
イネのアントラニル酸シンターゼの第2アイソザイムのαサブユニットをコードする遺伝子に関連するDNA
【課題】イネのアントラニル酸シンターゼ(ASA)の第2アイソザイムのαサブユニットをコードするDNAを提供する。
【解決手段】イネのアントラニル酸シンターゼ(ASA)の第1アイソザイムのαサブユニットであるタンパク質をコードできるDNA配列と、ASAの第2アイソザイムのαサブユニットであるタンパク質をコードできるDNA。前記のDNA配列を担うところのDNA断片をプロモーターの下流に組込まれて成る組換えベクターを構築し、これを導入した植物細胞を培養し、高いトリプトファン含量を有する形質転換植物を再生する。
【解決手段】イネのアントラニル酸シンターゼ(ASA)の第1アイソザイムのαサブユニットであるタンパク質をコードできるDNA配列と、ASAの第2アイソザイムのαサブユニットであるタンパク質をコードできるDNA。前記のDNA配列を担うところのDNA断片をプロモーターの下流に組込まれて成る組換えベクターを構築し、これを導入した植物細胞を培養し、高いトリプトファン含量を有する形質転換植物を再生する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、イネのアントラニル酸シンターゼの2つのアイソザイム(イソ酵素)、すなわち第1アイソザイムと第2アイソザイムとのうちの第2アイソザイムのαサブユニットをコードする遺伝子のDNAと、該遺伝子に関連するDNAに関する。詳しく言えば、本発明はイネ植物のトリプトファンの生合成に関与するアントラニル酸シンダーゼ(anthoranilate synthase)の2つのアイソザイム、すなわち第1アイソザイムと第2アイソザイムの各々のαサブユニットであるタンパク質をそれぞれコードする新規な2つのDNAのうちの、第2アイソザイムのαサブユニットのタンパク質をコードするDNAに関する。
【0002】
また、本発明は、アントラニル酸シンターゼの第2アイソザイムのαサブユニットのタンパク質をコードする新規なDNAが組み込まれた新規な組換えベクターに関する。さらに本発明はその新規なDNAで形質転換された大腸菌、あるいはその新規なDNAで形質転換された植物および種子も包含する。
【0003】
さらに、本発明は、その新規DNAを用いることによる植物のトリプトファン含有量の増加方法に関する。
【背景技術】
【0004】
イネ、トウモロコシ、コムギなどの穀物種子は、人や家畜等にとって重要な栄養源である。しかし、これらの種子は必須アミノ酸の一つであるトリプトファンの含量が低く、栄養価が劣っている。トリプトファン含量の高くて栄養価に優れた穀物種子を産生できる新しい品種の植物の作出が望まれている。
【0005】
植物体中のトリプトファンの生合成経路において、コリスミン酸からアントラニル酸が生合成されるが、アントラニル酸の生成はアントラニル酸シンターゼ (以下、「ASA」 と略記することがある) の触媒作用が関与して、これによってアントラニル酸が生成し、さらにアントラニル酸から6段階の酵素反応によりインドールを経てトリプトファンが生成することが知られている (非特許文献1:生化学実験講座、第11巻、652頁〜653頁、1976年、東京化学同人より刊行)。
【0006】
植物の従来既知のアントラニル酸シンターゼは、複数のサブユニットから構成されていることが知られている。例えばアラビドプシス (和名:シロイヌナズナ;学名Arabidopsis thaliana) のアントラニル酸シンターゼは2種のアイソザイムからなり、またその第1アイソザイムも第2アイソザイムもそれぞれαサブユニットとβサブユニットからなる2量体であることが知られている。アラビドプシスのアントラニル酸シンターゼの第1アイソザイム (ASA1と略記される) のαサブユニットをコードする遺伝子 (asa1) と、第2アイソザイム (ASA2と略記される) のαサブユニットをコードする遺伝子 (asa2) とは単離され、それら遺伝子のDNAの塩基配列が解明されている (非特許文献2:The Plant Cell, 4巻, 721頁〜733頁, 1992年)。
【0007】
他方、先に、本発明者らは、イネのトリプトファン生合成の調節に重要な機能領域を有することが示唆されるアントラニル酸シンターゼαサブユニットに着目して、トリプトファンと植物ホルモンIAAの生合成調節機構についての知見を得ることを目的として、アントラニル酸シンターゼ・タンパク質をコードする遺伝子を単離する研究を1996年に行った。この研究の報告の要約として、本発明者らは、イネ (農林8号) の幼植物から、mRNAとゲノミックDNAを抽出し、cDNAライブラリーとゲノミックDNAライブラリーを作製し、これらライブラリーを用いて且つシロイヌナズナのasa遺伝子のcDNA断片をプローブとして用いて、ゲノミックサザン解析とライブラリーのスクリーニングを行い、これによってイネのアントラニル酸シンターゼのasa遺伝子に相当すると考えられるDNAを収得したことを発表した (非特許文献3:「育種学雑誌」 46巻、別冊2、28頁、1996年)。この文献に発表された研究報告の要約には、イネのアントラニル酸シンターゼのasa遺伝子に相当するDNA断片を収得したことを報告したが、そのDNA断片の収得に用いた具体的な手法を本発明者らは開示されていず、また前記のDNA断片の塩基配列も未だ決定されていないことを報告した。また、前記の研究報告の要約には、イネのアントラニル酸シンターゼをコードする遺伝子に相当すると考えられるDNAには、2種類のDNAが存在することに言及した(前記の 「育種学雑誌」 46巻、別冊2、28頁、1996年)。
【0008】
また、アラビドプシスのASAの第1アイソザイムのαサブユニットのASA遺伝子のDNAと、該DNAを改変したDNA断片とを、タバコ植物体に導入し、その遺伝子の機能をタバコで発現させることに関する報告がされている (非特許文献4:Massachusetts Institute of Technology, Cambridge, MA, 1993年)。
【非特許文献1】「生化学実験講座」第11巻、652頁〜653頁、(1976年)
【非特許文献2】「The Plant Cell」4巻、721頁〜733頁、(1992年)
【非特許文献3】「育種学雑誌」46巻、別冊2、28頁、(1996年)
【非特許文献4】「Massachusetts Institute of Technology,Cambridge, MA」(1993年) しかしながら、本発明者の知る限りでは、イネのASAのアイソザイムのαサブユニットであるタンパク質のアミノ酸配列を解析したこと、およびイネのASAのアイソザイムのαサブユニットをコードする遺伝子を取得できた具体的な方法を報告する文献は知られていない。また、該遺伝子の発現に関わるプロモーター配列も知られていない。またイネのASAをコードする遺伝子を利用することを報告する文献も知られていない。
【発明の開示】
【発明が解決しようとする課題】
【0009】
本発明の一つの目的は、イネのASAに関連する遺伝子、詳しくはイネのASAの第2アイソザイムのαサブユニットをコードする新規なDNAをイネ植物から収得することにあり、また、本発明の他の目的はこのDNAの塩基配列を決定することにある。
【0010】
本発明の別の目的は、その新規DNAによって、トウモロコシ、イネ、ダイズ、コムギ、オオムギ、トマト、ジャガイモなどの有用植物を形質転換することであり、また高いトリプトファン含量を持つ種子を産生できる有用植物の新しい形質転換品種を提供することである。本発明のその他の目的は、後記の説明から明らかになるであろう。
【課題を解決するための手段】
【0011】
上記の諸目的を達成するために、本発明者らは一連の種々研究を行った。先づ、イネ植物からASAの2つのアイソザイムの各々のαサブユニットをコードする遺伝子を取得するための研究を行った。この研究の結果、イネの幼植物体の組織、例えば緑色の茎葉の破砕物から全RNAを遺伝子工学技術で知られる方法により抽出し、その抽出された全RNAから全mRNAを常法により単離し、その全mRNAから、市販の全cDNA合成キットによりイネの全cDNAを収得することに成功した。得られた全cDNAは、λgt11ファージベクターのEcoRI切断片の末端を仔ウシ小腸由来アルカリホスファターゼで処理したファージベクター (STRATAGEN社製の市販品) に連結すると、組換えベクターを作成できること、さらに作成された組換えベクターをラムダ-ファージにパッケージすると、複製可能な組換えラムダ-ファージを構築できることが多くの試行錯誤の結果により知見された。
【0012】
その組換えラムダ-ファージを大腸菌Y1088に感染させ、インキュベートすると、多数のプラーク (溶菌斑) として組換えλファージの多数が得られること、また得られた多数のプラークに含まれる組換えλファージ群は、イネ由来の全cDNAを含有する各種多様なファージであって、これはイネのcDNAライブラリーとして利用できることが知見された。
【0013】
他方、前記の非特許文献2:「The Plant Cell」4巻、721頁〜733頁 (1992年) に記載されたアラビドプシスのASAの第1アイソザイムのαサブユニットであるタンパク質のアミノ酸配列と、これから推認される該タンパク質をコードする遺伝子の塩基配列と、アラビドプシスのASAの第2アイソザイムのαサブユニットであるタンパク質のアミノ酸配列と、これから推認される該タンパク質をコードする遺伝子の塩基配列とを参照しながら、PCR法のプライマーとして適すると考えられた21個のヌクレオチドからなる第1のオリゴヌクレオチドと、24個のヌクレオチドからなる第2のオリゴヌクレオチドとを本発明者は、化学合成により作製した。
【0014】
前記の第1のオリゴヌクレオチドと第2のオリゴヌクレオチドと市販されているアラビドプシスのcDNAライブラリー(テンプレートとして利用される) との混合物を用いてPCR法増幅反応を行うと、第1および第2のオリゴヌクレオチドは、PCR法で所要なプライマー (相補的DNA) として働くことができ、そして、アラビドプシスのASAの2つのアイソザイムのそれぞれのαサブユニットをコードする遺伝子に相当するDNA配列のうちの一部分を構成しているDNA断片が増幅できることが認められた。そしてPCR増幅反応液からアラビドプシスのASAの第1および第2アイソザイムの各々のαサブユニットをコードする遺伝子の一部分の増幅生成物を、プローブDNAとして回収することに成功した。
【0015】
このように収得された上記のプローブDNAを利用することによって、ファージプラーク-ハイブリダイゼーション法によって、先に得られたイネのcDNAライブラリー (すなわち前記の組換えλファージの30万個のプラーク) から、多くの試行錯誤の試験の結果として、イネのASAの第1アイソザイムのαサブユニットをコードする遺伝子と第2アイソザイムのαサブユニットをコードする遺伝子を組込まれた組換えλファージのプラークの8個を単離することに幸にも成功した。それら8個のプラークの組換えλファージを別々に増幅した後に、各々のλファージDNAを常法で単離した。
【0016】
上記のようにして得られた、イネのASAの第1および第2アイソザイムの各々のαサブユニットをコードする遺伝子に相当すると認められるDNA配列を内部に含有する組換えファージDNAを、制限酵素EcoRIで切断して得られたDNA断片は、これを次に、市販の既知のプラスミドベクターp Bluescript II SK(+)のEcoRI切断部位にDNAリゲーションキットにより挿入、連結することができた。こうして得られた組換えプラスミドベクターで大腸菌XL1-Blue MRF'を形質転換することができた。得られた大腸菌形質転換体をインキュベートして多数の菌体を得ることができた。得られた菌体からプラスミドDNAを採取した。このように採取したプラスミドDNAを、DNAの塩基配列の解析にかけることによって、このDNA断片に含有されて且つイネのASAの第1および第2アイソザイムの各々のαサブユニットをコードする遺伝子に相当すると判定された2種のDNA配列のうちの第1のDNA配列の塩基配列は、後記の配列表の配列番号1に記載の塩基配列を有するものであると今回、確認された。また、前記の2種のDNA配列のうちの第2のDNA配列の塩基配列は配列表の配列番号10に記載の塩基配列を有するものであると今回、確認された。
【0017】
さらに、本発明者らの知る限りでは、配列表の配列番号1に記載の塩基配列を有するDNAと、配列番号10に記載の塩基配列を有するDNAとは、何れの文献にも記載されていない新規なDNA配列であると認められた。
【0018】
配列表の配列番号1の塩基配列を有するDNAでコードされるタンパク質は配列表の配列番号2に記載のアミノ酸配列を有するタンパク質であると認められ、またこのタンパク質はイネのASAの第1アイソザイムのαサブユニットを構成するタンパク質であると認められる。
【0019】
従って、本明細書に開示される第1の発明(本件分割出願の原願である特願2000-508808号の請求項1の発明)においては、配列表の配列番号2に示すアミノ酸配列を有するタンパク質であって、イネのアントラニル酸シンターゼの第1アイソザイムのαサブユニットをコードするDNAが提供される。
【0020】
第1の発明による上記のDNAは、具体的には、配列表の配列番号1に記載の塩基配列を有するDNAであることができる。
【0021】
第1の発明のDNAは、イネのASAの第1アイソザイムのαサブユニットであるタンパク質をコードするDNAである。この第1の発明のDNAは、本発明者らの研究を行うに際しては、前述のように、イネのcDNAライブラリーから遺伝子工学の技法で取得されたものである。しかし、そのDNAの塩基配列が本発明で明らかにされたので、配列表の配列番号1の塩基配列を参照してヌクレオチドから化学合成によっても取得することができる。また、前記の配列番号1の塩基配列を参照して合成ヌクレオチドをプローブとして作製して用いるか、もしくはその合成オリゴヌクレオチドをプライマーとして用いる公知の方法によってイネ染色体のDNAライブラリーから、ポリメラーゼ・チェイン・リアクション (PCR) の方法又はハイブリダイゼーションによって第1の本発明のDNAを取得することもできる。
【0022】
さらに、配列表の配列番号10の塩基配列を有するDNAでコードされるタンパク質は配列表の配列番号11に記載のアミノ酸配列を有するタンパク質であると認められ、またこのタンパク質はイネのASAの第2アイソザイムのαサブユニットを構成するタンパク質であると認められる。
【0023】
従って、第2の本発明(本件特許請求の範囲の請求項1の発明)においては、配列表の配列番号11に示すアミノ酸配列を有するタンパク質であって、イネのアントラニル酸シンターゼの第2アイソザイムのαサブユニットをコードするDNAが提供される。
【0024】
第2の本発明による上記のDNAは、具体的には、配列表の配列番号10に記載の塩基配列を有するDNAであることができる(請求項2)。
【0025】
第2の本発明(本件特許請求の範囲の請求項1または2の発明)のDNAは、イネのASAの第2アイソザイムのαサブユニットであるタンパク質をコードするDNAである。この第2の本発明のDNAは、本発明者らの研究を行うに際しては、前述のように、イネのcDNAライブラリーから遺伝子工学の技法で取得されたものである。しかし、そのDNAの塩基配列が本発明で明らかにされたので、配列表の配列番号10の塩基配列を参照してヌクレオチドから化学合成によっても取得することができる。また、前記の配列番号10の塩基配列を参照して合成ヌクレオチドをプローブとして作製して用いるか、もしくはその合成オリゴヌクレオチドをプライマーとして用いる公知の方法によってイネ染色体のDNAライブラリーから、ポリメラーゼ・チェイン・リアクション(PCR)の方法又はハイブリダイゼーションによって第2の本発明のDNAを取得することもできる。
【0026】
以下に、イネの茎葉から遺伝子工学の技法によって、前記した第1の発明によるDNAおよび第2の本発明によるDNAを取得する方法を概略的に説明する。
【0027】
(1) イネmRNAの調製およびイネのcDNAライブラリーの構築
イネ (Oriza sativa) の種々な組織、例えば茎葉、根、カルスなどの組織、好ましくは緑色の茎葉から、常法により全RNAを抽出する。抽出された全RNAから蛋白質などの夾雑物を除いた後、さらにオリゴdTセルロースの充填カラムに通してpoly(A)+RNAを精製することによって、イネの全mRNAを得ることができる。
【0028】
次に、全mRNAから市販のcDNAを合成キットによりイネの全cDNAを合成する。合成された全cDNAをファージベクター、例えばλgt11ベクター又はλZAPIIベクターなどに連結する。得られた組換えベクターをラムダファージに組み込み、多数の組換えファージを得る。それら組換えファージを宿主として大腸菌に感染させ、インキュベートすると、プラークとして多数の組換えファージを得ることができる。これら一連の操作は、市販のcDNAクローニングキットを使用して実施できる。
【0029】
このように宿主大腸菌の溶菌のプラークとして得られた多数の組換えファージは、イネ由来の全cDNAを含有する多種多様のファージから成るものであるから、イネのcDNAライブラリーとして利用できる。
【0030】
(2) PCR法用のプライマーの構築
アラビドプシスのASAの第1アイソザイムのαサブユニットの遺伝子の既知の塩基配列と第2アイソザイムのαサブユニットの遺伝子の既知の塩基配列との間に共通に保存されている塩基配列 (オンラインデータベースEMBL: M92353) を参照して且つアラビドプシスのASAの第1アイソザイムが植物体でより強い発現力をもつことを勘案して、本発明者らは2種類のオリゴヌクレオチド (後記の配列表の配列番号8および9のオリゴヌクレオチド) をPCR法用のプライマー (相補的DNA) として化学合成により作製して構築した。
【0031】
(3) プローブDNAの作製
先に多数の組換えファージからなるプラークとして得られたイネのcDNAライブラリーの中で、所望のイネのASAのαサブユニットをコードするDNAを選択的に採取するのに用いられるプローブDNAを作製する。このプローブDNAの作製のために、前記の合成オリゴヌクレオチドよりなるプライマーを使用し、それで、PCR法によりテンプレートとしてのアラビドプシスのcDNAライブラリーからアラビドプシスのASAの第1および第2アイソザイムのαサブユニットの遺伝子の一部分をコードするDNAを増幅する。
【0032】
増幅反応をPCR法によって反復した後に、アラビドプシスのASAのαサブユニットの遺伝子に相当するDNA配列のうちの一部分であるDNA断片の増幅生成物を、増幅反応液から、所望なプローブDNAとして採取する。
【0033】
(4) イネcDNAライブラリーからのイネのASAのαサブユニットの遺伝子のDNAの選択
先にイネのcDNAライブラリーとして得られた組換えファージの多数のプラークの中から、上記のプローブDNAをスクリーニング用に利用するファージプラーク-ハイブリダイゼーション法により、目的のイネのASAのαサブユニットの遺伝子に全体的に対応するDNA配列を組込まれた組換えファージよりなる数個のプラークを選抜する。
【0034】
これによって選抜されたプラークとして採取された組換えファージは、イネのASAのαサブユニットの遺伝子のDNAに相当する目的のDNA配列を内部に含有するDNA断片が組込まれている組換えファージの形のものである。
【0035】
すなわち、詳しく言えば、上記のようなプラーク・ハイブリダイゼーションで選抜されたプラークのファージを採取して、さらに採取ファージからファージDNAを回収する。回収したファージDNAをジデオキシ法などで処理することにより、そのファージDNAの内部に存在するイネ由来の挿入DNA断片の塩基配列を決定することができる。そのイネ由来の挿入DNA配列の塩基配列中のタンパク質コード領域 (オープンリーディングフレーム) から規定されるアミノ酸配列を、アラビドプシスのASAのαサブユニットであるタンパク質の既知のアミノ酸配列に対して比較すると、相同性を判定することにより、上記で回収されたファージDNAがイネのASAのαサブユニットの遺伝子に対応するDNA配列を内部に含有するDNA断片であることを特定できる。
【0036】
従って、イネのASAのαサブユニットの遺伝子に相当するDNA配列を内部に含有すると判定された挿入DNA断片は、上記のように選抜された採取ファージのファージDNAから制限酵素で切出して収得できる。
【0037】
(5) イネのASAのαサブユニットの遺伝子に相当するcDNAのクローニング
上記のようにイネのASAのαサブユニットの遺伝子に相当するDNA配列を内部に含有するDNA断片として、ファージから切出して収得されたDNAは、これをプラスミドベクターp Bluescript II SK(+)のEcoRI切断部位に挿入、連結して組換えプラスミドベクターを構築する。このように構築された組換えプラスミドベクターで大腸菌XL1-Blue MRF'を形質転換する。こうして得られた大腸菌形質転換体を培養して増殖させると、イネのASAのαサブユニット遺伝子に相当するDNA配列を含有するDNA断片を担う前記の組換えプラスミドをクローニングできる。従って、イネのASAのαサブユニットの遺伝子に対応するDNA配列を含有するDNA断片がクローニングできる。
【0038】
本明細書に記載の発明において、上記のようにプラスミドベクターp Bluescript II SK (+) に連結されて大腸菌中でクローニングされて得られて且つイネのASAのαサブユニットの遺伝子に対応するDNA配列を内部に含有するDNA断片として、塩基数の異なる2種類のDNA断片が得られた。ここで得た小さいサイズのDNA断片をDNA断片Xとここで仮称し、また大きいサイズのDNA断片を、DNA断片Yとここで仮称する。
【0039】
(6) クローニングされたDNAのシークエンス解析
(i) 上記のクローニングされた組換えプラスミドから制限酵素EcoRIによりイネのASAのαサブユニットの遺伝子に該当のDNA配列を内部に含有する2種類のDNA断片として、DNA断片XとDNA断片Yとを別々に切り出す。切り出されたDNA断片XおよびDNA断片Yをそれぞれに市販の塩基配列決定キットで処理すると、イネのASAのαサブユニットの遺伝子に該当するDNA配列を内部に含有するDNA断片XまたはDNA断片Yにおける全体の塩基配列を決定できる。上記のDNA断片Xについて、このように決定された塩基配列をもち且つイネのASAの第1アイソザイムのαサブユニットをコードするDNA配列は、後記の配列表の配列番号1に記載された塩基配列を有し、1734個の塩基から成る。このDNA配列を配列OSASA-1配列と命名する。このDNA配列OSASA-1は、第1の本発明によるDNAの一例である。
【0040】
なお、後記の実施例1で得られたところの配列表の配列番号1に記載の塩基配列を有して且つイネのASAの第1アイソザイムのαサブユニットの遺伝子に相当するDNA配列を内部に含有する前記のDNA断片Xは、p Bluescript II SK (+)プラスミドベクター (STRATGENE社製) のEcoRI切断部位に挿入、連結された。このように得られた組換えプラスミドベクター (ベクターpOSASA-1と命名された) の導入により形質転換された大腸菌XL1-Blue MRF'はエシェリヒア・コリ (Escherichia coli) XL1-Blue MRF' (OS-asa1) と命名され、日本の茨城県、つくば市の工業技術院生命工学工業技術研究所に1997年8月18日に、FERM P-16388の受託番号で寄託されている。さらに、エシエリヒア・コリOS-asa1は、1998年8月7日以降、ブダペスト条約の規約下でFERM BP-6453の受託番号で前記の研究所に寄託されてある。
【0041】
前記された第1の発明によるDNAは、本発明で解明された該DNAの塩基配列を指針として用いることによって、イネのASAの第1アイソザイムのαサブユニットであるタンパク質を化学合成により多量に収得することを可能にしている点で有用であり、そしてイネのASAの第1アイソザイムのαサブユニットであるタンパク質の酵素学的研究を進めるのに貢献できるものである。
【0042】
(ii) さらに、前記のDNA断片Yについて、前記のように決定された塩基配列をもち且つイネのASAの第2アイソザイムのαサブユニットをコードするDNA配列は、配列表の配列番号10に記載された塩基配列を有し、1821個の塩基から成り、このDNA配列はOSASA-2配列と命名された。このDNA配列OSASA-2は、第2の本発明(本件特許請求の範囲の請求項1または2に記載の発明)によるDNAの一例である。
【0043】
なお、後記の実施例1で得られたところの、配列表の配列番号10に記載の塩基配列を有して且つイネのASAの第2アイソザイムのαサブユニットの遺伝子に相当するDNA配列を内部に含有する前記のDNA断片Yは、p Bluescript II SK (+)プラスミドベクターのEcoRI切断部位に挿入、連結された。ここで得られた組換えプラスミドベクター (ベクターpOSASA-2と命名される) の導入により形質転換された大腸菌XL1-Blue MRF'は、エシエリヒア・コリXL1-Blue MRF' (Os-asa2) と命名され、工業技術院生命工学工業技術研究所に1998年6月18日にFERM P-16853の受託番号で寄託され、また1998年8月7日以降、ブダペスト条約の規約下にFERM BP-6454の受託番号で寄託されてある。
【0044】
第3の発明(本件特許願の特許請求の範囲から除外されてあるが、特願2004-257204号の請求項1の発明)においては、配列表の配列番号13に記載のアミノ酸配列を有するタンパク質であって、且つイネのアントラニル酸シンターゼの第1アイソザイムのαサブユニットの活性を有するが但しトリプトファンによるフィードバック抑制に対して非感受性であるタンパク質をコードするDNAが得られている。
【0045】
前記した第3の発明による上記のDNAの具体的な例には、後記の実施例2に記載された方法で作製されて実施例2で改変D配列と命名され且つ配列表の配列番号12に記載の塩基配列を有するDNA配列がある。
【0046】
なお、配列表の配列番号12に記載の塩基配列を有して改変D配列と命名された上記のDNAは、イネのASAの第1アイソザイムのαサブユニットの活性を有するけれどもトリプトファンによるフィードバック抑制に対して非感受性である上記タンパク質をコードするDNAである。
【0047】
第3の発明による新規なDNAの具体的な例は、前述したように、後記の実施例2に記載された方法で作製されて改変D配列と命名されたDNA配列がある。
【0048】
その改変D配列と命名されたDNA配列は、配列表の配列番号12に記載の塩基配列を有するDNAである。この改変D配列は、配列表の配列番号1に記載の塩基配列を有する第1の本発明のDNAの中にある967番目、968番目、969番目のGAC配列 (アスパラギン酸をコードするコドン) での967番目の塩基G (グアニン) 1個をA (アラニン) 1個に置換して、塩基配列の967番目に配列 (アスパラギンをコードするコドン) が在るように改変されたDNAに相当する。この改変D配列でコードされるタンパク質は、配列表の配列番号13に記載のアミノ酸配列を有するものであり、イネのASAの第1アイソザイムのαサブユニットの活性を有する。
【0049】
また、第3の発明のDNAの具体例である上記の改変D配列でコードされるタンパク質は、トリプトファンの生合成経路に関与するASAが生合成生成物としてのトリプトファンによる酵素活性のフィードバック阻害を受けなくなるように酵素活性を改変された新規なタンパク質である。この新規なタンパク質をコードする改変DNAは、後記されるように、植物のトリプトファン含量を増加させる目的で植物の形質転換に使用される。
【0050】
なお、一般的には、DNA配列の一部分の領域中にある塩基配列を改変する方法として、(Kunkel法 「Methods in Enzymology」 154巻、367号)、およびオリゴヌクレオチド−ダイレクト デュアルアンバー法、さらにその他の方法が知られている。
【0051】
第1の本発明のDNAでコードされるASAの第1アイソザイムαサブユニットを、トリプトファンによる酵素活性のフィードバック阻害を受けなくなるように改変するために、前記した第1の発明のDNAの塩基配列の一部を改変する方針として、文献 「Plant Physiology」 第110巻、51頁〜59頁、1996年に記載されるトリプトファンアナログ体に耐性を示す突然変異アラビドプシスの公知のASA遺伝子に関する報告を参考にしながら、本発明者らは、配列表の配列番号1に記載のDNA (OSASA-1配列) の塩基配列の967番目の塩基、グアニン (G) をアデニン (A) に取換えることによって作製された改変DNAが所期の目的に有効であろうとの予想的な着想を得た。
【0052】
この着想に基づいて、本発明者らは、種々の研究を行ったが、多くの試行錯誤の結果として、イネのASAの第1アイソザイムのαサブユニットの遺伝子に対応するDNA配列 (すなわちOSASA-1配列) を含有するDNA断片を、プラスミドベクターp Bluescript II SK (+) のEcoRI切断部位の間にリゲーションキットにより挿入、連結してなる組換えプラスミドベクター (以下、pOSASA-1と称する) を作成し、これが前記した第3の発明での目的の新規な改変DNAの作製のための出発材料として適することを認めた。
【0053】
このような目的で新規な改変DNAを、前記の出発材料である組換えプラスミドベクターpOSASA-1から、PCR法に準じて作製するために本発明者らは研究した。そのPCR法のために適当に使用できるプライマーとして、4種類のプライマー、すなわち、後記の配列表の配列番号16に記載の塩基配列を有するオリゴヌクレオチドよりなるプライマーOSASN1と、配列番号17に記載の塩基配列を有するオリゴヌクレオチドよりなるプライマーOSASN2と、配列番号18に記載の塩基配列を有するオリゴヌクレオチドよりなるプライマーOSASC1と、配列番号19に記載の塩基配列を有するオリゴヌクレオチドよりなるプライマーOSASC2とを化学合成により作製した。
【0054】
上記の組換えプラスミドベクターpOSASA-1と、上記の4種の合成オリゴヌクレオチドよりなるプライマーとを利用して、後記の実施例2に詳述される方法を実施すると、第3の発明における改変DNAの具体例である配列番号12に記載のDNA、すなわち改変 「D配列」 を収得することに成功できたのである。
【0055】
次に、前記した第3の発明によるDNAの具体例である上記の改変D配列を含有するDNA断片を作製するのに好適に使用でき且つ後記の実施例2において例示される手順よりなるDNAの改変方法をここに要約的に説明する。
【0056】
(1) 前記した第1の発明のDNAのクローニング
配列表の配列番号1に記載される1734個の塩基からなる第1の発明のDNA、すなわち前記のOSASA-1配列を含有するDNA断片を、DNAリゲーションキットの利用により、ベクターp Bluescript II SK (+) のEcoRI切断部位の間に挿入、連結することによって、前記の組換えプラスミドベクターpOSASA-1を得る。このベクターpOSASA-1を大腸菌XLl-Blue MRF'に導入し、これで得られた形質転換大腸菌を増殖する。増殖された大腸菌細胞から、常法で抽出することにより組換えプラスミドベクターpOSASA-1を大量に収得する。この操作により第1の発明によるDNA配列、すなわちOSASA-1配列をクローニングする。
【0057】
(2) CR用プライマーの構築
DNA合成装置 (Model-391、アプライド バイオシステムズ社製) を利用して、下記の塩基配列を有する4種類のオリゴヌクレオチドをプライマーとして合成する。すなわち、下記のとおり、配列表の配列番号16に記載の塩基配列を有するオリゴヌクレオチドよりなるプライマーOSASN1と、配列番号17に記載の塩基配列を有するオリゴヌクレオチドよりなるプライマーOSASN2と、配列番号18に記載の塩基配列を有するオリゴヌクレオチドよりなるプライマーOSASC1と、配列番号19に記載の塩基配列を有するオリゴヌクレオチドよりなるプライマーOSASC2とを化学合成により作製する。
【0058】
(i) プライマーOSASAN1 (配列表の配列番号16に記載され且つ下記される塩基配列をもつプライマー):
5'-GAGTCAGTTGACGAAGCGTATGAGG-3'
(ii) プライマーOSASAN2 (配列表の配列番号17に記載され且つ下記される塩基配列をもつプライマー):
5'-GTACATTTGCTAACCCCTTTGAGG-3'
(iii) プライマーOSASAC1 (配列表の配列番号18に記載され且つ下記される塩基配列をもつプライマー):
5'-CAAAGGGGTTAGCAAATGTACGC-3'
(iv) プライマーOSASAC2 (配列表の配列番号19に記載され且つ下記される塩基配列をもつプライマー):
5'-GTTCAACGTTCATCAGTTTCTCCACC-3'
(3) 所要なDNA断片のPCR法による増殖と回収
所要なDNA断片を増殖して得るために、PCR法の第一段階として2つの反応、すなわち下記の (A) 反応と (B)反応とを行う。
【0059】
その(A)反応は、テンプレートとして用いられる前記の組換えプラスミドベクターpOSASA-1と、プライマーとして用いられる前記のプライマーOSASAN1 (配列番号16の合成オリゴヌクレオチド) と、プライマーOSASAC1 (配列番号18の合成オリゴヌクレオチドであり、5'-末端から8〜10番目のGTTが改変D配列の改変部AACを誘導できるプライマー) とを、PCR法用の通常の反応液 (Tris-HCl、MgCl2、KCl、4種のデオキシヌクチオチド三リン酸 (dNTP)、およびLaTaqDNAポリメラーゼを含有) に加え、次いで増殖反応を行うことから成る。
【0060】
この(A)反応によって、前記の改変D配列のうちの或る領域における塩基配列をもつDNA断片 (DNA断片-Aと称す) が増殖反応により生成される。
【0061】
また (B) 反応は、テンプレートとして用いられる前記の組換えプラスミドベクターpOSASA-1と、プライマーとして用いられる前記のプライマーOSASAC2 (配列番号19の合成オリゴヌクレオチド) と、プライマーOSASAN2 (配列番号17の合成オリゴヌクレオチドであり、5'-末端から12〜14番目のAACが改変D配列の改変AACを誘導できるプライマー) とを、(A) 反応で用いたと同じPCR法用の通常の反応液に加え、次いで増殖反応を行うことから成る。
【0062】
この(B)反応によって、前記の改変D配列のうちの或る領域における塩基配列を含有するDNA断片 (DNA断片-Bと称す) が増殖反応により生成される。PCR法による上記の増殖反応は、市販のPCR反応装置を用いて実施できる。
【0063】
増殖反応の終了後に、上記の (A) 反応の増殖反応液を低融点アガロース電気泳動により分画して、増殖生成物としての268bp (ベースペア) のDNA断片-Aを含むバンドをアガロースゲルから切出す。また、上記の (B) 反応の増殖反応液を同様に低融点アガロース電気泳動により分画し、そしてそのアガロースゲルから336bp (ベースペア) のDNA断片-Bを含むバンドを切出す。
【0064】
次いで、それら2つのゲル切片をDNA精製キット、例えばGenclean II (フナコシ社製) により精製することにより、DNA断片-Aの精製品と、DNA断片-Bの精製品とをそれぞれ回収する。
【0065】
さらに、PCR法の第2段階として、配列表の配列番号1に記載の塩基配列のうちの967番目のグアニンをアデニンに置換してなる塩基配列をもつDNA配列 (すなわち配列番号12の塩基配列をもつ第3の発明による改変D配列断片に相当する配列) の部分領域に相当する583bp (ベースペア) のDNA断片 (C断片) を作製する工程を行う。
【0066】
このためには、テンプレートとして用いられる前記の (A) 反応の増副生成物であるDNA断片-Aの精製品 (配列長268bp) と、(B) 反応の増副生成物であるDNA断片-Bの精製品 (配列長336bp) とを、PCR法用の通常の増殖反応液 (Tris-HCl、MgCl2、KCl、4種のdNTP、およびLa Taq DNAポリメラーゼを含有) に加え、次いで増殖反応を行う。その反応終了後に、その反応液を低融点アガロース電気泳動により分画し、そのアガロースゲルから、配列長583bpの所期のDNA断片 (DNA断片-Cと称す) を含有するバンドを切出す。
【0067】
そのゲル切片を、DNA精製キット、例えばGenclean IIキット (フナコシ社製) により精製して、DNA断片-Cの精製品を得る。このDNA断片-Cは、第3の発明による改変D配列のDNA断片の部分領域に相当する塩基配列をもち、且つ制限酵素AflIIおよびBglIIの切断部位を内部に含有する構造をもつものである。
【0068】
また、このDNA断片-Cは、制限酵素AflIIおよびBglIIで切断することにより目的とする塩基配列の置換部位を内部に含み、5'-末端に制限酵素AflII切断部位を、3'-末端に制限酵素BglII切断部位をもつ、全長288bpのDNA断片-αを得ることができる。
【0069】
(4) 改変D配列を含有するDNA断片のクローニング
次に、上記の(3)項で得られたDNA断片-Cを利用して、所望の改変D配列を含有するDNA断片を収得する。
【0070】
そのために、先ず、上記のDNA断片-Cを制限酵素AflIIと制限酵素BglIIで処理する。これによって、DNA断片-Cから、5'-側にAflII切断部位を、3'-側にBglII切断部位を有する改変D配列の一部分を有するDNA断片を切り取る。このようにして、目的の改変D配列に相当するDNA配列を含有するDNA断片の試料(i) が得られる。
