
アロステリック部位を同定するための方法
本発明は、タンパク質標的中の外側部位およびこれらの外側部位を同定するための方法に関する。本発明は、アロステリック部位を同定するための方法およびこのアロステリック部位に結合する化合物を同定するための方法に関する。アロステリック部位を同定するための方法であって、以下の工程:a)第一の結合部位、第二の結合部位および該第二の結合部位またはその近傍に化学反応基を備える標的を提供する工程;b)該標的を、該化学反応基と共有結合を形成し得る化合物と接触させる工程;c)該標的と該化合物との間に共有結合を形成し、それによって、標的−化合物結合体を形成する工程;d)該標的−化合物結合体が、該標的と比較して、主な結合部位中に変化を有するか否かを決定する工程、を包含する。
【発明の詳細な説明】
【技術分野】
【0001】
(背景)
(発明の分野)
薬物発見標的は、しばしば、タンパク質、特に、酵素である。ほとんどの薬物発明の努力は、リード化合物を同定するための大量の機能的スクリーニングに依存するので、強力な活性部位インヒビターがしばしば同定されるが、これらが薬物候補となるのはまれである。実に低い成功率についての理由は、変動するが、特異性の欠如および毒性は、それらがない場合より高い頻度で役割を果たす。
【0002】
酵素標的は、通常、関連する酵素のファミリーの1つのメンバーである。驚くべきことではないが、関連する酵素はしばしば、互いに類似する三次元構造を共有し、そして、その活性部位領域は、最も保存されている。標的化される部位は、ファミリーメンバーの中でも最も類似する酵素の領域であるので、1つのメンバーに対する選択性を達成することが、非常に困難なことであることは驚くべきことではない。特異性の欠如は、しばしば、意図しない標的の阻害に由来する毒性を生じる。固有の問題にもかかわらず、多くの最も有望な薬物標的は、大きな酵素ファミリー(例えば、プロテアーゼ(例えば、アスパルチルプロテアーゼ、システインプロテアーゼ、およびセリンプロテアーゼ)、キナーゼならびにホスファターゼ)のメンバーである。結果として、薬物発見のための新規な方法が、特異性を増強するこれらの型の標的に対して必要とされる。本発明は、このような方法を提供する。
【発明の開示】
【課題を解決するための手段】
【0003】
(好ましい実施形態の説明)
本発明は、本明細書で「外部部位(exosite)」として言及される、タンパク質上の新規な結合部位を同定するための方法およびそこに結合するリガンドを同定するための方法を提供する。本発明の方法によって同定されたリガンド自体は、例えば、新規な治療薬物、酵素インヒビター、標識化合物、診断試薬、タンパク質精製のための親和性試薬などの開発のためのリード化合物としての用途を見出す。
【0004】
他に規定されない限り、本明細書で使用される技術用語および科学用語は、本発明が属する分野の当業者に一般的に理解される意味と同じ意味を有する。参考文献(例えば、Singletonら、Dicitionary of Microbiology and Molecular Biology 第2版、J.Wiley&Sons(New York、NY 1994)および3月、Advanced Organic Reactions、Mechanisms and Structure 第4版、John Wiley&Sons(New York 1992))は、本発明の適用において使用される多くの用語に対する一般的指針を当業者に提供する。
【0005】
(定義)
本明細書で使用される用語の定義としては、以下が挙げられる:
用語「脂肪族」または「非置換脂肪族」とは、直鎖、分枝鎖、環状、または多環式の炭化水素をいい、アルキル部分、アルケニル部分、アルキニル部分、シクロアルキル部分、シクロアルケニル部分、およびシクロアルキニル部分が挙げられる。
【0006】
用語「アルキル」または「非置換アルキル」とは、飽和炭化水素をいう。
【0007】
用語「アルケニル」または「非置換アルケニル」とは、少なくとも1つの炭素−炭素二重結合を有する炭化水素をいう。
【0008】
用語「アルキニル」または「非置換アルキニル」とは、少なくとも1つの炭素−炭素三重結合を有する炭化水素をいう。
【0009】
用語「芳香族」または「非置換芳香族」とは、少なくとも1つのアリール基を有する部分をいう。この用語はまた、脂肪族修飾アリール(例えば、アルキルアリールなど)を含む。
【0010】
用語「アリール」または「非置換アリール」とは、少なくとも1つの芳香環を有する単環または多環の不飽和部分をいう。この用語は、少なくとも1つの芳香環内に1つ以上のヘテロ原子を含むヘテロアリールを含む。アリールの例示的な例としては、以下が挙げられる:フェニル、ナフチル、テトラヒドロナフチル、インダニル、インデニル、ピリジル、ピラジニル、ピリミジニル、ピロリル、ピラゾリル、イミダゾリル、チアゾリル、オキサゾリル、イソオキサゾリル、チアジアゾリル、オキサジアゾリル、チオフェニル、フラニル、キノリニル、イソキノリニルなど。
【0011】
部分を改変するために使用される場合、用語「置換(された)」は、その部分の置換されたバージョンをいい、ここで、少なくとも1つの水素原子は、以下が挙げられるが、これらに限定されない別の基により置換される:脂肪族、アリール、アルキルアリール、F、Cl、I、Br、−OH;−NO2;−CN;−CF3;−CH2CF3;−CH2Cl;−CH2OH;−CH2CH2OH;−CH2NH2;−CH2SO2CH3;−ORX;−C(O)RX;−COORX;−C(O)N(RX)2;−OC(O)RX;−OCOORX;−OC(O)N(Rx)2;−N(RX)2;−S(O)2RX;および−NRXC(O)RX。ここで、各RXの出現は、独立して、水素、置換脂肪族、非置換脂肪族、置換アリール、または非置換アリールである。さらに、部分上の隣接基の置換基は、一緒になって環状基を形成し得る。
【0012】
用語「アロステリック部位」とは、外部部位をいい、ここで、リガンドのこの部位への結合が、タンパク質の活性または機能を調節する。
【0013】
用語「アンタゴニスト」は、最も広い意味で使用され、標的(例えば、TBM)によって示される生物学的活性を部分的または完全にブロック、阻害または中和する任意のリガンドを含む。類似の様式において、用語「アゴニスト」は、最も広い意味で使用され、標的(例えば、TBM)によって示される生物学的活性を、例えば、このようなTBMの機能もしくは発現、またはこのようなTBMを介するシグナル伝達の効率を特異的に変化させ、それにより、既に存在する生物学的活性を変更(増大もしくは阻害)するか、または新たな生物学的活性を誘発することによって模倣する、任意のリガンドを含む。
【0014】
用語「外部部位」は、その主な結合部位でない、タンパク質上の結合部位である。例えば、酵素上の主な結合部位は、活性部位である。レセプター上の主な結合部位は、リガンド結合部位である。
【0015】
用語「リガンド候補」または「候補リガンド」とは、標的上の相補的または適合性の反応基と共有結合を形成し得る反応基を保有するか、または、保有するように改変された化合物をいう。リガンド候補上または標的上のいずれかの反応基は、例えば、保護基でマスクされ得る。
【0016】
用語「ポリヌクレオチド」は、単数または複数で使用される場合、任意のポリリボヌクレオチドまたはポリデオキシリボヌクレオチドを一般にいい、これらは、非改変RNAもしくは非改変DNA、または改変RNAもしくは改変DNAであり得る。従って、例えば、本明細書で定義されるポリヌクレオチドとしては、限定されないが、一本鎖DNAおよび二本鎖DNA、一本鎖領域および二本鎖領域を含むDNA、一本鎖RNAおよび二本鎖RNA、一本鎖領域および二本鎖領域を含むRNA、一本鎖、もしくはより代表的には二本鎖であり得るか、または一本鎖領域および二本鎖領域を含むDNAおよびRNAを含むハイブリッド分子が挙げられる。さらに、用語「ポリヌクレオチド」は、本明細書で使用される場合、RNAあるいはDNAまたはRNAおよびDNAの両方を含む三本鎖領域をいう。このような領域における鎖は、同じ分子由来であっても、異なる分子由来であってもよい。これらの領域は、1つ以上の分子の全てを含み得るが、より代表的には、幾つかの分子の1つの領域のみを包含する。三重らせん領域の分子の1つは、しばしば、オリゴヌクレオチドである。用語「ポリヌクレオチド」は、特に、1つ以上の改変された塩基を含むDNAおよびRNAを含む。従って、安定性または他の理由のために改変された骨格を有するDNAまたはRNAは、その用語が本明細書で意図されるような「ポリヌクレオチド」である。さらに、通常でない塩基(例えば、イノシン)または改変された塩基(例えば、トリチル化塩基)を含むDNAまたはRNAは、本明細書で定義されるような「ポリヌクレオチド」との用語に含まれる。一般的に、用語「ポリヌクレオチド」は、非改変のポリヌクレオチドの全ての化学的、酵素的および/または代謝的に改変された形態、ならびにウイルスおよび細胞(単純細胞および複雑細胞を含む)の特徴であるDNAおよびRNAの化学的形態を含む。
【0017】
語句「保護されたチオール」または「マスク(マスキング)されたチオール」とは、本明細書で使用される場合、基または分子と反応して、より反応性が低くされた共有結合を形成しているチオールをいい、これは、脱保護されて、遊離のチオールを再生し得る。
【0018】
語句「可逆的な共有結合」とは、本明細書で使用される場合、好ましくは、標的を変性させない条件下で分解され得る共有結合をいう。例としては、ジスルフィド、シッフ塩基、チオエステル、配位錯体、ボロン酸エステルなどが挙げられるが、これらに限定されない。
【0019】
語句「反応基」は、存在する場合、適合性または相補的な反応基との共有結合が形成され得る部位を提供する化学的な基または部分である。例証的な例は、以下である:−SHであり、これは、別の−SHまたは−SS−と反応して、それぞれジスルフィドまたはジスルフィド交換を形成し得る;−NH2であり、これは、活性化された−COOHと反応して、アミドを形成し得る;−NH2であり、これは、アルデヒドまたはケトンと反応して、シッフ塩基などを形成し得る。
【0020】
語句「反応性求核試薬」は、本明細書で使用される場合、標的を変性も損傷も条件下で、別の分子上の適合性の官能基と共有結合を形成し得る求核試薬をいう。最も適切な求核試薬は、チオール、アルコール、およびアミンである。同様に、語句「反応性求電子試薬」とは、本明細書で使用される場合、好ましくは、標的を変性も損傷もしない条件下で、別の分子上の適合性の官能基と共有結合を形成し得る求電子試薬をいう。最も適切な求電子試薬は、ハロゲン化アルキル、イミン、カルボニル、エポキシド、アジリジン(aziridie)、スルホネート、ジスルフィド、活性化エステル、活性化カルボニル、およびヘミアセタールである。
【0021】
語句「目的の部位」とは、リガンドが結合し得る、標的上の任意の部位をいう。本明細書で使用される場合、目的の部位は、タンパク質の主な結合部位の外側にある任意の部位である。例えば、標的が酵素である場合、目的の部位は、活性部位でない部位である。標的がレセプターである場合、目的の部位は、レセプターのリガンドの結合部位でない部位である。
【0022】
用語「標的」、「標的分子」および「TM」は、交換可能に、最も広い意味で使用され、リガンドの結合が標的の機能に対して影響を有する化学的または生物学的な実体をいう。標的は、分子、分子の一部、または分子の凝集体であり得る。リガンドの結合は、可逆的でも不可逆的でもよい。標的分子の特定の例としては、以下が挙げられる:ポリペプチドまたはタンパク質(例えば、酵素およびレセプター)、転写因子、増殖因子およびサイトカインのような、レセプターに対するリガンド、免疫グロブリン、核タンパク質、シグナル伝達成分(例えば、キナーゼ、ホスファターゼ)、ポリヌクレオチド、糖質、糖タンパク質、糖脂質、および他の高分子(例えば、核酸−タンパク質複合体、クロマチンまたはリボソーム)、資質二重層含有構造(例えば、膜)、または膜から誘導される構造(例えば、ベシクル)。この定義は、特に、以下に定義されるような標的生物学的分子(Target Biological Molecule)(「TBM」)を含む。
【0023】
「標的生物学的分子」または「TBM」とは、本明細書で使用される場合、低分子アゴニストまたは低分子アンタゴニストがTBMの機能に対する効果を有する、互いに生物学的に関連する複合体を形成し得る、単数または複数の生物学的分子をいう。好ましい実施形態において、TBMは、2つ以上のアミノ酸を含むタンパク質またはその一部であり、これらは、相補的な反応基を有する化合物と共有結合を形成し得る反応基を保有するか、または保有するように改変され得る。好ましいTBMとしては、以下が挙げられる:細胞表面レセプターおよび可溶性レセプターならびにそれらのリガンド;ステロイドレセプター;ホルモン;免疫グロブリン;凝血因子;核タンパク質;転写因子;シグナル伝達分子;細胞接着分子、同時刺激性分子、ケモカイン、アポトーシスの媒介に関与する分子、酵素、ならびにDNAおよび/またはRNA合成または分解に関連するタンパク質。
【0024】
多くのTBMは、レセプター−リガンド結合相互作用に関与し、レセプター−リガンド対のいずれかのメンバーであり得る分子である。増殖因子およびそれらのそれぞれのレセプターの例証的な例としては、以下に対するものが挙げられる:エリスロポエチン(EPO)、トロンボポエチン(TPO)、アンジオポエチン(ANG)、顆粒球コロニー刺激因子(G−CSF)、顆粒球マクロファージコロニー刺激因子(GM−CSF)、上皮増殖因子(EGF)、ヘレグリン−a(heregulin−a)およびヘレグリン−b、脈管内皮細胞増殖因子(VEGF)、胎盤増殖因子(PLGF)、トランスフォーミング増殖因子(TGF−aおよびTGF−b)、神経成長因子(NGF)、ニューロトロフィン、線維芽細胞増殖因子(FGF)、血小板由来増殖因子(PDGF)、骨形態形成タンパク質(BMP)、結合組織増殖因子(CTGF)、肝細胞増殖因子(HGF)、ならびにインスリン様増殖因子1(IGF−1)。ホルモンおよびそれらのそれぞれのレセプターの例証的な例としては、以下に対するもの挙げられる:成長ホルモン、プロラクチン、胎盤ラクトゲン(LPL)、インスリン、卵胞刺激ホルモン(FSH)、黄体形成ホルモン(LH)、ならびにニューロキニン−1。サイトカインおよびそれらのそれぞれのレセプターの例証的な例としては、以下に対するものが挙げられる:毛様体神経栄養因子(CNTF)、オンコスタチンM(OSM)、TNF−a;CD40L、幹細胞因子(SCF);インターロイキン−1、インターロイキン−2、インターロイキン−4、インターロイキン−5、インターロイキン−6、インターロイキン−8、インターロイキン−9、インターロイキン−13、ならびにインターロイキン−18。
【0025】
他のTBMとしては以下が挙げられる:細胞接着分子(例えば、CD2、CD11a、LFA−1、LFA−3、ICAM−5、VCAM−1、VCAM−5およびVLA−4);同時刺激分子(例えば、CD28、CTLA−4、B7−1;B7−2、ICOSおよびB7RP−1);ケモカイン(例えば、RANTESおよびMIP1b);アポトーシス因子(例えば、APAF−1、p53、bax、bak、bad、bidおよびc−abl);抗アポトーシス因子(例えば、bcl2、bcl−x(L)およびmdm2);転写調節因子(例えば、AP−1およびAP−2);シグナル伝達タンパク質(例えば、TRAF−1、TRAF−2、TRAF−3、TRAF−4、TRAF−5およびTRAF−6);およびアダプタータンパク質(例えば、grb2、cbl、shc、nckおよびcrk)。
【0026】
酵素は、別の分類の好ましいTBMであり、以下を含む多くの方法で分類され得る:アロステリック酵素;細菌性酵素(イソロイシルtRNAシンターゼ、ペプチドデホルミラーゼ、DNAジャイレースなど);真菌性酵素(チミジル酸合成酵素など);ウイルス酵素(HIVインテグラーゼ、HSVプロテアーゼ、C型肝炎Cヘリカーゼ、C型肝炎プロテアーゼ、ライノウイルスプロテアーゼなど);キナーゼ(セリン/スレオニン、チロシンおよび二重特異性(dual specificity));ホスファターゼ(セリン/スレオニン、チロシンおよび二重特異性);およびプロテアーゼ(アスパルチルプロテアーゼ、システインプロテアーゼ、メタロプロテアーゼおよびセリンプロテアーゼ)。酵素の注目すべき下位分類としては、以下が挙げられる:キナーゼ(例えば、Lck、Syk、Zap−70、JAK、FAK、ITK、BTK、MEK、MEKK、GSK−3、Raf、tgf−b−活性化キナーゼ−1(TAK−1)、PAK−1、cdk4、Akt、PKCq、IKKb、IKK−2、PDK、ask、nik、MAPKAPK、p90rsk、p70s6kおよびPI3−K(p85およびp110サブユニット));ホスファターゼ(例えば、CD45、LAR、RPTP−a、RPTP−m、Cdc25A、キナーゼ関連ホスファターゼ、mapキナーゼホスファターゼ−1、PTP−1B、TC−PTP、PTP−PEST、SHP−1およびSHP−2;カスパーゼ(例えば、カスパーゼ−1、カスパーゼ−3、カスパーゼ−7、カスパーゼ−8、カスパーゼ−9およびカスパーゼ−11);およびカテプシン(例えば、カテプシンB、カテプシンF、カテプシンK、カテプシンL、カテプシンSおよびカテプシンV)。他の酵素標的としては、以下が挙げられる:BACE、TACE、細胞質ホスホリパーゼA2(cPLA2)、PARP、PDE I〜VII、Rac−2、CD26、イノシン1リン酸デヒドロゲナーゼ、15−リポキシゲナーゼ、アセチルCoAカルボキシラーゼ、アデノシルメチオニンデカルボキシラーゼ、ジヒドロオロテートデヒドロゲナーゼ、ロイコトリエンA4ヒドロラーゼ、および一酸化窒素シンターゼ。
【0027】
(外部部位)
本発明は、本明細書中で「外部部位(exosite)」と呼ばれるタンパク質上の新規の結合部位を同定するための方法を提供する。これらの外部部位は、特定の標的タンパク質の一次結合領域とは異なる標的タンパク質上の結合部位である。例えば、酵素上の外部部位は、活性部位でない任意の結合部位である。同様に、レセプター上の外部部位は、レセプターのリガンドの結合部位ではない任意の結合部位である。
【0028】
1つの実施形態において、目的の外部部位は、タンパク質の適応性の結合部位である。用語「適応性」は、活性部位のようなよく定義されたポケットとは異なり、適応性の結合部位は、リガンドの不在下では明らかでないため、これらの部位をさすために使用される。リガンドの存在は、タンパク質の1つ以上の鎖の立体配座の変化を誘導し、リガンドが最終的に結合し得る結合部位を生じる。
【0029】
別の実施形態において、目的の外部部位はアロステリック部位である。言い換えると、標的タンパク質のこのような部位へのリガンドの結合は、その標的タンパク質の機能を調節する。この調節は、負ならびに正の両方であり得る。例えば、調節が負である場合、外部部位へのリガンドの結合は、標的の機能を阻害する。調節が正である場合、外部部位のリガンドへの結合は、標的の機能を増強(または増幅)する。アロステリック部位は、しばしば、アクセサリータンパク質および/または調節タンパク質に対する認識部位である。
【0030】
なお別の実施形態において、目的の外部部位は適応性の結合部位とアロステリック部位との両方である。
【0031】
(係留方法)
係留は、標的上の反応基と潜在的なリガンド上の相補的な反応基との間の共有結合の形成に依存するリガンドの同定方法である。係留方法は、発明者ら(Daniel Erlanson、Andrew BraistedおよびJames Wells)による米国特許第6,335,155号;PCT公報番号WO00/00823およびWO02/42773;METHODS FOR LIGAND DISCOVERYとの表題の米国番号10/121,216(対応するPCT出願番号US02/13061);およびErlansonら、Proc.Nat.Acad.Sci USA 97:9367−9372(2000)(これらは、全てが参考として援用される)に記載される。得られた共有複合体は、標的−リガンド結合体と称される。共有結合が標的(例えば、ネイティブまたは非ネイティブのシステイン)の所定の部位で形成されるので、化学量論および結合位置は、この方法によって同定されるリガンドについて公知である。
【0032】
一旦形成されると、標的−リガンド結合体のリガンド部分が多数の方法を使用して同定され得る。好ましい実施形態において、質量分析法が使用される。質量分析法は、質量/電荷比(m/z)に基づいて分子を検出し、そして、それらのサイズに基づいて分子を分離し得る(Yates,Trends Genet.16:5−8[2000]において総説される)。標的−リガンド結合体は、質量分析計において直接検出され得るか、または検出の前にフラグメント化され得る。あるいは、化合物は、質量分光光度計内で遊離され、引き続いて同定され得る。さらに、質量分析法を単独でかまたは標的に共有結合する化合物を検出もしくは同定するための他の手段と組み合せて使用し得る。さらに、質量分析法技術の説明としては、FitzgeraldおよびSiuzdak,Chemstry & Biology 3:707−715[1996];Chuら,J.Am.Chem.Soc.118:7827−7835[1996];Siudzak,Proc.Natl.Acad.Sci.USA 91:11290−11297[1994];Burlingameら,Anal.Chem.68:599R−651R[1996];Wuら,Chemistry & Biology 4:653−657[1997];ならびにLooら,Am.Reports Med.Chem.31:319−325[1996])が挙げられる。
【0033】
あるいは、標的−リガンド結合体は、他の手段を用いて同定され得る。例えば、反応混合物の成分の分離のために種々のクロマトグラフィー技術(例えば、液体クロマトグラフィー、薄層クロマトグラフィーなど)を使用し得、その結果、共有結合分子を同定する能力を高める。このようなクロマトグラフィー技術は、質量分析法と組み合せてかまたは質量分析法とは別に使用され得る。また、標識化プローブ(蛍光、放射線または他の手段で)を遊離した化合物に結合させ、それによって上記の技術のいずれかを用いる同定を容易にし得る。なお別の実施形態において、新しい結合の形成は、標識化プローブを遊離させ、次いで、モニタリングされ得る。単純な機能的アッセイ(例えば、ELISAまたは酵素アッセイ)がまた、アッセイが測定するのに必須の領域において結合が生じるときに結合を検出するのに使用され得る。標的分子に結合する有機化合物を同定するための用途が見出され得る他の技術としては、例えば、核磁気共鳴分析法(NMR)、表面プラスモン共鳴(例えば、BIACORE)、キャピラリー電気泳動、X線結晶解析、などが挙げられ、これらは全て、当業者に周知である。
【0034】
標的がタンパク質であり、かつ共有結合がジスルフィドである係留方法の1つの実施形態の略図を図1に示す。チオール含有タンパク質を、複数のリガンド候補と反応させる。この実施形態において、リガンド候補は、式−SSR1のジスルフィドの形態でマスクされたチオールを保有し、ここでR1は非置換のC1〜C10アルキル、置換C1〜C10アルキル、非置換のアリールまたは置換アリールである。特定の実施形態において、R1は潜在的なリガンド候補の溶解性を高めるように選択される。示すように、標的に対して固有の結合親和性を保有するリガンド候補が同定され、ジスルフィド部分を含まない対応するリガンドが、同定された結合決定基(円で表される)から作製される。
図2は、2つの代表的な係留実験を示す。ここで、標的酵素、E.coliチミジル酸シンターゼは以下の式
【0035】
【化3】

