J. Gen. Virol. 78: 2813-2820, 1997; Kato, A. et al., EMBO J. 16: 578-587, 1997; Yu, D. et al., Genes Cells 2: 457-466, 1997; Tokusumi, T. et al., Virus Res. 2002: 86; 33-38, 1997; Li, H.-O. et al., J. Virol., 74: 6564-6569, 2000。
【0083】
マイナス鎖RNAウイルスベクターを生成する核酸を動物に接種する場合、Aβをコードするマイナス鎖RNAウイルスのゲノムRNAまたはその相補鎖をコードする核酸、ならびにN、P、およびLタンパク質をコードする核酸を、5:0.5:0.5:2の重量比で動物に導入してもよい。核酸の比は適切に調整してよい。例えば発現プラスミドを用いる場合、AβをコードするウイルスゲノムRNA(プラス鎖またはマイナス鎖)をコードするプラスミド5μg〜1000μg、ならびにN発現プラスミド0.5μg〜200μg、P発現プラスミド0.5μg〜200μg、およびL発現プラスミド2μg〜500μgを用いて投与を実施することができる。核酸の投与は、例えば裸DNAを直接、またはトランスフェクション試薬と混合した後、注入することによって実施することができる。トランスフェクション試薬には、例えばリポフェクトアミンおよびポリカチオニックリポソームが含まれる。具体的には、DOTMA(Roche)、Superfect(QIAGEN #301305)、DOTAP、DOPE、DOSPER(Roche #1811169)、TransIT-LT1(Mirus, Product No. MIR 2300)などが利用できる。
【0084】
本発明のベクターの投与量は、疾患の種類、患者の体重、年齢、性別、および症状、投与目的、導入する組成物の投与形態、投与方法、導入する遺伝子のタイプなどにより異なる。当業者は適切な投与量を決定することが可能である。投与経路は適切に選択することができ、例えば経皮、鼻腔内、経気管支、筋内、腹腔内、静脈内、および皮下の経路を含むがそれらに限定されない。特に筋肉内投与(例えば腓腹筋)、皮下投与、点鼻投与またはスプレー、手掌または足蹠皮内投与、脾臓直接投与、腹腔内投与などが好ましい。接種部位は、一ヵ所または複数ヵ所、例えば2〜15ヶ所であってよい。接種量は、対象動物、接種部位、および接種回数などに応じて適切に調整してよい。ベクターは、約105〜約1011 CIU/ml、より好ましくは約107〜約109 CIU/ml、最も好ましくは約1×108〜約5×108 CIU/mlの範囲内の量で、薬学上許容される担体と組み合わせて投与することが好ましい。ヒト対する1回あたりの投与量は、ウイルス力価に換算して、1×104 CIU〜5×1011 CIU(細胞感染単位)、および好ましくは2×105 CIU〜2×1010 CIUである。投与回数は、1回、または副作用が臨床上許容される範囲内である限り複数回可能である。1日の投与回数についても同様に決定してもよい。単回投与だけでも有意な効果を発揮できるが、ベクターを2回以上導入することにより、より強い効果が得られる。
【0085】
複数回投与する場合、初回投与から例えば約1年後に再度投与してもよい。好ましい投与経路は、例えば鼻腔内投与または筋肉内投与である。より強い効果のために、投与を繰り返してよい。複数回投与には、好ましくは、Aβをコードする核酸を保持する上記のマイナス鎖RNAウイルスベクター、ウイルスベクターを生成する核酸、ウイルスベクターまたはベクターを生成する核酸が導入された細胞、または細胞のライセートの接種を用いてもよい。
【0086】
細胞を介して接種(エクスビボ投与)する場合、例えばヒト細胞、好ましくは自己の細胞にマイナス鎖RNAウイルスベクターを感染させ、104〜109細胞、および好ましくは105〜108細胞、またはそのライセートを用いることができる。このベクターは非ヒト動物に接種するのに用いることもでき、投与量は、例えば目的の動物とヒトとの体重または投与標的部位の容積比(例えば平均値)に基づき、上記の投与量を換算することができる。本発明のベクターを含む組成物の投与対象として、免疫系を有する所望の脊椎動物(ヒトおよび非ヒト脊椎動物)が含まれ、好ましくは鳥類および哺乳動物であり、より好ましくは哺乳動物(ヒトおよび非ヒト哺乳動物を含む)である。具体的には、ヒト、サルなどの非ヒト霊長類、マウスおよびラットなどのげっ歯類、ウサギ、ヤギ、ヒツジ、ブタ、ウシ、およびイヌなどその他の全ての哺乳動物が含まれる。投与対象となる動物は、例えばアルツハイマー病に罹患した個体、Aβレベルが亢進している個体、Aβ沈着が亢進している個体、アルツハイマー型変異遺伝子を持つ個体などである。
【0087】
