
イオン液体およびイオン液体修飾基材
【課題】様々な目的化合物をその性質を改変することなく基材表面へ導入できるイオン液体およびイオン液体修飾基材を提供する。
【解決手段】イオン液体として、基板表面に化学結合されたイオン液体中に目的化合物を導入するだけの空間的な隙間を作り出すことができる有機リン型もしくは4級アミン型のイオン液体を用いる。
【解決手段】イオン液体として、基板表面に化学結合されたイオン液体中に目的化合物を導入するだけの空間的な隙間を作り出すことができる有機リン型もしくは4級アミン型のイオン液体を用いる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、常温で液体であるイオン化合物である「イオン液体」を基材表面に修飾する技術およびその技術により得られた「イオン液体修飾基材」上に様々な化合物を好適に導入する技術に関するものである。
【背景技術】
【0002】
電極などの基材表面を合目的に化合物で修飾する技術は、これまでに様々な方法が提案されている。特に自己組織化単分子膜を利用した基材表面への化合物の修飾方法は、その簡便性などから非常に多くの研究が行われている。ただし、この方法では、化合物自身に何らかの結合性の官能基を導入することが必要であり、それら官能基を通して直接基材表面と、あるいは基材表面に別途修飾した化合物と結合させることで、化合物の基材表面への固定化を実現している。そのため、基材表面に修飾したい分子に官能基を導入する必要があり、そのため、化合物構造の再設計や、官能基の導入による性質が大きく変化することがしばしば報告されている。発明者等の研究グル−プにおいても、酸素の活性化が可能な化合物を電極上に修飾し、その酸素活性化能を以前に検討したが、電極表面へ化学結合により修飾したために、その性質が大きく変わり、通常の状態に比べてその酸素活性化能力は大きく損なわれてしまった(非特許文献1参照)。
【0003】
そこで、この対策として、溶液に近い状態で基材表面に目的の化合物を固定化するためにイオン液体の利用を考えた。イオン液体は常温で液体の分子性液体であり、近年、様々な分野で応用研究が進められている化合物である。イオン液体はこれまでに様々な構造のものが報告・市販されており、分子構造をかなり自由に制御することができる。また、イオン液体を電極に修飾したイオン液体修飾電極に関する研究例も近年幾つか報告されている。さらに、イオン液体修飾電極へ化合物を導入する試みも行われているが、現在の所、イミダゾ−ル型イオン液体を用いたものが殆どであり、イオン液体の対陰イオンを他の陰イオンに交換するイオン交換法によるものが殆どである(例えば、非特許文献2参照)。
【先行技術文献】
【非特許文献】
【0004】
【非特許文献1】猪股智彦、篠崎数馬、林裕也、有井秀和、舩橋靖博、小澤智宏、増田秀樹、「ケミカルコミュニケ−ションズ」(英国)、2008年、p.392
【非特許文献2】チ−(Y.Shik.Chi)、ファン(S.Hwang)、リ−(B.S.Lee)、クォク(J.Kwak)、チョイ(I.S.Choi)、リ−(S.Lee)、「ラングミュア」、(米国)、2005年、21巻、p.4268
【発明の概要】
【発明が解決しようとする課題】
【0005】
上記の通り、従来法によるイオン液体修飾電極への外来性の化合物の導入は、静電的相互作用を利用したイオン交換法であり、そのため導入可能な化合物は、基材表面に修飾されたイオン液体に対して反対の電荷を持つ分子に限られるという欠点があった。そのため、様々な目的化合物を基材表面上へ固定化するための要素技術としては不十分であった。
【0006】
本発明は上記点に鑑みて、基材表面に目的化合物を修飾するためのイオン液体およびそのイオン液体が修飾されたイオン液体修飾基材であって、様々な目的化合物をその性質を改変することなく基材表面へ導入できるイオン液体およびイオン液体修飾基材を提供することを目的とする。
【課題を解決するための手段】
【0007】
本発明者等は、従来のイオン液体修飾電極やこれまでに報告されているイオン液体の構造・性質を幅広く検討した結果、本発明に到達した。
【0008】
すなわち、本発明の第1の特徴は、基材表面に目的化合物を修飾するためのイオン液体であって、一般式(I)で表される構造単位を含む有機リン型もしくは4級アミン型のイオン液体であり、本発明の第2の特徴は、このイオン液体が基材表面に修飾されたイオン液体修飾基材である。
【0009】
【化1】

【0010】
(一般式(I)中、MはP元素またはN元素である。一般式(I)中、R1〜R4は、それぞれ独立に、炭素数1〜30のアルキル基、炭素数2〜30のアルケニル基、炭素数2〜30のアルキニル基、炭素数1〜30のアルコキシアルキル基、炭素数1〜30のアミノアルキル基、炭素数1〜30のパ−フルオロアルキル基、炭素数6〜30のアリ−ル基、炭素数7〜30のアラルキル基、またはカルボニル基を有するアルキル基、アルケニル基、アリ−ル基もしくはアラルキル基を表し、またはRnとRn+1(nは1〜3の整数)が結合して環状構造を有していても良い。ただし、一般式(I)中のR1〜R4の少なくとも1つは、少なくとも1つの結合性官能基(−SH基、−SS−基、−S−基、−COOH基、−NH2基、シラノ−ル基、リン酸基、アルケニル基、アルキニル基、またはアジ基)を有する。一般式(I)中、X−は対陰イオンを表す。)
対陰イオンX−は一価あるいはそれ以上の価数を有する陰イオンであり、各種ハロゲンイオン,BF4−,PF6−,CF3SO3−(略称TfO−),(CF3SO2)2N−(略称Tf2N−)などが好適に用いられる。
【0011】
本発明のイオン液体修飾基材では、イオン液体が一般式(I)で表される構造単位を有するので、従来型のイオン液体修飾電極とは異なり、基材表面に修飾されたイオン液体において、目的化合物を導入するだけの空間的な隙間を作り出すことができる。すなわち、この空間的な隙間に目的化合物を導入することができる(後述の実施例18〜22参照)。このため、本発明によれば、イオン液体に対して反対の電荷を持つ分子(目的化合物)に限らず、様々な目的化合物をその性質を改変することなく基材表面へ導入できる。
【0012】
なお、目的化合物の性質が改変されないのは、このときの目的化合物はイオン液体に溶け込んだ状態であり、目的化合物がイオン液体との間での相互作用によって固定され、化学結合によって固定されるものではないからである。
【0013】
上記の条件を満足する一般式(I)で表される化合物(イオン液体)として、下記の化合物を例示できるが、これらに限定されるものではない。なお、下記の化合物は、一般式(I)中のR1に、「−SS−基」もしくは「−S−基」を有している。
【0014】
【化2】
【図面の簡単な説明】
【0015】
【図1】本発明の実施例1における化合物1の1H−NMRスペクトルを示す図である。
【図2】本発明の実施例2における化合物2のFT−IRスペクトルを示す図である。
【図3】本発明の実施例3における化合物3の1H−NMRスペクトルを示す図である。
【図4】本発明の実施例4における化合物4の1H−NMRスペクトルを示す図である。
【図5】本発明の実施例5における化合物5の1H−NMRスペクトルを示す図である。
【図6】本発明の実施例6における化合物6の1H−NMRスペクトルを示す図である。
【図7】本発明の実施例7における化合物7の1H−NMRスペクトルを示す図である。
【図8】本発明の実施例8における化合物8の1H−NMRスペクトルを示す図である。
【図9】本発明の実施例9における化合物9の1H−NMRスペクトルを示す図である。
【図10】本発明の実施例10における化合物10の1H−NMRスペクトルを示す図である。
【図11】本発明の実施例11における化合物11の1H−NMRスペクトルを示す図である。
【図12】本発明の実施例12における化合物12の1H−NMRスペクトルを示す図である。
【図13】本発明の実施例14における化合物14の1H−NMRスペクトルを示す図である。
【図14】本発明の実施例15における化合物15のORTEP図を示す図である。
【図15】本発明の実施例17におけるイオン液体修飾基板(上)およびイオン液体(下)のFT−IRスペクトルを示す図である。
【図16】本発明の実施例18における鉄複核錯体が導入されたイオン液体修飾基板のサイクリックボルタモグラムを示す図である。
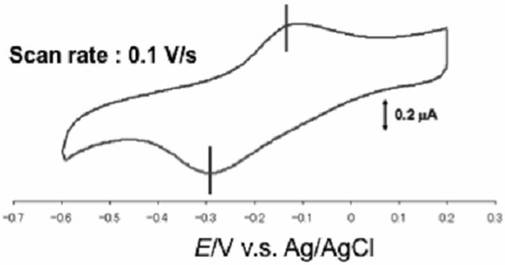
【図17】本発明の実施例19におけるコバルト単核錯体が導入されたイオン液体修飾基板のサイクリックボルタモグラムを示す図である。
【図18】本発明の実施例20におけるイオン液体修飾電極(左)およびフェロセンが導入されたイオン液体修飾基板(右)のサイクリックボルタモグラムを示す図である。
【図19】本発明の実施例21におけるヘキサシアノ鉄(III)錯体が導入されたイオン液体修飾基板のサイクリックボルタモグラムを示す図である。
【図20】本発明の実施例22におけるヘキサアンミンルテニウム(II)錯体が導入されたイオン液体修飾基板のサイクリックボルタモグラムを示す図である。
【発明を実施するための形態】
【0016】
本発明における上述のイオン液体の基材表面への修飾法について説明する。
【0017】
まず、修飾する基材としては、金属、金属酸化物、ガラス、シリコン、ゼオライトやFSMなどの無機細孔材料などが好適に用いられる。イオン液体に含まれる結合性官能基の種類により、例えば、−SH基、−SS−基、−S−基には金、銀、銅などの金属基板が好適に用いられる。リン酸基を含むイオン液体については、酸化ジルコニウムなどが好適に用いられる。またシラノ−ル基を含むイオン液体については、ガラス基板やシリコン基板、あるいはシリカ材料などが好適に用いられる。これらの基板はそのままでも特に問題なくイオン液体を結合することができるが、好ましくは清浄化などの表面処理をしておくことが望ましい。好ましくは濃硝酸、ピランハ溶液、あるいはフッ化水素酸による基板処理をイオン液体修飾前に行うことが望ましい。
【0018】
