
イミド変性エラストマーの製造方法
【課題】 着色が低減され、耐溶剤性が向上したイミド変性エラストマーを得ることができ、製造時の生産効率が高く、環境に配慮し、反応工程が短く、コストが低減された、イミド変性エラストマーの製造方法を提供することにある。
【解決手段】 ウレタンプレポリマーと、ジアミン化合物と、テトラカルボン酸二無水物とを、溶媒非存在下で加熱下に混合して重合させる第一の工程、及び
ジアミン化合物及びテトラカルボン酸二無水物の固体を粉砕して、平均粒子径が10μm以下の微粒子として、前記微粒子を含む混合物を生成する第二の工程、
を含むポリウレタンアミック酸合成工程、並びに
前記ポリウレタンアミック酸合成工程で得られたポリウレタンアミック酸を加熱処理することにより閉環反応を起こさせるポリウレタンアミック酸閉環工程を含み、
イミド変性エラストマーを得ることを特徴とするイミド変性エラストマーの製造方法、を提供する。
【解決手段】 ウレタンプレポリマーと、ジアミン化合物と、テトラカルボン酸二無水物とを、溶媒非存在下で加熱下に混合して重合させる第一の工程、及び
ジアミン化合物及びテトラカルボン酸二無水物の固体を粉砕して、平均粒子径が10μm以下の微粒子として、前記微粒子を含む混合物を生成する第二の工程、
を含むポリウレタンアミック酸合成工程、並びに
前記ポリウレタンアミック酸合成工程で得られたポリウレタンアミック酸を加熱処理することにより閉環反応を起こさせるポリウレタンアミック酸閉環工程を含み、
イミド変性エラストマーを得ることを特徴とするイミド変性エラストマーの製造方法、を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、イミド変性エラストマーの製造方法に関する。
【背景技術】
【0002】
柔軟な低い弾性率を有するゴム状物で、かつ高強度及び高耐熱性を有する新規なイミド変性エラストマーの製造方法として、ジイソシアネートとポリオールから得た分子両末端にイソシアネート基を有するウレタンプレポリマーを、ジアミン化合物でウレア結合により鎖延長し、さらにテトラカルボン酸二無水物との反応によりポリウレタンアミック酸(PUA)溶液を得て、これを遠心成形後、加熱処理により閉環反応(脱水縮合反応)させることにより、ウレア結合部にイミドユニットを導入したブロック共重合体であるイミド変性エラストマーを合成する製造方法が知られている(特許文献1)。
【0003】
【特許文献1】特開2008−101195号公報
【発明の概要】
【発明が解決しようとする課題】
【0004】
しかしながら、上記製造方法では、PUA溶液を得るために、あらかじめ重合したウレタンプレポリマーをN−メチル−2−ピロリドン(NMP)等の有機溶媒に溶解させる必要がある。この場合、ポリマー量の3〜5倍量の溶媒を使用するため、製造時の生産効率が低く、コストが増大し、環境への影響が懸念されるという問題があった。さらに、反応後に溶媒を除去する必要があるため、反応工程が長く、この点でもコストが増大するという問題があった。
【0005】
また、上記製造方法によって得られるイミド変性エラストマーは、着色の程度が強く、カラーリングをする用途には不向きであるという問題があった。さらに、上記製造方法によって得られるイミド変性エラストマーは、耐溶剤性についても充分ではないという問題もあった。
【0006】
上記製造方法では、PUA溶液を得て、これを遠心成形後、加熱処理するのに対して、溶媒を用いずに塊状重合させた場合には、反応が進まないため、得られるPUAは粘調体となる。そのため、PUAを長時間の熱処理(100℃程度で約12時間)によって粘度を上げて粘土状にしてから成形する必要があった。また、得られるイミド変性エラストマー中に溶媒に不溶な凝集物が残存して製品の品質を損ねるという問題があった。このように、単に溶媒を用いずに塊状重合させれば良いというものでもないのである。
【0007】
従って、本発明の目的は、着色が低減され、耐溶剤性が向上したイミド変性エラストマーを得ることができ、製造時の生産効率が高く、環境に配慮し、反応工程が短く、コストが低減された、イミド変性エラストマーの製造方法を提供することにある。
【課題を解決するための手段】
【0008】
そこで、本発明者らが、上記課題を解決するために鋭意検討した結果、テトラカルボン酸二無水物を添加する際に、テトラカルボン酸二無水物を含む微粒子の平均粒子径を10μm以下とすることにより、溶媒を用いずに塊状重合させても凝集物が生じず、しかも、得られるPUAの粘度が高く粘土状であるため、長時間の熱処理をせずに成形できることを見いだし、本発明を完成させた。
【0009】
すなわち、本発明は、
下記式(c)で表されるウレタンプレポリマー(c)と、下記式(d)で表されるジアミン化合物(d)と、下記式(f)で表されるテトラカルボン酸二無水物(f)とを、溶媒非存在下で加熱下に混合して重合させる第一の工程、及び
ジアミン化合物(d)及びテトラカルボン酸二無水物(f)の固体を粉砕して、平均粒子径が10μm以下の微粒子として、前記微粒子を含む混合物を生成する第二の工程、
を含むポリウレタンアミック酸合成工程、並びに
前記ポリウレタンアミック酸合成工程で得られたポリウレタンアミック酸を加熱処理することにより閉環反応を起こさせるポリウレタンアミック酸閉環工程を含み、
下記式(I)で表されるイミド変性エラストマー(I)を得ることを特徴とするイミド変性エラストマーの製造方法、を提供する。
【化1】

[式中、R1は、芳香族環又は脂肪族環を含む2価の有機基を示す。R2は、分子量100〜10,000の2価の有機基を示す。R3は、芳香族環、脂肪族環又は脂肪族鎖を含む2価の有機基を示す。R4は、4個以上の炭素原子を含む4価の有機基を示す。nは1〜100の整数を示す。]
【化2】
[式中、R1、R2、R3、R4、nは上記と同じ。mは5〜200の整数を示す。]
【0010】
前記ポリウレタンアミック酸合成工程において、第二の工程を第一の工程後に行い、且つ、
前記ポリウレタンアミック酸合成工程が、前記第二の工程の後に、前記微粒子を含む混合物をさらに加熱下に混合して重合させる第三の工程をさらに含むことが好ましい。
【0011】
