
アルファ放出複合体
【課題】
【解決手段】本発明は、組織標的部位、オクタデンテートヒドロキシピリジノンリガンドおよびアルファ放出トリウム放射性核種のイオンを含む組織標的複合体を提供する。本発明は、さらにそのような複合体を用いた治療方法、その製造方法および使用方法、ならびにそのような複合体を含むキットおよび薬剤を提供する。

【解決手段】本発明は、組織標的部位、オクタデンテートヒドロキシピリジノンリガンドおよびアルファ放出トリウム放射性核種のイオンを含む組織標的複合体を提供する。本発明は、さらにそのような複合体を用いた治療方法、その製造方法および使用方法、ならびにそのような複合体を含むキットおよび薬剤を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、トリウム同位体の複合体に関し、とくに特定のオクタデンテートリガンドを有するトリウム−227の複合体に関する。本発明はまた、病気の治療に関する、特に腫瘍性の病気に関し、そのような複合体を投与することを含む。
【背景技術】
【0002】
特定の細胞の死滅させることは、哺乳類のさまざまな病気の有効な治療に基本となり得る。この典型的な例は、悪性の病気、たとえば肉腫およびガン種の治療における。しかしながら、特定の細胞のタイプの選択的除去は、他の病気、特に増殖性のおよび腫瘍性の病気、の治療においても重要な役割を果たす。
【0003】
選択的な治療の最も普通の方法は、最近の外科、化学療法および外部ビーム照射である。標的化された放射性核種の治療法は、しかしながら、将来性があり、発展途上の領域であり、望ましくない細胞のタイプに高い細胞毒性の放射をする潜在力を有する。放射性医薬品の最も普通の形態は、最近ではベータ放出および/またはガンマ放出放射性核種を導入することにより、人間に用いることが許可されている。しかしながら、治療におけるアルファ放出放射性核種の使用にも関心がある。なぜなら、細胞を死滅させる滞在力が大きいからである。
【0004】
生理学的な環境における典型的なアルファ放出器の放射範囲は、通常100マイクロメータ未満であり、いくつかの細胞の直径にのみ等しい。このことは、これらの源をミクロ転移を含む腫瘍の治療に好適とする。なぜなら、もしそれらが好適に標的化されるのであれば、標的細胞を超えて通過する放射エネルギーはほとんどないことになる。こうして周りの健康な組織の損傷を小さくすることができる(Feinendegenら、Radiat Res 148:195−201(1997)参照)。対照的に、ベータ粒子は、水中で1mm以上の範囲である(Wilbur,抗体免疫放射性 4:85−96(1991)参照)。
【0005】
アルファ粒子放射のエネルギーは、ベータ粒子、ガンマ線およびX線によるエネルギーに比較して高い。典型的には5−8MeVで、ベータ粒子のエネルギーの5〜10倍であり、ガンマ線のエネルギーの20倍以上である。こうして、非常に短い距離に渡って大量のエネルギーが堆積することが、α−放射に、ガンマ放射およびベータ放射に比較して、予期できないほど高い線エネルギー付与(LET)、高い生物学的効果比(RBE),低い酸素増感比(OER)を与える(Hall,「放射線医師のための放射線生物学」、5版、Lippincott Williams & Wilkins, Philadelphia PA, USA, 2000参照)。このことは、アルファ放出放射線核種の細胞毒性の例外を説明し、そのような同位体の生物学的標的化および制御のレベルおよび受け入れがたい副作用を避けるために必要であるアルファ放出放射線核種の分布の研究に厳しい要求を強いる。
【0006】
下記の表1は、今までのところ、治療の効果を有すると考えられるとして文献において広く提案されているアルファ放出体の物理学的崩壊特性を表す。
【0007】
【表1】
【0008】
今までのところ、放射線免疫治療に関して、主な関心は、211At、213Biおよび225Acに注がれ、これらの3種の核種は臨床の免疫試験において研究されてきた。
【0009】
いくつかの提案されている放射線核種は寿命が短い、たとえば半減期が12時間未満である。そのような短い半減期は、これらの放射線核種に基づく放射性医薬品の商業的な製造および流通を困難としている。寿命の短い核種を投与することはまた放射線量の割合を増大させる、なぜなら標的部位に達成する前に体外に放出されるからである。
【0010】
アルファ放出の反動エネルギーは、多くの場合親の放射線崩壊の位置から娘核種の放出を生じる。この反動エネルギーは、親を保持してよい化学的環境、たとえば親がキレート剤のようなリガンドにより複合化されたような環境から多くの娘核を取り出すのに十分なものである。このことは、娘が同じリガンドに化学的に適合する場合すなわち複合化されることさえも生じる。同様に、娘核種がガスである場合、特にラドンのような希ガスである場合、または化学的にリガンドと適合しない場合、この放出効果はさらに大きくなる。娘核種が数秒以上の半減期を有する場合、それらは血液系に拡散することができ、親を保持する複合体に抑制されることがない。これらの自由な放射活性娘は望ましくない全身の毒性を生じる。
【0011】
223Ra娘同位体が制御する状況でのトリウム−227(T1/2=18.7日)の使用が数年前に提案された(WO01/60417およびWO02/05859参照)。これは、キャリアーシステムが用いられた状況におけるもので、その状況は娘核種が閉じられた環境によって保持されることを許容する。第1のケースにおいて、放射性核種はリポソーム内で処置され、リポソームの実質的な大きさ(反動距離に対して)は娘核種をリポソーム内に保持することを助ける。第2のケースにおいて、向骨性の放射性核種の複合体は、骨マトリクスへ組み入れるために用いられる、それゆえ娘核種の放出を制限する。これらは、潜在的に非常に有利な方法である、しかしながらリポソームの投与は、いくつかの状況では望ましくない、そして、娘同位体を保持することができるように放射性核種が石灰化したマトリクスに囲まれることができないたくさんの柔らかい組織の病気がある。
【0012】
最も最近、227Thの崩壊によって放出される223Ra娘核の毒性が、比較できる核の前もった試験から予想されるよりも、哺乳類の体でかなりの程度にまで耐えうる方法が確立された。上記で述べたトリウム−227のラジウム娘を保持する特定の方法なくしては、ラジウムの毒性に関し公然と得られる情報は、トリウム−227を治療的な薬剤として用いることは不可能であることを明らかにした。なぜならトリウム−227の崩壊から治療効果を得るのに必要な投与量は、非常に毒性があり、致死量となりうるラジウム娘の崩壊からの放射となる、すなわち治療的な開口はない。
【0013】
WO04/091668は、標的化されたトリウム−227の放射性核種の治療に効果的な量を対象(典型的には哺乳類)に受け入れることのできない骨髄毒性を生じるのに十分なラジウム−223の量を生じることなく投与することができるという治療的な処置の開口が確かに存在するという予期できない発見について述べている。このことは、それゆえ骨および柔らかい組織のサイトのいずれにもおけるすべてのタイプの病気の治療および予防に用いることができる。
【0014】
上記発展によって、今やアルファ放出トリウム−227核を、223Raを生じることによる致死の骨髄毒性を生じることなく内部放射性核治療に用いることが可能である。それにもかかわらず、治療の開口は狭いままであり、対象に対するアルファ放出放射性同位体を投与することが好ましい全てのケースにおいて、絶対的に必要であるとは言えない、この新しい治療の開口の有用な開発は、それゆえアルファ放出トリウム−227核が複合化されているかどうかおよび高い信頼性で標的化されるかどうかによって非常に高められるであろう。
【0015】
放射性核種は常に崩壊するため、単離から対象に投与されるまでの間の材料を取り扱う時間が非常に重要である。アルファ放出トリウム核が、調製するためにすばやく便利な形態、好ましくはほとんどステップを必要としない、短いインキュベイション期間および/または標的とする実体の特性に不可逆的に影響を与えない温度で、複合化され、標的化され、および/または投与されるいるか否かということもまた、かなりの価値があるであろう。
【0016】
本発明の発明者らは、標的部位に結合したオクタデンテートヒドロキシピリジノン(HOPO)タイプのリガンドで複合化された4+トリウム−227イオンがトリウム−227イオンをめざましい程度で制御することを予想外に見出した。さらに、そのような複合体は、ここで述べる方法によって、相対的にすばやく容易に調製することができる。
【先行技術文献】
【特許文献】
【0017】
【特許文献1】国際公開第01/60417号
【特許文献2】国際公開第02/05859号
【特許文献3】国際公開第04/091668号
【非特許文献】
【0018】
【非特許文献1】Feinendegenら、Radiat Res 148:195−201(1997)
【非特許文献2】Wilbur,抗体免疫放射性 4:85−96(1991)
【非特許文献3】Hall,「放射線医師のための放射線生物学」、5版、Lippincott Williams & Wilkins, Philadelphia PA, USA, 2000
【発明の概要】
【課題を解決するための手段】
【0019】
本発明の一つの観点は、組織標的部位、オクタデンテートヒドロキシピリジノンリガンドおよびアルファ放出トリウム放射性核種のイオンを含む組織標的複合体を提供する。特に好ましい観点は、少なくとも1つの3,2−ヒドロキシピリジノン部位を含むオクタデンテートリガンドに共有結合し、そのようなリガンドは227Thのようなアルファ放出トリウム放射性核種の4+イオンに複合化しているポリペプチド組織標的部位を含む組織標的複合体である。
【0020】
本発明のさらなる観点は、増殖性または腫瘍性の病気、ここで述べる病気は何でも含むが、を治療するための薬剤の製造における、組織標的部位、オクタデンテートヒドロキシピリジノンリガンドおよびアルファ放出トリウム放射性核種のイオンを含む組織標的複合体(ここで述べるそのような複合体は何でも含む)の使用を提供する。
【0021】
対応する観点において、本発明は、少なくとも1つの組織標的部位、オクタデンテートヒドロキシピリジノンリガンドおよびアルファ放出トリウム放射性核種のイオンを含む組織標的複合体(ここで述べるそのような複合体は何でも含む)の投与を含む人間および人間でない動物(特にそれを必要としているもの)の治療方法を提供する。そのような方法は、好ましくはここで述べる病気は何でも含む増殖性または腫瘍性の病気の治療である。
【0022】
本発明のさらなる観点は、組織標的部位、オクタデンテートヒドロキシピリジノンリガンドおよびアルファ放出トリウム放射性核種のイオンを含む組織標的複合体(ここで述べるそのような複合体は何でも含む)を少なくとも1つの薬剤のキャリアまたは賦形剤とともに合わせて含む薬剤を提供することである。
【0023】
もっとも天然に豊富に生じるトリウム同位体、すなわちトリウム−232(半減期1010年であり、事実上放射性活性がない)、のトリウム複合体から区別するために、ここで定義するトリウム複合体およびその組成物は、天然に相対的に豊富なものよりも、アルファ放出トリウム放射性同位体(すなわち、少なくとも半減期が103年未満のトリウムの1つの同位体、たとえばトリウム−227)を多く含む、たとえば少なくとも20%多い。このことは本発明の方法の定義に影響を与えることを必要としない、それは放射性トリウム、たとえばトリウム−227の治療的に効果のある量が明確に必要とされるからである、しかしながら、それはすべての観点から好ましい場合である。
【0024】
本発明のさらなる観点は本発明に関する方法に用いられるキットを提供することである、前記キットは、オクタデンテートヒドロキシピリジノン含有リガンドに接合されたまたは接合可能な組織標的部位を含む。すべての結合部位およびリガンドは、好ましくはここで述べたものである、そのようなキットは任意におよび好ましくアルファ放出トリウム放射性核種、たとえば227Thを含む。
【0025】
本発明はさらに添付の図面によって説明される。
【図面の簡単な説明】
【0026】
【図1】図1は、232Th4+(HNO3)および1mg/mlのDMSO中のALG−DD−NCS(理論比2:3)を室温で15分反応させた後の反応混合物のクロマトグラムを示す。かなりの量の複合体232Th−ALG−DD−NCS(tR=5.74分;m/z1328.5)があり、および自由ALG−DD−NCSリガンド(tR=5.36)の痕跡はなかった。
【図2】図2は、前記232Th−ALG−DD−NCS複合体(m/z1328.3949)の質量スペクトルを示す。
【図3】図3は、Fe3+(HNO3)および1mg/mlのDMSO中のALG−DD−NCS(理論比2:3)を室温で15分反応させた後の反応混合物のクロマトグラムを示す。かなりの量の複合体Fe−ALG−DD−NCS(tR=5.81分;m/z1153.5)があり、および自由ALG−DD−NCSリガンド(tR=5.36分)の痕跡はなかった。
【図4】図4は、前記Fe−ALG−DD−NCS複合体(m/z1153.3002)の質量スペクトルを示す。
【図5】図5は、In3+(HCl)および1mg/mlのDMSO中のALG−DD−NCS(理論比2:3)を室温で15分反応させた後の反応混合物のクロマトグラムを示す。かなりの量の複合体In−ALG−DD−NCS(tR=5.95分;m/z1212.5)があり、および自由ALG−DD−NCSリガンド(tR=5.36分)の痕跡はなかった。
【図6】図6は、前記In−ALG−DD−NCS複合体(m/z1212.2666)の質量スペクトルを示す。
【図7】図7は、232Th4+(HNO3)および1mg/mlのpH5.5のアセテート緩衝液中のALG1005−38を室温で10分反応させた後の反応混合物(理論比1:3)のクロマトグラムを示す。かなりの量の複合体232Th−ALG1005−38(tR=3.56;m/z1184.5)があり、および自由ALG1005−38リガンド(tR=3.20分)の残渣があった。
【図8】図8は、前記232Th−ALG1005−38複合体(m/z1184.4662)の質量スペクトルを示す。
【図9】図9は、Fe3+(HNO3)および1mg/mlのpH5.5のアセテート緩衝液中のALG1005−38を室温で10分反応させた後の反応混合物(理論比1:3)のクロマトグラムを示す。かなりの量の複合体Fe−ALG1005−38(tR=3.70;m/z1009.5)があり、および自由ALG1005−38リガンド(tR=3.22分)の残渣があった。
【図10】図10は、前記Fe−ALG1005−38複合体(m/z1009.3727)の質量スペクトルを示す。
【図11】図11は、In3+(HCl)および1mg/mlのpH5.5のアセテート緩衝液中のALG1005−38を室温で10分反応させた後の反応混合物(理論比1:3)のクロマトグラムを示す。かなりの量の複合体In−ALG1005−38(tR=3.72;m/z1068.5)があり、および自由ALG1005−38リガンド(tR=3.21分)が痕跡量あった。
【図12】図12は、前記In−ALG1005−38複合体(m/z1068.3485)の質量スペクトルを示す。
【図13】図13は、Ga3+(HCl)および1mg/mlのpH5.5のアセテート緩衝液中のALG1005−38を室温で10分反応させた反応混合物(理論比1:3)のクロマトグラムを示す。かなりの量の複合体Ga−ALG1005−38(tR=3.65;m/z1022.5)があり、および自由ALG1005−38リガンド(tR=3.21)の残渣があった。
【図14】図14は、前記Ga−ALG1005−38複合体(m/z1022.3571)の質量スペクトルを示す。
【図15】図15は、232Th4+(HCl)および1mg/mlのBb−1−HOPO−1−DEBN(理論比2:3)を室温で15分反応させた後の反応混合物のクロマトグラムを示す。かなりの量の複合体232Th−Bb−1−HOPO−1−DEBN(tR=3.52)があった。質量クロマトグラムは、m/z 1222.5である、低い痕跡:UV330nm。
【図16】図16は、前記232Th−Bb−1−HOPO−1−DEBN複合体(m/z1222.5)の質量スペクトルを示す。
【図17】図17は、227Th−ALG−DD−NCS−trastuzumabおよび自由227Thが0−7日でSK−OV−3細胞ペレットに結合する計算量であって、補正された227Th−放射能の崩壊によって表わす。
【図18】図18は、(1)正規の培地(対照);(2)20μg/mLのtrastuzumab;(3)20kBq/mLの227Th−ALG−DD−NCS−trastuzumab(10166Bq/μg);および(4)20kBq/mLの自由227Thで処理されたSK−OV−3細胞の標準化された発光値を示す。
【図19】図19は、(1)正規の培地(対照);(2)trastuzumab;(3)自由227Th;(4)227Th−ALG−DD−NCS−trastuzumab(10166Bq/μg)(Mean±SD;n=3)で処理された1000SK−OV−3細胞のクローン形成を示す。
【図20】図20は、(1)正規の培地(対照);(2)trastuzumab;(3)自由227Th;(4)227Th−ALG−DD−NCS−trastuzumab(10166Bq/μg)で処理された3000SK−OV−3細胞のクローン形成を示す。
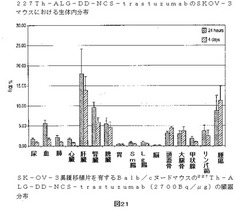
【図21】図21は、4日と24時間生体内分布をした後のSK−OV−3異種移植片を有するBalb/cヌードマウスの227Th−ALG−DD−NCS−trastuzumab(2700Bq/μg)の臓器分布を示す(Mean±SD;n=5)。
【発明を実施するための形態】
【0027】
本発明において、「組織標的」とは対象となる物質を示す(特にトリウム複合体に接合している形態のとき)、選択的にそれが存在する(たとえば、放射線崩壊をもたらすために)少なくとも1つの組織の部位に、自身の位置を特定する役割を果たすこと(そして特にどのような接合したトリウム複合体を特定すること)が望まれる。標識化した部位は、例えば、細胞表面のマーカー(たとえば、レセプター、輸送タンパク質、細胞接着分子)に結合し、病気の細胞または病気の細胞に近接した細胞上に存在してもよい。そのような細胞表面のマーカーは、健康な細胞の表面の上よりも病気の細胞の表面の上により大量に発現されるタンパク質または休眠の段階よりも成長もしくは複製の期間の細胞の表面の上により大量に発現されるタンパク質を含む。本発明のどのような態様においても、標的細胞、組織もしくはその結合物に近接して存在する構成は、治療のために標的において利用されてもよい。たとえば、標的細胞または組織の回りのマトリクスに存在するまたは放出される構成は、存在する場合には標的化に用いられてもよい、形成または濃縮は、前記領域を健康な組織から区別することを可能にする。この例はテネイシンのようなマトリクス抗原が挙げられる、それは脳腫瘍と関連するが、細胞の間のマトリクス中に発現する。そのようなマトリクス抗原は、ここで述べるような単一のまたは複合の標的部位によって、標的とされることができる。
【0028】
前記組織標的部位は合計で2つ以上の成分を含んでいてもよく、トリウム複合体を所望の組織へ標的化する効果を有する。このことは、たとえば、1つの成分が初めに投与され、特定の組織、腫瘍または細胞型に結合し、次におよび/またはさらに成分(連結剤)を同時に好ましくは続いて投与することにより、in vivoで組織結合剤に結合する。前記結合剤は直接または間接的に複合α放射トリウムに接合され、こうして組織結合および連結剤は合わさって組織標的部分を形成する。適切な特定の結合ペアが組織結合および連結剤に相互の親和性を供給するのに適していることは周知である(たとえば、ビオチンとアビジンまたはストレプトアビジン)。
【0029】
ここで述べる本発明の様々な態様は、病気の治療に関する、特に病気の組織の選択的標的化に関し、同様に複合体、接合、薬剤、処方、キットなどそのような方法に役に立つものに関する。すべての態様において、前記病気の組織は体の1つのサイトにおいて存在していてもよく(たとえば、局部的な固体の腫瘍の場合が挙げられる)、多くのサイトにおいて存在していてもよい(たとえば、いくつかの接合が関節炎の影響を受けている場合またはガン性の病気が広がったり転移した場合が挙げられる)。
【0030】
標的化される病気の組織は、柔らかい組織のサイトにある可能性もあり、石灰化された組織のサイトのある可能性もあり、全てが柔らかい組織中のたくさんの組織、石灰化された組織中のたくさんの組織にある可能性もあり、少なくとも1つの柔らかい組織のサイトおよび/または少なくとも1つの石灰化された組織のサイトも含む可能性がある。前記標的化されたサイトおよび病気の根源のサイトは同じ可能性もあるが、その代わりに異なる可能性もある、1つ以上のサイトが含まれている場合、これは根源のサイトを含む場合があり、またはたくさんの二次的なサイトである場合がある。
【0031】
「柔らかい組織」とは、ここでは「固い」石灰化したマトリクスを有さない組織を示す。特に、ここで用いられる柔らかい組織は、骨格的な組織でない組織であればなんでもよい。それに応じて、ここで用いられる「柔らかい組織の病気」とは、ここで用いられる柔らかい組織に起きる病気を示す。本発明は特にガンおよび「柔らかい組織の病気」の治療に適する、したがってどのような「柔らかい」(すなわち、非石灰化された)組織に生じた癌腫、肉腫、骨髄腫、白血病、リンパ腫および混合タイプのガン、同様にそのような組織のその他の非ガン性の病気に適する。ガン性の「柔らかい組織の病気」は、柔らかい組織に生じた固体の腫瘍、同様に転移したおよび微小転移した腫瘍を含む。実際、前記柔らかい組織の病気は、柔らかい組織の一次の固体の腫瘍および同じ患者の柔らかい組織の少なくとも1つの転移した腫瘍を含む、言い換えれば、「柔らかい組織の病気」は固体の腫瘍のみ、または骨格の病気の1次的な腫瘍を伴う転移のみからなってもよい。
【0032】
近日の主要な発見は、特定のアルファー放射性トリウム同位体(たとえば227Th)は治療的に効果があり、かつ耐えられない骨髄毒性を生じない量で投与されることが可能であることである。ここで、「許容される非骨髄毒性」は、最も重要に、投与されたトリウム−227の放射性同位体の崩壊により生じたラジウム−223の量は、通常対象者を直接致死とするのに十分でない量を示す。当業者には、しかしながら、そのような治療の副作用として許容可能な骨髄を損傷する量(および致死反応の可能性)は、対象者の治療を受ける病気のタイプ、治療計画の目的、およびに予後より著しくさまざまであることは明らかであろう。本発明の好ましい対象は人間であるが、その他の哺乳類、特に犬は、本発明を用いる恩恵を受けるであろうし、許容可能な骨髄を損傷するレベルもまた対象の種類を反映するであろう。前記許容可能な骨髄を損傷するレベルは、一般的に悪質でない病気よりも悪質な病気の治療のほうが高い。1つのよく知られている骨髄毒性の測定は、好中球の数を数えることであり、本発明において223Raの許容可能な骨髄を損傷する量は、典型的に治療前に好中球フラクションの最も低いポイントを10%以上のカウントすることにより制御された量である。好ましくは、223Raの許容可能な骨髄を損傷しない量は、好中球のフラクションは、一番低い値で少なくても20%、より好ましくは少なくとも30%である。一番低い値の好中球のフラクションは、少なくとも40%が最も好ましい。
【0033】
それに加えて、放射性トリウム(たとえば227Th)を含む化合物は、高い放射線の治療に用いられ、その治療では生じたラジウム(たとえば223Ra)の骨髄毒性は、幹細胞サポートまたは同等の回復方法が含まれる場合、普通は耐えられない。そのような場合、好中球のカウントは、一番低い値で10%以下まで減じてもよく、例外的に5%まで、必要であれば5%以下まで減じてよく、そうして適切な予防が行われ、次の幹細胞サポートが与えられる。そのような技術は周知である。
【0034】
本発明において特に興味があるのが、トリウム−227である、トリウム−227は、背景が許容するように、ここでのすべてのトリウムに対して好ましい同位体である。トリウム−227は相対的に製造が容易で、中性子照射した226Raから間接的に製造することができ、227Thの母核種、すなわち227Ac(T1/2=22年)を含むものである。アクチニウム−227は前記226Raターゲットから容易に分離することができ、227Thの発生器として用いられる、このプロセスは、必要であれば工業スケールとすることもできる、こうして多くの他のアルファ放出体にみられる供給問題は、分子を標的化した放射線治療の候補から除かれる可能性がある。トリウム−227はラジウム−223を経て崩壊する。この場合、前記最初の娘は、半減期が11.4日である。純粋な227Th源から、適度な量のラジウムのみが最初の2,3日の間に作られる。しかしながら、223Raの滞在的な毒性は、227Thのものより高い、それはアルファ粒子の223Raからの放出に、数分以内に短寿命の娘からの3つのさらなるアルファ粒子が続くからである(下記の表2参照、表2はトリウム−227の崩壊系列を提示した)。
【0035】
【表2】
【0036】
滞在的に毒性のある崩壊生成物を生成することも部分的な理由として、トリウム−227(T1/2=18.7日)はアルファ粒子治療のために広く検討されてこなかった。
【0037】
トリウム−227は、骨髄の耐えられないような状態を生ずるような多量のラジウム−223を生ずることなく、所望の治療効果を提供するのに十分な量を投与することもできる。標的の領域において娘同位体を保持すると、その崩壊によりさらなる治療効果が得られるため望ましい。しかしながら、受け入れがたい骨髄毒性を誘導することなく有用な治療効果を得るために、トリウムの崩壊生成物の制御を保つことは必要ない。
【0038】
ガン細胞を殺す効果が主にトリウム−227から得られ、娘から得られないと仮定すると、この同位体の適した治療のための放射量は、他のアルファ放出体に比較して定められる。たとえば、アスタチン−221は、動物における治療効果のある放射量は典型的に2−10MBq/kgである。半減期とエネルギーを補正することによって、トリウム−227の対応する放射線量は少なくても体重の36−200kBqとなるであろう。これは227Thの量の下限を定めることになり、それは治療効果を予想して、役立つように投与されることができるであろう。この計算はアスタチンおよびトリウムの同等の保持を仮定したものである。明らかに、しかしながら前記トリウムの18,7日の半減期は、この同位体が崩壊前により大きい放出をする結果になる傾向があるであろう。この計算された放射線量は、それゆえ、普通は最少の効果の量と考えられるべきである。十分に保持された227Th(たとえば、体から放出されたものでない227Th)の観点から表現された前記治療効果のある放射線量は、典型的には少なくとも18または25kBq/kgであり、好ましくは少なくとも36kBq/kgであり、より好ましくは少なくとも75kBq/kgである、たとえば100kBq/kg以上である。トリウムの量がより多い程、より治療効果が高いことが期待されるが、耐えられない副作用が生じるのであれば投与できない。同じように、もしトリウムが短い生物学的半減期(たとえば、なおトリウムを有している体から放射される前の半減期)を有する形態で投与されれば、治療の効果を得るためには、より多い量の放射性同位体が必要とされる、なぜなら、前記トリウムの多くの量が崩壊前に放出されるからである。しかしながら、ラジウム−223の対応する減少が生じる。投与されるトリウム−227の上記量は、同位体が十分に保持されるとき、より短い生物学的な半減期を伴う等量の放射線量に容易に関連付けられる。そのような計算は、周知であり、WO04/091668(たとえば実施例1および2の文中)に開示されている。