本発明の結合性官能基を含むイオン液体を目的の基材表面へ化学結合させる方法としては、例えば、有機溶媒にイオン液体を溶解させた溶液に基材表面を浸漬する方法、あるいは溶液を基材表面にスプレ−コ−トやスピンコ−トなどにより塗布する方法により、目的のイオン液体修飾基材を形成することができる。またイオン液体そのものに基材を浸漬、あるいはイオン液体を上記方法により塗布することでも目的のイオン液体修飾基材を得ることができる。浸漬する時間はイオン液体が基材表面に固定されれば特に制限されることはないが、好ましくは5分〜60時間、より好ましくは1〜24時間である。また必要に応じて浸漬あるいは塗布する際に基材や溶液を加熱しても良い。溶液にする場合、イオン液体の濃度としては、0.01mmol/L〜100mmol/L、好ましくは0.1mmol/L〜50mmol/L程度である。溶媒としては、アルコ−ル類、エ−テル類、ニトリル類、エステル類、ケトン類、炭化水素、クロロホルムなどを用いることができる。
【0019】
上記により得られたイオン液体修飾基材への目的化合物の導入方法としては、例えば、目的化合物の溶液にイオン液体修飾基材を浸漬する方法、あるいは溶液をイオン液体修飾基材にスプレ−コ−トやスピンコ−トなどにより塗布する方法などが挙げられる.浸漬する時間は目的化合物がイオン液体修飾基材に導入されれば特に制限されることはないが、好ましくは5分〜60時間、より好ましくは1〜24時間である。また必要に応じて浸漬あるいは塗布する際にイオン液体修飾基材や溶液を加熱しても良い。溶液にする場合、目的化合物の濃度としては、0.01mmol/L〜100mmol/L、好ましくは0.1mmol/L〜50mmol/L程度である。溶媒としては、アルコ−ル類、エ−テル類、ニトリル類、エステル類、ケトン類、炭化水素、クロロホルム、水などを用いることができる。
【0020】
また、他の導入方法として、基材表面へのイオン液体修飾の際に、導入したい化合物を共存させた溶液を調整し、一度にイオン液体と目的化合物を基材表面上に修飾・固定することも可能である。その際の溶液を浸漬する時間は目的化合物およびイオン液体が基材上に導入されれば特に制限されることはないが、好ましくは5分〜60時間、より好ましくは1〜24時間である。また必要に応じて浸漬する際に基材や溶液を加熱しても良い。溶液にする場合、目的化合物の濃度としては、0.01mmol/L〜100mmol/L、好ましくは0.1mmol/L〜50mmol/L程度である。溶媒としては、アルコ−ル類、エ−テル類、ニトリル類、エステル類、ケトン類、炭化水素、クロロホルムなどを用いることができる。
【0021】
更に、別の導入方法として、イオン液体修飾基材を電極とし、電気化学測定装置を用いて、目的化合物を含んだ電解質溶液中で電気化学測定、好ましくはサイクリックボルタンメトリ−などのポテンシオメトリ−測定を行うことにより、イオン液体修飾電極中に電解質溶液中の化合物を導入することが可能である。測定の際の各種条件(濃度、温度、溶媒、測定時間、用いる電解質など)は目的化合物がイオン液体修飾電極中に導入されれば特に制限されることはないが、導入する目的化合物の濃度は好ましくは0.05mmol/L〜10mmol/L、より好ましくは0.1mmol/L〜5mmol/Lである。また測定する際の温度は好ましくは-10℃〜100℃、より好ましくは0℃〜30℃である。溶媒としては、アルコ−ル類、エ−テル類、ニトリル類、エステル類、ケトン類、炭化水素、クロロホルム、水などを用いることができる。測定時間は好ましくは1分〜2時間程度、より好ましくは5〜30分程度である。電解質に関しては、通常の電気化学測定に使用する電解質であれば特に制限はなく、溶媒が水系であれば、過塩素酸リチウムや過塩素酸ナトリウム、有機溶媒であればテトラアルキルアンモニウムのテトラボレ−ト塩やヘキサフルオロリン酸塩、あるいは過塩素酸塩が好適に用いられる。
【0022】
本発明のイオン液体修飾基材に導入可能な化合物としては、どのような分子・材料も使用可能であるが、好ましくは有機化合物、有機金属、金属錯体、金属酸化物、ゼオライトやFSMなどの無機細孔材料、有機高分子、DNAやタンパク質などの生体高分子などを導入することが可能である。より好ましくは修飾されたイオン液体に可溶な分子や無機材料、高分子を用いることができる。また電荷を有する化合物も好適にイオン液体修飾基材に導入することが可能である。空気中で不安定な化合物や材料をイオン液体修飾基材へ導入する場合は、上記の導入操作をグロ−ブボックス内で行うことで、目的の化合物が導入された修飾基材を好適に得ることができる。
【実施例】
【0023】
下記の実施例1〜22のうち、実施例5がイオン液体(化合物5)の合成例であり、実施例1〜4が化合物5を合成するための化合物の合成例である。また、実施例15が目的化合物(化合物15)の合成例であり、実施例6〜14が化合物15を合成するための化合物の合成例である。また、実施例16が基板の作製例であり、実施例17がイオン液体修飾基板の作製例である。また、実施例18〜実施例22のそれぞれが、実施例17のイオン液体修飾基板への様々な目的化合物の導入例である。
(実施例1)
<化合物1の合成>
Ar雰囲気下において200ml三口ナスフラスコに1、12−ジブロモドデカン(2.00g、6.10mmol)とトルエン10mlを加え、1、12−ジブロモドデカンが溶解するまで撹拌した。得られた溶液にトルエン10mlに溶解したトリヘキシルホスフィン(1.75g、6.11mmol)を滴下漏斗を用いて3時間かけて滴下した。得られた混合物を室温、Ar雰囲気下で92時間撹拌後、真空ラインにより乾固させた。得られた白色固体をシリカゲルカラム(酢酸エチル:メタノ−ル=9:1)によって分離した。目的物の含まれたフラクションをエバポレ−タ−によって濃縮乾燥し、薄黄色透明の粘性のある液体を得た。収量1.82g(48.6%)。なお、化合物1は1H−NMR、ESI−MS及びFT−IRスペクトルにより同定した。図1に化合物1の1H−NMRスペクトルを示す。
【0024】
【化3】
【0025】
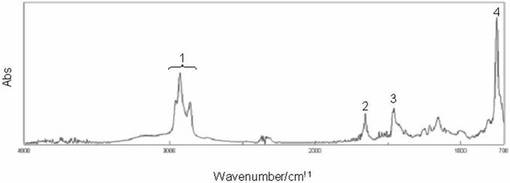
(実施例2)
<化合物2の合成>
チオ尿素(0.13g、1.71mmol)および化合物1(1.09g、1.77mmol)を100ml二口ナスフラスコに入れ、エタノ−ル25mlに溶解し、90℃で44時間還流した。その後、溶液を減圧濃縮することで得られた残渣をクロロホルム100mlに溶解させた。水(50ml)で2回、飽和食塩水(50ml)で2回洗浄後、有機相に硫酸ナトリウムを加えて乾燥させた。硫酸マグネシウムを濾去し、減圧乾燥することで薄黄色透明の粘性のある液体を得た。収量1.17g(100%)。なお、化合物2はESI−MS及びFT−IRスペクトルにより同定した。図2に化合物2のFT−IRスペクトルを示す。
【0026】
【化4】
【0027】
(実施例3)
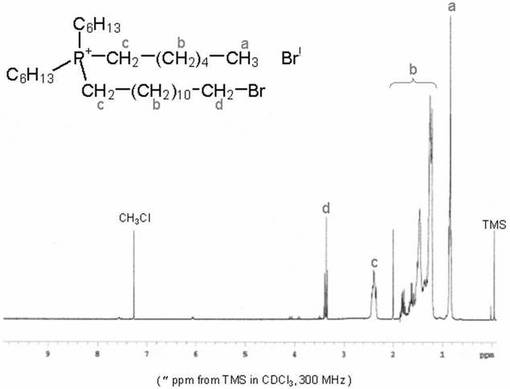
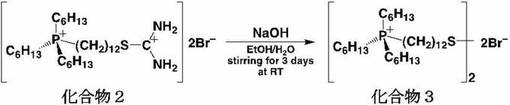
<化合物3の合成>
200mlナスフラスコ中に化合物2(0.63g、0.91mmol)を加え、エタノ−ル(15ml)に溶解した。そこへ小過剰の水酸化ナトリウムを加え、室温で3日間撹拌した。その後、反応溶液をクロロホルム(40ml)により3回抽出した。続いて有機相を水(60ml)で2回洗浄し、硫酸ナトリウムを加えて脱水した。硫酸ナトリウムを吸引濾過によって濾去し、エバポレ−タ−で濃縮することにより、黄色透明の粘性のある液体を得た。収量0.41g(80.4%)。化合物3は1H−NMR、ESI−MS及びFT−IRスペクトルにより同定した。図3に化合物3の1H−NMRスペクトルを示す。
【0028】
【化5】
【0029】
(実施例4)
<化合物4の合成>
100mlナスフラスコに化合物3(0.35g、0.31mmol)およびクロロホルム20mlを加えた。続いてビス(トリフルオロメタンスルホニル)イミドリチウム(0.215g、0.773mmol)のエタノ−ル溶液(10ml)を加え、室温で2時間撹拌した。得られた反応溶液をエバポレ−タ−により濃縮乾固した。ここへクロロホルム50mlを加え、得られた懸濁液を洗液中にBr−イオンが含まれなくなるまで水で洗浄した。有機相を硫酸ナトリウムで脱水後、硫酸ナトリウムを濾去し、有機相をエバポレ−タ−により濃縮乾固した。薄黄色透明の粘性のある液体を得た。収量0.43g(90.9%)。化合物4は1H−NMR、ESI−MS及びFT−IRスペクトルにより同定した。図4に化合物4の1H−NMRスペクトルを示す。
【0030】
【化6】
【0031】
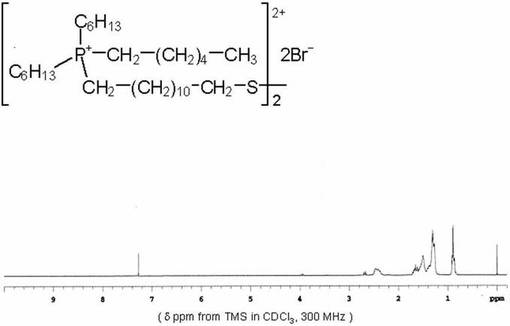
(実施例5)
<化合物5の合成>
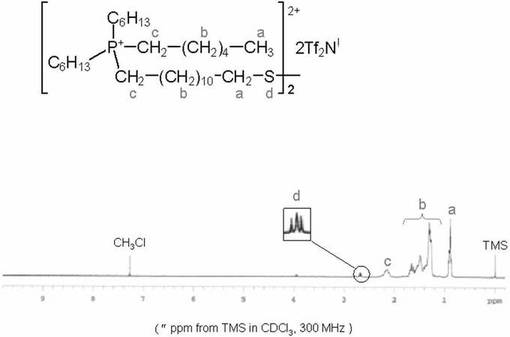
200mlナスフラスコに化合物3(0.65g、0.57mmol)を入れ、クロロホルム35ml加えた。その後トリフルオロメタンスルホン酸ナトリウム(0.238g、1.38mmol)のエタノ−ル溶液(15ml)を加え、室温で6時間撹拌した。得られた反応溶液を洗浄液中にBr−が含まれなくなるまで水で洗浄した。有機相を硫酸ナトリウムで脱水後、濾去し、エバポレ−タ−及び真空ラインを用いて濃縮乾固した。薄黄色透明の粘性のある液体を得た。収量0.61g(84.7%)。化合物5は1H−NMR、ESI−MS及びFT−IRスペクトルにより同定した。図5に化合物5の1H−NMRスペクトルを示す。
【0032】
【化7】
【0033】
なお、化合物5は、一般式(I)において、MがP原子、R1が結合性官能基として−SS−基を有する−(CH2)12SS(CH2)12−基、R2〜R4がC6H13−基、X−がCF3SO3−(TfO−)である。
(実施例6)
<鉄複核錯体の合成>
<化合物6の合成>
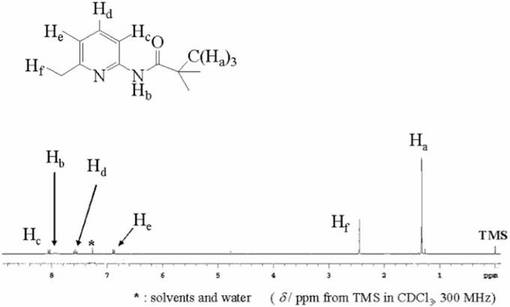
