
イソシアヌレート化合物
【課題】光硬化性樹脂の原料として有用な、新規イソシアヌレート化合物の提供。
【解決手段】化学式(I)で示されるイソシアヌレート化合物。

【解決手段】化学式(I)で示されるイソシアヌレート化合物。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規なイソシアヌレート化合物に関するものである。
【背景技術】
【0002】
本発明に類似する物質として、例えば特許文献1には、化1の化学式(II)で示されるイソシアヌレート化合物が開示されている。
化学式(II):
【0003】
【化1】
【0004】
この化合物は、トリス(メタ)アクリロキシエチルイソシアヌレートあるいはイソシアヌル酸エチレンオキシド変性トリ(メタ)アクリレート等と呼ばれている物質であり、紫外線や電子線の照射により硬化する樹脂(以下、単に光硬化性樹脂と云うことがある)の原料として広く使用されている。
【0005】
また、特許文献2には、化2の化学式(III)で示されるイソシアヌレート化合物が開示されている。
【0006】
【化2】
【0007】
この化合物は、カーボンナノチューブを含む撚糸に電子線を照射し、隣接するカーボンナノチューブの炭素原子間に新たな結合を生成させて、カーボンナノチューブを含む撚糸の強度を向上させる作用を高めるための架橋剤として提案されている。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】特開昭52−128387号公報
【特許文献2】特開2011−153392号公報
【発明の概要】
【発明が解決しようとする課題】
【0009】
本発明は、光硬化性樹脂の原料としての用途が期待される、新規なイソシアヌレート化合物を提供することを目的とする。
【課題を解決するための手段】
【0010】
本発明者らは、前記の課題を解決するために鋭意研究を重ねた結果、化3の化学式(I)で示されるイソシアヌレート化合物を合成し得ることを認め、本発明を完成するに至ったものである。
化学式(I):
【0011】
【化3】
【発明の効果】
【0012】
本発明のイソシアヌレート化合物は、熱可塑性樹脂や熱硬化性樹脂の改質剤としての他、光硬化性樹脂の原料としての用途が期待されるが、エポキシ樹脂やシリコーン樹脂で改質することによって、光硬化性樹脂の耐湿性、接着性、電気的特性、機械的特性等の向上が見込まれる。
【発明を実施するための形態】
【0013】
以下、本発明を詳細に説明する。
本発明は、前記の化学式(I)で示されるイソシアヌレート化合物であり、具体的には、
1,3−ジメチル−5−(2−ヒドロキシエチル)イソシアヌレート、
1,3−ジメチル−5−(2−ビニルカルボニルオキシエチル)イソシアヌレート、
1,3−ジメチル−5−[2−(2−プロペニル)カルボニルオキシエチル]イソシアヌレート、
1,3−ビス(オキシラニルメチル)−5−(2−ヒドロキシエチル)イソシアヌレート、
1,3−ビス(オキシラニルメチル)−5−(2−ビニルカルボニルオキシエチル)イソシアヌレートおよび
1,3−ビス(オキシラニルメチル)−5−[2−(2−プロペニル)カルボニルオキシエチル]イソシアヌレートである。
【0014】
本発明の1,3−ジメチル−5−(2−ヒドロキシエチル)イソシアヌレートは、例えば適量の反応溶媒中で、1,3−ジメチルイソシアヌレートと、酸化エチレンを反応させることにより合成することができる。
【0015】
この反応における反応温度については、110〜140℃とすることが好ましく、同反応時間については1〜3時間とすることが好ましい。なお、常温で気体の酸化エチレンが原料である都合上、耐圧性の反応容器であるオートクレーブを使用することが好ましい。
また、前記の反応溶媒としては、例えば、N,N−ジメチルホルムアミドやジメチルスルホキシドの如き非プロトン性極性溶媒を好ましく使用することができる。
【0016】
この1,3−ジメチル−5−(2−ヒドロキシエチル)イソシアヌレートの合成においては、例えば、反応終了後の反応液から反応溶媒を留去し、メタノールやイソプロパノールの如きアルコール溶媒中で再結晶させて精製し、目的物を得ることができる。
【0017】
本発明の1,3−ジメチル−5−(2−ビニルカルボニルオキシエチル)イソシアヌレート及び1,3−ジメチル−5−[2−(2−プロペニル)カルボニルオキシエチル]イソシアヌレートは、例えば適量の反応溶媒中で、1,3−ジメチル−5−(2−ヒドロキシエチル)イソシアヌレートと、アクリル酸またはメタクリル酸とを脱水エステル化反応させることにより合成することができる。
この反応においては、硫酸、パラトルエンスルホン酸やメタンスルホン酸等の酸性触媒を使用する。また、ハイドロキノン、メトキシハイドロキノン、t−ブチルカテコール、t−ブチルハイドロキノン、フェノチアジン等や、塩化銅、硫酸銅等の重合禁止剤を使用することが好ましい。
【0018】
前記の脱水エステル化反応における反応温度については、100〜140℃とすることが好ましく、同反応時間については、9〜15時間とすることが好ましい。