【0039】
もし放射線で標識化された化合物が娘核種を放出すれば、もし当てはまれば、どのような放射性の娘核種の宿命を知っておくことが重要である。227Thとともに主娘生成物は、226Raであり、その向骨性のため、それは臨床の評価のもとにある。ラジウム−223は非常にすばやく血液を飛び越えて骨に濃縮されるか、または腸および腎臓のルートを経由して排出される(Larsen,J NuclMed 43(5 補足):160P(2002)参照)。ラジウム−223は、in vivoで227Thから放出され、それゆえ、健康な柔らかい組織にそれほど影響を与えない。Mueller in Int.J.Radiat.Biol.20:233−243(1971)による227Thを溶解されたクエン酸塩とした分布の研究によれば、柔らかい組織中で227Thから生じた226Raは、直ちに骨に再分布されるか、排出される。アルファ放出ラジウムの公知の毒性は、特に骨髄に対するものであり、トリウムの投与の問題点である。
【0040】
実際、少なくとも200kBq/kgの放射線量の223Raが人間に投与することができ、耐えられることを最初に確立したのはWO04/091668である。これらのデータは、前記公報中に存在する。それゆえ、極めて予想できなかったことに、対象が深刻なまたは致死の骨髄毒性の受け入れがたい危険性をこうむることが予想されることなく、治療に効果のある量の227Th(たとえば36kBq/kgより大きい量)を哺乳類の動物に投与することができることが今や明らかになった。それにもかかわらず、この治療効果のある方法を最善に使用することは非常に重要であり、それゆえ、放射性のあるトリウムはすばやく効率的に複合体とすることおよび放射線量の可能な限り最大の割合が標的のサイトに届けられるように非常に高い親和性を有することが不可欠である。
【0041】
227Thの薬剤から生じる223Raの量は、放射性標識化された化合物の生物学的な半減期に依存する。理想的な状況は、すばやく腫瘍を取り込む複合体を用いることであり、腫瘍細胞に内面化すること、強く腫瘍を保持することおよび正常の組織中において短い生物学的半減期とすることを含む。生物学的半減期が理想よりも短い複合体は、しかしながら、223Raを耐えうるレベルで維持する量である限りは有用である。in vivoで生じたラジウム−223の量は、投与されるトリウムの量およびトリウム複合体の生物学的保持時間の因子である。どのようなケースで生じたラジウム−223の量も、当業者であれば容易に計算できる。227Thの最大の投与可能な量は、in vivoで生じたラジウムの量により定められ、耐えることのできないレベルの副作用、特に骨髄毒性を生じる量未満でなければならない。その量は一般的に300kBq/kg未満であり、好ましくは200kBq/kg未満である、より好ましくは170kBq/kg未満である(たとえば130kBq/kg未満である)。最小の効果的な放射線量は、トリウムの細胞毒性、生じたアルファ照射に対する病気の組織の影響の受けやすさ、およびトリウムが標的複合体(この場合リガンドおよび標的の部位の組み合わせ)によって効果的に結合し、保持されおよび届けられる程度によって定められる。
【0042】
本発明の方法において、トリウム複合体は望ましくはトリウム−227放射線量が18〜400kBq/kg体重、好ましくは36〜200kBq/kg(たとえば50〜200kBq/kg)、より好ましくは75〜170kBq/kg、特には100〜130kBq/kgにおいて投与される。それに応じて、1回の放射線量が、たとえば30〜150Kg、好ましくは40〜100Kgといった適切な体重によってこれらの範囲のどのような外延も含んでよい(たとえば放射線量あたり540kBq〜4000kBq他)。前記トリウムの放射線量、複合化剤および投与経路は、in vivoで生じたラジウム−223の放射線量が、300kBq/kg未満であり、好ましくは200kBq/kg未満であり、より好ましくは150kBq/kg未満であり、特に好ましくは100kBq/kg未満であることがさらに望ましい。さらにこのことは、示された体重のいずれにもよって拡大された範囲へ223Raへの露出を提供する。上記放射線量のレベルは、好ましくは227Thの十分に保持された放射線量であり、しかしながら、いくらかの227Thが崩壊前に体から消えることを考慮した投与された放射線量であってもよい。
【0043】
227Th複合体の生物学的半減期は、物理学的半減期(たとえば7日未満、特に3日未満)に較べて短い場合は、等しく保持された放射線量を提供するために特により多くの放射線量を投与することが必要とされるかもしれない。こうして、たとえば、150kBq/kgの十分に保持された放射線量は、5日半減期の複合体を711kBq/kgの放射線量で投与したのに等しい。どのような適切に保持された放射線量に対して等しく投与された放射線量は、周知の方法を用いて複合体の生物学的クリアランス速度によって計算されてもよい。
【0044】
1つの227Thの核の崩壊は1つの223Ra原子を提供するため、227Thの保持および治療効果のある活性は、患者が受ける223Raの放射線量に直接関連する。どのような状況で生じた223Raの量も、周知の方法で計算される。
【0045】
好ましい態様においては、本発明はそれゆえ、哺乳類(ここで述べた)の病気を治療するための方法を提供する。前記方法は組織標的部位を含む接合、オクタデンテートリガンド(特にここで述べたどのようなものも)、および放射性トリウム同位体(たとえばトリウム−227)の治療効果のある量を前記対象に投与することを含む。
【0046】
この特性が有用である限り、明らかに対象を223Raの娘の同位体に露出することを最小限にすることは望ましい。特にin vivoで生じるラジウム−223の量は、典型的には40kBq/kgを超える、たとえば60kBq/kgを超える。いくつかのケースにおいて、in vivoで生じる223Raは、80kBq/kgを超える、たとえば100または115kBq/kgを超える。
【0047】
適切なキャリア溶媒中のトリウム−227で標識化した接合体は、静脈内、膣内(たとえば腹腔内)、皮下、口内または局所に、1回または分割した方法により投与されてもよい。好ましくは、標的の部位に接合した複合体は、非経口(たとえば経皮)経路によって、特に、静脈内または膣内の経路によって投与される。好ましくは、本発明の組成物は、非経口投与のため無菌の溶液として作られる。
【0048】
本発明の方法および生成物におけるトリウム−227は、単独でまたは外科、体外照射療法、化学療法、その他の放射線核種、または組織温調節を含む他の治療法と組み合わせて用いてもよい。
【0049】
これはさらに本発明の好ましい態様の方法、製剤/薬剤が、他の放射性物質または化学療法の物質のようなさらなる少なくとも1つの治療活性物質を含んでいてもよいことを示す。
【0050】
1つの特に好ましい態様においては、対象は幹細胞治療および/または他の対症的な治療法を行って、ラジウム−223が誘発した骨髄毒性を減じる。
【0051】
本発明によれば、227Thは、標的複合化薬剤によって複合化されてもよい。典型的な標的部位は100g/molから数百万g/mol(特に100g/molから100万g/mol)の分子量を有し、好ましくは病気の関連したレセプターに直接的に親和性と有し、および/または投与前に病気を標的とした分子に結合する適切な前投与バインダー(たとえば、ビオチンまたはアビジン)を含む。適切な標的部位は、ポリ−およびオリゴ−ペプチド、プロテイン、DNAおよびRNAフラグメント、アプタマーなどを含み、好ましくは、たとえばアビジン、ストレプトアビジン、ポリクローナルまたはモノクローナル抗体(たとえば、IgGおよびIgMタイプの抗体)といったプロテイン、またはプロテインの混合物、またはプロテインのフラグメントまたは構築物を含む。抗体、抗体構築物、抗体のフラグメント(たとえばFABフラグメントまたは少なくとも抗原結合した領域、フラグメントの構築物(たとえば単鎖抗体)またはそれらの混合物が特に好ましい。
【0052】
本発明において用いるのに適しているものとしては、さらに、ペプチド、アミノ酸、ステロイド性のまたは非ステロイド性のホルモン、葉酸塩、エストロゲン、テストステロン、ビオチン、または他の典型的に10000g/mol以下の分子量を有する特異的な結合をする化合物を伴う複合化された227Thの治療的接合が挙げられる。
【0053】
一般的に、オクタデンテートリガンドは、直接的にまたは間接的に(たとえばリンカー部位を経由して)、標的部位に接合している。このタイプの一般的構築物は、たとえば活性(治療上または診断上の活性)金属−複合体部位−任意のリンカー部位−標的部位が、放射性医薬品および標的化された造影剤の分野でよく知られている。しかしながら、トリウム4+イオンの特定の使用に用いられるさまざまなリガンドの適性を評価する研究はほとんどない。この点で「造影剤の標的化された投与のハンドブック」Ed.Torchilin,CRC Press,1995は参照される。
【0054】
従来知られていたトリウムのキレーターはポリアミノポリアシッドキレーターを含む、それは鎖状、環状またな分枝状の骨格の窒素に酸基(カルボキシアルキル)を有するポリアザアルカン骨格を含む。そのようなキレーターはDOTA誘導体、たとえばp−イソチオシアナートベンジル−1,4,7,10−テトラアザシシクロドデカン−1,4,7,10−テトラ酢酸(p−SCN−Bz−DOTA)およびDTPA誘導体たとえばp−イソチオシアナートベンジル−ジエチレントリアミンペンタ酢酸(p−SCN−Bz−DTPA)、最初は環状のキレーターであり後から鎖状のキレーターになるもの、が挙げられる。
【0055】
1,4,7,10−テトラアザシシクロドデカン−1,4,7,10−テトラ酢酸の誘導体は、従来実証されてきた、しかしながら、標準的な方法はDOTA誘導体を有するキレートトリウムに容易に用いることはできない。金属を有するDOTA誘導体を加熱することは効果的にキレートを提供するが、多くの場合収率が低い。少なくてもリガンドの一部は、生成の過程で不可逆的に変性するという傾向がある。さらに、不可逆的な変性に相対的に高い影響を受ける性質により、全ての加熱過程が完了するまで通常標的部位の結合物を避けなければならない。このことはさらなる化学的なステップ(加えて全ての必要な検査および分離)を加えることになり、そのステップはアルファ放出トリウム同位体の崩壊半減期の間に行わなければならない。明らかにアルファ放出材料はこの方法である扱わないことが好ましく、必要性よりもより多くの無駄を生じる。さらに接合体を生成する全ての時間は、トリウムが準備期間に崩壊するので、トリウムの一部を無駄にする。
【0056】
本発明のすべての観点においてアルファ放出トリウムおよびオクタデンテートリガンドの複合体が形成されること、または60℃以上に加熱することなく(例えば50℃以上に加熱することなく)、好ましくは38℃以上に加熱することなく、最も好ましくは25℃以上に加熱することなく形成されることが好ましい。
【0057】
加えて好ましいのは標的部位およびオクタデンテートリガンドの接合がアルファ放出トリウム同位体(たとえば227Th4+イオン)の付加よりも前に生成されることである。本発明の生成物は、こうして、オクタデンテートリガンドおよび組織標的部位の接合によるアルファ放出トリウム同位体(たとえば227Th4+イオン)の複合化によって好ましく形成され、または形成されうる。
【0058】
前記キレーターは、非ホスホン酸分子であってもよく、本発明の一態様では、227Thはどのようなホスホン酸または他の骨を標的とした基に付着しないし、そのような材料とともに投与されない。
【0059】
標的化合物のタイプは、オクタデンテートキレーター(ここで述べたようにカップリング部位を含む)を経由してトリウム(たとえばトリウム−227)に結合していてもよい。標的部位は、公知の標的基から選ばれてもよく、モノクローナルまたはポリクローナル抗体、成長因子、ペプチド、ホルモンおよびホルモン類似体、葉酸および葉酸誘導体、ビオチン、アビジンおよびストレプトアビジンまたはその類似物を含む。他の可能なキャリアはRNA、DNA、もしくはそのフラグメント、オリゴヌクレオチド、炭水化物、脂質またはそのようなグループをタンパク質とともに、またはタンパク質を伴わないで結合した化合物であってもよい。
【0060】
組織標的部位は、一つの態様において、抗体または抗体のフラグメントに接合した向骨性物質、リポゾームおよび葉酸を含まなくてもよい。もしくは、そのような部位は含まれていてもよい。
【0061】
本発明の前記トリウム(たとえばトリウム−227)で標識化した分子は、病気の関連したレセプターを標的化することにより、ガン性のまたは非ガン性の病気の治療に用いられてもよい。典型的には、そのような227Thの医学的使用は、ガン性のまたは非ガン性の病気の治療のために、抗体、抗体フラグメント、または抗体もしくは抗体のフラグメントの構築物へのキレーターによる227Thの結合に基づく放射線免疫治療によってもよい。本発明による方法および薬剤における227Thの使用は特に癌腫、肉腫、リンパ腫、白血病を含むどのような形態のガン、特に肺、胸、前立腺、膀胱、腎臓、胃、すい臓、食道、脳、卵巣および子宮のガン、口頭ガン、大腸がん、黒色腫、複合骨髄腫およびホジキンリンパ腫を含む。
【0062】
227Thを有する分子がin vivoで短い生物学的な保持半減期を有する場合、放出される223Raの量は減少させることができる、なぜなら放射性核種はたいてい227Thの多くの割合が崩壊して223Raになる前に、排出されるからである。しかしながら、本発明によれば、227Thの量は、治療効果を保持するために増やされる必要がある。もし複合剤が、標的の細胞の内部へ227Thを運ぶように選択されるのであれば、腫瘍サイトにおける娘同位体の少なくとも部分的な保持により、さらに骨髄毒性を上昇させ、放射性娘の全身性独作用を減少させるであろう。これらいずれもの特徴は、227Thの治療方法を拡大し、本発明の好ましい態様となる。
【0063】
本発明のさらなる態様において、軟らかい組織および骨格の病気を有する患者は、トリウムを投与することにより、227Thおよびin vivoで生じた223Raによって治療されてもよい。この特に有利な観点において、治療にさらなる治療的な成分は、骨格の病気を標的化することにより受容可能に非骨髄毒性量の223Raから由来する。この治療方法において、227Thは典型的に柔らかい組織の初期のおよび/または転移性のガンを適切に標的化するのに利用される。そして227Thの崩壊から生じた223Raは、同じ対象の関連する骨格の病気の治療に利用される。この骨格の病気は初期の柔らかい組織のガンから生じた骨格への転移であってもよい、または柔らかい組織の治療が転移性のガンを攻撃するためのものである初期の病気であってもよい。場合によっては、前記柔らかい組織および骨格の病気は関連していないかもしれない(たとえば、リウマチの柔らかい組織の病気を有する患者における骨格の病気のさらなる治療が挙げられる)。
【0064】
すべての観点における本発明の主な観点は、オクタデンテートリガンドを用いること、とくにオクタデンテートヒドロキシピリジノン含有リガンドを用いることである。そのようなリガンドは典型的に以下の置換されたピリジン構造(I)の少なくとも1つのキレート基を含むであろう。
【0065】
【化1】
【0066】
式中R1は、任意のN−置換基であり、なくてもよいし、ヒドロカルビル基、OH基、O−ヒドロカルビル基、SH基、およびS−ヒドロカルビル基から選ばれてもよい、前記ヒドロカルビル部位のいずれもまたはそれぞれは、独立に、短いヒドロカルビル基、たとえば、C1〜C8アルキル基、アルケニル基またはアルキニル基を含むC1〜C8のヒドロカルビル基から選ばれ、OHまたはO−ヒドロカルビル基であってもよい。R1は、下記に示すようにリンカー部位もまた含んでいてよい、および/または下記に示すようにカップリング部位を含んでいてもよい。
【0067】
式Iにおいて、置換基R2〜R6はそれぞれ独立にH、OH、=O、短いヒドロカルビル(ここで述べたように)、リンカー部位(ここで述べたように)、および/またはカップリング部位(ここで述べたように)から選ばれてもよい。通常、R1〜R6の少なくとも1つは、OHである。通常、R2〜R6の少なくとも1つは、=Oである。通常、R1〜R6の少なくとも1つは、リンカー部位(ここで述べたように)である。残りのR1〜R6は、ここで述べた部位のいすれであってもよく、好ましくはHである。リンカー部位またはさらなるリンカー、リンカー部位に付いているテンプレートまたはキレート基は、カップリング部位を含まず、R1〜R6の1つは、好ましくはカップリング部位(ここで述べたように)である。
【0068】
好ましい態様において、R1〜R6の1つはOHであって、R2〜R6の1つは=Oであって、OHおよび=O基は、環における隣接した原子である。こうして、好ましい態様において、OHおよび=Oは、(予想されたように、窒素から数えた)1,2;2,3;3,2;3,4;または4,3のそれぞれの原子上にあるであろう。オクタデンテートリガンドは、OHおよび=O基が3および2の位置に其々存在する少なくとも1つのキレート部位であることが、非常に好ましい。前記オクタデンテートリガンドは、2、3または4つのキレート基を有することが好ましく、2または4のキレート基を有することが非常に好ましい。
【0069】
好適なキレート部位は周知の方法により形成することができる、その方法はUS5,624,901(たとえば実施例1および2)およびWO2008/063721に記載されている(いずれも参考として組み込む)。
【0070】
ここで用いられるように、「リンカー部位」(式IIにおけるRL)という用語は、オクタデンテートリガンドにおいて、少なくとも2つのキレート基を結合する化学物質を示すために用いられる、それは本発明のさまざまな点において主要の成分を形成する。典型的に、それぞれのキレート基(たとえば上記式Iおよび/または下記式II)は、ビ−デンテートであり、そのため4つのキレート基である、その少なくとも1つは式Iである、典型的にリガンド中に存在する。そのようなキレート基はそのリンカー部位によって互いに結合しあう。こうして、リンカー部位(たとえば下記のRL基)は、式Iおよび/またはIIの1を超えるキレート基の間に共有されます。前記リンカー部位は、オクタデンテートリガンドの複合化部位と標的部位との間の連結部位として働く。そのような場合、少なくとも1つのリンカー部位は、カップリング部位(RC)に結合する。適切なリンカー部位は、すべてのトポロジーの中のメチル基、エチル基、プロピル基、ブチル基、ペンチル基および/またはヘキシル基を含むC1〜C12アルキル基、アルケニル基、またはアルキニル基を含むC1〜C12ヒドロカルビル基のような短いヒドロカルビル基を含む。
【0071】
リンカー部位はまた、その他の適切な強固な化学結合であっても、または含んでいてもよい、たとえばエステル基、エーテル基、アミン基および/またはアミド基を含む。2つのキレート部位を結合する(1を超える経路がある場合は、もっとも短い経路を数える)原子の総数は通常限定される、キレート部位が複合体の形成のために適切な配置となるように。こうしてリンカー部位は、典型的にキレート部位の間に15原子以下、好ましくは1〜12原子、より好ましくは1〜10原子となるように選択される。リンカー部位が2つのキレート部位に直接的に結合している場合、前記リンカーは長さで1〜12原子、好ましくは2〜10原子(たとえばエチル、プロピル、n−ブチルなど)である。前記リンカーが中心のテンプレートに結合している場合(下記参照)、それぞれのリンカーはキレート部位を結合する2つの分離したリンカーを有し、より短い。1〜8原子の長さ、好ましくは1〜6原子の長さのリンカーが、この場合好ましい(メチル、エチルおよびプロピルが適している、同様にそれらが片方または両端にエステル、エーテルおよびアミド結合を有する基も適している)。
【0072】
リンカー部位は、第1にオクタデンテートリガンドのさまざまなキレート基を互いに、または中心のテンプレートに結合させるように働くが、リンカー部位に加えて、前記オクタデンテートは好ましくは、さらに「カップリング部位」(RC)を含む。前記カップリング部位の機能は、オクタデンテートリガンドを標的部位に結合させることである。これは、共有結合または非共有結合のいずれによっても達成される(たとえば、ビオチン/アビジン(ストレプトアビジン)のような特異の結合によって達成される)。好ましくは、カップリング部位は、キレート基の一つに直接共有結合していることによるか、より典型的にはリンカー部位またはテンプレートに結合することによりキレート基に共有結合で結合している。2以上のカップリング部位が用いられるならば、それぞれは、たとえばどのようなテンプレート、リンカーまたはキレート基でも適切なサイトであればいずれにも結合することができる。
【0073】
一つの態様において、カップリング部位は、下記の構造を有していてもよい。
【0074】
【化2】
【0075】
式中、R7は結合部位であり、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換ヘテロシクロアルキル、置換または非置換アリル、置換または非置換ヘテロアリルからなる群から選ばれ、Xは標的部位または反応性の官能基である。好ましい結合部位は、ここで好ましいリンカー部位として述べた全ての基を含む。好ましい標的部位は、ここで述べた全てのものを含み、好ましい反応性のX基は、標的部位と共有結合を形成可能などのような基も含み、たとえばCOOH、OH、SH、NHRおよびCOH基を含む、ここでNHRのRはHまたはここで述べた短いヒドロカルビル基であればなんでもよい。標的部位への結合のための非常に好ましい基は、リジン残基のイプシロンアミンおよびシステイン残基のチオール基が挙げられる。好適な反応性のX基の例としては、制限されることなく、N−ヒドロキシスクシミジルエステル、イミドエステル、アクリルハライド、N−マレイミド、アルファ−ハロアセチルおよびイソチオシアネートが挙げられる、ここで、後ろから3つはチオールとの反応に好適である。
【0076】
生じた結合されたオクタデンテートリガンドが安定な金属イオン複合体を形成することができるように、カップリング部位は好ましくは結合される。前記カップリング部位はこうして好ましくは、リンカー、テンプレートまたはキレート部位の複合化をさほど妨げないサイトにおいて結合する。そのようなサイトは、好ましくはリンカーまたはテンプレート上にあり、より好ましくは標的に結合した表面から離れた位置にある。
【0077】
好ましいキレート基は、下記の式IIで表わされる基を含む。
【0078】
【化3】
【0079】
上記式IIにおいて、=O部位は、ピリジン環のどのような炭素に結合したものであってもよいケト基を表わし、−OHは、ピリジン環のどのような原子に結合したものであってもよいヒドロキシ部位を表わし、−RLは、オクタデンテートリガンド全体を形成するために、ヒドロキシピリジノン部位を他の複合化部位に結合するリンカー部位を表わす。ここで述べるどのようなリンカー部位もRLとして適切であり、短いヒドロカルビル基、たとえば全てのトポロジーのメチル基、エチル基、プロピル基、ブチル基、ペンチル基および/またはヘキシル基を含むたとえばC1〜C8アルキル基、アルケニル基、アルキニル基を含むC1〜C8ヒドロカルビル基、を含む。RLは、ピリジン環のどのような原子、たとえば炭素または窒素原子において式IIの環に結合していてもよい。前記RL基は、同様に他のキレート部位、他のリンカー基および/または中心の原子または基、たとえば環または他のテンプレート(ここで述べたように)に直接結合していてもよい。リンカー、キレート基および任意のテンプレート部位は、適切なオクタデンテートリガンドを形成するために選択される。
【0080】
1つの好ましい態様において、式IIの前記−OHおよび=O部位は、ピリジン環の隣接する原子に存在する、たとえば1,2−;2,3−,3,2−;4,3−および3,4−ヒドロキシピリジノン誘導体がすべてにおいて非常に好適である。
【0081】
ここで、前記OH基またはリンカー部位RLはピリジン環の窒素の上に存在する場合、RNは通常存在しない。しかしながら、存在する場合は、RNは、置換または非置換のヒドロカルビル基、特にここで述べた短いヒドロカルビル基を含むどのような好適な部位であってもよい。
【0082】
1つの好適な態様においては、少なくとも1つの3,2−ヒドロキシピリジノン部位はオクタデンテートリガンド構造中に存在する。これは明らかに、ここで述べたさまざまな置換部位であればどのようなものによっても置換されていてもよい。
【0083】
式IIのそれぞれの部位は、2つの滞在的に複合化した酸素を有する、本発明の1つの好ましい態様は、オクタデンテートリガンドが少なくとも2、好ましくは少なくとも3そして最も好ましくは4の独立に選ばれた式Iの部位を有する。式IIのそれぞれの部位は、独立した置換形式を有する、しかしながら、1つの好ましい態様においては、少なくとも1つの部位が3,2−ヒドロキシピリジノン部位である。前記リガンドは、2、3または4つの3,2−ヒドロキシピリジノン部位(適切に置換されたもの、ここで述べたように)を含んでいてもよい。
【0084】
オクタデンテートリガンドの式IまたはIIのそれぞれの部位は、ここで述べたどのような適切なリンカー基のどのような適切なトポロジーによって、前記リガンドの残りに結合されていてもよい。たとえば、式Iの4つの基は鎖状のリガンドを形成することができるように、そのリンカー基によって骨格に結合されていてもよい。または「オリゴマー」タイプの構造を形成するためにリンカー基によって橋かけされていてもよく、それは鎖状であっても、環状であってもよい。その代わりに、式IまたはIIのリガンド部位は、「交差」または「星状」トポグラフィの態様で、それぞれがリンカー(たとえば「RL」部位)によって、中心の原子または基に結合されていてもよい。リンカー(RL)部位は、単独で、炭素−炭素結合を介して結合していてもよい、または互いに、キレート基に、骨格に、テンプレート基に、カップリング部位に、または他のリンカーに、適切な強い基の官能基、たとえばアミン、アミド、エステル、チオエーテルまたはジスルフィド結合によって結合していてもよい。
【0085】
「星状の」配置は、下記式IIIに表わされる。
【0086】
【化4】
【0087】