【0071】
次に、配列表の配列番号1に記載のDNA (すなわちOSASA-1配列) を含むプラスミドベクターpOSASA-1を制限酵素AflIIと制限酵素BglIIで処理することにより、5'-側にBglII切断部位を有し且つ3'-側にAflII切断部位を有して、しかも配列番号1に記載のDNAの1番目の塩基から933番目までの部分領域と1220番目の塩基から1734番目の塩基までの部分領域を含むプラスミド断片 (ii) が選られる。
【0072】
ここで得られたAflII-BglII-プラスミド断片 (ii) を前記の改変D配列に相当するDNA配列を含有するDNA断片の試料 (i) と混合し、ついでDNAリゲーション・キットで連結反応にかけると、配列番号12の改変D配列を含有する組換えプラスミド (以下では、pBluescript-DNA-Dプラスミドと称す) を作製できる。
【0073】
この組換えプラスミドすなわちpBluescript-DNA-Dプラスミドを大腸菌XL1-Blue MRF'に導入して形質転換し、こうして得られた形質転換大腸菌 (エシエリヒア・コリXLl-Blue MRF'/pBluescript-DNA-Dと称する) (寄託番号 FERM-6451)を液体培地中で培養して増殖させる。大量に増殖された大腸菌細胞は、前記の組換えプラスミド、すなわちpBluescript-DNA-Dプラスミドのコッピイを含有する。このようにして改変D配列はクローニングできる。その大腸菌培養細胞から、常法により改変D配列を含有するプラスミドを抽出して取る。
【0074】
(5) 改変D配列のDNA断片の回収
前項 (4) で得られた改変D配列を含有するプラスミドを、次いで制限酵素EcoRIで処理することによって消化する。これによってEcoRI切断部位に隣接する5'-側端に塩基配列ATGを有し且つ3'-側端にもEcoRI切断部位を有する延伸部を有する改変D配列を含有するDNA断片を含有する消化反応液を得ることができる。
【0075】
この消化反応液を、低融点アガロース電気泳動により分画して、前記のDNA断片を含むバンドをアガロースゲルから切出す。切出されたアガロースゲル片をTEバッファーに溶解し、得られた溶液をフェノールで抽出すると、フェノール抽出液に前記のDNA断片が回収される。前記のDNA断片を含むフェノール抽出液を、3M酢酸ナトリウム水溶液およびエタノールを混合し、その混合物を20℃で約6時間放置し、さらに低温で遠心分離すると、前記のDNA断片が沈殿する。これを減圧下に乾燥すると、目的の改変D配列を内部に含有するDNA断片が粉末として得られる。この改変D配列を含有するDNA断片の粉末は水に可溶性である。
【0076】
以上の説明においては、第3の発明によるDNAの具体例である改変D配列を含有するDNA断片は、遺伝子工学の技法を利用する方法で作製された。しかしながら、配列表の配列番号12に記載の塩基配列を参照しながら、従来知られているポリヌクレオチド化学合成法によっても製造できる。
【0077】
なお、第1の発明による配列番号1のDNAの塩基配列のうちの967番目のグアニンをアデニンに置換して改変する場合について、第3の発明の実施法を説明した。しかしながら、第1の発明による配列番号1に記載のDNAを、テンプレートとして用い且つ適当に工夫された塩基配列を有する合成オリゴヌクレオチドの数種類を組み合わせてプライマーとして用いるならば、第1の発明による配列番号1のDNAの967番目の場所とは別の場所における塩基を別の塩基で取代えて改変された別の改変DNAを作製することが可能である。
【0078】
更に、本発明者らは別段の研究を進めた。その結果、植物体に外来遺伝子を導入して植物体を形質転換され、得られたトランスジェニック植物体で外来遺伝子を発現させるための従来知られたバイオテクノロジー技法を利用することによって、第1の発明によるイネのASAの第1アイソザイムのαサブユニットをコードする新規DNA(配列番号1)も、第2の発明によるイネASAの第2アイソザイムのαサブユニットをコードする新規DNA(配列番号10)も、第3の発明によるところの、イネのASAの第1アイソザイムのαサブユニットをコードするDNAから作成された前記の新規な改変DNA(配列番号12)も、組換えベクターに組込んだ後に植物体に導入することができ、しかも植物体内で発現できることが知見された。
【0079】
従って、第4の発明(本件特許請求の範囲の請求項3の発明)においては、イネのアントラニル酸シンターゼの第2アイソザイムのαサブユニットをコードする配列表の配列番号10のDNAを担持する組換えベクターを導入されて形質転換された植物細胞を有し、そして導入された前記DNAが発現可能であることを特徴とする、形質転換植物が提供される。
【0080】
しかも、前記の第4の発明による形質転換された植物体がこれを裁培すると種子を結実できる植物である場合、その植物を通常の条件で裁培して植物の種子を収獲できることが知見された。
【0081】
従って、第5の発明(本件特許請求の範囲の請求項4の発明)においては、イネのアントラニル酸シンターゼの第2アイソザイムのαサブユニットをコードする配列表の配列番号10のDNAを担持する組換えベクターを植物細胞に導入することによって得られ且つ前記のDNAが発現可能である形質転換植物を裁培し、さらにその裁培により結実した植物から採取された種子であることを特徴とする、形質転換植物の種子が提供される。
【0082】
また、第6の発明(本件特許請求の範囲の請求項5の発明)によると、配列表の配列番号10に記載の塩基配列を有するDNA配列を担うDNA断片が組み込まれた組換えベクターであって、宿主細胞中で該DNA配列を発現することができる組換えベクターが提供される。
【0083】
さらに、第7の発明(本件特許請求の範囲の請求項6の発明)においては、配列表の配列番号10に記載の塩基配列を有するDNA配列を担うDNA断片が組み込まれた組換えベクターであって、宿主細胞中で該DNA配列を発現することができる組換えベクターで形質転換された大腸菌が新規微生物として提供される。
【0084】
第7の発明による形質転換大腸菌を増殖させると、その中に含有された組換えベクターを大量にクローニングできる。第7の発明による大腸菌の例には、ブタベスト条約の規約下に受託番号FERM BP-6454で寄託された前記の大腸菌XL1-Blue MRF'(OS-asa-2) がある。
【0085】
前記の改変D配列(配列番号12)を担うDNA断片を保有する組換えベクターを導入された形質転換大腸菌の例は、ブダペスト条約の規約下に受託番号FERM BP-6451で寄託されてある前記の大腸菌XL1-Blue MRF'/p Bluescript-DNA-Dであることができる。
【0086】
前記した第1の発明によるDNA(配列番号1)、あるいは第2の発明によるDNA(配列番号10)を外来遺伝子として用いて植物を形質転換する場合には、形質転換できる植物の種類は多種多様であり、また植物の形質転換のために外来遺伝子としての本発明DNAを植物に導入する方法は、その目的で行われる従来公知のバイオテクノロジーの種々な技法であることができる。
【0087】
前記した第1の発明のDNAまたは第2の発明のDNAを外来遺伝子としてイネ植物に導入するために好適に実施できる且つ後記の実施例3に例示される方法を以下に要約的に説明する。
【0088】
(a) 外来遺伝子の導入用の組換えベクターの作成
従来知られているトウモロコシのユビキチンプロモーター、1stイントロンおよびNOSターミネーターと、ホスフィノスリシン耐性遺伝子、さらに微生物にのみに効果の発現するアンピシリン耐性遺伝子とを含有する既知のプラスミドベクターpUBA(Plant Molecular Biology, 第18巻4号、675頁-689頁、1992年) をバッファー中で制限酵素BamHIおよびSacIで処理すると、ユビキチンプロモーターの下流で、1stイントロンの下流のBamHI切断部位で切断され、且つNOSターミネーターの上流のSacI切断部位で切断された約4.8Kbのサイズのベクター断片を得ることができる。
【0089】
そのベクターDNA断片の水溶液を、本発明のDNAを含有するDNA断片の水溶液と混合し、その混合物をDNAリゲーションキットで処理して連結反応を行う。これによると、ベクターDNA断片のユビキチンプロモーターとNOSターミネーター領域の間に本発明のDNAを含有するDNA断片が挿入、連結されてある組換えベクターを得ることができる。
【0090】
選られた組換えベクターを大腸菌JM109に導入して、形質転換大腸菌を得る。
【0091】
得られた形質転換大腸菌を、抗生物質アンピシリン含有培地に接種、培養することにより、アンピシリン耐性の大腸菌コロニーを数個を得る。これらのコロニーをさらにアンピシリン含有の培地中でそれぞれ別々に増殖させる。
【0092】
各コロニーごとの増殖されたアンピシリン耐性の大腸菌から、それぞれにプラスミドを回収する。回収されたプラスミドは、挿入DNAの向きが相異なる種々なプラスミドを含む。各コロニーごとに回収されたプラスミドを、適当な制限酵素で消化して切り出された種々のDNA断片を含む消化反応液をアガロースゲル電気泳動にかけて、それらDNA断片の大きさ及び塩基配列を解析すると、組換えプラスミドのユビキチンプロモーターの下流に本発明のDNAを正常な向きで挿入、連結されている適当なプラスミド(約6.5Kbのサイズ) を選抜できる。
【0093】
前記した第1の発明の配列表の配列番号1のDNAが組み込まれているプラスミドをベクターpUBdW1と称し、第2の発明の配列表の配列番号10のDNA(本件特許請求の範囲の請求項2のDNA)が組み込まれているプラスミドをベクターpUBdW2と称し、さらには、第3の発明の配列表の配列番号12の改変D配列のDNAが組み込まれているプラスミドをベクターpUBdDと称する。
【0094】
また、アグロバクテリウム法に遺伝子導入用組換えベクターとしてハイグロマイシン耐性遺伝子を含有する既知のプラスミドベクターpIG121-Hm(Plant Cell Phisiol, 第31巻805頁〜813頁、1990年) をバッファー中で制限酵素PmeIおよびSacIで処理すると、約9.8Kbのサイズのベクター断片を得ることができる。
【0095】
また、上記したプラスミドベクターpUBdW1, pUBdW2またはpUBdDをバッファー中でSphIおよびSacIで処理し、SphI切断部位を平滑化処理することによりユビキチンプロモーター、1stイントロンの下流に本発明のDNAが連結したベクター断片を得ることができる。
【0096】
これらの本発明DNAが連結したベクター断片の各々を用いると、上記の方法と同じ方法により、連結反応、形質転換大腸菌の作出、プラスミドの回収を行うことにより本発明のDNAが正常な向きで挿入連結されているアグロバクテリウム法による遺伝子導入用の組換えベクターを得ることができる。
【0097】
第1の発明の配列表の配列番号1のDNAが組み込まれているプラスミドを、ベクターpUb-OSASAW1と称し、第2の発明の配列表の配列番号10のDNAが組み込まれているプラスミドを、ベクターpUb-OSASAW2と称し、さらには、第3の発明の配列表の配列番号12の改変D配列の断片のDNAが組み込まれているプラスミドを、ベクターpUb-OSASA1Dと称する。
【0098】
(b) イネのカルスの調製
イネの完熟種子から籾殻を除き、得られた外皮付きコメ種子をエタノール溶液で殺菌、次いで次亜塩素酸ナトリウムの希水溶液で殺菌し、さらに滅菌水で洗浄する。
【0099】
MS培地にショ糖、植物ホルモンとしての2,4-PA、寒天を加えてなる組成のカルス形成用培地に、前の外皮付きコメ種子を置く。28℃で1日当り15〜18時間にわたり、1500〜2500ルックスの大陽光を照射しながら40〜50日間、培養すると、イネのカルスが形成される。そのカルスを種子の胚乳部分から切取りカルスを得る。
【0100】
(c) イネ・カルス細胞への外来遺伝子の導入
前項(a) に記載したように作製された、本発明DNAを正常に挿入、連結してある組換えベクター(すなわち、配列番号1のDNAを保有する前記のベクターpUb-OSASAW1、もしくは配列番号10のDNAを保有するベクターpUb-OSASAW2または配列番号12のDNAを保有するベクターpUb-OSASA1D) を、公知のアグロバクテリウム法によりイネのカルス細胞へ導入するために、まず宿主であるアグロバクテリウム・ツメファシエンス(Agrobacterium tumefaciens) への組み込みを行う。組み込みは公知のエレクトロポレーション法(植物組織培養、第10巻2号、194頁-196頁、1993年)により行う。
【0101】
前記(b) に示されたようにして得られたイネのカルス細胞と、このようにして本発明DNAを組み込んで得られたアグロバクテリウム細菌とを公知の方法(細胞工学別冊、モデル植物の実験プロトコール、93頁〜98頁、1996年、秀潤社発行) にしたがって、共存培養を行うと、イネカルス細胞への本発明のDNAを導入できる。そして、ハイグロマイシンに耐性で、本発明のDNAが形質転換されたイネ植物細胞が得られる。
【0102】
(d) 形質転換植物細胞の再選抜
上記のように得られたハイグロマイシン耐性の形質転換イネ植物細胞の中から、本発明DNAを外来遺伝子として十分に有効な量で含有する形質転換イネ植物細胞のみを再選抜する。
【0103】
このために、前者の形質転換細胞を、N6培地にショ糖と、2,4-PAと、ゲルライトと、トリプトファンのアナログ体である、5MT(すなわち5-メチルトリプトファン) とを添加してなる再選抜用培地上に移植する。
【0104】
移植された細胞を、1日当たり16時間、2000ルックスの光で照射しながら25〜28℃で25〜30日間培養する。
【0105】
本発明DNAを外来遺伝子として十分に有効な量で含有する形質転換イネ植物細胞は、培地中に細胞増殖阻害剤として作用する5MTが存在していても5MT耐性であって、生育できる。5MT添加の上記培地で生育できた5MT耐性の培養植物細胞を選抜する。
【0106】
(e) 再選抜された5MT耐性の形質転換イネ植物細胞からの植物体の再生
上記のように再選抜された5HT耐性の培養植物細胞を、植物組織培養用のMS培地にショ糖、植物ホルモンとしてのベンジルアデニン、ナフタレン酢酸、ゲルライトを添加してなる植物体再生用の分化培地に移植する。
【0107】
移植された細胞を、2000ルックスの光を1日当り16時間照射しながら25〜28℃で25〜30日間培養する。このようにすると、培養された形質転換イネ植物細胞から、分化により、芽と根が再生できる。
【0108】
再生した芽と根を含有する幼芽が長さ10〜30mmに生育した後に、その幼芽を、MS培地にショ糖とゲルライトを添加してなる順化用の培地に移植する。さらに、1日当り16時間、2000ルックスの光を照射しながら25〜28℃で18〜20日間育成する。
【0109】
このようにして、形質転換イネ植物が再生できる。こうして得られた植物体は、温室内で土壌に移植して通常の条件下で裁培すると、順長に生育して3〜6ケ月の裁培でイネ種子を結実できる。
【0110】
(f) 導入した外来遺伝子の確認
上記のように再生された形質転換イネ植物から緑色の葉を採取する。採取された葉を液体窒素で凍結した後に破砕する。その葉破砕物から、J.Sambrookらの方法(Molecular Cloning、第2版、Cold Spring Habor Laboratory Press、1989年に記載) に準じてDNAを抽出する。
【0111】
他方、後記の配列表の配列番号14に記載される塩基配列を有するオリゴヌクレオチドと、配列番号15に記載の塩基配列を有するオリゴヌクレオチドを化学合成して、プライマー用に用いる。
【0112】
再生イネ植物体から上記のように抽出されたDNAをテンプレートとして用い且つ上記の2種の合成オリゴヌクレオチドをプライマーとして用いて、公知のPCR法によって前記DNAを増幅する。得られた増幅反応液を常法によりアガロース電気泳動にかけて分画し、再生イネ植物から抽出されたDNAの種々のDNA分画のうちで、導入された外来遺伝子のDNAに相当するDNA断片を含むゲルバンドを採る。
【0113】
このバンドに含有されたDNA断片の塩基配列を公知のサザン法によって解析すると、本発明DNAに相当するか否かを確認できる。
【0114】
植物中のトリプトファンの抽出および含有量の測定は、公知の方法(Hopkins-Cole法、生化学実験講座第11巻、東京化学同人社刊) によって行うことができるが、後記の実施例に挙げたようなHPLCを用いる方法によっても行うことができる。このトリプトファンの抽出および含有量の測定は、対象とする植物種、対象とする植物の部位、対象とする植物の生育ステージに併せて、適宜変更することができる。
【0115】
次に、本発明のDNAの導入により形質転換された植物細胞の選抜方法および形質転換植物の作成方法を説明する。
【0116】
(a) 選抜用の組換えベクターの作成
形質転換植物の作出のために、植物細胞への直接的導入法に用いられる組換えベクターとして、従来知られているカリフラワーモザイクウイルスの35Sプロモーターと、NOSターミネーターと、微生物にのみに効果の発現するアンピシリン耐性遺伝子とを含有する既知のプラスミドベクターpBI221(Clontech社製) を、バッファー中で制限酵素XbaIおよびSacIで処理すると、35Sプロモーターの下流のXbaI切断部位で切断され、且つNOSターミネーターの上流のSacI切断部位で切断された約3.8Kbのサイズのベクター断片を得ることができる。
【0117】
そのベクターDNA断片の水溶液を、第3の発明のDNA(改変D配列) を含有するDNA断片の水溶液と混合し、その混合物をDNAリゲーションキットで処理して連結反応を行う。これによると、ベクターDNA断片の35SプロモーターとNOSターミネーター領域の間に第3の発明のDNA(改変D配列) を含有するDNA断片が挿入、連結されてある組換えベクターを得ることができる。
【0118】
選られた組換えベクターを大腸菌JM109に導入して、形質転換大腸菌を得る。
【0119】
得られた形質転換大腸菌を、抗生物質アンピシリン含有培地に接種、培養することにより、アンピシリン耐性の大腸菌コロニーを数個を得る。これらのコロニーをさらにアンピシリン含有の培地中でそれぞれ別々に増殖させる。
【0120】
各コロニーごとの増殖されたアンピシリン耐性の大腸菌から、それぞれにプラスミドを回収する。回収されたプラスミドは、挿入DNAの向きが相異なる種々なプラスミドを含む。各コロニーごとに回収されたプラスミドを、適当な制限酵素で消化して切り出された種々のDNAを含む消化反応液をアガロースゲル電気泳動にかけて、それらDNA断片の大きさ及び塩基配列を解析すると、組換えプラスミドの35Sプロモーターの下流に第3の発明のDNA(改変D配列) を正常な向きで挿入、連結されている適当なプラスミド(約5.6Kbのサイズ) を選抜できる。また、形質転換植物の作出のために、植物細胞への直接的導入法に用いられる組換えベクターとして前記のベクターpUBdW1あるいはpUBdW2も、またはアグロバクテリウム法に用いられる組換えベクターとして前記のベクターpUb-OSASA1Dも選抜用の組換えベクターとして用いることができる。
【0121】
(b) イネのカルスの調製
イネの完熟種子から籾殻を除き、得られた外皮付きコメ種子をエタノール溶液で殺菌、次いで次亜塩素酸ナトリウムの希水溶液で殺菌し、さらに滅菌水で洗浄する。
【0122】
MS培地にショ糖、植物ホルモンとしての2,4-PA、寒天を加えてなる組成のカルス形成用培地に、前の外皮付きコメ種子を置く。28℃で1日当り15〜18時間にわたり、1500〜2500ルックスの大陽光を照射しながら40〜50日間、培養すると、イネのカルスが形成される。そのカルスを種子の胚乳部分から切取りカルスを得る。
【0123】
(c) イネ・カルス細胞への選抜用組換えベクターの導入
前項(a) に記載したように作製された本発明DNAを正常に挿入、連結してある選抜用組換えベクターのpUBdW1あるいはpUBdW2を公知のウイスカーを用いる直接的導入法によりカルス細胞へ導入を行う。また、選抜用組換えベクターのpUb-OSASA1Dを公知のアグロバクテリウム法(細胞工学別冊、モデル植物の実験プロトコール、93頁〜98頁、1996年、秀潤社発行) によりカルス細胞へ導入を行う。
【0124】
(d) 形質転換イネ植物細胞の選抜
上記のように得られた選抜用組換えベクターpUBdW1またはpUBdW2を含有するカルス細胞を、N6培地にショ糖、2,4-PA、ゲルライトおよび選抜薬剤としてトリプトファン類縁化合物を10mg/L〜200mg/L〜200mg/L、好ましくは30mg/L〜50mg/Lを添加してなる選抜用培地の上に均一に広げて加える。さらに、暗所もしくは2000ルックスの光を1日当たり16時間照射しながら25〜28℃で20〜60日間、好ましくは25〜30日間培養する。
【0125】
こうしてトリプトファン類縁化合物に耐性であり且つ選抜用組換えベクターで形質転換された植物細胞が選抜される。
【0126】
(e) 形質転換植物の選抜
上記のように得られたトリプトファン類縁化合物耐性の形質転換イネ植物細胞から、形質転換イネ植物を得るために、前者の形質転換細胞を、MS培地にショ糖、植物ホルモンとしてベンジルアデニン、ナフタレン酢酸、ゲルライトおよび選抜薬剤としてトリプトファン類縁化合物を10mg/L〜200mg/L、好ましくは30mg/L〜50mg/L添加してなる植物体再生用の選抜分化培地に移植する。
【0127】
移植された細胞を、2000ルックスの光を1日当り16時間照射しながら25〜28℃で25〜30日間培養する。このようにすると、培養された形質転換植物細胞から、分化により、芽と根が再生できる。
【0128】
再生した芽と根を含有する幼芽が長さ10〜30mmに生育した後に、その幼芽を、MS培地にショ糖とゲルライトを添加してなる順化用の培地に移植する。さらに、1日当り16時間、2000ルックスの光を照射しながら25〜28℃で18〜20日間育成する。このようにして、形質転換イネ植物が再生できる。
【0129】
本発明におけるDNAは、外来遺伝子として前記したイネ植物のみならず、他の種類の植物に導入して植物の形質転換を行うことに使用できる。
【0130】
また、本発明のDNAすなわち、前記の第1の発明のDNA(配列番号1のDNA)あるいは第2の発明のDNA(配列番号10のDNA)は、前記したイネ植物のトリプトファン含有量の増加および選抜のみならず、他の種類の植物に対しても使用できる。
【0131】
従って、本発明のDNAを一般の植物体に導入できる方法、またトリプトファン含有量を増加させる方法、さらに形質転換細胞および形質転換植物を選抜する方法を次に一般的に説明する。
【0132】
本発明のDNAを導入できる植物は、特に限定される種類のものではなく、例えば、イネ、トウモロコシ、コムギ、オオムギ、およびその他の単子葉植物、ならびにタバコ、ダイズ、ワタ、トマト、ハクサイ、キュウリ、レタスなどの双子葉植物などが挙げられる。先づ、これらの植物から培養細胞を調製し、得られた培養細胞に本発明DNAを外来遺伝子として導入するのが都合よい。
【0133】
本発明のDNA導入に用いられる培養細胞の作製は、植物由来のいかなる外殖物であってもよく、例えば、胚盤、生長点、花粉、葯、葉身、茎、葉柄、根由来の外殖物を使用できる。
【0134】
上記した外殖片を、カルス形成用の培地、例えばMS培地(Murashigeら、「Physiologia Plantarum」、1962年、第15巻、473頁〜497頁)、またはR2培地(Ojimaら、「Plant and Cell Physiology」、1973年、第14巻、1113〜1121頁、あるいはN6培地(Chuら、1978年、「In Proc. Symp. Plant Tissue Culture, Science Press Peking」、43頁〜50頁などの如く、必須成分としての無機塩成分およびビタミン類を含む植物組織培養用の培地に、植物ホルモンとして、例えば、2,4-PA(2,4-dichlorophenoxyacetic acid) 0.1〜5mg/リットル、ならびに、炭素源として、例えば、ショ糖10〜60g/リットル、ゲルライト1〜5g/リットルを添加してなる培地に置床して培養することにより得られる培養細胞を用いて、これに本発明DNAを導入するのが便利である。
【0135】
本発明のDNAの導入の対象となる植物細胞は、例えば、カルスや懸濁細胞などの脱分化した培養細胞あるいは不定胚、苗条原基などの培養細胞、もしくは植物組織例えば葉、根、茎、胚、生長点などの細胞から作られたカルス細胞および懸濁細胞であるのが好ましい。
【0136】
本発明のDNAの導入用の培養細胞を調製するために、カルス形成用培地に外殖片を置床する場合、本発明DNAの導入用の培養細胞が得られるまでの培養期間は特に限定されるものではない。しかし、形質転換植物を再生することが必要であるから、当該の培養細胞からの植物体再生が可能であること、つまり、当該植物細胞が植物体再生能力を保有している期間内に得られた培養細胞を用いることが重要である。
【0137】
また、本発明のDNAの導入用の培養細胞は、植物体再生能力を保有している培養細胞であれば、液体培地中で培養された懸濁細胞であってもよい。
【0138】
本発明のDNAを植物細胞に導入するためには、先づ、本発明のDNAを発現ベクターに組込んだ組換えベクターを調製することが必要である。その組換えベクターは、植物体に導入後の本発明DNAが植物体中で発現するように、発現プロモーターの下流に本発明DNAを配し、さらに本発明のDNAの下流にターミネーターを配するように調整された組換えベクターであることを要する。この目的に用いられる組換えベクターは、植物への導入方法の種類に応じて、通常の植物形質転換に利用される各種のベクターであることができる。例えば、エレクトロポレーション法、パーティクルガン法、ウイスカー法などによる植物細胞への直接的なDNA導入法を用いる場合は、pUC系またはpBR322系などの大腸菌で増殖可能であるプラスミドベクターを利用するのがよく、またアグロバクテリウム法を用いる場合はplan系などのプラスミドベクターを利用するのがよい。
【0139】
組換えベクター中で本発明DNAの上流に配されるプロモーターは、例えばカリフラワーモザイクウイルス由来のCaMV35S「The EMBO Journal」、第6巻、第3901頁〜3907頁、1987年、または、特開平6-315381号公報」、トウモロコシのユビキチンプロモーター[特開平2-79983号公報]、ファゼオリンプロモーター「Plant Cell、第1巻、第839頁〜853頁、1989年] であることができる。また、本発明DNAの下流に配されるターミネーターは、例えばカリフラワーモザイクウイルス由来のターミネーター、あるいはノパリン合成酵素遺伝子由来のターミネーター[The EMBO J.第6巻、第3901頁〜3907頁、1987年]であるのがよい。しかし、植物体中で機能するプロモーターやターミネーターであれば、何れでもよい。
【0140】
また、本発明DNAの導入で形質転換された植物細胞を効率的に選択するためには、用いる上記の組換えベクターは、適当な選抜マーカー遺伝子を含有するプラスミドベクターと共に植物細胞に導入されることが好ましい。この目的に使用する選抜マーカー遺伝子は、抗生物質ハイグロマイシンに耐性であるハイグロマイシンフォスフォトランスフェラーゼ遺伝子、あるいはカナマイシン、ゲンタマイシンに耐性であるネオマイシンフォスフォトランスフェラーゼ遺伝子、もしくは除草剤フォスフィノスリシンに耐性であるアセチルトランスフェラーゼ遺伝子[The EMBO Journal、第6巻、第2513頁〜2518頁、1987年、または特開平2-171188号公報] であることができる。
【0141】
本発明DNAを外来遺伝子として植物に導入する方法は、アグロバクテリウム法[Bio/technology、第6巻、第915頁〜922頁、1988年]、エレクトロポレーション法[Plant cell Rep., 第10巻、第106頁〜110頁、1991年]、パーティクルガン法[Theor. Appl. Genet., 第79巻、第337頁〜341頁、1990年]、ウイスカー法であることができ、しかし特にこれらに限定されない。
【0142】
次に、本発明DNAを含有する組換えベクターを十分に有効な量で含有する形質転換細胞を効率よく再選抜するために、細胞増殖阻害剤であるトリプトファンのアナログ体を添加した培地上で培養する。
【0143】
トリプトファンアナログとしては、例えば5-メチルトリプトファン(5MT) を、10mg/l〜1000mg/l、好ましくは20mg/l〜100mg/lの濃度で再選抜用培地に添加できる。
【0144】
前記のように再選抜された本発明DNAを外来遺伝子として組込まれた組換えベクターと選抜マーカー用のベクターとを含有する形質転換植物細胞から、次に、植物体を再生する。植物体の再生は公知の要領で行うことができる。例えば、前記のように再選抜された形質転換植物細胞を、公知の植物体再生用培地に置床して培養することにより植物体の再生を行うことができる。
【0145】
植物体再生用の培地に置床された形質転換細胞は15〜30℃、好ましくは、20〜28℃の温度で500〜2000ルックス、好ましくは、800〜1000ルックスの光を照射しながら培養期間20〜60日間、好ましくは30〜40日間で培養する。
【0146】
これによって、個々の植物細胞から本発明DNAよりなる外来遺伝子を担う組換えベクターが導入されて形質転換された植物体が再生できる。
【0147】
形質転換細胞から再生された植物体は次に順化用培地で培養する。その後、順化された再生植物体を、温室内で通常の裁培条件下で育成すると、3〜6カ月の裁培で植物体は成熟し結実して種子を得ることができる。
【0148】
なお、このように再生され且つ裁培された形質転換植物体中の導入された外来遺伝子の存在は、公知のPCR法とサザン法(Southern, 「J. Mol. Biol.,」 98巻503-517頁、1975年) によって、植物体中のDNAの塩基配列を解析することによって確認できる。
【0149】
この場合、形質転換植物体からのDNAの抽出は、公知のJ. Sambrookらの方法(「Molecular Cloning」第2版、Cold Spring Habor Laboratory Press, 1989年) に準じて実施できる。
【0150】
再生された植物体中に存在する本発明DNAよりなる外来遺伝子をPCR法を用いて解析する場合には、上記のように再生植物体から抽出されたDNAを、テンプレートとして用い本発明によるDNA、あるいは本発明による改変DNAの塩基配列に従って適当に選択された塩基配列をもつ合成したオリゴヌクレオチドをプライマーとして用い、これらを混合してPCR法用反応液中に加えて増幅反応を行う。この増幅反応においては、DNAの変性、アニーリング、伸張反応を数十回繰り返えすと、本発明DNA配列を含むDNA断片の増幅生成物を得ることができる。
【0151】
増幅生成物を含む反応液を例えばアガロース電気泳動にかけると、増幅された各種のDNA断片が分画される。導入した外来遺伝子である本発明DNAに相当するDNA配列を含有すると認められるDNA断片を含むバンドアガロースゲル片を切出す。切出されたゲル片内に含有されるDNA断片のDNA配列の塩基配列をサザン法で解析すると、そのDNA配列が本発明DNAに対応するか確認できる。
【0152】
本発明の形質転換細胞の選抜および形質転換植物の選抜に用いることのできる選抜用薬剤としてトリプトファン類縁化合物が挙げられるが、具体的には、5-メチルトリプトファン(5MT)、4-メチルトリプトファン(4MT)、6-メチルトリプトファン(6MT)、7-メチルトリプトファン(7MT)、6-メチルアントラニル酸(6MA)、5-メチルアントラニル酸(5MA)、3-メチルアントラニル酸(3MA)、5-フルオロアントラニル酸(5FA)、6-フルオロアントラニル酸(6FA) などのトリプトファン類縁化合物の生合成中間体も使用できる。
【0153】
前述したように、第1の発明または第2の発明による新規なDNAは、外来遺伝子として、植物または植物細胞に導入してトリプトファン含量の増強された植物または植物細胞の形質転換に利用できる。
【0154】
従って、第8の発明(本件特許請求の範囲の請求項7の発明)では、アントラニル酸シンターゼの第1または第2アイソザイムのαサブユニットのタンパク質をコードして且つ配列表の配列番号1または配列番号10の塩基配列を有するDNAを担持する組換えベクターであって、しかも該DNAが植物中で発現可能である組換えベクターを、植物細胞カルスに導入し、このことによって、該DNAで形質転換された植物細胞カルスを収得し、さらに該カルスを植物体に再生することからなる、植物のトリプトファン含有量の増加方法が提供される。
【0155】
なお更に、本発明者らは、イネのASAの第1アイソザイムのαサブユニットをコードする遺伝子の発現のために有用なプロモーターDNAを単離する目的で別途の研究を行った。その研究の結果、下記に説明される手順によって、イネのASAの第1アイソザイムのαサブユニットの遺伝子DNA、もしくはその改変DNAを発現するのに有効なプロモーターDNA配列を内部に含有するDNA断片を収得することに成功した。該DNA断片を収得する操作手順を以下に簡略に説明する(手順の詳細は後記の実施例5を参照)。
【0156】
(a) イネのゲノミックDNAの調製
イネ(Oriza sativa)の茎葉、根、カルスなどの組織、好ましくは茎葉またはカルスから、常法によりゲノミックDNAを抽出する。抽出したゲノミックDNAから蛋白質などの夾雑物を除き、さらに超遠心分離によりゲノミックDNAの精製品を得る。
【0157】
(b) イネのゲノミックDNA断片の作成
上記で得られたゲノミックDNA (精製品) を制限酵素 EcoRIで部分消化する。得られたDNAの部分消化物をアガロースゲルにて電気泳動する。電気泳動により分画されたDNA断片をナイロン膜ハイボンドNに転写する。転写されたDNAは変性処理により、ナイロン膜に固定する。
【0158】
このようにナイロン膜に固定されたDNAの各分画に対して、後記の実施例1の(5) でアラビドプシスから作られたDIGラベルされた標識プロープDNAをハイブリダイゼーション反応させる。ナイロン膜上のDNAのサイズ6kb付近に、シグナルを発するDNA分画が検定できる。このシグナルを発したDNA分画に相当するアガロースゲル中のDNA分画を、上記のアガロース中で制限酵素RcoRIで部分消化してから、該ゲルから切出す。切出されたDNAを精製し、得られたDNA断片 (精製品) をTEバッファーに溶解すると、こうして、分画ゲノミックDNAが得られる。
【0159】
(c) イネの分画ゲノミックDNAライブラリーの構築
得られたゲノミックDNAをファージベクターに連結し、得られた組換えベクターをλファージにパッケージする。こうして得られた組換えλファージを大腸菌に感染させて増殖させると、前記の組換えλファージの多量が得られる。この多量の組換えλファージをイネの分画ゲノミックDNAライブラリーとして利用する。
【0160】
(d) イネのゲノミックDNAライブラリーからのプロモーター遺伝子の選択
上記で構築されたイネのゲノミックDNAライブラリーである組換えファージを用い、また実施例1の(5)でアラビドプシスから作られたDIGラベルした標識プローブDNAを利用することによりプラーク・ハイブリダイゼーション法によって、イネのASA遺伝子のためのプロモーターの遺伝子に相当するDNA配列を含有する組換えファージを、前記ゲノミックDNAライブラリーからスクリーニングできる。このスクリーニングにより、該ライブラリーの10万個のファージプラークから、ASA遺伝子のプロモーター遺伝子が組込れたと判定できたファージプラーク3個を単離することに幸にも成功した。
【0161】
これら3個のプラークを別々に制限酵素RcoRIで消化して、DNAのEcoRI-DNA断片を含む消化液を得る。
【0162】
(e) ゲノミックDNAのクローニング
上記で得られたEcoRI-DNA断片を、プラスミドベクターp Bluecsript II SK(+)のEcoRI切断部位に連結する。得られた組換えプラスミドベクターを大腸菌に導入して、クローニングする。大腸菌から組換えプラスミド・ベクターのクローンを分離する。
【0163】
(f) クローニングされたプラスミドDNAのシークエンス解析
上記のように得た組換えプラスミド・ベクターのクローンを制限酵素RcoRIと制限酵素BamHIで消化する。こうして得たEccoRI-BamHI断片を塩基配列の解析にかける。
【0164】
上記のようにして得られたゲノミックDNAのクローンから、イネのASAの第1アイソザイムのαサブユニットの遺伝子の発現のためのプロモーター領域のDNA配列を内部に含有するDNA断片(DNA断片Zとここで仮称する) を、DNA断片の塩基配列を参考にして、分離できた。
【0165】
このようにして得られたプロモーターDNAを内部に含有するDNA断片Zの全体の塩基配列を、常用のシークエンシング・キットにより決定し、その決定された全体の塩基配列は、配列表の配列番号3に記載の塩基配列であると認められた。このDNA断片の内部に含有されたプロモーターDNAは、アビドプシスの第1アイソザイムのαサブユニットの遺伝子のプロモーター領域のDNAについて既知である塩基配列を参考にして、配列表の配列番号7に記載された塩基配列を有すると認められた。
【0166】
このDNA断片Zの塩基配列の全部またはそのうちの一部は、後記の実施例5の試験からプロモーター活性を有することが確認されている。
【0167】
