
イソブタノールを生産するための欠失突然変異体
大腸菌(E.coli)宿主株が改変され、ここでは遺伝子adhE、ldhA、frdB、およびpflBが破壊され、かつアクロモバクター・キシロスオキシダンス(Achromobacter xylosoxidans)由来の新規なブタノールデヒドロゲナーゼ遺伝子sadBが加えられ、イソブタノール生産宿主が生産された。

【発明の詳細な説明】
【技術分野】
【0001】
関連出願の相互参照
本願は、2008年6月4日に出願された米国仮特許出願第61/058568号明細書の便益性を主張し、その開示内容はその全体が本明細書によって援用される。
【0002】
本発明は、微生物学および分子生物学の分野に関する。より詳細には、本発明は、イソブタノールに対して向上した生成能を有する腸内の欠失突然変異体について記載する。
【背景技術】
【0003】
ブタノールは、燃料添加剤として、プラスチック産業における化学原料として、さらに食品および香料産業における食品等級の抽出剤として有用な重要な産業化学物質である。毎年、100〜120億ポンドのブタノールが石油化学的手段によって生産されており、この汎用化学物質に対する需要は高まる可能性が高い。石油原料を介したイソブタノールの既知の化学合成は、高価であり、環境に優しくないが、植物由来の原料からのイソブタノールの生成であれば、温室効果ガス排出を最小限にし、当該技術分野において進歩性を示すこととなる。
【0004】
イソブタノールは、酵母発酵の間、アミノ酸、具体的にはL−バリンの不完全代謝の副生成物として生物学的に生成される。窒素源としてのL−バリンのアミン基の代謝後、得られるα−ケト酸は、Ehrlich経路を介して、脱カルボキシル化され、非常に低い収率であってもイソブタノールに還元される。例えば、ビール発酵において生成されるイソブタノールの濃度は、100万分の16未満である。
【0005】
糖類からの直接的なイソブタノールの経済的な生合成であれば、単一の生成物としてのイソブタノールの生成を目的とした、環境的な責任がある費用効果的なプロセスを示すことになる。同時係属中の共同所有される特許文献1では、グルコースのイソブタノールへの変換に必要なあらゆる酵素活性を過剰発現する株が開示され、低濃度(0.3〜10mM)でのイソブタノールの生成が示された。
【0006】
最近、非特許文献1は、イソブタノールを最大で300mMの濃度で生成する組換え大腸菌(E.coli)株の開発について記載した。この組換え大腸菌(E.coli)では、遺伝子adhE、ldhA、frdBC、fnr、ptaおよびpflB、ならびに2つの共同所有された同時係属中の特許文献1中に記載の場合と同様のイソブタノール生合成経路を担持するプラスミドが破壊された。これらのプラスミドは、アセト乳酸シンターゼ、アセトヒドロキシ酸レダクトイソメラーゼ、アセトヒドロキシ酸デヒドラターゼ、2−ケト酸デカルボキシラーゼおよびアルコールデヒドロゲナーゼを有する。Atsumiら(上記)の見解は、イソブタノール生合成経路を有する宿主細胞が、競合する炭素経路に対して重要な遺伝子が破壊される場合の改善されたイソブタノール生成を得る可能性があることを意味している。しかし、Atsumiら(上記)の宿主細胞は、改善されたイソブタノール生成を得るのに必要とされる場合よりはるかに多い数の遺伝子組換えを有するように思われる。基本的な内因性炭素経路における遺伝子組換えの数が増えると、宿主細胞代謝が低下する可能性が高まり、それにより、最終的には細胞の生産宿主としての使用に支障をきたすことになる。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】米国特許出願公開第20070092957号明細書
【0008】
【非特許文献1】Atsumi S.ら、Nature 451:86−90頁、2008年
【発明の概要】
【発明が解決しようとする課題】
【0009】
したがって、イソブタノールを生成するための内因性炭素経路における最小数の遺伝子組換えを有する宿主細胞に対して需要がある。出願人は、上述の課題を解決しており、イソブタノールの収率向上をもたらす、内因性炭素経路内の4つの遺伝子のみに破壊を有する腸内細菌宿主細胞について本明細書中に記載する。
【課題を解決するための手段】
【0010】
本発明は、イソブタノールを生成するための腸内細菌生産宿主について記載する。宿主細胞は、好ましくは、ブタノールの生成の最終ステップにおいてブタノールデヒドロゲナーゼ(二次アルコールデヒドロゲナーゼ、sadB)を用いるイソブタノール生合成経路を有し、かつ、内因性炭素経路において遺伝子組換えを有し、これにより、細胞は、次の酵素活性、すなわち、1)ピルビン酸・ギ酸リアーゼ(EC2.3.1.54)、2)フマル酸レダクターゼ酵素複合体(EC1.3.99.1)、3)アルコールデヒドロゲナーゼ(EC1.2.1.10−アセトアルデヒドデヒドロゲナーゼおよびEC1.1.1.1−アルコールデヒドロゲナーゼ)、および4)乳酸デヒドロゲナーゼ(EC1.1.1.28)の少なくとも1つを含まない状態になる。これらの酵素活性における破壊を有する腸内宿主は、イソブタノールの速度が、これらの破壊を有しない類似宿主に対して改善されることを示す。
【0011】
したがって、本発明は、ブタノールデヒドロゲナーゼ活性を有するポリペプチドをコードする少なくとも1つの遺伝子を含む、イソブタノールを生成するための腸内生産宿主を提供し、ここで宿主は、イソブタノールを生成し、かつ、以下の酵素活性、すなわち、
a)ピルビン酸・ギ酸リアーゼ(EC2.3.1.54)
b)フマル酸レダクターゼ酵素複合体(EC1.3.99.1)
c)アルコールデヒドロゲナーゼ(EC1.2.1.10/EC1.1.1.1)
d)乳酸デヒドロゲナーゼ(EC1.1.1.28)
の少なくとも1つを実質的に有しない。
【0012】
別の実施形態では、本発明は、本発明の宿主細胞が、ポリペプチドが、スコアリングマトリックスBLOSUM62、期待値カットオフ(expect cutoff)10およびワードサイズ3と、ギャップオープニングペナルティ(gap opening penalty)11およびギャップ伸長(gap extension)1とを伴うBLASTを用いると、348アミノ酸長を超える配列番号10で示されるアミノ酸配列に対して少なくとも90%の同一性を有する場合のブタノールデヒドロゲナーゼ活性を有するポリペプチドをコードする少なくとも1つの遺伝子を含むことを提供する。
【0013】
別の実施形態では、本発明は、
a)ピルビン酸塩をアセト乳酸塩に変換するためのEC番号2.2.1.69を有するアセト乳酸シンターゼをコードする少なくとも1つの遺伝子、
b)アセト乳酸塩を2,3−ジヒドロキシイソ吉草酸に変換するためのアセトヒドロキシ酸イソメロレダクターゼEC番号1.1.1.86をコードする少なくとも1つの遺伝子、
c)2,3−ジヒドロキシイソ吉草酸をα−ケトイソ吉草酸に変換するためのアセトヒドロキシ酸デヒドラターゼEC番号4.2.1.9をコードする少なくとも1つの遺伝子、
d)α−ケトイソ吉草酸をイソブチルアルデヒドに変換するための分岐鎖ケト酸デカルボキシラーゼEC番号4.1.1.72をコードする少なくとも1つの遺伝子、
e)A.キシロスオキシダンス(A.xylosoxidans)から単離される、ブタノールデヒドロゲナーゼポリペプチドをコードする少なくとも1つの遺伝子
を含むイソブタノール生合成経路を含む宿主細胞を提供する。
【0014】
別の実施形態では、本発明は、本発明の生産宿主を、イソブタノールが生成される条件下で、炭素基質を含む発酵培地中で成長させるステップを含む、イソブタノールを生成するための方法を含む。
【0015】
本発明は、本願の一部を形成する、以下の詳細な説明、図面、および添付の配列記述からより十分に理解されうる。
【図面の簡単な説明】
【0016】
【図1A】「a」、「b」、「c」、「d」、および「e」と称されるステップからなる本発明のイソブタノール生合成経路を表し、下記の生成物変換に対する基質を表す。「f」〜「i」の反応は、イソブタノール合成における可用性を低減する副反応におけるピルビン酸塩の消費を阻止するための、本開示において破壊されている特定の4つの反応を表す。
【図1B】「a」、「b」、「c」、「d」、および「e」と称されるステップからなる本発明のイソブタノール生合成経路を表し、下記の生成物変換に対する基質を表す。「f」〜「i」の反応は、イソブタノール合成における可用性を低減する副反応におけるピルビン酸塩の消費を阻止するための、本開示において破壊されている特定の4つの反応を表す。
【0017】
以下の配列は、米国特許施行規則第1.821−1.825条(「ヌクレオチド配列および/またはアミノ酸配列開示を有する特許出願の要件−配列の規則(Requirements for Patent Applications Containing Nucleotide Sequences and/or Amino Acid Sequence Disclosures−the Sequence Rules)」)に従い、世界知的所有権機関(World Intellectual Property Organization)(WIPO)基準ST.25(1998年)、ならびにEPOおよびPCTの配列表の要件(規則5.2および49.5(aの2)、ならびに実施細則の第208節および付録C)に一致する。ヌクレオチドおよびアミノ酸配列データにおいて用いられる記号および形式は、米国特許施行規則第1.822条に示される規則に従う。
【0018】
本発明のヌクレオチドおよびアミノ酸配列は、下記の表1および2中にあげられる。
【0019】
【表1】
【0020】
【表2】
【0021】
【表3】
【0022】
【表4】
【0023】
【表5】
【発明を実施するための形態】
【0024】
本開示では、抽出発酵条件下で予想外の高レベルのイソブタノール(例えば35g/L)を生成する、一連の経路因子および欠失を組み合わせた新規な生産宿主の開発について記載する。本開示では、遺伝子adhE、ldhA、frdB、およびpflBが破壊された大腸菌(E.coli)株について記載する。共同所有された米国特許出願第20070092957号明細書中に記載のpTrc99A::budB−ilvC−ilvD−kivDプラスミドは、アクロモバクター・キシロスオキシダンス(Achromobacter xylosoxidans)由来のブタノールデヒドロゲナーゼsadBの付加によって修飾され、イソブタノール生産宿主が生成された。本開示は、複数の商業的および産業的需要を満たす。ブタノールは、種々の用途を有する重要な産業用商品の化学物質であり、その場合、燃料または燃料添加剤としてのその可能性は特に有意である。ブタノールは、4炭素アルコールにすぎなくても、ガソリンの含量に類似のエネルギー含量を有し、任意の化石燃料と混和されうる。ブタノールは、標準の内燃エンジン内で燃焼される場合にCO2のみを生成し、SOXまたはNOXをほとんど生成しないことから燃料または燃料添加剤として好まれる。さらにブタノールは、今日まで最も好ましい燃料添加剤であるエタノールよりも腐食性が少ない。
【0025】
さらに、本開示では、植物由来の炭素源からのイソブタノールの生成について記載し、ブタノール生成のための標準の石油化学プロセスに関連した負の環境影響を回避する。
【0026】
次の定義および略語は、特許請求の範囲および本明細書の解釈を意図して使用されるべきである。
【0027】
本明細書で使用される用語「comprises」、「comprising」、「includes」、「including」、「has」、「having」、「contains」または「containing」、または任意の他のこれらの変形は、非排他的な包含(non−exclusive inclusion)を網羅するように意図される。たとえば、要素のリストを含む組成物、混合物、プロセス、方法、物品、または装置は、必ずしもそれらの要素のみに限定されないが、明示的に列挙されないかまたはかかる組成物、混合物、プロセス、方法、物品、または装置に固有の他の要素を含みうる。さらに、相反する明示的な記載がない限り、「または」は、「包含的なまたは(inclusive or)」を示し、「限定的なまたは(exclusive or)」を示さない。たとえば、条件AまたはBは、Aは真であり(または存在し)かつBは偽である(存在しない)、Aは偽であり(存在せず)かつBは真である(または存在する)、およびAとBの双方が真である(または存在する)のいずれか1つによって満足される。
【0028】
また、本発明の要素または成分に先行する不定冠詞「a」および「an」は、要素または成分の事例(すなわち出現)の数について非制限的であることが意図される。したがって、「a」または「an」は、1つまたは少なくとも1つを含むように解釈されるべきであり、要素または成分の単数の語形はまた、数が明らかに単数であることを意味しない限り、複数を含む。
【0029】
用いられる本発明の成分または反応物の量を修飾する、本明細書で使用される用語「約」は、たとえば、実地での濃縮物の作製または溶液の使用に用いられる典型的な測定および液体処理方法を通じて;これらの方法における不測の誤りを通じて;組成物を作製するかまたはこれらの方法を実施するために使用される成分の製造、供給源、または純度における差異を通じて生じうる数量における変動を示す。用語「約」はまた、特定の初期混合物から生じる組成物に対する、異なる平衡条件によって異なる量を包含する。用語「約」によって修飾されるか否かに無関係に、特許請求の範囲は、量に対する等価物を含む。一実施形態では、用語「約」は、報告される数値の10%以内、好ましくは報告される数値の5%以内を意味する。
【0030】
用語「発明」または「本発明」は、本明細書で使用される場合、非限定的用語であり、特定の発明の任意の単一の実施形態を示すように意図されていないが、本明細書および特許請求の範囲において記載される、考えられるあらゆる実施形態を包含する。
【0031】
用語「イソブタノール生合成経路」は、イソブタノールを生成するための酵素経路を示す。典型的なイソブタノール生合成経路は、共同所有された同時係属中の米国特許出願公開第20070092957A1号明細書(その全体が参照により本明細書中に援用される)中で考察され、説明されている。
【0032】
用語「ノックアウト」は、その特定の遺伝子を機能不全にするための、プラスミドまたは微生物内での特定の遺伝子の破壊を示す。本開示では、遺伝子adhE、ldhA、frdB、およびpflBは、イソブタノール生成のため、宿主株においてノックアウトされた。
【0033】
用語「pflB」は、ピルビン酸塩をギ酸塩に変換するピルビン酸・ギ酸リアーゼ酵素をコードする遺伝子を示す。
【0034】
用語「frdABCD」は、コハク酸塩をフマル酸塩に変換するフマル酸レダクターゼ酵素複合体をコードするオペロンを示す。
【0035】
用語「ldhA」は、乳酸デヒドロゲナーゼ酵素をコードする遺伝子を示し、ピルビン酸塩を乳酸塩に変換する。
【0036】
用語「adhE」は、アセチル−CoAをエタノールに変換するピルビン酸・ギ酸リアーゼ酵素をコードする遺伝子を示す。
【0037】
用語「アセト乳酸シンターゼ」および「アセト乳酸シンテターゼ」は、ピルビン酸塩のアセト乳酸塩およびCO2への変換を触媒する酵素を示すように、本明細書中で交換可能に使用される。好ましいアセト乳酸シンターゼは、EC番号2.2.1.69(Enzyme Nomenclature 1992年、Academic Press,San Diego)により既知である。これらの酵素は、限定はされないが、枯草菌(Bacillus subtilis)(GenBank番号:それぞれ、CAB15618(配列番号11)、Z99122(配列番号12)、NCBI(National Center for Biotechnology Information)アミノ酸配列、NCBIヌクレオチド配列)、クレブシエラ・ニューモニエ(Klebsiella pneumoniae)(GenBank番号:AAA25079(配列番号2)、M73842(配列番号1)、およびラクトコッカス・ラクティス(Lactococcus lactis)(GenBank番号:AAA25161(配列番号13)、L16975(配列番号14)を含む複数の供給源から入手可能である。
【0038】
用語「アセトヒドロキシ酸イソメロレダクターゼ」および「アセトヒドロキシ酸レダクトイソメラーゼ」は、電子供与体としてNADPH(還元型ニコチンアミドアデニンジヌクレオチドリン酸)を使用する、アセト乳酸塩の2,3−ジヒドロキシイソ吉草酸への変換を触媒する酵素を示すように、本明細書中で交換可能に使用される。好ましいアセトヒドロキシ酸イソメロレダクターゼは、EC番号1.1.1.86により既知であり、配列は、限定はされないが、大腸菌(Escherichia coli)(GenBank番号:NP_418222(配列番号4)、NC_000913(配列番号3))、サッカロミセス・セレビシエ(Saccharomyces cerevisiae)(GenBank番号:NP_013459(配列番号15)、NC_001144(配列番号16))、メタノコッカス・マリパルディス(Methanococcus maripaludis)(GenBank番号:CAF30210(配列番号17)、BX957220(配列番号18))、および枯草菌(Bacillus subtilis)(GenBank番号:CAB14789(配列番号19)、Z99118(配列番号20))を含む非常に多数の微生物から入手可能である。
【0039】
用語「アセトヒドロキシ酸デヒドラターゼ」は、2,3−ジヒドロキシイソ吉草酸のα−ケトイソ吉草酸への変換を触媒する酵素を示す。好ましいアセトヒドロキシ酸デヒドラターゼは、EC番号4.2.1.9により既知である。これらの酵素は、限定はされないが、大腸菌(E.coli)(GenBank番号:YP_026248(配列番号6)、NC_000913(配列番号5))、S.セレビシアエ(S.cerevisiae)(GenBank番号:NP_012550(配列番号21)、NC_001142(配列番号22))、M.マリパルディス(M.maripaludis)(GenBank番号:CAF29874(配列番号23)、BX957219(配列番号24))、および枯草菌(B.subtilis)(GenBank番号:CAB14105(配列番号25)、Z99115(配列番号26))を含む非常に多数の微生物から入手可能である。
【0040】
用語「分岐鎖α−ケト酸デカルボキシラーゼ」は、α−ケトイソ吉草酸のイソブチルアルデヒドおよびCO2への変換を触媒する酵素を示す。好ましい分岐鎖α−ケト酸デカルボキシラーゼは、EC番号4.1.1.72により既知であり、限定はされないが、ラクトコッカス・ラクティス(Lactococcus lactis)(GenBank番号:AAS49166(配列番号27)、AY548760(配列番号28);CAG34226(配列番号8)、AJ746364(配列番号29))、ネズミチフス菌(Salmonella typhimurium)(インドールピルビン酸デカルボキシラーゼとしても既知)(GenBank番号:NP_461346(配列番号30)、NC_003197(配列番号31))、およびクロストリジウム・アセトブチリクム(Clostridium acetobutylicum)(ピルビン酸デカルボキシラーゼとしても既知)(GenBank番号:NP_149189(配列番号32)、NC_001988(配列番号33))を含む非常に多数の微生物から入手可能である。
【0041】
用語「ブタノールデヒドロゲナーゼ」および「二次アルコールデヒドロゲナーゼ」は、ここでは交換可能に使用され、かつ多数の微生物内で生じ、NAD+のNADHへの還元とともにアルコールとアルデヒドまたはケトンとの間の相互変換を促進する酵素を示す。かかる酵素の好ましい例は、アクロモバクター・キシロスオキシダンス(Achromobacter xylosoxidans)由来のブタノールデヒドロゲナーゼである(ヌクレオチド配列番号9およびアミノ酸配列番号10)。A.キシロスオキシダンス(A.xylosoxidans)sadB酵素は、イソブチルアルデヒドのイソブタノールへの変換を触媒する。
【0042】
用語「炭素基質」または「発酵性炭素基質」は、本発明の宿主微生物によって代謝可能な炭素源、特に、単糖、例えばグルコースまたはフルクトース;二糖、例えば乳糖またはスクロース;オリゴ糖;多糖、例えばデンプンまたはセルロース;1つの炭素基質;およびそれらの混合物からなる群から選択される炭素源を示す。
【0043】
「遺伝子」という用語は、場合によりコード配列の上流(5’非コード配列)および下流(3’非コード配列)の調節配列を含む、特定のタンパク質として発現可能な核酸断片を示す。「天然遺伝子」は、それ自体の調節配列を有する天然に見出される遺伝子を示す。「キメラ遺伝子」は、天然遺伝子でない任意の遺伝子を示し、天然に見出されることのない調節およびコード配列を含む。したがって、キメラ遺伝子は、異なる供給源に由来する調節配列およびコード配列または同じ供給源に由来する調節配列およびコード配列を含みうるが、天然に見出されるものとは異なる様式で配列されている。「内因性遺伝子」は、微生物のゲノム内のその天然の位置における天然遺伝子を示す。「外来遺伝子」または「異種遺伝子」は、通常は宿主微生物内に見出されることはないが宿主微生物に遺伝子導入によって導入される遺伝子を示す。外来遺伝子は、非天然の微生物に導入される天然遺伝子またはキメラ遺伝子を含みうる。「トランス遺伝子」は、形質転換手順によりゲノムに導入されている遺伝子である。
【0044】
本明細書で用いられる「単離核酸断片」または「単離核酸分子」または「遺伝的構造物」は同義に用いられ、かつ一本鎖または二本鎖のRNAまたはDNAの高分子を意味し、場合により合成、非天然または改変ヌクレオチド塩基を有することになる。DNAの高分子の形態の単離核酸断片については、cDNA、ゲノムDNAまたは合成DNAのうちの1つもしくは複数のセグメントから構成されうる。
【0045】
本明細書で使用される用語「コード配列」は、特定のアミノ酸配列をコードするDNA配列を示す。「好適な調節配列」は、コード配列の上流(5’非コード配列)、内部、または下流(3’非コード配列)に位置し、関連コード配列の転写、RNAプロセシングまたは安定性、または翻訳に影響を与えるヌクレオチド配列を示す。調節配列は、プロモーター、翻訳リーダー配列、イントロン、ポリアデニル化認識配列、RNAプロセシング部位、エフェクター結合部位およびステムループ構造を含んでもよい。
【0046】
用語「プロモーター」は、コード配列または機能RNAの発現を制御することが可能なDNA配列を示す。一般に、コード配列は、プロモーター配列の3’側に位置する。プロモーターは、その全体において天然遺伝子に由来するか、または天然に見出される異なるプロモーターに由来する異なる要素から構成されるか、または合成DNAセグメントをさらに含む場合がある。異なるプロモーターが、異なる組織または細胞種において、または発現の異なる段階で、または異なる環境的または生理的状態に応答して、遺伝子の発現を誘導することが可能であることは当業者によって理解されている。ほとんどの時期に大部分の細胞種において遺伝子の発現を引き起こすプロモーターは、一般に「構成的プロモーター」と称される。ほとんどの場合、調節配列の正確な境界が完全には限定されていないことから、異なる長さのDNA断片が同一のプロモーター活性を有しうることはさらに理解されている。
【0047】
用語「発現」は、本明細書で使用される場合、本発明の核酸断片に由来するセンス(mRNA)またはアンチセンスRNAの転写および安定な蓄積を示す。発現はまた、mRNAのポリペプチドへの翻訳を示しうる。
【0048】
本明細書で使用される用語「形質転換」は、遺伝的に安定な遺伝的形質をもたらす、核酸断片の宿主微生物への転移を示す。形質転換核酸断片を有する宿主微生物は、「トランスジェニック」または「組換え」または「形質転換」微生物と称される。
【0049】
用語「プラスミド」は、細胞の中央代謝の一部でない遺伝子を有することが多く、通常は環状二本鎖DNA分子の形態をなす余分な染色体要素を示す。かかる要素は、任意の供給源に由来する線状または環状の一本鎖または二本鎖のDNAまたはRNAにおける自動複製配列、ゲノム組み込み(integrating)配列、ファージまたはヌクレオチド配列である場合があり、ここでは複数のヌクレオチド配列が、発現カセットを細胞に導入可能な固有の作成物に連結されるかまたは組換えられており、ここで前記発現カセットは、選択される遺伝子のコード配列と、選択される遺伝子産物の発現にとって必要とされるコード配列に先行する調節配列(5’非コード配列)および後続する調節配列(3’非コード配列)とを含む。
【0050】
用語「コドン最適化された」は、さまざまな宿主の形質転換における核酸分子の遺伝子またはコード領域を示す場合、DNAによってコードされるポリペプチドを改変することなく宿主微生物の典型的なコドン使用を反映させるための、核酸分子の遺伝子またはコード領域におけるコドンの改変を示す。
【0051】
本明細書で使用される用語「形質導入」および「普遍形質導入」は、交換可能に使用され、細菌DNAが、細菌DNAを有するファージ粒子により、ある細菌細胞(ドナ−)から別の(レシピエント)へ転移される現象を示す。
【0052】
用語「P1ドナー細胞」および「ドナー細胞」は、交換可能に使用され、バクテリオファージまたはウイルスによって感染を受けやすく、また形質導入粒子にパッケージ化される核酸断片における供給源としての役割を果たす細菌株を示す。典型的には、ドナー細胞の遺伝子構成(genetic make up)は、ドナー細胞によって生成される形質導入ファージまたはウイルスを含有するP1溶解物を受け取る役割を果たす「レシピエント細胞」と類似または同一である。
【0053】
用語「P1レシピエント細胞」および「レシピエント細胞」は、交換可能に使用され、バクテリオファージまたはウイルスによって感染を受けやすく、またドナー細胞によって生成される形質導入ファージまたはウイルスを含有する溶解物を受け取る役割を果たす細菌株を示す。
【0054】
用語「カオトロピック剤」は、タンパク質、DNA、またはRNAなどの高分子中の三次元構造を破壊する物質を意味する。
【0055】
用語「共沸剤」は、組成が単なる蒸留によって変化を受ける可能性がないような比での2つ以上の純粋な化学物質の混合物を示す。
【0056】
用語「パーベーパレイション」は、無孔質膜または多孔質膜を通る部分的蒸発による、液体の混合物を分離するための方法を示す。
【0057】
用語「親水性」は、水素結合を介して水(H2O)と一時的に結合可能な分子の物理的特性を示す。
【0058】
用語「実質的に有しない」は、本イソブタノール経路と競合する炭素経路内での酵素活性(例えば、ピルビン酸・ギ酸リアーゼ、フマル酸レダクターゼ、アルコールデヒドロゲナーゼおよび乳酸デヒドロゲナーゼ)の存在または不在に関連して使用される場合、酵素のレベルが、実質的に野生型宿主内での同じ酵素のレベル未満であること(野生型レベルの50%未満が好ましく、かつ野生型レベルの約90%未満が最も好ましい場合)を意味する。
【0059】
当該技術分野で既知のように、用語「同一性(%)」は、配列の比較による判定からの、2つ以上のポリペプチド配列または2つ以上のポリヌクレオチド配列の間の関係性である。当該技術分野では、「同一性」は、場合によっては、かかる配列の文字列の間の一致による判定からの、ポリペプチドまたはポリヌクレオチド配列の間の配列の関連性の程度も意味する。「同一性」および「類似性」は、限定はされないが、1)Computational Molecular Biology(Lesk A.M.編) Oxford University Press:NY(1988年);2)Biocomputing:Informatics and Genome Projects(Smith D.W.編) Academic Press:NY(1993年);3)Computer Analysis of Sequence Data、Part I(Griffin A.M.およびGriffin H.G.編) Humania Press:NJ(1994年);4)Sequence Analysis in Molecular Biology(von Heinje G.編) Academic Press(1987年);ならびに5)Sequence Analysis Primer(Gribskov M.およびDevereux J.編) Stockton:NY(1991年)に記載の方法を含む既知の方法により容易に計算されうる。
【0060】
同一性を判定するための好ましい方法は、試験される配列間に最適な一致を与えるように設計される。同一性および類似性を判定するため方法は、公的に利用可能なコンピュータープログラム内にコード化される。配列アラインメントおよび同一性パーセントの計算は、LASERGENE bioinformatics computing suiteのMegAlign(商標)プログラム(DNASTAR Inc.,Madison,WI)を用いて行われうる。配列の複数のアラインメントは、HigginsおよびSharp(CABIOS.5:151−153頁、1989年);およびHiggins D.G.ら、(Comput.Appl.Biosci.、8:189−191頁、1992年)によって記載された、Clustal Vと称されるアラインメント法に対応する「アラインメントのClustal V法」を含む数種類のアルゴリズムを包含する「アラインメントのClustal法」を用いて行われ、かつLASERGENE bioinformatics computing suiteのMegAlign(商標)プログラム(DNASTAR Inc.)において見出される。複数のアラインメントにおいては、デフォルト値はギャップペナルティ=10およびギャップ長ペナルティ=10に対応する。Clustal法を用いる、タンパク質配列のペアワイズアラインメントおよび同一性パーセントの計算におけるデフォルトパラメータは、KTUPLE=1、ギャップペナルティ=3、ウインドウ(WINDOW)=5およびダイアゴナルズセイブド(DIAGONALS SAVED)=5である。核酸においては、これらのパラメータは、KTUPLE=2、ギャップペナルティ=5、ウインドウ=4およびダイアゴナルズセイブド=4である。Clustal Vプログラムを用いての配列のアラインメント後、同じプログラム内の「配列距離(sequence distances)」表を見ることにより「同一性パーセント」を得ることが可能である。さらに、「Clustal Wのアラインメント法」が利用可能あり、HigginsおよびSharp(上記);Higgins D.G.ら、(上記)によって記載されたClustal Wと称されるアラインメント法に対応し、LASERGENE bioinformatics computing suiteのMegAlign(商標)v6.1プログラム(DNASTAR Inc.)において見出される。複数のアラインメントにおけるデフォルトパラメータ(ギャップペナルティ=10、ギャップ長ペナルティ=0.2、ディレイ・ディバージェン配列(Delay Divergen Seqs)(%)=30、DNAトランジション・ウェイト(Transition Weight)=0.5、タンパク質重み行列(Protein Weight Matrix)=Gonnetシリーズ(Series)、DNA重み行列(Weight Matrix)=IUB)。Clustal Wプログラムを用いての配列のアラインメント後、同じプログラム内の「配列距離」表を見ることにより「同一性パーセント」を得ることが可能である。
【0061】
配列同一性の多数のレベルが、同一または類似の機能または活性を有するポリペプチドを他の種から同定するのに有用であることが当業者により十分に理解されている。パーセント同一性の有用な例として、限定はされないが、24%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、または95%があげられるか、または24%〜100%の任意の整数の百分率、例えば、25%、26%、27%、28%、29%、30%、31%、32%、33%、34%、35%、36%、37%、38%、39%、40%、41%、42%、43%、44%、45%、46%、47%、48%、49%、50%、51%、52%、53%、54%、55%、56%、57%、58%、59%、60%、61%、62%、63%、64%、65%、66%、67%、68%、69%、70%、71%、72%、73%、74%、75%、76%、77%、78%、79%、80%、81%、82%、83%、84%、85%、86%、87%、88%、89%、90%、91%、92%、93%、94%、95%、96%、97%、98%または99%は、本発明の記載において有用でありうる。好適な核酸断片は、上記の同一性を有するだけでなく、典型的には、少なくとも50アミノ酸、好ましくは少なくとも100アミノ酸、より好ましくは少なくとも150アミノ酸、さらにより好ましくは少なくとも200アミノ酸、および最も好ましくは少なくとも250アミノ酸を有するポリペプチドをコードする。