2−アミノ−6−メチルピコリン(0.54mol)とトリエチルアミン(0.54mol)をジクロロメタン(300ml)に溶かし、撹拌しながらピコリン酸クロリド(0.54mol)のジクロロメタン溶液(100ml)を滴下した。反応の様子をTLC(展開溶媒;ヘキサン:酢酸エチル=4:1)により追跡し、原料のスポットが消失したことで反応終了を確認した。その後、反応に伴って析出した白色粉末を濾去し、ジクロロメタン溶液に対して抽出操作を行った。まず有機層を水で3回洗浄し、続いて1Nの塩酸、0.5Nの炭酸水素ナトリウム水溶液、飽和食塩水の順に洗浄した後、無水硫酸マグネシウムを加えて数時間放置した。無水硫酸マグネシウムを濾去し、ロ−タリ−エバポレ−タ−で減圧濃縮して得られた粗結晶をジエチルエ−テルに溶解させ、数日放置することにより析出した無色柱状結晶を濾過して集めることにより、目的物を得た。収量62.6g(59.7%).化合物6は1H−NMRにより同定した。図6に化合物6の1H−NMRスペクトルを示す。
【0034】
【化8】
【0035】
(実施例7)
<化合物7の合成>
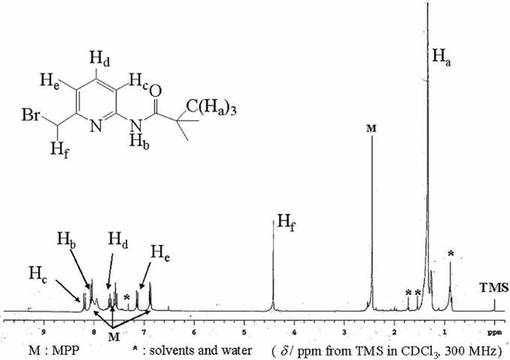
化合物6(0.16mol)を四塩化炭素(300ml)に溶かし、撹拌しながらN−ブロモスクシンイミド(NBS;5.3x10−2mol)と2、2’−アゾビスイソブチロニトリル(AIBN;3.0x10−3mol)を加え、80℃で還流を開始した。開始2時間後にNBS(5.3x10−2mol)とAIBN(3.0x10−3mol)を追加し、次いで、NBS(2.7x10−2mol)とAIBN(2.0x10−3mol)を1時間毎に2回加え、更に1時間還流した後、放冷し、濾過した四塩化炭素溶液に対して洗浄操作を行った。まず有機層を0.5Nの炭酸水素ナトリウム水溶液、飽和食塩水の順で洗浄し、無水硫酸マグネシウムを加えて数時間放置した。無水硫酸マグネシウムを濾去し、ロ−タリ−エバポレ−タ−で減圧濃縮して得られた褐色油状物をシリカゲルカラム(溶離液;ヘキサン:酢酸エチル=10:1)により精製し、目的物を含む褐色油状物(20.75g)を得た。この褐色油状物は目的物の化合物7だけでなく原料である化合物6との混合物であったため、化合物7の含有量は1H−NMRスペクトルにより計算した。収量13.3g(30.8%)。化合物7は1H−NMRにより同定した。図7に化合物7の1H−NMRスペクトルを示す。
【0036】
【化9】
【0037】
(実施例8)
<化合物8の合成>
化合物7(4.9x10−2mol)をN、N−ジメチルホルムアミド(50ml)に溶かし、撹拌しながらフタルイミドカリウム(6.26x10−2mol)を加え130℃で2時間還流した。目的物の生成はTLC(展開溶媒;ヘキサン:酢酸エチル=4:1)により確認した。反応終了を原料である化合物7のTLCスポットが消失したことで確認した後、放冷し、濾過した。N、N−ジメチルホルムアミド溶液に水(50ml)を加えて、クロロホルムで抽出操作を行った。まず目的物を有機層に抽出した後、有機物を飽和食塩水で洗浄し、無水硫酸マグネシウムを加えて数時間放置した。無水硫酸マグネシウムを濾去し、ロ−タリ−エバポレ−タ−で減圧濃縮して得られる褐色粉末をクロロホルム(100ml)に溶解させ、ジエチルエ−テル(200ml)を加えて冷蔵庫にて24時間放置することにより、褐色結晶が析出した。収量(43.3%).化合物8は1H−NMRにより同定した。図8に化合物8の1H−NMRスペクトルを示す。
【0038】
【化10】
【0039】
(実施例9)
<化合物9の合成>
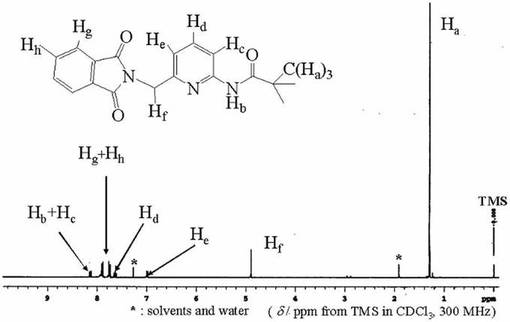
化合物8(2.12x10−2mol)をエタノ−ル(100ml)に完全に溶解させた後、撹拌しながらヒドラジン・一水和物(4.24x10−2mol)を加え、80℃で4時間還流した。目的物の生成はTLC(展開溶媒;ヘキサン:酢酸エチル=4:1)およびニンヒドリン呈色反応により確認した。反応終了を原料である化合物8のTLCスポットが消失したことで確認した後、放冷し、濾過後、濾液をロ−タリ−エバポレ−タ−で減圧濃縮して得られた白色粉末をクロロホルム(50ml)に溶解させた。不溶なフタルヒドラジドを濾別し、濾液を減圧濃縮して得られた白色粉末を再度クロロホルム(50ml)より冷蔵庫内にて再結晶させた。得られた結晶を濾取史し、数時間真空乾燥させることにより化合物9を得た。収量2.70g(69.3%)。化合物9は1H−NMRにより同定した。図9に化合物9の1H−NMRスペクトルを示す。
【0040】
【化11】
【0041】
(実施例10)
<化合物10の合成>
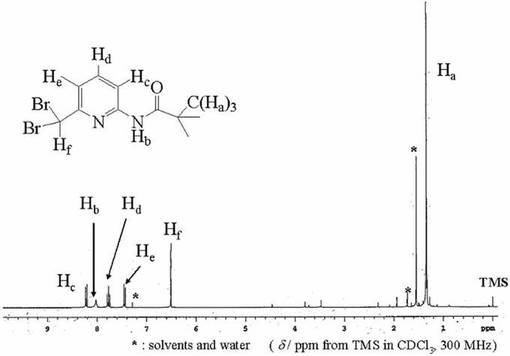
化合物9(0.10mol)を四塩化炭素(250ml)に溶かし、撹拌しながらNBS(3.0x10−2mol)とAIBN(6.0x10−3mol)を加え80℃で還流した。2時間後に更にNBS(3.0x10−2mol)とAIBN(6.0x10−3mol)を加え、その後1時間ごとにNBS(3.0x10−2mol)とAIBN(6.0x10−3mol)を計6回加えた。12時間後、反応溶液を放冷後、濾過し、濾液を減圧濃縮して得られた赤褐色油状物をメタノ−ル(100ml)溶解した。この溶液に水酸化カリウム(0.24mol)を含む水溶液(50ml)を加え、2時間撹拌した。TLC(展開溶媒;ヘキサン:酢酸エチル=4:1)にて化合物7のスポットが消失し、それより下に新たなスポットが出現したことで反応収量を確認した。この溶液を減圧濃縮後、ヘキサン(100ml)に溶かし、有機層を6M塩酸により洗浄し、水層に不純物を抽出した。続いて0.5Mの炭酸水素ナトリウム水溶液を塩基性になるまで加え、水層を取り除いた後、有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムを加えて乾燥させた。硫酸マグネシウムを濾別後、ロ−タリ−エバポレ−タ−で減圧濃縮することにより白色結晶が析出したので、これを数時間真空乾燥させた。収量12.0g(46.0%).化合物10は1H−NMRにより同定した。図10に化合物10の1H−NMRスペクトルを示す。
【0042】
【化12】
【0043】
(実施例11)
<化合物11の合成>
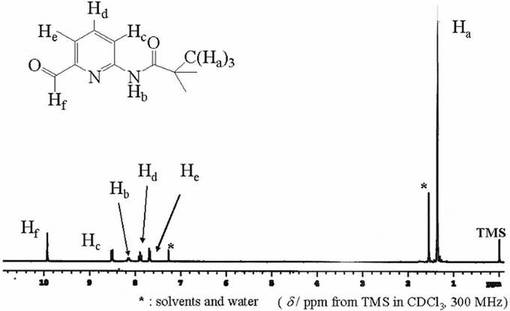
化合物10(3.42x10−2mol)をアセトン(150ml)に溶かし、硝酸銀(11.37x10−2mol)を含む水溶液(40ml)を加え、室温・遮光条件下にして16時間撹拌した。目的物の生成をTLC(展開溶媒;ヘキサン:酢酸エチル=4:1)で確認した。不純物を濾別した後、濾液をロ−タリ−エバポレ−タ−により減圧濃縮し、得られた黄色油状物にジクロロメタンを加えた。まず有機層を水で洗浄し、0.5N炭酸水素ナトリウム水溶液を塩基性になるまで加えた。有機相を抽出後、飽和食塩水で洗浄し、無水硫酸マグネシウムを加えて乾燥させた。硫酸マグネシウムを濾別し、ロ−タリ−エバポレ−タ−により減圧濃縮後、数時間真空乾燥することで黄色油状物を得た。収量(71.6%).化合物11は1H−NMRにより同定した。図11に化合物11の1H−NMRスペクトルを示す。
【0044】
【化13】
【0045】
(実施例12)
<化合物12の合成>
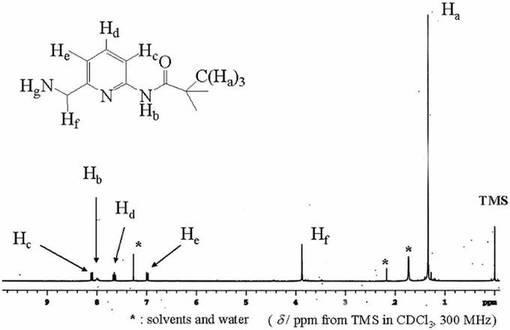
化合物10(1.20x10−2mol)を脱水メタノ−ル(50ml)に溶かし、そこに化合物7(1.29x10−2mol)を加えて耐圧ビンに入れ、酸化白金(10mg)を触媒として、水素圧3atm、室温で接触水素化反応を行った。目的物の生成はTLC(展開溶媒;クロロホルム:メタノ−ル=7:1)およびニンヒドリン呈色反応により確認した。濾過により酸化白金を取り除いた後、濾液をロ−タリ−エバポレ−タ−で減圧濃縮することにより黄色油状物を得た。これをシリカゲルカラム(溶離液;クロロホルム)により精製し、目的物を含む橙色油状物を得た。これをアセトン(50ml)に溶解し、ゆっくりと溶媒を蒸発させることにより析出した無色結晶を濾過し、数時間減圧乾燥させることにより目的物を得た。収量1.47g(30.9%).化合物12は1H−NMRにより同定した。図12に化合物12の1H−NMRスペクトルを示す。
【0046】
【化14】
【0047】
(実施例13)
<化合物13の合成>
2、6−ビス(ヒドロキシメチル)パラクレゾ−ル酢酸(1.8x10−3)と少量のピリジンをジクロロメタン(15ml)に溶かし、塩化チオニル(2.1x10−2)を含むジクロロメタン溶液(30ml)を室温下で撹拌しながらゆっくりと滴下した。得られた溶液を30〜35℃で撹拌し、ジクロロメタンを揮発させることで、目的の黄白色粉末を得た。収量3.68g(100%)。化合物13は1H−NMRにより同定した。
【0048】
【化15】
【0049】
(実施例14)
<化合物14の合成>
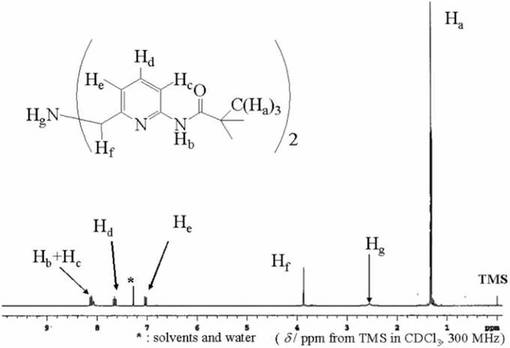
化合物13(7.4x10−3mol)をテトラヒドロフラン(20ml)に溶かし、窒素雰囲気下、氷浴中で撹拌しながら、化合物12(1.8x10−2mol)、トリエチルアミン(1.8x10−2mol)を含むテトラヒドロフラン溶液(70ml)をゆっくりと滴下した。得られた溶液は室温で3日間撹拌した。生成したトリエチルアミン塩酸塩を濾別し、ロ−タリ−エバポレ−タ−により減圧濃縮することにより、黄色油状物を得た。得られた油状物をジクロロメタンに溶かし、水、飽和食塩水を用いて洗浄し、無水硫酸マグネシウムで乾燥させた。硫酸マグネシウムを濾別後、減圧濃縮し、得られた黄色油状物をシリカゲルカラムにより、化合物13(溶離液;ヘキサン:酢酸エチル=4:1)と目的物(溶離液;ヘキサン:酢酸エチル=4:1)に分離・精製し、黄白色粉末を得た。収量5.76g(84%)。化合物14は1H−NMRにより同定した。図13に化合物14の1H−NMRスペクトルを示す。
【0050】
【化16】
【0051】
(実施例15)
<化合物15の合成>
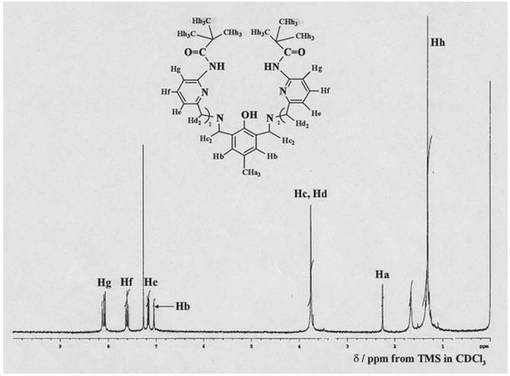
アルゴン雰囲気下、化合物14(3.6x10−5mol)をジクロロメタン及び少量のメタノ−ル混合溶液に溶かし、Fe(CF3SO3)2(7.1x10−5mol)、安息香酸ナトリウム (7.1x10−5mol)を含むメタノ−ル溶液を添加した。続いて、この溶液にトリエチルアミン(3.6x10−5mol)を加え、数時間放置した後、ゆっくりと減圧濃縮した。得られた残渣を少量のメタノ−ルに溶かし、溶媒をゆっくりと蒸発させることで、化合物15の黄色板状結晶を得た。収量10mg(19%)。化合物15はIR、UV/vis、ESI−TOF MSスペクトル、およびX線結晶構造解析により同定した。図14に化合物15の結晶構造のORTEP図を示す。
