
イソピマル酸製造プロセス
次のステップを具備するイソピマル酸製造プロセスが記載されている。第1のステップ:熱的に異性化されたロジンをアセトンに溶解させ、沈殿物を形成するために、アセトンに、イソブタノールアミンの溶液を滴下にて添加し、放置し、フィルターし、エタノールで洗浄し、イソピマル酸の粗アンモニウム塩を得るために乾燥し、イソピマル酸の粗アンモニウム塩を、マルチ・再結晶化によって、再結晶化し、イソピマル酸のアンモニウム塩の精製された結晶を得るために乾燥する。第2のステップ:第1のステップで得られたイソピマル酸のアンモニウム塩の前記精製された結晶をエーテルに溶解させ、塩酸を、イソピマル酸のアンモニウム塩の前記結晶が消滅するまで、部分的に添加し、水の層を取り除き、中性になるように、エーテルの層を洗浄し、エーテルを蒸発させ、その残渣を、アセトンに溶解させ、結晶が成長するのをやめるまで、水を、滴下で、前記溶液にゆっくりと添加し、フィルターし、精製されたイソピマル酸を得るために乾燥させる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ロジンまたは松脂の重要な構成要素であるイソピマル酸製造プロセスに関する。
【背景技術】
【0002】
イソピマル酸は、ロジンまたは松脂の重要な構成要素である。エリオッティ松・ロジンにおけるその含有量は、およそ20%である。イソピマル酸の安定なフェナントレン環スケルトン構造および特有の環外二重結合には、化学反応の多重特性が授けられている。従って、典型的な松脂またはロジンからイソピマル酸を分離して精製することは、ピマル・タイプの樹脂酸の開発および利用にとって重要である。
【0003】
3つの周知のイソピマル酸分離および精製方法がある、すなわち、「ピペリジン沈殿方法」、「無水マレイン酸添加/イソブタノールアミン沈殿の方法」および「蒸留方法」である。Loeblich他のレポートでは、イソピマル酸が、直接的なピペリジン沈殿方法によって分離されており、イソピマル酸の収率は4.7%である(Loeblich V M、Lawrence R V:松脂およびロジンからイソデキストロピマル酸を分離する新しい方法[ジャーナル]:J Org Chem、1958、23(1):25〜26)。この方法は、イソピマル酸のアンモニウム塩をロジンの原材料から直接分離しており、操作が簡単である。その欠点は、ピペリジンがイソピマル酸にとって選択的であるが、発生されたイソピマル酸のアンモニウム塩の結晶化比が非常に低く、生産サイクルが長くて収率が低いことである。Harris他は、無水マレイン酸のディールス・アルダー添加反応を介して、アビエチン酸型の樹脂酸を除去し、その後、イソブタノールアミンの選択沈殿を介して、イソピマル酸を得ている。イソピマル酸の収率は8%である(Harris G C、Sanderson T F:ロジン酸(III)・・・デキストロピマル酸、新しいピマルータイプの酸、イソデキストロピマル酸の単離[ジャーナル]:J Am Chem Soc、1948、70(1):2079〜2085)。この方法では、マレオピマル酸と未反応の樹脂酸(ピマルータイプの樹脂酸、脱水素アビエチン酸および完全には反応されていない少量のアビエチン酸タイプの樹脂酸を含む)を分離することは困難であり、分離された未反応の樹脂酸は、マレオピマル酸を含むだろう。Harris他は、また、ピマル酸とイソピマル酸を直接蒸発させるための蒸留方法を採用し、その後、イソピマル酸を分離するためにイソブタノールアミンを使用した。ロジンに関連する収率は4%である(Harris G C、Sanderson T F:ロジン酸(III)・・・デキストロピマル酸、新しいピマルータイプの酸、イソデキストロピマル酸の単離[ジャーナル]:J Am Chem Soc、1948、70(1):2079〜2085)。この方法では、分留条件への高度な要求があり、操作が容易でなく、イソピマル酸の収率が低い。
【0004】
現在、イソピマル酸の分離および精製の研究についての国内報告書は少なくて、産業グレードの高純度のイソピマル酸製品はない。従って、イソピマル酸を高い効率、高い収率および高い経済的な実現可能性で分離する方法についての探査は、医療、生物、材料および他の分野において、イソピマル酸の適用を加速するであろう。
【発明の開示】
【0005】
イソピマル酸製造において高コストおよび低収率である、先行技術の欠点を解決するために、本発明は、イソピマル酸製造プロセスを提供する。そのプロセスは、低製造コスト、高い収率および95%と同じくらい高いイソピマル酸製品の高い純度に特徴付けられる。
【0006】
本発明では、主に次の技術ルートが採用される:
【表A】

【0007】
本発明の技術的スキームは、次のようなステップを有するイソピマル酸製造プロセスである。
【0008】
ステップ1:熱的に異性化されたロジンを1倍以上10倍以下の質量を有するアセトンに溶解させ、沈殿物を形成するために、熱的に異性化されたロジンの質量の1%以上40%以下と同等の質量を有するイソブタノールアミンのアセトン溶液(アセトン/イソブタノールアミン=1:1mL/g)を滴下にて添加し、放置し、フィルターし、50%(v/v)エタノールで洗浄し、イソピマル酸の粗アンモニウム塩を得るために乾燥し、イソピマル酸の粗アンモニウム塩の各グループを、マルチ・再結晶化によって、5回再結晶化し、イソピマル酸のアンモニウム塩の精製された結晶を得るために乾燥するステップであり、マルチ・再結晶化方法において使用される溶媒が、95%(v/v)エタノール、メチルアセテート、メチルアセテート・無水エタノール、無水エタノールおよびメタノールのグループから選択されるいずれかである。
【0009】
ステップ2:第1のステップで得られたイソピマル酸のアンモニウム塩の前記精製された結晶を、1倍以上20倍以下の質量を有するエーテルに溶解させ、1wt%以上20wt%以下の塩酸を、イソピマル酸のアンモニウム塩の前記結晶が消滅するまで、何回かに分けて添加し、水の層を取り除き、中性になるように、エーテルの層を洗浄し、1気圧および30℃以上50℃以下で、エーテルを蒸発させ、その残渣を、0.5倍以上5倍以下の質量を有するアセトンに溶解させ、沈殿された結晶が成長するのをやめるまで、水を、滴下にて、前記溶液にゆっくりと添加し、フィルターし、精製されたイソピマル酸を得るために乾燥させる。
【0010】
前記熱的に異性化されたロジンが、次の方法で製造されることを特徴とする。すなわち、イソピマル酸を含む松脂から、1気圧および90℃以上114℃以下で、少量の水を蒸発させ、その後、10kPa以上50kPa以下および140℃以上170℃以下で、テレビン油を蒸発させ、その後、1時間以上3時間以下の異性化反応を取得するために、温度を150℃以上180℃以下へと上昇させ、熱的に異性化されたロジンを得るために、フィルターし、室温へと冷却する方法。イソピマル酸を含む前記松脂が、エリオッティ松・松脂、カリバエ松・松脂、カシヤ松・松脂およびマソン松・松脂のグループのいずれかであり、そこで最も好適なのは、エリオッティ松・松脂である。前記ロジンは、ガムロジン、パーマイトまたは木材のロジンだろうし、そこでは、ガムロジンの中では、エリオッティ松・ロジン、カリバエ松・ロジン、カシヤ松・ロジンまたはマソン松・ロジンが好適であり、最も好適なのは、エリオッティ松・ロジンである。
