
アルファ−2アドレナリン受容体アゴニスト、および、エンドセリン受容体アンタゴニストを用いて疼痛を治療する方法
本願発明は、一般的には、疼痛の治療に関し、アルファ-2(α2)アドレナリン受容体アゴニストとエンドセリン受容体アンタゴニストとを投与することを含み、これら薬剤の投与によって、対象動物において鎮痛作用を発現し、また、疼痛を緩和する。
【発明の詳細な説明】
【技術分野】
【0001】
本願発明は、一般的には、疼痛の治療および鎮痛の増強に関し、対象動物に対して、イミダゾリン活性を有する、または、イミダゾリン活性を欠いたアルファ-2(α2)アドレナリン受容体アゴニストと、エンドセリン受容体アンタゴニストとの組み合わせを投与する、ことを含む。
【背景技術】
【0002】
鎮痛薬とは、好ましくは、意識を攪乱したり、あるいは、その他の感覚機能に影響を及ぼさずに、中枢に作用して疼痛閾値を高めることによって疼痛を緩和する薬剤である。 鎮痛薬が、疼痛を鈍らせる(すなわち、疼痛閾値を高める)作用機序は定式化されている。
【0003】
国立健康統計センター(2006)は、米国では、20歳以上の国民の四分の一以上(26%)、つまり、7650万人以上の国民が、24時間以上にわたって持続するタイプの疼痛に患わされており、また、1億9100万人以上が、急性疼痛を経験している、という報告を行っている。 オピオイドは、急性および慢性の疼痛の臨床管理において最も一般的に用いられている鎮痛薬である。 オピオイドを長期使用することで、効果的な疼痛緩和を妨げる耐性の出現などの副作用を招くこととなる。 鎮痛作用を高め、また、効果的に疼痛に対処する目的で策定された、非ステロイド性抗炎症剤(NSAIDS)、その他のオピオイド、それに、非オピオイドとオピオイド療法との組み合わせなどの幾つかの治療方法がある。 これら手法によって症状の緩和には至るものの、耐性の出現に関与する作用機序に対する効果はほとんど無く、また、毒性、依存性、および、中毒性を得るに至る危険性は非常に大きい。
【0004】
アルファ-2(α2)アドレナリンアゴニストであるクロニジンは、マウスやラットにおいて顕著な鎮痛作用を示すことが実証されており[31]、また、クロニジンを反復して(7日間にわたって毎日2回)投与することで、耐性が出現するのに対して、クロニジンを急性投与すると、モルヒネの鎮痛効果が改善することも知られている[30]。 クロニジンは、モルヒネの鎮痛効果を高めるが、クロニジンの鎮痛作用とモルヒネの鎮痛作用との間には、交差耐性がない[38]。 間脳室周囲灰白質、背側縫線核、および、中脳水道周囲灰白質でのアドレナリン作動性およびアヘン製剤抗侵害受容メカニズムの活性化に対してクロニジンが関与しているかもしれないとの考え方も出されている[39]。 クロニジンは、アヘン製剤離脱を抑制するためにも使用されている。 クロニジンの奏するこれら二つの特性は、オピオイドに対して耐性を示す患者において、好適な鎮痛剤代替物の提供を理論的に可能にしている[27]。 クロニジンは、モルヒネの鎮痛作用のみならず、ペンタゾシン[13]、フェンタニル[9]、および、ブピバカイン[19]の鎮痛作用をも改善することが実証されている。
【0005】
エンドセリン(ETs)は、血管収縮剤の中でも最も強力な血管収縮剤として知られている内皮由来ポリペプチドのファミリーで構成されている。 主要なエンドセリン受容体のサブタイプ(ETAおよびETB)は、血管収縮を司る血管平滑筋において発現する。 内皮細胞にあるETBサブタイプは、血管弛緩を司っているものと考えられている。 ETA受容体アンタゴニストは、ラットとマウスの双方において、モルヒネに対する抗侵害受容応答を顕著に高める、ことが報告されている[5、7、16]。 モルヒネと共にETA受容体アンタゴニストBQ123を慢性注射投与することで、モルヒネに対する耐性の出現が予防された[6]。 ETA受容体アンタゴニストを単回投与することで、マウスとラットにおいて、モルヒネの鎮痛作用に対する耐性が逆になった[4、6、7]。 したがって、ETA受容体アンタゴニストによって耐性が予防および逆転する現象は、ETが、モルヒネ耐性に関与していることを実証するものである[6、7、33、34]。 さらに、ETAアンタゴニストは、強硬症作用[5]や消化管通過[26]などのモルヒネの副作用に関して効果を示さなかった。 したがって、ETA受容体アンタゴニストは、モルヒネの副作用[16]を増大せずに、モルヒネの鎮痛作用を改善するのである。
【0006】
スルフィソキサゾールとは、中耳炎の治療や細菌感染症の治療において広く利用されており、また、好ましい抗菌活性を有している。 スルフィソキサゾールの化学構造は、4-アミノ-N-(3,4-ジメチルオキサゾール-5-イル)-ベンゼンスルホンアミドである。 スルフィソキサゾールは、易溶性である。 スルフィソキサゾールを経口投与すると、迅速に吸収され、また、迅速に排泄され、そして、可溶性にも富んでいるので、スルホンアミドの使用に伴って認められる腎臓毒性も軽減される[25]。 ETAおよびETB受容体に対するET-1の結合に関するスルファニルアミドの阻害効果を、膜調製物において決定をした。 スルフィソキサゾールが、最も活性なスルファニルアミドであり、ETAおよびETB受容体に対するIC50は、それぞれ、0.60μMおよび22μMであった[8]。
【0007】
交感神経系が関与している心臓血管系作用でのクロニジンとETとの間の相互作用についての報告がされている[15、17、18、28]。 エンドセリン-1(ET-1)が、クロニジンによって引き起こされた低血圧症を悪化させることも知られている。 この効果は、一酸化窒素メカニズムを媒介しているのかもしれない[23]。 鎮痛に関するクロニジンとETA受容体アンタゴニストとの間の相互作用を決定するための研究は行われていない。
【0008】
したがって、オピオイド系疼痛緩和剤に対する耐性を軽減し、疼痛症状を緩和し、そして、効果的な非オピオイド鎮痛薬として機能できる薬剤または薬剤の組み合わせを同定することが、当該技術分野において必要とされているのである。
【発明の開示】
【0009】
本願発明は、対象動物の疼痛を治療するための鎮痛剤としてのアドレナリンアゴニストとエンドセリンアンタゴニストの使用に関する。 さらに、本願発明において、アルファ2(α2)アドレナリンアゴニストとエンドセリンA(ETA)アンタゴニストとの組み合わせが、オピオイド鎮痛薬の鎮痛効果を高め、また、その組み合わせが、鎮痛作用の発現に関与していることを知見するに至ったのである。
【0010】
ある実施態様によれば、本願発明は、疼痛を治療または予防する方法であって、治療または予防の必要がある哺乳動物に対して、治療有効量のアルファ-2(α2)アドレナリン受容体アゴニスト、および、治療有効量のエンドセリン受容体アンタゴニストを投与する、ことを含む方法を提供する。
【0011】
ある実施態様によれば、α2アドレナリンアゴニストとは、イミダゾリン活性を有する、または、イミダゾリン活性を欠いたα2アドレナリンアゴニストである。 さらに他の実施態様によれば、α2アドレナリンアゴニストは、デクスメデトミジン、デトミジン、ST-91、メデトミジン、ブリモニジン、チザニジン、ミバゼロール、グアナベンズ、グアンファシン、ヨードクロニジン、キシラジン、リルメニジン、ロフェキシジン、アゼペキソール、アルファ-メチルドパ、および、アルファ-メチルノルアドレナリン、あるいは、これらの誘導体、塩類、または、構造的類似体からなるグループから選択される。 関連する実施態様において、α2アドレナリンアゴニストとは、クロニジンである。
【0012】
さらに別の実施態様によれば、エンドセリン受容体アンタゴニストとして、エンドセリン受容体A(ETA)アンタゴニストを意図している。 別の実施態様によれば、ETAアンタゴニストは、スルフィソキサゾール、アトラセンタン、テゾセンタン、ボセンタン、シタキスセンタン、エンラセンタン、BMS207940、BMS193884、BMS182874、J 104132、VML 588/Ro 61 1790、T-O115、TAK 044、BQ 788、TBC2576、TBC3214、PD180988、ABT 546、SB247083、RPR118031A、および、BQ123からなるグループから選択される。 さらに別の実施態様によれば、ETAアンタゴニストは、スルフィソキサゾールである。
【0013】
ある実施態様によれば、α2アドレナリンアゴニストおよびエンドセリン受容体アンタゴニストは、単一の組成物で投与される。 関連する実施態様によれば、α2アドレナリンアゴニストおよびエンドセリン受容体アンタゴニストは、個別の組成物で投与される。
【0014】
ある実施態様によれば、これら組成物は、同時に投与される。 関連する実施態様によれば、これら組成物は、別個に投与される。 さらに別の実施態様によれば、α2アドレナリンアゴニストおよびエンドセリン受容体アンタゴニスト分子は、約24時間以内に連続投与される。
【0015】
さらに別の実施態様によれば、これら組成物は、医薬担体または医薬賦形剤をさらに含む。
【0016】
さらに他の実施態様によれば、α2アドレナリンアゴニストおよびエンドセリン受容体アンタゴニストは、経口的、経頬的、吸入的、舌下、経直腸的、経膣的、嚢内、関節内、経尿道的、経鼻的、経皮的、静脈内、筋肉内、または、皮下に投与される。
【0017】
ある実施態様によれば、クロニジンは、約10μg〜約300μgの範囲の用量で投与される。 関連する実施態様によれば、スルフィソキサゾールは、約0.1g〜約3gの範囲の用量で投与される。 また、投与されたα2アドレナリンアゴニストと投与されたエンドセリン受容体アンタゴニストとの比率を、1:500〜1:50,000、1:500〜1:20,000、1:500〜1:10,000、1:500〜1:5,000、1:500〜1:2,500、1:100〜1:1000、または、1:100〜1:500の範囲とすることも意図している。
【0018】
その他の実施態様によれば、本願発明は、疼痛を治療または予防する方法であって、対象動物に対して、一つまたはそれ以上のアルファ-2(α2)アドレナリン受容体アゴニストと、一つまたはそれ以上のエンドセリン受容体アンタゴニストとの相乗的組み合わせを投与する、ことを含む方法を提供する。 ある実施態様によれば、その組み合わせに用いられたアゴニスト分子およびアンタゴニスト分子が、約24時間以内に連続投与されるか、あるいは、同時投与される。 薬剤の投与を、30分間〜約1日(24時間)までの範囲で行うことも意図されている。
【0019】
本願発明は、一つまたはそれ以上のアルファ-2(α2)アドレナリンアゴニストと、一つまたはそれ以上のエンドセリン受容体アンタゴニストとの相乗的組み合わせを含む、疼痛を治療または予防するための組成物をさらに提供する。 ある実施態様によれば、この組成物は、低用量のα2アドレナリンアゴニストと低用量のエンドセリン受容体アンタゴニストを含む。 関連する実施態様によれば、α2アドレナリンアゴニストは、クロニジンであり、そして、エンドセリン受容体アンタゴニストは、スルフィソキサゾールである。 さらに別の実施態様によれば、この組成物には、約10μg〜約300μgの範囲のクロニジンと、約0.1g〜約3gの範囲のスルフィソキサゾールが含まれている。
【0020】
さらに他の実施態様によれば、その組成物は、薬学的に許容可能な担体をさらに含んでいる。
【0021】
さらに別の実施態様によれば、本願発明は、対象動物での疼痛を治療するための薬剤を製造するためのアルファ-2(α2)アドレナリンアゴニストとエンドセリン受容体アンタゴニストを含む組成物の使用も提供する。
【0022】
本願発明は、哺乳動物を治療対象とすることを意図している。 ある実施態様によれば、治療対象となる哺乳動物として、ヒト、または、ヒトに関する医学研究のための非ヒト動物モデル、あるいは、家畜として重要な動物、あるいは、例えば、愛玩動物のようなペットがある。 関連する実施態様によれば、対象動物は、ヒトである。
【0023】
ある実施態様によれば、治療の対象となる疼痛は、慢性疼痛または急性疼痛である。 関連する実施態様によれば、疼痛は、灼熱痛、接触性アロディニア、神経因性疼痛、痛覚過敏症、痛感過敏症、炎症性疼痛、術後疼痛、慢性腰痛、群発頭痛、帯状疱疹後神経痛、幻肢痛および断端痛、中心性疼痛、歯痛、神経因性疼痛、オピオイド耐性疼痛、内臓痛、術後疼痛、骨損傷痛、糖尿病性神経障害疼痛、術後または外傷性神経症疼痛、末梢神経障害疼痛、エントラップメント神経障害疼痛、アルコール依存症に起因する神経障害、HIV感染、多発性硬化症、甲状腺機能低下症に起因する疼痛、または、抗癌化学療法に起因する疼痛、分娩中の疼痛、日焼けを含む火傷に起因する疼痛、産後疼痛、偏頭痛、咽喉痛、および、膀胱炎などの尿生殖路関連疼痛からなるグループから選択される。
【0024】
さらに別の実施態様によれば、一つまたはそれ以上のアルファ-2(α2)アドレナリンアゴニスト、または、一つまたはそれ以上のエンドセリン受容体アンタゴニストは、麻薬性鎮痛薬の鎮痛作用を高める上で有用である。 ある実施態様によれば、一つまたはそれ以上のアルファ-2(α2)アドレナリンアゴニストと一つまたはそれ以上のエンドセリンアンタゴニストとの組み合わせは、麻薬性鎮痛薬の鎮痛作用を高める上で有用である。 このように、本願発明は、疼痛を治療または予防する方法であって、治療または予防の必要がある哺乳動物に対して、治療有効量の麻薬性鎮痛薬、および、一つまたはそれ以上のアルファ-2(α2)アドレナリンアゴニストを含む治療有効量の組成物を投与する、ことを含む方法を提供する。 関連する実施態様によれば、本願発明は、疼痛を治療または予防する方法であって、治療または予防の必要がある哺乳動物に対して、治療有効量の麻薬性鎮痛薬、一つまたはそれ以上のアルファ-2(α2)アドレナリンアゴニストを含む治療有効量の組成物、および、一つまたはそれ以上のエンドセリン受容体アンタゴニストを含む治療有効量の組成物を投与する、ことを含む方法を意図している。 さらに別の実施態様によれば、本願発明は、疼痛を治療または予防する方法であって、治療または予防の必要がある哺乳動物に対して、治療有効量の麻薬性鎮痛薬、および、一つまたはそれ以上のアルファ-2(α2)アドレナリンアゴニストと、一つまたはそれ以上のエンドセリン受容体アンタゴニストとを含む治療有効量の組成物を投与する、ことを含む方法を提供する。
【0025】
ある実施態様によれば、一つまたはそれ以上のアルファ-2(α2)アドレナリンアゴニストは、デクスメデトミジン、デトミジン、ST-91、メデトミジン、ブリモニジン、チザニジン、ミバゼロール、グアナベンズ、グアンファシン、ヨードクロニジン、キシラジン、リルメニジン、ロフェキシジン、アゼペキソール、アルファ-メチルドパ、および、アルファ-メチルノルアドレナリン、または、これらの誘導体、塩類、または、構造類似体からなるグループから選択される。 関連する実施態様によれば、一つまたはそれ以上のエンドセリンアンタゴニストは、スルフィソキサゾール、アトラセンタン、テゾセンタン、ボセンタン、シタキスセンタン、エンラセンタン、BMS207940、BMS193884、BMS182874、J 104132、VML 588/Ro 61 1790、T-O115、TAK 044、BQ 788、TBC2576、TBC3214、PD180988、ABT 546、SB247083、RPR118031A、および、BQ123からなるグループから選択されるETAである。
【0026】
さらに別の実施態様によれば、麻薬性鎮痛薬は、モルヒネ、硫酸モルヒネ、コデイン、ジアセチルモルヒネ、デキストロメトルファン、ヒドロコドン、ヒドロモルフォン、ヒドロモルフォン、レボルファノール、オキシモルホン、オキシコドン、レバロルファン、および、これらの塩類からなるグループから選択される。
【0027】
ある実施態様によれば、麻薬性鎮痛薬、および、一つまたはそれ以上のアルファ-2(α2)アドレナリンアゴニスト、および/または、一つまたはそれ以上のエンドセリンアンタゴニストは、同時に投与される。 関連する実施態様によれば、麻薬性鎮痛薬、および、一つまたはそれ以上のアルファ-2(α2)アドレナリンアゴニスト、および/または、一つまたはそれ以上のエンドセリンアンタゴニストは、単一の組成物または別個の組成物から投与される。 さらに別の実施態様によれば、麻薬性鎮痛薬、および、一つまたはそれ以上のアルファ-2(α2)アドレナリンアゴニスト、および/または、一つまたはそれ以上のエンドセリンアンタゴニストは、連続して投与される。
【0028】
さらに他の実施態様によれば、麻薬性鎮痛薬は、一つまたはそれ以上のアルファ-2(α2)アドレナリンアゴニスト、および/または、一つまたはそれ以上のエンドセリンアンタゴニストに先駆けて投与され、あるいは、、一つまたはそれ以上のアルファ-2(α2)アドレナリンアゴニスト、および/または、一つまたはそれ以上のエンドセリンアンタゴニストに続いて投与される。
【0029】
本明細書に記載の活性成分の用量、および、本明細書に記載の投薬計画は、本願発明において意図しているすべての方法に対して有用である。
【0030】
その他の実施態様によれば、本願発明は、アルファ-2(α2)アドレナリンアゴニストとエンドセリン受容体アンタゴニスト、それに、本願発明の方法を示したラベルを含む製造品も意図している。
【0031】
さらに別の実施態様によれば、本願発明は、疼痛を治療または予防するためのキットであって、アルファ-2(α2)アドレナリンアゴニストとエンドセリン受容体アンタゴニストを含む組成物、および、そのキットを用いて疼痛を治療するためのプロトコル、を含むキットが提供される。 ある実施態様によれば、その組成物は、薬学的に許容可能な担体をさらに含む。 関連する実施態様によれば、このキットは、麻薬性鎮痛薬をさらに含む。 関連する実施態様によれば、麻薬性鎮痛薬は、薬学的に許容可能な担体または賦形剤と共にある。
【発明を実施するための最良の形態】
【0032】
本願発明は、顕著な鎮痛作用を示し、また、疼痛刺激も顕著に緩和する、α2アドレナリンアゴニストとエンドセリン受容体アンタゴニストとの組み合わせを用いて疼痛を治療する方法に関する。 具体的には、本願発明は、ETAアンタゴニストとアルファ-2 アドレナリンアゴニストとを併用することで、オピオイド疼痛緩和剤に対する耐性が顕著に減少し、また、疼痛症状も消失する、との発見に関するものである。
【0033】
本願発明の方法は、強力な鎮痛作用を呈するα2アドレナリンアゴニスト(例えば、クロニジン)とETA受容体アンタゴニスト(例えば、スルフィソキサゾール)との組み合わせを利用するものである。 ある実施態様によれば、本願発明の方法は、マウスでのテイルフリック潜時によって実証されている通り、スルフィソキサゾールとクロニジンによって増強された鎮痛作用を利用している。 本願発明の方法にしたがってクロニジンとスルフィソキサゾールとを併用して得られる鎮痛作用は、高用量のモルヒネが奏する鎮痛作用に匹敵するものであるので、特筆すべき事項といえる。
【0034】
本明細書で用いる「治療」の用語は、疼痛を予防、軽減、あるいは、改善することを指す。 そのようなものとして、「治療」の用語は、必要に応じて、内科治療的投与、および/または、予防的投与の双方を含んでいる。 疼痛症状の治療および緩和は、当該技術分野で周知の疼痛評価スケールを用いて評価することができる[例えば、3、5、9を参照されたい]。 評価手順の例として、客観的疼痛閾値(視覚的アナログ尺度)や客観的侵害受容屈曲反射(R III)閾値の測定がある。
【0035】
本明細書で用いる「疼痛」の用語は、すべてのタイプの疼痛を指す。 ある実施態様によれば、この用語は、急性疼痛と慢性疼痛を指す。 疼痛の例として、灼熱痛、接触性アロディニア、神経因性疼痛、痛覚過敏症、痛感過敏症、炎症性疼痛、術後疼痛、慢性腰痛、群発頭痛、帯状疱疹後神経痛、幻肢痛および断端痛、中心性疼痛、歯痛、神経因性疼痛、オピオイド耐性疼痛、内臓痛、術後疼痛、骨損傷痛、糖尿病性神経障害疼痛、術後または外傷性神経症疼痛、末梢神経障害疼痛、エントラップメント神経障害疼痛、アルコール依存症に起因する神経障害、HIV感染、多発性硬化症、甲状腺機能低下症に起因する疼痛、または、抗癌化学療法に起因する疼痛、分娩中の疼痛、日焼けを含む火傷に起因する疼痛、産後疼痛、偏頭痛、咽喉痛、および、膀胱炎などの尿生殖路関連疼痛などがあるが、これらに限定されない。
【0036】
本明細書で用いる「鎮痛剤」の用語は、対象動物での疼痛を緩和する活性成分のことを指す。 「麻薬性鎮痛薬」または「オピオイド鎮痛剤」の用語は、例えば、麻酔剤の補助として、あるいは、疼痛を緩和するために用いられる麻薬性鎮痛剤を指す。 「非麻薬性鎮痛薬」の用語は、疼痛の治療に適応される非麻酔剤を指す。
【0037】
「治療有効量」とは、所望の効果を奏する活性成分または薬剤の量のことを指す。 それら活性成分の毒性や治療効果は、細胞培養物や実験動物を用いた標準的な製剤手法、例えば、 LD50(個体群の50%に対する致死用量)やED50(個体群の50%に対する治療効果用量)によって決定される。 毒性と治療効果との間の用量比は、LD50とED50との間の比率として表される治療指数である。 治療指数は、大きい方が望ましい。 これらデータから取得されるデータは、ヒトに対して用いる用量の範囲を決定するために用いられている。 ある実施態様によれば、活性成分の用量は、ED50を含む循環濃度の範囲内にあり、その毒性は皆無か、あるいは、ほとんどない。 これら用量は、使用する剤形や、利用した投与経路に応じて、この範囲内で変化する。
【0038】
α2アドレナリンアゴニストとエンドセリン受容体アンタゴニストの「相乗的組み合わせ」とは、その活性成分だけを投与した場合に得られる効果の合計よりも大きな効果を示す組み合わせのことを指す。
【0039】
本明細書で用いる「増強する」または「増強作用」という用語は、鎮痛剤の効果を高めたり、あるいは、鎮痛剤と共に相乗的に作用して、例えば、生化学的効果や生理学的効果を増強するアルファ-2(α2)アドレナリンアゴニストまたはエンドセリンアンタゴニストの性質のことを指す。 ある実施態様によれば、この増強作用によって、所望の疼痛緩和効果を得るに必要な鎮痛剤の用量が効果的に減量している。
【0040】
「同時投与」、「組み合わせで投与した」、「同時の投与」、または、それらと同様の表現は、二つまたはそれ以上の薬剤を含む組成物が、治療を受けている対象動物に対して同時に投与されることを意味している。 「同時」の表現は、各薬剤が、同時に投与されること、あるいは、所定の時間内に任意の順序で連続して投与されること、を意味している。 しかしながら、同時に投与されない場合であっても、ある実施態様によれば、所望のレベルの治療効果を引き出すために、所定の時間内において、ごく短時間の内に投与が行われる。 薬剤の適切な投与間隔と投与順序は、当業者に自明の事項である。 二つまたはそれ以上の薬剤を、個別の組成物を用いて投与することも意図しており、ある実施態様によれば、一方の薬剤は、他方の薬剤を投与する以前または以後に投与される。 事前投与とは、治療を行う1日(24時間)前から治療を行う30分前までの間に薬剤を投与することを指す。 さらに、一方の薬剤を、他方の薬剤を投与した後に投与することも意図している。 事後投与とは、第一薬剤を投与して30分以後の投与から、第一薬剤の投与から1日(24時間)後の投与までを指す。 30分から24時間の範囲とは、30分、1、2、3、4、5、6、7、8、9、10、11、12、16、20、または、24時間の時点での投与を含むものである。
【0041】
本明細書で用いる「低用量」という用語は、組成物に含まれる活性成分の用量のことを指し、組成物に含まれる活性成分の量は、対象動物の治療において一般的に用いられる量よりも少ない。 例えば、低用量の活性成分を、第二の活性成分と組み合わせて投与して、活性成分の相乗効果を引き出すことができ、また、組み合わせ療法で用いる各活性成分の用量を、第二の活性成分と組み合わせずに薬剤を投与する場合に必要な用量よりも少なくすることもできる。 ある実施態様によれば、低用量のクロニジンは、10μg〜約300μgの範囲にある。 関連する実施態様において、低用量のスルフィソキサゾールは、0.1g〜約3.0gの範囲にある。
【0042】
アルファ-2(α2)アドレナリンアゴニスト
アルファ-2(α2)アドレナリン作動性受容体(アドレナリン受容体)は、神経系や身体のその他の各系統の双方において遍在して分布をしている。 α2アドレナリン受容体は、三つの異なる受容体サブタイプ(サブタイプA、BおよびCと称する)を含んでおり、これらは、非選択的内因性アドレナリンアゴニストアドレナリンおよびノルアドレナリンによって活性化され、また、その他の6個のアドレナリン受容体サブタイプをも活性化する(米国特許第6,562,855号)。 臨床研究によって、α2アゴニストが、強力な鎮痛作用を示すことが明らかになっている。 クロニジンやデクスメデトミジンのようなα2アゴニストの全身投与および脊髄投与は、ヒトや動物モデルでの疼痛を緩和する。 α2アゴニストは、脊柱作用ならびに局所脊髄麻酔作用によって鎮痛効果を奏する(本明細書の一部を構成するものとしてその内容を援用するGuo et al, Anesthesiology 1991; 75: 252-6、米国特許第6,562,855号)。 α2アドレナリンアゴニストに関するその他の知見は、本明細書の一部を構成するものとしてその内容を援用する米国特許第6,562,855号、第5,605,911号、および、第5,980,927号に記載されている。
【0043】
α2アドレナリンアゴニストであるクロニジンは、マウスやラットで顕著な鎮痛作用を呈することが実証されている[31]。 また、クロニジンを継続して(毎日2回、7日間)投与することで、耐性が出現することも知られている。 その一方で、クロニジンの急性投与によって、モルヒネの鎮痛効果が高まることも知られている[30]。 他の研究によれば、クロニジンが、モルヒネの鎮痛作用を増強するものの、クロニジンとモルヒネの鎮痛作用との間には交差耐性が認められなかったと報告されている[38]。 ホルマリン試験による疼痛感受性の評価結果は、クロニジンが、鎮痛作用を呈すること、そして、その効果が、ナロキソン(2mg/kg、腹腔内投与)によって阻害されることを示しており、また、クロニジン効果に関わるナロキソン感受性成分は、β-エンドルフィン様オピオイドの放出に起因するものであると推察されている[22]。 クロニジンは、アヘン製剤離脱を鎮静するためにも使用されている。 これら二つの特性が故に、クロニジンは、オピオイドに対して耐性を示す患者での魅力的な鎮痛剤代替物となっている[27]。 クロニジンが、モルヒネの鎮痛作用のみならず、ペンタゾシン[13]、フェンタニル[9]、および、ブピバカイン[19]の鎮痛作用をも高めることが実証されている。 さらに、クロニジンの鎮痛作用は、アミトリプチリンによっても高められることが知られている[1]。
【0044】
動物でのクロニジンの鎮痛作用が、健常な志願者が参加した交差式・二重盲検式・プラシーボ対照式試験法によって研究がされており、この試験では、偽薬、クロニジン(0.2mg、経口投与)、または、クロニジンおよびナロキソン(2.8mg、5時間にわたる点滴)が被験者に対して投与された。 経皮電気刺激を与えた後に、主観的疼痛閾値(視覚的アナログ尺度)と客観的侵害作用屈曲反射(R III)閾値の計測を行って、鎮痛作用を評価した。 主観的閾値と客観的閾値との間で相関が認められた(r:0.78)。 クロニジン単独を経口投与することで、あるいは、クロニジンとナロキソンとの組み合わせを経口投与することで、少なくとも4時間にわたって、主観的疼痛閾値と客観的疼痛閾値が増大した(0.01未満のP値、ANOVA). ナロキソンは、クロニジンの鎮痛作用を増強する傾向にあった。 適度で、良好な耐容性の副作用だけが認められた[32]。
【0045】
成人を対象とした研究において、術前、術中、または、術後に、クロニジンを投与することで、顕著な鎮痛効果が認められ、また、副作用の発現が少なくなる、ことが明らかにされている。 小児患者でのクロニジンの効果を決定するための研究も行われている。 無作為に選んだ1〜7歳の45名の小児患者にクロニジンを硬膜外投与したところ、術後のブピバカインの仙骨麻酔の効果が、1μg/kgのクロニジンを加えることで、顕著に改善したことが報告されている[20]。 身体の78%に第二度および第三度熱傷をおった11歳の少年に対して、大量のモルヒネを投与したところ重度の副作用が認められたが、低用量のクロニジンを静脈投与することで、その副作用は顕著に軽減された[24]。 しかしながら、小児患者に関する研究において、術前の鎮静状態下では、クロニジンの経口投与よりも、ミダゾラムの方が有効であるが、得られる鎮痛作用は、小児麻酔においても有効であると考えられる[29]。
【0046】
様々な薬剤が、補助鎮痛薬として使用されているが、それらは、もともも疼痛以外の主症状に対応するために開発されたものである。 これら薬剤は、特定の条件下で鎮痛作用を高めるために用いられており、また、それらの内の幾つかは、主に、鎮痛薬として用いられている[21]。 アミトリプチリン、ノルトリプチリン、および、デシプラミンのような三環系抗鬱薬は、たいていの神経因性疼痛に対して有効である[36]。 ブプロピオン、ベンラファクシン、および、デュロキセチンも、神経因性疼痛管理において有効であることが知られている[35、37]。 抗癲癇薬は、神経因性疼痛の管理に最も適した薬剤になりつつあり、ガバペンチンとプレガバリンの双方について、神経因性疼痛に対する効果が確認されている[2、12]。 クロニジンは、鎮痛剤として主作用の他に、オピオイドと併用することで、抗侵害受容効果をも相乗的に奏する[14]。 チザニジンは、比較的に短時間作用型のα2アドレナリンアゴニストであり、また、降圧作用がクロニジンに比べてかなり小さいが、疼痛障害において利用可能である[11]。 NMDAアンタゴニストデキストロメトルファン、メタドン、メマンチン、アマンタジン、および、ケタミンは、痛覚過敏神経症状態に対して効果的であると思われる[3]。
【0047】
抗侵害受容特性を有する非アヘン製剤であるクロニジンは、術後に鎮痛作用を示さないという副作用を有するオピオイドの代替物となるかもしれない。 脊椎固定術を行った直後の50名の患者を、二つのグループに無作為に振り分け、そして、クロニジン(最初の1時間に5μg/kgを点滴し、次いで、11時間かけて、0.3μg/kg/時間を点滴した)または偽薬のいずれかを無分別に投与をして、術後期間に静脈投与をしたクロニジンの鎮痛作用を決定するための研究が行われた。 クロニジンまたは偽薬を投与する以前(T0)、負荷用量の終了時(T1)、それに、その2時間ごと(T3、T5、T7、T9およびT11)に、疼痛を評価するために、視覚的アナログ尺度を用いて、0(無疼痛)〜100mmに等級付けをした。 スコアが50mmを超えた場合には、各々の疼痛測定の後に、モルヒネ(0.1 mg/kg)を、筋肉内に投与した。 T0の時点では、モルヒネは与えていない。 疼痛スコアを測定した各時点において、血行動態、血液ガス、および、血漿クロニジン濃度を測定した。 クロニジン投与グループでは、疼痛スコアが、42+/-5〜26+/-3mm(平均値+/-標準誤差)にまで減少したが、偽薬投与グループでは、モルヒネ要求量が多かったにもかかわらず(各患者に対する用量:3.8+/-1に対して10.8+/-1.2mg)、疼痛スコアに変化は認められなかった。 クロニジンは、疼痛の発症を遅らせるので、最初のモルヒネ注射の時期も遅れることとなる。 クロニジン投与グループでは、累積液量P1が顕著に増大するにもかかわらず、平均動脈圧は、74+/-2mmHg(T11での偽薬投与グループにおける-15+/-2%に対して、-26+/-2)にまで減少した。
【0048】
腹式子宮摘出術を受けた13名の患者に対してクロニジンを硬膜外投与した後の効果を決定するための研究が行われている。 血圧および口頭式類推疼痛スコアにおいて、顕著な減少が認められた。 硬膜外にクロニジン(150μg)を投与した後の鎮痛の程度は適度なものであり、また、作用は長続きはしなかったが、硬膜外の空間から血液へのクロニジンの吸収は非常に迅速であった[66]。 ある実験によれば、クロニジンとグアンファシンとの間に明確な血液動態的差異が認められないにもかかわらず、グアンファシンの方が、長時間にわたって痛覚抑制(グアンファシンの8時間に対して、クロニジンの5.5時間)を呈することが認められている。 長時間にわたる作用の持続性と呼吸抑制が少ない点に鑑みて、硬膜外へのグアンファシンの投与は、術後鎮痛と慢性疼痛症候群に対して有利であると言うことができる[67]。
【0049】
人工股関節全置換術を受けた患者を対象とした二重盲検式比較試験において、術後鎮痛に関するクロニジンの硬膜外投与の効果と、モルヒネの硬膜外投与に関するクロニジンの効果が検証されている。 二つの用量の硬膜外クロニジン(25μg/時間、または、50μg/時間)、低用量の硬膜外モルヒネ、または、モルヒネとクロニジンとの組み合わせのいずれかを無作為に、患者に対して投与した。 術後の最初1時間でのモルヒネを投与したグループの疼痛スコアは、クロニジン投与グループ(0.01未満のP値)と組み合わせ投与グループ(0.05未満のP値)のスコアと比べて非常に大きかった。 モルヒネ投与グループと低用量クロニジングループ(0.05未満のP値)と比較して、組み合わせ投与グループと多用量クロニジングループは、全身鎮静の必要性が最も少なく、また、最初のボーラス用量の持続効果も非常に大きかった。 クロニジングループでの臨床的低血圧症の兆候は顕著ではなかったが、動脈圧の減少が認められた。 嘔吐症状または尿閉に関して、グループ間での顕著な相違は認められなかった[70]。
【0050】
手術の1時間前および12時間後に、クロニジン(300μg)を経口投与したところ、モルヒネ要求性の改善はわずかであったが、腹部の大手術を受けた患者の心拍数を十分に下げ、また、鎮静状態を顕著に改善した。
【0051】
帝王切開分娩をした患者が参加した無作為化二重盲検式用量依存性試験で、硬膜外モルヒネに対してクロニジンを添加して得られる効果を調べた。 低用量の75μgのクロニジンが奏する鎮痛作用の持続期間が、2mgのモルヒネが示す持続期間の倍であり、また、術後でのモルヒネ要求性が、クロニジンを添加することで顕著に軽減されることが明らかとなった[68]。
【0052】
膝の整形外科的大手術を受けた成人患者での術後の鎮痛作用およびPCAモルヒネ要求性に関する術中のクロニジンの経口投与の効果が評価されている。 クロニジンは、偽薬と比較して、術後の吐き気と嘔吐の症状を顕著に軽減し、また、PCAモルヒネ要求性も顕著に軽減した[65]。 脊髄内モルヒネ麻酔を施して腹式単純子宮全摘術を受けた37〜60歳の26名の患者に対して、クロニジン(5μg/kg)を経口的に前投与したところ、モルヒネの副作用を増強せずに、術後のモルヒネの鎮痛作用が増大した[63]。 クロニジン(4μg/kg)を経口投与することで、帝王切開をした後でも、胎児や新生児の状態を危険にさらすことなく、PCAモルヒネ要求性を軽減することが明らかになった[64]。
【0053】
下腹部手術を受けた患者に対して、無作為化二重盲検式比較試験への参加を募った。 グループCの外科処置の最後の段階で、4μg/kg/20分のクロニジンの点滴と、20μgのPCAクロニジンと1mgのモルヒネのボーラス投与を行った。 グループMには、生理食塩水の点滴と、1mgのPCAモルヒネのボーラス投与を行った。 疼痛状態、鎮静状態、それに、吐き気と嘔吐を、12、24および36時間後に評価を行い、また、鎮痛効果に関する満足度を、36時間の時点で評価をした。 グループCでの疼痛スコアは、0時間と12時間との間では非常に小さかったが、その後は、大差がなかった。 24〜36時間に至るまでの双方のグループでのモルヒネ消費量は同じであった。 グループCでの0時間と24時間との間において、吐き気と嘔吐は、顕著に軽減されていた。 クロニジンを用いたグループの患者は、疼痛が緩和されたことを非常に喜んでいた[62]。
【0054】
椎間板手術後の疼痛緩和のために用いた硬膜外投与された低用量モルヒネとクロニジンとの組み合わせの効果と副作用についての研究が行われている。 モルヒネとクロニジンの硬膜外投与が、ブピバカインとクロニジンの硬膜外投与と比較して、術後疼痛を顕著に緩和し、そして、ピリトラミドの消費量も減少していたことが明らかになった[61]。
【0055】
冠動脈バイパス移植手術を受けた45名の患者を、自己調節鎮痛(PCA)モルヒネ(ボーラス、1mg;ロックアウト時間、7分間)だけを点滴で投与するグループ(コントロールグループ)、4μg/kgのモルヒネを髄腔内にさらに投与するグループ、あるいは、4μg/kgのモルヒネと1μg/kgのクロニジンの双方を髄腔内にさらに投与するグループへと無作為に分けて、無作為化二重盲検比較試験をした。 全身麻酔を行う以前に、髄腔内注射を行った。 視覚的アナログ尺度(VAS)を用いて、術後に疼痛の評価を行った。 モルヒネの用量[平均値(第一〜第三四分位数)]は、モルヒネとクロニジンを髄腔内投与された患者での最初の24時間の用量[7(0〜37)mg]の方が、その他の患者[モルヒネを髄腔内投与したグループでの40.5(15〜61.5)mg、および、モルヒネを点滴で投与したグループでの37(30.5〜51)mg]よりも、少なかった。 VASスコアは、コントロールグループと比較して、モルヒネとクロニジンを髄腔内投与した後の方が小さかった。 点滴でモルヒネを投与したグループと比較して、モルヒネとクロニジンを髄腔内投与した後の方が、抜管に要する時間が短かった[330(300〜360)分に対して225(195〜330)分、P<0.05]。 モルヒネとクロニジンの髄腔内投与を行うことで、冠動脈バイパス移植手術の術後において効果的な鎮痛効果を与えることができ、また、抜管の作業が迅速に行える[60]。
【0056】
クロニジン(120μg)とフェンタニル(50μg)との組み合わせは、分娩第一期に用いられている0.25%ブピバカインと同等の硬膜外鎮痛作用を呈し、また、好ましくない神経学的副作用も軽減する[59]。 しかしながら、クロニジンとモルヒネの組み合わせの硬膜外への投与は、ブピバカインとスフェンタニルを併用する自己調節療法と比較して、鎮痛作用が劣り、また、副作用も大きい[58]。 その他の研究によれば、硬膜外投与されたブトルファノールにクロニジンを加えても、その鎮痛作用に変化はなく、むしろ、開腹手術を受けた患者に悪影響を及ぼすとの報告もされている[57]。 健常者にクロニジンを経口的に前投与すると、感覚運動ブロックの長期化を招かずに、望ましい鎮静状態、不安緩解、それに、安定した血行動態をもたらす。 5μg/kg用量のクロニジンだけに副作用が認められ、2.5μg/kg用量のクロニジンでの副作用は、最小のものでしかなかった[56]。 クロニジン(0.75μg/kg)の使用は、臨床的に副作用が非常に小さいが故に、フェンタニル(0.5μg/kg)よりも望ましいことが証明されている[55]。 クロニジン(5μg/kg)を経口的に前投与すると、プロポフォールの導入量を減らすことができるが、プロポフォール麻酔の効果の出現が遅くなる[54]。
【0057】
関節鏡視下膝手術の術後に、クロニジン(150μg)を関節内に投与したところ、5mgのモルヒネと比較して、疼痛緩和が長期にわたって持続をした[53]。
【0058】
イミダゾリン活性を有する、または、イミダゾリン活性を欠いている、本願発明での使用が意図されている、その他のα2アドレナリンアゴニストとして、デクスメデトミジン、デトミジン、ST-91、メデトミジン、ブリモニジン、チザニジン、ミバゼロール、グアナベンズ、グアンファシン、ヨードクロニジン、キシラジン、リルメニジン、ロフェキシジン、アゼペキソール、アルファ-メチルドパ、および、アルファ-メチルノルアドレナリン、または、これらの誘導体、塩類、または、構造類似体などがあるが、これらに限定されない。
【0059】
エンドセリン受容体アンタゴニスト
ETAおよびETBの双方に結合するエンドセリン(例えば、ET-1、ET-2、および、ET-3)は、強力な血管収縮剤である。 エンドセリンは、プレプロホルモンとして合成され、そして、翻訳後プロセシングによって、活性ペプチドへと変化する。 ET-1プロセシングの特徴は周知であり、212個のアミノ酸のペプチド(プレプロET-1)から始まり、次いで、エンドペプチダーゼの作用を受けて、タンパク質分解的に開裂して巨大ET-1(プロET-1)となる。 39個のアミノ酸のプロET-1は、金属エンドプロテアーゼエンドセリン変換酵素(ECE)の作用を受けて開裂し、その結果、強力な生物学的機能を具備した21個のアミノ酸からなるタンパク質となる(Fagan et al., Respiratory Research 2:90-101, 2001)。
【0060】
本明細書で使用する「エンドセリンアンタゴニスト」および「エンドセリン受容体アンタゴニスト」の用語は、互換的に用いられている。 エンドセリン受容体アンタゴニストは、急性心不全、鬱血性/慢性心不全、肺動脈高血圧症、肺水腫、くも膜下出血、慢性閉塞性肺疾患、心筋梗塞、急性脳虚血、急性冠不全症候群、急性腎不全、肝臓手術の術後状態、および、前立腺癌を治療するために用いられている。
【0061】
幾つかの研究によって、構造的に異なるETA受容体アンタゴニストであるBQ123(ペプチド)とBMS182874(非ペプチド)が、マウスとラットの双方において、モルヒネの鎮痛反応を増強する、ことが明らかにされている[5〜7、16、26、33]。 交感神経系に関与する心臓血管系作用でのクロニジンとETとの間における相互作用についても報告がされている[15、17、18、28]。 エンドセリン-1(ET-1)が、クロニジンに起因する低血圧症を悪化させることも知られている[23]。 しかしながら、鎮痛作用を示すクロニジンに関するETA受容体アンタゴニストの効果に関する報告は皆無である。 スルフィソキサゾールが、ETAおよびETB受容体のそれぞれに対して、0.60μMおよび22μMのIC50値を示す最も活性に富んだスルファニルアミドである、ことが明らかにされている[8]。 したがって、ここでの研究は、クロニジンを併用して投与した場合における、鎮痛作用に関するスルフィソキサゾールの効果を決定するために行ったものである。 これら研究の結果は、クロニジンとスルフィソキサゾールの組み合わせが、高用量のモルヒネと同等の強力な鎮痛作用を示すことを実証している(実施例5を参照されたい)。
【0062】
弱いエンドセリンAアンタゴニストであるスルフィソキサゾールは、血漿タンパク質に広く結合し、そして、2〜4gの用量で経口投与をして2〜4時間後には、110〜250μg/mlというピーク値の血漿濃度に達する。 尿中のスルフィソキサゾールの濃度は、血中のスルフィソキサゾールの濃度よりも高く、また、脳脊髄液でのスルフィソキサゾールの濃度は、平均して、血中のスルフィソキサゾールの濃度の約1/3である。 大抵(95%)の薬剤は、24時間以内に、腎臓から尿中に排出される(Goodman Gilmans 1990 8th Edition).
