
アントラサイクリン誘導体
本発明は、新しいアントラサイクリン誘導体と、癌の治療および診断へのそれらの使用に関する。これらのアントラサイクリン誘導体を放射性標識し、癌の診断での造影剤として使用できる。放射性標識されたアントラサイクリン誘導体は、固形腫瘍および播種性腫瘍を治療するための、特に二段階ターゲッティング戦略を含む薬物送達システムと共に使用することもできる。これらの薬物送達システムを乳癌の治療および診断に有利に使用できる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新しいアントラサイクリン誘導体ならびに癌の治療および癌の診断へのそれらの使用とに関する。
【背景技術】
【0002】
DOXと省略され、アドリアマイシンまたはアドリアブラスチン等の他の名前でも公知であるドキソルビシン(C27H29NO11;MW:543.53)は抗生物質であり、アントラサイクリンファミリーの抗腫瘍薬である(構造は下記参照)。DOXは、もともと水中細菌ストレプトミセスペウセティウス変種コエシウス(streptomyces peucetius var. coesius)から単離され、1970年代前半以来、アントラサイクリン、特にドキソルビシンおよびダウノルビシンならびにアルキル化剤(シクロホスファミド、メルファラン等)が、今日臨床で最も多方面で最も頻繁に使用されている化学療法剤である[1]。アントラサイクリンは、キノン−ヒドロキノン官能基および親水性アミノ糖部分を有する疎水性アグリコン複素環からなる両親媒性分子である[2,3]。
【0003】
ダウノルビシンおよびドキソルビシンの化学構造
【化1】

【0004】
ドキソルビシンは骨肉腫および軟部組織肉腫ならびに肺、乳房、甲状腺、膀胱、卵巣、精巣、頭部、および頚部の癌腫の治療に広く使用されている[1、4]。ドキソルビシンは、白血病およびリンパ腫に対しても使用されるが、ダウノルビシンは急性白血病に対する一次治療である。ドキソルビシンに関する全奏効率は、甲状腺癌では45%、リンパ腫では41%、膀胱癌では33%、肉腫では26%、卵巣癌では25%、白血病では24%である[5]。
【0005】
ドキソルビシンは多様な作用機序を有するが、ドキソルビシンおよび他のアントラサイクリンの主な抗腫瘍活性は、それらがDNAにインターカレートする結果として、DNA、RNA、およびタンパク質の合成の遮断を招く能力に由来する。アントラサイクリンはトポイソメラーゼIIも阻害し、DNA修復を障害する[1、5]。キノン−ヒドロキノン官能基が原因で、アントラサイクリンはDNA損傷に至るフリーラジカルの発生に関与すると考えられている[2]。アントラサイクリンは、心ミトコンドリアおよび悪性細胞の膜に高濃度にみられるリン脂質であるカルジオリピンに特異的に結合し、これによって、ドキソルビシンの心毒性副作用を説明できる[1]。用量が少なすぎると、アントラサイクリンは効果を有さず、用量が多すぎると、重度の副作用が起こるおそれがあることから、この薬物は狭い治療指数を有する。すなわち、投与量は狭い範囲内に入らなければならない。ドキソルビシンの急性の用量制限毒性は、骨髄抑制、白血球減少、および投与患者の80%で起こる口内炎である。他の副作用には、脱毛(100%)、悪心、嘔吐(20〜55%)、心毒性、すなわち上室性不整脈、心ブロック、心室頻拍、および患者の1〜10%で生じるうっ血性心不全さえある。
【0006】
アントラサイクリン誘導体は、以前に例えばPribe(2003)Chemico-Biological Interactions 145:349〜358、US−4948880、US−6673907、およびWO00/56267に開示された。これらの誘導体は、癌の治療で細胞毒性作用を有する。
【0007】
放射性核種治療は、癌の治療に比較的小さいが重要な役割を有し、現在ますます大きな注目を集めている。放射性核種治療は、核放射線を使用して悪性細胞を根絶させる。その放射線は、中性子活性化の後の安定核種、例えば10Bおよび157Gによって、または放射性核種によって発生させることができる。特に甲状腺癌に対する最も一般に使用されている治療用放射性核種は、中距離(800μm)β照射体の131Iであるが、131Iの投与は健常組織にかなりの照射損傷を引き起こす[6]。しかし、これらの副作用により、125I等の短距離低エネルギーオージェ電子放射体の治療上の潜在能力が広く認められてきている。抗癌治療に有効であるためには、125Iは直接的かつ選択的に腫瘍細胞の核に送達しなければならない。それは、125IがDNAから数ナノメーター以内に入らない限り毒性ではないからである[7]。このように、125Iによる治療は核への特異的な送達法を必要とし、その方法は、125Iで標識したヌクレオシド、オリゴヌクレオチド、ステロイドホルモン、および成長因子を用いて以前は実現されたが、改善の必要が認識されていた[7]。
【0008】
Murali D.およびDeJesus.O,Bioorganic&Medicinal Chemistry letters 8 (1998) 3419〜3422は、ドキソルビシンに比べて改善された細胞毒性を有する放射性標識ダウノルビシン誘導体を記載している。この論文に結果は提示されておらず、この研究の継続は発表されていない。
【0009】
リポソームを介した標的を定めた薬物送達は、膜結合型小胞(リポソーム)に細胞毒性剤を封入することおよびリポソーム膜に腫瘍特異的抗体を結合すること(標的を定めたリポソーム)によって、骨髄抑制、粘膜炎、心毒性、神経毒性、および腎毒性等の通常の癌化学療法の用量規制副作用を最小にする[1]。血中にある標的を定めたリポソームは、標的となる表面マーカーを過剰発現している腫瘍細胞によって能動的かつ選択的に取り込まれる。しかし、いくらか前進したにもかかわらず、この戦略は化学療法の大きな進歩を今のところもたらしていない[8]。
【0010】
このように、より強力な治療剤および治療戦略の大きな必要性が存在する。
【発明の開示】
【発明が解決しようとする課題】
【0011】
(発明の概要)
本発明は、DNAにインターカレートする性質を有し、核種を結合させることができる診断剤および治療剤に関する。本発明はさらに、これらの診断剤または治療剤に関する薬物送達システムならびに前記薬剤または薬物送達システムを使用した治療法および診断法に関する。本発明は、癌の診断および治療を意図する。本発明の目的は、核種を腫瘍細胞の核に特異的に送達することであり、したがって、健常組織での細胞毒性副作用を最小にする単一の二段階ターゲッティング法で、放射性核種治療、化学療法、および標的を定めたリポソーム薬物送達の利益を合せることである。このように、本発明は新薬を用いた新しい治療戦略を提供し、その新薬は以前より公知であった化学療法薬よりも潜在的に強力である。
【課題を解決するための手段】
【0012】
アントラサイクリンはそれ自体で強力な抗癌薬である。本出願において、本発明者らは、ダウノルビシンのアミノ−ベンジル誘導体(薬物前駆体)を合成した。ダウノルビシンは、商業的に成功しているアントラサイクリンであるドキソルビシンおよびダウノルビシンに類似した細胞毒性プロフィールを有する。本発明者らがこの薬物前駆体を125Iでヨウ化したとき、培養腫瘍細胞に対してさらに有効な放射性治療剤が得られた。健常組織を保護し、悪性癌細胞に放射性核種を選択的に送達するために、標的を定めたリポソームに125Iと結合したダウノルビシン誘導体を封入することができ、そのリポソームは腫瘍細胞に対する特異的薬物送達小胞として働く。
【0013】
本明細書に記載した実験の結果は、標的を定めたリポソームに本発明による治療剤をうまく封入することができ、その薬物が実験の調製条件およびアッセイ条件で十分に保持されることを確認する。腫瘍細胞と共にインキュベートした後で、上に挙げた薬剤は細胞核に達して、ドキソルビシンと同様の親和性で結合する。DNA結合すると、その放射性治療剤はDNAの断片化を引き起こし、今日臨床で最も成功した化学療法剤の2つであるドキソルビシンおよびダウノルビシンによって起こるよりも数桁高い腫瘍細胞成長阻害に導く。
【0014】
本発明による薬物前駆体または放射性治療薬のどれも、先行技術で以前に記載されていない。
【0015】
本発明の一態様によると、放射性治療剤の前駆分子として働き、以下に薬物前駆体と呼ぶアントラサイクリン誘導体が提供される。これらの薬物前駆体は、DNAにインターカレートし、細胞分裂抑制性を有する。このように、これらの薬物前駆体は、細胞または組織に標的を定めた癌治療に単独で使用することができる。アントラサイクリン誘導体(薬物前駆体)は請求項1〜3に定義する。
【0016】
本発明のさらなる態様によると、細胞または組織に標的を定めた放射性治療に使用できる放射性治療薬が提供される。これらの放射性治療剤は、核種または核種を含む化学基に薬物前駆体を結合することによって、それらの前駆体から生成する。そのような核種は、放射性核種、安定核種、または中性子もしくは光子に曝露することによって活性化できる核種、例えば(closo-カルボランであるo−、m−またはp−C2H12B10等のホウ素リッチケージ(boron-rich cage)化合物の一部としての)10Bであってよく、以下に核種と呼ぶ。放射性治療薬は請求項4から5に定義する。
【0017】
本発明のさらなる態様によると、癌診断の画像化ツールとしてこれらの放射性治療薬を使用することもできる。画像化ツールとしての使用は請求項6に定義する。
【0018】
本発明のさらなる態様によると、薬物前駆体または放射性治療薬をDNAターゲッティング剤、すなわち請求項7に定義するようなDNA相互作用剤として使用することもできる。
【0019】
本発明のさらなる態様によると、薬物送達システムも提供される。前記システムは、(DNAにインターカレートする性質を有する)薬物前駆体または放射性治療薬を封入することができる担体を含み、標的となる細胞集団に特異的または優先的に薬物を案内する。結果として、細胞および組織を損傷する作用は、標的となる細胞および組織に優先的に影響することになる。そのような薬物送達システムは、一段階または多段階ターゲッティング戦略を含むことがある。薬物前駆体がDNAにインターカレートする性質は、それらの細胞毒性効果を核に向けるか、または、それらの核種が薬物前駆体に結合しているときは、それらの核種から放出された放射能を細胞核に局在化するDNAターゲッティング段階の基本である。以下にDNAターゲッティング段階と呼ぶこのターゲッティング段階は、このように放射性治療薬の治療効果を劇的に増やし、健康な細胞および組織に対する損傷を減少させる。
【0020】
悪性組織と健常組織とを区別するために、放射性治療薬は、二段階ターゲッティング戦略における細胞ターゲッティング段階として、表面に腫瘍細胞特異的ターゲッティング剤を示す薬物送達システムを使用して癌細胞または癌組織に向けられる。その段階は以下に、細胞ターゲッティング段階と呼ぶ。薬物担体は、放射性治療薬を包有または結合し、全身投与後に、その放射性治療薬の標的細胞の膜への、好ましくはその膜を通過する輸送を指令できる。放射性治療薬の細胞毒性および/または放射性毒性作用は、結果として標的となる細胞集団に局在化するであろう。薬物送達システムは請求項8から13に定義されている。
【0021】
本発明のさらなる態様によると、癌を診断または治療する方法も提供され、その方法は、それを必要とする患者に前駆体薬物または放射性治療薬を含む前記薬物送達システムを投与することを含む。固形腫瘍に対する治療としての使用に加え、本発明は、原発腫瘍除去後の全身循環における転移腫瘍細胞に対する治療としても予想される。その治療は、播種性乳癌だけではなく、播種性卵巣癌、前立腺癌、および結腸直腸癌にも特に有益であり得る。
【0022】
腫瘍の位置が分かっている場合は、前記治療法をそれに続く腫瘍照射と組み合わせて使用することもできる。安定核種と、中性子または光子を用いた外部照射による活性化とを使用することによって、局所放射線治療を実現できる。
【0023】
本発明は、生体異物に対する膜結合型外向き排出ポンプであるP−糖タンパク質(PGP)を過剰発現している多剤耐性(MDR)腫瘍の有効な治療でもあり得る。この場合、放射性治療薬はアントラサイクリンと結合することが知られているP−糖タンパク質を標的として放射性損傷するであろう。この治療法を請求項14から17に定義する。
【発明の効果】
【0024】
本明細書に記載する薬物送達は、強力な抗癌放射性治療薬または診断用画像化ツールとしての、薬物前駆体の形態または核種と結合した形態の癌治療としての使用を意図する。
【0025】
しかし、それらの有用性は、健常組織に対する無差別の細胞毒性によって損なわれるおそれがある。この問題は、リポソームまたは代替の薬物担体に薬物を封入して、リポソームまたは代替の薬物担体の表面に腫瘍特異的標的薬剤を加えることにより腫瘍細胞を選択的に標的とすることによって最小となるであろう。このように、これらの薬物前駆体または放射性治療薬は、米国特許第6562316号に開示されたような、標的を定めた二段階薬物送達システムと組み合わせた使用を意図する。そのような薬物送達システムは、細胞ターゲッティング段階において薬物送達システムを標的となる細胞集団または組織に特異的に向けるための、細胞ターゲッティング剤と結合した担体を含む。DNAターゲッティング段階では、封入された核種は、DNAにインターカレートする性質を有する分子に結合することによって細胞核に向けられるであろう。本明細書に記載した、薬物前駆体または放射性治療薬は、DNAにインターカレートする性質を有するそのようなDNAターゲッティング剤として働くであろう。
