
アンダーフィル剤およびそれを用いた半導体装置
【課題】高靭性と粘度特性に優れたアンダーフィル剤を提供すること。
【解決手段】(a)重量平均分子量3万未満のポリフェニルスルホン樹脂と、(b)エポキシ樹脂を含有し、(c)溶剤の含有量が1重量%以下であることを特徴とするアンダーフィル剤。
【解決手段】(a)重量平均分子量3万未満のポリフェニルスルホン樹脂と、(b)エポキシ樹脂を含有し、(c)溶剤の含有量が1重量%以下であることを特徴とするアンダーフィル剤。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、アンダーフィル剤に関する。より詳しくは、半導体素子と基板との接合に好ましく用いられるアンダーフィル剤に関する。
【背景技術】
【0002】
近年、半導体素子の大型化が急速に進み、半導体素子と基板との間に大きな熱応力が発生し、問題となっている。従来より、半導体素子と基板との接合の後、空隙にエポキシ系アンダーフィル剤を充填することにより、応力を分散させて、半導体素子/基板界面の接合信頼性を高める手法が知られている。アンダーフィル剤に求められる特性としては、低粘度性であり、半導体素子と基板との隙間に素早く進入し、ボイドが生じないこと、硬化後の熱的または物理的応力に対しての接続信頼性、耐熱性、靭性が高いことや短時間・低温硬化性が挙げられる。エポキシ樹脂は硬くて脆い性質を有するため、機械的・熱的ストレスをより吸収させるために、各種熱可塑性樹脂をエポキシ樹脂に混合する技術が提案されている。
【0003】
例えば、強靭性に優れた熱硬化性樹脂組成物として、ポリエーテルスルホン樹脂、エポキシ樹脂、多価フェノール系硬化剤を含有する樹脂組成物や、ポリエーテルスルホン樹脂とビスマレイミド化合物とジアミン化合物の共重合体、エポキシ樹脂、硬化剤を含有する樹脂組成物が提案されている(例えば、特許文献1、2参照)。また、ポリイミド樹脂、エポキシ樹脂、硬化剤を含有し、25℃の溶液粘度が500センチポイズ以下であるアンダーフィル剤が提案されている(例えば、特許文献3参照)。これらの材料は、強靭化剤或いは低応力化剤として配合されている樹脂を、トリグライムやジメチルアセトアミドなどの高沸点溶媒に溶解させるものであり、これをアンダーフィル剤として用いた場合、接合後に溶媒の飛散によりボイドが発生して信頼性が低下したりするなど、実用的ではなかった。実質的に無溶剤の液状アンダーフィル剤として、エポキシ変性シリコーン樹脂、エポキシ樹脂、硬化剤を含有し、25℃の溶液粘度が18〜72Pa・s以下であるアンダーフィル剤が提案されている(例えば、特許文献4参照)。これらの材料は弾性率を下げる効果はあるが、アンダーフィル剤として用いた場合、強靭性に劣るなど、実用的ではなかった。
【0004】
また機械特性や靭性に優れるビフェニル基を有するポリエーテルスルホン樹脂として、重量平均分子量が約40,000〜約60,000のポリフェニルスルホン樹脂が知られている(例えば、特許文献5参照)。しかし、無溶剤系液状アンダーフィル材料に適用すると、エポキシ樹脂に不溶であることから、不均一溶液となり、塗布性が不十分であったり、分子量が高いため、溶液が高粘度になるなど実用的でなかった。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開2001−72833号公報(特許請求の範囲)
【特許文献2】特開2009−155354号公報(特許請求の範囲)
【特許文献3】特許第3719018号公報(特許請求の範囲)
【特許文献4】特開2004−346232号公報(特許請求の範囲)
【特許文献5】特表2002−525406号公報(特許請求の範囲)
【発明の概要】
【発明が解決しようとする課題】
【0006】
このように、従来技術では実質的に無溶剤で低粘度性と高靭性を両立したアンダーフィル剤は得られていなかった。本発明は、上記課題を解決すべく、低粘度性と高靭性に優れたアンダーフィル剤を提供することを目的とする。
【課題を解決するための手段】
【0007】
本発明は、(a)重量平均分子量3万未満のポリフェニルスルホン樹脂と、(b)エポキシ樹脂を含有し、(c)溶剤の含有量が1重量%以下であることを特徴とするアンダーフィル剤である。
【発明の効果】
【0008】
本発明によれば、優れた高靭性と粘度特性を有するアンダーフィル剤を得ることができる。
【図面の簡単な説明】
【0009】
【図1】実施例2のアンダーフィル剤のDSC曲線
【発明を実施するための形態】
【0010】
本発明のアンダーフィル剤は(a)重量平均分子量3万未満のポリフェニルスルホン樹脂と、(b)エポキシ樹脂を含有し、(c)溶剤の含有量が1重量%以下であることを特徴とする。
【0011】
本発明において溶剤とは、粘度を調整したり塗布性を向上させる目的で加えられる揮発性の有機または無機の液体を指し、アンダーフィル剤を塗布した後のエポキシ樹脂を硬化させるまでの工程で蒸発または飛散するものを指す。アンダーフィル剤中の溶剤の含有量は、例えば熱重量分析により25℃から300℃まで5℃/分で昇温させた際の減量率から求めることができる。
【0012】
本発明のアンダーフィル剤は実質的に無溶剤であるため、溶剤飛散によるボイドの発生を抑制し、接合後の絶縁信頼性を高めることができる。またポリフェニルスルホン樹脂はその構造中にビフェニル基を含有した剛直な構造を有し、硬化後の強靭性に優れ、またガラス転移温度が200℃以上と非常に高い特徴を有する。そのためポリフェニルスルホン樹脂をエポキシ樹脂に含有することにより、得られるアンダーフィル剤の硬化後の靭性と耐熱性(ガラス転移温度)を向上させることができる。
【0013】
また(a)ポリフェニルスルホン樹脂は、重量平均分子量3万未満にすることで(b)エポキシ樹脂と25℃、1.013×105Paの条件下で溶解し、均一な液状アンダーフィル剤を得ることができる。このような溶液を有することで、溶液中に樹脂の残渣がなく、したがって、本発明のアンダーフィル剤は基板への塗布性や浸透性に優れる。
【0014】
本発明のアンダーフィル剤において、(a)成分のポリフェニルスルホン樹脂は、(b)エポキシ樹脂と溶解するために、重量平均分子量が3万未満であることが重要である。重量平均分子量が3万以上であると、(b)エポキシ樹脂に溶解せず、均一な液状溶液を得ることが困難となる。粘度特性をより適切な範囲に調整し、低粘度にして浸透性をより向上させるために、重量平均分子量が2万以下が好ましい。一方硬化後の強靭性の観点からは、(a)成分のポリフェニルスルホン樹脂の重量平均分子量は2000以上が好ましく、5000以上がより好ましい。ポリフェニルスルホン樹脂を2種以上含有してもよく、各樹脂の重量平均分子量がいずれも3万未満であることが好ましい。
【0015】
本発明における重量平均分子量は、ゲルパーミエーションクロマトグラフィー(GPC)法により、NMP/H3PO4の混合溶媒を用いてポリフェニルスルホン樹脂の分子量を測定し、標準ポリスチレンの校正曲線を用いて算出した値を指す。
【0016】
ポリフェニルスルホン樹脂は、フェノール性水酸基とアルカリ金属炭酸塩とを反応させてフェノール化合物のアルカリ金属塩を生成し、これにジハロゲノジフェニルスルホンを反応させることによって得ることができる。例えば、反応系でフェノール化合物のアルカリ金属塩を合成し、またはフェノール化合物のアルカリ金属塩を予め合成しておいて、ジハロゲノジフェニルスルホンを有機溶剤中で反応させた後、ジクロロメタンを用いて末端停止反応を行う技術が知られている。例えば、特開昭53−12991号公報、特開昭53―16098号公報には、4,4’−ヒドロキシジフェニルスルホンと、4,4’−ジクロロジフェニルスルホン、無水炭酸カリウムを用いて、芳香族ポリフェニルスルホンを製造し、反応終了後、クロロメタンを吹き込む技術が開示されている。また、特表2002−525406号公報には、溶媒で重合する際の固形分濃度を規定して、改良された多分散度をもち、かつ低分子量オリゴマーの濃度が低いポリフェニルスルホン樹脂の製造方法が記載されている。
【0017】
本発明で用いられるポリフェニルスルホン樹脂の合成に使用されるジハロゲノジフェニルスルホンとしては、例えば、4,4’−ジクロロジフェニルスルホン、4,4’−ジフルオロジフェニスルホン、4−クロロ−4’−フルオロジフェニルスルホン、4,4’−ジブロモジフェニルスルホン、1,4−ビス(4−クロロベンゼンスルホニル)ベンゼン、ビス(4’−クロロベンゼンスルホニル)エーテル、1,3−ビス(4’−クロロベンゼンスルホニル)ベンゼン、ビス(3’−クロロベンゼンスルホニル)エーテル、1,2−ビス(4’−クロロベンゼンスルホニル)ベンゼン、ビス(2’−クロロベンゼンスルホニル)エーテル、4,4’−ビス(4−クロロフェニルスルホニル)ジフェニルなどが挙げられる。
【0018】
本発明で用いられるポリフェニルスルホン樹脂の合成に使用されるフェノール化合物としては、例えば、3,3’−ジヒドロキシジフェニル、4,4’−ジヒドロキシジフェニル、3,4’−ジヒドロキシジフェニルなどのビフェニル基を有する2価のフェノール化合物が挙げられる。
【0019】
また、これ以外のフェノール化合物を、重量平均分子量3万未満を損なわない範囲で共重合してもよい。4,4,4’−トリヒドロキシトリフェニルメタン、2,2’,4,4’−テトラヒドロキシベンゾフェノン、α,α,α−トリス(4−ヒドロキシフェニル)−1−エチル−4−イソプロピルベンゼンなどの多価のフェノール化合物や、4,4’−ジヒドロキシジフェニルスルフィド、4,4’−ジヒドロキシベンゾフェノン、4,4’−ジヒドロキシジフェニルエーテル、9,9’−ビス(4−ヒドロキシフェニル)フルオレン、α,α’−ビス(4−ヒドロキシ−3,5−ジメチルフェニル)−1,4−ジイソプロピルベンゼン、2,2’−ビス(4’−ヒドロキシフェニル)プロパン、2,2−ビス(4’−ヒドロキシ−3’,5’−ジメチルフェニル)プロパン、ビス(4’−ジヒドロキシフェニル)プロパン、ビス(4’−ヒドロキシフェニル)メタン、ビス(4’−ヒドロキシフェニル)シクロヘキサン、1,1−ビス(4’−ヒドロキシフェニル)エタン、1,4−ビス(4’−ヒドロキシフェノキシ)ベンゼン、1,3−ビス(4’−ヒドロキシフェノキシ)ベンゼン、1,4−ビス(4’−ヒドロキシベンゾイル)ベンゼン、1,3−ビス(4’−ヒドロキシベンゼンスルホニル)ベンゼンなどの2価のフェノール化合物や、フェノール、o−クレゾール、m−クレゾール、p−クレゾール、2−アミノフェノール、3−アミノフェノール、4−アミノフェノール、2−アミノチオフェノール、3−アミノチオフェノール、4−アミノチオフェノール、4−ヒドロキシ安息香酸、3−ヒドロキシ安息香酸、o−エチルフェノール、m−エチルフェノール、p−エチルフェノール、2,3−ジメチルフェノール、2,4−ジメチルフェノール、2,5−ジメチルフェノール、2,6−ジメチルフェノール、3,4−ジメチルフェノール、3,5−ジメチルフェノール、o−メトキシフェノール、m−メトキシフェノール、p−メトキシフェノール、o−エトキシフェノール、m−エトキシフェノール、p−エトキシフェノール、o−n−プロピルフェノール、o−イソプロピルフェノール、m−n−プロピルフェノール、m−イソプロピルフェノール、p−n−プロピルフェノール、p−イソプロピルフェノール、o−n−ブチルフェノール、o−sec−ブチルフェノール、o−tert−ブチルフェノール、m−n−ブチルフェノール、m−sec−ブチルフェノール、m−tert−ブチルフェノール、p−n−ブチルフェノール、p−sec−ブチルフェノール、p−tert−ブチルフェノール、o−フェニルフェノール、m−フェニルフェノール、p−フェニルフェノール、2,3,5−トリメチルフェノール、2,3,6−トリメチルフェノール、2,4,6−トリメチルフェノール、o−ベンジルフェノール、m−ベンジルフェノール、p−ベンジルフェノール、o−ブトキシフェノール、m−ブトキシフェノール、p−ブトキシフェノール、6−tert−ブチル−p−キシレノール、2−tert−ブチル−p−キシレノール、2−tert−ブチル−p−クレゾール、o−シクロヘキシルフェノール、m−シクロヘキシルフェノール、p−シクロヘキシルフェノール、2,6−ジ−tert−ブチル−p−クレゾール、4,6−ジ−tert−ブチル−4−ヒドロキシメチルフェノール、2,4−ジメトキシフェノール、2,6−ジメトキシフェノール、バニリン、クミルフェノールなどの1価のフェノール化合物などが挙げられる。
【0020】
ポリフェニルスルホン樹脂の重量平均分子量を3万未満にするためには、ジハロゲノジフェニルスルホンを2価のフェノール化合物に対して0.95〜0.75当量使用することが好ましい。
【0021】
本発明で用いられるポリフェニルスルホン樹脂の合成に使用されるアルカリ金属塩のアルカリ金属としては、リチウム、ナトリウム、カリウム、ルビジウムが挙げられ、カリウムまたはナトリウムが好ましい。ジアルカリ塩を形成させるために用いられる金属化合物としては、水酸化物、炭酸塩、炭酸水素塩が挙げられ、水酸化物または炭酸塩が好ましい。したがって、ビスフェノール類のジアルカリ塩を形成させるためには、水酸化カリウム、水酸化ナトリウム、炭酸カリウムまたは炭酸ナトリウムが好ましく、これらを2種以上用いてもよい。これらアルカリ金属塩の量はその種類によって差があるが、2価のフェノール化合物を基準にその1倍モル以上が好ましく、2倍モル以上がより好ましい。この範囲であれば、ビスフェノールのアルカリ金属塩を十分生成させることができ、重量平均分子量3万未満の高分子量の重合体を容易に得ることができる。一方、副反応を抑制する観点からは、8倍モル以下が好ましく、4倍モル以下がより好ましい。
【0022】
重合温度は100〜300℃の範囲が好ましい。温度を100℃以上とすることで、未反応のモノマーのない重合体を容易に得ることができる。また、300℃以下とすることで、ポリマーの副反応を抑制することができる。重合時間は2〜4時間程度が好ましい。
【0023】
反応溶媒として用いる有機溶媒は、非プロトン性極性溶媒が好ましい。例えば、ジメチルスルホキシド、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチル−2−ピロリドン、1,4−ジメチルベンダゾリジノン、ヘキサメチルトリアミド、1,3−ジメチルイミダゾリジノンなどが挙げられる。また、これらにトルエン、キシレンなどの芳香族炭化水素系溶媒、メチルエチルケトン、メチルイソブチルケトン、シクロヘキサノンなどのケトン系溶媒、プロピレングリコールモノメチルエーテルアセテート、メチル−メトキシブタノールアセテートなどのエステル系溶媒などを加えることもできる。
【0024】
重縮合で使用される溶媒の量は、全モノマーの重量に対して0.5倍以上が好ましく、2倍以上がより好ましい。溶媒の量を全モノマーの重量に対して0.5倍以上とすることにより、撹拌などの操作が容易となり、重縮合反応が順調に進行し易くなる。一方、20倍以下が好ましく、8倍以下がより好ましい。20倍以下とすることによって、溶媒中のモノマー濃度が高くなり重合速度が向上するため、未反応のモノマーがなく重量平均分子量3万未満の重合体を容易に得ることができる。
【0025】
上記方法により得られるポリフェニルスルホン樹脂溶液は、重合後に水、メタノール、エタノール、イソプロピルアルコール、アセトン、トルエン、キシレンなどの貧溶媒中に投入してポリフェニルスルホン樹脂を析出させることが望ましい。貧溶媒の使用量に制限はないが、好ましくは重合に使用した溶媒重量の5〜100倍、より好ましくは10〜50倍である。析出したポリフェニルスルホン樹脂粉末は、本発明のアンダーフィル剤では無溶剤系とするため、溶濾過、洗浄し、乾燥して用いることが好ましい。乾燥条件は、大気圧条件下で50℃〜100℃の温度範囲で5〜50時間乾燥させるのが好ましく、またより乾燥を十分にさせるべく真空圧条件下での乾燥工程も好ましく挙げられる。
【0026】
重量平均分子量が3万未満のポリフェニルスルホン樹脂は、前記のように公知の技術で重合することもできるが、市販のポリエーテルスルホン樹脂および、ポリフェニルスルホン樹脂に2価のフェノール化合物と金属塩を有機溶剤中で反応させることによって、重合製造することができる。
【0027】
ポリエーテルスルホン樹脂の市販品としては、“スミカエクセル(登録商標)”3600P、“スミカエクセル”4100P、“スミカエクセル”4800P、“スミカエクセル”5200P、“スミカエクセル”5003P(以上、商品名、住友化学工業(株)製)、“Ultrazon(登録商標)”E2010、“Ultrazon”E3010、“Ultrazon”E6020P(以上、商品名、BASFジャパン(株)製)、“VERADEL(登録商標)”(以上、商品名、ソルベイアドバンストポリマーズ)などが挙げられ、またポリフェニルスルホン樹脂としては“Ultrazon”P3010(以上、商品名、BASFジャパン(株)製)、“RADEL(登録商標)”, “ACUDEL(登録商標)”(以上、商品名、ソルベイアドバンストポリマーズ)が挙げられ、これらのポリエーテルスルホン樹脂および、ポリフェニルスルホン樹脂を2種以上含有してもよい。
【0028】
また、本発明において、(a)成分のポリフェニルスルホン樹脂の含有量は、5重量%以上が好ましく、8重量%以上がより好ましい。5重量%以上とすることで、エポキシ樹脂が本来有するもろさや硬さを改善することができる。一方、粘度をより低減して浸透性を向上させる観点から、20重量%以下が好ましく、15重量%以下がより好ましい。
【0029】
また、本発明において、(a)成分のポリフェニルスルホン樹脂は末端にフェノール性水酸基、フルオロ基、クロロ基、ブロモ基、メトキシ基、エトキシ基などを有してもよく、これらの基を2種以上有してもよい。これらの中でも、フェノール性水酸基を有することが望ましい。フェノール性水酸基を有することでエポキシ樹脂のオキシラン環またはエポキシ樹脂の反応性基と反応し、密度の高い網目構造を形成するため、得られる硬化物は、強靭性を発現する。
【0030】
末端のフェノール性水酸基量は、核磁気共鳴(1H−NMR)法から析出したピークの積分値を求めることによって測定することができる。樹脂中の繰り返し単位を除く末端官能基量中、フェノール性水酸基を5モル%以上含有することが好ましく、30モル%以上がより好ましい。フェノール性水酸基を5モル%以上有することにより、エポキシ樹脂との親和性が向上し、硬化後の耐熱性や強靭性が向上する。一方、90モル%以下が好ましく、80モル%以下がより好ましい。90モル%以下とすることで経時による粘度上昇がなく、保存安定性がより良好となる。
【0031】
本発明で用いるポリフェニルスルホン樹脂は、フレーク状またはパウダー状のものが好ましく、パウダー状のものがより好ましい。パウダー状のポリフェニルスルホン樹脂は、ペレット状またはフレーク状の樹脂を凍結粉砕することで容易に得ることができる。パウダー状のポリフェニルスルホン樹脂の粒子径は100μm以下が好ましく、30μm以下がより好ましい。100μm以下とすることで、ポリフェニルスルホン樹脂の粒子とエポキシ樹脂を、容易に溶解させることができる。
【0032】
本発明のアンダーフィル剤は、(b)エポキシ樹脂を含有する。エポキシ樹脂は、他の化合物との熱反応性が良好であり、硬化後の接着強度が高く、熱収縮の少ない成型物を与えることができる。
【0033】
本発明における(b)エポキシ樹脂の好ましい態様の他の一つとして、SP値が1.60×104〜1.80×104(J/m3)1/2であるエポキシ樹脂と、SP値が2.00×104〜2.60×104(J/m3)1/2であるエポキシ樹脂とをそれぞれ一種以上含有することが挙げられる。本発明では(a)重量平均分子量3万未満のポリフェニルスルホン樹脂と、上記のエポキシ樹脂を用いることによって、硬化後の強靭性を向上させることができる。
【0034】
SP値(溶解性パラメーター値)の求め方は各種あるが、本発明においては、文献「ポリマー・エンジニアリング・アンド・サイエンス(R.F.Fedors,Polymer.Eng.,14,(2)147−154(1974)」に記載された方法により求める。すなわち、求める化合物の構造式において、原子および原子団の蒸発エネルギーとモル体積のデータより次式にて計算する。
【0035】
【数1】

【0036】
上記式において、Δeiは原子または基に帰属する25℃における蒸発エネルギー、Δviは25℃におけるモル体積を示し、分子中のi個の原子および基に与えられた一定の数値である。代表的な原子および基のΔeiおよびΔviは、前記文献に記載された値を用いる。
【0037】
強靭性を向上させるための一つの方法として、硬化後の樹脂組成物が海島構造を有することが知られている。SP値が2.00×104〜2.60×104(J/m3)1/2であるエポキシ樹脂は、本発明における(a)成分の分子量3万未満のポリフェニルスルホン樹脂と相溶性が高く、硬化後にエポキシ樹脂とポリフェニルスルホン樹脂とが均一な相を形成しやすい。一方で、SP値が1.60×104〜1.80×104(J/m3)1/2であるエポキシ樹脂は、本発明の(a)成分の分子量3万未満のポリフェニルスルホン樹脂との相溶性が低く、硬化後にエポキシ樹脂とポリフェニルスルホン樹脂が相分離構造を形成しやすい。このため硬化後にポリフェニルスルホン樹脂とエポキシ樹脂間で相分離構造が形成されやすく、硬化後の強靭性を向上させることができる。さらに、前述のSP値が2.00×104〜2.60×104(J/m3)1/2であるエポキシ樹脂は、耐熱性を向上させることができ、前述のSP値が1.60×104〜1.80×104(J/m3)1/2であるエポキシ樹脂は、反応希釈剤として容易に低粘度特性を向上させることができる。以上のことから、これらのエポキシ樹脂をそれぞれ一種以上含有することにより、硬化後の強靭性と耐熱性を向上させることができ、また、より容易に低粘度特性を得ることができる。
【0038】