【0052】
【化17】
【0053】
(実施例16)
<金蒸着基板の作製>
まず天然雲母片(mica)を14×14mm2四方に切り出し、セロテ−プ(登録商標)などによりmica表面を剥がすことで、清浄で平滑な面を露出させた。続いてφ1mmのAu線(99.999%)を適量切り出し、アセトンで湿らせたキムワイプで汚れを落とした後に、適当な大きさに丸めて蒸着装置内のバスケットに設置した。切り出したmicaをAu線が入ったバスケット上部のサンプルホルダ−に設置した。続いて蒸着装置内を〜10−5Paまで減圧し、ランプヒ−タ−によりmicaを300℃で3時間加熱した。続いてバスケット内のAu線を加熱し、蒸着速度約0.9As−1で金薄膜が1000Aになるまで蒸着した。得られた金基板は使用前に水素炎によるアニ−ル処理を行った。
(実施例17)
<イオン液体修飾金基板の作製>
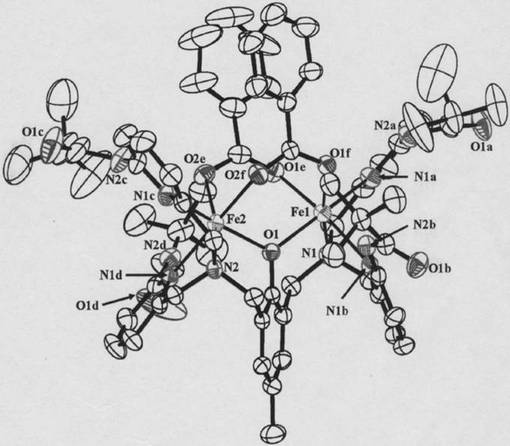
化合物5の1mMエタノ−ルまたはアセトニトリル溶液を調製し、あらかじめエタノ−ルまたはアセトニトリルで表面をリンスした蒸着金基板上をこの溶液中に1日間金基板を浸漬させた。続いて金基板をエタノ−ルまたはアセトニトリルでリンスし、数時間風乾させることで修飾基板1を得た。得られたイオン液体修飾基板は、FT−IRスペクトル(RAS法)、およびサイクリックボルタンメトリ−によりイオン液体の修飾を確認した。図15に修飾基板のFT−IRスペクトルを示す。FT−IRスペクトルにより、金基板上に修飾されたイオン液体に特徴的なC−H結合に由来する伸縮振動および変角振動を2940、1464cm−1に、対陰イオンに由来する伸縮振動を1265、1154、1029cm−1に観測した。またサイクリックボルタンメトリ−測定において、バックグラウンド電流が小さくなったこと、および0.5M KOH水溶液中において−0.94V(vs.Ag/AgCl)にAu−S結合の還元脱離反応に由来する還元波を観測したことから、化合物5は金電極上に−SS−結合によって化学修飾されていることが示唆された。また、この還元脱離波の電流値の解析から、化合物5は9.1(±4.3)×10−11mol・cm−2の被覆率で表面に修飾されていることが判明した。一般的な長鎖アルカンチオ−ル(C18H37SH)を用いて同様の実験を行ったところ,その表面被覆率は5.4×10−10mol・cm−2となり、それに比べると被覆率は1/6程度であった。従って,イオン液体1分子あたり、アルカンチオ−ルの大きさに換算して6分子分の空間が電極表面に存在していることが示唆された。
【0054】
【化18】
【0055】
(実施例18)
<イオン液体修飾金基板への鉄複核錯体の導入法>
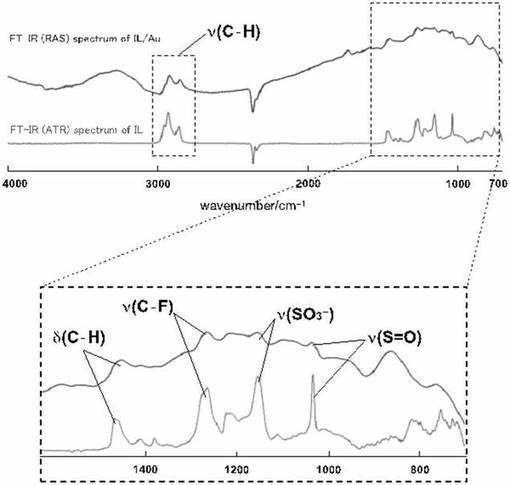
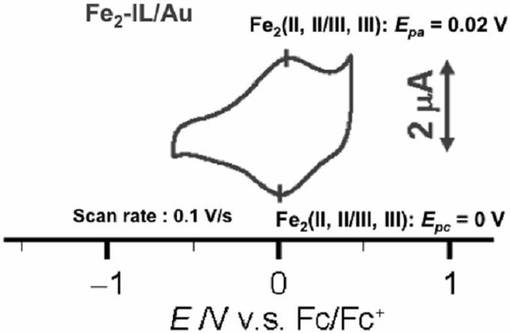
イオン液体修飾基板上に、アルゴン雰囲気下において、10mMの鉄複核錯体(化合物15)を含むメタノ−ル溶液を滴下し、数日間放置した。その後、電極表面を塩化メチレン、およびアセトンを用いて洗浄後、基板を真空乾燥させた。イオン液体修飾基板への鉄複核錯体の導入は、FT−IRスペクトル(RAS法)、およびサイクリックボルタンメトリ−によりイオン液体修飾基板への鉄複核錯体の導入を確認した。図16に鉄複核錯体が導入されたイオン液体修飾基板のサイクリックボルタモグラムを示す。
【0056】
【化19】
【0057】
FT−IRスペクトルでは、導入された化合物15に由来する特徴的な吸収(アミド部位のC=O伸縮振動およびC−N伸縮振動)が1526cm−1および1109cm−1に観測された。サイクリックボルタモグラムではイオン液体修飾電極に導入された化合物15に由来する酸化還元波が0.01V(vs.Fc・Fc+)に観測された。以上の結果より化合物15はイオン液体修飾電極上に固定化されたことが示唆された。
(比較例1)
非特許文献1において、鉄複核錯体を化学結合により電極上に修飾した場合の電気化学測定から、錯体の酸化還元波は−0.78V(vs.Fc・Fc+)に観測された。また均一溶液中では、本鉄複核錯体は0.23V(vs.Fc・Fc+)に酸化還元波が観測された。その差は約1.01Vである。一方、実施例18では、酸化還元波は0.01V(vs.Fc・Fc+)に観測され、その差は約0.22Vである。従って、直接化学結合を利用して鉄複核錯体を修飾した場合に比べて、イオン液体修飾電極を利用した場合は、溶液中のものにかなり近い状態で存在していることが示唆された。
(実施例19)
<イオン液体修飾金基板へのコバルト単核錯体の導入法>
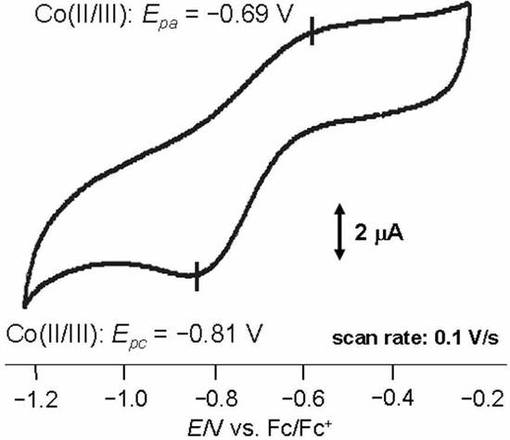
イオン液体修飾基板上に、10mMのコバルト単核錯体を含む水溶液を滴下し、数日間放置した。その後、電極表面をMilli−Q水を用いて洗浄後、基板を真空乾燥させた。イオン液体修飾基板へのコバルト単核錯体の導入は、FT−IRスペクトル(RAS法)、およびサイクリックボルタンメトリ−によりイオン液体修飾基板へのコバルト単核錯体の導入を確認した。図17にコバルト単核錯体が導入されたイオン液体修飾基板のサイクリックボルタモグラムを示す。
【0058】
【化20】
【0059】
FT−IRスペクトルでは、導入されたコバルト錯体に由来する特徴的な吸収(アミド部位のC=O伸縮振動)が1537cm−1に観測された。サイクリックボルタモグラムではイオン液体修飾電極に導入されたコバルト錯体に由来する酸化還元波が−0.70V(vs.Fc・Fc+)に観測された。以上の結果から、コバルト錯体はイオン液体修飾電極上に固定化されたことが示唆された。均一溶液中でのコバルト錯体の酸化還元波は−0.79V(vs.Fc・Fc+)に観測されるが、実施例19との差は殆どないことから、コバルト錯体の性質の改変は無いことが示唆される。
(実施例20)
<イオン液体修飾金基板へのフェロセンの導入法>
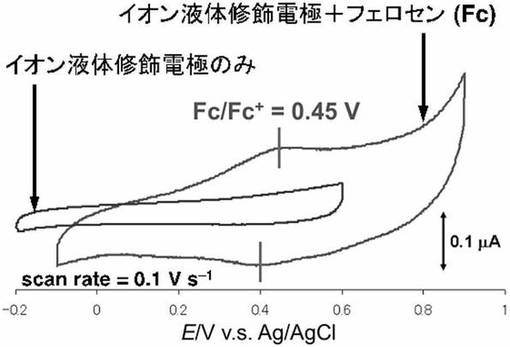
化合物5(0.16mmol)に市販のフェロセン(0.16mmol)を溶解し、得られた溶液を金基板上に滴下し、数日間放置した。続いて、基板表面をクロロホルム、アセトン、Milli−Q水で洗浄後、風乾させた。イオン液体の修飾、およびフェロセンの導入は、サイクリックボルタンメトリ−により確認した。図18にフェロセンが導入されたイオン液体修飾基板のサイクリックボルタモグラムを示す。
【0060】
【化21】
【0061】
サイクリックボルタモグラムではイオン液体修飾電極に導入されたフェロセンに由来する酸化還元波が0.42V(vs.Ag/AgCl)に観測され、フェロセンはイオン液体修飾電極上に固定化されたことが示唆された。均一溶液中でのフェロセンの酸化還元波は0.42V(vs.Ag・AgCl)に観測されるが、実施例20との差は殆どないことから、フェロセンの性質の改変は無いことが示唆される。
(実施例21)
<イオン液体修飾金基板へのヘキサシアノ鉄(III)錯体の導入法>
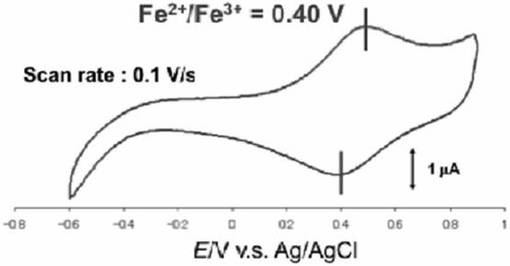
0.1MのNaClO4電解質溶液に市販のヘキサシアノ鉄(III)錯体(K3[Fe(CN)6])を0.5mMとなるように溶解させた。続いて、イオン液体修飾基板を作用極、対極を白金線、参照極を銀/塩化銀電極として、この溶液のサイクリックボルタンメトリ−測定を数回繰り返し行った。測定終了後にイオン液体修飾基板をミリQ水で洗浄し、目的の基板を得た。イオン液体修飾電極へのヘキサシアノ鉄(III)錯体の導入は、サイクリックボルタンメトリ−測定により確認した。図19にヘキサシアノ鉄(III)錯体が導入されたイオン液体修飾基板のサイクリックボルタモグラムを示す。
【0062】
【化22】
【0063】
サイクリックボルタモグラムではイオン液体修飾電極に導入されたヘキサシアノ鉄(III)錯体に由来する酸化還元波が0.40V(vs.Ag/AgCl)に観測され、ヘキサシアノ鉄(III)錯体はイオン液体修飾電極上に固定化されたことが示唆された。均一溶液中でのヘキサシアノ鉄(III)錯体の酸化還元波は0.20V(vs.Ag・AgCl)に観測されるが、実施例21との差は殆どないことから、ヘキサシアノ鉄(III)錯体の性質の改変は無いことが示唆される。
(実施例22)
<イオン液体修飾金基板へのヘキサアンミンルテニウム(II)錯体の導入法>
0.1MのNaClO4電解質溶液に市販のヘキサアンミンルテニウム(II)錯体([Ru(NH3)6]Cl2)を0.5mMとなるように溶解させた。続いて、イオン液体修飾基板を作用極、対極を白金線、参照極を銀/塩化銀電極として、この溶液のサイクリックボルタンメトリ−測定を数回繰り返し行った。測定終了後にイオン液体修飾基板をミリQ水で洗浄し、目的の基板を得た。イオン液体修飾電極へのヘキサアンミンルテニウム(II)錯体の導入は、サイクリックボルタンメトリ−測定により確認した。図20にヘキサアンミンルテニウム(II)錯体が導入されたイオン液体修飾基板のサイクリックボルタモグラムを示す。
【0064】
【化23】
【0065】
サイクリックボルタモグラムではイオン液体修飾電極に導入されたヘキサアンミンルテニウム(II)錯体に由来する酸化還元波が−0.20V(vs.Ag/AgCl)に観測され、フェロセンはイオン液体修飾電極上に固定化されたことが示唆された。均一溶液中でのヘキサアンミンルテニウム(II)錯体の酸化還元波は−0.13V(vs.Ag・AgCl)に観測されるが、実施例22との差は殆どないことから、ヘキサアンミンルテニウム(II)錯体の性質の改変は無いことが示唆される。
【技術分野】
【0001】
本発明は、常温で液体であるイオン化合物である「イオン液体」を基材表面に修飾する技術およびその技術により得られた「イオン液体修飾基材」上に様々な化合物を好適に導入する技術に関するものである。
【背景技術】
【0002】