【0026】
記載された二段階ターゲッティングシステムは、健常組織での核種、薬物前駆体、または放射性治療薬の細胞毒性を最小にするであろう。二段階ターゲッティングシステムのさらなる利点は、転移および/または多剤耐性腫瘍細胞の治療に潜在能力を有することである。
【0027】
薬物担体は、薬学的に活性な薬剤、すなわち薬物前駆体または放射性治療薬を結合または包有できる分子、凝集体、または粒子であり得る。リポソームは現在好ましい薬物担体であるが、ミクロゲル、または脂質/高分子複合材料粒子等の高分子薬物担体も、ある種の適用に等しく、またはそれ以上に適することがある。
【0028】
二段階ターゲッティング戦略の細胞ターゲッティング段階のためのターゲッティング剤は、腫瘍細胞に選択的に高親和性で結合する。理想的には、腫瘍特異的標的は、腫瘍細胞表面にのみ存在する分子である。しかし、全ての癌細胞に存在するが、正常細胞に存在しない一般的な腫瘍特異的細胞マーカーはまだ発見されておらず、腫瘍細胞と正常細胞との間の類似性は、それらの細胞の相違よりもはるかに数が多い。しかし、ある種の腫瘍細胞で特異的に過剰発現するいくつかの細胞表面マーカーが存在している。EGF(上皮成長因子)受容体は、例えば正常細胞に比べて脳、膀胱、乳房、および肺の腫瘍細胞で過剰発現する。このように、腫瘍中のEGF受容体を標的に定めて、EGFと結合した放射性核種または安定核種について高い選択性を達成することができる。標的腫瘍細胞に対するモノクローナル抗体は、腫瘍のターゲッティングに有効であることも実証された。
【0029】
細胞ターゲッティング段階のための薬剤は、このようにリガンド、抗体、または抗体断片を含む群から好ましくは選択され、上皮成長因子(EGF)、腫瘍特異的突然変異EGF受容体に結合する分子も含むことがある。
【0030】
リポソーム薬物送達システム、第1段階のターゲッティング剤、およびそれらそれぞれの調製は、上に挙げた米国特許第6562316号に記載されている。高分子担体または脂質/高分子担体の調製は本特許に開示されていないが、当業者は文献から関連する調製プロトコールを容易に入手するであろう。
【発明を実施するための最良の形態】
【0031】
薬物前駆体(DNAターゲッティング剤)
DNAターゲッティング剤として使用されるアントラサイクリン誘導体(薬物前駆体)は、DNAと相互作用する性質または静電結合性を有しなければならない。DNAインターカレーターはDNAターゲッティング剤として特に適する。それは、その分子のDNAへの挿入自体によって、治療活性が生じるからである。
【0032】
本発明による一般式Iのアントラサイクリン誘導体は、下記:
【化2】
[式中、Rは、二重結合酸素原子または両方の立体異性体のヒドロキシル基のいずれかであり、また
R1は、CH3またはCH2OHのいずれかである、
R2は、Y−Ar−Z基であり、ここで、
Yは、−(CH2)n−等のスペーサー分子または式−(CH2CH2O)n−(ここで、nは1〜8である)を有するポリエチレングリコール鎖のいずれかであり;
Arは、通常の単環式芳香族基または安定な芳香族ホウ素ケージ化合物であり、ここで、その通常の芳香族残基は、求電子芳香族置換を用いて直接放射性標識できる(水素原子等の)置換基または活性化基(例えば、トリメチルスタニルまたはトリブチルスタニル基等のトリアルキルスタニル基)を含み、その活性化基は、放射性核種に交換できるか、または、その芳香族残基がハロゲン−ハロゲン交換反応を受けることができるようにハロゲン(例えばBrまたはI)を含み;かつ
Zは、任意に、糖基等の親水性を増加させるための化学基である]
およびその塩である。
【0033】
塩の例は、塩酸塩、臭化水素塩、ギ酸塩、および他のカルボン酸塩等の塩である。
【0034】
通常の単環式芳香族基は、好ましくはフェニルまたはピリジン基であり、安定な芳香族ホウ素ケージ化合物は、好ましくはC2H12B10等の安定なcloso-カルボランである。
【0035】
本発明は、(放射性治療薬の基本となる)前記薬物前駆体の立体異性体混合物および個別の立体異性体に関する。
【0036】
DNAターゲッティング段階のための薬剤は、
・核DNAに対して高い親和性を有し、
・担体への効率的な負荷を許容する性質を有し、
・担体に包有(または結合)された場合に、生理的なpHおよびイオン強度で最小の漏出(または放出)を示し、
・担体からの放出後に薬剤を核DNAに到達させて結合できるようにする性質を有する。
【0037】
本発明による薬物前駆体および放射性治療薬は、これらの特性を表す。
【0038】
薬物前駆体の具体的な例を下式に示す:
【化3】
【0039】
薬物前駆体の合成
薬物前駆体の合成は標準法に従い、当業者はこれを行うことができる。薬物前駆体の合成は、意図した保護範囲の一部ではない。
【0040】
放射性治療薬
本発明による放射性治療薬、すなわち核種と結合した薬物前駆体を使用する場合、多量の核種が腫瘍細胞に送達され、これらの核種は核DNAに到達および結合するであろう。各放射性崩壊は、核DNAの損傷を招くであろう。このように、放射性治療薬は、同濃度の薬物前駆体よりも強力であろう。放射性核種が細胞核外側に存在する状況に比べて、放射性核種が細胞核内部に局在する場合、DNA損傷の量は少なくとも10倍高いであろう。よって、腫瘍細胞への多量の放射性核種の送達は、姑息的から治癒的に治療の範囲を広げることができる。(DNAのインターカレートなしの)通常の細胞一段階ターゲッティング戦略が使用されるならば、姑息的治療だけが可能と思われる。
【0041】
各放射性核種は、半減期等の別個のセットの性質と、別個の種類の放出された放射線とを有する。本発明は、特定の種類の癌または特定の臨床問題に適した核種を選択的に選ぶことを可能にする。選ばれた放射性核種の物理半減期を薬物前駆体の生物学的半減期と合致させなければならない。したがって、放射性核種の放射性質が特定の腫瘍の大きさおよび位置と合致することが重要である。90Y等の高エネルギーベータ放射体は、大きな腫瘍の治療に適することがある。131I等の他の核種は、低エネルギーベータ粒子を放出し、結果としてより短距離の放射線を有し、それによってこれらの核種はより小さい腫瘍または単一の腫瘍細胞にさえもさらに適するようになる。125Iおよび123I等のオージェ電子放射体は、約1〜2μmしか進行しない粒子を放出し、よって、DNA損傷を引き起こすには癌細胞の核内に局在しなければならない。アルファ粒子放射体の飛程は概して50から70μmの間であり、多量のエネルギーの局所蓄積を引き起こす。
【0042】
放射性ハロゲンは、広範囲の物理的半減期および何種類かの放出された放射線を有し、これは放射性ハロゲンの適用の範囲を広げている。ベータ放射体である131Iは、核種治療で最も一般的に使用されている放射性核種の1つである。陽電子放出トモグラフィー(PET)では、18F、76Br、および124I等の陽電子を放出する放射性ハロゲンを診断目的に使用できる。それらの放射性ハロゲンは組織の放射線量測定およびPETスキャンを介した腫瘍体積の変化の監視を可能にする。
【0043】
放射性ハロゲンである211Atは比較的短い半減期(7.2時間)を有し、α粒子の放射によって崩壊する。これは、極めて細胞毒性が高く、よって標的となる細胞集団に特異的に送達されるならば有効な治療剤であり得る。
【0044】
オージェ電子放射体は、細胞核に組み入れられると細胞を効果的に殺滅する。125I(半減期60日)、123I(半減期13.2時間)、および77Br(半減期56時間)は電子捕獲によって崩壊し、それに続いてオージェ電子が放射される。本特許出願で論じる放射性治療薬は、125Iとの結合に特によく適する。125Iは比較的安価であり、商業的に広く入手でき、その相対的に長い半減期はin vitro適用に適する。125Iは物理的半減期が短く、特にオージェ電子以外に画像化を可能にするガンマ線も放出することから、治療応用への魅力的な候補でもあり得る。
【0045】
短い放射飛程を有する放射性核種、例えば125I(オージェ放射)および211At(アルファ粒子)は高い局所イオン化密度を発生し、単一細胞に標的を定めるために現在最も適すると思われ、周囲の健康な細胞または組織に最小の損傷しか起こさないであろう。
【0046】
放射性治療薬には、中性子または光子によって活性化できる安定核種が含まれ得る。安定核種である10Bは細胞毒性を示さない。しかし、10Bに富む化合物が腫瘍細胞に選択的に局在化するならば、細胞は次に無毒低エネルギー中性子を外部照射されることがある。これらの中性子は、10B原子に捕捉され、励起状態の11B原子を発生し、その原子は2つの高細胞毒性粒子であるα粒子および7Li3+イオンに即座に瞬間的に解体する。組織中のこれらのイオンの飛程は、それぞれ約9および5μmであり、これは細胞1個の直径に近い。157Gdも中性子活性化に供することができる。他の代替物は、ヨウ素または臭素の安定同位体であり、これらの同位体を光子によって活性化することができる。さらに、これらの同位体を長距離β放出放射性核種、すなわち131I、32P、67Cu、90Y、または189Reと組み合わせて、大きな腫瘍細胞凝集体に適した十字砲火放射を生み出すことができる。これらの中性子または光子で活性化された核種を、例えば閉じたカルボラン、例えばcloso-カルボランであるo−、m−、またはp−C2H12B10等のホウ素リッチケージ化合物を使用して安定化できる。
【0047】
本特許出願に記載した放射性薬物中の放射性核種は、好ましくは123−125I、131I、18F、76−77Br、211At、90Y、32P、67Cu、または189Reであり、125Iが特に好ましく、安定核種は好ましくは10Bおよび157Gdである。
【0048】
薬物前駆体の放射性標識
放射性ヨウ化のための全ての薬物前駆体は、芳香族残基を含み、クロラミンT法等の通常法を使用して125Iで標識される。芳香環の直接求電子置換または芳香環内のトリアルキルスタニル、例えばトリメチルスタニルまたはトリブチルスタニル基等の活性化基の交換のいずれかによって放射性標識を行った。これらの方法は標準法に従い、当業者は実施することができる。
【実施例】
【0049】
本発明による薬物前駆体または放射性治療薬を薬物送達システムに使用できるかどうかを試験するために以下の実験を行った。
【0050】
実施例1.リポソームの薬物保持
リポソームの調製
リポソームは、モル比57:40:3の、1,2−ジステアロイル−sn−グリセロ−3−ホスファチジルコリン(DSPC)、コレステロール、1,2−ジステアロイル−sn−グリセロ−3−ホスファチジルエタノールアミンーN−[(メトキシ)−(ポリエチレングリコール)−2000(DSPE−PEG2000)から構成された。脂質薄膜水和法によってリポソームを調製した[9]。簡潔には、コレステロールおよび脂質をクロロホルムに溶解させた。溶媒を穏やかな窒素気流下で蒸発させ、得られた脂質薄膜を減圧下で一晩乾燥させた。その脂質薄膜を脂質濃度20mM、温度60℃で300mMクエン酸緩衝液(pH4)で1時間、途中でボルテックスミキサーで混合して水和させた。リポソームを液体窒素中で凍結させ、温度60℃で解凍することを5回繰り返してから押出した。Avanti Mini押出機(Avanti Polar Lipids Inc.、Alabaster、アラバマ州)を使用して、リポソームに孔径100nmを有する2枚の重層したポリカーボネート濾紙(Whatman Inc.Nucleopore、Newton、マサチューセッツ州)を室温で10回通過させた。
【0051】
薬物の封入
化合物1(3’−N−(4−ヒドロキシ−3−ヨードベンジル)−13−(R/S)−ジヒドロダウノルビシン、立体異性体混合物)および化合物2(3’−N−(4−ヒドロキシ−3−ヨードベンジル)ダウノルビシン)(構造は下記)、またはドキソルビシン(Sigma Aldrich、St Louis、ミズーリ州、米国)をMayerらによるpH勾配駆動負荷プロトコールを使用してリポソームに封入した。Sephadex G−50カラムで小胞外の300mMクエン酸緩衝液(pH4)を20mM N−[2−ヒドロキシエチル]ピペラジン−N’−[2−エタンスルホン酸](HEPES)緩衝150mM食塩水(HBS)(pH7.5)に交換することによって、リポソーム膜を介したpH勾配を発生させた。ドキソルビシンを1mMの濃度でHBSに溶かした。化合物1を0.5mMの濃度で10%(wt/vol)スクロース溶液に溶かした。化合物2は10%(wt/vol)スクロース溶液に0.5mMの濃度で完全には溶解しなかったが、リポソームを加えてその薬物をリポソームに封入すると溶解した。予熱した薬物溶液を薬物対脂質のモル比0.2でリポソームに加えた。ボルテックス装置を使用して時々混合しながら、得られた混合物を温度60℃で15分間インキュベートした結果、100%の薬物封入がもたらされた。