SP値が1.60×104〜1.80×104(J/m3)1/2であるエポキシ樹脂としては、2官能型の脂肪族あるいは脂環式グリシジルエーテル型エポキシ樹脂が好ましく、EX−212L(SP値=8.25(cal/cm3)1/2=1.69×104(J/m3))1/2)、EX−214L(SP値=8.28(cal/cm3)1/2=1.69×104(J/m3)1/2)、EX−216L(SP値=7.93(cal/cm3)1/2=1.62×104(J/m3)1/2)、EX−850L(SP値=8.42(cal/cm3)1/2=1.72×104(J/m3)1/2)(以上、商品名、ナガセケムテックス(株)製)、“エピクロン(登録商標)”705(SP値=8.10(cal/cm3)1/2=1.66×104(J/m3)1/2)、“エピクロン”707(SP値=7.98(cal/cm3)1/2=1.63×104(J/m3)1/2)、“エピクロン”720(SP値=8.45(cal/cm3)1/2=1.73×104(J/m3)1/2)、“エピクロン”726(SP値=8.25(cal/cm3)1/2=1.69×104(J/m3)1/2)、EXA−4880(SP値=8.35(cal/cm3)1/2=1.71×104(J/m3)1/2)、EXA−4882(SP値=7.98(cal/cm3)1/2=1.63×104(J/m3)1/2)(以上、商品名、DIC(株)製)、エポライト40E(SP値=8.33(cal/cm3)1/2=1.70×104(J/m3)1/2)、エポライト100E(SP値=8.42(cal/cm3)1/2=1.72×104(J/m3)1/2)、エポライト200E(SP値=8.50(cal/cm3)1/2=1.74×104(J/m3)1/2)、エポライト400E(SP値=8.58(cal/cm3)1/2=1.76×104(J/m3)1/2)、エポライト70P(SP値=8.13(cal/cm3)1/2=1.66×104(J/m3)1/2)、エポライト200P(SP値=8.10(cal/cm3)1/2=1.66×104(J/m3)1/2)、エポライト400P(SP値=7.98(cal/cm3)1/2=1.63×104(J/m3)1/2)、エポライト1500NP(SP値=8.45(cal/cm3)1/2=1.73×104(J/m3)1/2)、エポライト1600(SP値=8.25(cal/cm3)1/2=1.69×104(J/m3)1/2)(以上、商品名、共栄社化学(株)製)、リカレジンDME−100(SP値=7.93(cal/cm3)1/2=1.62×104(J/m3)1/2)、(商品名、新日本理化(株)製)、YED216(SP値=8.25(cal/cm3)1/2=1.69×104(J/m3)1/2)、YED216D(SP値=8.25(cal/cm3)1/2=1.69×104(J/m3)1/2)、YED216M(SP値=8.25(cal/cm3)1/2=1.69×104(J/m3)1/2)(以上、商品名、ジャパンエポキシレジン(株)製)などが挙げられる。これらを2種以上含有してもよい。
【0039】
SP値が2.00×104〜2.60×104(J/m3)1/2であるエポキシ樹脂としては、低粘度性を有する観点から2官能型のビスフェノールF型、ビスフェノールA型エポキシ樹脂が好ましい。また、耐熱性の観点からは、フェノールノボラック型、クレゾールノボラック型エポキシ樹脂、ナフタレン型エポキシ樹脂、ビフェニル型エポキシ樹脂が好ましい。
【0040】
2官能型のビスフェノールF型エポキシ樹脂としては、“エピクロン(登録商標)”830(SP値=11.47(cal/cm3)1/2=2.35×104(J/m3)1/2)(以上、商品名、DIC(株)製)、RE−303S(SP値=11.47(cal/cm3)1/2=2.35×104(J/m3)1/2)(以上、商品名、日本化薬(株)製)、“jER(登録商標)”806(SP値=11.50(cal/cm3)1/2=2.35×104(J/m3)1/2)、“jER”807(SP値=11.50(cal/cm3)1/2=2.35×104(J/m3)1/2)(以上、商品名、ジャパンエポキシレジン(株)製)、2官能型のビスフェノールA型エポキシ樹脂としては、840(SP値=12.31(cal/cm3)1/2=2.52×104(J/m3)1/2)、850(SP値=12.31(cal/cm3)1/2=2.52×104(J/m3)1/2)、860(SP値=12.31(cal/cm3)1/2=2.52×104(J/m3)1/2)((以上、商品名、DIC(株)製)、RE−310S(SP値=12.31(cal/cm3)1/2=2.52×104(J/m3)1/2)(以上、商品名、日本化薬(株)製)、“jER”827(SP値=12.31(cal/cm3)1/2=2.52×104(J/m3)1/2)、“jER”828(SP値=12.31(cal/cm3)1/2=2.52×104(J/m3)1/2)、“jER”834(SP値=12.35(cal/cm3)1/2=2.53×104(J/m3)1/2)(以上、商品名、ジャパンエポキシレジン(株)製)、フェノールノボラック型エポキシ樹脂としては、“エピクロン”N−540(SP値=11.26(cal/cm3)1/2=2.30×104(J/m3)1/2)、N−740、N−770(いずれもSP値=11.30(cal/cm3)1/2=2.30×104(J/m3)1/2)、(以上、商品名、DIC(株)製)、EPPN−201(SP値=11.26(cal/cm3)1/2=2.30×104(J/m3)1/2)(以上、商品名、日本化薬(株)製)、“jER”152、154(いずれもSP値=11.30(cal/cm3)1/2=2.30×104(J/m3)1/2)、クレゾールノボラック型エポキシ樹脂としては、“エピクロン”N−660、N−665、N−670、N−680、N−690(いずれもSP値=10.28(cal/cm3)1/2=2.10×104(J/m3)1/2)(以上、商品名、DIC(株)製)、EOCN−1020(SP値=10.40(cal/cm3)1/2=2.13×104(J/m3)1/2)、EOCN−102S(SP値=10.44(cal/cm3)1/2=2.14×104(J/m3)1/2)、EOCN−103S(SP値=10.48(cal/cm3)1/2=2.14×104(J/m3)1/2)、EOCN−104S(SP値=10.52(cal/cm3)1/2=2.15×104(J/m3)1/2)、(以上、商品名、日本化薬(株)製)、ナフタレン型エポキシ樹脂としては、“エピクロン”HP4700、EXA4710、HP4770(いずれもSP値=10.09(cal/cm3)1/2=2.06×104(J/m3)1/2)、(以上、商品名、DIC(株)製)、ビフェニル型エポキシ樹脂としては“jER”YX4000(SP値=9.89(cal/cm3)1/2=2.02×104(J/m3)1/2)、YL6121H(SP値=11.44(cal/cm3)1/2=2.34×104(J/m3)1/2)、(以上、商品名、ジャパンエポキシレジン(株)製)、NC3000、NC3000H(いずれもSP値=11.23(cal/cm3)1/2=2.30×104(J/m3)1/2)、(以上、商品名、日本化薬(株)製)などが挙げられる。これらを2種以上含有してもよい。
【0041】
さらに、(b)エポキシ樹脂に加えて、オキセタン樹脂を含有することができる。オキセタン樹脂を含有することで、熱安定性が向上するため、硬化後の耐熱性を向上させることができる。このようなオキセタン樹脂のうち、1官能性オキセタン樹脂としては、エタナコール(登録商標)”EHO、“エタナコール”OXMA、(以上、商品名、宇部興産(株)製)、OXT−101、OXT−211、OXT−212、OXT−610(以上、商品名、東亜合成(株)製)、3−エチル−3−(シクロヘキシロキシ)メチルオキセタンが挙げられ、2官能性オキセタン樹脂としては、“エタナコール“OXBP、“エタナコール”OXTP、“エタナコール”OXIPA(以上、商品名、宇部興産(株)製)、OXT−121、OXT−221(以上、商品名、東亜合成(株)製)、3つ以上のオキセタニル基を有する樹脂としては、オキセタン化フェノール樹脂や、オキセタニルシリケート、フェノールノボラック型オキセタン樹脂が挙げられる。これらを2種以上含有してもよい。
【0042】
本発明のアンダーフィル剤は、粘度を調節したり、塗布性を向上する目的で、(c)溶剤を1重量%以下含有することもできる。溶剤含有量を1重量%以下とすることで、熱処理時の溶媒飛散によるボイドの発生を抑制することができる。
【0043】
溶剤としては、例えば、ケトン系溶剤のアセトン、メチルエチルケトン、メチルイソブチルケトン、シクロペンタノン、シクロヘキサノン、エーテル系溶剤の1,4−ジオキサン、テトラヒドロフラン、ジグライム、グリコールエーテル系溶剤のメチルセロソルブ、エチルセロソルブ、プロピレングリコールモノメチルエーテル、プロピレングリコールモノエチルエーテル、プロピレングリコールモノブチルエーテル、ジエチレングリコールメチルエチルエーテル、芳香族炭化水素系溶剤のベンゼン、キシレン、トルエン、その他ベンジルアルコール、プロパノール、N−メチルピロリドン、γ−ブチロラクトン、酢酸エチル、N,N−ジメチルホルムアミドなどが挙げられる。これらを2種以上含有してもよい。特に大気圧下沸点が120℃以下であるものを用いると、低温、短時間で脱溶媒できるため好ましい。
【0044】
本発明のアンダーフィル剤は、(d)シリカ、チタニア、ジルコニア、窒化ケイ素、アルミナ、セリア、タルクおよび炭酸カルシウムからなる群より選ばれる少なくとも一種の無機微粒子を含有することができる。これらの無機微粒子を含有することにより、熱伝導性を付与したり、硬化後の熱線膨張係数を低減することができる。熱伝導性を付与する場合は、炭酸カルシウム、シリカ、アルミナ、チタニア、シリカ−チタニア複合粒子が好ましい。さらに、無機微粒子とエポキシ樹脂の界面を強固に結合するために、無機微粒子表面をシラン系、チタン系、アルミニウム系などの各種カップリング剤、脂肪酸、リン酸エステルなどで処理したものや、ロジン処理、酸性処理、塩基性処理を施したものも好適に用いられる。特にエポキシ基含有シランカップリング剤は、強靭性を高める点から好ましく用いられる。シランカップリング剤の好ましい例としては、γ−グリシドキシトリメトトキシシラン、γ−グリシドキシトリエトトキシシラン、γ−グリシドキシトリプロポキシシラン、2−(3,4−エポキシシクロヘキシル)エチルトリメトキシシラン、2−(3,4−エポキシシクロヘキシル)エチルトリエトキシシラン、2−(3,4−エポキシシクロヘキシル)エチルトリプロポキシシラン、3−アミノプロピルトリメトキシシラン、3−アミノプロピルトリエトキシシラン、3−アミノプロピルトリプロポキシシランなどが挙げられる。これらを2種以上用いてもよい。
【0045】
これら(d)無機微粒子の含有量は、(a)成分のポリフェニルスルホン樹脂と(b)成分のエポキシ樹脂との総量100重量部に対して10〜5000重量部が好ましい。無機微粒子の含有量が10重量部以上であることで、熱伝導性を向上させたり、熱線膨張係数をより低減したりすることができ、5000重量部以下とすることでアンダーフィル剤の粘度上昇を防ぐことができる。より好ましくは20重量部以上500重量部以下である。
【0046】
また、無機微粒子の平均粒子径は5nm〜30μmが好ましく、さらに好ましくは5nm〜10μmである。ここで、本発明における平均粒子径は、超薄切片法を用いて調製した厚さ20μmのコーティング被膜の断面を、透過型電子顕微鏡(日立製作所(株)製、H−7100FA型)を用いて、加速電圧100kVの条件にて観察倍率200000倍で観察し、得られた像から個々の粒子単一の粒子径を測定し、それらの数平均値とする。測定に用いる粒子の個数は数百から数千個である。平均粒子径が5nm以上であることで、適度な粘性を付与すると同時に安定した粒子分散性を得ることができ、30μm以下とすることで、低粘度特性を向上させ、無機微粒子の沈降を防ぐことができる。
【0047】
本発明のアンダーフィル剤は、(e)ポリイミド、ポリアミド、ポリアミドイミド、ポリスチレン、ポリアクリロニトリル、ポリフェニレンエーテル、ポリエステルおよびポリカーボネートからなる群より選ばれる少なくとも一種の有機微粒子を含有することができる。これらの有機微粒子を含有することにより、耐熱性や低吸湿性を付与することができる。
【0048】
これら(e)有機微粒子の含有量は、(a)成分のポリフェニルスルホン樹脂と(b)成分のエポキシ樹脂との総量100重量部に対して5〜5000重量部が好ましい。有機微粒子の含有量が5重量部以上であることで、耐熱性や低吸湿性をより容易に付与することができ、5000重量部以下とすることで樹脂組成物の粘度上昇を防ぐことができる。
【0049】
また、有機微粒子の平均粒子径は5nm〜10μmが好ましく、さらに好ましくは5nm〜5μmである。有機微粒子の平均粒子径の測定方法は、上記の無機微粒子の平均粒子径の測定方法と同様である。
【0050】
その他にフェノキシ樹脂、ポリウレタン、ポリプロピレン、アクリロニトリル−ブタジエン共重合体(NBR)、スチレン−ブタジエン共重合体、(SBR)、アクリロニトリル−ブタジエン−メタクリル酸共重合体、アクリロニトリル−ブタジエン−アクリル酸共重合体などを含有してもよい。
【0051】
本発明のアンダーフィル剤は、さらに硬化剤や硬化促進剤を含有することができる。ここで、硬化剤とは、単独で硬化する作用を有するものをいい、硬化促進剤とは、硬化剤とともに用いて反応を促進する作用を有するものをいう。本発明のアンダーフィル剤に用いることのできる硬化剤または硬化促進剤としては、脂肪族ポリアミン、脂環式ポリアミン、芳香族ポリアミン、酸無水物、フェノール樹脂、ジシアンジアミドとその誘導体、イミダゾール類とその誘導体、アミン誘導体とホルムアルデヒドの縮合体(尿素ホルムアルデヒド、メラミンホルムアルデヒド)、3級アミン化合物、有機金属錯体、ポリチオール類、オニウム塩類などが挙げられる。これらを2種以上含有してもよい。
【0052】
具体的には、脂肪族ポリアミンとしては、ジエチルトリアミン、トリエチレンテトラミン、キシレンジアミンなど、脂環式ポリアミンとしては、イソホロンジアミン、1,3−ビスアミノメチルシクロヘキサン、ノルボルネンジアミンなど、芳香族ポリアミンとしては、メタフェニレンジアミン、ジアミノジフェニルメタン、ジアミノジフェニルスルホン、ビスアミノフェノキシベンゼンなどのフェノキシ系ジアミンなどが挙げられる。また、酸無水物としては、無水こはく酸、イタコン酸無水物、無水フタール酸、ピロメリット酸二無水物、ベンゾフェノンテトラカルボン酸二無水物、ビフェニルテトラカルボン酸二無水物、メチルヘキサヒドロフタル酸無水物、ヘキサハイドロフタル酸無水物、メチルテトラハイドロフタル酸無水物、“アデカハードナー(登録商標)”EH−3326、“アデカハードナー”EH−703、“アデカハードナー”EH−705A(以上、商品名、旭電化工業(株)製)、“エピクロン”B−570、“エピクロン”B−650(以上、商品名、大日本インキ化学(株)製)、“リカシッド(登録商標)”MH−700(商品名、新日本理化(株)製)などが挙げられる。フェノール樹脂としては、各種フェノールノボラック樹脂やクレゾールノボラック樹脂、MEH−7500、MEH−7800、MEH−7851、MEH−8000H(以上、商品名、明和化成(株)製)などが挙げられる。ジシアンジアミドとその誘導体としては、DICY7、DICY15、DICY50(以上、商品名、ジャパンエポキシレジン(株)製)、“アミキュア(登録商標)”AH−154、“アミキュア”AH−162(以上、商品名、味の素ファインテクノ(株)製)などが挙げられる。イミダゾール類とその誘導体としては、イミダゾール、2−メチルイミダゾール、2−ウンデシルイミダゾール、2−エチル−4−メチルイミダゾール、2−フェニル−4−メチルイミダゾール、2−フェニル−4−メチル−5−ヒドロキシイミダゾール、イミダゾール、2−フェニルイミダゾール、1−ベンジル−2−メチルイミダゾール、1−ベンジル−2−フェニルイミダゾール、1−シアノエチル−2−メチルイミダゾール、1−シアノエチル−2−エチル−4−メチルイミダゾール、1−シアノエチル−2−フェニルイミダゾール、1−シアノエチル−2−フェニルイミダゾリウムトリメリテイトや、IS−1000、IS−1000D、IM−1000、SP−1000、IA−100A、IA−100P、IA−100F(以上、商品名、日鉱マテリアルズ(株)製)などのイミダゾールシランなどが挙げられる。アミン誘導体とホルムアルデヒドの縮合体(尿素ホルムアルデヒド、メラミンホルムアルデヒド)としては、4−クロロ−フェニル−N,N−ジメチル尿素、3,4−ジクロロフェニル−N,N−ジメチル尿素(DCMU)などが挙げられる。3級アミン化合物としては、1,8−ジアザビシクロ(5,4,0)−ウンデセン−7(DBU)、1,5−ジアザビシクロ(4,3,0)−ノネン−5(DBN)およびその塩が挙げられ、具体的には、U−CATSA1(DBU−フェノール塩)、U−CAT SA102(DBU−オクチル酸塩)、U−CATSA603(DBU−蟻酸塩)、U−CAT SA810(DBU−オルソフタル酸塩)、U−CATSA841(DBU−フェノールノボラック樹脂塩)、U−CAT SA881(DBN−フェノールノボラック樹脂塩)(以上、商品名、サンアプロ(株)製)などが挙げられる。有機金属錯体としては、トリフェニルホスフィン、トリフェニルホスホニウムトリフェニルボレートなど、ポリチオール類としては、脂肪族ポリチオエーテル、脂肪族ポリチオエステル、芳香族環含有ポリチオエーテルなどが挙げられる。オニウム塩類としては、スルホニウムやヨードニウムなどのオニウム塩が挙げられ、オニウム塩型のジフェニルヨードニウムヘキサフロロホスフェート、トリフェニルスルホニウムヘキサフロロホスフェート、サイラキュアーUVI−6992、サイラキュアーUVI−6974(以上、商品名、ダウ・ケミカル日本(株)製)、アデカオプトマーSP150、アデカオプトマーSP170(以上、商品名、旭電化工業(株)製)、サンエイドSI−60L、SI−80L、SI−100L、SI−150L(以上、商品名、三新化学工業(株)製)などが挙げられる。
【0053】
その他に、エポキシ樹脂に混合した状態で長時間保存でき、熱・光・圧力・湿気などの刺激を与えると硬化反応を開始する潜在性硬化剤の例としては、加熱硬化型潜在性硬化剤、マイクロカプセル型潜在性硬化剤、アミンアダクト型潜在性硬化剤などが挙げられる。加熱硬化型潜在性硬化剤の具体例としては、ジシアンジアミド、3,4−ジクロロフェニル−N,N−ジメチル尿素(DCMU)が挙げられる。マイクロカプセル型潜在性硬化剤は、コア(芯物質)/シェル(カプセル膜)構造を有する硬化剤である。コアとしては、種々のイミダゾール化合物やトリフェニルホスフィンなど、シェルとしては、有機ポリマーや無機化合物などが挙げられる。具体的には、“ノバキュア(登録商標)”HX−3941HP、“ノバキュア”HXA3922HP、“ノバキュア”HXA3932HP、“ノバキュア”HXA3042HP(以上商品名、旭化成ケミカルズ(株)製)などが挙げられる。
【0054】
アミンアダクト型潜在性硬化剤は、イミダゾール化合物、3級アミノ基含有化合物またはヒドラジド化合物をエポキシ樹脂やイソシアネート化合物などと反応させて高分子量化したものを微粉砕化したものであり、常温での溶解度が低く、潜在性を示す。具体的には、“アミキュア”PN−23、“アミキュア”PN−40、“アミキュア”MY−24、“アミキュア”MY−H(以上、商品名、味の素ファインテクノ(株)製)、“フジキュア(登録商標)”FXR−1030(商品名、富士化成(株)製)、“アミキュア”VDH、“アミキュア”UDH(以上、商品名、味の素ファインテクノ(株)製)などが挙げられる。
【0055】
本発明においては、アンダーフィル剤への硬化性と保存安定性が良好な点から、芳香族ポリアミンがより好ましい。このような芳香族ポリアミンの例としては、メタフェニレンジアミン、パラフェニレンジアミン、2,4−トルエンジアミン、2,5−トルエンジアミン、ジメチルトルエンジアミン、ジエチルトルエンジアミン、4,4’−ジアミノジフェニルメタン、4,4’−ジアミノ−3,3’−ジメチル−ジフェニルメタン、3,3’−ジアミノ−4,4’−ジメチル−ジフェニルメタン、4,4’−ジアミノ−3,3’−ジクロロ−ジフェニルメタン、3,3’−ジアミノ−4,4’−ジクロロ−ジフェニルメタン、4,4’−ジアミノ−3,3’,5,5’−テトラメチル−ジフェニルメタン、4,4’−ジアミノ−3,3’−ジエチル−ジフェニルメタン、4,4’−ジアミノ−3,3’,5,5’−テトラエチル−ジフェニルメタン、3,3’−ジアミノジフェニルスルホン、4,4’−ジアミノジフェニルスルホン、3,3’−ジアミノジフェニルスルフィド、4,4’−ジアミノジフェニルスルフィド、ビス(p−(m−アミノフェノキシ)フェニル)スルホン、ビス(p−(p−アミノフェノキシ)フェニル)スルホン、3,3’−ビス(p−アミノフェノキシ)ビフェニル、2,2’−ビス(p−アミノフェノキシ)フェニル)−1,1,1,3,3,3−ヘキサフルオロプロパン、4,4’−ビス(m−アミノフェノキシ)ビフェニル、m−ビス(p−アミノフェノキシ)ベンゼン、p−ビス(p−アミノフェノキシ)ベンゼン、m−ビス(m−アミノフェノキシ)ベンゼン、1,3−ビス(4−アミノ−3−ヒドロキシフェノキシ)ベンゼン、p−ビス(p−アミノフェノキシ)ジフェニルエーテル、p−ビス(m−アミノフェノキシ)ジフェニルエーテル、ビス(p−(p−アミノフェノキシ)フェノキシ)ベンゼンなどが挙げられる。この中でも高い融点を持ち、保存安定性が良好な点から芳香族スルホン系ジアミンが好ましく用いられる。芳香族スルホン系ジアミンとしては、3,3’−ジアミノジフェニルスルホン、4,4’−ジアミノジフェニルスルホンが挙げられ、市販品としてはセイカキュアS(商品名、セイカ(株)製)が挙げられる。
【0056】
さらに好適な硬化剤としては、フェノキシ系ジアミンが挙げられる。フェノキシ系ジアミンは、エーテル結合を有し、硬化後の樹脂の靭性を向上させることができる。さらに、下記一般式(1)で表されるフェノキシ系ジアミンは、室温および高温におけるアンダーフィル剤の粘度を大きく下げる作用を有する。
【0057】
【化1】
【0058】
上記一般式(1)中、nは2〜4の整数を表す。R1〜R4は水素原子、水酸基、ハロゲン原子、炭素数1〜3のアルキル基、アルコキシ基、フルオロアルキル基またはアリール基を表し、同じでも異なっていてもよい。アルキル基は鎖状でも環状でもよい。
【0059】
前記一般式(1)で表されるフェノキシ系ジアミンは、特に、SP値が1.60×104〜1.80×104(J/m3)1/2であるエポキシ樹脂と、SP値が2.00×104〜2.60×104(J/m3)1/2であるエポキシ樹脂からそれぞれ一種類以上選ばれてなるエポキシ樹脂と組み合わせて用いることが好ましく、硬化後の強靭性を向上させることができる。前記一般式(1)で表されるフェノキシ系ジアミンとしては、m−ビス(p−アミノフェノキシ)ベンゼン、p−ビス(p−アミノフェノキシ)ベンゼン、m−ビス(m−アミノフェノキシ)ベンゼン、3,3’−ビス(3”−アミノフェノキシ)ジフェニルエーテル、4,4’−ビス(3”−アミノフェノキシ)ジフェニルエーテル、ビス(p−(p−アミノフェノキシ)フェノキシ)ベンゼン、ビス(m−(m−アミノフェノキシ)フェノキシ)ベンゼン、1,3−ビス(4−アミノ−3−ヒドロキシフェノキシ)ベンゼンなどが挙げられ、市販品として、APB、APB−N、APB−NP、APB−5(以上、商品名、三井化学(株)製)、TPE−R、TPE−Q(以上、商品名、和歌山精化工業(株)製)、4−APB、3,4’−DAPE、3−APB、AHPB(以上、商品名、日本純良薬品(株)製)などが挙げられる。
【0060】
その他に本発明で好ましく用いられる硬化促進剤としては、ポリフェニルスルホン樹脂とエポキシ樹脂に常温で非相溶で混合できることから、加熱硬化型潜在性硬化剤である3−フェニル−N、N−ジメチル尿素、4−クロロフェニル−N,N−ジメチル尿素、3,4−ジクロロフェニル−N,N−ジメチル尿素、3−クロロ−4−メチルフェニル−N,N−ジメチル尿素などが挙げられる。
【0061】