配列表の配列番号7に記載の塩基配列をもつプロモーターDNAを内部に含有するDNA断片ZをpBluescript II SK(+) プラスミドベクターにクローニングされ、こうして得られた組換えベクターを導入した大腸菌XLl-Blue MRF'は、Escherichia coli(Os-asa#7) と命名され、工業技術院生命工学工業技術研究所に1997年8月18日より、受託番号FERM P-16387として寄託されている。また、1998年8月7日以降はブダペスト条約の規約下にFERM BP-6452の受託番号で寄託されている。
【0168】
(g) プロモーター活性の検定
前記の得られたイネのゲノミックDNAクローンから分離された前記のDNA断片Zに含有されるところの、ASAの第1アイソザイムのαサブユニットの遺伝子のプロモーター領域について、そのプロモーターとしての機能を確認するために、その活性の検定試験を行った。すなわち、前記のDNA断片Zを、レポーター遺伝子であるβ-グルクロニダーゼ(GUS) 遺伝子を担持する市販のpBI101プラスミドベクター(Clontech社製) の制限酵素切断部位に挿入することによって、組換えプラスミドベクターを作成した。この組換えプラスミドベクターを常法によって植物細胞、例えばイネ培養細胞へ導入して、市販のGUS活性測定キットにより、該プロモーター領域がGUS活性の発現に有効であることを確認することができる。
【0169】
第1の発明によるイネASAのアイソザイムのαサブユニットの遺伝子のDNA、ならびにそのプロモーター配列は、本発明を完成するに際しては、上記のようにしてcDNAライブラリー又はゲノミックDNAライブラリーから単離されたものである。本発明により、それらプロモーターのDNAの塩基配列が明らかにされたので、後記の配列表に示す塩基配列に基づいて化学合成することによっても取得できる。また、前記の塩基配列に基づいて作製した合成オリゴヌクレオチドプライマーを用い、公知の方法でイネのcDNAライブラリー又は染色体DNAライブラリーから、PCRにより取得することもできる。
【発明を実施するための最良の形態】
【0170】
以下に、本発明を実施例を参照して具体的に説明するが、本発明はこれら実施例に限定されるものではない。
【0171】
なお、以下の実施例で行われる実験操作の手順は、特に記述しない限り、「Molecular Cloning」 第2版 (J. Sambrookら、Cold Spring Habor Laboratory press, 1989年発行)に記載される方法に従った。
【0172】
実施例1
本実施例はイネのアントラニル酸シンターゼ(ASA) の第1のアイソザイム(ASA1) のαサブユニットをコードする遺伝子と、第2のアイソザイム(ASA2) のαサブユニットをコードする遺伝子(すなわち、本件特許請求の範囲の請求項1または2のDNA)とを単離する方法を例示する。
【0173】
(1) イネのmRNAの調製
イネ(品種: 日本晴) の種子を播種した。その後、栽培7日目の幼植物体の茎葉2gを液体窒素で凍結した。凍結した茎葉を乳鉢内で粉砕した。その粉砕物から、公知のAGPC法(Acid Guanidinium thiocyanate-phenol-chloroformを使用する方法)(実験医学、Vol. 9, No. 15 (11月号)、第99頁〜第102頁、1991年) に従い、全RNAの約2mgを抽出した。次に、上記で得られた全RNAから、Pharmacia Biotech社製のmRNA精製キット(mRNA Purification Kit) を用いてmRNAを単離した。こうしてイネの全mRNAの約30μgを得た。
【0174】
(2) イネのcDNAライブラリーの構築
上記(1) で得られたイネの全mRNAから、DNA合成キット(Pharmacia Biotech社製のTimeSaver cDNA Synthesis Kit) によりイネの全cDNAを得た。
【0175】
この全cDNAを、EcoRI切断末端が仔ウシ小腸由来アルカリホスファターゼで処理されたλgt11ファージ・ベクター(Lambda gtll/EcoRI/CIAP-Treated Vector Kit (STRATGENE社製)) であるファージベクターに連結した。こうして得られた組換えベクターを、イン・ビトロパッケージングキット(Gigapack II Gold Packaging Extract) によりラムダファージにパッケイジングした。
【0176】
組換えベクターをパッケージした組換えラムダファージを大腸菌Y1088に感染させて増殖させた。増殖された大腸菌の溶菌プラークとして、多数の前記組換えラムダファージが得られた。該プラーク中の組換えラムダファージは、イネ由来の全cDNAを含有する多種多様のファージから成るものであり、これら多種多様のファージをイネのcDNAライブラリーとして利用した。
【0177】
(3) PCR法用のプライマーの構築
イネのアントラニル酸シンターゼ(ASA) の第1のアイソザイム(ASA1) と第2のアイソザイム(ASA2) のそれぞれのαサブユニットをコードするcDNA断片をクローニングするのに用いるDNAプローブをPCR法用に作製する。このために、まずプライマーの設計を行った。そのためには、まずアラビドプシス(和名: シロイヌナズナ) のアントラニル酸シンターゼの第1のアイソザイムと第2のアイソザイムとのそれぞれのαサブユニットをコードする遺伝子の既知の塩基配列および既知のアミノ酸配列を参考にして、下記に示す塩基配列を有する2種類のオリゴヌクレオチドを、プライマーNo.1とプライマーNo.2として化学合成により作製した。
【0178】
プライマーNo.1(配列表の配列番号8) に示す塩基配列を有するオリゴヌクレオチド):
5'-CATATGTCTTCCTCTATGAAC-3'
プライマーNo.2(配列表の配列番号9) に示す塩基配列を有するオリゴヌクレオチド):
5'-GGATCCTCATTTTTTCACAAATGC-3'
なお、上記の2種類のオリゴヌクレオチドの作製は、DNA合成装置(Model 391、アプライドバイオシステムズ社製) を用いてオリゴヌクレオチドを合成し、さらにその合成品をイオン交換HPLCで精製することにより行った。
【0179】
(4) プローブDNAの作製
次に、上記のように構築された2種類の合成オリゴヌクレオチドの各10p.Mを第1のプライマーおよび第2のプライマーとして用い、またアラビドプシスの市販されるcDNAライブラリー(STRATAGENE社製) の1μlをテンプレイトとして用いた。第1および第2プライマーならびにアラビドプシスのcDNAライブラリーを、PCR法用の増幅反応液(10mMのTris-HCl(pH 8.3)、1.5mMのMgCl2、50mMのKCl、0.001%ゼラチン、pH 8.3; 4種のヌクレオチドdNTPの各2.5mMの混合物、およびDNAポリメラーゼTakara Ex Taq、2.5単位) 50μlに加えて、DNAの増幅反応を行った。ここで使用される増幅反応液は、PCRキット(PCR Amplification Kit(宝酒造(株)社製) により調製した。
【0180】
PCR法によるDNAの上記の増幅反応は、PCR反応装置(PERKIN ELMER社製、DNA、Thermal Cycler 480) を用いて、変性を94℃、30秒間、アニーリングを55℃、1分間、また伸張(extention) を72℃、2分間行う3つの反応操作を35回繰り返すことによって実施した。
【0181】
上記のようにすると、アラビドプシスASAの2つのアイソザイムのそれぞれのαサブユニットをコードする遺伝子に相当するDNA配列のうちの一部分を構成しているDNA断片の増幅生成物が生成される。これらDNA断片の増幅生成物をプローブDNAとして採取する。そして、該プローブDNAをイネのcDNAライブラリーのクローニングに下記の通りに用いた。
【0182】
(5) イネのcDNAライブラリーから、イネのASAの2つのアイソザイム(ASA1およびASA2) のαサブユニットをコードする遺伝子のDNAの選択
上記(2)で構築されたイネのcDNAライブラリーである組換えラムダファージから、上記(4)でアラビドプシスのDNAライブラリーから作製したプローブDNAの利用により、イネのASAの2つのアイソザイムのαサブユニットをコードする遺伝子に対応するDNA配列を組込まれた組換えラムダファージをプラーク・ハイブリダイゼーション法により、スクリーニングにより採取する。
【0183】
このスクリーニングの目的のためには、上記(2)で得られたイネcDNAライブラリーである組換えラムダファージのプラークを、複数のシャーレ内の1.5%寒天培地上に形成させた。それらファージプラークを複数枚のナイロン膜(アマシャム社製) ハイボンドNに転写した。こうしてナイロン膜に転写されたファージのプラークに含まれているファージDNAは、アルカリ変性液(1.5M NaCl、2.0M NaOH) および中和液(1.0M Tris-HCl pH5、2.0M NaCl) でそれぞれ10分間処理し、その後にUV照射によりナイロン膜上に固定した。
【0184】
次に、上記(4)でアラビドプシスのcDNAライブラリーから得られたプローブDNAをジゴキシゲニン(DIG)でラベルして標識プローブDNAを得た。その標識プローブを前記のファージDNA(すなわち、イネのcDNAライブラリー) を固定したナイロン膜に対してプラーク・ハイブリダイゼーションさせた。該プローブDNAの前記ラベリングはDIG-ELISA DNA Labeling & Detection Kit(ベーリンガー・マンハイム社製) により行った。
【0185】
このプラーク・ハイブリダイゼーションの場合、前記のファージDNAが固定されたナイロン膜をハイブリダイゼーション溶液(500mM Na-Piバッファー、pH7.2、7%SDS、1mM EDTA) に浸し、さらに65℃、10分間、浸漬処理を行った。次に前記のDIGラベルした標識プローブDNA(10ng/ml)を加え、65℃で、15時間インキュベートすると前記のプラーク・ハイブリダイゼーション反応が行われた。
【0186】
その反応終了後に、洗浄液(40mM Na-Piバッファー、pH7.2、1%SDS) で前記のナイロン膜を20分間ずつ3回洗浄した。その後、上記のDIG-ELISA Labeling & Detection Kitを用いて、イネのASAのαサブユニットをコードするDNAを含有した目的の組換えファージの検出を行った。ハイブリダイズしてX線フィルムで強くシグナルを発している8個の組換えファージプラーク、すなわちイネのASAをコードする遺伝子が組み込まれていると思われる8個のファージプラークを30万個のファージプラークから、ナイロン膜上で探知することができた。そして、イネのASA遺伝子が組み込まれている前記の8個の組換えファージプラークを単離して選抜できた。
【0187】
これら採取された8個のプラークの組換えファージから、λDNA単離キット(Lambda DNA Purification Kit(STRATAGENE社製)) により、それぞれにファージプラークの8個ごとに別々に、λDNAを単離した。
【0188】
この際の上記のλDNAの単離は次のようにして行った。すなわち、8個のプラークの各々のファージをそれぞれ別々に大量に増殖させたそれぞれの培養液5mlに、DNaseI(20mg/ml) 50μl、RNaseA(2mg/ml) 200μlを加え、室温で15分間、放置した。得られたファージ増殖溶液を15000rpm、4℃で10分間遠心分離した。得られた上清に80%DEAE-セルロース 25ml加え、室温で10分間インキュベートした。このようにインキュベートされた混合液を遠心分離し、得られた上清に0.5M EDTA 2ml、Pronase(50mg/ml) 770μlを加え、37℃で15分間放置した。さらに、5%CTAB溶液(1%CTAB (Cetyltrimethyl- ammonium bromide)、50mM Tris-HCl、pH8.0、10mM EDTA) 1.5mlを加えた。得られた混合物を65℃、3分間処理した後、氷中で5分間放置した。こうして得られたそれぞれの反応液に1/10容の3M酢酸ナトリウム、2倍容のエタノールを加え、−20℃で約6時間放置し、その後、得られた溶液を遠心分離し、それぞれの溶液から沈澱したファージDNAを別々に乾燥させ、5mlの水にそれぞれ溶解して保存した。
【0189】
上記の操作により単離された8種のファージDNAが得られた。これらDNAの各々5μlを、Hバッファー中で制限酵素EcoRIの10ユニットで別々に消化して、得られた消化反応液を別々に分析した。
【0190】
こうして、イネのASAの2つのアイソザイムのαサブユニットをコードする遺伝子に相当するDNA配列を含有すると判定されたところの、ファージDNA中の挿入DNA断片は、上記のように選抜された採取ファージのファージDNAから該ファージDNAを制限酵素EcoRIで上記のように消化して切出すことによって収得できた。
【0191】
(6) イネのASAの2つのアイソザイム(ASA1およびASA2) のそれぞれのαサブユニットをコードする遺伝子に相当するDNA配列を含有するcDNAのクローニング
上記のように、イネのASAの2つのアイソザイムのαサブユニットをコードする遺伝子に相当するDNA配列を含有すると判定されたDNA断片として、前記の8種の各々の組換えファージから切出して収得されたDNA断片は、8種である。このようなDNA断片の各1種づつをプラスミドベクター p Bluescript II SK(+) のEcoRI切断部位にDNAリゲーション・キットにより挿入、連結することにより、組換えプラスミドベクターを構築した。このように構築された組換えプラスミドベクターの導入で大腸菌XLl-Blue MRF' を形質転換した。
【0192】
上記のように形質転換の目的で、構築された組換えプラスミドベクターを大腸菌XL1-Blue MRF' に導入する操作は、詳しくは次のように行った。すなわち、上記で得られた採取ファージの組換えファージDNAの10μgを、先づHバッファー中で、制限酵素EcoRIの10ユニットで消化して消化反応液を得た。また他方、プラスミドベクターp Bluescript II SK(+)の10μgを同様にEcoR Iで消化して消化反応液を得た。上記のようにして得られたそれぞれのDNA消化反応液に対して別々に1/10容の3M酢酸ナトリウム、2倍容のエタノールを加え、−20℃で約6時間放置し、その後に、それぞれの得られたDNA溶液を遠心分離し、それぞれの溶液から沈殿したDNAを乾燥させ、5μlの水に各々溶解した。こうして得られたファージ由来のDNAの水溶液と、プラスミド由来のDNA水溶液の各々5μlを混合し、得られた混合物をDNA Ligation kit(宝酒造(株)製) で処理して、それら2種のDNAを連結させた。得られたDNA連結反応液に10分の1容量の3M酢酸ナトリウム、2倍容のエタノールを加え、−20℃に約6時間放置した。得られた混合液を遠心分離し、沈殿したDNAを乾燥させた。こうして連結ベクターDNAが採取された。この連結ベクターDNAを10μlの水に溶解した。
【0193】
こうして得られた連結ベクターDNAの水溶液10μl (DNA 10ng)と、市販の大腸菌XL1-Blue MRF'コンピテントセル(STRATAGENE社製) 100μlとを、1.5ml容チューブに入れ、氷中で30分間、42℃で30秒間、そして再び氷中で2分間インキュベートした。その後に、インキュベートされた混合物に対してSOC液体培地(2% バクト-トリプトン、0.5%バクト-酵母抽出物、10mM NaCl、2.5mM KCl、10mM MgSO4、10mM MgCl2、20mM Glucoseを含有) 900μlを加えてから、37℃で1時間大腸菌を振とう培養した。
【0194】
得られた大腸菌の培養液の100μlを、アンピシリンを50mg/lの濃度、X-gal(5-Bromo-4-Chloro-3-Indolyl-b-D-Galactoside)を20mg/lの濃度、IPTG
(Isopropyl-b-D-thiogalactopyranoside)を20mg/lの濃度で添加されてあるLB寒天培地(1%バクト−トリプトン、0.5%バクト−酵母抽出物、0.5% NaCl、0.1% Glucose、pH 7.5、寒天1.5%含有) にプレーティングした。37℃で16時間、大腸菌を培養後、白色に発色してない大腸菌コロニーから、白色に発色している大腸菌コロニーを、前記の組換えファージのDNA断片を連結された組換えプラスミドベクターDNAで形質転換されてある大腸菌として、分離した。
【0195】
このようにして分離された白色を発色している且つアンピシリン耐性である形質転換大腸菌のコロニー10個を、アンピシリン50mg/lを含む液体培地で増殖させ、さらに増殖された大腸菌から、プラスミド精製キット(QIA filter Plasmid Midi Kit, QIAGEN社製) によりプラスミドDNAを分離し且つ精製した。この精製により、上記で得られたアンピシリン耐性コロニーの形質転換大腸菌から50μg(50μl) の組換えプラスミドのDNAを得た。
【0196】
上記で単離された各々の組換えファージ(計8種) から、クローニングを上記のように行うと、組換えプラスミドのDNA8種が得られた。これら8種のDNAの塩基配列を解析するために次の操作を行った。
【0197】
(7) クローニングされたDNAのシークエンス解析
上記のクローニングされた8種のDNAである組換えプラスミドDNAをそれぞれに市販の塩基配列決定キットで処理すると、それらのDNA断片の全体の塩基配列を決定できた。そして、それらのDNA断片の内部に含有されたイネのASA1のαサブユニットをコードする遺伝子に該当するDNAの塩基配列、ならびにイネのASA2のαサブユニットをコードする遺伝子に該当するDNAの塩基配列を判定できた。
【0198】
なお、上記のDNAの塩基配列の決定を行うに当っては、塩基配列決定キット(Autoread Sequencing Kit (Pharmacia Biotech社製) を用いて、変性処理を行った後に自動DNAシーケンサALF DNA Sequencer II(ファルマシア社製)で前記DNA断片の塩基配列を決定した。
【0199】
このようにして、上記のDNA断片の塩基配列を判定した結果、イネのASAのαサブユニットをコードするDNA配列は2種類の相異なる塩基配列を有するものであることが解った。従って、イネのASAは2つのアイソザイム(イソ酵素) から成ること、またその第1のアイソザイム(ASA1) のαサブユニットをコードするDNAは、その第2のアイソザイム(ASA2)のαサブユニットをコードするDNAと相異なる塩基配列を有することが知見された。
【0200】
そして、イネのASA1のαサブユニットをコードする遺伝子に相当するDNAは、上記のように判定されて、後記の配列表の配列番号1に記載されてある塩基配列を有すると今回、認められた。
【0201】
また、イネのASA2のαサブユニットをコードする遺伝子に相当するDNAは、上記のように判定されて、後記の配列表の配列番号10に記載されてある塩基配列を有すると今回、認められた。
【0202】
第1の発明のイネのASA1のαサブユニットをコードする遺伝子のDNAの塩基配列は、配列表の配列番号1に記載の単一のオープンリーディングフレーム内にある1734個の塩基からなる。また、配列表の配列番号1に示されたイネのASA1のαサブユニットをコードする遺伝子のDNAの塩基配列は、配列番号2に記載の577個のアミノ酸残基からなるタンパクをコードしている。この配列番号2のアミノ酸配列をアラビドプシスのASA1のαサブユニットのタンパク質の既知のアミノ酸配列(The Plant Cell, 第4巻, 721頁〜733頁, 1992年) と比較すると、68%の相同性が認められるが、他の領域ではアラビドプシスのASA1のαサブユニットとは明らかに異なり、イネ特有の配列である。
【0203】
第2の発明のイネのASA2のαサブユニットをコードする遺伝子のDNAの塩基配列は、配列表の配列番号10に記載の単一のオープンリーディングフレーム内にある1821個の塩基からなる。また、配列番号10に示されたイネのASA2のαサブユニットをコードする遺伝子のDNAの塩基配列は、配列番号11に記載の606個のアミノ酸残基からなるタンパクをコードしている。この配列番号11のアミノ酸配列をアラビドプシスのASA2のαサブユニットのタンパク質の既知のアミノ酸配列(The Plant Cell, 第4巻, 721頁〜733頁, 1992年) と比較すると、72%の相同性が認められるが、他の領域ではアラビドプシスのASA2のαサブユニットとは明らかに異なり、イネ特有の配列である。
【0204】
(8) イネのASAαサブユニット遺伝子を導入された大腸菌形質転換株の寄託
前項(6)の操作により、最終的に組換えプラスミドDNAの8種が得られた。これら8種の組換えプラスミドDNA断片の各々のDNA断片全体の塩基配列が前項(7)で塩基配列決定キットにより決定された。
【0205】
(i) 次に、配列表の配列番号1に記載された1734個の塩基からなるDNA配列(DNA配列OSASA-W1と命名される)(すなわち、イネのASA1のαサブユニットをコードするDNA) を内部に含有すると判定された組換えプラスミドDNA断片を、前記の8種の組換えプラスミドDNA断片のうちから選択した。この選択された組換えプラスミドDNA断片を、制限酵素EcoRIで消化した。その消化反応液から、DNA配列OSASA-W1を内部に含有すると判定されたDNA断片(DNA配列OSASA-1と命名する) を低融点アガロース電気泳動法により分画して採取した。このように採取されたDNA配列OSASA-1を、プラスミドベクターp Bluescript II Sk(+)のEcoRI切断部位にDNAリゲーション・キット(宝酒造(株)製) により連結して、組換えプラスミドベクターpOSASA-1を構築した。
【0206】
このように構築したプラスミドベクターpOSASA-1を、前項(6)に記載された方法により、大腸菌XLl-Blue MRF'に導入した。プラスミドベクターpOSASA-1の導入により形質転換された大腸菌株をエシエリヒア・コリ(Escherichia coli) XLl-Blue MRF'(Os-asa1) と命名した。エシエリヒア・コリXLl-Blue MRF'(Os-asa1) は、日本の茨城県、つくば市の工業技術院生命工学工業技術研究所に1997年8月18日に、FERM P-16388の受託番号で寄託されている。さらに、1998年8月7日以降、ブタペスト条約の規約下でFERM BP-6453の受託番号で前記の研究所に寄託されてある。
【0207】
(ii) また、配列表の配列番号10に記載された1821個の塩基からなるDNA配列(DNA配列OSASA-W2と命名される)(すなわち、イネのASA2のαサブユニットをコードするDNA) を内部に含有すると判定された組換えプラスミドDNA断片を、前記の8種の組換えプラスミドDNA断片のうちから選択した。この選択された組換えプラスミドDNA断片を、制限酵素EcoRIで消化した。その消化反応液から、DNA配列OSASA-2を内部に含有すると判定されたDNA断片(DNA配列OSASA-2と命名する) を低融点アガロース電気泳動法により採取した。このように採取されたDNA配列OSASA-W2を、プラスミドベクターp Bluescript II Sk(+)のEcoRI切断部位にDNAリゲーション・キット(宝酒造(株)製) により連結して、組換えプラスミドベクターpOSASA-2を構築した。
【0208】
このように構築したプラスミドベクターpOSASA-2を、前項(6)に記載された方法により、大腸菌XLl-Blue MRF'に導入した。プラスミドベクターpOSASA-2の導入により形質転換された大腸菌株をエシエリヒア・コリ(Escherichia coli) XLl-Blue MRF'(Os-asa2)と命名した。エシエリヒア・コリXLl-Blue MRF'(Os-asa2) は、日本の茨城県、つくば市の工業技術院生命工学工業技術研究所に1998年6月18日に、FERM P-16853の受託番号で寄託されている。さらに、1998年8月7日以降、ブタペスト条約の規約下でFERM BP-6454の受託番号で前記の研究所に寄託されてある。
【0209】
実施例2
本例は、第3の発明(本件特許請求の範囲から除外されてあるが、特願2004-257204号の請求項1の発明)により得られた改変D配列と命名されたDNA配列(すなわち、配列表の配列番号12に記載された1734個の塩基からなる塩基配列を有するDNA配列) を含有するDNA断片の製造を例示する。
【0210】
(1) PCR用のプライマーとしての合成オリゴヌクレオチドの構築
DNA合成装置(Model-391、アプライドバイオシステムズ社製) を利用して、4種のプライマーを合成した。すなわち、配列表の配列番号16に記載の塩基配列を有するオリゴヌクレオチドであるプライマーOSASN1と、配列番号17に記載の塩基配列を有するプライマーOSASN2と、配列番号18に記載の塩基配列を有するプライマーOSASC1と、配列番号19に記載の塩基配列を有するプライマーOSASC2とを化学合成により構築した。
【0211】
(2) PCR法用のテンプレートの作製
実施例1の(8)項において、イネのASA1のαサブユニットの遺伝子に相当するDNA配列すなわち前記のDNA配列OSASA-W1を含有すると判定されるDNA断片として、組換えファージから制限酵素EcoRIで切出して収得されたDNA断片OSASA-1は、プラスミドベクターp Bluescript II SK(+)のEcoRI切断部位にDNAリゲーション・キットにより連結されて、組換えプラスミドベクターpOSASA-1と命名)を構築した。このプラスミドベクターpOSASA-1が下記のPCR法でテンプレートとして利用された。
【0212】
(3) 所要なDNA断片のPCR法による増幅と回収
(i) PCR法の第1回段階
所要なDNA断片をPCR法による増幅反応により収得するために、PCR法の第1回段階として下記の(A)反応と(B)反応とを行った。
【0213】
すなわち、その(A)反応では、テンプレートとして用いられる前記の組換えプラスミドベクターpOSASA-1の5μlと、前記のプライマーOSASAN1の1μMと、プライマーOSASAC1の1μMとを、増幅反応液(10mMのTris-HCl(pH8.3)、1mMのMgCl2、50mMのKCl、4種のヌクレオチドdNTPの各0.2mMの混合物、LA Taq DNAポリメラーゼの2.5ユニットを含有)の100μlに加えて、増幅反応を行った。この(A)反応で得られたDNAの増副生成物を、DNA断片-Aと称する。
【0214】
また、(B)反応では、テンプレートとして用いられる前記プラスミドベクターpOSASA-1の5μlと、プライマーOSASAC2の1μMと、プライマーOSASAN2の1μMとを、(A)反応で用いたと同じ組成の増幅反応液に加えて、増幅反応を行った。この(B)反応で得られたDNAの増幅生成物をDNA断片-Bと称する。
【0215】
なお、前記の増幅反応は、PCR反応装置(Promram Temp Control System PC-700(アステック社製))内で、変性を94℃で30秒間、アニーリングを55℃で30秒間、伸張を72℃で1分間行う3つの反応操作を20回繰り返すことにより実施された。
【0216】
増幅反応の終了後、(A)反応の増幅反応液を低融点アガロース電気泳動により分画し、増幅DNA生成物としての268bpのDNA断片-Aを含むバンドをアガロースゲルから切出した。また、(B)反応の増幅反応液を低融点アガロース電気泳動にかけて分画し、増幅DNA生成物としての336bpのDNA断片-Bを含むバンドをアガロースゲルから切出した。
【0217】
これら2つのゲル切片から、それぞれに、GENECLEAN II Kit(フナコシ社製) により分離して、DNA断片-Aの精製品とDNA断片-Bの精製品を得た。
【0218】
(ii) PCR法の第2回段階
次に、PCR法の第2回段階として、前記の(A)反応および(B)反応で増幅したDNA断片-Aの精製品の1μlとDNA断片-Bの精製品の1μlを、増幅反応液(10mMのTris-HCl(pH8.3)、1mMのMgCl2、50mMのKCl, dNTPの各0.2mMの混合物およびLA Taq DNA ポリメラーゼの2.5ユニットを含有) 100μlに加えて、増幅反応を行った。この場合の増幅反応は、変性を94℃、5分間、アニーリングを65℃、10分と55℃、10分行い、また、伸張を72℃、1分行う3つの反応操作を1回実施した。
【0219】
この増幅反応の終了後、その増幅反応液を低融点アガロース電気泳動により分画し、増幅DNA生成物として583bpの所期のDNA断片-Cを含有するバンドをゲルから切出した。このゲル切片からGENECLEAN II Kit(フナコシ社製) によりDNA断片-Cを分離して且つDNA断片-Cの精製品を得た。
【0220】
(4) 上記で得られたDNA断片-Cを利用して、目的の改変D配列を含有するDNA断片をクローニングにより十分な量で収得する操作を次に行う。
【0221】
(i) 先づ、上記で得られたDNA断片-Cの10μgを、Hバッファー(宝酒造(株)社製) 中で制限酵素Afl IIの10ユニットとBgl IIの10ユニットで消化した。この消化で得られたDNA断片をDNA断片-α称する。他方、前記のプラスミドベクターpOSASA-1の10μgをHバッファー(宝酒造(株)社製) 中で、Afl IIの10ユニットとBgl IIの10ユニットで消化して短縮プラスミドを得た。DNA断片-αを含む消化反応液と、前記の短縮プラスミドを含む消化反応液とに、それぞれに別々に1/10容の3M酢酸ナトリウム、2倍容のエタノールを加え、−20℃に約6時間インキュベートした。その後、インキュベートされた溶液を遠心分離し、それぞれの溶液から沈殿したDNAを乾燥させ、5μlの水に溶解した。
【0222】
こうして得られたDNA断片-αの水溶液の5μlと、該短縮プラスミドDNAの水溶液の5μlとを混合した。次に、得られた混合物(10μl) に含まれる各々のDNAを、DNA Ligation kit(宝酒造(株)社製) により連結反応させた。その連結反応液に1/10容の3M酢酸ナトリウム、2倍容のエタノールを加え、−20℃に約6時間インキュベートした。インキュベートされた反応溶液を遠心分離し、沈殿したDNA連結物を乾燥させ、5μlの水に溶解して水溶液にした。
【0223】
この水溶液に含まれるDNA連結物は、前記のDNA断片-αと、プラスミドベクターpOSASA-1のAfl II及びBgl II切断で得られた前記の短縮プラスミドとが互いに連結して作られた環状2本鎖の組換えプラスミド(前記のp Bluescript-DNA-Dプラスミドである) であり、そのプラスミドDNA中の挿入DNA領域内に、前記の改変D配列が含有された。
【0224】
(ii) このp Bluescript-DNA-Dプラスミドを大腸菌XL1-Blue MRF′に導入することにより大腸菌XL1-Blue MRF′を形質転換した。
【0225】
このように形質転換された大腸菌をエシエリヒア・コリXLl-Blue MRF'/p Bluescript-DNA-Dと命名し、前記の生命工学工業技術研究所に1998年8月7日以下、ブダペスト条約の規約下にFERM BP-6451の受託番号で寄託した。この形質転換された大腸菌細胞を次に液体培地中で培養した。
【0226】
培養された大腸菌細胞から、さらにプラスミドを抽出した。こうして改変D配列を含有する組換えプラスミドp Bluescript-DNA-Dがクローニングできた。
【0227】
(5) 改変D配列を含有するDNA断片の回収
得られたプラスミドp Bluescript-DNA-DであるプラスミドDNAの10μlを、Hバッファー(宝酒造(株)社製)中でEcoRIの10ユニットで消化した。その消化反応液を低融点アガロース電気泳動により分画した。DNA配列OSASA-W1配列(1734 bpのサイズ) を内部に含有するDNA断片(DNA断片-βと称する) をアガロースから切出した。
【0228】
このDNA断片-βを含有するアガロース切片にTEバッファー(10mMのTris-HCl、1mMのEDTAを含有、pH8)を等量加え、68℃、20分間加温することによりアガロースを溶解した。得られた溶液を飽和フェノールで2回抽出することによりアガロースを除いた。得られたDNAを含むフェノール抽出液に対して、それの10分の1容の3M酢酸ナトリウム、2倍容のエタノールを加え、−20℃に約6時間放置しインキュベートした。インキュベートされた溶液を、15000rpm、4℃で10分間遠心分離した。得られたDNA沈殿を減圧下で乾燥させ、得られたDNA粉末を10μlの水に溶解させた。
【0229】
そのDNA粉末は、目的の改変D配列を含有するDNA断片-βからなるものであった。
【0230】
なお、実施例2の上記(5)で得られた前記のDNA断片-βを、実施例1の(7)項に記載されたと同様にして、塩基配列決定キットを用いる自動DNAシーケンサ、ALF DNA sequencer IIにかけた。DNA断片-βは配列表の配列番号12に記載される1734個の塩基からなる塩基配列をもつ改変D配列を内部に含有するDNA断片であると確認できた。
【0231】
実施例3
本例は、配列表の配列番号1に示すDNA配列を外来遺伝子として、または配列番号12に示す塩基配列をもつ改変DNA配列を外来遺伝子として、イネ植物にアグロバクテリウム法で導入することからなるイネ植物の形質転換方法を例示する。また、本例で形質転換された植物のトリプトファン含量が増加されたことが例証された。
【0232】
(1) イネ植物への外来遺伝子の導入用の組換えベクターpUBdDの作成
(i) トウモロコシのユビキチンプロモーターと、1stイントロンと、NOSターミネーターと、アンピシリン耐性遺伝子とを含有する5.6kbのサイズの既知のプラスミドベクターpUBAのDNA(10μg) を、Kバッファー(宝酒造(株)製) 中で制限酵素BamHIの10ユニットで消化した。消化反応液からDNAを沈殿させ、遠心分離してから乾燥させた。このDNA乾燥物を10μlの滅菌水で溶解した後、DNAブランティング・キット(宝酒造社製)で平滑化した後、末端平滑化されたDNAを沈殿させ、遠心分離してから乾燥させた。この乾燥物を10μlの滅菌水で溶解した後、Lバッファー(宝酒造(株)製) 中で制限酵素SacIの10ユニットで消化した。消化反応液からDNAを沈殿させ、遠心分離してから乾燥させた。このようにして、ユビキチンプロモーターと、1stイントロンと、NOSターミネーターとアンピシリン耐性遺伝子を保有する約4.8kbのサイズのベクター断片を得た(添付図面の図1の左、参照)。
【0233】
(ii) さらに別に、第3の発明のDNAである改変されたD配列を内部に含有する組換えプラスミドpBluescript-DNA-D(1.7kbのサイズ) のDNA(10μg) を、Hバッファー(宝酒造(株)製) 中で制限酵素EcoRVの10ユニットで消化し、その消化反応液からDNAを沈殿させ、遠心分離してから乾燥させた。このDNA乾燥物を10μlの滅菌水で溶解した後、Lバッファー(宝酒造社製) 中で制限酵素SacIの10ユニットで消化した。消化反応液から上記と同様にしてDNAを乾燥させてpBluescript-DNA-Dベクター断片を得た(添付図面の図1の右、参照)。
【0234】
(iii) 上記の(i)で得た約4.8kbのサイズのベクター断片と、(ii)で得られたベクター断片を5μlの滅菌水に溶解した。そのベクター断片の水溶液の5μlをそれぞれ混合した。得られた混合物をDNAリゲーション・キット(宝酒造(株)製) で処理して、DNA連結反応を行った。こうしてプラスミドベクターpUBAの切断ベクター断片に対して、前記のpBluescript-DNA-Dの切断ベクター断片を連結してなる環状の組換えベクター(6.5kbのサイズ) を作成した。この組換えベクターをpUBdDと称する(図1の下、参照)。ベクターpUBdDは、ユビキチンプロモーター領域の下流に1stイントロン領域を有し、1stイントロン領域とNOSターミネーター領域との間に、第3の発明の改変D配列が挿入、連結されてあり、且つアンピシリン耐性遺伝子を含有する構造を有し、6.5kbのサイズを有した。
【0235】
(2) イネ植物への外来遺伝子の導入用の組換えベクターpUBdD-W1の作成
(i) トウモロコシのユビキチンプロモーターと、1stイントロンと、NOSターミネーターと、アンピシリン耐性遺伝子とを含有する5.6kbのサイズの既知のプラスミドベクターpUBAのDNA(10μg) から、前記の(i)および(ii)と同様にして、ユビキチンプロモーターと、1stイントロンと、NOSターミネーターとアンピシリン耐性遺伝子とを保有する約4.8kbのサイズのベクター断片を得た(図1の左、参照)。
【0236】
(ii) さらに別に、第1の発明による配列番号1のDNA配列を内部に含有する組換えプラスミドpOSASA-W1(4.7kbのサイズ) のDNA(10μg) を、Hバッファー(宝酒造(株)製) 中で制限酵素EcoRVの10ユニットで消化し、その消化反応液からDNAを沈殿させ、遠心分離してから乾燥させた。この乾燥物を10μlの滅菌水で溶解した後、Lバッファー(宝酒造社製) 中で制限酵素SacIの10ユニットで消化した。消化反応液から上記と同様にしてDNAを乾燥させた。
【0237】
(iii) 上記の(i)で得た約4.8kbのサイズのベクター断片と上記の(ii)で得られたベクター断片を5μlの滅菌水に溶解した。そのベクター断片の水溶液の5μlをそれぞれ混合した。得られた混合物をDNAリゲーション・キット(宝酒造(株)製) で処理して、DNA連結反応を行った。こうしてpUBAの切断ベクター断片に対して、前記のpOSASA-W1の切断ベクター断片を連結してなる環状の組換えベクターを作成した。この組換えベクターをpUBdD-W1と称する。ベクターpUBdD-W1は、ユビキチンプロモーター領域の下流に1stイントロン領域を有し、1stイントロン領域とNOSターミネーター領域との間に、配列OSASA-W1が挿入、連結されてあり、且つアンピシリン耐性遺伝子を含有する構造を有する。
