
イオン液体担持合成
本発明は、化学用途に用いるための、かつ溶媒及び液体担体の二重の機能を果たしうるイオン液体に関する。このイオン液体は、オリゴペプチド、オリゴサッカライド、及びオリゴヌクレオチドからなる群から選択されるオリゴマーの合成法にそれ自体が役立ち、この方法は、イオン液体が結合したモノマー単位をもたらす反応条件においてイオン液体と第一のモノマーを接触させるステップ;及び、イオン液体が結合した2〜30のモノマー単位を含むオリゴマーをもたらす反応条件において、前記イオン液体が結合したモノマー単位を、少なくとも1つのさらなるモノマー単位と接触させるステップを含む。本方法はそれ自体が、オリゴペプチド、オリゴサッカライド、及びオリゴヌクレオチドの大量製造に役立つ。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、イオン液体担持合成に関する。より詳細には、本発明は、イオン液体担持オリゴペプチド、オリゴサッカライド、及びオリゴヌクレオチド合成に関する。なおさらに詳細には、本発明は、生物活性オリゴペプチドのイオン液体担持合成に関する。
【背景技術】
【0002】
オリゴペプチド、オリゴサッカライド、及びオリゴヌクレオチドの効率的合成は、いくつかの新規なアプローチの開発へと導いている現課題である。溶液相合成を用いた、構造的に明らかにされたオリゴサッカライドの入手方法は、労力がかかり、かつ各段階の後でクロマトグラフィーによる反応生成物の精製を必要とする1。
【0003】
合成オリゴヌクレオチドに対する科学者グループの要求は、過去数十年にわたって指数的に増大してきている。幸いなことに、DNAオリゴヌクレオチドプライマーの豊富な資源は、遺伝子配列解析努力、機能ゲノム科学、及びポリメラーゼ連鎖反応(PCR)に基づく検出方法の膨大な要求を満たしている。さらに、オリゴヌクレオチドは、チップベースDNAマイクロアレイを含めた、治療及び診断用途の開発に広く用いられている、構造生物学及び構造生化学における顕著な進歩が、DNA及びRNA化学での同時に起こっている進歩を通じて達成されている2-6。例えば、リボザイム及びsiRNA研究における当分野の現状は、結晶構造を含め、RNA合成において同時に進歩がなければ可能とはならなかっただろう4。
【0004】
Merrifield7とLetsinger8,9らがオリゴペプチド及びオリゴヌクレオチドそれぞれの合成のための高分子担体の使用を導入して以来、不溶性担体の使用は、有機合成、特にオリゴヌクレオチド、ペプチド、及びさらに最近では炭水化物10-12などの生体高分子の合成において、重要な手段となっている。濾過による過剰な試薬と副生成物の除去を含む容易な精製方法は、簡単な生成物分離を可能にし、自動化を可能にする。大きな成功にもかかわらず、不溶性高分子による不均一性のために、固相合成は、固体担体自体の高コストに加えて不均一な反応条件に典型的に伴う欠点を残しており、これらの化合物の大規模合成を非常にコストのかかるものにしている。
【0005】
高分子担持固相合成における最近の進歩13-15は、複雑なオリゴサッカライドを組み立てるための簡単な方法を提供しており、なぜなら、その方法は、樹脂を単に洗浄することによる過剰な試薬の除去を可能にし、それにより、必要なクロマトグラフィーステップの数を最小にするからである16,17。これは、うまい自動化合成のめざましい例をもたらしている14,18。しかし、固相合成の欠点は、不均一反応を完結させるためにしばしば大過剰の高価な炭化水素単位を必要とし、しかも通常の特性分析方法、例えば、TLC、NMR、及びマススペクトルによって糖−糖カップリング工程を監視することが一層困難であることである。さらに、固相方法によってもたらされる多くの利点にもかかわらず、不溶性高分子と反応条件の不均一な特性は、非線形反応速度論、反応部位の及び/又は反応部位へのアクセスの不均一な分布、溶媒和の問題、及び非効率なカップリング速度、を含めた一連の問題をしばしばもたらす。
【0006】
均一反応条件の再構築と固相合成の欠点のいくつかを克服する目的による代替方法の探索は、可溶性高分子担体の開発へと導いている。近年、可溶性高分子担体の使用がかなりの注目を集めているが、これは、そのような「液相」合成は従来の溶液化学の利点の多くを残すと同時に、生成物の容易な精製の利点をなおも可能にするからである。可溶性のポリエチレングリコール(PEG)、ポリビニルアルコール、及びその他の高分子は全て、オリゴペプチド19及びヌクレオチド11の合成に首尾良く用いられている。さらに、可溶性のポリエチレングリコール(PEG)ポリマーは、オリゴサッカライド12,20-23及び小分子合成24のための担体としても用いられている。しかし、可溶性高分子担体の使用は、低い負荷容量、オリゴサッカライドが結合したポリマーの選択的沈殿の難しさ、より長いペプチドの合成時の貧弱な溶解性、低い水溶性、エーテル溶媒中での溶解性の欠如24、及び精製に必要とされるエネルギーを大量に消費する冷却、の欠点がある。
【0007】
さらに最近、フッ素化(フッ素)可溶性担体に基づく新しい溶液相合成が提唱されている25。このアプローチはフッ素系溶媒(すなわち、パーフルオロアルカン類)中でのフッ素系担体とフッ素化された試薬との好ましい可溶性に基づいている。非フッ素化試薬は、フッ素系有機溶媒分配26a−d又はフッ素系シリカゲルに基づく固相抽出(SPE)26e−hによって、担持された生成物から容易に分離されうる。このアプローチは、通常容易に入手できないフッ素化化合物の使用を必要とする。精製は、予め混和性のフッ素系溶媒と有機溶媒との間の相分離を引き起こす温度切り替えによって達成され、それによって分離を促進する。有機合成のためのフッ素系相方法の使用は、オリゴペプチド27、オリゴサッカライド28、及び小分子25について実証されている。オリゴサッカライド合成の場合は、フッ素系溶媒へのサッカライド結合担体の溶解性は、サッカライド単位の数が増加するに従って低下するフッ素含量に左右されることが実証されている。パーフルオロアルカン溶媒のコスト、特殊なフッ素化された試薬の必要性、及び温度切り替えに伴うエネルギーコストは、フッ素系相有機合成の広い応用を阻害する潜在的制限である。
【0008】
イオン液体(Ionic liquid(IL))は、有機反応のための環境にやさしい反応媒体として近年大きな注目を集めている29。イオン液体の現実的な定義は、水の沸点より低い、しばしば非常に低い融点を有する塩、というものである。イオン液体の共通の性質は、多くは有機カチオンと無機アニオンとを有することである。イオン液体の非限定的例は、ハロゲン化物、テトラフルオロボレート、及びヘキサフルオロホスフェートの、アルキルイミダゾリウム及びピリジニウム塩である。多くの化学反応が、いくつかの酵素反応も含めて、イオン液体中で実施されうる30。室温イオン液体は、電気化学技術31、化学抽出32、及びその他の工業的方法33のための媒体として広く検討されてもいる。これは、イオン液体のいくつかの興味ある化学的及び物理的性質:すなわち、高い熱的及び化学的安定性、不燃性、測定可能な蒸気圧の欠如、及び高い負荷特性、によるものである。多くの場合に、イオン液体は容易にリサイクルできる。カチオン又はアニオンの構造を修正することによって、イオン液体の溶解性は容易に変えることができ、それにより、それらが有機媒体並びに無機媒体から相分離し、分離及び精製を容易にするようにできる。このことが、イオン液体が有機合成のための実施可能な可溶性機能性担体として機能しうる可能性を提供している。基質の溶解度も変えることができる34。最近の報告は、小分子35,37,38及び小ペプチド39のためのILSS(Ionic Liquid Supported Synthesis;イオン液体担持合成)の有効性を首尾良く実証している。回収可能且つリサイクル可能なイオン液体担持触媒を開発する可能性もまた、探索されている36。
【0009】
有機反応のためのマトリクス(すなわち、溶媒)としてのイオン液体の使用は、以前、Vaultierら(WO2004/029004)によって記載されている。この有機反応は、可溶性担体として働く「オニウム塩」として公知の官能化された塩類を用いて行われる。このイオン液体は、官能化された塩の可溶化を確実にし、反応が均一条件下で行われうる。Vaultierら(WO2005/005345)は、以前、有機反応のための可溶性担体として、「オニウム塩担持有機合成(Onium Salt Supported Organic Synthesis)」と名付けられた、少なくとも1種の有機溶媒中に溶解したオニウム塩(すなわち、イオン液体)の使用、を記載している。
【特許文献1】国際公開WO2004/029004号パンフレット
【特許文献2】国際公開WO2005/005345号パンフレット
【発明の開示】
【発明が解決しようとする課題】
【0010】
したがって、以下のものに限定されないが、オリゴペプチド、オリゴサッカライド、及びオリゴヌクレオチドを含めたオリゴマーの合成のための改良方法に対する必要性が依然として存在し、この方法は、固相及び溶液相合成の利点を結合させ、均一溶液相条件を含み、且つ生成物の容易な精製を可能にするものである。
【0011】
本発明は、これら及びその他の必要性を満たすことを目的とする。
【0012】
本明細書の記載は多くの文献を参照し、その内容は、その全体を参照により本明細書に援用する。
【課題を解決するための手段】
【0013】
[発明のまとめ]
本発明は、イオン液体(すなわちイオン性液体)担持合成に関する。
【0014】
ある態様では、本発明は、イオン液体によって担持された、オリゴペプチド、オリゴサッカライド、及びオリゴヌクレオチド、の合成に関する。
【0015】
ある態様では、本発明は、オリゴペプチド、オリゴサッカライド、及びオリゴヌクレオチドを含むがこれらに限定されないオリゴマーの合成のためのイオン液体担持方法に関する。
【0016】
ある態様では、本発明は、オリゴペプチド、オリゴサッカライド、及びオリゴヌクレオチドからなる群から選択されるオリゴマーの合成方法に関するものであり、この方法は、イオン液体に結合したモノマー単位をもたらす反応条件下において、第一のモノマー単位をイオン液体と接触させるステップ;及び2〜30のモノマー単位を含むイオン液体結合オリゴマーをもたらす反応条件下において、前記イオン液体に結合したモノマー単位を、少なくとも1つのさらなるモノマー単位と接触させるステップを含む。次に、このイオン液体結合オリゴマーはイオン液体から切り離されて、遊離したオリゴマーをもたらす。
【0017】
さらなる態様では、本発明は、オリゴペプチド、オリゴサッカライド、及びオリゴヌクレオチドからなる群から選択されるオリゴマーの合成方法に関し、本方法は、モノマー単位に対してイオン液体を用いて、2〜30のモノマー単位を含むオリゴマーをもたらすことを含む。
【0018】
さらなる態様では、本発明は、化学合成に用いるための、且つ溶媒及び液体担体の両方の二重機能を果たしうる液体担体に関する。具体的態様では、イオン液体は、ヘテロ環式又は置換ヘテロ環式第四級窒素含有有機カチオン、ヘテロ環式又は置換ヘテロ環式第四級ホスホニウム含有有機カチオン、又はヘテロ環式又は置換ヘテロ環式の三価のスルホニウム含有有機カチオン;及び、前記有機カチオンの電荷と釣り合うアニオン、を含む有機塩である。より詳細な態様では、有機カチオンは、N−置換ピリジン及び1,3−ジ置換イミダゾールからなる群から選択される。有機カチオン上の電荷と釣り合うアニオンは、Cl−、Br−、BF4−、PF6−、SbF6−、CuCl2−、及びAlCl4−からなる群から選択できる。その他の適切なアニオンも使用でき、充分に当業者の能力の範囲内である。
【0019】
具体的な態様では、本発明は、オリゴペプチドのイオン液体担持合成に関する。なお、さらに具体的な態様では、本発明は、ペンタペプチドLeu5−エンケファリンの合成に関し、その構造は以下に示す。
【化1】
【0020】
本発明の、上述の及びその他の目的、利点、並びに特性は、付属の図面を参照にして、参考のみの例として提供する本発明の説明のための態様の以下の非制限的説明を読むことで、さらに明らかになろう。
【0021】
[実例態様の詳細な説明]
本明細書中で用いる用語の明確かつ一貫した理解を提供するために、以下に多くの定義を示す。さらに、他に定義されていない限り、本明細書で用いる全ての技術的及び科学的用語は、本発明が関係する技術分野の当業者に通常理解されているものと同じ意味を有する。
【0022】
請求項及び/又は明細書中の「含む、含有する」の用語との関連で用いられる用語は、単数を意味することができるが、「1以上」、「少なくとも1」、及び「1又は1より多く」の意味ともなる。同様に、「別の」の用語は、少なくとも第二の、もしくはそれ以上のものを意味することができる。
【0023】
本明細書及び請求項で用いるように、「含む、含有する」(及び、含む、含有する、の全て活用形)、「有する」(及び、有する、の全ての活用形)、「包含する」(及び、包含する、の全ての活用形)、の用語は、包含的、すなわち限定のないものであり、追加の記載されていない要素又は方法ステップを排除するものではない。
【0024】
「約」の用語は、その値が、その値を測定するために用いた装置又は方法についての内在する誤差の変動を含んでいることを意味するために用いる。
【0025】
「アミノ酸(類)」の用語は、本明細書で用いるように、天然アミノ酸のL及びD異性体の両方、並びにペプチドの合成アナログを調製するためにペプチド化学で用いられるその他の非タンパク質関連アミノ酸のL及びD異性体の両方を含むものとして理解される。天然アミノ酸の例には、グリシン、アラニン、バリン、ロイシン、イソロイシン、セリン、及びトレオニンが含まれるが、これらに限定されない。非タンパク質関連アミノ酸の例には、ノルロイシン、ノルバリン、シクロヘキシルアラニン、ビフェニルアラニン、ホモフェニルアラニン、ナフチルアラニン、ピリジルアラニン、及び置換フェニルアラニン類(アルコキシ、ハロゲン、及びニトロ基を含むがこれらに限定されない1以上の置換基で置換されたもの)が含まれるが、これらに限定されない。ベータ及びガンマアミノ酸も、「アミノ酸」の用語の範囲内である。ペプチド合成で用いられる標準の保護基で保護されたアミノ酸もまた、「アミノ酸」の用語の範囲内である。これらの化合物は、ペプチド化学分野の当業者に公知である。
【0026】
「ヌクレオチド」の用語は、本明細書で用いるように、個別のヌクレオチド又はヌクレオチドの変種を言うものとして理解され、プリン又はピリミジン、リボース又はデオキシリボース糖残基、及びホスフェート基又は(オリゴヌクレオチド又はポリヌクレオチド内のヌクレオチドの場合は)ホスホジエステル結合を含む分子又は大きな核酸分子内の個々の単位を意味する。「ヌクレオチド」の用語はまた、(a)別の連結基、(b)プリンの類似形、(c)ピリミジンの類似形、又は(d)類似の糖、から選択される少なくとも1つの修飾を含む「修飾ヌクレオチド」も包含する。オリゴヌクレオチド合成に通常用いられる標準保護基で保護されたヌクレオチドも、「ヌクレオチド」の用語の範囲内である。これらの化合物は、ヌクレオチド化学分野の当業者に公知である。
【0027】
「サッカライド」の用語は、本明細書で用いるように、実験式(CH2O)n(式中、nは整数、典型的には3より大きな整数である)を有する、ポリヒドロキシアルデヒドもしくはポリヒドロキシケトン又はそれらの誘導体である炭水化物を言うものとして理解される。モノサッカライド、すなわち単糖類は、単一のポリヒドロキシアルデヒド又はポリヒドロキシケトン単位からなる。モノサッカライドには、リボース、2-デオキシ-リボース、グルコース、マンノース、キシロース、ガラクトース、フコース、フルクトースなどが含まれるがこれらに限定されない。ジサッカライドは、グリコシド結合によって結合された2つのモノサッカライド単位を含む。ジサッカライドには、例えば、スクロース、ラクトース、マルトース、セロビオースなどが含まれる。オリゴサッカライドは、典型的には、グリコシド結合で結合された2〜10のモノサッカライド単位を含む。ポリサッカライド(グリカン)は、典型的には10より多くのそのような単位を含み、ヘパリン、ヘパラン硫酸、コンドロイチン硫酸、デルマタン硫酸、及びそれらのポリサッカライド誘導体が含まれるがこれらに限定されない。「糖」の用語は、一般に、モノ−、ジ−、又はオリゴサッカライドをいう。サッカライドは、例えば、置換された、グルコサミン、ガラクトサミン、アセチルグルコース、アセチルガラクトース、N−アセチルグルコサミン、N−アセチル−ガラクトサミン、ガラクトシル−N−アセチルグルコサミン、N−アセチルノイラミン酸(シアリン酸)などであってもよい。サッカライドは、例えば、ヌクレオシド、ヌクレオチド、ポリヌクレオチド、DNA、RNAなどのサッカライド残基のように、より大きな分子の構成要素として存在することもできる。本明細書で用いるように、「サッカライド」の用語は、修飾されたサッカライド、例えば、(a)H、NH2、ハロゲン、アルキル、アリールを含むがこれらに限定されない置換基による、1つ以上のOH基の置換;(b)アルデヒド、ケトン、酸、エステル、及びそれらの誘導基への1つ以上のOH基の酸化、から選択される少なくとも1種の修飾を含むもの、を包含するものとしても理解される。オリゴサッカライド合成に通常用いられる標準保護基によって保護されたサッカライドも、「サッカライド」の用語の範囲内である。これらの化合物は、当業者に公知である。
【0028】
「伸長(する)オリゴペプチド鎖」、「伸長(する)オリゴサッカライド鎖」、及び「伸長(する)オリゴヌクレオチド鎖」の用語は本明細書でも用いるように、任意で適切に保護されていてもよいアミノ酸、サッカライド、又はヌクレオチドの順次の付加によって調製される鎖をいう。各反応サイクルの後で、この伸長オリゴペプチド鎖、伸長オリゴサッカライド鎖、又は伸長オリゴヌクレオチド鎖は、少なくとも1個のアミノ酸、サッカライド、又はヌクレオチドだけ長さが増大し、次の反応サイクルのための出発物質となる。本明細書で用いるように、この用語は、出発物質又は生成物のいずれをもいうことができ、具体的な前後関係においてこの用語によって意図されるものを当業者は理解しよう。
【0029】
本明細書で用いるとおり、「アルキル基」の用語は、飽和で一価の非分岐又は分岐した炭化水素鎖をいうものとして理解される。アルキル基の例には、(C1〜C6)アルキル基が含まれるが、これに限定されない。(C1〜C6)アルキル基の例には、メチル、エチル、プロピル、イソプロピル、2-メチル-1-プロピル、2-メチル-2-プロピル、2-メチル-1-ブチル、3-メチル-1-ブチル、2-メチル-3-ブチル、2,2-ジメチル-1-プロピル、2-メチル-1-ペンチル、3-メチル-1-ペンチル、4-メチル-1-ペンチル、2-メチル-2-ペンチル、3-メチル-2-ペンチル、4-メチル-2-ペンチル、2,2-ジメチル-1-ブチル、3,3-ジメチル-1-ブチル、2-エチル-1-ブチル、ブチル、イソブチル、t-ブチル、ペンチル、イソペンチル、ネオペンチル、及びヘキシル、が含まれるがこれらに限定されない。
【0030】
本明細書で用いるとおり、「アリール基」の用語は、環内に0〜4個のヘテロ原子を含んでいてもよい、5−、6−、及び7−員環芳香族基をいい、例えば、フェニル、ピロリル、フリル、チオフェニル、イミダゾリル、オキサゾリル、チアゾリル、トリアゾリル、ピラゾリル、ピリジル、ピラジニル、ピリダジニル、及びピリミジニルなどをいう。環構造中にヘテロ原子を有するこれらのアリール基は「アリールヘテロ環」又は「ヘテロ芳香族」ともいうことができる。この芳香族環は、1以上の環上位置で置換されていることができる。アリール基はまた、多環式基の一部であることもできる。例えば、アリール基には、ナフチル、アントラセニル、キノリル、インドリル、などの縮合芳香族残基が含まれる。
【0031】
本明細書で用いるように、「ハロゲン」の用語は、フッ素、塩素、臭素、又はヨウ素をいうものとして理解される。したがって、用語「ハロ」は、フルオロ、クロロ、ブロモ、及びヨードを包含するものとして理解される。
【0032】
本発明における保護基は、オリゴペプチド合成、オリゴヌクレオチド合成、及びオリゴサッカライド合成に関連して用いられる。保護基は、アミノ酸、サッカライド、又はヌクレオチドのいずれかのモノマーの反応性末端をブロックする。化学合成の性質は、どの反応性基が保護基を必要とするかを決定するであろう。具体的使用にかかわらず、保護基は、別の試薬と反応することから分子上の残基を保護するために用いられる。本発明で用いられる保護基は、以下の性質を有する:保護基は、選択した試薬が、保護基が結合している基を修飾することを防止する;保護基はその合成反応条件に対して安定である(すなわち、保護基は分子に結合したまま残る);及び、保護基は、残りの構造に悪影響を及ぼすことのない条件下で除去可能である。適切な保護基の選択は、もちろん、モノマー単位及びオリゴマーの化学的性質、並びに保護基がそれに対して保護すべき具体的試薬に左右される。所定の反応スキームに対して適切な保護基を選択することは、当業者の能力内である。
【0033】
本明細書の記載は、当業者によって用いられる多数の化学用語と略号に言及している。それでもなお、選択した用語の定義を、明確性及び一貫性のために提供した。
【0034】
略号:
DCC:ジシクロヘキシルカルボジイミド; DMAP:4-(N,N−ジメチルアミノ)ピリジン; EDC:1-(3-ジメチルアミノプロピル)-3-エチルカルボジイミド塩酸塩; HOBt:1-ヒドロキシベンゾトリアゾール水和物; HATU:O-(7-アザベンゾトリアゾール-1-イル) N, N, N’, N’-テトラメチルウロニウムヘキサフルオロホスフェート; PyBOP:(ベンゾトリアゾール-1-イルオキシ)トリピロリジノホスホニウムヘキサフルオロホスフェート; DCU:ジシクロヘキシルウレア; DIPEA(ヒューニッヒ塩基(Hunig’ base)):ジイソプロピルエチルアミン; DCI:4,5-ジシアノイミダゾール; DMSO:ジメチルスルホキシド; DMSO−d6:重水素化ジメチルスルホキシド; DCM:ジクロロメタン; NMI:N-メチルイミダゾール; TEA:トリエチルアミン; TEAA:トリエチルアンモニウムアセテート; THF:テトラヒドロフラン; CE:シアノエチル; PEG:ポリエチレングリコール; Py:ピリジン; Succ:スクシニル; CPG:細孔制御されたガラス; IL:イオン液体; ILSS:イオン液体担持合成; DMT:ジメトキシトリチル; LCAA:長鎖アルキルアミン; EEDQ:2-エトキシ-1-エトキシカルボニル-1,2-ジヒドロキノリン; IIDQ:2-イソブトキシ-1-イソブトキシカルボニル-1,2-ジヒドロキノリン; TFA:トリフルオロ酢酸; AcOH:酢酸; NMR:核磁気共鳴; MS:マススペクトル; m/z:電荷に対する質量比; m.p.:融点; FAB:高速原子衝撃; TLC:薄層クロマトグラフィー; ESI:エレクトロスプレーイオン化; FTMS:フーリエ変換マススペクトル; LCMS:液体クロマトグラフィー−マススペクトル; J:結合定数; s:単一線(シングレット); t:三重線(トリプレット); m:多重線(マルチプレット); P−III:三価のリン; P−V:五価のリン。
【0035】
本発明によるイオン液体担持合成の一般概念を図1及び2に示す。イオン液体残基上に固定された基質(反応剤)は極性有機溶媒に可溶性であり、液相反応をすることができる。反応の終了及び溶媒の留去後、イオン液体に固定された生成物が溶けない極性の低い有機溶媒によって過剰な試薬を除去しうる。無機試薬及び/又は副生成物は、沈殿により、又は水溶液で洗浄することにより除去しうる。この反応順を繰り返して、より複雑な構造を得ることができる。最後に、生成物を切り離し、有機溶媒抽出によってイオン液体残基から分離することができる。イオン液体担体に固定された基質は、従来の溶液系反応と同様の反応性を大部分保っていると予想される。イオン液体担体上で行う反応の進行は、標準の分光学的手法によって容易に監視及び分析される。
【0036】
イオン液体によって担持された、オリゴペプチド、オリゴサッカライド、及びオリゴヌクレオチドの合成についての新規な溶液相アプローチを説明する。
【0037】
ある態様では、本発明は、オリゴペプチド、オリゴサッカライド、及びオリゴヌクレオチドを含むがこれらに限定されないオリゴマーの合成のためのイオン液体担持方法に関する。さらなる態様では、本発明は、オリゴペプチド、オリゴサッカライド、及びオリゴヌクレオチドを含むがこれらに限定されないオリゴマーの化学合成に用いるためのイオン液体に関するものであり、このイオン液体は、溶媒及び液体担体の両方の二重の機能を果たしうる。このイオン液体は、有機合成に一般に適用される様々な合成方法に適合性である。さらに詳細には、イオン液体は、オリゴペプチド、オリゴサッカライド、及びオリゴヌクレオチドを含むがこれらに限定されないオリゴマーの合成に一般に適用される様々な合成方法に適合性である。さらに、特に、しかしこれに限定されないが、オリゴペプチド合成の場合には、イオン液体は、その本来のイオン的性質によって、ペプチドビルディングブロックのラセミ化又はエピ化(エピマー化)を起こしてはならない。さらに、イオン液体担体の溶解性が伸長オリゴペプチド鎖、伸長オリゴサッカライド鎖、又は伸長オリゴヌクレオチド鎖によって影響を受け、分離及び精製手段が過度に複雑になってはならない。分離及び精製手段は、非常に簡単化され、通常は水性溶媒及び有機溶媒での洗浄ステップを含む。
【0038】
本発明のある態様では、イオン液体は、ヘテロ環式又は置換ヘテロ環式第四級窒素含有有機カチオン、及び前記有機カチオンの電荷と釣り合うアニオン、を含む有機塩である。より具体的な態様では、有機カチオンは、N−置換ピリジン及び1,3−ジ置換イミダゾールからなる群から選択され、かつ、アニオンは、Cl−、Br−、BF4−、PF6−、SbF6−、CuCl2−、及びAlCl4−からなる群から選択される。その他のイオン性液体が当分野で公知であり、当業者の能力の範囲内である。さらに、前記アニオンが有機アニオンであってもよいことも当業者の能力の範囲内であり、有機アニオンの制限されない例には、CH3CO2−、CF3CO2−、CH3SO4−、及びCF3SO2−が含まれる。
【0039】
以下の、例示の態様の非限定的説明によって、さらに詳細に本発明を説明する。
【実施例】
【0040】
[実施例1]イオン液体担体への結合とラセミ化の試験(スキーム1)
3-ヒドロキシエチル-(1-メチルイミダゾリウム)-テトラフルオロボレート(1)(1-メチルイミダゾールと2-ブロモエタノール[35a,d]の反応によって容易に入手できる)を、本発明で意図するイオン液体担持オリゴペプチド合成を説明するための、適切なイオン液体担体の非限定的な例として選択した。本発明にしたがって使用することができるその他のイオン液体担体は、当分野で公知であり、当業者の能力のうちである。
【0041】
【化2】
【0042】
化合物1上への第一のアミノ酸、Boc−ロイシンの担持のために、様々なカップリング反応条件を試験した。これらのカップリング反応の有効性は、以下に、表1中に示す。
【0043】
【表1】
【0044】
探索したカップリング条件のなかでは、DCC/DMAPの組み合わせ(試験8)が最良の結果をもたらした。その他の試験したカップリング条件には、以下の組み合わせ:EDC/HOBt、HATU/HOBt、PyBOP/DIPEA、EEDQ、又はIIDQが含まれる。しかし、後者の条件群は、カップリング反応(すなわち、エステル化反応)を定量的転換にまでにするためには充分ではなかった。DCC/DMAP経路に対する代替かつ有用なカップリング経路は、追加のカップリングステップを含むけれども、未反応の化合物1を無水酢酸でキャッピングすることを含む。順次のエーテル洗浄及び酸水溶液洗浄によって、過剰なBoc−ロイシン、DMAP、及びDCU副生成物を全て除去した。イオン液体担持Boc−ロイシン2が高い単離収率(91%)かつNMRスペクトルによって検証した高純度で得られた。このカップリング反応は、L−及びD−Leu−Bocの両方を用いて行い、それぞれ、2a及び2bが得られた。
【0045】
2a及び2bの様々なBoc−保護反応条件を試し、その結果を以下の表2に示す。テトラフルオロホウ酸及びトリフルオロ酢酸の両方が、1H NMRスペクトルによって示されるとおり、非常にきれいな脱保護を起こさせることができた。効率的ではないが、Boc脱保護は、Dowex(登録商標)50Wx8-100又はAmberlyst(登録商標)15などの酸性イオン交換樹脂を用いて行うこともできる。その他の脱保護法、例えば「Protective Groups in Organic Synthesis」(Greeneら, John Wiley & Sons, Inc, NY, 1991)中に記載されている方法を本発明にしたがって用いることができ、当業者の能力の範囲内である。
【0046】
【表2】
【0047】
次のアミノ酸ビルディングブロック、すなわちBoc−Phe−OHのカップリング(スキーム2)は、先に中和ステップを行うことが好ましい(表2)。Boc保護に先に用いた過剰のTFA又はHBF4は、NaHCO3、Et3N、DIPEA、又はDowex(登録商標)550A OHなどの塩基性イオン交換樹脂、で中和しうる。本発明に従って使用できるその他の適切な中和剤は当分野で公知であり、当業者の能力の範囲内である。驚くべきことに、次のアミノ酸ビルディングブロック、すなわち、Boc−Phe−OHのカップリング反応を、PyBOP/DIPEA条件を用いて行う場合は、脱保護/中和と次のカップリング反応との両方を、中間体分離及び精製ステップなしに順次行うことができる。
【0048】
【化3】
【0049】
ペプチド4aは、実質的に定量的収率で得られた40。本発明のイオン液体担持方法の利点は、大過剰の試薬を必要としないことである。このカップリング反応は、わずか1.2当量のPyBOPとBoc−Phe−OHで達成しうる。
【0050】
イオン液体担持合成時の、より特にはDCC/DMAP条件を用いて担持するステップでの、ラセミ化又はエピ化の可能性を評価するために、一連のイオン液体担持ペプチド4a〜dを合成した。このペプチドを次にアンモニア/メタノールを用いて開裂させてジペプチド5a〜dを得た。5a(L,L)及び5b(D,L)の引き続いてのHPLC分析は、検出可能な量のその他の異性体がない(<0.5%)ことを明らかにし、担持ステップ及び続いてのジペプチド合成時に、いかなる有意なラセミ化又はエピ化も起こらないことを示した。さらに、この結果は、塩基条件(アンモニア/メタノール)を用いる開裂ステップ時のラセミ化又はエピ化の不存在をも示唆している。
【0051】
[実施例2]オリゴペプチド合成及び、イオン液体担体からの切り離し
上記伸長ペプチド鎖4aに、2つの連続するグリシン残基、次にチロシン残基(全てその保護されたBoc形で)を、先に開発したカップリング及び脱保護条件を用いて次にカップリングさせ、イオン液体担持ペンタペプチド11を泡状の淡黄色固体として得た(スキーム3)。この合成スキームにおいて、Boc保護形(7、9、11)又は脱保護形(6、8、10)のいずれかの全ての中間体を、当業者に周知の標準的手順で単離し且つ精製した。そのような手順は、一般に、順次の有機溶媒(1種以上)及び水での洗浄を含み、その非制限的実施例を本明細書に記載する。
【0052】
【化4】
【0053】
構造の確認と純度分析は、従来の分光学的方法、例えば、1H、13C NMR、及びMSによって実現した。1H NMRスペクトルにおけるBoc基のt−ブチル基の存在又は不存在が、カップリング又は脱保護ステップのそれぞれの特徴であった。イオン液体担持オリゴペプチド類(2a、4a、7、9、及び11)のマススペクトルは、構造の特性解析をさらに助けた(イオン液体担持オリゴペプチドのカチオンに相当する最も強いシグナル)。
【0054】
ペンタペプチド生成物であるLeu5−エンケファリン14をイオン液体担体から切り離すための2つの経路を評価した(スキーム3)。第一の経路は、塩基性水性条件下でイオン液体担体からペンタペプチド14を切り離し、次にTFA/アニソールを用いてBoc及びt−Bu保護基を除去することを含む。第二の経路は、最初にTFA/アニソールを用いてBoc及びt−Bu保護基を除去し、次に塩基性水性条件下でイオン液体担体からペンタペプチド14を切り離すことを含む。第一の経路は、イオン液体担体からの開裂と反応媒体の酸性化に続いて、保護されたペンタペプチド12を良好な収率(84%収率)及び純度で溶液から沈殿させた。
【0055】
このように調製したLeu5−エンケファリンペンタペプチド14の純度を分析した。ペンタペプチド14は、いかなる再結晶又はクロマトグラフィー手段なしに、その対応するTFA塩として、化合物1から50%の全収率で得られた[m.p. 148〜152℃、及び[α]20D = 25.0(c:0.8、95%AcOH)]。14の真正サンプルは150〜153℃の範囲の融点をもち、比旋光度[α]20D = 25.6(c:0.9、95%AcOH)]を有する41。14の1H NMRスペクトルは、真正サンプルを用いて得られたスペクトルと実質的に同じだった(図3)。Leu5−エンケファリンペンタペプチド14の純度は、HPLC分析によって実証されたように約90%純度を超えるものであることが測定された(図4)。このレベルの純度は、クロマトグラフィー精製前に固相ペプチド合成によって一般的に得られるものよりも優れている42。