【0062】
「配列分析ソフトウェア」という用語は、ヌクレオチドまたはアミノ酸配列の分析にとって有用な任意のコンピュータアルゴリズムまたはソフトウェアプログラムを示す。「配列分析ソフトウェア」は商業的に利用可能であるかまたは個別に開発される場合がある。典型的な配列分析ソフトウェアは、限定はされないが、1)プログラムのGCGスイート(Wisconsin Packageバージョン9.0、Genetics Computer Group(GCG),Madison,WI);2)BLASTP、BLASTN、BLASTX(Altschulら、J.Mol.Biol.、215:403−410頁(1990年));3)DNASTAR(DNASTAR Inc.,Madison,WI);4)Sequencher(Gene Codes Corporation,Ann Arbor,MI);および5)Smith−Watermanアルゴリズム(W.R.Pearson、Comput.Meth.Gen.Res.、[Proc.Int.Symp.]、会合日1992年、111−120頁、1994年、Suhai編、Sandor.Plenum Press,New York,NY)を組み入れたFASTAプログラムを含むことになる。本願の文脈の中で、配列分析ソフトウェアが分析に用いられる場合、分析の結果が、他に規定がない限り、参照されるプログラムの「デフォルト値」に基づくことが理解されるであろう。本明細書で用いられる「デフォルト値」は、最初に初期化される際に最初にソフトウェアが読み込む任意の値またはパラメータのセットを意味することになる。
【0063】
核酸断片については、ある一本鎖形態の核酸断片が温度および溶液のイオン強度の適切な条件下で他方の核酸断片にアニール可能である場合、別の核酸断片、例えばcDNA、ゲノムDNA、またはRNA分子に対して「ハイブリダイズ可能」である。ハイブリダイゼーションおよび洗浄の条件については周知であり、Sambrook J.、Fritsch E.F.およびManiatis T.Molecular Cloning:A Laboratory Manual、第2版、Cold Spring Harbor Laboratory(Cold Spring Harbor,NY)(1989年)、特に第11章およびその中の表11.1(全体として参照により本明細書中に援用される)に例示されている。温度およびイオン強度の条件はハイブリダイゼーションの「ストリンジェンシー」を決定する。ストリンジェンシー条件は、中程度に類似の断片(遠縁の微生物由来の相同配列など)から高度に類似の断片(近縁の微生物由来の機能酵素を複製する遺伝子など)にかけてスクリーニングするように調整されうる。
【0064】
ハイブリダイゼーション後の洗浄がストリンジェンシー条件を決定する。1つの好ましい条件のセットでは、室温で15分間の6×SSC、0.5% SDSの場合から開始し、次いで45℃で30分間の2×SSC、0.5% SDSの場合を繰り返し、次いで50℃で30分間の0.2×SSC、0.5% SDSの場合を2回繰り返すという一連の洗浄が用いられる。より好ましいストリンジェントな条件のセットではより高温が用いられ、ここでの洗浄は終わりの2回の0.2×SSC、0.5% SDSにおける30分の洗浄で温度が60℃に高められた点を除いて上記の洗浄と同一である。別の好ましい高度にストリンジェントな条件のセットでは、65℃での0.1×SSC、0.1% SDSにおける2回の最終の洗浄が用いられる。さらなるストリンジェントな条件のセットでは、例えば0.1×SSC、0.1% SDS、65℃でのハイブリダイゼーションおよび2×SSC、0.1% SDSとそれに続く0.1×SSC、0.1% SDSの場合での洗浄が含まれる。
【0065】
ハイブリダイゼーションでは、塩基間の不一致がハイブリダイゼーションのストリンジェンシーによりありうるとしても2つの核酸が相補配列を有することが必要である。核酸をハイブリダイズするのに適するストリンジェンシーは、当該技術分野での周知の変数である核酸の長さおよび相補性の程度に依存している。2つのヌクレオチド配列間の類似性または相同性の程度が大きくなると、それらの配列を有する核酸のハイブリッドにおけるTmの値が増加する。核酸ハイブリダイゼーションの(より高いTmに対応する)相対的安定性は、以下の順、すなわちRNA:RNA、DNA:RNA、DNA:DNAで低下する。100ヌクレオチド長を超えるハイブリッドにおいては、Tmを計算するための方程式が導かれている(Sambrookら、上記、9.50〜9.51を参照)。より短い核酸すなわちオリゴヌクレオチドの場合のハイブリダイゼーションにおいては、不一致の位置がより重要になり、オリゴヌクレオチド長がその特異性を決定する(Sambrookら、上記、11.7〜11.8を参照)。一実施形態では、ハイブリダイズ可能な核酸における長さは少なくとも約10ヌクレオチド長である。好ましくは、ハイブリダイズ可能な核酸における最小の長さは少なくとも約15ヌクレオチド長、より好ましくは少なくとも約20ヌクレオチド長、および最も好ましくは長さは少なくとも約30ヌクレオチド長である。さらに、当業者は、温度および洗浄溶液の塩濃度がプローブ長などの要素による必要性に応じて調整可能であることを理解するであろう。
【0066】
アミノ酸またはヌクレオチド配列の「大部分」は、ポリペプチドのアミノ酸配列または遺伝子のヌクレオチド配列を、当業者による配列の人手による評価、またはBLAST(Altschul S.F.ら、上記)などのアルゴリズムを使用するコンピュータで自動化された配列比較および同定のいずれかにより、ポリペプチドまたは遺伝子を推定的に同定する程度に十分に含む部分である。一般に、ポリペプチドまたは核酸配列を公知のタンパク質または遺伝子に対して相同なものとして推定的に同定するため、10個以上の隣接アミノ酸または30個以上のヌクレオチドの配列が必要である。さらに、ヌクレオチド配列に関しては、20〜30個の隣接ヌクレオチドを含む遺伝子に特異的なオリゴヌクレオチドプローブは、遺伝子同定(たとえば、サザンハイブリダイゼーション)および単離(たとえば、細菌コロニーまたはバクテリオファージプラークのインサイチュハイブリダイゼーション)の配列に依存した方法において使用することが可能である。さらに、12〜15塩基の短いオリゴヌクレオチドは、プライマーを含む特定の核酸断片を得るため、PCRにおける増幅プライマーとして使用することが可能である。したがって、ヌクレオチド配列の「大部分」は、配列を、配列を含む核酸断片を特異的に同定および/または単離する程度に十分に含む。本明細書は、特定の真菌タンパク質をコードする完全なアミノ酸およびヌクレオチド配列について教示する。本明細書で報告されるような配列の利点を有する当業者は、当業者に公知の目的のため、開示される配列のすべてまたは大部分をここで使用することができる。したがって、本発明は、添付の配列表において報告されるような完全な配列、ならびに上で定義されるような配列の大部分を含む。
【0067】
ここで用いられる標準の組換えDNAおよび分子クローニング技術は、当該技術分野で周知であり、Sambrook J.、Fritsch E.F.およびManiatis T.、Molecular Cloning:A Laboratory Manual、第2版、Cold Spring Harbor Laboratory Press,Cold Spring Harbor,NY(1989年)(以後Maniatis);Silhavy T.J.、Bennan M.L.、およびEnquist L.W.、Experiments with Gene Fusions、Cold Spring Harbor Laboratory Press,Cold Spring Harbor,NY(1984年);ならびにAusubel F.M.ら、Current Protocols in Molecular Biology、Greene Publishing Assoc. and Wiley−Interscience発行(1987年)に記載されている。
【0068】
本発明は、二次アルコールデヒドロゲナーゼ活性を有するポリペプチドをコードする少なくとも1つの遺伝子を含む、イソブタノールを生成するための腸内生産宿主を提供し、ここで宿主は、イソブタノールを生成し、かつ、次の酵素活性、すなわちピルビン酸・ギ酸リアーゼ、フマル酸レダクターゼ酵素複合体、アルコールデヒドロゲナーゼおよび乳酸デヒドロゲナーゼ(図1A、反応「f」、「g」、「h」、「i」を参照)のうちの少なくとも1つを実質的に有しない。生産宿主の二次アルコールデヒドロゲナーゼは、イソブチルアルデヒドのイソブタノールへの変換において特に有効である。
【0069】
(IdhAによってコードされる)乳酸デヒドロゲナーゼの欠失は、乳酸塩の生成におけるピルビン酸塩の変換(図1A、反応「f」)を阻止する。
【0070】
イソブタノールを生成するための微生物宿主
イソブタノールの生成のために選択される微生物宿主は、炭水化物をイソブタノールに変換できる必要がある。好適な微生物宿主の選択における基準は、次のもの、すなわち高率のグルコース利用、遺伝子操作のための遺伝的ツールの利用可能性、および安定な染色体改変をもたらす能力を含む。
【0071】
大部分の微生物は、炭水化物を利用することができる。しかし、特定の環境微生物は、炭水化物を高効率で利用することができず、それ故、好適な宿主とはならない。
【0072】
宿主を遺伝的に修飾する能力は、任意の組換え微生物の生成にとって必須である。遺伝子導入技術の様式は、エレクトロポレーション、複合、形質導入または天然形質転換によるものであってもよく、当該技術分野で周知である。広範囲宿主の複合プラスミドおよび薬剤耐性マーカーが使用可能である。クローニングベクターは、宿主微生物に対し、その宿主内で機能可能な抗生物質耐性マーカーの性質に基づいて適合され、当該技術分野で周知である。
【0073】
微生物宿主はまた、本明細書中の下記におけるさまざまな遺伝子を欠失させることによって炭素流における競合経路を不活性化するように操作してもよい。上記の基準に基づき、好ましい宿主は、エシェリキア(Escherichia)属、サルモネラ(Salmonella)属、クレブシエラ(Klebsiella)属、セラチア(Serratia)属、エルウィニア(Erwinia)属およびシゲラ(Shigella)属のさまざまな種を含む。
【0074】
また、微生物に対するイソブタノールの毒性により、宿主株を同定または改変して、それがイソブタノールに対してより耐性を示すのであれば望ましいことになる。かかる耐性宿主の選択については、同時係属中の共同所有された米国特許出願公開第20070259411号明細書中で開示されている。
【0075】
イソブタノールを蓄積するためのノックアウト突然変異体の生成
糖基質を代謝する微生物は、混合酸発酵において種々の副生成物を生成する(Moat A.G.ら、Microbial Physiology、第4版、John Wiley Publishers,N.Y.、2002年)。混合酸発酵の典型的な生成物は、ギ酸、乳酸およびコハク酸などの酸ならびにエタノールである。イソブタノール発酵中におけるこれらの副生成物の形成は、イソブタノールの潜在的収率を低下させうる。イソブタノールの収率低下を阻止するため、副生成物の形成に対応する酵素活性は低下しうる。副生成物の形成に関与する酵素は、限定はされないが、次のものを含む。1)ピルビン酸塩をギ酸塩およびアセチル−補酵素Aに代謝する、pflB遺伝子によってコードされるピルビン酸・ギ酸リアーゼ(EC2.3.1.54)(アミノ酸配列番号46;DNA配列番号47)。この酵素の欠失は、ピルビン酸塩がギ酸塩およびアセチル−CoAを形成する場合の競合を除外する(図1A、反応「g」);2)フマル酸塩のコハク酸塩への還元を触媒し、NADHを必要とする、frdABCDオペロンによってコードされるフマル酸レダクターゼ酵素複合体(EC1.3.99.1);FrdA(アミノ酸配列番号54;DNA配列番号55)サブユニットは、共有結合されたフラビンアデニンジヌクレオチドを有する;FrdBは酵素(アミノ酸配列番号48;DNA配列番号49)の鉄−硫黄中心を有する;FrdC(アミノ酸配列番号56;DNA配列番号57)およびFrdD(アミノ酸配列番号58;DNA配列番号59)は、触媒FrdABドメインを細胞質膜に結合させるインテグラル(integral)膜タンパク質である。フマル酸レダクターゼの機能は、frdA、B、C、またはDのサブユニットの任意の1つの欠失によって除外される場合があり、ここではfrdBの欠失が好ましい。この活性の欠失は、ピルビン酸塩におけるそのフマル酸塩への変換に対する誘引(draw)を除外する(図1A、反応「i」);3)2段階反応(両反応はadhEによって触媒され、両反応はNADHを必要とする)でエタノールをアセチル−CoAから合成する(図1A、反応「h」)、adhE遺伝子によってコードされるアルコールデヒドロゲナーゼ(EC1.2.1.10−アセトアルデヒドデヒドロゲナーゼおよびEC1.1.1.1−アルコールデヒドロゲナーゼ)(アミノ酸配列番号52;DNA配列番号53);ならびに
4)NADHの酸化とともにピルビン酸塩を乳酸塩に還元する、ldhA遺伝子によってコードされる乳酸デヒドロゲナーゼ(EC1.1.1.28)(アミノ酸配列番号50;DNA配列番号51)。この酵素の欠失は、この酵素によるピルビン酸塩に対する競合を除外し、そのギ酸塩およびアセチル−CoAへの変換を遮断する(図1A、反応「g」)。遺伝子突然変異をもたらすための方法は、一般的であり、当該技術分野で周知であり、(ピルビン酸・ギ酸リアーゼをコードする)pflB、(フマル酸レダクターゼのサブユニットをコードする)frdB、(乳酸デヒドロゲナーゼをコードする)ldhAおよび(アルコールデヒドロゲナーゼをコードする)adhEの欠損突然変異体を生成する実験に適用してもよい。Miller J.H.(1992年、A Short Course in Bacterial Genetics. Cold Spring Harbor Press,Plainview,NY.)にレビューされた、一般に用いられるランダム遺伝子組換え(random genetic modification)法は、自発的突然変異誘発、ミューテーター遺伝子によって引き起こされる突然変異誘発、化学的突然変異誘発、UVまたはX線の照射、およびトランスポゾン挿入を含む。トランスポゾンは、
1.ファージ媒介性形質導入(両方の種に特異的でかつ異種間の場合に利用されている)
2.複合(同一種または異種のメンバー間でありうる)
3.形質転換(化学的に補助され、電気的ショックに媒介されるDNAの取り込みが利用可能)
を含む種々の方法で細菌に導入されている。
【0076】
これらの方法では、トランスポゾンは、レシピエントにおいて、移入DNAからレシピエントゲノムへの遺伝子ホッピングを触媒するトランスポーゼースを発現する。トランスポゾンDNAは、裸でファージまたはプラスミド核酸内に取り込まれるか、またはトランスポーゼースと複合されうる。ほとんどの場合、トランスポゾンを有する移入DNAの複製および/または維持は阻止されることから、トランスポゾンに対するマーカー(たいていは抗生物質耐性)における遺伝子選択は、各組換えが移入DNA分子からレシピエントゲノムへのトランスポゾンの移動の結果であることを保証する。他の方法は、染色体DNA、その1つ以上の断片の場合にインビトロで転移がなされ、次いで生成されている新規な挿入対立遺伝子がレシピエント細胞に導入される(この場合、それは相同組換えによって内在性(resident)対立遺伝子を置換する)という方法である。トランスポゾン挿入は、多くの誘導方法を介して、KlecknerおよびBotstein(J.Mol.Biol.、116:125−159頁、1977年)に記載または上記のように行ってもよい。
【0077】
pflB、frdB、ldhA、adhE遺伝子の欠失はまた、細菌染色体において直接作成してもよい。改変された染色体セグメントは、腸内細菌標的宿主染色体の内因性遺伝子の部位に挿入され、内因性領域を置換する。改変された染色体セグメントの挿入は、当業者に既知の任意の方法、例えばファージ形質導入、複合、あるいはプラスミド導入または非プラスミド二本鎖もしくは一本鎖DNA導入とその後の相同組換えによって行ってもよい。バクテリオファージ形質導入では、形質導入のための標準の遺伝学的方法が用いられ、それは当該技術分野で周知であり、Miller J.H.(Experiments in Molecular Genetics、Cold Spring Harbor Laboratory Press,Cold Spring Harbor,N.Y.、1972年)で記載されている。細菌染色体内で作成されている改変された染色体セグメントは、ファージ内にパーケージ化され、次いでファージ感染を通じて標的宿主細胞に導入され、その後、相同組換えにより、改変された染色体セグメントが標的宿主細胞染色体に挿入される。
【0078】
DNA断片は、改変された染色体セグメントを担持する細菌染色体から、細菌染色体内でこの染色体セグメントに天然に隣接する配列を含む方法によって調製し、相同組換えが生じることになる場合の配列を提供してもよい。隣接する相同配列は、Lloyd R.G.およびK.B.Low(Homologous recombination、2236−2255頁;F.C.Neidhardt編、Escherichia coli and Salmonella:Cellular and Molecular Biology、1996年、ASM Press,Washington,DC)に記載のように、相同組換えを支持するのに十分である。典型的には、相同組換えに使用される相同配列は、1kb長を超えるが、せいぜい50もしくは100塩基対程度であってもよい。改変された染色体セグメントおよび隣接する相同配列を有するDNA断片は、例えば制限消化により、または超音波処理など、ランダムな末端を生成する方法を用いて、画定された末端を用いて調製してもよい。いずれの場合においても、改変された染色体セグメントを有するDNA断片は、例えばエレクトロポレーション、凍結−解凍法を含む任意のDNA取り込み法により、または化学的にコンピテントな細胞を使用して、標的宿主細胞に導入してもよい。DNA断片が相同組換えを受ける結果、標的宿主の内因性染色体領域は改変された染色体セグメントと置換される。
【0079】
プラスミドを使用し、改変された染色体セグメントを挿入のために標的宿主細胞内に運んでもよい。典型的には、非複製プラスミドを使用し、組込みが促進される。改変された染色体セグメントは、細菌の標的宿主ゲノム内でこの染色体セグメントに天然に隣接するDNA配列によってプラスミド内に隣接され、相同組換えが生じることになる配列が提供される。隣接する相同配列は上記の通りであり、プラスミドDNAの導入は上記の通りである。
【0080】
これらの方法のいずれかを用いて、バクテリオファージ相同組換え系、例えばバクテリオファージλRed系の使用により、相同組換えを促進してもよい((DatsenkoおよびWanner,Proc.Natl.Acad.Sci.USA,97:6640−6645頁、2000年)および(Ellisら、Proc.Natl.Acad.Sci.USA、98:6742−6746頁、2001年)またはRacファージRecE/RecT系(Zhangら、Nature Biotechnol.、18:1314−1317頁、2000年))。
【0081】
これらの方法のいずれかにおいては、相同組換えは、標的宿主の内因性染色体領域と改変された染色体セグメントとの置換をもたらす。
【0082】
改変された染色体セグメントの挿入に奏功したレシピエント株は、マーカーを使用して同定してもよい。スクリーニングまたは選択マーカーのいずれかを使用してもよく、ここでは選択マーカーが特に有用である。例えば、抗生物質耐性マーカーは、改変された染色体セグメント内に存在しうることから、それが新しい宿主に移される場合、改変された染色体セグメントを受ける細胞は、対応する抗生物質上での成長によって容易に同定することが可能である。あるいは、スクリーニングマーカー(容易に検出される生成物の生成をもたらすもの)を使用してもよい。マーカーがレシピエント株中に残存しないことが所ましい場合、その後、それを例えば部位特異的組換えを用いて除去してもよい。この場合、部位特異的組換え部位は、リコンビナーゼの発現がマーカーの欠失を引き起こすように、マーカーDNA配列の5’および3’側に位置する。一旦、突然変異が生じていると、細胞は、これらの特異的遺伝子の不在についてスクリーニングされる必要がある。この目的のため、複数の方法を用いて分析してもよい。
【0083】
pflB、frdB、ldhAおよびadhEとして同定される任意の細菌遺伝子は、イソブタノールの生成を目的とした本発明の株を作製するための、対応する微生物における組換えを意図した標的である。大腸菌(E.coli)、サルモネラ(Salmonella)、セラチア(Serratia)、エルウィニア(Erwinia)、シゲラ(Shigella)などのさまざまな腸内微生物由来の遺伝子および遺伝子産物は、本明細書中に記載のように、ハイブリダイゼーション、インフォマティクスまたは相同体によって同定してもよい。
【0084】
相同体の単離
本明細書中に記載の、配列番号9、46、48、50および52などの本発明における目的の遺伝子をコードする核酸分子、またはイソブタノール生合成経路において挙げられる配列のいずれかを用いて、同じまたは他の微生物種から、この核酸断片に対して少なくとも70%〜75%、75%〜80%、80%〜85%、85%〜90%、90%〜95%、もしくは95%〜100%の配列同一性を有する、相同タンパク質をコードする核酸分子を単離してもよい。配列依存性プロトコルを用いる相同体の単離は、当該技術分野で周知である。配列依存性プロトコルの例は、限定はされないが、核酸ハイブリダイゼーションの方法、ならびに、核酸増幅技術(たとえば、ポリメラーゼ連鎖反応(PCR)、Mullisら、米国特許第4,683,202号明細書;リガーゼ連鎖反応(LCR)、Tabor S.ら(Proc.Natl.Acad.Sci.USA、82:1074、1985年);または鎖置換増幅(SDA)、Walkerら、Proc.Natl.Acad.Sci.USA、89:392頁、1992年)のさまざまな使用によって例示されるDNAおよびRNA増幅の方法を含む。
【0085】
たとえば、本発明の核酸断片は、当業者に周知の方法を用い、DNAハイブリダイゼーションプローブとして配列番号9、46、48、50および52の核酸断片の全部または一部を使用し、任意の所望される細菌からライブラリをスクリーニングすることによって直接単離することが可能である。配列番号9、46、48、50および52に基づく特異的なオリゴヌクレオチドプローブは、当該技術分野で公知の方法(Maniatis、上記)によって設計し、合成することが可能である。さらに、配列を直接使用し、DNAプローブを、ランダムプライマーDNAラベリング、ニックトランスレーション、または末端標識技術などの当業者に公知の方法により、またはRNAプローブを、利用可能なインビトロ転写系を使用し、合成することが可能である。さらに、特異的なプライマーを設計し、使用し、配列番号9、46、48、50および52の相同体の一部または完全長を増幅することが可能である。得られる増幅産物は、増幅反応の間に直接標識するかまたは増幅反応後に標識し、プローブとして使用し、完全長DNA断片を好適なストリンジェントな条件下で単離することが可能である。
【0086】
典型的には、PCRタイプ増幅技術においては、プライマーは、異なる配列を有し、互いに相補的でない。所望される試験条件に依存し、プライマーの配列は、標的核酸の効率的かつ忠実な複製を提供するように設計される必要がある。PCRプライマー設計の方法は、一般的であり、当該技術分野で周知である(TheinおよびWallace、“The use of oligonucleotide as specific hybridization probes in the Diagnosis of Genetic Disorders”,in Human Genetic Diseases:A Practical Approach、K.E.Davis編(1986年)、33−50頁、IRL Press(Herndon,VA));Rychlik W.、(1993年)、In White B.A.(編)、Methods in Molecular Biology、第15巻、31−39頁、PCR Protocols:Current Methods and Applications.、Humania Press,Inc.(Totowa,NJ))。
【0087】
一般に、本核酸配列の2つの短いセグメントを使用し、ポリメラーゼ連鎖反応プロトコルにおける使用を意図したプライマーを設計し、DNAまたはRNAから相同コード領域をコードするより長い核酸断片を増幅することが可能である。PCRは、鋳型として配列番号9、46、48、50および52に相同な核酸配列を有する任意のDNA、たとえば鋳型としてゲノムDNA、cDNAまたはプラスミドDNAを使用して実施してもよい。クローン化cDNAのライブラリを使用する場合、一方のプライマーの配列は配列番号9、46、48、50および52に由来し、他方のプライマーの配列は微生物遺伝子をコードするmRNA前駆体の3’末端でのポリアデニル酸トラクト(tracts)の存在を利用する。あるいは、第2のプライマー配列は、クローン化ベクターに由来する配列に基づく場合がある。たとえば、当業者は、鋳型としてmRNAを使用するRACEプロトコルに従い(Frohmanら、Proc.Natl.Acad.Sci.USA、85:8998頁、1988年)、PCRを用いてcDNAを生成し、転写産物における単一点(single point)と3’または5’末端の間の領域のコピーを増幅することができる。3’および5’方向に方向づけられるプライマーは、本核酸配列から設計することが可能である。市販の3’RACEまたは5’RACEシステム(Life Technologies(Rockville,MD))を使用し、特異的な3’または5’cDNA断片を単離することが可能である(Oharaら、Proc.Natl.Acad.Sci.USA、86:5673頁、1989年);およびLohら、Science、243:217頁、1989年)。
【0088】
ハイブリダイゼーション
あるいは、配列番号9、46、48、54、56、50および52の核酸分子またはそれらの相補体は、相同体の同定のためのイブリダイゼーション試薬として使用してもよい。核酸ハイブリダイゼーション試験の基本的要素は、プローブ、目的の遺伝子または遺伝子断片を含有すると思われる試料、および特異的ハイブリダイゼーション法を含む。本発明のプローブは、典型的には、検出されるべき核酸配列に対して相補的な一本鎖核酸配列である。プローブは、検出されるべき核酸配列に対して「ハイブリダイズ可能」である。プローブ長は、5塩基から数万塩基まで可変であり、実施されるべき特定の試験に依存することになる。典型的には、約15塩基〜約30塩基のプローブ長が好適である。検出されるべき核酸配列に対して相補的である必要があるプローブ分子はほんの一部に限られる。さらに、プローブと標的配列の間の相補性は完全である必要はない。ハイブリダイゼーションは、相補性が不完全な分子間では生じず、その結果として、ハイブリダイズされる領域内の塩基の特定の画分は適切な相補的塩基と対合しない。
【0089】
ハイブリダイゼーション方法は十分に確立されている。典型的には、プローブおよび試料は、核酸ハイブリダイゼーションを可能にする条件下で混合されなければならない。これは、適切な濃度および温度条件下、無機または有機塩の存在下で、プローブと試料を接触させることを含む。プローブと試料核酸は、プローブと試料核酸の間の任意の可能なハイブリダイゼーションが生じうるような十分に長い期間接触状態でなければならない。混合物中のプローブまたは標的の濃度によって、ハイブリダイゼーションが生じるのに必要な時間が決まることになる。プローブまたは標的濃度が高まると、必要とされるハイブリダイゼーションのインキュベーション時間が短くなる。場合によって、カオトロピック剤を加えてもよい。カオトロピック剤は、ヌクレアーゼ活性を阻害することにより、核酸を安定化する。さらに、カオトロピック剤は、室温での短いオリゴヌクレオチドプローブの高感度かつストリンジェントなハイブリダイゼーションを可能にする(Van NessおよびChen,Nucl.Acid Res.19:5143−5151頁、1991年)。好適なカオトロピック剤は、特に、塩化グアニジン、チオシアン酸グアニジン、チオシアン酸ナトリウム、テトラクロロ酢酸リチウム、過塩素酸ナトリウム、テトラクロロ酢酸ルビジウム、ヨウ化カリウム、およびトリフルオロ酢酸セシウムを含む。典型的には、カオトロピック剤は、約3Mの最終濃度で存在することになる。必要に応じて、ハイブリダイゼーション混合物にホルムアミドを、典型的には30〜50%(v/v)加えてもよい。
【0090】
さまざまなハイブリダイゼーション溶液を使用してもよい。典型的には、これらは、約20〜60%容量、好ましくは30%の極性有機溶媒を含む。一般的なハイブリダイゼーション溶液では、約30〜50% v/vのホルムアミド、約0.15〜1Mの塩化ナトリウム、約0.05〜0.1Mの緩衝液、たとえばクエン酸ナトリウム、トリス−HCl、PIPESまたはHEPES(約6〜9のpH範囲)、約0.05〜0.2%の洗剤、たとえばドデシル硫酸ナトリウム、または0.5〜20mMのEDTA、FICOLL(Pharmacia Inc.)(約300〜500kD)、ポリビニルピロリドン(約250〜500kD)、および血清アルブミンが使用される。また、典型的なハイブリダイゼーション溶液中に、約0.1〜5mg/mLの未標識キャリア核酸、断片化核DNA(nucleic DNA)、たとえば仔ウシ胸腺またはサケ精液DNA、または酵母RNA、および場合によって約0.5〜2% w/vのグリシンが含まれることになる。また、他の添加剤、たとえば体積排除剤(volume exclusion agent)を含めてもよく、それは、種々の極性水溶性剤または膨潤剤(swellable agent)、たとえばポリエチレングリコール、ポリアクリル酸塩またはポリアクリル酸メチルなどのアニオンポリマー、およびアニオンサッカライドポリマー(anionic saccharidic polymer)、たとえば硫酸デキストランを含む。
【0091】
核酸ハイブリダイゼーションは、種々のアッセイフォーマットに適合可能である。最も好適なものの1つが、サンドイッチアッセイフォーマットである。サンドイッチアッセイは、特に、非変性条件下でのハイブリダイゼーションに適合可能である。サンドイッチ型アッセイの主要素が固体支持体である。固体支持体は、それに吸着しているか、またはそれに未標識でかつ配列の一部に相補的である固定化核酸プローブを共有結合させている。
【0092】
さらに、微生物ゲノムの配列が急速に公的に利用可能になりつつあることから、相同体は、バイオインフォマティクスのみを用いて同定してもよい。
【0093】
したがって、本発明は、組換え腸内細菌細胞を提供し、ここで遺伝的修飾は、イソブタノール生成に向けての炭素の集中流を可能にするため、特定のpflB、frdB、ldhAおよびadhE遺伝子の欠失をもたらす。
【0094】
イソブタノール生合成経路
