
イノシトール系分子輸送体及びその製造方法

【課題】 治療物質の生体膜透過輸送を効果的に向上させるイノシトール系分子輸送体及びその製造方法を提供する。
【解決手段】 本発明に基づくイノシトール誘導体は細胞膜、核膜、血液脳関門等のような生体膜の透過性に優れ、制限された生体膜通過性のため医薬品としての開発が困難な薬物又は診断試薬等を細胞、組織、臓器内に効果的に輸送することができる。
【解決手段】 本発明に基づくイノシトール誘導体は細胞膜、核膜、血液脳関門等のような生体膜の透過性に優れ、制限された生体膜通過性のため医薬品としての開発が困難な薬物又は診断試薬等を細胞、組織、臓器内に効果的に輸送することができる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、イノシトール系分子輸送体及びその製造方法に関する。
【背景技術】
【0002】
細胞の細胞膜(plasma membrane)は細胞内小器官が存在している細胞質(cytoplasm)を外部環境から保護する役割を果たし、リン脂質二重層及びこれらの内部にまたは表面に結合されている蛋白質で構成されている。一般的に細胞膜は特定物質のみを細胞内外に出入りさせる門番(gate−keeper)の役割をする。しかし、このような細胞膜の選択的透過性のために有用な治療剤の細胞膜透過が制限され、特に親水性分子、多荷電分子、およびペプチド、オリゴヌクレオチド(例:核酸及び遺伝子)等のような巨大分子等は細胞膜を通過しにくい。従って、このような薬物及び巨大分子達を細胞内へ効率的に輸送するための方法について研究が進んでいる。
【0003】
今まで多くの輸送体分子が様々な種類の薬物及び巨大分子等を細胞内へ輸送するため開発されており、例として陽電荷を帯びた脂質、ポリ−リジンのような陽電荷の重合体及び陽電荷を帯びたデンドリマーなどがある。しかし、一般的にこのような脂質、重合体及びデンドリマーは細胞内で不溶性を示したり生分解されないので沈殿を形成し、毒性を引き起こすなどの問題があった。
【0004】
一方、HIVウィルス複製に必須の転写活性因子(trans−activator)として知られているTat(transacting transcriptional activator)蛋白質の塩基性領域(即ち、49−57番目アミノ酸)がペプチドの細胞膜透過に決定的な役割を果たすことが報告されている。また、このようなTATの塩基性領域と類似した細胞膜透過機能を行う蛋白質変換領域(PTD、protein transduction domain)を有する蛋白質として、下記表1のようなAntpホメオドメイン(Antennapedia homeodomain)蛋白質、ヘルペス(Herpes)ウィルス蛋白質VP22、NLS(nuclear localization signal)序列等が報告されている。
【表1】

【0005】
前記蛋白質は細胞に存在する特定受容体(receptor)や輸送体(transporter)の助けを受けずに生体膜を透過でき、これらPTDは主に塩基性アミノ酸残基、特にアルギニン及びリジンで構成されるという共通点を有している。
【0006】
これら蛋白質の細胞膜透過メカニズムに対しては様々な意見があるが、最近の研究によるとPTDを有する細胞透過性蛋白質はエンドサイトーシス(endocytosis)により細胞内に通過する可能性が高いと提案されている。Lebleuなどは蛍光活性化細胞選別(FACS)分析を通じてHIV−1Tat(48−60番目アミノ酸)及びアルギニン9量体(Arg9)がエンドサイトーシスによって細胞内に移動することを報告したが、その詳しいメカニズムは未だ究明されていない(Lebleu,B.ら、Biol.Chem.,278,585,(2003))。
【0007】
また、高い細胞膜透過性を付与するため、複数のアルギニンを有する様々な形態のオリゴメールを合成する研究が試みられてきた。例えば、1991年マン(Mann)らはTat蛋白質がこれらに結合されている分子の生体膜透過輸送には効果的であるが、水不溶性でありしかも細胞膜に過剰に強く結合することにより凝集を起こすので実質的に細胞内に移動するTat蛋白質の数には限界があることを報告した(Mann,D.A.ら、EMBO J.,10、1733(1991))。
【0008】
バルソウム(Barsoum)らはTat蛋白質の短い断片(1−72番目アミノ酸)及び塩基性領域(37−58番目アミノ酸)を含む他の断片もまたそれに結合された分子の生体膜透過輸送を効果的に向上させると報告した。このような研究は塩基性領域を有する小さいペプチドもそれらに結合された分子の生体膜透過輸送を行うことを示すものである(Barsoum、J.ら、Proc. Natl. Acad. Sci. U.S.A.,91,664(1994))。
【0009】
フタキ(Futaki)らは複数のアルギニン残基を有する様々なペプチドに蛍光物質を付着し、これらのマウスマクロファージRAW264.7細胞に対する膜透過性を調査した結果、アルギニン豊富なペプチドがTat蛋白質(49−57番目アミノ酸)と類似した膜透過性を現すことと、その中、8個のアルギニン残基を有するオリゴメールがこれらに結合された分子の生体膜透過輸送に最も効果的であることを確認した(Futaki,S.ら、J.Biol.Chem.,276,5836(2001))。このような研究はアルギニンのグアニジニウム基が生体膜透過輸送において重要な要素であることを示す。
【0010】
ウェンダー(Wender)らは生体膜透過性がペプチド内グアニジニウム基数、リンカーチェーン(linker chain)の長さ及びキラリティ等に依存するという事実に基づいてペプトイド(peptoid)分子輸送体を製作し、ジャーカット(Jurkat)細胞に対するFACS分析を行った結果、L−アルギニン9量体がTat蛋白質(49−57番目アミノ酸)より生体膜透過輸送において20倍程高効率を示し、D−アルギニン9量体も又遥かに高い透過輸送効率を示すことを確認した。それにより、グアニジニウム基を有するペプトイドの膜透過性はアミノ酸のキラリティによって大きく影響されないことが分かる(米国特許第6,495,663号;及びWender,P.A.ら、Proc. Natl. Acad. Sci. U.S.A.,97、13003(2000))。しかし、ポリアルギニンペプチド又はペプトイド分子は生体内で毒性を示すだけでなく、短時間内に代謝された後、肝と腎臓を通じて早期に除去されるという問題がある。
【0011】
そこで本発明者らは、ミオ(myo)−イノシトール及び複数の陽電荷を帯びたグアニジニウム基から製造したイノシトール誘導体がこれらに結合された様々な治療物質の生体膜透過輸送に効果的であることを発見し、本発明に至った。
【特許文献1】米国特許第6,495,663号
【非特許文献1】Lebleu, B.ら著、Biol.Chem., 278, 585、2003年発行
【非特許文献2】Mann, D. A.ら著、EMBO J.,10, 1733、1991年発行
【非特許文献3】Barsoum, J.ら著、Proc. Natl. Acad. Sci. U.S.A., 91, 664、1994年発行
【非特許文献4】Futaki, S.ら著、J. Biol. Chem., 276, 5836、2001年発行
【非特許文献5】Wender, P. A.ら著、Proc. Natl. Acad. Sci. U.S.A.,97,13003、2000年発行
【発明の開示】
【発明が解決しようとする課題】
【0012】
本発明の目的は、治療物質の生体膜透過輸送を効果的に向上させるイノシトール系分子輸送体及びその製造方法を提供することである。
【0013】
本発明の他の目的は、前記イノシトール系分子輸送体を含む細胞内治療物質輸送用組成物を提供することである。
【課題を解決するための手段】
【0014】
本発明の一実施形態によって、本発明は、下記化学式(I)のイノシトール誘導体を提供する:
【化9】
【0015】
前記式で、
R1は
【化10】
【0016】
であり、ここでnは1〜12範囲の整数であり;
R2及びR3はそれぞれ独立的にH、アルキル、アリールアルキル、シクロアルキル、ヘテロアルキル、−(CH2)mNHR’、−(CH2)lCO2R’’、−COR’’’又は−SO2R’’’’であり、その際R’、R’’、R’’’及びR’’’’はそれぞれアルキルであり、mは2〜5範囲の整数で、lは1〜5範囲の整数であり;
pは0〜2範囲の整数であり;
X及びX’はそれぞれ独立的に−O−CO−O−、−O−CO−NH−(CH2)m−O−、−O−CO−(CH2)l−O−又は−O−(CH2)l−CO−NH−(CH2)m−O−であり、ここでmは2〜5範囲の整数、lは1〜5範囲の整数である。
【0017】
本発明の他の実施形態によって、本発明は、
(a)myo−又はscyllo−イノシトールの水酸基を保護して中間体を得る段階;
(b)段階(a)で得た中間体を2つ以上カップリングしてイノシトール重合体を得る段階;
(c)アシル化反応を行って段階(b)で得たイノシトール重合体に一つ以上のアミノ酸を導入する段階;及び
(d)段階(c)で得たイノシトール重合体のアミノ酸N−末端にグアニジニウム基を導入する段階を含む前記化学式(I)のイノシトール誘導体の製造方法を提供する。
【発明の効果】
【0018】
本発明によれば、イノシトール骨格構造にグアニジニウム基を導入して製造された本発明によるイノシトール誘導体は、細胞膜、核膜、血液−脳関門等のような生体膜に対する透過性が優れ、制限された生体膜通過のため医薬品として開発が困難であった薬物又は診断試薬等を細胞、組織、臓器内へ輸送できる分子輸送体として活用することができる。
【発明を実施するための最良の形態】
【0019】
本発明による化学式(I)のイノシトール誘導体は側鎖に多数のグアニジニウム基を有するイノシトール骨格構造を示し、これらの作用基の位置関係によって様々な異性体(isomer)として存在し得る。また、本発明の化学式(I)のイノシトール誘導体は、イノシトール2量体(dimer)(p=0の場合)、イノシトール3量体(trimer)(p=1)又はイノシトール4量体(tetramer)(p=2)であるか、イノシトール2量体及び3量体が好ましい。
【0020】
本発明の好ましい実施例としては、nが3〜8範囲の整数である式(II)の誘導体であり、このようなn値を有する化合物はこれらに結合された分子らの生体膜透過輸送を向上させる効果を提供する。
【0021】
本発明による好ましいイノシトール誘導体としては下記化学式(XV)の化合物である:
【化11】
【0022】
前記式で、R1、R2、R3、X、X’及びpは前記で定義した通りである。
【0023】
化学式(I)の化合物中、好ましいイノシトール2量体(p=0)としては下記化学式(II)〜(IV)の化合物である:
【化12】
【0024】
前記式で、R1、R2、R3及びXは前記で定義した通りである。
【0025】
前記化学式(II)〜(IV)のイノシトール誘導体における置換基R2及びR3は、作用基を通じて本発明のイノシトール誘導体に結合できる薬物及び診断試薬のような治療物質であり得る。このような物質は分子量が100〜1,500g/molである薬物、または、ペプチド及び核酸等のような重合体化合物であり得る。
【0026】
本発明の好ましい実施例としては、イノシトール誘導体が多様な鎖長のグアニジニウム側鎖を有しながら炭酸塩、カルバマート、エステル又はアミド基で繋がった形のイノシトール2量体でもあり得る。
【0027】
本発明によるイノシトール誘導体は、これらに結合された薬物、診断試薬又は蛍光物質等のような様々な種類の物質を細胞膜、核膜及び血液−脳関門(blood−brain barrier)などのような生体膜を透過させて容易に輸送することができる。
【0028】
前記化学式(II)〜(IV)のイノシトール誘導体は、
(a)myo−又はscyllo−イノシトールの水酸基を保護する反応を行って中間体を得る段階;
(b)段階(a)で得た中間体を2つ以上カップリングしてイノシトール重合体を得る段階;
(c)段階(b)で得たイノシトール重合体のアシル化反応を行い一つ以上のアミノ酸を導入する段階;及び
(d)段階(c)で得たイノシトール重合体のN−末端にグアニジニウム基を導入する段階を含む方法で製造することができる。
【0029】
具体的には、段階(a)では、公知の方法、例えば文献[Chung、Sung−Keeら、Daewoo Science Book series、Natural Science 122,Minumsa(1998)]に記載された方法に基づいて、myo−又はscyllo−イノシトールの水酸基に位置選択的にそれぞれ適切な保護基を導入することができ、このように得られた好ましい中間体の例としては下記化学式(V)〜(XIII)の化合物がある:
【化13】
【0030】
前記式でR’、R’’、l及びmは前記で定義した通りであり、Bnはベンジル、PMBはp−メトキシベンジルである。
【0031】
前記化学式(V)〜(VII)の化合物は本発明の化学式(II)の化合物を製造するための中間体であり、それらのうち、化学式(V)及び(VI)の化合物は下記反応式1に示したように、myo−イノシトールを出発物質として2,3:5,6−ジ−O−イソプロピリデン−myo−イノシトールを合成し、該化合物の1−OH又は4−OHに位置選択的にそれぞれ適切な保護基を導入して製造することができる。
【化14】
【0032】
前記式で、TBDMSはtert−ブチルジメチルシリル、PMBはp−メトキシベンジル、Bnはベンジルである。
【0033】
化学式(II)の化合物の製造において出発物質として使われるmyo−イノシトールは価格が比較的安く、かつ工程を単純化させ得る。しかし、前記反応式1に示したようにmyo−イノシトールから製造された化学式(II)の化合物は立体異性体(diastereomer)の混合物であるため一つの形態の異性体として得るためには化学式(III)及び(IV)の化合物の一つを利用して化学式(I)の化合物を製造することができる。
【0034】
前記化学式(VIII)〜(X)の化合物は化学式(III)の化合物を製造するための中間体であり、それらのうち、化学式(VIII)及び(IX)の化合物らは、下記反応式2に示したように、myo−イノシトールを出発物質として1,6:3,4−ジ−O−イソプロピリデン−myo−イノシトールを合成した後、該化合物の2−OH又は5−OHに位置選択的にそれぞれ適切な保護基を導入して製造することができる。
【化15】
【0035】
前記式でBzはベンゾイルでありPMB及びBnは反応式1で定義した通りである。
【0036】
また、化学式(XI)〜(XIII)の化合物は本発明の化学式(IV)の化合物を製造するための中間体であって、それらのうち、化学式(XI)及び(XII)の化合物は、下記反応式3に示したように、ミツノブ反応(Mitsunobu reaction)を通じてmyo−イノシトールの2−OHの立体構造を反転させてから、ここから1,6:3,4−ジ−O−イソプロピリデン−scyllo−イノシトールを合成した後、該化合物の2−OH又は5−OHに位置選択的にそれぞれ適切な保護基を導入して製造することができる。
【化16】
【0037】
前記式でBz、Bn及びPMBは前記反応式1及び2に定義された通りである。
【0038】
前記反応式2又は3により、純粋な立体異性体(stereoisomer)を得ることができる。
【0039】
また、カルボキシラート基及びアミン基を含む化学式(VII)、(X)及び(XIII)の化合物は、それぞれ1つの水酸基を有する化学式(V)、(VI)、(VIII)、(IX)、(XI)又は(XII)の化合物を位置選択的にアルキル化させて製造することができ、その例として、化学式(XIII)の化合物の製造工程を下記反応式4に示した。化学式(VII)及び(X)の化合物もそれぞれ下記反応式4と類似する工程で製造することができる。
【化17】
【0040】
前記式で、R’、R’’、l、m、Bn及びBzは前記で定義された通りである。
【0041】
段階(b)では文献[The Chemical Synthesis of Peptides、J.Jones、Clarendon Press、Oxford、1991]に記載された方法に基づき、段階(a)で得た2つ以上の中間体を様々な作用基(X及びX’)を利用してカップリングさせてから、アセトニド保護基を取り除くことによりイノシトール重合体を製造することができる。前記作用基(X及びX’)はイノシトール誘導体をカップリングさせるためのリンカーとして作用し、本発明では通常的に使われる様々なリンカーがX及びX’として使用可能であるが、特に−O−CO−O−、−O−CO−NH−(CH2)m−O−、−O−CO−(CH2)l−O−又は−O−(CH2)l−CO−NH−(CH2)m−O−(mは2〜5範囲の整数であり、lは1〜5範囲の整数である)を使うことが好ましい。
【0042】
段階(c)では、ジシクロヘキシルカルボジイミド又は1−[3−(ジメチルアミノ)プロピル)]−エチルカルボジイミド塩酸塩のような縮合剤(condensing agent)を使用して多様な鎖長を有するアミノ酸を段階(b)で得たイノシトール重合体に導入することができる。
【0043】
本発明で使われる多様な鎖長を有するアミノ酸は商業的に利用可能なω−アミノ酸から得ることができ、好ましくは文献[Protective Groups in Organic Synthesis 3rd Ed. T. W. Greene and P. G. M. Wuts、Wiley−Interscience,1999]に記載されたように、アミノ保護基で保護された下記化学式(XIV)のアミノアルカン酸(aminoalkanoic acid)を使うことができる。
【化18】
【0044】
前記アシル化反応は、段階(b)で得たイノシトール重合体1当量当り化学式(XIV)の化合物10〜20当量を用いて0〜60℃範囲の温度で5〜96時間行うことができる。
【0045】
段階(d)では、段階(c)で得たイノシトール重合体のアミノ酸N−末端に存在するt−Boc(tert−ブチルオキシカルボニル)のようなアミノ酸保護基を取り除いた後、有機溶媒中で塩基存在下でN,N−ジ−Boc−N’−トリフロロメタンスルホニルグアニジン(trifluoromethanesulfonylguanidine)と反応させグアニジニウム基を導入することができる。前記N,N−ジ−Boc−N’−トリフロロメタンスルホニルグアニジンは、製造例10および11に示したように、文献[T.T.Bakerら、J.Org.Chem.,65,9054,(2000)]に記載された方法に基づいて製造することができる。
【0046】
前記段階(d)で使える有機溶媒としては、ジメチルホルムアミド、クロロホルム及び酢酸エチル等があり、塩基はトリエチルアミン等があり得る。また、前記反応は0〜60℃で12〜120時間行うことができる。
【0047】
前記段階(b)〜(d)において、化学式(V)〜(XIII)の中間体は多様な組合せでカップリングされて化学式(II)〜(IV)の化合物に変換することができる。本発明によるイノシトール誘導体の製造方法の一例を下記反応式5に示す。
【化19】
【0048】
前記式で、R1、R2及びnは前記で定義した通りである。
【0049】
前記反応式5に示したように、炭酸塩(X=−O−CO−O−)リンカーを用いてカップリングされたイノシトール2量体形態である、グアニジニウム基を有するイノシトール誘導体は化学式(V)及び(VI)の化合物を縮合剤の存在下でカップリングさせて製造することができる。具体的に言えば、化学式(VI)の化合物と化学式(V)の化合物をカップリングさせ、それからアセトニド保護基を除去する。このように得たイノシトール2量体にt−Boc(tert−butyloxycarbonyl)基で保護された多様な鎖長(R1に当たる)を有するアミノ酸をアシル化(acylation)反応を通じて導入し、PMB保護基を除去してからダンシル(dansyl,5−ジメチルアミノ−1−ナフタレンスルホニル(5−dimethylamino−1−naphthalenesulfonyl))基(R2)のような蛍光マーカー(fluorescent tag)を導入する。それからアミノ酸N−末端のt−Boc保護基を取り除き、そこにグアニジニウム基を導入して本発明のイノシトール誘導体を製造することができる。
【0050】
さらに、前記で得られた蛍光物質で表示されたイノシトール誘導体のベンジル保護基を取り除いた後、治療物質(薬物)又は診断試薬(R3に当たる)等を結合させることができる。
【0051】
エステル(X=−O−CO−(CH2)l−O−)、アミド(X=−O−(CH2)l−CO−NH−(CH2)m−O−)及びカルバマート(X=−O−CO−NH−(CH2)m−O−)基を通じて繋がったイノシトール単位を有するイノシトール重合体は文献[The Chemical Synthesis of Peptides、J. Jones、Clarendon Press、Oxford、1991]に記載された方法に基づいて製造することができる。
【0052】
本発明の化学式(I)のイノシトール誘導体は、前記例示した中間体、すなわち化学式(V)〜(XIII)の化合物ばかりでなく、他の中間体からも前記カップリング及びグアニジニウム基を有するアミノ酸の導入工程を含む方法と類似した方法で製造することができる。
【0053】
本発明によるイノシトール誘導体は、薬物及び診断試薬のような治療用物質との結合体でもあり得、本発明によるイノシトール誘導体は様々な治療用物質が細胞膜、核膜及び血管−脳関門等の生体膜を透過して移動することを効果的に向上させる。
【実施例】
【0054】
以下、本発明を下記実施例によって更に詳細に説明する。但し、これらは本発明を例示するためのものであり、本発明の範囲を制限しない。
【0055】
[製造例1]:(±)−1,4−ジ−O−ベンゾイル−2,3:5,6−ジ−O−イソプロピリデン−myo−イノシトールの製造
myo−イノシトール(50g、278mmol)、2,2−ジメトキシプロパン(150ml、1.2mol)及びp−トルエンスルホン酸(1g、5.2mmol)をN,N−ジメチルホルムアミド(200ml)に溶解してから100℃で21時間還流させた。この反応物を常温に冷やしてからトリエチルアミン(10ml)を加え、沈殿物を濾過した後、濾過液にトルエン(25ml)を加えて減圧蒸発させた。得られた残留物をピリジン(150ml)に溶解し、これに塩化ベンゾイル(200ml、1.74mol)を0℃で30分間シリンジで滴加した。その後、反応液を常温で2時間攪拌してから濾過し、沈殿物をピリジン、水、アセトン及びジエチルエーテルで順次洗滌して白色固体状の表題化合物(35.55g)を得た。
【0056】
m.p.=322−325℃
1H−NMR(CDCl3):1.30、1.43、1.50、1.63(4s、3H)、3.73(dd、J=9.6Hz、11.1Hz、1H)、4.36(dd、J=9.6Hz、10.6Hz、1H)、4.41(dd、J=4.5Hz、9.6Hz、1H)、4.78(dd、J=4.5Hz、4.5Hz、1H)、5.42(dd、J=4.5Hz、10.6Hz、1H)、5.60(dd、J=9.6Hz、11.1Hz、1H)、7.45(m、5H)。
【0057】
[製造例2]:(±)−2,3:5,6−ジ−O−イソプロピリデン−myo−イノシトールの製造
製造例1で得た(±)−1,4−ジ−O−ベンゾイル−2,3:5,6−ジ−O−イソプロピリデン−myo−イノシトール(35.5g、75.8mmol)及びNaOCH3(2.41g、45.48mmol)をCH3OH(500ml)に溶解し16時間還流させた。この反応物を常温に冷やしてから減圧蒸発させ、残留物にCH2Cl2(700ml)を加えてシリカベット(silica bed)に通して濾過し、濾過液を濃縮して白色固体状の表題化合物(18.21g)を得た。
【0058】