【0019】
また、この反応においては、反応溶媒として、エステル化反応で生成する水の溶解度が低い有機溶媒を使用し、水を共沸させながら脱水を促進することが好ましい。この場合の好ましい有機溶媒としては、例えばトルエン、ベンゼン及びキシレン等の芳香族炭化水素、ヘキサン及びヘプタン等の脂肪族炭化水素、メチルエチルケトン及びシクロヘキサノン等のケトン類や、四塩化炭素、ジクロロエタン及びトリクロロエタン等のハロゲン化炭化水素が挙げられる。
【0020】
本発明の1,3−ジメチル−5−(2−ビニルカルボニルオキシエチル)イソシアヌレート及び1,3−ジメチル−5−[2−(2−プロペニル)カルボニルオキシエチル]イソシアヌレートの合成においては、反応終了後の反応液に、水酸化ナトリウム等のアルカリ水溶液を添加することにより、未反応の(メタ)アクリル酸や、触媒として添加した酸性触媒を中和して、続いて、分離した有機層から反応溶媒を除去し、シリカゲルクロマトグラフィーによる精製を行い、目的物を得ることができる。
【0021】
本発明の1,3−ビス(オキシラニルメチル)−5−(2−ヒドロキシエチル)イソシアヌレート、1,3−ビス(オキシラニルメチル)−5−(2−ビニルカルボニルオキシエチル)イソシアヌレート及び1,3−ビス(オキシラニルメチル)−5−[2−(2−プロペニル)カルボニルオキシエチル]イソシアヌレートは、例えば適量の反応溶媒中で、それぞれ1,3−ジアリル−5−(2−ヒドロキシエチル)イソシアヌレート、1,3−ジアリル−5−(2−ビニルカルボニルオキシエチル)イソシアヌレート及び1,3−ジアリル−5−[2−(2−プロペニル)カルボニルオキシエチル]イソシアヌレートを酸化反応させることにより合成することができる。この反応においては、メタクロロ過安息香酸や過酢酸等の酸化剤を使用する。
【0022】
前記の酸化反応における反応温度については、0℃〜室温とすることが好ましく、同反応時間については、6〜12時間とすることが好ましい。
【0023】
前記の反応溶媒としては、ジクロロメタン、ジクロロエタン及びクロロホルム等のハロゲン化炭化水素を好ましく使用することができる。
【0024】
本発明の1,3−ビス(オキシラニルメチル)−5−(2−ヒドロキシエチル)イソシアヌレート、1,3−ビス(オキシラニルメチル)−5−(2−ビニルカルボニルオキシエチル)イソシアヌレート及び1,3−ビス(オキシラニルメチル)−5−[2−(2−プロペニル)カルボニルオキシエチル]イソシアヌレートの合成においては、反応終了後の反応液に、亜硫酸ナトリウム等の水溶液を添加することにより、未反応の酸化剤を不活性化し、続いて、分離した有機層から反応溶媒を除去し、シリカゲルクロマトグラフィーによる精製を行い、目的物を得ることができる。
【0025】
本発明のこれらのイソシアヌレート化合物を原料とする樹脂は、プリント配線板や電子部品用の塗料、光学レンズ、接着剤、レジストインク等の他、木工用塗料、光ファイバーやプラスチック、缶の表面を保護するためのコーティング剤等への利用が期待される。
【実施例】
【0026】
以下、本発明を実施例に示した合成試験によって具体的に説明する。
なお、原料の1,3−ジメチルイソシアヌレートについては、「Edwin M. Smolin;Lorence Rapoport.“Isocyanuric acid and derivatives”.
The chemistry of heterocyclic compounds. s-Triazines and derivatives.,
INTERSCIENCE PUBLISHERS, INC., 1959, p.389-422.」に記載された方法に従って合成した。
また、同1,3−ジアリル−5−(2−ヒドロキシエチル)イソシアヌレートについては、英国特許第961624号公開公報に記載された方法に従って合成し、同1,3−ジアリル−5−(2−ビニルカルボニルオキシエチル)イソシアヌレートについては、欧州特許第156710号公開公報に記載された方法に従って合成した。
【0027】
〔実施例1〕
<1,3−ジメチル−5−(2−ヒドロキシエチル)イソシアヌレートの合成>
加熱用ジャケット、攪拌機、ガス導入管および排出管を備えた1500mlのオートクレーブに、1,3−ジメチルイソシアヌレート100g(0.6mol)とN,N−ジメチルホルムアミド200gを仕込んだ。容器を密閉後、圧力が1.3MPaになるまで酸化エチレン(0.7mol)を注入した。内温が120℃になるよう設定し、撹拌を行いながら2時間反応させた。
反応液を減圧下で濃縮して得られた粗結晶98gを、イソプロパノール50gで再結晶して、白色結晶61.0g(収率48%、純度97%)を得た。
【0028】
得られた結晶の融点、マススペクトルデータおよび1H−NMRスペクトルデータは、以下のとおりであった。
・融点:113-116℃
・MS:201(M+)
・1H-NMR(DMSO-d6) δ:4.73(t,1H),3.82(t,2H),3.52(dt,2H),3.17(s,3H).
