
アルツハイマー病動物モデル、それを得るための方法およびその使用
本発明は、トランスジェニック非ヒト動物に関し、このトランスジェニック非ヒト動物は、アルツハイマー病(AD)を研究するための非ヒト動物モデルとして使用することができ、このトランスジェニック非ヒト動物は、そのゲノムに挿入され、完全ヒトAPP遺伝子のヌクレオチド配列をその調節配列とともに含む、異種ポリヌクレオチド(トランスジーン)を含有すること;そしてヒトにおけるhAPP遺伝子と同様の内因性発現パターンを有すること:を特徴とする。本発明のモデルは、ADを研究するために、そしてADの予防および/または治療のために潜在的に有用な化合物のスクリーニングにおいて使用することができる。
【発明の詳細な説明】
【発明の詳細な説明】
【0001】
発明の属する技術分野
本発明は、バイオテクノロジー分野および薬学分野に含まれる:本発明は、特に非ヒトアルツハイマー病動物モデルとして使用するためのトランスジェニック非ヒト動物の開発およびその使用に関する。
【0002】
発明の背景
1907年に、Alois Alzheimerが初めて認知症と認識能力の進行性低下を特徴とする神経病理学的症候群を報告した。彼はまた、認知記憶の低下と、剖検で観察された神経病理学的病変と、患者の病歴との関連性も最初に証明した。アルツハイマー病(AD)の臨床的特徴は、幻覚、鬱病、失見当および攻撃行動を伴う、記憶、言語(失語)、運動機能(失行)および知覚(失認)の進行性消失である。
【0003】
Alzheimerによって行われた2つの神経学的所見は、ADを説明するために現在使用されている:(i)神経原繊維変化(無定形のニューロン内で見られる緻密な繊維のもつれであり、その主要なサブユニットは微小管関連タンパク質Tauである)および(ii)老人斑(細胞外斑であり、その主要成分はβ−アミロイドペプチド前駆体タンパク質(APP)と呼ばれる前駆体タンパク質由来のβ−アミロイド(Aβ)と呼ばれるペプチドである)。
【0004】
ヒトAPP遺伝子は21q21.3領域の第21染色体に位置し、300キロベース(kb)にわたって分布する18のエキソンによって形成される。その発現は特定組織に限定されず、内皮、脳のグリアおよびニューロンを含む多くの細胞種および組織で発現される。メッセンジャーRNA(mRNA)は成熟し、3つの主要なアイソフォームを産生するための代替法を受ける可能性がある。これらの3つの主要なアイソフォームにはそれらを形成しているアミノ酸の数により名前が付けられている(APP770、APP751およびAPP695)。最長のアイソフォームは完全遺伝子配列を含むAPP770であり;アイソフォームAPP751は最も多量にかつ普遍的に発現されるアイソフォームであり;そしてアイソフォームAPP695は脳において主要なアイソフォームであり、皮質での3つのmRNA間の比率はAPP770:APP751:APP695、1:10:20であり、そのため、アイソフォームAPP695がADに関連して最も研究されたアイソフォームであった。
【0005】
ADの発症に関与する、APP遺伝子の異なる突然変異が1990〜1992年の間に発見され、その伝達は、浸透度がほぼ100%の常染色体優性であることを確認することができた。1995年には、ADに関連する2つの他の遺伝子が発見され、それらの遺伝子はプレセニリン1(PS1)およびプレセニリン2(PS2)と呼ばれている。
【0006】
ADは、その原因に基づいて2つのタイプに分けることができる:
(i)家族性アルツハイマー病(FAD)(APP、PS1およびPS2遺伝子の突然変異に関連し、発症年齢が早期であり、アルツハイマー病患者のおよそ1%を占める);および
(ii)孤発性アルツハイマー病(SAD)(晩期発症型(通常60歳以後)であり、原因不明である(この疾患に対する罹患素因は一連の感受性遺伝子および環境因子によって決まるようであり、その罹患素因により複雑な多因子疾患となり、主な危険因子は老齢であることと家族歴があることである);SADはアルツハイマー病患者のおよそ99%を占める)。
【0007】
両疾患(FADおよびSAD)は臨床症状によっても神経病理学によっても区別することができない。
【0008】
知られているとおり、実験神経系疾患モデルの開発は生物医学研究に極めて重要である。ヒトにおける神経変性疾患の特徴によく似たモデルの開発には、大きな方法論的進歩が必要であった。ADの場合、主な進歩はFADに関与するタンパク質を同定することに関係していた。
【0009】
90年代の初めに、APP遺伝子におけるいくつかのFAD関連突然変異が発見され、それらの突然変異は、遺伝子導入技術によって得られる進歩とともに、ADを研究するためのモデルを開発する可能性をもたらした。遺伝子導入を用いて、病原型APP遺伝子を再現可能な、より適した方法で、脳の異なる領域において発現させることが可能であり、それにより、野生型APP、APP断片、突然変異APPタンパク質および他のAD関連遺伝子を発現するトランスジェニックマウスに大きな真実性が与えられた。
【0010】
ADのためにマウスにおいて開発されたモデルとしては、以下が挙げられる:
【0011】
NSEAPP:このマウスを作製するために、ニューロン特異的エノラーゼ(NSE)プロモーターおよびAPP遺伝子アイソフォーム751cDNAを有する構築物を調製した。この構築物の過剰発現は、月齢22ヶ月のときにびまん性Aβ斑を発生させた。これらの斑は、マウスの5%でのみ、アルツハイマー病患者に見られる斑によく似たより成熟した外観を呈し、Bielschowskyおよびチオフラビン−S染色液で陽性に染色された。
【0012】
APP YAC:多量のDNAを挿入することができる、トランスジェニックマウスを作製するための担体として酵母人工染色体(YAC)を使用することにより、3つのグループによってほぼ同時に、全APP遺伝子コード領域と該遺伝子の3’および5’の両方にある広範な領域を含む650kb YAC(B142F9 Washington University YAC library)を有するトランスジェニックマウスの作製が公開された。これらのマウスでは、ヒト遺伝子の発現は、マウスにおける内因性発現と同様に、斑の形成または神経原繊維変化を観察することなく行われた。ヒトゲノム配列決定では、これらのマウスを作製するのに用いられるYACは、APP遺伝子に加えて、望ましくない表現型を生じる可能性があるGABPA転写因子をコードする遺伝子も含むことが分かった。
【0013】
PDAPP:このトランスジェニックマウスは、1995年に報告されており、多様なAD神経病理を発現する初めてのマウスであり、「Indiana」V717F突然変異を含むAPP遺伝子cDNAを過剰発現させる構築物を用いて得られた;それらのマウスの脳では異栄養性神経突起(dystrophic neurites)に囲まれた神経炎性斑、星状細胞増加症、小膠細胞症およびシナプスの喪失を観察することができたが、神経原繊維変化は観察されなかった。皮質領域では斑の存在は50%占有に達した。この割合は、6%〜12%間である傾向があるアルツハイマー病患者のものと比較するならば、極めて高い割合である。脳においてこのように多量の斑が存在するにもかかわらず、これらのマウスにおいて神経変性は示されなかった。
【0014】
Tg2576:このマウスは、1996年に報告されており、おそらく、ADを研究するために最もよく用いられているモデルである;トランスジーンは、脳における高発現を確実にするプリオンタンパク質プロモーターによって形成され、遺伝的C57BL/6J//SJLバックグラウンドにおいて「Swedish」K670N/M671L突然変異を含むアイソフォームAPP695 cDNAを有する。Tg2576マウスは、脳におけるマウスの内因性発現レベルの5.6倍であるヒトAPP(hAPP)発現レベルを有する。それらのマウスは、月齢9〜12ヶ月のときには、すでに内側嗅領に神経炎性斑があり、後にその斑は海馬および皮質にまで広がった。
【0015】
APP23:ADを研究するためのこの新しいネズミモデルは、1997年に、Thy1プロモーターと、内因性発現の7倍を上回る発現が行われる、「Swedish」K670N/M671L突然変異を有するhAPP751 cDNAの過剰発現を用いて作製され、月齢6ヶ月のときに斑の発生を起こした。これらのマウスの最も重要な特徴は、月齢14〜18ヶ月のときに海馬のCA1領域における神経細胞死がようやく報告されたということである。このモデルのもう1つの重要な特徴は、ADを研究するための優れたモデルにする脳アミロイド血管症の発生である。これらのマウスの行動解析では、結果において大きな差があり、月齢18ヶ月からの学習において有意差があることが示された。
【0016】
今日までの作製された動物モデルでは、総てのAD病理を作製することができなかった。これらのモデルの作製は、野生型またはFAD関連突然変異を有するAPPタンパク質の過剰発現に基づいたものであり、そのようなモデルは、SAD患者では起こらない状況から出発するため、FADの研究に限定される。これらのモデルでは、マウス脳において脳領域の20〜50%を占めるアミロイド生成性斑(amyloidogenic plaques)の発生が報告されているが、一方、ヒト患者では斑の存在は12%を超えない。
【0017】
本明細書に記載のモデル、ヒトAPP YACマウス「hAPPy」では、APP遺伝子発現は、APP YACを用いて行われた先行研究に記載されているとおり、マウスの内因性発現とほぼ同じように行われた(Buxbaum, J. D., et al. (1993). "Expression of APP in brains of transgenic mice containing the entire human APP gene." Biochem Biophys Res Commun 197(2): 639-45; Lamb, B. T., et al. (1993). " Introduction and expression of the 400 kilobase amyloid precursor protein gene in transgenic mice [補正済み]. "Nat Genet 5(1): 22-30; Pearson, B. E. and T. K. Choi (1993). "Expression of the human beta-amyloid precursor protein gene from a yeast artificial chromosome in transgenic mice." Proc Nati Acad Sci USA 90(22): 10578-82)が、「hAPPy」マウスでは、これまでYAC中に存在していたGABPA遺伝子の存在が消失したという違いがあった(Lamb, B. T., et al. (1997). "Altered metabolism of familial Alzheimer's disease-linked amyloid precursor protein variants in yeast artificial chromosome transgenic mice." Hum Mol Genet 6(9): 1535-41.)。ADモデルの作製には、好適なAPP遺伝子発現と外部刺激によるその発現における考えられる応答が極めて重要である。
【0018】
これまで行われた、多くのAD関連神経病理を作成し、さらにFADをよりよく理解するという努力にもかかわらず、得られたモデルは、SADの原因やその発症を理解するためにそれらを使用することが難しいほど現実から離れている。このため、既存のモデルの欠点を克服するAD動物モデルを提供する必要性がまだある。
【0019】
ヒトAPPタンパク質発現、ひいてはヒトAβペプチド発現は、ネズミタンパク質はその配列が異なることによりアミロイド斑を形成することができないという事実によって、このモデルにおいてアミロイド斑の形成を可能にする。よって、異なる環境因子または病原因子を「hAPPy」に適用することで、APP遺伝子の正確な応答を可能にし、アミロイド斑の形成を可能にする。その疾患に大きく関与すると考えられるこれらの環境因子をそのモデルに適用する可能性により、そのモデルはSADを研究するための極めて重要なツールとなる。
【0020】
発明の詳細な説明
本発明は、一般に、アルツハイマー病(AD)を研究するための非ヒト動物モデルとして使用することができるトランスジェニック非ヒト動物に関する。前記トランスジェニック動物は、そのゲノムに挿入されており、完全ヒトAPP遺伝子(hAPP)のヌクレオチド配列をその調節配列とともに含む、異種ポリヌクレオチド(トランスジーン)を含む(該トランスジーンはhAPP遺伝子の自然環境に存在する他の遺伝子、例えばGABPA、ATPJおよびCYYR1遺伝子を欠いている)こと、そして該トランスジーンの発現がトランスジェニック動物において、ヒトにおけるこの同じhAPP遺伝子の内因性発現パターンと同様の内因性発現パターンを有することを特徴とする。本明細書において、表現「ヒトにおけるhAPP遺伝子の内因性発現パターンと同様のトランスジェニック動物におけるhAPPトランスジーンの内因性発現パターン」などは、該hAPPトランスジーンを含むトランスジェニック動物がhAPP遺伝子を、そのhAPP遺伝子がヒトにおいて発現される同じ組織において実質的に同じ割合で発現するという事実による。
【0021】
本発明によって提供されるトランスジェニック非ヒト動物は完全hAPP遺伝子だけをその調節配列とともに含むトランスジーンを含み、hAPP遺伝子の自然環境に存在する、その発現を妨げる可能性のある他の遺伝子(例えば、GABPA、ATPJおよびCYYR1遺伝子)の干渉を受けないという事実により、ヒトにおけるhAPP遺伝子の内因性発現パターンと同様の内因性発現パターンでの該トランスジェニック動物における該トランスジーンの発現を可能にする(すなわち該トランスジェニック動物はヒトにおける状況をかなりの信頼性で再現する)ため、該トランスジェニック動物は、AD、その進行および発症を研究するためだけでなく、それを引き起こしている要因および刺激を研究するためや該疾患の予防および/または治療のために潜在的に有用な化合物を同定するための非ヒト動物モデルとして使用することができる。
【0022】
よって、一態様では、本発明は、ヒト第21染色体(NCBI35:21:26064934:26642665:1)のヌクレオチド26,064,934とヌクレオチド26,642,665との間に含まれるヌクレオチド配列からなる単離ポリヌクレオチドまたは完全hAPP遺伝子をその調節配列とともに含む該ポリヌクレオチドの断片(以下本発明のポリヌクレオチド)に関する。前記ポリヌクレオチドは完全hAPP遺伝子のヌクレオチド配列をその調節配列とともに含み、hAPP遺伝子の該ヌクレオチド配列の末端に隣接する(flanking)それぞれの近接(adjacent)領域は固有の組換え配列を含み、完全hAPP遺伝子のヌクレオチド配列をその調節配列とともに単離するのにその固有の組換え配列を使用することができる。
【0023】
特定の一実施形態では、前記完全hAPP遺伝子は、その調節配列とともに、ヒト第21染色体(NCBI35:21:26174733:26464809:1)のヌクレオチド26,174,733とヌクレオチド26,464,809との間に含まれる。
【0024】
もう1つの特定の実施形態では、本発明のポリヌクレオチドは、ヒト第21染色体(NCBI35:21:26064934:26573344:1)のヌクレオチド26,064,934とヌクレオチド26,573,344との間に含まれるヌクレオチド配列からなる。前記ポリヌクレオチドは、完全hAPP遺伝子のヌクレオチド配列をその調節配列とともに含み、それらに少なくとも1つの固有の組換え配列を含む領域に隣接される。
【0025】
よって、もう1つの特定の実施形態では、図1に示されるように、前記hAPP遺伝子は、その5’末端において(APP遺伝子転写の始点および方向に関連)第1のヌクレオチド配列に隣接され(flanked)、その3’末端において(APP遺伝子転写の終点および方向に関連)第2のヌクレオチド配列に隣接され、ここで:
該第1のヌクレオチド配列は、ヒト第21染色体(NCBI35:21:26464810:26642665:l)のヌクレオチド26,464,810とヌクレオチド26,642,665との間に含まれるヌクレオチド配列、または少なくとも1つの固有の配列を含むその断片からなり、かつ
該第2のヌクレオチド配列は、ヒト第21染色体(NCBI35:21:26064934:26174732:1)のヌクレオチド26,064,934とヌクレオチド26,174,732との間に含まれるヌクレオチド配列、または少なくとも1つの固有の配列を含むその断片からなる。
【0026】
具体的な一実施形態では、前記第1のヌクレオチド配列は、ヒト第21染色体(NCBI35:21:26464810:26573344:l)のヌクレオチド26,464,810とヌクレオチド26,573,344との間に含まれるヌクレオチド配列、または少なくとも1つの固有の配列を含むその断片を含み、かつ前記第2のヌクレオチド配列は、ヒト第21染色体(NCBI35:21:26064934:26174732:1)のヌクレオチド26,064,934とヌクレオチド26,174,732との間に含まれるヌクレオチド配列、または少なくとも1つの固有の配列を含むその断片を含む。
【0027】
上述の第1および第2のヌクレオチド配列に含まれる(非反復)固有の配列は、本明細書に含まれる実施例に記載のとおり、相同組換えを利用して、その環境から完全hAPP遺伝子をその調節配列とともに単離するのに利用することができる。前記固有の配列は、当技術分野の水準において記載された当業者に公知のプロセスに従って同定することができる。例として、前記プロセスには一般にRepeatMasterプログラムなどの公的バイオコンピューター資源が含まれる[http://woody.embl-heidelberg.of/repeatmask/;Chenna Ramu et al., (2000) cgi-model: CGI Programming Made Easy with Python. LINUX Journal, July, 142-149; Smit, AFA and Green, P. RepeatMasker at http://repeatmasker.genome.washington.edu/cgi-bin/RM2_req.pl]; Jurka, J. 2000 Repbase Update: a database and an electronic journal of repetitive elements. Trend in Genetics 16(9): 418-420)]。
【0028】
特定の一実施形態では、前記第1のヌクレオチド配列は、ヒト第21染色体(NCBI35:21:26571876:26573344:1)のヌクレオチド26,571,876とヌクレオチド26,573,344との間に含まれる固有の配列(本発明ではSeq2と呼ぶ)を含む。
【0029】
もう1つの特定の実施形態では、前記第2のヌクレオチド配列は、ヒト第21染色体(NCBI35:21:26064934:26067221:1)のヌクレオチド26,064,934とヌクレオチド26,067,221との間に含まれる固有の配列(本発明ではSeqBと呼ぶ)を含む。
【0030】
当業者ならば、完全hAPP遺伝子とその調節配列との末端に隣接する前記第1および第2のヌクレオチド配列内に含まれるいずれの固有の配列も同じ目的(完全hAPP遺伝子とその調節配列のそれらの環境からの単離)に利用することができることは分かる。よって、本発明は、本明細書に記載の固有の配列(本発明の特定の一実施形態を構成する)に限定されるのみならず、前記第1および第2のヌクレオチド配列内に含まれる、当技術分野の水準の一部をなす当業者に公知のバイオコンピューターツールを利用して位置づけることができる総ての固有の配列を含む。
【0031】
本発明のポリヌクレオチドは好適なベクター内に含めることができる。よって、もう1つの態様では、本発明は、本発明のポリヌクレオチドを含むベクター(以下本発明のベクター)に関する。前記ベクターは、それゆえ、完全hAPP遺伝子のヌクレオチド配列をその調節配列とともに含み、それらに本発明に記載の固有の配列に隣接される。ベクターの選択は、そのベクターをその後に導入する宿主細胞によって決まる。一例として、前記ポリヌクレオチドを導入するベクターは酵母人工染色体(YAC)、細菌人工染色体(BAC)またはP1由来人工染色体(PAC)であってよい。よって、特定の一実施形態では、本発明のベクターは、形質転換されるゲスト細胞に応じて、YAC、BACまたはPACからなる群から選択される。
【0032】
特定の一実施形態では、本発明のベクターが導入される細胞が酵母である場合、好ましいベクターは、それぞれのアームにその機能を果たすための総ての好適な構造要素、例えばテロメア、セントロメア、自己複製配列および栄養要求性選択マーカーを含むYACである。本明細書に含まれる実施例では、酵母細胞を形質転換するのに用いられる上述の構造要素を有するYACの獲得について記載する。
【0033】
もう1つの特定の実施形態では、本発明のベクターが導入される細胞が細菌である場合には、好ましいベクターは、必要最小限の要素が複製起点とコピー数およびBAC分裂を制御する遺伝子であるBACである。
【0034】
YAC、BACおよびPACの特徴は当業者には公知である。前記種類のベクターについての詳細な情報は、例えば、Giraldo および Montoliuによって提供されている[Giraldo, P. & Montoliu L., 2001 Size matters: use of YACs, BACs and PACs in transgenic animals, Transgenic Research 10(2): 83-110]。
【0035】
よって、特定の一実施形態では、本発明は、その一方の末端において第3のヌクレオチド配列に隣接され、もう一方の末端(反対端)において第4のヌクレオチド配列に隣接される本発明のポリヌクレオチドを含む酵母を形質転換するのに有用なベクターに関し、この場合:
−前記第3のヌクレオチド配列は、(i)酵母細胞において機能的なテロメアおよび少なくとも(ii)1つの栄養要求性選択マーカーを含み、かつ
−前記第4のヌクレオチド配列は、(i)酵母細胞において機能的なセントロメア、酵母細胞において機能的なテロメアおよび少なくとも1つの栄養要求性選択マーカーを含む。
【0036】
酵母において機能的な、事実上いかなるテロメアおよびセントロメアも本発明において用いることができる。酵母において機能的な前記テロメアおよびセントロメアの限定されない例示的な例には、本明細書に含まれる実施例に記載されているものが含まれる。
【0037】
同様に、事実上いかなる栄養要求性選択マーカーも本発明において用いることができる。前記栄養要求性選択マーカーの限定されない例示的な例には、TRP1、LEU2、LYS2、HIS3、HIS5、TRP1およびURA3が含まれる。
【0038】
本発明のベクターは、当業者に公知の従来の方法によって得ることができる[Sambrok et al., 1989 "Molecular cloning, a Laboratory Manual", 2nd ed., Cold Spring Harbor Laboratory Press, N.Y. Vol 1-3]。
【0039】
本発明のベクターは、例えば、酵母細胞、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)などの真核細胞、または例えば、細菌、大腸菌(Escherichia coli)などの原核細胞を形質転換するのに用いることができる。
【0040】
よって、もう1つの態様では、本発明は、本発明のポリヌクレオチド、または本発明のベクターを含む細胞(以下本発明の細胞)に関する。このような細胞は、当業者に公知の従来の方法によって得ることができる[Sambrok et al., 1989 "Molecular cloning, a Laboratory Manual", 2nd ed., Cold Spring Harbor Laboratory Press, N.Y. Vol 1-3]。
【0041】
記載する本発明のポリヌクレオチド、ベクターまたは細胞は、そのゲノムに、完全hAPP遺伝子のヌクレオチド配列がその調節配列とともに挿入されたトランスジェニック非ヒト動物を得るのに用いることができる。
【0042】
よって、もう1つの態様では、本発明は、そのゲノムに挿入された本発明のポリヌクレオチドを含むトランスジェニック非ヒト動物(以下本発明のトランスジェニック非ヒト動物)に関する。結果として、本発明のトランスジェニック非ヒト動物のhAPP遺伝子の内因性発現パターンは、この同じhAPP遺伝子がヒトにおいて示す発現パターンと同様である。いずれかの理論に拘束される訳ではないが、これは、主に、その遺伝子がそれ自体の調節配列により調節されるという事実によるものと思われる。このことは、本発明のトランスジェニック非ヒト動物におけるhAPP遺伝子発現を、ヒトにおいてその発現を誘導する同じ因子によって誘導することを可能にし、そのトランスジェニック非ヒト動物をヒトにおいて起こるADの現象および特徴を確実に再現する動物モデルにする。
【0043】
本発明の特定の一実施形態では、本発明のトランスジェニック非ヒト動物は、哺乳類、好ましくは齧歯類、より好ましくはマウスまたはラットである。
【0044】
本発明のトランスジェニック非ヒト動物は、当業者による技術水準において公知のものの任意の遺伝的背景を有していてよい、すなわち該トランスジェニック非ヒト動物は、野生型(wt)動物由来のものであってよく、またはADと直接または間接的に関係している任意の分子マーカーを組み込んでいる遺伝的背景を有する非ヒト動物由来のものであってよい。よって、特定の一実施形態では、本発明のトランスジェニック非ヒト動物は、hAPP遺伝子の他に、ADの発症と直接または間接的に関係している1つ以上の遺伝子を発現することができる。ADの発症と直接または間接的に関係している事実上いかなる分子マーカーも用いることができる。ADの分子マーカーとして認識されている前記遺伝子の限定されない例示的な例には、プレセニリン1(PS1)遺伝子、プレセニリン2(PS2)遺伝子およびアポリポタンパク質E(APOE)遺伝子のε4対立遺伝子が含まれる。
【0045】
よって、特定の一実施形態では、本発明のトランスジェニック非ヒト動物のゲノムは、本発明のポリヌクレオチドの他に、1つ以上のAD分子マーカーを含む。限定されない例として、前記AD分子マーカーは、PS1、PS2遺伝子、およびAPOE遺伝子のイプシロン4(ε4)対立遺伝子からなる群から選択される。
【0046】
ADを発症しやすい素因に関連する他の遺伝因子には、α2−マクログロブリン遺伝子、超低密度リポタンパク質受容体遺伝子、リポタンパク質受容体関連タンパク質LRPの遺伝子、Tauタンパク質の遺伝子、ブチルコリンエステラーゼKの遺伝子、カテプシンDの遺伝子、インターロイキンの遺伝子およびコリンアセチルトランスフェラーゼの遺伝子が含まれる。
【0047】
本発明の非ヒトマウスの遺伝的背景は、前記AD分子マーカーの対立遺伝子変異体、突然変異体および多型をさらに含み得る。
【0048】
完全ヒトAPP(hAPP)遺伝子のヌクレオチド配列がその調節配列とともに本発明のトランスジェニック非ヒト動物のゲノム中に存在することにより、該遺伝子を、この同じ遺伝子がヒトにおいて示す発現パターンと同様の内因性発現パターンで発現させることを可能にするため、該トランスジェニック動物を、ADを研究するためのトランスジェニック非ヒト動物モデルとして使用することができる。前述のとおり、本発明のトランスジェニック非ヒト動物は、必要に応じて、さらなるAD分子マーカーも有していてよい。この総てにより、一般にはADだけでなく、異なる異型アルツハイマー病、例えばFADおよびSADを研究することも可能にする。
【0049】
よって、もう1つの態様では、本発明は、ADの研究における本発明のトランスジェニック非ヒト動物の使用に関する。特定の一実施形態では、ADはFADおよびSADから選択される。
【0050】
もう1つの態様では、本発明は、該疾患の病因、原因、誘発因子、その疾患の確立につながる環境的および/または生理病理学的攻撃または傷害を研究するためのADモデルとしての本発明のトランスジェニック非ヒト動物の使用に関する。
【0051】
その限定されない例示的な例には、年齢、頭蓋外傷、女性、ヘルペス型(HSV1)ウイルスおよび細菌、細菌、例えば、クラミジア・ニューモニエ(Chlamydia pneumoniae)などのような病原体の作用、脳内でのアルミニウム、亜鉛型金属の蓄積などが含まれる。
【0052】
もう1つの態様では、本発明は、ADの治療および/または予防のために潜在的に有用な化合物をスクリーニングし、発見し、同定し、評価し、および妥当性を立証するための本発明のトランスジェニック非ヒト動物の使用に向けられる。前記化合物は、いかなる性質の化合物であってもよく、例えば、単離されたまたは1種以上の異なる化合物と混合された化学的、生物学的、微生物学的化合物などであってよく、既知または未知組成および構造を有する化合物、任意の既知治療用途を有する医薬製剤、生物学的製剤、微生物学的製剤など、例えば、有機または無機化学化合物、ペプチド、タンパク質、核酸、抽出物などが含まれる。
【0053】
本発明のトランスジェニック非ヒト動物、例えば、マウスは、それらの先祖の遺伝子型および表現型特徴を有する非ヒト動物を得るために、同じ種の他の非ヒト動物との交配を用いてさらに使用することができる。本発明の非ヒト動物と交配させることができる非ヒト動物の例には:
−APP遺伝子、あるいはADと直接または間接的に関係している他の遺伝子についてのノックアウト非ヒト動物;
−ADに関与する遺伝子の対立遺伝子変異体、多型または突然変異体を発現する非ヒト動物、あるいは
−ADモデルとして使用することができる他の非ヒト動物
が含まれる。
【0054】
特定の一実施形態では、ADと直接または間接的に関係している遺伝子は、PS1、PS2遺伝子、およびAPOE遺伝子のイプシロン4(ε4)対立遺伝子、またはその疾患に関連する他の遺伝的素因因子、例えばα2−マクログロブリン遺伝子、超低密度リポタンパク質受容体遺伝子、リポタンパク質受容体関連タンパク質LRPの遺伝子、Tauタンパク質の遺伝子、ブチルコリンエステラーゼKの遺伝子、カテプシンDの遺伝子、インターロイキンの遺伝子およびコリンアセチルトランスフェラーゼの遺伝子からなる群から選択される。
【0055】
もう1つの特定の実施形態では、本発明のトランスジェニック非ヒトマウスは、数回の交配後に、hAPPトランスジーンだけを発現するマウスが得られるように、ネズミAPP(mAPP)遺伝子についてのノックアウトマウスと交配させることができる。
【0056】
よって、もう1つの態様では、本発明は、hAPPトランスジーンだけを発現し、mAPP遺伝子を発現しないトランスジェニック非ヒト動物を得るための本発明によるトランスジェニック非ヒト動物の使用に関する。
【0057】
もう1つの態様では、本発明は、ADの治療および/または予防のために潜在的に有用な化合物を同定するための方法であって、
a)本発明のトランスジェニック非ヒト動物に候補化合物を投与すること;
b)工程(a)の該トランスジェニック非ヒト動物における、hAPP遺伝子の発現パターンおよび/またはhAPPタンパク質産生および/またはβ−アミロイド(Aβ)ペプチド産生を決定すること、および
c)該トランスジェニック非ヒト動物において(i)神経組織におけるhAPP遺伝子発現レベルの低下、(ii)hAPPタンパク質産生および/もしくは蓄積の減少、ならびに/または(iii)Aβペプチド産生および/もしくは蓄積の減少を引き起こす該候補化合物を選択すること
を含む方法に関する。
【0058】
神経組織におけるhAPP遺伝子発現レベル、hAPPタンパク質産生および/もしくは蓄積レベル、ならびにAβペプチド産生および/もしくは蓄積レベルは、定量RT−PCR、ノーザンブロット、ウエスタンブロットなどのような当業者に公知の従来の技術によって測定することができる。hAPP遺伝子発現レベルまたはhAPPタンパク質蓄積レベルの好ましい範囲は、それぞれ、図24および図26で見ることができる。
【0059】
特定の一実施形態では、対照レベルと比べて、hAPP遺伝子発現レベルの低下、またはhAPPタンパク質産生および/もしくは蓄積レベルの減少、またはAβペプチド産生および/もしくは蓄積の減少、を引き起こす前記候補化合物が選択される。前記化合物は、上記化合物のいずれであってもよい。
【0060】