ここで、すべての置換基および位置は上述の通りであり、「T」は、さらに中心の原子またはテンプレート基、たとえば、炭素原子、ヒドロカルビル鎖(上記で述べたものはどのようなものでもよい)、脂肪族または芳香環(ヘテロ環を含む)または縮合環系である。もっとも基本的なテンプレートは、1炭素であろう、それはその結合基によってキレート部位のそれぞれに結合するであろう、長い鎖、たとえば、エチルまたはプロピルは、テンプレートのいずれの端にも結合して、2つのキレート部位とともに等しく存在することができる。明らかに、どのような適した強い結合、それは炭素−炭素結合、エステル、エーテル、アミン、アミド、エステル、チオエーテルまたはジスルフィド結合を含む、をテンプレートおよびリンカー部位を結合するために用いてもよい。
【0088】
式IIIにおいて、RNおよびRLは同じ基であり、そうしてキレート部位がテンプレートTにピリジン環の窒素原子を介して結合することができる。これは下記のIIIb型の構造を提供する:
【0089】
【化5】
【0090】
明らかに、式II,III,IIIb,IVおよびIVbの構造において異なる方法では置換されてない(たとえば、リンカーまたはカップリング部位によって)、それらのピリジン環の位置は、適切な態様で、式IにおけるR1〜R5の置換基を有していてもよい。特に、小さいアルキル置換基、たとえばメチル基、エチル基またはプロピル基がどのような位置にあってもよい。
【0091】
前記オクタデンテートリガンドは通常、さらに上記で述べた少なくとも1つのカップリング部位を含む。これは、ここで述べたどのような好適な構造であってもよく、末端が標的部位、特定のバインダー、またはそのような標的部位または特異的なバインダーに結合可能な特定のバインダーまたは官能基であってもよい。
【0092】
カップリング部位は、前記リンカー、テンプレートまたはキレート部位のどのような好適な位置に結合していてもよい、たとえば式III中で述べられたa)、b)および/またはc)の位置が挙げられる。カップリング部位の結合は、どのような強い結合によるものであってもよい、たとえば炭素−炭素結合、エステル、エーテル、アミン、アミド、チオエーテルまたはジスルフィド結合が挙げられる。同様に、標的部位にそのような結合を形成することが可能な基はカップリング部位の官能端に適し、その部位は標的部位に結合している時に、そのような基を終結させる。
【0093】
代わりに、「骨格」タイプの構造は、式IVで表わされる。
【0094】
【化6】
【0095】
ここで、上記式で表わされる全ての置換基および位置は、上記の通りである、および「RB」はさらに骨格部位である、それは典型的に、ここで述べたいずれのリンカーに対しても同じ構造および機能であり、こうして、状況が許す限り、リンカー部位のどのような定義も骨格部位に適用される。好適な骨格部位は、キレート部位がそのリンカー基によって結合するための足場を形成する。通常、3または4つの骨格部位が必要とされる。典型的に、鎖状の骨格には3つであり、環状の骨格には4つである。特に好ましい骨格部位は、ヘテロ原子または官能部位を片端または両端に任意に有する短い炭化水素鎖(たとえば、ここで述べたもの)を含む。アミン基およびアミド基はこの観点から好適である。
【0096】
前記カップリング基は、前記リンカー、骨格またはキレート部位のどのような好適な位置に結合していてもよい、たとえば式IV中で述べられたa)、b)および/またはc’)の位置が挙げられる。カップリング部位の結合は、どのような強い結合によるものであってもよい、たとえば炭素−炭素結合、エステル、エーテル、アミン、アミド、チオエーテルまたはジスルフィド結合が挙げられる。同様に、標的部位にそのような結合を形成することが可能な基はカップリング部位の官能基に適し、その部位は標的部位に結合している時に、そのような基を終結させる。
【0097】
式IIIに関し、式IVの構造において、RNおよびRLは同じ基であって、そのためピリジン環の窒素原子を介してキレート部位が骨格RBに結合する。このことにより、下記タイプIVBの構造となる。
【0098】
【化7】
【0099】
「骨格」タイプのオクタデンテートリガンドであって、2つの1,2および3,2−HOPOキレート部位を有し、骨格にアミドリンカー基で結合するものの例は、下記の式Vで表わされる。
【0100】
【化8】
【0101】
「テンプレート化された」オクタデンテートリガンドであって、エチルアミド基によって、エチルおよびプロピルジアミンに結合している4つの3,2−HOPOキレート部位を有するものを、それぞれ下記式VIおよびVIIに示す。
【0102】
【化9】
【0103】
上記式に表わされるように、3,2−HOPO部位の自由窒素は、メチル基および小さいヒドロカルビル基、たとえばメチルまたはエチルは、この位置が好ましい(式IのR1または式IIのRN)。
【0104】
「テンプレート化された」オクタデンテートリガンドであって、HOPO環における窒素によって、アミンシクランに結合している4つの3,2−HOPOキレート部位を有するものを、下記式VIIIに示す。
【0105】
【化10】
【0106】
上記で述べたように、オクタデンテートリガンドは典型的にリガンドの残りとどのような位置でも結合可能なカップリング部位を含む。カップリング基を終結させる官能部位を有する例示的化合物は、この態様によれば、下記構造IXである。
【0107】
【化11】
【0108】
ここで参照した全ての文献は、参照として組み込む、その中には、Gordon AEVら、プルトニウムおよび他のアクチニドの封鎖薬の合理的設計.Chem.Rev.2003,103,4207−4282,PCT特許出願WO2008/063721 A2およびT.N.Lambertら、Tetrahedron Letters 43(2002)7379−7383が含まれる。
【0109】
本発明は、以下の実施例において表わされるが、特に制限されることはない。実施例に例示された全ての化合物は本発明の好ましい態様である(好ましい中間体および前駆体を含む)そして、文脈に沿う限りどのような観点においても単独で、または組み合わせて用いられることができる。こうして、たとえば、実施例2の化合物2〜4のそれぞれおよびすべて、実施例3の化合物10ならびに実施例4の化合物7が、さまざまなタイプの中で好ましい態様を形成する。
【実施例】
【0110】
以下、実施例に基づいて本発明をさらに具体的に説明するが、本発明はこれらの実施例に限定されるものではない。
【0111】
[実施例1]
<純トリウム−227の単離>
トリウム−227は、アクチニウム−227カウ(cow)から単離される。アクチニウム−227は、ラジウム−226の熱中性子照射をした後、続いてラジウム−227(t1/2=42.2m)のアクチニウム−227への放射線崩壊することにより製造される。トリウム−227は、アクチニウム−227の放射線崩壊混合物の8MのHNO3溶液をアニオン交換クロマトグラフィーを通過させると選択的に保持される。カラムは、内径が2mmであり、長さが30mmであり、AG(登録商標)1−X8樹脂(200−400メッシュ、硝酸塩の形態)を70mg含有するものを用いた。アクチニウム−227、ラジウム−223および娘はカラムから溶出された後、トリウム−227は、12MのHCl溶液でカラムから抽出された。トリウム−227を含む抽出液は、エバポレートして乾燥され、残渣は0.01M HClで再度けん濁された。
【0112】
[実施例2]
<ALG−DD−NCSの合成>
【0113】
【化12】
【0114】
3−ベンゾイルオキシ−1−メチル−4−(2−チオキソチアゾリジン−1−イル)カルボニル−2(1H)−ピリジノン(Raymond,K.; Xu, J.,.US5,624,901に従って合成した)(2.22mmol,0.8g)、トリエチルアミン(0.31ml,2.22mmol)およびDMAP(5mg)を[5−アミノ−6−((2−アミノ−エチル)−{2−[ビス−(2−アミノ−エチル)−アミノ]−エチル}−アミノ)−ヘキシル]−カルバミン酸−t−ブチルエステル(BocLys−H(2,2)amine)(Raymond,K.; Corneillie,T.M.;Xu.J.WO2008/063721に従って合成した)(0.204g,0.505mmol)のジクロロメタン(80ml)溶液に加えた。前記混合物を室温で一晩攪拌し、それからエバポレートして乾燥した。残渣はジクロロメタンに溶解し、フラッシュシリカゲルカラムにかけ、ジクロロメタン中の2−8%メタノール勾配により溶出する。適切なフラクションを集め、エバポレートして乾燥し、ALG−001(1)(〜0.4g)を薄いベージュ色の粘性油として得た。
MS(ESI,pos):m/z 1369[M+H]+,m/z 1391[M+Na]+
ALG−001(1)(280mg,0.205mmol)を氷冷した酢酸(20ml)に溶解し、20% Pd(OH)2および炭触媒(60mg)を加えた、そして、前記混合物を40−45psi、室温で一晩水素添加した。濾過後、ロータリーエバポレーションによりALG−DEBN(2)(〜260mg,痕跡量の酢酸を含む)を赤色の粘性油として得た。
MS(ESI,pos):m/z 1007[M+H]+,m/z 1029[M+Na]+
ALG−DEBN(2)(70mg)を室温で2:1のMeOH/ジクロロメタン(15ml)に溶解した。それから精製された0.5gのAmberlyst−15樹脂を加え、前記混合物を穏やかに一晩攪拌した。前記樹脂はその後濾過により除去し、ヘキサン(10ml)、テトラヒドロフラン(10ml)およびメタノール(10ml)で順に洗浄した。このアミンが結合した樹脂は、4Mのアンモニアメタノール溶液(20ml)に移し、穏やかに50分攪拌した。テトラヒドロフラン(10ml)を加え、すべての脱保護生成物を溶解した。前記樹脂はそれから濾過により除かれ、前記溶液はエバポレートされ、ALG−DEBN−DEBOC(3)(37mg)を得た。
MS(ESI,pos):m/z 909[M+H]+,m/z 931[M+Na]+
40mgのALG−DEBN−DEBOC(3)の6:1イソプロパノール−水(v/v,7ml)懸濁液を1,4−フェニレンジイソチオシアネート(4.1当量)のクロロホルム(2.5ml)溶液と反応させた。試料をMS分析にかけるとき、反応物質は、室温で30分攪拌されていた。予想された質量(1100に相当するピークが観測された。真空下揮発性物質を除去した後、約95mgの明るい茶色の固体が単離された。明るい茶色の固体はアセトニトリルに懸濁し、前記混合物を15分間還流下加熱した。前記混合物は室温まで冷却し、続いて茶色の固体として26mgのALG−DD−NCS(4)を濾過して単離した。
[実施例3]
<対称な3,2−HOPO 含有キレート剤の合成>
【0115】
【化13】
【0116】
水素化ナトリウム(ミネラルオイル中の60%分散液,60.1g,1.5mol,5当量)をフラスコに入れ、テトラヒドロフラン(1L)を加えた。マロン酸ジメチル(172ml,1.5mol,5当量)を1.5時間かけて滴下した。反応混合物の温度は、+10℃以下に保った。反応混合物は、それからテトラヒドロフラン(400ml)で希釈した。4−ニトロベンジルブロミド(65.0g,0.3mol,1当量)のテトラヒドロフラン(170ml)溶液を上記作成した混合物に激しく振りながら30分をかけてゆっくりと加えた。30分0℃で攪拌した後、前記反応混合物は、塩水(1Lの飽和食塩水)に注ぎ、室温で一晩攪拌して、白い沈殿物を得た、前記混合物は、メチルtert−ブチルエーテルで希釈し、前記沈殿物を濾過して温エタノールに溶解した。前記エタノール混合物を濾過して、ろ液を濃縮して体積を少なくし、そこから24.8g(31%)のジメチル(4−ニトロベンジル)プロパンジオネート(1)が白色の結晶状の固体として沈殿した。
LC純度:>90%(254nm)
MS(APCl,pos):m/z 285.1[M+NH4]+
化合物1(24.8g,92.7mmol)をテトラヒドロフラン(230mL)に溶解し、ボラン−ジメチルスルフィド複合体(28.5ml,301ml,3.3当量)に加えた。前記反応混合物を24時間還流し、それから室温で一晩置いた。0℃でメタノール(250ml)を加え、塩水(600ml)に注ぎ、酢酸エチルで抽出した。前記合わせた有機層をNa2SO4の上で乾燥させ、真空下で濃縮した。残渣はメタノールと蒸発した。粗生成物は、フラッシュクロマトグラフィーで精製し、14.5g(74%)の2−(4−ニトロベンジル)プロパン−1,3−ジオール(2)を黄色の油として得た。
LC純度:>92%(254nm)
化合物2(10.0mg,47.3mmol)をジクロロメタン(100ml)に溶解し、トリエチルアミン(14.5ml,104mmol,2当量)を加えた。前記反応混合物は、氷浴で冷やし、メタンスルホニルクロライド(8.0ml,104mmol,2当量)を少しずつ加えた。最終混合物は、室温になるまで一晩置いた。それから、さらなるトリエチルアミン(1.5ml)およびメタンスルホニルクロライド(0.8ml)を加え、30分攪拌した。それから前記反応混合物は、ジクロロメタンに溶解し、生成した沈殿物を濾過して除去した。前記ジクロロメタンのろ液は飽和NaHCO3水溶液、0.5MのHCl水溶液および塩水で洗浄し、Na2SO4の上で乾燥させ、真空下で濃縮し、14.5g(83%)の2−(4−ニトロベンジル)プロパン−1,3−ジイルジメタンスルフォネート(3)をオレンジ色の油として得た。化合物(3)は、さらに精製することなく次の段階で用いた。
LC純度:>87%(254nm)
MS(APClpos):m/z 385.3[M+NH4]+
化合物3(14.5g,39.5mmol)は、メチルエチルケトン(100ml)、ヨウ化ナトリウム(16.0g,107mmol,2.7当量)を加え、前記混合物を95℃で1時間加熱した。得られた白色の沈殿物は濾過して除去し、メチルエチルケトンで洗い、前記ろ液は真空下で濃縮した。前記粗生成物はメチルtert−ブチルエーテルから砕いて、フラッシュクロマトグラフィーで精製して、10.6g(63%)の1−[3−ヨード−2−(ヨードメチル)プロピル]−4−ニトロベンゼン(4)を黄色の固体として得た。
LC純度:>90%(254nm)
MS(APClpos):m/z 431.1[M+H]+
ジエチレントリアミン(10.8ml,100mmol)およびトリエチルアミン(42ml,300mmol,3当量)をテトラヒドロフラン(500ml)に溶解し、氷浴で冷やした。それからBoc−ON(49.5g,200mmol,2当量)をテトラヒドロフラン(190ml)で溶解した溶液を2.5時間かけて滴下した。前記反応混合物は、さらなる1時間0℃で攪拌し、その後室温になるまで一晩置いた。それから前記反応混合物は、真空下濃縮した。残渣はジクロロメタンに溶解し、1MのNaOH水溶液で洗った。前記有機層はNa2SO4の上で乾燥させ、真空下で濃縮した。粗生成物はフラッシュクロマトグラフィーで精製し、20.0g(66%)のジ−tert−ブチル(イミノジエタン−2,1−ジイル)ビスカーバメート(5)を黄色の粘性のある油として得た。
MS(APClpos):m/z 304.3[M+H]+
化合物5(26.0g,86mmol,4当量)をドライトルエン(40ml)に溶解し、N−エチルモルフォリン(5.4ml,42mmol,2当量)を加えた。化合物4(9.2g,21mmol)をドライトルエン(35ml)に溶解し、前記反応混合物に加えた。前記混合物は、圧力反応器で105℃で5日間保温した。それから、前記反応混合物は、室温に冷却し、酢酸エチルで希釈し、飽和NaHCO3水溶液で洗浄した。前記合わせた有機層を水と塩水で洗い、Na2SO4の上で乾燥させ、真空下で濃縮した。前記粗生成物はフラッシュクロマトグラフィーで精製し、11.4g(70%)の化合物6を黄色の堅い泡として得た。
LC純度:>96%(210nm)
MS(APCl,pos)m/z 782.8,[M+H]+;682.7[M+H−Boc]+;582.6[M+H−2Boc]+;382.1[M+H−4Boc]+
化合物6(2.0g,2.6mmol)をジオキサン(16ml)に溶解し、9MのHCLのジオキサン(22ml)溶液を加えた。前記反応混合物は、室温で一晩攪拌し、真空下で乾燥した状態まで濃縮し、1.9gの化合物7のHCL塩をベージュ色の固体として得た。
MS(APClpos)m/z 382.5[M+H]+
化合物7(1.9g,2.6mmol)をDMPU(1,3−ジメチル−3,4,5,6−テトラヒドロ−2(1H)−ピリミジノン)(9.5ml)および3−(ベンゾイルオキシ)−1−メチル−4[(2−チオキソ−1,3−チアゾリジン−3−イル)カルボニル]ピリジン−2−(1H)−オン(3.7g,10.3mmol,4当量)に加えた。それから、DBU(2.3ml,15.5mmol,6当量)をDMPU(4.7ml)に溶解した溶液をゆっくり40分間で滴下した。一晩室温で攪拌を続けた。前記反応混合物はジクロロメタンで希釈して飽和NaHCO3水溶液で洗浄した。前記合わせた有機層を水と半飽和塩水で洗い、Na2SO4の上で乾燥させ、真空下で濃縮した。前記粗生成物はフラッシュクロマトグラフィーで精製して、4.0gの化合物8を黄色の油として得た。化合物8は更なる精製をすることなく次の段階で用いた。
MS(APCl,pos)m/z 1347.7[M+H]+
化合物8(4.0g,2.6mmol)を酢酸(200ml)に溶解し、Pd(OH)2(炭素上20% w/w, 50%湿潤)(800mg,10% w/w)を加えた。前記反応混合物は、Parr圧力反応器で30barの水素圧、室温で一晩攪拌した。前記反応混合物は、Celite(登録商標)のパッドを通過させて濾過し、ろ液を真空下で濃縮した。粗生成物はフラッシュクロマトグラフィーで精製し、化合物9をベージュの泡状の固体として得た。
【0117】
化合物9(996mg)を酢酸(5.5ml)に溶解し、続いて6MのHCl水溶液および5%のPd/C(99mg,10% w/w)を加えた。前記反応混合物は、圧力反応器で8barの水素圧で一晩攪拌し、それからCelite(登録商標)のパッドを通過させて濾過した。前記ろ液は真空下で濃縮し、トルエンおよびメタノールとともにエバポレートした。前記残渣はメタノールに溶解し、ジエチルエーテルを加えて沈殿させた。形成したベージュの沈殿物は濾過して除き、真空下で一晩乾燥し、700mgの化合物10のHCL塩である、ALG1005−38を得た。
MS(APCl pos)m/z 956.7[M+H]+
[実施例4]
<別の対称な3,2−HOPO 含有キレート剤の合成>
【0118】
【化14】
【0119】
無水Boc(24.4g,119mmol)をイミダゾール(8.0g,117mmol)の50mlジクロロメタン溶液に少しずつ加えた。前記反応混合物を1時間かけて攪拌した。前記反応混合物を50mlの水で2回洗浄し、Na2SO4の上で乾燥させ、濾過し真空下で濃縮した。前記残渣は、25mlのトルエンおよびトリス(2−アミノエチル)アミン(8.19g,56mmol)を加えた。前記反応混合物は、60℃で2時間攪拌して、真空下で容量を減らした。前記残渣は125mlのジクロロメタンに溶解し、水で洗い(50mlで4回)、Na2SO4の上で乾燥させ、濾過し真空下で濃縮した。ドライフラッシュクロマトグラフィー(1%のトリエチルアミンを加えた0〜15%のメタノールのジクロロメタン溶液)をして、ジ−tert−ブチル(((2−アミノエチル)アザンジイル)ビス(エタン−2,1−ジイル))ジカーバメート(1)(13.51g,39.0mmol,70%)を薄い色のない油として得た。
【0120】
分光データは、Frullanoらによって報告されたデータ(Chem.Eur.J.2004,10,5205−17)に従う。
【0121】
N−Cbz−β−アラニン(8.04g,36mmol)、4−(ジメチルアミノ)ピリジン(4.40g,36mmol)およびEDC・HCL(6.90g,36mmol)を25mlのテトラヒドロフランに溶解し、10分間攪拌した後、化合物1(10.4g,30mmol)を75mlのテトラヒドロフランに溶解したものを加えた、前記反応混合物は、4時間攪拌し、真空下で容量を減らした。ドライフラッシュクロマトグラフィー(0〜40%のテトラヒドロフランのジクロロメタン溶液)により精製し、ジ−tert−ブチル(((2−(3−(Cbz−アミノ)プロパンアミド)エチル)アザンジイル)ビス(エタン−2,1−ジイル))ジカーバメート(2)(15.11g,27.4mmol,91%)を非常に粘性の高いわずかに黄色の油とした得た。
1H−NMR(300MHz,CDCl3):1.43(s,18H),2.42−2.56(m,8H),3.04−3.17(m,4H),3.19−3.32(m,2H),3.41−3.57(m,2H),5.08(s,2H),5.22(bs,2H),5.83(bs,1H),7.23(bs,1H),7.26−7.43(m,5H)
MS(ESL,pos)m/z 574.3[M+Na]+
ジ−tert−ブチル(((2−(3−(Cbz−アミノ)プロパンアミド)エチル)アザンジイル)ビス(エタン−2,1−ジイル)ジカーバメート(10.30g,18.7mmol)を100mlのメタノールに溶解し、アセチルクロライド(25ml,0.35mol)を滴下した。前記反応混合物は、室温で1時間攪拌した。前記反応混合物は、真空下でおおよそ1/3の体積にしてから、100mlのエーテルを加え、無色の固体を沈殿させた。前記固体は濾過し、エーテルで洗い、真空下で乾燥し、ベンジル(3−((2−(ビス(2−アミノエチル)アミノ)エチル)アミノ)−3−オキソプロピル)カーバメートトリヒドロクロライド(3)(8.70g,18.7mmol,〜100%)を無色の固体として得た。
1H−NMR(300MHz,MeOD):3.19−3.28(m,2H),3.38−3.62(m,14H),5.10(s,2H),7.29−7.39(m,5H)
MS(ESL,pos)m/z 352.2[M+H]+
化合物3(10.56g,11.4mmol)の320mlのテトラヒドロフランの懸濁液および320mlのN,N−ジメチルホルムアミドにトリエチルアミン(6.75ml,48.4mmol)を室温で加えた。N−boc−2−アミノアセトアルデヒド(16.0g,100.5mmol)およびトリアセトキシボロヒドリドナトリウム(32.00g,151mmol)を加えた。前記反応混合物は、20時間攪拌した。200mlの塩水および500mlのクロロホルムを加え、相を分離し、前記水層を100mlのクロロホルムで3回抽出した。前記合わせた有機層は100mlの食塩水で洗い、Na2SO4の上で乾燥させ、濾過し、真空下で体積を減らした。フラッシュクロマトグラフィー(0〜10%のメタノールのジクロロメタン溶液)により、テトラ−tert−ブチル(((((2−3−Cbz−アミノプロパンアミド)エチル)アザンジイル)ビス(エタン−2,1−ジイル))ビス(アザントリイル)テトラキス(エタン−2,1−ジイル)テトラカーバメート(4)(3.40g,3.7mmol,32%)を黄色の固体とした得た。
MS(ESL,pos):m/z 946.7[M+Na]+
化合物4(3.40g,3.7mmol)を60mlのメタノールに溶解し、アセチルクロライド(12ml,0.17mol)を滴下した。前記反応混合物を室温で1時間攪拌した。前記反応混合物を、真空下でおおよそ10mlの体積にしてから、50mlのアクリロニトリルを加え、沈殿させた。前記固体は濾過し、アクリロニトリルで洗い、真空下で乾燥し、ベンジル(3−((2−(ビス(2−アミノエチル)アミノ)エチル)アミノ)−3−オキソプロピル)カーバメートヘプタヒドロクロライド(5)(2.88g,3.7mmol,〜100%)を無色の固体として得た。
1H−NMR(300MHz,MeOD):2.44−2.61(m,2H),2.68−3.00(m,4H),3.08−3.40(m,16H),3.44−3.52(m,4H),3.54−3.77(m,6H),5.12(s,2H),7.30−7.47(m,5H)
MS(ESL,pos):m/z 524.5[M+H]+
トリエチルアミン(2.52ml,18.1mmol)および4−(ジメチルアミノ)ピリジン(12mg,触媒)を化合物5(0.81g,1.04mmol)および3−(ベンジルオキシ)−1−メチル−4−(2−チオキソチアゾリジン−3−カルボニル)ピリジン−2(1H)−オン(1.50g,4.16mmol)の180mlのジクロロメタンの懸濁液に加えた。前記反応混合物は一晩攪拌し、真空下で体積を減らした。フラッシュクロマトグラフィー(0〜10%のメタノールのジクロロメタン溶液)により、ベンジル(1−(3−ベンジルオキシ)−1−メチル−2−オキソ−1,2−ジヒドロピリジン−4−イル)−5−(2−(3−(ベンジルオキシ)−1−メチル−2−オキソ−1,2−ジヒドロピリジン−4−カルボキサミド)エチル)−8−(2−(ビス(2−(3−ベンジルオキシ)−1−メチル−2−オキソ−1,2−ジヒドロピリジン−4−カルボキシアミド)エチル)アミノ)エチル)−1,12−ジオキソ−2,5,8,11−テトラアザテトラデカン−14−イル)カーバメート(6)(673mg,0.45mmol,43%)を明るい茶色の固体として得た。
【0122】
1H−NMR(300MHz,CDCl3):2.10−2.40(m,20H),3.02−3.21(m,10H),3.32−3.46(m,2H),3.50(s,12H),4.99(s,2H),5.25(s,8H),5.95(bs,1H),6.62(d,7.17Hz,4H),7.01(d,7.17Hz,4H),7.15−7.40(m,25H),7.89(bs,4H),
13C−NMR(75MHz,CDCl3):11.48,35.71,37.18,37.35,37.50,37.66,46.18,51.55,52.15,52.82,53.28,66.34,74.64,74.75,77.43,104.63,127.90,127.94,128.40,128.65,128.75,128.94,130.81,132.32,136.27,136.76,146.19,156.45,159.51,163.30,171.31
MS(ESL,pos):m/z 766.9[M+2Na]2+
化合物6は、基本的に実施例2に記載されているように、溶解され、脱ベンジル化し、Bb−1−HOPO−1−DEBN(7)を得た。
[実施例5]
<ALG−DD−NCSとのキレート化実験>
固体のALG−DD−NCS(実施例2の4)をDMSO(バイオテックグレード溶媒99.8%)に1mg/mlとなるように溶かし、反応溶液を調製した。
【0123】
異なる金属とのキレート化反応は1mg/mlの反応溶液を選択された金属溶液と混合し、リガンド対金属が3:2とすることにより行った。試験のための金属イオンは以下のようにして得た:232Thの2%HNO3(パーキンエルマーピュアプラス)溶液、Fe3+の2%HNO3(パーキンエルマーピュアプラス)溶液およびInCl3(無水粉末99.999+%,アルドリッチ)を0.01MのHClに溶解した。前記試薬は室温で15分反応させてから、5μmの反応混合物をLC−MSに入れ分析した。
【0124】
結果物は、金属−ALG−DD−NCS錯体が232Th4+、Fe3+およびIn3+の金属に対して定量的に形成された。232Th−ALG−DD−NCS、Fe−ALG−DD−NCSおよびIn−−ALG−DD−NCS錯体のLC−MSクロマトグラムおよびスペクトルを図1−6に示す。
【0125】