本発明のアンダーフィル剤において、硬化剤の含有量は(a)成分のポリフェニルスルホン樹脂と(b)エポキシ樹脂との総量100重量部に対して0.1〜60重量部が好ましい。硬化剤の含有量を0.1重量部以上とすることでエポキシ樹脂の硬化を効果的に行い、60重量部以下とすることで硬化物の高粘度化を防ぐことができる。より好ましくは20〜40重量部である。
【0062】
硬化剤の形状は、液状、固体状でも特に限定されないが、固体状の場合は、分散性や溶解性の点から粉末状が好ましい。
【0063】
本発明の熱硬化性樹脂組成物は、さらに熱架橋剤を含有してもよい。熱架橋剤としては、例えば、エチニル基、ビニル基、メチロール基、メトキシメチロール基などを有する化合物や、ベンゾオキサジン化合物を挙げることができる。さらに、無水マレイン酸、エチニルフタール酸無水物、エチニルアニリン、ビニルアニリンなどのように熱架橋基とエポキシ樹脂の硬化機能を有する化合物を含有してもよい。
【0064】
そのほか、本発明のアンダーフィル剤は、ノニオン性、カチオン性、アニオン性の界面活性剤、多価カルボン酸などの湿潤剤、両親和性物質、高立体障害の置換基を有する樹脂などを含有してもよい。また、必要に応じて、安定化剤、分散剤、沈降防止剤、可塑剤、酸化防止剤などを含有してもよい。
【0065】
一般的にアンダーフィル剤として使用する場合、基板温度である90〜140℃の温度範囲内で、粘度の最低値が1.0×10Pa・s以下であると、半導体素子と基板の隙間に速やかに充填することができる。このため、本発明のアンダーフィル剤は、動的粘弾性測定(昇温速度2℃/分)により得られる粘弾性−温度曲線において、粘度の最低値η*が1.0×102Pa・s以下であり、かつ粘度の最低値を示す温度が90〜140℃であることが好ましい。さらに粘度の最低値η*が1.0Pa・s以下であることがより好ましく、また粘度の最低値を示す温度が100〜120℃であることがより好ましい。
【0066】
アンダーフィル剤の粘度は、平行円板型の測定システムを用いた動的粘弾性測定により求める。平行円板型の場合、両円板の間に試料を満たし、一方の円板を一定の周波数で振動させ、その際に他方の円板に生じるトルクと位相差から剛性率、粘度、tanδを求める。ここでいう粘度とは、測定によって得られる複素粘性率を示し、単位はPa・sである。本発明においては、振幅0.5deg、角周波数3s−1、周波数0.5Hz、測定温度25℃〜170℃、昇温速度2℃/分、N2ガス気流中の条件で測定する。
【0067】
このような粘度特性を有するアンダーフィル剤を得るためには、(a)成分のポリフェニルスルホン樹脂の含有量を20重量%以下にすることが好ましく、10重量%以下がより好ましい。また、反応希釈剤として、低粘度性を有するエポキシ樹脂を用いることが好ましい。低粘度性を有するエポキシ樹脂は、25℃、1.013×105Paにおける粘度が200mPa・s未満であることが好ましい。25℃、1.013×105Paにおける粘度が200mPa・s未満のエポキシ樹脂の具体例としては、YED216M,YED216D,YED111N、YED122N(以上、商品名、ジャパンエポキシレジン(株)製)、エポライト40E、エポライト100E、エポライト200E、エポライト400E、エポライト70P、エポライト200P、エポライト400P、エポライト1500NP、エポライト1600、エポライト80MF、エポライト100MF(以上、商品名、共栄社化学(株)製)、EX−214L、EX−212L、EX−216L、EX−850L(以上、商品名、ナガセケムテックス(株)製)、“エピクロン(登録商標)”EXA−4880、“エピクロン”520、“エピクロン”703、“エピクロン”705、“エピクロン”725、“エピクロン”727、(以上、商品名、DIC(株)製)が挙げられる。この中でも具体的にはSP値が1.60×104〜1.80×104(J/m3)1/2であるエポキシ樹脂が好ましい。また、ヒドロキシメチルオキセタン、エチルオキセタンメタノール、メチルオキセタンメタノールなどの1価のオキセタン樹脂を含有することによっても粘度を低下させることができる。
【0068】
アンダーフィル剤の粘度を上記範囲に調整するその他の手法としては、硬化剤として前記一般式(1)で表されるフェノキシ系ジアミンを用いる方法が挙げられ、室温から高温にかけて粘度を大きく下げることができる。
【0069】
アンダーフィル剤の反応開始温度を90℃付近にするためには、硬化剤として脂肪族系ポリアミンや2級アミンなどのアミン類、イミダゾール類が好適に用いられる。また反応開始温度を90〜140℃付近にするためには、3級アミン、芳香族系アミン、マイクロカプセル型潜在性硬化剤が好適に用いられる。さらに140℃を越えて硬化反応を開始させたい場合は、DCMU、DCMU型潜在性硬化剤、DICY(ジシアンジアミド)、DICY型潜在性硬化剤などが好適に用いられる。また、硬化剤を2種以上含有したり、硬化促進剤と組み合わせることにより、硬化速度を調節してもよい。
【0070】
本発明のアンダーフィル剤は、硬化後の耐熱性の指標として、硬化後のガラス転移温度が90℃以上であることが好ましく、140℃以上がより好ましい。かかるガラス転移温度は、(a)重量平均分子量3万未満のポリフェニルスルホン樹脂の高いガラス転移温度や、エポキシ樹脂および硬化剤の反応基数に起因する。
【0071】
本発明のアンダーフィル剤の硬化後の破壊靭性値K1cは2.0MPa・m1/2以上であることが好ましく、さらに好ましくは2.5MPa・m1/2以上である。硬化後の破壊靭性値K1cが2.0MPa・m1/2以上であれば、本発明のアンダーフィル剤の硬化後の半導体素子と基板間の接続信頼性や耐熱衝撃性を高めることができる。
【0072】
破壊靭性値の測定・算出方法は、ASTMD5045−93[プラスチック材料の平面歪み破壊靭性及び歪みエネルギー解放率](=ISO13586)により規定されている。本発明においては、厚み6mm×幅12mm×長さ52.8mmの試験片に予備クラックを入れ、テンシロン万能試験機(エーアンドデイ(株)製)を用いて、試験温度23℃、クロスヘッド速度10mm/分、エッジスパン間隔48mmで3点曲げ試験を行い、き裂長さおよび最大破壊荷重から破壊靭性値K1cを求める。K1cの値を求める式は次の式で表され、単位はMPa・m1/2である。
K1c=PmaxSf(a/W)/BW3/2
ただし、Pmaxは最大破壊荷重[kN]、Sはスパン間距離(治具ローラー間距離)[cm]、aはき裂長さ[cm]、Wは試験片幅[cm]、Bは試験片厚さ[cm]である。またf(a/W)は、a/W=X、S/W=4.0とし、f(X)は次の式で求められる。
【0073】
【数2】
【0074】
このような硬化後の破壊靭性値を有するアンダーフィル剤は、エポキシ樹脂の強靭化剤として、ポリフェニルスルホン樹脂を含有しており、ポリフェニルスルホン樹脂の末端がエポキシ基との反応性基を有することが好ましく、より好ましくはフェノール性水酸基が挙げられる。また、強靭性と低粘度特性をより高いレベルで両立するためには、ポリフェニルスルホン樹脂の重量平均分子量が3万未満であり、さらに好ましくは重量平均分子量が2万以下、5000以上である。また(b)成分のエポキシ樹脂が、SP値が1.60×104〜1.80×104(J/m3)1/2であるエポキシ樹脂と、SP値が2.00×104〜2.60×104(J/m3)1/2であるエポキシ樹脂とをそれぞれ一種以上含有し、さらに硬化剤として、前記一般式(1)で表されるフェノキシ系ジアミンを含有することが好適である。
【0075】
本発明のアンダーフィル剤は、例えば、(a)成分のポリフェニルスルホン樹脂と(b)成分のエポキシ樹脂を混合し、均一な溶液にすることで得ることができる。
【0076】
(b)エポキシ樹脂には液状と固体状の2種類があり、固体状のエポキシ樹脂の場合は、液状のエポキシ樹脂にあらかじめ溶解させるのがよい。この場合、各固体状エポキシ樹脂の軟化点および融点を目処に、軟化点および融点の−10℃〜+30℃の温度範囲で熱溶解させることが好ましい。
(b)以外のエポキシ樹脂、有機/無機微粒子、硬化剤、硬化促進剤などを含有する場合は、前記の(a)成分のポリフェニルスルホン樹脂と(b)成分のエポキシ樹脂を混合し、溶解させた後でこれらを添加し、混合することが好ましい。
【0077】
硬化剤および硬化促進剤は液状と固体状の2種類があり、固体状の硬化剤および硬化促進剤の場合は、液状の硬化剤および硬化促進剤に溶解させても良いし、液状のエポキシ樹脂に溶解させてもよい。この場合、溶解方法は特に限定されないが、熱溶解する場合、30℃〜180℃の温度範囲で、0.5時間〜8時間撹拌することが好ましく、より好ましくは50℃〜80℃の範囲で1時間〜2時間である。
【0078】
また、硬化剤および硬化促進剤の添加順序は特に限定されないが、反応性が高い硬化剤および硬化促進剤は保存安定性を考慮して最後に添加することが好ましい。一方、反応性が低い硬化剤および硬化促進剤はあらかじめエポキシ樹脂に添加し、溶解させて用いてもよい。
【0079】
無機微粒子または有機微粒子を含有する場合、分散方法は特に限定されないが、特に分散性の点でボールミル、ホモジナイザーを用いることが好ましい。
【0080】
無機微粒子の分散性を向上させるために、例えば、分散剤や界面活性剤、溶剤を添加してもよい。分散剤の例としては、リン酸、カルボン酸、脂肪酸、およびそれらのエステル類などの酸基を有する分散剤などが挙げられる。特に、無機微粒子表面の水酸基と反応し、粒子表面を覆うことができることから、リン酸化合物が好ましく用いられる。
【0081】
本発明のアンダーフィル剤は、半導体素子と基板を接合するための接着剤(アンダーフィル剤)として好適に用いられる。その他にも、本発明のアンダーフィル剤を硬化して得られる膜を、半導体素子の保護膜、高密度実装用多層配線用の層間絶縁膜、ビルドアッププリント配線板用の層間絶縁膜、回路基板の配線保護絶縁膜などの用途に用いることもできる。
【0082】
本発明のアンダーフィル剤の充填方法としては、半導体素子と基板の空隙にアンダーフィル剤を滴下する方法などが一般的であり、その他ディスペンサー法や印刷法で塗布する方法が挙げられる。また塗布後のアンダーフィル剤の充填速度は好ましくは0.5cm/分以上、より好ましくは1.0cm/分以上である。
【0083】
本発明のアンダーフィル剤を保存する際の温度は−10〜25℃の範囲が好ましく、より好ましくは9℃以下、さらに好ましくは5℃以下である。
【0084】
本発明のアンダーフィル剤は、塗布前に予め呼び加熱処理しておき、粘度を下げてから塗布しても差し支えない。加熱温度は30℃〜60℃が好ましい。60℃を超えると加熱によりエポキシ樹脂の硬化反応が始まり粘度が上昇する場合がある。また、30℃を下回ると、粘度が下がらない場合がある。また過熱処理後は保存安定性の観点から0.1秒〜30分の間に塗布することが好ましい。
【0085】
本発明のアンダーフィル剤を半導体素子と基板の空隙に充填し、硬化させる。アンダーフィル剤の硬化条件としては、通常は室温以上250℃以下の温度で0.5分間〜10時間の処理を行うが、他の電子部品などの損傷防止およびラインタクト短縮のために、低温硬化性および短時間化が望ましい。本発明のアンダーフィル剤の硬化条件は160℃〜250℃の温度範囲で30分間〜5時間が好ましい。これ以外の硬化条件としては50〜160℃の低温から中温で1〜100時間硬化する方法や、250℃〜300℃の高温で1秒〜3時間硬化する方法も挙げられ、また予備加熱として50〜160℃の低温〜中温で1分〜1時間行ってもよい。
【0086】
本発明のアンダーフィル剤を用いた半導体装置において、アンダーフィル剤は半導体素子と基板間に位置する。半導体装置の好ましい製造例としては、バンプを有する基板素子を、配線基板表面と接合させ、接合と同時、または接合後にアンダーフィル剤を半導体素子と基板間の空隙に充填し、熱硬化して半導体素子と基板が固着された半導体装置を得る方法が挙げられる。その他にアンダーフィル剤を先に補給する製造例としては、まず配線基板表面にアンダーフィル剤をディスペンサー法または印刷法などで塗布し、その後バンプを有する半導体素子を基板と接合させ、接合と同時、または接合後にアンダーフィル剤を熱硬化して半導体素子と基板が固着された半導体装置を得る方法が挙げられる。
【実施例】
【0087】
以下実施例および技術をあげて本発明を説明するが、本発明はこれらの例によって限定されるものではない。なお、実施例中の測定、評価は以下の方法により行った。
【0088】
(1)ポリフェニルスルホン樹脂およびポリエーテルスルホン樹脂の分子量の測定
ゲルパーミエーションクロマトグラフィー(GPC)法により、システムコントローラーWaters 2690(ウォーターズ(株)製)を用いて、NMP(LiCl(0.05mol/L))/H3PO4(0.05mol/L)=10/1(重量比)の展開溶媒を0.4mL/分の条件で用いてポリフェニルスルホン樹脂およびポリエーテルスルホン樹脂の分子量を測定し、標準ポリスチレンの校正曲線を用いて重量平均分子量(Mw)を算出した。
【0089】
(2)ガラス転移温度の測定
熱硬化前の樹脂組成物を“テフロン(登録商標)”製の鋳型に流しこみ、イナートオーブンに投入し、昇温速度1.5℃/分で170℃の硬化温度まで上昇させ、170℃で120分間加熱処理を行った。その後、オーブン内が50℃以下になるまで徐冷し、樹脂硬化物を得た。厚み4mm×幅4mm×長さ7mmの試料片を得た後、熱機械分析装置SS-6100を用いて、圧縮モード、温度範囲5〜300℃、昇温速度5℃/分、初期荷重0.5gの条件で成型体の圧縮量を測定した。得られた測定結果からdTMA/dtの極大点における温度をガラス転移温度とした。
【0090】
(3)末端水酸基含有率の測定
核磁気共鳴スペクトル(1H−NMR)測定法により、測定機器は日本電子EX−270型(270MHz)を用いた。測定溶媒にはCDCl3、DMSOを用いた。CDCl3を用いた場合は化学シフトσ7.26PPMをリファレンスとし、DMSOを用いた場合は化学シフトσ2.5PPMをリファレンスとした。サンプルを10mg秤量し、サンプルチューブに投入し、さらに測定溶媒を0.75mL投入し測定用サンプルとした。化学シフトがσ8.00〜σ6.00PPMにシフトされるポリマーユニット由来の芳香族環の水素原子と末端フェノール性水酸基と末端Cl基の検出ピークの積分強度比から末端水酸基含有率(モル%)を求めた。
末端水酸基含有率(モル%)=水酸基末端量/(水酸基末端量+Cl末端量)×100(モル%)
(4)粘度特性の測定
樹脂組成物を直径25mmの円板型アルミニウム製ディスポーザブルパラレルプレート二枚の間隔が0.5mmになるように二枚の平行円板の間に充填し、ついでAR−G2レオメーター(ティーエーインスツルメント(株)製)にて昇温速度2℃/分、角周波数3s−1(周波数は0.5Hz)、振幅0.5deg、窒素ガス気流中での測定条件で、25〜170℃における粘弾性−温度曲線を測定した。
【0091】
(5)溶解性の評価
樹脂組成物をガラスのスクリュー管に入れ、各サンプルを23℃で6時間静置後肉眼で観察し、アンダーフィル剤の溶解性を評価した。全体的に非相溶であり、均一にポリマーが分散されているものを非相溶、所々でポリマーがやや不均一に溶解され、一部凝集がみられるものを部分相溶、全体的に均一にポリマーが溶解されているものを溶解とした。
【0092】
(6)硬化開始温度の測定
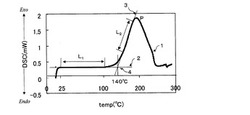
樹脂組成物10mgをアルミニウムセルに入れ、シールしたものを測定用サンプルとした。示差走査熱量計DSC−50(島津製作所(株)製)を用いて、窒素流量20ml/分の条件で、昇温速度5℃/分、20℃〜300℃の温度範囲で熱量の測定を行った。得られたチャートを用いて硬化開始温度を次のようにして求めた。例えば、図1は、実施例2のアンダーフィル剤B−1のDSC曲線である。図1においてDSC曲線1は25〜100℃の温度範囲に認められる直線部分L1、立ち上がり後ピークPに到達するまでに認められる直線部分L2を有する。L1の延長線である直線2と、L2の延長線である直線3との交点4をアンダーフィル剤B−1の硬化開始温度とした。
【0093】
(7)破壊靭性値の測定
ASTM D5045に準じて破壊靭性値K1cの測定を行った。すなわち、アンダーフィル剤を“テフロン(登録商標)”製の鋳型に流し込み、イナートオーブン(光洋サーモシステム(株)製INH−21CD)に投入し、170℃の硬化温度まで80分間かけて上昇させ、170℃で120分間加熱処理を行った。その後、オーブン内が50℃以下になるまで徐冷し、硬化物を得た。得られた硬化物をオートカッターで切り出し、厚み6mm×幅12mm×長さ52.8mmの試料片を得た後、さらに予備クラックを入れ、テンシロン万能試験機(エーアンドデイ(株)製)を用いて、温度23℃、クロスヘッド速度10mm/分、エッジスパン間隔48mmの条件で3点曲げ試験を行い、測定したき裂長さおよび最大破壊荷重から、破壊靭性値K1c(MPa・m1/2)の値を求めた。K1cの値を求める式は次の式で表される。
K1c=PmaxSf(a/W)/BW3/2
ただし、Pmaxは最大破壊荷重[kN]、Sはスパン間距離(治具ローラー間距離)[cm]、aはき裂長さ[cm]、W=試験片幅[cm]、B=試験片厚さ[cm]である。またf(a/W)は、a/W=X、S/W=4.0とし、f(X)は次の式で求められる。
【0094】
【数3】
【0095】
(8)ボイド評価
銅回路が形成されたポリイミド基板と、該基板上に搭載された半導体素子との空隙に、アンダーフィル剤を充填し、その後170℃で1時間加熱処理して硬化させた。直径3μm以上のボイドの有無を半導体検査装置C−SAM(SONIX(株)製)を用いて調
べた。測定範囲は3mm×3mmとした。
【0096】
(9)平均熱線膨張係数の測定
熱硬化性樹脂組成物をNMP溶液で固形分濃度70重量%に調製した後、これを新品のシリコンウェハ上に塗布、さらに上記(2)に記載の方法で硬化させ、膜厚が約10μmのシリコンウェハ付きキュア膜を作製した。次にシリコンウェハ付きキュア膜を室温でフッ化水素酸に約7分間浸漬後、水道水で洗浄しながらキュア膜を手作業でシリコンウェハから剥離した。得られたキュア膜を3mm×17mmに切り出し、熱機械分析装置SS−6100(セイコーインスツルメント(株)製)を用いて、引っ張りモード、温度範囲25〜170℃、昇温速度5℃/分、初期荷重0.5g、チャック間15mmの条件でキュア膜の伸びを測定した。得られた測定結果から下記の計算式を用いてT1〜T2℃の平均熱線膨張係数を算出した。ここでLT1はT1℃でのサンプル長、LT2はT2℃でのサンプル長である。
平均熱線膨張係数=(1/LT1)[(LT2−LT1)/(T2−T1)]
(10)SP値の算出
SP値は、文献「ポリマー・エンジニアリング・アンド・サイエンス(R.F.Fedors,Polymer.Eng.,14,(2)147−154(1974)」に記載された方法により求めた。すなわち、求める化合物の構造式において、原子および原子団の蒸発エネルギーとモル体積のデータより次式にて算出した。
【0097】
【数4】
【0098】
ただし、Δeiは原子または基に帰属する25℃における蒸発エネルギー、Δviは25℃におけるモル体積を示し、分子中のi個の原子及び基に与えられた一定の数値である。代表的な原子および基のΔeiおよびΔviは、前記文献に記載された値を用いた。
【0099】
合成例1:ポリフェニルスルホン樹脂Aの合成(直接重合方法)
窒素導入管、撹拌棒、温度計を取り付けた500mLの3つ口フラスコに乾燥窒素気流下、4,4’−ジクロロジフェニルスルホン27.28g(95ミリモル、和光純薬工業(株)製)と、4,4’−ジヒドロキシジフェニルスルホン18.62g(100ミリモル、和光純薬工業(株)製)と、無水炭酸カリウム17.25g(0.125モル、東京化成工業(株)製)をNMP(三菱化学(株)製)140gとトルエン60g(東京化成工業(株)製)に溶解させ、溶液の温度を170℃で8時間反応させた。その後冷却を行い、溶液の温度が室温にまで低下したら、メタノール3Lに溶液を投入し、白色のポリマー固体を得た。このポリマー固体をろ過で集め、さらにメタノール洗浄を2回行った後、水で3回洗浄を行った。洗浄後、集めたポリマー固体を90℃の真空乾燥機で72時間乾燥しポリフェニルスルホン樹脂Aを得た。得られた粉体を、KBr法による赤外吸収スペクトルで測定した。KBr法とはKBr粉末と測定するポリマーを混合、圧縮成型し得られたペレット状サンプルの赤外吸収スペクトルを測定する方法である。その結果ポリフェニルスルホンの構造に起因するピークが1578cm−1付近、1484cm−1、1238cm−1、1149cm−1、1103cm−1付近に検出された。このようにして得られたポリマーの重量平均分子量(Mw)を算出したところ、28600であった。また末端水酸基含有率は20モル%であった。また得られた粉体を用いてDSC法を用いてガラス転移温度を測定したところ、220℃であった。
【0100】
合成例2:ポリフェニルスルホン樹脂Bの合成(直接重合方法)
窒素導入管、撹拌棒、温度計を取り付けた500mLの3つ口フラスコに乾燥窒素気流下、4,4’−ジクロロジフェニルスルホン25.84g(90ミリモル、和光純薬工業(株)製)と、4,4’−ジヒドロキシジフェニルスルホン18.62g(100ミリモル、和光純薬工業(株)製)と、無水炭酸カリウム17.25g(0.125モル、東京化成工業(株)製)をNMP(三菱化学(株)製)140gとトルエン60g(東京化成工業(株)製)に溶解させ、溶液の温度を170℃で8時間反応させた。その後冷却を行い、溶液の温度が室温にまで低下したら、メタノール3Lに溶液を投入し、白色のポリマー固体を得た。このポリマー固体をろ過で集め、さらにメタノール洗浄を2回行った後、水で3回洗浄を行った。洗浄後、集めたポリマー固体を90℃の真空乾燥機で72時間乾燥しポリフェニルスルホン樹脂Bを得た。得られた粉体を、KBr法による赤外吸収スペクトルで測定したところ、ポリフェニルスルホンの構造に起因するピークが1578cm−1付近、1484cm−1、1238cm−1、1149cm−1、1103cm−1付近に検出された。このようにして得られたポリマーの重量平均分子量(Mw)を算出したところ、13800であった。また末端水酸基含有率は20モル%であった。また得られた粉体を用いてDSC法を用いてガラス転移温度を測定したところ、210℃であった。
【0101】
合成例3:ポリフェニルスルホン樹脂Cの合成(直接重合方法)
