
アルデヒドデヒドロゲナーゼ遺伝子
本発明は、アルデヒドデヒドロゲナーゼ(SNDH)をコードするDNA、前記DNAを含む発現ベクター、および前記DNAを含む組換え微生物に関する。さらに、本発明は、組換えアルデヒドデヒドロゲナーゼタンパク質を産生するプロセス、および、組換えアルデヒドデヒドロゲナーゼタンパク質または発現ベクターを含む組換え微生物を使用することにより、L−ソルボソンからL−アスコルビン酸(ビタミンC)および/または2−ケト−L−グロン酸(2−KGA)を産生するプロセスにも関する。また、前記アルデヒドデヒドロゲナーゼをコードする遺伝子が破壊されている微生物を用いて、2−KGAを産生するプロセスも提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、Gluconobacter oxydansDSM4025から得られるアルデヒドデヒドロゲナーゼ(SNDH)をコードする新規DNA、前記DNAを含む発現ベクター、および前記発現ベクターを含む組換え微生物に関する。さらに、本発明は、組換えアルデヒドデヒドロゲナーゼタンパク質を産生するプロセス、および、組換えアルデヒドデヒドロゲナーゼタンパク質または前記発現ベクターを含む組換え微生物を使用することにより、L−ソルボソンからL−アスコルビン酸(ビタミンC)および/または2−ケト−L−グロン酸(2−KGA)を産生するプロセスに関する。
【0002】
ビタミンCはヒトにとって不可欠な栄養因子の1つであり、約60年間の間、ライヒシュタインプロセスにより商業的に合成されてきた。家畜動物はビタミンCを自身の体内で合成できるとしても、合成ビタミンCはまた動物食餌中にも使用されている。ライヒシュタインプロセスは工業的ビタミンC産生において多くの有益な点を有するが、高いエネルギー消費量ならびにかなりの量の有機溶媒および無機溶媒の使用などの望ましくない問題を有する。それ故、過去数十年間かけて、酵素変換を使用してビタミンCを製造するアプローチが数多く研究されており、このアプローチはより経済的かつ環境に優しいであろう。
【0003】
本発明は、配列番号1のヌクレオチド配列に少なくとも95%同一であるポリヌクレオチドを含むアルデヒドデヒドロゲナーゼをコードする単離核酸分子に関する。
【0004】
本明細書に使用したような「SNDH」はアルデヒドデヒドロゲナーゼを意味する。
【0005】
本明細書に使用したような「核酸分子」は、DNAおよびRNAの両方を含み、特記しない限り、二本鎖核酸、一本鎖核酸、およびそのヌクレオシドを含む。DNA−RNAハイブリッド、DNA−RNA−タンパク質ハイブリッド、RNA−タンパク質ハイブリッド、およびDNA−タンパク質ハイブリッドなどのハイブリッドも含まれる。
【0006】
本明細書に使用したような「変異」は、対象のヌクレオチド配列の1塩基対の変化、挿入、または欠失を意味する。
【0007】
本明細書に使用したような「突然変異生成」は、変異がDNAに生じるプロセスを意味する。「無作為」突然変異生成では、変異の正確な部位は予測不可能であり、微生物の染色体の何処でも起こり、突然変異は、照射または化学的処理などの要因により引き起こされる物理的傷害の結果としてもたらされる。
【0008】
本明細書に使用したような「プロモーター」は、開始コドンの近くに位置する、遺伝子の5’領域として一般に記載されるDNA配列を意味する。隣接遺伝子(群)の転写は、プロモーター領域で開始する。プロモーターが誘導性プロモーターである場合、転写速度は、誘導剤に応答して増加する。これに対し、プロモーターが構成性プロモーターである場合、転写速度は、誘導剤により調節されない。
【0009】
本明細書に使用したような「同一率」は、以下に例示したような配列解析プログラムにより、比較するヌクレオチドまたはアミノ酸配列における同一ヌクレオチドまたはアミノ酸と一致した、対象ヌクレオチドまたはアミノ酸配列のヌクレオチドまたはアミノ酸の比率を意味する。
【0010】
本発明は、(a)配列番号1のヌクレオチド258〜2084、(b)配列番号1のヌクレオチド351〜2084、(c)配列番号1のヌクレオチド258〜1955、および(d)配列番号1のヌクレオチド351〜1955からなる群より選択されるポリヌクレオチドに少なくとも95%同一であるポリヌクレオチドを含むアルデヒドデヒドロゲナーゼをコードする単離核酸分子を含む。
【0011】
本発明の別の態様は、(a)配列番号2のアミノ酸配列を有するポリペプチドをコードするポリヌクレオチド、(b)配列番号2のアミノ酸32〜609からなるポリペプチドをコードするポリヌクレオチド、(c)配列番号2のアミノ酸1〜566からなるポリペプチドをコードするポリヌクレオチド、および(d)配列番号2のアミノ酸32〜566からなるポリペプチドをコードするポリヌクレオチドからなる群より選択されるポリヌクレオチドを含むアルデヒドデヒドロゲナーゼをコードする単離核酸分子を提供することである。SNDH活性を有し、前記したアミノ酸配列中の1つ以上のアミノ酸(群)の置換、欠失、挿入、または付加により前記したタンパク質から得られるタンパク質も含む。
【0012】
本発明の別の態様としての機能的誘導体は、本発明のアミノ酸配列に基づいて、かかる配列の1つ以上のアミノ酸残基の付加、挿入、欠失、および/または置換により定義され、このような誘導体は、当分野で既知のまたは本明細書で明記したアッセイにより測定したSNDH活性を依然として有する。このような機能的誘導体は、当分野で既知の方法により本明細書に開示したようなDNA配列に基づいて、当分野で既知の化学的ペプチド合成により、または、組換え技術により作製できる。このような分子の活性を一般に変化させないタンパク質およびペプチド中のアミノ酸の交換は、当分野で既知である。
【0013】
本発明の特定の実施形態において、対象の保存的置換は以下のように起こる:例として以下の置換、AlaからVal/Leu/Ile、ArgからLys/Gln/Asn、AsnからGln/His/Lys/Arg、AspからGlu、CysからSer、GlnからAsn、GluからAsp、GlyからPro/Ala、HisからAsn/Gln/Lys/Arg、IleからLeu/Val/Met/Ala/Phe/norLeu、LysからArg/Gln/Asn、MetからLeu/Phe/Ile、PheからLeu/Val/Ile/Ala/Tyr、ProからAla、SerからThr、ThrからSer、TrpからTyr/Phe、TyrからTrp/Phe/Thr/Ser、およびValからIle/Leu/Met/Phe/Ala/norLeuが妥当である。好ましい例として、AlaからVal、ArgからLys、AsnからGln、AspからGlu、CysからSer、GlnからAsn、GluからAsp、GlyからAla、HisからArg、IleからLeu、LeuからIle、LysからArg、MetからLeu、PheからLeu、ProからAla、SerからThr、ThrからSer、TrpからTyr、TyrからPhe、およびValからLeuが妥当である。このような置換により生物活性が変化した場合、より実質的な変化、前記した表示した例示的置換を導入し、産物をスクリーニングする。
【0014】
さらに、本発明は、配列番号2として配列表に開示したようなSNDH活性を有するポリペプチドをコードするポリヌクレオチドおよびその相補鎖、または、これらの配列、そのDNA配列またはその断片を含むもの、および、このような配列と標準的な条件下でハイブリダイズするが、正に同じアミノ酸配列を有するポリペプチドをコードするDNA配列に関する。
【0015】
従って、本発明は、アルデヒドデヒドロゲナーゼ活性を有するポリペプチドをコードする単離核酸分子を提供し、前記核酸分子の相補体は、前記したような核酸分子と標準的な条件下でハイブリダイズする。本発明の1つの態様は、アルデヒドデヒドロゲナーゼ活性を有するポリペプチドをコードする単離核酸分子を提供することであり、前記核酸分子は、(i)配列番号1のヌクレオチド配列に少なくとも95%同一であるポリヌクレオチドを含むアルデヒドデヒドロゲナーゼ;(ii)(a)配列番号1のヌクレオチド258〜2084、(b)配列番号1のヌクレオチド351〜2084、(c)配列番号1のヌクレオチド258〜1955、および(d)配列番号1のヌクレオチド351〜1955からなる群より選択されるポリヌクレオチドに少なくとも95%同一であるポリヌクレオチドを含むアルデヒドデヒドロゲナーゼ;および(iii)(a)配列番号2のアミノ酸配列を有するポリペプチドをコードするポリヌクレオチド、(b)配列番号2のアミノ酸32〜609からなるポリペプチドをコードするポリヌクレオチド、(c)配列番号2のアミノ酸1〜566からなるポリペプチドをコードするポリヌクレオチド、および(d)配列番号2のアミノ酸32〜566からなるポリペプチドをコードするポリヌクレオチドからなる群より選択されるポリヌクレオチドを含むアルデヒドデヒドロゲナーゼをコードする核酸分子の相補鎖と標準的な条件下でハイブリダイズする。
【0016】
ハイブリダイゼーションの「標準的条件」は、この脈絡では、特異的ハイブリダイゼーションシグナルを検出するために当業者が一般に使用する条件、または好ましくはいわゆる当業者が使用するストリンジェントなハイブリダイゼーション条件を意味する。
【0017】
従って、本明細書に使用したような「ストリンジェントなハイブリダイゼーション条件」は、配列間に95%、好ましくは少なくとも97%の同一性が存在した場合にハイブリダイゼーションが起こることを意味する。ストリンジェントなハイブリダイゼーション条件は、例えば、50%ホルムアミド、5×SSC(150mM NaCl、15mMクエン酸三ナトリウム)、0.2%ドデシル硫酸ナトリウム、0.1%N−ラウロイルサルコシン、および2%遮断剤(Roche Diagnostics GmbH)を含む溶液中で、ジゴキシゲニン(DIG)標識DNAプローブ(DIGラベリングシステムを使用することにより作製;Roche Diagnostics GmbH、68298 Mannheim、ドイツ)を使用して42℃で一晩インキュベートし、その後、約60℃で0.1×SSC中でフィルターを洗浄する条件である。
【0018】
本発明はまた、前記したような核酸分子を含む組換えベクター、すなわち発現ベクターに関する。本発明の発現ベクターは、適切な宿主細胞で機能するものである。本発明の核酸分子の発現に好ましいベクターは、pQE、pUC、pBluescriptII、pACYC177、pACYC184、pVK100、およびRSF1010からなる群より選択されるベクターまたはその誘導体である。
【0019】
本発明のヌクレオチド配列の発現に適切な宿主細胞は、細菌、酵母、および植物細胞からなる群より選択される組換え微生物である。好ましくは、微生物は、Gluconobacter、Acetobacter、Pseudomonas、Acinetobacter、Klebsiella、およびEscherichiaからなる群より選択される。このような好ましい微生物の例はE.coliである。より好ましい宿主細胞は、Gluconobacter oxydans、最も好ましくはG.oxydansDSM4025(FERM BP−3812)に属し、これは1987年3月17日、ブダペスト条約の条件下でDeutsche Sammlung von Mikroorganismen und Zellkulturen GmbH、Braunschweig、ドイツに寄託された。
【0020】
微生物「Gluconobacter oxydans」はまた、国際原核生物命名規約により定義されたような、同じ物理化学的特性を有するこのような種の同義語またはバソニム(basonyms)を含む。
【0021】
従って、本発明は、前記したような発現ベクターで形質転換されるか、または、その染色体DNAに組み込まれた前記したような核酸分子を含む、組換え微生物に関する。
【0022】
多種多様な宿主/ベクターの組合せを、本発明の二本鎖ヌクレオチド配列のクローニングに使用し得る。E.coliが好ましい宿主細胞であるので、E.coliに通常使用される任意のベクターが本発明に有用である。このようなベクターには、Hisタグ化組換えタンパク質を発現できるpQEベクター(QIAGEN K.K.、東京、日本)、pUC18およびpBluescriptII(Stratagene Cloning Systems、カリフォルニア、米国)を含むpBR322またはその誘導体、pACYC177およびpACYC184およびその誘導体、および、RK2およびRSF1010などの広宿主域プラスミドから得られるベクターが含まれるがこれに限定されない。従って、本発明に使用する発現ベクターは、pQE−プラスミド、pUC−プラスミド、pBluescriptII、pACYC177、pACYC184、およびその誘導体プラスミド、および、pVK100およびRSF1010などの広宿主域プラスミドから得られる。
【0023】
本明細書に使用したような「発現ベクター」は、適切な宿主への形質転換後に、クローニングベクター中にクローニングした遺伝子の発現を増強できるクローニングベクターを意味する。クローニングされた遺伝子は、プロモーター配列などの特定の制御配列の制御下(すなわちそれに作動可能に連結)に置かれる。プロモーター配列は構成性でも誘導性でもよい。
【0024】
本明細書に使用したような「クローニングベクター」は、宿主細胞中で自律的に複製でき、1つまたは少数の制限エンドヌクレアーゼ認識部位により特徴づけられる、プラスミドもしくはファージDNAまたは他のDNA配列を意味し、ここでの制限エンドヌクレアーゼ認識部位において、ベクターの本質的な生物学的機能が損なわれることなく決まった様式でこのようなDNA配列が切断され得、ここにDNA断片が導入されてその複製およびクローニングがなされ得る。クローニングベクターはさらに、クローニングベクターで形質転換された細胞の同定に使用するに適切なマーカーを含む。このようなマーカーは、例えば、テトラサイクリンまたはアンピシリン耐性を付与する。
【0025】
本明細書に使用したような「組換えベクター」は、望ましいクローニングした遺伝子(群)を含む任意のクローニングベクターまたは発現ベクターを含む。