のリガンド候補と接触し、ここで、Rcは、ライブラリメンバーのこのプールの中で可変の部分であり、そして、非置換の脂肪族、置換脂肪族、非置換のアリール、または置換アリールである。全てのTS酵素のと同様に、E.Coli TSは係留に使用され得る活性な部位システイン(Cys146)を有する。E.Coli TSはまた4つの他のシステインを含むが、これらのシステインは変化し、係留実験において反応性でないことが見出された。例えば、最初の実験において、野生型のE.coli TSおよびC146S変異体(146位のシステインがセリンに変異している)をシスタミン(H2NCH2CH2SSCH2CH2NH2)と接触させた。野生型のTS酵素は、シスタミンの1つの等価物ときちんと反応したが、その一方で、変異体TSは反応せず、これは、シスタミンがCys146と反応し、Cys146に選択的であったことを示す。
【0036】
図2Aは、TSとTSに対してほとんど結合親和性を有さないかまたは全く結合親和性を有さない10の異なるリガンド候補のプールとの反応の逆重畳質量スペクトルである。任意の結合相互作用の不在下で、TSと個々のリガンド候補との間のジスルフィド交換反応の平衡は改変されていない酵素に向う。このことを以下の式によって模式的に示す。
【0037】
【化4】
予想した通り、改変されていない酵素に対応するピークは、スペクトルのうち2つの最も突出したピークのうちの1つである。もう一方の突出したピークは、TSであり、ここでは、Cys146のチオールがシステアミンで改変されている。この種は任意の個々のライブラリメンバーについて有意な程度まで形成されないが、このピークはライブラリプールの各メンバーについての平衡反応の蓄積効果に起因する。反応がチオール含有還元剤(例えば、2−メルカプトエタノール)の存在下で実行される場合、活性部位のシステインはまた、還元剤で改変され得る。システアミンと2−メルカプトエタノールは類似した分子量を有するので、これらのそれぞれのジスルフィド結合化TS酵素は、この実験で用いた条件下においては区別できない。右側の小さなピークは、目立たないライブラリメンバーに対応する。特に、これらのピークはいずれもが非常に顕著でない。図2Aは、リガンド候補のいずれもが標的に対して固有の結合親和性を保有しないスペクトルの特徴である。
【0038】
図2Bは、リガンド候補の1つが酵素に対して固有の結合親和性を保有する、10の異なるリガンド候補のプールとのTSの反応の逆重畳質量スペクトルである。見られ得るように、最も突出したピークはTSに対応するものであり、ここでは、Cys146のチオールがN−トシル−D−プロリン化合物で改変されている。このピークは、改変されていない酵素およびTSに対応するピークを含む全ての他のピークを矮化し、ここではCys146のチオールがシステアミンで改変されている。図2Bは、係留が所望の部位に対して強い固有の結合親和性を保有する部分を有する、質量スペクトルの例である。
【0039】
(係留を用いた外部部位同定)
本発明の1つの局面において、タンパク質標的表面上の外部部位を同定するための方法が提供される。一般に、この方法は以下の工程を包含する:
a)主な(primary)結合部位、および主な結合部位以外の部位またはその付近の部位に化学反応基を有する標的を提供する工程;
b)上記の化学反応基と共有結合を形成し得る化合物に標的を接触させる工程;
c)標的と化合物との間に共有結合を形成し、それによって標的−化合物結合体を形成する工程;ならびに
d)標的との共有結合の不在下で、上記部位における標的に化合物が結合するか否かを決定する工程。
【0040】
多くの場合、潜在的な外部部位が標的の凹部領域に位置される。他の場合において、潜在的な外部部位は、明らかでない。なぜならば、この部位はリガンドの存在がタンパク質の1つ以上の側鎖の立体配置の変化を誘導し、リガンドが最終的には結合し得る結合部位を作成する、適応性の結合部位であるからである。
【0041】
標的が酵素である場合、主な結合部位は活性部位である。標的がレセプターである場合、主な結合部位はレセプターのリガンドが結合する部位である。
【0042】
化学反応基が、この結合部位を含む残基の任意の原子から10Å以下である場合、この基は、結合部位の近くであると考えられる。別の実施形態において、化学反応基が、この結合部位を含む残基の任意の原子から5Å以下である場合、この基は結合部位の近くであると考えられる。
【0043】
別の実施形態において、この方法は以下の工程を包含する:
a)第1の結合部位、第2の結合部位および第2の結合部位または第2の結合部位の近くの化学反応基を有する標的を提供する工程;
b)化学反応基と共有結合を形成し得る第1の化合物に標的を接触させる工程;
c)標的と第1の化合物との間に共有結合を形成させ、それによって標的−化合物結合体を形成する工程;
d)標的を第2の化合物に接触させる工程であって、第2の化合物が化学反応基を欠失した第1の化合物の変形である、工程;ならびに
e)第2の結合部位に対して非共有結合するための第2の化合物の親和性を決定する工程。
【0044】
本発明の別の局面において、アロステリック外側部位を同定するための方法が提供される。概して、この方法は、以下の工程を包含する:
a)主な結合部位および該主な結合部位以外の部位またはその近傍の化学反応基を備える標的を提供する工程;
b)該標的を、該化学反応基と共有結合を形成し得る化合物と接触させる工程;
c)該標的と該化合物との間の共有結合を形成し、それによって、標的−化合物結合体を形成する工程;
d)該標的−化合物結合体が、該標的と比較して、主な結合部位中に変化を有するか否かを決定する工程。
【0045】
この方法によって同定されるアロステリック部位は、ネガティブおよびポジティブの両方で標的タンパク質の機能を調節し得る。例えば、この調節はネガティブである場合、外側部位へのリガンドの結合は、この標的の機能を阻害する。この調節がポジティブである場合、外側部位へのリガンドの結合は、この標的の機能を促進(または増幅)させる。
【0046】
1実施形態において、主な結合部位における変化は、構造的変化であり、そして主な結合部位の三次元構造における変更である。三次元構造における変更は、少なくとも1Åまでの、活性部位残基の少なくとも1つのヘテロ原子の動きとして規定される。構造における変化は、x線結晶学、NMR、円偏光二色性などを含む多くの任意の方法によって検出される。別の実施形態において、この主な結合部位における変化は、機能的変化である。標的が酵素である場合、その機能は、阻害または促進され得る。この標的がレセプターである場合、レセプターリガンド結合部位への、レセプターリガンドの結合は、阻害または促進され得る。
【0047】
別の実施形態において、この方法は、以下の工程を包含する:
a)第一の結合部位、第二の結合部位およびこの第二の結合部位またはその近傍の化学反応基を備える標的を提供する工程;
b)この標的を、この化学反応基と共有結合を形成し得る化合物と接触させる工程;
c)この標的と化合物との間の共有結合を形成し、それによって、標的−化合物結合体を形成する工程;
d)この標的−化合物結合体が、標的の第一の結合部位と比較して、第一結合部位中に変化を有するか否かを決定する工程。
【0048】
本方法の特定の実施形態において、この化学反応基は、システイン残基のチオールであり、そしてこの化合物は、−SH基を有する。他の実施形態において、この化合物は、マスキングされたチオールを有する。他の実施形態において、この化合物は、式−SSR1のジスルフィドの形態でマスキングされたチオールを有するリガンド候補であり、ここで、R1は、非置換C1〜C10脂肪族、置換C1〜C10脂肪族、非置換アリールまたは置換アリールである。他の実施形態において、このリガンド候補は、式−SSR2R3のジスルフィドのようにマスキングされたチオールを有し、ここで、R2は、C1〜C5アルキル(好ましくは、−CH2−、−CH2CH2−、または−CH2CH2CH2−)であり、そしてR3は、NH2、OH、またはCOOHである。リガンド候補の例示的な例としては、以下が挙げられる:
【0049】
【化5】
ここで、RおよびR’は、各々独立して非置換C1〜C20脂肪族、置換C1〜C20脂肪族、非置換アリールまたは置換アリールであり;
mは0、1または2であり;そして
nは1または2である。
【0050】
他の実施形態において、この標的は、還元剤の存在下においてジスルフィド結合を形成し得る化合物と接触される。適切な還元剤の例示的な例としては、以下が挙げられるが、これらに限定されない:システイン、システアミン、ジチオスレイトール、ジチオエリスリトール、グルタチオン、2−メルカプトエタノール、3−メルカプトプロピオン酸、ホスフィン(例えば、トリス−(2−カルボキシ−ホスフィン)(「TCEP」)、または水素化ホウ素ナトリウム。1実施形態において、この還元剤は、2−メルカプトエタノールである。別の実施形態において、この還元剤は、システアミンである。別の実施形態において、この還元剤は、グルタチオンである。別の実施形態において、この還元剤はシステインである。
【0051】
外側部位を同定する係留方法の概略図を、図3に示す。この実施形態において、主な結合部位は、活性部位として示される。図3Aは、外側部位が適応性結合部位である状況を示す。理解され得るように、この外側部位は、リガンドの存在によって誘導される。しかし、この外側部位へのリガンドの結合は、活性部位のコンフォメーションも、標的の機能も変更しない。対照的に、図3Bは、アロステリック部位でもある外側部位が同定される状況を示す。示されるように、アロステリック外側部位へのリガンドの結合は、活性部位のコンフォメーションを変更し、その結果、標的の機能を阻害する。両方の場合において、この標的−化合物結合体は、必要に応じて、標的を再生するために還元剤と接触され得る。この外側部位がアロステリック外側部位である場合、リガンドの除去は、このアロステリック外側部位へのリガンドの結合から生じる変化を逆転する。
【0052】
図3に示される方法は、所望の標的のシステイン変異体を作製することにより適用される。システイン残基は、タンパク質標的上の、目的の部位またはその近傍に導入される。1実施形態において、目的の部位は、標的上の位置が系統的に調査されるように選択される。別の実施形態において、標的が複数のサブユニットから構成される場合、目的の部位は境界面領域の近傍にある。これらのサブユニットは、同じポリペプチド(例えば、ホモダイマー)または異なるポリペプチド(例えば、へテロダイマー)から構成され得る。システインは、目的の部位から10Å内、好ましくは、目的の部位から5Å内に位置される場合、目的の部位の近傍にあるとみなされる。標的が、目的の部位の外側の天然に存在するシステインを含む場合、それらは、必要に応じて二重標識の可能性を排除するために、アラニンのような別の残基に変異され得る。
【0053】
概して、システイン残基に変異される残基は、溶媒接近可能である。溶媒接近可能性は、標準的な数値(Lee,B.&Richards,F.M.J.Mol.Biol 55:379−400(1971);Shrake,A.&Rupley,J.A.J.Mol.Biol.79:351−371(1973))または分析(Connolly,M.L.Science 221:709−713(1983);Richmond,T.J.J.Mol.Biol.178:63−89(1984))法を使用する構造モデルから計算され得る。例えば、潜在的なシステイン改変体は、LeeおよびRichardsの方法(Lee,B.&Richards,F.M.J.MoL Biol 55:379−400(1971))によって計算されるとき、炭素−β(CB)、または硫黄−γ(SG)の合わせた表面領域が、20Å2より大きい場合に、溶媒接近可能であるとみなされる。この値は、Creamerら(Creamer,T.P.ら、Biochemistry 34:16245−16250(1995))によって記載されるような、システイン側鎖に接近可能な理論上の表面領域の約33%を示す。
【0054】
また、システイン、または別のチオール含有アミノ酸残基へ変異される残基が、骨格原子との水素結合に関与しないこと、あるいは、多くとも1つの水素結合のみによって骨格と相互作用することが好ましい。側鎖が他の側鎖との複数(>1)の水素結合に関与する野生型残基はまた、あまり好ましくない。全ての標準的な回転異性体(−60°、60°、または180°のchi1角度)が他の任意の残基のN原子、CA原子、C原子、O原子、またはCB原子との、都合の悪い立体的接触を誘導し得る改変体もまた、あまり好ましくない。都合の悪い接触は、関与している原子のファンデルワールス半径の合計の80%未満である原子間距離として規定される。目的の部位が凹部領域である特定の実施形態において、このような部位の端(例えば、隆線または近接した凹部領域)に見出される残基は、システイン残基に変異するのにより好ましい。凸性および凹性は、表面ベクトル(vector)(Duncan,B.S.&Olson,A.J.Biopolymers 33:219−229(1993))に基づいて計算され得るか、または分子表面に沿って配置される水プローブの接近可能性を決定すること(Nicholls,A.らProteins 11:281−296(1991);Brady,G.P.,Jr.& Stouten,P.F.J.Comput.Aided Mol.Des.14:383−401(2000))によって計算され得る。L−アミノ酸について通常禁じられた骨格コンフォメーションを有する残基(Ramachandran,G.N.ら、J.Mol.Biol.7:95−99(1963);Ramachandran,G.N.&Sasisekharahn,V.Adv.Prot.Chem.23:283−437(1968))は、システインの改変についてのあまり好ましい標的ではない。禁じられたコンフォメーションは、φ角度の正の値を一般的に特徴とする。
【0055】
他の好ましい改変体は、システインに変異され、かつ−Cys−SSR1を含むように係留される場合、R1の原子を目的の部位に向かって指向するコンホメーションを有するものである。2つの一般的手順が使用されて、これらの好ましい改変体が同定され得る。第1の手順において、独特の構造(Hobohm,U.ら.Protein Science 1:409−417(1992))に関してProtein Databank(Berman,H.M.ら Nucleic Acids Research 28:235−242(2000))において検索が行われて、j位においてジスルフィド結合されたシステインを含む構造フラグメントが同定される。この位置において、フラグメントの残基j−1、j、およびj+1のその骨格原子は、0.75平方Å未満のRMSDと、標的分子の残基i−1、i、およびi+1の骨格原子上で重ね合わせられ得る。フラグメントが、残基iのCβ原子(システインに変異された場合)よりも、目的の部位の何れの原子にもより近いj位においてシステインにジスルフィド結合される残基のCβ原子が位置すると同定される場合、i位が好ましいと考えられる。代替的手順において、i位の残基は、システインへと計算により「変異」され、ジスルフィド結合を介してS−メチル基でキャップされる。
【0056】
種々の組換え技術、化学的技術、合成技術および/または他の技術が、標的が係留に利用可能な所望の数の遊離チオール基を有するように標的を改変するために使用され得る。このような技術としては、例えば、標的ポリペプチドをコードする核酸配列の部位特異的変異誘発が挙げられ、その結果、この核酸配列は、異なる数のシステイン残基を有するポリペプチドをコードする。特に好ましいのは、ポリメラーゼ連鎖反応(PCR)増幅を使用する部位特異的変異誘発である(例えば、米国特許第4,683,195号(1987年7月28日発行);およびCurrent Protocols In Molecular Biology,第15章(Ausubelら編,1991)を参照のこと)。他の部位指向性変異誘発技術もまた、当該分野で周知であり、例えば、以下の刊行物において記載されている:Ausubelら,前出,第8章;Molecular Cloning:A Laboratory Manual.,第2版(Sambrookら,1989);Zollerら,Methods Enzymol.100:468−500(1983);Zoller&Smith,DNA 3:479−488(1984);Zollerら,Nucl.Acids Res.,10:6487(1987);Brakeら,Proc.Natl.Acad.Sci.USA 81:4642−4646(1984);Botsteinら,Science 229:1193(1985);Kunkelら,Methods Enzymol.154:367−82(1987)、Adelmanら,DNA 2:183(1983);ならびにCarterら,Nucl.Acids Res.,13:4331(1986)。カセット変異誘発(Wellsら,Gene,34:315[1985])、および制限選択変異誘発(Wellsら,Philos.Trans.R.Soc.London SerA,317:415[1986])もまた、使用され得る。
【0057】
1つを超えるアミノ酸置換を有するアミノ酸配列改変体は、いくつかの方法のうちの1つで生成され得る。そのアミノ酸がポリペプチド鎖中でともに近くに位置する場合、それらは、所望のアミノ酸置換の全てをコードする1つのオリゴヌクレオチドを用いて、同時に変異され得る。しかし、そのアミノ酸が互いにいくらか距離が離れて位置する(例えば、10アミノ酸を超えて分離されている)場合、所望の変化の全てをコードする単一のオリゴヌクレオチドを生成することはより困難である。代わりに、2つの代替的方法のうちの1つが、使用され得る。第1の方法において、別個のオリゴヌクレオチドが、置換されるべき各アミノ酸について生成される。次いで、これらのオリゴヌクレオチドは、一本鎖テンプレートDNAに同時にアニールされ、そのテンプレートから合成される第2鎖のDNAは、所望のアミノ酸置換の全てをコードする。代替的方法は、所望の変異を生成するために2回を超える変異誘発を含む。
【0058】
本発明の別の局面において、アロステリックインヒビターを同定するための方法が提供され、この方法は、以下:
a)第一の係留実験を実施する工程、および
b)第二の係留実験を実施する工程、
を包含し、ここで、両方の係留実験が、
i)第一の結合部位、第二の結合部位およびこの第二の結合部位またはその近傍の化学反応基を含む標的を提供する工程;
ii)この標的を、この化学反応基と共有結合を形成し得る化合物と、接触させる工程;
iii)この標的とこの化合物との間の共有結合を形成し、それによって、標的−化合物結合体を形成する工程;および
iv)この標的−化合物結合体を同定する工程、
を包含し、ここで、
この第一の係留実験が、この第一の結合部位に結合するリガンドの存在下で実施され、そしてこの第二の係留実験が、この第一の結合部位に結合するリガンドの非存在下で実施される。
【0059】
その基質またはリガンドの非存在下で標的−化合物結合体を形成するが、その基質またはリガンドの存在下では標的−化合物結合体を形成しない化合物は、アロステリックインヒビターの候補である。一実施形態において、その標的は酵素であり、その第1の結合部位に結合するリガンドは、既知の競合的インヒビターである。別の実施形態において、その共有結合は、ジスルフィドであり、その化合物は、マスキングされたジスルフィドを有するリガンド候補である。
【0060】
本発明の別の局面において、アロステリック調節をし得る標的においてアロステリックインヒビターを同定するための方法が提供される。この方法は、リガンドのアロステリック部位への結合が、もはや標的を阻害しないように、アロステリック部位を無能にすることに依存する。この方法は、以下:
a)アロステリック調節をし得る標的およびアロステリック調節をし得ないその変異体を提供する工程;
b)この標的を化合物と接触させる工程;
c)この変異体をこの化合物と接触させる工程;ならびに
d)この標的に対するこの化合物の活性を、変異体に対するこの化合物の活性と比較する工程、
を包含する。
【0061】
一実施形態において、このアロステリック無能化変異体(allosterically disabled mutant)は、アロステリック部位を含む、少なくとも1つの残基において変異を有する。別の実施形態において、このアロステリック無能化変異体は、アロステリック部位を含む、少なくとも2つの残基において変異を有する。なお別の実施形態において、このアロステリック無能化変異体は、アロステリック部位を含む、少なくとも3つの残基において変異を有する。
【0062】
(カスパーゼ)
カスパーゼ(システイニルアスパラギン酸特異的プロテアーゼ)は、サイトカイン成熟およびアポトーシスの両方において中心的(pivital)な役割を果たす細胞内システインプロテアーゼのファミリーである。多くの他のプロテアーゼと同様に、カスパーゼは、不活性チモーゲンとして合成される。これらのチモーゲンは、N末端プロドメイン、および切断されると、大きなサブユニットドメインおよび小さなサブユニットドメインを生じる切断部位を含む。一般に、Asp−X結合の内部切断は、活性なプロテアーゼのアセンブリおよびそれ自体のプロドメインの切断を可能にする、短いC末端の小さなサブユニットを分離する。これらの酵素の活性形態は、2つの大きなサブユニットおよび2つの小さなサブユニットから構成されるヘテロテトラマーである。しかし、この大きさサブユニットおよび小さなサブユニットが同じポリペプチドに由来するので、この活性形態は、しばしば、(本明細書中でいわれるように)ホモダイマーといわれる。
【0063】
本明細書中に記載される方法を使用して、新規なアロステリック部位が、カスパーゼのダイマー境界面において同定されている。この部位は、カスパーゼ−3において初めて同定され、カスパーゼファミリーの酵素内の顕著な構造類似性に起因して、他のカスパーゼにも存在すると考えられた。例えば、カスパーゼ−3と、カスパーゼ−1(29%同一性)およびカスパーゼ−9(24%同一性)との間の相対的に低い配列同一性にも拘わらず、この3つの酵素は、高い程度の構造類似性を共有し、本質的に、互いに重ね合わせることが可能である。Mittlら,J.Biol Chem 272:6539;Rotondaら,Nat Struct Biol 3:619;Chaiら,Proc.Natl.Acad.Sci(USA),98:14250−14255;ならびにWattら,Structure 7:1135−1143を参照のこと。さらに記載されるように、このカスパーゼのアロステリック部位が、他のカスパーゼにおいて同定されている。しかし、このカスパーゼのアロステリック部位は、カスパーゼ−3において初めて特徴付けられたので、カスパーゼのアロステリック部位を含む残基は、カスパーゼ−3の番号付けスキームを用いて記載される。
【0064】
カスパーゼ−3と選択された代表的なカスパーゼとの配列アライメントを図4に示す。このアライメントを、示されたカスパーゼについて以下のアミノ酸を使用して作製した:XP_054686(カスパーゼ−3);NP_150634(カスパーゼ−1);NP−116764(カスパーゼ−2);AAH15799(カスパーゼ−7);およびAAH02452(カスパーゼ−9)。カスパーゼ−3配列とカスパーゼ−1配列、カスパーゼ−2配列、カスパーゼ−7配列、およびカスパーゼ−9配列とのそれぞれの間の整列している残基は、互いに対応していると言われる。例えば、Cys−264は、カスパーゼ−3の264番目のアミノ酸残基であり、カスパーゼ−1のスレオニン、カスパーゼ−2のチロシン、カスパーゼ−7のシステイン、およびカスパーゼ−9のグリシンに対応する。
【0065】
他のカスパーゼは、図4に示されるアライメントを参照して整列され得る。あるいは、配列は、Clustal W(1.81)(http://www2.ebi.ac.uk/clustalw/)のような標準的なアライメントソフトウェアを用いて整列され得る。
【0066】
1つの実施形態において、カスパーゼアロステリック部位は、カスパーゼ−3のCys−264に対応する残基の5Å以内であるカスパーゼの残基を含む。残基は、その原子のいずれかが、カスパーゼ−3のCys−264に対応する残基のいずれかの原子から5Å以下である場合、5Å以内であると言われる。別の実施形態において、カスパーゼアロステリック部位は、カスパーゼ−3のCys−264に対応する残基のいずれかの原子から3Å以下であるカスパーゼの残基を含む。
【0067】