スルフィソキサゾールの大半は、細胞外空間に存在しており、そして、脳脊髄液での含有濃度は、血中濃度の10〜80%の範囲の濃度で対応している(Goodman Gilmans 1990 8th Edition)。 血中濃度が50〜150μg/mlの遊離スルフィソキサゾールは、大抵の感染症に対して治療効果があるものと考えられており、また、血中濃度が120〜150μg/mlにまでなると、重篤感染症に対して好適であると考えられている。 200μg/mlの濃度を超えると副作用が頻繁に認められるようになるので、血中での最高濃度は、200μg/mlとすべきである(PDR 59th Edition 2005)。 1日に4度、健常者に対して、500mgを経口的に繰り返し投与をしたところ、定常状態でのスルフィソキサゾールの平均血漿濃度は、49.9〜88.8μg/mlの範囲であった(平均値63.4μg/ml)(Oie et al., J Pharmacokinetics Biopharm 1982: 10: 157-172)。
【0063】
プロポフォールとスルフィソキサゾールとの間の相互作用が、マウスにおいて認められている。 プロポフォールによる立ち直り反射の障害は、スルフィソキサゾールを用いて前処理することでさらに強くなった。 スルフィソキサゾールそれ自体は、何らの効果も示さず、また、プロポフォールの総血漿濃度およびタンパク質結合についても何らの効果も示さなかった[51]。
【0064】
スルフィソキサゾールが、一酸化窒素上昇を抑え、そして、リポ多糖類(LPS)に起因するGABA含有ニューロンの数を減らすことで、緑内障に認められる虚血性傷害から網膜を保護することが知られている[52]。 クロニジンの局所投与によって、α2-アドレナリン受容体が刺激を受けて、ラットの網膜が、虚血/再灌流から保護されることが知られており、この保護効果は、α2-アドレナリン受容体の関与を確認するヨヒンビンまたはラウオルシンによって選択的に弱められる[50]。
【0065】
幾つかのETA受容体アンタゴニストについて、肺動脈高血圧症、鬱血性心不全、脳卒中、バルーン血管形成後の冠動脈の再閉鎖、ならびに、高血圧症の治療、および、癌性疼痛管理での使用に向けた評価が行われているので、クロニジンとスルフィソキサゾールに関する研究は臨床的に重要である。 本明細書で示した結果は、弱いETA受容体アンタゴニストであるスルフィソキサゾールを、α2アドレナリンアゴニストであるクロニジンと共に用いることで、予期せぬ鎮痛作用が得られることを示す証拠に他ならない。 その他のETA受容体アンタゴニストとクロニジンなどのα2アドレナリンアゴニストとを併用した研究も行って、これら薬剤の組み合わせが、鎮痛作用を増大するかどうか、そして、これら組み合わせが、安全な非麻薬性鎮痛薬として使用できるかどうかを見極めることも意図している。 本明細書に記載の結果は、痛覚を変化させることのできる新たな標的を提供するのみならず、患者の慢性疼痛の新たな管理手法をもたらすかもしれない。
【0066】
本願発明において有用なエンドセリン受容体アンタゴニストの例として、スルフィソキサゾール、アトラセンタン、テゾセンタン、ボセンタン、シタキスセンタン、エンラセンタン、BMS207940(ブリストル・マイヤーズ スクイブ)、BMS193884、BMS182874、J 104132(萬有製薬)、VML 588/Ro 61 1790(バンガード・メディカ)、T-0115(田辺製薬)、TAK 044(タケダ)、BQ 788、TBC2576、TBC3214、PD180988、ABT 546、SB247083、RPR118O31A、および、BQ123などがあるが、これらに限定されない。 BQ123は、エンドセリンAアンタゴニストの具体例であって、このものは、シクロ(D-Trp-D-Asp-Pro-D-Val-Leu)のナトリウム塩である。 その他の有用なエンドセリンアンタゴニストとして、YM 598、LU 135252、PD 145065、A 127722、ABT 627、A 192621、A 182086、TBC3711、BSF208075、S 0139、および、SB209670という名称のものがあるが、これらに限定されない。 その他の有用なエンドセリンAアンタゴニストは、本明細書の一部を構成するものとしてそれらの内容を援用する米国公開特許公報第US 2002/0082285 Al号および米国特許出願第10/659,579号に開示されている。
【0067】
従来のエンドセリン受容体アンタゴニストに加えて、内因性エンドセリンの形成を阻害する化合物も、本願発明のエンドセリン受容体アンタゴニストとして使用することができる。 これら化合物は、エンドセリンの形成を妨げ、それにより、エンドセリン受容体の活性を低下させるので、有用な化合物であると言える。 それら化合物のクラスの一つに、エンドセリン変換酵素(ECE)阻害剤がある。
【0068】
有用なECE阻害剤として、[DVal22、Phe33]巨大エンドセリン-1(16-38)、ヒト(すなわち、His-Leu-Asp-Ile-Ile-Trp-DVal-Asn-Thr-Pro-Glu-His-Val-Val-Pro-Tyr-Gly-Phe-Gly-Ser-Pro-Arg-Ser);[DVal22]巨大エンドセリン(16-38)、ヒト(すなわち、His-Leu-Asp-Ile-Ile-Trp-DVal-Asn-Thr-Pro-Glu-His-Val-Val-Pro-Try-Gly-Leu-Gly-Ser-Pro-Arg-Ser);[Phe22]巨大エンドセリン-1(19-37)、ヒト(すなわち、Ile-Ile-Trp-Phe-Asn-Thr-Pro-Glu-His-Val-Val-Pro-Tyr-Gly-Leu-Gly-Ser-Pro-Arg);および、ホスホラミドン(すなわち、N-(a-ラムノピラノシルオキシヒドロホスフィニル)-Leu-Trp)などがあるが、これらに限定されない。
【0069】
麻薬性鎮痛薬
入手可能なアヘン製剤およびオピオイド鎮痛薬は、五つの化合物グループ(すなわち、 フェナントレン、フェニルヘプチルアミン、フェニルピペリジン、モルフィナン、および、ベンゾモルファン)の誘導体である。 薬理学的には、アヘン製剤と非アヘン製剤とは、活性が顕著に異なる。 幾つかは、強力なアゴニスト(モルヒネ)であるが、その他は、中程度〜軽度のアゴニスト(コデイン)である。 これに対して、幾つかのアヘン製剤誘導体は、アゴニストとアンタゴニストの混合活性を示す(ナルブフィン)のに対して、その他の誘導体は、アヘン製剤アンタゴニスト(ナロキソン)である。 モルヒネは、アヘン製剤およびオピオイド鎮痛薬の原型であり、そのすべてが、中枢神経系に対して同様の作用を及ぼす。
【0070】
モルヒネは、アヘンから化学的に誘導される。 ヘロインなどのその他の薬物は、モルヒネまたはコデインから加工される。 このようなアヘン製剤は、何世紀にもわたって、医学的におよび非医学的に用いられてきた。 19世紀初頭まで、モルヒネは、溶解に適した純粋な形で抽出されていた。 皮下注射針の普及に伴い、モルヒネ溶液の注射投与が、一般的な投与方法となった。 アヘンに含まれる20ものアルカロイドの内、コデインとモルヒネだけが、臨床的に広く利用されている。
【0071】
それらの内でも、麻薬のアヘンは、最も強力に作用する薬剤であり、中枢神経系を抑制するなど臨床的に有用な薬剤でもある。 このグループに属する薬剤は、基本的には、鎮痛薬として用いられるが、その他にも無数の有用な特性を具備している。 例えば、モルヒネは、疼痛を緩和し、疼痛時に睡眠を促し、下痢を止め、咳を鎮め、呼吸を楽にし、そして、麻酔の効果を助長するために使われている。
【0072】
モルヒネやその関連化合物を長期間にわたって投与をすると、鎮痛作用に対する耐性が出現し、そして、同様の疼痛緩和効果を得るためには、定期的に用量を増量しなくてはならなくなる。 そして、最終的には、陶酔感と共に耐性や身体的依存性が強くなり、薬物の過剰摂取を経て、過敏な人格を有する中毒患者となってしまう。 このような理由から、モルヒネやその誘導体は、医師の処方のみに従って使用しなくてはならず(すなわち、処方された以上の用量、頻度、または、期間を超えて使用してはならない)、また、他の鎮痛剤で十分に対処できる場合には、それらを疼痛を治療する目的で使用すべきではない。
【0073】
一つまたはそれ以上のアルファ-2(α2)アドレナリンアゴニスト、および/または、一つまたはそれ以上のエンドセリンアンタゴニストは、麻薬性鎮痛薬の鎮痛作用を増強する上で有用であり、また、そうすることも意図している。 さらに、一つまたはそれ以上のアルファ-2(α2)アドレナリンアゴニストと一つまたはそれ以上のエンドセリンアンタゴニストとの組み合わせは、麻薬性鎮痛薬の鎮痛作用を増強する上で有用であり、また、そうすることも意図している。
【0074】
麻薬性鎮痛薬として、(a)アヘン;(b)モルヒネ、硫酸モルヒネ、コデイン、リン酸コデイン、硫酸コデイン、ジアセチルモルヒネ、塩酸モルヒネ、酒石酸モルヒネ、および、塩酸ジアセチルモルヒネなどのアヘンアルカロイド;および、(c)臭化水素酸デキストロメトルファン、酒石酸水素ヒドロコドン、ヒドロモルフォン、塩酸ヒドロモルフォン、酒石酸レボルファノール、塩酸オキシモルホン、および、塩酸オキシコドンなどの半合成麻薬性鎮痛薬などがあるが、これらに限定されない。
【0075】
医薬化合物の製剤
本願発明の活性成分、すなわち、本明細書に記載のα2アドレナリンアゴニストとエンドセリン受容体アンタゴニストは、それら単独で投与することができ、あるいは、意図している投与経路および標準的な薬務にしたがって選択した医薬担体との混合物として投与することができる。 よって、本願発明にしたがって用いられる医薬組成物は、薬学的に使用可能な調製物への活性成分の加工処理を円滑にする賦形剤や助剤を含む一つまたはそれ以上の生理学的に許容可能な担体を用いて、従来の方法によって、製剤することができる。
【0076】
これら医薬組成物は、従来の加工プロセス、例えば、混合、溶解、造粒、糖衣錠調製法、乳化、カプセル化、封入、または、凍結乾燥などのプロセスを用いて、従来の方法で製造することができる。 選択した投与経路に応じて、適切な製剤が調製される。
【0077】
本明細書に記載の活性成分を含む医薬組成物を意図しており、ある実施態様によれば、それら化合物は、薬学的に許容可能な希釈剤、補助剤、賦形剤、および/または、担体を用いて製剤される。 「薬学的または薬理学的に許容可能」という表現は、例えば、吸入噴霧、経膣的、経直腸的、または、頭蓋内注射によって、経口的、局所的、経皮的、非経口的に、動物、または、ヒトに対して投与をした時に、副作用、アレルギー反応、または、その他の有害な作用を招かない化合物および組成物のことを指す。 本明細書で用いる非経口の用語は、皮下注射、静脈投与、筋内、嚢内注射、または、点滴などを含む。 同様に、静脈投与、皮内、筋内、乳房内、腹腔内、関節内、髄腔内への投与、球後注射、肺内注射などによる投与、あるいは、特定部位への外科的移植をも意図している。 一般的に、調製された組成物は、発熱物質を含んでおらず、また、ヒトや動物に対して有害なその他の不純物も含んでいない。 「薬学的に許容可能な担体」の用語は、一部およびすべての溶剤、分散媒、コーティング剤、抗菌剤、および、抗真菌剤、等張剤、および、吸収遅延剤などを含む。 このような医薬活性物質のための媒体および薬剤は、当該技術分野において周知である。
【0078】
前述した本願発明の方法で用いる医薬組成物は、例えば、錠剤、トローチ、ドロップ、水性懸濁液または油性懸濁液、分散性粉末または分散性顆粒、エマルジョン、硬質カプセルまたは軟質カプセル、または、シロップ、または、エリキシル剤などの経口投与に適した形態にすることができる。 経口投与用組成物は、公知の方法で調製することができ、また、薬学的に好ましく、かつ、口当たりの良い調製物を提供すべく、それら組成物は、甘味料、着香料、着色料、および、保存料からなるグループから選択される一つまたはそれ以上の物質を含むこともできる。 錠剤には、その製造に適した非毒性の薬学的に許容可能な賦形剤と共に混合した活性成分を含めることができる。 これら賦形剤として、不活性希釈剤、例えば、炭酸カルシウム、炭酸ナトリウム、ラクトース、リン酸カルシウム、または、リン酸ナトリウムなど、造粒剤および崩壊剤、例えば、コーンスターチ、または、アルギン酸など、結合剤、例えば、澱粉、ゼラチン、または、アラビアゴムなど、滑剤、例えば、ステアリン酸マグネシウム、ステアリン酸、または、滑石などがある。 錠剤にはコーティングを施さなくともよく、あるいは、消化管での崩壊と吸収を遅らせて、それにより、長期間にわたって作用を持続させるために、公知の方法で錠剤にコーティングを施してもよい。 例えば、モノステアリン酸グリセリン、または、ジステアリン酸グリセリンなどの遅延物質を用いることができる。 また、放出制御のための浸透圧性治療用錠剤を形成するために、米国特許第4,256,108号、第4,166,452号、および、第4,265,874号に記載の技術でコーティングを施すこともできる。
【0079】
経口投与製剤は、活性成分が、不活性固体希釈剤、例えば、炭酸カルシウム、リン酸カルシウム、または、カオリンと共に混合されている硬ゼラチンカプセルとして、あるいは、活性成分が、水性媒体または油性媒体、液体パラフィン、または、オリーブオイルと共に混合されている軟ゼラチンカプセルとして、提供することもできる。 錠剤の形態で投与する場合には、この組成物に、ゼラチンなどの固体担体、あるいは、補助剤をさらに含ませることができる。 錠剤、カプセル、および、粉末は、本願発明の活性成分を、それらの約5%〜約95%の量で含み、好ましくは、それらの約25%〜約90%は、本願発明の化合物である。 液体の形態で投与する場合には、水、石油、または、動物または植物に由来する油脂などの液体担体を含ませることができる。 液体形態の組成物は、生理学的食塩水、デキストロース、または、その他の糖溶液、あるいは、グリコールをさらに含むことができる。 液体の形態で投与する場合には、この組成物は、約0.5重量%〜約90重量%の活性成分、好ましくは、約1重量%〜約50重量%の活性成分を含む。
【0080】
水性懸濁液は、水性懸濁液の製造に適した賦形剤と共に混合した活性成分を含むことができる。 賦形剤とは、懸濁化剤、例えば、カルボキシメチルセルロースナトリウム、メチルセルロース、ヒドロキシプロピルメチルセルロース、アルギン酸ナトリウム、ポリビニルピロリドン、トラガカントゴム、および、アラビアゴムなどであり、分散剤または湿潤剤は、天然リン脂質、例えば、レシチンなど、脂肪酸を有するアルキレンオキシドの縮合産物、例えば、ステアリン酸ポリオキシエチレンなど、または、長鎖脂肪族アルコールを有するエチレンオキシドの縮合産物、例えば、ヘプタデカエチレンオキシセタノールなど、または、ポリオキシエチレンソルビタンモノオレアートなどの脂肪酸およびヘキシトールから誘導された部分エステルを有するエチレンオキシドの縮合産物、または、脂肪酸および無水ヘキシトールから誘導された部分エステルを有するエチレンオキシドの縮合産物、例えば、ポリエチレンソルビタンモノオレアートなどである。 この水性懸濁液は、一つまたはそれ以上の保存料、例えば、エチル、または、n-プロピル、p-ヒドロキシ安息香酸など、一つまたはそれ以上の着色料、一つまたはそれ以上の着香料、および、ショ糖またはサッカリンなどの一つまたはそれ以上の甘味料なども含むことができる。
【0081】
油性懸濁液は、植物油、例えば、落花生油、オリーブ油、胡麻油、または、椰子油など、あるいは、液体パラフィンなどの鉱油において、活性成分を懸濁させて調製することができる。 この油性懸濁液は、増粘剤、例えば、蜜蝋、固形パラフィン、または、セチルアルコールを含むことができる。 良好な口当たりを呈する調製物を製造するために、前述した甘味料や着香料を加えることもできる。 これら組成物は、アスコルビン酸などの抗酸化剤を加えて保存することができる。
【0082】
加水して水性懸濁液を調製するのに適した分散可能な粉末および顆粒は、分散剤、または、湿潤剤、懸濁化剤、および、一つまたはそれ以上の保存料を伴った活性化合物を提供する。 好適な分散剤、または、湿潤剤、および、懸濁化剤として、先に例示したものなどがある。 その他の賦形剤、例えば、甘味料、着香料、および、着色料も加えることができる。
【0083】
本願発明において有用な医薬組成物は、水中油型エマルションの形態とすることもできる。 油相には、植物油、例えば、オリーブ油、または、落花生油、あるいは、鉱油、例えば、液体パラフィン、または、これらの混合物を用いることができる。 好適な乳化剤として、天然ゴム、例えば、アラビアゴム、または、トラガカントゴムなど、天然リン脂質、例えば、大豆、レシチン、および、脂肪酸から誘導されたエステルまたは部分エステルなど、無水ヘキシトール、例えば、ソルビタンオレイン酸モノエステルなど、および、エチレンオキシドを有する前記部分エステルの縮合産物、例えば、ポリオキシエチレンソルビタンオレイン酸モノエステルなどがある。 このエマルジョンに、甘味料や着香料も含めることができる。
【0084】
シロップ剤、および、エリキシル剤は、甘味料、例えば、グリセロール、プロピレングリコール、ソルビトール、または、ショ糖などを用いて調製することができる。 このような製剤は、鎮痛薬、保存料、および、着香料および/または着色料なども含むことができる。 これら組成物は、直腸に投与する坐薬の形態とすることもできる。 これら組成物は、常温では固体であるが、直腸温度では液状となり、つまり、直腸内で溶解して薬剤を放出するという好適な非刺激性賦形剤と薬剤とを混合して調製することができる。 そのような賦形剤として、例えば、ココアバターやポリエチレングリコールなどがある。
【0085】
この医薬組成物は、滅菌済の注射可能な水性または油性の懸濁液の形態とすることができる。 この懸濁液は、前述した好適な分散剤、または、湿潤剤、および、懸濁化剤を用いて、当該技術分野で周知の方法で調製をすることができる。 滅菌済の注射可能な調製物は、非毒性の非経口的に許容可能な希釈剤または溶剤、例えば、1,3-ブタンジオールの溶液などを用いて、滅菌済の注射可能な溶液または懸濁液とすることもできる。 利用することができる許容可能な賦形剤や溶剤として、水、リンガー溶液、生理食塩水などがある。 加えて、滅菌済の固定油が、従来より、溶剤または懸濁媒体として用いられている。 本願発明の目的を鑑みれば、合成モノグリセリド、または、合成ジグリセリドなどの無刺激性の固定油を用いることができる。 さらに、オレイン酸などの脂肪酸が、注射剤の調製において利用できることも知られている。
【0086】
注射に適した医薬品形態として、注射可能な滅菌済溶液または分散液を即座に調製するための滅菌済水性溶液または分散液、および、滅菌済粉末などがある。 すべての事例において、医薬品形態は、滅菌処理を行っていなければならず、また、容易に注射できる程度の流動性を有していなくてはならない。 また、この医薬品形態は、製造および貯蔵の条件下で安定でなくてはならず、また、細菌や黴などの微生物による汚染を受けないようにして保存しなければならない。 そして、例えば、水、エタノール、ポリオール(例えば、グリセロール、プロピレングリコール、および、液体ポリエチレングリコールなど)、これらの好適な混合物、および、植物油を含む溶剤または分散媒を、担体とすることができる。 また、例えば、レシチンなどのコーティング剤を使用したり、分散液の場合には所望の粒径を維持したり、そして、界面活性剤を使用することで、適切な流動性を維持することができる。 また、様々な抗生物質や抗真菌剤、例えば、パラベン、クロロブタノール、フェノール、ソルビン酸、チメロサールなどを用いることで、微生物の影響を回避することができる。 多くの事例において、等張剤、例えば、糖類や塩化ナトリウムなどを用いることが好ましい。 そして、吸収遅延剤、例えば、モノステアリン酸アルミニウムやゼラチンなどの組成物を用いることで、注射可能な組成物に対して吸収持続性を付与することができる。
【0087】
投与および投薬
本願発明の医薬組成物には、所定の治療目的を達成する治療有効量の活性成分を含んだ組成物が含まれている。 治療有効量の決定は、とりわけ、本明細書での開示を参照し、かつ、目的とする所望の薬効、投与経路、および、投与を受ける患者の病態などを考慮すれば、当業者が十分に対処できる事項である。
【0088】
正確な処方、投与経路、および、用量は、患者の病態を考慮して、個々の医師によって決定される。 投与量と投与間隔は、治療効果または予防効果を維持するに十分なレベルの活性成分を提供するために、個別に調整をすることができる。
【0089】
本明細書に記載の方法を用いた治療での対象動物を、哺乳動物とすることを意図している。 この哺乳動物として、ヒトの医学研究のために用いる非ヒト動物モデル、あるいは、家畜の他に、例えば、愛玩動物などのペットとして重要な動物などがある。
【0090】
医薬組成物の投与は、疼痛が発症する以前、発症している最中、または、発症した後に行うことができる。
【0091】
これら活性成分は、好適な経路、例えば、経口的、経頬的、吸入的、舌下、経直腸的、経膣的、腰椎穿刺による嚢内、経尿道的、経鼻的、経皮的、すなわち、経皮的または非経口的(静脈、筋内、皮下、および、冠動脈内を含む)経路をもってして投与される。 非経口的投与は、注射針やシリンジ、あるいは、POWDERJECT(商品名)のような高圧技術を用いて行うことができる。
【0092】
本願発明の活性成分は、それ単独で投与することができ、また、意図している投与経路や標準的な薬務に鑑みて選択をした医薬担体と混合してから投与することもできる。 よって、本願発明の用途に供する医薬組成物は、薬学的に使用可能な調製物への活性成分の取り込みを促す賦形剤や補助剤を含む一つまたはそれ以上の生理学的に許容可能な担体を用いて、従来の方法で、製剤をすることができる。 医薬組成物は、本明細書に記載され、かつ、当該技術分野で周知の従来の方法でもってして製造をすることができる。
【0093】
医薬組成物の投与量は、治療を受ける対象動物、対象動物の体重、疼痛の程度、投与方法、それに、処方医師の判断によって定まることとなる。
【0094】
獣医学的用途については、活性成分は、通常の獣医学的手順にしたがって、好適な許容可能な製剤として投与される。 獣医師であれば、個々の動物に対して最適の投与計画と投与経路を容易に決定することができる。
【0095】
ある実施態様によれば、ヒトの疼痛を治療または予防するために投与する場合、α2アドレナリンアゴニスト、および、エンドセリン受容体アンタゴニストそれぞれの経口投与量は、一般的には、平均的な成人の患者(70kg)で、毎日、約10〜約200mgであり、通常は、一日当たりの用量を、2〜3回に分けて投与する。 一般的な成人患者に対して、これら活性成分を麻薬性鎮痛薬と共に投与する場合、それぞれの錠剤またはカプセルは、薬学的に許容可能な好適な賦形剤または担体と共に、約0.1〜約200mgのオピオイド鎮痛剤、約5μg/kgまたは300μgのα2アドレナリンアゴニスト、および/または、約0.1〜約50mgのエンドセリンアンタゴニストを含んでおり、それらは、単一用量または複数回用量で、一日当たり一回または数回かけて投与がされる。 静脈投与、経頬投与、または、舌下投与のために必要な用量は、通常は、約0.1〜約10mg/kg/単一用量である。 実際には、医師が、個々の患者に対して最適な投与計画を決定するものであって、その用量は、個々の患者の年齢、体重、および、反応状況によって変化する。 上記した用量は、平均的な事例を例示したものでしかなく、個々の事例によっては、用量を増減させた方が好都合な場合も当然あり、そのような事例も、本願発明の範疇に含まれる。
【0096】
ある実施態様によれば、クロニジン、スルフィソキサゾール、または、クロニジン、および、スルフィソキサゾールを含む調製物は、錠剤またはカプセルの形態で調製される。
【0097】
クロニジンは、鎮痛作用を促す補助剤として、5μg/kgの用量、または、300μgの総量で経口的に用いられている。 概して、クロニジンを用いた臨床研究は、1〜5μg/kgの用量のクロニジンが、効果的に鎮痛作用をもたらすことを示している。 本明細書に記載のラットを用いた実施例は、2mg/kgのクロニジンと1000mg/kgのスルフィソキサゾールの組み合わせが、顕著な鎮痛作用を得る上で効果的であったことを示している。 スルフィソキサゾールは、2〜4グラムの用量で経口的に用いられている。
【0098】
ある実施態様によれば、α2アドレナリンアゴニストは、約50μg、約110μg、約150μg、約200μg、約250μg、約300μg、約350μg、約400μg、約450μg、約500μg、約750μg、または、約1000μgの用量で投与される。 関連する実施態様によれば、クロニジンは、 約10〜約500μg、約10μg〜約300μg、約75〜約300μg、または、約100〜約250μgの範囲の用量で投与される。
【0099】
さらに別の実施態様によれば、エンドセリン受容体アンタゴニストは、約0.1、約0.5、約1、約2、約3、約4、約5、約10、約15、約20、約25、約30、約35、約40、約45、または、約50mgの用量、あるいは、約0.1mg〜約50mgの範囲の用量で投与される。 関連する実施態様によれば、スルフィソキサゾールは、2〜4グラム、750mg〜1.5g、1〜2グラム、0.1〜3グラムの範囲の用量で投与される。
【0100】
疼痛を効果的に治療するために対象動物に投与するα2アドレナリンアゴニスト:エンドセリンアンタゴニストの比率を、1:500〜1:50,000の範囲にすることを意図している。 ある実施態様によれば、クロニジン:スルフィソキサゾールの比率は、鎮痛作用を効果的に得るためには、1:500〜1:50,000にすべきとされている。 さらに強力なエンドセリン受容体アンタゴニストを用いる場合には、クロニジン:エンドセリンアンタゴニストの比率を、1:100〜1:1000に変化させておくことを意図している。 本願発明は、アルファ-2 アドレナリンアゴニスト:エンドセリンアンタゴニストとの比率を、 1:500〜1:50,000、1:500〜1:20,000、1:500〜1:10,000、1:500〜1:5,000、1:500〜1:2,500、1:100〜1:1000、および、1:100〜1:500の範囲とした組成物の投与を行う。
【0101】
α2アドレナリンアゴニストとエンドセリンアンタゴニストを、同時または別個のいずれかで投与することを意図している。 さらに、α2アドレナリンアゴニストとエンドセリンアンタゴニストは、単一の組成物、または、別個の組成物で投与することができる。
【0102】
同時投与にあっては、一つまたはそれ以上のα2アドレナリンアゴニスト、および/または、一つまたはそれ以上のエンドセリン受容体アンタゴニストを含む組成物が、各成分が、同時に、あるいは、任意の順序で異なる時点で連続して投与されるように投与が行われる。 しかしながら、同時に投与が行われないにしても、ある実施態様によれば、所望の治療効果を得るべく、時間をあまり開けずに、両者の投与が行われている。 一つまたはそれ以上のα2アドレナリンアゴニスト、および/または、一つまたはそれ以上のエンドセリン受容体アンタゴニストを、別個の組成物で投与する場合において、ある実施態様によれば、最初の薬剤の投与の前または後に、他方の組成物を投与することも意図されている。 先行投与とは、処置の1日前(24時間前)から処置の30分前の範囲で、薬剤を投与することを指す。 さらに、他方の薬剤の投与に続いて、一方の薬剤を投与することも意図している。 後続投与とは、最初の薬剤を投与してから30分後から、最初の薬剤を投与してから1日後(24時間後)までに投与を行うことを指す。 30分後から24時間後の範囲には、30分、1、2、3、4、5、6、7、8、9、10、11、12、16、20、または、24時間の時点での投与を含むことができる。
【0103】
組成物の投与は、患者の疼痛症状に応じて行うことができる。 ある実施態様によれば、これら薬剤は、毎日2回、毎日、48時間毎、3日毎、4日毎、7日毎、または、14日毎に、何週間にもわたって、投与がされる。 さらに、必要に応じて、複数の箇所に投与を行うことができ、また、疼痛の箇所に対して全身的または局所的に投与することもできる。
【0104】
ある実施態様によれば、エンドセリン受容体アンタゴニストは、α2アドレナリンアゴニストよりも先に投与することが意図されている。 関連する実施態様によれば、α2アドレナリンアゴニストは、エンドセリン受容体アンタゴニストよりも先に投与がされる。
【0105】
一つまたはそれ以上のα2アドレナリンアゴニスト、および/または、一つまたはそれ以上のエンドセリンアンタゴニストが、麻薬性鎮痛薬と共に投与される場合、その麻薬性鎮痛薬は、α2アドレナリンアゴニストおよびエンドセリン受容体アンタゴニストよりも先に投与され、α2アドレナリンアゴニストおよびエンドセリン受容体アンタゴニストよりも後に投与され、あるいは、最初の薬剤の投与の後で、かつ、他方の薬剤が投与される以前に投与されるものであって、α2アドレナリンアゴニストまたはエンドセリン受容体アンタゴニストは、麻薬性鎮痛薬よりも先または後に投与がされる、ことも意図をしている。
【0106】
これら化合物の好ましい投与間隔と投与量は、当業者にとって自明の事項である。
【0107】
疼痛の治療方法
本願発明は、疼痛を経験している対象動物で認められる症状を軽減および治療するための方法を提供する。 ある実施態様によれば、本願発明は、治療有効量のα2アドレナリンアゴニストと治療有効量のエンドセリン受容体アンタゴニストを哺乳動物に対して投与することを含む、疼痛を治療または予防する方法を提供する。 関連する実施態様によれば、疼痛の治療は、α2アドレナリンアゴニストとエンドセリン受容体アンタゴニストに加えて、麻薬性鎮痛薬の投与をさらに含む。
【0108】
疼痛の原因として、炎症、負傷、疾病、筋肉の痙攣、および、神経障害または神経障害症候群の発症などがあるが、これらに限定されない。 急性疼痛は、通常、自然に治癒するものであるが、慢性疼痛は、一般的には、三ヶ月またはそれ以上の期間にわたって疼痛が持続するので、患者の性格、生活様式、機能的能力、それに、全体的な生活の質に変化を及ぼすことがある。 疼痛が効果的に治療できないと、身体機能が制限され、運動機能が減退し、不眠になり、そして、一般的な生活の質が損なわれるなどの体験を経て、患者に悪影響が及ぶこととなる。
【0109】
外科手術や、有害な物理的、化学的、または、熱的現象、あるいは、生物製剤による感染などに起因して組織が損傷を受けると、炎症性(侵害性)の疼痛が起こる。 神経因性疼痛は、神経系、末梢神経、後根神経節、または、後根、あるいは、中枢神経系の損傷に起因する持続性または慢性の疼痛症候群である。 神経因性疼痛症候群として、異痛、そして、ヘルペス後神経痛および三叉神経痛などの様々な神経痛、幻想痛、それに、反射性交感神経性ジストロフィーや灼熱痛などの複合性局所疼痛症候群がある。 灼熱痛は、痛覚過敏症と異痛を伴う不随意的な灼熱痛を特徴としている。 痛覚過敏症は、疼痛性刺激に対する異常過敏を特徴としている(Meller et al., Neuropharmacol. 33:1471-8, 1994)。
【0110】
この病態は、内臓器官に疼痛の感覚をもたらす内臓痛覚過敏症を含んでいる。 また、神経因性疼痛は、通常は無害な刺激が長期間にわたって与えられた場合に、激しい疼痛を引き起こす痛感過敏症も含んでいる。
【0111】
ある実施態様によれば、治療をする疼痛は、急性疼痛または慢性疼痛である。 関連する実施態様によれば、疼痛は、灼熱痛、接触性アロディニア、神経因性疼痛、痛覚過敏症、痛感過敏症、炎症性疼痛、術後疼痛、慢性腰痛、群発頭痛、帯状疱疹後神経痛、幻肢痛および断端痛、中心性疼痛、歯痛、神経因性疼痛、オピオイド耐性疼痛、内臓痛、術後疼痛、骨損傷痛、糖尿病性神経障害疼痛、術後または外傷性神経症疼痛、末梢神経障害疼痛、エントラップメント神経障害疼痛、アルコール依存症に起因する神経障害、HIV感染、多発性硬化症、甲状腺機能低下症に起因する疼痛、または、抗癌化学療法に起因する疼痛、分娩中の疼痛、日焼けを含む火傷に起因する疼痛、産後疼痛、偏頭痛、咽喉痛、および、膀胱炎などの尿生殖路関連疼痛からなるグループから選択される。
【0112】
α2アドレナリンアゴニストとエンドセリン受容体アンタゴニストとを組み合わせることで、各化合物をそれぞれ単独で用いた場合と比較して、各化合物の用量の減少という相乗効果を意図している。 さらに、α2アドレナリンアゴニストまたはエンドセリン受容体アンタゴニストのいずれか一方だけを投与することで、オピオイド鎮痛剤の効果を強めることができ、それにより、疼痛の症状を効果的に治療するために必要なオピオイドの用量を減少させることを意図している。 また、α2アドレナリンアゴニストとエンドセリン受容体アンタゴニストの双方を、麻薬性鎮痛薬と共に投与しても、オピオイドの効果を強めることができ、そして、疼痛を緩和するために必要なオピオイドの用量を減少させることを意図している。
【0113】
ヒト患者での慢性疼痛の治療は、一般的には、米国特許第6,372,226号に記載されているようにして行われている。 ある実施態様によれば、急性炎症性疼痛、神経因性疼痛、痙攣状態、または、障害に起因するその他の慢性疼痛を経験している患者は、髄腔内投与、例えば、本願発明の方法で用いる適切な用量の本明細書に記載の組成物を腰部に脊椎穿刺して投与することで治療がされている。 その他の実施例では、対象動物が関節炎やその他の関節疼痛を患っている場合に、組成物を、関節内に投与している。 具体的な用量や注射する部位、それに、投与頻度などは、様々な要素を勘案して決められるものであって、それらは、治療にあたる医師の裁量の範囲である。
【0114】
疼痛症状の改善度は、視覚的アナログ尺度(VAS)、口頭式評価スケール(VRS)、および、数値化スケール(NRS)(Williamson et al, J Clin Nurs. 14:798-804, 2005; Carlsson, A., Pain. 1983 16:87-101, 1983)などの当該技術分野で周知の方法を利用して検査をした。 視覚的アナログ尺度、口頭式評価スケール、および、数値化スケールにあっては、一般的に、疼痛刺激を与える前後に、疼痛に関する数値について、患者に対して質問がされる。 慢性疼痛は、神経障害の症状および兆候に関する疼痛スケールであるリーズ評価法(LANSS)(Bennett, M. Pain. 92:147-157, 2001)などの客観的スケール試験によっても評価がされる。 α2アドレナリンアゴニストとエンドセリン受容体アンタゴニストを含む組成物で治療をした後に、疼痛刺激に対する過敏性が抑制されていることは、それらの活性が影響を及ぼしていること、そして、α2アドレナリン受容体、および/または、エンドセリン受容体が、慢性疼痛に関連する症状を緩和していることを指し示すものである。 本願発明のその他の実施態様によれば、本明細書に記載の組成物は、前述したその他の鎮痛薬と組み合わせて投与がされており、そのような治療を行うことで、慢性疼痛の症状の緩和において相乗効果が認められている。
【0115】
鎮痛剤を投与した後の様々な時点において疼痛の改善についての検査を行い、そして、検査結果に基づいた疼痛の解消度を評価した。 ある実施態様によれば、疼痛の症状の評価を、1、2、3、4、5、6または8週目の各週に行い、あるいは、担当医師が評価を行う。 ある実施態様によれば、対象動物での疼痛症状は、治療を行う以前の疼痛症状の評価結果と比較して、当該技術分野で周知の疼痛スケールを用いて決定したところでは、少なくとも10%、少なくとも20%、少なくとも25%、少なくとも30%、少なくとも35%、少なくとも40%、少なくとも45%、少なくとも50%、少なくとも55%、少なくとも60%、少なくとも65%、少なくとも70%、少なくとも75%、少なくとも80%、少なくとも85%、少なくとも90%、少なくとも95%、または、100%にまで改善することができる。
【0116】
キット
さらに別の実施態様として、本願発明は、本願発明の方法の実施にあたって円滑に利用可能に包装された一つまたはそれ以上の化合物または組成物を含むキットに関する。 最も単純な実施態様によれば、このキットは、密閉したボトルや瓶などの容器に包装された本願発明の方法の実施にあたって有用な本明細書に記載の化合物または組成物(例えば、α2アドレナリンアゴニストを含む組成物、および、エンドセリン受容体アンタゴニストを含む組成物、または、α2アドレナリンアゴニストとエンドセリン受容体アンタゴニストの双方を含む組成物)と、本願発明の方法を実施するにあたっての化合物または組成物の使用についての記載がされ、かつ、容器に貼着されているか、あるいは、キットに収容されているラベルを具備している。 好ましくは、本願発明の化合物または組成物は、単位投与用量で包装されている。 このキットは、好ましい投与経路で組成物の投与を行うために、それに適した器具をさらに具備することができる。 その他の実施態様によれば、このキットは、一つまたはそれ以上のオピオイド鎮痛薬を具備することができる。
【0117】
本願発明のその他の態様や詳細は、本願発明の限定を意図したものではなく、その例示を目的とした以下の実施例から自明であろう。
【実施例】
【0118】
実施例1:試料および方法
動物: 25〜30gの体重のオスのスイス・ウェブスターマウス(ハーラン社、インディアナポリス、インディアナ州)を用いた。 この動物を、室温(23±1℃)、湿度(50±10%)、および、12時間明暗サイクル(午前6時〜午後6時)が調節された部屋で、5匹/カゴずつ収容をした。 食餌と水は、自由に摂らせた。 この環境に動物が順応するまで、少なくとも4日間が過ぎてから、実験を開始した。 実験用の動物の飼育と使用については、動物実験委員会の許可を得た。 すべての麻酔処置と外科処置は、動物実験委員会のガイドラインを遵守して行った。
【0119】
薬剤: 硫酸モルヒネ(マリンクロットケミカル社、セントルイス、ミズーリ州)を、発熱物質を含んでいない蒸溜・脱イオン水に溶解し、そして、皮下(s.c.)に注射をした。 4-アミノ-N-(3,4-ジメチルオキサゾール-5-イル)-ベンゼンスルホンアミド(シグマケミカル社、セントルイス、ミズーリ州)であるスルフィソキサゾールを、カルボキシメチルセルロースに溶解し、そして、経口投与をした。 N-(2,6-ジクロロフェニル)-4,5-ジヒドロ-1H-イミダゾ−ル-2-アミン(シグマケミカル社、セントルイス、ミズーリ州)であるクロニジンを、滅菌生理食塩水に溶解し、そして、腹腔内(i.p.)に注射をした。
【0120】
統計値: すべてのデータ値は、平均値±SEMで示してある。 グループ内およびグループ間の差異を検証するために、ANOVAに続いて、ポストホックテスト(ボンフェローニの検定)を用いた。 P<0.05のレベルを、有意とした。
【0121】
実施例2:テイルフリック潜時の決定
モルヒネに対する抗侵害受容反応については、ダムールとスミスのテイルフリック潜時試験法[10]によって決定を行った。 動物の尻尾に熱刺激(収束光線)を与えると、即座に俊敏な動きで尻尾を引っ込める動作を示した。 この動作の反応時間を、無痛覚計を用いて検査して、テイルフリック潜時として記録をした。 熱刺激(収束光線)に対するテイルフリック潜時を、モルヒネまたは生理食塩水の注射を行う前と、注射を行ってから、30、60、90、120、180、および、240分後に決定をした。 尻尾に損傷を与えないようにすべく、カットオフ時間を、10秒間に設定した。 テイルフリック潜時から基礎潜時を差し引き、そして、その差異を、曲線下の面積(AUC)を算出するために用いた。 各マウスで得られた抗侵害受容反応値を、AUC0→240分に変換し、そして、平均±標準誤差として表した。
【0122】
実施例3:モルヒネ痛覚抑制に関するクロニジンの効果の決定
モルヒネが示す痛覚抑制に関するクロニジンの効果を決定するために、マウスを、次の4つのグループに分けた。 グループ1:賦形剤(生理食塩水、腹腔内投与)+賦形剤(生理食塩水、皮下投与)、グループ2:賦形剤+モルヒネ(4mg/kg、皮下投与)、グループ3;クロニジン(2mg/kg、腹腔内投与)+賦形剤(生理食塩水、皮下投与)、および、グループ4;クロニジン(2mg/kg、腹腔内投与)+モルヒネ(4mg/kg、皮下投与)。 モルヒネまたは賦形剤は、クロニジンを投与して30分後に投与をした。
【0123】
薬剤の投与を行わないテイルフリック基礎潜時値は、1.5〜2.3秒であった。 コントロールグループ(賦形剤+賦形剤)では、テイルフリック潜時は、4時間にわたって、基礎潜時から変化しなかった。 しかしながら、モルヒネ(4mg/kg、皮下投与)は、テイルフリック潜時を顕著に増大した。 クロニジン(2mg/kg、腹腔内投与)は、テイルフリック潜時を増大し、また、クロニジンで治療をしたマウスにモルヒネを投与したところ、モルヒネと比較をして、テイルフリック潜時をさらに増大した(図1A)。 モルヒネ(4mg/kg、皮下投与)で治療をしたグループで認められたAUCは、88.4±12.4であった。 クロニジンは、鎮痛作用を示し、そして、68.4±12.3のAUCを示したが、この数値は、コントロールが示す数値よりも遙かに大きな数値であった。 クロニジンで前処理をしたところ、モルヒネの鎮痛作用が顕著に増大し、また、125.4± 12.3のAUCが認められた。 クロニジンは、モルヒネがもたらす痛覚抑制を顕著に改善した(42%)(図1B)。
【0124】
実施例4:モルヒネ痛覚抑制に関するスルフィソキサゾールの効果の決定
モルヒネが示す痛覚抑制に関するスルフィソキサゾールの効果を決定するために、マウスを、次の4つのグループに分けた。 グループ1:賦形剤(カルボキシメチルセルロース(CMC)、経口投与)+賦形剤(生理食塩水、皮下投与)、グループ2:賦形剤(CMC)+モルヒネ(4mg/kg、皮下投与)、グループ3:スルフィソキサゾール(500mg/kg、経口投与)+賦形剤(生理食塩水、皮下投与)、および、グループ4:スルフィソキサゾール(500mg/kg、経口投与)+モルヒネ(4mg/kg、皮下投与)。 モルヒネまたは賦形剤は、スルフィソキサゾールを投与して30分後に投与をした。
【0125】
薬剤の投与を行わないテイルフリック基礎潜時値は、2.0〜2.4秒であった。 コントロールグループ(賦形剤+賦形剤)では、テイルフリック潜時は、4時間にわたって、基礎潜時から変化しなかった。 しかしながら、モルヒネ(4mg/kg、皮下投与)は、テイルフリック潜時を顕著に増大した。 スルフィソキサゾール(500mg/kg、経口投与)は、テイルフリック潜時を増大した。 しかしながら、スルフィソキサゾール(500 mg/kg、経口投与)で前処理を行っても、マウスにおいてテイルフリック潜時を増大するモルヒネの効果には影響が及ばなかったことは、500mg/kgの用量のスルフィソキサゾールを経口投与しても、モルヒネの鎮痛作用を増強しないことを示すものである(図2A)。 スルフィソキサゾール(56.0±9.1)が、コントロール(0.3±6.4)と比較してAUCを増大していることは、スルフィソキサゾールが、軽度の鎮痛作用を呈することを示すものである。 モルヒネ(4mg/kg、皮下投与)は、顕著な鎮痛作用を示し、また、そのAUCは、91.0±10.8であった。 しかしながら、スルフィソキサゾールは、モルヒネの鎮痛作用を実質的に増大するまでには至らず(AUC 103.8±11.3)、モルヒネが呈する痛覚抑制を、わずかに14%だけ増強したに過ぎなかった(図2B)。
【0126】
実施例5:モルヒネ痛覚抑制に関するクロニジンとスルフィソキサゾールの併用効果の決定
モルヒネが示す痛覚抑制に関するクロニジンとスルフィソキサゾールとの組み合わせが奏する効果を決定するために、マウスを、次のグループに分けた。
【0127】
試験1。 マウスを、次の三つのグループに分けた。 グループ1:クロニジン(2mg/kg、腹腔内投与)+スルフィソキサゾール(500mg/kg、経口投与)+賦形剤(V)(生理食塩水、皮下投与)、グループ2:クロニジン(2mg/kg、腹腔内投与)+スルフィソキサゾール(500mg/kg、経口投与)+モルヒネ(8mg/kg、皮下投与)、グループ3:賦形剤(生理食塩水、腹腔内投与)+賦形剤(カルボキシメチルセルロース、経口投与)+モルヒネ(8mg/kg、皮下投与)、および、グループ4:賦形剤(生理食塩水、腹腔内投与)+賦形剤(カルボキシメチルセルロース、経口投与)+賦形剤(生理食塩水、皮下投与)。 モルヒネまたは賦形剤は、クロニジンとスルフィソキサゾールを投与して30分後に投与をした。
【0128】
試験2。 マウスを、次の六つのグループに分けた。 グループ1:賦形剤(生理食塩水、腹腔内投与)+賦形剤(カルボキシメチルセルロース(CMC))+賦形剤(生理食塩水、皮下投与)、グループ2:クロニジン(2mg/kg、腹腔内投与)+賦形剤(CMC)+賦形剤(生理食塩水、皮下投与)、グループ3:スルフィソキサゾール(1000 mg/kg、経口投与)+賦形剤(生理食塩水、腹腔内投与)+賦形剤(生理食塩水、皮下投与)、グループ4:賦形剤(生理食塩水、腹腔内投与)+賦形剤(CMC)+モルヒネ(8mg/kg、皮下投与)、グループ5:クロニジン(2mg/kg、腹腔内投与)+スルフィソキサゾール(1000mg/kg、経口投与)+賦形剤(生理食塩水、皮下投与)、および、グループ6:クロニジン(2mg/kg、腹腔内投与)+スルフィソキサゾール(1000mg/kg、経口投与)+モルヒネ(8mg/kg、皮下投与)。 モルヒネまたは賦形剤は、クロニジンとスルフィソキサゾールを投与して30分後に投与をした。
【0129】
薬剤の投与を行わないテイルフリック基礎潜時値は、1.5〜2.0秒であった。 コントロールグループ(賦形剤+賦形剤)では、テイルフリック潜時は、4時間にわたって、基礎潜時から変化しなかった。 しかしながら、スルフィソキサゾール(500mg/kg、経口投与)とクロニジン(2mg/kg、腹腔内投与)とを併用すると、モルヒネが無くとも、テイルフリック潜時を顕著に増大した。 スルフィソキサゾール(500mg/kg、経口投与)とクロニジン(2mg/kg、腹腔内投与)とを併用して投与しても、モルヒネの効果に変化はなく、また、テイルフリック潜時にも変化は認められなかった(図3)。 カットオフ時間が10秒間に設定されており、また、本実施例での技術では、10秒を超えた後には何らの変化も認められなかったので、さらに別の増強作用を認めることは不可能であった。 クロニジンとスルフィソキサゾールという二つの非アヘン製剤を一緒に投与したところ、高用量のモルヒネに匹敵する鎮痛作用を呈したということは、非常に興味深いことであった。 さらに、高用量のスルフィソキサゾール(1000mg/kg、経口投与)を単独で、あるいは、クロニジン(2mg/kg、腹腔内投与)と組み合わせて利用した研究も行った。 コントロールのマウスと比較して、クロニジン(2mg/kg、腹腔内投与)は、テイルフリック潜時のAUCを増大し、同様に、コントロールのマウスと比較して、スルフィソキサゾール(1000mg/kg、経口投与)も、テイルフリック潜時のAUCを増大していた。 モルヒネ(8mg/kg、皮下投与)は、テイルフリック潜時を顕著に増大しており、AUCは、130.9±19.2であった。
【0130】
スルフィソキサゾール(1000mg/kg、経口投与)とクロニジン(2mg/kg、腹腔内投与)を併用したところ、テイルフリック潜時の顕著な増大が認められたことは興味深いことであり、また、このグループで認められたAUC(119.1±8.9)は、8mg/kgという高用量のモルヒネでのAUCと同等の数値であった。 クロニジンとスルフィソキサゾールを同時に投与した場合、クロニジンまたはスルフィソキサゾールのいずれか一方だけを投与した場合と比較して、AUCが、167%増大していた。 これら知見は、クロニジンとスルフィソキサゾールという二つの非アヘン製剤を一緒に投与することで、モルヒネに匹敵する痛覚抑制が実現可能となる、ことを確認するものであった(図4)。
【0131】
実施例6:鎮痛作用に関するクロニジンの用量応答効果
鎮痛作用に関するクロニジンの用量応答効果を決定するために、0.3mg/ml、1.0mg/ml、および、3.0mg/mlのクロニジンをマウスに投与し、そして、鎮痛作用に関する効果の評価を行った。
【0132】