【0026】
本明細書に使用したような「発現」は、ポリペプチドが構造遺伝子から産生されるプロセスを意味する。このプロセスには、遺伝子からmRNAへの転写、および、このようなmRNAからポリペプチド(群)への翻訳が含まれる。
【0027】
本明細書に使用したような「組換え微生物」は、発現ベクターまたはクローニングベクター上に望ましいクローニング遺伝子(群)を含む任意の原核細胞または真核細胞であり得る組換え宿主を含む。この用語はまた、前記微生物の染色体またはゲノムに望ましい遺伝子(群)を含むように遺伝子操作された原核細胞または真核細胞も含む。
【0028】
本明細書に使用したような「宿主」は、複製可能な発現ベクターまたはクローニングベクターのレシピエントである任意の原核細胞または真核細胞を含む。「宿主」は、この用語が本明細書で使用されているように、その染色体またはゲノム上に望ましい遺伝子(群)を含むように公知の技術により遺伝子操作することのできる原核細胞または真核細胞も含む。かかる宿主の例は当業者には周知である。
【0029】
組換えDNA、例えば組換えベクターを有する組換え微生物を作製するために、形質転換、形質導入、接合、または電気穿孔を含むがこれに限定されない種々の遺伝子導入法を使用できる。これらの方法は分子生物学の分野では公知である。慣用的な形質転換システムを、Gluconobacter、Acetobacter、Pseudomonas、Acinetobacter、Klebsiella、またはEscherichiaに使用できる。形質導入システムはまた、E.coliにも使用できる。接合システムは、E.coli、P.putida、およびGluconobacterを含むグラム陽性およびグラム陰性細菌に広く使用できる。接合の例はWO89/06,688号に開示されている。接合は、液体培地または固体表面上で起こり得る。SNDH産生のための適切なレシピエントの例には、Gluconobacter、Acetobacter、Pseudomonas、Acinetobacter、Klebsiella、またはEscherichiaの微生物が含まれる。接合用のレシピエントに、選択マーカー、例えばナリジクス酸またはリファンピシンに対する耐性を付加し得る。天然淘汰も使用でき、例えば、ポリミキシンBに対する耐性は、多くのGluconobacterに有用である。
【0030】
本発明に有用である好ましいベクターは、広宿主域ベクター、例えばpVK100およびその誘導体およびRSF1010のようなコスミドベクターである。ベクターのコピー数および安定性は、クローニングされた核酸分子の安定かつ効率的な発現のため、および、前記のクローニングした分子を有する宿主細胞の効率的な培養のために注意深く考慮すべきである。Tn5などの転位可能な要素を含む核酸分子を使用して、望ましいDNAを、好ましい宿主に、特に染色体上に導入することもできる。好ましい宿主から単離した任意のDNAを本発明のヌクレオチド配列と共に含む核酸分子も、本発明のヌクレオチド配列を、好ましい宿主細胞、特に染色体上に導入するのに有用である。このような核酸分子は、宿主細胞および核酸分子の性質を考慮しながら、当分野で公知である、例えば、形質転換、形質導入、接合、または電気穿孔などのいずれかの慣用的な方法を適用することにより、好ましい宿主に導入することができる。
【0031】
本発明で提供されるSNDH遺伝子を含むヌクレオチド配列を、当分野で公知の方法を用いて、前記した宿主細胞で作動可能なプロモーター、リボソーム結合部位、および転写ターミネーターなどの調節領域を含む適切なベクターに連結して、適切な発現ベクターを作製する。
【0032】
G.oxydansDSM4025から単離した望ましい遺伝子/ヌクレオチド配列を効率的に発現するために、種々のプロモーター、例えば遺伝子の元々のプロモーター、Tn5のカナマイシン耐性遺伝子、pBR322のアンピシリン耐性遺伝子などの抗生物質耐性遺伝子のプロモーター、およびE.coli(lac)のβ−ガラクトシダーゼ、trp−、tac−、trc−プロモーター、λファージのプロモーター、および、宿主細胞で機能的である任意のプロモーターを使用することができる。この目的では、宿主細胞は、細菌、酵母、および植物細胞からなる群より選択できる。好ましくは、宿主細胞は、Gluconobacter、Acetobacter、Pseudomonas、Acinetobacter、Klebsiella、またはEscherichia属に属する。
【0033】
発現において、宿主細胞(これにコード配列を導入して本発明の組換え細胞を得る)で作動可能であるシャイン・ダルガーノ(SD)配列(例えば、宿主細胞で作動可能である天然および合成配列を含むAGGAGG)および転写ターミネーター(宿主細胞で作動可能である任意の天然および合成配列を含む逆方向反復構造)などの、他の調節要素を、前記のプロモーターと共に使用できる。
【0034】
本発明のSNDHタンパク質のようにペリプラスム空間に位置するポリペプチドの発現では、通常15〜50アミノ酸残基を含み全体的に疎水性であるシグナルペプチドが好ましくは会合している。シグナルペプチドをコードするDNAは、望ましい宿主細胞で作動可能である任意の天然および合成配列から選択できる。配列番号2のアミノ酸残基1〜31を含む推定シグナルペプチドはまた、本発明のSNDH遺伝子により発現されるタンパク質にも見出された(配列番号4)。
【0035】
特記しない限り、本明細書の精製SNDHタンパク質をシークエンスすることにより決定された全てのアミノ酸配列は、自動アミノ酸シークエンサー(例えばモデル470A、パーキンエルマー・アプライド・バイオシステムズ)を使用して決定した。
【0036】
特記しない限り、本明細書のDNA分子をシークエンスすることにより決定された全てのヌクレオチド配列は、自動DNAシークエンサー(例えばモデルALFエクスプレスII、アマシャム・ファルマシア・バイオテック)を使用して決定し、本明細書で決定したDNA分子によりコードされるポリペプチドの全てのアミノ酸配列は、前記のように決定したDNA配列の翻訳により予測された。それ故、この自動アプローチにより決定された任意のDNA配列について当分野で既知であるように、本明細書で決定された任意のヌクレオチド配列はいくつかのエラーを含み得る。自動化により決定されたヌクレオチド配列は、典型的には、シークエンスしたDNA分子の実際のヌクレオチド配列と少なくとも約90%同一、より典型的には少なくとも約95%から少なくとも約99.9%同一である。実際の配列は、当分野で公知のマニュアルDNAシークエンス法を含む他のアプローチにより、より正確に決定することができる。これも当分野で既知であるように、実際の配列と比べて、決定されたヌクレオチド配列における1つの挿入または欠失により、ヌクレオチド配列の翻訳においてフレームシフトが引き起こされ、決定されたヌクレオチド配列によりコードされる予測アミノ酸配列は、このような挿入点または欠失点から始まって、シークエンスされたDNA分子により実際にコードされるアミノ酸配列とは完全に異なるであろう。
【0037】
本発明は、酵素(SNDH)をコードする単離核酸分子を提供する。単離核酸分子を操作するように設計された方法および技術は、当分野で公知である。核酸分子を、単離、精製およびクローニングする方法、ならびに、真核および原核宿主細胞の使用ならびにそこでの核酸およびタンパク質発現を記載した方法および技術は、当業者に既知である。
【0038】
簡潔には、SNDH遺伝子、前記遺伝子を含むDNA分子、組換え発現ベクター、および本発明で使用する組換え微生物は、以下の工程により得ることができる:
(1)G.oxydansDSM4025から染色体DNAを単離し、例えばE.coliなどの適切な宿主細胞において染色体DNAを用いて遺伝子ライブラリーを作製し;
(2)コロニーハイブリダイゼーション、プラークハイブリダイゼーション、またはサザンハイブリダイゼーション、PCR(ポリメラーゼ連鎖反応)クローニング、ウェスタンブロット解析または当分野で既知の他の技術により染色体DNAからSNDH遺伝子をクローニングし;
(3)慣用的な方法により前記のようにして得られたSNDH遺伝子のヌクレオチド配列を決定して前記SNDH遺伝子を含むDNA分子を選択し、SNDH遺伝子を効率的に発現できる組換え発現ベクターを作製し;
(4)宿主細胞にDNAを導入する適切な方法、例えば形質転換、形質導入、接合および/または電気穿孔によりSNDH遺伝子を有する組換え微生物を作製し、ここでの宿主細胞は、よって、本発明の組換え微生物となる。
【0039】
本発明の前記態様に使用した物質および技術は、以下のように詳細に例示する。
【0040】
全染色体DNAは、当分野で公知の手法により精製できる。望ましい遺伝子を、典型的には以下の例示的方法のいずれかにより、全染色体DNAから、プラスミドまたはファージベクターにクローニングできる:
(i)部分的アミノ酸配列を、精製タンパク質またはそのペプチド断片から決定する。このような全タンパク質またはペプチド断片は、このような全タンパク質の単離により、または、SDSポリアクリルアミドゲル電気泳動後のゲルからのペプチダーゼ処理により調製できる。このようにして得られたタンパク質またはその断片を、アプライド・バイオシステムズ自動ガス相シークエンサー470Aなどのタンパク質シークエンサーに適用する。アミノ酸配列を使用して、アプライド・バイオシステムズ自動DNAシークエンサー381AなどのDNA合成機を用いてオリゴヌクレオチドプローブおよび/またはプライマーを設計および調製できる。プローブは、サザンハイブリダイゼーション、コロニーハイブリダイゼーション、またはプラークハイブリダイゼーションを用いて、標的遺伝子を有する株の遺伝子ライブラリーから、標的遺伝子を有するクローンを単離するために使用できる。
(ii)または、遺伝子ライブラリーから標的タンパク質を発現するクローンを選択する目的で、標的タンパク質に対して調製した抗体を用いる免疫学的方法を適用できる。
(iii)標的遺伝子のDNA断片を、1セットのプライマー、すなわち、前記のように決定したアミノ酸配列に従って合成した2つのオリゴヌクレオチドを用いて、PCR法により、全染色体DNAから増幅できる。その後、標的全遺伝子を有するクローンを、プローブとして前記で得られたPCR産物を用いて、サザンハイブリダイゼーション、コロニーハイブリダイゼーション、またはプラークハイブリダイゼーションにより、例えばE.coliにおいて作製した遺伝子ライブラリーから単離できる。
【0041】
当分野で既知の方法により本明細書に開示したDNA配列に基づいて設計したプライマーを使用することによりPCRにより作製できるDNA配列もまた、本発明の目的である。
【0042】
前記した抗体は、抗原としての、精製SNDHタンパク質、E.coliにおいて発現されるHisタグ化SNDHなどの精製組換えSNDHタンパク質、またはそのペプチド断片を用いて調製できる。
【0043】
一旦、望む遺伝子を有するクローンが得られると、標的遺伝子のヌクレオチド配列を、M13ファージを用いたジデオキシ鎖終結法などの公知の方法により決定することができる。
【0044】
本発明の遺伝子は、図2に示したような31アミノ酸残基の推定シグナルペプチド(配列番号2のアミノ酸1〜31からなる配列番号4)と共に、578アミノ酸残基(配列番号2のアミノ酸32〜609からなる配列番号5)のSNDH酵素をコードする。ヌクレオチド配列に関して、SNDH遺伝子のコード領域は、配列番号1のヌクレオチド258〜2087を包含し、推定シグナルペプチド(配列番号1のヌクレオチド258〜350)および終止コドン(配列番号1のヌクレオチド2085〜2087)のコード配列を含む。従って、終止コドンを含まないヌクレオチド配列は、配列番号1の258〜2084位のヌクレオチド配列であり、さらにシグナル配列を含まない場合にはヌクレオチド配列は、配列番号1のヌクレオチド351〜2084を包含する。
【0045】
本発明に開示したような核酸分子は、2−KGAおよび/またはビタミンCの産生のために組換え微生物において使用できる。Gluconobacter、Acetobacter、Peudomonas、Acinetobacter、Klebsiella、およびEscherichiaから選択される組換え微生物を、好気的条件下で適切な栄養を補充した水性培地中で培養し得る。培養は、4.0〜9.0、好ましくは6.0〜8.0のpHで実施し得る。培養期間は、使用するpH、温度および栄養培地により変更し、好ましくは1〜5日間である。培養を実施するに好ましい温度範囲は、約13℃から約36℃、好ましくは約18℃から約33℃である。培養培地が、同化可能な炭素源としてこのような栄養、例えば、グリセロール、D−マンニトール、D−ソルビトール、エリトリトール、リビトール、キシリトール、アラビトール、イノシトール、ズルシトール、D−リボース、D−フルクトース、D−グルコース、およびスクロース、好ましくはD−ソルビトール、D−マンニトール、およびグリセロール;および、消化可能な窒素源、例えば有機物質、例えばペプトン、酵母抽出液、パン酵母、尿素、アミノ酸、およびコーンスティープリカーを含むことが通常必要である。種々の無機物質、例えば硝酸塩およびアンモニウム塩なども窒素源として使用し得る。さらに、培養培地は、通常、無機塩、例えば硫酸マグネシウム、リン酸カリウム、および炭酸カルシウムを含む。培養は、ジャー発酵器、フラスコ、またはチューブなどの適切な装置中で実施する。組換え微生物は、前記のような核酸分子を含む発現ベクターで形質転換されているか、または、その染色体DNAに組み込まれた本発明の核酸分子を含む。
【0046】
2−KGAおよび/またはビタミンCの産生用の基質として使用する適切な糖化合物は、L−ソルボソンである。2−KGAおよびビタミンCの代謝経路は、D−ソルビトールからL−ソルボースを介してL−ソルボソンへと進行し、これはその後、2−KGAおよび/またはビタミンCに変換される。従って、両方の産物の直接的な基質は、L−ソルボソンである。
【0047】