【0052】
化合物1および2の化学構造
【化4】
【0053】
薬物負荷リポソームに対するターゲッティングリガンドであるトラスツズマブの添付
a)DSPE−PEG3400に対する125I−トラスツズマブの結合
1,2−ジステアロイル−sn−グリセロ−3−ホスファチジルエタノールアミン−N−[メトキシ(ポリエチレングリコール)−3400のN−ヒドロキシスクリンイミジルエステル(NHS−DSPE−PEG3400)を125I−トラスツズマブ溶液と共にモル比0.1:6で温度60℃で5分間水和させた。放射性標識トラスツズマブを使用してリポソームを追跡した。NHS−DSPE−PEG3400の濃度は約0.2mMであった。得られた混合物を撹拌しながら室温で1時間インキュベートした。HBS(pH7.4)を用いたSephacryl S−300カラムのゲル濾過によって、未結合のトラスツズマブを125I−トラスツズマブ−DSPE−PEG3400から除いた。
【0054】
b)リポソームへの125I−トラスツズマブ−DSPE−PEG3400の移行
125I−トラスツズマブ−DSPE−PEG3400を薬物負荷リポソームと1:33のモル比で温度60℃で1時間混合した。HBS(pH7.4)を用いたSepharose CL−4Bカラムのゲル濾過によって、取り込まれていない125I−トラスツズマブ−DSPE−PEG3400をリポソームから除いた。
【0055】
薬物保持の決定
リポソームを温度37℃または60℃でインキュベートした。選択した時間間隔で、3分取分の部分試料を回収し、HBS(pH7.5)を用いてSephadex G−50ミニカラムで680gの遠心場で2分間封入されていない薬物を除いた。溶出液の体積をHBSで1mlに調整し、1%Triton X−100溶液1mlを加えた。試料を温度90℃に加熱し、室温に冷却した。試料の蛍光強度を励起波長468nmおよび発光波長589nmで決定した。インキュベーション前に封入された薬物の量に対するインキュベーション後に封入された薬物の率を決定した。
【0056】
負荷されたリポソームの極低温透過型電子顕微鏡観察(Cryo−TEM)
簡潔には、注文製造した環境チャンバーの中で制御した温度(25℃)で水の蒸発を最小にする湿度条件で、有孔高分子薄膜を被覆した銅格子上にリポソーム試料を移行させた。濾紙に吸引することによって過剰の試料を除いた。薄い(100〜500nm)試料薄膜を温度−165℃で液体エタンに沈めることによってガラス化し、窒素雰囲気で温度−165℃でZeiss EM920A透過型電子顕微鏡(Carl Zeiss Inc.、Oberkochen、ドイツ)に移行させた。試料を電子密度5〜15e−/Å2に曝露し、ゼロロス明視野モードで加速電圧80kVで画像を取得した。
【0057】
結果および考察
温度37℃で24時間培地中でインキュベートしたとき、約80%のドキソルビシンおよび90%を超える化合物1がリポソームに封入されたままであった(図1)。これらの結果は、リポソーム調製および細胞培養に関して記載された条件で、ドキソルビシンおよび化合物1はリポソームからあまり放出されないであろうことを示している。
【0058】
125I−トラスツズマブ−DSPE−PEG3400を含有し、化合物1を負荷したリポソームのcryo−TEM画像(図2)から、化合物1がリポソーム内で結晶状態であることが確認される。
【0059】
実施例2.DNAの結合
実施例2a
細胞培養
過剰発現しているヒト培養腫瘍細胞を10%ウシ胎仔血清、グルタミン(2mM)、ストレプトマイシン(100μg/ml)、およびペニシリン(100IU/ml)を含有するハムのF−10培地(Biochrom AG、Berlin、ドイツ)を用いて温度37℃で加湿した5%CO2インキュベーター中で単層培養として成長させた。
【0060】
DNAの結合
培養ヒト腫瘍A431細胞(扁平上皮癌)をスライドガラス上に成長させ、温度4℃に冷却し、リン酸緩衝液で洗浄した。細胞を温度−20℃で15分間メタノールに入れて不活性化させてから、温度4℃でリン酸緩衝液で速やかに洗浄した。次に、温度4℃で10秒間アセトン処理することによって細胞を透過性にした。乾燥させてから細胞を室温で1時間125I−化合物1と共にインキュベートし、洗浄し、ガンマカウンター(1480 Wallac Wizard、Perkin Elmer、Wellesley、マサチューセッツ州、米国)を用いて放射能について分析した。さらに、細胞を蛍光顕微鏡で検査した。
【0061】
結果および考察
透過性にした培養ヒト腫瘍A431細胞(扁平上皮癌)を125I−化合物1と共に室温で1時間インキュベートしたとき、細胞に結合した化合物の比放射能は、バックグラウンドを減算した後で細胞105個あたり21.1±3.7×103cpmと決定された。過剰のドキソルビシンを使用することによって結合を遮断できた。
【0062】
蛍光顕微鏡観察から、化合物1が細胞核に結合して細胞質内部に局在しなかったことが明らかとなった。ヒト膀胱癌T24細胞およびヒト神経膠腫U343細胞を用いて蛍光顕微鏡実験を繰り返し、同様の結果を得た。
【0063】
実施例2b
アガロースプラグ
InCertアガロース(BioWhittaker Molecular Applications、Rockland、メイン州)を無血清培地に終濃度1%になるように溶かした。アガロース溶液1mlをU−343細胞1.5×106個と混合し、プラグ20μlをプラスチックの型で固めて温度4℃で30分間冷却した。プラグを溶解緩衝液(1mg/mlプロテイナーゼK、0.5M Na3−EDTA(pH8.0)10mlに溶かした2%サルコシル)に温度50℃で一晩沈め、純粋なDNAを得た。溶解後にプラグを0.5M Na3−EDTAで2回洗浄して細胞の破片を除いた。プラグを0.5M Na3−EDTAに入れて温度4℃で保存した。DNAを有さないプラグを対照として供した。アガロースプラグを125I−化合物1を含有する溶液600μlと共に2回の繰り返しで氷上で3時間インキュベートした。対照プラグは濃度3×10−5Mの過剰量のドキソルビシンを含有した。125I−化合物1の終濃度は4×10−7M(dH2O溶液)であった。インキュベーションの後、氷上で洗浄することによってプラグに残った放射能をガンマカウンターを使用して決定した。
【0064】
オートラジオグラフィー
培養SKBR−3乳癌細胞を培地1mlあたり0.1μgの濃度の125I−化合物1(0.3kBq/ng)と共に温度37℃で1時間インキュベートした。インキュベーション後に細胞を無血清培地で6回洗浄し、トリプシン/EDTA(PBS溶液で0.25%/0.02%、Biochrome、Berlin、ドイツ)1mlを用いて10分間剥離させた。細胞を培地14mlで再懸濁して遠心分離管に移した。細胞を1200rpmで5分間遠心分離して細胞ペレットを得た。細胞ペレットをホルマリン緩衝液(0.01Mリン酸緩衝ホルムアルデヒド(4%)、Histolab Products AB, Goteborg, スウェーデン)で4℃で1週間固定した。その後、ペレットを以下の手順で脱水した:70%EtOH 2×15分、90%EtOH 30分、95%EtOH 2×15分、99%EtOH 2×15分、およびHistoresin浸透液(Leica Instruments Gmbh、Heidelberg、ドイツ)3×20分。活性化剤を用いてHistoresinに一晩包埋した後で、ペレットの4μm切片を切り出し、スライドガラスに移行させた。スライドガラスを暗条件でKodak NTB感光乳剤(Eastman Kodak Company、Rochester、ニュ−ヨーク州、米国)に浸し、乾燥してから4℃で3日保存した。そのスライドガラスをKodak D19液を用いて3分間現像してから、0.1%酢酸に10秒間移行させ、Kodak固定液を用いて5分間固定した。これら全てを暗条件で行った。水で大規模に洗浄した後で、細胞核をマイアーヘマトキシリン(Histolab Products AB、Goteborg、スウェーデン)で3分間染色した。Pertex(Histolab Products AB、Goteborg、スウェーデン)でマウントする前に、スライドガラスを水で5分間洗浄して風乾させた。細胞を顕微鏡で検査して、代表的な細胞の画像を取得した。
【0065】
結果および考察
125Iの染色パターンがヘマトキシリン染色細胞核と共局在していることから、細胞核に対する化合物1の結合を125I−化合物1のオートラジオグラフィーによって確認できた(図3)。
【0066】
U−343細胞DNAを含有するアガロースプラグを125I−化合物1溶液と共に氷上で3時間インキュベートすると、125I−化合物1はアガロースプラグ中に蓄積された。DNAに対する125I−化合物1の蓄積と、よって結合とを過剰量のドキソルビシンによって遮断された。結果は、DNAに対する化合物1の親和性を確認している。DNAに対する125I−化合物1の結合をドキソルビシンによって置換できたという事実(図4)は、化合物1とドキソルビシンとの両方が同じDNA結合部位を占有することを示唆している。
【0067】
実施例2c
パルスフィールドゲル電気泳動(PFGE)
125I−化合物1がDNA損傷を誘導する効率を検討するために、パルスフィールドゲル電気泳動(PFGE、Pharmacia Biotech、Uppsala、スウェーデン)で二本鎖切断(dsb)の誘導を分析した。上記のようにアガロースプラグを125I−化合物1と共にインキュベートした。洗浄後に、プラグを温度4℃で8日間保持した。次にプラグを0.8%(w/v)アガロースゲル(Seakem Gold粉末アガロース、Cambrex Bio Science Rockland Inc.、Rockland、メイン州、米国)に負荷した。DNAの断片化を以下のプロトコールにより2V/cmで45時間のパルスフィールドゲル電気泳動で分析した:10分のパルス(すなわち10分ごとに場にパルスを与える)3時間、20分のパルス5時間20分、30分のパルス8時間、40分のパルス9時間20分、および1時間のパルス20時間。ゲルを泳動させた後で臭化エチジウム(0.5μg/ml)で8時間染色してから、dH2Oで一晩脱色させた。分子量マーカーとして、シゾサッカロミセス ポンベ(Scizosaccharomyces pombe)メガ塩基DNA標準(Cambrex Bio Science Rockland Inc.、Rockland、メイン州、米国)を使用した。各レーンを、それぞれ5.7Mbp以下および5.7Mbp以上のサイズを有するDNA断片に対応する2つのブロックに切り出した。各ブロックをバイアルに入れ、以前に挙げた自動ガンマカウンターで放射能を測定した。5.7Mbp未満のDNAの割合を決定して、次にBlocherの式を用いた計算に使用した:
F<k=1−e−rk/n(1+rk/n(1−k/n)) (Int J Rad Biol 57, 7〜12、1990)
F<kはk塩基対よりも小さいDNAの割合であり、rはdsb/染色体の平均数であり、nは平均サイズの染色体1本に存在する塩基対の総数である。k=このアッセイでは5.7Mbpを使用した。平均的なヒト染色体ではn=130Mbpである。
【0068】
二本鎖切断数(r)を得るためには、式が数値的に解かれなければならない。
【0069】
結果および考察
125I−化合物1を神経膠腫細胞DNAと共にインキュベートした後の二本鎖切断(dsb)の数(図5)を崩壊1回あたり約0.4dsbと決定した。このように、化合物Iと結合すると、125IはDNAに十分近く位置してDNAの断片化を引き起こす。それは、DNAに結合していない125IはDNAの断片化をもたらさないと思われるからである。比較として、DNA鎖への核種の直接組み入れを招く125I標識DNA前駆分子の使用は、崩壊1回あたり約1dsbのdsb値を与える。dsb値の差は比較的小さく、これは、(放射線が作用を有するために)その核種がDNAから非常に短い距離内に入らなければならず、DNAのインターカレーションが起こったことを示している。
【0070】
実施例2d
1型ウシ胸腺DNAナトリウム塩(Sigma Aldrich Co.、St Louis、ミズーリ州、米国)を約2mg/mlの濃度でBPES緩衝液(6mM Na2HPO4、2mM NaH2PO4、1mM EDTA、185mM NaCl、pH7)で水和させ、MSE Soniprep150超音波粉砕機(Integrated Services TCP Inc.、Palisades Park、ニュージャージー州、米国)に入れて氷冷した水浴中で30分間約8μAで超音波処理した。次に、Slide-A-Lyzer(登録商標)透析カセット(10000MWCO)(Pierce、Rockford、イリノイ州、米国)を使用してBPESに対して試料を48時間透析した。HP8453分光光度計(Hewlett-Packard Company、Houston、テキサス州、米国)で吸光係数12824M(bp)−1cm−1を用いて波長260nmでの吸光度定量により、DNAの終濃度を決定した。