m.p.=169−171℃
1H−NMR(CDCl3):1.38、1.46、1.48、1.54(4s、3H)、2.36(d、J=8.8Hz、1H)、2.45(d、J=2.9Hz、1H)、3.32(dd、J=9.4Hz、10.5Hz、1H)、3.83(dd、J=9.4Hz、9.4Hz、1H)、3.90(ddd、J=2.9Hz、6.4Hz、10.5Hz、1H)、4.02(ddd、J=4.8Hz、8.8Hz、9.4Hz、1H)、4.08(dd、J=4.8Hz、6.4Hz、1H)、4.48(dd、J=4.8Hz、4.8Hz、1H)。
【0059】
[製造例3]:(±)−1−O−t−ブチルジメチルシリル−2,3:5,6−ジ−O−イソプロピリデン−myo−イノシトールの製造
製造例2で得た(±)−2,3:5,6−ジ−O−イソプロピリデン−myo−イノシトール(6g、23mmol)及びイミダゾール(5.2g、76mmol)をN,N−ジメチルホルムアミド(70ml)に溶解し、それに0℃でtert−ブチルジメチルシリルクロライド(3.8g、25mmol)を添加して常温で14時間攪拌させた。反応混合物を酢酸エチルで抽出した後、NaClとNaHCO3飽和水溶液で洗滌し、MgSO4で乾燥させてから減圧濃縮させた。濃縮物をカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:10〜1:1)で精製して白色固体状の表題化合物(5.84g)を得た。
【0060】
m.p.=148−150℃
1H−NMR(CDCl3):0.14(s、6H)、0.93(s、9H)、1.35、1.42、1.44、1.53(4s、3H)、2.87(brs、1H)、3.26(dd、J=9.4Hz、10.4Hz、1H)、3.87(dd、J=6.4Hz、10.4Hz、1H)、3.90(dd、J=9.4Hz、10.4Hz、1H)、3.97(dd、J=4.4Hz、4.6Hz、1H)、4.03(dd、J=4.4Hz、10.4Hz、1H)、4.29(dd、J=4.4Hz、4.4Hz、1H)。
【0061】
[製造例4]:(±)−4−O−ベンジル−2,3:5,6−ジ−O−イソプロピリデン−1−O−t−ブチルジメチルシリル−myo−イノシトールの製造
製造例3で得た(±)−1−O−t−ブチルジメチルシリル−2,3:5,6−ジ−O−イソプロピリデン−myo−イノシトール(5.76g、15mmol)をCH2Cl2(100ml)に溶解してから酸化銀(I)(10.7g、46mmol)、ベンジルブロマイド(5.47ml、46mmol)及びテトラブチルアンモニウム・ヨウ化物(0.55g、1.5mmol)を加えて常温で2時間攪拌させた。得られた溶液をセライトで濾過させ濾過液をCH2Cl2で洗滌した後減圧濃縮させ、濃縮物をカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:40)で精製して白色固体状の表題化合物(4.91g)を得た。
【0062】
m.p.=89℃
1H−NMR(CDCl3):0.12(s、6H)、0.90(s、9H)、1.31、1.35、1.40、1.41(4s、3H)、3.31(t、J=9.7Hz、1H)、3.65(dd、J=6.6Hz、10.4Hz、1H)、3.85(t、J=9.3Hz、1H)、3.98(dd、J=4.3Hz、10.1Hz、1H)、4.07(t、J=5.6Hz、1H)、4.27(t、J=5.6Hz、1H)、4.80(s、2H)、7.21−7.40(m、5H)
MS(FAB)m/z465.21(M++H)。
【0063】
[製造例5]:(±)−4−O−ベンジル−2,3:5,6−ジ−O−イソプロピリデン−myo−イノシトール(化学式(V)の化合物)の製造
製造例4で得た(±)−4−O−ベンジル−2,3:5,6−ジ−O−イソプロピリデン−1−O−t−ブチルジメチルシリル−myo−イノシトール(4.6g、9.9mmol)をテトラヒドロフランに溶解し、これにフッ化テトラブチルアンモニウムをTHFに溶解して作った1.0M溶液(29.7ml、29.7mmol)を加えて常温で7時間攪拌させた。得られた溶液を酢酸エチルで抽出した後、NaCl飽和水溶液で洗滌し、有機層をMgSO4で乾燥させた後、減圧蒸発させ濃縮させてからヘキサンで再結晶して白色固体状の表題化合物(3.7g)を得た。
【0064】
m.p.=131℃
1H−NMR(CDCl3):1.33、1.36、1.43、1.46(4s、3H)、2.40(d、J=8.4Hz、1H)、3.39(t、J=9.6Hz、1H)、3.65(dd、J=6.4Hz、10.4Hz、1H)、3.78(t、J=9.8Hz、1H)、3.94−4.02(m、1H)、4.18(t、J=5.6Hz、1H)、4.24(t、J=4.9Hz、1H)、4.80(s、2H)、7.22−7.40(m、5H)。
【0065】
[製造例6]:(±)−1−O−p−メトキシベンジル−2,3:5,6−ジ−O−イソプロピリデン−myo−イノシトール(化学式(VI)の化合物)の製造
製造例2で得た(±)−2,3:5,6−ジ−O−イソプロピリデン−myo−イノシトール(5.59g、20mmol)をN,N−ジメチルホルムアミド(90ml)に溶解してからNaH(0.96g、40mmol)及びp−メトキシベンジルクロライド(3.2ml、24mmol)を加えて常温で攪拌させた。10時間後反応が完結したら、0℃で水を加えて反応を中断させた。得られた溶液をCH2Cl2で抽出した後、NaHCO3飽和水溶液で洗滌し有機層をMgSO4で乾燥させてから減圧蒸発させて濃縮し、それをカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:2〜1:1)で精製して白色固体状の表題化合物(2.2g)を得た。
【0066】
m.p.=153−154℃
1H−NMR(CDCl3):1.32、1.44、1.46、1.52(4s、3H)、2.27(d、J=2.5Hz)、3.24(dd、J=10.1Hz、10.1Hz、1H)、3.75(dd、J=4.3Hz、10.1Hz、1H)、3.79(s、3H)、3.83−3.92(m、2H)、4.01(dd、J=10.1Hz、10.1Hz、1H)、4.27(dd、J=4.3Hz、4.3Hz、1H)、4.71(dd、J=12.1Hz、1H)、4.82(d、J=12.1Hz、1H)、6.86(d、J=8.6Hz、2H)、7.32(d、J=8.6Hz、2H)。
【0067】
[製造例7]:4−Boc−アミノブタン酸(4−Boc−aminobutanoic acid)の製造
4−アミノブタン酸(1g、9.7mmol)をtert−ブチルアルコール(15ml)に溶解してから5N NaOH(1.93ml、9.7mmol)を加えて常温で10分間攪拌させた。これにtert−ブチルアルコール(17ml)に溶解したジ−tert−ブチル−ジカーボネート(di−tert−butyl−dicarbonate)(2.33g、10.7mmol)を加えて常温で24時間攪拌させた。これに水(12ml)を加え減圧濃縮させた後0℃に冷却させ、反応溶液のpHが2になるまで2N H2SO4を滴加した。得られた溶液を酢酸エチルで抽出して蒸留水(H2O)で洗滌した後有機層をMgSO4で乾燥させ、これを減圧濃縮させて白色固体状の表題化合物(2.1g)を得た。
【0068】
m.p.=57−58℃
1H−NMR(CDCl3):1.45(s、9H)、3.19(m、2H)、4.70(brs、1H)。
【0069】
[製造例8]:6−Boc−アミノヘキサン酸(6−Boc−aminohexanoic acid)の製造
6−アミノカプロン酸(6−aminocaproic acid)(10g、76mmol)をtert−ブチルアルコール(100ml)に溶解してから5N NaOH(15ml、76mmol)を加えて常温で10分間攪拌させた。前記反応液にtert−ブチルアルコール(100ml)に溶解したジ−tert−ブチル−ジカーボネート(18.3g、83mmol)を加えた後、常温で24時間攪拌させた。これに蒸留水(100ml)を加えた後、減圧蒸発させ濃縮させてから0℃に冷却させ溶液のpHが2になるまで2N H2SO4を滴加した。引き続き、得られた溶液を酢酸エチルで抽出し蒸留水で洗滌した後有機層をMgSO4で乾燥させてから減圧蒸発させ濃縮させ、カラムクロマトグラフィー(酢酸エチル:トルエン:酢酸=20:10:0.3)で精製して白色固体状の前記表題化合物(17.9g)を得た。
【0070】
m.p.=38−39℃
1H−NMR(CDCl3):1.30(m、2H)、1.40(s、9H)、1.50(m、2H)、2.28(t、J=7.4Hz、2H)、3.01(m、2H)。
【0071】
[製造例9]:8−Boc−アミノオクタン酸(8−Boc−aminooctanoic acid)の製造
8−アミノカプリン酸(8−aminocaprylic acid)(970.3mg、6.1mmol)をtert−ブチルアルコール(14.7ml)に溶解してから5N NaOH(1.44ml、6.1mmol)を加え常温で10分間攪拌させた。前記反応液にtert−ブチルアルコール(10ml)に溶解したジ−tert−ブチル−ジカーボネート(1.46g、6.7mmol)を加えて常温で24時間攪拌させた。これに蒸留水(10ml)を加えた後減圧蒸発・濃縮させてから0℃に冷却させ溶液のpHが2になるまで2N H2SO4を滴加した。引き続き、得られた溶液を酢酸エチルで抽出し蒸留水で洗滌した後有機層をMgSO4で乾燥させてから減圧揮発して濃縮させ、カラムクロマトグラフィー(酢酸エチル:トルエン:酢酸=20:10:0.3)で精製して白色固体状の前記表題化合物(1.78g)を得た。
【0072】
m.p.=57−58℃
1H−NMR(CDCl3):1.30(m、6H)、1.45(s、9H)、1.46(m、2H)、1.63(m、2H)、2.34(t、J=7.4Hz、2H)、3.10(m、2H)。
【0073】
[製造例10]:N,N−ビス−Boc−グアニジンの製造
グアニジン塩酸塩(guanidine hydrochloride)(4g、42mmol)及び4N NaOH(42ml、0.1mol)を1,4−ジオキサン(80ml)に溶解し0℃に冷却させた。これにジ−tert−ブチル−ジカーボネート(20g、92.1mol)を加えた後、常温に加熱し18時間攪拌させた。元の反応物容積の1/3になるよう減圧蒸発させた後、水を加えて酢酸エチルで抽出した。得られた溶液を10%クエン酸、蒸留水及びNaCl飽和水溶液順に洗滌しMgSO4で乾燥させた後、減圧蒸発させて濃縮させ、カラムクロマトグラフィー(CH2Cl2:MeOH=20:1)で精製して前記表題化合物(6.47g)を得た。
【0074】
m.p.=144−145℃
1H−NMR(DMSO−d6):1.39(s、18H)、8.47(brs、2H)、10.42(brs、1H)。
【0075】
[製造例11]:N,N−ジ−Boc−N’−トリフロロメタンスルホニルグアニジン(N,N−di−Boc−N’−trifluoromethanesulfonylguanidine)の製造
製造例10で得たN,N−ビス−Boc−グアニジン(6.05g、23mmol)をCH2Cl2(40ml)に溶解してからトリエチルアミン(4.8ml、34.5mmol)を滴加し、−78℃でトリフリックアンハイドライド(triflic anhydride)(3.9ml、25.3mmol)を注射器で滴加した。得られた溶液を4時間常温に加熱した後、常温で3時間攪拌させた。反応生成物を2N NaSO4及び蒸留水で洗滌し有機層をNa2SO4で乾燥させた後減圧蒸発・濃縮させてカラムクロマトグラフィー(CH2Cl2)で精製した。これをヘキサンで再結晶して白色固体状の前記表題化合物(7.09g)を得た。
【0076】
m.p.=114−115℃
1H−NMR(DMSO−d6):1.45(s、18H)、11.45(brs、2H)。
【0077】
[製造例12]:2−O−ベンゾイル−myo−イノシトールオルトギ酸塩(2−O−benzoyl−myo−inositol orthoformate)の製造
myo−イノシトール(50g、278mmol)、オルトギ酸トリメチル(trimethyl orthoformate)(63ml、570mmol)及びp−トルエンスルホン酸(2.0g)をN,N−ジメチルホルムアミド(200ml)に溶解してから120℃で7時間攪拌した。得られた溶液を50℃で2時間減圧蒸留した後、ピリジン(100ml)を加え0℃で塩化ベンゾイル(35ml、299mmol)を滴加した。得られた溶液を常温で一日攪拌した後、蒸留水(10ml)を加え30分間攪拌してから反応液を酢酸エチルで希釈しNaHSO4、NaHCO3及びNaCl飽和水溶液で洗滌した。引き続き、有機層をMgSO4で乾燥してから濾過し濾過液を濃縮した後、酢酸エチルで再結晶して白色固体状の前記表題化合物(35.5g)を得た。
【0078】
m.p.=206−207℃
1H−NMR(acetone−d6、with drops of D2O):δ4.26(m、1H)、4.37(m、2H)、4.50(t、J=3.8Hz、2H)、5.52(d、J=1.3Hz、1H)、5.57(app q,J=1.9Hz、1H)、7.50−8.11(m、5H)。
【0079】
[製造例13]:2−O−ベンゾイル−myo−イノシトールの製造
製造例12で得た2−O−ベンゾイル−myo−イノシトールオルトギ酸塩(10g、34mmol)をCH3OH(200ml)に溶解しp−トルエンスルホン酸(646mg、3.4mmol)を加えてから、60℃で3時間攪拌した。その後、反応液の温度を常温に下げ一日放置し結晶化した後、生成された固体を濾過しCH3OH及び酢酸エチルで洗滌して白色固体状の前記表題化合物(8g)を得た。
【0080】
m.p.=240−242℃
1H−NMR(CD3OD−DMSO−d6):δ3.30(t、J=8.9Hz、1H)、3.66(dd、J=2.6、9.7Hz、2H)、3.73(dd、J=8.9Hz、9.7Hz、2H)、5.69(t、J=2.6Hz、1H)、7.51−8.09(m、5H)。
【0081】
[製造例14]:2−O−ベンゾイル−1,6:3,4−ジ−O−イソプロピリデン−myo−イノシトールの製造
製造例13で得た2−O−ベンゾイル−myo−イノシトール(5.0g、17.6mmol)及び2−メトキシプロペン(10.1ml、105.5mmol)をN,N−ジメチルホルムアミド(100ml)に溶解し0℃でp−トルエンスルホン酸(335mg、1.76mmol)を少しずつ分けて加えた。常温で20時間攪拌した後、反応液をNaHCO3飽和水溶液(200ml)に注ぎ入れて酢酸エチル(400ml)で抽出した後、抽出した有機層をNaCl飽和水溶液(400ml)で洗滌した。引き続き有機層を乾燥(MgSO4)・濃縮してカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:3〜1:1)及び分別結晶(酢酸エチル及びn−ヘキサン)を行い白色固体状の前記表題化合物(2g)を得た。
【0082】
m.p.=181−183℃
1H−NMR(CDCl3):δ1.40、1.48(2s、12H)、2.97(d、J=1.5Hz、1H)、3.83(dd、J=2.1Hz、8.4Hz、2H)、4.09−4.15(m、3H)、6.06(t、J=2.1Hz、1H)、7.47−8.09(m、5H)
MS(FAB)m/z=387(M++Na)、365(M++H)。
【0083】
[製造例15]:(±)−2,3−O−イソプロピリデン−myo−イノシトールの製造
myo−イノシトール(50g、277mmol)、2,2−ジメトキシプロパン(85ml、691mmol)及びp−トルエンスルホン酸(500mg、2.6mmol)をメチルスルホキシド(methyl sulfoxide)(160ml)に溶解してから90〜100℃で1時間攪拌させた。反応溶液を20℃に冷却させた後、エチルアルコール(200ml)及びエーテル(1L)を加えて2時間攪拌させてからトリエチルアミン(10ml)を加えて4時間攪拌させた。反応生成物を濾過した後、CH3OH/エーテル(1:5、240ml)で洗滌し、エチルアルコールで再結晶して白色固体状の前記表題化合物(51g)を得た。
【0084】
m.p.=165−166℃
1H−NMR(CDCl3):δ1.32、1.47(2s、6H)、3.09(app.t、J=9.1Hz、1H)、3.53(dd、J=9Hz、9.3Hz、2H)、3.90(app.t、J=8.7Hz、1H)、4.34(app.t、J=9.3Hz、1H)。
【0085】
[製造例16]:(±)−1,4,5,6−テトラ−O−ベンジル−2,3−O−イソプロピリデン−myo−イノシトールの製造
製造例15で得た(±)−2,3−O−イソプロピリデン−myo−イノシトール(20g、90.8mmol)及びNaH(47.5g、1090mmol)をN,N−ジメチルホルムアミド(600ml)に溶解してから0℃でベンジルブロマイド(108.02ml、908mmol)を入れて2時間攪拌した後、室温に上げさらに20時間攪拌させた。反応生成物を酢酸エチルで抽出しNaHCO3とNaCl飽和水溶液で洗滌した後、MgSO4で乾燥させ、これを減圧蒸発させカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:9)で精製してシロップ状の前記表題化合物(45g)を得た。
【0086】
1H−NMR(CDCl3):δ1.37、1.55(2s、6H)、3.42(t、J=8.7Hz、1H)、3.88(dd、J=3.7Hz、8.8Hz、1H)、3.79(dd、J=7.0Hz、9.5Hz、1H)、3.99(app.t、J=8.5Hz、1H)、4.10(app.t、J=6.2Hz、1H)、4.27(dd、J=3.8Hz、5.5Hz、1H)、4.72−4.90(m、8H)、7.22−7.40(m、20H)。
【0087】
[製造例17]:(±)−1,4,5,6−テトラ−O−ベンジル−myo−イノシトールの製造
製造例16で得た(±)−1,4,5,6−テトラ−O−ベンジル−2,3−O−イソプロピリデン−myo−イノシトール(42g、72.38mmol)を80%酢酸水溶液(290ml)に溶解し、100℃で3時間攪拌した。反応生成物を減圧蒸発・濃縮させた後、酢酸エチル−ヘキサン(1:2)溶液で再結晶し、精製された白色固体状の前記表題化合物(36.5g)を得た。
【0088】
m.p.=126.5−127℃
1H−NMR(CDCl3):δ2.40(d、4.5Hz、1H)、2.45(s、1H)、3.44−3.51(m、3H)、3.85(app.t、J=9.5Hz、1H)、4.0(app.t、J=9.5Hz、1H)、4.21(brs、1H)、4.7−4.97(m、8H)、7.25−7.40(m、20H)。
【0089】
[製造例18]:(±)−1−O−ベンゾイル−2,3,4,5−テトラ−O−ベンジル−scyllo−イノシトールの製造
製造例17で得た(±)−1,4,5,6−テトラ−O−ベンジル−myo−イノシトール(30g、55.53mmol)、トリフェニルホスフィン(triphenyl
phosphine)(17.5g、66.63mmol)、安息香酸(8.1g、66.63mmol)及びジエチルアゾジカルボキシラート(diethyl azodicarboxylate)(10.5ml、66.63mmol)をトルエン(300ml)に溶解して80〜85℃で4時間攪拌させた。生成された溶液を濾過した後、濾過液を減圧蒸発させカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:2)で精製して固体状の前記表題化合物(30.8g)を得た。
【0090】
m.p.=135−136℃
1H−NMR(CDCl3):δ2.45(s、1H)、3.55−3.77(m、5H)、4.7−5.0(m、8H)、5.39(app.t、J=9.8Hz、1H)、7.10−8.05(m、25H)。
【0091】
[製造例19]:1−O−ベンゾイル−scyllo−イノシトールの製造
製造例18で得た(±)−1−O−ベンゾイル−2,3,4,5−テトラ−O−ベンジル−scyllo−イノシトール(8g、12.4mmol)および20%Pd(OH)2/C(4.3g)をCH2Cl2/CH3OH(1:2、210ml)混合溶液に加えてH2(50psi)下で24時間混合させた。得られた溶液を濾過し減圧蒸発させた後、CH3OHに再結晶して固体状の前記表題化合物(3.6g)を得た。
【0092】
m.p.=245−247℃
1H−NMR(CD3OD):δ3.30−3.40(m、3H)、3.55(app.t、J=9.2Hz、2H)、5.1(t、J=9.7Hz、1H)、7.45−8.1(m、5H)。
【0093】
[製造例20]:1−O−ベンゾイル−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトールの製造
製造例19で得た1−O−ベンゾイル−scyllo−イノシトール(6g、21.11mmol)及びp−トルエンスルホン酸(400mg、2.1mmol)をN,N−ジメチルホルムアミド(100ml)に溶解し、これに室温で2−メトキシプロペン(10ml、105.6mmol)をゆっくり30分間滴加した。得られた溶液を2時間攪拌させた後、2−メトキシプロペン(10ml、105.6mmol)を滴加し、8時間後にNaHCO3飽和水溶液を入れ攪拌させ、これを酢酸エチルで抽出してMgSO4で乾燥させた後減圧蒸発させた。反応生成物を再結晶(酢酸エチル:核酸2:1)して立体異性体を互いに分離させてからカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:1)で精製して前記表題化合物(2.1g)を得た。
【0094】
m.p.=284−286℃
1H−NMR(CDCl3):δ1.44、1.48(6H)、2.46(d、J=2.8Hz、1H)、3.76(app.t、J=9.4Hz、2H)、3.88(app.t、J=9.4Hz、2H)、4.13(dt、J=2.8Hz、8.8Hz)、5.7(t、J=9.3Hz、1H)、7.27−8.1(m、5H)。
【0095】
[製造例21]:4−O−(1’−イミダゾリルカルボニルオキシ)−2,3:5,6−ジ−O−イソプロピリデン−1−O−p−メトキシベンジル−myo−イノシトールの製造
製造例6で得た1−O−p−メトキシベンジル−2,3:5,6−ジ−O−イソプロピリデン−myo−イノシトール(2.2g、5.8mmol)をトルエン(40ml)に溶解してからCaH2(0.59g、13.3mmol)およびカルボニルジイミダゾール(carbonyldiimidazole)(2.35g、15.4mmol)を加えて11時間攪拌させた。生成された溶液を濾過した後濾過液を減圧蒸発・濃縮させ、カラムクロマトグラフィー(酢酸エチル:ヘキサン=1:1)で精製して白色固体状の前記表題化合物(2.6g)を得た。
【0096】
m.p.=144−145℃
1H−NMR(CDCl3):1.35、1.46、1.45、1.63(4s、3H)、3.49(dd、J=9.3Hz、11.4Hz、1H)、3.80(s、3H)、3.84(d、J=4.2Hz、1H)、4.17(dd、J=2.4Hz、5.4Hz、1H)、4.21(d、J=9.6Hz、1H)、4.32(dd、J=4.5Hz、1H)、4.74(d、J=12.0Hz、1H)、4.86(d、J=12.0Hz、1H)、5.35(dd、J=6.9Hz、11.1Hz、1H)、6.89(d、J=8.7Hz、2H)、7.05(dd、J=0.6Hz、1.5Hz、1H)、7.35(d、J=8.7Hz、2H)、7.42(dd、J=1.5Hz、1H)、8.12(s、1H)
MS(FAB)m/z497(M++Na)。
【0097】
[製造例22]:2−O−ベンゾイル−1,6:3,4−ジ−O−イソプロピリデン−5−O−p−メトキシベンジル−myo−イノシトールの製造