従って、本発明の1つの態様は、(a)本発明で形質転換されているか、または、その染色体DNAに組み込まれた本発明の核酸分子を含む組換え微生物を、適切な培養培地中で増殖または培養し、(b)前記の培養培地から2−KGAおよび/またはビタミンCを回収および分離することを含む、L−ソルボソンから2−KGAおよび/またはビタミンCを産生するプロセスを提供することである。
【0048】
1つの実施形態は、(a)適切な培養培地中で組換え微生物を繁殖し、ここで本発明の核酸分子は前記微生物に異種的に導入されており、(b)前記培養培地からビタミンCおよび/または2−KGAを回収および分離することを含む、L−ソルボソンからビタミンCおよび/または2−KGAを産生するプロセスを提供することである。
【0049】
本発明は、組換えSNDHを提供する。本発明により提供されるSNDH遺伝子を、G.oxydansDSM4025を含む宿主細胞に導入することにより、SNDH酵素の産生収率を向上することができる。また、本発明のSNDH遺伝子を使用することにより、Gluconobacter、Acetobacter、Pseudomonas、Acinetobacter、Klebsiella、およびEsherichiaからなる群より選択される宿主細胞において、SNDHタンパク質をより効率的に産生することができる。微生物は前記したように培養し得る。
【0050】
培養後に微生物から組換えSNDHを単離および精製するための実施形態は、本明細書で以後に簡潔に記載している:細胞を、遠心分離またはろ過により液体培養ブロスから収集する。収集した細胞を、水、生理的食塩水、または適切なpHを有する緩衝溶液で洗浄する。洗浄した細胞を、緩衝溶液に懸濁し、ホモジナイザー、ソニケーター、もしくはフレンチプレスを用いて、または、リゾチームでの処理により破壊して、破壊細胞の溶液を得る。組換えSNDHを、破壊細胞の細胞非含有抽出液から、好ましくは微生物のサイトゾル画分から単離および精製する。組換えSNDHは、固相酵素反応のために固相担体上に固定できる。
【0051】
本発明はさらに、(a)適切な培養培地中でGluconobacter oxydansDSN4025に属する微生物を培養し、ここで前記微生物中の配列番号2により示されるアルデヒドデヒドロゲナーゼをコードする遺伝子は破壊されており、(b)前記培養培地から2−KGAを回収および分離することを含む、L−ソルボソンから2−KGAを産生するプロセスに関する。破壊は、遺伝子中のどの場所でも起こり得、その結果、コードされている酵素は機能しない。
【0052】
従って、(a)適切な培養培地中でGluconobacter oxydansDMS4025に属する微生物を繁殖することを含む、適切な糖化合物からL−ソルボソンを介して2−KGAを産生するプロセスを提供し、ここで前記微生物中のアルデヒドデヒドロゲナーゼをコードする遺伝子は破壊されており、前記アルデヒドデヒドロゲナーゼは、(i)配列番号1のヌクレオチド配列に少なくとも95%同一であるポリヌクレオチド;(ii)(a)配列番号1のヌクレオチド258〜2084、(b)配列番号1のヌクレオチド351〜2084、(c)配列番号1のヌクレオチド258〜1955、および(d)配列番号1のヌクレオチド351〜1955からなる群より選択されるポリヌクレオチドに少なくとも95%同一であるポリヌクレオチド;および(iii)(a)配列番号2のアミノ酸配列を有するポリペプチドをコードするポリヌクレオチド、(b)配列番号2のアミノ酸32〜609からなるポリペプチドをコードするポリヌクレオチド、(c)配列番号2のアミノ酸1〜566からなるポリペプチドをコードするポリヌクレオチド、および(d)配列番号2のアミノ酸32〜566からなるポリペプチドをコードするポリヌクレオチドからなる群より選択されたポリヌクレオチドによりコードされている。得られた2−KGAは、前記培養培地からさらに回収され単離される。
【0053】
1つの実施形態において、本発明は、インビボならびにインビトロにおける、UV照射、または任意の変異試薬、例えばN−メチル−N’−ニトロ−N−ニトロソグアニジン(NTG)、ICR170、またはアクリジンオレンジなどによる化学的処理などの要因による古典的な変異誘発による、SNDH遺伝子の破壊のためのプロセスを提供する。
【0054】
別の実施形態において、本発明は、インビボならびにインビトロにおける、トランスポゾン挿入またはPCRによる部位特異的変異誘発などのDNA組換え技術により、SNDH遺伝子を破壊するプロセスを提供する。
【0055】
別の実施形態において、本発明は、L−ソルボソン、D−グルコース、D−ソルビトール、およびL−ソルボースからなる群より選択される適切な基質、すなわち糖化合物からの発酵により、前記したような破壊物を使用して2−KGAを産生するプロセスを提供する。プロセスは、ジャー発酵器、フラスコ、またはチューブなどの適切な装置中で行なう。さらに、本発明は、バイオリアクターなどの適切な装置中で、適切な基質、例えばL−ソルボソン、D−グルコース、L−ソルボース、およびD−ソルビトールから、インキュベートにより、前記した破壊物の細胞非含有抽出液を使用して、2−KGAを産生するプロセスを提供する。
【0056】
本発明は、組換えSNDHを提供する。さらに、(a)(i)配列番号1のヌクレオチド配列に少なくとも95%同一であるポリヌクレオチド;(ii)(a)配列番号1のヌクレオチド258〜2084、(b)配列番号1のヌクレオチド351〜2084、(c)配列番号1のヌクレオチド258〜1955、および(d)配列番号1のヌクレオチド351〜1955からなる群より選択されるポリヌクレオチドに少なくとも95%同一であるポリヌクレオチド;および(iii)(a)配列番号2のアミノ酸配列を有するポリペプチドをコードするポリヌクレオチド、(b)配列番号2のアミノ酸32〜609からなるポリペプチドをコードするポリヌクレオチド、(c)配列番号2のアミノ酸1〜566からなるポリペプチドをコードするポリヌクレオチド、および(d)配列番号2のアミノ酸32〜566からなるポリペプチドをコードするポリヌクレオチドからなる群より選択されるポリヌクレオチドを含む、アルデヒドデヒドロゲナーゼをコードする核酸分子を含む組換え微生物を培養し;ここで前記微生物は適切な培養培地中で培養し、(b)前記アルデヒドデヒドロゲナーゼを前記培養培地から回収および分離することを含む、アルデヒドデヒドロゲナーゼの産生プロセスに関する。
【0057】
実施例1:SNDHのN末端からのアミノ酸シークエンス
SNDHタンパク質のN末端75kDaサブユニットの部分アミノ酸配列を決定した。75kDaサブユニットからなる約10μgのSDS処理精製SNDHを、SDS−PAGEにかけ、タンパク質バンドをPVDFメンブランにエレクトロブロットした。メンブラン上にブロットされたタンパク質を、消化緩衝液(100mMリン酸カリウム緩衝液、5mMジチオトレイトール、10mM EDTA、pH8.0)に浸漬し、30℃で24時間、5.04μgのピログルタメートアミノペプチダーゼ(シグマ、米国)と共にインキュベートした。インキュベート後、メンブランを脱イオン水で洗浄し、自動アミノ酸シークエンサー(ABIモデル490、パーキンエルマー社、コネチカット州、米国)を使用したN末端アミノ酸シークエンスにかけた。結果として、配列番号3で示したようなN末端アミノ酸配列の14残基が得られた。
【0058】
実施例2:PCRによる部分SNDH遺伝子のクローニング
部分SNDH遺伝子断片の増幅を、G.oxydansDSM4025の染色体DNA(FERM BP−3812)ならびに縮重オリゴヌクレオチドDNAプライマーのP11(配列番号6)およびP12(配列番号7)を用いてPCRにより実施した。両方のプライマーが、Gluconobacterコドン使用に偏りを有する縮重DNA混合物であった。PCRは、熱安定tapポリメラーゼ(TAKARA ExTap(商標)、宝酒造株式会社、日本520−2193滋賀県大津瀬田3−4−1)を用いて、サーマルサイクラー(GeneAmpPCRシステム2400−R、PEバイオシステムズ、850リンカーン・センター・ドライブ、フォスターシティ、CA94404、米国)を使用して実施した。反応混合物(25μl)は、200μMのdNTP、50pmolの各プライマー(24〜48回縮重)、5ngの染色体DNA、および業者から提供された緩衝液中の1.25単位のDNAポリメラーゼから構成されていた。反応は、94℃で30秒間の変性工程、37℃で30秒間のアニーリング工程、70℃で1分間の合成工程を5サイクル、その後、94℃で30秒間の変性工程、50℃で30秒間のアニーリング工程、70℃で1分間の合成工程を25サイクルで実施した。結果として、41bpのDNA断片が特異的に増幅され、ベクターpCR2.1−TOPO(Invitrogen、1600ファラデイ・アベニュー・カールスバッド、カリフォルニア92008、米国)にクローニングし、組換えプラスミドpMTSN2を得た。成熟SNDHタンパク質のN末端部分アミノ酸配列をコードするクローニングされた41bpのDNA断片のヌクレオチド配列は、ジデオキシ鎖終結法(F. Sangerら、Proc. Natl .Acad. Sci. USA、74、5463〜5467、1977)により確認した。
【0059】
実施例3:SNDH遺伝子の完全クローニング
(1)G.oxydansDSM4025の遺伝子ライブラリーの作製
G.oxydansDMS4025の染色体DNAを、4日間27℃で、5%D−マンニトール、1.75%コーンスティープリカー、5%パン酵母、0.25%MgSO4・7H2O、0.5%CaCO3(実践用等級)、0.5%尿素、および2.0%寒天(pH7.0)を含むM寒天培地上で増殖させた細胞から調製した。染色体DNA(4μg)を、20μlの反応混合物中の4単位のEcoRIで部分的に消化した。部分的に消化したDNA断片を含む試料の一部(8μl)を、1%アガロースゲルを使用して電気泳動により分離した。15〜35kbの範囲の断片を切り出し、QIAEXII(QIAGEN社、28159アベニュー・スタンフォード、バレンシア、CA91355、米国)を使用して化学的に融かして回収した。回収した目的のDNA断片を、H2O中に懸濁した。一方、2μgのコスミドベクターpVK100を、EcoRIで完全に消化し、細菌アルカリホスファターゼ(E.coliC75)(宝酒造)での処理により5’末端を脱リン酸化した。処理したpVK100(220ng)を、36μlの反応混合物中において、ライゲーションキット(宝酒造)を使用して、15〜35kbのEcoRI断片(1μg)とライゲートした。エタノール沈降し適切な量のTE緩衝液(10mMトリス−HClpH8.0、1mM EDTA)に溶かしておいたライゲートしたDNAを、インビトロでのパッケージングに使用し(GigapackIII Gold Packaging Extract、Stratagene、11011ノース・トーリー・パイン・ロード、ラ・ホーヤ、CA92037、米国)、ゲノムライブラリーの宿主株であるE.coliVCS257に感染させた。結果として、約25kbの挿入DNA断片を含む合計400,000〜670,000個のクローンを得た。
【0060】
(2)コロニーハイブリダイゼーションによるSNDH遺伝子の完全クローニング
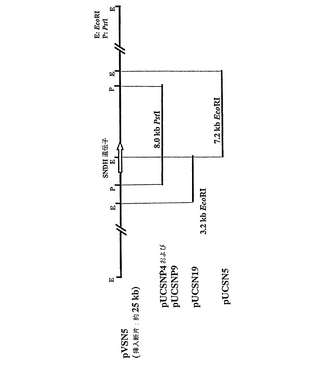
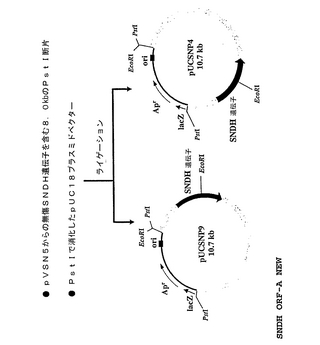
コロニーハイブリダイゼーション法により完全SNDH遺伝子を有するクローンを検出するための、前記したコスミドライブラリーのスクリーニングに使用するプローブを作製した。SNDHのN末端アミノ酸配列をコードする41bpDNA断片を増幅し、PCR−DIGラベリング法(Roche Molecular Systems社、1145アトランティック・アベニュー、アラバマ、CA94501、米国)により標識した。鋳型としてのプラスミドpMTSN2DNAおよびオリゴヌクレオチドDNAプライマーP13(配列番号8)およびP14(配列番号9)を用いてのPCRを、熱安定taqポリメラーゼ(TAKARA Ex Taq(商標)、宝酒造株式会社)を用いて、サーマルサイクラー(Gene Amp PCR System2400−R、PEバイオシステムズ)を使用して実施した。反応は、94℃で30秒間の変性工程、55℃で30秒間のアニーリング工程、70℃で1分間の合成工程を25サイクルで実施した。DIG標識プローブを使用して、コロニーハイブリダイゼーションによるコスミドライブラリーのスクリーニング(約1000クローン)および業者(Roche Molecular Systems Inc.、米国)により提供された方法に従っての化学ルミネセンス検出を実施した。結果として、3つの陽性クローンが単離され、その中の1つをpVSN5と称し、これはpVK100ベクター中に約25kbの挿入断片DNAを有していた。これから、異なるサイズの25kbDNA挿入断片をさらに、pUC18ベクター(図2)にサブクローニングした:(1)SNDH遺伝子の上流部分(N末端部分)を含む3.2kbのEcoRI断片によりpUCSN19が得られ、(2)SNDH遺伝子の下流部分(C末端部分)を含む7.2kbEcoRI断片によりpUCSN5が得られ、(3)無傷または完全SNDH遺伝子を含む1.8kbPstI断片によりそれぞれpUCSNP4およびpUCSNP9が得られた。pUSNP4およびpUSNP9における挿入断片は同じであるが逆の方向であることを注記する。
【0061】
(3)SNDH遺伝子のヌクレオチドシークエンス
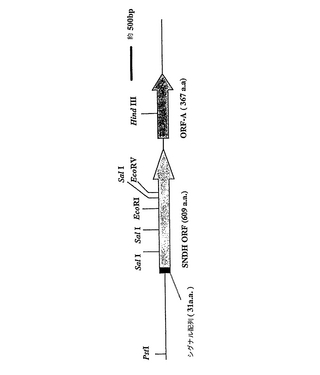
プラスミドpUCSN19、pUCSN5、およびpUCSNP4を、SNDH遺伝子または遺伝子断片を含む領域のヌクレオチドシークエンスに使用した。決定されたヌクレオチド配列(3,408bpを有する配列番号1)により、SNDH遺伝子のORF(1,827bp;配列番号1のヌクレオチド258〜2084)は、609アミノ酸残基(配列番号2)のポリペプチドをコードしていることが判明した。他のOPFであるORF−Aは、図1に示したように、SNDH ORFの下流に見られた。ORF−A(1,101bp;配列番号1のヌクレオチド2214〜3314)は367アミノ酸のポリペプチドをコードしていた。