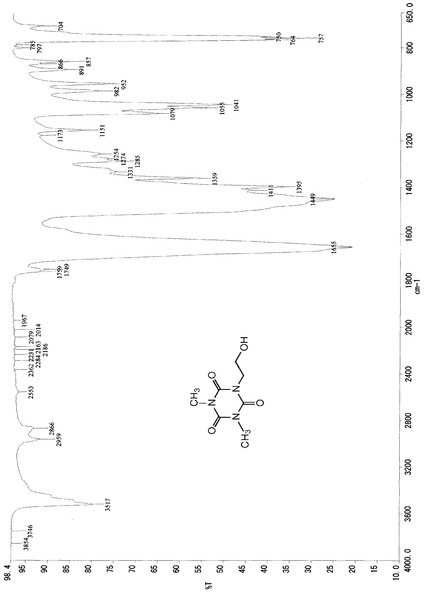
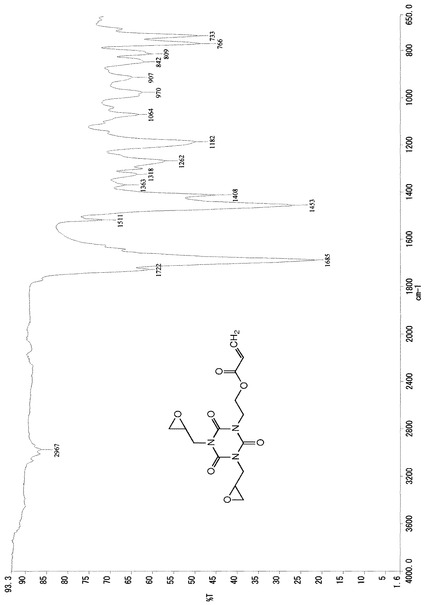
また、この結晶のIRスペクトルデータは、図1に示したチャートのとおりであった。
これらのスペクトルデータより、得られた生成物は、化4の化学式(IV)で示される1,3−ジメチル−5−(2−ヒドロキシエチル)イソシアヌレートであるものと同定した。
化学式(IV):
【0029】
【化4】
【0030】
〔実施例2〕
<1,3−ジメチル−5−(2−ビニルカルボニルオキシエチル)イソシアヌレートの合成>
水分離機付冷却器および温度計を備えた100mLフラスコに、1,3−ジメチル−5−(2−ヒドロキシエチル)イソシアヌレート10.0g(49.7mmol)、トルエン20.0g、パラトルエンスルホン酸0.2g(1.0mmol)、メトキシハイドロキノン0.01g(0.1mmol)、アクリル酸3.9g(54.7mmol)を仕込んだ。
フラスコを110℃に設定したオイルバスに浸漬して、反応液を加熱撹拌し、発生した水分を反応系外に除去しながら12時間反応させた。
反応終了後、反応液中の不溶物を濾去し、濾液を10%炭酸水素ナトリウム水溶液20gで2回洗浄し、次いで水20gで1回洗浄し、分離した有機層を減圧下で乾固させた。
得られた乾固物をシリカゲルクロマトグラフィー(酢酸エチル/ヘキサン=5/1、v/v)により精製し、白色結晶6.7g(収率52%、純度97%)を得た。
【0031】
得られた結晶の融点、マススペクトルデータおよび1H−NMRスペクトルデータは、以下のとおりであった。
・融点:85-93℃
・MS:255(M+)
・1H-NMR(DMSO-d6) δ:6.30(d,1H),6.11(dd,1H),5.94(d,1H),4.28(t,2H),4.05(t,2H),3.16(s,6H).
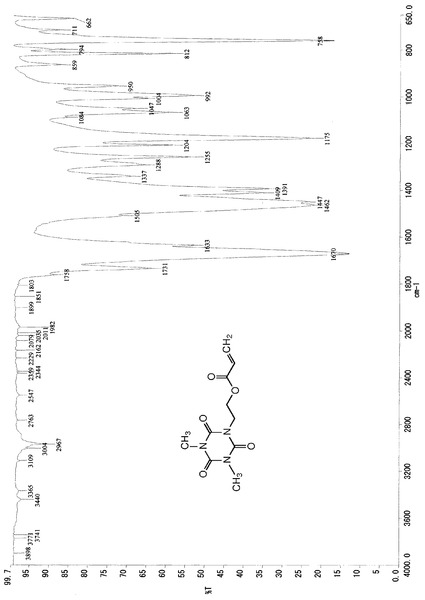
また、この結晶のIRスペクトルデータは、図2に示したチャートのとおりであった。
これらのスペクトルデータより、得られた生成物は、化5の化学式(V)で示される1,3−ジメチル−5−(2−ビニルカルボニルオキシエチル)イソシアヌレートであるものと同定した。
化学式(V):
【0032】
【化5】
【0033】
〔実施例3〕
<1,3−ジメチル−5−[2−(2−プロペニル)カルボニルオキシエチル]イソシアヌレートの合成>
水分離機付冷却器および温度計を備えた100mLフラスコに、1,3−ジメチル−5−(2−ヒドロキシエチル)イソシアヌレート10.0g(49.7mmol)、トルエン20.0g、パラトルエンスルホン酸0.2g(1.0mmol)、メトキシハイドロキノン0.01g(0.1mmol)、メタクリル酸4.7g(54.7mmol)を仕込んだ。
フラスコを110℃に設定したオイルバスに浸漬して加熱撹拌し、発生した水分を反応系外に除去しながら12時間反応させた。
反応終了後、反応液中の不溶物を濾去し、濾液を10%炭酸水素ナトリウム水溶液10gで2回洗浄し、次いで水10gで1回洗浄し、分離した有機層を減圧下で乾固させた。
得られた乾固物をシリカゲルクロマトグラフィー(酢酸エチル/ヘキサン=5/1、v/v)により精製し、白色結晶8.0g(収率60%、純度98%)を得た。
【0034】
得られた結晶の融点、マススペクトルデータおよび1H−NMRスペクトルデータは、以下のとおりであった。
・融点:68-70℃
・MS:269(M+)
・1H-NMR (DMSO-d6) δ:5.99(s,1H),5.65(s,1H),4.26(t,2H),4.06(t,2H),3.16(s,6H),1.83(s,3H).