【0011】
本発明の技術上の効果は、次のとおりである。
【0012】
1.松脂に、テレビン油の蒸留およびロジンの熱的異性化を行うと、レボピマール酸のほとんどが、アビエチン酸型の樹脂酸内に異性化される。レボピマール酸とイソピマル酸の両方が、イソブタノールアミンまたはピペリジンと反応するだろうにつれて、熱的異性化の後に、イソピマル酸のみが、保持されて、反応収率が上昇される。本発明で提供されるプロセスにおける、熱的に異性化されたロジンに対する収率は、10.1%に達するだろう。
【0013】
2.イソピマル酸のアンモニウム塩は、溶媒として、95%(v/v)エタノールを採用する。マルチ・再結晶化によって、結晶の各グループは、5回再結晶化される。この手順は、イソピマル酸のアンモニウム塩に含まれた他の樹脂酸、特にアビエチン酸を効果的に取り除くことができる。このようにして、他の樹脂酸の干渉と、イソピマル酸のアンモニウム塩の損失が、この上なく最小化され、イソピマル酸の純度は、95%を超えて上昇される。
【0014】
3.ピペリジン方法と比較して、本発明で提供されるプロセスにおける、イソピマル酸に対するイソブタノールアミンの結晶化比は、イソピマル酸に対するピペリジンの結晶化比よりも高いので、このプロセスは、分離時間を著しく低減し、有機アミンを節約するし、無水マレイン酸添加/イソブタノールアミン沈殿の方法と比較して、このプロセスは、ロジンからイソピマル酸を直接分離し、中間ステップに起因する損失を低減し、イソピマル酸の収率を上昇させ、操作ステップを簡単にするし、蒸留方法と比較して、この方法は、操作条件への要求が低く、より高い収率を有する。
【発明を実施するための最良の形態】
【0015】
本発明は、実施例に関連して、さらに記載される。
【0016】
実施例1
イソピマル酸製造プロセスで次のステップを具備する。
【0017】
ステップ1:熱的に異性化されたロジンを1倍以上10倍以下の質量を有するアセトンに溶解させ、沈殿物を形成するために、熱的に異性化されたロジンの質量の1%以上40%以下と同等の質量を有するイソブタノールアミンのアセトン溶液(アセトン/イソブタノールアミン=1:1mL/g)を滴下にて添加し、放置し、フィルターし、50%(v/v)エタノールで洗浄し、イソピマル酸の粗アンモニウム塩を得るために乾燥し、イソピマル酸の粗アンモニウム塩の各グループを、マルチ・再結晶化によって、5回再結晶化し、イソピマル酸のアンモニウム塩の精製された結晶を得るために乾燥するステップであり、マルチ・再結晶化方法において使用される溶媒が、95%(v/v)エタノール、メチルアセテート、メチルアセテート・無水エタノール、無水エタノールおよびメタノールのグループから選択されるいずれかである。アセトンの量は、熱的に異性化されたロジンの質量の1倍、3倍または10倍だろう。滴下にて添加されたイソブタノールアミンの量は、熱的に異性化されたロジンの質量の1%、22%または40%だろう。
【0018】
ステップ2:第1のステップで得られたイソピマル酸のアンモニウム塩の前記精製された結晶を、1倍以上20倍以下の質量を有するエーテルに溶解させ、1wt%以上20wt%以下の塩酸を、イソピマル酸のアンモニウム塩の前記結晶が消滅するまで、何回かに分けて添加し、水の層を取り除き、中性になるように、エーテルの層を洗浄し、1気圧および30℃以上50℃以下で、エーテルを蒸発させ、その残渣を、0.5倍以上5倍以下の質量を有するアセトンに溶解させ、沈殿された結晶が成長するのをやめるまで、水を、滴下にて、前記溶液にゆっくりと添加し、フィルターし、精製されたイソピマル酸を得るために乾燥させる。エーテルの量は、イソピマル酸のアンモニウム塩の質量の1倍、11倍または20倍だろう。塩酸の濃度は、1wt%、10wt%または20wt%だろう。
【0019】
実施例2
600gのエリオッティ松・松脂を1Lの四口フラスコに投入し、温度計、ト字管、Y尾管および受けフラスコを設置し、窒素を投入し、1気圧および90℃以上114℃以下でエリオッティ松・松脂に含まれる少量の水を蒸発させ、10kPa以上50kPa以下および140℃以上170℃以下でテレビン油を蒸発させ、1時間以上3時間以下の異性化反応を取得するために、再び温度を150℃以上180℃以下に上昇させ、478gの熱的に異性化されたロジンを得るために、フィルターし、室温へと冷却する。サンプルを取って、メチルエステル化を受けて、その後、GC分析する。熱的に異性化されたロジンにおいて、イソピマル酸の質量割合は、23.75%である。
【0020】
実施例3
実施例2におけるエリオッティ松・松脂を、カリバエ松・松脂に変更し、その他は変更しない。
【0021】
実施例4
実施例2におけるエリオッティ松・松脂を、カシヤ松・松脂に変更し、その他は変更しない。
【0022】
実施例5
実施例2おけるエリオッティ松・松脂を、マソン松・松脂に変更し、その他は変更しない。
【0023】
実施例6
エリオッティ松・ロジン、カリバエ松・ロジン、カシヤ松・ロジンまたはマソン松・ロジンであろうガムロジン200gを150℃以上180℃以下で1時間以上3時間以下異性化し、熱的に異性化されたロジンを得るために、フィルターし、室温へと冷却する。
【0024】
実施例7
実施例2において得られた200gの熱的に異性化されたロジンを同じ質量を有するアセトンに溶解させ、沈殿物を形成するために、熱的に異性化されたロジンの質量の15%と同等の質量を有するイソブタノールアミンのアセトン溶液(イソブタノールアミン/アセトン=1:1mL/g)を滴下にて添加し、2時間放置し、フィルターし、50%(v/v)エタノールで3回洗浄し、イソピマル酸の粗アンモニウム塩を得るために乾燥する。マルチ・再結晶化によって5回イソピマル酸の粗アンモニウム塩の各グループを再結晶化する。各回のイソピマル酸のアンモニウム塩の溶解の期間に、1倍以上3倍以下の容量の95%(v/v)エタノールが添加される。ろ過と結晶化の期間に、50%(v/v)エタノールが洗浄のために使用される。乾燥の後に、イソピマル酸のアンモニウム塩の25.7gの精製された結晶が得られる。
【0025】
イソピマル酸の精製されたアンモニウム塩の全てを、同じ質量を有するエーテルに溶解させ、1wt%塩酸を、イソピマル酸のアンモニウム塩の結晶が消滅するまで、何回かに分けて添加し、水の層を取り除き、中性になるように、エーテルの層を洗浄し、1気圧および30℃で、エーテルを蒸発させ、その残渣を、0.5倍の質量を有するアセトンに溶解させ、沈殿された結晶が成長するのをやめるまで、水を、滴下にて、溶液にゆっくりと添加し、フィルターし、精製されたイソピマル酸20.1gを得るために乾燥させる。熱的に異性化されたロジンに対する収率は10.1%である。