【0062】
SNDH遺伝子のORFでは、シグナルペプチド様の配列(31アミノ酸を有する配列番号4)が、(i)多くの疎水性残基、(ii)N末端に近い正に荷電した残基、および(iii)シグナル配列の切断部位のためのAla−Xaa−Alaモチーフを含む、推定アミノ酸配列におそらく含まれている。SNDH遺伝子の推定リボソーム結合部位(シャインダルガーノ、SD、配列)は、開始コドンの6bp上流に位置していた(配列番号1のヌクレオチド位置247〜252のAGGAGA)。さらに、ヘムc結合部位として定義されたモチーフ(Cys−Xaa−Xaa−Cys−His)は、配列番号2の位置530〜534に見出された。前記に示したような遺伝子解析から、SNDHタンパク質はキノヘモタンパク質の1つであると考えられた。
【0063】
GCG(Genetics Computer Group、マディソン、WI、米国)のFASTAプログラムを使用したSNDH遺伝子の相同性探索により、配列番号5のArg227、Asn228、Gln230、Gly246、およびAsp251が、Oubrieら[J.Mol.Biol.289: 319〜333(1999)]により記載のA.calcoaceticusGDH−Bタンパク質の推定活性部位におけるいくつかの高度に保存された残基に対応することが判明した。
【0064】
実施例4:E.coliにおけるSNDH遺伝子の発現
無傷の、すなわち完全なSNDH遺伝子を有する8.0kbのPstI断片を含むプラスミドpUCSNP4およびpUCSNP9(図3)を、E.coliJM109に形質転換し、SNDHタンパク質の発現および活性を確認した。酵素活性として産生されるビタミンCの量を、UV検出器(TOSOH UV8000;Tosoh、日本)、デュアルポンプ(TOSOH CCPE;Tosoh)、積分器(島津C−R6A;島津、日本)およびカラム(YMC−Packポリアミン−II;内径[i.d.]4.6mm×15cm、YMC、米国)から構成される高速液体クロマトグラフィーシステム(HPLC)により264nmの波長で測定した。
【0065】
組換えE.coliのサイトゾル画分を使用することによるL−ソルボソンからビタミンCへの変換活性を試験した(表1)。細胞を、場合により10μlのPQQおよび1.0mMのCaCl2を補充したLB培地中で培養した。サイトゾル画分を、50mMリン酸カリウム緩衝液(pH7.0)中での細胞非含有抽出液の超遠心分離(100,000×g)45分間)により調製した。反応混合物(100μl)は、125μgの組換えE.coliのサイトゾル画分、50mMのL−ソルボソン、1.0mMのフェナジンメソスルフェート(PMS)、場合に応じて、補因子として1.0μMのPQQおよび1.0mMのCaCl2を添加したまたは添加しないものから構成された。酵素反応は、30℃で30分間実施した。10μMのPQQおよび1.0mMのCaCl2を含むLB培地中で培養した細胞のホロ−SNDHにより、PQQおよびCaCl2の補因子を添加しない規定の反応条件下で、明確にビタミンCが産生された。補因子の添加により、pUCSNP4およびpUCSNP9で発現されたアポ−SNDHは、ホロ酵素の活性とほぼ同じ活性を示した。
【0066】
【表1】

【0067】
実施例5:G.oxydansのSNDH遺伝子破壊株の作製および培養
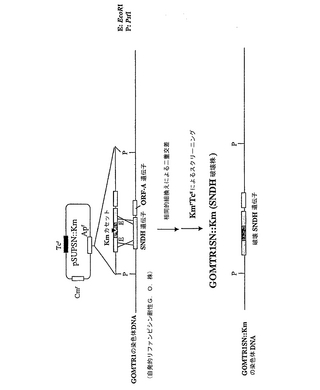
図4は、SNDH遺伝子標的化ベクター、GOMTR1SN::Km(SNDH破壊物)の作製のスキームを示す。最初に、プラスミドpSUPSNを、プラスミドpUCSNP4からのSNDH遺伝子を含む8.0kbのPstI断片を、自殺ベクターpSUP202とライゲートすることにより作製した(参照については、Simonet et., Abroad host range mobilization system for in vitro genetic engineering: transposon mutagenesis in Gram negative bacteria、Biotechnology、1、784〜791、1983を参照のこと)。第二に、カナマイシン耐性遺伝子カセット(Kmカセット)を、プラスミドpSUPSNにクローニングしたSNDH遺伝子のEcoRI部位に挿入して、プラスミドpSUPSN::Km(KmrTcr)を得た。その後、プラスミドpSUPSN::Kmを、リファンピシン(Rif)耐性で野生型G.oxydansDSM4025株から自発的に派生したGOMTR1に導入し、SNDH−ヌル変異体(KmrRifrTcs)を得た。
【0068】
GOMTR1は、30g/lのトリプチカーゼソイブロス(BBL;Becton Dickinson and Company、コッキーズビル、MD21030、米国)および3g/lの酵母抽出液(ディフコ;Becton Dickinson Microbiology Systems、Becton Dickinson and Company、スパークス、MD21152、米国)と、100μg/mlのリファンピシンからなる50mlのTブロスを含む200mlのフラスコ中で30℃で一晩培養した。E.coliHB101(pRK2013)[D.H.Figurski、Proc.Natl.Acad.Sci.USA、76、1648〜1652、1979]およびE.coliJM109(pSUPSN::Km)を、50μg/mlのカナマイシンを含む2mlのLB培地を含む試験管中で30℃で一晩培養した。GOMTR1、E.coliHB101(pRK2013)、およびE.coliJM109(pSUPSN::Km)の培養細胞を、遠心分離により別々に回収し、LB培地中の各細胞懸濁液を、それぞれ10:1:1の比で混合した。その後、これらの細胞懸濁液を、同じ容量で混合し、E.coliドナーからの自殺プラスミドの、レシピエントであるGOMTR1への接合導入のために、5.0%マンニトール、0.25%MgSO4・7H2O、1.75%コーンスティープリカー、5.0%パン酵母、0.5%尿素、0.5%CaCO3、および2.0%寒天から構成される寒天培地上にのせた0.45μmニトロセルロースメンブラン(PROTRAN、Schleicher & SchuellGmbH、Postfach4、D-37582 Dassel、ドイツ)に混合物を蒔いた。27℃で1日間培養した後、トランスコンジュガント(transconjugant)を含む細胞を懸濁し、Tブロスで適切に希釈し、100μg/mlのリファンピシンおよび50μg/mlのカナマイシンを含むスクリーニング寒天プレート上に蒔いた。最後に、Kmカセットにより破壊されたSNDH遺伝子を有するいくつかのトランスコンジュガント(KmrRifTcs)を得た。
【0069】
GOMTR1および破壊株GOMTR1SN::Kmを、8.0%のL−ソルボース、0.25%MgSO4・7H2O、1.75%コーンスティープリカー、5.0%パン酵母、0.5%尿素、0.5%CaCO3、および2.0%寒天を含む寒天プレート上で27℃で4日間増殖させた。白金棒でひとかきした細胞を、500mlエーレンマイヤーフラスコ中の、4%のD−ソルビトール、0.4%酵母抽出液、0.05%グリセロール、0.25%MgSO4・7H2O、1.75%コーンスティープリカー、0.1%尿素、および1.5%CaCO3を含む50mlの種培養培地(pH6.0)に接種し、30℃で回転振とう機で180rpmで1日間培養した。このようにして調製した種培養液を、500mlエーレンマイヤーフラスコ中の、12.0%L−ソルボース、2.0%尿素、0.05%グリセロール、0.25%MgSO4・7H2O、3.0%コーンスティープリカー、0.4%酵母抽出液、および1.5%CaCO3からなる50mlの主培養培地への接種に使用した。培養は30℃で180rpmで4日間行なった。活性アッセイは実施例4に記載のように実施した。酵素活性として産生される2−KGAの量は、UV検出器(TOSOH UV8000;Tosoh)、デュアルポンプ(TOSOH CCPE;Tosoh)、積分器(島津C−R6A;島津)およびカラム(YMC−PackProC18、YMC)で構成されるHPLCシステムにより340nmの波長で測定した。表2に示したように、SNDH遺伝子破壊株の2−KGA産生効率は、親株のGOMTR1の効率より高かった。1モルのL−ソルボースあたりの2−KGAへの変換率の差は、約3%であった。
【0070】
【表2】
【0071】
実施例6:G.oxydansDMS4025のSNDH遺伝子破壊株への、SNDH遺伝子を有するプラスミドの導入
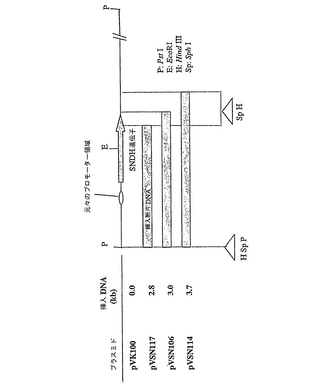
広宿主域ベクターpVK100を使用した数種類のSNDH発現プラスミドを、図5に示したように作製した。このようなプラスミドは、以下のように記載したpVK100のHindIII部位において、異なる挿入断片DNAを有していた:pVSN117は、配列番号5のGly535(配列番号2のアミノ酸残基566)で終結するポリペプチドをコードする不完全なSNDH遺伝子、すなわち、僅か55kDaのタンパク質を発現するC末端欠失SNDH遺伝子を含む、挿入断片DNAを有する。プラスミドpVSN106およびpVSN114は、それぞれ、完全なSNDH遺伝子を含む挿入断片DNAを有していた。このようなプラスミドは、接合導入法によりGOMTR1SN::Km株に導入した。
【0072】
図5に示したプラスミドを有するトランスコンジュガントを、10.0%のL−ソルボース、0.25%MgSO4・7H2O、1.75%コーンスティープリカー、5.0%パン酵母、0.5%尿素、0.5%CaCO3、および2.0%寒天を含む寒天プレート上で27℃で4日間増殖させた。酵素反応混合物は、組換えGluconobacter株の細胞非含有抽出液80μg、25mMリン酸カリウム緩衝液(pH7.0)、50mMのL−ソルボソン、および0.05mMのPMSから構成された。酵素反応は、1,000rpmで振とうしながら、30℃で30分間実施した。活性アッセイは、実施例4に従って実施した。結果を表3に示した。
【0073】
【表3】
【図面の簡単な説明】
【0074】
【図1】本発明のアルデヒドデヒドロゲナーゼ活性を有するタンパク質をコードする遺伝子を示した図である。SNDH遺伝子およびORF−A遺伝子の制限地図が示されており、ここでORFは、オープンリーディングフレームを意味し、シグナル配列は、SNDH遺伝子の推定シグナルペプチド配列を意味する。
【図2】コスミドpVSN5にクローニングしたSNDHおよびORF−A遺伝子の制限地図、ならびに、pUCプラスミド、pUCSNP4、pUCSNP9、pUCSN19、およびpUCSN5への異なるサイズの挿入断片DNAのクローニングを示した図である。pVSN5の物理的地図において、灰色の矢印は、SNDH遺伝子を示す。
【図3】pVSN5からの無傷SNDH遺伝子を含む8.0kbのPstI断片を、pUC18ベクターにクローニングする戦略を示し、その結果、pUCSNP4およびpUCSNP9が得られる。pUCSNP4およびpUCSNP9は、挿入断片の方向を除いては同一であることを注記する。
【図4】カナマイシンカセット(Km)を用いて破壊したSNDH遺伝子を有する自殺ベクタープラスミドを使用することによる、GOMTR1SN::Km(SNDH破壊株)の作製を示した図である。対応する領域における、ベクタープラスミドと、親株であるGOMTR1の染色体DNAとの間の相同的組換えにより、破壊株が得られる。「G.O.」は、Gluconobacter oxydansを意味する。
【図5】pVSN117、pVSN106、およびpVSN114の挿入断片DNAの物理的地図を示した図である。プラスミドpVSN117は、C末端欠失SNDH遺伝子をコードする挿入断片DNAを有し(配列番号1のヌクレオチド258〜1955;すなわち、配列番号2のアミノ酸1〜566)、これは僅か55kDaのタンパク質を発現する。プラスミドpVSN106およびpVSN114は、無傷SNDH遺伝子をコードする挿入断片DNAを有する。
【技術分野】
【0001】
本発明は、Gluconobacter oxydansDSM4025から得られるアルデヒドデヒドロゲナーゼ(SNDH)をコードする新規DNA、前記DNAを含む発現ベクター、および前記発現ベクターを含む組換え微生物に関する。さらに、本発明は、組換えアルデヒドデヒドロゲナーゼタンパク質を産生するプロセス、および、組換えアルデヒドデヒドロゲナーゼタンパク質または前記発現ベクターを含む組換え微生物を使用することにより、L−ソルボソンからL−アスコルビン酸(ビタミンC)および/または2−ケト−L−グロン酸(2−KGA)を産生するプロセスに関する。
【0002】
ビタミンCはヒトにとって不可欠な栄養因子の1つであり、約60年間の間、ライヒシュタインプロセスにより商業的に合成されてきた。家畜動物はビタミンCを自身の体内で合成できるとしても、合成ビタミンCはまた動物食餌中にも使用されている。ライヒシュタインプロセスは工業的ビタミンC産生において多くの有益な点を有するが、高いエネルギー消費量ならびにかなりの量の有機溶媒および無機溶媒の使用などの望ましくない問題を有する。それ故、過去数十年間かけて、酵素変換を使用してビタミンCを製造するアプローチが数多く研究されており、このアプローチはより経済的かつ環境に優しいであろう。
【0003】
本発明は、配列番号1のヌクレオチド配列に少なくとも95%同一であるポリヌクレオチドを含むアルデヒドデヒドロゲナーゼをコードする単離核酸分子に関する。
【0004】
本明細書に使用したような「SNDH」はアルデヒドデヒドロゲナーゼを意味する。
【0005】
本明細書に使用したような「核酸分子」は、DNAおよびRNAの両方を含み、特記しない限り、二本鎖核酸、一本鎖核酸、およびそのヌクレオシドを含む。DNA−RNAハイブリッド、DNA−RNA−タンパク質ハイブリッド、RNA−タンパク質ハイブリッド、およびDNA−タンパク質ハイブリッドなどのハイブリッドも含まれる。
【0006】