前記LC−MSの条件は以下の通りである:分離は1.7μm、2.1×50mmのAcquity UPLC BEH C18カラム、50℃、0.05%のギ酸水溶液を移動相Aおよび0.05%のギ酸のアクリルニトリル溶液を移動相Bとして用いた。前記傾斜組成および流量を表3に示す。
【0126】
【表3】
【0127】
マススペクトル分析は、Xevo−ToF−1で、取得質量範囲は〜490−1950Da、質量ES+、3.0kVキャピラリー、コーン電圧20Vで1000秒以上スキャンした。
[実施例6]
<ALG10058−38とのキレート化実験>
ALG1005−38(実施例3の10)をメタノール(LC−MSグレード)に溶解して10mg/mlとして原液を調製した。前記10mg/ml原液は、メタノールおよび0.5MのNaOAc−緩衝液(pH5.5)で1:8:1の割合で希釈し、1mg/mlのALG1005−38反応溶液を得た。異なる金属とのキレート化反応は実施例5と同様に1mg/mlの反応溶液を選択された金属溶液と混合し、さらにGaCl3(無水ビーズ 99.99%、アルドリッチ)を0.01M Hclに溶解したものを加え、リガンド対金属が3:1〜1:2とした。前記試薬は室温で10分反応させた。最後に前記反応混合物は10倍のギ酸(0.1%)で希釈してから、5μmの反応混合物をLC−MSに入れ分析した。前記LC−MS分離および分析は、コーン電圧を35Vとして以外は、実施例5で述べたように行った。
【0128】
結果物は、金属−ALG1005−38錯体が232Th4+、Fe3+、In3+およびGa3+の金属に対してかなりの量で形成された。232Th−ALG1005−38、Fe−ALG1005−38、In−ALG1005−38およびGa−ALG1005−38錯体のLC−MSクロマトグラムおよびスペクトルを図7−14に示す。
[実施例7]
<Bb−1−HOPO−1−DEBNとのキレート化実験>
Bb−1−HOPO−1−DEBN(実施例4で製造したもの)をDMSO(バイオテックグレード溶媒99.8%)に溶解して1mg/mlとして原液を調製した。キレート化実験は前記1mg/mlの溶液を実施例5で述べた232Th0.01M HCl(パーキンエルマー)溶液と混合することにより行った。前記LC−MS分離および分析は、実施例6で述べたように行った。
【0129】
結果物は、予想された232Th−Bb−1−HOPO−1−DEBN複合体がすばやく形成された、図15および16に示す。
[実施例8]
<ALG−DD−NCSのtrastuzumabへの接合、トリウム−227で接合の標識化を行い、標的の結合が保持されていることを確認した。>
接合
trastuzumabのモノクローナル抗体の薬剤グレード(Herceptin(登録商標),ロッシェ)を用いた。前記抗体は、0.9%のNaClに緩衝液交換をして6.3mg/mlに濃縮した。ALG−DD−NCSの原液はDMSOに溶解して1mg/mlに調製した(純度は考慮しない)。DMSO原液は、キレーター対抗体のモル比が6.5:1以下に相当する抗体溶液に加えた。0.07Mのホウ砂溶液を反応溶液に加え、pH9にし、一晩37℃で培養した。得られた接合を精製し、Amicon Ultra−4(30k MWCO)遠心分離フィルターユニットで緩衝液交換をした。1mgを100μlの0.9%のNaClに分注し、凍らせ、使用するまで−18℃で保管した。
標識化
100μlの0.5MのNaOAc−緩衝液(pH5.5)を1mgのALG−DD−NCS−trastuzumab接合の小瓶に加え標識化に適切なpHとした。この溶液を0.01M HCl中の4.2−16.4MBqの227Thのと混合し、そのpHをpH紙で確認した。エッペンチューブキャップをアルミニウムホイルでくるみ、前記チューブを室温でサーモミキサー中に置き15分穏やかにかき混ぜた。
【0130】
前記チューブは10μlの飽和DTPAのMF−H2Oに加えた後、さらに室温で5分間培養し、保持されている自由トリウム−227をとらえた。
PBSを用いて標識化されたALG−DD−NCS−trastuzumab接合を希釈して、結果物をNAP−5カラムで精製した、そして自由放射性核種(トリウム−227および娘核種)およびDTPA−トリウムキレートをの大多数はカラムに残されていた。
【0131】
前記カラムおよび希釈されたフラクションはHPGe−検出器 GEM(15)で測定して、生成物の反応収率および特定の活性を測定した。前記生成物のフラクションは濾過され4℃で一晩保管した。
前記生成物フラクションの第2のNAP−5カラムによる精製を使用前に行った。
免疫反応性フラクションの測定
SK−OV−3細胞(ATCC)を標準状態で培養した。固定されたSK−OV−3細胞は、調製され、使用前にフローサイトメトリーにより分析されてその表面にHER2が存在することを確認した(データは開示しない)。正規の培地は、McCoyの5A培地(GIBCO)に10%FBS(PAA)および1%Pen/Strep(BioChrom)を加えて調製した。細胞の培養は、T25(25cm2)およびT75(75cm2)細胞フラスコを用いて、37℃および5%CO2でCO2培養器で行った。
【0132】
それぞれの実験は2回行った。固定したSK−OV−3細胞はPBSに懸濁し、エッペンチューブに移した(10000,000細胞/チューブ)。非接合trastuzumab(Herceptin(登録商標),原液から10μl)を2つの参照チューブに加え、37℃で30分間、阻害反応を行った。同量の227Th−ALG−DD−NCS−trastuzumab(ca 500cpm)をそれぞれのチューブに加えてから、37℃で25時間培養した。前記試料はPBSで希釈してから、遠心分離して上澄みの半分を新しいエッペンチューブに移した。すべてのチューブは、ウィザードガンマカウンターで5分間測定し、トリウム−227の量を227Th−プロトコルを適用して測定した。
【0133】
結合の%は、以下のように計算した:100*[(P+1/2S)−(1/2S)][(P+1/2S)+(1/2S)]、ここでPおよびSは、それぞれ前記細胞ペレットおよび前記上澄み中で測定された活性を示す。前記計算は、全体の結合(%)を求めるために、非阻害試料に関して行い、非特異結合(%)を求めるために、阻害試料に関し行った。そしてIRFは以下のように計算した:IRF(%)=全体の結合(%)−非特異結合(%)
この品質管理からの前記データは、表4にまとめ、227Th−ALG−DD−NCS−trastuzumabが標識細胞に結合していることを表わしている。
【0134】
【表4】
【0135】
i. 収率(%)は、反応混合物のNAP−5精製の後計算した。
ii. 回収(%)は、生成物フラクションの新たなNAP−5精製の後計算した。
iii. 特異活性(Bq/μg)は、生成物フラクション中に見つけられた227Th(Bq)の量を反応混合物中に用いられたALG−DD−NCS−trastuzumab(μg)の量に基づいて割ったものを用いて計算した。
iv. IRFは標準の手順に従って見積もった。
[実施例9]
<トリウム−227で標識化されたALG−DD−NCS−trastuzumabのin vitro安定性および有効性の研究>
時間をかけた結合の評価
227Th−ALG−DD−NCS−trastuzumabのSK−OV−3細胞への結合の安定性は、SK−OV−3細胞ペレットと関連づけられた227Th−活性の量を7日を超えて測定して評価した。前記結果は自由227Thとともに培養したデータと比較した。
【0136】
実施例8で述べたように培養したSK−OV−3細胞は最初に700kBqの227Th−ALG−DD−NCS−trastuzumabまたは700kBqの自由227Thで扱い、1時間培養した後(HPGe−検出器 GEM(50))細胞ペレット上に、それぞれ77kBq(11%)および7kBq(1%)227Th−活性を生じた。前記SK−OV−3細胞上の227Th−活性は、毎日ウィザードガンマカウンター(227Th−プロトコル)で測定した。前記測定した放射能は、崩壊に対する補正をした。前記結果は図17に示すように1時間後にペレットに残されたたった20%の活性が、次の7日間で消滅した、それに対して、自由227Thで扱った細胞は加えられた活性の小さなフラクションにのみ結合し、その多くが(ca95%)続く24時間の間に消失した。こうして、自由227ThのSK−OV−3細胞への特異結合はなく、一方で227Th−ALG−DD−NCS−trastuzumabは効果的に保持された。
細胞毒性の評価
腫瘍細胞の成長の効果を2つの相補的な分析で研究した。最初の実験で、細胞中のメタボリック活性はATP分子に結合した発光試薬を用いて測定した。このようにして、発光シグナルの減少は、メタボリック活性の消失を示す。第2の実験で、形成された多くのコロニーの数が数えられた。
【0137】
発光は、毎日9日間測定した、そしてその結果227Th−ALG−DD−NCS−trastuzumabへの被爆からの結果をtrastuzumabおよび自由227Thのそれぞれへ被爆したSK−OV−3細胞の発光に比較した。SK−OV−3細胞の正規の培地での成長は、参照として用いた。
0日目
正規の培地中の35mlの500000SK−OV−3細胞/mlを4 T75細胞フラスコに移した。これらの細胞フラスコは以下の試料溶液を調製するために用いた:(1)正規の培地、(2)20μg/mlのtrastuzumab、(3)20kBq/mlの227Th−ALG−DD−NCS−trastuzumab(10166Bq/μg)、(4)20kBq/mlの自由227Th。前記フラスコは、CO2培養器で1時間培養した。前記上澄みを捨て、前記細胞を0.25%のトリプシン−EDTA溶液でトリプシン処理し、正規の媒体で洗い遠心分離した。細胞ペレットは、新鮮な正規の媒体に懸濁し、ウィザードガンマカウンター(227Th−プロトコル)で測定した。それぞれの培養溶液は、新しい7つのT25細胞フラスコに分配し、CO2培養器で培養した。
1日目および2日目
4つの試料溶液のそれぞれの1つのフラスコからの前記上澄みは、捨てた。前記細胞ペレットは、0日目と同様に調製して、放射能はウィザードガンマカウンター(227Th−プロトコル)およびHPGe−検出器 GEM(50)で測定した。全ての試料は、正規の媒体で4倍に希釈し、それぞれの懸濁出来の5×100μlを5wellのView Plate−96に移した。5つのwellは100μmの正規の媒体(細胞を含まない)を加え、背景発光の測定をした。最後に、100μLのCell Titer−Glo 試薬をそれぞれのwellに加え、前記溶液は溶解を誘導するためにオービタルシェーカーで混合した。前記発光シグナルは、室温で10分間安定させた、それから発光は、LUM−シングルプログラムを用いて、エンビジョンマルチプルプレートリーダーで3回測定した。
3日目
細胞は、1日目および2日目で述べたように調製して、ウィザードガンマカウンターおよびエンビジョンマルチプルプレートリーダーで測定した。それから、細胞の成長で濃くなっているため、それぞれの細胞の懸濁液を希釈した;3/4の細胞の懸濁液は新しいT75細胞フラスコに移し、正規の媒体を加え、CO2培養器で一晩培養した。
4〜9日目のそれぞれ
4つのT75細胞フラスコを0.25%のトリプシン−EDTA溶液でトリプシン処理し、正規の媒体で洗浄し、遠心分離した。細胞ペレットは4倍の正規の媒体で希釈し、100μlを取り出し、上述のように測定した。それから3/4の細胞の懸濁液は正規の媒体で希釈し、新しいT75細胞フラスコに移し、CO2培養器で一晩培養した。
【0138】
図18は、227Th−ALG−DD−NCS−trastuzumab(10166Bq/μg)、trastuzumabおよび自由227Thで処理したSK−OV−3細胞の発光測定を示す、それは正規の媒体(参照)中で育てられたSK−OV−3細胞について得られた発光値に標準化されたものである。前記227Th−ALG−DD−NCS−trastuzumabで処理したSK−OV−3細胞の発光シグナルは、1日目〜9日目にかけて徐々に減少し、一方他の処理をしたものは、この期間の間ほぼ一定の発光シグナルと成長を示した。227Th−ALG−DD−NCS−trastuzumabで処理したSK−OV−3細胞のメタボリック活性は、その期間の間減少し、9日目にはほとんど全ての細胞が死滅した、この結果は、227Th−ALG−DD−NCS−trastuzumabの重要な維持された細胞毒性を示す。
【0139】
一つの細胞を滞在的に細胞毒性の状況にさらした後のコロニーの成長を評価するために、クローン形成を行ったSK−OV−3細胞は、異なる濃度の227Th−ALG−DD−NCS−trastuzumab、trastuzumabまたは自由227Th中で1時間培養し、続いてコロニーの成長を誘導するために12日間培養した。
【0140】
細胞の調製は、上記のようにして行った。そして試料ごとに5mlの正規の媒体中の12000細胞を、T25フラスコに移動した。227Th−ALG−DD−NCS−trastuzumabおよび227Thの5、10および20kBq/mlならびにtrastuzumabの5、10および20μg/ml(最終濃度)のそれぞれについて、それぞれの試薬の3種の量の効果を調べた。1つの参照フラスコを調製した、全ての10のT25細胞フラスコは、CO2培養器で一時間培養した。前記上澄みは捨て、前記細胞は正規の媒体で洗浄し、0.25%のトリプシン−EDTA溶液でトリプシン処理し、正規の媒体で洗浄し、遠心分離した。細胞ペレットは正規の媒体に懸濁し、10の細胞の懸濁液のそれぞれを、6つの新しいT25細胞フラスコに分けた、3つの細胞フラスコは1000細胞であり3つの細胞フラスコは3000細胞である、えられた60のT25細胞フラスコは、CO2培養器でコロニーが目に見える大きさになるまで培養した(ca 50細胞/コロニー)。12日間培養した後、前記媒体は除去した、細胞はPBSを用いて洗い、エタノールで還流し、PBS中の0.25%トリパンブルーで染め、水道水で洗い、最後に45℃で一晩乾燥させた。コロニーは手作業で数え、それぞれの培養におけるコロニーの平均数は、グラフに表した。
【0141】
図19および20に示された結果は、227Th−ALG−DD−NCS−trastuzumabを用いた1時間の培養は、効果的にSK−OV−3細胞を殺すことを示している。自由227Thを用いた培養は、いくらかの細胞を殺す効果があったが、たくさんの数の細胞を用いた最初の細胞毒性分析では明らかでなかった。trastuzumabを用いた培養は、細胞の成長に否定的な効果はなかった。
[実施例10]
<トリウム−227で標識化されたALG−DD−NCS−trastuzumabのin vivo腫瘍標的>
100μl(15kBq)の細菌濾過した227Th−ALG−DD−NCS−trastuzumabをSK−OV−3の異種移植片を有する10匹のBalb/cヌードマウスの尾の側面の血管に注入した。5匹のマウスは24時間後に犠牲となり、残りの5匹は4日後に犠牲となった。そして実験対象のマウスの組織を切除して重量を測った。全ての試料および10% ID/gを含む3つの標準溶液は5分間ウィザードガンマカウンター(227Th−プロトコル)で測定した。結果はグラム組織(% ID/g)当たりの注入量の%で表わした。
【0142】
前記結果は図21に示すように、227Th−ALG−DD−NCS−trastuzumabは、特異的に腫瘍に結合する。取り込み値は、適度であるが、24時間と4日の間にいくらか上昇している(8.7% ID/g〜11.3% ID/g)、一方で血液中の濃度は1%ID/gあたりに減っている。
【0143】
頭骸骨および大腿骨における取り込み量もまた、いくらか24時間および4日の間に上昇した、しかしながら、これらの値はほとんどの場合低く(2.8−3.2%、24時間)、系中を循環する自由227Thはほとんどない、そして227Th−ALG−DD−NCS−trastuzumab複合体の安定性の高い度合いを示している。さらにこの結論をサポートするものとして、頭骸骨および大腿骨において測定される活性フラクションは223Raの娘核種を取り込むことにより生じるという高い可能性がある。この223Raのいくらかは、ウィザードガンマカウンターの227Th−測定器のための窓の中に検出されることもある。こうして頭骸骨および大腿骨の計算された% ID/g−値は、これらの組織に蓄積した227Th−ALG−DD−NCS−trastuzumabの量を課題評価することもある。
[実施例11]
<キレート部位の合成>
3−ベンジルオキシ−1−メチル−4−(2−チオキソチアゾリジン−1−イル)カルボニル−2(1H)−ピリジノン(構造A)
ステップ1)−1−メチル−3−ヒドロキシ−2(1H)−ピリジノン
3−ヒドロキシ−2(1H)−ピリジノン(34.44g,0.31mol)およびヨードメタン(75g、0.53mol)を80mlの蓋をしたテフロン(登録商標)の入れ物に入れた。そして、Parr bomb中で48−60時間150℃で加熱した。冷却した前記bombを開け、過剰なヨードメタンを静かに注いだ。得られた薄い黒みがかった油を亜硫酸ナトリウム(64g,0.5mol)と混合し、300mlの水に溶解し、薄い茶色の溶液を形成した、前記溶液は、pH7−8に中和し、不溶の不純物を濾過して除去した。前記ろ液はそれからメチレンクロライド(4×100ml)。合わせた抽出物は、それから乾燥させ、フラッシュシリカゲルプラグ(6cm×8cm)をし、4%エタノールのメチレンクロライド溶液で希釈した。前記溶媒は、上記標題の化合物(24.3g,62.6%)を無色の結晶として得た。
【0144】
ステップ2)−4−カルボキシル−1−メチル−3−ヒドロキシ−2(1H)−ピリジノン
1−メチル−3−ヒドロキシ−2(1H)−ピリジノン(1)(6.25g,50mmol)を無水炭酸カリウム(36g,0.26mol)と混合し、真空下で乾燥した、前記混合物は、それからParr bomb中で二酸化炭素ガス(850psi)下で175−185℃で3日間加熱した。前記冷却したbombを開け、結果物の薄い黄色の固体を氷で冷やした水に溶解し、6N のHClで酸性にし、ベージュの結晶性の生成物を得た。
【0145】
ステップ3)−3−ベンジルオキシ−4−カルボキシ−1−メチル−2(1H)−ピリジノン
4−カルボキシル−1−メチル−3−ヒドロキシ−2(1H)−ピリジノン(6.8g,0.04mol)をベンジルクロライド(12.1g,0.088mol)および無水ジメチル−ホルムアミド(DMF)(120ml)中の無水炭酸カリウム(13.8g,0.1mol)と混合した。前記混合物は、暗闇で、窒素の存在下、75−80℃で16時間加熱した。前記混合物は、濾過し、溶媒をエバポレートして暗い色の油を得た。前記油は、シリカゲルプラグ(6cm×8cm)にかけ、4%メタノールのメチレンクロライド溶液で希釈し、3−ベンジルオキシ−4−ベンジルオキシカルボニル−1−メチル−2(1H)−ピリジノンを薄い黄色の、粘度の高い油として得た。これをメタノール(50ml)および6MのNaOH溶液(10ml)に採り、前記混合物を室温で4時間攪拌し、エバポレートして乾燥させた。前記残渣は水(100ml)に溶解し、6M HCl溶液で酸性にしてpH2とし、上記標題の化合物(9.3g,88.7%)を白色の結晶として得た。
【0146】
ステップ4)−3−ベンジルオキシ−1−メチル−4−(2−チオキソチアゾリジン−1−イル)カルボニル−2(1H)−ピリジノン
3−ベンジルオキシ−4−カルボキシ−1−メチル−2(1H)−ピリジノン(1.05g,4mmol),2−メルカプトチアゾリン(0.50g,4.2mmol)および触媒量の4−ジメチルアミノピリジン(DMAP)のドライジクロロメタン(50ml)溶液に、N,N’−ジシクロヘキシルカルボジイミド(DCC)(0.86g,4.2mmol)を加えた。4時間攪拌後、ジシクロヘキシルウレア(DCU)の固体を濾過により除去した。黄色のろ液をロータリーエバポレートして黄色の固体を得た。イソプロパノールメチレンクロライドからの再結晶により、標題の化合物(構造A,1.16g,80.4%)を明るい黄色の結晶板として得た。
【0147】
【化15】
【0148】
[実施例12]
<式VIIIで表わされる3,2−HOPO前駆体の生成>
環状塩(構造5)の生成は、下記式にしたがって行った。
【0149】
【化16】
【0150】
アクリルニトリルを還流させながら、フッ化セシウム(10mol%)により、2,3−ジヒドロキシピリジン(2,3−DHP)をエチルアクリレートとマイケル付加反応させ、対応するエステル2を良好な収率で得た。続く2のO−ベンジル化は標準の状態(K2CO3/アクリロニトリル/還流)で行い、続けてエステル部位の還元(BH3・THF、室温)を行い、クロマトグラフィーを行って、アルコール3を90%の収率で得た。アルコール3をトリエチルアミンの存在下、ジクロロメタン中のメタンスルホン酸無水物で処理し、中間体であるメシラート4(〜10−15%)を伴う目的とする環状塩5(85−90%)を直接形成したことを1H NMR分析で確認した。環状塩5への完全な変換は、室温でのクロロホルム中のメシル化からの粗生成物の攪拌によって達成される。温エチルアセテートで粉砕した後、前記塩5は、薄く白い固体として92%の収率で高い純度で得られた。
[実施例13]
<式VIIIで表わされる化合物の生成>
塩5(0.2g)をアクリロニトリル(3ml)中トリエチルアミンの存在下1.2当量のシクレン(1,4,7,10−テトラアザシクロドデカン)に加え、窒素下60℃で2日間加熱した。前記反応はその後ジクロロメタン(50ml)で希釈し飽和NaHCO3(50ml)で洗った。前記水層はもう一度ジクロロメタン(25ml)で抽出した。合わせた有機層は硫酸ナトリウムで乾燥させ、前記溶媒は真空下除去し、過剰なN−メチルベンジルアミンは真空蒸留で除去し、VIIIを得た。
[実施例14]
<式IXで表わされる化合物の生成>
3−ベンジルオキシ−1−メチル−4−(2−チオキソチアゾリジン−1−イル)カルボニル−2(1H)−ピリジノンのメチレンクロライド溶液に対応する官能化されたN,N,N’,N’−テトラキス(2−アミノエチル)エチレンジアミン(構造B、Zは保護基を表わす)を加えた。4時間攪拌後、前記混合物は濾過され、乾燥された。
【0151】
【化17】
【0152】
前記適切なベンジル−保護前駆体生成物をメチレンクロライド中のプロパノールを用いて、フラッシュシリカカラムで反応混合物から単離した。前記構造IXの最終生成物は、酸による水酸基の脱保護により得られる。
[実施例15]
構造VIIIは、当業者にとって標準の方法で標的の分子と接合させる。例えばN−ヒドロキシスクシンイミド(NHS)エステルは、タンパク質やペプチドへ標準の状態で接合させることで生成され、得られた接合されたプロテインはゲル濾過により単離される。
[実施例16]
トリウム−227による接合されたタンパク質の標識化
適切な緩衝液、例えば0.5MのNaOAc−緩衝液(pH5.5)中に接合されたタンパク質(オクタデンテートリガンドを結合部位によって付けている)の溶液10mg/mlを作る、そして精製されたトリウム−227で25℃で1時間標識化し、続いてゲル濾過カラムで精製した。
【技術分野】
【0001】
本発明は、トリウム同位体の複合体に関し、とくに特定のオクタデンテートリガンドを有するトリウム−227の複合体に関する。本発明はまた、病気の治療に関する、特に腫瘍性の病気に関し、そのような複合体を投与することを含む。
【背景技術】
【0002】
特定の細胞の死滅させることは、哺乳類のさまざまな病気の有効な治療に基本となり得る。この典型的な例は、悪性の病気、たとえば肉腫およびガン種の治療における。しかしながら、特定の細胞のタイプの選択的除去は、他の病気、特に増殖性のおよび腫瘍性の病気、の治療においても重要な役割を果たす。
【0003】
選択的な治療の最も普通の方法は、最近の外科、化学療法および外部ビーム照射である。標的化された放射性核種の治療法は、しかしながら、将来性があり、発展途上の領域であり、望ましくない細胞のタイプに高い細胞毒性の放射をする潜在力を有する。放射性医薬品の最も普通の形態は、最近ではベータ放出および/またはガンマ放出放射性核種を導入することにより、人間に用いることが許可されている。しかしながら、治療におけるアルファ放出放射性核種の使用にも関心がある。なぜなら、細胞を死滅させる滞在力が大きいからである。
【0004】
生理学的な環境における典型的なアルファ放出器の放射範囲は、通常100マイクロメータ未満であり、いくつかの細胞の直径にのみ等しい。このことは、これらの源をミクロ転移を含む腫瘍の治療に好適とする。なぜなら、もしそれらが好適に標的化されるのであれば、標的細胞を超えて通過する放射エネルギーはほとんどないことになる。こうして周りの健康な組織の損傷を小さくすることができる(Feinendegenら、Radiat Res 148:195−201(1997)参照)。対照的に、ベータ粒子は、水中で1mm以上の範囲である(Wilbur,抗体免疫放射性 4:85−96(1991)参照)。
【0005】
アルファ粒子放射のエネルギーは、ベータ粒子、ガンマ線およびX線によるエネルギーに比較して高い。典型的には5−8MeVで、ベータ粒子のエネルギーの5〜10倍であり、ガンマ線のエネルギーの20倍以上である。こうして、非常に短い距離に渡って大量のエネルギーが堆積することが、α−放射に、ガンマ放射およびベータ放射に比較して、予期できないほど高い線エネルギー付与(LET)、高い生物学的効果比(RBE),低い酸素増感比(OER)を与える(Hall,「放射線医師のための放射線生物学」、5版、Lippincott Williams & Wilkins, Philadelphia PA, USA, 2000参照)。このことは、アルファ放出放射線核種の細胞毒性の例外を説明し、そのような同位体の生物学的標的化および制御のレベルおよび受け入れがたい副作用を避けるために必要であるアルファ放出放射線核種の分布の研究に厳しい要求を強いる。
【0006】
下記の表1は、今までのところ、治療の効果を有すると考えられるとして文献において広く提案されているアルファ放出体の物理学的崩壊特性を表す。
【0007】
【表1】
【0008】
今までのところ、放射線免疫治療に関して、主な関心は、211At、213Biおよび225Acに注がれ、これらの3種の核種は臨床の免疫試験において研究されてきた。
【0009】
いくつかの提案されている放射線核種は寿命が短い、たとえば半減期が12時間未満である。そのような短い半減期は、これらの放射線核種に基づく放射性医薬品の商業的な製造および流通を困難としている。寿命の短い核種を投与することはまた放射線量の割合を増大させる、なぜなら標的部位に達成する前に体外に放出されるからである。
【0010】
アルファ放出の反動エネルギーは、多くの場合親の放射線崩壊の位置から娘核種の放出を生じる。この反動エネルギーは、親を保持してよい化学的環境、たとえば親がキレート剤のようなリガンドにより複合化されたような環境から多くの娘核を取り出すのに十分なものである。このことは、娘が同じリガンドに化学的に適合する場合すなわち複合化されることさえも生じる。同様に、娘核種がガスである場合、特にラドンのような希ガスである場合、または化学的にリガンドと適合しない場合、この放出効果はさらに大きくなる。娘核種が数秒以上の半減期を有する場合、それらは血液系に拡散することができ、親を保持する複合体に抑制されることがない。これらの自由な放射活性娘は望ましくない全身の毒性を生じる。