【0056】
Leu5−エンケファリンペンタペプチド14の合成によって具体化されたように、イオン液体担持合成は、ペプチド合成のこれまでの方法の多くに対していくつかの見込まれる利点を提供する(表3)。
【0057】
【表3】
【0058】
第一に、その他の溶液相法と同じに、固相ペプチド合成(SPPS)とは対照的に、大過剰の反応試薬の使用を避けることができる。非天然型アミノ酸(例えば、D−アミノ酸)を含むペプチドを合成することを計画した場合、又は大スケールでのペプチド合成が必要な場合に、このことは考慮すべき重要な要因である。第二に、イオン液体担持合成の場合、各中間体は、繰り返しの溶媒洗浄によって容易に精製することができる(液/液相分離)。この面で、ILSSは、FPPS(フッ素系相ペプチド合成)並びにSPSPS(可溶性ポリマー担持ペプチド合成)に類似している。
【0059】
ILSSの場合、分離工程は、固体/液体相分離を含むSPPSに対する場合と同様には容易に自動化されない。しかし、わずか約1モル当量の低分子量イオン液体1しか通常は必要とされないので、ILSSに対する負荷容量はSPPSよりも大きい。その上、イオン液体担体のコストは、フッ素系担体のコスト、又は固体ポリマー担体のコストよりも通常は低く、このことは、大スケールのペプチド合成を計画する場合にさらなる重要な考慮事項である。最後に、ILSSの場合、合成中の各中間体の構造及び純度が、通常のスペクトル法(すなわち、1H、13C、及びMS)によって容易に検証可能であった。
【0060】
[実施例3]イオン液体担持オリゴサッカライド合成
以下のスキーム4に図示したように、イオン液体担持法を用いるオリゴサッカライドの合成は、簡単な相分離を用いて、生成物精製のためのクロマトグラフィー法を必要とすることなく、簡便に達成された。
【0061】
【化5】
【0062】
フェニル2,3,4,6-O-アセチル-1-チオ-β-O-グルコピラノシドから出発して文献記載の方法43の修正法によって調製したβ-チオグリコシド15を、室温でCH2Cl2中、ブロモ酢酸/DCC/DMAPでアシル化し、ブロモアセテート16a(X=Br)を得た(スキーム4)。
【0063】
アセトン中で、室温における、16a(X=Br)と1-メチルイミダゾール及びナトリウムテトラフルオロボレートとの反応は、イオン液体に固定されたサッカライド16b(X=[mim][BF4])を98%収率で与えた。化合物16bは、最初に粗生成物をジエチルエーテルで洗浄して過剰な1-メチルイミダゾールを除去して精製した。生成物は次にCH2Cl2に溶かし、反応で生じた不溶性の無機NaBr塩を濾過によって簡便に除去した。16bの構造は、NMR(1H、13C、COSY、及びHMQC)スペクトルによって確認し、これはイミダゾリウム残基とβ-アノマーとしてのアノメリック炭素を明らかに示した。16bの電子スプレーイオン化マススペクトル(ESI−MS)は、唯一の分子イオンとしてのカチオンの存在を示した。
【0064】
次に、モノサッカライド16bを、CH2Cl2中で、−78℃で20分間、m-クロロ過安息香酸によって酸化して、97%収率で、ジアステレオマー(スルホキシドの光学不斉による)の混合物としてスルホキシド17b(X=[mim][BF4])を得た。反応の他方の生成物である、m-クロロ安息香酸は、ジイソプロピルエーテルで洗浄することによって粗生成物から除去した。
【0065】
次に、スルホキシドグリコシレーション反応を、グリコシル供与体としての17bと受容体としての15(3当量)を用いた糖−糖組み立て法44−46として用いた。グリコシル供与体及び受容体の両方とも低温でジクロロメタン中に良く溶けるので、2,6-ジ-tert-ブチル-4-メチルピリジン(4当量)とTf2O(1当量)を用いるカップリング反応を無水CH2Cl2中、−78℃で行った。本反応過程はTLCによって観察した。
【0066】
過剰なグリコシルアルコール15、酸スカベンジャー塩基、及びその他の副生成物をn-ペンタンで、次にジイソプロピルエーテルで洗浄除去することによって、生成物ジサッカライド18b(X=[mim][BF4])を得た。CH2Cl2中の粗製18bの溶液にn-ペンタンを添加し、次に溶液を冷却することによって溶液から17bを相分離させることで、プロトン化した酸スカベンジャー、2,6-ジ-tert-ブチル-4-メチルピリジニウムトリフレートを18bから除去した。イオン液体に結合したジサッカライド18bを、2つのアノメリック炭素を示すHMQC NMRスペクトルによって示されるように高純度で得た。さらに、EIS−MSはジサッカライド構造を裏付けるだけでなく、未反応のモノサッカライドの不存在をも示した。同じ一連の反応の繰り返しで、スルホキシド19bが得られ、次にイオン液体に結合したトリサッカライド20b(X=[mim][BF4])を得た。このトリサッカライドの形成は、3つのアノメリック炭素の存在を示しているHMQC NMRスペクトルと、モノ又はジサッカライドのいかなる混入もないトリサッカライドカチオンを明確に示しているESI−MSによって確かめた。
【0067】
イオン液体残基と連結基は、メタノール中で1当量のCs2CO3を用いることによって容易に20bから切り離されて、定量的に生成物21を与える。トリサッカライド21は、メタノール溶媒の蒸発、及び、粗生成物をCH2Cl2中に溶かし、次に濾過によってイミダゾリウム塩を除去することによって容易に単離された。クロマトグラフィー又はその他の生成方法なしにそのように得られたトリサッカライド21は、NMR及びTLCでみて純粋であることがわかった。そのHMQC NMRスペクトルとESI−MS(M+Na+=1429.7)は、16c(X=H)から出発する古典的な溶液相合成によって、スキーム4に図示したとおりの同じ反応順序にしたがい、しかし、反応ステップのそれぞれに続くクロマトグラフィー精製を行って別個に調製した21の真正サンプルと同じであることがわかった。16cを通っての古典的溶液相合成と比較してILSSアプローチによって得られた21の良好な純度は、古典的な溶液相合成のために開発されたカップリング条件及び立体選択性が、ILSSにそのまま移されうることを示唆している。この例はまた、固相担体からの切り離し後、最終生成物を精製するためにクロマトグラフィーが一般に必要な固相合成47との対照をも示している。
【0068】
[実施例4]イオン液体担持オリゴヌクレオチド合成
ホスホルアミダイト法2−6を用いるオリゴヌクレオチドの固相合成は反復法であり、その方法においては、結合されたヌクレオシドをもつ固体担体が、不安定な保護基を除去することによって末端で脱保護され、それによって求核性水酸基を自由にする。この末端求核性基が次に活性化剤の存在下で、保護されたホスホルアミダイトモノマーに結合される。この新しく作られたホスファイトトリエステル連結基が次に酸化されて、所望し且つより安定なホスフェートトリエステルをもたらす。この方法は、所望する長さ及び組成のオリゴマーが得られるまで繰り返される。可溶性担体を用いた場合に、同様の反復法が用いられうる。
【0069】
3-ヒドロキシエチル-(1-メチルイミダゾリウム)-テトラフルオロボレート(1)(1-メチルイミダゾールと2-ブロモエタノールの反応で容易に入手可能である[35a,d])を、本発明によって意図されるイオン液体担持オリゴヌクレオチド合成を説明するための好適なイオン液体担体の非制限的な例として選択した。本発明に従って用いることができるその他のイオン液体担体は当分野で公知であり、当業者の能力の範囲内である。
【0070】
スクシニル化5’-MMT-デオキシヌクレオシド2348を、アセトニトリル中、ジシクロヘキシルカルボジイミド(DCC)と触媒量の4-ジメチルアミノピリジン(DMAP)を用いてイオン液体1と結合し、イオン液体担持ヌクレオシド24(スキーム5)を得る。反応混合物を濾過して、反応中に形成した不溶性のジシクロヘキシルウレア(DCU)副生物を除去した。次に、反応混合物を、撹拌したジエチルエーテル−酢酸エチル溶液に滴下して加えることによって、化合物24を単離し且つ精製する。誘導体化されていないイオン液体1と所望する生成物24からなる得られる沈殿物を、次にクロロホルム中に溶かし、水で抽出する。イオン液体1は水相とともに除去され、一方、所望する生成物24は、チミジンの5’位に結合したモノメトキシトリチル保護基の疎水性のために有機相に留まる。5’-モノメトキシトリチル(MMT)チミジン誘導体をこの場合には用いたけれども、より一般的に用いられる5’-ジメトキシトリチル(DMT)チミジン誘導体は、このステップで容易に置換されうる。溶媒の除去により、妥当な収率でイオン液体誘導体化ヌクレオシド24が得られる。24の構造は、1H NMRと、電子スプレーイオン化マススペクトル(ESI−MS)によって確かめられた。
【0071】
化合物24の脱トリチル化は、アセトニトリル中での3%のトリフルオロ酢酸の添加と室温での20分間の撹拌によって達成される。反応混合物を、撹拌したエチルエーテル−酢酸エチル溶液に滴下して加え、沈殿物を集めることによって、生成物は速やかに単離且つ精製される。時々、酸溶液中への再溶解と二回目の沈殿が、脱保護を完了させ、残存するトリタノールを完全に除去するために必要とされる。化合物25は、96%の収率で、淡褐色泡状物として得られ、その構造と純度は1H NMRとESI−MSによって検証される。
【0072】
【化6】
【0073】
THF又はアセトニトリル中で4,5-ジシアノイミダゾール(DCI)を活性化剤として用いて、1〜2時間撹拌して、イオン液体担持ヌクレオシド25を1.5倍過剰の適切なホスホルアミダイト誘導体(26a〜d)と反応させることによって、ジヌクレオシドホスホトリエステル類 ApT、CpT、GpT、及びTpT(27a〜d)49(スキーム6)を250μmolスケールで調製した。反応が完了した後、過剰な活性化ホスホルアミダイトを過剰の無水エタノールの添加とさらに10分間の撹拌で失活させる。ホスホルアミダイトを失活させることは精製時にそれらの除去を容易にする。ジヌクレオシドホスファイトトリエステル中間体は、次に、酸化の前に、室温で酢酸エチル:エチルエーテル(1:9)から沈殿させることによって簡単に単離される。失活した過剰のモノヌクレオシド3’-O-ホスホルアミダイトが、カップリング生成物よりも酢酸エチル/エーテル中にずっと可溶性であるので、このことがジヌクレオシドホスファイトトリエステルからのモノヌクレオシド3’-O-ホスホルアミダイトの除去を可能にしている。通常、沈殿物は、アセトニトリル又はメタノールを用いてフィルターから直接溶かし出され、所望の生成物の純度を高めるために、1:9の酢酸エチル:エチルエーテルから再び沈殿させる。
【0074】
ホスファイトトリエステル中間体の酸化を行うために、集めた沈殿物を少量のアセトニトリルに再び溶かし、次に少過剰のピリジンと、2:1のテトラヒドロフラン(THF):水中のヨウ素の0.1M溶液の大過剰量とを添加する。ひとたび(ヨウ素による)色が持続したら、亜硫酸水素ナトリウム水溶液(5%)を添加して過剰のヨウ素を減らす。次に、反応混合物をクロロホルムで希釈し、水で抽出した。水層は、カップリングしていないイオン液体担持ヌクレオシド(ヌクレオチド)(なぜなら、それは末端のトリチル塩を欠き、それに水溶性を付与するからである)に加えて、生じた塩(NaI、Na2SO4、過剰の亜硫酸水素塩、ヨウ化ピリジニウムなど)を含む。減圧下での有機溶媒の除去により、良好な収率(表4)と高純度で淡褐色泡状物として生成物(27a〜d)が得られ、キャッピング段階の必要はない。27a〜dの脱トリチル化は、ジクロロメタン又はアセトニトリル中の3%のトリフルオロ酢酸の添加、20分間の撹拌、及び酢酸エチル:エチルエーテル(1:9)中での沈殿によって達成される。この時点で得られる物質は、このステップの間に確立された平衡(ROH+DMT+=DMT−OR)から生じる最大でも5%のトリチル化出発原料を含みうる。DMTカチオンをトラップするためのエタノールでの失活は、この問題を取り除くとは思われなかった。しかし、TFA処理を繰り返す場合(すなわち、3%トリフルオロ酢酸溶液中への沈殿の再溶解と、続いての第二の沈殿)は、純度は顕著に高くなる(低分解能ESI−MSによって観測されるいかなるトリチル化生成物もバルク生成物中にない)。精製した生成物は単純に濾過して、灰白色から淡黄色の粉末状固体を高収率で生じる(表4)。集めた固体(28a〜d)はカップリングの別のラウンド、又は所望する配列が完了している場合は脱保護に、そのまま用いられる。
【0075】
【化7】
【0076】
上述したダイマー28a〜dに加えて、チミジントリマー(29、30)及びテトラマー(31、32)も、50〜100μmolスケールで合成した。イオン液体担持化合物の同一性は、31P−NMR並びに低及び高分解能ESI−MSの両方によって確かめた(表4)。ダイマー種についての31P NMRデータ(表4)は、2種の可能なRp及びSpジアステレオマーホスホトリエステル類に対応する2つのシグナルを示している。β-シアノエチルホスフェート保護基が除去される場合、ヌクレオチド間の架橋がアキラルになり、単一のシグナルのみが観測される。同様に、トリマー種に対する31P-NMRデータ(表4)は、トリチル保護基がなお結合している場合は、8つのシグナルを示す(4つのジアステレオマーそれぞれが、2つのシグナルを示す)。ひとたびトリチル基が除去されると、シグナルはもはや分解され得ない。このことはテトラマーに対しても同様であり、この場合ジアステレオマーの予期される数は8(23×3=24ピーク)である。単離された化合物がジアステレオマーのペアとして存在するという事実は、1H NMRピークの帰属を複雑にもする。しかし、31Pに調整したCIGAR実験装置を用いることで、ホスホトリエステル連結基を通した予測される3’-5’連結を観測することが可能だった50。
【0077】
【表4】
【0078】
上で合成されたオリゴヌクレオチドを、同じ配列を用いて、一般に用いられる固体担体である細孔制御ガラス(CPG;1μmスケール)上で合成したものと比較した。2つのシステムは平行して脱保護した。所望するオリゴヌクレオチドの完全な脱保護は、それらを、室温で48時間、又は60℃で16時間、濃水酸化アンモニウム/エタノールで処理することによって行った。これらの条件は、シアノエチル保護基、イオン液体残基、塩基(Ade、Cyt、及びGua)の環外アミンの全ての保護、及びモノスクシネート連結基の完全な開裂を確実にする。オリゴヌクレオチドは、真空下でのエタノールと水酸化アンモニウム溶液の除去、水への再溶解(固体担体は、CPG担持オリゴマーに対する遠心によって簡単に沈殿させられる)、及び次にイオンペア逆相HPLC、アニオン交換HPLC、又はポリアクリルアミドゲル電気泳動によるクロマトグラフ精製、によって単離された。ILSS法の生成物を、標準的な遺伝子の機械合成法によって得られた生成物と、LCMSによって比較した。図7は、イオン液体(IL)担体上で成長させたオリゴマーと、CPG上で成長させたオリゴマーとの比較を示す。LCMSのデータは、以下の表5にまとめた。
【0079】
【表5】
【0080】
全ての場合において、ILで生成したオリゴヌクレオチド物質の保持時間は、CPGで生成したオリゴマーと良く一致する。ILオリゴマーに対する主ピークの面積パーセントは、CPG誘導オリゴマーのものより常に数%上である。しかし、イオン液体自身が、オリゴマーを測定するために用いる波長において弱い吸収(保持時間11.2分;λmax217nm)をすることを考慮に入れるといくらか誤っている。イオン液体由来ピークによる面積を除くことで、IL誘導オリゴマーの値が、CPG上で合成したオリゴマーについて得られた値と一致する。HPLC線図のいくつかに見られるチミジンヌクレオシドは、不完全なカップリング又は最初のキャッピング段階より前での固体単体の部分的脱トリチル化によって生じ、約19分に溶出される。CPG上で合成した物質中には通常存在するが、後酸化抽出は、ILが介在する生成物からこの物質をかなり除去する(図1)。先に記載したように、トリマー及びテトラマー中の誤った配列の存在(いわゆる「n−1」ピーク)は、不完全なカップリングによるものではなく、むしろカップリングの前の不完全な脱トリチル化によるようである。この物質は抽出時には除去されずに、カップリングに続く脱トリチル化の後で脱保護される(すなわち、トリマー合成後、脱トリチル化は所望するトリマーと、前の段階から持ち込まれた望まないダイマーとを生成する)。したがって、各カップリングステップの前に完全な脱トリチル化を確実にすることが重要である。
【0081】
[実施例]
一般
全ての試薬は市販品を入手し、他に注意事項のない限り、さらに精製することなく用いた。溶媒は試薬等級のものであり、必要な場合は、標準手法によって乾燥した。反応は、他に特に指示のある場合を除いて無水条件下で行い、シリカゲル60F−254の背面ポリエステルのプレート(250μm厚)上での薄層クロマトグラフィー(TLC)によって、UV光(254nm)下での検出又は過マンガン酸カリウム水溶液で炭化させて観察した。1H NMR及び13C NMRスペクトルはVarian Mercury-300(300MHz)又はMercury-400(400MHz)分光計で20℃において記録した(オリゴペプチド合成)。1H及び13C NMR、HMQC NMR、COSY NMRスペクトルは、Sunワークステーションを備えたUnity 500、Varian Mercury 400、及びVarian Mercury 300分光計で記録した(オリゴサッカライド合成)。化学シフトは、1H NMRについては、δ7.24ppmの残留CHCl3、4.67ppmの水、2.06ppmのアセトン、又は3.30ppmのメタノールを基準にし、13C NMRについては、77.0ppmのCHCl3、49.0ppmのメタノール、及び29.8ppmのアセトンを基準にした、δスケール上の百万分の一(ppm)で報告した。プロトンとカーボンの帰属は、標準のgHMQC及びgCOSY試験によって行った。融点は、温度計を較正することなく、融点装置で測定した。電子スプレーイオン化(ESI)マススペクトルは、KRATOS MS25RFA質量分析計で記録した。高分解能質量分析は、VG MICROMASS ZAB 2F HS分光計(FAB)で行った。オリゴヌクレオチド高分解能マススペクトル測定は、IonSpec 7.0 tesla FTMSで、陽イオン電気スプレーモードで行い、ポリエチレングリコール300、600、及び1000で較正した。旋光度は、DIP-140(JASCO)デジタル旋光計を使用して測定した。ラセミ化の程度及び最終生成物の純度は、溶離液としてヘキサン中2−プロパノールを用いてのDiacel Chiralcel OD-H, OD-Jキラル分析カラムと、溶離システムとしてCH3CN/H2O/TFAを用いてのZorbax SB-CN半予備カラムとを使用して、分析用HPLCによって測定した。
【0082】
〔オリゴヌクレオチド合成、精製、及び同定〕
オリゴヌクレオチドは、その各担体から切り離した後、逆相高速液体クロマトグラフィー(HPLC)によって精製した。分離は、60℃に加熱したPolymerex 10μ RP-1カラム(Phenomenex、10 mm x 250 mm, 10μパッキング)を使用し、Waters 1525 バイナリーHPLCポンプによって作りだした1mL/分の移動相流速で行った。最初の移動相は、100mMの酢酸トリエチルアンモニウム緩衝液(pH7.0、80:20(v/v)の水:メタノール)の2分間の定組成流と、続いての35分間にわたる100mMの酢酸トリエチルアンモニウム緩衝液(pH7.0、70:30(v/v)の水:メタノール)への傾斜シフトと、さらに続いての1分間の定組成流と、その後の初期条件への14分間の傾斜を付けた戻りと、最後に5分間の定組成流から構成した。溶出は、Waters 2487二重吸光度検出器で、260nmと217nmで観察した。溶出ピークの画分を集め、移動相を真空下で除去した。サンプルを10%メタノール−水に再溶解し、続いて分子量を、ThermoQuest Finnigan LCQ Duo質量分析計での低解像度電子スプレーイオン化マススペクトルによって、マイナスイオンモードにおいて測定した。オリゴヌクレオチドの固相合成を、先の記載したように、標準条件6を用いて、Applied Biosystems 3400 DNA/RNA合成装置を使用して、500Åの長鎖アルキルアミンで被覆され且つ制御された細孔ガラス(LCAA−CPG)上で、1μmolスケールで行った。
【0083】
エステル化(ローディング)ステップ
無水アセトニトリル(25mL)中の、イオン液体1(1.07g、5mmol)、Boc-ロイシン(L-又はD-形 2.31g、10mmol)、及びジメチルアミノピリジン(0.25g、2mmol)の混合物に、ジシクロヘキシルカルボジイミド(DCC、CH2Cl2中に1M、10mL、10mmol)を添加した。この混合物を18時間、室温で、窒素下で激しく撹拌し、Celite(登録商標)の栓を通して濾過した。Celite(登録商標)の栓をアセトニトリルで濯ぎ、合わせた有機相を真空下で濃縮した。粗製残渣を最初にエーテルで洗浄し(20mL×3)、次にCH2Cl2に溶解し、2M HCl(10mL×3)で洗浄した。有機相をNa2SO4上で乾燥させ、濃縮して、イオン液体担持Boc-ロイシン 2a(L-異性体)又は2b(D-異性体)1.95g(91%)を淡黄色オイルとして得た。
1H NMR (300 MHz, Acetone-d6) δ 9.08 (s, 1H), 7.80 (s, 1H), 7.71 (s, 1H), 6.41 (d, br, J = 7.8 Hz, 1H), 4.66-4.64 (m, 2H), 4.60-4.43 (m, 2H), 4.20-4.13 (m, 1 H), 4.04 (s, 3H), 1.68-1.53 (m, 3H), 1.39 (s, 9H), 0.91 (d, J = 6.0 Hz, 3H), 0.89 (d, J = 6.0 Hz, 3H); 13C NMR (100 MHz, Acetone-d6) δ 173.2, 156.4, 138.0, 124.4, 123.6, 79.3, 63.4, 53.0, 49.3, 40.7, 36.6, 28.5 (3C), 25.4, 23.1, 21.6; HRMS (FAB) C17H30N3O4 (M+) に対する計算値340.2237, 測定値 340.2236。
【0084】
ペプチド形成;カップリング及び脱保護ステップ
CH2Cl2(10mL)中のイオン液体担持Boc−ペプチド(2mmol)の溶液に、TFA(10mL)を添加した。反応混合物を室温で窒素下にて0.5時間撹拌した。減圧下で濃縮したら、残渣をエーテルで2回洗浄し、真空下で乾燥させて、脱保護したイオン液体担持ペプチドをTFA塩として得た。次に、残渣を、その次のBocで保護したアミノ酸(3mmol)及びPyBOP(3mmol)とともに、CH3CN(35mL)中に溶かした。DIPEA(6mmol)を滴下して加え、得られた反応混合物を35℃で窒素雰囲気下にて8時間撹拌した。溶媒を真空下で除去し、残渣を最初にエーテル(20mL×3)で洗浄し、次にCH2Cl2中に溶かし、水(10mL×3)で洗った。有機相をNa2SO4上で乾燥させ、濃縮して、イオン液体担持Boc−ペプチド(90%収率)を得て、これはペプチド合成の次のサイクルに直接用いるために充分な純度であった。この生成物は、所望する場合には、この化合物をアセトン中に溶かし、シリカゲルの短い充填物を通して濾過し、溶媒を蒸発させることによって、さらに精製することができる。
【0085】
イオン液体担持ロイシンTFA塩 3a: 1H NMR (400 MHz, Acetone-d6) δ 9.20 (s, 0.55H), 9.12 (s, 0.45H), 7.85 (s, 1H), 7.68 (s, 1H), 5.02-4.98 (m, 0.45H), 4.75-4.58 (m, 4H), 4.27-4.25 (m, 0.55H), 4.05 (s, 3H), 1.90-1.87 (m, 2H), 1.73-1.70 (m, 1 H), 0.95 (d, J = 6.0 Hz, 3H), 0.93 (d, J = 6.0 Hz, 3H); 13C NMR (100 MHz, Acetone-d6) δ 169.4 (0.55C), 167.7 (0.45C), 138.0 (0.55C), 137.8 (0.45C), 124.4, 123.6 (0.45C), 123.5 (0.55C), 65.1 (0.45C), 64.9 (0.55C), 52.2, 48.9, 39.8 (0.55C), 39.4 (0.45C), 36.4, 25.1 (0.45C), 24.8 (0.55C), 22.7 (0.45C), 22.3 (0.55C), 22.1 (0.55C), 21.6 (0.45C); HRMS (FAB) C24H44N6O4BF4 (2M++BF4-) に対する計算値567.3450, 測定値 567.3453。
【0086】
イオン液体担持Leu-Boc-フェニルアラニン 4a:泡状淡黄色固体;1H NMR (400 MHz, Acetone-d6) δ 9.04 (s, 1H), 7.81 (s, 1H), 7.70 (s, 1 H), 7.67 (d, br, J = 6.8 Hz, 1 H), 7.29-7.20 (m, 5H), 6.20 (d, br, J = 8.0 Hz, 1H), 4.69-4.66 (m, 2H), 4.64-4.52 (m, 2H), 4.44-4.37 (m, 2H), 4.08 (s, 3H), 3.19-2.91 (m, 2H), 1.72-1.58 (m, 3H), 1.35 (s, 9H), 0.92 (d, J = 6.0 Hz, 3H), 0.89 (d, J = 6.0 Hz, 3H); 13C NMR (100 MHz, Acetone-d6) δ 172.9, 172.4, 156.5, 138.2, 132.6, 129.9 (2C), 128.8 (2C), 127.0, 124.5, 123.5, 79.4, 63.4, 56.6, 51.6, 49.4, 40.4, 38.3, 36.7, 28.4 (3C), 25.2, 23.2, 21.6; HRMS (FAB) C26H39N4O5 (M+) に対する計算値487.2920, 測定値 487.2920。
トリフルオロ酢酸による処理で、イオン液体担持Leu-フェニルアラニンTFA塩6が得られた: 1H NMR (300 MHz, Acetone-d6) δ 9.33 (s, br, 0.6H), 9.25 (s, br, 0.4H), 7.87 (s, br, 1H), 7.72 (s, br, 0.6H), 7.67 (s, br, 0.4H), 7.28-7.25 (m, 5H), 5.38-5.33 (m, 0.4H), 4.70-4.50 (m, 4H), 4.40-4.34 (m, 1.6H), 4.07 (s, 1.8H), 4.00 (s, 1.2H), 3.55-3.26 (m, 2H), 1.82-1.70 (m, 2H), 1.58-1.53 (m, 1 H), 0.92 (d, J = 6.0 Hz, 3H), 0.87 (d, J = 6.0 Hz, 3H); 13C NMR (75 MHz, Acetone-d6) δ 172.2, 172.0, 138.7 (0.6C), 138.5 (0.4C), 136.5 (0.6C), 135.6 (0.4C),
130.5 (0.8C), 130.4 (1.2C), 129.3 (1.2C), 129.2 (0.8C), 128.0 (0.6C), 127.9 (0.4C), 124.5, 123.6 (0.4C), 123.5 (0.6C), 63.6 (0.4C), 63.5 (0.6C), 54.9, 52.1 (0.6C), 52.0 (0.4C), 49.5 (0.6C), 49.4 (0.4C), 40.3 (0.4C), 39.9 (0.6C), 38.2 (0.6C), 37.4 (0.4C), 36.6, 25.2 (0.6C), 25.1 (0.4C), 23.1, 21.6 (0.4C), 21.5 (0.6C); HRMS (FAB) C21H31N4O3 (M+)に対する計算値 387.2395, 測定値 387.2396。
【0087】
イオン液体担持Leu-Phe-Boc-グリシン 7:泡状淡黄色固体; 1H NMR (300 MHz, Acetone-d6) δ 9.04 (s, 1H), 7.78 (t, J = 1.8 Hz, 1H), 7.66 (t, J = 1.8 Hz, 1H), 7.62 (d, J = 7.5 Hz, 1H), 7.55 (d, J = 8.1 Hz, 1H), 7.30-7.18 (m, 5H), 6.46 (m, 1H), 4.73-4.65 (m, 3H), 4.54-4.51 (m, 2H), 4.45-4.38 (m, 1H), 4.06 (s, 3H), 3.78-3.61 (m, 2H), 3.24-3.00 (m, 2H), 1.71-1.56 (m, 3H), 1.39 (s, 9H), 0.90 (d, J = 6.0 Hz, 3H), 0.88 (d, J = 6.0 Hz, 3H); 13C NMR (75 MHz,
Acetone-d6) δ 171.7 (2C), 170.3, 156.7, 137.7, 137.5, 129.4 (2C), 128.6 (2C), 126.8, 124.1, 123.0, 79.3, 63.1, 54.9, 51.3, 48.9, 44.4, 39.9, 37.4, 36.2, 28.0 (3C), 24.7, 22.7, 21.2; HRMS (FAB) C28H42N5O6 (M+)に対する計算値 544.3134, 測定値 544.3135。
トリフルオロ酢酸による処理で、イオン液体担持Leu-Phe-グリシンTFA塩 8が得られた: 1H NMR (300 MHz, Acetone-d6) δ 9.13 (s, 1 H), 7.80 (s, 1H), 7.68 (s, 1H), 7.32-7.20 (m, 5H), 4.66-4.50 (m, 7H), 4.41-4.39 (m, 1H), 4.06 (s, 3H), 3.25-2.96 (m, 2H), 1.77-1.68 (m, 2H), 1.59-1.54 (m, 1H), 0.92 (d, J = 6.0 Hz, 3H), 0.87 (d, J = 6.0 Hz, 3H); 13C NMR (75 MHz, Acetone-d6) δ 172.4, 172.3, 172.2, 138.4 (0.5C), 138.3 (0.5C), 138.2 (0.5C), 138.0 (0.5C), 130.0 (2C), 128.94 (1C), 128.91 (1C), 127.22 (0.5C), 127.17 (0.5C), 124.57 (0.5C), 124.54 (0.5C), 123.44 (0.5C), 123.40 (0.5C), 63.4, 56.8 (0.5C), 56.6 (0.5C), 51.6 (0.5C), 51.5 (0.5C), 49.5 (0.5C), 49.3 (0.5C), 42.8, 39.9 (0.5C), 39.8 (0.5C), 38.4 (0.5C), 38.3 (0.5C), 36.6, 25.2 (0.5C), 25.16 (0.5C), 23.17 (0.5C), 23.15 (0.5C), 21.5 (0.5C), 21.4 (0.5C); HRMS (FAB) C23H34N5O4 (M+) に対する計算値444.2609, 測定値 444.2611。
【0088】
イオン液体担持Leu-Phe-Gly-Boc-グリシン 9:泡状淡黄色固体; 1H NMR (300 MHz, Acetone-d6) δ 9.09 (s, 1H), 8.12 (m, br, 1H), 7.80 (t, J = 1.8 Hz, 1H), 7.68 (t, J = 1.8 Hz, 1H), 7.64 (m, 1 H), 7.61 (m, 1 H), 7.30-7.21 (m, 5H), 6.57 (m, 1H), 4.76-4.59 (m, 3H), 4.54-4.51 (m, 2H), 4.40-4.33 (m, 1H), 4.08 (s, 3H), 3.82-3.75 (m, 4H), 3.33-2.95 (m, 2H), 1.89-1.55 (m, 3H), 1.43 (s, 9H), 0.94 (d, J = 6.0 Hz, 3H), 0.89 (d, J = 6.0 Hz, 3H); 13C NMR (100 MHz, Acetone-d6) δ 171.9, 171.8, 171.6, 169.8, 156.9, 138.0, 137.8, 129.1 (2C), 128.6 (2C), 126.7, 124.1, 123.0, 79.4, 63.1, 55.6, 51.4, 48.9, 44.6, 43.8, 39.9, 37.3, 36.3, 28.2 (3C), 24.7, 22.9, 21.2; HRMS (FAB) C30H45N6O7 (M+) に対する計算値601.3348, 測定値 601.3350。