さらに、前記イミド変性エラストマーの製造方法は、下記の反応工程式(A)に示すように、下記式(a)で表されるジイソシアネート(a)と下記式(b)で表されるポリオール(b)とを混合することにより反応させて、下記式(c)で表されるウレタンプレポリマー(c)を得るウレタンプレポリマー合成工程(A)を含み、ウレタンプレポリマー合成工程(A)によって得られたウレタンプレポリマー(c)を前記第一の工程に用いることが好ましい。
【化3】
[式中、R1、R2、nは上記と同じ。]
【発明の効果】
【0012】
本発明のイミド変性エラストマーの製造方法によれば、溶媒を使用せずに済むため、環境に与える影響が少なく、製造時の生産効率が高くなり、コストが低減できる。また、溶媒を除去したり、成形前に長時間の熱処理をしたりする必要等も無く、反応工程の短縮化が図られるため、この点でもコストが低減できる。さらに、着色が低減され、耐溶剤性が向上したイミド変性エラストマーが得られる。
【図面の簡単な説明】
【0013】
【図1】本発明のイミド変性エラストマーの製造方法によって得られたイミド変性エラストマーのFT−IR試験の結果を示すIRスペクトルの解析結果である。
【図2】本発明のイミド変性エラストマーの製造方法によって得られたイミド変性エラストマーの引張り試験の結果を示すグラフである。
【図3】本発明のイミド変性エラストマーの製造方法によって得られたイミド変性エラストマーの動的粘弾性試験の結果を示すグラフである。
【図4】実施例1における、乳鉢で混合したモノマー微粒子を示す顕微鏡写真である。
【図5】比較例1における、モノマーを含む凝集物を示す顕微鏡写真である。
【発明を実施するための形態】
【0014】
以下に、ポリウレタンアミック酸合成工程及びポリウレタンアミック酸閉環工程の各工程を詳細に説明する。
【0015】
[1.ポリウレタンアミック酸合成工程の第一の工程]
第一の工程は、上記式(c)で表されるウレタンプレポリマー(c)と、上記式(d)で表されるジアミン化合物(d)と、上記式(f)で表されるテトラカルボン酸二無水物(f)とを、溶媒非存在下で加熱下に混合して重合させる工程であれば良く、特に限定されない。
【0016】
(1−1.混合方法)
混合方法としては、特に限定されないが、まず、ウレタンプレポリマー(c)とジアミン化合物(d)とを混合し、その後に、テトラカルボン酸二無水物(f)を添加して混合することが好ましい。
【0017】
前記混合の際には、ジアミン化合物(d)及びテトラカルボン酸二無水物(f)の固体を破砕しながら混合することが好ましい。破砕しながら混合する際には、テトラカルボン酸二無水物(f)の固体の平均粒子径を、例えば、30μm以下となるように破砕することが考えられるが、20μm以下となるように破砕することが好ましく、15μm以下となるように破砕することがより好ましい。
【0018】
混合の条件としては、特に限定されないが、混練機を用いて混合することが好ましい。前記混練機としては、高せん断ミキサーを用いることが好ましい。特に、まず、ウレタンプレポリマー(c)とジアミン化合物(d)とを混合し、その後に、テトラカルボン酸二無水物(f)を添加して混合する場合に、テトラカルボン酸二無水物(f)の添加後に、混練機を用いて混合することが好ましく、高せん断ミキサーを用いることがより好ましい。混練機を用いる場合の混合条件としては、例えば、5〜200rpm程度であり、好ましくは10〜100rpm程度である。
【0019】
混合時の温度等の条件としては、特に限定されないが、窒素ガス、アルゴンガス等の不活性ガス雰囲気下、25〜150℃、好ましくは50〜100℃において、1〜5時間程度攪拌させればよい。
【0020】
(1−2.混合比)
ウレタンプレポリマー(c)、ジアミン化合物(d)、及びテトラカルボン酸二無水物(f)の混合比は、特に限定されない。しかし、ウレタンプレポリマー(c)に対して、ジアミン化合物(d)を、NCO/NH2(モル比)が0.9〜1.1程度の割合で混合することが好ましく、0.95〜1.05程度の割合で混合することがより好ましい。また、ウレタンプレポリマー(c)に対して、テトラカルボン酸二無水物(f)を、混合比(モル)が、ジアミン化合物(d):テトラカルボン酸二無水物(f)=1:2〜1:2.02の範囲となる割合で混合するのが好ましい。これにより、確実にウレア結合部にイミドユニットを導入することができる。
【0021】
次に、ウレタンプレポリマー(c)、ジアミン化合物(d)、及びテトラカルボン酸二無水物(f)のそれぞれについて説明する。
【0022】
(1−3.ウレタンプレポリマー(c))
ウレタンプレポリマー(c)としては、式(c)で表されるウレタンプレポリマー(c)であれば、特に制限されないが、下記反応工程式(A)で示す反応により得られるウレタンプレポリマー(c)であることが好ましい。具体的には、式(a)で表されるジイソシアネート(a)と式(b)で表されるポリオール(b)とを混合することにより反応させて、式(c)で表されるウレタンプレポリマー(c)が得られる。
【0023】
[反応工程式(A)]
【化4】
[式中、R1、R2、nは、前記と同じである。]
【0024】
上記反応工程式(A)では、ジイソシアネート(a)とポリオール(b)から、分子両末端にイソシアネート基を有するウレタンプレポリマー(c)を得る。このウレタンプレポリマー(c)をエラストマー成分とした場合、ゴム状領域(室温付近)の弾性率が低くなり、よりエラスティックにすることができると共に、このウレタンプレポリマー(c)の重量平均分子量を制御することにより、主鎖に連続した2つのイミドユニットを、その分布を制御しつつ所望の割合で導入することが可能となる。
【0025】
