
アンモニア分解用触媒及びそれを用いたアンモニア分解方法、並びに該アンモニア分解用触媒の再生方法
【課題】アンモニア分解反応に使用することで劣化した場合に、再度、触媒性能を再生できるアンモニア分解用触媒を提供することを目的とする。
【解決手段】本発明のアンモニア分解用触媒は、希土類、アルカリ金属及びアルカリ土類金属よりなる群から選ばれる少なくとも一種の元素(A成分)の酸化物と、Co、Ni及びFeよりなる群から選ばれる少なくとも一種の元素(B成分)の金属微粒子とを含有する触媒であって、前記A成分と前記B成分とで形成されるペロブスカイト構造を有する酸化物を、還元処理して得られたことを特徴とする。
【解決手段】本発明のアンモニア分解用触媒は、希土類、アルカリ金属及びアルカリ土類金属よりなる群から選ばれる少なくとも一種の元素(A成分)の酸化物と、Co、Ni及びFeよりなる群から選ばれる少なくとも一種の元素(B成分)の金属微粒子とを含有する触媒であって、前記A成分と前記B成分とで形成されるペロブスカイト構造を有する酸化物を、還元処理して得られたことを特徴とする。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、アンモニア分解用触媒に関するものであり、特に、アンモニア分解反応に使用することで、触媒成分の粒子径が増大し触媒性能が劣化した際に、触媒成分を再分散させることができるアンモニア分解用触媒に関するものである。
【背景技術】
【0002】
従来、アンモニアを分解して水素を製造する技術が提案されている。このようなアンモニアの分解に使用される触媒として、例えば、特許文献1には、触媒成分としてコバルト、ニッケル等の特定成分を含む触媒が提案されており、具体例として、ペロブスカイト構造を有するランタン−コバルト複合酸化物触媒、ペロブスカイト構造を有するニッケル−ランタン複合酸化物触媒が記載されている(特許文献1(段落[0025]、[0026])参照)。特許文献2には、アンモニア燃焼触媒とアンモニア分解触媒とを含み、アンモニア燃焼触媒としてペロブスカイト構造を有するマンガン−ランタン酸化物を含有する触媒が提案されている(特許文献2(請求項6)参照)。また、特許文献3には、ペロブスカイト構造を有するランタン−コバルト複合酸化物触媒、ペロブスカイト構造を有するランタン−ニッケル複合酸化物触媒(特許文献3(段落[0037]、[0039])参照)が提案されている。
なお、これらの特許文献に記載された触媒は、いずれもペロブスカイト構造を維持したものである。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】特開2010−240644号公報
【特許文献2】特開2010−240646号公報
【特許文献3】特開2010−241675号公報
【発明の概要】
【発明が解決しようとする課題】
【0004】
上記特許文献1〜3に記載されたような、ペロブスカイト構造を維持した触媒では、アンモニア分解反応に使用した際に、アンモニアによって触媒が還元されて触媒成分となる金属粒子が生成し、触媒性能を発揮することとなる。しかしながら、このようなペロブスカイト構造を維持した触媒をそのまま使用した場合、触媒の還元が充分でなく、触媒性能を充分に発揮できていなかった。
また、アンモニア分解用触媒は、アンモニア分解反応に使用すると、触媒成分となる金属粒子の粒子径が増大し、触媒成分の比表面積が低下するため、触媒性能が低下する。このように触媒性能が低下した触媒は、もはや使用できないため、定期的にアンモニア分解触媒を交換する必要があった。
本発明は上記事情に鑑みてなされたものであり、アンモニア分解反応に使用することで劣化した場合に、触媒性能を再生できるアンモニア分解用触媒を提供することを目的とする。また、本発明は、上記アンモニア分解用触媒を用いたアンモニアの分解方法、及び、上記アンモニア分解用触媒の再生方法を提供することも目的とする。
【課題を解決するための手段】
【0005】
本発明者らは、アンモニア分解反応に使用することで触媒成分である金属粒子の粒子径が増大した場合に、この金属粒子を再度、微粒子状に分散させる方法について検討を進めた。その結果、ペロブスカイト構造を還元処理により崩壊させて金属微粒子を生成させた場合には、金属粒子の粒子径が増大した場合であっても、再度ペロブスカイト構造を構築、崩壊させることにより、金属粗粒子を再度微粒子に分散できることを見出し、本発明を完成した。
【0006】
本発明のアンモニア分解用触媒は、希土類、アルカリ金属及びアルカリ土類金属よりなる群から選ばれる少なくとも一種の元素(A成分)の酸化物と、Co、Ni及びFeよりなる群から選ばれる少なくとも一種の元素(B成分)の金属微粒子とを含有する触媒であって、前記A成分と前記B成分とで形成されるペロブスカイト構造を有する酸化物を、還元処理して得られたことを特徴とする。前記B成分の金属微粒子のX線回折法で測定される結晶子径は、25nm以下であることが好ましい。アンモニアの分解反応に用いた使用済み触媒を、酸化処理して、前記A成分と前記B成分とで形成されるペロブスカイト構造を有する酸化物を再構築し、これを還元処理して得られたものも好ましい態様である。
【0007】
本発明には、前記アンモニア分解用触媒を用いるアンモニアの分解方法も含まれる。アンモニア分解方法としては、前記アンモニア分解用触媒を用いてアンモニアを分解する分解工程;アンモニア分解反応後のアンモニア分解用触媒を酸化処理する酸化工程;及び、アンモニア分解用触媒の酸化処理物を、還元処理する還元工程とを含む態様が好ましい。また、本発明には、アンモニアの分解反応に用いた使用済み触媒を、酸化処理した後、さらに還元処理するアンモニア分解用触媒の再生方法も含まれる。
【発明の効果】
【0008】
本発明のアンモニア分解用触媒は、アンモニア分解反応に使用して劣化した場合であっても、使用済みの触媒に酸化処理、還元処理を施すことにより、触媒性能を再生できるため、長期間にわたって使用することができる。
また、上記アンモニア分解用触媒を用いたアンモニアの分解方法は、触媒が劣化しても、酸化工程、還元工程を経ることで、触媒性能を再生できるため、触媒を交換することなく長期間にわたってアンモニア分解反応を行うことができ、作業性が向上する。
【図面の簡単な説明】
【0009】
【図1】触媒前駆体5−1〜5−6のX線回折測定結果を示す図である。
【図2】触媒5−1〜5−6のアンモニア分解率を示す図である。
【図3】触媒前駆体6−1〜6−5のX線回折測定結果を示す図である。
【図4】触媒6−1〜6−5のアンモニア分解率を示す図である。
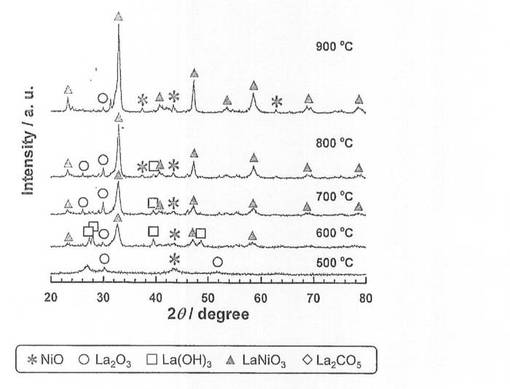
【図5】触媒前駆体7−1〜7−5のX線回折測定結果を示す図である。
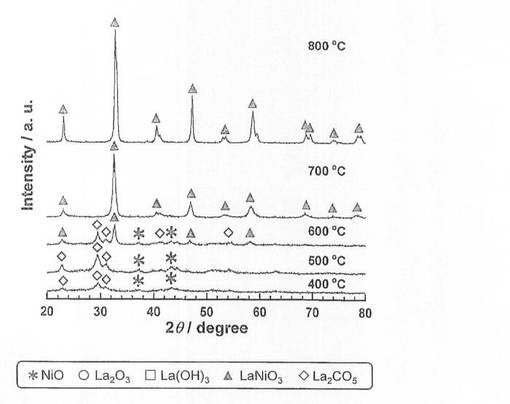
【図6】触媒前駆体8−1〜8−5のX線回折測定結果を示す図である。
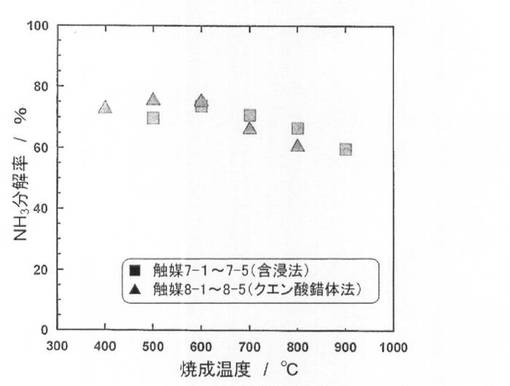
【図7】触媒7−1〜7−5、8−1〜8−5のアンモニア分解率、Niの比表面積を示す図である。
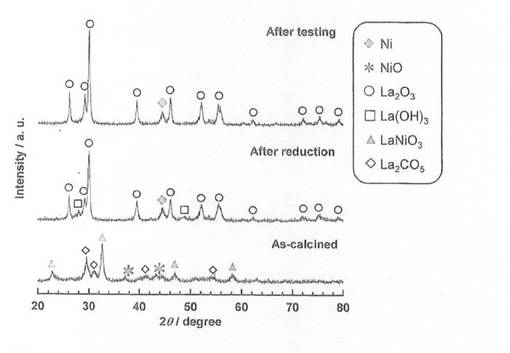
【図8】触媒前駆体8−3、触媒8−3、アンモニア分解反応に使用した後の触媒8−3のX線回折測定結果を示す図である。
【図9】触媒前駆体9−1〜9−5のX線回折測定結果を示す図である。
【図10】触媒9−1〜9−5のX線回折測定結果を示す図である。
【図11】触媒10−1〜10−5のX線回折測定結果を示す図である。
【図12】触媒9−1〜9−5、10−1〜10−5のアンモニア分解率を示す図である。
【発明を実施するための形態】
【0010】
本発明のアンモニア分解用触媒(以下、単に「触媒」と称する場合がある。)は、希土類、アルカリ金属及びアルカリ土類金属よりなる群から選ばれる少なくとも一種の元素(A成分)と、Co、Ni及びFeよりなる群から選ばれる少なくとも一種の元素(B成分)とで形成されるペロブスカイト構造を有する酸化物を、還元処理して得られたことを特徴とする。
【0011】
A成分とB成分とで形成されるペロブスカイト構造を構築した後、これを崩壊させてB成分の金属微粒子を生成させた場合、A成分とB成分との元素の配置は、再度ペロブスカイト構造を構築し易い配置となる。よって、本発明の触媒は、酸化処理を施すことにより、A成分とB成分とで形成されるペロブスカイト構造を容易に再構築することができる。
そのため、本発明の触媒では、B成分の金属粒子の粒子径が増大した場合でも、酸化処理を施しペロブスカイト構造を再構築すれば、B成分が各結晶構造中に取り込まれるため、B成分を原子レベルにまで細分化することができる。そして、さらに還元処理を施し、再構築したペロブスカイト構造を再崩壊させることで、B成分の金属微粒子を再度生成させることができる。よって、本発明の触媒は、アンモニア分解反応に使用して劣化した場合であっても、このような劣化した使用済みの触媒に酸化処理、還元処理を施すことにより、触媒性能を再生できる。
【0012】
本発明の触媒は、希土類、アルカリ金属及びアルカリ土類金属よりなる群から選ばれる少なくとも一種の元素(A成分)の酸化物と、Co、Ni及びFeよりなる群から選ばれる少なくとも一種の元素(B成分)の金属微粒子とを含有する。B成分の金属微粒子が触媒として作用する。
【0013】
前記A成分としては、具体的には、スカンジウム、イットリウム、ランタン、セリウム、プラセオジム、ネオジム、プロメチウム、サマリウム、ユウロピウム、ガドリニウム、テルビウム、ジスプロシウム、ホルミウム、エルビウム、ツリウム、イッテルビウム、ルテチウム等の希土類元素;リチウム、ナトリウム、カリウム、ルビジウム、セシウム等のアルカリ金属元素;マグネシウム、カルシウム、ストロンチウム、バリウム等のアルカリ土類金属が挙げられる。これらのA成分は単独で使用してもよいし、2種以上を併用してもよい。これらの中でも、A成分としては、希土類元素、アルカリ土類金属元素が好ましく、より好ましくはランタン、カルシウム、ストロンチウム、バリウムである。
前記A成分の酸化物としては、具体的には、酸化ランタン、酸化カルシウム、酸化ストロンチウム、酸化バリウム等が挙げられる。
【0014】
前記B成分としては、Co、Ni、Feを単独で使用してもよいし、2種以上を併用してもよい。これらの中でもCo、Niが好ましい。