窒素導入管、撹拌棒、温度計を取り付けた500mLの3つ口フラスコに乾燥窒素気流下、4,4’−ジクロロジフェニルスルホン24.87g(78ミリモル、和光純薬工業(株)製)と、4,4’−ジヒドロキシジフェニルスルホン18.62g(100ミリモル、和光純薬工業(株)製)と、無水炭酸カリウム17.25g(0.125モル、東京化成工業(株)製)をNMP(三菱化学(株)製)140gとトルエン60g(東京化成工業(株)製)に溶解させ、溶液の温度を170℃で8時間反応させた。その後冷却を行い、溶液の温度が室温にまで低下したら、メタノール3Lに溶液を投入し、白色のポリマー固体を得た。このポリマー固体をろ過で集め、さらにメタノール洗浄を2回行った後、水で3回洗浄を行った。洗浄後、集めたポリマー固体を90℃の真空乾燥機で72時間乾燥しポリフェニルスルホン樹脂Cを得た。得られた粉体を、KBr法による赤外吸収スペクトルで測定したところ、ポリフェニルスルホンの構造に起因するピークが1578cm−1付近、1484cm−1、1238cm−1、1149cm−1、1103cm−1付近に検出された。このようにして得られたポリマーの重量平均分子量(Mw)を算出したところ、5500であった。また末端水酸基含有率は20モル%であった。また得られた粉体を用いてDSC法を用いてガラス転移温度を測定したところ、172℃であった。
【0102】
合成例4:ポリフェニルスルホン樹脂Dの合成(解重合方法)
窒素導入管、撹拌棒、温度計を取り付けた500mLの3つ口フラスコに乾燥窒素気流下、スミカエクセル4800P67g(0.288モル、住友化学工業(株)製)と、4,4’−ジヒドロキシジフェニル2.83g(0.015モル、和光純薬工業(株)製)と、無水炭酸カリウム3.57g(0.0258モル、東京化成工業(株)製)をNMP540gとトルエン60gに溶解させ、溶液の温度を150℃で5時間反応させた。その後冷却を行い、溶液の温度が室温にまで低下したら、メタノール3Lに溶液を投入し、白色のポリマー固体を得た。このポリマー固体をろ過で集め、さらにメタノール洗浄を2回行った後、水で3回洗浄を行った。洗浄後、集めたポリマー固体を90℃の真空乾燥機で72時間乾燥しポリフェニルスルホン樹脂Dを得た。得られた粉体を、KBr法による赤外吸収スペクトルで測定したところ、ポリフェニルスルホンの構造に起因するピークが1578cm−1付近、1484cm−1、1238cm−1、1149cm−1、1103cm−1付近に検出された。このようにして得られたポリマーの重量平均分子量(Mw)を算出したところ、20100であった。また末端水酸基含有率は20モル%であった。また得られたポリマー粉体を用いてDSC法を用いてガラス転移温度を測定したところ、210℃であった。
【0103】
合成例5:ポリフェニルスルホン樹脂Eの合成(解重合方法)
窒素導入管、撹拌棒、温度計を取り付けた500mLの3つ口フラスコに乾燥窒素気流下、スミカエクセル4800P60g(0.258モル、住友化学工業(株)製)と、4,4’−ジヒドロキシジフェニル8.48g(0.0455モル、和光純薬工業(株)製)と、無水炭酸カリウム3.57g(0.0258モル、東京化成工業(株)製)をNMP540gとトルエン60gに溶解させ、溶液の温度を150℃で5時間反応させた。その後冷却を行い、溶液の温度が室温にまで低下したら、メタノール3Lに溶液を投入し、白色のポリマー固体を得た。このポリマー固体をろ過で集め、さらにメタノール洗浄を2回行った後、水で3回洗浄を行った。洗浄後、集めたポリマー固体を90℃の真空乾燥機で72時間乾燥しポリフェニルスルホン樹脂Eを得た。得られた粉体を、KBr法による赤外吸収スペクトルで測定したところ、ポリフェニルスルホンの構造に起因するピークが1578cm−1付近、1484cm−1、1238cm−1、1149cm−1、1103cm−1付近に検出された。このようにして得られたポリマーの重量平均分子量(Mw)を算出したところ、7185であった。また末端水酸基含有率は30モル%であった。また得られたポリマー粉体を用いてDSC法を用いてガラス転移温度を測定したところ、150℃であった。
【0104】
合成例6:ポリフェニルスルホン樹脂Fの合成(直接重合方法)
窒素導入管、撹拌棒、温度計を取り付けた500mLの3つ口フラスコに乾燥窒素気流下、4,4’−ジクロロジフェニルスルホン28.72g(0.1モル、和光純薬工業(株)製)と、4,4’−ジヒドロキシジフェニルスルホン18.62g(0.1モル、和光純薬工業(株)製)と、無水炭酸カリウム17.25g(0.125モル、東京化成工業(株)製)をNMP(三菱化学(株)製)140gとトルエン60g(東京化成工業(株)製)に溶解させ、溶液の温度を170℃で8時間反応させた。その後冷却を行い、溶液の温度が室温にまで低下したら、メタノール3Lに溶液を投入し、白色のポリマー固体を得た。このポリマー固体をろ過で集め、さらにメタノール洗浄を2回行った後、水で3回洗浄を行った。洗浄後、集めたポリマー固体を90℃の真空乾燥機で72時間乾燥しポリフェニルスルホン樹脂Fを得た。得られた粉体を、KBr法による赤外吸収スペクトルで測定したところ、ポリフェニルスルホンの構造に起因するピークが1578cm−1付近、1484cm−1、1238cm−1、1149cm−1、1103cm−1付近に検出された。このようにして得られたポリマーの重量平均分子量(Mw)を算出したところ、69893であった。また末端水酸基含有率は15モル%であった。また得られた粉体を用いてDSC法を用いてガラス転移温度を測定したところ、231℃であった。
【0105】
合成例7:ポリイミド化合物の合成
冷却管および撹拌装置付きの1Lセパラブルフラスコに、2,3,3’,4’−ビフェニルテトラカルボン酸二無水物411.9g(1.40mol)、ビス(3−カルボキシ−4−アミノフェニル)メタン71.58g(0.25mol)、1,3−ビス(3−アミノプロピル)テトラメチルジシロキサン186g(0.75モル)をトリエチレングリコールジメチルエーテル(商品名、丸善石油化学(株)製)溶媒中で温度160℃で6時間撹拌してポリイミド溶液(固形分濃度56.5%)を得た。撹拌終了し、室温まで冷却後、溶液を水3Lに投入して白色沈殿を得た。この沈殿をろ過して回収し、水で3回洗浄した後、真空乾燥機を用いて80℃、20時間乾燥し、白色粉末のポリイミド化合物を得た。
【0106】
各実施例および比較例で使用した原料を以下に示す。
【0107】
ポリエーテルスルホン樹脂:
スミカエクセル5003P(商品名、住友化学工業(株)製:重量平均分子量67250、末端フェノール性水酸基含有率46モル%、ガラス転移温度230℃)
スミカエクセル4800P(商品名、住友化学工業(株)製:重量平均分子量66700、末端フェノール性水酸基無し、ガラス転移温度225℃)
ポリフェニルスルホン樹脂:
ポリフェニルスルホン樹脂“Ultrazon”P3010(以上、商品名、BASFジャパン(株)製、重量平均分子量=55675)
ポリイミド化合物:合成例7で得られたポリイミド化合物
“E2020”(商品名、プリンテック(株)製)芳香族ビスマレイミドと芳香族ジアミンの共重合体
エポキシ変性シリコーン樹脂:アルケニル基含有フェノールノボラック型樹脂とポリジメチルシロキサンの共重合体
SP値が2.00×104〜2.60×104(J/m3)1/2であるエポキシ樹脂:
“jER”828(商品名、ジャパンエポキシレジン(株)製):エポキシ当量187g/eq、SP値=12.31(cal/cm3)1/2=2.52×104(J/m3)1/2
N660(商品名、DIC(株)製:クレゾールノボラック型固形エポキシ樹脂:エポキシ当量206g/eq、SP値=10.11(cal/cm3)1/2=2.07×104(J/m)1/2)
EXA4710(商品名、DIC(株)製:ナフタレン型固形エポキシ樹脂:エポキシ当量170g/eq、SP値=10.07(cal/cm3)1/2=2.06×104(J/m)1/2))
SP値が1.60×104〜1.80×104(J/m3)1/2であるエポキシ樹脂:
エポライト400E(商品名、共栄社化学(株)製):ポリエチレングリコールジグリシジルエーテル、エポキシ当量264〜290g/eq、SP値=8.58(cal/cm3)1/2=1.76×104(J/m3)1/2
YED216M(商品名、ジャパンエポキシレジン(株)製):1,6−ヘキサンジオールジグリシジルエーテル、エポキシ当量148g/eq、SP値=8.25(cal/cm3)1/2=1.69×104(J/m3)1/2
比較例で用いたエポキシ樹脂
エポキシ樹脂A=“jER”1032H60(商品名、ジャパンエポキシレジン(株)製):トリスヒドロキシフェニルメタン型エポキシ樹脂
エポキシ樹脂C=“jER”807(商品名、ジャパンエポキシレジン(株)製):ビスフェノールF型液状エポキシ樹脂)
オキセタン化合物:
POX(略号)=OXT−211(商品名、ジャパンエポキシレジン(株)製):3−エチル−3−(フェノキシメチル)オキセタン
OXIPA(略号)=“エタナコール”OXIPA(商品名、宇部興産(株)製)
硬化剤および硬化促進剤:
硬化剤A:DDS:芳香族ジアミン(4,4’−ジアミノジフェニルスルホンと3,3’−ジアミノジフェニルスルホンの1対1混合物(和光純薬工業(株)製))
硬化剤A−2:APB−N:芳香族ジアミン:m−ビス(m−アミノフェノキシ)ベンゼン(三井化学(株)製))
硬化剤A−3:TPE−R:芳香族ジアミン:m−ビス(p−アミノフェノキシ)ベンゼン(和歌山精化工業(株)製))
硬化剤A−4:APB−5:芳香族ジアミン:ビス(m−(m−アミノフェノキシ)フェノキシ)ベンゼン(三井化学(株)製)
硬化促進剤:3,4−ジクロロフェニル−N,N−ジメチル尿素(東京化成工業(株)製)
比較例で用いた硬化剤
硬化剤B:EXB9889(商品名、DIC(株)製、トリアジン骨格含有フェノールノボラック樹脂)
硬化剤C:2E4MZ(2−エチル−4−メチルイミダゾール)(商品名、四国化成工業(株)製、非水溶性)
硬化剤D:KAYAHARD−AA(商品名、日本化薬(株)製、3,3’−ジエチル−4,4‘−ジアミノジフェニルメタン)
ラジカル重合開始剤:パークミルD(商品名、日本油脂(株)製)
無機微粒子および有機微粒子:
球状シリカ微粒子:アドマファインGRJ(商品名、アドマテックス(株)製:表面型修飾シリカ微粒子、表面修飾粒子、平均粒子径0.5μm)
球状スチレン微粒子:ガンツパール(商品名、ガンツ化成(株)製、:ポリスチレン系有機微粒子、平均粒子径2.5μm)
溶剤:
溶剤A:NMP(略号)=:N−メチルピロリドン(商品名、東京化成工業(株)製、沸点202−204℃)
比較例で用いた溶剤
溶剤B:トリグライム(略号)=:トリエチレングリコールジメチルエーテル(商品名、丸善石油化学(株)製、沸点216℃)
溶剤C:DMAc(略号)=:N,N’−ジメチルアセトアミド(商品名、東京化成工業(株)製、沸点166℃)
溶剤D::2−ブトキシエチルアセテート(商品名、東京化成工業(株)製、沸点188℃)
実施例1
窒素導入管、撹拌棒、温度計を取り付けた100mLの3つ口フラスコにN660 30gとYED216M 30gを投入し、乾燥窒素気流下、80℃で1時間撹拌して溶解させた。さらに硬化剤A 21.6gを投入し、乾燥窒素気流下、60℃で2時間撹拌して溶解させ、室温まで冷却した。その後、合成例1で得られたポリフェニルスルホン樹脂A 6gを添加し、遊星式撹拌脱泡機(マゼルスター(クラボウ製))を用いて25℃で30分間撹拌して混合させた後、最後に脱泡を行い、アンダーフィル剤A−1を得た。得られたアンダーフィル剤A−1のガラス転移温度、粘度特性、溶解性、硬化開始温度、破壊靭性値、ボイドの有無を、前記方法により測定および評価した。
【0108】
実施例2
ポリフェニルスルホン樹脂A 6gに代えて合成例2で得られたポリフェニルスルホン樹脂B 6gを用いた以外は実施例1と同様にして、アンダーフィル剤B−1を得た。得られたアンダーフィル剤B−1のガラス転移温度、粘度特性、溶解性、硬化開始温度、破壊靭性値、平均熱線膨張係数、ボイドの有無を、前記方法により測定および評価した。平均熱線膨張係数の測定温度範囲はT1=25℃、T2=100℃とした。
【0109】
実施例3
ポリフェニルスルホン樹脂A 6gに代えて合成例3で得られたポリフェニルスルホン樹脂C 6gを用いた以外は実施例1と同様にして、アンダーフィル剤C−1を得、実施例1と同様に測定および評価した。
【0110】
実施例4
ポリフェニルスルホン樹脂A 6gに代えて合成例4で得られたポリフェニルスルホン樹脂D 6gを用いた以外は実施例1と同様にして、アンダーフィル剤D−1を得、実施例1と同様に測定および評価した。
【0111】
実施例5
ポリフェニルスルホン樹脂A 6gに代えて合成例5で得られたポリフェニルスルホン樹脂E6g用いた以外は実施例1と同様にして、アンダーフィル剤E−1を得、実施例1と同様に測定および評価した。
【0112】
実施例6
ポリフェニルスルホン樹脂B 6gを3gに代えて用いた以外は実施例2と同様にして、アンダーフィル剤F−1を得、実施例1と同様に測定および評価した。
【0113】
実施例7
ポリフェニルスルホン樹脂B 6gを10gに代えて用いた以外は実施例2と同様にして、アンダーフィル剤G−1を得、実施例1と同様に測定および評価した。
【0114】
実施例8
窒素導入管、撹拌棒、温度計を取り付けた100mLの3つ口フラスコにjER828 30gとYED216M 30gを投入し、硬化剤A 22.5gを投入し、乾燥窒素気流下、60℃で2時間撹拌して溶解させ、室温まで冷却した。その後、合成例2で得られたポリフェニルスルホン樹脂B 6gを添加し、遊星式撹拌脱泡機(マゼルスター(クラボウ製))を用いて25℃で30分間撹拌して混合させた後、最後に脱泡を行い、アンダーフィル剤H−1を得、実施例1と同様に測定および評価した。
【0115】
実施例9
N660をEXA4710に代えて、硬化剤A 21.6gを23.5gに代えて用いた以外は実施例2と同様にして、アンダーフィル剤I−1を得、実施例1と同様に測定および評価した。
【0116】
実施例10
YED216Mをエポライト400Eに代えて、硬化剤A 21.6gを19.3gに代えて用いた以外は実施例2と同様にして、アンダーフィル剤J−1を得、実施例1と同様に測定および評価した。
【0117】
実施例11
窒素導入管、撹拌棒、温度計を取り付けた100mLの3つ口フラスコにjER828 60gを投入し、さらに硬化剤A 19.9gを投入し、乾燥窒素気流下、60℃で2時間撹拌して溶解させ、室温まで冷却した。その後、合成例2で得られたポリフェニルスルホン樹脂B 6gを添加し、遊星式撹拌脱泡機(マゼルスター(クラボウ製))を用いて25℃で30分間撹拌して混合させた後、最後に脱泡を行い、アンダーフィル剤K−1を得、実施例1と同様に測定および評価した。
【0118】
実施例12
窒素導入管、撹拌棒、温度計を取り付けた100mLの3つ口フラスコにN660 30gとYED216M 30gを投入し、乾燥窒素気流下、80℃で1時間撹拌して溶解させた。さらに硬化剤A 21.6gを投入し、乾燥窒素気流下、60℃で2時間撹拌して溶解させ、室温まで冷却した。その後、合成例2で得られたポリフェニルスルホン樹脂B 6g、OXIPA 1gを添加し、遊星式撹拌脱泡機(マゼルスター(クラボウ製))を用いて25℃で30分間撹拌して混合させた後、最後に脱泡を行い、アンダーフィル剤L−1を得、実施例1と同様に測定および評価した。
【0119】
実施例13
OXIPAをPOXに代えて用いた以外は実施例11と同様にして、アンダーフィル剤M−1を得、実施例1と同様に測定および評価した。
【0120】
実施例14
硬化剤A21.6gを硬化剤A−2 25.5gに代えて用いた以外は実施例2と同様にして、アンダーフィル剤N−1を得、実施例1と同様に測定および評価した。
【0121】
実施例15
硬化剤A21.6gを硬化剤A−3 25.5gに代えて用いた以外は実施例2と同様にして、アンダーフィル剤O−1を得、実施例1と同様に測定および評価した。
【0122】
実施例16
硬化剤A21.6gを硬化剤A−4 39gに代えて用いた以外は実施例2と同様にして、アンダーフィル剤P−1を得、実施例1と同様に測定および評価した。
【0123】
実施例17
窒素導入管、撹拌棒、温度計を取り付けた100mLの3つ口フラスコにN660 30gとYED216M30gを投入し、乾燥窒素気流下、80℃で1時間撹拌して溶解させた。さらに硬化剤A21.6gを投入し、乾燥窒素気流下、60℃で2時間撹拌して溶解させ、室温まで冷却した。その後、合成例2で得られたポリフェニルスルホン樹脂B 6g、硬化促進剤9.7gを添加し、遊星式撹拌脱泡機(マゼルスター(クラボウ製))を用いて25℃で30分間撹拌して混合させた後、最後に脱泡を行い、アンダーフィル剤Q−1を得、実施例1と同様に測定および評価した。
【0124】
実施例18
窒素導入管、撹拌棒、温度計を取り付けた100mLの3つ口フラスコにN660 30gとYED216M 30gを投入し、乾燥窒素気流下、80℃で1時間撹拌して溶解させた。さらに硬化剤A 21.6gを投入し、乾燥窒素気流下、60℃で2時間撹拌して溶解させ、室温まで冷却した。その後、合成例2で得られたポリフェニルスルホン樹脂B 6gを添加し、遊星式撹拌脱泡機(マゼルスター(クラボウ製))を用いて25℃で30分間撹拌して混合させた後、球状シリカ微粒子を8.8g加えて、30分間撹拌して混合させた後,3本ロールで混練した。その後さらに30分間撹拌して混合させ、最後に脱泡を行い、アンダーフィル剤R−1を得、実施例2と同様に測定および評価した。
【0125】
実施例19
球状シリカ微粒子8.8gを21.9gに代えて用いた以外は実施例17と同様にして、アンダーフィル剤S−1を得、実施例2と同様に測定および評価した。
【0126】
実施例20
球状シリカ微粒子8.8gを球状スチレン微粒子9.7gに代えて用いた以外は実施例18と同様にして、アンダーフィル剤T−1を得、実施例1と同様に測定および評価した。
【0127】
実施例21
窒素導入管、撹拌棒、温度計を取り付けた100mLの3つ口フラスコにN660 30gとYED216M 30gを投入し、乾燥窒素気流下、80℃で1時間撹拌して溶解させた。さらに硬化剤A 21.6gを投入し、乾燥窒素気流下、60℃で2時間撹拌して溶解させ、室温まで冷却した。その後、合成例2で得られたポリフェニルスルホン樹脂B 6gを添加し、遊星式撹拌脱泡機(マゼルスター(クラボウ製))を用いて25℃で30分間撹拌して混合させた。最後に溶剤Aを0.3g添加し、さらに30分間撹拌して混合させ、脱泡を行い、アンダーフィルU−1を得、実施例1と同様に測定および評価した。
【0128】
比較例1
溶剤A 0.5gを40gに代えた以外は実施例20と同様にして、アンダーフィル剤A’−1を得、実施例1と同様に測定および評価した。
【0129】
比較例2
エポキシ樹脂を用いなかった以外は実施例2と同様にして、アンダーフィル剤A’−2を得たが、固体粉体の状態であった。
【0130】
比較例3
ポリフェニルスルホン樹脂を用いなかった以外は実施例2と同様にして、アンダーフィル剤A’−3を得、実施例1と同様に測定および評価した。
【0131】
比較例4
ポリフェニルスルホン樹脂Bに代えて合成例6で得られたポリフェニルスルホン樹脂Fを用いた以外は実施例2と同様にして、アンダーフィル剤A’−4を得、実施例1と同様に測定および評価した。
【0132】
比較例5
ポリフェニルスルホン樹脂Bに代えて市販のポリフェニルスルホン樹脂“Ultrazon”P3010(以上、商品名、BASFジャパン(株)製、重量平均分子量=55675)を用いた以外は実施例2と同様にして、アンダーフィル剤A’−5を得、実施例1と同様に測定および評価した。
【0133】
比較例6
E2020 20g、スミカエクセル5003P 10g、エポキシ樹脂C 15g、溶剤C 48g、硬化剤B 8g、ラジカル重合開始剤 0.16gを遊星式撹拌脱泡機(マゼルスター(クラボウ製))を用いて25℃で60分間撹拌して混合させ、最後に脱泡を行い、アンダーフィルA’−6を得、実施例1と同様に測定および評価した。
【0134】
比較例7
エポキシ樹脂A 15g、合成例7で得られたポリイミド化合物 57gを遊星式撹拌脱泡機(マゼルスター(クラボウ製))を用いて25℃で60分間撹拌して混合させた後、硬化剤Cを0.3g添加し、さらに30分間撹拌して混合させた。最後に溶剤Bを43g添加し、さらに30分間撹拌して混合させ、脱泡を行い、アンダーフィルA’−7を得、実施例1と同様に測定および評価した。
【0135】
比較例8
エポキシ樹脂C 63g、エポキシ変性シリコーン樹脂4g、球状シリカ微粒子150gを遊星式撹拌脱泡機(マゼルスター(クラボウ製))を用いて25℃で60分間撹拌して混合させた後、3本ロールで混練した。その後硬化剤Dを30g添加し、さらに溶剤Dを2g添加して30分間撹拌して混合させ、最後に脱泡を行い、アンダーフィルA’−8を得、実施例1と同様に測定および評価した。
各実施例および比較例の組成を表1〜3に、評価結果を表4〜6に示す。
【0136】
【表1】
【0137】
【表2】
【0138】
【表3】
【0139】
【表4】
【0140】
【表5】
【0141】
【表6】
【符号の説明】
【0142】
1:DSC曲線
2:L1の延長線
3:L2の延長線
4:交点
P:ピーク
L1:直線部分
L2:立ち上がり後ピークに到達するまでに認められる直線部分
【技術分野】
【0001】
本発明は、アンダーフィル剤に関する。より詳しくは、半導体素子と基板との接合に好ましく用いられるアンダーフィル剤に関する。
【背景技術】
【0002】
近年、半導体素子の大型化が急速に進み、半導体素子と基板との間に大きな熱応力が発生し、問題となっている。従来より、半導体素子と基板との接合の後、空隙にエポキシ系アンダーフィル剤を充填することにより、応力を分散させて、半導体素子/基板界面の接合信頼性を高める手法が知られている。アンダーフィル剤に求められる特性としては、低粘度性であり、半導体素子と基板との隙間に素早く進入し、ボイドが生じないこと、硬化後の熱的または物理的応力に対しての接続信頼性、耐熱性、靭性が高いことや短時間・低温硬化性が挙げられる。エポキシ樹脂は硬くて脆い性質を有するため、機械的・熱的ストレスをより吸収させるために、各種熱可塑性樹脂をエポキシ樹脂に混合する技術が提案されている。
【0003】
例えば、強靭性に優れた熱硬化性樹脂組成物として、ポリエーテルスルホン樹脂、エポキシ樹脂、多価フェノール系硬化剤を含有する樹脂組成物や、ポリエーテルスルホン樹脂とビスマレイミド化合物とジアミン化合物の共重合体、エポキシ樹脂、硬化剤を含有する樹脂組成物が提案されている(例えば、特許文献1、2参照)。また、ポリイミド樹脂、エポキシ樹脂、硬化剤を含有し、25℃の溶液粘度が500センチポイズ以下であるアンダーフィル剤が提案されている(例えば、特許文献3参照)。これらの材料は、強靭化剤或いは低応力化剤として配合されている樹脂を、トリグライムやジメチルアセトアミドなどの高沸点溶媒に溶解させるものであり、これをアンダーフィル剤として用いた場合、接合後に溶媒の飛散によりボイドが発生して信頼性が低下したりするなど、実用的ではなかった。実質的に無溶剤の液状アンダーフィル剤として、エポキシ変性シリコーン樹脂、エポキシ樹脂、硬化剤を含有し、25℃の溶液粘度が18〜72Pa・s以下であるアンダーフィル剤が提案されている(例えば、特許文献4参照)。これらの材料は弾性率を下げる効果はあるが、アンダーフィル剤として用いた場合、強靭性に劣るなど、実用的ではなかった。
【0004】