【0238】
(3) イネ植物への外来遺伝子の導入用の組換えベクターpUb-OSASA1Dまたは組換えベクターpUb-OSASAW1の作成
(i) 上記の(2)(iii)で得られたプラスミドベクターpUBdD-W1のDNA(10μg) を、Hバッファー(宝酒造(株)製) 中で制限酵素SphIの10ユニットで消化し、その消化反応液からDNAを沈殿させ、遠心分離してから乾燥させた。得られたベクター断片を10μlの滅菌水に溶解した(添付図面の第2図の右、参照)。そのベクター断片の水溶液の10μlをDNAブランティング・キット(宝酒造(株)製) で処理して、制限酵素切断部位の平滑化反応を行った。その反応液からDNAを沈殿させ、遠心分離してから乾燥させた後、10μlの滅菌水に溶解した。このベクター断片のDNA(10μg) をLバッファー(宝酒造(株)製) 中で制限酵素SacIの10ユニットで消化した後、消化反応液からDNAを沈殿させ、遠心分離してから乾燥させた。このようにして、ユビキチンプロモーターおよび第3の発明による配列番号12の改変D配列を保有するベクター断片を得た(添付図面の図2の右の第3番目の図、参照)。
【0239】
(ii) ハイグロマイシン耐性遺伝子を含有する15kbサイズの公知のプラスミドベクターplG121-HmのDNA(10μg) を、Lバッファー(宝酒造(株)製) 中で制限酵素SacIの10ユニットで消化し、その消化反応液からDNAを沈殿させ、遠心分離してから乾燥させた。この乾燥物を10μlの滅菌水で溶解した後、NEB4バッファー(ニューイングランド・バイオラブ社製) 中で制限酵素PmeIの10ユニットで消化した。消化反応液から上記の(1)(i)と同様にして、DNAを乾燥させた。このようにしてハイグロマイシン耐性遺伝子を保有する約9.8kbのサイズのベクター断片を得た(図2の左、参照)。
【0240】
(iii) 上記の(ii)で得られた約9.8kbのサイズのベクター断片と、上記の(i)で得られた改変D配列含有ベクター断片をそれぞれ5μlの滅菌水に溶解した。そのベクター断片の水溶液の5μlを、混合した後、DNAリゲーション・キット(宝酒造(株)製) で処理して、DNA連結反応を行った。このようにしてプラスミドベクターpl121-HmのPmeI-SacI切断ベクター断片に対して、前記の改変D配列断片を連結してなる環状の組換えベクターを作成した。この組換えベクターをpUb-OSASA1Dと称する(図2の下、参照)。ベクターpUb-OSASA1Dは、ユビキチンプロモーター領域の下流に1stイントロン領域を有し、1stイントロン領域とNOSターミネーター領域との間に、改変D配列が挿入、連結されてあり、植物中で発現するハイグロマイシン耐性遺伝子、且つカナマイシン耐性遺伝子を含有する構造を有し、11.5kbのサイズを有した。
【0241】
さらに別に、上記の改変D配列断片の代わりに、第1の発明による配列番号1の塩基配列を有するOSASAW1断片を前記と同様に上記の(ii)で得られた約9.8kbのサイズのベクター断片に対して連結してなる環状の組換えベクターを作成した。この組換えベクターをpUb-OSASAW1と称する。
【0242】
(4) アグロバクテリウム細菌の調製
LB培地(バクトトリプトン10g/l、バクトイーストエクストラクト5g/l、NaCl 5μl pH7) の液体培地300mlに、アグロバクテリウム(Agrobacterium tumefaciens LBA4404 (Clontech社から購入)) を移植し、30℃で16時間振とう培養した後、培溶液を4℃で10分間冷却した後、遠心分離により細菌の沈殿を得た。細菌の沈殿物を氷冷した10%グリセロールで洗浄操作と遠心分離を3回繰り返した後、沈殿物を10mlの10グリセロールに溶解した。
【0243】
そのうちの40μlをエッペンドルフチューブに移し、このチューブに上記した組換えベクターpUb-OSASA1D(配列番号12の改変DNAを保有)あるいはベクターpUb-OSASAW1(第1の発明による配列番号1のDNAを保有)の溶解液(40ng) 5μlをそれぞれ添加し、よく混合した。これらを氷上に3分間置いた後、エレクトロポレーション用のキュベット(電極間0.2cm) に移し、エレクトロポレーション装置(ジーン・パルサー(バイオラッド社製)) で、電圧12.5kV/cm、コンデンサー容量25μF、抵抗値600Ωの条件でエレクトロポレーションを行った。キュベット内にSOC培地(GIBCO BRL社製) を0.8ml添加し、けんだく液を2ml容の試験管に移し、30℃で1時間振とう培養を行った。この培養液をLB 培地にカナマイシン50mg/lを添加してなる寒天培地に塗布し、30℃で36時間培養して、生育してきたコロニーを組換えベクターが導入された形質転換アグロバクテリウムとして得た。
【0244】
組換えベクターpUb-OSASA1Dで形質転換されているアグロバクテリウム細菌をpUb-OSASA1D/LBA4404と称する。ベクターpUb-OSASAW1で形質転換されているアグロバクテリウム細菌をpUb-OSASAW1/LBA4404と称する。
【0245】
(5) イネのカルスの調製
イネ(品種: 日本晴)の完熟種子から籾穀を脱穀した。得られた外皮付きの種子を70%エタノール溶液に60秒間、ついで有効塩素約1%の次亜塩素酸ナトリウム溶液に6分間浸漬して種子を殺菌処理した。さらに種子を滅菌水で洗浄した。
【0246】
MS培地の無機成分組成にショ糖30g/l、植物ホルモンとして2,4-PA 2mg/l、寒天8g/lを添加してなる培地に、上記で殺菌したイネ種子を置床した。28℃で21日間、2000ルックスの光を1日当たり16時間照明しながらインキュベートした。カルスが形成された。それらカルスを胚乳部分から切り出し、そのカルスを上記と同じ組成の培地に移植して、3日間培養を行った。
【0247】
(6) イネのカルスへの外来遺伝子の導入操作
上記のようにして得られた、遺伝子導入用の形質転換アグロバクテリウム(pUb-OSASA1D/LBA4404、あるいはpUb-OSASAW1/LBA4404) をAA培地の無機塩組成にショ糖20g/l、2,4-PA 2mg/l、カイネチン0.2mg/lおよびアセトシリンゴン10mg/lを添加してなる液体培地30mlにけんだくし菌液を得た。このけんだく液を9cmのシャーレに入れ、上記(5)で得られたイネのカルスを100個入れた後、5分間浸せきした。浸せき後、ペーパータオルで余分な菌液を除去した後、N6培地の無機塩組成にショ糖30g/l、グルコース10g/l、2,4-PA 2mg/l、ゲルライト2g/lおよびアセトシリンゴン10mg/lを添加してなる固体培地に、上記のしんせきしたカルスを各シャーレ当たり20個ずつ置床した。28℃で3日間、暗黒下で培養し、イネのカルスへのアグロバクテリウムの感染を行った。
【0248】
(7) 導入ベクターで形質転換されたカルスの選抜
上記(6) で組換えベクターが導入された形質転換カルスを、500mg/lカルベニシリンを添加してなる滅菌水で洗浄してアグロバクテリウム細菌を除去した。カルスから余分な水分を除いた後、カルスをN6培地の無機塩組成にショ糖30g/l、2,4-PA 2mg/l、カルベニシリン500mg/l、ハイグロマイシン50mg/l、ゲルライト2mg/lを添加してなる固体培地上に各シャーレ当たり20個ずつ移植した。25℃で21日間、2000ルックスの光を1日当たり16時間照明しながら培養して、ハイグロマイシン耐性の形質転換カルスを得た。
【0249】
(8) ベクターpUb-OSASA1Dによる形質転換カルスの再選抜
上記により得たハイグロマイシン耐性である形質転換カルスのうち、ベクターpUb-OSASA1Dに含有されて植物中で発現される機能を有する改変D配列が外来遺伝子として十分導入されて形質転換されたカルスのみを再選抜する。そのためには、形質転換カルス15個(直径5mm)を、N6培地の無機塩組成にショ糖30g/l、2,4-PA 2mg/l、カルベニシリン250mg/l、ゲルライト2g/lと細胞増殖阻害剤として働く5-メチルトリプトファン(以下、5MTという) (トリプトファンのアナログ体) の3×10−4Mを添加してなる固体培地上に移植した。25℃で21日間、2000ルックスの光を1日当たり16時間照明しながら培養した。前記の5MT添加培地で生育できた植物中で発現される機能を有する改変D配列含有の形質転換カルスをこのようにして再選抜した。
【0250】
(9) 形質転換カルス細胞からの植物体の再生
上記により得たハイグロマイシン耐性および5MT耐性である形質転換カルス培養の14〜15個を、MS培地の無機塩組成にショ糖の30g/l、ソルビトール30g/l、カサミノ酸の2g/l、NAA 1mg/l、BA 2mg/l、ハイグロマイシン50mg/l、ゲルライト4g/lを添加してなる固体培地に移植した。25℃で30日間、2000ルックスの光を1日当たり16時間照射しながら培養すると、形質転換カルスから芽と根が再生できた。再生した幼芽(長さ10〜30mmに生育した芽)を、ショ糖の30g/l、ゲルライトの2g/lを含むMS培地を入れた試験管(直径45mm、長さ25cm)に移植した。移植された幼芽を20日間培養して形質転換したイネ植物を得た。
【0251】
また、上記の方法により、第1の発明のDNAである配列表の配列番号1のDNAを保有する前記のOSASAW1配列を外来遺伝子として含有する形質転換カルス15個から、10個のイネ植物が再生できた。
【0252】
また、第3の発明による配列番号12の改変D配列を外来遺伝子として含有する5MT耐性の形質転換カルス14個から10個のイネ植物が再生できた。
【0253】
(10) 形質転換イネ植物の再生体の遺伝子解析
上記のように再生されたイネ植物体に存在するASAをコードするDNAの解析をPCR法で次の手順により行った。
【0254】
(i) 前項(9)で得た再生イネ植物から葉を採取した。その葉50mgを1.5ml容のマイクロチューブに入れ、10mM EDTAを含む20mM Tris-HCl緩衝液(pH 7.5) 300μlを添加し、これを磨砕した。磨砕物に20%SDSを20ml加えて、65℃で10分間加温した。得られた混合物に5M酢酸カリウムを100μl加え、氷中に20分間置いた後、1,7000xgの遠心加速度で20分間遠心分離した。得られた上清にイソプロパノール200μlを加え、転倒攪拌し、これを再び1,7000xgの遠心加速度で20分間遠心分離した。得られたDNAの沈殿を減圧下で乾燥した。100μlのTE緩衝液にそのDNAを溶解した。
【0255】
(ii) PCR用のプライマーとして、配列表の配列番号14に示す塩基配列を有する前記オリゴヌクレオチドと、配列番号15に示す塩基配列を有する前記オリゴヌクレオチドとを用いた。
【0256】
次に、これら2種類のオリゴヌクレオチドの各1μMをプライマーとして用い且つ上記再生イネ植物体由来のDNAの5μlをテンプレイトとして用い、これらを増幅用反応液(10mM Tris-HCl(pH8.3)、1.0mM MgCl2、50mM KCl、0.01%ゼラチン、pH 8.3、dNTPの各0.2mMの混合物およびTaq DNAポリメタラーゼ2.5ユニットを含有) 100μlに加えた。そして増幅反応を行った。用いた増幅反応液は、PCRキット(PCR Amplification Kit(宝酒造(株)製)で調製した。
【0257】
増幅反応は、PCR反応装置(Program Temp Control System PC-700(アズテック社製)) 内で、変性94℃、1分、アニーリング60℃、30秒、伸張(extention) 72℃、1分の3つの反応操作を30回繰り返すことによって実施した。
【0258】
(iii) 得られたPCR反応液を、常法によりアガロース電気泳動にかけて分析したところ、再生イネ植物から抽出したDNAから増幅された各種のDNAのバンドが確認された。
【0259】
さらに、それらの各種のDNA配列を、塩基配列決定キットにより解析したところ、第1の発明のよるDNA配列 (配列番号1) を保有するOSASA-W1配列または第3の発明による改変D配列(配列番号12) に相当するDNA断片が再生イネ植物から上記のように抽出された各種のDNA断片の中に存在することが確認できた。
【0260】
上記のようにPCR法による遺伝子解析を行うことにより、導入した外来遺伝子が確認できた再生イネ植物体は、各々を培養土を入れたポットに移植して栽培した。これらの再生イネ植物体は正常に生育したので自殖種子を得ることができた。
【0261】
(11) 形質転換イネ植物の再生体中のトリプトファン含量の測定
前項(10)で得られた、第1の発明によるDNAを保有するOSASA-W1配列を含有する組換えベクターpUb-OSASAW1を導入された植物細胞を有する再生された形質転換イネ植物体と、第3の発明による改変D配列を含有する組換えベクターpUb-OSASA1Dを導入された植物細胞を有する再生された形質転換イネ植物体とから、夫々に緑色の葉を採取した。
【0262】
このようにそれぞれに採取された葉を1gづつ取り、それらの葉の100mgを、1.5ml容の第1のチューブに入れ、さらに50%アセトニトリル1mlを添加し、磨砕した。磨砕混合物を新たらしい1.5ml容の第2のチューブに移し替え、17000Xgの遠心加速度で20分間遠心分離した。得られた上清を、再び新たらしい第3のチユーブに移し入れ、その第3のチューブ内に50%アセトニトリル1mlを加え、十分に転倒攪拌し、再び遠心分離した。得られた上清を第1のチューブに加え、この操作を3回繰り返した。このようにして、葉から抽出されたトリプトファンを含有したアセトニトリル抽出液を得た。
【0263】
このアセトニトリル抽出液を減圧下で乾固させた後、蒸留水1mlを加えて水溶液を得た。その水溶液を17000Xgの遠心加速度で20分間遠心分離により、上清の0.5mlを得た。その上清のうち100μlに5mM DNFB(2,4-ジニトロ-1-フルオロベンゼン)100μlを混合した。得られた混合物を一晩反応させた。この反応液に200μlのアセトニトリルを添加し、攪拌した後、17000xgの遠心加速度で20分間遠心分離した。上清液として、トリプトファンの抽出液を得た。これを、高速液体クロマトグラフィー(HPLC)装置(東ソー(株)製、8020型)にかけて遊離トリプトファン含量を測定した。このHPLCに用いたカラムは、CAPCELL PAK-C18(資生堂(株)製)であり、展開溶媒はアセトニトリル-水をアセトニトリルの60%〜72%濃度勾配で用い、流量は0.8ml/分として、350nmの光の吸光度を測定して、トリプトファン含量を評定した。
【0264】
対照のイネ植物として、形質転換されてない通常のイネ(品種: 日本晴)の植物体を用いた。トリプトファン含量について得られた測定結果を次の表1に示す。
【表1】

【0265】
上記の表1に示された結果から明らかなように、第1の発明による新規DNA配列を外来遺伝子として用い且つ植物細胞中で発現できるプロモーターを有する組換えベクターを利用すると、第1の発明による新規DNA配列を導入することにより、イネ植物体中に生成される遊離トリプトファン含量が増加できることが確認された。
【0266】
実施例4
本例は、第3の発明による配列番号12のDNA配列を、外来遺伝子としてイネ植物に導入することからなる形質転換細胞の選抜方法と形質転換植物の作成方法を例示する。
【0267】
(1) 外来遺伝子の導入用の組換えベクターの作成
外来遺伝子の導入用の組換えベクターとして、実施例3、(3)(iii)に記載の組換えベクターpUb-OSASA1Dまたは実施例3、(1)(iii)に示された組換えベクターpUBdDを用いた。
【0268】
(2) アグロバクテリウム細菌法またはウイスカー法によるイネのカルス細胞への外来遺伝子の導入と形質転換細胞の選抜の操作
(i) アグロバクテリウム細菌法
上記の(1)に示した組換えベクターpUb-OSASA1Dを用いて実施例3の(4)と同様の方法によりアグロバクテリウム細菌の調製を行い、また実施例3の(5)と同様の方法によりイネのカルス細胞の調製を行い、実施例3の(6)と同様の方法によりイネのカルス細胞への導入操作を行った。
【0269】
上記で組換えベクターが導入された形質転換カルスを、500mg/lカルベニシリンを添加してなる滅菌水で洗浄してアグロバクテリウム細菌を除去した。カルスから余分な水分を除いた後、カルスをN6培地の無機塩細成にショ糖30g/l、2,4-PA 2mg/l、カルベニシリン500mg/l、ゲルライト2g/lおよび選抜用薬剤として5MT3×10−4Mを添加してなる固体培地上に各シャーレ当たり20個ずつ、合計500個を移殖した。25℃で1か月間、2000ルックスの光を1日当たり16時間照明しながら培養した。
【0270】
1か月培養の後に、形質転換カルスが選抜されて得られたカルスの個数の計数した結果を後記の表2に示す。なお、対照区には、5MTの代わりにハイグロマイシンの50mg/lを添加した固体培地を用いた。
【0271】
上記の培養により5MT耐性を基準にして選抜されて得られた形質転換カルスを、MS培地の無機塩組成にショ糖30g/l、ソルビトール30g/l、カサミノ酸2g/l、NAA 1mg/l、BA 2mg/l、ゲルライト4g/lおよび5MT3×10−4Mを添加してなる固体再生培地に移殖した。25℃で30日間、2000ルックスの光を1日当たり16時間照明しながら培養した。形質転換カルスから再生された形質転換植物体の再生数を計数した結果を後記の表2に示す。なお、対照区には、5MTの代わりにハイグロマイシンの50mg/lを添加した固体培地を用いた。
【0272】
(ii) ウイスカによる直接導入法
イネ(品種:日本晴) の完熟種子から籾穀を脱穀した。得られた外皮付きの種子を70%エタノール溶液に60秒間、ついで有効塩素約1%の次亜塩素酸ナトリウム溶液に6分間浸漬して種子を殺菌処理した。さらに種子を滅菌水で洗浄した。
【0273】
MS培地の無機成分組成にショ糖30g/l、植物ホルモンとして2,4-PA 2mg/l、寒天8g/lを添加してなる培地に、上記で殺菌したイネ種子を置床した。28℃で45日間、2000ルックスの光を1日当たり16時間照明しながらインキュベートした。カルスが形成された。それらカルスを胚乳部分から切り出し、孔1mmのステンレスメッシュの篩を用いて、1mm以下のカルスをPCV(Packed Cell Volume; 圧縮細胞量) として3mlの量で得た。
【0274】
チタン酸カリウム製ウイスカ(チタン工業(株)製、LS20) 5gを1.5ml容のチューブに入れ、エタノールを0.5ml加えて、一晩放置した。エタノールを完全に蒸発させて、殺菌されたウイスカを得た。このウイスカの入ったチューブに滅菌水1mlを入れ、よく攪拌した。ウイスカと滅菌水を遠心分離し、上清の水を捨てた。このようにしてウイスカを洗浄した。このウイスカ洗浄操作を3回行った。その後、同チューブ内にR2液体培地の0.5mlを加えてウイスカ懸濁液を得た。
【0275】
上記で得られたウイスカ懸濁液の入ったチューブに1mm以下のカルスを250μl入れて、攪拌した。得られた混合物を1000rpm/分で10秒間遠心分離し、カルスとウイスカを沈殿させた。上清を捨てた。カルスとウイスカとの混合物が得られた。
【0276】
カルスとウイスカとの混合物を入れたチューブに、組換えベクター(すなわち、前記の組換えベクターpUBdD)の10μl(DNAの10μg含有)とを加えた。十分振り混ぜた均質な混合物を得た。
【0277】
次にこの均質な混合物の入ったチューブを18000 x gで5分間遠心分離した。遠心分離された混合物を再度振り混ぜた。この遠心分離の操作と再度振り混ぜる操作を3回反復した。
【0278】
上記のようにして得られた、カルス細胞と、ウイスカと、第3の本発明の改変DNA配列を含有する組換えベクターを収容しているチューブを、超音波発生機の浴槽にチューブが十分浸かるように設置した。周波数40kHzの超音波を強度0.25W/cm2で1分間照射した。照射後、30分間、4℃で超音波処理された混合物をインキュベートした。
【0279】
このように超音波処理した混合物をR2液体培地で洗浄し、組換えベクターpUBdDを導入した目的の形質転換カルス細胞を得た。
【0280】
上記で組換えベクターを導入して得た形質転換細胞を有するカルスを、3.5cmのシャーレに入れた。さらにR2培地の無機成分組成にショ糖30g/l、2,4-PA 2mg/lを添加して得た液体培地を3ml加えた。その後に、28℃で2000ルックスの光を1日当たり16時間照射しながらロータリーシェーカー(50rpm) 上でカルス細胞を培養し、分裂細胞を得た。
【0281】
培養3日目に、得られた分裂細胞の懸濁液3ml(分裂細胞数2000個) を、N6培地の無機成分組成に、ショ糖30g/l、2,4-PA 2mg/l、ゲルライト3g/lおよび選抜用薬剤として5MT 10−4Mを添加してなる培地上に均一に広げた。培地上の細胞を、28℃で2000ルックスの光を1日当たり16時間照射しながら1か月間培養した。
【0282】
1か月後培養の後に、形質転換細胞よりなるカルスが選抜できた。こうして得られたカルスの個数の計算した結果を表2に示す。なお、対照区には、5MTの代わりにハイグロマイシンの50mg/lを添加した固体培地を用いた。
【0283】
また上記の培養により5MT耐性を基準にして選抜されて得られた形質転換細胞よりなるカルスを、MS培地の無機塩組成にショ糖30g/l、ソルビトール30g/l、カサミノ酸2g/l、NAA 1mg/l、BA 2mg/l、ゲルライト4g/lおよび5MT3×10−4Mを添加してなる固体再生培地に移殖した。25℃で30日間、2000ルックスの光を1日当り16時間照明しながら培養した。形質転換カルスから再生された形質転換植物体が得られた。その形質転換植物の再生数を計数した結果を表2に示す。なお、対照区には、5MTの代わりにハイグロマイシンの50mg/lを添加した固体培地を用いた。
【表2】
【0284】
実施例5
本例は、イネのASA遺伝子のプロモーターの遺伝子に相当するDNAを収得する方法を例示する。
【0285】
(1) イネのゲノミックDNAの調製
イネ(品種:日本晴) のカルス2gから、公知のCTAB法(クローニングとシークエンス: 1989年、262頁〜264頁、農村文化社) に従い、ゲノミックDNAの2mgを抽出した。
【0286】
(2) イネのゲノミックDNAライブラリーの構築
10マイクログラムの上記ゲノミックDNAを制限酵素EcoRIで部分消化した後、得られたDNA断片を0.8%アガロースゲルにて電気泳動した。電気泳動により分画したDNA断片をナイロン膜(アマシャム社製)ハイボンドNに転写した。このようにしてナイロン膜に転写したDNAはアルカリ変性液(1.5M NaCl、2.0M NaOH) および中和液(1.0 M Tris-HCl pH5、2.0M NaCl) でそれぞれ10分間処理し、その後に80℃、2時間処理してDNAをナイロン膜に固定した。
【0287】
次に、実施例1の(5)で得られたDIGでラベルして得られた標識プローブDNAを用いた。この標識プローブDNAを上記のナイロン膜に対してハイブリダイゼーションさせた。この操作は実施例1の(5)と同様の方法で行った。
【0288】
その結果、ナイロン膜上のDNAサイズ6kb付近に、X線フィルムに強くシグナルを発するDNAが検出された。そこで、それらDNAを分離し、そのDNAを実施例1の(5) と同様の方法で制限酵素EcoRIで部分消化し、電気泳動したDNA断片の6kb付近をゲルから切り出し、DNA精製キット(Gene Clean II Kit (Bio 101社製)) を用いて精製した。得られたDNA断片(精製品) を20μlのTEバッファー(10mM Tris-HCl(pH7.5)、1mM EDTA) に溶解した。このようにして、分画したイネのゲノミックDNAが得られた。これをクローニングキット(Lambda ZAPII/EcoRI/CIP Cloning Kit(STRATGENE社製)) を用いてベクターにライゲーションした。得られた組換えベクターを、GigapackII Gold Packaging Extractを用いてラムダファージにパッケイジングを行った。得られた組換えλファージを大腸菌XL1-Blue MRF'に感染させて、前記の組み換えファージを多数得た。これら得られた組換えファージをイネの分画ゲノミックDNAライブラリーとした。
【0289】
(3) イネのゲノミックDNAライブラリーからのプロモーター遺伝子の選択
上記(2)で作製したイネのゲノミックDNAライブラリーである組換えファージから、実施例1の(5)で作製した標識プローブDNAの利用により、イネのASA遺伝子の発現のためのプロモーターの遺伝子に相当するDNA配列を組込まれた組換えファージをスクリーニングする。
【0290】
このスクリーニングの目的のために、実施例1 (5)と同様の方法で、組換えファージをナイロン膜に固定した後、標識プローブDNAをハイブリダイゼーションさせた。こうして目的のDNA配列が組み込まれたファージを選択した。その結果、ASA遺伝子のプロモーター遺伝子が組み込まれていると思われるファージのプラークの3個を、10万個のファージプラークから、単離選抜した。これら3個の組換えファージから、実施例1 (6)と同様の方法で各々別々に3種のλDNAを単離した。
【0291】
上記のようにして単離された3種のファージDNAを各々別々に用いて、それの5μlを制限酵素EcoRIの10ユニットで消化して、その消化液を分析した。単離された3種のファージDNAのRcoRI-DNA断片は同じ塩基配列を有すると認められた。
【0292】
(4) イネのASA遺伝子のプロモーター遺伝子に相当するDNA配列を含有するゲノミックDNAのクローニング
上記のようにイネのASA遺伝子に相当するDNA配列を含有すると判定されたDNA断片として、3種のDNAのうちの各々のファージから制限酵素EcoRIで切り出されて収得された6.2kbのサイズのDNA断片は、これをプラスミドベクターpBluewscriptIISK(+)のEcoRI切断部位にDNAリゲーション・キットにより挿入、連結して組換えプラスミドベクターを構築した。このように構築された組換えプラスミドベクターの導入で大腸菌XL1-BlueMRF'を形質転換した。得られた形質転換株はエシエリヒア・コリXL1-Blue MRF'(Os-asa#7) と命名し、これは生命工学工業技術研究所に1997年8月18日にFERM P-16387として寄託され、また1998年8月7日以降はブダペスト条約の規約下にFERM BP-6452の受託番号で寄託されてある。
【0293】
上記における組換えプラスミドベクターの構築および大腸菌への導入の操作は、実施例1(6) の方法と同様にして行った。
【0294】
組換えプラスミドベクターを導入した大腸菌から、プラスミド精製キット(QIA filter Plasmid Midi Kit, QIAGEN社製) によりプラスミドを分離且つ精製した。この精製により、上記で得られた形質転換大腸菌から50μg(50μl)のプラスミドDNAの3種を得た。
【0295】
このように各々の単離された組換えファージからクローニングされて得た前記のプラスミド3種類に含まれてあり且つ制限酵素EcoRIとBamHIの切断部位に挟まれた約1.5kbのDNAは、その塩基配列を解析するために次の操作を行った。
【0296】
(5) クローニングされたDNAのシークエンス解析
上記のクローニングされた3種類のDNAである組換えプラスミドを市販の塩基配列決定キットで処理すると、そのDNA断片の全体の塩基配列を決定できる。そして、そのDNA断片内部に含有されたイネのASA遺伝子のプロモーター遺伝子に該当するDNA配列を判定できる。上記の塩基配列の決定は、実施例1(7) の方法と同様に行った。
【0297】
このようにして上記のDNAの塩基配列を判定した結果、クローニングされた3種類のDNAである組換えプラスミドはすべて同一であることがわかった。
【0298】
こうして得られた、ASA遺伝子のプロモーターのDNAは、後記の配列表の配列番号7に記載されてある塩基配列を有すると今回認められた。ここで得られた配列番号7の塩基配列は、イントロンを含むものであり、またそれのタンパク質をコードしている部分がcDNAクローンと完全に一致するものであった。得られた組換えプラスミドのDNAについて決定した塩基配列のうち、ASA遺伝子のプロモーター部分及びそのエクソン部分およびイントロン部分のDNAを含むDNA配列の塩基配列を配列番号3に示す。
【0299】
なお、配列表の配列番号3は、イネのASAの第1アイソザイムのαサブユニットであるタンパク質の部分アミノ配列も併記してある。また、配列表の配列番号4は、配列番号3に示された塩基配列のうちのアミノ酸コード部分のアミノ酸配列を示す。さらに、配列番号5は、配列番号3のDNAのうちのエクソン−1の塩基配列を示し、配列番号6は、配列番号3のDNAのうちのエクソン−2の部分配列を示す。
【0300】
(6) プロモーター活性の検定
(i) 検定用の組換えベクターの作成
上記で得られたプロモーターDNA(配列番号7) のプロモーター活性を調べるため、このDNA(前記の(4) で得たEcoRI-BamHI断片) を植物細胞形質転換用ベクターpBI101(Clontech社製) のβ-グルクロニダーゼ(GUS)遺伝子の上流部分に組み込み形質転換用ベクターpGUS#3を得た。
【0301】
形質転換用ベクターの作製においては、GUS遺伝子のNOSターミネーターとカナマイシン耐性遺伝子とを含有する12.2kbのサイズのプラスミドベクターpBI101(Clontech社製) のDNA(10μg)を、Mバッファー中で制限酵素HindIIIの10ユニットで消化した。その消化反応液からDNAを沈殿させ、遠心分離してから乾燥させた。得られた乾燥物を10μlの滅菌水で溶解した後、続いて、切断部位を平滑化するために、DNAプランティング・キット(宝酒造(株)製) を用いて処理した後、DNA精製キット(Gene Clean II Kit (Bio 101社製)) を用いて精製した。その精製DNAを10μlの滅菌水に溶解した。この溶解液を、Kバッファー中で制限酵素BamHIの10ユニットで消化した。その消化反応液からDNAを沈殿させ、遠心分離してから乾燥させた。このようにして12.2kbのサイズを有して且つ平滑末端およびBamHI切断部位を両端に持つ、ベクター断片を得た(添付図面の図3の左、参照)。
【0302】
一方、上記したイネのASA遺伝子のプロモーターDNA断片を含むプラスミドベクターDNA(10μg)を、Hバッファー中で制限酵素EcoRIの10ユニットで消化した。その消化反応液からDNAを沈殿させ、遠心分離してから乾燥させた。得られた乾燥物を10μlの滅菌水で溶解した後、続いて、切断部位を平滑化するために、DNAプランティング・キット(宝酒造(株)製) を用いて処理した後、DNA精製キット(Gene Clean II Kit (Bio 101社製)) を用いて精製した。その精製DNAを10μlの滅菌水に溶解した。この溶解液を、Kバッファー中で制限酵素BamHIの10ユニットで消化した。その消化液をアガロース電気泳動により約1.5kb付近をゲルから切り出し、DNA精製キットを用いて精製した後、10μlの滅菌水に溶解した。このようにして1.5kbのサイズを有して且つ平滑末端およびBamHI切断部位を両端に持つ、プロモーターDNA断片を得た(図3の右、参照)。
【0303】
上記したプラスミドベクター断片溶液の5μlと上記のプロモーターDNA断片溶液の5μlとを混合した後、この混合液をDNAリゲーション・キットで処理してDNAの連結を行った。
【0304】
このようにしてプラスミドベクターpBI101の平滑末端−BamHI−切断ベクター断片に対して、前記のイネのASA遺伝子のプロモーターDNAの平滑末端−BamHI−断片を連結してなる環状の組換えベクターを作製した。こ組換えベクターをpGUS#3と称する(図3の下、参照)。
【0305】
ベクターpGUS#3は、イネのASA遺伝子のプロモーターDNAの下流に、GUS遺伝子を持ち、さらに下流のNOSターミネーターが連結されてあり、且つカナマイシン耐性遺伝子を含有する構造を有し、13.7kbのサイズを有した。
【0306】
(ii) イネのカルス細胞への検定用組換えベクターの導入
上記で得られた組換えベクターpGUS#3を実施例3 (6)と同様の方法によりイネのカルス細胞への導入を行った。このようにして、組換えベクターpGUS#3が形質転換されているイネのカルス細胞を得た。
【0307】
(iii) GUS活性の測定
上記で得られた組換えベクターpGUS#3が形質転換されたイネのカルスを、28℃で3日間培養した後、カルスの約0.5gを乳鉢に入れ、500μlの抽出用バッファー50mM NaPO4(pH7)、10mM EDTA、0.1%TritonX100、0.1%Sarkosyl、10mM β-メルカプトエタノール) 中で添加し、磨砕した。磨砕液を1.5ml容のチューブに移し、17000×gの遠心速度で20分間遠心分離した。得られた上清のうち、10μlを新しいチューブに移し、基質である1mMの4-メチルアンペリフェリルグルクロナイド(4-MUG) 溶液の100μlを加えて混合した。得られた混合物を37℃で3時間反応させた。その反応液に3%炭酸水素ナトリウム溶液の1.8mlを添加して、混合した。それを蛍光分光光度計(日立社製、F2000型) にかけて蛍光吸収測定を行った。その際の365nm励起光、455nm発光の条件でGUS活性の評定を行った。
【0308】
GUS活性を測定した結果を下記の表3に示す。数値はカルスから抽出したタンパク質1g当たり、1時間にGUS酵素の作用により生成する4-メチルアンベリフェロン(4-MU)の量を示す。なお、対照区には、プロモーターを組み込まれていないpBI101ベクターを導入したカルスを用いた。
【表3】
【0309】
上記のようにGUS活性が発現されたことにより、塩基配列の解析の結果から確認されたプロモーター領域がプロモーターとして機能したことが証明される。
【産業上の利用可能性】
【0310】
本発明により提供されるイネの第2ASAのαサブユニットをコードする遺伝子DNAは、単独又は他の遺伝子と組み合わせて植物体に導入することにより、植物体中の必須アミノ酸であるトリプトファンの含量を高めることができ、栄養価種の高い植物の育種に有用である。また、本明細書に記載されるイネのASA遺伝子のプロモーターDNAは、植物体中で外来の有用遺伝子の発現を行う際のプロモーターとして利用することができ、形質転換植物の作出するのに有効である。
【図面の簡単な説明】
【0311】
【図1】図1は、イネのカルス細胞を、ウイスカー法による外来遺伝子の直接導入法により形質転換するのに用いる外来遺伝子の導入用の組換えベクターであって、配列表の配列番号12の塩基配列をもつ第3の発明の改変D配列を内部に含有する組換えベクターpUBdDを、ベクターpUBAから作成するのに実施例3の(1) で用いた手順を図解的に示す。
【図2】図2は、イネのカルス細胞を、アグロバクテリウム法による外来遺伝子の導入法により形質転換するのに用いる外来遺伝子の導入用の組換えベクターであって、第3の発明の改変D配列を内部に含有するベクターpUb-OSASA1Dを、ベクターp1G121-Hmから作成するのに実施例3の(3)で用いた手順を図解的に示す。
【図3】図3は、イネのASAのためのプロモーター配列の活性の検定の試験において、イネのカルス細胞を形質転換するのに用いるべき外来遺伝子として該プロモーター配列を含有する組換えベクターであって、配列表の配列番号3に示された塩基配列を有するプロモーター配列を内部に含有するDNA断片#3が連結された組換えベクターpGUS#3を、ベクターpBI101から作成するのに実施例5の(6) で用いた手順を図解的に示す。
【技術分野】
【0001】
本発明は、イネのアントラニル酸シンターゼの2つのアイソザイム(イソ酵素)、すなわち第1アイソザイムと第2アイソザイムとのうちの第2アイソザイムのαサブユニットをコードする遺伝子のDNAと、該遺伝子に関連するDNAに関する。詳しく言えば、本発明はイネ植物のトリプトファンの生合成に関与するアントラニル酸シンダーゼ(anthoranilate synthase)の2つのアイソザイム、すなわち第1アイソザイムと第2アイソザイムの各々のαサブユニットであるタンパク質をそれぞれコードする新規な2つのDNAのうちの、第2アイソザイムのαサブユニットのタンパク質をコードするDNAに関する。
【0002】
また、本発明は、アントラニル酸シンターゼの第2アイソザイムのαサブユニットのタンパク質をコードする新規なDNAが組み込まれた新規な組換えベクターに関する。さらに本発明はその新規なDNAで形質転換された大腸菌、あるいはその新規なDNAで形質転換された植物および種子も包含する。
【0003】
さらに、本発明は、その新規DNAを用いることによる植物のトリプトファン含有量の増加方法に関する。
【背景技術】
【0004】
イネ、トウモロコシ、コムギなどの穀物種子は、人や家畜等にとって重要な栄養源である。しかし、これらの種子は必須アミノ酸の一つであるトリプトファンの含量が低く、栄養価が劣っている。トリプトファン含量の高くて栄養価に優れた穀物種子を産生できる新しい品種の植物の作出が望まれている。
【0005】
植物体中のトリプトファンの生合成経路において、コリスミン酸からアントラニル酸が生合成されるが、アントラニル酸の生成はアントラニル酸シンターゼ (以下、「ASA」 と略記することがある) の触媒作用が関与して、これによってアントラニル酸が生成し、さらにアントラニル酸から6段階の酵素反応によりインドールを経てトリプトファンが生成することが知られている (非特許文献1:生化学実験講座、第11巻、652頁〜653頁、1976年、東京化学同人より刊行)。