前記B成分の金属微粒子の結晶子径は、25nm以下が好ましく、より好ましくは20nm以下、さらに好ましくは18nm以下である。前記結晶子径が25nm以下であれば、再生処理において酸化処理を施した際に、ペロブスカイト構造が構築され易くなるため、B成分の再分散効果がより良好となり、触媒性能の回復率が向上する。前記結晶子径の下限は特に限定されないが、通常5nm程度である。なお、本発明において金属微粒子の結晶子径は、X線回折法で測定される値であり、測定方法は後述する。
【0015】
触媒中の前記B成分の含有量は、5質量%以上が好ましく、より好ましくは15質量%以上、さらに好ましくは25質量%以上であり、80質量%以下が好ましく、より好ましくは60質量%以下、さらに好ましくは50質量%以下である。前記B成分の含有量が5質量%以上であれば触媒性能がより向上し、80質量%以下であれば、B成分が過剰とならず、再生処理において酸化処理を施した際にB成分の大部分がペロブスカイト構造に取り込まれることとなり、B成分の再分散効果がより良好となる。
【0016】
触媒中の前記B成分と前記A成分とのモル比(A成分/B成分)は、0.1以上が好ましく、より好ましくは0.5以上、さらに好ましくは0.8以上であり、10以下が好ましく、より好ましくは3以下、さらに好ましくは1.5以下である。前記モル比が上記範囲内であれば、再生処理において酸化処理を施した際に構築されるペロブスカイト構造の量が多くなり、B成分の再分散効果がより良好となる。
【0017】
本発明の触媒の製造方法の一例について説明する。本発明の触媒は、A成分とB成分とを含有する複合物を作製し、この複合物を酸化性雰囲気で焼成して、A成分とB成分とで形成されるペロブスカイト構造を有する酸化物(以下、「触媒前駆体」と称する場合がある。)を作製し、この触媒前駆体を還元処理することで得られる。
【0018】
前記複合物を作製する方法は特に限定されず、例えば、共沈法、含浸法、クエン酸錯体法等が挙げられる。
共沈法としては、例えば、B成分の水溶性塩とA成分の水溶性塩を溶解させた水溶液と、アルカリ性化合物(好ましくは水溶液)とを混合し、A成分を含有する微粒子とB成分を含有する微粒子からなる沈殿物を生成させ、これをろ過により取り出し、水洗、乾燥する方法が挙げられる。含浸法としては、例えば、B成分の水溶性塩を溶解させた溶液を、A成分の酸化物に含浸させ、水分を蒸発させる方法が挙げられる。クエン酸錯体法としては、例えば、A成分の水溶性塩とB成分の水溶性塩とを溶解させた水溶液に、クエン酸水和物を加えて攪拌し、溶液がゲル状になったところで、加熱して熱分解する方法が挙げられる。
【0019】
前記A成分の水溶性塩としては、例えば、硝酸塩、酢酸塩、塩化物等が挙げられる。前記A成分の水溶性塩としては、例えば、硝酸コバルト六水和物、酢酸コバルト四水和物、塩化コバルト六水和物等のコバルトの水溶性塩;硝酸ニッケル六水和物、二酢酸ニッケル四水和物等のニッケルの水溶性塩;硝酸鉄(II)六水和物、硝酸鉄(III)九水和物等の鉄の水溶性塩;等が挙げられる。
【0020】
B成分の水溶性塩としては、例えば、硝酸塩、酢酸塩、塩化物、硫酸塩等が挙げられる。前記B成分の水溶性塩としては、例えば、硝酸ランタン六水和物等のランタンの水溶性塩;硝酸マグネシウム六水和物等のマグネシウムの水溶性塩;硝酸セリウム六水和物等のセリウムの水溶性塩;等が挙げられる。
【0021】
前記アルカリ性化合物としては、例えば、アンモニア、炭酸アンモニウム、水酸化テトラメチルアンモニウム等のアンモニア系化合物;水酸化カリウム等のアルカリ金属水酸化物;等が挙げられる。
【0022】
前記複合物を酸化性雰囲気で焼成することにより、触媒前駆体を得る。前記酸化性雰囲気としては、例えば、空気雰囲気が挙げられる。前記複合物を焼成する温度は、550℃以上が好ましく、より好ましくは650℃以上、さらに好ましくは750℃以上であり、950℃以下が好ましく、より好ましくは900℃以下である。焼成時間は、0.5時間以上が好ましく、より好ましくは2時間以上であり、48時間以下が好ましく、より好ましくは24時間以下、さらに好ましくは12時間以下である。
【0023】
また、触媒前駆体中のペロブスカイト構造の含有量を増加させるために、焼成を2段階の温度で行うことも好ましい。この場合、第一焼成の焼成温度は300℃以上が好ましく、より好ましくは350℃以上であり、550℃以下が好ましく、より好ましくは450℃以下が好ましい。第一焼成の焼成時間は0.5時間以上が好ましく、より好ましくは2時間以上であり、24時間以下が好ましく、より好ましくは12時間以下である。また、第二焼成の焼成温度は550℃以上が好ましく、より好ましくは650℃以上、さらに好ましくは750℃以上であり、950℃以下が好ましく、より好ましくは900℃以下が好ましい。第二焼成の焼成時間は0.5時間以上が好ましく、より好ましくは2時間以上であり、48時間以下が好ましく、より好ましくは24時間以下、さらに好ましくは12時間以下である。
【0024】
前記触媒前駆体を還元処理することにより、本発明の触媒が得られる。前記還元処理の方法としては、還元性雰囲気(例えば、水素等の還元性ガスを含む還元性ガス雰囲気)中で熱処理する方法が挙げられる。前記触媒前駆体を焼成する温度は、300℃以上が好ましく、より好ましくは400℃以上であり、800℃以下が好ましく、より好ましくは700℃以下である。焼成時間は、0.5時間以上が好ましく、より好ましくは2時間以上であり、24時間以下が好ましく、より好ましくは12時間以下である。
【0025】
上述したように、本発明の触媒は、ペロブスカイト構造を再構築、再崩壊させることにより、触媒性能を再生することができる。本発明の触媒の再生方法としては、例えば、アンモニアの分解反応に用いた使用済み触媒を、酸化処理した後、さらに還元処理する方法が挙げられる。本発明の触媒には、アンモニアの分解反応に用いた使用済み触媒を、酸化処理して、前記A成分と前記B成分とで形成されるペロブスカイト構造を有する酸化物を再構築し、これを還元処理して得られたものも含まれる。
【0026】
前記酸化処理の方法としては、酸化性雰囲気(例えば、空気雰囲気)中で熱処理する方法が挙げられる。熱処理温度は、550℃以上が好ましく、より好ましくは650℃以上、さらに好ましくは750℃以上であり、950℃以下が好ましく、より好ましくは900℃以下である。熱処理時間は、0.5時間以上が好ましく、より好ましくは2時間以上であり、48時間以下が好ましく、より好ましくは24時間以下、さらに好ましくは12時間以下である。触媒の再生方法における還元処理は、前記触媒製造方法における還元処理と同様に行えばよい。
【0027】
本発明の触媒を用いたアンモニアの分解方法について説明する。
本発明の触媒を用いたアンモニアの分解方法としては、アンモニアを分解する分解工程;アンモニア分解反応後のアンモニア分解用触媒を酸化処理する酸化工程;及び、アンモニア分解用触媒の酸化処理物を、還元処理する還元工程とを含む態様が好ましい。
【0028】
前記分解工程では、加熱下で触媒とアンモニアとを接触させることにより、アンモニアを水素と窒素に分解する。
アンモニアの分解反応に使用する原料ガスは、アンモニアガスであるが、分解反応を阻害しないものであれば、他のガスを加えることができる。他のガスとしては、例えば、窒素、アルゴン、ヘリウム、一酸化炭素、酸素が挙げられる。特に、原料ガスが酸素を含む場合、アンモニアガスやアンモニア分解反応で生成した水素の一部を燃焼し、その燃焼熱をアンモニア分解反応の反応熱として使用するオートサーマルリフォーマーによるアンモニア分解を行うことができる。この場合、アンモニアに対する酸素のモル比(酸素/アンモニア)は、0.75未満とする必要がある。また、アンモニア分解により得られる水素量と、燃焼反応による燃焼熱とを両立させる観点から、モル比(酸素/アンモニア)は0.05以上が好ましく、より好ましくは0.1以上、さらに好ましくは0.12以上であり、0.5以下が好ましく、より好ましくは0.3以下である。
【0029】
アンモニア分解反応の反応温度(例えば、触媒を加熱するための電気炉の設定温度)は、300℃以上が好ましく、より好ましくは400℃以上であり、900℃以下が好ましく、より好ましくは700℃以下である。反応圧力は、絶対圧で、0.002MPa以上が好ましく、より好ましくは0.004MPa以上であり、2MPa以下が好ましく、より好ましくは1MPa以下である。反応ガス(原料ガス)導入時の空間速度(SV)は1,000h-1以上が好ましく、より好ましくは2,000h-1以上であり、500,000h-1以下が好ましく、より好ましくは200,000h-1以下である。
【0030】
前記酸化工程では、アンモニア分解反応に使用した後の使用済み触媒に酸化処理を施し、ペロブスカイト構造を再構築させる。前記酸化処理の方法としては、前記触媒再生方法における酸化処理と同様に行えばよい。前記還元工程では、触媒の酸化処理物に還元処理を施し、酸化工程で構築されたペロブスカイト構造を崩壊させ、B成分を再分散させる。前記還元処理の方法としては、前記触媒再生方法における還元処理と同様に行えばよい。
【0031】
本発明の触媒を用いたアンモニアの分解方法では、触媒が劣化しても、酸化工程、還元工程を経ることで、触媒性能を再生できるため、触媒を交換することなく長期間にわたってアンモニア分解反応を行うことができ、作業性が向上する。なお、上述したようにB成分の金属微粒子の結晶子径が小さい状態で、酸化処理及び還元処理を施したほうが、B成分の再分散効果が高くなる。そのため、上記分解方法では、触媒のB成分の金属微粒子の結晶子径が25nm(より好ましくは20nm、さらに好ましくは18nm)以下の状態で酸化工程を行うことが好ましい。
【0032】
触媒の劣化速度、すなわちB成分の金属微粒子の結晶子径が増大する速度は、アンモニア分解反応の反応温度、反応時間、アンモニア導入量等により決まる。そのため、実際に分解反応を行う条件で予備試験を行い、触媒の劣化速度を確認しておくことで、分解工程から酸化工程に移行する時期を決定すればよい。
【実施例】
【0033】
以下に実施例を挙げて本発明をより具体的に説明するが、本発明は、下記実施例によって限定されるものではなく、前・後記の趣旨に適合しうる範囲で適宜変更して実施することも可能であり、それらはいずれも本発明の技術的範囲に包含される。
【0034】
<評価方法>
B金属微粒子の結晶子径
B金属微粒子の結晶子径の測定は、X線回折測定の結果について、結晶構造の帰属を行い、最大強度を示すピークの半値幅から下記のシェラー式を用いて算出した。
結晶子径(nm) = Kλ/βcosθ
ここで、Kは形状ファクター(球状として0.9を代入)、λは測定X線波長(CuKα:0.154nm)、βは半値幅(rad)、θはブラッグ角(回折角2θの半分;deg)である。