本発明のトランスジェニック非ヒト動物は、当業者に公知の任意の遺伝子導入法例えばマイクロインジェクション、エレクトロポレーション、粒子衝撃、細胞形質転換とその後のクローニング(形質転換に成功した細胞の細胞核を除核卵に移し、受容体女性に移植する)、生殖体形質転換(卵母細胞または精母細胞に遺伝子を導入し、形質転換された生殖体を受精に使用し、完全動物を作り出すこと)および/または卵細胞質内精子注入法(ICSI)によって得ることができる。
【0061】
特定の一実施形態では、本明細書に含まれる実施例に記載のとおり、用いられる遺伝子導入法は卵細胞質内精子注入法に基づく。前記技術については国際特許出願WO 2005/098010に詳細に記載されている。
【0062】
これまでに記載された遺伝子導入法によって得られる動物は、本発明のトランスジェニック非ヒト動物の特徴的な遺伝子型および表現型を有し、それらは、それらのゲノムに挿入された、完全hAPP遺伝子のヌクレオチド配列とその調節配列とを含み、該hAPP遺伝子は、この同じ遺伝子がヒトにおいて示す発現パターンと同様の内因性発現パターンを有する。
【0063】
本発明のトランスジェニック非ヒト動物のように、これまでに記載された遺伝子導入法によって得られるトランスジェニック非ヒト動物もまた、それらの先祖の遺伝子型および表現型特徴を有するトランスジェニック非ヒト動物を得るために、同じ種の他の非ヒト動物と交配させ、ADを研究するためのモデルとして使用することができる異なる非ヒト動物を生み出すことができる。
【0064】
加えて、本発明に記載の遺伝子導入法によって得られるトランスジェニック非ヒト動物の子孫も、ヒトにおけるhAPP遺伝子タンパク質のパターンと同様のhAPP遺伝子の内因性発現パターンを有し得る。
【0065】
本発明のもう1つの態様は、それゆえ、本発明に記載の遺伝子導入法に従って得ることができる、ヒトにおける前記遺伝子の発現パターンと同様のまたは同等の発現パターンでヒトAPP遺伝子を内因的に発現するトランスジェニック非ヒト動物の子孫に関する。
【0066】
次の実施例により本発明を説明するが、本発明を限定する意味に解してはならない。
【実施例】
【0067】
完全ヒトAPP遺伝子を、ヒトにおける該遺伝子の発現パターンと同等の発現パターンで内因性発現するトランスジェニック非ヒト動物の獲得
【0068】
材料および方法
1.プラスミドの作製
1.1 一般的なクローニング
通常の分子生物学技術(Sambroock, J., 1989, 上記; Ausubel FM, B. R., Kingston RE, Moore DD, Seidman JG, Smith JA and struhl, K. (1999). Current Protocols in Molecular Biology, John Wiley and Sons.)と本発明者らの研究室で改良した技術(Montoliu, 1997 Lab protocols. Generation of a transgenic mouse)を用いて、この研究に必要なベクターを作製した。
【0069】
1.2 DNAの電気泳動
アガロースゲルでの電気泳動を用いて、異なるDNA断片の分離、同定および定量を実施した。100bp〜12kbの様々な分子を分離することができる、0.5〜2%間に含まれる濃度のUltra Pure Agarose(GIBCO-BRL)を用いた。
【0070】
水平型Ecogenセル(6.5x8cmおよび11x15cm)を使用し、それらのセル内でSegainvex電源を用いて電流量5V/cmを適用した。ゲルは、TAEバッファー(40mM Tris−Acetate、2mM EDTA(エチレンジアミン四酢酸) pH8(Merck))中に入れ、臭化エチジウム(Roche)を0.5μg/mlで加えて調製した。
【0071】
1.3 DNAの定量
必要な精度および予測濃度に応じて、異なる方法を用いてDNAの定量を実施した。予測濃度範囲が0.05〜2μg/μl間に及ぶサンプルにおけるDNA濃度の推定を、波長260nmでのDNA吸光度の分光光度測定に基づき、SHIMADZU社製UV−1601型分光光度計において行った。測定は会社の使用説明書に従って実施し、波長260nmでの光学濃度(OD260)の一単位は50μg/mlの二本鎖DNAに相当することを考慮して、DNA濃度を次の方程式に従って算出した:
【数1】

【0072】
Hoefer DyNA Quant 200 Fluorometer(Amersham Pharmacia Biotech)を使用して、濃度が0.01〜0.05μg/μl間であると推定されるサンプルを評価した。この方法では、ビスベンズアミド(Hoeschst 33258)の蛍光を測定し、このビスベンズアミドの分子は二本鎖DNAと結合する能力を有し、365nmの紫外線を用いてそれを励起すると蛍光光度計により検出することができる蛍光を発する。この方法では、遊離ヌクレオチドまたはRNAによるバックグラウンドが抑制されることから、小濃度範囲で測定するのに非常に有用である。これら総ての測定をnanodrop(NanoDrop、ND−1000分光光度計)を用いて検証した。
【0073】
1.4 ポリメラーゼ鎖増幅反応(PCR)
組換えを解析するため、さらにトランスジェニックマウスへのその組込みを確認するための特異的YAC領域のクローニングおよび増幅用のDNA断片を、PCRを用いて得た。
【0074】
PCRでは各増幅断片に特異的な条件が必要である。混合物の反応濃度は、温度、増幅時間およびサイクルと同様に、それぞれの反応によって変化する。総ての反応についての標準的な出発条件は以下のとおりである:1.5mM MgCl2(Biotools)、200μMデオキシヌクレオチド[dATP、dCTP、dTTPおよびdGTP(Roche)]、1μMの特異的オリゴヌクレオチド対(Isogen)、1x反応バッファー(Biotools)および1単位のTaqポリメラーゼ(Biotools)、合計量25μl。反応はMJ Researchys社製PCT−100モデルサーマルサイクラーにおいて0.2mlチューブ(MJ Research)中で行った。使用するプログラムは増幅する断片に応じて変更した。それらのプログラムは、一般に、95℃で5分間の最初のDNA変性、その後3温度のサイクル、95℃で30秒、オリゴヌクレオチドハイブリダイゼーション温度に応じて55〜65℃間で30秒および72℃で1分の35回の反復で構成された。断片を完全増幅するために、それらを最後に72℃で10分に供した。
【0075】
1.4.1 酵母コロニーのPCR
異なる相同組換えを研究するために、酵母コロニーを解析した。コロニーの直接PCRを行うために、コロニーの増殖を防ぎ、そこからDNAを抽出して、コロニーをPCRチューブ(0.2ml)に移し、そのPCRチューブ中でそれを50μlのザイモリアーゼ(20U/ml)中に再懸濁し、室温で10分間インキュベートした。それらのチューブをEppendorf社製5417c卓上遠心機で5,000r.p.mにて遠心分離し、上清を除去し、細胞を95℃で5分間加熱した。その沈殿物を15μlの水中に再懸濁し、5μlを用いて、PCRを行った。
【0076】
1.5 アガロースゲルからのDNAの精製および抽出
10kb未満のDNA断片を、TAEバッファー中のアガロースゲルを用いて精製した。ゲル中で断片を分離した後、それらを紫外線灯(Upland)を用いて確認した。その後、滅菌ナイフでバンドを切り取り、Eppendorf社製チューブに入れた。QIAagen社製Qiaquick gel extraction kitを、そのプロトコールに記載の使用説明書に従って用いてゲルからDNAを抽出した。
【0077】
1.6 原核細胞
1.6.1 細菌株および培養
組換えプラスミドを市販の大腸菌株中で増幅した。サイズが約5kbであるプラスミドを増幅し、DH5α細胞(Gibco)に形質転換し、一方、10kbに近いサイズのプラスミドを増幅し、TOP10F’細胞(Stratagen)に形質転換した。
異なるプラスミドの増幅のための細菌増殖は、LB(Luria−Bertani)液体培地中、37℃で少なくとも8時間、攪拌しながら(250r.p.m.)、アンピシリン(Sigma)の存在下で、終濃度50μg/mlで行った。コロニーのスクリーニングは、形質転換体を寒天およびLB培地を入れたペトリ皿で、アンピシリンの存在下で増殖させた後、カラースクリーニングを実施し得る場合にはX−galおよびIPTGを用いて、実施した。
【0078】
1.6.2 コンピテント細菌の調製および形質転換
コンピテントDH5α細胞を、塩化カルシウム法を用いて調製し、熱ショック法を用いて形質転換した。
【0079】
大型プラスミドは市販のTOP10F’株(Stratagen)を用いて形質転換した。エレクトロポレーションは、予め氷冷しておいた直径2mmのエレクトロポレーション用セル(Bio Rad)中で、Bio-Rad社製Gene Pulserを会社の指示に従って用いて、25μF、2.5KVおよび20Ωの条件でパルスを与えて実施した。
【0080】
1.6.3 プラスミドDNAの精製
小規模プラスミドDNA精製を、抗生物質を含むLB培地2mlに問題のコロニーを植菌して行った。これらの培養物を37℃で少なくとも8時間、攪拌しながら(250r.p.m.)増殖させた。プラスミドDNAはアルカリ溶菌法を用いて抽出した。より高い濃度および量でのプラスミドDNAの調製は、大量細菌(100〜500ml)培養を利用して、市販のQiagen maxi kits(Qiagen)とFlexiprep Kits(Amersham Pharmacia)を各キットの具体的なプロトコールに従って用いて行った。
【0081】
2.オリゴヌクレオチド
YACにおけるAPP遺伝子のフランキング5’および3’領域と、最近コード遺伝子領域までのそれらの距離の範囲を定めるのに用いられるオリゴヌクレオチドは:
APP3'-B-U 5'-atcctaagaggaagggatcttaagg-3' [配列番号:1]
APP3'-B-L 5'-gggagaacaaacatacctcactagc-3' [配列番号:2]
APP3'-A-U 5'-ccagcatgttttccaaagtatgag-3' [配列番号:3]
APP3'-A-L 5'-tgaaatgtccccacactgatcactg-3' [配列番号:4]
APP3'-1-U 5'-gtatagtatggatggttcaaacagagccta-3' [配列番号:5]
APP3'-1-L 5'-aaggtaaagctctagaatctatcagtgcct-3' [配列番号:6]
APP3'-2-U 5'-tttagaagagctcagtaagaatccacattt-3' [配列番号:7]
APP3'-2-L 5'-cattttgtcgttactagtaccattggtttc-3' [配列番号:8]
APP5'-1-U 5'-ctcaaaaacagcaacacagatgtgc-3' [配列番号:9]
APP5'-1-L 5'-gagcttaaacaccttcctctgc-3' [配列番号:10]
APP5'-2-U 5'-tggggagaggaatggaatttggagc-3' [配列番号:11]
APP5'-2-L 5'-gtcatttccagaaaaagcccactgc-3' [配列番号:12]
APP5'-3-U 5'-tcaaggacatggagattgggcaggc-3' [配列番号:13]
APP5'-3-L 5'-ggggtaacaggttgccggtcatatg-3' [配列番号:14]
APP5'-4-U 5'-caccagaccatgatgtctcagtagc-3' [配列番号:15]
APP5'-4-L 5'-ctgcaaaacagccctcatattctgc-3' [配列番号:16]
APP5'-5-U 5'-tagcatcctttgctaagccagttgc-3' [配列番号:17]
APP5'-5-L 5'-gcttgttattggaggttccagcacg-3' [配列番号:18]
APP5'-6-U 5'-tccaacgggggagtgagtgaaaggc-3' [配列番号:19]
APP5'-6-L 5'-gcctcactcctgcaaacgtgcccaa-3' [配列番号:20]
APP5'-7-U 5'-tctttcttccaccttggtatcctgc-3' [配列番号:21]
APP5'-7-L 5'-caggatgtctctggatttttactcg-3' [配列番号:22]
である。
【0082】
APP遺伝子のエキソン、ならびにGABPA遺伝子の存在を確認するのに用いられるオリゴヌクレオチドは以下である:
APP-Ex1-U 5'-agtttcctcggcagcggtagg-3' [配列番号:23]
APP-Ex1-L 5'-ccagcaggagcagtgccaaac-3' [配列番号:24]
APP-Ex2-U 5'-aagaccgggctgattcctaa-3' [配列番号:25]
APP-Ex2-L 5'-tccaacgtgaattgctagcc-3' [配列番号:26]
APP-Ex3-U 5'-cccaagcattttggataagg-3' [配列番号:27]
APP-Ex3-L 5'-cctctttttcttccctcaag-3' [配列番号:28]
APP-Ex4-U 5'-ttgattgggttgcttaggca-3' [配列番号:29]
APP-Ex4-L 5'-tgttgcctcaaaatacccct-3' [配列番号:30]
APP-Ex5-U 5'-ctaccactcactgttttctc-3' [配列番号:31]
APP-Ex5-L 5'-gcagagaccttttcagtgat-3' [配列番号:32]
APP-Ex6-U 5'-tgccaaaattccatatggacg-3' [配列番号:33]
APP-Ex6-L 5'-gggatttgccaagcagcatat-3' [配列番号:34]
APP-Ex7-U 5'-ccactgggaggattaaaaga-3' [配列番号:35]
APP-Ex7-L 5'-gagaagtggacagaaatgtg-3' [配列番号:36]
APP-Ex8-U 5'-tgtcagtggactcgtgcatt-3' [配列番号:37]
APP-Ex8-L 5'-catctcaagctgtctggcaa-3' [配列番号:38]
APP-Ex9-U 5'-catgtcttcagcaccaactg-3' [配列番号:39]
APP-Ex9-L 5'-caaactgtgcccacacagta-3' [配列番号:40]
APP-Ex10-U 5'-cagataggaaggggtatgta-3' [配列番号:41]
APP-Ex10-L 5'-ggagcaaatataaggcagga-3' [配列番号:42]
APP-Ex11-U 5'-tgatgagggttggagagtgca-3' [配列番号:43]
APP-Ex11-L 5'-caagatggaatggacaggggt-3' [配列番号:44]
APP-Ex12-13-U 5'-cctcgtcacgtgttcaatat-3' [配列番号:45]
APP-Ex12-13-L 5'-caacttcatcctgaatctcc-3' [配列番号:46]
APP-Ex14-U 5'-cacgtgaaagcagttgaagt-3' [配列番号:47]
APP-Ex14-L 5'-ttgccacctatacaatggag-3' [配列番号:48]
APP-Ex15-U 5'-ctggcacatcaatagcgata-3' [配列番号:49]
APP-Ex15-L 5'-actcggaacttgggaaatga-3' [配列番号:50]
APP-Ex16-U 5'-taaaggcagcagaagcctta-3' [配列番号:51]
APP-Ex16-L 5'-gctcagcctagcctatttat-3' [配列番号:52]
APP-Ex17-U 5'-accagttgggcagagaatat-3' [配列番号:53]
APP-Ex17-L 5'-cttgagcagaatattcacgg-3' [配列番号:54]
APP-Ex18-U 5'-cttggtaattgaagaccagc-3' [配列番号:55]
APP-Ex18-L 5'-cttttgacagctgtgctgta-3' [配列番号:56]
GABPA-Ex-U 5'-tattattacgatggggacat-3' [配列番号:57]
GABPA-Ex-L 5'-gtaaagcaacatctaaagca-3' [配列番号:58]
組換えに用いた異なるクローニングベクターの配列決定に用いられるオリゴヌクレオチドは:
SV40A-2 5'-gtgaaggaaccttacttctgtggtg-3' [配列番号:59]
Sp6プロモーター 5'-atttaggtgacactata-3' [配列番号:60]
T7プロモーター 5'-taatacgactcactataggg-3' [配列番号:61]
である。
【0083】
酵母URA遺伝子のクローニングに用いられるオリゴヌクレオチドは:
URA3-U 5'-gcccagtattcttaacccaaactgca-3' [配列番号:62]
URA3-L 5'-cttccacccatgtctctttgagcaa-3' [配列番号:63]
である。
【0084】
栄養要求性YAC配列を検出するのに用いられるオリゴヌクレオチドは:
TRP1 5'-gcccaatagaaagagaacaattgacc-3' [配列番号:64]
TRP2 5'-acacctccgcttacatcaacacc-3' [配列番号:65]
LYS1 5'-accaagccagcatctgtatcacc-3' [配列番号:66]
LYS2 5'-gccaaatccatccacttctcatc-3' [配列番号:67]
である。
【0085】
サザンブロット解析用のプローブを作製するのに用いられるオリゴヌクレオチドは:
プローブAPP-seq2-U 5'-tgaatcttggcatttgggcc-3' [配列番号:68]
プローブAPP-seq2-L 5'-cctgtaacttgccctcatag-3' [配列番号:69]
プローブAPP-seqA-U 5'-gaaacgagttgaaaggcacag-3' [配列番号:70]
プローブAPP-seqA-L 5'-tttacccttccatgaagtccc-3' [配列番号:71]
プローブAPP-seqB-U 5'-ggcttgtaaattaaactctgaagc-3' [配列番号:72]
プローブAPP-seqB-L 5'-cctcagtggctgttttgagagcat-3' [配列番号:73]
である。
【0086】
3.酵母人工染色体の修飾
3.1 真核細胞
酵母において実施した総ての試験はS.セレビシエ AB1380株において実施し、その代謝特性は:Mata、ura3−52、trp1、ade2−1、lys2−1、his5およびcan1−100であった。使用したオリジナルクローンは、Washington Universityで購入し(Human Yeast Artificial chromosome Clone)、そのクローンはYAC B142F9を含むものであった(完全ヒトAPP遺伝子を含む650KbのYAC)。
【0087】
YAC担体酵母は、時々起こるYAC消失を防ぐために独立栄養セレクター(autotrophic selectors)を加えた液体および固体培地で増殖させた。液体培養物を30℃で2〜3日間、攪拌しながら(200r.p.m.)増殖させ、一方、酵母を30℃で4〜5日間増殖させて、プレートでコロニーを得た。
【0088】
選択培地はD+Glucose(Merck)、YNB(Yeast Nitrogen Base w/o Amino Acids)(Difco)および対応アミノ酸(Serva)を加えて調製した。
【0089】
3.2 酵母における相同組換え
酵母細胞の形質転換を2つの方法により実施し、それらの方法ではそれぞれ非常に異なる結果を生じた:酢酸リチウム(LiAc)法(Markie, D. (1995). YAC Protocols_[Please complete the reference]; Alison, A. G., D; Kaiser, C; Stearns, T (1997). Methods in Yeast Genetics, Cold Spring Harbor Laboratory Press.)および酵母細胞エレクトロポレーション(Becker, D. M. and L. Guarente (1991). "High-efficiency transformation of yeast by electroporation." Methods Enzymol 194: 182-7)。
【0090】
3.3 DNA−DNAハイブリダイゼーション(サザンブロット)を利用した解析
サザンブロット技術を利用して陽性クローンの解析を実施した。2〜4μg間のゲノムDNAを、4mMスペルミジンを含む1x消化バッファー中40〜60Uの対応酵素[Eco RV、Sca I、Bgl II、Bam HI、Hind III、Sal IおよびEco RI]で消化した。その消化物を酵素切断温度で12〜14時間インキュベートした。それらの消化産物を水平型Horizonセル(20x25cm)(Life Technologies)内で0.8%アガロースゲルにおいて電気泳動により分離した。
【0091】
キャピラリーを用いてDNAをHybond(商標)−Nナイロン膜(Amersham)に転写した。そのためには、SSC 20Xトランスファーバッファー[3M NaCl、0.3Mクエン酸ナトリウム]中で14〜16時間を要した。ゲルは、DNAの脱プリン化のための0.25N HClでの洗浄、そしてDNAの中和および変性のための0.4Mでの15分洗浄2回を行って予め処理した。
【0092】
一度、DNAが膜に転写されたら、CL−1000 Ultraviolet Crosslink(UVP-Stratagen)を用いて紫外線照射により、パルス70,000mJ/cm2を2回与えてそれを固定した。
【0093】
酵素制限によりまたはPCRを用いて単一コピープローブを得、50ngのDNA、25μCiのα−32P−dCTP(Amersham)およびHigh Prime kit(Roche)を会社の使用説明書に従って用いて放射能標識した。標識プローブから遊離ヌクレオチドを除去するために、かかるプローブをProbe Quant G50カラム(Amersham)に通して精製した。プローブの活性をW 1414シンチレーションカウンター(Beckmann)で定量し、およそ1x106cpm/μlの活性を得た。
【0094】
ハイブリダイゼーションを行い、膜を対応するプローブ(配列番号68、配列番号69、配列番号72および配列番号73)とともに65℃で14〜16時間インキュベートした。
【0095】
ハイブリダイズされたフィルターを、増感スクリーン(Miersham)と写真用フィルム(X-Omat, Kodak)の入った露光カセット内で−80℃で数日間露光した。あるいは、フィルターをPhosphorlmagerのカセット(Molecular Dynamics)内で室温で1〜2日間露光し、そのシグナルをIQMac v 1.2プログラム(Molecular Dynamics)を用いて解析した。
【0096】
3.4 酵母細胞を含むアガロースブロックの小規模調製
YACの修飾を解析するために、全細胞を含むアガロースブロックを調製し、そうすることによって、染色体を無傷のまま保つこと、そして染色体によって受けるサイズ変化をパルスフィールド電気泳動を利用して確認できることを実現した。次のプロトコールに従った:
【0097】
10mlの選択培地に、解析する酵母クローンを接種し、それを30℃で攪拌しながら(200r.p.m.)飽和まで増殖させた。その培養物を3,000r.p.mにて室温で5分間遠心分離し、上清を廃棄し、沈殿物を10mlの50mM EDTA pH8中に再懸濁し、再度3,000r.p.mにて室温で5分間遠心分離した。この際、その沈殿物を300μlの溶液I(1Mソルビトール、20mM EDTA pH8、14mM β−メルカプトエタノールおよび2mg/mlのzymolyase 20T)中に再懸濁し、40℃に温度調整した。一度、温度調整したら、10mg/mlのzymolyase 20T溶液50μlを加え、その後、40℃に温度調整した500μlの溶液II(2%のアガロースを含有する溶液I、Sea Plaque)を加えた。それを、先端を切った青色チップをつけたピペットを用いて混合し、それを、先端を切った黄色チップをつけた200μlピペットを用いて型に分配した。各型には80μlを加えた。アガロースが固化し得るようにそれらを氷中で10分間インキュベートし、一度固化したら、それらのキューブをまた、4mlの溶液III(1Mソルビトール、20mM EDTA pH8、10mM Tris pH7.5、14mM β−メルカプトエタノールおよび2mg/mlのzymolyase 20T)を入れた10mlチューブに移し、37℃で1〜3時間、ゆっくりと攪拌しながらインキュベートした。その後、溶液IIIを5mlの溶液IV(1%ラウリル硫酸塩、100mM EDTA pH8、10mM Tris pH8)と交換し、それを再度37℃で1時間インキュベートした。続いて、溶液IVを新しい溶液と置き換え、37℃で一晩インキュベートした。翌日、キューブを5mlの20%NDS(0.5M EDTA、1mM Trizma base、34mM N−ラウリルサルコシン、pH9)で室温で2時間洗浄し、それらを新しい溶液と交換した。それらは4℃で保存し得る。
【0098】
3.5 パルスフィールドゲル電気泳動(PFGE)
総てのゲルを0.5xTBEバッファー(100mM Tris−Borate、1mM EDTA pH8.3)中1%のアガロース LMP Sea Plaqueに調製した。パルスフィールド電気泳動システムは、Gene Navigator System(Amersham)のものであった。酵母マーカー PFGEマーカーII(Roche)を分子量マーカーとして使用し、それらのマーカーはS.セレビシエ YPH 755のアガロースキューブに対応する(染色体 90kb、220kb、280kb、360kb、450kb、560kb、610kb、700kb、770kb、810kb、850kb、920kb、960kb、1,010kb、1,100kb、1,600kbおよび2,200kb)。
【0099】
最初のYACはもともとサイズがおよそ650kbであったが、修飾後の最終的なサイズは520kbであった。そのため、実施した総てのパルスフィールド電気泳動ではこのサイズの染色体を最適に分離するプログラムを使用した。このプログラムは200Vで26時間続き:最初の12時間は50秒のパルス、残る14時間は100秒のパルスであった。一度、ゲルが泳動し終えたら、それを臭化エチジウム(EtBr)(0.5μg/ml)を含む電気泳動バッファー1〜2l中で30分間染色した。
【0100】
得られたYACを解析するために、染色体をHybond(商標)−Nナイロン膜(Amersham)に転写して、解析した。転写は、いくつかのわずかな違いを除いて、サザンブロットに関してすでに記載した転写と同様に行った:大型DNA分子を破壊するために、転写の前にゲルを紫外線(254nm)での5分の処置に供した。続いて、それらのそれぞれを0.25N HCLとともに20分間で2回、次いで、0.4M NaOHとともに30分間で1回インキュベートした。キャピラリーによる転写状態を少なくとも48時間保った。固定とハイブリダイゼーションの両方を、第(3.3)節ですでに記載したとおりに行った。
【0101】
4.マイクロインジェクションのためのYACの調製
4.1 酵母細胞を含むアガロースブロックの大規模調製
十分な量のDNAを単離し、本発明のトランスジェニックマウスを作製するために、高細胞密度のアガロースキューブを調製した。そのためには、次のプロセスに従った:
【0102】
200mlの選択培地に、YACを有する酵母を接種して、飽和まで増殖させた。それを600gにて5分間遠心分離し、残留培地を除去した。それを、オリジナル培養物100mlにつき40mlの50mM EDTA(pH8)中に再懸濁し、それを再度遠心分離し、上清を廃棄した。酵母沈殿物の重量を測定し、密度1と仮定すれば、酵母を体積50%として溶液Iを加え、その混合物を37℃に温度調整した。一度、温度調整したら(約30秒)、同じ量の溶液IIを加え、それをゆっくりと攪拌し、4℃で冷却されていた型に80μlを分配した。それを10分間ゲル化させた。それらのキューブを、1mlのキューブにつき8mlの溶液IIIを入れたチューブに入れかえ、それを37℃で2時間、攪拌しながらインキュベートした。溶液IIIを同量の溶液IVと置き換え、それを37℃で1時間インキュベートした。次いで、それを新しい溶液と置き換える必要があり、それを37℃で一晩放置した。翌日、それらのキューブを、1mlのキューブにつき8mlの20%NDSで室温で2時間洗浄した。洗浄を繰り返し、それらをロードする準備ができた。
【0103】
4.2 パルス磁場を利用したYACの精製
この場合の精製産物はトランスジェニックマウスを作製するのに直接使用されることから、YACの精製のためのパルス磁場の調製では一連の検討が必要であった。常に滅菌済みの使い捨て材料を使用して、高度無菌操作条件を用いた;EtBrは高い突然変異誘発能力を有するため、DNAが抽出されるゲルの領域がEtBrと接触しないようにし、YACを含む任意の溶液を、滅菌チップをつけてピペットで取った。滅菌チップは先端で切り、DNA分子の破損を防いだ。
【0104】
9つのキューブが入るように、パルス磁場の7つの通常のウェルを合わせることによって大型ウェルを調製した。マーカーIIおよび1/2、1/4、1/8および1/16キューブ画分を連続したウェルにロードした。
【0105】
このサイズ範囲での分離のために選択したプログラム(200ボルトにおいて26時間;最初の12時間は50秒のパルス、残る時間は100秒のパルス)を用いてゲルを泳動した。
一度、ゲルを泳動したら、そのゲルを、ゲルの泳動方向に滅菌メスで3つに切り、それによって大型ウェル全体を含む中央の部分と、画分およびマーカーを含む両側の部分とを作った。これらの2つの部分はEtBr(0.5μg/ml)で染色し、一方、中央の部分は泳動バッファー中に保った。染色された部分を用いて、染色されなかった断片において修飾YACを位置づけた。一度、そのYACが位置づけられたら、それを含むアガロースブロックを切り取り、50mlの平衡バッファー(10nM Bis−Tris−HCl pH7.5、0.1mM EDTA、100mM NaCl、0.03mMスペルミンおよび0.07mMスペルミジン)中で4℃で少なくとも16時間平衡化した。次に、修飾YACを含むアガロースブロックを4つに分け、それらを1.5mlチューブに移し、70℃で10分間加熱し、続いて、45℃に温度調整することにより液化して、アガラーゼ(GELase(商標) Epicentre)、100mgのアガロースにつき5単位を加え、それを45℃で3時間インキュベートした。溶けたアガロースを、次に、Millipore社製超遠心分離用カラム(Ultra free-(登録商標)MC Millipore)に移し、連続した6,000r.p.mにて2分間の遠心分離により総てのDNAをカラム内に濃縮した。この操作を最終量100μlが得られるまで繰り返し、DNAが再懸濁するように、カラムを一晩4℃で放置した。続いて、40mlのマイクロインジェクションバッファー(10mM Tris−HCl pH7.5、0.1mM EDTA pH8、100mM NaCl、0.03mMスペルミン、0.07mMスペルミジンおよび滅菌水)を入れたペトリ皿に透析フィルター(Millipore)を入れ、それを2または3時間透析させた。この後、修飾YACを含む液を回収した。
【0106】
5.卵細胞質内精子注入法(ICSI)(精子はYAC DNAとともにインキュベートされる)を利用したトランスジェニックマウスの作製
修飾YACを含むトランスジェニックマウスの作製は、特許出願WO2005/098010ならびにMoreira, 2003 (Moreira, P. N., et al., 2003, "Mouse ICSI with frozen-thawed sperm: the impact of sperm freezing procedure and sperm donor strain", Mol Reprod Dev. 66(1) : 98-103)およびMoreira, 2004 (Moreira, P. N., et al., 2004, "Efficient generation of transgenic mice with yeast artificial chromosomes by intracytoplasmic sperm injection", Biol Reprod. 71(6) : 1943-7)に詳細に記載されているプロトコールに従って行った。