また機械特性や靭性に優れるビフェニル基を有するポリエーテルスルホン樹脂として、重量平均分子量が約40,000〜約60,000のポリフェニルスルホン樹脂が知られている(例えば、特許文献5参照)。しかし、無溶剤系液状アンダーフィル材料に適用すると、エポキシ樹脂に不溶であることから、不均一溶液となり、塗布性が不十分であったり、分子量が高いため、溶液が高粘度になるなど実用的でなかった。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開2001−72833号公報(特許請求の範囲)
【特許文献2】特開2009−155354号公報(特許請求の範囲)
【特許文献3】特許第3719018号公報(特許請求の範囲)
【特許文献4】特開2004−346232号公報(特許請求の範囲)
【特許文献5】特表2002−525406号公報(特許請求の範囲)
【発明の概要】
【発明が解決しようとする課題】
【0006】
このように、従来技術では実質的に無溶剤で低粘度性と高靭性を両立したアンダーフィル剤は得られていなかった。本発明は、上記課題を解決すべく、低粘度性と高靭性に優れたアンダーフィル剤を提供することを目的とする。
【課題を解決するための手段】
【0007】
本発明は、(a)重量平均分子量3万未満のポリフェニルスルホン樹脂と、(b)エポキシ樹脂を含有し、(c)溶剤の含有量が1重量%以下であることを特徴とするアンダーフィル剤である。
【発明の効果】
【0008】
本発明によれば、優れた高靭性と粘度特性を有するアンダーフィル剤を得ることができる。
【図面の簡単な説明】
【0009】
【図1】実施例2のアンダーフィル剤のDSC曲線
【発明を実施するための形態】
【0010】
本発明のアンダーフィル剤は(a)重量平均分子量3万未満のポリフェニルスルホン樹脂と、(b)エポキシ樹脂を含有し、(c)溶剤の含有量が1重量%以下であることを特徴とする。
【0011】
本発明において溶剤とは、粘度を調整したり塗布性を向上させる目的で加えられる揮発性の有機または無機の液体を指し、アンダーフィル剤を塗布した後のエポキシ樹脂を硬化させるまでの工程で蒸発または飛散するものを指す。アンダーフィル剤中の溶剤の含有量は、例えば熱重量分析により25℃から300℃まで5℃/分で昇温させた際の減量率から求めることができる。
【0012】
本発明のアンダーフィル剤は実質的に無溶剤であるため、溶剤飛散によるボイドの発生を抑制し、接合後の絶縁信頼性を高めることができる。またポリフェニルスルホン樹脂はその構造中にビフェニル基を含有した剛直な構造を有し、硬化後の強靭性に優れ、またガラス転移温度が200℃以上と非常に高い特徴を有する。そのためポリフェニルスルホン樹脂をエポキシ樹脂に含有することにより、得られるアンダーフィル剤の硬化後の靭性と耐熱性(ガラス転移温度)を向上させることができる。
【0013】
また(a)ポリフェニルスルホン樹脂は、重量平均分子量3万未満にすることで(b)エポキシ樹脂と25℃、1.013×105Paの条件下で溶解し、均一な液状アンダーフィル剤を得ることができる。このような溶液を有することで、溶液中に樹脂の残渣がなく、したがって、本発明のアンダーフィル剤は基板への塗布性や浸透性に優れる。
【0014】
本発明のアンダーフィル剤において、(a)成分のポリフェニルスルホン樹脂は、(b)エポキシ樹脂と溶解するために、重量平均分子量が3万未満であることが重要である。重量平均分子量が3万以上であると、(b)エポキシ樹脂に溶解せず、均一な液状溶液を得ることが困難となる。粘度特性をより適切な範囲に調整し、低粘度にして浸透性をより向上させるために、重量平均分子量が2万以下が好ましい。一方硬化後の強靭性の観点からは、(a)成分のポリフェニルスルホン樹脂の重量平均分子量は2000以上が好ましく、5000以上がより好ましい。ポリフェニルスルホン樹脂を2種以上含有してもよく、各樹脂の重量平均分子量がいずれも3万未満であることが好ましい。
【0015】
本発明における重量平均分子量は、ゲルパーミエーションクロマトグラフィー(GPC)法により、NMP/H3PO4の混合溶媒を用いてポリフェニルスルホン樹脂の分子量を測定し、標準ポリスチレンの校正曲線を用いて算出した値を指す。
【0016】
ポリフェニルスルホン樹脂は、フェノール性水酸基とアルカリ金属炭酸塩とを反応させてフェノール化合物のアルカリ金属塩を生成し、これにジハロゲノジフェニルスルホンを反応させることによって得ることができる。例えば、反応系でフェノール化合物のアルカリ金属塩を合成し、またはフェノール化合物のアルカリ金属塩を予め合成しておいて、ジハロゲノジフェニルスルホンを有機溶剤中で反応させた後、ジクロロメタンを用いて末端停止反応を行う技術が知られている。例えば、特開昭53−12991号公報、特開昭53―16098号公報には、4,4’−ヒドロキシジフェニルスルホンと、4,4’−ジクロロジフェニルスルホン、無水炭酸カリウムを用いて、芳香族ポリフェニルスルホンを製造し、反応終了後、クロロメタンを吹き込む技術が開示されている。また、特表2002−525406号公報には、溶媒で重合する際の固形分濃度を規定して、改良された多分散度をもち、かつ低分子量オリゴマーの濃度が低いポリフェニルスルホン樹脂の製造方法が記載されている。
【0017】
本発明で用いられるポリフェニルスルホン樹脂の合成に使用されるジハロゲノジフェニルスルホンとしては、例えば、4,4’−ジクロロジフェニルスルホン、4,4’−ジフルオロジフェニスルホン、4−クロロ−4’−フルオロジフェニルスルホン、4,4’−ジブロモジフェニルスルホン、1,4−ビス(4−クロロベンゼンスルホニル)ベンゼン、ビス(4’−クロロベンゼンスルホニル)エーテル、1,3−ビス(4’−クロロベンゼンスルホニル)ベンゼン、ビス(3’−クロロベンゼンスルホニル)エーテル、1,2−ビス(4’−クロロベンゼンスルホニル)ベンゼン、ビス(2’−クロロベンゼンスルホニル)エーテル、4,4’−ビス(4−クロロフェニルスルホニル)ジフェニルなどが挙げられる。
【0018】
本発明で用いられるポリフェニルスルホン樹脂の合成に使用されるフェノール化合物としては、例えば、3,3’−ジヒドロキシジフェニル、4,4’−ジヒドロキシジフェニル、3,4’−ジヒドロキシジフェニルなどのビフェニル基を有する2価のフェノール化合物が挙げられる。
【0019】
また、これ以外のフェノール化合物を、重量平均分子量3万未満を損なわない範囲で共重合してもよい。4,4,4’−トリヒドロキシトリフェニルメタン、2,2’,4,4’−テトラヒドロキシベンゾフェノン、α,α,α−トリス(4−ヒドロキシフェニル)−1−エチル−4−イソプロピルベンゼンなどの多価のフェノール化合物や、4,4’−ジヒドロキシジフェニルスルフィド、4,4’−ジヒドロキシベンゾフェノン、4,4’−ジヒドロキシジフェニルエーテル、9,9’−ビス(4−ヒドロキシフェニル)フルオレン、α,α’−ビス(4−ヒドロキシ−3,5−ジメチルフェニル)−1,4−ジイソプロピルベンゼン、2,2’−ビス(4’−ヒドロキシフェニル)プロパン、2,2−ビス(4’−ヒドロキシ−3’,5’−ジメチルフェニル)プロパン、ビス(4’−ジヒドロキシフェニル)プロパン、ビス(4’−ヒドロキシフェニル)メタン、ビス(4’−ヒドロキシフェニル)シクロヘキサン、1,1−ビス(4’−ヒドロキシフェニル)エタン、1,4−ビス(4’−ヒドロキシフェノキシ)ベンゼン、1,3−ビス(4’−ヒドロキシフェノキシ)ベンゼン、1,4−ビス(4’−ヒドロキシベンゾイル)ベンゼン、1,3−ビス(4’−ヒドロキシベンゼンスルホニル)ベンゼンなどの2価のフェノール化合物や、フェノール、o−クレゾール、m−クレゾール、p−クレゾール、2−アミノフェノール、3−アミノフェノール、4−アミノフェノール、2−アミノチオフェノール、3−アミノチオフェノール、4−アミノチオフェノール、4−ヒドロキシ安息香酸、3−ヒドロキシ安息香酸、o−エチルフェノール、m−エチルフェノール、p−エチルフェノール、2,3−ジメチルフェノール、2,4−ジメチルフェノール、2,5−ジメチルフェノール、2,6−ジメチルフェノール、3,4−ジメチルフェノール、3,5−ジメチルフェノール、o−メトキシフェノール、m−メトキシフェノール、p−メトキシフェノール、o−エトキシフェノール、m−エトキシフェノール、p−エトキシフェノール、o−n−プロピルフェノール、o−イソプロピルフェノール、m−n−プロピルフェノール、m−イソプロピルフェノール、p−n−プロピルフェノール、p−イソプロピルフェノール、o−n−ブチルフェノール、o−sec−ブチルフェノール、o−tert−ブチルフェノール、m−n−ブチルフェノール、m−sec−ブチルフェノール、m−tert−ブチルフェノール、p−n−ブチルフェノール、p−sec−ブチルフェノール、p−tert−ブチルフェノール、o−フェニルフェノール、m−フェニルフェノール、p−フェニルフェノール、2,3,5−トリメチルフェノール、2,3,6−トリメチルフェノール、2,4,6−トリメチルフェノール、o−ベンジルフェノール、m−ベンジルフェノール、p−ベンジルフェノール、o−ブトキシフェノール、m−ブトキシフェノール、p−ブトキシフェノール、6−tert−ブチル−p−キシレノール、2−tert−ブチル−p−キシレノール、2−tert−ブチル−p−クレゾール、o−シクロヘキシルフェノール、m−シクロヘキシルフェノール、p−シクロヘキシルフェノール、2,6−ジ−tert−ブチル−p−クレゾール、4,6−ジ−tert−ブチル−4−ヒドロキシメチルフェノール、2,4−ジメトキシフェノール、2,6−ジメトキシフェノール、バニリン、クミルフェノールなどの1価のフェノール化合物などが挙げられる。
【0020】
ポリフェニルスルホン樹脂の重量平均分子量を3万未満にするためには、ジハロゲノジフェニルスルホンを2価のフェノール化合物に対して0.95〜0.75当量使用することが好ましい。
【0021】
本発明で用いられるポリフェニルスルホン樹脂の合成に使用されるアルカリ金属塩のアルカリ金属としては、リチウム、ナトリウム、カリウム、ルビジウムが挙げられ、カリウムまたはナトリウムが好ましい。ジアルカリ塩を形成させるために用いられる金属化合物としては、水酸化物、炭酸塩、炭酸水素塩が挙げられ、水酸化物または炭酸塩が好ましい。したがって、ビスフェノール類のジアルカリ塩を形成させるためには、水酸化カリウム、水酸化ナトリウム、炭酸カリウムまたは炭酸ナトリウムが好ましく、これらを2種以上用いてもよい。これらアルカリ金属塩の量はその種類によって差があるが、2価のフェノール化合物を基準にその1倍モル以上が好ましく、2倍モル以上がより好ましい。この範囲であれば、ビスフェノールのアルカリ金属塩を十分生成させることができ、重量平均分子量3万未満の高分子量の重合体を容易に得ることができる。一方、副反応を抑制する観点からは、8倍モル以下が好ましく、4倍モル以下がより好ましい。
【0022】
重合温度は100〜300℃の範囲が好ましい。温度を100℃以上とすることで、未反応のモノマーのない重合体を容易に得ることができる。また、300℃以下とすることで、ポリマーの副反応を抑制することができる。重合時間は2〜4時間程度が好ましい。
【0023】
反応溶媒として用いる有機溶媒は、非プロトン性極性溶媒が好ましい。例えば、ジメチルスルホキシド、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチル−2−ピロリドン、1,4−ジメチルベンダゾリジノン、ヘキサメチルトリアミド、1,3−ジメチルイミダゾリジノンなどが挙げられる。また、これらにトルエン、キシレンなどの芳香族炭化水素系溶媒、メチルエチルケトン、メチルイソブチルケトン、シクロヘキサノンなどのケトン系溶媒、プロピレングリコールモノメチルエーテルアセテート、メチル−メトキシブタノールアセテートなどのエステル系溶媒などを加えることもできる。
【0024】
重縮合で使用される溶媒の量は、全モノマーの重量に対して0.5倍以上が好ましく、2倍以上がより好ましい。溶媒の量を全モノマーの重量に対して0.5倍以上とすることにより、撹拌などの操作が容易となり、重縮合反応が順調に進行し易くなる。一方、20倍以下が好ましく、8倍以下がより好ましい。20倍以下とすることによって、溶媒中のモノマー濃度が高くなり重合速度が向上するため、未反応のモノマーがなく重量平均分子量3万未満の重合体を容易に得ることができる。
【0025】
上記方法により得られるポリフェニルスルホン樹脂溶液は、重合後に水、メタノール、エタノール、イソプロピルアルコール、アセトン、トルエン、キシレンなどの貧溶媒中に投入してポリフェニルスルホン樹脂を析出させることが望ましい。貧溶媒の使用量に制限はないが、好ましくは重合に使用した溶媒重量の5〜100倍、より好ましくは10〜50倍である。析出したポリフェニルスルホン樹脂粉末は、本発明のアンダーフィル剤では無溶剤系とするため、溶濾過、洗浄し、乾燥して用いることが好ましい。乾燥条件は、大気圧条件下で50℃〜100℃の温度範囲で5〜50時間乾燥させるのが好ましく、またより乾燥を十分にさせるべく真空圧条件下での乾燥工程も好ましく挙げられる。
【0026】
重量平均分子量が3万未満のポリフェニルスルホン樹脂は、前記のように公知の技術で重合することもできるが、市販のポリエーテルスルホン樹脂および、ポリフェニルスルホン樹脂に2価のフェノール化合物と金属塩を有機溶剤中で反応させることによって、重合製造することができる。
【0027】
ポリエーテルスルホン樹脂の市販品としては、“スミカエクセル(登録商標)”3600P、“スミカエクセル”4100P、“スミカエクセル”4800P、“スミカエクセル”5200P、“スミカエクセル”5003P(以上、商品名、住友化学工業(株)製)、“Ultrazon(登録商標)”E2010、“Ultrazon”E3010、“Ultrazon”E6020P(以上、商品名、BASFジャパン(株)製)、“VERADEL(登録商標)”(以上、商品名、ソルベイアドバンストポリマーズ)などが挙げられ、またポリフェニルスルホン樹脂としては“Ultrazon”P3010(以上、商品名、BASFジャパン(株)製)、“RADEL(登録商標)”, “ACUDEL(登録商標)”(以上、商品名、ソルベイアドバンストポリマーズ)が挙げられ、これらのポリエーテルスルホン樹脂および、ポリフェニルスルホン樹脂を2種以上含有してもよい。
【0028】
また、本発明において、(a)成分のポリフェニルスルホン樹脂の含有量は、5重量%以上が好ましく、8重量%以上がより好ましい。5重量%以上とすることで、エポキシ樹脂が本来有するもろさや硬さを改善することができる。一方、粘度をより低減して浸透性を向上させる観点から、20重量%以下が好ましく、15重量%以下がより好ましい。
【0029】
また、本発明において、(a)成分のポリフェニルスルホン樹脂は末端にフェノール性水酸基、フルオロ基、クロロ基、ブロモ基、メトキシ基、エトキシ基などを有してもよく、これらの基を2種以上有してもよい。これらの中でも、フェノール性水酸基を有することが望ましい。フェノール性水酸基を有することでエポキシ樹脂のオキシラン環またはエポキシ樹脂の反応性基と反応し、密度の高い網目構造を形成するため、得られる硬化物は、強靭性を発現する。
【0030】
末端のフェノール性水酸基量は、核磁気共鳴(1H−NMR)法から析出したピークの積分値を求めることによって測定することができる。樹脂中の繰り返し単位を除く末端官能基量中、フェノール性水酸基を5モル%以上含有することが好ましく、30モル%以上がより好ましい。フェノール性水酸基を5モル%以上有することにより、エポキシ樹脂との親和性が向上し、硬化後の耐熱性や強靭性が向上する。一方、90モル%以下が好ましく、80モル%以下がより好ましい。90モル%以下とすることで経時による粘度上昇がなく、保存安定性がより良好となる。
【0031】
本発明で用いるポリフェニルスルホン樹脂は、フレーク状またはパウダー状のものが好ましく、パウダー状のものがより好ましい。パウダー状のポリフェニルスルホン樹脂は、ペレット状またはフレーク状の樹脂を凍結粉砕することで容易に得ることができる。パウダー状のポリフェニルスルホン樹脂の粒子径は100μm以下が好ましく、30μm以下がより好ましい。100μm以下とすることで、ポリフェニルスルホン樹脂の粒子とエポキシ樹脂を、容易に溶解させることができる。
【0032】
本発明のアンダーフィル剤は、(b)エポキシ樹脂を含有する。エポキシ樹脂は、他の化合物との熱反応性が良好であり、硬化後の接着強度が高く、熱収縮の少ない成型物を与えることができる。
【0033】
本発明における(b)エポキシ樹脂の好ましい態様の他の一つとして、SP値が1.60×104〜1.80×104(J/m3)1/2であるエポキシ樹脂と、SP値が2.00×104〜2.60×104(J/m3)1/2であるエポキシ樹脂とをそれぞれ一種以上含有することが挙げられる。本発明では(a)重量平均分子量3万未満のポリフェニルスルホン樹脂と、上記のエポキシ樹脂を用いることによって、硬化後の強靭性を向上させることができる。
【0034】
SP値(溶解性パラメーター値)の求め方は各種あるが、本発明においては、文献「ポリマー・エンジニアリング・アンド・サイエンス(R.F.Fedors,Polymer.Eng.,14,(2)147−154(1974)」に記載された方法により求める。すなわち、求める化合物の構造式において、原子および原子団の蒸発エネルギーとモル体積のデータより次式にて計算する。
【0035】
【数1】
【0036】
上記式において、Δeiは原子または基に帰属する25℃における蒸発エネルギー、Δviは25℃におけるモル体積を示し、分子中のi個の原子および基に与えられた一定の数値である。代表的な原子および基のΔeiおよびΔviは、前記文献に記載された値を用いる。
【0037】
強靭性を向上させるための一つの方法として、硬化後の樹脂組成物が海島構造を有することが知られている。SP値が2.00×104〜2.60×104(J/m3)1/2であるエポキシ樹脂は、本発明における(a)成分の分子量3万未満のポリフェニルスルホン樹脂と相溶性が高く、硬化後にエポキシ樹脂とポリフェニルスルホン樹脂とが均一な相を形成しやすい。一方で、SP値が1.60×104〜1.80×104(J/m3)1/2であるエポキシ樹脂は、本発明の(a)成分の分子量3万未満のポリフェニルスルホン樹脂との相溶性が低く、硬化後にエポキシ樹脂とポリフェニルスルホン樹脂が相分離構造を形成しやすい。このため硬化後にポリフェニルスルホン樹脂とエポキシ樹脂間で相分離構造が形成されやすく、硬化後の強靭性を向上させることができる。さらに、前述のSP値が2.00×104〜2.60×104(J/m3)1/2であるエポキシ樹脂は、耐熱性を向上させることができ、前述のSP値が1.60×104〜1.80×104(J/m3)1/2であるエポキシ樹脂は、反応希釈剤として容易に低粘度特性を向上させることができる。以上のことから、これらのエポキシ樹脂をそれぞれ一種以上含有することにより、硬化後の強靭性と耐熱性を向上させることができ、また、より容易に低粘度特性を得ることができる。
【0038】
SP値が1.60×104〜1.80×104(J/m3)1/2であるエポキシ樹脂としては、2官能型の脂肪族あるいは脂環式グリシジルエーテル型エポキシ樹脂が好ましく、EX−212L(SP値=8.25(cal/cm3)1/2=1.69×104(J/m3))1/2)、EX−214L(SP値=8.28(cal/cm3)1/2=1.69×104(J/m3)1/2)、EX−216L(SP値=7.93(cal/cm3)1/2=1.62×104(J/m3)1/2)、EX−850L(SP値=8.42(cal/cm3)1/2=1.72×104(J/m3)1/2)(以上、商品名、ナガセケムテックス(株)製)、“エピクロン(登録商標)”705(SP値=8.10(cal/cm3)1/2=1.66×104(J/m3)1/2)、“エピクロン”707(SP値=7.98(cal/cm3)1/2=1.63×104(J/m3)1/2)、“エピクロン”720(SP値=8.45(cal/cm3)1/2=1.73×104(J/m3)1/2)、“エピクロン”726(SP値=8.25(cal/cm3)1/2=1.69×104(J/m3)1/2)、EXA−4880(SP値=8.35(cal/cm3)1/2=1.71×104(J/m3)1/2)、EXA−4882(SP値=7.98(cal/cm3)1/2=1.63×104(J/m3)1/2)(以上、商品名、DIC(株)製)、エポライト40E(SP値=8.33(cal/cm3)1/2=1.70×104(J/m3)1/2)、エポライト100E(SP値=8.42(cal/cm3)1/2=1.72×104(J/m3)1/2)、エポライト200E(SP値=8.50(cal/cm3)1/2=1.74×104(J/m3)1/2)、エポライト400E(SP値=8.58(cal/cm3)1/2=1.76×104(J/m3)1/2)、エポライト70P(SP値=8.13(cal/cm3)1/2=1.66×104(J/m3)1/2)、エポライト200P(SP値=8.10(cal/cm3)1/2=1.66×104(J/m3)1/2)、エポライト400P(SP値=7.98(cal/cm3)1/2=1.63×104(J/m3)1/2)、エポライト1500NP(SP値=8.45(cal/cm3)1/2=1.73×104(J/m3)1/2)、エポライト1600(SP値=8.25(cal/cm3)1/2=1.69×104(J/m3)1/2)(以上、商品名、共栄社化学(株)製)、リカレジンDME−100(SP値=7.93(cal/cm3)1/2=1.62×104(J/m3)1/2)、(商品名、新日本理化(株)製)、YED216(SP値=8.25(cal/cm3)1/2=1.69×104(J/m3)1/2)、YED216D(SP値=8.25(cal/cm3)1/2=1.69×104(J/m3)1/2)、YED216M(SP値=8.25(cal/cm3)1/2=1.69×104(J/m3)1/2)(以上、商品名、ジャパンエポキシレジン(株)製)などが挙げられる。これらを2種以上含有してもよい。
【0039】
SP値が2.00×104〜2.60×104(J/m3)1/2であるエポキシ樹脂としては、低粘度性を有する観点から2官能型のビスフェノールF型、ビスフェノールA型エポキシ樹脂が好ましい。また、耐熱性の観点からは、フェノールノボラック型、クレゾールノボラック型エポキシ樹脂、ナフタレン型エポキシ樹脂、ビフェニル型エポキシ樹脂が好ましい。
【0040】