【0035】
X線回折測定条件
X線回折装置(製品名「RINT−TTRIII」、株式会社リガク製)を用いた。X線源には、CuKα(0.154nm)を用い、測定条件として、X線出力50kV、300mA、サンプリング幅0.02°、測定温度25℃であり、測定範囲は測定すべき物質に応じて適宜選択して実施した。
【0036】
製造例1−1
硝酸ニッケル六水和物(Ni(NO3)2・6H2O)23.26g、硝酸ランタン六水和物(La(NO3)3・6H2O)34.64gを純水430mLに投入し、ニッケル−ランタン混合水溶液を調製した。25質量%TMAH(水酸化テトラメチルアンモニウム)水溶液175gに純水を追加して液量約600mLにした希釈TMAH水溶液を調製した。
この希釈TMAH水溶液を激しく撹拌した状態で、ここに前記ニッケル−ランタン混合水溶液を1時間かけてゆっくりと滴下した。滴下終了後、1時間程度撹拌を継続することで熟成を行った。ブフナー漏斗を用いてろ過し、ろ取物を純水で水洗し、110℃で乾燥した。乾燥物を粉砕後、空気雰囲気中、400℃で1時間、更に昇温して850℃で2時間焼成して、ペロブスカイト構造を有するニッケル−ランタン複合酸化物(触媒前駆体1)を得た。
触媒前駆体1を管状炉に充填して、10体積%水素ガス(窒素希釈)を流通させながら、600℃で1時間熱処理(還元処理)し、酸化ランタン−ニッケル金属微粒子触媒(触媒1)を得た。触媒1のニッケルの結晶子径の値を表1に示した。
【0037】
製造例2−1
硝酸コバルト六水和物23.28g、硝酸ランタン六水和物34.64gを純水430mLに投入し、コバルト−ランタン混合水溶液を調製した。25質量%TMAH水溶液175gに純水を追加して液量約700mLにした希釈TMAH水溶液を調製した。
この希釈TMAH水溶液を激しく撹拌した状態で、ここに前記コバルト−ランタン混合水溶液を1時間かけてゆっくりと滴下した。滴下終了後、1時間程度撹拌を継続することで熟成を行った。ブフナー漏斗を用いてろ過し、ろ取物を純水で水洗し、110℃で乾燥した。乾燥物を粉砕後、空気雰囲気中、400℃で1時間、更に昇温して850℃で2時間焼成して、ペロブスカイト構造を有するランタン−コバルト複合酸化物(触媒前駆体2)を得た。
触媒前駆体2を管状炉に充填して、10体積%水素ガス(窒素希釈)を流通させながら、600℃で1時間熱処理(還元処理)し、酸化ランタン−コバルト金属微粒子触媒(触媒2)を得た。触媒2のコバルトの結晶子径の値を表1に示した。
【0038】
製造例3−1
硝酸コバルト六水和物11.64g、硝酸ランタン六水和物10.39g、硝酸ストロンチウム(Sr(NO3)2)3.39gを純水300mLに投入し、コバルト−ランタン−ストロンチウム混合水溶液を調製した。7.7質量%TMAH水溶液33.5gに純水を追加して液量約400mLにした希釈TMAH水溶液を調製した。
この希釈TMAH水溶液を激しく撹拌した状態で、ここに前記コバルト−ランタン−ストロンチウム混合水溶液を1時間かけてゆっくりと滴下した。滴下終了後、1時間程度撹拌を継続することで熟成を行った。ブフナー漏斗を用いてろ過し、ろ取物を純水で水洗し、110℃で乾燥した。乾燥物を粉砕後、空気雰囲気中、400℃で1時間、更に昇温して650℃で2時間焼成して、ペロブスカイト構造を有するストロンチウム−ランタン−コバルト複合酸化物(触媒前駆体3)を得た。
触媒前駆体3を管状炉に充填して、10体積%水素ガス(窒素希釈)を流通させながら、600℃で1時間熱処理(還元処理)し、酸化ストロンチウム−酸化ランタン−コバルト金属微粒子触媒(触媒3)を得た。触媒3のコバルトの結晶子径の値を表1に示した。
【0039】
【表1】

【0040】
製造例1−1、2−1、3−1で得られた触媒1〜3を用いて、99.9体積%以上の純度のアンモニアガスについて、アンモニア分解反応を行った(常圧下、SV=6,000h-1)。反応温度を変更してアンモニア分解率(以下、分解率)を測定した。結果を表2に示した。なお、分解率は以下の式で求めた。
【0041】
【数1】
【0042】
【表2】
【0043】
製造例1−2a、2−2a
上記製造例1−1、2−1のアンモニア分解反応後の使用済みの触媒1及び2を管状炉に充填して、10体積%水素ガス(窒素希釈)を流通させながら750℃で1時間熱処理(還元処理)した。還元処理後のコバルト又はニッケルの結晶子径の値を表3に示した。還元後の触媒1及び2は、いずれもB金属微粒子の結晶子径が増大していた。続いて製造例1−1、2−1と同様にアンモニア分解反応を行い、反応温度を変更して分解率を測定した。結果を表3に示した。
【0044】
製造例1−3a、2−3a
上記製造例1−2a、2−2aのアンモニア分解反応後の使用済みの触媒1及び2を管状炉に充填して、体積比10/90の空気/窒素ガスを流通させながら、650℃で3時間熱処理(酸化処理(再生処理))した後、10体積%水素ガス(窒素希釈)を流通させながら、600℃で1時間熱処理(還元処理)した。還元処理後のコバルトまたはニッケルの結晶子径の値を表3に示した。続いて製造例1−1、2−1と同様にアンモニア分解反応を行い、反応温度を変更して分解率を測定した。結果を表3に示した。製造例1−2a、2−2aと製造例1−3a、2−3aとの比較により、再生処理によって分解率の回復が見られた。
【0045】
【表3】
【0046】
製造例1−2b、2−2b
上記製造例1−1、2−1のアンモニア分解反応後の使用済みの触媒1及び2を管状炉に充填して、10体積%水素ガス(窒素希釈)を流通させながら、850℃で1時間熱処理(還元処理)した。還元処理後のコバルト又はニッケルの結晶子径の値を表4に示した。還元後の触媒1及び2は、いずれもB金属微粒子の結晶子径が25nm超に増大していた。続いて製造例1−1、2−1と同様にアンモニア分解反応を行い、反応温度を変更して分解率を測定した。結果は表4に示した。
【0047】
製造例1−3b、2−3b
上記製造例1−2b、2−2bのアンモニア分解反応後の使用済みの触媒1及び2を管状炉に充填して、体積比10/90の空気/窒素ガスを流通させながら、650℃で3時間熱処理(酸化処理(再生処理))した後、10体積%水素ガス(窒素希釈)を流通させながら、600℃で1時間熱処理(還元処理)した。還元処理後のコバルト又はニッケルの結晶子径の値を表4に示した。続いて製造例1−1、2−1と同様にアンモニア分解反応を行い、反応温度を変更して分解率を測定した。結果は表4に示した。製造例1−2b、2−2bと製造例1−3b、2−3bとの比較により、再生処理によって分解率の回復が見られた。なお、分解率の回復の度合いは、前記製造例1−3a、2−3aよりも小さいものとなった。これは、再生処理前のB金属微粒子の結晶子径が25nm超であり、酸化処理において再構築されるペロブスカイト構造が少なくなり、再分散されるB金属微粒子が少なくなったためと考えられる。
【0048】
【表4】
【0049】
比較例1−1
120℃で一晩乾燥させたγ−アルミナ(Strem Chemicals Inc.製)10.02gに、硝酸ニッケル六水和物12.39gを蒸留水5.00gに溶解させた水溶液を滴下して混合した。この混合物を密閉して1時間静置した後、湯浴上で乾燥させた。この乾燥した混合物を、窒素気流下、350℃で5時間焼成した後、空気気流下、500℃で3時間焼成した。この焼成物を管状炉に充填し、10体積%水素ガス(窒素希釈)を流通させながら、450℃で5時間熱処理(還元処理)して、触媒4を得た。なお、触媒4のニッケル担持量は、20質量%であった。触媒4のニッケルの結晶子径の値を表5に示した。
触媒4を用いて、99.9体積%以上の純度のアンモニアについて、アンモニア分解反応を行った(常圧下、SV=6,000h-1)。反応温度を変更してアンモニア分解率(以下、分解率)を測定した。結果は表5に示した。
【0050】
比較例1−2
上記比較例1−1のアンモニア分解反応後の使用済みの触媒4を管状炉に充填して、10体積%水素ガス(窒素希釈)を流通させながら、750℃で1時間熱処理(還元処理)した。還元処理後のニッケルの結晶子径の値を表5に示した。続いて比較例1−1と同様にアンモニア分解反応を行い、反応温度を変更して分解率を測定した。結果は表5に示した。
【0051】
比較例1−3
上記比較例1−2のアンモニア分解反応後の使用済みの触媒4を管状炉に充填して、体積比10/90の空気/窒素ガスを流通させながら、650℃で3時間熱処理(酸化処理(再生処理))した後、10体積%水素ガス(窒素希釈)を流通させながら、600℃で1時間還元処理した。還元処理後のニッケルの結晶子径の値を表5に示した。続いて比較例1−1と同様にアンモニア分解反応を行い、反応温度を変更して分解率を測定した。結果は表5に示した。比較例1−2と比較例1−3との比較により、再生処理によって分解率の回復は見られなかった。
【0052】
【表5】
【0053】
製造例4
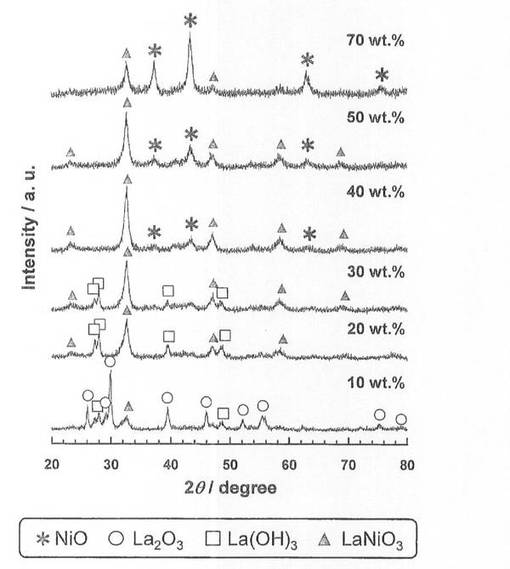
硝酸ニッケル六水和物(和光純薬工業社製、純度99.9質量%以上)を純水に溶解させた水溶液に、酸化ランタン(信越化学工業社製、純度99.99質量%以上)を加え、80℃に設定したスチームバス上で、蒸発、乾固した。乾固物を、空気雰囲気中、600℃で5時間焼成し、触媒前駆体5−1〜5−6を得た。各触媒前駆体のNi担持量は、触媒前駆体5−1〜5−6の順に、それぞれ10、20、30、40、50、70質量%となるように調整した。得られた触媒前駆体についてX線回折(XRD)測定を行い、結果を図1に示した。図1に示すように、Ni含有量が10質量%から40質量%では、Ni含有量が増加するにしたがい、ペロブスカイト構造であるLaNiO3のピーク強度が増大し、La2O3及びLa(OH)のピーク強度が低下している。一方、Ni含有量が40質量%から70質量%では、Ni含有量が増加するにしたがい、LaNiO3のピーク強度が低下し、NiOのピーク強度が増大している。
【0054】
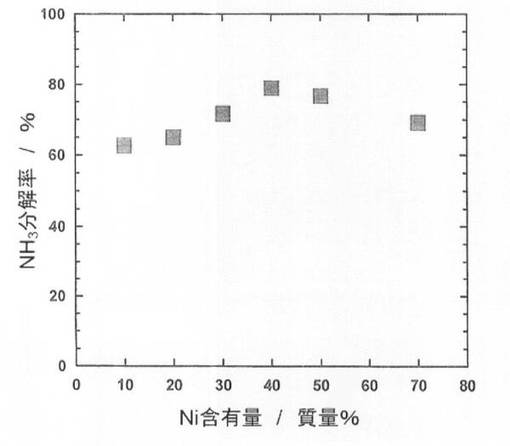
続いて、前記触媒前駆体5−1〜5−6を、電気炉を用いて、体積比50/50の水素/ヘリウム混合ガスを流通させながら(100ml/min)、600℃で2時間熱処理(還元処理)し、触媒5−1〜5−6を得た。得られた触媒を用いて、純度100体積%のアンモニアガスについてアンモニア分解反応を行った。なお、アンモニアガスの流通量は、30ml/min、SV=6,000lkg-1h-1、反応温度は550℃とした。結果を図2に示した。
【0055】