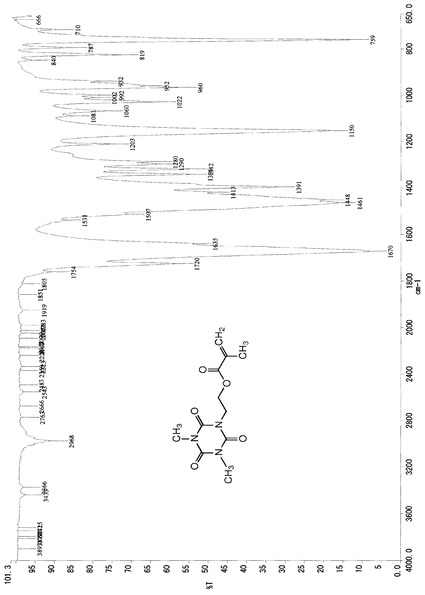
また、この結晶のIRスペクトルデータは、図3に示したチャートのとおりであった。
これらのスペクトルデータより、得られた生成物は、化6の化学式(VI)で示される1,3−ジメチル−5−[2−(2−プロペニル)カルボニルオキシエチル]イソシアヌレートであるものと同定した。
化学式(VI):
【0035】
【化6】
【0036】
〔実施例4〕
<1,3−ビス(オキシラニルメチル)−5−(2−ヒドロキシエチル)イソシアヌレートの合成>
温度計を備えた50mLフラスコに、1,3−ジアリル−5−(2−ヒドロキシエチル)イソシアヌレート760mg(3.0mmol)、ジクロロメタン15.0mlを仕込んだ。
氷冷下にて、65%メタクロロ過安息香酸4.8g(18.0mmol)を添加し、1時間攪拌した後、室温にて12時間攪拌した。
反応終了後、クロロホルム30ml及び10%亜硫酸ナトリウム水溶液50mlを添加し、分離した有機層を減圧下で濃縮した。
得られた濃縮物をシリカゲルクロマトグラフィー(クロロホルム/メタノール=10/1、v/v)により精製し、無色液体650mg(収率76%,純度99%)を得た。
【0037】
得られた液体の1H−NMRスペクトルデータは、以下のとおりであった。
・1H-NMR (CDCl3) δ:4.10-4.21(m,4H),4.00-4.06(m,2H),3.87-3.91(m,4H),3.24-3.28(m,2H),2.83(t,2H),2.68-2.71(m,2H).
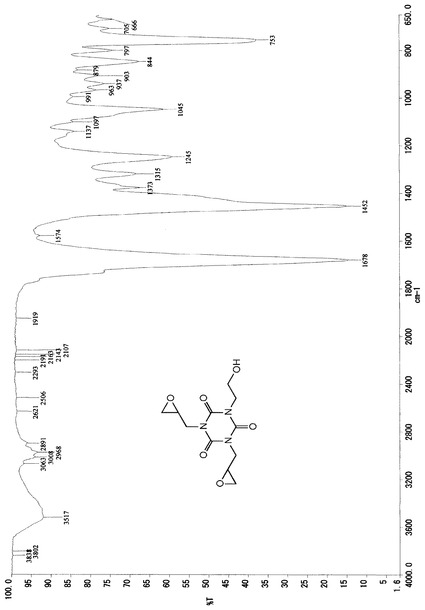
また、この液体のIRスペクトルデータは、図4に示したチャートのとおりであった。
これらのスペクトルデータより、得られた生成物は、化7の化学式(VII)で示される1,3−ビス(オキシラニルメチル)−5−(2−ヒドロキシエチル)イソシアヌレートであるものと同定した。
化学式(VII):
【0038】
【化7】
【0039】
〔実施例5〕
<1,3−ビス(オキシラニルメチル)−5−(2−ビニルカルボニルオキシエチル)イソシアヌレートの合成>
温度計を備えた50mLフラスコに、1,3−ジアリル−5−(2−ビニルカルボニルオキシエチル)イソシアヌレート307mg(1.0mmol)、ジクロロメタン5.0mlを仕込んだ。
氷冷下にて、65%メタクロロ過安息香酸1.6g(6.0mmol)を添加し、1時間攪拌した後、室温にて9時間攪拌した。
反応終了後、クロロホルム10ml及び10%亜硫酸ナトリウム水溶液30mlを添加し、分離した有機層を減圧下で濃縮した。
得られた濃縮物をシリカゲルクロマトグラフィー(酢酸エチル/ヘキサン=1/3、v/v)により精製し、無色液体268.0mg(収率79%,純度99%)を得た。
【0040】
得られた液体の1H−NMRスペクトルデータは、以下のとおりであった。
・1H-NMR (DMSO-d6) δ:6.42(dd,1H),6.07(dd,1H),5.86(dd,1H),4.43(t,2H)4.26(t,2H),4.18(ddd,2H),4.03(dd,2H),3.22-3.26(m,2H),2.82(t,2H),2.69(m,2H).