【0071】
SPEX1680 Fluorolog蛍光光度計(SPEX Industries Inc.、Edison、ニュージャージー州、米国)で室温でλex=480nm(スリッド(slid)幅2.5mm)およびλem=592nm(スリッド幅2.5mm)で蛍光滴定実験を行った。初発遊離薬物濃度は1μMであった。次式により、DNAの不在下(I0)および存在下(I)で被験化合物の蛍光強度比を決定することによって遊離薬物濃度(Cf(M))が計算した:
Cf=CT(I/I0−P)/(1−P) (Biopolymers 6、1225〜1235、1968)
式中、CT(M)は初発薬物濃度であり、Pは完全に結合した薬物の蛍光強度の観察された量子収量(Imin)と、遊離薬物の量子収量との間の比である(P=Imin/I0)。CTとCfとの間の差によって、結合した薬物の濃度を計算した。rに対してr/Cfをプロットすること(スキャッチャードプロット、Scatchard plot)によって結合定数(Ki)および排除パラメータ(n)を計算した。ここで、rはDNA塩基対1モルあたり結合した薬物のモル数である。隣接排除モデルに関する理論曲線を次のアルゴリズムを使用して計算した:
r/Cf=Ki(1−nr)[(1−nr)/[1−(n−1)r]]n−1 (J.Mol.Biol. 86、469〜489、1974)
式中、Ki(M−1)は固有の結合定数であり、n(塩基対)は排除パラメータである。実験データに最も適合する理論曲線を発生するように、パラメータKiおよびnを変動させた。
【0072】
表1(下記)は、ウシ胸腺DNA(CT DNA)に対するダウノルビシン、ドキソルビシン、化合物1、および化合物2の結合定数(Ki)および排除パラメータ(n)を示す。カッコ内の値は、平均値の標準誤差(SEM)(n=3)を示す。a=ダウノルビシンに対してp<0.05。b=化合物2に対してp<0.05。平均値の間の差を一元配置分散分析の後にStudent Newman-Keuls検定により検定した。
【0073】
文献値
ダウノルビシンのKi:0.7×106M−1±0.07、n:3.5塩基対±0.35(Chairesら(1982)Biochemistry 21、3933〜3940)。ドキソルビシンのKi:3.3×106M−1、n:3.8塩基対(Messoriら(2001)Bioorg.Med.Chem. 9、1815〜1825)。
【0074】
表1:ドキソルビシン、ダウノルビシン、化合物1、および化合物2の、結合定数および排除パラメータ
【表1】
【0075】
結果および考察
化合物1および化合物2のCT DNAとの結合定数および排除パラメータがドキソルビシンの場合と類似していることから、ダウノルビシンの誘導体化がDNA結合性の喪失を起こさなかったことが確認される(表1)。逆に、化合物1および化合物2の結合定数は親化合物のダウノルビシンの結合定数よりも高い値であった?。
【0076】
実施例3.毒性
成長曲線125I−化合物1、化合物1、ドキソルビシン、またはダウノルビシンを0.5ng/mlの濃度に培地に溶解させた。125I−化合物1の比放射能は100kBq/ngであった。SKBR−3細胞を、60mmプラスチックペトリ皿中で3回の繰り返しで2.5時間薬物と共にインキュベートした。対照細胞を通常の培地と共にインキュベートした。インキュベートの後で、培地を全ての皿から除き、細胞を無血清培地で6回洗浄した。トリプシン/EDTA0.5mlを温度37℃で10分間加えることによって細胞を皿から剥離させた。培地1mlに再懸濁した後で、細胞をコールターカウンター(Z2コールターカウンター、Beckman Coulter)を使用して計数し、細胞105個まで継代培養して成長曲線を作製した。継代培養毎に細胞の損失に関して成長曲線を補正した。
【0077】
結果および考察
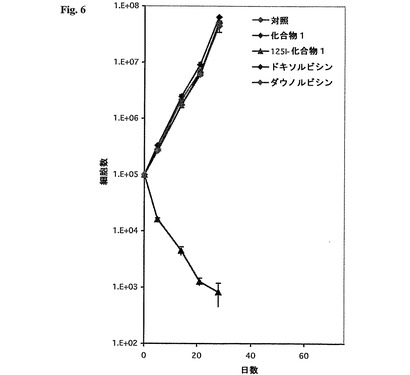
培養SKBR−3細胞単層の成長曲線は、濃度わずか0.5ng/mlの125I−化合物1の細胞毒性作用が化合物1、ドキソルビシン、またはダウノルビシンとインキュベートした後に比べて数桁大きかったことを明らかにした(図6)。後者の化合物類のどれも有意な細胞毒性作用を有さなかった。よって、125I−化合物1によって表された細胞毒性作用は化合物1に結合した核種125I単独によって引き起こされる。
【0078】
参照文献
[1]B.A.Chabner,C.E.Meyers(1982) ”Cancer: Principles and practice of oncology” (DeVita Jr.V.T., S.HellmanおよびS.A.Rosenberg編), 156〜197頁, J.B.Lippincott Company, Philadelphia, Toronto.
[2]H.G.Keizer, H.M.Pinedo, G.J.Schuurhuis, H.Joenje, ドキソルビシン(アドリアマイシン):細胞毒性の遊離ラジカル依存性メカニズムの決定的な総説、Pharmaceutics and Therapeutics 47(1990)219〜231.
[3]J.Bouma, J.H.Beijnen, A.Bult, W.J.M.Underberg, アントラサイクリン系抗腫瘍剤、Pharmaceutisch Weekblad Scientific Edition 8(1986)109〜133.
[4]C.P.Association, Compendium of pharmaceuticals and specialities, 第13巻、CK Productions, Toronto、1995.
[5]R.H.Blum, S.K.Carter, アドリアマイシン。重大な臨床活性を有する新しい抗癌薬,Ann Intern Med 80(1974)249〜59.
[6]M.N.Gaze, 臨床業務における標的を定めた放射線治療の現状,Phys Med Biol 41(1996)1895〜903.
[7]J.A.O’Donoghue, T.E.Wheldon, オージェ電子放射体を用いた標的を定めた放射線治療、Phys Med Biol 41(1996)1973〜92.
[8]S.Kim, 癌化学療法の担体としてのリポソーム。現状と将来展望、Drugs46(1993)618〜38.
[9](M.J.Hope, M.B.Bally, G.Webb, およびP.R.Cullis (1985) Biochim. Biophys. Acta 812: 55〜65).
【図面の簡単な説明】
【0079】
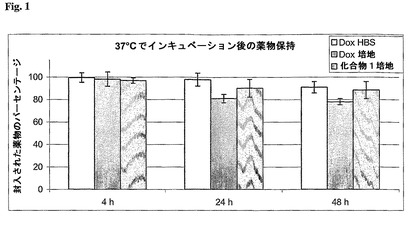
【図1】温度37℃での緩衝液または培地中のドキソルビシンと、緩衝液中の化合物1との保持を示す図である。
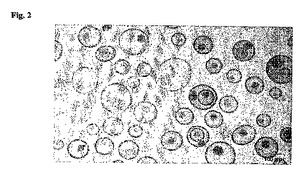
【図2】化合物1を負荷したリポソームの極低温透過型顕微鏡画像を示す図である。球体はリポソームを表し、リポソーム内部の斑点は結晶状の化合物1を表す。
【図3】腫瘍細胞と共に37℃で1時間インキュベートした後の125I−化合物1のオートラジオグラフィーを示す図である。
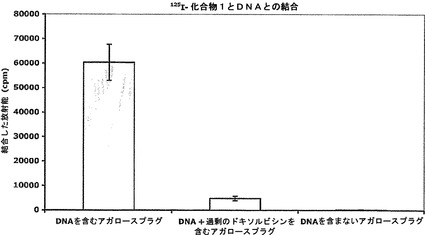
【図4】氷上で2.5時間インキュベートした後の遊離DNAに対する125I−化合物1の結合を示す図である。
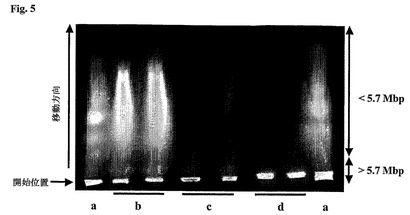
【図5】U−343MGaC12:6神経膠腫細胞DNAを含有するプラグを負荷したアガロースゲルでの125I−化合物1の結合後のDNA断片化を示す図である。図中、a)分子量マーカー(シゾサッカロミセスポンベ、メガ塩基DNA標準)、b)125I−化合物1と共にインキュベートしたDNA、c)125I−化合物1および過剰のドキソルビシンと共にインキュベートしたDNA、ならびにd)125I−化合物1不在下でインキュベートしたDNAを有する対照。
【図6】0.5ng/mlドキソルビシン、ダウノルビシン、化合物1、または125I−化合物1で処理した腫瘍細胞の成長曲線を示す図である。
【技術分野】
【0001】
本発明は、新しいアントラサイクリン誘導体ならびに癌の治療および癌の診断へのそれらの使用とに関する。
【背景技術】
【0002】
DOXと省略され、アドリアマイシンまたはアドリアブラスチン等の他の名前でも公知であるドキソルビシン(C27H29NO11;MW:543.53)は抗生物質であり、アントラサイクリンファミリーの抗腫瘍薬である(構造は下記参照)。DOXは、もともと水中細菌ストレプトミセスペウセティウス変種コエシウス(streptomyces peucetius var. coesius)から単離され、1970年代前半以来、アントラサイクリン、特にドキソルビシンおよびダウノルビシンならびにアルキル化剤(シクロホスファミド、メルファラン等)が、今日臨床で最も多方面で最も頻繁に使用されている化学療法剤である[1]。アントラサイクリンは、キノン−ヒドロキノン官能基および親水性アミノ糖部分を有する疎水性アグリコン複素環からなる両親媒性分子である[2,3]。
【0003】
ダウノルビシンおよびドキソルビシンの化学構造
【化1】
【0004】
ドキソルビシンは骨肉腫および軟部組織肉腫ならびに肺、乳房、甲状腺、膀胱、卵巣、精巣、頭部、および頚部の癌腫の治療に広く使用されている[1、4]。ドキソルビシンは、白血病およびリンパ腫に対しても使用されるが、ダウノルビシンは急性白血病に対する一次治療である。ドキソルビシンに関する全奏効率は、甲状腺癌では45%、リンパ腫では41%、膀胱癌では33%、肉腫では26%、卵巣癌では25%、白血病では24%である[5]。
【0005】
ドキソルビシンは多様な作用機序を有するが、ドキソルビシンおよび他のアントラサイクリンの主な抗腫瘍活性は、それらがDNAにインターカレートする結果として、DNA、RNA、およびタンパク質の合成の遮断を招く能力に由来する。アントラサイクリンはトポイソメラーゼIIも阻害し、DNA修復を障害する[1、5]。キノン−ヒドロキノン官能基が原因で、アントラサイクリンはDNA損傷に至るフリーラジカルの発生に関与すると考えられている[2]。アントラサイクリンは、心ミトコンドリアおよび悪性細胞の膜に高濃度にみられるリン脂質であるカルジオリピンに特異的に結合し、これによって、ドキソルビシンの心毒性副作用を説明できる[1]。用量が少なすぎると、アントラサイクリンは効果を有さず、用量が多すぎると、重度の副作用が起こるおそれがあることから、この薬物は狭い治療指数を有する。すなわち、投与量は狭い範囲内に入らなければならない。ドキソルビシンの急性の用量制限毒性は、骨髄抑制、白血球減少、および投与患者の80%で起こる口内炎である。他の副作用には、脱毛(100%)、悪心、嘔吐(20〜55%)、心毒性、すなわち上室性不整脈、心ブロック、心室頻拍、および患者の1〜10%で生じるうっ血性心不全さえある。
【0006】
アントラサイクリン誘導体は、以前に例えばPribe(2003)Chemico-Biological Interactions 145:349〜358、US−4948880、US−6673907、およびWO00/56267に開示された。これらの誘導体は、癌の治療で細胞毒性作用を有する。
【0007】
放射性核種治療は、癌の治療に比較的小さいが重要な役割を有し、現在ますます大きな注目を集めている。放射性核種治療は、核放射線を使用して悪性細胞を根絶させる。その放射線は、中性子活性化の後の安定核種、例えば10Bおよび157Gによって、または放射性核種によって発生させることができる。特に甲状腺癌に対する最も一般に使用されている治療用放射性核種は、中距離(800μm)β照射体の131Iであるが、131Iの投与は健常組織にかなりの照射損傷を引き起こす[6]。しかし、これらの副作用により、125I等の短距離低エネルギーオージェ電子放射体の治療上の潜在能力が広く認められてきている。抗癌治療に有効であるためには、125Iは直接的かつ選択的に腫瘍細胞の核に送達しなければならない。それは、125IがDNAから数ナノメーター以内に入らない限り毒性ではないからである[7]。このように、125Iによる治療は核への特異的な送達法を必要とし、その方法は、125Iで標識したヌクレオシド、オリゴヌクレオチド、ステロイドホルモン、および成長因子を用いて以前は実現されたが、改善の必要が認識されていた[7]。
【0008】
Murali D.およびDeJesus.O,Bioorganic&Medicinal Chemistry letters 8 (1998) 3419〜3422は、ドキソルビシンに比べて改善された細胞毒性を有する放射性標識ダウノルビシン誘導体を記載している。この論文に結果は提示されておらず、この研究の継続は発表されていない。
【0009】