別の実施形態において、カスパーゼアロステリック部位は、カスパーゼ−3の残基に対応する少なくとも2つの残基を含む:Glu−124;Gly−125;Lys−135;Leu−136;Lys−137,Lys−138;Ile−139;Thr−140;Leu−157;Phe−158;Ile−159;Phe−193,Leu−194;Tyr−195;Ala−196;Tyr−197;Ala−200;Pro−201;Gly−202;Cys−264;Ile−265;Val−266;Ser−267;Met−268;およびLeu−269。これらのカスパーゼ−3の残基ならびにカスパーゼ−1、カスパーゼ−2、カスパーゼ−7、およびカスパーゼ−9の対応する残基を、図4で囲む。別の実施形態において、カスパーゼアロステリック部位は、カスパーゼ−3の残基に対応する少なくとも2つの残基を含む:Cys−264;Ile−265;Val−266;Ser−267;Met−268;およびLeu−269。
【0068】
(カスパーゼ−3)
カスパーゼ−3は、クローニングされ、そしてシステイン残基がタンパク質の全体にわたる種々の位置で導入されている変異体が作製された。実施例1は、例示的なセットのシステイン変異体についてのクローニングおよび変異誘発をより詳細に記載する。クローニンされたタンパク質および変異体タンパク質を、実施例2に記載されるテトラペプチド酵素アッセイを使用して特徴付けた。
【0069】
係留(tethering)実験を、式
【0070】
【化6】
のリガンド候補のライブラリーを用いて、実施例3に記載されるように、カスパーゼ−3およびそのシステイン変異体を使用して、実施した。ここで、Rcは、先に規定された通りである。これらの係留実験の過程の間、カスパーゼ−3のアロステリック部位は、小さなサブユニット中の天然に存在するシステイン(Cys−264)の近位において発見された。
【0071】
選択されたリガンド候補によって改変された天然に存在するシステインとしてのCys264の同定を、実施例4に記載する。
【0072】
2つの選択されたリガンド候補は、化合物1および2であった:
【0073】
【化7】
示された部分は、Cys264とジスルフィド結合を形成するリガンド候補のRcC(=O)NHCH2CH2S−部分に対応する。図5は、カスパーゼ−3の小さなサブユニットとジスルフィド結合を形成するが、大きなサブユニットとは結合を形成しない化合物1を示す代表的な係留実験である。
【0074】
化合物1および2は、強く選択され、これらの化合物がアロステリック部位に対して固有の結合親和性を有することを示した。さらに、構造−活性関係を係留実験から観察した。例えば、化合物1が強く選択されるものの、化合物3および4は、そうではない。
【0075】
【化8】
実施例5に記載されるアッセイを使用して、化合物1および2をさらに特徴付け、そして化学量論的な様式でカスパーゼ−3活性を阻害することを示した。図6において示され得るように、阻害%を、化合物1または2とジスルフィド結合を形成するカスパーゼ−3の%を用いて追跡する。注目すべきことに、この阻害は、カスパーゼ−3と化合物2との間のジスルフィド結合の減少の際の酵素活性の回復で実証されるように、可逆である。
【0076】
これらの化合物が活性ホモダイマーの形成を破壊する可能性を調査しそして排除した。活性ホモダイマーからモノマーへの変換に続くサイジングクロマトグラフィーの溶出プロフィールは、化合物1または2の非存在および存在の両方において、カスパーゼ−3について本質的に同一であった。
【0077】
代わりに、構造実験によって実証されるように、作用の機構は、アロステリック部位への化合物1または2の結合の際の活性部位の再配列に起因する。注目すべきことに、アロステリック外側部位(exosite)への化合物1または2の結合は、活性部位での基質の結合を防止する。興味深いことに、逆もまた真実である。活性部位への基質の結合は、アロステリック外側部位への化合物1または2の結合を防止する。言い換えると、活性部位およびアロステリック部位への結合事象は、相互に排他的である。
【0078】
(カスパーゼ−7)
カスパーゼ−3との53%の配列同一性およびCys−264として対応する部位に位置するシステインに基づいて、カスパーゼ−7のアロステリック部位が、カスパーゼ−3のアロステリック部位と同様に挙動することが予期された。化合物1および2が、カスパーゼ−3と同様な様式でカスパーゼ−7を阻害することを確認した後に、カスパーゼ−7は、pro形態、活性apo形態および活性阻害された形態で結晶化された唯一のカスパーゼであるので、カスパーゼアロステリック部位の構造研究のために選択した。
【0079】
実施例6は、化合物1および化合物2と複合体化されたカスパーゼ−7の構造研究のためのクローニングおよび結晶化手順を記載する。これらの化合物は、ダイマー界面における深いポケットに結合する。このポケットが化合物1または2の非存在下においてさえ識別可能であるので、カスパーゼアロステリック部位は、アロステリックインヒビターの存在によって誘導されなかった。化合物1または2を用いるカスパーゼ−7の構造研究は、これらの化合物が、アロステリック部位に特異的に結合することを確認し、そしてアロステリック阻害の背後にある潜在的機構を明らかにする。
【0080】
カスパーゼの活性形態がホモダイマーであるので、アロステリックインヒビターの2つの分子が、活性複合体に結合する(各小さなサブユニットについて1つの分子)。2つの小さなサブユニットの各々によって形成されるアロステリック結合部位は、互いに空間的に隣接する。
【0081】
化合物1の2つの分子は、それぞれの部位に結合し、そしてアンチパラレルの方向で互いに面する。2つの分子の間の2つの最も近い原子がそれぞれのカルボニルであり、そして7Åの距離で分離されているので、これらは、互いに相互作用するようではない。実際に、化合物1の2つの分子の間にも、化合物1のいずれかの分子とタンパク質との間にも、直接的な水素結合相互作用の証拠は無かった。しかし、5つの潜在的な水媒介性の水素結合が、化合物1の2つの分子とタンパク質との間で同定された。
【0082】
対照的に、化合物2の2つの分子は、1つの分子のインドール窒素と他の分子のカルボニル酸素との間で1つの分子内水素結合を形成する縁部−縁部(edge−to−edge)様式で、互いに相互作用する。さらに、3つの直接的な水素結合が、化合物2とタンパク質との間でなされるようである。
【0083】
2つの異なる結合様式は、化合物1および化合物2が活性部位に対してそれぞれの効果を及ぼす異なる方法で相関するようである。化合物1の場合、この化合物は、その化合物が阻害する活性部位と同じポリペプチドによって形成されるアロステリック部位に結合する。化合物2の場合、この化合物は、1つのポリペプチドによって形成されるアロステリック部位に結合し、そして他のポリペプチドによって形成される活性部位を阻害する。
【0084】
それにもかかわらず、化合物1または化合物2の結合が活性部位において阻害を達成する機構は、同じであるようである。カスパーゼの活性化は、pro−ペプチドの切断、および大きなサブユニットと小さなサブユニットとの間の切断の両方を必要とする。これらの切断がタンパク質の「活性」形態を与えるものの、得られるカスパーゼは、ペプチド結合溝の構造的再配列が生じるまで、触媒的に適格ではない。
【0085】
結合された基質の非存在下において、カスパーゼのいわゆる「活性」形態は、触媒的に不活性なコンフォメーションのままである。図7Aに示されるように、Arg−164(カスパーゼ−3の番号付けを使用する)(活性部位システインにすぐに隣接する残基)は、活性部位にまで突出し、その結果、ペプチド結合溝は、基質に結合するのに適切なコンフォメーションではない。基質が結合した場合、多くの構造的変化が誘導される。Arg−164は、タンパク質のコア内に押し込められ、そしてペプチド結合溝は、基質に適合するようになり(図7Bを参照のこと)、これらの両方が、酵素の触媒的に適格な形態に必要とされる。
【0086】
化合物1または2のいずれかに結合されたカスパーゼ−7の構造において、Tyr−197(カスパーゼ−3の番号付けを使用する)は、タンパク質のコアにおけるArg−164の埋没(burial)を防止するように、変位する。結果として、Arg−164は、非結合構造において見られるように、上に突出する(図7Cを参照のこと)。さらに、ペプチド結合溝は、乱れる。
【0087】
(カスパーゼ−9)
カスパーゼ−9はまた、アロステリック部位の証拠について研究された。カスパーゼ−7とは異なり、カスパーゼ−9は、カスパーゼ−3と24%のみの配列同一性を共有する。さらに、カスパーゼ−3のCys−264に対応する残基は、グリシンであり、システインではない。しかし、天然に存在するシステインはまた、アロステリックポケット内に存在し、カスパーゼ−3のIle−265に対応する。
【0088】
実施例7は、カスパーゼ−9のクローニングおよびアッセイのために使用される手順を記載する。カスパーゼ−3と同様に、係留実験は、小さなサブユニットに結合するとして、以下:
【0089】
【化9】
を含むいくつかのリガンド候補を同定した。カスパーゼ−9が小さなサブユニットにおいてただ1つの天然に存在するシステインを含むので、それは、カスパーゼ−3のIle−265に対応する対応物として容易に同定された。注目すべきことに、この残基は、カスパーゼ−3のCys−264とほとんど同一の位置である。
【0090】
カスパーゼ−3の場合のように、これらの化合物がアロステリック部位に特異的に結合するという証拠には、識別可能な構造−活性関係が存在した。例えば、構造
【0091】
【化10】
を有する化合物8のリガンド候補もまた選択した。この化合物は、さらなるカルボキシル基の存在によって、化合物5と一部異なる。しかし、他のリガンド候補を試験し、選択されないことが見出された。試験されたビアリールエーテルにおいて置換された特定の官能基は、さらなるヒドロキシル基、ニトロ基などを有する化合物を含むか、またはこのカルボキシル基が、以下に示されるような異なる位置で置換される:
【0092】
【化11】
(PTP−1B)
カスパーゼに加えて、別の以前に未知であったアロステリック部位が、PTP−1B(最近の数年において、糖尿病および肥満のような種々の代謝障害の処置についての非常に確証された標的になったホスファターゼ)において同定された。
【0093】
ヒトPTP−1Bは、435アミノ酸タンパク質である。しかし、全長タンパク質が細菌において十分に発現しないので、PTP−1Bの研究は、代表的に、タンパク質の最初の321アミノ酸または最初の298アミノ酸に対応する形態のような短縮形態を使用して実施される。実施例8は、PTP−1Bの短縮型およびその変異体を作製するためのプロトコルを記載する。
【0094】
種々のアロステリックインヒビターの存在および非存在下でのPTP−1Bの結晶学的研究は、カスパーゼにおいて同定されたアロステリック部位とは異なり、PTP−1Bのアロステリック部位が、適合可能であることを示した。言い換えると、アロステリック結合部位は、適切なリガンドの非存在下で識別不可能である。
【0095】
PTP−1Bのアロステリックインヒビターの例示的な例は、以下:
【0096】
【化12】
に示される化合物15である。
【0097】
この化合物は、約30μMのIC50で非競合的にPTP−1Bに結合し、そして阻害する。PTP−1Bの活性を決定するための例示的なプロトコルを、実施例9に記載する。化合物15のようなアロステリックインヒビターの存在下において、以下の残基によって作製された凹部が形成される:Glu−186;Ser−187;Pro−188;Ala−189;Leu−192;Asn−193;Phe−196;Lys−197;Glu−200;Leu−272;Glu−276;Gly−277;Lys−279;Phe−280;Ile−281;およびMet−282。これらの残基は、化合物が結合する接触表面を形成する。しかし、アロステリックインヒビターの存在下において、この部位の大部分は、残基283〜298によって形成されるヘリックスの存在によって塞がれ、そして上記残基の大部分が、もはや接近可能ではない。
【0098】
アロステリックインヒビターの存在下で生じる大きなコンフォメーションの変化は、少なくとも3つの残基(Tyr−152、Asn−193、およびTryp−291)の相互作用によって媒介され、そしてPTP−1Bについての調節機構の一部であると考えられる。アロステリックインヒビターの非存在下において、Asn−193のNδ2(アロステリック部位形成残基の1つ)は、Tyr−152のOηと水素結合を形成し、そして残基283〜298によって形成されるヘリックスは、少なくとも一部、Trp−291のインドール環とPhe−280およびPhe−196(これらの両方がまたアロステリック部位形成残基である)のフェニル環との非結合相互作用により、適所に維持される。第4の残基(Lys−197(別のアロステリック部位形成残基))もまた、Asn−193のNδ2とTyr−152のOηとの間の水素結合相互作用を維持する際に関与すると考えられる。
【0099】
化合物15のようなアロステリックインヒビターの存在下において、残基283〜298によって形成される螺旋は、変位され、そして/または乱される。化合物15の場合、ベンゾフラン部分は、Trp−291のインドール環を変位する。化合物15のカルボニル酸素は、Asn−193のNδ2と水素結合を形成し、その結果、Asn−193のNδ2は、Trp−152のOηに対する水素結合にもはや利用可能ではない。Asn−193とTyr−152との間の水素結合の破壊は、一部、Tyr−152のフェノール環のコンフォメーションの変化を媒介する。Tyr−152のフェノール環の回転は、酵素を機能的に不活性化するPTP−1Bの活性部位におけるコンフォメーションの変化を広げる。
【0100】
アロステリック形成残基(Asn−193、Phe−196、およびPhe−280)の重要性は、変異誘発実験を使用して一部確認された。変異体PTP−1B(Asn−193がアラニンに変異され、Phe−196がアルギニンに変異され、そしてPhe−280がシステインに変異される)が作製された場合、アロステリック機構は無効になり、化合物15のようなアロステリックインヒビターは、もはや酵素を阻害し得ない。
【0101】
本発明は、さらに、以下の非制限的な実施例によって例示される。
【0102】
(実施例1 ヒトカスパーゼ−3のクローニング)
ヒトのカスパーゼ−3(Yama、CPP32ベータとしても公知)を、ジャーカット細胞(クローンE6−1;ATCC)から直接クローニングした。手短には、総RNAを、Tri−試薬(シグマ社)を用いて、37℃、5%CO2で増殖したジャーカット細胞から精製した。ポリメラーゼ連鎖反応(PCR)によって、カスパーゼ−3/Yama/CPP32のラージ(大きい)サブユニットおよびスモール(小さい)サブユニットをコードするDNAの増殖を可能にするように、オリゴヌクレオチドプライマーを設計した。手短には、Ready−To−Go−PCRビーズ(Amersham/Pharmacia)および以下のオリゴヌクレオチド:
5’−TTCCATATGTCTGGAATATCCCTGGACAACAGTTA−3’(配列番号1)および
5’−AAGGAATTCTTAGTCTGTCTCAATGCCACAGTCCAG−3’(配列番号2)
を用いて、1μgの総RNAから、アミノ酸28−175(ラージサブユニットのほとんどを含む)をコードするDNAを直接増幅した。Ready−To−Go−PCRビーズ(Amersham/Pharmacia)および以下のオリゴヌクレオチド:
5’−TTCCATATGAGTGGTGTTGATGATGACATGGCG−3’(配列番号3)および
5’−AAGGAATTCTTAGTGATAAAAATAGAGTTCTTTTGTGAG−3’(配列番号4)
を用いて、1μgの総RNAから、アミノ酸176−277(スモールサブユニットのほとんどを含む)をコードするDNAを直接増幅した。
【0103】
次に、カスパーゼ−3のラージサブユニットまたはスモールサブユニットのいずれかに対応する増幅したDNAを、制限酵素EcoRIおよびNdeIで切断し、標準的な分子生物学の技術を用いて、EcoRIおよびNdeIで消化したpRSET−b(インビトロジェン社)中に直接クローニングした。[例えば、Tewari M,Quan LT,O’Rourke K,Desnoyers S,Zeng Z,Beidler DR,Poirier GG,Salvesen GSおよびDixit VM.Yama/CPP32 beta,a mammalian homolog of CED−3,is a CrmA−inhibitable protease that cleaves the death substrate poly (ADP−ribose) polymerase、Cell 81 (5), 801−809 (1995)を参照のこと]。
【0104】
スモールサブユニットについて2つの報告されたタンパク質配列があり、そして各々は、1アミノ酸が異なり、アミノ酸190位(活性部位のシステインを163位とした相対位置)に、アスパラギン酸(GenBankアクセッション番号P42574)またはグルタミン酸(GenBankアクセッション番号XP_054686)のいずれかを有する。両方の形態のクローニング、発現、および精製が成功しており、両者は、機能的に区別できない。
【0105】
(一本鎖DNAの調製)
カスパーゼ−3のラージサブユニットまたはスモールサブユニットのいずれかをコードするDNAを含むプラスミドを、別々にE.coli K12 CJ236細胞(ニューイングランドバイオラブス)中に形質転換し、各々の構築物を含む細胞を、アンピシリン含有寒天プレート上での増殖能力によって選択した。ラージサブユニットおよびスモールサブユニットのオーバーナイト培養物を、別々に2YT(100μg/mLのアンピシリンを含む)中で、37℃で増殖させた。各々の培養物を、1:100で希釈し、そしてA600=0.3−0.6まで増殖させた。各培養物の1.5mLのサンプルを取り出し、そして10μLのファージVCS−M13(ストラタジーン社)で感染し、37℃で60分間振盪し、そして各々のオーバーナイト培養物を、100μg/mLのアンピシリンと20μg/mLのクロラムフェニコールを含む2YT中に1:100で希釈した感染培養物1mLを用いて調製し、そして37℃で増殖させた。細胞を、3000rcfで10分間遠心し、そして1/5容量の20% PEG/2.5M NaClを上清に添加した。サンプルを室温にて10分間インキュベートし、次に、4000rcfで15分間遠心分離した。ファージのペレットを、PBS中に再懸濁し、15Krpmで10分間遠心して、残りの微粒子状の物質を取り除いた。上清を保持し、一本鎖DNAを、QIAプレップスピンM13キット(キアゲン社)の手順に従って、上清から精製した。
【0106】
(システイン残基に改変する残基の同定)
システイン残基に改変したアミノ酸残基の選択は、カスパーゼ−3の三次元結晶構造を考察することによって、なされた。9つの異なるアミノ酸残基を、システイン残基に改変するために選択した。システイン変異を含む各カスパーゼ−3を、E.coli細胞中で、高レベルで発現した(一般的に1mg/Lより多い)。1例以外の全ての場合において、本発明者らは、正確にリフォールディングした四量体タンパク質の精製に成功し得た(Uno−5 Qクロマトグラフィーによって精製される能力によって評価)。しかし、ラージサブユニットのアミノ酸121においてヒスチジンのシステインへの変異を含むカスパーゼ−3タンパク質は、通常のクロマトグラフィーでは精製することができなかった。本発明者らは、個々のサブユニットを別々に精製することができたので、本発明者らは、カスパーゼ−3のこの改変体が正確に四量体を形成する(すなわち、ラージサブユニットがスモールサブユニットとリフォールディングする)ことができないことに起因する可能性が高いことが原因であると結論付けた。本発明者らはまた、新たなシステイン残基を有するカスパーゼ−3の全てのバージョンが、触媒活性を有するわけではないことを見出した。例えば、Y204Cは触媒活性を有さない。
【0107】
(一本鎖変異誘発)
スモールサブユニット内のシステイン変異体の例示的な例としては、F256C;S209C;S251C;W214C;およびY204Cが挙げられる。これらの変異体は、以下のプライマー:
F256C(5’−CTT TGC ATG ACA AGT AGC GTC−3’)、(配列番号5)
S209C(5’−GCC ATC CTT ACA ATT TCG CCA−3’)、(配列番号6)
S251C(5’−AGC GTC AAA GCA AAA GGA CTC−3’)、(配列番号7)
W214C(5’−CTG GAT GAA ACA GGA GCC ATC−3’)、(配列番号8)および
Y204C(5’−TCG CCA AGA ACA ATA ACC AGG−3’)、(配列番号9)
を用いて作製された。
【0108】
ラージサブユニット内のシステイン変異体の例示的な例としては、H121C;L168C;M61C;およびS65Cが挙げられる。これらの変異体は、以下のプライマー:
H121C(5’−TTC TTC ACC ACA GCT CAG AAG−3’)、(配列番号10)
L168C(5’−GCC ACA GTC ACA TTC TGT ACC−3’)、(配列番号11)
M61C (5’−CCG AGA TGT ACA TCC AGT GCT−3’)、(配列番号12)および
S65C (5’−ATC TGT ACC ACA CCG AGA TGT−3’)、(配列番号13)
を用いて作製された。
【0109】
約100pmolの各プラスミドを、1×TM緩衝液(0.5M Tris、pH 7.5、0.1M MgCl2)、lmM ATP、5mM DTT、および5単位のT4キナーゼ(NEB)を含む緩衝液中で、37℃で60分間インキュベートすることによってリン酸化した。キナーゼ処理したプライマーを、20μLの反応容量(〜50ngのキナーゼ処理したプライマー、1×TM緩衝液、および10〜50ng一本鎖DNA)中で、85℃で2分間、50℃で5分間、次に4℃で30〜60分間インキュベートすることによって、鋳型DNAとアニーリングした。伸長カクテル(2mM ATP、5mM dNTPs、30mM DTT、T4 DNAリガーゼ(NEB)、およびT7ポリメラーゼ(NEB))を、各アニーリング反応物に添加し、そして室温で3時間インキュベートした。変異誘発したDNAをE.coli XL−1 Blue細胞中に形質転換し、プラスミドDNAを含むコロニーを100μg/mlのアンピシリンを含むLB寒天プレートでの増殖によって選択した。DNA配列決定を用いて、適切な変異を含むプラスミドを同定した。
【0110】
(タンパク質発現および精製)
ラージサブユニット中のシステイン変異をコードするプラスミドDNAを、Codon Plus BL21細胞中に形質転換し、そしてスモールサブユニット中のシステイン変異をコードするプラスミドDNAを、BL21(DE3)pLysS細胞中に形質転換した。野生型ラージサブユニットおよびシステイン変異型ラージサブユニットをコードするプラスミドを含むCodon Plus BL21細胞を、150μg/mLのアンピシリンを含む2YT中で一晩、37℃で増殖させ、すぐに収集した。野生型スモールサブユニットおよびシステイン変異型スモールサブユニットをコードするプラスミドを含むBL21pLysS細胞を、150μg/mLのアンピシリンを含む2YT中でA600=0.6まで、37℃で増殖させた。その後、培養物を1mMのIPTGを用いて誘導し、さらに3〜4時間、37℃で増殖させた。4Krpmで10分間の遠心によって細胞を収集した後、細胞のペレットを、Tris−HCl(pH8.0)/5mM EDTA中に再懸濁し、2回、ミクロ流動化した(micro fluidize)。封入体を、9Krpmで10分間の遠心によって単離し、次に、6Mのグアニジン塩酸中に再懸濁した。変性したサブユニットを迅速かつ均一に、100μg/mLの濃度で再生緩衝液(l00mM Tris/KOH(pH 8.0)、10%スクロース、0.1% CHAPS、0.15M NaCl、およびl0mM DTT)中に希釈し、室温で60分間、ゆっくりと攪拌しながら、インキュベートすることによって、再生させた。