トリフルオロ酢酸による処理で、イオン液体担持Leu-Phe-Gly-グリシンTFA塩 10が得られた: 1H NMR (400 MHz, Acetone-d6) δ 9.10 (s, 1H), 7.81 (s, 1H), 7.70 (s, 1H), 7.28-7.20 (m, 5H), 4.75-4.67 (m, 5H), 4.58-4.50 (m, 2H), 4.43-4.38 (m, 1H), 4.08 (s, 3H), 3.89-2.87 (m, 2H), 3.23-3.04 (m, 2H), 1.65-1.58 (m, 3H), 0.92 (d, J = 6.0 Hz, 3H), 0.87 (d, J = 6.0 Hz, 3H); 13C NMR (100 MHz, Acetone-d6) δ 171.9, 171.7, 137.9, 129.4, 129.3 (2C), 128.4 (2C), 126.7, 124.0, 123.0, 63.0, 55.8, 50.8, 49.3, 48.9, 43.5, 39.7, 37.3, 36.3, 24.8, 22.7, 21.2.; HRMS (FAB) C25H37N6O5 (M+) に対する計算値501.2826, 測定値501.2825。
【0089】
イオン液体担持Leu-Phe-Gly-Gly-Boc-チロシン 11:泡状淡黄色固体; 1H NMR (400 MHz, Acetone-d6) δ 9.08 (s, 1H), 8.21 (s, br, 1H), 8.07 (s, br, 1H), 7.80 (s, 1H), 7.68 (d, J = 7.2 Hz, 1H), 7.67 (s, 1H), 7.56 (d, J = 7.2 Hz, 1H), 7.34-7.20 (m, 5H), 7.15 (d, J = 8.0 Hz, 2H), 6.90 (d, J = 8.0 Hz, 2H), 6.49 (d, J = 7.2 Hz, 1H), 4.76-4.56 (m, 3H), 4.54-4.51 (m, 2H), 4.40-4.33 (m, 2H), 4.08 (s, 3H), 3.96-3.66 (m, 4H), 3.31-2.91 (m, 4H), 1.85-1.70 (m, 2H), 1.62-1.55 (m, 1H), 1.36 (s, 9H), 1.30 (s, 9H), 0.94 (d, J = 6.0 Hz, 3H), 0.89 (d, J = 6.0 Hz, 3H); 13C NMR (100 MHz, Acetone-d6) δ 174.0, 171.9, 171.7, 171.3, 169.8, 156.4, 154.5, 138.1, 137.8, 132.2, 129.9(2C), 129.3 (2C), 128.6 (2C), 126.7, 124.1, 124.0 (2C), 122.9, 79.6, 77.9, 63.2, 57.2, 56.0, 51.4, 48.5, 44.0, 43.8, 39.8, 37.4, 37.0, 36.3, 28.6 (3C), 28.1 (3C), 24.8, 22.9, 21.2; HRMS (FAB) C43H62N7O9 (M+) に対する計算値820.4605, 測定値 820.4609。
【0090】
〔担持及びペプチド形成ステップにおけるラセミ化の程度の測定(それがある場合)〕
イオン液体担持Leu-Phe-Boc 4a〜dの調製: 4a〜dは、上に記載したとおりの一般手順を用いて、イオン液体担持ロイシン2a〜b(L-及びD-エナンチオマー)と、Boc-L-フェニルアラニン及びBoc-DL-フェニルアラニンとの反応で調製した。4a:L,L-エピマー、4b:D,L-エピマー、4c:L,DL-エピマー、及び4d:D,DL-エピマー。
【0091】
メチルN-(tert-ブチルオキシカルボニル)-フェニルアラニル-ロイシネート5a〜d:
先のステップからの4a(0.1mmol)を、アンモニアで飽和させたメタノール(2mL)中に溶かした。得られた溶液を、次に、室温で夜通し撹拌した。減圧下で溶媒を除去した後、残渣をフラッシュカラムクロマトグラフィー(シリカゲル230〜400メッシュ、ヘキサン−酢酸エチル勾配)で精製してジペプチド生成物MeO-Leu(L)-Phe(L)-Boc5a-(L,L)を得た。同様にして、ジペプチドMeO-Leu(D)-Phe(L)-Boc 5b-(D,L)、MeO-Leu(L)-Phe(DL)-Boc 5c-(L,DL)、及びMeO-Leu(D)-Phe(DL)-Boc 5d-(D,DL)も調製した。HPLC分析:Diacel Chiralcel OD-Hカラム、ヘキサン中5%イソプロパノール、0.5mL/分、λ=220nm、tR(L,L-エピマー):13.3分、tR(D,L-エピマー):13.4分;tR(L,D-エピマー):15.7分;tR(D,D-エピマー):22.2分; Diacel Chiralcel OD-Jカラム、ヘキサン中5%イソプロパノール、0.3mL/分、λ=220nm、tR(L,L-エピマー):16.0分、tR(D,L-エピマー):17.4分。
【0092】
MeO-Leu(L)-Phe(L)-Boc 5a-(L,L)51:白色固体、m.p. 101-103 °C (文献17: m.p. 101-104 °C). 1H NMR (400 MHz, Acetone-d6) δ 7.52 (d, J = 7.6 Hz,
1H), 7.26-7.18 (m, 5H), 6.07 (d, J = 8.0 Hz, 1H), 4.56-4.50 (m, 1H), 4.42-4.38 (m, 1H), 3.68 (s, 3H), 3.20-2.88 (m, 2H), 1.75-1.68 (m, 1H), 1.62-1.58 (m, 2H), 1.35 (s, 9H), 0.92 (d, J = 6.4 Hz, 3H), 0.91 (d, J = 6.4 Hz, 3H)。 HPLC分析では、いかなる検出可能な量のD,L-エピマー、L,D-エピマー、及びD,D-エピマーも示さなかった。
【0093】
MeO-Leu(D)-Phe(L)-Boc 5b-(D,L)51:白色結晶、m.p.136-138 °C. 1H NMR (400 MHz, Acetone-d6) δ 7.44 (d, J = 7.2 Hz, 1 H), 7.28-7.18 (m, 5H), 6.06 (d, J = 8.0 Hz, 1H), 4.48-4.42 (m, 1H), 4.41-4.35 (m, 1H), 3.66 (s, 3H), 3.11-2.93 (m, 2H), 1.55-1.52 (m, 3H), 1.36 (s, 9H), 0.88 (d, J = 6.4 Hz, 3H), 0.87 (d, J = 6.4 Hz, 3H)。HPLC分析ではいかなる検出可能な量のL,L-エピマー、L,D-エピマー、及びD,D-エピマーも示さなかった。
【0094】
〔ペプチド遊離(切り離し)ステップ〕
THF/H2O(1:2、v/v、3.0mL)中のイオン液体担持ペンタペプチド11(180mg、0.2mmol)の混合物に、1M NaOH水溶液(0.2mL)を加えた。混合物を室温で5時間撹拌した。揮発性成分を減圧下で除去し、残った溶液をpH5〜6に酸性化した。沈殿物を蒸留水で2回洗浄し、真空下で乾燥して120mg(85%)のBoc-ペンタペプチド12を泡状淡黄色固体として得た。1H NMR (400 MHz, Acetone-d6) δ 8.01 (s, br, 1H), 7.80 (s, br, 1H), 7.65 (d, J = 6.8 Hz, 2H), 7.30-7.15 (m, 5H),
7.17 (d, J = 8.0 Hz, 2H), 6.89 (d, J = 8.0 Hz, 2H), 6.43 (d, J = 7.2 Hz, 1H), 4.76-4.74 (m, 1H), 4.52-4.47 (m, 1H), 4.42-4.39 (m, 1H), 4.01-3.74 (m, 4H), 3.28-2.90 (m, 4H), 1.79-1.65 (m, 3H), 1.35 (s, 9H), 1.30 (s, 9H), 0.94 (d, J = 6.0 Hz, 3H), 0.92 (d, J = 6.0 Hz, 3H); 13C NMR (100 MHz, Acetone-d6) δ 173.5, 173.2, 171.4, 170.0, 169.0, 156.1, 154.3, 138.0, 132,6, 130.0 (2C), 129.6 (2C), 128.4 (2C), 126.5, 124.0 (2C), 79.2, 77.8, 56.8, 54.9, 51.1, 43.5, 43.1, 40.8, 37.9, 37.3, 28.7 (3C), 28.2 (3C), 25.0, 23.0, 21.5; HRMS (FAB) C37H53N5O9Na (M+Na) に対する計算値734.3741, 測定値 734.3742。
【0095】
Boc-ペンタペプチド12(117mg、0.16mmol)をCH2Cl2(1.5mL)中に溶かし、次にTFA(1.5mL)とアニソールを数滴添加した。反応混合物を常温において窒素雰囲気下で0.5時間撹拌した。蒸発とエーテルでの洗浄によって、Leu5-エンケファリンをTFA塩14として、淡黄色固体として得た(109mg、99%)。m.p. 148-152 °C (真正サンプルのm.p. 150-153 °C); [α]20D = 25.0 (c 0.8, 95% AcOH), 真正サンプル[α]20D = 25.6 (c 0.9, 95% AcOH); HPLC tR = 16.0 min, (真正サンプル tR = 15.7 min), Agilent Zorbax SB-CN Semi-prepカラム; 溶離液, 0-5 min, H20-0.05%TFA (v/v), 6-15 min, 0-60% CH3CN/H2O-0.05%TFA (v/v/v), 16-23 min, 60-100% CH3CN/H2O-0.05%TFA (v/v/v); ポスト時間, 7分; 流速, 3.0 ml/min; 1H NMR (400 MHz, Methanol-d4) δ 7.24-7.21 (m, 4H), 7.18-7.15 (m, 1H), 7.08 (d, J = 8.4 Hz, 2H), 6.77 (d, J = 8.4 Hz, 2H), 4.71-4,67 (m, 1H), 4.42-4.39 (m, 1H), 4.07 (t, J= 7.2 Hz, 1H), 3.96-3.72 (m, 4H), 3.19-2.92 (m, 4H), 1.71-1.61 (m, 3H), 0.94 (d, J = 6.0 Hz, 3H), 0.90 (d, J = 6.0 Hz, 3H); 13C NMR (100 MHz, Methanol-d4) δ 175.1, 173.1, 171.0, 170.6, 170.4, 158.0, 138.0, 131.3 (2C), 130.2 (2C), 129.2 (2C), 127.5, 125.8, 116.8 (2C), 56.2, 55.6, 52.3, 43.9, 43.4, 41.7, 39.0, 37.7, 26.0, 23.2, 22.0; HRMS (FAB) C28H37N5O7Na (M+Na) に対する計算値578.2592, 測定値 578.2591。
【0096】
フェニル2,3,4-トリ-O-ベンジル-1-チオ-β-D-グルコピラノシド(15):
フェニル2,3,4-トリ-O-ベンジル-6-O-トリメチルアセチル-1-チオ-β-D-グルコピラノシド(3.02g、4.82mmol)を入れたフラスコに、NaOCH3/HOCH3溶液(50mL)とCH2Cl2(15mL)(pH=13)を添加した。溶液を室温で48時間撹拌し、蒸発により溶媒を除去し、真空中で乾燥させた。無水CH2Cl2を粗精製物残渣に添加し、不溶性塩を、濾剤であるセライト(登録商標)521を詰めたブフナーロートで濾別した。濾液を集め、溶媒をロータリーエバポレーターで除去し、真空中で乾燥させて生成物を白色固体として得た(2.61g、収率100%)。m.p.127-129 °C; 1H NMR (400MHz, CDCl3), δ 7.51 (m, 2H), 7.40-7.27 (m, 18H), 4.94-4.85 (m, 4H), 4.77 (d, J=l0Hz, 1H), 4.73 (d, J=9.6Hz, 1H), 4.66 (d, J=10.8Hz, 1H), 3.89 (dd, J=2.8Hz, 14.8Hz, H6a, 1H), 3.77-3.69 (m, 2H), 3.59 (t, J=8.8Hz, 1H), 3.50 (t, J=8.8Hz, 1H), 3.40 (m, 1H); 13C NMR (400MHz, CDCl3), δ 138.49, 138.09, 138.02, 133.67, 132.06, 129.25, 128.73-127.89 (m), 87.82, 86.84, 81.41, 79.61, 77.90, 76.13, 75.85, 75.43, 62.48; ESI-MS, C33H34SO5 (M+Na+)に対する計算値: m/z=565, 測定値: 565。
【0097】
フェニル2,3,4-トリ-O-ベンジル-6-O-ブロモアセチル-1-チオ-β-D-グルコピラノシド(16a):
CH2Cl2(40mL)中のフェニル2,3,4-トリ-O-ベンジル-1-チオ-β-D-グルコピラノシド(15)(0.988g、1.82mmol)及びブロモ酢酸(0.308g、2.21mmol)の溶液に、CH2Cl2中のDCCの1.0M溶液(2.20mL、2.20mmol)及び少量のDMAP(糖に対して<5%当量)を添加した。室温で2時間撹拌した後、混合物を濾過して副生成物の1,3-ジシクロヘキシルウレアを除去した。乾燥するまで濾液を蒸発させ、溶離液としてヘキサンと酢酸エチル(7:1、V/V)を用いて、残渣をフラッシュカラムクロマトグラフィー(シリカゲル60A、230〜400メッシュ)にかけて純粋な生成物を得た(1.08g、収率89%)。
m.p. 85-87 °C; 1H NMR (400MHz, CDCl3), δ 7.54 (m, 2H), 7.41-7.25 (m, 18H), 4.95-4.84 (m, 4H), 4.75 (d, J=l0Hz, 1H), 4.67 (d, J=9.6Hz, 1H), 4.61 (d, J=10.8Hz, 1H), 4.45 (dd, J=11.2Hz, 0.8Hz, 1H), 4.28 (m, 1H), 3.79 (s, 2H), 3.73 (m, 1H), 3.55 (m, 2H), 3.51 (t, J=18.4Hz, 1H); 13C NMR (300MHz, CDCl3), δ 166.91, 138.29, 138.02, 137.70, 133.51, 132.39, 129.14, 128.76-127.94 (m), 87.81, 86.96, 81.06, 77.49, 77.36, 76.19, 75.79, 75.38, 65.07, 25.90; ESI-MS, C35H35SO6Br (M+Na+)に対する計算値: m/z=685, 測定値: 685。
【0098】
2,3,4-トリ-O-ベンジル-6-O-[2-(3-メチル-イミダゾリウム)]アセチル-1-チオ-β-D-グルコピラノシドテトラフルオロボレート(16b):
アセトン(25mL)中のフェニル2,3,4-トリ-O-ベンジル-6-O-ブロモアセチル-1-チオ-β-D-グルコピラノシド(16a)(0.890g、1.34mmol)に、CH2Cl2中の1-メチルイミダゾールの1.0M溶液(1.61mL、1.61mmol)を加え、さらにNaBF4(0.182g、1.66mmol)を加えた。室温での72時間の撹拌後、沈殿物を濾別し、乾燥するまで濾液を蒸発させ、真空中で夜通し乾燥させた。生成物残渣をジエチルエーテル(15mL)で3回洗浄し、上澄みをデカンテーションした。固体を真空中で乾燥させ、15mLの無水CH2Cl2を添加し、沈殿物を濾過することにより、残渣をさらに精製した。乾燥するまで濾液を蒸発させ、真空中で乾燥させて、白色泡状物として純粋な生成物を得た(0.989g、収率98%)。
m.p. 42-44 °C; 1H NMR (400MHz, CDCl3), δ 8.74 (s, 1H), 7.46 (m, 2H), 7.36-7.23 (m, 18H), 7.17 (m, 2H), 5.01-4.82 (m, 6H), 4.72 (d, J=10.0Hz, 1H), 4.71(d, J=9.6Hz, 1H), 4.58 (d, J=11.2Hz, 1H), 4.50 (dd, J=11.6Hz, 2.4Hz, 1H), 4.24 (dd, J=6.4Hz, 12Hz, 1H), 3.82 (s, 3H), 3.73 (m, 1H), 3.58 (m, 1H), 3.52-3.43 (m, 2H); 13C NMR (300MHz, CDCl3), δ 165.90, 138.33, 138.07, 138.02, 137.83, 133.65, 131.58, 129.37, 128.76-127.92 (m), 123.69, 123.25, 87.41, 86.79, 81.02, 77.53, 76.74, 76.01, 75.70, 75.15, 65.51, 50.03, 36.81; ESI-MS C39H41SO6N2 (M+)カチオンに対する計算値: m/z = 665, 測定値: 665。
【0099】
フェニル2,3,4-トリ-O-ベンジル-6-O-[2-(3-メチル-イミダゾリウム)]アセチル-1-チオ-β-D-グルコピラノシルスルホキシドテトラフルオロボレート(17b):
−78℃で、CH2Cl2(2mL)中の16b(50.0mg、0.066mmol)の溶液に、CH2Cl2(1mL)中のm-クロロ過安息香酸(m-CPBA)(15.0mg、0.066mmol)を5分にわたって滴下して加えた。−78℃で20分撹拌後、混合物を15mLのCH2Cl2とともにNaHCO3飽和水溶液(10mL)中に注いだ。有機相を分離し、飽和NaHCO3水溶液(10mL)で洗浄し、無水Na2SO4上で乾燥させた。溶媒を減圧下にてロータリーエバポレーターで除去した。生成物残渣を、ヘキサンとイソプロピルエーテルで洗浄することによってさらに精製(TLCで観察)して、2つのジアステレオマーを含む白色固体としてグリコシルスルホキシドを得た(49.5mg;収率97%;NMRによって測定した2つのジアステレオマーの比は60:40)。
1H NMR (400MHz, CDCl3), δ 9.08 (s, 1H), 7.54 (m, 2H), 7.40-7.16 (m, 20H), 5.11-4.20 (m, 11H), 3.97 (m, 2H), 3.91 (s, 2H), 3.90 (s, 1H), 3.85-3.26 (m, 2H); ESI-MS C39H41SO7N2 (M+)カチオンに対する計算値: m/z = 681, 測定値: 681。
【0100】
フェニル2,3,4-トリ-O-ベンジル-6-O-[2-(3-メチル-イミダゾリウム)]アセチル-β-D-グルコピラノシル-(1→6)-2,3,4-トリ-O-ベンジル-1-チオ-β-D-グルコピラノシド テトラフルオロボレート(18b):
−78℃で、CH2Cl2(5mL)中の、イオン液体に結合したモノサッカライドスルホキシド(51.8mg、0.067mmol;グリコシル供与体)、フェニル2,3,4-トリ-O-ベンジル-1-チオ-β-D-グルコピラノシド(15、114mg、0.21mmol;グリコシル受容体)、及び2,6-ジ-tert-ブチル-4-メチルピリジン(58.8mg、0.286mmol;酸除去剤)の溶液に、Tf2O(12μL、0.067mmol)をシリンジから10分間にわたって徐々に添加した。−78℃で20分間撹拌後、混合物を1時間にわたって0℃までゆっくり温めた。白色沈殿物の出現をもたらすn-ペンタン(5mL)を添加することによって反応を止めた。混合物を−78℃に冷却し、白色沈殿物をすぐに遠心分離した。溶液相にn-ペンタンのさらなる一部(5mL)を加え、次に−78℃に冷却した。得られた沈殿物を再び遠心分離した。溶液相を蒸発させ、残渣を数回n-ペンタンで洗い、次にTLC分析が、残渣が純粋であることを示すまで、イソプロピルエーテルでさらに洗った。白色固体として生成物を得た(39mg、収率50%)。
m.p. 45-47°C; 1H NMR (400MHz, CDCl3), δ 9.10 (s, 1 H), 7.51 (m, 2H), 7.41-7.13 (m, 35H), 5.10 (d, J=10.4Hz, 1H), 5.00-4.52 (m, 15H), 4.41 (m, 1H), 4.19 (dd, J=12.4Hz, 4.0Hz, 1H), 3.99 (t, J=8.8Hz, 1H), 3.90-3.75 (m, 6H), 3.73-3.62 (m, 2H), 3.55 (m, 2H), 3.42 (t, J=8.8Hz,
1H), 3.29 (t, 1H); 13C NMR (400MHz, CDCl3), δ=165.79, 138.78, 138.60, 138.41, 138.38, 138.20, 138.11, 138.03, 134.18, 131.78, 129.32, 128.73-127.69 (m), 123.65, 123.24, 97.30, 88.25, 86.88, 81.82, 81.31, 80.30, 79.06, 77.76, 77.57, 75.99, 75.89, 75.81, 75.25, 75.09, 72.83, 68.68, 66.89, 65.49, 50.51, 37.16; ESI-MS C66H69SO11N2 (M+)カチオンに対する計算値: m/z= 1097, 測定値: 1097。
【0101】
フェニル2,3,4-トリ-O-ベンジル-6-O-[2-(3-メチル-イミダゾリウム)]アセチル-β-D-グルコピラノシル-(1→6)-2,3,4-トリ-O-ベンジル-1-チオ-β-D-グルコピラノシルスルホキシド テトラフルオロボレート(19b):
−78℃で、CH2Cl2(2.5mL)中のジサッカライドスルフィド18b(56.1mg、0.047mmol)溶液に、5分間にわたって、CH2Cl2(1mL)中のm-クロロ過安息香酸(m-CPBA)(8.1mg、0.047mmol)を5分間にわたって滴下して加えた。窒素雰囲気保護下、−78℃で20分間撹拌後、混合物をCH2Cl2(15mL)とともに飽和NaHCO3水溶液(10mL)中に注いだ。有機相を分離し、再度、飽和NaHCO3水溶液(10mL)で洗い、無水Na2SO4上で乾燥させた。溶媒を、減圧下でロータリーエバポレーターによって除去した。生成物残渣をヘキサンとイソプロピルエーテルで洗浄すること(TLCでモニターした)によって、さらに精製して、2つのジアステレオマーを含む白色固体として所望するグリコシルスルホキシドを得た(54.0mg、収率95%、NMRで測定した2つのジアステレオマーの比は60:40)。
1H NMR (400MHz, CDCl3), δ 9.48 (s, 1H), 7.58-7.13 (m, 37H), 5.00-4.40 (m, 18H), 3.95-3.25 (m, 13H); ESI-MS C66H69SO12N2 (M+)カチオンに対する計算値: m/z = 1113, 測定値: 1113。
【0102】
フェニル2,3,4-トリ-O-ベンジル-6-O-[2-(3-メチル-イミダゾリウム)]アセチル-β-D-グルコピラノシル-(1→6)-2,3,4-トリ-O-ベンジル-β-D-グルコピラノシル-(1→6)-2,3,4-トリ-O-ベンジル-1-チオ-β-D-グルコピラノシド テトラフルオロボレート(20b):
イオン液体に結合したジサッカライドスルホキシド(19b;45.1mg;0.038mmol;グリコシル供与体)、フェニル2,3,4-トリ-O-ベンジル-1-チオ-β-D-グルコピラノシド(15;62.1mg;0.114mmol;グリコシル受容体)、及び2,6-ジ-tert-ブチル-4-メチルピリジン(30.8mg;0.150mmol;酸除去剤)のCH2Cl2(5mL)中の溶液に、−78℃で、シリンジから10分間にわたってTf2O(6μL、0.036mmol)を徐々に添加した。−78℃で20分間撹拌後、混合物を1時間にわたってゆっくりと0℃まで温めた。白色沈殿の出現をもたらすn-ペンタン(5mL)を添加することによって、反応混合物を失活させた。反応混合物を−78℃に冷却し、沈殿物を直ちに遠心分離した。溶液相にn-ペンタンの別の一部(5mL)を加え、次に−78℃に冷却した。得られた沈殿物を再び遠心分離によって除去した。溶液相を蒸発させ、残渣を数回n-ペンタンで洗い、次にTLC分析が、残渣が純粋であることを示すまで、イソプロピルエーテルでさらに洗った。白色固体として所望の生成物を得た(34mg、収率56%)。
1H NMR (400MHz, CDCl3), δ 9.04 (s, 1H), 7.51 (m, 2H), 7.42-7.17 (m, 48H), 7.05 (d, J=10.2Hz, 2H), 5.00-4.50 (m, 25H), 4.39 (d, J=8.0Hz, 1H), 4.15 (m, 1H), 4.00 (m, 2H), 3.89-3.72 (m, 6H), 3.70-3.30 (m, 8H), 3.25 (t, J=8.8Hz, 1H); 13C NMR (400MHz, CDCl3), δ 165.77, 138.95-138.09 (m), 134.22, 131.87, 131.31, 129.29, 128.74-127.36 (m), 123.72, 123.76, 123.01, 103.91,97.47, 97.26, 88.33, 87.74, 86.85, 84.88, 82.66-80.14 (m), 79.21, 79.06, 78.67, 77.54, 75.93-75.85 (m), 75.22-75.00 (m), 72.79, 72.72, 72.51, 70.86, 68.90, 69.58, 68.31, 66.44, 65.81, 65.53, 50.01, 36.90; ESI-MS C93H97SO16N2 (M+)カチオンに対する計算値: m/z= 1529, 測定値: 1529。
【0103】
トリサッカライド21を得るための、20bからのイオン液体残基の切り離し:
イオン液体に結合したトリサッカライド(20b、30.0mg、0.018mmol)を入れたフラスコに、Cs2CO3(6.1mg、0.018mmol)とメタノール(5mL)を添加した。室温での12時間撹拌後に、溶媒をロータリーエバポレーターによって除去し、残渣を減圧下で乾燥させた。この残渣に無水CH2Cl2(3mL)を添加し、不溶性塩を濾過により分離した。濾液を乾燥するまで蒸発させ、減圧下で乾燥させて所望の生成物21を得た(26.1mg、収率100%)。
1H NMR (400MHz, CDCl3), δ 7.54 (m, 2H), 7.43-7.14 (m, 48H), 5.16 (d, J=3.2Hz, 1H), 5.00-4.93 (m, 3H), 4.91-4.86 (m, 4H), 4.85- 4.74 (m, 5H), 4.71-4.53 (m, 9H), 4.40 (d, J=8.0Hz, 1H), 4.12 (d, J=10.0Hz, 1H), 3.99 (m, 2H), 3.86-3.38 (m, 12H), 3.26 (m, 1H); 13C NMR (400MHz, CDCl3), δ 138.94-138.22 (m), 134.33, 132.01, 131.55, 129.19, 129.10, 128.59-127.44 (m), 103.91, 97.54, 97.31, 88.31, 87.79, 86.93, 84.81, 82.62, 81.95, 81.85, 81.75, 81.44, 81.33, 80.73, 80.63, 80.49, 79.23, 79.00, 78.12, 77.91, 77.75, 77.51, 75.95-75.80 (m), 75.45-75.25 (m), 72.75,
72.61, 72.57, 71.24, 68.65, 66.02, 65.52, 62.30; ESI-MS C87H90SO15Na
(M+Na+)に対する計算値: m/z= 1429, 測定値: 1429。
【0104】
フェニル2,3,4-トリ-O-ベンジル-6-O-アセチル-1-チオ-β-D-グルコピラノシド(16c):
フェニル2,3,4-トリ-O-ベンジル-1-チオ-β-D-グルコピラノシド(15)(0.801g、1.48mmol)、無水ピリジン(0.45mL)、及びDMAP(糖に対して5当量%未満)のCH2Cl2(9mL)中の溶液に、無水酢酸(0.21mL、2.22mmol)を添加した。窒素雰囲気の保護下で、室温にて4時間撹拌した後、反応混合物を150mLのCH2Cl2中に移し、0.2M HCl水溶液(30mL)、H2O(30mL)、飽和NaHCO3水溶液(30mL)、そして次に食塩水(30mL)で、それぞれ洗った。有機相を無水Na2SO4上で乾燥させ、溶媒をロータリーエバポレーターによって除去して、粗生成物残渣を得、これを溶離液としてヘキサン及び酢酸エチル(5:1、V/V)を用いてフラッシュカラムクロマトグラフィー(シリカゲル60A、230〜400メッシュ)にかけて純粋な生成物(0.845g、収率98%)を得た。
1H NMR (400MHz, CDCl3), δ 7.55 (m, 2H), 7.40 (m, 2H), 7.36-7.25 (m, 16H), 4.95-4.84 (m, 4H), 4.75 (d, J=10Hz, 1H), 4.67 (d, J=9.6Hz, 1H), 4.58 (d, J=10.8, 1H), 4.37 (d, J=11.6Hz, 1H), 4.22 (m, 1H), 3.73 (t, J=8.4 Hz, 1H), 3.55 (m, 2H), 3.50 (t, J=9.6Hz, 1H), 2.11(s, 3H); ESI-MS C35H36SO6Na (M+Na+)に対する計算値: m/z=607, 測定値: 607。
【0105】
フェニル2,3,4-トリ-O-ベンジル-6-O-アセチル-β-D-グルコピラノシルスルホキシド(17c):
−78℃で、CH2Cl2(5mL)中の16c(1.01g、1.73mmol)の溶液に、CH2Cl2(7mL)中のm-クロロ過安息香酸(m-CPBA)(0.387g、1.73mmol)を5分にわたって滴下して加えた。窒素雰囲気の保護下で−78℃にて20分撹拌後、混合物をCH2Cl2(50mL)とともにNaHCO3飽和水溶液(100mL)中に注いだ。有機相を分離し、飽和NaHCO3水溶液(50mL)で再度洗浄し、無水Na2SO4上で乾燥させた。溶媒を減圧下にてロータリーエバポレーターで除去して粗生成物残渣を得、これをヘキサン及び酢酸エチル(3:1、V/V)を用いるフラッシュカラムクロマトグラフィー(シリカゲル60A、230〜400メッシュ)にかけて、2つのジアステレオマーの混合物として、純粋な生成物を得た(0.984g、収率95%、NMRによって測定した2つのジアステレオマーの比は3:1)。
1H NMR (400MHz, CDCl3), δ 7.62-7.16 (m, 20H), 5.06-4.78 (m, 6H), 4.59-4.36 (m, 2H), 4.19-3.30 (m, 8H); SI-MS C35H36SO7Na (M+Na+)に対する計算値: m/z=623, 測定値: 623。
【0106】
フェニル2,3,4-トリ-O-ベンジル-6-O-アセチル-β-D-グルコピラノシル-(1→6)-2,3,4-トリ-O-ベンジル-1-チオ-β-D-グルコピラノシド(18c):