【0107】
6.創始(founder)マウスおよびそれらの子孫の解析
6.1 PCRを利用した創始マウスの解析
PCR技術は、高度に特異的であり、トランスジーンの同定について最速のものであるという事実から、トランスジーンの解析を実施するためにこの技術を選択した。このマウスを研究するために、APP遺伝子の異なるエキソンについての17のPCRと、YACアームについての2つのPCRと、非遺伝子コード領域についてのさらに4つのPCRとを設計した。上記(1.3)のプロトコールに従って、生まれた総てのマウスについての23のPCRを行い、総てのPCRについて陽性であるものだけが全トランスジーンの担体マウス(carrier mice)と考えられた。
【0108】
6.2 生殖細胞系への伝達
トランスジーンの担体と考えられるマウスをC57BL/6Jマウスと交配させ、それらの子孫を解析した。伝達の解析は、2つのYACアーム(LYS2およびTRP1)についてのPCRと、APP遺伝子の2つのエキソン(エキソン6および11)についてのPCRとを利用して実施した。
【0109】
7.タンパク質の特性評価
7.1 タンパク質濃度の測定
タンパク質濃度を、標準としてウシ血清アルブミン(BSA)を用いてBradford法により定量した。
【0110】
7.2 SDSの存在下でのポリアクリルアミドゲル中でのタンパク質の電気泳動(SDS−PAGE)
3%濃縮ゲルと、解析するタンパク質の分子量に従って様々なパーセンテージ(7.5%、8%、10%または15%)の分離ポリアクリルアミドゲルとを使用した。サンプルバッファーとして25mM Tris−HCl pH6.3、10%グリセロール、2%SDS、5%β−メルカプトエタノールおよび0.01%ブロモフェノールブルーを使用した。
【0111】
分子量標準としてAmershamm Pharmacia社製の高分子量Rainbow(商標)カラー分子量標準(220kDa、97.4kDa、66kDa、46kDa、30kDa、21.5kDaおよび14.3kDa)を使用した。
【0112】
7.3 ウエスタンブロット
簡潔には、上記節に記載のように、ポリアクリルアミド−SDSゲルで決定されたタンパク質を、セミドライ式電気泳動転写法を用いて、孔径0.45μmのニトロセルロースフィルター(Bio-Rad)に電流量1.3mA/cm2で90分間転写した。
【0113】
ポンソーSレッドでの染色を利用して、転写されたタンパク質を観察した後、非特異的結合をブロックするために、その膜を3%BSAおよび0.2%Tween−20を含むPBS中で4℃で一晩飽和させた。
【0114】
次いで、それらの膜を0.05%Tween−20(PBST)および1%BSAを含むPBSで希釈した異なる特異的抗体とともに室温で2時間インキュベートした。それらの膜をPBSTで3回洗浄し、1%BSAを含むPBSTで1:50,000希釈したペルオキシダーゼ結合二次抗体とともに室温で2時間インキュベートした。最後に、発現させるために化学発光法(この方法ではペルオキシダーゼがH2O2の存在下でルミナル試薬(the luminal reagent)の酸化を触媒する)を用いた(Amersham社の「ECL」)。
【0115】
それらのバンドを濃度測定(GS−710 Calibrated Imaging Densitometer、Bio-Rad)を用いて定量した。
【0116】
7.4 抗体
6E10.6E10はヒトAβ配列の残基1〜17を認識する精製マウスモノクローナル抗体(Signet)である。6E10は電気泳動後の終濃度5μg/mlでの免疫標識に使用された。
【0117】
抗α−チュブリン:抗α−チュブリン(クローンB−5−1−2)はチュブリンのアイソフォームαのC末端エピトープを認識するマウスモノクローナル抗体(Sigma)である。抗α−チュブリンは電気泳動後の終濃度1:10,000での免疫標識に使用された。
【0118】
ビオチン結合抗マウス.ウマにおいて作製された精製抗体;ビオチン結合抗マウスはVector Laboratoriesから入手した。
【0119】
8.異なる組織におけるトランスジーン発現の解析
8.1 全RNAの単離
異なるマウス組織サンプルを機械的破壊によりホモジナイズし、全RNAを、市販の「High Pure RNA Isolation」 kit(Roche diagnostic 1 828 665)を製造業者によって示されるプロトコールに従って用いて抽出した。得られたRNAの量の評価はNanodrop装置(NanoDrop. ND−1000分光光度計)によって実施した。
【0120】
8.2 レトロ転写(retrotranscription)によるRNAからDNAへの移行
レトロ転写は「High Capacity cDNA Archive Kit」(Applied Biosystem)を用いて行い、RNAサンプル各1μgに対して、4単位の逆転写酵素「MultiScribe Reverse Transcriptase」、デオキシヌクレオチド(dNTP)、特異的バッファーを使用し、ランダムヘキサマーを製造業者によって示される条件の指標として最終量100μlで使用した。反応混合物を入れたチューブをGeneAmp PCR system 2400または9600サーマルサイクラー(Perkin-Elmer)により次の温度サイクルに供した:
−25℃で10分間のプレインキュベーション
−37℃で120分間のインキュベーション。
【0121】
8.3 384−ウェルプレートでの定量PCR
調製した総ての組織サンプルについて定量PCRを行い、それらを三連でアッセイし、その場合、50ngのオリジナルRNAに相当する5μlのcDNAを、Taqman Universal PCR Master Mix、no. AmpErase Ung(Applied Biosystem)によって記載された一般的な条件を用いて使用した。用いたプローブは:ヒトAPP遺伝子、onDemand hAPP Hs00169098_ml assay;ネズミAPP遺伝子、onDemand mAPP Mn00431827_ml assay;および内部対照遺伝子(ハウスキーピング)gapdh、onDemand Mn99999915_g1 assayであった。
【0122】
サンプルと反応混合物とを入れたプレートを、リアルタイムでのサンプルの解析を可能にするABI PRISM 7900HT Sequence Detection System(Applied Biosystem)を用いて、製造業者によって指定される温度サイクルに供した。得られたデータはSDS 2.1 ABI PRISMプログラム(Applied Biosystem)を用いて解析した。
【0123】
9.光学顕微鏡での観察:アミロイド斑の検出
組織学的研究のためのサンプルを得る前に、マウスを潅流に供し、続いて、その動物を解剖し、脳を摘出した。
【0124】
Condo red染色を、Accustain amyloid stain kit(コンゴレッド.Sigma diagnostic)を製造業者によって推奨されるプロトコールを用いて使用して行った。アミロイドの染色は赤色またはピンクがかった赤色で観察することができ、細胞核は青色、弾性繊維は明るい赤色である。サンプルを複屈折検査すると、それらは緑黄色で観察することができる。
【0125】
結果
ヒトAPP遺伝子を内因的に発現するトランスジェニックマウスの作製については、次の工程を通じて開発された:第一に、YAC b142f9およびその近接領域のコンピューター研究を実施する工程、次に、未知の配列またはYACに存在する他の遺伝子に対応する配列を削除する工程、修飾YACを得る工程、続いて、全修飾YAC構築物の組込みを達成するために遺伝子導入を実施する工程、それが確実に次の世代に伝達されるようにする工程、最後に、マウス組織におけるトランスジーン発現およびそのレベルを確認する工程。
【0126】
1.APP遺伝子およびその環境についてのコンピューター解析
ヒトAPP遺伝子は21q21.3領域の第21染色体に位置し、5’ではCYYR1遺伝子に隣接され、3’ではGABPAおよびATP5J遺伝子に隣接される(図2)。この全領域の配列はENSEMBLデータベース(http://www.ensembl.org/)から入手した。このデータベースでは、ヒト配列の他に、ハツカネズミ配列などの異なる生物の配列も入手可能である。APP遺伝子のハツカネズミにおけるホルトロゴウス領域(the hortologous region)は染色体16c3.3に位置し、ヒトの場合と同じ遺伝子に隣接され(図2)、このことはその配列が完全に保存されるという推測につながる。
【0127】
ヒトAPP遺伝子配列およびその環境を、相同性マトリックスを用いてネズミ遺伝子のものと比較している(図3)。両種間の1MbのDNA配列をMacVector 7.1プログラム(Accelrys, Burlington, MA, USA)を用いて比較すると、APP遺伝子のプロモーター領域のような、コード領域をコードする領域は高度の相同性を示すことが認められる。近接領域に関しては、APP遺伝子の5’領域ではコード領域より観察される保存は少ないが、ゲノム構造は維持され、一方、その遺伝子の3’領域では、その約60kbにおいて、ヒト領域に関連したネズミ領域で一連の再配列が起こっている。この特定のゲノム構造は、YACを修飾するのに用いる領域を定義するのに理想的であり、その境界を5’ではSeq2配列に相当する領域まで、そして3’ではGABPA遺伝子のコード領域の開始点までに定める[図1]。
【0128】
2.異なるベクターにおける組換えおよびクローニングのための相同配列の選択
2.1 反復領域の研究
酵母の相同組換えに用いる配列を選択するときには2つの条件が重要であると考えられている:一方では、サイズ、そしてもう一方では、その特異な状況。選択する配列のサイズに関しては、1〜2kb間であることが好適であると通常考えられる。その特異な状況に関しては、その適合性は、組換えが所望の場所で起こるという事実による。反復配列を位置づけ、それらを避けるために、RepeatMaskerプログラム(A. F. A. Smith & P. Green, 未公開; http://ftp.genome.washington.edu/cgi-bin/repeatmasker/)を用いて、1メガベースのAPP遺伝子環境を解析した。反復配列を排除しておいたこの配列においてAPP遺伝子の3’および5’の両方における1〜2kbの領域を選択し、PCRによってクローニングし、酵母における相同組換えに用いた(図4)。
【0129】
2.2 組換え配列のクローニング
APP遺伝子を含むYAC(b142f9)の異なるPCRを、第2節に記載のオリゴヌクレオチドを用いて行った(図4)。YACの伸長はこれらのPCRにより確認し、APP遺伝子の5’領域において配列2(sequence2)と配列3(sequence3)の間に位置しており、配列AおよびB(sequenceAおよびB)はAPP遺伝子の3’領域においてGABPA遺伝子の前に位置していた。配列2は、YAC中にある、遺伝子転写開始部位から128,354kbのところに位置する、APP遺伝子から最も離れた5’における配列であることから、この配列を5’での相同組換えに使用した。3’における領域では、配列AおよびBがAPP遺伝子の末端からそれぞれ100kbおよび80kbの距離のところに位置し、GABPA遺伝子の開始点の前にあることから、配列AおよびBの両方をクローニングした。クローニングでは、配列2、AおよびBのPCRを行い(図5)、選択した配列に対応するバンドを精製し、PCRIIクローニングベクター(Invitrogen)にクローニングして、PCRII−Seq2、PCRII−SeqAおよびPCRII−SeqBを得た。
【0130】
2.3 組換えベクター pB1R−Seq2の作製
図6に図式に示すように、プラスミド PCRII−Seq2およびプラスミド pB1Rの変異体を用いて、プラスミド pB1R−Seq2を作製した(Lewis, B. C, N. P. Shah, B. S. Braun and C. T. Denny (1992). "Creation of a yeast artificial chromosome fragmentation vector based on lysine-2." Genet Anal Tech Appl 9(3): 86-90)。このYACが一度修飾を受けたら、必要に応じて他のタイプの培養に使用できるように、オリジナルプラスミド pB1Rにネオマイシン選択遺伝子を加えた。クローニングベクターの配列2、PCRII Seq2を、EcoRI酵素での消化により抽出した。
【0131】
この断片の末端を、それらのその後のクローニングのためにクレノウ酵素を用いて平滑末端にした。ベクター pB1R−NeoをSacI酵素での消化により線状化した。T4リガーゼを用いて、Seq2断片をベクター pB1R−Neoと結合し、後に酵素消化によりベクターにおけるその配列の正確な方向性を確認した。前記ベクターを酵母における第1回目の相同組換えに使用することができるように、そのベクターをSalIおよびSacI酵素での消化により線状化した(図6)。
【0132】
2.4.組換えベクター pYAC4 3’−SeqAおよびpYAC4 3’−seqBの作製
これら2つの組換えベクターの作製を図7に要約している(2配列についてクローニングを並行して実施した)。組換え配列をプラスミド PCRII−SeqAおよびPCRII−seqBから得た。抽出では、それらを最初にXhoI酵素で消化し、末端をクレノウ酵素で平滑末端にし、続いて、それらの配列が平滑末端とさらに付着末端を有するようにそれらをBamHIで消化した。プラスミド pYAC4 3’はチロシナーゼ遺伝子の調節配列を研究するために研究室で作製されたpYAC4の誘導体である(Burke, D. T., G. F. Carle and M. V. Olson (1992). "Cloning of large segments of exogenous DNA into yeast by means of artificial chromosome vectors. 1987." Biotechnology 24: 172-8、およびBurke, D. T. and M. V. Olson (1991). "Preparation of clone libraries in yeast artificial-chromosome vectors." Methods Enzymol 194: 251-70)。配列AおよびBを、ベクター pYAC4 3’−SeqAおよびpYAC4 3’−SeqBが生成する配向方法で結合し得るように、消化を利用して、最初にEcoRIを用いて、それらの末端を再び平滑末端にし、後にBamHIで消化して、目的のものではない領域を削除し、そのベクターを線状化した。
【0133】
しかしながら、これらのベクターを第2回目の相同組換えに使用するためには、新規栄養要求性マーカーの存在が必要であった。そのために、最初にNotIでの消化によってベクター pYAC4 3’−SeqAおよびpYAC4 3’−SeqBを線状化して(この場合もクレノウ酵素を用いて、それらの末端を平滑末端にした)、プラスミド YDp−UのURA3マーカーをクローニングし(Alani, E., L. Cao and N. Kleckner (1987). "A method for gene disruption that allows repeated use of URA3 selection in the construction of multiply disrupted yeast strains." Genetics 116(4): 541-5)、続いてBamHIでの消化によってプラスミド YDp−UのURA3遺伝子を得た、正確なクローニングのためにその末端も平滑末端にした。T4リガーゼを用いて、その断片をベクターにクローニングした。YACの第2回目の組換えにそれらを正しく使用するためのベクター pYAC4 3’−SeqA URA3およびpYAC4 3’−SeqB URA3の線状化を、BamHIおよびSpeIでの酵素消化によって行った(図7)。
【0134】
3.YACの5’領域における相同組換え
本発明では染色体断片化プロセスを5’および3’領域を修飾するために用いた;このプロセスはオリジナルYACの末端にある2つの領域を削除することからなり、それらの領域うちの一方が3’におけるGABPA遺伝子を含む領域である。
【0135】
3.1 APP遺伝子の5’領域のYAC b142f9における相同組換え
100〜1,000ng間で変化するDNA量を用いて、LiAc法による酵母の形質転換によって合計40のコロニーを得た。これらの40コロニーを、PCRを利用して解析し、陽性の可能性があるクローンとして3つのクローンを得た。
次に、酵母のエレクトロポレーションでは合計96のコロニーが生じ、これらのコロニーを、PCR技術を利用して解析した。これらの中で陽性の可能性があるクローンとしての正確なパターンを示したものはなかった。
【0136】
3.2 PCRを利用した、相同組換えによって生じるクローンの解析
相同組換えによって得られたクローンの解析を、用いる2つのオリゴヌクレオチドのうちの1つが組換えプラスミド中に位置し(YACには存在しない領域内)、一方、もう1つのオリゴヌクレオチドがYAC組換え領域に近接する領域中に位置することを特徴とするハイブリッドPCRを設計することによって実施した。よって、このPCRでは、組換えが予測される部位で起こった場合にのみ断片を増幅することができる。陽性対照として両方の領域を含む別のベクターを作製した。このPCRについて陽性であったのは、解析した136のコロニーのうちの3つのもののみ、13、14およびb7の番号のついたコロニーであった。
【0137】
これらのクローンを用いた解析を、別の3つのPCRを用いることによって行い、これによってそれらについてのより多くの情報を提供することができた。最初の2つのPCRは、APP遺伝子の部分を維持するクローンがそのエキソン6および11のものであるかどうかを確認するために行った(図8)。用いた3つ目のPCRは組換え領域の5’に位置する配列のものであり、相同組換えが正確であったとすれば陰性となる(図8)。解析した3つのクローンは、図8に示されるように、エキソン6および11のPCRについて陽性であり、5’領域について陰性であった。
【0138】
3.3 パルスフィールド電気泳動を用いることによるYACの断片化の判定
YACにおける正確な相同組換えはそのサイズの減少を伴い、そのことはその電気泳動移動度を調べることにより見ることができる。酵母ゲノムはパルスフィールド電気泳動技術(PFGE)を用いてゲル中で分離することができる。この技術に従って、解析する3つのクローンを、オリジナルクローン、同じ酵母株のクローン(YACを全く含まない)および分子量マーカーとともに泳動した。図9ではEtBrで染色した泳動後のゲルを示している。オリジナルYACクローンにおいて680kbマーカーと580kbマーカーとの間に、YACを含まない酵母クローンには存在しないバンドが存在することを観察することができる。このバンドは、クローン13およびB7では維持されているように見えるが、一方、クローン14では消失しているように見える。
【0139】
続いて、このゲルをニトロセルロース膜に転写して、APP遺伝子 cDNAプローブとのDNA−DNAハイブリダイゼーションを行い、これによれば電気泳動移動度の変化が極めて正確に確認される。図9では、クローン14が、正確な組換え後に予測されるYACサイズの約50kbの変化をどのように受けたかを示している。
【0140】
3.4 サザンブロット技術を利用した組換え領域の解析
相同組換えによるYACの断片化は、組換え領域のDNA配列において一連の修飾をもたらす。組換えクローン配列とオリジナルクローン配列との間の違いについては、サザンブロット技術を利用して解析することができる。BamHI、ScaI、EcoRV、BglIIおよびHindIIIの消化パターンは2つのクローンで異なっていた。
【0141】
加えて、APP遺伝子のオリジナルクローンとクローン14について実施したサザンブロットにおいてバンドサイズの変化が得られ(図10)、このことにより組換えが正確な相同部位で起こったことが分かり、PCR解析とパルスフィールド電気泳動の両方で得られた結果と合わせて、APP遺伝子の5’領域において組換えが成功したこと、そしてクローン14を次の組換えのベースとして使用可能なことを示している。
【0142】
3.5 クローン14のコード領域の解析
クローン14がAPP遺伝子のコード領域の欠失を受けていないことを確認するために、サザンブロットの比較を実施した。このために、オリジナルYACと修飾YACをBamHI制限酵素で消化し、続いて、APP遺伝子 cDNAとハイブリダイズした。このハイブリダイゼーションでは異なる遺伝子コード領域に対応するバンドのラダーが生じるが、これはオリジナルクローンとクローン14の両方で同じであるにちがいない。MacVectorプログラムを使用して、BamHI酵素での300kbのAPP遺伝子の消化パターンを得た。次いで、遺伝子コード領域の任意のものを含む断片を確認した。図11では、このサザンブロットの理論パターンと、それがオリジナルクローンb142f9と組換えクローン14で得られたバンドのラダーと一致していることを示している。オリジナルクローンb142f9と組換えクローン14で得られたバンドパターンの保存は、組換え後にコード領域は大きな修飾または再配列を受けなかったということを示している。
【0143】
4.YACの3’領域における相同組換え
4.1 クローン14のYACにおける相同組換え.GABPA遺伝子削減
第1回目の組換え後に修飾YACにおいてAPP遺伝子の3’領域における相同組換えを行い、これには形質転換に用いるコンピテント酵母を調製するためにクローン14を用いることを必要とした。この組換えには2つの異なるベクター、pYAC4 3’−SeqA URA3およびpYAC4 3’−SeqB URA3(図7)を用い、これらのベクターは組換え配列が異なるだけのものである。前記配列は20kb相互に離れており、遺伝子の末端から80kbより遠く離れている(図4)。これらの組換え配列を用いることによってGABPA遺伝子を含む、YACの40〜60kbを削除した(図12)。
【0144】
LiAc法とエレクトロポレーション法の形質転換効率の違いを考えると、明らかに前者が有利であり(前節の第3.1節参照)、第2回目の形質転換を行うために使用することを決めた。第1回目の組換えとは異なり、dual PCRを利用したこの組換えでは2,317のコロニーを解析する必要があり、APP遺伝子のエキソン6で行われる第1回目のPCRで陽性を示し、GABPA遺伝子で行われる第2回目のPCRでは陰性を示すと、このことによりGABPA遺伝子に対応する配列が削除されたことが示される。
【0145】
解析した2,317のコロニーのうちの1,720のものは配列Aに対応し、597のものは配列Bに対応した。異なるコロニーのPCR解析後、1720のうちの90のものと、597のうちの5のものだけをPFGEおよびサザンブロットによって解析した。PFGEおよびサザンブロットを用いたこれらのクローンの研究により、配列Aを用いて行われた相同組換えが高い確実性で異なる領域で起こり、その結果、見慣れない組換えパターンをもたらし、いくつかの場合では、組換えYACが予想をかなり上回る再配列プロセスおよび断片化を受けて、説明することが非常に難しいパターンが生じたことが明らかになった。
【0146】
配列Bを用いた相同組換えについて解析した5つのクローンは、トランスジェニックマウスの作製に使用するための必要要件を満たすクローンを与えた。このクローンにおいて実施した様々な検証を以下に記載する。
【0147】
4.2 PCR技術を利用した、3’における組換えによって生じたクローンの解析
酵母形質転換によって得られた異なるクローンの解析を、dual PCRを用いて実施した。図13では、配列Aを用いた組換えについてのいくつかのクローンと配列Bについての5つの陽性のものとにおいて実施したdual PCRを示している。観察できるように、クローンb106、b108、b109、b111およびb113はそれらの後の研究用の候補として反応している。APP遺伝子のエキソン6のPCRではそれらの総てが陽性であり(このことは、それらのクローンはAPP遺伝子とともにYACを維持していることを示す)、その一方で、GABPA遺伝子のPCRでは陰性である(このことは、これらのクローンはGABPA遺伝子配列を失った可能性があることを示唆する)。以下に示すように、これら5つのクローンのうちクローンb106だけが正確な相同組換えパターンを示した。
【0148】
PCRを用いてクローンb106の研究を行うために、配列BとGABPA遺伝子との間:配列AおよびA’との間に位置する2つの領域についてのPCRを行った。この研究では、GABPA遺伝子とエキソン6のPCRを繰り返し、APP遺伝子のエキソン11のPCRも含めた。図14から分かるように、組換え領域に関して3’位置でのPCRで陽性であったものはなく、クローンb106にはこれらの配列が存在しないことが示された。2つのエキソンのPCRは陽性対照の場合ほど明らかではなく、それらを何度か繰り返したが、常に同じ陽性結果が得られた。
【0149】
4.3 PFGEを利用したYACの電気泳動移動度の変化の判定
YACの電気泳動移動度は、第1回目の相同組換え後と同じように、この第2回目の組換え後にも変化し、得られるYACの最終的なサイズは520kbである、すなわちYACを含むクローン14よりも40kb小さい。図15では、異なるクローンを泳動した1%アガロースゲルを示している:分子量マーカー、オリジナルクローンb142f9、クローン14およびクローンb106。前記ゲルをニトロセルロース膜に転写し、APP遺伝子 cDNAとハイブリダイズした。異なるレーンのYACの観察が得られ、クローンb106におけるYACのサイズの変化はオリジナルYACおよびクローン14に関して容易に観察された。
【0150】
4.4 クローンb106のYACとGABPA遺伝子 cDNAとのハイブリダイゼーション
YAC b106とGABPA遺伝子 cDNAとのハイブリダイゼーションを、該YACにおいてGABPA遺伝子のコード領域が削除されたことを確認するために用いた。
【0151】
図15では、前節で用いたパルスフィールドアガロースゲルを示しており、このパルスフィールドアガロースゲルに他のレーンを加え、前記レーンの場合と同じサンプルをこれらのレーンにロードしている。この場合には、ゲルの半分をAPP遺伝子 cDNAプローブとハイブリダイズし、残りの半分をGABPA遺伝子 cDNAプローブとハイブリダイズした。その図から分かるように、クローンb106のAPP遺伝子を含むYACはGABPA遺伝子配列とはハイブリダイズしないが、一方、オリジナル酵母のYAC、およびクローン14のものは同じYACにおいて両方の遺伝子を有している。
【0152】
4.5 サザンブロット技術を利用した3’における組換え領域の解析
組換えベクターがYACに正確に組み込まれていることを、サザンブロット技術を用いて確認した。図16に示されるパターンは、オリジナルYACとクローン14では共通しているが、一方、クローンb106は異なった、予測ダイアグラムに従うバンドパターンを有する。ゲルハイブリダイゼーションでは、YAC b106に対応するバンドは前者のクローンのものとは異なるサイズを有することが分かり、このことは、プローブがハイブリダイズする、組換え配列の周辺領域にあり、後者の5’に位置する領域において、異なるクローン間で配列の違いがあることを明確に示している(図16)。
【0153】
4.6 クローンb106のコード領域の解析
クローンb106の妥当性を確認するために、サザンブロットの比較を実施した。クローンb106のYACが、コード領域ではなく、YACの3’領域で修飾を受けただけならば、3つのYAC(オリジナルYAC、クローン14のYACおよびクローンb106のYAC)は、図11に示された同じラダーのバンドを有するはずである。実施したサザンブロットを示している図17を観察すると、3つのクローンのバンドパターンは全く同じであり、そして、それが図11に示されるものとは明らかに異なるということが分かる。しかしながら、これらの違いは、実際には、より清浄なDNAを、より高い濃度で使用することによって最後のゲルにおいてより良好な分解能が得られたという事実によるものにすぎない。図17のゲルのバンドパターンを観察すると、いくつかの二重および三重バンドはより明確に見える。
【0154】
いずれにせよ、APP遺伝子を含むYACはクローンb106において重要なものであった。クローンに用いた同じ命名法を用いて、このYACに名前を付け、それをYAC b106と呼んだ。
【0155】
5.YAC B106の精製
本プロセスにより3回の連続したDNA抽出を実施した。材料および方法の第4.2節に記載のプロトコールに従って実施した3回の精製のうちの1つを示している図18から分かるように、EtBrで染色したゲルのエッジを用いて、YAC b106の位置を決定し、染まらなかったゲルの部分からYAC b106を抽出した。抽出後にゲルの他の部分を染色することによって、YAC b106の総てのバンドが採取されたことを確認することができた(図18)。
【0156】
3つの異なる技術によって3回のDNA抽出を測定した。まず第一に、アガロースゲルでの評価と既知量の参照YACとの比較を利用する。得られた結果を、蛍光光度計とnanodropを使用することによって調べた。その結果、抽出I、IIおよびIIIで得られた濃度はそれぞれ4ng/μl、7.1ng/μlおよび10ng/μlであった。
【0157】
6.卵細胞質内精子注入法を介した遺伝子導入(精子はYAC b106とともにインキュベートされる)
遺伝子導入を実施するために、異なるマイクロインジェクションセッションを実施し、プロセスが終わった後に生まれた合計102匹の動物を得た。遺伝子導入に用いた卵母細胞のドナーマウス系はCD1とC57BL/6Jとの混合系であり、それによって純系を使用するよりも高い効率が得られる。創始マウスが生まれてくるときに、それらが75%C57BL/6J遺伝的背景を有するように、用いた精子はC57BL/6J系のものである。
【0158】
7.創始マウスの解析
PCR技術を用いて、細胞質注入後に得られた102匹のマウスの解析を実施した。520Kbのトランスジーンの挿入を正確に確認する手段として総てのYAC領域に対してスキャンを実施した。この目的のために次のPCRを設計した:APP遺伝子のエキソンについての17のPCR(それらのうちの1つはエキソン12とエキソン13の両方を含む)と、YACの2つのアームについての2つのPCR(LYS2遺伝子の選択遺伝子およびTRP1のものの両方に関する)と、コード領域とYACのアームとの間に位置する配列についてのさらに4つのPCR(図19)。
【0159】
得られた102匹のマウスについての23のPCRを行った(図20)。それらのうちの17においてある程度の遺伝子導入が得られ、これはそれらがYACの外因性DNAを完全にまたは部分的に組み込んだことを意味する。このDNAの組込みは、15のケースで起こったように、部分的なものであり得るが、それらのうちの2つ、マウス35および雌マウス64においては完全なものであり、YAC b106に関するこの調査で得られる第1のトランスジェニックマウスが生成したため、その主要目的の1つに適合する。
【0160】
8.雑種第一代(F1)におけるトランスジーンの伝達
2つのトランスジェニック創始マウス、マウス「35」およびマウス「64」の作製を確認したので、それらをC57BL/6Jマウスと交配させて、それらの子孫へのトランスジーンの伝達を調べ、それらにおけるそのようなトランスジーンの発現を解析した。YAC b106の伝達を研究するために、それらの子孫で4つのPCRを行った:YACのアーム、すなわちLYS2およびTRP1遺伝子についての2つのPCRと、APP遺伝子のエキソン6および11についてのPCR。この結果、完全YACが伝達されたこと、そしてそのためYACの部分的組込みが起こらなかったことを確認することができた。本明細書の作成まで41匹のF1マウスをマウス35の子孫について解析し;18匹のトランスジェニックマウスを確認し、子孫の44%への伝達がなされていた。雌マウス64の場合では、32匹のF1子孫を解析して、その子孫中18匹のトランスジェニックマウスを得、56%伝達がなされていた。要するに、2つのケースでは、創始マウスの第一代子孫へのトランスジーン伝達指数は、Mendelianのパターンによって予測されるものに一致して約50%である。図21では、マウス35のいくつかの子孫についての例示的なPCRを示している。