電極などの基材表面を合目的に化合物で修飾する技術は、これまでに様々な方法が提案されている。特に自己組織化単分子膜を利用した基材表面への化合物の修飾方法は、その簡便性などから非常に多くの研究が行われている。ただし、この方法では、化合物自身に何らかの結合性の官能基を導入することが必要であり、それら官能基を通して直接基材表面と、あるいは基材表面に別途修飾した化合物と結合させることで、化合物の基材表面への固定化を実現している。そのため、基材表面に修飾したい分子に官能基を導入する必要があり、そのため、化合物構造の再設計や、官能基の導入による性質が大きく変化することがしばしば報告されている。発明者等の研究グル−プにおいても、酸素の活性化が可能な化合物を電極上に修飾し、その酸素活性化能を以前に検討したが、電極表面へ化学結合により修飾したために、その性質が大きく変わり、通常の状態に比べてその酸素活性化能力は大きく損なわれてしまった(非特許文献1参照)。
【0003】
そこで、この対策として、溶液に近い状態で基材表面に目的の化合物を固定化するためにイオン液体の利用を考えた。イオン液体は常温で液体の分子性液体であり、近年、様々な分野で応用研究が進められている化合物である。イオン液体はこれまでに様々な構造のものが報告・市販されており、分子構造をかなり自由に制御することができる。また、イオン液体を電極に修飾したイオン液体修飾電極に関する研究例も近年幾つか報告されている。さらに、イオン液体修飾電極へ化合物を導入する試みも行われているが、現在の所、イミダゾ−ル型イオン液体を用いたものが殆どであり、イオン液体の対陰イオンを他の陰イオンに交換するイオン交換法によるものが殆どである(例えば、非特許文献2参照)。
【先行技術文献】
【非特許文献】
【0004】
【非特許文献1】猪股智彦、篠崎数馬、林裕也、有井秀和、舩橋靖博、小澤智宏、増田秀樹、「ケミカルコミュニケ−ションズ」(英国)、2008年、p.392
【非特許文献2】チ−(Y.Shik.Chi)、ファン(S.Hwang)、リ−(B.S.Lee)、クォク(J.Kwak)、チョイ(I.S.Choi)、リ−(S.Lee)、「ラングミュア」、(米国)、2005年、21巻、p.4268
【発明の概要】
【発明が解決しようとする課題】
【0005】
上記の通り、従来法によるイオン液体修飾電極への外来性の化合物の導入は、静電的相互作用を利用したイオン交換法であり、そのため導入可能な化合物は、基材表面に修飾されたイオン液体に対して反対の電荷を持つ分子に限られるという欠点があった。そのため、様々な目的化合物を基材表面上へ固定化するための要素技術としては不十分であった。
【0006】
本発明は上記点に鑑みて、基材表面に目的化合物を修飾するためのイオン液体およびそのイオン液体が修飾されたイオン液体修飾基材であって、様々な目的化合物をその性質を改変することなく基材表面へ導入できるイオン液体およびイオン液体修飾基材を提供することを目的とする。
【課題を解決するための手段】
【0007】
本発明者等は、従来のイオン液体修飾電極やこれまでに報告されているイオン液体の構造・性質を幅広く検討した結果、本発明に到達した。
【0008】
すなわち、本発明の第1の特徴は、基材表面に目的化合物を修飾するためのイオン液体であって、一般式(I)で表される構造単位を含む有機リン型もしくは4級アミン型のイオン液体であり、本発明の第2の特徴は、このイオン液体が基材表面に修飾されたイオン液体修飾基材である。
【0009】
【化1】
【0010】
(一般式(I)中、MはP元素またはN元素である。一般式(I)中、R1〜R4は、それぞれ独立に、炭素数1〜30のアルキル基、炭素数2〜30のアルケニル基、炭素数2〜30のアルキニル基、炭素数1〜30のアルコキシアルキル基、炭素数1〜30のアミノアルキル基、炭素数1〜30のパ−フルオロアルキル基、炭素数6〜30のアリ−ル基、炭素数7〜30のアラルキル基、またはカルボニル基を有するアルキル基、アルケニル基、アリ−ル基もしくはアラルキル基を表し、またはRnとRn+1(nは1〜3の整数)が結合して環状構造を有していても良い。ただし、一般式(I)中のR1〜R4の少なくとも1つは、少なくとも1つの結合性官能基(−SH基、−SS−基、−S−基、−COOH基、−NH2基、シラノ−ル基、リン酸基、アルケニル基、アルキニル基、またはアジ基)を有する。一般式(I)中、X−は対陰イオンを表す。)
対陰イオンX−は一価あるいはそれ以上の価数を有する陰イオンであり、各種ハロゲンイオン,BF4−,PF6−,CF3SO3−(略称TfO−),(CF3SO2)2N−(略称Tf2N−)などが好適に用いられる。
【0011】
本発明のイオン液体修飾基材では、イオン液体が一般式(I)で表される構造単位を有するので、従来型のイオン液体修飾電極とは異なり、基材表面に修飾されたイオン液体において、目的化合物を導入するだけの空間的な隙間を作り出すことができる。すなわち、この空間的な隙間に目的化合物を導入することができる(後述の実施例18〜22参照)。このため、本発明によれば、イオン液体に対して反対の電荷を持つ分子(目的化合物)に限らず、様々な目的化合物をその性質を改変することなく基材表面へ導入できる。
【0012】
なお、目的化合物の性質が改変されないのは、このときの目的化合物はイオン液体に溶け込んだ状態であり、目的化合物がイオン液体との間での相互作用によって固定され、化学結合によって固定されるものではないからである。
【0013】
上記の条件を満足する一般式(I)で表される化合物(イオン液体)として、下記の化合物を例示できるが、これらに限定されるものではない。なお、下記の化合物は、一般式(I)中のR1に、「−SS−基」もしくは「−S−基」を有している。
【0014】
【化2】
【図面の簡単な説明】
【0015】
【図1】本発明の実施例1における化合物1の1H−NMRスペクトルを示す図である。
【図2】本発明の実施例2における化合物2のFT−IRスペクトルを示す図である。
【図3】本発明の実施例3における化合物3の1H−NMRスペクトルを示す図である。
【図4】本発明の実施例4における化合物4の1H−NMRスペクトルを示す図である。
【図5】本発明の実施例5における化合物5の1H−NMRスペクトルを示す図である。
【図6】本発明の実施例6における化合物6の1H−NMRスペクトルを示す図である。
【図7】本発明の実施例7における化合物7の1H−NMRスペクトルを示す図である。
【図8】本発明の実施例8における化合物8の1H−NMRスペクトルを示す図である。
【図9】本発明の実施例9における化合物9の1H−NMRスペクトルを示す図である。
【図10】本発明の実施例10における化合物10の1H−NMRスペクトルを示す図である。
【図11】本発明の実施例11における化合物11の1H−NMRスペクトルを示す図である。
【図12】本発明の実施例12における化合物12の1H−NMRスペクトルを示す図である。
【図13】本発明の実施例14における化合物14の1H−NMRスペクトルを示す図である。
【図14】本発明の実施例15における化合物15のORTEP図を示す図である。
【図15】本発明の実施例17におけるイオン液体修飾基板(上)およびイオン液体(下)のFT−IRスペクトルを示す図である。
【図16】本発明の実施例18における鉄複核錯体が導入されたイオン液体修飾基板のサイクリックボルタモグラムを示す図である。
【図17】本発明の実施例19におけるコバルト単核錯体が導入されたイオン液体修飾基板のサイクリックボルタモグラムを示す図である。
【図18】本発明の実施例20におけるイオン液体修飾電極(左)およびフェロセンが導入されたイオン液体修飾基板(右)のサイクリックボルタモグラムを示す図である。
【図19】本発明の実施例21におけるヘキサシアノ鉄(III)錯体が導入されたイオン液体修飾基板のサイクリックボルタモグラムを示す図である。
【図20】本発明の実施例22におけるヘキサアンミンルテニウム(II)錯体が導入されたイオン液体修飾基板のサイクリックボルタモグラムを示す図である。
【発明を実施するための形態】
【0016】
本発明における上述のイオン液体の基材表面への修飾法について説明する。
【0017】
まず、修飾する基材としては、金属、金属酸化物、ガラス、シリコン、ゼオライトやFSMなどの無機細孔材料などが好適に用いられる。イオン液体に含まれる結合性官能基の種類により、例えば、−SH基、−SS−基、−S−基には金、銀、銅などの金属基板が好適に用いられる。リン酸基を含むイオン液体については、酸化ジルコニウムなどが好適に用いられる。またシラノ−ル基を含むイオン液体については、ガラス基板やシリコン基板、あるいはシリカ材料などが好適に用いられる。これらの基板はそのままでも特に問題なくイオン液体を結合することができるが、好ましくは清浄化などの表面処理をしておくことが望ましい。好ましくは濃硝酸、ピランハ溶液、あるいはフッ化水素酸による基板処理をイオン液体修飾前に行うことが望ましい。
【0018】
本発明の結合性官能基を含むイオン液体を目的の基材表面へ化学結合させる方法としては、例えば、有機溶媒にイオン液体を溶解させた溶液に基材表面を浸漬する方法、あるいは溶液を基材表面にスプレ−コ−トやスピンコ−トなどにより塗布する方法により、目的のイオン液体修飾基材を形成することができる。またイオン液体そのものに基材を浸漬、あるいはイオン液体を上記方法により塗布することでも目的のイオン液体修飾基材を得ることができる。浸漬する時間はイオン液体が基材表面に固定されれば特に制限されることはないが、好ましくは5分〜60時間、より好ましくは1〜24時間である。また必要に応じて浸漬あるいは塗布する際に基材や溶液を加熱しても良い。溶液にする場合、イオン液体の濃度としては、0.01mmol/L〜100mmol/L、好ましくは0.1mmol/L〜50mmol/L程度である。溶媒としては、アルコ−ル類、エ−テル類、ニトリル類、エステル類、ケトン類、炭化水素、クロロホルムなどを用いることができる。
【0019】