図5Aおよび図5Bには、鎮痛作用に関するクロニジンの用量応答効果(テイルフリック潜時)と、賦形剤(生理食塩水)、0.3mg/ml、1.0mg/ml、および、3.0mg/mlのクロニジンのいずれかを投与したマウスの体温が示されている。 実験結果は、3.0mg/mlのクロニジンのテイルフリック潜時が、賦形剤について計測をしたすべての時点で得られたテイルフリック潜時の約5倍の数値であったことを示している。 1.0mg/mlのクロニジンのテイルフリック潜時は、刺激を与えて約3時間までは、約5倍の数値を示していたが、約6時間の時点では、賦形剤について得られたテイルフリック潜時の3倍の数値にまで減少していた。 0.3mg/mlのクロニジンのテイルフリック潜時は、驚くべきことに、刺激を与えた後は、約4倍の数値にまで増大をしていたが、約3時間の時点では、賦形剤について得られたテイルフリック潜時の約3倍の数値にまで減少し、そして、約5時間の時点では、2.5倍の数値にまで減少していた。 これらの結果は、鎮痛作用に関するクロニジンの効果は、用量依存的であり、そして、0.3mg/kgという低用量のクロニジンでも、鎮痛作用を得る上で効果的であることを実証している。
【0133】
用量応答に関するこの研究は、すべての用量のクロニジンが、治療を施した動物の体温を同様に下げていることを示している(図5B)。
【0134】
実施例7:鎮痛作用に関するクロニジンとスルフィソキサゾールの用量応答効果
前述したように、クロニジンについては、治療を施した動物での鎮痛作用に関する用量応答効果が実証されている。 治療計画にスルフィソキサゾールを加えても用量応答効果が得られるかどうかを決定するために、0.3mg/kgのクロニジンを、様々な用量のスルフィソキサゾールと共に投与をした。 0.3mg/kgのクロニジンと、0.5%CMC(10ml/kg)、あるいは、250、500、または、1000mg/kgのスルフィソキサゾールのいずれかとで、マウスの治療にあたった。 その結果、クロニジンと、0.5%CMC、または、いずれかの用量のスルフィソキサゾールとを用いた場合に、3時間後には、体温が、37℃から約32℃にまで同様に低下し(図6B)、そして、図5Bに示したように、すべての用量のクロニジンに匹敵するような体温の下げ方が認められた。 鎮痛作用に関するテイルフリック潜時検査において、0.3mg/kgのクロニジンとCMCの組み合わせは、刺激を与えて2時間後には、テイルフリック潜時を5倍にまで増大したが、6時間を経過するまでには、賦形剤で治療を施した動物でのテイルフリック潜時と大差のないレベルにまで減少した。 クロニジンが誘発した鎮痛作用に関するスルフィソキサゾールの効果は、用量依存的ではなかった(図6A)。 250、500、または、1000mg/kgのいずれかの濃度のスルフィソキサゾールを、0.3mg/kgのクロニジンと共に投与すると、刺激を与えて2時間後には、テイルフリック潜時は5倍にまで増大し、そして、この鎮痛作用のレベルは、刺激を与えて6時間後もなお維持されていた。
【0135】
これら結果は、クロニジンの鎮痛作用は、スルフィソキサゾールによって増強されるものであって、この効果は、最低用量(250mg/kg)のスルフィソキサゾールであっても認められるものである、ことを示している。
【0136】
クロニジンの鎮痛作用を増強することができるスルフィソキサゾールの最低用量を決定するために、別の研究を行った。 クロニジン(0.3mg/kg)と、0.5%CMC、または、25、75、または、225mg/kgの濃度のスルフィソキサゾールのいずれかとを、マウスに対して投与をした。 その結果、スルフィソキサゾールは、クロニジンが誘発する体温低下に対して何らの影響も及ぼさなかった(図7B)が、スルフィソキサゾールは、クロニジンが誘発する鎮痛作用に対しては用量依存効果を示す(図7A)、ことが明らかとなった。 225mg/kgの濃度のスルフィソキサゾールは、刺激を与えてから1.5時間後には、テイルフリック潜時を4〜5倍にまで増大しており、この鎮痛作用のレベルは、刺激を与えてから6時間後もなお維持されていた。 75mg/kgのスルフィソキサゾールのテイルフリック潜時は、刺激を与えて約3時間までは、約4倍の数値を示していたが、6時間の時点では、賦形剤で治療した動物に関して得られたテイルフリック潜時の約3倍の数値にまで減少していた。 25mg/kgのスルフィソキサゾールは、クロニジンが誘発する鎮痛作用を、賦形剤で治療したマウスと比較して、約5倍のレベルにまで増強したが、このレベルは徐々に減少していき、治療が終了して6時間後には、約2倍のレベルにまでなっていた。 スルフィソキサゾールの用量を変えても、体温には何らの影響も無かった。
【0137】
これらの結果は、低用量のスルフィソキサゾール(225および75mg/kg)が、低用量のクロニジン(0.3mg/kg)の鎮痛作用を増強する、ことを指し示すものである。 よって、これら二つの薬剤の組み合わせは、モルヒネと同様の鎮痛作用をもたらすが、それら薬剤は、クロニジンだけ、あるいは、スルフィソキサゾールだけを用いる場合の高投与量の用量よりも低用量ですむこととなる。
【0138】
実施例8:クロニジンの鎮痛作用のメカニズム
鎮痛作用に関係するクロニジンとスルフィソキサゾールが奏する作用のメカニズムを決定するために、異なる疼痛受容体、例えば、アルファ-2アドレナリン受容体、イミダゾリンアドレナリン受容体、または、オピオイド受容体などに作用する幾つかの異なる薬剤を、クロニジンと共に投与した。
【0139】
α2アドレナリン受容体が、クロニジンの鎮痛作用を媒介するかどうかを決定するために、α2アドレナリン作動性受容体アンタゴニストであるヨヒンビンを、クロニジンとCMCの組み合わせ、または、クロニジンとスルフィソキサゾールの組み合わせと共に投与をし、そして、テイルフリック潜時と体温の検査を行った。 マウスを、次の5つのグループに分けた。 グループ1:賦形剤+賦形剤(生理食塩水、腹腔内投与)+賦形剤(生理食塩水、腹腔内投与)、グループ2:賦形剤+CMC(経口投与)+賦形剤(生理食塩水、腹腔内投与)、グループ3:ヨヒンビン(2mg/kg、腹腔内投与)+CMC(経口投与)+賦形剤(生理食塩水、腹腔内投与)、グループ4:ヨヒンビン(2mg/kg、腹腔内投与)+CMC(経口投与)+クロニジン(0.3mg/kg、腹腔内投与)、グループ5:ヨヒンビン(2mg/kg、腹腔内投与)+スルフィソキサゾール(250mg/kg、経口投与)+クロニジン(0.3mg/kg、腹腔内投与)。
【0140】
ヨヒンビンは、クロニジンが誘発した体温低下を若干緩和していた(図8B)。 ヨヒンビン、クロニジン、および、スルフィソキサゾールを投与したところ、ヨヒンビンは、クロニジンとスルフィソキサゾールとの組み合わせが奏する鎮痛作用に対しては効果を示さなかった(図8A)。 これら研究結果は、クロニジンとスルフィソキサゾールとの組み合わせが奏する鎮痛作用が、α2アドレナリン受容体を媒介したものではない、ことを指し示すものである。
【0141】
イミダゾリンアドレナリン受容体が、クロニジンの鎮痛作用を媒介するかどうかを決定するために、イミダゾリン受容体アンタゴニストであるイダゾキサンを、クロニジンとCMCの組み合わせ、または、クロニジンとスルフィソキサゾールの組み合わせと共に投与をし、そして、テイルフリック潜時と体温の検査を行った。 マウスを、次の5つのグループに分けた。 グループ1:賦形剤+賦形剤(生理食塩水、腹腔内投与)+賦形剤(CMC、経口投与)、グループ2:イダゾキサン(2mg/kg、腹腔内投与)+CMC(経口投与)+賦形剤(生理食塩水、腹腔内投与)、グループ3:イダゾキサン(2mg/kg、腹腔内投与)+CMC(経口投与)+賦形剤(生理食塩水、腹腔内投与)、グループ4:イダゾキサン(2mg/kg、腹腔内投与)+CMC(経口投与)+クロニジン(0.3mg/kg、腹腔内投与)、グループ5:イダゾキサン(2mg/kg、腹腔内投与)+スルフィソキサゾール(250mg/kg、経口投与)+クロニジン(0.3mg/kg、腹腔内投与)。 イダゾキサンは、クロニジンが誘発した体温低下を若干緩和していた(図9B)。 クロニジンとスルフィソキサゾールの投与を受けたマウスに対してイダゾキサンを投与したところ、クロニジンとスルフィソキサゾールとの組み合わせが奏する鎮痛作用に対して効果が認められた(図9A)。 これら研究結果は、クロニジンとスルフィソキサゾールとの組み合わせが奏する鎮痛作用の一部が、イミダゾリンアドレナリン受容体を媒介したものである、ことを指し示すものである。
【0142】
オピオイドアドレナリン受容体が、クロニジンの鎮痛作用を媒介するかどうかを決定するために、オピオイド受容体アンタゴニストであるナロキソンを、クロニジンとCMCの組み合わせ、または、クロニジンとスルフィソキサゾールの組み合わせと共に投与をし、そして、テイルフリック潜時と体温の検査を行った。 マウスを、次の5つのグループに分けた。 グループ1:ナロキソン(1.0mg/kg、腹腔内投与)+CMC(経口投与)+賦形剤(生理食塩水、腹腔内投与)、グループ2:ナロキソン(1.0mg/kg、腹腔内投与)+CMC(経口投与)+クロニジン(0.3mg/kg、腹腔内投与)、グループ3:ナロキソン(1.0mg/kg、腹腔内投与)+スルフィソキサゾール(250mg/kg、経口投与)+クロニジン(0.3mg/kg、腹腔内投与)。 しかしながら、ナロキソンは、クロニジンが誘発した体温低下には作用を示さなかった(図10B)。 その一方で、ナロキソンを投与することで、クロニジンとスルフィソキサゾールの組み合わせが奏する鎮痛作用を抑制し、約5倍にまで増大したテイルフリック潜時(図5Aを参照)は、治療を終えて4時間後には、わずか2倍の増大に止まるだけであった(図10A)。 これら研究結果は、クロニジンとスルフィソキサゾールとの組み合わせが奏する鎮痛作用が、オピオイド受容体を媒介したものである、ことを指し示すものである。
【0143】
実施例9:クロニジンの鎮痛作用に関するその他のエンドセリンAアンタゴニストの効果
クロニジンの鎮痛作用に関するスルフィソキサゾールの効果が、分子に対して特異的であるものかどうか、また、その他のETAアンタゴニストも、クロニジンの鎮痛作用を増強するものかどうか、について決定をするために、クロニジンとETAアンタゴニストのBMS182874を投与したマウスにおいて、テイルフリック潜時と体温の評価を行った。
【0144】
マウスを、次の3つの治療グループに分けた。 グループ1:クロニジン(0.3mg/kg、腹腔内投与)+BMS182874(2.0μg/kg、脳室内投与)、グループ2:クロニジン(0.3mg/kg、腹腔内投与)+BMS182874(10.0μg/kg、脳室内投与)、グループ3:クロニジン(0.3mg/kg、腹腔内投与)+BMS182874(50.0μg/kg、脳室内投与)。 その結果、低用量および高用量のBMS182874は、クロニジンの鎮痛作用を適度に増強していたが、中用量(10μg/kg)でも、クロニジンの鎮痛作用の増強が認められており(図11A)、このことは、治療を終えて1時間後および2時間後には、テイルフリック潜時が5倍にまで増大し、また、治療を終えて4時間後には、テイルフリック潜時が約3倍のレベルにまで減少し、そして、治療を終えて6時間後には、テイルフリック潜時が2倍のレベルにまで減少している、ことから明らかとなった(図5Aでの賦形剤の結果と図11Aとの比較)。 BMS182874は、クロニジンが引き起こす体温低下に対しては何らの影響も及ぼさなかった(図11B)。
【0145】
これら結果は、ETA受容体が、クロニジンの鎮痛作用の増強に関与しているかもしれないこと、そして、最適な用量を決定する際に用いられるその他のETAアンタゴニストが、クロニジンが奏する鎮痛作用を増強することができること、を指し示すものである。
【0146】
実施例10:オピオイドの鎮痛作用に関するクロニジンまたはBMS182874の効果
モルヒネの鎮痛作用とオキシコドンの鎮痛作用との間の差異について、報告がされている[40、41、42]。 オキシコドンは、1917年から臨床で使用されてきている[43]。 オキシコドンは、μ-オピオイド受容体アゴニスト[44]であり、μ-オピオイド受容体に対するKi値は18nMであり、δ-オピオイド受容体に対するKi値は958nMであり、そして、κ-オピオイド受容体に対するKi値は677nMである[45]。 しかしながら、オキシコドンのμ-オピオイド受容体に対する親和性は、モルヒネの1/20にも満たない。 モルヒネが、雌のラットよりも雄のラットに対して大きな鎮痛作用を示すのに対して、オキシコドンが、雌と雄の双方のラットに対して同様の鎮痛作用を示しており、このことが、モルヒネとオキシコドンとの間の鎮痛作用の差異を示す別の根拠となっております[46]。 α2アドレナリン作動性受容体阻害剤であるイダゾキサンでのα2アドレナリン受容体に対するKi値は3.6nMであり、また、I1-イミダゾリン受容体に対するKi値は186nMであり[47]、このことは、イダゾキサンが、α2アドレナリン受容体に対して作用し、それゆえに、クロニジンによるモルヒネの鎮痛作用の増強をブロックすること、を指し示すものである。 クロニジンでのα2アドレナリン作動性受容体部位に対するKi値は3.8nMであり、また、I1-イミダゾリン受容体に対するKi値は1.0nMである[48]。
【0147】
オピオイド鎮痛作用の増強に関するクロニジンとその他のETAアンタゴニストの効果を決定するために、クロニジン、BMS182874、および、オピオイドを投与したマウスのテイルフリック潜時を、イダゾキサンの存在下または非存在下で評価をした。
【0148】
モルヒネの鎮痛作用に関するクロニジンまたはBMS182874の効果、および、イダゾキサンによるそれらの阻害
準備した動物を、次のグループ(n=6/グループ)に分けた。 グループ1:賦形剤(1ml/kg、腹腔内投与)+賦形剤(5μl、脳室内投与)+モルヒネ(8mg/kg、皮下投与)、グループ2:賦形剤(1ml/kg、腹腔内投与)+クロニジン(1mg/kg、腹腔内投与)+賦形剤(1ml/kg、皮下投与)、グループ3:賦形剤(1ml/kg、腹腔内投与)+クロニジン(1mg/kg、腹腔内投与)+モルヒネ(8mg/kg、皮下投与)、グループ4:イダゾキサン(2mg/kg、腹腔内投与)+クロニジン(1mg/kg、腹腔内投与)+モルヒネ(8mg/kg、皮下投与)、グループ5:賦形剤(1ml/kg、腹腔内投与)+賦形剤(5μl、脳室内投与)+モルヒネ(8mg/kg、皮下投与)、グループ6:賦形剤(1ml/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+賦形剤(1ml/kg、皮下投与)、グループ7:賦形剤(1ml/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+モルヒネ(8mg/kg、皮下投与)、グループ8:イダゾキサン(2mg/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+モルヒネ(8mg/kg、皮下投与)。
【0149】
7,8-ジデヒドロ-4,5α-エポキシ-17-メチルモルフィナン-3,6α-ジオール硫酸塩(マリンクロットケミカル社、セントルイス、ミズーリ州)である硫酸モルヒネを、滅菌済生理食塩水に溶解し、そして、皮下(s.c.)に注射をした。 N-(2,6-ジクロロフェニル)-4,5-ジヒドロ-1H-イミダゾ−ル-2-アミン(シグマケミカル社、セントルイス、ミズーリ州)であるクロニジンを、滅菌済生理食塩水に溶解し、そして、腹腔内(i.p.)に注射をした。 5-(ジメチルアミノ)-N-(3,4-ジメチル-5-イソキサゾリル)-1-ナフタレンスルホンアミド(トクリスファーマシューティカルズ社、エリスヴィル、ミズーリ州)であるBMS182874を、20%DMSOに溶解し、そして、脳室内(i.c.v.)に注射をした。
【0150】
2-(1,4-ベンゾジオキサン-2-イル)-2-イミダゾリン(シグマケミカル社、セントルイス、ミズーリ州)であるイダゾキサンを、滅菌済生理食塩水に溶解し、そして、腹腔内(i.p.)に注射をした。 実験を行うにあたって、クロニジンまたはBMS182874で治療を行う15分前にイダゾキサンを投与し、また、クロニジンまたはBMS182874で治療を行った30分後にモルヒネを投与した。
【0151】
モルヒネ(8mg/kg、皮下投与)は、顕著な鎮痛作用を示しており、AUC0→360は、21.23±3.18秒.分であった(図12B)。 クロニジン(1mg/kg、腹腔内投与)も鎮痛作用を示しており、AUC0→360は、16.62±1.61秒.分であった(図12B)。 クロニジン(1mg/kg、腹腔内投与)とモルヒネ(8mg/kg、皮下投与)の組み合わせで治療をしたラットでは、モルヒネ(P=0.015)またはクロニジン(P=0.001)のいずれかだけで治療をしたラットと比較して、遙かに大きな鎮痛作用が認められた。 イダゾキサン(2mg/kg、腹腔内投与)は、クロニジンとモルヒネの組み合わせを投与したラットで認められた鎮痛作用に対しては、何らの影響も及ぼさなかった(図12A)。
【0152】
BMS182874(50μg、脳室内投与)だけを投与しても鎮痛作用は認められず、AUC0→360は、4.47±1.49秒.分であった。 しかしながら、BMS182874(50μg、脳室内投与)とモルヒネ(8mg/kg、皮下投与)の組み合わせで治療をしたラットでは、モルヒネ(P=0.009)またはBMS182874(P=0.0006)のいずれかだけで治療をしたラットと比較して、遙かに大きな鎮痛作用が認められた(図12C)。 イダゾキサン(2mg/kg、腹腔内投与)は、BMS182874とモルヒネの組み合わせを投与したラットで認められた鎮痛作用に対しては、何らの影響も及ぼさなかった(図12A)。 これら結果は、クロニジン(P=0.015)とBMS182874(P=0.009)が、モルヒネの鎮痛作用を顕著に増強することを指し示すものであった。
【0153】
オキシコドンの鎮痛作用に関するクロニジンまたはBMS182874の効果、および、イダゾキサンによるそれらの阻害
オキシコドンの鎮痛作用に関するクロニジンまたはBMS182874の効果を評価するために、治療グループを、次のようにして分けた(n=6/グループ)。 グループ1:賦形剤(1ml/kg、腹腔内投与)+賦形剤(1ml/kg、腹腔内投与)+オキシコドン(4mg/kg、皮下投与)、グループ2:賦形剤(1ml/kg、腹腔内投与)+クロニジン(1mg/kg、腹腔内投与)+賦形剤(1ml/kg、皮下投与)、グループ3:賦形剤(1ml/kg、腹腔内投与)+クロニジン(1mg/kg、腹腔内投与)+オキシコドン(4mg/kg、皮下投与)、グループ4:イダゾキサン(2mg/kg、腹腔内投与)+クロニジン(1mg/kg、腹腔内投与)+オキシコドン(4mg/kg、皮下投与)、グループ5:賦形剤(1ml/kg、腹腔内投与)+賦形剤(5μl、脳室内投与)+オキシコドン(4mg/kg、皮下投与)、グループ6:賦形剤(1ml/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+賦形剤(1ml/kg、皮下投与)、グループ7:賦形剤(1ml/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+オキシコドン(4mg/kg、皮下投与)、グループ8:イダゾキサン(2mg/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+オキシコドン(4mg/kg、皮下投与)。
【0154】
クロニジン、BMS182874、および、イダゾキサンは、前述した手順で調製をした。 4,5α-エポキシ-14-ヒドロキシ-3-メトキシ-17-メチルモルフィナン-6-オン塩酸塩(スペクトラムケミカル社、サンガーデナ、カリフォルニア州)である塩酸オキシコドンを、滅菌済生理食塩水に溶解し、そして、皮下(s.c.)に注射をした。 クロニジンまたはBMS182874で治療を行った30分後にオキシコドンを投与した。 クロニジンまたはBMS182874で治療を行う15分前にイダゾキサンを投与した。
【0155】
その結果、オキシコドン(4mg/kg、皮下投与)は、顕著な鎮痛作用を示しており、AUC0→360は、18.23±2.32秒.分であった。 クロニジン(1mg/kg、腹腔内投与)も鎮痛作用を示していた。 クロニジン(1mg/kg、腹腔内投与)とオキシコドン(4mg/kg、皮下投与)の組み合わせで治療をしたラットでは、オキシコドン(P=0.025)またはクロニジン(P=0.016)のいずれかだけで治療をしたラットと比較して、遙かに大きな鎮痛作用が認められた(図13B)。 イダゾキサン(2mg/kg、腹腔内投与)は、クロニジンとオキシコドンの組み合わせを投与したラットで認められた鎮痛作用をブロックした(図13A)。
【0156】
オキシコドン(4mg/kg、皮下投与)は、顕著な鎮痛作用を示したが、BMS182874(50μg、脳室内投与)は、鎮痛作用は全く示さなかった(図13A)。 しかしながら、BMS182874 (50μg、脳室内投与)とオキシコドン(4mg/kg、皮下投与)の組み合わせで治療をしたラットでは、オキシコドン(P=0.012)またはBMS182874(P=0.0004)のいずれかだけで治療をしたラットと比較して、遙かに大きな鎮痛作用が認められた(図13C)。 イダゾキサン(2mg/kg、腹腔内投与)は、BMS182874とオキシコドンの組み合わせを投与したラットで認められた鎮痛作用を顕著に(P=0.003)ブロックした(図13A、13C)。 これら結果は、クロニジン(P=0.025)とBMS182874(P=0.012)が、オキシコドンが示す鎮痛作用を増強することを指し示すものであった。
【0157】
概して、I1-イミダゾリンであり、かつ、α2アドレナリン作動性受容体アンタゴニストでもあるイダゾキサンは、オキシコドンの鎮痛作用が、クロニジンまたはBMS182874によって増強されることをブロックするが、その一方で、イダゾキサンは、モルヒネの鎮痛作用が、クロニジンまたはBMS182874によって増強されることに対しては何らの影響も与えないことが明らかとなった。 この知見は、オキシコドンの鎮痛作用でのイミダゾリン受容体の関与を示すものであって、つまりは、I1-イミダゾリン受容体が、オキシコドンの鎮痛作用の増強には関与しているものの、クロニジンまたはBMS182874によるモルヒネの鎮痛作用の増強には関与していない、ことを示すものである。 本実施例は、オキシコドンの鎮痛作用を増強するクロニジンまたはBMS182874が、I1-イミダゾリン受容体とは関連があるものの、モルヒネの鎮痛作用とは関連が認められない、ことを初めて報告するものである。
【0158】
実施例11:モルヒネまたはオキシコドンの鎮痛作用を増強したクロニジンに関するヨヒンビンの効果
α2アドレナリン作動性受容体の関与を決定するために、選択的アンタゴニストであるヨヒンビンを用いた。 ヨヒンビンは、非常に選択性に富んだα2アドレナリン作動性受容体アンタゴニストであって、α2アドレナリン受容体に対するKi値は22nMであり、そして、I1-イミダゾリン受容体に対するKi値は21,810nMである[49]。 ヨヒンビンは、イミダゾリン環を有しておらず、また、イミダゾリン受容体に対して作用もしない。
【0159】
治療グループを、次のようにして分けた。 グループ1:賦形剤(1ml/kg、腹腔内投与)+賦形剤(5μl、脳室内投与)+モルヒネ(4mg/kg、皮下投与)、グループ2:賦形剤(1ml/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+モルヒネ(4mg/kg、皮下投与)、グループ3:ヨヒンビン(2mg/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+モルヒネ(4mg/kg、皮下投与)、グループ4:賦形剤(1ml/kg、腹腔内投与)+賦形剤(5μl、脳室内投与)+オキシコドン(4mg/kg、皮下投与)、グループ5:賦形剤(1ml/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+オキシコドン(4mg/kg、皮下投与)、グループ 6:ヨヒンビン(2mg/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+オキシコドン(4mg/kg、皮下投与)。
【0160】
17α-ヒドロキシ-ヨヒンバン-16α-カルボキシレート塩酸塩(シグマケミカル社、セントルイス、ミズーリ州)である塩酸ヨヒンビンを、アルコール(1部)と滅菌生理食塩水(9部)に溶解し、そして、 腹腔内(i.p.)に注射をした。 BMS182874で治療を行う15分前にヨヒンビンを投与し、また、BMS182874で治療を行った30分後にモルヒネまたはオキシコドンを投与した。
【0161】
その結果、クロニジンが、モルヒネ(P=0.0366)ならびにオキシコドン(P=0.0587)の鎮痛作用を増強し、また、ヨヒンビンが、モルヒネ(P=0.058)またはオキシコドン(P=0.0167)の鎮痛作用を増強するというクロニジンの作用をブロックすることが明らかとなった(図14A−14C)。 この拮抗作用については、モルヒネでの重要性は不明確であるが、オキシコドンの鎮痛作用においては非常に重要であった。 モルヒネ(8mg/kg、皮下投与)の鎮痛作用の改善は、ヨヒンビンを用いて前処理することによって阻害された(P=0.0355)(図14B)。 同様に、オキシコドン(4mg/kg、皮下投与)が奏する鎮痛作用をクロニジンが増強し、その鎮痛作用が、ヨヒンビンを用いて前処理することによって顕著にブロックされる、ことが明らかとなった(図14C)。
【0162】
モルヒネの鎮痛作用、および、オキシコドンの鎮痛作用を増強するBMS182874に対するα2アドレナリン作動性受容体の関与を決定するために、これら薬剤が投与された動物に対して、α2アドレナリン作動性選択的アンタゴニストであるヨヒンビンを投与した。 治療グループを、次のようにして分けた。 グループ1:賦形剤(1ml/kg、腹腔内投与)+賦形剤(5μl、脳室内投与)+モルヒネ(4mg/kg、皮下投与)、グループ2:賦形剤(1ml/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+モルヒネ(4mg/kg、皮下投与)、グループ3:ヨヒンビン(2mg/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+モルヒネ(4mg/kg、皮下投与)、グループ4:賦形剤(1ml/kg、腹腔内投与)+賦形剤(5μl、脳室内投与)+オキシコドン(4mg/kg、皮下投与)、グループ5:賦形剤(1ml/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+オキシコドン(4mg/kg、皮下投与)、グループ 6:ヨヒンビン(2mg/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+オキシコドン(4mg/kg、皮下投与)。
【0163】
BMS182874で治療を行う15分前にヨヒンビンを投与した。 BMS182874で治療を行った30分後にモルヒネまたはオキシコドンを投与した。
【0164】
前述したように、BMS182874は、オキシコドン(4mg/kg、皮下投与)が奏する鎮痛作用を増強し(P=0.0001)、また、モルヒネ(8mg/kg、皮下投与)が奏する鎮痛作用も増大させている(P=0.003)(図15Aおよび15B)。 BMS182874は、モルヒネの鎮痛作用を増強し(P=0.003)、また、オキシコドンの鎮痛作用も増強する(P=0.0001)(図15Bおよび15C)。 BMS182874で治療を施した動物に対してヨヒンビンを投与したところ、BMS182874で治療を施したラットにおいてモルヒネまたはオキシコドンが示した鎮痛作用は、ヨヒンビンを用いて前処理を行っても、何らの影響も受けなかった(P=0.156)(図15C)。
【0165】
これらの結果は、モルヒネおよびオキシコドンの鎮痛作用をクロニジンが増強するという作用が、選択的α2アドレナリン作動性アンタゴニストであるヨヒンビンによってブロックされていることを示すものであって、つまり、モルヒネおよびオキシコドンの鎮痛作用を増強するというクロニジンの作用に対してα2アドレナリン受容体が関与している、ことを指し示すものである。 また、これら知見は、モルヒネおよびオキシコドンの鎮痛作用を増強するというBMS182874およびクロニジンの作用が、異なるメカニズムを介したものであることも示している。
【0166】
実施例12:BMS182874で治療を施したラットでのモルヒネおよびオキシコドンの鎮痛作用に関するクロニジンの用量応答効果
先に見てきたように、クロニジンまたはスルフィソキサゾールの用量は、治療を受けた動物で認められる鎮痛レベルと相関をしていた。 オピオイドの鎮痛作用に関するクロニジンとETAアンタゴニストのBMS182874の用量効果を決定するために、用量反応実験を行った。
【0167】
治療グループを、次のようにして分けた(n=6/グループ)。 グループ1:賦形剤(1ml/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+モルヒネ(4mg/kg、皮下投与)、グループ2:クロニジン(0.1mg/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+モルヒネ(4mg/kg、皮下投与)、グループ3:クロニジン(0.3mg/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+モルヒネ(4mg/kg、皮下投与)、グループ4:クロニジン(1.0mg/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+モルヒネ(4mg/kg、皮下投与)、グループ5:賦形剤(1ml/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+オキシコドン (4mg/kg、皮下投与)、グループ6:クロニジン(0.1mg/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+オキシコドン(4mg/kg、皮下投与)、グループ7:クロニジン(0.3mg/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+オキシコドン(4mg/kg、皮下投与)、グループ8:クロニジン(1.0mg/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+オキシコドン(4mg/kg、皮下投与)。
【0168】
BMS182874で治療を行う15分前にヨヒンビンを投与し、そして、BMS182874で治療を行った30分後にモルヒネまたはオキシコドンを投与した。 その他の活性成分は、前述した手順に従って、調製および投与を行った。
【0169】
モルヒネ(4mg/kg、皮下投与)は、BMS182874で治療を施したラットにおいて顕著な鎮痛作用を示した。 クロニジンは、BMS182874で治療を施したラットにおいて、モルヒネの鎮痛作用を用量依存的に増大した(図16A)。 0.1mg/kgの用量でクロニジンを腹腔内投与したところ、鎮痛作用の改善は認められなかったが、0.3mg/kgおよび1.0mg/kgの用量でクロニジンを腹腔内投与したところ、BMS182874で治療を施したラットにおいて、モルヒネの鎮痛作用を顕著に増強した(順にP=0.016とP=0.011であった)(図16Aおよび16B)。
【0170】
オキシコドン(4mg/kg、皮下投与)は、BMS182874で治療を施したラットにおいて顕著な鎮痛作用を示した。 クロニジンは、BMS182874で治療を施したラットにおいて、オキシコドンの鎮痛作用を用量依存的に増大した(図16C)。 0.1mg/kgの用量でクロニジンを腹腔内投与したところ、鎮痛作用の改善は認められなかったが、0.3mg/kgおよび1.0mg/kgの用量でクロニジンを腹腔内投与したところ、BMS182874で治療を施したラットにおいて、オキシコドンの鎮痛作用を顕著に増強した(順にP=0.02とP=0.031であった)(図16Cおよび16D)。
【0171】
実施例13:オピオイドの鎮痛作用に関するクロニジン、BMS182874、および、クロニジンとBMS182874との組み合わせの効果
クロニジンおよびBMS182874の併用に起因するモルヒネまたはオキシコドンの鎮痛効果の増強作用を確認するために、モルヒネ(8mg/kg、皮下投与)とオキシコドン(4mg/kg、皮下投与)で治療を施したラットにおいて、クロニジン(1mg/kg、腹腔内投与)、BMS182874(50μg、脳室内投与)、および、クロニジン(1mg/kg、腹腔内投与)とBMS182874(50μg、脳室内投与)との組み合わせの効果を決定した。
【0172】
治療グループを、次のようにして分けた(n=6/グループ)。 グループ1:賦形剤(1ml/kg、腹腔内投与)+クロニジン(1mg/kg、腹腔内投与)+モルヒネ(8mg/kg、皮下投与)、グループ2:賦形剤(1ml/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+モルヒネ (8mg/kg、皮下投与)、グループ3:クロニジン(1.0mg/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+モルヒネ(8mg/kg、皮下投与)、グループ 4:賦形剤(1ml/kg、腹腔内投与)+クロニジン(1mg/kg、腹腔内投与)+オキシコドン(4mg/kg、皮下投与)、グループ5:賦形剤(1ml/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+オキシコドン (4mg/kg、皮下投与)、グループ6:クロニジン(1.0mg/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+オキシコドン(4mg/kg、皮下投与)。
【0173】
BMS182874で治療を行う15分前にクロニジンを投与し、そして、BMS182874またはクロニジンで治療を行った30分後にモルヒネまたはオキシコドンを投与した。
【0174】
その結果、クロニジンとBMS182874との組み合わせで治療をしたラットでのモルヒネの鎮痛作用(図17A)が、クロニジン(P=0.09)またはBMS182874(P=0.07)のいずれかだけで治療を行った場合(図17B)との比較において、さらに増強されていることは認められなかった。 同様に、クロニジンとBMS182874との組み合わせで治療をしたラットでのオキシコドンの鎮痛作用が、クロニジン(P=0.18)またはBMS182874(P=0.11)のいずれかだけで治療を行った場合(図17C)との比較において、さらに増強されていることも認められなかった。
【0175】
実施例14:クロニジンの鎮痛作用に関するBMS182874の効果
クロニジン(1mg/kg、腹腔内投与)の鎮痛作用が、BMS182874(50μg、脳室内投与)によって増強されるかどうかを決定するために、BMS182874、クロニジン、および、クロニジンとBMS182874との組み合わせの効果を、ラットを用いて決定をした。
【0176】
治療グループを、次のようにして分けた(n=6/グループ)。 グループ1:BMS182874(50μg、脳室内投与)+賦形剤(1ml/kg、腹腔内投与)、グループ2:賦形剤(5μl、脳室内投与)+クロニジン(1.0mg/kg、腹腔内投与)、グループ3:BMS182874(50μg、脳室内投与)+クロニジン(1.0mg/kg、腹腔内投与)。 クロニジンで治療を行う15分前にBMS182874を投与した。
【0177】
その結果、BMS182874は、鎮痛作用を示さなかったが、クロニジンは、BMS182874と比較して顕著な鎮痛作用を示す(P=0.00086)ことが明らかとなった(図18A)。 しかしながら、BMS182874は、クロニジンの鎮痛作用に影響を及ぼさなかった(P=0.645)(図18B)。
【0178】
以下の表には、様々な薬剤が奏する鎮痛作用がまとめられている。
【0179】
【表1】

【0180】
クロニジンとBMS182874は、異なるメカニズムを介して作用していることが、上記結果によってさらに確認されたとの結論は、モルヒネの鎮痛作用とオキシコドンの鎮痛作用を増強するクロニジンの作用が、BMS182874を用いた治療を行っても影響を受けないことを指し示すものである。 同様に、モルヒネの鎮痛作用とオキシコドンの鎮痛作用を増強するBMS182874の作用は、クロニジンを用いた治療を行っても影響を受けない。 さらに、クロニジンは、鎮痛作用を示すが、この作用は、BMS182874を用いた治療を行っても影響を受けない(図18)。 これらすべての知見は、クロニジンとBMS182874が、異なるメカニズムを介して、モルヒネの鎮痛作用とオキシコドンの鎮痛作用を増強することを指し示している。
【0181】
さらに、ヨヒンビンは、モルヒネの鎮痛作用を増強するクロニジンの作用をブロックするが、イダゾキサンは、モルヒネの鎮痛作用を増強するクロニジンの作用には影響を及ぼさないので、α2アドレナリン受容体は、クロニジンの増強作用に関与はするが、I1-イミダゾリン受容体は、モルヒネの鎮痛作用を増強するクロニジンの作用に関与はしていない、と結論付けることができる。
【0182】
α2アドレナリンアゴニストおよびエンドセリンAアンタゴニストが、オピオイドの鎮痛作用を効果的に増強できる、という本明細書に記載の知見は、これら薬剤の組み合わせが、治療を施す動物での疼痛を軽減するのに必要なオピオイドの用量を効果的に減少させるであろう、ということを示唆するものである。 さらに、本明細書に記載の実験は、α2アドレナリンアゴニストとエンドセリンAアンタゴニストとを組み合わせることで、高用量のモルヒネと同様の鎮痛作用を相乗的に奏することを実証している。 したがって、α2アドレナリンアゴニストとエンドセリンAアンタゴニストとの組み合わせを含む組成物、または、一方の薬剤だけを含み、一斉に投与がされる複数の組成物は、耐性のリスクも無く、また、オピオイド鎮痛薬の追加を必要とせずに、疼痛の症状を治療する上で有用である。
【0183】
これまでに説明してきた本願発明の実施例を考慮して、当業者が、本願発明に対して数多くの修正や変更を加えることが予想される。 従って、特許請求の範囲の欄に記載の限定事項のみが、本願発明に付加されるべきである。
【0184】
参照文献
[1] Adithan, C., Sivagnanam, G., Swain, R., Shashindran, C.H. and Bapna, J. S., Potentiation of clonidine analgesia by amitriptyline, Indian J Exp Biol, 24 (1986) 256-8.
[2] Backonja, M., Beydoun, A., Edwards, K.R., Schwartz, S.L., Fonseca, V., Hes, M., LaMoreaux, L. and Garofalo, E., Gabapentin for the symptomatic treatment of painful neuropathy in patients with diabetes mellitus: a randomized controlled trial, Jama, 280 (1998) 1831-6.
[3] Bennett, G.J., Update on the neurophysiology of pain transmission and modulation: focus on the NMDA-receptor, J Pain Symptom Manage, 19 (2000) S2-6.
[4] Bhalla, S., Ciaccio, N., Wang, Z.J. and Gulati, A., Involvement of endothelin in morphine tolerance in neuroblastoma (SH-SY5Y) cells, Exp Biol Med (Maywood), 231 (2006) 1152-6.
[5] Bhalla, S., Matwyshyn, G. and Gulati, A., Potentiation of morphine analgesia by BQ 123, an endothelin antagonist, Peptides, 23 (2002) 1837-45.
[6] Bhalla, S., Matwyshyn, G. and Gulati, A., Endothelin receptor antagonists restore morphine analgesia in morphine tolerant rats, Peptides, 24 (2003) 553-61.
[7] Bhalla, S., Matwyshyn, G. and Gulati, A., Morphine tolerance does not develop in mice treated with endothelin-A receptor antagonists, Brain Res, 1064 (2005) 126-35.
[8] Chan, M.F., Okun, L., Stavros, F.L., Hwang, E., Wolff, M.E. and Balaji, V.N., Identification of a new class of ETA selective endothelin antagonists by pharmacophore directed screening, Biochem Biophys Res Commun, 201 (1994) 228- 34.
[9] Constant, L., Gall, O., Gouyet, L., Chauvin, M. and Murat, L., Addition of clonidine or fentanyl to local anaesthetics prolongs the duration of surgical analgesia after single shot caudal block in children, Br J Anaesth, 80 (1998) 294-8.
[10] D'Amour, F.E., Smith, D.L., A method for determining loss of pain sensation, J Pharmacol Exp Ther, 71 (1941) 74-79.
[11] Fromm, G.H., Aumentado, D. and Terrence, C.F., A clinical and experimental investigation of the effects of tizanidine in trigeminal neuralgia, Pain, 53 (1993) 265-71.
[12] Gilron, L., Bailey, J.M., Tu, D., Holden, R.R., Weaver, D.F. and Houlden, R.L., Morphine, gabapentin, or their combination for neuropathic pain, N Engl J Med, 352 (2005) 1324-34.
[13] Gordon, N.C., Heller, P.H. and Levine, J.D., Enhancement of pentazocine analgesia by clonidine, Pain, 48 (1992) 167-9.
[14] Goudas, L.C. and Carr, D.B., Is there a place for intrathecal clonidine for clinical analgesia? Anesth Analg, 83 (1996) 891.
[15] Gulati, A., Evidence for antagonistic activity of endothelin for clonidine induced hypotension and bradycardia, Life Sci, 50 (1992) 153-60.
[16] Gulati, A., Bhalla, S. and Matwyshyn, G., A Novel Combination of Opiates and Endothelin Antagonists to Manage Pain Without Any Tolerance Development, J Cardiovasc Pharmacol, 44 (2004) S129-S131.
[17] Gulati, A., Rebello, S. and Kumar, A., Role of sympathetic nervous system in cardiovascular effects of centrally administered endothelin-1 in rats, Am J Physiol, 273 (1997) Hl 177-86.
[18] Gulati, A. and Srimal, R.C., Endothelin antagonizes the hypotension and potentiates the hypertension induced by clonidine, Eur J Pharmacol, 230 (1993) 293-300.
[19] Hutschala, D., Mascher, H., Schmetterer, L., Klimscha, W., Fleck, T., Eichler, H.G. and Tschemko, E.M., Clonidine added to bupivacaine enhances and prolongs analgesia after brachial plexus block via a local mechanism in healthy volunteers, Eur J Anaesthesiol, 21 (2004) 198-204.
[20] Jamali, S., Monin, S., Begon, C, Dubousset, A.M. and Ecoffey, C, Clonidine in pediatric caudal anesthesia, Anesth Analg, 78 (1994) 663-6.
[21] Knotkova, H. and Pappagallo, M., Adjuvant analgesics, Med Clin North Am, 91 (2007) 113-24.
[22] Kunos, G., Mosqueda-Garcia, R., Mastrianni, J.A. and Abbott, F.V., Endorphinergic mechanism in the central cardiovascular and analgesic effects of clonidine, Can J Physiol Pharmacol, 65 (1987) 1624-32.
[23] Lim, D.Y., Heo, K., Choi, C.H. and Lee, E.W., Influence of endothelin-1 on clonidine-induced cardiovascular effects in anesthetized rabbits, J Cardiovasc Pharmacol, 31 Suppl 1 (1998) S 122-5.
[24] Lyons, B., Casey, W., Doherty, P., McHugh, M. and Moore, K.P., Pain relief with low-dose intravenous clonidine in a child with severe burns, Intensive Care Med, 22 (1996) 249-51.