炭水化物利用微生物は、中央代謝経路として、Embden−Meyerhof−Parnas(EMP)経路、Entner−Doudoroff経路およびペントースリン酸サイクルを利用し、成長および維持のためのエネルギーおよび細胞前駆体を提供する。これらの経路は、中間体のグリセルアルデヒド−3−リン酸を共有し、ピルビン酸塩は、最終的には直接にまたはEMP経路と併せて形成される。それに続き、ピルビン酸塩は、種々の手段を介してアセチル−補酵素A(アセチル−CoA)に形質転換される。アセチル−CoAは、例えば脂肪酸、アミノ酸および二次代謝物質を生成する上での主要な中間体としての役割を果たす。糖のピルビン酸塩への変換の複合反応により、エネルギーが生成され(例えば、アデノシン−5’−三リン酸、ATP)、還元等価物が得られる(例えば、還元型ニコチンアミドアデニンジヌクレオチド、NADH、および還元型ニコチンアミドアデニンジヌクレオチドリン酸、NADPH)。NADHおよびNADPHは、それらの酸化形態(それぞれNAD+およびNADP+)に再生されなければならない。無機電子受容体(例えばO2、NO3−およびSO42−)の存在下で、還元等価物を使用し、エネルギープールを増大させるか、あるいは還元型炭素副産物を形成してもよい。
【0095】
本発明の腸内宿主は、イソブタノールを生成する。典型的には、イソブタノール生合成経路は、図1Aおよび1Bに示されるように、イソブタノールの炭水化物からの生成を可能にする宿主細胞に改変されることになる。1つの経路は、以下の基質の生成物への変換、すなわち、
a)アセト乳酸シンターゼによって触媒される、ピルビン酸塩のアセト乳酸塩への変換、
b)アセトヒドロキシ酸イソメロレダクターゼによって触媒される、アセト乳酸塩の2,3−ジヒドロキシイソ吉草酸への変換、
c)アセトヒドロキシ酸デヒドラターゼによって触媒される、2,3−ジヒドロキシイソ吉草酸のα−ケトイソ吉草酸への変換、
d)分岐鎖ケト酸デカルボキシラーゼによって触媒される、α−ケトイソ吉草酸のイソブチルアルデヒドへの変換、および、
e)ブタノールデヒドロゲナーゼ(二次アルコールデヒドロゲナーゼ)によって触媒される、イソブチルアルデヒドのイソブタノールへの変換
を含む。
【0096】
この経路は、バリン生合成(ピルビン酸塩からα−ケトイソ吉草酸塩へ)およびバリン異化(α−ケトイソ吉草酸からイソブチルアルデヒドへ)における十分に特徴づけられた経路に関与することが知られる酵素と、最終ステップでの新規ブタノールデヒドロゲナーゼを組み合わせる。別のイソブタノール経路は、共同所有された同時係属中の米国特許出願公開第20070092957号明細書(参照により本明細書中に援用される)中に記載されている。
【0097】
多数のバリン生合成酵素はまた、イソロイシン生合成経路における類似反応を触媒することから、基質特異性は遺伝子源を選択する場合での主な検討事項である。したがって、アセト乳酸シンターゼ酵素における主な目的の遺伝子は、バチルス(Bacillus)(alsS)およびクレブシエラ(Klebsiella)(budB)に由来するものである。これらの特定のアセト乳酸シンターゼは、これらの微生物におけるブタンジオール発酵に関与することが知られ、ケト酪酸塩よりピルビン酸塩に対して親和性が高まることを示す(Gollopら、J.Bacteriol.172:3444−3449頁、1990年);Holtzclawら、J.Bacteriol.121:917−922頁、1975年)。第2および第3経路ステップは、それぞれアセトヒドロキシ酸レダクトイソメラーゼおよびデヒドラターゼによって触媒される。これらの酵素は、複数の供給源、例えば大腸菌(E.coli)などから特徴づけられている(Chunduruら、Biochemistry 28:486−493頁、1989年;およびFlintら、J.Biol.Chem.268:14732−14742頁、1993年)。好ましいイソブタノール経路の最終の2つのステップは、酵母内で生じることが知られ、そこでは窒素源としてバリンが使用され、同プロセスではイソブタノールが分泌されうる。α−ケトイソ吉草酸は、複数のケト酸デカルボキシラーゼ酵素、例えばピルビン酸デカルボキシラーゼなどにより、イソブチルアルデヒドに変換されうる。イソブタノール生成から外れたピルビン酸塩の誤誘導を阻止するため、ピルビン酸塩に対する親和性が低下したデカルボキシラーゼが所望される。これまで、当該技術分野で公知の2つのかかる酵素が存在する(Smitら、Appl.Environ.Microbiol.71:303−311頁、2005年;およびde la Plazaら、FEMS Microbiol.Lett.238:367−374頁、2004年)。両酵素は、ラクトコッカス・ラクティス(Lactococcus lactis)の株に由来し、ケトイソ吉草酸においてピルビン酸塩より50〜200倍の優位性を有する。最終的に、複数のアルデヒドレダクターゼが酵母内で同定されており、その多くは基質特異性が重複している。分岐鎖基質がアセトアルデヒドより好ましいことで公知のものとして、限定はされないが、アルコールデヒドロゲナーゼVI(ADH6)およびYpr1p(Larroyら、Biochem.J.361:163−172頁、2002年;およびFordら、Yeast 19:1087−1096頁、2002年)が挙げられ、それらの双方は電子供与体としてNADPHを使用する。分岐鎖基質の場合に活性があるNADPH依存性レダクターゼYqhDはまた、最近、大腸菌(E.coli)において同定されている(Sulzenbacherら、J.Mol.Biol.342:489−502頁、2004年)。
【0098】
本開示のイソブタノール経路においては、アクロモバクター・キシロスオキシダンス(Achromobacter xylosoxidans)由来の新規なブタノールデヒドロゲナーゼが使用され、それは本明細書中に記載される。
【0099】
アクロモバクター・キシロスオキシダンス(Achromobacter xylosoxidans)のブタノールデヒドロゲナーゼ活性
1−ブタノールを含有する培地上で連続的に培養することによって環境汚泥試料を富化することを通じて、唯一の炭素源として1−ブタノールを利用することができる微生物が単離された。1つの単離物が、細菌種アクロモバクター・キシロスオキシダンス(Achromobacter xylosoxidans)に属するものとして、その16SrRNA配列によって同定された。この単離物は、ブチルアルデヒドと1−ブタノールを相互変換したブタノールデヒドロゲナーゼ酵素活性を有する。予想外にも、このブタノールデヒドロゲナーゼ酵素活性がまた、イソブチルアルデヒドとイソブタノールの相互変換、ならびに2−ブタノンと2−ブタノールの相互変換を触媒することが見出された。意外なことに、この酵素は、集積培地中で使用される1−ブタノール基質の場合に匹敵するかまたはより優れた代替基質における動的定数を有した。これらの結果は、このアクロモバクター・キシロスオキシダンス(Achromobacter xylosoxidans)のブタノールデヒドロゲナーゼが、1−ブタノール、イソブタノール、または2−ブタノールを、それぞれブチルアルデヒド、イソブチルアルデヒドまたは2−ブタノン基質の供給源を有する組換え微生物宿主細胞内で生成するために使用可能であることを示した。
【0100】
ブタノールデヒドロゲナーゼのタンパク質およびコード配列
ブタノールデヒドロゲナーゼ活性を有する酵素をコードする、アクロモバクター・キシロスオキシダンス(Achromobacter xylosoxidans)において同定されるヌクレオチド配列は、配列番号9として与えられる。全タンパク質のアミノ酸配列は、配列番号10として与えられる。このアミノ酸配列と公的データベース内の配列との比較によると、このタンパク質が公知のアルコールデヒドロゲナーゼに対して極めて低い類似性を有することが示された。最も類似した公知の配列は、スコアリングマトリックスBLOSUM62、期待値カットオフ10およびワードサイズ3を伴うBLASTを用いると、その348アミノ酸長を超える配列番号10のアミノ酸配列に対して67%同一である。ギャップオープニングペナルティ11およびギャップ伸長1を用いた。見出された最も近い類似性は、髄膜炎菌(Neisseria meningitides)MC58のZn含有アルコールデヒドロゲナーゼ(登録番号AAF41759.1)に対して67%のアミノ酸同一性およびマイコプラズマ・アガラクティアエ(Mycoplasma agalactiae)のZn含有アルコールデヒドロゲナーゼ(登録番号A5IY63)に対して67%のアミノ酸同一性であった。したがって、好ましいブタノールデヒドロゲナーゼ(sadB)は、スコアリングマトリックスBLOSUM62、期待値カットオフ10およびワードサイズ3ならびにギャップオープニングペナルティ11およびギャップ伸長1を伴うBLASTを用いると、その348アミノ酸長を超える配列番号10に対して少なくとも約70%〜75%、約75%〜80%、約80%〜85%、85%〜90%、もしくは90%〜95%同一であるものである。
【0101】
ブタノールデヒドロゲナーゼ活性
配列番号10に対して少なくとも約70%以上のアミノ酸同一性を有し、かつブタノールデヒドロゲナーゼ活性を有するタンパク質が、本発明では特に有用である。本発明の核酸分子は、ブタノールデヒドロゲナーゼ活性を有する配列番号10に対して少なくとも約70%以上のアミノ酸同一性を有するタンパク質をコードする。当業者は、タンパク質におけるブタノールデヒドロゲナーゼ活性を容易に評価することができる。タンパク質は、下記のように微生物細胞内で発現され、細胞抽出物、粗酵素調製物、または精製酵素調製物中でのブタノールデヒドロゲナーゼ活性についてアッセイされる。たとえば、精製酵素および粗酵素調製物のアッセイは、本明細書中、実施例1で説明される。1−ブタノールデヒドロゲナーゼ活性についてのアッセイでは、50mMのブチルアルデヒドおよび0.2mMのNADHを含有する50mMのリン酸カリウム緩衝液(pH6.2、35℃)中の酵素を適量で使用し、NADHの消失が340nmで分光測定的にモニタリングされる。アルコール基質を用いる代替アッセイが、3mMのNAD+およびさまざまな濃度のアルコールを含有するトリス緩衝液(pH8.5)において35℃で実施されるか、あるいはケトンまたはアルデヒド基質を用いる場合は、200μMのNADHおよびさまざまな濃度のケトンまたはアルデヒドを含有する50mMのMES緩衝液(pH6.0)において35℃で実施される。これらまたは他の容易に実施可能なアッセイによると、ブタノールデヒドロゲナーゼの機能は、単離核酸分子によってコードされる同定されたタンパク質の構造に関連性がある(そのいずれもが同定された配列を有する)。
【0102】
生産宿主の作成
発酵性炭素基質をイソブタノールに変換するための酵素経路をコードするのに必要な遺伝子を有する組換え微生物は、当該技術分野で周知の技術を用いて作成してもよい。本発明のイソブタノール生合成経路の酵素、すなわちアセト乳酸シンターゼ、ケトール酸レダクトイソメラーゼ、アセトヒドロキシ酸デヒドラターゼ、分岐鎖α−ケト酸デカルボキシラーゼ、および分岐鎖アルコールデヒドロゲナーゼをコードする遺伝子は、上記のように、さまざまな供給源から単離してもよい。
【0103】
本願中に開示される操作のために使用されるイソブタノール生成株の作成は、共同所有された同時係属中の米国特許出願公開第20070092957号明細書の特に実施例1、2、9、10、11、12、13および14(参照により本明細書中に援用される)において開示されている。
【0104】
分子生物学の当該技術分野で周知の微生物ゲノムから所望される遺伝子を得る方法。例えば、遺伝子の配列が公知である場合、好適なゲノムライブラリは、制限エンドヌクレアーゼ消化によって生成され、所望される遺伝子配列に対して相補的なプローブでスクリーニングされうる。一旦、配列が単離されると、DNAは、ポリメラーゼ連鎖反応などの標準プライマーに特異的な増幅方法(米国特許第4,683,202号明細書)を用いて増幅され、適切なベクターを使用して、形質転換に適したDNAの量を得ることが可能である。異種宿主内での発現のためのコドン最適化用のツールは、容易に利用可能である。コドン最適化用のいくつかのツールは、宿主微生物のGC含量に基づいて利用可能である。
【0105】
一旦、関連経路遺伝子が同定され、単離されると、それらは、当該技術分野で周知の手段により、好適な発現宿主に形質転換することが可能である。種々の宿主細胞の形質転換にとって有用なベクターまたはカセットについては一般的であり、EPICENTRE(登録商標)(Madison,WI)、Invitrogen Corp.(Carlsbad,CA)、Stratagene(La Jolla,CA)、およびNew England Biolabs,Inc.(Beverly,MA)などの企業から市販されている。典型的には、ベクターまたはカセットは、関連遺伝子の転写および翻訳を誘導する配列、選択可能なマーカー、および自律的複製または染色体組込みを可能にする配列を有する。好適なベクターは、転写開始制御を有する遺伝子の5’領域およびを制御する転写終結DNA断片の3’領域を含む。両方の制御領域は形質転換された宿主細胞に対して相同な遺伝子から誘導されうるが、かかる制御領域が生産宿主として選択される特定の種に対して天然でない遺伝子からも誘導されることが理解されるべきである。
【0106】
所望される宿主細胞内での関連経路コード領域の発現を駆動するのに有用な開始制御領域またはプロモーターは極めて多く、当業者に周知である。これらの遺伝因子を駆動可能な仮想的に任意のプロモーター、限定はされないが、(大腸菌(Escherichia coli)および他の腸内細菌(Enterobacteriaceae)内での発現にとって有用な)lac、ara、tet、trp、lPL、lPR、T7、tac、およびtrcなどは、本発明に適している。
【0107】
終結制御領域についても好ましい宿主に対して天然の様々な遺伝子から誘導されうる。場合により終結部位は不要でありうるが、含められる場合が最も好ましい。
【0108】
特定のベクターが広範囲の宿主細菌内で複製可能であり、かつ複合により導入可能である。pRK404および3つの関連ベクター:pRK437、pRK442、およびpRK442(H)の完全でかつアノテートされた配列が利用可能である。これらの誘導体はグラム陰性菌における遺伝子操作にとって有用なツールであることが判明している(スコット(Scott)ら、Plasmid 50:74−79頁、2003年)。広範な宿主範囲のInc P4プラスミドRSF1010における数種のプラスミド誘導体についても、グラム陰性菌の範囲内で機能しうるプロモーターとともに利用可能である。プラスミドpAYC36およびpAYC37が複数のクローニング部位とともに活性プロモーターを有し、グラム陰性菌内での異種の遺伝子発現が可能である。
【0109】
さまざまな好ましい微生物宿主内でのイソブタノール生合成経路の発現は、下記にさらに詳述される。
【0110】
大腸菌(E.coli)内でのイソブタノール生合成経路の発現
大腸菌(E.coli)の形質転換にとって有用なベクターまたはカセットは、一般的であり、上掲の企業から市販されている。例えば、イソブタノール生合成経路の遺伝子は、さまざまな供給源から単離し、修飾pUC19ベクターにクローン化し、大腸菌(E.coli)に形質転換することが可能である。
【0111】
腸内細菌(Enterobacteriaceae)科におけるイソブタノール生合成経路の発現
本発明における使用に適した腸内細菌の例として、限定はされないが、セラチア(Serratia)属、エルウィニア(Erwinia)属、エシェリキア(Escherichia)属、クレブシエラ(Klebsiella)属、サルモネラ(Salmonella)属、およびシゲラ(Shigella)属のメンバーがあげられる。腸内細菌(Enterobacteriaceae)における遺伝子発現および突然変異の生成のための方法もまた、当該技術分野で周知である。例えば、イソブタノール生合成経路の遺伝子は、共同所有された同時係属中の米国特許出願公開第20070092957号明細書の実施例1、2、9、19、11、12、13および14に記載のように、さまざまな供給源から単離し、さまざまなベクターにクローン化してもよい。本発明において特に好適なのは、細菌の腸内クラスのメンバーである。腸内細菌は、腸内細菌科(Enterobacteriaceae)のメンバーであり、エシェリキア(Escherichia)、サルモネラ(Salmonella)、およびシゲラ(Shigella)などのメンバーを含む。それらは、グラム陰性の0.3〜1.0×1.0〜6.0mmの直鎖状桿菌(straight rod)であり、周毛性鞭毛によって運動性であり(テイタメラ(Tatumella)を除く)、または非運動性である。それらは酸素の存在および不在下で成長し、ペプトン、肉抽出物、および(通常は)マッコンキー培地などのさまざまな培地上で良好に成長する。いくつかは唯一の炭素源としてD−グルコース上で成長する一方、その他はビタミンおよび/またはミネラルを必要とする。それらは呼吸性および発酵性代謝を有する化学有機栄養性であるが、好塩性ではなく、D−グルコース、その他の炭水化物、およびポリヒドロキシルアルコールの発酵中に、酸や、しばしば可視性ガスが生成する。それらはオキシダーゼ陰性であり、赤痢菌(Shigella dysenteriae)0グループ1およびゼノラブダス・ネマトフィルス(Xenorhabdus nematophilus)を除いて、カタラーゼ陽性である。(エルウィニア(Erwinia)およびエルシニア(Yersina)のいくつかの株を除いて)硝酸塩は亜硝酸塩に還元される。DNAのG+C含量は、38〜60mol%(Tm、Bd)である。大部分の属内の種からのDNAは、互いに、また科の基準種である大腸菌(Escherichia coli)に少なくとも20%の関連性がある。注目すべき例外は、そのDNAがその他の属からの種と10〜20%の関連性がある、エルシニア(Yersina)、プロテウス(Proteus)、プロビデンシア(Providenica)、ハフニア(Hafnia)、およびエドワードシエラ(Edwardsiella)の種である。エルウィニア・クリサンテミ(Erwinia chrysanthemi)を除いて、試験したすべての種は腸内細菌共通抗原を有する(「ベルジーの系統的細菌学マニュアル(Bergy’s Manual of Systematic Bacteriology)」D.H.Bergyら、Williams and Wilkins Press,Baltimore、1984年)。
【0112】
発酵性炭素基質
本発明の組換え微生物生産宿主は、好適な炭素基質を含有しなければならない。適切な炭素基質が、限定はされないが、グルコースおよびフルクトースなどの単糖、乳糖またはスクロースなどのオリゴ糖、デンプンまたはセルロースなどの多糖、あるいはそれらの混合物、ならびに乳清透過液、コーンスティープリカー(cornsteep liquor)、砂糖液(sugar beet molasses)、および大麦モルト(barley malt)などの再生可能な原料由来の未精製混合物を含みうる。
【0113】
上記の炭素基質およびその混合物のすべてが本発明において好適であると考えられるが、好ましい炭素基質は、グルコース、フルクトース、およびスクロースである。スクロースは、サトウキビ、サトウダイコン、キャッサバ、サトウモロコシ、およびこれらの混合物などの再生可能な糖供給源に由来しうる。グルコース(デキストロース)は、トウモロコシ、小麦、ライ麦、大麦、オート麦、およびこれらの混合物などの穀物を含むデンプンに基づく供給原料の糖化を通じて、再生可能な穀物供給源に由来しうる。さらに、発酵性糖は、たとえば共同所有および同時係属中の米国特許出願公開第20070031918A1号明細書(参照によって本明細書中に援用される)中に記載のように、前処理および糖化のプロセスを通じて、再生可能なセルロース系またはリグノセルロース系バイオマスに由来しうる。バイオマスは、任意のセルロース系またはリグノセルロース系の原料を示し、セルロースを含有し、かつ場合により、ヘミセルロース、リグニン、デンプン、オリゴ糖および/または単糖をさらに含有する原料を含む。バイオマスは、追加成分、例えばタンパク質および/または脂質も含有しうる。バイオマスは単一の供給源から誘導されうるか、またはバイオマスは2つ以上の供給源から誘導される混合物を含有しうる。バイオマスは、例えばトウモロコシ穂軸(corn cob)およびコーンストーバーの混合物または草および葉の混合物を含有しうる。バイオマスは、限定はされないが、バイオエネルギー作物、農業残渣、地方自治体の固体廃棄物、産業固体廃棄物、製紙から排出される汚泥、庭ごみ、廃材および林業廃棄物を含む。バイオマスの例として、限定はされないが、トウモロコシ粒、トウモロコシ穂軸、トウモロコシの皮などのトウモロコシ残渣、コーンストーバー、草、小麦、麦かん(wheat straw)、大麦、麦かん(barley straw)、干し草、稲わら、スイッチグラス、紙くず、サトウキビバガス、サトウモロコシ、大豆、穀物、木、枝、根、葉、木片、おがくず、シュラブおよびブッシュのミリングから得られる成分、野菜、果物、花、家畜糞尿ならびにそれらの混合物が挙げられる。
【0114】
発酵培地は、適切な炭素基質に加え、イソブタノールの生成に必要な培養物の成長および酵素経路の促進に適する、当業者に既知の、適切なミネラル、塩、共同因子、緩衝液および他の成分を含有する必要がある。
【0115】
培養条件
典型的には、細胞は、適切な培地中で約25℃〜約40℃の範囲内の温度で成長する。本発明における好適な成長培地は、Luria Bertani(LB)培養液などの一般的な商業的に調製された培地である。他の限定または合成された成長培地が用いられる場合があり、かつ特定の微生物の成長に適した培地について微生物学または発酵科学に関する当業者は知っているであろう。異化代謝産物抑制を直接的または間接的に調節することで知られる作用物質、例えば環状アデノシン2’:3’一リン酸の使用についても発酵培地内に取り込まれうる。
【0116】
酵母の発酵に適するpH範囲はpH5.0〜pH9.0であり、初期条件としてはpH6.0〜pH8.0が好ましい。
【0117】
発酵は好気性または嫌気性条件下で実施可能であり、嫌気性または微好気性条件が好ましい。
【0118】
発酵培地中で生成されるイソブタノールの量は、当該技術分野で既知の複数の方法、例えば、高性能液体クロマトグラフィー(HPLC)またはガスクロマトグラフィー(GC)を用いて測定することが可能である。
【0119】
工業用バッチおよび連続発酵
発酵のバッチ方法が使用されうる。従来のバッチ発酵は、培地の組成物が発酵開始時に設定され、発酵の間に人工的な改変が施されることがない閉鎖系である。したがって、発酵開始時に培地に望ましい微生物が接種され、系に何も添加しなくても発酵の生成が可能である。しかし、典型的には「バッチ」発酵は炭素源の添加に関連したバッチであり、pHおよび酸素濃度などの要素を制御する試みがなされることが多い。バッチシステムでは、システムの代謝産物およびバイオマス組成物は、最大で発酵が停止する時間まで常時変化する。バッチ培養物内では、細胞が、変化のない誘導期(lag phase)から高成長の対数期(log phase)、最終的に成長率が減少または停止する定常期(stationay phase)にかけて抑制される。定常期における細胞が、未処理の場合、最終的に死滅することになる。対数期における細胞が、一般に最終生成物または中間体の生成の大部分に関与する。
【0120】
標準のバッチシステムに対する変形がフェドバッチシステムである。フェドバッチ発酵プロセスは本発明においても適切であり、基質が発酵プロセスごとに添加されること以外では典型的なバッチシステムを含む。フェドバッチシステムは、異化代謝産物抑制が細胞の代謝を阻害する傾向がある場合や培地内に限られた量の基質を有することが望ましい場合に有用である。フェドバッチシステム内での実際の基質濃度の測定は困難であることから、それはpH、溶解酸素およびCO2などの排ガスの分圧などの測定可能な要素の変化に基づいて評価される。バッチおよびフェドバッチ発酵は一般的で、当該技術分野で周知であり、例がThomas D.、Brock(Biotechnology:A Textbook of Industrial Microbiology、第2版、1989年、Sinauer Associates,Inc.,Sunderland,MA)またはDeshpande,Mukund V.、(Appl.Biochem.Biotechnol.、36:227頁、1992年)(参照により本明細書中に援用される)において見出されうる。
【0121】
本発明はバッチモードで実施されるが、本方法であれば連続発酵方法に適合可能であると考えられる。連続発酵は、限定された発酵培地がバイオリアクターに連続的に添加されかつ等量の条件培地が処理と同時に除去される場合の開放系である。連続発酵では、一般に細胞が主に対数増殖期にある一定の高密度で培養物が維持される。
【0122】
連続発酵では、細胞成長または最終生成物濃度に作用する1つの要素またはいくつかの要素の調節が可能になる。例えば、1つの方法では、炭素源などの限られた栄養素または一定速度での窒素レベルが維持され、あらゆる他のパラメータの抑制が可能になる。他のシステムでは、培地の濁度により測定される細胞濃度が一定に保持される間、成長に作用する多数の要素を連続的に改変することが可能である。連続システムでは、恒常的成長条件を維持するように試みることで、培地の除去(drawn off)に起因する細胞欠損を発酵における細胞成長率に対して均衡させなければならない。連続発酵プロセス用の栄養素および成長因子を調節する方法ならびに生成物形成の速度を最大化するための技術については産業微生物学における当該技術分野で周知であり、かつ種々の方法がBrock、上記で詳述されている。
【0123】
本発明は、バッチ、フェドバッチまたは連続プロセスのいずれかを用いて実施可能であり、かつ既知の発酵モードであればいずれも適するものと考えられる。さらに、細胞は、全細胞触媒としての基質上に固定化され、イソブタノール生成用の発酵条件に従いうると考えられる。
【0124】
発酵培地からイソブタノールを単離するための方法
生物学的に生成されたイソブタノールは、当該技術分野で既知の方法を用いて発酵培地から単離してもよい。例えば、固体は、遠心分離、濾過、デカンテーション、または同様のものにより、発酵培地から除去してもよい。次いで、イソブタノールは、蒸留、液体−液体抽出、または膜に基づく分離などの方法を用いて、上記のように固体を除去するように処理されている発酵培地から単離してもよい。イソブタノールが水と低沸点の共沸混合物を形成することから、単に蒸留を用い、混合物をその共沸組成物に至るまで分離してもよい。蒸留を、別の分離方法と併用し、共沸混合物についての分離を得てもよい。イソブタノールを単離し、精製するための、蒸留と併用可能な方法は、限定はされないが、デカンテーション、液体−液体抽出、吸着、および膜に基づく技術を含む。さらに、イソブタノールは、エントレーナを使用する共沸蒸留を用いて単離してもよい(例えば、DohertyおよびMalone、Conceptual Design of Distillation Systems、McGraw Hill,New York、2001年を参照)。
【0125】
イソブタノール−水混合物は異種共沸混合物を形成し、それから蒸留をデカンテーションと併用し、イソブタノールを単離し、精製してもよい。この方法では、イソブタノールを含有する発酵ブロスは、ほぼ共沸組成物に至るまで蒸留される。次いで、共沸混合物は濃縮され、イソブタノールはデカンテーションによって発酵培地から分離される。デカントされた水相は、還流としての第1の蒸留カラムに戻してもよい。イソブタノールに富むデカントされた有機相は、第2の蒸留カラム内で、蒸留によってさらに精製してもよい。
【0126】
イソブタノールはまた、蒸留と併用し、液体−液体抽出を用いて発酵培地から単離してもよい。この方法では、イソブタノールは、好適な溶媒による液体−液体抽出を用いて、発酵ブロスから抽出される。次いで、イソブタノール含有有機相は蒸留され、イソブタノールは溶媒から分離される。
【0127】
また、蒸留を吸着と併用し、イソブタノールを発酵培地から単離してもよい。この方法では、イソブタノールを含有する発酵ブロスは、ほぼ共沸組成物に至るまで蒸留され、次いで残留水は、吸着剤、例えば分子ふるいの使用によって除去される(Adenら、Lignocellulosic Biomass to Ethanol Process Design and Economics Utilizing Co−Current Dilute Acid Prehydrolysis and Enzymatic Hydrolysis for Corn Stover,Report NREL/TP−510−32438,National Renewable Energy Laboratory、2002年6月)。
【0128】
さらに、蒸留をパーベーパレイションと併用し、イソブタノールを発酵培地から単離し、精製してもよい。この方法では、イソブタノールを含有する発酵ブロスは、ほぼ共沸組成物に至るまで蒸留され、次いで残留水は、親水性膜を通したパーベーパレイションによって除去される(Guoら、J.Membr.Sci.245、199−210頁(2004年))。
【実施例】
【0129】
本発明はさらに以下の実施例にて定義される。これらの実施例が本発明の好ましい実施形態を示す一方であくまで例示目的で与えられることは理解されるべきである。上記の考察およびこれらの実施例から、当業者は、本発明の本質的な特性により、その趣旨および範囲から逸脱することなく本発明に対してさまざまな変更および改良を行うことで、本発明のさまざまな利用および条件への適合が可能であることが確認できる。
【0130】
一般的方法
実施例において用いられる標準の組換えDNAおよび分子クローニング技術は、当該技術分野で周知であり、Sambrook J.ら、Molecular Cloning:A Laboratory Manual(Cold Spring Harbor Laboratory Press,Cold Spring Harbor,NY、1989年、Maniatisとしても公知)およびT.J.Silhavy、M.L.Bennan、およびL.W.Enquist、Experiments with Gene Fusions(Cold Spring Harbor Laboratory Press,Cold Spring Harbor,N.Y.、1984年)およびAusubel F.M.ら、Current Protocols in Molecular Biology(Greene Publishing Assoc.およびWiley−Interscience,N.Y.、1987年)によって記載されている。
【0131】
細菌培養物の維持および成長に適した材料および方法は、当該技術分野で周知である。以下の実施例における使用に適した技術は、Manual of Methods for General Bacteriology(Phillipp Gerhardt編、American Society for Microbiology,Washington,DC.、1994年)またはThomas D.、Brock in Biotechnology:A Textbook of Industrial Microbiology(第2版、Sinauer Associates,Inc.,Sunderland,MA、1989年)に示されるように見出されうる。細菌細胞の成長および維持のために使用されるすべての試薬、制限酵素および材料は、他に特に規定がなければ、Aldrich Chemicals(Milwaukee,WI)、BD Diagnostic Systems(Sparks,MD)、Life Technologies(Rockville,MD)、またはSigma Chemical Company(St.Louis,MO)から入手した。
【0132】
微生物株は、特に断りのない限り、The American Type Culture Collection(ATCC)(Manassas,VA)から入手した。
【0133】
P1形質導入