図2に示すように、Ni含有量が10質量%から40質量%では、Ni含有量が増加するにしたがい、アンモニア分解率が上昇し、一方、Ni含有量が40質量%から70質量%では、Ni含有量が増加するにしたがい、アンモニア分解率が減少している。
【0056】
製造例5
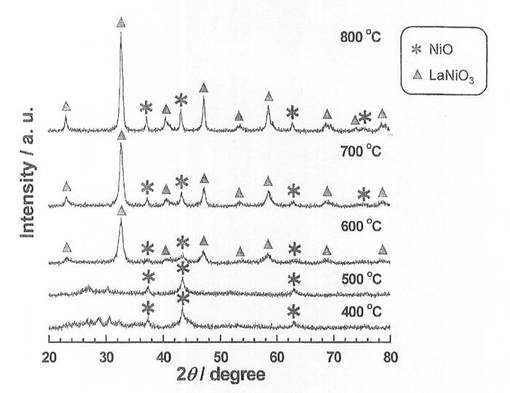
硝酸ニッケル六水和物(和光純薬工業社製、純度99.9質量%以上)を純水に溶解させた水溶液に、酸化ランタン(信越化学工業社製、純度99.99質量%以上)を加え、80℃に設定したスチームバス上で、蒸発、乾固した。なお、触媒前駆体のNi担持量は、40質量%となるように調整した。乾固物を、空気雰囲気中、5時間焼成し、触媒前駆体6−1〜6−5を得た。各触媒前駆体の焼成温度は、触媒前駆体6−1〜6−5の順に、それぞれ400、500、600、700、800℃とした。得られた触媒前駆体についてX線回折(XRD)測定を行い、結果を図3に示した。図3に示すように、焼成温度600、700、800℃では、LaNiO3のピークが確認され、且つ焼成温度が高くなるほど、強度が増大している。一方、焼成温度400、500℃では、LaNiO3のピークが確認されなかった。
【0057】
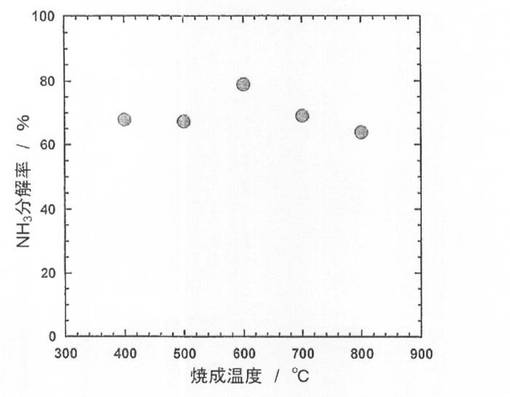
続いて、前記触媒前駆体6−1〜6−5を、前記製造例4と同様にして還元処理し、触媒6−1〜6−5を得た。得られた触媒を用いて、前記製造例4と同様にしてアンモニア分解反応を行い、分解率を測定した。結果を図4に示した。図4に示すように、触媒前駆体においてLaNiO3のピークが確認されなかった触媒6−1、6−2はアンモニア分解率が低いのに対して、触媒前駆体においてLaNiO3のピークが確認された触媒6−3ではアンモニア分解率が上昇している。一方、触媒前駆体においてLaNiO3のピークが確認された触媒6−3〜6−5を比較すると、触媒前駆体におけるLaNiO3のピーク強度が増大するほど、触媒性能が低下していることがわかる。このことから、あまり結晶化させすぎても触媒性能が向上しないことがわかる。
【0058】
製造例6
硝酸ニッケル六水和物(和光純薬工業社製、純度99.9質量%以上)を純水に溶解させた水溶液に、酸化ランタン(信越化学工業社製、純度99.99質量%以上)を加え、80℃に設定したスチームバス上で、蒸発、乾固した。なお、触媒前駆体のNi担持量は、26.5質量%となるように調整した。乾固物を、空気雰囲気中、5時間焼成し、触媒前駆体7−1〜7−5を得た。各触媒前駆体の焼成温度は、触媒前駆体7−1〜7−5の順に、それぞれ500、600、700、800、900℃とした。得られた触媒前駆体についてX線回折(XRD)測定を行い、結果を図5に示した。図5に示すように、焼成温度600、700、800、900℃では、LaNiO3のピークが確認され、且つ焼成温度が高くなるほど、強度が増大している。一方、焼成温度500℃では、LaNiO3のピークが確認されなかった。
【0059】
続いて、前記触媒前駆体7−1〜7−5を、前記製造例4と同様にして還元処理し、触媒7−1〜7−5を得た。得られた触媒を用いて、前記製造例4と同様にしてアンモニア分解反応を行い、分解率を測定した。結果を図7に示した。
【0060】
製造例7
モル比でNi:La=1:1となるように硝酸ニッケル六水和物(和光純薬工業社製、純度99.9質量%以上)と硝酸ランタン六水和物(和光純薬工業社製、純度99.9質量%以上)を秤量して、純水に溶解させた。この水溶液を60℃で1時間攪拌した後、総カチオン量の1.5倍(モル比)のクエン酸水和物を加え、さらに攪拌を続けた。溶液がゲル状になったところで、液温を350℃まで段階的に昇温して、熱分解した。得られた粉末試料を空気中、350℃で24時間熱処理した後、空気雰囲気中で、5時間焼成し、触媒前駆体8−1〜8−5を得た。各触媒前駆体の焼成温度は、触媒前駆体8−1〜8−5の順に、それぞれ400、500、600、700、800℃とした。なお、各触媒前駆体のNi担持量は、26.5質量%である。得られた触媒前駆体についてX線回折(XRD)測定を行い、結果を図6に示した。図6に示すように、焼成温度600、700、800℃では、LaNiO3のピークが確認され、且つ焼成温度が高くなるほど、強度が増大している。一方、焼成温度400、500℃では、LaNiO3のピークが確認されなかった。
【0061】
続いて、前記触媒前駆体8−1〜8−5を、前記製造例4と同様にして還元処理し、触媒8−1〜8−5を得た。得られた触媒を用いて、前記製造例4と同様にしてアンモニア分解反応を行い、分解率を測定した。結果を図7に示した。
【0062】
また、製造例7における焼成温度600℃の触媒前駆体8−3、この触媒前駆体8−3を600℃で2時間還元処理した触媒8−3、並びに、アンモニア分解反応に使用した後の触媒8−3について、X線回折(XRD)測定を行い、結果を図8に示した。図8より、還元処理を施すことにより、触媒前駆体で見られたLaNiO3のピークが消滅し、Ni、La2O3及びLa(OH)3のピークが現れている。また、アンモニア分解反応後には、La(OH)3のピークが消滅し、NiとLa2O3のピークのみとなっている。
【0063】
製造例8
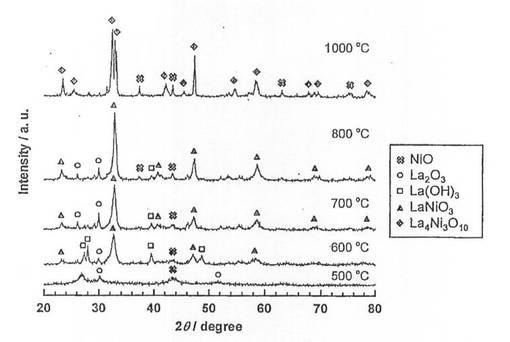
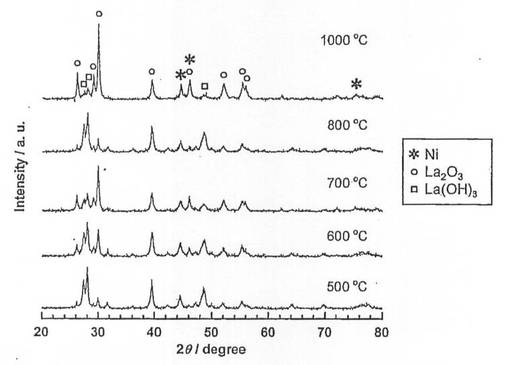
硝酸ニッケル六水和物(和光純薬工業社製、純度99.9質量%以上)を純水に溶解させた水溶液に、酸化ランタン(信越化学工業社製、純度99.99質量%以上)を加え、80℃に設定したスチームバス上で、蒸発、乾固した。なお、触媒前駆体のNi担持量は、26.5質量%となるように調整した。乾固物を、空気雰囲気中、5時間焼成し、触媒前駆体9−1〜9−5を得た。各触媒前駆体の焼成温度は、触媒前駆体9−1〜9−5の順に、それぞれ500、600、700、800、1000℃とした。得られた触媒前駆体についてX線回折(XRD)測定を行い、結果を図9に示した。図9に示すように、焼成温度600、700、800℃では、LaNiO3のピークが確認され、且つ焼成温度が高くなるほど、強度が増大している。一方、焼成温度500℃では、LaNiO3のピークが確認されなかった。また、焼成温度1000℃では、ペロブスカイト構造とは異なる構造(La4Ni3O10)となっていた。
【0064】
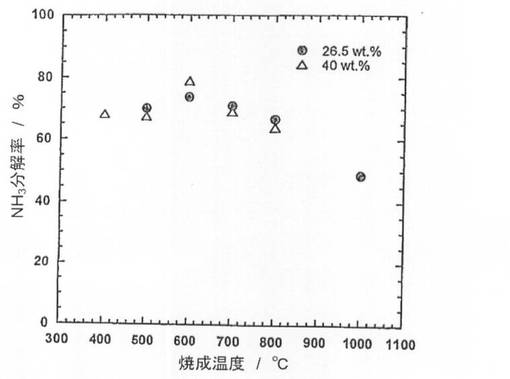
続いて、前記触媒前駆体9−1〜9−5を、電気炉を用いて、体積比50/50の水素/ヘリウム混合ガスを流通させながら(100ml/min)、600℃で2時間熱処理(還元処理)し、触媒9−1〜9−5を得た。得られた触媒についてX線回折(XRD)測定を行い、結果を図10に示した。また、XRD測定結果よりNi結晶子径を求め、表6に示した。図10より、還元処理を施すことにより、触媒9−2〜9−5では、触媒前駆体で見られたLaNiO3のピークが消滅し、Ni、La2O3及びLa(OH)3のピークが現れていることがわかる。つまり、還元処理により、LaNiO3が、Ni及びLa2O3に分解されていることがわかる。なお、La(OH)3のピークが現れているが、これはLa2O3が空気中の水分と反応して生成したものと考えられる。また、焼成温度500、600、700℃の触媒9−1、9−2、9−3では、Niの結晶子径が17〜18nmであり、ほとんど差は見られなかった。これに対して、焼成温度800、1000℃の触媒9−4、9−5では、Niの結晶子径が22.1nm、27.8nmとなっており、焼成温度700℃以上では、焼成温度の上昇に伴い、結晶子径が増大することがわかる。
【0065】
得られた触媒を用いて、純度100体積%のアンモニアガスについてアンモニア分解反応を行った。なお、アンモニアガスの流通量は、30ml/min、SV=6,000lkg-1h-1、反応温度は550℃とした。結果を図12に示した。
【0066】
製造例9
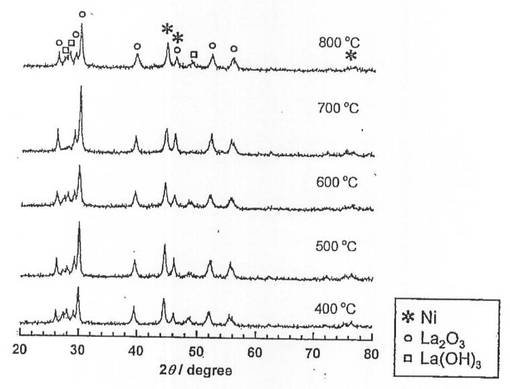
硝酸ニッケル六水和物(和光純薬工業社製、純度99.9質量%以上)を純水に溶解させた水溶液に、酸化ランタン(信越化学工業社製、純度99.99質量%以上)を加え、80℃に設定したスチームバス上で、蒸発、乾固した。なお、触媒前駆体のNi担持量は、40質量%となるように調整した。乾固物を、空気雰囲気中、5時間焼成し、触媒前駆体10−1〜10−5を得た。各触媒前駆体の焼成温度は、触媒前駆体10−1〜10−5の順に、それぞれ400、500、600、700、800℃とした。
【0067】
続いて、前記触媒前駆体10−1〜10−5を、電気炉を用いて、体積比50/50の水素/ヘリウム混合ガスを流通させながら(100ml/min)、600℃で2時間熱処理(還元処理)し、触媒10−1〜10−5を得た。得られた触媒についてX線回折(XRD)測定を行い、結果を図11に示した。また、XRD測定結果よりNi結晶子径を求め、表6に示した。また得られた触媒を用いて、純度100体積%のアンモニアガスについてアンモニア分解反応を行った。なお、アンモニアガスの流通量は、30ml/min、SV=6,000lkg-1h-1、反応温度は550℃とした。結果を図12に示した。
【0068】
【表6】
【産業上の利用可能性】
【0069】
本発明の自己分散型アンモニア触媒を使用すれば、アンモニアから水素を効率よく得ることができる。本発明は、水素製造技術に関して広く応用することができるものである。
【技術分野】
【0001】
本発明は、アンモニア分解用触媒に関するものであり、特に、アンモニア分解反応に使用することで、触媒成分の粒子径が増大し触媒性能が劣化した際に、触媒成分を再分散させることができるアンモニア分解用触媒に関するものである。
【背景技術】
【0002】
従来、アンモニアを分解して水素を製造する技術が提案されている。このようなアンモニアの分解に使用される触媒として、例えば、特許文献1には、触媒成分としてコバルト、ニッケル等の特定成分を含む触媒が提案されており、具体例として、ペロブスカイト構造を有するランタン−コバルト複合酸化物触媒、ペロブスカイト構造を有するニッケル−ランタン複合酸化物触媒が記載されている(特許文献1(段落[0025]、[0026])参照)。特許文献2には、アンモニア燃焼触媒とアンモニア分解触媒とを含み、アンモニア燃焼触媒としてペロブスカイト構造を有するマンガン−ランタン酸化物を含有する触媒が提案されている(特許文献2(請求項6)参照)。また、特許文献3には、ペロブスカイト構造を有するランタン−コバルト複合酸化物触媒、ペロブスカイト構造を有するランタン−ニッケル複合酸化物触媒(特許文献3(段落[0037]、[0039])参照)が提案されている。