[25] Mandell, G. and Sande, M., Antimicrobial Agents, In Goodman and Gilman's The Pharmacological Basis of Therapeutics, Eighth Edition, Pergamon Press, New York, 1991, 1047-1064 pp.
[26] Matwyshyn, G.A., Bhalla, S. and Gulati, A., Endothelin ETA receptor blockade potentiates morphine analgesia but does not affect gastrointestinal transit in mice, Eur J Pharmacol, 543 (2006) 48-53.
[27] Milne, B., Cervenko, F. W., Jhamandas, K. and Sutak, M., Intrathecal clonidine: analgesia and effect on opiate withdrawal in the rat, Anesthesiology, 62 (1985) 34-8.
[28] Mutafova-Yambolieva, V., Petkov, O., Staneva-Stoytcheva, D. and Lasova, L., Interactions between the effects of endothelin-1, clonidine and yohimbine on electrically-induced contractions in rat vas deferens, Gen Pharmacol, 23 (1992) 529-34.
[29] Nishina, K. and Mikawa, K., Clonidine in paediatric anaesthesia, Curr Opin Anaesthesiol, 15 (2002) 309-16.
[30] Paalzow, G., Development of tolerance to the analgesic effect of clonidine in rats. Cross-tolerance to morphine, Naunyn Schmiedebergs Arch Pharmacol, 304 (1978) 1-4.
[31] Paalzow, L., Analgesia produced by clonidine in mice and rats, J Pharm Pharmacol, 26 (1974) 361-3.
[32] Porchet, H.C., Piletta, P. and Dayer, P., Objective assessment of clonidine analgesia in man and influence of naloxone, Life Sci, 46 (1990) 991-8.
[33] Puppala, B. L., Matwyshyn, G., Bhalla, S. and Gulati, A., Evidence that morphine tolerance may be regulated by endothelin in the neonatal rat. Biol Neonate, 86 (2004) 138-44.
[34] Puppala, B.L., Matwyshyn, G., Bhalla, S. and Gulati, A., Role of Endothelin in Neonatal Morphine Tolerance, J Cardiovasc Pharmacol, 44 (2004) S383-S385.
[35] Rowbotham, M.C., Goli, V., Kunz, N.R. and Lei, D., Venlafaxine extended release in the treatment of painful diabetic neuropathy: a double-blind, placebo- controlled study, Pain, 110 (2004) 697-706.
[36] Saarto, T. and Wiffen, P.J., Antidepressants for neuropathic pain, Cochrane Database Syst Rev (2005) CD005454.
[37] Semenchuk, M.R., Sherman, S. and Davis, B., Double-blind, randomized trial of bupropion SR for the treatment of neuropathic pain, Neurology, 57 (2001) 1583-8.
[38] Spaulding, T.C., Fielding, S., Venafro, J.J. and LaI, H., Antinociceptive activity of clonidine and its potentiation of morphine analgesia, Eur J Pharmacol, 58 (1979) 19-25.
[39] Wang, Y.C., Su, CF. and Lin, M.T., The site and the mode of analgesic actions exerted by clonidine in monkeys, Exp Neurol, 90 (1985) 479-88.
[40] Ordonez Gallego A, Gonzalez Baron M, Espinosa Arranz E: Oxycodone: A pharmacological and clinical review. Clin Transl Oncol 2007;9:298-307.
[41] Nielsen CK, Ross FB, Lotfipour S, Saini KS, Edwards SR, Smith MT: Oxycodone and morphine have distinctly different pharmacological profiles: Radioligand binding and behavioural studies in two rat models of neuropathic pain. Pain 2007; 132:289-300.
[42] Nozaki C, Kamei J: Involvement of mul-opioid receptor on oxycodone-induced antinociception in diabetic mice. Eur J Pharmacol 2007;560: 160-162.
[43] Falk E: Eukodal ein neues narkotikum [eukodal, a new narcotic]. Muenchener Med Wochenschr 1917;64:381-384.
[44] Yoburn BC, Shah S, Chan K, Duttaroy A, Davis T: Supersensitivity to opioid analgesics following chronic opioid antagonist treatment: Relationship to receptor selectivity. Pharmacol Biochem Behav 1995 ;51:535-539.
[45J Monory K, Greiner E, Sartania N, Sallai L, Pouille Y, Schmidhammer H, Hanoune J, Borsodi A: Opioid binding profiles of new hydrazone, oxime, carbazone and semicarbazone derivatives of 14-alkoxymorphinans. Life Sci 1999;64:2011-2020.
[46] Peckham EM, Traynor JR: Comparison of the antinociceptive response to morphine and morphine-like compounds in male and female sprague-dawley rats. J Pharmacol Exp Ther 2006;316: 1195-1201.
[47] Ernsberger P, Giuliano R, Willette RN, Reis DJ: Role of imidazole receptors in the vasodepressor response to clonidine analogs in the rostral ventrolateral medulla. J Pharmacol Exp Ther 1990;253:408-418.
[48] Ernsberger P, Damon TH, Graff LM, Schafer SG, Christen MO: Moxonidine, a centrally acting antihypertensive agent, is a selective ligand for il -imidazoline sites. J Pharmacol Exp Ther 1993;264:172-182.
[49] Hamilton CA, Yakubu MA, Jardine E, Reid JL: Imidazole binding sites in rabbit kidney and forebrain membranes. Journal of autonomic pharmacology 1991;l l:277-283.
[50] Chao, H.M. and Osborne, N.N., Topically applied clonidine protects the rat retina from ischaemia/reperfusion by stimulating alpha(2)-adrenoceptors and not by an action on imidazoline receptors, Brain Res, 904 (2001) 126-36.
[51] Costela, J.L., Carlos, R., Zamacona, M.K., Jimenez, R. and Calvo, R., Interaction between propofol and sulfisoxazole in mice an in vivo and in vitro study, Res Commun MoI Pathol Pharmacol, 93 (1996) 89-100.
[52] Syed, H., Safa, R., Chidlow, G. and Osborne, N.N., Sulfisoxazole, an endothelin receptor antagonist, protects retinal neurones from insults of ischemia/reperfusion or lipopolysaccharide, Neurochem Int, 48 (2006) 708-17.
[53] Iqbal, J., Wig, J., Bhardwaj, N. and Dhillon, M.S., Intra-articular clonidine vs. morphine for post-operative analgesia following arthroscopic knee surgery (a comparative evaluation), Knee, 7 (2000) 109-113.
[54] Goyagi, T., Tanaka, M. and Nishikawa, T., Oral clonidine premedication reduces induction dose and prolongs awakening time from propofol-nitrous oxide anesthesia, Can J Anaesth, 46 (1999) 894-6.
[55] Constant, L, Gall, O., Gouyet, L., Chauvin, M. and Murat, L, Addition of clonidine or fentanyl to local anaesthetics prolongs the duration of surgical analgesia after single shot caudal block in children, Br J Anaesth, 80 (1998) 294-8.
[56] Ezri, T., Szmuk, P., Shklar, B., Katz, J. and Geva, D., Oral clonidine premedication does not prolong analgesia after herniorrhaphy under subarachnoid anesthesia, J Clin Anesth, 10 (1998) 474-81.
[57] Tan, P.H., Chou, A.K., Perng, J.S., Chung, H.C., Lee, C.C. and Mok, M.S., Comparison of epidural butorphanol plus clonidine with butorphanol alone for postoperative pain relief, Acta Anaesthesiol Sin, 35 (1997) 91-6.
[58] Rockemann, M.G., Seeling, W., Duschek, S., Reinelt, H., Steffen, P. and Georgieff, M., Epidural bolus clonidine/morphine versus epidural patient-controlled bupivacaine/sufentanil: quality of postoperative analgesia and cost- identification analysis, Anesth Analg, 85 (1997) 864-9.
[59] Buggy, D.J. and MacDowell, C, Extradural analgesia with clonidine and fentanyl compared with 0.25% bupivacaine in the first stage of labour, Br J Anaesth, 76 (1996) 319-21.
[60] Lena, P., Balarac, N., Arnulf, JJ., Teboul, J. and Bonnet, F., Intrathecal morphine and clonidine for coronary artery bypass grafting, Br J Anaesth, 90 (2003) 300-3.
[61] Bonhomme, V., Doll, A., Dewandre, P. Y., Brichant, J.F., Ghassempour, K. and Hans, P., Epidural administration of low-dose morphine combined with clonidine for postoperative analgesia after lumbar disc surgery, J Neurosurg Anesthesiol, 14 (2002) 1-6.
[62] Jeffs, S. A., Hall, J. E. and Morris, S., Comparison of morphine alone with morphine plus clonidine for postoperative patient-controlled analgesia, Br J Anaesth, 89 (2002) 424-7.
[63] Goyagi, T. and Nishikawa, T., Oral clonidine premedication enhances the quality of postoperative analgesia by intrathecal morphine, Anesth Analg, 82 (1996) 1192-6.
[64] Yanagidate, F., Hamaya, Y. and Dohi, S., Clonidine premedication reduces maternal requirement for intravenous morphine after cesarean delivery without affecting newborn's outcome, Reg Anesth Pain Med, 26 (2001) 461-7.
[65] Park, J., Forrest, J., Kolesar, R., Bhola, D., Beattie, S. and Chu, C, Oral clonidine reduces postoperative PCA morphine requirements, Can J Anaesth, 43 (1996) 900-6.
[66] van Essen, EJ., Bovill, J.G., Ploeger, EJ. and Houben, JJ., Pharmacokinetics of clonidine after epidural administration in surgical patients. Lack of correlation between plasma concentration and analgesia and blood pressure changes, Acta Anaesthesiol Scand, 36 (1992) 300-4).
[67] Smith, B. D., Baudendistel, LJ., Gibbons, JJ. and Schweiss, J.F., A comparison of two epidural alpha 2-agonists, guanfacine and clonidine, in regard to duration of antinociception, and ventilatory and hemodynamic effects in goats, Anesth Analg, IA (1992) 712-8.)
[68] Capogna, G., Celleno, D., Zangrillo, A., Costantino, P. and Foresta, S., Addition of clonidine to epidural morphine enhances postoperative analgesia after cesarean delivery, Reg Anesth, 20 (1995) 57-61.
[69] Benhamou, D., Narchi, P., Hamza, J., Marx, M., Peyrol, M.T. and Sembeil, F., Addition of oral clonidine to postoperative patient-controlled analgesia with i.v. morphine, Br J Anaesth, 72 (1994) 537-40.
[70] Carabine, U. A., Milligan, K.R., Mulholland, D. and Moore, J., Extradural clonidine infusions for analgesia after total hip replacement, Br J Anaesth, 68 (1992) 338-43.
【図面の簡単な説明】
【0185】
【図1】モルヒネ(4mg/kg、腹腔内投与)の存在下および非存在下でのテイルフリック潜時に関するクロニジン(2mg/kg、腹腔内投与)の効果を示している。 マウスを4つのグループに分けた。 グループ1には、賦形剤(生理食塩水、腹腔内投与)+賦形剤(生理食塩水、皮下投与)を投与し、グループ2には、賦形剤+モルヒネ(4mg/kg、皮下投与)を投与し、グループ3には、クロニジン(2mg/kg、腹腔内投与)+賦形剤(生理食塩水、皮下投与)を投与し、そして、グループ4には、クロニジン(2mg/kg、腹腔内投与)+モルヒネ(4mg/kg、皮下投与)を投与した。 クロニジンを投与して30分後に、モルヒネまたは賦形剤を投与した。 図1Aは、様々な時点での数秒間のテイルフリック潜時データを示している。 図1Bは、テイルフリック潜時のデータ値から決定したAUC0→240分によって表された痛覚抑制を示している。 数値は、平均値±標準誤差であり、Nは、6/グループである。 *P<0.05 クロニジンを、対照グループと比較をした。 #P<0.05 クロニジン+モルヒネを、賦形剤+モルヒネのグループと比較をした。
【図2】モルヒネ(4mg/kg、皮下投与)の存在下および非存在下でのテイルフリック潜時に関するスルフィソキサゾール(500mg/kg、経口投与)の効果を示している。 マウスを4つのグループに分けた。 グループ1には、賦形剤(カルボキシメチルセルロース(CMC)、経口投与)+賦形剤(生理食塩水、皮下投与)を投与し、グループ2には、賦形剤(CMC)+モルヒネ(4mg/kg、皮下投与)を投与し、グループ3には、スルフィソキサゾール(500mg/kg、経口投与)+賦形剤(生理食塩水、皮下投与)を投与し、そして、グループ4には、スルフィソキサゾール(500mg/kg、経口投与)+モルヒネ(4mg/kg、皮下投与)を投与した。 スルフィソキサゾールを投与して30分後に、モルヒネまたは賦形剤を投与した。 図2Aは、様々な時点での数秒間のテイルフリック潜時データを示している。 図2Bは、テイルフリック潜時のデータ値から決定したAUC0→240分によって表された痛覚抑制を示している。 数値は、平均値±標準誤差であり、Nは、6/グループである。 *P<0.05 スルフィソキサゾールを、対照グループと比較をした。
【図3】モルヒネ(M)(8mg/kg、皮下投与)の存在下および非存在下でのテイルフリック潜時に関するクロニジン(C)(2mg/kg、腹腔内投与)とスルフィソキサゾール(S)(500mg/kg、経口投与)との組み合わせの効果を示している。 マウスを3つのグループに分けた。 グループ1には、クロニジン(2mg/kg、腹腔内投与)とスルフィソキサゾール(500mg/kg、経口投与)との組み合わせ+賦形剤(V)(生理食塩水、皮下投与)を投与し、グループ2には、クロニジン(2mg/kg、腹腔内投与)とスルフィソキサゾール(500mg/kg、経口投与)との組み合わせ+モルヒネ(8mg/kg、皮下投与)を投与し、グループ3には、賦形剤(生理食塩水、腹腔内投与)と賦形剤(カルボキシメチルセルロース、経口投与)との組み合わせ+モルヒネ(M)(8mg/kg、皮下投与)を投与し、そして、グループ4には、賦形剤(生理食塩水、腹腔内投与)と賦形剤(カルボキシメチルセルロース、経口投与)+賦形剤(生理食塩水、皮下投与)を投与した。 クロニジンとスルフィソキサゾールとの組み合わせを投与して30分後に、モルヒネまたは賦形剤を投与した。 クロニジン(2mg/kg、腹腔内投与)とスルフィソキサゾールとの組み合わせが、痛覚抑制を顕著に改善していた。 数値は、平均値±標準誤差であり、Nは、6/グループである。 *P<0.05 クロニジンとスルフィソキサゾールとの組み合わせを、対照(賦形剤)グループと比較をした。
【図4】モルヒネ(8mg/kg、腹腔内投与)の存在下および非存在下でのテイルフリック潜時のデータ値から決定したAUC0→240分によって表された痛覚抑制に関するクロニジン(2mg/kg、腹腔内投与)とスルフィソキサゾール(1000mg/kg、経口投与)との組み合わせの効果を示している。 マウスを6つのグループに分けた。 グループ1には、賦形剤(生理食塩水、腹腔内投与)と賦形剤(カルボキシメチルセルロース(CMC))との組み合わせ+賦形剤(生理食塩水、皮下投与)を投与し、グループ2には、クロニジン(2mg/kg、腹腔内投与)と賦形剤(CMC)との組み合わせ+賦形剤(生理食塩水、皮下投与)を投与し、グループ3には、スルフィソキサゾール(1000mg/kg、経口投与)と賦形剤(生理食塩水、腹腔内投与)との組み合わせ+賦形剤(生理食塩水、皮下投与)を投与し、グループ4には、賦形剤(生理食塩水、腹腔内投与)と賦形剤(CMC)との組み合わせ+モルヒネ(8mg/kg、皮下投与)を投与し、グループ5には、クロニジン(2mg/kg、腹腔内投与)とスルフィソキサゾール(1000mg/kg、経口投与)との組み合わせ+賦形剤(生理食塩水、皮下投与)を投与し、および、グループ6には、クロニジン(2mg/kg、腹腔内投与)とスルフィソキサゾール(1000mg/kg、経口投与)との組み合わせ+モルヒネ(8mg/kg、皮下投与)を投与した。 クロニジンとスルフィソキサゾールとの組み合わせを投与して30分後に、モルヒネまたは賦形剤を投与した。 クロニジンとスルフィソキサゾールとの組み合わせが、痛覚抑制を顕著に改善していた。 数値は、平均値±標準誤差であり、Nは、6/グループである。 *P<0.05 モルヒネを、賦形剤グループと比較をした。 #P<0.05 クロニジンとスルフィソキサゾールとの組み合わせを、クロニジンだけのグループ、または、スルフィソキサゾールだけのグループと比較をした。
【図5】マウスでの鎮痛作用(テイルフリック潜時)(図5A)とマウスの体温(図5B)に関するクロニジンの用量応答効果を示している。 グループ1:賦形剤(生理食塩水、腹腔内投与)、グループ2:クロニジン(0.3mg/kg、腹腔内投与)、グループ3:クロニジン(1.0mg/kg、腹腔内投与)、グループ4:クロニジン(3.0mg/kg、腹腔内投与)。
【図6】マウスでの鎮痛作用(図6A)とマウスの体温(図6B)に関するクロニジンとスルフィソキサゾールとの組み合わせの効果を示している。 グループ1:0.5%カルボキシメチルセルロース(CMC、経口投与)+賦形剤(生理食塩水、腹腔内投与)、グループ2:CMC(経口投与)+クロニジン(0.3mg/kg、腹腔内投与)、グループ3:スルフィソキサゾール(250mg/kg、経口投与)+クロニジン(0.3mg/kg、腹腔内投与)、グループ4:スルフィソキサゾール(500mg/kg、経口投与)+クロニジン(0.3mg/kg、腹腔内投与)、グループ5:スルフィソキサゾール(1000mg/kg、経口投与)+クロニジン(0.3mg/kg、腹腔内投与)。
【図7】マウスでの鎮痛作用(図7A)とマウスの体温(図7B)に関するクロニジンとスルフィソキサゾールとの組み合わせの効果を示している。 グループ1:0.5%カルボキシメチルセルロース(CMC、経口投与)+賦形剤(生理食塩水、腹腔内投与)、グループ2:CMC(経口投与)+クロニジン(0.3mg/kg、腹腔内投与)、グループ3:スルフィソキサゾール(25mg/kg、経口投与)+クロニジン(0.3mg/kg、腹腔内投与)、グループ4:スルフィソキサゾール(75mg/kg、経口投与)+クロニジン(0.3mg/kg、腹腔内投与)、グループ5:スルフィソキサゾール(225mg/kg、経口投与)+クロニジン(0.3mg/kg、腹腔内投与)。
【図8】マウスでの鎮痛作用(図8A)とマウスの体温(図8B)に関するクロニジン、および、クロニジンとスルフィソキサゾールとの組み合わせに関するヨヒンビンの効果を示している。 グループ1:賦形剤+賦形剤(生理食塩水、腹腔内投与)+賦形剤(生理食塩水、腹腔内投与)、グループ2:賦形剤+CMC(経口投与)+賦形剤(生理食塩水、腹腔内投与)、グループ3:ヨヒンビン(2mg/kg、腹腔内投与)+CMC(経口投与)+賦形剤(生理食塩水、腹腔内投与)、グループ4:ヨヒンビン(2mg/kg、腹腔内投与)+CMC(経口投与)+クロニジン(0.3mg/kg、腹腔内投与)、グループ5:ヨヒンビン(2mg/kg、腹腔内投与)+スルフィソキサゾール(250mg/kg、経口投与)+クロニジン(0.3mg/kg、腹腔内投与)。
【図9】マウスでの鎮痛作用(図9A)とマウスの体温(図9B)に関するクロニジン、および、クロニジンとスルフィソキサゾールとの組み合わせに関するイダゾキサンの効果を示している。 グループ1:賦形剤+賦形剤(生理食塩水、腹腔内投与)+賦形剤(CMC)、経口投与)、グループ2:イダゾキサン(2mg/kg、腹腔内投与)+CMC(経口投与)+賦形剤(生理食塩水、腹腔内投与)、グループ3:イダゾキサン(2mg/kg、腹腔内投与)+CMC(経口投与)+賦形剤(生理食塩水、腹腔内投与)、グループ4:イダゾキサン(2mg/kg、腹腔内投与)+CMC(経口投与)+クロニジン(0.3mg/kg、腹腔内投与)、グループ5:イダゾキサン(2mg/kg、腹腔内投与)+スルフィソキサゾール(250mg/kg、経口投与)+クロニジン(0.3mg/kg、腹腔内投与)。
【図10】マウスでの鎮痛作用(図10A)とマウスの体温(図10B)に関するクロニジン、および、クロニジンとスルフィソキサゾールとの組み合わせに関するナロキソンの効果を示している。 グループ1:ナロキソン(1.0mg/kg、腹腔内投与)+CMC(経口投与)+賦形剤(生理食塩水、腹腔内投与)、グループ2:ナロキソン(1.0mg/kg、腹腔内投与)+CMC(経口投与)+クロニジン(0.3mg/kg、腹腔内投与)、グループ3:ナロキソン(1.0mg/kg、腹腔内投与)+スルフィソキサゾール(250mg/kg、経口投与)+クロニジン(0.3mg/kg、腹腔内投与)。
【図11】マウスでの鎮痛作用(図11A)とマウスの体温(図11B)に関するクロニジンとスルフィソキサゾールとの組み合わせによる効果と、その他のETAアンタゴニスト(BMS182874)による効果の比較について示している。 グループ1:クロニジン(0.3mg/kg、腹腔内投与)+BMS182874(2.0μg/kg、脳室内投与)、グループ2:クロニジン(0.3mg/kg、腹腔内投与)+BMS182874(10.0μg/kg、脳室内投与)、グループ3:クロニジン(0.3mg/kg、腹腔内投与)+BMS182874(50.0μg/kg、脳室内投与)。
【図12】モルヒネ(8mg/kg、皮下投与)の鎮痛作用に関するクロニジン(1mg/kg、腹腔内投与)とBMS182874(50μg、脳室内投与)の効果を示している。 様々な時点においてテイルフリック潜時を計測し(図12A)、そして、各ラットにおける抗侵害受容応答を、AUC0→360minに変換し(図12B、クロニジン;図12C、BMS182874)、次いで、得られた数値を、平均±標準誤差(V=賦形剤;B=BMS182874;I=イダゾキサン;および、M=モルヒネ)として表した。
【図13】オキシコドン(4mg/kg、皮下投与)の鎮痛作用に関するクロニジン(1mg/kg、腹腔内投与)とBMS182874(50μg、脳室内投与)の効果を示している。 様々な時点においてテイルフリック潜時を計測し(図13A)、そして、各ラットにおける抗侵害受容応答を、AUC0→360minに変換し(図13B、クロニジン;図13C、BMS182874)、次いで、得られた数値を、平均±標準誤差(V=賦形剤;B=BMS182874;I=イダゾキサン;および、O=オキシコドン)として表した。
【図14】モルヒネ(8mg/kg、皮下投与)の鎮痛作用、または、オキシコドン(4mg/kg、皮下投与)の鎮痛作用を高めるクロニジン(1mg/kg、腹腔内投与)に関するヨヒンビン(2mg/kg、腹腔内投与)の効果を示している。 様々な時点においてテイルフリック潜時を計測し(図14A)、そして、各ラットにおける抗侵害受容応答を、AUC0→360minに変換し(図14B、モルヒネ;図14C、オキシコドン)、次いで、得られた数値を、平均±標準誤差(V=賦形剤;C=クロニジン;Y=ヨヒンビン;O=オキシコドン;および、M=モルヒネ)として表した。
【図15】モルヒネ(8mg/kg、皮下投与)の鎮痛作用、または、オキシコドン(4mg/kg、皮下投与)の鎮痛作用を高めるBMS182874(50μg、脳室内投与)に関するヨヒンビン(2mg/kg、腹腔内投与)の効果を示している。 様々な時点においてテイルフリック潜時を計測し(図15A)、そして、各ラットにおける抗侵害受容応答を、AUC0→360minに変換し(図15B、モルヒネ;図15C、オキシコドン)、次いで、得られた数値を、平均±標準誤差(V=賦形剤;B=BMS182874;Y=ヨヒンビン;O=オキシコドン;および、M=モルヒネ)として表した。
【図16A−B】BMS182874(50μg、脳室内投与)の存在下でのモルヒネ(8mg/kg、皮下投与)の鎮痛作用、または、オキシコドン(4mg/kg、皮下投与)の鎮痛作用に関するクロニジン [0(C0)、0.1(C1)、0.3(C2)、および、1.0(C3)mg/kg、腹腔内投与]の効果を示している。 様々な時点においてテイルフリック潜時を計測し(図16A、モルヒネ;図16C、オキシコドン)、そして、各ラットにおける抗侵害受容応答を、AUC0→360minに変換し(図16B、モルヒネ;図16C、オキシコドン)、次いで、得られた数値を、平均±標準誤差(C=クロニジン;B=BMS182874;O=オキシコドン;および、M=モルヒネ)として表した。
【図16C−D】BMS182874(50μg、脳室内投与)の存在下でのモルヒネ(8mg/kg、皮下投与)の鎮痛作用、または、オキシコドン(4mg/kg、皮下投与)の鎮痛作用に関するクロニジン [0(C0)、0.1(C1)、0.3(C2)、および、1.0(C3)mg/kg、腹腔内投与]の効果を示している。 様々な時点においてテイルフリック潜時を計測し(図16A、モルヒネ;図16C、オキシコドン)、そして、各ラットにおける抗侵害受容応答を、AUC0→360minに変換し(図16B、モルヒネ;図16C、オキシコドン)、次いで、得られた数値を、平均±標準誤差(C=クロニジン;B=BMS182874;O=オキシコドン;および、M=モルヒネ)として表した。
【図17】モルヒネ(8mg/kg、皮下投与)の鎮痛作用、または、オキシコドン(4mg/kg、皮下投与)の鎮痛作用に関するクロニジン(1mg/kg、腹腔内投与)単体、および、BMS182874(50μg、脳室内投与)単体、および、クロニジンとBMS182874との組み合わせの効果を示している。 様々な時点においてテイルフリック潜時を計測し(図17A)、そして、各ラットにおける抗侵害受容応答を、AUC0→360minに変換し(図17B、モルヒネ;図17C、オキシコドン)、次いで、得られた数値を、平均±標準誤差(V=賦形剤;C=クロニジン;B=BMS182874;O=オキシコドン;および、M=モルヒネ)として表した。
【図18】鎮痛作用に関するBMS182874(50μg、脳室内投与)単体、および、クロニジン(1mg/kg、腹腔内投与)単体、および、BMS182874(50μg、脳室内投与)とクロニジン(1mg/kg、腹腔内投与)との組み合わせの効果を示している。 様々な時点においてテイルフリック潜時を計測し(図18A)、そして、各ラットにおける抗侵害受容応答を、AUC0→360minに変換し(図18B)、次いで、得られた数値を、平均±標準誤差(V=賦形剤;C=クロニジン;B=BMS182874)として表した。
【技術分野】
【0001】
本願発明は、一般的には、疼痛の治療および鎮痛の増強に関し、対象動物に対して、イミダゾリン活性を有する、または、イミダゾリン活性を欠いたアルファ-2(α2)アドレナリン受容体アゴニストと、エンドセリン受容体アンタゴニストとの組み合わせを投与する、ことを含む。
【背景技術】
【0002】
鎮痛薬とは、好ましくは、意識を攪乱したり、あるいは、その他の感覚機能に影響を及ぼさずに、中枢に作用して疼痛閾値を高めることによって疼痛を緩和する薬剤である。 鎮痛薬が、疼痛を鈍らせる(すなわち、疼痛閾値を高める)作用機序は定式化されている。
【0003】
国立健康統計センター(2006)は、米国では、20歳以上の国民の四分の一以上(26%)、つまり、7650万人以上の国民が、24時間以上にわたって持続するタイプの疼痛に患わされており、また、1億9100万人以上が、急性疼痛を経験している、という報告を行っている。 オピオイドは、急性および慢性の疼痛の臨床管理において最も一般的に用いられている鎮痛薬である。 オピオイドを長期使用することで、効果的な疼痛緩和を妨げる耐性の出現などの副作用を招くこととなる。 鎮痛作用を高め、また、効果的に疼痛に対処する目的で策定された、非ステロイド性抗炎症剤(NSAIDS)、その他のオピオイド、それに、非オピオイドとオピオイド療法との組み合わせなどの幾つかの治療方法がある。 これら手法によって症状の緩和には至るものの、耐性の出現に関与する作用機序に対する効果はほとんど無く、また、毒性、依存性、および、中毒性を得るに至る危険性は非常に大きい。
【0004】
アルファ-2(α2)アドレナリンアゴニストであるクロニジンは、マウスやラットにおいて顕著な鎮痛作用を示すことが実証されており[31]、また、クロニジンを反復して(7日間にわたって毎日2回)投与することで、耐性が出現するのに対して、クロニジンを急性投与すると、モルヒネの鎮痛効果が改善することも知られている[30]。 クロニジンは、モルヒネの鎮痛効果を高めるが、クロニジンの鎮痛作用とモルヒネの鎮痛作用との間には、交差耐性がない[38]。 間脳室周囲灰白質、背側縫線核、および、中脳水道周囲灰白質でのアドレナリン作動性およびアヘン製剤抗侵害受容メカニズムの活性化に対してクロニジンが関与しているかもしれないとの考え方も出されている[39]。 クロニジンは、アヘン製剤離脱を抑制するためにも使用されている。 クロニジンの奏するこれら二つの特性は、オピオイドに対して耐性を示す患者において、好適な鎮痛剤代替物の提供を理論的に可能にしている[27]。 クロニジンは、モルヒネの鎮痛作用のみならず、ペンタゾシン[13]、フェンタニル[9]、および、ブピバカイン[19]の鎮痛作用をも改善することが実証されている。
【0005】
エンドセリン(ETs)は、血管収縮剤の中でも最も強力な血管収縮剤として知られている内皮由来ポリペプチドのファミリーで構成されている。 主要なエンドセリン受容体のサブタイプ(ETAおよびETB)は、血管収縮を司る血管平滑筋において発現する。 内皮細胞にあるETBサブタイプは、血管弛緩を司っているものと考えられている。 ETA受容体アンタゴニストは、ラットとマウスの双方において、モルヒネに対する抗侵害受容応答を顕著に高める、ことが報告されている[5、7、16]。 モルヒネと共にETA受容体アンタゴニストBQ123を慢性注射投与することで、モルヒネに対する耐性の出現が予防された[6]。 ETA受容体アンタゴニストを単回投与することで、マウスとラットにおいて、モルヒネの鎮痛作用に対する耐性が逆になった[4、6、7]。 したがって、ETA受容体アンタゴニストによって耐性が予防および逆転する現象は、ETが、モルヒネ耐性に関与していることを実証するものである[6、7、33、34]。 さらに、ETAアンタゴニストは、強硬症作用[5]や消化管通過[26]などのモルヒネの副作用に関して効果を示さなかった。 したがって、ETA受容体アンタゴニストは、モルヒネの副作用[16]を増大せずに、モルヒネの鎮痛作用を改善するのである。
【0006】
スルフィソキサゾールとは、中耳炎の治療や細菌感染症の治療において広く利用されており、また、好ましい抗菌活性を有している。 スルフィソキサゾールの化学構造は、4-アミノ-N-(3,4-ジメチルオキサゾール-5-イル)-ベンゼンスルホンアミドである。 スルフィソキサゾールは、易溶性である。 スルフィソキサゾールを経口投与すると、迅速に吸収され、また、迅速に排泄され、そして、可溶性にも富んでいるので、スルホンアミドの使用に伴って認められる腎臓毒性も軽減される[25]。 ETAおよびETB受容体に対するET-1の結合に関するスルファニルアミドの阻害効果を、膜調製物において決定をした。 スルフィソキサゾールが、最も活性なスルファニルアミドであり、ETAおよびETB受容体に対するIC50は、それぞれ、0.60μMおよび22μMであった[8]。
【0007】
交感神経系が関与している心臓血管系作用でのクロニジンとETとの間の相互作用についての報告がされている[15、17、18、28]。 エンドセリン-1(ET-1)が、クロニジンによって引き起こされた低血圧症を悪化させることも知られている。 この効果は、一酸化窒素メカニズムを媒介しているのかもしれない[23]。 鎮痛に関するクロニジンとETA受容体アンタゴニストとの間の相互作用を決定するための研究は行われていない。
【0008】
したがって、オピオイド系疼痛緩和剤に対する耐性を軽減し、疼痛症状を緩和し、そして、効果的な非オピオイド鎮痛薬として機能できる薬剤または薬剤の組み合わせを同定することが、当該技術分野において必要とされているのである。
【発明の開示】
【0009】
本願発明は、対象動物の疼痛を治療するための鎮痛剤としてのアドレナリンアゴニストとエンドセリンアンタゴニストの使用に関する。 さらに、本願発明において、アルファ2(α2)アドレナリンアゴニストとエンドセリンA(ETA)アンタゴニストとの組み合わせが、オピオイド鎮痛薬の鎮痛効果を高め、また、その組み合わせが、鎮痛作用の発現に関与していることを知見するに至ったのである。
【0010】
ある実施態様によれば、本願発明は、疼痛を治療または予防する方法であって、治療または予防の必要がある哺乳動物に対して、治療有効量のアルファ-2(α2)アドレナリン受容体アゴニスト、および、治療有効量のエンドセリン受容体アンタゴニストを投与する、ことを含む方法を提供する。
【0011】
ある実施態様によれば、α2アドレナリンアゴニストとは、イミダゾリン活性を有する、または、イミダゾリン活性を欠いたα2アドレナリンアゴニストである。 さらに他の実施態様によれば、α2アドレナリンアゴニストは、デクスメデトミジン、デトミジン、ST-91、メデトミジン、ブリモニジン、チザニジン、ミバゼロール、グアナベンズ、グアンファシン、ヨードクロニジン、キシラジン、リルメニジン、ロフェキシジン、アゼペキソール、アルファ-メチルドパ、および、アルファ-メチルノルアドレナリン、あるいは、これらの誘導体、塩類、または、構造的類似体からなるグループから選択される。 関連する実施態様において、α2アドレナリンアゴニストとは、クロニジンである。
【0012】
さらに別の実施態様によれば、エンドセリン受容体アンタゴニストとして、エンドセリン受容体A(ETA)アンタゴニストを意図している。 別の実施態様によれば、ETAアンタゴニストは、スルフィソキサゾール、アトラセンタン、テゾセンタン、ボセンタン、シタキスセンタン、エンラセンタン、BMS207940、BMS193884、BMS182874、J 104132、VML 588/Ro 61 1790、T-O115、TAK 044、BQ 788、TBC2576、TBC3214、PD180988、ABT 546、SB247083、RPR118031A、および、BQ123からなるグループから選択される。 さらに別の実施態様によれば、ETAアンタゴニストは、スルフィソキサゾールである。
【0013】
ある実施態様によれば、α2アドレナリンアゴニストおよびエンドセリン受容体アンタゴニストは、単一の組成物で投与される。 関連する実施態様によれば、α2アドレナリンアゴニストおよびエンドセリン受容体アンタゴニストは、個別の組成物で投与される。
【0014】
ある実施態様によれば、これら組成物は、同時に投与される。 関連する実施態様によれば、これら組成物は、別個に投与される。 さらに別の実施態様によれば、α2アドレナリンアゴニストおよびエンドセリン受容体アンタゴニスト分子は、約24時間以内に連続投与される。
【0015】
さらに別の実施態様によれば、これら組成物は、医薬担体または医薬賦形剤をさらに含む。
【0016】
さらに他の実施態様によれば、α2アドレナリンアゴニストおよびエンドセリン受容体アンタゴニストは、経口的、経頬的、吸入的、舌下、経直腸的、経膣的、嚢内、関節内、経尿道的、経鼻的、経皮的、静脈内、筋肉内、または、皮下に投与される。
【0017】
ある実施態様によれば、クロニジンは、約10μg〜約300μgの範囲の用量で投与される。 関連する実施態様によれば、スルフィソキサゾールは、約0.1g〜約3gの範囲の用量で投与される。 また、投与されたα2アドレナリンアゴニストと投与されたエンドセリン受容体アンタゴニストとの比率を、1:500〜1:50,000、1:500〜1:20,000、1:500〜1:10,000、1:500〜1:5,000、1:500〜1:2,500、1:100〜1:1000、または、1:100〜1:500の範囲とすることも意図している。
【0018】
その他の実施態様によれば、本願発明は、疼痛を治療または予防する方法であって、対象動物に対して、一つまたはそれ以上のアルファ-2(α2)アドレナリン受容体アゴニストと、一つまたはそれ以上のエンドセリン受容体アンタゴニストとの相乗的組み合わせを投与する、ことを含む方法を提供する。 ある実施態様によれば、その組み合わせに用いられたアゴニスト分子およびアンタゴニスト分子が、約24時間以内に連続投与されるか、あるいは、同時投与される。 薬剤の投与を、30分間〜約1日(24時間)までの範囲で行うことも意図されている。
【0019】
本願発明は、一つまたはそれ以上のアルファ-2(α2)アドレナリンアゴニストと、一つまたはそれ以上のエンドセリン受容体アンタゴニストとの相乗的組み合わせを含む、疼痛を治療または予防するための組成物をさらに提供する。 ある実施態様によれば、この組成物は、低用量のα2アドレナリンアゴニストと低用量のエンドセリン受容体アンタゴニストを含む。 関連する実施態様によれば、α2アドレナリンアゴニストは、クロニジンであり、そして、エンドセリン受容体アンタゴニストは、スルフィソキサゾールである。 さらに別の実施態様によれば、この組成物には、約10μg〜約300μgの範囲のクロニジンと、約0.1g〜約3gの範囲のスルフィソキサゾールが含まれている。
【0020】
さらに他の実施態様によれば、その組成物は、薬学的に許容可能な担体をさらに含んでいる。
【0021】
さらに別の実施態様によれば、本願発明は、対象動物での疼痛を治療するための薬剤を製造するためのアルファ-2(α2)アドレナリンアゴニストとエンドセリン受容体アンタゴニストを含む組成物の使用も提供する。
【0022】
本願発明は、哺乳動物を治療対象とすることを意図している。 ある実施態様によれば、治療対象となる哺乳動物として、ヒト、または、ヒトに関する医学研究のための非ヒト動物モデル、あるいは、家畜として重要な動物、あるいは、例えば、愛玩動物のようなペットがある。 関連する実施態様によれば、対象動物は、ヒトである。
【0023】
ある実施態様によれば、治療の対象となる疼痛は、慢性疼痛または急性疼痛である。 関連する実施態様によれば、疼痛は、灼熱痛、接触性アロディニア、神経因性疼痛、痛覚過敏症、痛感過敏症、炎症性疼痛、術後疼痛、慢性腰痛、群発頭痛、帯状疱疹後神経痛、幻肢痛および断端痛、中心性疼痛、歯痛、神経因性疼痛、オピオイド耐性疼痛、内臓痛、術後疼痛、骨損傷痛、糖尿病性神経障害疼痛、術後または外傷性神経症疼痛、末梢神経障害疼痛、エントラップメント神経障害疼痛、アルコール依存症に起因する神経障害、HIV感染、多発性硬化症、甲状腺機能低下症に起因する疼痛、または、抗癌化学療法に起因する疼痛、分娩中の疼痛、日焼けを含む火傷に起因する疼痛、産後疼痛、偏頭痛、咽喉痛、および、膀胱炎などの尿生殖路関連疼痛からなるグループから選択される。
【0024】
さらに別の実施態様によれば、一つまたはそれ以上のアルファ-2(α2)アドレナリンアゴニスト、または、一つまたはそれ以上のエンドセリン受容体アンタゴニストは、麻薬性鎮痛薬の鎮痛作用を高める上で有用である。 ある実施態様によれば、一つまたはそれ以上のアルファ-2(α2)アドレナリンアゴニストと一つまたはそれ以上のエンドセリンアンタゴニストとの組み合わせは、麻薬性鎮痛薬の鎮痛作用を高める上で有用である。 このように、本願発明は、疼痛を治療または予防する方法であって、治療または予防の必要がある哺乳動物に対して、治療有効量の麻薬性鎮痛薬、および、一つまたはそれ以上のアルファ-2(α2)アドレナリンアゴニストを含む治療有効量の組成物を投与する、ことを含む方法を提供する。 関連する実施態様によれば、本願発明は、疼痛を治療または予防する方法であって、治療または予防の必要がある哺乳動物に対して、治療有効量の麻薬性鎮痛薬、一つまたはそれ以上のアルファ-2(α2)アドレナリンアゴニストを含む治療有効量の組成物、および、一つまたはそれ以上のエンドセリン受容体アンタゴニストを含む治療有効量の組成物を投与する、ことを含む方法を意図している。 さらに別の実施態様によれば、本願発明は、疼痛を治療または予防する方法であって、治療または予防の必要がある哺乳動物に対して、治療有効量の麻薬性鎮痛薬、および、一つまたはそれ以上のアルファ-2(α2)アドレナリンアゴニストと、一つまたはそれ以上のエンドセリン受容体アンタゴニストとを含む治療有効量の組成物を投与する、ことを含む方法を提供する。