本明細書に使用したような「変異」は、対象のヌクレオチド配列の1塩基対の変化、挿入、または欠失を意味する。
【0007】
本明細書に使用したような「突然変異生成」は、変異がDNAに生じるプロセスを意味する。「無作為」突然変異生成では、変異の正確な部位は予測不可能であり、微生物の染色体の何処でも起こり、突然変異は、照射または化学的処理などの要因により引き起こされる物理的傷害の結果としてもたらされる。
【0008】
本明細書に使用したような「プロモーター」は、開始コドンの近くに位置する、遺伝子の5’領域として一般に記載されるDNA配列を意味する。隣接遺伝子(群)の転写は、プロモーター領域で開始する。プロモーターが誘導性プロモーターである場合、転写速度は、誘導剤に応答して増加する。これに対し、プロモーターが構成性プロモーターである場合、転写速度は、誘導剤により調節されない。
【0009】
本明細書に使用したような「同一率」は、以下に例示したような配列解析プログラムにより、比較するヌクレオチドまたはアミノ酸配列における同一ヌクレオチドまたはアミノ酸と一致した、対象ヌクレオチドまたはアミノ酸配列のヌクレオチドまたはアミノ酸の比率を意味する。
【0010】
本発明は、(a)配列番号1のヌクレオチド258〜2084、(b)配列番号1のヌクレオチド351〜2084、(c)配列番号1のヌクレオチド258〜1955、および(d)配列番号1のヌクレオチド351〜1955からなる群より選択されるポリヌクレオチドに少なくとも95%同一であるポリヌクレオチドを含むアルデヒドデヒドロゲナーゼをコードする単離核酸分子を含む。
【0011】
本発明の別の態様は、(a)配列番号2のアミノ酸配列を有するポリペプチドをコードするポリヌクレオチド、(b)配列番号2のアミノ酸32〜609からなるポリペプチドをコードするポリヌクレオチド、(c)配列番号2のアミノ酸1〜566からなるポリペプチドをコードするポリヌクレオチド、および(d)配列番号2のアミノ酸32〜566からなるポリペプチドをコードするポリヌクレオチドからなる群より選択されるポリヌクレオチドを含むアルデヒドデヒドロゲナーゼをコードする単離核酸分子を提供することである。SNDH活性を有し、前記したアミノ酸配列中の1つ以上のアミノ酸(群)の置換、欠失、挿入、または付加により前記したタンパク質から得られるタンパク質も含む。
【0012】
本発明の別の態様としての機能的誘導体は、本発明のアミノ酸配列に基づいて、かかる配列の1つ以上のアミノ酸残基の付加、挿入、欠失、および/または置換により定義され、このような誘導体は、当分野で既知のまたは本明細書で明記したアッセイにより測定したSNDH活性を依然として有する。このような機能的誘導体は、当分野で既知の方法により本明細書に開示したようなDNA配列に基づいて、当分野で既知の化学的ペプチド合成により、または、組換え技術により作製できる。このような分子の活性を一般に変化させないタンパク質およびペプチド中のアミノ酸の交換は、当分野で既知である。
【0013】
本発明の特定の実施形態において、対象の保存的置換は以下のように起こる:例として以下の置換、AlaからVal/Leu/Ile、ArgからLys/Gln/Asn、AsnからGln/His/Lys/Arg、AspからGlu、CysからSer、GlnからAsn、GluからAsp、GlyからPro/Ala、HisからAsn/Gln/Lys/Arg、IleからLeu/Val/Met/Ala/Phe/norLeu、LysからArg/Gln/Asn、MetからLeu/Phe/Ile、PheからLeu/Val/Ile/Ala/Tyr、ProからAla、SerからThr、ThrからSer、TrpからTyr/Phe、TyrからTrp/Phe/Thr/Ser、およびValからIle/Leu/Met/Phe/Ala/norLeuが妥当である。好ましい例として、AlaからVal、ArgからLys、AsnからGln、AspからGlu、CysからSer、GlnからAsn、GluからAsp、GlyからAla、HisからArg、IleからLeu、LeuからIle、LysからArg、MetからLeu、PheからLeu、ProからAla、SerからThr、ThrからSer、TrpからTyr、TyrからPhe、およびValからLeuが妥当である。このような置換により生物活性が変化した場合、より実質的な変化、前記した表示した例示的置換を導入し、産物をスクリーニングする。
【0014】
さらに、本発明は、配列番号2として配列表に開示したようなSNDH活性を有するポリペプチドをコードするポリヌクレオチドおよびその相補鎖、または、これらの配列、そのDNA配列またはその断片を含むもの、および、このような配列と標準的な条件下でハイブリダイズするが、正に同じアミノ酸配列を有するポリペプチドをコードするDNA配列に関する。
【0015】
従って、本発明は、アルデヒドデヒドロゲナーゼ活性を有するポリペプチドをコードする単離核酸分子を提供し、前記核酸分子の相補体は、前記したような核酸分子と標準的な条件下でハイブリダイズする。本発明の1つの態様は、アルデヒドデヒドロゲナーゼ活性を有するポリペプチドをコードする単離核酸分子を提供することであり、前記核酸分子は、(i)配列番号1のヌクレオチド配列に少なくとも95%同一であるポリヌクレオチドを含むアルデヒドデヒドロゲナーゼ;(ii)(a)配列番号1のヌクレオチド258〜2084、(b)配列番号1のヌクレオチド351〜2084、(c)配列番号1のヌクレオチド258〜1955、および(d)配列番号1のヌクレオチド351〜1955からなる群より選択されるポリヌクレオチドに少なくとも95%同一であるポリヌクレオチドを含むアルデヒドデヒドロゲナーゼ;および(iii)(a)配列番号2のアミノ酸配列を有するポリペプチドをコードするポリヌクレオチド、(b)配列番号2のアミノ酸32〜609からなるポリペプチドをコードするポリヌクレオチド、(c)配列番号2のアミノ酸1〜566からなるポリペプチドをコードするポリヌクレオチド、および(d)配列番号2のアミノ酸32〜566からなるポリペプチドをコードするポリヌクレオチドからなる群より選択されるポリヌクレオチドを含むアルデヒドデヒドロゲナーゼをコードする核酸分子の相補鎖と標準的な条件下でハイブリダイズする。
【0016】
ハイブリダイゼーションの「標準的条件」は、この脈絡では、特異的ハイブリダイゼーションシグナルを検出するために当業者が一般に使用する条件、または好ましくはいわゆる当業者が使用するストリンジェントなハイブリダイゼーション条件を意味する。
【0017】
従って、本明細書に使用したような「ストリンジェントなハイブリダイゼーション条件」は、配列間に95%、好ましくは少なくとも97%の同一性が存在した場合にハイブリダイゼーションが起こることを意味する。ストリンジェントなハイブリダイゼーション条件は、例えば、50%ホルムアミド、5×SSC(150mM NaCl、15mMクエン酸三ナトリウム)、0.2%ドデシル硫酸ナトリウム、0.1%N−ラウロイルサルコシン、および2%遮断剤(Roche Diagnostics GmbH)を含む溶液中で、ジゴキシゲニン(DIG)標識DNAプローブ(DIGラベリングシステムを使用することにより作製;Roche Diagnostics GmbH、68298 Mannheim、ドイツ)を使用して42℃で一晩インキュベートし、その後、約60℃で0.1×SSC中でフィルターを洗浄する条件である。
【0018】
本発明はまた、前記したような核酸分子を含む組換えベクター、すなわち発現ベクターに関する。本発明の発現ベクターは、適切な宿主細胞で機能するものである。本発明の核酸分子の発現に好ましいベクターは、pQE、pUC、pBluescriptII、pACYC177、pACYC184、pVK100、およびRSF1010からなる群より選択されるベクターまたはその誘導体である。
【0019】
本発明のヌクレオチド配列の発現に適切な宿主細胞は、細菌、酵母、および植物細胞からなる群より選択される組換え微生物である。好ましくは、微生物は、Gluconobacter、Acetobacter、Pseudomonas、Acinetobacter、Klebsiella、およびEscherichiaからなる群より選択される。このような好ましい微生物の例はE.coliである。より好ましい宿主細胞は、Gluconobacter oxydans、最も好ましくはG.oxydansDSM4025(FERM BP−3812)に属し、これは1987年3月17日、ブダペスト条約の条件下でDeutsche Sammlung von Mikroorganismen und Zellkulturen GmbH、Braunschweig、ドイツに寄託された。
【0020】
微生物「Gluconobacter oxydans」はまた、国際原核生物命名規約により定義されたような、同じ物理化学的特性を有するこのような種の同義語またはバソニム(basonyms)を含む。
【0021】
従って、本発明は、前記したような発現ベクターで形質転換されるか、または、その染色体DNAに組み込まれた前記したような核酸分子を含む、組換え微生物に関する。
【0022】
多種多様な宿主/ベクターの組合せを、本発明の二本鎖ヌクレオチド配列のクローニングに使用し得る。E.coliが好ましい宿主細胞であるので、E.coliに通常使用される任意のベクターが本発明に有用である。このようなベクターには、Hisタグ化組換えタンパク質を発現できるpQEベクター(QIAGEN K.K.、東京、日本)、pUC18およびpBluescriptII(Stratagene Cloning Systems、カリフォルニア、米国)を含むpBR322またはその誘導体、pACYC177およびpACYC184およびその誘導体、および、RK2およびRSF1010などの広宿主域プラスミドから得られるベクターが含まれるがこれに限定されない。従って、本発明に使用する発現ベクターは、pQE−プラスミド、pUC−プラスミド、pBluescriptII、pACYC177、pACYC184、およびその誘導体プラスミド、および、pVK100およびRSF1010などの広宿主域プラスミドから得られる。
【0023】
本明細書に使用したような「発現ベクター」は、適切な宿主への形質転換後に、クローニングベクター中にクローニングした遺伝子の発現を増強できるクローニングベクターを意味する。クローニングされた遺伝子は、プロモーター配列などの特定の制御配列の制御下(すなわちそれに作動可能に連結)に置かれる。プロモーター配列は構成性でも誘導性でもよい。
【0024】
本明細書に使用したような「クローニングベクター」は、宿主細胞中で自律的に複製でき、1つまたは少数の制限エンドヌクレアーゼ認識部位により特徴づけられる、プラスミドもしくはファージDNAまたは他のDNA配列を意味し、ここでの制限エンドヌクレアーゼ認識部位において、ベクターの本質的な生物学的機能が損なわれることなく決まった様式でこのようなDNA配列が切断され得、ここにDNA断片が導入されてその複製およびクローニングがなされ得る。クローニングベクターはさらに、クローニングベクターで形質転換された細胞の同定に使用するに適切なマーカーを含む。このようなマーカーは、例えば、テトラサイクリンまたはアンピシリン耐性を付与する。
【0025】
本明細書に使用したような「組換えベクター」は、望ましいクローニングした遺伝子(群)を含む任意のクローニングベクターまたは発現ベクターを含む。
【0026】
本明細書に使用したような「発現」は、ポリペプチドが構造遺伝子から産生されるプロセスを意味する。このプロセスには、遺伝子からmRNAへの転写、および、このようなmRNAからポリペプチド(群)への翻訳が含まれる。
【0027】
本明細書に使用したような「組換え微生物」は、発現ベクターまたはクローニングベクター上に望ましいクローニング遺伝子(群)を含む任意の原核細胞または真核細胞であり得る組換え宿主を含む。この用語はまた、前記微生物の染色体またはゲノムに望ましい遺伝子(群)を含むように遺伝子操作された原核細胞または真核細胞も含む。
【0028】
本明細書に使用したような「宿主」は、複製可能な発現ベクターまたはクローニングベクターのレシピエントである任意の原核細胞または真核細胞を含む。「宿主」は、この用語が本明細書で使用されているように、その染色体またはゲノム上に望ましい遺伝子(群)を含むように公知の技術により遺伝子操作することのできる原核細胞または真核細胞も含む。かかる宿主の例は当業者には周知である。
【0029】
組換えDNA、例えば組換えベクターを有する組換え微生物を作製するために、形質転換、形質導入、接合、または電気穿孔を含むがこれに限定されない種々の遺伝子導入法を使用できる。これらの方法は分子生物学の分野では公知である。慣用的な形質転換システムを、Gluconobacter、Acetobacter、Pseudomonas、Acinetobacter、Klebsiella、またはEscherichiaに使用できる。形質導入システムはまた、E.coliにも使用できる。接合システムは、E.coli、P.putida、およびGluconobacterを含むグラム陽性およびグラム陰性細菌に広く使用できる。接合の例はWO89/06,688号に開示されている。接合は、液体培地または固体表面上で起こり得る。SNDH産生のための適切なレシピエントの例には、Gluconobacter、Acetobacter、Pseudomonas、Acinetobacter、Klebsiella、またはEscherichiaの微生物が含まれる。接合用のレシピエントに、選択マーカー、例えばナリジクス酸またはリファンピシンに対する耐性を付加し得る。天然淘汰も使用でき、例えば、ポリミキシンBに対する耐性は、多くのGluconobacterに有用である。
【0030】