なお、これらの特許文献に記載された触媒は、いずれもペロブスカイト構造を維持したものである。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】特開2010−240644号公報
【特許文献2】特開2010−240646号公報
【特許文献3】特開2010−241675号公報
【発明の概要】
【発明が解決しようとする課題】
【0004】
上記特許文献1〜3に記載されたような、ペロブスカイト構造を維持した触媒では、アンモニア分解反応に使用した際に、アンモニアによって触媒が還元されて触媒成分となる金属粒子が生成し、触媒性能を発揮することとなる。しかしながら、このようなペロブスカイト構造を維持した触媒をそのまま使用した場合、触媒の還元が充分でなく、触媒性能を充分に発揮できていなかった。
また、アンモニア分解用触媒は、アンモニア分解反応に使用すると、触媒成分となる金属粒子の粒子径が増大し、触媒成分の比表面積が低下するため、触媒性能が低下する。このように触媒性能が低下した触媒は、もはや使用できないため、定期的にアンモニア分解触媒を交換する必要があった。
本発明は上記事情に鑑みてなされたものであり、アンモニア分解反応に使用することで劣化した場合に、触媒性能を再生できるアンモニア分解用触媒を提供することを目的とする。また、本発明は、上記アンモニア分解用触媒を用いたアンモニアの分解方法、及び、上記アンモニア分解用触媒の再生方法を提供することも目的とする。
【課題を解決するための手段】
【0005】
本発明者らは、アンモニア分解反応に使用することで触媒成分である金属粒子の粒子径が増大した場合に、この金属粒子を再度、微粒子状に分散させる方法について検討を進めた。その結果、ペロブスカイト構造を還元処理により崩壊させて金属微粒子を生成させた場合には、金属粒子の粒子径が増大した場合であっても、再度ペロブスカイト構造を構築、崩壊させることにより、金属粗粒子を再度微粒子に分散できることを見出し、本発明を完成した。
【0006】
本発明のアンモニア分解用触媒は、希土類、アルカリ金属及びアルカリ土類金属よりなる群から選ばれる少なくとも一種の元素(A成分)の酸化物と、Co、Ni及びFeよりなる群から選ばれる少なくとも一種の元素(B成分)の金属微粒子とを含有する触媒であって、前記A成分と前記B成分とで形成されるペロブスカイト構造を有する酸化物を、還元処理して得られたことを特徴とする。前記B成分の金属微粒子のX線回折法で測定される結晶子径は、25nm以下であることが好ましい。アンモニアの分解反応に用いた使用済み触媒を、酸化処理して、前記A成分と前記B成分とで形成されるペロブスカイト構造を有する酸化物を再構築し、これを還元処理して得られたものも好ましい態様である。
【0007】
本発明には、前記アンモニア分解用触媒を用いるアンモニアの分解方法も含まれる。アンモニア分解方法としては、前記アンモニア分解用触媒を用いてアンモニアを分解する分解工程;アンモニア分解反応後のアンモニア分解用触媒を酸化処理する酸化工程;及び、アンモニア分解用触媒の酸化処理物を、還元処理する還元工程とを含む態様が好ましい。また、本発明には、アンモニアの分解反応に用いた使用済み触媒を、酸化処理した後、さらに還元処理するアンモニア分解用触媒の再生方法も含まれる。
【発明の効果】
【0008】
本発明のアンモニア分解用触媒は、アンモニア分解反応に使用して劣化した場合であっても、使用済みの触媒に酸化処理、還元処理を施すことにより、触媒性能を再生できるため、長期間にわたって使用することができる。
また、上記アンモニア分解用触媒を用いたアンモニアの分解方法は、触媒が劣化しても、酸化工程、還元工程を経ることで、触媒性能を再生できるため、触媒を交換することなく長期間にわたってアンモニア分解反応を行うことができ、作業性が向上する。
【図面の簡単な説明】
【0009】
【図1】触媒前駆体5−1〜5−6のX線回折測定結果を示す図である。
【図2】触媒5−1〜5−6のアンモニア分解率を示す図である。
【図3】触媒前駆体6−1〜6−5のX線回折測定結果を示す図である。
【図4】触媒6−1〜6−5のアンモニア分解率を示す図である。
【図5】触媒前駆体7−1〜7−5のX線回折測定結果を示す図である。
【図6】触媒前駆体8−1〜8−5のX線回折測定結果を示す図である。
【図7】触媒7−1〜7−5、8−1〜8−5のアンモニア分解率、Niの比表面積を示す図である。
【図8】触媒前駆体8−3、触媒8−3、アンモニア分解反応に使用した後の触媒8−3のX線回折測定結果を示す図である。
【図9】触媒前駆体9−1〜9−5のX線回折測定結果を示す図である。
【図10】触媒9−1〜9−5のX線回折測定結果を示す図である。
【図11】触媒10−1〜10−5のX線回折測定結果を示す図である。
【図12】触媒9−1〜9−5、10−1〜10−5のアンモニア分解率を示す図である。
【発明を実施するための形態】
【0010】
本発明のアンモニア分解用触媒(以下、単に「触媒」と称する場合がある。)は、希土類、アルカリ金属及びアルカリ土類金属よりなる群から選ばれる少なくとも一種の元素(A成分)と、Co、Ni及びFeよりなる群から選ばれる少なくとも一種の元素(B成分)とで形成されるペロブスカイト構造を有する酸化物を、還元処理して得られたことを特徴とする。
【0011】
A成分とB成分とで形成されるペロブスカイト構造を構築した後、これを崩壊させてB成分の金属微粒子を生成させた場合、A成分とB成分との元素の配置は、再度ペロブスカイト構造を構築し易い配置となる。よって、本発明の触媒は、酸化処理を施すことにより、A成分とB成分とで形成されるペロブスカイト構造を容易に再構築することができる。
そのため、本発明の触媒では、B成分の金属粒子の粒子径が増大した場合でも、酸化処理を施しペロブスカイト構造を再構築すれば、B成分が各結晶構造中に取り込まれるため、B成分を原子レベルにまで細分化することができる。そして、さらに還元処理を施し、再構築したペロブスカイト構造を再崩壊させることで、B成分の金属微粒子を再度生成させることができる。よって、本発明の触媒は、アンモニア分解反応に使用して劣化した場合であっても、このような劣化した使用済みの触媒に酸化処理、還元処理を施すことにより、触媒性能を再生できる。
【0012】
本発明の触媒は、希土類、アルカリ金属及びアルカリ土類金属よりなる群から選ばれる少なくとも一種の元素(A成分)の酸化物と、Co、Ni及びFeよりなる群から選ばれる少なくとも一種の元素(B成分)の金属微粒子とを含有する。B成分の金属微粒子が触媒として作用する。
【0013】
前記A成分としては、具体的には、スカンジウム、イットリウム、ランタン、セリウム、プラセオジム、ネオジム、プロメチウム、サマリウム、ユウロピウム、ガドリニウム、テルビウム、ジスプロシウム、ホルミウム、エルビウム、ツリウム、イッテルビウム、ルテチウム等の希土類元素;リチウム、ナトリウム、カリウム、ルビジウム、セシウム等のアルカリ金属元素;マグネシウム、カルシウム、ストロンチウム、バリウム等のアルカリ土類金属が挙げられる。これらのA成分は単独で使用してもよいし、2種以上を併用してもよい。これらの中でも、A成分としては、希土類元素、アルカリ土類金属元素が好ましく、より好ましくはランタン、カルシウム、ストロンチウム、バリウムである。
前記A成分の酸化物としては、具体的には、酸化ランタン、酸化カルシウム、酸化ストロンチウム、酸化バリウム等が挙げられる。
【0014】
前記B成分としては、Co、Ni、Feを単独で使用してもよいし、2種以上を併用してもよい。これらの中でもCo、Niが好ましい。
前記B成分の金属微粒子の結晶子径は、25nm以下が好ましく、より好ましくは20nm以下、さらに好ましくは18nm以下である。前記結晶子径が25nm以下であれば、再生処理において酸化処理を施した際に、ペロブスカイト構造が構築され易くなるため、B成分の再分散効果がより良好となり、触媒性能の回復率が向上する。前記結晶子径の下限は特に限定されないが、通常5nm程度である。なお、本発明において金属微粒子の結晶子径は、X線回折法で測定される値であり、測定方法は後述する。
【0015】
触媒中の前記B成分の含有量は、5質量%以上が好ましく、より好ましくは15質量%以上、さらに好ましくは25質量%以上であり、80質量%以下が好ましく、より好ましくは60質量%以下、さらに好ましくは50質量%以下である。前記B成分の含有量が5質量%以上であれば触媒性能がより向上し、80質量%以下であれば、B成分が過剰とならず、再生処理において酸化処理を施した際にB成分の大部分がペロブスカイト構造に取り込まれることとなり、B成分の再分散効果がより良好となる。
【0016】
触媒中の前記B成分と前記A成分とのモル比(A成分/B成分)は、0.1以上が好ましく、より好ましくは0.5以上、さらに好ましくは0.8以上であり、10以下が好ましく、より好ましくは3以下、さらに好ましくは1.5以下である。前記モル比が上記範囲内であれば、再生処理において酸化処理を施した際に構築されるペロブスカイト構造の量が多くなり、B成分の再分散効果がより良好となる。
【0017】
本発明の触媒の製造方法の一例について説明する。本発明の触媒は、A成分とB成分とを含有する複合物を作製し、この複合物を酸化性雰囲気で焼成して、A成分とB成分とで形成されるペロブスカイト構造を有する酸化物(以下、「触媒前駆体」と称する場合がある。)を作製し、この触媒前駆体を還元処理することで得られる。
【0018】
前記複合物を作製する方法は特に限定されず、例えば、共沈法、含浸法、クエン酸錯体法等が挙げられる。
共沈法としては、例えば、B成分の水溶性塩とA成分の水溶性塩を溶解させた水溶液と、アルカリ性化合物(好ましくは水溶液)とを混合し、A成分を含有する微粒子とB成分を含有する微粒子からなる沈殿物を生成させ、これをろ過により取り出し、水洗、乾燥する方法が挙げられる。含浸法としては、例えば、B成分の水溶性塩を溶解させた溶液を、A成分の酸化物に含浸させ、水分を蒸発させる方法が挙げられる。クエン酸錯体法としては、例えば、A成分の水溶性塩とB成分の水溶性塩とを溶解させた水溶液に、クエン酸水和物を加えて攪拌し、溶液がゲル状になったところで、加熱して熱分解する方法が挙げられる。
【0019】