また、この液体のIRスペクトルデータは、図5に示したチャートのとおりであった。
これらのスペクトルデータより、得られた生成物は、化8の化学式(VIII)で示される1,3−ビス(オキシラニルメチル)−5−(2−ビニルカルボニルオキシエチル)イソシアヌレートであるものと同定した。
化学式(VIII):
【0041】
【化8】
【図面の簡単な説明】
【0042】
【図1】実施例1で得られた結晶のIRスペクトルチャートである。
【図2】実施例2で得られた結晶のIRスペクトルチャートである。
【図3】実施例3で得られた結晶のIRスペクトルチャートである。
【図4】実施例4で得られた液体のIRスペクトルチャートである。
【図5】実施例5で得られた液体のIRスペクトルチャートである。
【産業上の利用可能性】
【0043】
本発明によれば、光硬化性樹脂の原料としての用途が期待されるイソシアヌレート化合物を提供することができる。
【技術分野】
【0001】
本発明は、新規なイソシアヌレート化合物に関するものである。
【背景技術】
【0002】
本発明に類似する物質として、例えば特許文献1には、化1の化学式(II)で示されるイソシアヌレート化合物が開示されている。
化学式(II):
【0003】
【化1】
【0004】
この化合物は、トリス(メタ)アクリロキシエチルイソシアヌレートあるいはイソシアヌル酸エチレンオキシド変性トリ(メタ)アクリレート等と呼ばれている物質であり、紫外線や電子線の照射により硬化する樹脂(以下、単に光硬化性樹脂と云うことがある)の原料として広く使用されている。
【0005】
また、特許文献2には、化2の化学式(III)で示されるイソシアヌレート化合物が開示されている。
【0006】
【化2】
【0007】
この化合物は、カーボンナノチューブを含む撚糸に電子線を照射し、隣接するカーボンナノチューブの炭素原子間に新たな結合を生成させて、カーボンナノチューブを含む撚糸の強度を向上させる作用を高めるための架橋剤として提案されている。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】特開昭52−128387号公報
【特許文献2】特開2011−153392号公報
【発明の概要】
【発明が解決しようとする課題】
【0009】
本発明は、光硬化性樹脂の原料としての用途が期待される、新規なイソシアヌレート化合物を提供することを目的とする。
【課題を解決するための手段】
【0010】
本発明者らは、前記の課題を解決するために鋭意研究を重ねた結果、化3の化学式(I)で示されるイソシアヌレート化合物を合成し得ることを認め、本発明を完成するに至ったものである。
化学式(I):
【0011】
【化3】
【発明の効果】
【0012】
本発明のイソシアヌレート化合物は、熱可塑性樹脂や熱硬化性樹脂の改質剤としての他、光硬化性樹脂の原料としての用途が期待されるが、エポキシ樹脂やシリコーン樹脂で改質することによって、光硬化性樹脂の耐湿性、接着性、電気的特性、機械的特性等の向上が見込まれる。
【発明を実施するための形態】
【0013】
以下、本発明を詳細に説明する。
本発明は、前記の化学式(I)で示されるイソシアヌレート化合物であり、具体的には、
1,3−ジメチル−5−(2−ヒドロキシエチル)イソシアヌレート、
1,3−ジメチル−5−(2−ビニルカルボニルオキシエチル)イソシアヌレート、
1,3−ジメチル−5−[2−(2−プロペニル)カルボニルオキシエチル]イソシアヌレート、
1,3−ビス(オキシラニルメチル)−5−(2−ヒドロキシエチル)イソシアヌレート、
1,3−ビス(オキシラニルメチル)−5−(2−ビニルカルボニルオキシエチル)イソシアヌレートおよび
1,3−ビス(オキシラニルメチル)−5−[2−(2−プロペニル)カルボニルオキシエチル]イソシアヌレートである。
【0014】
本発明の1,3−ジメチル−5−(2−ヒドロキシエチル)イソシアヌレートは、例えば適量の反応溶媒中で、1,3−ジメチルイソシアヌレートと、酸化エチレンを反応させることにより合成することができる。
【0015】
この反応における反応温度については、110〜140℃とすることが好ましく、同反応時間については1〜3時間とすることが好ましい。なお、常温で気体の酸化エチレンが原料である都合上、耐圧性の反応容器であるオートクレーブを使用することが好ましい。
また、前記の反応溶媒としては、例えば、N,N−ジメチルホルムアミドやジメチルスルホキシドの如き非プロトン性極性溶媒を好ましく使用することができる。
【0016】