【0026】
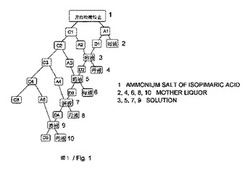
マルチ・再結晶化方法の操作プロセスは、図1に示される。Cは、結晶化を指しており、Aは、ろ過と結晶化後に残され、融合され、次の結晶化プロセスで使用される「有用な母液」を指しており、母液は、もはや使用されずに廃棄されるであろう部分を指している。得られたイソピマル酸の粗アンモニウム塩は、有機溶媒に溶解される。最初の結晶化において得られる結晶はC1であり、母液はA1である。A1は、結晶D1を得るために、1/2容量に濃縮される。母液は廃棄される。C1は、結晶C2を得るために、溶解され、再結晶化される。母液はA2である。結晶D1は、A2に溶解され、その後、結晶D2を得るために、1/2容量に濃縮される。母液は廃棄される。結晶C2は、溶解され、結晶C3を得るために、再結晶化される。母液はA3である。結晶D2は、A3に溶解され、その後、結晶D3を得るために、1/2容量に濃縮される。母液は廃棄される。結晶C3は、溶解され、結晶C4を得るために、再結晶化される。母液はA5である。結晶D3は、A4に溶解され、その後、結晶D4を得るために、1/2容量に濃縮される。母液は廃棄される。結晶C4は、溶解され、結晶C5を得るために、再結晶化される。母液はA5である。結晶D4は、A5に溶解され、その後、結晶D5を得るために、1/2容量に濃縮される。母液は廃棄される。C5とD5は、イソピマル酸の精製されたアンモニウム塩を得るために、融合される。
【0027】
実施例8
実施例2において得られた、200gの熱的に異性化されたロジンを11倍の質量を有するアセトンに溶解させ、沈殿物を形成するために、熱的に異性化されたロジンの質量の21%と同等の質量を有するイソブタノールアミンのアセトン溶液(イソブタノールアミン/アセトン=1:1mL/g)を滴下にて添加し、2時間放置し、フィルターし、50%(v/v)エタノールで3回洗浄し、イソピマル酸の粗アンモニウム塩を得るために乾燥させ、5回のマルチ・再結晶化によってイソピマル酸の粗アンモニウム塩の各グループを再結晶化する。各回のイソピマル酸のアンモニウム塩の溶解の期間に、2倍の容量の95%(v/v)エタノールが添加される。ろ過と結晶化の期間に、50%(v/v)エタノールが洗浄のために使用される。乾燥の後に、イソピマル酸のアンモニウム塩の精製された結晶が得られる。
【0028】
イソピマル酸の精製されたアンモニウム塩の全てを、11倍の質量を有するエーテルに溶解させ、11wt%塩酸を、イソピマル酸のアンモニウム塩の結晶が消滅するまで、何回かに分けて添加し、水の層を取り除き、中性になるように、エーテルの層を洗浄し、1気圧および40℃で、エーテルを蒸発させ、その残渣を、3.5倍の質量を有するアセトンに溶解させ沈殿された結晶が成長するのをやめるまで、水を、滴下にて、溶液にゆっくりと添加し、フィルターし、精製されたイソピマル酸を得るために乾燥させる。
【0029】
実施例9
実施例2において得られた熱的に異性化されたロジンの200gを10倍の質量を有するアセトンに溶解させ、沈殿物を形成するために、熱的に異性化されたロジンの質量の40%と同等のイソブタノールアミンのアセトン溶液(イソブタノールアミン/アセトン=1:1mL/g)を滴下にて添加し、2時間放置し、フィルターし、50%(v/v)エタノールで3回洗浄し、イソピマル酸の粗アンモニウム塩を得るために乾燥させ、5回のマルチ・再結晶化によってイソピマル酸の粗アンモニウム塩の各グループを再結晶化する。各回のイソピマル酸のアンモニウム塩の溶解の期間に、1倍以上3倍以下の容量の95%(v/v)エタノールが添加される。ろ過と結晶化の期間に、50%(v/v)エタノールが洗浄のために使用される。乾燥の後に、イソピマル酸のアンモニウム塩の精製された結晶が得られる。
【0030】
イソピマル酸の精製されたアンモニウム塩の全てを、20倍の質量を有するエーテルに溶解させ、20wt%塩酸を、イソピマル酸のアンモニウム塩の結晶が消滅するまで、何回かに分けて添加し、水の層を取り除き、中性になるように、エーテルの層を洗浄し、1気圧および50℃で、エーテルを蒸発させ、その残渣を、5倍の質量を有するアセトンに溶解させ、沈殿された結晶が成長するのをやめるまで、水を、滴下にて、溶液にゆっくりと添加し、フィルターし、精製されたイソピマル酸を得るために乾燥させる。
【0031】
実施例10
実施例6において得られた200gの熱的に異性化されたロジンを同じ質量を有するアセトンに溶解させ、沈殿物を形成するために、熱的に異性化されたロジンの質量の12%と同等の質量を有するアセトンイソブタノールアミン溶液(イソブタノールアミン/アセトン=1:1g/mL)を滴下にて添加し、2時間放置し、フィルターし、50%(v/v)エタノールで3回洗浄し、イソピマル酸の粗アンモニウム塩を得るために乾燥させ、5回のマルチ・再結晶化によってイソピマル酸の粗アンモニウム塩の各グループを再結晶化する。各回のイソピマル酸のアンモニウム塩の溶解の期間に、1倍以上3倍以下の容量の95%(v/v)エタノールが添加される。ろ過と結晶化の期間に、50%(v/v)エタノールが洗浄のために使用される。乾燥の後に、イソピマル酸のアンモニウム塩の24.8gの精製された結晶が得られる。
【0032】
イソピマル酸の精製されたアンモニウム塩の全てを、同じ質量を有するエーテルに溶解させ、1wt%塩酸を、イソピマル酸のアンモニウム塩の結晶が消滅するまで、何回かに分けて添加し、水の層を取り除き、中性になるように、エーテルの層を洗浄し、1気圧および30℃で、エーテルを蒸発させ、その残渣を、0.5倍の質量を有するアセトンに溶解させ、沈殿された結晶が成長するのをやめるまで、水を、滴下にて、溶液にゆっくりと添加し、フィルターし、精製されたイソピマル酸19.8gを得るために乾燥させる。熱的に異性化されたロジンに対する収率は、9.9%である。
【0033】
実施例11
実施例7と実施例10において得られたイソピマル酸の製品の融点は、162℃以上164℃以下であり、比旋光度〔α〕20Dは、0°である(溶媒は95%エタノールで、質量割合は2%である)。ガスクロマトグラフィーにより決定される製品におけるイソピマル酸の質量割合は、95.4%である。
【0034】
HR−MSのイオンピークの決定された質量:〔M−H〕は、301.2174であり、〔C20H29O2〕−の算出された値は、301.2168、△=0.66ppmである。決定された分子質量は、302.2248である。与えられた分子式は、C20H30O2(DBE=6)であり、算出された値は、302.2246である。
【0035】
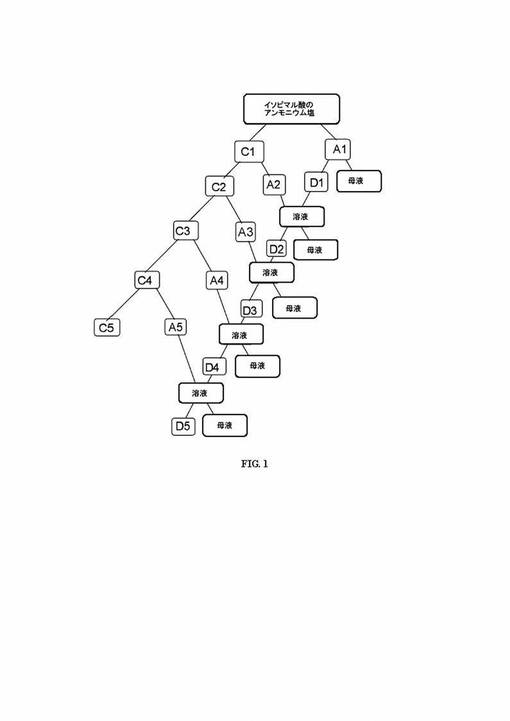