本発明の方法により、Aβに対する免疫反応が誘導される。Aβに対する免疫反応は、抗Aβ抗体の産生、脳組織内のAβ量の減少、およびAβ沈着の減少などにより確認することができる。抗Aβ抗体の産生は、血中の抗Aβ抗体の検出によって調べてもよい。抗体レベルは、ELISA(enzyme-linked immunosorbent assay)およびオークタロニー法により測定することができる。ELISA法は、例えばマイクロプレートに抗原を吸着させ、抗血清を調製し、調製した抗血清を2倍段階希釈(開始溶液1:1000)し、希釈した抗血清プレートに加え抗原抗体反応を行わせることによって実施する。次いで発色のために、免疫動物の抗体をペルオキシダーゼ酵素標識した異種抗体と反応させ、二次抗体とする。吸光度が最大発色吸光度の1/2である場合、抗体の希釈倍率に基づいて、抗体価を算出することができる。または、オークタロニー法において、寒天ゲル内に抗原および抗体が拡散し、免疫沈降反応の結果として白い沈降線が形成される。沈降線は、免疫沈降反応が生じた場合の抗血清の希釈倍率である抗体価を測定するのに用いることができる。脳組織におけるAβレベルは、例えば脳組織の抽出物およびBiosource ELISA kitなどを用いて測定することができる。
【0088】
Aβ沈着(老人斑)の減少効果は、例えば以下の手順によって測定することができる:脳組織切片を70%ギ酸で処理し、5%H2O2で内因性のペルオキシダーゼを失活させた後、抗Aβ抗体と切片を反応させ、ペルオキシダーゼ標識二次抗体を用いてDAB染色を行う。染色後、顕微鏡での観察によりAβ蓄積部分の面積を測定することができる。本発明のベクターを投与しない場合に比べ、蓄積部分の面積の割合が減少すれば、Aβ沈着レベルが減少したと判断できる。または、1-フルオロ-2,5-ビス-(3-ヒドロキシカルボニル-4-ヒドロキシ)スチリルベンゼン(FSB)などのアミロイドに親和性の化合物を静注投与した後、MRIを用いて、生きた対象において老人斑を観察することができる(Higuchi M et al., Nat. Neurosci. 8(4):527-33, 2005; Sato, K. et al., Eur. J. Med. Chem. 39: 573, 2004; Klunk, W. E. et al., Ann. Neurol. 55(3):306-19, 2004)。このような非侵襲的なアミロイドイメージング技術により、本発明のベクターの効果を確認することができる。
【0089】
本発明はまた、以下の工程を含む、アミロイドβに対する免疫反応を測定する方法に関する:本発明のベクターまたはベクターを含む組成物を、アミロイドβ沈着を有する対象に導入する工程、および対象における抗アミロイドβ抗体を検出する工程。本発明はまた、以下の工程を含む、アミロイドβ沈着を測定する方法に関する:本発明のベクターまたはベクターを含む組成物を、アミロイドβ沈着を有する対象に投与する工程、および対象におけるアミロイドβ沈着のレベルを検出する工程。これらの方法により、アミロイドβに対する免疫反応および/またはアミロイドβ沈着の減少効果のモニタリングが可能である。
【0090】
本発明を実施するための最良の形態
以下実施例により本発明を具体的に説明するが、これらに限定されるとは解釈されない。本明細書中に引用された文献は、参照により組み込まれる。
【0091】
[実施例1]Aβ遺伝子を保持したマイナス鎖RNAウイルスゲノムcDNAの構築
(1−1)NotI断片の構築(マイナス鎖RNAウイルスの転写シグナルの付加)
鋳型として、ヒトAβペプチド1〜43(配列番号:27)にアミロイド前駆体タンパク質(APP:アクセッション番号AF282245)の分泌シグナル(1〜18アミノ酸)を付加することによって分泌型としたAβペプチド(配列番号:28)をコードする遺伝子(特開2005-21149;配列番号:1)、ならびにphAbeta-NnotI(配列番号:19)およびphAbeta-CnotI(配列番号:20)の2種のプライマーを用いてPCRを行った。得られたPCR産物を、NotIで消化し、次いでpBluescript(商標)II KS(Stratagene)にサブクローニングし、センダイウイルスの転写シグナルを含むAβペプチド遺伝子のNotI断片(配列番号:21)を構築した。マウスIL-10(mIL10)についても同様に、鋳型としてmIL10 cDNA(アクセッション番号NM_010548;配列番号:2)、ならびにpmIL10-N(配列番号:22)およびpmIL10-C(配列番号:23)の2種のプライマーを用いてPCRを行った。得られたPCR産物を、NotIで消化し、次いでpBluescript(商標)II KS(Stratagene)にサブクローニングし、センダイウイルスの転写シグナルを含むmIL10遺伝子のNotI断片(配列番号:24)を構築した。
【0092】
配列番号:1
ATGCTGCCCGGTTTGGCACTGCTCCTGCTGGCCGCCTGGACGGCTCGGGCGCTTGATGCAGAATTCCGACATGACTCAGGATATGAAGTTCATCATCAAAAATTGGTGTTCTTTGCAGAAGATGTGGGTTCAAACAAAGGTGCAATCATTGGACTCATGGTGGGCGGTGTTGTCATAGCGACTTAA
配列番号:19
ATTGCGGCCGCCAAGGTTCACTTATGCTGCCCGGTTTGGCACTGCTCCTG
配列番号:20
ATTGCGGCCGCGATGAACTTTCACCCTAAGTTTTTCTTACTACGGTTAAGTCGCTATGACAACACCGCCCACCATGAGTCC
配列番号:21