【0011】
223Ra娘同位体が制御する状況でのトリウム−227(T1/2=18.7日)の使用が数年前に提案された(WO01/60417およびWO02/05859参照)。これは、キャリアーシステムが用いられた状況におけるもので、その状況は娘核種が閉じられた環境によって保持されることを許容する。第1のケースにおいて、放射性核種はリポソーム内で処置され、リポソームの実質的な大きさ(反動距離に対して)は娘核種をリポソーム内に保持することを助ける。第2のケースにおいて、向骨性の放射性核種の複合体は、骨マトリクスへ組み入れるために用いられる、それゆえ娘核種の放出を制限する。これらは、潜在的に非常に有利な方法である、しかしながらリポソームの投与は、いくつかの状況では望ましくない、そして、娘同位体を保持することができるように放射性核種が石灰化したマトリクスに囲まれることができないたくさんの柔らかい組織の病気がある。
【0012】
最も最近、227Thの崩壊によって放出される223Ra娘核の毒性が、比較できる核の前もった試験から予想されるよりも、哺乳類の体でかなりの程度にまで耐えうる方法が確立された。上記で述べたトリウム−227のラジウム娘を保持する特定の方法なくしては、ラジウムの毒性に関し公然と得られる情報は、トリウム−227を治療的な薬剤として用いることは不可能であることを明らかにした。なぜならトリウム−227の崩壊から治療効果を得るのに必要な投与量は、非常に毒性があり、致死量となりうるラジウム娘の崩壊からの放射となる、すなわち治療的な開口はない。
【0013】
WO04/091668は、標的化されたトリウム−227の放射性核種の治療に効果的な量を対象(典型的には哺乳類)に受け入れることのできない骨髄毒性を生じるのに十分なラジウム−223の量を生じることなく投与することができるという治療的な処置の開口が確かに存在するという予期できない発見について述べている。このことは、それゆえ骨および柔らかい組織のサイトのいずれにもおけるすべてのタイプの病気の治療および予防に用いることができる。
【0014】
上記発展によって、今やアルファ放出トリウム−227核を、223Raを生じることによる致死の骨髄毒性を生じることなく内部放射性核治療に用いることが可能である。それにもかかわらず、治療の開口は狭いままであり、対象に対するアルファ放出放射性同位体を投与することが好ましい全てのケースにおいて、絶対的に必要であるとは言えない、この新しい治療の開口の有用な開発は、それゆえアルファ放出トリウム−227核が複合化されているかどうかおよび高い信頼性で標的化されるかどうかによって非常に高められるであろう。
【0015】
放射性核種は常に崩壊するため、単離から対象に投与されるまでの間の材料を取り扱う時間が非常に重要である。アルファ放出トリウム核が、調製するためにすばやく便利な形態、好ましくはほとんどステップを必要としない、短いインキュベイション期間および/または標的とする実体の特性に不可逆的に影響を与えない温度で、複合化され、標的化され、および/または投与されるいるか否かということもまた、かなりの価値があるであろう。
【0016】
本発明の発明者らは、標的部位に結合したオクタデンテートヒドロキシピリジノン(HOPO)タイプのリガンドで複合化された4+トリウム−227イオンがトリウム−227イオンをめざましい程度で制御することを予想外に見出した。さらに、そのような複合体は、ここで述べる方法によって、相対的にすばやく容易に調製することができる。
【先行技術文献】
【特許文献】
【0017】
【特許文献1】国際公開第01/60417号
【特許文献2】国際公開第02/05859号
【特許文献3】国際公開第04/091668号
【非特許文献】
【0018】
【非特許文献1】Feinendegenら、Radiat Res 148:195−201(1997)
【非特許文献2】Wilbur,抗体免疫放射性 4:85−96(1991)
【非特許文献3】Hall,「放射線医師のための放射線生物学」、5版、Lippincott Williams & Wilkins, Philadelphia PA, USA, 2000
【発明の概要】
【課題を解決するための手段】
【0019】
本発明の一つの観点は、組織標的部位、オクタデンテートヒドロキシピリジノンリガンドおよびアルファ放出トリウム放射性核種のイオンを含む組織標的複合体を提供する。特に好ましい観点は、少なくとも1つの3,2−ヒドロキシピリジノン部位を含むオクタデンテートリガンドに共有結合し、そのようなリガンドは227Thのようなアルファ放出トリウム放射性核種の4+イオンに複合化しているポリペプチド組織標的部位を含む組織標的複合体である。
【0020】
本発明のさらなる観点は、増殖性または腫瘍性の病気、ここで述べる病気は何でも含むが、を治療するための薬剤の製造における、組織標的部位、オクタデンテートヒドロキシピリジノンリガンドおよびアルファ放出トリウム放射性核種のイオンを含む組織標的複合体(ここで述べるそのような複合体は何でも含む)の使用を提供する。
【0021】
対応する観点において、本発明は、少なくとも1つの組織標的部位、オクタデンテートヒドロキシピリジノンリガンドおよびアルファ放出トリウム放射性核種のイオンを含む組織標的複合体(ここで述べるそのような複合体は何でも含む)の投与を含む人間および人間でない動物(特にそれを必要としているもの)の治療方法を提供する。そのような方法は、好ましくはここで述べる病気は何でも含む増殖性または腫瘍性の病気の治療である。
【0022】
本発明のさらなる観点は、組織標的部位、オクタデンテートヒドロキシピリジノンリガンドおよびアルファ放出トリウム放射性核種のイオンを含む組織標的複合体(ここで述べるそのような複合体は何でも含む)を少なくとも1つの薬剤のキャリアまたは賦形剤とともに合わせて含む薬剤を提供することである。
【0023】
もっとも天然に豊富に生じるトリウム同位体、すなわちトリウム−232(半減期1010年であり、事実上放射性活性がない)、のトリウム複合体から区別するために、ここで定義するトリウム複合体およびその組成物は、天然に相対的に豊富なものよりも、アルファ放出トリウム放射性同位体(すなわち、少なくとも半減期が103年未満のトリウムの1つの同位体、たとえばトリウム−227)を多く含む、たとえば少なくとも20%多い。このことは本発明の方法の定義に影響を与えることを必要としない、それは放射性トリウム、たとえばトリウム−227の治療的に効果のある量が明確に必要とされるからである、しかしながら、それはすべての観点から好ましい場合である。
【0024】
本発明のさらなる観点は本発明に関する方法に用いられるキットを提供することである、前記キットは、オクタデンテートヒドロキシピリジノン含有リガンドに接合されたまたは接合可能な組織標的部位を含む。すべての結合部位およびリガンドは、好ましくはここで述べたものである、そのようなキットは任意におよび好ましくアルファ放出トリウム放射性核種、たとえば227Thを含む。
【0025】
本発明はさらに添付の図面によって説明される。
【図面の簡単な説明】
【0026】
【図1】図1は、232Th4+(HNO3)および1mg/mlのDMSO中のALG−DD−NCS(理論比2:3)を室温で15分反応させた後の反応混合物のクロマトグラムを示す。かなりの量の複合体232Th−ALG−DD−NCS(tR=5.74分;m/z1328.5)があり、および自由ALG−DD−NCSリガンド(tR=5.36)の痕跡はなかった。
【図2】図2は、前記232Th−ALG−DD−NCS複合体(m/z1328.3949)の質量スペクトルを示す。
【図3】図3は、Fe3+(HNO3)および1mg/mlのDMSO中のALG−DD−NCS(理論比2:3)を室温で15分反応させた後の反応混合物のクロマトグラムを示す。かなりの量の複合体Fe−ALG−DD−NCS(tR=5.81分;m/z1153.5)があり、および自由ALG−DD−NCSリガンド(tR=5.36分)の痕跡はなかった。
【図4】図4は、前記Fe−ALG−DD−NCS複合体(m/z1153.3002)の質量スペクトルを示す。
【図5】図5は、In3+(HCl)および1mg/mlのDMSO中のALG−DD−NCS(理論比2:3)を室温で15分反応させた後の反応混合物のクロマトグラムを示す。かなりの量の複合体In−ALG−DD−NCS(tR=5.95分;m/z1212.5)があり、および自由ALG−DD−NCSリガンド(tR=5.36分)の痕跡はなかった。
【図6】図6は、前記In−ALG−DD−NCS複合体(m/z1212.2666)の質量スペクトルを示す。
【図7】図7は、232Th4+(HNO3)および1mg/mlのpH5.5のアセテート緩衝液中のALG1005−38を室温で10分反応させた後の反応混合物(理論比1:3)のクロマトグラムを示す。かなりの量の複合体232Th−ALG1005−38(tR=3.56;m/z1184.5)があり、および自由ALG1005−38リガンド(tR=3.20分)の残渣があった。
【図8】図8は、前記232Th−ALG1005−38複合体(m/z1184.4662)の質量スペクトルを示す。
【図9】図9は、Fe3+(HNO3)および1mg/mlのpH5.5のアセテート緩衝液中のALG1005−38を室温で10分反応させた後の反応混合物(理論比1:3)のクロマトグラムを示す。かなりの量の複合体Fe−ALG1005−38(tR=3.70;m/z1009.5)があり、および自由ALG1005−38リガンド(tR=3.22分)の残渣があった。
【図10】図10は、前記Fe−ALG1005−38複合体(m/z1009.3727)の質量スペクトルを示す。
【図11】図11は、In3+(HCl)および1mg/mlのpH5.5のアセテート緩衝液中のALG1005−38を室温で10分反応させた後の反応混合物(理論比1:3)のクロマトグラムを示す。かなりの量の複合体In−ALG1005−38(tR=3.72;m/z1068.5)があり、および自由ALG1005−38リガンド(tR=3.21分)が痕跡量あった。
【図12】図12は、前記In−ALG1005−38複合体(m/z1068.3485)の質量スペクトルを示す。
【図13】図13は、Ga3+(HCl)および1mg/mlのpH5.5のアセテート緩衝液中のALG1005−38を室温で10分反応させた反応混合物(理論比1:3)のクロマトグラムを示す。かなりの量の複合体Ga−ALG1005−38(tR=3.65;m/z1022.5)があり、および自由ALG1005−38リガンド(tR=3.21)の残渣があった。
【図14】図14は、前記Ga−ALG1005−38複合体(m/z1022.3571)の質量スペクトルを示す。
【図15】図15は、232Th4+(HCl)および1mg/mlのBb−1−HOPO−1−DEBN(理論比2:3)を室温で15分反応させた後の反応混合物のクロマトグラムを示す。かなりの量の複合体232Th−Bb−1−HOPO−1−DEBN(tR=3.52)があった。質量クロマトグラムは、m/z 1222.5である、低い痕跡:UV330nm。
【図16】図16は、前記232Th−Bb−1−HOPO−1−DEBN複合体(m/z1222.5)の質量スペクトルを示す。
【図17】図17は、227Th−ALG−DD−NCS−trastuzumabおよび自由227Thが0−7日でSK−OV−3細胞ペレットに結合する計算量であって、補正された227Th−放射能の崩壊によって表わす。
【図18】図18は、(1)正規の培地(対照);(2)20μg/mLのtrastuzumab;(3)20kBq/mLの227Th−ALG−DD−NCS−trastuzumab(10166Bq/μg);および(4)20kBq/mLの自由227Thで処理されたSK−OV−3細胞の標準化された発光値を示す。
【図19】図19は、(1)正規の培地(対照);(2)trastuzumab;(3)自由227Th;(4)227Th−ALG−DD−NCS−trastuzumab(10166Bq/μg)(Mean±SD;n=3)で処理された1000SK−OV−3細胞のクローン形成を示す。
【図20】図20は、(1)正規の培地(対照);(2)trastuzumab;(3)自由227Th;(4)227Th−ALG−DD−NCS−trastuzumab(10166Bq/μg)で処理された3000SK−OV−3細胞のクローン形成を示す。
【図21】図21は、4日と24時間生体内分布をした後のSK−OV−3異種移植片を有するBalb/cヌードマウスの227Th−ALG−DD−NCS−trastuzumab(2700Bq/μg)の臓器分布を示す(Mean±SD;n=5)。
【発明を実施するための形態】
【0027】
本発明において、「組織標的」とは対象となる物質を示す(特にトリウム複合体に接合している形態のとき)、選択的にそれが存在する(たとえば、放射線崩壊をもたらすために)少なくとも1つの組織の部位に、自身の位置を特定する役割を果たすこと(そして特にどのような接合したトリウム複合体を特定すること)が望まれる。標識化した部位は、例えば、細胞表面のマーカー(たとえば、レセプター、輸送タンパク質、細胞接着分子)に結合し、病気の細胞または病気の細胞に近接した細胞上に存在してもよい。そのような細胞表面のマーカーは、健康な細胞の表面の上よりも病気の細胞の表面の上により大量に発現されるタンパク質または休眠の段階よりも成長もしくは複製の期間の細胞の表面の上により大量に発現されるタンパク質を含む。本発明のどのような態様においても、標的細胞、組織もしくはその結合物に近接して存在する構成は、治療のために標的において利用されてもよい。たとえば、標的細胞または組織の回りのマトリクスに存在するまたは放出される構成は、存在する場合には標的化に用いられてもよい、形成または濃縮は、前記領域を健康な組織から区別することを可能にする。この例はテネイシンのようなマトリクス抗原が挙げられる、それは脳腫瘍と関連するが、細胞の間のマトリクス中に発現する。そのようなマトリクス抗原は、ここで述べるような単一のまたは複合の標的部位によって、標的とされることができる。
【0028】
前記組織標的部位は合計で2つ以上の成分を含んでいてもよく、トリウム複合体を所望の組織へ標的化する効果を有する。このことは、たとえば、1つの成分が初めに投与され、特定の組織、腫瘍または細胞型に結合し、次におよび/またはさらに成分(連結剤)を同時に好ましくは続いて投与することにより、in vivoで組織結合剤に結合する。前記結合剤は直接または間接的に複合α放射トリウムに接合され、こうして組織結合および連結剤は合わさって組織標的部分を形成する。適切な特定の結合ペアが組織結合および連結剤に相互の親和性を供給するのに適していることは周知である(たとえば、ビオチンとアビジンまたはストレプトアビジン)。
【0029】
ここで述べる本発明の様々な態様は、病気の治療に関する、特に病気の組織の選択的標的化に関し、同様に複合体、接合、薬剤、処方、キットなどそのような方法に役に立つものに関する。すべての態様において、前記病気の組織は体の1つのサイトにおいて存在していてもよく(たとえば、局部的な固体の腫瘍の場合が挙げられる)、多くのサイトにおいて存在していてもよい(たとえば、いくつかの接合が関節炎の影響を受けている場合またはガン性の病気が広がったり転移した場合が挙げられる)。
【0030】
標的化される病気の組織は、柔らかい組織のサイトにある可能性もあり、石灰化された組織のサイトのある可能性もあり、全てが柔らかい組織中のたくさんの組織、石灰化された組織中のたくさんの組織にある可能性もあり、少なくとも1つの柔らかい組織のサイトおよび/または少なくとも1つの石灰化された組織のサイトも含む可能性がある。前記標的化されたサイトおよび病気の根源のサイトは同じ可能性もあるが、その代わりに異なる可能性もある、1つ以上のサイトが含まれている場合、これは根源のサイトを含む場合があり、またはたくさんの二次的なサイトである場合がある。
【0031】
「柔らかい組織」とは、ここでは「固い」石灰化したマトリクスを有さない組織を示す。特に、ここで用いられる柔らかい組織は、骨格的な組織でない組織であればなんでもよい。それに応じて、ここで用いられる「柔らかい組織の病気」とは、ここで用いられる柔らかい組織に起きる病気を示す。本発明は特にガンおよび「柔らかい組織の病気」の治療に適する、したがってどのような「柔らかい」(すなわち、非石灰化された)組織に生じた癌腫、肉腫、骨髄腫、白血病、リンパ腫および混合タイプのガン、同様にそのような組織のその他の非ガン性の病気に適する。ガン性の「柔らかい組織の病気」は、柔らかい組織に生じた固体の腫瘍、同様に転移したおよび微小転移した腫瘍を含む。実際、前記柔らかい組織の病気は、柔らかい組織の一次の固体の腫瘍および同じ患者の柔らかい組織の少なくとも1つの転移した腫瘍を含む、言い換えれば、「柔らかい組織の病気」は固体の腫瘍のみ、または骨格の病気の1次的な腫瘍を伴う転移のみからなってもよい。
【0032】
近日の主要な発見は、特定のアルファー放射性トリウム同位体(たとえば227Th)は治療的に効果があり、かつ耐えられない骨髄毒性を生じない量で投与されることが可能であることである。ここで、「許容される非骨髄毒性」は、最も重要に、投与されたトリウム−227の放射性同位体の崩壊により生じたラジウム−223の量は、通常対象者を直接致死とするのに十分でない量を示す。当業者には、しかしながら、そのような治療の副作用として許容可能な骨髄を損傷する量(および致死反応の可能性)は、対象者の治療を受ける病気のタイプ、治療計画の目的、およびに予後より著しくさまざまであることは明らかであろう。本発明の好ましい対象は人間であるが、その他の哺乳類、特に犬は、本発明を用いる恩恵を受けるであろうし、許容可能な骨髄を損傷するレベルもまた対象の種類を反映するであろう。前記許容可能な骨髄を損傷するレベルは、一般的に悪質でない病気よりも悪質な病気の治療のほうが高い。1つのよく知られている骨髄毒性の測定は、好中球の数を数えることであり、本発明において223Raの許容可能な骨髄を損傷する量は、典型的に治療前に好中球フラクションの最も低いポイントを10%以上のカウントすることにより制御された量である。好ましくは、223Raの許容可能な骨髄を損傷しない量は、好中球のフラクションは、一番低い値で少なくても20%、より好ましくは少なくとも30%である。一番低い値の好中球のフラクションは、少なくとも40%が最も好ましい。
【0033】
それに加えて、放射性トリウム(たとえば227Th)を含む化合物は、高い放射線の治療に用いられ、その治療では生じたラジウム(たとえば223Ra)の骨髄毒性は、幹細胞サポートまたは同等の回復方法が含まれる場合、普通は耐えられない。そのような場合、好中球のカウントは、一番低い値で10%以下まで減じてもよく、例外的に5%まで、必要であれば5%以下まで減じてよく、そうして適切な予防が行われ、次の幹細胞サポートが与えられる。そのような技術は周知である。
【0034】
本発明において特に興味があるのが、トリウム−227である、トリウム−227は、背景が許容するように、ここでのすべてのトリウムに対して好ましい同位体である。トリウム−227は相対的に製造が容易で、中性子照射した226Raから間接的に製造することができ、227Thの母核種、すなわち227Ac(T1/2=22年)を含むものである。アクチニウム−227は前記226Raターゲットから容易に分離することができ、227Thの発生器として用いられる、このプロセスは、必要であれば工業スケールとすることもできる、こうして多くの他のアルファ放出体にみられる供給問題は、分子を標的化した放射線治療の候補から除かれる可能性がある。トリウム−227はラジウム−223を経て崩壊する。この場合、前記最初の娘は、半減期が11.4日である。純粋な227Th源から、適度な量のラジウムのみが最初の2,3日の間に作られる。しかしながら、223Raの滞在的な毒性は、227Thのものより高い、それはアルファ粒子の223Raからの放出に、数分以内に短寿命の娘からの3つのさらなるアルファ粒子が続くからである(下記の表2参照、表2はトリウム−227の崩壊系列を提示した)。
【0035】
【表2】
【0036】
滞在的に毒性のある崩壊生成物を生成することも部分的な理由として、トリウム−227(T1/2=18.7日)はアルファ粒子治療のために広く検討されてこなかった。
【0037】
トリウム−227は、骨髄の耐えられないような状態を生ずるような多量のラジウム−223を生ずることなく、所望の治療効果を提供するのに十分な量を投与することもできる。標的の領域において娘同位体を保持すると、その崩壊によりさらなる治療効果が得られるため望ましい。しかしながら、受け入れがたい骨髄毒性を誘導することなく有用な治療効果を得るために、トリウムの崩壊生成物の制御を保つことは必要ない。
【0038】
ガン細胞を殺す効果が主にトリウム−227から得られ、娘から得られないと仮定すると、この同位体の適した治療のための放射量は、他のアルファ放出体に比較して定められる。たとえば、アスタチン−221は、動物における治療効果のある放射量は典型的に2−10MBq/kgである。半減期とエネルギーを補正することによって、トリウム−227の対応する放射線量は少なくても体重の36−200kBqとなるであろう。これは227Thの量の下限を定めることになり、それは治療効果を予想して、役立つように投与されることができるであろう。この計算はアスタチンおよびトリウムの同等の保持を仮定したものである。明らかに、しかしながら前記トリウムの18,7日の半減期は、この同位体が崩壊前により大きい放出をする結果になる傾向があるであろう。この計算された放射線量は、それゆえ、普通は最少の効果の量と考えられるべきである。十分に保持された227Th(たとえば、体から放出されたものでない227Th)の観点から表現された前記治療効果のある放射線量は、典型的には少なくとも18または25kBq/kgであり、好ましくは少なくとも36kBq/kgであり、より好ましくは少なくとも75kBq/kgである、たとえば100kBq/kg以上である。トリウムの量がより多い程、より治療効果が高いことが期待されるが、耐えられない副作用が生じるのであれば投与できない。同じように、もしトリウムが短い生物学的半減期(たとえば、なおトリウムを有している体から放射される前の半減期)を有する形態で投与されれば、治療の効果を得るためには、より多い量の放射性同位体が必要とされる、なぜなら、前記トリウムの多くの量が崩壊前に放出されるからである。しかしながら、ラジウム−223の対応する減少が生じる。投与されるトリウム−227の上記量は、同位体が十分に保持されるとき、より短い生物学的な半減期を伴う等量の放射線量に容易に関連付けられる。そのような計算は、周知であり、WO04/091668(たとえば実施例1および2の文中)に開示されている。
【0039】
もし放射線で標識化された化合物が娘核種を放出すれば、もし当てはまれば、どのような放射性の娘核種の宿命を知っておくことが重要である。227Thとともに主娘生成物は、226Raであり、その向骨性のため、それは臨床の評価のもとにある。ラジウム−223は非常にすばやく血液を飛び越えて骨に濃縮されるか、または腸および腎臓のルートを経由して排出される(Larsen,J NuclMed 43(5 補足):160P(2002)参照)。ラジウム−223は、in vivoで227Thから放出され、それゆえ、健康な柔らかい組織にそれほど影響を与えない。Mueller in Int.J.Radiat.Biol.20:233−243(1971)による227Thを溶解されたクエン酸塩とした分布の研究によれば、柔らかい組織中で227Thから生じた226Raは、直ちに骨に再分布されるか、排出される。アルファ放出ラジウムの公知の毒性は、特に骨髄に対するものであり、トリウムの投与の問題点である。
【0040】
実際、少なくとも200kBq/kgの放射線量の223Raが人間に投与することができ、耐えられることを最初に確立したのはWO04/091668である。これらのデータは、前記公報中に存在する。それゆえ、極めて予想できなかったことに、対象が深刻なまたは致死の骨髄毒性の受け入れがたい危険性をこうむることが予想されることなく、治療に効果のある量の227Th(たとえば36kBq/kgより大きい量)を哺乳類の動物に投与することができることが今や明らかになった。それにもかかわらず、この治療効果のある方法を最善に使用することは非常に重要であり、それゆえ、放射性のあるトリウムはすばやく効率的に複合体とすることおよび放射線量の可能な限り最大の割合が標的のサイトに届けられるように非常に高い親和性を有することが不可欠である。
【0041】
227Thの薬剤から生じる223Raの量は、放射性標識化された化合物の生物学的な半減期に依存する。理想的な状況は、すばやく腫瘍を取り込む複合体を用いることであり、腫瘍細胞に内面化すること、強く腫瘍を保持することおよび正常の組織中において短い生物学的半減期とすることを含む。生物学的半減期が理想よりも短い複合体は、しかしながら、223Raを耐えうるレベルで維持する量である限りは有用である。in vivoで生じたラジウム−223の量は、投与されるトリウムの量およびトリウム複合体の生物学的保持時間の因子である。どのようなケースで生じたラジウム−223の量も、当業者であれば容易に計算できる。227Thの最大の投与可能な量は、in vivoで生じたラジウムの量により定められ、耐えることのできないレベルの副作用、特に骨髄毒性を生じる量未満でなければならない。その量は一般的に300kBq/kg未満であり、好ましくは200kBq/kg未満である、より好ましくは170kBq/kg未満である(たとえば130kBq/kg未満である)。最小の効果的な放射線量は、トリウムの細胞毒性、生じたアルファ照射に対する病気の組織の影響の受けやすさ、およびトリウムが標的複合体(この場合リガンドおよび標的の部位の組み合わせ)によって効果的に結合し、保持されおよび届けられる程度によって定められる。
【0042】
本発明の方法において、トリウム複合体は望ましくはトリウム−227放射線量が18〜400kBq/kg体重、好ましくは36〜200kBq/kg(たとえば50〜200kBq/kg)、より好ましくは75〜170kBq/kg、特には100〜130kBq/kgにおいて投与される。それに応じて、1回の放射線量が、たとえば30〜150Kg、好ましくは40〜100Kgといった適切な体重によってこれらの範囲のどのような外延も含んでよい(たとえば放射線量あたり540kBq〜4000kBq他)。前記トリウムの放射線量、複合化剤および投与経路は、in vivoで生じたラジウム−223の放射線量が、300kBq/kg未満であり、好ましくは200kBq/kg未満であり、より好ましくは150kBq/kg未満であり、特に好ましくは100kBq/kg未満であることがさらに望ましい。さらにこのことは、示された体重のいずれにもよって拡大された範囲へ223Raへの露出を提供する。上記放射線量のレベルは、好ましくは227Thの十分に保持された放射線量であり、しかしながら、いくらかの227Thが崩壊前に体から消えることを考慮した投与された放射線量であってもよい。
【0043】