【0111】
再生したタンパク質を、10mM Tris(pH8.5)、10mM DTT、および0.1mM DETAを含む緩衝液中で一晩透析した。9Krpmで15分間遠心し、0.22μmの硝酸セルロースフィルターを通して上清をろ過することによって、沈殿物を取り除いた。次に、上清をアニオン交換カラム(Uno5 Q−カラム(バイオラッド))にロードし、正確に折りたたまれたカスパーゼ−3タンパク質を0−0.25M NaCl勾配を3mL/分で用いて、溶出した。各画分のアリコートを、変性ポリアクリルアミドゲル上で電気泳動して、カスパーゼ−3タンパク質を含む画分をプールした。
【0112】
(実施例2)
本実施例は、カスパーゼ−3の酵素活性を特徴付ける1つの方法を記載する。
【0113】
カスパーゼ−3についての最適なテトラペプチド認識モチーフを組み込んだクマリンベースの蛍光発生的な基質を、Alexis Biochemicals社より購入した。カスパーゼ−3を、1×反応緩衝液(25mM HEPES pH 7.4、0.1% CHAPS、50mM KClおよび5mM β−メルカプトエタノール)に添加して、最終濃度を、〜1.6nMとした。テトラペプチド基質(Ac−Asp−Glu−Val−Asp−AFC)を、添加して、最終濃度を、5μMとし、最終的な容量を50μLとした。アッセイを黒色の96ウェル平底ポリスチレンプレート(コーニング社)中で行い、そしてカスパーゼ活性を、365nmの励起波長および495nmの発光波長を用いて、Molecular Devices社のMicroplate Spectrofluorometer Gemini XSを用いて、モニターした。速度論データを、室温での15分間にわたるアッセイによって収集した。
【0114】
(実施例3)
本実施例は、カスパーゼ−3またはそのシステイン変異体を使用する、つなぎとめ(tethering)実験の1つの実施形態を記載する。つなぎとめスクリーニングは、典型的には、50μLの容量中で、最終濃度1〜5μMのカスパーゼ−3、1〜20mMのβ−メルカプトエタノール、および1〜2mMのリガンド候補(プール中の全てのリガンド候補の総濃度;従って、〜10のプール中の各リガンド候補は、100〜200μMの最終濃度を有する)を用いて、TE緩衝液(10mM Tris、1mM EDTA、pH8.0)中で行われた。質量分析によって分析する前に、平衡まで反応を進ませた(1時間以上)。反応混合物を、Finnegan LCQ2またはLCQ3 LCMSにロードし、分離手順に依存して、各々2〜5分間分析した。逆重畳の後、ラージサブユニットおよび/またはスモールサブユニットを公知の分子量に基づいて同定した。
【0115】
(実施例4)
本実施例は、化合物1および2がカスパーゼ−3とジスルフィド結合を形成する、天然に存在するシステインとしてのCys264の同定を記載する。カスパーゼ−3のスモールサブユニットは、3つのシステイン(Cysl84、Cys220、およびCys264)を含む。これら3つの中で、Cys220が、埋もれており、従って、可能性として除外された。
【0116】
1つの実験において、適切なDNAプライマー(C184S 5’−TAT TTT ATG AGA CGC CAT GTC−3’ (配列番号14);C264S 5’−GGA AAC AAT CGA TGG AAT CTG−3’ (配列番号15)、下線を引いたトリプレットは、導入されたセリン残基を示す)を用いて、184位および264位のシステインのいずれかを、セリンに変異した変異体を作製した。その後、クローンを、DNA配列分析によって確認した。C184S変異体は、化合物1および2と、標的−化合物結合体を形成し得たが、C264S変異体は、形成し得なかった。
【0117】
システイン残基としてのCys264の同定を、化合物1との標的−化合物結合体のペプチドマッピングによって、さらに確認した。TE緩衝液(pH8.0)中のカスパーゼ−3(4μM)を、化合物1(200μM)およびβメルカプトエタノール(1mM)の存在下で、1時間、25℃でインキュベートした。100%のスモールサブユニットが化合物1によって改変した時(LC/MSによって決定した)に、過剰な化合物1および還元剤を、セファデックスG−25カラムでのサイズ排除クロマトグラフィーによって除去した。微量の還元剤または未結合の化合物1が除去されることを確実にするために、タンパク質をTE緩衝液中に10倍に希釈し、そして、ミリポア5,000MWCOろ過デバイスによって再度濃縮した。このプロセスを、3回繰り返した。改変されたカスパーゼ−3を、98℃ 1分間熱変性し、次に、溶液が室温になるまで、氷上でインキュベートした。熱処理後、約70%のカスパーゼ−3が、標的−化合物結合体として残った。標的−化合物結合体を、500mMの酢酸アンモニウム緩衝液(pH4.0)中のエンドプロテイナーゼGlu−c(20ng/μL)によって、20時間 室温で消化した。ペプチドの質量を、Q−STAR装置においてLC/MS分析した。化合物1と共有結合したCys264を含むペプチドを含む、予測された消化フラグメントの各々の質量を観察した。化合物1に共有結合したCys184またはCys220に対応するペプチド質量は、観察されなかった。このことを、MS/MSによってフラグメント化した後の、トリペプチドP263C264I265+化合物1の観察によって、確認した。
【0118】
(実施例5)
本実施例は、ジスルフィド形成の程度(標的−リガンド結合体の形成)と、カスパーゼ−3酵素活性の阻害の程度とを相関付ける1つの実施形態を記載する。
【0119】
一定濃度のタンパク質(1〜5μM)および化合物(典型的には200μM)を用いて、反応物中のβ−メルカプトエタノールの濃度を変化させ、標的−リガンド結合体の形成の程度を変化させた。約1時間後、サンプルを、LC/MSによって試験して、各β−メルカプトエタノール濃度において、化合物1によって改変されたスモールサブユニットの割合を決定した。同時に、1μLのサンプルを取り出し、そして199μLの1×反応緩衝液(5μMのAc−Asp−Glu−Val−Asp−AFCを含む、25mM HEPES pH 7.4、0.1% CHAPS、50mM KClおよび5mM β−メルカプトエタノール)に添加した。化合物1の分析のために、200μMの化合物および種々の濃度のβ−メルカプトエタノールもまた添加し、その結果、つなぎとめ反応において、β−メルカプトエタノールの最終濃度を同一にした。希釈後、カスパーゼ−3酵素の最終濃度は、〜5nMであった。カスパーゼ活性を、365nmの励起波長および495nmの発光波長を用いて、Molecular Devices社のMicroplate Spectrofluorometer Gemini XSを用いて、モニターした。種々の量の化合物1を用いて改変したカスパーゼ−3の相対活性を、化合物1の不存在下での同様の反応条件下で観察された活性と比較した。化合物2を、同様に試験した。
【0120】
(実施例6)
本実施例は、化合物1および2と複合体化したカスパーゼ−7の構造研究のためのクローニングおよび結晶化手順を記載する。
【0121】
表1において詳細に記載されるように、カスパーゼ−7のラージサブユニット発現のための一連のプラスミドを、カスパーゼ−7の残基50−198または57−198のコード配列を、pRSET(ampr、インビトロジェン)またはpET3a(ampr、ノバジェン)中にサブクローニングすることによって、作製した。
【0122】
【表1】
スモールサブユニットの発現のために、カスパーゼ−7残基199−303+アミノ酸QLHHHHHH、または同様の添加残基を有する残基210−303のコード配列を、pBB75(kanr)中に連結した(Batchelor、Piperら、1998)。変異D192Aもまた、ラージサブユニット中に導入し、異質性を最小限とした。プラスミドpJH02、03、05、 08、09または11(表2)を、pJH06または07と組み合わせて形質転換し、過剰発現試験を行った。これらの組み合わせの中で、pJH07および08、またはpJH07および09が、最も高く過剰発現し、そしてChaiら、Cell 104:769−80(2001)およびChaiら、Cell 107:399−407に記載される方法を用いて、容易に精製された。
【0123】
pJH07およびpJH08(10μM)から発現されたカスパーゼ−7(D192A)を、化合物1(100μM)によって、500μMのβ−メルカプトエタノールを含むTE緩衝液(10mM Tris、pH8.0、1mM EDTA)中で1週間室温にてインキュベーションすることによって、標識した。スモールサブユニットの標識は、数時間後、98%完全であったが(質量分析計によって決定された)、100%完全な標識を得るために、より長時間行った。化合物2(50μM)によるカスパーゼ−7(D192A)の標識化を、TE緩衝液中の250μMのβ−メルカプトエタノール存在下で行った。標識は、4時間後に60%完全であり、1週間後に100%完全であった。これらのタンパク質をNAP−5カラム(アマシャムファルマシアバイオテクAB)中での緩衝液交換によって、結晶化のための100mM NaCl、10mM Tris pH8.0を含む緩衝液中に移した。標識されたタンパク質を、ミリポア5K MWCO濃縮デバイス中で12mg/mLに濃縮した。
【0124】
1μLのタンパク質および2μLの母液溶液(100mM シトレート緩衝液(pH5.8)、1M LiSO4、1M NaCl)を含む液滴から、4℃でのハンギングドロップ拡散蒸着によって1週間で形成されたカスパーゼ−7(D192A)/化合物1の結晶。カスパーゼ−7/化合物2の結晶は、1μLのタンパク質および1μLの母液である液滴から成長した。いずれかの化合物とのカスパーゼ−7(D192A)の結晶を、20%グリセロールを含む成長母液の液滴に移し、4℃で一晩インキュベートした。次いで、この結晶を、液体窒素中でフラッシュ凍結した。
【0125】
化合物1との複合体についてのデータを、Raxis−4検出器を備えるRigakuジェネレータで収集した。データを、D*trekを用いて処理した。化合物2との複合体についてのデータを、Quantum−315 CCDカメラ(ADSC)で、SSRL Beamline 9−1にて収集した。データを、Project,C.C.,Acta Crysta.D.50:760−763(1994)に記載されるように、CCP4−mosflmおよびスカラーを用いて処理した。これらの構造を、活性カスパーゼ−7(1K86.pdb)の構造およびCCP4−amoreにおける剛性本体の精製を使用して、直接的分子置換によって解析した。これらの構造を、反復回の、Oにおける分子再構築(Jonesら、Acta Crystallogr A 47:110−119(1991))およびCCP4−refmacにおけるエネルギー最小化によって、精緻化した。最終データ統計を、表2に示す。
【0126】
(表2)
【0127】
【表2】
両方の複合体についての電子密度マップは、このタンパク質のコアと相互作用する化合物1および化合物2の方向を明確に明らかにした。これらの化合物の順序付けられた性質は、これらの係留化合物が特異的様式で結合していることを確認する。これらの化合物についての温度要因は、タンパク質自体由来の周辺原子と同じくらい低いかまたはそれよりも低く、インヒビター分子が、非常によく順序付けられ、そしてランダム様式または擬似様式で結合していないことを示す。
【0128】
(実施例7)
ヒトカスパーゼ−9を、刊行された手順(Garcia−Calvo,Mら、1998.Inhibition of Human Caspases by Peptide−based and Macromolecular Inhibitors,JBC 273(49):32608−32613;Thornberry,NAら、A combinatorial Approach Defines Specificities of Members of the Caspase Family and Granzyme B,JBC 272(29):17907−17911)に従って、クローニング、発現および精製し、次いで、適切な酵素活性について試験した。カスパーゼ−9酵素を、1×反応緩衝液(100mM MES(pH6.5)、10%グルコース、0.1% CHAPS、10mM DTTおよび100mM NaCl)に添加した。200μMの最終濃度までの基質の添加(Ac−Leu−Glu−His−Asp−AFC)により反応を開始し、そして最終反応容量を50μLにした。アッセイを、黒色96ウェル平底ポリスチレンプレート(Corning)中で実施した。カスパーゼ活性を、365nmの励起波長および495nmの発光波長でMolecular Devices’ Microplate Spectrofluorometer Gemini XSを使用してモニタリングした。反応速度論的データを、室温での15分間の実行時間の間に収集した。
【0129】
(実施例8)
この実施例は、野生型ヒトPTP−1Bの短縮バージョンを作製するための1つの実施形態を記載する。ヒトPTP−1Bの最初の321アミノ酸をコードするcDNAを、ヒト胎児心臓総RNA(Clontech)から単離した。PTP−1B cDNA(Genbank M31724.1、Chernoff、1990)のヌクレオチド91〜114(For)に対応するオリゴヌクレオチドプライマーおよびヌクレオチド1030〜1053(Rev)に相補的なオリゴヌクレオチドプライマーを合成し、そしてポリメラーゼ連鎖反応を使用してDNAを生成するために使用した。
【0130】
プライマーForwardは、最初のATGコドンでNdeI制限部位を組み込み、そしてプライマーRevは、ヌクレオチド1053の後ろに、UAAコドンとその後ろのEcoRI制限部位とを挿入する。cDNAを、標準的な分子生物学的技術を用いて、制限ヌクレアーゼNdeIおよびEcoRIで消化し、そしてpRSETc(Invitrogen)にクローニングした。単離されたcDNAの正体を、DNA配列決定分析によって確認した。
【0131】
アミノ酸残基1〜298をコードする、より短いcDNAであるPTP−1B 298を、ポリメラーゼ連鎖反応において、オリゴヌクレオチドプライマーForwardおよびRev2、ならびにテンプレートとして上記のクローンを使用して生成した。
【0132】
【化13】
ヒトPTP−lBの321アミノ酸形態は、以下の配列番号17である:
【0133】
【化14】
変異体を、以下のように作製した。pRSETc(Invitrogen)中のPTP−1B 321を、テンプレートとして使用し、そしてT7プライマーおよびRSETrevプライマーを、「外側」プライマーとして使用した。変異誘発プライマーは、以下であった:
【0134】
【化15】
PTP−1B[321;N193A;F196R;F280C]を、残基1〜215に対応する、PTP−1B[321;N193A;F196R]由来のNdeI−PstIフラグメントを、残基216〜321に対応する、PTP−1B[321;F280C]由来のPstI−EcoRIフラグメントと連結することによって、生成した。
【0135】
PTP−1B[298;N193A;F196R;F280C]を、テンプレートとしてPTP−1B[321;N193A;F196R;F280C]を使用するPCRによって生成した。T7ベクタープライマーを順方向プライマーとして使用して、残基298での短縮を、プライマー
【0136】
【化16】
を使用して生成した。
【0137】
PTP−lB[298;C215S]を、Kunkel変異誘発およびテンプレートとしてのPTP−1B[298]を使用して生成した。変異誘発プライマーは、以下であった:
【0138】
【化17】
(実施例9)
この実施例は、PTP−1Bに対する本発明の化合物のIC50を決定するための1つの例示的な方法を記載する。基質であるpNPP(Sigma)を、1×HN緩衝液(50mM HEPES(pH7.0);100mM NaCl;1mM DTT)中に4mMで溶解させ、そして83μlを2μlのDMSOまたは2μlのDMSO中化合物と混合した。この反応を、15μlの1×HN緩衝液中のPTP−1B(標準的なアッセイ条件で750ηg)の添加によって開始した。産物形成の速度(OD405nm−(マイナス)OD655nm、BioRad BenchmarkまたはMolecular Devices Spectramax 190)を、25℃で15分間にわたって30秒毎に測定し、データを線形回帰によって分析した。終点アッセイのために、この反応を、50μl 3M NaOHを用いて15分後に停止させ、そしてOD405nm−OD655nmを測定した。IC50決定のために、非阻害コントロールに対して正規化した速度を、化合物濃度に対してプロットし、そして4パラメータの非線形回帰曲線当てはめ(
【0139】
【数1】
、Spectramax Software package)を使用して当てはめた。
【0140】
本明細書中で引用される全ての参考文献は、本明細書中で参考として明示的に援用される。本発明は、その特定の実施形態を参照して記載されているが、本発明の真の精神および範囲から逸脱することなしに、種々の変化がなされ得、そして等価物が置換され得ることが、当業者に理解されるべきである。さらに、多くの改変が、特定の状況、材料、物の組成、プロセスなどを適合させるためになされ得る。全てのこのような改変は、添付の特許請求の範囲の範囲内である。
【図面の簡単な説明】
【0141】
【図1】図1は、係留方法の1つの実施形態の模式図である。チオール含有タンパク質は、複数のリガンド候補と反応される。標的に対して固有の結合親和性を保有するリガンド候補が同定され、ジスルフィド部分を含まない同定された結合決定基(円によって表される)を含むリガンドが、作製される。
【図2】図2は、2つの係留実験の代表的な例である。図2Aは、チミジル酸シンターゼ(「TS」)に対する結合親和性をほとんど有さないか、または全く有さない10個の異なるリガンド候補のプールとTSとの反応の逆重畳質量分析スペクトルである。図2Bは、リガンド候補のうちの1つがTSに対して固有の結合親和性を保有する10個の異なるリガンド候補のプールとこの酵素との反応の逆重畳質量分析スペクトルである。
【図3】図3は、係留実験の模式図であり、ここで、チオールは、2つの異なる外部部位か、またはその付近に位置する。図3Aは、リガンドの外部部位への結合が、標的の機能に影響しない状況を示す。図3Bは、リガンドの外部部位への結合が、標的の機能に影響する状況を示す。この場合において、リガンドの外部部位への結合は、活性部位のコンフォメーションを変更し、その結果、これは、標的の機能を阻害する。図3Bは、外部部位がまた、アロステリック部位である例である。
【図4】図4は、選択されたカスパーゼの配列整列である。アロステリック部位を含む残基は、四角で囲んである。数字は、カスパーゼ−3における番号付けに対応する。
【図5】図5は、係留実験の結果であり、化合物1が、カスパーゼ−3の小さなサブユニットとジスルフィド結合を形成するが、大きなサブユニットとはジスルフィド結合を形成しないことを示す。
【図6】図6は、カスパーゼ−3活性の阻害と、カスパーゼ−3と化合物1または2との間のジスルフィド形成の度合いとを相関させた実験の結果である。
【図7】図7は、カスパーゼ−7の未結合形態(A)、基質結合形態(B)、およびアロステリック的に阻害された形態(C)を示す。
【配列表】
【技術分野】
【0001】
(背景)
(発明の分野)
薬物発見標的は、しばしば、タンパク質、特に、酵素である。ほとんどの薬物発明の努力は、リード化合物を同定するための大量の機能的スクリーニングに依存するので、強力な活性部位インヒビターがしばしば同定されるが、これらが薬物候補となるのはまれである。実に低い成功率についての理由は、変動するが、特異性の欠如および毒性は、それらがない場合より高い頻度で役割を果たす。
【0002】
酵素標的は、通常、関連する酵素のファミリーの1つのメンバーである。驚くべきことではないが、関連する酵素はしばしば、互いに類似する三次元構造を共有し、そして、その活性部位領域は、最も保存されている。標的化される部位は、ファミリーメンバーの中でも最も類似する酵素の領域であるので、1つのメンバーに対する選択性を達成することが、非常に困難なことであることは驚くべきことではない。特異性の欠如は、しばしば、意図しない標的の阻害に由来する毒性を生じる。固有の問題にもかかわらず、多くの最も有望な薬物標的は、大きな酵素ファミリー(例えば、プロテアーゼ(例えば、アスパルチルプロテアーゼ、システインプロテアーゼ、およびセリンプロテアーゼ)、キナーゼならびにホスファターゼ)のメンバーである。結果として、薬物発見のための新規な方法が、特異性を増強するこれらの型の標的に対して必要とされる。本発明は、このような方法を提供する。
【発明の開示】
【課題を解決するための手段】
【0003】
(好ましい実施形態の説明)
本発明は、本明細書で「外部部位(exosite)」として言及される、タンパク質上の新規な結合部位を同定するための方法およびそこに結合するリガンドを同定するための方法を提供する。本発明の方法によって同定されたリガンド自体は、例えば、新規な治療薬物、酵素インヒビター、標識化合物、診断試薬、タンパク質精製のための親和性試薬などの開発のためのリード化合物としての用途を見出す。
【0004】
他に規定されない限り、本明細書で使用される技術用語および科学用語は、本発明が属する分野の当業者に一般的に理解される意味と同じ意味を有する。参考文献(例えば、Singletonら、Dicitionary of Microbiology and Molecular Biology 第2版、J.Wiley&Sons(New York、NY 1994)および3月、Advanced Organic Reactions、Mechanisms and Structure 第4版、John Wiley&Sons(New York 1992))は、本発明の適用において使用される多くの用語に対する一般的指針を当業者に提供する。
【0005】
(定義)
本明細書で使用される用語の定義としては、以下が挙げられる:
用語「脂肪族」または「非置換脂肪族」とは、直鎖、分枝鎖、環状、または多環式の炭化水素をいい、アルキル部分、アルケニル部分、アルキニル部分、シクロアルキル部分、シクロアルケニル部分、およびシクロアルキニル部分が挙げられる。
【0006】
用語「アルキル」または「非置換アルキル」とは、飽和炭化水素をいう。
【0007】
用語「アルケニル」または「非置換アルケニル」とは、少なくとも1つの炭素−炭素二重結合を有する炭化水素をいう。
【0008】
用語「アルキニル」または「非置換アルキニル」とは、少なくとも1つの炭素−炭素三重結合を有する炭化水素をいう。
【0009】
用語「芳香族」または「非置換芳香族」とは、少なくとも1つのアリール基を有する部分をいう。この用語はまた、脂肪族修飾アリール(例えば、アルキルアリールなど)を含む。
【0010】
用語「アリール」または「非置換アリール」とは、少なくとも1つの芳香環を有する単環または多環の不飽和部分をいう。この用語は、少なくとも1つの芳香環内に1つ以上のヘテロ原子を含むヘテロアリールを含む。アリールの例示的な例としては、以下が挙げられる:フェニル、ナフチル、テトラヒドロナフチル、インダニル、インデニル、ピリジル、ピラジニル、ピリミジニル、ピロリル、ピラゾリル、イミダゾリル、チアゾリル、オキサゾリル、イソオキサゾリル、チアジアゾリル、オキサジアゾリル、チオフェニル、フラニル、キノリニル、イソキノリニルなど。
【0011】