gcggccgccaaggttcacttATGCTGCCCGGTTTGGCACTGCTCCTGCTGGCCGCCTGGACGGCTCGGGCGCTTGATGCAGAATTCCGACATGACTCAGGATATGAAGTTCATCATCAAAAATTGGTGTTCTTTGCAGAAGATGTGGGTTCAAACAAAGGTGCAATCATTGGACTCATGGTGGGCGGTGTTGTCATAGCGACTTAAccgtagtaagaaaaacttagggtgaaagttcatcgcggccgc
配列番号:2
ATGCCTGGCTCAGCACTGCTATGCTGCCTGCTCTTACTGACTGGCATGAGGATCAGCAGGGGCCAGTACAGCCGGGAAGACAATAACTGCACCCACTTCCCAGTCGGCCAGAGCCACATGCTCCTAGAGCTGCGGACTGCCTTCAGCCAGGTGAAGACTTTCTTTCAAACAAAGGACCAGCTGGACAACATACTGCTAACCGACTCCTTAATGCAGGACTTTAAGGGTTACTTGGGTTGCCAAGCCTTATCGGAAATGATCCAGTTTTACCTGGTAGAAGTGATGCCCCAGGCAGAGAAGCATGGCCCAGAAATCAAGGAGCATTTGAATTCCCTGGGTGAGAAGCTGAAGACCCTCAGGATGCGGCTGAGGCGCTGTCATCGATTTCTCCCCTGTGAAAATAAGAGCAAGGCAGTGGAGCAGGTGAAGAGTGATTTTAATAAGCTCCAAGACCAAGGTGTCTACAAGGCCATGAATGAATTTGACATCTTCATCAACTGCATAGAAGCATACATGATGATCAAAATGAAAAGCTAA
配列番号:22
ACTTGCGGCCGCCAAAGTTCAATGCCTGGCTCAGCACTGCTATGCTGCCTG
配列番号:23
ATCCGCGGCCGCGATGAACTTTCACCCTAAGTTTTTCTTACTACGGTTAGCTTTTCATTTTGATCATCATGTATGCTTC
配列番号:24
gcggccgccaaagttcaATGCCTGGCTCAGCACTGCTATGCTGCCTGCTCTTACTGACTGGCATGAGGATCAGCAGGGGCCAGTACAGCCGGGAAGACAATAACTGCACCCACTTCCCAGTCGGCCAGAGCCACATGCTCCTAGAGCTGCGGACTGCCTTCAGCCAGGTGAAGACTTTCTTTCAAACAAAGGACCAGCTGGACAACATACTGCTAACCGACTCCTTAATGCAGGACTTTAAGGGTTACTTGGGTTGCCAAGCCTTATCGGAAATGATCCAGTTTTACCTGGTAGAAGTGATGCCCCAGGCAGAGAAGCATGGCCCAGAAATCAAGGAGCATTTGAATTCCCTGGGTGAGAAGCTGAAGACCCTCAGGATGCGGCTGAGGCGCTGTCATCGATTTCTCCCCTGTGAAAATAAGAGCAAGGCAGTGGAGCAGGTGAAGAGTGATTTTAATAAGCTCCAAGACCAAGGTGTCTACAAGGCCATGAATGAATTTGACATCTTCATCAACTGCATAGAAGCATACATGATGATCAAAATGAAAAGCTAAccgtagtaagaaaaacttagggtgaaagttcatcgcggccgc
【0093】
(1−2)F遺伝子欠失部位へのmIL10 NotI断片の挿入
9つの点でアミノ酸変異(Mタンパク質、G69E、T116A、およびA183S、HNタンパク質;A262T、G264R、およびK461G、Pタンパク質;L511F、Lタンパク質;N1197SおよびK1795E)を導入したF遺伝子欠失型SeVベクター(WO 2003/025570)のcDNA(pSeV18+NotI/TSΔF)を、SalIおよびNheIで消化し、Litmus38(New England Biolabs)へサブクローニングした。次いで、MおよびHN遺伝子を含む、得られたLitmus TSΔFプラスミドのF遺伝子欠失部位に、NotI切断部位を挿入するための変異を導入した。鋳型としてLitmus TSΔF、ならびにF_lit3008_NotI(配列番号:25)およびR_lit3020_NotI(配列番号:26)の2種のプライマーを用いたPCRにより、NotI切断部位を挿入した(図1上)。このLitmus TSΔF NotIをNotIで消化後、mIL10遺伝子NotI断片を挿入した(図1下)。
配列番号25:AAGCGGCCGCCTCAAACAAGCACAGATCATGGATG
配列番号26:AGGCGGCCGCTTAATTAACCAAGCACTCACAAGG
【0094】
(1−3)Aβ遺伝子を保持したF遺伝子欠失型SeV cDNAの構築
NotIで消化したpSeV18+NotI/TSΔF上のNotI部位に、Aβペプチド遺伝子NotI断片を挿入し、Aβ遺伝子を保持したF遺伝子欠失型SeV cDNA(pSeV18+Aβ/TSΔF)を構築した(図2)。
【0095】
(1−4)Aβ遺伝子およびIL-10を両方保持したF遺伝子欠失型SeVゲノムcDNAの構築
SalIおよびNheIでpSeV18+Aβ/TSΔFを消化した後、MおよびHN遺伝子を含まない断片を回収した。一方、Litmus TSΔF-mIL10をSalIおよびNheIで消化し、MおよびHN遺伝子を含む断片を調製した。この2つの断片をライゲーションし、Aβ遺伝子およびIL-10遺伝子を両方保持したF遺伝子欠失型SeVゲノムcDNA(pSeV18+Aβ/TSΔF-mIL10)を構築した(図3)。