製造例14で得た2−O−ベンゾイル−1,6:3,4−ジ−O−イソプロピリデン−myo−イノシトール(1g、2.74mmol)、Ag2O(1.27g、5.49mmol)、p−メトキシベンジルクロライド(744μl、5.49mmol)、分子体(molecular sieve)4Å(粉末、1g)及びテトラブチルアンモニウムヨージド(tetrabutylammonium iodide)(101mg、274μmol)をCH2Cl2(10ml)に溶解して常温で30時間攪拌した。反応生成物をカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:2)で精製して結晶化(酢酸エチル及びn−ヘキサン)し、白色固体状の前記表題化合物(837mg)を得た。
【0098】
m.p.=150−152℃
1H−NMR(CDCl3):δ1.35、1.44(2s、12H)、3.77(dd、J=9.5Hz、2.0Hz、2H)、3.80(s、3H)、3.87(t、J=9.1Hz、1H)、4.15(t、J=9.3Hz、1H)、4.80(s、2H)、6.0(t、J=1.9Hz、1H)、6.87(d、J=8.8Hz、2H)、7.35−7.57(m、5H)、8.0(d、J=8.3Hz、2H)
MS(FAB)m/z=507(M++Na)、485(M++H)。
【0099】
[製造例23]:1,6:3,4−ジ−O−イソプロピリデン−5−O−p−メトキシベンジル−myo−イノシトール(化学式(VIII)の化合物)の製造
製造例22で得た2−O−ベンゾイル−1,6:3,4−ジ−O−イソプロピリデン−5−O−p−メトキシベンジル−myo−イノシトール(700mg、1.45mmol)をCH3OH(15ml)に溶解してからNaOCH3(66μl、25%inCH3OH,0.29mmol)を加えて2時間加熱還流した。反応生成物の温度を常温に下げて一日間放置し結晶化した後、生成された固体を濾過してCH3OH及び酢酸エチルで洗滌して白色固体状の前記表題化合物(484mg)を得た。
【0100】
m.p.=191−192℃
1H−NMR(CDCl3):δ1.48、1.50(2s、12H)、2.22(brs、1H)、3.59(dd、J=9.4Hz、2.1Hz、2H)、3.82(s、3H)、3.82(t、J=9.4Hz、1H)、4.12(t、J=9.3Hz、2H)、4.58(broadd、J=1.9Hz、1H)、4.80(s、2H)、6.89(d、J=8.6Hz、2H)、7.37(d、J=8.6Hz、2H)
MS(FAB)m/z=403(M++Na)。
【0101】
[製造例24]:2−O−ベンジル−1,6:3,4−ジ−O−イソプロピリデン−5−O−p−メトキシベンジル−myo−イノシトールの製造
製造例23で得た1,6:3,4−ジ−O−イソプロピリデン−5−O−p−メトキシベンジル−myo−イノシトール(280mg、0.736mmol)をN,N−ジメチルホルムアミド(5ml)に溶解した後、0℃でNaH(70mg、ミネラルオイル中55%、1.47mmol)を加え、常温で30分間攪拌した。これにベンジルブロマイド(256mg、1.47mmol)を加え、常温で1時間攪拌し、これにNaHCO3飽和水溶液(50ml)を加えて反応を終結し、酢酸エチル(300ml)で希釈した。これをNaHCO3(50ml)とNaCl(50ml)飽和水溶液で洗滌した後有機層を乾燥(Na2SO4)・濃縮してカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:2)で精製して白色固体状の前記表題化合物(320mg)を得た。
【0102】
m.p.=180−181℃
1H−NMR(CDCl3):δ1.47、1.48(2s、12H)、3.62(dd、J=9.5Hz、2.0Hz、2H)、3.80(t、J=9.2Hz、1H)、3.82(s、3H)、4.20(t、J=9.3Hz、2H)、4.37(t、J=2.0Hz、1H)、4.80、4.85(2s、4H)、6.90(dm、J=6.7Hz、2H)、7.27−7.39(m、8H)
MS(FAB)m/z=493(M++Na)。
【0103】
[製造例25]:2−O−ベンジル−1,6:3,4−ジ−O−イソプロピリデン−myo−イノシトール(化学式(IX)の化合物)の製造
製造例24で得た2−O−ベンジル−1,6:3,4−ジ−O−イソプロピリデン−5−O−p−メトキシベンジル−myo−イノシトール(283mg、0.60mmol)をCH2Cl2に溶解してから常温でMn(OAc)3(449mg、1.8mmol)及び2,3−ジクロロ−5,6−ジシアノ−1,4−ベンゾキノン(147mg、0.65mmol)を加え12時間攪拌した。得られた反応液にNaHCO3飽和水溶液(30ml)をゆっくり加え、酢酸エチル(50ml)で希釈した後有機層を蒸留水(30ml×2回)及び飽和NaCl水溶液で洗滌した。洗滌した有機層は乾燥(Na2SO4)後濃縮して酢酸エチル:ヘキサン=1:2〜1:1の混合溶液で精製し、白色固体状の前記表題化合物(176mg)を得た。
【0104】
m.p.=190−191℃
1H−NMR(CDCl3):δ1.48、1.49(2s、12H)、2.44(brs、1H)、3.64(dd、J=9.1Hz、1.9Hz、2H)、4.02(m、1H)、4.13(t、J=9.4Hz、2H)、4.40(t、J=2.0Hz、1H)、4.85(s、2H)、6.90(dm、J=6.7Hz、2H)、7.27−7.40(m、5H)
MS(FAB)m/z=373(M++Na)。
【0105】
[製造例26]:2−O−(1’−イミダゾール−イル−カルボニルオキシ)−5−O−p−メトキシベンジル−1,6:3,4−ジ−O−イソプロピリデン−myo−イノシトールの製造
製造例23で得た1,6:3,4−ジ−O−イソプロピリデン−5−O−p−メトキシベンジル−myo−イノシトール(150mg、0.39mmol)をトルエン(3ml)に溶解し常温でCaH2(41mg、0.9mmol)を加えて20分間攪拌した。これにカルボニルジイミダゾール(170mg、0.98mmol)を加えて36時間攪拌し、反応生成物を濾過した濾過液を濃縮した後、カラムクロマトグラフィーで精製して白色固体状の前記表題化合物(184mg)を得た。
【0106】
m.p.=159−162℃
1H−NMR(CDCl3):δ1.41、1.49(2s、12H,2CMe2)、3.80(dd、J=9.5、2.3Hz、2H,H−1&H−3)、3.83(s、3H,OCH3)、3.90(t、J=Hz、9.0Hz、1H,H−5)、4.05(t、J=9.3Hz、2H,H−4&H−6)、4.82(s、2H,PhCH2)、5.92(t、J=2.2Hz、1H,H−2)、6.91(dt、J=8.7、2.9Hz、2H,Ph)、7.13(dd、J=1.54、0.7Hz、1H,imidazole)、7.38(dt、J=8.65、2.7Hz、2H,Ph)7.44(t、J=1.4Hz、1H,imidazole)、8.17(s、1H,imidazole)
MS(FAB)m/z=475(M++1)。
【0107】
[製造例27]:1−O−ベンゾイル−4−O−ベンジル−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトールの製造
製造例20で得た1−O−ベンゾイル−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトール(200mg、0.549mmol)、酸化銀(silver(I)oxide)(381mg、1.64mmol)及びテトラブチルアンモニウムヨージド(40mg、0.109mmol)をCH2Cl2に溶解してから室温でベンジルブロマイド(0.19ml、1.64mmol)を滴加した。2時間後、反応液をセライトで濾過した後、CH2Cl2で数回洗滌した。洗滌した濾過液をNaHCO3とNaCl飽和水溶液で洗滌した後有機層をMgSO4で乾燥させて減圧蒸発させた後、カラムクロマトグラフィー(酢酸エチル:ヘキサン=1:9)で精製して白色固体状の前記表題化合物(200mg)を得た。
【0108】
m.p.=175−176℃
1H−NMR(CDCl3):δ1.44、1.46(s、6H)、3.81−3.9(m、5H)、4.86(s、2H)、5.58(app.t、J=9.3Hz、1H)、7.25−7.56(m、7H)、8.06(d、J=8.7Hz、2H)
MS(FAB)m/z478(M++Na)。
【0109】
[製造例28]:1−O−ベンジル−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトール(化学式(XII)2の化合物)の製造
製造例27で得た1−O−ベンゾイル−4−O−ベンジル−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトール(200mg、0.44mmol)をCH3OH(15ml)に溶解してからNaOCH3(0.04ml、0.17mmol、25% w/v in CH3OH)を滴加して3時間還流させた。得られた溶液を室温でシリカゲルを通じて濾過させてから、濾過液を濃縮し5%酢酸エチルで洗滌してメチルベンゾエイトを取り除くことにより白色固体状の前記表題化合物(150mg)を得た。
【0110】
m.p.=215−216℃
1H−NMR(CDCl3):δ1.46(s、12H)、2.41(d、J=2.7Hz、1H)、3.63(t、J=9.1Hz、4H)、3.71(t、J=9Hz、4H)、3.87(t、J=8.9Hz、1H)、4.1(app.t、J=9.1Hz、1H)、4.83(s、2H)、26−7.39(m、5H)
MS(FAB)m/z373(M++Na)。
【0111】
[製造例29]:1−O−p−メトキシベンジル−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトール(化学式(XI)1の化合物)の製造
製造例20で得た1−O−ベンゾイル−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトール(300mg、0.85mmol)をN,N−ジメチルホルムアミド(10ml)に溶解してから0℃でNaH(57.8mg、1.44mmol)を加え、p−メトキシベンジルクロライド(p−methoxybenzyl chloride)(0.12ml、0.93mmol)をゆっくり滴加した。これにテトラブチルアンモニウムヨージドを触媒量だけ滴加し、室温で4時間攪拌した。反応生成物を酢酸エチルで抽出してNaHCO3とNaCl飽和水溶液で洗滌した。得られた有機層をMgSO4で乾燥させて減圧蒸発させた後、CH3OH(15ml)及びNaOCH3(0.05ml、0.057mmol、25% w/v in CH3OH)を加えて3時間還流させた。反応生成物を室温に冷却させシリカゲルを通じて濾過した後、濾過液を減圧蒸発させカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:3)で精製して前記表題化合物(210mg)を得た。
【0112】
m.p.=198−200℃
1H−NMR(CDCl3):δ1.47(s、12H)、2.41(brs、1H)、3.59−3.63(m、5H)、3.65(s、3H)、3.80(app.t、J=9.1Hz、1H)、4.76(s、2H)、6.87(d、J=9Hz、2H)、7.33(d、J=9.1Hz、2H)
MS(FAB)m/z403(M++Na)。
【0113】
[製造例30]:1−O−ベンジル−4−(1−イミダゾール−イル−カルボニルオキシ)−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトールの製造
製造例28で得た1−O−ベンジル−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトール(70mg、0.19mmol)をトルエン(6ml)に溶解し常温でCaH2(21mg、0.49mmol)を加え、20分間攪拌した。これにカルボニルジイミダゾール(81mg、0.49mmol)を加えて18時間攪拌し、反応生成物を濾過し、得られた濾過液を濃縮した後カラムクロマトグラフィー(酢酸エチル:ヘキサン=1:1)で精製して白色固体状の前記表題化合物(87mg)を得た。
【0114】
m.p.=214−216℃
1H−NMR(CDCl3):δ1.45、1.47(2s、12H,2CMe2)、3.80−3.87(m、5H)、4.85(s、2H,PhCH2)、5.45(app.t、J=1.9Hz、1H,H−1)、7.07(s、1H)、7.29−7.42(m、6H)、8.16(s、1H)
MS(FAB)m/z=445(M++1)、467(M++1)。
【0115】
[製造例31]:4−O−アリール−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトールの製造
製造例20で得た1−O−ベンゾイル−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトール(500mg、1.37mmol)をN,N−ジメチルホルムアミド(10ml)に溶解してから0℃でNaH(9.8mg、2.47mmol)を加え、10分後アリールブロマイド(0.18ml、2.05mmol)を加えて室温で20時間攪拌させた。得られた反応液を酢酸エチルで抽出してNaCl飽和水溶液で洗滌し、これをMgSO4で乾燥させてから減圧揮発し、カラムクロマトグラフィー(酢酸エチル:ヘキサン=3:7)で精製して4−O−アリール−1−O−ベンゾイル−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトール(450mg)を得た。得られた4−O−アリール−1−O−ベンゾイル−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトール(400mg、0.989mmol)をCH3OH(12ml)に溶解してからNaOCH3(0.09ml、0.395mmol、25% w/v in CH3OH)を滴加して3時間還流させた。反応生成物を室温に冷却させシリカゲルを通じて濾過した後、濾過液を濃縮した後5%酢酸エチルで洗滌してメチルベンゾエイト(methylbenzoate)を取り除くことにより白色固体状の前記表題化合物(290mg)を得た。
【0116】
m.p.=200−202℃
1H−NMR(CDCl3):δ1.46(s、12H)、2.46(d、J=2.7Hz、1H)、3.57−3.68(m、4H)、3.83(t、J=8.1Hz、1H)、4.05(t、J=6.9Hz、1H)、4.28(d、J=5.7Hz、2H)、5.21(d、J=10.2Hz、1H)、5.33(dd、J=18.1Hz、1.5Hz1H)、5.95(m、1H)
MS(FAB)m/z301(M++1)、323(M++Na)。
【0117】
[製造例32]:4−O−(2−N,N−ジベンジルアミノエチル)−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトールの製造
製造例31で得た4−O−アリール−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトール(250mg、0.832mmol)及びNaHCO3(139mg、1.66mmol)をCH2Cl2/CH3OHの混合溶液(6:1、50ml)に溶解し、−78℃で反応溶液の色が青になるまでO3を滴加した。再び溶液の青色が消えてしまうまでN2を滴加した後、トリフェニルホスフィン(327mg、12.49mmol)を滴加した。ヒドロペルオキシドの還元反応が終結すると、カラムクロマトグラフィーで精製して4−O−エタナール−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトール(235mg)を得た。これ(150mg、0.496mmol)をジクロロエタン(15ml)に溶解し0〜5℃でジベンジルアミン(dibenzylamine)(0.04ml、0.744mmol)を加える。10分後、これにソジウムトリアセトキシボロハイドライド(sodium triacetoxyborohydride)(265mg、1.24mmol)を加えて0〜5℃で1時間攪拌させた後、室温で12時間攪拌した。反応生成物をCH2Cl2(60ml)で抽出した後、1N NaOH(30ml)溶液で洗滌し、蒸留水で5回洗滌した。得られた有機層をMgSO4で乾燥させた後、減圧蒸発・濃縮させ、カラムクロマトグラフィー(酢酸エチル:ヘキサン=7:3)で精製して白色固体状の前記表題化合物(180mg)を得た。
【0118】
m.p.=144−145℃
1H−NMR(CDCl3):δ1.44(s、12H)、2.70(brs、1H)、2.74(t、J=6Hz、2H)、3.54−3.59(m、4H)、3.64(s、4H)、3.74(t、J=6.2Hz、1H)、3.87(t、J=6.2Hz、2H)、4.0(t、J=6.3Hz、1H)、7.19−7.39(m、10H)
MS(FAB)m/z484(M++1)、506(M++Na)。
【0119】
[製造例33]:4−O−(2−N,N−ジベンジルアミノエチル)−1−O−(メチルオキシカルボニルメチル)−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトール(化学式(XIII)の化合物)の製造
製造例32で得た4−O−(2−N,N−ジベンジルアミノエチル)−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトール(125mg、0.258mmol)をN,N−ジメチルホルムアミド(10ml)に溶解してから0℃でNaH(20mg、0.51mmol)を滴加した。反応液を0℃で20分間攪拌させた後、エチルブロモアセテート(ethyl bromoacetate)(0.05ml、0.51mmol)を滴加して常温で10時間攪拌させた。反応生成物を酢酸エチルで抽出しNaCl飽和水溶液及び蒸留水で洗滌した後MgSO4で乾燥させ、これを減圧蒸発させカラムクロマトグラフィー(酢酸エチル:ヘキサン=3:7)で精製して前記表題化合物(110mg)を得た。
【0120】
m.p.=130−132℃
1H−NMR(CDCl3):δ1.41(s、12H)、2.72(t、J=6Hz、2H)、3.58(t、J=6.3Hz、2H)、3.63−3.89(m、13H)、4.36(s、2H)、7.12−7.40(m、10H)
MS(FAB)m/z556(M++1)、579(M++Na)。
【0121】
[製造例34]:4−O−(2−アミノエチル)−1−O−(メチルオキシカルボニルメチル)−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトールの製造
製造例33で得た4−O−(2−N,N−ジベンジルアミノエチル)−1−O−(メチルオキシカルボニルメチル)−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトール(20mg、0.035mmol)をCH2Cl2−メタノール混合溶媒(1:2、12ml)に溶解し、これに水酸化パラジウム(palladium hydroxide)(20% on carbon、9.7mg)を加え、40psiの水素大気下で1時間攪拌した。反応生成物をセライトを通じて濾し、濾過液を減圧蒸発・濃縮した後、メタノール溶媒下で再結晶して白色固体状の前記表題化合物(14mg)を得た。
【0122】
m.p.=193−195℃
1H−NMR(CD3OD):δ1.41(s、12H)、3.10(t、J=5.9Hz、2H)、3.27(dd、J=6.5、1.8Hz、2H)、3.69−3.90(m、11H)、4.28(s、2H)
MS (FAB) m/z 376 (M++1)。
【0123】
[製造例35]:4−O−(2−N,N−ジベンジルアミノエチル)−1−O−(カルボキシメチル)−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトールの製造
製造例33で得た4−O−(2−N,N−ジベンジルアミノエチル)−1−O−(メチルオキシカルボニルメチル)−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトール(20mg、0.035mmol)をメタノール(3ml)に溶解しNaOHペレット(4.3mg、0.107mmol)を加えて常温で10時間攪拌した。反応生成物を減圧蒸発させ濃縮した後蒸留水(5ml)を加え、得られた水層をジエチルエーテル(2mlずつ2回)で洗滌した。これに5%AcOH水溶液を加えてpHが6.5になるようにした後、CH2Cl2(4x4ml)で抽出し有機層を集め乾燥(Na2SO4)・減圧蒸発させて白色固体状の前記表題化合物(13mg)を得た。
【0124】
1H−NMR(CDCl3):δ1.41&1.44(2s、12H)、2.77(t、J=5.8Hz、2H)、3.35−3.91(m、12H)、4.37(s、2H)、7.20−7.40(m、10H)。
【0125】
[実施例1]:化学式(II)(x=O−CO−O、n=5の場合)の化合物の製造
1−1)myo−イノシトール二量体化合物の製造
製造例21で得た化合物(1.62g、3.4mmol)をトルエン(21ml)に溶解してから製造例5で得た化合物(1.8g、5mmol)及び1,8−ジアゾビシクロ[5.4.0]ウンデカ−7−エン(1,8−diazobicyclo[5.4.0]undec−7−ene)(0.05ml、0.34mmol)を加え、常温で18時間攪拌させた。この反応生成物をCH2Cl2で抽出し、MgSO4で乾燥してから減圧蒸発させて濃縮させ、ヘキサン/酢酸エチル(5:1)を用いて再結晶して白色固体状のカップリングされた二量体形の化合物(2.23g)を得た。
【0126】
m.p.=225−226℃
1H−NMR(CDCl3):1.10−1.72(m、24H)、3.35&3.36(dd、J=9.3Hz、11.0&9.5Hz、11.1Hz、2H)、3.49(t、J=9.3&10.8Hz、2H)、3.73(dd&t、J=2.6Hz、5.8&1.4Hz、2H)、4.00−4.15(m、6H)、4.12(dd、J=5.9Hz、9.1&2.6Hz、5.9Hz、4H)、4.69(t、J=2.5&4.4Hz、4H)、4.82(s、8H)、4.95&4.96(dd、J=4.1Hz、10.5&4.1Hz、10.5Hz、2H)、5.04&5.05(dd、J=6.9Hz、11.1&7.0Hz、11.0Hz、2H)、6.89(d、J=8.7Hz、4H)、7.24−7.42(m、14H)
IR(KBr):1748cm−1(CO)
MS(FAB)m/z779.82(M++Na)。
【0127】
1−2)myo−イノシトール二量体のアセトニド保護基除去
段階1−1)で得たmyo−イノシトール二量体化合物(2.21g)をCH2Cl2(25ml)に溶解し、これにCH3OH(24.3ml)及びp−トルエンスルホン酸(286.1mg、1.46mmol)を加え、常温で14時間攪拌させた。この反応生成物を濾過して白色固体状のアセトニド保護基が除去されたmyo−イノシトール二量体化合物(1.45g)を得た。
【0128】
m.p.=169℃
1H−NMR(DMSO):3.11−3.24(m、4H)、3.35−3.48(m、5H)、3.49(dd、J=9.6Hz、21.2Hz、4H)、3.74(s、3H)、3.90−3.97(m、3H)、4.25−4.33(m、2H)、4.53(dd、J=1.8Hz、24.9Hz、4H)、4.70−4.74(m、12H)、4.77−4.81(m、2H)、6.89(d、J=8.6Hz、2H)、7.21−7.35(m、5H)、7.43(d、J=7.1Hz、2H)