【0006】
植物の従来既知のアントラニル酸シンターゼは、複数のサブユニットから構成されていることが知られている。例えばアラビドプシス (和名:シロイヌナズナ;学名Arabidopsis thaliana) のアントラニル酸シンターゼは2種のアイソザイムからなり、またその第1アイソザイムも第2アイソザイムもそれぞれαサブユニットとβサブユニットからなる2量体であることが知られている。アラビドプシスのアントラニル酸シンターゼの第1アイソザイム (ASA1と略記される) のαサブユニットをコードする遺伝子 (asa1) と、第2アイソザイム (ASA2と略記される) のαサブユニットをコードする遺伝子 (asa2) とは単離され、それら遺伝子のDNAの塩基配列が解明されている (非特許文献2:The Plant Cell, 4巻, 721頁〜733頁, 1992年)。
【0007】
他方、先に、本発明者らは、イネのトリプトファン生合成の調節に重要な機能領域を有することが示唆されるアントラニル酸シンターゼαサブユニットに着目して、トリプトファンと植物ホルモンIAAの生合成調節機構についての知見を得ることを目的として、アントラニル酸シンターゼ・タンパク質をコードする遺伝子を単離する研究を1996年に行った。この研究の報告の要約として、本発明者らは、イネ (農林8号) の幼植物から、mRNAとゲノミックDNAを抽出し、cDNAライブラリーとゲノミックDNAライブラリーを作製し、これらライブラリーを用いて且つシロイヌナズナのasa遺伝子のcDNA断片をプローブとして用いて、ゲノミックサザン解析とライブラリーのスクリーニングを行い、これによってイネのアントラニル酸シンターゼのasa遺伝子に相当すると考えられるDNAを収得したことを発表した (非特許文献3:「育種学雑誌」 46巻、別冊2、28頁、1996年)。この文献に発表された研究報告の要約には、イネのアントラニル酸シンターゼのasa遺伝子に相当するDNA断片を収得したことを報告したが、そのDNA断片の収得に用いた具体的な手法を本発明者らは開示されていず、また前記のDNA断片の塩基配列も未だ決定されていないことを報告した。また、前記の研究報告の要約には、イネのアントラニル酸シンターゼをコードする遺伝子に相当すると考えられるDNAには、2種類のDNAが存在することに言及した(前記の 「育種学雑誌」 46巻、別冊2、28頁、1996年)。
【0008】
また、アラビドプシスのASAの第1アイソザイムのαサブユニットのASA遺伝子のDNAと、該DNAを改変したDNA断片とを、タバコ植物体に導入し、その遺伝子の機能をタバコで発現させることに関する報告がされている (非特許文献4:Massachusetts Institute of Technology, Cambridge, MA, 1993年)。
【非特許文献1】「生化学実験講座」第11巻、652頁〜653頁、(1976年)
【非特許文献2】「The Plant Cell」4巻、721頁〜733頁、(1992年)
【非特許文献3】「育種学雑誌」46巻、別冊2、28頁、(1996年)
【非特許文献4】「Massachusetts Institute of Technology,Cambridge, MA」(1993年) しかしながら、本発明者の知る限りでは、イネのASAのアイソザイムのαサブユニットであるタンパク質のアミノ酸配列を解析したこと、およびイネのASAのアイソザイムのαサブユニットをコードする遺伝子を取得できた具体的な方法を報告する文献は知られていない。また、該遺伝子の発現に関わるプロモーター配列も知られていない。またイネのASAをコードする遺伝子を利用することを報告する文献も知られていない。
【発明の開示】
【発明が解決しようとする課題】
【0009】
本発明の一つの目的は、イネのASAに関連する遺伝子、詳しくはイネのASAの第2アイソザイムのαサブユニットをコードする新規なDNAをイネ植物から収得することにあり、また、本発明の他の目的はこのDNAの塩基配列を決定することにある。
【0010】
本発明の別の目的は、その新規DNAによって、トウモロコシ、イネ、ダイズ、コムギ、オオムギ、トマト、ジャガイモなどの有用植物を形質転換することであり、また高いトリプトファン含量を持つ種子を産生できる有用植物の新しい形質転換品種を提供することである。本発明のその他の目的は、後記の説明から明らかになるであろう。
【課題を解決するための手段】
【0011】
上記の諸目的を達成するために、本発明者らは一連の種々研究を行った。先づ、イネ植物からASAの2つのアイソザイムの各々のαサブユニットをコードする遺伝子を取得するための研究を行った。この研究の結果、イネの幼植物体の組織、例えば緑色の茎葉の破砕物から全RNAを遺伝子工学技術で知られる方法により抽出し、その抽出された全RNAから全mRNAを常法により単離し、その全mRNAから、市販の全cDNA合成キットによりイネの全cDNAを収得することに成功した。得られた全cDNAは、λgt11ファージベクターのEcoRI切断片の末端を仔ウシ小腸由来アルカリホスファターゼで処理したファージベクター (STRATAGEN社製の市販品) に連結すると、組換えベクターを作成できること、さらに作成された組換えベクターをラムダ-ファージにパッケージすると、複製可能な組換えラムダ-ファージを構築できることが多くの試行錯誤の結果により知見された。
【0012】
その組換えラムダ-ファージを大腸菌Y1088に感染させ、インキュベートすると、多数のプラーク (溶菌斑) として組換えλファージの多数が得られること、また得られた多数のプラークに含まれる組換えλファージ群は、イネ由来の全cDNAを含有する各種多様なファージであって、これはイネのcDNAライブラリーとして利用できることが知見された。
【0013】
他方、前記の非特許文献2:「The Plant Cell」4巻、721頁〜733頁 (1992年) に記載されたアラビドプシスのASAの第1アイソザイムのαサブユニットであるタンパク質のアミノ酸配列と、これから推認される該タンパク質をコードする遺伝子の塩基配列と、アラビドプシスのASAの第2アイソザイムのαサブユニットであるタンパク質のアミノ酸配列と、これから推認される該タンパク質をコードする遺伝子の塩基配列とを参照しながら、PCR法のプライマーとして適すると考えられた21個のヌクレオチドからなる第1のオリゴヌクレオチドと、24個のヌクレオチドからなる第2のオリゴヌクレオチドとを本発明者は、化学合成により作製した。
【0014】
前記の第1のオリゴヌクレオチドと第2のオリゴヌクレオチドと市販されているアラビドプシスのcDNAライブラリー(テンプレートとして利用される) との混合物を用いてPCR法増幅反応を行うと、第1および第2のオリゴヌクレオチドは、PCR法で所要なプライマー (相補的DNA) として働くことができ、そして、アラビドプシスのASAの2つのアイソザイムのそれぞれのαサブユニットをコードする遺伝子に相当するDNA配列のうちの一部分を構成しているDNA断片が増幅できることが認められた。そしてPCR増幅反応液からアラビドプシスのASAの第1および第2アイソザイムの各々のαサブユニットをコードする遺伝子の一部分の増幅生成物を、プローブDNAとして回収することに成功した。
【0015】
このように収得された上記のプローブDNAを利用することによって、ファージプラーク-ハイブリダイゼーション法によって、先に得られたイネのcDNAライブラリー (すなわち前記の組換えλファージの30万個のプラーク) から、多くの試行錯誤の試験の結果として、イネのASAの第1アイソザイムのαサブユニットをコードする遺伝子と第2アイソザイムのαサブユニットをコードする遺伝子を組込まれた組換えλファージのプラークの8個を単離することに幸にも成功した。それら8個のプラークの組換えλファージを別々に増幅した後に、各々のλファージDNAを常法で単離した。
【0016】
上記のようにして得られた、イネのASAの第1および第2アイソザイムの各々のαサブユニットをコードする遺伝子に相当すると認められるDNA配列を内部に含有する組換えファージDNAを、制限酵素EcoRIで切断して得られたDNA断片は、これを次に、市販の既知のプラスミドベクターp Bluescript II SK(+)のEcoRI切断部位にDNAリゲーションキットにより挿入、連結することができた。こうして得られた組換えプラスミドベクターで大腸菌XL1-Blue MRF'を形質転換することができた。得られた大腸菌形質転換体をインキュベートして多数の菌体を得ることができた。得られた菌体からプラスミドDNAを採取した。このように採取したプラスミドDNAを、DNAの塩基配列の解析にかけることによって、このDNA断片に含有されて且つイネのASAの第1および第2アイソザイムの各々のαサブユニットをコードする遺伝子に相当すると判定された2種のDNA配列のうちの第1のDNA配列の塩基配列は、後記の配列表の配列番号1に記載の塩基配列を有するものであると今回、確認された。また、前記の2種のDNA配列のうちの第2のDNA配列の塩基配列は配列表の配列番号10に記載の塩基配列を有するものであると今回、確認された。
【0017】
さらに、本発明者らの知る限りでは、配列表の配列番号1に記載の塩基配列を有するDNAと、配列番号10に記載の塩基配列を有するDNAとは、何れの文献にも記載されていない新規なDNA配列であると認められた。
【0018】
配列表の配列番号1の塩基配列を有するDNAでコードされるタンパク質は配列表の配列番号2に記載のアミノ酸配列を有するタンパク質であると認められ、またこのタンパク質はイネのASAの第1アイソザイムのαサブユニットを構成するタンパク質であると認められる。
【0019】
従って、本明細書に開示される第1の発明(本件分割出願の原願である特願2000-508808号の請求項1の発明)においては、配列表の配列番号2に示すアミノ酸配列を有するタンパク質であって、イネのアントラニル酸シンターゼの第1アイソザイムのαサブユニットをコードするDNAが提供される。
【0020】
第1の発明による上記のDNAは、具体的には、配列表の配列番号1に記載の塩基配列を有するDNAであることができる。
【0021】
第1の発明のDNAは、イネのASAの第1アイソザイムのαサブユニットであるタンパク質をコードするDNAである。この第1の発明のDNAは、本発明者らの研究を行うに際しては、前述のように、イネのcDNAライブラリーから遺伝子工学の技法で取得されたものである。しかし、そのDNAの塩基配列が本発明で明らかにされたので、配列表の配列番号1の塩基配列を参照してヌクレオチドから化学合成によっても取得することができる。また、前記の配列番号1の塩基配列を参照して合成ヌクレオチドをプローブとして作製して用いるか、もしくはその合成オリゴヌクレオチドをプライマーとして用いる公知の方法によってイネ染色体のDNAライブラリーから、ポリメラーゼ・チェイン・リアクション (PCR) の方法又はハイブリダイゼーションによって第1の本発明のDNAを取得することもできる。
【0022】
さらに、配列表の配列番号10の塩基配列を有するDNAでコードされるタンパク質は配列表の配列番号11に記載のアミノ酸配列を有するタンパク質であると認められ、またこのタンパク質はイネのASAの第2アイソザイムのαサブユニットを構成するタンパク質であると認められる。
【0023】
従って、第2の本発明(本件特許請求の範囲の請求項1の発明)においては、配列表の配列番号11に示すアミノ酸配列を有するタンパク質であって、イネのアントラニル酸シンターゼの第2アイソザイムのαサブユニットをコードするDNAが提供される。
【0024】
第2の本発明による上記のDNAは、具体的には、配列表の配列番号10に記載の塩基配列を有するDNAであることができる(請求項2)。
【0025】
第2の本発明(本件特許請求の範囲の請求項1または2の発明)のDNAは、イネのASAの第2アイソザイムのαサブユニットであるタンパク質をコードするDNAである。この第2の本発明のDNAは、本発明者らの研究を行うに際しては、前述のように、イネのcDNAライブラリーから遺伝子工学の技法で取得されたものである。しかし、そのDNAの塩基配列が本発明で明らかにされたので、配列表の配列番号10の塩基配列を参照してヌクレオチドから化学合成によっても取得することができる。また、前記の配列番号10の塩基配列を参照して合成ヌクレオチドをプローブとして作製して用いるか、もしくはその合成オリゴヌクレオチドをプライマーとして用いる公知の方法によってイネ染色体のDNAライブラリーから、ポリメラーゼ・チェイン・リアクション(PCR)の方法又はハイブリダイゼーションによって第2の本発明のDNAを取得することもできる。
【0026】
以下に、イネの茎葉から遺伝子工学の技法によって、前記した第1の発明によるDNAおよび第2の本発明によるDNAを取得する方法を概略的に説明する。
【0027】
(1) イネmRNAの調製およびイネのcDNAライブラリーの構築
イネ (Oriza sativa) の種々な組織、例えば茎葉、根、カルスなどの組織、好ましくは緑色の茎葉から、常法により全RNAを抽出する。抽出された全RNAから蛋白質などの夾雑物を除いた後、さらにオリゴdTセルロースの充填カラムに通してpoly(A)+RNAを精製することによって、イネの全mRNAを得ることができる。
【0028】
次に、全mRNAから市販のcDNAを合成キットによりイネの全cDNAを合成する。合成された全cDNAをファージベクター、例えばλgt11ベクター又はλZAPIIベクターなどに連結する。得られた組換えベクターをラムダファージに組み込み、多数の組換えファージを得る。それら組換えファージを宿主として大腸菌に感染させ、インキュベートすると、プラークとして多数の組換えファージを得ることができる。これら一連の操作は、市販のcDNAクローニングキットを使用して実施できる。
【0029】
このように宿主大腸菌の溶菌のプラークとして得られた多数の組換えファージは、イネ由来の全cDNAを含有する多種多様のファージから成るものであるから、イネのcDNAライブラリーとして利用できる。
【0030】
(2) PCR法用のプライマーの構築
アラビドプシスのASAの第1アイソザイムのαサブユニットの遺伝子の既知の塩基配列と第2アイソザイムのαサブユニットの遺伝子の既知の塩基配列との間に共通に保存されている塩基配列 (オンラインデータベースEMBL: M92353) を参照して且つアラビドプシスのASAの第1アイソザイムが植物体でより強い発現力をもつことを勘案して、本発明者らは2種類のオリゴヌクレオチド (後記の配列表の配列番号8および9のオリゴヌクレオチド) をPCR法用のプライマー (相補的DNA) として化学合成により作製して構築した。
【0031】
(3) プローブDNAの作製
先に多数の組換えファージからなるプラークとして得られたイネのcDNAライブラリーの中で、所望のイネのASAのαサブユニットをコードするDNAを選択的に採取するのに用いられるプローブDNAを作製する。このプローブDNAの作製のために、前記の合成オリゴヌクレオチドよりなるプライマーを使用し、それで、PCR法によりテンプレートとしてのアラビドプシスのcDNAライブラリーからアラビドプシスのASAの第1および第2アイソザイムのαサブユニットの遺伝子の一部分をコードするDNAを増幅する。
【0032】
増幅反応をPCR法によって反復した後に、アラビドプシスのASAのαサブユニットの遺伝子に相当するDNA配列のうちの一部分であるDNA断片の増幅生成物を、増幅反応液から、所望なプローブDNAとして採取する。
【0033】
(4) イネcDNAライブラリーからのイネのASAのαサブユニットの遺伝子のDNAの選択
先にイネのcDNAライブラリーとして得られた組換えファージの多数のプラークの中から、上記のプローブDNAをスクリーニング用に利用するファージプラーク-ハイブリダイゼーション法により、目的のイネのASAのαサブユニットの遺伝子に全体的に対応するDNA配列を組込まれた組換えファージよりなる数個のプラークを選抜する。
【0034】
これによって選抜されたプラークとして採取された組換えファージは、イネのASAのαサブユニットの遺伝子のDNAに相当する目的のDNA配列を内部に含有するDNA断片が組込まれている組換えファージの形のものである。
【0035】
すなわち、詳しく言えば、上記のようなプラーク・ハイブリダイゼーションで選抜されたプラークのファージを採取して、さらに採取ファージからファージDNAを回収する。回収したファージDNAをジデオキシ法などで処理することにより、そのファージDNAの内部に存在するイネ由来の挿入DNA断片の塩基配列を決定することができる。そのイネ由来の挿入DNA配列の塩基配列中のタンパク質コード領域 (オープンリーディングフレーム) から規定されるアミノ酸配列を、アラビドプシスのASAのαサブユニットであるタンパク質の既知のアミノ酸配列に対して比較すると、相同性を判定することにより、上記で回収されたファージDNAがイネのASAのαサブユニットの遺伝子に対応するDNA配列を内部に含有するDNA断片であることを特定できる。
【0036】
従って、イネのASAのαサブユニットの遺伝子に相当するDNA配列を内部に含有すると判定された挿入DNA断片は、上記のように選抜された採取ファージのファージDNAから制限酵素で切出して収得できる。
【0037】
(5) イネのASAのαサブユニットの遺伝子に相当するcDNAのクローニング
上記のようにイネのASAのαサブユニットの遺伝子に相当するDNA配列を内部に含有するDNA断片として、ファージから切出して収得されたDNAは、これをプラスミドベクターp Bluescript II SK(+)のEcoRI切断部位に挿入、連結して組換えプラスミドベクターを構築する。このように構築された組換えプラスミドベクターで大腸菌XL1-Blue MRF'を形質転換する。こうして得られた大腸菌形質転換体を培養して増殖させると、イネのASAのαサブユニット遺伝子に相当するDNA配列を含有するDNA断片を担う前記の組換えプラスミドをクローニングできる。従って、イネのASAのαサブユニットの遺伝子に対応するDNA配列を含有するDNA断片がクローニングできる。
【0038】
本明細書に記載の発明において、上記のようにプラスミドベクターp Bluescript II SK (+) に連結されて大腸菌中でクローニングされて得られて且つイネのASAのαサブユニットの遺伝子に対応するDNA配列を内部に含有するDNA断片として、塩基数の異なる2種類のDNA断片が得られた。ここで得た小さいサイズのDNA断片をDNA断片Xとここで仮称し、また大きいサイズのDNA断片を、DNA断片Yとここで仮称する。
【0039】
(6) クローニングされたDNAのシークエンス解析
(i) 上記のクローニングされた組換えプラスミドから制限酵素EcoRIによりイネのASAのαサブユニットの遺伝子に該当のDNA配列を内部に含有する2種類のDNA断片として、DNA断片XとDNA断片Yとを別々に切り出す。切り出されたDNA断片XおよびDNA断片Yをそれぞれに市販の塩基配列決定キットで処理すると、イネのASAのαサブユニットの遺伝子に該当するDNA配列を内部に含有するDNA断片XまたはDNA断片Yにおける全体の塩基配列を決定できる。上記のDNA断片Xについて、このように決定された塩基配列をもち且つイネのASAの第1アイソザイムのαサブユニットをコードするDNA配列は、後記の配列表の配列番号1に記載された塩基配列を有し、1734個の塩基から成る。このDNA配列を配列OSASA-1配列と命名する。このDNA配列OSASA-1は、第1の本発明によるDNAの一例である。
【0040】
なお、後記の実施例1で得られたところの配列表の配列番号1に記載の塩基配列を有して且つイネのASAの第1アイソザイムのαサブユニットの遺伝子に相当するDNA配列を内部に含有する前記のDNA断片Xは、p Bluescript II SK (+)プラスミドベクター (STRATGENE社製) のEcoRI切断部位に挿入、連結された。このように得られた組換えプラスミドベクター (ベクターpOSASA-1と命名された) の導入により形質転換された大腸菌XL1-Blue MRF'はエシェリヒア・コリ (Escherichia coli) XL1-Blue MRF' (OS-asa1) と命名され、日本の茨城県、つくば市の工業技術院生命工学工業技術研究所に1997年8月18日に、FERM P-16388の受託番号で寄託されている。さらに、エシエリヒア・コリOS-asa1は、1998年8月7日以降、ブダペスト条約の規約下でFERM BP-6453の受託番号で前記の研究所に寄託されてある。
【0041】
前記された第1の発明によるDNAは、本発明で解明された該DNAの塩基配列を指針として用いることによって、イネのASAの第1アイソザイムのαサブユニットであるタンパク質を化学合成により多量に収得することを可能にしている点で有用であり、そしてイネのASAの第1アイソザイムのαサブユニットであるタンパク質の酵素学的研究を進めるのに貢献できるものである。
【0042】
(ii) さらに、前記のDNA断片Yについて、前記のように決定された塩基配列をもち且つイネのASAの第2アイソザイムのαサブユニットをコードするDNA配列は、配列表の配列番号10に記載された塩基配列を有し、1821個の塩基から成り、このDNA配列はOSASA-2配列と命名された。このDNA配列OSASA-2は、第2の本発明(本件特許請求の範囲の請求項1または2に記載の発明)によるDNAの一例である。
【0043】
なお、後記の実施例1で得られたところの、配列表の配列番号10に記載の塩基配列を有して且つイネのASAの第2アイソザイムのαサブユニットの遺伝子に相当するDNA配列を内部に含有する前記のDNA断片Yは、p Bluescript II SK (+)プラスミドベクターのEcoRI切断部位に挿入、連結された。ここで得られた組換えプラスミドベクター (ベクターpOSASA-2と命名される) の導入により形質転換された大腸菌XL1-Blue MRF'は、エシエリヒア・コリXL1-Blue MRF' (Os-asa2) と命名され、工業技術院生命工学工業技術研究所に1998年6月18日にFERM P-16853の受託番号で寄託され、また1998年8月7日以降、ブダペスト条約の規約下にFERM BP-6454の受託番号で寄託されてある。
【0044】
第3の発明(本件特許願の特許請求の範囲から除外されてあるが、特願2004-257204号の請求項1の発明)においては、配列表の配列番号13に記載のアミノ酸配列を有するタンパク質であって、且つイネのアントラニル酸シンターゼの第1アイソザイムのαサブユニットの活性を有するが但しトリプトファンによるフィードバック抑制に対して非感受性であるタンパク質をコードするDNAが得られている。
【0045】
前記した第3の発明による上記のDNAの具体的な例には、後記の実施例2に記載された方法で作製されて実施例2で改変D配列と命名され且つ配列表の配列番号12に記載の塩基配列を有するDNA配列がある。
【0046】
なお、配列表の配列番号12に記載の塩基配列を有して改変D配列と命名された上記のDNAは、イネのASAの第1アイソザイムのαサブユニットの活性を有するけれどもトリプトファンによるフィードバック抑制に対して非感受性である上記タンパク質をコードするDNAである。
【0047】
第3の発明による新規なDNAの具体的な例は、前述したように、後記の実施例2に記載された方法で作製されて改変D配列と命名されたDNA配列がある。
【0048】
その改変D配列と命名されたDNA配列は、配列表の配列番号12に記載の塩基配列を有するDNAである。この改変D配列は、配列表の配列番号1に記載の塩基配列を有する第1の本発明のDNAの中にある967番目、968番目、969番目のGAC配列 (アスパラギン酸をコードするコドン) での967番目の塩基G (グアニン) 1個をA (アラニン) 1個に置換して、塩基配列の967番目に配列 (アスパラギンをコードするコドン) が在るように改変されたDNAに相当する。この改変D配列でコードされるタンパク質は、配列表の配列番号13に記載のアミノ酸配列を有するものであり、イネのASAの第1アイソザイムのαサブユニットの活性を有する。
【0049】
また、第3の発明のDNAの具体例である上記の改変D配列でコードされるタンパク質は、トリプトファンの生合成経路に関与するASAが生合成生成物としてのトリプトファンによる酵素活性のフィードバック阻害を受けなくなるように酵素活性を改変された新規なタンパク質である。この新規なタンパク質をコードする改変DNAは、後記されるように、植物のトリプトファン含量を増加させる目的で植物の形質転換に使用される。
【0050】
なお、一般的には、DNA配列の一部分の領域中にある塩基配列を改変する方法として、(Kunkel法 「Methods in Enzymology」 154巻、367号)、およびオリゴヌクレオチド−ダイレクト デュアルアンバー法、さらにその他の方法が知られている。
【0051】
第1の本発明のDNAでコードされるASAの第1アイソザイムαサブユニットを、トリプトファンによる酵素活性のフィードバック阻害を受けなくなるように改変するために、前記した第1の発明のDNAの塩基配列の一部を改変する方針として、文献 「Plant Physiology」 第110巻、51頁〜59頁、1996年に記載されるトリプトファンアナログ体に耐性を示す突然変異アラビドプシスの公知のASA遺伝子に関する報告を参考にしながら、本発明者らは、配列表の配列番号1に記載のDNA (OSASA-1配列) の塩基配列の967番目の塩基、グアニン (G) をアデニン (A) に取換えることによって作製された改変DNAが所期の目的に有効であろうとの予想的な着想を得た。
【0052】
この着想に基づいて、本発明者らは、種々の研究を行ったが、多くの試行錯誤の結果として、イネのASAの第1アイソザイムのαサブユニットの遺伝子に対応するDNA配列 (すなわちOSASA-1配列) を含有するDNA断片を、プラスミドベクターp Bluescript II SK (+) のEcoRI切断部位の間にリゲーションキットにより挿入、連結してなる組換えプラスミドベクター (以下、pOSASA-1と称する) を作成し、これが前記した第3の発明での目的の新規な改変DNAの作製のための出発材料として適することを認めた。
【0053】
このような目的で新規な改変DNAを、前記の出発材料である組換えプラスミドベクターpOSASA-1から、PCR法に準じて作製するために本発明者らは研究した。そのPCR法のために適当に使用できるプライマーとして、4種類のプライマー、すなわち、後記の配列表の配列番号16に記載の塩基配列を有するオリゴヌクレオチドよりなるプライマーOSASN1と、配列番号17に記載の塩基配列を有するオリゴヌクレオチドよりなるプライマーOSASN2と、配列番号18に記載の塩基配列を有するオリゴヌクレオチドよりなるプライマーOSASC1と、配列番号19に記載の塩基配列を有するオリゴヌクレオチドよりなるプライマーOSASC2とを化学合成により作製した。
【0054】
上記の組換えプラスミドベクターpOSASA-1と、上記の4種の合成オリゴヌクレオチドよりなるプライマーとを利用して、後記の実施例2に詳述される方法を実施すると、第3の発明における改変DNAの具体例である配列番号12に記載のDNA、すなわち改変 「D配列」 を収得することに成功できたのである。
【0055】
次に、前記した第3の発明によるDNAの具体例である上記の改変D配列を含有するDNA断片を作製するのに好適に使用でき且つ後記の実施例2において例示される手順よりなるDNAの改変方法をここに要約的に説明する。
【0056】
(1) 前記した第1の発明のDNAのクローニング
配列表の配列番号1に記載される1734個の塩基からなる第1の発明のDNA、すなわち前記のOSASA-1配列を含有するDNA断片を、DNAリゲーションキットの利用により、ベクターp Bluescript II SK (+) のEcoRI切断部位の間に挿入、連結することによって、前記の組換えプラスミドベクターpOSASA-1を得る。このベクターpOSASA-1を大腸菌XLl-Blue MRF'に導入し、これで得られた形質転換大腸菌を増殖する。増殖された大腸菌細胞から、常法で抽出することにより組換えプラスミドベクターpOSASA-1を大量に収得する。この操作により第1の発明によるDNA配列、すなわちOSASA-1配列をクローニングする。
【0057】
(2) CR用プライマーの構築
DNA合成装置 (Model-391、アプライド バイオシステムズ社製) を利用して、下記の塩基配列を有する4種類のオリゴヌクレオチドをプライマーとして合成する。すなわち、下記のとおり、配列表の配列番号16に記載の塩基配列を有するオリゴヌクレオチドよりなるプライマーOSASN1と、配列番号17に記載の塩基配列を有するオリゴヌクレオチドよりなるプライマーOSASN2と、配列番号18に記載の塩基配列を有するオリゴヌクレオチドよりなるプライマーOSASC1と、配列番号19に記載の塩基配列を有するオリゴヌクレオチドよりなるプライマーOSASC2とを化学合成により作製する。
【0058】
(i) プライマーOSASAN1 (配列表の配列番号16に記載され且つ下記される塩基配列をもつプライマー):
5'-GAGTCAGTTGACGAAGCGTATGAGG-3'
(ii) プライマーOSASAN2 (配列表の配列番号17に記載され且つ下記される塩基配列をもつプライマー):
5'-GTACATTTGCTAACCCCTTTGAGG-3'
(iii) プライマーOSASAC1 (配列表の配列番号18に記載され且つ下記される塩基配列をもつプライマー):
5'-CAAAGGGGTTAGCAAATGTACGC-3'
(iv) プライマーOSASAC2 (配列表の配列番号19に記載され且つ下記される塩基配列をもつプライマー):
5'-GTTCAACGTTCATCAGTTTCTCCACC-3'
(3) 所要なDNA断片のPCR法による増殖と回収
所要なDNA断片を増殖して得るために、PCR法の第一段階として2つの反応、すなわち下記の (A) 反応と (B)反応とを行う。
【0059】
その(A)反応は、テンプレートとして用いられる前記の組換えプラスミドベクターpOSASA-1と、プライマーとして用いられる前記のプライマーOSASAN1 (配列番号16の合成オリゴヌクレオチド) と、プライマーOSASAC1 (配列番号18の合成オリゴヌクレオチドであり、5'-末端から8〜10番目のGTTが改変D配列の改変部AACを誘導できるプライマー) とを、PCR法用の通常の反応液 (Tris-HCl、MgCl2、KCl、4種のデオキシヌクチオチド三リン酸 (dNTP)、およびLaTaqDNAポリメラーゼを含有) に加え、次いで増殖反応を行うことから成る。
【0060】
この(A)反応によって、前記の改変D配列のうちの或る領域における塩基配列をもつDNA断片 (DNA断片-Aと称す) が増殖反応により生成される。
【0061】
また (B) 反応は、テンプレートとして用いられる前記の組換えプラスミドベクターpOSASA-1と、プライマーとして用いられる前記のプライマーOSASAC2 (配列番号19の合成オリゴヌクレオチド) と、プライマーOSASAN2 (配列番号17の合成オリゴヌクレオチドであり、5'-末端から12〜14番目のAACが改変D配列の改変AACを誘導できるプライマー) とを、(A) 反応で用いたと同じPCR法用の通常の反応液に加え、次いで増殖反応を行うことから成る。
【0062】
この(B)反応によって、前記の改変D配列のうちの或る領域における塩基配列を含有するDNA断片 (DNA断片-Bと称す) が増殖反応により生成される。PCR法による上記の増殖反応は、市販のPCR反応装置を用いて実施できる。
【0063】
増殖反応の終了後に、上記の (A) 反応の増殖反応液を低融点アガロース電気泳動により分画して、増殖生成物としての268bp (ベースペア) のDNA断片-Aを含むバンドをアガロースゲルから切出す。また、上記の (B) 反応の増殖反応液を同様に低融点アガロース電気泳動により分画し、そしてそのアガロースゲルから336bp (ベースペア) のDNA断片-Bを含むバンドを切出す。
【0064】
次いで、それら2つのゲル切片をDNA精製キット、例えばGenclean II (フナコシ社製) により精製することにより、DNA断片-Aの精製品と、DNA断片-Bの精製品とをそれぞれ回収する。
【0065】
さらに、PCR法の第2段階として、配列表の配列番号1に記載の塩基配列のうちの967番目のグアニンをアデニンに置換してなる塩基配列をもつDNA配列 (すなわち配列番号12の塩基配列をもつ第3の発明による改変D配列断片に相当する配列) の部分領域に相当する583bp (ベースペア) のDNA断片 (C断片) を作製する工程を行う。
【0066】
このためには、テンプレートとして用いられる前記の (A) 反応の増副生成物であるDNA断片-Aの精製品 (配列長268bp) と、(B) 反応の増副生成物であるDNA断片-Bの精製品 (配列長336bp) とを、PCR法用の通常の増殖反応液 (Tris-HCl、MgCl2、KCl、4種のdNTP、およびLa Taq DNAポリメラーゼを含有) に加え、次いで増殖反応を行う。その反応終了後に、その反応液を低融点アガロース電気泳動により分画し、そのアガロースゲルから、配列長583bpの所期のDNA断片 (DNA断片-Cと称す) を含有するバンドを切出す。
【0067】
そのゲル切片を、DNA精製キット、例えばGenclean IIキット (フナコシ社製) により精製して、DNA断片-Cの精製品を得る。このDNA断片-Cは、第3の発明による改変D配列のDNA断片の部分領域に相当する塩基配列をもち、且つ制限酵素AflIIおよびBglIIの切断部位を内部に含有する構造をもつものである。
【0068】
また、このDNA断片-Cは、制限酵素AflIIおよびBglIIで切断することにより目的とする塩基配列の置換部位を内部に含み、5'-末端に制限酵素AflII切断部位を、3'-末端に制限酵素BglII切断部位をもつ、全長288bpのDNA断片-αを得ることができる。
【0069】
(4) 改変D配列を含有するDNA断片のクローニング
次に、上記の(3)項で得られたDNA断片-Cを利用して、所望の改変D配列を含有するDNA断片を収得する。
【0070】
そのために、先ず、上記のDNA断片-Cを制限酵素AflIIと制限酵素BglIIで処理する。これによって、DNA断片-Cから、5'-側にAflII切断部位を、3'-側にBglII切断部位を有する改変D配列の一部分を有するDNA断片を切り取る。このようにして、目的の改変D配列に相当するDNA配列を含有するDNA断片の試料(i) が得られる。
【0071】
次に、配列表の配列番号1に記載のDNA (すなわちOSASA-1配列) を含むプラスミドベクターpOSASA-1を制限酵素AflIIと制限酵素BglIIで処理することにより、5'-側にBglII切断部位を有し且つ3'-側にAflII切断部位を有して、しかも配列番号1に記載のDNAの1番目の塩基から933番目までの部分領域と1220番目の塩基から1734番目の塩基までの部分領域を含むプラスミド断片 (ii) が選られる。
【0072】
ここで得られたAflII-BglII-プラスミド断片 (ii) を前記の改変D配列に相当するDNA配列を含有するDNA断片の試料 (i) と混合し、ついでDNAリゲーション・キットで連結反応にかけると、配列番号12の改変D配列を含有する組換えプラスミド (以下では、pBluescript-DNA-Dプラスミドと称す) を作製できる。
【0073】
この組換えプラスミドすなわちpBluescript-DNA-Dプラスミドを大腸菌XL1-Blue MRF'に導入して形質転換し、こうして得られた形質転換大腸菌 (エシエリヒア・コリXLl-Blue MRF'/pBluescript-DNA-Dと称する) (寄託番号 FERM-6451)を液体培地中で培養して増殖させる。大量に増殖された大腸菌細胞は、前記の組換えプラスミド、すなわちpBluescript-DNA-Dプラスミドのコッピイを含有する。このようにして改変D配列はクローニングできる。その大腸菌培養細胞から、常法により改変D配列を含有するプラスミドを抽出して取る。
【0074】
(5) 改変D配列のDNA断片の回収
前項 (4) で得られた改変D配列を含有するプラスミドを、次いで制限酵素EcoRIで処理することによって消化する。これによってEcoRI切断部位に隣接する5'-側端に塩基配列ATGを有し且つ3'-側端にもEcoRI切断部位を有する延伸部を有する改変D配列を含有するDNA断片を含有する消化反応液を得ることができる。
【0075】
この消化反応液を、低融点アガロース電気泳動により分画して、前記のDNA断片を含むバンドをアガロースゲルから切出す。切出されたアガロースゲル片をTEバッファーに溶解し、得られた溶液をフェノールで抽出すると、フェノール抽出液に前記のDNA断片が回収される。前記のDNA断片を含むフェノール抽出液を、3M酢酸ナトリウム水溶液およびエタノールを混合し、その混合物を20℃で約6時間放置し、さらに低温で遠心分離すると、前記のDNA断片が沈殿する。これを減圧下に乾燥すると、目的の改変D配列を内部に含有するDNA断片が粉末として得られる。この改変D配列を含有するDNA断片の粉末は水に可溶性である。
【0076】
以上の説明においては、第3の発明によるDNAの具体例である改変D配列を含有するDNA断片は、遺伝子工学の技法を利用する方法で作製された。しかしながら、配列表の配列番号12に記載の塩基配列を参照しながら、従来知られているポリヌクレオチド化学合成法によっても製造できる。