【0096】
[実施例2]センダイウイルスベクターの再構成と増幅
ウイルスの再構成および増幅は、Liらの報告した方法(Li, H.-O. et al., J. Virology 74: 6564-6569, 2000; WO 00/70070)、およびその改良法(PCT/JP2005/00705)に従って行った。ウイルスベクターがF遺伝子欠失型であるため、Fタンパク質を供給したヘルパー細胞を利用した。このヘルパー細胞は、Cre/loxP誘導発現システムを用いて調製した。このシステムは、Cre DNA リコンビナーゼにより遺伝子産物を誘導発現するように設計されたプラスミドpCALNdLw(Arai, T. et al., J. Virol. 72: 1115-1121, 1988)を利用して、pCALNdLwプラスミドで形質転換したトランスフォーマントに、Cre DNAリコンビナーゼを発現する組換えアデノウイルス(AxCANCre)を、Saitoらの方法(Saito, I. et al., Nucl. Acid Res. 23: 3816-3821, 1995; Arai, T. et al., J. Virol. 72: 1115-1121, 1998)に従って感染させた。
【0097】
上記の方法によって、Aβ遺伝子を保持したF遺伝子欠失型SeVベクター(SeV18+Aβ/TSΔF)、ならびにAβ遺伝子およびIL-10遺伝子を両方保持したF遺伝子欠失型SeVベクター(SeV18+Aβ/TSΔF-mIL10)を調製した。SeV18+Aβ/TSΔF-mIL10のプラス鎖をコードするDNA配列を配列番号:4に、ゲノムRNA(マイナス鎖)のヌクレオチド配列を配列番号:5に示す。コントロールとして、Aβ遺伝子およびIL-10遺伝子の代わりに、F遺伝子欠失部位にLacZ遺伝子を保持するSeVベクター(SeV18+ LacZ /TSΔF;「SeV LacZ」とも称す)を調製した。
【0098】
[実施例3]アルツハイマー病動物モデルにおけるSeV-Aβ1-43/mIL10の鼻腔内投与効果
(3−1)動物および投与方法
24〜25ヶ月齢のAPPトランスジェニックマウス(Tg2576)(Hsiao, K., et al., Science 274:99-102, 1996)を用い、本発明のSeV18+Aβ/TSΔF-mIL10(以下、「SeV-Aβ1-43/mIL-10」とも記載)の効果を検討した。マウスを4匹ずつ2群に分け、一方を治療群とし、もう一方をコントロール群とした。治療群には、SeV-Aβ1-43/mIL-10(1匹あたり5×106 CIU)を、コントロール群には、SeV LacZ(1匹あたり5×106 CIU)を鼻腔内投与した。
【0099】
(3−2)血清中の抗Aβ抗体の測定
上記処置4週間後および8週間後、マウスから血液を採取し、血清中の抗Aβ42抗体量を測定した。Aβ1-42ペプチド(5μg/mL)を、96ウェルプレート(Nunc, MaxiSorp)の各ウェルに吸着させた。5%脱脂乳/TBS-T緩衝液でブロックした後、マウス血清を加え(500倍希釈)、ペルオキシダーゼ標識抗マウスIgG抗体で検出した。抗体価の評価は、ELISAリーダーでの吸光度測定(O.D.450)により行った。
【0100】
結果を図4に示す。コントロール群と比較し、治療群の全個体において抗体価の顕著な増加が観察された。
【0101】
(3−3)SeV-Aβ1-43/mIL-10の老人斑消失効果
上記センダイウイルスベクターを鼻腔内投与したマウスを、投与後8週間(26または27ヶ月齢)で解剖し、前頭葉皮質、頭頂葉、および海馬の皮質の脳組織切片を調製した。上記組織の凍結切片を、以下のように処理した。Aβタンパク質または老人斑を検出するために、組織切片を70%ギ酸で処理し、5% H2O2で内因性のペルオキシダーゼを失活させた。次いで組織切片をラビット抗pan-Aβ抗体(1000倍希釈)と反応させ、ペルオキシダーゼ標識二次抗体を加え、DAB染色を行った。染色した切片を、顕微鏡に連結させた3CCDカメラを用いて観察し、各領域におけるAβ蓄積部分の面積を測定し、面積率を計算した。
【0102】
切片の染色結果を図5(前頭葉)、図6(頭頂葉)、および図7(海馬)に示す。Aβ沈着面積率を図8に示す。前頭葉、頭頂葉、および海馬のいずれにおいても、SeV-Aβ1-43/mIL-10投与により、老人斑がコントロールの10〜20%に顕著に減少したことが確認された。
【0103】
(3−4)SeV-Aβ1-43/mIL投与の安全性の検討
上記(3-3)と同様の手順で、投与後8週間(26〜27ヶ月齢)の治療群およびコントロール群から、前頭葉および海馬組織の凍結切片を調製した。抗CD3抗体(T細胞)、抗Iba-1抗体(ミクログリア)を用いて、凍結切片をABC法にて染色し、中枢神経系におけるリンパ球の浸潤、およびミクログリアの変化が生じているか確認した。図9に示すように、Tリンパ球(pan T細胞マーカーであるCD3陽性の細胞)の浸潤は、治療群またはコントロール群のいずれにおいても、前頭葉および海馬の両方で観察されなかった。従って、本ベクターを治療に用いることにより、中枢神経系の炎症を起こす可能性が非常に低いことが示された。さらに、ミクログリアマーカーであるIBA-1陽性細胞の量は、コントロール群と治療群の間で同程度であり、治療群における極端な活性化(蓄積)は生じないことが確認された。