【0025】
ある実施態様によれば、一つまたはそれ以上のアルファ-2(α2)アドレナリンアゴニストは、デクスメデトミジン、デトミジン、ST-91、メデトミジン、ブリモニジン、チザニジン、ミバゼロール、グアナベンズ、グアンファシン、ヨードクロニジン、キシラジン、リルメニジン、ロフェキシジン、アゼペキソール、アルファ-メチルドパ、および、アルファ-メチルノルアドレナリン、または、これらの誘導体、塩類、または、構造類似体からなるグループから選択される。 関連する実施態様によれば、一つまたはそれ以上のエンドセリンアンタゴニストは、スルフィソキサゾール、アトラセンタン、テゾセンタン、ボセンタン、シタキスセンタン、エンラセンタン、BMS207940、BMS193884、BMS182874、J 104132、VML 588/Ro 61 1790、T-O115、TAK 044、BQ 788、TBC2576、TBC3214、PD180988、ABT 546、SB247083、RPR118031A、および、BQ123からなるグループから選択されるETAである。
【0026】
さらに別の実施態様によれば、麻薬性鎮痛薬は、モルヒネ、硫酸モルヒネ、コデイン、ジアセチルモルヒネ、デキストロメトルファン、ヒドロコドン、ヒドロモルフォン、ヒドロモルフォン、レボルファノール、オキシモルホン、オキシコドン、レバロルファン、および、これらの塩類からなるグループから選択される。
【0027】
ある実施態様によれば、麻薬性鎮痛薬、および、一つまたはそれ以上のアルファ-2(α2)アドレナリンアゴニスト、および/または、一つまたはそれ以上のエンドセリンアンタゴニストは、同時に投与される。 関連する実施態様によれば、麻薬性鎮痛薬、および、一つまたはそれ以上のアルファ-2(α2)アドレナリンアゴニスト、および/または、一つまたはそれ以上のエンドセリンアンタゴニストは、単一の組成物または別個の組成物から投与される。 さらに別の実施態様によれば、麻薬性鎮痛薬、および、一つまたはそれ以上のアルファ-2(α2)アドレナリンアゴニスト、および/または、一つまたはそれ以上のエンドセリンアンタゴニストは、連続して投与される。
【0028】
さらに他の実施態様によれば、麻薬性鎮痛薬は、一つまたはそれ以上のアルファ-2(α2)アドレナリンアゴニスト、および/または、一つまたはそれ以上のエンドセリンアンタゴニストに先駆けて投与され、あるいは、、一つまたはそれ以上のアルファ-2(α2)アドレナリンアゴニスト、および/または、一つまたはそれ以上のエンドセリンアンタゴニストに続いて投与される。
【0029】
本明細書に記載の活性成分の用量、および、本明細書に記載の投薬計画は、本願発明において意図しているすべての方法に対して有用である。
【0030】
その他の実施態様によれば、本願発明は、アルファ-2(α2)アドレナリンアゴニストとエンドセリン受容体アンタゴニスト、それに、本願発明の方法を示したラベルを含む製造品も意図している。
【0031】
さらに別の実施態様によれば、本願発明は、疼痛を治療または予防するためのキットであって、アルファ-2(α2)アドレナリンアゴニストとエンドセリン受容体アンタゴニストを含む組成物、および、そのキットを用いて疼痛を治療するためのプロトコル、を含むキットが提供される。 ある実施態様によれば、その組成物は、薬学的に許容可能な担体をさらに含む。 関連する実施態様によれば、このキットは、麻薬性鎮痛薬をさらに含む。 関連する実施態様によれば、麻薬性鎮痛薬は、薬学的に許容可能な担体または賦形剤と共にある。
【発明を実施するための最良の形態】
【0032】
本願発明は、顕著な鎮痛作用を示し、また、疼痛刺激も顕著に緩和する、α2アドレナリンアゴニストとエンドセリン受容体アンタゴニストとの組み合わせを用いて疼痛を治療する方法に関する。 具体的には、本願発明は、ETAアンタゴニストとアルファ-2 アドレナリンアゴニストとを併用することで、オピオイド疼痛緩和剤に対する耐性が顕著に減少し、また、疼痛症状も消失する、との発見に関するものである。
【0033】
本願発明の方法は、強力な鎮痛作用を呈するα2アドレナリンアゴニスト(例えば、クロニジン)とETA受容体アンタゴニスト(例えば、スルフィソキサゾール)との組み合わせを利用するものである。 ある実施態様によれば、本願発明の方法は、マウスでのテイルフリック潜時によって実証されている通り、スルフィソキサゾールとクロニジンによって増強された鎮痛作用を利用している。 本願発明の方法にしたがってクロニジンとスルフィソキサゾールとを併用して得られる鎮痛作用は、高用量のモルヒネが奏する鎮痛作用に匹敵するものであるので、特筆すべき事項といえる。
【0034】
本明細書で用いる「治療」の用語は、疼痛を予防、軽減、あるいは、改善することを指す。 そのようなものとして、「治療」の用語は、必要に応じて、内科治療的投与、および/または、予防的投与の双方を含んでいる。 疼痛症状の治療および緩和は、当該技術分野で周知の疼痛評価スケールを用いて評価することができる[例えば、3、5、9を参照されたい]。 評価手順の例として、客観的疼痛閾値(視覚的アナログ尺度)や客観的侵害受容屈曲反射(R III)閾値の測定がある。
【0035】
本明細書で用いる「疼痛」の用語は、すべてのタイプの疼痛を指す。 ある実施態様によれば、この用語は、急性疼痛と慢性疼痛を指す。 疼痛の例として、灼熱痛、接触性アロディニア、神経因性疼痛、痛覚過敏症、痛感過敏症、炎症性疼痛、術後疼痛、慢性腰痛、群発頭痛、帯状疱疹後神経痛、幻肢痛および断端痛、中心性疼痛、歯痛、神経因性疼痛、オピオイド耐性疼痛、内臓痛、術後疼痛、骨損傷痛、糖尿病性神経障害疼痛、術後または外傷性神経症疼痛、末梢神経障害疼痛、エントラップメント神経障害疼痛、アルコール依存症に起因する神経障害、HIV感染、多発性硬化症、甲状腺機能低下症に起因する疼痛、または、抗癌化学療法に起因する疼痛、分娩中の疼痛、日焼けを含む火傷に起因する疼痛、産後疼痛、偏頭痛、咽喉痛、および、膀胱炎などの尿生殖路関連疼痛などがあるが、これらに限定されない。
【0036】
本明細書で用いる「鎮痛剤」の用語は、対象動物での疼痛を緩和する活性成分のことを指す。 「麻薬性鎮痛薬」または「オピオイド鎮痛剤」の用語は、例えば、麻酔剤の補助として、あるいは、疼痛を緩和するために用いられる麻薬性鎮痛剤を指す。 「非麻薬性鎮痛薬」の用語は、疼痛の治療に適応される非麻酔剤を指す。
【0037】
「治療有効量」とは、所望の効果を奏する活性成分または薬剤の量のことを指す。 それら活性成分の毒性や治療効果は、細胞培養物や実験動物を用いた標準的な製剤手法、例えば、 LD50(個体群の50%に対する致死用量)やED50(個体群の50%に対する治療効果用量)によって決定される。 毒性と治療効果との間の用量比は、LD50とED50との間の比率として表される治療指数である。 治療指数は、大きい方が望ましい。 これらデータから取得されるデータは、ヒトに対して用いる用量の範囲を決定するために用いられている。 ある実施態様によれば、活性成分の用量は、ED50を含む循環濃度の範囲内にあり、その毒性は皆無か、あるいは、ほとんどない。 これら用量は、使用する剤形や、利用した投与経路に応じて、この範囲内で変化する。
【0038】
α2アドレナリンアゴニストとエンドセリン受容体アンタゴニストの「相乗的組み合わせ」とは、その活性成分だけを投与した場合に得られる効果の合計よりも大きな効果を示す組み合わせのことを指す。
【0039】
本明細書で用いる「増強する」または「増強作用」という用語は、鎮痛剤の効果を高めたり、あるいは、鎮痛剤と共に相乗的に作用して、例えば、生化学的効果や生理学的効果を増強するアルファ-2(α2)アドレナリンアゴニストまたはエンドセリンアンタゴニストの性質のことを指す。 ある実施態様によれば、この増強作用によって、所望の疼痛緩和効果を得るに必要な鎮痛剤の用量が効果的に減量している。
【0040】
「同時投与」、「組み合わせで投与した」、「同時の投与」、または、それらと同様の表現は、二つまたはそれ以上の薬剤を含む組成物が、治療を受けている対象動物に対して同時に投与されることを意味している。 「同時」の表現は、各薬剤が、同時に投与されること、あるいは、所定の時間内に任意の順序で連続して投与されること、を意味している。 しかしながら、同時に投与されない場合であっても、ある実施態様によれば、所望のレベルの治療効果を引き出すために、所定の時間内において、ごく短時間の内に投与が行われる。 薬剤の適切な投与間隔と投与順序は、当業者に自明の事項である。 二つまたはそれ以上の薬剤を、個別の組成物を用いて投与することも意図しており、ある実施態様によれば、一方の薬剤は、他方の薬剤を投与する以前または以後に投与される。 事前投与とは、治療を行う1日(24時間)前から治療を行う30分前までの間に薬剤を投与することを指す。 さらに、一方の薬剤を、他方の薬剤を投与した後に投与することも意図している。 事後投与とは、第一薬剤を投与して30分以後の投与から、第一薬剤の投与から1日(24時間)後の投与までを指す。 30分から24時間の範囲とは、30分、1、2、3、4、5、6、7、8、9、10、11、12、16、20、または、24時間の時点での投与を含むものである。
【0041】
本明細書で用いる「低用量」という用語は、組成物に含まれる活性成分の用量のことを指し、組成物に含まれる活性成分の量は、対象動物の治療において一般的に用いられる量よりも少ない。 例えば、低用量の活性成分を、第二の活性成分と組み合わせて投与して、活性成分の相乗効果を引き出すことができ、また、組み合わせ療法で用いる各活性成分の用量を、第二の活性成分と組み合わせずに薬剤を投与する場合に必要な用量よりも少なくすることもできる。 ある実施態様によれば、低用量のクロニジンは、10μg〜約300μgの範囲にある。 関連する実施態様において、低用量のスルフィソキサゾールは、0.1g〜約3.0gの範囲にある。
【0042】
アルファ-2(α2)アドレナリンアゴニスト
アルファ-2(α2)アドレナリン作動性受容体(アドレナリン受容体)は、神経系や身体のその他の各系統の双方において遍在して分布をしている。 α2アドレナリン受容体は、三つの異なる受容体サブタイプ(サブタイプA、BおよびCと称する)を含んでおり、これらは、非選択的内因性アドレナリンアゴニストアドレナリンおよびノルアドレナリンによって活性化され、また、その他の6個のアドレナリン受容体サブタイプをも活性化する(米国特許第6,562,855号)。 臨床研究によって、α2アゴニストが、強力な鎮痛作用を示すことが明らかになっている。 クロニジンやデクスメデトミジンのようなα2アゴニストの全身投与および脊髄投与は、ヒトや動物モデルでの疼痛を緩和する。 α2アゴニストは、脊柱作用ならびに局所脊髄麻酔作用によって鎮痛効果を奏する(本明細書の一部を構成するものとしてその内容を援用するGuo et al, Anesthesiology 1991; 75: 252-6、米国特許第6,562,855号)。 α2アドレナリンアゴニストに関するその他の知見は、本明細書の一部を構成するものとしてその内容を援用する米国特許第6,562,855号、第5,605,911号、および、第5,980,927号に記載されている。
【0043】
α2アドレナリンアゴニストであるクロニジンは、マウスやラットで顕著な鎮痛作用を呈することが実証されている[31]。 また、クロニジンを継続して(毎日2回、7日間)投与することで、耐性が出現することも知られている。 その一方で、クロニジンの急性投与によって、モルヒネの鎮痛効果が高まることも知られている[30]。 他の研究によれば、クロニジンが、モルヒネの鎮痛作用を増強するものの、クロニジンとモルヒネの鎮痛作用との間には交差耐性が認められなかったと報告されている[38]。 ホルマリン試験による疼痛感受性の評価結果は、クロニジンが、鎮痛作用を呈すること、そして、その効果が、ナロキソン(2mg/kg、腹腔内投与)によって阻害されることを示しており、また、クロニジン効果に関わるナロキソン感受性成分は、β-エンドルフィン様オピオイドの放出に起因するものであると推察されている[22]。 クロニジンは、アヘン製剤離脱を鎮静するためにも使用されている。 これら二つの特性が故に、クロニジンは、オピオイドに対して耐性を示す患者での魅力的な鎮痛剤代替物となっている[27]。 クロニジンが、モルヒネの鎮痛作用のみならず、ペンタゾシン[13]、フェンタニル[9]、および、ブピバカイン[19]の鎮痛作用をも高めることが実証されている。 さらに、クロニジンの鎮痛作用は、アミトリプチリンによっても高められることが知られている[1]。
【0044】
動物でのクロニジンの鎮痛作用が、健常な志願者が参加した交差式・二重盲検式・プラシーボ対照式試験法によって研究がされており、この試験では、偽薬、クロニジン(0.2mg、経口投与)、または、クロニジンおよびナロキソン(2.8mg、5時間にわたる点滴)が被験者に対して投与された。 経皮電気刺激を与えた後に、主観的疼痛閾値(視覚的アナログ尺度)と客観的侵害作用屈曲反射(R III)閾値の計測を行って、鎮痛作用を評価した。 主観的閾値と客観的閾値との間で相関が認められた(r:0.78)。 クロニジン単独を経口投与することで、あるいは、クロニジンとナロキソンとの組み合わせを経口投与することで、少なくとも4時間にわたって、主観的疼痛閾値と客観的疼痛閾値が増大した(0.01未満のP値、ANOVA). ナロキソンは、クロニジンの鎮痛作用を増強する傾向にあった。 適度で、良好な耐容性の副作用だけが認められた[32]。
【0045】
成人を対象とした研究において、術前、術中、または、術後に、クロニジンを投与することで、顕著な鎮痛効果が認められ、また、副作用の発現が少なくなる、ことが明らかにされている。 小児患者でのクロニジンの効果を決定するための研究も行われている。 無作為に選んだ1〜7歳の45名の小児患者にクロニジンを硬膜外投与したところ、術後のブピバカインの仙骨麻酔の効果が、1μg/kgのクロニジンを加えることで、顕著に改善したことが報告されている[20]。 身体の78%に第二度および第三度熱傷をおった11歳の少年に対して、大量のモルヒネを投与したところ重度の副作用が認められたが、低用量のクロニジンを静脈投与することで、その副作用は顕著に軽減された[24]。 しかしながら、小児患者に関する研究において、術前の鎮静状態下では、クロニジンの経口投与よりも、ミダゾラムの方が有効であるが、得られる鎮痛作用は、小児麻酔においても有効であると考えられる[29]。
【0046】
様々な薬剤が、補助鎮痛薬として使用されているが、それらは、もともも疼痛以外の主症状に対応するために開発されたものである。 これら薬剤は、特定の条件下で鎮痛作用を高めるために用いられており、また、それらの内の幾つかは、主に、鎮痛薬として用いられている[21]。 アミトリプチリン、ノルトリプチリン、および、デシプラミンのような三環系抗鬱薬は、たいていの神経因性疼痛に対して有効である[36]。 ブプロピオン、ベンラファクシン、および、デュロキセチンも、神経因性疼痛管理において有効であることが知られている[35、37]。 抗癲癇薬は、神経因性疼痛の管理に最も適した薬剤になりつつあり、ガバペンチンとプレガバリンの双方について、神経因性疼痛に対する効果が確認されている[2、12]。 クロニジンは、鎮痛剤として主作用の他に、オピオイドと併用することで、抗侵害受容効果をも相乗的に奏する[14]。 チザニジンは、比較的に短時間作用型のα2アドレナリンアゴニストであり、また、降圧作用がクロニジンに比べてかなり小さいが、疼痛障害において利用可能である[11]。 NMDAアンタゴニストデキストロメトルファン、メタドン、メマンチン、アマンタジン、および、ケタミンは、痛覚過敏神経症状態に対して効果的であると思われる[3]。
【0047】
抗侵害受容特性を有する非アヘン製剤であるクロニジンは、術後に鎮痛作用を示さないという副作用を有するオピオイドの代替物となるかもしれない。 脊椎固定術を行った直後の50名の患者を、二つのグループに無作為に振り分け、そして、クロニジン(最初の1時間に5μg/kgを点滴し、次いで、11時間かけて、0.3μg/kg/時間を点滴した)または偽薬のいずれかを無分別に投与をして、術後期間に静脈投与をしたクロニジンの鎮痛作用を決定するための研究が行われた。 クロニジンまたは偽薬を投与する以前(T0)、負荷用量の終了時(T1)、それに、その2時間ごと(T3、T5、T7、T9およびT11)に、疼痛を評価するために、視覚的アナログ尺度を用いて、0(無疼痛)〜100mmに等級付けをした。 スコアが50mmを超えた場合には、各々の疼痛測定の後に、モルヒネ(0.1 mg/kg)を、筋肉内に投与した。 T0の時点では、モルヒネは与えていない。 疼痛スコアを測定した各時点において、血行動態、血液ガス、および、血漿クロニジン濃度を測定した。 クロニジン投与グループでは、疼痛スコアが、42+/-5〜26+/-3mm(平均値+/-標準誤差)にまで減少したが、偽薬投与グループでは、モルヒネ要求量が多かったにもかかわらず(各患者に対する用量:3.8+/-1に対して10.8+/-1.2mg)、疼痛スコアに変化は認められなかった。 クロニジンは、疼痛の発症を遅らせるので、最初のモルヒネ注射の時期も遅れることとなる。 クロニジン投与グループでは、累積液量P1が顕著に増大するにもかかわらず、平均動脈圧は、74+/-2mmHg(T11での偽薬投与グループにおける-15+/-2%に対して、-26+/-2)にまで減少した。
【0048】
腹式子宮摘出術を受けた13名の患者に対してクロニジンを硬膜外投与した後の効果を決定するための研究が行われている。 血圧および口頭式類推疼痛スコアにおいて、顕著な減少が認められた。 硬膜外にクロニジン(150μg)を投与した後の鎮痛の程度は適度なものであり、また、作用は長続きはしなかったが、硬膜外の空間から血液へのクロニジンの吸収は非常に迅速であった[66]。 ある実験によれば、クロニジンとグアンファシンとの間に明確な血液動態的差異が認められないにもかかわらず、グアンファシンの方が、長時間にわたって痛覚抑制(グアンファシンの8時間に対して、クロニジンの5.5時間)を呈することが認められている。 長時間にわたる作用の持続性と呼吸抑制が少ない点に鑑みて、硬膜外へのグアンファシンの投与は、術後鎮痛と慢性疼痛症候群に対して有利であると言うことができる[67]。
【0049】
人工股関節全置換術を受けた患者を対象とした二重盲検式比較試験において、術後鎮痛に関するクロニジンの硬膜外投与の効果と、モルヒネの硬膜外投与に関するクロニジンの効果が検証されている。 二つの用量の硬膜外クロニジン(25μg/時間、または、50μg/時間)、低用量の硬膜外モルヒネ、または、モルヒネとクロニジンとの組み合わせのいずれかを無作為に、患者に対して投与した。 術後の最初1時間でのモルヒネを投与したグループの疼痛スコアは、クロニジン投与グループ(0.01未満のP値)と組み合わせ投与グループ(0.05未満のP値)のスコアと比べて非常に大きかった。 モルヒネ投与グループと低用量クロニジングループ(0.05未満のP値)と比較して、組み合わせ投与グループと多用量クロニジングループは、全身鎮静の必要性が最も少なく、また、最初のボーラス用量の持続効果も非常に大きかった。 クロニジングループでの臨床的低血圧症の兆候は顕著ではなかったが、動脈圧の減少が認められた。 嘔吐症状または尿閉に関して、グループ間での顕著な相違は認められなかった[70]。
【0050】
手術の1時間前および12時間後に、クロニジン(300μg)を経口投与したところ、モルヒネ要求性の改善はわずかであったが、腹部の大手術を受けた患者の心拍数を十分に下げ、また、鎮静状態を顕著に改善した。
【0051】
帝王切開分娩をした患者が参加した無作為化二重盲検式用量依存性試験で、硬膜外モルヒネに対してクロニジンを添加して得られる効果を調べた。 低用量の75μgのクロニジンが奏する鎮痛作用の持続期間が、2mgのモルヒネが示す持続期間の倍であり、また、術後でのモルヒネ要求性が、クロニジンを添加することで顕著に軽減されることが明らかとなった[68]。
【0052】
膝の整形外科的大手術を受けた成人患者での術後の鎮痛作用およびPCAモルヒネ要求性に関する術中のクロニジンの経口投与の効果が評価されている。 クロニジンは、偽薬と比較して、術後の吐き気と嘔吐の症状を顕著に軽減し、また、PCAモルヒネ要求性も顕著に軽減した[65]。 脊髄内モルヒネ麻酔を施して腹式単純子宮全摘術を受けた37〜60歳の26名の患者に対して、クロニジン(5μg/kg)を経口的に前投与したところ、モルヒネの副作用を増強せずに、術後のモルヒネの鎮痛作用が増大した[63]。 クロニジン(4μg/kg)を経口投与することで、帝王切開をした後でも、胎児や新生児の状態を危険にさらすことなく、PCAモルヒネ要求性を軽減することが明らかになった[64]。
【0053】
下腹部手術を受けた患者に対して、無作為化二重盲検式比較試験への参加を募った。 グループCの外科処置の最後の段階で、4μg/kg/20分のクロニジンの点滴と、20μgのPCAクロニジンと1mgのモルヒネのボーラス投与を行った。 グループMには、生理食塩水の点滴と、1mgのPCAモルヒネのボーラス投与を行った。 疼痛状態、鎮静状態、それに、吐き気と嘔吐を、12、24および36時間後に評価を行い、また、鎮痛効果に関する満足度を、36時間の時点で評価をした。 グループCでの疼痛スコアは、0時間と12時間との間では非常に小さかったが、その後は、大差がなかった。 24〜36時間に至るまでの双方のグループでのモルヒネ消費量は同じであった。 グループCでの0時間と24時間との間において、吐き気と嘔吐は、顕著に軽減されていた。 クロニジンを用いたグループの患者は、疼痛が緩和されたことを非常に喜んでいた[62]。
【0054】
椎間板手術後の疼痛緩和のために用いた硬膜外投与された低用量モルヒネとクロニジンとの組み合わせの効果と副作用についての研究が行われている。 モルヒネとクロニジンの硬膜外投与が、ブピバカインとクロニジンの硬膜外投与と比較して、術後疼痛を顕著に緩和し、そして、ピリトラミドの消費量も減少していたことが明らかになった[61]。
【0055】
冠動脈バイパス移植手術を受けた45名の患者を、自己調節鎮痛(PCA)モルヒネ(ボーラス、1mg;ロックアウト時間、7分間)だけを点滴で投与するグループ(コントロールグループ)、4μg/kgのモルヒネを髄腔内にさらに投与するグループ、あるいは、4μg/kgのモルヒネと1μg/kgのクロニジンの双方を髄腔内にさらに投与するグループへと無作為に分けて、無作為化二重盲検比較試験をした。 全身麻酔を行う以前に、髄腔内注射を行った。 視覚的アナログ尺度(VAS)を用いて、術後に疼痛の評価を行った。 モルヒネの用量[平均値(第一〜第三四分位数)]は、モルヒネとクロニジンを髄腔内投与された患者での最初の24時間の用量[7(0〜37)mg]の方が、その他の患者[モルヒネを髄腔内投与したグループでの40.5(15〜61.5)mg、および、モルヒネを点滴で投与したグループでの37(30.5〜51)mg]よりも、少なかった。 VASスコアは、コントロールグループと比較して、モルヒネとクロニジンを髄腔内投与した後の方が小さかった。 点滴でモルヒネを投与したグループと比較して、モルヒネとクロニジンを髄腔内投与した後の方が、抜管に要する時間が短かった[330(300〜360)分に対して225(195〜330)分、P<0.05]。 モルヒネとクロニジンの髄腔内投与を行うことで、冠動脈バイパス移植手術の術後において効果的な鎮痛効果を与えることができ、また、抜管の作業が迅速に行える[60]。
【0056】
クロニジン(120μg)とフェンタニル(50μg)との組み合わせは、分娩第一期に用いられている0.25%ブピバカインと同等の硬膜外鎮痛作用を呈し、また、好ましくない神経学的副作用も軽減する[59]。 しかしながら、クロニジンとモルヒネの組み合わせの硬膜外への投与は、ブピバカインとスフェンタニルを併用する自己調節療法と比較して、鎮痛作用が劣り、また、副作用も大きい[58]。 その他の研究によれば、硬膜外投与されたブトルファノールにクロニジンを加えても、その鎮痛作用に変化はなく、むしろ、開腹手術を受けた患者に悪影響を及ぼすとの報告もされている[57]。 健常者にクロニジンを経口的に前投与すると、感覚運動ブロックの長期化を招かずに、望ましい鎮静状態、不安緩解、それに、安定した血行動態をもたらす。 5μg/kg用量のクロニジンだけに副作用が認められ、2.5μg/kg用量のクロニジンでの副作用は、最小のものでしかなかった[56]。 クロニジン(0.75μg/kg)の使用は、臨床的に副作用が非常に小さいが故に、フェンタニル(0.5μg/kg)よりも望ましいことが証明されている[55]。 クロニジン(5μg/kg)を経口的に前投与すると、プロポフォールの導入量を減らすことができるが、プロポフォール麻酔の効果の出現が遅くなる[54]。
【0057】
関節鏡視下膝手術の術後に、クロニジン(150μg)を関節内に投与したところ、5mgのモルヒネと比較して、疼痛緩和が長期にわたって持続をした[53]。
【0058】
イミダゾリン活性を有する、または、イミダゾリン活性を欠いている、本願発明での使用が意図されている、その他のα2アドレナリンアゴニストとして、デクスメデトミジン、デトミジン、ST-91、メデトミジン、ブリモニジン、チザニジン、ミバゼロール、グアナベンズ、グアンファシン、ヨードクロニジン、キシラジン、リルメニジン、ロフェキシジン、アゼペキソール、アルファ-メチルドパ、および、アルファ-メチルノルアドレナリン、または、これらの誘導体、塩類、または、構造類似体などがあるが、これらに限定されない。
【0059】
エンドセリン受容体アンタゴニスト
ETAおよびETBの双方に結合するエンドセリン(例えば、ET-1、ET-2、および、ET-3)は、強力な血管収縮剤である。 エンドセリンは、プレプロホルモンとして合成され、そして、翻訳後プロセシングによって、活性ペプチドへと変化する。 ET-1プロセシングの特徴は周知であり、212個のアミノ酸のペプチド(プレプロET-1)から始まり、次いで、エンドペプチダーゼの作用を受けて、タンパク質分解的に開裂して巨大ET-1(プロET-1)となる。 39個のアミノ酸のプロET-1は、金属エンドプロテアーゼエンドセリン変換酵素(ECE)の作用を受けて開裂し、その結果、強力な生物学的機能を具備した21個のアミノ酸からなるタンパク質となる(Fagan et al., Respiratory Research 2:90-101, 2001)。
【0060】
本明細書で使用する「エンドセリンアンタゴニスト」および「エンドセリン受容体アンタゴニスト」の用語は、互換的に用いられている。 エンドセリン受容体アンタゴニストは、急性心不全、鬱血性/慢性心不全、肺動脈高血圧症、肺水腫、くも膜下出血、慢性閉塞性肺疾患、心筋梗塞、急性脳虚血、急性冠不全症候群、急性腎不全、肝臓手術の術後状態、および、前立腺癌を治療するために用いられている。
【0061】
幾つかの研究によって、構造的に異なるETA受容体アンタゴニストであるBQ123(ペプチド)とBMS182874(非ペプチド)が、マウスとラットの双方において、モルヒネの鎮痛反応を増強する、ことが明らかにされている[5〜7、16、26、33]。 交感神経系に関与する心臓血管系作用でのクロニジンとETとの間における相互作用についても報告がされている[15、17、18、28]。 エンドセリン-1(ET-1)が、クロニジンに起因する低血圧症を悪化させることも知られている[23]。 しかしながら、鎮痛作用を示すクロニジンに関するETA受容体アンタゴニストの効果に関する報告は皆無である。 スルフィソキサゾールが、ETAおよびETB受容体のそれぞれに対して、0.60μMおよび22μMのIC50値を示す最も活性に富んだスルファニルアミドである、ことが明らかにされている[8]。 したがって、ここでの研究は、クロニジンを併用して投与した場合における、鎮痛作用に関するスルフィソキサゾールの効果を決定するために行ったものである。 これら研究の結果は、クロニジンとスルフィソキサゾールの組み合わせが、高用量のモルヒネと同等の強力な鎮痛作用を示すことを実証している(実施例5を参照されたい)。
【0062】
弱いエンドセリンAアンタゴニストであるスルフィソキサゾールは、血漿タンパク質に広く結合し、そして、2〜4gの用量で経口投与をして2〜4時間後には、110〜250μg/mlというピーク値の血漿濃度に達する。 尿中のスルフィソキサゾールの濃度は、血中のスルフィソキサゾールの濃度よりも高く、また、脳脊髄液でのスルフィソキサゾールの濃度は、平均して、血中のスルフィソキサゾールの濃度の約1/3である。 大抵(95%)の薬剤は、24時間以内に、腎臓から尿中に排出される(Goodman Gilmans 1990 8th Edition).
スルフィソキサゾールの大半は、細胞外空間に存在しており、そして、脳脊髄液での含有濃度は、血中濃度の10〜80%の範囲の濃度で対応している(Goodman Gilmans 1990 8th Edition)。 血中濃度が50〜150μg/mlの遊離スルフィソキサゾールは、大抵の感染症に対して治療効果があるものと考えられており、また、血中濃度が120〜150μg/mlにまでなると、重篤感染症に対して好適であると考えられている。 200μg/mlの濃度を超えると副作用が頻繁に認められるようになるので、血中での最高濃度は、200μg/mlとすべきである(PDR 59th Edition 2005)。 1日に4度、健常者に対して、500mgを経口的に繰り返し投与をしたところ、定常状態でのスルフィソキサゾールの平均血漿濃度は、49.9〜88.8μg/mlの範囲であった(平均値63.4μg/ml)(Oie et al., J Pharmacokinetics Biopharm 1982: 10: 157-172)。
【0063】
プロポフォールとスルフィソキサゾールとの間の相互作用が、マウスにおいて認められている。 プロポフォールによる立ち直り反射の障害は、スルフィソキサゾールを用いて前処理することでさらに強くなった。 スルフィソキサゾールそれ自体は、何らの効果も示さず、また、プロポフォールの総血漿濃度およびタンパク質結合についても何らの効果も示さなかった[51]。
【0064】
スルフィソキサゾールが、一酸化窒素上昇を抑え、そして、リポ多糖類(LPS)に起因するGABA含有ニューロンの数を減らすことで、緑内障に認められる虚血性傷害から網膜を保護することが知られている[52]。 クロニジンの局所投与によって、α2-アドレナリン受容体が刺激を受けて、ラットの網膜が、虚血/再灌流から保護されることが知られており、この保護効果は、α2-アドレナリン受容体の関与を確認するヨヒンビンまたはラウオルシンによって選択的に弱められる[50]。
【0065】
幾つかのETA受容体アンタゴニストについて、肺動脈高血圧症、鬱血性心不全、脳卒中、バルーン血管形成後の冠動脈の再閉鎖、ならびに、高血圧症の治療、および、癌性疼痛管理での使用に向けた評価が行われているので、クロニジンとスルフィソキサゾールに関する研究は臨床的に重要である。 本明細書で示した結果は、弱いETA受容体アンタゴニストであるスルフィソキサゾールを、α2アドレナリンアゴニストであるクロニジンと共に用いることで、予期せぬ鎮痛作用が得られることを示す証拠に他ならない。 その他のETA受容体アンタゴニストとクロニジンなどのα2アドレナリンアゴニストとを併用した研究も行って、これら薬剤の組み合わせが、鎮痛作用を増大するかどうか、そして、これら組み合わせが、安全な非麻薬性鎮痛薬として使用できるかどうかを見極めることも意図している。 本明細書に記載の結果は、痛覚を変化させることのできる新たな標的を提供するのみならず、患者の慢性疼痛の新たな管理手法をもたらすかもしれない。
【0066】
本願発明において有用なエンドセリン受容体アンタゴニストの例として、スルフィソキサゾール、アトラセンタン、テゾセンタン、ボセンタン、シタキスセンタン、エンラセンタン、BMS207940(ブリストル・マイヤーズ スクイブ)、BMS193884、BMS182874、J 104132(萬有製薬)、VML 588/Ro 61 1790(バンガード・メディカ)、T-0115(田辺製薬)、TAK 044(タケダ)、BQ 788、TBC2576、TBC3214、PD180988、ABT 546、SB247083、RPR118O31A、および、BQ123などがあるが、これらに限定されない。 BQ123は、エンドセリンAアンタゴニストの具体例であって、このものは、シクロ(D-Trp-D-Asp-Pro-D-Val-Leu)のナトリウム塩である。 その他の有用なエンドセリンアンタゴニストとして、YM 598、LU 135252、PD 145065、A 127722、ABT 627、A 192621、A 182086、TBC3711、BSF208075、S 0139、および、SB209670という名称のものがあるが、これらに限定されない。 その他の有用なエンドセリンAアンタゴニストは、本明細書の一部を構成するものとしてそれらの内容を援用する米国公開特許公報第US 2002/0082285 Al号および米国特許出願第10/659,579号に開示されている。
【0067】
従来のエンドセリン受容体アンタゴニストに加えて、内因性エンドセリンの形成を阻害する化合物も、本願発明のエンドセリン受容体アンタゴニストとして使用することができる。 これら化合物は、エンドセリンの形成を妨げ、それにより、エンドセリン受容体の活性を低下させるので、有用な化合物であると言える。 それら化合物のクラスの一つに、エンドセリン変換酵素(ECE)阻害剤がある。
【0068】
有用なECE阻害剤として、[DVal22、Phe33]巨大エンドセリン-1(16-38)、ヒト(すなわち、His-Leu-Asp-Ile-Ile-Trp-DVal-Asn-Thr-Pro-Glu-His-Val-Val-Pro-Tyr-Gly-Phe-Gly-Ser-Pro-Arg-Ser);[DVal22]巨大エンドセリン(16-38)、ヒト(すなわち、His-Leu-Asp-Ile-Ile-Trp-DVal-Asn-Thr-Pro-Glu-His-Val-Val-Pro-Try-Gly-Leu-Gly-Ser-Pro-Arg-Ser);[Phe22]巨大エンドセリン-1(19-37)、ヒト(すなわち、Ile-Ile-Trp-Phe-Asn-Thr-Pro-Glu-His-Val-Val-Pro-Tyr-Gly-Leu-Gly-Ser-Pro-Arg);および、ホスホラミドン(すなわち、N-(a-ラムノピラノシルオキシヒドロホスフィニル)-Leu-Trp)などがあるが、これらに限定されない。
【0069】
麻薬性鎮痛薬
入手可能なアヘン製剤およびオピオイド鎮痛薬は、五つの化合物グループ(すなわち、 フェナントレン、フェニルヘプチルアミン、フェニルピペリジン、モルフィナン、および、ベンゾモルファン)の誘導体である。 薬理学的には、アヘン製剤と非アヘン製剤とは、活性が顕著に異なる。 幾つかは、強力なアゴニスト(モルヒネ)であるが、その他は、中程度〜軽度のアゴニスト(コデイン)である。 これに対して、幾つかのアヘン製剤誘導体は、アゴニストとアンタゴニストの混合活性を示す(ナルブフィン)のに対して、その他の誘導体は、アヘン製剤アンタゴニスト(ナロキソン)である。 モルヒネは、アヘン製剤およびオピオイド鎮痛薬の原型であり、そのすべてが、中枢神経系に対して同様の作用を及ぼす。
【0070】
モルヒネは、アヘンから化学的に誘導される。 ヘロインなどのその他の薬物は、モルヒネまたはコデインから加工される。 このようなアヘン製剤は、何世紀にもわたって、医学的におよび非医学的に用いられてきた。 19世紀初頭まで、モルヒネは、溶解に適した純粋な形で抽出されていた。 皮下注射針の普及に伴い、モルヒネ溶液の注射投与が、一般的な投与方法となった。 アヘンに含まれる20ものアルカロイドの内、コデインとモルヒネだけが、臨床的に広く利用されている。
【0071】
それらの内でも、麻薬のアヘンは、最も強力に作用する薬剤であり、中枢神経系を抑制するなど臨床的に有用な薬剤でもある。 このグループに属する薬剤は、基本的には、鎮痛薬として用いられるが、その他にも無数の有用な特性を具備している。 例えば、モルヒネは、疼痛を緩和し、疼痛時に睡眠を促し、下痢を止め、咳を鎮め、呼吸を楽にし、そして、麻酔の効果を助長するために使われている。
【0072】
モルヒネやその関連化合物を長期間にわたって投与をすると、鎮痛作用に対する耐性が出現し、そして、同様の疼痛緩和効果を得るためには、定期的に用量を増量しなくてはならなくなる。 そして、最終的には、陶酔感と共に耐性や身体的依存性が強くなり、薬物の過剰摂取を経て、過敏な人格を有する中毒患者となってしまう。 このような理由から、モルヒネやその誘導体は、医師の処方のみに従って使用しなくてはならず(すなわち、処方された以上の用量、頻度、または、期間を超えて使用してはならない)、また、他の鎮痛剤で十分に対処できる場合には、それらを疼痛を治療する目的で使用すべきではない。
【0073】
一つまたはそれ以上のアルファ-2(α2)アドレナリンアゴニスト、および/または、一つまたはそれ以上のエンドセリンアンタゴニストは、麻薬性鎮痛薬の鎮痛作用を増強する上で有用であり、また、そうすることも意図している。 さらに、一つまたはそれ以上のアルファ-2(α2)アドレナリンアゴニストと一つまたはそれ以上のエンドセリンアンタゴニストとの組み合わせは、麻薬性鎮痛薬の鎮痛作用を増強する上で有用であり、また、そうすることも意図している。
【0074】
麻薬性鎮痛薬として、(a)アヘン;(b)モルヒネ、硫酸モルヒネ、コデイン、リン酸コデイン、硫酸コデイン、ジアセチルモルヒネ、塩酸モルヒネ、酒石酸モルヒネ、および、塩酸ジアセチルモルヒネなどのアヘンアルカロイド;および、(c)臭化水素酸デキストロメトルファン、酒石酸水素ヒドロコドン、ヒドロモルフォン、塩酸ヒドロモルフォン、酒石酸レボルファノール、塩酸オキシモルホン、および、塩酸オキシコドンなどの半合成麻薬性鎮痛薬などがあるが、これらに限定されない。
【0075】
医薬化合物の製剤
本願発明の活性成分、すなわち、本明細書に記載のα2アドレナリンアゴニストとエンドセリン受容体アンタゴニストは、それら単独で投与することができ、あるいは、意図している投与経路および標準的な薬務にしたがって選択した医薬担体との混合物として投与することができる。 よって、本願発明にしたがって用いられる医薬組成物は、薬学的に使用可能な調製物への活性成分の加工処理を円滑にする賦形剤や助剤を含む一つまたはそれ以上の生理学的に許容可能な担体を用いて、従来の方法によって、製剤することができる。
【0076】
これら医薬組成物は、従来の加工プロセス、例えば、混合、溶解、造粒、糖衣錠調製法、乳化、カプセル化、封入、または、凍結乾燥などのプロセスを用いて、従来の方法で製造することができる。 選択した投与経路に応じて、適切な製剤が調製される。
【0077】
本明細書に記載の活性成分を含む医薬組成物を意図しており、ある実施態様によれば、それら化合物は、薬学的に許容可能な希釈剤、補助剤、賦形剤、および/または、担体を用いて製剤される。 「薬学的または薬理学的に許容可能」という表現は、例えば、吸入噴霧、経膣的、経直腸的、または、頭蓋内注射によって、経口的、局所的、経皮的、非経口的に、動物、または、ヒトに対して投与をした時に、副作用、アレルギー反応、または、その他の有害な作用を招かない化合物および組成物のことを指す。 本明細書で用いる非経口の用語は、皮下注射、静脈投与、筋内、嚢内注射、または、点滴などを含む。 同様に、静脈投与、皮内、筋内、乳房内、腹腔内、関節内、髄腔内への投与、球後注射、肺内注射などによる投与、あるいは、特定部位への外科的移植をも意図している。 一般的に、調製された組成物は、発熱物質を含んでおらず、また、ヒトや動物に対して有害なその他の不純物も含んでいない。 「薬学的に許容可能な担体」の用語は、一部およびすべての溶剤、分散媒、コーティング剤、抗菌剤、および、抗真菌剤、等張剤、および、吸収遅延剤などを含む。 このような医薬活性物質のための媒体および薬剤は、当該技術分野において周知である。
【0078】
前述した本願発明の方法で用いる医薬組成物は、例えば、錠剤、トローチ、ドロップ、水性懸濁液または油性懸濁液、分散性粉末または分散性顆粒、エマルジョン、硬質カプセルまたは軟質カプセル、または、シロップ、または、エリキシル剤などの経口投与に適した形態にすることができる。 経口投与用組成物は、公知の方法で調製することができ、また、薬学的に好ましく、かつ、口当たりの良い調製物を提供すべく、それら組成物は、甘味料、着香料、着色料、および、保存料からなるグループから選択される一つまたはそれ以上の物質を含むこともできる。 錠剤には、その製造に適した非毒性の薬学的に許容可能な賦形剤と共に混合した活性成分を含めることができる。 これら賦形剤として、不活性希釈剤、例えば、炭酸カルシウム、炭酸ナトリウム、ラクトース、リン酸カルシウム、または、リン酸ナトリウムなど、造粒剤および崩壊剤、例えば、コーンスターチ、または、アルギン酸など、結合剤、例えば、澱粉、ゼラチン、または、アラビアゴムなど、滑剤、例えば、ステアリン酸マグネシウム、ステアリン酸、または、滑石などがある。 錠剤にはコーティングを施さなくともよく、あるいは、消化管での崩壊と吸収を遅らせて、それにより、長期間にわたって作用を持続させるために、公知の方法で錠剤にコーティングを施してもよい。 例えば、モノステアリン酸グリセリン、または、ジステアリン酸グリセリンなどの遅延物質を用いることができる。 また、放出制御のための浸透圧性治療用錠剤を形成するために、米国特許第4,256,108号、第4,166,452号、および、第4,265,874号に記載の技術でコーティングを施すこともできる。
【0079】
経口投与製剤は、活性成分が、不活性固体希釈剤、例えば、炭酸カルシウム、リン酸カルシウム、または、カオリンと共に混合されている硬ゼラチンカプセルとして、あるいは、活性成分が、水性媒体または油性媒体、液体パラフィン、または、オリーブオイルと共に混合されている軟ゼラチンカプセルとして、提供することもできる。 錠剤の形態で投与する場合には、この組成物に、ゼラチンなどの固体担体、あるいは、補助剤をさらに含ませることができる。 錠剤、カプセル、および、粉末は、本願発明の活性成分を、それらの約5%〜約95%の量で含み、好ましくは、それらの約25%〜約90%は、本願発明の化合物である。 液体の形態で投与する場合には、水、石油、または、動物または植物に由来する油脂などの液体担体を含ませることができる。 液体形態の組成物は、生理学的食塩水、デキストロース、または、その他の糖溶液、あるいは、グリコールをさらに含むことができる。 液体の形態で投与する場合には、この組成物は、約0.5重量%〜約90重量%の活性成分、好ましくは、約1重量%〜約50重量%の活性成分を含む。
【0080】
水性懸濁液は、水性懸濁液の製造に適した賦形剤と共に混合した活性成分を含むことができる。 賦形剤とは、懸濁化剤、例えば、カルボキシメチルセルロースナトリウム、メチルセルロース、ヒドロキシプロピルメチルセルロース、アルギン酸ナトリウム、ポリビニルピロリドン、トラガカントゴム、および、アラビアゴムなどであり、分散剤または湿潤剤は、天然リン脂質、例えば、レシチンなど、脂肪酸を有するアルキレンオキシドの縮合産物、例えば、ステアリン酸ポリオキシエチレンなど、または、長鎖脂肪族アルコールを有するエチレンオキシドの縮合産物、例えば、ヘプタデカエチレンオキシセタノールなど、または、ポリオキシエチレンソルビタンモノオレアートなどの脂肪酸およびヘキシトールから誘導された部分エステルを有するエチレンオキシドの縮合産物、または、脂肪酸および無水ヘキシトールから誘導された部分エステルを有するエチレンオキシドの縮合産物、例えば、ポリエチレンソルビタンモノオレアートなどである。 この水性懸濁液は、一つまたはそれ以上の保存料、例えば、エチル、または、n-プロピル、p-ヒドロキシ安息香酸など、一つまたはそれ以上の着色料、一つまたはそれ以上の着香料、および、ショ糖またはサッカリンなどの一つまたはそれ以上の甘味料なども含むことができる。
【0081】
油性懸濁液は、植物油、例えば、落花生油、オリーブ油、胡麻油、または、椰子油など、あるいは、液体パラフィンなどの鉱油において、活性成分を懸濁させて調製することができる。 この油性懸濁液は、増粘剤、例えば、蜜蝋、固形パラフィン、または、セチルアルコールを含むことができる。 良好な口当たりを呈する調製物を製造するために、前述した甘味料や着香料を加えることもできる。 これら組成物は、アスコルビン酸などの抗酸化剤を加えて保存することができる。
【0082】
加水して水性懸濁液を調製するのに適した分散可能な粉末および顆粒は、分散剤、または、湿潤剤、懸濁化剤、および、一つまたはそれ以上の保存料を伴った活性化合物を提供する。 好適な分散剤、または、湿潤剤、および、懸濁化剤として、先に例示したものなどがある。 その他の賦形剤、例えば、甘味料、着香料、および、着色料も加えることができる。
【0083】
本願発明において有用な医薬組成物は、水中油型エマルションの形態とすることもできる。 油相には、植物油、例えば、オリーブ油、または、落花生油、あるいは、鉱油、例えば、液体パラフィン、または、これらの混合物を用いることができる。 好適な乳化剤として、天然ゴム、例えば、アラビアゴム、または、トラガカントゴムなど、天然リン脂質、例えば、大豆、レシチン、および、脂肪酸から誘導されたエステルまたは部分エステルなど、無水ヘキシトール、例えば、ソルビタンオレイン酸モノエステルなど、および、エチレンオキシドを有する前記部分エステルの縮合産物、例えば、ポリオキシエチレンソルビタンオレイン酸モノエステルなどがある。 このエマルジョンに、甘味料や着香料も含めることができる。
【0084】
シロップ剤、および、エリキシル剤は、甘味料、例えば、グリセロール、プロピレングリコール、ソルビトール、または、ショ糖などを用いて調製することができる。 このような製剤は、鎮痛薬、保存料、および、着香料および/または着色料なども含むことができる。 これら組成物は、直腸に投与する坐薬の形態とすることもできる。 これら組成物は、常温では固体であるが、直腸温度では液状となり、つまり、直腸内で溶解して薬剤を放出するという好適な非刺激性賦形剤と薬剤とを混合して調製することができる。 そのような賦形剤として、例えば、ココアバターやポリエチレングリコールなどがある。
【0085】
この医薬組成物は、滅菌済の注射可能な水性または油性の懸濁液の形態とすることができる。 この懸濁液は、前述した好適な分散剤、または、湿潤剤、および、懸濁化剤を用いて、当該技術分野で周知の方法で調製をすることができる。 滅菌済の注射可能な調製物は、非毒性の非経口的に許容可能な希釈剤または溶剤、例えば、1,3-ブタンジオールの溶液などを用いて、滅菌済の注射可能な溶液または懸濁液とすることもできる。 利用することができる許容可能な賦形剤や溶剤として、水、リンガー溶液、生理食塩水などがある。 加えて、滅菌済の固定油が、従来より、溶剤または懸濁媒体として用いられている。 本願発明の目的を鑑みれば、合成モノグリセリド、または、合成ジグリセリドなどの無刺激性の固定油を用いることができる。 さらに、オレイン酸などの脂肪酸が、注射剤の調製において利用できることも知られている。
【0086】