MS(添付の図2参照)、m/z(ピーク強度、%):316(M+・、イソピマル酸メチルエステルの分子イオンピーク、C21H32O2、35.5)、301(M−Me、21)、287(M−(CH2=CH2+H)、20)、257(M−Me-CO2、59.7)、256(m−CO2Me、50.7)、242(25.3)、241(100、M−Me−HCO2CH3)、227(20)、187(30.2)、119(26.3)、121(25.8)、105(30.3)、91(25.7)。MSデータベースの検出結果は、イソピマル酸の標準スペクトルと一致する。
【0036】
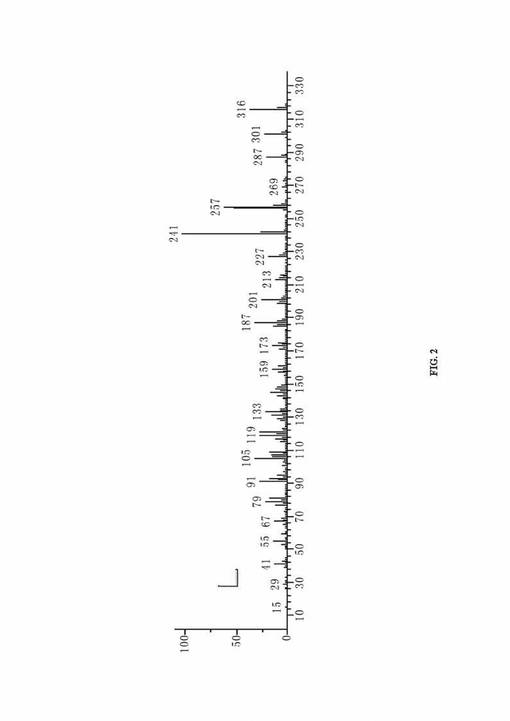
IR(KBrペレット法、スペクトルは図3に示される)、(cm−1):3432(中位の強さ、広いピーク、OH)、3079(C=C−H)、2940(C−H)、2656、2528、1694(強い、C=O)、1463、1387(CH3)、1277(C−O)、1190、906(C=C−H)、658。1HNMR(図4参照、CDCl3)、δ(ppm):0.8620(s、3H、C10−CH3)、0.9068(s、3H、C13−CH3)、1.2673(s、3H、C4−CH3)、1.085〜1.146(m、1H)、1.3216〜1.4138(m、2H)、1.4716〜1.4891(m、1H)、1.5250〜1.5800(m、3H)、4.8651(dd、1H、J1=10.70Hz、J3=1.25Hz、CH)、4.9251(dd、1H、J2=17.50Hz、J3=1.25Hz、CH)、5.322(dd、J=7.45Hz、1H、C=CH)、5.801(dd、1H、J1=10.70Hz、J2=17.50Hz、CH)、11.6〜12.2(広いs、1H、COOH)。
【0037】
この製品の物性、MS、IRおよびPNMRの分析結果を介して、イソピマル酸の化学構造が正確に識別される。
【0038】
比較1
200gのエリオッティ松・ロジンを400mLのn−ヘプタンに溶解させ、15gのピペリジンを添加し、溶液を攪拌し、室温へと冷却し、それを冷蔵庫で2時間冷却し、それが常温に回復する時は溶液を攪拌し続け、混合物を冷蔵庫に入れ、アンモニウム塩を分離するためにそれを一晩凍結し、溶媒として95%エタノールを使用し、そして、25gのアンモニウム塩を得るために、5回のマルチ・再結晶化によって、イソピマル酸のアンモニウム塩を再結晶化させる。
【0039】
アンモニウム塩を100mLエーテルに溶解させ、イソピマル酸のアンモニウム塩の結晶が消滅するまで、1wt%以上20wt%以下の塩酸を添加する。水の層を取り除き、中性になるように、エーテルの層を洗浄し、エーテルを1気圧および30℃以上50℃以下で蒸発させ、残渣を0.5倍以上5倍以下の質量を有するアセトン内で溶解させ、沈殿された結晶が成長するのをやめるまで、蒸留された水を、滴下にて、溶液にゆっくりと添加し、フィルターし、8.2gの純粋イソピマル酸を得るために乾燥する。収率は4.1%である。ガスクロマトグラフィーにより決定されるイソピマル酸の純度は87%である。
【0040】
実施例12
実施例2における方法が採用され、そこでは、イソブタノールアミンはピペリジンにて置き換えられる。イソブタノールアミンとピペリジンの理論量が、それぞれ同等量のロジンに加えられる。イソブタノールアミンが使用される時は、アセトンは再結晶化溶媒であり、ピペリジが使用される時は、n−ヘプタンが再結晶化溶媒である。結果は表1に示されている。
【0041】
【表1】
【0042】
実施例13
実施例2における方法が採用され、そこでは、イソピマル酸の粗アンモニウム塩の95%エタノールマルチ・再結晶化は、95%(v/v)エタノール、メチルアセテート、メチルアセテート・無水エタノール(2:3、V/V)、無水エタノール、メタノールまたは他の溶媒での3回の再結晶化にて置き換えられる。実施例1に記載された方法により同じ処置が、精製されたイソピマル酸を得るために採用される。異なる再結晶化溶媒の中での再結晶化効果の比較は、表2に示される。
【0043】
【表2】
【図面の簡単な説明】
【0044】
【図1】図1は、本発明におけるマルチ・結晶化方法の動作フローチャートである。
【図2】図2は、イソピマル酸メチルエステルの製品のMS分析スペクトル写真である。
【図3】図3は、イソピマル酸の製品のIR分析スペクトル写真である。
【図4】図4は、イソピマル酸の製品のPNMR分析スペクトル写真である。
【技術分野】
【0001】
本発明は、ロジンまたは松脂の重要な構成要素であるイソピマル酸製造プロセスに関する。
【背景技術】
【0002】
イソピマル酸は、ロジンまたは松脂の重要な構成要素である。エリオッティ松・ロジンにおけるその含有量は、およそ20%である。イソピマル酸の安定なフェナントレン環スケルトン構造および特有の環外二重結合には、化学反応の多重特性が授けられている。従って、典型的な松脂またはロジンからイソピマル酸を分離して精製することは、ピマル・タイプの樹脂酸の開発および利用にとって重要である。
【0003】
3つの周知のイソピマル酸分離および精製方法がある、すなわち、「ピペリジン沈殿方法」、「無水マレイン酸添加/イソブタノールアミン沈殿の方法」および「蒸留方法」である。Loeblich他のレポートでは、イソピマル酸が、直接的なピペリジン沈殿方法によって分離されており、イソピマル酸の収率は4.7%である(Loeblich V M、Lawrence R V:松脂およびロジンからイソデキストロピマル酸を分離する新しい方法[ジャーナル]:J Org Chem、1958、23(1):25〜26)。この方法は、イソピマル酸のアンモニウム塩をロジンの原材料から直接分離しており、操作が簡単である。その欠点は、ピペリジンがイソピマル酸にとって選択的であるが、発生されたイソピマル酸のアンモニウム塩の結晶化比が非常に低く、生産サイクルが長くて収率が低いことである。Harris他は、無水マレイン酸のディールス・アルダー添加反応を介して、アビエチン酸型の樹脂酸を除去し、その後、イソブタノールアミンの選択沈殿を介して、イソピマル酸を得ている。イソピマル酸の収率は8%である(Harris G C、Sanderson T F:ロジン酸(III)・・・デキストロピマル酸、新しいピマルータイプの酸、イソデキストロピマル酸の単離[ジャーナル]:J Am Chem Soc、1948、70(1):2079〜2085)。この方法では、マレオピマル酸と未反応の樹脂酸(ピマルータイプの樹脂酸、脱水素アビエチン酸および完全には反応されていない少量のアビエチン酸タイプの樹脂酸を含む)を分離することは困難であり、分離された未反応の樹脂酸は、マレオピマル酸を含むだろう。Harris他は、また、ピマル酸とイソピマル酸を直接蒸発させるための蒸留方法を採用し、その後、イソピマル酸を分離するためにイソブタノールアミンを使用した。ロジンに関連する収率は4%である(Harris G C、Sanderson T F:ロジン酸(III)・・・デキストロピマル酸、新しいピマルータイプの酸、イソデキストロピマル酸の単離[ジャーナル]:J Am Chem Soc、1948、70(1):2079〜2085)。この方法では、分留条件への高度な要求があり、操作が容易でなく、イソピマル酸の収率が低い。