この1,3−ジメチル−5−(2−ヒドロキシエチル)イソシアヌレートの合成においては、例えば、反応終了後の反応液から反応溶媒を留去し、メタノールやイソプロパノールの如きアルコール溶媒中で再結晶させて精製し、目的物を得ることができる。
【0017】
本発明の1,3−ジメチル−5−(2−ビニルカルボニルオキシエチル)イソシアヌレート及び1,3−ジメチル−5−[2−(2−プロペニル)カルボニルオキシエチル]イソシアヌレートは、例えば適量の反応溶媒中で、1,3−ジメチル−5−(2−ヒドロキシエチル)イソシアヌレートと、アクリル酸またはメタクリル酸とを脱水エステル化反応させることにより合成することができる。
この反応においては、硫酸、パラトルエンスルホン酸やメタンスルホン酸等の酸性触媒を使用する。また、ハイドロキノン、メトキシハイドロキノン、t−ブチルカテコール、t−ブチルハイドロキノン、フェノチアジン等や、塩化銅、硫酸銅等の重合禁止剤を使用することが好ましい。
【0018】
前記の脱水エステル化反応における反応温度については、100〜140℃とすることが好ましく、同反応時間については、9〜15時間とすることが好ましい。
【0019】
また、この反応においては、反応溶媒として、エステル化反応で生成する水の溶解度が低い有機溶媒を使用し、水を共沸させながら脱水を促進することが好ましい。この場合の好ましい有機溶媒としては、例えばトルエン、ベンゼン及びキシレン等の芳香族炭化水素、ヘキサン及びヘプタン等の脂肪族炭化水素、メチルエチルケトン及びシクロヘキサノン等のケトン類や、四塩化炭素、ジクロロエタン及びトリクロロエタン等のハロゲン化炭化水素が挙げられる。
【0020】
本発明の1,3−ジメチル−5−(2−ビニルカルボニルオキシエチル)イソシアヌレート及び1,3−ジメチル−5−[2−(2−プロペニル)カルボニルオキシエチル]イソシアヌレートの合成においては、反応終了後の反応液に、水酸化ナトリウム等のアルカリ水溶液を添加することにより、未反応の(メタ)アクリル酸や、触媒として添加した酸性触媒を中和して、続いて、分離した有機層から反応溶媒を除去し、シリカゲルクロマトグラフィーによる精製を行い、目的物を得ることができる。
【0021】
本発明の1,3−ビス(オキシラニルメチル)−5−(2−ヒドロキシエチル)イソシアヌレート、1,3−ビス(オキシラニルメチル)−5−(2−ビニルカルボニルオキシエチル)イソシアヌレート及び1,3−ビス(オキシラニルメチル)−5−[2−(2−プロペニル)カルボニルオキシエチル]イソシアヌレートは、例えば適量の反応溶媒中で、それぞれ1,3−ジアリル−5−(2−ヒドロキシエチル)イソシアヌレート、1,3−ジアリル−5−(2−ビニルカルボニルオキシエチル)イソシアヌレート及び1,3−ジアリル−5−[2−(2−プロペニル)カルボニルオキシエチル]イソシアヌレートを酸化反応させることにより合成することができる。この反応においては、メタクロロ過安息香酸や過酢酸等の酸化剤を使用する。
【0022】
前記の酸化反応における反応温度については、0℃〜室温とすることが好ましく、同反応時間については、6〜12時間とすることが好ましい。
【0023】
前記の反応溶媒としては、ジクロロメタン、ジクロロエタン及びクロロホルム等のハロゲン化炭化水素を好ましく使用することができる。
【0024】
本発明の1,3−ビス(オキシラニルメチル)−5−(2−ヒドロキシエチル)イソシアヌレート、1,3−ビス(オキシラニルメチル)−5−(2−ビニルカルボニルオキシエチル)イソシアヌレート及び1,3−ビス(オキシラニルメチル)−5−[2−(2−プロペニル)カルボニルオキシエチル]イソシアヌレートの合成においては、反応終了後の反応液に、亜硫酸ナトリウム等の水溶液を添加することにより、未反応の酸化剤を不活性化し、続いて、分離した有機層から反応溶媒を除去し、シリカゲルクロマトグラフィーによる精製を行い、目的物を得ることができる。
【0025】
本発明のこれらのイソシアヌレート化合物を原料とする樹脂は、プリント配線板や電子部品用の塗料、光学レンズ、接着剤、レジストインク等の他、木工用塗料、光ファイバーやプラスチック、缶の表面を保護するためのコーティング剤等への利用が期待される。
【実施例】
【0026】
以下、本発明を実施例に示した合成試験によって具体的に説明する。
なお、原料の1,3−ジメチルイソシアヌレートについては、「Edwin M. Smolin;Lorence Rapoport.“Isocyanuric acid and derivatives”.