【0104】
(3−5)脳組織におけるAβの測定
マウス大脳および小脳を正中裂にそって切断し、半球を-80℃に急速冷凍して保存した。脳半球を1mlのTBS溶液中でホモジェナイズし、卓上超遠心機で100,000 g、1時間遠心した。可溶性画分(TBS画分)を保存した。不溶画分を2%SDSに溶解しホモジェナイズ後、さらに100,000 g、1時間遠心した。得られた可溶性画分(2%SDS画分)を保存した。不溶画分を70%ギ酸に溶解しホモジェナイズ後、再び100,000 g、1時間遠心した。得られた可溶性画分(ギ酸画分)を保存した。脳組織中のAβ40および42は、Biosource ELISA kitを用いて測定した。TBS画分を4倍希釈し;2%SDS画分は400〜2000倍希釈し;ギ酸画分は1M Tris溶液で1000倍希釈した。次いで希釈した画分は、測定のため、ELISA希釈液でさらに希釈(2〜10倍)した。
【0105】
図10に示すように、SDS画分、ギ酸画分、およびTBS画分の全てにおいて、SeV-Aβ1-43/mIL-10投与による脳組織中のAβレベルの顕著な減少が確認された。
【0106】
[実施例4]アルツハイマー病動物モデルにおけるSeV-Aβ1-43/mIL-10筋肉内投与効果
(4−1)動物および投与方法
投与経路以外、上記鼻腔内投与の場合と全く同様に行った。5×106 CIUの投与量で、SeV-Aβ1-43/mIL-10ベクターを後肢大腿筋に投与した。
【0107】
(4−2)血清中の抗Aβ抗体の測定
上記鼻腔内投与の場合と同様の手法によって、SeV-Aβ1-43/mIL-10ベクターを筋肉内投与したマウスの血清中の抗Aβ抗体量を測定した。結果を図11に示す。
【0108】
(4−3)SeV-Aβ1-43/mILの老人斑消失効果
上記鼻腔内投与の場合と同様の方法によって、SeV-Aβ1-43/mIL-10ベクターの筋肉内投与後の前頭葉、頭頂葉、および海馬における老人斑消失効果を検討した。結果を、図12(前頭葉)、図13(頭頂葉)、および図14(海馬)に示す。Aβ沈着面積率を図15に示す。各領域は、一定のAβ沈着減少を示した。
【0109】
(4−4)SeV-Aβ1-43/mIL-10投与の安全性の検討
上記鼻腔内投与の場合と同様の方法によって、SeV-Aβ1-43/mIL-10ベクターの筋肉内投与後の中枢神経系におけるリンパ球の浸潤の存在を確認した。図16に示すように、コントロール群または治療群のいずれも、前頭葉におけるTリンパ球の浸潤は観察されなかった。
【0110】
(4−5)脳組織におけるAβの測定
上記鼻腔内投与の場合と同様の方法によって、SeV-Aβ1-43/mIL-10ベクターの筋肉内投与後の脳組織中のAβ40およびAβ42を測定した。結果を図17に示す。いくつかの可溶性画分においてAβレベルの減少が確認された。
【0111】
[実施例5]正常マウスにおけるSeV-Aβ1-43/hIL-10の鼻腔内投与による抗体価の上昇
(5−1)動物および投与方法
本発明のSeV18+Aβ1-43/TSΔF-hIL10(以下「SeV-Aβ1-43/hIL-10」とも称する)の効果を、6週齢C57BL/6Nマウスを用いて試験した。マウスを、各6匹のマウスからなる3つの群に分けた。SeV-Aβ1-43/hIL-10(群1:5×105 CIU/匹、群2:5×106 CIU/匹、群3:5×107 CIU/匹)を、各マウスに鼻腔内投与した。
【0112】
(5−2)血漿中の抗Aβ抗体の測定
投与前ならびに投与の2、4、および8週間後、マウスから血液を採取し、血漿中に存在する抗Aβ42抗体量を測定した。Aβ1-42ペプチド(4μg/mL)を、96ウェルプレート(Nunc, MaxiSorp)の各ウェルに吸着させた。1%BSA/2%ヤギ血清/TBS-T緩衝液でブロックした後、マウス血漿(300倍希釈)を加え、ペルオキシダーゼ標識抗マウスIgG抗体で検出した。抗体価の評価は、ELISAリーダーでの吸光度測定(O.D.450)により行った。
【0113】
結果を図18に示す。抗Aβ抗体量は、3つの群全てで経時的に増加した。ベクター投与によって、投与量依存性の様式で、抗体量は増加したが、群2(5×106 CIU/匹)および群3(5×107 CIU/匹)の間で抗体量の有意な差は観察されなかった。結果は、本発明の試薬(SeV-Aβ1-43/hIL-10)の投与による抗Aβ抗体の誘導は、アルツハイマー病モデルマウス(APP Tgマウス)に特異的ではないが、正常マウスにおいても十分達成されることを示す。
【0114】
[実施例6]正常マウスへの鼻腔内投与におけるアルツハイマー病に対する治療的ベクターをコードするサイトカインの効果
(6−1)動物および投与方法
いくつかのサイトカインをコードする遺伝子を、上記のアルツハイマー病に対する治療用ベクターに挿入した。これらのセンダイウイルスベクターの効果は、6週齢C57BL/6Nマウスを用いて試験した。マウスを、各6匹のマウスからなる3つの群に分けた。(Aβペプチドをコードする遺伝子を保持する、ならびにマウスIL-4、IL-5、またはIL-6をコードする遺伝子を保持する;それぞれSeV-Aβ1-42/mIL-4、SeV-Aβ1-42/mIL-5、またはSeV-Aβ1-42/mIL-6と称される)センダイウイルスベクターを、各マウスに鼻腔内投与した(5×106 CIU/10μl)。