P1vir形質導入は、いくつかの修飾を伴い、Millerによる記載のように行われた(Miller J.H.、1992年、A Short Course in Bacterial Genetics. Cold Spring Harbor Laboratory,Cold Spring Harbor,N.Y)。つまり、形質導入溶解物を調製するため、ドナー株の細胞を、振とうしながら、Luriaブロス(LB)培地中、37℃で一晩成長させた。これらの細胞の一晩成長を、0.005M CaCl2を含有するLB培地中に継代培養し、通気のない37℃の水槽内に入れた。ファージ添加の1時間前、細胞を、振とうしながら37℃下に置いた。細胞の最終成長後、一定分量1.0mLの培養物を14mlのFalconチューブに分注し、約107個のP1virファージを加えた。これらのチューブを、2.5mLの0.8%LB上層寒天を各チューブに加える前に37℃の水槽内で20分間インキュベートし、内容物を、LB寒天プレート上に展開し、37℃でインキュベートした。翌日、軟質寒天層をこすり、遠心管内に入れた。プレートの表面をLB培地で洗浄し、チューブをボルテックスミキサーを使用して激しく撹拌する前に、遠心管に加え、次いでCHCl3数滴を加えた。4,000rpmで10分間の遠心分離後、P1vir溶解物を含有する上清を回収した。
【0134】
形質導入においては、レシピエント株を、1〜2mLのLB培地中で、振とうしながら。37℃で一晩成長させた。培養物を、10,000rpm、室温で1分間のEppendorf微量遠心管での遠心分離によってペレット状にした。細胞ペレットを、等しい容量のMC緩衝液(0.1M MgSO4、0.005M CaCl2)中に再懸濁し、0.1mLの一定分量でチューブに分注し、0.1mLおよび0.01mLのP1vir溶解物を加えた。P1vir溶解物を全く含有しない対照チューブも含めた。0.2mLの0.1Mクエン酸ナトリウムを加えてP1の感染の停止する前、チューブを37℃で20分間インキュベートした。チューブを37℃で1時間インキュベートする前、1mLのLB培地を各チューブに加えた。インキュベーション後、細胞を上記のようにペレット状にし、25μg/mLのカナマイシンを含有するLBプレート上に展開する前に50〜200μLのLB中に再懸濁し、37℃で一晩インキュベートした。形質導入体を、カナマイシンマーカー挿入物の上流および下流の領域に隣接する染色体に特異的なプライマーを使用するコロニーPCRによってスクリーニングした。
【0135】
染色体からのカナマイシンマーカーの除去が、カナマイシン−耐性株をプラスミドpCP20で形質転換し(Cherepanov P.P.およびWackernagel W.、Gene、158:9−14頁、1995年)、次いでLBアンピシリン(100μg/mL)プレート上に展開し、30℃でインキュベートすることによって得られた。pCP20プラスミドは、λPRプロモーターの制御下で酵母FLPリコンビナーゼを有する。このプロモーターからの発現は、プラスミド上に存在するcI857温度感受性リプレッサーによって制御される。pCP20の複製の複製起点もまた、温度感受性である。アンピシリン耐性コロニーを、LB寒天プレート上にストリークし、42℃でインキュベートした。インキュベーション温度が高くなると同時にFLPリコンビナーゼの発現が誘発され、pCP20プラスミドが細胞から回復された。単離したコロニーを、カナマイシン(25μg/mL)を含有するLBプレートと、LBアンピシリン(100μg/mL)プレートおよびLBプレート上のグリッドにパッチした。得られたカナマイシン感受性、アンピシリン感受性コロニーを、コロニーPCRによってスクリーニングし、カナマイシンマーカーの染色体からの除去を確認した。
【0136】
コロニーPCR増幅においては、HotStarTaq Master Mix(Qiagen(Valencia,CA);カタログ番号71805−3)を、製造業者のプロトコルに従って使用した。0.2μMの各染色体に特異的なPCRプライマーを含有する25μLのMaster Mix反応物に、少量のコロニーを加えた。増幅を、DNA Thermocycler GeneAmp 9700(PE Applied Biosystems(Foster City,CA))で行った。典型的なコロニーPCR条件は、95℃で15分.;95℃で30秒間を30サイクル、50〜58℃の範囲の30秒間のアニーリング温度、約1分/DNAのkbの伸長時間、72℃で伸長されるプライマー;次いで72℃で10分間、次いで4℃で保持であった。PCR産物サイズを、公知の分子量標準との比較により、ゲル電気泳動によって測定した。
【0137】
制限酵素、T4 DNAリガーゼおよびPhusion High Fidelity DNAポリメラーゼ(New England Biolabs(Beverely,MA))を、製造業者の推奨に従って使用した。
【0138】
ゲル電気泳動を、製造業者のプロトコルに従い、プレキャストReliant(登録商標)1%アガロースゲル(Lonza Rockland,Inc.(Rockland,ME))を有するRunOne電気泳動システム(Embi Tec(San Diego,CA))を使用して行った。ゲルは、典型的にはTBE緩衝液(Invitrogen、カタログ番号15581−044)中を走る。
【0139】
形質転換においては、大腸菌(E.coli)のエレクトロコンピテント細胞を、Ausubel F.M.ら、(Current Protocols in Molecular Biology、1987年、Wiley−Interscience)で記載のように調製した。細胞を、25〜50mLのLB培地中、30〜37℃で成長させ、10,000rpmで10分間の遠心分離により、0.5〜0.7のOD600で収穫した。これらの細胞を、培養物の元の開始容量に等しい容量の無菌氷冷水で2回洗浄する。最終洗浄後、細胞を、滅菌水中に再懸濁し、形質転換されるべきDNAを加えた。細胞およびDNAを、冷却したキュベットに移し、製造業者の使用説明書に従い、Bio−Rad Gene Pulser IIでエレクトロポレートした(Bio−Rad Laboratories,Inc.(Hercules,CA))。
【0140】
次の実施例で使用するオリゴヌクレオチドプライマーを表2に示す。すべてのオリゴヌクレオチドプライマーは、Sigma−Genosys(Woodlands,TX)によって合成された。
【0141】
培地中でイソブタノール濃度を測定するための方法
水相および有機相中でのイソブタノールの濃度を、Agilent Technologies(Santa Clara,CA)製のHP−InnoWaxカラム(30m×0.32mm ID、0.25μmのフィルム)を使用するガスクロマトグラフィー(GC)によって測定した。キャリアガスは、一定の上部圧力、150℃での測定された1mL/分の流速のヘリウムであり、インジェクタスプリット比は200℃で1:10であり、オーブン温度は45℃で1分間、10℃/分で45℃〜230℃、および230℃で30秒間であった。フレームイオン化検出を、40mL/分のヘリウム補給気体(makeup gas)を使用して260℃で用いた。培養液試料を、GCへの注入前に、0.2μmの回転フィルタを通して濾過した。所望される分析感度に依存し、0.1μLまたは0.5μLのいずれかの注入容量を使用した。較正標準曲線を、次の化合物、すなわちエタノール、イソブタノール、アセトイン、メソ−2,3−ブタンジオール、および(2S,3S)−2,3−ブタンジオールについて生成した。また、分析標準を使用し、イソブチルアルデヒド、イソ酪酸、およびイソアミルアルコールにおける保持時間を同定した。これらの条件下で、イソブタノール保持時間は、約5.33分であった。
【0142】
略語の意味は以下の通りである。「m」はメートルを意味し、「mm」はミリメートルを意味し、「μm」はミクロンまたはマイクロメートルを意味し、「sec」は秒を意味し、「min」は分を意味し、「hr」は時間を意味し、「nm」はナノメートルを意味し、「μL」はマイクロリットルを意味し、「mL」はミリリットルを意味し、「rpm」は回毎分を意味し、「L」はリットルを意味し、「mm」はミリメートルを意味し、「nm」はナノメートルを意味し、「mM」はミリモル濃度を意味し、「μg/mL」はマイクログラム/ミリリットルを意味し、「mmol/分/mg」は、ミリモル/分/ミリグラムを意味し、「μM」はマイクロモルを意味し、「M」はモル濃度を意味し、「mmol」はミリモルを意味し、「μmol」はマイクロモルを意味し、「g」はグラムを意味し、「μg」はマイクログラムを意味し、「PCR」はポリメラーゼ連鎖反応を意味し、「OD」は光学密度を意味し、「OD600」は600nmの波長で測定された光学密度を意味し、「kD」はキロダルトンを意味し、「bp」は塩基対を意味し、「kb」はキロ塩基対を意味し、「%」はパーセントを意味し、「%w/v」は重量/容量パーセントを意味し、「%v/v」は容量/容量パーセントを意味し、「IPTG」はイソプロピル−β−D−チオガラクトピラノシドを意味し、「wt%」は重量パーセントを意味し、「RBS」はリボソーム結合部位を意味し、「HPLC」は高性能液体クロマトグラフィーを意味し、「GC」はガスクロマトグラフィーを意味し、「g/L」はグラム/リットルを意味し、「g/L/h」はグラム/リットル/時間を意味し、「g/g」はグラム/グラムを意味し、「mL/分」はミリリットル/分を意味し、「℃/分」は摂氏度/分を意味し、「vvm」は容量/容量/分を意味し、「v/v」は容量/容量を意味し、「vol%」は容量パーセントを意味し、「ID」は内径を意味する。
【0143】
実施例1
pflB、frdB、ldhA、およびadhE遺伝子の欠失を有する大腸菌(E.coli)株の作成
本実施例では、4つの遺伝子が不活性化された大腸菌(E.coli)株の改変について記載する。大腸菌(E.coli)株のKeio collection(Babaら、Mol.Syst.Biol.、2:1−11頁、2006年)を、4KO大腸菌(E.coli)(4ノックアウト)の生成に使用した。Keio collectionは、DatsenkoおよびWannerの方法(Datsenko K.A.およびWanner B.L.、Proc Natl Acad Sci.、USA、97:6640−6645頁、2000年)により、大腸菌(E.coli)株BW25113中で生成される単一の遺伝子ノックアウトのライブラリである。collection中では、各欠失遺伝子は、Flpリコンビナーゼによって除去可能なFRTに隣接したカナマイシンマーカーと置換された。4KO大腸菌(E.coli)株は、ノックアウト−カナマイシンマーカーを、P1形質導入により、Keioドナー株からレシピエント株に移すことによって作成した。ノックアウトを生成するための各P1形質導入後、カナマイシンマーカーをFlpリコンビナーゼによって除去した。このマーカーレス株は、次のP1形質導入のための新しいドナー株としての機能を果たした。
【0144】
4KO大腸菌(E.coli)株は、JW0886に加え、3つのKeio株から調製したP1ファージ溶解物でのP1vir形質導入により、Keio株JW0886中で作成した。使用したKeio株を次に挙げる。
−JW0886:kanマーカーはpflBに挿入される
−JW4114:kanマーカーはfrdBに挿入される
−JW1375:kanマーカーはldhAに挿入される
−JW1228:kanマーカーはadhEに挿入される
【0145】
染色体からのカナマイシンマーカーの除去を、カナマイシン耐性株をアンピシリン耐性プラスミドpCP20(CherepanovおよびWackernagel、上記)で形質転換することによって行った。形質転換体を、100μg/mLのアンピシリンを含有するLBプレート上に展開した。プラスミドpCP20は、λPRプロモーターの制御下で酵母FLPリコンビナーゼを有し、このプロモーターからの発現は、プラスミド上に存在するcI857温度感受性リプレッサーによって制御される。pCP20の複製の複製起点はまた、温度感受性である。
【0146】
JW0886株(ΔpflB::kan)を、プラスミドpCP20で形質転換し、100μg/mLのアンピシリンを含有するLBプレート上に30℃で展開した。次いで、アンピシリン耐性形質転換体を選択し、LBプレート上にストリークし、42℃で成長させた。単離したコロニーを、アンピシリンおよびカナマイシン選択培地プレートならびにLBプレート上にパッチした。カナマイシン感受性およびアンピシリン感受性コロニーを、プライマーpflB CkUp(配列番号34)およびpflB CkDn(配列番号35)を伴うコロニーPCRによってスクリーニングした。PCR反応ミックスの一定分量10μLを、ゲル電気泳動によって分析した。予想された約0.4kbのPCR産物が認められ、それからマーカーの除去が確認され、「JW0886マーカーレス」株が生成された。この株は、pflB遺伝子の欠失を有する。
【0147】
「JW0886マーカーレス」株を、JW4114(frdB::kan)由来のP1vir溶解物で形質導入し、25μg/mLのカナマイシンを含有するLBプレート上にストリークした。カナマイシン耐性形質導入体を、プライマーfrdB CkUp(配列番号36)およびfrdB CkDn(配列番号37)を伴うコロニーPCRによってスクリーニングした。予想された約1.6kbのPCR産物を生成するコロニーを、エレクトロコンピテントにし、上記のようにマーカーの除去のため、pCP20で形質転換した。形質転換体をまず、100μg/mLのアンピシリンを含有するLBプレート上に30℃で展開し、次いでアンピシリン耐性形質転換体を選択し、LBプレート上にストリークし、42℃で成長させた。単離したコロニーを、アンピシリンおよびカナマイシン選択培地プレートならびにLBプレート上にパッチした。カナマイシン選択、アンピシリン選択コロニーを、プライマーfrdB CkUp(配列番号36)およびfrdB CkDn(配列番号37)を伴うPCRによってスクリーニングした。予想された約0.4kbのPCR産物が認められ、それからマーカーの除去が確認され、二重ノックアウト株「ΔpflB frdB」が生成された。
【0148】
二重ノックアウト株を、JW1375(ΔldhA::kan)由来のP1vir溶解物で形質導入し、25μg/mLのカナマイシンを含有するLBプレート上に展開した。カナマイシン耐性形質導入体を、プライマーldhA CkUp(配列番号38)およびldhA CkDn(配列番号39)を伴うコロニーPCRによってスクリーニングした。予想された1.1kbのPCR産物を生成するクローンを、エレクトロコンピテントにし、上記のようにマーカーの除去のため、pCP20で形質転換した。形質転換体を、100μg/mLのアンピシリンを含有するLBプレート上に30℃で展開し、アンピシリン耐性形質転換体をLBプレート上にストリークし、42℃で成長させた。単離したコロニーを、アンピシリンおよびカナマイシン選択培地プレートならびにLBプレート上にパッチした。カナマイシン選択、アンピシリン選択コロニーを、0.3kbの産物について、プライマーldhA CkUp(配列番号38)およびldhA CkDn(配列番号39)を伴うPCRによってスクリーニングした。予想された約0.3kbのPCR産物を生成するクローンにより、マーカーの除去が確認され、「3KO」(ΔpflB frdB ldhA)と称される三重ノックアウト株が生成された。
【0149】
「3KO」株を、JW1228(ΔadhE::kan)由来のP1vir溶解物で形質導入し、25μg/mLのカナマイシンを含有するLBプレート上に展開した。カナマイシン耐性形質導入体を、プライマーadhE CkUp(配列番号40)およびadhE CkDn(配列番号41)を伴うコロニーPCRによってスクリーニングした。予想された1.6kbのPCR産物を生成するクローンを、エレクトロコンピテントにし、マーカーの除去のため、pCP20で形質転換した。形質転換体を、100μg/mLのアンピシリンを含有するLBプレート上に30℃で展開した。アンピシリン耐性形質転換体をLBプレート上にストリークし、42℃で成長させた。単離したコロニーを、アンピシリンおよびカナマイシン選択プレートならびにLBプレート上にパッチした。カナマイシン選択、アンピシリン選択コロニーを、プライマーadhE CkUp(配列番号40)およびadhE CkDn(配列番号41)を伴うPCRによってスクリーニングした。予想された約0.4kbのPCR産物を生成するクローンを、「4KO」(ΔpflB frdB ldhA adhE)と称した。
【0150】
実施例2
イソブタノール生合成経路ならびにpflB、frdB、ldhA、およびadhE遺伝子の欠失を有する大腸菌(E.coli)生産宿主の作成
アクロモバクター・キシロスオキシダンス(Achromobacter xylosoxidans)由来のブタノールデヒドロゲナーゼをコードするDNA断片(DNA配列番号9;タンパク質配列番号10)を、標準条件を使用して、A.キシロスオキシダンス(A.xylosoxidans)ゲノムDNAから増幅した。DNAを、Gentra Puregeneキット(Gentra Systems,Inc.(Minneapolis,MN);カタログ番号D−5500A)を使用し、グラム陰性微生物における推奨プロトコルに従って調製した。PCR増幅を、フォワードおよびリバースプライマーのN473およびN469(配列番号44および45)を使用し、それぞれPhusion high Fidelity DNAポリメラーゼ(New England Biolabs(Beverly,MA))を使用して行った。PCR産物を、pCR4 BLUNT(Invitrogen)にTOPO−Bluntクローニングを行い、pCR4Blunt::sadBを生成し、それを大腸菌(E.coli)Mach−1細胞に形質転換した。それに続き、プラスミドを4つのクローンから単離し、配列を検証した。
【0151】
次いで、sadBコード領域を、ベクターpTrc99a(Amannら、Gene 69:301−315頁、1988年)にクローン化した。pCR4Blunt::sadBをEcoRIで消化し、sadB断片を放出し、それをEcoRIで消化されたpTrc99aとライゲートし、pTrc99a::sadBを生成した。このプラスミドを、大腸菌(E.coli)Mach1細胞に形質転換し、得られた形質転換体をMach1/pTrc99a::sadBと称した。これらの細胞におけるsadB遺伝子から発現される酵素の活性は、標準としてイソブチルアルデヒドを使用して分析すると、無細胞抽出液中で3.5mmol/分/mgのタンパク質であることが判定された。
【0152】
次いで、sadB遺伝子を、下記のように、pTrc99A::budB−ilvC−ilvD−kivDにサブクローン化した。pTrc99A::budB−ilvC−ilvD−kivDは、(同時係属中の共同所有された米国特許出願公開第20070092957号明細書(参照により本明細書中に援用される)の実施例9〜14に記載の)イソブタノール発現のためのオペロンを有するpTrc−99a発現ベクターである。pTrc99A::budB−ilvC−ilvD−kivDイソブタノールオペロン内の最初の遺伝子はクレブシエラ・ニューモニエ(Klebsiella pneumoniae)ATCC25955由来のアセト乳酸シンターゼをコードするbudBであり、次いで大腸菌(E.coli)由来のアセトヒドロキシ酸レダクトイソメラーゼをコードするilvC遺伝子である。これに、大腸菌(E.coli)由来のアセトヒドロキシ酸デヒドラターゼをコードするilvDが続き、最後はL.ラクティス(L.lactis)由来の分岐鎖ケト酸デカルボキシラーゼをコードするkivD遺伝子である。
【0153】
sadBコード領域を、プライマーN695A(配列番号42)およびN696A(配列番号43)を使用し、Phusion High Fidelity DNAポリメラーゼ(New England Biolabs(Beverly,MA))を使用し、pTrc99a::sadBから増幅した。増幅を、98℃で1分間の初期変性、次いで98℃で10秒間の変性の30サイクル、62℃で30秒間のアニーリング、72℃で20秒間の伸長および72℃で5分間の最終伸長サイクル、次いで4℃で保持の場合に行った。プライマーN695Aは、クローニングにおけるAvrII制限部位と、sadBコード領域のATG開始コドンの上流にRBSを有した。N696Aプライマーは、クローニングにおけるXbaI部位を含んだ。1.1kbのPCR産物を、AvrIIおよびXbaI(New England Biolabs(Beverly,MA))で消化し、Qiaquick Gel Extraction Kit(Qiagen Inc.(Valencia,CA))を使用してゲル精製した。精製断片を、T4 DNAリガーゼ(New England Biolabs(Beverly,MA))を使用し、同じ制限酵素で切断済みのpTrc99A::budB−ilvC−ilvD−kivDとライゲートした。ライゲーション混合物を、16℃で一晩インキュベートし、次いで製造業者のプロトコルに従い、大腸菌(E.coli)Mach1(商標)コンピテント細胞(Invitrogen)に形質転換した。形質転換体を、100μg/mlのアンピシリンを含有するLB寒天上で成長させた後に得た。形質転換体由来のプラスミドDNAを、QIAprep Spin Miniprep Kit(Qiagen Inc.(Valencia,CA))を使用し、製造業者のプロトコルに従って調製した。得られたプラスミドを、pTrc99A::budB−ilvC−ilvD−kivD−sadBと称した。エレクトロコンピテント4KO細胞を、上記のように調製し、pTrc99A::budB−ilvC−ilvD−kivD−sadBで形質転換した。形質転換体を、100μg/mLのアンピシリンを含有するLB寒天プレート上にストリークした。4KOを伴うプラスミドpTrc99A::budB−ilvC−ilvD−kivD−sadBを有する得られた株(NGCI−031株と称する)を、実施例3で概説される発酵試験において使用した。
【0154】
実施例3
抽出発酵を使用する、組換え大腸菌(E.coli)によるイソブタノールの生成
本実施例の目的は、本明細書中で上記のように作成した大腸菌(E.coli)株NGCI−031によるイソブタノールの生成を示すことである。接種材料の調製のため、すべてのシード培養物を、選択抗生物質としてアンピシリン(100mg/L)を含有するLB培地中で成長させた。この発酵用に使用した半合成培地の組成物および使用した微量金属の調合物を、下記の表3および4に示す。
【0155】
【表6】
【0156】
【表7】
【0157】
表3からの成分1〜10を、所定濃度で水に加え、発酵槽内での最終容量を1.5Lにし、発酵槽の内容物をオートクレーブによって滅菌した。オートクレーブ培地を冷却した上で、成分11〜13を、混合し、濾過滅菌し、次いで発酵槽に加えた。発酵培地(水相)の全最終容量は約1.6Lであった。
【0158】
2.0Lの作動容量を有する3LのBiostat−B DCU−3発酵槽(Braun Biotech International(Melesungen,Germany))を発酵に使用する一方、温度を30℃で維持し、水酸化アンモニウムを使用して
pHを6.8に維持した。シード培養物(2〜10vol%)を培地に接種後、発酵槽を、撹拌速度(rpm)を自動的に制御しながら、0.5vvmの気流を伴う30%の溶解酸素(DO)の設定点で好気的に作動させた。一旦、OD600が10に達すると、培養物を、0.4〜0.5mMのIPTGで誘導し、イソブタノール経路を過剰発現させた。誘導の4時間後、発酵条件を、撹拌器速度を200rpmに低下させることにより、微好気性に変更した。微好気的条件への変更により、バイオマスを生成するための炭素の取り込みを最小化しながらイソブタノールの生成が開始され、それによりバイオマスの形成とイソブタノールの生成が脱共役された。オレイルアルコール(約780mL)をイソブタノール生成段階中に添加し、水相中でのイソブタノールの形成に起因する生成物誘発性の阻害を緩和した。グルコースを発酵槽にボーラス(50wt%のストック溶液)として加え、グルコースレベルを30g/L〜2g/Lの間で保持した。
【0159】
イソブタノールの効率的生成には、生合成経路内でのレドックス均衡を可能にする微好気的条件が要求されることから、空気を発酵槽に0.5vvmで連続的に供給した。連続的吸気により、発酵槽の水相からのイソブタノールの有意なストリッピングが生じた。ストリッピングに起因するイソブタノールの損失を測定するため、発酵槽からの排気を冷却(6.5℃)水トラップを通してスパージし、イソブタノールを濃縮し、次いでそれを、Prima dB質量分析計(Thermo Electron Corp.(Madison,WI))を使用する質量分析を用いて定量した。74または42の質量対電荷比でのイソブタノールのピークを使用し、存在するイソブタノールの量を測定した。
【0160】
水相中のグルコースおよび有機酸を、BioProfile(登録商標)300 Analyzer(Nova Biomedical(Waltham,MA))を使用する発酵中に定期的に監視した。グルコースについても、グルコース分析器(YSI,Inc.(Yellow Springs,OH))を使用して監視した。水相中のイソブタノールおよびオレイルアルコール相中のイソブタノールを、下記のガスクロマトグラフィー(GC)を用いて監視した。2つの相を、遠心分離によって分離した。GC分析を上記のように実施した。イソブタノールの生成における有効力価、速度、および収率(ストリッピングに起因するイソブタノールの損失に対して補正された)は、それぞれ35g/L、0.40g/L/h、および0.33g/gであった。イソブタノールの生成を目的とした抽出発酵におけるオレイルアルコールの使用は、発酵培地および宿主株からの毒性イソブタノール生成物の抽出故に、結果として有効力価、速度、および収率が有意に高まる。
【0161】
実施例4
本実施例の目的は、大腸菌(E.coli)宿主内でのピルビン酸・ギ酸リアーゼ、フマル酸レダクターゼ、アルコールデヒドロゲナーゼおよび乳酸デヒドロゲナーゼをコードする遺伝子における欠失のイソブタノールの生成に対する効果を、これらの欠失を有しない宿主の場合と比較することである。
【0162】
(pflB、frdB、ldhA、およびadhE遺伝子における欠失を含む)大腸菌(E.coli)株NGCI−031によるイソブタノールの生成を、pflB、frdB、ldhA、およびadhE遺伝子に対する欠失を有しない大腸菌(E.coli)株の場合と比較するため、大腸菌(E.coli)株MG1655(ATCC47076)を、プラスミドpTrc99A::budB−ilvC−ilvD−kivD−sadBで形質転換し、大腸菌(E.coli)株MG1655/pTrc99A::budB−ilvC−ilvD−kivD−sadBを生成した。発酵は、本質的に上記のように行ったが、オレイルアルコールを伴わなかった。NGCI−031株におけるイソブタノールの生成における有効力価、速度、および収率(ストリッピングに起因するイソブタノールの損失に対して補正された)は、それぞれ11g/L、0.23g/L/h、および0.25g/gであった一方、MG1655/pTrc99A::budB−ilvC−ilvD−kivD−sadB株におけるイソブタノールの生成における有効力価、速度、および収率(ストリッピングに起因するイソブタノールの損失に対して補正された)は、それぞれ14g/L、0.18g/L/h、および0.12g/gであった。pflB、frdB、ldhA、およびadhEにおける欠失により、pflB、frdB、ldhA、およびadhEにおける欠失を伴わない株に対して有意に改善された速度および収率が得られ、pflB、frdB、ldhA、およびadhEが欠失した株における力価の低下は、発酵時間が短縮した結果であった。
【技術分野】
【0001】
関連出願の相互参照
本願は、2008年6月4日に出願された米国仮特許出願第61/058568号明細書の便益性を主張し、その開示内容はその全体が本明細書によって援用される。
【0002】
本発明は、微生物学および分子生物学の分野に関する。より詳細には、本発明は、イソブタノールに対して向上した生成能を有する腸内の欠失突然変異体について記載する。
【背景技術】
【0003】
ブタノールは、燃料添加剤として、プラスチック産業における化学原料として、さらに食品および香料産業における食品等級の抽出剤として有用な重要な産業化学物質である。毎年、100〜120億ポンドのブタノールが石油化学的手段によって生産されており、この汎用化学物質に対する需要は高まる可能性が高い。石油原料を介したイソブタノールの既知の化学合成は、高価であり、環境に優しくないが、植物由来の原料からのイソブタノールの生成であれば、温室効果ガス排出を最小限にし、当該技術分野において進歩性を示すこととなる。
【0004】
イソブタノールは、酵母発酵の間、アミノ酸、具体的にはL−バリンの不完全代謝の副生成物として生物学的に生成される。窒素源としてのL−バリンのアミン基の代謝後、得られるα−ケト酸は、Ehrlich経路を介して、脱カルボキシル化され、非常に低い収率であってもイソブタノールに還元される。例えば、ビール発酵において生成されるイソブタノールの濃度は、100万分の16未満である。
【0005】
糖類からの直接的なイソブタノールの経済的な生合成であれば、単一の生成物としてのイソブタノールの生成を目的とした、環境的な責任がある費用効果的なプロセスを示すことになる。同時係属中の共同所有される特許文献1では、グルコースのイソブタノールへの変換に必要なあらゆる酵素活性を過剰発現する株が開示され、低濃度(0.3〜10mM)でのイソブタノールの生成が示された。
【0006】
最近、非特許文献1は、イソブタノールを最大で300mMの濃度で生成する組換え大腸菌(E.coli)株の開発について記載した。この組換え大腸菌(E.coli)では、遺伝子adhE、ldhA、frdBC、fnr、ptaおよびpflB、ならびに2つの共同所有された同時係属中の特許文献1中に記載の場合と同様のイソブタノール生合成経路を担持するプラスミドが破壊された。これらのプラスミドは、アセト乳酸シンターゼ、アセトヒドロキシ酸レダクトイソメラーゼ、アセトヒドロキシ酸デヒドラターゼ、2−ケト酸デカルボキシラーゼおよびアルコールデヒドロゲナーゼを有する。Atsumiら(上記)の見解は、イソブタノール生合成経路を有する宿主細胞が、競合する炭素経路に対して重要な遺伝子が破壊される場合の改善されたイソブタノール生成を得る可能性があることを意味している。しかし、Atsumiら(上記)の宿主細胞は、改善されたイソブタノール生成を得るのに必要とされる場合よりはるかに多い数の遺伝子組換えを有するように思われる。基本的な内因性炭素経路における遺伝子組換えの数が増えると、宿主細胞代謝が低下する可能性が高まり、それにより、最終的には細胞の生産宿主としての使用に支障をきたすことになる。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】米国特許出願公開第20070092957号明細書
【0008】
【非特許文献1】Atsumi S.ら、Nature 451:86−90頁、2008年
【発明の概要】
【発明が解決しようとする課題】
【0009】
したがって、イソブタノールを生成するための内因性炭素経路における最小数の遺伝子組換えを有する宿主細胞に対して需要がある。出願人は、上述の課題を解決しており、イソブタノールの収率向上をもたらす、内因性炭素経路内の4つの遺伝子のみに破壊を有する腸内細菌宿主細胞について本明細書中に記載する。
【課題を解決するための手段】
【0010】
本発明は、イソブタノールを生成するための腸内細菌生産宿主について記載する。宿主細胞は、好ましくは、ブタノールの生成の最終ステップにおいてブタノールデヒドロゲナーゼ(二次アルコールデヒドロゲナーゼ、sadB)を用いるイソブタノール生合成経路を有し、かつ、内因性炭素経路において遺伝子組換えを有し、これにより、細胞は、次の酵素活性、すなわち、1)ピルビン酸・ギ酸リアーゼ(EC2.3.1.54)、2)フマル酸レダクターゼ酵素複合体(EC1.3.99.1)、3)アルコールデヒドロゲナーゼ(EC1.2.1.10−アセトアルデヒドデヒドロゲナーゼおよびEC1.1.1.1−アルコールデヒドロゲナーゼ)、および4)乳酸デヒドロゲナーゼ(EC1.1.1.28)の少なくとも1つを含まない状態になる。これらの酵素活性における破壊を有する腸内宿主は、イソブタノールの速度が、これらの破壊を有しない類似宿主に対して改善されることを示す。
【0011】
したがって、本発明は、ブタノールデヒドロゲナーゼ活性を有するポリペプチドをコードする少なくとも1つの遺伝子を含む、イソブタノールを生成するための腸内生産宿主を提供し、ここで宿主は、イソブタノールを生成し、かつ、以下の酵素活性、すなわち、