227Th複合体の生物学的半減期は、物理学的半減期(たとえば7日未満、特に3日未満)に較べて短い場合は、等しく保持された放射線量を提供するために特により多くの放射線量を投与することが必要とされるかもしれない。こうして、たとえば、150kBq/kgの十分に保持された放射線量は、5日半減期の複合体を711kBq/kgの放射線量で投与したのに等しい。どのような適切に保持された放射線量に対して等しく投与された放射線量は、周知の方法を用いて複合体の生物学的クリアランス速度によって計算されてもよい。
【0044】
1つの227Thの核の崩壊は1つの223Ra原子を提供するため、227Thの保持および治療効果のある活性は、患者が受ける223Raの放射線量に直接関連する。どのような状況で生じた223Raの量も、周知の方法で計算される。
【0045】
好ましい態様においては、本発明はそれゆえ、哺乳類(ここで述べた)の病気を治療するための方法を提供する。前記方法は組織標的部位を含む接合、オクタデンテートリガンド(特にここで述べたどのようなものも)、および放射性トリウム同位体(たとえばトリウム−227)の治療効果のある量を前記対象に投与することを含む。
【0046】
この特性が有用である限り、明らかに対象を223Raの娘の同位体に露出することを最小限にすることは望ましい。特にin vivoで生じるラジウム−223の量は、典型的には40kBq/kgを超える、たとえば60kBq/kgを超える。いくつかのケースにおいて、in vivoで生じる223Raは、80kBq/kgを超える、たとえば100または115kBq/kgを超える。
【0047】
適切なキャリア溶媒中のトリウム−227で標識化した接合体は、静脈内、膣内(たとえば腹腔内)、皮下、口内または局所に、1回または分割した方法により投与されてもよい。好ましくは、標的の部位に接合した複合体は、非経口(たとえば経皮)経路によって、特に、静脈内または膣内の経路によって投与される。好ましくは、本発明の組成物は、非経口投与のため無菌の溶液として作られる。
【0048】
本発明の方法および生成物におけるトリウム−227は、単独でまたは外科、体外照射療法、化学療法、その他の放射線核種、または組織温調節を含む他の治療法と組み合わせて用いてもよい。
【0049】
これはさらに本発明の好ましい態様の方法、製剤/薬剤が、他の放射性物質または化学療法の物質のようなさらなる少なくとも1つの治療活性物質を含んでいてもよいことを示す。
【0050】
1つの特に好ましい態様においては、対象は幹細胞治療および/または他の対症的な治療法を行って、ラジウム−223が誘発した骨髄毒性を減じる。
【0051】
本発明によれば、227Thは、標的複合化薬剤によって複合化されてもよい。典型的な標的部位は100g/molから数百万g/mol(特に100g/molから100万g/mol)の分子量を有し、好ましくは病気の関連したレセプターに直接的に親和性と有し、および/または投与前に病気を標的とした分子に結合する適切な前投与バインダー(たとえば、ビオチンまたはアビジン)を含む。適切な標的部位は、ポリ−およびオリゴ−ペプチド、プロテイン、DNAおよびRNAフラグメント、アプタマーなどを含み、好ましくは、たとえばアビジン、ストレプトアビジン、ポリクローナルまたはモノクローナル抗体(たとえば、IgGおよびIgMタイプの抗体)といったプロテイン、またはプロテインの混合物、またはプロテインのフラグメントまたは構築物を含む。抗体、抗体構築物、抗体のフラグメント(たとえばFABフラグメントまたは少なくとも抗原結合した領域、フラグメントの構築物(たとえば単鎖抗体)またはそれらの混合物が特に好ましい。
【0052】
本発明において用いるのに適しているものとしては、さらに、ペプチド、アミノ酸、ステロイド性のまたは非ステロイド性のホルモン、葉酸塩、エストロゲン、テストステロン、ビオチン、または他の典型的に10000g/mol以下の分子量を有する特異的な結合をする化合物を伴う複合化された227Thの治療的接合が挙げられる。
【0053】
一般的に、オクタデンテートリガンドは、直接的にまたは間接的に(たとえばリンカー部位を経由して)、標的部位に接合している。このタイプの一般的構築物は、たとえば活性(治療上または診断上の活性)金属−複合体部位−任意のリンカー部位−標的部位が、放射性医薬品および標的化された造影剤の分野でよく知られている。しかしながら、トリウム4+イオンの特定の使用に用いられるさまざまなリガンドの適性を評価する研究はほとんどない。この点で「造影剤の標的化された投与のハンドブック」Ed.Torchilin,CRC Press,1995は参照される。
【0054】
従来知られていたトリウムのキレーターはポリアミノポリアシッドキレーターを含む、それは鎖状、環状またな分枝状の骨格の窒素に酸基(カルボキシアルキル)を有するポリアザアルカン骨格を含む。そのようなキレーターはDOTA誘導体、たとえばp−イソチオシアナートベンジル−1,4,7,10−テトラアザシシクロドデカン−1,4,7,10−テトラ酢酸(p−SCN−Bz−DOTA)およびDTPA誘導体たとえばp−イソチオシアナートベンジル−ジエチレントリアミンペンタ酢酸(p−SCN−Bz−DTPA)、最初は環状のキレーターであり後から鎖状のキレーターになるもの、が挙げられる。
【0055】
1,4,7,10−テトラアザシシクロドデカン−1,4,7,10−テトラ酢酸の誘導体は、従来実証されてきた、しかしながら、標準的な方法はDOTA誘導体を有するキレートトリウムに容易に用いることはできない。金属を有するDOTA誘導体を加熱することは効果的にキレートを提供するが、多くの場合収率が低い。少なくてもリガンドの一部は、生成の過程で不可逆的に変性するという傾向がある。さらに、不可逆的な変性に相対的に高い影響を受ける性質により、全ての加熱過程が完了するまで通常標的部位の結合物を避けなければならない。このことはさらなる化学的なステップ(加えて全ての必要な検査および分離)を加えることになり、そのステップはアルファ放出トリウム同位体の崩壊半減期の間に行わなければならない。明らかにアルファ放出材料はこの方法である扱わないことが好ましく、必要性よりもより多くの無駄を生じる。さらに接合体を生成する全ての時間は、トリウムが準備期間に崩壊するので、トリウムの一部を無駄にする。
【0056】
本発明のすべての観点においてアルファ放出トリウムおよびオクタデンテートリガンドの複合体が形成されること、または60℃以上に加熱することなく(例えば50℃以上に加熱することなく)、好ましくは38℃以上に加熱することなく、最も好ましくは25℃以上に加熱することなく形成されることが好ましい。
【0057】
加えて好ましいのは標的部位およびオクタデンテートリガンドの接合がアルファ放出トリウム同位体(たとえば227Th4+イオン)の付加よりも前に生成されることである。本発明の生成物は、こうして、オクタデンテートリガンドおよび組織標的部位の接合によるアルファ放出トリウム同位体(たとえば227Th4+イオン)の複合化によって好ましく形成され、または形成されうる。
【0058】
前記キレーターは、非ホスホン酸分子であってもよく、本発明の一態様では、227Thはどのようなホスホン酸または他の骨を標的とした基に付着しないし、そのような材料とともに投与されない。
【0059】
標的化合物のタイプは、オクタデンテートキレーター(ここで述べたようにカップリング部位を含む)を経由してトリウム(たとえばトリウム−227)に結合していてもよい。標的部位は、公知の標的基から選ばれてもよく、モノクローナルまたはポリクローナル抗体、成長因子、ペプチド、ホルモンおよびホルモン類似体、葉酸および葉酸誘導体、ビオチン、アビジンおよびストレプトアビジンまたはその類似物を含む。他の可能なキャリアはRNA、DNA、もしくはそのフラグメント、オリゴヌクレオチド、炭水化物、脂質またはそのようなグループをタンパク質とともに、またはタンパク質を伴わないで結合した化合物であってもよい。
【0060】
組織標的部位は、一つの態様において、抗体または抗体のフラグメントに接合した向骨性物質、リポゾームおよび葉酸を含まなくてもよい。もしくは、そのような部位は含まれていてもよい。
【0061】
本発明の前記トリウム(たとえばトリウム−227)で標識化した分子は、病気の関連したレセプターを標的化することにより、ガン性のまたは非ガン性の病気の治療に用いられてもよい。典型的には、そのような227Thの医学的使用は、ガン性のまたは非ガン性の病気の治療のために、抗体、抗体フラグメント、または抗体もしくは抗体のフラグメントの構築物へのキレーターによる227Thの結合に基づく放射線免疫治療によってもよい。本発明による方法および薬剤における227Thの使用は特に癌腫、肉腫、リンパ腫、白血病を含むどのような形態のガン、特に肺、胸、前立腺、膀胱、腎臓、胃、すい臓、食道、脳、卵巣および子宮のガン、口頭ガン、大腸がん、黒色腫、複合骨髄腫およびホジキンリンパ腫を含む。
【0062】
227Thを有する分子がin vivoで短い生物学的な保持半減期を有する場合、放出される223Raの量は減少させることができる、なぜなら放射性核種はたいてい227Thの多くの割合が崩壊して223Raになる前に、排出されるからである。しかしながら、本発明によれば、227Thの量は、治療効果を保持するために増やされる必要がある。もし複合剤が、標的の細胞の内部へ227Thを運ぶように選択されるのであれば、腫瘍サイトにおける娘同位体の少なくとも部分的な保持により、さらに骨髄毒性を上昇させ、放射性娘の全身性独作用を減少させるであろう。これらいずれもの特徴は、227Thの治療方法を拡大し、本発明の好ましい態様となる。
【0063】
本発明のさらなる態様において、軟らかい組織および骨格の病気を有する患者は、トリウムを投与することにより、227Thおよびin vivoで生じた223Raによって治療されてもよい。この特に有利な観点において、治療にさらなる治療的な成分は、骨格の病気を標的化することにより受容可能に非骨髄毒性量の223Raから由来する。この治療方法において、227Thは典型的に柔らかい組織の初期のおよび/または転移性のガンを適切に標的化するのに利用される。そして227Thの崩壊から生じた223Raは、同じ対象の関連する骨格の病気の治療に利用される。この骨格の病気は初期の柔らかい組織のガンから生じた骨格への転移であってもよい、または柔らかい組織の治療が転移性のガンを攻撃するためのものである初期の病気であってもよい。場合によっては、前記柔らかい組織および骨格の病気は関連していないかもしれない(たとえば、リウマチの柔らかい組織の病気を有する患者における骨格の病気のさらなる治療が挙げられる)。
【0064】
すべての観点における本発明の主な観点は、オクタデンテートリガンドを用いること、とくにオクタデンテートヒドロキシピリジノン含有リガンドを用いることである。そのようなリガンドは典型的に以下の置換されたピリジン構造(I)の少なくとも1つのキレート基を含むであろう。
【0065】
【化1】
【0066】
式中R1は、任意のN−置換基であり、なくてもよいし、ヒドロカルビル基、OH基、O−ヒドロカルビル基、SH基、およびS−ヒドロカルビル基から選ばれてもよい、前記ヒドロカルビル部位のいずれもまたはそれぞれは、独立に、短いヒドロカルビル基、たとえば、C1〜C8アルキル基、アルケニル基またはアルキニル基を含むC1〜C8のヒドロカルビル基から選ばれ、OHまたはO−ヒドロカルビル基であってもよい。R1は、下記に示すようにリンカー部位もまた含んでいてよい、および/または下記に示すようにカップリング部位を含んでいてもよい。
【0067】
式Iにおいて、置換基R2〜R6はそれぞれ独立にH、OH、=O、短いヒドロカルビル(ここで述べたように)、リンカー部位(ここで述べたように)、および/またはカップリング部位(ここで述べたように)から選ばれてもよい。通常、R1〜R6の少なくとも1つは、OHである。通常、R2〜R6の少なくとも1つは、=Oである。通常、R1〜R6の少なくとも1つは、リンカー部位(ここで述べたように)である。残りのR1〜R6は、ここで述べた部位のいすれであってもよく、好ましくはHである。リンカー部位またはさらなるリンカー、リンカー部位に付いているテンプレートまたはキレート基は、カップリング部位を含まず、R1〜R6の1つは、好ましくはカップリング部位(ここで述べたように)である。
【0068】
好ましい態様において、R1〜R6の1つはOHであって、R2〜R6の1つは=Oであって、OHおよび=O基は、環における隣接した原子である。こうして、好ましい態様において、OHおよび=Oは、(予想されたように、窒素から数えた)1,2;2,3;3,2;3,4;または4,3のそれぞれの原子上にあるであろう。オクタデンテートリガンドは、OHおよび=O基が3および2の位置に其々存在する少なくとも1つのキレート部位であることが、非常に好ましい。前記オクタデンテートリガンドは、2、3または4つのキレート基を有することが好ましく、2または4のキレート基を有することが非常に好ましい。
【0069】
好適なキレート部位は周知の方法により形成することができる、その方法はUS5,624,901(たとえば実施例1および2)およびWO2008/063721に記載されている(いずれも参考として組み込む)。
【0070】
ここで用いられるように、「リンカー部位」(式IIにおけるRL)という用語は、オクタデンテートリガンドにおいて、少なくとも2つのキレート基を結合する化学物質を示すために用いられる、それは本発明のさまざまな点において主要の成分を形成する。典型的に、それぞれのキレート基(たとえば上記式Iおよび/または下記式II)は、ビ−デンテートであり、そのため4つのキレート基である、その少なくとも1つは式Iである、典型的にリガンド中に存在する。そのようなキレート基はそのリンカー部位によって互いに結合しあう。こうして、リンカー部位(たとえば下記のRL基)は、式Iおよび/またはIIの1を超えるキレート基の間に共有されます。前記リンカー部位は、オクタデンテートリガンドの複合化部位と標的部位との間の連結部位として働く。そのような場合、少なくとも1つのリンカー部位は、カップリング部位(RC)に結合する。適切なリンカー部位は、すべてのトポロジーの中のメチル基、エチル基、プロピル基、ブチル基、ペンチル基および/またはヘキシル基を含むC1〜C12アルキル基、アルケニル基、またはアルキニル基を含むC1〜C12ヒドロカルビル基のような短いヒドロカルビル基を含む。
【0071】
リンカー部位はまた、その他の適切な強固な化学結合であっても、または含んでいてもよい、たとえばエステル基、エーテル基、アミン基および/またはアミド基を含む。2つのキレート部位を結合する(1を超える経路がある場合は、もっとも短い経路を数える)原子の総数は通常限定される、キレート部位が複合体の形成のために適切な配置となるように。こうしてリンカー部位は、典型的にキレート部位の間に15原子以下、好ましくは1〜12原子、より好ましくは1〜10原子となるように選択される。リンカー部位が2つのキレート部位に直接的に結合している場合、前記リンカーは長さで1〜12原子、好ましくは2〜10原子(たとえばエチル、プロピル、n−ブチルなど)である。前記リンカーが中心のテンプレートに結合している場合(下記参照)、それぞれのリンカーはキレート部位を結合する2つの分離したリンカーを有し、より短い。1〜8原子の長さ、好ましくは1〜6原子の長さのリンカーが、この場合好ましい(メチル、エチルおよびプロピルが適している、同様にそれらが片方または両端にエステル、エーテルおよびアミド結合を有する基も適している)。
【0072】
リンカー部位は、第1にオクタデンテートリガンドのさまざまなキレート基を互いに、または中心のテンプレートに結合させるように働くが、リンカー部位に加えて、前記オクタデンテートは好ましくは、さらに「カップリング部位」(RC)を含む。前記カップリング部位の機能は、オクタデンテートリガンドを標的部位に結合させることである。これは、共有結合または非共有結合のいずれによっても達成される(たとえば、ビオチン/アビジン(ストレプトアビジン)のような特異の結合によって達成される)。好ましくは、カップリング部位は、キレート基の一つに直接共有結合していることによるか、より典型的にはリンカー部位またはテンプレートに結合することによりキレート基に共有結合で結合している。2以上のカップリング部位が用いられるならば、それぞれは、たとえばどのようなテンプレート、リンカーまたはキレート基でも適切なサイトであればいずれにも結合することができる。
【0073】
一つの態様において、カップリング部位は、下記の構造を有していてもよい。
【0074】
【化2】
【0075】
式中、R7は結合部位であり、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換ヘテロシクロアルキル、置換または非置換アリル、置換または非置換ヘテロアリルからなる群から選ばれ、Xは標的部位または反応性の官能基である。好ましい結合部位は、ここで好ましいリンカー部位として述べた全ての基を含む。好ましい標的部位は、ここで述べた全てのものを含み、好ましい反応性のX基は、標的部位と共有結合を形成可能などのような基も含み、たとえばCOOH、OH、SH、NHRおよびCOH基を含む、ここでNHRのRはHまたはここで述べた短いヒドロカルビル基であればなんでもよい。標的部位への結合のための非常に好ましい基は、リジン残基のイプシロンアミンおよびシステイン残基のチオール基が挙げられる。好適な反応性のX基の例としては、制限されることなく、N−ヒドロキシスクシミジルエステル、イミドエステル、アクリルハライド、N−マレイミド、アルファ−ハロアセチルおよびイソチオシアネートが挙げられる、ここで、後ろから3つはチオールとの反応に好適である。
【0076】
生じた結合されたオクタデンテートリガンドが安定な金属イオン複合体を形成することができるように、カップリング部位は好ましくは結合される。前記カップリング部位はこうして好ましくは、リンカー、テンプレートまたはキレート部位の複合化をさほど妨げないサイトにおいて結合する。そのようなサイトは、好ましくはリンカーまたはテンプレート上にあり、より好ましくは標的に結合した表面から離れた位置にある。
【0077】
好ましいキレート基は、下記の式IIで表わされる基を含む。
【0078】
【化3】
【0079】
上記式IIにおいて、=O部位は、ピリジン環のどのような炭素に結合したものであってもよいケト基を表わし、−OHは、ピリジン環のどのような原子に結合したものであってもよいヒドロキシ部位を表わし、−RLは、オクタデンテートリガンド全体を形成するために、ヒドロキシピリジノン部位を他の複合化部位に結合するリンカー部位を表わす。ここで述べるどのようなリンカー部位もRLとして適切であり、短いヒドロカルビル基、たとえば全てのトポロジーのメチル基、エチル基、プロピル基、ブチル基、ペンチル基および/またはヘキシル基を含むたとえばC1〜C8アルキル基、アルケニル基、アルキニル基を含むC1〜C8ヒドロカルビル基、を含む。RLは、ピリジン環のどのような原子、たとえば炭素または窒素原子において式IIの環に結合していてもよい。前記RL基は、同様に他のキレート部位、他のリンカー基および/または中心の原子または基、たとえば環または他のテンプレート(ここで述べたように)に直接結合していてもよい。リンカー、キレート基および任意のテンプレート部位は、適切なオクタデンテートリガンドを形成するために選択される。
【0080】
1つの好ましい態様において、式IIの前記−OHおよび=O部位は、ピリジン環の隣接する原子に存在する、たとえば1,2−;2,3−,3,2−;4,3−および3,4−ヒドロキシピリジノン誘導体がすべてにおいて非常に好適である。
【0081】
ここで、前記OH基またはリンカー部位RLはピリジン環の窒素の上に存在する場合、RNは通常存在しない。しかしながら、存在する場合は、RNは、置換または非置換のヒドロカルビル基、特にここで述べた短いヒドロカルビル基を含むどのような好適な部位であってもよい。
【0082】
1つの好適な態様においては、少なくとも1つの3,2−ヒドロキシピリジノン部位はオクタデンテートリガンド構造中に存在する。これは明らかに、ここで述べたさまざまな置換部位であればどのようなものによっても置換されていてもよい。
【0083】
式IIのそれぞれの部位は、2つの滞在的に複合化した酸素を有する、本発明の1つの好ましい態様は、オクタデンテートリガンドが少なくとも2、好ましくは少なくとも3そして最も好ましくは4の独立に選ばれた式Iの部位を有する。式IIのそれぞれの部位は、独立した置換形式を有する、しかしながら、1つの好ましい態様においては、少なくとも1つの部位が3,2−ヒドロキシピリジノン部位である。前記リガンドは、2、3または4つの3,2−ヒドロキシピリジノン部位(適切に置換されたもの、ここで述べたように)を含んでいてもよい。
【0084】
オクタデンテートリガンドの式IまたはIIのそれぞれの部位は、ここで述べたどのような適切なリンカー基のどのような適切なトポロジーによって、前記リガンドの残りに結合されていてもよい。たとえば、式Iの4つの基は鎖状のリガンドを形成することができるように、そのリンカー基によって骨格に結合されていてもよい。または「オリゴマー」タイプの構造を形成するためにリンカー基によって橋かけされていてもよく、それは鎖状であっても、環状であってもよい。その代わりに、式IまたはIIのリガンド部位は、「交差」または「星状」トポグラフィの態様で、それぞれがリンカー(たとえば「RL」部位)によって、中心の原子または基に結合されていてもよい。リンカー(RL)部位は、単独で、炭素−炭素結合を介して結合していてもよい、または互いに、キレート基に、骨格に、テンプレート基に、カップリング部位に、または他のリンカーに、適切な強い基の官能基、たとえばアミン、アミド、エステル、チオエーテルまたはジスルフィド結合によって結合していてもよい。
【0085】
「星状の」配置は、下記式IIIに表わされる。
【0086】
【化4】
【0087】
ここで、すべての置換基および位置は上述の通りであり、「T」は、さらに中心の原子またはテンプレート基、たとえば、炭素原子、ヒドロカルビル鎖(上記で述べたものはどのようなものでもよい)、脂肪族または芳香環(ヘテロ環を含む)または縮合環系である。もっとも基本的なテンプレートは、1炭素であろう、それはその結合基によってキレート部位のそれぞれに結合するであろう、長い鎖、たとえば、エチルまたはプロピルは、テンプレートのいずれの端にも結合して、2つのキレート部位とともに等しく存在することができる。明らかに、どのような適した強い結合、それは炭素−炭素結合、エステル、エーテル、アミン、アミド、エステル、チオエーテルまたはジスルフィド結合を含む、をテンプレートおよびリンカー部位を結合するために用いてもよい。
【0088】
式IIIにおいて、RNおよびRLは同じ基であり、そうしてキレート部位がテンプレートTにピリジン環の窒素原子を介して結合することができる。これは下記のIIIb型の構造を提供する:
【0089】
【化5】
【0090】
明らかに、式II,III,IIIb,IVおよびIVbの構造において異なる方法では置換されてない(たとえば、リンカーまたはカップリング部位によって)、それらのピリジン環の位置は、適切な態様で、式IにおけるR1〜R5の置換基を有していてもよい。特に、小さいアルキル置換基、たとえばメチル基、エチル基またはプロピル基がどのような位置にあってもよい。
【0091】
前記オクタデンテートリガンドは通常、さらに上記で述べた少なくとも1つのカップリング部位を含む。これは、ここで述べたどのような好適な構造であってもよく、末端が標的部位、特定のバインダー、またはそのような標的部位または特異的なバインダーに結合可能な特定のバインダーまたは官能基であってもよい。
【0092】
カップリング部位は、前記リンカー、テンプレートまたはキレート部位のどのような好適な位置に結合していてもよい、たとえば式III中で述べられたa)、b)および/またはc)の位置が挙げられる。カップリング部位の結合は、どのような強い結合によるものであってもよい、たとえば炭素−炭素結合、エステル、エーテル、アミン、アミド、チオエーテルまたはジスルフィド結合が挙げられる。同様に、標的部位にそのような結合を形成することが可能な基はカップリング部位の官能端に適し、その部位は標的部位に結合している時に、そのような基を終結させる。
【0093】
代わりに、「骨格」タイプの構造は、式IVで表わされる。
【0094】
【化6】
【0095】
ここで、上記式で表わされる全ての置換基および位置は、上記の通りである、および「RB」はさらに骨格部位である、それは典型的に、ここで述べたいずれのリンカーに対しても同じ構造および機能であり、こうして、状況が許す限り、リンカー部位のどのような定義も骨格部位に適用される。好適な骨格部位は、キレート部位がそのリンカー基によって結合するための足場を形成する。通常、3または4つの骨格部位が必要とされる。典型的に、鎖状の骨格には3つであり、環状の骨格には4つである。特に好ましい骨格部位は、ヘテロ原子または官能部位を片端または両端に任意に有する短い炭化水素鎖(たとえば、ここで述べたもの)を含む。アミン基およびアミド基はこの観点から好適である。
【0096】
前記カップリング基は、前記リンカー、骨格またはキレート部位のどのような好適な位置に結合していてもよい、たとえば式IV中で述べられたa)、b)および/またはc’)の位置が挙げられる。カップリング部位の結合は、どのような強い結合によるものであってもよい、たとえば炭素−炭素結合、エステル、エーテル、アミン、アミド、チオエーテルまたはジスルフィド結合が挙げられる。同様に、標的部位にそのような結合を形成することが可能な基はカップリング部位の官能基に適し、その部位は標的部位に結合している時に、そのような基を終結させる。
【0097】
式IIIに関し、式IVの構造において、RNおよびRLは同じ基であって、そのためピリジン環の窒素原子を介してキレート部位が骨格RBに結合する。このことにより、下記タイプIVBの構造となる。
【0098】
【化7】
【0099】
「骨格」タイプのオクタデンテートリガンドであって、2つの1,2および3,2−HOPOキレート部位を有し、骨格にアミドリンカー基で結合するものの例は、下記の式Vで表わされる。
【0100】
【化8】
【0101】
「テンプレート化された」オクタデンテートリガンドであって、エチルアミド基によって、エチルおよびプロピルジアミンに結合している4つの3,2−HOPOキレート部位を有するものを、それぞれ下記式VIおよびVIIに示す。
【0102】
【化9】
【0103】
上記式に表わされるように、3,2−HOPO部位の自由窒素は、メチル基および小さいヒドロカルビル基、たとえばメチルまたはエチルは、この位置が好ましい(式IのR1または式IIのRN)。
【0104】
「テンプレート化された」オクタデンテートリガンドであって、HOPO環における窒素によって、アミンシクランに結合している4つの3,2−HOPOキレート部位を有するものを、下記式VIIIに示す。
【0105】
【化10】
【0106】
上記で述べたように、オクタデンテートリガンドは典型的にリガンドの残りとどのような位置でも結合可能なカップリング部位を含む。カップリング基を終結させる官能部位を有する例示的化合物は、この態様によれば、下記構造IXである。