【0161】
9.トランスジェニックマウスにおけるYAC b106構造の保存
創始マウスを交配することによって得られたトランスジェニックマウスのゲノム研究を、YAC b106におけるAPP遺伝子コード領域の構造を確認することにより行い、そのために2つの創始マウスの子孫についてのサザンブロットの比較を実施した。
【0162】
図22では、創始マウス35および64のF1、すなわち第一代子孫において得られたバンドパターンを示しており、そのバンドパターンは各創始マウスにつき2匹の同腹子(それらの一方のものはトランスジェニックであり、もう一方のものは非トランスジェニックである)に対応している。パターンは図11で得られたものと同じであることを観察することができ、そのことによりマウスゲノムにおいてトランスジーン構造が保存されていることが示される。
【0163】
10.トランスジェニックマウス系統におけるヒトAPP遺伝子発現の解析
トランスジェニック系統35および64におけるヒトAPP遺伝子の伝達を立証したら、異なるマウス組織におけるRNAおよびタンパク質両方の発現レベルを特徴づける必要があった。
【0164】
この解析には系統35の第一代に相当するマウスを使用し、合計4匹の2ヶ月齢雄マウスを用いて、それらをそれらの同腹子と比較した。この研究の主な焦点は、APPタンパク質の高い発現が起こる組織であることから中枢および末梢神経系であった。異なるマウス体器官における発現もまた解析した、全体的に以下を解析した:脳(皮質、脳室、小脳および中脳と呼ばれる(それらの解剖学的内容と完全に一致する)4つの領域に細分される);脊髄、肝臓;肺;膵臓;脾臓;小腸;筋肉;精巣;血液;心臓および腎臓。
【0165】
10.1.APP遺伝子メッセンジャーRNAの発現の解析
トランスジェニック系統35における正確なヒトAPP遺伝子発現を確認するための第1の工程は、RT−PCRを利用して異なるマウス組織におけるヒトAPP遺伝子の転写を調べることであった。そのためには、異なるマウス組織からメッセンジャーRNAを抽出し、そのRNAの転写レベルを、リアルタイムRT−PCRを利用して、ヒトAPP遺伝子に向けられるTaqmanプローブを用いて測定した。この試験の内部対照としてネズミGAPDH遺伝子を用いた。この研究を行うために、ネズミAPP遺伝子RNAレベルも測定し、それらをGAPDHに対して標準化すれば、両方の遺伝子の発現レベルを異なる組織間で比較することができるようにした。
【0166】
図23では、解析した異なる組織で得られたヒトAPP遺伝子メッセンジャーRNAレベルを示している。発現レベルを相対対数単位で表し、データをGAPDH遺伝子に対して標準化している。この遺伝子は主として神経組織において発現され;その値は、若干低い小脳の場合を除いて、それらの総てにおいてよく似ていることが認められる。他の研究した組織に関しては、精巣、血液、心臓および腎臓で検出可能な発現レベルに達しただけであり、いずれの場合においても、神経系で検出されたものより一桁低い。
【0167】
図24では、トランスジェニックマウスおよびそれらの同腹子に関するヒト遺伝子およびネズミ遺伝子の両方についてのAPP遺伝子メッセンジャーRNA発現を示している。前のグラフ同様に、データはネズミGAPDH遺伝子に対して標準化している。示した結果により、神経組織では遺伝子発現レベルが高いが、小脳では低下していることが分かる。他の解析した器官に関しては、肺、精巣、血液、心臓および腎臓などの組織において発現を示しているが、一方、肝臓、膵臓、腸、脾臓および筋肉などの組織ではそのレベルはかなり低くなっており、極めて重要な変化を示している。
【0168】
10.2 タンパク質レベルの解析
マウスにおけるヒトAPPタンパク質の発現によって、マウスにおける正確なYAC b106の転写および翻訳を確認する。タンパク質レベルを、ポリアクリルアミドゲル中での電気泳動後の免疫標識技術(ウエスタンブロット)を用いて研究した。この研究では、APPタンパク質を認識したが、ヒト型とネズミ型とを区別することができない抗体を使用した[Lamb, B. T., et al. (1997). "Altered metabolism of familial Alzheimer's disease-linked amyloid precursor protein variants in yeast artificial chromosome transgenic mice. "Hum Mol Genet 6(9): 1535-41.; Lehman, E. J., et al. (2003). "Genetic background regulates beta-amyloid precursor protein processing and beta-amyloid deposition in the mouse." Hum Mol Genet 12(22): 2949-56]ため、発現レベルを比較することができるように、結果をアクチンに対して標準化する必要がある。
【0169】
神経系の異なる組織では免疫反応性が欠如しており、低いAPP遺伝子発現レベルを示す。図25では、系統35および野生マウスの両方に関する代表的なゲルを示し、発現の評価を、アクチンに対して標準化して、下のグラフに示している。そのグラフでは、系統35が野生マウスの少なくとも2倍の量のタンパク質を有することがはっきりと分かる。この割合はこの研究において解析した異なる脳領域間でわずかに変化するが、その水準を維持している。
【0170】
10.3 マウス脳の組織学的研究
本発明のマウスになければならない特徴の1つは、正確なAPPタンパク質発現およびプロセッシングであるが、その一方で、不正確な発現または誤りのあるプロセッシングの形跡があると、脳においてこのタンパク質(Aβペプチド)由来のペプチドの蓄積が起こる。この研究を行うために、月齢2ヶ月の2つのマウス系統の脳の切開を行った。これらの切片をコンゴレッドで染色して、アミロイドを検出した。2系統またはそれらの同腹子ではいずれのAPPタンパク質誘導体も蓄積は検出されない(図26および27)が、1年齢の対照トランスジェニックマウス(Tg 2576)ではそれらを検出することができる。
【0171】
考察
1.酵母の相同組換えによるYAC b106の構築
アルツハイマー病の調査を可能にする、この調査で開発されたトランスジェニックマウスを設計する際に利用できたのは、YAC b142f9(APP遺伝子を含むことは分かっているが、場合によってはその後の使用において摂動を導入し得る他の遺伝子も含むかどうかは不明である、600kbを超えるサイズの人工染色体)だけであった。ヒトゲノム配列が公開され、YAC b142f9で実施された他の具体的な研究が発表されたことで、該YAC中にはAPP遺伝子が単独で存在するのではなく、GABPA遺伝子が同時に存在することの証明が可能になった。そのため、そのGABPA遺伝子を削除し、これまでに報告された総てのAPP遺伝子コード領域および調節領域を尊重して、YAC b142f9をその5’および3’領域において修飾する必要が生じる。そのようにして修飾したYACを、アルツハイマー病に関する調査のためのモデルとして使用することができるトランスジェニックマウスの作製において使用し得るようにするために、このことがこの調査の最初の目標となった。
【0172】
5’で行われる相同組換えはYACの40kbの削除を伴った(図12および図9)。それでもなお、この削除後、APP遺伝子の5’領域において120kbがまだ保存されており、調査の現状に従ってその正確な発現を可能にするには十分なものであった。実際、APP遺伝子の調節エレメントはその遺伝子の近位プロモーターにおいて報告されているだけであったが、距離が離れている調節エレメントの存在については議論の余地がないと考えられている。そのため、正確な時空間的遺伝子発現を成し遂げるために、進化的に保存されるエレメントを存在させる組換えを実施した[マウス遺伝子のものとの比較ゲノミクスによる配列解析後(図3)]。従って、本調査ではそれを確実にするために端で十分なスペースが保存された。
【0173】
YACにおける相同組換えによって5’領域の境界を定めた後、染色体断片化によってGABPA遺伝子を直接削除する第2回目の組換えを実施した。GABPA遺伝子は転写因子をコードし、トランスジェニックマウスにおいてそれが発現することによって、トランスジェニック表現型の研究に干渉し得る未知の方法でマウスの正確な開発に影響を及ぼす可能性があり、どの遺伝子が該表現型に関与するのかを確認することができなくなるため、このYACを動物モデルの作製に用いようとする場合には、その削除は必須である。
【0174】
APP遺伝子の3’領域における相同組換えは、5’において実施されるものよりもずっと複雑であるが、これは3’領域に二次DNA構造が存在することにより、この構造が組換え配列へのアクセスをより複雑にしている可能性がある。
【0175】
第1回目の組換えの場合と同じように、正確な相同組換えとGABPA遺伝子の削除を正確に確認するために一連の試験を設計した。第1回目のコロニースクリーニングを、dual PCRを用いて実施した。この方法は得られた数百ものコロニーを区別すること、そしてこの数を一桁低減することを可能にした。GABPA遺伝子を有していなかったがエキソン6は有していたコロニーは適していると考えられるようだが、PFGEを利用した後の解析ではいつもそういうわけではないことが分かった。
【0176】
この意味で、PFGEと、続いての、フィルターのAPP遺伝子cDNAおよびGABPA遺伝子cDNAとのハイブリダイゼーションを利用した解析は、中でも最も信頼性の高い最終的な解析であった。これは;実際には、YAC b106の電気泳動移動度の変化がクローン14のYACの断片化によるもののみであり得るためである。GABPA遺伝子cDNAとのハイブリダイゼーションは、総ての遺伝子コード領域の認識を可能にするが、一方、PCRはエキソンの1つを対象にするだけであった。YAC b106のGABPA遺伝子をコードする配列の消失についてのハイブリダイゼーションによる確認では、予測される電気泳動移動度の変化とともに、このコロニーが計画された組換えを受けたという確認を必要とした。それでもなお、サザンブロットによる確認および異なるPCRは、研究を行い、その結果、トランスジェニックマウスを作製するためのプロセスに移るために有用であった。
【0177】
2.ICSIを利用したトランスジェニックマウスの作製
520kbのYAC b106を有するトランスジェニックマウスを、WO2005/098010およびMoreira, 2003および2004(上記)に記載のプロセスに従って、ICSIを利用して得た。生まれた102匹のマウスから、合計17匹のトランスジェニックマウス、すなわちYAC b106Ofの任意のDNA断片の担体を得、解析した。
【0178】
520kb構築物が完全に組み込まれたことを確認するには、配列全体にわたる入念な解析が必要であり、そのために、各マウスにおいてYAC全体にわたって23のPCRを実施した。YACの完全組込みを確認するために設計されたPCR群(図19)では、マウス35および64の場合だけ陽性であり、このことからこれらのマウスを新規動物モデルの候補創始マウスであると特定した。
【0179】
しかしながら、創始マウスにおいてトランスジーンが存在するからといって、その後の使用に最も重要な条件である生殖細胞系においてそれが存在することが確実とはならないため、次の世代でその伝達を確認しなければならない。それを調べるために、クローンをC57BL/6Jマウスと交配させ、2つのトランスジェニック系統(図21)から生まれた総ての同腹子をPCRによって解析し、第一代、すなわちF1においておよそ50%の伝達が得られた(結果の第8節)。生殖細胞系におけるトランスジーンの伝達(図21)ならびにかかるトランスジーンの発現(図23)を確認することにより、YACの組込みを確認するために設計されたPCR群が適切な手法であったことが証明された。
【0180】
3.マウスにおけるヒトAPP遺伝子の発現
マウスにおけるヒトAPP遺伝子の発現を特徴づけるための第1の工程を、遺伝子RNA発現を解析することによって実施した。そのためには、定量プレートPCR技術を用いて、異なる組織間のそのレベルを比較した。既存の参照によれば、この遺伝子の発現はいくつかの組織において起こるが、これらレベルは脳において高い。実際には、図23から分かるように、系統35の発現レベルは、解析した異なる領域中で脳において最も高い値を示している。脊髄で表れた高い遺伝子発現指数を強調する必要があるが、脊髄はこのタンパク質を産生する細胞系統が存在する神経組織によって形成されていることから、このデータは当然のことである。よって、マウスにおけるヒトAPP遺伝子の発現に関するこの調査の結果は、脳においてRNAが最高発現レベルにあるという、同様のマウスに関してこれまでに報告されているものと十分に一致している。
【0181】
異なる器官におけるヒトAPP遺伝子の発現はネズミAPP遺伝子の発現と非常に似ており(図24)、神経組織だけでなく、心臓、血液、精巣および腎臓においても発現パターンが保存されている。しかしながら、ネズミAPP遺伝子発現は、肝臓、肺、膵臓、腸および筋肉で検出され、心臓、血液、精巣および腎臓のものと同様の発現が起こっている肺の場合を除いて、他の器官よりもずっと低い値である(一桁低い(one order or magnitude lower))が、ヒトAPP遺伝子ではそのようなことは起こらず、すでに指摘したようにその発現はそれらの器官では検出されなかった。
【0182】
一度、異なるマウス組織において遺伝子発現が確認されたら、そのRNAのタンパク質への翻訳を確認する必要があった。この研究のために、RT−PCRを用いて解析した異なる組織中に存在するタンパク質の量についてのウエスタンブロット解析を実施した。この研究には、最初はヒトタンパク質に特異的な6E10抗体を用いることによって対応していたが、この調査の試験においてその特異性を再現することができなかったため、ヒトタンパク質とネズミタンパク質の両方を認識する22C11抗体を使用する決定をし、その結果、タンパク質の総量を評価することができた。
【0183】
神経組織ではタンパク質発現が高いことから、起こり得るかかるタンパク質の同定および評価を行ったが、一方、他の組織では、タンパク質レベルの表示はずっと低く、その同定は不可能であった(図25)。神経組織におけるタンパク質レベルを評価し、それらをアクチン値に対して標準化した後、トランスジェニックマウスおよびそれらの非トランスジェニック同胞種のタンパク質レベルを比較し、前者の場合に、特定組織に応じて、値が内因性タンパク質のものに対して2〜2.5倍高いことを観察した。これを受けて、皮質、脳室および脊髄では、タンパク質レベルは内因性タンパク質に対して2倍を少し上回っており、一方、中脳および小脳では、比率が上昇し、2.5倍に達していた。トランスジェニックマウスではヒトタンパク質とネズミタンパク質の両方が検出されることを考えなければならないため、総APPタンパク質発現が2倍というのは、同量のヒトタンパク質とネズミタンパク質が発現されているということを意味する。2つの遺伝子間でのこれらのような発現差はこれまでに記載されているもの(皮質領域でのみ測定され、内因性遺伝子のものの1.7〜2倍間の発現レベルを示す)より少し大きいが、その違いは使用したマウス系に起因する可能性があり、そのマウス系の遺伝的特徴はそのレベルとそのプロセッシングの両方に影響を及ぼす可能性がある。
【0184】
最後に、マウス脳において予備研究を実施し、本疾患の典型的な神経病理学のいくつかの検出を試みた。2ヶ月齢マウスでのこのような予備研究では、現在のところアミロイドペプチドの沈着の兆候は検出されず、これらの特徴を有するマウスでの過去の研究と一致している。
【図面の簡単な説明】
【0185】
【図1】ヒト第21染色体(NCBI build 35)におけるAPP遺伝子の位置を示す図である。SeqB領域は全GABPA遺伝子コード領域を排除するが、その3’UTR領域内で始まる。
【図2】APP遺伝子の染色体上の位置を示す図である;A.ENSEMBLにおいて下方に示した近接遺伝子に隣接する、第21染色体におけるヒトAPP遺伝子の位置。B.ENSEMBLにおいて下方に示した隣接遺伝子に隣接する、第16染色体におけるネズミAPP遺伝子の位置。
【図3】ヒトおよびネズミAPP遺伝子配列の相同性マトリックスを示す図である。ヒトAPP遺伝子を含むヒト第21染色体配列メガベースを、ネズミAPP遺伝子も含まれる第16染色体メガベースと比較する。
【図4】APP遺伝子環境のPCRのダイアグラムを示す。PCRの位置を利用して、その遺伝子の5’および3’の両方において、その遺伝子コード領域までの最近距離を有するYAC b142f9のサイズの範囲を定める。
【図5】PCRによるYAC b142f9の伸長の境界を示すダイアグラムを示す。配列A、B、1および2のPCRに関する1%アガロースゲル。レーン1は分子量マーカーに相当する。各配列に対して、陰性対照、YAC、および陽性対照それぞれを示す B.配列3、4、5および6のPCRに関する1%ゲル。レーン1は分子量マーカーに相当する。各配列に対して、陽性対照、YAC、および陰性対照それぞれを示す。
【図6】組換えプラスミド pB1R Seq2の作製および線状化において従う工程のダイアグラムを示す。
【図7】組換えプラスミドpYAC4 3’SeqAおよびpYAC4 3’SeqBの作製および線状化において従う工程のダイアグラムを示す。
【図8】エキソン6および11ならびに配列2’のPCRを示す図である。APP遺伝子のエキソン6、エキソン11およびいくつかの解析した酵母クローンの配列2’のPCR。M、分子量マーカー;G、ゲノムDNA;c+、陽性対照およびc−、陰性対照。
【図9】PFGEによる酵母染色体の分離およびAPP遺伝子cDNAとのハイブリダイゼーションを示す図である。A.臭化エチジウムで染色したPFGE。B.APP遺伝子cDNAとハイブリダイズした膜。レーン:M 分子量マーカー、YAC b142f9、AB1380(YACフリー酵母)、クローン13、クローン14、クローンB7。
【図10】YAC b142f9およびクローン14の5’領域のサザンブロットを示す図である。オリジナルクローンb142f9(APP)とクローン14とのサザンブロットバンドパターンの違い。
【図11】YAC b142f9およびクローン14のサザンブロットの比較を示す図である。クローンB142F9とクローン14の両方のDNAをBam HI酵素で消化した。アガロースゲルで分離し、APP遺伝子cDNAとハイブリダイズした。M、分子量マーカー;APP、クローンB142F9;14、クローン14。右側、B.300kbのAPP遺伝子をBam HIで消化した後に得られたバンドパターン、遺伝子コード領域を含む断片には網がけしている。
【図12】2連続ラウンドの酵母における相同組換えを利用したYAC b142f9の断片化のために提供されるスキームを示す。
【図13】クローンb106のdual PCR解析を示す図である。SeqAおよびSeqBの両方について、異なるクローンで行ったPCR。第1回目のPCRはAPP遺伝子のエキソン6で行い、一方、第2回目のPCRはGABPA遺伝子で行った。第1回目のPCRに関して陽性であり、第2回目のPCRに関して陰性であるクローンにはアスタリスクをつけて示している。C+、陽性対照;C−、陰性対照。
【図14】組換え環境のPCR解析を示す図である。PCRダイアグラム、陰影マークはSeqBの場所を示している。B.GABPA遺伝子、APP遺伝子のエキソン11、エキソン6、SeqA’およびSeqAのPCR。各PCRに対して、陽性対照、クローンb106および陰性対照に対応する3つのレーンが存在する。
【図15】APP遺伝子cDNAとGABPA遺伝子cDNAとを比較したPFGEについてのハイブリダイゼーションを示す図である。A.臭化エチジウムで染色したPFGEゲル。1.マーカー。2および5 b142f9。3および6 クローン14。4および7 クローンb106。B.レーン1〜4のAPP遺伝子cDNAとのハイブリダイゼーション。レーン5〜7のGABPA遺伝子cDNAとのハイブリダイゼーション。
【図16】3’における組換えについてのサザンブロットダイアグラムおよびゲルを示す。A.3’における組換えについてのサザンブロットダイアグラム、上部にはオリジナルYAC、下部には組み換えられたYAC。Reco Bの前の陰影四角は使用したプローブの位置を示している。B.3つのYAC、b142f9(APP)、クローン14およびクローンb106のEco RIおよびSca Iでの消化についてのサザンブロット。
【図17】クローンb142f9、14およびb106のサザンブロットの比較を示す図である。Bam HIで消化し、APP遺伝子cDNAプローブとハイブリダイズしたYAC b142f9(APP)、クローン14およびクローンb106のサザンブロットの比較。3つのYAC間ではバンドパターンは保存されている。M、マーカー。
【図18】YAC b106の抽出についてのPFGEを示す図である。YAC b106に対応するバンドの抽出。ゲルにおけるYACの位置を決めるためにまずサイドを染色した。YAC b106に対応するアガロースの一部分が中央部でなくなっている。
【図19】創始マウスを解析するために用いたPCRのダイアグラムを示す。
【図20】創始マウスについての代表的なPCRを示す図である。トランスジェニックマウスで行った23のPCRについてのサンプル、マウス2、12、35、37、41および46で行ったものを示している、c− 陰性対照、c+ 陽性対照。
【図21】マウス35のいくつかの子孫についての例示的なPCRおよびAPP遺伝子の生殖細胞系への伝達を示す図である。マウス35の最初の7子孫で行った4つのPCRについての画像。TRP1、エキソン6、エキソン11およびLYS2のPCRを行った。子孫1、4、5および7がトランスジーンを受け継いでいることを観察することができる。
【図22】創始マウスのF1のサザンブロットの比較を示す図である。非トランスジェニック同腹子と比較したマウス35および64のフィンガープリント。YAC b106で得られたものと同一のバンドパターンを観察することができる。
【図23】ヒトAPP遺伝子発現を示す棒グラフを示す。系統35にけるヒトAPP遺伝子発現についての相対単位での対数表現。97%信頼区間。中枢および末梢神経組織において高い発現が検出される。精巣、血液、心臓および腎臓ではレベルがより低い。
【図24】ネズミAPP遺伝子発現を示す棒グラフを示す。マウスにおけるネズミAPP遺伝子発現についての相対単位での表現。このグラフは、95%信頼区間を用いて、脾臓における発現(最も低い発現)に対して標準化している。凡例では各棒が対応している組織を示している。
【図25】APPタンパク質発現および評価を示す図である。A.APPタンパク質およびアクチンについてのウエスタンブロット解析の代表的なゲル。組織:1 皮質、2 脳室、3 小脳、4 中脳、5 脊髄、6 肝臓、7 肺、8 膵臓、9 脾臓、10 小腸、11 筋肉、12 精巣、13 心臓および14 腎臓。B.APPレベルのグラフ表現。青色の棒、野生マウス、赤色の棒、トランスジェニックマウス。組織:1 皮質、2 脳室、3 中脳、4 小脳および5 脊髄。
【図26】系統35のトランスジェニックマウスの組織学的解析を示す図である。マウス脳のコンゴレッド染色。A 野生マウスの皮質。B 野生マウスの海馬。C 系統35の皮質。D 系統35の海馬。E 1年齢Tg2576マウスの海馬、ここではアミロイド生成性斑が観察される。
【図27】系統64のトランスジェニックマウスの組織学的解析を示す図である。マウス脳のコンゴレッド染色。A 野生マウスの皮質。B 野生マウスの海馬。C 系統64の皮質。D 系統64の海馬。E 1年齢Tg2576マウスの海馬、ここではアミロイド生成性斑が観察される。
【発明の詳細な説明】
【0001】
発明の属する技術分野
本発明は、バイオテクノロジー分野および薬学分野に含まれる:本発明は、特に非ヒトアルツハイマー病動物モデルとして使用するためのトランスジェニック非ヒト動物の開発およびその使用に関する。
【0002】
発明の背景
1907年に、Alois Alzheimerが初めて認知症と認識能力の進行性低下を特徴とする神経病理学的症候群を報告した。彼はまた、認知記憶の低下と、剖検で観察された神経病理学的病変と、患者の病歴との関連性も最初に証明した。アルツハイマー病(AD)の臨床的特徴は、幻覚、鬱病、失見当および攻撃行動を伴う、記憶、言語(失語)、運動機能(失行)および知覚(失認)の進行性消失である。
【0003】
Alzheimerによって行われた2つの神経学的所見は、ADを説明するために現在使用されている:(i)神経原繊維変化(無定形のニューロン内で見られる緻密な繊維のもつれであり、その主要なサブユニットは微小管関連タンパク質Tauである)および(ii)老人斑(細胞外斑であり、その主要成分はβ−アミロイドペプチド前駆体タンパク質(APP)と呼ばれる前駆体タンパク質由来のβ−アミロイド(Aβ)と呼ばれるペプチドである)。
【0004】
ヒトAPP遺伝子は21q21.3領域の第21染色体に位置し、300キロベース(kb)にわたって分布する18のエキソンによって形成される。その発現は特定組織に限定されず、内皮、脳のグリアおよびニューロンを含む多くの細胞種および組織で発現される。メッセンジャーRNA(mRNA)は成熟し、3つの主要なアイソフォームを産生するための代替法を受ける可能性がある。これらの3つの主要なアイソフォームにはそれらを形成しているアミノ酸の数により名前が付けられている(APP770、APP751およびAPP695)。最長のアイソフォームは完全遺伝子配列を含むAPP770であり;アイソフォームAPP751は最も多量にかつ普遍的に発現されるアイソフォームであり;そしてアイソフォームAPP695は脳において主要なアイソフォームであり、皮質での3つのmRNA間の比率はAPP770:APP751:APP695、1:10:20であり、そのため、アイソフォームAPP695がADに関連して最も研究されたアイソフォームであった。
【0005】
ADの発症に関与する、APP遺伝子の異なる突然変異が1990〜1992年の間に発見され、その伝達は、浸透度がほぼ100%の常染色体優性であることを確認することができた。1995年には、ADに関連する2つの他の遺伝子が発見され、それらの遺伝子はプレセニリン1(PS1)およびプレセニリン2(PS2)と呼ばれている。
【0006】
ADは、その原因に基づいて2つのタイプに分けることができる:
(i)家族性アルツハイマー病(FAD)(APP、PS1およびPS2遺伝子の突然変異に関連し、発症年齢が早期であり、アルツハイマー病患者のおよそ1%を占める);および
(ii)孤発性アルツハイマー病(SAD)(晩期発症型(通常60歳以後)であり、原因不明である(この疾患に対する罹患素因は一連の感受性遺伝子および環境因子によって決まるようであり、その罹患素因により複雑な多因子疾患となり、主な危険因子は老齢であることと家族歴があることである);SADはアルツハイマー病患者のおよそ99%を占める)。
【0007】
両疾患(FADおよびSAD)は臨床症状によっても神経病理学によっても区別することができない。
【0008】
知られているとおり、実験神経系疾患モデルの開発は生物医学研究に極めて重要である。ヒトにおける神経変性疾患の特徴によく似たモデルの開発には、大きな方法論的進歩が必要であった。ADの場合、主な進歩はFADに関与するタンパク質を同定することに関係していた。
【0009】
90年代の初めに、APP遺伝子におけるいくつかのFAD関連突然変異が発見され、それらの突然変異は、遺伝子導入技術によって得られる進歩とともに、ADを研究するためのモデルを開発する可能性をもたらした。遺伝子導入を用いて、病原型APP遺伝子を再現可能な、より適した方法で、脳の異なる領域において発現させることが可能であり、それにより、野生型APP、APP断片、突然変異APPタンパク質および他のAD関連遺伝子を発現するトランスジェニックマウスに大きな真実性が与えられた。
【0010】
ADのためにマウスにおいて開発されたモデルとしては、以下が挙げられる:
【0011】
NSEAPP:このマウスを作製するために、ニューロン特異的エノラーゼ(NSE)プロモーターおよびAPP遺伝子アイソフォーム751cDNAを有する構築物を調製した。この構築物の過剰発現は、月齢22ヶ月のときにびまん性Aβ斑を発生させた。これらの斑は、マウスの5%でのみ、アルツハイマー病患者に見られる斑によく似たより成熟した外観を呈し、Bielschowskyおよびチオフラビン−S染色液で陽性に染色された。
【0012】
APP YAC:多量のDNAを挿入することができる、トランスジェニックマウスを作製するための担体として酵母人工染色体(YAC)を使用することにより、3つのグループによってほぼ同時に、全APP遺伝子コード領域と該遺伝子の3’および5’の両方にある広範な領域を含む650kb YAC(B142F9 Washington University YAC library)を有するトランスジェニックマウスの作製が公開された。これらのマウスでは、ヒト遺伝子の発現は、マウスにおける内因性発現と同様に、斑の形成または神経原繊維変化を観察することなく行われた。ヒトゲノム配列決定では、これらのマウスを作製するのに用いられるYACは、APP遺伝子に加えて、望ましくない表現型を生じる可能性があるGABPA転写因子をコードする遺伝子も含むことが分かった。
【0013】
PDAPP:このトランスジェニックマウスは、1995年に報告されており、多様なAD神経病理を発現する初めてのマウスであり、「Indiana」V717F突然変異を含むAPP遺伝子cDNAを過剰発現させる構築物を用いて得られた;それらのマウスの脳では異栄養性神経突起(dystrophic neurites)に囲まれた神経炎性斑、星状細胞増加症、小膠細胞症およびシナプスの喪失を観察することができたが、神経原繊維変化は観察されなかった。皮質領域では斑の存在は50%占有に達した。この割合は、6%〜12%間である傾向があるアルツハイマー病患者のものと比較するならば、極めて高い割合である。脳においてこのように多量の斑が存在するにもかかわらず、これらのマウスにおいて神経変性は示されなかった。
【0014】
Tg2576:このマウスは、1996年に報告されており、おそらく、ADを研究するために最もよく用いられているモデルである;トランスジーンは、脳における高発現を確実にするプリオンタンパク質プロモーターによって形成され、遺伝的C57BL/6J//SJLバックグラウンドにおいて「Swedish」K670N/M671L突然変異を含むアイソフォームAPP695 cDNAを有する。Tg2576マウスは、脳におけるマウスの内因性発現レベルの5.6倍であるヒトAPP(hAPP)発現レベルを有する。それらのマウスは、月齢9〜12ヶ月のときには、すでに内側嗅領に神経炎性斑があり、後にその斑は海馬および皮質にまで広がった。
【0015】
APP23:ADを研究するためのこの新しいネズミモデルは、1997年に、Thy1プロモーターと、内因性発現の7倍を上回る発現が行われる、「Swedish」K670N/M671L突然変異を有するhAPP751 cDNAの過剰発現を用いて作製され、月齢6ヶ月のときに斑の発生を起こした。これらのマウスの最も重要な特徴は、月齢14〜18ヶ月のときに海馬のCA1領域における神経細胞死がようやく報告されたということである。このモデルのもう1つの重要な特徴は、ADを研究するための優れたモデルにする脳アミロイド血管症の発生である。これらのマウスの行動解析では、結果において大きな差があり、月齢18ヶ月からの学習において有意差があることが示された。
【0016】
今日までの作製された動物モデルでは、総てのAD病理を作製することができなかった。これらのモデルの作製は、野生型またはFAD関連突然変異を有するAPPタンパク質の過剰発現に基づいたものであり、そのようなモデルは、SAD患者では起こらない状況から出発するため、FADの研究に限定される。これらのモデルでは、マウス脳において脳領域の20〜50%を占めるアミロイド生成性斑(amyloidogenic plaques)の発生が報告されているが、一方、ヒト患者では斑の存在は12%を超えない。
【0017】