本発明に有用である好ましいベクターは、広宿主域ベクター、例えばpVK100およびその誘導体およびRSF1010のようなコスミドベクターである。ベクターのコピー数および安定性は、クローニングされた核酸分子の安定かつ効率的な発現のため、および、前記のクローニングした分子を有する宿主細胞の効率的な培養のために注意深く考慮すべきである。Tn5などの転位可能な要素を含む核酸分子を使用して、望ましいDNAを、好ましい宿主に、特に染色体上に導入することもできる。好ましい宿主から単離した任意のDNAを本発明のヌクレオチド配列と共に含む核酸分子も、本発明のヌクレオチド配列を、好ましい宿主細胞、特に染色体上に導入するのに有用である。このような核酸分子は、宿主細胞および核酸分子の性質を考慮しながら、当分野で公知である、例えば、形質転換、形質導入、接合、または電気穿孔などのいずれかの慣用的な方法を適用することにより、好ましい宿主に導入することができる。
【0031】
本発明で提供されるSNDH遺伝子を含むヌクレオチド配列を、当分野で公知の方法を用いて、前記した宿主細胞で作動可能なプロモーター、リボソーム結合部位、および転写ターミネーターなどの調節領域を含む適切なベクターに連結して、適切な発現ベクターを作製する。
【0032】
G.oxydansDSM4025から単離した望ましい遺伝子/ヌクレオチド配列を効率的に発現するために、種々のプロモーター、例えば遺伝子の元々のプロモーター、Tn5のカナマイシン耐性遺伝子、pBR322のアンピシリン耐性遺伝子などの抗生物質耐性遺伝子のプロモーター、およびE.coli(lac)のβ−ガラクトシダーゼ、trp−、tac−、trc−プロモーター、λファージのプロモーター、および、宿主細胞で機能的である任意のプロモーターを使用することができる。この目的では、宿主細胞は、細菌、酵母、および植物細胞からなる群より選択できる。好ましくは、宿主細胞は、Gluconobacter、Acetobacter、Pseudomonas、Acinetobacter、Klebsiella、またはEscherichia属に属する。
【0033】
発現において、宿主細胞(これにコード配列を導入して本発明の組換え細胞を得る)で作動可能であるシャイン・ダルガーノ(SD)配列(例えば、宿主細胞で作動可能である天然および合成配列を含むAGGAGG)および転写ターミネーター(宿主細胞で作動可能である任意の天然および合成配列を含む逆方向反復構造)などの、他の調節要素を、前記のプロモーターと共に使用できる。
【0034】
本発明のSNDHタンパク質のようにペリプラスム空間に位置するポリペプチドの発現では、通常15〜50アミノ酸残基を含み全体的に疎水性であるシグナルペプチドが好ましくは会合している。シグナルペプチドをコードするDNAは、望ましい宿主細胞で作動可能である任意の天然および合成配列から選択できる。配列番号2のアミノ酸残基1〜31を含む推定シグナルペプチドはまた、本発明のSNDH遺伝子により発現されるタンパク質にも見出された(配列番号4)。
【0035】
特記しない限り、本明細書の精製SNDHタンパク質をシークエンスすることにより決定された全てのアミノ酸配列は、自動アミノ酸シークエンサー(例えばモデル470A、パーキンエルマー・アプライド・バイオシステムズ)を使用して決定した。
【0036】
特記しない限り、本明細書のDNA分子をシークエンスすることにより決定された全てのヌクレオチド配列は、自動DNAシークエンサー(例えばモデルALFエクスプレスII、アマシャム・ファルマシア・バイオテック)を使用して決定し、本明細書で決定したDNA分子によりコードされるポリペプチドの全てのアミノ酸配列は、前記のように決定したDNA配列の翻訳により予測された。それ故、この自動アプローチにより決定された任意のDNA配列について当分野で既知であるように、本明細書で決定された任意のヌクレオチド配列はいくつかのエラーを含み得る。自動化により決定されたヌクレオチド配列は、典型的には、シークエンスしたDNA分子の実際のヌクレオチド配列と少なくとも約90%同一、より典型的には少なくとも約95%から少なくとも約99.9%同一である。実際の配列は、当分野で公知のマニュアルDNAシークエンス法を含む他のアプローチにより、より正確に決定することができる。これも当分野で既知であるように、実際の配列と比べて、決定されたヌクレオチド配列における1つの挿入または欠失により、ヌクレオチド配列の翻訳においてフレームシフトが引き起こされ、決定されたヌクレオチド配列によりコードされる予測アミノ酸配列は、このような挿入点または欠失点から始まって、シークエンスされたDNA分子により実際にコードされるアミノ酸配列とは完全に異なるであろう。
【0037】
本発明は、酵素(SNDH)をコードする単離核酸分子を提供する。単離核酸分子を操作するように設計された方法および技術は、当分野で公知である。核酸分子を、単離、精製およびクローニングする方法、ならびに、真核および原核宿主細胞の使用ならびにそこでの核酸およびタンパク質発現を記載した方法および技術は、当業者に既知である。
【0038】
簡潔には、SNDH遺伝子、前記遺伝子を含むDNA分子、組換え発現ベクター、および本発明で使用する組換え微生物は、以下の工程により得ることができる:
(1)G.oxydansDSM4025から染色体DNAを単離し、例えばE.coliなどの適切な宿主細胞において染色体DNAを用いて遺伝子ライブラリーを作製し;
(2)コロニーハイブリダイゼーション、プラークハイブリダイゼーション、またはサザンハイブリダイゼーション、PCR(ポリメラーゼ連鎖反応)クローニング、ウェスタンブロット解析または当分野で既知の他の技術により染色体DNAからSNDH遺伝子をクローニングし;
(3)慣用的な方法により前記のようにして得られたSNDH遺伝子のヌクレオチド配列を決定して前記SNDH遺伝子を含むDNA分子を選択し、SNDH遺伝子を効率的に発現できる組換え発現ベクターを作製し;
(4)宿主細胞にDNAを導入する適切な方法、例えば形質転換、形質導入、接合および/または電気穿孔によりSNDH遺伝子を有する組換え微生物を作製し、ここでの宿主細胞は、よって、本発明の組換え微生物となる。
【0039】
本発明の前記態様に使用した物質および技術は、以下のように詳細に例示する。
【0040】
全染色体DNAは、当分野で公知の手法により精製できる。望ましい遺伝子を、典型的には以下の例示的方法のいずれかにより、全染色体DNAから、プラスミドまたはファージベクターにクローニングできる:
(i)部分的アミノ酸配列を、精製タンパク質またはそのペプチド断片から決定する。このような全タンパク質またはペプチド断片は、このような全タンパク質の単離により、または、SDSポリアクリルアミドゲル電気泳動後のゲルからのペプチダーゼ処理により調製できる。このようにして得られたタンパク質またはその断片を、アプライド・バイオシステムズ自動ガス相シークエンサー470Aなどのタンパク質シークエンサーに適用する。アミノ酸配列を使用して、アプライド・バイオシステムズ自動DNAシークエンサー381AなどのDNA合成機を用いてオリゴヌクレオチドプローブおよび/またはプライマーを設計および調製できる。プローブは、サザンハイブリダイゼーション、コロニーハイブリダイゼーション、またはプラークハイブリダイゼーションを用いて、標的遺伝子を有する株の遺伝子ライブラリーから、標的遺伝子を有するクローンを単離するために使用できる。
(ii)または、遺伝子ライブラリーから標的タンパク質を発現するクローンを選択する目的で、標的タンパク質に対して調製した抗体を用いる免疫学的方法を適用できる。
(iii)標的遺伝子のDNA断片を、1セットのプライマー、すなわち、前記のように決定したアミノ酸配列に従って合成した2つのオリゴヌクレオチドを用いて、PCR法により、全染色体DNAから増幅できる。その後、標的全遺伝子を有するクローンを、プローブとして前記で得られたPCR産物を用いて、サザンハイブリダイゼーション、コロニーハイブリダイゼーション、またはプラークハイブリダイゼーションにより、例えばE.coliにおいて作製した遺伝子ライブラリーから単離できる。
【0041】
当分野で既知の方法により本明細書に開示したDNA配列に基づいて設計したプライマーを使用することによりPCRにより作製できるDNA配列もまた、本発明の目的である。
【0042】
前記した抗体は、抗原としての、精製SNDHタンパク質、E.coliにおいて発現されるHisタグ化SNDHなどの精製組換えSNDHタンパク質、またはそのペプチド断片を用いて調製できる。
【0043】
一旦、望む遺伝子を有するクローンが得られると、標的遺伝子のヌクレオチド配列を、M13ファージを用いたジデオキシ鎖終結法などの公知の方法により決定することができる。
【0044】
本発明の遺伝子は、図2に示したような31アミノ酸残基の推定シグナルペプチド(配列番号2のアミノ酸1〜31からなる配列番号4)と共に、578アミノ酸残基(配列番号2のアミノ酸32〜609からなる配列番号5)のSNDH酵素をコードする。ヌクレオチド配列に関して、SNDH遺伝子のコード領域は、配列番号1のヌクレオチド258〜2087を包含し、推定シグナルペプチド(配列番号1のヌクレオチド258〜350)および終止コドン(配列番号1のヌクレオチド2085〜2087)のコード配列を含む。従って、終止コドンを含まないヌクレオチド配列は、配列番号1の258〜2084位のヌクレオチド配列であり、さらにシグナル配列を含まない場合にはヌクレオチド配列は、配列番号1のヌクレオチド351〜2084を包含する。
【0045】
本発明に開示したような核酸分子は、2−KGAおよび/またはビタミンCの産生のために組換え微生物において使用できる。Gluconobacter、Acetobacter、Peudomonas、Acinetobacter、Klebsiella、およびEscherichiaから選択される組換え微生物を、好気的条件下で適切な栄養を補充した水性培地中で培養し得る。培養は、4.0〜9.0、好ましくは6.0〜8.0のpHで実施し得る。培養期間は、使用するpH、温度および栄養培地により変更し、好ましくは1〜5日間である。培養を実施するに好ましい温度範囲は、約13℃から約36℃、好ましくは約18℃から約33℃である。培養培地が、同化可能な炭素源としてこのような栄養、例えば、グリセロール、D−マンニトール、D−ソルビトール、エリトリトール、リビトール、キシリトール、アラビトール、イノシトール、ズルシトール、D−リボース、D−フルクトース、D−グルコース、およびスクロース、好ましくはD−ソルビトール、D−マンニトール、およびグリセロール;および、消化可能な窒素源、例えば有機物質、例えばペプトン、酵母抽出液、パン酵母、尿素、アミノ酸、およびコーンスティープリカーを含むことが通常必要である。種々の無機物質、例えば硝酸塩およびアンモニウム塩なども窒素源として使用し得る。さらに、培養培地は、通常、無機塩、例えば硫酸マグネシウム、リン酸カリウム、および炭酸カルシウムを含む。培養は、ジャー発酵器、フラスコ、またはチューブなどの適切な装置中で実施する。組換え微生物は、前記のような核酸分子を含む発現ベクターで形質転換されているか、または、その染色体DNAに組み込まれた本発明の核酸分子を含む。
【0046】
2−KGAおよび/またはビタミンCの産生用の基質として使用する適切な糖化合物は、L−ソルボソンである。2−KGAおよびビタミンCの代謝経路は、D−ソルビトールからL−ソルボースを介してL−ソルボソンへと進行し、これはその後、2−KGAおよび/またはビタミンCに変換される。従って、両方の産物の直接的な基質は、L−ソルボソンである。
【0047】
従って、本発明の1つの態様は、(a)本発明で形質転換されているか、または、その染色体DNAに組み込まれた本発明の核酸分子を含む組換え微生物を、適切な培養培地中で増殖または培養し、(b)前記の培養培地から2−KGAおよび/またはビタミンCを回収および分離することを含む、L−ソルボソンから2−KGAおよび/またはビタミンCを産生するプロセスを提供することである。
【0048】
1つの実施形態は、(a)適切な培養培地中で組換え微生物を繁殖し、ここで本発明の核酸分子は前記微生物に異種的に導入されており、(b)前記培養培地からビタミンCおよび/または2−KGAを回収および分離することを含む、L−ソルボソンからビタミンCおよび/または2−KGAを産生するプロセスを提供することである。
【0049】
本発明は、組換えSNDHを提供する。本発明により提供されるSNDH遺伝子を、G.oxydansDSM4025を含む宿主細胞に導入することにより、SNDH酵素の産生収率を向上することができる。また、本発明のSNDH遺伝子を使用することにより、Gluconobacter、Acetobacter、Pseudomonas、Acinetobacter、Klebsiella、およびEsherichiaからなる群より選択される宿主細胞において、SNDHタンパク質をより効率的に産生することができる。微生物は前記したように培養し得る。
【0050】
培養後に微生物から組換えSNDHを単離および精製するための実施形態は、本明細書で以後に簡潔に記載している:細胞を、遠心分離またはろ過により液体培養ブロスから収集する。収集した細胞を、水、生理的食塩水、または適切なpHを有する緩衝溶液で洗浄する。洗浄した細胞を、緩衝溶液に懸濁し、ホモジナイザー、ソニケーター、もしくはフレンチプレスを用いて、または、リゾチームでの処理により破壊して、破壊細胞の溶液を得る。組換えSNDHを、破壊細胞の細胞非含有抽出液から、好ましくは微生物のサイトゾル画分から単離および精製する。組換えSNDHは、固相酵素反応のために固相担体上に固定できる。
【0051】
本発明はさらに、(a)適切な培養培地中でGluconobacter oxydansDSN4025に属する微生物を培養し、ここで前記微生物中の配列番号2により示されるアルデヒドデヒドロゲナーゼをコードする遺伝子は破壊されており、(b)前記培養培地から2−KGAを回収および分離することを含む、L−ソルボソンから2−KGAを産生するプロセスに関する。破壊は、遺伝子中のどの場所でも起こり得、その結果、コードされている酵素は機能しない。
【0052】