ジイソシアネート(a)としては、特に限定されないが、例えば、2,4−トリレンジイソシアネート(TDI)、2,6−トリレンジイソシアネート(TDI)、4,4’−ジフェニルメタンジイソシアネート(MDI)、ポリメリックMDI(Cr.MDI)、ジアニシジンジイソシアネート(DADL)、ジフェニルエーテルジイソシアネート(PEDI)、ピトリレンジイソシアネート(TODI)、ナフタレンジイソシアネート(NDI)、ヘキサメチレンジイソシアネート(HMDI)、イソホロンジイソシアネート(IPDI)、リジンジイソシアネートメチルエステル(LDI)、メタキシリレンジイソシアネート(MXDI)、2,2,4−トリメチルヘキサメチレンジイソシアネート(TMDI)、2,4,4−トリメチルヘキサメチレンジイソシアネート(TMDI)、ダイマー酸ジイソシアネート(DDI)、イソプロピリデンビス(4−シクロヘキシルイソシアネート)(IPCI)、シクロヘキシルメタンジイソシアネート(水添MDI)、メチルシクロヘキサンジイソシアネート(水添TDI)、TDI2量体(TT)等が挙げられるが、減圧蒸留したものを用いるのが好ましい。これらは1種又は2種以上を混合して用いてもよい。
【0026】
ポリオール(b)としては、特に限定されないが、例えば、ポリプロピレングリコール(PPG)、ポリオキシテトラメチレングリコール(PTMG)、ポリマーポリオール等のポリエーテルポリオール;アジペート系ポリオール(縮合ポリエステルポリオール)、ポリカプロラクトン系ポリオール等のポリエステルポリオール;ポリカーボネートポリオール(PCD);ポリブタジエンポリオール;アクリルポリオール等が挙げられる。これらは1種又は2種以上を混合して用いてもよい。
【0027】
ポリオール(b)は、70〜90℃、1〜5mmHg、30分〜30時間程度の条件で減圧乾燥したものを用いるのが好ましい。また、ポリオール(b)の重量平均分子量は100〜10,000、好ましくは300〜5,000である。前記重量平均分子量は、ポリオール(b)をゲルパーミエーションクロマトグラフィー(GPC)で測定し、得られた測定値をポリスチレン換算した値である。
【0028】
反応は、上記で例示したジイソシアネート(a)とポリオール(b)とを所定の割合で混合した後、窒素ガス、アルゴンガス等の不活性ガス雰囲気下、室温〜100℃で1時間〜5時間程度反応させればよい。ジイソシアネート(a)とポリオール(b)との混合比(モル)は、ジイソシアネート(a):ポリオール(b)=1.01:1〜2:1の範囲にするのが好ましい。これにより、得られるウレタンプレポリマー(c)の重量平均分子量を、下記で説明する所定の値にすることができる。
【0029】
すなわち、得られるウレタンプレポリマー(c)の重量平均分子量は、300〜50,000、好ましくは500〜45,000であるのがよい。この範囲内でウレタンプレポリマー(c)の重量平均分子量を制御して、イミドユニットを所望の割合で導入すると、柔軟な低い弾性率を有するゴム状物で、かつ高強度及び高耐熱性を有するイミド変性エラストマー(I)を得ることができる。
【0030】
より具体的には、ウレタンプレポリマー(c)の重量平均分子量を上記所定の範囲にすると、イミド変性エラストマー(I)のイミド分率(イミド成分含有率)を5〜45重量%、好ましくは5〜40重量%にすることができる。該イミド分率は、イミド変性エラストマー中のイミド成分の割合を意味している。該イミド分率が前記所定の範囲にあると、主鎖に導入される連続した2つのイミドユニットの分布及び割合が最適化され、その結果、イミド変性エラストマー(I)が、柔軟な低い弾性率を有するゴム状物で、かつ高強度及び高耐熱性を有するようになる。これに対し、前記イミド分率が5重量%より低いと、強度や耐熱性が低下するおそれがあり、45重量%を超えると、柔軟性が低下するおそれがある。前記イミド分率は、原料、すなわちジイソシアネート(a)、ポリオール(b)、後述するジアミン化合物(d)及びテトラカルボン酸二無水物(f)の仕込み量から算出される値であり、より具体的には、下記式(α)から算出される値である。
イミド分率(%)=[(Wa'+Wc+Wd)/Wtotal]×100・・・(α)
Wtotal:Wa+Wb+Wc+Wd
Wa:ジイソシアネート仕込量(モル)×ジイソシアネート式量
Wb:ポリオール仕込量(モル)×ポリオール式量
Wc:ジアミン化合物仕込量(モル)×ジアミン化合物式量
Wd:テトラカルボン酸二無水物仕込量(モル)×テトラカルボン酸二無水物式量
Wa':[ジイソシアネート仕込量(モル)−ポリオール仕込量(モル)]×ジイソシアネート式量
【0031】
また、ウレタンプレポリマー(c)の重量平均分子量が、前記範囲内において小さいほど、ハードなイミド変性エラストマー(I)を得ることができる。これに対し、前記分子量が300より小さいと、イミド変性エラストマー(I)がハードになりすぎ、柔軟性が低下するおそれがある。また、50,000より大きいと、イミド変性エラストマー(I)がソフトになりすぎ、強度や耐熱性が低下するおそれがあるので好ましくない。前記重量平均分子量は、ウレタンプレポリマー(c)をGPCで測定し、得られた測定値をポリスチレン換算した値である。
【0032】
(1−4.ジアミン化合物(d))
ジアミン化合物(d)としては、特に限定されないが、例えば、1,4−ジアミノベンゼン(別名:p−フェニレンジアミン、略称:PPD)、1,3−ジアミノベンゼン(別名:m−フェニレンジアミン、略称:MPD)、2,4−ジアミノトルエン(別名:2,4−トルエンジアミン、略称:2、4−TDA)、4,4’−ジアミノジフェニルメタン(別名:4,4’−メチレンジアニリン、略称:MDA)、4,4’−ジアミノジフェニルエーテル(別名:4,4’−オキシジアニリン、略称:ODA、DPE)、3,4’−ジアミノジフェニルエーテル(別名:3,4’−オキシジアニリン、略称:3,4’−DPE)、3,3’−ジメチル−4,4’−ジアミノビフェニル(別名:o−トリジン、略称:TB)、2,2’−ジメチル−4,4’−ジアミノビフェニル(別名:m−トリジン、略称:m−TB)、2,2’−ビス(トリフルオロメチル)−4,4’−ジアミノビフェニル(略称:TFMB)、3,7−ジアミノ−ジメチルジベンゾチオフェン−5,5−ジオキシド(別名:o−トリジンスルホン、略称:TSN)、4,4’−ジアミノベンゾフェノン、3,3’−ジアミノベンゾフェノン、4,4’−ビス(4−アミノフェニル)スルフィド(別名:4,4’−チオジアニリン、略称:ASD)、4,4’−ジアミノジフェニルスルホン(別名:4,4’−スルホニルジアニリン、略称:ASN)、4,4’−ジアミノベンズアニリド(略称:DABA)、1,n−ビス(4−アミノフェノキシ)アルカン(n=3,4,5、略称:DAnMG)、1,3−ビス(4−アミノフェノキシ)−2,2−ジメチルプロパン(略称:DANPG)、1,2−ビス[2−(4−アミノフェノキシ)エトキシ]エタン(略称:DA3EG)、9,9−ビス(4−アミノフェニル)フルオレン(略称:FDA)、5(6)−アミノ−1−(4−アミノメチル)−1,3,3−トリメチルインダン、1,4−ビス(4−アミノフェノキシ)ベンゼン(略称:TPE−Q)、1,3−ビス(4−アミノフェノキシ)ベンゼン(別名:レゾルシンオキシジアニリン、略称:TPE−R)、1,3−ビス(3−アミノフェノキシ)ベンゼン(略称:APB)、4,4’−ビス(4−アミノフェノキシ)ビフェニル(略称:BAPB)、4,4’−ビス(3−アミノフェノキシ)ビフェニル、2,2−ビス(4−アミノフェノキシフェニル)プロパン(略称:BAPP)、ビス[4−(4−アミノフェノキシ)フェニル]スルホン(略称:BAPS)、ビス[4−(3−アミノフェノキシ)フェニル]スルホン(略称:BAPS−M)、2,2−ビス[4−(4−アミノフェノキシ)フェニル]ヘキサフルオロプロパン(略称:HFBAPP)、3,3’−ジカルボキシ−4,4’−ジアミノジフェニルメタン(略称:MBAA)、4,6−ジヒドロキシ−1,3−フェニレンジアミン(別名:4,6−ジアミノレゾルシン)、3,3’−ジヒドロキシ−4,4’−ジアミノビフェニル(別名:3,3’−ジヒドロキシベンジジン、略称:HAB)、3,3’,4,4’−テトラアミノビフェニル(別名:3,3’−ジアミノベンジジン、略称:TAB)等の炭素数6〜27の芳香族ジアミン化合物;1,6−ヘキサメチレンジアミン(HMDA)、1,8−オクタメチレンジアミン(OMDA)、1,9−ノナメチレンジアミン、1,12−ドデカメチレンジアミン(DMDA)、1−アミノ−3−アミノメチル−3,5,5−トリメチルシクロヘキサン(別名:イソホロンジアミン)、4,4’−ジシクロヘキシルメタンジアミン、シクロヘキサンジアミン等の炭素数6〜24の脂肪族又は脂環式ジアミン化合物;1,3−ビス(3−アミノプロピル)−1,1,3,3−テトラメチルジシロキサン等のシリコーン系ジアミン化合物等が挙げられ、これらは1種又は2種以上を混合して用いてもよい。特に、1,6−ヘキサメチレンジアミン(HMDA)を用いると、強度に優れるイミド変性エラストマー(I)を得ることができる。1,4−ビス(4−アミノフェノキシ)ベンゼン(TPE−Q)、1,3−ビス(4−アミノフェノキシ)ベンゼン(TPE−R)を用いると、耐溶剤性に優れるイミド変性エラストマー(I)を得ることができる。
【0033】
(1−5.テトラカルボン酸二無水物(f))
テトラカルボン酸二無水物(f)としては、特に限定されないが、例えば、無水ピロメリット酸(PMDA)、オキシジフタル酸二無水物(ODPA)、ベンゾフェノン−3,4,3’,4’−テトラカルボン酸二無水物(BTDA)、ビフェニル−3,4,3’,4’−テトラカルボン酸二無水物(BPDA)、ジフェニルスルホン−3,4,3’,4’−テトラカルボン酸二無水物(DSDA)、4,4’−(2,2−ヘキサフルオロイソプロピリデン)ジフタル酸二無水物(6FDA)、m(p)−ターフェニル−3,4,3’,4’−テトラカルボン酸二無水物、シクロブタン−1,2,3,4−テトラカルボン酸二無水物、1−カルボキシメチル−2,3,5−シクロペンタントリカルボン酸−2,6:3,5−二無水物等が挙げられる。なかでも、耐熱性、溶剤性、加工性の観点から、無水ピロメリット酸(PMDA)、オキシジフタル酸二無水物(ODPA)、ベンゾフェノン−3,4,3’,4’−テトラカルボン酸二無水物(BTDA)が好ましい。これらは1種又は2種以上を混合して用いてもよい。
【0034】
[2.ポリウレタンアミック酸合成工程の第二の工程]
第二の工程は、ジアミン化合物(d)及びテトラカルボン酸二無水物(f)の固体を粉砕して、平均粒子径が10μm以下の微粒子として、前記微粒子を含む混合物を生成する工程であれば、特に制限されないが、微粒子の平均粒子径を10μm以下とすることがより好ましく、5μm以下とすることがさらに好ましい。
【0035】
第二の工程は、第一の工程の前若しくは後、又は第一の工程とともに行ってもよいが、第一の工程後に行うことが好ましい。第二の工程を、第一の工程後に行い、さらに第三の工程を行った場合、反応物を混合することがより容易になるという利点がある。
【0036】
本願発明によるイミド変性エラストマーの製造方法は、第二の工程を含むことにより、溶媒を用いずに反応を行った場合にイミド変性エラストマー(I)中に見られる溶媒不溶の凝集物が低減されるため、製品の品質を損ねることがなくなる。
【0037】
ジアミン化合物(d)及びテトラカルボン酸二無水物(f)の固体を粉砕して、平均粒子径が10μm以下である微粒子とする方法としては、特に限定されない。具体的には、例えば、乳鉢、三本ロール等を用いることが考えられ、乳鉢を用いることがより好ましい。
【0038】
[3.ポリウレタンアミック酸合成工程の第三の工程]
第三の工程は、前記第二の工程の後に、前記微粒子を含む混合物をさらに加熱下に混合して重合させる工程であれば良く、特に限定されない。また、ポリウレタンアミック酸合成工程は、必ずしも第三の工程を含む必要は無い。
【0039】
上述のように、第二の工程を、第一の工程後に行い、さらに第三の工程を行った場合、反応物を混合することがより容易になるという利点がある。また、第二の工程でジアミン化合物(d)及びテトラカルボン酸二無水物(f)の固体が粉砕され、重合反応に供されることによって、未反応のジアミン化合物(d)及びテトラカルボン酸二無水物(f)が大幅に減少するという利点もある。
【0040】
混合方法としては、特に限定されないが、混練機を用いて混合することが好ましい。前記混練機としては、密閉式混練機を用いることが好ましく、小型密閉式混練機を用いることがより好ましい。また、1軸押出し機や2軸押出し機を用いて連続混合することも好ましい。また、前記混練機としては、加温機能を有する混練機であることが好ましい。また、混練機を用いる場合の混合条件としては、例えば、10〜200rpm程度であり、好ましくは20〜100rpm程度である。