【0115】
(6−2)血漿中の抗Aβ抗体の測定
投与前ならびに投与の1および2週間後、マウスから血液を採取し、血漿中に存在する抗Aβ抗体量を測定した。Aβ1-42ペプチド(4μg/mL)を、96ウェルプレート(Nunc, MaxiSorp)の各ウェルに吸着させた。1%BSA/2%ヤギ血清/TBS-T緩衝液でブロックした後、マウス血漿(300倍希釈)を加え、ペルオキシダーゼ標識抗マウスIgG抗体で検出した。抗体価の評価は、ELISAリーダーでの吸光度測定(O.D.450)により行った。
【0116】
結果を図19に示す。抗Aβ抗体量は、3つの群全てで経時的に増加した。結果は、IL-4、IL-5、およびIL-6が抗Aβ抗体を誘導するのに効果的で、Aβ抗原をコードする核酸、ならびにIL-4、IL-5、およびIL-6より選択される1つまたは複数のサイトカインの両方を保持するセンダイウイルスベクターを含む製剤が、アルツハイマー病に対する治療において、抗Aβ抗体を誘導するのに好ましいことを示す。
【0117】
産業上の利用可能性
本発明によって、アミロイドβ沈着レベルを有効に減少させることのできる新規マイナス鎖RNAウイルスベクターが提供された。本発明のベクターは、アルツハイマー病の治療に有用である。
【図面の簡単な説明】
【0118】
【図1】Litmus SalI/NheIfrg-MtsHNtsPLmut-dF mIL10(以下、Litmus TS-dF mIL10と略す)の構築過程を示す図である。Litmus TS-dFのF遺伝子欠失部位に変異を導入してNotI部位を作製し、そこにNotIを用いて切り出したmIL-10遺伝子を挿入した。
【図2】pSeV18+Aβ/MtsHNtsP511Lmut-dF(以下、pSeV18+Aβ/TS-dFと略す)の構築過程を示す図である。pBluescript(商標)のNotI部位に挿入されているAβ遺伝子を切り出し、pSeV18+NotI/TS-dFのNotI部位に挿入した。
【図3】pSeV18+Aβ/MtsHNtsP511Lmut-dF mIL10 (以下、pSeV18+Aβ/TS-dF mIL10と略す)の構築過程を示す図である。pSeV18+Aβ/TS-dFとLitmus TS-dF mIL10から、SalIおよびNheIを用いて、必要な断片を切り出し、交換した。
【図4】SeV-Aβ1-43/mIL-10またはコントロールベクターの鼻腔内投与の4および8週間後のAPPトランスジェニックマウスの血清中の抗Aβ抗体レベルを示す図である。
【図5】SeV-Aβ1-43/mIL-10 またはコントロールベクターの鼻腔内投与8週間後のAPPトランスジェニックマウスの前頭葉における老人斑を示す図である。
【図6】SeV-Aβ1-43/mIL-10またはコントロールベクターの鼻腔内投与8週間後のAPPトランスジェニックマウスの頭頂葉における老人斑を示す図である。
【図7】SeV-Aβ1-43/mIL-10またはコントロールベクターの鼻腔内投与8週間後のAPPトランスジェニックマウスの海馬における老人斑を示す図である。
【図8】SeV-Aβ1-43/mIL-10またはコントロールベクターの鼻腔内投与8週間後のAPPトランスジェニックマウスにおける老人斑(前頭葉、頭頂葉、および海馬)を定量化した結果を示す図である。
【図9】SeV-Aβ1-43/mIL-10またはコントロールベクターの鼻腔内投与8週間後のAPPトランスジェニックマウスの中枢神経系におけるリンパ球浸潤を検討した結果を示す。前頭葉におけるT細胞の浸潤を、CD3を指標として検出した。海馬におけるミクログリアの変化を、Iba-1を指標として検出した。
【図10】SeV-Aβ1-43/mIL-10またはコントロールベクターの鼻腔内投与8週間後のAPPトランスジェニックマウスの脳組織におけるAβ量を示す図のセットである。
【図11】SeV-Aβ1-43/mIL-10またはコントロールベクターの筋肉内投与4および8週間後のAPPトランスジェニックマウスの血清中のAβ抗体レベルを示す図である。
【図12】SeV-Aβ1-43/mIL-10またはコントロールベクターの筋肉内投与8週間後のAPPトランスジェニックマウスの前頭葉における老人斑を示す写真である。
【図13】SeV-Aβ1-43/mIL-10またはコントロールベクターの筋肉内投与8週間後のAPPトランスジェニックマウスの頭頂葉における老人斑を示す写真である。
【図14】SeV-Aβ1-43/mIL-10またはコントロールベクターの筋肉内投与8週間後のAPPトランスジェニックマウスの海馬における老人斑を示す写真である。
【図15】SeV-Aβ1-43/mIL-10またはコントロールベクターの筋肉内投与8週間後のAPPトランスジェニックマウスにおける老人斑(前頭葉、頭頂葉、および海馬)を定量化した結果を示す図である。
【図16】SeV-Aβ1-43/mIL-10またはコントロールベクターの筋肉内投与8週間後の前頭葉におけるリンパ球浸潤を検討した結果を示す写真である。
【図17】SeV-Aβ1-43/mIL-10またはコントロールベクターの筋肉内投与8週間後の脳組織におけるAβ量を示す図のセットである。