部分を改変するために使用される場合、用語「置換(された)」は、その部分の置換されたバージョンをいい、ここで、少なくとも1つの水素原子は、以下が挙げられるが、これらに限定されない別の基により置換される:脂肪族、アリール、アルキルアリール、F、Cl、I、Br、−OH;−NO2;−CN;−CF3;−CH2CF3;−CH2Cl;−CH2OH;−CH2CH2OH;−CH2NH2;−CH2SO2CH3;−ORX;−C(O)RX;−COORX;−C(O)N(RX)2;−OC(O)RX;−OCOORX;−OC(O)N(Rx)2;−N(RX)2;−S(O)2RX;および−NRXC(O)RX。ここで、各RXの出現は、独立して、水素、置換脂肪族、非置換脂肪族、置換アリール、または非置換アリールである。さらに、部分上の隣接基の置換基は、一緒になって環状基を形成し得る。
【0012】
用語「アロステリック部位」とは、外部部位をいい、ここで、リガンドのこの部位への結合が、タンパク質の活性または機能を調節する。
【0013】
用語「アンタゴニスト」は、最も広い意味で使用され、標的(例えば、TBM)によって示される生物学的活性を部分的または完全にブロック、阻害または中和する任意のリガンドを含む。類似の様式において、用語「アゴニスト」は、最も広い意味で使用され、標的(例えば、TBM)によって示される生物学的活性を、例えば、このようなTBMの機能もしくは発現、またはこのようなTBMを介するシグナル伝達の効率を特異的に変化させ、それにより、既に存在する生物学的活性を変更(増大もしくは阻害)するか、または新たな生物学的活性を誘発することによって模倣する、任意のリガンドを含む。
【0014】
用語「外部部位」は、その主な結合部位でない、タンパク質上の結合部位である。例えば、酵素上の主な結合部位は、活性部位である。レセプター上の主な結合部位は、リガンド結合部位である。
【0015】
用語「リガンド候補」または「候補リガンド」とは、標的上の相補的または適合性の反応基と共有結合を形成し得る反応基を保有するか、または、保有するように改変された化合物をいう。リガンド候補上または標的上のいずれかの反応基は、例えば、保護基でマスクされ得る。
【0016】
用語「ポリヌクレオチド」は、単数または複数で使用される場合、任意のポリリボヌクレオチドまたはポリデオキシリボヌクレオチドを一般にいい、これらは、非改変RNAもしくは非改変DNA、または改変RNAもしくは改変DNAであり得る。従って、例えば、本明細書で定義されるポリヌクレオチドとしては、限定されないが、一本鎖DNAおよび二本鎖DNA、一本鎖領域および二本鎖領域を含むDNA、一本鎖RNAおよび二本鎖RNA、一本鎖領域および二本鎖領域を含むRNA、一本鎖、もしくはより代表的には二本鎖であり得るか、または一本鎖領域および二本鎖領域を含むDNAおよびRNAを含むハイブリッド分子が挙げられる。さらに、用語「ポリヌクレオチド」は、本明細書で使用される場合、RNAあるいはDNAまたはRNAおよびDNAの両方を含む三本鎖領域をいう。このような領域における鎖は、同じ分子由来であっても、異なる分子由来であってもよい。これらの領域は、1つ以上の分子の全てを含み得るが、より代表的には、幾つかの分子の1つの領域のみを包含する。三重らせん領域の分子の1つは、しばしば、オリゴヌクレオチドである。用語「ポリヌクレオチド」は、特に、1つ以上の改変された塩基を含むDNAおよびRNAを含む。従って、安定性または他の理由のために改変された骨格を有するDNAまたはRNAは、その用語が本明細書で意図されるような「ポリヌクレオチド」である。さらに、通常でない塩基(例えば、イノシン)または改変された塩基(例えば、トリチル化塩基)を含むDNAまたはRNAは、本明細書で定義されるような「ポリヌクレオチド」との用語に含まれる。一般的に、用語「ポリヌクレオチド」は、非改変のポリヌクレオチドの全ての化学的、酵素的および/または代謝的に改変された形態、ならびにウイルスおよび細胞(単純細胞および複雑細胞を含む)の特徴であるDNAおよびRNAの化学的形態を含む。
【0017】
語句「保護されたチオール」または「マスク(マスキング)されたチオール」とは、本明細書で使用される場合、基または分子と反応して、より反応性が低くされた共有結合を形成しているチオールをいい、これは、脱保護されて、遊離のチオールを再生し得る。
【0018】
語句「可逆的な共有結合」とは、本明細書で使用される場合、好ましくは、標的を変性させない条件下で分解され得る共有結合をいう。例としては、ジスルフィド、シッフ塩基、チオエステル、配位錯体、ボロン酸エステルなどが挙げられるが、これらに限定されない。
【0019】
語句「反応基」は、存在する場合、適合性または相補的な反応基との共有結合が形成され得る部位を提供する化学的な基または部分である。例証的な例は、以下である:−SHであり、これは、別の−SHまたは−SS−と反応して、それぞれジスルフィドまたはジスルフィド交換を形成し得る;−NH2であり、これは、活性化された−COOHと反応して、アミドを形成し得る;−NH2であり、これは、アルデヒドまたはケトンと反応して、シッフ塩基などを形成し得る。
【0020】
語句「反応性求核試薬」は、本明細書で使用される場合、標的を変性も損傷も条件下で、別の分子上の適合性の官能基と共有結合を形成し得る求核試薬をいう。最も適切な求核試薬は、チオール、アルコール、およびアミンである。同様に、語句「反応性求電子試薬」とは、本明細書で使用される場合、好ましくは、標的を変性も損傷もしない条件下で、別の分子上の適合性の官能基と共有結合を形成し得る求電子試薬をいう。最も適切な求電子試薬は、ハロゲン化アルキル、イミン、カルボニル、エポキシド、アジリジン(aziridie)、スルホネート、ジスルフィド、活性化エステル、活性化カルボニル、およびヘミアセタールである。
【0021】
語句「目的の部位」とは、リガンドが結合し得る、標的上の任意の部位をいう。本明細書で使用される場合、目的の部位は、タンパク質の主な結合部位の外側にある任意の部位である。例えば、標的が酵素である場合、目的の部位は、活性部位でない部位である。標的がレセプターである場合、目的の部位は、レセプターのリガンドの結合部位でない部位である。
【0022】
用語「標的」、「標的分子」および「TM」は、交換可能に、最も広い意味で使用され、リガンドの結合が標的の機能に対して影響を有する化学的または生物学的な実体をいう。標的は、分子、分子の一部、または分子の凝集体であり得る。リガンドの結合は、可逆的でも不可逆的でもよい。標的分子の特定の例としては、以下が挙げられる:ポリペプチドまたはタンパク質(例えば、酵素およびレセプター)、転写因子、増殖因子およびサイトカインのような、レセプターに対するリガンド、免疫グロブリン、核タンパク質、シグナル伝達成分(例えば、キナーゼ、ホスファターゼ)、ポリヌクレオチド、糖質、糖タンパク質、糖脂質、および他の高分子(例えば、核酸−タンパク質複合体、クロマチンまたはリボソーム)、資質二重層含有構造(例えば、膜)、または膜から誘導される構造(例えば、ベシクル)。この定義は、特に、以下に定義されるような標的生物学的分子(Target Biological Molecule)(「TBM」)を含む。
【0023】
「標的生物学的分子」または「TBM」とは、本明細書で使用される場合、低分子アゴニストまたは低分子アンタゴニストがTBMの機能に対する効果を有する、互いに生物学的に関連する複合体を形成し得る、単数または複数の生物学的分子をいう。好ましい実施形態において、TBMは、2つ以上のアミノ酸を含むタンパク質またはその一部であり、これらは、相補的な反応基を有する化合物と共有結合を形成し得る反応基を保有するか、または保有するように改変され得る。好ましいTBMとしては、以下が挙げられる:細胞表面レセプターおよび可溶性レセプターならびにそれらのリガンド;ステロイドレセプター;ホルモン;免疫グロブリン;凝血因子;核タンパク質;転写因子;シグナル伝達分子;細胞接着分子、同時刺激性分子、ケモカイン、アポトーシスの媒介に関与する分子、酵素、ならびにDNAおよび/またはRNA合成または分解に関連するタンパク質。
【0024】
多くのTBMは、レセプター−リガンド結合相互作用に関与し、レセプター−リガンド対のいずれかのメンバーであり得る分子である。増殖因子およびそれらのそれぞれのレセプターの例証的な例としては、以下に対するものが挙げられる:エリスロポエチン(EPO)、トロンボポエチン(TPO)、アンジオポエチン(ANG)、顆粒球コロニー刺激因子(G−CSF)、顆粒球マクロファージコロニー刺激因子(GM−CSF)、上皮増殖因子(EGF)、ヘレグリン−a(heregulin−a)およびヘレグリン−b、脈管内皮細胞増殖因子(VEGF)、胎盤増殖因子(PLGF)、トランスフォーミング増殖因子(TGF−aおよびTGF−b)、神経成長因子(NGF)、ニューロトロフィン、線維芽細胞増殖因子(FGF)、血小板由来増殖因子(PDGF)、骨形態形成タンパク質(BMP)、結合組織増殖因子(CTGF)、肝細胞増殖因子(HGF)、ならびにインスリン様増殖因子1(IGF−1)。ホルモンおよびそれらのそれぞれのレセプターの例証的な例としては、以下に対するもの挙げられる:成長ホルモン、プロラクチン、胎盤ラクトゲン(LPL)、インスリン、卵胞刺激ホルモン(FSH)、黄体形成ホルモン(LH)、ならびにニューロキニン−1。サイトカインおよびそれらのそれぞれのレセプターの例証的な例としては、以下に対するものが挙げられる:毛様体神経栄養因子(CNTF)、オンコスタチンM(OSM)、TNF−a;CD40L、幹細胞因子(SCF);インターロイキン−1、インターロイキン−2、インターロイキン−4、インターロイキン−5、インターロイキン−6、インターロイキン−8、インターロイキン−9、インターロイキン−13、ならびにインターロイキン−18。
【0025】
他のTBMとしては以下が挙げられる:細胞接着分子(例えば、CD2、CD11a、LFA−1、LFA−3、ICAM−5、VCAM−1、VCAM−5およびVLA−4);同時刺激分子(例えば、CD28、CTLA−4、B7−1;B7−2、ICOSおよびB7RP−1);ケモカイン(例えば、RANTESおよびMIP1b);アポトーシス因子(例えば、APAF−1、p53、bax、bak、bad、bidおよびc−abl);抗アポトーシス因子(例えば、bcl2、bcl−x(L)およびmdm2);転写調節因子(例えば、AP−1およびAP−2);シグナル伝達タンパク質(例えば、TRAF−1、TRAF−2、TRAF−3、TRAF−4、TRAF−5およびTRAF−6);およびアダプタータンパク質(例えば、grb2、cbl、shc、nckおよびcrk)。
【0026】
酵素は、別の分類の好ましいTBMであり、以下を含む多くの方法で分類され得る:アロステリック酵素;細菌性酵素(イソロイシルtRNAシンターゼ、ペプチドデホルミラーゼ、DNAジャイレースなど);真菌性酵素(チミジル酸合成酵素など);ウイルス酵素(HIVインテグラーゼ、HSVプロテアーゼ、C型肝炎Cヘリカーゼ、C型肝炎プロテアーゼ、ライノウイルスプロテアーゼなど);キナーゼ(セリン/スレオニン、チロシンおよび二重特異性(dual specificity));ホスファターゼ(セリン/スレオニン、チロシンおよび二重特異性);およびプロテアーゼ(アスパルチルプロテアーゼ、システインプロテアーゼ、メタロプロテアーゼおよびセリンプロテアーゼ)。酵素の注目すべき下位分類としては、以下が挙げられる:キナーゼ(例えば、Lck、Syk、Zap−70、JAK、FAK、ITK、BTK、MEK、MEKK、GSK−3、Raf、tgf−b−活性化キナーゼ−1(TAK−1)、PAK−1、cdk4、Akt、PKCq、IKKb、IKK−2、PDK、ask、nik、MAPKAPK、p90rsk、p70s6kおよびPI3−K(p85およびp110サブユニット));ホスファターゼ(例えば、CD45、LAR、RPTP−a、RPTP−m、Cdc25A、キナーゼ関連ホスファターゼ、mapキナーゼホスファターゼ−1、PTP−1B、TC−PTP、PTP−PEST、SHP−1およびSHP−2;カスパーゼ(例えば、カスパーゼ−1、カスパーゼ−3、カスパーゼ−7、カスパーゼ−8、カスパーゼ−9およびカスパーゼ−11);およびカテプシン(例えば、カテプシンB、カテプシンF、カテプシンK、カテプシンL、カテプシンSおよびカテプシンV)。他の酵素標的としては、以下が挙げられる:BACE、TACE、細胞質ホスホリパーゼA2(cPLA2)、PARP、PDE I〜VII、Rac−2、CD26、イノシン1リン酸デヒドロゲナーゼ、15−リポキシゲナーゼ、アセチルCoAカルボキシラーゼ、アデノシルメチオニンデカルボキシラーゼ、ジヒドロオロテートデヒドロゲナーゼ、ロイコトリエンA4ヒドロラーゼ、および一酸化窒素シンターゼ。
【0027】
(外部部位)
本発明は、本明細書中で「外部部位(exosite)」と呼ばれるタンパク質上の新規の結合部位を同定するための方法を提供する。これらの外部部位は、特定の標的タンパク質の一次結合領域とは異なる標的タンパク質上の結合部位である。例えば、酵素上の外部部位は、活性部位でない任意の結合部位である。同様に、レセプター上の外部部位は、レセプターのリガンドの結合部位ではない任意の結合部位である。
【0028】
1つの実施形態において、目的の外部部位は、タンパク質の適応性の結合部位である。用語「適応性」は、活性部位のようなよく定義されたポケットとは異なり、適応性の結合部位は、リガンドの不在下では明らかでないため、これらの部位をさすために使用される。リガンドの存在は、タンパク質の1つ以上の鎖の立体配座の変化を誘導し、リガンドが最終的に結合し得る結合部位を生じる。
【0029】
別の実施形態において、目的の外部部位はアロステリック部位である。言い換えると、標的タンパク質のこのような部位へのリガンドの結合は、その標的タンパク質の機能を調節する。この調節は、負ならびに正の両方であり得る。例えば、調節が負である場合、外部部位へのリガンドの結合は、標的の機能を阻害する。調節が正である場合、外部部位のリガンドへの結合は、標的の機能を増強(または増幅)する。アロステリック部位は、しばしば、アクセサリータンパク質および/または調節タンパク質に対する認識部位である。
【0030】
なお別の実施形態において、目的の外部部位は適応性の結合部位とアロステリック部位との両方である。
【0031】
(係留方法)
係留は、標的上の反応基と潜在的なリガンド上の相補的な反応基との間の共有結合の形成に依存するリガンドの同定方法である。係留方法は、発明者ら(Daniel Erlanson、Andrew BraistedおよびJames Wells)による米国特許第6,335,155号;PCT公報番号WO00/00823およびWO02/42773;METHODS FOR LIGAND DISCOVERYとの表題の米国番号10/121,216(対応するPCT出願番号US02/13061);およびErlansonら、Proc.Nat.Acad.Sci USA 97:9367−9372(2000)(これらは、全てが参考として援用される)に記載される。得られた共有複合体は、標的−リガンド結合体と称される。共有結合が標的(例えば、ネイティブまたは非ネイティブのシステイン)の所定の部位で形成されるので、化学量論および結合位置は、この方法によって同定されるリガンドについて公知である。
【0032】
一旦形成されると、標的−リガンド結合体のリガンド部分が多数の方法を使用して同定され得る。好ましい実施形態において、質量分析法が使用される。質量分析法は、質量/電荷比(m/z)に基づいて分子を検出し、そして、それらのサイズに基づいて分子を分離し得る(Yates,Trends Genet.16:5−8[2000]において総説される)。標的−リガンド結合体は、質量分析計において直接検出され得るか、または検出の前にフラグメント化され得る。あるいは、化合物は、質量分光光度計内で遊離され、引き続いて同定され得る。さらに、質量分析法を単独でかまたは標的に共有結合する化合物を検出もしくは同定するための他の手段と組み合せて使用し得る。さらに、質量分析法技術の説明としては、FitzgeraldおよびSiuzdak,Chemstry & Biology 3:707−715[1996];Chuら,J.Am.Chem.Soc.118:7827−7835[1996];Siudzak,Proc.Natl.Acad.Sci.USA 91:11290−11297[1994];Burlingameら,Anal.Chem.68:599R−651R[1996];Wuら,Chemistry & Biology 4:653−657[1997];ならびにLooら,Am.Reports Med.Chem.31:319−325[1996])が挙げられる。
【0033】
あるいは、標的−リガンド結合体は、他の手段を用いて同定され得る。例えば、反応混合物の成分の分離のために種々のクロマトグラフィー技術(例えば、液体クロマトグラフィー、薄層クロマトグラフィーなど)を使用し得、その結果、共有結合分子を同定する能力を高める。このようなクロマトグラフィー技術は、質量分析法と組み合せてかまたは質量分析法とは別に使用され得る。また、標識化プローブ(蛍光、放射線または他の手段で)を遊離した化合物に結合させ、それによって上記の技術のいずれかを用いる同定を容易にし得る。なお別の実施形態において、新しい結合の形成は、標識化プローブを遊離させ、次いで、モニタリングされ得る。単純な機能的アッセイ(例えば、ELISAまたは酵素アッセイ)がまた、アッセイが測定するのに必須の領域において結合が生じるときに結合を検出するのに使用され得る。標的分子に結合する有機化合物を同定するための用途が見出され得る他の技術としては、例えば、核磁気共鳴分析法(NMR)、表面プラスモン共鳴(例えば、BIACORE)、キャピラリー電気泳動、X線結晶解析、などが挙げられ、これらは全て、当業者に周知である。
【0034】
標的がタンパク質であり、かつ共有結合がジスルフィドである係留方法の1つの実施形態の略図を図1に示す。チオール含有タンパク質を、複数のリガンド候補と反応させる。この実施形態において、リガンド候補は、式−SSR1のジスルフィドの形態でマスクされたチオールを保有し、ここでR1は非置換のC1〜C10アルキル、置換C1〜C10アルキル、非置換のアリールまたは置換アリールである。特定の実施形態において、R1は潜在的なリガンド候補の溶解性を高めるように選択される。示すように、標的に対して固有の結合親和性を保有するリガンド候補が同定され、ジスルフィド部分を含まない対応するリガンドが、同定された結合決定基(円で表される)から作製される。
図2は、2つの代表的な係留実験を示す。ここで、標的酵素、E.coliチミジル酸シンターゼは以下の式
【0035】
【化3】
のリガンド候補と接触し、ここで、Rcは、ライブラリメンバーのこのプールの中で可変の部分であり、そして、非置換の脂肪族、置換脂肪族、非置換のアリール、または置換アリールである。全てのTS酵素のと同様に、E.Coli TSは係留に使用され得る活性な部位システイン(Cys146)を有する。E.Coli TSはまた4つの他のシステインを含むが、これらのシステインは変化し、係留実験において反応性でないことが見出された。例えば、最初の実験において、野生型のE.coli TSおよびC146S変異体(146位のシステインがセリンに変異している)をシスタミン(H2NCH2CH2SSCH2CH2NH2)と接触させた。野生型のTS酵素は、シスタミンの1つの等価物ときちんと反応したが、その一方で、変異体TSは反応せず、これは、シスタミンがCys146と反応し、Cys146に選択的であったことを示す。
【0036】
図2Aは、TSとTSに対してほとんど結合親和性を有さないかまたは全く結合親和性を有さない10の異なるリガンド候補のプールとの反応の逆重畳質量スペクトルである。任意の結合相互作用の不在下で、TSと個々のリガンド候補との間のジスルフィド交換反応の平衡は改変されていない酵素に向う。このことを以下の式によって模式的に示す。
【0037】
【化4】
予想した通り、改変されていない酵素に対応するピークは、スペクトルのうち2つの最も突出したピークのうちの1つである。もう一方の突出したピークは、TSであり、ここでは、Cys146のチオールがシステアミンで改変されている。この種は任意の個々のライブラリメンバーについて有意な程度まで形成されないが、このピークはライブラリプールの各メンバーについての平衡反応の蓄積効果に起因する。反応がチオール含有還元剤(例えば、2−メルカプトエタノール)の存在下で実行される場合、活性部位のシステインはまた、還元剤で改変され得る。システアミンと2−メルカプトエタノールは類似した分子量を有するので、これらのそれぞれのジスルフィド結合化TS酵素は、この実験で用いた条件下においては区別できない。右側の小さなピークは、目立たないライブラリメンバーに対応する。特に、これらのピークはいずれもが非常に顕著でない。図2Aは、リガンド候補のいずれもが標的に対して固有の結合親和性を保有しないスペクトルの特徴である。
【0038】
図2Bは、リガンド候補の1つが酵素に対して固有の結合親和性を保有する、10の異なるリガンド候補のプールとのTSの反応の逆重畳質量スペクトルである。見られ得るように、最も突出したピークはTSに対応するものであり、ここでは、Cys146のチオールがN−トシル−D−プロリン化合物で改変されている。このピークは、改変されていない酵素およびTSに対応するピークを含む全ての他のピークを矮化し、ここではCys146のチオールがシステアミンで改変されている。図2Bは、係留が所望の部位に対して強い固有の結合親和性を保有する部分を有する、質量スペクトルの例である。
【0039】
(係留を用いた外部部位同定)
本発明の1つの局面において、タンパク質標的表面上の外部部位を同定するための方法が提供される。一般に、この方法は以下の工程を包含する:
a)主な(primary)結合部位、および主な結合部位以外の部位またはその付近の部位に化学反応基を有する標的を提供する工程;
b)上記の化学反応基と共有結合を形成し得る化合物に標的を接触させる工程;
c)標的と化合物との間に共有結合を形成し、それによって標的−化合物結合体を形成する工程;ならびに
d)標的との共有結合の不在下で、上記部位における標的に化合物が結合するか否かを決定する工程。
【0040】
多くの場合、潜在的な外部部位が標的の凹部領域に位置される。他の場合において、潜在的な外部部位は、明らかでない。なぜならば、この部位はリガンドの存在がタンパク質の1つ以上の側鎖の立体配置の変化を誘導し、リガンドが最終的には結合し得る結合部位を作成する、適応性の結合部位であるからである。
【0041】
標的が酵素である場合、主な結合部位は活性部位である。標的がレセプターである場合、主な結合部位はレセプターのリガンドが結合する部位である。
【0042】
化学反応基が、この結合部位を含む残基の任意の原子から10Å以下である場合、この基は、結合部位の近くであると考えられる。別の実施形態において、化学反応基が、この結合部位を含む残基の任意の原子から5Å以下である場合、この基は結合部位の近くであると考えられる。
【0043】
別の実施形態において、この方法は以下の工程を包含する:
a)第1の結合部位、第2の結合部位および第2の結合部位または第2の結合部位の近くの化学反応基を有する標的を提供する工程;
b)化学反応基と共有結合を形成し得る第1の化合物に標的を接触させる工程;
c)標的と第1の化合物との間に共有結合を形成させ、それによって標的−化合物結合体を形成する工程;
d)標的を第2の化合物に接触させる工程であって、第2の化合物が化学反応基を欠失した第1の化合物の変形である、工程;ならびに
e)第2の結合部位に対して非共有結合するための第2の化合物の親和性を決定する工程。
【0044】
本発明の別の局面において、アロステリック外側部位を同定するための方法が提供される。概して、この方法は、以下の工程を包含する:
a)主な結合部位および該主な結合部位以外の部位またはその近傍の化学反応基を備える標的を提供する工程;
b)該標的を、該化学反応基と共有結合を形成し得る化合物と接触させる工程;
c)該標的と該化合物との間の共有結合を形成し、それによって、標的−化合物結合体を形成する工程;
d)該標的−化合物結合体が、該標的と比較して、主な結合部位中に変化を有するか否かを決定する工程。
【0045】