【0107】
【化11】
【0108】
ここで参照した全ての文献は、参照として組み込む、その中には、Gordon AEVら、プルトニウムおよび他のアクチニドの封鎖薬の合理的設計.Chem.Rev.2003,103,4207−4282,PCT特許出願WO2008/063721 A2およびT.N.Lambertら、Tetrahedron Letters 43(2002)7379−7383が含まれる。
【0109】
本発明は、以下の実施例において表わされるが、特に制限されることはない。実施例に例示された全ての化合物は本発明の好ましい態様である(好ましい中間体および前駆体を含む)そして、文脈に沿う限りどのような観点においても単独で、または組み合わせて用いられることができる。こうして、たとえば、実施例2の化合物2〜4のそれぞれおよびすべて、実施例3の化合物10ならびに実施例4の化合物7が、さまざまなタイプの中で好ましい態様を形成する。
【実施例】
【0110】
以下、実施例に基づいて本発明をさらに具体的に説明するが、本発明はこれらの実施例に限定されるものではない。
【0111】
[実施例1]
<純トリウム−227の単離>
トリウム−227は、アクチニウム−227カウ(cow)から単離される。アクチニウム−227は、ラジウム−226の熱中性子照射をした後、続いてラジウム−227(t1/2=42.2m)のアクチニウム−227への放射線崩壊することにより製造される。トリウム−227は、アクチニウム−227の放射線崩壊混合物の8MのHNO3溶液をアニオン交換クロマトグラフィーを通過させると選択的に保持される。カラムは、内径が2mmであり、長さが30mmであり、AG(登録商標)1−X8樹脂(200−400メッシュ、硝酸塩の形態)を70mg含有するものを用いた。アクチニウム−227、ラジウム−223および娘はカラムから溶出された後、トリウム−227は、12MのHCl溶液でカラムから抽出された。トリウム−227を含む抽出液は、エバポレートして乾燥され、残渣は0.01M HClで再度けん濁された。
【0112】
[実施例2]
<ALG−DD−NCSの合成>
【0113】
【化12】
【0114】
3−ベンゾイルオキシ−1−メチル−4−(2−チオキソチアゾリジン−1−イル)カルボニル−2(1H)−ピリジノン(Raymond,K.; Xu, J.,.US5,624,901に従って合成した)(2.22mmol,0.8g)、トリエチルアミン(0.31ml,2.22mmol)およびDMAP(5mg)を[5−アミノ−6−((2−アミノ−エチル)−{2−[ビス−(2−アミノ−エチル)−アミノ]−エチル}−アミノ)−ヘキシル]−カルバミン酸−t−ブチルエステル(BocLys−H(2,2)amine)(Raymond,K.; Corneillie,T.M.;Xu.J.WO2008/063721に従って合成した)(0.204g,0.505mmol)のジクロロメタン(80ml)溶液に加えた。前記混合物を室温で一晩攪拌し、それからエバポレートして乾燥した。残渣はジクロロメタンに溶解し、フラッシュシリカゲルカラムにかけ、ジクロロメタン中の2−8%メタノール勾配により溶出する。適切なフラクションを集め、エバポレートして乾燥し、ALG−001(1)(〜0.4g)を薄いベージュ色の粘性油として得た。
MS(ESI,pos):m/z 1369[M+H]+,m/z 1391[M+Na]+
ALG−001(1)(280mg,0.205mmol)を氷冷した酢酸(20ml)に溶解し、20% Pd(OH)2および炭触媒(60mg)を加えた、そして、前記混合物を40−45psi、室温で一晩水素添加した。濾過後、ロータリーエバポレーションによりALG−DEBN(2)(〜260mg,痕跡量の酢酸を含む)を赤色の粘性油として得た。
MS(ESI,pos):m/z 1007[M+H]+,m/z 1029[M+Na]+
ALG−DEBN(2)(70mg)を室温で2:1のMeOH/ジクロロメタン(15ml)に溶解した。それから精製された0.5gのAmberlyst−15樹脂を加え、前記混合物を穏やかに一晩攪拌した。前記樹脂はその後濾過により除去し、ヘキサン(10ml)、テトラヒドロフラン(10ml)およびメタノール(10ml)で順に洗浄した。このアミンが結合した樹脂は、4Mのアンモニアメタノール溶液(20ml)に移し、穏やかに50分攪拌した。テトラヒドロフラン(10ml)を加え、すべての脱保護生成物を溶解した。前記樹脂はそれから濾過により除かれ、前記溶液はエバポレートされ、ALG−DEBN−DEBOC(3)(37mg)を得た。
MS(ESI,pos):m/z 909[M+H]+,m/z 931[M+Na]+
40mgのALG−DEBN−DEBOC(3)の6:1イソプロパノール−水(v/v,7ml)懸濁液を1,4−フェニレンジイソチオシアネート(4.1当量)のクロロホルム(2.5ml)溶液と反応させた。試料をMS分析にかけるとき、反応物質は、室温で30分攪拌されていた。予想された質量(1100に相当するピークが観測された。真空下揮発性物質を除去した後、約95mgの明るい茶色の固体が単離された。明るい茶色の固体はアセトニトリルに懸濁し、前記混合物を15分間還流下加熱した。前記混合物は室温まで冷却し、続いて茶色の固体として26mgのALG−DD−NCS(4)を濾過して単離した。
[実施例3]
<対称な3,2−HOPO 含有キレート剤の合成>
【0115】
【化13】
【0116】
水素化ナトリウム(ミネラルオイル中の60%分散液,60.1g,1.5mol,5当量)をフラスコに入れ、テトラヒドロフラン(1L)を加えた。マロン酸ジメチル(172ml,1.5mol,5当量)を1.5時間かけて滴下した。反応混合物の温度は、+10℃以下に保った。反応混合物は、それからテトラヒドロフラン(400ml)で希釈した。4−ニトロベンジルブロミド(65.0g,0.3mol,1当量)のテトラヒドロフラン(170ml)溶液を上記作成した混合物に激しく振りながら30分をかけてゆっくりと加えた。30分0℃で攪拌した後、前記反応混合物は、塩水(1Lの飽和食塩水)に注ぎ、室温で一晩攪拌して、白い沈殿物を得た、前記混合物は、メチルtert−ブチルエーテルで希釈し、前記沈殿物を濾過して温エタノールに溶解した。前記エタノール混合物を濾過して、ろ液を濃縮して体積を少なくし、そこから24.8g(31%)のジメチル(4−ニトロベンジル)プロパンジオネート(1)が白色の結晶状の固体として沈殿した。
LC純度:>90%(254nm)
MS(APCl,pos):m/z 285.1[M+NH4]+
化合物1(24.8g,92.7mmol)をテトラヒドロフラン(230mL)に溶解し、ボラン−ジメチルスルフィド複合体(28.5ml,301ml,3.3当量)に加えた。前記反応混合物を24時間還流し、それから室温で一晩置いた。0℃でメタノール(250ml)を加え、塩水(600ml)に注ぎ、酢酸エチルで抽出した。前記合わせた有機層をNa2SO4の上で乾燥させ、真空下で濃縮した。残渣はメタノールと蒸発した。粗生成物は、フラッシュクロマトグラフィーで精製し、14.5g(74%)の2−(4−ニトロベンジル)プロパン−1,3−ジオール(2)を黄色の油として得た。
LC純度:>92%(254nm)
化合物2(10.0mg,47.3mmol)をジクロロメタン(100ml)に溶解し、トリエチルアミン(14.5ml,104mmol,2当量)を加えた。前記反応混合物は、氷浴で冷やし、メタンスルホニルクロライド(8.0ml,104mmol,2当量)を少しずつ加えた。最終混合物は、室温になるまで一晩置いた。それから、さらなるトリエチルアミン(1.5ml)およびメタンスルホニルクロライド(0.8ml)を加え、30分攪拌した。それから前記反応混合物は、ジクロロメタンに溶解し、生成した沈殿物を濾過して除去した。前記ジクロロメタンのろ液は飽和NaHCO3水溶液、0.5MのHCl水溶液および塩水で洗浄し、Na2SO4の上で乾燥させ、真空下で濃縮し、14.5g(83%)の2−(4−ニトロベンジル)プロパン−1,3−ジイルジメタンスルフォネート(3)をオレンジ色の油として得た。化合物(3)は、さらに精製することなく次の段階で用いた。
LC純度:>87%(254nm)
MS(APClpos):m/z 385.3[M+NH4]+
化合物3(14.5g,39.5mmol)は、メチルエチルケトン(100ml)、ヨウ化ナトリウム(16.0g,107mmol,2.7当量)を加え、前記混合物を95℃で1時間加熱した。得られた白色の沈殿物は濾過して除去し、メチルエチルケトンで洗い、前記ろ液は真空下で濃縮した。前記粗生成物はメチルtert−ブチルエーテルから砕いて、フラッシュクロマトグラフィーで精製して、10.6g(63%)の1−[3−ヨード−2−(ヨードメチル)プロピル]−4−ニトロベンゼン(4)を黄色の固体として得た。
LC純度:>90%(254nm)
MS(APClpos):m/z 431.1[M+H]+
ジエチレントリアミン(10.8ml,100mmol)およびトリエチルアミン(42ml,300mmol,3当量)をテトラヒドロフラン(500ml)に溶解し、氷浴で冷やした。それからBoc−ON(49.5g,200mmol,2当量)をテトラヒドロフラン(190ml)で溶解した溶液を2.5時間かけて滴下した。前記反応混合物は、さらなる1時間0℃で攪拌し、その後室温になるまで一晩置いた。それから前記反応混合物は、真空下濃縮した。残渣はジクロロメタンに溶解し、1MのNaOH水溶液で洗った。前記有機層はNa2SO4の上で乾燥させ、真空下で濃縮した。粗生成物はフラッシュクロマトグラフィーで精製し、20.0g(66%)のジ−tert−ブチル(イミノジエタン−2,1−ジイル)ビスカーバメート(5)を黄色の粘性のある油として得た。
MS(APClpos):m/z 304.3[M+H]+
化合物5(26.0g,86mmol,4当量)をドライトルエン(40ml)に溶解し、N−エチルモルフォリン(5.4ml,42mmol,2当量)を加えた。化合物4(9.2g,21mmol)をドライトルエン(35ml)に溶解し、前記反応混合物に加えた。前記混合物は、圧力反応器で105℃で5日間保温した。それから、前記反応混合物は、室温に冷却し、酢酸エチルで希釈し、飽和NaHCO3水溶液で洗浄した。前記合わせた有機層を水と塩水で洗い、Na2SO4の上で乾燥させ、真空下で濃縮した。前記粗生成物はフラッシュクロマトグラフィーで精製し、11.4g(70%)の化合物6を黄色の堅い泡として得た。
LC純度:>96%(210nm)
MS(APCl,pos)m/z 782.8,[M+H]+;682.7[M+H−Boc]+;582.6[M+H−2Boc]+;382.1[M+H−4Boc]+
化合物6(2.0g,2.6mmol)をジオキサン(16ml)に溶解し、9MのHCLのジオキサン(22ml)溶液を加えた。前記反応混合物は、室温で一晩攪拌し、真空下で乾燥した状態まで濃縮し、1.9gの化合物7のHCL塩をベージュ色の固体として得た。
MS(APClpos)m/z 382.5[M+H]+
化合物7(1.9g,2.6mmol)をDMPU(1,3−ジメチル−3,4,5,6−テトラヒドロ−2(1H)−ピリミジノン)(9.5ml)および3−(ベンゾイルオキシ)−1−メチル−4[(2−チオキソ−1,3−チアゾリジン−3−イル)カルボニル]ピリジン−2−(1H)−オン(3.7g,10.3mmol,4当量)に加えた。それから、DBU(2.3ml,15.5mmol,6当量)をDMPU(4.7ml)に溶解した溶液をゆっくり40分間で滴下した。一晩室温で攪拌を続けた。前記反応混合物はジクロロメタンで希釈して飽和NaHCO3水溶液で洗浄した。前記合わせた有機層を水と半飽和塩水で洗い、Na2SO4の上で乾燥させ、真空下で濃縮した。前記粗生成物はフラッシュクロマトグラフィーで精製して、4.0gの化合物8を黄色の油として得た。化合物8は更なる精製をすることなく次の段階で用いた。
MS(APCl,pos)m/z 1347.7[M+H]+
化合物8(4.0g,2.6mmol)を酢酸(200ml)に溶解し、Pd(OH)2(炭素上20% w/w, 50%湿潤)(800mg,10% w/w)を加えた。前記反応混合物は、Parr圧力反応器で30barの水素圧、室温で一晩攪拌した。前記反応混合物は、Celite(登録商標)のパッドを通過させて濾過し、ろ液を真空下で濃縮した。粗生成物はフラッシュクロマトグラフィーで精製し、化合物9をベージュの泡状の固体として得た。
【0117】
化合物9(996mg)を酢酸(5.5ml)に溶解し、続いて6MのHCl水溶液および5%のPd/C(99mg,10% w/w)を加えた。前記反応混合物は、圧力反応器で8barの水素圧で一晩攪拌し、それからCelite(登録商標)のパッドを通過させて濾過した。前記ろ液は真空下で濃縮し、トルエンおよびメタノールとともにエバポレートした。前記残渣はメタノールに溶解し、ジエチルエーテルを加えて沈殿させた。形成したベージュの沈殿物は濾過して除き、真空下で一晩乾燥し、700mgの化合物10のHCL塩である、ALG1005−38を得た。
MS(APCl pos)m/z 956.7[M+H]+
[実施例4]
<別の対称な3,2−HOPO 含有キレート剤の合成>
【0118】
【化14】
【0119】
無水Boc(24.4g,119mmol)をイミダゾール(8.0g,117mmol)の50mlジクロロメタン溶液に少しずつ加えた。前記反応混合物を1時間かけて攪拌した。前記反応混合物を50mlの水で2回洗浄し、Na2SO4の上で乾燥させ、濾過し真空下で濃縮した。前記残渣は、25mlのトルエンおよびトリス(2−アミノエチル)アミン(8.19g,56mmol)を加えた。前記反応混合物は、60℃で2時間攪拌して、真空下で容量を減らした。前記残渣は125mlのジクロロメタンに溶解し、水で洗い(50mlで4回)、Na2SO4の上で乾燥させ、濾過し真空下で濃縮した。ドライフラッシュクロマトグラフィー(1%のトリエチルアミンを加えた0〜15%のメタノールのジクロロメタン溶液)をして、ジ−tert−ブチル(((2−アミノエチル)アザンジイル)ビス(エタン−2,1−ジイル))ジカーバメート(1)(13.51g,39.0mmol,70%)を薄い色のない油として得た。
【0120】
分光データは、Frullanoらによって報告されたデータ(Chem.Eur.J.2004,10,5205−17)に従う。
【0121】
N−Cbz−β−アラニン(8.04g,36mmol)、4−(ジメチルアミノ)ピリジン(4.40g,36mmol)およびEDC・HCL(6.90g,36mmol)を25mlのテトラヒドロフランに溶解し、10分間攪拌した後、化合物1(10.4g,30mmol)を75mlのテトラヒドロフランに溶解したものを加えた、前記反応混合物は、4時間攪拌し、真空下で容量を減らした。ドライフラッシュクロマトグラフィー(0〜40%のテトラヒドロフランのジクロロメタン溶液)により精製し、ジ−tert−ブチル(((2−(3−(Cbz−アミノ)プロパンアミド)エチル)アザンジイル)ビス(エタン−2,1−ジイル))ジカーバメート(2)(15.11g,27.4mmol,91%)を非常に粘性の高いわずかに黄色の油とした得た。
1H−NMR(300MHz,CDCl3):1.43(s,18H),2.42−2.56(m,8H),3.04−3.17(m,4H),3.19−3.32(m,2H),3.41−3.57(m,2H),5.08(s,2H),5.22(bs,2H),5.83(bs,1H),7.23(bs,1H),7.26−7.43(m,5H)
MS(ESL,pos)m/z 574.3[M+Na]+
ジ−tert−ブチル(((2−(3−(Cbz−アミノ)プロパンアミド)エチル)アザンジイル)ビス(エタン−2,1−ジイル)ジカーバメート(10.30g,18.7mmol)を100mlのメタノールに溶解し、アセチルクロライド(25ml,0.35mol)を滴下した。前記反応混合物は、室温で1時間攪拌した。前記反応混合物は、真空下でおおよそ1/3の体積にしてから、100mlのエーテルを加え、無色の固体を沈殿させた。前記固体は濾過し、エーテルで洗い、真空下で乾燥し、ベンジル(3−((2−(ビス(2−アミノエチル)アミノ)エチル)アミノ)−3−オキソプロピル)カーバメートトリヒドロクロライド(3)(8.70g,18.7mmol,〜100%)を無色の固体として得た。
1H−NMR(300MHz,MeOD):3.19−3.28(m,2H),3.38−3.62(m,14H),5.10(s,2H),7.29−7.39(m,5H)
MS(ESL,pos)m/z 352.2[M+H]+
化合物3(10.56g,11.4mmol)の320mlのテトラヒドロフランの懸濁液および320mlのN,N−ジメチルホルムアミドにトリエチルアミン(6.75ml,48.4mmol)を室温で加えた。N−boc−2−アミノアセトアルデヒド(16.0g,100.5mmol)およびトリアセトキシボロヒドリドナトリウム(32.00g,151mmol)を加えた。前記反応混合物は、20時間攪拌した。200mlの塩水および500mlのクロロホルムを加え、相を分離し、前記水層を100mlのクロロホルムで3回抽出した。前記合わせた有機層は100mlの食塩水で洗い、Na2SO4の上で乾燥させ、濾過し、真空下で体積を減らした。フラッシュクロマトグラフィー(0〜10%のメタノールのジクロロメタン溶液)により、テトラ−tert−ブチル(((((2−3−Cbz−アミノプロパンアミド)エチル)アザンジイル)ビス(エタン−2,1−ジイル))ビス(アザントリイル)テトラキス(エタン−2,1−ジイル)テトラカーバメート(4)(3.40g,3.7mmol,32%)を黄色の固体とした得た。
MS(ESL,pos):m/z 946.7[M+Na]+
化合物4(3.40g,3.7mmol)を60mlのメタノールに溶解し、アセチルクロライド(12ml,0.17mol)を滴下した。前記反応混合物を室温で1時間攪拌した。前記反応混合物を、真空下でおおよそ10mlの体積にしてから、50mlのアクリロニトリルを加え、沈殿させた。前記固体は濾過し、アクリロニトリルで洗い、真空下で乾燥し、ベンジル(3−((2−(ビス(2−アミノエチル)アミノ)エチル)アミノ)−3−オキソプロピル)カーバメートヘプタヒドロクロライド(5)(2.88g,3.7mmol,〜100%)を無色の固体として得た。
1H−NMR(300MHz,MeOD):2.44−2.61(m,2H),2.68−3.00(m,4H),3.08−3.40(m,16H),3.44−3.52(m,4H),3.54−3.77(m,6H),5.12(s,2H),7.30−7.47(m,5H)
MS(ESL,pos):m/z 524.5[M+H]+
トリエチルアミン(2.52ml,18.1mmol)および4−(ジメチルアミノ)ピリジン(12mg,触媒)を化合物5(0.81g,1.04mmol)および3−(ベンジルオキシ)−1−メチル−4−(2−チオキソチアゾリジン−3−カルボニル)ピリジン−2(1H)−オン(1.50g,4.16mmol)の180mlのジクロロメタンの懸濁液に加えた。前記反応混合物は一晩攪拌し、真空下で体積を減らした。フラッシュクロマトグラフィー(0〜10%のメタノールのジクロロメタン溶液)により、ベンジル(1−(3−ベンジルオキシ)−1−メチル−2−オキソ−1,2−ジヒドロピリジン−4−イル)−5−(2−(3−(ベンジルオキシ)−1−メチル−2−オキソ−1,2−ジヒドロピリジン−4−カルボキサミド)エチル)−8−(2−(ビス(2−(3−ベンジルオキシ)−1−メチル−2−オキソ−1,2−ジヒドロピリジン−4−カルボキシアミド)エチル)アミノ)エチル)−1,12−ジオキソ−2,5,8,11−テトラアザテトラデカン−14−イル)カーバメート(6)(673mg,0.45mmol,43%)を明るい茶色の固体として得た。
【0122】
1H−NMR(300MHz,CDCl3):2.10−2.40(m,20H),3.02−3.21(m,10H),3.32−3.46(m,2H),3.50(s,12H),4.99(s,2H),5.25(s,8H),5.95(bs,1H),6.62(d,7.17Hz,4H),7.01(d,7.17Hz,4H),7.15−7.40(m,25H),7.89(bs,4H),
13C−NMR(75MHz,CDCl3):11.48,35.71,37.18,37.35,37.50,37.66,46.18,51.55,52.15,52.82,53.28,66.34,74.64,74.75,77.43,104.63,127.90,127.94,128.40,128.65,128.75,128.94,130.81,132.32,136.27,136.76,146.19,156.45,159.51,163.30,171.31
MS(ESL,pos):m/z 766.9[M+2Na]2+
化合物6は、基本的に実施例2に記載されているように、溶解され、脱ベンジル化し、Bb−1−HOPO−1−DEBN(7)を得た。
[実施例5]
<ALG−DD−NCSとのキレート化実験>
固体のALG−DD−NCS(実施例2の4)をDMSO(バイオテックグレード溶媒99.8%)に1mg/mlとなるように溶かし、反応溶液を調製した。
【0123】
異なる金属とのキレート化反応は1mg/mlの反応溶液を選択された金属溶液と混合し、リガンド対金属が3:2とすることにより行った。試験のための金属イオンは以下のようにして得た:232Thの2%HNO3(パーキンエルマーピュアプラス)溶液、Fe3+の2%HNO3(パーキンエルマーピュアプラス)溶液およびInCl3(無水粉末99.999+%,アルドリッチ)を0.01MのHClに溶解した。前記試薬は室温で15分反応させてから、5μmの反応混合物をLC−MSに入れ分析した。
【0124】
結果物は、金属−ALG−DD−NCS錯体が232Th4+、Fe3+およびIn3+の金属に対して定量的に形成された。232Th−ALG−DD−NCS、Fe−ALG−DD−NCSおよびIn−−ALG−DD−NCS錯体のLC−MSクロマトグラムおよびスペクトルを図1−6に示す。
【0125】
前記LC−MSの条件は以下の通りである:分離は1.7μm、2.1×50mmのAcquity UPLC BEH C18カラム、50℃、0.05%のギ酸水溶液を移動相Aおよび0.05%のギ酸のアクリルニトリル溶液を移動相Bとして用いた。前記傾斜組成および流量を表3に示す。
【0126】
【表3】
【0127】
マススペクトル分析は、Xevo−ToF−1で、取得質量範囲は〜490−1950Da、質量ES+、3.0kVキャピラリー、コーン電圧20Vで1000秒以上スキャンした。
[実施例6]
<ALG10058−38とのキレート化実験>
ALG1005−38(実施例3の10)をメタノール(LC−MSグレード)に溶解して10mg/mlとして原液を調製した。前記10mg/ml原液は、メタノールおよび0.5MのNaOAc−緩衝液(pH5.5)で1:8:1の割合で希釈し、1mg/mlのALG1005−38反応溶液を得た。異なる金属とのキレート化反応は実施例5と同様に1mg/mlの反応溶液を選択された金属溶液と混合し、さらにGaCl3(無水ビーズ 99.99%、アルドリッチ)を0.01M Hclに溶解したものを加え、リガンド対金属が3:1〜1:2とした。前記試薬は室温で10分反応させた。最後に前記反応混合物は10倍のギ酸(0.1%)で希釈してから、5μmの反応混合物をLC−MSに入れ分析した。前記LC−MS分離および分析は、コーン電圧を35Vとして以外は、実施例5で述べたように行った。
【0128】
結果物は、金属−ALG1005−38錯体が232Th4+、Fe3+、In3+およびGa3+の金属に対してかなりの量で形成された。232Th−ALG1005−38、Fe−ALG1005−38、In−ALG1005−38およびGa−ALG1005−38錯体のLC−MSクロマトグラムおよびスペクトルを図7−14に示す。
[実施例7]
<Bb−1−HOPO−1−DEBNとのキレート化実験>
Bb−1−HOPO−1−DEBN(実施例4で製造したもの)をDMSO(バイオテックグレード溶媒99.8%)に溶解して1mg/mlとして原液を調製した。キレート化実験は前記1mg/mlの溶液を実施例5で述べた232Th0.01M HCl(パーキンエルマー)溶液と混合することにより行った。前記LC−MS分離および分析は、実施例6で述べたように行った。
【0129】
結果物は、予想された232Th−Bb−1−HOPO−1−DEBN複合体がすばやく形成された、図15および16に示す。
[実施例8]
<ALG−DD−NCSのtrastuzumabへの接合、トリウム−227で接合の標識化を行い、標的の結合が保持されていることを確認した。>
接合
trastuzumabのモノクローナル抗体の薬剤グレード(Herceptin(登録商標),ロッシェ)を用いた。前記抗体は、0.9%のNaClに緩衝液交換をして6.3mg/mlに濃縮した。ALG−DD−NCSの原液はDMSOに溶解して1mg/mlに調製した(純度は考慮しない)。DMSO原液は、キレーター対抗体のモル比が6.5:1以下に相当する抗体溶液に加えた。0.07Mのホウ砂溶液を反応溶液に加え、pH9にし、一晩37℃で培養した。得られた接合を精製し、Amicon Ultra−4(30k MWCO)遠心分離フィルターユニットで緩衝液交換をした。1mgを100μlの0.9%のNaClに分注し、凍らせ、使用するまで−18℃で保管した。
標識化
100μlの0.5MのNaOAc−緩衝液(pH5.5)を1mgのALG−DD−NCS−trastuzumab接合の小瓶に加え標識化に適切なpHとした。この溶液を0.01M HCl中の4.2−16.4MBqの227Thのと混合し、そのpHをpH紙で確認した。エッペンチューブキャップをアルミニウムホイルでくるみ、前記チューブを室温でサーモミキサー中に置き15分穏やかにかき混ぜた。
【0130】
前記チューブは10μlの飽和DTPAのMF−H2Oに加えた後、さらに室温で5分間培養し、保持されている自由トリウム−227をとらえた。
PBSを用いて標識化されたALG−DD−NCS−trastuzumab接合を希釈して、結果物をNAP−5カラムで精製した、そして自由放射性核種(トリウム−227および娘核種)およびDTPA−トリウムキレートをの大多数はカラムに残されていた。
【0131】
前記カラムおよび希釈されたフラクションはHPGe−検出器 GEM(15)で測定して、生成物の反応収率および特定の活性を測定した。前記生成物のフラクションは濾過され4℃で一晩保管した。
前記生成物フラクションの第2のNAP−5カラムによる精製を使用前に行った。
免疫反応性フラクションの測定
SK−OV−3細胞(ATCC)を標準状態で培養した。固定されたSK−OV−3細胞は、調製され、使用前にフローサイトメトリーにより分析されてその表面にHER2が存在することを確認した(データは開示しない)。正規の培地は、McCoyの5A培地(GIBCO)に10%FBS(PAA)および1%Pen/Strep(BioChrom)を加えて調製した。細胞の培養は、T25(25cm2)およびT75(75cm2)細胞フラスコを用いて、37℃および5%CO2でCO2培養器で行った。
【0132】