注射に適した医薬品形態として、注射可能な滅菌済溶液または分散液を即座に調製するための滅菌済水性溶液または分散液、および、滅菌済粉末などがある。 すべての事例において、医薬品形態は、滅菌処理を行っていなければならず、また、容易に注射できる程度の流動性を有していなくてはならない。 また、この医薬品形態は、製造および貯蔵の条件下で安定でなくてはならず、また、細菌や黴などの微生物による汚染を受けないようにして保存しなければならない。 そして、例えば、水、エタノール、ポリオール(例えば、グリセロール、プロピレングリコール、および、液体ポリエチレングリコールなど)、これらの好適な混合物、および、植物油を含む溶剤または分散媒を、担体とすることができる。 また、例えば、レシチンなどのコーティング剤を使用したり、分散液の場合には所望の粒径を維持したり、そして、界面活性剤を使用することで、適切な流動性を維持することができる。 また、様々な抗生物質や抗真菌剤、例えば、パラベン、クロロブタノール、フェノール、ソルビン酸、チメロサールなどを用いることで、微生物の影響を回避することができる。 多くの事例において、等張剤、例えば、糖類や塩化ナトリウムなどを用いることが好ましい。 そして、吸収遅延剤、例えば、モノステアリン酸アルミニウムやゼラチンなどの組成物を用いることで、注射可能な組成物に対して吸収持続性を付与することができる。
【0087】
投与および投薬
本願発明の医薬組成物には、所定の治療目的を達成する治療有効量の活性成分を含んだ組成物が含まれている。 治療有効量の決定は、とりわけ、本明細書での開示を参照し、かつ、目的とする所望の薬効、投与経路、および、投与を受ける患者の病態などを考慮すれば、当業者が十分に対処できる事項である。
【0088】
正確な処方、投与経路、および、用量は、患者の病態を考慮して、個々の医師によって決定される。 投与量と投与間隔は、治療効果または予防効果を維持するに十分なレベルの活性成分を提供するために、個別に調整をすることができる。
【0089】
本明細書に記載の方法を用いた治療での対象動物を、哺乳動物とすることを意図している。 この哺乳動物として、ヒトの医学研究のために用いる非ヒト動物モデル、あるいは、家畜の他に、例えば、愛玩動物などのペットとして重要な動物などがある。
【0090】
医薬組成物の投与は、疼痛が発症する以前、発症している最中、または、発症した後に行うことができる。
【0091】
これら活性成分は、好適な経路、例えば、経口的、経頬的、吸入的、舌下、経直腸的、経膣的、腰椎穿刺による嚢内、経尿道的、経鼻的、経皮的、すなわち、経皮的または非経口的(静脈、筋内、皮下、および、冠動脈内を含む)経路をもってして投与される。 非経口的投与は、注射針やシリンジ、あるいは、POWDERJECT(商品名)のような高圧技術を用いて行うことができる。
【0092】
本願発明の活性成分は、それ単独で投与することができ、また、意図している投与経路や標準的な薬務に鑑みて選択をした医薬担体と混合してから投与することもできる。 よって、本願発明の用途に供する医薬組成物は、薬学的に使用可能な調製物への活性成分の取り込みを促す賦形剤や補助剤を含む一つまたはそれ以上の生理学的に許容可能な担体を用いて、従来の方法で、製剤をすることができる。 医薬組成物は、本明細書に記載され、かつ、当該技術分野で周知の従来の方法でもってして製造をすることができる。
【0093】
医薬組成物の投与量は、治療を受ける対象動物、対象動物の体重、疼痛の程度、投与方法、それに、処方医師の判断によって定まることとなる。
【0094】
獣医学的用途については、活性成分は、通常の獣医学的手順にしたがって、好適な許容可能な製剤として投与される。 獣医師であれば、個々の動物に対して最適の投与計画と投与経路を容易に決定することができる。
【0095】
ある実施態様によれば、ヒトの疼痛を治療または予防するために投与する場合、α2アドレナリンアゴニスト、および、エンドセリン受容体アンタゴニストそれぞれの経口投与量は、一般的には、平均的な成人の患者(70kg)で、毎日、約10〜約200mgであり、通常は、一日当たりの用量を、2〜3回に分けて投与する。 一般的な成人患者に対して、これら活性成分を麻薬性鎮痛薬と共に投与する場合、それぞれの錠剤またはカプセルは、薬学的に許容可能な好適な賦形剤または担体と共に、約0.1〜約200mgのオピオイド鎮痛剤、約5μg/kgまたは300μgのα2アドレナリンアゴニスト、および/または、約0.1〜約50mgのエンドセリンアンタゴニストを含んでおり、それらは、単一用量または複数回用量で、一日当たり一回または数回かけて投与がされる。 静脈投与、経頬投与、または、舌下投与のために必要な用量は、通常は、約0.1〜約10mg/kg/単一用量である。 実際には、医師が、個々の患者に対して最適な投与計画を決定するものであって、その用量は、個々の患者の年齢、体重、および、反応状況によって変化する。 上記した用量は、平均的な事例を例示したものでしかなく、個々の事例によっては、用量を増減させた方が好都合な場合も当然あり、そのような事例も、本願発明の範疇に含まれる。
【0096】
ある実施態様によれば、クロニジン、スルフィソキサゾール、または、クロニジン、および、スルフィソキサゾールを含む調製物は、錠剤またはカプセルの形態で調製される。
【0097】
クロニジンは、鎮痛作用を促す補助剤として、5μg/kgの用量、または、300μgの総量で経口的に用いられている。 概して、クロニジンを用いた臨床研究は、1〜5μg/kgの用量のクロニジンが、効果的に鎮痛作用をもたらすことを示している。 本明細書に記載のラットを用いた実施例は、2mg/kgのクロニジンと1000mg/kgのスルフィソキサゾールの組み合わせが、顕著な鎮痛作用を得る上で効果的であったことを示している。 スルフィソキサゾールは、2〜4グラムの用量で経口的に用いられている。
【0098】
ある実施態様によれば、α2アドレナリンアゴニストは、約50μg、約110μg、約150μg、約200μg、約250μg、約300μg、約350μg、約400μg、約450μg、約500μg、約750μg、または、約1000μgの用量で投与される。 関連する実施態様によれば、クロニジンは、 約10〜約500μg、約10μg〜約300μg、約75〜約300μg、または、約100〜約250μgの範囲の用量で投与される。
【0099】
さらに別の実施態様によれば、エンドセリン受容体アンタゴニストは、約0.1、約0.5、約1、約2、約3、約4、約5、約10、約15、約20、約25、約30、約35、約40、約45、または、約50mgの用量、あるいは、約0.1mg〜約50mgの範囲の用量で投与される。 関連する実施態様によれば、スルフィソキサゾールは、2〜4グラム、750mg〜1.5g、1〜2グラム、0.1〜3グラムの範囲の用量で投与される。
【0100】
疼痛を効果的に治療するために対象動物に投与するα2アドレナリンアゴニスト:エンドセリンアンタゴニストの比率を、1:500〜1:50,000の範囲にすることを意図している。 ある実施態様によれば、クロニジン:スルフィソキサゾールの比率は、鎮痛作用を効果的に得るためには、1:500〜1:50,000にすべきとされている。 さらに強力なエンドセリン受容体アンタゴニストを用いる場合には、クロニジン:エンドセリンアンタゴニストの比率を、1:100〜1:1000に変化させておくことを意図している。 本願発明は、アルファ-2 アドレナリンアゴニスト:エンドセリンアンタゴニストとの比率を、 1:500〜1:50,000、1:500〜1:20,000、1:500〜1:10,000、1:500〜1:5,000、1:500〜1:2,500、1:100〜1:1000、および、1:100〜1:500の範囲とした組成物の投与を行う。
【0101】
α2アドレナリンアゴニストとエンドセリンアンタゴニストを、同時または別個のいずれかで投与することを意図している。 さらに、α2アドレナリンアゴニストとエンドセリンアンタゴニストは、単一の組成物、または、別個の組成物で投与することができる。
【0102】
同時投与にあっては、一つまたはそれ以上のα2アドレナリンアゴニスト、および/または、一つまたはそれ以上のエンドセリン受容体アンタゴニストを含む組成物が、各成分が、同時に、あるいは、任意の順序で異なる時点で連続して投与されるように投与が行われる。 しかしながら、同時に投与が行われないにしても、ある実施態様によれば、所望の治療効果を得るべく、時間をあまり開けずに、両者の投与が行われている。 一つまたはそれ以上のα2アドレナリンアゴニスト、および/または、一つまたはそれ以上のエンドセリン受容体アンタゴニストを、別個の組成物で投与する場合において、ある実施態様によれば、最初の薬剤の投与の前または後に、他方の組成物を投与することも意図されている。 先行投与とは、処置の1日前(24時間前)から処置の30分前の範囲で、薬剤を投与することを指す。 さらに、他方の薬剤の投与に続いて、一方の薬剤を投与することも意図している。 後続投与とは、最初の薬剤を投与してから30分後から、最初の薬剤を投与してから1日後(24時間後)までに投与を行うことを指す。 30分後から24時間後の範囲には、30分、1、2、3、4、5、6、7、8、9、10、11、12、16、20、または、24時間の時点での投与を含むことができる。
【0103】
組成物の投与は、患者の疼痛症状に応じて行うことができる。 ある実施態様によれば、これら薬剤は、毎日2回、毎日、48時間毎、3日毎、4日毎、7日毎、または、14日毎に、何週間にもわたって、投与がされる。 さらに、必要に応じて、複数の箇所に投与を行うことができ、また、疼痛の箇所に対して全身的または局所的に投与することもできる。
【0104】
ある実施態様によれば、エンドセリン受容体アンタゴニストは、α2アドレナリンアゴニストよりも先に投与することが意図されている。 関連する実施態様によれば、α2アドレナリンアゴニストは、エンドセリン受容体アンタゴニストよりも先に投与がされる。
【0105】
一つまたはそれ以上のα2アドレナリンアゴニスト、および/または、一つまたはそれ以上のエンドセリンアンタゴニストが、麻薬性鎮痛薬と共に投与される場合、その麻薬性鎮痛薬は、α2アドレナリンアゴニストおよびエンドセリン受容体アンタゴニストよりも先に投与され、α2アドレナリンアゴニストおよびエンドセリン受容体アンタゴニストよりも後に投与され、あるいは、最初の薬剤の投与の後で、かつ、他方の薬剤が投与される以前に投与されるものであって、α2アドレナリンアゴニストまたはエンドセリン受容体アンタゴニストは、麻薬性鎮痛薬よりも先または後に投与がされる、ことも意図をしている。
【0106】
これら化合物の好ましい投与間隔と投与量は、当業者にとって自明の事項である。
【0107】
疼痛の治療方法
本願発明は、疼痛を経験している対象動物で認められる症状を軽減および治療するための方法を提供する。 ある実施態様によれば、本願発明は、治療有効量のα2アドレナリンアゴニストと治療有効量のエンドセリン受容体アンタゴニストを哺乳動物に対して投与することを含む、疼痛を治療または予防する方法を提供する。 関連する実施態様によれば、疼痛の治療は、α2アドレナリンアゴニストとエンドセリン受容体アンタゴニストに加えて、麻薬性鎮痛薬の投与をさらに含む。
【0108】
疼痛の原因として、炎症、負傷、疾病、筋肉の痙攣、および、神経障害または神経障害症候群の発症などがあるが、これらに限定されない。 急性疼痛は、通常、自然に治癒するものであるが、慢性疼痛は、一般的には、三ヶ月またはそれ以上の期間にわたって疼痛が持続するので、患者の性格、生活様式、機能的能力、それに、全体的な生活の質に変化を及ぼすことがある。 疼痛が効果的に治療できないと、身体機能が制限され、運動機能が減退し、不眠になり、そして、一般的な生活の質が損なわれるなどの体験を経て、患者に悪影響が及ぶこととなる。
【0109】
外科手術や、有害な物理的、化学的、または、熱的現象、あるいは、生物製剤による感染などに起因して組織が損傷を受けると、炎症性(侵害性)の疼痛が起こる。 神経因性疼痛は、神経系、末梢神経、後根神経節、または、後根、あるいは、中枢神経系の損傷に起因する持続性または慢性の疼痛症候群である。 神経因性疼痛症候群として、異痛、そして、ヘルペス後神経痛および三叉神経痛などの様々な神経痛、幻想痛、それに、反射性交感神経性ジストロフィーや灼熱痛などの複合性局所疼痛症候群がある。 灼熱痛は、痛覚過敏症と異痛を伴う不随意的な灼熱痛を特徴としている。 痛覚過敏症は、疼痛性刺激に対する異常過敏を特徴としている(Meller et al., Neuropharmacol. 33:1471-8, 1994)。
【0110】
この病態は、内臓器官に疼痛の感覚をもたらす内臓痛覚過敏症を含んでいる。 また、神経因性疼痛は、通常は無害な刺激が長期間にわたって与えられた場合に、激しい疼痛を引き起こす痛感過敏症も含んでいる。
【0111】
ある実施態様によれば、治療をする疼痛は、急性疼痛または慢性疼痛である。 関連する実施態様によれば、疼痛は、灼熱痛、接触性アロディニア、神経因性疼痛、痛覚過敏症、痛感過敏症、炎症性疼痛、術後疼痛、慢性腰痛、群発頭痛、帯状疱疹後神経痛、幻肢痛および断端痛、中心性疼痛、歯痛、神経因性疼痛、オピオイド耐性疼痛、内臓痛、術後疼痛、骨損傷痛、糖尿病性神経障害疼痛、術後または外傷性神経症疼痛、末梢神経障害疼痛、エントラップメント神経障害疼痛、アルコール依存症に起因する神経障害、HIV感染、多発性硬化症、甲状腺機能低下症に起因する疼痛、または、抗癌化学療法に起因する疼痛、分娩中の疼痛、日焼けを含む火傷に起因する疼痛、産後疼痛、偏頭痛、咽喉痛、および、膀胱炎などの尿生殖路関連疼痛からなるグループから選択される。
【0112】
α2アドレナリンアゴニストとエンドセリン受容体アンタゴニストとを組み合わせることで、各化合物をそれぞれ単独で用いた場合と比較して、各化合物の用量の減少という相乗効果を意図している。 さらに、α2アドレナリンアゴニストまたはエンドセリン受容体アンタゴニストのいずれか一方だけを投与することで、オピオイド鎮痛剤の効果を強めることができ、それにより、疼痛の症状を効果的に治療するために必要なオピオイドの用量を減少させることを意図している。 また、α2アドレナリンアゴニストとエンドセリン受容体アンタゴニストの双方を、麻薬性鎮痛薬と共に投与しても、オピオイドの効果を強めることができ、そして、疼痛を緩和するために必要なオピオイドの用量を減少させることを意図している。
【0113】
ヒト患者での慢性疼痛の治療は、一般的には、米国特許第6,372,226号に記載されているようにして行われている。 ある実施態様によれば、急性炎症性疼痛、神経因性疼痛、痙攣状態、または、障害に起因するその他の慢性疼痛を経験している患者は、髄腔内投与、例えば、本願発明の方法で用いる適切な用量の本明細書に記載の組成物を腰部に脊椎穿刺して投与することで治療がされている。 その他の実施例では、対象動物が関節炎やその他の関節疼痛を患っている場合に、組成物を、関節内に投与している。 具体的な用量や注射する部位、それに、投与頻度などは、様々な要素を勘案して決められるものであって、それらは、治療にあたる医師の裁量の範囲である。
【0114】
疼痛症状の改善度は、視覚的アナログ尺度(VAS)、口頭式評価スケール(VRS)、および、数値化スケール(NRS)(Williamson et al, J Clin Nurs. 14:798-804, 2005; Carlsson, A., Pain. 1983 16:87-101, 1983)などの当該技術分野で周知の方法を利用して検査をした。 視覚的アナログ尺度、口頭式評価スケール、および、数値化スケールにあっては、一般的に、疼痛刺激を与える前後に、疼痛に関する数値について、患者に対して質問がされる。 慢性疼痛は、神経障害の症状および兆候に関する疼痛スケールであるリーズ評価法(LANSS)(Bennett, M. Pain. 92:147-157, 2001)などの客観的スケール試験によっても評価がされる。 α2アドレナリンアゴニストとエンドセリン受容体アンタゴニストを含む組成物で治療をした後に、疼痛刺激に対する過敏性が抑制されていることは、それらの活性が影響を及ぼしていること、そして、α2アドレナリン受容体、および/または、エンドセリン受容体が、慢性疼痛に関連する症状を緩和していることを指し示すものである。 本願発明のその他の実施態様によれば、本明細書に記載の組成物は、前述したその他の鎮痛薬と組み合わせて投与がされており、そのような治療を行うことで、慢性疼痛の症状の緩和において相乗効果が認められている。
【0115】
鎮痛剤を投与した後の様々な時点において疼痛の改善についての検査を行い、そして、検査結果に基づいた疼痛の解消度を評価した。 ある実施態様によれば、疼痛の症状の評価を、1、2、3、4、5、6または8週目の各週に行い、あるいは、担当医師が評価を行う。 ある実施態様によれば、対象動物での疼痛症状は、治療を行う以前の疼痛症状の評価結果と比較して、当該技術分野で周知の疼痛スケールを用いて決定したところでは、少なくとも10%、少なくとも20%、少なくとも25%、少なくとも30%、少なくとも35%、少なくとも40%、少なくとも45%、少なくとも50%、少なくとも55%、少なくとも60%、少なくとも65%、少なくとも70%、少なくとも75%、少なくとも80%、少なくとも85%、少なくとも90%、少なくとも95%、または、100%にまで改善することができる。
【0116】
キット
さらに別の実施態様として、本願発明は、本願発明の方法の実施にあたって円滑に利用可能に包装された一つまたはそれ以上の化合物または組成物を含むキットに関する。 最も単純な実施態様によれば、このキットは、密閉したボトルや瓶などの容器に包装された本願発明の方法の実施にあたって有用な本明細書に記載の化合物または組成物(例えば、α2アドレナリンアゴニストを含む組成物、および、エンドセリン受容体アンタゴニストを含む組成物、または、α2アドレナリンアゴニストとエンドセリン受容体アンタゴニストの双方を含む組成物)と、本願発明の方法を実施するにあたっての化合物または組成物の使用についての記載がされ、かつ、容器に貼着されているか、あるいは、キットに収容されているラベルを具備している。 好ましくは、本願発明の化合物または組成物は、単位投与用量で包装されている。 このキットは、好ましい投与経路で組成物の投与を行うために、それに適した器具をさらに具備することができる。 その他の実施態様によれば、このキットは、一つまたはそれ以上のオピオイド鎮痛薬を具備することができる。
【0117】
本願発明のその他の態様や詳細は、本願発明の限定を意図したものではなく、その例示を目的とした以下の実施例から自明であろう。
【実施例】
【0118】
実施例1:試料および方法
動物: 25〜30gの体重のオスのスイス・ウェブスターマウス(ハーラン社、インディアナポリス、インディアナ州)を用いた。 この動物を、室温(23±1℃)、湿度(50±10%)、および、12時間明暗サイクル(午前6時〜午後6時)が調節された部屋で、5匹/カゴずつ収容をした。 食餌と水は、自由に摂らせた。 この環境に動物が順応するまで、少なくとも4日間が過ぎてから、実験を開始した。 実験用の動物の飼育と使用については、動物実験委員会の許可を得た。 すべての麻酔処置と外科処置は、動物実験委員会のガイドラインを遵守して行った。
【0119】
薬剤: 硫酸モルヒネ(マリンクロットケミカル社、セントルイス、ミズーリ州)を、発熱物質を含んでいない蒸溜・脱イオン水に溶解し、そして、皮下(s.c.)に注射をした。 4-アミノ-N-(3,4-ジメチルオキサゾール-5-イル)-ベンゼンスルホンアミド(シグマケミカル社、セントルイス、ミズーリ州)であるスルフィソキサゾールを、カルボキシメチルセルロースに溶解し、そして、経口投与をした。 N-(2,6-ジクロロフェニル)-4,5-ジヒドロ-1H-イミダゾ−ル-2-アミン(シグマケミカル社、セントルイス、ミズーリ州)であるクロニジンを、滅菌生理食塩水に溶解し、そして、腹腔内(i.p.)に注射をした。
【0120】
統計値: すべてのデータ値は、平均値±SEMで示してある。 グループ内およびグループ間の差異を検証するために、ANOVAに続いて、ポストホックテスト(ボンフェローニの検定)を用いた。 P<0.05のレベルを、有意とした。
【0121】
実施例2:テイルフリック潜時の決定
モルヒネに対する抗侵害受容反応については、ダムールとスミスのテイルフリック潜時試験法[10]によって決定を行った。 動物の尻尾に熱刺激(収束光線)を与えると、即座に俊敏な動きで尻尾を引っ込める動作を示した。 この動作の反応時間を、無痛覚計を用いて検査して、テイルフリック潜時として記録をした。 熱刺激(収束光線)に対するテイルフリック潜時を、モルヒネまたは生理食塩水の注射を行う前と、注射を行ってから、30、60、90、120、180、および、240分後に決定をした。 尻尾に損傷を与えないようにすべく、カットオフ時間を、10秒間に設定した。 テイルフリック潜時から基礎潜時を差し引き、そして、その差異を、曲線下の面積(AUC)を算出するために用いた。 各マウスで得られた抗侵害受容反応値を、AUC0→240分に変換し、そして、平均±標準誤差として表した。
【0122】
実施例3:モルヒネ痛覚抑制に関するクロニジンの効果の決定
モルヒネが示す痛覚抑制に関するクロニジンの効果を決定するために、マウスを、次の4つのグループに分けた。 グループ1:賦形剤(生理食塩水、腹腔内投与)+賦形剤(生理食塩水、皮下投与)、グループ2:賦形剤+モルヒネ(4mg/kg、皮下投与)、グループ3;クロニジン(2mg/kg、腹腔内投与)+賦形剤(生理食塩水、皮下投与)、および、グループ4;クロニジン(2mg/kg、腹腔内投与)+モルヒネ(4mg/kg、皮下投与)。 モルヒネまたは賦形剤は、クロニジンを投与して30分後に投与をした。
【0123】
薬剤の投与を行わないテイルフリック基礎潜時値は、1.5〜2.3秒であった。 コントロールグループ(賦形剤+賦形剤)では、テイルフリック潜時は、4時間にわたって、基礎潜時から変化しなかった。 しかしながら、モルヒネ(4mg/kg、皮下投与)は、テイルフリック潜時を顕著に増大した。 クロニジン(2mg/kg、腹腔内投与)は、テイルフリック潜時を増大し、また、クロニジンで治療をしたマウスにモルヒネを投与したところ、モルヒネと比較をして、テイルフリック潜時をさらに増大した(図1A)。 モルヒネ(4mg/kg、皮下投与)で治療をしたグループで認められたAUCは、88.4±12.4であった。 クロニジンは、鎮痛作用を示し、そして、68.4±12.3のAUCを示したが、この数値は、コントロールが示す数値よりも遙かに大きな数値であった。 クロニジンで前処理をしたところ、モルヒネの鎮痛作用が顕著に増大し、また、125.4± 12.3のAUCが認められた。 クロニジンは、モルヒネがもたらす痛覚抑制を顕著に改善した(42%)(図1B)。
【0124】
実施例4:モルヒネ痛覚抑制に関するスルフィソキサゾールの効果の決定
モルヒネが示す痛覚抑制に関するスルフィソキサゾールの効果を決定するために、マウスを、次の4つのグループに分けた。 グループ1:賦形剤(カルボキシメチルセルロース(CMC)、経口投与)+賦形剤(生理食塩水、皮下投与)、グループ2:賦形剤(CMC)+モルヒネ(4mg/kg、皮下投与)、グループ3:スルフィソキサゾール(500mg/kg、経口投与)+賦形剤(生理食塩水、皮下投与)、および、グループ4:スルフィソキサゾール(500mg/kg、経口投与)+モルヒネ(4mg/kg、皮下投与)。 モルヒネまたは賦形剤は、スルフィソキサゾールを投与して30分後に投与をした。
【0125】
薬剤の投与を行わないテイルフリック基礎潜時値は、2.0〜2.4秒であった。 コントロールグループ(賦形剤+賦形剤)では、テイルフリック潜時は、4時間にわたって、基礎潜時から変化しなかった。 しかしながら、モルヒネ(4mg/kg、皮下投与)は、テイルフリック潜時を顕著に増大した。 スルフィソキサゾール(500mg/kg、経口投与)は、テイルフリック潜時を増大した。 しかしながら、スルフィソキサゾール(500 mg/kg、経口投与)で前処理を行っても、マウスにおいてテイルフリック潜時を増大するモルヒネの効果には影響が及ばなかったことは、500mg/kgの用量のスルフィソキサゾールを経口投与しても、モルヒネの鎮痛作用を増強しないことを示すものである(図2A)。 スルフィソキサゾール(56.0±9.1)が、コントロール(0.3±6.4)と比較してAUCを増大していることは、スルフィソキサゾールが、軽度の鎮痛作用を呈することを示すものである。 モルヒネ(4mg/kg、皮下投与)は、顕著な鎮痛作用を示し、また、そのAUCは、91.0±10.8であった。 しかしながら、スルフィソキサゾールは、モルヒネの鎮痛作用を実質的に増大するまでには至らず(AUC 103.8±11.3)、モルヒネが呈する痛覚抑制を、わずかに14%だけ増強したに過ぎなかった(図2B)。
【0126】
実施例5:モルヒネ痛覚抑制に関するクロニジンとスルフィソキサゾールの併用効果の決定
モルヒネが示す痛覚抑制に関するクロニジンとスルフィソキサゾールとの組み合わせが奏する効果を決定するために、マウスを、次のグループに分けた。
【0127】
試験1。 マウスを、次の三つのグループに分けた。 グループ1:クロニジン(2mg/kg、腹腔内投与)+スルフィソキサゾール(500mg/kg、経口投与)+賦形剤(V)(生理食塩水、皮下投与)、グループ2:クロニジン(2mg/kg、腹腔内投与)+スルフィソキサゾール(500mg/kg、経口投与)+モルヒネ(8mg/kg、皮下投与)、グループ3:賦形剤(生理食塩水、腹腔内投与)+賦形剤(カルボキシメチルセルロース、経口投与)+モルヒネ(8mg/kg、皮下投与)、および、グループ4:賦形剤(生理食塩水、腹腔内投与)+賦形剤(カルボキシメチルセルロース、経口投与)+賦形剤(生理食塩水、皮下投与)。 モルヒネまたは賦形剤は、クロニジンとスルフィソキサゾールを投与して30分後に投与をした。
【0128】
試験2。 マウスを、次の六つのグループに分けた。 グループ1:賦形剤(生理食塩水、腹腔内投与)+賦形剤(カルボキシメチルセルロース(CMC))+賦形剤(生理食塩水、皮下投与)、グループ2:クロニジン(2mg/kg、腹腔内投与)+賦形剤(CMC)+賦形剤(生理食塩水、皮下投与)、グループ3:スルフィソキサゾール(1000 mg/kg、経口投与)+賦形剤(生理食塩水、腹腔内投与)+賦形剤(生理食塩水、皮下投与)、グループ4:賦形剤(生理食塩水、腹腔内投与)+賦形剤(CMC)+モルヒネ(8mg/kg、皮下投与)、グループ5:クロニジン(2mg/kg、腹腔内投与)+スルフィソキサゾール(1000mg/kg、経口投与)+賦形剤(生理食塩水、皮下投与)、および、グループ6:クロニジン(2mg/kg、腹腔内投与)+スルフィソキサゾール(1000mg/kg、経口投与)+モルヒネ(8mg/kg、皮下投与)。 モルヒネまたは賦形剤は、クロニジンとスルフィソキサゾールを投与して30分後に投与をした。
【0129】
薬剤の投与を行わないテイルフリック基礎潜時値は、1.5〜2.0秒であった。 コントロールグループ(賦形剤+賦形剤)では、テイルフリック潜時は、4時間にわたって、基礎潜時から変化しなかった。 しかしながら、スルフィソキサゾール(500mg/kg、経口投与)とクロニジン(2mg/kg、腹腔内投与)とを併用すると、モルヒネが無くとも、テイルフリック潜時を顕著に増大した。 スルフィソキサゾール(500mg/kg、経口投与)とクロニジン(2mg/kg、腹腔内投与)とを併用して投与しても、モルヒネの効果に変化はなく、また、テイルフリック潜時にも変化は認められなかった(図3)。 カットオフ時間が10秒間に設定されており、また、本実施例での技術では、10秒を超えた後には何らの変化も認められなかったので、さらに別の増強作用を認めることは不可能であった。 クロニジンとスルフィソキサゾールという二つの非アヘン製剤を一緒に投与したところ、高用量のモルヒネに匹敵する鎮痛作用を呈したということは、非常に興味深いことであった。 さらに、高用量のスルフィソキサゾール(1000mg/kg、経口投与)を単独で、あるいは、クロニジン(2mg/kg、腹腔内投与)と組み合わせて利用した研究も行った。 コントロールのマウスと比較して、クロニジン(2mg/kg、腹腔内投与)は、テイルフリック潜時のAUCを増大し、同様に、コントロールのマウスと比較して、スルフィソキサゾール(1000mg/kg、経口投与)も、テイルフリック潜時のAUCを増大していた。 モルヒネ(8mg/kg、皮下投与)は、テイルフリック潜時を顕著に増大しており、AUCは、130.9±19.2であった。
【0130】
スルフィソキサゾール(1000mg/kg、経口投与)とクロニジン(2mg/kg、腹腔内投与)を併用したところ、テイルフリック潜時の顕著な増大が認められたことは興味深いことであり、また、このグループで認められたAUC(119.1±8.9)は、8mg/kgという高用量のモルヒネでのAUCと同等の数値であった。 クロニジンとスルフィソキサゾールを同時に投与した場合、クロニジンまたはスルフィソキサゾールのいずれか一方だけを投与した場合と比較して、AUCが、167%増大していた。 これら知見は、クロニジンとスルフィソキサゾールという二つの非アヘン製剤を一緒に投与することで、モルヒネに匹敵する痛覚抑制が実現可能となる、ことを確認するものであった(図4)。
【0131】
実施例6:鎮痛作用に関するクロニジンの用量応答効果
鎮痛作用に関するクロニジンの用量応答効果を決定するために、0.3mg/ml、1.0mg/ml、および、3.0mg/mlのクロニジンをマウスに投与し、そして、鎮痛作用に関する効果の評価を行った。
【0132】
図5Aおよび図5Bには、鎮痛作用に関するクロニジンの用量応答効果(テイルフリック潜時)と、賦形剤(生理食塩水)、0.3mg/ml、1.0mg/ml、および、3.0mg/mlのクロニジンのいずれかを投与したマウスの体温が示されている。 実験結果は、3.0mg/mlのクロニジンのテイルフリック潜時が、賦形剤について計測をしたすべての時点で得られたテイルフリック潜時の約5倍の数値であったことを示している。 1.0mg/mlのクロニジンのテイルフリック潜時は、刺激を与えて約3時間までは、約5倍の数値を示していたが、約6時間の時点では、賦形剤について得られたテイルフリック潜時の3倍の数値にまで減少していた。 0.3mg/mlのクロニジンのテイルフリック潜時は、驚くべきことに、刺激を与えた後は、約4倍の数値にまで増大をしていたが、約3時間の時点では、賦形剤について得られたテイルフリック潜時の約3倍の数値にまで減少し、そして、約5時間の時点では、2.5倍の数値にまで減少していた。 これらの結果は、鎮痛作用に関するクロニジンの効果は、用量依存的であり、そして、0.3mg/kgという低用量のクロニジンでも、鎮痛作用を得る上で効果的であることを実証している。
【0133】
用量応答に関するこの研究は、すべての用量のクロニジンが、治療を施した動物の体温を同様に下げていることを示している(図5B)。
【0134】
実施例7:鎮痛作用に関するクロニジンとスルフィソキサゾールの用量応答効果
前述したように、クロニジンについては、治療を施した動物での鎮痛作用に関する用量応答効果が実証されている。 治療計画にスルフィソキサゾールを加えても用量応答効果が得られるかどうかを決定するために、0.3mg/kgのクロニジンを、様々な用量のスルフィソキサゾールと共に投与をした。 0.3mg/kgのクロニジンと、0.5%CMC(10ml/kg)、あるいは、250、500、または、1000mg/kgのスルフィソキサゾールのいずれかとで、マウスの治療にあたった。 その結果、クロニジンと、0.5%CMC、または、いずれかの用量のスルフィソキサゾールとを用いた場合に、3時間後には、体温が、37℃から約32℃にまで同様に低下し(図6B)、そして、図5Bに示したように、すべての用量のクロニジンに匹敵するような体温の下げ方が認められた。 鎮痛作用に関するテイルフリック潜時検査において、0.3mg/kgのクロニジンとCMCの組み合わせは、刺激を与えて2時間後には、テイルフリック潜時を5倍にまで増大したが、6時間を経過するまでには、賦形剤で治療を施した動物でのテイルフリック潜時と大差のないレベルにまで減少した。 クロニジンが誘発した鎮痛作用に関するスルフィソキサゾールの効果は、用量依存的ではなかった(図6A)。 250、500、または、1000mg/kgのいずれかの濃度のスルフィソキサゾールを、0.3mg/kgのクロニジンと共に投与すると、刺激を与えて2時間後には、テイルフリック潜時は5倍にまで増大し、そして、この鎮痛作用のレベルは、刺激を与えて6時間後もなお維持されていた。
【0135】
これら結果は、クロニジンの鎮痛作用は、スルフィソキサゾールによって増強されるものであって、この効果は、最低用量(250mg/kg)のスルフィソキサゾールであっても認められるものである、ことを示している。
【0136】
クロニジンの鎮痛作用を増強することができるスルフィソキサゾールの最低用量を決定するために、別の研究を行った。 クロニジン(0.3mg/kg)と、0.5%CMC、または、25、75、または、225mg/kgの濃度のスルフィソキサゾールのいずれかとを、マウスに対して投与をした。 その結果、スルフィソキサゾールは、クロニジンが誘発する体温低下に対して何らの影響も及ぼさなかった(図7B)が、スルフィソキサゾールは、クロニジンが誘発する鎮痛作用に対しては用量依存効果を示す(図7A)、ことが明らかとなった。 225mg/kgの濃度のスルフィソキサゾールは、刺激を与えてから1.5時間後には、テイルフリック潜時を4〜5倍にまで増大しており、この鎮痛作用のレベルは、刺激を与えてから6時間後もなお維持されていた。 75mg/kgのスルフィソキサゾールのテイルフリック潜時は、刺激を与えて約3時間までは、約4倍の数値を示していたが、6時間の時点では、賦形剤で治療した動物に関して得られたテイルフリック潜時の約3倍の数値にまで減少していた。 25mg/kgのスルフィソキサゾールは、クロニジンが誘発する鎮痛作用を、賦形剤で治療したマウスと比較して、約5倍のレベルにまで増強したが、このレベルは徐々に減少していき、治療が終了して6時間後には、約2倍のレベルにまでなっていた。 スルフィソキサゾールの用量を変えても、体温には何らの影響も無かった。
【0137】
これらの結果は、低用量のスルフィソキサゾール(225および75mg/kg)が、低用量のクロニジン(0.3mg/kg)の鎮痛作用を増強する、ことを指し示すものである。 よって、これら二つの薬剤の組み合わせは、モルヒネと同様の鎮痛作用をもたらすが、それら薬剤は、クロニジンだけ、あるいは、スルフィソキサゾールだけを用いる場合の高投与量の用量よりも低用量ですむこととなる。
【0138】
実施例8:クロニジンの鎮痛作用のメカニズム
鎮痛作用に関係するクロニジンとスルフィソキサゾールが奏する作用のメカニズムを決定するために、異なる疼痛受容体、例えば、アルファ-2アドレナリン受容体、イミダゾリンアドレナリン受容体、または、オピオイド受容体などに作用する幾つかの異なる薬剤を、クロニジンと共に投与した。
【0139】
α2アドレナリン受容体が、クロニジンの鎮痛作用を媒介するかどうかを決定するために、α2アドレナリン作動性受容体アンタゴニストであるヨヒンビンを、クロニジンとCMCの組み合わせ、または、クロニジンとスルフィソキサゾールの組み合わせと共に投与をし、そして、テイルフリック潜時と体温の検査を行った。 マウスを、次の5つのグループに分けた。 グループ1:賦形剤+賦形剤(生理食塩水、腹腔内投与)+賦形剤(生理食塩水、腹腔内投与)、グループ2:賦形剤+CMC(経口投与)+賦形剤(生理食塩水、腹腔内投与)、グループ3:ヨヒンビン(2mg/kg、腹腔内投与)+CMC(経口投与)+賦形剤(生理食塩水、腹腔内投与)、グループ4:ヨヒンビン(2mg/kg、腹腔内投与)+CMC(経口投与)+クロニジン(0.3mg/kg、腹腔内投与)、グループ5:ヨヒンビン(2mg/kg、腹腔内投与)+スルフィソキサゾール(250mg/kg、経口投与)+クロニジン(0.3mg/kg、腹腔内投与)。
【0140】
ヨヒンビンは、クロニジンが誘発した体温低下を若干緩和していた(図8B)。 ヨヒンビン、クロニジン、および、スルフィソキサゾールを投与したところ、ヨヒンビンは、クロニジンとスルフィソキサゾールとの組み合わせが奏する鎮痛作用に対しては効果を示さなかった(図8A)。 これら研究結果は、クロニジンとスルフィソキサゾールとの組み合わせが奏する鎮痛作用が、α2アドレナリン受容体を媒介したものではない、ことを指し示すものである。
【0141】
イミダゾリンアドレナリン受容体が、クロニジンの鎮痛作用を媒介するかどうかを決定するために、イミダゾリン受容体アンタゴニストであるイダゾキサンを、クロニジンとCMCの組み合わせ、または、クロニジンとスルフィソキサゾールの組み合わせと共に投与をし、そして、テイルフリック潜時と体温の検査を行った。 マウスを、次の5つのグループに分けた。 グループ1:賦形剤+賦形剤(生理食塩水、腹腔内投与)+賦形剤(CMC、経口投与)、グループ2:イダゾキサン(2mg/kg、腹腔内投与)+CMC(経口投与)+賦形剤(生理食塩水、腹腔内投与)、グループ3:イダゾキサン(2mg/kg、腹腔内投与)+CMC(経口投与)+賦形剤(生理食塩水、腹腔内投与)、グループ4:イダゾキサン(2mg/kg、腹腔内投与)+CMC(経口投与)+クロニジン(0.3mg/kg、腹腔内投与)、グループ5:イダゾキサン(2mg/kg、腹腔内投与)+スルフィソキサゾール(250mg/kg、経口投与)+クロニジン(0.3mg/kg、腹腔内投与)。 イダゾキサンは、クロニジンが誘発した体温低下を若干緩和していた(図9B)。 クロニジンとスルフィソキサゾールの投与を受けたマウスに対してイダゾキサンを投与したところ、クロニジンとスルフィソキサゾールとの組み合わせが奏する鎮痛作用に対して効果が認められた(図9A)。 これら研究結果は、クロニジンとスルフィソキサゾールとの組み合わせが奏する鎮痛作用の一部が、イミダゾリンアドレナリン受容体を媒介したものである、ことを指し示すものである。
【0142】
オピオイドアドレナリン受容体が、クロニジンの鎮痛作用を媒介するかどうかを決定するために、オピオイド受容体アンタゴニストであるナロキソンを、クロニジンとCMCの組み合わせ、または、クロニジンとスルフィソキサゾールの組み合わせと共に投与をし、そして、テイルフリック潜時と体温の検査を行った。 マウスを、次の5つのグループに分けた。 グループ1:ナロキソン(1.0mg/kg、腹腔内投与)+CMC(経口投与)+賦形剤(生理食塩水、腹腔内投与)、グループ2:ナロキソン(1.0mg/kg、腹腔内投与)+CMC(経口投与)+クロニジン(0.3mg/kg、腹腔内投与)、グループ3:ナロキソン(1.0mg/kg、腹腔内投与)+スルフィソキサゾール(250mg/kg、経口投与)+クロニジン(0.3mg/kg、腹腔内投与)。 しかしながら、ナロキソンは、クロニジンが誘発した体温低下には作用を示さなかった(図10B)。 その一方で、ナロキソンを投与することで、クロニジンとスルフィソキサゾールの組み合わせが奏する鎮痛作用を抑制し、約5倍にまで増大したテイルフリック潜時(図5Aを参照)は、治療を終えて4時間後には、わずか2倍の増大に止まるだけであった(図10A)。 これら研究結果は、クロニジンとスルフィソキサゾールとの組み合わせが奏する鎮痛作用が、オピオイド受容体を媒介したものである、ことを指し示すものである。
【0143】
実施例9:クロニジンの鎮痛作用に関するその他のエンドセリンAアンタゴニストの効果
クロニジンの鎮痛作用に関するスルフィソキサゾールの効果が、分子に対して特異的であるものかどうか、また、その他のETAアンタゴニストも、クロニジンの鎮痛作用を増強するものかどうか、について決定をするために、クロニジンとETAアンタゴニストのBMS182874を投与したマウスにおいて、テイルフリック潜時と体温の評価を行った。
【0144】
マウスを、次の3つの治療グループに分けた。 グループ1:クロニジン(0.3mg/kg、腹腔内投与)+BMS182874(2.0μg/kg、脳室内投与)、グループ2:クロニジン(0.3mg/kg、腹腔内投与)+BMS182874(10.0μg/kg、脳室内投与)、グループ3:クロニジン(0.3mg/kg、腹腔内投与)+BMS182874(50.0μg/kg、脳室内投与)。 その結果、低用量および高用量のBMS182874は、クロニジンの鎮痛作用を適度に増強していたが、中用量(10μg/kg)でも、クロニジンの鎮痛作用の増強が認められており(図11A)、このことは、治療を終えて1時間後および2時間後には、テイルフリック潜時が5倍にまで増大し、また、治療を終えて4時間後には、テイルフリック潜時が約3倍のレベルにまで減少し、そして、治療を終えて6時間後には、テイルフリック潜時が2倍のレベルにまで減少している、ことから明らかとなった(図5Aでの賦形剤の結果と図11Aとの比較)。 BMS182874は、クロニジンが引き起こす体温低下に対しては何らの影響も及ぼさなかった(図11B)。
【0145】
これら結果は、ETA受容体が、クロニジンの鎮痛作用の増強に関与しているかもしれないこと、そして、最適な用量を決定する際に用いられるその他のETAアンタゴニストが、クロニジンが奏する鎮痛作用を増強することができること、を指し示すものである。
【0146】
実施例10:オピオイドの鎮痛作用に関するクロニジンまたはBMS182874の効果
モルヒネの鎮痛作用とオキシコドンの鎮痛作用との間の差異について、報告がされている[40、41、42]。 オキシコドンは、1917年から臨床で使用されてきている[43]。 オキシコドンは、μ-オピオイド受容体アゴニスト[44]であり、μ-オピオイド受容体に対するKi値は18nMであり、δ-オピオイド受容体に対するKi値は958nMであり、そして、κ-オピオイド受容体に対するKi値は677nMである[45]。 しかしながら、オキシコドンのμ-オピオイド受容体に対する親和性は、モルヒネの1/20にも満たない。 モルヒネが、雌のラットよりも雄のラットに対して大きな鎮痛作用を示すのに対して、オキシコドンが、雌と雄の双方のラットに対して同様の鎮痛作用を示しており、このことが、モルヒネとオキシコドンとの間の鎮痛作用の差異を示す別の根拠となっております[46]。 α2アドレナリン作動性受容体阻害剤であるイダゾキサンでのα2アドレナリン受容体に対するKi値は3.6nMであり、また、I1-イミダゾリン受容体に対するKi値は186nMであり[47]、このことは、イダゾキサンが、α2アドレナリン受容体に対して作用し、それゆえに、クロニジンによるモルヒネの鎮痛作用の増強をブロックすること、を指し示すものである。 クロニジンでのα2アドレナリン作動性受容体部位に対するKi値は3.8nMであり、また、I1-イミダゾリン受容体に対するKi値は1.0nMである[48]。
【0147】
オピオイド鎮痛作用の増強に関するクロニジンとその他のETAアンタゴニストの効果を決定するために、クロニジン、BMS182874、および、オピオイドを投与したマウスのテイルフリック潜時を、イダゾキサンの存在下または非存在下で評価をした。
【0148】
モルヒネの鎮痛作用に関するクロニジンまたはBMS182874の効果、および、イダゾキサンによるそれらの阻害
準備した動物を、次のグループ(n=6/グループ)に分けた。 グループ1:賦形剤(1ml/kg、腹腔内投与)+賦形剤(5μl、脳室内投与)+モルヒネ(8mg/kg、皮下投与)、グループ2:賦形剤(1ml/kg、腹腔内投与)+クロニジン(1mg/kg、腹腔内投与)+賦形剤(1ml/kg、皮下投与)、グループ3:賦形剤(1ml/kg、腹腔内投与)+クロニジン(1mg/kg、腹腔内投与)+モルヒネ(8mg/kg、皮下投与)、グループ4:イダゾキサン(2mg/kg、腹腔内投与)+クロニジン(1mg/kg、腹腔内投与)+モルヒネ(8mg/kg、皮下投与)、グループ5:賦形剤(1ml/kg、腹腔内投与)+賦形剤(5μl、脳室内投与)+モルヒネ(8mg/kg、皮下投与)、グループ6:賦形剤(1ml/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+賦形剤(1ml/kg、皮下投与)、グループ7:賦形剤(1ml/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+モルヒネ(8mg/kg、皮下投与)、グループ8:イダゾキサン(2mg/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+モルヒネ(8mg/kg、皮下投与)。
【0149】
7,8-ジデヒドロ-4,5α-エポキシ-17-メチルモルフィナン-3,6α-ジオール硫酸塩(マリンクロットケミカル社、セントルイス、ミズーリ州)である硫酸モルヒネを、滅菌済生理食塩水に溶解し、そして、皮下(s.c.)に注射をした。 N-(2,6-ジクロロフェニル)-4,5-ジヒドロ-1H-イミダゾ−ル-2-アミン(シグマケミカル社、セントルイス、ミズーリ州)であるクロニジンを、滅菌済生理食塩水に溶解し、そして、腹腔内(i.p.)に注射をした。 5-(ジメチルアミノ)-N-(3,4-ジメチル-5-イソキサゾリル)-1-ナフタレンスルホンアミド(トクリスファーマシューティカルズ社、エリスヴィル、ミズーリ州)であるBMS182874を、20%DMSOに溶解し、そして、脳室内(i.c.v.)に注射をした。
【0150】
2-(1,4-ベンゾジオキサン-2-イル)-2-イミダゾリン(シグマケミカル社、セントルイス、ミズーリ州)であるイダゾキサンを、滅菌済生理食塩水に溶解し、そして、腹腔内(i.p.)に注射をした。 実験を行うにあたって、クロニジンまたはBMS182874で治療を行う15分前にイダゾキサンを投与し、また、クロニジンまたはBMS182874で治療を行った30分後にモルヒネを投与した。
【0151】
モルヒネ(8mg/kg、皮下投与)は、顕著な鎮痛作用を示しており、AUC0→360は、21.23±3.18秒.分であった(図12B)。 クロニジン(1mg/kg、腹腔内投与)も鎮痛作用を示しており、AUC0→360は、16.62±1.61秒.分であった(図12B)。 クロニジン(1mg/kg、腹腔内投与)とモルヒネ(8mg/kg、皮下投与)の組み合わせで治療をしたラットでは、モルヒネ(P=0.015)またはクロニジン(P=0.001)のいずれかだけで治療をしたラットと比較して、遙かに大きな鎮痛作用が認められた。 イダゾキサン(2mg/kg、腹腔内投与)は、クロニジンとモルヒネの組み合わせを投与したラットで認められた鎮痛作用に対しては、何らの影響も及ぼさなかった(図12A)。
【0152】
BMS182874(50μg、脳室内投与)だけを投与しても鎮痛作用は認められず、AUC0→360は、4.47±1.49秒.分であった。 しかしながら、BMS182874(50μg、脳室内投与)とモルヒネ(8mg/kg、皮下投与)の組み合わせで治療をしたラットでは、モルヒネ(P=0.009)またはBMS182874(P=0.0006)のいずれかだけで治療をしたラットと比較して、遙かに大きな鎮痛作用が認められた(図12C)。 イダゾキサン(2mg/kg、腹腔内投与)は、BMS182874とモルヒネの組み合わせを投与したラットで認められた鎮痛作用に対しては、何らの影響も及ぼさなかった(図12A)。 これら結果は、クロニジン(P=0.015)とBMS182874(P=0.009)が、モルヒネの鎮痛作用を顕著に増強することを指し示すものであった。
【0153】
オキシコドンの鎮痛作用に関するクロニジンまたはBMS182874の効果、および、イダゾキサンによるそれらの阻害
オキシコドンの鎮痛作用に関するクロニジンまたはBMS182874の効果を評価するために、治療グループを、次のようにして分けた(n=6/グループ)。 グループ1:賦形剤(1ml/kg、腹腔内投与)+賦形剤(1ml/kg、腹腔内投与)+オキシコドン(4mg/kg、皮下投与)、グループ2:賦形剤(1ml/kg、腹腔内投与)+クロニジン(1mg/kg、腹腔内投与)+賦形剤(1ml/kg、皮下投与)、グループ3:賦形剤(1ml/kg、腹腔内投与)+クロニジン(1mg/kg、腹腔内投与)+オキシコドン(4mg/kg、皮下投与)、グループ4:イダゾキサン(2mg/kg、腹腔内投与)+クロニジン(1mg/kg、腹腔内投与)+オキシコドン(4mg/kg、皮下投与)、グループ5:賦形剤(1ml/kg、腹腔内投与)+賦形剤(5μl、脳室内投与)+オキシコドン(4mg/kg、皮下投与)、グループ6:賦形剤(1ml/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+賦形剤(1ml/kg、皮下投与)、グループ7:賦形剤(1ml/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+オキシコドン(4mg/kg、皮下投与)、グループ8:イダゾキサン(2mg/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+オキシコドン(4mg/kg、皮下投与)。
【0154】