この方法によって同定されるアロステリック部位は、ネガティブおよびポジティブの両方で標的タンパク質の機能を調節し得る。例えば、この調節はネガティブである場合、外側部位へのリガンドの結合は、この標的の機能を阻害する。この調節がポジティブである場合、外側部位へのリガンドの結合は、この標的の機能を促進(または増幅)させる。
【0046】
1実施形態において、主な結合部位における変化は、構造的変化であり、そして主な結合部位の三次元構造における変更である。三次元構造における変更は、少なくとも1Åまでの、活性部位残基の少なくとも1つのヘテロ原子の動きとして規定される。構造における変化は、x線結晶学、NMR、円偏光二色性などを含む多くの任意の方法によって検出される。別の実施形態において、この主な結合部位における変化は、機能的変化である。標的が酵素である場合、その機能は、阻害または促進され得る。この標的がレセプターである場合、レセプターリガンド結合部位への、レセプターリガンドの結合は、阻害または促進され得る。
【0047】
別の実施形態において、この方法は、以下の工程を包含する:
a)第一の結合部位、第二の結合部位およびこの第二の結合部位またはその近傍の化学反応基を備える標的を提供する工程;
b)この標的を、この化学反応基と共有結合を形成し得る化合物と接触させる工程;
c)この標的と化合物との間の共有結合を形成し、それによって、標的−化合物結合体を形成する工程;
d)この標的−化合物結合体が、標的の第一の結合部位と比較して、第一結合部位中に変化を有するか否かを決定する工程。
【0048】
本方法の特定の実施形態において、この化学反応基は、システイン残基のチオールであり、そしてこの化合物は、−SH基を有する。他の実施形態において、この化合物は、マスキングされたチオールを有する。他の実施形態において、この化合物は、式−SSR1のジスルフィドの形態でマスキングされたチオールを有するリガンド候補であり、ここで、R1は、非置換C1〜C10脂肪族、置換C1〜C10脂肪族、非置換アリールまたは置換アリールである。他の実施形態において、このリガンド候補は、式−SSR2R3のジスルフィドのようにマスキングされたチオールを有し、ここで、R2は、C1〜C5アルキル(好ましくは、−CH2−、−CH2CH2−、または−CH2CH2CH2−)であり、そしてR3は、NH2、OH、またはCOOHである。リガンド候補の例示的な例としては、以下が挙げられる:
【0049】
【化5】
ここで、RおよびR’は、各々独立して非置換C1〜C20脂肪族、置換C1〜C20脂肪族、非置換アリールまたは置換アリールであり;
mは0、1または2であり;そして
nは1または2である。
【0050】
他の実施形態において、この標的は、還元剤の存在下においてジスルフィド結合を形成し得る化合物と接触される。適切な還元剤の例示的な例としては、以下が挙げられるが、これらに限定されない:システイン、システアミン、ジチオスレイトール、ジチオエリスリトール、グルタチオン、2−メルカプトエタノール、3−メルカプトプロピオン酸、ホスフィン(例えば、トリス−(2−カルボキシ−ホスフィン)(「TCEP」)、または水素化ホウ素ナトリウム。1実施形態において、この還元剤は、2−メルカプトエタノールである。別の実施形態において、この還元剤は、システアミンである。別の実施形態において、この還元剤は、グルタチオンである。別の実施形態において、この還元剤はシステインである。
【0051】
外側部位を同定する係留方法の概略図を、図3に示す。この実施形態において、主な結合部位は、活性部位として示される。図3Aは、外側部位が適応性結合部位である状況を示す。理解され得るように、この外側部位は、リガンドの存在によって誘導される。しかし、この外側部位へのリガンドの結合は、活性部位のコンフォメーションも、標的の機能も変更しない。対照的に、図3Bは、アロステリック部位でもある外側部位が同定される状況を示す。示されるように、アロステリック外側部位へのリガンドの結合は、活性部位のコンフォメーションを変更し、その結果、標的の機能を阻害する。両方の場合において、この標的−化合物結合体は、必要に応じて、標的を再生するために還元剤と接触され得る。この外側部位がアロステリック外側部位である場合、リガンドの除去は、このアロステリック外側部位へのリガンドの結合から生じる変化を逆転する。
【0052】
図3に示される方法は、所望の標的のシステイン変異体を作製することにより適用される。システイン残基は、タンパク質標的上の、目的の部位またはその近傍に導入される。1実施形態において、目的の部位は、標的上の位置が系統的に調査されるように選択される。別の実施形態において、標的が複数のサブユニットから構成される場合、目的の部位は境界面領域の近傍にある。これらのサブユニットは、同じポリペプチド(例えば、ホモダイマー)または異なるポリペプチド(例えば、へテロダイマー)から構成され得る。システインは、目的の部位から10Å内、好ましくは、目的の部位から5Å内に位置される場合、目的の部位の近傍にあるとみなされる。標的が、目的の部位の外側の天然に存在するシステインを含む場合、それらは、必要に応じて二重標識の可能性を排除するために、アラニンのような別の残基に変異され得る。
【0053】
概して、システイン残基に変異される残基は、溶媒接近可能である。溶媒接近可能性は、標準的な数値(Lee,B.&Richards,F.M.J.Mol.Biol 55:379−400(1971);Shrake,A.&Rupley,J.A.J.Mol.Biol.79:351−371(1973))または分析(Connolly,M.L.Science 221:709−713(1983);Richmond,T.J.J.Mol.Biol.178:63−89(1984))法を使用する構造モデルから計算され得る。例えば、潜在的なシステイン改変体は、LeeおよびRichardsの方法(Lee,B.&Richards,F.M.J.MoL Biol 55:379−400(1971))によって計算されるとき、炭素−β(CB)、または硫黄−γ(SG)の合わせた表面領域が、20Å2より大きい場合に、溶媒接近可能であるとみなされる。この値は、Creamerら(Creamer,T.P.ら、Biochemistry 34:16245−16250(1995))によって記載されるような、システイン側鎖に接近可能な理論上の表面領域の約33%を示す。
【0054】
また、システイン、または別のチオール含有アミノ酸残基へ変異される残基が、骨格原子との水素結合に関与しないこと、あるいは、多くとも1つの水素結合のみによって骨格と相互作用することが好ましい。側鎖が他の側鎖との複数(>1)の水素結合に関与する野生型残基はまた、あまり好ましくない。全ての標準的な回転異性体(−60°、60°、または180°のchi1角度)が他の任意の残基のN原子、CA原子、C原子、O原子、またはCB原子との、都合の悪い立体的接触を誘導し得る改変体もまた、あまり好ましくない。都合の悪い接触は、関与している原子のファンデルワールス半径の合計の80%未満である原子間距離として規定される。目的の部位が凹部領域である特定の実施形態において、このような部位の端(例えば、隆線または近接した凹部領域)に見出される残基は、システイン残基に変異するのにより好ましい。凸性および凹性は、表面ベクトル(vector)(Duncan,B.S.&Olson,A.J.Biopolymers 33:219−229(1993))に基づいて計算され得るか、または分子表面に沿って配置される水プローブの接近可能性を決定すること(Nicholls,A.らProteins 11:281−296(1991);Brady,G.P.,Jr.& Stouten,P.F.J.Comput.Aided Mol.Des.14:383−401(2000))によって計算され得る。L−アミノ酸について通常禁じられた骨格コンフォメーションを有する残基(Ramachandran,G.N.ら、J.Mol.Biol.7:95−99(1963);Ramachandran,G.N.&Sasisekharahn,V.Adv.Prot.Chem.23:283−437(1968))は、システインの改変についてのあまり好ましい標的ではない。禁じられたコンフォメーションは、φ角度の正の値を一般的に特徴とする。
【0055】
他の好ましい改変体は、システインに変異され、かつ−Cys−SSR1を含むように係留される場合、R1の原子を目的の部位に向かって指向するコンホメーションを有するものである。2つの一般的手順が使用されて、これらの好ましい改変体が同定され得る。第1の手順において、独特の構造(Hobohm,U.ら.Protein Science 1:409−417(1992))に関してProtein Databank(Berman,H.M.ら Nucleic Acids Research 28:235−242(2000))において検索が行われて、j位においてジスルフィド結合されたシステインを含む構造フラグメントが同定される。この位置において、フラグメントの残基j−1、j、およびj+1のその骨格原子は、0.75平方Å未満のRMSDと、標的分子の残基i−1、i、およびi+1の骨格原子上で重ね合わせられ得る。フラグメントが、残基iのCβ原子(システインに変異された場合)よりも、目的の部位の何れの原子にもより近いj位においてシステインにジスルフィド結合される残基のCβ原子が位置すると同定される場合、i位が好ましいと考えられる。代替的手順において、i位の残基は、システインへと計算により「変異」され、ジスルフィド結合を介してS−メチル基でキャップされる。
【0056】
種々の組換え技術、化学的技術、合成技術および/または他の技術が、標的が係留に利用可能な所望の数の遊離チオール基を有するように標的を改変するために使用され得る。このような技術としては、例えば、標的ポリペプチドをコードする核酸配列の部位特異的変異誘発が挙げられ、その結果、この核酸配列は、異なる数のシステイン残基を有するポリペプチドをコードする。特に好ましいのは、ポリメラーゼ連鎖反応(PCR)増幅を使用する部位特異的変異誘発である(例えば、米国特許第4,683,195号(1987年7月28日発行);およびCurrent Protocols In Molecular Biology,第15章(Ausubelら編,1991)を参照のこと)。他の部位指向性変異誘発技術もまた、当該分野で周知であり、例えば、以下の刊行物において記載されている:Ausubelら,前出,第8章;Molecular Cloning:A Laboratory Manual.,第2版(Sambrookら,1989);Zollerら,Methods Enzymol.100:468−500(1983);Zoller&Smith,DNA 3:479−488(1984);Zollerら,Nucl.Acids Res.,10:6487(1987);Brakeら,Proc.Natl.Acad.Sci.USA 81:4642−4646(1984);Botsteinら,Science 229:1193(1985);Kunkelら,Methods Enzymol.154:367−82(1987)、Adelmanら,DNA 2:183(1983);ならびにCarterら,Nucl.Acids Res.,13:4331(1986)。カセット変異誘発(Wellsら,Gene,34:315[1985])、および制限選択変異誘発(Wellsら,Philos.Trans.R.Soc.London SerA,317:415[1986])もまた、使用され得る。
【0057】
1つを超えるアミノ酸置換を有するアミノ酸配列改変体は、いくつかの方法のうちの1つで生成され得る。そのアミノ酸がポリペプチド鎖中でともに近くに位置する場合、それらは、所望のアミノ酸置換の全てをコードする1つのオリゴヌクレオチドを用いて、同時に変異され得る。しかし、そのアミノ酸が互いにいくらか距離が離れて位置する(例えば、10アミノ酸を超えて分離されている)場合、所望の変化の全てをコードする単一のオリゴヌクレオチドを生成することはより困難である。代わりに、2つの代替的方法のうちの1つが、使用され得る。第1の方法において、別個のオリゴヌクレオチドが、置換されるべき各アミノ酸について生成される。次いで、これらのオリゴヌクレオチドは、一本鎖テンプレートDNAに同時にアニールされ、そのテンプレートから合成される第2鎖のDNAは、所望のアミノ酸置換の全てをコードする。代替的方法は、所望の変異を生成するために2回を超える変異誘発を含む。
【0058】
本発明の別の局面において、アロステリックインヒビターを同定するための方法が提供され、この方法は、以下:
a)第一の係留実験を実施する工程、および
b)第二の係留実験を実施する工程、
を包含し、ここで、両方の係留実験が、
i)第一の結合部位、第二の結合部位およびこの第二の結合部位またはその近傍の化学反応基を含む標的を提供する工程;
ii)この標的を、この化学反応基と共有結合を形成し得る化合物と、接触させる工程;
iii)この標的とこの化合物との間の共有結合を形成し、それによって、標的−化合物結合体を形成する工程;および
iv)この標的−化合物結合体を同定する工程、
を包含し、ここで、
この第一の係留実験が、この第一の結合部位に結合するリガンドの存在下で実施され、そしてこの第二の係留実験が、この第一の結合部位に結合するリガンドの非存在下で実施される。
【0059】
その基質またはリガンドの非存在下で標的−化合物結合体を形成するが、その基質またはリガンドの存在下では標的−化合物結合体を形成しない化合物は、アロステリックインヒビターの候補である。一実施形態において、その標的は酵素であり、その第1の結合部位に結合するリガンドは、既知の競合的インヒビターである。別の実施形態において、その共有結合は、ジスルフィドであり、その化合物は、マスキングされたジスルフィドを有するリガンド候補である。
【0060】
本発明の別の局面において、アロステリック調節をし得る標的においてアロステリックインヒビターを同定するための方法が提供される。この方法は、リガンドのアロステリック部位への結合が、もはや標的を阻害しないように、アロステリック部位を無能にすることに依存する。この方法は、以下:
a)アロステリック調節をし得る標的およびアロステリック調節をし得ないその変異体を提供する工程;
b)この標的を化合物と接触させる工程;
c)この変異体をこの化合物と接触させる工程;ならびに
d)この標的に対するこの化合物の活性を、変異体に対するこの化合物の活性と比較する工程、
を包含する。
【0061】
一実施形態において、このアロステリック無能化変異体(allosterically disabled mutant)は、アロステリック部位を含む、少なくとも1つの残基において変異を有する。別の実施形態において、このアロステリック無能化変異体は、アロステリック部位を含む、少なくとも2つの残基において変異を有する。なお別の実施形態において、このアロステリック無能化変異体は、アロステリック部位を含む、少なくとも3つの残基において変異を有する。
【0062】
(カスパーゼ)
カスパーゼ(システイニルアスパラギン酸特異的プロテアーゼ)は、サイトカイン成熟およびアポトーシスの両方において中心的(pivital)な役割を果たす細胞内システインプロテアーゼのファミリーである。多くの他のプロテアーゼと同様に、カスパーゼは、不活性チモーゲンとして合成される。これらのチモーゲンは、N末端プロドメイン、および切断されると、大きなサブユニットドメインおよび小さなサブユニットドメインを生じる切断部位を含む。一般に、Asp−X結合の内部切断は、活性なプロテアーゼのアセンブリおよびそれ自体のプロドメインの切断を可能にする、短いC末端の小さなサブユニットを分離する。これらの酵素の活性形態は、2つの大きなサブユニットおよび2つの小さなサブユニットから構成されるヘテロテトラマーである。しかし、この大きさサブユニットおよび小さなサブユニットが同じポリペプチドに由来するので、この活性形態は、しばしば、(本明細書中でいわれるように)ホモダイマーといわれる。
【0063】
本明細書中に記載される方法を使用して、新規なアロステリック部位が、カスパーゼのダイマー境界面において同定されている。この部位は、カスパーゼ−3において初めて同定され、カスパーゼファミリーの酵素内の顕著な構造類似性に起因して、他のカスパーゼにも存在すると考えられた。例えば、カスパーゼ−3と、カスパーゼ−1(29%同一性)およびカスパーゼ−9(24%同一性)との間の相対的に低い配列同一性にも拘わらず、この3つの酵素は、高い程度の構造類似性を共有し、本質的に、互いに重ね合わせることが可能である。Mittlら,J.Biol Chem 272:6539;Rotondaら,Nat Struct Biol 3:619;Chaiら,Proc.Natl.Acad.Sci(USA),98:14250−14255;ならびにWattら,Structure 7:1135−1143を参照のこと。さらに記載されるように、このカスパーゼのアロステリック部位が、他のカスパーゼにおいて同定されている。しかし、このカスパーゼのアロステリック部位は、カスパーゼ−3において初めて特徴付けられたので、カスパーゼのアロステリック部位を含む残基は、カスパーゼ−3の番号付けスキームを用いて記載される。
【0064】
カスパーゼ−3と選択された代表的なカスパーゼとの配列アライメントを図4に示す。このアライメントを、示されたカスパーゼについて以下のアミノ酸を使用して作製した:XP_054686(カスパーゼ−3);NP_150634(カスパーゼ−1);NP−116764(カスパーゼ−2);AAH15799(カスパーゼ−7);およびAAH02452(カスパーゼ−9)。カスパーゼ−3配列とカスパーゼ−1配列、カスパーゼ−2配列、カスパーゼ−7配列、およびカスパーゼ−9配列とのそれぞれの間の整列している残基は、互いに対応していると言われる。例えば、Cys−264は、カスパーゼ−3の264番目のアミノ酸残基であり、カスパーゼ−1のスレオニン、カスパーゼ−2のチロシン、カスパーゼ−7のシステイン、およびカスパーゼ−9のグリシンに対応する。
【0065】
他のカスパーゼは、図4に示されるアライメントを参照して整列され得る。あるいは、配列は、Clustal W(1.81)(http://www2.ebi.ac.uk/clustalw/)のような標準的なアライメントソフトウェアを用いて整列され得る。
【0066】
1つの実施形態において、カスパーゼアロステリック部位は、カスパーゼ−3のCys−264に対応する残基の5Å以内であるカスパーゼの残基を含む。残基は、その原子のいずれかが、カスパーゼ−3のCys−264に対応する残基のいずれかの原子から5Å以下である場合、5Å以内であると言われる。別の実施形態において、カスパーゼアロステリック部位は、カスパーゼ−3のCys−264に対応する残基のいずれかの原子から3Å以下であるカスパーゼの残基を含む。
【0067】
別の実施形態において、カスパーゼアロステリック部位は、カスパーゼ−3の残基に対応する少なくとも2つの残基を含む:Glu−124;Gly−125;Lys−135;Leu−136;Lys−137,Lys−138;Ile−139;Thr−140;Leu−157;Phe−158;Ile−159;Phe−193,Leu−194;Tyr−195;Ala−196;Tyr−197;Ala−200;Pro−201;Gly−202;Cys−264;Ile−265;Val−266;Ser−267;Met−268;およびLeu−269。これらのカスパーゼ−3の残基ならびにカスパーゼ−1、カスパーゼ−2、カスパーゼ−7、およびカスパーゼ−9の対応する残基を、図4で囲む。別の実施形態において、カスパーゼアロステリック部位は、カスパーゼ−3の残基に対応する少なくとも2つの残基を含む:Cys−264;Ile−265;Val−266;Ser−267;Met−268;およびLeu−269。
【0068】
(カスパーゼ−3)
カスパーゼ−3は、クローニングされ、そしてシステイン残基がタンパク質の全体にわたる種々の位置で導入されている変異体が作製された。実施例1は、例示的なセットのシステイン変異体についてのクローニングおよび変異誘発をより詳細に記載する。クローニンされたタンパク質および変異体タンパク質を、実施例2に記載されるテトラペプチド酵素アッセイを使用して特徴付けた。
【0069】
係留(tethering)実験を、式
【0070】
【化6】
のリガンド候補のライブラリーを用いて、実施例3に記載されるように、カスパーゼ−3およびそのシステイン変異体を使用して、実施した。ここで、Rcは、先に規定された通りである。これらの係留実験の過程の間、カスパーゼ−3のアロステリック部位は、小さなサブユニット中の天然に存在するシステイン(Cys−264)の近位において発見された。
【0071】
選択されたリガンド候補によって改変された天然に存在するシステインとしてのCys264の同定を、実施例4に記載する。
【0072】
2つの選択されたリガンド候補は、化合物1および2であった:
【0073】
【化7】
示された部分は、Cys264とジスルフィド結合を形成するリガンド候補のRcC(=O)NHCH2CH2S−部分に対応する。図5は、カスパーゼ−3の小さなサブユニットとジスルフィド結合を形成するが、大きなサブユニットとは結合を形成しない化合物1を示す代表的な係留実験である。
【0074】
化合物1および2は、強く選択され、これらの化合物がアロステリック部位に対して固有の結合親和性を有することを示した。さらに、構造−活性関係を係留実験から観察した。例えば、化合物1が強く選択されるものの、化合物3および4は、そうではない。
【0075】
【化8】
実施例5に記載されるアッセイを使用して、化合物1および2をさらに特徴付け、そして化学量論的な様式でカスパーゼ−3活性を阻害することを示した。図6において示され得るように、阻害%を、化合物1または2とジスルフィド結合を形成するカスパーゼ−3の%を用いて追跡する。注目すべきことに、この阻害は、カスパーゼ−3と化合物2との間のジスルフィド結合の減少の際の酵素活性の回復で実証されるように、可逆である。
【0076】
これらの化合物が活性ホモダイマーの形成を破壊する可能性を調査しそして排除した。活性ホモダイマーからモノマーへの変換に続くサイジングクロマトグラフィーの溶出プロフィールは、化合物1または2の非存在および存在の両方において、カスパーゼ−3について本質的に同一であった。
【0077】
代わりに、構造実験によって実証されるように、作用の機構は、アロステリック部位への化合物1または2の結合の際の活性部位の再配列に起因する。注目すべきことに、アロステリック外側部位(exosite)への化合物1または2の結合は、活性部位での基質の結合を防止する。興味深いことに、逆もまた真実である。活性部位への基質の結合は、アロステリック外側部位への化合物1または2の結合を防止する。言い換えると、活性部位およびアロステリック部位への結合事象は、相互に排他的である。
【0078】
(カスパーゼ−7)
カスパーゼ−3との53%の配列同一性およびCys−264として対応する部位に位置するシステインに基づいて、カスパーゼ−7のアロステリック部位が、カスパーゼ−3のアロステリック部位と同様に挙動することが予期された。化合物1および2が、カスパーゼ−3と同様な様式でカスパーゼ−7を阻害することを確認した後に、カスパーゼ−7は、pro形態、活性apo形態および活性阻害された形態で結晶化された唯一のカスパーゼであるので、カスパーゼアロステリック部位の構造研究のために選択した。
【0079】
実施例6は、化合物1および化合物2と複合体化されたカスパーゼ−7の構造研究のためのクローニングおよび結晶化手順を記載する。これらの化合物は、ダイマー界面における深いポケットに結合する。このポケットが化合物1または2の非存在下においてさえ識別可能であるので、カスパーゼアロステリック部位は、アロステリックインヒビターの存在によって誘導されなかった。化合物1または2を用いるカスパーゼ−7の構造研究は、これらの化合物が、アロステリック部位に特異的に結合することを確認し、そしてアロステリック阻害の背後にある潜在的機構を明らかにする。
【0080】