2官能型のビスフェノールF型エポキシ樹脂としては、“エピクロン(登録商標)”830(SP値=11.47(cal/cm3)1/2=2.35×104(J/m3)1/2)(以上、商品名、DIC(株)製)、RE−303S(SP値=11.47(cal/cm3)1/2=2.35×104(J/m3)1/2)(以上、商品名、日本化薬(株)製)、“jER(登録商標)”806(SP値=11.50(cal/cm3)1/2=2.35×104(J/m3)1/2)、“jER”807(SP値=11.50(cal/cm3)1/2=2.35×104(J/m3)1/2)(以上、商品名、ジャパンエポキシレジン(株)製)、2官能型のビスフェノールA型エポキシ樹脂としては、840(SP値=12.31(cal/cm3)1/2=2.52×104(J/m3)1/2)、850(SP値=12.31(cal/cm3)1/2=2.52×104(J/m3)1/2)、860(SP値=12.31(cal/cm3)1/2=2.52×104(J/m3)1/2)((以上、商品名、DIC(株)製)、RE−310S(SP値=12.31(cal/cm3)1/2=2.52×104(J/m3)1/2)(以上、商品名、日本化薬(株)製)、“jER”827(SP値=12.31(cal/cm3)1/2=2.52×104(J/m3)1/2)、“jER”828(SP値=12.31(cal/cm3)1/2=2.52×104(J/m3)1/2)、“jER”834(SP値=12.35(cal/cm3)1/2=2.53×104(J/m3)1/2)(以上、商品名、ジャパンエポキシレジン(株)製)、フェノールノボラック型エポキシ樹脂としては、“エピクロン”N−540(SP値=11.26(cal/cm3)1/2=2.30×104(J/m3)1/2)、N−740、N−770(いずれもSP値=11.30(cal/cm3)1/2=2.30×104(J/m3)1/2)、(以上、商品名、DIC(株)製)、EPPN−201(SP値=11.26(cal/cm3)1/2=2.30×104(J/m3)1/2)(以上、商品名、日本化薬(株)製)、“jER”152、154(いずれもSP値=11.30(cal/cm3)1/2=2.30×104(J/m3)1/2)、クレゾールノボラック型エポキシ樹脂としては、“エピクロン”N−660、N−665、N−670、N−680、N−690(いずれもSP値=10.28(cal/cm3)1/2=2.10×104(J/m3)1/2)(以上、商品名、DIC(株)製)、EOCN−1020(SP値=10.40(cal/cm3)1/2=2.13×104(J/m3)1/2)、EOCN−102S(SP値=10.44(cal/cm3)1/2=2.14×104(J/m3)1/2)、EOCN−103S(SP値=10.48(cal/cm3)1/2=2.14×104(J/m3)1/2)、EOCN−104S(SP値=10.52(cal/cm3)1/2=2.15×104(J/m3)1/2)、(以上、商品名、日本化薬(株)製)、ナフタレン型エポキシ樹脂としては、“エピクロン”HP4700、EXA4710、HP4770(いずれもSP値=10.09(cal/cm3)1/2=2.06×104(J/m3)1/2)、(以上、商品名、DIC(株)製)、ビフェニル型エポキシ樹脂としては“jER”YX4000(SP値=9.89(cal/cm3)1/2=2.02×104(J/m3)1/2)、YL6121H(SP値=11.44(cal/cm3)1/2=2.34×104(J/m3)1/2)、(以上、商品名、ジャパンエポキシレジン(株)製)、NC3000、NC3000H(いずれもSP値=11.23(cal/cm3)1/2=2.30×104(J/m3)1/2)、(以上、商品名、日本化薬(株)製)などが挙げられる。これらを2種以上含有してもよい。
【0041】
さらに、(b)エポキシ樹脂に加えて、オキセタン樹脂を含有することができる。オキセタン樹脂を含有することで、熱安定性が向上するため、硬化後の耐熱性を向上させることができる。このようなオキセタン樹脂のうち、1官能性オキセタン樹脂としては、エタナコール(登録商標)”EHO、“エタナコール”OXMA、(以上、商品名、宇部興産(株)製)、OXT−101、OXT−211、OXT−212、OXT−610(以上、商品名、東亜合成(株)製)、3−エチル−3−(シクロヘキシロキシ)メチルオキセタンが挙げられ、2官能性オキセタン樹脂としては、“エタナコール“OXBP、“エタナコール”OXTP、“エタナコール”OXIPA(以上、商品名、宇部興産(株)製)、OXT−121、OXT−221(以上、商品名、東亜合成(株)製)、3つ以上のオキセタニル基を有する樹脂としては、オキセタン化フェノール樹脂や、オキセタニルシリケート、フェノールノボラック型オキセタン樹脂が挙げられる。これらを2種以上含有してもよい。
【0042】
本発明のアンダーフィル剤は、粘度を調節したり、塗布性を向上する目的で、(c)溶剤を1重量%以下含有することもできる。溶剤含有量を1重量%以下とすることで、熱処理時の溶媒飛散によるボイドの発生を抑制することができる。
【0043】
溶剤としては、例えば、ケトン系溶剤のアセトン、メチルエチルケトン、メチルイソブチルケトン、シクロペンタノン、シクロヘキサノン、エーテル系溶剤の1,4−ジオキサン、テトラヒドロフラン、ジグライム、グリコールエーテル系溶剤のメチルセロソルブ、エチルセロソルブ、プロピレングリコールモノメチルエーテル、プロピレングリコールモノエチルエーテル、プロピレングリコールモノブチルエーテル、ジエチレングリコールメチルエチルエーテル、芳香族炭化水素系溶剤のベンゼン、キシレン、トルエン、その他ベンジルアルコール、プロパノール、N−メチルピロリドン、γ−ブチロラクトン、酢酸エチル、N,N−ジメチルホルムアミドなどが挙げられる。これらを2種以上含有してもよい。特に大気圧下沸点が120℃以下であるものを用いると、低温、短時間で脱溶媒できるため好ましい。
【0044】
本発明のアンダーフィル剤は、(d)シリカ、チタニア、ジルコニア、窒化ケイ素、アルミナ、セリア、タルクおよび炭酸カルシウムからなる群より選ばれる少なくとも一種の無機微粒子を含有することができる。これらの無機微粒子を含有することにより、熱伝導性を付与したり、硬化後の熱線膨張係数を低減することができる。熱伝導性を付与する場合は、炭酸カルシウム、シリカ、アルミナ、チタニア、シリカ−チタニア複合粒子が好ましい。さらに、無機微粒子とエポキシ樹脂の界面を強固に結合するために、無機微粒子表面をシラン系、チタン系、アルミニウム系などの各種カップリング剤、脂肪酸、リン酸エステルなどで処理したものや、ロジン処理、酸性処理、塩基性処理を施したものも好適に用いられる。特にエポキシ基含有シランカップリング剤は、強靭性を高める点から好ましく用いられる。シランカップリング剤の好ましい例としては、γ−グリシドキシトリメトトキシシラン、γ−グリシドキシトリエトトキシシラン、γ−グリシドキシトリプロポキシシラン、2−(3,4−エポキシシクロヘキシル)エチルトリメトキシシラン、2−(3,4−エポキシシクロヘキシル)エチルトリエトキシシラン、2−(3,4−エポキシシクロヘキシル)エチルトリプロポキシシラン、3−アミノプロピルトリメトキシシラン、3−アミノプロピルトリエトキシシラン、3−アミノプロピルトリプロポキシシランなどが挙げられる。これらを2種以上用いてもよい。
【0045】
これら(d)無機微粒子の含有量は、(a)成分のポリフェニルスルホン樹脂と(b)成分のエポキシ樹脂との総量100重量部に対して10〜5000重量部が好ましい。無機微粒子の含有量が10重量部以上であることで、熱伝導性を向上させたり、熱線膨張係数をより低減したりすることができ、5000重量部以下とすることでアンダーフィル剤の粘度上昇を防ぐことができる。より好ましくは20重量部以上500重量部以下である。
【0046】
また、無機微粒子の平均粒子径は5nm〜30μmが好ましく、さらに好ましくは5nm〜10μmである。ここで、本発明における平均粒子径は、超薄切片法を用いて調製した厚さ20μmのコーティング被膜の断面を、透過型電子顕微鏡(日立製作所(株)製、H−7100FA型)を用いて、加速電圧100kVの条件にて観察倍率200000倍で観察し、得られた像から個々の粒子単一の粒子径を測定し、それらの数平均値とする。測定に用いる粒子の個数は数百から数千個である。平均粒子径が5nm以上であることで、適度な粘性を付与すると同時に安定した粒子分散性を得ることができ、30μm以下とすることで、低粘度特性を向上させ、無機微粒子の沈降を防ぐことができる。
【0047】
本発明のアンダーフィル剤は、(e)ポリイミド、ポリアミド、ポリアミドイミド、ポリスチレン、ポリアクリロニトリル、ポリフェニレンエーテル、ポリエステルおよびポリカーボネートからなる群より選ばれる少なくとも一種の有機微粒子を含有することができる。これらの有機微粒子を含有することにより、耐熱性や低吸湿性を付与することができる。
【0048】
これら(e)有機微粒子の含有量は、(a)成分のポリフェニルスルホン樹脂と(b)成分のエポキシ樹脂との総量100重量部に対して5〜5000重量部が好ましい。有機微粒子の含有量が5重量部以上であることで、耐熱性や低吸湿性をより容易に付与することができ、5000重量部以下とすることで樹脂組成物の粘度上昇を防ぐことができる。
【0049】
また、有機微粒子の平均粒子径は5nm〜10μmが好ましく、さらに好ましくは5nm〜5μmである。有機微粒子の平均粒子径の測定方法は、上記の無機微粒子の平均粒子径の測定方法と同様である。
【0050】
その他にフェノキシ樹脂、ポリウレタン、ポリプロピレン、アクリロニトリル−ブタジエン共重合体(NBR)、スチレン−ブタジエン共重合体、(SBR)、アクリロニトリル−ブタジエン−メタクリル酸共重合体、アクリロニトリル−ブタジエン−アクリル酸共重合体などを含有してもよい。
【0051】
本発明のアンダーフィル剤は、さらに硬化剤や硬化促進剤を含有することができる。ここで、硬化剤とは、単独で硬化する作用を有するものをいい、硬化促進剤とは、硬化剤とともに用いて反応を促進する作用を有するものをいう。本発明のアンダーフィル剤に用いることのできる硬化剤または硬化促進剤としては、脂肪族ポリアミン、脂環式ポリアミン、芳香族ポリアミン、酸無水物、フェノール樹脂、ジシアンジアミドとその誘導体、イミダゾール類とその誘導体、アミン誘導体とホルムアルデヒドの縮合体(尿素ホルムアルデヒド、メラミンホルムアルデヒド)、3級アミン化合物、有機金属錯体、ポリチオール類、オニウム塩類などが挙げられる。これらを2種以上含有してもよい。
【0052】
具体的には、脂肪族ポリアミンとしては、ジエチルトリアミン、トリエチレンテトラミン、キシレンジアミンなど、脂環式ポリアミンとしては、イソホロンジアミン、1,3−ビスアミノメチルシクロヘキサン、ノルボルネンジアミンなど、芳香族ポリアミンとしては、メタフェニレンジアミン、ジアミノジフェニルメタン、ジアミノジフェニルスルホン、ビスアミノフェノキシベンゼンなどのフェノキシ系ジアミンなどが挙げられる。また、酸無水物としては、無水こはく酸、イタコン酸無水物、無水フタール酸、ピロメリット酸二無水物、ベンゾフェノンテトラカルボン酸二無水物、ビフェニルテトラカルボン酸二無水物、メチルヘキサヒドロフタル酸無水物、ヘキサハイドロフタル酸無水物、メチルテトラハイドロフタル酸無水物、“アデカハードナー(登録商標)”EH−3326、“アデカハードナー”EH−703、“アデカハードナー”EH−705A(以上、商品名、旭電化工業(株)製)、“エピクロン”B−570、“エピクロン”B−650(以上、商品名、大日本インキ化学(株)製)、“リカシッド(登録商標)”MH−700(商品名、新日本理化(株)製)などが挙げられる。フェノール樹脂としては、各種フェノールノボラック樹脂やクレゾールノボラック樹脂、MEH−7500、MEH−7800、MEH−7851、MEH−8000H(以上、商品名、明和化成(株)製)などが挙げられる。ジシアンジアミドとその誘導体としては、DICY7、DICY15、DICY50(以上、商品名、ジャパンエポキシレジン(株)製)、“アミキュア(登録商標)”AH−154、“アミキュア”AH−162(以上、商品名、味の素ファインテクノ(株)製)などが挙げられる。イミダゾール類とその誘導体としては、イミダゾール、2−メチルイミダゾール、2−ウンデシルイミダゾール、2−エチル−4−メチルイミダゾール、2−フェニル−4−メチルイミダゾール、2−フェニル−4−メチル−5−ヒドロキシイミダゾール、イミダゾール、2−フェニルイミダゾール、1−ベンジル−2−メチルイミダゾール、1−ベンジル−2−フェニルイミダゾール、1−シアノエチル−2−メチルイミダゾール、1−シアノエチル−2−エチル−4−メチルイミダゾール、1−シアノエチル−2−フェニルイミダゾール、1−シアノエチル−2−フェニルイミダゾリウムトリメリテイトや、IS−1000、IS−1000D、IM−1000、SP−1000、IA−100A、IA−100P、IA−100F(以上、商品名、日鉱マテリアルズ(株)製)などのイミダゾールシランなどが挙げられる。アミン誘導体とホルムアルデヒドの縮合体(尿素ホルムアルデヒド、メラミンホルムアルデヒド)としては、4−クロロ−フェニル−N,N−ジメチル尿素、3,4−ジクロロフェニル−N,N−ジメチル尿素(DCMU)などが挙げられる。3級アミン化合物としては、1,8−ジアザビシクロ(5,4,0)−ウンデセン−7(DBU)、1,5−ジアザビシクロ(4,3,0)−ノネン−5(DBN)およびその塩が挙げられ、具体的には、U−CATSA1(DBU−フェノール塩)、U−CAT SA102(DBU−オクチル酸塩)、U−CATSA603(DBU−蟻酸塩)、U−CAT SA810(DBU−オルソフタル酸塩)、U−CATSA841(DBU−フェノールノボラック樹脂塩)、U−CAT SA881(DBN−フェノールノボラック樹脂塩)(以上、商品名、サンアプロ(株)製)などが挙げられる。有機金属錯体としては、トリフェニルホスフィン、トリフェニルホスホニウムトリフェニルボレートなど、ポリチオール類としては、脂肪族ポリチオエーテル、脂肪族ポリチオエステル、芳香族環含有ポリチオエーテルなどが挙げられる。オニウム塩類としては、スルホニウムやヨードニウムなどのオニウム塩が挙げられ、オニウム塩型のジフェニルヨードニウムヘキサフロロホスフェート、トリフェニルスルホニウムヘキサフロロホスフェート、サイラキュアーUVI−6992、サイラキュアーUVI−6974(以上、商品名、ダウ・ケミカル日本(株)製)、アデカオプトマーSP150、アデカオプトマーSP170(以上、商品名、旭電化工業(株)製)、サンエイドSI−60L、SI−80L、SI−100L、SI−150L(以上、商品名、三新化学工業(株)製)などが挙げられる。
【0053】
その他に、エポキシ樹脂に混合した状態で長時間保存でき、熱・光・圧力・湿気などの刺激を与えると硬化反応を開始する潜在性硬化剤の例としては、加熱硬化型潜在性硬化剤、マイクロカプセル型潜在性硬化剤、アミンアダクト型潜在性硬化剤などが挙げられる。加熱硬化型潜在性硬化剤の具体例としては、ジシアンジアミド、3,4−ジクロロフェニル−N,N−ジメチル尿素(DCMU)が挙げられる。マイクロカプセル型潜在性硬化剤は、コア(芯物質)/シェル(カプセル膜)構造を有する硬化剤である。コアとしては、種々のイミダゾール化合物やトリフェニルホスフィンなど、シェルとしては、有機ポリマーや無機化合物などが挙げられる。具体的には、“ノバキュア(登録商標)”HX−3941HP、“ノバキュア”HXA3922HP、“ノバキュア”HXA3932HP、“ノバキュア”HXA3042HP(以上商品名、旭化成ケミカルズ(株)製)などが挙げられる。
【0054】
アミンアダクト型潜在性硬化剤は、イミダゾール化合物、3級アミノ基含有化合物またはヒドラジド化合物をエポキシ樹脂やイソシアネート化合物などと反応させて高分子量化したものを微粉砕化したものであり、常温での溶解度が低く、潜在性を示す。具体的には、“アミキュア”PN−23、“アミキュア”PN−40、“アミキュア”MY−24、“アミキュア”MY−H(以上、商品名、味の素ファインテクノ(株)製)、“フジキュア(登録商標)”FXR−1030(商品名、富士化成(株)製)、“アミキュア”VDH、“アミキュア”UDH(以上、商品名、味の素ファインテクノ(株)製)などが挙げられる。
【0055】
本発明においては、アンダーフィル剤への硬化性と保存安定性が良好な点から、芳香族ポリアミンがより好ましい。このような芳香族ポリアミンの例としては、メタフェニレンジアミン、パラフェニレンジアミン、2,4−トルエンジアミン、2,5−トルエンジアミン、ジメチルトルエンジアミン、ジエチルトルエンジアミン、4,4’−ジアミノジフェニルメタン、4,4’−ジアミノ−3,3’−ジメチル−ジフェニルメタン、3,3’−ジアミノ−4,4’−ジメチル−ジフェニルメタン、4,4’−ジアミノ−3,3’−ジクロロ−ジフェニルメタン、3,3’−ジアミノ−4,4’−ジクロロ−ジフェニルメタン、4,4’−ジアミノ−3,3’,5,5’−テトラメチル−ジフェニルメタン、4,4’−ジアミノ−3,3’−ジエチル−ジフェニルメタン、4,4’−ジアミノ−3,3’,5,5’−テトラエチル−ジフェニルメタン、3,3’−ジアミノジフェニルスルホン、4,4’−ジアミノジフェニルスルホン、3,3’−ジアミノジフェニルスルフィド、4,4’−ジアミノジフェニルスルフィド、ビス(p−(m−アミノフェノキシ)フェニル)スルホン、ビス(p−(p−アミノフェノキシ)フェニル)スルホン、3,3’−ビス(p−アミノフェノキシ)ビフェニル、2,2’−ビス(p−アミノフェノキシ)フェニル)−1,1,1,3,3,3−ヘキサフルオロプロパン、4,4’−ビス(m−アミノフェノキシ)ビフェニル、m−ビス(p−アミノフェノキシ)ベンゼン、p−ビス(p−アミノフェノキシ)ベンゼン、m−ビス(m−アミノフェノキシ)ベンゼン、1,3−ビス(4−アミノ−3−ヒドロキシフェノキシ)ベンゼン、p−ビス(p−アミノフェノキシ)ジフェニルエーテル、p−ビス(m−アミノフェノキシ)ジフェニルエーテル、ビス(p−(p−アミノフェノキシ)フェノキシ)ベンゼンなどが挙げられる。この中でも高い融点を持ち、保存安定性が良好な点から芳香族スルホン系ジアミンが好ましく用いられる。芳香族スルホン系ジアミンとしては、3,3’−ジアミノジフェニルスルホン、4,4’−ジアミノジフェニルスルホンが挙げられ、市販品としてはセイカキュアS(商品名、セイカ(株)製)が挙げられる。
【0056】
さらに好適な硬化剤としては、フェノキシ系ジアミンが挙げられる。フェノキシ系ジアミンは、エーテル結合を有し、硬化後の樹脂の靭性を向上させることができる。さらに、下記一般式(1)で表されるフェノキシ系ジアミンは、室温および高温におけるアンダーフィル剤の粘度を大きく下げる作用を有する。
【0057】
【化1】
【0058】
上記一般式(1)中、nは2〜4の整数を表す。R1〜R4は水素原子、水酸基、ハロゲン原子、炭素数1〜3のアルキル基、アルコキシ基、フルオロアルキル基またはアリール基を表し、同じでも異なっていてもよい。アルキル基は鎖状でも環状でもよい。
【0059】
前記一般式(1)で表されるフェノキシ系ジアミンは、特に、SP値が1.60×104〜1.80×104(J/m3)1/2であるエポキシ樹脂と、SP値が2.00×104〜2.60×104(J/m3)1/2であるエポキシ樹脂からそれぞれ一種類以上選ばれてなるエポキシ樹脂と組み合わせて用いることが好ましく、硬化後の強靭性を向上させることができる。前記一般式(1)で表されるフェノキシ系ジアミンとしては、m−ビス(p−アミノフェノキシ)ベンゼン、p−ビス(p−アミノフェノキシ)ベンゼン、m−ビス(m−アミノフェノキシ)ベンゼン、3,3’−ビス(3”−アミノフェノキシ)ジフェニルエーテル、4,4’−ビス(3”−アミノフェノキシ)ジフェニルエーテル、ビス(p−(p−アミノフェノキシ)フェノキシ)ベンゼン、ビス(m−(m−アミノフェノキシ)フェノキシ)ベンゼン、1,3−ビス(4−アミノ−3−ヒドロキシフェノキシ)ベンゼンなどが挙げられ、市販品として、APB、APB−N、APB−NP、APB−5(以上、商品名、三井化学(株)製)、TPE−R、TPE−Q(以上、商品名、和歌山精化工業(株)製)、4−APB、3,4’−DAPE、3−APB、AHPB(以上、商品名、日本純良薬品(株)製)などが挙げられる。
【0060】
その他に本発明で好ましく用いられる硬化促進剤としては、ポリフェニルスルホン樹脂とエポキシ樹脂に常温で非相溶で混合できることから、加熱硬化型潜在性硬化剤である3−フェニル−N、N−ジメチル尿素、4−クロロフェニル−N,N−ジメチル尿素、3,4−ジクロロフェニル−N,N−ジメチル尿素、3−クロロ−4−メチルフェニル−N,N−ジメチル尿素などが挙げられる。
【0061】
本発明のアンダーフィル剤において、硬化剤の含有量は(a)成分のポリフェニルスルホン樹脂と(b)エポキシ樹脂との総量100重量部に対して0.1〜60重量部が好ましい。硬化剤の含有量を0.1重量部以上とすることでエポキシ樹脂の硬化を効果的に行い、60重量部以下とすることで硬化物の高粘度化を防ぐことができる。より好ましくは20〜40重量部である。
【0062】
硬化剤の形状は、液状、固体状でも特に限定されないが、固体状の場合は、分散性や溶解性の点から粉末状が好ましい。
【0063】
本発明の熱硬化性樹脂組成物は、さらに熱架橋剤を含有してもよい。熱架橋剤としては、例えば、エチニル基、ビニル基、メチロール基、メトキシメチロール基などを有する化合物や、ベンゾオキサジン化合物を挙げることができる。さらに、無水マレイン酸、エチニルフタール酸無水物、エチニルアニリン、ビニルアニリンなどのように熱架橋基とエポキシ樹脂の硬化機能を有する化合物を含有してもよい。
【0064】
そのほか、本発明のアンダーフィル剤は、ノニオン性、カチオン性、アニオン性の界面活性剤、多価カルボン酸などの湿潤剤、両親和性物質、高立体障害の置換基を有する樹脂などを含有してもよい。また、必要に応じて、安定化剤、分散剤、沈降防止剤、可塑剤、酸化防止剤などを含有してもよい。
【0065】
一般的にアンダーフィル剤として使用する場合、基板温度である90〜140℃の温度範囲内で、粘度の最低値が1.0×10Pa・s以下であると、半導体素子と基板の隙間に速やかに充填することができる。このため、本発明のアンダーフィル剤は、動的粘弾性測定(昇温速度2℃/分)により得られる粘弾性−温度曲線において、粘度の最低値η*が1.0×102Pa・s以下であり、かつ粘度の最低値を示す温度が90〜140℃であることが好ましい。さらに粘度の最低値η*が1.0Pa・s以下であることがより好ましく、また粘度の最低値を示す温度が100〜120℃であることがより好ましい。
【0066】
アンダーフィル剤の粘度は、平行円板型の測定システムを用いた動的粘弾性測定により求める。平行円板型の場合、両円板の間に試料を満たし、一方の円板を一定の周波数で振動させ、その際に他方の円板に生じるトルクと位相差から剛性率、粘度、tanδを求める。ここでいう粘度とは、測定によって得られる複素粘性率を示し、単位はPa・sである。本発明においては、振幅0.5deg、角周波数3s−1、周波数0.5Hz、測定温度25℃〜170℃、昇温速度2℃/分、N2ガス気流中の条件で測定する。
【0067】