クロニジン、BMS182874、および、イダゾキサンは、前述した手順で調製をした。 4,5α-エポキシ-14-ヒドロキシ-3-メトキシ-17-メチルモルフィナン-6-オン塩酸塩(スペクトラムケミカル社、サンガーデナ、カリフォルニア州)である塩酸オキシコドンを、滅菌済生理食塩水に溶解し、そして、皮下(s.c.)に注射をした。 クロニジンまたはBMS182874で治療を行った30分後にオキシコドンを投与した。 クロニジンまたはBMS182874で治療を行う15分前にイダゾキサンを投与した。
【0155】
その結果、オキシコドン(4mg/kg、皮下投与)は、顕著な鎮痛作用を示しており、AUC0→360は、18.23±2.32秒.分であった。 クロニジン(1mg/kg、腹腔内投与)も鎮痛作用を示していた。 クロニジン(1mg/kg、腹腔内投与)とオキシコドン(4mg/kg、皮下投与)の組み合わせで治療をしたラットでは、オキシコドン(P=0.025)またはクロニジン(P=0.016)のいずれかだけで治療をしたラットと比較して、遙かに大きな鎮痛作用が認められた(図13B)。 イダゾキサン(2mg/kg、腹腔内投与)は、クロニジンとオキシコドンの組み合わせを投与したラットで認められた鎮痛作用をブロックした(図13A)。
【0156】
オキシコドン(4mg/kg、皮下投与)は、顕著な鎮痛作用を示したが、BMS182874(50μg、脳室内投与)は、鎮痛作用は全く示さなかった(図13A)。 しかしながら、BMS182874 (50μg、脳室内投与)とオキシコドン(4mg/kg、皮下投与)の組み合わせで治療をしたラットでは、オキシコドン(P=0.012)またはBMS182874(P=0.0004)のいずれかだけで治療をしたラットと比較して、遙かに大きな鎮痛作用が認められた(図13C)。 イダゾキサン(2mg/kg、腹腔内投与)は、BMS182874とオキシコドンの組み合わせを投与したラットで認められた鎮痛作用を顕著に(P=0.003)ブロックした(図13A、13C)。 これら結果は、クロニジン(P=0.025)とBMS182874(P=0.012)が、オキシコドンが示す鎮痛作用を増強することを指し示すものであった。
【0157】
概して、I1-イミダゾリンであり、かつ、α2アドレナリン作動性受容体アンタゴニストでもあるイダゾキサンは、オキシコドンの鎮痛作用が、クロニジンまたはBMS182874によって増強されることをブロックするが、その一方で、イダゾキサンは、モルヒネの鎮痛作用が、クロニジンまたはBMS182874によって増強されることに対しては何らの影響も与えないことが明らかとなった。 この知見は、オキシコドンの鎮痛作用でのイミダゾリン受容体の関与を示すものであって、つまりは、I1-イミダゾリン受容体が、オキシコドンの鎮痛作用の増強には関与しているものの、クロニジンまたはBMS182874によるモルヒネの鎮痛作用の増強には関与していない、ことを示すものである。 本実施例は、オキシコドンの鎮痛作用を増強するクロニジンまたはBMS182874が、I1-イミダゾリン受容体とは関連があるものの、モルヒネの鎮痛作用とは関連が認められない、ことを初めて報告するものである。
【0158】
実施例11:モルヒネまたはオキシコドンの鎮痛作用を増強したクロニジンに関するヨヒンビンの効果
α2アドレナリン作動性受容体の関与を決定するために、選択的アンタゴニストであるヨヒンビンを用いた。 ヨヒンビンは、非常に選択性に富んだα2アドレナリン作動性受容体アンタゴニストであって、α2アドレナリン受容体に対するKi値は22nMであり、そして、I1-イミダゾリン受容体に対するKi値は21,810nMである[49]。 ヨヒンビンは、イミダゾリン環を有しておらず、また、イミダゾリン受容体に対して作用もしない。
【0159】
治療グループを、次のようにして分けた。 グループ1:賦形剤(1ml/kg、腹腔内投与)+賦形剤(5μl、脳室内投与)+モルヒネ(4mg/kg、皮下投与)、グループ2:賦形剤(1ml/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+モルヒネ(4mg/kg、皮下投与)、グループ3:ヨヒンビン(2mg/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+モルヒネ(4mg/kg、皮下投与)、グループ4:賦形剤(1ml/kg、腹腔内投与)+賦形剤(5μl、脳室内投与)+オキシコドン(4mg/kg、皮下投与)、グループ5:賦形剤(1ml/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+オキシコドン(4mg/kg、皮下投与)、グループ 6:ヨヒンビン(2mg/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+オキシコドン(4mg/kg、皮下投与)。
【0160】
17α-ヒドロキシ-ヨヒンバン-16α-カルボキシレート塩酸塩(シグマケミカル社、セントルイス、ミズーリ州)である塩酸ヨヒンビンを、アルコール(1部)と滅菌生理食塩水(9部)に溶解し、そして、 腹腔内(i.p.)に注射をした。 BMS182874で治療を行う15分前にヨヒンビンを投与し、また、BMS182874で治療を行った30分後にモルヒネまたはオキシコドンを投与した。
【0161】
その結果、クロニジンが、モルヒネ(P=0.0366)ならびにオキシコドン(P=0.0587)の鎮痛作用を増強し、また、ヨヒンビンが、モルヒネ(P=0.058)またはオキシコドン(P=0.0167)の鎮痛作用を増強するというクロニジンの作用をブロックすることが明らかとなった(図14A−14C)。 この拮抗作用については、モルヒネでの重要性は不明確であるが、オキシコドンの鎮痛作用においては非常に重要であった。 モルヒネ(8mg/kg、皮下投与)の鎮痛作用の改善は、ヨヒンビンを用いて前処理することによって阻害された(P=0.0355)(図14B)。 同様に、オキシコドン(4mg/kg、皮下投与)が奏する鎮痛作用をクロニジンが増強し、その鎮痛作用が、ヨヒンビンを用いて前処理することによって顕著にブロックされる、ことが明らかとなった(図14C)。
【0162】
モルヒネの鎮痛作用、および、オキシコドンの鎮痛作用を増強するBMS182874に対するα2アドレナリン作動性受容体の関与を決定するために、これら薬剤が投与された動物に対して、α2アドレナリン作動性選択的アンタゴニストであるヨヒンビンを投与した。 治療グループを、次のようにして分けた。 グループ1:賦形剤(1ml/kg、腹腔内投与)+賦形剤(5μl、脳室内投与)+モルヒネ(4mg/kg、皮下投与)、グループ2:賦形剤(1ml/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+モルヒネ(4mg/kg、皮下投与)、グループ3:ヨヒンビン(2mg/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+モルヒネ(4mg/kg、皮下投与)、グループ4:賦形剤(1ml/kg、腹腔内投与)+賦形剤(5μl、脳室内投与)+オキシコドン(4mg/kg、皮下投与)、グループ5:賦形剤(1ml/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+オキシコドン(4mg/kg、皮下投与)、グループ 6:ヨヒンビン(2mg/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+オキシコドン(4mg/kg、皮下投与)。
【0163】
BMS182874で治療を行う15分前にヨヒンビンを投与した。 BMS182874で治療を行った30分後にモルヒネまたはオキシコドンを投与した。
【0164】
前述したように、BMS182874は、オキシコドン(4mg/kg、皮下投与)が奏する鎮痛作用を増強し(P=0.0001)、また、モルヒネ(8mg/kg、皮下投与)が奏する鎮痛作用も増大させている(P=0.003)(図15Aおよび15B)。 BMS182874は、モルヒネの鎮痛作用を増強し(P=0.003)、また、オキシコドンの鎮痛作用も増強する(P=0.0001)(図15Bおよび15C)。 BMS182874で治療を施した動物に対してヨヒンビンを投与したところ、BMS182874で治療を施したラットにおいてモルヒネまたはオキシコドンが示した鎮痛作用は、ヨヒンビンを用いて前処理を行っても、何らの影響も受けなかった(P=0.156)(図15C)。
【0165】
これらの結果は、モルヒネおよびオキシコドンの鎮痛作用をクロニジンが増強するという作用が、選択的α2アドレナリン作動性アンタゴニストであるヨヒンビンによってブロックされていることを示すものであって、つまり、モルヒネおよびオキシコドンの鎮痛作用を増強するというクロニジンの作用に対してα2アドレナリン受容体が関与している、ことを指し示すものである。 また、これら知見は、モルヒネおよびオキシコドンの鎮痛作用を増強するというBMS182874およびクロニジンの作用が、異なるメカニズムを介したものであることも示している。
【0166】
実施例12:BMS182874で治療を施したラットでのモルヒネおよびオキシコドンの鎮痛作用に関するクロニジンの用量応答効果
先に見てきたように、クロニジンまたはスルフィソキサゾールの用量は、治療を受けた動物で認められる鎮痛レベルと相関をしていた。 オピオイドの鎮痛作用に関するクロニジンとETAアンタゴニストのBMS182874の用量効果を決定するために、用量反応実験を行った。
【0167】
治療グループを、次のようにして分けた(n=6/グループ)。 グループ1:賦形剤(1ml/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+モルヒネ(4mg/kg、皮下投与)、グループ2:クロニジン(0.1mg/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+モルヒネ(4mg/kg、皮下投与)、グループ3:クロニジン(0.3mg/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+モルヒネ(4mg/kg、皮下投与)、グループ4:クロニジン(1.0mg/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+モルヒネ(4mg/kg、皮下投与)、グループ5:賦形剤(1ml/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+オキシコドン (4mg/kg、皮下投与)、グループ6:クロニジン(0.1mg/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+オキシコドン(4mg/kg、皮下投与)、グループ7:クロニジン(0.3mg/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+オキシコドン(4mg/kg、皮下投与)、グループ8:クロニジン(1.0mg/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+オキシコドン(4mg/kg、皮下投与)。
【0168】
BMS182874で治療を行う15分前にヨヒンビンを投与し、そして、BMS182874で治療を行った30分後にモルヒネまたはオキシコドンを投与した。 その他の活性成分は、前述した手順に従って、調製および投与を行った。
【0169】
モルヒネ(4mg/kg、皮下投与)は、BMS182874で治療を施したラットにおいて顕著な鎮痛作用を示した。 クロニジンは、BMS182874で治療を施したラットにおいて、モルヒネの鎮痛作用を用量依存的に増大した(図16A)。 0.1mg/kgの用量でクロニジンを腹腔内投与したところ、鎮痛作用の改善は認められなかったが、0.3mg/kgおよび1.0mg/kgの用量でクロニジンを腹腔内投与したところ、BMS182874で治療を施したラットにおいて、モルヒネの鎮痛作用を顕著に増強した(順にP=0.016とP=0.011であった)(図16Aおよび16B)。
【0170】
オキシコドン(4mg/kg、皮下投与)は、BMS182874で治療を施したラットにおいて顕著な鎮痛作用を示した。 クロニジンは、BMS182874で治療を施したラットにおいて、オキシコドンの鎮痛作用を用量依存的に増大した(図16C)。 0.1mg/kgの用量でクロニジンを腹腔内投与したところ、鎮痛作用の改善は認められなかったが、0.3mg/kgおよび1.0mg/kgの用量でクロニジンを腹腔内投与したところ、BMS182874で治療を施したラットにおいて、オキシコドンの鎮痛作用を顕著に増強した(順にP=0.02とP=0.031であった)(図16Cおよび16D)。
【0171】
実施例13:オピオイドの鎮痛作用に関するクロニジン、BMS182874、および、クロニジンとBMS182874との組み合わせの効果
クロニジンおよびBMS182874の併用に起因するモルヒネまたはオキシコドンの鎮痛効果の増強作用を確認するために、モルヒネ(8mg/kg、皮下投与)とオキシコドン(4mg/kg、皮下投与)で治療を施したラットにおいて、クロニジン(1mg/kg、腹腔内投与)、BMS182874(50μg、脳室内投与)、および、クロニジン(1mg/kg、腹腔内投与)とBMS182874(50μg、脳室内投与)との組み合わせの効果を決定した。
【0172】
治療グループを、次のようにして分けた(n=6/グループ)。 グループ1:賦形剤(1ml/kg、腹腔内投与)+クロニジン(1mg/kg、腹腔内投与)+モルヒネ(8mg/kg、皮下投与)、グループ2:賦形剤(1ml/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+モルヒネ (8mg/kg、皮下投与)、グループ3:クロニジン(1.0mg/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+モルヒネ(8mg/kg、皮下投与)、グループ 4:賦形剤(1ml/kg、腹腔内投与)+クロニジン(1mg/kg、腹腔内投与)+オキシコドン(4mg/kg、皮下投与)、グループ5:賦形剤(1ml/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+オキシコドン (4mg/kg、皮下投与)、グループ6:クロニジン(1.0mg/kg、腹腔内投与)+BMS182874(50μg、脳室内投与)+オキシコドン(4mg/kg、皮下投与)。
【0173】
BMS182874で治療を行う15分前にクロニジンを投与し、そして、BMS182874またはクロニジンで治療を行った30分後にモルヒネまたはオキシコドンを投与した。
【0174】
その結果、クロニジンとBMS182874との組み合わせで治療をしたラットでのモルヒネの鎮痛作用(図17A)が、クロニジン(P=0.09)またはBMS182874(P=0.07)のいずれかだけで治療を行った場合(図17B)との比較において、さらに増強されていることは認められなかった。 同様に、クロニジンとBMS182874との組み合わせで治療をしたラットでのオキシコドンの鎮痛作用が、クロニジン(P=0.18)またはBMS182874(P=0.11)のいずれかだけで治療を行った場合(図17C)との比較において、さらに増強されていることも認められなかった。
【0175】
実施例14:クロニジンの鎮痛作用に関するBMS182874の効果
クロニジン(1mg/kg、腹腔内投与)の鎮痛作用が、BMS182874(50μg、脳室内投与)によって増強されるかどうかを決定するために、BMS182874、クロニジン、および、クロニジンとBMS182874との組み合わせの効果を、ラットを用いて決定をした。
【0176】
治療グループを、次のようにして分けた(n=6/グループ)。 グループ1:BMS182874(50μg、脳室内投与)+賦形剤(1ml/kg、腹腔内投与)、グループ2:賦形剤(5μl、脳室内投与)+クロニジン(1.0mg/kg、腹腔内投与)、グループ3:BMS182874(50μg、脳室内投与)+クロニジン(1.0mg/kg、腹腔内投与)。 クロニジンで治療を行う15分前にBMS182874を投与した。
【0177】
その結果、BMS182874は、鎮痛作用を示さなかったが、クロニジンは、BMS182874と比較して顕著な鎮痛作用を示す(P=0.00086)ことが明らかとなった(図18A)。 しかしながら、BMS182874は、クロニジンの鎮痛作用に影響を及ぼさなかった(P=0.645)(図18B)。
【0178】
以下の表には、様々な薬剤が奏する鎮痛作用がまとめられている。
【0179】
【表1】
【0180】
クロニジンとBMS182874は、異なるメカニズムを介して作用していることが、上記結果によってさらに確認されたとの結論は、モルヒネの鎮痛作用とオキシコドンの鎮痛作用を増強するクロニジンの作用が、BMS182874を用いた治療を行っても影響を受けないことを指し示すものである。 同様に、モルヒネの鎮痛作用とオキシコドンの鎮痛作用を増強するBMS182874の作用は、クロニジンを用いた治療を行っても影響を受けない。 さらに、クロニジンは、鎮痛作用を示すが、この作用は、BMS182874を用いた治療を行っても影響を受けない(図18)。 これらすべての知見は、クロニジンとBMS182874が、異なるメカニズムを介して、モルヒネの鎮痛作用とオキシコドンの鎮痛作用を増強することを指し示している。
【0181】
さらに、ヨヒンビンは、モルヒネの鎮痛作用を増強するクロニジンの作用をブロックするが、イダゾキサンは、モルヒネの鎮痛作用を増強するクロニジンの作用には影響を及ぼさないので、α2アドレナリン受容体は、クロニジンの増強作用に関与はするが、I1-イミダゾリン受容体は、モルヒネの鎮痛作用を増強するクロニジンの作用に関与はしていない、と結論付けることができる。
【0182】
α2アドレナリンアゴニストおよびエンドセリンAアンタゴニストが、オピオイドの鎮痛作用を効果的に増強できる、という本明細書に記載の知見は、これら薬剤の組み合わせが、治療を施す動物での疼痛を軽減するのに必要なオピオイドの用量を効果的に減少させるであろう、ということを示唆するものである。 さらに、本明細書に記載の実験は、α2アドレナリンアゴニストとエンドセリンAアンタゴニストとを組み合わせることで、高用量のモルヒネと同様の鎮痛作用を相乗的に奏することを実証している。 したがって、α2アドレナリンアゴニストとエンドセリンAアンタゴニストとの組み合わせを含む組成物、または、一方の薬剤だけを含み、一斉に投与がされる複数の組成物は、耐性のリスクも無く、また、オピオイド鎮痛薬の追加を必要とせずに、疼痛の症状を治療する上で有用である。
【0183】
これまでに説明してきた本願発明の実施例を考慮して、当業者が、本願発明に対して数多くの修正や変更を加えることが予想される。 従って、特許請求の範囲の欄に記載の限定事項のみが、本願発明に付加されるべきである。
【0184】
参照文献
[1] Adithan, C., Sivagnanam, G., Swain, R., Shashindran, C.H. and Bapna, J. S., Potentiation of clonidine analgesia by amitriptyline, Indian J Exp Biol, 24 (1986) 256-8.
[2] Backonja, M., Beydoun, A., Edwards, K.R., Schwartz, S.L., Fonseca, V., Hes, M., LaMoreaux, L. and Garofalo, E., Gabapentin for the symptomatic treatment of painful neuropathy in patients with diabetes mellitus: a randomized controlled trial, Jama, 280 (1998) 1831-6.
[3] Bennett, G.J., Update on the neurophysiology of pain transmission and modulation: focus on the NMDA-receptor, J Pain Symptom Manage, 19 (2000) S2-6.
[4] Bhalla, S., Ciaccio, N., Wang, Z.J. and Gulati, A., Involvement of endothelin in morphine tolerance in neuroblastoma (SH-SY5Y) cells, Exp Biol Med (Maywood), 231 (2006) 1152-6.
[5] Bhalla, S., Matwyshyn, G. and Gulati, A., Potentiation of morphine analgesia by BQ 123, an endothelin antagonist, Peptides, 23 (2002) 1837-45.
[6] Bhalla, S., Matwyshyn, G. and Gulati, A., Endothelin receptor antagonists restore morphine analgesia in morphine tolerant rats, Peptides, 24 (2003) 553-61.
[7] Bhalla, S., Matwyshyn, G. and Gulati, A., Morphine tolerance does not develop in mice treated with endothelin-A receptor antagonists, Brain Res, 1064 (2005) 126-35.
[8] Chan, M.F., Okun, L., Stavros, F.L., Hwang, E., Wolff, M.E. and Balaji, V.N., Identification of a new class of ETA selective endothelin antagonists by pharmacophore directed screening, Biochem Biophys Res Commun, 201 (1994) 228- 34.
[9] Constant, L., Gall, O., Gouyet, L., Chauvin, M. and Murat, L., Addition of clonidine or fentanyl to local anaesthetics prolongs the duration of surgical analgesia after single shot caudal block in children, Br J Anaesth, 80 (1998) 294-8.
[10] D'Amour, F.E., Smith, D.L., A method for determining loss of pain sensation, J Pharmacol Exp Ther, 71 (1941) 74-79.
[11] Fromm, G.H., Aumentado, D. and Terrence, C.F., A clinical and experimental investigation of the effects of tizanidine in trigeminal neuralgia, Pain, 53 (1993) 265-71.
[12] Gilron, L., Bailey, J.M., Tu, D., Holden, R.R., Weaver, D.F. and Houlden, R.L., Morphine, gabapentin, or their combination for neuropathic pain, N Engl J Med, 352 (2005) 1324-34.
[13] Gordon, N.C., Heller, P.H. and Levine, J.D., Enhancement of pentazocine analgesia by clonidine, Pain, 48 (1992) 167-9.
[14] Goudas, L.C. and Carr, D.B., Is there a place for intrathecal clonidine for clinical analgesia? Anesth Analg, 83 (1996) 891.
[15] Gulati, A., Evidence for antagonistic activity of endothelin for clonidine induced hypotension and bradycardia, Life Sci, 50 (1992) 153-60.
[16] Gulati, A., Bhalla, S. and Matwyshyn, G., A Novel Combination of Opiates and Endothelin Antagonists to Manage Pain Without Any Tolerance Development, J Cardiovasc Pharmacol, 44 (2004) S129-S131.
[17] Gulati, A., Rebello, S. and Kumar, A., Role of sympathetic nervous system in cardiovascular effects of centrally administered endothelin-1 in rats, Am J Physiol, 273 (1997) Hl 177-86.
[18] Gulati, A. and Srimal, R.C., Endothelin antagonizes the hypotension and potentiates the hypertension induced by clonidine, Eur J Pharmacol, 230 (1993) 293-300.
[19] Hutschala, D., Mascher, H., Schmetterer, L., Klimscha, W., Fleck, T., Eichler, H.G. and Tschemko, E.M., Clonidine added to bupivacaine enhances and prolongs analgesia after brachial plexus block via a local mechanism in healthy volunteers, Eur J Anaesthesiol, 21 (2004) 198-204.
[20] Jamali, S., Monin, S., Begon, C, Dubousset, A.M. and Ecoffey, C, Clonidine in pediatric caudal anesthesia, Anesth Analg, 78 (1994) 663-6.
[21] Knotkova, H. and Pappagallo, M., Adjuvant analgesics, Med Clin North Am, 91 (2007) 113-24.
[22] Kunos, G., Mosqueda-Garcia, R., Mastrianni, J.A. and Abbott, F.V., Endorphinergic mechanism in the central cardiovascular and analgesic effects of clonidine, Can J Physiol Pharmacol, 65 (1987) 1624-32.
[23] Lim, D.Y., Heo, K., Choi, C.H. and Lee, E.W., Influence of endothelin-1 on clonidine-induced cardiovascular effects in anesthetized rabbits, J Cardiovasc Pharmacol, 31 Suppl 1 (1998) S 122-5.
[24] Lyons, B., Casey, W., Doherty, P., McHugh, M. and Moore, K.P., Pain relief with low-dose intravenous clonidine in a child with severe burns, Intensive Care Med, 22 (1996) 249-51.
[25] Mandell, G. and Sande, M., Antimicrobial Agents, In Goodman and Gilman's The Pharmacological Basis of Therapeutics, Eighth Edition, Pergamon Press, New York, 1991, 1047-1064 pp.
[26] Matwyshyn, G.A., Bhalla, S. and Gulati, A., Endothelin ETA receptor blockade potentiates morphine analgesia but does not affect gastrointestinal transit in mice, Eur J Pharmacol, 543 (2006) 48-53.
[27] Milne, B., Cervenko, F. W., Jhamandas, K. and Sutak, M., Intrathecal clonidine: analgesia and effect on opiate withdrawal in the rat, Anesthesiology, 62 (1985) 34-8.
[28] Mutafova-Yambolieva, V., Petkov, O., Staneva-Stoytcheva, D. and Lasova, L., Interactions between the effects of endothelin-1, clonidine and yohimbine on electrically-induced contractions in rat vas deferens, Gen Pharmacol, 23 (1992) 529-34.
[29] Nishina, K. and Mikawa, K., Clonidine in paediatric anaesthesia, Curr Opin Anaesthesiol, 15 (2002) 309-16.
[30] Paalzow, G., Development of tolerance to the analgesic effect of clonidine in rats. Cross-tolerance to morphine, Naunyn Schmiedebergs Arch Pharmacol, 304 (1978) 1-4.
[31] Paalzow, L., Analgesia produced by clonidine in mice and rats, J Pharm Pharmacol, 26 (1974) 361-3.
[32] Porchet, H.C., Piletta, P. and Dayer, P., Objective assessment of clonidine analgesia in man and influence of naloxone, Life Sci, 46 (1990) 991-8.
[33] Puppala, B. L., Matwyshyn, G., Bhalla, S. and Gulati, A., Evidence that morphine tolerance may be regulated by endothelin in the neonatal rat. Biol Neonate, 86 (2004) 138-44.
[34] Puppala, B.L., Matwyshyn, G., Bhalla, S. and Gulati, A., Role of Endothelin in Neonatal Morphine Tolerance, J Cardiovasc Pharmacol, 44 (2004) S383-S385.
[35] Rowbotham, M.C., Goli, V., Kunz, N.R. and Lei, D., Venlafaxine extended release in the treatment of painful diabetic neuropathy: a double-blind, placebo- controlled study, Pain, 110 (2004) 697-706.
[36] Saarto, T. and Wiffen, P.J., Antidepressants for neuropathic pain, Cochrane Database Syst Rev (2005) CD005454.
[37] Semenchuk, M.R., Sherman, S. and Davis, B., Double-blind, randomized trial of bupropion SR for the treatment of neuropathic pain, Neurology, 57 (2001) 1583-8.
[38] Spaulding, T.C., Fielding, S., Venafro, J.J. and LaI, H., Antinociceptive activity of clonidine and its potentiation of morphine analgesia, Eur J Pharmacol, 58 (1979) 19-25.
[39] Wang, Y.C., Su, CF. and Lin, M.T., The site and the mode of analgesic actions exerted by clonidine in monkeys, Exp Neurol, 90 (1985) 479-88.
[40] Ordonez Gallego A, Gonzalez Baron M, Espinosa Arranz E: Oxycodone: A pharmacological and clinical review. Clin Transl Oncol 2007;9:298-307.
[41] Nielsen CK, Ross FB, Lotfipour S, Saini KS, Edwards SR, Smith MT: Oxycodone and morphine have distinctly different pharmacological profiles: Radioligand binding and behavioural studies in two rat models of neuropathic pain. Pain 2007; 132:289-300.
[42] Nozaki C, Kamei J: Involvement of mul-opioid receptor on oxycodone-induced antinociception in diabetic mice. Eur J Pharmacol 2007;560: 160-162.
[43] Falk E: Eukodal ein neues narkotikum [eukodal, a new narcotic]. Muenchener Med Wochenschr 1917;64:381-384.
[44] Yoburn BC, Shah S, Chan K, Duttaroy A, Davis T: Supersensitivity to opioid analgesics following chronic opioid antagonist treatment: Relationship to receptor selectivity. Pharmacol Biochem Behav 1995 ;51:535-539.
[45J Monory K, Greiner E, Sartania N, Sallai L, Pouille Y, Schmidhammer H, Hanoune J, Borsodi A: Opioid binding profiles of new hydrazone, oxime, carbazone and semicarbazone derivatives of 14-alkoxymorphinans. Life Sci 1999;64:2011-2020.
[46] Peckham EM, Traynor JR: Comparison of the antinociceptive response to morphine and morphine-like compounds in male and female sprague-dawley rats. J Pharmacol Exp Ther 2006;316: 1195-1201.
[47] Ernsberger P, Giuliano R, Willette RN, Reis DJ: Role of imidazole receptors in the vasodepressor response to clonidine analogs in the rostral ventrolateral medulla. J Pharmacol Exp Ther 1990;253:408-418.
[48] Ernsberger P, Damon TH, Graff LM, Schafer SG, Christen MO: Moxonidine, a centrally acting antihypertensive agent, is a selective ligand for il -imidazoline sites. J Pharmacol Exp Ther 1993;264:172-182.
[49] Hamilton CA, Yakubu MA, Jardine E, Reid JL: Imidazole binding sites in rabbit kidney and forebrain membranes. Journal of autonomic pharmacology 1991;l l:277-283.
[50] Chao, H.M. and Osborne, N.N., Topically applied clonidine protects the rat retina from ischaemia/reperfusion by stimulating alpha(2)-adrenoceptors and not by an action on imidazoline receptors, Brain Res, 904 (2001) 126-36.
[51] Costela, J.L., Carlos, R., Zamacona, M.K., Jimenez, R. and Calvo, R., Interaction between propofol and sulfisoxazole in mice an in vivo and in vitro study, Res Commun MoI Pathol Pharmacol, 93 (1996) 89-100.
[52] Syed, H., Safa, R., Chidlow, G. and Osborne, N.N., Sulfisoxazole, an endothelin receptor antagonist, protects retinal neurones from insults of ischemia/reperfusion or lipopolysaccharide, Neurochem Int, 48 (2006) 708-17.
[53] Iqbal, J., Wig, J., Bhardwaj, N. and Dhillon, M.S., Intra-articular clonidine vs. morphine for post-operative analgesia following arthroscopic knee surgery (a comparative evaluation), Knee, 7 (2000) 109-113.
[54] Goyagi, T., Tanaka, M. and Nishikawa, T., Oral clonidine premedication reduces induction dose and prolongs awakening time from propofol-nitrous oxide anesthesia, Can J Anaesth, 46 (1999) 894-6.
[55] Constant, L, Gall, O., Gouyet, L., Chauvin, M. and Murat, L, Addition of clonidine or fentanyl to local anaesthetics prolongs the duration of surgical analgesia after single shot caudal block in children, Br J Anaesth, 80 (1998) 294-8.
[56] Ezri, T., Szmuk, P., Shklar, B., Katz, J. and Geva, D., Oral clonidine premedication does not prolong analgesia after herniorrhaphy under subarachnoid anesthesia, J Clin Anesth, 10 (1998) 474-81.
[57] Tan, P.H., Chou, A.K., Perng, J.S., Chung, H.C., Lee, C.C. and Mok, M.S., Comparison of epidural butorphanol plus clonidine with butorphanol alone for postoperative pain relief, Acta Anaesthesiol Sin, 35 (1997) 91-6.
[58] Rockemann, M.G., Seeling, W., Duschek, S., Reinelt, H., Steffen, P. and Georgieff, M., Epidural bolus clonidine/morphine versus epidural patient-controlled bupivacaine/sufentanil: quality of postoperative analgesia and cost- identification analysis, Anesth Analg, 85 (1997) 864-9.
[59] Buggy, D.J. and MacDowell, C, Extradural analgesia with clonidine and fentanyl compared with 0.25% bupivacaine in the first stage of labour, Br J Anaesth, 76 (1996) 319-21.
[60] Lena, P., Balarac, N., Arnulf, JJ., Teboul, J. and Bonnet, F., Intrathecal morphine and clonidine for coronary artery bypass grafting, Br J Anaesth, 90 (2003) 300-3.
[61] Bonhomme, V., Doll, A., Dewandre, P. Y., Brichant, J.F., Ghassempour, K. and Hans, P., Epidural administration of low-dose morphine combined with clonidine for postoperative analgesia after lumbar disc surgery, J Neurosurg Anesthesiol, 14 (2002) 1-6.
[62] Jeffs, S. A., Hall, J. E. and Morris, S., Comparison of morphine alone with morphine plus clonidine for postoperative patient-controlled analgesia, Br J Anaesth, 89 (2002) 424-7.
[63] Goyagi, T. and Nishikawa, T., Oral clonidine premedication enhances the quality of postoperative analgesia by intrathecal morphine, Anesth Analg, 82 (1996) 1192-6.
[64] Yanagidate, F., Hamaya, Y. and Dohi, S., Clonidine premedication reduces maternal requirement for intravenous morphine after cesarean delivery without affecting newborn's outcome, Reg Anesth Pain Med, 26 (2001) 461-7.
[65] Park, J., Forrest, J., Kolesar, R., Bhola, D., Beattie, S. and Chu, C, Oral clonidine reduces postoperative PCA morphine requirements, Can J Anaesth, 43 (1996) 900-6.
[66] van Essen, EJ., Bovill, J.G., Ploeger, EJ. and Houben, JJ., Pharmacokinetics of clonidine after epidural administration in surgical patients. Lack of correlation between plasma concentration and analgesia and blood pressure changes, Acta Anaesthesiol Scand, 36 (1992) 300-4).
[67] Smith, B. D., Baudendistel, LJ., Gibbons, JJ. and Schweiss, J.F., A comparison of two epidural alpha 2-agonists, guanfacine and clonidine, in regard to duration of antinociception, and ventilatory and hemodynamic effects in goats, Anesth Analg, IA (1992) 712-8.)
[68] Capogna, G., Celleno, D., Zangrillo, A., Costantino, P. and Foresta, S., Addition of clonidine to epidural morphine enhances postoperative analgesia after cesarean delivery, Reg Anesth, 20 (1995) 57-61.
[69] Benhamou, D., Narchi, P., Hamza, J., Marx, M., Peyrol, M.T. and Sembeil, F., Addition of oral clonidine to postoperative patient-controlled analgesia with i.v. morphine, Br J Anaesth, 72 (1994) 537-40.
[70] Carabine, U. A., Milligan, K.R., Mulholland, D. and Moore, J., Extradural clonidine infusions for analgesia after total hip replacement, Br J Anaesth, 68 (1992) 338-43.