本明細書に記載のモデル、ヒトAPP YACマウス「hAPPy」では、APP遺伝子発現は、APP YACを用いて行われた先行研究に記載されているとおり、マウスの内因性発現とほぼ同じように行われた(Buxbaum, J. D., et al. (1993). "Expression of APP in brains of transgenic mice containing the entire human APP gene." Biochem Biophys Res Commun 197(2): 639-45; Lamb, B. T., et al. (1993). " Introduction and expression of the 400 kilobase amyloid precursor protein gene in transgenic mice [補正済み]. "Nat Genet 5(1): 22-30; Pearson, B. E. and T. K. Choi (1993). "Expression of the human beta-amyloid precursor protein gene from a yeast artificial chromosome in transgenic mice." Proc Nati Acad Sci USA 90(22): 10578-82)が、「hAPPy」マウスでは、これまでYAC中に存在していたGABPA遺伝子の存在が消失したという違いがあった(Lamb, B. T., et al. (1997). "Altered metabolism of familial Alzheimer's disease-linked amyloid precursor protein variants in yeast artificial chromosome transgenic mice." Hum Mol Genet 6(9): 1535-41.)。ADモデルの作製には、好適なAPP遺伝子発現と外部刺激によるその発現における考えられる応答が極めて重要である。
【0018】
これまで行われた、多くのAD関連神経病理を作成し、さらにFADをよりよく理解するという努力にもかかわらず、得られたモデルは、SADの原因やその発症を理解するためにそれらを使用することが難しいほど現実から離れている。このため、既存のモデルの欠点を克服するAD動物モデルを提供する必要性がまだある。
【0019】
ヒトAPPタンパク質発現、ひいてはヒトAβペプチド発現は、ネズミタンパク質はその配列が異なることによりアミロイド斑を形成することができないという事実によって、このモデルにおいてアミロイド斑の形成を可能にする。よって、異なる環境因子または病原因子を「hAPPy」に適用することで、APP遺伝子の正確な応答を可能にし、アミロイド斑の形成を可能にする。その疾患に大きく関与すると考えられるこれらの環境因子をそのモデルに適用する可能性により、そのモデルはSADを研究するための極めて重要なツールとなる。
【0020】
発明の詳細な説明
本発明は、一般に、アルツハイマー病(AD)を研究するための非ヒト動物モデルとして使用することができるトランスジェニック非ヒト動物に関する。前記トランスジェニック動物は、そのゲノムに挿入されており、完全ヒトAPP遺伝子(hAPP)のヌクレオチド配列をその調節配列とともに含む、異種ポリヌクレオチド(トランスジーン)を含む(該トランスジーンはhAPP遺伝子の自然環境に存在する他の遺伝子、例えばGABPA、ATPJおよびCYYR1遺伝子を欠いている)こと、そして該トランスジーンの発現がトランスジェニック動物において、ヒトにおけるこの同じhAPP遺伝子の内因性発現パターンと同様の内因性発現パターンを有することを特徴とする。本明細書において、表現「ヒトにおけるhAPP遺伝子の内因性発現パターンと同様のトランスジェニック動物におけるhAPPトランスジーンの内因性発現パターン」などは、該hAPPトランスジーンを含むトランスジェニック動物がhAPP遺伝子を、そのhAPP遺伝子がヒトにおいて発現される同じ組織において実質的に同じ割合で発現するという事実による。
【0021】
本発明によって提供されるトランスジェニック非ヒト動物は完全hAPP遺伝子だけをその調節配列とともに含むトランスジーンを含み、hAPP遺伝子の自然環境に存在する、その発現を妨げる可能性のある他の遺伝子(例えば、GABPA、ATPJおよびCYYR1遺伝子)の干渉を受けないという事実により、ヒトにおけるhAPP遺伝子の内因性発現パターンと同様の内因性発現パターンでの該トランスジェニック動物における該トランスジーンの発現を可能にする(すなわち該トランスジェニック動物はヒトにおける状況をかなりの信頼性で再現する)ため、該トランスジェニック動物は、AD、その進行および発症を研究するためだけでなく、それを引き起こしている要因および刺激を研究するためや該疾患の予防および/または治療のために潜在的に有用な化合物を同定するための非ヒト動物モデルとして使用することができる。
【0022】
よって、一態様では、本発明は、ヒト第21染色体(NCBI35:21:26064934:26642665:1)のヌクレオチド26,064,934とヌクレオチド26,642,665との間に含まれるヌクレオチド配列からなる単離ポリヌクレオチドまたは完全hAPP遺伝子をその調節配列とともに含む該ポリヌクレオチドの断片(以下本発明のポリヌクレオチド)に関する。前記ポリヌクレオチドは完全hAPP遺伝子のヌクレオチド配列をその調節配列とともに含み、hAPP遺伝子の該ヌクレオチド配列の末端に隣接する(flanking)それぞれの近接(adjacent)領域は固有の組換え配列を含み、完全hAPP遺伝子のヌクレオチド配列をその調節配列とともに単離するのにその固有の組換え配列を使用することができる。
【0023】
特定の一実施形態では、前記完全hAPP遺伝子は、その調節配列とともに、ヒト第21染色体(NCBI35:21:26174733:26464809:1)のヌクレオチド26,174,733とヌクレオチド26,464,809との間に含まれる。
【0024】
もう1つの特定の実施形態では、本発明のポリヌクレオチドは、ヒト第21染色体(NCBI35:21:26064934:26573344:1)のヌクレオチド26,064,934とヌクレオチド26,573,344との間に含まれるヌクレオチド配列からなる。前記ポリヌクレオチドは、完全hAPP遺伝子のヌクレオチド配列をその調節配列とともに含み、それらに少なくとも1つの固有の組換え配列を含む領域に隣接される。
【0025】
よって、もう1つの特定の実施形態では、図1に示されるように、前記hAPP遺伝子は、その5’末端において(APP遺伝子転写の始点および方向に関連)第1のヌクレオチド配列に隣接され(flanked)、その3’末端において(APP遺伝子転写の終点および方向に関連)第2のヌクレオチド配列に隣接され、ここで:
該第1のヌクレオチド配列は、ヒト第21染色体(NCBI35:21:26464810:26642665:l)のヌクレオチド26,464,810とヌクレオチド26,642,665との間に含まれるヌクレオチド配列、または少なくとも1つの固有の配列を含むその断片からなり、かつ
該第2のヌクレオチド配列は、ヒト第21染色体(NCBI35:21:26064934:26174732:1)のヌクレオチド26,064,934とヌクレオチド26,174,732との間に含まれるヌクレオチド配列、または少なくとも1つの固有の配列を含むその断片からなる。
【0026】
具体的な一実施形態では、前記第1のヌクレオチド配列は、ヒト第21染色体(NCBI35:21:26464810:26573344:l)のヌクレオチド26,464,810とヌクレオチド26,573,344との間に含まれるヌクレオチド配列、または少なくとも1つの固有の配列を含むその断片を含み、かつ前記第2のヌクレオチド配列は、ヒト第21染色体(NCBI35:21:26064934:26174732:1)のヌクレオチド26,064,934とヌクレオチド26,174,732との間に含まれるヌクレオチド配列、または少なくとも1つの固有の配列を含むその断片を含む。
【0027】
上述の第1および第2のヌクレオチド配列に含まれる(非反復)固有の配列は、本明細書に含まれる実施例に記載のとおり、相同組換えを利用して、その環境から完全hAPP遺伝子をその調節配列とともに単離するのに利用することができる。前記固有の配列は、当技術分野の水準において記載された当業者に公知のプロセスに従って同定することができる。例として、前記プロセスには一般にRepeatMasterプログラムなどの公的バイオコンピューター資源が含まれる[http://woody.embl-heidelberg.of/repeatmask/;Chenna Ramu et al., (2000) cgi-model: CGI Programming Made Easy with Python. LINUX Journal, July, 142-149; Smit, AFA and Green, P. RepeatMasker at http://repeatmasker.genome.washington.edu/cgi-bin/RM2_req.pl]; Jurka, J. 2000 Repbase Update: a database and an electronic journal of repetitive elements. Trend in Genetics 16(9): 418-420)]。
【0028】
特定の一実施形態では、前記第1のヌクレオチド配列は、ヒト第21染色体(NCBI35:21:26571876:26573344:1)のヌクレオチド26,571,876とヌクレオチド26,573,344との間に含まれる固有の配列(本発明ではSeq2と呼ぶ)を含む。
【0029】
もう1つの特定の実施形態では、前記第2のヌクレオチド配列は、ヒト第21染色体(NCBI35:21:26064934:26067221:1)のヌクレオチド26,064,934とヌクレオチド26,067,221との間に含まれる固有の配列(本発明ではSeqBと呼ぶ)を含む。
【0030】
当業者ならば、完全hAPP遺伝子とその調節配列との末端に隣接する前記第1および第2のヌクレオチド配列内に含まれるいずれの固有の配列も同じ目的(完全hAPP遺伝子とその調節配列のそれらの環境からの単離)に利用することができることは分かる。よって、本発明は、本明細書に記載の固有の配列(本発明の特定の一実施形態を構成する)に限定されるのみならず、前記第1および第2のヌクレオチド配列内に含まれる、当技術分野の水準の一部をなす当業者に公知のバイオコンピューターツールを利用して位置づけることができる総ての固有の配列を含む。
【0031】
本発明のポリヌクレオチドは好適なベクター内に含めることができる。よって、もう1つの態様では、本発明は、本発明のポリヌクレオチドを含むベクター(以下本発明のベクター)に関する。前記ベクターは、それゆえ、完全hAPP遺伝子のヌクレオチド配列をその調節配列とともに含み、それらに本発明に記載の固有の配列に隣接される。ベクターの選択は、そのベクターをその後に導入する宿主細胞によって決まる。一例として、前記ポリヌクレオチドを導入するベクターは酵母人工染色体(YAC)、細菌人工染色体(BAC)またはP1由来人工染色体(PAC)であってよい。よって、特定の一実施形態では、本発明のベクターは、形質転換されるゲスト細胞に応じて、YAC、BACまたはPACからなる群から選択される。
【0032】
特定の一実施形態では、本発明のベクターが導入される細胞が酵母である場合、好ましいベクターは、それぞれのアームにその機能を果たすための総ての好適な構造要素、例えばテロメア、セントロメア、自己複製配列および栄養要求性選択マーカーを含むYACである。本明細書に含まれる実施例では、酵母細胞を形質転換するのに用いられる上述の構造要素を有するYACの獲得について記載する。
【0033】
もう1つの特定の実施形態では、本発明のベクターが導入される細胞が細菌である場合には、好ましいベクターは、必要最小限の要素が複製起点とコピー数およびBAC分裂を制御する遺伝子であるBACである。
【0034】
YAC、BACおよびPACの特徴は当業者には公知である。前記種類のベクターについての詳細な情報は、例えば、Giraldo および Montoliuによって提供されている[Giraldo, P. & Montoliu L., 2001 Size matters: use of YACs, BACs and PACs in transgenic animals, Transgenic Research 10(2): 83-110]。
【0035】
よって、特定の一実施形態では、本発明は、その一方の末端において第3のヌクレオチド配列に隣接され、もう一方の末端(反対端)において第4のヌクレオチド配列に隣接される本発明のポリヌクレオチドを含む酵母を形質転換するのに有用なベクターに関し、この場合:
−前記第3のヌクレオチド配列は、(i)酵母細胞において機能的なテロメアおよび少なくとも(ii)1つの栄養要求性選択マーカーを含み、かつ
−前記第4のヌクレオチド配列は、(i)酵母細胞において機能的なセントロメア、酵母細胞において機能的なテロメアおよび少なくとも1つの栄養要求性選択マーカーを含む。
【0036】
酵母において機能的な、事実上いかなるテロメアおよびセントロメアも本発明において用いることができる。酵母において機能的な前記テロメアおよびセントロメアの限定されない例示的な例には、本明細書に含まれる実施例に記載されているものが含まれる。
【0037】
同様に、事実上いかなる栄養要求性選択マーカーも本発明において用いることができる。前記栄養要求性選択マーカーの限定されない例示的な例には、TRP1、LEU2、LYS2、HIS3、HIS5、TRP1およびURA3が含まれる。
【0038】
本発明のベクターは、当業者に公知の従来の方法によって得ることができる[Sambrok et al., 1989 "Molecular cloning, a Laboratory Manual", 2nd ed., Cold Spring Harbor Laboratory Press, N.Y. Vol 1-3]。
【0039】
本発明のベクターは、例えば、酵母細胞、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)などの真核細胞、または例えば、細菌、大腸菌(Escherichia coli)などの原核細胞を形質転換するのに用いることができる。
【0040】
よって、もう1つの態様では、本発明は、本発明のポリヌクレオチド、または本発明のベクターを含む細胞(以下本発明の細胞)に関する。このような細胞は、当業者に公知の従来の方法によって得ることができる[Sambrok et al., 1989 "Molecular cloning, a Laboratory Manual", 2nd ed., Cold Spring Harbor Laboratory Press, N.Y. Vol 1-3]。
【0041】
記載する本発明のポリヌクレオチド、ベクターまたは細胞は、そのゲノムに、完全hAPP遺伝子のヌクレオチド配列がその調節配列とともに挿入されたトランスジェニック非ヒト動物を得るのに用いることができる。
【0042】
よって、もう1つの態様では、本発明は、そのゲノムに挿入された本発明のポリヌクレオチドを含むトランスジェニック非ヒト動物(以下本発明のトランスジェニック非ヒト動物)に関する。結果として、本発明のトランスジェニック非ヒト動物のhAPP遺伝子の内因性発現パターンは、この同じhAPP遺伝子がヒトにおいて示す発現パターンと同様である。いずれかの理論に拘束される訳ではないが、これは、主に、その遺伝子がそれ自体の調節配列により調節されるという事実によるものと思われる。このことは、本発明のトランスジェニック非ヒト動物におけるhAPP遺伝子発現を、ヒトにおいてその発現を誘導する同じ因子によって誘導することを可能にし、そのトランスジェニック非ヒト動物をヒトにおいて起こるADの現象および特徴を確実に再現する動物モデルにする。
【0043】
本発明の特定の一実施形態では、本発明のトランスジェニック非ヒト動物は、哺乳類、好ましくは齧歯類、より好ましくはマウスまたはラットである。
【0044】
本発明のトランスジェニック非ヒト動物は、当業者による技術水準において公知のものの任意の遺伝的背景を有していてよい、すなわち該トランスジェニック非ヒト動物は、野生型(wt)動物由来のものであってよく、またはADと直接または間接的に関係している任意の分子マーカーを組み込んでいる遺伝的背景を有する非ヒト動物由来のものであってよい。よって、特定の一実施形態では、本発明のトランスジェニック非ヒト動物は、hAPP遺伝子の他に、ADの発症と直接または間接的に関係している1つ以上の遺伝子を発現することができる。ADの発症と直接または間接的に関係している事実上いかなる分子マーカーも用いることができる。ADの分子マーカーとして認識されている前記遺伝子の限定されない例示的な例には、プレセニリン1(PS1)遺伝子、プレセニリン2(PS2)遺伝子およびアポリポタンパク質E(APOE)遺伝子のε4対立遺伝子が含まれる。
【0045】
よって、特定の一実施形態では、本発明のトランスジェニック非ヒト動物のゲノムは、本発明のポリヌクレオチドの他に、1つ以上のAD分子マーカーを含む。限定されない例として、前記AD分子マーカーは、PS1、PS2遺伝子、およびAPOE遺伝子のイプシロン4(ε4)対立遺伝子からなる群から選択される。
【0046】
ADを発症しやすい素因に関連する他の遺伝因子には、α2−マクログロブリン遺伝子、超低密度リポタンパク質受容体遺伝子、リポタンパク質受容体関連タンパク質LRPの遺伝子、Tauタンパク質の遺伝子、ブチルコリンエステラーゼKの遺伝子、カテプシンDの遺伝子、インターロイキンの遺伝子およびコリンアセチルトランスフェラーゼの遺伝子が含まれる。
【0047】
本発明の非ヒトマウスの遺伝的背景は、前記AD分子マーカーの対立遺伝子変異体、突然変異体および多型をさらに含み得る。
【0048】
完全ヒトAPP(hAPP)遺伝子のヌクレオチド配列がその調節配列とともに本発明のトランスジェニック非ヒト動物のゲノム中に存在することにより、該遺伝子を、この同じ遺伝子がヒトにおいて示す発現パターンと同様の内因性発現パターンで発現させることを可能にするため、該トランスジェニック動物を、ADを研究するためのトランスジェニック非ヒト動物モデルとして使用することができる。前述のとおり、本発明のトランスジェニック非ヒト動物は、必要に応じて、さらなるAD分子マーカーも有していてよい。この総てにより、一般にはADだけでなく、異なる異型アルツハイマー病、例えばFADおよびSADを研究することも可能にする。
【0049】
よって、もう1つの態様では、本発明は、ADの研究における本発明のトランスジェニック非ヒト動物の使用に関する。特定の一実施形態では、ADはFADおよびSADから選択される。
【0050】
もう1つの態様では、本発明は、該疾患の病因、原因、誘発因子、その疾患の確立につながる環境的および/または生理病理学的攻撃または傷害を研究するためのADモデルとしての本発明のトランスジェニック非ヒト動物の使用に関する。
【0051】
その限定されない例示的な例には、年齢、頭蓋外傷、女性、ヘルペス型(HSV1)ウイルスおよび細菌、細菌、例えば、クラミジア・ニューモニエ(Chlamydia pneumoniae)などのような病原体の作用、脳内でのアルミニウム、亜鉛型金属の蓄積などが含まれる。
【0052】
もう1つの態様では、本発明は、ADの治療および/または予防のために潜在的に有用な化合物をスクリーニングし、発見し、同定し、評価し、および妥当性を立証するための本発明のトランスジェニック非ヒト動物の使用に向けられる。前記化合物は、いかなる性質の化合物であってもよく、例えば、単離されたまたは1種以上の異なる化合物と混合された化学的、生物学的、微生物学的化合物などであってよく、既知または未知組成および構造を有する化合物、任意の既知治療用途を有する医薬製剤、生物学的製剤、微生物学的製剤など、例えば、有機または無機化学化合物、ペプチド、タンパク質、核酸、抽出物などが含まれる。
【0053】
本発明のトランスジェニック非ヒト動物、例えば、マウスは、それらの先祖の遺伝子型および表現型特徴を有する非ヒト動物を得るために、同じ種の他の非ヒト動物との交配を用いてさらに使用することができる。本発明の非ヒト動物と交配させることができる非ヒト動物の例には:
−APP遺伝子、あるいはADと直接または間接的に関係している他の遺伝子についてのノックアウト非ヒト動物;
−ADに関与する遺伝子の対立遺伝子変異体、多型または突然変異体を発現する非ヒト動物、あるいは
−ADモデルとして使用することができる他の非ヒト動物
が含まれる。
【0054】
特定の一実施形態では、ADと直接または間接的に関係している遺伝子は、PS1、PS2遺伝子、およびAPOE遺伝子のイプシロン4(ε4)対立遺伝子、またはその疾患に関連する他の遺伝的素因因子、例えばα2−マクログロブリン遺伝子、超低密度リポタンパク質受容体遺伝子、リポタンパク質受容体関連タンパク質LRPの遺伝子、Tauタンパク質の遺伝子、ブチルコリンエステラーゼKの遺伝子、カテプシンDの遺伝子、インターロイキンの遺伝子およびコリンアセチルトランスフェラーゼの遺伝子からなる群から選択される。
【0055】
もう1つの特定の実施形態では、本発明のトランスジェニック非ヒトマウスは、数回の交配後に、hAPPトランスジーンだけを発現するマウスが得られるように、ネズミAPP(mAPP)遺伝子についてのノックアウトマウスと交配させることができる。
【0056】
よって、もう1つの態様では、本発明は、hAPPトランスジーンだけを発現し、mAPP遺伝子を発現しないトランスジェニック非ヒト動物を得るための本発明によるトランスジェニック非ヒト動物の使用に関する。
【0057】
もう1つの態様では、本発明は、ADの治療および/または予防のために潜在的に有用な化合物を同定するための方法であって、
a)本発明のトランスジェニック非ヒト動物に候補化合物を投与すること;
b)工程(a)の該トランスジェニック非ヒト動物における、hAPP遺伝子の発現パターンおよび/またはhAPPタンパク質産生および/またはβ−アミロイド(Aβ)ペプチド産生を決定すること、および
c)該トランスジェニック非ヒト動物において(i)神経組織におけるhAPP遺伝子発現レベルの低下、(ii)hAPPタンパク質産生および/もしくは蓄積の減少、ならびに/または(iii)Aβペプチド産生および/もしくは蓄積の減少を引き起こす該候補化合物を選択すること
を含む方法に関する。
【0058】
神経組織におけるhAPP遺伝子発現レベル、hAPPタンパク質産生および/もしくは蓄積レベル、ならびにAβペプチド産生および/もしくは蓄積レベルは、定量RT−PCR、ノーザンブロット、ウエスタンブロットなどのような当業者に公知の従来の技術によって測定することができる。hAPP遺伝子発現レベルまたはhAPPタンパク質蓄積レベルの好ましい範囲は、それぞれ、図24および図26で見ることができる。
【0059】
特定の一実施形態では、対照レベルと比べて、hAPP遺伝子発現レベルの低下、またはhAPPタンパク質産生および/もしくは蓄積レベルの減少、またはAβペプチド産生および/もしくは蓄積の減少、を引き起こす前記候補化合物が選択される。前記化合物は、上記化合物のいずれであってもよい。
【0060】
本発明のトランスジェニック非ヒト動物は、当業者に公知の任意の遺伝子導入法例えばマイクロインジェクション、エレクトロポレーション、粒子衝撃、細胞形質転換とその後のクローニング(形質転換に成功した細胞の細胞核を除核卵に移し、受容体女性に移植する)、生殖体形質転換(卵母細胞または精母細胞に遺伝子を導入し、形質転換された生殖体を受精に使用し、完全動物を作り出すこと)および/または卵細胞質内精子注入法(ICSI)によって得ることができる。
【0061】
特定の一実施形態では、本明細書に含まれる実施例に記載のとおり、用いられる遺伝子導入法は卵細胞質内精子注入法に基づく。前記技術については国際特許出願WO 2005/098010に詳細に記載されている。
【0062】
これまでに記載された遺伝子導入法によって得られる動物は、本発明のトランスジェニック非ヒト動物の特徴的な遺伝子型および表現型を有し、それらは、それらのゲノムに挿入された、完全hAPP遺伝子のヌクレオチド配列とその調節配列とを含み、該hAPP遺伝子は、この同じ遺伝子がヒトにおいて示す発現パターンと同様の内因性発現パターンを有する。
【0063】
本発明のトランスジェニック非ヒト動物のように、これまでに記載された遺伝子導入法によって得られるトランスジェニック非ヒト動物もまた、それらの先祖の遺伝子型および表現型特徴を有するトランスジェニック非ヒト動物を得るために、同じ種の他の非ヒト動物と交配させ、ADを研究するためのモデルとして使用することができる異なる非ヒト動物を生み出すことができる。
【0064】
加えて、本発明に記載の遺伝子導入法によって得られるトランスジェニック非ヒト動物の子孫も、ヒトにおけるhAPP遺伝子タンパク質のパターンと同様のhAPP遺伝子の内因性発現パターンを有し得る。
【0065】
本発明のもう1つの態様は、それゆえ、本発明に記載の遺伝子導入法に従って得ることができる、ヒトにおける前記遺伝子の発現パターンと同様のまたは同等の発現パターンでヒトAPP遺伝子を内因的に発現するトランスジェニック非ヒト動物の子孫に関する。
【0066】
次の実施例により本発明を説明するが、本発明を限定する意味に解してはならない。
【実施例】
【0067】
完全ヒトAPP遺伝子を、ヒトにおける該遺伝子の発現パターンと同等の発現パターンで内因性発現するトランスジェニック非ヒト動物の獲得
【0068】
材料および方法
1.プラスミドの作製
1.1 一般的なクローニング
通常の分子生物学技術(Sambroock, J., 1989, 上記; Ausubel FM, B. R., Kingston RE, Moore DD, Seidman JG, Smith JA and struhl, K. (1999). Current Protocols in Molecular Biology, John Wiley and Sons.)と本発明者らの研究室で改良した技術(Montoliu, 1997 Lab protocols. Generation of a transgenic mouse)を用いて、この研究に必要なベクターを作製した。
【0069】
1.2 DNAの電気泳動
アガロースゲルでの電気泳動を用いて、異なるDNA断片の分離、同定および定量を実施した。100bp〜12kbの様々な分子を分離することができる、0.5〜2%間に含まれる濃度のUltra Pure Agarose(GIBCO-BRL)を用いた。
【0070】
水平型Ecogenセル(6.5x8cmおよび11x15cm)を使用し、それらのセル内でSegainvex電源を用いて電流量5V/cmを適用した。ゲルは、TAEバッファー(40mM Tris−Acetate、2mM EDTA(エチレンジアミン四酢酸) pH8(Merck))中に入れ、臭化エチジウム(Roche)を0.5μg/mlで加えて調製した。
【0071】
1.3 DNAの定量
必要な精度および予測濃度に応じて、異なる方法を用いてDNAの定量を実施した。予測濃度範囲が0.05〜2μg/μl間に及ぶサンプルにおけるDNA濃度の推定を、波長260nmでのDNA吸光度の分光光度測定に基づき、SHIMADZU社製UV−1601型分光光度計において行った。測定は会社の使用説明書に従って実施し、波長260nmでの光学濃度(OD260)の一単位は50μg/mlの二本鎖DNAに相当することを考慮して、DNA濃度を次の方程式に従って算出した:
【数1】
【0072】
Hoefer DyNA Quant 200 Fluorometer(Amersham Pharmacia Biotech)を使用して、濃度が0.01〜0.05μg/μl間であると推定されるサンプルを評価した。この方法では、ビスベンズアミド(Hoeschst 33258)の蛍光を測定し、このビスベンズアミドの分子は二本鎖DNAと結合する能力を有し、365nmの紫外線を用いてそれを励起すると蛍光光度計により検出することができる蛍光を発する。この方法では、遊離ヌクレオチドまたはRNAによるバックグラウンドが抑制されることから、小濃度範囲で測定するのに非常に有用である。これら総ての測定をnanodrop(NanoDrop、ND−1000分光光度計)を用いて検証した。
【0073】
1.4 ポリメラーゼ鎖増幅反応(PCR)
組換えを解析するため、さらにトランスジェニックマウスへのその組込みを確認するための特異的YAC領域のクローニングおよび増幅用のDNA断片を、PCRを用いて得た。
【0074】
PCRでは各増幅断片に特異的な条件が必要である。混合物の反応濃度は、温度、増幅時間およびサイクルと同様に、それぞれの反応によって変化する。総ての反応についての標準的な出発条件は以下のとおりである:1.5mM MgCl2(Biotools)、200μMデオキシヌクレオチド[dATP、dCTP、dTTPおよびdGTP(Roche)]、1μMの特異的オリゴヌクレオチド対(Isogen)、1x反応バッファー(Biotools)および1単位のTaqポリメラーゼ(Biotools)、合計量25μl。反応はMJ Researchys社製PCT−100モデルサーマルサイクラーにおいて0.2mlチューブ(MJ Research)中で行った。使用するプログラムは増幅する断片に応じて変更した。それらのプログラムは、一般に、95℃で5分間の最初のDNA変性、その後3温度のサイクル、95℃で30秒、オリゴヌクレオチドハイブリダイゼーション温度に応じて55〜65℃間で30秒および72℃で1分の35回の反復で構成された。断片を完全増幅するために、それらを最後に72℃で10分に供した。
【0075】
1.4.1 酵母コロニーのPCR
異なる相同組換えを研究するために、酵母コロニーを解析した。コロニーの直接PCRを行うために、コロニーの増殖を防ぎ、そこからDNAを抽出して、コロニーをPCRチューブ(0.2ml)に移し、そのPCRチューブ中でそれを50μlのザイモリアーゼ(20U/ml)中に再懸濁し、室温で10分間インキュベートした。それらのチューブをEppendorf社製5417c卓上遠心機で5,000r.p.mにて遠心分離し、上清を除去し、細胞を95℃で5分間加熱した。その沈殿物を15μlの水中に再懸濁し、5μlを用いて、PCRを行った。
【0076】
1.5 アガロースゲルからのDNAの精製および抽出
10kb未満のDNA断片を、TAEバッファー中のアガロースゲルを用いて精製した。ゲル中で断片を分離した後、それらを紫外線灯(Upland)を用いて確認した。その後、滅菌ナイフでバンドを切り取り、Eppendorf社製チューブに入れた。QIAagen社製Qiaquick gel extraction kitを、そのプロトコールに記載の使用説明書に従って用いてゲルからDNAを抽出した。
【0077】
1.6 原核細胞
1.6.1 細菌株および培養
組換えプラスミドを市販の大腸菌株中で増幅した。サイズが約5kbであるプラスミドを増幅し、DH5α細胞(Gibco)に形質転換し、一方、10kbに近いサイズのプラスミドを増幅し、TOP10F’細胞(Stratagen)に形質転換した。
異なるプラスミドの増幅のための細菌増殖は、LB(Luria−Bertani)液体培地中、37℃で少なくとも8時間、攪拌しながら(250r.p.m.)、アンピシリン(Sigma)の存在下で、終濃度50μg/mlで行った。コロニーのスクリーニングは、形質転換体を寒天およびLB培地を入れたペトリ皿で、アンピシリンの存在下で増殖させた後、カラースクリーニングを実施し得る場合にはX−galおよびIPTGを用いて、実施した。
【0078】
1.6.2 コンピテント細菌の調製および形質転換
コンピテントDH5α細胞を、塩化カルシウム法を用いて調製し、熱ショック法を用いて形質転換した。
【0079】
大型プラスミドは市販のTOP10F’株(Stratagen)を用いて形質転換した。エレクトロポレーションは、予め氷冷しておいた直径2mmのエレクトロポレーション用セル(Bio Rad)中で、Bio-Rad社製Gene Pulserを会社の指示に従って用いて、25μF、2.5KVおよび20Ωの条件でパルスを与えて実施した。
【0080】
1.6.3 プラスミドDNAの精製
小規模プラスミドDNA精製を、抗生物質を含むLB培地2mlに問題のコロニーを植菌して行った。これらの培養物を37℃で少なくとも8時間、攪拌しながら(250r.p.m.)増殖させた。プラスミドDNAはアルカリ溶菌法を用いて抽出した。より高い濃度および量でのプラスミドDNAの調製は、大量細菌(100〜500ml)培養を利用して、市販のQiagen maxi kits(Qiagen)とFlexiprep Kits(Amersham Pharmacia)を各キットの具体的なプロトコールに従って用いて行った。