リポソームを介した標的を定めた薬物送達は、膜結合型小胞(リポソーム)に細胞毒性剤を封入することおよびリポソーム膜に腫瘍特異的抗体を結合すること(標的を定めたリポソーム)によって、骨髄抑制、粘膜炎、心毒性、神経毒性、および腎毒性等の通常の癌化学療法の用量規制副作用を最小にする[1]。血中にある標的を定めたリポソームは、標的となる表面マーカーを過剰発現している腫瘍細胞によって能動的かつ選択的に取り込まれる。しかし、いくらか前進したにもかかわらず、この戦略は化学療法の大きな進歩を今のところもたらしていない[8]。
【0010】
このように、より強力な治療剤および治療戦略の大きな必要性が存在する。
【発明の開示】
【発明が解決しようとする課題】
【0011】
(発明の概要)
本発明は、DNAにインターカレートする性質を有し、核種を結合させることができる診断剤および治療剤に関する。本発明はさらに、これらの診断剤または治療剤に関する薬物送達システムならびに前記薬剤または薬物送達システムを使用した治療法および診断法に関する。本発明は、癌の診断および治療を意図する。本発明の目的は、核種を腫瘍細胞の核に特異的に送達することであり、したがって、健常組織での細胞毒性副作用を最小にする単一の二段階ターゲッティング法で、放射性核種治療、化学療法、および標的を定めたリポソーム薬物送達の利益を合せることである。このように、本発明は新薬を用いた新しい治療戦略を提供し、その新薬は以前より公知であった化学療法薬よりも潜在的に強力である。
【課題を解決するための手段】
【0012】
アントラサイクリンはそれ自体で強力な抗癌薬である。本出願において、本発明者らは、ダウノルビシンのアミノ−ベンジル誘導体(薬物前駆体)を合成した。ダウノルビシンは、商業的に成功しているアントラサイクリンであるドキソルビシンおよびダウノルビシンに類似した細胞毒性プロフィールを有する。本発明者らがこの薬物前駆体を125Iでヨウ化したとき、培養腫瘍細胞に対してさらに有効な放射性治療剤が得られた。健常組織を保護し、悪性癌細胞に放射性核種を選択的に送達するために、標的を定めたリポソームに125Iと結合したダウノルビシン誘導体を封入することができ、そのリポソームは腫瘍細胞に対する特異的薬物送達小胞として働く。
【0013】
本明細書に記載した実験の結果は、標的を定めたリポソームに本発明による治療剤をうまく封入することができ、その薬物が実験の調製条件およびアッセイ条件で十分に保持されることを確認する。腫瘍細胞と共にインキュベートした後で、上に挙げた薬剤は細胞核に達して、ドキソルビシンと同様の親和性で結合する。DNA結合すると、その放射性治療剤はDNAの断片化を引き起こし、今日臨床で最も成功した化学療法剤の2つであるドキソルビシンおよびダウノルビシンによって起こるよりも数桁高い腫瘍細胞成長阻害に導く。
【0014】
本発明による薬物前駆体または放射性治療薬のどれも、先行技術で以前に記載されていない。
【0015】
本発明の一態様によると、放射性治療剤の前駆分子として働き、以下に薬物前駆体と呼ぶアントラサイクリン誘導体が提供される。これらの薬物前駆体は、DNAにインターカレートし、細胞分裂抑制性を有する。このように、これらの薬物前駆体は、細胞または組織に標的を定めた癌治療に単独で使用することができる。アントラサイクリン誘導体(薬物前駆体)は請求項1〜3に定義する。
【0016】
本発明のさらなる態様によると、細胞または組織に標的を定めた放射性治療に使用できる放射性治療薬が提供される。これらの放射性治療剤は、核種または核種を含む化学基に薬物前駆体を結合することによって、それらの前駆体から生成する。そのような核種は、放射性核種、安定核種、または中性子もしくは光子に曝露することによって活性化できる核種、例えば(closo-カルボランであるo−、m−またはp−C2H12B10等のホウ素リッチケージ(boron-rich cage)化合物の一部としての)10Bであってよく、以下に核種と呼ぶ。放射性治療薬は請求項4から5に定義する。
【0017】
本発明のさらなる態様によると、癌診断の画像化ツールとしてこれらの放射性治療薬を使用することもできる。画像化ツールとしての使用は請求項6に定義する。
【0018】
本発明のさらなる態様によると、薬物前駆体または放射性治療薬をDNAターゲッティング剤、すなわち請求項7に定義するようなDNA相互作用剤として使用することもできる。
【0019】
本発明のさらなる態様によると、薬物送達システムも提供される。前記システムは、(DNAにインターカレートする性質を有する)薬物前駆体または放射性治療薬を封入することができる担体を含み、標的となる細胞集団に特異的または優先的に薬物を案内する。結果として、細胞および組織を損傷する作用は、標的となる細胞および組織に優先的に影響することになる。そのような薬物送達システムは、一段階または多段階ターゲッティング戦略を含むことがある。薬物前駆体がDNAにインターカレートする性質は、それらの細胞毒性効果を核に向けるか、または、それらの核種が薬物前駆体に結合しているときは、それらの核種から放出された放射能を細胞核に局在化するDNAターゲッティング段階の基本である。以下にDNAターゲッティング段階と呼ぶこのターゲッティング段階は、このように放射性治療薬の治療効果を劇的に増やし、健康な細胞および組織に対する損傷を減少させる。
【0020】
悪性組織と健常組織とを区別するために、放射性治療薬は、二段階ターゲッティング戦略における細胞ターゲッティング段階として、表面に腫瘍細胞特異的ターゲッティング剤を示す薬物送達システムを使用して癌細胞または癌組織に向けられる。その段階は以下に、細胞ターゲッティング段階と呼ぶ。薬物担体は、放射性治療薬を包有または結合し、全身投与後に、その放射性治療薬の標的細胞の膜への、好ましくはその膜を通過する輸送を指令できる。放射性治療薬の細胞毒性および/または放射性毒性作用は、結果として標的となる細胞集団に局在化するであろう。薬物送達システムは請求項8から13に定義されている。
【0021】
本発明のさらなる態様によると、癌を診断または治療する方法も提供され、その方法は、それを必要とする患者に前駆体薬物または放射性治療薬を含む前記薬物送達システムを投与することを含む。固形腫瘍に対する治療としての使用に加え、本発明は、原発腫瘍除去後の全身循環における転移腫瘍細胞に対する治療としても予想される。その治療は、播種性乳癌だけではなく、播種性卵巣癌、前立腺癌、および結腸直腸癌にも特に有益であり得る。
【0022】
腫瘍の位置が分かっている場合は、前記治療法をそれに続く腫瘍照射と組み合わせて使用することもできる。安定核種と、中性子または光子を用いた外部照射による活性化とを使用することによって、局所放射線治療を実現できる。
【0023】
本発明は、生体異物に対する膜結合型外向き排出ポンプであるP−糖タンパク質(PGP)を過剰発現している多剤耐性(MDR)腫瘍の有効な治療でもあり得る。この場合、放射性治療薬はアントラサイクリンと結合することが知られているP−糖タンパク質を標的として放射性損傷するであろう。この治療法を請求項14から17に定義する。
【発明の効果】
【0024】
本明細書に記載する薬物送達は、強力な抗癌放射性治療薬または診断用画像化ツールとしての、薬物前駆体の形態または核種と結合した形態の癌治療としての使用を意図する。
【0025】
しかし、それらの有用性は、健常組織に対する無差別の細胞毒性によって損なわれるおそれがある。この問題は、リポソームまたは代替の薬物担体に薬物を封入して、リポソームまたは代替の薬物担体の表面に腫瘍特異的標的薬剤を加えることにより腫瘍細胞を選択的に標的とすることによって最小となるであろう。このように、これらの薬物前駆体または放射性治療薬は、米国特許第6562316号に開示されたような、標的を定めた二段階薬物送達システムと組み合わせた使用を意図する。そのような薬物送達システムは、細胞ターゲッティング段階において薬物送達システムを標的となる細胞集団または組織に特異的に向けるための、細胞ターゲッティング剤と結合した担体を含む。DNAターゲッティング段階では、封入された核種は、DNAにインターカレートする性質を有する分子に結合することによって細胞核に向けられるであろう。本明細書に記載した、薬物前駆体または放射性治療薬は、DNAにインターカレートする性質を有するそのようなDNAターゲッティング剤として働くであろう。
【0026】
記載された二段階ターゲッティングシステムは、健常組織での核種、薬物前駆体、または放射性治療薬の細胞毒性を最小にするであろう。二段階ターゲッティングシステムのさらなる利点は、転移および/または多剤耐性腫瘍細胞の治療に潜在能力を有することである。
【0027】
薬物担体は、薬学的に活性な薬剤、すなわち薬物前駆体または放射性治療薬を結合または包有できる分子、凝集体、または粒子であり得る。リポソームは現在好ましい薬物担体であるが、ミクロゲル、または脂質/高分子複合材料粒子等の高分子薬物担体も、ある種の適用に等しく、またはそれ以上に適することがある。
【0028】
二段階ターゲッティング戦略の細胞ターゲッティング段階のためのターゲッティング剤は、腫瘍細胞に選択的に高親和性で結合する。理想的には、腫瘍特異的標的は、腫瘍細胞表面にのみ存在する分子である。しかし、全ての癌細胞に存在するが、正常細胞に存在しない一般的な腫瘍特異的細胞マーカーはまだ発見されておらず、腫瘍細胞と正常細胞との間の類似性は、それらの細胞の相違よりもはるかに数が多い。しかし、ある種の腫瘍細胞で特異的に過剰発現するいくつかの細胞表面マーカーが存在している。EGF(上皮成長因子)受容体は、例えば正常細胞に比べて脳、膀胱、乳房、および肺の腫瘍細胞で過剰発現する。このように、腫瘍中のEGF受容体を標的に定めて、EGFと結合した放射性核種または安定核種について高い選択性を達成することができる。標的腫瘍細胞に対するモノクローナル抗体は、腫瘍のターゲッティングに有効であることも実証された。
【0029】
細胞ターゲッティング段階のための薬剤は、このようにリガンド、抗体、または抗体断片を含む群から好ましくは選択され、上皮成長因子(EGF)、腫瘍特異的突然変異EGF受容体に結合する分子も含むことがある。
【0030】
リポソーム薬物送達システム、第1段階のターゲッティング剤、およびそれらそれぞれの調製は、上に挙げた米国特許第6562316号に記載されている。高分子担体または脂質/高分子担体の調製は本特許に開示されていないが、当業者は文献から関連する調製プロトコールを容易に入手するであろう。
【発明を実施するための最良の形態】
【0031】
薬物前駆体(DNAターゲッティング剤)
DNAターゲッティング剤として使用されるアントラサイクリン誘導体(薬物前駆体)は、DNAと相互作用する性質または静電結合性を有しなければならない。DNAインターカレーターはDNAターゲッティング剤として特に適する。それは、その分子のDNAへの挿入自体によって、治療活性が生じるからである。
【0032】
本発明による一般式Iのアントラサイクリン誘導体は、下記:
【化2】
[式中、Rは、二重結合酸素原子または両方の立体異性体のヒドロキシル基のいずれかであり、また
R1は、CH3またはCH2OHのいずれかである、
R2は、Y−Ar−Z基であり、ここで、
Yは、−(CH2)n−等のスペーサー分子または式−(CH2CH2O)n−(ここで、nは1〜8である)を有するポリエチレングリコール鎖のいずれかであり;
Arは、通常の単環式芳香族基または安定な芳香族ホウ素ケージ化合物であり、ここで、その通常の芳香族残基は、求電子芳香族置換を用いて直接放射性標識できる(水素原子等の)置換基または活性化基(例えば、トリメチルスタニルまたはトリブチルスタニル基等のトリアルキルスタニル基)を含み、その活性化基は、放射性核種に交換できるか、または、その芳香族残基がハロゲン−ハロゲン交換反応を受けることができるようにハロゲン(例えばBrまたはI)を含み;かつ
Zは、任意に、糖基等の親水性を増加させるための化学基である]
およびその塩である。
【0033】
塩の例は、塩酸塩、臭化水素塩、ギ酸塩、および他のカルボン酸塩等の塩である。
【0034】
通常の単環式芳香族基は、好ましくはフェニルまたはピリジン基であり、安定な芳香族ホウ素ケージ化合物は、好ましくはC2H12B10等の安定なcloso-カルボランである。
【0035】
本発明は、(放射性治療薬の基本となる)前記薬物前駆体の立体異性体混合物および個別の立体異性体に関する。
【0036】
DNAターゲッティング段階のための薬剤は、
・核DNAに対して高い親和性を有し、
・担体への効率的な負荷を許容する性質を有し、
・担体に包有(または結合)された場合に、生理的なpHおよびイオン強度で最小の漏出(または放出)を示し、
・担体からの放出後に薬剤を核DNAに到達させて結合できるようにする性質を有する。
【0037】
本発明による薬物前駆体および放射性治療薬は、これらの特性を表す。
【0038】
薬物前駆体の具体的な例を下式に示す:
【化3】
【0039】
薬物前駆体の合成
薬物前駆体の合成は標準法に従い、当業者はこれを行うことができる。薬物前駆体の合成は、意図した保護範囲の一部ではない。
【0040】
放射性治療薬