【図18】SeV-Aβ1-43/mIL-10の鼻腔内投与後の正常マウスの血清抗Aβ抗体レベルを示す図である。
【図19】サイトカインをコードするセンダイウイルス治療用ベクターの鼻腔内投与後の正常マウスの血清抗Aβ抗体レベルを示す図である。
【特許請求の範囲】
【請求項1】
アミロイドβをコードする核酸を含む、マイナス鎖RNAウイルスベクター。
【請求項2】
Th2サイトカインまたはその部分ペプチドをコードする核酸をさらに含む、請求項1記載のマイナス鎖RNAウイルスベクター。
【請求項3】
Th2サイトカインまたはその部分ペプチドが、IL-10またはその部分ペプチドである、請求項2記載のマイナス鎖RNAウイルスベクター。
【請求項4】
マイナス鎖RNAウイルスベクターがパラミクソウイルスベクターである、請求項1記載のベクター。
【請求項5】
パラミクソウイルスベクターがセンダイウイルスベクターである、請求項4記載のベクター。
【請求項6】
アミロイドβがAβ43またはその部分ペプチドである、請求項1記載のベクター。
【請求項7】
アミロイドβがヒト由来である、請求項1記載のベクター。
【請求項8】
IL-10がマウスまたはヒト由来である、請求項3記載のベクター。
【請求項9】
アミロイドβが、配列番号:1のヌクレオチド配列がコードするポリペプチドまたはその部分である、請求項1記載のベクター。
【請求項10】
IL-10が、配列番号:2または3のヌクレオチド配列がコードするポリペプチドである、請求項3記載のベクター。
【請求項11】
配列番号:5の核酸を含む、請求項1記載のマイナス鎖RNAウイルスベクター。
【請求項12】
請求項1〜11のいずれか一項記載のベクターおよび薬学的に許容される担体を含む組成物。
【請求項13】
(i)Th2サイトカインもしくはその部分ペプチドを発現可能様式でコードする核酸、(ii)Th2サイトカインもしくはその部分ペプチド、または(iii)Th2アジュバントをさらに含む、請求項12記載の組成物。
【請求項14】
Th2サイトカイン、その部分ペプチド、またはTh2アジュバントを含む、請求項13記載の組成物。
【請求項15】
Th2サイトカインまたはその部分ペプチドを、発現可能様式でコードする核酸を含む、請求項13記載の組成物。
【請求項16】
Th2サイトカインが、アミロイドβをコードする核酸を含むマイナス鎖RNAウイルスベクターによってコードされる、請求項15記載の組成物。
【請求項17】
Th2サイトカインが、アミロイドβをコードする核酸を含むマイナス鎖RNAウイルスベクター以外のベクターによってコードされる、請求項15記載の組成物。
【請求項18】
薬学的組成物である、請求項12記載の組成物。
【請求項19】
請求項1〜11のいずれか一項記載のベクターを含む、アルツハイマー病を治療または予防するために用いられる薬学的組成物。
【請求項20】
(i)Th2サイトカインもしくはその部分ペプチドを発現可能様式でコードする核酸、(ii)Th2サイトカインもしくはその部分ペプチド、または(iii)Th2アジュバントをさらに含む、請求項19記載の組成物。
【請求項21】
鼻腔内投与に用いるための、請求項18または19記載の薬学的組成物。
【請求項22】
請求項1〜11のいずれか一項記載のベクター、または該ベクターを含む組成物を投与する段階を含む、アミロイドβの沈着を減少させる方法。
【請求項23】
請求項1〜11のいずれか一項記載のベクターまたは該ベクターを含む組成物を投与する段階を含む、アルツハイマー病を治療または予防する方法。
【請求項24】
投与が鼻腔内投与である、請求項22または23記載の方法。
【請求項25】
投与が筋肉内投与である、請求項22または23記載の方法。
【請求項26】
Th2サイトカイン、その部分ペプチド、Th2アジュバント、またはTh2サイトカインもしくはその部分ペプチドをコードするベクターを投与する段階をさらに含む、請求項22または23記載の方法。
【請求項27】
Th2サイトカイン、その部分ペプチド、またはTh2アジュバントを投与する、請求項26記載の方法。
【請求項28】
Th2サイトカインまたはその部分ペプチドをコードするベクターを投与する、請求項26記載の組成物。
【請求項1】
アミロイドβをコードする核酸を含む、マイナス鎖RNAウイルスベクター。
【請求項2】
Th2サイトカインまたはその部分ペプチドをコードする核酸をさらに含む、請求項1記載のマイナス鎖RNAウイルスベクター。
【請求項3】
Th2サイトカインまたはその部分ペプチドが、IL-10またはその部分ペプチドである、請求項2記載のマイナス鎖RNAウイルスベクター。
【請求項4】
マイナス鎖RNAウイルスベクターがパラミクソウイルスベクターである、請求項1記載のベクター。
【請求項5】
パラミクソウイルスベクターがセンダイウイルスベクターである、請求項4記載のベクター。
【請求項6】
アミロイドβがAβ43またはその部分ペプチドである、請求項1記載のベクター。
【請求項7】
アミロイドβがヒト由来である、請求項1記載のベクター。
【請求項8】
IL-10がマウスまたはヒト由来である、請求項3記載のベクター。
【請求項9】
アミロイドβが、配列番号:1のヌクレオチド配列がコードするポリペプチドまたはその部分である、請求項1記載のベクター。
【請求項10】