The chemistry of heterocyclic compounds. s-Triazines and derivatives.,
INTERSCIENCE PUBLISHERS, INC., 1959, p.389-422.」に記載された方法に従って合成した。
また、同1,3−ジアリル−5−(2−ヒドロキシエチル)イソシアヌレートについては、英国特許第961624号公開公報に記載された方法に従って合成し、同1,3−ジアリル−5−(2−ビニルカルボニルオキシエチル)イソシアヌレートについては、欧州特許第156710号公開公報に記載された方法に従って合成した。
【0027】
〔実施例1〕
<1,3−ジメチル−5−(2−ヒドロキシエチル)イソシアヌレートの合成>
加熱用ジャケット、攪拌機、ガス導入管および排出管を備えた1500mlのオートクレーブに、1,3−ジメチルイソシアヌレート100g(0.6mol)とN,N−ジメチルホルムアミド200gを仕込んだ。容器を密閉後、圧力が1.3MPaになるまで酸化エチレン(0.7mol)を注入した。内温が120℃になるよう設定し、撹拌を行いながら2時間反応させた。
反応液を減圧下で濃縮して得られた粗結晶98gを、イソプロパノール50gで再結晶して、白色結晶61.0g(収率48%、純度97%)を得た。
【0028】
得られた結晶の融点、マススペクトルデータおよび1H−NMRスペクトルデータは、以下のとおりであった。
・融点:113-116℃
・MS:201(M+)
・1H-NMR(DMSO-d6) δ:4.73(t,1H),3.82(t,2H),3.52(dt,2H),3.17(s,3H).
また、この結晶のIRスペクトルデータは、図1に示したチャートのとおりであった。
これらのスペクトルデータより、得られた生成物は、化4の化学式(IV)で示される1,3−ジメチル−5−(2−ヒドロキシエチル)イソシアヌレートであるものと同定した。
化学式(IV):
【0029】
【化4】
【0030】
〔実施例2〕
<1,3−ジメチル−5−(2−ビニルカルボニルオキシエチル)イソシアヌレートの合成>
水分離機付冷却器および温度計を備えた100mLフラスコに、1,3−ジメチル−5−(2−ヒドロキシエチル)イソシアヌレート10.0g(49.7mmol)、トルエン20.0g、パラトルエンスルホン酸0.2g(1.0mmol)、メトキシハイドロキノン0.01g(0.1mmol)、アクリル酸3.9g(54.7mmol)を仕込んだ。
フラスコを110℃に設定したオイルバスに浸漬して、反応液を加熱撹拌し、発生した水分を反応系外に除去しながら12時間反応させた。
反応終了後、反応液中の不溶物を濾去し、濾液を10%炭酸水素ナトリウム水溶液20gで2回洗浄し、次いで水20gで1回洗浄し、分離した有機層を減圧下で乾固させた。
得られた乾固物をシリカゲルクロマトグラフィー(酢酸エチル/ヘキサン=5/1、v/v)により精製し、白色結晶6.7g(収率52%、純度97%)を得た。
【0031】
得られた結晶の融点、マススペクトルデータおよび1H−NMRスペクトルデータは、以下のとおりであった。
・融点:85-93℃
・MS:255(M+)
・1H-NMR(DMSO-d6) δ:6.30(d,1H),6.11(dd,1H),5.94(d,1H),4.28(t,2H),4.05(t,2H),3.16(s,6H).
また、この結晶のIRスペクトルデータは、図2に示したチャートのとおりであった。
これらのスペクトルデータより、得られた生成物は、化5の化学式(V)で示される1,3−ジメチル−5−(2−ビニルカルボニルオキシエチル)イソシアヌレートであるものと同定した。
化学式(V):
【0032】
【化5】
【0033】
〔実施例3〕
<1,3−ジメチル−5−[2−(2−プロペニル)カルボニルオキシエチル]イソシアヌレートの合成>
水分離機付冷却器および温度計を備えた100mLフラスコに、1,3−ジメチル−5−(2−ヒドロキシエチル)イソシアヌレート10.0g(49.7mmol)、トルエン20.0g、パラトルエンスルホン酸0.2g(1.0mmol)、メトキシハイドロキノン0.01g(0.1mmol)、メタクリル酸4.7g(54.7mmol)を仕込んだ。
フラスコを110℃に設定したオイルバスに浸漬して加熱撹拌し、発生した水分を反応系外に除去しながら12時間反応させた。
反応終了後、反応液中の不溶物を濾去し、濾液を10%炭酸水素ナトリウム水溶液10gで2回洗浄し、次いで水10gで1回洗浄し、分離した有機層を減圧下で乾固させた。
得られた乾固物をシリカゲルクロマトグラフィー(酢酸エチル/ヘキサン=5/1、v/v)により精製し、白色結晶8.0g(収率60%、純度98%)を得た。
【0034】
得られた結晶の融点、マススペクトルデータおよび1H−NMRスペクトルデータは、以下のとおりであった。
・融点:68-70℃
・MS:269(M+)
・1H-NMR (DMSO-d6) δ:5.99(s,1H),5.65(s,1H),4.26(t,2H),4.06(t,2H),3.16(s,6H),1.83(s,3H).