【0041】
混合時の温度としては、特に限定されないが、25〜200℃、好ましくは50〜150℃において攪拌させればよい。混合の時間としては、特に限定されないが、トルクが上昇し極大となった後、低下して安定となった時点で、混合を停止することが好ましい。
【0042】
[4.ポリウレタンアミック酸閉環工程]
ポリウレタンアミック酸閉環工程は、前記ポリウレタンアミック酸合成工程で得られたポリウレタンアミック酸(PUA)を加熱処理することにより閉環反応を起こさせる工程であれば良く、特に限定されない。
【0043】
PUAを加熱処理に供することにより、閉環反応(脱水縮合反応)が起こり、前記式(I)で表されるイミド変性エラストマー(I)が得られる。
【0044】
前記加熱処理における加熱条件としては、特に限定されないが、例えば、減圧条件下であり、例えば150〜450℃、好ましくは150〜250℃であり、例えば1時間〜5時間程度である。これらの条件下では、PUAがより熱分解しにくいという点で好ましい。
【0045】
前記ポリウレタンアミック酸合成工程で得られたポリウレタンアミック酸(PUA)を成形した後に、成形したポリウレタンアミック酸(PUA)について前記加熱処理を行っても良いし、前記ポリウレタンアミック酸合成工程で得られたポリウレタンアミック酸(PUA)を成形する前に、前記加熱処理を行っても良い。前記成形の方法としては、特に限定されないが、例えば、1軸押出し機や2軸押出し機での連続混合後に押出し成型する方法、熱盤プレスのようなプレス成形、又は、オープンロール練りのようなロール練りであることが好ましい。
【0046】
前記成形によりPUAをどのような形状とするかについては、特に限定されないが、例えば、シート状、フィルム状又は板状に成形することができ、シート状に成形することが
好ましい。
【0047】
PUAを成形して、シート状、フィルム状又は板状とした場合に得られるイミド変性エラストマー(I)(又はPUI)の厚みとしては、例えば、0.1〜10mm程度が挙げられる。
【0048】
[5.本願発明の製造方法における化学反応]
本願発明の製造方法における化学反応においては、下記反応工程式(D)で示すように、下記式(c)で表されるウレタンプレポリマー(c)が、まずジアミン化合物(d)と反応して、下記式(e)で表されるポリウレタン−ウレア化合物(e)が得られ、その後に、ポリウレタン−ウレア化合物(e)がテトラカルボン酸二無水物(f)と反応することにより、下記式(I)で表されるイミド変性エラストマー(I)が得られると考えられる。
【0049】
【化5】
[式中、R1、R2、R3、R4、n、mは、前記と同じである。]
【0050】
以下、本願発明における化学反応について、ウレタンプレポリマー(c)とジアミン化合物(d)が反応してポリウレタン−ウレア化合物(e)が生じるまでの反応と、ポリウレタン−ウレア化合物(e)とテトラカルボン酸二無水物(f)が反応してイミド変性エラストマー(I)が生じるまでの反応とに分けて説明する。
【0051】
(5−1.ポリウレタン−ウレア化合物(e)が生じるまでの反応)
ポリウレタン−ウレア化合物(e)が生じるまでの反応としては、下記反応工程式(B)で示すように、ウレタンプレポリマー(c)をジアミン化合物(d)でウレア結合により鎖延長してポリウレタン−ウレア化合物(e)を得る。
【0052】
[反応工程式(B)]
【化6】
[式中、R1、R2、R3、n、mは、前記と同じである。]
【0053】
上記反応は、主に第一の工程中に、溶媒非存在下で起こっているものと推測される。
【0054】
(5−2.イミド変性エラストマー(I)が生じるまでの反応)
イミド変性エラストマー(I)が生じるまでの反応としては、下記反応工程式(C)で示すように、ポリウレタン−ウレア化合物(e)と、テトラカルボン酸二無水物(f)とが反応して、下記式(I)で表されるイミド変性エラストマー(I)が生じる。すなわち、テトラカルボン酸二無水物(f)により、ウレア結合部にイミドユニットを導入してブロック共重合体であるイミド変性エラストマー(I)が得られる。下記式(I)で表されるイミド変性エラストマー(I)のポリマー鎖の左側の末端は、通常、NCO基であり、右側の末端は、通常、NCO基、NH2基、(CO)2O基のいずれかである。
【0055】
[反応工程式(C)]
【化7】
[式中、R1、R2、R3、R4、n、mは、前記と同じである。]
【0056】
反応工程式(C)で示す反応は、ポリウレタン−ウレア化合物(e)とテトラカルボン酸二無水物(f)とのイミド化反応である。
【0057】
前記イミド化反応としては、まず、ポリウレタン−ウレア化合物(e)と、上記で例示したテトラカルボン酸二無水物(f)とが反応して、下記式(g)で表されるポリウレタンアミック酸(PUA)が得られる。上記反応は、主にポリウレタンアミック酸合成工程中に、溶媒非存在下で起こっているものと推測される。
【0058】
【化8】
【0059】
前記イミド化反応後のPUAは、粘土状であり、既に充分な粘度を有しているので、長時間の熱処理をすることなく、そのまま成形することができる。上述したように、単に溶媒を用いずに塊状重合させた従来の場合には、得られるPUAは粘調体であるため、PUAを長時間熱処理して粘土状にしてから成形する必要があった。そのため、本発明の方法によれば、反応工程が短くなり、この点でもコストが低減されることになる。上記反応後のPUAが既に充分な粘度を有している理由については、初期段階での均一混合により、反応の効率が向上したためであると推定される。
【0060】
(5−3.イミド変性エラストマー(I))
本発明のイミド変性エラストマーは、前記一般式(I)で表される。この式中において前記R1としては、例えば、2価の脂肪族炭化水素基、2価の芳香族炭化水素基、並びに、エーテル結合(−O−)、エステル結合(−C(=O)O−)、チオエーテル結合(−S−)、アミド結合(−C(=O)−NH2)、カルボニル基(−C(=O)−)、スルフィニル基(−SO−)、又はスルホニル基(−SO2−)を介して、又は介することなく、これらが2以上結合した炭化水素基等が挙げられる。