カスパーゼの活性形態がホモダイマーであるので、アロステリックインヒビターの2つの分子が、活性複合体に結合する(各小さなサブユニットについて1つの分子)。2つの小さなサブユニットの各々によって形成されるアロステリック結合部位は、互いに空間的に隣接する。
【0081】
化合物1の2つの分子は、それぞれの部位に結合し、そしてアンチパラレルの方向で互いに面する。2つの分子の間の2つの最も近い原子がそれぞれのカルボニルであり、そして7Åの距離で分離されているので、これらは、互いに相互作用するようではない。実際に、化合物1の2つの分子の間にも、化合物1のいずれかの分子とタンパク質との間にも、直接的な水素結合相互作用の証拠は無かった。しかし、5つの潜在的な水媒介性の水素結合が、化合物1の2つの分子とタンパク質との間で同定された。
【0082】
対照的に、化合物2の2つの分子は、1つの分子のインドール窒素と他の分子のカルボニル酸素との間で1つの分子内水素結合を形成する縁部−縁部(edge−to−edge)様式で、互いに相互作用する。さらに、3つの直接的な水素結合が、化合物2とタンパク質との間でなされるようである。
【0083】
2つの異なる結合様式は、化合物1および化合物2が活性部位に対してそれぞれの効果を及ぼす異なる方法で相関するようである。化合物1の場合、この化合物は、その化合物が阻害する活性部位と同じポリペプチドによって形成されるアロステリック部位に結合する。化合物2の場合、この化合物は、1つのポリペプチドによって形成されるアロステリック部位に結合し、そして他のポリペプチドによって形成される活性部位を阻害する。
【0084】
それにもかかわらず、化合物1または化合物2の結合が活性部位において阻害を達成する機構は、同じであるようである。カスパーゼの活性化は、pro−ペプチドの切断、および大きなサブユニットと小さなサブユニットとの間の切断の両方を必要とする。これらの切断がタンパク質の「活性」形態を与えるものの、得られるカスパーゼは、ペプチド結合溝の構造的再配列が生じるまで、触媒的に適格ではない。
【0085】
結合された基質の非存在下において、カスパーゼのいわゆる「活性」形態は、触媒的に不活性なコンフォメーションのままである。図7Aに示されるように、Arg−164(カスパーゼ−3の番号付けを使用する)(活性部位システインにすぐに隣接する残基)は、活性部位にまで突出し、その結果、ペプチド結合溝は、基質に結合するのに適切なコンフォメーションではない。基質が結合した場合、多くの構造的変化が誘導される。Arg−164は、タンパク質のコア内に押し込められ、そしてペプチド結合溝は、基質に適合するようになり(図7Bを参照のこと)、これらの両方が、酵素の触媒的に適格な形態に必要とされる。
【0086】
化合物1または2のいずれかに結合されたカスパーゼ−7の構造において、Tyr−197(カスパーゼ−3の番号付けを使用する)は、タンパク質のコアにおけるArg−164の埋没(burial)を防止するように、変位する。結果として、Arg−164は、非結合構造において見られるように、上に突出する(図7Cを参照のこと)。さらに、ペプチド結合溝は、乱れる。
【0087】
(カスパーゼ−9)
カスパーゼ−9はまた、アロステリック部位の証拠について研究された。カスパーゼ−7とは異なり、カスパーゼ−9は、カスパーゼ−3と24%のみの配列同一性を共有する。さらに、カスパーゼ−3のCys−264に対応する残基は、グリシンであり、システインではない。しかし、天然に存在するシステインはまた、アロステリックポケット内に存在し、カスパーゼ−3のIle−265に対応する。
【0088】
実施例7は、カスパーゼ−9のクローニングおよびアッセイのために使用される手順を記載する。カスパーゼ−3と同様に、係留実験は、小さなサブユニットに結合するとして、以下:
【0089】
【化9】
を含むいくつかのリガンド候補を同定した。カスパーゼ−9が小さなサブユニットにおいてただ1つの天然に存在するシステインを含むので、それは、カスパーゼ−3のIle−265に対応する対応物として容易に同定された。注目すべきことに、この残基は、カスパーゼ−3のCys−264とほとんど同一の位置である。
【0090】
カスパーゼ−3の場合のように、これらの化合物がアロステリック部位に特異的に結合するという証拠には、識別可能な構造−活性関係が存在した。例えば、構造
【0091】
【化10】
を有する化合物8のリガンド候補もまた選択した。この化合物は、さらなるカルボキシル基の存在によって、化合物5と一部異なる。しかし、他のリガンド候補を試験し、選択されないことが見出された。試験されたビアリールエーテルにおいて置換された特定の官能基は、さらなるヒドロキシル基、ニトロ基などを有する化合物を含むか、またはこのカルボキシル基が、以下に示されるような異なる位置で置換される:
【0092】
【化11】
(PTP−1B)
カスパーゼに加えて、別の以前に未知であったアロステリック部位が、PTP−1B(最近の数年において、糖尿病および肥満のような種々の代謝障害の処置についての非常に確証された標的になったホスファターゼ)において同定された。
【0093】
ヒトPTP−1Bは、435アミノ酸タンパク質である。しかし、全長タンパク質が細菌において十分に発現しないので、PTP−1Bの研究は、代表的に、タンパク質の最初の321アミノ酸または最初の298アミノ酸に対応する形態のような短縮形態を使用して実施される。実施例8は、PTP−1Bの短縮型およびその変異体を作製するためのプロトコルを記載する。
【0094】
種々のアロステリックインヒビターの存在および非存在下でのPTP−1Bの結晶学的研究は、カスパーゼにおいて同定されたアロステリック部位とは異なり、PTP−1Bのアロステリック部位が、適合可能であることを示した。言い換えると、アロステリック結合部位は、適切なリガンドの非存在下で識別不可能である。
【0095】
PTP−1Bのアロステリックインヒビターの例示的な例は、以下:
【0096】
【化12】
に示される化合物15である。
【0097】
この化合物は、約30μMのIC50で非競合的にPTP−1Bに結合し、そして阻害する。PTP−1Bの活性を決定するための例示的なプロトコルを、実施例9に記載する。化合物15のようなアロステリックインヒビターの存在下において、以下の残基によって作製された凹部が形成される:Glu−186;Ser−187;Pro−188;Ala−189;Leu−192;Asn−193;Phe−196;Lys−197;Glu−200;Leu−272;Glu−276;Gly−277;Lys−279;Phe−280;Ile−281;およびMet−282。これらの残基は、化合物が結合する接触表面を形成する。しかし、アロステリックインヒビターの存在下において、この部位の大部分は、残基283〜298によって形成されるヘリックスの存在によって塞がれ、そして上記残基の大部分が、もはや接近可能ではない。
【0098】
アロステリックインヒビターの存在下で生じる大きなコンフォメーションの変化は、少なくとも3つの残基(Tyr−152、Asn−193、およびTryp−291)の相互作用によって媒介され、そしてPTP−1Bについての調節機構の一部であると考えられる。アロステリックインヒビターの非存在下において、Asn−193のNδ2(アロステリック部位形成残基の1つ)は、Tyr−152のOηと水素結合を形成し、そして残基283〜298によって形成されるヘリックスは、少なくとも一部、Trp−291のインドール環とPhe−280およびPhe−196(これらの両方がまたアロステリック部位形成残基である)のフェニル環との非結合相互作用により、適所に維持される。第4の残基(Lys−197(別のアロステリック部位形成残基))もまた、Asn−193のNδ2とTyr−152のOηとの間の水素結合相互作用を維持する際に関与すると考えられる。
【0099】
化合物15のようなアロステリックインヒビターの存在下において、残基283〜298によって形成される螺旋は、変位され、そして/または乱される。化合物15の場合、ベンゾフラン部分は、Trp−291のインドール環を変位する。化合物15のカルボニル酸素は、Asn−193のNδ2と水素結合を形成し、その結果、Asn−193のNδ2は、Trp−152のOηに対する水素結合にもはや利用可能ではない。Asn−193とTyr−152との間の水素結合の破壊は、一部、Tyr−152のフェノール環のコンフォメーションの変化を媒介する。Tyr−152のフェノール環の回転は、酵素を機能的に不活性化するPTP−1Bの活性部位におけるコンフォメーションの変化を広げる。
【0100】
アロステリック形成残基(Asn−193、Phe−196、およびPhe−280)の重要性は、変異誘発実験を使用して一部確認された。変異体PTP−1B(Asn−193がアラニンに変異され、Phe−196がアルギニンに変異され、そしてPhe−280がシステインに変異される)が作製された場合、アロステリック機構は無効になり、化合物15のようなアロステリックインヒビターは、もはや酵素を阻害し得ない。
【0101】
本発明は、さらに、以下の非制限的な実施例によって例示される。
【0102】
(実施例1 ヒトカスパーゼ−3のクローニング)
ヒトのカスパーゼ−3(Yama、CPP32ベータとしても公知)を、ジャーカット細胞(クローンE6−1;ATCC)から直接クローニングした。手短には、総RNAを、Tri−試薬(シグマ社)を用いて、37℃、5%CO2で増殖したジャーカット細胞から精製した。ポリメラーゼ連鎖反応(PCR)によって、カスパーゼ−3/Yama/CPP32のラージ(大きい)サブユニットおよびスモール(小さい)サブユニットをコードするDNAの増殖を可能にするように、オリゴヌクレオチドプライマーを設計した。手短には、Ready−To−Go−PCRビーズ(Amersham/Pharmacia)および以下のオリゴヌクレオチド:
5’−TTCCATATGTCTGGAATATCCCTGGACAACAGTTA−3’(配列番号1)および
5’−AAGGAATTCTTAGTCTGTCTCAATGCCACAGTCCAG−3’(配列番号2)
を用いて、1μgの総RNAから、アミノ酸28−175(ラージサブユニットのほとんどを含む)をコードするDNAを直接増幅した。Ready−To−Go−PCRビーズ(Amersham/Pharmacia)および以下のオリゴヌクレオチド:
5’−TTCCATATGAGTGGTGTTGATGATGACATGGCG−3’(配列番号3)および
5’−AAGGAATTCTTAGTGATAAAAATAGAGTTCTTTTGTGAG−3’(配列番号4)
を用いて、1μgの総RNAから、アミノ酸176−277(スモールサブユニットのほとんどを含む)をコードするDNAを直接増幅した。
【0103】
次に、カスパーゼ−3のラージサブユニットまたはスモールサブユニットのいずれかに対応する増幅したDNAを、制限酵素EcoRIおよびNdeIで切断し、標準的な分子生物学の技術を用いて、EcoRIおよびNdeIで消化したpRSET−b(インビトロジェン社)中に直接クローニングした。[例えば、Tewari M,Quan LT,O’Rourke K,Desnoyers S,Zeng Z,Beidler DR,Poirier GG,Salvesen GSおよびDixit VM.Yama/CPP32 beta,a mammalian homolog of CED−3,is a CrmA−inhibitable protease that cleaves the death substrate poly (ADP−ribose) polymerase、Cell 81 (5), 801−809 (1995)を参照のこと]。
【0104】
スモールサブユニットについて2つの報告されたタンパク質配列があり、そして各々は、1アミノ酸が異なり、アミノ酸190位(活性部位のシステインを163位とした相対位置)に、アスパラギン酸(GenBankアクセッション番号P42574)またはグルタミン酸(GenBankアクセッション番号XP_054686)のいずれかを有する。両方の形態のクローニング、発現、および精製が成功しており、両者は、機能的に区別できない。
【0105】
(一本鎖DNAの調製)
カスパーゼ−3のラージサブユニットまたはスモールサブユニットのいずれかをコードするDNAを含むプラスミドを、別々にE.coli K12 CJ236細胞(ニューイングランドバイオラブス)中に形質転換し、各々の構築物を含む細胞を、アンピシリン含有寒天プレート上での増殖能力によって選択した。ラージサブユニットおよびスモールサブユニットのオーバーナイト培養物を、別々に2YT(100μg/mLのアンピシリンを含む)中で、37℃で増殖させた。各々の培養物を、1:100で希釈し、そしてA600=0.3−0.6まで増殖させた。各培養物の1.5mLのサンプルを取り出し、そして10μLのファージVCS−M13(ストラタジーン社)で感染し、37℃で60分間振盪し、そして各々のオーバーナイト培養物を、100μg/mLのアンピシリンと20μg/mLのクロラムフェニコールを含む2YT中に1:100で希釈した感染培養物1mLを用いて調製し、そして37℃で増殖させた。細胞を、3000rcfで10分間遠心し、そして1/5容量の20% PEG/2.5M NaClを上清に添加した。サンプルを室温にて10分間インキュベートし、次に、4000rcfで15分間遠心分離した。ファージのペレットを、PBS中に再懸濁し、15Krpmで10分間遠心して、残りの微粒子状の物質を取り除いた。上清を保持し、一本鎖DNAを、QIAプレップスピンM13キット(キアゲン社)の手順に従って、上清から精製した。
【0106】
(システイン残基に改変する残基の同定)
システイン残基に改変したアミノ酸残基の選択は、カスパーゼ−3の三次元結晶構造を考察することによって、なされた。9つの異なるアミノ酸残基を、システイン残基に改変するために選択した。システイン変異を含む各カスパーゼ−3を、E.coli細胞中で、高レベルで発現した(一般的に1mg/Lより多い)。1例以外の全ての場合において、本発明者らは、正確にリフォールディングした四量体タンパク質の精製に成功し得た(Uno−5 Qクロマトグラフィーによって精製される能力によって評価)。しかし、ラージサブユニットのアミノ酸121においてヒスチジンのシステインへの変異を含むカスパーゼ−3タンパク質は、通常のクロマトグラフィーでは精製することができなかった。本発明者らは、個々のサブユニットを別々に精製することができたので、本発明者らは、カスパーゼ−3のこの改変体が正確に四量体を形成する(すなわち、ラージサブユニットがスモールサブユニットとリフォールディングする)ことができないことに起因する可能性が高いことが原因であると結論付けた。本発明者らはまた、新たなシステイン残基を有するカスパーゼ−3の全てのバージョンが、触媒活性を有するわけではないことを見出した。例えば、Y204Cは触媒活性を有さない。
【0107】
(一本鎖変異誘発)
スモールサブユニット内のシステイン変異体の例示的な例としては、F256C;S209C;S251C;W214C;およびY204Cが挙げられる。これらの変異体は、以下のプライマー:
F256C(5’−CTT TGC ATG ACA AGT AGC GTC−3’)、(配列番号5)
S209C(5’−GCC ATC CTT ACA ATT TCG CCA−3’)、(配列番号6)
S251C(5’−AGC GTC AAA GCA AAA GGA CTC−3’)、(配列番号7)
W214C(5’−CTG GAT GAA ACA GGA GCC ATC−3’)、(配列番号8)および
Y204C(5’−TCG CCA AGA ACA ATA ACC AGG−3’)、(配列番号9)
を用いて作製された。
【0108】
ラージサブユニット内のシステイン変異体の例示的な例としては、H121C;L168C;M61C;およびS65Cが挙げられる。これらの変異体は、以下のプライマー:
H121C(5’−TTC TTC ACC ACA GCT CAG AAG−3’)、(配列番号10)
L168C(5’−GCC ACA GTC ACA TTC TGT ACC−3’)、(配列番号11)
M61C (5’−CCG AGA TGT ACA TCC AGT GCT−3’)、(配列番号12)および
S65C (5’−ATC TGT ACC ACA CCG AGA TGT−3’)、(配列番号13)
を用いて作製された。
【0109】
約100pmolの各プラスミドを、1×TM緩衝液(0.5M Tris、pH 7.5、0.1M MgCl2)、lmM ATP、5mM DTT、および5単位のT4キナーゼ(NEB)を含む緩衝液中で、37℃で60分間インキュベートすることによってリン酸化した。キナーゼ処理したプライマーを、20μLの反応容量(〜50ngのキナーゼ処理したプライマー、1×TM緩衝液、および10〜50ng一本鎖DNA)中で、85℃で2分間、50℃で5分間、次に4℃で30〜60分間インキュベートすることによって、鋳型DNAとアニーリングした。伸長カクテル(2mM ATP、5mM dNTPs、30mM DTT、T4 DNAリガーゼ(NEB)、およびT7ポリメラーゼ(NEB))を、各アニーリング反応物に添加し、そして室温で3時間インキュベートした。変異誘発したDNAをE.coli XL−1 Blue細胞中に形質転換し、プラスミドDNAを含むコロニーを100μg/mlのアンピシリンを含むLB寒天プレートでの増殖によって選択した。DNA配列決定を用いて、適切な変異を含むプラスミドを同定した。
【0110】
(タンパク質発現および精製)
ラージサブユニット中のシステイン変異をコードするプラスミドDNAを、Codon Plus BL21細胞中に形質転換し、そしてスモールサブユニット中のシステイン変異をコードするプラスミドDNAを、BL21(DE3)pLysS細胞中に形質転換した。野生型ラージサブユニットおよびシステイン変異型ラージサブユニットをコードするプラスミドを含むCodon Plus BL21細胞を、150μg/mLのアンピシリンを含む2YT中で一晩、37℃で増殖させ、すぐに収集した。野生型スモールサブユニットおよびシステイン変異型スモールサブユニットをコードするプラスミドを含むBL21pLysS細胞を、150μg/mLのアンピシリンを含む2YT中でA600=0.6まで、37℃で増殖させた。その後、培養物を1mMのIPTGを用いて誘導し、さらに3〜4時間、37℃で増殖させた。4Krpmで10分間の遠心によって細胞を収集した後、細胞のペレットを、Tris−HCl(pH8.0)/5mM EDTA中に再懸濁し、2回、ミクロ流動化した(micro fluidize)。封入体を、9Krpmで10分間の遠心によって単離し、次に、6Mのグアニジン塩酸中に再懸濁した。変性したサブユニットを迅速かつ均一に、100μg/mLの濃度で再生緩衝液(l00mM Tris/KOH(pH 8.0)、10%スクロース、0.1% CHAPS、0.15M NaCl、およびl0mM DTT)中に希釈し、室温で60分間、ゆっくりと攪拌しながら、インキュベートすることによって、再生させた。
【0111】
再生したタンパク質を、10mM Tris(pH8.5)、10mM DTT、および0.1mM DETAを含む緩衝液中で一晩透析した。9Krpmで15分間遠心し、0.22μmの硝酸セルロースフィルターを通して上清をろ過することによって、沈殿物を取り除いた。次に、上清をアニオン交換カラム(Uno5 Q−カラム(バイオラッド))にロードし、正確に折りたたまれたカスパーゼ−3タンパク質を0−0.25M NaCl勾配を3mL/分で用いて、溶出した。各画分のアリコートを、変性ポリアクリルアミドゲル上で電気泳動して、カスパーゼ−3タンパク質を含む画分をプールした。
【0112】
(実施例2)
本実施例は、カスパーゼ−3の酵素活性を特徴付ける1つの方法を記載する。
【0113】
カスパーゼ−3についての最適なテトラペプチド認識モチーフを組み込んだクマリンベースの蛍光発生的な基質を、Alexis Biochemicals社より購入した。カスパーゼ−3を、1×反応緩衝液(25mM HEPES pH 7.4、0.1% CHAPS、50mM KClおよび5mM β−メルカプトエタノール)に添加して、最終濃度を、〜1.6nMとした。テトラペプチド基質(Ac−Asp−Glu−Val−Asp−AFC)を、添加して、最終濃度を、5μMとし、最終的な容量を50μLとした。アッセイを黒色の96ウェル平底ポリスチレンプレート(コーニング社)中で行い、そしてカスパーゼ活性を、365nmの励起波長および495nmの発光波長を用いて、Molecular Devices社のMicroplate Spectrofluorometer Gemini XSを用いて、モニターした。速度論データを、室温での15分間にわたるアッセイによって収集した。
【0114】
(実施例3)
本実施例は、カスパーゼ−3またはそのシステイン変異体を使用する、つなぎとめ(tethering)実験の1つの実施形態を記載する。つなぎとめスクリーニングは、典型的には、50μLの容量中で、最終濃度1〜5μMのカスパーゼ−3、1〜20mMのβ−メルカプトエタノール、および1〜2mMのリガンド候補(プール中の全てのリガンド候補の総濃度;従って、〜10のプール中の各リガンド候補は、100〜200μMの最終濃度を有する)を用いて、TE緩衝液(10mM Tris、1mM EDTA、pH8.0)中で行われた。質量分析によって分析する前に、平衡まで反応を進ませた(1時間以上)。反応混合物を、Finnegan LCQ2またはLCQ3 LCMSにロードし、分離手順に依存して、各々2〜5分間分析した。逆重畳の後、ラージサブユニットおよび/またはスモールサブユニットを公知の分子量に基づいて同定した。
【0115】
(実施例4)
本実施例は、化合物1および2がカスパーゼ−3とジスルフィド結合を形成する、天然に存在するシステインとしてのCys264の同定を記載する。カスパーゼ−3のスモールサブユニットは、3つのシステイン(Cysl84、Cys220、およびCys264)を含む。これら3つの中で、Cys220が、埋もれており、従って、可能性として除外された。
【0116】
1つの実験において、適切なDNAプライマー(C184S 5’−TAT TTT ATG AGA CGC CAT GTC−3’ (配列番号14);C264S 5’−GGA AAC AAT CGA TGG AAT CTG−3’ (配列番号15)、下線を引いたトリプレットは、導入されたセリン残基を示す)を用いて、184位および264位のシステインのいずれかを、セリンに変異した変異体を作製した。その後、クローンを、DNA配列分析によって確認した。C184S変異体は、化合物1および2と、標的−化合物結合体を形成し得たが、C264S変異体は、形成し得なかった。
【0117】
システイン残基としてのCys264の同定を、化合物1との標的−化合物結合体のペプチドマッピングによって、さらに確認した。TE緩衝液(pH8.0)中のカスパーゼ−3(4μM)を、化合物1(200μM)およびβメルカプトエタノール(1mM)の存在下で、1時間、25℃でインキュベートした。100%のスモールサブユニットが化合物1によって改変した時(LC/MSによって決定した)に、過剰な化合物1および還元剤を、セファデックスG−25カラムでのサイズ排除クロマトグラフィーによって除去した。微量の還元剤または未結合の化合物1が除去されることを確実にするために、タンパク質をTE緩衝液中に10倍に希釈し、そして、ミリポア5,000MWCOろ過デバイスによって再度濃縮した。このプロセスを、3回繰り返した。改変されたカスパーゼ−3を、98℃ 1分間熱変性し、次に、溶液が室温になるまで、氷上でインキュベートした。熱処理後、約70%のカスパーゼ−3が、標的−化合物結合体として残った。標的−化合物結合体を、500mMの酢酸アンモニウム緩衝液(pH4.0)中のエンドプロテイナーゼGlu−c(20ng/μL)によって、20時間 室温で消化した。ペプチドの質量を、Q−STAR装置においてLC/MS分析した。化合物1と共有結合したCys264を含むペプチドを含む、予測された消化フラグメントの各々の質量を観察した。化合物1に共有結合したCys184またはCys220に対応するペプチド質量は、観察されなかった。このことを、MS/MSによってフラグメント化した後の、トリペプチドP263C264I265+化合物1の観察によって、確認した。