上記により得られたイオン液体修飾基材への目的化合物の導入方法としては、例えば、目的化合物の溶液にイオン液体修飾基材を浸漬する方法、あるいは溶液をイオン液体修飾基材にスプレ−コ−トやスピンコ−トなどにより塗布する方法などが挙げられる.浸漬する時間は目的化合物がイオン液体修飾基材に導入されれば特に制限されることはないが、好ましくは5分〜60時間、より好ましくは1〜24時間である。また必要に応じて浸漬あるいは塗布する際にイオン液体修飾基材や溶液を加熱しても良い。溶液にする場合、目的化合物の濃度としては、0.01mmol/L〜100mmol/L、好ましくは0.1mmol/L〜50mmol/L程度である。溶媒としては、アルコ−ル類、エ−テル類、ニトリル類、エステル類、ケトン類、炭化水素、クロロホルム、水などを用いることができる。
【0020】
また、他の導入方法として、基材表面へのイオン液体修飾の際に、導入したい化合物を共存させた溶液を調整し、一度にイオン液体と目的化合物を基材表面上に修飾・固定することも可能である。その際の溶液を浸漬する時間は目的化合物およびイオン液体が基材上に導入されれば特に制限されることはないが、好ましくは5分〜60時間、より好ましくは1〜24時間である。また必要に応じて浸漬する際に基材や溶液を加熱しても良い。溶液にする場合、目的化合物の濃度としては、0.01mmol/L〜100mmol/L、好ましくは0.1mmol/L〜50mmol/L程度である。溶媒としては、アルコ−ル類、エ−テル類、ニトリル類、エステル類、ケトン類、炭化水素、クロロホルムなどを用いることができる。
【0021】
更に、別の導入方法として、イオン液体修飾基材を電極とし、電気化学測定装置を用いて、目的化合物を含んだ電解質溶液中で電気化学測定、好ましくはサイクリックボルタンメトリ−などのポテンシオメトリ−測定を行うことにより、イオン液体修飾電極中に電解質溶液中の化合物を導入することが可能である。測定の際の各種条件(濃度、温度、溶媒、測定時間、用いる電解質など)は目的化合物がイオン液体修飾電極中に導入されれば特に制限されることはないが、導入する目的化合物の濃度は好ましくは0.05mmol/L〜10mmol/L、より好ましくは0.1mmol/L〜5mmol/Lである。また測定する際の温度は好ましくは-10℃〜100℃、より好ましくは0℃〜30℃である。溶媒としては、アルコ−ル類、エ−テル類、ニトリル類、エステル類、ケトン類、炭化水素、クロロホルム、水などを用いることができる。測定時間は好ましくは1分〜2時間程度、より好ましくは5〜30分程度である。電解質に関しては、通常の電気化学測定に使用する電解質であれば特に制限はなく、溶媒が水系であれば、過塩素酸リチウムや過塩素酸ナトリウム、有機溶媒であればテトラアルキルアンモニウムのテトラボレ−ト塩やヘキサフルオロリン酸塩、あるいは過塩素酸塩が好適に用いられる。
【0022】
本発明のイオン液体修飾基材に導入可能な化合物としては、どのような分子・材料も使用可能であるが、好ましくは有機化合物、有機金属、金属錯体、金属酸化物、ゼオライトやFSMなどの無機細孔材料、有機高分子、DNAやタンパク質などの生体高分子などを導入することが可能である。より好ましくは修飾されたイオン液体に可溶な分子や無機材料、高分子を用いることができる。また電荷を有する化合物も好適にイオン液体修飾基材に導入することが可能である。空気中で不安定な化合物や材料をイオン液体修飾基材へ導入する場合は、上記の導入操作をグロ−ブボックス内で行うことで、目的の化合物が導入された修飾基材を好適に得ることができる。
【実施例】
【0023】
下記の実施例1〜22のうち、実施例5がイオン液体(化合物5)の合成例であり、実施例1〜4が化合物5を合成するための化合物の合成例である。また、実施例15が目的化合物(化合物15)の合成例であり、実施例6〜14が化合物15を合成するための化合物の合成例である。また、実施例16が基板の作製例であり、実施例17がイオン液体修飾基板の作製例である。また、実施例18〜実施例22のそれぞれが、実施例17のイオン液体修飾基板への様々な目的化合物の導入例である。
(実施例1)
<化合物1の合成>
Ar雰囲気下において200ml三口ナスフラスコに1、12−ジブロモドデカン(2.00g、6.10mmol)とトルエン10mlを加え、1、12−ジブロモドデカンが溶解するまで撹拌した。得られた溶液にトルエン10mlに溶解したトリヘキシルホスフィン(1.75g、6.11mmol)を滴下漏斗を用いて3時間かけて滴下した。得られた混合物を室温、Ar雰囲気下で92時間撹拌後、真空ラインにより乾固させた。得られた白色固体をシリカゲルカラム(酢酸エチル:メタノ−ル=9:1)によって分離した。目的物の含まれたフラクションをエバポレ−タ−によって濃縮乾燥し、薄黄色透明の粘性のある液体を得た。収量1.82g(48.6%)。なお、化合物1は1H−NMR、ESI−MS及びFT−IRスペクトルにより同定した。図1に化合物1の1H−NMRスペクトルを示す。
【0024】
【化3】
【0025】
(実施例2)
<化合物2の合成>
チオ尿素(0.13g、1.71mmol)および化合物1(1.09g、1.77mmol)を100ml二口ナスフラスコに入れ、エタノ−ル25mlに溶解し、90℃で44時間還流した。その後、溶液を減圧濃縮することで得られた残渣をクロロホルム100mlに溶解させた。水(50ml)で2回、飽和食塩水(50ml)で2回洗浄後、有機相に硫酸ナトリウムを加えて乾燥させた。硫酸マグネシウムを濾去し、減圧乾燥することで薄黄色透明の粘性のある液体を得た。収量1.17g(100%)。なお、化合物2はESI−MS及びFT−IRスペクトルにより同定した。図2に化合物2のFT−IRスペクトルを示す。
【0026】
【化4】
【0027】
(実施例3)
<化合物3の合成>
200mlナスフラスコ中に化合物2(0.63g、0.91mmol)を加え、エタノ−ル(15ml)に溶解した。そこへ小過剰の水酸化ナトリウムを加え、室温で3日間撹拌した。その後、反応溶液をクロロホルム(40ml)により3回抽出した。続いて有機相を水(60ml)で2回洗浄し、硫酸ナトリウムを加えて脱水した。硫酸ナトリウムを吸引濾過によって濾去し、エバポレ−タ−で濃縮することにより、黄色透明の粘性のある液体を得た。収量0.41g(80.4%)。化合物3は1H−NMR、ESI−MS及びFT−IRスペクトルにより同定した。図3に化合物3の1H−NMRスペクトルを示す。
【0028】
【化5】
【0029】
(実施例4)
<化合物4の合成>
100mlナスフラスコに化合物3(0.35g、0.31mmol)およびクロロホルム20mlを加えた。続いてビス(トリフルオロメタンスルホニル)イミドリチウム(0.215g、0.773mmol)のエタノ−ル溶液(10ml)を加え、室温で2時間撹拌した。得られた反応溶液をエバポレ−タ−により濃縮乾固した。ここへクロロホルム50mlを加え、得られた懸濁液を洗液中にBr−イオンが含まれなくなるまで水で洗浄した。有機相を硫酸ナトリウムで脱水後、硫酸ナトリウムを濾去し、有機相をエバポレ−タ−により濃縮乾固した。薄黄色透明の粘性のある液体を得た。収量0.43g(90.9%)。化合物4は1H−NMR、ESI−MS及びFT−IRスペクトルにより同定した。図4に化合物4の1H−NMRスペクトルを示す。
【0030】
【化6】
【0031】
(実施例5)
<化合物5の合成>
200mlナスフラスコに化合物3(0.65g、0.57mmol)を入れ、クロロホルム35ml加えた。その後トリフルオロメタンスルホン酸ナトリウム(0.238g、1.38mmol)のエタノ−ル溶液(15ml)を加え、室温で6時間撹拌した。得られた反応溶液を洗浄液中にBr−が含まれなくなるまで水で洗浄した。有機相を硫酸ナトリウムで脱水後、濾去し、エバポレ−タ−及び真空ラインを用いて濃縮乾固した。薄黄色透明の粘性のある液体を得た。収量0.61g(84.7%)。化合物5は1H−NMR、ESI−MS及びFT−IRスペクトルにより同定した。図5に化合物5の1H−NMRスペクトルを示す。
【0032】
【化7】
【0033】
なお、化合物5は、一般式(I)において、MがP原子、R1が結合性官能基として−SS−基を有する−(CH2)12SS(CH2)12−基、R2〜R4がC6H13−基、X−がCF3SO3−(TfO−)である。
(実施例6)
<鉄複核錯体の合成>
<化合物6の合成>
2−アミノ−6−メチルピコリン(0.54mol)とトリエチルアミン(0.54mol)をジクロロメタン(300ml)に溶かし、撹拌しながらピコリン酸クロリド(0.54mol)のジクロロメタン溶液(100ml)を滴下した。反応の様子をTLC(展開溶媒;ヘキサン:酢酸エチル=4:1)により追跡し、原料のスポットが消失したことで反応終了を確認した。その後、反応に伴って析出した白色粉末を濾去し、ジクロロメタン溶液に対して抽出操作を行った。まず有機層を水で3回洗浄し、続いて1Nの塩酸、0.5Nの炭酸水素ナトリウム水溶液、飽和食塩水の順に洗浄した後、無水硫酸マグネシウムを加えて数時間放置した。無水硫酸マグネシウムを濾去し、ロ−タリ−エバポレ−タ−で減圧濃縮して得られた粗結晶をジエチルエ−テルに溶解させ、数日放置することにより析出した無色柱状結晶を濾過して集めることにより、目的物を得た。収量62.6g(59.7%).化合物6は1H−NMRにより同定した。図6に化合物6の1H−NMRスペクトルを示す。
【0034】
【化8】
【0035】
(実施例7)
<化合物7の合成>
化合物6(0.16mol)を四塩化炭素(300ml)に溶かし、撹拌しながらN−ブロモスクシンイミド(NBS;5.3x10−2mol)と2、2’−アゾビスイソブチロニトリル(AIBN;3.0x10−3mol)を加え、80℃で還流を開始した。開始2時間後にNBS(5.3x10−2mol)とAIBN(3.0x10−3mol)を追加し、次いで、NBS(2.7x10−2mol)とAIBN(2.0x10−3mol)を1時間毎に2回加え、更に1時間還流した後、放冷し、濾過した四塩化炭素溶液に対して洗浄操作を行った。まず有機層を0.5Nの炭酸水素ナトリウム水溶液、飽和食塩水の順で洗浄し、無水硫酸マグネシウムを加えて数時間放置した。無水硫酸マグネシウムを濾去し、ロ−タリ−エバポレ−タ−で減圧濃縮して得られた褐色油状物をシリカゲルカラム(溶離液;ヘキサン:酢酸エチル=10:1)により精製し、目的物を含む褐色油状物(20.75g)を得た。この褐色油状物は目的物の化合物7だけでなく原料である化合物6との混合物であったため、化合物7の含有量は1H−NMRスペクトルにより計算した。収量13.3g(30.8%)。化合物7は1H−NMRにより同定した。図7に化合物7の1H−NMRスペクトルを示す。