【0004】
現在、イソピマル酸の分離および精製の研究についての国内報告書は少なくて、産業グレードの高純度のイソピマル酸製品はない。従って、イソピマル酸を高い効率、高い収率および高い経済的な実現可能性で分離する方法についての探査は、医療、生物、材料および他の分野において、イソピマル酸の適用を加速するであろう。
【発明の開示】
【0005】
イソピマル酸製造において高コストおよび低収率である、先行技術の欠点を解決するために、本発明は、イソピマル酸製造プロセスを提供する。そのプロセスは、低製造コスト、高い収率および95%と同じくらい高いイソピマル酸製品の高い純度に特徴付けられる。
【0006】
本発明では、主に次の技術ルートが採用される:
【表A】
【0007】
本発明の技術的スキームは、次のようなステップを有するイソピマル酸製造プロセスである。
【0008】
ステップ1:熱的に異性化されたロジンを1倍以上10倍以下の質量を有するアセトンに溶解させ、沈殿物を形成するために、熱的に異性化されたロジンの質量の1%以上40%以下と同等の質量を有するイソブタノールアミンのアセトン溶液(アセトン/イソブタノールアミン=1:1mL/g)を滴下にて添加し、放置し、フィルターし、50%(v/v)エタノールで洗浄し、イソピマル酸の粗アンモニウム塩を得るために乾燥し、イソピマル酸の粗アンモニウム塩の各グループを、マルチ・再結晶化によって、5回再結晶化し、イソピマル酸のアンモニウム塩の精製された結晶を得るために乾燥するステップであり、マルチ・再結晶化方法において使用される溶媒が、95%(v/v)エタノール、メチルアセテート、メチルアセテート・無水エタノール、無水エタノールおよびメタノールのグループから選択されるいずれかである。
【0009】
ステップ2:第1のステップで得られたイソピマル酸のアンモニウム塩の前記精製された結晶を、1倍以上20倍以下の質量を有するエーテルに溶解させ、1wt%以上20wt%以下の塩酸を、イソピマル酸のアンモニウム塩の前記結晶が消滅するまで、何回かに分けて添加し、水の層を取り除き、中性になるように、エーテルの層を洗浄し、1気圧および30℃以上50℃以下で、エーテルを蒸発させ、その残渣を、0.5倍以上5倍以下の質量を有するアセトンに溶解させ、沈殿された結晶が成長するのをやめるまで、水を、滴下にて、前記溶液にゆっくりと添加し、フィルターし、精製されたイソピマル酸を得るために乾燥させる。
【0010】
前記熱的に異性化されたロジンが、次の方法で製造されることを特徴とする。すなわち、イソピマル酸を含む松脂から、1気圧および90℃以上114℃以下で、少量の水を蒸発させ、その後、10kPa以上50kPa以下および140℃以上170℃以下で、テレビン油を蒸発させ、その後、1時間以上3時間以下の異性化反応を取得するために、温度を150℃以上180℃以下へと上昇させ、熱的に異性化されたロジンを得るために、フィルターし、室温へと冷却する方法。イソピマル酸を含む前記松脂が、エリオッティ松・松脂、カリバエ松・松脂、カシヤ松・松脂およびマソン松・松脂のグループのいずれかであり、そこで最も好適なのは、エリオッティ松・松脂である。前記ロジンは、ガムロジン、パーマイトまたは木材のロジンだろうし、そこでは、ガムロジンの中では、エリオッティ松・ロジン、カリバエ松・ロジン、カシヤ松・ロジンまたはマソン松・ロジンが好適であり、最も好適なのは、エリオッティ松・ロジンである。
【0011】
本発明の技術上の効果は、次のとおりである。
【0012】
1.松脂に、テレビン油の蒸留およびロジンの熱的異性化を行うと、レボピマール酸のほとんどが、アビエチン酸型の樹脂酸内に異性化される。レボピマール酸とイソピマル酸の両方が、イソブタノールアミンまたはピペリジンと反応するだろうにつれて、熱的異性化の後に、イソピマル酸のみが、保持されて、反応収率が上昇される。本発明で提供されるプロセスにおける、熱的に異性化されたロジンに対する収率は、10.1%に達するだろう。
【0013】
2.イソピマル酸のアンモニウム塩は、溶媒として、95%(v/v)エタノールを採用する。マルチ・再結晶化によって、結晶の各グループは、5回再結晶化される。この手順は、イソピマル酸のアンモニウム塩に含まれた他の樹脂酸、特にアビエチン酸を効果的に取り除くことができる。このようにして、他の樹脂酸の干渉と、イソピマル酸のアンモニウム塩の損失が、この上なく最小化され、イソピマル酸の純度は、95%を超えて上昇される。
【0014】
3.ピペリジン方法と比較して、本発明で提供されるプロセスにおける、イソピマル酸に対するイソブタノールアミンの結晶化比は、イソピマル酸に対するピペリジンの結晶化比よりも高いので、このプロセスは、分離時間を著しく低減し、有機アミンを節約するし、無水マレイン酸添加/イソブタノールアミン沈殿の方法と比較して、このプロセスは、ロジンからイソピマル酸を直接分離し、中間ステップに起因する損失を低減し、イソピマル酸の収率を上昇させ、操作ステップを簡単にするし、蒸留方法と比較して、この方法は、操作条件への要求が低く、より高い収率を有する。
【発明を実施するための最良の形態】
【0015】
本発明は、実施例に関連して、さらに記載される。
【0016】
実施例1
イソピマル酸製造プロセスで次のステップを具備する。
【0017】
ステップ1:熱的に異性化されたロジンを1倍以上10倍以下の質量を有するアセトンに溶解させ、沈殿物を形成するために、熱的に異性化されたロジンの質量の1%以上40%以下と同等の質量を有するイソブタノールアミンのアセトン溶液(アセトン/イソブタノールアミン=1:1mL/g)を滴下にて添加し、放置し、フィルターし、50%(v/v)エタノールで洗浄し、イソピマル酸の粗アンモニウム塩を得るために乾燥し、イソピマル酸の粗アンモニウム塩の各グループを、マルチ・再結晶化によって、5回再結晶化し、イソピマル酸のアンモニウム塩の精製された結晶を得るために乾燥するステップであり、マルチ・再結晶化方法において使用される溶媒が、95%(v/v)エタノール、メチルアセテート、メチルアセテート・無水エタノール、無水エタノールおよびメタノールのグループから選択されるいずれかである。アセトンの量は、熱的に異性化されたロジンの質量の1倍、3倍または10倍だろう。滴下にて添加されたイソブタノールアミンの量は、熱的に異性化されたロジンの質量の1%、22%または40%だろう。
【0018】
ステップ2:第1のステップで得られたイソピマル酸のアンモニウム塩の前記精製された結晶を、1倍以上20倍以下の質量を有するエーテルに溶解させ、1wt%以上20wt%以下の塩酸を、イソピマル酸のアンモニウム塩の前記結晶が消滅するまで、何回かに分けて添加し、水の層を取り除き、中性になるように、エーテルの層を洗浄し、1気圧および30℃以上50℃以下で、エーテルを蒸発させ、その残渣を、0.5倍以上5倍以下の質量を有するアセトンに溶解させ、沈殿された結晶が成長するのをやめるまで、水を、滴下にて、前記溶液にゆっくりと添加し、フィルターし、精製されたイソピマル酸を得るために乾燥させる。エーテルの量は、イソピマル酸のアンモニウム塩の質量の1倍、11倍または20倍だろう。塩酸の濃度は、1wt%、10wt%または20wt%だろう。