IL-10が、配列番号:2または3のヌクレオチド配列がコードするポリペプチドである、請求項3記載のベクター。
【請求項11】
配列番号:5の核酸を含む、請求項1記載のマイナス鎖RNAウイルスベクター。
【請求項12】
請求項1〜11のいずれか一項記載のベクターおよび薬学的に許容される担体を含む組成物。
【請求項13】
(i)Th2サイトカインもしくはその部分ペプチドを発現可能様式でコードする核酸、(ii)Th2サイトカインもしくはその部分ペプチド、または(iii)Th2アジュバントをさらに含む、請求項12記載の組成物。
【請求項14】
Th2サイトカイン、その部分ペプチド、またはTh2アジュバントを含む、請求項13記載の組成物。
【請求項15】
Th2サイトカインまたはその部分ペプチドを、発現可能様式でコードする核酸を含む、請求項13記載の組成物。
【請求項16】
Th2サイトカインが、アミロイドβをコードする核酸を含むマイナス鎖RNAウイルスベクターによってコードされる、請求項15記載の組成物。
【請求項17】
Th2サイトカインが、アミロイドβをコードする核酸を含むマイナス鎖RNAウイルスベクター以外のベクターによってコードされる、請求項15記載の組成物。
【請求項18】
薬学的組成物である、請求項12記載の組成物。
【請求項19】
請求項1〜11のいずれか一項記載のベクターを含む、アルツハイマー病を治療または予防するために用いられる薬学的組成物。
【請求項20】
(i)Th2サイトカインもしくはその部分ペプチドを発現可能様式でコードする核酸、(ii)Th2サイトカインもしくはその部分ペプチド、または(iii)Th2アジュバントをさらに含む、請求項19記載の組成物。
【請求項21】
鼻腔内投与に用いるための、請求項18または19記載の薬学的組成物。
【請求項22】
請求項1〜11のいずれか一項記載のベクター、または該ベクターを含む組成物を投与する段階を含む、アミロイドβの沈着を減少させる方法。
【請求項23】
請求項1〜11のいずれか一項記載のベクターまたは該ベクターを含む組成物を投与する段階を含む、アルツハイマー病を治療または予防する方法。
【請求項24】
投与が鼻腔内投与である、請求項22または23記載の方法。
【請求項25】
投与が筋肉内投与である、請求項22または23記載の方法。
【請求項26】
Th2サイトカイン、その部分ペプチド、Th2アジュバント、またはTh2サイトカインもしくはその部分ペプチドをコードするベクターを投与する段階をさらに含む、請求項22または23記載の方法。
【請求項27】
Th2サイトカイン、その部分ペプチド、またはTh2アジュバントを投与する、請求項26記載の方法。
【請求項28】
Th2サイトカインまたはその部分ペプチドをコードするベクターを投与する、請求項26記載の組成物。
【図1】


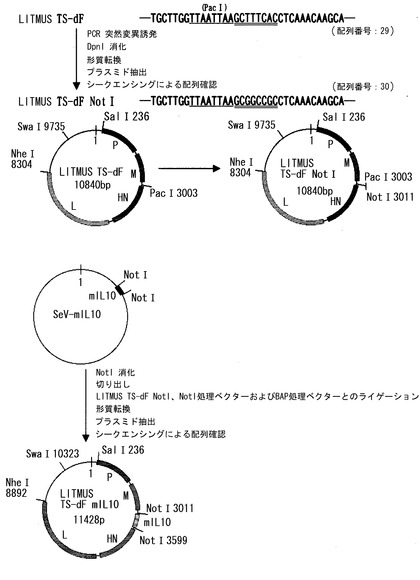
【図2】

【図3】
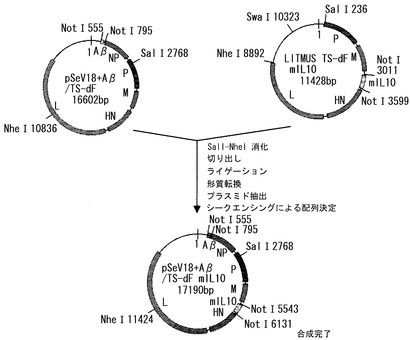
【図4】
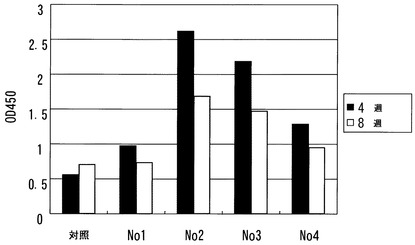
【図5】

【図6】

【図7】

【図8】
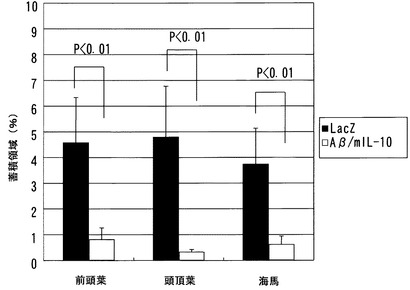
【図9】
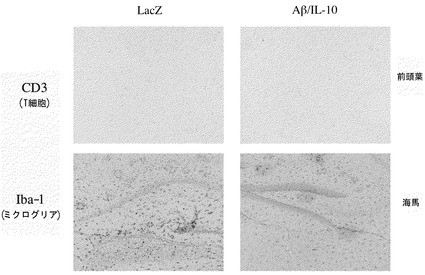
【図10】
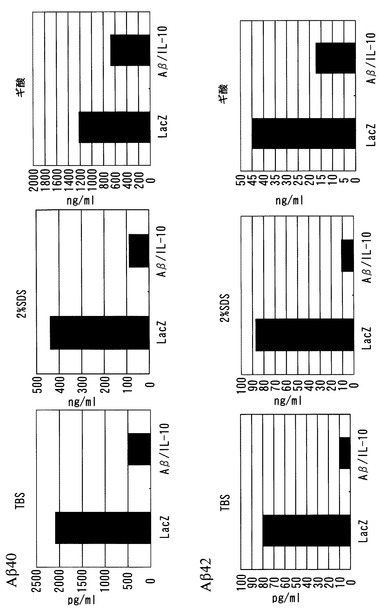
【図11】
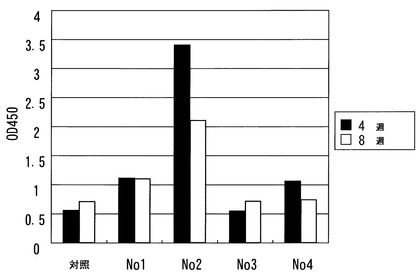
【図12】

【図13】

【図14】

【図15】
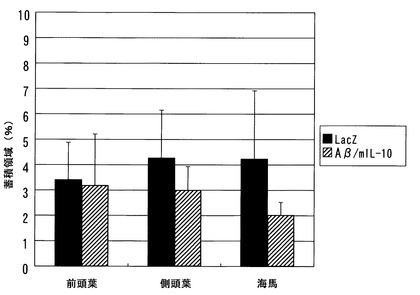
【図16】

【図17】
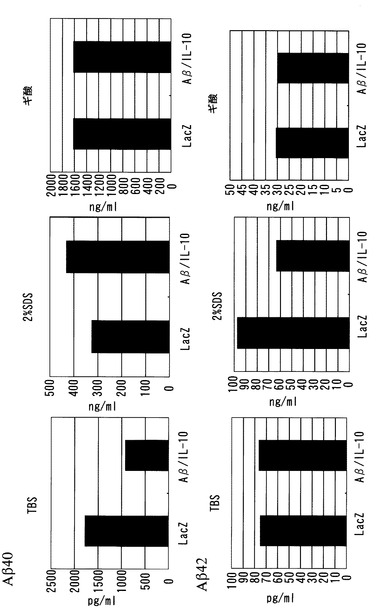
【図18】
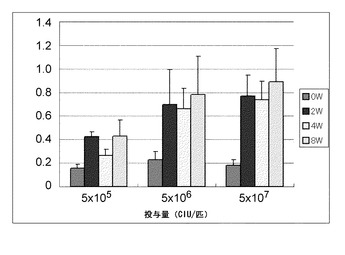
【図19】
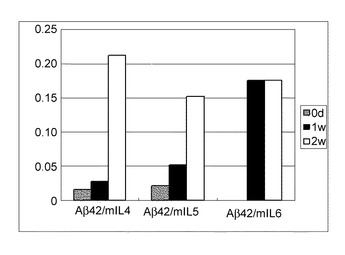
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【公表番号】特表2008−536476(P2008−536476A)
【公表日】平成20年9月11日(2008.9.11)
【国際特許分類】
【出願番号】特願2007−549619(P2007−549619)
【出願日】平成18年4月20日(2006.4.20)
【国際出願番号】PCT/JP2006/308913
【国際公開番号】WO2006/112553
【国際公開日】平成18年10月26日(2006.10.26)
【出願人】(505048482)ディナベック株式会社 (5)
【出願人】(501304319)国立長寿医療センター総長 (9)
【Fターム(参考)】
【公表日】平成20年9月11日(2008.9.11)
【国際特許分類】
【出願日】平成18年4月20日(2006.4.20)
【国際出願番号】PCT/JP2006/308913
【国際公開番号】WO2006/112553
【国際公開日】平成18年10月26日(2006.10.26)
【出願人】(505048482)ディナベック株式会社 (5)
【出願人】(501304319)国立長寿医療センター総長 (9)
【Fターム(参考)】
[ Back to top ]