スルホキシド 17c(0.302g、0.503mmol)、フェニル2,3,4-トリ-O-ベンジル-1-チオ-β-D-グルコピラノシド(15、0.406g、0.754mmol)及び2,6-ジ-tert-ブチル-4-メチルピリジン(0.315g、1.53mmol)のCH2Cl2(10mL)中の溶液に、−78℃で、CH2Cl2(5mL)中のTf2O溶液(0.09mL、0.503mmol)をシリンジから10分間にわたって徐々に添加した。−78℃で20分間撹拌後、混合物を1.5時間にわたって−5℃までゆっくり温めた。飽和NaHCO3水溶液(6mL)を添加することによって反応物を失活させ、CH2Cl2(35mL)と飽和NaHCO3水溶液(20mL)との間で分配させた。有機相を分離し、飽和NaHCO3水溶液(40mL)でさらに洗った。無水Na2SO4上で乾燥させた後、有機相を乾燥するまで蒸発させ、残渣を、溶離液としてヘキサン及び酢酸エチル(6:1、5:1、4:1、V/V)を用いるフラッシュカラムクロマトグラフィーにかけて、無色シロップとして純粋な生成物を得た(0.414g、収率81%)。
1H NMR (400MHz, CDCl3), δ 7.54 (m, 2H), 7.42 (m, 2H), 7.38-7.24 (m, 31 H), 5.05 (m, 2H), 4.94-4.56 (m, 12H), 4.25 (m, 2H), 4.02 (t, J=9.2Hz, 1H), 3.92 (m, 1H), 3.86 (d, J=4.8Hz, 1H), 3.80 (m, 1H), 3.75 (m, 1H), 3.70 (t, 1H), 3.62-3.46 (m, 3H), 3.28 (t, J=8.4Hz, 1H), 2.03 (s, 3H); ESI-MS C62H64SO11Na (M+Na+)に対する計算値: m/z=1039, 測定値: 1039。
【0107】
フェニル2,3,4-トリ-O-ベンジル-6-O-アセチル-β-D-グルコピラノシル-(1→6)-2,3,4-トリ-O-ベンジル-β-D-グルコピラノシルスルホキシド(19c):
ジサッカライドスルフィド18c(0.202g、0.199mmol)のCH2Cl2(12mL)中の溶液に、−78℃で、5分間にわたって、CH2Cl2(6mL)中のm-クロロ過安息香酸(m-CPBA)(0.045g、0.199mmol)を5分間にわたって滴下して加えた。窒素雰囲気保護下、−78℃で20分間撹拌後、混合物をCH2Cl2(60mL)とともに飽和NaHCO3水溶液(30mL)中に注いだ。有機相を分離し、再度、飽和NaHCO3水溶液(30mL)で洗い、無水Na2SO4上で乾燥させた。溶媒を、減圧下でロータリーエバポレーターによって除去して粗生成物残渣を得、これを、溶離システムとしてヘキサン及び酢酸エチル(2:1、V/V)を用いるフラッシュカラムクロマトグラフィー(シリカゲル60A、230〜400メッシュ)にかけて、2つのジアステレオマーの混合物として生成物を得た(0.197g、全収率96%、NMRで測定した2つのジアステレオマーの比は4:1)。
第一の異性体、Rf=0.27(2:1、ヘキサン/酢酸エチル、V/V);1H NMR (400MHz, CDCl3), δ7.60 (m, 2H), 7.46 (m, 4H), 7.42-7.22 (m, 29H), 5.05 (d, J=3.6Hz, 1H), 5.00-4.66 (m, 12H), 4.60 (d, J=10.8Hz, 1H), 4.27 (m, 2H), 3.87 (m, 3H), 3.74 (m, 4H), 3.59-3.47 (m, 3H), 3.35 (m, 1H) 2.07 (s, 3H);
第二の異性体、Rf=0.12(2:1、ヘキサン/酢酸エチル、V/V);1H NMR (400MHz, CDCl3), δ 7.51 (m, 2H), 7.39-7.19 (m, 31H), 7.06 (m, 2H), 4.98-4.88 (m, 3H), 4.82-4.52 (m, 10H), 4.43 (d, J=8.8Hz, 2H), 4.25 (m, 2H), 4.39 (t, J=9.6Hz, 1H), 3.90-3.69 (m, 6H), 3.60 (m, 1H), 3.49 (m, 2H), 2.05 (s, 3H). ESI-MS C62H64SO12Na (M+Na+)に対する計算値: m/z=1055, 測定値: 1055。
【0108】
フェニル2,3,4-トリ-O-ベンジル-6-O-アセチル-β-D-グルコピラノシル-(1→6)-2,3,4-トリ-O-ベンジル-β-D-グルコピラノシル-(1→6)-2,3,4-トリ-O-ベンジル-1-チオ-β-D-グルコピラノシド(20c):
スルホキシド19c(0.110g、0.107mmol)、フェニル2,3,4-トリ-O-ベンジル-1-チオ-β-D-グルコピラノシド(15、0.088g、0.161mmol)及び2,6-ジ-tert-ブチル-4-メチルピリジン(0.070g、0.34mmol)のCH2Cl2(6mL)中の溶液に、−78℃で、シリンジから10分間にわたってCH2Cl2(3mL)中のTf2O(0.018mL、0.107mmol)を徐々に添加した。−78℃で20分間撹拌後、混合物を1時間にわたってゆっくりと−15℃まで温めた。飽和NaHCO3水溶液(6mL)を添加することによって反応物を失活させ、CH2Cl2(30mL)と飽和NaHCO3水溶液(15mL)との間で分配させた。有機相を分離し、飽和NaHCO3水溶液(20mL)でさらに洗った。無水Na2SO4上で乾燥させた後、有機相を乾燥するまで蒸発させ、粗製物残渣を、溶離システムとしてヘキサン及び酢酸エチル(4:1、V/V)を用いるフラッシュカラムクロマトグラフィー(シリカゲル60A、230〜400メッシュ)にかけて、無色シロップとして生成物を得た(0.132g、収率85%)。
1H NMR (400MHz, CDCl3), δ 7.53 (m, 2H), 7.44-7.14 (m, 48H), 5.12 (d, J=3.2Hz, 1H), 5.04-4.52 (m, 21H), 4.40 (m, 1H), 4.24-4.12 (m, 2H), 4.00 (m, 11-1), 3.90-3.36 (m, 12H), 3.28 (m, 1H), 2.00 (s, 3H); 13C NMR (400MHz, CDCl3), δ 170.85, 138.99-138.14 (m), 134.27, 134.17,
132.06, 131.66, 129.24, 129.15, 128.64, 128.61-127.48 (m), 103.74, 97.51, 97.23, 88.37, 87.77, 86.91, 86.86, 84.79, 82.59, 81.95, 81.86, 81.41, 81.23, 80.67, 80.36, 80.25, 79.16, 78.99, 78.02 77.56, 75.88 (m), 75.25 (m), 72.71, 72.49, 72.37, 71.02, 69.04, 68.30, 66.39, 65.94, 65.67, 63.47, 63.36, 21.34; ESI-MS C89H92SO16Na (M+Na+)に対する計算値: m/z= 1471, 測定値: 1471。
【0109】
フェニル2,3,4-トリ-O-ベンジル-β-D-グルコピラノシル-(1→6)-2,3,4-トリ-O-ベンジル-β-D-グルコピラノシル-(1→6)-2,3,4-トリ-O-ベンジル-1-チオ-β-D-グルコピラノシド(21):
フェニル2,3,4-トリ-O-ベンジル-6-O-アセチル-β-D-グルコピラノシル-(1→6)-2,3,4-トリ-O-ベンジル-β-D-グルコピラノシル-(1→6)-2,3,4-トリ-O-ベンジル-1-チオ-β-D-グルコピラノシド 20c(101mg、0.069mmol)を入れたフラスコに、Cs2CO3(22.4mg、0.069mmol)とメタノール(6mL)を添加した。室温で14時間撹拌した後、溶媒をロータリーエバポレーターで除去し、未精製残渣を減圧下で乾燥させた。次に、この残渣にCH2Cl2(3mL)を添加し、不溶性塩を濾過により分離した。濾液を乾燥するまで蒸発させ、減圧下で乾燥させて純粋な生成物を得た(96.1mg、収率98%)。Rf=0.28(2:1、ヘキサン/酢酸エチル、V/V);1H NMR (400MHz, CDCl3), δ 7.54 (m, 2H), 7.43-7.14 (m, 48H), 5.16 (d, J=3.2Hz, 1H), 5.00-4.93 (m, 3H), 4.91-4.86 (m, 4H), 4.85- 4.74 (m, 5H), 4.71-4.53 (m, 9H), 4.40 (d, J=8.0Hz, 1H), 4.12 (d, J=10.0Hz, 1H), 3.99 (m, 2H), 3.86-3.38 (m, 12H), 3.26 (m, 1H); 13C NMR (400MHz, CDCl3), δ 138.94-138.22 (m), 134.33, 132.01, 131.55, 129.19, 129.10, 128.59-127.44 (m), 103.91, 97.54, 97.31, 88.31, 87.79, 86.93, 84.81, 82.62, 81.95, 81.85, 81.75, 81.44, 81.33, 80.73, 80.63, 80.49, 79.23, 79.00, 78.12, 77.91, 77.75, 77.51, 75.95-75.80 (m), 75.45-75.25 (m), 72.75, 72.61, 72.57, 71.24, 68.65, 66.02, 65.52, 62.30; マススペクトル用の試料はシリカゲルで濾過してセシウムイオンを除去した; ESI-MS C87H90SO15Na (M+Na+)に対する計算値: m/z=1429, 測定値: 1429。
【0110】
5’-O-モノメトキシトリチル-3’-O-スクシニル-チミジン(23):
化合物22(2.86g、5.56mmol)、無水コハク酸(1.67g、16.7mmol)、及びDMAP(0.34g、2.78mmol)を、オーブンで乾燥してアルゴン置換した50mLフラスコ中で、無水ピリジン(20mL)に溶かした。2日間、室温で撹拌して反応させた。ジクロロメタン:メタノール(9:1)のTLC分析は、出発物質の完全な消失と、単一のより極性の高い生成物の形成を示した。ピリジンを減圧下で除去した。暗褐色の残渣を次にCH2Cl2(100mL)中に溶かし、飽和食塩水で洗った。有機相をNa2SO4上で乾燥させ、次に減圧下で溶媒を除去した。生成物を淡褐色泡状物として高収率で得た(3.14g、92%収率)。
1H NMR (DMSO-d6) δ 11.39 & 8.30 (1H, s, NH), 7.50 (1H, s, CH), 7.37-7.21 (14H, m, Ar-H), 6.18 (1 H, t, J = 8 Hz, CH, H1'), 5.28 (1H, d, J = 6 Hz, CH, H3'), 4.03 (1H, broad s, CH), 3.73 (3H, s, CH3), 3.32-3.19 (2H, m, CH2, H5'&5"), 2.62-2.61 (4H, 分離されないm, 2CH2), 2.51-2.27 (2H, m, CH2), 1.41 (3H, s, CH3); C34H34N2O9Na+ 低分解能ESI-MS 計算値637.2, 測定値: 637.1。
【0111】
MMTTSucc−IL(24):
化合物23(1g、1.63mmol)、置換イミダゾリウムテトラフルオロボレート1(0.38g、1.76mmol)、及びDMAP(0.052g、0.41mmol)を、乾燥し、窒素置換した100mL丸底フラスコに入れた。この混合物に、ジシクロヘキシルカルボジイミド(DCC)(0.68g、3.3mmol)を添加し、次に無水アセトニトリル(20mL)を添加した。反応混合物を3日間、室温で撹拌した。9:1のクロロホルム:メタノールでのTLC分析は、ベースラインから移動しないより極性の高い生成物の形成を示した。反応時に、副生成物であるジシクロヘキシルウレア(DCU)が白色固体として生じた。反応を停止させたときに、DCUは沈殿し、反応混合物を焼結ガラスロートを通して濾過し、数回アセトニトリルで洗った。濾過した溶液を、撹拌したジエチルエーテル−酢酸エチル溶液に滴下して加えることによって、さらなる精製を行った。誘導体化されていないイオン液体1と所望の生成物24からなる、得られた沈殿物は、次にクロロホルムに溶かし、水で抽出した。有機相を硫酸ナトリウム上で乾燥させ、溶媒を蒸発させて、生成物を減圧下で乾燥させた。生成物24を淡褐色泡状物として得た(1.1g、83%収率)。
1H NMR (アセトン-d6) δ 10.03 (1H, s, NH), 9.10 (1H, s, CH), 7.83 (1H, s, CH), 7.71
(1H, s, CH), 7.60 (1H, s, CH), 7.50-6.92 (14H, m, Ar-H), 6.33 (1 H, t, J = 8 Hz, CH, H1'), 5.50 (1H, d, J = 6 Hz, CH, H3'), 4.55-4.51 (2H, m, CH2), 4.70-4.66 (2H, m, CH2), 4.15 (1 H, broad s, CH), 4.05 (3, s, CH3), 3.80 (3H, s, CH3), 3.50-3.40 (2H, m, CH2, H5'&5"), 2.70 (4H, broad s, 2 CH2), 2.61-2.40 (2H, m, CH2), 1.42 (3H, s, CH3); C40H43N4O9+。低分解能ESI-MS 計算値 723.3, 測定値: 723.4。
【0112】
HOTSucc−IL(25):
24(2.96g、3.65mmol)のジクロロメタン(200mL)中の溶液に、ジクロロメタン又はアセトニトリル(100mL)中の3%トリフルオロ酢酸を添加した。TFAの添加時に、溶液は赤橙色になり、撹拌を20分間続けた。生成物を10%酢酸エチル/ジエチルエーテルから沈殿させ、濾過し、最少量の上記酸溶液に再溶解し、再度沈殿させて濾過した。沈殿物を10%酢酸エチル/ジエチルエーテルで濯ぎ、アセトニトリル中に溶かすことによってフィルターから回収し、減圧下で溶媒留去し、化合物25を淡褐色泡状物として得た(1.88g、96%収率)。
1H NMR (DMSO-d6) δ 11.40及び11.39 (合計1H, 2 s, NH), 9.10 (1H, s, CH), 7.76 (1H, s, CH), 7.72及び7.45 (1H, s, CH), 7.70 (1H, s, CH), 6.15 (1H, m, CH, H1'), 5.27及び5.19 (1H, m, CH, H3'), 4.60及び3.60 (2H, m, CH2, H5'及び5"), 4.45 (2H, broad s, CH2), 4.39 (2H, broad s, CH2), 4.24-3.93 (1H, m, CH), 3.86 (3, s, CH3), 2.61-2.60 (4H, 分離されないm, 2 CH2), 2.40-2.16 (2H, m, CH2), 1.77 (3H, s, CH3); C20H27N4O8+ 高分解能ESI-MS 計算値 451.18289, 測定値 451.18234。
【0113】
DMTApTSucc−IL(27a):
化合物25(0.12g、0.22mmol)、アデノシンホスホルアミダイト26a(0.33g、0.38mmol)、及びジシアノイミダゾール(0.33g、2.8mmol)を、50mLの、オーブンで乾燥して窒素置換した丸底フラスコに入れた。この混合物に、無水THF又はアセトニトリル(5mL)を加え、得られた溶液を室温で1〜2時間撹拌した。生成物を、10%酢酸エチル/ジエチルエーテルから2回沈殿させた。この時点で、沈殿物を、アセトニトリル中に再溶解し、2,4,6−コリジン又はピリジン(約300μL)を添加し、次に水性ヨウ素溶液(THF/水(2:1)中0.1M、過剰量)を添加してホスファイトトリエステル中間体を酸化した。5分後、反応混合物を重硫酸ナトリウム水溶液(9mL)で失活させ、90mLのCHCl3で希釈し、50mLの水で抽出した。有機相を無水Na2SO4上で乾燥させ、減圧下で溶媒留去して27aの泡状物を得た(0.259g、89%)。
31P NMR (アセトニトリル-d3) δ -1.324, -1.477; C61H64N10O16P+ 低分解能ESI-MS 計算値 1223.4, 測定値 1223.4。
【0114】
DMTCpTSucc−IL(27b):
化合物25(0.11g、0.20mmol)、シチジンホスホルアミダイト26b(0.35g、0.41mmol)、及びジシアノイミダゾール(0.37g、3.1mmol)を、50mLの、オーブンで乾燥して窒素置換した丸底フラスコに入れた。溶媒の不存在が、反応が起こることを妨げた。無水THF又はアセトニトリル(5mL)をフラスコ中に注入することによって反応を開始させ、その溶液を室温で1〜2時間撹拌した。27aの場合と同じ処理をして、生成物を高収率で27bの泡状物として得た(0.234g、91%)。
31P NMR (アセトニトリル-d3) δ -1.545, -1.754; C60H64N8O17P+ 低分解能ESI-MS 計算値 1199.4, 測定値 1199.4。
【0115】
DMTGpTSucc−IL(27c):
化合物25(0.13g、0.24mmol)、グアノシンホスホルアミダイト26c(0.35g、0.42mmol)及びジシアノイミダゾール(0.38g、3.2mmol)を、50mLの、オーブンで乾燥して窒素置換した丸底フラスコに入れた。溶媒の不存在が、反応が起こることを妨げた。無水THF又はアセトニトリル(5mL)をフラスコ中に注入することによって反応を開始させ、その溶液を室温で1〜2時間撹拌した。27aの場合と同じ処理をして、生成物を高収率で27cの泡状物として得た(0.214g、90%)。
31P NMR (アセトニトリル-d3) δ -1.149, -1.194; C58H66N10O17P+ 低分解能ESI-MS 計算値 1205.4, 測定値 1205.6。
【0116】
DMTTpTSucc−IL(27d):
化合物25(0.24g、0.45mmol)、チミジンホスホルアミダイト26d(0.57g、0.77mmol)、及びジシアノイミダゾール(0.66g、5.6mmol)を、50mLの、オーブンで乾燥して窒素置換した丸底フラスコに入れた。溶媒の不存在が、反応が起こることを妨げた。無水THF又はアセトニトリル(5mL)をフラスコ中に注入することによって反応を開始させ、その溶液を室温で1〜2時間撹拌した。27aの場合と同じ処理をして、生成物を高収率で27dの泡状物として得た(0.492g、91%)。
31P NMR (アセトニトリル-d3) δ -1.494, -1.584; C54H61N7O17P+ 低解像度ESI-MS 計算値 1110.4, 測定値 1110.4。
【0117】
HOApTSucc−IL(28a):
化合物27a(0.26g、0.20mmol)をアセトニトリル(1〜2mL)に溶かし、アセトニトリル(2〜3mL)中の3%トリフルオロ酢酸を添加した。反応混合物を25に対してと同じ方法に従って処理し、泡状物として高収率で28aが得られた(0.185g、93%)。
1H NMR (アセトニトリル-d3) δ 6.47-6.51 (Ade H1'), 6.17-6.22 (Thy H1'), 5.25-5.30 (Ade and Thy H3'), 2.82-3.09 (Thy H5'&5"); 31P NMR (アセトニトリル-d3) δ -1.176, -1.516; C40H46N10O14P+, 高分解能ESI-MS 計算値 921.29326, 測定値 921.29271。
【0118】
HOCpTSucc−IL(28b):
アセトニトリル(1〜2mL)中の化合物27b(0.23g、0.18mmol)の溶液に、アセトニトリル(2〜3mL)中の3%トリフルオロ酢酸を添加して28bを生成させた。反応混合物を25に対してと同じ方法に従って処理し、泡状物として高収率で28bが得られた(0.167g、95%)。
1H NMR (アセトニトリル-d3) δ 6.09-6.24 (Cyt 及び Thy H1'), 5.21-5.33 (Thy H3'), 5.06-5.14 (Cyt H3'), 2.82-2.88 (Thy H5'及び5"); 31P NMR (アセトニトリル-d3) δ -1.381, -1.613; C39H46N8O15P+ 高分解能 ESI-MS 計算値 897.28203, 測定値 897.28148。
【0119】
HOGpTSucc−IL(28c):
アセトニトリル(1〜2mL)中の化合物27c(0.21g、0.17mmol)の溶液に、アセトニトリル(2〜3mL)中の3%トリフルオロ酢酸を添加した。反応混合物を25に対してと同じ方法に従って処理し、泡状物として高収率で28cが得られた(0.157g、96%)。
1H NMR (アセトニトリル-d3) δ 6.08-6.25 (Gua及び Thy H1'), 5.26-5.30 (Gua H3'), 5.17-5.22 (Thy H3'), 2.81-3.05 (Thy H5'及び5"); 31P NMR (アセトニトリル-d3) δ -1.047, -1.064; C37H48N10O15P+ 高分解能 ESI-MS 計算値 903.30382, 測定値 903.30328。
【0120】
HOTpTSucc−IL(28d):
アセトニトリル(1〜2mL)中の化合物27d(0.21g、0.18mmol)の溶液に、アセトニトリル(2〜3mL)中の3%トリフルオロ酢酸を添加した。反応混合物を25に対してと同じ方法に従って処理し、泡状物として高収率で28dが得られた(0.123g、78%)。
1H NMR (アセトニトリル-d3) δ 6.16-6.24 (5'-Thy及び 3'-Thy H1'), 5.22-5.30 (3'-Thy H3'), 5.07-5.12 (5'-Thy H3'), 2.30-2.86 (3'-Thy H5'及び5"); 31P NMR (アセトニトリル-d3) δ -1.188, -1.284; C33H43N7O15P+ 高分解能ESI-MS 計算値 808.25548, 測定値 808.25493。
【0121】
DMTTpTpTSucc−IL(29):
化合物28d(0.065g、0.073mmol)を、3’-ホスホルアミダイト26d(0.233g、0.31mmol)及びジシアノイミダゾール(0.31g、2.6mmol)と、無水アセトニトリル(5mL)中で室温にて混合した。2時間撹拌した後、化合物27a〜dに対しての方法と同じ方法で、生成物を10%酢酸エチル/ジエチルエーテルから2回沈殿させ、酸化し、抽出して、化合物29(104mg、92%収率)を高純度で得た。
31P NMR (アセトニトリル-d3) -1.081, -1.194, -1.233, -1.262, -1.381, -1.403, -1.448, -1.477; C67H77N10O24P2+ 低分解能ESI-MS 計算値 1467.5, 測定値1467.8.
【0122】
HOTpTpTSucc−IL(30):
アセトニトリル(1〜2mL)中の化合物29(97mg、0.06mmol)の溶液に、アセトニトリル(2〜3mL)中の3%トリフルオロ酢酸を添加した。反応混合物を25に対してと同じ方法に従って処理し、ガラス状固体として30が得られた(77mg、98%収率)。
31P NMR (アセトニトリル-d3) -1.157〜-1.531 (ピークの広い重なり); C46H59N10O22P2+ 高分解能ESI-MS 計算値 1165.32807, 測定値1165.32752。
【0123】
DMTTpTpTpSucc−IL(31):
化合物30(170mg、0.136mmol)を、3’-ホスホルアミダイト26d(0.160g、0.215mmol)及びジシアノイミダゾール(0.21g、1.8mmol)と、無水アセトニトリル(5mL)中で室温にて混合した。2時間撹拌した後、化合物27a〜dに対しての方法と同じ方法で、生成物を10%酢酸エチル/ジエチルエーテルから2回沈殿させ、酸化し、抽出して、化合物31(231mg、89%収率)を高純度で得た。
31P NMR (アセトニトリル-d3) -1.169〜-1.859 (ピークの広い重なり); C80H93N13O31P3+ 低分解能ESI-MS 計算値 1824.5, 測定値 1824.2。
【0124】
HOTpTpTSucc−IL(32):
アセトニトリル(1〜2mL)中の化合物31(190mg、0.122mmol)の溶液に、アセトニトリル(2〜3mL)中の3%トリフルオロ酢酸を添加した。反応混合物を25に対してと同じ方法に従って処理し、化合物32が得られた(160mg、99.9%収率)。
31P NMR (アセトニトリル-d3) -1.142〜-1.51 (ピークの広い重なり); C59H75N13O29P3+ 高分解能ESI-MS 計算値 1522.40066, 測定値 1522.40011。
【0125】
オリゴヌクレオチド(33a〜f)の脱保護:
シアノエチル保護基、塩基保護基(a〜cについて)、及びイオン液体担体へのスクシニル架橋基の除去は、化合物28a〜d並びに化合物30〜32の1〜5mgを、1.5mLの濃水酸化アンモニウムと無水エタノール(3:1)で、60℃にて16時間又は室温にて48時間処理して達成される。サンプルを冷却した後、アンモニア溶液を蒸発させ、残渣を水に溶かし、イオンペア逆相HPLCによって精製して、33a〜fを得た。
【0126】
本発明は、上述した構成及び部分の詳細にその応用が限定されないことが理解されるべきである。本発明は、その他の態様であることができ、様々なやり方で実施されうる。本明細書で使用した語法及び術語は、説明のために用いたものであって限定されるものではないことも理解されよう。したがって、本発明を、本発明の実例を用いて上に説明しているが、付属の請求項で定義されるように、本発明の精神、範囲及び特質から離れることなく変更することができる。
【0127】
〔参考文献〕
【表6A】
【表6B】
【表6C】
【表6D】
【表6E】
【図面の簡単な説明】
【0128】
【図1】図1は、本発明によるイオン液体担持合成の一般的概念のある態様の説明図である。
【図2】図2は、本発明によるイオン液体担持合成の一般的概念のさらなる態様の説明図である。
【図3】図3は、真正のLeu5−エンケファリン41と合成したLeu5−エンケファリン14のTFA塩の1H NMRスペクトルの図である。
【図4】図4は、ILSSによって合成した粗製Leu5−エンケファリン14のHPLC分析の図である。42
【図5】図5は、イオン液体担持ペプチド2a、4a、7、9、及び11の1H NMRスペクトルの図である。
【図6】図6は、イオン液体担持ペプチド2a、4a、7、9、及び11のマススペクトルの図である。
【図7】図7は、脱保護したオリゴヌクレオチドの様々なHPLCクロマトグラフィーの図である。
【技術分野】
【0001】
本発明は、イオン液体担持合成に関する。より詳細には、本発明は、イオン液体担持オリゴペプチド、オリゴサッカライド、及びオリゴヌクレオチド合成に関する。なおさらに詳細には、本発明は、生物活性オリゴペプチドのイオン液体担持合成に関する。
【背景技術】
【0002】
オリゴペプチド、オリゴサッカライド、及びオリゴヌクレオチドの効率的合成は、いくつかの新規なアプローチの開発へと導いている現課題である。溶液相合成を用いた、構造的に明らかにされたオリゴサッカライドの入手方法は、労力がかかり、かつ各段階の後でクロマトグラフィーによる反応生成物の精製を必要とする1。
【0003】
合成オリゴヌクレオチドに対する科学者グループの要求は、過去数十年にわたって指数的に増大してきている。幸いなことに、DNAオリゴヌクレオチドプライマーの豊富な資源は、遺伝子配列解析努力、機能ゲノム科学、及びポリメラーゼ連鎖反応(PCR)に基づく検出方法の膨大な要求を満たしている。さらに、オリゴヌクレオチドは、チップベースDNAマイクロアレイを含めた、治療及び診断用途の開発に広く用いられている、構造生物学及び構造生化学における顕著な進歩が、DNA及びRNA化学での同時に起こっている進歩を通じて達成されている2-6。例えば、リボザイム及びsiRNA研究における当分野の現状は、結晶構造を含め、RNA合成において同時に進歩がなければ可能とはならなかっただろう4。
【0004】
Merrifield7とLetsinger8,9らがオリゴペプチド及びオリゴヌクレオチドそれぞれの合成のための高分子担体の使用を導入して以来、不溶性担体の使用は、有機合成、特にオリゴヌクレオチド、ペプチド、及びさらに最近では炭水化物10-12などの生体高分子の合成において、重要な手段となっている。濾過による過剰な試薬と副生成物の除去を含む容易な精製方法は、簡単な生成物分離を可能にし、自動化を可能にする。大きな成功にもかかわらず、不溶性高分子による不均一性のために、固相合成は、固体担体自体の高コストに加えて不均一な反応条件に典型的に伴う欠点を残しており、これらの化合物の大規模合成を非常にコストのかかるものにしている。
【0005】
高分子担持固相合成における最近の進歩13-15は、複雑なオリゴサッカライドを組み立てるための簡単な方法を提供しており、なぜなら、その方法は、樹脂を単に洗浄することによる過剰な試薬の除去を可能にし、それにより、必要なクロマトグラフィーステップの数を最小にするからである16,17。これは、うまい自動化合成のめざましい例をもたらしている14,18。しかし、固相合成の欠点は、不均一反応を完結させるためにしばしば大過剰の高価な炭化水素単位を必要とし、しかも通常の特性分析方法、例えば、TLC、NMR、及びマススペクトルによって糖−糖カップリング工程を監視することが一層困難であることである。さらに、固相方法によってもたらされる多くの利点にもかかわらず、不溶性高分子と反応条件の不均一な特性は、非線形反応速度論、反応部位の及び/又は反応部位へのアクセスの不均一な分布、溶媒和の問題、及び非効率なカップリング速度、を含めた一連の問題をしばしばもたらす。
【0006】
均一反応条件の再構築と固相合成の欠点のいくつかを克服する目的による代替方法の探索は、可溶性高分子担体の開発へと導いている。近年、可溶性高分子担体の使用がかなりの注目を集めているが、これは、そのような「液相」合成は従来の溶液化学の利点の多くを残すと同時に、生成物の容易な精製の利点をなおも可能にするからである。可溶性のポリエチレングリコール(PEG)、ポリビニルアルコール、及びその他の高分子は全て、オリゴペプチド19及びヌクレオチド11の合成に首尾良く用いられている。さらに、可溶性のポリエチレングリコール(PEG)ポリマーは、オリゴサッカライド12,20-23及び小分子合成24のための担体としても用いられている。しかし、可溶性高分子担体の使用は、低い負荷容量、オリゴサッカライドが結合したポリマーの選択的沈殿の難しさ、より長いペプチドの合成時の貧弱な溶解性、低い水溶性、エーテル溶媒中での溶解性の欠如24、及び精製に必要とされるエネルギーを大量に消費する冷却、の欠点がある。
【0007】
さらに最近、フッ素化(フッ素)可溶性担体に基づく新しい溶液相合成が提唱されている25。このアプローチはフッ素系溶媒(すなわち、パーフルオロアルカン類)中でのフッ素系担体とフッ素化された試薬との好ましい可溶性に基づいている。非フッ素化試薬は、フッ素系有機溶媒分配26a−d又はフッ素系シリカゲルに基づく固相抽出(SPE)26e−hによって、担持された生成物から容易に分離されうる。このアプローチは、通常容易に入手できないフッ素化化合物の使用を必要とする。精製は、予め混和性のフッ素系溶媒と有機溶媒との間の相分離を引き起こす温度切り替えによって達成され、それによって分離を促進する。有機合成のためのフッ素系相方法の使用は、オリゴペプチド27、オリゴサッカライド28、及び小分子25について実証されている。オリゴサッカライド合成の場合は、フッ素系溶媒へのサッカライド結合担体の溶解性は、サッカライド単位の数が増加するに従って低下するフッ素含量に左右されることが実証されている。パーフルオロアルカン溶媒のコスト、特殊なフッ素化された試薬の必要性、及び温度切り替えに伴うエネルギーコストは、フッ素系相有機合成の広い応用を阻害する潜在的制限である。
【0008】
イオン液体(Ionic liquid(IL))は、有機反応のための環境にやさしい反応媒体として近年大きな注目を集めている29。イオン液体の現実的な定義は、水の沸点より低い、しばしば非常に低い融点を有する塩、というものである。イオン液体の共通の性質は、多くは有機カチオンと無機アニオンとを有することである。イオン液体の非限定的例は、ハロゲン化物、テトラフルオロボレート、及びヘキサフルオロホスフェートの、アルキルイミダゾリウム及びピリジニウム塩である。多くの化学反応が、いくつかの酵素反応も含めて、イオン液体中で実施されうる30。室温イオン液体は、電気化学技術31、化学抽出32、及びその他の工業的方法33のための媒体として広く検討されてもいる。これは、イオン液体のいくつかの興味ある化学的及び物理的性質:すなわち、高い熱的及び化学的安定性、不燃性、測定可能な蒸気圧の欠如、及び高い負荷特性、によるものである。多くの場合に、イオン液体は容易にリサイクルできる。カチオン又はアニオンの構造を修正することによって、イオン液体の溶解性は容易に変えることができ、それにより、それらが有機媒体並びに無機媒体から相分離し、分離及び精製を容易にするようにできる。このことが、イオン液体が有機合成のための実施可能な可溶性機能性担体として機能しうる可能性を提供している。基質の溶解度も変えることができる34。最近の報告は、小分子35,37,38及び小ペプチド39のためのILSS(Ionic Liquid Supported Synthesis;イオン液体担持合成)の有効性を首尾良く実証している。回収可能且つリサイクル可能なイオン液体担持触媒を開発する可能性もまた、探索されている36。