a)ピルビン酸・ギ酸リアーゼ(EC2.3.1.54)
b)フマル酸レダクターゼ酵素複合体(EC1.3.99.1)
c)アルコールデヒドロゲナーゼ(EC1.2.1.10/EC1.1.1.1)
d)乳酸デヒドロゲナーゼ(EC1.1.1.28)
の少なくとも1つを実質的に有しない。
【0012】
別の実施形態では、本発明は、本発明の宿主細胞が、ポリペプチドが、スコアリングマトリックスBLOSUM62、期待値カットオフ(expect cutoff)10およびワードサイズ3と、ギャップオープニングペナルティ(gap opening penalty)11およびギャップ伸長(gap extension)1とを伴うBLASTを用いると、348アミノ酸長を超える配列番号10で示されるアミノ酸配列に対して少なくとも90%の同一性を有する場合のブタノールデヒドロゲナーゼ活性を有するポリペプチドをコードする少なくとも1つの遺伝子を含むことを提供する。
【0013】
別の実施形態では、本発明は、
a)ピルビン酸塩をアセト乳酸塩に変換するためのEC番号2.2.1.69を有するアセト乳酸シンターゼをコードする少なくとも1つの遺伝子、
b)アセト乳酸塩を2,3−ジヒドロキシイソ吉草酸に変換するためのアセトヒドロキシ酸イソメロレダクターゼEC番号1.1.1.86をコードする少なくとも1つの遺伝子、
c)2,3−ジヒドロキシイソ吉草酸をα−ケトイソ吉草酸に変換するためのアセトヒドロキシ酸デヒドラターゼEC番号4.2.1.9をコードする少なくとも1つの遺伝子、
d)α−ケトイソ吉草酸をイソブチルアルデヒドに変換するための分岐鎖ケト酸デカルボキシラーゼEC番号4.1.1.72をコードする少なくとも1つの遺伝子、
e)A.キシロスオキシダンス(A.xylosoxidans)から単離される、ブタノールデヒドロゲナーゼポリペプチドをコードする少なくとも1つの遺伝子
を含むイソブタノール生合成経路を含む宿主細胞を提供する。
【0014】
別の実施形態では、本発明は、本発明の生産宿主を、イソブタノールが生成される条件下で、炭素基質を含む発酵培地中で成長させるステップを含む、イソブタノールを生成するための方法を含む。
【0015】
本発明は、本願の一部を形成する、以下の詳細な説明、図面、および添付の配列記述からより十分に理解されうる。
【図面の簡単な説明】
【0016】
【図1A】「a」、「b」、「c」、「d」、および「e」と称されるステップからなる本発明のイソブタノール生合成経路を表し、下記の生成物変換に対する基質を表す。「f」〜「i」の反応は、イソブタノール合成における可用性を低減する副反応におけるピルビン酸塩の消費を阻止するための、本開示において破壊されている特定の4つの反応を表す。
【図1B】「a」、「b」、「c」、「d」、および「e」と称されるステップからなる本発明のイソブタノール生合成経路を表し、下記の生成物変換に対する基質を表す。「f」〜「i」の反応は、イソブタノール合成における可用性を低減する副反応におけるピルビン酸塩の消費を阻止するための、本開示において破壊されている特定の4つの反応を表す。
【0017】
以下の配列は、米国特許施行規則第1.821−1.825条(「ヌクレオチド配列および/またはアミノ酸配列開示を有する特許出願の要件−配列の規則(Requirements for Patent Applications Containing Nucleotide Sequences and/or Amino Acid Sequence Disclosures−the Sequence Rules)」)に従い、世界知的所有権機関(World Intellectual Property Organization)(WIPO)基準ST.25(1998年)、ならびにEPOおよびPCTの配列表の要件(規則5.2および49.5(aの2)、ならびに実施細則の第208節および付録C)に一致する。ヌクレオチドおよびアミノ酸配列データにおいて用いられる記号および形式は、米国特許施行規則第1.822条に示される規則に従う。
【0018】
本発明のヌクレオチドおよびアミノ酸配列は、下記の表1および2中にあげられる。
【0019】
【表1】
【0020】
【表2】
【0021】
【表3】
【0022】
【表4】
【0023】
【表5】
【発明を実施するための形態】
【0024】
本開示では、抽出発酵条件下で予想外の高レベルのイソブタノール(例えば35g/L)を生成する、一連の経路因子および欠失を組み合わせた新規な生産宿主の開発について記載する。本開示では、遺伝子adhE、ldhA、frdB、およびpflBが破壊された大腸菌(E.coli)株について記載する。共同所有された米国特許出願第20070092957号明細書中に記載のpTrc99A::budB−ilvC−ilvD−kivDプラスミドは、アクロモバクター・キシロスオキシダンス(Achromobacter xylosoxidans)由来のブタノールデヒドロゲナーゼsadBの付加によって修飾され、イソブタノール生産宿主が生成された。本開示は、複数の商業的および産業的需要を満たす。ブタノールは、種々の用途を有する重要な産業用商品の化学物質であり、その場合、燃料または燃料添加剤としてのその可能性は特に有意である。ブタノールは、4炭素アルコールにすぎなくても、ガソリンの含量に類似のエネルギー含量を有し、任意の化石燃料と混和されうる。ブタノールは、標準の内燃エンジン内で燃焼される場合にCO2のみを生成し、SOXまたはNOXをほとんど生成しないことから燃料または燃料添加剤として好まれる。さらにブタノールは、今日まで最も好ましい燃料添加剤であるエタノールよりも腐食性が少ない。
【0025】
さらに、本開示では、植物由来の炭素源からのイソブタノールの生成について記載し、ブタノール生成のための標準の石油化学プロセスに関連した負の環境影響を回避する。
【0026】
次の定義および略語は、特許請求の範囲および本明細書の解釈を意図して使用されるべきである。
【0027】
本明細書で使用される用語「comprises」、「comprising」、「includes」、「including」、「has」、「having」、「contains」または「containing」、または任意の他のこれらの変形は、非排他的な包含(non−exclusive inclusion)を網羅するように意図される。たとえば、要素のリストを含む組成物、混合物、プロセス、方法、物品、または装置は、必ずしもそれらの要素のみに限定されないが、明示的に列挙されないかまたはかかる組成物、混合物、プロセス、方法、物品、または装置に固有の他の要素を含みうる。さらに、相反する明示的な記載がない限り、「または」は、「包含的なまたは(inclusive or)」を示し、「限定的なまたは(exclusive or)」を示さない。たとえば、条件AまたはBは、Aは真であり(または存在し)かつBは偽である(存在しない)、Aは偽であり(存在せず)かつBは真である(または存在する)、およびAとBの双方が真である(または存在する)のいずれか1つによって満足される。
【0028】
また、本発明の要素または成分に先行する不定冠詞「a」および「an」は、要素または成分の事例(すなわち出現)の数について非制限的であることが意図される。したがって、「a」または「an」は、1つまたは少なくとも1つを含むように解釈されるべきであり、要素または成分の単数の語形はまた、数が明らかに単数であることを意味しない限り、複数を含む。
【0029】
用いられる本発明の成分または反応物の量を修飾する、本明細書で使用される用語「約」は、たとえば、実地での濃縮物の作製または溶液の使用に用いられる典型的な測定および液体処理方法を通じて;これらの方法における不測の誤りを通じて;組成物を作製するかまたはこれらの方法を実施するために使用される成分の製造、供給源、または純度における差異を通じて生じうる数量における変動を示す。用語「約」はまた、特定の初期混合物から生じる組成物に対する、異なる平衡条件によって異なる量を包含する。用語「約」によって修飾されるか否かに無関係に、特許請求の範囲は、量に対する等価物を含む。一実施形態では、用語「約」は、報告される数値の10%以内、好ましくは報告される数値の5%以内を意味する。
【0030】
用語「発明」または「本発明」は、本明細書で使用される場合、非限定的用語であり、特定の発明の任意の単一の実施形態を示すように意図されていないが、本明細書および特許請求の範囲において記載される、考えられるあらゆる実施形態を包含する。
【0031】
用語「イソブタノール生合成経路」は、イソブタノールを生成するための酵素経路を示す。典型的なイソブタノール生合成経路は、共同所有された同時係属中の米国特許出願公開第20070092957A1号明細書(その全体が参照により本明細書中に援用される)中で考察され、説明されている。
【0032】
用語「ノックアウト」は、その特定の遺伝子を機能不全にするための、プラスミドまたは微生物内での特定の遺伝子の破壊を示す。本開示では、遺伝子adhE、ldhA、frdB、およびpflBは、イソブタノール生成のため、宿主株においてノックアウトされた。
【0033】
用語「pflB」は、ピルビン酸塩をギ酸塩に変換するピルビン酸・ギ酸リアーゼ酵素をコードする遺伝子を示す。
【0034】
用語「frdABCD」は、コハク酸塩をフマル酸塩に変換するフマル酸レダクターゼ酵素複合体をコードするオペロンを示す。
【0035】
用語「ldhA」は、乳酸デヒドロゲナーゼ酵素をコードする遺伝子を示し、ピルビン酸塩を乳酸塩に変換する。
【0036】
用語「adhE」は、アセチル−CoAをエタノールに変換するピルビン酸・ギ酸リアーゼ酵素をコードする遺伝子を示す。
【0037】
用語「アセト乳酸シンターゼ」および「アセト乳酸シンテターゼ」は、ピルビン酸塩のアセト乳酸塩およびCO2への変換を触媒する酵素を示すように、本明細書中で交換可能に使用される。好ましいアセト乳酸シンターゼは、EC番号2.2.1.69(Enzyme Nomenclature 1992年、Academic Press,San Diego)により既知である。これらの酵素は、限定はされないが、枯草菌(Bacillus subtilis)(GenBank番号:それぞれ、CAB15618(配列番号11)、Z99122(配列番号12)、NCBI(National Center for Biotechnology Information)アミノ酸配列、NCBIヌクレオチド配列)、クレブシエラ・ニューモニエ(Klebsiella pneumoniae)(GenBank番号:AAA25079(配列番号2)、M73842(配列番号1)、およびラクトコッカス・ラクティス(Lactococcus lactis)(GenBank番号:AAA25161(配列番号13)、L16975(配列番号14)を含む複数の供給源から入手可能である。
【0038】
用語「アセトヒドロキシ酸イソメロレダクターゼ」および「アセトヒドロキシ酸レダクトイソメラーゼ」は、電子供与体としてNADPH(還元型ニコチンアミドアデニンジヌクレオチドリン酸)を使用する、アセト乳酸塩の2,3−ジヒドロキシイソ吉草酸への変換を触媒する酵素を示すように、本明細書中で交換可能に使用される。好ましいアセトヒドロキシ酸イソメロレダクターゼは、EC番号1.1.1.86により既知であり、配列は、限定はされないが、大腸菌(Escherichia coli)(GenBank番号:NP_418222(配列番号4)、NC_000913(配列番号3))、サッカロミセス・セレビシエ(Saccharomyces cerevisiae)(GenBank番号:NP_013459(配列番号15)、NC_001144(配列番号16))、メタノコッカス・マリパルディス(Methanococcus maripaludis)(GenBank番号:CAF30210(配列番号17)、BX957220(配列番号18))、および枯草菌(Bacillus subtilis)(GenBank番号:CAB14789(配列番号19)、Z99118(配列番号20))を含む非常に多数の微生物から入手可能である。
【0039】
用語「アセトヒドロキシ酸デヒドラターゼ」は、2,3−ジヒドロキシイソ吉草酸のα−ケトイソ吉草酸への変換を触媒する酵素を示す。好ましいアセトヒドロキシ酸デヒドラターゼは、EC番号4.2.1.9により既知である。これらの酵素は、限定はされないが、大腸菌(E.coli)(GenBank番号:YP_026248(配列番号6)、NC_000913(配列番号5))、S.セレビシアエ(S.cerevisiae)(GenBank番号:NP_012550(配列番号21)、NC_001142(配列番号22))、M.マリパルディス(M.maripaludis)(GenBank番号:CAF29874(配列番号23)、BX957219(配列番号24))、および枯草菌(B.subtilis)(GenBank番号:CAB14105(配列番号25)、Z99115(配列番号26))を含む非常に多数の微生物から入手可能である。
【0040】
用語「分岐鎖α−ケト酸デカルボキシラーゼ」は、α−ケトイソ吉草酸のイソブチルアルデヒドおよびCO2への変換を触媒する酵素を示す。好ましい分岐鎖α−ケト酸デカルボキシラーゼは、EC番号4.1.1.72により既知であり、限定はされないが、ラクトコッカス・ラクティス(Lactococcus lactis)(GenBank番号:AAS49166(配列番号27)、AY548760(配列番号28);CAG34226(配列番号8)、AJ746364(配列番号29))、ネズミチフス菌(Salmonella typhimurium)(インドールピルビン酸デカルボキシラーゼとしても既知)(GenBank番号:NP_461346(配列番号30)、NC_003197(配列番号31))、およびクロストリジウム・アセトブチリクム(Clostridium acetobutylicum)(ピルビン酸デカルボキシラーゼとしても既知)(GenBank番号:NP_149189(配列番号32)、NC_001988(配列番号33))を含む非常に多数の微生物から入手可能である。
【0041】
用語「ブタノールデヒドロゲナーゼ」および「二次アルコールデヒドロゲナーゼ」は、ここでは交換可能に使用され、かつ多数の微生物内で生じ、NAD+のNADHへの還元とともにアルコールとアルデヒドまたはケトンとの間の相互変換を促進する酵素を示す。かかる酵素の好ましい例は、アクロモバクター・キシロスオキシダンス(Achromobacter xylosoxidans)由来のブタノールデヒドロゲナーゼである(ヌクレオチド配列番号9およびアミノ酸配列番号10)。A.キシロスオキシダンス(A.xylosoxidans)sadB酵素は、イソブチルアルデヒドのイソブタノールへの変換を触媒する。
【0042】
用語「炭素基質」または「発酵性炭素基質」は、本発明の宿主微生物によって代謝可能な炭素源、特に、単糖、例えばグルコースまたはフルクトース;二糖、例えば乳糖またはスクロース;オリゴ糖;多糖、例えばデンプンまたはセルロース;1つの炭素基質;およびそれらの混合物からなる群から選択される炭素源を示す。
【0043】
「遺伝子」という用語は、場合によりコード配列の上流(5’非コード配列)および下流(3’非コード配列)の調節配列を含む、特定のタンパク質として発現可能な核酸断片を示す。「天然遺伝子」は、それ自体の調節配列を有する天然に見出される遺伝子を示す。「キメラ遺伝子」は、天然遺伝子でない任意の遺伝子を示し、天然に見出されることのない調節およびコード配列を含む。したがって、キメラ遺伝子は、異なる供給源に由来する調節配列およびコード配列または同じ供給源に由来する調節配列およびコード配列を含みうるが、天然に見出されるものとは異なる様式で配列されている。「内因性遺伝子」は、微生物のゲノム内のその天然の位置における天然遺伝子を示す。「外来遺伝子」または「異種遺伝子」は、通常は宿主微生物内に見出されることはないが宿主微生物に遺伝子導入によって導入される遺伝子を示す。外来遺伝子は、非天然の微生物に導入される天然遺伝子またはキメラ遺伝子を含みうる。「トランス遺伝子」は、形質転換手順によりゲノムに導入されている遺伝子である。
【0044】
本明細書で用いられる「単離核酸断片」または「単離核酸分子」または「遺伝的構造物」は同義に用いられ、かつ一本鎖または二本鎖のRNAまたはDNAの高分子を意味し、場合により合成、非天然または改変ヌクレオチド塩基を有することになる。DNAの高分子の形態の単離核酸断片については、cDNA、ゲノムDNAまたは合成DNAのうちの1つもしくは複数のセグメントから構成されうる。
【0045】
本明細書で使用される用語「コード配列」は、特定のアミノ酸配列をコードするDNA配列を示す。「好適な調節配列」は、コード配列の上流(5’非コード配列)、内部、または下流(3’非コード配列)に位置し、関連コード配列の転写、RNAプロセシングまたは安定性、または翻訳に影響を与えるヌクレオチド配列を示す。調節配列は、プロモーター、翻訳リーダー配列、イントロン、ポリアデニル化認識配列、RNAプロセシング部位、エフェクター結合部位およびステムループ構造を含んでもよい。
【0046】
用語「プロモーター」は、コード配列または機能RNAの発現を制御することが可能なDNA配列を示す。一般に、コード配列は、プロモーター配列の3’側に位置する。プロモーターは、その全体において天然遺伝子に由来するか、または天然に見出される異なるプロモーターに由来する異なる要素から構成されるか、または合成DNAセグメントをさらに含む場合がある。異なるプロモーターが、異なる組織または細胞種において、または発現の異なる段階で、または異なる環境的または生理的状態に応答して、遺伝子の発現を誘導することが可能であることは当業者によって理解されている。ほとんどの時期に大部分の細胞種において遺伝子の発現を引き起こすプロモーターは、一般に「構成的プロモーター」と称される。ほとんどの場合、調節配列の正確な境界が完全には限定されていないことから、異なる長さのDNA断片が同一のプロモーター活性を有しうることはさらに理解されている。
【0047】
用語「発現」は、本明細書で使用される場合、本発明の核酸断片に由来するセンス(mRNA)またはアンチセンスRNAの転写および安定な蓄積を示す。発現はまた、mRNAのポリペプチドへの翻訳を示しうる。
【0048】
本明細書で使用される用語「形質転換」は、遺伝的に安定な遺伝的形質をもたらす、核酸断片の宿主微生物への転移を示す。形質転換核酸断片を有する宿主微生物は、「トランスジェニック」または「組換え」または「形質転換」微生物と称される。
【0049】
用語「プラスミド」は、細胞の中央代謝の一部でない遺伝子を有することが多く、通常は環状二本鎖DNA分子の形態をなす余分な染色体要素を示す。かかる要素は、任意の供給源に由来する線状または環状の一本鎖または二本鎖のDNAまたはRNAにおける自動複製配列、ゲノム組み込み(integrating)配列、ファージまたはヌクレオチド配列である場合があり、ここでは複数のヌクレオチド配列が、発現カセットを細胞に導入可能な固有の作成物に連結されるかまたは組換えられており、ここで前記発現カセットは、選択される遺伝子のコード配列と、選択される遺伝子産物の発現にとって必要とされるコード配列に先行する調節配列(5’非コード配列)および後続する調節配列(3’非コード配列)とを含む。
【0050】
用語「コドン最適化された」は、さまざまな宿主の形質転換における核酸分子の遺伝子またはコード領域を示す場合、DNAによってコードされるポリペプチドを改変することなく宿主微生物の典型的なコドン使用を反映させるための、核酸分子の遺伝子またはコード領域におけるコドンの改変を示す。
【0051】
本明細書で使用される用語「形質導入」および「普遍形質導入」は、交換可能に使用され、細菌DNAが、細菌DNAを有するファージ粒子により、ある細菌細胞(ドナ−)から別の(レシピエント)へ転移される現象を示す。
【0052】
用語「P1ドナー細胞」および「ドナー細胞」は、交換可能に使用され、バクテリオファージまたはウイルスによって感染を受けやすく、また形質導入粒子にパッケージ化される核酸断片における供給源としての役割を果たす細菌株を示す。典型的には、ドナー細胞の遺伝子構成(genetic make up)は、ドナー細胞によって生成される形質導入ファージまたはウイルスを含有するP1溶解物を受け取る役割を果たす「レシピエント細胞」と類似または同一である。
【0053】
用語「P1レシピエント細胞」および「レシピエント細胞」は、交換可能に使用され、バクテリオファージまたはウイルスによって感染を受けやすく、またドナー細胞によって生成される形質導入ファージまたはウイルスを含有する溶解物を受け取る役割を果たす細菌株を示す。
【0054】
用語「カオトロピック剤」は、タンパク質、DNA、またはRNAなどの高分子中の三次元構造を破壊する物質を意味する。
【0055】
用語「共沸剤」は、組成が単なる蒸留によって変化を受ける可能性がないような比での2つ以上の純粋な化学物質の混合物を示す。
【0056】
用語「パーベーパレイション」は、無孔質膜または多孔質膜を通る部分的蒸発による、液体の混合物を分離するための方法を示す。
【0057】
用語「親水性」は、水素結合を介して水(H2O)と一時的に結合可能な分子の物理的特性を示す。
【0058】
用語「実質的に有しない」は、本イソブタノール経路と競合する炭素経路内での酵素活性(例えば、ピルビン酸・ギ酸リアーゼ、フマル酸レダクターゼ、アルコールデヒドロゲナーゼおよび乳酸デヒドロゲナーゼ)の存在または不在に関連して使用される場合、酵素のレベルが、実質的に野生型宿主内での同じ酵素のレベル未満であること(野生型レベルの50%未満が好ましく、かつ野生型レベルの約90%未満が最も好ましい場合)を意味する。
【0059】
当該技術分野で既知のように、用語「同一性(%)」は、配列の比較による判定からの、2つ以上のポリペプチド配列または2つ以上のポリヌクレオチド配列の間の関係性である。当該技術分野では、「同一性」は、場合によっては、かかる配列の文字列の間の一致による判定からの、ポリペプチドまたはポリヌクレオチド配列の間の配列の関連性の程度も意味する。「同一性」および「類似性」は、限定はされないが、1)Computational Molecular Biology(Lesk A.M.編) Oxford University Press:NY(1988年);2)Biocomputing:Informatics and Genome Projects(Smith D.W.編) Academic Press:NY(1993年);3)Computer Analysis of Sequence Data、Part I(Griffin A.M.およびGriffin H.G.編) Humania Press:NJ(1994年);4)Sequence Analysis in Molecular Biology(von Heinje G.編) Academic Press(1987年);ならびに5)Sequence Analysis Primer(Gribskov M.およびDevereux J.編) Stockton:NY(1991年)に記載の方法を含む既知の方法により容易に計算されうる。
【0060】
同一性を判定するための好ましい方法は、試験される配列間に最適な一致を与えるように設計される。同一性および類似性を判定するため方法は、公的に利用可能なコンピュータープログラム内にコード化される。配列アラインメントおよび同一性パーセントの計算は、LASERGENE bioinformatics computing suiteのMegAlign(商標)プログラム(DNASTAR Inc.,Madison,WI)を用いて行われうる。配列の複数のアラインメントは、HigginsおよびSharp(CABIOS.5:151−153頁、1989年);およびHiggins D.G.ら、(Comput.Appl.Biosci.、8:189−191頁、1992年)によって記載された、Clustal Vと称されるアラインメント法に対応する「アラインメントのClustal V法」を含む数種類のアルゴリズムを包含する「アラインメントのClustal法」を用いて行われ、かつLASERGENE bioinformatics computing suiteのMegAlign(商標)プログラム(DNASTAR Inc.)において見出される。複数のアラインメントにおいては、デフォルト値はギャップペナルティ=10およびギャップ長ペナルティ=10に対応する。Clustal法を用いる、タンパク質配列のペアワイズアラインメントおよび同一性パーセントの計算におけるデフォルトパラメータは、KTUPLE=1、ギャップペナルティ=3、ウインドウ(WINDOW)=5およびダイアゴナルズセイブド(DIAGONALS SAVED)=5である。核酸においては、これらのパラメータは、KTUPLE=2、ギャップペナルティ=5、ウインドウ=4およびダイアゴナルズセイブド=4である。Clustal Vプログラムを用いての配列のアラインメント後、同じプログラム内の「配列距離(sequence distances)」表を見ることにより「同一性パーセント」を得ることが可能である。さらに、「Clustal Wのアラインメント法」が利用可能あり、HigginsおよびSharp(上記);Higgins D.G.ら、(上記)によって記載されたClustal Wと称されるアラインメント法に対応し、LASERGENE bioinformatics computing suiteのMegAlign(商標)v6.1プログラム(DNASTAR Inc.)において見出される。複数のアラインメントにおけるデフォルトパラメータ(ギャップペナルティ=10、ギャップ長ペナルティ=0.2、ディレイ・ディバージェン配列(Delay Divergen Seqs)(%)=30、DNAトランジション・ウェイト(Transition Weight)=0.5、タンパク質重み行列(Protein Weight Matrix)=Gonnetシリーズ(Series)、DNA重み行列(Weight Matrix)=IUB)。Clustal Wプログラムを用いての配列のアラインメント後、同じプログラム内の「配列距離」表を見ることにより「同一性パーセント」を得ることが可能である。
【0061】
配列同一性の多数のレベルが、同一または類似の機能または活性を有するポリペプチドを他の種から同定するのに有用であることが当業者により十分に理解されている。パーセント同一性の有用な例として、限定はされないが、24%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、または95%があげられるか、または24%〜100%の任意の整数の百分率、例えば、25%、26%、27%、28%、29%、30%、31%、32%、33%、34%、35%、36%、37%、38%、39%、40%、41%、42%、43%、44%、45%、46%、47%、48%、49%、50%、51%、52%、53%、54%、55%、56%、57%、58%、59%、60%、61%、62%、63%、64%、65%、66%、67%、68%、69%、70%、71%、72%、73%、74%、75%、76%、77%、78%、79%、80%、81%、82%、83%、84%、85%、86%、87%、88%、89%、90%、91%、92%、93%、94%、95%、96%、97%、98%または99%は、本発明の記載において有用でありうる。好適な核酸断片は、上記の同一性を有するだけでなく、典型的には、少なくとも50アミノ酸、好ましくは少なくとも100アミノ酸、より好ましくは少なくとも150アミノ酸、さらにより好ましくは少なくとも200アミノ酸、および最も好ましくは少なくとも250アミノ酸を有するポリペプチドをコードする。
【0062】
「配列分析ソフトウェア」という用語は、ヌクレオチドまたはアミノ酸配列の分析にとって有用な任意のコンピュータアルゴリズムまたはソフトウェアプログラムを示す。「配列分析ソフトウェア」は商業的に利用可能であるかまたは個別に開発される場合がある。典型的な配列分析ソフトウェアは、限定はされないが、1)プログラムのGCGスイート(Wisconsin Packageバージョン9.0、Genetics Computer Group(GCG),Madison,WI);2)BLASTP、BLASTN、BLASTX(Altschulら、J.Mol.Biol.、215:403−410頁(1990年));3)DNASTAR(DNASTAR Inc.,Madison,WI);4)Sequencher(Gene Codes Corporation,Ann Arbor,MI);および5)Smith−Watermanアルゴリズム(W.R.Pearson、Comput.Meth.Gen.Res.、[Proc.Int.Symp.]、会合日1992年、111−120頁、1994年、Suhai編、Sandor.Plenum Press,New York,NY)を組み入れたFASTAプログラムを含むことになる。本願の文脈の中で、配列分析ソフトウェアが分析に用いられる場合、分析の結果が、他に規定がない限り、参照されるプログラムの「デフォルト値」に基づくことが理解されるであろう。本明細書で用いられる「デフォルト値」は、最初に初期化される際に最初にソフトウェアが読み込む任意の値またはパラメータのセットを意味することになる。
【0063】
核酸断片については、ある一本鎖形態の核酸断片が温度および溶液のイオン強度の適切な条件下で他方の核酸断片にアニール可能である場合、別の核酸断片、例えばcDNA、ゲノムDNA、またはRNA分子に対して「ハイブリダイズ可能」である。ハイブリダイゼーションおよび洗浄の条件については周知であり、Sambrook J.、Fritsch E.F.およびManiatis T.Molecular Cloning:A Laboratory Manual、第2版、Cold Spring Harbor Laboratory(Cold Spring Harbor,NY)(1989年)、特に第11章およびその中の表11.1(全体として参照により本明細書中に援用される)に例示されている。温度およびイオン強度の条件はハイブリダイゼーションの「ストリンジェンシー」を決定する。ストリンジェンシー条件は、中程度に類似の断片(遠縁の微生物由来の相同配列など)から高度に類似の断片(近縁の微生物由来の機能酵素を複製する遺伝子など)にかけてスクリーニングするように調整されうる。
【0064】
ハイブリダイゼーション後の洗浄がストリンジェンシー条件を決定する。1つの好ましい条件のセットでは、室温で15分間の6×SSC、0.5% SDSの場合から開始し、次いで45℃で30分間の2×SSC、0.5% SDSの場合を繰り返し、次いで50℃で30分間の0.2×SSC、0.5% SDSの場合を2回繰り返すという一連の洗浄が用いられる。より好ましいストリンジェントな条件のセットではより高温が用いられ、ここでの洗浄は終わりの2回の0.2×SSC、0.5% SDSにおける30分の洗浄で温度が60℃に高められた点を除いて上記の洗浄と同一である。別の好ましい高度にストリンジェントな条件のセットでは、65℃での0.1×SSC、0.1% SDSにおける2回の最終の洗浄が用いられる。さらなるストリンジェントな条件のセットでは、例えば0.1×SSC、0.1% SDS、65℃でのハイブリダイゼーションおよび2×SSC、0.1% SDSとそれに続く0.1×SSC、0.1% SDSの場合での洗浄が含まれる。
【0065】
ハイブリダイゼーションでは、塩基間の不一致がハイブリダイゼーションのストリンジェンシーによりありうるとしても2つの核酸が相補配列を有することが必要である。核酸をハイブリダイズするのに適するストリンジェンシーは、当該技術分野での周知の変数である核酸の長さおよび相補性の程度に依存している。2つのヌクレオチド配列間の類似性または相同性の程度が大きくなると、それらの配列を有する核酸のハイブリッドにおけるTmの値が増加する。核酸ハイブリダイゼーションの(より高いTmに対応する)相対的安定性は、以下の順、すなわちRNA:RNA、DNA:RNA、DNA:DNAで低下する。100ヌクレオチド長を超えるハイブリッドにおいては、Tmを計算するための方程式が導かれている(Sambrookら、上記、9.50〜9.51を参照)。より短い核酸すなわちオリゴヌクレオチドの場合のハイブリダイゼーションにおいては、不一致の位置がより重要になり、オリゴヌクレオチド長がその特異性を決定する(Sambrookら、上記、11.7〜11.8を参照)。一実施形態では、ハイブリダイズ可能な核酸における長さは少なくとも約10ヌクレオチド長である。好ましくは、ハイブリダイズ可能な核酸における最小の長さは少なくとも約15ヌクレオチド長、より好ましくは少なくとも約20ヌクレオチド長、および最も好ましくは長さは少なくとも約30ヌクレオチド長である。さらに、当業者は、温度および洗浄溶液の塩濃度がプローブ長などの要素による必要性に応じて調整可能であることを理解するであろう。