本発明による放射性治療薬、すなわち核種と結合した薬物前駆体を使用する場合、多量の核種が腫瘍細胞に送達され、これらの核種は核DNAに到達および結合するであろう。各放射性崩壊は、核DNAの損傷を招くであろう。このように、放射性治療薬は、同濃度の薬物前駆体よりも強力であろう。放射性核種が細胞核外側に存在する状況に比べて、放射性核種が細胞核内部に局在する場合、DNA損傷の量は少なくとも10倍高いであろう。よって、腫瘍細胞への多量の放射性核種の送達は、姑息的から治癒的に治療の範囲を広げることができる。(DNAのインターカレートなしの)通常の細胞一段階ターゲッティング戦略が使用されるならば、姑息的治療だけが可能と思われる。
【0041】
各放射性核種は、半減期等の別個のセットの性質と、別個の種類の放出された放射線とを有する。本発明は、特定の種類の癌または特定の臨床問題に適した核種を選択的に選ぶことを可能にする。選ばれた放射性核種の物理半減期を薬物前駆体の生物学的半減期と合致させなければならない。したがって、放射性核種の放射性質が特定の腫瘍の大きさおよび位置と合致することが重要である。90Y等の高エネルギーベータ放射体は、大きな腫瘍の治療に適することがある。131I等の他の核種は、低エネルギーベータ粒子を放出し、結果としてより短距離の放射線を有し、それによってこれらの核種はより小さい腫瘍または単一の腫瘍細胞にさえもさらに適するようになる。125Iおよび123I等のオージェ電子放射体は、約1〜2μmしか進行しない粒子を放出し、よって、DNA損傷を引き起こすには癌細胞の核内に局在しなければならない。アルファ粒子放射体の飛程は概して50から70μmの間であり、多量のエネルギーの局所蓄積を引き起こす。
【0042】
放射性ハロゲンは、広範囲の物理的半減期および何種類かの放出された放射線を有し、これは放射性ハロゲンの適用の範囲を広げている。ベータ放射体である131Iは、核種治療で最も一般的に使用されている放射性核種の1つである。陽電子放出トモグラフィー(PET)では、18F、76Br、および124I等の陽電子を放出する放射性ハロゲンを診断目的に使用できる。それらの放射性ハロゲンは組織の放射線量測定およびPETスキャンを介した腫瘍体積の変化の監視を可能にする。
【0043】
放射性ハロゲンである211Atは比較的短い半減期(7.2時間)を有し、α粒子の放射によって崩壊する。これは、極めて細胞毒性が高く、よって標的となる細胞集団に特異的に送達されるならば有効な治療剤であり得る。
【0044】
オージェ電子放射体は、細胞核に組み入れられると細胞を効果的に殺滅する。125I(半減期60日)、123I(半減期13.2時間)、および77Br(半減期56時間)は電子捕獲によって崩壊し、それに続いてオージェ電子が放射される。本特許出願で論じる放射性治療薬は、125Iとの結合に特によく適する。125Iは比較的安価であり、商業的に広く入手でき、その相対的に長い半減期はin vitro適用に適する。125Iは物理的半減期が短く、特にオージェ電子以外に画像化を可能にするガンマ線も放出することから、治療応用への魅力的な候補でもあり得る。
【0045】
短い放射飛程を有する放射性核種、例えば125I(オージェ放射)および211At(アルファ粒子)は高い局所イオン化密度を発生し、単一細胞に標的を定めるために現在最も適すると思われ、周囲の健康な細胞または組織に最小の損傷しか起こさないであろう。
【0046】
放射性治療薬には、中性子または光子によって活性化できる安定核種が含まれ得る。安定核種である10Bは細胞毒性を示さない。しかし、10Bに富む化合物が腫瘍細胞に選択的に局在化するならば、細胞は次に無毒低エネルギー中性子を外部照射されることがある。これらの中性子は、10B原子に捕捉され、励起状態の11B原子を発生し、その原子は2つの高細胞毒性粒子であるα粒子および7Li3+イオンに即座に瞬間的に解体する。組織中のこれらのイオンの飛程は、それぞれ約9および5μmであり、これは細胞1個の直径に近い。157Gdも中性子活性化に供することができる。他の代替物は、ヨウ素または臭素の安定同位体であり、これらの同位体を光子によって活性化することができる。さらに、これらの同位体を長距離β放出放射性核種、すなわち131I、32P、67Cu、90Y、または189Reと組み合わせて、大きな腫瘍細胞凝集体に適した十字砲火放射を生み出すことができる。これらの中性子または光子で活性化された核種を、例えば閉じたカルボラン、例えばcloso-カルボランであるo−、m−、またはp−C2H12B10等のホウ素リッチケージ化合物を使用して安定化できる。
【0047】
本特許出願に記載した放射性薬物中の放射性核種は、好ましくは123−125I、131I、18F、76−77Br、211At、90Y、32P、67Cu、または189Reであり、125Iが特に好ましく、安定核種は好ましくは10Bおよび157Gdである。
【0048】
薬物前駆体の放射性標識
放射性ヨウ化のための全ての薬物前駆体は、芳香族残基を含み、クロラミンT法等の通常法を使用して125Iで標識される。芳香環の直接求電子置換または芳香環内のトリアルキルスタニル、例えばトリメチルスタニルまたはトリブチルスタニル基等の活性化基の交換のいずれかによって放射性標識を行った。これらの方法は標準法に従い、当業者は実施することができる。
【実施例】
【0049】
本発明による薬物前駆体または放射性治療薬を薬物送達システムに使用できるかどうかを試験するために以下の実験を行った。
【0050】
実施例1.リポソームの薬物保持
リポソームの調製
リポソームは、モル比57:40:3の、1,2−ジステアロイル−sn−グリセロ−3−ホスファチジルコリン(DSPC)、コレステロール、1,2−ジステアロイル−sn−グリセロ−3−ホスファチジルエタノールアミンーN−[(メトキシ)−(ポリエチレングリコール)−2000(DSPE−PEG2000)から構成された。脂質薄膜水和法によってリポソームを調製した[9]。簡潔には、コレステロールおよび脂質をクロロホルムに溶解させた。溶媒を穏やかな窒素気流下で蒸発させ、得られた脂質薄膜を減圧下で一晩乾燥させた。その脂質薄膜を脂質濃度20mM、温度60℃で300mMクエン酸緩衝液(pH4)で1時間、途中でボルテックスミキサーで混合して水和させた。リポソームを液体窒素中で凍結させ、温度60℃で解凍することを5回繰り返してから押出した。Avanti Mini押出機(Avanti Polar Lipids Inc.、Alabaster、アラバマ州)を使用して、リポソームに孔径100nmを有する2枚の重層したポリカーボネート濾紙(Whatman Inc.Nucleopore、Newton、マサチューセッツ州)を室温で10回通過させた。
【0051】
薬物の封入
化合物1(3’−N−(4−ヒドロキシ−3−ヨードベンジル)−13−(R/S)−ジヒドロダウノルビシン、立体異性体混合物)および化合物2(3’−N−(4−ヒドロキシ−3−ヨードベンジル)ダウノルビシン)(構造は下記)、またはドキソルビシン(Sigma Aldrich、St Louis、ミズーリ州、米国)をMayerらによるpH勾配駆動負荷プロトコールを使用してリポソームに封入した。Sephadex G−50カラムで小胞外の300mMクエン酸緩衝液(pH4)を20mM N−[2−ヒドロキシエチル]ピペラジン−N’−[2−エタンスルホン酸](HEPES)緩衝150mM食塩水(HBS)(pH7.5)に交換することによって、リポソーム膜を介したpH勾配を発生させた。ドキソルビシンを1mMの濃度でHBSに溶かした。化合物1を0.5mMの濃度で10%(wt/vol)スクロース溶液に溶かした。化合物2は10%(wt/vol)スクロース溶液に0.5mMの濃度で完全には溶解しなかったが、リポソームを加えてその薬物をリポソームに封入すると溶解した。予熱した薬物溶液を薬物対脂質のモル比0.2でリポソームに加えた。ボルテックス装置を使用して時々混合しながら、得られた混合物を温度60℃で15分間インキュベートした結果、100%の薬物封入がもたらされた。
【0052】
化合物1および2の化学構造
【化4】
【0053】
薬物負荷リポソームに対するターゲッティングリガンドであるトラスツズマブの添付
a)DSPE−PEG3400に対する125I−トラスツズマブの結合
1,2−ジステアロイル−sn−グリセロ−3−ホスファチジルエタノールアミン−N−[メトキシ(ポリエチレングリコール)−3400のN−ヒドロキシスクリンイミジルエステル(NHS−DSPE−PEG3400)を125I−トラスツズマブ溶液と共にモル比0.1:6で温度60℃で5分間水和させた。放射性標識トラスツズマブを使用してリポソームを追跡した。NHS−DSPE−PEG3400の濃度は約0.2mMであった。得られた混合物を撹拌しながら室温で1時間インキュベートした。HBS(pH7.4)を用いたSephacryl S−300カラムのゲル濾過によって、未結合のトラスツズマブを125I−トラスツズマブ−DSPE−PEG3400から除いた。
【0054】
b)リポソームへの125I−トラスツズマブ−DSPE−PEG3400の移行
125I−トラスツズマブ−DSPE−PEG3400を薬物負荷リポソームと1:33のモル比で温度60℃で1時間混合した。HBS(pH7.4)を用いたSepharose CL−4Bカラムのゲル濾過によって、取り込まれていない125I−トラスツズマブ−DSPE−PEG3400をリポソームから除いた。
【0055】
薬物保持の決定
リポソームを温度37℃または60℃でインキュベートした。選択した時間間隔で、3分取分の部分試料を回収し、HBS(pH7.5)を用いてSephadex G−50ミニカラムで680gの遠心場で2分間封入されていない薬物を除いた。溶出液の体積をHBSで1mlに調整し、1%Triton X−100溶液1mlを加えた。試料を温度90℃に加熱し、室温に冷却した。試料の蛍光強度を励起波長468nmおよび発光波長589nmで決定した。インキュベーション前に封入された薬物の量に対するインキュベーション後に封入された薬物の率を決定した。
【0056】
負荷されたリポソームの極低温透過型電子顕微鏡観察(Cryo−TEM)
簡潔には、注文製造した環境チャンバーの中で制御した温度(25℃)で水の蒸発を最小にする湿度条件で、有孔高分子薄膜を被覆した銅格子上にリポソーム試料を移行させた。濾紙に吸引することによって過剰の試料を除いた。薄い(100〜500nm)試料薄膜を温度−165℃で液体エタンに沈めることによってガラス化し、窒素雰囲気で温度−165℃でZeiss EM920A透過型電子顕微鏡(Carl Zeiss Inc.、Oberkochen、ドイツ)に移行させた。試料を電子密度5〜15e−/Å2に曝露し、ゼロロス明視野モードで加速電圧80kVで画像を取得した。
【0057】
結果および考察
温度37℃で24時間培地中でインキュベートしたとき、約80%のドキソルビシンおよび90%を超える化合物1がリポソームに封入されたままであった(図1)。これらの結果は、リポソーム調製および細胞培養に関して記載された条件で、ドキソルビシンおよび化合物1はリポソームからあまり放出されないであろうことを示している。
【0058】
125I−トラスツズマブ−DSPE−PEG3400を含有し、化合物1を負荷したリポソームのcryo−TEM画像(図2)から、化合物1がリポソーム内で結晶状態であることが確認される。
【0059】
実施例2.DNAの結合
実施例2a
細胞培養
過剰発現しているヒト培養腫瘍細胞を10%ウシ胎仔血清、グルタミン(2mM)、ストレプトマイシン(100μg/ml)、およびペニシリン(100IU/ml)を含有するハムのF−10培地(Biochrom AG、Berlin、ドイツ)を用いて温度37℃で加湿した5%CO2インキュベーター中で単層培養として成長させた。
【0060】
DNAの結合
培養ヒト腫瘍A431細胞(扁平上皮癌)をスライドガラス上に成長させ、温度4℃に冷却し、リン酸緩衝液で洗浄した。細胞を温度−20℃で15分間メタノールに入れて不活性化させてから、温度4℃でリン酸緩衝液で速やかに洗浄した。次に、温度4℃で10秒間アセトン処理することによって細胞を透過性にした。乾燥させてから細胞を室温で1時間125I−化合物1と共にインキュベートし、洗浄し、ガンマカウンター(1480 Wallac Wizard、Perkin Elmer、Wellesley、マサチューセッツ州、米国)を用いて放射能について分析した。さらに、細胞を蛍光顕微鏡で検査した。
【0061】
結果および考察
透過性にした培養ヒト腫瘍A431細胞(扁平上皮癌)を125I−化合物1と共に室温で1時間インキュベートしたとき、細胞に結合した化合物の比放射能は、バックグラウンドを減算した後で細胞105個あたり21.1±3.7×103cpmと決定された。過剰のドキソルビシンを使用することによって結合を遮断できた。
【0062】
蛍光顕微鏡観察から、化合物1が細胞核に結合して細胞質内部に局在しなかったことが明らかとなった。ヒト膀胱癌T24細胞およびヒト神経膠腫U343細胞を用いて蛍光顕微鏡実験を繰り返し、同様の結果を得た。
【0063】
実施例2b
アガロースプラグ