【0036】
【化9】
【0037】
(実施例8)
<化合物8の合成>
化合物7(4.9x10−2mol)をN、N−ジメチルホルムアミド(50ml)に溶かし、撹拌しながらフタルイミドカリウム(6.26x10−2mol)を加え130℃で2時間還流した。目的物の生成はTLC(展開溶媒;ヘキサン:酢酸エチル=4:1)により確認した。反応終了を原料である化合物7のTLCスポットが消失したことで確認した後、放冷し、濾過した。N、N−ジメチルホルムアミド溶液に水(50ml)を加えて、クロロホルムで抽出操作を行った。まず目的物を有機層に抽出した後、有機物を飽和食塩水で洗浄し、無水硫酸マグネシウムを加えて数時間放置した。無水硫酸マグネシウムを濾去し、ロ−タリ−エバポレ−タ−で減圧濃縮して得られる褐色粉末をクロロホルム(100ml)に溶解させ、ジエチルエ−テル(200ml)を加えて冷蔵庫にて24時間放置することにより、褐色結晶が析出した。収量(43.3%).化合物8は1H−NMRにより同定した。図8に化合物8の1H−NMRスペクトルを示す。
【0038】
【化10】
【0039】
(実施例9)
<化合物9の合成>
化合物8(2.12x10−2mol)をエタノ−ル(100ml)に完全に溶解させた後、撹拌しながらヒドラジン・一水和物(4.24x10−2mol)を加え、80℃で4時間還流した。目的物の生成はTLC(展開溶媒;ヘキサン:酢酸エチル=4:1)およびニンヒドリン呈色反応により確認した。反応終了を原料である化合物8のTLCスポットが消失したことで確認した後、放冷し、濾過後、濾液をロ−タリ−エバポレ−タ−で減圧濃縮して得られた白色粉末をクロロホルム(50ml)に溶解させた。不溶なフタルヒドラジドを濾別し、濾液を減圧濃縮して得られた白色粉末を再度クロロホルム(50ml)より冷蔵庫内にて再結晶させた。得られた結晶を濾取史し、数時間真空乾燥させることにより化合物9を得た。収量2.70g(69.3%)。化合物9は1H−NMRにより同定した。図9に化合物9の1H−NMRスペクトルを示す。
【0040】
【化11】
【0041】
(実施例10)
<化合物10の合成>
化合物9(0.10mol)を四塩化炭素(250ml)に溶かし、撹拌しながらNBS(3.0x10−2mol)とAIBN(6.0x10−3mol)を加え80℃で還流した。2時間後に更にNBS(3.0x10−2mol)とAIBN(6.0x10−3mol)を加え、その後1時間ごとにNBS(3.0x10−2mol)とAIBN(6.0x10−3mol)を計6回加えた。12時間後、反応溶液を放冷後、濾過し、濾液を減圧濃縮して得られた赤褐色油状物をメタノ−ル(100ml)溶解した。この溶液に水酸化カリウム(0.24mol)を含む水溶液(50ml)を加え、2時間撹拌した。TLC(展開溶媒;ヘキサン:酢酸エチル=4:1)にて化合物7のスポットが消失し、それより下に新たなスポットが出現したことで反応収量を確認した。この溶液を減圧濃縮後、ヘキサン(100ml)に溶かし、有機層を6M塩酸により洗浄し、水層に不純物を抽出した。続いて0.5Mの炭酸水素ナトリウム水溶液を塩基性になるまで加え、水層を取り除いた後、有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムを加えて乾燥させた。硫酸マグネシウムを濾別後、ロ−タリ−エバポレ−タ−で減圧濃縮することにより白色結晶が析出したので、これを数時間真空乾燥させた。収量12.0g(46.0%).化合物10は1H−NMRにより同定した。図10に化合物10の1H−NMRスペクトルを示す。
【0042】
【化12】
【0043】
(実施例11)
<化合物11の合成>
化合物10(3.42x10−2mol)をアセトン(150ml)に溶かし、硝酸銀(11.37x10−2mol)を含む水溶液(40ml)を加え、室温・遮光条件下にして16時間撹拌した。目的物の生成をTLC(展開溶媒;ヘキサン:酢酸エチル=4:1)で確認した。不純物を濾別した後、濾液をロ−タリ−エバポレ−タ−により減圧濃縮し、得られた黄色油状物にジクロロメタンを加えた。まず有機層を水で洗浄し、0.5N炭酸水素ナトリウム水溶液を塩基性になるまで加えた。有機相を抽出後、飽和食塩水で洗浄し、無水硫酸マグネシウムを加えて乾燥させた。硫酸マグネシウムを濾別し、ロ−タリ−エバポレ−タ−により減圧濃縮後、数時間真空乾燥することで黄色油状物を得た。収量(71.6%).化合物11は1H−NMRにより同定した。図11に化合物11の1H−NMRスペクトルを示す。
【0044】
【化13】
【0045】
(実施例12)
<化合物12の合成>
化合物10(1.20x10−2mol)を脱水メタノ−ル(50ml)に溶かし、そこに化合物7(1.29x10−2mol)を加えて耐圧ビンに入れ、酸化白金(10mg)を触媒として、水素圧3atm、室温で接触水素化反応を行った。目的物の生成はTLC(展開溶媒;クロロホルム:メタノ−ル=7:1)およびニンヒドリン呈色反応により確認した。濾過により酸化白金を取り除いた後、濾液をロ−タリ−エバポレ−タ−で減圧濃縮することにより黄色油状物を得た。これをシリカゲルカラム(溶離液;クロロホルム)により精製し、目的物を含む橙色油状物を得た。これをアセトン(50ml)に溶解し、ゆっくりと溶媒を蒸発させることにより析出した無色結晶を濾過し、数時間減圧乾燥させることにより目的物を得た。収量1.47g(30.9%).化合物12は1H−NMRにより同定した。図12に化合物12の1H−NMRスペクトルを示す。
【0046】
【化14】
【0047】
(実施例13)
<化合物13の合成>
2、6−ビス(ヒドロキシメチル)パラクレゾ−ル酢酸(1.8x10−3)と少量のピリジンをジクロロメタン(15ml)に溶かし、塩化チオニル(2.1x10−2)を含むジクロロメタン溶液(30ml)を室温下で撹拌しながらゆっくりと滴下した。得られた溶液を30〜35℃で撹拌し、ジクロロメタンを揮発させることで、目的の黄白色粉末を得た。収量3.68g(100%)。化合物13は1H−NMRにより同定した。
【0048】
【化15】
【0049】
(実施例14)
<化合物14の合成>
化合物13(7.4x10−3mol)をテトラヒドロフラン(20ml)に溶かし、窒素雰囲気下、氷浴中で撹拌しながら、化合物12(1.8x10−2mol)、トリエチルアミン(1.8x10−2mol)を含むテトラヒドロフラン溶液(70ml)をゆっくりと滴下した。得られた溶液は室温で3日間撹拌した。生成したトリエチルアミン塩酸塩を濾別し、ロ−タリ−エバポレ−タ−により減圧濃縮することにより、黄色油状物を得た。得られた油状物をジクロロメタンに溶かし、水、飽和食塩水を用いて洗浄し、無水硫酸マグネシウムで乾燥させた。硫酸マグネシウムを濾別後、減圧濃縮し、得られた黄色油状物をシリカゲルカラムにより、化合物13(溶離液;ヘキサン:酢酸エチル=4:1)と目的物(溶離液;ヘキサン:酢酸エチル=4:1)に分離・精製し、黄白色粉末を得た。収量5.76g(84%)。化合物14は1H−NMRにより同定した。図13に化合物14の1H−NMRスペクトルを示す。
【0050】
【化16】
【0051】
(実施例15)
<化合物15の合成>
アルゴン雰囲気下、化合物14(3.6x10−5mol)をジクロロメタン及び少量のメタノ−ル混合溶液に溶かし、Fe(CF3SO3)2(7.1x10−5mol)、安息香酸ナトリウム (7.1x10−5mol)を含むメタノ−ル溶液を添加した。続いて、この溶液にトリエチルアミン(3.6x10−5mol)を加え、数時間放置した後、ゆっくりと減圧濃縮した。得られた残渣を少量のメタノ−ルに溶かし、溶媒をゆっくりと蒸発させることで、化合物15の黄色板状結晶を得た。収量10mg(19%)。化合物15はIR、UV/vis、ESI−TOF MSスペクトル、およびX線結晶構造解析により同定した。図14に化合物15の結晶構造のORTEP図を示す。
【0052】
【化17】
【0053】
(実施例16)
<金蒸着基板の作製>
まず天然雲母片(mica)を14×14mm2四方に切り出し、セロテ−プ(登録商標)などによりmica表面を剥がすことで、清浄で平滑な面を露出させた。続いてφ1mmのAu線(99.999%)を適量切り出し、アセトンで湿らせたキムワイプで汚れを落とした後に、適当な大きさに丸めて蒸着装置内のバスケットに設置した。切り出したmicaをAu線が入ったバスケット上部のサンプルホルダ−に設置した。続いて蒸着装置内を〜10−5Paまで減圧し、ランプヒ−タ−によりmicaを300℃で3時間加熱した。続いてバスケット内のAu線を加熱し、蒸着速度約0.9As−1で金薄膜が1000Aになるまで蒸着した。得られた金基板は使用前に水素炎によるアニ−ル処理を行った。
(実施例17)
<イオン液体修飾金基板の作製>
化合物5の1mMエタノ−ルまたはアセトニトリル溶液を調製し、あらかじめエタノ−ルまたはアセトニトリルで表面をリンスした蒸着金基板上をこの溶液中に1日間金基板を浸漬させた。続いて金基板をエタノ−ルまたはアセトニトリルでリンスし、数時間風乾させることで修飾基板1を得た。得られたイオン液体修飾基板は、FT−IRスペクトル(RAS法)、およびサイクリックボルタンメトリ−によりイオン液体の修飾を確認した。図15に修飾基板のFT−IRスペクトルを示す。FT−IRスペクトルにより、金基板上に修飾されたイオン液体に特徴的なC−H結合に由来する伸縮振動および変角振動を2940、1464cm−1に、対陰イオンに由来する伸縮振動を1265、1154、1029cm−1に観測した。またサイクリックボルタンメトリ−測定において、バックグラウンド電流が小さくなったこと、および0.5M KOH水溶液中において−0.94V(vs.Ag/AgCl)にAu−S結合の還元脱離反応に由来する還元波を観測したことから、化合物5は金電極上に−SS−結合によって化学修飾されていることが示唆された。また、この還元脱離波の電流値の解析から、化合物5は9.1(±4.3)×10−11mol・cm−2の被覆率で表面に修飾されていることが判明した。一般的な長鎖アルカンチオ−ル(C18H37SH)を用いて同様の実験を行ったところ,その表面被覆率は5.4×10−10mol・cm−2となり、それに比べると被覆率は1/6程度であった。従って,イオン液体1分子あたり、アルカンチオ−ルの大きさに換算して6分子分の空間が電極表面に存在していることが示唆された。