従って、(a)適切な培養培地中でGluconobacter oxydansDMS4025に属する微生物を繁殖することを含む、適切な糖化合物からL−ソルボソンを介して2−KGAを産生するプロセスを提供し、ここで前記微生物中のアルデヒドデヒドロゲナーゼをコードする遺伝子は破壊されており、前記アルデヒドデヒドロゲナーゼは、(i)配列番号1のヌクレオチド配列に少なくとも95%同一であるポリヌクレオチド;(ii)(a)配列番号1のヌクレオチド258〜2084、(b)配列番号1のヌクレオチド351〜2084、(c)配列番号1のヌクレオチド258〜1955、および(d)配列番号1のヌクレオチド351〜1955からなる群より選択されるポリヌクレオチドに少なくとも95%同一であるポリヌクレオチド;および(iii)(a)配列番号2のアミノ酸配列を有するポリペプチドをコードするポリヌクレオチド、(b)配列番号2のアミノ酸32〜609からなるポリペプチドをコードするポリヌクレオチド、(c)配列番号2のアミノ酸1〜566からなるポリペプチドをコードするポリヌクレオチド、および(d)配列番号2のアミノ酸32〜566からなるポリペプチドをコードするポリヌクレオチドからなる群より選択されたポリヌクレオチドによりコードされている。得られた2−KGAは、前記培養培地からさらに回収され単離される。
【0053】
1つの実施形態において、本発明は、インビボならびにインビトロにおける、UV照射、または任意の変異試薬、例えばN−メチル−N’−ニトロ−N−ニトロソグアニジン(NTG)、ICR170、またはアクリジンオレンジなどによる化学的処理などの要因による古典的な変異誘発による、SNDH遺伝子の破壊のためのプロセスを提供する。
【0054】
別の実施形態において、本発明は、インビボならびにインビトロにおける、トランスポゾン挿入またはPCRによる部位特異的変異誘発などのDNA組換え技術により、SNDH遺伝子を破壊するプロセスを提供する。
【0055】
別の実施形態において、本発明は、L−ソルボソン、D−グルコース、D−ソルビトール、およびL−ソルボースからなる群より選択される適切な基質、すなわち糖化合物からの発酵により、前記したような破壊物を使用して2−KGAを産生するプロセスを提供する。プロセスは、ジャー発酵器、フラスコ、またはチューブなどの適切な装置中で行なう。さらに、本発明は、バイオリアクターなどの適切な装置中で、適切な基質、例えばL−ソルボソン、D−グルコース、L−ソルボース、およびD−ソルビトールから、インキュベートにより、前記した破壊物の細胞非含有抽出液を使用して、2−KGAを産生するプロセスを提供する。
【0056】
本発明は、組換えSNDHを提供する。さらに、(a)(i)配列番号1のヌクレオチド配列に少なくとも95%同一であるポリヌクレオチド;(ii)(a)配列番号1のヌクレオチド258〜2084、(b)配列番号1のヌクレオチド351〜2084、(c)配列番号1のヌクレオチド258〜1955、および(d)配列番号1のヌクレオチド351〜1955からなる群より選択されるポリヌクレオチドに少なくとも95%同一であるポリヌクレオチド;および(iii)(a)配列番号2のアミノ酸配列を有するポリペプチドをコードするポリヌクレオチド、(b)配列番号2のアミノ酸32〜609からなるポリペプチドをコードするポリヌクレオチド、(c)配列番号2のアミノ酸1〜566からなるポリペプチドをコードするポリヌクレオチド、および(d)配列番号2のアミノ酸32〜566からなるポリペプチドをコードするポリヌクレオチドからなる群より選択されるポリヌクレオチドを含む、アルデヒドデヒドロゲナーゼをコードする核酸分子を含む組換え微生物を培養し;ここで前記微生物は適切な培養培地中で培養し、(b)前記アルデヒドデヒドロゲナーゼを前記培養培地から回収および分離することを含む、アルデヒドデヒドロゲナーゼの産生プロセスに関する。
【0057】
実施例1:SNDHのN末端からのアミノ酸シークエンス
SNDHタンパク質のN末端75kDaサブユニットの部分アミノ酸配列を決定した。75kDaサブユニットからなる約10μgのSDS処理精製SNDHを、SDS−PAGEにかけ、タンパク質バンドをPVDFメンブランにエレクトロブロットした。メンブラン上にブロットされたタンパク質を、消化緩衝液(100mMリン酸カリウム緩衝液、5mMジチオトレイトール、10mM EDTA、pH8.0)に浸漬し、30℃で24時間、5.04μgのピログルタメートアミノペプチダーゼ(シグマ、米国)と共にインキュベートした。インキュベート後、メンブランを脱イオン水で洗浄し、自動アミノ酸シークエンサー(ABIモデル490、パーキンエルマー社、コネチカット州、米国)を使用したN末端アミノ酸シークエンスにかけた。結果として、配列番号3で示したようなN末端アミノ酸配列の14残基が得られた。
【0058】
実施例2:PCRによる部分SNDH遺伝子のクローニング
部分SNDH遺伝子断片の増幅を、G.oxydansDSM4025の染色体DNA(FERM BP−3812)ならびに縮重オリゴヌクレオチドDNAプライマーのP11(配列番号6)およびP12(配列番号7)を用いてPCRにより実施した。両方のプライマーが、Gluconobacterコドン使用に偏りを有する縮重DNA混合物であった。PCRは、熱安定tapポリメラーゼ(TAKARA ExTap(商標)、宝酒造株式会社、日本520−2193滋賀県大津瀬田3−4−1)を用いて、サーマルサイクラー(GeneAmpPCRシステム2400−R、PEバイオシステムズ、850リンカーン・センター・ドライブ、フォスターシティ、CA94404、米国)を使用して実施した。反応混合物(25μl)は、200μMのdNTP、50pmolの各プライマー(24〜48回縮重)、5ngの染色体DNA、および業者から提供された緩衝液中の1.25単位のDNAポリメラーゼから構成されていた。反応は、94℃で30秒間の変性工程、37℃で30秒間のアニーリング工程、70℃で1分間の合成工程を5サイクル、その後、94℃で30秒間の変性工程、50℃で30秒間のアニーリング工程、70℃で1分間の合成工程を25サイクルで実施した。結果として、41bpのDNA断片が特異的に増幅され、ベクターpCR2.1−TOPO(Invitrogen、1600ファラデイ・アベニュー・カールスバッド、カリフォルニア92008、米国)にクローニングし、組換えプラスミドpMTSN2を得た。成熟SNDHタンパク質のN末端部分アミノ酸配列をコードするクローニングされた41bpのDNA断片のヌクレオチド配列は、ジデオキシ鎖終結法(F. Sangerら、Proc. Natl .Acad. Sci. USA、74、5463〜5467、1977)により確認した。
【0059】
実施例3:SNDH遺伝子の完全クローニング
(1)G.oxydansDSM4025の遺伝子ライブラリーの作製
G.oxydansDMS4025の染色体DNAを、4日間27℃で、5%D−マンニトール、1.75%コーンスティープリカー、5%パン酵母、0.25%MgSO4・7H2O、0.5%CaCO3(実践用等級)、0.5%尿素、および2.0%寒天(pH7.0)を含むM寒天培地上で増殖させた細胞から調製した。染色体DNA(4μg)を、20μlの反応混合物中の4単位のEcoRIで部分的に消化した。部分的に消化したDNA断片を含む試料の一部(8μl)を、1%アガロースゲルを使用して電気泳動により分離した。15〜35kbの範囲の断片を切り出し、QIAEXII(QIAGEN社、28159アベニュー・スタンフォード、バレンシア、CA91355、米国)を使用して化学的に融かして回収した。回収した目的のDNA断片を、H2O中に懸濁した。一方、2μgのコスミドベクターpVK100を、EcoRIで完全に消化し、細菌アルカリホスファターゼ(E.coliC75)(宝酒造)での処理により5’末端を脱リン酸化した。処理したpVK100(220ng)を、36μlの反応混合物中において、ライゲーションキット(宝酒造)を使用して、15〜35kbのEcoRI断片(1μg)とライゲートした。エタノール沈降し適切な量のTE緩衝液(10mMトリス−HClpH8.0、1mM EDTA)に溶かしておいたライゲートしたDNAを、インビトロでのパッケージングに使用し(GigapackIII Gold Packaging Extract、Stratagene、11011ノース・トーリー・パイン・ロード、ラ・ホーヤ、CA92037、米国)、ゲノムライブラリーの宿主株であるE.coliVCS257に感染させた。結果として、約25kbの挿入DNA断片を含む合計400,000〜670,000個のクローンを得た。
【0060】
(2)コロニーハイブリダイゼーションによるSNDH遺伝子の完全クローニング
コロニーハイブリダイゼーション法により完全SNDH遺伝子を有するクローンを検出するための、前記したコスミドライブラリーのスクリーニングに使用するプローブを作製した。SNDHのN末端アミノ酸配列をコードする41bpDNA断片を増幅し、PCR−DIGラベリング法(Roche Molecular Systems社、1145アトランティック・アベニュー、アラバマ、CA94501、米国)により標識した。鋳型としてのプラスミドpMTSN2DNAおよびオリゴヌクレオチドDNAプライマーP13(配列番号8)およびP14(配列番号9)を用いてのPCRを、熱安定taqポリメラーゼ(TAKARA Ex Taq(商標)、宝酒造株式会社)を用いて、サーマルサイクラー(Gene Amp PCR System2400−R、PEバイオシステムズ)を使用して実施した。反応は、94℃で30秒間の変性工程、55℃で30秒間のアニーリング工程、70℃で1分間の合成工程を25サイクルで実施した。DIG標識プローブを使用して、コロニーハイブリダイゼーションによるコスミドライブラリーのスクリーニング(約1000クローン)および業者(Roche Molecular Systems Inc.、米国)により提供された方法に従っての化学ルミネセンス検出を実施した。結果として、3つの陽性クローンが単離され、その中の1つをpVSN5と称し、これはpVK100ベクター中に約25kbの挿入断片DNAを有していた。これから、異なるサイズの25kbDNA挿入断片をさらに、pUC18ベクター(図2)にサブクローニングした:(1)SNDH遺伝子の上流部分(N末端部分)を含む3.2kbのEcoRI断片によりpUCSN19が得られ、(2)SNDH遺伝子の下流部分(C末端部分)を含む7.2kbEcoRI断片によりpUCSN5が得られ、(3)無傷または完全SNDH遺伝子を含む1.8kbPstI断片によりそれぞれpUCSNP4およびpUCSNP9が得られた。pUSNP4およびpUSNP9における挿入断片は同じであるが逆の方向であることを注記する。
【0061】
(3)SNDH遺伝子のヌクレオチドシークエンス
プラスミドpUCSN19、pUCSN5、およびpUCSNP4を、SNDH遺伝子または遺伝子断片を含む領域のヌクレオチドシークエンスに使用した。決定されたヌクレオチド配列(3,408bpを有する配列番号1)により、SNDH遺伝子のORF(1,827bp;配列番号1のヌクレオチド258〜2084)は、609アミノ酸残基(配列番号2)のポリペプチドをコードしていることが判明した。他のOPFであるORF−Aは、図1に示したように、SNDH ORFの下流に見られた。ORF−A(1,101bp;配列番号1のヌクレオチド2214〜3314)は367アミノ酸のポリペプチドをコードしていた。
【0062】
SNDH遺伝子のORFでは、シグナルペプチド様の配列(31アミノ酸を有する配列番号4)が、(i)多くの疎水性残基、(ii)N末端に近い正に荷電した残基、および(iii)シグナル配列の切断部位のためのAla−Xaa−Alaモチーフを含む、推定アミノ酸配列におそらく含まれている。SNDH遺伝子の推定リボソーム結合部位(シャインダルガーノ、SD、配列)は、開始コドンの6bp上流に位置していた(配列番号1のヌクレオチド位置247〜252のAGGAGA)。さらに、ヘムc結合部位として定義されたモチーフ(Cys−Xaa−Xaa−Cys−His)は、配列番号2の位置530〜534に見出された。前記に示したような遺伝子解析から、SNDHタンパク質はキノヘモタンパク質の1つであると考えられた。
【0063】
GCG(Genetics Computer Group、マディソン、WI、米国)のFASTAプログラムを使用したSNDH遺伝子の相同性探索により、配列番号5のArg227、Asn228、Gln230、Gly246、およびAsp251が、Oubrieら[J.Mol.Biol.289: 319〜333(1999)]により記載のA.calcoaceticusGDH−Bタンパク質の推定活性部位におけるいくつかの高度に保存された残基に対応することが判明した。
【0064】
実施例4:E.coliにおけるSNDH遺伝子の発現
無傷の、すなわち完全なSNDH遺伝子を有する8.0kbのPstI断片を含むプラスミドpUCSNP4およびpUCSNP9(図3)を、E.coliJM109に形質転換し、SNDHタンパク質の発現および活性を確認した。酵素活性として産生されるビタミンCの量を、UV検出器(TOSOH UV8000;Tosoh、日本)、デュアルポンプ(TOSOH CCPE;Tosoh)、積分器(島津C−R6A;島津、日本)およびカラム(YMC−Packポリアミン−II;内径[i.d.]4.6mm×15cm、YMC、米国)から構成される高速液体クロマトグラフィーシステム(HPLC)により264nmの波長で測定した。
【0065】
組換えE.coliのサイトゾル画分を使用することによるL−ソルボソンからビタミンCへの変換活性を試験した(表1)。細胞を、場合により10μlのPQQおよび1.0mMのCaCl2を補充したLB培地中で培養した。サイトゾル画分を、50mMリン酸カリウム緩衝液(pH7.0)中での細胞非含有抽出液の超遠心分離(100,000×g)45分間)により調製した。反応混合物(100μl)は、125μgの組換えE.coliのサイトゾル画分、50mMのL−ソルボソン、1.0mMのフェナジンメソスルフェート(PMS)、場合に応じて、補因子として1.0μMのPQQおよび1.0mMのCaCl2を添加したまたは添加しないものから構成された。酵素反応は、30℃で30分間実施した。10μMのPQQおよび1.0mMのCaCl2を含むLB培地中で培養した細胞のホロ−SNDHにより、PQQおよびCaCl2の補因子を添加しない規定の反応条件下で、明確にビタミンCが産生された。補因子の添加により、pUCSNP4およびpUCSNP9で発現されたアポ−SNDHは、ホロ酵素の活性とほぼ同じ活性を示した。