【0081】
2.オリゴヌクレオチド
YACにおけるAPP遺伝子のフランキング5’および3’領域と、最近コード遺伝子領域までのそれらの距離の範囲を定めるのに用いられるオリゴヌクレオチドは:
APP3'-B-U 5'-atcctaagaggaagggatcttaagg-3' [配列番号:1]
APP3'-B-L 5'-gggagaacaaacatacctcactagc-3' [配列番号:2]
APP3'-A-U 5'-ccagcatgttttccaaagtatgag-3' [配列番号:3]
APP3'-A-L 5'-tgaaatgtccccacactgatcactg-3' [配列番号:4]
APP3'-1-U 5'-gtatagtatggatggttcaaacagagccta-3' [配列番号:5]
APP3'-1-L 5'-aaggtaaagctctagaatctatcagtgcct-3' [配列番号:6]
APP3'-2-U 5'-tttagaagagctcagtaagaatccacattt-3' [配列番号:7]
APP3'-2-L 5'-cattttgtcgttactagtaccattggtttc-3' [配列番号:8]
APP5'-1-U 5'-ctcaaaaacagcaacacagatgtgc-3' [配列番号:9]
APP5'-1-L 5'-gagcttaaacaccttcctctgc-3' [配列番号:10]
APP5'-2-U 5'-tggggagaggaatggaatttggagc-3' [配列番号:11]
APP5'-2-L 5'-gtcatttccagaaaaagcccactgc-3' [配列番号:12]
APP5'-3-U 5'-tcaaggacatggagattgggcaggc-3' [配列番号:13]
APP5'-3-L 5'-ggggtaacaggttgccggtcatatg-3' [配列番号:14]
APP5'-4-U 5'-caccagaccatgatgtctcagtagc-3' [配列番号:15]
APP5'-4-L 5'-ctgcaaaacagccctcatattctgc-3' [配列番号:16]
APP5'-5-U 5'-tagcatcctttgctaagccagttgc-3' [配列番号:17]
APP5'-5-L 5'-gcttgttattggaggttccagcacg-3' [配列番号:18]
APP5'-6-U 5'-tccaacgggggagtgagtgaaaggc-3' [配列番号:19]
APP5'-6-L 5'-gcctcactcctgcaaacgtgcccaa-3' [配列番号:20]
APP5'-7-U 5'-tctttcttccaccttggtatcctgc-3' [配列番号:21]
APP5'-7-L 5'-caggatgtctctggatttttactcg-3' [配列番号:22]
である。
【0082】
APP遺伝子のエキソン、ならびにGABPA遺伝子の存在を確認するのに用いられるオリゴヌクレオチドは以下である:
APP-Ex1-U 5'-agtttcctcggcagcggtagg-3' [配列番号:23]
APP-Ex1-L 5'-ccagcaggagcagtgccaaac-3' [配列番号:24]
APP-Ex2-U 5'-aagaccgggctgattcctaa-3' [配列番号:25]
APP-Ex2-L 5'-tccaacgtgaattgctagcc-3' [配列番号:26]
APP-Ex3-U 5'-cccaagcattttggataagg-3' [配列番号:27]
APP-Ex3-L 5'-cctctttttcttccctcaag-3' [配列番号:28]
APP-Ex4-U 5'-ttgattgggttgcttaggca-3' [配列番号:29]
APP-Ex4-L 5'-tgttgcctcaaaatacccct-3' [配列番号:30]
APP-Ex5-U 5'-ctaccactcactgttttctc-3' [配列番号:31]
APP-Ex5-L 5'-gcagagaccttttcagtgat-3' [配列番号:32]
APP-Ex6-U 5'-tgccaaaattccatatggacg-3' [配列番号:33]
APP-Ex6-L 5'-gggatttgccaagcagcatat-3' [配列番号:34]
APP-Ex7-U 5'-ccactgggaggattaaaaga-3' [配列番号:35]
APP-Ex7-L 5'-gagaagtggacagaaatgtg-3' [配列番号:36]
APP-Ex8-U 5'-tgtcagtggactcgtgcatt-3' [配列番号:37]
APP-Ex8-L 5'-catctcaagctgtctggcaa-3' [配列番号:38]
APP-Ex9-U 5'-catgtcttcagcaccaactg-3' [配列番号:39]
APP-Ex9-L 5'-caaactgtgcccacacagta-3' [配列番号:40]
APP-Ex10-U 5'-cagataggaaggggtatgta-3' [配列番号:41]
APP-Ex10-L 5'-ggagcaaatataaggcagga-3' [配列番号:42]
APP-Ex11-U 5'-tgatgagggttggagagtgca-3' [配列番号:43]
APP-Ex11-L 5'-caagatggaatggacaggggt-3' [配列番号:44]
APP-Ex12-13-U 5'-cctcgtcacgtgttcaatat-3' [配列番号:45]
APP-Ex12-13-L 5'-caacttcatcctgaatctcc-3' [配列番号:46]
APP-Ex14-U 5'-cacgtgaaagcagttgaagt-3' [配列番号:47]
APP-Ex14-L 5'-ttgccacctatacaatggag-3' [配列番号:48]
APP-Ex15-U 5'-ctggcacatcaatagcgata-3' [配列番号:49]
APP-Ex15-L 5'-actcggaacttgggaaatga-3' [配列番号:50]
APP-Ex16-U 5'-taaaggcagcagaagcctta-3' [配列番号:51]
APP-Ex16-L 5'-gctcagcctagcctatttat-3' [配列番号:52]
APP-Ex17-U 5'-accagttgggcagagaatat-3' [配列番号:53]
APP-Ex17-L 5'-cttgagcagaatattcacgg-3' [配列番号:54]
APP-Ex18-U 5'-cttggtaattgaagaccagc-3' [配列番号:55]
APP-Ex18-L 5'-cttttgacagctgtgctgta-3' [配列番号:56]
GABPA-Ex-U 5'-tattattacgatggggacat-3' [配列番号:57]
GABPA-Ex-L 5'-gtaaagcaacatctaaagca-3' [配列番号:58]
組換えに用いた異なるクローニングベクターの配列決定に用いられるオリゴヌクレオチドは:
SV40A-2 5'-gtgaaggaaccttacttctgtggtg-3' [配列番号:59]
Sp6プロモーター 5'-atttaggtgacactata-3' [配列番号:60]
T7プロモーター 5'-taatacgactcactataggg-3' [配列番号:61]
である。
【0083】
酵母URA遺伝子のクローニングに用いられるオリゴヌクレオチドは:
URA3-U 5'-gcccagtattcttaacccaaactgca-3' [配列番号:62]
URA3-L 5'-cttccacccatgtctctttgagcaa-3' [配列番号:63]
である。
【0084】
栄養要求性YAC配列を検出するのに用いられるオリゴヌクレオチドは:
TRP1 5'-gcccaatagaaagagaacaattgacc-3' [配列番号:64]
TRP2 5'-acacctccgcttacatcaacacc-3' [配列番号:65]
LYS1 5'-accaagccagcatctgtatcacc-3' [配列番号:66]
LYS2 5'-gccaaatccatccacttctcatc-3' [配列番号:67]
である。
【0085】
サザンブロット解析用のプローブを作製するのに用いられるオリゴヌクレオチドは:
プローブAPP-seq2-U 5'-tgaatcttggcatttgggcc-3' [配列番号:68]
プローブAPP-seq2-L 5'-cctgtaacttgccctcatag-3' [配列番号:69]
プローブAPP-seqA-U 5'-gaaacgagttgaaaggcacag-3' [配列番号:70]
プローブAPP-seqA-L 5'-tttacccttccatgaagtccc-3' [配列番号:71]
プローブAPP-seqB-U 5'-ggcttgtaaattaaactctgaagc-3' [配列番号:72]
プローブAPP-seqB-L 5'-cctcagtggctgttttgagagcat-3' [配列番号:73]
である。
【0086】
3.酵母人工染色体の修飾
3.1 真核細胞
酵母において実施した総ての試験はS.セレビシエ AB1380株において実施し、その代謝特性は:Mata、ura3−52、trp1、ade2−1、lys2−1、his5およびcan1−100であった。使用したオリジナルクローンは、Washington Universityで購入し(Human Yeast Artificial chromosome Clone)、そのクローンはYAC B142F9を含むものであった(完全ヒトAPP遺伝子を含む650KbのYAC)。
【0087】
YAC担体酵母は、時々起こるYAC消失を防ぐために独立栄養セレクター(autotrophic selectors)を加えた液体および固体培地で増殖させた。液体培養物を30℃で2〜3日間、攪拌しながら(200r.p.m.)増殖させ、一方、酵母を30℃で4〜5日間増殖させて、プレートでコロニーを得た。
【0088】
選択培地はD+Glucose(Merck)、YNB(Yeast Nitrogen Base w/o Amino Acids)(Difco)および対応アミノ酸(Serva)を加えて調製した。
【0089】
3.2 酵母における相同組換え
酵母細胞の形質転換を2つの方法により実施し、それらの方法ではそれぞれ非常に異なる結果を生じた:酢酸リチウム(LiAc)法(Markie, D. (1995). YAC Protocols_[Please complete the reference]; Alison, A. G., D; Kaiser, C; Stearns, T (1997). Methods in Yeast Genetics, Cold Spring Harbor Laboratory Press.)および酵母細胞エレクトロポレーション(Becker, D. M. and L. Guarente (1991). "High-efficiency transformation of yeast by electroporation." Methods Enzymol 194: 182-7)。
【0090】
3.3 DNA−DNAハイブリダイゼーション(サザンブロット)を利用した解析
サザンブロット技術を利用して陽性クローンの解析を実施した。2〜4μg間のゲノムDNAを、4mMスペルミジンを含む1x消化バッファー中40〜60Uの対応酵素[Eco RV、Sca I、Bgl II、Bam HI、Hind III、Sal IおよびEco RI]で消化した。その消化物を酵素切断温度で12〜14時間インキュベートした。それらの消化産物を水平型Horizonセル(20x25cm)(Life Technologies)内で0.8%アガロースゲルにおいて電気泳動により分離した。
【0091】
キャピラリーを用いてDNAをHybond(商標)−Nナイロン膜(Amersham)に転写した。そのためには、SSC 20Xトランスファーバッファー[3M NaCl、0.3Mクエン酸ナトリウム]中で14〜16時間を要した。ゲルは、DNAの脱プリン化のための0.25N HClでの洗浄、そしてDNAの中和および変性のための0.4Mでの15分洗浄2回を行って予め処理した。
【0092】
一度、DNAが膜に転写されたら、CL−1000 Ultraviolet Crosslink(UVP-Stratagen)を用いて紫外線照射により、パルス70,000mJ/cm2を2回与えてそれを固定した。
【0093】
酵素制限によりまたはPCRを用いて単一コピープローブを得、50ngのDNA、25μCiのα−32P−dCTP(Amersham)およびHigh Prime kit(Roche)を会社の使用説明書に従って用いて放射能標識した。標識プローブから遊離ヌクレオチドを除去するために、かかるプローブをProbe Quant G50カラム(Amersham)に通して精製した。プローブの活性をW 1414シンチレーションカウンター(Beckmann)で定量し、およそ1x106cpm/μlの活性を得た。
【0094】
ハイブリダイゼーションを行い、膜を対応するプローブ(配列番号68、配列番号69、配列番号72および配列番号73)とともに65℃で14〜16時間インキュベートした。
【0095】
ハイブリダイズされたフィルターを、増感スクリーン(Miersham)と写真用フィルム(X-Omat, Kodak)の入った露光カセット内で−80℃で数日間露光した。あるいは、フィルターをPhosphorlmagerのカセット(Molecular Dynamics)内で室温で1〜2日間露光し、そのシグナルをIQMac v 1.2プログラム(Molecular Dynamics)を用いて解析した。
【0096】
3.4 酵母細胞を含むアガロースブロックの小規模調製
YACの修飾を解析するために、全細胞を含むアガロースブロックを調製し、そうすることによって、染色体を無傷のまま保つこと、そして染色体によって受けるサイズ変化をパルスフィールド電気泳動を利用して確認できることを実現した。次のプロトコールに従った:
【0097】
10mlの選択培地に、解析する酵母クローンを接種し、それを30℃で攪拌しながら(200r.p.m.)飽和まで増殖させた。その培養物を3,000r.p.mにて室温で5分間遠心分離し、上清を廃棄し、沈殿物を10mlの50mM EDTA pH8中に再懸濁し、再度3,000r.p.mにて室温で5分間遠心分離した。この際、その沈殿物を300μlの溶液I(1Mソルビトール、20mM EDTA pH8、14mM β−メルカプトエタノールおよび2mg/mlのzymolyase 20T)中に再懸濁し、40℃に温度調整した。一度、温度調整したら、10mg/mlのzymolyase 20T溶液50μlを加え、その後、40℃に温度調整した500μlの溶液II(2%のアガロースを含有する溶液I、Sea Plaque)を加えた。それを、先端を切った青色チップをつけたピペットを用いて混合し、それを、先端を切った黄色チップをつけた200μlピペットを用いて型に分配した。各型には80μlを加えた。アガロースが固化し得るようにそれらを氷中で10分間インキュベートし、一度固化したら、それらのキューブをまた、4mlの溶液III(1Mソルビトール、20mM EDTA pH8、10mM Tris pH7.5、14mM β−メルカプトエタノールおよび2mg/mlのzymolyase 20T)を入れた10mlチューブに移し、37℃で1〜3時間、ゆっくりと攪拌しながらインキュベートした。その後、溶液IIIを5mlの溶液IV(1%ラウリル硫酸塩、100mM EDTA pH8、10mM Tris pH8)と交換し、それを再度37℃で1時間インキュベートした。続いて、溶液IVを新しい溶液と置き換え、37℃で一晩インキュベートした。翌日、キューブを5mlの20%NDS(0.5M EDTA、1mM Trizma base、34mM N−ラウリルサルコシン、pH9)で室温で2時間洗浄し、それらを新しい溶液と交換した。それらは4℃で保存し得る。
【0098】
3.5 パルスフィールドゲル電気泳動(PFGE)
総てのゲルを0.5xTBEバッファー(100mM Tris−Borate、1mM EDTA pH8.3)中1%のアガロース LMP Sea Plaqueに調製した。パルスフィールド電気泳動システムは、Gene Navigator System(Amersham)のものであった。酵母マーカー PFGEマーカーII(Roche)を分子量マーカーとして使用し、それらのマーカーはS.セレビシエ YPH 755のアガロースキューブに対応する(染色体 90kb、220kb、280kb、360kb、450kb、560kb、610kb、700kb、770kb、810kb、850kb、920kb、960kb、1,010kb、1,100kb、1,600kbおよび2,200kb)。
【0099】
最初のYACはもともとサイズがおよそ650kbであったが、修飾後の最終的なサイズは520kbであった。そのため、実施した総てのパルスフィールド電気泳動ではこのサイズの染色体を最適に分離するプログラムを使用した。このプログラムは200Vで26時間続き:最初の12時間は50秒のパルス、残る14時間は100秒のパルスであった。一度、ゲルが泳動し終えたら、それを臭化エチジウム(EtBr)(0.5μg/ml)を含む電気泳動バッファー1〜2l中で30分間染色した。
【0100】
得られたYACを解析するために、染色体をHybond(商標)−Nナイロン膜(Amersham)に転写して、解析した。転写は、いくつかのわずかな違いを除いて、サザンブロットに関してすでに記載した転写と同様に行った:大型DNA分子を破壊するために、転写の前にゲルを紫外線(254nm)での5分の処置に供した。続いて、それらのそれぞれを0.25N HCLとともに20分間で2回、次いで、0.4M NaOHとともに30分間で1回インキュベートした。キャピラリーによる転写状態を少なくとも48時間保った。固定とハイブリダイゼーションの両方を、第(3.3)節ですでに記載したとおりに行った。
【0101】
4.マイクロインジェクションのためのYACの調製
4.1 酵母細胞を含むアガロースブロックの大規模調製
十分な量のDNAを単離し、本発明のトランスジェニックマウスを作製するために、高細胞密度のアガロースキューブを調製した。そのためには、次のプロセスに従った:
【0102】
200mlの選択培地に、YACを有する酵母を接種して、飽和まで増殖させた。それを600gにて5分間遠心分離し、残留培地を除去した。それを、オリジナル培養物100mlにつき40mlの50mM EDTA(pH8)中に再懸濁し、それを再度遠心分離し、上清を廃棄した。酵母沈殿物の重量を測定し、密度1と仮定すれば、酵母を体積50%として溶液Iを加え、その混合物を37℃に温度調整した。一度、温度調整したら(約30秒)、同じ量の溶液IIを加え、それをゆっくりと攪拌し、4℃で冷却されていた型に80μlを分配した。それを10分間ゲル化させた。それらのキューブを、1mlのキューブにつき8mlの溶液IIIを入れたチューブに入れかえ、それを37℃で2時間、攪拌しながらインキュベートした。溶液IIIを同量の溶液IVと置き換え、それを37℃で1時間インキュベートした。次いで、それを新しい溶液と置き換える必要があり、それを37℃で一晩放置した。翌日、それらのキューブを、1mlのキューブにつき8mlの20%NDSで室温で2時間洗浄した。洗浄を繰り返し、それらをロードする準備ができた。
【0103】
4.2 パルス磁場を利用したYACの精製
この場合の精製産物はトランスジェニックマウスを作製するのに直接使用されることから、YACの精製のためのパルス磁場の調製では一連の検討が必要であった。常に滅菌済みの使い捨て材料を使用して、高度無菌操作条件を用いた;EtBrは高い突然変異誘発能力を有するため、DNAが抽出されるゲルの領域がEtBrと接触しないようにし、YACを含む任意の溶液を、滅菌チップをつけてピペットで取った。滅菌チップは先端で切り、DNA分子の破損を防いだ。
【0104】
9つのキューブが入るように、パルス磁場の7つの通常のウェルを合わせることによって大型ウェルを調製した。マーカーIIおよび1/2、1/4、1/8および1/16キューブ画分を連続したウェルにロードした。
【0105】
このサイズ範囲での分離のために選択したプログラム(200ボルトにおいて26時間;最初の12時間は50秒のパルス、残る時間は100秒のパルス)を用いてゲルを泳動した。
一度、ゲルを泳動したら、そのゲルを、ゲルの泳動方向に滅菌メスで3つに切り、それによって大型ウェル全体を含む中央の部分と、画分およびマーカーを含む両側の部分とを作った。これらの2つの部分はEtBr(0.5μg/ml)で染色し、一方、中央の部分は泳動バッファー中に保った。染色された部分を用いて、染色されなかった断片において修飾YACを位置づけた。一度、そのYACが位置づけられたら、それを含むアガロースブロックを切り取り、50mlの平衡バッファー(10nM Bis−Tris−HCl pH7.5、0.1mM EDTA、100mM NaCl、0.03mMスペルミンおよび0.07mMスペルミジン)中で4℃で少なくとも16時間平衡化した。次に、修飾YACを含むアガロースブロックを4つに分け、それらを1.5mlチューブに移し、70℃で10分間加熱し、続いて、45℃に温度調整することにより液化して、アガラーゼ(GELase(商標) Epicentre)、100mgのアガロースにつき5単位を加え、それを45℃で3時間インキュベートした。溶けたアガロースを、次に、Millipore社製超遠心分離用カラム(Ultra free-(登録商標)MC Millipore)に移し、連続した6,000r.p.mにて2分間の遠心分離により総てのDNAをカラム内に濃縮した。この操作を最終量100μlが得られるまで繰り返し、DNAが再懸濁するように、カラムを一晩4℃で放置した。続いて、40mlのマイクロインジェクションバッファー(10mM Tris−HCl pH7.5、0.1mM EDTA pH8、100mM NaCl、0.03mMスペルミン、0.07mMスペルミジンおよび滅菌水)を入れたペトリ皿に透析フィルター(Millipore)を入れ、それを2または3時間透析させた。この後、修飾YACを含む液を回収した。
【0106】
5.卵細胞質内精子注入法(ICSI)(精子はYAC DNAとともにインキュベートされる)を利用したトランスジェニックマウスの作製
修飾YACを含むトランスジェニックマウスの作製は、特許出願WO2005/098010ならびにMoreira, 2003 (Moreira, P. N., et al., 2003, "Mouse ICSI with frozen-thawed sperm: the impact of sperm freezing procedure and sperm donor strain", Mol Reprod Dev. 66(1) : 98-103)およびMoreira, 2004 (Moreira, P. N., et al., 2004, "Efficient generation of transgenic mice with yeast artificial chromosomes by intracytoplasmic sperm injection", Biol Reprod. 71(6) : 1943-7)に詳細に記載されているプロトコールに従って行った。
【0107】
6.創始(founder)マウスおよびそれらの子孫の解析
6.1 PCRを利用した創始マウスの解析
PCR技術は、高度に特異的であり、トランスジーンの同定について最速のものであるという事実から、トランスジーンの解析を実施するためにこの技術を選択した。このマウスを研究するために、APP遺伝子の異なるエキソンについての17のPCRと、YACアームについての2つのPCRと、非遺伝子コード領域についてのさらに4つのPCRとを設計した。上記(1.3)のプロトコールに従って、生まれた総てのマウスについての23のPCRを行い、総てのPCRについて陽性であるものだけが全トランスジーンの担体マウス(carrier mice)と考えられた。
【0108】
6.2 生殖細胞系への伝達
トランスジーンの担体と考えられるマウスをC57BL/6Jマウスと交配させ、それらの子孫を解析した。伝達の解析は、2つのYACアーム(LYS2およびTRP1)についてのPCRと、APP遺伝子の2つのエキソン(エキソン6および11)についてのPCRとを利用して実施した。
【0109】
7.タンパク質の特性評価
7.1 タンパク質濃度の測定
タンパク質濃度を、標準としてウシ血清アルブミン(BSA)を用いてBradford法により定量した。
【0110】
7.2 SDSの存在下でのポリアクリルアミドゲル中でのタンパク質の電気泳動(SDS−PAGE)
3%濃縮ゲルと、解析するタンパク質の分子量に従って様々なパーセンテージ(7.5%、8%、10%または15%)の分離ポリアクリルアミドゲルとを使用した。サンプルバッファーとして25mM Tris−HCl pH6.3、10%グリセロール、2%SDS、5%β−メルカプトエタノールおよび0.01%ブロモフェノールブルーを使用した。
【0111】
分子量標準としてAmershamm Pharmacia社製の高分子量Rainbow(商標)カラー分子量標準(220kDa、97.4kDa、66kDa、46kDa、30kDa、21.5kDaおよび14.3kDa)を使用した。
【0112】
7.3 ウエスタンブロット
簡潔には、上記節に記載のように、ポリアクリルアミド−SDSゲルで決定されたタンパク質を、セミドライ式電気泳動転写法を用いて、孔径0.45μmのニトロセルロースフィルター(Bio-Rad)に電流量1.3mA/cm2で90分間転写した。
【0113】
ポンソーSレッドでの染色を利用して、転写されたタンパク質を観察した後、非特異的結合をブロックするために、その膜を3%BSAおよび0.2%Tween−20を含むPBS中で4℃で一晩飽和させた。
【0114】
次いで、それらの膜を0.05%Tween−20(PBST)および1%BSAを含むPBSで希釈した異なる特異的抗体とともに室温で2時間インキュベートした。それらの膜をPBSTで3回洗浄し、1%BSAを含むPBSTで1:50,000希釈したペルオキシダーゼ結合二次抗体とともに室温で2時間インキュベートした。最後に、発現させるために化学発光法(この方法ではペルオキシダーゼがH2O2の存在下でルミナル試薬(the luminal reagent)の酸化を触媒する)を用いた(Amersham社の「ECL」)。
【0115】
それらのバンドを濃度測定(GS−710 Calibrated Imaging Densitometer、Bio-Rad)を用いて定量した。
【0116】
7.4 抗体
6E10.6E10はヒトAβ配列の残基1〜17を認識する精製マウスモノクローナル抗体(Signet)である。6E10は電気泳動後の終濃度5μg/mlでの免疫標識に使用された。
【0117】
抗α−チュブリン:抗α−チュブリン(クローンB−5−1−2)はチュブリンのアイソフォームαのC末端エピトープを認識するマウスモノクローナル抗体(Sigma)である。抗α−チュブリンは電気泳動後の終濃度1:10,000での免疫標識に使用された。
【0118】
ビオチン結合抗マウス.ウマにおいて作製された精製抗体;ビオチン結合抗マウスはVector Laboratoriesから入手した。
【0119】
8.異なる組織におけるトランスジーン発現の解析
8.1 全RNAの単離
異なるマウス組織サンプルを機械的破壊によりホモジナイズし、全RNAを、市販の「High Pure RNA Isolation」 kit(Roche diagnostic 1 828 665)を製造業者によって示されるプロトコールに従って用いて抽出した。得られたRNAの量の評価はNanodrop装置(NanoDrop. ND−1000分光光度計)によって実施した。
【0120】
8.2 レトロ転写(retrotranscription)によるRNAからDNAへの移行
レトロ転写は「High Capacity cDNA Archive Kit」(Applied Biosystem)を用いて行い、RNAサンプル各1μgに対して、4単位の逆転写酵素「MultiScribe Reverse Transcriptase」、デオキシヌクレオチド(dNTP)、特異的バッファーを使用し、ランダムヘキサマーを製造業者によって示される条件の指標として最終量100μlで使用した。反応混合物を入れたチューブをGeneAmp PCR system 2400または9600サーマルサイクラー(Perkin-Elmer)により次の温度サイクルに供した:
−25℃で10分間のプレインキュベーション
−37℃で120分間のインキュベーション。
【0121】
8.3 384−ウェルプレートでの定量PCR
調製した総ての組織サンプルについて定量PCRを行い、それらを三連でアッセイし、その場合、50ngのオリジナルRNAに相当する5μlのcDNAを、Taqman Universal PCR Master Mix、no. AmpErase Ung(Applied Biosystem)によって記載された一般的な条件を用いて使用した。用いたプローブは:ヒトAPP遺伝子、onDemand hAPP Hs00169098_ml assay;ネズミAPP遺伝子、onDemand mAPP Mn00431827_ml assay;および内部対照遺伝子(ハウスキーピング)gapdh、onDemand Mn99999915_g1 assayであった。
【0122】
サンプルと反応混合物とを入れたプレートを、リアルタイムでのサンプルの解析を可能にするABI PRISM 7900HT Sequence Detection System(Applied Biosystem)を用いて、製造業者によって指定される温度サイクルに供した。得られたデータはSDS 2.1 ABI PRISMプログラム(Applied Biosystem)を用いて解析した。
【0123】
9.光学顕微鏡での観察:アミロイド斑の検出
組織学的研究のためのサンプルを得る前に、マウスを潅流に供し、続いて、その動物を解剖し、脳を摘出した。
【0124】
Condo red染色を、Accustain amyloid stain kit(コンゴレッド.Sigma diagnostic)を製造業者によって推奨されるプロトコールを用いて使用して行った。アミロイドの染色は赤色またはピンクがかった赤色で観察することができ、細胞核は青色、弾性繊維は明るい赤色である。サンプルを複屈折検査すると、それらは緑黄色で観察することができる。
【0125】
結果
ヒトAPP遺伝子を内因的に発現するトランスジェニックマウスの作製については、次の工程を通じて開発された:第一に、YAC b142f9およびその近接領域のコンピューター研究を実施する工程、次に、未知の配列またはYACに存在する他の遺伝子に対応する配列を削除する工程、修飾YACを得る工程、続いて、全修飾YAC構築物の組込みを達成するために遺伝子導入を実施する工程、それが確実に次の世代に伝達されるようにする工程、最後に、マウス組織におけるトランスジーン発現およびそのレベルを確認する工程。
【0126】
1.APP遺伝子およびその環境についてのコンピューター解析
ヒトAPP遺伝子は21q21.3領域の第21染色体に位置し、5’ではCYYR1遺伝子に隣接され、3’ではGABPAおよびATP5J遺伝子に隣接される(図2)。この全領域の配列はENSEMBLデータベース(http://www.ensembl.org/)から入手した。このデータベースでは、ヒト配列の他に、ハツカネズミ配列などの異なる生物の配列も入手可能である。APP遺伝子のハツカネズミにおけるホルトロゴウス領域(the hortologous region)は染色体16c3.3に位置し、ヒトの場合と同じ遺伝子に隣接され(図2)、このことはその配列が完全に保存されるという推測につながる。
【0127】
ヒトAPP遺伝子配列およびその環境を、相同性マトリックスを用いてネズミ遺伝子のものと比較している(図3)。両種間の1MbのDNA配列をMacVector 7.1プログラム(Accelrys, Burlington, MA, USA)を用いて比較すると、APP遺伝子のプロモーター領域のような、コード領域をコードする領域は高度の相同性を示すことが認められる。近接領域に関しては、APP遺伝子の5’領域ではコード領域より観察される保存は少ないが、ゲノム構造は維持され、一方、その遺伝子の3’領域では、その約60kbにおいて、ヒト領域に関連したネズミ領域で一連の再配列が起こっている。この特定のゲノム構造は、YACを修飾するのに用いる領域を定義するのに理想的であり、その境界を5’ではSeq2配列に相当する領域まで、そして3’ではGABPA遺伝子のコード領域の開始点までに定める[図1]。