前記A成分の水溶性塩としては、例えば、硝酸塩、酢酸塩、塩化物等が挙げられる。前記A成分の水溶性塩としては、例えば、硝酸コバルト六水和物、酢酸コバルト四水和物、塩化コバルト六水和物等のコバルトの水溶性塩;硝酸ニッケル六水和物、二酢酸ニッケル四水和物等のニッケルの水溶性塩;硝酸鉄(II)六水和物、硝酸鉄(III)九水和物等の鉄の水溶性塩;等が挙げられる。
【0020】
B成分の水溶性塩としては、例えば、硝酸塩、酢酸塩、塩化物、硫酸塩等が挙げられる。前記B成分の水溶性塩としては、例えば、硝酸ランタン六水和物等のランタンの水溶性塩;硝酸マグネシウム六水和物等のマグネシウムの水溶性塩;硝酸セリウム六水和物等のセリウムの水溶性塩;等が挙げられる。
【0021】
前記アルカリ性化合物としては、例えば、アンモニア、炭酸アンモニウム、水酸化テトラメチルアンモニウム等のアンモニア系化合物;水酸化カリウム等のアルカリ金属水酸化物;等が挙げられる。
【0022】
前記複合物を酸化性雰囲気で焼成することにより、触媒前駆体を得る。前記酸化性雰囲気としては、例えば、空気雰囲気が挙げられる。前記複合物を焼成する温度は、550℃以上が好ましく、より好ましくは650℃以上、さらに好ましくは750℃以上であり、950℃以下が好ましく、より好ましくは900℃以下である。焼成時間は、0.5時間以上が好ましく、より好ましくは2時間以上であり、48時間以下が好ましく、より好ましくは24時間以下、さらに好ましくは12時間以下である。
【0023】
また、触媒前駆体中のペロブスカイト構造の含有量を増加させるために、焼成を2段階の温度で行うことも好ましい。この場合、第一焼成の焼成温度は300℃以上が好ましく、より好ましくは350℃以上であり、550℃以下が好ましく、より好ましくは450℃以下が好ましい。第一焼成の焼成時間は0.5時間以上が好ましく、より好ましくは2時間以上であり、24時間以下が好ましく、より好ましくは12時間以下である。また、第二焼成の焼成温度は550℃以上が好ましく、より好ましくは650℃以上、さらに好ましくは750℃以上であり、950℃以下が好ましく、より好ましくは900℃以下が好ましい。第二焼成の焼成時間は0.5時間以上が好ましく、より好ましくは2時間以上であり、48時間以下が好ましく、より好ましくは24時間以下、さらに好ましくは12時間以下である。
【0024】
前記触媒前駆体を還元処理することにより、本発明の触媒が得られる。前記還元処理の方法としては、還元性雰囲気(例えば、水素等の還元性ガスを含む還元性ガス雰囲気)中で熱処理する方法が挙げられる。前記触媒前駆体を焼成する温度は、300℃以上が好ましく、より好ましくは400℃以上であり、800℃以下が好ましく、より好ましくは700℃以下である。焼成時間は、0.5時間以上が好ましく、より好ましくは2時間以上であり、24時間以下が好ましく、より好ましくは12時間以下である。
【0025】
上述したように、本発明の触媒は、ペロブスカイト構造を再構築、再崩壊させることにより、触媒性能を再生することができる。本発明の触媒の再生方法としては、例えば、アンモニアの分解反応に用いた使用済み触媒を、酸化処理した後、さらに還元処理する方法が挙げられる。本発明の触媒には、アンモニアの分解反応に用いた使用済み触媒を、酸化処理して、前記A成分と前記B成分とで形成されるペロブスカイト構造を有する酸化物を再構築し、これを還元処理して得られたものも含まれる。
【0026】
前記酸化処理の方法としては、酸化性雰囲気(例えば、空気雰囲気)中で熱処理する方法が挙げられる。熱処理温度は、550℃以上が好ましく、より好ましくは650℃以上、さらに好ましくは750℃以上であり、950℃以下が好ましく、より好ましくは900℃以下である。熱処理時間は、0.5時間以上が好ましく、より好ましくは2時間以上であり、48時間以下が好ましく、より好ましくは24時間以下、さらに好ましくは12時間以下である。触媒の再生方法における還元処理は、前記触媒製造方法における還元処理と同様に行えばよい。
【0027】
本発明の触媒を用いたアンモニアの分解方法について説明する。
本発明の触媒を用いたアンモニアの分解方法としては、アンモニアを分解する分解工程;アンモニア分解反応後のアンモニア分解用触媒を酸化処理する酸化工程;及び、アンモニア分解用触媒の酸化処理物を、還元処理する還元工程とを含む態様が好ましい。
【0028】
前記分解工程では、加熱下で触媒とアンモニアとを接触させることにより、アンモニアを水素と窒素に分解する。
アンモニアの分解反応に使用する原料ガスは、アンモニアガスであるが、分解反応を阻害しないものであれば、他のガスを加えることができる。他のガスとしては、例えば、窒素、アルゴン、ヘリウム、一酸化炭素、酸素が挙げられる。特に、原料ガスが酸素を含む場合、アンモニアガスやアンモニア分解反応で生成した水素の一部を燃焼し、その燃焼熱をアンモニア分解反応の反応熱として使用するオートサーマルリフォーマーによるアンモニア分解を行うことができる。この場合、アンモニアに対する酸素のモル比(酸素/アンモニア)は、0.75未満とする必要がある。また、アンモニア分解により得られる水素量と、燃焼反応による燃焼熱とを両立させる観点から、モル比(酸素/アンモニア)は0.05以上が好ましく、より好ましくは0.1以上、さらに好ましくは0.12以上であり、0.5以下が好ましく、より好ましくは0.3以下である。
【0029】
アンモニア分解反応の反応温度(例えば、触媒を加熱するための電気炉の設定温度)は、300℃以上が好ましく、より好ましくは400℃以上であり、900℃以下が好ましく、より好ましくは700℃以下である。反応圧力は、絶対圧で、0.002MPa以上が好ましく、より好ましくは0.004MPa以上であり、2MPa以下が好ましく、より好ましくは1MPa以下である。反応ガス(原料ガス)導入時の空間速度(SV)は1,000h-1以上が好ましく、より好ましくは2,000h-1以上であり、500,000h-1以下が好ましく、より好ましくは200,000h-1以下である。
【0030】
前記酸化工程では、アンモニア分解反応に使用した後の使用済み触媒に酸化処理を施し、ペロブスカイト構造を再構築させる。前記酸化処理の方法としては、前記触媒再生方法における酸化処理と同様に行えばよい。前記還元工程では、触媒の酸化処理物に還元処理を施し、酸化工程で構築されたペロブスカイト構造を崩壊させ、B成分を再分散させる。前記還元処理の方法としては、前記触媒再生方法における還元処理と同様に行えばよい。
【0031】
本発明の触媒を用いたアンモニアの分解方法では、触媒が劣化しても、酸化工程、還元工程を経ることで、触媒性能を再生できるため、触媒を交換することなく長期間にわたってアンモニア分解反応を行うことができ、作業性が向上する。なお、上述したようにB成分の金属微粒子の結晶子径が小さい状態で、酸化処理及び還元処理を施したほうが、B成分の再分散効果が高くなる。そのため、上記分解方法では、触媒のB成分の金属微粒子の結晶子径が25nm(より好ましくは20nm、さらに好ましくは18nm)以下の状態で酸化工程を行うことが好ましい。
【0032】
触媒の劣化速度、すなわちB成分の金属微粒子の結晶子径が増大する速度は、アンモニア分解反応の反応温度、反応時間、アンモニア導入量等により決まる。そのため、実際に分解反応を行う条件で予備試験を行い、触媒の劣化速度を確認しておくことで、分解工程から酸化工程に移行する時期を決定すればよい。
【実施例】
【0033】
以下に実施例を挙げて本発明をより具体的に説明するが、本発明は、下記実施例によって限定されるものではなく、前・後記の趣旨に適合しうる範囲で適宜変更して実施することも可能であり、それらはいずれも本発明の技術的範囲に包含される。
【0034】
<評価方法>
B金属微粒子の結晶子径
B金属微粒子の結晶子径の測定は、X線回折測定の結果について、結晶構造の帰属を行い、最大強度を示すピークの半値幅から下記のシェラー式を用いて算出した。
結晶子径(nm) = Kλ/βcosθ
ここで、Kは形状ファクター(球状として0.9を代入)、λは測定X線波長(CuKα:0.154nm)、βは半値幅(rad)、θはブラッグ角(回折角2θの半分;deg)である。
【0035】
X線回折測定条件
X線回折装置(製品名「RINT−TTRIII」、株式会社リガク製)を用いた。X線源には、CuKα(0.154nm)を用い、測定条件として、X線出力50kV、300mA、サンプリング幅0.02°、測定温度25℃であり、測定範囲は測定すべき物質に応じて適宜選択して実施した。
【0036】
製造例1−1
硝酸ニッケル六水和物(Ni(NO3)2・6H2O)23.26g、硝酸ランタン六水和物(La(NO3)3・6H2O)34.64gを純水430mLに投入し、ニッケル−ランタン混合水溶液を調製した。25質量%TMAH(水酸化テトラメチルアンモニウム)水溶液175gに純水を追加して液量約600mLにした希釈TMAH水溶液を調製した。
この希釈TMAH水溶液を激しく撹拌した状態で、ここに前記ニッケル−ランタン混合水溶液を1時間かけてゆっくりと滴下した。滴下終了後、1時間程度撹拌を継続することで熟成を行った。ブフナー漏斗を用いてろ過し、ろ取物を純水で水洗し、110℃で乾燥した。乾燥物を粉砕後、空気雰囲気中、400℃で1時間、更に昇温して850℃で2時間焼成して、ペロブスカイト構造を有するニッケル−ランタン複合酸化物(触媒前駆体1)を得た。
触媒前駆体1を管状炉に充填して、10体積%水素ガス(窒素希釈)を流通させながら、600℃で1時間熱処理(還元処理)し、酸化ランタン−ニッケル金属微粒子触媒(触媒1)を得た。触媒1のニッケルの結晶子径の値を表1に示した。
【0037】
製造例2−1
硝酸コバルト六水和物23.28g、硝酸ランタン六水和物34.64gを純水430mLに投入し、コバルト−ランタン混合水溶液を調製した。25質量%TMAH水溶液175gに純水を追加して液量約700mLにした希釈TMAH水溶液を調製した。
この希釈TMAH水溶液を激しく撹拌した状態で、ここに前記コバルト−ランタン混合水溶液を1時間かけてゆっくりと滴下した。滴下終了後、1時間程度撹拌を継続することで熟成を行った。ブフナー漏斗を用いてろ過し、ろ取物を純水で水洗し、110℃で乾燥した。乾燥物を粉砕後、空気雰囲気中、400℃で1時間、更に昇温して850℃で2時間焼成して、ペロブスカイト構造を有するランタン−コバルト複合酸化物(触媒前駆体2)を得た。
触媒前駆体2を管状炉に充填して、10体積%水素ガス(窒素希釈)を流通させながら、600℃で1時間熱処理(還元処理)し、酸化ランタン−コバルト金属微粒子触媒(触媒2)を得た。触媒2のコバルトの結晶子径の値を表1に示した。
【0038】
製造例3−1