【0066】
アミノ酸またはヌクレオチド配列の「大部分」は、ポリペプチドのアミノ酸配列または遺伝子のヌクレオチド配列を、当業者による配列の人手による評価、またはBLAST(Altschul S.F.ら、上記)などのアルゴリズムを使用するコンピュータで自動化された配列比較および同定のいずれかにより、ポリペプチドまたは遺伝子を推定的に同定する程度に十分に含む部分である。一般に、ポリペプチドまたは核酸配列を公知のタンパク質または遺伝子に対して相同なものとして推定的に同定するため、10個以上の隣接アミノ酸または30個以上のヌクレオチドの配列が必要である。さらに、ヌクレオチド配列に関しては、20〜30個の隣接ヌクレオチドを含む遺伝子に特異的なオリゴヌクレオチドプローブは、遺伝子同定(たとえば、サザンハイブリダイゼーション)および単離(たとえば、細菌コロニーまたはバクテリオファージプラークのインサイチュハイブリダイゼーション)の配列に依存した方法において使用することが可能である。さらに、12〜15塩基の短いオリゴヌクレオチドは、プライマーを含む特定の核酸断片を得るため、PCRにおける増幅プライマーとして使用することが可能である。したがって、ヌクレオチド配列の「大部分」は、配列を、配列を含む核酸断片を特異的に同定および/または単離する程度に十分に含む。本明細書は、特定の真菌タンパク質をコードする完全なアミノ酸およびヌクレオチド配列について教示する。本明細書で報告されるような配列の利点を有する当業者は、当業者に公知の目的のため、開示される配列のすべてまたは大部分をここで使用することができる。したがって、本発明は、添付の配列表において報告されるような完全な配列、ならびに上で定義されるような配列の大部分を含む。
【0067】
ここで用いられる標準の組換えDNAおよび分子クローニング技術は、当該技術分野で周知であり、Sambrook J.、Fritsch E.F.およびManiatis T.、Molecular Cloning:A Laboratory Manual、第2版、Cold Spring Harbor Laboratory Press,Cold Spring Harbor,NY(1989年)(以後Maniatis);Silhavy T.J.、Bennan M.L.、およびEnquist L.W.、Experiments with Gene Fusions、Cold Spring Harbor Laboratory Press,Cold Spring Harbor,NY(1984年);ならびにAusubel F.M.ら、Current Protocols in Molecular Biology、Greene Publishing Assoc. and Wiley−Interscience発行(1987年)に記載されている。
【0068】
本発明は、二次アルコールデヒドロゲナーゼ活性を有するポリペプチドをコードする少なくとも1つの遺伝子を含む、イソブタノールを生成するための腸内生産宿主を提供し、ここで宿主は、イソブタノールを生成し、かつ、次の酵素活性、すなわちピルビン酸・ギ酸リアーゼ、フマル酸レダクターゼ酵素複合体、アルコールデヒドロゲナーゼおよび乳酸デヒドロゲナーゼ(図1A、反応「f」、「g」、「h」、「i」を参照)のうちの少なくとも1つを実質的に有しない。生産宿主の二次アルコールデヒドロゲナーゼは、イソブチルアルデヒドのイソブタノールへの変換において特に有効である。
【0069】
(IdhAによってコードされる)乳酸デヒドロゲナーゼの欠失は、乳酸塩の生成におけるピルビン酸塩の変換(図1A、反応「f」)を阻止する。
【0070】
イソブタノールを生成するための微生物宿主
イソブタノールの生成のために選択される微生物宿主は、炭水化物をイソブタノールに変換できる必要がある。好適な微生物宿主の選択における基準は、次のもの、すなわち高率のグルコース利用、遺伝子操作のための遺伝的ツールの利用可能性、および安定な染色体改変をもたらす能力を含む。
【0071】
大部分の微生物は、炭水化物を利用することができる。しかし、特定の環境微生物は、炭水化物を高効率で利用することができず、それ故、好適な宿主とはならない。
【0072】
宿主を遺伝的に修飾する能力は、任意の組換え微生物の生成にとって必須である。遺伝子導入技術の様式は、エレクトロポレーション、複合、形質導入または天然形質転換によるものであってもよく、当該技術分野で周知である。広範囲宿主の複合プラスミドおよび薬剤耐性マーカーが使用可能である。クローニングベクターは、宿主微生物に対し、その宿主内で機能可能な抗生物質耐性マーカーの性質に基づいて適合され、当該技術分野で周知である。
【0073】
微生物宿主はまた、本明細書中の下記におけるさまざまな遺伝子を欠失させることによって炭素流における競合経路を不活性化するように操作してもよい。上記の基準に基づき、好ましい宿主は、エシェリキア(Escherichia)属、サルモネラ(Salmonella)属、クレブシエラ(Klebsiella)属、セラチア(Serratia)属、エルウィニア(Erwinia)属およびシゲラ(Shigella)属のさまざまな種を含む。
【0074】
また、微生物に対するイソブタノールの毒性により、宿主株を同定または改変して、それがイソブタノールに対してより耐性を示すのであれば望ましいことになる。かかる耐性宿主の選択については、同時係属中の共同所有された米国特許出願公開第20070259411号明細書中で開示されている。
【0075】
イソブタノールを蓄積するためのノックアウト突然変異体の生成
糖基質を代謝する微生物は、混合酸発酵において種々の副生成物を生成する(Moat A.G.ら、Microbial Physiology、第4版、John Wiley Publishers,N.Y.、2002年)。混合酸発酵の典型的な生成物は、ギ酸、乳酸およびコハク酸などの酸ならびにエタノールである。イソブタノール発酵中におけるこれらの副生成物の形成は、イソブタノールの潜在的収率を低下させうる。イソブタノールの収率低下を阻止するため、副生成物の形成に対応する酵素活性は低下しうる。副生成物の形成に関与する酵素は、限定はされないが、次のものを含む。1)ピルビン酸塩をギ酸塩およびアセチル−補酵素Aに代謝する、pflB遺伝子によってコードされるピルビン酸・ギ酸リアーゼ(EC2.3.1.54)(アミノ酸配列番号46;DNA配列番号47)。この酵素の欠失は、ピルビン酸塩がギ酸塩およびアセチル−CoAを形成する場合の競合を除外する(図1A、反応「g」);2)フマル酸塩のコハク酸塩への還元を触媒し、NADHを必要とする、frdABCDオペロンによってコードされるフマル酸レダクターゼ酵素複合体(EC1.3.99.1);FrdA(アミノ酸配列番号54;DNA配列番号55)サブユニットは、共有結合されたフラビンアデニンジヌクレオチドを有する;FrdBは酵素(アミノ酸配列番号48;DNA配列番号49)の鉄−硫黄中心を有する;FrdC(アミノ酸配列番号56;DNA配列番号57)およびFrdD(アミノ酸配列番号58;DNA配列番号59)は、触媒FrdABドメインを細胞質膜に結合させるインテグラル(integral)膜タンパク質である。フマル酸レダクターゼの機能は、frdA、B、C、またはDのサブユニットの任意の1つの欠失によって除外される場合があり、ここではfrdBの欠失が好ましい。この活性の欠失は、ピルビン酸塩におけるそのフマル酸塩への変換に対する誘引(draw)を除外する(図1A、反応「i」);3)2段階反応(両反応はadhEによって触媒され、両反応はNADHを必要とする)でエタノールをアセチル−CoAから合成する(図1A、反応「h」)、adhE遺伝子によってコードされるアルコールデヒドロゲナーゼ(EC1.2.1.10−アセトアルデヒドデヒドロゲナーゼおよびEC1.1.1.1−アルコールデヒドロゲナーゼ)(アミノ酸配列番号52;DNA配列番号53);ならびに
4)NADHの酸化とともにピルビン酸塩を乳酸塩に還元する、ldhA遺伝子によってコードされる乳酸デヒドロゲナーゼ(EC1.1.1.28)(アミノ酸配列番号50;DNA配列番号51)。この酵素の欠失は、この酵素によるピルビン酸塩に対する競合を除外し、そのギ酸塩およびアセチル−CoAへの変換を遮断する(図1A、反応「g」)。遺伝子突然変異をもたらすための方法は、一般的であり、当該技術分野で周知であり、(ピルビン酸・ギ酸リアーゼをコードする)pflB、(フマル酸レダクターゼのサブユニットをコードする)frdB、(乳酸デヒドロゲナーゼをコードする)ldhAおよび(アルコールデヒドロゲナーゼをコードする)adhEの欠損突然変異体を生成する実験に適用してもよい。Miller J.H.(1992年、A Short Course in Bacterial Genetics. Cold Spring Harbor Press,Plainview,NY.)にレビューされた、一般に用いられるランダム遺伝子組換え(random genetic modification)法は、自発的突然変異誘発、ミューテーター遺伝子によって引き起こされる突然変異誘発、化学的突然変異誘発、UVまたはX線の照射、およびトランスポゾン挿入を含む。トランスポゾンは、
1.ファージ媒介性形質導入(両方の種に特異的でかつ異種間の場合に利用されている)
2.複合(同一種または異種のメンバー間でありうる)
3.形質転換(化学的に補助され、電気的ショックに媒介されるDNAの取り込みが利用可能)
を含む種々の方法で細菌に導入されている。
【0076】
これらの方法では、トランスポゾンは、レシピエントにおいて、移入DNAからレシピエントゲノムへの遺伝子ホッピングを触媒するトランスポーゼースを発現する。トランスポゾンDNAは、裸でファージまたはプラスミド核酸内に取り込まれるか、またはトランスポーゼースと複合されうる。ほとんどの場合、トランスポゾンを有する移入DNAの複製および/または維持は阻止されることから、トランスポゾンに対するマーカー(たいていは抗生物質耐性)における遺伝子選択は、各組換えが移入DNA分子からレシピエントゲノムへのトランスポゾンの移動の結果であることを保証する。他の方法は、染色体DNA、その1つ以上の断片の場合にインビトロで転移がなされ、次いで生成されている新規な挿入対立遺伝子がレシピエント細胞に導入される(この場合、それは相同組換えによって内在性(resident)対立遺伝子を置換する)という方法である。トランスポゾン挿入は、多くの誘導方法を介して、KlecknerおよびBotstein(J.Mol.Biol.、116:125−159頁、1977年)に記載または上記のように行ってもよい。
【0077】
pflB、frdB、ldhA、adhE遺伝子の欠失はまた、細菌染色体において直接作成してもよい。改変された染色体セグメントは、腸内細菌標的宿主染色体の内因性遺伝子の部位に挿入され、内因性領域を置換する。改変された染色体セグメントの挿入は、当業者に既知の任意の方法、例えばファージ形質導入、複合、あるいはプラスミド導入または非プラスミド二本鎖もしくは一本鎖DNA導入とその後の相同組換えによって行ってもよい。バクテリオファージ形質導入では、形質導入のための標準の遺伝学的方法が用いられ、それは当該技術分野で周知であり、Miller J.H.(Experiments in Molecular Genetics、Cold Spring Harbor Laboratory Press,Cold Spring Harbor,N.Y.、1972年)で記載されている。細菌染色体内で作成されている改変された染色体セグメントは、ファージ内にパーケージ化され、次いでファージ感染を通じて標的宿主細胞に導入され、その後、相同組換えにより、改変された染色体セグメントが標的宿主細胞染色体に挿入される。
【0078】
DNA断片は、改変された染色体セグメントを担持する細菌染色体から、細菌染色体内でこの染色体セグメントに天然に隣接する配列を含む方法によって調製し、相同組換えが生じることになる場合の配列を提供してもよい。隣接する相同配列は、Lloyd R.G.およびK.B.Low(Homologous recombination、2236−2255頁;F.C.Neidhardt編、Escherichia coli and Salmonella:Cellular and Molecular Biology、1996年、ASM Press,Washington,DC)に記載のように、相同組換えを支持するのに十分である。典型的には、相同組換えに使用される相同配列は、1kb長を超えるが、せいぜい50もしくは100塩基対程度であってもよい。改変された染色体セグメントおよび隣接する相同配列を有するDNA断片は、例えば制限消化により、または超音波処理など、ランダムな末端を生成する方法を用いて、画定された末端を用いて調製してもよい。いずれの場合においても、改変された染色体セグメントを有するDNA断片は、例えばエレクトロポレーション、凍結−解凍法を含む任意のDNA取り込み法により、または化学的にコンピテントな細胞を使用して、標的宿主細胞に導入してもよい。DNA断片が相同組換えを受ける結果、標的宿主の内因性染色体領域は改変された染色体セグメントと置換される。
【0079】
プラスミドを使用し、改変された染色体セグメントを挿入のために標的宿主細胞内に運んでもよい。典型的には、非複製プラスミドを使用し、組込みが促進される。改変された染色体セグメントは、細菌の標的宿主ゲノム内でこの染色体セグメントに天然に隣接するDNA配列によってプラスミド内に隣接され、相同組換えが生じることになる配列が提供される。隣接する相同配列は上記の通りであり、プラスミドDNAの導入は上記の通りである。
【0080】
これらの方法のいずれかを用いて、バクテリオファージ相同組換え系、例えばバクテリオファージλRed系の使用により、相同組換えを促進してもよい((DatsenkoおよびWanner,Proc.Natl.Acad.Sci.USA,97:6640−6645頁、2000年)および(Ellisら、Proc.Natl.Acad.Sci.USA、98:6742−6746頁、2001年)またはRacファージRecE/RecT系(Zhangら、Nature Biotechnol.、18:1314−1317頁、2000年))。
【0081】
これらの方法のいずれかにおいては、相同組換えは、標的宿主の内因性染色体領域と改変された染色体セグメントとの置換をもたらす。
【0082】
改変された染色体セグメントの挿入に奏功したレシピエント株は、マーカーを使用して同定してもよい。スクリーニングまたは選択マーカーのいずれかを使用してもよく、ここでは選択マーカーが特に有用である。例えば、抗生物質耐性マーカーは、改変された染色体セグメント内に存在しうることから、それが新しい宿主に移される場合、改変された染色体セグメントを受ける細胞は、対応する抗生物質上での成長によって容易に同定することが可能である。あるいは、スクリーニングマーカー(容易に検出される生成物の生成をもたらすもの)を使用してもよい。マーカーがレシピエント株中に残存しないことが所ましい場合、その後、それを例えば部位特異的組換えを用いて除去してもよい。この場合、部位特異的組換え部位は、リコンビナーゼの発現がマーカーの欠失を引き起こすように、マーカーDNA配列の5’および3’側に位置する。一旦、突然変異が生じていると、細胞は、これらの特異的遺伝子の不在についてスクリーニングされる必要がある。この目的のため、複数の方法を用いて分析してもよい。
【0083】
pflB、frdB、ldhAおよびadhEとして同定される任意の細菌遺伝子は、イソブタノールの生成を目的とした本発明の株を作製するための、対応する微生物における組換えを意図した標的である。大腸菌(E.coli)、サルモネラ(Salmonella)、セラチア(Serratia)、エルウィニア(Erwinia)、シゲラ(Shigella)などのさまざまな腸内微生物由来の遺伝子および遺伝子産物は、本明細書中に記載のように、ハイブリダイゼーション、インフォマティクスまたは相同体によって同定してもよい。
【0084】
相同体の単離
本明細書中に記載の、配列番号9、46、48、50および52などの本発明における目的の遺伝子をコードする核酸分子、またはイソブタノール生合成経路において挙げられる配列のいずれかを用いて、同じまたは他の微生物種から、この核酸断片に対して少なくとも70%〜75%、75%〜80%、80%〜85%、85%〜90%、90%〜95%、もしくは95%〜100%の配列同一性を有する、相同タンパク質をコードする核酸分子を単離してもよい。配列依存性プロトコルを用いる相同体の単離は、当該技術分野で周知である。配列依存性プロトコルの例は、限定はされないが、核酸ハイブリダイゼーションの方法、ならびに、核酸増幅技術(たとえば、ポリメラーゼ連鎖反応(PCR)、Mullisら、米国特許第4,683,202号明細書;リガーゼ連鎖反応(LCR)、Tabor S.ら(Proc.Natl.Acad.Sci.USA、82:1074、1985年);または鎖置換増幅(SDA)、Walkerら、Proc.Natl.Acad.Sci.USA、89:392頁、1992年)のさまざまな使用によって例示されるDNAおよびRNA増幅の方法を含む。
【0085】
たとえば、本発明の核酸断片は、当業者に周知の方法を用い、DNAハイブリダイゼーションプローブとして配列番号9、46、48、50および52の核酸断片の全部または一部を使用し、任意の所望される細菌からライブラリをスクリーニングすることによって直接単離することが可能である。配列番号9、46、48、50および52に基づく特異的なオリゴヌクレオチドプローブは、当該技術分野で公知の方法(Maniatis、上記)によって設計し、合成することが可能である。さらに、配列を直接使用し、DNAプローブを、ランダムプライマーDNAラベリング、ニックトランスレーション、または末端標識技術などの当業者に公知の方法により、またはRNAプローブを、利用可能なインビトロ転写系を使用し、合成することが可能である。さらに、特異的なプライマーを設計し、使用し、配列番号9、46、48、50および52の相同体の一部または完全長を増幅することが可能である。得られる増幅産物は、増幅反応の間に直接標識するかまたは増幅反応後に標識し、プローブとして使用し、完全長DNA断片を好適なストリンジェントな条件下で単離することが可能である。
【0086】
典型的には、PCRタイプ増幅技術においては、プライマーは、異なる配列を有し、互いに相補的でない。所望される試験条件に依存し、プライマーの配列は、標的核酸の効率的かつ忠実な複製を提供するように設計される必要がある。PCRプライマー設計の方法は、一般的であり、当該技術分野で周知である(TheinおよびWallace、“The use of oligonucleotide as specific hybridization probes in the Diagnosis of Genetic Disorders”,in Human Genetic Diseases:A Practical Approach、K.E.Davis編(1986年)、33−50頁、IRL Press(Herndon,VA));Rychlik W.、(1993年)、In White B.A.(編)、Methods in Molecular Biology、第15巻、31−39頁、PCR Protocols:Current Methods and Applications.、Humania Press,Inc.(Totowa,NJ))。
【0087】
一般に、本核酸配列の2つの短いセグメントを使用し、ポリメラーゼ連鎖反応プロトコルにおける使用を意図したプライマーを設計し、DNAまたはRNAから相同コード領域をコードするより長い核酸断片を増幅することが可能である。PCRは、鋳型として配列番号9、46、48、50および52に相同な核酸配列を有する任意のDNA、たとえば鋳型としてゲノムDNA、cDNAまたはプラスミドDNAを使用して実施してもよい。クローン化cDNAのライブラリを使用する場合、一方のプライマーの配列は配列番号9、46、48、50および52に由来し、他方のプライマーの配列は微生物遺伝子をコードするmRNA前駆体の3’末端でのポリアデニル酸トラクト(tracts)の存在を利用する。あるいは、第2のプライマー配列は、クローン化ベクターに由来する配列に基づく場合がある。たとえば、当業者は、鋳型としてmRNAを使用するRACEプロトコルに従い(Frohmanら、Proc.Natl.Acad.Sci.USA、85:8998頁、1988年)、PCRを用いてcDNAを生成し、転写産物における単一点(single point)と3’または5’末端の間の領域のコピーを増幅することができる。3’および5’方向に方向づけられるプライマーは、本核酸配列から設計することが可能である。市販の3’RACEまたは5’RACEシステム(Life Technologies(Rockville,MD))を使用し、特異的な3’または5’cDNA断片を単離することが可能である(Oharaら、Proc.Natl.Acad.Sci.USA、86:5673頁、1989年);およびLohら、Science、243:217頁、1989年)。
【0088】
ハイブリダイゼーション
あるいは、配列番号9、46、48、54、56、50および52の核酸分子またはそれらの相補体は、相同体の同定のためのイブリダイゼーション試薬として使用してもよい。核酸ハイブリダイゼーション試験の基本的要素は、プローブ、目的の遺伝子または遺伝子断片を含有すると思われる試料、および特異的ハイブリダイゼーション法を含む。本発明のプローブは、典型的には、検出されるべき核酸配列に対して相補的な一本鎖核酸配列である。プローブは、検出されるべき核酸配列に対して「ハイブリダイズ可能」である。プローブ長は、5塩基から数万塩基まで可変であり、実施されるべき特定の試験に依存することになる。典型的には、約15塩基〜約30塩基のプローブ長が好適である。検出されるべき核酸配列に対して相補的である必要があるプローブ分子はほんの一部に限られる。さらに、プローブと標的配列の間の相補性は完全である必要はない。ハイブリダイゼーションは、相補性が不完全な分子間では生じず、その結果として、ハイブリダイズされる領域内の塩基の特定の画分は適切な相補的塩基と対合しない。
【0089】
ハイブリダイゼーション方法は十分に確立されている。典型的には、プローブおよび試料は、核酸ハイブリダイゼーションを可能にする条件下で混合されなければならない。これは、適切な濃度および温度条件下、無機または有機塩の存在下で、プローブと試料を接触させることを含む。プローブと試料核酸は、プローブと試料核酸の間の任意の可能なハイブリダイゼーションが生じうるような十分に長い期間接触状態でなければならない。混合物中のプローブまたは標的の濃度によって、ハイブリダイゼーションが生じるのに必要な時間が決まることになる。プローブまたは標的濃度が高まると、必要とされるハイブリダイゼーションのインキュベーション時間が短くなる。場合によって、カオトロピック剤を加えてもよい。カオトロピック剤は、ヌクレアーゼ活性を阻害することにより、核酸を安定化する。さらに、カオトロピック剤は、室温での短いオリゴヌクレオチドプローブの高感度かつストリンジェントなハイブリダイゼーションを可能にする(Van NessおよびChen,Nucl.Acid Res.19:5143−5151頁、1991年)。好適なカオトロピック剤は、特に、塩化グアニジン、チオシアン酸グアニジン、チオシアン酸ナトリウム、テトラクロロ酢酸リチウム、過塩素酸ナトリウム、テトラクロロ酢酸ルビジウム、ヨウ化カリウム、およびトリフルオロ酢酸セシウムを含む。典型的には、カオトロピック剤は、約3Mの最終濃度で存在することになる。必要に応じて、ハイブリダイゼーション混合物にホルムアミドを、典型的には30〜50%(v/v)加えてもよい。
【0090】
さまざまなハイブリダイゼーション溶液を使用してもよい。典型的には、これらは、約20〜60%容量、好ましくは30%の極性有機溶媒を含む。一般的なハイブリダイゼーション溶液では、約30〜50% v/vのホルムアミド、約0.15〜1Mの塩化ナトリウム、約0.05〜0.1Mの緩衝液、たとえばクエン酸ナトリウム、トリス−HCl、PIPESまたはHEPES(約6〜9のpH範囲)、約0.05〜0.2%の洗剤、たとえばドデシル硫酸ナトリウム、または0.5〜20mMのEDTA、FICOLL(Pharmacia Inc.)(約300〜500kD)、ポリビニルピロリドン(約250〜500kD)、および血清アルブミンが使用される。また、典型的なハイブリダイゼーション溶液中に、約0.1〜5mg/mLの未標識キャリア核酸、断片化核DNA(nucleic DNA)、たとえば仔ウシ胸腺またはサケ精液DNA、または酵母RNA、および場合によって約0.5〜2% w/vのグリシンが含まれることになる。また、他の添加剤、たとえば体積排除剤(volume exclusion agent)を含めてもよく、それは、種々の極性水溶性剤または膨潤剤(swellable agent)、たとえばポリエチレングリコール、ポリアクリル酸塩またはポリアクリル酸メチルなどのアニオンポリマー、およびアニオンサッカライドポリマー(anionic saccharidic polymer)、たとえば硫酸デキストランを含む。
【0091】
核酸ハイブリダイゼーションは、種々のアッセイフォーマットに適合可能である。最も好適なものの1つが、サンドイッチアッセイフォーマットである。サンドイッチアッセイは、特に、非変性条件下でのハイブリダイゼーションに適合可能である。サンドイッチ型アッセイの主要素が固体支持体である。固体支持体は、それに吸着しているか、またはそれに未標識でかつ配列の一部に相補的である固定化核酸プローブを共有結合させている。
【0092】
さらに、微生物ゲノムの配列が急速に公的に利用可能になりつつあることから、相同体は、バイオインフォマティクスのみを用いて同定してもよい。
【0093】
したがって、本発明は、組換え腸内細菌細胞を提供し、ここで遺伝的修飾は、イソブタノール生成に向けての炭素の集中流を可能にするため、特定のpflB、frdB、ldhAおよびadhE遺伝子の欠失をもたらす。
【0094】
イソブタノール生合成経路
炭水化物利用微生物は、中央代謝経路として、Embden−Meyerhof−Parnas(EMP)経路、Entner−Doudoroff経路およびペントースリン酸サイクルを利用し、成長および維持のためのエネルギーおよび細胞前駆体を提供する。これらの経路は、中間体のグリセルアルデヒド−3−リン酸を共有し、ピルビン酸塩は、最終的には直接にまたはEMP経路と併せて形成される。それに続き、ピルビン酸塩は、種々の手段を介してアセチル−補酵素A(アセチル−CoA)に形質転換される。アセチル−CoAは、例えば脂肪酸、アミノ酸および二次代謝物質を生成する上での主要な中間体としての役割を果たす。糖のピルビン酸塩への変換の複合反応により、エネルギーが生成され(例えば、アデノシン−5’−三リン酸、ATP)、還元等価物が得られる(例えば、還元型ニコチンアミドアデニンジヌクレオチド、NADH、および還元型ニコチンアミドアデニンジヌクレオチドリン酸、NADPH)。NADHおよびNADPHは、それらの酸化形態(それぞれNAD+およびNADP+)に再生されなければならない。無機電子受容体(例えばO2、NO3−およびSO42−)の存在下で、還元等価物を使用し、エネルギープールを増大させるか、あるいは還元型炭素副産物を形成してもよい。
【0095】
本発明の腸内宿主は、イソブタノールを生成する。典型的には、イソブタノール生合成経路は、図1Aおよび1Bに示されるように、イソブタノールの炭水化物からの生成を可能にする宿主細胞に改変されることになる。1つの経路は、以下の基質の生成物への変換、すなわち、
a)アセト乳酸シンターゼによって触媒される、ピルビン酸塩のアセト乳酸塩への変換、
b)アセトヒドロキシ酸イソメロレダクターゼによって触媒される、アセト乳酸塩の2,3−ジヒドロキシイソ吉草酸への変換、
c)アセトヒドロキシ酸デヒドラターゼによって触媒される、2,3−ジヒドロキシイソ吉草酸のα−ケトイソ吉草酸への変換、
d)分岐鎖ケト酸デカルボキシラーゼによって触媒される、α−ケトイソ吉草酸のイソブチルアルデヒドへの変換、および、
e)ブタノールデヒドロゲナーゼ(二次アルコールデヒドロゲナーゼ)によって触媒される、イソブチルアルデヒドのイソブタノールへの変換
を含む。
【0096】
この経路は、バリン生合成(ピルビン酸塩からα−ケトイソ吉草酸塩へ)およびバリン異化(α−ケトイソ吉草酸からイソブチルアルデヒドへ)における十分に特徴づけられた経路に関与することが知られる酵素と、最終ステップでの新規ブタノールデヒドロゲナーゼを組み合わせる。別のイソブタノール経路は、共同所有された同時係属中の米国特許出願公開第20070092957号明細書(参照により本明細書中に援用される)中に記載されている。
【0097】
多数のバリン生合成酵素はまた、イソロイシン生合成経路における類似反応を触媒することから、基質特異性は遺伝子源を選択する場合での主な検討事項である。したがって、アセト乳酸シンターゼ酵素における主な目的の遺伝子は、バチルス(Bacillus)(alsS)およびクレブシエラ(Klebsiella)(budB)に由来するものである。これらの特定のアセト乳酸シンターゼは、これらの微生物におけるブタンジオール発酵に関与することが知られ、ケト酪酸塩よりピルビン酸塩に対して親和性が高まることを示す(Gollopら、J.Bacteriol.172:3444−3449頁、1990年);Holtzclawら、J.Bacteriol.121:917−922頁、1975年)。第2および第3経路ステップは、それぞれアセトヒドロキシ酸レダクトイソメラーゼおよびデヒドラターゼによって触媒される。これらの酵素は、複数の供給源、例えば大腸菌(E.coli)などから特徴づけられている(Chunduruら、Biochemistry 28:486−493頁、1989年;およびFlintら、J.Biol.Chem.268:14732−14742頁、1993年)。好ましいイソブタノール経路の最終の2つのステップは、酵母内で生じることが知られ、そこでは窒素源としてバリンが使用され、同プロセスではイソブタノールが分泌されうる。α−ケトイソ吉草酸は、複数のケト酸デカルボキシラーゼ酵素、例えばピルビン酸デカルボキシラーゼなどにより、イソブチルアルデヒドに変換されうる。イソブタノール生成から外れたピルビン酸塩の誤誘導を阻止するため、ピルビン酸塩に対する親和性が低下したデカルボキシラーゼが所望される。これまで、当該技術分野で公知の2つのかかる酵素が存在する(Smitら、Appl.Environ.Microbiol.71:303−311頁、2005年;およびde la Plazaら、FEMS Microbiol.Lett.238:367−374頁、2004年)。両酵素は、ラクトコッカス・ラクティス(Lactococcus lactis)の株に由来し、ケトイソ吉草酸においてピルビン酸塩より50〜200倍の優位性を有する。最終的に、複数のアルデヒドレダクターゼが酵母内で同定されており、その多くは基質特異性が重複している。分岐鎖基質がアセトアルデヒドより好ましいことで公知のものとして、限定はされないが、アルコールデヒドロゲナーゼVI(ADH6)およびYpr1p(Larroyら、Biochem.J.361:163−172頁、2002年;およびFordら、Yeast 19:1087−1096頁、2002年)が挙げられ、それらの双方は電子供与体としてNADPHを使用する。分岐鎖基質の場合に活性があるNADPH依存性レダクターゼYqhDはまた、最近、大腸菌(E.coli)において同定されている(Sulzenbacherら、J.Mol.Biol.342:489−502頁、2004年)。
【0098】
本開示のイソブタノール経路においては、アクロモバクター・キシロスオキシダンス(Achromobacter xylosoxidans)由来の新規なブタノールデヒドロゲナーゼが使用され、それは本明細書中に記載される。