【0061】
前記2価の脂肪族炭化水素基、及び前記2価の芳香族炭化水素基は、置換基を有していても良い。前記置換基としては、例えば、アルキル基、アラルキル基、アリール基、アルコキシ基、水酸基、アミノ基、カルボキシル基、ハロゲン原子等が挙げられる。前記アルキル基としては、例えば、メチル基、エチル基、プロピル基等の炭素数1〜6のアルキル基が挙げられる。前記アラルキル基としては、例えば、ベンジル基、フェニルエチル基、フェニルプロピル基等の炭素数6〜12のアラルキル基が挙げられる。前記アリール基としては、例えば、フェニル基、トリル基、キシリル基、ビフェニリル基、ナフチル基、アントリル基、フェナントリル基等の炭素数6〜20のアリール基が挙げられる。前記アルコキシ基としては、例えば、メトキシ基、エトキシ基、プロポキシ基、ブトキシ基等の炭素数1〜6のアルコキシ基が挙げられる。前記ハロゲン原子としては、フッ素原子、塩素原子、臭素原子、ヨウ素原子等が挙げられる。
【0062】
前記2価の脂肪族炭化水素基、及び前記2価の芳香族炭化水素基は、ヘテロ原子を有していても良い。前記ヘテロ原子としては、例えば、窒素原子、酸素原子、イオウ原子等が挙げられる。
【0063】
前記2価の脂肪族炭化水素基としては、例えば、アルキレン基、アルケニレン基、アルキニレン基、アラルキレン基等が挙げられ、脂肪族環を有するものが好ましい。前記2価の脂肪族炭化水素基としては、炭素数1〜30(特に、炭素数2〜20)のものが好ましい。
【0064】
前記2価の芳香族炭化水素基としては、例えば、フェニレン基、ビフェニレン基、ナフチレン基等が挙げられる。前記2価の芳香族炭化水素基としては、炭素数1〜30(特に、炭素数6〜20)のものが好ましい。
【0065】
また、前記R1としては、上述した反応工程式(A)に従ってウレタンプレポリマー(c)を合成する際に用いるジイソシアネート(a)の残基等が挙げられる。中でも、ジイソシアネート(a)のうち、4,4’−ジフェニルメタンジイソシアネート(MDI)又はトリレンジイソシアネートの残基等が好ましい。
【0066】
前記R2は、分子量100〜10,000、好ましくは300〜5,000の2価の有機基を示すものである。該有機基としては、例えば、飽和又は不飽和の直鎖状又は分岐鎖状の多価の脂肪族炭化水素基、多価の脂環式炭化水素基、多価の芳香族炭化水素基からなる群から選択される化学基、又はこれらが2以上結合した炭化水素基等が挙げられる。前記炭化水素基は、ヘテロ原子及び/又は置換基を有していてもよく、ヘテロ原子、置換基として具体的に挙げられるものは、R1の場合と同様である。
【0067】
飽和又は不飽和の直鎖状又は分岐鎖状の多価の脂肪族炭化水素基としては、例えば、アルキレン基、アルケニレン基、アルカントリイル基、アルケントリイル基等が挙げられる。多価の脂環式炭化水素基としては、例えば、1,2−シクロペンチレン基、1,3−シクロペンチレン基、1,2−シクロヘキシレン基、1,3−シクロヘキシレン基、1,4−シクロヘキシレン基等のシクロアルキレン基;シクロヘキシリデン基等のシクロアルキリデン基;ノルボルナン−2,3−ジイル基、ノルボルナン−2,5−ジイル基、ノルボルナン−2,6−ジイル基等の多価の橋かけ環式炭化水素基等が挙げられる。多価の芳香族炭化水素基としては、例えば、1,2−フェニレン基、1,3−フェニレン基、1,4−フェニレン基等のフェニレン基等のアリレン基等が挙げられる。
【0068】
また、前記R2としては、上述した反応工程式(A)に従ってウレタンプレポリマー(c)を合成する際に用いるポリオール(b)の残基等が挙げられる。中でも、ポリオール(b)の残基のうち、ポリオキシテトラメチレングリコール(PTMG)又はポリプロピレングリコール(PPG)の残基等が好ましい。
【0069】
前記R3は、芳香族環、脂肪族環又は脂肪族鎖を含む2価の有機基を示すものである。前記有機基としては、例えば、2価の脂肪族炭化水素基、2価の芳香族炭化水素基、並びに、エーテル結合(−O−)、エステル結合(−C(=O)O−)、チオエーテル結合(−S−)、アミド結合(−C(=O)−NH2)、カルボニル基(−C(=O)−)、スルフィニル基(−SO−)、又はスルホニル基(−SO2−)を介して、又は介することなく、これらが2以上結合した炭化水素基等が挙げられる。前記炭化水素基は、ヘテロ原子及び/又は置換基を有していてもよく、ヘテロ原子、置換基として具体的に挙げられるものは、R1の場合と同様である。
【0070】
前記2価の脂肪族炭化水素基としては、例えば、アルキレン基、アルケニレン基、アルキニレン基、アラルキレン基等が挙げられる。前記2価の脂肪族炭化水素基としては、炭素数1のものも含み、炭素数1〜30のものが好ましいが、炭素数6〜24であることがより好ましい。1,6−ヘキサメチレン基であると、強度に優れるイミド変性エラストマー(I)を得ることができ、特に好ましい。
【0071】
前記2価の芳香族炭化水素基としては、例えば、フェニレン基、ビフェニレン基、ナフチレン基等が挙げられる。前記2価の芳香族炭化水素基としては、炭素数1のものも含み、炭素数1〜30のものが好ましいが、炭素数6〜27であることがより好ましい。
【0072】
前記これらが2以上結合した炭化水素基としては、例えば、メチルフェニレン基、メチルビフェニレン基、ジフェニルエーテル基等が挙げられる。
【0073】
前記R3としては、例えば、上述した反応工程式(B)に従ってポリウレタン−ウレア化合物(e)を合成する際に用いるジアミン化合物(d)の残基等が挙げられる。該残基としては、具体的には、例えば、ジアミン化合物(d)の残基のうち、4,4’−ジアミノジフェニルメタン、1,4−ビス(4−アミノフェノキシ)ベンゼン(TPE−Q)、1,3−ビス(4−アミノフェノキシ)ベンゼン(TPE−R)の残基等が、耐溶剤性に優れるイミド変性エラストマー(I)を得ることができる点で、特に好ましい。
【0074】