【0009】
有機反応のためのマトリクス(すなわち、溶媒)としてのイオン液体の使用は、以前、Vaultierら(WO2004/029004)によって記載されている。この有機反応は、可溶性担体として働く「オニウム塩」として公知の官能化された塩類を用いて行われる。このイオン液体は、官能化された塩の可溶化を確実にし、反応が均一条件下で行われうる。Vaultierら(WO2005/005345)は、以前、有機反応のための可溶性担体として、「オニウム塩担持有機合成(Onium Salt Supported Organic Synthesis)」と名付けられた、少なくとも1種の有機溶媒中に溶解したオニウム塩(すなわち、イオン液体)の使用、を記載している。
【特許文献1】国際公開WO2004/029004号パンフレット
【特許文献2】国際公開WO2005/005345号パンフレット
【発明の開示】
【発明が解決しようとする課題】
【0010】
したがって、以下のものに限定されないが、オリゴペプチド、オリゴサッカライド、及びオリゴヌクレオチドを含めたオリゴマーの合成のための改良方法に対する必要性が依然として存在し、この方法は、固相及び溶液相合成の利点を結合させ、均一溶液相条件を含み、且つ生成物の容易な精製を可能にするものである。
【0011】
本発明は、これら及びその他の必要性を満たすことを目的とする。
【0012】
本明細書の記載は多くの文献を参照し、その内容は、その全体を参照により本明細書に援用する。
【課題を解決するための手段】
【0013】
[発明のまとめ]
本発明は、イオン液体(すなわちイオン性液体)担持合成に関する。
【0014】
ある態様では、本発明は、イオン液体によって担持された、オリゴペプチド、オリゴサッカライド、及びオリゴヌクレオチド、の合成に関する。
【0015】
ある態様では、本発明は、オリゴペプチド、オリゴサッカライド、及びオリゴヌクレオチドを含むがこれらに限定されないオリゴマーの合成のためのイオン液体担持方法に関する。
【0016】
ある態様では、本発明は、オリゴペプチド、オリゴサッカライド、及びオリゴヌクレオチドからなる群から選択されるオリゴマーの合成方法に関するものであり、この方法は、イオン液体に結合したモノマー単位をもたらす反応条件下において、第一のモノマー単位をイオン液体と接触させるステップ;及び2〜30のモノマー単位を含むイオン液体結合オリゴマーをもたらす反応条件下において、前記イオン液体に結合したモノマー単位を、少なくとも1つのさらなるモノマー単位と接触させるステップを含む。次に、このイオン液体結合オリゴマーはイオン液体から切り離されて、遊離したオリゴマーをもたらす。
【0017】
さらなる態様では、本発明は、オリゴペプチド、オリゴサッカライド、及びオリゴヌクレオチドからなる群から選択されるオリゴマーの合成方法に関し、本方法は、モノマー単位に対してイオン液体を用いて、2〜30のモノマー単位を含むオリゴマーをもたらすことを含む。
【0018】
さらなる態様では、本発明は、化学合成に用いるための、且つ溶媒及び液体担体の両方の二重機能を果たしうる液体担体に関する。具体的態様では、イオン液体は、ヘテロ環式又は置換ヘテロ環式第四級窒素含有有機カチオン、ヘテロ環式又は置換ヘテロ環式第四級ホスホニウム含有有機カチオン、又はヘテロ環式又は置換ヘテロ環式の三価のスルホニウム含有有機カチオン;及び、前記有機カチオンの電荷と釣り合うアニオン、を含む有機塩である。より詳細な態様では、有機カチオンは、N−置換ピリジン及び1,3−ジ置換イミダゾールからなる群から選択される。有機カチオン上の電荷と釣り合うアニオンは、Cl−、Br−、BF4−、PF6−、SbF6−、CuCl2−、及びAlCl4−からなる群から選択できる。その他の適切なアニオンも使用でき、充分に当業者の能力の範囲内である。
【0019】
具体的な態様では、本発明は、オリゴペプチドのイオン液体担持合成に関する。なお、さらに具体的な態様では、本発明は、ペンタペプチドLeu5−エンケファリンの合成に関し、その構造は以下に示す。
【化1】
【0020】
本発明の、上述の及びその他の目的、利点、並びに特性は、付属の図面を参照にして、参考のみの例として提供する本発明の説明のための態様の以下の非制限的説明を読むことで、さらに明らかになろう。
【0021】
[実例態様の詳細な説明]
本明細書中で用いる用語の明確かつ一貫した理解を提供するために、以下に多くの定義を示す。さらに、他に定義されていない限り、本明細書で用いる全ての技術的及び科学的用語は、本発明が関係する技術分野の当業者に通常理解されているものと同じ意味を有する。
【0022】
請求項及び/又は明細書中の「含む、含有する」の用語との関連で用いられる用語は、単数を意味することができるが、「1以上」、「少なくとも1」、及び「1又は1より多く」の意味ともなる。同様に、「別の」の用語は、少なくとも第二の、もしくはそれ以上のものを意味することができる。
【0023】
本明細書及び請求項で用いるように、「含む、含有する」(及び、含む、含有する、の全て活用形)、「有する」(及び、有する、の全ての活用形)、「包含する」(及び、包含する、の全ての活用形)、の用語は、包含的、すなわち限定のないものであり、追加の記載されていない要素又は方法ステップを排除するものではない。
【0024】
「約」の用語は、その値が、その値を測定するために用いた装置又は方法についての内在する誤差の変動を含んでいることを意味するために用いる。
【0025】
「アミノ酸(類)」の用語は、本明細書で用いるように、天然アミノ酸のL及びD異性体の両方、並びにペプチドの合成アナログを調製するためにペプチド化学で用いられるその他の非タンパク質関連アミノ酸のL及びD異性体の両方を含むものとして理解される。天然アミノ酸の例には、グリシン、アラニン、バリン、ロイシン、イソロイシン、セリン、及びトレオニンが含まれるが、これらに限定されない。非タンパク質関連アミノ酸の例には、ノルロイシン、ノルバリン、シクロヘキシルアラニン、ビフェニルアラニン、ホモフェニルアラニン、ナフチルアラニン、ピリジルアラニン、及び置換フェニルアラニン類(アルコキシ、ハロゲン、及びニトロ基を含むがこれらに限定されない1以上の置換基で置換されたもの)が含まれるが、これらに限定されない。ベータ及びガンマアミノ酸も、「アミノ酸」の用語の範囲内である。ペプチド合成で用いられる標準の保護基で保護されたアミノ酸もまた、「アミノ酸」の用語の範囲内である。これらの化合物は、ペプチド化学分野の当業者に公知である。
【0026】
「ヌクレオチド」の用語は、本明細書で用いるように、個別のヌクレオチド又はヌクレオチドの変種を言うものとして理解され、プリン又はピリミジン、リボース又はデオキシリボース糖残基、及びホスフェート基又は(オリゴヌクレオチド又はポリヌクレオチド内のヌクレオチドの場合は)ホスホジエステル結合を含む分子又は大きな核酸分子内の個々の単位を意味する。「ヌクレオチド」の用語はまた、(a)別の連結基、(b)プリンの類似形、(c)ピリミジンの類似形、又は(d)類似の糖、から選択される少なくとも1つの修飾を含む「修飾ヌクレオチド」も包含する。オリゴヌクレオチド合成に通常用いられる標準保護基で保護されたヌクレオチドも、「ヌクレオチド」の用語の範囲内である。これらの化合物は、ヌクレオチド化学分野の当業者に公知である。
【0027】
「サッカライド」の用語は、本明細書で用いるように、実験式(CH2O)n(式中、nは整数、典型的には3より大きな整数である)を有する、ポリヒドロキシアルデヒドもしくはポリヒドロキシケトン又はそれらの誘導体である炭水化物を言うものとして理解される。モノサッカライド、すなわち単糖類は、単一のポリヒドロキシアルデヒド又はポリヒドロキシケトン単位からなる。モノサッカライドには、リボース、2-デオキシ-リボース、グルコース、マンノース、キシロース、ガラクトース、フコース、フルクトースなどが含まれるがこれらに限定されない。ジサッカライドは、グリコシド結合によって結合された2つのモノサッカライド単位を含む。ジサッカライドには、例えば、スクロース、ラクトース、マルトース、セロビオースなどが含まれる。オリゴサッカライドは、典型的には、グリコシド結合で結合された2〜10のモノサッカライド単位を含む。ポリサッカライド(グリカン)は、典型的には10より多くのそのような単位を含み、ヘパリン、ヘパラン硫酸、コンドロイチン硫酸、デルマタン硫酸、及びそれらのポリサッカライド誘導体が含まれるがこれらに限定されない。「糖」の用語は、一般に、モノ−、ジ−、又はオリゴサッカライドをいう。サッカライドは、例えば、置換された、グルコサミン、ガラクトサミン、アセチルグルコース、アセチルガラクトース、N−アセチルグルコサミン、N−アセチル−ガラクトサミン、ガラクトシル−N−アセチルグルコサミン、N−アセチルノイラミン酸(シアリン酸)などであってもよい。サッカライドは、例えば、ヌクレオシド、ヌクレオチド、ポリヌクレオチド、DNA、RNAなどのサッカライド残基のように、より大きな分子の構成要素として存在することもできる。本明細書で用いるように、「サッカライド」の用語は、修飾されたサッカライド、例えば、(a)H、NH2、ハロゲン、アルキル、アリールを含むがこれらに限定されない置換基による、1つ以上のOH基の置換;(b)アルデヒド、ケトン、酸、エステル、及びそれらの誘導基への1つ以上のOH基の酸化、から選択される少なくとも1種の修飾を含むもの、を包含するものとしても理解される。オリゴサッカライド合成に通常用いられる標準保護基によって保護されたサッカライドも、「サッカライド」の用語の範囲内である。これらの化合物は、当業者に公知である。
【0028】
「伸長(する)オリゴペプチド鎖」、「伸長(する)オリゴサッカライド鎖」、及び「伸長(する)オリゴヌクレオチド鎖」の用語は本明細書でも用いるように、任意で適切に保護されていてもよいアミノ酸、サッカライド、又はヌクレオチドの順次の付加によって調製される鎖をいう。各反応サイクルの後で、この伸長オリゴペプチド鎖、伸長オリゴサッカライド鎖、又は伸長オリゴヌクレオチド鎖は、少なくとも1個のアミノ酸、サッカライド、又はヌクレオチドだけ長さが増大し、次の反応サイクルのための出発物質となる。本明細書で用いるように、この用語は、出発物質又は生成物のいずれをもいうことができ、具体的な前後関係においてこの用語によって意図されるものを当業者は理解しよう。
【0029】
本明細書で用いるとおり、「アルキル基」の用語は、飽和で一価の非分岐又は分岐した炭化水素鎖をいうものとして理解される。アルキル基の例には、(C1〜C6)アルキル基が含まれるが、これに限定されない。(C1〜C6)アルキル基の例には、メチル、エチル、プロピル、イソプロピル、2-メチル-1-プロピル、2-メチル-2-プロピル、2-メチル-1-ブチル、3-メチル-1-ブチル、2-メチル-3-ブチル、2,2-ジメチル-1-プロピル、2-メチル-1-ペンチル、3-メチル-1-ペンチル、4-メチル-1-ペンチル、2-メチル-2-ペンチル、3-メチル-2-ペンチル、4-メチル-2-ペンチル、2,2-ジメチル-1-ブチル、3,3-ジメチル-1-ブチル、2-エチル-1-ブチル、ブチル、イソブチル、t-ブチル、ペンチル、イソペンチル、ネオペンチル、及びヘキシル、が含まれるがこれらに限定されない。
【0030】
本明細書で用いるとおり、「アリール基」の用語は、環内に0〜4個のヘテロ原子を含んでいてもよい、5−、6−、及び7−員環芳香族基をいい、例えば、フェニル、ピロリル、フリル、チオフェニル、イミダゾリル、オキサゾリル、チアゾリル、トリアゾリル、ピラゾリル、ピリジル、ピラジニル、ピリダジニル、及びピリミジニルなどをいう。環構造中にヘテロ原子を有するこれらのアリール基は「アリールヘテロ環」又は「ヘテロ芳香族」ともいうことができる。この芳香族環は、1以上の環上位置で置換されていることができる。アリール基はまた、多環式基の一部であることもできる。例えば、アリール基には、ナフチル、アントラセニル、キノリル、インドリル、などの縮合芳香族残基が含まれる。
【0031】
本明細書で用いるように、「ハロゲン」の用語は、フッ素、塩素、臭素、又はヨウ素をいうものとして理解される。したがって、用語「ハロ」は、フルオロ、クロロ、ブロモ、及びヨードを包含するものとして理解される。
【0032】
本発明における保護基は、オリゴペプチド合成、オリゴヌクレオチド合成、及びオリゴサッカライド合成に関連して用いられる。保護基は、アミノ酸、サッカライド、又はヌクレオチドのいずれかのモノマーの反応性末端をブロックする。化学合成の性質は、どの反応性基が保護基を必要とするかを決定するであろう。具体的使用にかかわらず、保護基は、別の試薬と反応することから分子上の残基を保護するために用いられる。本発明で用いられる保護基は、以下の性質を有する:保護基は、選択した試薬が、保護基が結合している基を修飾することを防止する;保護基はその合成反応条件に対して安定である(すなわち、保護基は分子に結合したまま残る);及び、保護基は、残りの構造に悪影響を及ぼすことのない条件下で除去可能である。適切な保護基の選択は、もちろん、モノマー単位及びオリゴマーの化学的性質、並びに保護基がそれに対して保護すべき具体的試薬に左右される。所定の反応スキームに対して適切な保護基を選択することは、当業者の能力内である。
【0033】
本明細書の記載は、当業者によって用いられる多数の化学用語と略号に言及している。それでもなお、選択した用語の定義を、明確性及び一貫性のために提供した。
【0034】
略号:
DCC:ジシクロヘキシルカルボジイミド; DMAP:4-(N,N−ジメチルアミノ)ピリジン; EDC:1-(3-ジメチルアミノプロピル)-3-エチルカルボジイミド塩酸塩; HOBt:1-ヒドロキシベンゾトリアゾール水和物; HATU:O-(7-アザベンゾトリアゾール-1-イル) N, N, N’, N’-テトラメチルウロニウムヘキサフルオロホスフェート; PyBOP:(ベンゾトリアゾール-1-イルオキシ)トリピロリジノホスホニウムヘキサフルオロホスフェート; DCU:ジシクロヘキシルウレア; DIPEA(ヒューニッヒ塩基(Hunig’ base)):ジイソプロピルエチルアミン; DCI:4,5-ジシアノイミダゾール; DMSO:ジメチルスルホキシド; DMSO−d6:重水素化ジメチルスルホキシド; DCM:ジクロロメタン; NMI:N-メチルイミダゾール; TEA:トリエチルアミン; TEAA:トリエチルアンモニウムアセテート; THF:テトラヒドロフラン; CE:シアノエチル; PEG:ポリエチレングリコール; Py:ピリジン; Succ:スクシニル; CPG:細孔制御されたガラス; IL:イオン液体; ILSS:イオン液体担持合成; DMT:ジメトキシトリチル; LCAA:長鎖アルキルアミン; EEDQ:2-エトキシ-1-エトキシカルボニル-1,2-ジヒドロキノリン; IIDQ:2-イソブトキシ-1-イソブトキシカルボニル-1,2-ジヒドロキノリン; TFA:トリフルオロ酢酸; AcOH:酢酸; NMR:核磁気共鳴; MS:マススペクトル; m/z:電荷に対する質量比; m.p.:融点; FAB:高速原子衝撃; TLC:薄層クロマトグラフィー; ESI:エレクトロスプレーイオン化; FTMS:フーリエ変換マススペクトル; LCMS:液体クロマトグラフィー−マススペクトル; J:結合定数; s:単一線(シングレット); t:三重線(トリプレット); m:多重線(マルチプレット); P−III:三価のリン; P−V:五価のリン。
【0035】
本発明によるイオン液体担持合成の一般概念を図1及び2に示す。イオン液体残基上に固定された基質(反応剤)は極性有機溶媒に可溶性であり、液相反応をすることができる。反応の終了及び溶媒の留去後、イオン液体に固定された生成物が溶けない極性の低い有機溶媒によって過剰な試薬を除去しうる。無機試薬及び/又は副生成物は、沈殿により、又は水溶液で洗浄することにより除去しうる。この反応順を繰り返して、より複雑な構造を得ることができる。最後に、生成物を切り離し、有機溶媒抽出によってイオン液体残基から分離することができる。イオン液体担体に固定された基質は、従来の溶液系反応と同様の反応性を大部分保っていると予想される。イオン液体担体上で行う反応の進行は、標準の分光学的手法によって容易に監視及び分析される。
【0036】
イオン液体によって担持された、オリゴペプチド、オリゴサッカライド、及びオリゴヌクレオチドの合成についての新規な溶液相アプローチを説明する。
【0037】
ある態様では、本発明は、オリゴペプチド、オリゴサッカライド、及びオリゴヌクレオチドを含むがこれらに限定されないオリゴマーの合成のためのイオン液体担持方法に関する。さらなる態様では、本発明は、オリゴペプチド、オリゴサッカライド、及びオリゴヌクレオチドを含むがこれらに限定されないオリゴマーの化学合成に用いるためのイオン液体に関するものであり、このイオン液体は、溶媒及び液体担体の両方の二重の機能を果たしうる。このイオン液体は、有機合成に一般に適用される様々な合成方法に適合性である。さらに詳細には、イオン液体は、オリゴペプチド、オリゴサッカライド、及びオリゴヌクレオチドを含むがこれらに限定されないオリゴマーの合成に一般に適用される様々な合成方法に適合性である。さらに、特に、しかしこれに限定されないが、オリゴペプチド合成の場合には、イオン液体は、その本来のイオン的性質によって、ペプチドビルディングブロックのラセミ化又はエピ化(エピマー化)を起こしてはならない。さらに、イオン液体担体の溶解性が伸長オリゴペプチド鎖、伸長オリゴサッカライド鎖、又は伸長オリゴヌクレオチド鎖によって影響を受け、分離及び精製手段が過度に複雑になってはならない。分離及び精製手段は、非常に簡単化され、通常は水性溶媒及び有機溶媒での洗浄ステップを含む。
【0038】
本発明のある態様では、イオン液体は、ヘテロ環式又は置換ヘテロ環式第四級窒素含有有機カチオン、及び前記有機カチオンの電荷と釣り合うアニオン、を含む有機塩である。より具体的な態様では、有機カチオンは、N−置換ピリジン及び1,3−ジ置換イミダゾールからなる群から選択され、かつ、アニオンは、Cl−、Br−、BF4−、PF6−、SbF6−、CuCl2−、及びAlCl4−からなる群から選択される。その他のイオン性液体が当分野で公知であり、当業者の能力の範囲内である。さらに、前記アニオンが有機アニオンであってもよいことも当業者の能力の範囲内であり、有機アニオンの制限されない例には、CH3CO2−、CF3CO2−、CH3SO4−、及びCF3SO2−が含まれる。
【0039】
以下の、例示の態様の非限定的説明によって、さらに詳細に本発明を説明する。
【実施例】
【0040】
[実施例1]イオン液体担体への結合とラセミ化の試験(スキーム1)
3-ヒドロキシエチル-(1-メチルイミダゾリウム)-テトラフルオロボレート(1)(1-メチルイミダゾールと2-ブロモエタノール[35a,d]の反応によって容易に入手できる)を、本発明で意図するイオン液体担持オリゴペプチド合成を説明するための、適切なイオン液体担体の非限定的な例として選択した。本発明にしたがって使用することができるその他のイオン液体担体は、当分野で公知であり、当業者の能力のうちである。
【0041】
【化2】
【0042】
化合物1上への第一のアミノ酸、Boc−ロイシンの担持のために、様々なカップリング反応条件を試験した。これらのカップリング反応の有効性は、以下に、表1中に示す。
【0043】
【表1】
【0044】
探索したカップリング条件のなかでは、DCC/DMAPの組み合わせ(試験8)が最良の結果をもたらした。その他の試験したカップリング条件には、以下の組み合わせ:EDC/HOBt、HATU/HOBt、PyBOP/DIPEA、EEDQ、又はIIDQが含まれる。しかし、後者の条件群は、カップリング反応(すなわち、エステル化反応)を定量的転換にまでにするためには充分ではなかった。DCC/DMAP経路に対する代替かつ有用なカップリング経路は、追加のカップリングステップを含むけれども、未反応の化合物1を無水酢酸でキャッピングすることを含む。順次のエーテル洗浄及び酸水溶液洗浄によって、過剰なBoc−ロイシン、DMAP、及びDCU副生成物を全て除去した。イオン液体担持Boc−ロイシン2が高い単離収率(91%)かつNMRスペクトルによって検証した高純度で得られた。このカップリング反応は、L−及びD−Leu−Bocの両方を用いて行い、それぞれ、2a及び2bが得られた。
【0045】
2a及び2bの様々なBoc−保護反応条件を試し、その結果を以下の表2に示す。テトラフルオロホウ酸及びトリフルオロ酢酸の両方が、1H NMRスペクトルによって示されるとおり、非常にきれいな脱保護を起こさせることができた。効率的ではないが、Boc脱保護は、Dowex(登録商標)50Wx8-100又はAmberlyst(登録商標)15などの酸性イオン交換樹脂を用いて行うこともできる。その他の脱保護法、例えば「Protective Groups in Organic Synthesis」(Greeneら, John Wiley & Sons, Inc, NY, 1991)中に記載されている方法を本発明にしたがって用いることができ、当業者の能力の範囲内である。
【0046】
【表2】
【0047】
次のアミノ酸ビルディングブロック、すなわちBoc−Phe−OHのカップリング(スキーム2)は、先に中和ステップを行うことが好ましい(表2)。Boc保護に先に用いた過剰のTFA又はHBF4は、NaHCO3、Et3N、DIPEA、又はDowex(登録商標)550A OHなどの塩基性イオン交換樹脂、で中和しうる。本発明に従って使用できるその他の適切な中和剤は当分野で公知であり、当業者の能力の範囲内である。驚くべきことに、次のアミノ酸ビルディングブロック、すなわち、Boc−Phe−OHのカップリング反応を、PyBOP/DIPEA条件を用いて行う場合は、脱保護/中和と次のカップリング反応との両方を、中間体分離及び精製ステップなしに順次行うことができる。
【0048】
【化3】
【0049】
ペプチド4aは、実質的に定量的収率で得られた40。本発明のイオン液体担持方法の利点は、大過剰の試薬を必要としないことである。このカップリング反応は、わずか1.2当量のPyBOPとBoc−Phe−OHで達成しうる。
【0050】
イオン液体担持合成時の、より特にはDCC/DMAP条件を用いて担持するステップでの、ラセミ化又はエピ化の可能性を評価するために、一連のイオン液体担持ペプチド4a〜dを合成した。このペプチドを次にアンモニア/メタノールを用いて開裂させてジペプチド5a〜dを得た。5a(L,L)及び5b(D,L)の引き続いてのHPLC分析は、検出可能な量のその他の異性体がない(<0.5%)ことを明らかにし、担持ステップ及び続いてのジペプチド合成時に、いかなる有意なラセミ化又はエピ化も起こらないことを示した。さらに、この結果は、塩基条件(アンモニア/メタノール)を用いる開裂ステップ時のラセミ化又はエピ化の不存在をも示唆している。
【0051】
[実施例2]オリゴペプチド合成及び、イオン液体担体からの切り離し
上記伸長ペプチド鎖4aに、2つの連続するグリシン残基、次にチロシン残基(全てその保護されたBoc形で)を、先に開発したカップリング及び脱保護条件を用いて次にカップリングさせ、イオン液体担持ペンタペプチド11を泡状の淡黄色固体として得た(スキーム3)。この合成スキームにおいて、Boc保護形(7、9、11)又は脱保護形(6、8、10)のいずれかの全ての中間体を、当業者に周知の標準的手順で単離し且つ精製した。そのような手順は、一般に、順次の有機溶媒(1種以上)及び水での洗浄を含み、その非制限的実施例を本明細書に記載する。
【0052】
【化4】
【0053】
構造の確認と純度分析は、従来の分光学的方法、例えば、1H、13C NMR、及びMSによって実現した。1H NMRスペクトルにおけるBoc基のt−ブチル基の存在又は不存在が、カップリング又は脱保護ステップのそれぞれの特徴であった。イオン液体担持オリゴペプチド類(2a、4a、7、9、及び11)のマススペクトルは、構造の特性解析をさらに助けた(イオン液体担持オリゴペプチドのカチオンに相当する最も強いシグナル)。
【0054】
ペンタペプチド生成物であるLeu5−エンケファリン14をイオン液体担体から切り離すための2つの経路を評価した(スキーム3)。第一の経路は、塩基性水性条件下でイオン液体担体からペンタペプチド14を切り離し、次にTFA/アニソールを用いてBoc及びt−Bu保護基を除去することを含む。第二の経路は、最初にTFA/アニソールを用いてBoc及びt−Bu保護基を除去し、次に塩基性水性条件下でイオン液体担体からペンタペプチド14を切り離すことを含む。第一の経路は、イオン液体担体からの開裂と反応媒体の酸性化に続いて、保護されたペンタペプチド12を良好な収率(84%収率)及び純度で溶液から沈殿させた。
【0055】
このように調製したLeu5−エンケファリンペンタペプチド14の純度を分析した。ペンタペプチド14は、いかなる再結晶又はクロマトグラフィー手段なしに、その対応するTFA塩として、化合物1から50%の全収率で得られた[m.p. 148〜152℃、及び[α]20D = 25.0(c:0.8、95%AcOH)]。14の真正サンプルは150〜153℃の範囲の融点をもち、比旋光度[α]20D = 25.6(c:0.9、95%AcOH)]を有する41。14の1H NMRスペクトルは、真正サンプルを用いて得られたスペクトルと実質的に同じだった(図3)。Leu5−エンケファリンペンタペプチド14の純度は、HPLC分析によって実証されたように約90%純度を超えるものであることが測定された(図4)。このレベルの純度は、クロマトグラフィー精製前に固相ペプチド合成によって一般的に得られるものよりも優れている42。
【0056】
Leu5−エンケファリンペンタペプチド14の合成によって具体化されたように、イオン液体担持合成は、ペプチド合成のこれまでの方法の多くに対していくつかの見込まれる利点を提供する(表3)。
【0057】
【表3】
【0058】
第一に、その他の溶液相法と同じに、固相ペプチド合成(SPPS)とは対照的に、大過剰の反応試薬の使用を避けることができる。非天然型アミノ酸(例えば、D−アミノ酸)を含むペプチドを合成することを計画した場合、又は大スケールでのペプチド合成が必要な場合に、このことは考慮すべき重要な要因である。第二に、イオン液体担持合成の場合、各中間体は、繰り返しの溶媒洗浄によって容易に精製することができる(液/液相分離)。この面で、ILSSは、FPPS(フッ素系相ペプチド合成)並びにSPSPS(可溶性ポリマー担持ペプチド合成)に類似している。
【0059】
ILSSの場合、分離工程は、固体/液体相分離を含むSPPSに対する場合と同様には容易に自動化されない。しかし、わずか約1モル当量の低分子量イオン液体1しか通常は必要とされないので、ILSSに対する負荷容量はSPPSよりも大きい。その上、イオン液体担体のコストは、フッ素系担体のコスト、又は固体ポリマー担体のコストよりも通常は低く、このことは、大スケールのペプチド合成を計画する場合にさらなる重要な考慮事項である。最後に、ILSSの場合、合成中の各中間体の構造及び純度が、通常のスペクトル法(すなわち、1H、13C、及びMS)によって容易に検証可能であった。
【0060】
[実施例3]イオン液体担持オリゴサッカライド合成
以下のスキーム4に図示したように、イオン液体担持法を用いるオリゴサッカライドの合成は、簡単な相分離を用いて、生成物精製のためのクロマトグラフィー法を必要とすることなく、簡便に達成された。
【0061】
【化5】
【0062】
フェニル2,3,4,6-O-アセチル-1-チオ-β-O-グルコピラノシドから出発して文献記載の方法43の修正法によって調製したβ-チオグリコシド15を、室温でCH2Cl2中、ブロモ酢酸/DCC/DMAPでアシル化し、ブロモアセテート16a(X=Br)を得た(スキーム4)。
【0063】
アセトン中で、室温における、16a(X=Br)と1-メチルイミダゾール及びナトリウムテトラフルオロボレートとの反応は、イオン液体に固定されたサッカライド16b(X=[mim][BF4])を98%収率で与えた。化合物16bは、最初に粗生成物をジエチルエーテルで洗浄して過剰な1-メチルイミダゾールを除去して精製した。生成物は次にCH2Cl2に溶かし、反応で生じた不溶性の無機NaBr塩を濾過によって簡便に除去した。16bの構造は、NMR(1H、13C、COSY、及びHMQC)スペクトルによって確認し、これはイミダゾリウム残基とβ-アノマーとしてのアノメリック炭素を明らかに示した。16bの電子スプレーイオン化マススペクトル(ESI−MS)は、唯一の分子イオンとしてのカチオンの存在を示した。
【0064】
次に、モノサッカライド16bを、CH2Cl2中で、−78℃で20分間、m-クロロ過安息香酸によって酸化して、97%収率で、ジアステレオマー(スルホキシドの光学不斉による)の混合物としてスルホキシド17b(X=[mim][BF4])を得た。反応の他方の生成物である、m-クロロ安息香酸は、ジイソプロピルエーテルで洗浄することによって粗生成物から除去した。
【0065】
次に、スルホキシドグリコシレーション反応を、グリコシル供与体としての17bと受容体としての15(3当量)を用いた糖−糖組み立て法44−46として用いた。グリコシル供与体及び受容体の両方とも低温でジクロロメタン中に良く溶けるので、2,6-ジ-tert-ブチル-4-メチルピリジン(4当量)とTf2O(1当量)を用いるカップリング反応を無水CH2Cl2中、−78℃で行った。本反応過程はTLCによって観察した。
【0066】
過剰なグリコシルアルコール15、酸スカベンジャー塩基、及びその他の副生成物をn-ペンタンで、次にジイソプロピルエーテルで洗浄除去することによって、生成物ジサッカライド18b(X=[mim][BF4])を得た。CH2Cl2中の粗製18bの溶液にn-ペンタンを添加し、次に溶液を冷却することによって溶液から17bを相分離させることで、プロトン化した酸スカベンジャー、2,6-ジ-tert-ブチル-4-メチルピリジニウムトリフレートを18bから除去した。イオン液体に結合したジサッカライド18bを、2つのアノメリック炭素を示すHMQC NMRスペクトルによって示されるように高純度で得た。さらに、EIS−MSはジサッカライド構造を裏付けるだけでなく、未反応のモノサッカライドの不存在をも示した。同じ一連の反応の繰り返しで、スルホキシド19bが得られ、次にイオン液体に結合したトリサッカライド20b(X=[mim][BF4])を得た。このトリサッカライドの形成は、3つのアノメリック炭素の存在を示しているHMQC NMRスペクトルと、モノ又はジサッカライドのいかなる混入もないトリサッカライドカチオンを明確に示しているESI−MSによって確かめた。
【0067】
イオン液体残基と連結基は、メタノール中で1当量のCs2CO3を用いることによって容易に20bから切り離されて、定量的に生成物21を与える。トリサッカライド21は、メタノール溶媒の蒸発、及び、粗生成物をCH2Cl2中に溶かし、次に濾過によってイミダゾリウム塩を除去することによって容易に単離された。クロマトグラフィー又はその他の生成方法なしにそのように得られたトリサッカライド21は、NMR及びTLCでみて純粋であることがわかった。そのHMQC NMRスペクトルとESI−MS(M+Na+=1429.7)は、16c(X=H)から出発する古典的な溶液相合成によって、スキーム4に図示したとおりの同じ反応順序にしたがい、しかし、反応ステップのそれぞれに続くクロマトグラフィー精製を行って別個に調製した21の真正サンプルと同じであることがわかった。16cを通っての古典的溶液相合成と比較してILSSアプローチによって得られた21の良好な純度は、古典的な溶液相合成のために開発されたカップリング条件及び立体選択性が、ILSSにそのまま移されうることを示唆している。この例はまた、固相担体からの切り離し後、最終生成物を精製するためにクロマトグラフィーが一般に必要な固相合成47との対照をも示している。
【0068】
[実施例4]イオン液体担持オリゴヌクレオチド合成