【0077】
なお、第1の発明による配列番号1のDNAの塩基配列のうちの967番目のグアニンをアデニンに置換して改変する場合について、第3の発明の実施法を説明した。しかしながら、第1の発明による配列番号1に記載のDNAを、テンプレートとして用い且つ適当に工夫された塩基配列を有する合成オリゴヌクレオチドの数種類を組み合わせてプライマーとして用いるならば、第1の発明による配列番号1のDNAの967番目の場所とは別の場所における塩基を別の塩基で取代えて改変された別の改変DNAを作製することが可能である。
【0078】
更に、本発明者らは別段の研究を進めた。その結果、植物体に外来遺伝子を導入して植物体を形質転換され、得られたトランスジェニック植物体で外来遺伝子を発現させるための従来知られたバイオテクノロジー技法を利用することによって、第1の発明によるイネのASAの第1アイソザイムのαサブユニットをコードする新規DNA(配列番号1)も、第2の発明によるイネASAの第2アイソザイムのαサブユニットをコードする新規DNA(配列番号10)も、第3の発明によるところの、イネのASAの第1アイソザイムのαサブユニットをコードするDNAから作成された前記の新規な改変DNA(配列番号12)も、組換えベクターに組込んだ後に植物体に導入することができ、しかも植物体内で発現できることが知見された。
【0079】
従って、第4の発明(本件特許請求の範囲の請求項3の発明)においては、イネのアントラニル酸シンターゼの第2アイソザイムのαサブユニットをコードする配列表の配列番号10のDNAを担持する組換えベクターを導入されて形質転換された植物細胞を有し、そして導入された前記DNAが発現可能であることを特徴とする、形質転換植物が提供される。
【0080】
しかも、前記の第4の発明による形質転換された植物体がこれを裁培すると種子を結実できる植物である場合、その植物を通常の条件で裁培して植物の種子を収獲できることが知見された。
【0081】
従って、第5の発明(本件特許請求の範囲の請求項4の発明)においては、イネのアントラニル酸シンターゼの第2アイソザイムのαサブユニットをコードする配列表の配列番号10のDNAを担持する組換えベクターを植物細胞に導入することによって得られ且つ前記のDNAが発現可能である形質転換植物を裁培し、さらにその裁培により結実した植物から採取された種子であることを特徴とする、形質転換植物の種子が提供される。
【0082】
また、第6の発明(本件特許請求の範囲の請求項5の発明)によると、配列表の配列番号10に記載の塩基配列を有するDNA配列を担うDNA断片が組み込まれた組換えベクターであって、宿主細胞中で該DNA配列を発現することができる組換えベクターが提供される。
【0083】
さらに、第7の発明(本件特許請求の範囲の請求項6の発明)においては、配列表の配列番号10に記載の塩基配列を有するDNA配列を担うDNA断片が組み込まれた組換えベクターであって、宿主細胞中で該DNA配列を発現することができる組換えベクターで形質転換された大腸菌が新規微生物として提供される。
【0084】
第7の発明による形質転換大腸菌を増殖させると、その中に含有された組換えベクターを大量にクローニングできる。第7の発明による大腸菌の例には、ブタベスト条約の規約下に受託番号FERM BP-6454で寄託された前記の大腸菌XL1-Blue MRF'(OS-asa-2) がある。
【0085】
前記の改変D配列(配列番号12)を担うDNA断片を保有する組換えベクターを導入された形質転換大腸菌の例は、ブダペスト条約の規約下に受託番号FERM BP-6451で寄託されてある前記の大腸菌XL1-Blue MRF'/p Bluescript-DNA-Dであることができる。
【0086】
前記した第1の発明によるDNA(配列番号1)、あるいは第2の発明によるDNA(配列番号10)を外来遺伝子として用いて植物を形質転換する場合には、形質転換できる植物の種類は多種多様であり、また植物の形質転換のために外来遺伝子としての本発明DNAを植物に導入する方法は、その目的で行われる従来公知のバイオテクノロジーの種々な技法であることができる。
【0087】
前記した第1の発明のDNAまたは第2の発明のDNAを外来遺伝子としてイネ植物に導入するために好適に実施できる且つ後記の実施例3に例示される方法を以下に要約的に説明する。
【0088】
(a) 外来遺伝子の導入用の組換えベクターの作成
従来知られているトウモロコシのユビキチンプロモーター、1stイントロンおよびNOSターミネーターと、ホスフィノスリシン耐性遺伝子、さらに微生物にのみに効果の発現するアンピシリン耐性遺伝子とを含有する既知のプラスミドベクターpUBA(Plant Molecular Biology, 第18巻4号、675頁-689頁、1992年) をバッファー中で制限酵素BamHIおよびSacIで処理すると、ユビキチンプロモーターの下流で、1stイントロンの下流のBamHI切断部位で切断され、且つNOSターミネーターの上流のSacI切断部位で切断された約4.8Kbのサイズのベクター断片を得ることができる。
【0089】
そのベクターDNA断片の水溶液を、本発明のDNAを含有するDNA断片の水溶液と混合し、その混合物をDNAリゲーションキットで処理して連結反応を行う。これによると、ベクターDNA断片のユビキチンプロモーターとNOSターミネーター領域の間に本発明のDNAを含有するDNA断片が挿入、連結されてある組換えベクターを得ることができる。
【0090】
選られた組換えベクターを大腸菌JM109に導入して、形質転換大腸菌を得る。
【0091】
得られた形質転換大腸菌を、抗生物質アンピシリン含有培地に接種、培養することにより、アンピシリン耐性の大腸菌コロニーを数個を得る。これらのコロニーをさらにアンピシリン含有の培地中でそれぞれ別々に増殖させる。
【0092】
各コロニーごとの増殖されたアンピシリン耐性の大腸菌から、それぞれにプラスミドを回収する。回収されたプラスミドは、挿入DNAの向きが相異なる種々なプラスミドを含む。各コロニーごとに回収されたプラスミドを、適当な制限酵素で消化して切り出された種々のDNA断片を含む消化反応液をアガロースゲル電気泳動にかけて、それらDNA断片の大きさ及び塩基配列を解析すると、組換えプラスミドのユビキチンプロモーターの下流に本発明のDNAを正常な向きで挿入、連結されている適当なプラスミド(約6.5Kbのサイズ) を選抜できる。
【0093】
前記した第1の発明の配列表の配列番号1のDNAが組み込まれているプラスミドをベクターpUBdW1と称し、第2の発明の配列表の配列番号10のDNA(本件特許請求の範囲の請求項2のDNA)が組み込まれているプラスミドをベクターpUBdW2と称し、さらには、第3の発明の配列表の配列番号12の改変D配列のDNAが組み込まれているプラスミドをベクターpUBdDと称する。
【0094】
また、アグロバクテリウム法に遺伝子導入用組換えベクターとしてハイグロマイシン耐性遺伝子を含有する既知のプラスミドベクターpIG121-Hm(Plant Cell Phisiol, 第31巻805頁〜813頁、1990年) をバッファー中で制限酵素PmeIおよびSacIで処理すると、約9.8Kbのサイズのベクター断片を得ることができる。
【0095】
また、上記したプラスミドベクターpUBdW1, pUBdW2またはpUBdDをバッファー中でSphIおよびSacIで処理し、SphI切断部位を平滑化処理することによりユビキチンプロモーター、1stイントロンの下流に本発明のDNAが連結したベクター断片を得ることができる。
【0096】
これらの本発明DNAが連結したベクター断片の各々を用いると、上記の方法と同じ方法により、連結反応、形質転換大腸菌の作出、プラスミドの回収を行うことにより本発明のDNAが正常な向きで挿入連結されているアグロバクテリウム法による遺伝子導入用の組換えベクターを得ることができる。
【0097】
第1の発明の配列表の配列番号1のDNAが組み込まれているプラスミドを、ベクターpUb-OSASAW1と称し、第2の発明の配列表の配列番号10のDNAが組み込まれているプラスミドを、ベクターpUb-OSASAW2と称し、さらには、第3の発明の配列表の配列番号12の改変D配列の断片のDNAが組み込まれているプラスミドを、ベクターpUb-OSASA1Dと称する。
【0098】
(b) イネのカルスの調製
イネの完熟種子から籾殻を除き、得られた外皮付きコメ種子をエタノール溶液で殺菌、次いで次亜塩素酸ナトリウムの希水溶液で殺菌し、さらに滅菌水で洗浄する。
【0099】
MS培地にショ糖、植物ホルモンとしての2,4-PA、寒天を加えてなる組成のカルス形成用培地に、前の外皮付きコメ種子を置く。28℃で1日当り15〜18時間にわたり、1500〜2500ルックスの大陽光を照射しながら40〜50日間、培養すると、イネのカルスが形成される。そのカルスを種子の胚乳部分から切取りカルスを得る。
【0100】
(c) イネ・カルス細胞への外来遺伝子の導入
前項(a) に記載したように作製された、本発明DNAを正常に挿入、連結してある組換えベクター(すなわち、配列番号1のDNAを保有する前記のベクターpUb-OSASAW1、もしくは配列番号10のDNAを保有するベクターpUb-OSASAW2または配列番号12のDNAを保有するベクターpUb-OSASA1D) を、公知のアグロバクテリウム法によりイネのカルス細胞へ導入するために、まず宿主であるアグロバクテリウム・ツメファシエンス(Agrobacterium tumefaciens) への組み込みを行う。組み込みは公知のエレクトロポレーション法(植物組織培養、第10巻2号、194頁-196頁、1993年)により行う。
【0101】
前記(b) に示されたようにして得られたイネのカルス細胞と、このようにして本発明DNAを組み込んで得られたアグロバクテリウム細菌とを公知の方法(細胞工学別冊、モデル植物の実験プロトコール、93頁〜98頁、1996年、秀潤社発行) にしたがって、共存培養を行うと、イネカルス細胞への本発明のDNAを導入できる。そして、ハイグロマイシンに耐性で、本発明のDNAが形質転換されたイネ植物細胞が得られる。
【0102】
(d) 形質転換植物細胞の再選抜
上記のように得られたハイグロマイシン耐性の形質転換イネ植物細胞の中から、本発明DNAを外来遺伝子として十分に有効な量で含有する形質転換イネ植物細胞のみを再選抜する。
【0103】
このために、前者の形質転換細胞を、N6培地にショ糖と、2,4-PAと、ゲルライトと、トリプトファンのアナログ体である、5MT(すなわち5-メチルトリプトファン) とを添加してなる再選抜用培地上に移植する。
【0104】
移植された細胞を、1日当たり16時間、2000ルックスの光で照射しながら25〜28℃で25〜30日間培養する。
【0105】
本発明DNAを外来遺伝子として十分に有効な量で含有する形質転換イネ植物細胞は、培地中に細胞増殖阻害剤として作用する5MTが存在していても5MT耐性であって、生育できる。5MT添加の上記培地で生育できた5MT耐性の培養植物細胞を選抜する。
【0106】
(e) 再選抜された5MT耐性の形質転換イネ植物細胞からの植物体の再生
上記のように再選抜された5HT耐性の培養植物細胞を、植物組織培養用のMS培地にショ糖、植物ホルモンとしてのベンジルアデニン、ナフタレン酢酸、ゲルライトを添加してなる植物体再生用の分化培地に移植する。
【0107】
移植された細胞を、2000ルックスの光を1日当り16時間照射しながら25〜28℃で25〜30日間培養する。このようにすると、培養された形質転換イネ植物細胞から、分化により、芽と根が再生できる。
【0108】
再生した芽と根を含有する幼芽が長さ10〜30mmに生育した後に、その幼芽を、MS培地にショ糖とゲルライトを添加してなる順化用の培地に移植する。さらに、1日当り16時間、2000ルックスの光を照射しながら25〜28℃で18〜20日間育成する。
【0109】
このようにして、形質転換イネ植物が再生できる。こうして得られた植物体は、温室内で土壌に移植して通常の条件下で裁培すると、順長に生育して3〜6ケ月の裁培でイネ種子を結実できる。
【0110】
(f) 導入した外来遺伝子の確認
上記のように再生された形質転換イネ植物から緑色の葉を採取する。採取された葉を液体窒素で凍結した後に破砕する。その葉破砕物から、J.Sambrookらの方法(Molecular Cloning、第2版、Cold Spring Habor Laboratory Press、1989年に記載) に準じてDNAを抽出する。
【0111】
他方、後記の配列表の配列番号14に記載される塩基配列を有するオリゴヌクレオチドと、配列番号15に記載の塩基配列を有するオリゴヌクレオチドを化学合成して、プライマー用に用いる。
【0112】
再生イネ植物体から上記のように抽出されたDNAをテンプレートとして用い且つ上記の2種の合成オリゴヌクレオチドをプライマーとして用いて、公知のPCR法によって前記DNAを増幅する。得られた増幅反応液を常法によりアガロース電気泳動にかけて分画し、再生イネ植物から抽出されたDNAの種々のDNA分画のうちで、導入された外来遺伝子のDNAに相当するDNA断片を含むゲルバンドを採る。
【0113】
このバンドに含有されたDNA断片の塩基配列を公知のサザン法によって解析すると、本発明DNAに相当するか否かを確認できる。
【0114】
植物中のトリプトファンの抽出および含有量の測定は、公知の方法(Hopkins-Cole法、生化学実験講座第11巻、東京化学同人社刊) によって行うことができるが、後記の実施例に挙げたようなHPLCを用いる方法によっても行うことができる。このトリプトファンの抽出および含有量の測定は、対象とする植物種、対象とする植物の部位、対象とする植物の生育ステージに併せて、適宜変更することができる。
【0115】
次に、本発明のDNAの導入により形質転換された植物細胞の選抜方法および形質転換植物の作成方法を説明する。
【0116】
(a) 選抜用の組換えベクターの作成
形質転換植物の作出のために、植物細胞への直接的導入法に用いられる組換えベクターとして、従来知られているカリフラワーモザイクウイルスの35Sプロモーターと、NOSターミネーターと、微生物にのみに効果の発現するアンピシリン耐性遺伝子とを含有する既知のプラスミドベクターpBI221(Clontech社製) を、バッファー中で制限酵素XbaIおよびSacIで処理すると、35Sプロモーターの下流のXbaI切断部位で切断され、且つNOSターミネーターの上流のSacI切断部位で切断された約3.8Kbのサイズのベクター断片を得ることができる。
【0117】
そのベクターDNA断片の水溶液を、第3の発明のDNA(改変D配列) を含有するDNA断片の水溶液と混合し、その混合物をDNAリゲーションキットで処理して連結反応を行う。これによると、ベクターDNA断片の35SプロモーターとNOSターミネーター領域の間に第3の発明のDNA(改変D配列) を含有するDNA断片が挿入、連結されてある組換えベクターを得ることができる。
【0118】
選られた組換えベクターを大腸菌JM109に導入して、形質転換大腸菌を得る。
【0119】
得られた形質転換大腸菌を、抗生物質アンピシリン含有培地に接種、培養することにより、アンピシリン耐性の大腸菌コロニーを数個を得る。これらのコロニーをさらにアンピシリン含有の培地中でそれぞれ別々に増殖させる。
【0120】
各コロニーごとの増殖されたアンピシリン耐性の大腸菌から、それぞれにプラスミドを回収する。回収されたプラスミドは、挿入DNAの向きが相異なる種々なプラスミドを含む。各コロニーごとに回収されたプラスミドを、適当な制限酵素で消化して切り出された種々のDNAを含む消化反応液をアガロースゲル電気泳動にかけて、それらDNA断片の大きさ及び塩基配列を解析すると、組換えプラスミドの35Sプロモーターの下流に第3の発明のDNA(改変D配列) を正常な向きで挿入、連結されている適当なプラスミド(約5.6Kbのサイズ) を選抜できる。また、形質転換植物の作出のために、植物細胞への直接的導入法に用いられる組換えベクターとして前記のベクターpUBdW1あるいはpUBdW2も、またはアグロバクテリウム法に用いられる組換えベクターとして前記のベクターpUb-OSASA1Dも選抜用の組換えベクターとして用いることができる。
【0121】
(b) イネのカルスの調製
イネの完熟種子から籾殻を除き、得られた外皮付きコメ種子をエタノール溶液で殺菌、次いで次亜塩素酸ナトリウムの希水溶液で殺菌し、さらに滅菌水で洗浄する。
【0122】
MS培地にショ糖、植物ホルモンとしての2,4-PA、寒天を加えてなる組成のカルス形成用培地に、前の外皮付きコメ種子を置く。28℃で1日当り15〜18時間にわたり、1500〜2500ルックスの大陽光を照射しながら40〜50日間、培養すると、イネのカルスが形成される。そのカルスを種子の胚乳部分から切取りカルスを得る。
【0123】
(c) イネ・カルス細胞への選抜用組換えベクターの導入
前項(a) に記載したように作製された本発明DNAを正常に挿入、連結してある選抜用組換えベクターのpUBdW1あるいはpUBdW2を公知のウイスカーを用いる直接的導入法によりカルス細胞へ導入を行う。また、選抜用組換えベクターのpUb-OSASA1Dを公知のアグロバクテリウム法(細胞工学別冊、モデル植物の実験プロトコール、93頁〜98頁、1996年、秀潤社発行) によりカルス細胞へ導入を行う。
【0124】
(d) 形質転換イネ植物細胞の選抜
上記のように得られた選抜用組換えベクターpUBdW1またはpUBdW2を含有するカルス細胞を、N6培地にショ糖、2,4-PA、ゲルライトおよび選抜薬剤としてトリプトファン類縁化合物を10mg/L〜200mg/L〜200mg/L、好ましくは30mg/L〜50mg/Lを添加してなる選抜用培地の上に均一に広げて加える。さらに、暗所もしくは2000ルックスの光を1日当たり16時間照射しながら25〜28℃で20〜60日間、好ましくは25〜30日間培養する。
【0125】
こうしてトリプトファン類縁化合物に耐性であり且つ選抜用組換えベクターで形質転換された植物細胞が選抜される。
【0126】
(e) 形質転換植物の選抜
上記のように得られたトリプトファン類縁化合物耐性の形質転換イネ植物細胞から、形質転換イネ植物を得るために、前者の形質転換細胞を、MS培地にショ糖、植物ホルモンとしてベンジルアデニン、ナフタレン酢酸、ゲルライトおよび選抜薬剤としてトリプトファン類縁化合物を10mg/L〜200mg/L、好ましくは30mg/L〜50mg/L添加してなる植物体再生用の選抜分化培地に移植する。
【0127】
移植された細胞を、2000ルックスの光を1日当り16時間照射しながら25〜28℃で25〜30日間培養する。このようにすると、培養された形質転換植物細胞から、分化により、芽と根が再生できる。
【0128】
再生した芽と根を含有する幼芽が長さ10〜30mmに生育した後に、その幼芽を、MS培地にショ糖とゲルライトを添加してなる順化用の培地に移植する。さらに、1日当り16時間、2000ルックスの光を照射しながら25〜28℃で18〜20日間育成する。このようにして、形質転換イネ植物が再生できる。
【0129】
本発明におけるDNAは、外来遺伝子として前記したイネ植物のみならず、他の種類の植物に導入して植物の形質転換を行うことに使用できる。
【0130】
また、本発明のDNAすなわち、前記の第1の発明のDNA(配列番号1のDNA)あるいは第2の発明のDNA(配列番号10のDNA)は、前記したイネ植物のトリプトファン含有量の増加および選抜のみならず、他の種類の植物に対しても使用できる。
【0131】
従って、本発明のDNAを一般の植物体に導入できる方法、またトリプトファン含有量を増加させる方法、さらに形質転換細胞および形質転換植物を選抜する方法を次に一般的に説明する。
【0132】
本発明のDNAを導入できる植物は、特に限定される種類のものではなく、例えば、イネ、トウモロコシ、コムギ、オオムギ、およびその他の単子葉植物、ならびにタバコ、ダイズ、ワタ、トマト、ハクサイ、キュウリ、レタスなどの双子葉植物などが挙げられる。先づ、これらの植物から培養細胞を調製し、得られた培養細胞に本発明DNAを外来遺伝子として導入するのが都合よい。
【0133】
本発明のDNA導入に用いられる培養細胞の作製は、植物由来のいかなる外殖物であってもよく、例えば、胚盤、生長点、花粉、葯、葉身、茎、葉柄、根由来の外殖物を使用できる。
【0134】
上記した外殖片を、カルス形成用の培地、例えばMS培地(Murashigeら、「Physiologia Plantarum」、1962年、第15巻、473頁〜497頁)、またはR2培地(Ojimaら、「Plant and Cell Physiology」、1973年、第14巻、1113〜1121頁、あるいはN6培地(Chuら、1978年、「In Proc. Symp. Plant Tissue Culture, Science Press Peking」、43頁〜50頁などの如く、必須成分としての無機塩成分およびビタミン類を含む植物組織培養用の培地に、植物ホルモンとして、例えば、2,4-PA(2,4-dichlorophenoxyacetic acid) 0.1〜5mg/リットル、ならびに、炭素源として、例えば、ショ糖10〜60g/リットル、ゲルライト1〜5g/リットルを添加してなる培地に置床して培養することにより得られる培養細胞を用いて、これに本発明DNAを導入するのが便利である。
【0135】
本発明のDNAの導入の対象となる植物細胞は、例えば、カルスや懸濁細胞などの脱分化した培養細胞あるいは不定胚、苗条原基などの培養細胞、もしくは植物組織例えば葉、根、茎、胚、生長点などの細胞から作られたカルス細胞および懸濁細胞であるのが好ましい。
【0136】
本発明のDNAの導入用の培養細胞を調製するために、カルス形成用培地に外殖片を置床する場合、本発明DNAの導入用の培養細胞が得られるまでの培養期間は特に限定されるものではない。しかし、形質転換植物を再生することが必要であるから、当該の培養細胞からの植物体再生が可能であること、つまり、当該植物細胞が植物体再生能力を保有している期間内に得られた培養細胞を用いることが重要である。
【0137】
また、本発明のDNAの導入用の培養細胞は、植物体再生能力を保有している培養細胞であれば、液体培地中で培養された懸濁細胞であってもよい。
【0138】
本発明のDNAを植物細胞に導入するためには、先づ、本発明のDNAを発現ベクターに組込んだ組換えベクターを調製することが必要である。その組換えベクターは、植物体に導入後の本発明DNAが植物体中で発現するように、発現プロモーターの下流に本発明DNAを配し、さらに本発明のDNAの下流にターミネーターを配するように調整された組換えベクターであることを要する。この目的に用いられる組換えベクターは、植物への導入方法の種類に応じて、通常の植物形質転換に利用される各種のベクターであることができる。例えば、エレクトロポレーション法、パーティクルガン法、ウイスカー法などによる植物細胞への直接的なDNA導入法を用いる場合は、pUC系またはpBR322系などの大腸菌で増殖可能であるプラスミドベクターを利用するのがよく、またアグロバクテリウム法を用いる場合はplan系などのプラスミドベクターを利用するのがよい。
【0139】
組換えベクター中で本発明DNAの上流に配されるプロモーターは、例えばカリフラワーモザイクウイルス由来のCaMV35S「The EMBO Journal」、第6巻、第3901頁〜3907頁、1987年、または、特開平6-315381号公報」、トウモロコシのユビキチンプロモーター[特開平2-79983号公報]、ファゼオリンプロモーター「Plant Cell、第1巻、第839頁〜853頁、1989年] であることができる。また、本発明DNAの下流に配されるターミネーターは、例えばカリフラワーモザイクウイルス由来のターミネーター、あるいはノパリン合成酵素遺伝子由来のターミネーター[The EMBO J.第6巻、第3901頁〜3907頁、1987年]であるのがよい。しかし、植物体中で機能するプロモーターやターミネーターであれば、何れでもよい。
【0140】
また、本発明DNAの導入で形質転換された植物細胞を効率的に選択するためには、用いる上記の組換えベクターは、適当な選抜マーカー遺伝子を含有するプラスミドベクターと共に植物細胞に導入されることが好ましい。この目的に使用する選抜マーカー遺伝子は、抗生物質ハイグロマイシンに耐性であるハイグロマイシンフォスフォトランスフェラーゼ遺伝子、あるいはカナマイシン、ゲンタマイシンに耐性であるネオマイシンフォスフォトランスフェラーゼ遺伝子、もしくは除草剤フォスフィノスリシンに耐性であるアセチルトランスフェラーゼ遺伝子[The EMBO Journal、第6巻、第2513頁〜2518頁、1987年、または特開平2-171188号公報] であることができる。
【0141】
本発明DNAを外来遺伝子として植物に導入する方法は、アグロバクテリウム法[Bio/technology、第6巻、第915頁〜922頁、1988年]、エレクトロポレーション法[Plant cell Rep., 第10巻、第106頁〜110頁、1991年]、パーティクルガン法[Theor. Appl. Genet., 第79巻、第337頁〜341頁、1990年]、ウイスカー法であることができ、しかし特にこれらに限定されない。
【0142】
次に、本発明DNAを含有する組換えベクターを十分に有効な量で含有する形質転換細胞を効率よく再選抜するために、細胞増殖阻害剤であるトリプトファンのアナログ体を添加した培地上で培養する。
【0143】
トリプトファンアナログとしては、例えば5-メチルトリプトファン(5MT) を、10mg/l〜1000mg/l、好ましくは20mg/l〜100mg/lの濃度で再選抜用培地に添加できる。
【0144】
前記のように再選抜された本発明DNAを外来遺伝子として組込まれた組換えベクターと選抜マーカー用のベクターとを含有する形質転換植物細胞から、次に、植物体を再生する。植物体の再生は公知の要領で行うことができる。例えば、前記のように再選抜された形質転換植物細胞を、公知の植物体再生用培地に置床して培養することにより植物体の再生を行うことができる。
【0145】
植物体再生用の培地に置床された形質転換細胞は15〜30℃、好ましくは、20〜28℃の温度で500〜2000ルックス、好ましくは、800〜1000ルックスの光を照射しながら培養期間20〜60日間、好ましくは30〜40日間で培養する。
【0146】
これによって、個々の植物細胞から本発明DNAよりなる外来遺伝子を担う組換えベクターが導入されて形質転換された植物体が再生できる。
【0147】
形質転換細胞から再生された植物体は次に順化用培地で培養する。その後、順化された再生植物体を、温室内で通常の裁培条件下で育成すると、3〜6カ月の裁培で植物体は成熟し結実して種子を得ることができる。
【0148】
なお、このように再生され且つ裁培された形質転換植物体中の導入された外来遺伝子の存在は、公知のPCR法とサザン法(Southern, 「J. Mol. Biol.,」 98巻503-517頁、1975年) によって、植物体中のDNAの塩基配列を解析することによって確認できる。
【0149】
この場合、形質転換植物体からのDNAの抽出は、公知のJ. Sambrookらの方法(「Molecular Cloning」第2版、Cold Spring Habor Laboratory Press, 1989年) に準じて実施できる。
【0150】
再生された植物体中に存在する本発明DNAよりなる外来遺伝子をPCR法を用いて解析する場合には、上記のように再生植物体から抽出されたDNAを、テンプレートとして用い本発明によるDNA、あるいは本発明による改変DNAの塩基配列に従って適当に選択された塩基配列をもつ合成したオリゴヌクレオチドをプライマーとして用い、これらを混合してPCR法用反応液中に加えて増幅反応を行う。この増幅反応においては、DNAの変性、アニーリング、伸張反応を数十回繰り返えすと、本発明DNA配列を含むDNA断片の増幅生成物を得ることができる。
【0151】
増幅生成物を含む反応液を例えばアガロース電気泳動にかけると、増幅された各種のDNA断片が分画される。導入した外来遺伝子である本発明DNAに相当するDNA配列を含有すると認められるDNA断片を含むバンドアガロースゲル片を切出す。切出されたゲル片内に含有されるDNA断片のDNA配列の塩基配列をサザン法で解析すると、そのDNA配列が本発明DNAに対応するか確認できる。
【0152】
本発明の形質転換細胞の選抜および形質転換植物の選抜に用いることのできる選抜用薬剤としてトリプトファン類縁化合物が挙げられるが、具体的には、5-メチルトリプトファン(5MT)、4-メチルトリプトファン(4MT)、6-メチルトリプトファン(6MT)、7-メチルトリプトファン(7MT)、6-メチルアントラニル酸(6MA)、5-メチルアントラニル酸(5MA)、3-メチルアントラニル酸(3MA)、5-フルオロアントラニル酸(5FA)、6-フルオロアントラニル酸(6FA) などのトリプトファン類縁化合物の生合成中間体も使用できる。
【0153】
前述したように、第1の発明または第2の発明による新規なDNAは、外来遺伝子として、植物または植物細胞に導入してトリプトファン含量の増強された植物または植物細胞の形質転換に利用できる。
【0154】
従って、第8の発明(本件特許請求の範囲の請求項7の発明)では、アントラニル酸シンターゼの第1または第2アイソザイムのαサブユニットのタンパク質をコードして且つ配列表の配列番号1または配列番号10の塩基配列を有するDNAを担持する組換えベクターであって、しかも該DNAが植物中で発現可能である組換えベクターを、植物細胞カルスに導入し、このことによって、該DNAで形質転換された植物細胞カルスを収得し、さらに該カルスを植物体に再生することからなる、植物のトリプトファン含有量の増加方法が提供される。
【0155】
なお更に、本発明者らは、イネのASAの第1アイソザイムのαサブユニットをコードする遺伝子の発現のために有用なプロモーターDNAを単離する目的で別途の研究を行った。その研究の結果、下記に説明される手順によって、イネのASAの第1アイソザイムのαサブユニットの遺伝子DNA、もしくはその改変DNAを発現するのに有効なプロモーターDNA配列を内部に含有するDNA断片を収得することに成功した。該DNA断片を収得する操作手順を以下に簡略に説明する(手順の詳細は後記の実施例5を参照)。
【0156】
(a) イネのゲノミックDNAの調製
イネ(Oriza sativa)の茎葉、根、カルスなどの組織、好ましくは茎葉またはカルスから、常法によりゲノミックDNAを抽出する。抽出したゲノミックDNAから蛋白質などの夾雑物を除き、さらに超遠心分離によりゲノミックDNAの精製品を得る。
【0157】
(b) イネのゲノミックDNA断片の作成
上記で得られたゲノミックDNA (精製品) を制限酵素 EcoRIで部分消化する。得られたDNAの部分消化物をアガロースゲルにて電気泳動する。電気泳動により分画されたDNA断片をナイロン膜ハイボンドNに転写する。転写されたDNAは変性処理により、ナイロン膜に固定する。
【0158】
このようにナイロン膜に固定されたDNAの各分画に対して、後記の実施例1の(5) でアラビドプシスから作られたDIGラベルされた標識プロープDNAをハイブリダイゼーション反応させる。ナイロン膜上のDNAのサイズ6kb付近に、シグナルを発するDNA分画が検定できる。このシグナルを発したDNA分画に相当するアガロースゲル中のDNA分画を、上記のアガロース中で制限酵素RcoRIで部分消化してから、該ゲルから切出す。切出されたDNAを精製し、得られたDNA断片 (精製品) をTEバッファーに溶解すると、こうして、分画ゲノミックDNAが得られる。
【0159】
(c) イネの分画ゲノミックDNAライブラリーの構築
得られたゲノミックDNAをファージベクターに連結し、得られた組換えベクターをλファージにパッケージする。こうして得られた組換えλファージを大腸菌に感染させて増殖させると、前記の組換えλファージの多量が得られる。この多量の組換えλファージをイネの分画ゲノミックDNAライブラリーとして利用する。
【0160】
(d) イネのゲノミックDNAライブラリーからのプロモーター遺伝子の選択
上記で構築されたイネのゲノミックDNAライブラリーである組換えファージを用い、また実施例1の(5)でアラビドプシスから作られたDIGラベルした標識プローブDNAを利用することによりプラーク・ハイブリダイゼーション法によって、イネのASA遺伝子のためのプロモーターの遺伝子に相当するDNA配列を含有する組換えファージを、前記ゲノミックDNAライブラリーからスクリーニングできる。このスクリーニングにより、該ライブラリーの10万個のファージプラークから、ASA遺伝子のプロモーター遺伝子が組込れたと判定できたファージプラーク3個を単離することに幸にも成功した。
【0161】
これら3個のプラークを別々に制限酵素RcoRIで消化して、DNAのEcoRI-DNA断片を含む消化液を得る。
【0162】
(e) ゲノミックDNAのクローニング
上記で得られたEcoRI-DNA断片を、プラスミドベクターp Bluecsript II SK(+)のEcoRI切断部位に連結する。得られた組換えプラスミドベクターを大腸菌に導入して、クローニングする。大腸菌から組換えプラスミド・ベクターのクローンを分離する。
【0163】
(f) クローニングされたプラスミドDNAのシークエンス解析
上記のように得た組換えプラスミド・ベクターのクローンを制限酵素RcoRIと制限酵素BamHIで消化する。こうして得たEccoRI-BamHI断片を塩基配列の解析にかける。
【0164】
上記のようにして得られたゲノミックDNAのクローンから、イネのASAの第1アイソザイムのαサブユニットの遺伝子の発現のためのプロモーター領域のDNA配列を内部に含有するDNA断片(DNA断片Zとここで仮称する) を、DNA断片の塩基配列を参考にして、分離できた。
【0165】
このようにして得られたプロモーターDNAを内部に含有するDNA断片Zの全体の塩基配列を、常用のシークエンシング・キットにより決定し、その決定された全体の塩基配列は、配列表の配列番号3に記載の塩基配列であると認められた。このDNA断片の内部に含有されたプロモーターDNAは、アビドプシスの第1アイソザイムのαサブユニットの遺伝子のプロモーター領域のDNAについて既知である塩基配列を参考にして、配列表の配列番号7に記載された塩基配列を有すると認められた。
【0166】
このDNA断片Zの塩基配列の全部またはそのうちの一部は、後記の実施例5の試験からプロモーター活性を有することが確認されている。
【0167】
配列表の配列番号7に記載の塩基配列をもつプロモーターDNAを内部に含有するDNA断片ZをpBluescript II SK(+) プラスミドベクターにクローニングされ、こうして得られた組換えベクターを導入した大腸菌XLl-Blue MRF'は、Escherichia coli(Os-asa#7) と命名され、工業技術院生命工学工業技術研究所に1997年8月18日より、受託番号FERM P-16387として寄託されている。また、1998年8月7日以降はブダペスト条約の規約下にFERM BP-6452の受託番号で寄託されている。
【0168】
(g) プロモーター活性の検定
前記の得られたイネのゲノミックDNAクローンから分離された前記のDNA断片Zに含有されるところの、ASAの第1アイソザイムのαサブユニットの遺伝子のプロモーター領域について、そのプロモーターとしての機能を確認するために、その活性の検定試験を行った。すなわち、前記のDNA断片Zを、レポーター遺伝子であるβ-グルクロニダーゼ(GUS) 遺伝子を担持する市販のpBI101プラスミドベクター(Clontech社製) の制限酵素切断部位に挿入することによって、組換えプラスミドベクターを作成した。この組換えプラスミドベクターを常法によって植物細胞、例えばイネ培養細胞へ導入して、市販のGUS活性測定キットにより、該プロモーター領域がGUS活性の発現に有効であることを確認することができる。
【0169】
第1の発明によるイネASAのアイソザイムのαサブユニットの遺伝子のDNA、ならびにそのプロモーター配列は、本発明を完成するに際しては、上記のようにしてcDNAライブラリー又はゲノミックDNAライブラリーから単離されたものである。本発明により、それらプロモーターのDNAの塩基配列が明らかにされたので、後記の配列表に示す塩基配列に基づいて化学合成することによっても取得できる。また、前記の塩基配列に基づいて作製した合成オリゴヌクレオチドプライマーを用い、公知の方法でイネのcDNAライブラリー又は染色体DNAライブラリーから、PCRにより取得することもできる。
【発明を実施するための最良の形態】
【0170】
以下に、本発明を実施例を参照して具体的に説明するが、本発明はこれら実施例に限定されるものではない。
【0171】
なお、以下の実施例で行われる実験操作の手順は、特に記述しない限り、「Molecular Cloning」 第2版 (J. Sambrookら、Cold Spring Habor Laboratory press, 1989年発行)に記載される方法に従った。
【0172】
実施例1
本実施例はイネのアントラニル酸シンターゼ(ASA) の第1のアイソザイム(ASA1) のαサブユニットをコードする遺伝子と、第2のアイソザイム(ASA2) のαサブユニットをコードする遺伝子(すなわち、本件特許請求の範囲の請求項1または2のDNA)とを単離する方法を例示する。
【0173】
(1) イネのmRNAの調製