MS(FAB)m/z619(M++Na)。
【0129】
1−3)アシル化反応を通したアミノ酸の導入
段階1−2)で得た化合物(200mg、0.34mmol)、製造例8で得た化合物(1.55g、6.7mmol)及びN,N−ジメチルアミノピリジン(62.3mg、0.51mmol)をN,N−ジメチルホルムアミド(4ml)に溶解し、これに1−[3−(ジメチルアミノ)プロピル]−エチルカルボジイミド塩酸(1−[3−(dimethyl amino)propyl]−ethylcarboiimide hydrochloride)(1.3g、6.7mmol)を加え常温で1日間攪拌した。この反応生成物をCH2Cl2で抽出した後、NaHCO3 飽和水溶液及び蒸留水で数回洗滌した。得られた有機層を分離してNa2SO4で乾燥し、濃縮して、骨格構造に8個のアミノ酸が導入された茶色固体状の目的化合物(776.6mg)を得た。
【0130】
1H−NMR(CDCl3):1.13−1.32(m、16H)、1.36(s、72H)、1.45−1.57(m、32H)、2.06−2.15(m、16H)、2.95−3.02(m、16H)、3.7(s、3H)、3.85−3.91(m、1H)、4.22(dd、J=1.9Hz、9.3Hz、1H)、4.48(d、J=11.8Hz、2H)、4.55(d、J=6.6Hz、2H)、4.60−5.68(m、10H)、6.76(d、J=8.7Hz、2H)、7.05(d、J=8.4Hz、2H)、7.10−7.26(m、5H)
MS(FAB)m/z2324.6(M++Na)。
【0131】
1−4)p−メトキシベンジル保護基の除去
段階1−3)で得た化合物(705.1mg、0.31mmol)をCH2Cl2/H2O(18:1)(5ml)に溶解し、0℃で2,3−ジクロロ−5,6−ジシアノ−1,4−ベンゾキノン(2,3−dichloro−5,6−dicyano−1,4−benzoquinone)(139.0mg、0.61mmol)を添加して常温で2日間攪拌させた。これにNaHCO3飽和水溶液を加えて反応を中断させ、蒸留水で洗滌した後、有機層をNa2SO4で乾燥し、濃縮してp−メトキシベンジル保護基が除去された茶色固体状の目的化合物(373.9mg)を得た。
【0132】
1H−NMR(CDCl3):1.17−1.26(m、16H)、1.37(s、72H)、1.41−1.53(m、32H)、2.94−3.05(m、16H)、2.07−2.41(m、16H)、3.86−3.92(m、2H)、4.31−5.56(m、24H)、7.11−7.27(m、5H)
MS(FAB)m/z2204.8(M++Na)。
【0133】
1−5)蛍光物質の導入
段階1−4)で得た化合物(284.1mg、0.13mmol)及びN,N−ジメチルアミノピリジン(47.6mg、0.38mmol)をアセトニトリル(4ml)に溶解し、これに5−ジメチルアミノ−1−ナフタレンスルホニルクロライド(5−dimethylamino−1−naphthalenesulfonyl chloride;70.2mg、0.26mmol)を加えて常温で2日間攪拌した。これにNH4Cl飽和水溶液(8.3ml)を加えて中和させてから酢酸エチルで抽出し、NaCl飽和水溶液で洗滌した。得られた有機層をNa2SO4で乾燥させた後、減圧蒸発で濃縮し、残留物をカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:1)で精製して蛍光物質が導入された黄色固体状の目的化合物(261.7mg)を得た。
【0134】
1H−NMR(CDCl3):1.21(s、16H)、1.38(s、72H)、1.53−1.59(m、32H)、1.99−2.33(m、16H)、2.83(s、6H)、2.94−3.04(m、16H)、3.89(t、J=9.8Hz、1H)、4.56(dd、J=12.0Hz、18.6Hz、2H)、4.69−5.54(m、11H)、7.11−7.29(m、6H)、7.50(dd、J=8.4Hz、16.5Hz、2H)、8.03(d、J=7.5Hz、1H)、8.18(d、J=7.2Hz、1H)、8.61(brs、1H)
MS(FAB)m/z2438.1(M++Na)。
【0135】
1−6)アミノ酸末端のt−Boc保護基の除去
段階1−5)で得た化合物(202.3mg、0.08mmol)をHClガスで飽和させた酢酸エチル(8ml)に溶解して常温で45分間攪拌した。この反応生成物を減圧蒸発させてアミノ酸末端のt−Boc保護基が除去された黄色固体状の目的化合物(180.4mg)を得た。
【0136】
1H−NMR(CD3OD):1.37(m、16H)、1.69(m、32H)、2.23(m、16H)、2.94(m、16H)、3.28−3.30(m、1H)、3.44(s、6H)、4.71(m、2H)、4.95−5.62(m、11H)、7.21−7.30(m、5H)、7.87−7.98(m、2H)、8.14(brs、1H)、8.53(dd、J=8.4Hz、15.0Hz、2H)、8.95(t、J=3.9Hz、1H)
MS(MALDI−TOF)m/z1636.2(M++Na)。
【0137】
1−7)グアニジニウム基の導入
段階1−6)で得た化合物(141.6mg、0.07mmol)をN,N−ジメチルホルムアミド(2.8ml)に溶解してからこれにトリエチルアミン(0.4ml、3mmol)及び製造例11で得たN,N−ジ−Boc−N’−トリフロロメタンスルホニルグアニジン(1.2g、3mmol)を加えて常温で2日間攪拌した。これに過量の酢酸エチルを加えて2N NaHSO4、飽和NaHCO3及び飽和NaCl水溶液で洗滌した後、有機層をNa2SO4で乾燥してから減圧蒸発で濃縮し、残留物をカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:2)で精製してグアニジニウム基が8つ導入された黄色固体状の目的化合物(93.1mg)を得た。
【0138】
1H−NMR(CDCl3):1.13−1.65(m、192H)、1.60−2.31(m、16H)、2.82(s、6H)、3.24−3.36(m、16H)、3.88(t、J=9.9Hz、1H)、4.56−5.51(m、13H)、7.11−7.29(m、6H)、7.51(dd、J=8.4Hz、16.2Hz、2H)、7.99(d、J=8.7Hz、1H)、8.17(dd、J=0.9Hz、7.5Hz、1H)、8.22−8.28(m、8H)、11.46(s、8H)。
【0139】
1−8)グアニジニウム基末端のt−Boc保護基の除去
段階1−7)で得た化合物(38mg、0.01mmol)をトリフロロ酢酸/CH2Cl2(1:1)(2ml)に溶解して常温で5時間攪拌した。得られた反応生成物を減圧蒸発によって濃縮し、残留物に蒸留水及び酢酸エチルを加えた後、水層を分離して減圧蒸発することによってグアニジニウム基末端のt−Boc保護基が除去された黄色固体状の目的化合物(27mg)を得た。
【0140】
1H−NMR(CD3OD):1.13−1.35(m、16H)、1.42−1.61(m、32H)、2.13−2.43(m、16H)、2.88(s、6H)、3.02−3.16(m、16H)、4.01(t、J=9.6H,1H)、4.64(dd、J=11.9Hz、18.7Hz、2H)、5.01−5.61(m、11H)、7.16−7.28(m、6H)、7.52−7.66(m、2H)、8.00(d、J=8.6Hz、1H)、8.27(d、J=7.3Hz、1H)、8.64(d、J=8.5Hz、1H)
MS(MALDI−TOF)m/z1973.1(M++Na)。
【0141】
1−9)ベンジル(Bn)保護基の除去
段階1−8)で得た化合物(10mg、3.5μmol)をメタノール(1ml)に溶解し、これに水酸化パラジウム(palladiumhydroxide)(20% on carbon、20mg)を加えて水素1気圧下で4時間攪拌した。得られた反応生成物をセライト(celite)に通して濾過し、濾液を減圧蒸発してベンジル保護基が取り除かれた黄色固体状の目的化合物(7mg)を得た。
【0142】
1H−NMR(CD3OD):1.1−1.6(m、48H)、2.1−2.5(m、16H)、2.90(s、6H)、3.0−3.26(m、16H)、3.97(t、J=9.9Hz、1H)、5.0−5.7(m、11H)、7.29(d、J=7.7Hz、1H)、7.57(t、J=8.1Hz、1H)、7.67(t、J=8.1Hz、1H),8.00(d、J=8.5Hz、1H)、8.30(d、J=7.5Hz、1H)、8.64(d、J=8.7Hz、1H)。
【0143】
[実施例2]:化学式(II)(x=O−CO−O、n=3及びn=7の場合)の化合物の製造
前記実施例1の段階1−3)で用いた6−Boc−アミノヘキサン酸の代わりに製造例7及び製造例9で得た4−Boc−アミノブタン酸及び8−Boc−アミノオクタン酸を用いてそれぞれn=3及びn=7である化学式(II)の化合物を製造した。
【0144】
[実施例3]:化学式(III)(x=O−CO−Oの場合)の化合物の製造
3−1)製造例25で得た化合物(42mg、0.12mmol)をテトラヒドロフラン(5ml)に溶解し、これに0℃でNaH(6mg、0.25mmol)を加えて常温で25分間攪拌した。この反応生成物の温度を0℃に下げた後、製造例26で得た化合物(48mg、0.10mmol)を加えて常温で2時間攪拌した。これを酢酸エチル(30ml)で希釈した後、NaHCO3飽和水溶液(30ml)と食塩水(brine)で洗滌し、得られた有機層を乾燥(Na2SO4)し、残留物をカラムクロマトグラフィーで精製し再結晶してカップリングされた二量体形態の化合物(29mg)を得た。
【0145】
m.p.=168−170℃
1H−NMR(CDCl3):δ1.45、1.46(2s、24H,4CMe2)、3.68(appdt、J=9.5、2.1Hz、4H,H−1、H−1’、H−3、H3’)、3.82(s、3H,OCH3)、3.82(t、J=9.1、1H,H−5’)、4.10(t、J=9.3Hz、2H,H−4’&H−6’)、4.23(t、J=9.6Hz、2H,H−4&H−6)、4.39(t、J=1.8Hz、1H)、4.79、4.85(2s、4H,2PhCH2)、5.17(t、J=9.6Hz、H−5)、5.58(t、J=2.0Hz、H−2’)、6.89(dd、J=6.7、1.9Hz、2H,Ph)、7.28−7.40(m、8H,Ph)
MS(FAB)m/z=779(M++Na)。
【0146】
3−2)前記カップリングされた二量体形態の化合物を用いて実施例1と同じ方法でアシル化反応を行ってアミノ酸を導入した後、アミノ酸が導入された化合物のアミノ酸N−末端にグアニジニウム基が導入された化学式(III)の化合物を製造した。
【0147】
[実施例4]:化学式(IV)(x=O−CO−Oの場合)の化合物の製造
4−1)製造例29で得た化合物(43mg、0.11mmol)をテトラヒドロフラン(2ml)に溶解し、これに0℃でNaH(1.2mg、0.05mmol)を加えて常温で25分間攪拌した。この反応生成物を製造例30で得た化合物(50mg、0.11mmol)のテトラヒドロフラン(3ml)溶液に0℃で加えて0〜5℃で1時間30分間攪拌し、これをCH2Cl2 (25ml)で希釈した後、NH4CO3飽和水溶液(30ml)で洗滌した。得られた有機層を乾燥(MgSO4)・濃縮し、カラムクロマトグラフィー(酢酸エチル:ヘキサン=1:3)で精製してカップリングされた二量体形態の化合物(41mg)を得た。
【0148】
m.p.=284−286℃
1H−NMR(CDCl3):δ1.43、1.47(2s、24H,4CMe2)、3.66−3.88(m、13H)、4.47(s、2H)、4.83(s、2H)、5.12(t、J=9.3Hz、2H,H−6、H−6’)、6.88(d、J=8.7Hz、2H)、7.27−7.42(m、7H)
MS(FAB)m/z=757(M++1)、780(M++Na)。
【0149】
4−2)前記カップリングされた二量体形態の化合物を用いて実施例1と同じ方法でアシル化反応を行いアミノ酸を導入した後、アミノ酸が導入された化合物のアミノ酸N−末端にグアニジニウム基が導入された化学式(IV)(x=O−CO−Oの場合)の化合物を製造した。
【0150】
[実施例5]:化学式(IV)(x=O−CH2−CO−NH−(CH2)2−Oの場合)の化合物の製造
5−1)製造例34で得た化合物(8.3mg、0.022mmol)と製造例35で得た化合物(12mg、0.022mmol)をN,N−ジメチルホルムアミド(2.5ml)に溶解し、これに常温でトリエチルアミン(3μl、0.035mmol)、1−ヒドロキシベンゾトリアゾール水和物(1−hydroxybenzotriazole hydrate、HOBT);(3.2mg、0.024mmol)及び1−[3−(ジメチルアミノ)プロピル]−エチルカルボイミド塩酸(1−[3−(dimethylamino)propyl]−ethylcarbodiimide hydrochloride);(4.6mg、0.024mmol)を順に加えて22時間攪拌した。得られた反応生成物を酢酸エチル(15ml)で希釈した後、有機層をNH4Cl水溶液(5ml)、NaHCO3飽和水溶液(10ml×2)、蒸留水(5ml×3)で洗滌し、乾燥(Na2SO4)してから減圧蒸発させ、残留物をカラムクロマトグラフィー(酢酸エチル:n−ヘキサン=1:1)で精製して白色固体状のカップリングされた二量体化合物(14mg)を得た。
【0151】
1H−NMR(CDCl3):δ1.40、1.43(2s、12HeacH,4CMe2)、2.72(t、J=6.1Hz、2H)、3.35−3.88(m、25H)、4.23(s、2H),4.36(s、2H),7.02(t、J=6Hz、1H),7.19−7.39(m、10H)。
【0152】
5−2)前記カップリングされた二量体形態の化合物を用いて実施例1と同様な方法でアシル化反応を行いアミノ酸を導入した後、アミノ酸が導入された化合物のアミノ酸N−末端にグアニジニウム基が導入された化学式(IV)(x=O−CH2−CO−NH−(CH2)2−Oの場合)の化合物を製造した。
【0153】
試験例1:細胞膜透過効率測定
前記実施例で製造された、ダンシル(dansyl)蛍光物質が付着された化合物の細胞膜透過性を次のように測定し、既存の生体膜透過性が優れていると知られたアルギニン9量体(dansyl−Arg9,d−Arg9)、及び対照群としての実施例1の段階1−6)で製造されたグアニジニウム基を有しない中間体と比較した。
【0154】
先ず、C12ウェルプレート(well plate)をカバーガラスを覆い、該プレート上でCOS7細胞(monkey kidney epithelial cell)を培養した。培地は10%FBSが含まれたDMEM(Dulbecco’s modified Eagle’s medium)を用いて24時間安定化させた後、該細胞を飢餓(starvation)させるため無血清培地(serum−free medium)で再び24時間培養した。その後、これらの細胞をダンシル−Arg9、実施例1の段階1−6)で製造された中間体、又は実施例1又は2で得た化合物で5分間処理(〜7μM)した後、カバーガラスを取り出してPBSで洗滌し、スライドにマウンティング(mounting)し、共焦点顕微鏡(confocal microscope)で細胞断面を観察した。その際、蛍光物質を観察するため、Arレーザー(波長455nm)を装備した顕微鏡を用いて対物レンズ40x・対眼レンズ10x、総400倍拡大して観察した。その結果を図1に示す。
【0155】
図1の(1)はダンシル−Arg9で処理したCOS7細胞の蛍光イメージ;(2)はグアニジニウム基を有しない中間体化合物(実施例1の段階1−6で得た中間体)で処理したCOS7細胞の蛍光イメージ;(3)、(4)及び(5)は本発明による化学式(II)の化合物で処理したCOS7細胞の蛍光イメージである。ここで緑色部分は蛍光であり、蛍光が多く見えるほど細胞内に該化合物が多く透過されていることを示す。
【0156】
図1に示したように、本発明の化合物((3)〜(5))はダンシル−Arg9((1))より高い細胞膜透過性を示し、グアニジニウム基を有しない中間体化合物((2))は細胞内に透過されないことを確認した。また、本発明の化合物のうち、n=5の鎖長を有する化合物((4))が最も高い細胞膜透過率を有することを確認した。
【0157】
試験例2:核膜透過性測定
前記実施例で製造されたダンシル(dansyl)蛍光物質が付着された化合物の核膜透過性を次のように測定し、既存の生体膜透過性が優れていると知られたアルギニン9量体(dansyl−Arg9、d−Arg9)及び対照群としての実施例1の段階1−6)で製造されたグアニジニウム基を有しない中間体と比較した。
【0158】
C12ウェルプレートをカバーガラスで覆い、該プレート上でマウスマクロファージRAW264.7細胞を培養した。培地は10%FBSが含まれているDMEMを用いて24時間安定化させた後、該細胞を飢餓させるため無血清培地で再び24時間培養した。該細胞を室温でリボヌクレアーゼ(RNase)(100μg/ml)で処理してから、常温(23〜25℃)で5分間ヨウ化プロピジウム(2μg/ml)で処理して核を染色した後、PBSで3回洗滌した。その後、該細胞をダンシル−Arg9、実施例1の段階1−6)で製造された中間体、又は実施例1又は2で得た化合物で5分間処理(〜7μM)した後、カバーガラスを取り出して洗滌し、これをスライドにマウンティングして共焦点顕微鏡で細胞断面を観察した。その際、蛍光物質を観察するためArレーザー(波長455nm)を装備した顕微鏡を用いて対物レンズ40x・対眼レンズ10x、総400倍拡大して観察した。その結果を図2に示す。
【0159】
図2の(1)はダンシル−Arg9で処理したRAW264.7細胞の蛍光イメージ;(2)はグアニジニウム基を有しない中間体化合物(実施例1の段階1−6)で得た化合物)で処理したRAW264.7細胞の蛍光イメージ;(3)〜(5)はそれぞれn=3、5及び7である本発明による化学式(II)の化合物で処理したRAW264.7細胞の蛍光イメージである。図2の左カラムのイメージの緑色はそれぞれの輸送体化合物が細胞内に透過されたことを示し、中央のカラムのイメージの赤色はヨウ化プロピジウムで核が染色されたことを示し、右カラムのイメージにおいては赤色と緑色が重なり黄色に近くなるほど化合物が核膜内に多く透過されていることを示す。
【0160】
図2に示したように、本発明による化合物((3)〜(5))は既存の分子輸送体であるダンシル−Arg9((1))より核膜透過性が高いことを確認した。また、本発明の化合物のうち、鎖長がn=3((3))又はn=7((5))である場合が核膜透過性が特に優れていることが分かる。
【0161】
また、蛍光物質が付着されたmyo−イノシトール燐酸[Prestwich,G. D.,Acc. Chem. Res.,29,503 (1996); Chung S. K. et al. J. Org. Chem.,67,5256(2002)]を蛍光表示されていない本発明の化学式(II)〜(IV)の化合物と混合して製造されたイオン結合体の組成物も優れた生体膜透過性を現すことを確認した。
【0162】
本発明に関する多くの実施形態を記述したが、本発明の範囲を外れない様々な変形が可能であることを理解できるであろう。したがって、他の実施形態も後述する請求項の範囲に含まれる。
【図面の簡単な説明】
【0163】
【図1】ダンシル(dansyl)−Arg9((1))、実施例1の段階1−6)で製造されたグアニジニウム基を有しない中間体((2))及び本発明の実施例1〜2に基づいて製造されたイノシトール誘導体ら((3)〜(5))の細胞膜透過程度を比較した結果を示す図である。
【図2】ダンシル(dansyl)−Arg9((1))、実施例1の段階1−6)で製造されたグアニジニウム基を有しない中間体((2)) 及び本発明の実施例1〜2に基づいて製造されたイノシトール誘導体ら((3)〜(5))の核膜透過程度を比較した結果を示す図である。
【技術分野】
【0001】
本発明は、イノシトール系分子輸送体及びその製造方法に関する。
【背景技術】
【0002】
細胞の細胞膜(plasma membrane)は細胞内小器官が存在している細胞質(cytoplasm)を外部環境から保護する役割を果たし、リン脂質二重層及びこれらの内部にまたは表面に結合されている蛋白質で構成されている。一般的に細胞膜は特定物質のみを細胞内外に出入りさせる門番(gate−keeper)の役割をする。しかし、このような細胞膜の選択的透過性のために有用な治療剤の細胞膜透過が制限され、特に親水性分子、多荷電分子、およびペプチド、オリゴヌクレオチド(例:核酸及び遺伝子)等のような巨大分子等は細胞膜を通過しにくい。従って、このような薬物及び巨大分子達を細胞内へ効率的に輸送するための方法について研究が進んでいる。
【0003】
今まで多くの輸送体分子が様々な種類の薬物及び巨大分子等を細胞内へ輸送するため開発されており、例として陽電荷を帯びた脂質、ポリ−リジンのような陽電荷の重合体及び陽電荷を帯びたデンドリマーなどがある。しかし、一般的にこのような脂質、重合体及びデンドリマーは細胞内で不溶性を示したり生分解されないので沈殿を形成し、毒性を引き起こすなどの問題があった。
【0004】
一方、HIVウィルス複製に必須の転写活性因子(trans−activator)として知られているTat(transacting transcriptional activator)蛋白質の塩基性領域(即ち、49−57番目アミノ酸)がペプチドの細胞膜透過に決定的な役割を果たすことが報告されている。また、このようなTATの塩基性領域と類似した細胞膜透過機能を行う蛋白質変換領域(PTD、protein transduction domain)を有する蛋白質として、下記表1のようなAntpホメオドメイン(Antennapedia homeodomain)蛋白質、ヘルペス(Herpes)ウィルス蛋白質VP22、NLS(nuclear localization signal)序列等が報告されている。
【表1】
【0005】
前記蛋白質は細胞に存在する特定受容体(receptor)や輸送体(transporter)の助けを受けずに生体膜を透過でき、これらPTDは主に塩基性アミノ酸残基、特にアルギニン及びリジンで構成されるという共通点を有している。
【0006】
これら蛋白質の細胞膜透過メカニズムに対しては様々な意見があるが、最近の研究によるとPTDを有する細胞透過性蛋白質はエンドサイトーシス(endocytosis)により細胞内に通過する可能性が高いと提案されている。Lebleuなどは蛍光活性化細胞選別(FACS)分析を通じてHIV−1Tat(48−60番目アミノ酸)及びアルギニン9量体(Arg9)がエンドサイトーシスによって細胞内に移動することを報告したが、その詳しいメカニズムは未だ究明されていない(Lebleu,B.ら、Biol.Chem.,278,585,(2003))。
【0007】
また、高い細胞膜透過性を付与するため、複数のアルギニンを有する様々な形態のオリゴメールを合成する研究が試みられてきた。例えば、1991年マン(Mann)らはTat蛋白質がこれらに結合されている分子の生体膜透過輸送には効果的であるが、水不溶性でありしかも細胞膜に過剰に強く結合することにより凝集を起こすので実質的に細胞内に移動するTat蛋白質の数には限界があることを報告した(Mann,D.A.ら、EMBO J.,10、1733(1991))。
【0008】