ホスホルアミダイト法2−6を用いるオリゴヌクレオチドの固相合成は反復法であり、その方法においては、結合されたヌクレオシドをもつ固体担体が、不安定な保護基を除去することによって末端で脱保護され、それによって求核性水酸基を自由にする。この末端求核性基が次に活性化剤の存在下で、保護されたホスホルアミダイトモノマーに結合される。この新しく作られたホスファイトトリエステル連結基が次に酸化されて、所望し且つより安定なホスフェートトリエステルをもたらす。この方法は、所望する長さ及び組成のオリゴマーが得られるまで繰り返される。可溶性担体を用いた場合に、同様の反復法が用いられうる。
【0069】
3-ヒドロキシエチル-(1-メチルイミダゾリウム)-テトラフルオロボレート(1)(1-メチルイミダゾールと2-ブロモエタノールの反応で容易に入手可能である[35a,d])を、本発明によって意図されるイオン液体担持オリゴヌクレオチド合成を説明するための好適なイオン液体担体の非制限的な例として選択した。本発明に従って用いることができるその他のイオン液体担体は当分野で公知であり、当業者の能力の範囲内である。
【0070】
スクシニル化5’-MMT-デオキシヌクレオシド2348を、アセトニトリル中、ジシクロヘキシルカルボジイミド(DCC)と触媒量の4-ジメチルアミノピリジン(DMAP)を用いてイオン液体1と結合し、イオン液体担持ヌクレオシド24(スキーム5)を得る。反応混合物を濾過して、反応中に形成した不溶性のジシクロヘキシルウレア(DCU)副生物を除去した。次に、反応混合物を、撹拌したジエチルエーテル−酢酸エチル溶液に滴下して加えることによって、化合物24を単離し且つ精製する。誘導体化されていないイオン液体1と所望する生成物24からなる得られる沈殿物を、次にクロロホルム中に溶かし、水で抽出する。イオン液体1は水相とともに除去され、一方、所望する生成物24は、チミジンの5’位に結合したモノメトキシトリチル保護基の疎水性のために有機相に留まる。5’-モノメトキシトリチル(MMT)チミジン誘導体をこの場合には用いたけれども、より一般的に用いられる5’-ジメトキシトリチル(DMT)チミジン誘導体は、このステップで容易に置換されうる。溶媒の除去により、妥当な収率でイオン液体誘導体化ヌクレオシド24が得られる。24の構造は、1H NMRと、電子スプレーイオン化マススペクトル(ESI−MS)によって確かめられた。
【0071】
化合物24の脱トリチル化は、アセトニトリル中での3%のトリフルオロ酢酸の添加と室温での20分間の撹拌によって達成される。反応混合物を、撹拌したエチルエーテル−酢酸エチル溶液に滴下して加え、沈殿物を集めることによって、生成物は速やかに単離且つ精製される。時々、酸溶液中への再溶解と二回目の沈殿が、脱保護を完了させ、残存するトリタノールを完全に除去するために必要とされる。化合物25は、96%の収率で、淡褐色泡状物として得られ、その構造と純度は1H NMRとESI−MSによって検証される。
【0072】
【化6】
【0073】
THF又はアセトニトリル中で4,5-ジシアノイミダゾール(DCI)を活性化剤として用いて、1〜2時間撹拌して、イオン液体担持ヌクレオシド25を1.5倍過剰の適切なホスホルアミダイト誘導体(26a〜d)と反応させることによって、ジヌクレオシドホスホトリエステル類 ApT、CpT、GpT、及びTpT(27a〜d)49(スキーム6)を250μmolスケールで調製した。反応が完了した後、過剰な活性化ホスホルアミダイトを過剰の無水エタノールの添加とさらに10分間の撹拌で失活させる。ホスホルアミダイトを失活させることは精製時にそれらの除去を容易にする。ジヌクレオシドホスファイトトリエステル中間体は、次に、酸化の前に、室温で酢酸エチル:エチルエーテル(1:9)から沈殿させることによって簡単に単離される。失活した過剰のモノヌクレオシド3’-O-ホスホルアミダイトが、カップリング生成物よりも酢酸エチル/エーテル中にずっと可溶性であるので、このことがジヌクレオシドホスファイトトリエステルからのモノヌクレオシド3’-O-ホスホルアミダイトの除去を可能にしている。通常、沈殿物は、アセトニトリル又はメタノールを用いてフィルターから直接溶かし出され、所望の生成物の純度を高めるために、1:9の酢酸エチル:エチルエーテルから再び沈殿させる。
【0074】
ホスファイトトリエステル中間体の酸化を行うために、集めた沈殿物を少量のアセトニトリルに再び溶かし、次に少過剰のピリジンと、2:1のテトラヒドロフラン(THF):水中のヨウ素の0.1M溶液の大過剰量とを添加する。ひとたび(ヨウ素による)色が持続したら、亜硫酸水素ナトリウム水溶液(5%)を添加して過剰のヨウ素を減らす。次に、反応混合物をクロロホルムで希釈し、水で抽出した。水層は、カップリングしていないイオン液体担持ヌクレオシド(ヌクレオチド)(なぜなら、それは末端のトリチル塩を欠き、それに水溶性を付与するからである)に加えて、生じた塩(NaI、Na2SO4、過剰の亜硫酸水素塩、ヨウ化ピリジニウムなど)を含む。減圧下での有機溶媒の除去により、良好な収率(表4)と高純度で淡褐色泡状物として生成物(27a〜d)が得られ、キャッピング段階の必要はない。27a〜dの脱トリチル化は、ジクロロメタン又はアセトニトリル中の3%のトリフルオロ酢酸の添加、20分間の撹拌、及び酢酸エチル:エチルエーテル(1:9)中での沈殿によって達成される。この時点で得られる物質は、このステップの間に確立された平衡(ROH+DMT+=DMT−OR)から生じる最大でも5%のトリチル化出発原料を含みうる。DMTカチオンをトラップするためのエタノールでの失活は、この問題を取り除くとは思われなかった。しかし、TFA処理を繰り返す場合(すなわち、3%トリフルオロ酢酸溶液中への沈殿の再溶解と、続いての第二の沈殿)は、純度は顕著に高くなる(低分解能ESI−MSによって観測されるいかなるトリチル化生成物もバルク生成物中にない)。精製した生成物は単純に濾過して、灰白色から淡黄色の粉末状固体を高収率で生じる(表4)。集めた固体(28a〜d)はカップリングの別のラウンド、又は所望する配列が完了している場合は脱保護に、そのまま用いられる。
【0075】
【化7】
【0076】
上述したダイマー28a〜dに加えて、チミジントリマー(29、30)及びテトラマー(31、32)も、50〜100μmolスケールで合成した。イオン液体担持化合物の同一性は、31P−NMR並びに低及び高分解能ESI−MSの両方によって確かめた(表4)。ダイマー種についての31P NMRデータ(表4)は、2種の可能なRp及びSpジアステレオマーホスホトリエステル類に対応する2つのシグナルを示している。β-シアノエチルホスフェート保護基が除去される場合、ヌクレオチド間の架橋がアキラルになり、単一のシグナルのみが観測される。同様に、トリマー種に対する31P-NMRデータ(表4)は、トリチル保護基がなお結合している場合は、8つのシグナルを示す(4つのジアステレオマーそれぞれが、2つのシグナルを示す)。ひとたびトリチル基が除去されると、シグナルはもはや分解され得ない。このことはテトラマーに対しても同様であり、この場合ジアステレオマーの予期される数は8(23×3=24ピーク)である。単離された化合物がジアステレオマーのペアとして存在するという事実は、1H NMRピークの帰属を複雑にもする。しかし、31Pに調整したCIGAR実験装置を用いることで、ホスホトリエステル連結基を通した予測される3’-5’連結を観測することが可能だった50。
【0077】
【表4】
【0078】
上で合成されたオリゴヌクレオチドを、同じ配列を用いて、一般に用いられる固体担体である細孔制御ガラス(CPG;1μmスケール)上で合成したものと比較した。2つのシステムは平行して脱保護した。所望するオリゴヌクレオチドの完全な脱保護は、それらを、室温で48時間、又は60℃で16時間、濃水酸化アンモニウム/エタノールで処理することによって行った。これらの条件は、シアノエチル保護基、イオン液体残基、塩基(Ade、Cyt、及びGua)の環外アミンの全ての保護、及びモノスクシネート連結基の完全な開裂を確実にする。オリゴヌクレオチドは、真空下でのエタノールと水酸化アンモニウム溶液の除去、水への再溶解(固体担体は、CPG担持オリゴマーに対する遠心によって簡単に沈殿させられる)、及び次にイオンペア逆相HPLC、アニオン交換HPLC、又はポリアクリルアミドゲル電気泳動によるクロマトグラフ精製、によって単離された。ILSS法の生成物を、標準的な遺伝子の機械合成法によって得られた生成物と、LCMSによって比較した。図7は、イオン液体(IL)担体上で成長させたオリゴマーと、CPG上で成長させたオリゴマーとの比較を示す。LCMSのデータは、以下の表5にまとめた。
【0079】
【表5】
【0080】
全ての場合において、ILで生成したオリゴヌクレオチド物質の保持時間は、CPGで生成したオリゴマーと良く一致する。ILオリゴマーに対する主ピークの面積パーセントは、CPG誘導オリゴマーのものより常に数%上である。しかし、イオン液体自身が、オリゴマーを測定するために用いる波長において弱い吸収(保持時間11.2分;λmax217nm)をすることを考慮に入れるといくらか誤っている。イオン液体由来ピークによる面積を除くことで、IL誘導オリゴマーの値が、CPG上で合成したオリゴマーについて得られた値と一致する。HPLC線図のいくつかに見られるチミジンヌクレオシドは、不完全なカップリング又は最初のキャッピング段階より前での固体単体の部分的脱トリチル化によって生じ、約19分に溶出される。CPG上で合成した物質中には通常存在するが、後酸化抽出は、ILが介在する生成物からこの物質をかなり除去する(図1)。先に記載したように、トリマー及びテトラマー中の誤った配列の存在(いわゆる「n−1」ピーク)は、不完全なカップリングによるものではなく、むしろカップリングの前の不完全な脱トリチル化によるようである。この物質は抽出時には除去されずに、カップリングに続く脱トリチル化の後で脱保護される(すなわち、トリマー合成後、脱トリチル化は所望するトリマーと、前の段階から持ち込まれた望まないダイマーとを生成する)。したがって、各カップリングステップの前に完全な脱トリチル化を確実にすることが重要である。
【0081】
[実施例]
一般
全ての試薬は市販品を入手し、他に注意事項のない限り、さらに精製することなく用いた。溶媒は試薬等級のものであり、必要な場合は、標準手法によって乾燥した。反応は、他に特に指示のある場合を除いて無水条件下で行い、シリカゲル60F−254の背面ポリエステルのプレート(250μm厚)上での薄層クロマトグラフィー(TLC)によって、UV光(254nm)下での検出又は過マンガン酸カリウム水溶液で炭化させて観察した。1H NMR及び13C NMRスペクトルはVarian Mercury-300(300MHz)又はMercury-400(400MHz)分光計で20℃において記録した(オリゴペプチド合成)。1H及び13C NMR、HMQC NMR、COSY NMRスペクトルは、Sunワークステーションを備えたUnity 500、Varian Mercury 400、及びVarian Mercury 300分光計で記録した(オリゴサッカライド合成)。化学シフトは、1H NMRについては、δ7.24ppmの残留CHCl3、4.67ppmの水、2.06ppmのアセトン、又は3.30ppmのメタノールを基準にし、13C NMRについては、77.0ppmのCHCl3、49.0ppmのメタノール、及び29.8ppmのアセトンを基準にした、δスケール上の百万分の一(ppm)で報告した。プロトンとカーボンの帰属は、標準のgHMQC及びgCOSY試験によって行った。融点は、温度計を較正することなく、融点装置で測定した。電子スプレーイオン化(ESI)マススペクトルは、KRATOS MS25RFA質量分析計で記録した。高分解能質量分析は、VG MICROMASS ZAB 2F HS分光計(FAB)で行った。オリゴヌクレオチド高分解能マススペクトル測定は、IonSpec 7.0 tesla FTMSで、陽イオン電気スプレーモードで行い、ポリエチレングリコール300、600、及び1000で較正した。旋光度は、DIP-140(JASCO)デジタル旋光計を使用して測定した。ラセミ化の程度及び最終生成物の純度は、溶離液としてヘキサン中2−プロパノールを用いてのDiacel Chiralcel OD-H, OD-Jキラル分析カラムと、溶離システムとしてCH3CN/H2O/TFAを用いてのZorbax SB-CN半予備カラムとを使用して、分析用HPLCによって測定した。
【0082】
〔オリゴヌクレオチド合成、精製、及び同定〕
オリゴヌクレオチドは、その各担体から切り離した後、逆相高速液体クロマトグラフィー(HPLC)によって精製した。分離は、60℃に加熱したPolymerex 10μ RP-1カラム(Phenomenex、10 mm x 250 mm, 10μパッキング)を使用し、Waters 1525 バイナリーHPLCポンプによって作りだした1mL/分の移動相流速で行った。最初の移動相は、100mMの酢酸トリエチルアンモニウム緩衝液(pH7.0、80:20(v/v)の水:メタノール)の2分間の定組成流と、続いての35分間にわたる100mMの酢酸トリエチルアンモニウム緩衝液(pH7.0、70:30(v/v)の水:メタノール)への傾斜シフトと、さらに続いての1分間の定組成流と、その後の初期条件への14分間の傾斜を付けた戻りと、最後に5分間の定組成流から構成した。溶出は、Waters 2487二重吸光度検出器で、260nmと217nmで観察した。溶出ピークの画分を集め、移動相を真空下で除去した。サンプルを10%メタノール−水に再溶解し、続いて分子量を、ThermoQuest Finnigan LCQ Duo質量分析計での低解像度電子スプレーイオン化マススペクトルによって、マイナスイオンモードにおいて測定した。オリゴヌクレオチドの固相合成を、先の記載したように、標準条件6を用いて、Applied Biosystems 3400 DNA/RNA合成装置を使用して、500Åの長鎖アルキルアミンで被覆され且つ制御された細孔ガラス(LCAA−CPG)上で、1μmolスケールで行った。
【0083】
エステル化(ローディング)ステップ
無水アセトニトリル(25mL)中の、イオン液体1(1.07g、5mmol)、Boc-ロイシン(L-又はD-形 2.31g、10mmol)、及びジメチルアミノピリジン(0.25g、2mmol)の混合物に、ジシクロヘキシルカルボジイミド(DCC、CH2Cl2中に1M、10mL、10mmol)を添加した。この混合物を18時間、室温で、窒素下で激しく撹拌し、Celite(登録商標)の栓を通して濾過した。Celite(登録商標)の栓をアセトニトリルで濯ぎ、合わせた有機相を真空下で濃縮した。粗製残渣を最初にエーテルで洗浄し(20mL×3)、次にCH2Cl2に溶解し、2M HCl(10mL×3)で洗浄した。有機相をNa2SO4上で乾燥させ、濃縮して、イオン液体担持Boc-ロイシン 2a(L-異性体)又は2b(D-異性体)1.95g(91%)を淡黄色オイルとして得た。
1H NMR (300 MHz, Acetone-d6) δ 9.08 (s, 1H), 7.80 (s, 1H), 7.71 (s, 1H), 6.41 (d, br, J = 7.8 Hz, 1H), 4.66-4.64 (m, 2H), 4.60-4.43 (m, 2H), 4.20-4.13 (m, 1 H), 4.04 (s, 3H), 1.68-1.53 (m, 3H), 1.39 (s, 9H), 0.91 (d, J = 6.0 Hz, 3H), 0.89 (d, J = 6.0 Hz, 3H); 13C NMR (100 MHz, Acetone-d6) δ 173.2, 156.4, 138.0, 124.4, 123.6, 79.3, 63.4, 53.0, 49.3, 40.7, 36.6, 28.5 (3C), 25.4, 23.1, 21.6; HRMS (FAB) C17H30N3O4 (M+) に対する計算値340.2237, 測定値 340.2236。
【0084】
ペプチド形成;カップリング及び脱保護ステップ
CH2Cl2(10mL)中のイオン液体担持Boc−ペプチド(2mmol)の溶液に、TFA(10mL)を添加した。反応混合物を室温で窒素下にて0.5時間撹拌した。減圧下で濃縮したら、残渣をエーテルで2回洗浄し、真空下で乾燥させて、脱保護したイオン液体担持ペプチドをTFA塩として得た。次に、残渣を、その次のBocで保護したアミノ酸(3mmol)及びPyBOP(3mmol)とともに、CH3CN(35mL)中に溶かした。DIPEA(6mmol)を滴下して加え、得られた反応混合物を35℃で窒素雰囲気下にて8時間撹拌した。溶媒を真空下で除去し、残渣を最初にエーテル(20mL×3)で洗浄し、次にCH2Cl2中に溶かし、水(10mL×3)で洗った。有機相をNa2SO4上で乾燥させ、濃縮して、イオン液体担持Boc−ペプチド(90%収率)を得て、これはペプチド合成の次のサイクルに直接用いるために充分な純度であった。この生成物は、所望する場合には、この化合物をアセトン中に溶かし、シリカゲルの短い充填物を通して濾過し、溶媒を蒸発させることによって、さらに精製することができる。
【0085】
イオン液体担持ロイシンTFA塩 3a: 1H NMR (400 MHz, Acetone-d6) δ 9.20 (s, 0.55H), 9.12 (s, 0.45H), 7.85 (s, 1H), 7.68 (s, 1H), 5.02-4.98 (m, 0.45H), 4.75-4.58 (m, 4H), 4.27-4.25 (m, 0.55H), 4.05 (s, 3H), 1.90-1.87 (m, 2H), 1.73-1.70 (m, 1 H), 0.95 (d, J = 6.0 Hz, 3H), 0.93 (d, J = 6.0 Hz, 3H); 13C NMR (100 MHz, Acetone-d6) δ 169.4 (0.55C), 167.7 (0.45C), 138.0 (0.55C), 137.8 (0.45C), 124.4, 123.6 (0.45C), 123.5 (0.55C), 65.1 (0.45C), 64.9 (0.55C), 52.2, 48.9, 39.8 (0.55C), 39.4 (0.45C), 36.4, 25.1 (0.45C), 24.8 (0.55C), 22.7 (0.45C), 22.3 (0.55C), 22.1 (0.55C), 21.6 (0.45C); HRMS (FAB) C24H44N6O4BF4 (2M++BF4-) に対する計算値567.3450, 測定値 567.3453。
【0086】
イオン液体担持Leu-Boc-フェニルアラニン 4a:泡状淡黄色固体;1H NMR (400 MHz, Acetone-d6) δ 9.04 (s, 1H), 7.81 (s, 1H), 7.70 (s, 1 H), 7.67 (d, br, J = 6.8 Hz, 1 H), 7.29-7.20 (m, 5H), 6.20 (d, br, J = 8.0 Hz, 1H), 4.69-4.66 (m, 2H), 4.64-4.52 (m, 2H), 4.44-4.37 (m, 2H), 4.08 (s, 3H), 3.19-2.91 (m, 2H), 1.72-1.58 (m, 3H), 1.35 (s, 9H), 0.92 (d, J = 6.0 Hz, 3H), 0.89 (d, J = 6.0 Hz, 3H); 13C NMR (100 MHz, Acetone-d6) δ 172.9, 172.4, 156.5, 138.2, 132.6, 129.9 (2C), 128.8 (2C), 127.0, 124.5, 123.5, 79.4, 63.4, 56.6, 51.6, 49.4, 40.4, 38.3, 36.7, 28.4 (3C), 25.2, 23.2, 21.6; HRMS (FAB) C26H39N4O5 (M+) に対する計算値487.2920, 測定値 487.2920。
トリフルオロ酢酸による処理で、イオン液体担持Leu-フェニルアラニンTFA塩6が得られた: 1H NMR (300 MHz, Acetone-d6) δ 9.33 (s, br, 0.6H), 9.25 (s, br, 0.4H), 7.87 (s, br, 1H), 7.72 (s, br, 0.6H), 7.67 (s, br, 0.4H), 7.28-7.25 (m, 5H), 5.38-5.33 (m, 0.4H), 4.70-4.50 (m, 4H), 4.40-4.34 (m, 1.6H), 4.07 (s, 1.8H), 4.00 (s, 1.2H), 3.55-3.26 (m, 2H), 1.82-1.70 (m, 2H), 1.58-1.53 (m, 1 H), 0.92 (d, J = 6.0 Hz, 3H), 0.87 (d, J = 6.0 Hz, 3H); 13C NMR (75 MHz, Acetone-d6) δ 172.2, 172.0, 138.7 (0.6C), 138.5 (0.4C), 136.5 (0.6C), 135.6 (0.4C),
130.5 (0.8C), 130.4 (1.2C), 129.3 (1.2C), 129.2 (0.8C), 128.0 (0.6C), 127.9 (0.4C), 124.5, 123.6 (0.4C), 123.5 (0.6C), 63.6 (0.4C), 63.5 (0.6C), 54.9, 52.1 (0.6C), 52.0 (0.4C), 49.5 (0.6C), 49.4 (0.4C), 40.3 (0.4C), 39.9 (0.6C), 38.2 (0.6C), 37.4 (0.4C), 36.6, 25.2 (0.6C), 25.1 (0.4C), 23.1, 21.6 (0.4C), 21.5 (0.6C); HRMS (FAB) C21H31N4O3 (M+)に対する計算値 387.2395, 測定値 387.2396。
【0087】
イオン液体担持Leu-Phe-Boc-グリシン 7:泡状淡黄色固体; 1H NMR (300 MHz, Acetone-d6) δ 9.04 (s, 1H), 7.78 (t, J = 1.8 Hz, 1H), 7.66 (t, J = 1.8 Hz, 1H), 7.62 (d, J = 7.5 Hz, 1H), 7.55 (d, J = 8.1 Hz, 1H), 7.30-7.18 (m, 5H), 6.46 (m, 1H), 4.73-4.65 (m, 3H), 4.54-4.51 (m, 2H), 4.45-4.38 (m, 1H), 4.06 (s, 3H), 3.78-3.61 (m, 2H), 3.24-3.00 (m, 2H), 1.71-1.56 (m, 3H), 1.39 (s, 9H), 0.90 (d, J = 6.0 Hz, 3H), 0.88 (d, J = 6.0 Hz, 3H); 13C NMR (75 MHz,
Acetone-d6) δ 171.7 (2C), 170.3, 156.7, 137.7, 137.5, 129.4 (2C), 128.6 (2C), 126.8, 124.1, 123.0, 79.3, 63.1, 54.9, 51.3, 48.9, 44.4, 39.9, 37.4, 36.2, 28.0 (3C), 24.7, 22.7, 21.2; HRMS (FAB) C28H42N5O6 (M+)に対する計算値 544.3134, 測定値 544.3135。
トリフルオロ酢酸による処理で、イオン液体担持Leu-Phe-グリシンTFA塩 8が得られた: 1H NMR (300 MHz, Acetone-d6) δ 9.13 (s, 1 H), 7.80 (s, 1H), 7.68 (s, 1H), 7.32-7.20 (m, 5H), 4.66-4.50 (m, 7H), 4.41-4.39 (m, 1H), 4.06 (s, 3H), 3.25-2.96 (m, 2H), 1.77-1.68 (m, 2H), 1.59-1.54 (m, 1H), 0.92 (d, J = 6.0 Hz, 3H), 0.87 (d, J = 6.0 Hz, 3H); 13C NMR (75 MHz, Acetone-d6) δ 172.4, 172.3, 172.2, 138.4 (0.5C), 138.3 (0.5C), 138.2 (0.5C), 138.0 (0.5C), 130.0 (2C), 128.94 (1C), 128.91 (1C), 127.22 (0.5C), 127.17 (0.5C), 124.57 (0.5C), 124.54 (0.5C), 123.44 (0.5C), 123.40 (0.5C), 63.4, 56.8 (0.5C), 56.6 (0.5C), 51.6 (0.5C), 51.5 (0.5C), 49.5 (0.5C), 49.3 (0.5C), 42.8, 39.9 (0.5C), 39.8 (0.5C), 38.4 (0.5C), 38.3 (0.5C), 36.6, 25.2 (0.5C), 25.16 (0.5C), 23.17 (0.5C), 23.15 (0.5C), 21.5 (0.5C), 21.4 (0.5C); HRMS (FAB) C23H34N5O4 (M+) に対する計算値444.2609, 測定値 444.2611。
【0088】
イオン液体担持Leu-Phe-Gly-Boc-グリシン 9:泡状淡黄色固体; 1H NMR (300 MHz, Acetone-d6) δ 9.09 (s, 1H), 8.12 (m, br, 1H), 7.80 (t, J = 1.8 Hz, 1H), 7.68 (t, J = 1.8 Hz, 1H), 7.64 (m, 1 H), 7.61 (m, 1 H), 7.30-7.21 (m, 5H), 6.57 (m, 1H), 4.76-4.59 (m, 3H), 4.54-4.51 (m, 2H), 4.40-4.33 (m, 1H), 4.08 (s, 3H), 3.82-3.75 (m, 4H), 3.33-2.95 (m, 2H), 1.89-1.55 (m, 3H), 1.43 (s, 9H), 0.94 (d, J = 6.0 Hz, 3H), 0.89 (d, J = 6.0 Hz, 3H); 13C NMR (100 MHz, Acetone-d6) δ 171.9, 171.8, 171.6, 169.8, 156.9, 138.0, 137.8, 129.1 (2C), 128.6 (2C), 126.7, 124.1, 123.0, 79.4, 63.1, 55.6, 51.4, 48.9, 44.6, 43.8, 39.9, 37.3, 36.3, 28.2 (3C), 24.7, 22.9, 21.2; HRMS (FAB) C30H45N6O7 (M+) に対する計算値601.3348, 測定値 601.3350。
トリフルオロ酢酸による処理で、イオン液体担持Leu-Phe-Gly-グリシンTFA塩 10が得られた: 1H NMR (400 MHz, Acetone-d6) δ 9.10 (s, 1H), 7.81 (s, 1H), 7.70 (s, 1H), 7.28-7.20 (m, 5H), 4.75-4.67 (m, 5H), 4.58-4.50 (m, 2H), 4.43-4.38 (m, 1H), 4.08 (s, 3H), 3.89-2.87 (m, 2H), 3.23-3.04 (m, 2H), 1.65-1.58 (m, 3H), 0.92 (d, J = 6.0 Hz, 3H), 0.87 (d, J = 6.0 Hz, 3H); 13C NMR (100 MHz, Acetone-d6) δ 171.9, 171.7, 137.9, 129.4, 129.3 (2C), 128.4 (2C), 126.7, 124.0, 123.0, 63.0, 55.8, 50.8, 49.3, 48.9, 43.5, 39.7, 37.3, 36.3, 24.8, 22.7, 21.2.; HRMS (FAB) C25H37N6O5 (M+) に対する計算値501.2826, 測定値501.2825。
【0089】
イオン液体担持Leu-Phe-Gly-Gly-Boc-チロシン 11:泡状淡黄色固体; 1H NMR (400 MHz, Acetone-d6) δ 9.08 (s, 1H), 8.21 (s, br, 1H), 8.07 (s, br, 1H), 7.80 (s, 1H), 7.68 (d, J = 7.2 Hz, 1H), 7.67 (s, 1H), 7.56 (d, J = 7.2 Hz, 1H), 7.34-7.20 (m, 5H), 7.15 (d, J = 8.0 Hz, 2H), 6.90 (d, J = 8.0 Hz, 2H), 6.49 (d, J = 7.2 Hz, 1H), 4.76-4.56 (m, 3H), 4.54-4.51 (m, 2H), 4.40-4.33 (m, 2H), 4.08 (s, 3H), 3.96-3.66 (m, 4H), 3.31-2.91 (m, 4H), 1.85-1.70 (m, 2H), 1.62-1.55 (m, 1H), 1.36 (s, 9H), 1.30 (s, 9H), 0.94 (d, J = 6.0 Hz, 3H), 0.89 (d, J = 6.0 Hz, 3H); 13C NMR (100 MHz, Acetone-d6) δ 174.0, 171.9, 171.7, 171.3, 169.8, 156.4, 154.5, 138.1, 137.8, 132.2, 129.9(2C), 129.3 (2C), 128.6 (2C), 126.7, 124.1, 124.0 (2C), 122.9, 79.6, 77.9, 63.2, 57.2, 56.0, 51.4, 48.5, 44.0, 43.8, 39.8, 37.4, 37.0, 36.3, 28.6 (3C), 28.1 (3C), 24.8, 22.9, 21.2; HRMS (FAB) C43H62N7O9 (M+) に対する計算値820.4605, 測定値 820.4609。
【0090】
〔担持及びペプチド形成ステップにおけるラセミ化の程度の測定(それがある場合)〕
イオン液体担持Leu-Phe-Boc 4a〜dの調製: 4a〜dは、上に記載したとおりの一般手順を用いて、イオン液体担持ロイシン2a〜b(L-及びD-エナンチオマー)と、Boc-L-フェニルアラニン及びBoc-DL-フェニルアラニンとの反応で調製した。4a:L,L-エピマー、4b:D,L-エピマー、4c:L,DL-エピマー、及び4d:D,DL-エピマー。
【0091】
メチルN-(tert-ブチルオキシカルボニル)-フェニルアラニル-ロイシネート5a〜d:
先のステップからの4a(0.1mmol)を、アンモニアで飽和させたメタノール(2mL)中に溶かした。得られた溶液を、次に、室温で夜通し撹拌した。減圧下で溶媒を除去した後、残渣をフラッシュカラムクロマトグラフィー(シリカゲル230〜400メッシュ、ヘキサン−酢酸エチル勾配)で精製してジペプチド生成物MeO-Leu(L)-Phe(L)-Boc5a-(L,L)を得た。同様にして、ジペプチドMeO-Leu(D)-Phe(L)-Boc 5b-(D,L)、MeO-Leu(L)-Phe(DL)-Boc 5c-(L,DL)、及びMeO-Leu(D)-Phe(DL)-Boc 5d-(D,DL)も調製した。HPLC分析:Diacel Chiralcel OD-Hカラム、ヘキサン中5%イソプロパノール、0.5mL/分、λ=220nm、tR(L,L-エピマー):13.3分、tR(D,L-エピマー):13.4分;tR(L,D-エピマー):15.7分;tR(D,D-エピマー):22.2分; Diacel Chiralcel OD-Jカラム、ヘキサン中5%イソプロパノール、0.3mL/分、λ=220nm、tR(L,L-エピマー):16.0分、tR(D,L-エピマー):17.4分。
【0092】
MeO-Leu(L)-Phe(L)-Boc 5a-(L,L)51:白色固体、m.p. 101-103 °C (文献17: m.p. 101-104 °C). 1H NMR (400 MHz, Acetone-d6) δ 7.52 (d, J = 7.6 Hz,
1H), 7.26-7.18 (m, 5H), 6.07 (d, J = 8.0 Hz, 1H), 4.56-4.50 (m, 1H), 4.42-4.38 (m, 1H), 3.68 (s, 3H), 3.20-2.88 (m, 2H), 1.75-1.68 (m, 1H), 1.62-1.58 (m, 2H), 1.35 (s, 9H), 0.92 (d, J = 6.4 Hz, 3H), 0.91 (d, J = 6.4 Hz, 3H)。 HPLC分析では、いかなる検出可能な量のD,L-エピマー、L,D-エピマー、及びD,D-エピマーも示さなかった。
【0093】