このような粘度特性を有するアンダーフィル剤を得るためには、(a)成分のポリフェニルスルホン樹脂の含有量を20重量%以下にすることが好ましく、10重量%以下がより好ましい。また、反応希釈剤として、低粘度性を有するエポキシ樹脂を用いることが好ましい。低粘度性を有するエポキシ樹脂は、25℃、1.013×105Paにおける粘度が200mPa・s未満であることが好ましい。25℃、1.013×105Paにおける粘度が200mPa・s未満のエポキシ樹脂の具体例としては、YED216M,YED216D,YED111N、YED122N(以上、商品名、ジャパンエポキシレジン(株)製)、エポライト40E、エポライト100E、エポライト200E、エポライト400E、エポライト70P、エポライト200P、エポライト400P、エポライト1500NP、エポライト1600、エポライト80MF、エポライト100MF(以上、商品名、共栄社化学(株)製)、EX−214L、EX−212L、EX−216L、EX−850L(以上、商品名、ナガセケムテックス(株)製)、“エピクロン(登録商標)”EXA−4880、“エピクロン”520、“エピクロン”703、“エピクロン”705、“エピクロン”725、“エピクロン”727、(以上、商品名、DIC(株)製)が挙げられる。この中でも具体的にはSP値が1.60×104〜1.80×104(J/m3)1/2であるエポキシ樹脂が好ましい。また、ヒドロキシメチルオキセタン、エチルオキセタンメタノール、メチルオキセタンメタノールなどの1価のオキセタン樹脂を含有することによっても粘度を低下させることができる。
【0068】
アンダーフィル剤の粘度を上記範囲に調整するその他の手法としては、硬化剤として前記一般式(1)で表されるフェノキシ系ジアミンを用いる方法が挙げられ、室温から高温にかけて粘度を大きく下げることができる。
【0069】
アンダーフィル剤の反応開始温度を90℃付近にするためには、硬化剤として脂肪族系ポリアミンや2級アミンなどのアミン類、イミダゾール類が好適に用いられる。また反応開始温度を90〜140℃付近にするためには、3級アミン、芳香族系アミン、マイクロカプセル型潜在性硬化剤が好適に用いられる。さらに140℃を越えて硬化反応を開始させたい場合は、DCMU、DCMU型潜在性硬化剤、DICY(ジシアンジアミド)、DICY型潜在性硬化剤などが好適に用いられる。また、硬化剤を2種以上含有したり、硬化促進剤と組み合わせることにより、硬化速度を調節してもよい。
【0070】
本発明のアンダーフィル剤は、硬化後の耐熱性の指標として、硬化後のガラス転移温度が90℃以上であることが好ましく、140℃以上がより好ましい。かかるガラス転移温度は、(a)重量平均分子量3万未満のポリフェニルスルホン樹脂の高いガラス転移温度や、エポキシ樹脂および硬化剤の反応基数に起因する。
【0071】
本発明のアンダーフィル剤の硬化後の破壊靭性値K1cは2.0MPa・m1/2以上であることが好ましく、さらに好ましくは2.5MPa・m1/2以上である。硬化後の破壊靭性値K1cが2.0MPa・m1/2以上であれば、本発明のアンダーフィル剤の硬化後の半導体素子と基板間の接続信頼性や耐熱衝撃性を高めることができる。
【0072】
破壊靭性値の測定・算出方法は、ASTMD5045−93[プラスチック材料の平面歪み破壊靭性及び歪みエネルギー解放率](=ISO13586)により規定されている。本発明においては、厚み6mm×幅12mm×長さ52.8mmの試験片に予備クラックを入れ、テンシロン万能試験機(エーアンドデイ(株)製)を用いて、試験温度23℃、クロスヘッド速度10mm/分、エッジスパン間隔48mmで3点曲げ試験を行い、き裂長さおよび最大破壊荷重から破壊靭性値K1cを求める。K1cの値を求める式は次の式で表され、単位はMPa・m1/2である。
K1c=PmaxSf(a/W)/BW3/2
ただし、Pmaxは最大破壊荷重[kN]、Sはスパン間距離(治具ローラー間距離)[cm]、aはき裂長さ[cm]、Wは試験片幅[cm]、Bは試験片厚さ[cm]である。またf(a/W)は、a/W=X、S/W=4.0とし、f(X)は次の式で求められる。
【0073】
【数2】
【0074】
このような硬化後の破壊靭性値を有するアンダーフィル剤は、エポキシ樹脂の強靭化剤として、ポリフェニルスルホン樹脂を含有しており、ポリフェニルスルホン樹脂の末端がエポキシ基との反応性基を有することが好ましく、より好ましくはフェノール性水酸基が挙げられる。また、強靭性と低粘度特性をより高いレベルで両立するためには、ポリフェニルスルホン樹脂の重量平均分子量が3万未満であり、さらに好ましくは重量平均分子量が2万以下、5000以上である。また(b)成分のエポキシ樹脂が、SP値が1.60×104〜1.80×104(J/m3)1/2であるエポキシ樹脂と、SP値が2.00×104〜2.60×104(J/m3)1/2であるエポキシ樹脂とをそれぞれ一種以上含有し、さらに硬化剤として、前記一般式(1)で表されるフェノキシ系ジアミンを含有することが好適である。
【0075】
本発明のアンダーフィル剤は、例えば、(a)成分のポリフェニルスルホン樹脂と(b)成分のエポキシ樹脂を混合し、均一な溶液にすることで得ることができる。
【0076】
(b)エポキシ樹脂には液状と固体状の2種類があり、固体状のエポキシ樹脂の場合は、液状のエポキシ樹脂にあらかじめ溶解させるのがよい。この場合、各固体状エポキシ樹脂の軟化点および融点を目処に、軟化点および融点の−10℃〜+30℃の温度範囲で熱溶解させることが好ましい。
(b)以外のエポキシ樹脂、有機/無機微粒子、硬化剤、硬化促進剤などを含有する場合は、前記の(a)成分のポリフェニルスルホン樹脂と(b)成分のエポキシ樹脂を混合し、溶解させた後でこれらを添加し、混合することが好ましい。
【0077】
硬化剤および硬化促進剤は液状と固体状の2種類があり、固体状の硬化剤および硬化促進剤の場合は、液状の硬化剤および硬化促進剤に溶解させても良いし、液状のエポキシ樹脂に溶解させてもよい。この場合、溶解方法は特に限定されないが、熱溶解する場合、30℃〜180℃の温度範囲で、0.5時間〜8時間撹拌することが好ましく、より好ましくは50℃〜80℃の範囲で1時間〜2時間である。
【0078】
また、硬化剤および硬化促進剤の添加順序は特に限定されないが、反応性が高い硬化剤および硬化促進剤は保存安定性を考慮して最後に添加することが好ましい。一方、反応性が低い硬化剤および硬化促進剤はあらかじめエポキシ樹脂に添加し、溶解させて用いてもよい。
【0079】
無機微粒子または有機微粒子を含有する場合、分散方法は特に限定されないが、特に分散性の点でボールミル、ホモジナイザーを用いることが好ましい。
【0080】
無機微粒子の分散性を向上させるために、例えば、分散剤や界面活性剤、溶剤を添加してもよい。分散剤の例としては、リン酸、カルボン酸、脂肪酸、およびそれらのエステル類などの酸基を有する分散剤などが挙げられる。特に、無機微粒子表面の水酸基と反応し、粒子表面を覆うことができることから、リン酸化合物が好ましく用いられる。
【0081】
本発明のアンダーフィル剤は、半導体素子と基板を接合するための接着剤(アンダーフィル剤)として好適に用いられる。その他にも、本発明のアンダーフィル剤を硬化して得られる膜を、半導体素子の保護膜、高密度実装用多層配線用の層間絶縁膜、ビルドアッププリント配線板用の層間絶縁膜、回路基板の配線保護絶縁膜などの用途に用いることもできる。
【0082】
本発明のアンダーフィル剤の充填方法としては、半導体素子と基板の空隙にアンダーフィル剤を滴下する方法などが一般的であり、その他ディスペンサー法や印刷法で塗布する方法が挙げられる。また塗布後のアンダーフィル剤の充填速度は好ましくは0.5cm/分以上、より好ましくは1.0cm/分以上である。
【0083】
本発明のアンダーフィル剤を保存する際の温度は−10〜25℃の範囲が好ましく、より好ましくは9℃以下、さらに好ましくは5℃以下である。
【0084】
本発明のアンダーフィル剤は、塗布前に予め呼び加熱処理しておき、粘度を下げてから塗布しても差し支えない。加熱温度は30℃〜60℃が好ましい。60℃を超えると加熱によりエポキシ樹脂の硬化反応が始まり粘度が上昇する場合がある。また、30℃を下回ると、粘度が下がらない場合がある。また過熱処理後は保存安定性の観点から0.1秒〜30分の間に塗布することが好ましい。
【0085】
本発明のアンダーフィル剤を半導体素子と基板の空隙に充填し、硬化させる。アンダーフィル剤の硬化条件としては、通常は室温以上250℃以下の温度で0.5分間〜10時間の処理を行うが、他の電子部品などの損傷防止およびラインタクト短縮のために、低温硬化性および短時間化が望ましい。本発明のアンダーフィル剤の硬化条件は160℃〜250℃の温度範囲で30分間〜5時間が好ましい。これ以外の硬化条件としては50〜160℃の低温から中温で1〜100時間硬化する方法や、250℃〜300℃の高温で1秒〜3時間硬化する方法も挙げられ、また予備加熱として50〜160℃の低温〜中温で1分〜1時間行ってもよい。
【0086】
本発明のアンダーフィル剤を用いた半導体装置において、アンダーフィル剤は半導体素子と基板間に位置する。半導体装置の好ましい製造例としては、バンプを有する基板素子を、配線基板表面と接合させ、接合と同時、または接合後にアンダーフィル剤を半導体素子と基板間の空隙に充填し、熱硬化して半導体素子と基板が固着された半導体装置を得る方法が挙げられる。その他にアンダーフィル剤を先に補給する製造例としては、まず配線基板表面にアンダーフィル剤をディスペンサー法または印刷法などで塗布し、その後バンプを有する半導体素子を基板と接合させ、接合と同時、または接合後にアンダーフィル剤を熱硬化して半導体素子と基板が固着された半導体装置を得る方法が挙げられる。
【実施例】
【0087】
以下実施例および技術をあげて本発明を説明するが、本発明はこれらの例によって限定されるものではない。なお、実施例中の測定、評価は以下の方法により行った。
【0088】
(1)ポリフェニルスルホン樹脂およびポリエーテルスルホン樹脂の分子量の測定
ゲルパーミエーションクロマトグラフィー(GPC)法により、システムコントローラーWaters 2690(ウォーターズ(株)製)を用いて、NMP(LiCl(0.05mol/L))/H3PO4(0.05mol/L)=10/1(重量比)の展開溶媒を0.4mL/分の条件で用いてポリフェニルスルホン樹脂およびポリエーテルスルホン樹脂の分子量を測定し、標準ポリスチレンの校正曲線を用いて重量平均分子量(Mw)を算出した。
【0089】
(2)ガラス転移温度の測定
熱硬化前の樹脂組成物を“テフロン(登録商標)”製の鋳型に流しこみ、イナートオーブンに投入し、昇温速度1.5℃/分で170℃の硬化温度まで上昇させ、170℃で120分間加熱処理を行った。その後、オーブン内が50℃以下になるまで徐冷し、樹脂硬化物を得た。厚み4mm×幅4mm×長さ7mmの試料片を得た後、熱機械分析装置SS-6100を用いて、圧縮モード、温度範囲5〜300℃、昇温速度5℃/分、初期荷重0.5gの条件で成型体の圧縮量を測定した。得られた測定結果からdTMA/dtの極大点における温度をガラス転移温度とした。
【0090】
(3)末端水酸基含有率の測定
核磁気共鳴スペクトル(1H−NMR)測定法により、測定機器は日本電子EX−270型(270MHz)を用いた。測定溶媒にはCDCl3、DMSOを用いた。CDCl3を用いた場合は化学シフトσ7.26PPMをリファレンスとし、DMSOを用いた場合は化学シフトσ2.5PPMをリファレンスとした。サンプルを10mg秤量し、サンプルチューブに投入し、さらに測定溶媒を0.75mL投入し測定用サンプルとした。化学シフトがσ8.00〜σ6.00PPMにシフトされるポリマーユニット由来の芳香族環の水素原子と末端フェノール性水酸基と末端Cl基の検出ピークの積分強度比から末端水酸基含有率(モル%)を求めた。
末端水酸基含有率(モル%)=水酸基末端量/(水酸基末端量+Cl末端量)×100(モル%)
(4)粘度特性の測定
樹脂組成物を直径25mmの円板型アルミニウム製ディスポーザブルパラレルプレート二枚の間隔が0.5mmになるように二枚の平行円板の間に充填し、ついでAR−G2レオメーター(ティーエーインスツルメント(株)製)にて昇温速度2℃/分、角周波数3s−1(周波数は0.5Hz)、振幅0.5deg、窒素ガス気流中での測定条件で、25〜170℃における粘弾性−温度曲線を測定した。
【0091】
(5)溶解性の評価
樹脂組成物をガラスのスクリュー管に入れ、各サンプルを23℃で6時間静置後肉眼で観察し、アンダーフィル剤の溶解性を評価した。全体的に非相溶であり、均一にポリマーが分散されているものを非相溶、所々でポリマーがやや不均一に溶解され、一部凝集がみられるものを部分相溶、全体的に均一にポリマーが溶解されているものを溶解とした。
【0092】
(6)硬化開始温度の測定
樹脂組成物10mgをアルミニウムセルに入れ、シールしたものを測定用サンプルとした。示差走査熱量計DSC−50(島津製作所(株)製)を用いて、窒素流量20ml/分の条件で、昇温速度5℃/分、20℃〜300℃の温度範囲で熱量の測定を行った。得られたチャートを用いて硬化開始温度を次のようにして求めた。例えば、図1は、実施例2のアンダーフィル剤B−1のDSC曲線である。図1においてDSC曲線1は25〜100℃の温度範囲に認められる直線部分L1、立ち上がり後ピークPに到達するまでに認められる直線部分L2を有する。L1の延長線である直線2と、L2の延長線である直線3との交点4をアンダーフィル剤B−1の硬化開始温度とした。
【0093】
(7)破壊靭性値の測定
ASTM D5045に準じて破壊靭性値K1cの測定を行った。すなわち、アンダーフィル剤を“テフロン(登録商標)”製の鋳型に流し込み、イナートオーブン(光洋サーモシステム(株)製INH−21CD)に投入し、170℃の硬化温度まで80分間かけて上昇させ、170℃で120分間加熱処理を行った。その後、オーブン内が50℃以下になるまで徐冷し、硬化物を得た。得られた硬化物をオートカッターで切り出し、厚み6mm×幅12mm×長さ52.8mmの試料片を得た後、さらに予備クラックを入れ、テンシロン万能試験機(エーアンドデイ(株)製)を用いて、温度23℃、クロスヘッド速度10mm/分、エッジスパン間隔48mmの条件で3点曲げ試験を行い、測定したき裂長さおよび最大破壊荷重から、破壊靭性値K1c(MPa・m1/2)の値を求めた。K1cの値を求める式は次の式で表される。
K1c=PmaxSf(a/W)/BW3/2
ただし、Pmaxは最大破壊荷重[kN]、Sはスパン間距離(治具ローラー間距離)[cm]、aはき裂長さ[cm]、W=試験片幅[cm]、B=試験片厚さ[cm]である。またf(a/W)は、a/W=X、S/W=4.0とし、f(X)は次の式で求められる。
【0094】
【数3】
【0095】
(8)ボイド評価
銅回路が形成されたポリイミド基板と、該基板上に搭載された半導体素子との空隙に、アンダーフィル剤を充填し、その後170℃で1時間加熱処理して硬化させた。直径3μm以上のボイドの有無を半導体検査装置C−SAM(SONIX(株)製)を用いて調
べた。測定範囲は3mm×3mmとした。
【0096】
(9)平均熱線膨張係数の測定
熱硬化性樹脂組成物をNMP溶液で固形分濃度70重量%に調製した後、これを新品のシリコンウェハ上に塗布、さらに上記(2)に記載の方法で硬化させ、膜厚が約10μmのシリコンウェハ付きキュア膜を作製した。次にシリコンウェハ付きキュア膜を室温でフッ化水素酸に約7分間浸漬後、水道水で洗浄しながらキュア膜を手作業でシリコンウェハから剥離した。得られたキュア膜を3mm×17mmに切り出し、熱機械分析装置SS−6100(セイコーインスツルメント(株)製)を用いて、引っ張りモード、温度範囲25〜170℃、昇温速度5℃/分、初期荷重0.5g、チャック間15mmの条件でキュア膜の伸びを測定した。得られた測定結果から下記の計算式を用いてT1〜T2℃の平均熱線膨張係数を算出した。ここでLT1はT1℃でのサンプル長、LT2はT2℃でのサンプル長である。
平均熱線膨張係数=(1/LT1)[(LT2−LT1)/(T2−T1)]
(10)SP値の算出
SP値は、文献「ポリマー・エンジニアリング・アンド・サイエンス(R.F.Fedors,Polymer.Eng.,14,(2)147−154(1974)」に記載された方法により求めた。すなわち、求める化合物の構造式において、原子および原子団の蒸発エネルギーとモル体積のデータより次式にて算出した。
【0097】
【数4】
【0098】
ただし、Δeiは原子または基に帰属する25℃における蒸発エネルギー、Δviは25℃におけるモル体積を示し、分子中のi個の原子及び基に与えられた一定の数値である。代表的な原子および基のΔeiおよびΔviは、前記文献に記載された値を用いた。
【0099】
合成例1:ポリフェニルスルホン樹脂Aの合成(直接重合方法)
窒素導入管、撹拌棒、温度計を取り付けた500mLの3つ口フラスコに乾燥窒素気流下、4,4’−ジクロロジフェニルスルホン27.28g(95ミリモル、和光純薬工業(株)製)と、4,4’−ジヒドロキシジフェニルスルホン18.62g(100ミリモル、和光純薬工業(株)製)と、無水炭酸カリウム17.25g(0.125モル、東京化成工業(株)製)をNMP(三菱化学(株)製)140gとトルエン60g(東京化成工業(株)製)に溶解させ、溶液の温度を170℃で8時間反応させた。その後冷却を行い、溶液の温度が室温にまで低下したら、メタノール3Lに溶液を投入し、白色のポリマー固体を得た。このポリマー固体をろ過で集め、さらにメタノール洗浄を2回行った後、水で3回洗浄を行った。洗浄後、集めたポリマー固体を90℃の真空乾燥機で72時間乾燥しポリフェニルスルホン樹脂Aを得た。得られた粉体を、KBr法による赤外吸収スペクトルで測定した。KBr法とはKBr粉末と測定するポリマーを混合、圧縮成型し得られたペレット状サンプルの赤外吸収スペクトルを測定する方法である。その結果ポリフェニルスルホンの構造に起因するピークが1578cm−1付近、1484cm−1、1238cm−1、1149cm−1、1103cm−1付近に検出された。このようにして得られたポリマーの重量平均分子量(Mw)を算出したところ、28600であった。また末端水酸基含有率は20モル%であった。また得られた粉体を用いてDSC法を用いてガラス転移温度を測定したところ、220℃であった。
【0100】
合成例2:ポリフェニルスルホン樹脂Bの合成(直接重合方法)
窒素導入管、撹拌棒、温度計を取り付けた500mLの3つ口フラスコに乾燥窒素気流下、4,4’−ジクロロジフェニルスルホン25.84g(90ミリモル、和光純薬工業(株)製)と、4,4’−ジヒドロキシジフェニルスルホン18.62g(100ミリモル、和光純薬工業(株)製)と、無水炭酸カリウム17.25g(0.125モル、東京化成工業(株)製)をNMP(三菱化学(株)製)140gとトルエン60g(東京化成工業(株)製)に溶解させ、溶液の温度を170℃で8時間反応させた。その後冷却を行い、溶液の温度が室温にまで低下したら、メタノール3Lに溶液を投入し、白色のポリマー固体を得た。このポリマー固体をろ過で集め、さらにメタノール洗浄を2回行った後、水で3回洗浄を行った。洗浄後、集めたポリマー固体を90℃の真空乾燥機で72時間乾燥しポリフェニルスルホン樹脂Bを得た。得られた粉体を、KBr法による赤外吸収スペクトルで測定したところ、ポリフェニルスルホンの構造に起因するピークが1578cm−1付近、1484cm−1、1238cm−1、1149cm−1、1103cm−1付近に検出された。このようにして得られたポリマーの重量平均分子量(Mw)を算出したところ、13800であった。また末端水酸基含有率は20モル%であった。また得られた粉体を用いてDSC法を用いてガラス転移温度を測定したところ、210℃であった。
【0101】
合成例3:ポリフェニルスルホン樹脂Cの合成(直接重合方法)
窒素導入管、撹拌棒、温度計を取り付けた500mLの3つ口フラスコに乾燥窒素気流下、4,4’−ジクロロジフェニルスルホン24.87g(78ミリモル、和光純薬工業(株)製)と、4,4’−ジヒドロキシジフェニルスルホン18.62g(100ミリモル、和光純薬工業(株)製)と、無水炭酸カリウム17.25g(0.125モル、東京化成工業(株)製)をNMP(三菱化学(株)製)140gとトルエン60g(東京化成工業(株)製)に溶解させ、溶液の温度を170℃で8時間反応させた。その後冷却を行い、溶液の温度が室温にまで低下したら、メタノール3Lに溶液を投入し、白色のポリマー固体を得た。このポリマー固体をろ過で集め、さらにメタノール洗浄を2回行った後、水で3回洗浄を行った。洗浄後、集めたポリマー固体を90℃の真空乾燥機で72時間乾燥しポリフェニルスルホン樹脂Cを得た。得られた粉体を、KBr法による赤外吸収スペクトルで測定したところ、ポリフェニルスルホンの構造に起因するピークが1578cm−1付近、1484cm−1、1238cm−1、1149cm−1、1103cm−1付近に検出された。このようにして得られたポリマーの重量平均分子量(Mw)を算出したところ、5500であった。また末端水酸基含有率は20モル%であった。また得られた粉体を用いてDSC法を用いてガラス転移温度を測定したところ、172℃であった。
【0102】
合成例4:ポリフェニルスルホン樹脂Dの合成(解重合方法)
窒素導入管、撹拌棒、温度計を取り付けた500mLの3つ口フラスコに乾燥窒素気流下、スミカエクセル4800P67g(0.288モル、住友化学工業(株)製)と、4,4’−ジヒドロキシジフェニル2.83g(0.015モル、和光純薬工業(株)製)と、無水炭酸カリウム3.57g(0.0258モル、東京化成工業(株)製)をNMP540gとトルエン60gに溶解させ、溶液の温度を150℃で5時間反応させた。その後冷却を行い、溶液の温度が室温にまで低下したら、メタノール3Lに溶液を投入し、白色のポリマー固体を得た。このポリマー固体をろ過で集め、さらにメタノール洗浄を2回行った後、水で3回洗浄を行った。洗浄後、集めたポリマー固体を90℃の真空乾燥機で72時間乾燥しポリフェニルスルホン樹脂Dを得た。得られた粉体を、KBr法による赤外吸収スペクトルで測定したところ、ポリフェニルスルホンの構造に起因するピークが1578cm−1付近、1484cm−1、1238cm−1、1149cm−1、1103cm−1付近に検出された。このようにして得られたポリマーの重量平均分子量(Mw)を算出したところ、20100であった。また末端水酸基含有率は20モル%であった。また得られたポリマー粉体を用いてDSC法を用いてガラス転移温度を測定したところ、210℃であった。
【0103】
合成例5:ポリフェニルスルホン樹脂Eの合成(解重合方法)
窒素導入管、撹拌棒、温度計を取り付けた500mLの3つ口フラスコに乾燥窒素気流下、スミカエクセル4800P60g(0.258モル、住友化学工業(株)製)と、4,4’−ジヒドロキシジフェニル8.48g(0.0455モル、和光純薬工業(株)製)と、無水炭酸カリウム3.57g(0.0258モル、東京化成工業(株)製)をNMP540gとトルエン60gに溶解させ、溶液の温度を150℃で5時間反応させた。その後冷却を行い、溶液の温度が室温にまで低下したら、メタノール3Lに溶液を投入し、白色のポリマー固体を得た。このポリマー固体をろ過で集め、さらにメタノール洗浄を2回行った後、水で3回洗浄を行った。洗浄後、集めたポリマー固体を90℃の真空乾燥機で72時間乾燥しポリフェニルスルホン樹脂Eを得た。得られた粉体を、KBr法による赤外吸収スペクトルで測定したところ、ポリフェニルスルホンの構造に起因するピークが1578cm−1付近、1484cm−1、1238cm−1、1149cm−1、1103cm−1付近に検出された。このようにして得られたポリマーの重量平均分子量(Mw)を算出したところ、7185であった。また末端水酸基含有率は30モル%であった。また得られたポリマー粉体を用いてDSC法を用いてガラス転移温度を測定したところ、150℃であった。