バルソウム(Barsoum)らはTat蛋白質の短い断片(1−72番目アミノ酸)及び塩基性領域(37−58番目アミノ酸)を含む他の断片もまたそれに結合された分子の生体膜透過輸送を効果的に向上させると報告した。このような研究は塩基性領域を有する小さいペプチドもそれらに結合された分子の生体膜透過輸送を行うことを示すものである(Barsoum、J.ら、Proc. Natl. Acad. Sci. U.S.A.,91,664(1994))。
【0009】
フタキ(Futaki)らは複数のアルギニン残基を有する様々なペプチドに蛍光物質を付着し、これらのマウスマクロファージRAW264.7細胞に対する膜透過性を調査した結果、アルギニン豊富なペプチドがTat蛋白質(49−57番目アミノ酸)と類似した膜透過性を現すことと、その中、8個のアルギニン残基を有するオリゴメールがこれらに結合された分子の生体膜透過輸送に最も効果的であることを確認した(Futaki,S.ら、J.Biol.Chem.,276,5836(2001))。このような研究はアルギニンのグアニジニウム基が生体膜透過輸送において重要な要素であることを示す。
【0010】
ウェンダー(Wender)らは生体膜透過性がペプチド内グアニジニウム基数、リンカーチェーン(linker chain)の長さ及びキラリティ等に依存するという事実に基づいてペプトイド(peptoid)分子輸送体を製作し、ジャーカット(Jurkat)細胞に対するFACS分析を行った結果、L−アルギニン9量体がTat蛋白質(49−57番目アミノ酸)より生体膜透過輸送において20倍程高効率を示し、D−アルギニン9量体も又遥かに高い透過輸送効率を示すことを確認した。それにより、グアニジニウム基を有するペプトイドの膜透過性はアミノ酸のキラリティによって大きく影響されないことが分かる(米国特許第6,495,663号;及びWender,P.A.ら、Proc. Natl. Acad. Sci. U.S.A.,97、13003(2000))。しかし、ポリアルギニンペプチド又はペプトイド分子は生体内で毒性を示すだけでなく、短時間内に代謝された後、肝と腎臓を通じて早期に除去されるという問題がある。
【0011】
そこで本発明者らは、ミオ(myo)−イノシトール及び複数の陽電荷を帯びたグアニジニウム基から製造したイノシトール誘導体がこれらに結合された様々な治療物質の生体膜透過輸送に効果的であることを発見し、本発明に至った。
【特許文献1】米国特許第6,495,663号
【非特許文献1】Lebleu, B.ら著、Biol.Chem., 278, 585、2003年発行
【非特許文献2】Mann, D. A.ら著、EMBO J.,10, 1733、1991年発行
【非特許文献3】Barsoum, J.ら著、Proc. Natl. Acad. Sci. U.S.A., 91, 664、1994年発行
【非特許文献4】Futaki, S.ら著、J. Biol. Chem., 276, 5836、2001年発行
【非特許文献5】Wender, P. A.ら著、Proc. Natl. Acad. Sci. U.S.A.,97,13003、2000年発行
【発明の開示】
【発明が解決しようとする課題】
【0012】
本発明の目的は、治療物質の生体膜透過輸送を効果的に向上させるイノシトール系分子輸送体及びその製造方法を提供することである。
【0013】
本発明の他の目的は、前記イノシトール系分子輸送体を含む細胞内治療物質輸送用組成物を提供することである。
【課題を解決するための手段】
【0014】
本発明の一実施形態によって、本発明は、下記化学式(I)のイノシトール誘導体を提供する:
【化9】
【0015】
前記式で、
R1は
【化10】
【0016】
であり、ここでnは1〜12範囲の整数であり;
R2及びR3はそれぞれ独立的にH、アルキル、アリールアルキル、シクロアルキル、ヘテロアルキル、−(CH2)mNHR’、−(CH2)lCO2R’’、−COR’’’又は−SO2R’’’’であり、その際R’、R’’、R’’’及びR’’’’はそれぞれアルキルであり、mは2〜5範囲の整数で、lは1〜5範囲の整数であり;
pは0〜2範囲の整数であり;
X及びX’はそれぞれ独立的に−O−CO−O−、−O−CO−NH−(CH2)m−O−、−O−CO−(CH2)l−O−又は−O−(CH2)l−CO−NH−(CH2)m−O−であり、ここでmは2〜5範囲の整数、lは1〜5範囲の整数である。
【0017】
本発明の他の実施形態によって、本発明は、
(a)myo−又はscyllo−イノシトールの水酸基を保護して中間体を得る段階;
(b)段階(a)で得た中間体を2つ以上カップリングしてイノシトール重合体を得る段階;
(c)アシル化反応を行って段階(b)で得たイノシトール重合体に一つ以上のアミノ酸を導入する段階;及び
(d)段階(c)で得たイノシトール重合体のアミノ酸N−末端にグアニジニウム基を導入する段階を含む前記化学式(I)のイノシトール誘導体の製造方法を提供する。
【発明の効果】
【0018】
本発明によれば、イノシトール骨格構造にグアニジニウム基を導入して製造された本発明によるイノシトール誘導体は、細胞膜、核膜、血液−脳関門等のような生体膜に対する透過性が優れ、制限された生体膜通過のため医薬品として開発が困難であった薬物又は診断試薬等を細胞、組織、臓器内へ輸送できる分子輸送体として活用することができる。
【発明を実施するための最良の形態】
【0019】
本発明による化学式(I)のイノシトール誘導体は側鎖に多数のグアニジニウム基を有するイノシトール骨格構造を示し、これらの作用基の位置関係によって様々な異性体(isomer)として存在し得る。また、本発明の化学式(I)のイノシトール誘導体は、イノシトール2量体(dimer)(p=0の場合)、イノシトール3量体(trimer)(p=1)又はイノシトール4量体(tetramer)(p=2)であるか、イノシトール2量体及び3量体が好ましい。
【0020】
本発明の好ましい実施例としては、nが3〜8範囲の整数である式(II)の誘導体であり、このようなn値を有する化合物はこれらに結合された分子らの生体膜透過輸送を向上させる効果を提供する。
【0021】
本発明による好ましいイノシトール誘導体としては下記化学式(XV)の化合物である:
【化11】
【0022】
前記式で、R1、R2、R3、X、X’及びpは前記で定義した通りである。
【0023】
化学式(I)の化合物中、好ましいイノシトール2量体(p=0)としては下記化学式(II)〜(IV)の化合物である:
【化12】
【0024】
前記式で、R1、R2、R3及びXは前記で定義した通りである。
【0025】
前記化学式(II)〜(IV)のイノシトール誘導体における置換基R2及びR3は、作用基を通じて本発明のイノシトール誘導体に結合できる薬物及び診断試薬のような治療物質であり得る。このような物質は分子量が100〜1,500g/molである薬物、または、ペプチド及び核酸等のような重合体化合物であり得る。
【0026】
本発明の好ましい実施例としては、イノシトール誘導体が多様な鎖長のグアニジニウム側鎖を有しながら炭酸塩、カルバマート、エステル又はアミド基で繋がった形のイノシトール2量体でもあり得る。
【0027】
本発明によるイノシトール誘導体は、これらに結合された薬物、診断試薬又は蛍光物質等のような様々な種類の物質を細胞膜、核膜及び血液−脳関門(blood−brain barrier)などのような生体膜を透過させて容易に輸送することができる。
【0028】
前記化学式(II)〜(IV)のイノシトール誘導体は、
(a)myo−又はscyllo−イノシトールの水酸基を保護する反応を行って中間体を得る段階;
(b)段階(a)で得た中間体を2つ以上カップリングしてイノシトール重合体を得る段階;
(c)段階(b)で得たイノシトール重合体のアシル化反応を行い一つ以上のアミノ酸を導入する段階;及び
(d)段階(c)で得たイノシトール重合体のN−末端にグアニジニウム基を導入する段階を含む方法で製造することができる。
【0029】
具体的には、段階(a)では、公知の方法、例えば文献[Chung、Sung−Keeら、Daewoo Science Book series、Natural Science 122,Minumsa(1998)]に記載された方法に基づいて、myo−又はscyllo−イノシトールの水酸基に位置選択的にそれぞれ適切な保護基を導入することができ、このように得られた好ましい中間体の例としては下記化学式(V)〜(XIII)の化合物がある:
【化13】
【0030】
前記式でR’、R’’、l及びmは前記で定義した通りであり、Bnはベンジル、PMBはp−メトキシベンジルである。
【0031】
前記化学式(V)〜(VII)の化合物は本発明の化学式(II)の化合物を製造するための中間体であり、それらのうち、化学式(V)及び(VI)の化合物は下記反応式1に示したように、myo−イノシトールを出発物質として2,3:5,6−ジ−O−イソプロピリデン−myo−イノシトールを合成し、該化合物の1−OH又は4−OHに位置選択的にそれぞれ適切な保護基を導入して製造することができる。
【化14】
【0032】
前記式で、TBDMSはtert−ブチルジメチルシリル、PMBはp−メトキシベンジル、Bnはベンジルである。
【0033】
化学式(II)の化合物の製造において出発物質として使われるmyo−イノシトールは価格が比較的安く、かつ工程を単純化させ得る。しかし、前記反応式1に示したようにmyo−イノシトールから製造された化学式(II)の化合物は立体異性体(diastereomer)の混合物であるため一つの形態の異性体として得るためには化学式(III)及び(IV)の化合物の一つを利用して化学式(I)の化合物を製造することができる。
【0034】
前記化学式(VIII)〜(X)の化合物は化学式(III)の化合物を製造するための中間体であり、それらのうち、化学式(VIII)及び(IX)の化合物らは、下記反応式2に示したように、myo−イノシトールを出発物質として1,6:3,4−ジ−O−イソプロピリデン−myo−イノシトールを合成した後、該化合物の2−OH又は5−OHに位置選択的にそれぞれ適切な保護基を導入して製造することができる。
【化15】
【0035】
前記式でBzはベンゾイルでありPMB及びBnは反応式1で定義した通りである。
【0036】
また、化学式(XI)〜(XIII)の化合物は本発明の化学式(IV)の化合物を製造するための中間体であって、それらのうち、化学式(XI)及び(XII)の化合物は、下記反応式3に示したように、ミツノブ反応(Mitsunobu reaction)を通じてmyo−イノシトールの2−OHの立体構造を反転させてから、ここから1,6:3,4−ジ−O−イソプロピリデン−scyllo−イノシトールを合成した後、該化合物の2−OH又は5−OHに位置選択的にそれぞれ適切な保護基を導入して製造することができる。
【化16】
【0037】
前記式でBz、Bn及びPMBは前記反応式1及び2に定義された通りである。
【0038】
前記反応式2又は3により、純粋な立体異性体(stereoisomer)を得ることができる。
【0039】
また、カルボキシラート基及びアミン基を含む化学式(VII)、(X)及び(XIII)の化合物は、それぞれ1つの水酸基を有する化学式(V)、(VI)、(VIII)、(IX)、(XI)又は(XII)の化合物を位置選択的にアルキル化させて製造することができ、その例として、化学式(XIII)の化合物の製造工程を下記反応式4に示した。化学式(VII)及び(X)の化合物もそれぞれ下記反応式4と類似する工程で製造することができる。
【化17】
【0040】
前記式で、R’、R’’、l、m、Bn及びBzは前記で定義された通りである。
【0041】
段階(b)では文献[The Chemical Synthesis of Peptides、J.Jones、Clarendon Press、Oxford、1991]に記載された方法に基づき、段階(a)で得た2つ以上の中間体を様々な作用基(X及びX’)を利用してカップリングさせてから、アセトニド保護基を取り除くことによりイノシトール重合体を製造することができる。前記作用基(X及びX’)はイノシトール誘導体をカップリングさせるためのリンカーとして作用し、本発明では通常的に使われる様々なリンカーがX及びX’として使用可能であるが、特に−O−CO−O−、−O−CO−NH−(CH2)m−O−、−O−CO−(CH2)l−O−又は−O−(CH2)l−CO−NH−(CH2)m−O−(mは2〜5範囲の整数であり、lは1〜5範囲の整数である)を使うことが好ましい。
【0042】
段階(c)では、ジシクロヘキシルカルボジイミド又は1−[3−(ジメチルアミノ)プロピル)]−エチルカルボジイミド塩酸塩のような縮合剤(condensing agent)を使用して多様な鎖長を有するアミノ酸を段階(b)で得たイノシトール重合体に導入することができる。
【0043】
本発明で使われる多様な鎖長を有するアミノ酸は商業的に利用可能なω−アミノ酸から得ることができ、好ましくは文献[Protective Groups in Organic Synthesis 3rd Ed. T. W. Greene and P. G. M. Wuts、Wiley−Interscience,1999]に記載されたように、アミノ保護基で保護された下記化学式(XIV)のアミノアルカン酸(aminoalkanoic acid)を使うことができる。
【化18】
【0044】
前記アシル化反応は、段階(b)で得たイノシトール重合体1当量当り化学式(XIV)の化合物10〜20当量を用いて0〜60℃範囲の温度で5〜96時間行うことができる。
【0045】
段階(d)では、段階(c)で得たイノシトール重合体のアミノ酸N−末端に存在するt−Boc(tert−ブチルオキシカルボニル)のようなアミノ酸保護基を取り除いた後、有機溶媒中で塩基存在下でN,N−ジ−Boc−N’−トリフロロメタンスルホニルグアニジン(trifluoromethanesulfonylguanidine)と反応させグアニジニウム基を導入することができる。前記N,N−ジ−Boc−N’−トリフロロメタンスルホニルグアニジンは、製造例10および11に示したように、文献[T.T.Bakerら、J.Org.Chem.,65,9054,(2000)]に記載された方法に基づいて製造することができる。
【0046】
前記段階(d)で使える有機溶媒としては、ジメチルホルムアミド、クロロホルム及び酢酸エチル等があり、塩基はトリエチルアミン等があり得る。また、前記反応は0〜60℃で12〜120時間行うことができる。
【0047】
前記段階(b)〜(d)において、化学式(V)〜(XIII)の中間体は多様な組合せでカップリングされて化学式(II)〜(IV)の化合物に変換することができる。本発明によるイノシトール誘導体の製造方法の一例を下記反応式5に示す。
【化19】
【0048】
前記式で、R1、R2及びnは前記で定義した通りである。
【0049】
前記反応式5に示したように、炭酸塩(X=−O−CO−O−)リンカーを用いてカップリングされたイノシトール2量体形態である、グアニジニウム基を有するイノシトール誘導体は化学式(V)及び(VI)の化合物を縮合剤の存在下でカップリングさせて製造することができる。具体的に言えば、化学式(VI)の化合物と化学式(V)の化合物をカップリングさせ、それからアセトニド保護基を除去する。このように得たイノシトール2量体にt−Boc(tert−butyloxycarbonyl)基で保護された多様な鎖長(R1に当たる)を有するアミノ酸をアシル化(acylation)反応を通じて導入し、PMB保護基を除去してからダンシル(dansyl,5−ジメチルアミノ−1−ナフタレンスルホニル(5−dimethylamino−1−naphthalenesulfonyl))基(R2)のような蛍光マーカー(fluorescent tag)を導入する。それからアミノ酸N−末端のt−Boc保護基を取り除き、そこにグアニジニウム基を導入して本発明のイノシトール誘導体を製造することができる。
【0050】
さらに、前記で得られた蛍光物質で表示されたイノシトール誘導体のベンジル保護基を取り除いた後、治療物質(薬物)又は診断試薬(R3に当たる)等を結合させることができる。
【0051】
エステル(X=−O−CO−(CH2)l−O−)、アミド(X=−O−(CH2)l−CO−NH−(CH2)m−O−)及びカルバマート(X=−O−CO−NH−(CH2)m−O−)基を通じて繋がったイノシトール単位を有するイノシトール重合体は文献[The Chemical Synthesis of Peptides、J. Jones、Clarendon Press、Oxford、1991]に記載された方法に基づいて製造することができる。
【0052】
本発明の化学式(I)のイノシトール誘導体は、前記例示した中間体、すなわち化学式(V)〜(XIII)の化合物ばかりでなく、他の中間体からも前記カップリング及びグアニジニウム基を有するアミノ酸の導入工程を含む方法と類似した方法で製造することができる。
【0053】
本発明によるイノシトール誘導体は、薬物及び診断試薬のような治療用物質との結合体でもあり得、本発明によるイノシトール誘導体は様々な治療用物質が細胞膜、核膜及び血管−脳関門等の生体膜を透過して移動することを効果的に向上させる。
【実施例】
【0054】
以下、本発明を下記実施例によって更に詳細に説明する。但し、これらは本発明を例示するためのものであり、本発明の範囲を制限しない。
【0055】
[製造例1]:(±)−1,4−ジ−O−ベンゾイル−2,3:5,6−ジ−O−イソプロピリデン−myo−イノシトールの製造
myo−イノシトール(50g、278mmol)、2,2−ジメトキシプロパン(150ml、1.2mol)及びp−トルエンスルホン酸(1g、5.2mmol)をN,N−ジメチルホルムアミド(200ml)に溶解してから100℃で21時間還流させた。この反応物を常温に冷やしてからトリエチルアミン(10ml)を加え、沈殿物を濾過した後、濾過液にトルエン(25ml)を加えて減圧蒸発させた。得られた残留物をピリジン(150ml)に溶解し、これに塩化ベンゾイル(200ml、1.74mol)を0℃で30分間シリンジで滴加した。その後、反応液を常温で2時間攪拌してから濾過し、沈殿物をピリジン、水、アセトン及びジエチルエーテルで順次洗滌して白色固体状の表題化合物(35.55g)を得た。
【0056】
m.p.=322−325℃
1H−NMR(CDCl3):1.30、1.43、1.50、1.63(4s、3H)、3.73(dd、J=9.6Hz、11.1Hz、1H)、4.36(dd、J=9.6Hz、10.6Hz、1H)、4.41(dd、J=4.5Hz、9.6Hz、1H)、4.78(dd、J=4.5Hz、4.5Hz、1H)、5.42(dd、J=4.5Hz、10.6Hz、1H)、5.60(dd、J=9.6Hz、11.1Hz、1H)、7.45(m、5H)。
【0057】
[製造例2]:(±)−2,3:5,6−ジ−O−イソプロピリデン−myo−イノシトールの製造
製造例1で得た(±)−1,4−ジ−O−ベンゾイル−2,3:5,6−ジ−O−イソプロピリデン−myo−イノシトール(35.5g、75.8mmol)及びNaOCH3(2.41g、45.48mmol)をCH3OH(500ml)に溶解し16時間還流させた。この反応物を常温に冷やしてから減圧蒸発させ、残留物にCH2Cl2(700ml)を加えてシリカベット(silica bed)に通して濾過し、濾過液を濃縮して白色固体状の表題化合物(18.21g)を得た。
【0058】
m.p.=169−171℃
1H−NMR(CDCl3):1.38、1.46、1.48、1.54(4s、3H)、2.36(d、J=8.8Hz、1H)、2.45(d、J=2.9Hz、1H)、3.32(dd、J=9.4Hz、10.5Hz、1H)、3.83(dd、J=9.4Hz、9.4Hz、1H)、3.90(ddd、J=2.9Hz、6.4Hz、10.5Hz、1H)、4.02(ddd、J=4.8Hz、8.8Hz、9.4Hz、1H)、4.08(dd、J=4.8Hz、6.4Hz、1H)、4.48(dd、J=4.8Hz、4.8Hz、1H)。
【0059】
[製造例3]:(±)−1−O−t−ブチルジメチルシリル−2,3:5,6−ジ−O−イソプロピリデン−myo−イノシトールの製造
製造例2で得た(±)−2,3:5,6−ジ−O−イソプロピリデン−myo−イノシトール(6g、23mmol)及びイミダゾール(5.2g、76mmol)をN,N−ジメチルホルムアミド(70ml)に溶解し、それに0℃でtert−ブチルジメチルシリルクロライド(3.8g、25mmol)を添加して常温で14時間攪拌させた。反応混合物を酢酸エチルで抽出した後、NaClとNaHCO3飽和水溶液で洗滌し、MgSO4で乾燥させてから減圧濃縮させた。濃縮物をカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:10〜1:1)で精製して白色固体状の表題化合物(5.84g)を得た。
【0060】
m.p.=148−150℃
1H−NMR(CDCl3):0.14(s、6H)、0.93(s、9H)、1.35、1.42、1.44、1.53(4s、3H)、2.87(brs、1H)、3.26(dd、J=9.4Hz、10.4Hz、1H)、3.87(dd、J=6.4Hz、10.4Hz、1H)、3.90(dd、J=9.4Hz、10.4Hz、1H)、3.97(dd、J=4.4Hz、4.6Hz、1H)、4.03(dd、J=4.4Hz、10.4Hz、1H)、4.29(dd、J=4.4Hz、4.4Hz、1H)。
【0061】
[製造例4]:(±)−4−O−ベンジル−2,3:5,6−ジ−O−イソプロピリデン−1−O−t−ブチルジメチルシリル−myo−イノシトールの製造
製造例3で得た(±)−1−O−t−ブチルジメチルシリル−2,3:5,6−ジ−O−イソプロピリデン−myo−イノシトール(5.76g、15mmol)をCH2Cl2(100ml)に溶解してから酸化銀(I)(10.7g、46mmol)、ベンジルブロマイド(5.47ml、46mmol)及びテトラブチルアンモニウム・ヨウ化物(0.55g、1.5mmol)を加えて常温で2時間攪拌させた。得られた溶液をセライトで濾過させ濾過液をCH2Cl2で洗滌した後減圧濃縮させ、濃縮物をカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:40)で精製して白色固体状の表題化合物(4.91g)を得た。
【0062】
m.p.=89℃
1H−NMR(CDCl3):0.12(s、6H)、0.90(s、9H)、1.31、1.35、1.40、1.41(4s、3H)、3.31(t、J=9.7Hz、1H)、3.65(dd、J=6.6Hz、10.4Hz、1H)、3.85(t、J=9.3Hz、1H)、3.98(dd、J=4.3Hz、10.1Hz、1H)、4.07(t、J=5.6Hz、1H)、4.27(t、J=5.6Hz、1H)、4.80(s、2H)、7.21−7.40(m、5H)
MS(FAB)m/z465.21(M++H)。
【0063】
[製造例5]:(±)−4−O−ベンジル−2,3:5,6−ジ−O−イソプロピリデン−myo−イノシトール(化学式(V)の化合物)の製造
製造例4で得た(±)−4−O−ベンジル−2,3:5,6−ジ−O−イソプロピリデン−1−O−t−ブチルジメチルシリル−myo−イノシトール(4.6g、9.9mmol)をテトラヒドロフランに溶解し、これにフッ化テトラブチルアンモニウムをTHFに溶解して作った1.0M溶液(29.7ml、29.7mmol)を加えて常温で7時間攪拌させた。得られた溶液を酢酸エチルで抽出した後、NaCl飽和水溶液で洗滌し、有機層をMgSO4で乾燥させた後、減圧蒸発させ濃縮させてからヘキサンで再結晶して白色固体状の表題化合物(3.7g)を得た。
【0064】
m.p.=131℃