【0118】
(実施例5)
本実施例は、ジスルフィド形成の程度(標的−リガンド結合体の形成)と、カスパーゼ−3酵素活性の阻害の程度とを相関付ける1つの実施形態を記載する。
【0119】
一定濃度のタンパク質(1〜5μM)および化合物(典型的には200μM)を用いて、反応物中のβ−メルカプトエタノールの濃度を変化させ、標的−リガンド結合体の形成の程度を変化させた。約1時間後、サンプルを、LC/MSによって試験して、各β−メルカプトエタノール濃度において、化合物1によって改変されたスモールサブユニットの割合を決定した。同時に、1μLのサンプルを取り出し、そして199μLの1×反応緩衝液(5μMのAc−Asp−Glu−Val−Asp−AFCを含む、25mM HEPES pH 7.4、0.1% CHAPS、50mM KClおよび5mM β−メルカプトエタノール)に添加した。化合物1の分析のために、200μMの化合物および種々の濃度のβ−メルカプトエタノールもまた添加し、その結果、つなぎとめ反応において、β−メルカプトエタノールの最終濃度を同一にした。希釈後、カスパーゼ−3酵素の最終濃度は、〜5nMであった。カスパーゼ活性を、365nmの励起波長および495nmの発光波長を用いて、Molecular Devices社のMicroplate Spectrofluorometer Gemini XSを用いて、モニターした。種々の量の化合物1を用いて改変したカスパーゼ−3の相対活性を、化合物1の不存在下での同様の反応条件下で観察された活性と比較した。化合物2を、同様に試験した。
【0120】
(実施例6)
本実施例は、化合物1および2と複合体化したカスパーゼ−7の構造研究のためのクローニングおよび結晶化手順を記載する。
【0121】
表1において詳細に記載されるように、カスパーゼ−7のラージサブユニット発現のための一連のプラスミドを、カスパーゼ−7の残基50−198または57−198のコード配列を、pRSET(ampr、インビトロジェン)またはpET3a(ampr、ノバジェン)中にサブクローニングすることによって、作製した。
【0122】
【表1】
スモールサブユニットの発現のために、カスパーゼ−7残基199−303+アミノ酸QLHHHHHH、または同様の添加残基を有する残基210−303のコード配列を、pBB75(kanr)中に連結した(Batchelor、Piperら、1998)。変異D192Aもまた、ラージサブユニット中に導入し、異質性を最小限とした。プラスミドpJH02、03、05、 08、09または11(表2)を、pJH06または07と組み合わせて形質転換し、過剰発現試験を行った。これらの組み合わせの中で、pJH07および08、またはpJH07および09が、最も高く過剰発現し、そしてChaiら、Cell 104:769−80(2001)およびChaiら、Cell 107:399−407に記載される方法を用いて、容易に精製された。
【0123】
pJH07およびpJH08(10μM)から発現されたカスパーゼ−7(D192A)を、化合物1(100μM)によって、500μMのβ−メルカプトエタノールを含むTE緩衝液(10mM Tris、pH8.0、1mM EDTA)中で1週間室温にてインキュベーションすることによって、標識した。スモールサブユニットの標識は、数時間後、98%完全であったが(質量分析計によって決定された)、100%完全な標識を得るために、より長時間行った。化合物2(50μM)によるカスパーゼ−7(D192A)の標識化を、TE緩衝液中の250μMのβ−メルカプトエタノール存在下で行った。標識は、4時間後に60%完全であり、1週間後に100%完全であった。これらのタンパク質をNAP−5カラム(アマシャムファルマシアバイオテクAB)中での緩衝液交換によって、結晶化のための100mM NaCl、10mM Tris pH8.0を含む緩衝液中に移した。標識されたタンパク質を、ミリポア5K MWCO濃縮デバイス中で12mg/mLに濃縮した。
【0124】
1μLのタンパク質および2μLの母液溶液(100mM シトレート緩衝液(pH5.8)、1M LiSO4、1M NaCl)を含む液滴から、4℃でのハンギングドロップ拡散蒸着によって1週間で形成されたカスパーゼ−7(D192A)/化合物1の結晶。カスパーゼ−7/化合物2の結晶は、1μLのタンパク質および1μLの母液である液滴から成長した。いずれかの化合物とのカスパーゼ−7(D192A)の結晶を、20%グリセロールを含む成長母液の液滴に移し、4℃で一晩インキュベートした。次いで、この結晶を、液体窒素中でフラッシュ凍結した。
【0125】
化合物1との複合体についてのデータを、Raxis−4検出器を備えるRigakuジェネレータで収集した。データを、D*trekを用いて処理した。化合物2との複合体についてのデータを、Quantum−315 CCDカメラ(ADSC)で、SSRL Beamline 9−1にて収集した。データを、Project,C.C.,Acta Crysta.D.50:760−763(1994)に記載されるように、CCP4−mosflmおよびスカラーを用いて処理した。これらの構造を、活性カスパーゼ−7(1K86.pdb)の構造およびCCP4−amoreにおける剛性本体の精製を使用して、直接的分子置換によって解析した。これらの構造を、反復回の、Oにおける分子再構築(Jonesら、Acta Crystallogr A 47:110−119(1991))およびCCP4−refmacにおけるエネルギー最小化によって、精緻化した。最終データ統計を、表2に示す。
【0126】
(表2)
【0127】
【表2】
両方の複合体についての電子密度マップは、このタンパク質のコアと相互作用する化合物1および化合物2の方向を明確に明らかにした。これらの化合物の順序付けられた性質は、これらの係留化合物が特異的様式で結合していることを確認する。これらの化合物についての温度要因は、タンパク質自体由来の周辺原子と同じくらい低いかまたはそれよりも低く、インヒビター分子が、非常によく順序付けられ、そしてランダム様式または擬似様式で結合していないことを示す。
【0128】
(実施例7)
ヒトカスパーゼ−9を、刊行された手順(Garcia−Calvo,Mら、1998.Inhibition of Human Caspases by Peptide−based and Macromolecular Inhibitors,JBC 273(49):32608−32613;Thornberry,NAら、A combinatorial Approach Defines Specificities of Members of the Caspase Family and Granzyme B,JBC 272(29):17907−17911)に従って、クローニング、発現および精製し、次いで、適切な酵素活性について試験した。カスパーゼ−9酵素を、1×反応緩衝液(100mM MES(pH6.5)、10%グルコース、0.1% CHAPS、10mM DTTおよび100mM NaCl)に添加した。200μMの最終濃度までの基質の添加(Ac−Leu−Glu−His−Asp−AFC)により反応を開始し、そして最終反応容量を50μLにした。アッセイを、黒色96ウェル平底ポリスチレンプレート(Corning)中で実施した。カスパーゼ活性を、365nmの励起波長および495nmの発光波長でMolecular Devices’ Microplate Spectrofluorometer Gemini XSを使用してモニタリングした。反応速度論的データを、室温での15分間の実行時間の間に収集した。
【0129】
(実施例8)
この実施例は、野生型ヒトPTP−1Bの短縮バージョンを作製するための1つの実施形態を記載する。ヒトPTP−1Bの最初の321アミノ酸をコードするcDNAを、ヒト胎児心臓総RNA(Clontech)から単離した。PTP−1B cDNA(Genbank M31724.1、Chernoff、1990)のヌクレオチド91〜114(For)に対応するオリゴヌクレオチドプライマーおよびヌクレオチド1030〜1053(Rev)に相補的なオリゴヌクレオチドプライマーを合成し、そしてポリメラーゼ連鎖反応を使用してDNAを生成するために使用した。
【0130】
プライマーForwardは、最初のATGコドンでNdeI制限部位を組み込み、そしてプライマーRevは、ヌクレオチド1053の後ろに、UAAコドンとその後ろのEcoRI制限部位とを挿入する。cDNAを、標準的な分子生物学的技術を用いて、制限ヌクレアーゼNdeIおよびEcoRIで消化し、そしてpRSETc(Invitrogen)にクローニングした。単離されたcDNAの正体を、DNA配列決定分析によって確認した。
【0131】
アミノ酸残基1〜298をコードする、より短いcDNAであるPTP−1B 298を、ポリメラーゼ連鎖反応において、オリゴヌクレオチドプライマーForwardおよびRev2、ならびにテンプレートとして上記のクローンを使用して生成した。
【0132】
【化13】
ヒトPTP−lBの321アミノ酸形態は、以下の配列番号17である:
【0133】
【化14】
変異体を、以下のように作製した。pRSETc(Invitrogen)中のPTP−1B 321を、テンプレートとして使用し、そしてT7プライマーおよびRSETrevプライマーを、「外側」プライマーとして使用した。変異誘発プライマーは、以下であった:
【0134】
【化15】
PTP−1B[321;N193A;F196R;F280C]を、残基1〜215に対応する、PTP−1B[321;N193A;F196R]由来のNdeI−PstIフラグメントを、残基216〜321に対応する、PTP−1B[321;F280C]由来のPstI−EcoRIフラグメントと連結することによって、生成した。
【0135】
PTP−1B[298;N193A;F196R;F280C]を、テンプレートとしてPTP−1B[321;N193A;F196R;F280C]を使用するPCRによって生成した。T7ベクタープライマーを順方向プライマーとして使用して、残基298での短縮を、プライマー
【0136】
【化16】
を使用して生成した。
【0137】
PTP−lB[298;C215S]を、Kunkel変異誘発およびテンプレートとしてのPTP−1B[298]を使用して生成した。変異誘発プライマーは、以下であった:
【0138】
【化17】
(実施例9)
この実施例は、PTP−1Bに対する本発明の化合物のIC50を決定するための1つの例示的な方法を記載する。基質であるpNPP(Sigma)を、1×HN緩衝液(50mM HEPES(pH7.0);100mM NaCl;1mM DTT)中に4mMで溶解させ、そして83μlを2μlのDMSOまたは2μlのDMSO中化合物と混合した。この反応を、15μlの1×HN緩衝液中のPTP−1B(標準的なアッセイ条件で750ηg)の添加によって開始した。産物形成の速度(OD405nm−(マイナス)OD655nm、BioRad BenchmarkまたはMolecular Devices Spectramax 190)を、25℃で15分間にわたって30秒毎に測定し、データを線形回帰によって分析した。終点アッセイのために、この反応を、50μl 3M NaOHを用いて15分後に停止させ、そしてOD405nm−OD655nmを測定した。IC50決定のために、非阻害コントロールに対して正規化した速度を、化合物濃度に対してプロットし、そして4パラメータの非線形回帰曲線当てはめ(
【0139】
【数1】
、Spectramax Software package)を使用して当てはめた。
【0140】
本明細書中で引用される全ての参考文献は、本明細書中で参考として明示的に援用される。本発明は、その特定の実施形態を参照して記載されているが、本発明の真の精神および範囲から逸脱することなしに、種々の変化がなされ得、そして等価物が置換され得ることが、当業者に理解されるべきである。さらに、多くの改変が、特定の状況、材料、物の組成、プロセスなどを適合させるためになされ得る。全てのこのような改変は、添付の特許請求の範囲の範囲内である。
【図面の簡単な説明】
【0141】
【図1】図1は、係留方法の1つの実施形態の模式図である。チオール含有タンパク質は、複数のリガンド候補と反応される。標的に対して固有の結合親和性を保有するリガンド候補が同定され、ジスルフィド部分を含まない同定された結合決定基(円によって表される)を含むリガンドが、作製される。
【図2】図2は、2つの係留実験の代表的な例である。図2Aは、チミジル酸シンターゼ(「TS」)に対する結合親和性をほとんど有さないか、または全く有さない10個の異なるリガンド候補のプールとTSとの反応の逆重畳質量分析スペクトルである。図2Bは、リガンド候補のうちの1つがTSに対して固有の結合親和性を保有する10個の異なるリガンド候補のプールとこの酵素との反応の逆重畳質量分析スペクトルである。
【図3】図3は、係留実験の模式図であり、ここで、チオールは、2つの異なる外部部位か、またはその付近に位置する。図3Aは、リガンドの外部部位への結合が、標的の機能に影響しない状況を示す。図3Bは、リガンドの外部部位への結合が、標的の機能に影響する状況を示す。この場合において、リガンドの外部部位への結合は、活性部位のコンフォメーションを変更し、その結果、これは、標的の機能を阻害する。図3Bは、外部部位がまた、アロステリック部位である例である。
【図4】図4は、選択されたカスパーゼの配列整列である。アロステリック部位を含む残基は、四角で囲んである。数字は、カスパーゼ−3における番号付けに対応する。
【図5】図5は、係留実験の結果であり、化合物1が、カスパーゼ−3の小さなサブユニットとジスルフィド結合を形成するが、大きなサブユニットとはジスルフィド結合を形成しないことを示す。
【図6】図6は、カスパーゼ−3活性の阻害と、カスパーゼ−3と化合物1または2との間のジスルフィド形成の度合いとを相関させた実験の結果である。
【図7】図7は、カスパーゼ−7の未結合形態(A)、基質結合形態(B)、およびアロステリック的に阻害された形態(C)を示す。
【配列表】
【特許請求の範囲】
【請求項1】
アロステリック部位を同定するための方法であって、該方法は、以下の工程:
a)第一の結合部位、第二の結合部位および該第二の結合部位またはその近傍に化学反応基を備える標的を提供する工程;
b)該標的を、該化学反応基と共有結合を形成し得る化合物と接触させる工程;
c)該標的と該化合物との間に共有結合を形成し、それによって、標的−化合物結合体を形成する工程;
d)該標的−化合物結合体が、該標的と比較して、主な結合部位中に変化を有するか否かを決定する工程、
を包含する、方法。
【請求項2】
前記化学反応基がチオールである、請求項1に記載の方法。
【請求項3】
前記化学反応基がマスキングされたチオールである、請求項1に記載の方法。
【請求項4】
前記マスキングされたチオールが、ジスルフィドの形態である、請求項3に記載の方法。
【請求項5】
前記共有結合がジスルフィド結合であり、そして前記接触させる工程が、還元剤の存在下で生じる、請求項2または3に記載の方法。
【請求項6】
前記化合物が、以下:
【化1】
からなる群より選択されるリガンド候補であり、
ここで、RおよびR’は、各々独立して非置換C1〜C20脂肪族、置換C1〜C20脂肪族、非置換アリールまたは置換アリールであり;
mは0、1または2であり;そして
nは1または2である、
請求項5に記載の方法。
【請求項7】
前記変化が、前記標的の活性における機能的変化である、請求項1に記載の方法。
【請求項8】
前記変化が、構造的変化である、請求項1に記載の方法。
【請求項9】
前記構造的変化が、x線結晶学を使用して決定される、請求項8に記載の方法。
【請求項10】
前記構造的変化が、NMRを使用して決定される、請求項8に記載の方法。
【請求項11】
前記構造的変化が、円偏光二色性を使用して決定される、請求項8に記載の方法。
【請求項12】
前記標的がプロテアーゼである、請求項1に記載の方法。
【請求項13】
前記標的がキナーゼである、請求項1に記載の方法。
【請求項14】
前記標的がホスファターゼである、請求項1に記載の方法。
【請求項15】
アロステリックインヒビターを同定する方法であって、該方法は、以下:
a)第一の係留実験を実施する工程、および
b)第二の係留実験を実施する工程、
を包含し、ここで、両方の係留実験が、以下:
i)第一の結合部位、第二の結合部位および該第二の結合部位またはその近傍に化学反応基を備える標的を提供する工程;
ii)該標的を、該化学反応基と共有結合を形成し得る化合物と接触させる工程;
iii)該標的と該化合物との間に共有結合を形成し、それによって、標的−化合物結合体を形成する工程;および
iv)該標的−化合物結合体を同定する工程、
を包含し、ここで、
該第一の係留実験が、該第一の結合部位に結合するリガンドの存在下で実施され、そして該第二の係留実験が、該第一の結合部位に結合するリガンドの非存在下で実施される、
方法。
【請求項16】
前記化学反応基が、チオールまたはマスキングされたチオールである、請求項15に記載の方法。
【請求項17】
前記共有結合がジスルフィド結合であり、そして前記接触させる工程が、還元剤の存在下で生じる、請求項16に記載の方法。
【請求項18】
前記化合物が、以下:
【化2】
からなる群より選択されるリガンド候補であり、
ここで、RおよびR’は、各々独立して非置換C1〜C20脂肪族、置換C1〜C20脂肪族、非置換アリールまたは置換アリールであり;
mは0、1または2であり;そして
nは1または2である、
請求項17に記載の方法。
【請求項19】
アロステリックインヒビターを同定する方法であって、該方法は、以下:
a)アロステリック調節をし得る標的およびアロステリック調節をし得ないその変異体を提供する工程;
b)該標的を化合物と接触させる工程;
c)該変異体を該化合物と接触させる工程;ならびに
d)該標的に対する該化合物の活性を、変異体に対する該化合物の活性と比較する工程、
を包含する、方法。
【請求項20】
カスパーゼのアロステリックインヒビター。
【請求項1】
アロステリック部位を同定するための方法であって、該方法は、以下の工程:
a)第一の結合部位、第二の結合部位および該第二の結合部位またはその近傍に化学反応基を備える標的を提供する工程;
b)該標的を、該化学反応基と共有結合を形成し得る化合物と接触させる工程;
c)該標的と該化合物との間に共有結合を形成し、それによって、標的−化合物結合体を形成する工程;
d)該標的−化合物結合体が、該標的と比較して、主な結合部位中に変化を有するか否かを決定する工程、
を包含する、方法。
【請求項2】
前記化学反応基がチオールである、請求項1に記載の方法。
【請求項3】
前記化学反応基がマスキングされたチオールである、請求項1に記載の方法。
【請求項4】
前記マスキングされたチオールが、ジスルフィドの形態である、請求項3に記載の方法。
【請求項5】
前記共有結合がジスルフィド結合であり、そして前記接触させる工程が、還元剤の存在下で生じる、請求項2または3に記載の方法。
【請求項6】
前記化合物が、以下:
【化1】
からなる群より選択されるリガンド候補であり、
ここで、RおよびR’は、各々独立して非置換C1〜C20脂肪族、置換C1〜C20脂肪族、非置換アリールまたは置換アリールであり;
mは0、1または2であり;そして
nは1または2である、
請求項5に記載の方法。
【請求項7】
前記変化が、前記標的の活性における機能的変化である、請求項1に記載の方法。
【請求項8】
前記変化が、構造的変化である、請求項1に記載の方法。
【請求項9】
前記構造的変化が、x線結晶学を使用して決定される、請求項8に記載の方法。
【請求項10】
前記構造的変化が、NMRを使用して決定される、請求項8に記載の方法。
【請求項11】
前記構造的変化が、円偏光二色性を使用して決定される、請求項8に記載の方法。
【請求項12】
前記標的がプロテアーゼである、請求項1に記載の方法。
【請求項13】
前記標的がキナーゼである、請求項1に記載の方法。
【請求項14】
前記標的がホスファターゼである、請求項1に記載の方法。
【請求項15】
アロステリックインヒビターを同定する方法であって、該方法は、以下:
a)第一の係留実験を実施する工程、および
b)第二の係留実験を実施する工程、
を包含し、ここで、両方の係留実験が、以下:
i)第一の結合部位、第二の結合部位および該第二の結合部位またはその近傍に化学反応基を備える標的を提供する工程;
ii)該標的を、該化学反応基と共有結合を形成し得る化合物と接触させる工程;
iii)該標的と該化合物との間に共有結合を形成し、それによって、標的−化合物結合体を形成する工程;および
iv)該標的−化合物結合体を同定する工程、
を包含し、ここで、
該第一の係留実験が、該第一の結合部位に結合するリガンドの存在下で実施され、そして該第二の係留実験が、該第一の結合部位に結合するリガンドの非存在下で実施される、
方法。
【請求項16】
前記化学反応基が、チオールまたはマスキングされたチオールである、請求項15に記載の方法。
【請求項17】
前記共有結合がジスルフィド結合であり、そして前記接触させる工程が、還元剤の存在下で生じる、請求項16に記載の方法。
【請求項18】
前記化合物が、以下:
【化2】
からなる群より選択されるリガンド候補であり、
ここで、RおよびR’は、各々独立して非置換C1〜C20脂肪族、置換C1〜C20脂肪族、非置換アリールまたは置換アリールであり;
mは0、1または2であり;そして
nは1または2である、
請求項17に記載の方法。
【請求項19】
アロステリックインヒビターを同定する方法であって、該方法は、以下:
a)アロステリック調節をし得る標的およびアロステリック調節をし得ないその変異体を提供する工程;
b)該標的を化合物と接触させる工程;
c)該変異体を該化合物と接触させる工程;ならびに
d)該標的に対する該化合物の活性を、変異体に対する該化合物の活性と比較する工程、
を包含する、方法。
【請求項20】
カスパーゼのアロステリックインヒビター。
【図1】

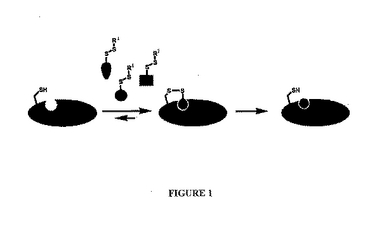
【図2】
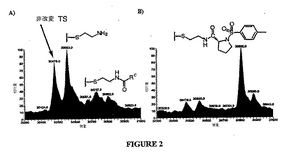
【図3】
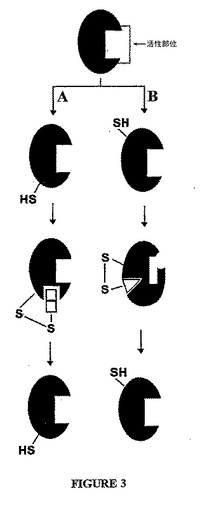
【図4】

【図5】
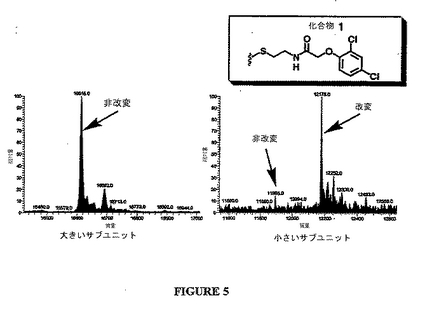
【図6】
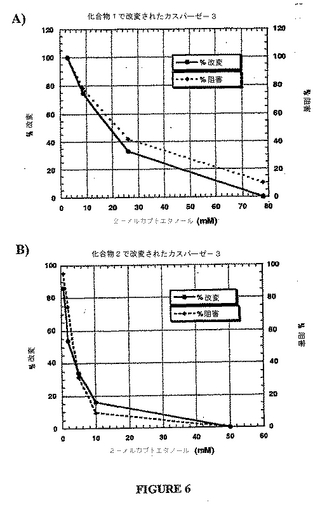
【図7】
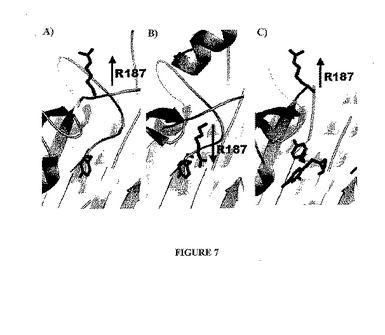
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公表番号】特表2006−503264(P2006−503264A)
【公表日】平成18年1月26日(2006.1.26)
【国際特許分類】
【出願番号】特願2003−584007(P2003−584007)
【出願日】平成15年4月8日(2003.4.8)
【国際出願番号】PCT/US2003/010831
【国際公開番号】WO2003/087051
【国際公開日】平成15年10月23日(2003.10.23)
【出願人】(500586635)サネシス ファーマシューティカルズ, インコーポレイテッド (29)
【Fターム(参考)】
【公表日】平成18年1月26日(2006.1.26)
【国際特許分類】
【出願日】平成15年4月8日(2003.4.8)
【国際出願番号】PCT/US2003/010831
【国際公開番号】WO2003/087051
【国際公開日】平成15年10月23日(2003.10.23)
【出願人】(500586635)サネシス ファーマシューティカルズ, インコーポレイテッド (29)
【Fターム(参考)】
[ Back to top ]