【0104】
合成例6:ポリフェニルスルホン樹脂Fの合成(直接重合方法)
窒素導入管、撹拌棒、温度計を取り付けた500mLの3つ口フラスコに乾燥窒素気流下、4,4’−ジクロロジフェニルスルホン28.72g(0.1モル、和光純薬工業(株)製)と、4,4’−ジヒドロキシジフェニルスルホン18.62g(0.1モル、和光純薬工業(株)製)と、無水炭酸カリウム17.25g(0.125モル、東京化成工業(株)製)をNMP(三菱化学(株)製)140gとトルエン60g(東京化成工業(株)製)に溶解させ、溶液の温度を170℃で8時間反応させた。その後冷却を行い、溶液の温度が室温にまで低下したら、メタノール3Lに溶液を投入し、白色のポリマー固体を得た。このポリマー固体をろ過で集め、さらにメタノール洗浄を2回行った後、水で3回洗浄を行った。洗浄後、集めたポリマー固体を90℃の真空乾燥機で72時間乾燥しポリフェニルスルホン樹脂Fを得た。得られた粉体を、KBr法による赤外吸収スペクトルで測定したところ、ポリフェニルスルホンの構造に起因するピークが1578cm−1付近、1484cm−1、1238cm−1、1149cm−1、1103cm−1付近に検出された。このようにして得られたポリマーの重量平均分子量(Mw)を算出したところ、69893であった。また末端水酸基含有率は15モル%であった。また得られた粉体を用いてDSC法を用いてガラス転移温度を測定したところ、231℃であった。
【0105】
合成例7:ポリイミド化合物の合成
冷却管および撹拌装置付きの1Lセパラブルフラスコに、2,3,3’,4’−ビフェニルテトラカルボン酸二無水物411.9g(1.40mol)、ビス(3−カルボキシ−4−アミノフェニル)メタン71.58g(0.25mol)、1,3−ビス(3−アミノプロピル)テトラメチルジシロキサン186g(0.75モル)をトリエチレングリコールジメチルエーテル(商品名、丸善石油化学(株)製)溶媒中で温度160℃で6時間撹拌してポリイミド溶液(固形分濃度56.5%)を得た。撹拌終了し、室温まで冷却後、溶液を水3Lに投入して白色沈殿を得た。この沈殿をろ過して回収し、水で3回洗浄した後、真空乾燥機を用いて80℃、20時間乾燥し、白色粉末のポリイミド化合物を得た。
【0106】
各実施例および比較例で使用した原料を以下に示す。
【0107】
ポリエーテルスルホン樹脂:
スミカエクセル5003P(商品名、住友化学工業(株)製:重量平均分子量67250、末端フェノール性水酸基含有率46モル%、ガラス転移温度230℃)
スミカエクセル4800P(商品名、住友化学工業(株)製:重量平均分子量66700、末端フェノール性水酸基無し、ガラス転移温度225℃)
ポリフェニルスルホン樹脂:
ポリフェニルスルホン樹脂“Ultrazon”P3010(以上、商品名、BASFジャパン(株)製、重量平均分子量=55675)
ポリイミド化合物:合成例7で得られたポリイミド化合物
“E2020”(商品名、プリンテック(株)製)芳香族ビスマレイミドと芳香族ジアミンの共重合体
エポキシ変性シリコーン樹脂:アルケニル基含有フェノールノボラック型樹脂とポリジメチルシロキサンの共重合体
SP値が2.00×104〜2.60×104(J/m3)1/2であるエポキシ樹脂:
“jER”828(商品名、ジャパンエポキシレジン(株)製):エポキシ当量187g/eq、SP値=12.31(cal/cm3)1/2=2.52×104(J/m3)1/2
N660(商品名、DIC(株)製:クレゾールノボラック型固形エポキシ樹脂:エポキシ当量206g/eq、SP値=10.11(cal/cm3)1/2=2.07×104(J/m)1/2)
EXA4710(商品名、DIC(株)製:ナフタレン型固形エポキシ樹脂:エポキシ当量170g/eq、SP値=10.07(cal/cm3)1/2=2.06×104(J/m)1/2))
SP値が1.60×104〜1.80×104(J/m3)1/2であるエポキシ樹脂:
エポライト400E(商品名、共栄社化学(株)製):ポリエチレングリコールジグリシジルエーテル、エポキシ当量264〜290g/eq、SP値=8.58(cal/cm3)1/2=1.76×104(J/m3)1/2
YED216M(商品名、ジャパンエポキシレジン(株)製):1,6−ヘキサンジオールジグリシジルエーテル、エポキシ当量148g/eq、SP値=8.25(cal/cm3)1/2=1.69×104(J/m3)1/2
比較例で用いたエポキシ樹脂
エポキシ樹脂A=“jER”1032H60(商品名、ジャパンエポキシレジン(株)製):トリスヒドロキシフェニルメタン型エポキシ樹脂
エポキシ樹脂C=“jER”807(商品名、ジャパンエポキシレジン(株)製):ビスフェノールF型液状エポキシ樹脂)
オキセタン化合物:
POX(略号)=OXT−211(商品名、ジャパンエポキシレジン(株)製):3−エチル−3−(フェノキシメチル)オキセタン
OXIPA(略号)=“エタナコール”OXIPA(商品名、宇部興産(株)製)
硬化剤および硬化促進剤:
硬化剤A:DDS:芳香族ジアミン(4,4’−ジアミノジフェニルスルホンと3,3’−ジアミノジフェニルスルホンの1対1混合物(和光純薬工業(株)製))
硬化剤A−2:APB−N:芳香族ジアミン:m−ビス(m−アミノフェノキシ)ベンゼン(三井化学(株)製))
硬化剤A−3:TPE−R:芳香族ジアミン:m−ビス(p−アミノフェノキシ)ベンゼン(和歌山精化工業(株)製))
硬化剤A−4:APB−5:芳香族ジアミン:ビス(m−(m−アミノフェノキシ)フェノキシ)ベンゼン(三井化学(株)製)
硬化促進剤:3,4−ジクロロフェニル−N,N−ジメチル尿素(東京化成工業(株)製)
比較例で用いた硬化剤
硬化剤B:EXB9889(商品名、DIC(株)製、トリアジン骨格含有フェノールノボラック樹脂)
硬化剤C:2E4MZ(2−エチル−4−メチルイミダゾール)(商品名、四国化成工業(株)製、非水溶性)
硬化剤D:KAYAHARD−AA(商品名、日本化薬(株)製、3,3’−ジエチル−4,4‘−ジアミノジフェニルメタン)
ラジカル重合開始剤:パークミルD(商品名、日本油脂(株)製)
無機微粒子および有機微粒子:
球状シリカ微粒子:アドマファインGRJ(商品名、アドマテックス(株)製:表面型修飾シリカ微粒子、表面修飾粒子、平均粒子径0.5μm)
球状スチレン微粒子:ガンツパール(商品名、ガンツ化成(株)製、:ポリスチレン系有機微粒子、平均粒子径2.5μm)
溶剤:
溶剤A:NMP(略号)=:N−メチルピロリドン(商品名、東京化成工業(株)製、沸点202−204℃)
比較例で用いた溶剤
溶剤B:トリグライム(略号)=:トリエチレングリコールジメチルエーテル(商品名、丸善石油化学(株)製、沸点216℃)
溶剤C:DMAc(略号)=:N,N’−ジメチルアセトアミド(商品名、東京化成工業(株)製、沸点166℃)
溶剤D::2−ブトキシエチルアセテート(商品名、東京化成工業(株)製、沸点188℃)
実施例1
窒素導入管、撹拌棒、温度計を取り付けた100mLの3つ口フラスコにN660 30gとYED216M 30gを投入し、乾燥窒素気流下、80℃で1時間撹拌して溶解させた。さらに硬化剤A 21.6gを投入し、乾燥窒素気流下、60℃で2時間撹拌して溶解させ、室温まで冷却した。その後、合成例1で得られたポリフェニルスルホン樹脂A 6gを添加し、遊星式撹拌脱泡機(マゼルスター(クラボウ製))を用いて25℃で30分間撹拌して混合させた後、最後に脱泡を行い、アンダーフィル剤A−1を得た。得られたアンダーフィル剤A−1のガラス転移温度、粘度特性、溶解性、硬化開始温度、破壊靭性値、ボイドの有無を、前記方法により測定および評価した。
【0108】
実施例2
ポリフェニルスルホン樹脂A 6gに代えて合成例2で得られたポリフェニルスルホン樹脂B 6gを用いた以外は実施例1と同様にして、アンダーフィル剤B−1を得た。得られたアンダーフィル剤B−1のガラス転移温度、粘度特性、溶解性、硬化開始温度、破壊靭性値、平均熱線膨張係数、ボイドの有無を、前記方法により測定および評価した。平均熱線膨張係数の測定温度範囲はT1=25℃、T2=100℃とした。
【0109】
実施例3
ポリフェニルスルホン樹脂A 6gに代えて合成例3で得られたポリフェニルスルホン樹脂C 6gを用いた以外は実施例1と同様にして、アンダーフィル剤C−1を得、実施例1と同様に測定および評価した。
【0110】
実施例4
ポリフェニルスルホン樹脂A 6gに代えて合成例4で得られたポリフェニルスルホン樹脂D 6gを用いた以外は実施例1と同様にして、アンダーフィル剤D−1を得、実施例1と同様に測定および評価した。
【0111】
実施例5
ポリフェニルスルホン樹脂A 6gに代えて合成例5で得られたポリフェニルスルホン樹脂E6g用いた以外は実施例1と同様にして、アンダーフィル剤E−1を得、実施例1と同様に測定および評価した。
【0112】
実施例6
ポリフェニルスルホン樹脂B 6gを3gに代えて用いた以外は実施例2と同様にして、アンダーフィル剤F−1を得、実施例1と同様に測定および評価した。
【0113】
実施例7
ポリフェニルスルホン樹脂B 6gを10gに代えて用いた以外は実施例2と同様にして、アンダーフィル剤G−1を得、実施例1と同様に測定および評価した。
【0114】
実施例8
窒素導入管、撹拌棒、温度計を取り付けた100mLの3つ口フラスコにjER828 30gとYED216M 30gを投入し、硬化剤A 22.5gを投入し、乾燥窒素気流下、60℃で2時間撹拌して溶解させ、室温まで冷却した。その後、合成例2で得られたポリフェニルスルホン樹脂B 6gを添加し、遊星式撹拌脱泡機(マゼルスター(クラボウ製))を用いて25℃で30分間撹拌して混合させた後、最後に脱泡を行い、アンダーフィル剤H−1を得、実施例1と同様に測定および評価した。
【0115】
実施例9
N660をEXA4710に代えて、硬化剤A 21.6gを23.5gに代えて用いた以外は実施例2と同様にして、アンダーフィル剤I−1を得、実施例1と同様に測定および評価した。
【0116】
実施例10
YED216Mをエポライト400Eに代えて、硬化剤A 21.6gを19.3gに代えて用いた以外は実施例2と同様にして、アンダーフィル剤J−1を得、実施例1と同様に測定および評価した。
【0117】
実施例11
窒素導入管、撹拌棒、温度計を取り付けた100mLの3つ口フラスコにjER828 60gを投入し、さらに硬化剤A 19.9gを投入し、乾燥窒素気流下、60℃で2時間撹拌して溶解させ、室温まで冷却した。その後、合成例2で得られたポリフェニルスルホン樹脂B 6gを添加し、遊星式撹拌脱泡機(マゼルスター(クラボウ製))を用いて25℃で30分間撹拌して混合させた後、最後に脱泡を行い、アンダーフィル剤K−1を得、実施例1と同様に測定および評価した。
【0118】
実施例12
窒素導入管、撹拌棒、温度計を取り付けた100mLの3つ口フラスコにN660 30gとYED216M 30gを投入し、乾燥窒素気流下、80℃で1時間撹拌して溶解させた。さらに硬化剤A 21.6gを投入し、乾燥窒素気流下、60℃で2時間撹拌して溶解させ、室温まで冷却した。その後、合成例2で得られたポリフェニルスルホン樹脂B 6g、OXIPA 1gを添加し、遊星式撹拌脱泡機(マゼルスター(クラボウ製))を用いて25℃で30分間撹拌して混合させた後、最後に脱泡を行い、アンダーフィル剤L−1を得、実施例1と同様に測定および評価した。
【0119】
実施例13
OXIPAをPOXに代えて用いた以外は実施例11と同様にして、アンダーフィル剤M−1を得、実施例1と同様に測定および評価した。
【0120】
実施例14
硬化剤A21.6gを硬化剤A−2 25.5gに代えて用いた以外は実施例2と同様にして、アンダーフィル剤N−1を得、実施例1と同様に測定および評価した。
【0121】
実施例15
硬化剤A21.6gを硬化剤A−3 25.5gに代えて用いた以外は実施例2と同様にして、アンダーフィル剤O−1を得、実施例1と同様に測定および評価した。
【0122】
実施例16
硬化剤A21.6gを硬化剤A−4 39gに代えて用いた以外は実施例2と同様にして、アンダーフィル剤P−1を得、実施例1と同様に測定および評価した。
【0123】
実施例17
窒素導入管、撹拌棒、温度計を取り付けた100mLの3つ口フラスコにN660 30gとYED216M30gを投入し、乾燥窒素気流下、80℃で1時間撹拌して溶解させた。さらに硬化剤A21.6gを投入し、乾燥窒素気流下、60℃で2時間撹拌して溶解させ、室温まで冷却した。その後、合成例2で得られたポリフェニルスルホン樹脂B 6g、硬化促進剤9.7gを添加し、遊星式撹拌脱泡機(マゼルスター(クラボウ製))を用いて25℃で30分間撹拌して混合させた後、最後に脱泡を行い、アンダーフィル剤Q−1を得、実施例1と同様に測定および評価した。
【0124】
実施例18
窒素導入管、撹拌棒、温度計を取り付けた100mLの3つ口フラスコにN660 30gとYED216M 30gを投入し、乾燥窒素気流下、80℃で1時間撹拌して溶解させた。さらに硬化剤A 21.6gを投入し、乾燥窒素気流下、60℃で2時間撹拌して溶解させ、室温まで冷却した。その後、合成例2で得られたポリフェニルスルホン樹脂B 6gを添加し、遊星式撹拌脱泡機(マゼルスター(クラボウ製))を用いて25℃で30分間撹拌して混合させた後、球状シリカ微粒子を8.8g加えて、30分間撹拌して混合させた後,3本ロールで混練した。その後さらに30分間撹拌して混合させ、最後に脱泡を行い、アンダーフィル剤R−1を得、実施例2と同様に測定および評価した。
【0125】
実施例19
球状シリカ微粒子8.8gを21.9gに代えて用いた以外は実施例17と同様にして、アンダーフィル剤S−1を得、実施例2と同様に測定および評価した。
【0126】
実施例20
球状シリカ微粒子8.8gを球状スチレン微粒子9.7gに代えて用いた以外は実施例18と同様にして、アンダーフィル剤T−1を得、実施例1と同様に測定および評価した。
【0127】
実施例21
窒素導入管、撹拌棒、温度計を取り付けた100mLの3つ口フラスコにN660 30gとYED216M 30gを投入し、乾燥窒素気流下、80℃で1時間撹拌して溶解させた。さらに硬化剤A 21.6gを投入し、乾燥窒素気流下、60℃で2時間撹拌して溶解させ、室温まで冷却した。その後、合成例2で得られたポリフェニルスルホン樹脂B 6gを添加し、遊星式撹拌脱泡機(マゼルスター(クラボウ製))を用いて25℃で30分間撹拌して混合させた。最後に溶剤Aを0.3g添加し、さらに30分間撹拌して混合させ、脱泡を行い、アンダーフィルU−1を得、実施例1と同様に測定および評価した。
【0128】
比較例1
溶剤A 0.5gを40gに代えた以外は実施例20と同様にして、アンダーフィル剤A’−1を得、実施例1と同様に測定および評価した。
【0129】
比較例2
エポキシ樹脂を用いなかった以外は実施例2と同様にして、アンダーフィル剤A’−2を得たが、固体粉体の状態であった。
【0130】
比較例3
ポリフェニルスルホン樹脂を用いなかった以外は実施例2と同様にして、アンダーフィル剤A’−3を得、実施例1と同様に測定および評価した。
【0131】
比較例4
ポリフェニルスルホン樹脂Bに代えて合成例6で得られたポリフェニルスルホン樹脂Fを用いた以外は実施例2と同様にして、アンダーフィル剤A’−4を得、実施例1と同様に測定および評価した。
【0132】
比較例5
ポリフェニルスルホン樹脂Bに代えて市販のポリフェニルスルホン樹脂“Ultrazon”P3010(以上、商品名、BASFジャパン(株)製、重量平均分子量=55675)を用いた以外は実施例2と同様にして、アンダーフィル剤A’−5を得、実施例1と同様に測定および評価した。
【0133】
比較例6
E2020 20g、スミカエクセル5003P 10g、エポキシ樹脂C 15g、溶剤C 48g、硬化剤B 8g、ラジカル重合開始剤 0.16gを遊星式撹拌脱泡機(マゼルスター(クラボウ製))を用いて25℃で60分間撹拌して混合させ、最後に脱泡を行い、アンダーフィルA’−6を得、実施例1と同様に測定および評価した。
【0134】
比較例7
エポキシ樹脂A 15g、合成例7で得られたポリイミド化合物 57gを遊星式撹拌脱泡機(マゼルスター(クラボウ製))を用いて25℃で60分間撹拌して混合させた後、硬化剤Cを0.3g添加し、さらに30分間撹拌して混合させた。最後に溶剤Bを43g添加し、さらに30分間撹拌して混合させ、脱泡を行い、アンダーフィルA’−7を得、実施例1と同様に測定および評価した。
【0135】
比較例8
エポキシ樹脂C 63g、エポキシ変性シリコーン樹脂4g、球状シリカ微粒子150gを遊星式撹拌脱泡機(マゼルスター(クラボウ製))を用いて25℃で60分間撹拌して混合させた後、3本ロールで混練した。その後硬化剤Dを30g添加し、さらに溶剤Dを2g添加して30分間撹拌して混合させ、最後に脱泡を行い、アンダーフィルA’−8を得、実施例1と同様に測定および評価した。
各実施例および比較例の組成を表1〜3に、評価結果を表4〜6に示す。
【0136】
【表1】
【0137】
【表2】
【0138】
【表3】
【0139】
【表4】
【0140】
【表5】
【0141】
【表6】
【符号の説明】
【0142】
1:DSC曲線
2:L1の延長線
3:L2の延長線
4:交点
P:ピーク
L1:直線部分
L2:立ち上がり後ピークに到達するまでに認められる直線部分
【特許請求の範囲】
【請求項1】
(a)重量平均分子量3万未満のポリフェニルスルホン樹脂と、(b)エポキシ樹脂を含有し、(c)溶剤の含有量が1重量%以下であることを特徴とするアンダーフィル剤。
【請求項2】
前記(a)重量平均分子量3万未満のポリフェニルスルホン樹脂を5〜20重量%含有することを特徴とする請求項1記載のアンダーフィル剤。
【請求項3】
前記(a)重量平均分子量3万未満のポリフェニルスルホン樹脂がフェノール性水酸基を有することを特徴とする請求項1または2記載のアンダーフィル剤。
【請求項4】
前記(b)エポキシ樹脂がSP値が1.60×104〜1.80×104(J/m3)1/2であるエポキシ樹脂と、SP値が2.00×104〜2.60×104(J/m3)1/2であるエポキシ樹脂からそれぞれ一種類以上選ばれてなることを特徴とする請求項1〜3いずれか記載のアンダーフィル剤。
【請求項5】
さらに(d)シリカ、チタニア、ジルコニア、窒化ケイ素、アルミナ、セリア、タルクおよび炭酸カルシウムからなる群より選ばれる少なくとも一種の無機微粒子を含有することを特徴とする請求項1〜4いずれか記載のアンダーフィル剤。
【請求項6】
さらに(e)ポリイミド、ポリアミド、ポリアミドイミド、ポリスチレン、ポリアクリロニトリル、ポリフェニレンエーテル、ポリエステルおよびポリカーボネートからなる群より選ばれる少なくとも一種の有機微粒子を含有することを特徴とする請求項1〜5いずれか記載のアンダーフィル剤。
【請求項7】
さらに硬化剤を含有し、硬化剤の含有量が、前記(a)成分のポリフェニルスルホン樹脂と前記(b)エポキシ樹脂との総量100重量部に対して0.1〜60重量部であることを特徴とする請求項1〜6いずれか記載のアンダーフィル剤。
【請求項8】
動的粘弾性測定(昇温速度2℃/分)により得られる粘弾性−温度曲線において、粘度の最低値η*が1.0×102Pa・s以下であり、かつ粘度の最低値を示す温度が100〜140℃であることを特徴とする請求項1〜7いずれか記載のアンダーフィル剤。
【請求項9】
硬化後の破壊靭性値K1cが2.0MPa・m1/2以上である請求項1〜8いずれか記載のアンダーフィル剤。
【請求項10】
請求項1〜9いずれか記載のアンダーフィル剤を用いた半導体装置。
【請求項1】
(a)重量平均分子量3万未満のポリフェニルスルホン樹脂と、(b)エポキシ樹脂を含有し、(c)溶剤の含有量が1重量%以下であることを特徴とするアンダーフィル剤。
【請求項2】
前記(a)重量平均分子量3万未満のポリフェニルスルホン樹脂を5〜20重量%含有することを特徴とする請求項1記載のアンダーフィル剤。
【請求項3】
前記(a)重量平均分子量3万未満のポリフェニルスルホン樹脂がフェノール性水酸基を有することを特徴とする請求項1または2記載のアンダーフィル剤。
【請求項4】
前記(b)エポキシ樹脂がSP値が1.60×104〜1.80×104(J/m3)1/2であるエポキシ樹脂と、SP値が2.00×104〜2.60×104(J/m3)1/2であるエポキシ樹脂からそれぞれ一種類以上選ばれてなることを特徴とする請求項1〜3いずれか記載のアンダーフィル剤。
【請求項5】
さらに(d)シリカ、チタニア、ジルコニア、窒化ケイ素、アルミナ、セリア、タルクおよび炭酸カルシウムからなる群より選ばれる少なくとも一種の無機微粒子を含有することを特徴とする請求項1〜4いずれか記載のアンダーフィル剤。
【請求項6】
さらに(e)ポリイミド、ポリアミド、ポリアミドイミド、ポリスチレン、ポリアクリロニトリル、ポリフェニレンエーテル、ポリエステルおよびポリカーボネートからなる群より選ばれる少なくとも一種の有機微粒子を含有することを特徴とする請求項1〜5いずれか記載のアンダーフィル剤。
【請求項7】
さらに硬化剤を含有し、硬化剤の含有量が、前記(a)成分のポリフェニルスルホン樹脂と前記(b)エポキシ樹脂との総量100重量部に対して0.1〜60重量部であることを特徴とする請求項1〜6いずれか記載のアンダーフィル剤。
【請求項8】
動的粘弾性測定(昇温速度2℃/分)により得られる粘弾性−温度曲線において、粘度の最低値η*が1.0×102Pa・s以下であり、かつ粘度の最低値を示す温度が100〜140℃であることを特徴とする請求項1〜7いずれか記載のアンダーフィル剤。
【請求項9】
硬化後の破壊靭性値K1cが2.0MPa・m1/2以上である請求項1〜8いずれか記載のアンダーフィル剤。
【請求項10】
請求項1〜9いずれか記載のアンダーフィル剤を用いた半導体装置。
【図1】

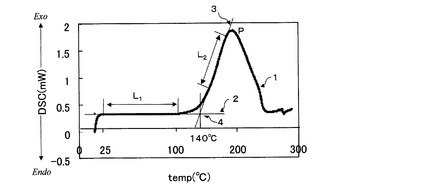
【公開番号】特開2012−25847(P2012−25847A)
【公開日】平成24年2月9日(2012.2.9)
【国際特許分類】
【出願番号】特願2010−165609(P2010−165609)
【出願日】平成22年7月23日(2010.7.23)
【出願人】(000003159)東レ株式会社 (7,677)
【Fターム(参考)】
【公開日】平成24年2月9日(2012.2.9)
【国際特許分類】
【出願日】平成22年7月23日(2010.7.23)
【出願人】(000003159)東レ株式会社 (7,677)
【Fターム(参考)】
[ Back to top ]