InCertアガロース(BioWhittaker Molecular Applications、Rockland、メイン州)を無血清培地に終濃度1%になるように溶かした。アガロース溶液1mlをU−343細胞1.5×106個と混合し、プラグ20μlをプラスチックの型で固めて温度4℃で30分間冷却した。プラグを溶解緩衝液(1mg/mlプロテイナーゼK、0.5M Na3−EDTA(pH8.0)10mlに溶かした2%サルコシル)に温度50℃で一晩沈め、純粋なDNAを得た。溶解後にプラグを0.5M Na3−EDTAで2回洗浄して細胞の破片を除いた。プラグを0.5M Na3−EDTAに入れて温度4℃で保存した。DNAを有さないプラグを対照として供した。アガロースプラグを125I−化合物1を含有する溶液600μlと共に2回の繰り返しで氷上で3時間インキュベートした。対照プラグは濃度3×10−5Mの過剰量のドキソルビシンを含有した。125I−化合物1の終濃度は4×10−7M(dH2O溶液)であった。インキュベーションの後、氷上で洗浄することによってプラグに残った放射能をガンマカウンターを使用して決定した。
【0064】
オートラジオグラフィー
培養SKBR−3乳癌細胞を培地1mlあたり0.1μgの濃度の125I−化合物1(0.3kBq/ng)と共に温度37℃で1時間インキュベートした。インキュベーション後に細胞を無血清培地で6回洗浄し、トリプシン/EDTA(PBS溶液で0.25%/0.02%、Biochrome、Berlin、ドイツ)1mlを用いて10分間剥離させた。細胞を培地14mlで再懸濁して遠心分離管に移した。細胞を1200rpmで5分間遠心分離して細胞ペレットを得た。細胞ペレットをホルマリン緩衝液(0.01Mリン酸緩衝ホルムアルデヒド(4%)、Histolab Products AB, Goteborg, スウェーデン)で4℃で1週間固定した。その後、ペレットを以下の手順で脱水した:70%EtOH 2×15分、90%EtOH 30分、95%EtOH 2×15分、99%EtOH 2×15分、およびHistoresin浸透液(Leica Instruments Gmbh、Heidelberg、ドイツ)3×20分。活性化剤を用いてHistoresinに一晩包埋した後で、ペレットの4μm切片を切り出し、スライドガラスに移行させた。スライドガラスを暗条件でKodak NTB感光乳剤(Eastman Kodak Company、Rochester、ニュ−ヨーク州、米国)に浸し、乾燥してから4℃で3日保存した。そのスライドガラスをKodak D19液を用いて3分間現像してから、0.1%酢酸に10秒間移行させ、Kodak固定液を用いて5分間固定した。これら全てを暗条件で行った。水で大規模に洗浄した後で、細胞核をマイアーヘマトキシリン(Histolab Products AB、Goteborg、スウェーデン)で3分間染色した。Pertex(Histolab Products AB、Goteborg、スウェーデン)でマウントする前に、スライドガラスを水で5分間洗浄して風乾させた。細胞を顕微鏡で検査して、代表的な細胞の画像を取得した。
【0065】
結果および考察
125Iの染色パターンがヘマトキシリン染色細胞核と共局在していることから、細胞核に対する化合物1の結合を125I−化合物1のオートラジオグラフィーによって確認できた(図3)。
【0066】
U−343細胞DNAを含有するアガロースプラグを125I−化合物1溶液と共に氷上で3時間インキュベートすると、125I−化合物1はアガロースプラグ中に蓄積された。DNAに対する125I−化合物1の蓄積と、よって結合とを過剰量のドキソルビシンによって遮断された。結果は、DNAに対する化合物1の親和性を確認している。DNAに対する125I−化合物1の結合をドキソルビシンによって置換できたという事実(図4)は、化合物1とドキソルビシンとの両方が同じDNA結合部位を占有することを示唆している。
【0067】
実施例2c
パルスフィールドゲル電気泳動(PFGE)
125I−化合物1がDNA損傷を誘導する効率を検討するために、パルスフィールドゲル電気泳動(PFGE、Pharmacia Biotech、Uppsala、スウェーデン)で二本鎖切断(dsb)の誘導を分析した。上記のようにアガロースプラグを125I−化合物1と共にインキュベートした。洗浄後に、プラグを温度4℃で8日間保持した。次にプラグを0.8%(w/v)アガロースゲル(Seakem Gold粉末アガロース、Cambrex Bio Science Rockland Inc.、Rockland、メイン州、米国)に負荷した。DNAの断片化を以下のプロトコールにより2V/cmで45時間のパルスフィールドゲル電気泳動で分析した:10分のパルス(すなわち10分ごとに場にパルスを与える)3時間、20分のパルス5時間20分、30分のパルス8時間、40分のパルス9時間20分、および1時間のパルス20時間。ゲルを泳動させた後で臭化エチジウム(0.5μg/ml)で8時間染色してから、dH2Oで一晩脱色させた。分子量マーカーとして、シゾサッカロミセス ポンベ(Scizosaccharomyces pombe)メガ塩基DNA標準(Cambrex Bio Science Rockland Inc.、Rockland、メイン州、米国)を使用した。各レーンを、それぞれ5.7Mbp以下および5.7Mbp以上のサイズを有するDNA断片に対応する2つのブロックに切り出した。各ブロックをバイアルに入れ、以前に挙げた自動ガンマカウンターで放射能を測定した。5.7Mbp未満のDNAの割合を決定して、次にBlocherの式を用いた計算に使用した:
F<k=1−e−rk/n(1+rk/n(1−k/n)) (Int J Rad Biol 57, 7〜12、1990)
F<kはk塩基対よりも小さいDNAの割合であり、rはdsb/染色体の平均数であり、nは平均サイズの染色体1本に存在する塩基対の総数である。k=このアッセイでは5.7Mbpを使用した。平均的なヒト染色体ではn=130Mbpである。
【0068】
二本鎖切断数(r)を得るためには、式が数値的に解かれなければならない。
【0069】
結果および考察
125I−化合物1を神経膠腫細胞DNAと共にインキュベートした後の二本鎖切断(dsb)の数(図5)を崩壊1回あたり約0.4dsbと決定した。このように、化合物Iと結合すると、125IはDNAに十分近く位置してDNAの断片化を引き起こす。それは、DNAに結合していない125IはDNAの断片化をもたらさないと思われるからである。比較として、DNA鎖への核種の直接組み入れを招く125I標識DNA前駆分子の使用は、崩壊1回あたり約1dsbのdsb値を与える。dsb値の差は比較的小さく、これは、(放射線が作用を有するために)その核種がDNAから非常に短い距離内に入らなければならず、DNAのインターカレーションが起こったことを示している。
【0070】
実施例2d
1型ウシ胸腺DNAナトリウム塩(Sigma Aldrich Co.、St Louis、ミズーリ州、米国)を約2mg/mlの濃度でBPES緩衝液(6mM Na2HPO4、2mM NaH2PO4、1mM EDTA、185mM NaCl、pH7)で水和させ、MSE Soniprep150超音波粉砕機(Integrated Services TCP Inc.、Palisades Park、ニュージャージー州、米国)に入れて氷冷した水浴中で30分間約8μAで超音波処理した。次に、Slide-A-Lyzer(登録商標)透析カセット(10000MWCO)(Pierce、Rockford、イリノイ州、米国)を使用してBPESに対して試料を48時間透析した。HP8453分光光度計(Hewlett-Packard Company、Houston、テキサス州、米国)で吸光係数12824M(bp)−1cm−1を用いて波長260nmでの吸光度定量により、DNAの終濃度を決定した。
【0071】
SPEX1680 Fluorolog蛍光光度計(SPEX Industries Inc.、Edison、ニュージャージー州、米国)で室温でλex=480nm(スリッド(slid)幅2.5mm)およびλem=592nm(スリッド幅2.5mm)で蛍光滴定実験を行った。初発遊離薬物濃度は1μMであった。次式により、DNAの不在下(I0)および存在下(I)で被験化合物の蛍光強度比を決定することによって遊離薬物濃度(Cf(M))が計算した:
Cf=CT(I/I0−P)/(1−P) (Biopolymers 6、1225〜1235、1968)
式中、CT(M)は初発薬物濃度であり、Pは完全に結合した薬物の蛍光強度の観察された量子収量(Imin)と、遊離薬物の量子収量との間の比である(P=Imin/I0)。CTとCfとの間の差によって、結合した薬物の濃度を計算した。rに対してr/Cfをプロットすること(スキャッチャードプロット、Scatchard plot)によって結合定数(Ki)および排除パラメータ(n)を計算した。ここで、rはDNA塩基対1モルあたり結合した薬物のモル数である。隣接排除モデルに関する理論曲線を次のアルゴリズムを使用して計算した:
r/Cf=Ki(1−nr)[(1−nr)/[1−(n−1)r]]n−1 (J.Mol.Biol. 86、469〜489、1974)
式中、Ki(M−1)は固有の結合定数であり、n(塩基対)は排除パラメータである。実験データに最も適合する理論曲線を発生するように、パラメータKiおよびnを変動させた。
【0072】
表1(下記)は、ウシ胸腺DNA(CT DNA)に対するダウノルビシン、ドキソルビシン、化合物1、および化合物2の結合定数(Ki)および排除パラメータ(n)を示す。カッコ内の値は、平均値の標準誤差(SEM)(n=3)を示す。a=ダウノルビシンに対してp<0.05。b=化合物2に対してp<0.05。平均値の間の差を一元配置分散分析の後にStudent Newman-Keuls検定により検定した。
【0073】
文献値
ダウノルビシンのKi:0.7×106M−1±0.07、n:3.5塩基対±0.35(Chairesら(1982)Biochemistry 21、3933〜3940)。ドキソルビシンのKi:3.3×106M−1、n:3.8塩基対(Messoriら(2001)Bioorg.Med.Chem. 9、1815〜1825)。
【0074】
表1:ドキソルビシン、ダウノルビシン、化合物1、および化合物2の、結合定数および排除パラメータ
【表1】
【0075】
結果および考察
化合物1および化合物2のCT DNAとの結合定数および排除パラメータがドキソルビシンの場合と類似していることから、ダウノルビシンの誘導体化がDNA結合性の喪失を起こさなかったことが確認される(表1)。逆に、化合物1および化合物2の結合定数は親化合物のダウノルビシンの結合定数よりも高い値であった?。
【0076】
実施例3.毒性
成長曲線125I−化合物1、化合物1、ドキソルビシン、またはダウノルビシンを0.5ng/mlの濃度に培地に溶解させた。125I−化合物1の比放射能は100kBq/ngであった。SKBR−3細胞を、60mmプラスチックペトリ皿中で3回の繰り返しで2.5時間薬物と共にインキュベートした。対照細胞を通常の培地と共にインキュベートした。インキュベートの後で、培地を全ての皿から除き、細胞を無血清培地で6回洗浄した。トリプシン/EDTA0.5mlを温度37℃で10分間加えることによって細胞を皿から剥離させた。培地1mlに再懸濁した後で、細胞をコールターカウンター(Z2コールターカウンター、Beckman Coulter)を使用して計数し、細胞105個まで継代培養して成長曲線を作製した。継代培養毎に細胞の損失に関して成長曲線を補正した。
【0077】
結果および考察
培養SKBR−3細胞単層の成長曲線は、濃度わずか0.5ng/mlの125I−化合物1の細胞毒性作用が化合物1、ドキソルビシン、またはダウノルビシンとインキュベートした後に比べて数桁大きかったことを明らかにした(図6)。後者の化合物類のどれも有意な細胞毒性作用を有さなかった。よって、125I−化合物1によって表された細胞毒性作用は化合物1に結合した核種125I単独によって引き起こされる。
【0078】
参照文献
[1]B.A.Chabner,C.E.Meyers(1982) ”Cancer: Principles and practice of oncology” (DeVita Jr.V.T., S.HellmanおよびS.A.Rosenberg編), 156〜197頁, J.B.Lippincott Company, Philadelphia, Toronto.
[2]H.G.Keizer, H.M.Pinedo, G.J.Schuurhuis, H.Joenje, ドキソルビシン(アドリアマイシン):細胞毒性の遊離ラジカル依存性メカニズムの決定的な総説、Pharmaceutics and Therapeutics 47(1990)219〜231.
[3]J.Bouma, J.H.Beijnen, A.Bult, W.J.M.Underberg, アントラサイクリン系抗腫瘍剤、Pharmaceutisch Weekblad Scientific Edition 8(1986)109〜133.