硝酸コバルト六水和物11.64g、硝酸ランタン六水和物10.39g、硝酸ストロンチウム(Sr(NO3)2)3.39gを純水300mLに投入し、コバルト−ランタン−ストロンチウム混合水溶液を調製した。7.7質量%TMAH水溶液33.5gに純水を追加して液量約400mLにした希釈TMAH水溶液を調製した。
この希釈TMAH水溶液を激しく撹拌した状態で、ここに前記コバルト−ランタン−ストロンチウム混合水溶液を1時間かけてゆっくりと滴下した。滴下終了後、1時間程度撹拌を継続することで熟成を行った。ブフナー漏斗を用いてろ過し、ろ取物を純水で水洗し、110℃で乾燥した。乾燥物を粉砕後、空気雰囲気中、400℃で1時間、更に昇温して650℃で2時間焼成して、ペロブスカイト構造を有するストロンチウム−ランタン−コバルト複合酸化物(触媒前駆体3)を得た。
触媒前駆体3を管状炉に充填して、10体積%水素ガス(窒素希釈)を流通させながら、600℃で1時間熱処理(還元処理)し、酸化ストロンチウム−酸化ランタン−コバルト金属微粒子触媒(触媒3)を得た。触媒3のコバルトの結晶子径の値を表1に示した。
【0039】
【表1】
【0040】
製造例1−1、2−1、3−1で得られた触媒1〜3を用いて、99.9体積%以上の純度のアンモニアガスについて、アンモニア分解反応を行った(常圧下、SV=6,000h-1)。反応温度を変更してアンモニア分解率(以下、分解率)を測定した。結果を表2に示した。なお、分解率は以下の式で求めた。
【0041】
【数1】
【0042】
【表2】
【0043】
製造例1−2a、2−2a
上記製造例1−1、2−1のアンモニア分解反応後の使用済みの触媒1及び2を管状炉に充填して、10体積%水素ガス(窒素希釈)を流通させながら750℃で1時間熱処理(還元処理)した。還元処理後のコバルト又はニッケルの結晶子径の値を表3に示した。還元後の触媒1及び2は、いずれもB金属微粒子の結晶子径が増大していた。続いて製造例1−1、2−1と同様にアンモニア分解反応を行い、反応温度を変更して分解率を測定した。結果を表3に示した。
【0044】
製造例1−3a、2−3a
上記製造例1−2a、2−2aのアンモニア分解反応後の使用済みの触媒1及び2を管状炉に充填して、体積比10/90の空気/窒素ガスを流通させながら、650℃で3時間熱処理(酸化処理(再生処理))した後、10体積%水素ガス(窒素希釈)を流通させながら、600℃で1時間熱処理(還元処理)した。還元処理後のコバルトまたはニッケルの結晶子径の値を表3に示した。続いて製造例1−1、2−1と同様にアンモニア分解反応を行い、反応温度を変更して分解率を測定した。結果を表3に示した。製造例1−2a、2−2aと製造例1−3a、2−3aとの比較により、再生処理によって分解率の回復が見られた。
【0045】
【表3】
【0046】
製造例1−2b、2−2b
上記製造例1−1、2−1のアンモニア分解反応後の使用済みの触媒1及び2を管状炉に充填して、10体積%水素ガス(窒素希釈)を流通させながら、850℃で1時間熱処理(還元処理)した。還元処理後のコバルト又はニッケルの結晶子径の値を表4に示した。還元後の触媒1及び2は、いずれもB金属微粒子の結晶子径が25nm超に増大していた。続いて製造例1−1、2−1と同様にアンモニア分解反応を行い、反応温度を変更して分解率を測定した。結果は表4に示した。
【0047】
製造例1−3b、2−3b
上記製造例1−2b、2−2bのアンモニア分解反応後の使用済みの触媒1及び2を管状炉に充填して、体積比10/90の空気/窒素ガスを流通させながら、650℃で3時間熱処理(酸化処理(再生処理))した後、10体積%水素ガス(窒素希釈)を流通させながら、600℃で1時間熱処理(還元処理)した。還元処理後のコバルト又はニッケルの結晶子径の値を表4に示した。続いて製造例1−1、2−1と同様にアンモニア分解反応を行い、反応温度を変更して分解率を測定した。結果は表4に示した。製造例1−2b、2−2bと製造例1−3b、2−3bとの比較により、再生処理によって分解率の回復が見られた。なお、分解率の回復の度合いは、前記製造例1−3a、2−3aよりも小さいものとなった。これは、再生処理前のB金属微粒子の結晶子径が25nm超であり、酸化処理において再構築されるペロブスカイト構造が少なくなり、再分散されるB金属微粒子が少なくなったためと考えられる。
【0048】
【表4】
【0049】
比較例1−1
120℃で一晩乾燥させたγ−アルミナ(Strem Chemicals Inc.製)10.02gに、硝酸ニッケル六水和物12.39gを蒸留水5.00gに溶解させた水溶液を滴下して混合した。この混合物を密閉して1時間静置した後、湯浴上で乾燥させた。この乾燥した混合物を、窒素気流下、350℃で5時間焼成した後、空気気流下、500℃で3時間焼成した。この焼成物を管状炉に充填し、10体積%水素ガス(窒素希釈)を流通させながら、450℃で5時間熱処理(還元処理)して、触媒4を得た。なお、触媒4のニッケル担持量は、20質量%であった。触媒4のニッケルの結晶子径の値を表5に示した。
触媒4を用いて、99.9体積%以上の純度のアンモニアについて、アンモニア分解反応を行った(常圧下、SV=6,000h-1)。反応温度を変更してアンモニア分解率(以下、分解率)を測定した。結果は表5に示した。
【0050】
比較例1−2
上記比較例1−1のアンモニア分解反応後の使用済みの触媒4を管状炉に充填して、10体積%水素ガス(窒素希釈)を流通させながら、750℃で1時間熱処理(還元処理)した。還元処理後のニッケルの結晶子径の値を表5に示した。続いて比較例1−1と同様にアンモニア分解反応を行い、反応温度を変更して分解率を測定した。結果は表5に示した。
【0051】
比較例1−3
上記比較例1−2のアンモニア分解反応後の使用済みの触媒4を管状炉に充填して、体積比10/90の空気/窒素ガスを流通させながら、650℃で3時間熱処理(酸化処理(再生処理))した後、10体積%水素ガス(窒素希釈)を流通させながら、600℃で1時間還元処理した。還元処理後のニッケルの結晶子径の値を表5に示した。続いて比較例1−1と同様にアンモニア分解反応を行い、反応温度を変更して分解率を測定した。結果は表5に示した。比較例1−2と比較例1−3との比較により、再生処理によって分解率の回復は見られなかった。
【0052】
【表5】
【0053】
製造例4
硝酸ニッケル六水和物(和光純薬工業社製、純度99.9質量%以上)を純水に溶解させた水溶液に、酸化ランタン(信越化学工業社製、純度99.99質量%以上)を加え、80℃に設定したスチームバス上で、蒸発、乾固した。乾固物を、空気雰囲気中、600℃で5時間焼成し、触媒前駆体5−1〜5−6を得た。各触媒前駆体のNi担持量は、触媒前駆体5−1〜5−6の順に、それぞれ10、20、30、40、50、70質量%となるように調整した。得られた触媒前駆体についてX線回折(XRD)測定を行い、結果を図1に示した。図1に示すように、Ni含有量が10質量%から40質量%では、Ni含有量が増加するにしたがい、ペロブスカイト構造であるLaNiO3のピーク強度が増大し、La2O3及びLa(OH)のピーク強度が低下している。一方、Ni含有量が40質量%から70質量%では、Ni含有量が増加するにしたがい、LaNiO3のピーク強度が低下し、NiOのピーク強度が増大している。
【0054】
続いて、前記触媒前駆体5−1〜5−6を、電気炉を用いて、体積比50/50の水素/ヘリウム混合ガスを流通させながら(100ml/min)、600℃で2時間熱処理(還元処理)し、触媒5−1〜5−6を得た。得られた触媒を用いて、純度100体積%のアンモニアガスについてアンモニア分解反応を行った。なお、アンモニアガスの流通量は、30ml/min、SV=6,000lkg-1h-1、反応温度は550℃とした。結果を図2に示した。
【0055】
図2に示すように、Ni含有量が10質量%から40質量%では、Ni含有量が増加するにしたがい、アンモニア分解率が上昇し、一方、Ni含有量が40質量%から70質量%では、Ni含有量が増加するにしたがい、アンモニア分解率が減少している。
【0056】
製造例5
硝酸ニッケル六水和物(和光純薬工業社製、純度99.9質量%以上)を純水に溶解させた水溶液に、酸化ランタン(信越化学工業社製、純度99.99質量%以上)を加え、80℃に設定したスチームバス上で、蒸発、乾固した。なお、触媒前駆体のNi担持量は、40質量%となるように調整した。乾固物を、空気雰囲気中、5時間焼成し、触媒前駆体6−1〜6−5を得た。各触媒前駆体の焼成温度は、触媒前駆体6−1〜6−5の順に、それぞれ400、500、600、700、800℃とした。得られた触媒前駆体についてX線回折(XRD)測定を行い、結果を図3に示した。図3に示すように、焼成温度600、700、800℃では、LaNiO3のピークが確認され、且つ焼成温度が高くなるほど、強度が増大している。一方、焼成温度400、500℃では、LaNiO3のピークが確認されなかった。
【0057】
続いて、前記触媒前駆体6−1〜6−5を、前記製造例4と同様にして還元処理し、触媒6−1〜6−5を得た。得られた触媒を用いて、前記製造例4と同様にしてアンモニア分解反応を行い、分解率を測定した。結果を図4に示した。図4に示すように、触媒前駆体においてLaNiO3のピークが確認されなかった触媒6−1、6−2はアンモニア分解率が低いのに対して、触媒前駆体においてLaNiO3のピークが確認された触媒6−3ではアンモニア分解率が上昇している。一方、触媒前駆体においてLaNiO3のピークが確認された触媒6−3〜6−5を比較すると、触媒前駆体におけるLaNiO3のピーク強度が増大するほど、触媒性能が低下していることがわかる。このことから、あまり結晶化させすぎても触媒性能が向上しないことがわかる。
【0058】
製造例6
硝酸ニッケル六水和物(和光純薬工業社製、純度99.9質量%以上)を純水に溶解させた水溶液に、酸化ランタン(信越化学工業社製、純度99.99質量%以上)を加え、80℃に設定したスチームバス上で、蒸発、乾固した。なお、触媒前駆体のNi担持量は、26.5質量%となるように調整した。乾固物を、空気雰囲気中、5時間焼成し、触媒前駆体7−1〜7−5を得た。各触媒前駆体の焼成温度は、触媒前駆体7−1〜7−5の順に、それぞれ500、600、700、800、900℃とした。得られた触媒前駆体についてX線回折(XRD)測定を行い、結果を図5に示した。図5に示すように、焼成温度600、700、800、900℃では、LaNiO3のピークが確認され、且つ焼成温度が高くなるほど、強度が増大している。一方、焼成温度500℃では、LaNiO3のピークが確認されなかった。
【0059】