1H−NMR(CDCl3):1.33、1.36、1.43、1.46(4s、3H)、2.40(d、J=8.4Hz、1H)、3.39(t、J=9.6Hz、1H)、3.65(dd、J=6.4Hz、10.4Hz、1H)、3.78(t、J=9.8Hz、1H)、3.94−4.02(m、1H)、4.18(t、J=5.6Hz、1H)、4.24(t、J=4.9Hz、1H)、4.80(s、2H)、7.22−7.40(m、5H)。
【0065】
[製造例6]:(±)−1−O−p−メトキシベンジル−2,3:5,6−ジ−O−イソプロピリデン−myo−イノシトール(化学式(VI)の化合物)の製造
製造例2で得た(±)−2,3:5,6−ジ−O−イソプロピリデン−myo−イノシトール(5.59g、20mmol)をN,N−ジメチルホルムアミド(90ml)に溶解してからNaH(0.96g、40mmol)及びp−メトキシベンジルクロライド(3.2ml、24mmol)を加えて常温で攪拌させた。10時間後反応が完結したら、0℃で水を加えて反応を中断させた。得られた溶液をCH2Cl2で抽出した後、NaHCO3飽和水溶液で洗滌し有機層をMgSO4で乾燥させてから減圧蒸発させて濃縮し、それをカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:2〜1:1)で精製して白色固体状の表題化合物(2.2g)を得た。
【0066】
m.p.=153−154℃
1H−NMR(CDCl3):1.32、1.44、1.46、1.52(4s、3H)、2.27(d、J=2.5Hz)、3.24(dd、J=10.1Hz、10.1Hz、1H)、3.75(dd、J=4.3Hz、10.1Hz、1H)、3.79(s、3H)、3.83−3.92(m、2H)、4.01(dd、J=10.1Hz、10.1Hz、1H)、4.27(dd、J=4.3Hz、4.3Hz、1H)、4.71(dd、J=12.1Hz、1H)、4.82(d、J=12.1Hz、1H)、6.86(d、J=8.6Hz、2H)、7.32(d、J=8.6Hz、2H)。
【0067】
[製造例7]:4−Boc−アミノブタン酸(4−Boc−aminobutanoic acid)の製造
4−アミノブタン酸(1g、9.7mmol)をtert−ブチルアルコール(15ml)に溶解してから5N NaOH(1.93ml、9.7mmol)を加えて常温で10分間攪拌させた。これにtert−ブチルアルコール(17ml)に溶解したジ−tert−ブチル−ジカーボネート(di−tert−butyl−dicarbonate)(2.33g、10.7mmol)を加えて常温で24時間攪拌させた。これに水(12ml)を加え減圧濃縮させた後0℃に冷却させ、反応溶液のpHが2になるまで2N H2SO4を滴加した。得られた溶液を酢酸エチルで抽出して蒸留水(H2O)で洗滌した後有機層をMgSO4で乾燥させ、これを減圧濃縮させて白色固体状の表題化合物(2.1g)を得た。
【0068】
m.p.=57−58℃
1H−NMR(CDCl3):1.45(s、9H)、3.19(m、2H)、4.70(brs、1H)。
【0069】
[製造例8]:6−Boc−アミノヘキサン酸(6−Boc−aminohexanoic acid)の製造
6−アミノカプロン酸(6−aminocaproic acid)(10g、76mmol)をtert−ブチルアルコール(100ml)に溶解してから5N NaOH(15ml、76mmol)を加えて常温で10分間攪拌させた。前記反応液にtert−ブチルアルコール(100ml)に溶解したジ−tert−ブチル−ジカーボネート(18.3g、83mmol)を加えた後、常温で24時間攪拌させた。これに蒸留水(100ml)を加えた後、減圧蒸発させ濃縮させてから0℃に冷却させ溶液のpHが2になるまで2N H2SO4を滴加した。引き続き、得られた溶液を酢酸エチルで抽出し蒸留水で洗滌した後有機層をMgSO4で乾燥させてから減圧蒸発させ濃縮させ、カラムクロマトグラフィー(酢酸エチル:トルエン:酢酸=20:10:0.3)で精製して白色固体状の前記表題化合物(17.9g)を得た。
【0070】
m.p.=38−39℃
1H−NMR(CDCl3):1.30(m、2H)、1.40(s、9H)、1.50(m、2H)、2.28(t、J=7.4Hz、2H)、3.01(m、2H)。
【0071】
[製造例9]:8−Boc−アミノオクタン酸(8−Boc−aminooctanoic acid)の製造
8−アミノカプリン酸(8−aminocaprylic acid)(970.3mg、6.1mmol)をtert−ブチルアルコール(14.7ml)に溶解してから5N NaOH(1.44ml、6.1mmol)を加え常温で10分間攪拌させた。前記反応液にtert−ブチルアルコール(10ml)に溶解したジ−tert−ブチル−ジカーボネート(1.46g、6.7mmol)を加えて常温で24時間攪拌させた。これに蒸留水(10ml)を加えた後減圧蒸発・濃縮させてから0℃に冷却させ溶液のpHが2になるまで2N H2SO4を滴加した。引き続き、得られた溶液を酢酸エチルで抽出し蒸留水で洗滌した後有機層をMgSO4で乾燥させてから減圧揮発して濃縮させ、カラムクロマトグラフィー(酢酸エチル:トルエン:酢酸=20:10:0.3)で精製して白色固体状の前記表題化合物(1.78g)を得た。
【0072】
m.p.=57−58℃
1H−NMR(CDCl3):1.30(m、6H)、1.45(s、9H)、1.46(m、2H)、1.63(m、2H)、2.34(t、J=7.4Hz、2H)、3.10(m、2H)。
【0073】
[製造例10]:N,N−ビス−Boc−グアニジンの製造
グアニジン塩酸塩(guanidine hydrochloride)(4g、42mmol)及び4N NaOH(42ml、0.1mol)を1,4−ジオキサン(80ml)に溶解し0℃に冷却させた。これにジ−tert−ブチル−ジカーボネート(20g、92.1mol)を加えた後、常温に加熱し18時間攪拌させた。元の反応物容積の1/3になるよう減圧蒸発させた後、水を加えて酢酸エチルで抽出した。得られた溶液を10%クエン酸、蒸留水及びNaCl飽和水溶液順に洗滌しMgSO4で乾燥させた後、減圧蒸発させて濃縮させ、カラムクロマトグラフィー(CH2Cl2:MeOH=20:1)で精製して前記表題化合物(6.47g)を得た。
【0074】
m.p.=144−145℃
1H−NMR(DMSO−d6):1.39(s、18H)、8.47(brs、2H)、10.42(brs、1H)。
【0075】
[製造例11]:N,N−ジ−Boc−N’−トリフロロメタンスルホニルグアニジン(N,N−di−Boc−N’−trifluoromethanesulfonylguanidine)の製造
製造例10で得たN,N−ビス−Boc−グアニジン(6.05g、23mmol)をCH2Cl2(40ml)に溶解してからトリエチルアミン(4.8ml、34.5mmol)を滴加し、−78℃でトリフリックアンハイドライド(triflic anhydride)(3.9ml、25.3mmol)を注射器で滴加した。得られた溶液を4時間常温に加熱した後、常温で3時間攪拌させた。反応生成物を2N NaSO4及び蒸留水で洗滌し有機層をNa2SO4で乾燥させた後減圧蒸発・濃縮させてカラムクロマトグラフィー(CH2Cl2)で精製した。これをヘキサンで再結晶して白色固体状の前記表題化合物(7.09g)を得た。
【0076】
m.p.=114−115℃
1H−NMR(DMSO−d6):1.45(s、18H)、11.45(brs、2H)。
【0077】
[製造例12]:2−O−ベンゾイル−myo−イノシトールオルトギ酸塩(2−O−benzoyl−myo−inositol orthoformate)の製造
myo−イノシトール(50g、278mmol)、オルトギ酸トリメチル(trimethyl orthoformate)(63ml、570mmol)及びp−トルエンスルホン酸(2.0g)をN,N−ジメチルホルムアミド(200ml)に溶解してから120℃で7時間攪拌した。得られた溶液を50℃で2時間減圧蒸留した後、ピリジン(100ml)を加え0℃で塩化ベンゾイル(35ml、299mmol)を滴加した。得られた溶液を常温で一日攪拌した後、蒸留水(10ml)を加え30分間攪拌してから反応液を酢酸エチルで希釈しNaHSO4、NaHCO3及びNaCl飽和水溶液で洗滌した。引き続き、有機層をMgSO4で乾燥してから濾過し濾過液を濃縮した後、酢酸エチルで再結晶して白色固体状の前記表題化合物(35.5g)を得た。
【0078】
m.p.=206−207℃
1H−NMR(acetone−d6、with drops of D2O):δ4.26(m、1H)、4.37(m、2H)、4.50(t、J=3.8Hz、2H)、5.52(d、J=1.3Hz、1H)、5.57(app q,J=1.9Hz、1H)、7.50−8.11(m、5H)。
【0079】
[製造例13]:2−O−ベンゾイル−myo−イノシトールの製造
製造例12で得た2−O−ベンゾイル−myo−イノシトールオルトギ酸塩(10g、34mmol)をCH3OH(200ml)に溶解しp−トルエンスルホン酸(646mg、3.4mmol)を加えてから、60℃で3時間攪拌した。その後、反応液の温度を常温に下げ一日放置し結晶化した後、生成された固体を濾過しCH3OH及び酢酸エチルで洗滌して白色固体状の前記表題化合物(8g)を得た。
【0080】
m.p.=240−242℃
1H−NMR(CD3OD−DMSO−d6):δ3.30(t、J=8.9Hz、1H)、3.66(dd、J=2.6、9.7Hz、2H)、3.73(dd、J=8.9Hz、9.7Hz、2H)、5.69(t、J=2.6Hz、1H)、7.51−8.09(m、5H)。
【0081】
[製造例14]:2−O−ベンゾイル−1,6:3,4−ジ−O−イソプロピリデン−myo−イノシトールの製造
製造例13で得た2−O−ベンゾイル−myo−イノシトール(5.0g、17.6mmol)及び2−メトキシプロペン(10.1ml、105.5mmol)をN,N−ジメチルホルムアミド(100ml)に溶解し0℃でp−トルエンスルホン酸(335mg、1.76mmol)を少しずつ分けて加えた。常温で20時間攪拌した後、反応液をNaHCO3飽和水溶液(200ml)に注ぎ入れて酢酸エチル(400ml)で抽出した後、抽出した有機層をNaCl飽和水溶液(400ml)で洗滌した。引き続き有機層を乾燥(MgSO4)・濃縮してカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:3〜1:1)及び分別結晶(酢酸エチル及びn−ヘキサン)を行い白色固体状の前記表題化合物(2g)を得た。
【0082】
m.p.=181−183℃
1H−NMR(CDCl3):δ1.40、1.48(2s、12H)、2.97(d、J=1.5Hz、1H)、3.83(dd、J=2.1Hz、8.4Hz、2H)、4.09−4.15(m、3H)、6.06(t、J=2.1Hz、1H)、7.47−8.09(m、5H)
MS(FAB)m/z=387(M++Na)、365(M++H)。
【0083】
[製造例15]:(±)−2,3−O−イソプロピリデン−myo−イノシトールの製造
myo−イノシトール(50g、277mmol)、2,2−ジメトキシプロパン(85ml、691mmol)及びp−トルエンスルホン酸(500mg、2.6mmol)をメチルスルホキシド(methyl sulfoxide)(160ml)に溶解してから90〜100℃で1時間攪拌させた。反応溶液を20℃に冷却させた後、エチルアルコール(200ml)及びエーテル(1L)を加えて2時間攪拌させてからトリエチルアミン(10ml)を加えて4時間攪拌させた。反応生成物を濾過した後、CH3OH/エーテル(1:5、240ml)で洗滌し、エチルアルコールで再結晶して白色固体状の前記表題化合物(51g)を得た。
【0084】
m.p.=165−166℃
1H−NMR(CDCl3):δ1.32、1.47(2s、6H)、3.09(app.t、J=9.1Hz、1H)、3.53(dd、J=9Hz、9.3Hz、2H)、3.90(app.t、J=8.7Hz、1H)、4.34(app.t、J=9.3Hz、1H)。
【0085】
[製造例16]:(±)−1,4,5,6−テトラ−O−ベンジル−2,3−O−イソプロピリデン−myo−イノシトールの製造
製造例15で得た(±)−2,3−O−イソプロピリデン−myo−イノシトール(20g、90.8mmol)及びNaH(47.5g、1090mmol)をN,N−ジメチルホルムアミド(600ml)に溶解してから0℃でベンジルブロマイド(108.02ml、908mmol)を入れて2時間攪拌した後、室温に上げさらに20時間攪拌させた。反応生成物を酢酸エチルで抽出しNaHCO3とNaCl飽和水溶液で洗滌した後、MgSO4で乾燥させ、これを減圧蒸発させカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:9)で精製してシロップ状の前記表題化合物(45g)を得た。
【0086】
1H−NMR(CDCl3):δ1.37、1.55(2s、6H)、3.42(t、J=8.7Hz、1H)、3.88(dd、J=3.7Hz、8.8Hz、1H)、3.79(dd、J=7.0Hz、9.5Hz、1H)、3.99(app.t、J=8.5Hz、1H)、4.10(app.t、J=6.2Hz、1H)、4.27(dd、J=3.8Hz、5.5Hz、1H)、4.72−4.90(m、8H)、7.22−7.40(m、20H)。
【0087】
[製造例17]:(±)−1,4,5,6−テトラ−O−ベンジル−myo−イノシトールの製造
製造例16で得た(±)−1,4,5,6−テトラ−O−ベンジル−2,3−O−イソプロピリデン−myo−イノシトール(42g、72.38mmol)を80%酢酸水溶液(290ml)に溶解し、100℃で3時間攪拌した。反応生成物を減圧蒸発・濃縮させた後、酢酸エチル−ヘキサン(1:2)溶液で再結晶し、精製された白色固体状の前記表題化合物(36.5g)を得た。
【0088】
m.p.=126.5−127℃
1H−NMR(CDCl3):δ2.40(d、4.5Hz、1H)、2.45(s、1H)、3.44−3.51(m、3H)、3.85(app.t、J=9.5Hz、1H)、4.0(app.t、J=9.5Hz、1H)、4.21(brs、1H)、4.7−4.97(m、8H)、7.25−7.40(m、20H)。
【0089】
[製造例18]:(±)−1−O−ベンゾイル−2,3,4,5−テトラ−O−ベンジル−scyllo−イノシトールの製造
製造例17で得た(±)−1,4,5,6−テトラ−O−ベンジル−myo−イノシトール(30g、55.53mmol)、トリフェニルホスフィン(triphenyl
phosphine)(17.5g、66.63mmol)、安息香酸(8.1g、66.63mmol)及びジエチルアゾジカルボキシラート(diethyl azodicarboxylate)(10.5ml、66.63mmol)をトルエン(300ml)に溶解して80〜85℃で4時間攪拌させた。生成された溶液を濾過した後、濾過液を減圧蒸発させカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:2)で精製して固体状の前記表題化合物(30.8g)を得た。
【0090】
m.p.=135−136℃
1H−NMR(CDCl3):δ2.45(s、1H)、3.55−3.77(m、5H)、4.7−5.0(m、8H)、5.39(app.t、J=9.8Hz、1H)、7.10−8.05(m、25H)。
【0091】
[製造例19]:1−O−ベンゾイル−scyllo−イノシトールの製造
製造例18で得た(±)−1−O−ベンゾイル−2,3,4,5−テトラ−O−ベンジル−scyllo−イノシトール(8g、12.4mmol)および20%Pd(OH)2/C(4.3g)をCH2Cl2/CH3OH(1:2、210ml)混合溶液に加えてH2(50psi)下で24時間混合させた。得られた溶液を濾過し減圧蒸発させた後、CH3OHに再結晶して固体状の前記表題化合物(3.6g)を得た。
【0092】
m.p.=245−247℃
1H−NMR(CD3OD):δ3.30−3.40(m、3H)、3.55(app.t、J=9.2Hz、2H)、5.1(t、J=9.7Hz、1H)、7.45−8.1(m、5H)。
【0093】
[製造例20]:1−O−ベンゾイル−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトールの製造
製造例19で得た1−O−ベンゾイル−scyllo−イノシトール(6g、21.11mmol)及びp−トルエンスルホン酸(400mg、2.1mmol)をN,N−ジメチルホルムアミド(100ml)に溶解し、これに室温で2−メトキシプロペン(10ml、105.6mmol)をゆっくり30分間滴加した。得られた溶液を2時間攪拌させた後、2−メトキシプロペン(10ml、105.6mmol)を滴加し、8時間後にNaHCO3飽和水溶液を入れ攪拌させ、これを酢酸エチルで抽出してMgSO4で乾燥させた後減圧蒸発させた。反応生成物を再結晶(酢酸エチル:核酸2:1)して立体異性体を互いに分離させてからカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:1)で精製して前記表題化合物(2.1g)を得た。
【0094】
m.p.=284−286℃
1H−NMR(CDCl3):δ1.44、1.48(6H)、2.46(d、J=2.8Hz、1H)、3.76(app.t、J=9.4Hz、2H)、3.88(app.t、J=9.4Hz、2H)、4.13(dt、J=2.8Hz、8.8Hz)、5.7(t、J=9.3Hz、1H)、7.27−8.1(m、5H)。
【0095】
[製造例21]:4−O−(1’−イミダゾリルカルボニルオキシ)−2,3:5,6−ジ−O−イソプロピリデン−1−O−p−メトキシベンジル−myo−イノシトールの製造
製造例6で得た1−O−p−メトキシベンジル−2,3:5,6−ジ−O−イソプロピリデン−myo−イノシトール(2.2g、5.8mmol)をトルエン(40ml)に溶解してからCaH2(0.59g、13.3mmol)およびカルボニルジイミダゾール(carbonyldiimidazole)(2.35g、15.4mmol)を加えて11時間攪拌させた。生成された溶液を濾過した後濾過液を減圧蒸発・濃縮させ、カラムクロマトグラフィー(酢酸エチル:ヘキサン=1:1)で精製して白色固体状の前記表題化合物(2.6g)を得た。
【0096】
m.p.=144−145℃
1H−NMR(CDCl3):1.35、1.46、1.45、1.63(4s、3H)、3.49(dd、J=9.3Hz、11.4Hz、1H)、3.80(s、3H)、3.84(d、J=4.2Hz、1H)、4.17(dd、J=2.4Hz、5.4Hz、1H)、4.21(d、J=9.6Hz、1H)、4.32(dd、J=4.5Hz、1H)、4.74(d、J=12.0Hz、1H)、4.86(d、J=12.0Hz、1H)、5.35(dd、J=6.9Hz、11.1Hz、1H)、6.89(d、J=8.7Hz、2H)、7.05(dd、J=0.6Hz、1.5Hz、1H)、7.35(d、J=8.7Hz、2H)、7.42(dd、J=1.5Hz、1H)、8.12(s、1H)
MS(FAB)m/z497(M++Na)。
【0097】
[製造例22]:2−O−ベンゾイル−1,6:3,4−ジ−O−イソプロピリデン−5−O−p−メトキシベンジル−myo−イノシトールの製造
製造例14で得た2−O−ベンゾイル−1,6:3,4−ジ−O−イソプロピリデン−myo−イノシトール(1g、2.74mmol)、Ag2O(1.27g、5.49mmol)、p−メトキシベンジルクロライド(744μl、5.49mmol)、分子体(molecular sieve)4Å(粉末、1g)及びテトラブチルアンモニウムヨージド(tetrabutylammonium iodide)(101mg、274μmol)をCH2Cl2(10ml)に溶解して常温で30時間攪拌した。反応生成物をカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:2)で精製して結晶化(酢酸エチル及びn−ヘキサン)し、白色固体状の前記表題化合物(837mg)を得た。
【0098】
m.p.=150−152℃
1H−NMR(CDCl3):δ1.35、1.44(2s、12H)、3.77(dd、J=9.5Hz、2.0Hz、2H)、3.80(s、3H)、3.87(t、J=9.1Hz、1H)、4.15(t、J=9.3Hz、1H)、4.80(s、2H)、6.0(t、J=1.9Hz、1H)、6.87(d、J=8.8Hz、2H)、7.35−7.57(m、5H)、8.0(d、J=8.3Hz、2H)
MS(FAB)m/z=507(M++Na)、485(M++H)。
【0099】
[製造例23]:1,6:3,4−ジ−O−イソプロピリデン−5−O−p−メトキシベンジル−myo−イノシトール(化学式(VIII)の化合物)の製造
製造例22で得た2−O−ベンゾイル−1,6:3,4−ジ−O−イソプロピリデン−5−O−p−メトキシベンジル−myo−イノシトール(700mg、1.45mmol)をCH3OH(15ml)に溶解してからNaOCH3(66μl、25%inCH3OH,0.29mmol)を加えて2時間加熱還流した。反応生成物の温度を常温に下げて一日間放置し結晶化した後、生成された固体を濾過してCH3OH及び酢酸エチルで洗滌して白色固体状の前記表題化合物(484mg)を得た。
【0100】
m.p.=191−192℃
1H−NMR(CDCl3):δ1.48、1.50(2s、12H)、2.22(brs、1H)、3.59(dd、J=9.4Hz、2.1Hz、2H)、3.82(s、3H)、3.82(t、J=9.4Hz、1H)、4.12(t、J=9.3Hz、2H)、4.58(broadd、J=1.9Hz、1H)、4.80(s、2H)、6.89(d、J=8.6Hz、2H)、7.37(d、J=8.6Hz、2H)
MS(FAB)m/z=403(M++Na)。
【0101】
[製造例24]:2−O−ベンジル−1,6:3,4−ジ−O−イソプロピリデン−5−O−p−メトキシベンジル−myo−イノシトールの製造
製造例23で得た1,6:3,4−ジ−O−イソプロピリデン−5−O−p−メトキシベンジル−myo−イノシトール(280mg、0.736mmol)をN,N−ジメチルホルムアミド(5ml)に溶解した後、0℃でNaH(70mg、ミネラルオイル中55%、1.47mmol)を加え、常温で30分間攪拌した。これにベンジルブロマイド(256mg、1.47mmol)を加え、常温で1時間攪拌し、これにNaHCO3飽和水溶液(50ml)を加えて反応を終結し、酢酸エチル(300ml)で希釈した。これをNaHCO3(50ml)とNaCl(50ml)飽和水溶液で洗滌した後有機層を乾燥(Na2SO4)・濃縮してカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:2)で精製して白色固体状の前記表題化合物(320mg)を得た。
【0102】
m.p.=180−181℃
1H−NMR(CDCl3):δ1.47、1.48(2s、12H)、3.62(dd、J=9.5Hz、2.0Hz、2H)、3.80(t、J=9.2Hz、1H)、3.82(s、3H)、4.20(t、J=9.3Hz、2H)、4.37(t、J=2.0Hz、1H)、4.80、4.85(2s、4H)、6.90(dm、J=6.7Hz、2H)、7.27−7.39(m、8H)
MS(FAB)m/z=493(M++Na)。
【0103】
[製造例25]:2−O−ベンジル−1,6:3,4−ジ−O−イソプロピリデン−myo−イノシトール(化学式(IX)の化合物)の製造