MeO-Leu(D)-Phe(L)-Boc 5b-(D,L)51:白色結晶、m.p.136-138 °C. 1H NMR (400 MHz, Acetone-d6) δ 7.44 (d, J = 7.2 Hz, 1 H), 7.28-7.18 (m, 5H), 6.06 (d, J = 8.0 Hz, 1H), 4.48-4.42 (m, 1H), 4.41-4.35 (m, 1H), 3.66 (s, 3H), 3.11-2.93 (m, 2H), 1.55-1.52 (m, 3H), 1.36 (s, 9H), 0.88 (d, J = 6.4 Hz, 3H), 0.87 (d, J = 6.4 Hz, 3H)。HPLC分析ではいかなる検出可能な量のL,L-エピマー、L,D-エピマー、及びD,D-エピマーも示さなかった。
【0094】
〔ペプチド遊離(切り離し)ステップ〕
THF/H2O(1:2、v/v、3.0mL)中のイオン液体担持ペンタペプチド11(180mg、0.2mmol)の混合物に、1M NaOH水溶液(0.2mL)を加えた。混合物を室温で5時間撹拌した。揮発性成分を減圧下で除去し、残った溶液をpH5〜6に酸性化した。沈殿物を蒸留水で2回洗浄し、真空下で乾燥して120mg(85%)のBoc-ペンタペプチド12を泡状淡黄色固体として得た。1H NMR (400 MHz, Acetone-d6) δ 8.01 (s, br, 1H), 7.80 (s, br, 1H), 7.65 (d, J = 6.8 Hz, 2H), 7.30-7.15 (m, 5H),
7.17 (d, J = 8.0 Hz, 2H), 6.89 (d, J = 8.0 Hz, 2H), 6.43 (d, J = 7.2 Hz, 1H), 4.76-4.74 (m, 1H), 4.52-4.47 (m, 1H), 4.42-4.39 (m, 1H), 4.01-3.74 (m, 4H), 3.28-2.90 (m, 4H), 1.79-1.65 (m, 3H), 1.35 (s, 9H), 1.30 (s, 9H), 0.94 (d, J = 6.0 Hz, 3H), 0.92 (d, J = 6.0 Hz, 3H); 13C NMR (100 MHz, Acetone-d6) δ 173.5, 173.2, 171.4, 170.0, 169.0, 156.1, 154.3, 138.0, 132,6, 130.0 (2C), 129.6 (2C), 128.4 (2C), 126.5, 124.0 (2C), 79.2, 77.8, 56.8, 54.9, 51.1, 43.5, 43.1, 40.8, 37.9, 37.3, 28.7 (3C), 28.2 (3C), 25.0, 23.0, 21.5; HRMS (FAB) C37H53N5O9Na (M+Na) に対する計算値734.3741, 測定値 734.3742。
【0095】
Boc-ペンタペプチド12(117mg、0.16mmol)をCH2Cl2(1.5mL)中に溶かし、次にTFA(1.5mL)とアニソールを数滴添加した。反応混合物を常温において窒素雰囲気下で0.5時間撹拌した。蒸発とエーテルでの洗浄によって、Leu5-エンケファリンをTFA塩14として、淡黄色固体として得た(109mg、99%)。m.p. 148-152 °C (真正サンプルのm.p. 150-153 °C); [α]20D = 25.0 (c 0.8, 95% AcOH), 真正サンプル[α]20D = 25.6 (c 0.9, 95% AcOH); HPLC tR = 16.0 min, (真正サンプル tR = 15.7 min), Agilent Zorbax SB-CN Semi-prepカラム; 溶離液, 0-5 min, H20-0.05%TFA (v/v), 6-15 min, 0-60% CH3CN/H2O-0.05%TFA (v/v/v), 16-23 min, 60-100% CH3CN/H2O-0.05%TFA (v/v/v); ポスト時間, 7分; 流速, 3.0 ml/min; 1H NMR (400 MHz, Methanol-d4) δ 7.24-7.21 (m, 4H), 7.18-7.15 (m, 1H), 7.08 (d, J = 8.4 Hz, 2H), 6.77 (d, J = 8.4 Hz, 2H), 4.71-4,67 (m, 1H), 4.42-4.39 (m, 1H), 4.07 (t, J= 7.2 Hz, 1H), 3.96-3.72 (m, 4H), 3.19-2.92 (m, 4H), 1.71-1.61 (m, 3H), 0.94 (d, J = 6.0 Hz, 3H), 0.90 (d, J = 6.0 Hz, 3H); 13C NMR (100 MHz, Methanol-d4) δ 175.1, 173.1, 171.0, 170.6, 170.4, 158.0, 138.0, 131.3 (2C), 130.2 (2C), 129.2 (2C), 127.5, 125.8, 116.8 (2C), 56.2, 55.6, 52.3, 43.9, 43.4, 41.7, 39.0, 37.7, 26.0, 23.2, 22.0; HRMS (FAB) C28H37N5O7Na (M+Na) に対する計算値578.2592, 測定値 578.2591。
【0096】
フェニル2,3,4-トリ-O-ベンジル-1-チオ-β-D-グルコピラノシド(15):
フェニル2,3,4-トリ-O-ベンジル-6-O-トリメチルアセチル-1-チオ-β-D-グルコピラノシド(3.02g、4.82mmol)を入れたフラスコに、NaOCH3/HOCH3溶液(50mL)とCH2Cl2(15mL)(pH=13)を添加した。溶液を室温で48時間撹拌し、蒸発により溶媒を除去し、真空中で乾燥させた。無水CH2Cl2を粗精製物残渣に添加し、不溶性塩を、濾剤であるセライト(登録商標)521を詰めたブフナーロートで濾別した。濾液を集め、溶媒をロータリーエバポレーターで除去し、真空中で乾燥させて生成物を白色固体として得た(2.61g、収率100%)。m.p.127-129 °C; 1H NMR (400MHz, CDCl3), δ 7.51 (m, 2H), 7.40-7.27 (m, 18H), 4.94-4.85 (m, 4H), 4.77 (d, J=l0Hz, 1H), 4.73 (d, J=9.6Hz, 1H), 4.66 (d, J=10.8Hz, 1H), 3.89 (dd, J=2.8Hz, 14.8Hz, H6a, 1H), 3.77-3.69 (m, 2H), 3.59 (t, J=8.8Hz, 1H), 3.50 (t, J=8.8Hz, 1H), 3.40 (m, 1H); 13C NMR (400MHz, CDCl3), δ 138.49, 138.09, 138.02, 133.67, 132.06, 129.25, 128.73-127.89 (m), 87.82, 86.84, 81.41, 79.61, 77.90, 76.13, 75.85, 75.43, 62.48; ESI-MS, C33H34SO5 (M+Na+)に対する計算値: m/z=565, 測定値: 565。
【0097】
フェニル2,3,4-トリ-O-ベンジル-6-O-ブロモアセチル-1-チオ-β-D-グルコピラノシド(16a):
CH2Cl2(40mL)中のフェニル2,3,4-トリ-O-ベンジル-1-チオ-β-D-グルコピラノシド(15)(0.988g、1.82mmol)及びブロモ酢酸(0.308g、2.21mmol)の溶液に、CH2Cl2中のDCCの1.0M溶液(2.20mL、2.20mmol)及び少量のDMAP(糖に対して<5%当量)を添加した。室温で2時間撹拌した後、混合物を濾過して副生成物の1,3-ジシクロヘキシルウレアを除去した。乾燥するまで濾液を蒸発させ、溶離液としてヘキサンと酢酸エチル(7:1、V/V)を用いて、残渣をフラッシュカラムクロマトグラフィー(シリカゲル60A、230〜400メッシュ)にかけて純粋な生成物を得た(1.08g、収率89%)。
m.p. 85-87 °C; 1H NMR (400MHz, CDCl3), δ 7.54 (m, 2H), 7.41-7.25 (m, 18H), 4.95-4.84 (m, 4H), 4.75 (d, J=l0Hz, 1H), 4.67 (d, J=9.6Hz, 1H), 4.61 (d, J=10.8Hz, 1H), 4.45 (dd, J=11.2Hz, 0.8Hz, 1H), 4.28 (m, 1H), 3.79 (s, 2H), 3.73 (m, 1H), 3.55 (m, 2H), 3.51 (t, J=18.4Hz, 1H); 13C NMR (300MHz, CDCl3), δ 166.91, 138.29, 138.02, 137.70, 133.51, 132.39, 129.14, 128.76-127.94 (m), 87.81, 86.96, 81.06, 77.49, 77.36, 76.19, 75.79, 75.38, 65.07, 25.90; ESI-MS, C35H35SO6Br (M+Na+)に対する計算値: m/z=685, 測定値: 685。
【0098】
2,3,4-トリ-O-ベンジル-6-O-[2-(3-メチル-イミダゾリウム)]アセチル-1-チオ-β-D-グルコピラノシドテトラフルオロボレート(16b):
アセトン(25mL)中のフェニル2,3,4-トリ-O-ベンジル-6-O-ブロモアセチル-1-チオ-β-D-グルコピラノシド(16a)(0.890g、1.34mmol)に、CH2Cl2中の1-メチルイミダゾールの1.0M溶液(1.61mL、1.61mmol)を加え、さらにNaBF4(0.182g、1.66mmol)を加えた。室温での72時間の撹拌後、沈殿物を濾別し、乾燥するまで濾液を蒸発させ、真空中で夜通し乾燥させた。生成物残渣をジエチルエーテル(15mL)で3回洗浄し、上澄みをデカンテーションした。固体を真空中で乾燥させ、15mLの無水CH2Cl2を添加し、沈殿物を濾過することにより、残渣をさらに精製した。乾燥するまで濾液を蒸発させ、真空中で乾燥させて、白色泡状物として純粋な生成物を得た(0.989g、収率98%)。
m.p. 42-44 °C; 1H NMR (400MHz, CDCl3), δ 8.74 (s, 1H), 7.46 (m, 2H), 7.36-7.23 (m, 18H), 7.17 (m, 2H), 5.01-4.82 (m, 6H), 4.72 (d, J=10.0Hz, 1H), 4.71(d, J=9.6Hz, 1H), 4.58 (d, J=11.2Hz, 1H), 4.50 (dd, J=11.6Hz, 2.4Hz, 1H), 4.24 (dd, J=6.4Hz, 12Hz, 1H), 3.82 (s, 3H), 3.73 (m, 1H), 3.58 (m, 1H), 3.52-3.43 (m, 2H); 13C NMR (300MHz, CDCl3), δ 165.90, 138.33, 138.07, 138.02, 137.83, 133.65, 131.58, 129.37, 128.76-127.92 (m), 123.69, 123.25, 87.41, 86.79, 81.02, 77.53, 76.74, 76.01, 75.70, 75.15, 65.51, 50.03, 36.81; ESI-MS C39H41SO6N2 (M+)カチオンに対する計算値: m/z = 665, 測定値: 665。
【0099】
フェニル2,3,4-トリ-O-ベンジル-6-O-[2-(3-メチル-イミダゾリウム)]アセチル-1-チオ-β-D-グルコピラノシルスルホキシドテトラフルオロボレート(17b):
−78℃で、CH2Cl2(2mL)中の16b(50.0mg、0.066mmol)の溶液に、CH2Cl2(1mL)中のm-クロロ過安息香酸(m-CPBA)(15.0mg、0.066mmol)を5分にわたって滴下して加えた。−78℃で20分撹拌後、混合物を15mLのCH2Cl2とともにNaHCO3飽和水溶液(10mL)中に注いだ。有機相を分離し、飽和NaHCO3水溶液(10mL)で洗浄し、無水Na2SO4上で乾燥させた。溶媒を減圧下にてロータリーエバポレーターで除去した。生成物残渣を、ヘキサンとイソプロピルエーテルで洗浄することによってさらに精製(TLCで観察)して、2つのジアステレオマーを含む白色固体としてグリコシルスルホキシドを得た(49.5mg;収率97%;NMRによって測定した2つのジアステレオマーの比は60:40)。
1H NMR (400MHz, CDCl3), δ 9.08 (s, 1H), 7.54 (m, 2H), 7.40-7.16 (m, 20H), 5.11-4.20 (m, 11H), 3.97 (m, 2H), 3.91 (s, 2H), 3.90 (s, 1H), 3.85-3.26 (m, 2H); ESI-MS C39H41SO7N2 (M+)カチオンに対する計算値: m/z = 681, 測定値: 681。
【0100】
フェニル2,3,4-トリ-O-ベンジル-6-O-[2-(3-メチル-イミダゾリウム)]アセチル-β-D-グルコピラノシル-(1→6)-2,3,4-トリ-O-ベンジル-1-チオ-β-D-グルコピラノシド テトラフルオロボレート(18b):
−78℃で、CH2Cl2(5mL)中の、イオン液体に結合したモノサッカライドスルホキシド(51.8mg、0.067mmol;グリコシル供与体)、フェニル2,3,4-トリ-O-ベンジル-1-チオ-β-D-グルコピラノシド(15、114mg、0.21mmol;グリコシル受容体)、及び2,6-ジ-tert-ブチル-4-メチルピリジン(58.8mg、0.286mmol;酸除去剤)の溶液に、Tf2O(12μL、0.067mmol)をシリンジから10分間にわたって徐々に添加した。−78℃で20分間撹拌後、混合物を1時間にわたって0℃までゆっくり温めた。白色沈殿物の出現をもたらすn-ペンタン(5mL)を添加することによって反応を止めた。混合物を−78℃に冷却し、白色沈殿物をすぐに遠心分離した。溶液相にn-ペンタンのさらなる一部(5mL)を加え、次に−78℃に冷却した。得られた沈殿物を再び遠心分離した。溶液相を蒸発させ、残渣を数回n-ペンタンで洗い、次にTLC分析が、残渣が純粋であることを示すまで、イソプロピルエーテルでさらに洗った。白色固体として生成物を得た(39mg、収率50%)。
m.p. 45-47°C; 1H NMR (400MHz, CDCl3), δ 9.10 (s, 1 H), 7.51 (m, 2H), 7.41-7.13 (m, 35H), 5.10 (d, J=10.4Hz, 1H), 5.00-4.52 (m, 15H), 4.41 (m, 1H), 4.19 (dd, J=12.4Hz, 4.0Hz, 1H), 3.99 (t, J=8.8Hz, 1H), 3.90-3.75 (m, 6H), 3.73-3.62 (m, 2H), 3.55 (m, 2H), 3.42 (t, J=8.8Hz,
1H), 3.29 (t, 1H); 13C NMR (400MHz, CDCl3), δ=165.79, 138.78, 138.60, 138.41, 138.38, 138.20, 138.11, 138.03, 134.18, 131.78, 129.32, 128.73-127.69 (m), 123.65, 123.24, 97.30, 88.25, 86.88, 81.82, 81.31, 80.30, 79.06, 77.76, 77.57, 75.99, 75.89, 75.81, 75.25, 75.09, 72.83, 68.68, 66.89, 65.49, 50.51, 37.16; ESI-MS C66H69SO11N2 (M+)カチオンに対する計算値: m/z= 1097, 測定値: 1097。
【0101】
フェニル2,3,4-トリ-O-ベンジル-6-O-[2-(3-メチル-イミダゾリウム)]アセチル-β-D-グルコピラノシル-(1→6)-2,3,4-トリ-O-ベンジル-1-チオ-β-D-グルコピラノシルスルホキシド テトラフルオロボレート(19b):
−78℃で、CH2Cl2(2.5mL)中のジサッカライドスルフィド18b(56.1mg、0.047mmol)溶液に、5分間にわたって、CH2Cl2(1mL)中のm-クロロ過安息香酸(m-CPBA)(8.1mg、0.047mmol)を5分間にわたって滴下して加えた。窒素雰囲気保護下、−78℃で20分間撹拌後、混合物をCH2Cl2(15mL)とともに飽和NaHCO3水溶液(10mL)中に注いだ。有機相を分離し、再度、飽和NaHCO3水溶液(10mL)で洗い、無水Na2SO4上で乾燥させた。溶媒を、減圧下でロータリーエバポレーターによって除去した。生成物残渣をヘキサンとイソプロピルエーテルで洗浄すること(TLCでモニターした)によって、さらに精製して、2つのジアステレオマーを含む白色固体として所望するグリコシルスルホキシドを得た(54.0mg、収率95%、NMRで測定した2つのジアステレオマーの比は60:40)。
1H NMR (400MHz, CDCl3), δ 9.48 (s, 1H), 7.58-7.13 (m, 37H), 5.00-4.40 (m, 18H), 3.95-3.25 (m, 13H); ESI-MS C66H69SO12N2 (M+)カチオンに対する計算値: m/z = 1113, 測定値: 1113。
【0102】
フェニル2,3,4-トリ-O-ベンジル-6-O-[2-(3-メチル-イミダゾリウム)]アセチル-β-D-グルコピラノシル-(1→6)-2,3,4-トリ-O-ベンジル-β-D-グルコピラノシル-(1→6)-2,3,4-トリ-O-ベンジル-1-チオ-β-D-グルコピラノシド テトラフルオロボレート(20b):
イオン液体に結合したジサッカライドスルホキシド(19b;45.1mg;0.038mmol;グリコシル供与体)、フェニル2,3,4-トリ-O-ベンジル-1-チオ-β-D-グルコピラノシド(15;62.1mg;0.114mmol;グリコシル受容体)、及び2,6-ジ-tert-ブチル-4-メチルピリジン(30.8mg;0.150mmol;酸除去剤)のCH2Cl2(5mL)中の溶液に、−78℃で、シリンジから10分間にわたってTf2O(6μL、0.036mmol)を徐々に添加した。−78℃で20分間撹拌後、混合物を1時間にわたってゆっくりと0℃まで温めた。白色沈殿の出現をもたらすn-ペンタン(5mL)を添加することによって、反応混合物を失活させた。反応混合物を−78℃に冷却し、沈殿物を直ちに遠心分離した。溶液相にn-ペンタンの別の一部(5mL)を加え、次に−78℃に冷却した。得られた沈殿物を再び遠心分離によって除去した。溶液相を蒸発させ、残渣を数回n-ペンタンで洗い、次にTLC分析が、残渣が純粋であることを示すまで、イソプロピルエーテルでさらに洗った。白色固体として所望の生成物を得た(34mg、収率56%)。
1H NMR (400MHz, CDCl3), δ 9.04 (s, 1H), 7.51 (m, 2H), 7.42-7.17 (m, 48H), 7.05 (d, J=10.2Hz, 2H), 5.00-4.50 (m, 25H), 4.39 (d, J=8.0Hz, 1H), 4.15 (m, 1H), 4.00 (m, 2H), 3.89-3.72 (m, 6H), 3.70-3.30 (m, 8H), 3.25 (t, J=8.8Hz, 1H); 13C NMR (400MHz, CDCl3), δ 165.77, 138.95-138.09 (m), 134.22, 131.87, 131.31, 129.29, 128.74-127.36 (m), 123.72, 123.76, 123.01, 103.91,97.47, 97.26, 88.33, 87.74, 86.85, 84.88, 82.66-80.14 (m), 79.21, 79.06, 78.67, 77.54, 75.93-75.85 (m), 75.22-75.00 (m), 72.79, 72.72, 72.51, 70.86, 68.90, 69.58, 68.31, 66.44, 65.81, 65.53, 50.01, 36.90; ESI-MS C93H97SO16N2 (M+)カチオンに対する計算値: m/z= 1529, 測定値: 1529。
【0103】
トリサッカライド21を得るための、20bからのイオン液体残基の切り離し:
イオン液体に結合したトリサッカライド(20b、30.0mg、0.018mmol)を入れたフラスコに、Cs2CO3(6.1mg、0.018mmol)とメタノール(5mL)を添加した。室温での12時間撹拌後に、溶媒をロータリーエバポレーターによって除去し、残渣を減圧下で乾燥させた。この残渣に無水CH2Cl2(3mL)を添加し、不溶性塩を濾過により分離した。濾液を乾燥するまで蒸発させ、減圧下で乾燥させて所望の生成物21を得た(26.1mg、収率100%)。
1H NMR (400MHz, CDCl3), δ 7.54 (m, 2H), 7.43-7.14 (m, 48H), 5.16 (d, J=3.2Hz, 1H), 5.00-4.93 (m, 3H), 4.91-4.86 (m, 4H), 4.85- 4.74 (m, 5H), 4.71-4.53 (m, 9H), 4.40 (d, J=8.0Hz, 1H), 4.12 (d, J=10.0Hz, 1H), 3.99 (m, 2H), 3.86-3.38 (m, 12H), 3.26 (m, 1H); 13C NMR (400MHz, CDCl3), δ 138.94-138.22 (m), 134.33, 132.01, 131.55, 129.19, 129.10, 128.59-127.44 (m), 103.91, 97.54, 97.31, 88.31, 87.79, 86.93, 84.81, 82.62, 81.95, 81.85, 81.75, 81.44, 81.33, 80.73, 80.63, 80.49, 79.23, 79.00, 78.12, 77.91, 77.75, 77.51, 75.95-75.80 (m), 75.45-75.25 (m), 72.75,
72.61, 72.57, 71.24, 68.65, 66.02, 65.52, 62.30; ESI-MS C87H90SO15Na
(M+Na+)に対する計算値: m/z= 1429, 測定値: 1429。
【0104】
フェニル2,3,4-トリ-O-ベンジル-6-O-アセチル-1-チオ-β-D-グルコピラノシド(16c):
フェニル2,3,4-トリ-O-ベンジル-1-チオ-β-D-グルコピラノシド(15)(0.801g、1.48mmol)、無水ピリジン(0.45mL)、及びDMAP(糖に対して5当量%未満)のCH2Cl2(9mL)中の溶液に、無水酢酸(0.21mL、2.22mmol)を添加した。窒素雰囲気の保護下で、室温にて4時間撹拌した後、反応混合物を150mLのCH2Cl2中に移し、0.2M HCl水溶液(30mL)、H2O(30mL)、飽和NaHCO3水溶液(30mL)、そして次に食塩水(30mL)で、それぞれ洗った。有機相を無水Na2SO4上で乾燥させ、溶媒をロータリーエバポレーターによって除去して、粗生成物残渣を得、これを溶離液としてヘキサン及び酢酸エチル(5:1、V/V)を用いてフラッシュカラムクロマトグラフィー(シリカゲル60A、230〜400メッシュ)にかけて純粋な生成物(0.845g、収率98%)を得た。
1H NMR (400MHz, CDCl3), δ 7.55 (m, 2H), 7.40 (m, 2H), 7.36-7.25 (m, 16H), 4.95-4.84 (m, 4H), 4.75 (d, J=10Hz, 1H), 4.67 (d, J=9.6Hz, 1H), 4.58 (d, J=10.8, 1H), 4.37 (d, J=11.6Hz, 1H), 4.22 (m, 1H), 3.73 (t, J=8.4 Hz, 1H), 3.55 (m, 2H), 3.50 (t, J=9.6Hz, 1H), 2.11(s, 3H); ESI-MS C35H36SO6Na (M+Na+)に対する計算値: m/z=607, 測定値: 607。
【0105】
フェニル2,3,4-トリ-O-ベンジル-6-O-アセチル-β-D-グルコピラノシルスルホキシド(17c):
−78℃で、CH2Cl2(5mL)中の16c(1.01g、1.73mmol)の溶液に、CH2Cl2(7mL)中のm-クロロ過安息香酸(m-CPBA)(0.387g、1.73mmol)を5分にわたって滴下して加えた。窒素雰囲気の保護下で−78℃にて20分撹拌後、混合物をCH2Cl2(50mL)とともにNaHCO3飽和水溶液(100mL)中に注いだ。有機相を分離し、飽和NaHCO3水溶液(50mL)で再度洗浄し、無水Na2SO4上で乾燥させた。溶媒を減圧下にてロータリーエバポレーターで除去して粗生成物残渣を得、これをヘキサン及び酢酸エチル(3:1、V/V)を用いるフラッシュカラムクロマトグラフィー(シリカゲル60A、230〜400メッシュ)にかけて、2つのジアステレオマーの混合物として、純粋な生成物を得た(0.984g、収率95%、NMRによって測定した2つのジアステレオマーの比は3:1)。
1H NMR (400MHz, CDCl3), δ 7.62-7.16 (m, 20H), 5.06-4.78 (m, 6H), 4.59-4.36 (m, 2H), 4.19-3.30 (m, 8H); SI-MS C35H36SO7Na (M+Na+)に対する計算値: m/z=623, 測定値: 623。
【0106】
フェニル2,3,4-トリ-O-ベンジル-6-O-アセチル-β-D-グルコピラノシル-(1→6)-2,3,4-トリ-O-ベンジル-1-チオ-β-D-グルコピラノシド(18c):
スルホキシド 17c(0.302g、0.503mmol)、フェニル2,3,4-トリ-O-ベンジル-1-チオ-β-D-グルコピラノシド(15、0.406g、0.754mmol)及び2,6-ジ-tert-ブチル-4-メチルピリジン(0.315g、1.53mmol)のCH2Cl2(10mL)中の溶液に、−78℃で、CH2Cl2(5mL)中のTf2O溶液(0.09mL、0.503mmol)をシリンジから10分間にわたって徐々に添加した。−78℃で20分間撹拌後、混合物を1.5時間にわたって−5℃までゆっくり温めた。飽和NaHCO3水溶液(6mL)を添加することによって反応物を失活させ、CH2Cl2(35mL)と飽和NaHCO3水溶液(20mL)との間で分配させた。有機相を分離し、飽和NaHCO3水溶液(40mL)でさらに洗った。無水Na2SO4上で乾燥させた後、有機相を乾燥するまで蒸発させ、残渣を、溶離液としてヘキサン及び酢酸エチル(6:1、5:1、4:1、V/V)を用いるフラッシュカラムクロマトグラフィーにかけて、無色シロップとして純粋な生成物を得た(0.414g、収率81%)。
1H NMR (400MHz, CDCl3), δ 7.54 (m, 2H), 7.42 (m, 2H), 7.38-7.24 (m, 31 H), 5.05 (m, 2H), 4.94-4.56 (m, 12H), 4.25 (m, 2H), 4.02 (t, J=9.2Hz, 1H), 3.92 (m, 1H), 3.86 (d, J=4.8Hz, 1H), 3.80 (m, 1H), 3.75 (m, 1H), 3.70 (t, 1H), 3.62-3.46 (m, 3H), 3.28 (t, J=8.4Hz, 1H), 2.03 (s, 3H); ESI-MS C62H64SO11Na (M+Na+)に対する計算値: m/z=1039, 測定値: 1039。
【0107】
フェニル2,3,4-トリ-O-ベンジル-6-O-アセチル-β-D-グルコピラノシル-(1→6)-2,3,4-トリ-O-ベンジル-β-D-グルコピラノシルスルホキシド(19c):
ジサッカライドスルフィド18c(0.202g、0.199mmol)のCH2Cl2(12mL)中の溶液に、−78℃で、5分間にわたって、CH2Cl2(6mL)中のm-クロロ過安息香酸(m-CPBA)(0.045g、0.199mmol)を5分間にわたって滴下して加えた。窒素雰囲気保護下、−78℃で20分間撹拌後、混合物をCH2Cl2(60mL)とともに飽和NaHCO3水溶液(30mL)中に注いだ。有機相を分離し、再度、飽和NaHCO3水溶液(30mL)で洗い、無水Na2SO4上で乾燥させた。溶媒を、減圧下でロータリーエバポレーターによって除去して粗生成物残渣を得、これを、溶離システムとしてヘキサン及び酢酸エチル(2:1、V/V)を用いるフラッシュカラムクロマトグラフィー(シリカゲル60A、230〜400メッシュ)にかけて、2つのジアステレオマーの混合物として生成物を得た(0.197g、全収率96%、NMRで測定した2つのジアステレオマーの比は4:1)。
第一の異性体、Rf=0.27(2:1、ヘキサン/酢酸エチル、V/V);1H NMR (400MHz, CDCl3), δ7.60 (m, 2H), 7.46 (m, 4H), 7.42-7.22 (m, 29H), 5.05 (d, J=3.6Hz, 1H), 5.00-4.66 (m, 12H), 4.60 (d, J=10.8Hz, 1H), 4.27 (m, 2H), 3.87 (m, 3H), 3.74 (m, 4H), 3.59-3.47 (m, 3H), 3.35 (m, 1H) 2.07 (s, 3H);
第二の異性体、Rf=0.12(2:1、ヘキサン/酢酸エチル、V/V);1H NMR (400MHz, CDCl3), δ 7.51 (m, 2H), 7.39-7.19 (m, 31H), 7.06 (m, 2H), 4.98-4.88 (m, 3H), 4.82-4.52 (m, 10H), 4.43 (d, J=8.8Hz, 2H), 4.25 (m, 2H), 4.39 (t, J=9.6Hz, 1H), 3.90-3.69 (m, 6H), 3.60 (m, 1H), 3.49 (m, 2H), 2.05 (s, 3H). ESI-MS C62H64SO12Na (M+Na+)に対する計算値: m/z=1055, 測定値: 1055。
【0108】
フェニル2,3,4-トリ-O-ベンジル-6-O-アセチル-β-D-グルコピラノシル-(1→6)-2,3,4-トリ-O-ベンジル-β-D-グルコピラノシル-(1→6)-2,3,4-トリ-O-ベンジル-1-チオ-β-D-グルコピラノシド(20c):
スルホキシド19c(0.110g、0.107mmol)、フェニル2,3,4-トリ-O-ベンジル-1-チオ-β-D-グルコピラノシド(15、0.088g、0.161mmol)及び2,6-ジ-tert-ブチル-4-メチルピリジン(0.070g、0.34mmol)のCH2Cl2(6mL)中の溶液に、−78℃で、シリンジから10分間にわたってCH2Cl2(3mL)中のTf2O(0.018mL、0.107mmol)を徐々に添加した。−78℃で20分間撹拌後、混合物を1時間にわたってゆっくりと−15℃まで温めた。飽和NaHCO3水溶液(6mL)を添加することによって反応物を失活させ、CH2Cl2(30mL)と飽和NaHCO3水溶液(15mL)との間で分配させた。有機相を分離し、飽和NaHCO3水溶液(20mL)でさらに洗った。無水Na2SO4上で乾燥させた後、有機相を乾燥するまで蒸発させ、粗製物残渣を、溶離システムとしてヘキサン及び酢酸エチル(4:1、V/V)を用いるフラッシュカラムクロマトグラフィー(シリカゲル60A、230〜400メッシュ)にかけて、無色シロップとして生成物を得た(0.132g、収率85%)。
1H NMR (400MHz, CDCl3), δ 7.53 (m, 2H), 7.44-7.14 (m, 48H), 5.12 (d, J=3.2Hz, 1H), 5.04-4.52 (m, 21H), 4.40 (m, 1H), 4.24-4.12 (m, 2H), 4.00 (m, 11-1), 3.90-3.36 (m, 12H), 3.28 (m, 1H), 2.00 (s, 3H); 13C NMR (400MHz, CDCl3), δ 170.85, 138.99-138.14 (m), 134.27, 134.17,
132.06, 131.66, 129.24, 129.15, 128.64, 128.61-127.48 (m), 103.74, 97.51, 97.23, 88.37, 87.77, 86.91, 86.86, 84.79, 82.59, 81.95, 81.86, 81.41, 81.23, 80.67, 80.36, 80.25, 79.16, 78.99, 78.02 77.56, 75.88 (m), 75.25 (m), 72.71, 72.49, 72.37, 71.02, 69.04, 68.30, 66.39, 65.94, 65.67, 63.47, 63.36, 21.34; ESI-MS C89H92SO16Na (M+Na+)に対する計算値: m/z= 1471, 測定値: 1471。
【0109】
フェニル2,3,4-トリ-O-ベンジル-β-D-グルコピラノシル-(1→6)-2,3,4-トリ-O-ベンジル-β-D-グルコピラノシル-(1→6)-2,3,4-トリ-O-ベンジル-1-チオ-β-D-グルコピラノシド(21):