【0054】
【化18】
【0055】
(実施例18)
<イオン液体修飾金基板への鉄複核錯体の導入法>
イオン液体修飾基板上に、アルゴン雰囲気下において、10mMの鉄複核錯体(化合物15)を含むメタノ−ル溶液を滴下し、数日間放置した。その後、電極表面を塩化メチレン、およびアセトンを用いて洗浄後、基板を真空乾燥させた。イオン液体修飾基板への鉄複核錯体の導入は、FT−IRスペクトル(RAS法)、およびサイクリックボルタンメトリ−によりイオン液体修飾基板への鉄複核錯体の導入を確認した。図16に鉄複核錯体が導入されたイオン液体修飾基板のサイクリックボルタモグラムを示す。
【0056】
【化19】
【0057】
FT−IRスペクトルでは、導入された化合物15に由来する特徴的な吸収(アミド部位のC=O伸縮振動およびC−N伸縮振動)が1526cm−1および1109cm−1に観測された。サイクリックボルタモグラムではイオン液体修飾電極に導入された化合物15に由来する酸化還元波が0.01V(vs.Fc・Fc+)に観測された。以上の結果より化合物15はイオン液体修飾電極上に固定化されたことが示唆された。
(比較例1)
非特許文献1において、鉄複核錯体を化学結合により電極上に修飾した場合の電気化学測定から、錯体の酸化還元波は−0.78V(vs.Fc・Fc+)に観測された。また均一溶液中では、本鉄複核錯体は0.23V(vs.Fc・Fc+)に酸化還元波が観測された。その差は約1.01Vである。一方、実施例18では、酸化還元波は0.01V(vs.Fc・Fc+)に観測され、その差は約0.22Vである。従って、直接化学結合を利用して鉄複核錯体を修飾した場合に比べて、イオン液体修飾電極を利用した場合は、溶液中のものにかなり近い状態で存在していることが示唆された。
(実施例19)
<イオン液体修飾金基板へのコバルト単核錯体の導入法>
イオン液体修飾基板上に、10mMのコバルト単核錯体を含む水溶液を滴下し、数日間放置した。その後、電極表面をMilli−Q水を用いて洗浄後、基板を真空乾燥させた。イオン液体修飾基板へのコバルト単核錯体の導入は、FT−IRスペクトル(RAS法)、およびサイクリックボルタンメトリ−によりイオン液体修飾基板へのコバルト単核錯体の導入を確認した。図17にコバルト単核錯体が導入されたイオン液体修飾基板のサイクリックボルタモグラムを示す。
【0058】
【化20】
【0059】
FT−IRスペクトルでは、導入されたコバルト錯体に由来する特徴的な吸収(アミド部位のC=O伸縮振動)が1537cm−1に観測された。サイクリックボルタモグラムではイオン液体修飾電極に導入されたコバルト錯体に由来する酸化還元波が−0.70V(vs.Fc・Fc+)に観測された。以上の結果から、コバルト錯体はイオン液体修飾電極上に固定化されたことが示唆された。均一溶液中でのコバルト錯体の酸化還元波は−0.79V(vs.Fc・Fc+)に観測されるが、実施例19との差は殆どないことから、コバルト錯体の性質の改変は無いことが示唆される。
(実施例20)
<イオン液体修飾金基板へのフェロセンの導入法>
化合物5(0.16mmol)に市販のフェロセン(0.16mmol)を溶解し、得られた溶液を金基板上に滴下し、数日間放置した。続いて、基板表面をクロロホルム、アセトン、Milli−Q水で洗浄後、風乾させた。イオン液体の修飾、およびフェロセンの導入は、サイクリックボルタンメトリ−により確認した。図18にフェロセンが導入されたイオン液体修飾基板のサイクリックボルタモグラムを示す。
【0060】
【化21】
【0061】
サイクリックボルタモグラムではイオン液体修飾電極に導入されたフェロセンに由来する酸化還元波が0.42V(vs.Ag/AgCl)に観測され、フェロセンはイオン液体修飾電極上に固定化されたことが示唆された。均一溶液中でのフェロセンの酸化還元波は0.42V(vs.Ag・AgCl)に観測されるが、実施例20との差は殆どないことから、フェロセンの性質の改変は無いことが示唆される。
(実施例21)
<イオン液体修飾金基板へのヘキサシアノ鉄(III)錯体の導入法>
0.1MのNaClO4電解質溶液に市販のヘキサシアノ鉄(III)錯体(K3[Fe(CN)6])を0.5mMとなるように溶解させた。続いて、イオン液体修飾基板を作用極、対極を白金線、参照極を銀/塩化銀電極として、この溶液のサイクリックボルタンメトリ−測定を数回繰り返し行った。測定終了後にイオン液体修飾基板をミリQ水で洗浄し、目的の基板を得た。イオン液体修飾電極へのヘキサシアノ鉄(III)錯体の導入は、サイクリックボルタンメトリ−測定により確認した。図19にヘキサシアノ鉄(III)錯体が導入されたイオン液体修飾基板のサイクリックボルタモグラムを示す。
【0062】
【化22】
【0063】
サイクリックボルタモグラムではイオン液体修飾電極に導入されたヘキサシアノ鉄(III)錯体に由来する酸化還元波が0.40V(vs.Ag/AgCl)に観測され、ヘキサシアノ鉄(III)錯体はイオン液体修飾電極上に固定化されたことが示唆された。均一溶液中でのヘキサシアノ鉄(III)錯体の酸化還元波は0.20V(vs.Ag・AgCl)に観測されるが、実施例21との差は殆どないことから、ヘキサシアノ鉄(III)錯体の性質の改変は無いことが示唆される。
(実施例22)
<イオン液体修飾金基板へのヘキサアンミンルテニウム(II)錯体の導入法>
0.1MのNaClO4電解質溶液に市販のヘキサアンミンルテニウム(II)錯体([Ru(NH3)6]Cl2)を0.5mMとなるように溶解させた。続いて、イオン液体修飾基板を作用極、対極を白金線、参照極を銀/塩化銀電極として、この溶液のサイクリックボルタンメトリ−測定を数回繰り返し行った。測定終了後にイオン液体修飾基板をミリQ水で洗浄し、目的の基板を得た。イオン液体修飾電極へのヘキサアンミンルテニウム(II)錯体の導入は、サイクリックボルタンメトリ−測定により確認した。図20にヘキサアンミンルテニウム(II)錯体が導入されたイオン液体修飾基板のサイクリックボルタモグラムを示す。
【0064】
【化23】
【0065】
サイクリックボルタモグラムではイオン液体修飾電極に導入されたヘキサアンミンルテニウム(II)錯体に由来する酸化還元波が−0.20V(vs.Ag/AgCl)に観測され、フェロセンはイオン液体修飾電極上に固定化されたことが示唆された。均一溶液中でのヘキサアンミンルテニウム(II)錯体の酸化還元波は−0.13V(vs.Ag・AgCl)に観測されるが、実施例22との差は殆どないことから、ヘキサアンミンルテニウム(II)錯体の性質の改変は無いことが示唆される。
【特許請求の範囲】
【請求項1】
基材表面に目的化合物を修飾するためのイオン液体であって、
下記の一般式(I)で表される構造単位を含む有機リン型もしくは4級アミン型のイオン液体。
【化24】
(一般式(I)中、MはP元素またはN元素である。一般式(I)中、R1〜R4は、それぞれ独立に、炭素数1〜30のアルキル基、炭素数2〜30のアルケニル基、炭素数2〜30のアルキニル基、炭素数1〜30のアルコキシアルキル基、炭素数1〜30のアミノアルキル基、炭素数1〜30のパ−フルオロアルキル基、炭素数6〜30のアリ−ル基、炭素数7〜30のアラルキル基、またはカルボニル基を有するアルキル基、アルケニル基、アリ−ル基もしくはアラルキル基を表し、またはRnとRn+1(nは1〜3の整数)が結合して環状構造を有していても良い。ただし、一般式(I)中のR1〜R4の少なくとも1つは、少なくとも1つの結合性官能基(−SH基、−SS−基、−S−基、−COOH基、−NH2基、シラノ−ル基、リン酸基、アルケニル基、アルキニル基、またはアジ基)を有する。一般式(I)中、X−は対陰イオンを表す。)
【請求項2】
請求項1に記載のイオン液体が基材表面に修飾されたイオン液体修飾基材。
【請求項1】
基材表面に目的化合物を修飾するためのイオン液体であって、
下記の一般式(I)で表される構造単位を含む有機リン型もしくは4級アミン型のイオン液体。
【化24】
(一般式(I)中、MはP元素またはN元素である。一般式(I)中、R1〜R4は、それぞれ独立に、炭素数1〜30のアルキル基、炭素数2〜30のアルケニル基、炭素数2〜30のアルキニル基、炭素数1〜30のアルコキシアルキル基、炭素数1〜30のアミノアルキル基、炭素数1〜30のパ−フルオロアルキル基、炭素数6〜30のアリ−ル基、炭素数7〜30のアラルキル基、またはカルボニル基を有するアルキル基、アルケニル基、アリ−ル基もしくはアラルキル基を表し、またはRnとRn+1(nは1〜3の整数)が結合して環状構造を有していても良い。ただし、一般式(I)中のR1〜R4の少なくとも1つは、少なくとも1つの結合性官能基(−SH基、−SS−基、−S−基、−COOH基、−NH2基、シラノ−ル基、リン酸基、アルケニル基、アルキニル基、またはアジ基)を有する。一般式(I)中、X−は対陰イオンを表す。)
【請求項2】
請求項1に記載のイオン液体が基材表面に修飾されたイオン液体修飾基材。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【公開番号】特開2012−167045(P2012−167045A)
【公開日】平成24年9月6日(2012.9.6)
【国際特許分類】
【出願番号】特願2011−28243(P2011−28243)
【出願日】平成23年2月14日(2011.2.14)
【出願人】(304021277)国立大学法人 名古屋工業大学 (784)
【Fターム(参考)】
【公開日】平成24年9月6日(2012.9.6)
【国際特許分類】
【出願日】平成23年2月14日(2011.2.14)
【出願人】(304021277)国立大学法人 名古屋工業大学 (784)
【Fターム(参考)】
[ Back to top ]