イネ(品種: 日本晴) の種子を播種した。その後、栽培7日目の幼植物体の茎葉2gを液体窒素で凍結した。凍結した茎葉を乳鉢内で粉砕した。その粉砕物から、公知のAGPC法(Acid Guanidinium thiocyanate-phenol-chloroformを使用する方法)(実験医学、Vol. 9, No. 15 (11月号)、第99頁〜第102頁、1991年) に従い、全RNAの約2mgを抽出した。次に、上記で得られた全RNAから、Pharmacia Biotech社製のmRNA精製キット(mRNA Purification Kit) を用いてmRNAを単離した。こうしてイネの全mRNAの約30μgを得た。
【0174】
(2) イネのcDNAライブラリーの構築
上記(1) で得られたイネの全mRNAから、DNA合成キット(Pharmacia Biotech社製のTimeSaver cDNA Synthesis Kit) によりイネの全cDNAを得た。
【0175】
この全cDNAを、EcoRI切断末端が仔ウシ小腸由来アルカリホスファターゼで処理されたλgt11ファージ・ベクター(Lambda gtll/EcoRI/CIAP-Treated Vector Kit (STRATGENE社製)) であるファージベクターに連結した。こうして得られた組換えベクターを、イン・ビトロパッケージングキット(Gigapack II Gold Packaging Extract) によりラムダファージにパッケイジングした。
【0176】
組換えベクターをパッケージした組換えラムダファージを大腸菌Y1088に感染させて増殖させた。増殖された大腸菌の溶菌プラークとして、多数の前記組換えラムダファージが得られた。該プラーク中の組換えラムダファージは、イネ由来の全cDNAを含有する多種多様のファージから成るものであり、これら多種多様のファージをイネのcDNAライブラリーとして利用した。
【0177】
(3) PCR法用のプライマーの構築
イネのアントラニル酸シンターゼ(ASA) の第1のアイソザイム(ASA1) と第2のアイソザイム(ASA2) のそれぞれのαサブユニットをコードするcDNA断片をクローニングするのに用いるDNAプローブをPCR法用に作製する。このために、まずプライマーの設計を行った。そのためには、まずアラビドプシス(和名: シロイヌナズナ) のアントラニル酸シンターゼの第1のアイソザイムと第2のアイソザイムとのそれぞれのαサブユニットをコードする遺伝子の既知の塩基配列および既知のアミノ酸配列を参考にして、下記に示す塩基配列を有する2種類のオリゴヌクレオチドを、プライマーNo.1とプライマーNo.2として化学合成により作製した。
【0178】
プライマーNo.1(配列表の配列番号8) に示す塩基配列を有するオリゴヌクレオチド):
5'-CATATGTCTTCCTCTATGAAC-3'
プライマーNo.2(配列表の配列番号9) に示す塩基配列を有するオリゴヌクレオチド):
5'-GGATCCTCATTTTTTCACAAATGC-3'
なお、上記の2種類のオリゴヌクレオチドの作製は、DNA合成装置(Model 391、アプライドバイオシステムズ社製) を用いてオリゴヌクレオチドを合成し、さらにその合成品をイオン交換HPLCで精製することにより行った。
【0179】
(4) プローブDNAの作製
次に、上記のように構築された2種類の合成オリゴヌクレオチドの各10p.Mを第1のプライマーおよび第2のプライマーとして用い、またアラビドプシスの市販されるcDNAライブラリー(STRATAGENE社製) の1μlをテンプレイトとして用いた。第1および第2プライマーならびにアラビドプシスのcDNAライブラリーを、PCR法用の増幅反応液(10mMのTris-HCl(pH 8.3)、1.5mMのMgCl2、50mMのKCl、0.001%ゼラチン、pH 8.3; 4種のヌクレオチドdNTPの各2.5mMの混合物、およびDNAポリメラーゼTakara Ex Taq、2.5単位) 50μlに加えて、DNAの増幅反応を行った。ここで使用される増幅反応液は、PCRキット(PCR Amplification Kit(宝酒造(株)社製) により調製した。
【0180】
PCR法によるDNAの上記の増幅反応は、PCR反応装置(PERKIN ELMER社製、DNA、Thermal Cycler 480) を用いて、変性を94℃、30秒間、アニーリングを55℃、1分間、また伸張(extention) を72℃、2分間行う3つの反応操作を35回繰り返すことによって実施した。
【0181】
上記のようにすると、アラビドプシスASAの2つのアイソザイムのそれぞれのαサブユニットをコードする遺伝子に相当するDNA配列のうちの一部分を構成しているDNA断片の増幅生成物が生成される。これらDNA断片の増幅生成物をプローブDNAとして採取する。そして、該プローブDNAをイネのcDNAライブラリーのクローニングに下記の通りに用いた。
【0182】
(5) イネのcDNAライブラリーから、イネのASAの2つのアイソザイム(ASA1およびASA2) のαサブユニットをコードする遺伝子のDNAの選択
上記(2)で構築されたイネのcDNAライブラリーである組換えラムダファージから、上記(4)でアラビドプシスのDNAライブラリーから作製したプローブDNAの利用により、イネのASAの2つのアイソザイムのαサブユニットをコードする遺伝子に対応するDNA配列を組込まれた組換えラムダファージをプラーク・ハイブリダイゼーション法により、スクリーニングにより採取する。
【0183】
このスクリーニングの目的のためには、上記(2)で得られたイネcDNAライブラリーである組換えラムダファージのプラークを、複数のシャーレ内の1.5%寒天培地上に形成させた。それらファージプラークを複数枚のナイロン膜(アマシャム社製) ハイボンドNに転写した。こうしてナイロン膜に転写されたファージのプラークに含まれているファージDNAは、アルカリ変性液(1.5M NaCl、2.0M NaOH) および中和液(1.0M Tris-HCl pH5、2.0M NaCl) でそれぞれ10分間処理し、その後にUV照射によりナイロン膜上に固定した。
【0184】
次に、上記(4)でアラビドプシスのcDNAライブラリーから得られたプローブDNAをジゴキシゲニン(DIG)でラベルして標識プローブDNAを得た。その標識プローブを前記のファージDNA(すなわち、イネのcDNAライブラリー) を固定したナイロン膜に対してプラーク・ハイブリダイゼーションさせた。該プローブDNAの前記ラベリングはDIG-ELISA DNA Labeling & Detection Kit(ベーリンガー・マンハイム社製) により行った。
【0185】
このプラーク・ハイブリダイゼーションの場合、前記のファージDNAが固定されたナイロン膜をハイブリダイゼーション溶液(500mM Na-Piバッファー、pH7.2、7%SDS、1mM EDTA) に浸し、さらに65℃、10分間、浸漬処理を行った。次に前記のDIGラベルした標識プローブDNA(10ng/ml)を加え、65℃で、15時間インキュベートすると前記のプラーク・ハイブリダイゼーション反応が行われた。
【0186】
その反応終了後に、洗浄液(40mM Na-Piバッファー、pH7.2、1%SDS) で前記のナイロン膜を20分間ずつ3回洗浄した。その後、上記のDIG-ELISA Labeling & Detection Kitを用いて、イネのASAのαサブユニットをコードするDNAを含有した目的の組換えファージの検出を行った。ハイブリダイズしてX線フィルムで強くシグナルを発している8個の組換えファージプラーク、すなわちイネのASAをコードする遺伝子が組み込まれていると思われる8個のファージプラークを30万個のファージプラークから、ナイロン膜上で探知することができた。そして、イネのASA遺伝子が組み込まれている前記の8個の組換えファージプラークを単離して選抜できた。
【0187】
これら採取された8個のプラークの組換えファージから、λDNA単離キット(Lambda DNA Purification Kit(STRATAGENE社製)) により、それぞれにファージプラークの8個ごとに別々に、λDNAを単離した。
【0188】
この際の上記のλDNAの単離は次のようにして行った。すなわち、8個のプラークの各々のファージをそれぞれ別々に大量に増殖させたそれぞれの培養液5mlに、DNaseI(20mg/ml) 50μl、RNaseA(2mg/ml) 200μlを加え、室温で15分間、放置した。得られたファージ増殖溶液を15000rpm、4℃で10分間遠心分離した。得られた上清に80%DEAE-セルロース 25ml加え、室温で10分間インキュベートした。このようにインキュベートされた混合液を遠心分離し、得られた上清に0.5M EDTA 2ml、Pronase(50mg/ml) 770μlを加え、37℃で15分間放置した。さらに、5%CTAB溶液(1%CTAB (Cetyltrimethyl- ammonium bromide)、50mM Tris-HCl、pH8.0、10mM EDTA) 1.5mlを加えた。得られた混合物を65℃、3分間処理した後、氷中で5分間放置した。こうして得られたそれぞれの反応液に1/10容の3M酢酸ナトリウム、2倍容のエタノールを加え、−20℃で約6時間放置し、その後、得られた溶液を遠心分離し、それぞれの溶液から沈澱したファージDNAを別々に乾燥させ、5mlの水にそれぞれ溶解して保存した。
【0189】
上記の操作により単離された8種のファージDNAが得られた。これらDNAの各々5μlを、Hバッファー中で制限酵素EcoRIの10ユニットで別々に消化して、得られた消化反応液を別々に分析した。
【0190】
こうして、イネのASAの2つのアイソザイムのαサブユニットをコードする遺伝子に相当するDNA配列を含有すると判定されたところの、ファージDNA中の挿入DNA断片は、上記のように選抜された採取ファージのファージDNAから該ファージDNAを制限酵素EcoRIで上記のように消化して切出すことによって収得できた。
【0191】
(6) イネのASAの2つのアイソザイム(ASA1およびASA2) のそれぞれのαサブユニットをコードする遺伝子に相当するDNA配列を含有するcDNAのクローニング
上記のように、イネのASAの2つのアイソザイムのαサブユニットをコードする遺伝子に相当するDNA配列を含有すると判定されたDNA断片として、前記の8種の各々の組換えファージから切出して収得されたDNA断片は、8種である。このようなDNA断片の各1種づつをプラスミドベクター p Bluescript II SK(+) のEcoRI切断部位にDNAリゲーション・キットにより挿入、連結することにより、組換えプラスミドベクターを構築した。このように構築された組換えプラスミドベクターの導入で大腸菌XLl-Blue MRF' を形質転換した。
【0192】
上記のように形質転換の目的で、構築された組換えプラスミドベクターを大腸菌XL1-Blue MRF' に導入する操作は、詳しくは次のように行った。すなわち、上記で得られた採取ファージの組換えファージDNAの10μgを、先づHバッファー中で、制限酵素EcoRIの10ユニットで消化して消化反応液を得た。また他方、プラスミドベクターp Bluescript II SK(+)の10μgを同様にEcoR Iで消化して消化反応液を得た。上記のようにして得られたそれぞれのDNA消化反応液に対して別々に1/10容の3M酢酸ナトリウム、2倍容のエタノールを加え、−20℃で約6時間放置し、その後に、それぞれの得られたDNA溶液を遠心分離し、それぞれの溶液から沈殿したDNAを乾燥させ、5μlの水に各々溶解した。こうして得られたファージ由来のDNAの水溶液と、プラスミド由来のDNA水溶液の各々5μlを混合し、得られた混合物をDNA Ligation kit(宝酒造(株)製) で処理して、それら2種のDNAを連結させた。得られたDNA連結反応液に10分の1容量の3M酢酸ナトリウム、2倍容のエタノールを加え、−20℃に約6時間放置した。得られた混合液を遠心分離し、沈殿したDNAを乾燥させた。こうして連結ベクターDNAが採取された。この連結ベクターDNAを10μlの水に溶解した。
【0193】
こうして得られた連結ベクターDNAの水溶液10μl (DNA 10ng)と、市販の大腸菌XL1-Blue MRF'コンピテントセル(STRATAGENE社製) 100μlとを、1.5ml容チューブに入れ、氷中で30分間、42℃で30秒間、そして再び氷中で2分間インキュベートした。その後に、インキュベートされた混合物に対してSOC液体培地(2% バクト-トリプトン、0.5%バクト-酵母抽出物、10mM NaCl、2.5mM KCl、10mM MgSO4、10mM MgCl2、20mM Glucoseを含有) 900μlを加えてから、37℃で1時間大腸菌を振とう培養した。
【0194】
得られた大腸菌の培養液の100μlを、アンピシリンを50mg/lの濃度、X-gal(5-Bromo-4-Chloro-3-Indolyl-b-D-Galactoside)を20mg/lの濃度、IPTG
(Isopropyl-b-D-thiogalactopyranoside)を20mg/lの濃度で添加されてあるLB寒天培地(1%バクト−トリプトン、0.5%バクト−酵母抽出物、0.5% NaCl、0.1% Glucose、pH 7.5、寒天1.5%含有) にプレーティングした。37℃で16時間、大腸菌を培養後、白色に発色してない大腸菌コロニーから、白色に発色している大腸菌コロニーを、前記の組換えファージのDNA断片を連結された組換えプラスミドベクターDNAで形質転換されてある大腸菌として、分離した。
【0195】
このようにして分離された白色を発色している且つアンピシリン耐性である形質転換大腸菌のコロニー10個を、アンピシリン50mg/lを含む液体培地で増殖させ、さらに増殖された大腸菌から、プラスミド精製キット(QIA filter Plasmid Midi Kit, QIAGEN社製) によりプラスミドDNAを分離し且つ精製した。この精製により、上記で得られたアンピシリン耐性コロニーの形質転換大腸菌から50μg(50μl) の組換えプラスミドのDNAを得た。
【0196】
上記で単離された各々の組換えファージ(計8種) から、クローニングを上記のように行うと、組換えプラスミドのDNA8種が得られた。これら8種のDNAの塩基配列を解析するために次の操作を行った。
【0197】
(7) クローニングされたDNAのシークエンス解析
上記のクローニングされた8種のDNAである組換えプラスミドDNAをそれぞれに市販の塩基配列決定キットで処理すると、それらのDNA断片の全体の塩基配列を決定できた。そして、それらのDNA断片の内部に含有されたイネのASA1のαサブユニットをコードする遺伝子に該当するDNAの塩基配列、ならびにイネのASA2のαサブユニットをコードする遺伝子に該当するDNAの塩基配列を判定できた。
【0198】
なお、上記のDNAの塩基配列の決定を行うに当っては、塩基配列決定キット(Autoread Sequencing Kit (Pharmacia Biotech社製) を用いて、変性処理を行った後に自動DNAシーケンサALF DNA Sequencer II(ファルマシア社製)で前記DNA断片の塩基配列を決定した。
【0199】
このようにして、上記のDNA断片の塩基配列を判定した結果、イネのASAのαサブユニットをコードするDNA配列は2種類の相異なる塩基配列を有するものであることが解った。従って、イネのASAは2つのアイソザイム(イソ酵素) から成ること、またその第1のアイソザイム(ASA1) のαサブユニットをコードするDNAは、その第2のアイソザイム(ASA2)のαサブユニットをコードするDNAと相異なる塩基配列を有することが知見された。
【0200】
そして、イネのASA1のαサブユニットをコードする遺伝子に相当するDNAは、上記のように判定されて、後記の配列表の配列番号1に記載されてある塩基配列を有すると今回、認められた。
【0201】
また、イネのASA2のαサブユニットをコードする遺伝子に相当するDNAは、上記のように判定されて、後記の配列表の配列番号10に記載されてある塩基配列を有すると今回、認められた。
【0202】
第1の発明のイネのASA1のαサブユニットをコードする遺伝子のDNAの塩基配列は、配列表の配列番号1に記載の単一のオープンリーディングフレーム内にある1734個の塩基からなる。また、配列表の配列番号1に示されたイネのASA1のαサブユニットをコードする遺伝子のDNAの塩基配列は、配列番号2に記載の577個のアミノ酸残基からなるタンパクをコードしている。この配列番号2のアミノ酸配列をアラビドプシスのASA1のαサブユニットのタンパク質の既知のアミノ酸配列(The Plant Cell, 第4巻, 721頁〜733頁, 1992年) と比較すると、68%の相同性が認められるが、他の領域ではアラビドプシスのASA1のαサブユニットとは明らかに異なり、イネ特有の配列である。
【0203】
第2の発明のイネのASA2のαサブユニットをコードする遺伝子のDNAの塩基配列は、配列表の配列番号10に記載の単一のオープンリーディングフレーム内にある1821個の塩基からなる。また、配列番号10に示されたイネのASA2のαサブユニットをコードする遺伝子のDNAの塩基配列は、配列番号11に記載の606個のアミノ酸残基からなるタンパクをコードしている。この配列番号11のアミノ酸配列をアラビドプシスのASA2のαサブユニットのタンパク質の既知のアミノ酸配列(The Plant Cell, 第4巻, 721頁〜733頁, 1992年) と比較すると、72%の相同性が認められるが、他の領域ではアラビドプシスのASA2のαサブユニットとは明らかに異なり、イネ特有の配列である。
【0204】
(8) イネのASAαサブユニット遺伝子を導入された大腸菌形質転換株の寄託
前項(6)の操作により、最終的に組換えプラスミドDNAの8種が得られた。これら8種の組換えプラスミドDNA断片の各々のDNA断片全体の塩基配列が前項(7)で塩基配列決定キットにより決定された。
【0205】
(i) 次に、配列表の配列番号1に記載された1734個の塩基からなるDNA配列(DNA配列OSASA-W1と命名される)(すなわち、イネのASA1のαサブユニットをコードするDNA) を内部に含有すると判定された組換えプラスミドDNA断片を、前記の8種の組換えプラスミドDNA断片のうちから選択した。この選択された組換えプラスミドDNA断片を、制限酵素EcoRIで消化した。その消化反応液から、DNA配列OSASA-W1を内部に含有すると判定されたDNA断片(DNA配列OSASA-1と命名する) を低融点アガロース電気泳動法により分画して採取した。このように採取されたDNA配列OSASA-1を、プラスミドベクターp Bluescript II Sk(+)のEcoRI切断部位にDNAリゲーション・キット(宝酒造(株)製) により連結して、組換えプラスミドベクターpOSASA-1を構築した。
【0206】
このように構築したプラスミドベクターpOSASA-1を、前項(6)に記載された方法により、大腸菌XLl-Blue MRF'に導入した。プラスミドベクターpOSASA-1の導入により形質転換された大腸菌株をエシエリヒア・コリ(Escherichia coli) XLl-Blue MRF'(Os-asa1) と命名した。エシエリヒア・コリXLl-Blue MRF'(Os-asa1) は、日本の茨城県、つくば市の工業技術院生命工学工業技術研究所に1997年8月18日に、FERM P-16388の受託番号で寄託されている。さらに、1998年8月7日以降、ブタペスト条約の規約下でFERM BP-6453の受託番号で前記の研究所に寄託されてある。
【0207】
(ii) また、配列表の配列番号10に記載された1821個の塩基からなるDNA配列(DNA配列OSASA-W2と命名される)(すなわち、イネのASA2のαサブユニットをコードするDNA) を内部に含有すると判定された組換えプラスミドDNA断片を、前記の8種の組換えプラスミドDNA断片のうちから選択した。この選択された組換えプラスミドDNA断片を、制限酵素EcoRIで消化した。その消化反応液から、DNA配列OSASA-2を内部に含有すると判定されたDNA断片(DNA配列OSASA-2と命名する) を低融点アガロース電気泳動法により採取した。このように採取されたDNA配列OSASA-W2を、プラスミドベクターp Bluescript II Sk(+)のEcoRI切断部位にDNAリゲーション・キット(宝酒造(株)製) により連結して、組換えプラスミドベクターpOSASA-2を構築した。
【0208】
このように構築したプラスミドベクターpOSASA-2を、前項(6)に記載された方法により、大腸菌XLl-Blue MRF'に導入した。プラスミドベクターpOSASA-2の導入により形質転換された大腸菌株をエシエリヒア・コリ(Escherichia coli) XLl-Blue MRF'(Os-asa2)と命名した。エシエリヒア・コリXLl-Blue MRF'(Os-asa2) は、日本の茨城県、つくば市の工業技術院生命工学工業技術研究所に1998年6月18日に、FERM P-16853の受託番号で寄託されている。さらに、1998年8月7日以降、ブタペスト条約の規約下でFERM BP-6454の受託番号で前記の研究所に寄託されてある。
【0209】
実施例2
本例は、第3の発明(本件特許請求の範囲から除外されてあるが、特願2004-257204号の請求項1の発明)により得られた改変D配列と命名されたDNA配列(すなわち、配列表の配列番号12に記載された1734個の塩基からなる塩基配列を有するDNA配列) を含有するDNA断片の製造を例示する。
【0210】
(1) PCR用のプライマーとしての合成オリゴヌクレオチドの構築
DNA合成装置(Model-391、アプライドバイオシステムズ社製) を利用して、4種のプライマーを合成した。すなわち、配列表の配列番号16に記載の塩基配列を有するオリゴヌクレオチドであるプライマーOSASN1と、配列番号17に記載の塩基配列を有するプライマーOSASN2と、配列番号18に記載の塩基配列を有するプライマーOSASC1と、配列番号19に記載の塩基配列を有するプライマーOSASC2とを化学合成により構築した。
【0211】
(2) PCR法用のテンプレートの作製
実施例1の(8)項において、イネのASA1のαサブユニットの遺伝子に相当するDNA配列すなわち前記のDNA配列OSASA-W1を含有すると判定されるDNA断片として、組換えファージから制限酵素EcoRIで切出して収得されたDNA断片OSASA-1は、プラスミドベクターp Bluescript II SK(+)のEcoRI切断部位にDNAリゲーション・キットにより連結されて、組換えプラスミドベクターpOSASA-1と命名)を構築した。このプラスミドベクターpOSASA-1が下記のPCR法でテンプレートとして利用された。
【0212】
(3) 所要なDNA断片のPCR法による増幅と回収
(i) PCR法の第1回段階
所要なDNA断片をPCR法による増幅反応により収得するために、PCR法の第1回段階として下記の(A)反応と(B)反応とを行った。
【0213】
すなわち、その(A)反応では、テンプレートとして用いられる前記の組換えプラスミドベクターpOSASA-1の5μlと、前記のプライマーOSASAN1の1μMと、プライマーOSASAC1の1μMとを、増幅反応液(10mMのTris-HCl(pH8.3)、1mMのMgCl2、50mMのKCl、4種のヌクレオチドdNTPの各0.2mMの混合物、LA Taq DNAポリメラーゼの2.5ユニットを含有)の100μlに加えて、増幅反応を行った。この(A)反応で得られたDNAの増副生成物を、DNA断片-Aと称する。
【0214】
また、(B)反応では、テンプレートとして用いられる前記プラスミドベクターpOSASA-1の5μlと、プライマーOSASAC2の1μMと、プライマーOSASAN2の1μMとを、(A)反応で用いたと同じ組成の増幅反応液に加えて、増幅反応を行った。この(B)反応で得られたDNAの増幅生成物をDNA断片-Bと称する。
【0215】
なお、前記の増幅反応は、PCR反応装置(Promram Temp Control System PC-700(アステック社製))内で、変性を94℃で30秒間、アニーリングを55℃で30秒間、伸張を72℃で1分間行う3つの反応操作を20回繰り返すことにより実施された。
【0216】
増幅反応の終了後、(A)反応の増幅反応液を低融点アガロース電気泳動により分画し、増幅DNA生成物としての268bpのDNA断片-Aを含むバンドをアガロースゲルから切出した。また、(B)反応の増幅反応液を低融点アガロース電気泳動にかけて分画し、増幅DNA生成物としての336bpのDNA断片-Bを含むバンドをアガロースゲルから切出した。
【0217】
これら2つのゲル切片から、それぞれに、GENECLEAN II Kit(フナコシ社製) により分離して、DNA断片-Aの精製品とDNA断片-Bの精製品を得た。
【0218】
(ii) PCR法の第2回段階
次に、PCR法の第2回段階として、前記の(A)反応および(B)反応で増幅したDNA断片-Aの精製品の1μlとDNA断片-Bの精製品の1μlを、増幅反応液(10mMのTris-HCl(pH8.3)、1mMのMgCl2、50mMのKCl, dNTPの各0.2mMの混合物およびLA Taq DNA ポリメラーゼの2.5ユニットを含有) 100μlに加えて、増幅反応を行った。この場合の増幅反応は、変性を94℃、5分間、アニーリングを65℃、10分と55℃、10分行い、また、伸張を72℃、1分行う3つの反応操作を1回実施した。
【0219】
この増幅反応の終了後、その増幅反応液を低融点アガロース電気泳動により分画し、増幅DNA生成物として583bpの所期のDNA断片-Cを含有するバンドをゲルから切出した。このゲル切片からGENECLEAN II Kit(フナコシ社製) によりDNA断片-Cを分離して且つDNA断片-Cの精製品を得た。
【0220】
(4) 上記で得られたDNA断片-Cを利用して、目的の改変D配列を含有するDNA断片をクローニングにより十分な量で収得する操作を次に行う。
【0221】
(i) 先づ、上記で得られたDNA断片-Cの10μgを、Hバッファー(宝酒造(株)社製) 中で制限酵素Afl IIの10ユニットとBgl IIの10ユニットで消化した。この消化で得られたDNA断片をDNA断片-α称する。他方、前記のプラスミドベクターpOSASA-1の10μgをHバッファー(宝酒造(株)社製) 中で、Afl IIの10ユニットとBgl IIの10ユニットで消化して短縮プラスミドを得た。DNA断片-αを含む消化反応液と、前記の短縮プラスミドを含む消化反応液とに、それぞれに別々に1/10容の3M酢酸ナトリウム、2倍容のエタノールを加え、−20℃に約6時間インキュベートした。その後、インキュベートされた溶液を遠心分離し、それぞれの溶液から沈殿したDNAを乾燥させ、5μlの水に溶解した。
【0222】
こうして得られたDNA断片-αの水溶液の5μlと、該短縮プラスミドDNAの水溶液の5μlとを混合した。次に、得られた混合物(10μl) に含まれる各々のDNAを、DNA Ligation kit(宝酒造(株)社製) により連結反応させた。その連結反応液に1/10容の3M酢酸ナトリウム、2倍容のエタノールを加え、−20℃に約6時間インキュベートした。インキュベートされた反応溶液を遠心分離し、沈殿したDNA連結物を乾燥させ、5μlの水に溶解して水溶液にした。
【0223】
この水溶液に含まれるDNA連結物は、前記のDNA断片-αと、プラスミドベクターpOSASA-1のAfl II及びBgl II切断で得られた前記の短縮プラスミドとが互いに連結して作られた環状2本鎖の組換えプラスミド(前記のp Bluescript-DNA-Dプラスミドである) であり、そのプラスミドDNA中の挿入DNA領域内に、前記の改変D配列が含有された。
【0224】
(ii) このp Bluescript-DNA-Dプラスミドを大腸菌XL1-Blue MRF′に導入することにより大腸菌XL1-Blue MRF′を形質転換した。
【0225】
このように形質転換された大腸菌をエシエリヒア・コリXLl-Blue MRF'/p Bluescript-DNA-Dと命名し、前記の生命工学工業技術研究所に1998年8月7日以下、ブダペスト条約の規約下にFERM BP-6451の受託番号で寄託した。この形質転換された大腸菌細胞を次に液体培地中で培養した。
【0226】
培養された大腸菌細胞から、さらにプラスミドを抽出した。こうして改変D配列を含有する組換えプラスミドp Bluescript-DNA-Dがクローニングできた。
【0227】
(5) 改変D配列を含有するDNA断片の回収
得られたプラスミドp Bluescript-DNA-DであるプラスミドDNAの10μlを、Hバッファー(宝酒造(株)社製)中でEcoRIの10ユニットで消化した。その消化反応液を低融点アガロース電気泳動により分画した。DNA配列OSASA-W1配列(1734 bpのサイズ) を内部に含有するDNA断片(DNA断片-βと称する) をアガロースから切出した。
【0228】
このDNA断片-βを含有するアガロース切片にTEバッファー(10mMのTris-HCl、1mMのEDTAを含有、pH8)を等量加え、68℃、20分間加温することによりアガロースを溶解した。得られた溶液を飽和フェノールで2回抽出することによりアガロースを除いた。得られたDNAを含むフェノール抽出液に対して、それの10分の1容の3M酢酸ナトリウム、2倍容のエタノールを加え、−20℃に約6時間放置しインキュベートした。インキュベートされた溶液を、15000rpm、4℃で10分間遠心分離した。得られたDNA沈殿を減圧下で乾燥させ、得られたDNA粉末を10μlの水に溶解させた。
【0229】
そのDNA粉末は、目的の改変D配列を含有するDNA断片-βからなるものであった。
【0230】
なお、実施例2の上記(5)で得られた前記のDNA断片-βを、実施例1の(7)項に記載されたと同様にして、塩基配列決定キットを用いる自動DNAシーケンサ、ALF DNA sequencer IIにかけた。DNA断片-βは配列表の配列番号12に記載される1734個の塩基からなる塩基配列をもつ改変D配列を内部に含有するDNA断片であると確認できた。
【0231】
実施例3
本例は、配列表の配列番号1に示すDNA配列を外来遺伝子として、または配列番号12に示す塩基配列をもつ改変DNA配列を外来遺伝子として、イネ植物にアグロバクテリウム法で導入することからなるイネ植物の形質転換方法を例示する。また、本例で形質転換された植物のトリプトファン含量が増加されたことが例証された。
【0232】
(1) イネ植物への外来遺伝子の導入用の組換えベクターpUBdDの作成
(i) トウモロコシのユビキチンプロモーターと、1stイントロンと、NOSターミネーターと、アンピシリン耐性遺伝子とを含有する5.6kbのサイズの既知のプラスミドベクターpUBAのDNA(10μg) を、Kバッファー(宝酒造(株)製) 中で制限酵素BamHIの10ユニットで消化した。消化反応液からDNAを沈殿させ、遠心分離してから乾燥させた。このDNA乾燥物を10μlの滅菌水で溶解した後、DNAブランティング・キット(宝酒造社製)で平滑化した後、末端平滑化されたDNAを沈殿させ、遠心分離してから乾燥させた。この乾燥物を10μlの滅菌水で溶解した後、Lバッファー(宝酒造(株)製) 中で制限酵素SacIの10ユニットで消化した。消化反応液からDNAを沈殿させ、遠心分離してから乾燥させた。このようにして、ユビキチンプロモーターと、1stイントロンと、NOSターミネーターとアンピシリン耐性遺伝子を保有する約4.8kbのサイズのベクター断片を得た(添付図面の図1の左、参照)。
【0233】
(ii) さらに別に、第3の発明のDNAである改変されたD配列を内部に含有する組換えプラスミドpBluescript-DNA-D(1.7kbのサイズ) のDNA(10μg) を、Hバッファー(宝酒造(株)製) 中で制限酵素EcoRVの10ユニットで消化し、その消化反応液からDNAを沈殿させ、遠心分離してから乾燥させた。このDNA乾燥物を10μlの滅菌水で溶解した後、Lバッファー(宝酒造社製) 中で制限酵素SacIの10ユニットで消化した。消化反応液から上記と同様にしてDNAを乾燥させてpBluescript-DNA-Dベクター断片を得た(添付図面の図1の右、参照)。
【0234】
(iii) 上記の(i)で得た約4.8kbのサイズのベクター断片と、(ii)で得られたベクター断片を5μlの滅菌水に溶解した。そのベクター断片の水溶液の5μlをそれぞれ混合した。得られた混合物をDNAリゲーション・キット(宝酒造(株)製) で処理して、DNA連結反応を行った。こうしてプラスミドベクターpUBAの切断ベクター断片に対して、前記のpBluescript-DNA-Dの切断ベクター断片を連結してなる環状の組換えベクター(6.5kbのサイズ) を作成した。この組換えベクターをpUBdDと称する(図1の下、参照)。ベクターpUBdDは、ユビキチンプロモーター領域の下流に1stイントロン領域を有し、1stイントロン領域とNOSターミネーター領域との間に、第3の発明の改変D配列が挿入、連結されてあり、且つアンピシリン耐性遺伝子を含有する構造を有し、6.5kbのサイズを有した。
【0235】
(2) イネ植物への外来遺伝子の導入用の組換えベクターpUBdD-W1の作成
(i) トウモロコシのユビキチンプロモーターと、1stイントロンと、NOSターミネーターと、アンピシリン耐性遺伝子とを含有する5.6kbのサイズの既知のプラスミドベクターpUBAのDNA(10μg) から、前記の(i)および(ii)と同様にして、ユビキチンプロモーターと、1stイントロンと、NOSターミネーターとアンピシリン耐性遺伝子とを保有する約4.8kbのサイズのベクター断片を得た(図1の左、参照)。
【0236】
(ii) さらに別に、第1の発明による配列番号1のDNA配列を内部に含有する組換えプラスミドpOSASA-W1(4.7kbのサイズ) のDNA(10μg) を、Hバッファー(宝酒造(株)製) 中で制限酵素EcoRVの10ユニットで消化し、その消化反応液からDNAを沈殿させ、遠心分離してから乾燥させた。この乾燥物を10μlの滅菌水で溶解した後、Lバッファー(宝酒造社製) 中で制限酵素SacIの10ユニットで消化した。消化反応液から上記と同様にしてDNAを乾燥させた。
【0237】
(iii) 上記の(i)で得た約4.8kbのサイズのベクター断片と上記の(ii)で得られたベクター断片を5μlの滅菌水に溶解した。そのベクター断片の水溶液の5μlをそれぞれ混合した。得られた混合物をDNAリゲーション・キット(宝酒造(株)製) で処理して、DNA連結反応を行った。こうしてpUBAの切断ベクター断片に対して、前記のpOSASA-W1の切断ベクター断片を連結してなる環状の組換えベクターを作成した。この組換えベクターをpUBdD-W1と称する。ベクターpUBdD-W1は、ユビキチンプロモーター領域の下流に1stイントロン領域を有し、1stイントロン領域とNOSターミネーター領域との間に、配列OSASA-W1が挿入、連結されてあり、且つアンピシリン耐性遺伝子を含有する構造を有する。
【0238】
(3) イネ植物への外来遺伝子の導入用の組換えベクターpUb-OSASA1Dまたは組換えベクターpUb-OSASAW1の作成
(i) 上記の(2)(iii)で得られたプラスミドベクターpUBdD-W1のDNA(10μg) を、Hバッファー(宝酒造(株)製) 中で制限酵素SphIの10ユニットで消化し、その消化反応液からDNAを沈殿させ、遠心分離してから乾燥させた。得られたベクター断片を10μlの滅菌水に溶解した(添付図面の第2図の右、参照)。そのベクター断片の水溶液の10μlをDNAブランティング・キット(宝酒造(株)製) で処理して、制限酵素切断部位の平滑化反応を行った。その反応液からDNAを沈殿させ、遠心分離してから乾燥させた後、10μlの滅菌水に溶解した。このベクター断片のDNA(10μg) をLバッファー(宝酒造(株)製) 中で制限酵素SacIの10ユニットで消化した後、消化反応液からDNAを沈殿させ、遠心分離してから乾燥させた。このようにして、ユビキチンプロモーターおよび第3の発明による配列番号12の改変D配列を保有するベクター断片を得た(添付図面の図2の右の第3番目の図、参照)。
【0239】
(ii) ハイグロマイシン耐性遺伝子を含有する15kbサイズの公知のプラスミドベクターplG121-HmのDNA(10μg) を、Lバッファー(宝酒造(株)製) 中で制限酵素SacIの10ユニットで消化し、その消化反応液からDNAを沈殿させ、遠心分離してから乾燥させた。この乾燥物を10μlの滅菌水で溶解した後、NEB4バッファー(ニューイングランド・バイオラブ社製) 中で制限酵素PmeIの10ユニットで消化した。消化反応液から上記の(1)(i)と同様にして、DNAを乾燥させた。このようにしてハイグロマイシン耐性遺伝子を保有する約9.8kbのサイズのベクター断片を得た(図2の左、参照)。
【0240】
(iii) 上記の(ii)で得られた約9.8kbのサイズのベクター断片と、上記の(i)で得られた改変D配列含有ベクター断片をそれぞれ5μlの滅菌水に溶解した。そのベクター断片の水溶液の5μlを、混合した後、DNAリゲーション・キット(宝酒造(株)製) で処理して、DNA連結反応を行った。このようにしてプラスミドベクターpl121-HmのPmeI-SacI切断ベクター断片に対して、前記の改変D配列断片を連結してなる環状の組換えベクターを作成した。この組換えベクターをpUb-OSASA1Dと称する(図2の下、参照)。ベクターpUb-OSASA1Dは、ユビキチンプロモーター領域の下流に1stイントロン領域を有し、1stイントロン領域とNOSターミネーター領域との間に、改変D配列が挿入、連結されてあり、植物中で発現するハイグロマイシン耐性遺伝子、且つカナマイシン耐性遺伝子を含有する構造を有し、11.5kbのサイズを有した。