前記R4は、4個以上の炭素原子を含む4価の有機基を示すものであり、例えば、4価の脂肪族炭化水素基、及び4価の芳香族炭化水素基等の炭化水素基が挙げられ、4価の芳香族炭化水素基が好ましい。前記炭化水素基は、ヘテロ原子及び/又は置換基を有していてもよく、ヘテロ原子、置換基として具体的に挙げられるものは、R1の場合と同様である。
【0075】
前記R4としては、例えば、上述した反応工程式(C)に従ってイミド変性エラストマー(I)を合成する際に用いるテトラカルボン酸二無水物(f)の残基等が挙げられる。該残基としては、具体的には、例えば、無水ピロメリット酸(PMDA)、オキシジフタル酸二無水物(ODPA)、ベンゾフェノン−3,4,3’,4’−テトラカルボン酸二無水物(BTDA)の残基等が好ましい。
【0076】
前記一般式(I)で表されるイミド変性エラストマー(以下、イミド変性エラストマー(I)とも言う。)の具体例としては、下記式(1)で表されるイミド変性エラストマー等が挙げられる。
【0077】
【化9】
[式中、n、mは、前記と同じである。xは5〜100の整数を示す。]
【0078】
本願発明の製造方法によって得られるイミド変性エラストマー(I)は、ジイソシアネートとポリオールから得た分子両末端にイソシアネート基を有するウレタンプレポリマーをジアミン化合物でウレア結合により鎖延長し、テトラカルボン酸二無水物でウレア結合部にイミドユニットを導入したブロック共重合体であるのが好ましい。
【0079】
本願発明の製造方法によって得られるイミド変性エラストマー(I)は、柔軟な低い弾性率を有するゴム状物で、高強度及び高耐熱性を有するだけでなく、重量平均分子量が高く、明度が高く、耐溶剤性にも優れる。
【0080】
具体的には、本願発明の製造方法によって得られるイミド変性エラストマー(I)の50℃での貯蔵弾性率E’は、5×106〜1×108Paであるのが好ましい。該貯蔵弾性率は、後述するように、動的粘弾性測定装置を用いて測定して得られる値である。このような柔軟な低い弾性率を有するイミド変性エラストマー(I)は、ガラス転移温度(Tg)が、通常−30〜−60℃であり、ゴム状弾性領域の温度範囲が広いものになる。イミド変性エラストマー(I)が、柔軟な低い弾性率を有するゴム状物で、かつ高強度及び高耐熱性を有するようになる理由としては、以下の理由が推察される。すなわち、上記で説明した通り、本発明のイミド変性エラストマー(I)は、主鎖に連続した2つのイミドユニットを、その分布を制御しつつ所望の割合(イミド分率)で導入することができるので、このイミドユニットからなるハードセグメントの凝集が均一かつ強固なものになる。このため、イミド変性エラストマー(I)は、より均一かつ強固なミクロ相分離構造を形成し、ガラス転移温度が低くなることで、ゴム状弾性領域の温度範囲が広くなる。その結果、ポリウレタンをエラストマー成分として含有し、柔軟な低い弾性率を有するゴム状物としても、高強度及び高耐熱性を有するようになる。
【0081】
本願発明の製造方法によって得られるイミド変性エラストマー(I)の重量平均分子量は10,000〜1,000,000、好ましくは50,000〜500,000、より好ましくは80,000〜100,000であるのがよい。これに対し、前記重量平均分子量が10,000より小さいと、耐溶剤性、強度、耐熱性が低下するおそれがあり、1,000,000より大きいと、成形性が低下するおそれがあるので好ましくない。前記重量平均分子量は、ポリウレタンアミック酸合成工程後の重合物をNMPに溶解して、GPC測定することにより測定した値である。
【0082】
上記重量平均分子量が、従来の溶媒を用いて溶液重合を行って得られるイミド変性エラストマー(I)よりも高くなる理由としては、以下の理由が推察される。すなわち、ウレタンプレポリマー(c)をジアミン化合物(d)でウレア結合により鎖延長させる工程後、ウレア結合が切断されることがあり、その切断されたウレア結合が再結合する頻度は、化合物の濃度によって決まると考えられる。この点、溶液重合の場合、比較的濃度が低いため、切断されたウレア結合が再結合する確率は低い。これに対して、本願発明の場合は溶媒を用いないため、濃度が高く、ウレア結合が再結合する確率が高くなり、その結果高い重量平均分子量が保持されると推定される。
【0083】
本願発明の製造方法によって得られるイミド変性エラストマー(I)の明度は70〜95、好ましくは80〜95である。これに対し、前記明度が70より小さいと、カラーリングがしにくくなり用途が限定されるおそれがあるので好ましくない。前記明度は、JIS Z8729に準拠して測定した値である。
【0084】
本願発明の製造方法によって得られるイミド変性エラストマー(I)の耐溶剤性は、従来の溶媒を用いて溶液重合を行って得られるイミド変性エラストマー(I)よりも優れている。前記耐溶剤性は、後述するように、イミド変性エラストマー(1)シートを、所定の溶媒に浸漬し、浸漬前後の重量増加を計量して膨潤率(%)を算出することにより評価される。
【0085】
本発明のイミド変性エラストマーの製造方法によれば、柔軟な低い弾性率を有するゴム状物で、かつ高強度、高耐熱性及び熱可塑性を有すると共に、重量平均分子量が高く、明度が高くカラーリングが容易であり、溶剤に対しても安定なイミド変性エラストマーを製造することができる。前記イミド変性エラストマーは、例えば、ガスケット、シート、ベルト、フィルム、チューブ、ホース、ロールギア、パッキング材、防音材、防振材、ブーツ、被覆材、パーベーパレーション用の分離膜、光学非線形材料、弾性繊維、圧電素子、アクチュエーター、その他の各種自動車部品、工業機械部品、スポーツ用品等に使用することができる。また、反応で得られるイミド変性エラストマー(I)を、プレス成型、ロール練り等に供して、シート状、フィルム状、又は板状に成形することができるため、特にガスケット、シート、ベルト等に有用であるが、これらの用途に限定されるものではない。
【実施例】
【0086】
以下、合成例及び実施例を挙げて本発明のイミド変性エラストマーを詳細に説明するが、本発明は以下の合成例及び実施例のみに限定されるものではない。
【0087】
[合成例]
(ウレタンプレポリマー(j)の合成)