【0128】
2.異なるベクターにおける組換えおよびクローニングのための相同配列の選択
2.1 反復領域の研究
酵母の相同組換えに用いる配列を選択するときには2つの条件が重要であると考えられている:一方では、サイズ、そしてもう一方では、その特異な状況。選択する配列のサイズに関しては、1〜2kb間であることが好適であると通常考えられる。その特異な状況に関しては、その適合性は、組換えが所望の場所で起こるという事実による。反復配列を位置づけ、それらを避けるために、RepeatMaskerプログラム(A. F. A. Smith & P. Green, 未公開; http://ftp.genome.washington.edu/cgi-bin/repeatmasker/)を用いて、1メガベースのAPP遺伝子環境を解析した。反復配列を排除しておいたこの配列においてAPP遺伝子の3’および5’の両方における1〜2kbの領域を選択し、PCRによってクローニングし、酵母における相同組換えに用いた(図4)。
【0129】
2.2 組換え配列のクローニング
APP遺伝子を含むYAC(b142f9)の異なるPCRを、第2節に記載のオリゴヌクレオチドを用いて行った(図4)。YACの伸長はこれらのPCRにより確認し、APP遺伝子の5’領域において配列2(sequence2)と配列3(sequence3)の間に位置しており、配列AおよびB(sequenceAおよびB)はAPP遺伝子の3’領域においてGABPA遺伝子の前に位置していた。配列2は、YAC中にある、遺伝子転写開始部位から128,354kbのところに位置する、APP遺伝子から最も離れた5’における配列であることから、この配列を5’での相同組換えに使用した。3’における領域では、配列AおよびBがAPP遺伝子の末端からそれぞれ100kbおよび80kbの距離のところに位置し、GABPA遺伝子の開始点の前にあることから、配列AおよびBの両方をクローニングした。クローニングでは、配列2、AおよびBのPCRを行い(図5)、選択した配列に対応するバンドを精製し、PCRIIクローニングベクター(Invitrogen)にクローニングして、PCRII−Seq2、PCRII−SeqAおよびPCRII−SeqBを得た。
【0130】
2.3 組換えベクター pB1R−Seq2の作製
図6に図式に示すように、プラスミド PCRII−Seq2およびプラスミド pB1Rの変異体を用いて、プラスミド pB1R−Seq2を作製した(Lewis, B. C, N. P. Shah, B. S. Braun and C. T. Denny (1992). "Creation of a yeast artificial chromosome fragmentation vector based on lysine-2." Genet Anal Tech Appl 9(3): 86-90)。このYACが一度修飾を受けたら、必要に応じて他のタイプの培養に使用できるように、オリジナルプラスミド pB1Rにネオマイシン選択遺伝子を加えた。クローニングベクターの配列2、PCRII Seq2を、EcoRI酵素での消化により抽出した。
【0131】
この断片の末端を、それらのその後のクローニングのためにクレノウ酵素を用いて平滑末端にした。ベクター pB1R−NeoをSacI酵素での消化により線状化した。T4リガーゼを用いて、Seq2断片をベクター pB1R−Neoと結合し、後に酵素消化によりベクターにおけるその配列の正確な方向性を確認した。前記ベクターを酵母における第1回目の相同組換えに使用することができるように、そのベクターをSalIおよびSacI酵素での消化により線状化した(図6)。
【0132】
2.4.組換えベクター pYAC4 3’−SeqAおよびpYAC4 3’−seqBの作製
これら2つの組換えベクターの作製を図7に要約している(2配列についてクローニングを並行して実施した)。組換え配列をプラスミド PCRII−SeqAおよびPCRII−seqBから得た。抽出では、それらを最初にXhoI酵素で消化し、末端をクレノウ酵素で平滑末端にし、続いて、それらの配列が平滑末端とさらに付着末端を有するようにそれらをBamHIで消化した。プラスミド pYAC4 3’はチロシナーゼ遺伝子の調節配列を研究するために研究室で作製されたpYAC4の誘導体である(Burke, D. T., G. F. Carle and M. V. Olson (1992). "Cloning of large segments of exogenous DNA into yeast by means of artificial chromosome vectors. 1987." Biotechnology 24: 172-8、およびBurke, D. T. and M. V. Olson (1991). "Preparation of clone libraries in yeast artificial-chromosome vectors." Methods Enzymol 194: 251-70)。配列AおよびBを、ベクター pYAC4 3’−SeqAおよびpYAC4 3’−SeqBが生成する配向方法で結合し得るように、消化を利用して、最初にEcoRIを用いて、それらの末端を再び平滑末端にし、後にBamHIで消化して、目的のものではない領域を削除し、そのベクターを線状化した。
【0133】
しかしながら、これらのベクターを第2回目の相同組換えに使用するためには、新規栄養要求性マーカーの存在が必要であった。そのために、最初にNotIでの消化によってベクター pYAC4 3’−SeqAおよびpYAC4 3’−SeqBを線状化して(この場合もクレノウ酵素を用いて、それらの末端を平滑末端にした)、プラスミド YDp−UのURA3マーカーをクローニングし(Alani, E., L. Cao and N. Kleckner (1987). "A method for gene disruption that allows repeated use of URA3 selection in the construction of multiply disrupted yeast strains." Genetics 116(4): 541-5)、続いてBamHIでの消化によってプラスミド YDp−UのURA3遺伝子を得た、正確なクローニングのためにその末端も平滑末端にした。T4リガーゼを用いて、その断片をベクターにクローニングした。YACの第2回目の組換えにそれらを正しく使用するためのベクター pYAC4 3’−SeqA URA3およびpYAC4 3’−SeqB URA3の線状化を、BamHIおよびSpeIでの酵素消化によって行った(図7)。
【0134】
3.YACの5’領域における相同組換え
本発明では染色体断片化プロセスを5’および3’領域を修飾するために用いた;このプロセスはオリジナルYACの末端にある2つの領域を削除することからなり、それらの領域うちの一方が3’におけるGABPA遺伝子を含む領域である。
【0135】
3.1 APP遺伝子の5’領域のYAC b142f9における相同組換え
100〜1,000ng間で変化するDNA量を用いて、LiAc法による酵母の形質転換によって合計40のコロニーを得た。これらの40コロニーを、PCRを利用して解析し、陽性の可能性があるクローンとして3つのクローンを得た。
次に、酵母のエレクトロポレーションでは合計96のコロニーが生じ、これらのコロニーを、PCR技術を利用して解析した。これらの中で陽性の可能性があるクローンとしての正確なパターンを示したものはなかった。
【0136】
3.2 PCRを利用した、相同組換えによって生じるクローンの解析
相同組換えによって得られたクローンの解析を、用いる2つのオリゴヌクレオチドのうちの1つが組換えプラスミド中に位置し(YACには存在しない領域内)、一方、もう1つのオリゴヌクレオチドがYAC組換え領域に近接する領域中に位置することを特徴とするハイブリッドPCRを設計することによって実施した。よって、このPCRでは、組換えが予測される部位で起こった場合にのみ断片を増幅することができる。陽性対照として両方の領域を含む別のベクターを作製した。このPCRについて陽性であったのは、解析した136のコロニーのうちの3つのもののみ、13、14およびb7の番号のついたコロニーであった。
【0137】
これらのクローンを用いた解析を、別の3つのPCRを用いることによって行い、これによってそれらについてのより多くの情報を提供することができた。最初の2つのPCRは、APP遺伝子の部分を維持するクローンがそのエキソン6および11のものであるかどうかを確認するために行った(図8)。用いた3つ目のPCRは組換え領域の5’に位置する配列のものであり、相同組換えが正確であったとすれば陰性となる(図8)。解析した3つのクローンは、図8に示されるように、エキソン6および11のPCRについて陽性であり、5’領域について陰性であった。
【0138】
3.3 パルスフィールド電気泳動を用いることによるYACの断片化の判定
YACにおける正確な相同組換えはそのサイズの減少を伴い、そのことはその電気泳動移動度を調べることにより見ることができる。酵母ゲノムはパルスフィールド電気泳動技術(PFGE)を用いてゲル中で分離することができる。この技術に従って、解析する3つのクローンを、オリジナルクローン、同じ酵母株のクローン(YACを全く含まない)および分子量マーカーとともに泳動した。図9ではEtBrで染色した泳動後のゲルを示している。オリジナルYACクローンにおいて680kbマーカーと580kbマーカーとの間に、YACを含まない酵母クローンには存在しないバンドが存在することを観察することができる。このバンドは、クローン13およびB7では維持されているように見えるが、一方、クローン14では消失しているように見える。
【0139】
続いて、このゲルをニトロセルロース膜に転写して、APP遺伝子 cDNAプローブとのDNA−DNAハイブリダイゼーションを行い、これによれば電気泳動移動度の変化が極めて正確に確認される。図9では、クローン14が、正確な組換え後に予測されるYACサイズの約50kbの変化をどのように受けたかを示している。
【0140】
3.4 サザンブロット技術を利用した組換え領域の解析
相同組換えによるYACの断片化は、組換え領域のDNA配列において一連の修飾をもたらす。組換えクローン配列とオリジナルクローン配列との間の違いについては、サザンブロット技術を利用して解析することができる。BamHI、ScaI、EcoRV、BglIIおよびHindIIIの消化パターンは2つのクローンで異なっていた。
【0141】
加えて、APP遺伝子のオリジナルクローンとクローン14について実施したサザンブロットにおいてバンドサイズの変化が得られ(図10)、このことにより組換えが正確な相同部位で起こったことが分かり、PCR解析とパルスフィールド電気泳動の両方で得られた結果と合わせて、APP遺伝子の5’領域において組換えが成功したこと、そしてクローン14を次の組換えのベースとして使用可能なことを示している。
【0142】
3.5 クローン14のコード領域の解析
クローン14がAPP遺伝子のコード領域の欠失を受けていないことを確認するために、サザンブロットの比較を実施した。このために、オリジナルYACと修飾YACをBamHI制限酵素で消化し、続いて、APP遺伝子 cDNAとハイブリダイズした。このハイブリダイゼーションでは異なる遺伝子コード領域に対応するバンドのラダーが生じるが、これはオリジナルクローンとクローン14の両方で同じであるにちがいない。MacVectorプログラムを使用して、BamHI酵素での300kbのAPP遺伝子の消化パターンを得た。次いで、遺伝子コード領域の任意のものを含む断片を確認した。図11では、このサザンブロットの理論パターンと、それがオリジナルクローンb142f9と組換えクローン14で得られたバンドのラダーと一致していることを示している。オリジナルクローンb142f9と組換えクローン14で得られたバンドパターンの保存は、組換え後にコード領域は大きな修飾または再配列を受けなかったということを示している。
【0143】
4.YACの3’領域における相同組換え
4.1 クローン14のYACにおける相同組換え.GABPA遺伝子削減
第1回目の組換え後に修飾YACにおいてAPP遺伝子の3’領域における相同組換えを行い、これには形質転換に用いるコンピテント酵母を調製するためにクローン14を用いることを必要とした。この組換えには2つの異なるベクター、pYAC4 3’−SeqA URA3およびpYAC4 3’−SeqB URA3(図7)を用い、これらのベクターは組換え配列が異なるだけのものである。前記配列は20kb相互に離れており、遺伝子の末端から80kbより遠く離れている(図4)。これらの組換え配列を用いることによってGABPA遺伝子を含む、YACの40〜60kbを削除した(図12)。
【0144】
LiAc法とエレクトロポレーション法の形質転換効率の違いを考えると、明らかに前者が有利であり(前節の第3.1節参照)、第2回目の形質転換を行うために使用することを決めた。第1回目の組換えとは異なり、dual PCRを利用したこの組換えでは2,317のコロニーを解析する必要があり、APP遺伝子のエキソン6で行われる第1回目のPCRで陽性を示し、GABPA遺伝子で行われる第2回目のPCRでは陰性を示すと、このことによりGABPA遺伝子に対応する配列が削除されたことが示される。
【0145】
解析した2,317のコロニーのうちの1,720のものは配列Aに対応し、597のものは配列Bに対応した。異なるコロニーのPCR解析後、1720のうちの90のものと、597のうちの5のものだけをPFGEおよびサザンブロットによって解析した。PFGEおよびサザンブロットを用いたこれらのクローンの研究により、配列Aを用いて行われた相同組換えが高い確実性で異なる領域で起こり、その結果、見慣れない組換えパターンをもたらし、いくつかの場合では、組換えYACが予想をかなり上回る再配列プロセスおよび断片化を受けて、説明することが非常に難しいパターンが生じたことが明らかになった。
【0146】
配列Bを用いた相同組換えについて解析した5つのクローンは、トランスジェニックマウスの作製に使用するための必要要件を満たすクローンを与えた。このクローンにおいて実施した様々な検証を以下に記載する。
【0147】
4.2 PCR技術を利用した、3’における組換えによって生じたクローンの解析
酵母形質転換によって得られた異なるクローンの解析を、dual PCRを用いて実施した。図13では、配列Aを用いた組換えについてのいくつかのクローンと配列Bについての5つの陽性のものとにおいて実施したdual PCRを示している。観察できるように、クローンb106、b108、b109、b111およびb113はそれらの後の研究用の候補として反応している。APP遺伝子のエキソン6のPCRではそれらの総てが陽性であり(このことは、それらのクローンはAPP遺伝子とともにYACを維持していることを示す)、その一方で、GABPA遺伝子のPCRでは陰性である(このことは、これらのクローンはGABPA遺伝子配列を失った可能性があることを示唆する)。以下に示すように、これら5つのクローンのうちクローンb106だけが正確な相同組換えパターンを示した。
【0148】
PCRを用いてクローンb106の研究を行うために、配列BとGABPA遺伝子との間:配列AおよびA’との間に位置する2つの領域についてのPCRを行った。この研究では、GABPA遺伝子とエキソン6のPCRを繰り返し、APP遺伝子のエキソン11のPCRも含めた。図14から分かるように、組換え領域に関して3’位置でのPCRで陽性であったものはなく、クローンb106にはこれらの配列が存在しないことが示された。2つのエキソンのPCRは陽性対照の場合ほど明らかではなく、それらを何度か繰り返したが、常に同じ陽性結果が得られた。
【0149】
4.3 PFGEを利用したYACの電気泳動移動度の変化の判定
YACの電気泳動移動度は、第1回目の相同組換え後と同じように、この第2回目の組換え後にも変化し、得られるYACの最終的なサイズは520kbである、すなわちYACを含むクローン14よりも40kb小さい。図15では、異なるクローンを泳動した1%アガロースゲルを示している:分子量マーカー、オリジナルクローンb142f9、クローン14およびクローンb106。前記ゲルをニトロセルロース膜に転写し、APP遺伝子 cDNAとハイブリダイズした。異なるレーンのYACの観察が得られ、クローンb106におけるYACのサイズの変化はオリジナルYACおよびクローン14に関して容易に観察された。
【0150】
4.4 クローンb106のYACとGABPA遺伝子 cDNAとのハイブリダイゼーション
YAC b106とGABPA遺伝子 cDNAとのハイブリダイゼーションを、該YACにおいてGABPA遺伝子のコード領域が削除されたことを確認するために用いた。
【0151】
図15では、前節で用いたパルスフィールドアガロースゲルを示しており、このパルスフィールドアガロースゲルに他のレーンを加え、前記レーンの場合と同じサンプルをこれらのレーンにロードしている。この場合には、ゲルの半分をAPP遺伝子 cDNAプローブとハイブリダイズし、残りの半分をGABPA遺伝子 cDNAプローブとハイブリダイズした。その図から分かるように、クローンb106のAPP遺伝子を含むYACはGABPA遺伝子配列とはハイブリダイズしないが、一方、オリジナル酵母のYAC、およびクローン14のものは同じYACにおいて両方の遺伝子を有している。
【0152】
4.5 サザンブロット技術を利用した3’における組換え領域の解析
組換えベクターがYACに正確に組み込まれていることを、サザンブロット技術を用いて確認した。図16に示されるパターンは、オリジナルYACとクローン14では共通しているが、一方、クローンb106は異なった、予測ダイアグラムに従うバンドパターンを有する。ゲルハイブリダイゼーションでは、YAC b106に対応するバンドは前者のクローンのものとは異なるサイズを有することが分かり、このことは、プローブがハイブリダイズする、組換え配列の周辺領域にあり、後者の5’に位置する領域において、異なるクローン間で配列の違いがあることを明確に示している(図16)。
【0153】
4.6 クローンb106のコード領域の解析
クローンb106の妥当性を確認するために、サザンブロットの比較を実施した。クローンb106のYACが、コード領域ではなく、YACの3’領域で修飾を受けただけならば、3つのYAC(オリジナルYAC、クローン14のYACおよびクローンb106のYAC)は、図11に示された同じラダーのバンドを有するはずである。実施したサザンブロットを示している図17を観察すると、3つのクローンのバンドパターンは全く同じであり、そして、それが図11に示されるものとは明らかに異なるということが分かる。しかしながら、これらの違いは、実際には、より清浄なDNAを、より高い濃度で使用することによって最後のゲルにおいてより良好な分解能が得られたという事実によるものにすぎない。図17のゲルのバンドパターンを観察すると、いくつかの二重および三重バンドはより明確に見える。
【0154】
いずれにせよ、APP遺伝子を含むYACはクローンb106において重要なものであった。クローンに用いた同じ命名法を用いて、このYACに名前を付け、それをYAC b106と呼んだ。
【0155】
5.YAC B106の精製
本プロセスにより3回の連続したDNA抽出を実施した。材料および方法の第4.2節に記載のプロトコールに従って実施した3回の精製のうちの1つを示している図18から分かるように、EtBrで染色したゲルのエッジを用いて、YAC b106の位置を決定し、染まらなかったゲルの部分からYAC b106を抽出した。抽出後にゲルの他の部分を染色することによって、YAC b106の総てのバンドが採取されたことを確認することができた(図18)。
【0156】
3つの異なる技術によって3回のDNA抽出を測定した。まず第一に、アガロースゲルでの評価と既知量の参照YACとの比較を利用する。得られた結果を、蛍光光度計とnanodropを使用することによって調べた。その結果、抽出I、IIおよびIIIで得られた濃度はそれぞれ4ng/μl、7.1ng/μlおよび10ng/μlであった。
【0157】
6.卵細胞質内精子注入法を介した遺伝子導入(精子はYAC b106とともにインキュベートされる)
遺伝子導入を実施するために、異なるマイクロインジェクションセッションを実施し、プロセスが終わった後に生まれた合計102匹の動物を得た。遺伝子導入に用いた卵母細胞のドナーマウス系はCD1とC57BL/6Jとの混合系であり、それによって純系を使用するよりも高い効率が得られる。創始マウスが生まれてくるときに、それらが75%C57BL/6J遺伝的背景を有するように、用いた精子はC57BL/6J系のものである。
【0158】
7.創始マウスの解析
PCR技術を用いて、細胞質注入後に得られた102匹のマウスの解析を実施した。520Kbのトランスジーンの挿入を正確に確認する手段として総てのYAC領域に対してスキャンを実施した。この目的のために次のPCRを設計した:APP遺伝子のエキソンについての17のPCR(それらのうちの1つはエキソン12とエキソン13の両方を含む)と、YACの2つのアームについての2つのPCR(LYS2遺伝子の選択遺伝子およびTRP1のものの両方に関する)と、コード領域とYACのアームとの間に位置する配列についてのさらに4つのPCR(図19)。
【0159】
得られた102匹のマウスについての23のPCRを行った(図20)。それらのうちの17においてある程度の遺伝子導入が得られ、これはそれらがYACの外因性DNAを完全にまたは部分的に組み込んだことを意味する。このDNAの組込みは、15のケースで起こったように、部分的なものであり得るが、それらのうちの2つ、マウス35および雌マウス64においては完全なものであり、YAC b106に関するこの調査で得られる第1のトランスジェニックマウスが生成したため、その主要目的の1つに適合する。
【0160】
8.雑種第一代(F1)におけるトランスジーンの伝達
2つのトランスジェニック創始マウス、マウス「35」およびマウス「64」の作製を確認したので、それらをC57BL/6Jマウスと交配させて、それらの子孫へのトランスジーンの伝達を調べ、それらにおけるそのようなトランスジーンの発現を解析した。YAC b106の伝達を研究するために、それらの子孫で4つのPCRを行った:YACのアーム、すなわちLYS2およびTRP1遺伝子についての2つのPCRと、APP遺伝子のエキソン6および11についてのPCR。この結果、完全YACが伝達されたこと、そしてそのためYACの部分的組込みが起こらなかったことを確認することができた。本明細書の作成まで41匹のF1マウスをマウス35の子孫について解析し;18匹のトランスジェニックマウスを確認し、子孫の44%への伝達がなされていた。雌マウス64の場合では、32匹のF1子孫を解析して、その子孫中18匹のトランスジェニックマウスを得、56%伝達がなされていた。要するに、2つのケースでは、創始マウスの第一代子孫へのトランスジーン伝達指数は、Mendelianのパターンによって予測されるものに一致して約50%である。図21では、マウス35のいくつかの子孫についての例示的なPCRを示している。
【0161】
9.トランスジェニックマウスにおけるYAC b106構造の保存
創始マウスを交配することによって得られたトランスジェニックマウスのゲノム研究を、YAC b106におけるAPP遺伝子コード領域の構造を確認することにより行い、そのために2つの創始マウスの子孫についてのサザンブロットの比較を実施した。
【0162】
図22では、創始マウス35および64のF1、すなわち第一代子孫において得られたバンドパターンを示しており、そのバンドパターンは各創始マウスにつき2匹の同腹子(それらの一方のものはトランスジェニックであり、もう一方のものは非トランスジェニックである)に対応している。パターンは図11で得られたものと同じであることを観察することができ、そのことによりマウスゲノムにおいてトランスジーン構造が保存されていることが示される。
【0163】
10.トランスジェニックマウス系統におけるヒトAPP遺伝子発現の解析
トランスジェニック系統35および64におけるヒトAPP遺伝子の伝達を立証したら、異なるマウス組織におけるRNAおよびタンパク質両方の発現レベルを特徴づける必要があった。
【0164】
この解析には系統35の第一代に相当するマウスを使用し、合計4匹の2ヶ月齢雄マウスを用いて、それらをそれらの同腹子と比較した。この研究の主な焦点は、APPタンパク質の高い発現が起こる組織であることから中枢および末梢神経系であった。異なるマウス体器官における発現もまた解析した、全体的に以下を解析した:脳(皮質、脳室、小脳および中脳と呼ばれる(それらの解剖学的内容と完全に一致する)4つの領域に細分される);脊髄、肝臓;肺;膵臓;脾臓;小腸;筋肉;精巣;血液;心臓および腎臓。
【0165】
10.1.APP遺伝子メッセンジャーRNAの発現の解析
トランスジェニック系統35における正確なヒトAPP遺伝子発現を確認するための第1の工程は、RT−PCRを利用して異なるマウス組織におけるヒトAPP遺伝子の転写を調べることであった。そのためには、異なるマウス組織からメッセンジャーRNAを抽出し、そのRNAの転写レベルを、リアルタイムRT−PCRを利用して、ヒトAPP遺伝子に向けられるTaqmanプローブを用いて測定した。この試験の内部対照としてネズミGAPDH遺伝子を用いた。この研究を行うために、ネズミAPP遺伝子RNAレベルも測定し、それらをGAPDHに対して標準化すれば、両方の遺伝子の発現レベルを異なる組織間で比較することができるようにした。
【0166】
図23では、解析した異なる組織で得られたヒトAPP遺伝子メッセンジャーRNAレベルを示している。発現レベルを相対対数単位で表し、データをGAPDH遺伝子に対して標準化している。この遺伝子は主として神経組織において発現され;その値は、若干低い小脳の場合を除いて、それらの総てにおいてよく似ていることが認められる。他の研究した組織に関しては、精巣、血液、心臓および腎臓で検出可能な発現レベルに達しただけであり、いずれの場合においても、神経系で検出されたものより一桁低い。
【0167】
図24では、トランスジェニックマウスおよびそれらの同腹子に関するヒト遺伝子およびネズミ遺伝子の両方についてのAPP遺伝子メッセンジャーRNA発現を示している。前のグラフ同様に、データはネズミGAPDH遺伝子に対して標準化している。示した結果により、神経組織では遺伝子発現レベルが高いが、小脳では低下していることが分かる。他の解析した器官に関しては、肺、精巣、血液、心臓および腎臓などの組織において発現を示しているが、一方、肝臓、膵臓、腸、脾臓および筋肉などの組織ではそのレベルはかなり低くなっており、極めて重要な変化を示している。
【0168】
10.2 タンパク質レベルの解析
マウスにおけるヒトAPPタンパク質の発現によって、マウスにおける正確なYAC b106の転写および翻訳を確認する。タンパク質レベルを、ポリアクリルアミドゲル中での電気泳動後の免疫標識技術(ウエスタンブロット)を用いて研究した。この研究では、APPタンパク質を認識したが、ヒト型とネズミ型とを区別することができない抗体を使用した[Lamb, B. T., et al. (1997). "Altered metabolism of familial Alzheimer's disease-linked amyloid precursor protein variants in yeast artificial chromosome transgenic mice. "Hum Mol Genet 6(9): 1535-41.; Lehman, E. J., et al. (2003). "Genetic background regulates beta-amyloid precursor protein processing and beta-amyloid deposition in the mouse." Hum Mol Genet 12(22): 2949-56]ため、発現レベルを比較することができるように、結果をアクチンに対して標準化する必要がある。
【0169】
神経系の異なる組織では免疫反応性が欠如しており、低いAPP遺伝子発現レベルを示す。図25では、系統35および野生マウスの両方に関する代表的なゲルを示し、発現の評価を、アクチンに対して標準化して、下のグラフに示している。そのグラフでは、系統35が野生マウスの少なくとも2倍の量のタンパク質を有することがはっきりと分かる。この割合はこの研究において解析した異なる脳領域間でわずかに変化するが、その水準を維持している。
【0170】
10.3 マウス脳の組織学的研究
本発明のマウスになければならない特徴の1つは、正確なAPPタンパク質発現およびプロセッシングであるが、その一方で、不正確な発現または誤りのあるプロセッシングの形跡があると、脳においてこのタンパク質(Aβペプチド)由来のペプチドの蓄積が起こる。この研究を行うために、月齢2ヶ月の2つのマウス系統の脳の切開を行った。これらの切片をコンゴレッドで染色して、アミロイドを検出した。2系統またはそれらの同腹子ではいずれのAPPタンパク質誘導体も蓄積は検出されない(図26および27)が、1年齢の対照トランスジェニックマウス(Tg 2576)ではそれらを検出することができる。
【0171】
考察
1.酵母の相同組換えによるYAC b106の構築
アルツハイマー病の調査を可能にする、この調査で開発されたトランスジェニックマウスを設計する際に利用できたのは、YAC b142f9(APP遺伝子を含むことは分かっているが、場合によってはその後の使用において摂動を導入し得る他の遺伝子も含むかどうかは不明である、600kbを超えるサイズの人工染色体)だけであった。ヒトゲノム配列が公開され、YAC b142f9で実施された他の具体的な研究が発表されたことで、該YAC中にはAPP遺伝子が単独で存在するのではなく、GABPA遺伝子が同時に存在することの証明が可能になった。そのため、そのGABPA遺伝子を削除し、これまでに報告された総てのAPP遺伝子コード領域および調節領域を尊重して、YAC b142f9をその5’および3’領域において修飾する必要が生じる。そのようにして修飾したYACを、アルツハイマー病に関する調査のためのモデルとして使用することができるトランスジェニックマウスの作製において使用し得るようにするために、このことがこの調査の最初の目標となった。
【0172】
5’で行われる相同組換えはYACの40kbの削除を伴った(図12および図9)。それでもなお、この削除後、APP遺伝子の5’領域において120kbがまだ保存されており、調査の現状に従ってその正確な発現を可能にするには十分なものであった。実際、APP遺伝子の調節エレメントはその遺伝子の近位プロモーターにおいて報告されているだけであったが、距離が離れている調節エレメントの存在については議論の余地がないと考えられている。そのため、正確な時空間的遺伝子発現を成し遂げるために、進化的に保存されるエレメントを存在させる組換えを実施した[マウス遺伝子のものとの比較ゲノミクスによる配列解析後(図3)]。従って、本調査ではそれを確実にするために端で十分なスペースが保存された。
【0173】
YACにおける相同組換えによって5’領域の境界を定めた後、染色体断片化によってGABPA遺伝子を直接削除する第2回目の組換えを実施した。GABPA遺伝子は転写因子をコードし、トランスジェニックマウスにおいてそれが発現することによって、トランスジェニック表現型の研究に干渉し得る未知の方法でマウスの正確な開発に影響を及ぼす可能性があり、どの遺伝子が該表現型に関与するのかを確認することができなくなるため、このYACを動物モデルの作製に用いようとする場合には、その削除は必須である。
【0174】
APP遺伝子の3’領域における相同組換えは、5’において実施されるものよりもずっと複雑であるが、これは3’領域に二次DNA構造が存在することにより、この構造が組換え配列へのアクセスをより複雑にしている可能性がある。
【0175】
第1回目の組換えの場合と同じように、正確な相同組換えとGABPA遺伝子の削除を正確に確認するために一連の試験を設計した。第1回目のコロニースクリーニングを、dual PCRを用いて実施した。この方法は得られた数百ものコロニーを区別すること、そしてこの数を一桁低減することを可能にした。GABPA遺伝子を有していなかったがエキソン6は有していたコロニーは適していると考えられるようだが、PFGEを利用した後の解析ではいつもそういうわけではないことが分かった。