【0099】
アクロモバクター・キシロスオキシダンス(Achromobacter xylosoxidans)のブタノールデヒドロゲナーゼ活性
1−ブタノールを含有する培地上で連続的に培養することによって環境汚泥試料を富化することを通じて、唯一の炭素源として1−ブタノールを利用することができる微生物が単離された。1つの単離物が、細菌種アクロモバクター・キシロスオキシダンス(Achromobacter xylosoxidans)に属するものとして、その16SrRNA配列によって同定された。この単離物は、ブチルアルデヒドと1−ブタノールを相互変換したブタノールデヒドロゲナーゼ酵素活性を有する。予想外にも、このブタノールデヒドロゲナーゼ酵素活性がまた、イソブチルアルデヒドとイソブタノールの相互変換、ならびに2−ブタノンと2−ブタノールの相互変換を触媒することが見出された。意外なことに、この酵素は、集積培地中で使用される1−ブタノール基質の場合に匹敵するかまたはより優れた代替基質における動的定数を有した。これらの結果は、このアクロモバクター・キシロスオキシダンス(Achromobacter xylosoxidans)のブタノールデヒドロゲナーゼが、1−ブタノール、イソブタノール、または2−ブタノールを、それぞれブチルアルデヒド、イソブチルアルデヒドまたは2−ブタノン基質の供給源を有する組換え微生物宿主細胞内で生成するために使用可能であることを示した。
【0100】
ブタノールデヒドロゲナーゼのタンパク質およびコード配列
ブタノールデヒドロゲナーゼ活性を有する酵素をコードする、アクロモバクター・キシロスオキシダンス(Achromobacter xylosoxidans)において同定されるヌクレオチド配列は、配列番号9として与えられる。全タンパク質のアミノ酸配列は、配列番号10として与えられる。このアミノ酸配列と公的データベース内の配列との比較によると、このタンパク質が公知のアルコールデヒドロゲナーゼに対して極めて低い類似性を有することが示された。最も類似した公知の配列は、スコアリングマトリックスBLOSUM62、期待値カットオフ10およびワードサイズ3を伴うBLASTを用いると、その348アミノ酸長を超える配列番号10のアミノ酸配列に対して67%同一である。ギャップオープニングペナルティ11およびギャップ伸長1を用いた。見出された最も近い類似性は、髄膜炎菌(Neisseria meningitides)MC58のZn含有アルコールデヒドロゲナーゼ(登録番号AAF41759.1)に対して67%のアミノ酸同一性およびマイコプラズマ・アガラクティアエ(Mycoplasma agalactiae)のZn含有アルコールデヒドロゲナーゼ(登録番号A5IY63)に対して67%のアミノ酸同一性であった。したがって、好ましいブタノールデヒドロゲナーゼ(sadB)は、スコアリングマトリックスBLOSUM62、期待値カットオフ10およびワードサイズ3ならびにギャップオープニングペナルティ11およびギャップ伸長1を伴うBLASTを用いると、その348アミノ酸長を超える配列番号10に対して少なくとも約70%〜75%、約75%〜80%、約80%〜85%、85%〜90%、もしくは90%〜95%同一であるものである。
【0101】
ブタノールデヒドロゲナーゼ活性
配列番号10に対して少なくとも約70%以上のアミノ酸同一性を有し、かつブタノールデヒドロゲナーゼ活性を有するタンパク質が、本発明では特に有用である。本発明の核酸分子は、ブタノールデヒドロゲナーゼ活性を有する配列番号10に対して少なくとも約70%以上のアミノ酸同一性を有するタンパク質をコードする。当業者は、タンパク質におけるブタノールデヒドロゲナーゼ活性を容易に評価することができる。タンパク質は、下記のように微生物細胞内で発現され、細胞抽出物、粗酵素調製物、または精製酵素調製物中でのブタノールデヒドロゲナーゼ活性についてアッセイされる。たとえば、精製酵素および粗酵素調製物のアッセイは、本明細書中、実施例1で説明される。1−ブタノールデヒドロゲナーゼ活性についてのアッセイでは、50mMのブチルアルデヒドおよび0.2mMのNADHを含有する50mMのリン酸カリウム緩衝液(pH6.2、35℃)中の酵素を適量で使用し、NADHの消失が340nmで分光測定的にモニタリングされる。アルコール基質を用いる代替アッセイが、3mMのNAD+およびさまざまな濃度のアルコールを含有するトリス緩衝液(pH8.5)において35℃で実施されるか、あるいはケトンまたはアルデヒド基質を用いる場合は、200μMのNADHおよびさまざまな濃度のケトンまたはアルデヒドを含有する50mMのMES緩衝液(pH6.0)において35℃で実施される。これらまたは他の容易に実施可能なアッセイによると、ブタノールデヒドロゲナーゼの機能は、単離核酸分子によってコードされる同定されたタンパク質の構造に関連性がある(そのいずれもが同定された配列を有する)。
【0102】
生産宿主の作成
発酵性炭素基質をイソブタノールに変換するための酵素経路をコードするのに必要な遺伝子を有する組換え微生物は、当該技術分野で周知の技術を用いて作成してもよい。本発明のイソブタノール生合成経路の酵素、すなわちアセト乳酸シンターゼ、ケトール酸レダクトイソメラーゼ、アセトヒドロキシ酸デヒドラターゼ、分岐鎖α−ケト酸デカルボキシラーゼ、および分岐鎖アルコールデヒドロゲナーゼをコードする遺伝子は、上記のように、さまざまな供給源から単離してもよい。
【0103】
本願中に開示される操作のために使用されるイソブタノール生成株の作成は、共同所有された同時係属中の米国特許出願公開第20070092957号明細書の特に実施例1、2、9、10、11、12、13および14(参照により本明細書中に援用される)において開示されている。
【0104】
分子生物学の当該技術分野で周知の微生物ゲノムから所望される遺伝子を得る方法。例えば、遺伝子の配列が公知である場合、好適なゲノムライブラリは、制限エンドヌクレアーゼ消化によって生成され、所望される遺伝子配列に対して相補的なプローブでスクリーニングされうる。一旦、配列が単離されると、DNAは、ポリメラーゼ連鎖反応などの標準プライマーに特異的な増幅方法(米国特許第4,683,202号明細書)を用いて増幅され、適切なベクターを使用して、形質転換に適したDNAの量を得ることが可能である。異種宿主内での発現のためのコドン最適化用のツールは、容易に利用可能である。コドン最適化用のいくつかのツールは、宿主微生物のGC含量に基づいて利用可能である。
【0105】
一旦、関連経路遺伝子が同定され、単離されると、それらは、当該技術分野で周知の手段により、好適な発現宿主に形質転換することが可能である。種々の宿主細胞の形質転換にとって有用なベクターまたはカセットについては一般的であり、EPICENTRE(登録商標)(Madison,WI)、Invitrogen Corp.(Carlsbad,CA)、Stratagene(La Jolla,CA)、およびNew England Biolabs,Inc.(Beverly,MA)などの企業から市販されている。典型的には、ベクターまたはカセットは、関連遺伝子の転写および翻訳を誘導する配列、選択可能なマーカー、および自律的複製または染色体組込みを可能にする配列を有する。好適なベクターは、転写開始制御を有する遺伝子の5’領域およびを制御する転写終結DNA断片の3’領域を含む。両方の制御領域は形質転換された宿主細胞に対して相同な遺伝子から誘導されうるが、かかる制御領域が生産宿主として選択される特定の種に対して天然でない遺伝子からも誘導されることが理解されるべきである。
【0106】
所望される宿主細胞内での関連経路コード領域の発現を駆動するのに有用な開始制御領域またはプロモーターは極めて多く、当業者に周知である。これらの遺伝因子を駆動可能な仮想的に任意のプロモーター、限定はされないが、(大腸菌(Escherichia coli)および他の腸内細菌(Enterobacteriaceae)内での発現にとって有用な)lac、ara、tet、trp、lPL、lPR、T7、tac、およびtrcなどは、本発明に適している。
【0107】
終結制御領域についても好ましい宿主に対して天然の様々な遺伝子から誘導されうる。場合により終結部位は不要でありうるが、含められる場合が最も好ましい。
【0108】
特定のベクターが広範囲の宿主細菌内で複製可能であり、かつ複合により導入可能である。pRK404および3つの関連ベクター:pRK437、pRK442、およびpRK442(H)の完全でかつアノテートされた配列が利用可能である。これらの誘導体はグラム陰性菌における遺伝子操作にとって有用なツールであることが判明している(スコット(Scott)ら、Plasmid 50:74−79頁、2003年)。広範な宿主範囲のInc P4プラスミドRSF1010における数種のプラスミド誘導体についても、グラム陰性菌の範囲内で機能しうるプロモーターとともに利用可能である。プラスミドpAYC36およびpAYC37が複数のクローニング部位とともに活性プロモーターを有し、グラム陰性菌内での異種の遺伝子発現が可能である。
【0109】
さまざまな好ましい微生物宿主内でのイソブタノール生合成経路の発現は、下記にさらに詳述される。
【0110】
大腸菌(E.coli)内でのイソブタノール生合成経路の発現
大腸菌(E.coli)の形質転換にとって有用なベクターまたはカセットは、一般的であり、上掲の企業から市販されている。例えば、イソブタノール生合成経路の遺伝子は、さまざまな供給源から単離し、修飾pUC19ベクターにクローン化し、大腸菌(E.coli)に形質転換することが可能である。
【0111】
腸内細菌(Enterobacteriaceae)科におけるイソブタノール生合成経路の発現
本発明における使用に適した腸内細菌の例として、限定はされないが、セラチア(Serratia)属、エルウィニア(Erwinia)属、エシェリキア(Escherichia)属、クレブシエラ(Klebsiella)属、サルモネラ(Salmonella)属、およびシゲラ(Shigella)属のメンバーがあげられる。腸内細菌(Enterobacteriaceae)における遺伝子発現および突然変異の生成のための方法もまた、当該技術分野で周知である。例えば、イソブタノール生合成経路の遺伝子は、共同所有された同時係属中の米国特許出願公開第20070092957号明細書の実施例1、2、9、19、11、12、13および14に記載のように、さまざまな供給源から単離し、さまざまなベクターにクローン化してもよい。本発明において特に好適なのは、細菌の腸内クラスのメンバーである。腸内細菌は、腸内細菌科(Enterobacteriaceae)のメンバーであり、エシェリキア(Escherichia)、サルモネラ(Salmonella)、およびシゲラ(Shigella)などのメンバーを含む。それらは、グラム陰性の0.3〜1.0×1.0〜6.0mmの直鎖状桿菌(straight rod)であり、周毛性鞭毛によって運動性であり(テイタメラ(Tatumella)を除く)、または非運動性である。それらは酸素の存在および不在下で成長し、ペプトン、肉抽出物、および(通常は)マッコンキー培地などのさまざまな培地上で良好に成長する。いくつかは唯一の炭素源としてD−グルコース上で成長する一方、その他はビタミンおよび/またはミネラルを必要とする。それらは呼吸性および発酵性代謝を有する化学有機栄養性であるが、好塩性ではなく、D−グルコース、その他の炭水化物、およびポリヒドロキシルアルコールの発酵中に、酸や、しばしば可視性ガスが生成する。それらはオキシダーゼ陰性であり、赤痢菌(Shigella dysenteriae)0グループ1およびゼノラブダス・ネマトフィルス(Xenorhabdus nematophilus)を除いて、カタラーゼ陽性である。(エルウィニア(Erwinia)およびエルシニア(Yersina)のいくつかの株を除いて)硝酸塩は亜硝酸塩に還元される。DNAのG+C含量は、38〜60mol%(Tm、Bd)である。大部分の属内の種からのDNAは、互いに、また科の基準種である大腸菌(Escherichia coli)に少なくとも20%の関連性がある。注目すべき例外は、そのDNAがその他の属からの種と10〜20%の関連性がある、エルシニア(Yersina)、プロテウス(Proteus)、プロビデンシア(Providenica)、ハフニア(Hafnia)、およびエドワードシエラ(Edwardsiella)の種である。エルウィニア・クリサンテミ(Erwinia chrysanthemi)を除いて、試験したすべての種は腸内細菌共通抗原を有する(「ベルジーの系統的細菌学マニュアル(Bergy’s Manual of Systematic Bacteriology)」D.H.Bergyら、Williams and Wilkins Press,Baltimore、1984年)。
【0112】
発酵性炭素基質
本発明の組換え微生物生産宿主は、好適な炭素基質を含有しなければならない。適切な炭素基質が、限定はされないが、グルコースおよびフルクトースなどの単糖、乳糖またはスクロースなどのオリゴ糖、デンプンまたはセルロースなどの多糖、あるいはそれらの混合物、ならびに乳清透過液、コーンスティープリカー(cornsteep liquor)、砂糖液(sugar beet molasses)、および大麦モルト(barley malt)などの再生可能な原料由来の未精製混合物を含みうる。
【0113】
上記の炭素基質およびその混合物のすべてが本発明において好適であると考えられるが、好ましい炭素基質は、グルコース、フルクトース、およびスクロースである。スクロースは、サトウキビ、サトウダイコン、キャッサバ、サトウモロコシ、およびこれらの混合物などの再生可能な糖供給源に由来しうる。グルコース(デキストロース)は、トウモロコシ、小麦、ライ麦、大麦、オート麦、およびこれらの混合物などの穀物を含むデンプンに基づく供給原料の糖化を通じて、再生可能な穀物供給源に由来しうる。さらに、発酵性糖は、たとえば共同所有および同時係属中の米国特許出願公開第20070031918A1号明細書(参照によって本明細書中に援用される)中に記載のように、前処理および糖化のプロセスを通じて、再生可能なセルロース系またはリグノセルロース系バイオマスに由来しうる。バイオマスは、任意のセルロース系またはリグノセルロース系の原料を示し、セルロースを含有し、かつ場合により、ヘミセルロース、リグニン、デンプン、オリゴ糖および/または単糖をさらに含有する原料を含む。バイオマスは、追加成分、例えばタンパク質および/または脂質も含有しうる。バイオマスは単一の供給源から誘導されうるか、またはバイオマスは2つ以上の供給源から誘導される混合物を含有しうる。バイオマスは、例えばトウモロコシ穂軸(corn cob)およびコーンストーバーの混合物または草および葉の混合物を含有しうる。バイオマスは、限定はされないが、バイオエネルギー作物、農業残渣、地方自治体の固体廃棄物、産業固体廃棄物、製紙から排出される汚泥、庭ごみ、廃材および林業廃棄物を含む。バイオマスの例として、限定はされないが、トウモロコシ粒、トウモロコシ穂軸、トウモロコシの皮などのトウモロコシ残渣、コーンストーバー、草、小麦、麦かん(wheat straw)、大麦、麦かん(barley straw)、干し草、稲わら、スイッチグラス、紙くず、サトウキビバガス、サトウモロコシ、大豆、穀物、木、枝、根、葉、木片、おがくず、シュラブおよびブッシュのミリングから得られる成分、野菜、果物、花、家畜糞尿ならびにそれらの混合物が挙げられる。
【0114】
発酵培地は、適切な炭素基質に加え、イソブタノールの生成に必要な培養物の成長および酵素経路の促進に適する、当業者に既知の、適切なミネラル、塩、共同因子、緩衝液および他の成分を含有する必要がある。
【0115】
培養条件
典型的には、細胞は、適切な培地中で約25℃〜約40℃の範囲内の温度で成長する。本発明における好適な成長培地は、Luria Bertani(LB)培養液などの一般的な商業的に調製された培地である。他の限定または合成された成長培地が用いられる場合があり、かつ特定の微生物の成長に適した培地について微生物学または発酵科学に関する当業者は知っているであろう。異化代謝産物抑制を直接的または間接的に調節することで知られる作用物質、例えば環状アデノシン2’:3’一リン酸の使用についても発酵培地内に取り込まれうる。
【0116】
酵母の発酵に適するpH範囲はpH5.0〜pH9.0であり、初期条件としてはpH6.0〜pH8.0が好ましい。
【0117】
発酵は好気性または嫌気性条件下で実施可能であり、嫌気性または微好気性条件が好ましい。
【0118】
発酵培地中で生成されるイソブタノールの量は、当該技術分野で既知の複数の方法、例えば、高性能液体クロマトグラフィー(HPLC)またはガスクロマトグラフィー(GC)を用いて測定することが可能である。
【0119】
工業用バッチおよび連続発酵
発酵のバッチ方法が使用されうる。従来のバッチ発酵は、培地の組成物が発酵開始時に設定され、発酵の間に人工的な改変が施されることがない閉鎖系である。したがって、発酵開始時に培地に望ましい微生物が接種され、系に何も添加しなくても発酵の生成が可能である。しかし、典型的には「バッチ」発酵は炭素源の添加に関連したバッチであり、pHおよび酸素濃度などの要素を制御する試みがなされることが多い。バッチシステムでは、システムの代謝産物およびバイオマス組成物は、最大で発酵が停止する時間まで常時変化する。バッチ培養物内では、細胞が、変化のない誘導期(lag phase)から高成長の対数期(log phase)、最終的に成長率が減少または停止する定常期(stationay phase)にかけて抑制される。定常期における細胞が、未処理の場合、最終的に死滅することになる。対数期における細胞が、一般に最終生成物または中間体の生成の大部分に関与する。
【0120】
標準のバッチシステムに対する変形がフェドバッチシステムである。フェドバッチ発酵プロセスは本発明においても適切であり、基質が発酵プロセスごとに添加されること以外では典型的なバッチシステムを含む。フェドバッチシステムは、異化代謝産物抑制が細胞の代謝を阻害する傾向がある場合や培地内に限られた量の基質を有することが望ましい場合に有用である。フェドバッチシステム内での実際の基質濃度の測定は困難であることから、それはpH、溶解酸素およびCO2などの排ガスの分圧などの測定可能な要素の変化に基づいて評価される。バッチおよびフェドバッチ発酵は一般的で、当該技術分野で周知であり、例がThomas D.、Brock(Biotechnology:A Textbook of Industrial Microbiology、第2版、1989年、Sinauer Associates,Inc.,Sunderland,MA)またはDeshpande,Mukund V.、(Appl.Biochem.Biotechnol.、36:227頁、1992年)(参照により本明細書中に援用される)において見出されうる。
【0121】
本発明はバッチモードで実施されるが、本方法であれば連続発酵方法に適合可能であると考えられる。連続発酵は、限定された発酵培地がバイオリアクターに連続的に添加されかつ等量の条件培地が処理と同時に除去される場合の開放系である。連続発酵では、一般に細胞が主に対数増殖期にある一定の高密度で培養物が維持される。
【0122】
連続発酵では、細胞成長または最終生成物濃度に作用する1つの要素またはいくつかの要素の調節が可能になる。例えば、1つの方法では、炭素源などの限られた栄養素または一定速度での窒素レベルが維持され、あらゆる他のパラメータの抑制が可能になる。他のシステムでは、培地の濁度により測定される細胞濃度が一定に保持される間、成長に作用する多数の要素を連続的に改変することが可能である。連続システムでは、恒常的成長条件を維持するように試みることで、培地の除去(drawn off)に起因する細胞欠損を発酵における細胞成長率に対して均衡させなければならない。連続発酵プロセス用の栄養素および成長因子を調節する方法ならびに生成物形成の速度を最大化するための技術については産業微生物学における当該技術分野で周知であり、かつ種々の方法がBrock、上記で詳述されている。
【0123】
本発明は、バッチ、フェドバッチまたは連続プロセスのいずれかを用いて実施可能であり、かつ既知の発酵モードであればいずれも適するものと考えられる。さらに、細胞は、全細胞触媒としての基質上に固定化され、イソブタノール生成用の発酵条件に従いうると考えられる。
【0124】
発酵培地からイソブタノールを単離するための方法
生物学的に生成されたイソブタノールは、当該技術分野で既知の方法を用いて発酵培地から単離してもよい。例えば、固体は、遠心分離、濾過、デカンテーション、または同様のものにより、発酵培地から除去してもよい。次いで、イソブタノールは、蒸留、液体−液体抽出、または膜に基づく分離などの方法を用いて、上記のように固体を除去するように処理されている発酵培地から単離してもよい。イソブタノールが水と低沸点の共沸混合物を形成することから、単に蒸留を用い、混合物をその共沸組成物に至るまで分離してもよい。蒸留を、別の分離方法と併用し、共沸混合物についての分離を得てもよい。イソブタノールを単離し、精製するための、蒸留と併用可能な方法は、限定はされないが、デカンテーション、液体−液体抽出、吸着、および膜に基づく技術を含む。さらに、イソブタノールは、エントレーナを使用する共沸蒸留を用いて単離してもよい(例えば、DohertyおよびMalone、Conceptual Design of Distillation Systems、McGraw Hill,New York、2001年を参照)。
【0125】
イソブタノール−水混合物は異種共沸混合物を形成し、それから蒸留をデカンテーションと併用し、イソブタノールを単離し、精製してもよい。この方法では、イソブタノールを含有する発酵ブロスは、ほぼ共沸組成物に至るまで蒸留される。次いで、共沸混合物は濃縮され、イソブタノールはデカンテーションによって発酵培地から分離される。デカントされた水相は、還流としての第1の蒸留カラムに戻してもよい。イソブタノールに富むデカントされた有機相は、第2の蒸留カラム内で、蒸留によってさらに精製してもよい。
【0126】
イソブタノールはまた、蒸留と併用し、液体−液体抽出を用いて発酵培地から単離してもよい。この方法では、イソブタノールは、好適な溶媒による液体−液体抽出を用いて、発酵ブロスから抽出される。次いで、イソブタノール含有有機相は蒸留され、イソブタノールは溶媒から分離される。
【0127】
また、蒸留を吸着と併用し、イソブタノールを発酵培地から単離してもよい。この方法では、イソブタノールを含有する発酵ブロスは、ほぼ共沸組成物に至るまで蒸留され、次いで残留水は、吸着剤、例えば分子ふるいの使用によって除去される(Adenら、Lignocellulosic Biomass to Ethanol Process Design and Economics Utilizing Co−Current Dilute Acid Prehydrolysis and Enzymatic Hydrolysis for Corn Stover,Report NREL/TP−510−32438,National Renewable Energy Laboratory、2002年6月)。
【0128】
さらに、蒸留をパーベーパレイションと併用し、イソブタノールを発酵培地から単離し、精製してもよい。この方法では、イソブタノールを含有する発酵ブロスは、ほぼ共沸組成物に至るまで蒸留され、次いで残留水は、親水性膜を通したパーベーパレイションによって除去される(Guoら、J.Membr.Sci.245、199−210頁(2004年))。
【実施例】
【0129】
本発明はさらに以下の実施例にて定義される。これらの実施例が本発明の好ましい実施形態を示す一方であくまで例示目的で与えられることは理解されるべきである。上記の考察およびこれらの実施例から、当業者は、本発明の本質的な特性により、その趣旨および範囲から逸脱することなく本発明に対してさまざまな変更および改良を行うことで、本発明のさまざまな利用および条件への適合が可能であることが確認できる。
【0130】
一般的方法
実施例において用いられる標準の組換えDNAおよび分子クローニング技術は、当該技術分野で周知であり、Sambrook J.ら、Molecular Cloning:A Laboratory Manual(Cold Spring Harbor Laboratory Press,Cold Spring Harbor,NY、1989年、Maniatisとしても公知)およびT.J.Silhavy、M.L.Bennan、およびL.W.Enquist、Experiments with Gene Fusions(Cold Spring Harbor Laboratory Press,Cold Spring Harbor,N.Y.、1984年)およびAusubel F.M.ら、Current Protocols in Molecular Biology(Greene Publishing Assoc.およびWiley−Interscience,N.Y.、1987年)によって記載されている。
【0131】
細菌培養物の維持および成長に適した材料および方法は、当該技術分野で周知である。以下の実施例における使用に適した技術は、Manual of Methods for General Bacteriology(Phillipp Gerhardt編、American Society for Microbiology,Washington,DC.、1994年)またはThomas D.、Brock in Biotechnology:A Textbook of Industrial Microbiology(第2版、Sinauer Associates,Inc.,Sunderland,MA、1989年)に示されるように見出されうる。細菌細胞の成長および維持のために使用されるすべての試薬、制限酵素および材料は、他に特に規定がなければ、Aldrich Chemicals(Milwaukee,WI)、BD Diagnostic Systems(Sparks,MD)、Life Technologies(Rockville,MD)、またはSigma Chemical Company(St.Louis,MO)から入手した。
【0132】
微生物株は、特に断りのない限り、The American Type Culture Collection(ATCC)(Manassas,VA)から入手した。
【0133】
P1形質導入
P1vir形質導入は、いくつかの修飾を伴い、Millerによる記載のように行われた(Miller J.H.、1992年、A Short Course in Bacterial Genetics. Cold Spring Harbor Laboratory,Cold Spring Harbor,N.Y)。つまり、形質導入溶解物を調製するため、ドナー株の細胞を、振とうしながら、Luriaブロス(LB)培地中、37℃で一晩成長させた。これらの細胞の一晩成長を、0.005M CaCl2を含有するLB培地中に継代培養し、通気のない37℃の水槽内に入れた。ファージ添加の1時間前、細胞を、振とうしながら37℃下に置いた。細胞の最終成長後、一定分量1.0mLの培養物を14mlのFalconチューブに分注し、約107個のP1virファージを加えた。これらのチューブを、2.5mLの0.8%LB上層寒天を各チューブに加える前に37℃の水槽内で20分間インキュベートし、内容物を、LB寒天プレート上に展開し、37℃でインキュベートした。翌日、軟質寒天層をこすり、遠心管内に入れた。プレートの表面をLB培地で洗浄し、チューブをボルテックスミキサーを使用して激しく撹拌する前に、遠心管に加え、次いでCHCl3数滴を加えた。4,000rpmで10分間の遠心分離後、P1vir溶解物を含有する上清を回収した。
【0134】
形質導入においては、レシピエント株を、1〜2mLのLB培地中で、振とうしながら。37℃で一晩成長させた。培養物を、10,000rpm、室温で1分間のEppendorf微量遠心管での遠心分離によってペレット状にした。細胞ペレットを、等しい容量のMC緩衝液(0.1M MgSO4、0.005M CaCl2)中に再懸濁し、0.1mLの一定分量でチューブに分注し、0.1mLおよび0.01mLのP1vir溶解物を加えた。P1vir溶解物を全く含有しない対照チューブも含めた。0.2mLの0.1Mクエン酸ナトリウムを加えてP1の感染の停止する前、チューブを37℃で20分間インキュベートした。チューブを37℃で1時間インキュベートする前、1mLのLB培地を各チューブに加えた。インキュベーション後、細胞を上記のようにペレット状にし、25μg/mLのカナマイシンを含有するLBプレート上に展開する前に50〜200μLのLB中に再懸濁し、37℃で一晩インキュベートした。形質導入体を、カナマイシンマーカー挿入物の上流および下流の領域に隣接する染色体に特異的なプライマーを使用するコロニーPCRによってスクリーニングした。
【0135】
染色体からのカナマイシンマーカーの除去が、カナマイシン−耐性株をプラスミドpCP20で形質転換し(Cherepanov P.P.およびWackernagel W.、Gene、158:9−14頁、1995年)、次いでLBアンピシリン(100μg/mL)プレート上に展開し、30℃でインキュベートすることによって得られた。pCP20プラスミドは、λPRプロモーターの制御下で酵母FLPリコンビナーゼを有する。このプロモーターからの発現は、プラスミド上に存在するcI857温度感受性リプレッサーによって制御される。pCP20の複製の複製起点もまた、温度感受性である。アンピシリン耐性コロニーを、LB寒天プレート上にストリークし、42℃でインキュベートした。インキュベーション温度が高くなると同時にFLPリコンビナーゼの発現が誘発され、pCP20プラスミドが細胞から回復された。単離したコロニーを、カナマイシン(25μg/mL)を含有するLBプレートと、LBアンピシリン(100μg/mL)プレートおよびLBプレート上のグリッドにパッチした。得られたカナマイシン感受性、アンピシリン感受性コロニーを、コロニーPCRによってスクリーニングし、カナマイシンマーカーの染色体からの除去を確認した。
【0136】
コロニーPCR増幅においては、HotStarTaq Master Mix(Qiagen(Valencia,CA);カタログ番号71805−3)を、製造業者のプロトコルに従って使用した。0.2μMの各染色体に特異的なPCRプライマーを含有する25μLのMaster Mix反応物に、少量のコロニーを加えた。増幅を、DNA Thermocycler GeneAmp 9700(PE Applied Biosystems(Foster City,CA))で行った。典型的なコロニーPCR条件は、95℃で15分.;95℃で30秒間を30サイクル、50〜58℃の範囲の30秒間のアニーリング温度、約1分/DNAのkbの伸長時間、72℃で伸長されるプライマー;次いで72℃で10分間、次いで4℃で保持であった。PCR産物サイズを、公知の分子量標準との比較により、ゲル電気泳動によって測定した。
【0137】
制限酵素、T4 DNAリガーゼおよびPhusion High Fidelity DNAポリメラーゼ(New England Biolabs(Beverely,MA))を、製造業者の推奨に従って使用した。
【0138】
ゲル電気泳動を、製造業者のプロトコルに従い、プレキャストReliant(登録商標)1%アガロースゲル(Lonza Rockland,Inc.(Rockland,ME))を有するRunOne電気泳動システム(Embi Tec(San Diego,CA))を使用して行った。ゲルは、典型的にはTBE緩衝液(Invitrogen、カタログ番号15581−044)中を走る。
【0139】
形質転換においては、大腸菌(E.coli)のエレクトロコンピテント細胞を、Ausubel F.M.ら、(Current Protocols in Molecular Biology、1987年、Wiley−Interscience)で記載のように調製した。細胞を、25〜50mLのLB培地中、30〜37℃で成長させ、10,000rpmで10分間の遠心分離により、0.5〜0.7のOD600で収穫した。これらの細胞を、培養物の元の開始容量に等しい容量の無菌氷冷水で2回洗浄する。最終洗浄後、細胞を、滅菌水中に再懸濁し、形質転換されるべきDNAを加えた。細胞およびDNAを、冷却したキュベットに移し、製造業者の使用説明書に従い、Bio−Rad Gene Pulser IIでエレクトロポレートした(Bio−Rad Laboratories,Inc.(Hercules,CA))。