【0019】
実施例2
600gのエリオッティ松・松脂を1Lの四口フラスコに投入し、温度計、ト字管、Y尾管および受けフラスコを設置し、窒素を投入し、1気圧および90℃以上114℃以下でエリオッティ松・松脂に含まれる少量の水を蒸発させ、10kPa以上50kPa以下および140℃以上170℃以下でテレビン油を蒸発させ、1時間以上3時間以下の異性化反応を取得するために、再び温度を150℃以上180℃以下に上昇させ、478gの熱的に異性化されたロジンを得るために、フィルターし、室温へと冷却する。サンプルを取って、メチルエステル化を受けて、その後、GC分析する。熱的に異性化されたロジンにおいて、イソピマル酸の質量割合は、23.75%である。
【0020】
実施例3
実施例2におけるエリオッティ松・松脂を、カリバエ松・松脂に変更し、その他は変更しない。
【0021】
実施例4
実施例2におけるエリオッティ松・松脂を、カシヤ松・松脂に変更し、その他は変更しない。
【0022】
実施例5
実施例2おけるエリオッティ松・松脂を、マソン松・松脂に変更し、その他は変更しない。
【0023】
実施例6
エリオッティ松・ロジン、カリバエ松・ロジン、カシヤ松・ロジンまたはマソン松・ロジンであろうガムロジン200gを150℃以上180℃以下で1時間以上3時間以下異性化し、熱的に異性化されたロジンを得るために、フィルターし、室温へと冷却する。
【0024】
実施例7
実施例2において得られた200gの熱的に異性化されたロジンを同じ質量を有するアセトンに溶解させ、沈殿物を形成するために、熱的に異性化されたロジンの質量の15%と同等の質量を有するイソブタノールアミンのアセトン溶液(イソブタノールアミン/アセトン=1:1mL/g)を滴下にて添加し、2時間放置し、フィルターし、50%(v/v)エタノールで3回洗浄し、イソピマル酸の粗アンモニウム塩を得るために乾燥する。マルチ・再結晶化によって5回イソピマル酸の粗アンモニウム塩の各グループを再結晶化する。各回のイソピマル酸のアンモニウム塩の溶解の期間に、1倍以上3倍以下の容量の95%(v/v)エタノールが添加される。ろ過と結晶化の期間に、50%(v/v)エタノールが洗浄のために使用される。乾燥の後に、イソピマル酸のアンモニウム塩の25.7gの精製された結晶が得られる。
【0025】
イソピマル酸の精製されたアンモニウム塩の全てを、同じ質量を有するエーテルに溶解させ、1wt%塩酸を、イソピマル酸のアンモニウム塩の結晶が消滅するまで、何回かに分けて添加し、水の層を取り除き、中性になるように、エーテルの層を洗浄し、1気圧および30℃で、エーテルを蒸発させ、その残渣を、0.5倍の質量を有するアセトンに溶解させ、沈殿された結晶が成長するのをやめるまで、水を、滴下にて、溶液にゆっくりと添加し、フィルターし、精製されたイソピマル酸20.1gを得るために乾燥させる。熱的に異性化されたロジンに対する収率は10.1%である。
【0026】
マルチ・再結晶化方法の操作プロセスは、図1に示される。Cは、結晶化を指しており、Aは、ろ過と結晶化後に残され、融合され、次の結晶化プロセスで使用される「有用な母液」を指しており、母液は、もはや使用されずに廃棄されるであろう部分を指している。得られたイソピマル酸の粗アンモニウム塩は、有機溶媒に溶解される。最初の結晶化において得られる結晶はC1であり、母液はA1である。A1は、結晶D1を得るために、1/2容量に濃縮される。母液は廃棄される。C1は、結晶C2を得るために、溶解され、再結晶化される。母液はA2である。結晶D1は、A2に溶解され、その後、結晶D2を得るために、1/2容量に濃縮される。母液は廃棄される。結晶C2は、溶解され、結晶C3を得るために、再結晶化される。母液はA3である。結晶D2は、A3に溶解され、その後、結晶D3を得るために、1/2容量に濃縮される。母液は廃棄される。結晶C3は、溶解され、結晶C4を得るために、再結晶化される。母液はA5である。結晶D3は、A4に溶解され、その後、結晶D4を得るために、1/2容量に濃縮される。母液は廃棄される。結晶C4は、溶解され、結晶C5を得るために、再結晶化される。母液はA5である。結晶D4は、A5に溶解され、その後、結晶D5を得るために、1/2容量に濃縮される。母液は廃棄される。C5とD5は、イソピマル酸の精製されたアンモニウム塩を得るために、融合される。
【0027】
実施例8
実施例2において得られた、200gの熱的に異性化されたロジンを11倍の質量を有するアセトンに溶解させ、沈殿物を形成するために、熱的に異性化されたロジンの質量の21%と同等の質量を有するイソブタノールアミンのアセトン溶液(イソブタノールアミン/アセトン=1:1mL/g)を滴下にて添加し、2時間放置し、フィルターし、50%(v/v)エタノールで3回洗浄し、イソピマル酸の粗アンモニウム塩を得るために乾燥させ、5回のマルチ・再結晶化によってイソピマル酸の粗アンモニウム塩の各グループを再結晶化する。各回のイソピマル酸のアンモニウム塩の溶解の期間に、2倍の容量の95%(v/v)エタノールが添加される。ろ過と結晶化の期間に、50%(v/v)エタノールが洗浄のために使用される。乾燥の後に、イソピマル酸のアンモニウム塩の精製された結晶が得られる。
【0028】
イソピマル酸の精製されたアンモニウム塩の全てを、11倍の質量を有するエーテルに溶解させ、11wt%塩酸を、イソピマル酸のアンモニウム塩の結晶が消滅するまで、何回かに分けて添加し、水の層を取り除き、中性になるように、エーテルの層を洗浄し、1気圧および40℃で、エーテルを蒸発させ、その残渣を、3.5倍の質量を有するアセトンに溶解させ沈殿された結晶が成長するのをやめるまで、水を、滴下にて、溶液にゆっくりと添加し、フィルターし、精製されたイソピマル酸を得るために乾燥させる。
【0029】
実施例9
実施例2において得られた熱的に異性化されたロジンの200gを10倍の質量を有するアセトンに溶解させ、沈殿物を形成するために、熱的に異性化されたロジンの質量の40%と同等のイソブタノールアミンのアセトン溶液(イソブタノールアミン/アセトン=1:1mL/g)を滴下にて添加し、2時間放置し、フィルターし、50%(v/v)エタノールで3回洗浄し、イソピマル酸の粗アンモニウム塩を得るために乾燥させ、5回のマルチ・再結晶化によってイソピマル酸の粗アンモニウム塩の各グループを再結晶化する。各回のイソピマル酸のアンモニウム塩の溶解の期間に、1倍以上3倍以下の容量の95%(v/v)エタノールが添加される。ろ過と結晶化の期間に、50%(v/v)エタノールが洗浄のために使用される。乾燥の後に、イソピマル酸のアンモニウム塩の精製された結晶が得られる。
【0030】