【0176】
この意味で、PFGEと、続いての、フィルターのAPP遺伝子cDNAおよびGABPA遺伝子cDNAとのハイブリダイゼーションを利用した解析は、中でも最も信頼性の高い最終的な解析であった。これは;実際には、YAC b106の電気泳動移動度の変化がクローン14のYACの断片化によるもののみであり得るためである。GABPA遺伝子cDNAとのハイブリダイゼーションは、総ての遺伝子コード領域の認識を可能にするが、一方、PCRはエキソンの1つを対象にするだけであった。YAC b106のGABPA遺伝子をコードする配列の消失についてのハイブリダイゼーションによる確認では、予測される電気泳動移動度の変化とともに、このコロニーが計画された組換えを受けたという確認を必要とした。それでもなお、サザンブロットによる確認および異なるPCRは、研究を行い、その結果、トランスジェニックマウスを作製するためのプロセスに移るために有用であった。
【0177】
2.ICSIを利用したトランスジェニックマウスの作製
520kbのYAC b106を有するトランスジェニックマウスを、WO2005/098010およびMoreira, 2003および2004(上記)に記載のプロセスに従って、ICSIを利用して得た。生まれた102匹のマウスから、合計17匹のトランスジェニックマウス、すなわちYAC b106Ofの任意のDNA断片の担体を得、解析した。
【0178】
520kb構築物が完全に組み込まれたことを確認するには、配列全体にわたる入念な解析が必要であり、そのために、各マウスにおいてYAC全体にわたって23のPCRを実施した。YACの完全組込みを確認するために設計されたPCR群(図19)では、マウス35および64の場合だけ陽性であり、このことからこれらのマウスを新規動物モデルの候補創始マウスであると特定した。
【0179】
しかしながら、創始マウスにおいてトランスジーンが存在するからといって、その後の使用に最も重要な条件である生殖細胞系においてそれが存在することが確実とはならないため、次の世代でその伝達を確認しなければならない。それを調べるために、クローンをC57BL/6Jマウスと交配させ、2つのトランスジェニック系統(図21)から生まれた総ての同腹子をPCRによって解析し、第一代、すなわちF1においておよそ50%の伝達が得られた(結果の第8節)。生殖細胞系におけるトランスジーンの伝達(図21)ならびにかかるトランスジーンの発現(図23)を確認することにより、YACの組込みを確認するために設計されたPCR群が適切な手法であったことが証明された。
【0180】
3.マウスにおけるヒトAPP遺伝子の発現
マウスにおけるヒトAPP遺伝子の発現を特徴づけるための第1の工程を、遺伝子RNA発現を解析することによって実施した。そのためには、定量プレートPCR技術を用いて、異なる組織間のそのレベルを比較した。既存の参照によれば、この遺伝子の発現はいくつかの組織において起こるが、これらレベルは脳において高い。実際には、図23から分かるように、系統35の発現レベルは、解析した異なる領域中で脳において最も高い値を示している。脊髄で表れた高い遺伝子発現指数を強調する必要があるが、脊髄はこのタンパク質を産生する細胞系統が存在する神経組織によって形成されていることから、このデータは当然のことである。よって、マウスにおけるヒトAPP遺伝子の発現に関するこの調査の結果は、脳においてRNAが最高発現レベルにあるという、同様のマウスに関してこれまでに報告されているものと十分に一致している。
【0181】
異なる器官におけるヒトAPP遺伝子の発現はネズミAPP遺伝子の発現と非常に似ており(図24)、神経組織だけでなく、心臓、血液、精巣および腎臓においても発現パターンが保存されている。しかしながら、ネズミAPP遺伝子発現は、肝臓、肺、膵臓、腸および筋肉で検出され、心臓、血液、精巣および腎臓のものと同様の発現が起こっている肺の場合を除いて、他の器官よりもずっと低い値である(一桁低い(one order or magnitude lower))が、ヒトAPP遺伝子ではそのようなことは起こらず、すでに指摘したようにその発現はそれらの器官では検出されなかった。
【0182】
一度、異なるマウス組織において遺伝子発現が確認されたら、そのRNAのタンパク質への翻訳を確認する必要があった。この研究のために、RT−PCRを用いて解析した異なる組織中に存在するタンパク質の量についてのウエスタンブロット解析を実施した。この研究には、最初はヒトタンパク質に特異的な6E10抗体を用いることによって対応していたが、この調査の試験においてその特異性を再現することができなかったため、ヒトタンパク質とネズミタンパク質の両方を認識する22C11抗体を使用する決定をし、その結果、タンパク質の総量を評価することができた。
【0183】
神経組織ではタンパク質発現が高いことから、起こり得るかかるタンパク質の同定および評価を行ったが、一方、他の組織では、タンパク質レベルの表示はずっと低く、その同定は不可能であった(図25)。神経組織におけるタンパク質レベルを評価し、それらをアクチン値に対して標準化した後、トランスジェニックマウスおよびそれらの非トランスジェニック同胞種のタンパク質レベルを比較し、前者の場合に、特定組織に応じて、値が内因性タンパク質のものに対して2〜2.5倍高いことを観察した。これを受けて、皮質、脳室および脊髄では、タンパク質レベルは内因性タンパク質に対して2倍を少し上回っており、一方、中脳および小脳では、比率が上昇し、2.5倍に達していた。トランスジェニックマウスではヒトタンパク質とネズミタンパク質の両方が検出されることを考えなければならないため、総APPタンパク質発現が2倍というのは、同量のヒトタンパク質とネズミタンパク質が発現されているということを意味する。2つの遺伝子間でのこれらのような発現差はこれまでに記載されているもの(皮質領域でのみ測定され、内因性遺伝子のものの1.7〜2倍間の発現レベルを示す)より少し大きいが、その違いは使用したマウス系に起因する可能性があり、そのマウス系の遺伝的特徴はそのレベルとそのプロセッシングの両方に影響を及ぼす可能性がある。
【0184】
最後に、マウス脳において予備研究を実施し、本疾患の典型的な神経病理学のいくつかの検出を試みた。2ヶ月齢マウスでのこのような予備研究では、現在のところアミロイドペプチドの沈着の兆候は検出されず、これらの特徴を有するマウスでの過去の研究と一致している。
【図面の簡単な説明】
【0185】
【図1】ヒト第21染色体(NCBI build 35)におけるAPP遺伝子の位置を示す図である。SeqB領域は全GABPA遺伝子コード領域を排除するが、その3’UTR領域内で始まる。
【図2】APP遺伝子の染色体上の位置を示す図である;A.ENSEMBLにおいて下方に示した近接遺伝子に隣接する、第21染色体におけるヒトAPP遺伝子の位置。B.ENSEMBLにおいて下方に示した隣接遺伝子に隣接する、第16染色体におけるネズミAPP遺伝子の位置。
【図3】ヒトおよびネズミAPP遺伝子配列の相同性マトリックスを示す図である。ヒトAPP遺伝子を含むヒト第21染色体配列メガベースを、ネズミAPP遺伝子も含まれる第16染色体メガベースと比較する。
【図4】APP遺伝子環境のPCRのダイアグラムを示す。PCRの位置を利用して、その遺伝子の5’および3’の両方において、その遺伝子コード領域までの最近距離を有するYAC b142f9のサイズの範囲を定める。
【図5】PCRによるYAC b142f9の伸長の境界を示すダイアグラムを示す。配列A、B、1および2のPCRに関する1%アガロースゲル。レーン1は分子量マーカーに相当する。各配列に対して、陰性対照、YAC、および陽性対照それぞれを示す B.配列3、4、5および6のPCRに関する1%ゲル。レーン1は分子量マーカーに相当する。各配列に対して、陽性対照、YAC、および陰性対照それぞれを示す。
【図6】組換えプラスミド pB1R Seq2の作製および線状化において従う工程のダイアグラムを示す。
【図7】組換えプラスミドpYAC4 3’SeqAおよびpYAC4 3’SeqBの作製および線状化において従う工程のダイアグラムを示す。
【図8】エキソン6および11ならびに配列2’のPCRを示す図である。APP遺伝子のエキソン6、エキソン11およびいくつかの解析した酵母クローンの配列2’のPCR。M、分子量マーカー;G、ゲノムDNA;c+、陽性対照およびc−、陰性対照。
【図9】PFGEによる酵母染色体の分離およびAPP遺伝子cDNAとのハイブリダイゼーションを示す図である。A.臭化エチジウムで染色したPFGE。B.APP遺伝子cDNAとハイブリダイズした膜。レーン:M 分子量マーカー、YAC b142f9、AB1380(YACフリー酵母)、クローン13、クローン14、クローンB7。
【図10】YAC b142f9およびクローン14の5’領域のサザンブロットを示す図である。オリジナルクローンb142f9(APP)とクローン14とのサザンブロットバンドパターンの違い。
【図11】YAC b142f9およびクローン14のサザンブロットの比較を示す図である。クローンB142F9とクローン14の両方のDNAをBam HI酵素で消化した。アガロースゲルで分離し、APP遺伝子cDNAとハイブリダイズした。M、分子量マーカー;APP、クローンB142F9;14、クローン14。右側、B.300kbのAPP遺伝子をBam HIで消化した後に得られたバンドパターン、遺伝子コード領域を含む断片には網がけしている。
【図12】2連続ラウンドの酵母における相同組換えを利用したYAC b142f9の断片化のために提供されるスキームを示す。
【図13】クローンb106のdual PCR解析を示す図である。SeqAおよびSeqBの両方について、異なるクローンで行ったPCR。第1回目のPCRはAPP遺伝子のエキソン6で行い、一方、第2回目のPCRはGABPA遺伝子で行った。第1回目のPCRに関して陽性であり、第2回目のPCRに関して陰性であるクローンにはアスタリスクをつけて示している。C+、陽性対照;C−、陰性対照。
【図14】組換え環境のPCR解析を示す図である。PCRダイアグラム、陰影マークはSeqBの場所を示している。B.GABPA遺伝子、APP遺伝子のエキソン11、エキソン6、SeqA’およびSeqAのPCR。各PCRに対して、陽性対照、クローンb106および陰性対照に対応する3つのレーンが存在する。
【図15】APP遺伝子cDNAとGABPA遺伝子cDNAとを比較したPFGEについてのハイブリダイゼーションを示す図である。A.臭化エチジウムで染色したPFGEゲル。1.マーカー。2および5 b142f9。3および6 クローン14。4および7 クローンb106。B.レーン1〜4のAPP遺伝子cDNAとのハイブリダイゼーション。レーン5〜7のGABPA遺伝子cDNAとのハイブリダイゼーション。
【図16】3’における組換えについてのサザンブロットダイアグラムおよびゲルを示す。A.3’における組換えについてのサザンブロットダイアグラム、上部にはオリジナルYAC、下部には組み換えられたYAC。Reco Bの前の陰影四角は使用したプローブの位置を示している。B.3つのYAC、b142f9(APP)、クローン14およびクローンb106のEco RIおよびSca Iでの消化についてのサザンブロット。
【図17】クローンb142f9、14およびb106のサザンブロットの比較を示す図である。Bam HIで消化し、APP遺伝子cDNAプローブとハイブリダイズしたYAC b142f9(APP)、クローン14およびクローンb106のサザンブロットの比較。3つのYAC間ではバンドパターンは保存されている。M、マーカー。
【図18】YAC b106の抽出についてのPFGEを示す図である。YAC b106に対応するバンドの抽出。ゲルにおけるYACの位置を決めるためにまずサイドを染色した。YAC b106に対応するアガロースの一部分が中央部でなくなっている。
【図19】創始マウスを解析するために用いたPCRのダイアグラムを示す。
【図20】創始マウスについての代表的なPCRを示す図である。トランスジェニックマウスで行った23のPCRについてのサンプル、マウス2、12、35、37、41および46で行ったものを示している、c− 陰性対照、c+ 陽性対照。
【図21】マウス35のいくつかの子孫についての例示的なPCRおよびAPP遺伝子の生殖細胞系への伝達を示す図である。マウス35の最初の7子孫で行った4つのPCRについての画像。TRP1、エキソン6、エキソン11およびLYS2のPCRを行った。子孫1、4、5および7がトランスジーンを受け継いでいることを観察することができる。
【図22】創始マウスのF1のサザンブロットの比較を示す図である。非トランスジェニック同腹子と比較したマウス35および64のフィンガープリント。YAC b106で得られたものと同一のバンドパターンを観察することができる。
【図23】ヒトAPP遺伝子発現を示す棒グラフを示す。系統35にけるヒトAPP遺伝子発現についての相対単位での対数表現。97%信頼区間。中枢および末梢神経組織において高い発現が検出される。精巣、血液、心臓および腎臓ではレベルがより低い。
【図24】ネズミAPP遺伝子発現を示す棒グラフを示す。マウスにおけるネズミAPP遺伝子発現についての相対単位での表現。このグラフは、95%信頼区間を用いて、脾臓における発現(最も低い発現)に対して標準化している。凡例では各棒が対応している組織を示している。
【図25】APPタンパク質発現および評価を示す図である。A.APPタンパク質およびアクチンについてのウエスタンブロット解析の代表的なゲル。組織:1 皮質、2 脳室、3 小脳、4 中脳、5 脊髄、6 肝臓、7 肺、8 膵臓、9 脾臓、10 小腸、11 筋肉、12 精巣、13 心臓および14 腎臓。B.APPレベルのグラフ表現。青色の棒、野生マウス、赤色の棒、トランスジェニックマウス。組織:1 皮質、2 脳室、3 中脳、4 小脳および5 脊髄。
【図26】系統35のトランスジェニックマウスの組織学的解析を示す図である。マウス脳のコンゴレッド染色。A 野生マウスの皮質。B 野生マウスの海馬。C 系統35の皮質。D 系統35の海馬。E 1年齢Tg2576マウスの海馬、ここではアミロイド生成性斑が観察される。
【図27】系統64のトランスジェニックマウスの組織学的解析を示す図である。マウス脳のコンゴレッド染色。A 野生マウスの皮質。B 野生マウスの海馬。C 系統64の皮質。D 系統64の海馬。E 1年齢Tg2576マウスの海馬、ここではアミロイド生成性斑が観察される。
【特許請求の範囲】
【請求項1】
ヒト第21染色体(NCBI35:21:26064934:26642665:1)のヌクレオチド26,064,934とヌクレオチド26,642,665との間に含まれるヌクレオチド配列からなる単離ポリヌクレオチド、または完全ヒトAPP遺伝子(hAPP)をその調節配列とともに含んでなる該ポリヌクレオチドの断片。
【請求項2】
前記完全hAPP遺伝子が、その調節配列とともに、ヒト第21染色体(NCBI35:21:26174733:26464809:1)のヌクレオチド26,174,733とヌクレオチド26,464,809との間に含まれる、請求項1に記載のポリヌクレオチド。
【請求項3】
ヒト第21染色体(NCBI35:21:26064934:26573344:1)のヌクレオチド26,064,934とヌクレオチド26,573,344との間に含まれるヌクレオチド配列からなる、請求項1に記載のポリヌクレオチド。
【請求項4】
前記hAPP遺伝子が、その5’末端において(APP遺伝子転写の始点および方向に関連)第1のヌクレオチド配列に隣接され、その3’末端において(APP遺伝子転写の終点および方向に関連)第2のヌクレオチド配列に隣接され、ここで:
前記第1のヌクレオチド配列が、ヒト第21染色体(NCBI35:21:26464810:26642665:l)のヌクレオチド26,464,810とヌクレオチド26,642,665との間に含まれるヌクレオチド配列、または少なくとも1つの固有の配列を含むその断片からなり、かつ
前記第2のヌクレオチド配列が、ヒト第21染色体(NCBI35:21:26064934:26174732:1)のヌクレオチド26,064,934とヌクレオチド26,174,732との間に含まれるヌクレオチド配列、または少なくとも1つの固有の配列を含むその断片からなる、請求項1に記載のポリヌクレオチド。
【請求項5】
前記第1のヌクレオチド配列が、ヒト第21染色体(NCBI35:21:26464810:26573344:1)のヌクレオチド26,464,810とヌクレオチド26,573,344との間に含まれるヌクレオチド配列、または少なくとも1つの固有の配列を含むその断片を含んでなり、かつ
前記第2のヌクレオチド配列が、ヒト第21染色体(NCBI35:21:26064934:26174732:1)のヌクレオチド26,064,934とヌクレオチド26,174,732との間に含まれるヌクレオチド配列、または少なくとも1つの固有の配列を含むその断片を含んでなる、請求項4に記載のポリヌクレオチド。
【請求項6】
前記第1のヌクレオチド配列が、ヒト第21染色体(NCBI35:21:26571876:26573344:l)のヌクレオチド26,571,876とヌクレオチド26,573,344との間に含まれるヌクレオチド配列を含んでなる、請求項4または5に記載のポリヌクレオチド。
【請求項7】
前記第2のヌクレオチド配列が、ヒト第21染色体(NCBI35:21:26064934:26067221:l)のヌクレオチド26,064,934とヌクレオチド26,067,221との間に含まれるヌクレオチド配列を含んでなる、請求項4または5に記載のポリヌクレオチド。
【請求項8】
請求項1〜7のいずれか一項に記載のポリヌクレオチドを含んでなる、ベクター。
【請求項9】
YAC(酵母人工染色体)、BAC(細菌人工染色体)またはPAC(P1由来人工染色体)からなる群から選択される、請求項8に記載のベクター。
【請求項10】
前記ポリヌクレオチドが、その一方の末端において第3のヌクレオチド配列に隣接され、もう一方の反対の末端において第4のヌクレオチド配列に隣接され、ここで:
前記第3のヌクレオチド配列が、(i)酵母細胞において機能的なテロメア、および少なくとも(ii)1つの栄養要求性選択マーカーを含んでなり、かつ
前記第4のヌクレオチド配列が、(i)酵母細胞において機能的なセントロメア、酵母細胞において機能的なテロメア、および少なくとも1つの栄養要求性選択マーカーを含んでなる、請求項8に記載のベクター。
【請求項11】
前記栄養要求性選択マーカーが、TRP1、LEU2、LYS2、HIS3、HIS5、TRP1、URA3からなる群から選択される、請求項10に記載のベクター。
【請求項12】
請求項1〜7のいずれか一項に記載のポリヌクレオチド、または請求項8〜11のいずれか一項に記載のベクターを含んでなる、細胞。
【請求項13】
トランスジェニック非ヒト動物であって、そのゲノムに挿入された請求項1〜7のいずれか一項に記載のポリヌクレオチドを含有する、トランスジェニック非ヒト動物。
【請求項14】
前記動物が哺乳類、好ましくは齧歯類、より好ましくはマウスまたはラットである、請求項13に記載のトランスジェニック動物。
【請求項15】
そのゲノムがアルツハイマー病の1つ以上の分子マーカーをさらに含有する、請求項13または請求項14に記載のトランスジェニック動物。
【請求項16】
前記アルツハイマー病の分子マーカーが、PS1、PS2遺伝子、およびAPOE遺伝子のイプシロン4(ε4)対立遺伝子からなる群から選択される、請求項15に記載のトランスジェニック動物。
【請求項17】
孤発性アルツハイマー病(SAD)と家族性アルツハイマー病(FAD)の両方のアルツハイマー病を研究するための動物モデルとしての、請求項13〜16のいずれか一項に記載のトランスジェニック非ヒト動物の使用。
【請求項18】
アルツハイマー病の病因、原因、誘発因子、その疾患の確立につながる環境的および/または生理病理学的攻撃または傷害を研究するためのアルツハイマー病モデルとしての、請求項13〜16のいずれか一項に記載のトランスジェニック非ヒト動物の使用。
【請求項19】
アルツハイマー病の治療および/または予防に潜在的に有用な化合物をスクリーニング、発見、同定、評価、および妥当性を立証するための、請求項13〜16のいずれか一項に記載のトランスジェニック非ヒト動物の使用。
【請求項20】
前記hAPP遺伝子だけを発現するトランスジェニック非ヒト動物を得るための、請求項13〜16のいずれか一項に記載のトランスジェニック非ヒト動物の使用。
【請求項21】
アルツハイマー病の治療および/または予防に潜在的に有用な化合物を同定するための方法であって、
a)請求項13〜16のいずれか一項に記載のトランスジェニック非ヒト動物に候補化合物を投与する工程;
b)工程(a)の該トランスジェニック非ヒト動物における、hAPP遺伝子の発現パターン、および/またはhAPPタンパク質の産生、および/またはβ−アミロイド(Aβ)ペプチド産生を判定する工程、および
c)該トランスジェニック非ヒト動物において(i)神経組織におけるhAPP遺伝子発現レベルの低下、(ii)hAPPタンパク質産生および/もしくは蓄積の減少、ならびに/または(iii)Aβペプチド産生および/もしくは蓄積の減少、を引き起こす該候補化合物を選択する工程
を含んでなる、方法。
【請求項22】
ヒトにおける前記遺伝子の発現パターンと同様のまたは同等の発現パターンでヒトAPP遺伝子を内因的に発現するトランスジェニック非ヒト動物の子孫。
【請求項1】
ヒト第21染色体(NCBI35:21:26064934:26642665:1)のヌクレオチド26,064,934とヌクレオチド26,642,665との間に含まれるヌクレオチド配列からなる単離ポリヌクレオチド、または完全ヒトAPP遺伝子(hAPP)をその調節配列とともに含んでなる該ポリヌクレオチドの断片。
【請求項2】
前記完全hAPP遺伝子が、その調節配列とともに、ヒト第21染色体(NCBI35:21:26174733:26464809:1)のヌクレオチド26,174,733とヌクレオチド26,464,809との間に含まれる、請求項1に記載のポリヌクレオチド。
【請求項3】
ヒト第21染色体(NCBI35:21:26064934:26573344:1)のヌクレオチド26,064,934とヌクレオチド26,573,344との間に含まれるヌクレオチド配列からなる、請求項1に記載のポリヌクレオチド。
【請求項4】
前記hAPP遺伝子が、その5’末端において(APP遺伝子転写の始点および方向に関連)第1のヌクレオチド配列に隣接され、その3’末端において(APP遺伝子転写の終点および方向に関連)第2のヌクレオチド配列に隣接され、ここで:
前記第1のヌクレオチド配列が、ヒト第21染色体(NCBI35:21:26464810:26642665:l)のヌクレオチド26,464,810とヌクレオチド26,642,665との間に含まれるヌクレオチド配列、または少なくとも1つの固有の配列を含むその断片からなり、かつ
前記第2のヌクレオチド配列が、ヒト第21染色体(NCBI35:21:26064934:26174732:1)のヌクレオチド26,064,934とヌクレオチド26,174,732との間に含まれるヌクレオチド配列、または少なくとも1つの固有の配列を含むその断片からなる、請求項1に記載のポリヌクレオチド。
【請求項5】
前記第1のヌクレオチド配列が、ヒト第21染色体(NCBI35:21:26464810:26573344:1)のヌクレオチド26,464,810とヌクレオチド26,573,344との間に含まれるヌクレオチド配列、または少なくとも1つの固有の配列を含むその断片を含んでなり、かつ
前記第2のヌクレオチド配列が、ヒト第21染色体(NCBI35:21:26064934:26174732:1)のヌクレオチド26,064,934とヌクレオチド26,174,732との間に含まれるヌクレオチド配列、または少なくとも1つの固有の配列を含むその断片を含んでなる、請求項4に記載のポリヌクレオチド。
【請求項6】
前記第1のヌクレオチド配列が、ヒト第21染色体(NCBI35:21:26571876:26573344:l)のヌクレオチド26,571,876とヌクレオチド26,573,344との間に含まれるヌクレオチド配列を含んでなる、請求項4または5に記載のポリヌクレオチド。
【請求項7】
前記第2のヌクレオチド配列が、ヒト第21染色体(NCBI35:21:26064934:26067221:l)のヌクレオチド26,064,934とヌクレオチド26,067,221との間に含まれるヌクレオチド配列を含んでなる、請求項4または5に記載のポリヌクレオチド。
【請求項8】
請求項1〜7のいずれか一項に記載のポリヌクレオチドを含んでなる、ベクター。
【請求項9】
YAC(酵母人工染色体)、BAC(細菌人工染色体)またはPAC(P1由来人工染色体)からなる群から選択される、請求項8に記載のベクター。
【請求項10】
前記ポリヌクレオチドが、その一方の末端において第3のヌクレオチド配列に隣接され、もう一方の反対の末端において第4のヌクレオチド配列に隣接され、ここで:
前記第3のヌクレオチド配列が、(i)酵母細胞において機能的なテロメア、および少なくとも(ii)1つの栄養要求性選択マーカーを含んでなり、かつ
前記第4のヌクレオチド配列が、(i)酵母細胞において機能的なセントロメア、酵母細胞において機能的なテロメア、および少なくとも1つの栄養要求性選択マーカーを含んでなる、請求項8に記載のベクター。
【請求項11】
前記栄養要求性選択マーカーが、TRP1、LEU2、LYS2、HIS3、HIS5、TRP1、URA3からなる群から選択される、請求項10に記載のベクター。
【請求項12】
請求項1〜7のいずれか一項に記載のポリヌクレオチド、または請求項8〜11のいずれか一項に記載のベクターを含んでなる、細胞。
【請求項13】
トランスジェニック非ヒト動物であって、そのゲノムに挿入された請求項1〜7のいずれか一項に記載のポリヌクレオチドを含有する、トランスジェニック非ヒト動物。
【請求項14】
前記動物が哺乳類、好ましくは齧歯類、より好ましくはマウスまたはラットである、請求項13に記載のトランスジェニック動物。
【請求項15】
そのゲノムがアルツハイマー病の1つ以上の分子マーカーをさらに含有する、請求項13または請求項14に記載のトランスジェニック動物。
【請求項16】
前記アルツハイマー病の分子マーカーが、PS1、PS2遺伝子、およびAPOE遺伝子のイプシロン4(ε4)対立遺伝子からなる群から選択される、請求項15に記載のトランスジェニック動物。
【請求項17】
孤発性アルツハイマー病(SAD)と家族性アルツハイマー病(FAD)の両方のアルツハイマー病を研究するための動物モデルとしての、請求項13〜16のいずれか一項に記載のトランスジェニック非ヒト動物の使用。
【請求項18】
アルツハイマー病の病因、原因、誘発因子、その疾患の確立につながる環境的および/または生理病理学的攻撃または傷害を研究するためのアルツハイマー病モデルとしての、請求項13〜16のいずれか一項に記載のトランスジェニック非ヒト動物の使用。
【請求項19】
アルツハイマー病の治療および/または予防に潜在的に有用な化合物をスクリーニング、発見、同定、評価、および妥当性を立証するための、請求項13〜16のいずれか一項に記載のトランスジェニック非ヒト動物の使用。
【請求項20】
前記hAPP遺伝子だけを発現するトランスジェニック非ヒト動物を得るための、請求項13〜16のいずれか一項に記載のトランスジェニック非ヒト動物の使用。
【請求項21】
アルツハイマー病の治療および/または予防に潜在的に有用な化合物を同定するための方法であって、
a)請求項13〜16のいずれか一項に記載のトランスジェニック非ヒト動物に候補化合物を投与する工程;
b)工程(a)の該トランスジェニック非ヒト動物における、hAPP遺伝子の発現パターン、および/またはhAPPタンパク質の産生、および/またはβ−アミロイド(Aβ)ペプチド産生を判定する工程、および
c)該トランスジェニック非ヒト動物において(i)神経組織におけるhAPP遺伝子発現レベルの低下、(ii)hAPPタンパク質産生および/もしくは蓄積の減少、ならびに/または(iii)Aβペプチド産生および/もしくは蓄積の減少、を引き起こす該候補化合物を選択する工程
を含んでなる、方法。
【請求項22】
ヒトにおける前記遺伝子の発現パターンと同様のまたは同等の発現パターンでヒトAPP遺伝子を内因的に発現するトランスジェニック非ヒト動物の子孫。
【図1】

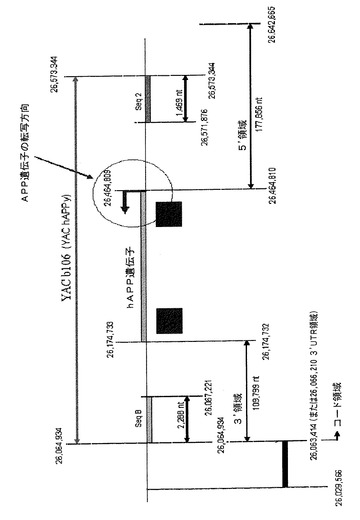
【図2】

【図3】
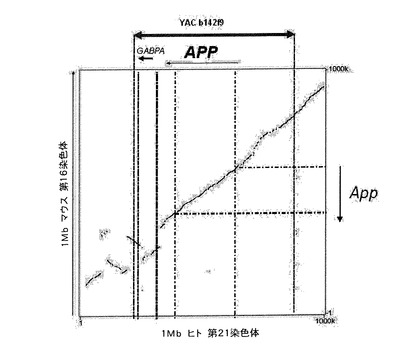
【図4】
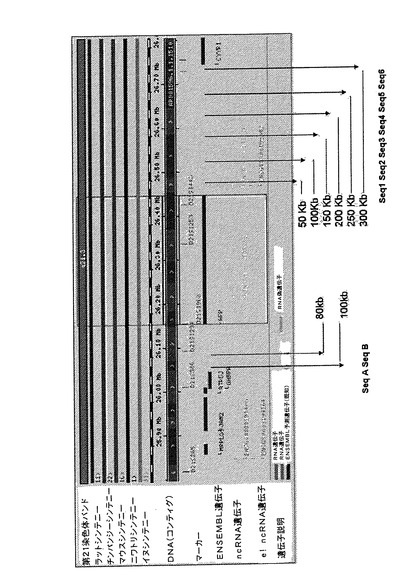
【図5】
【図6】
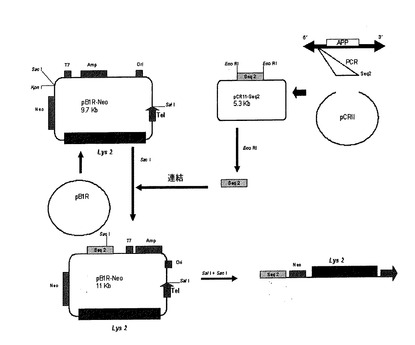
【図7】
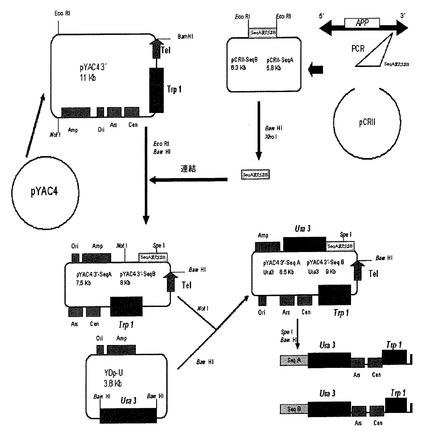
【図9】
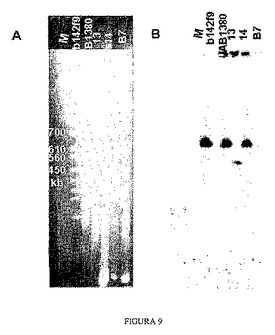
【図10】
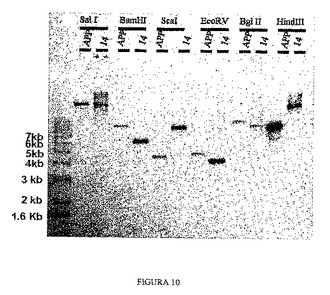
【図12】
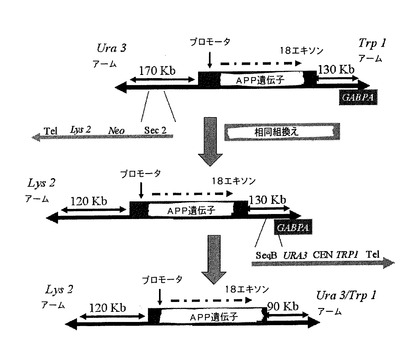
【図15】
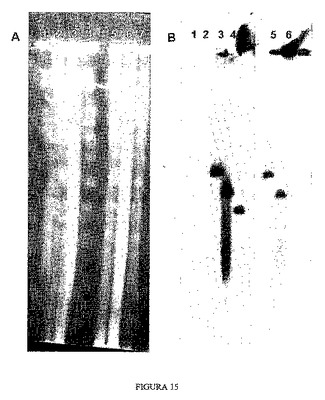
【図16】
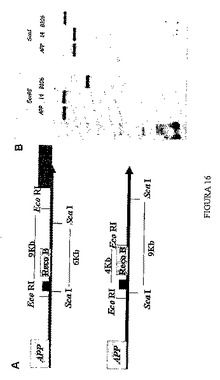
【図18】
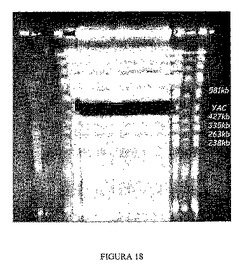
【図19】
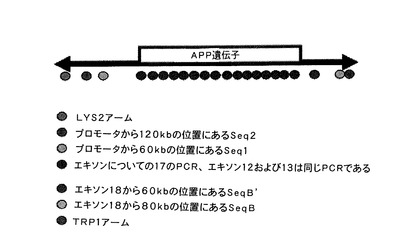
【図23】
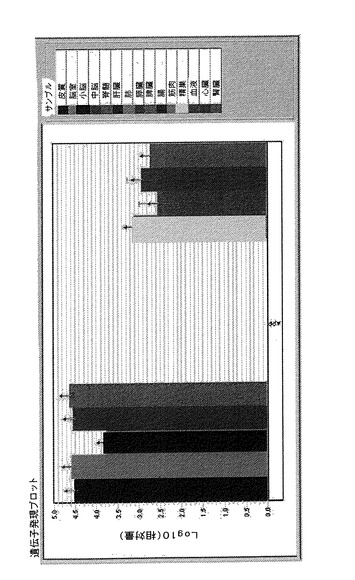
【図24】

【図26】
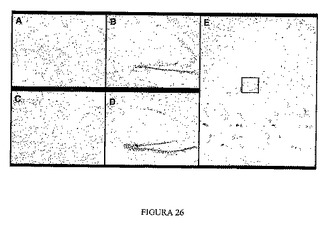
【図27】

【図8】

【図11】
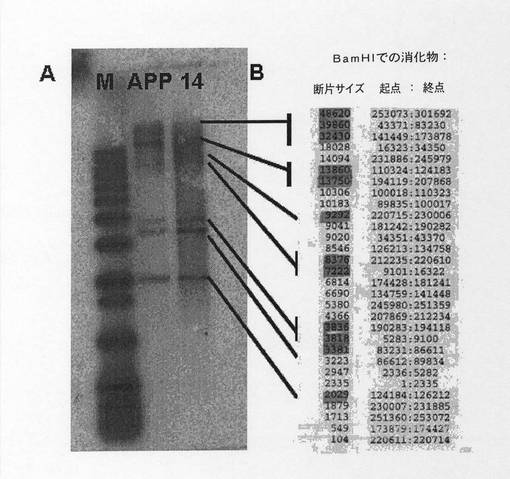
【図13】

【図14】
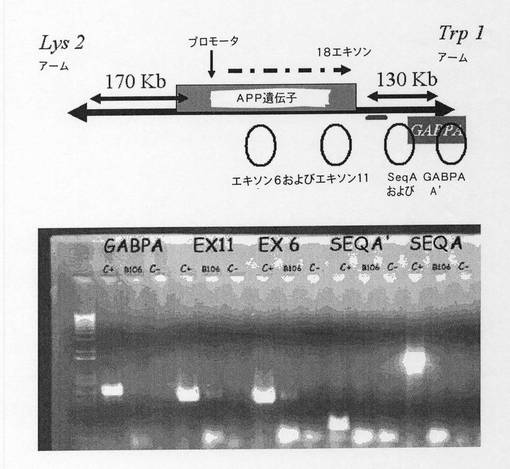
【図17】
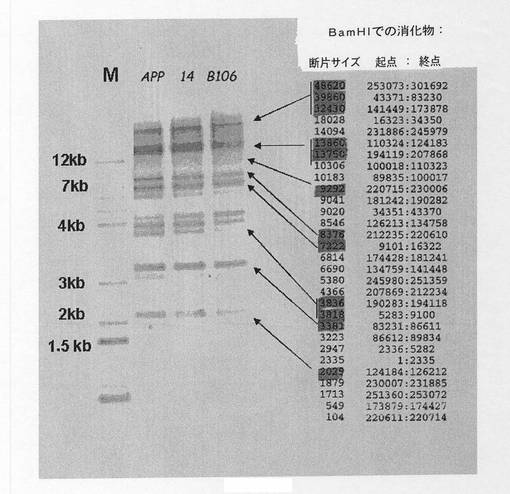
【図20】
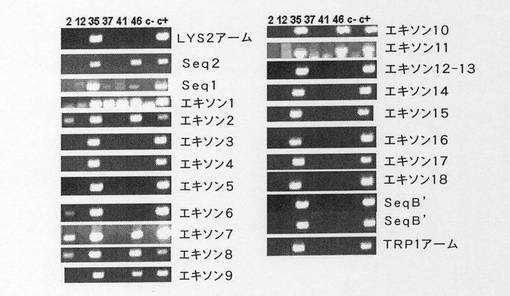
【図21】
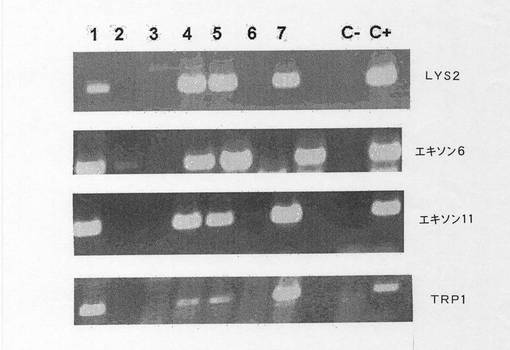
【図22】
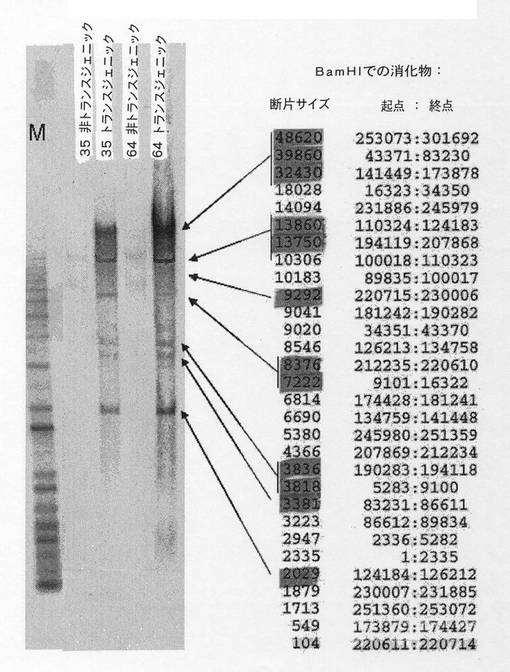
【図25】
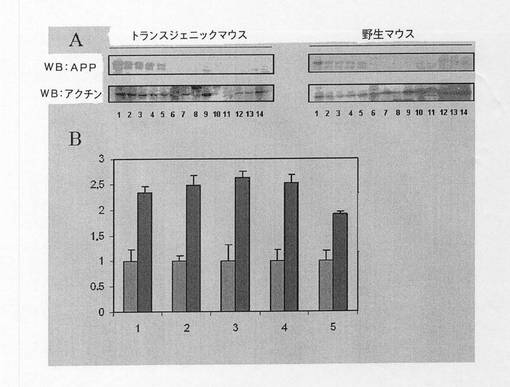
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図9】
【図10】
【図12】
【図15】
【図16】
【図18】
【図19】
【図23】
【図24】
【図26】
【図27】
【図8】
【図11】
【図13】
【図14】
【図17】
【図20】
【図21】
【図22】
【図25】
【公表番号】特表2009−517066(P2009−517066A)
【公表日】平成21年4月30日(2009.4.30)
【国際特許分類】
【出願番号】特願2008−542783(P2008−542783)
【出願日】平成18年12月1日(2006.12.1)
【国際出願番号】PCT/ES2006/000673
【国際公開番号】WO2007/063160
【国際公開日】平成19年6月7日(2007.6.7)
【出願人】(504434051)ウニベルシダッド・アウトノマ・デ・マドリッド (7)
【出願人】(593005895)コンセホ・スペリオール・デ・インベスティガシオネス・シエンティフィカス (67)
【氏名又は名称原語表記】CONSEJO SUPERIOR DE INVESTIGACIONES CIENTIFICAS
【Fターム(参考)】
【公表日】平成21年4月30日(2009.4.30)
【国際特許分類】
【出願日】平成18年12月1日(2006.12.1)
【国際出願番号】PCT/ES2006/000673
【国際公開番号】WO2007/063160
【国際公開日】平成19年6月7日(2007.6.7)
【出願人】(504434051)ウニベルシダッド・アウトノマ・デ・マドリッド (7)
【出願人】(593005895)コンセホ・スペリオール・デ・インベスティガシオネス・シエンティフィカス (67)
【氏名又は名称原語表記】CONSEJO SUPERIOR DE INVESTIGACIONES CIENTIFICAS
【Fターム(参考)】
[ Back to top ]