それぞれの実験は2回行った。固定したSK−OV−3細胞はPBSに懸濁し、エッペンチューブに移した(10000,000細胞/チューブ)。非接合trastuzumab(Herceptin(登録商標),原液から10μl)を2つの参照チューブに加え、37℃で30分間、阻害反応を行った。同量の227Th−ALG−DD−NCS−trastuzumab(ca 500cpm)をそれぞれのチューブに加えてから、37℃で25時間培養した。前記試料はPBSで希釈してから、遠心分離して上澄みの半分を新しいエッペンチューブに移した。すべてのチューブは、ウィザードガンマカウンターで5分間測定し、トリウム−227の量を227Th−プロトコルを適用して測定した。
【0133】
結合の%は、以下のように計算した:100*[(P+1/2S)−(1/2S)][(P+1/2S)+(1/2S)]、ここでPおよびSは、それぞれ前記細胞ペレットおよび前記上澄み中で測定された活性を示す。前記計算は、全体の結合(%)を求めるために、非阻害試料に関して行い、非特異結合(%)を求めるために、阻害試料に関し行った。そしてIRFは以下のように計算した:IRF(%)=全体の結合(%)−非特異結合(%)
この品質管理からの前記データは、表4にまとめ、227Th−ALG−DD−NCS−trastuzumabが標識細胞に結合していることを表わしている。
【0134】
【表4】
【0135】
i. 収率(%)は、反応混合物のNAP−5精製の後計算した。
ii. 回収(%)は、生成物フラクションの新たなNAP−5精製の後計算した。
iii. 特異活性(Bq/μg)は、生成物フラクション中に見つけられた227Th(Bq)の量を反応混合物中に用いられたALG−DD−NCS−trastuzumab(μg)の量に基づいて割ったものを用いて計算した。
iv. IRFは標準の手順に従って見積もった。
[実施例9]
<トリウム−227で標識化されたALG−DD−NCS−trastuzumabのin vitro安定性および有効性の研究>
時間をかけた結合の評価
227Th−ALG−DD−NCS−trastuzumabのSK−OV−3細胞への結合の安定性は、SK−OV−3細胞ペレットと関連づけられた227Th−活性の量を7日を超えて測定して評価した。前記結果は自由227Thとともに培養したデータと比較した。
【0136】
実施例8で述べたように培養したSK−OV−3細胞は最初に700kBqの227Th−ALG−DD−NCS−trastuzumabまたは700kBqの自由227Thで扱い、1時間培養した後(HPGe−検出器 GEM(50))細胞ペレット上に、それぞれ77kBq(11%)および7kBq(1%)227Th−活性を生じた。前記SK−OV−3細胞上の227Th−活性は、毎日ウィザードガンマカウンター(227Th−プロトコル)で測定した。前記測定した放射能は、崩壊に対する補正をした。前記結果は図17に示すように1時間後にペレットに残されたたった20%の活性が、次の7日間で消滅した、それに対して、自由227Thで扱った細胞は加えられた活性の小さなフラクションにのみ結合し、その多くが(ca95%)続く24時間の間に消失した。こうして、自由227ThのSK−OV−3細胞への特異結合はなく、一方で227Th−ALG−DD−NCS−trastuzumabは効果的に保持された。
細胞毒性の評価
腫瘍細胞の成長の効果を2つの相補的な分析で研究した。最初の実験で、細胞中のメタボリック活性はATP分子に結合した発光試薬を用いて測定した。このようにして、発光シグナルの減少は、メタボリック活性の消失を示す。第2の実験で、形成された多くのコロニーの数が数えられた。
【0137】
発光は、毎日9日間測定した、そしてその結果227Th−ALG−DD−NCS−trastuzumabへの被爆からの結果をtrastuzumabおよび自由227Thのそれぞれへ被爆したSK−OV−3細胞の発光に比較した。SK−OV−3細胞の正規の培地での成長は、参照として用いた。
0日目
正規の培地中の35mlの500000SK−OV−3細胞/mlを4 T75細胞フラスコに移した。これらの細胞フラスコは以下の試料溶液を調製するために用いた:(1)正規の培地、(2)20μg/mlのtrastuzumab、(3)20kBq/mlの227Th−ALG−DD−NCS−trastuzumab(10166Bq/μg)、(4)20kBq/mlの自由227Th。前記フラスコは、CO2培養器で1時間培養した。前記上澄みを捨て、前記細胞を0.25%のトリプシン−EDTA溶液でトリプシン処理し、正規の媒体で洗い遠心分離した。細胞ペレットは、新鮮な正規の媒体に懸濁し、ウィザードガンマカウンター(227Th−プロトコル)で測定した。それぞれの培養溶液は、新しい7つのT25細胞フラスコに分配し、CO2培養器で培養した。
1日目および2日目
4つの試料溶液のそれぞれの1つのフラスコからの前記上澄みは、捨てた。前記細胞ペレットは、0日目と同様に調製して、放射能はウィザードガンマカウンター(227Th−プロトコル)およびHPGe−検出器 GEM(50)で測定した。全ての試料は、正規の媒体で4倍に希釈し、それぞれの懸濁出来の5×100μlを5wellのView Plate−96に移した。5つのwellは100μmの正規の媒体(細胞を含まない)を加え、背景発光の測定をした。最後に、100μLのCell Titer−Glo 試薬をそれぞれのwellに加え、前記溶液は溶解を誘導するためにオービタルシェーカーで混合した。前記発光シグナルは、室温で10分間安定させた、それから発光は、LUM−シングルプログラムを用いて、エンビジョンマルチプルプレートリーダーで3回測定した。
3日目
細胞は、1日目および2日目で述べたように調製して、ウィザードガンマカウンターおよびエンビジョンマルチプルプレートリーダーで測定した。それから、細胞の成長で濃くなっているため、それぞれの細胞の懸濁液を希釈した;3/4の細胞の懸濁液は新しいT75細胞フラスコに移し、正規の媒体を加え、CO2培養器で一晩培養した。
4〜9日目のそれぞれ
4つのT75細胞フラスコを0.25%のトリプシン−EDTA溶液でトリプシン処理し、正規の媒体で洗浄し、遠心分離した。細胞ペレットは4倍の正規の媒体で希釈し、100μlを取り出し、上述のように測定した。それから3/4の細胞の懸濁液は正規の媒体で希釈し、新しいT75細胞フラスコに移し、CO2培養器で一晩培養した。
【0138】
図18は、227Th−ALG−DD−NCS−trastuzumab(10166Bq/μg)、trastuzumabおよび自由227Thで処理したSK−OV−3細胞の発光測定を示す、それは正規の媒体(参照)中で育てられたSK−OV−3細胞について得られた発光値に標準化されたものである。前記227Th−ALG−DD−NCS−trastuzumabで処理したSK−OV−3細胞の発光シグナルは、1日目〜9日目にかけて徐々に減少し、一方他の処理をしたものは、この期間の間ほぼ一定の発光シグナルと成長を示した。227Th−ALG−DD−NCS−trastuzumabで処理したSK−OV−3細胞のメタボリック活性は、その期間の間減少し、9日目にはほとんど全ての細胞が死滅した、この結果は、227Th−ALG−DD−NCS−trastuzumabの重要な維持された細胞毒性を示す。
【0139】
一つの細胞を滞在的に細胞毒性の状況にさらした後のコロニーの成長を評価するために、クローン形成を行ったSK−OV−3細胞は、異なる濃度の227Th−ALG−DD−NCS−trastuzumab、trastuzumabまたは自由227Th中で1時間培養し、続いてコロニーの成長を誘導するために12日間培養した。
【0140】
細胞の調製は、上記のようにして行った。そして試料ごとに5mlの正規の媒体中の12000細胞を、T25フラスコに移動した。227Th−ALG−DD−NCS−trastuzumabおよび227Thの5、10および20kBq/mlならびにtrastuzumabの5、10および20μg/ml(最終濃度)のそれぞれについて、それぞれの試薬の3種の量の効果を調べた。1つの参照フラスコを調製した、全ての10のT25細胞フラスコは、CO2培養器で一時間培養した。前記上澄みは捨て、前記細胞は正規の媒体で洗浄し、0.25%のトリプシン−EDTA溶液でトリプシン処理し、正規の媒体で洗浄し、遠心分離した。細胞ペレットは正規の媒体に懸濁し、10の細胞の懸濁液のそれぞれを、6つの新しいT25細胞フラスコに分けた、3つの細胞フラスコは1000細胞であり3つの細胞フラスコは3000細胞である、えられた60のT25細胞フラスコは、CO2培養器でコロニーが目に見える大きさになるまで培養した(ca 50細胞/コロニー)。12日間培養した後、前記媒体は除去した、細胞はPBSを用いて洗い、エタノールで還流し、PBS中の0.25%トリパンブルーで染め、水道水で洗い、最後に45℃で一晩乾燥させた。コロニーは手作業で数え、それぞれの培養におけるコロニーの平均数は、グラフに表した。
【0141】
図19および20に示された結果は、227Th−ALG−DD−NCS−trastuzumabを用いた1時間の培養は、効果的にSK−OV−3細胞を殺すことを示している。自由227Thを用いた培養は、いくらかの細胞を殺す効果があったが、たくさんの数の細胞を用いた最初の細胞毒性分析では明らかでなかった。trastuzumabを用いた培養は、細胞の成長に否定的な効果はなかった。
[実施例10]
<トリウム−227で標識化されたALG−DD−NCS−trastuzumabのin vivo腫瘍標的>
100μl(15kBq)の細菌濾過した227Th−ALG−DD−NCS−trastuzumabをSK−OV−3の異種移植片を有する10匹のBalb/cヌードマウスの尾の側面の血管に注入した。5匹のマウスは24時間後に犠牲となり、残りの5匹は4日後に犠牲となった。そして実験対象のマウスの組織を切除して重量を測った。全ての試料および10% ID/gを含む3つの標準溶液は5分間ウィザードガンマカウンター(227Th−プロトコル)で測定した。結果はグラム組織(% ID/g)当たりの注入量の%で表わした。
【0142】
前記結果は図21に示すように、227Th−ALG−DD−NCS−trastuzumabは、特異的に腫瘍に結合する。取り込み値は、適度であるが、24時間と4日の間にいくらか上昇している(8.7% ID/g〜11.3% ID/g)、一方で血液中の濃度は1%ID/gあたりに減っている。
【0143】
頭骸骨および大腿骨における取り込み量もまた、いくらか24時間および4日の間に上昇した、しかしながら、これらの値はほとんどの場合低く(2.8−3.2%、24時間)、系中を循環する自由227Thはほとんどない、そして227Th−ALG−DD−NCS−trastuzumab複合体の安定性の高い度合いを示している。さらにこの結論をサポートするものとして、頭骸骨および大腿骨において測定される活性フラクションは223Raの娘核種を取り込むことにより生じるという高い可能性がある。この223Raのいくらかは、ウィザードガンマカウンターの227Th−測定器のための窓の中に検出されることもある。こうして頭骸骨および大腿骨の計算された% ID/g−値は、これらの組織に蓄積した227Th−ALG−DD−NCS−trastuzumabの量を課題評価することもある。
[実施例11]
<キレート部位の合成>
3−ベンジルオキシ−1−メチル−4−(2−チオキソチアゾリジン−1−イル)カルボニル−2(1H)−ピリジノン(構造A)
ステップ1)−1−メチル−3−ヒドロキシ−2(1H)−ピリジノン
3−ヒドロキシ−2(1H)−ピリジノン(34.44g,0.31mol)およびヨードメタン(75g、0.53mol)を80mlの蓋をしたテフロン(登録商標)の入れ物に入れた。そして、Parr bomb中で48−60時間150℃で加熱した。冷却した前記bombを開け、過剰なヨードメタンを静かに注いだ。得られた薄い黒みがかった油を亜硫酸ナトリウム(64g,0.5mol)と混合し、300mlの水に溶解し、薄い茶色の溶液を形成した、前記溶液は、pH7−8に中和し、不溶の不純物を濾過して除去した。前記ろ液はそれからメチレンクロライド(4×100ml)。合わせた抽出物は、それから乾燥させ、フラッシュシリカゲルプラグ(6cm×8cm)をし、4%エタノールのメチレンクロライド溶液で希釈した。前記溶媒は、上記標題の化合物(24.3g,62.6%)を無色の結晶として得た。
【0144】
ステップ2)−4−カルボキシル−1−メチル−3−ヒドロキシ−2(1H)−ピリジノン
1−メチル−3−ヒドロキシ−2(1H)−ピリジノン(1)(6.25g,50mmol)を無水炭酸カリウム(36g,0.26mol)と混合し、真空下で乾燥した、前記混合物は、それからParr bomb中で二酸化炭素ガス(850psi)下で175−185℃で3日間加熱した。前記冷却したbombを開け、結果物の薄い黄色の固体を氷で冷やした水に溶解し、6N のHClで酸性にし、ベージュの結晶性の生成物を得た。
【0145】
ステップ3)−3−ベンジルオキシ−4−カルボキシ−1−メチル−2(1H)−ピリジノン
4−カルボキシル−1−メチル−3−ヒドロキシ−2(1H)−ピリジノン(6.8g,0.04mol)をベンジルクロライド(12.1g,0.088mol)および無水ジメチル−ホルムアミド(DMF)(120ml)中の無水炭酸カリウム(13.8g,0.1mol)と混合した。前記混合物は、暗闇で、窒素の存在下、75−80℃で16時間加熱した。前記混合物は、濾過し、溶媒をエバポレートして暗い色の油を得た。前記油は、シリカゲルプラグ(6cm×8cm)にかけ、4%メタノールのメチレンクロライド溶液で希釈し、3−ベンジルオキシ−4−ベンジルオキシカルボニル−1−メチル−2(1H)−ピリジノンを薄い黄色の、粘度の高い油として得た。これをメタノール(50ml)および6MのNaOH溶液(10ml)に採り、前記混合物を室温で4時間攪拌し、エバポレートして乾燥させた。前記残渣は水(100ml)に溶解し、6M HCl溶液で酸性にしてpH2とし、上記標題の化合物(9.3g,88.7%)を白色の結晶として得た。
【0146】
ステップ4)−3−ベンジルオキシ−1−メチル−4−(2−チオキソチアゾリジン−1−イル)カルボニル−2(1H)−ピリジノン
3−ベンジルオキシ−4−カルボキシ−1−メチル−2(1H)−ピリジノン(1.05g,4mmol),2−メルカプトチアゾリン(0.50g,4.2mmol)および触媒量の4−ジメチルアミノピリジン(DMAP)のドライジクロロメタン(50ml)溶液に、N,N’−ジシクロヘキシルカルボジイミド(DCC)(0.86g,4.2mmol)を加えた。4時間攪拌後、ジシクロヘキシルウレア(DCU)の固体を濾過により除去した。黄色のろ液をロータリーエバポレートして黄色の固体を得た。イソプロパノールメチレンクロライドからの再結晶により、標題の化合物(構造A,1.16g,80.4%)を明るい黄色の結晶板として得た。
【0147】
【化15】
【0148】
[実施例12]
<式VIIIで表わされる3,2−HOPO前駆体の生成>
環状塩(構造5)の生成は、下記式にしたがって行った。
【0149】
【化16】
【0150】
アクリルニトリルを還流させながら、フッ化セシウム(10mol%)により、2,3−ジヒドロキシピリジン(2,3−DHP)をエチルアクリレートとマイケル付加反応させ、対応するエステル2を良好な収率で得た。続く2のO−ベンジル化は標準の状態(K2CO3/アクリロニトリル/還流)で行い、続けてエステル部位の還元(BH3・THF、室温)を行い、クロマトグラフィーを行って、アルコール3を90%の収率で得た。アルコール3をトリエチルアミンの存在下、ジクロロメタン中のメタンスルホン酸無水物で処理し、中間体であるメシラート4(〜10−15%)を伴う目的とする環状塩5(85−90%)を直接形成したことを1H NMR分析で確認した。環状塩5への完全な変換は、室温でのクロロホルム中のメシル化からの粗生成物の攪拌によって達成される。温エチルアセテートで粉砕した後、前記塩5は、薄く白い固体として92%の収率で高い純度で得られた。
[実施例13]
<式VIIIで表わされる化合物の生成>
塩5(0.2g)をアクリロニトリル(3ml)中トリエチルアミンの存在下1.2当量のシクレン(1,4,7,10−テトラアザシクロドデカン)に加え、窒素下60℃で2日間加熱した。前記反応はその後ジクロロメタン(50ml)で希釈し飽和NaHCO3(50ml)で洗った。前記水層はもう一度ジクロロメタン(25ml)で抽出した。合わせた有機層は硫酸ナトリウムで乾燥させ、前記溶媒は真空下除去し、過剰なN−メチルベンジルアミンは真空蒸留で除去し、VIIIを得た。
[実施例14]
<式IXで表わされる化合物の生成>
3−ベンジルオキシ−1−メチル−4−(2−チオキソチアゾリジン−1−イル)カルボニル−2(1H)−ピリジノンのメチレンクロライド溶液に対応する官能化されたN,N,N’,N’−テトラキス(2−アミノエチル)エチレンジアミン(構造B、Zは保護基を表わす)を加えた。4時間攪拌後、前記混合物は濾過され、乾燥された。
【0151】
【化17】
【0152】
前記適切なベンジル−保護前駆体生成物をメチレンクロライド中のプロパノールを用いて、フラッシュシリカカラムで反応混合物から単離した。前記構造IXの最終生成物は、酸による水酸基の脱保護により得られる。
[実施例15]
構造VIIIは、当業者にとって標準の方法で標的の分子と接合させる。例えばN−ヒドロキシスクシンイミド(NHS)エステルは、タンパク質やペプチドへ標準の状態で接合させることで生成され、得られた接合されたプロテインはゲル濾過により単離される。
[実施例16]
トリウム−227による接合されたタンパク質の標識化
適切な緩衝液、例えば0.5MのNaOAc−緩衝液(pH5.5)中に接合されたタンパク質(オクタデンテートリガンドを結合部位によって付けている)の溶液10mg/mlを作る、そして精製されたトリウム−227で25℃で1時間標識化し、続いてゲル濾過カラムで精製した。
【特許請求の範囲】
【請求項1】
組織標的部位、オクタデンテートヒドロキシピリジノン含有リガンドおよびアルファ放出トリウム放射性核種のイオンを含む組織標的複合体。
【請求項2】
ポリペプチド組織標的部位を含む請求項1に記載の複合体。
【請求項3】
少なくとも1つの式Iのキレート部位を含むオクタデンテートリガンドを含む請求項1または2に記載の複合体、
【化1】
式中R1は、任意のN−置換基であり、なくてもよいし、ヒドロカルビル基、OH基、O−ヒドロカルビル基、SH基、およびS−ヒドロカルビル基であってもよく、リンカー部位および/またはカップリング部位を含む;R2〜R6はそれぞれ独立にH、OH、=O、短いヒドロカルビル基、リンカー部位および/またはカップリング部位である。
【請求項4】
R1〜R6の少なくとも1つが、OHであり、R2〜R6の少なくとも1つが、=Oである請求項3に記載の複合体。
【請求項5】
R1〜R6の少なくとも1つがリンカー部位である請求項3または4に記載の複合体。
【請求項6】
少なくとも1つの3,2−ヒドロキシピリジノン部位を有する請求項3〜5のいずれか1項に記載の複合体。
【請求項7】
前記アルファ放出放射性核種が227Thのようなアルファ放出トリウム放射性核種の4+イオンである請求項1〜6のいずれか1項に記載の複合体。
【請求項8】
増殖性または腫瘍性の病気を治療するための薬剤の製造における、組織標的部位、オクタデンテートヒドロキシピリジノン含有リガンドおよびアルファ放出トリウム放射性核種のイオンを含む組織標的複合体の使用。
【請求項9】
前記組織標的複合体が請求項1〜7のいずれか1項に記載の複合体である請求項8に記載の使用。
【請求項10】
前記病気がガン、肉腫、骨髄腫、白血病、リンパ腫または混合タイプのガンである請求項8または9に記載の使用。
【請求項11】
組織標的部位、オクタデンテートヒドロキシピリジノン含有リガンドおよびアルファ放出トリウム放射性核種のイオンを含む少なくとも1つの組織標的複合体の投与を含む人間および人間でない動物(特にそれを必要としているもの)の治療方法。
【請求項12】
前記組織標的複合体が請求項1〜7のいずれか1項に記載の複合体である請求項11に記載の治療方法。
【請求項13】
ガン、肉腫、骨髄腫、白血病、リンパ腫または混合タイプのガンといった増殖性または腫瘍性の病気の治療に用いられる請求項11または12に記載の治療方法。
【請求項14】
組織標的部位、オクタデンテートヒドロキシピリジノン含有リガンドおよびアルファ放出トリウム放射性核種のイオンを含む組織標的複合体を少なくとも1つの薬剤のキャリアまたは賦形剤とともに合わせて含む薬剤。
【請求項15】
前記組織標的複合体が請求項1〜7のいずれか1項に記載の複合体である請求項14に記載の薬剤。
【請求項16】
オクタデンテートヒドロキシピリジノンを含有リガンドに接合されたまたは接合可能な組織標的部位を含み、任意におよび好ましくは227Thのようなアルファ放出トリウム放射性核種を含む請求項11〜13のいずれか1項に記載の方法に用いられるキット。
【請求項17】
増殖性または腫瘍性の病気を治療するために用いられる組織標的部位、オクタデンテートヒドロキシピリジノン含有リガンドおよびアルファ放出トリウム放射性核種のイオンを含む組織標的複合体。
【請求項18】
前記組織標的複合体が請求項1〜7のいずれか1項に記載の複合体である請求項17に記載の組織標的複合体。
【請求項19】
前記病気がガン、肉腫、骨髄腫、白血病、リンパ腫または混合タイプのガンである請求項17または18に記載の組織標的複合体。
【請求項1】
組織標的部位、オクタデンテートヒドロキシピリジノン含有リガンドおよびアルファ放出トリウム放射性核種のイオンを含む組織標的複合体。
【請求項2】
ポリペプチド組織標的部位を含む請求項1に記載の複合体。
【請求項3】
少なくとも1つの式Iのキレート部位を含むオクタデンテートリガンドを含む請求項1または2に記載の複合体、
【化1】
式中R1は、任意のN−置換基であり、なくてもよいし、ヒドロカルビル基、OH基、O−ヒドロカルビル基、SH基、およびS−ヒドロカルビル基であってもよく、リンカー部位および/またはカップリング部位を含む;R2〜R6はそれぞれ独立にH、OH、=O、短いヒドロカルビル基、リンカー部位および/またはカップリング部位である。
【請求項4】
R1〜R6の少なくとも1つが、OHであり、R2〜R6の少なくとも1つが、=Oである請求項3に記載の複合体。
【請求項5】
R1〜R6の少なくとも1つがリンカー部位である請求項3または4に記載の複合体。
【請求項6】
少なくとも1つの3,2−ヒドロキシピリジノン部位を有する請求項3〜5のいずれか1項に記載の複合体。
【請求項7】
前記アルファ放出放射性核種が227Thのようなアルファ放出トリウム放射性核種の4+イオンである請求項1〜6のいずれか1項に記載の複合体。
【請求項8】
増殖性または腫瘍性の病気を治療するための薬剤の製造における、組織標的部位、オクタデンテートヒドロキシピリジノン含有リガンドおよびアルファ放出トリウム放射性核種のイオンを含む組織標的複合体の使用。
【請求項9】
前記組織標的複合体が請求項1〜7のいずれか1項に記載の複合体である請求項8に記載の使用。
【請求項10】
前記病気がガン、肉腫、骨髄腫、白血病、リンパ腫または混合タイプのガンである請求項8または9に記載の使用。
【請求項11】
組織標的部位、オクタデンテートヒドロキシピリジノン含有リガンドおよびアルファ放出トリウム放射性核種のイオンを含む少なくとも1つの組織標的複合体の投与を含む人間および人間でない動物(特にそれを必要としているもの)の治療方法。
【請求項12】
前記組織標的複合体が請求項1〜7のいずれか1項に記載の複合体である請求項11に記載の治療方法。
【請求項13】
ガン、肉腫、骨髄腫、白血病、リンパ腫または混合タイプのガンといった増殖性または腫瘍性の病気の治療に用いられる請求項11または12に記載の治療方法。
【請求項14】
組織標的部位、オクタデンテートヒドロキシピリジノン含有リガンドおよびアルファ放出トリウム放射性核種のイオンを含む組織標的複合体を少なくとも1つの薬剤のキャリアまたは賦形剤とともに合わせて含む薬剤。
【請求項15】
前記組織標的複合体が請求項1〜7のいずれか1項に記載の複合体である請求項14に記載の薬剤。
【請求項16】
オクタデンテートヒドロキシピリジノンを含有リガンドに接合されたまたは接合可能な組織標的部位を含み、任意におよび好ましくは227Thのようなアルファ放出トリウム放射性核種を含む請求項11〜13のいずれか1項に記載の方法に用いられるキット。
【請求項17】
増殖性または腫瘍性の病気を治療するために用いられる組織標的部位、オクタデンテートヒドロキシピリジノン含有リガンドおよびアルファ放出トリウム放射性核種のイオンを含む組織標的複合体。
【請求項18】
前記組織標的複合体が請求項1〜7のいずれか1項に記載の複合体である請求項17に記載の組織標的複合体。
【請求項19】
前記病気がガン、肉腫、骨髄腫、白血病、リンパ腫または混合タイプのガンである請求項17または18に記載の組織標的複合体。
【図1】

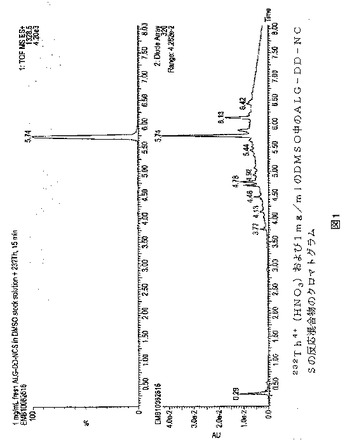
【図2】

【図3】
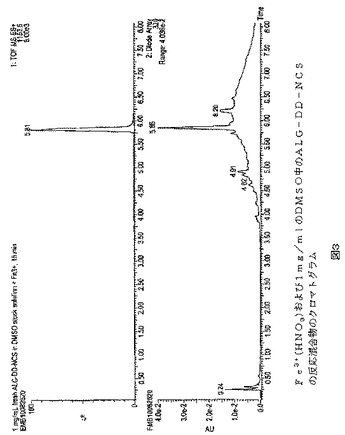
【図4】

【図5】

【図6】

【図7】
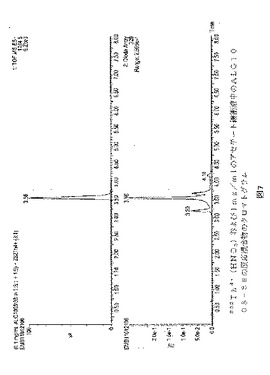
【図8】

【図9】

【図10】

【図11】

【図12】

【図13】

【図14】

【図15】
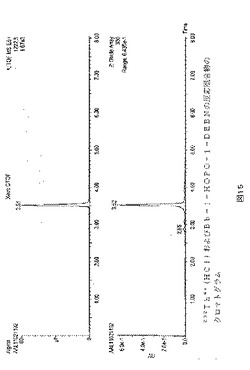
【図16】
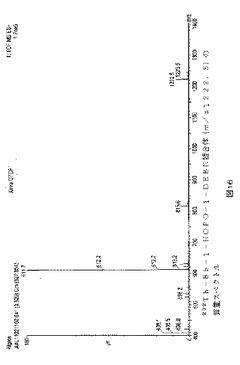
【図17】
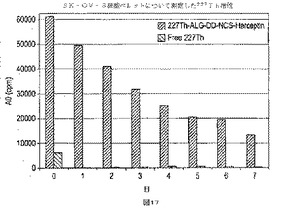
【図18】

【図19】
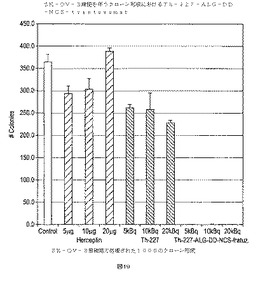
【図20】
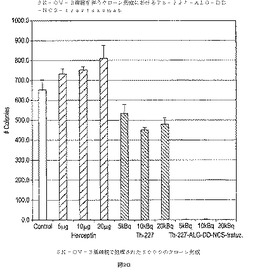
【図21】
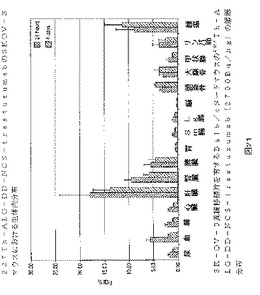
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【公表番号】特表2013−519658(P2013−519658A)
【公表日】平成25年5月30日(2013.5.30)
【国際特許分類】
【出願番号】特願2012−552424(P2012−552424)
【出願日】平成23年2月14日(2011.2.14)
【国際出願番号】PCT/EP2011/052158
【国際公開番号】WO2011/098611
【国際公開日】平成23年8月18日(2011.8.18)
【出願人】(505385723)アルゲッタ エイエスエイ (5)
【Fターム(参考)】
【公表日】平成25年5月30日(2013.5.30)
【国際特許分類】
【出願日】平成23年2月14日(2011.2.14)
【国際出願番号】PCT/EP2011/052158
【国際公開番号】WO2011/098611
【国際公開日】平成23年8月18日(2011.8.18)
【出願人】(505385723)アルゲッタ エイエスエイ (5)
【Fターム(参考)】
[ Back to top ]