製造例24で得た2−O−ベンジル−1,6:3,4−ジ−O−イソプロピリデン−5−O−p−メトキシベンジル−myo−イノシトール(283mg、0.60mmol)をCH2Cl2に溶解してから常温でMn(OAc)3(449mg、1.8mmol)及び2,3−ジクロロ−5,6−ジシアノ−1,4−ベンゾキノン(147mg、0.65mmol)を加え12時間攪拌した。得られた反応液にNaHCO3飽和水溶液(30ml)をゆっくり加え、酢酸エチル(50ml)で希釈した後有機層を蒸留水(30ml×2回)及び飽和NaCl水溶液で洗滌した。洗滌した有機層は乾燥(Na2SO4)後濃縮して酢酸エチル:ヘキサン=1:2〜1:1の混合溶液で精製し、白色固体状の前記表題化合物(176mg)を得た。
【0104】
m.p.=190−191℃
1H−NMR(CDCl3):δ1.48、1.49(2s、12H)、2.44(brs、1H)、3.64(dd、J=9.1Hz、1.9Hz、2H)、4.02(m、1H)、4.13(t、J=9.4Hz、2H)、4.40(t、J=2.0Hz、1H)、4.85(s、2H)、6.90(dm、J=6.7Hz、2H)、7.27−7.40(m、5H)
MS(FAB)m/z=373(M++Na)。
【0105】
[製造例26]:2−O−(1’−イミダゾール−イル−カルボニルオキシ)−5−O−p−メトキシベンジル−1,6:3,4−ジ−O−イソプロピリデン−myo−イノシトールの製造
製造例23で得た1,6:3,4−ジ−O−イソプロピリデン−5−O−p−メトキシベンジル−myo−イノシトール(150mg、0.39mmol)をトルエン(3ml)に溶解し常温でCaH2(41mg、0.9mmol)を加えて20分間攪拌した。これにカルボニルジイミダゾール(170mg、0.98mmol)を加えて36時間攪拌し、反応生成物を濾過した濾過液を濃縮した後、カラムクロマトグラフィーで精製して白色固体状の前記表題化合物(184mg)を得た。
【0106】
m.p.=159−162℃
1H−NMR(CDCl3):δ1.41、1.49(2s、12H,2CMe2)、3.80(dd、J=9.5、2.3Hz、2H,H−1&H−3)、3.83(s、3H,OCH3)、3.90(t、J=Hz、9.0Hz、1H,H−5)、4.05(t、J=9.3Hz、2H,H−4&H−6)、4.82(s、2H,PhCH2)、5.92(t、J=2.2Hz、1H,H−2)、6.91(dt、J=8.7、2.9Hz、2H,Ph)、7.13(dd、J=1.54、0.7Hz、1H,imidazole)、7.38(dt、J=8.65、2.7Hz、2H,Ph)7.44(t、J=1.4Hz、1H,imidazole)、8.17(s、1H,imidazole)
MS(FAB)m/z=475(M++1)。
【0107】
[製造例27]:1−O−ベンゾイル−4−O−ベンジル−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトールの製造
製造例20で得た1−O−ベンゾイル−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトール(200mg、0.549mmol)、酸化銀(silver(I)oxide)(381mg、1.64mmol)及びテトラブチルアンモニウムヨージド(40mg、0.109mmol)をCH2Cl2に溶解してから室温でベンジルブロマイド(0.19ml、1.64mmol)を滴加した。2時間後、反応液をセライトで濾過した後、CH2Cl2で数回洗滌した。洗滌した濾過液をNaHCO3とNaCl飽和水溶液で洗滌した後有機層をMgSO4で乾燥させて減圧蒸発させた後、カラムクロマトグラフィー(酢酸エチル:ヘキサン=1:9)で精製して白色固体状の前記表題化合物(200mg)を得た。
【0108】
m.p.=175−176℃
1H−NMR(CDCl3):δ1.44、1.46(s、6H)、3.81−3.9(m、5H)、4.86(s、2H)、5.58(app.t、J=9.3Hz、1H)、7.25−7.56(m、7H)、8.06(d、J=8.7Hz、2H)
MS(FAB)m/z478(M++Na)。
【0109】
[製造例28]:1−O−ベンジル−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトール(化学式(XII)2の化合物)の製造
製造例27で得た1−O−ベンゾイル−4−O−ベンジル−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトール(200mg、0.44mmol)をCH3OH(15ml)に溶解してからNaOCH3(0.04ml、0.17mmol、25% w/v in CH3OH)を滴加して3時間還流させた。得られた溶液を室温でシリカゲルを通じて濾過させてから、濾過液を濃縮し5%酢酸エチルで洗滌してメチルベンゾエイトを取り除くことにより白色固体状の前記表題化合物(150mg)を得た。
【0110】
m.p.=215−216℃
1H−NMR(CDCl3):δ1.46(s、12H)、2.41(d、J=2.7Hz、1H)、3.63(t、J=9.1Hz、4H)、3.71(t、J=9Hz、4H)、3.87(t、J=8.9Hz、1H)、4.1(app.t、J=9.1Hz、1H)、4.83(s、2H)、26−7.39(m、5H)
MS(FAB)m/z373(M++Na)。
【0111】
[製造例29]:1−O−p−メトキシベンジル−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトール(化学式(XI)1の化合物)の製造
製造例20で得た1−O−ベンゾイル−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトール(300mg、0.85mmol)をN,N−ジメチルホルムアミド(10ml)に溶解してから0℃でNaH(57.8mg、1.44mmol)を加え、p−メトキシベンジルクロライド(p−methoxybenzyl chloride)(0.12ml、0.93mmol)をゆっくり滴加した。これにテトラブチルアンモニウムヨージドを触媒量だけ滴加し、室温で4時間攪拌した。反応生成物を酢酸エチルで抽出してNaHCO3とNaCl飽和水溶液で洗滌した。得られた有機層をMgSO4で乾燥させて減圧蒸発させた後、CH3OH(15ml)及びNaOCH3(0.05ml、0.057mmol、25% w/v in CH3OH)を加えて3時間還流させた。反応生成物を室温に冷却させシリカゲルを通じて濾過した後、濾過液を減圧蒸発させカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:3)で精製して前記表題化合物(210mg)を得た。
【0112】
m.p.=198−200℃
1H−NMR(CDCl3):δ1.47(s、12H)、2.41(brs、1H)、3.59−3.63(m、5H)、3.65(s、3H)、3.80(app.t、J=9.1Hz、1H)、4.76(s、2H)、6.87(d、J=9Hz、2H)、7.33(d、J=9.1Hz、2H)
MS(FAB)m/z403(M++Na)。
【0113】
[製造例30]:1−O−ベンジル−4−(1−イミダゾール−イル−カルボニルオキシ)−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトールの製造
製造例28で得た1−O−ベンジル−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトール(70mg、0.19mmol)をトルエン(6ml)に溶解し常温でCaH2(21mg、0.49mmol)を加え、20分間攪拌した。これにカルボニルジイミダゾール(81mg、0.49mmol)を加えて18時間攪拌し、反応生成物を濾過し、得られた濾過液を濃縮した後カラムクロマトグラフィー(酢酸エチル:ヘキサン=1:1)で精製して白色固体状の前記表題化合物(87mg)を得た。
【0114】
m.p.=214−216℃
1H−NMR(CDCl3):δ1.45、1.47(2s、12H,2CMe2)、3.80−3.87(m、5H)、4.85(s、2H,PhCH2)、5.45(app.t、J=1.9Hz、1H,H−1)、7.07(s、1H)、7.29−7.42(m、6H)、8.16(s、1H)
MS(FAB)m/z=445(M++1)、467(M++1)。
【0115】
[製造例31]:4−O−アリール−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトールの製造
製造例20で得た1−O−ベンゾイル−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトール(500mg、1.37mmol)をN,N−ジメチルホルムアミド(10ml)に溶解してから0℃でNaH(9.8mg、2.47mmol)を加え、10分後アリールブロマイド(0.18ml、2.05mmol)を加えて室温で20時間攪拌させた。得られた反応液を酢酸エチルで抽出してNaCl飽和水溶液で洗滌し、これをMgSO4で乾燥させてから減圧揮発し、カラムクロマトグラフィー(酢酸エチル:ヘキサン=3:7)で精製して4−O−アリール−1−O−ベンゾイル−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトール(450mg)を得た。得られた4−O−アリール−1−O−ベンゾイル−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトール(400mg、0.989mmol)をCH3OH(12ml)に溶解してからNaOCH3(0.09ml、0.395mmol、25% w/v in CH3OH)を滴加して3時間還流させた。反応生成物を室温に冷却させシリカゲルを通じて濾過した後、濾過液を濃縮した後5%酢酸エチルで洗滌してメチルベンゾエイト(methylbenzoate)を取り除くことにより白色固体状の前記表題化合物(290mg)を得た。
【0116】
m.p.=200−202℃
1H−NMR(CDCl3):δ1.46(s、12H)、2.46(d、J=2.7Hz、1H)、3.57−3.68(m、4H)、3.83(t、J=8.1Hz、1H)、4.05(t、J=6.9Hz、1H)、4.28(d、J=5.7Hz、2H)、5.21(d、J=10.2Hz、1H)、5.33(dd、J=18.1Hz、1.5Hz1H)、5.95(m、1H)
MS(FAB)m/z301(M++1)、323(M++Na)。
【0117】
[製造例32]:4−O−(2−N,N−ジベンジルアミノエチル)−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトールの製造
製造例31で得た4−O−アリール−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトール(250mg、0.832mmol)及びNaHCO3(139mg、1.66mmol)をCH2Cl2/CH3OHの混合溶液(6:1、50ml)に溶解し、−78℃で反応溶液の色が青になるまでO3を滴加した。再び溶液の青色が消えてしまうまでN2を滴加した後、トリフェニルホスフィン(327mg、12.49mmol)を滴加した。ヒドロペルオキシドの還元反応が終結すると、カラムクロマトグラフィーで精製して4−O−エタナール−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトール(235mg)を得た。これ(150mg、0.496mmol)をジクロロエタン(15ml)に溶解し0〜5℃でジベンジルアミン(dibenzylamine)(0.04ml、0.744mmol)を加える。10分後、これにソジウムトリアセトキシボロハイドライド(sodium triacetoxyborohydride)(265mg、1.24mmol)を加えて0〜5℃で1時間攪拌させた後、室温で12時間攪拌した。反応生成物をCH2Cl2(60ml)で抽出した後、1N NaOH(30ml)溶液で洗滌し、蒸留水で5回洗滌した。得られた有機層をMgSO4で乾燥させた後、減圧蒸発・濃縮させ、カラムクロマトグラフィー(酢酸エチル:ヘキサン=7:3)で精製して白色固体状の前記表題化合物(180mg)を得た。
【0118】
m.p.=144−145℃
1H−NMR(CDCl3):δ1.44(s、12H)、2.70(brs、1H)、2.74(t、J=6Hz、2H)、3.54−3.59(m、4H)、3.64(s、4H)、3.74(t、J=6.2Hz、1H)、3.87(t、J=6.2Hz、2H)、4.0(t、J=6.3Hz、1H)、7.19−7.39(m、10H)
MS(FAB)m/z484(M++1)、506(M++Na)。
【0119】
[製造例33]:4−O−(2−N,N−ジベンジルアミノエチル)−1−O−(メチルオキシカルボニルメチル)−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトール(化学式(XIII)の化合物)の製造
製造例32で得た4−O−(2−N,N−ジベンジルアミノエチル)−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトール(125mg、0.258mmol)をN,N−ジメチルホルムアミド(10ml)に溶解してから0℃でNaH(20mg、0.51mmol)を滴加した。反応液を0℃で20分間攪拌させた後、エチルブロモアセテート(ethyl bromoacetate)(0.05ml、0.51mmol)を滴加して常温で10時間攪拌させた。反応生成物を酢酸エチルで抽出しNaCl飽和水溶液及び蒸留水で洗滌した後MgSO4で乾燥させ、これを減圧蒸発させカラムクロマトグラフィー(酢酸エチル:ヘキサン=3:7)で精製して前記表題化合物(110mg)を得た。
【0120】
m.p.=130−132℃
1H−NMR(CDCl3):δ1.41(s、12H)、2.72(t、J=6Hz、2H)、3.58(t、J=6.3Hz、2H)、3.63−3.89(m、13H)、4.36(s、2H)、7.12−7.40(m、10H)
MS(FAB)m/z556(M++1)、579(M++Na)。
【0121】
[製造例34]:4−O−(2−アミノエチル)−1−O−(メチルオキシカルボニルメチル)−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトールの製造
製造例33で得た4−O−(2−N,N−ジベンジルアミノエチル)−1−O−(メチルオキシカルボニルメチル)−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトール(20mg、0.035mmol)をCH2Cl2−メタノール混合溶媒(1:2、12ml)に溶解し、これに水酸化パラジウム(palladium hydroxide)(20% on carbon、9.7mg)を加え、40psiの水素大気下で1時間攪拌した。反応生成物をセライトを通じて濾し、濾過液を減圧蒸発・濃縮した後、メタノール溶媒下で再結晶して白色固体状の前記表題化合物(14mg)を得た。
【0122】
m.p.=193−195℃
1H−NMR(CD3OD):δ1.41(s、12H)、3.10(t、J=5.9Hz、2H)、3.27(dd、J=6.5、1.8Hz、2H)、3.69−3.90(m、11H)、4.28(s、2H)
MS (FAB) m/z 376 (M++1)。
【0123】
[製造例35]:4−O−(2−N,N−ジベンジルアミノエチル)−1−O−(カルボキシメチル)−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトールの製造
製造例33で得た4−O−(2−N,N−ジベンジルアミノエチル)−1−O−(メチルオキシカルボニルメチル)−2,3:5,6−ジ−O−イソプロピリデン−scyllo−イノシトール(20mg、0.035mmol)をメタノール(3ml)に溶解しNaOHペレット(4.3mg、0.107mmol)を加えて常温で10時間攪拌した。反応生成物を減圧蒸発させ濃縮した後蒸留水(5ml)を加え、得られた水層をジエチルエーテル(2mlずつ2回)で洗滌した。これに5%AcOH水溶液を加えてpHが6.5になるようにした後、CH2Cl2(4x4ml)で抽出し有機層を集め乾燥(Na2SO4)・減圧蒸発させて白色固体状の前記表題化合物(13mg)を得た。
【0124】
1H−NMR(CDCl3):δ1.41&1.44(2s、12H)、2.77(t、J=5.8Hz、2H)、3.35−3.91(m、12H)、4.37(s、2H)、7.20−7.40(m、10H)。
【0125】
[実施例1]:化学式(II)(x=O−CO−O、n=5の場合)の化合物の製造
1−1)myo−イノシトール二量体化合物の製造
製造例21で得た化合物(1.62g、3.4mmol)をトルエン(21ml)に溶解してから製造例5で得た化合物(1.8g、5mmol)及び1,8−ジアゾビシクロ[5.4.0]ウンデカ−7−エン(1,8−diazobicyclo[5.4.0]undec−7−ene)(0.05ml、0.34mmol)を加え、常温で18時間攪拌させた。この反応生成物をCH2Cl2で抽出し、MgSO4で乾燥してから減圧蒸発させて濃縮させ、ヘキサン/酢酸エチル(5:1)を用いて再結晶して白色固体状のカップリングされた二量体形の化合物(2.23g)を得た。
【0126】
m.p.=225−226℃
1H−NMR(CDCl3):1.10−1.72(m、24H)、3.35&3.36(dd、J=9.3Hz、11.0&9.5Hz、11.1Hz、2H)、3.49(t、J=9.3&10.8Hz、2H)、3.73(dd&t、J=2.6Hz、5.8&1.4Hz、2H)、4.00−4.15(m、6H)、4.12(dd、J=5.9Hz、9.1&2.6Hz、5.9Hz、4H)、4.69(t、J=2.5&4.4Hz、4H)、4.82(s、8H)、4.95&4.96(dd、J=4.1Hz、10.5&4.1Hz、10.5Hz、2H)、5.04&5.05(dd、J=6.9Hz、11.1&7.0Hz、11.0Hz、2H)、6.89(d、J=8.7Hz、4H)、7.24−7.42(m、14H)
IR(KBr):1748cm−1(CO)
MS(FAB)m/z779.82(M++Na)。
【0127】
1−2)myo−イノシトール二量体のアセトニド保護基除去
段階1−1)で得たmyo−イノシトール二量体化合物(2.21g)をCH2Cl2(25ml)に溶解し、これにCH3OH(24.3ml)及びp−トルエンスルホン酸(286.1mg、1.46mmol)を加え、常温で14時間攪拌させた。この反応生成物を濾過して白色固体状のアセトニド保護基が除去されたmyo−イノシトール二量体化合物(1.45g)を得た。
【0128】
m.p.=169℃
1H−NMR(DMSO):3.11−3.24(m、4H)、3.35−3.48(m、5H)、3.49(dd、J=9.6Hz、21.2Hz、4H)、3.74(s、3H)、3.90−3.97(m、3H)、4.25−4.33(m、2H)、4.53(dd、J=1.8Hz、24.9Hz、4H)、4.70−4.74(m、12H)、4.77−4.81(m、2H)、6.89(d、J=8.6Hz、2H)、7.21−7.35(m、5H)、7.43(d、J=7.1Hz、2H)
MS(FAB)m/z619(M++Na)。
【0129】
1−3)アシル化反応を通したアミノ酸の導入
段階1−2)で得た化合物(200mg、0.34mmol)、製造例8で得た化合物(1.55g、6.7mmol)及びN,N−ジメチルアミノピリジン(62.3mg、0.51mmol)をN,N−ジメチルホルムアミド(4ml)に溶解し、これに1−[3−(ジメチルアミノ)プロピル]−エチルカルボジイミド塩酸(1−[3−(dimethyl amino)propyl]−ethylcarboiimide hydrochloride)(1.3g、6.7mmol)を加え常温で1日間攪拌した。この反応生成物をCH2Cl2で抽出した後、NaHCO3 飽和水溶液及び蒸留水で数回洗滌した。得られた有機層を分離してNa2SO4で乾燥し、濃縮して、骨格構造に8個のアミノ酸が導入された茶色固体状の目的化合物(776.6mg)を得た。
【0130】
1H−NMR(CDCl3):1.13−1.32(m、16H)、1.36(s、72H)、1.45−1.57(m、32H)、2.06−2.15(m、16H)、2.95−3.02(m、16H)、3.7(s、3H)、3.85−3.91(m、1H)、4.22(dd、J=1.9Hz、9.3Hz、1H)、4.48(d、J=11.8Hz、2H)、4.55(d、J=6.6Hz、2H)、4.60−5.68(m、10H)、6.76(d、J=8.7Hz、2H)、7.05(d、J=8.4Hz、2H)、7.10−7.26(m、5H)
MS(FAB)m/z2324.6(M++Na)。
【0131】
1−4)p−メトキシベンジル保護基の除去
段階1−3)で得た化合物(705.1mg、0.31mmol)をCH2Cl2/H2O(18:1)(5ml)に溶解し、0℃で2,3−ジクロロ−5,6−ジシアノ−1,4−ベンゾキノン(2,3−dichloro−5,6−dicyano−1,4−benzoquinone)(139.0mg、0.61mmol)を添加して常温で2日間攪拌させた。これにNaHCO3飽和水溶液を加えて反応を中断させ、蒸留水で洗滌した後、有機層をNa2SO4で乾燥し、濃縮してp−メトキシベンジル保護基が除去された茶色固体状の目的化合物(373.9mg)を得た。
【0132】
1H−NMR(CDCl3):1.17−1.26(m、16H)、1.37(s、72H)、1.41−1.53(m、32H)、2.94−3.05(m、16H)、2.07−2.41(m、16H)、3.86−3.92(m、2H)、4.31−5.56(m、24H)、7.11−7.27(m、5H)
MS(FAB)m/z2204.8(M++Na)。
【0133】
1−5)蛍光物質の導入
段階1−4)で得た化合物(284.1mg、0.13mmol)及びN,N−ジメチルアミノピリジン(47.6mg、0.38mmol)をアセトニトリル(4ml)に溶解し、これに5−ジメチルアミノ−1−ナフタレンスルホニルクロライド(5−dimethylamino−1−naphthalenesulfonyl chloride;70.2mg、0.26mmol)を加えて常温で2日間攪拌した。これにNH4Cl飽和水溶液(8.3ml)を加えて中和させてから酢酸エチルで抽出し、NaCl飽和水溶液で洗滌した。得られた有機層をNa2SO4で乾燥させた後、減圧蒸発で濃縮し、残留物をカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:1)で精製して蛍光物質が導入された黄色固体状の目的化合物(261.7mg)を得た。
【0134】
1H−NMR(CDCl3):1.21(s、16H)、1.38(s、72H)、1.53−1.59(m、32H)、1.99−2.33(m、16H)、2.83(s、6H)、2.94−3.04(m、16H)、3.89(t、J=9.8Hz、1H)、4.56(dd、J=12.0Hz、18.6Hz、2H)、4.69−5.54(m、11H)、7.11−7.29(m、6H)、7.50(dd、J=8.4Hz、16.5Hz、2H)、8.03(d、J=7.5Hz、1H)、8.18(d、J=7.2Hz、1H)、8.61(brs、1H)
MS(FAB)m/z2438.1(M++Na)。
【0135】
1−6)アミノ酸末端のt−Boc保護基の除去
段階1−5)で得た化合物(202.3mg、0.08mmol)をHClガスで飽和させた酢酸エチル(8ml)に溶解して常温で45分間攪拌した。この反応生成物を減圧蒸発させてアミノ酸末端のt−Boc保護基が除去された黄色固体状の目的化合物(180.4mg)を得た。