【0066】
【表1】
【0067】
実施例5:G.oxydansのSNDH遺伝子破壊株の作製および培養
図4は、SNDH遺伝子標的化ベクター、GOMTR1SN::Km(SNDH破壊物)の作製のスキームを示す。最初に、プラスミドpSUPSNを、プラスミドpUCSNP4からのSNDH遺伝子を含む8.0kbのPstI断片を、自殺ベクターpSUP202とライゲートすることにより作製した(参照については、Simonet et., Abroad host range mobilization system for in vitro genetic engineering: transposon mutagenesis in Gram negative bacteria、Biotechnology、1、784〜791、1983を参照のこと)。第二に、カナマイシン耐性遺伝子カセット(Kmカセット)を、プラスミドpSUPSNにクローニングしたSNDH遺伝子のEcoRI部位に挿入して、プラスミドpSUPSN::Km(KmrTcr)を得た。その後、プラスミドpSUPSN::Kmを、リファンピシン(Rif)耐性で野生型G.oxydansDSM4025株から自発的に派生したGOMTR1に導入し、SNDH−ヌル変異体(KmrRifrTcs)を得た。
【0068】
GOMTR1は、30g/lのトリプチカーゼソイブロス(BBL;Becton Dickinson and Company、コッキーズビル、MD21030、米国)および3g/lの酵母抽出液(ディフコ;Becton Dickinson Microbiology Systems、Becton Dickinson and Company、スパークス、MD21152、米国)と、100μg/mlのリファンピシンからなる50mlのTブロスを含む200mlのフラスコ中で30℃で一晩培養した。E.coliHB101(pRK2013)[D.H.Figurski、Proc.Natl.Acad.Sci.USA、76、1648〜1652、1979]およびE.coliJM109(pSUPSN::Km)を、50μg/mlのカナマイシンを含む2mlのLB培地を含む試験管中で30℃で一晩培養した。GOMTR1、E.coliHB101(pRK2013)、およびE.coliJM109(pSUPSN::Km)の培養細胞を、遠心分離により別々に回収し、LB培地中の各細胞懸濁液を、それぞれ10:1:1の比で混合した。その後、これらの細胞懸濁液を、同じ容量で混合し、E.coliドナーからの自殺プラスミドの、レシピエントであるGOMTR1への接合導入のために、5.0%マンニトール、0.25%MgSO4・7H2O、1.75%コーンスティープリカー、5.0%パン酵母、0.5%尿素、0.5%CaCO3、および2.0%寒天から構成される寒天培地上にのせた0.45μmニトロセルロースメンブラン(PROTRAN、Schleicher & SchuellGmbH、Postfach4、D-37582 Dassel、ドイツ)に混合物を蒔いた。27℃で1日間培養した後、トランスコンジュガント(transconjugant)を含む細胞を懸濁し、Tブロスで適切に希釈し、100μg/mlのリファンピシンおよび50μg/mlのカナマイシンを含むスクリーニング寒天プレート上に蒔いた。最後に、Kmカセットにより破壊されたSNDH遺伝子を有するいくつかのトランスコンジュガント(KmrRifTcs)を得た。
【0069】
GOMTR1および破壊株GOMTR1SN::Kmを、8.0%のL−ソルボース、0.25%MgSO4・7H2O、1.75%コーンスティープリカー、5.0%パン酵母、0.5%尿素、0.5%CaCO3、および2.0%寒天を含む寒天プレート上で27℃で4日間増殖させた。白金棒でひとかきした細胞を、500mlエーレンマイヤーフラスコ中の、4%のD−ソルビトール、0.4%酵母抽出液、0.05%グリセロール、0.25%MgSO4・7H2O、1.75%コーンスティープリカー、0.1%尿素、および1.5%CaCO3を含む50mlの種培養培地(pH6.0)に接種し、30℃で回転振とう機で180rpmで1日間培養した。このようにして調製した種培養液を、500mlエーレンマイヤーフラスコ中の、12.0%L−ソルボース、2.0%尿素、0.05%グリセロール、0.25%MgSO4・7H2O、3.0%コーンスティープリカー、0.4%酵母抽出液、および1.5%CaCO3からなる50mlの主培養培地への接種に使用した。培養は30℃で180rpmで4日間行なった。活性アッセイは実施例4に記載のように実施した。酵素活性として産生される2−KGAの量は、UV検出器(TOSOH UV8000;Tosoh)、デュアルポンプ(TOSOH CCPE;Tosoh)、積分器(島津C−R6A;島津)およびカラム(YMC−PackProC18、YMC)で構成されるHPLCシステムにより340nmの波長で測定した。表2に示したように、SNDH遺伝子破壊株の2−KGA産生効率は、親株のGOMTR1の効率より高かった。1モルのL−ソルボースあたりの2−KGAへの変換率の差は、約3%であった。
【0070】
【表2】
【0071】
実施例6:G.oxydansDMS4025のSNDH遺伝子破壊株への、SNDH遺伝子を有するプラスミドの導入
広宿主域ベクターpVK100を使用した数種類のSNDH発現プラスミドを、図5に示したように作製した。このようなプラスミドは、以下のように記載したpVK100のHindIII部位において、異なる挿入断片DNAを有していた:pVSN117は、配列番号5のGly535(配列番号2のアミノ酸残基566)で終結するポリペプチドをコードする不完全なSNDH遺伝子、すなわち、僅か55kDaのタンパク質を発現するC末端欠失SNDH遺伝子を含む、挿入断片DNAを有する。プラスミドpVSN106およびpVSN114は、それぞれ、完全なSNDH遺伝子を含む挿入断片DNAを有していた。このようなプラスミドは、接合導入法によりGOMTR1SN::Km株に導入した。
【0072】
図5に示したプラスミドを有するトランスコンジュガントを、10.0%のL−ソルボース、0.25%MgSO4・7H2O、1.75%コーンスティープリカー、5.0%パン酵母、0.5%尿素、0.5%CaCO3、および2.0%寒天を含む寒天プレート上で27℃で4日間増殖させた。酵素反応混合物は、組換えGluconobacter株の細胞非含有抽出液80μg、25mMリン酸カリウム緩衝液(pH7.0)、50mMのL−ソルボソン、および0.05mMのPMSから構成された。酵素反応は、1,000rpmで振とうしながら、30℃で30分間実施した。活性アッセイは、実施例4に従って実施した。結果を表3に示した。
【0073】
【表3】
【図面の簡単な説明】
【0074】
【図1】本発明のアルデヒドデヒドロゲナーゼ活性を有するタンパク質をコードする遺伝子を示した図である。SNDH遺伝子およびORF−A遺伝子の制限地図が示されており、ここでORFは、オープンリーディングフレームを意味し、シグナル配列は、SNDH遺伝子の推定シグナルペプチド配列を意味する。
【図2】コスミドpVSN5にクローニングしたSNDHおよびORF−A遺伝子の制限地図、ならびに、pUCプラスミド、pUCSNP4、pUCSNP9、pUCSN19、およびpUCSN5への異なるサイズの挿入断片DNAのクローニングを示した図である。pVSN5の物理的地図において、灰色の矢印は、SNDH遺伝子を示す。
【図3】pVSN5からの無傷SNDH遺伝子を含む8.0kbのPstI断片を、pUC18ベクターにクローニングする戦略を示し、その結果、pUCSNP4およびpUCSNP9が得られる。pUCSNP4およびpUCSNP9は、挿入断片の方向を除いては同一であることを注記する。
【図4】カナマイシンカセット(Km)を用いて破壊したSNDH遺伝子を有する自殺ベクタープラスミドを使用することによる、GOMTR1SN::Km(SNDH破壊株)の作製を示した図である。対応する領域における、ベクタープラスミドと、親株であるGOMTR1の染色体DNAとの間の相同的組換えにより、破壊株が得られる。「G.O.」は、Gluconobacter oxydansを意味する。
【図5】pVSN117、pVSN106、およびpVSN114の挿入断片DNAの物理的地図を示した図である。プラスミドpVSN117は、C末端欠失SNDH遺伝子をコードする挿入断片DNAを有し(配列番号1のヌクレオチド258〜1955;すなわち、配列番号2のアミノ酸1〜566)、これは僅か55kDaのタンパク質を発現する。プラスミドpVSN106およびpVSN114は、無傷SNDH遺伝子をコードする挿入断片DNAを有する。
【特許請求の範囲】
【請求項1】
配列番号1のヌクレオチド配列に少なくとも95%同一であるポリヌクレオチドを含むアルデヒドデヒドロゲナーゼをコードする単離核酸分子。
【請求項2】
(a)配列番号1のヌクレオチド258〜2084;(b)配列番号1のヌクレオチド351〜2084;(c)配列番号1のヌクレオチド258〜1955、および(d)配列番号1のヌクレオチド351〜1955からなる群より選択されるポリヌクレオチドに少なくとも95%同一であるポリヌクレオチドを含むアルデヒドデヒドロゲナーゼをコードする単離核酸分子。
【請求項3】
(a)配列番号2のアミノ酸配列を有するポリペプチドをコードするポリヌクレオチド、(b)配列番号2のアミノ酸32〜609からなるポリペプチドをコードするポリヌクレオチド、(c)配列番号2のアミノ酸1〜566からなるポリペプチドをコードするポリヌクレオチド、および(d)配列番号2のアミノ酸32〜566からなるポリペプチドをコードするポリヌクレオチドからなる群より選択されるポリヌクレオチドを含むアルデヒドデヒドロゲナーゼをコードする単離核酸分子。
【請求項4】
アルデヒドデヒドロゲナーゼ活性を有するポリペプチドをコードする単離核酸分子であって、前記核酸分子は、標準的な条件下で請求項1〜3のいずれか一項の核酸分子の相補鎖にハイブリダイズする、前記単離核酸分子。
【請求項5】
請求項1〜4のいずれか一項の核酸分子を含む発現ベクター。
【請求項6】
ベクターが、pQE、pUC、pBluescriptII、pACYC177、pACYC184、pVK100、およびRSF1010からなる群より選択されるベクターまたはその誘導体から選択される、請求項5記載の発現ベクター。
【請求項7】
請求項5記載の発現ベクターで形質転換されている組換え微生物。
【請求項8】
その染色体DNAに組み込まれている請求項1〜4のいずれか一項記載の核酸分子を含む、組換え微生物。
【請求項9】
微生物が、細菌、酵母、および植物細胞からなる群より選択される、請求項7または8の組換え微生物。
【請求項1】
配列番号1のヌクレオチド配列に少なくとも95%同一であるポリヌクレオチドを含むアルデヒドデヒドロゲナーゼをコードする単離核酸分子。
【請求項2】
(a)配列番号1のヌクレオチド258〜2084;(b)配列番号1のヌクレオチド351〜2084;(c)配列番号1のヌクレオチド258〜1955、および(d)配列番号1のヌクレオチド351〜1955からなる群より選択されるポリヌクレオチドに少なくとも95%同一であるポリヌクレオチドを含むアルデヒドデヒドロゲナーゼをコードする単離核酸分子。
【請求項3】
(a)配列番号2のアミノ酸配列を有するポリペプチドをコードするポリヌクレオチド、(b)配列番号2のアミノ酸32〜609からなるポリペプチドをコードするポリヌクレオチド、(c)配列番号2のアミノ酸1〜566からなるポリペプチドをコードするポリヌクレオチド、および(d)配列番号2のアミノ酸32〜566からなるポリペプチドをコードするポリヌクレオチドからなる群より選択されるポリヌクレオチドを含むアルデヒドデヒドロゲナーゼをコードする単離核酸分子。
【請求項4】
アルデヒドデヒドロゲナーゼ活性を有するポリペプチドをコードする単離核酸分子であって、前記核酸分子は、標準的な条件下で請求項1〜3のいずれか一項の核酸分子の相補鎖にハイブリダイズする、前記単離核酸分子。
【請求項5】
請求項1〜4のいずれか一項の核酸分子を含む発現ベクター。
【請求項6】
ベクターが、pQE、pUC、pBluescriptII、pACYC177、pACYC184、pVK100、およびRSF1010からなる群より選択されるベクターまたはその誘導体から選択される、請求項5記載の発現ベクター。
【請求項7】
請求項5記載の発現ベクターで形質転換されている組換え微生物。
【請求項8】
その染色体DNAに組み込まれている請求項1〜4のいずれか一項記載の核酸分子を含む、組換え微生物。
【請求項9】
微生物が、細菌、酵母、および植物細胞からなる群より選択される、請求項7または8の組換え微生物。
【図1】

【図2】
【図3】
【図4】
【図5】
【図2】
【図3】
【図4】
【図5】
【公表番号】特表2006−500052(P2006−500052A)
【公表日】平成18年1月5日(2006.1.5)
【国際特許分類】
【出願番号】特願2004−538969(P2004−538969)
【出願日】平成15年9月22日(2003.9.22)
【国際出願番号】PCT/EP2003/010498
【国際公開番号】WO2004/029235
【国際公開日】平成16年4月8日(2004.4.8)
【出願人】(503220392)ディーエスエム アイピー アセッツ ビー.ブイ. (873)
【Fターム(参考)】
【公表日】平成18年1月5日(2006.1.5)
【国際特許分類】
【出願日】平成15年9月22日(2003.9.22)
【国際出願番号】PCT/EP2003/010498
【国際公開番号】WO2004/029235
【国際公開日】平成16年4月8日(2004.4.8)
【出願人】(503220392)ディーエスエム アイピー アセッツ ビー.ブイ. (873)
【Fターム(参考)】
[ Back to top ]