イソピマル酸の精製されたアンモニウム塩の全てを、20倍の質量を有するエーテルに溶解させ、20wt%塩酸を、イソピマル酸のアンモニウム塩の結晶が消滅するまで、何回かに分けて添加し、水の層を取り除き、中性になるように、エーテルの層を洗浄し、1気圧および50℃で、エーテルを蒸発させ、その残渣を、5倍の質量を有するアセトンに溶解させ、沈殿された結晶が成長するのをやめるまで、水を、滴下にて、溶液にゆっくりと添加し、フィルターし、精製されたイソピマル酸を得るために乾燥させる。
【0031】
実施例10
実施例6において得られた200gの熱的に異性化されたロジンを同じ質量を有するアセトンに溶解させ、沈殿物を形成するために、熱的に異性化されたロジンの質量の12%と同等の質量を有するアセトンイソブタノールアミン溶液(イソブタノールアミン/アセトン=1:1g/mL)を滴下にて添加し、2時間放置し、フィルターし、50%(v/v)エタノールで3回洗浄し、イソピマル酸の粗アンモニウム塩を得るために乾燥させ、5回のマルチ・再結晶化によってイソピマル酸の粗アンモニウム塩の各グループを再結晶化する。各回のイソピマル酸のアンモニウム塩の溶解の期間に、1倍以上3倍以下の容量の95%(v/v)エタノールが添加される。ろ過と結晶化の期間に、50%(v/v)エタノールが洗浄のために使用される。乾燥の後に、イソピマル酸のアンモニウム塩の24.8gの精製された結晶が得られる。
【0032】
イソピマル酸の精製されたアンモニウム塩の全てを、同じ質量を有するエーテルに溶解させ、1wt%塩酸を、イソピマル酸のアンモニウム塩の結晶が消滅するまで、何回かに分けて添加し、水の層を取り除き、中性になるように、エーテルの層を洗浄し、1気圧および30℃で、エーテルを蒸発させ、その残渣を、0.5倍の質量を有するアセトンに溶解させ、沈殿された結晶が成長するのをやめるまで、水を、滴下にて、溶液にゆっくりと添加し、フィルターし、精製されたイソピマル酸19.8gを得るために乾燥させる。熱的に異性化されたロジンに対する収率は、9.9%である。
【0033】
実施例11
実施例7と実施例10において得られたイソピマル酸の製品の融点は、162℃以上164℃以下であり、比旋光度〔α〕20Dは、0°である(溶媒は95%エタノールで、質量割合は2%である)。ガスクロマトグラフィーにより決定される製品におけるイソピマル酸の質量割合は、95.4%である。
【0034】
HR−MSのイオンピークの決定された質量:〔M−H〕は、301.2174であり、〔C20H29O2〕−の算出された値は、301.2168、△=0.66ppmである。決定された分子質量は、302.2248である。与えられた分子式は、C20H30O2(DBE=6)であり、算出された値は、302.2246である。
【0035】
MS(添付の図2参照)、m/z(ピーク強度、%):316(M+・、イソピマル酸メチルエステルの分子イオンピーク、C21H32O2、35.5)、301(M−Me、21)、287(M−(CH2=CH2+H)、20)、257(M−Me-CO2、59.7)、256(m−CO2Me、50.7)、242(25.3)、241(100、M−Me−HCO2CH3)、227(20)、187(30.2)、119(26.3)、121(25.8)、105(30.3)、91(25.7)。MSデータベースの検出結果は、イソピマル酸の標準スペクトルと一致する。
【0036】
IR(KBrペレット法、スペクトルは図3に示される)、(cm−1):3432(中位の強さ、広いピーク、OH)、3079(C=C−H)、2940(C−H)、2656、2528、1694(強い、C=O)、1463、1387(CH3)、1277(C−O)、1190、906(C=C−H)、658。1HNMR(図4参照、CDCl3)、δ(ppm):0.8620(s、3H、C10−CH3)、0.9068(s、3H、C13−CH3)、1.2673(s、3H、C4−CH3)、1.085〜1.146(m、1H)、1.3216〜1.4138(m、2H)、1.4716〜1.4891(m、1H)、1.5250〜1.5800(m、3H)、4.8651(dd、1H、J1=10.70Hz、J3=1.25Hz、CH)、4.9251(dd、1H、J2=17.50Hz、J3=1.25Hz、CH)、5.322(dd、J=7.45Hz、1H、C=CH)、5.801(dd、1H、J1=10.70Hz、J2=17.50Hz、CH)、11.6〜12.2(広いs、1H、COOH)。
【0037】
この製品の物性、MS、IRおよびPNMRの分析結果を介して、イソピマル酸の化学構造が正確に識別される。
【0038】
比較1
200gのエリオッティ松・ロジンを400mLのn−ヘプタンに溶解させ、15gのピペリジンを添加し、溶液を攪拌し、室温へと冷却し、それを冷蔵庫で2時間冷却し、それが常温に回復する時は溶液を攪拌し続け、混合物を冷蔵庫に入れ、アンモニウム塩を分離するためにそれを一晩凍結し、溶媒として95%エタノールを使用し、そして、25gのアンモニウム塩を得るために、5回のマルチ・再結晶化によって、イソピマル酸のアンモニウム塩を再結晶化させる。
【0039】
アンモニウム塩を100mLエーテルに溶解させ、イソピマル酸のアンモニウム塩の結晶が消滅するまで、1wt%以上20wt%以下の塩酸を添加する。水の層を取り除き、中性になるように、エーテルの層を洗浄し、エーテルを1気圧および30℃以上50℃以下で蒸発させ、残渣を0.5倍以上5倍以下の質量を有するアセトン内で溶解させ、沈殿された結晶が成長するのをやめるまで、蒸留された水を、滴下にて、溶液にゆっくりと添加し、フィルターし、8.2gの純粋イソピマル酸を得るために乾燥する。収率は4.1%である。ガスクロマトグラフィーにより決定されるイソピマル酸の純度は87%である。
【0040】
実施例12
実施例2における方法が採用され、そこでは、イソブタノールアミンはピペリジンにて置き換えられる。イソブタノールアミンとピペリジンの理論量が、それぞれ同等量のロジンに加えられる。イソブタノールアミンが使用される時は、アセトンは再結晶化溶媒であり、ピペリジが使用される時は、n−ヘプタンが再結晶化溶媒である。結果は表1に示されている。
【0041】
【表1】
【0042】
実施例13
実施例2における方法が採用され、そこでは、イソピマル酸の粗アンモニウム塩の95%エタノールマルチ・再結晶化は、95%(v/v)エタノール、メチルアセテート、メチルアセテート・無水エタノール(2:3、V/V)、無水エタノール、メタノールまたは他の溶媒での3回の再結晶化にて置き換えられる。実施例1に記載された方法により同じ処置が、精製されたイソピマル酸を得るために採用される。異なる再結晶化溶媒の中での再結晶化効果の比較は、表2に示される。
【0043】
【表2】
【図面の簡単な説明】
【0044】
【図1】図1は、本発明におけるマルチ・結晶化方法の動作フローチャートである。
【図2】図2は、イソピマル酸メチルエステルの製品のMS分析スペクトル写真である。