【図面の簡単な説明】
【0185】
【図1】モルヒネ(4mg/kg、腹腔内投与)の存在下および非存在下でのテイルフリック潜時に関するクロニジン(2mg/kg、腹腔内投与)の効果を示している。 マウスを4つのグループに分けた。 グループ1には、賦形剤(生理食塩水、腹腔内投与)+賦形剤(生理食塩水、皮下投与)を投与し、グループ2には、賦形剤+モルヒネ(4mg/kg、皮下投与)を投与し、グループ3には、クロニジン(2mg/kg、腹腔内投与)+賦形剤(生理食塩水、皮下投与)を投与し、そして、グループ4には、クロニジン(2mg/kg、腹腔内投与)+モルヒネ(4mg/kg、皮下投与)を投与した。 クロニジンを投与して30分後に、モルヒネまたは賦形剤を投与した。 図1Aは、様々な時点での数秒間のテイルフリック潜時データを示している。 図1Bは、テイルフリック潜時のデータ値から決定したAUC0→240分によって表された痛覚抑制を示している。 数値は、平均値±標準誤差であり、Nは、6/グループである。 *P<0.05 クロニジンを、対照グループと比較をした。 #P<0.05 クロニジン+モルヒネを、賦形剤+モルヒネのグループと比較をした。
【図2】モルヒネ(4mg/kg、皮下投与)の存在下および非存在下でのテイルフリック潜時に関するスルフィソキサゾール(500mg/kg、経口投与)の効果を示している。 マウスを4つのグループに分けた。 グループ1には、賦形剤(カルボキシメチルセルロース(CMC)、経口投与)+賦形剤(生理食塩水、皮下投与)を投与し、グループ2には、賦形剤(CMC)+モルヒネ(4mg/kg、皮下投与)を投与し、グループ3には、スルフィソキサゾール(500mg/kg、経口投与)+賦形剤(生理食塩水、皮下投与)を投与し、そして、グループ4には、スルフィソキサゾール(500mg/kg、経口投与)+モルヒネ(4mg/kg、皮下投与)を投与した。 スルフィソキサゾールを投与して30分後に、モルヒネまたは賦形剤を投与した。 図2Aは、様々な時点での数秒間のテイルフリック潜時データを示している。 図2Bは、テイルフリック潜時のデータ値から決定したAUC0→240分によって表された痛覚抑制を示している。 数値は、平均値±標準誤差であり、Nは、6/グループである。 *P<0.05 スルフィソキサゾールを、対照グループと比較をした。
【図3】モルヒネ(M)(8mg/kg、皮下投与)の存在下および非存在下でのテイルフリック潜時に関するクロニジン(C)(2mg/kg、腹腔内投与)とスルフィソキサゾール(S)(500mg/kg、経口投与)との組み合わせの効果を示している。 マウスを3つのグループに分けた。 グループ1には、クロニジン(2mg/kg、腹腔内投与)とスルフィソキサゾール(500mg/kg、経口投与)との組み合わせ+賦形剤(V)(生理食塩水、皮下投与)を投与し、グループ2には、クロニジン(2mg/kg、腹腔内投与)とスルフィソキサゾール(500mg/kg、経口投与)との組み合わせ+モルヒネ(8mg/kg、皮下投与)を投与し、グループ3には、賦形剤(生理食塩水、腹腔内投与)と賦形剤(カルボキシメチルセルロース、経口投与)との組み合わせ+モルヒネ(M)(8mg/kg、皮下投与)を投与し、そして、グループ4には、賦形剤(生理食塩水、腹腔内投与)と賦形剤(カルボキシメチルセルロース、経口投与)+賦形剤(生理食塩水、皮下投与)を投与した。 クロニジンとスルフィソキサゾールとの組み合わせを投与して30分後に、モルヒネまたは賦形剤を投与した。 クロニジン(2mg/kg、腹腔内投与)とスルフィソキサゾールとの組み合わせが、痛覚抑制を顕著に改善していた。 数値は、平均値±標準誤差であり、Nは、6/グループである。 *P<0.05 クロニジンとスルフィソキサゾールとの組み合わせを、対照(賦形剤)グループと比較をした。
【図4】モルヒネ(8mg/kg、腹腔内投与)の存在下および非存在下でのテイルフリック潜時のデータ値から決定したAUC0→240分によって表された痛覚抑制に関するクロニジン(2mg/kg、腹腔内投与)とスルフィソキサゾール(1000mg/kg、経口投与)との組み合わせの効果を示している。 マウスを6つのグループに分けた。 グループ1には、賦形剤(生理食塩水、腹腔内投与)と賦形剤(カルボキシメチルセルロース(CMC))との組み合わせ+賦形剤(生理食塩水、皮下投与)を投与し、グループ2には、クロニジン(2mg/kg、腹腔内投与)と賦形剤(CMC)との組み合わせ+賦形剤(生理食塩水、皮下投与)を投与し、グループ3には、スルフィソキサゾール(1000mg/kg、経口投与)と賦形剤(生理食塩水、腹腔内投与)との組み合わせ+賦形剤(生理食塩水、皮下投与)を投与し、グループ4には、賦形剤(生理食塩水、腹腔内投与)と賦形剤(CMC)との組み合わせ+モルヒネ(8mg/kg、皮下投与)を投与し、グループ5には、クロニジン(2mg/kg、腹腔内投与)とスルフィソキサゾール(1000mg/kg、経口投与)との組み合わせ+賦形剤(生理食塩水、皮下投与)を投与し、および、グループ6には、クロニジン(2mg/kg、腹腔内投与)とスルフィソキサゾール(1000mg/kg、経口投与)との組み合わせ+モルヒネ(8mg/kg、皮下投与)を投与した。 クロニジンとスルフィソキサゾールとの組み合わせを投与して30分後に、モルヒネまたは賦形剤を投与した。 クロニジンとスルフィソキサゾールとの組み合わせが、痛覚抑制を顕著に改善していた。 数値は、平均値±標準誤差であり、Nは、6/グループである。 *P<0.05 モルヒネを、賦形剤グループと比較をした。 #P<0.05 クロニジンとスルフィソキサゾールとの組み合わせを、クロニジンだけのグループ、または、スルフィソキサゾールだけのグループと比較をした。
【図5】マウスでの鎮痛作用(テイルフリック潜時)(図5A)とマウスの体温(図5B)に関するクロニジンの用量応答効果を示している。 グループ1:賦形剤(生理食塩水、腹腔内投与)、グループ2:クロニジン(0.3mg/kg、腹腔内投与)、グループ3:クロニジン(1.0mg/kg、腹腔内投与)、グループ4:クロニジン(3.0mg/kg、腹腔内投与)。
【図6】マウスでの鎮痛作用(図6A)とマウスの体温(図6B)に関するクロニジンとスルフィソキサゾールとの組み合わせの効果を示している。 グループ1:0.5%カルボキシメチルセルロース(CMC、経口投与)+賦形剤(生理食塩水、腹腔内投与)、グループ2:CMC(経口投与)+クロニジン(0.3mg/kg、腹腔内投与)、グループ3:スルフィソキサゾール(250mg/kg、経口投与)+クロニジン(0.3mg/kg、腹腔内投与)、グループ4:スルフィソキサゾール(500mg/kg、経口投与)+クロニジン(0.3mg/kg、腹腔内投与)、グループ5:スルフィソキサゾール(1000mg/kg、経口投与)+クロニジン(0.3mg/kg、腹腔内投与)。
【図7】マウスでの鎮痛作用(図7A)とマウスの体温(図7B)に関するクロニジンとスルフィソキサゾールとの組み合わせの効果を示している。 グループ1:0.5%カルボキシメチルセルロース(CMC、経口投与)+賦形剤(生理食塩水、腹腔内投与)、グループ2:CMC(経口投与)+クロニジン(0.3mg/kg、腹腔内投与)、グループ3:スルフィソキサゾール(25mg/kg、経口投与)+クロニジン(0.3mg/kg、腹腔内投与)、グループ4:スルフィソキサゾール(75mg/kg、経口投与)+クロニジン(0.3mg/kg、腹腔内投与)、グループ5:スルフィソキサゾール(225mg/kg、経口投与)+クロニジン(0.3mg/kg、腹腔内投与)。
【図8】マウスでの鎮痛作用(図8A)とマウスの体温(図8B)に関するクロニジン、および、クロニジンとスルフィソキサゾールとの組み合わせに関するヨヒンビンの効果を示している。 グループ1:賦形剤+賦形剤(生理食塩水、腹腔内投与)+賦形剤(生理食塩水、腹腔内投与)、グループ2:賦形剤+CMC(経口投与)+賦形剤(生理食塩水、腹腔内投与)、グループ3:ヨヒンビン(2mg/kg、腹腔内投与)+CMC(経口投与)+賦形剤(生理食塩水、腹腔内投与)、グループ4:ヨヒンビン(2mg/kg、腹腔内投与)+CMC(経口投与)+クロニジン(0.3mg/kg、腹腔内投与)、グループ5:ヨヒンビン(2mg/kg、腹腔内投与)+スルフィソキサゾール(250mg/kg、経口投与)+クロニジン(0.3mg/kg、腹腔内投与)。
【図9】マウスでの鎮痛作用(図9A)とマウスの体温(図9B)に関するクロニジン、および、クロニジンとスルフィソキサゾールとの組み合わせに関するイダゾキサンの効果を示している。 グループ1:賦形剤+賦形剤(生理食塩水、腹腔内投与)+賦形剤(CMC)、経口投与)、グループ2:イダゾキサン(2mg/kg、腹腔内投与)+CMC(経口投与)+賦形剤(生理食塩水、腹腔内投与)、グループ3:イダゾキサン(2mg/kg、腹腔内投与)+CMC(経口投与)+賦形剤(生理食塩水、腹腔内投与)、グループ4:イダゾキサン(2mg/kg、腹腔内投与)+CMC(経口投与)+クロニジン(0.3mg/kg、腹腔内投与)、グループ5:イダゾキサン(2mg/kg、腹腔内投与)+スルフィソキサゾール(250mg/kg、経口投与)+クロニジン(0.3mg/kg、腹腔内投与)。
【図10】マウスでの鎮痛作用(図10A)とマウスの体温(図10B)に関するクロニジン、および、クロニジンとスルフィソキサゾールとの組み合わせに関するナロキソンの効果を示している。 グループ1:ナロキソン(1.0mg/kg、腹腔内投与)+CMC(経口投与)+賦形剤(生理食塩水、腹腔内投与)、グループ2:ナロキソン(1.0mg/kg、腹腔内投与)+CMC(経口投与)+クロニジン(0.3mg/kg、腹腔内投与)、グループ3:ナロキソン(1.0mg/kg、腹腔内投与)+スルフィソキサゾール(250mg/kg、経口投与)+クロニジン(0.3mg/kg、腹腔内投与)。
【図11】マウスでの鎮痛作用(図11A)とマウスの体温(図11B)に関するクロニジンとスルフィソキサゾールとの組み合わせによる効果と、その他のETAアンタゴニスト(BMS182874)による効果の比較について示している。 グループ1:クロニジン(0.3mg/kg、腹腔内投与)+BMS182874(2.0μg/kg、脳室内投与)、グループ2:クロニジン(0.3mg/kg、腹腔内投与)+BMS182874(10.0μg/kg、脳室内投与)、グループ3:クロニジン(0.3mg/kg、腹腔内投与)+BMS182874(50.0μg/kg、脳室内投与)。
【図12】モルヒネ(8mg/kg、皮下投与)の鎮痛作用に関するクロニジン(1mg/kg、腹腔内投与)とBMS182874(50μg、脳室内投与)の効果を示している。 様々な時点においてテイルフリック潜時を計測し(図12A)、そして、各ラットにおける抗侵害受容応答を、AUC0→360minに変換し(図12B、クロニジン;図12C、BMS182874)、次いで、得られた数値を、平均±標準誤差(V=賦形剤;B=BMS182874;I=イダゾキサン;および、M=モルヒネ)として表した。
【図13】オキシコドン(4mg/kg、皮下投与)の鎮痛作用に関するクロニジン(1mg/kg、腹腔内投与)とBMS182874(50μg、脳室内投与)の効果を示している。 様々な時点においてテイルフリック潜時を計測し(図13A)、そして、各ラットにおける抗侵害受容応答を、AUC0→360minに変換し(図13B、クロニジン;図13C、BMS182874)、次いで、得られた数値を、平均±標準誤差(V=賦形剤;B=BMS182874;I=イダゾキサン;および、O=オキシコドン)として表した。
【図14】モルヒネ(8mg/kg、皮下投与)の鎮痛作用、または、オキシコドン(4mg/kg、皮下投与)の鎮痛作用を高めるクロニジン(1mg/kg、腹腔内投与)に関するヨヒンビン(2mg/kg、腹腔内投与)の効果を示している。 様々な時点においてテイルフリック潜時を計測し(図14A)、そして、各ラットにおける抗侵害受容応答を、AUC0→360minに変換し(図14B、モルヒネ;図14C、オキシコドン)、次いで、得られた数値を、平均±標準誤差(V=賦形剤;C=クロニジン;Y=ヨヒンビン;O=オキシコドン;および、M=モルヒネ)として表した。
【図15】モルヒネ(8mg/kg、皮下投与)の鎮痛作用、または、オキシコドン(4mg/kg、皮下投与)の鎮痛作用を高めるBMS182874(50μg、脳室内投与)に関するヨヒンビン(2mg/kg、腹腔内投与)の効果を示している。 様々な時点においてテイルフリック潜時を計測し(図15A)、そして、各ラットにおける抗侵害受容応答を、AUC0→360minに変換し(図15B、モルヒネ;図15C、オキシコドン)、次いで、得られた数値を、平均±標準誤差(V=賦形剤;B=BMS182874;Y=ヨヒンビン;O=オキシコドン;および、M=モルヒネ)として表した。
【図16A−B】BMS182874(50μg、脳室内投与)の存在下でのモルヒネ(8mg/kg、皮下投与)の鎮痛作用、または、オキシコドン(4mg/kg、皮下投与)の鎮痛作用に関するクロニジン [0(C0)、0.1(C1)、0.3(C2)、および、1.0(C3)mg/kg、腹腔内投与]の効果を示している。 様々な時点においてテイルフリック潜時を計測し(図16A、モルヒネ;図16C、オキシコドン)、そして、各ラットにおける抗侵害受容応答を、AUC0→360minに変換し(図16B、モルヒネ;図16C、オキシコドン)、次いで、得られた数値を、平均±標準誤差(C=クロニジン;B=BMS182874;O=オキシコドン;および、M=モルヒネ)として表した。
【図16C−D】BMS182874(50μg、脳室内投与)の存在下でのモルヒネ(8mg/kg、皮下投与)の鎮痛作用、または、オキシコドン(4mg/kg、皮下投与)の鎮痛作用に関するクロニジン [0(C0)、0.1(C1)、0.3(C2)、および、1.0(C3)mg/kg、腹腔内投与]の効果を示している。 様々な時点においてテイルフリック潜時を計測し(図16A、モルヒネ;図16C、オキシコドン)、そして、各ラットにおける抗侵害受容応答を、AUC0→360minに変換し(図16B、モルヒネ;図16C、オキシコドン)、次いで、得られた数値を、平均±標準誤差(C=クロニジン;B=BMS182874;O=オキシコドン;および、M=モルヒネ)として表した。
【図17】モルヒネ(8mg/kg、皮下投与)の鎮痛作用、または、オキシコドン(4mg/kg、皮下投与)の鎮痛作用に関するクロニジン(1mg/kg、腹腔内投与)単体、および、BMS182874(50μg、脳室内投与)単体、および、クロニジンとBMS182874との組み合わせの効果を示している。 様々な時点においてテイルフリック潜時を計測し(図17A)、そして、各ラットにおける抗侵害受容応答を、AUC0→360minに変換し(図17B、モルヒネ;図17C、オキシコドン)、次いで、得られた数値を、平均±標準誤差(V=賦形剤;C=クロニジン;B=BMS182874;O=オキシコドン;および、M=モルヒネ)として表した。
【図18】鎮痛作用に関するBMS182874(50μg、脳室内投与)単体、および、クロニジン(1mg/kg、腹腔内投与)単体、および、BMS182874(50μg、脳室内投与)とクロニジン(1mg/kg、腹腔内投与)との組み合わせの効果を示している。 様々な時点においてテイルフリック潜時を計測し(図18A)、そして、各ラットにおける抗侵害受容応答を、AUC0→360minに変換し(図18B)、次いで、得られた数値を、平均±標準誤差(V=賦形剤;C=クロニジン;B=BMS182874)として表した。
【特許請求の範囲】
【請求項1】
疼痛を治療または予防する方法であって、治療または予防の必要がある哺乳動物に対して、治療有効量のアルファ-2(α2)アドレナリン受容体アゴニスト、および、治療有効量のエンドセリン受容体アンタゴニストを投与する、ことを含む方法。
【請求項2】
前記α2アドレナリンアゴニストが、クロニジンである請求項1に記載の方法。
【請求項3】
前記エンドセリン受容体アンタゴニストが、エンドセリン受容体A(ETA)アンタゴニストである請求項1に記載の方法。
【請求項4】
前記ETAアンタゴニストが、スルフィソキサゾール、アトラセンタン、テゾセンタン、ボセンタン、シタキスセンタン、エンラセンタン、BMS207940、BMS193884、BMS182874、J 104132、VML 588/Ro 61 1790、T-0115、TAK 044、BQ 788、TBC2576、TBC3214、PD180988、ABT 546、SB247083、RPR118031A、および、BQ123からなるグループから選択される請求項3に記載の方法。
【請求項5】
前記ETAアンタゴニストが、スルフィソキサゾールである請求項4に記載の方法。
【請求項6】
前記α2アドレナリンアゴニストおよびエンドセリンアンタゴニストが、単一の組成物で投与される請求項1に記載の方法。
【請求項7】
前記α2アドレナリンアゴニストおよびエンドセリンアンタゴニストが、個別の組成物で投与される請求項1に記載の方法。
【請求項8】
前記組成物が、同時に投与される請求項7に記載の方法。
【請求項9】
前記組成物が、別個に投与される請求項7に記載の方法。
【請求項10】
前記組成物が、医薬担体または医薬賦形剤をさらに含む請求項7乃至9のいずれかに記載の方法。
【請求項11】
前記α2アドレナリンアゴニストおよびエンドセリンアンタゴニストが、経口的、経頬的、吸入的、舌下、経直腸的、経膣的、嚢内、関節内、経尿道的、経鼻的、経皮的、静脈内、筋肉内、または、皮下に投与される請求項1に記載の方法。
【請求項12】
前記クロニジンが、10μg〜約300μgの範囲の用量で投与される請求項2に記載の方法。
【請求項13】
前記スルフィソキサゾールが、0.1g〜約3gの範囲の用量で投与される請求項4に記載の方法。
【請求項14】
投与された前記α2アドレナリンアゴニストと投与された前記エンドセリン受容体アンタゴニストとの比率が、1:500〜1:50,000、1:500〜1:20,000、1:500〜1:10,000、1:500〜1:5,000、1:500〜1:2,500、1:100〜1:1000、または、1:100〜1:500の範囲である請求項1に記載の方法。
【請求項15】
疼痛を治療または予防する方法であって、対象動物に対して、一つまたはそれ以上のアルファ-2(α2)アドレナリン受容体アゴニストと、一つまたはそれ以上のエンドセリン受容体アンタゴニストとの相乗的組み合わせを投与する、ことを含む方法。
【請求項16】
前記組み合わせに用いられたアゴニスト分子およびアンタゴニスト分子が、約24時間以内に連続投与されるか、あるいは、同時投与される請求項1または15に記載の方法。
【請求項17】
疼痛を治療または予防する方法であって、治療または予防の必要がある哺乳動物に対して、治療有効量の麻薬性鎮痛薬、および、一つまたは一つまたはそれ以上のアルファ-2(α2)アドレナリン受容体アゴニストを含む治療有効量の組成物を投与する、ことを含む方法。
【請求項18】
疼痛を治療または予防する方法であって、治療または予防の必要がある哺乳動物に対して、治療有効量の麻薬性鎮痛薬、一つまたは一つまたはそれ以上のアルファ-2(α2)アドレナリン受容体アゴニストを含む治療有効量の組成物、および、一つまたは一つまたはそれ以上のエンドセリン受容体アンタゴニストを含む治療有効量の組成物を投与する、ことを含む方法。
【請求項19】
疼痛を治療または予防する方法であって、治療または予防の必要がある哺乳動物に対して、治療有効量の麻薬性鎮痛薬、および、一つまたは一つまたはそれ以上のアルファ-2(α2)アドレナリン受容体アゴニストと、一つまたは一つまたはそれ以上のエンドセリン受容体アンタゴニストとを含む治療有効量の組成物を投与する、ことを含む方法。
【請求項20】
前記α2アドレナリンアゴニストが、クロニジンである請求項17乃至19のいずれかに記載の方法。
【請求項21】
前記エンドセリン受容体アンタゴニストが、エンドセリン受容体A(ETA)アンタゴニストである請求項17乃至19のいずれかに記載の方法。
【請求項22】
前記ETAアンタゴニストが、スルフィソキサゾール、アトラセンタン、テゾセンタン、ボセンタン、シタキスセンタン、エンラセンタン、BMS207940、BMS193884、BMS182874、J 104132、VML 588/Ro 61 1790、T-0115、TAK 044、BQ 788、TBC2576、TBC3214、PD180988、ABT 546、SB247083、RPR118031A、および、BQ123からなるグループから選択される請求項17乃至19のいずれかに記載の方法。
【請求項23】
前記麻薬性鎮痛薬が、モルヒネ、硫酸モルヒネ、コデイン、ジアセチルモルヒネ、デキストロメトルファン、ヒドロコドン、ヒドロモルフォン、ヒドロモルフォン、レボルファノール、オキシモルホン、オキシコドン、レバロルファン、および、それらの塩からなるグループから選択される請求項17乃至19のいずれかに記載の方法。
【請求項24】
一つまたはそれ以上のアルファ-2(α2)アドレナリン受容体アゴニストと、一つまたはそれ以上のエンドセリン受容体アンタゴニストとの相乗的組み合わせを含む、疼痛を治療または予防するための組成物。
【請求項25】
低用量のα2アドレナリン受容体アゴニストと、低用量のエンドセリン受容体アンタゴニストとを含む請求項17に記載の組成物。
【請求項26】
前記α2アドレナリン受容体アゴニストが、クロニジンであり、および、前記エンドセリン受容体アンタゴニストが、スルフィソキサゾールである請求項18に記載の組成物。
【請求項27】
前記組成物でのクロニジンが、10μg〜約300μgの範囲であり、および、前記組成物でのスルフィソキサゾールが、約0.1g〜約3gの範囲である請求項19に記載の組成物。
【請求項28】
薬学的に許容可能な担体をさらに含む請求項25乃至27のいずれかに記載の組成物。
【請求項29】
対象動物での疼痛を治療するための薬剤を製造するためのアルファ-2(α2)アドレナリン受容体アゴニストとエンドセリンアンタゴニストを含む組成物の使用。
【請求項30】
前記対象動物が、哺乳動物である請求項1乃至23のいずれかに記載の方法または請求項29に記載の使用。
【請求項31】
前記対象動物が、ヒトである請求項30に記載の方法。
【請求項32】
前記疼痛が、慢性疼痛である請求項1乃至23のいずれかに記載の方法または請求項29に記載の使用。
【請求項33】
前記疼痛が、急性疼痛である請求項1乃至23のいずれかに記載の方法または請求項29に記載の使用。
【請求項34】
アルファ-2(α2)アドレナリン受容体アゴニスト、エンドセリンアンタゴニスト、および、請求項1または15乃至19のいずれかに記載の方法を示したラベルを含む製造品。
【請求項35】
疼痛を治療または予防するためのキットであって、
(a) 請求項24乃至28のいずれかに記載の組成物、および
(b) 当該キットを用いて疼痛を治療するためのプロトコル、を含むキット。
【請求項36】
麻薬性鎮痛薬をさらに含む請求項35に記載のキット。
【請求項1】
疼痛を治療または予防する方法であって、治療または予防の必要がある哺乳動物に対して、治療有効量のアルファ-2(α2)アドレナリン受容体アゴニスト、および、治療有効量のエンドセリン受容体アンタゴニストを投与する、ことを含む方法。
【請求項2】
前記α2アドレナリンアゴニストが、クロニジンである請求項1に記載の方法。
【請求項3】
前記エンドセリン受容体アンタゴニストが、エンドセリン受容体A(ETA)アンタゴニストである請求項1に記載の方法。
【請求項4】
前記ETAアンタゴニストが、スルフィソキサゾール、アトラセンタン、テゾセンタン、ボセンタン、シタキスセンタン、エンラセンタン、BMS207940、BMS193884、BMS182874、J 104132、VML 588/Ro 61 1790、T-0115、TAK 044、BQ 788、TBC2576、TBC3214、PD180988、ABT 546、SB247083、RPR118031A、および、BQ123からなるグループから選択される請求項3に記載の方法。
【請求項5】
前記ETAアンタゴニストが、スルフィソキサゾールである請求項4に記載の方法。
【請求項6】
前記α2アドレナリンアゴニストおよびエンドセリンアンタゴニストが、単一の組成物で投与される請求項1に記載の方法。
【請求項7】
前記α2アドレナリンアゴニストおよびエンドセリンアンタゴニストが、個別の組成物で投与される請求項1に記載の方法。
【請求項8】
前記組成物が、同時に投与される請求項7に記載の方法。
【請求項9】
前記組成物が、別個に投与される請求項7に記載の方法。
【請求項10】
前記組成物が、医薬担体または医薬賦形剤をさらに含む請求項7乃至9のいずれかに記載の方法。
【請求項11】
前記α2アドレナリンアゴニストおよびエンドセリンアンタゴニストが、経口的、経頬的、吸入的、舌下、経直腸的、経膣的、嚢内、関節内、経尿道的、経鼻的、経皮的、静脈内、筋肉内、または、皮下に投与される請求項1に記載の方法。
【請求項12】
前記クロニジンが、10μg〜約300μgの範囲の用量で投与される請求項2に記載の方法。
【請求項13】
前記スルフィソキサゾールが、0.1g〜約3gの範囲の用量で投与される請求項4に記載の方法。
【請求項14】
投与された前記α2アドレナリンアゴニストと投与された前記エンドセリン受容体アンタゴニストとの比率が、1:500〜1:50,000、1:500〜1:20,000、1:500〜1:10,000、1:500〜1:5,000、1:500〜1:2,500、1:100〜1:1000、または、1:100〜1:500の範囲である請求項1に記載の方法。
【請求項15】
疼痛を治療または予防する方法であって、対象動物に対して、一つまたはそれ以上のアルファ-2(α2)アドレナリン受容体アゴニストと、一つまたはそれ以上のエンドセリン受容体アンタゴニストとの相乗的組み合わせを投与する、ことを含む方法。
【請求項16】
前記組み合わせに用いられたアゴニスト分子およびアンタゴニスト分子が、約24時間以内に連続投与されるか、あるいは、同時投与される請求項1または15に記載の方法。
【請求項17】
疼痛を治療または予防する方法であって、治療または予防の必要がある哺乳動物に対して、治療有効量の麻薬性鎮痛薬、および、一つまたは一つまたはそれ以上のアルファ-2(α2)アドレナリン受容体アゴニストを含む治療有効量の組成物を投与する、ことを含む方法。
【請求項18】
疼痛を治療または予防する方法であって、治療または予防の必要がある哺乳動物に対して、治療有効量の麻薬性鎮痛薬、一つまたは一つまたはそれ以上のアルファ-2(α2)アドレナリン受容体アゴニストを含む治療有効量の組成物、および、一つまたは一つまたはそれ以上のエンドセリン受容体アンタゴニストを含む治療有効量の組成物を投与する、ことを含む方法。
【請求項19】
疼痛を治療または予防する方法であって、治療または予防の必要がある哺乳動物に対して、治療有効量の麻薬性鎮痛薬、および、一つまたは一つまたはそれ以上のアルファ-2(α2)アドレナリン受容体アゴニストと、一つまたは一つまたはそれ以上のエンドセリン受容体アンタゴニストとを含む治療有効量の組成物を投与する、ことを含む方法。
【請求項20】
前記α2アドレナリンアゴニストが、クロニジンである請求項17乃至19のいずれかに記載の方法。
【請求項21】
前記エンドセリン受容体アンタゴニストが、エンドセリン受容体A(ETA)アンタゴニストである請求項17乃至19のいずれかに記載の方法。
【請求項22】
前記ETAアンタゴニストが、スルフィソキサゾール、アトラセンタン、テゾセンタン、ボセンタン、シタキスセンタン、エンラセンタン、BMS207940、BMS193884、BMS182874、J 104132、VML 588/Ro 61 1790、T-0115、TAK 044、BQ 788、TBC2576、TBC3214、PD180988、ABT 546、SB247083、RPR118031A、および、BQ123からなるグループから選択される請求項17乃至19のいずれかに記載の方法。
【請求項23】
前記麻薬性鎮痛薬が、モルヒネ、硫酸モルヒネ、コデイン、ジアセチルモルヒネ、デキストロメトルファン、ヒドロコドン、ヒドロモルフォン、ヒドロモルフォン、レボルファノール、オキシモルホン、オキシコドン、レバロルファン、および、それらの塩からなるグループから選択される請求項17乃至19のいずれかに記載の方法。
【請求項24】
一つまたはそれ以上のアルファ-2(α2)アドレナリン受容体アゴニストと、一つまたはそれ以上のエンドセリン受容体アンタゴニストとの相乗的組み合わせを含む、疼痛を治療または予防するための組成物。
【請求項25】
低用量のα2アドレナリン受容体アゴニストと、低用量のエンドセリン受容体アンタゴニストとを含む請求項17に記載の組成物。
【請求項26】
前記α2アドレナリン受容体アゴニストが、クロニジンであり、および、前記エンドセリン受容体アンタゴニストが、スルフィソキサゾールである請求項18に記載の組成物。
【請求項27】
前記組成物でのクロニジンが、10μg〜約300μgの範囲であり、および、前記組成物でのスルフィソキサゾールが、約0.1g〜約3gの範囲である請求項19に記載の組成物。
【請求項28】
薬学的に許容可能な担体をさらに含む請求項25乃至27のいずれかに記載の組成物。
【請求項29】
対象動物での疼痛を治療するための薬剤を製造するためのアルファ-2(α2)アドレナリン受容体アゴニストとエンドセリンアンタゴニストを含む組成物の使用。
【請求項30】
前記対象動物が、哺乳動物である請求項1乃至23のいずれかに記載の方法または請求項29に記載の使用。
【請求項31】
前記対象動物が、ヒトである請求項30に記載の方法。
【請求項32】
前記疼痛が、慢性疼痛である請求項1乃至23のいずれかに記載の方法または請求項29に記載の使用。
【請求項33】
前記疼痛が、急性疼痛である請求項1乃至23のいずれかに記載の方法または請求項29に記載の使用。
【請求項34】
アルファ-2(α2)アドレナリン受容体アゴニスト、エンドセリンアンタゴニスト、および、請求項1または15乃至19のいずれかに記載の方法を示したラベルを含む製造品。
【請求項35】
疼痛を治療または予防するためのキットであって、
(a) 請求項24乃至28のいずれかに記載の組成物、および
(b) 当該キットを用いて疼痛を治療するためのプロトコル、を含むキット。
【請求項36】
麻薬性鎮痛薬をさらに含む請求項35に記載のキット。
【図1】

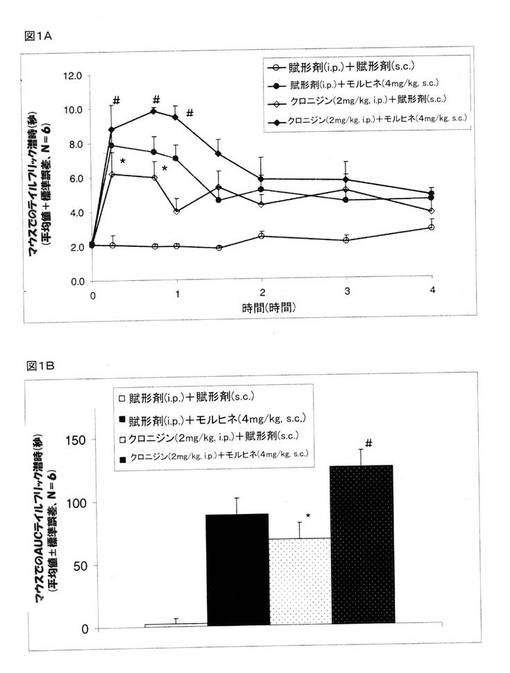
【図2】
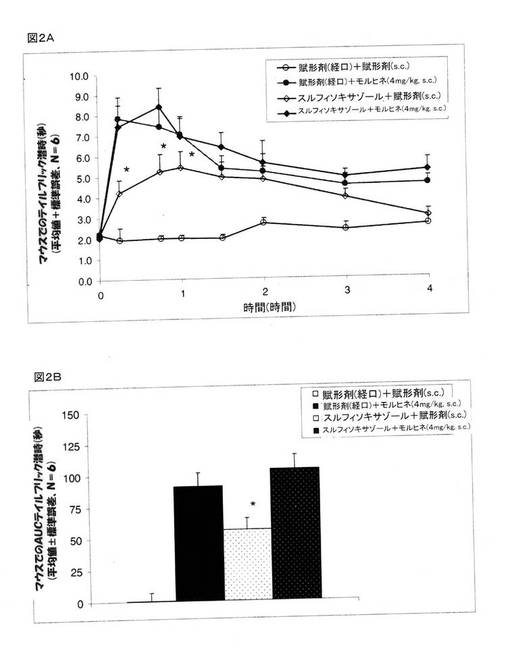
【図3】
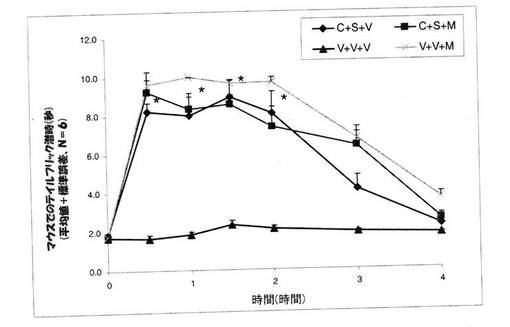
【図4】
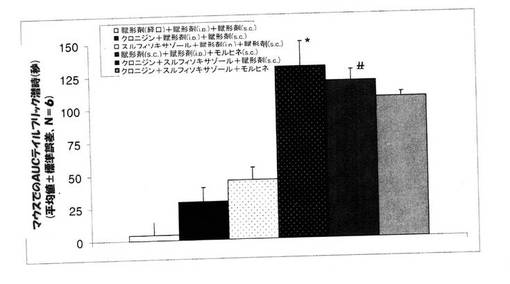
【図5】
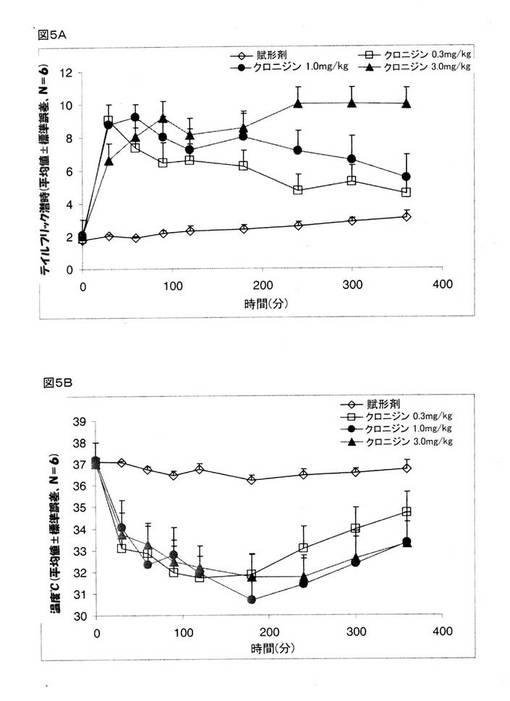
【図6】
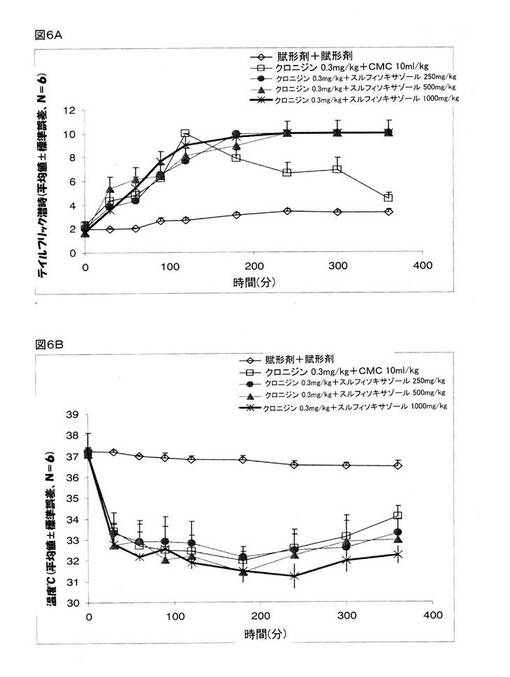
【図7】
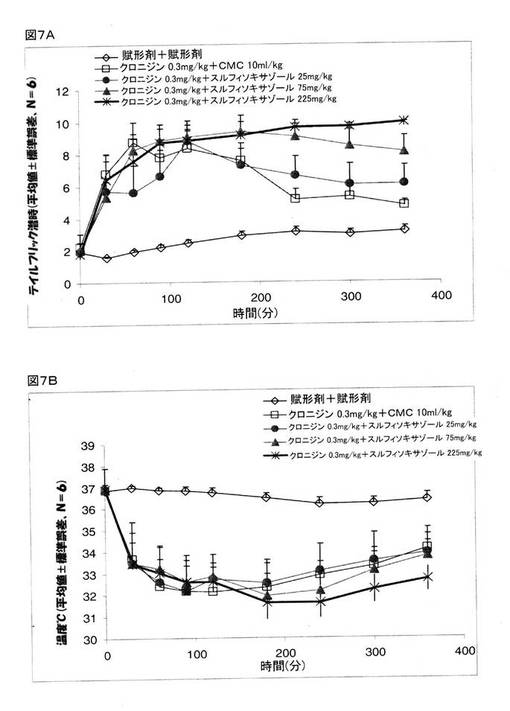
【図8】
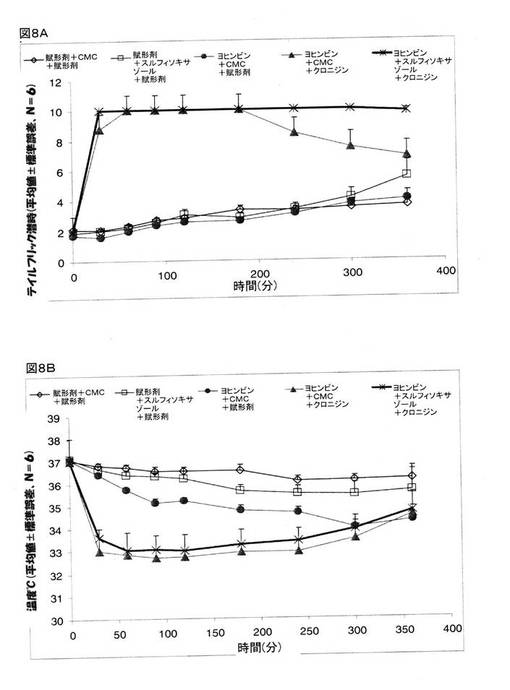
【図9】
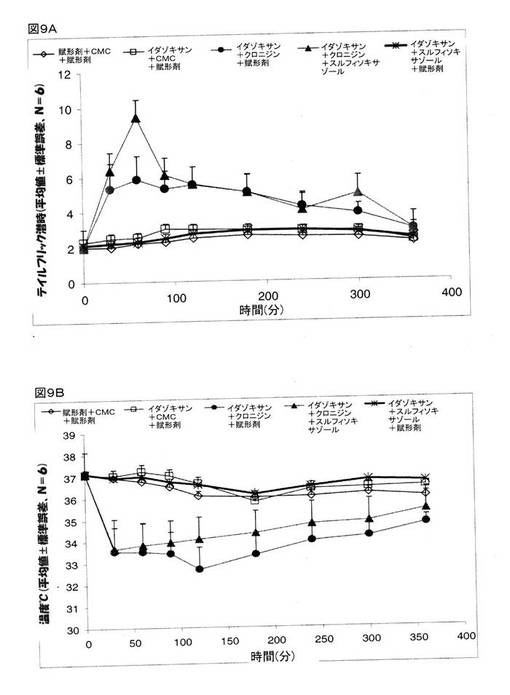
【図10】
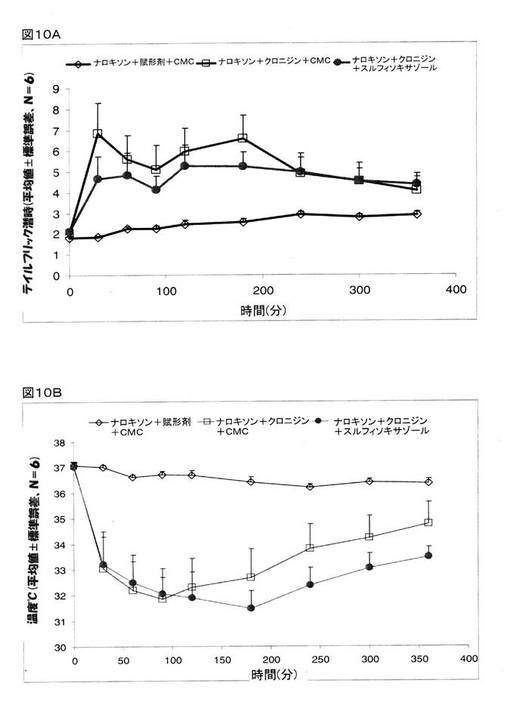
【図11】
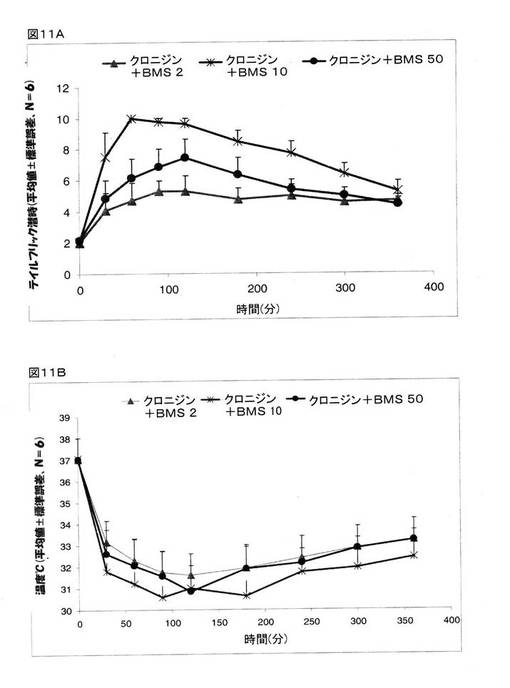
【図12】
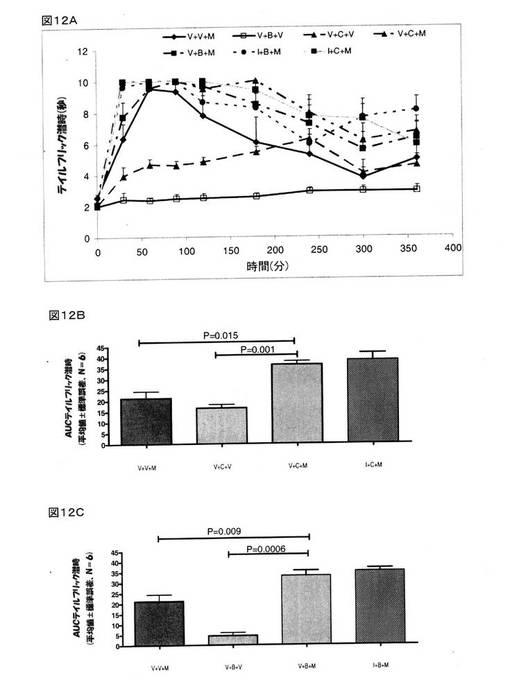
【図13】
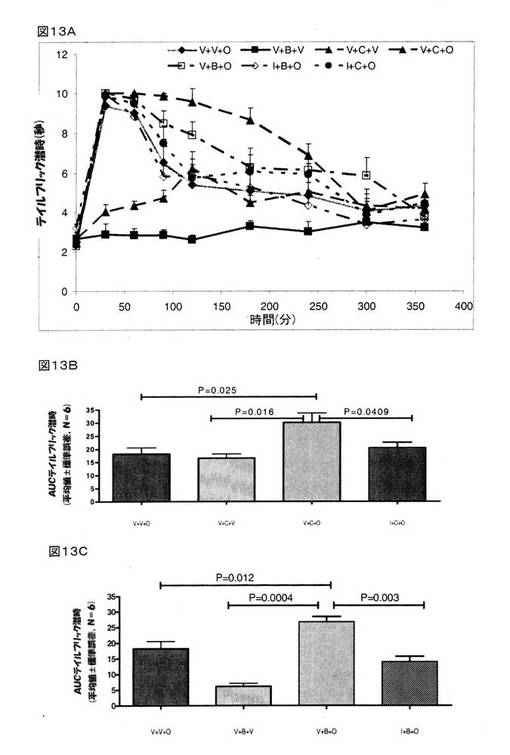
【図14】
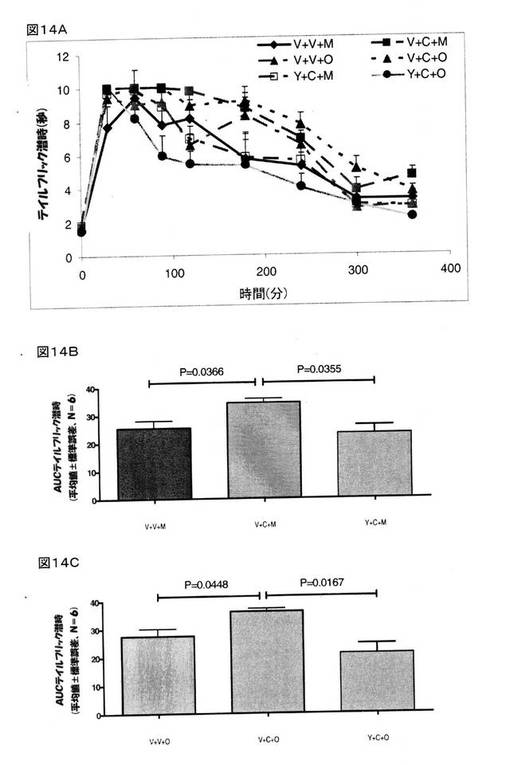
【図15】
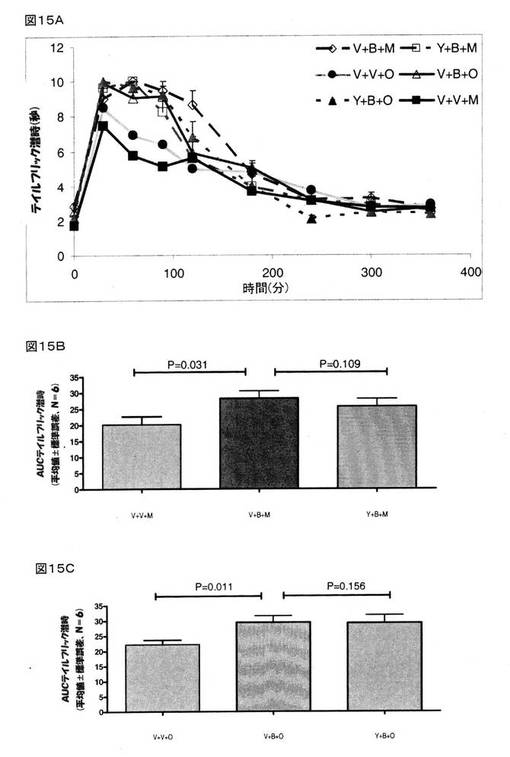
【図16A−B】
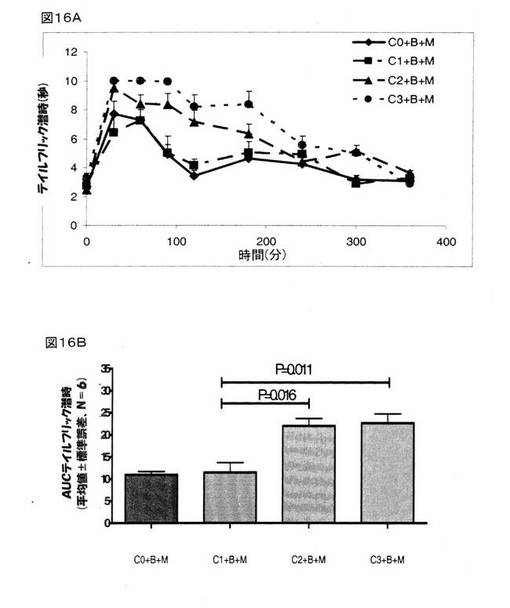
【図16C−D】
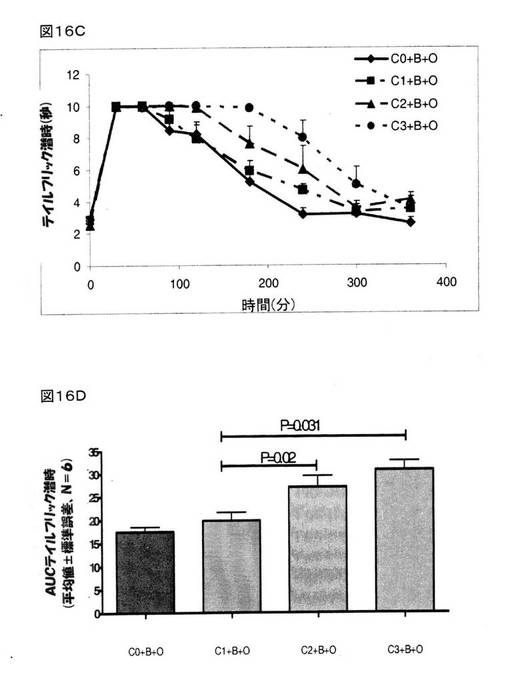
【図17】
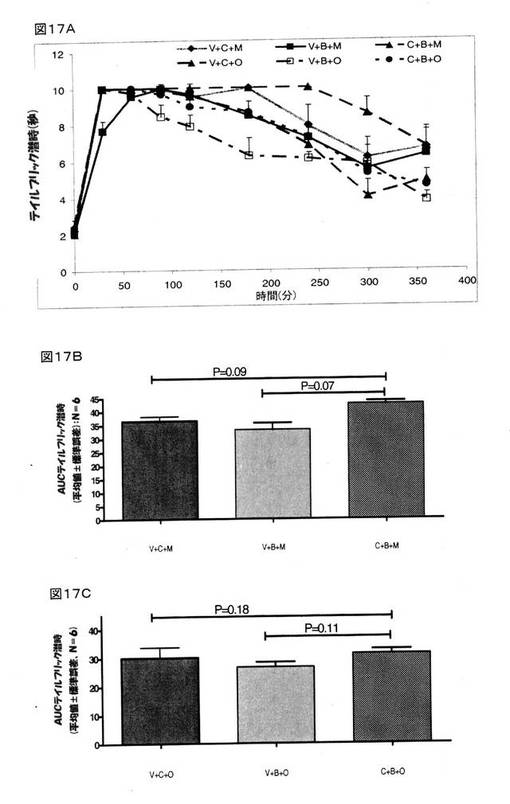
【図18】
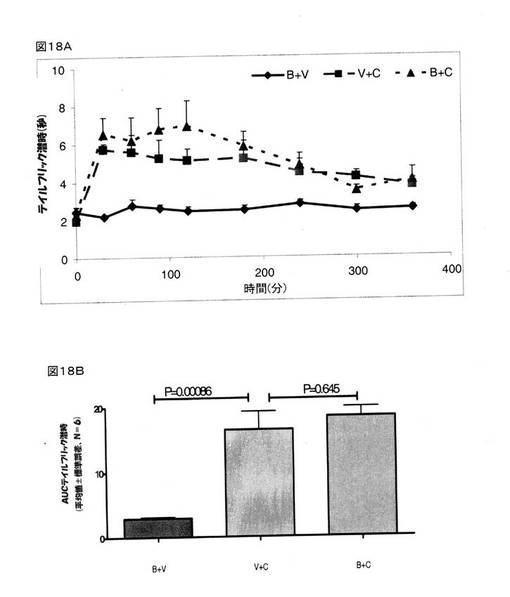
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16A−B】
【図16C−D】
【図17】
【図18】
【公表番号】特表2011−529490(P2011−529490A)
【公表日】平成23年12月8日(2011.12.8)
【国際特許分類】
【出願番号】特願2011−521070(P2011−521070)
【出願日】平成20年7月28日(2008.7.28)
【国際出願番号】PCT/US2008/071330
【国際公開番号】WO2010/014076
【国際公開日】平成22年2月4日(2010.2.4)
【出願人】(511024805)エンドジェンエックス, インコーポレイテッド (1)
【Fターム(参考)】
【公表日】平成23年12月8日(2011.12.8)
【国際特許分類】
【出願日】平成20年7月28日(2008.7.28)
【国際出願番号】PCT/US2008/071330
【国際公開番号】WO2010/014076
【国際公開日】平成22年2月4日(2010.2.4)
【出願人】(511024805)エンドジェンエックス, インコーポレイテッド (1)
【Fターム(参考)】
[ Back to top ]