また、この結晶のIRスペクトルデータは、図3に示したチャートのとおりであった。
これらのスペクトルデータより、得られた生成物は、化6の化学式(VI)で示される1,3−ジメチル−5−[2−(2−プロペニル)カルボニルオキシエチル]イソシアヌレートであるものと同定した。
化学式(VI):
【0035】
【化6】
【0036】
〔実施例4〕
<1,3−ビス(オキシラニルメチル)−5−(2−ヒドロキシエチル)イソシアヌレートの合成>
温度計を備えた50mLフラスコに、1,3−ジアリル−5−(2−ヒドロキシエチル)イソシアヌレート760mg(3.0mmol)、ジクロロメタン15.0mlを仕込んだ。
氷冷下にて、65%メタクロロ過安息香酸4.8g(18.0mmol)を添加し、1時間攪拌した後、室温にて12時間攪拌した。
反応終了後、クロロホルム30ml及び10%亜硫酸ナトリウム水溶液50mlを添加し、分離した有機層を減圧下で濃縮した。
得られた濃縮物をシリカゲルクロマトグラフィー(クロロホルム/メタノール=10/1、v/v)により精製し、無色液体650mg(収率76%,純度99%)を得た。
【0037】
得られた液体の1H−NMRスペクトルデータは、以下のとおりであった。
・1H-NMR (CDCl3) δ:4.10-4.21(m,4H),4.00-4.06(m,2H),3.87-3.91(m,4H),3.24-3.28(m,2H),2.83(t,2H),2.68-2.71(m,2H).
また、この液体のIRスペクトルデータは、図4に示したチャートのとおりであった。
これらのスペクトルデータより、得られた生成物は、化7の化学式(VII)で示される1,3−ビス(オキシラニルメチル)−5−(2−ヒドロキシエチル)イソシアヌレートであるものと同定した。
化学式(VII):
【0038】
【化7】
【0039】
〔実施例5〕
<1,3−ビス(オキシラニルメチル)−5−(2−ビニルカルボニルオキシエチル)イソシアヌレートの合成>
温度計を備えた50mLフラスコに、1,3−ジアリル−5−(2−ビニルカルボニルオキシエチル)イソシアヌレート307mg(1.0mmol)、ジクロロメタン5.0mlを仕込んだ。
氷冷下にて、65%メタクロロ過安息香酸1.6g(6.0mmol)を添加し、1時間攪拌した後、室温にて9時間攪拌した。
反応終了後、クロロホルム10ml及び10%亜硫酸ナトリウム水溶液30mlを添加し、分離した有機層を減圧下で濃縮した。
得られた濃縮物をシリカゲルクロマトグラフィー(酢酸エチル/ヘキサン=1/3、v/v)により精製し、無色液体268.0mg(収率79%,純度99%)を得た。
【0040】
得られた液体の1H−NMRスペクトルデータは、以下のとおりであった。
・1H-NMR (DMSO-d6) δ:6.42(dd,1H),6.07(dd,1H),5.86(dd,1H),4.43(t,2H)4.26(t,2H),4.18(ddd,2H),4.03(dd,2H),3.22-3.26(m,2H),2.82(t,2H),2.69(m,2H).
また、この液体のIRスペクトルデータは、図5に示したチャートのとおりであった。
これらのスペクトルデータより、得られた生成物は、化8の化学式(VIII)で示される1,3−ビス(オキシラニルメチル)−5−(2−ビニルカルボニルオキシエチル)イソシアヌレートであるものと同定した。
化学式(VIII):
【0041】
【化8】
【図面の簡単な説明】
【0042】
【図1】実施例1で得られた結晶のIRスペクトルチャートである。
【図2】実施例2で得られた結晶のIRスペクトルチャートである。
【図3】実施例3で得られた結晶のIRスペクトルチャートである。
【図4】実施例4で得られた液体のIRスペクトルチャートである。
【図5】実施例5で得られた液体のIRスペクトルチャートである。
【産業上の利用可能性】
【0043】
本発明によれば、光硬化性樹脂の原料としての用途が期待されるイソシアヌレート化合物を提供することができる。
【特許請求の範囲】
【請求項1】
化1の化学式(I)で示されるイソシアヌレート化合物。
化学式(I):
【化1】
【請求項1】
化1の化学式(I)で示されるイソシアヌレート化合物。
化学式(I):
【化1】
【図1】

【図2】
【図3】
【図4】
【図5】
【図2】
【図3】
【図4】
【図5】
【公開番号】特開2013−49657(P2013−49657A)
【公開日】平成25年3月14日(2013.3.14)
【国際特許分類】
【出願番号】特願2011−196994(P2011−196994)
【出願日】平成23年9月9日(2011.9.9)
【出願人】(000180302)四国化成工業株式会社 (167)
【Fターム(参考)】
【公開日】平成25年3月14日(2013.3.14)
【国際特許分類】
【出願日】平成23年9月9日(2011.9.9)
【出願人】(000180302)四国化成工業株式会社 (167)
【Fターム(参考)】
[ Back to top ]