【図3】図3は、イソピマル酸の製品のIR分析スペクトル写真である。
【図4】図4は、イソピマル酸の製品のPNMR分析スペクトル写真である。
【特許請求の範囲】
【請求項1】
次のステップを具備するイソピマル酸製造プロセス。
ステップ1:熱的に異性化されたロジンを1倍以上10倍以下の質量を有するアセトンに溶解させ、沈殿物を形成するために、熱的に異性化されたロジンの1%以上40%以下と同等の質量を有するイソブタノールアミンのアセトン溶液(アセトン/イソブタノールアミン=1:1mL/g)を滴下にて添加し、放置し、フィルターし、50%(v/v)エタノールで洗浄し、イソピマル酸の粗アンモニウム塩を得るために乾燥し、イソピマル酸の粗アンモニウム塩の各グループを、マルチ・再結晶化によって、5回再結晶化し、イソピマル酸のアンモニウム塩の精製された結晶を得るために乾燥する。
ステップ2:イソピマル酸のアンモニウム塩の前記精製された結晶を、1倍以上20倍以下の質量を有するエーテルに溶解させ、1wt%以上20wt%以下塩酸を、イソピマル酸のアンモニウム塩の前記結晶が消滅するまで、何回かに分けて添加し、水の層を取り除き、中性になるように、エーテルの層を洗浄し、1気圧および30℃以上50℃以下で、エーテルを蒸発させ、その残渣を、0.5倍以上5倍以下の質量を有するアセトンに溶解させ、沈殿された結晶が成長するのをやめるまで、水を、滴下にて、前記溶液にゆっくりと添加し、フィルターし、精製されたイソピマル酸を得るために乾燥させる。
【請求項2】
前記熱的に異性化されたロジンが、次の方法で製造されることを特徴とする請求項1記載のイソピマル酸製造プロセス。
イソピマル酸を含む松脂から、1気圧および90℃以上114℃以下で、少量の水を蒸発させ、その後、10kPa以上50kPa以下および140℃以上170℃以下で、テレビン油を蒸発させ、その後、1時間以上3時間以下の異性化反応を取得するために、温度を150℃以上180℃以下へと上昇させ、熱的に異性化されたロジンを得るために、フィルターし、室温へと冷却する方法。
【請求項3】
イソピマル酸を含む前記松脂が、エリオッティ松・松脂、カリバエ松・松脂、カシヤ松・松脂、または、マソン松・松脂であることを特徴とする請求項2記載のイソピマル酸製造プロセス。
【請求項4】
前記熱的に異性化されたロジンが、ロジンを150℃以上180℃以下で1時間以上3時間以下異性化し、フィルターし、室温へと冷却することで得られることを特徴とする請求項1記載のイソピマル酸製造プロセス。
【請求項5】
前記ロジンが、エリオッティ松・ロジン、カリバエ松・ロジン、カシヤ松・ロジン、または、マソン松・ロジンであることを特徴とする請求項4記載のイソピマル酸製造プロセス。
【請求項6】
ステップ1で記述したようなマルチ・再結晶化方法において使用される溶媒が、95%(v/v)エタノール、メチルアセテート、メチルアセテート・無水エタノール、無水エタノール、または、メタノールであることを特徴とする請求項1記載のイソピマル酸製造プロセス。
【請求項1】
次のステップを具備するイソピマル酸製造プロセス。
ステップ1:熱的に異性化されたロジンを1倍以上10倍以下の質量を有するアセトンに溶解させ、沈殿物を形成するために、熱的に異性化されたロジンの1%以上40%以下と同等の質量を有するイソブタノールアミンのアセトン溶液(アセトン/イソブタノールアミン=1:1mL/g)を滴下にて添加し、放置し、フィルターし、50%(v/v)エタノールで洗浄し、イソピマル酸の粗アンモニウム塩を得るために乾燥し、イソピマル酸の粗アンモニウム塩の各グループを、マルチ・再結晶化によって、5回再結晶化し、イソピマル酸のアンモニウム塩の精製された結晶を得るために乾燥する。
ステップ2:イソピマル酸のアンモニウム塩の前記精製された結晶を、1倍以上20倍以下の質量を有するエーテルに溶解させ、1wt%以上20wt%以下塩酸を、イソピマル酸のアンモニウム塩の前記結晶が消滅するまで、何回かに分けて添加し、水の層を取り除き、中性になるように、エーテルの層を洗浄し、1気圧および30℃以上50℃以下で、エーテルを蒸発させ、その残渣を、0.5倍以上5倍以下の質量を有するアセトンに溶解させ、沈殿された結晶が成長するのをやめるまで、水を、滴下にて、前記溶液にゆっくりと添加し、フィルターし、精製されたイソピマル酸を得るために乾燥させる。
【請求項2】
前記熱的に異性化されたロジンが、次の方法で製造されることを特徴とする請求項1記載のイソピマル酸製造プロセス。
イソピマル酸を含む松脂から、1気圧および90℃以上114℃以下で、少量の水を蒸発させ、その後、10kPa以上50kPa以下および140℃以上170℃以下で、テレビン油を蒸発させ、その後、1時間以上3時間以下の異性化反応を取得するために、温度を150℃以上180℃以下へと上昇させ、熱的に異性化されたロジンを得るために、フィルターし、室温へと冷却する方法。
【請求項3】
イソピマル酸を含む前記松脂が、エリオッティ松・松脂、カリバエ松・松脂、カシヤ松・松脂、または、マソン松・松脂であることを特徴とする請求項2記載のイソピマル酸製造プロセス。
【請求項4】
前記熱的に異性化されたロジンが、ロジンを150℃以上180℃以下で1時間以上3時間以下異性化し、フィルターし、室温へと冷却することで得られることを特徴とする請求項1記載のイソピマル酸製造プロセス。
【請求項5】
前記ロジンが、エリオッティ松・ロジン、カリバエ松・ロジン、カシヤ松・ロジン、または、マソン松・ロジンであることを特徴とする請求項4記載のイソピマル酸製造プロセス。
【請求項6】
ステップ1で記述したようなマルチ・再結晶化方法において使用される溶媒が、95%(v/v)エタノール、メチルアセテート、メチルアセテート・無水エタノール、無水エタノール、または、メタノールであることを特徴とする請求項1記載のイソピマル酸製造プロセス。
【図1】

【図2】
【図3】
【図4】

【図2】
【図3】
【図4】
【公表番号】特表2011−521980(P2011−521980A)
【公表日】平成23年7月28日(2011.7.28)
【国際特許分類】
【出願番号】特願2011−511964(P2011−511964)
【出願日】平成21年6月5日(2009.6.5)
【国際出願番号】PCT/CN2009/072160
【国際公開番号】WO2009/146659
【国際公開日】平成21年12月10日(2009.12.10)
【出願人】(510311241)中国林▲業▼科学研究院林▲産▼化学工▲業▼研究所 (2)
【Fターム(参考)】
【公表日】平成23年7月28日(2011.7.28)
【国際特許分類】
【出願日】平成21年6月5日(2009.6.5)
【国際出願番号】PCT/CN2009/072160
【国際公開番号】WO2009/146659
【国際公開日】平成21年12月10日(2009.12.10)
【出願人】(510311241)中国林▲業▼科学研究院林▲産▼化学工▲業▼研究所 (2)
【Fターム(参考)】
[ Back to top ]