フェニル2,3,4-トリ-O-ベンジル-6-O-アセチル-β-D-グルコピラノシル-(1→6)-2,3,4-トリ-O-ベンジル-β-D-グルコピラノシル-(1→6)-2,3,4-トリ-O-ベンジル-1-チオ-β-D-グルコピラノシド 20c(101mg、0.069mmol)を入れたフラスコに、Cs2CO3(22.4mg、0.069mmol)とメタノール(6mL)を添加した。室温で14時間撹拌した後、溶媒をロータリーエバポレーターで除去し、未精製残渣を減圧下で乾燥させた。次に、この残渣にCH2Cl2(3mL)を添加し、不溶性塩を濾過により分離した。濾液を乾燥するまで蒸発させ、減圧下で乾燥させて純粋な生成物を得た(96.1mg、収率98%)。Rf=0.28(2:1、ヘキサン/酢酸エチル、V/V);1H NMR (400MHz, CDCl3), δ 7.54 (m, 2H), 7.43-7.14 (m, 48H), 5.16 (d, J=3.2Hz, 1H), 5.00-4.93 (m, 3H), 4.91-4.86 (m, 4H), 4.85- 4.74 (m, 5H), 4.71-4.53 (m, 9H), 4.40 (d, J=8.0Hz, 1H), 4.12 (d, J=10.0Hz, 1H), 3.99 (m, 2H), 3.86-3.38 (m, 12H), 3.26 (m, 1H); 13C NMR (400MHz, CDCl3), δ 138.94-138.22 (m), 134.33, 132.01, 131.55, 129.19, 129.10, 128.59-127.44 (m), 103.91, 97.54, 97.31, 88.31, 87.79, 86.93, 84.81, 82.62, 81.95, 81.85, 81.75, 81.44, 81.33, 80.73, 80.63, 80.49, 79.23, 79.00, 78.12, 77.91, 77.75, 77.51, 75.95-75.80 (m), 75.45-75.25 (m), 72.75, 72.61, 72.57, 71.24, 68.65, 66.02, 65.52, 62.30; マススペクトル用の試料はシリカゲルで濾過してセシウムイオンを除去した; ESI-MS C87H90SO15Na (M+Na+)に対する計算値: m/z=1429, 測定値: 1429。
【0110】
5’-O-モノメトキシトリチル-3’-O-スクシニル-チミジン(23):
化合物22(2.86g、5.56mmol)、無水コハク酸(1.67g、16.7mmol)、及びDMAP(0.34g、2.78mmol)を、オーブンで乾燥してアルゴン置換した50mLフラスコ中で、無水ピリジン(20mL)に溶かした。2日間、室温で撹拌して反応させた。ジクロロメタン:メタノール(9:1)のTLC分析は、出発物質の完全な消失と、単一のより極性の高い生成物の形成を示した。ピリジンを減圧下で除去した。暗褐色の残渣を次にCH2Cl2(100mL)中に溶かし、飽和食塩水で洗った。有機相をNa2SO4上で乾燥させ、次に減圧下で溶媒を除去した。生成物を淡褐色泡状物として高収率で得た(3.14g、92%収率)。
1H NMR (DMSO-d6) δ 11.39 & 8.30 (1H, s, NH), 7.50 (1H, s, CH), 7.37-7.21 (14H, m, Ar-H), 6.18 (1 H, t, J = 8 Hz, CH, H1'), 5.28 (1H, d, J = 6 Hz, CH, H3'), 4.03 (1H, broad s, CH), 3.73 (3H, s, CH3), 3.32-3.19 (2H, m, CH2, H5'&5"), 2.62-2.61 (4H, 分離されないm, 2CH2), 2.51-2.27 (2H, m, CH2), 1.41 (3H, s, CH3); C34H34N2O9Na+ 低分解能ESI-MS 計算値637.2, 測定値: 637.1。
【0111】
MMTTSucc−IL(24):
化合物23(1g、1.63mmol)、置換イミダゾリウムテトラフルオロボレート1(0.38g、1.76mmol)、及びDMAP(0.052g、0.41mmol)を、乾燥し、窒素置換した100mL丸底フラスコに入れた。この混合物に、ジシクロヘキシルカルボジイミド(DCC)(0.68g、3.3mmol)を添加し、次に無水アセトニトリル(20mL)を添加した。反応混合物を3日間、室温で撹拌した。9:1のクロロホルム:メタノールでのTLC分析は、ベースラインから移動しないより極性の高い生成物の形成を示した。反応時に、副生成物であるジシクロヘキシルウレア(DCU)が白色固体として生じた。反応を停止させたときに、DCUは沈殿し、反応混合物を焼結ガラスロートを通して濾過し、数回アセトニトリルで洗った。濾過した溶液を、撹拌したジエチルエーテル−酢酸エチル溶液に滴下して加えることによって、さらなる精製を行った。誘導体化されていないイオン液体1と所望の生成物24からなる、得られた沈殿物は、次にクロロホルムに溶かし、水で抽出した。有機相を硫酸ナトリウム上で乾燥させ、溶媒を蒸発させて、生成物を減圧下で乾燥させた。生成物24を淡褐色泡状物として得た(1.1g、83%収率)。
1H NMR (アセトン-d6) δ 10.03 (1H, s, NH), 9.10 (1H, s, CH), 7.83 (1H, s, CH), 7.71
(1H, s, CH), 7.60 (1H, s, CH), 7.50-6.92 (14H, m, Ar-H), 6.33 (1 H, t, J = 8 Hz, CH, H1'), 5.50 (1H, d, J = 6 Hz, CH, H3'), 4.55-4.51 (2H, m, CH2), 4.70-4.66 (2H, m, CH2), 4.15 (1 H, broad s, CH), 4.05 (3, s, CH3), 3.80 (3H, s, CH3), 3.50-3.40 (2H, m, CH2, H5'&5"), 2.70 (4H, broad s, 2 CH2), 2.61-2.40 (2H, m, CH2), 1.42 (3H, s, CH3); C40H43N4O9+。低分解能ESI-MS 計算値 723.3, 測定値: 723.4。
【0112】
HOTSucc−IL(25):
24(2.96g、3.65mmol)のジクロロメタン(200mL)中の溶液に、ジクロロメタン又はアセトニトリル(100mL)中の3%トリフルオロ酢酸を添加した。TFAの添加時に、溶液は赤橙色になり、撹拌を20分間続けた。生成物を10%酢酸エチル/ジエチルエーテルから沈殿させ、濾過し、最少量の上記酸溶液に再溶解し、再度沈殿させて濾過した。沈殿物を10%酢酸エチル/ジエチルエーテルで濯ぎ、アセトニトリル中に溶かすことによってフィルターから回収し、減圧下で溶媒留去し、化合物25を淡褐色泡状物として得た(1.88g、96%収率)。
1H NMR (DMSO-d6) δ 11.40及び11.39 (合計1H, 2 s, NH), 9.10 (1H, s, CH), 7.76 (1H, s, CH), 7.72及び7.45 (1H, s, CH), 7.70 (1H, s, CH), 6.15 (1H, m, CH, H1'), 5.27及び5.19 (1H, m, CH, H3'), 4.60及び3.60 (2H, m, CH2, H5'及び5"), 4.45 (2H, broad s, CH2), 4.39 (2H, broad s, CH2), 4.24-3.93 (1H, m, CH), 3.86 (3, s, CH3), 2.61-2.60 (4H, 分離されないm, 2 CH2), 2.40-2.16 (2H, m, CH2), 1.77 (3H, s, CH3); C20H27N4O8+ 高分解能ESI-MS 計算値 451.18289, 測定値 451.18234。
【0113】
DMTApTSucc−IL(27a):
化合物25(0.12g、0.22mmol)、アデノシンホスホルアミダイト26a(0.33g、0.38mmol)、及びジシアノイミダゾール(0.33g、2.8mmol)を、50mLの、オーブンで乾燥して窒素置換した丸底フラスコに入れた。この混合物に、無水THF又はアセトニトリル(5mL)を加え、得られた溶液を室温で1〜2時間撹拌した。生成物を、10%酢酸エチル/ジエチルエーテルから2回沈殿させた。この時点で、沈殿物を、アセトニトリル中に再溶解し、2,4,6−コリジン又はピリジン(約300μL)を添加し、次に水性ヨウ素溶液(THF/水(2:1)中0.1M、過剰量)を添加してホスファイトトリエステル中間体を酸化した。5分後、反応混合物を重硫酸ナトリウム水溶液(9mL)で失活させ、90mLのCHCl3で希釈し、50mLの水で抽出した。有機相を無水Na2SO4上で乾燥させ、減圧下で溶媒留去して27aの泡状物を得た(0.259g、89%)。
31P NMR (アセトニトリル-d3) δ -1.324, -1.477; C61H64N10O16P+ 低分解能ESI-MS 計算値 1223.4, 測定値 1223.4。
【0114】
DMTCpTSucc−IL(27b):
化合物25(0.11g、0.20mmol)、シチジンホスホルアミダイト26b(0.35g、0.41mmol)、及びジシアノイミダゾール(0.37g、3.1mmol)を、50mLの、オーブンで乾燥して窒素置換した丸底フラスコに入れた。溶媒の不存在が、反応が起こることを妨げた。無水THF又はアセトニトリル(5mL)をフラスコ中に注入することによって反応を開始させ、その溶液を室温で1〜2時間撹拌した。27aの場合と同じ処理をして、生成物を高収率で27bの泡状物として得た(0.234g、91%)。
31P NMR (アセトニトリル-d3) δ -1.545, -1.754; C60H64N8O17P+ 低分解能ESI-MS 計算値 1199.4, 測定値 1199.4。
【0115】
DMTGpTSucc−IL(27c):
化合物25(0.13g、0.24mmol)、グアノシンホスホルアミダイト26c(0.35g、0.42mmol)及びジシアノイミダゾール(0.38g、3.2mmol)を、50mLの、オーブンで乾燥して窒素置換した丸底フラスコに入れた。溶媒の不存在が、反応が起こることを妨げた。無水THF又はアセトニトリル(5mL)をフラスコ中に注入することによって反応を開始させ、その溶液を室温で1〜2時間撹拌した。27aの場合と同じ処理をして、生成物を高収率で27cの泡状物として得た(0.214g、90%)。
31P NMR (アセトニトリル-d3) δ -1.149, -1.194; C58H66N10O17P+ 低分解能ESI-MS 計算値 1205.4, 測定値 1205.6。
【0116】
DMTTpTSucc−IL(27d):
化合物25(0.24g、0.45mmol)、チミジンホスホルアミダイト26d(0.57g、0.77mmol)、及びジシアノイミダゾール(0.66g、5.6mmol)を、50mLの、オーブンで乾燥して窒素置換した丸底フラスコに入れた。溶媒の不存在が、反応が起こることを妨げた。無水THF又はアセトニトリル(5mL)をフラスコ中に注入することによって反応を開始させ、その溶液を室温で1〜2時間撹拌した。27aの場合と同じ処理をして、生成物を高収率で27dの泡状物として得た(0.492g、91%)。
31P NMR (アセトニトリル-d3) δ -1.494, -1.584; C54H61N7O17P+ 低解像度ESI-MS 計算値 1110.4, 測定値 1110.4。
【0117】
HOApTSucc−IL(28a):
化合物27a(0.26g、0.20mmol)をアセトニトリル(1〜2mL)に溶かし、アセトニトリル(2〜3mL)中の3%トリフルオロ酢酸を添加した。反応混合物を25に対してと同じ方法に従って処理し、泡状物として高収率で28aが得られた(0.185g、93%)。
1H NMR (アセトニトリル-d3) δ 6.47-6.51 (Ade H1'), 6.17-6.22 (Thy H1'), 5.25-5.30 (Ade and Thy H3'), 2.82-3.09 (Thy H5'&5"); 31P NMR (アセトニトリル-d3) δ -1.176, -1.516; C40H46N10O14P+, 高分解能ESI-MS 計算値 921.29326, 測定値 921.29271。
【0118】
HOCpTSucc−IL(28b):
アセトニトリル(1〜2mL)中の化合物27b(0.23g、0.18mmol)の溶液に、アセトニトリル(2〜3mL)中の3%トリフルオロ酢酸を添加して28bを生成させた。反応混合物を25に対してと同じ方法に従って処理し、泡状物として高収率で28bが得られた(0.167g、95%)。
1H NMR (アセトニトリル-d3) δ 6.09-6.24 (Cyt 及び Thy H1'), 5.21-5.33 (Thy H3'), 5.06-5.14 (Cyt H3'), 2.82-2.88 (Thy H5'及び5"); 31P NMR (アセトニトリル-d3) δ -1.381, -1.613; C39H46N8O15P+ 高分解能 ESI-MS 計算値 897.28203, 測定値 897.28148。
【0119】
HOGpTSucc−IL(28c):
アセトニトリル(1〜2mL)中の化合物27c(0.21g、0.17mmol)の溶液に、アセトニトリル(2〜3mL)中の3%トリフルオロ酢酸を添加した。反応混合物を25に対してと同じ方法に従って処理し、泡状物として高収率で28cが得られた(0.157g、96%)。
1H NMR (アセトニトリル-d3) δ 6.08-6.25 (Gua及び Thy H1'), 5.26-5.30 (Gua H3'), 5.17-5.22 (Thy H3'), 2.81-3.05 (Thy H5'及び5"); 31P NMR (アセトニトリル-d3) δ -1.047, -1.064; C37H48N10O15P+ 高分解能 ESI-MS 計算値 903.30382, 測定値 903.30328。
【0120】
HOTpTSucc−IL(28d):
アセトニトリル(1〜2mL)中の化合物27d(0.21g、0.18mmol)の溶液に、アセトニトリル(2〜3mL)中の3%トリフルオロ酢酸を添加した。反応混合物を25に対してと同じ方法に従って処理し、泡状物として高収率で28dが得られた(0.123g、78%)。
1H NMR (アセトニトリル-d3) δ 6.16-6.24 (5'-Thy及び 3'-Thy H1'), 5.22-5.30 (3'-Thy H3'), 5.07-5.12 (5'-Thy H3'), 2.30-2.86 (3'-Thy H5'及び5"); 31P NMR (アセトニトリル-d3) δ -1.188, -1.284; C33H43N7O15P+ 高分解能ESI-MS 計算値 808.25548, 測定値 808.25493。
【0121】
DMTTpTpTSucc−IL(29):
化合物28d(0.065g、0.073mmol)を、3’-ホスホルアミダイト26d(0.233g、0.31mmol)及びジシアノイミダゾール(0.31g、2.6mmol)と、無水アセトニトリル(5mL)中で室温にて混合した。2時間撹拌した後、化合物27a〜dに対しての方法と同じ方法で、生成物を10%酢酸エチル/ジエチルエーテルから2回沈殿させ、酸化し、抽出して、化合物29(104mg、92%収率)を高純度で得た。
31P NMR (アセトニトリル-d3) -1.081, -1.194, -1.233, -1.262, -1.381, -1.403, -1.448, -1.477; C67H77N10O24P2+ 低分解能ESI-MS 計算値 1467.5, 測定値1467.8.
【0122】
HOTpTpTSucc−IL(30):
アセトニトリル(1〜2mL)中の化合物29(97mg、0.06mmol)の溶液に、アセトニトリル(2〜3mL)中の3%トリフルオロ酢酸を添加した。反応混合物を25に対してと同じ方法に従って処理し、ガラス状固体として30が得られた(77mg、98%収率)。
31P NMR (アセトニトリル-d3) -1.157〜-1.531 (ピークの広い重なり); C46H59N10O22P2+ 高分解能ESI-MS 計算値 1165.32807, 測定値1165.32752。
【0123】
DMTTpTpTpSucc−IL(31):
化合物30(170mg、0.136mmol)を、3’-ホスホルアミダイト26d(0.160g、0.215mmol)及びジシアノイミダゾール(0.21g、1.8mmol)と、無水アセトニトリル(5mL)中で室温にて混合した。2時間撹拌した後、化合物27a〜dに対しての方法と同じ方法で、生成物を10%酢酸エチル/ジエチルエーテルから2回沈殿させ、酸化し、抽出して、化合物31(231mg、89%収率)を高純度で得た。
31P NMR (アセトニトリル-d3) -1.169〜-1.859 (ピークの広い重なり); C80H93N13O31P3+ 低分解能ESI-MS 計算値 1824.5, 測定値 1824.2。
【0124】
HOTpTpTSucc−IL(32):
アセトニトリル(1〜2mL)中の化合物31(190mg、0.122mmol)の溶液に、アセトニトリル(2〜3mL)中の3%トリフルオロ酢酸を添加した。反応混合物を25に対してと同じ方法に従って処理し、化合物32が得られた(160mg、99.9%収率)。
31P NMR (アセトニトリル-d3) -1.142〜-1.51 (ピークの広い重なり); C59H75N13O29P3+ 高分解能ESI-MS 計算値 1522.40066, 測定値 1522.40011。
【0125】
オリゴヌクレオチド(33a〜f)の脱保護:
シアノエチル保護基、塩基保護基(a〜cについて)、及びイオン液体担体へのスクシニル架橋基の除去は、化合物28a〜d並びに化合物30〜32の1〜5mgを、1.5mLの濃水酸化アンモニウムと無水エタノール(3:1)で、60℃にて16時間又は室温にて48時間処理して達成される。サンプルを冷却した後、アンモニア溶液を蒸発させ、残渣を水に溶かし、イオンペア逆相HPLCによって精製して、33a〜fを得た。
【0126】
本発明は、上述した構成及び部分の詳細にその応用が限定されないことが理解されるべきである。本発明は、その他の態様であることができ、様々なやり方で実施されうる。本明細書で使用した語法及び術語は、説明のために用いたものであって限定されるものではないことも理解されよう。したがって、本発明を、本発明の実例を用いて上に説明しているが、付属の請求項で定義されるように、本発明の精神、範囲及び特質から離れることなく変更することができる。
【0127】
〔参考文献〕
【表6A】
【表6B】
【表6C】
【表6D】
【表6E】
【図面の簡単な説明】
【0128】
【図1】図1は、本発明によるイオン液体担持合成の一般的概念のある態様の説明図である。
【図2】図2は、本発明によるイオン液体担持合成の一般的概念のさらなる態様の説明図である。
【図3】図3は、真正のLeu5−エンケファリン41と合成したLeu5−エンケファリン14のTFA塩の1H NMRスペクトルの図である。
【図4】図4は、ILSSによって合成した粗製Leu5−エンケファリン14のHPLC分析の図である。42
【図5】図5は、イオン液体担持ペプチド2a、4a、7、9、及び11の1H NMRスペクトルの図である。
【図6】図6は、イオン液体担持ペプチド2a、4a、7、9、及び11のマススペクトルの図である。
【図7】図7は、脱保護したオリゴヌクレオチドの様々なHPLCクロマトグラフィーの図である。
【特許請求の範囲】
【請求項1】
以下の:
a)イオン液体が結合したモノマー単位をもたらす反応条件において、第一のモノマー単位をイオン液体と接触させるステップ;及び
b)2〜30のモノマー単位を含みイオン液体が結合したオリゴマーをもたらす反応条件下において、前記イオン液体が結合したモノマー単位を、少なくとも1つのさらなるモノマー単位と接触させるステップ、
を含む、オリゴペプチド、オリゴサッカライド、及びオリゴヌクレオチドからなる群から選択されるオリゴマーの合成方法。
【請求項2】
所望する、イオン液体に結合したオリゴペプチド、イオン液体に結合したオリゴサッカライド、又はイオン液体に結合したオリゴヌクレオチドを製造するために、選択した回数でステップb)を繰り返すことをさらに含む、請求項1に記載の合成方法。
【請求項3】
前記イオン液体から前記オリゴマーを単離することをさらに含む、請求項2に記載の合成方法。
【請求項4】
前記イオン液体が、ヘテロ環式もしくは置換ヘテロ環式第四級窒素含有有機カチオン、ヘテロ環式もしくは置換ヘテロ環式第四級ホスホニウム含有有機カチオン、又はヘテロ環式もしくは置換ヘテロ環式三価スルホニウム含有有機カチオンと、前記有機カチオン上の電荷と釣り合うアニオンとを含む有機塩である、請求項1記載の合成方法。
【請求項5】
前記有機カチオンが、N−置換ピリジン及び1,3−ジ置換イミダゾールからなる群から選択される、請求項4に記載の合成方法。
【請求項6】
前記アニオンが、Cl−、Br−、BF4−、PF6−、SbF6−、CuCl2−、及びAlCl4−からなる群から選択される、請求項4に記載の合成方法。
【請求項7】
前記モノマー単位がアミノ酸である、請求項1記載の合成方法。
【請求項8】
前記モノマー単位がヌクレオチドである、請求項1記載の合成方法。
【請求項9】
前記モノマー単位がサッカライドである、請求項1記載の合成方法。
【請求項10】
前記オリゴマーが2〜15のモノマー単位を含む、請求項7〜9のいずれか一項に記載の合成方法。
【請求項11】
前記オリゴマーが2〜10のモノマー単位を含む、請求項7〜9のいずれか一項に記載の合成方法。
【請求項12】
前記オリゴマーが2〜5のモノマー単位を含む、請求項7〜9のいずれか一項に記載の合成方法。
【請求項13】
前記イオン液体が、溶媒と可溶性担体の両方である、請求項1〜12のいずれか一項に記載の合成方法。
【請求項14】
オリゴペプチド、オリゴサッカライド、及びオリゴヌクレオチドからなる群から選択されるオリゴマーの合成方法であって、前記方法が、モノマー単位に対してイオン液体を用いて、2〜30のモノマー単位を含むオリゴマーにすることを含む、合成方法。
【請求項15】
イオン液体が結合したモノマー単位をもたらす反応条件下において、第一のモノマー単位を前記イオン液体と接触させるステップ;及びイオン液体に結合したオリゴマーをもたらす反応条件下において、前記イオン液体が結合したモノマー単位を、少なくとも1つのさらなるモノマー単位と接触させるステップ、
を含む、請求項14に記載の合成方法。
【請求項16】
1つのさらなるモノマー単位と接触させるステップを、選択した回数繰り返して、所望のイオン液体に結合したオリゴペプチド、オリゴサッカライド、又はオリゴヌクレオチドを製造する、請求項15に記載の合成方法。
【請求項17】
前記イオン液体から前記オリゴマーを単離するステップをさらに含む、請求項16に記載の合成方法。
【請求項18】
前記イオン液体が、ヘテロ環式もしくは置換ヘテロ環式第四級窒素含有有機カチオン、ヘテロ環式もしくは置換ヘテロ環式第四級ホスホニウム含有有機カチオン、又はヘテロ環式もしくは置換ヘテロ環式三価スルホニウム含有有機カチオンと、前記有機カチオン上の電荷と釣り合うアニオンとを含む有機塩である、請求項14に記載の合成方法。
【請求項19】
前記有機カチオンが、N−置換ピリジン及び1,3−ジ置換イミダゾールからなる群から選択される、請求項18に記載の合成方法。
【請求項20】
前記アニオンが、Cl−、Br−、BF4−、PF6−、SbF6−、CuCl2−、及びAlCl4−からなる群から選択される、請求項18に記載の合成方法。
【請求項21】
前記モノマーがアミノ酸である、請求項14に記載の合成方法。
【請求項22】
前記モノマーがヌクレオチドである、請求項14に記載の合成方法。
【請求項23】
前記モノマーがサッカライドである、請求項14に記載の合成方法。
【請求項24】
前記オリゴマーが2〜15のモノマー単位を含む、請求項21〜23のいずれか一項に記載の製造方法。
【請求項25】
前記オリゴマーが2〜10のモノマー単位を含む、請求項21〜23のいずれか一項に記載の製造方法。
【請求項26】
前記オリゴマーが2〜5のモノマー単位を含む、請求項21〜23のいずれか一項に記載の製造方法。
【請求項27】
前記イオン液体が溶媒と可溶性担体の両方である、請求項14〜26のいずれか一項に記載の製造方法。
【請求項28】
請求項4又は18において定義した少なくとも1種のイオン液体と、アミノ酸、サッカライド、及びヌクレオチドから選択されるモノマー単位とを含むキット。
【請求項29】
少なくとも2つのモノマー単位を含むオリゴペプチド、オリゴサッカライド、又はオリゴヌクレオチドを製造するのに適した、請求項28に記載のキット。
【請求項30】
請求項4又は18で定義した少なくとも1種のイオン液体を含み、アミノ酸、サッカライド、及びヌクレオチドから選択されるモノマー単位をさらに含む製品。
【請求項31】
少なくとも2つのモノマー単位を含む、オリゴペプチド、オリゴサッカライド、又はオリゴヌクレオチドを製造するために適した、請求項30に記載の製品。
【請求項1】
以下の:
a)イオン液体が結合したモノマー単位をもたらす反応条件において、第一のモノマー単位をイオン液体と接触させるステップ;及び
b)2〜30のモノマー単位を含みイオン液体が結合したオリゴマーをもたらす反応条件下において、前記イオン液体が結合したモノマー単位を、少なくとも1つのさらなるモノマー単位と接触させるステップ、
を含む、オリゴペプチド、オリゴサッカライド、及びオリゴヌクレオチドからなる群から選択されるオリゴマーの合成方法。
【請求項2】
所望する、イオン液体に結合したオリゴペプチド、イオン液体に結合したオリゴサッカライド、又はイオン液体に結合したオリゴヌクレオチドを製造するために、選択した回数でステップb)を繰り返すことをさらに含む、請求項1に記載の合成方法。
【請求項3】
前記イオン液体から前記オリゴマーを単離することをさらに含む、請求項2に記載の合成方法。
【請求項4】
前記イオン液体が、ヘテロ環式もしくは置換ヘテロ環式第四級窒素含有有機カチオン、ヘテロ環式もしくは置換ヘテロ環式第四級ホスホニウム含有有機カチオン、又はヘテロ環式もしくは置換ヘテロ環式三価スルホニウム含有有機カチオンと、前記有機カチオン上の電荷と釣り合うアニオンとを含む有機塩である、請求項1記載の合成方法。
【請求項5】
前記有機カチオンが、N−置換ピリジン及び1,3−ジ置換イミダゾールからなる群から選択される、請求項4に記載の合成方法。
【請求項6】
前記アニオンが、Cl−、Br−、BF4−、PF6−、SbF6−、CuCl2−、及びAlCl4−からなる群から選択される、請求項4に記載の合成方法。
【請求項7】
前記モノマー単位がアミノ酸である、請求項1記載の合成方法。
【請求項8】
前記モノマー単位がヌクレオチドである、請求項1記載の合成方法。
【請求項9】
前記モノマー単位がサッカライドである、請求項1記載の合成方法。
【請求項10】
前記オリゴマーが2〜15のモノマー単位を含む、請求項7〜9のいずれか一項に記載の合成方法。
【請求項11】
前記オリゴマーが2〜10のモノマー単位を含む、請求項7〜9のいずれか一項に記載の合成方法。
【請求項12】
前記オリゴマーが2〜5のモノマー単位を含む、請求項7〜9のいずれか一項に記載の合成方法。
【請求項13】
前記イオン液体が、溶媒と可溶性担体の両方である、請求項1〜12のいずれか一項に記載の合成方法。
【請求項14】
オリゴペプチド、オリゴサッカライド、及びオリゴヌクレオチドからなる群から選択されるオリゴマーの合成方法であって、前記方法が、モノマー単位に対してイオン液体を用いて、2〜30のモノマー単位を含むオリゴマーにすることを含む、合成方法。
【請求項15】
イオン液体が結合したモノマー単位をもたらす反応条件下において、第一のモノマー単位を前記イオン液体と接触させるステップ;及びイオン液体に結合したオリゴマーをもたらす反応条件下において、前記イオン液体が結合したモノマー単位を、少なくとも1つのさらなるモノマー単位と接触させるステップ、
を含む、請求項14に記載の合成方法。
【請求項16】
1つのさらなるモノマー単位と接触させるステップを、選択した回数繰り返して、所望のイオン液体に結合したオリゴペプチド、オリゴサッカライド、又はオリゴヌクレオチドを製造する、請求項15に記載の合成方法。
【請求項17】
前記イオン液体から前記オリゴマーを単離するステップをさらに含む、請求項16に記載の合成方法。
【請求項18】
前記イオン液体が、ヘテロ環式もしくは置換ヘテロ環式第四級窒素含有有機カチオン、ヘテロ環式もしくは置換ヘテロ環式第四級ホスホニウム含有有機カチオン、又はヘテロ環式もしくは置換ヘテロ環式三価スルホニウム含有有機カチオンと、前記有機カチオン上の電荷と釣り合うアニオンとを含む有機塩である、請求項14に記載の合成方法。
【請求項19】
前記有機カチオンが、N−置換ピリジン及び1,3−ジ置換イミダゾールからなる群から選択される、請求項18に記載の合成方法。
【請求項20】
前記アニオンが、Cl−、Br−、BF4−、PF6−、SbF6−、CuCl2−、及びAlCl4−からなる群から選択される、請求項18に記載の合成方法。
【請求項21】
前記モノマーがアミノ酸である、請求項14に記載の合成方法。
【請求項22】
前記モノマーがヌクレオチドである、請求項14に記載の合成方法。
【請求項23】
前記モノマーがサッカライドである、請求項14に記載の合成方法。
【請求項24】
前記オリゴマーが2〜15のモノマー単位を含む、請求項21〜23のいずれか一項に記載の製造方法。
【請求項25】
前記オリゴマーが2〜10のモノマー単位を含む、請求項21〜23のいずれか一項に記載の製造方法。
【請求項26】
前記オリゴマーが2〜5のモノマー単位を含む、請求項21〜23のいずれか一項に記載の製造方法。
【請求項27】
前記イオン液体が溶媒と可溶性担体の両方である、請求項14〜26のいずれか一項に記載の製造方法。
【請求項28】
請求項4又は18において定義した少なくとも1種のイオン液体と、アミノ酸、サッカライド、及びヌクレオチドから選択されるモノマー単位とを含むキット。
【請求項29】
少なくとも2つのモノマー単位を含むオリゴペプチド、オリゴサッカライド、又はオリゴヌクレオチドを製造するのに適した、請求項28に記載のキット。
【請求項30】
請求項4又は18で定義した少なくとも1種のイオン液体を含み、アミノ酸、サッカライド、及びヌクレオチドから選択されるモノマー単位をさらに含む製品。
【請求項31】
少なくとも2つのモノマー単位を含む、オリゴペプチド、オリゴサッカライド、又はオリゴヌクレオチドを製造するために適した、請求項30に記載の製品。
【図1】

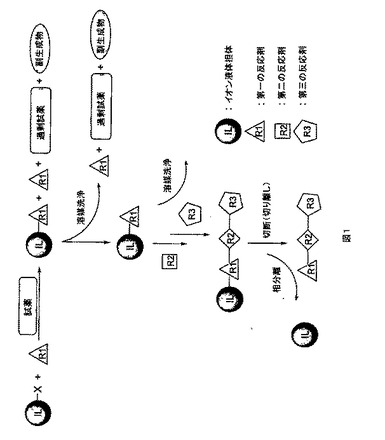
【図2】
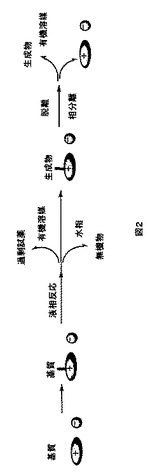
【図3】
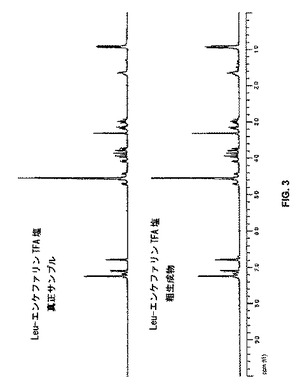
【図4】
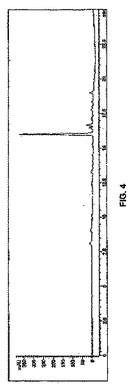
【図5】
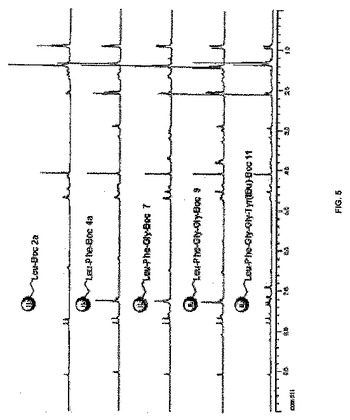
【図6】
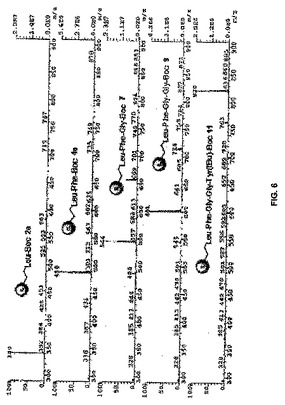
【図7】
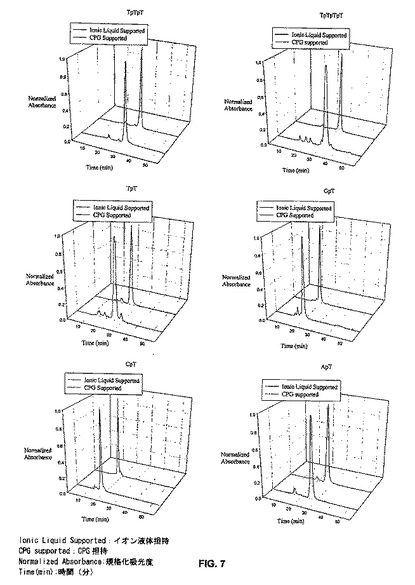
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公表番号】特表2008−537733(P2008−537733A)
【公表日】平成20年9月25日(2008.9.25)
【国際特許分類】
【出願番号】特願2008−501121(P2008−501121)
【出願日】平成18年3月15日(2006.3.15)
【国際出願番号】PCT/CA2006/000355
【国際公開番号】WO2006/096963
【国際公開日】平成18年9月21日(2006.9.21)
【出願人】(500539088)マクギル ユニバーシティー (3)
【出願人】(507308979)ザ・ホンコン・ポリテクニック・ユニヴァーシティ (1)
【Fターム(参考)】
【公表日】平成20年9月25日(2008.9.25)
【国際特許分類】
【出願日】平成18年3月15日(2006.3.15)
【国際出願番号】PCT/CA2006/000355
【国際公開番号】WO2006/096963
【国際公開日】平成18年9月21日(2006.9.21)
【出願人】(500539088)マクギル ユニバーシティー (3)
【出願人】(507308979)ザ・ホンコン・ポリテクニック・ユニヴァーシティ (1)
【Fターム(参考)】
[ Back to top ]