【0136】
1H−NMR(CD3OD):1.37(m、16H)、1.69(m、32H)、2.23(m、16H)、2.94(m、16H)、3.28−3.30(m、1H)、3.44(s、6H)、4.71(m、2H)、4.95−5.62(m、11H)、7.21−7.30(m、5H)、7.87−7.98(m、2H)、8.14(brs、1H)、8.53(dd、J=8.4Hz、15.0Hz、2H)、8.95(t、J=3.9Hz、1H)
MS(MALDI−TOF)m/z1636.2(M++Na)。
【0137】
1−7)グアニジニウム基の導入
段階1−6)で得た化合物(141.6mg、0.07mmol)をN,N−ジメチルホルムアミド(2.8ml)に溶解してからこれにトリエチルアミン(0.4ml、3mmol)及び製造例11で得たN,N−ジ−Boc−N’−トリフロロメタンスルホニルグアニジン(1.2g、3mmol)を加えて常温で2日間攪拌した。これに過量の酢酸エチルを加えて2N NaHSO4、飽和NaHCO3及び飽和NaCl水溶液で洗滌した後、有機層をNa2SO4で乾燥してから減圧蒸発で濃縮し、残留物をカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:2)で精製してグアニジニウム基が8つ導入された黄色固体状の目的化合物(93.1mg)を得た。
【0138】
1H−NMR(CDCl3):1.13−1.65(m、192H)、1.60−2.31(m、16H)、2.82(s、6H)、3.24−3.36(m、16H)、3.88(t、J=9.9Hz、1H)、4.56−5.51(m、13H)、7.11−7.29(m、6H)、7.51(dd、J=8.4Hz、16.2Hz、2H)、7.99(d、J=8.7Hz、1H)、8.17(dd、J=0.9Hz、7.5Hz、1H)、8.22−8.28(m、8H)、11.46(s、8H)。
【0139】
1−8)グアニジニウム基末端のt−Boc保護基の除去
段階1−7)で得た化合物(38mg、0.01mmol)をトリフロロ酢酸/CH2Cl2(1:1)(2ml)に溶解して常温で5時間攪拌した。得られた反応生成物を減圧蒸発によって濃縮し、残留物に蒸留水及び酢酸エチルを加えた後、水層を分離して減圧蒸発することによってグアニジニウム基末端のt−Boc保護基が除去された黄色固体状の目的化合物(27mg)を得た。
【0140】
1H−NMR(CD3OD):1.13−1.35(m、16H)、1.42−1.61(m、32H)、2.13−2.43(m、16H)、2.88(s、6H)、3.02−3.16(m、16H)、4.01(t、J=9.6H,1H)、4.64(dd、J=11.9Hz、18.7Hz、2H)、5.01−5.61(m、11H)、7.16−7.28(m、6H)、7.52−7.66(m、2H)、8.00(d、J=8.6Hz、1H)、8.27(d、J=7.3Hz、1H)、8.64(d、J=8.5Hz、1H)
MS(MALDI−TOF)m/z1973.1(M++Na)。
【0141】
1−9)ベンジル(Bn)保護基の除去
段階1−8)で得た化合物(10mg、3.5μmol)をメタノール(1ml)に溶解し、これに水酸化パラジウム(palladiumhydroxide)(20% on carbon、20mg)を加えて水素1気圧下で4時間攪拌した。得られた反応生成物をセライト(celite)に通して濾過し、濾液を減圧蒸発してベンジル保護基が取り除かれた黄色固体状の目的化合物(7mg)を得た。
【0142】
1H−NMR(CD3OD):1.1−1.6(m、48H)、2.1−2.5(m、16H)、2.90(s、6H)、3.0−3.26(m、16H)、3.97(t、J=9.9Hz、1H)、5.0−5.7(m、11H)、7.29(d、J=7.7Hz、1H)、7.57(t、J=8.1Hz、1H)、7.67(t、J=8.1Hz、1H),8.00(d、J=8.5Hz、1H)、8.30(d、J=7.5Hz、1H)、8.64(d、J=8.7Hz、1H)。
【0143】
[実施例2]:化学式(II)(x=O−CO−O、n=3及びn=7の場合)の化合物の製造
前記実施例1の段階1−3)で用いた6−Boc−アミノヘキサン酸の代わりに製造例7及び製造例9で得た4−Boc−アミノブタン酸及び8−Boc−アミノオクタン酸を用いてそれぞれn=3及びn=7である化学式(II)の化合物を製造した。
【0144】
[実施例3]:化学式(III)(x=O−CO−Oの場合)の化合物の製造
3−1)製造例25で得た化合物(42mg、0.12mmol)をテトラヒドロフラン(5ml)に溶解し、これに0℃でNaH(6mg、0.25mmol)を加えて常温で25分間攪拌した。この反応生成物の温度を0℃に下げた後、製造例26で得た化合物(48mg、0.10mmol)を加えて常温で2時間攪拌した。これを酢酸エチル(30ml)で希釈した後、NaHCO3飽和水溶液(30ml)と食塩水(brine)で洗滌し、得られた有機層を乾燥(Na2SO4)し、残留物をカラムクロマトグラフィーで精製し再結晶してカップリングされた二量体形態の化合物(29mg)を得た。
【0145】
m.p.=168−170℃
1H−NMR(CDCl3):δ1.45、1.46(2s、24H,4CMe2)、3.68(appdt、J=9.5、2.1Hz、4H,H−1、H−1’、H−3、H3’)、3.82(s、3H,OCH3)、3.82(t、J=9.1、1H,H−5’)、4.10(t、J=9.3Hz、2H,H−4’&H−6’)、4.23(t、J=9.6Hz、2H,H−4&H−6)、4.39(t、J=1.8Hz、1H)、4.79、4.85(2s、4H,2PhCH2)、5.17(t、J=9.6Hz、H−5)、5.58(t、J=2.0Hz、H−2’)、6.89(dd、J=6.7、1.9Hz、2H,Ph)、7.28−7.40(m、8H,Ph)
MS(FAB)m/z=779(M++Na)。
【0146】
3−2)前記カップリングされた二量体形態の化合物を用いて実施例1と同じ方法でアシル化反応を行ってアミノ酸を導入した後、アミノ酸が導入された化合物のアミノ酸N−末端にグアニジニウム基が導入された化学式(III)の化合物を製造した。
【0147】
[実施例4]:化学式(IV)(x=O−CO−Oの場合)の化合物の製造
4−1)製造例29で得た化合物(43mg、0.11mmol)をテトラヒドロフラン(2ml)に溶解し、これに0℃でNaH(1.2mg、0.05mmol)を加えて常温で25分間攪拌した。この反応生成物を製造例30で得た化合物(50mg、0.11mmol)のテトラヒドロフラン(3ml)溶液に0℃で加えて0〜5℃で1時間30分間攪拌し、これをCH2Cl2 (25ml)で希釈した後、NH4CO3飽和水溶液(30ml)で洗滌した。得られた有機層を乾燥(MgSO4)・濃縮し、カラムクロマトグラフィー(酢酸エチル:ヘキサン=1:3)で精製してカップリングされた二量体形態の化合物(41mg)を得た。
【0148】
m.p.=284−286℃
1H−NMR(CDCl3):δ1.43、1.47(2s、24H,4CMe2)、3.66−3.88(m、13H)、4.47(s、2H)、4.83(s、2H)、5.12(t、J=9.3Hz、2H,H−6、H−6’)、6.88(d、J=8.7Hz、2H)、7.27−7.42(m、7H)
MS(FAB)m/z=757(M++1)、780(M++Na)。
【0149】
4−2)前記カップリングされた二量体形態の化合物を用いて実施例1と同じ方法でアシル化反応を行いアミノ酸を導入した後、アミノ酸が導入された化合物のアミノ酸N−末端にグアニジニウム基が導入された化学式(IV)(x=O−CO−Oの場合)の化合物を製造した。
【0150】
[実施例5]:化学式(IV)(x=O−CH2−CO−NH−(CH2)2−Oの場合)の化合物の製造
5−1)製造例34で得た化合物(8.3mg、0.022mmol)と製造例35で得た化合物(12mg、0.022mmol)をN,N−ジメチルホルムアミド(2.5ml)に溶解し、これに常温でトリエチルアミン(3μl、0.035mmol)、1−ヒドロキシベンゾトリアゾール水和物(1−hydroxybenzotriazole hydrate、HOBT);(3.2mg、0.024mmol)及び1−[3−(ジメチルアミノ)プロピル]−エチルカルボイミド塩酸(1−[3−(dimethylamino)propyl]−ethylcarbodiimide hydrochloride);(4.6mg、0.024mmol)を順に加えて22時間攪拌した。得られた反応生成物を酢酸エチル(15ml)で希釈した後、有機層をNH4Cl水溶液(5ml)、NaHCO3飽和水溶液(10ml×2)、蒸留水(5ml×3)で洗滌し、乾燥(Na2SO4)してから減圧蒸発させ、残留物をカラムクロマトグラフィー(酢酸エチル:n−ヘキサン=1:1)で精製して白色固体状のカップリングされた二量体化合物(14mg)を得た。
【0151】
1H−NMR(CDCl3):δ1.40、1.43(2s、12HeacH,4CMe2)、2.72(t、J=6.1Hz、2H)、3.35−3.88(m、25H)、4.23(s、2H),4.36(s、2H),7.02(t、J=6Hz、1H),7.19−7.39(m、10H)。
【0152】
5−2)前記カップリングされた二量体形態の化合物を用いて実施例1と同様な方法でアシル化反応を行いアミノ酸を導入した後、アミノ酸が導入された化合物のアミノ酸N−末端にグアニジニウム基が導入された化学式(IV)(x=O−CH2−CO−NH−(CH2)2−Oの場合)の化合物を製造した。
【0153】
試験例1:細胞膜透過効率測定
前記実施例で製造された、ダンシル(dansyl)蛍光物質が付着された化合物の細胞膜透過性を次のように測定し、既存の生体膜透過性が優れていると知られたアルギニン9量体(dansyl−Arg9,d−Arg9)、及び対照群としての実施例1の段階1−6)で製造されたグアニジニウム基を有しない中間体と比較した。
【0154】
先ず、C12ウェルプレート(well plate)をカバーガラスを覆い、該プレート上でCOS7細胞(monkey kidney epithelial cell)を培養した。培地は10%FBSが含まれたDMEM(Dulbecco’s modified Eagle’s medium)を用いて24時間安定化させた後、該細胞を飢餓(starvation)させるため無血清培地(serum−free medium)で再び24時間培養した。その後、これらの細胞をダンシル−Arg9、実施例1の段階1−6)で製造された中間体、又は実施例1又は2で得た化合物で5分間処理(〜7μM)した後、カバーガラスを取り出してPBSで洗滌し、スライドにマウンティング(mounting)し、共焦点顕微鏡(confocal microscope)で細胞断面を観察した。その際、蛍光物質を観察するため、Arレーザー(波長455nm)を装備した顕微鏡を用いて対物レンズ40x・対眼レンズ10x、総400倍拡大して観察した。その結果を図1に示す。
【0155】
図1の(1)はダンシル−Arg9で処理したCOS7細胞の蛍光イメージ;(2)はグアニジニウム基を有しない中間体化合物(実施例1の段階1−6で得た中間体)で処理したCOS7細胞の蛍光イメージ;(3)、(4)及び(5)は本発明による化学式(II)の化合物で処理したCOS7細胞の蛍光イメージである。ここで緑色部分は蛍光であり、蛍光が多く見えるほど細胞内に該化合物が多く透過されていることを示す。
【0156】
図1に示したように、本発明の化合物((3)〜(5))はダンシル−Arg9((1))より高い細胞膜透過性を示し、グアニジニウム基を有しない中間体化合物((2))は細胞内に透過されないことを確認した。また、本発明の化合物のうち、n=5の鎖長を有する化合物((4))が最も高い細胞膜透過率を有することを確認した。
【0157】
試験例2:核膜透過性測定
前記実施例で製造されたダンシル(dansyl)蛍光物質が付着された化合物の核膜透過性を次のように測定し、既存の生体膜透過性が優れていると知られたアルギニン9量体(dansyl−Arg9、d−Arg9)及び対照群としての実施例1の段階1−6)で製造されたグアニジニウム基を有しない中間体と比較した。
【0158】
C12ウェルプレートをカバーガラスで覆い、該プレート上でマウスマクロファージRAW264.7細胞を培養した。培地は10%FBSが含まれているDMEMを用いて24時間安定化させた後、該細胞を飢餓させるため無血清培地で再び24時間培養した。該細胞を室温でリボヌクレアーゼ(RNase)(100μg/ml)で処理してから、常温(23〜25℃)で5分間ヨウ化プロピジウム(2μg/ml)で処理して核を染色した後、PBSで3回洗滌した。その後、該細胞をダンシル−Arg9、実施例1の段階1−6)で製造された中間体、又は実施例1又は2で得た化合物で5分間処理(〜7μM)した後、カバーガラスを取り出して洗滌し、これをスライドにマウンティングして共焦点顕微鏡で細胞断面を観察した。その際、蛍光物質を観察するためArレーザー(波長455nm)を装備した顕微鏡を用いて対物レンズ40x・対眼レンズ10x、総400倍拡大して観察した。その結果を図2に示す。
【0159】
図2の(1)はダンシル−Arg9で処理したRAW264.7細胞の蛍光イメージ;(2)はグアニジニウム基を有しない中間体化合物(実施例1の段階1−6)で得た化合物)で処理したRAW264.7細胞の蛍光イメージ;(3)〜(5)はそれぞれn=3、5及び7である本発明による化学式(II)の化合物で処理したRAW264.7細胞の蛍光イメージである。図2の左カラムのイメージの緑色はそれぞれの輸送体化合物が細胞内に透過されたことを示し、中央のカラムのイメージの赤色はヨウ化プロピジウムで核が染色されたことを示し、右カラムのイメージにおいては赤色と緑色が重なり黄色に近くなるほど化合物が核膜内に多く透過されていることを示す。
【0160】
図2に示したように、本発明による化合物((3)〜(5))は既存の分子輸送体であるダンシル−Arg9((1))より核膜透過性が高いことを確認した。また、本発明の化合物のうち、鎖長がn=3((3))又はn=7((5))である場合が核膜透過性が特に優れていることが分かる。
【0161】
また、蛍光物質が付着されたmyo−イノシトール燐酸[Prestwich,G. D.,Acc. Chem. Res.,29,503 (1996); Chung S. K. et al. J. Org. Chem.,67,5256(2002)]を蛍光表示されていない本発明の化学式(II)〜(IV)の化合物と混合して製造されたイオン結合体の組成物も優れた生体膜透過性を現すことを確認した。
【0162】
本発明に関する多くの実施形態を記述したが、本発明の範囲を外れない様々な変形が可能であることを理解できるであろう。したがって、他の実施形態も後述する請求項の範囲に含まれる。
【図面の簡単な説明】
【0163】
【図1】ダンシル(dansyl)−Arg9((1))、実施例1の段階1−6)で製造されたグアニジニウム基を有しない中間体((2))及び本発明の実施例1〜2に基づいて製造されたイノシトール誘導体ら((3)〜(5))の細胞膜透過程度を比較した結果を示す図である。
【図2】ダンシル(dansyl)−Arg9((1))、実施例1の段階1−6)で製造されたグアニジニウム基を有しない中間体((2)) 及び本発明の実施例1〜2に基づいて製造されたイノシトール誘導体ら((3)〜(5))の核膜透過程度を比較した結果を示す図である。
【特許請求の範囲】
【請求項1】
下記化学式(I)のイノシトール誘導体:
【化1】
前記式で、
R1 は
【化2】
(nは1〜12範囲の整数)であり;
R2及びR3はそれぞれ独立的にH、アルキル、アリールアルキル、シクロアルキル、ヘテロアルキル、−(CH2)mNHR’、−(CH2)lCO2R’’、−COR’’’又は−SO2R’’’’であり(ここで、R’、R’’、R’’’及びR’’’’はそれぞれアルキル、mは2〜5範囲の整数、lは1〜5範囲の整数);
Pは0〜2範囲の整数であり;
X及びX’はそれぞれ独立的に−O−CO−O−、−O−CO−NH−(CH2)m−O−、−O−CO−(CH2)l−O−又は−O−(CH2)l−CO−NH−(CH2)m−O−であり、ここで、mは2〜5範囲の整数、lは1〜5範囲の整数である。
【請求項2】
下記化学式(XV)で表現される請求項1に記載のイノシトール誘導体:
【化3】
前記式で、
R1、R2、R3、X、X’及びpは請求項1で定義された通りである。
【請求項3】
前記pが0又は1であることを特徴とする請求項1に記載のイノシトール誘導体。
【請求項4】
前記nが3〜8範囲の整数であることを特徴とする請求項1に記載のイノシトール誘導体。
【請求項5】
下記化学式(II)、(III)又は(IV)に表現される請求項1に記載のイノシトール誘導体:
【化4】
前記式で、
R1、R2、R3 及びXは請求項1で定義された通りである。
【請求項6】
(a)myo−又はscyllo−イノシトールの水酸基を保護する反応を行って中間体を得る段階;
(b)段階(a)で得た中間体を2つ以上カップリングしてイノシトール重合体を得る段階;
(c)アシル化反応を行って段階(b)で得たイノシトール重合体に一つ以上のアミノ酸を導入する段階;及び
(d)段階(c)で得たイノシトール重合体のN−末端にグアニジニウム基を導入する段階を含む、下記化学式(I)のイノシトール誘導体を製造する方法:
【化5】
前記式で、
R1、R2、R3、X、X’及びpは請求項1で定義された通りである。
【請求項7】
段階(a)で得た中間体が下記化学式(V)〜(XIII)によって表される化合物から選択されることを特徴とする請求項6に記載の製造方法。
【化6】
前記式で、R’、R’’、l及びmは請求項1で定義された通りであり、Bnはベンジル、PMBはp−メトキシベンジルである。
【請求項8】
下記化学式(I)のイノシトール誘導体を含む、生体膜を通過して細胞又は核内に薬物又は診断試薬を輸送するため組成物:
【化7】
前記式で、
R1、R2、R3、X、X’及びpは請求項1で定義された通りである。
【請求項9】
前記薬物又は診断試薬が100〜1500g/mol範囲の分子量を有する有機化合物であることを特徴とする請求項8に記載の組成物。
【請求項10】
前記薬物又は診断試薬がペプチド及び核酸から選択された重合体化合物であることを特徴とする請求項8に記載の組成物。
【請求項11】
化学式(I)のイノシトール誘導体が前記薬物又は診断試薬と共有結合を通じて結合体(conjugate)を形成することを特徴とする請求項8に記載の組成物。
【請求項12】
化学式(I)のイノシトール誘導体が前記薬物又は診断試薬とイオン結合を通じてイオン結合体(ion complex)を形成することを特徴とする請求項8に記載の組成物。
【請求項13】
下記化学式(I)のイノシトール誘導体を分子輸送体に用いて薬物又は診断試薬を生体膜に透過させ細胞又は核内に伝達する方法:
【化8】
前記式で、
R1、R2、R3、X、X’及びpは請求項1で定義された通りである。
【請求項1】
下記化学式(I)のイノシトール誘導体:
【化1】
前記式で、
R1 は
【化2】
(nは1〜12範囲の整数)であり;
R2及びR3はそれぞれ独立的にH、アルキル、アリールアルキル、シクロアルキル、ヘテロアルキル、−(CH2)mNHR’、−(CH2)lCO2R’’、−COR’’’又は−SO2R’’’’であり(ここで、R’、R’’、R’’’及びR’’’’はそれぞれアルキル、mは2〜5範囲の整数、lは1〜5範囲の整数);
Pは0〜2範囲の整数であり;
X及びX’はそれぞれ独立的に−O−CO−O−、−O−CO−NH−(CH2)m−O−、−O−CO−(CH2)l−O−又は−O−(CH2)l−CO−NH−(CH2)m−O−であり、ここで、mは2〜5範囲の整数、lは1〜5範囲の整数である。
【請求項2】
下記化学式(XV)で表現される請求項1に記載のイノシトール誘導体:
【化3】
前記式で、
R1、R2、R3、X、X’及びpは請求項1で定義された通りである。
【請求項3】
前記pが0又は1であることを特徴とする請求項1に記載のイノシトール誘導体。
【請求項4】
前記nが3〜8範囲の整数であることを特徴とする請求項1に記載のイノシトール誘導体。
【請求項5】
下記化学式(II)、(III)又は(IV)に表現される請求項1に記載のイノシトール誘導体:
【化4】
前記式で、
R1、R2、R3 及びXは請求項1で定義された通りである。
【請求項6】
(a)myo−又はscyllo−イノシトールの水酸基を保護する反応を行って中間体を得る段階;
(b)段階(a)で得た中間体を2つ以上カップリングしてイノシトール重合体を得る段階;
(c)アシル化反応を行って段階(b)で得たイノシトール重合体に一つ以上のアミノ酸を導入する段階;及び
(d)段階(c)で得たイノシトール重合体のN−末端にグアニジニウム基を導入する段階を含む、下記化学式(I)のイノシトール誘導体を製造する方法:
【化5】
前記式で、
R1、R2、R3、X、X’及びpは請求項1で定義された通りである。
【請求項7】
段階(a)で得た中間体が下記化学式(V)〜(XIII)によって表される化合物から選択されることを特徴とする請求項6に記載の製造方法。
【化6】
前記式で、R’、R’’、l及びmは請求項1で定義された通りであり、Bnはベンジル、PMBはp−メトキシベンジルである。
【請求項8】
下記化学式(I)のイノシトール誘導体を含む、生体膜を通過して細胞又は核内に薬物又は診断試薬を輸送するため組成物:
【化7】
前記式で、
R1、R2、R3、X、X’及びpは請求項1で定義された通りである。
【請求項9】
前記薬物又は診断試薬が100〜1500g/mol範囲の分子量を有する有機化合物であることを特徴とする請求項8に記載の組成物。
【請求項10】
前記薬物又は診断試薬がペプチド及び核酸から選択された重合体化合物であることを特徴とする請求項8に記載の組成物。
【請求項11】
化学式(I)のイノシトール誘導体が前記薬物又は診断試薬と共有結合を通じて結合体(conjugate)を形成することを特徴とする請求項8に記載の組成物。
【請求項12】
化学式(I)のイノシトール誘導体が前記薬物又は診断試薬とイオン結合を通じてイオン結合体(ion complex)を形成することを特徴とする請求項8に記載の組成物。
【請求項13】
下記化学式(I)のイノシトール誘導体を分子輸送体に用いて薬物又は診断試薬を生体膜に透過させ細胞又は核内に伝達する方法:
【化8】
前記式で、
R1、R2、R3、X、X’及びpは請求項1で定義された通りである。
【図1】


【図2】
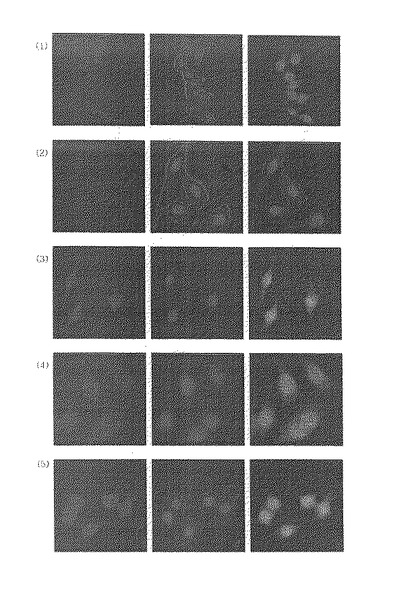
【図2】
【公表番号】特表2007−523889(P2007−523889A)
【公表日】平成19年8月23日(2007.8.23)
【国際特許分類】
【出願番号】特願2006−546800(P2006−546800)
【出願日】平成16年8月6日(2004.8.6)
【国際出願番号】PCT/KR2004/001982
【国際公開番号】WO2005/085159
【国際公開日】平成17年9月15日(2005.9.15)
【出願人】(500345478)ポステック・ファウンデーション (25)
【Fターム(参考)】
【公表日】平成19年8月23日(2007.8.23)
【国際特許分類】
【出願日】平成16年8月6日(2004.8.6)
【国際出願番号】PCT/KR2004/001982
【国際公開番号】WO2005/085159
【国際公開日】平成17年9月15日(2005.9.15)
【出願人】(500345478)ポステック・ファウンデーション (25)
【Fターム(参考)】
[ Back to top ]