[4]C.P.Association, Compendium of pharmaceuticals and specialities, 第13巻、CK Productions, Toronto、1995.
[5]R.H.Blum, S.K.Carter, アドリアマイシン。重大な臨床活性を有する新しい抗癌薬,Ann Intern Med 80(1974)249〜59.
[6]M.N.Gaze, 臨床業務における標的を定めた放射線治療の現状,Phys Med Biol 41(1996)1895〜903.
[7]J.A.O’Donoghue, T.E.Wheldon, オージェ電子放射体を用いた標的を定めた放射線治療、Phys Med Biol 41(1996)1973〜92.
[8]S.Kim, 癌化学療法の担体としてのリポソーム。現状と将来展望、Drugs46(1993)618〜38.
[9](M.J.Hope, M.B.Bally, G.Webb, およびP.R.Cullis (1985) Biochim. Biophys. Acta 812: 55〜65).
【図面の簡単な説明】
【0079】
【図1】温度37℃での緩衝液または培地中のドキソルビシンと、緩衝液中の化合物1との保持を示す図である。
【図2】化合物1を負荷したリポソームの極低温透過型顕微鏡画像を示す図である。球体はリポソームを表し、リポソーム内部の斑点は結晶状の化合物1を表す。
【図3】腫瘍細胞と共に37℃で1時間インキュベートした後の125I−化合物1のオートラジオグラフィーを示す図である。
【図4】氷上で2.5時間インキュベートした後の遊離DNAに対する125I−化合物1の結合を示す図である。
【図5】U−343MGaC12:6神経膠腫細胞DNAを含有するプラグを負荷したアガロースゲルでの125I−化合物1の結合後のDNA断片化を示す図である。図中、a)分子量マーカー(シゾサッカロミセスポンベ、メガ塩基DNA標準)、b)125I−化合物1と共にインキュベートしたDNA、c)125I−化合物1および過剰のドキソルビシンと共にインキュベートしたDNA、ならびにd)125I−化合物1不在下でインキュベートしたDNAを有する対照。
【図6】0.5ng/mlドキソルビシン、ダウノルビシン、化合物1、または125I−化合物1で処理した腫瘍細胞の成長曲線を示す図である。
【特許請求の範囲】
【請求項1】
一般式Iを有するアントラサイクリン誘導体(薬物前駆体)
【化1】
[式中、Rは、二重結合酸素原子または両方の立体異性体のヒドロキシル基のいずれかであり、または
R1は、CH3またはCH2OHのいずれかである。
R2はY−Ar−Z基であり、
ここで、Yは、−(CH2)n−等のスペーサー分子または式−(CH2CH2O)n−(ここで、nは1〜8である)を有するポリエチレングリコール鎖のいずれかであり;
Arは、通常の単環式芳香族基または安定な芳香族ホウ素ケージ(boron cage)化合物であり、ここで、前記通常の芳香族残基は、求電子芳香族置換を用いて直接放射性標識できる(水素原子等の)置換基または活性化基(例えばトリメチルスタニルまたはトリブチルスタニル基等のトリアルキルスタニル基)を含み、ここで、前記活性化基は、放射性核種に交換できるか、または前記芳香族残基がハロゲン−ハロゲン交換反応を受けることができるようにハロゲン(例えばBrまたはI)を含み;かつ
Zは、任意に糖基等の親水性を増加させる化学基である]
およびその塩。
【請求項2】
前記通常の単環式芳香族基がフェニルまたはピリジン基であり、前記安定な芳香族ホウ素ケージ化合物が、C2H12B10等の安定なcloso-カルボランである、請求項1に記載のアントラサイクリン誘導体。
【請求項3】
前記誘導体が、
【化2】
である、請求項1〜2のいずれかに記載のアントラサイクリン誘導体。
【請求項4】
請求項1〜3のいずれかに記載のアントラサイクリン誘導体を含む放射性治療薬であって、前記アントラサイクリン誘導体が、放射性核種か、または中性子もしくは光子を用いた外部照射によって活性化できる安定核種を含む、放射性治療薬。
【請求項5】
前記放射性核種が123−125I、131I、18F、76−77Br、211At、90Y、32P、67Cu、および189Reからなる群から選択され、前記安定核種が10Bおよび157Gdからなる群から選択される、請求項4に記載の放射性治療薬。
【請求項6】
癌診断のための造影剤としての、請求項4〜5のいずれかに記載の放射性治療薬の使用。
【請求項7】
DNAターゲッティング剤、すなわちDNA相互作用剤としての、請求項1〜3のいずれかに記載のアントラサイクリン誘導体または請求項4〜5のいずれかに記載の放射性治療薬の使用。
【請求項8】
DNA相互作用剤を含む薬物送達システムであって、前記DNA相互作用剤が請求項1〜3のいずれかに記載のアントラサイクリン誘導体または請求項4〜5のいずれかに記載の放射性治療薬である薬物送達システム。
【請求項9】
前記DNA相互作用剤を含めた薬学的に活性な薬剤を包有または結合できる担体を含む、請求項8に記載の薬物送達システム。
【請求項10】
前記担体がリポソーム等の脂質担体、ミクロゲル等の高分子担体、または脂質/高分子複合材料粒子である、請求項9に記載の薬物送達システム。
【請求項11】
(a)担体上に用意されて、特異的な細胞または組織に標的を定める細胞ターゲッティング剤と、
(b)細胞核に放射性核種の標的を定めるために、前記担体の内部に用意されたか、または前記担体に結合したDNAターゲッティング剤(DNA相互作用剤)と
を含む二段階ターゲッティングを含む、請求項10に記載の薬物送達システム。
【請求項12】
前記細胞ターゲッティング剤が、リガンド、抗体、および抗体断片からなる群から選択される、請求項11に記載の薬物送達システム。
【請求項13】
前記細胞ターゲッティング剤が、上皮成長因子(EGF)を含むか、または腫瘍特異的な突然変異EGF受容体に結合する分子を含む、請求項12に記載の薬物送達システム。
【請求項14】
癌の治療および/または診断のための方法であって、それを必要とする対象に治療有効量の請求項8〜13のいずれかに記載の薬物送達システムを投与する段階と、腫瘍が局在しているときは続いて前記腫瘍の領域を任意に照射する段階とを含む方法。
【請求項15】
乳癌の治療のための方法であって、それを必要とする対象に治療有効量の請求項8〜13のいずれかに記載の薬物送達システムを投与する段階と、腫瘍が局在しているときは続いて前記腫瘍の領域を任意に照射する段階とを含む方法。
【請求項16】
播種性癌の癌治療のための方法であって、それを必要とする対象に治療有効量の請求項8〜13のいずれかに記載の薬物送達システムを投与する段階と、腫瘍が局在しているときは続いて前記腫瘍の領域を任意に照射する段階とを含む方法。
【請求項17】
P−糖タンパク質(PGP)を過剰発現している多剤耐性(MDR)腫瘍の癌治療のための方法であって、それを必要とする対象に治療有効量の請求項8〜13のいずれかに記載の薬物送達システムを投与する段階と、前記腫瘍が局在しているときは続いて前記腫瘍の領域を任意に照射する段階とを含む方法。
【請求項1】
一般式Iを有するアントラサイクリン誘導体(薬物前駆体)
【化1】
[式中、Rは、二重結合酸素原子または両方の立体異性体のヒドロキシル基のいずれかであり、または
R1は、CH3またはCH2OHのいずれかである。
R2はY−Ar−Z基であり、
ここで、Yは、−(CH2)n−等のスペーサー分子または式−(CH2CH2O)n−(ここで、nは1〜8である)を有するポリエチレングリコール鎖のいずれかであり;
Arは、通常の単環式芳香族基または安定な芳香族ホウ素ケージ(boron cage)化合物であり、ここで、前記通常の芳香族残基は、求電子芳香族置換を用いて直接放射性標識できる(水素原子等の)置換基または活性化基(例えばトリメチルスタニルまたはトリブチルスタニル基等のトリアルキルスタニル基)を含み、ここで、前記活性化基は、放射性核種に交換できるか、または前記芳香族残基がハロゲン−ハロゲン交換反応を受けることができるようにハロゲン(例えばBrまたはI)を含み;かつ
Zは、任意に糖基等の親水性を増加させる化学基である]
およびその塩。
【請求項2】
前記通常の単環式芳香族基がフェニルまたはピリジン基であり、前記安定な芳香族ホウ素ケージ化合物が、C2H12B10等の安定なcloso-カルボランである、請求項1に記載のアントラサイクリン誘導体。
【請求項3】
前記誘導体が、
【化2】
である、請求項1〜2のいずれかに記載のアントラサイクリン誘導体。
【請求項4】
請求項1〜3のいずれかに記載のアントラサイクリン誘導体を含む放射性治療薬であって、前記アントラサイクリン誘導体が、放射性核種か、または中性子もしくは光子を用いた外部照射によって活性化できる安定核種を含む、放射性治療薬。
【請求項5】
前記放射性核種が123−125I、131I、18F、76−77Br、211At、90Y、32P、67Cu、および189Reからなる群から選択され、前記安定核種が10Bおよび157Gdからなる群から選択される、請求項4に記載の放射性治療薬。
【請求項6】
癌診断のための造影剤としての、請求項4〜5のいずれかに記載の放射性治療薬の使用。
【請求項7】
DNAターゲッティング剤、すなわちDNA相互作用剤としての、請求項1〜3のいずれかに記載のアントラサイクリン誘導体または請求項4〜5のいずれかに記載の放射性治療薬の使用。
【請求項8】
DNA相互作用剤を含む薬物送達システムであって、前記DNA相互作用剤が請求項1〜3のいずれかに記載のアントラサイクリン誘導体または請求項4〜5のいずれかに記載の放射性治療薬である薬物送達システム。
【請求項9】
前記DNA相互作用剤を含めた薬学的に活性な薬剤を包有または結合できる担体を含む、請求項8に記載の薬物送達システム。
【請求項10】
前記担体がリポソーム等の脂質担体、ミクロゲル等の高分子担体、または脂質/高分子複合材料粒子である、請求項9に記載の薬物送達システム。
【請求項11】
(a)担体上に用意されて、特異的な細胞または組織に標的を定める細胞ターゲッティング剤と、
(b)細胞核に放射性核種の標的を定めるために、前記担体の内部に用意されたか、または前記担体に結合したDNAターゲッティング剤(DNA相互作用剤)と
を含む二段階ターゲッティングを含む、請求項10に記載の薬物送達システム。
【請求項12】
前記細胞ターゲッティング剤が、リガンド、抗体、および抗体断片からなる群から選択される、請求項11に記載の薬物送達システム。
【請求項13】
前記細胞ターゲッティング剤が、上皮成長因子(EGF)を含むか、または腫瘍特異的な突然変異EGF受容体に結合する分子を含む、請求項12に記載の薬物送達システム。
【請求項14】
癌の治療および/または診断のための方法であって、それを必要とする対象に治療有効量の請求項8〜13のいずれかに記載の薬物送達システムを投与する段階と、腫瘍が局在しているときは続いて前記腫瘍の領域を任意に照射する段階とを含む方法。
【請求項15】
乳癌の治療のための方法であって、それを必要とする対象に治療有効量の請求項8〜13のいずれかに記載の薬物送達システムを投与する段階と、腫瘍が局在しているときは続いて前記腫瘍の領域を任意に照射する段階とを含む方法。
【請求項16】
播種性癌の癌治療のための方法であって、それを必要とする対象に治療有効量の請求項8〜13のいずれかに記載の薬物送達システムを投与する段階と、腫瘍が局在しているときは続いて前記腫瘍の領域を任意に照射する段階とを含む方法。
【請求項17】
P−糖タンパク質(PGP)を過剰発現している多剤耐性(MDR)腫瘍の癌治療のための方法であって、それを必要とする対象に治療有効量の請求項8〜13のいずれかに記載の薬物送達システムを投与する段階と、前記腫瘍が局在しているときは続いて前記腫瘍の領域を任意に照射する段階とを含む方法。
【図1】

【図2】
【図3】

【図4】
【図5】
【図6】
【図2】
【図3】
【図4】
【図5】
【図6】
【公表番号】特表2007−533736(P2007−533736A)
【公表日】平成19年11月22日(2007.11.22)
【国際特許分類】
【出願番号】特願2007−509425(P2007−509425)
【出願日】平成17年4月22日(2005.4.22)
【国際出願番号】PCT/SE2005/000596
【国際公開番号】WO2005/103065
【国際公開日】平成17年11月3日(2005.11.3)
【出願人】(500252671)
【出願人】(500252693)
【出願人】(500252682)
【出願人】(506353253)
【Fターム(参考)】
【公表日】平成19年11月22日(2007.11.22)
【国際特許分類】
【出願日】平成17年4月22日(2005.4.22)
【国際出願番号】PCT/SE2005/000596
【国際公開番号】WO2005/103065
【国際公開日】平成17年11月3日(2005.11.3)
【出願人】(500252671)
【出願人】(500252693)
【出願人】(500252682)
【出願人】(506353253)
【Fターム(参考)】
[ Back to top ]