【0241】
さらに別に、上記の改変D配列断片の代わりに、第1の発明による配列番号1の塩基配列を有するOSASAW1断片を前記と同様に上記の(ii)で得られた約9.8kbのサイズのベクター断片に対して連結してなる環状の組換えベクターを作成した。この組換えベクターをpUb-OSASAW1と称する。
【0242】
(4) アグロバクテリウム細菌の調製
LB培地(バクトトリプトン10g/l、バクトイーストエクストラクト5g/l、NaCl 5μl pH7) の液体培地300mlに、アグロバクテリウム(Agrobacterium tumefaciens LBA4404 (Clontech社から購入)) を移植し、30℃で16時間振とう培養した後、培溶液を4℃で10分間冷却した後、遠心分離により細菌の沈殿を得た。細菌の沈殿物を氷冷した10%グリセロールで洗浄操作と遠心分離を3回繰り返した後、沈殿物を10mlの10グリセロールに溶解した。
【0243】
そのうちの40μlをエッペンドルフチューブに移し、このチューブに上記した組換えベクターpUb-OSASA1D(配列番号12の改変DNAを保有)あるいはベクターpUb-OSASAW1(第1の発明による配列番号1のDNAを保有)の溶解液(40ng) 5μlをそれぞれ添加し、よく混合した。これらを氷上に3分間置いた後、エレクトロポレーション用のキュベット(電極間0.2cm) に移し、エレクトロポレーション装置(ジーン・パルサー(バイオラッド社製)) で、電圧12.5kV/cm、コンデンサー容量25μF、抵抗値600Ωの条件でエレクトロポレーションを行った。キュベット内にSOC培地(GIBCO BRL社製) を0.8ml添加し、けんだく液を2ml容の試験管に移し、30℃で1時間振とう培養を行った。この培養液をLB 培地にカナマイシン50mg/lを添加してなる寒天培地に塗布し、30℃で36時間培養して、生育してきたコロニーを組換えベクターが導入された形質転換アグロバクテリウムとして得た。
【0244】
組換えベクターpUb-OSASA1Dで形質転換されているアグロバクテリウム細菌をpUb-OSASA1D/LBA4404と称する。ベクターpUb-OSASAW1で形質転換されているアグロバクテリウム細菌をpUb-OSASAW1/LBA4404と称する。
【0245】
(5) イネのカルスの調製
イネ(品種: 日本晴)の完熟種子から籾穀を脱穀した。得られた外皮付きの種子を70%エタノール溶液に60秒間、ついで有効塩素約1%の次亜塩素酸ナトリウム溶液に6分間浸漬して種子を殺菌処理した。さらに種子を滅菌水で洗浄した。
【0246】
MS培地の無機成分組成にショ糖30g/l、植物ホルモンとして2,4-PA 2mg/l、寒天8g/lを添加してなる培地に、上記で殺菌したイネ種子を置床した。28℃で21日間、2000ルックスの光を1日当たり16時間照明しながらインキュベートした。カルスが形成された。それらカルスを胚乳部分から切り出し、そのカルスを上記と同じ組成の培地に移植して、3日間培養を行った。
【0247】
(6) イネのカルスへの外来遺伝子の導入操作
上記のようにして得られた、遺伝子導入用の形質転換アグロバクテリウム(pUb-OSASA1D/LBA4404、あるいはpUb-OSASAW1/LBA4404) をAA培地の無機塩組成にショ糖20g/l、2,4-PA 2mg/l、カイネチン0.2mg/lおよびアセトシリンゴン10mg/lを添加してなる液体培地30mlにけんだくし菌液を得た。このけんだく液を9cmのシャーレに入れ、上記(5)で得られたイネのカルスを100個入れた後、5分間浸せきした。浸せき後、ペーパータオルで余分な菌液を除去した後、N6培地の無機塩組成にショ糖30g/l、グルコース10g/l、2,4-PA 2mg/l、ゲルライト2g/lおよびアセトシリンゴン10mg/lを添加してなる固体培地に、上記のしんせきしたカルスを各シャーレ当たり20個ずつ置床した。28℃で3日間、暗黒下で培養し、イネのカルスへのアグロバクテリウムの感染を行った。
【0248】
(7) 導入ベクターで形質転換されたカルスの選抜
上記(6) で組換えベクターが導入された形質転換カルスを、500mg/lカルベニシリンを添加してなる滅菌水で洗浄してアグロバクテリウム細菌を除去した。カルスから余分な水分を除いた後、カルスをN6培地の無機塩組成にショ糖30g/l、2,4-PA 2mg/l、カルベニシリン500mg/l、ハイグロマイシン50mg/l、ゲルライト2mg/lを添加してなる固体培地上に各シャーレ当たり20個ずつ移植した。25℃で21日間、2000ルックスの光を1日当たり16時間照明しながら培養して、ハイグロマイシン耐性の形質転換カルスを得た。
【0249】
(8) ベクターpUb-OSASA1Dによる形質転換カルスの再選抜
上記により得たハイグロマイシン耐性である形質転換カルスのうち、ベクターpUb-OSASA1Dに含有されて植物中で発現される機能を有する改変D配列が外来遺伝子として十分導入されて形質転換されたカルスのみを再選抜する。そのためには、形質転換カルス15個(直径5mm)を、N6培地の無機塩組成にショ糖30g/l、2,4-PA 2mg/l、カルベニシリン250mg/l、ゲルライト2g/lと細胞増殖阻害剤として働く5-メチルトリプトファン(以下、5MTという) (トリプトファンのアナログ体) の3×10−4Mを添加してなる固体培地上に移植した。25℃で21日間、2000ルックスの光を1日当たり16時間照明しながら培養した。前記の5MT添加培地で生育できた植物中で発現される機能を有する改変D配列含有の形質転換カルスをこのようにして再選抜した。
【0250】
(9) 形質転換カルス細胞からの植物体の再生
上記により得たハイグロマイシン耐性および5MT耐性である形質転換カルス培養の14〜15個を、MS培地の無機塩組成にショ糖の30g/l、ソルビトール30g/l、カサミノ酸の2g/l、NAA 1mg/l、BA 2mg/l、ハイグロマイシン50mg/l、ゲルライト4g/lを添加してなる固体培地に移植した。25℃で30日間、2000ルックスの光を1日当たり16時間照射しながら培養すると、形質転換カルスから芽と根が再生できた。再生した幼芽(長さ10〜30mmに生育した芽)を、ショ糖の30g/l、ゲルライトの2g/lを含むMS培地を入れた試験管(直径45mm、長さ25cm)に移植した。移植された幼芽を20日間培養して形質転換したイネ植物を得た。
【0251】
また、上記の方法により、第1の発明のDNAである配列表の配列番号1のDNAを保有する前記のOSASAW1配列を外来遺伝子として含有する形質転換カルス15個から、10個のイネ植物が再生できた。
【0252】
また、第3の発明による配列番号12の改変D配列を外来遺伝子として含有する5MT耐性の形質転換カルス14個から10個のイネ植物が再生できた。
【0253】
(10) 形質転換イネ植物の再生体の遺伝子解析
上記のように再生されたイネ植物体に存在するASAをコードするDNAの解析をPCR法で次の手順により行った。
【0254】
(i) 前項(9)で得た再生イネ植物から葉を採取した。その葉50mgを1.5ml容のマイクロチューブに入れ、10mM EDTAを含む20mM Tris-HCl緩衝液(pH 7.5) 300μlを添加し、これを磨砕した。磨砕物に20%SDSを20ml加えて、65℃で10分間加温した。得られた混合物に5M酢酸カリウムを100μl加え、氷中に20分間置いた後、1,7000xgの遠心加速度で20分間遠心分離した。得られた上清にイソプロパノール200μlを加え、転倒攪拌し、これを再び1,7000xgの遠心加速度で20分間遠心分離した。得られたDNAの沈殿を減圧下で乾燥した。100μlのTE緩衝液にそのDNAを溶解した。
【0255】
(ii) PCR用のプライマーとして、配列表の配列番号14に示す塩基配列を有する前記オリゴヌクレオチドと、配列番号15に示す塩基配列を有する前記オリゴヌクレオチドとを用いた。
【0256】
次に、これら2種類のオリゴヌクレオチドの各1μMをプライマーとして用い且つ上記再生イネ植物体由来のDNAの5μlをテンプレイトとして用い、これらを増幅用反応液(10mM Tris-HCl(pH8.3)、1.0mM MgCl2、50mM KCl、0.01%ゼラチン、pH 8.3、dNTPの各0.2mMの混合物およびTaq DNAポリメタラーゼ2.5ユニットを含有) 100μlに加えた。そして増幅反応を行った。用いた増幅反応液は、PCRキット(PCR Amplification Kit(宝酒造(株)製)で調製した。
【0257】
増幅反応は、PCR反応装置(Program Temp Control System PC-700(アズテック社製)) 内で、変性94℃、1分、アニーリング60℃、30秒、伸張(extention) 72℃、1分の3つの反応操作を30回繰り返すことによって実施した。
【0258】
(iii) 得られたPCR反応液を、常法によりアガロース電気泳動にかけて分析したところ、再生イネ植物から抽出したDNAから増幅された各種のDNAのバンドが確認された。
【0259】
さらに、それらの各種のDNA配列を、塩基配列決定キットにより解析したところ、第1の発明のよるDNA配列 (配列番号1) を保有するOSASA-W1配列または第3の発明による改変D配列(配列番号12) に相当するDNA断片が再生イネ植物から上記のように抽出された各種のDNA断片の中に存在することが確認できた。
【0260】
上記のようにPCR法による遺伝子解析を行うことにより、導入した外来遺伝子が確認できた再生イネ植物体は、各々を培養土を入れたポットに移植して栽培した。これらの再生イネ植物体は正常に生育したので自殖種子を得ることができた。
【0261】
(11) 形質転換イネ植物の再生体中のトリプトファン含量の測定
前項(10)で得られた、第1の発明によるDNAを保有するOSASA-W1配列を含有する組換えベクターpUb-OSASAW1を導入された植物細胞を有する再生された形質転換イネ植物体と、第3の発明による改変D配列を含有する組換えベクターpUb-OSASA1Dを導入された植物細胞を有する再生された形質転換イネ植物体とから、夫々に緑色の葉を採取した。
【0262】
このようにそれぞれに採取された葉を1gづつ取り、それらの葉の100mgを、1.5ml容の第1のチューブに入れ、さらに50%アセトニトリル1mlを添加し、磨砕した。磨砕混合物を新たらしい1.5ml容の第2のチューブに移し替え、17000Xgの遠心加速度で20分間遠心分離した。得られた上清を、再び新たらしい第3のチユーブに移し入れ、その第3のチューブ内に50%アセトニトリル1mlを加え、十分に転倒攪拌し、再び遠心分離した。得られた上清を第1のチューブに加え、この操作を3回繰り返した。このようにして、葉から抽出されたトリプトファンを含有したアセトニトリル抽出液を得た。
【0263】
このアセトニトリル抽出液を減圧下で乾固させた後、蒸留水1mlを加えて水溶液を得た。その水溶液を17000Xgの遠心加速度で20分間遠心分離により、上清の0.5mlを得た。その上清のうち100μlに5mM DNFB(2,4-ジニトロ-1-フルオロベンゼン)100μlを混合した。得られた混合物を一晩反応させた。この反応液に200μlのアセトニトリルを添加し、攪拌した後、17000xgの遠心加速度で20分間遠心分離した。上清液として、トリプトファンの抽出液を得た。これを、高速液体クロマトグラフィー(HPLC)装置(東ソー(株)製、8020型)にかけて遊離トリプトファン含量を測定した。このHPLCに用いたカラムは、CAPCELL PAK-C18(資生堂(株)製)であり、展開溶媒はアセトニトリル-水をアセトニトリルの60%〜72%濃度勾配で用い、流量は0.8ml/分として、350nmの光の吸光度を測定して、トリプトファン含量を評定した。
【0264】
対照のイネ植物として、形質転換されてない通常のイネ(品種: 日本晴)の植物体を用いた。トリプトファン含量について得られた測定結果を次の表1に示す。
【表1】
【0265】
上記の表1に示された結果から明らかなように、第1の発明による新規DNA配列を外来遺伝子として用い且つ植物細胞中で発現できるプロモーターを有する組換えベクターを利用すると、第1の発明による新規DNA配列を導入することにより、イネ植物体中に生成される遊離トリプトファン含量が増加できることが確認された。
【0266】
実施例4
本例は、第3の発明による配列番号12のDNA配列を、外来遺伝子としてイネ植物に導入することからなる形質転換細胞の選抜方法と形質転換植物の作成方法を例示する。
【0267】
(1) 外来遺伝子の導入用の組換えベクターの作成
外来遺伝子の導入用の組換えベクターとして、実施例3、(3)(iii)に記載の組換えベクターpUb-OSASA1Dまたは実施例3、(1)(iii)に示された組換えベクターpUBdDを用いた。
【0268】
(2) アグロバクテリウム細菌法またはウイスカー法によるイネのカルス細胞への外来遺伝子の導入と形質転換細胞の選抜の操作
(i) アグロバクテリウム細菌法
上記の(1)に示した組換えベクターpUb-OSASA1Dを用いて実施例3の(4)と同様の方法によりアグロバクテリウム細菌の調製を行い、また実施例3の(5)と同様の方法によりイネのカルス細胞の調製を行い、実施例3の(6)と同様の方法によりイネのカルス細胞への導入操作を行った。
【0269】
上記で組換えベクターが導入された形質転換カルスを、500mg/lカルベニシリンを添加してなる滅菌水で洗浄してアグロバクテリウム細菌を除去した。カルスから余分な水分を除いた後、カルスをN6培地の無機塩細成にショ糖30g/l、2,4-PA 2mg/l、カルベニシリン500mg/l、ゲルライト2g/lおよび選抜用薬剤として5MT3×10−4Mを添加してなる固体培地上に各シャーレ当たり20個ずつ、合計500個を移殖した。25℃で1か月間、2000ルックスの光を1日当たり16時間照明しながら培養した。
【0270】
1か月培養の後に、形質転換カルスが選抜されて得られたカルスの個数の計数した結果を後記の表2に示す。なお、対照区には、5MTの代わりにハイグロマイシンの50mg/lを添加した固体培地を用いた。
【0271】
上記の培養により5MT耐性を基準にして選抜されて得られた形質転換カルスを、MS培地の無機塩組成にショ糖30g/l、ソルビトール30g/l、カサミノ酸2g/l、NAA 1mg/l、BA 2mg/l、ゲルライト4g/lおよび5MT3×10−4Mを添加してなる固体再生培地に移殖した。25℃で30日間、2000ルックスの光を1日当たり16時間照明しながら培養した。形質転換カルスから再生された形質転換植物体の再生数を計数した結果を後記の表2に示す。なお、対照区には、5MTの代わりにハイグロマイシンの50mg/lを添加した固体培地を用いた。
【0272】
(ii) ウイスカによる直接導入法
イネ(品種:日本晴) の完熟種子から籾穀を脱穀した。得られた外皮付きの種子を70%エタノール溶液に60秒間、ついで有効塩素約1%の次亜塩素酸ナトリウム溶液に6分間浸漬して種子を殺菌処理した。さらに種子を滅菌水で洗浄した。
【0273】
MS培地の無機成分組成にショ糖30g/l、植物ホルモンとして2,4-PA 2mg/l、寒天8g/lを添加してなる培地に、上記で殺菌したイネ種子を置床した。28℃で45日間、2000ルックスの光を1日当たり16時間照明しながらインキュベートした。カルスが形成された。それらカルスを胚乳部分から切り出し、孔1mmのステンレスメッシュの篩を用いて、1mm以下のカルスをPCV(Packed Cell Volume; 圧縮細胞量) として3mlの量で得た。
【0274】
チタン酸カリウム製ウイスカ(チタン工業(株)製、LS20) 5gを1.5ml容のチューブに入れ、エタノールを0.5ml加えて、一晩放置した。エタノールを完全に蒸発させて、殺菌されたウイスカを得た。このウイスカの入ったチューブに滅菌水1mlを入れ、よく攪拌した。ウイスカと滅菌水を遠心分離し、上清の水を捨てた。このようにしてウイスカを洗浄した。このウイスカ洗浄操作を3回行った。その後、同チューブ内にR2液体培地の0.5mlを加えてウイスカ懸濁液を得た。
【0275】
上記で得られたウイスカ懸濁液の入ったチューブに1mm以下のカルスを250μl入れて、攪拌した。得られた混合物を1000rpm/分で10秒間遠心分離し、カルスとウイスカを沈殿させた。上清を捨てた。カルスとウイスカとの混合物が得られた。
【0276】
カルスとウイスカとの混合物を入れたチューブに、組換えベクター(すなわち、前記の組換えベクターpUBdD)の10μl(DNAの10μg含有)とを加えた。十分振り混ぜた均質な混合物を得た。
【0277】
次にこの均質な混合物の入ったチューブを18000 x gで5分間遠心分離した。遠心分離された混合物を再度振り混ぜた。この遠心分離の操作と再度振り混ぜる操作を3回反復した。
【0278】
上記のようにして得られた、カルス細胞と、ウイスカと、第3の本発明の改変DNA配列を含有する組換えベクターを収容しているチューブを、超音波発生機の浴槽にチューブが十分浸かるように設置した。周波数40kHzの超音波を強度0.25W/cm2で1分間照射した。照射後、30分間、4℃で超音波処理された混合物をインキュベートした。
【0279】
このように超音波処理した混合物をR2液体培地で洗浄し、組換えベクターpUBdDを導入した目的の形質転換カルス細胞を得た。
【0280】
上記で組換えベクターを導入して得た形質転換細胞を有するカルスを、3.5cmのシャーレに入れた。さらにR2培地の無機成分組成にショ糖30g/l、2,4-PA 2mg/lを添加して得た液体培地を3ml加えた。その後に、28℃で2000ルックスの光を1日当たり16時間照射しながらロータリーシェーカー(50rpm) 上でカルス細胞を培養し、分裂細胞を得た。
【0281】
培養3日目に、得られた分裂細胞の懸濁液3ml(分裂細胞数2000個) を、N6培地の無機成分組成に、ショ糖30g/l、2,4-PA 2mg/l、ゲルライト3g/lおよび選抜用薬剤として5MT 10−4Mを添加してなる培地上に均一に広げた。培地上の細胞を、28℃で2000ルックスの光を1日当たり16時間照射しながら1か月間培養した。
【0282】
1か月後培養の後に、形質転換細胞よりなるカルスが選抜できた。こうして得られたカルスの個数の計算した結果を表2に示す。なお、対照区には、5MTの代わりにハイグロマイシンの50mg/lを添加した固体培地を用いた。
【0283】
また上記の培養により5MT耐性を基準にして選抜されて得られた形質転換細胞よりなるカルスを、MS培地の無機塩組成にショ糖30g/l、ソルビトール30g/l、カサミノ酸2g/l、NAA 1mg/l、BA 2mg/l、ゲルライト4g/lおよび5MT3×10−4Mを添加してなる固体再生培地に移殖した。25℃で30日間、2000ルックスの光を1日当り16時間照明しながら培養した。形質転換カルスから再生された形質転換植物体が得られた。その形質転換植物の再生数を計数した結果を表2に示す。なお、対照区には、5MTの代わりにハイグロマイシンの50mg/lを添加した固体培地を用いた。
【表2】
【0284】
実施例5
本例は、イネのASA遺伝子のプロモーターの遺伝子に相当するDNAを収得する方法を例示する。
【0285】
(1) イネのゲノミックDNAの調製
イネ(品種:日本晴) のカルス2gから、公知のCTAB法(クローニングとシークエンス: 1989年、262頁〜264頁、農村文化社) に従い、ゲノミックDNAの2mgを抽出した。
【0286】
(2) イネのゲノミックDNAライブラリーの構築
10マイクログラムの上記ゲノミックDNAを制限酵素EcoRIで部分消化した後、得られたDNA断片を0.8%アガロースゲルにて電気泳動した。電気泳動により分画したDNA断片をナイロン膜(アマシャム社製)ハイボンドNに転写した。このようにしてナイロン膜に転写したDNAはアルカリ変性液(1.5M NaCl、2.0M NaOH) および中和液(1.0 M Tris-HCl pH5、2.0M NaCl) でそれぞれ10分間処理し、その後に80℃、2時間処理してDNAをナイロン膜に固定した。
【0287】
次に、実施例1の(5)で得られたDIGでラベルして得られた標識プローブDNAを用いた。この標識プローブDNAを上記のナイロン膜に対してハイブリダイゼーションさせた。この操作は実施例1の(5)と同様の方法で行った。
【0288】
その結果、ナイロン膜上のDNAサイズ6kb付近に、X線フィルムに強くシグナルを発するDNAが検出された。そこで、それらDNAを分離し、そのDNAを実施例1の(5) と同様の方法で制限酵素EcoRIで部分消化し、電気泳動したDNA断片の6kb付近をゲルから切り出し、DNA精製キット(Gene Clean II Kit (Bio 101社製)) を用いて精製した。得られたDNA断片(精製品) を20μlのTEバッファー(10mM Tris-HCl(pH7.5)、1mM EDTA) に溶解した。このようにして、分画したイネのゲノミックDNAが得られた。これをクローニングキット(Lambda ZAPII/EcoRI/CIP Cloning Kit(STRATGENE社製)) を用いてベクターにライゲーションした。得られた組換えベクターを、GigapackII Gold Packaging Extractを用いてラムダファージにパッケイジングを行った。得られた組換えλファージを大腸菌XL1-Blue MRF'に感染させて、前記の組み換えファージを多数得た。これら得られた組換えファージをイネの分画ゲノミックDNAライブラリーとした。
【0289】
(3) イネのゲノミックDNAライブラリーからのプロモーター遺伝子の選択
上記(2)で作製したイネのゲノミックDNAライブラリーである組換えファージから、実施例1の(5)で作製した標識プローブDNAの利用により、イネのASA遺伝子の発現のためのプロモーターの遺伝子に相当するDNA配列を組込まれた組換えファージをスクリーニングする。
【0290】
このスクリーニングの目的のために、実施例1 (5)と同様の方法で、組換えファージをナイロン膜に固定した後、標識プローブDNAをハイブリダイゼーションさせた。こうして目的のDNA配列が組み込まれたファージを選択した。その結果、ASA遺伝子のプロモーター遺伝子が組み込まれていると思われるファージのプラークの3個を、10万個のファージプラークから、単離選抜した。これら3個の組換えファージから、実施例1 (6)と同様の方法で各々別々に3種のλDNAを単離した。
【0291】
上記のようにして単離された3種のファージDNAを各々別々に用いて、それの5μlを制限酵素EcoRIの10ユニットで消化して、その消化液を分析した。単離された3種のファージDNAのRcoRI-DNA断片は同じ塩基配列を有すると認められた。
【0292】
(4) イネのASA遺伝子のプロモーター遺伝子に相当するDNA配列を含有するゲノミックDNAのクローニング
上記のようにイネのASA遺伝子に相当するDNA配列を含有すると判定されたDNA断片として、3種のDNAのうちの各々のファージから制限酵素EcoRIで切り出されて収得された6.2kbのサイズのDNA断片は、これをプラスミドベクターpBluewscriptIISK(+)のEcoRI切断部位にDNAリゲーション・キットにより挿入、連結して組換えプラスミドベクターを構築した。このように構築された組換えプラスミドベクターの導入で大腸菌XL1-BlueMRF'を形質転換した。得られた形質転換株はエシエリヒア・コリXL1-Blue MRF'(Os-asa#7) と命名し、これは生命工学工業技術研究所に1997年8月18日にFERM P-16387として寄託され、また1998年8月7日以降はブダペスト条約の規約下にFERM BP-6452の受託番号で寄託されてある。
【0293】
上記における組換えプラスミドベクターの構築および大腸菌への導入の操作は、実施例1(6) の方法と同様にして行った。
【0294】
組換えプラスミドベクターを導入した大腸菌から、プラスミド精製キット(QIA filter Plasmid Midi Kit, QIAGEN社製) によりプラスミドを分離且つ精製した。この精製により、上記で得られた形質転換大腸菌から50μg(50μl)のプラスミドDNAの3種を得た。
【0295】
このように各々の単離された組換えファージからクローニングされて得た前記のプラスミド3種類に含まれてあり且つ制限酵素EcoRIとBamHIの切断部位に挟まれた約1.5kbのDNAは、その塩基配列を解析するために次の操作を行った。
【0296】
(5) クローニングされたDNAのシークエンス解析
上記のクローニングされた3種類のDNAである組換えプラスミドを市販の塩基配列決定キットで処理すると、そのDNA断片の全体の塩基配列を決定できる。そして、そのDNA断片内部に含有されたイネのASA遺伝子のプロモーター遺伝子に該当するDNA配列を判定できる。上記の塩基配列の決定は、実施例1(7) の方法と同様に行った。
【0297】
このようにして上記のDNAの塩基配列を判定した結果、クローニングされた3種類のDNAである組換えプラスミドはすべて同一であることがわかった。
【0298】
こうして得られた、ASA遺伝子のプロモーターのDNAは、後記の配列表の配列番号7に記載されてある塩基配列を有すると今回認められた。ここで得られた配列番号7の塩基配列は、イントロンを含むものであり、またそれのタンパク質をコードしている部分がcDNAクローンと完全に一致するものであった。得られた組換えプラスミドのDNAについて決定した塩基配列のうち、ASA遺伝子のプロモーター部分及びそのエクソン部分およびイントロン部分のDNAを含むDNA配列の塩基配列を配列番号3に示す。
【0299】
なお、配列表の配列番号3は、イネのASAの第1アイソザイムのαサブユニットであるタンパク質の部分アミノ配列も併記してある。また、配列表の配列番号4は、配列番号3に示された塩基配列のうちのアミノ酸コード部分のアミノ酸配列を示す。さらに、配列番号5は、配列番号3のDNAのうちのエクソン−1の塩基配列を示し、配列番号6は、配列番号3のDNAのうちのエクソン−2の部分配列を示す。
【0300】
(6) プロモーター活性の検定
(i) 検定用の組換えベクターの作成
上記で得られたプロモーターDNA(配列番号7) のプロモーター活性を調べるため、このDNA(前記の(4) で得たEcoRI-BamHI断片) を植物細胞形質転換用ベクターpBI101(Clontech社製) のβ-グルクロニダーゼ(GUS)遺伝子の上流部分に組み込み形質転換用ベクターpGUS#3を得た。
【0301】
形質転換用ベクターの作製においては、GUS遺伝子のNOSターミネーターとカナマイシン耐性遺伝子とを含有する12.2kbのサイズのプラスミドベクターpBI101(Clontech社製) のDNA(10μg)を、Mバッファー中で制限酵素HindIIIの10ユニットで消化した。その消化反応液からDNAを沈殿させ、遠心分離してから乾燥させた。得られた乾燥物を10μlの滅菌水で溶解した後、続いて、切断部位を平滑化するために、DNAプランティング・キット(宝酒造(株)製) を用いて処理した後、DNA精製キット(Gene Clean II Kit (Bio 101社製)) を用いて精製した。その精製DNAを10μlの滅菌水に溶解した。この溶解液を、Kバッファー中で制限酵素BamHIの10ユニットで消化した。その消化反応液からDNAを沈殿させ、遠心分離してから乾燥させた。このようにして12.2kbのサイズを有して且つ平滑末端およびBamHI切断部位を両端に持つ、ベクター断片を得た(添付図面の図3の左、参照)。
【0302】
一方、上記したイネのASA遺伝子のプロモーターDNA断片を含むプラスミドベクターDNA(10μg)を、Hバッファー中で制限酵素EcoRIの10ユニットで消化した。その消化反応液からDNAを沈殿させ、遠心分離してから乾燥させた。得られた乾燥物を10μlの滅菌水で溶解した後、続いて、切断部位を平滑化するために、DNAプランティング・キット(宝酒造(株)製) を用いて処理した後、DNA精製キット(Gene Clean II Kit (Bio 101社製)) を用いて精製した。その精製DNAを10μlの滅菌水に溶解した。この溶解液を、Kバッファー中で制限酵素BamHIの10ユニットで消化した。その消化液をアガロース電気泳動により約1.5kb付近をゲルから切り出し、DNA精製キットを用いて精製した後、10μlの滅菌水に溶解した。このようにして1.5kbのサイズを有して且つ平滑末端およびBamHI切断部位を両端に持つ、プロモーターDNA断片を得た(図3の右、参照)。
【0303】
上記したプラスミドベクター断片溶液の5μlと上記のプロモーターDNA断片溶液の5μlとを混合した後、この混合液をDNAリゲーション・キットで処理してDNAの連結を行った。
【0304】
このようにしてプラスミドベクターpBI101の平滑末端−BamHI−切断ベクター断片に対して、前記のイネのASA遺伝子のプロモーターDNAの平滑末端−BamHI−断片を連結してなる環状の組換えベクターを作製した。こ組換えベクターをpGUS#3と称する(図3の下、参照)。
【0305】
ベクターpGUS#3は、イネのASA遺伝子のプロモーターDNAの下流に、GUS遺伝子を持ち、さらに下流のNOSターミネーターが連結されてあり、且つカナマイシン耐性遺伝子を含有する構造を有し、13.7kbのサイズを有した。
【0306】
(ii) イネのカルス細胞への検定用組換えベクターの導入
上記で得られた組換えベクターpGUS#3を実施例3 (6)と同様の方法によりイネのカルス細胞への導入を行った。このようにして、組換えベクターpGUS#3が形質転換されているイネのカルス細胞を得た。
【0307】
(iii) GUS活性の測定
上記で得られた組換えベクターpGUS#3が形質転換されたイネのカルスを、28℃で3日間培養した後、カルスの約0.5gを乳鉢に入れ、500μlの抽出用バッファー50mM NaPO4(pH7)、10mM EDTA、0.1%TritonX100、0.1%Sarkosyl、10mM β-メルカプトエタノール) 中で添加し、磨砕した。磨砕液を1.5ml容のチューブに移し、17000×gの遠心速度で20分間遠心分離した。得られた上清のうち、10μlを新しいチューブに移し、基質である1mMの4-メチルアンペリフェリルグルクロナイド(4-MUG) 溶液の100μlを加えて混合した。得られた混合物を37℃で3時間反応させた。その反応液に3%炭酸水素ナトリウム溶液の1.8mlを添加して、混合した。それを蛍光分光光度計(日立社製、F2000型) にかけて蛍光吸収測定を行った。その際の365nm励起光、455nm発光の条件でGUS活性の評定を行った。
【0308】
GUS活性を測定した結果を下記の表3に示す。数値はカルスから抽出したタンパク質1g当たり、1時間にGUS酵素の作用により生成する4-メチルアンベリフェロン(4-MU)の量を示す。なお、対照区には、プロモーターを組み込まれていないpBI101ベクターを導入したカルスを用いた。
【表3】
【0309】
上記のようにGUS活性が発現されたことにより、塩基配列の解析の結果から確認されたプロモーター領域がプロモーターとして機能したことが証明される。
【産業上の利用可能性】
【0310】
本発明により提供されるイネの第2ASAのαサブユニットをコードする遺伝子DNAは、単独又は他の遺伝子と組み合わせて植物体に導入することにより、植物体中の必須アミノ酸であるトリプトファンの含量を高めることができ、栄養価種の高い植物の育種に有用である。また、本明細書に記載されるイネのASA遺伝子のプロモーターDNAは、植物体中で外来の有用遺伝子の発現を行う際のプロモーターとして利用することができ、形質転換植物の作出するのに有効である。
【図面の簡単な説明】
【0311】
【図1】図1は、イネのカルス細胞を、ウイスカー法による外来遺伝子の直接導入法により形質転換するのに用いる外来遺伝子の導入用の組換えベクターであって、配列表の配列番号12の塩基配列をもつ第3の発明の改変D配列を内部に含有する組換えベクターpUBdDを、ベクターpUBAから作成するのに実施例3の(1) で用いた手順を図解的に示す。
【図2】図2は、イネのカルス細胞を、アグロバクテリウム法による外来遺伝子の導入法により形質転換するのに用いる外来遺伝子の導入用の組換えベクターであって、第3の発明の改変D配列を内部に含有するベクターpUb-OSASA1Dを、ベクターp1G121-Hmから作成するのに実施例3の(3)で用いた手順を図解的に示す。
【図3】図3は、イネのASAのためのプロモーター配列の活性の検定の試験において、イネのカルス細胞を形質転換するのに用いるべき外来遺伝子として該プロモーター配列を含有する組換えベクターであって、配列表の配列番号3に示された塩基配列を有するプロモーター配列を内部に含有するDNA断片#3が連結された組換えベクターpGUS#3を、ベクターpBI101から作成するのに実施例5の(6) で用いた手順を図解的に示す。
【特許請求の範囲】
【請求項1】
配列表の配列番号11に示すアミノ酸配列を有するタンパク質であって、イネのアントラニル酸シンターゼの第2アイソザイムのαサブユニットであるタンパク質をコードするDNA。
【請求項2】
配列表の配列番号10に記載の塩基配列を有するDNAである、請求項1に記載のDNA。
【請求項3】
イネのアントラニル酸シンターゼの第2アイソザイムのαサブユニットをコードするところの、配列表の配列番号10の塩基配列をもつ請求項2に記載のDNAを担持する組換えベクターを導入されて形質転換された植物細胞を有し、そして導入された前記DNAが発現可能であることを特徴とする、形質転換植物。
【請求項4】
請求項3に記載の形質転換植物から採取された種子。
【請求項5】
配列表の配列番号10に記載の塩基配列を有するDNA配列を担うDNA断片が組み込まれた組換えベクターであって、宿主細胞中で該DNA配列を発現することができる組換えベクター。
【請求項6】
配列表の配列番号10に記載の塩基配列を有するDNA配列を担うDNA断片が組み込まれた組換えベクターであって、宿主細胞中で該DNA配列を発現することができる組換えベクターで形質転換された大腸菌。
【請求項7】
アントラニル酸シンターゼの第1または第2アイソザイムのαサブユニットのタンパク質をコードして且つ配列表の配列番号1または配列番号10の塩基配列を有するDNAを担持する組換えベクターであって、しかも該DNAが植物中で発現可能である組換えベクターを、植物細胞カルスに導入し、このことによって、該DNAで形質転換された植物細胞カルスを収得し、さらに該カルスを植物体に再生することからなる、植物のトリプトファン含有量の増加方法。
【請求項1】
配列表の配列番号11に示すアミノ酸配列を有するタンパク質であって、イネのアントラニル酸シンターゼの第2アイソザイムのαサブユニットであるタンパク質をコードするDNA。
【請求項2】
配列表の配列番号10に記載の塩基配列を有するDNAである、請求項1に記載のDNA。
【請求項3】
イネのアントラニル酸シンターゼの第2アイソザイムのαサブユニットをコードするところの、配列表の配列番号10の塩基配列をもつ請求項2に記載のDNAを担持する組換えベクターを導入されて形質転換された植物細胞を有し、そして導入された前記DNAが発現可能であることを特徴とする、形質転換植物。
【請求項4】
請求項3に記載の形質転換植物から採取された種子。
【請求項5】
配列表の配列番号10に記載の塩基配列を有するDNA配列を担うDNA断片が組み込まれた組換えベクターであって、宿主細胞中で該DNA配列を発現することができる組換えベクター。
【請求項6】
配列表の配列番号10に記載の塩基配列を有するDNA配列を担うDNA断片が組み込まれた組換えベクターであって、宿主細胞中で該DNA配列を発現することができる組換えベクターで形質転換された大腸菌。
【請求項7】
アントラニル酸シンターゼの第1または第2アイソザイムのαサブユニットのタンパク質をコードして且つ配列表の配列番号1または配列番号10の塩基配列を有するDNAを担持する組換えベクターであって、しかも該DNAが植物中で発現可能である組換えベクターを、植物細胞カルスに導入し、このことによって、該DNAで形質転換された植物細胞カルスを収得し、さらに該カルスを植物体に再生することからなる、植物のトリプトファン含有量の増加方法。
【図1】

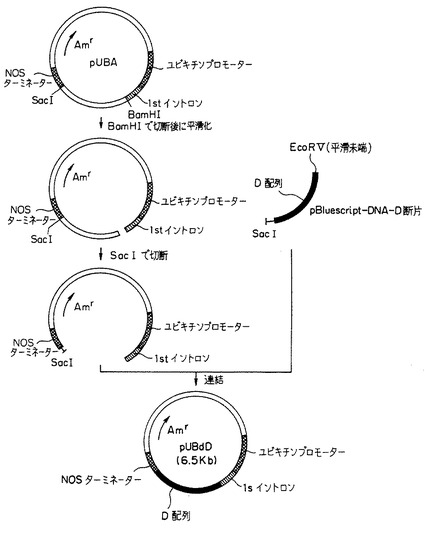
【図2】
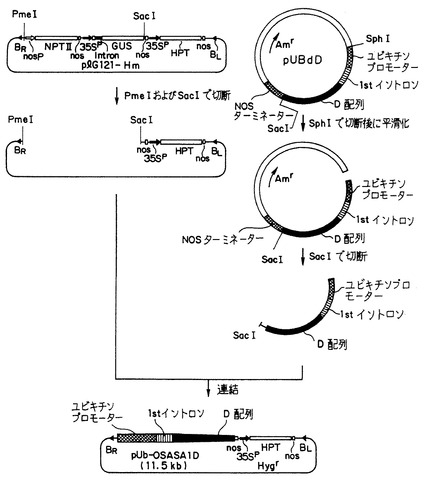
【図3】
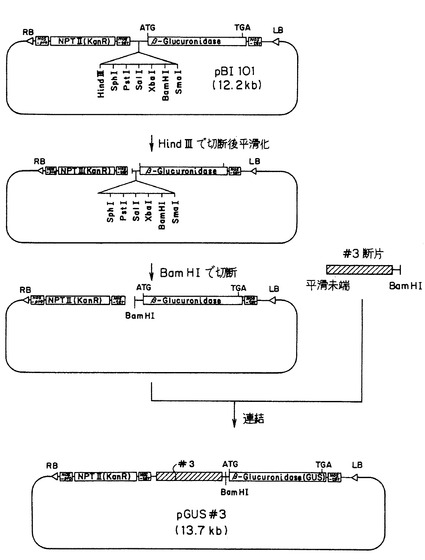
【図2】
【図3】
【公開番号】特開2008−167758(P2008−167758A)
【公開日】平成20年7月24日(2008.7.24)
【国際特許分類】
【出願番号】特願2008−23802(P2008−23802)
【出願日】平成20年2月4日(2008.2.4)
【分割の表示】特願2000−508808(P2000−508808)の分割
【原出願日】平成10年8月31日(1998.8.31)
【出願人】(000242002)北興化学工業株式会社 (182)
【出願人】(501203344)独立行政法人農業・食品産業技術総合研究機構 (827)
【Fターム(参考)】
【公開日】平成20年7月24日(2008.7.24)
【国際特許分類】
【出願日】平成20年2月4日(2008.2.4)
【分割の表示】特願2000−508808(P2000−508808)の分割
【原出願日】平成10年8月31日(1998.8.31)
【出願人】(000242002)北興化学工業株式会社 (182)
【出願人】(501203344)独立行政法人農業・食品産業技術総合研究機構 (827)
【Fターム(参考)】
[ Back to top ]