続いて、前記触媒前駆体7−1〜7−5を、前記製造例4と同様にして還元処理し、触媒7−1〜7−5を得た。得られた触媒を用いて、前記製造例4と同様にしてアンモニア分解反応を行い、分解率を測定した。結果を図7に示した。
【0060】
製造例7
モル比でNi:La=1:1となるように硝酸ニッケル六水和物(和光純薬工業社製、純度99.9質量%以上)と硝酸ランタン六水和物(和光純薬工業社製、純度99.9質量%以上)を秤量して、純水に溶解させた。この水溶液を60℃で1時間攪拌した後、総カチオン量の1.5倍(モル比)のクエン酸水和物を加え、さらに攪拌を続けた。溶液がゲル状になったところで、液温を350℃まで段階的に昇温して、熱分解した。得られた粉末試料を空気中、350℃で24時間熱処理した後、空気雰囲気中で、5時間焼成し、触媒前駆体8−1〜8−5を得た。各触媒前駆体の焼成温度は、触媒前駆体8−1〜8−5の順に、それぞれ400、500、600、700、800℃とした。なお、各触媒前駆体のNi担持量は、26.5質量%である。得られた触媒前駆体についてX線回折(XRD)測定を行い、結果を図6に示した。図6に示すように、焼成温度600、700、800℃では、LaNiO3のピークが確認され、且つ焼成温度が高くなるほど、強度が増大している。一方、焼成温度400、500℃では、LaNiO3のピークが確認されなかった。
【0061】
続いて、前記触媒前駆体8−1〜8−5を、前記製造例4と同様にして還元処理し、触媒8−1〜8−5を得た。得られた触媒を用いて、前記製造例4と同様にしてアンモニア分解反応を行い、分解率を測定した。結果を図7に示した。
【0062】
また、製造例7における焼成温度600℃の触媒前駆体8−3、この触媒前駆体8−3を600℃で2時間還元処理した触媒8−3、並びに、アンモニア分解反応に使用した後の触媒8−3について、X線回折(XRD)測定を行い、結果を図8に示した。図8より、還元処理を施すことにより、触媒前駆体で見られたLaNiO3のピークが消滅し、Ni、La2O3及びLa(OH)3のピークが現れている。また、アンモニア分解反応後には、La(OH)3のピークが消滅し、NiとLa2O3のピークのみとなっている。
【0063】
製造例8
硝酸ニッケル六水和物(和光純薬工業社製、純度99.9質量%以上)を純水に溶解させた水溶液に、酸化ランタン(信越化学工業社製、純度99.99質量%以上)を加え、80℃に設定したスチームバス上で、蒸発、乾固した。なお、触媒前駆体のNi担持量は、26.5質量%となるように調整した。乾固物を、空気雰囲気中、5時間焼成し、触媒前駆体9−1〜9−5を得た。各触媒前駆体の焼成温度は、触媒前駆体9−1〜9−5の順に、それぞれ500、600、700、800、1000℃とした。得られた触媒前駆体についてX線回折(XRD)測定を行い、結果を図9に示した。図9に示すように、焼成温度600、700、800℃では、LaNiO3のピークが確認され、且つ焼成温度が高くなるほど、強度が増大している。一方、焼成温度500℃では、LaNiO3のピークが確認されなかった。また、焼成温度1000℃では、ペロブスカイト構造とは異なる構造(La4Ni3O10)となっていた。
【0064】
続いて、前記触媒前駆体9−1〜9−5を、電気炉を用いて、体積比50/50の水素/ヘリウム混合ガスを流通させながら(100ml/min)、600℃で2時間熱処理(還元処理)し、触媒9−1〜9−5を得た。得られた触媒についてX線回折(XRD)測定を行い、結果を図10に示した。また、XRD測定結果よりNi結晶子径を求め、表6に示した。図10より、還元処理を施すことにより、触媒9−2〜9−5では、触媒前駆体で見られたLaNiO3のピークが消滅し、Ni、La2O3及びLa(OH)3のピークが現れていることがわかる。つまり、還元処理により、LaNiO3が、Ni及びLa2O3に分解されていることがわかる。なお、La(OH)3のピークが現れているが、これはLa2O3が空気中の水分と反応して生成したものと考えられる。また、焼成温度500、600、700℃の触媒9−1、9−2、9−3では、Niの結晶子径が17〜18nmであり、ほとんど差は見られなかった。これに対して、焼成温度800、1000℃の触媒9−4、9−5では、Niの結晶子径が22.1nm、27.8nmとなっており、焼成温度700℃以上では、焼成温度の上昇に伴い、結晶子径が増大することがわかる。
【0065】
得られた触媒を用いて、純度100体積%のアンモニアガスについてアンモニア分解反応を行った。なお、アンモニアガスの流通量は、30ml/min、SV=6,000lkg-1h-1、反応温度は550℃とした。結果を図12に示した。
【0066】
製造例9
硝酸ニッケル六水和物(和光純薬工業社製、純度99.9質量%以上)を純水に溶解させた水溶液に、酸化ランタン(信越化学工業社製、純度99.99質量%以上)を加え、80℃に設定したスチームバス上で、蒸発、乾固した。なお、触媒前駆体のNi担持量は、40質量%となるように調整した。乾固物を、空気雰囲気中、5時間焼成し、触媒前駆体10−1〜10−5を得た。各触媒前駆体の焼成温度は、触媒前駆体10−1〜10−5の順に、それぞれ400、500、600、700、800℃とした。
【0067】
続いて、前記触媒前駆体10−1〜10−5を、電気炉を用いて、体積比50/50の水素/ヘリウム混合ガスを流通させながら(100ml/min)、600℃で2時間熱処理(還元処理)し、触媒10−1〜10−5を得た。得られた触媒についてX線回折(XRD)測定を行い、結果を図11に示した。また、XRD測定結果よりNi結晶子径を求め、表6に示した。また得られた触媒を用いて、純度100体積%のアンモニアガスについてアンモニア分解反応を行った。なお、アンモニアガスの流通量は、30ml/min、SV=6,000lkg-1h-1、反応温度は550℃とした。結果を図12に示した。
【0068】
【表6】
【産業上の利用可能性】
【0069】
本発明の自己分散型アンモニア触媒を使用すれば、アンモニアから水素を効率よく得ることができる。本発明は、水素製造技術に関して広く応用することができるものである。
【特許請求の範囲】
【請求項1】
希土類、アルカリ金属及びアルカリ土類金属よりなる群から選ばれる少なくとも一種の元素(A成分)の酸化物と、
Co、Ni及びFeよりなる群から選ばれる少なくとも一種の元素(B成分)の金属微粒子とを含有する触媒であって、
前記A成分と前記B成分とで形成されるペロブスカイト構造を有する酸化物を、還元処理して得られたことを特徴とするアンモニア分解用触媒。
【請求項2】
前記B成分の金属微粒子のX線回折法で測定される結晶子径が、25nm以下である請求項1記載のアンモニア分解用触媒。
【請求項3】
アンモニアの分解反応に用いた使用済み触媒を、酸化処理して、前記A成分と前記B成分とで形成されるペロブスカイト構造を有する酸化物を再構築し、これを還元処理して得られたものである請求項1又は2に記載のアンモニア分解用触媒。
【請求項4】
請求項1〜3のいずれか1項に記載のアンモニア分解用触媒を用いることを特徴とするアンモニアの分解方法。
【請求項5】
前記アンモニア分解用触媒を用いてアンモニアを分解する分解工程;
アンモニア分解反応後のアンモニア分解用触媒を酸化処理する酸化工程;及び、
アンモニア分解用触媒の酸化処理物を、還元処理する還元工程とを含む請求項4に記載のアンモニアの分解方法。
【請求項6】
請求項1〜3のいずれか1項に記載のアンモニア分解用触媒の再生方法であって、
アンモニアの分解反応に用いた使用済み触媒を、酸化処理した後、さらに還元処理することを特徴とするアンモニア分解用触媒の再生方法。
【請求項1】
希土類、アルカリ金属及びアルカリ土類金属よりなる群から選ばれる少なくとも一種の元素(A成分)の酸化物と、
Co、Ni及びFeよりなる群から選ばれる少なくとも一種の元素(B成分)の金属微粒子とを含有する触媒であって、
前記A成分と前記B成分とで形成されるペロブスカイト構造を有する酸化物を、還元処理して得られたことを特徴とするアンモニア分解用触媒。
【請求項2】
前記B成分の金属微粒子のX線回折法で測定される結晶子径が、25nm以下である請求項1記載のアンモニア分解用触媒。
【請求項3】
アンモニアの分解反応に用いた使用済み触媒を、酸化処理して、前記A成分と前記B成分とで形成されるペロブスカイト構造を有する酸化物を再構築し、これを還元処理して得られたものである請求項1又は2に記載のアンモニア分解用触媒。
【請求項4】
請求項1〜3のいずれか1項に記載のアンモニア分解用触媒を用いることを特徴とするアンモニアの分解方法。
【請求項5】
前記アンモニア分解用触媒を用いてアンモニアを分解する分解工程;
アンモニア分解反応後のアンモニア分解用触媒を酸化処理する酸化工程;及び、
アンモニア分解用触媒の酸化処理物を、還元処理する還元工程とを含む請求項4に記載のアンモニアの分解方法。
【請求項6】
請求項1〜3のいずれか1項に記載のアンモニア分解用触媒の再生方法であって、
アンモニアの分解反応に用いた使用済み触媒を、酸化処理した後、さらに還元処理することを特徴とするアンモニア分解用触媒の再生方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【公開番号】特開2012−254419(P2012−254419A)
【公開日】平成24年12月27日(2012.12.27)
【国際特許分類】
【出願番号】特願2011−129582(P2011−129582)
【出願日】平成23年6月9日(2011.6.9)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成23年3月18日に触媒学会発行の「第107回触媒討論会 討論会A予稿集 ISSN 1343−9936」において発表
【出願人】(504132272)国立大学法人京都大学 (1,269)
【出願人】(000004628)株式会社日本触媒 (2,292)
【Fターム(参考)】
【公開日】平成24年12月27日(2012.12.27)
【国際特許分類】
【出願日】平成23年6月9日(2011.6.9)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成23年3月18日に触媒学会発行の「第107回触媒討論会 討論会A予稿集 ISSN 1343−9936」において発表
【出願人】(504132272)国立大学法人京都大学 (1,269)
【出願人】(000004628)株式会社日本触媒 (2,292)
【Fターム(参考)】
[ Back to top ]