【0140】
次の実施例で使用するオリゴヌクレオチドプライマーを表2に示す。すべてのオリゴヌクレオチドプライマーは、Sigma−Genosys(Woodlands,TX)によって合成された。
【0141】
培地中でイソブタノール濃度を測定するための方法
水相および有機相中でのイソブタノールの濃度を、Agilent Technologies(Santa Clara,CA)製のHP−InnoWaxカラム(30m×0.32mm ID、0.25μmのフィルム)を使用するガスクロマトグラフィー(GC)によって測定した。キャリアガスは、一定の上部圧力、150℃での測定された1mL/分の流速のヘリウムであり、インジェクタスプリット比は200℃で1:10であり、オーブン温度は45℃で1分間、10℃/分で45℃〜230℃、および230℃で30秒間であった。フレームイオン化検出を、40mL/分のヘリウム補給気体(makeup gas)を使用して260℃で用いた。培養液試料を、GCへの注入前に、0.2μmの回転フィルタを通して濾過した。所望される分析感度に依存し、0.1μLまたは0.5μLのいずれかの注入容量を使用した。較正標準曲線を、次の化合物、すなわちエタノール、イソブタノール、アセトイン、メソ−2,3−ブタンジオール、および(2S,3S)−2,3−ブタンジオールについて生成した。また、分析標準を使用し、イソブチルアルデヒド、イソ酪酸、およびイソアミルアルコールにおける保持時間を同定した。これらの条件下で、イソブタノール保持時間は、約5.33分であった。
【0142】
略語の意味は以下の通りである。「m」はメートルを意味し、「mm」はミリメートルを意味し、「μm」はミクロンまたはマイクロメートルを意味し、「sec」は秒を意味し、「min」は分を意味し、「hr」は時間を意味し、「nm」はナノメートルを意味し、「μL」はマイクロリットルを意味し、「mL」はミリリットルを意味し、「rpm」は回毎分を意味し、「L」はリットルを意味し、「mm」はミリメートルを意味し、「nm」はナノメートルを意味し、「mM」はミリモル濃度を意味し、「μg/mL」はマイクログラム/ミリリットルを意味し、「mmol/分/mg」は、ミリモル/分/ミリグラムを意味し、「μM」はマイクロモルを意味し、「M」はモル濃度を意味し、「mmol」はミリモルを意味し、「μmol」はマイクロモルを意味し、「g」はグラムを意味し、「μg」はマイクログラムを意味し、「PCR」はポリメラーゼ連鎖反応を意味し、「OD」は光学密度を意味し、「OD600」は600nmの波長で測定された光学密度を意味し、「kD」はキロダルトンを意味し、「bp」は塩基対を意味し、「kb」はキロ塩基対を意味し、「%」はパーセントを意味し、「%w/v」は重量/容量パーセントを意味し、「%v/v」は容量/容量パーセントを意味し、「IPTG」はイソプロピル−β−D−チオガラクトピラノシドを意味し、「wt%」は重量パーセントを意味し、「RBS」はリボソーム結合部位を意味し、「HPLC」は高性能液体クロマトグラフィーを意味し、「GC」はガスクロマトグラフィーを意味し、「g/L」はグラム/リットルを意味し、「g/L/h」はグラム/リットル/時間を意味し、「g/g」はグラム/グラムを意味し、「mL/分」はミリリットル/分を意味し、「℃/分」は摂氏度/分を意味し、「vvm」は容量/容量/分を意味し、「v/v」は容量/容量を意味し、「vol%」は容量パーセントを意味し、「ID」は内径を意味する。
【0143】
実施例1
pflB、frdB、ldhA、およびadhE遺伝子の欠失を有する大腸菌(E.coli)株の作成
本実施例では、4つの遺伝子が不活性化された大腸菌(E.coli)株の改変について記載する。大腸菌(E.coli)株のKeio collection(Babaら、Mol.Syst.Biol.、2:1−11頁、2006年)を、4KO大腸菌(E.coli)(4ノックアウト)の生成に使用した。Keio collectionは、DatsenkoおよびWannerの方法(Datsenko K.A.およびWanner B.L.、Proc Natl Acad Sci.、USA、97:6640−6645頁、2000年)により、大腸菌(E.coli)株BW25113中で生成される単一の遺伝子ノックアウトのライブラリである。collection中では、各欠失遺伝子は、Flpリコンビナーゼによって除去可能なFRTに隣接したカナマイシンマーカーと置換された。4KO大腸菌(E.coli)株は、ノックアウト−カナマイシンマーカーを、P1形質導入により、Keioドナー株からレシピエント株に移すことによって作成した。ノックアウトを生成するための各P1形質導入後、カナマイシンマーカーをFlpリコンビナーゼによって除去した。このマーカーレス株は、次のP1形質導入のための新しいドナー株としての機能を果たした。
【0144】
4KO大腸菌(E.coli)株は、JW0886に加え、3つのKeio株から調製したP1ファージ溶解物でのP1vir形質導入により、Keio株JW0886中で作成した。使用したKeio株を次に挙げる。
−JW0886:kanマーカーはpflBに挿入される
−JW4114:kanマーカーはfrdBに挿入される
−JW1375:kanマーカーはldhAに挿入される
−JW1228:kanマーカーはadhEに挿入される
【0145】
染色体からのカナマイシンマーカーの除去を、カナマイシン耐性株をアンピシリン耐性プラスミドpCP20(CherepanovおよびWackernagel、上記)で形質転換することによって行った。形質転換体を、100μg/mLのアンピシリンを含有するLBプレート上に展開した。プラスミドpCP20は、λPRプロモーターの制御下で酵母FLPリコンビナーゼを有し、このプロモーターからの発現は、プラスミド上に存在するcI857温度感受性リプレッサーによって制御される。pCP20の複製の複製起点はまた、温度感受性である。
【0146】
JW0886株(ΔpflB::kan)を、プラスミドpCP20で形質転換し、100μg/mLのアンピシリンを含有するLBプレート上に30℃で展開した。次いで、アンピシリン耐性形質転換体を選択し、LBプレート上にストリークし、42℃で成長させた。単離したコロニーを、アンピシリンおよびカナマイシン選択培地プレートならびにLBプレート上にパッチした。カナマイシン感受性およびアンピシリン感受性コロニーを、プライマーpflB CkUp(配列番号34)およびpflB CkDn(配列番号35)を伴うコロニーPCRによってスクリーニングした。PCR反応ミックスの一定分量10μLを、ゲル電気泳動によって分析した。予想された約0.4kbのPCR産物が認められ、それからマーカーの除去が確認され、「JW0886マーカーレス」株が生成された。この株は、pflB遺伝子の欠失を有する。
【0147】
「JW0886マーカーレス」株を、JW4114(frdB::kan)由来のP1vir溶解物で形質導入し、25μg/mLのカナマイシンを含有するLBプレート上にストリークした。カナマイシン耐性形質導入体を、プライマーfrdB CkUp(配列番号36)およびfrdB CkDn(配列番号37)を伴うコロニーPCRによってスクリーニングした。予想された約1.6kbのPCR産物を生成するコロニーを、エレクトロコンピテントにし、上記のようにマーカーの除去のため、pCP20で形質転換した。形質転換体をまず、100μg/mLのアンピシリンを含有するLBプレート上に30℃で展開し、次いでアンピシリン耐性形質転換体を選択し、LBプレート上にストリークし、42℃で成長させた。単離したコロニーを、アンピシリンおよびカナマイシン選択培地プレートならびにLBプレート上にパッチした。カナマイシン選択、アンピシリン選択コロニーを、プライマーfrdB CkUp(配列番号36)およびfrdB CkDn(配列番号37)を伴うPCRによってスクリーニングした。予想された約0.4kbのPCR産物が認められ、それからマーカーの除去が確認され、二重ノックアウト株「ΔpflB frdB」が生成された。
【0148】
二重ノックアウト株を、JW1375(ΔldhA::kan)由来のP1vir溶解物で形質導入し、25μg/mLのカナマイシンを含有するLBプレート上に展開した。カナマイシン耐性形質導入体を、プライマーldhA CkUp(配列番号38)およびldhA CkDn(配列番号39)を伴うコロニーPCRによってスクリーニングした。予想された1.1kbのPCR産物を生成するクローンを、エレクトロコンピテントにし、上記のようにマーカーの除去のため、pCP20で形質転換した。形質転換体を、100μg/mLのアンピシリンを含有するLBプレート上に30℃で展開し、アンピシリン耐性形質転換体をLBプレート上にストリークし、42℃で成長させた。単離したコロニーを、アンピシリンおよびカナマイシン選択培地プレートならびにLBプレート上にパッチした。カナマイシン選択、アンピシリン選択コロニーを、0.3kbの産物について、プライマーldhA CkUp(配列番号38)およびldhA CkDn(配列番号39)を伴うPCRによってスクリーニングした。予想された約0.3kbのPCR産物を生成するクローンにより、マーカーの除去が確認され、「3KO」(ΔpflB frdB ldhA)と称される三重ノックアウト株が生成された。
【0149】
「3KO」株を、JW1228(ΔadhE::kan)由来のP1vir溶解物で形質導入し、25μg/mLのカナマイシンを含有するLBプレート上に展開した。カナマイシン耐性形質導入体を、プライマーadhE CkUp(配列番号40)およびadhE CkDn(配列番号41)を伴うコロニーPCRによってスクリーニングした。予想された1.6kbのPCR産物を生成するクローンを、エレクトロコンピテントにし、マーカーの除去のため、pCP20で形質転換した。形質転換体を、100μg/mLのアンピシリンを含有するLBプレート上に30℃で展開した。アンピシリン耐性形質転換体をLBプレート上にストリークし、42℃で成長させた。単離したコロニーを、アンピシリンおよびカナマイシン選択プレートならびにLBプレート上にパッチした。カナマイシン選択、アンピシリン選択コロニーを、プライマーadhE CkUp(配列番号40)およびadhE CkDn(配列番号41)を伴うPCRによってスクリーニングした。予想された約0.4kbのPCR産物を生成するクローンを、「4KO」(ΔpflB frdB ldhA adhE)と称した。
【0150】
実施例2
イソブタノール生合成経路ならびにpflB、frdB、ldhA、およびadhE遺伝子の欠失を有する大腸菌(E.coli)生産宿主の作成
アクロモバクター・キシロスオキシダンス(Achromobacter xylosoxidans)由来のブタノールデヒドロゲナーゼをコードするDNA断片(DNA配列番号9;タンパク質配列番号10)を、標準条件を使用して、A.キシロスオキシダンス(A.xylosoxidans)ゲノムDNAから増幅した。DNAを、Gentra Puregeneキット(Gentra Systems,Inc.(Minneapolis,MN);カタログ番号D−5500A)を使用し、グラム陰性微生物における推奨プロトコルに従って調製した。PCR増幅を、フォワードおよびリバースプライマーのN473およびN469(配列番号44および45)を使用し、それぞれPhusion high Fidelity DNAポリメラーゼ(New England Biolabs(Beverly,MA))を使用して行った。PCR産物を、pCR4 BLUNT(Invitrogen)にTOPO−Bluntクローニングを行い、pCR4Blunt::sadBを生成し、それを大腸菌(E.coli)Mach−1細胞に形質転換した。それに続き、プラスミドを4つのクローンから単離し、配列を検証した。
【0151】
次いで、sadBコード領域を、ベクターpTrc99a(Amannら、Gene 69:301−315頁、1988年)にクローン化した。pCR4Blunt::sadBをEcoRIで消化し、sadB断片を放出し、それをEcoRIで消化されたpTrc99aとライゲートし、pTrc99a::sadBを生成した。このプラスミドを、大腸菌(E.coli)Mach1細胞に形質転換し、得られた形質転換体をMach1/pTrc99a::sadBと称した。これらの細胞におけるsadB遺伝子から発現される酵素の活性は、標準としてイソブチルアルデヒドを使用して分析すると、無細胞抽出液中で3.5mmol/分/mgのタンパク質であることが判定された。
【0152】
次いで、sadB遺伝子を、下記のように、pTrc99A::budB−ilvC−ilvD−kivDにサブクローン化した。pTrc99A::budB−ilvC−ilvD−kivDは、(同時係属中の共同所有された米国特許出願公開第20070092957号明細書(参照により本明細書中に援用される)の実施例9〜14に記載の)イソブタノール発現のためのオペロンを有するpTrc−99a発現ベクターである。pTrc99A::budB−ilvC−ilvD−kivDイソブタノールオペロン内の最初の遺伝子はクレブシエラ・ニューモニエ(Klebsiella pneumoniae)ATCC25955由来のアセト乳酸シンターゼをコードするbudBであり、次いで大腸菌(E.coli)由来のアセトヒドロキシ酸レダクトイソメラーゼをコードするilvC遺伝子である。これに、大腸菌(E.coli)由来のアセトヒドロキシ酸デヒドラターゼをコードするilvDが続き、最後はL.ラクティス(L.lactis)由来の分岐鎖ケト酸デカルボキシラーゼをコードするkivD遺伝子である。
【0153】
sadBコード領域を、プライマーN695A(配列番号42)およびN696A(配列番号43)を使用し、Phusion High Fidelity DNAポリメラーゼ(New England Biolabs(Beverly,MA))を使用し、pTrc99a::sadBから増幅した。増幅を、98℃で1分間の初期変性、次いで98℃で10秒間の変性の30サイクル、62℃で30秒間のアニーリング、72℃で20秒間の伸長および72℃で5分間の最終伸長サイクル、次いで4℃で保持の場合に行った。プライマーN695Aは、クローニングにおけるAvrII制限部位と、sadBコード領域のATG開始コドンの上流にRBSを有した。N696Aプライマーは、クローニングにおけるXbaI部位を含んだ。1.1kbのPCR産物を、AvrIIおよびXbaI(New England Biolabs(Beverly,MA))で消化し、Qiaquick Gel Extraction Kit(Qiagen Inc.(Valencia,CA))を使用してゲル精製した。精製断片を、T4 DNAリガーゼ(New England Biolabs(Beverly,MA))を使用し、同じ制限酵素で切断済みのpTrc99A::budB−ilvC−ilvD−kivDとライゲートした。ライゲーション混合物を、16℃で一晩インキュベートし、次いで製造業者のプロトコルに従い、大腸菌(E.coli)Mach1(商標)コンピテント細胞(Invitrogen)に形質転換した。形質転換体を、100μg/mlのアンピシリンを含有するLB寒天上で成長させた後に得た。形質転換体由来のプラスミドDNAを、QIAprep Spin Miniprep Kit(Qiagen Inc.(Valencia,CA))を使用し、製造業者のプロトコルに従って調製した。得られたプラスミドを、pTrc99A::budB−ilvC−ilvD−kivD−sadBと称した。エレクトロコンピテント4KO細胞を、上記のように調製し、pTrc99A::budB−ilvC−ilvD−kivD−sadBで形質転換した。形質転換体を、100μg/mLのアンピシリンを含有するLB寒天プレート上にストリークした。4KOを伴うプラスミドpTrc99A::budB−ilvC−ilvD−kivD−sadBを有する得られた株(NGCI−031株と称する)を、実施例3で概説される発酵試験において使用した。
【0154】
実施例3
抽出発酵を使用する、組換え大腸菌(E.coli)によるイソブタノールの生成
本実施例の目的は、本明細書中で上記のように作成した大腸菌(E.coli)株NGCI−031によるイソブタノールの生成を示すことである。接種材料の調製のため、すべてのシード培養物を、選択抗生物質としてアンピシリン(100mg/L)を含有するLB培地中で成長させた。この発酵用に使用した半合成培地の組成物および使用した微量金属の調合物を、下記の表3および4に示す。
【0155】
【表6】
【0156】
【表7】
【0157】
表3からの成分1〜10を、所定濃度で水に加え、発酵槽内での最終容量を1.5Lにし、発酵槽の内容物をオートクレーブによって滅菌した。オートクレーブ培地を冷却した上で、成分11〜13を、混合し、濾過滅菌し、次いで発酵槽に加えた。発酵培地(水相)の全最終容量は約1.6Lであった。
【0158】
2.0Lの作動容量を有する3LのBiostat−B DCU−3発酵槽(Braun Biotech International(Melesungen,Germany))を発酵に使用する一方、温度を30℃で維持し、水酸化アンモニウムを使用して
pHを6.8に維持した。シード培養物(2〜10vol%)を培地に接種後、発酵槽を、撹拌速度(rpm)を自動的に制御しながら、0.5vvmの気流を伴う30%の溶解酸素(DO)の設定点で好気的に作動させた。一旦、OD600が10に達すると、培養物を、0.4〜0.5mMのIPTGで誘導し、イソブタノール経路を過剰発現させた。誘導の4時間後、発酵条件を、撹拌器速度を200rpmに低下させることにより、微好気性に変更した。微好気的条件への変更により、バイオマスを生成するための炭素の取り込みを最小化しながらイソブタノールの生成が開始され、それによりバイオマスの形成とイソブタノールの生成が脱共役された。オレイルアルコール(約780mL)をイソブタノール生成段階中に添加し、水相中でのイソブタノールの形成に起因する生成物誘発性の阻害を緩和した。グルコースを発酵槽にボーラス(50wt%のストック溶液)として加え、グルコースレベルを30g/L〜2g/Lの間で保持した。
【0159】
イソブタノールの効率的生成には、生合成経路内でのレドックス均衡を可能にする微好気的条件が要求されることから、空気を発酵槽に0.5vvmで連続的に供給した。連続的吸気により、発酵槽の水相からのイソブタノールの有意なストリッピングが生じた。ストリッピングに起因するイソブタノールの損失を測定するため、発酵槽からの排気を冷却(6.5℃)水トラップを通してスパージし、イソブタノールを濃縮し、次いでそれを、Prima dB質量分析計(Thermo Electron Corp.(Madison,WI))を使用する質量分析を用いて定量した。74または42の質量対電荷比でのイソブタノールのピークを使用し、存在するイソブタノールの量を測定した。
【0160】
水相中のグルコースおよび有機酸を、BioProfile(登録商標)300 Analyzer(Nova Biomedical(Waltham,MA))を使用する発酵中に定期的に監視した。グルコースについても、グルコース分析器(YSI,Inc.(Yellow Springs,OH))を使用して監視した。水相中のイソブタノールおよびオレイルアルコール相中のイソブタノールを、下記のガスクロマトグラフィー(GC)を用いて監視した。2つの相を、遠心分離によって分離した。GC分析を上記のように実施した。イソブタノールの生成における有効力価、速度、および収率(ストリッピングに起因するイソブタノールの損失に対して補正された)は、それぞれ35g/L、0.40g/L/h、および0.33g/gであった。イソブタノールの生成を目的とした抽出発酵におけるオレイルアルコールの使用は、発酵培地および宿主株からの毒性イソブタノール生成物の抽出故に、結果として有効力価、速度、および収率が有意に高まる。
【0161】
実施例4
本実施例の目的は、大腸菌(E.coli)宿主内でのピルビン酸・ギ酸リアーゼ、フマル酸レダクターゼ、アルコールデヒドロゲナーゼおよび乳酸デヒドロゲナーゼをコードする遺伝子における欠失のイソブタノールの生成に対する効果を、これらの欠失を有しない宿主の場合と比較することである。
【0162】
(pflB、frdB、ldhA、およびadhE遺伝子における欠失を含む)大腸菌(E.coli)株NGCI−031によるイソブタノールの生成を、pflB、frdB、ldhA、およびadhE遺伝子に対する欠失を有しない大腸菌(E.coli)株の場合と比較するため、大腸菌(E.coli)株MG1655(ATCC47076)を、プラスミドpTrc99A::budB−ilvC−ilvD−kivD−sadBで形質転換し、大腸菌(E.coli)株MG1655/pTrc99A::budB−ilvC−ilvD−kivD−sadBを生成した。発酵は、本質的に上記のように行ったが、オレイルアルコールを伴わなかった。NGCI−031株におけるイソブタノールの生成における有効力価、速度、および収率(ストリッピングに起因するイソブタノールの損失に対して補正された)は、それぞれ11g/L、0.23g/L/h、および0.25g/gであった一方、MG1655/pTrc99A::budB−ilvC−ilvD−kivD−sadB株におけるイソブタノールの生成における有効力価、速度、および収率(ストリッピングに起因するイソブタノールの損失に対して補正された)は、それぞれ14g/L、0.18g/L/h、および0.12g/gであった。pflB、frdB、ldhA、およびadhEにおける欠失により、pflB、frdB、ldhA、およびadhEにおける欠失を伴わない株に対して有意に改善された速度および収率が得られ、pflB、frdB、ldhA、およびadhEが欠失した株における力価の低下は、発酵時間が短縮した結果であった。
【特許請求の範囲】
【請求項1】
ブタノールデヒドロゲナーゼ活性を有するポリペプチドをコードする少なくとも1つの遺伝子を含む、イソブタノールを生産するための腸内生産宿主であって、ここで、イソブタノールを生産し、次の酵素の活性:
a)ピルビン酸・ギ酸リアーゼ(EC2.3.1.54)
b)フマル酸レダクターゼ(EC1.3.99.1)、
c)アルコールデヒドロゲナーゼ(EC1.2.1.10/EC1.1.1.1)
d)乳酸デヒドロゲナーゼ(EC1.1.1.28)
の少なくとも1つを実質的に有しない、上記腸内生産宿主。
【請求項2】
ブタノールデヒドロゲナーゼ活性を有するポリペプチドをコードする少なくとも1つの遺伝子は、A.キシロスオキシダンス(A.xylosoxidans)から単離される、請求項1に記載の腸内生産宿主。
【請求項3】
ブタノールデヒドロゲナーゼを有するポリペプチドをコードする少なくとも1つの遺伝子は、スコアリングマトリックスBLOSUM62、期待値カットオフ値10およびワードサイズ3ならびにギャップオープニングペナルティ11およびギャップ伸長1でBLASTを用いると、348アミノ酸長を超える、配列番号10で示されるアミノ酸配列に対して少なくとも90%の同一性を有するポリペプチドをコードする、請求項2に記載の腸内生産宿主。
【請求項4】
宿主細胞は、エシェリキア、サルモネラ、エルウィニア、シゲラ、クレブシエラ、セラチアからなる群から選択される属のメンバーである、請求項1に記載の腸内生産宿主。
【請求項5】
宿主は、大腸菌である、請求項4に記載の腸内生産宿主。
【請求項6】
ピルビン酸・ギ酸リアーゼ(EC2.3.1.54)、フマル酸レダクターゼ(EC1.3.99.1)、アルコールデヒドロゲナーゼ(EC1.2.1.10/EC1.1.1.1)、および乳酸デヒドロゲナーゼ(EC1.1.1.28)からなる群から選択される酵素または酵素の一部をコードする少なくとも1つの内因性遺伝子に欠失を含む、請求項5に記載の腸内生産宿主。
【請求項7】
ピルビン酸・ギ酸リアーゼは、配列番号46で示されるアミノ酸配列を有する、請求項6に記載の腸内生産宿主。
【請求項8】
フマル酸レダクターゼは、配列番号54、48、56、および58からなる群から選択されるアミノ酸を有する、請求項6に記載の腸内生産宿主。
【請求項9】
アルコールデヒドロゲナーゼは、配列番号52で示されるアミノ酸配列を有する、請求項6に記載の腸内生産宿主。
【請求項10】
乳酸デヒドロゲナーゼは、配列番号50で示されるアミノ酸配列を有する、請求項6に記載の腸内生産宿主。
【請求項11】
a)ピルビン酸をアセト乳酸に変換するためのEC番号2.2.1.69を有するアセト乳酸シンターゼをコードする少なくとも1つの遺伝子、
b)アセト乳酸を2,3−ジヒドロキシイソ吉草酸に変換するためのEC番号1.1.1.86を有するアセトヒドロキシ酸イソメロレダクターゼをコードする少なくとも1つの遺伝子、
c)2,3−ジヒドロキシイソ吉草酸をα−ケトイソ吉草酸に変換するためのEC番号4.2.1.9を有するアセトヒドロキシ酸デヒドラターゼをコードする少なくとも1つの遺伝子、
d)α−ケトイソ吉草酸をイソブチルアルデヒドに変換するためのEC番号4.1.1.72を有する分岐鎖ケト酸デカルボキシラーゼをコードする少なくとも1つの遺伝子、
e)イソブチルアルデヒドのイソブタノールへの反応を触媒するように機能する、請求項3に記載のブタノールデヒドロゲナーゼであるブタノールデヒドロゲナーゼポリペプチドをコードする少なくとも1つの遺伝子
を含むイソブタノール生合成経路を有する、請求項1または6に記載の腸内生産宿主。
【請求項12】
アセト乳酸シンターゼは、配列番号11、配列番号2、および配列番号13からなる群から選択されるアミノ酸配列を有する、請求項11に記載の腸内生産宿主。
【請求項13】
アセトヒドロキシ酸イソメロレダクターゼは、配列番号15、17および19からなる群から選択されるアミノ酸配列を有する、請求項11に記載の腸内生産宿主。
【請求項14】
アセトヒドロキシ酸デヒドラターゼは、配列番号6、21、23、および25からなる群から選択されるアミノ酸配列を有する、請求項11に記載の腸内生産宿主。
【請求項15】
分岐鎖ケト酸デカルボキシラーゼは、配列番号27、8、30、および32からなる群から選択されるアミノ酸配列を有する、請求項11に記載の腸内生産宿主。
【請求項16】
請求項1または6に記載の生産宿主を、イソブタノールが生産される条件下で、炭素基質を含む発酵培地中で増殖させるステップを含む、イソブタノールを生産する方法。
【請求項1】
ブタノールデヒドロゲナーゼ活性を有するポリペプチドをコードする少なくとも1つの遺伝子を含む、イソブタノールを生産するための腸内生産宿主であって、ここで、イソブタノールを生産し、次の酵素の活性:
a)ピルビン酸・ギ酸リアーゼ(EC2.3.1.54)
b)フマル酸レダクターゼ(EC1.3.99.1)、
c)アルコールデヒドロゲナーゼ(EC1.2.1.10/EC1.1.1.1)
d)乳酸デヒドロゲナーゼ(EC1.1.1.28)
の少なくとも1つを実質的に有しない、上記腸内生産宿主。
【請求項2】
ブタノールデヒドロゲナーゼ活性を有するポリペプチドをコードする少なくとも1つの遺伝子は、A.キシロスオキシダンス(A.xylosoxidans)から単離される、請求項1に記載の腸内生産宿主。
【請求項3】
ブタノールデヒドロゲナーゼを有するポリペプチドをコードする少なくとも1つの遺伝子は、スコアリングマトリックスBLOSUM62、期待値カットオフ値10およびワードサイズ3ならびにギャップオープニングペナルティ11およびギャップ伸長1でBLASTを用いると、348アミノ酸長を超える、配列番号10で示されるアミノ酸配列に対して少なくとも90%の同一性を有するポリペプチドをコードする、請求項2に記載の腸内生産宿主。
【請求項4】
宿主細胞は、エシェリキア、サルモネラ、エルウィニア、シゲラ、クレブシエラ、セラチアからなる群から選択される属のメンバーである、請求項1に記載の腸内生産宿主。
【請求項5】
宿主は、大腸菌である、請求項4に記載の腸内生産宿主。
【請求項6】
ピルビン酸・ギ酸リアーゼ(EC2.3.1.54)、フマル酸レダクターゼ(EC1.3.99.1)、アルコールデヒドロゲナーゼ(EC1.2.1.10/EC1.1.1.1)、および乳酸デヒドロゲナーゼ(EC1.1.1.28)からなる群から選択される酵素または酵素の一部をコードする少なくとも1つの内因性遺伝子に欠失を含む、請求項5に記載の腸内生産宿主。
【請求項7】
ピルビン酸・ギ酸リアーゼは、配列番号46で示されるアミノ酸配列を有する、請求項6に記載の腸内生産宿主。
【請求項8】
フマル酸レダクターゼは、配列番号54、48、56、および58からなる群から選択されるアミノ酸を有する、請求項6に記載の腸内生産宿主。
【請求項9】
アルコールデヒドロゲナーゼは、配列番号52で示されるアミノ酸配列を有する、請求項6に記載の腸内生産宿主。
【請求項10】
乳酸デヒドロゲナーゼは、配列番号50で示されるアミノ酸配列を有する、請求項6に記載の腸内生産宿主。
【請求項11】
a)ピルビン酸をアセト乳酸に変換するためのEC番号2.2.1.69を有するアセト乳酸シンターゼをコードする少なくとも1つの遺伝子、
b)アセト乳酸を2,3−ジヒドロキシイソ吉草酸に変換するためのEC番号1.1.1.86を有するアセトヒドロキシ酸イソメロレダクターゼをコードする少なくとも1つの遺伝子、
c)2,3−ジヒドロキシイソ吉草酸をα−ケトイソ吉草酸に変換するためのEC番号4.2.1.9を有するアセトヒドロキシ酸デヒドラターゼをコードする少なくとも1つの遺伝子、
d)α−ケトイソ吉草酸をイソブチルアルデヒドに変換するためのEC番号4.1.1.72を有する分岐鎖ケト酸デカルボキシラーゼをコードする少なくとも1つの遺伝子、
e)イソブチルアルデヒドのイソブタノールへの反応を触媒するように機能する、請求項3に記載のブタノールデヒドロゲナーゼであるブタノールデヒドロゲナーゼポリペプチドをコードする少なくとも1つの遺伝子
を含むイソブタノール生合成経路を有する、請求項1または6に記載の腸内生産宿主。
【請求項12】
アセト乳酸シンターゼは、配列番号11、配列番号2、および配列番号13からなる群から選択されるアミノ酸配列を有する、請求項11に記載の腸内生産宿主。
【請求項13】
アセトヒドロキシ酸イソメロレダクターゼは、配列番号15、17および19からなる群から選択されるアミノ酸配列を有する、請求項11に記載の腸内生産宿主。
【請求項14】
アセトヒドロキシ酸デヒドラターゼは、配列番号6、21、23、および25からなる群から選択されるアミノ酸配列を有する、請求項11に記載の腸内生産宿主。
【請求項15】
分岐鎖ケト酸デカルボキシラーゼは、配列番号27、8、30、および32からなる群から選択されるアミノ酸配列を有する、請求項11に記載の腸内生産宿主。
【請求項16】
請求項1または6に記載の生産宿主を、イソブタノールが生産される条件下で、炭素基質を含む発酵培地中で増殖させるステップを含む、イソブタノールを生産する方法。
【図1A】

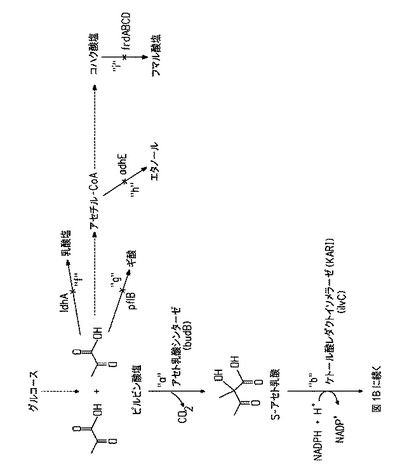
【図1B】
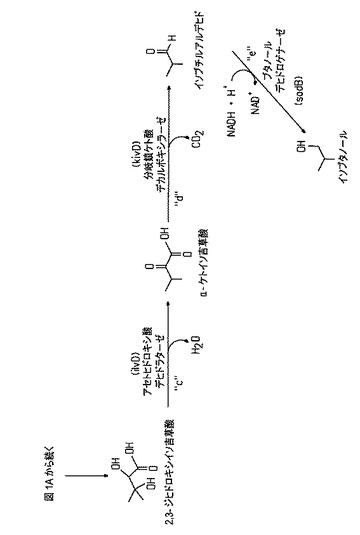
【図1B】
【公表番号】特表2011−522541(P2011−522541A)
【公表日】平成23年8月4日(2011.8.4)
【国際特許分類】
【出願番号】特願2011−512637(P2011−512637)
【出願日】平成21年6月4日(2009.6.4)
【国際出願番号】PCT/US2009/046221
【国際公開番号】WO2009/149240
【国際公開日】平成21年12月10日(2009.12.10)
【出願人】(310011310)ビュータマックス・アドバンスド・バイオフューエルズ・エルエルシー (24)
【Fターム(参考)】
【公表日】平成23年8月4日(2011.8.4)
【国際特許分類】
【出願日】平成21年6月4日(2009.6.4)
【国際出願番号】PCT/US2009/046221
【国際公開番号】WO2009/149240
【国際公開日】平成21年12月10日(2009.12.10)
【出願人】(310011310)ビュータマックス・アドバンスド・バイオフューエルズ・エルエルシー (24)
【Fターム(参考)】
[ Back to top ]