ウレタンプレポリマー(j)を下記反応工程式に基づいて合成した。具体的には、4,4’−ジフェニルメタンジイソシアネート(MDI)(h)[日本ポリウレタン工業社製]を減圧蒸留した。また、ポリオキシテトラメチレングリコール(PTMG)(i)[保土谷化学社製の商品名「PTMG1000」、重量平均分子量:1,000]を80℃、2〜3mmHg、24時間の条件で減圧乾燥した。
【0088】
【化10】
[式中、n、xは、前記と同じである。]
【0089】
次に、PTMG(i)69.54gを、500mlセパラブルフラスコに加え、真空条件下、80℃で1時間脱気した後、さらに、上記MDI(h)30.46gを加え、窒素雰囲気下、80℃で2時間半、180rpmで攪拌して、分子両末端にイソシアネート基を有するウレタンプレポリマー(j)を得た。
【0090】
[実施例1]
上記で得たウレタンプレポリマー(j)から、イミド変性エラストマー(1)を下記反応工程式に基づいて合成した。具体的には、ウレタンプレポリマー(j)100gを高せん断ミキサー(商品名「T.K.ハイビスミックス」、プライミクス社製)に投入し、80℃で撹絆しながら、4,4’−ジアミノジフェニルメタン(MDA)(k)を10.34g投入し、さらに1時間撹絆した。続いて、無水ピロメリット酸(PMDA)(m)を22.75g投入し、さらに1時間撹絆した。生成物を取り出し、モノマー微粒子が目視で確認できない程度(平均粒子径が約5μm)に分散するまで、乳鉢で混合した。混合後、生成物を取り出し、小型密閉式混練機(商品名「ラボプラストミル」、東洋精機製作所社製)に投入し、100℃、50rpmにて混合し、トルクが上昇し極大後、低下して安定となった時点で、混合を停止して、生成物を取り出した。得た生成物を金型に仕込み、180℃で熱盤プレスした後、200℃オーブン中で熱処理を2時間行って、厚さ0.5mmのシート状のイミド変性エラストマー(1)を得た。
【0091】
なお、モノマー微粒子を乳鉢で混合した結果、平均粒子径が約5μmになったことは、顕微鏡(商品名「デジタルマイクロスコープVHX−900」、キーエンス社製)を用いて確認した。その写真を図4に示す。
【0092】
【化11】
[式中、n、m、xは上記と同じ。]
【0093】
[比較例1]
乳鉢での混合を行わなかった結果、モノマーを含む凝集物の平均粒子径が約300μmであった以外は、実施例1と同様の実験を行い、厚さ0.5mmのシート状のイミド変性エラストマー(1)を得た。
【0094】
モノマーを含む凝集物の平均粒子径が約300μmであったことは、顕微鏡(商品名「デジタルマイクロスコープVHX−900」、キーエンス社製)を用いて確認した。その写真を図5に示す。
【0095】
[比較例2]
以下のように溶液重合により反応を行い、同様に厚さ0.5mmのシート状のイミド変性エラストマー(1)を得た。
【0096】
まず、下記反応工程式のように、実施例1と同様の方法で得たウレタンプレポリマー(j)と4,4’−ジアミノジフェニルメタン(MDA)(k)とを反応させて、下記式(l)で表されるポリウレタン−ウレア化合物(l)を得た。具体的には、まず、ウレタンプレポリマー(j)100gを脱水処理したN−メチル−2−ピロリドン(NMP)200mlに溶解させたものと、4,4’−ジアミノジフェニルメタン(MDA)(k)10.34gを脱水処理したNMP50mlに溶解させたものとを、500mlの四つ口セパラブルフラスコにそれぞれ加え、アルゴン雰囲気下、室温(23℃)で24時間攪拌して、ポリウレタン−ウレア化合物(l)の溶液を得た。
【化12】
[式中、n、xは上記と同じ。m’は2〜100の整数を示す。]
【0097】
次に、下記反応工程式のように、上記で得たポリウレタン−ウレア化合物(l)と無水ピロメリット酸(PMDA)(m)とを反応させて、イミド変性エラストマー(1)のシートを得た。具体的には、まず、上記で得たポリウレタン−ウレア化合物(l)の溶液中に、無水ピロメリット酸(PMDA)(m)22.75gを加え、アルゴンガス雰囲気下、150℃で2時間攪拌して、ポリウレタンアミック酸(PUA)溶液を得た。ついで、該PUA溶液を遠心成形機に流し込み、150℃で1,000rpm、1時間遠心成形してPUAシートを得た。このPUAシートを減圧デシケータ内で200℃、2時間加熱処理(脱水縮合反応)して、厚さ0.5mmのシート状のイミド変性エラストマー(1)を得た。
【化13】
[式中、n、m、xは上記と同じ。]
【0098】
[FT−IR試験]
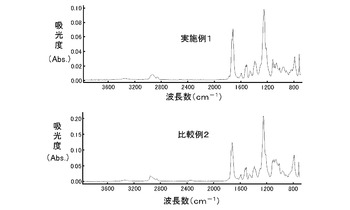
得られたイミド変性エラストマー(1)について、IRスペクトルを測定した。IRスペクトルの測定には、分光計(商品名「FTS−3000」、VALIAN社製)にてATR治具(PIKE社製)を用いた。また、IRスペクトルの測定は、サンプル押し付けによるATR法測定で、積算回数32回で行った。その結果を図1に示す。図1のグラフの横軸は波長数(cm-1)を示し、縦軸は吸光度(Abs.)を示す。
【0099】
図1に示した結果から、実施例1及び比較例2のいずれの場合でも、1780cm-1、1720cm-1及び1380cm-1にイミド環に由来する吸収が観察され、これらのイミド変性エラストマー(1)シートは同一の化学的構造を有することが示された。
【0100】
[引張り試験]
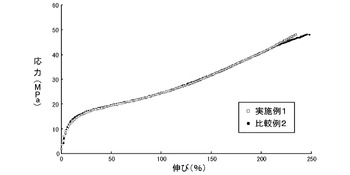
得られたイミド変性エラストマー(1)シートを3号ダンベルで打ち抜き、標線間20mm、引張り速度500mm/分の条件で、JIS K6251に準拠し、オートグラフ(商品名「AGS−G」、島津製作所社製)を用いて、応力、伸びをそれぞれ測定した。その結果を図2に示す。図2のグラフにおいて、縦軸は応力(MPa)、横軸は伸び(%)を示す。
【0101】
図2に示した結果から、実施例1及び比較例2(溶液重合)のいずれの場合でも、イミド変性エラストマー(1)シートの応力や柔軟性には差は無いことが示された。
【0102】
[動的粘弾性試験]
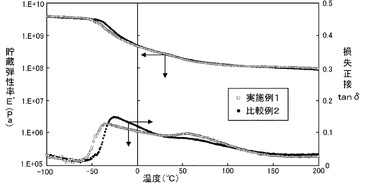
得られたイミド変性エラストマー(1)シートについて、動的粘弾性試験を行った。動的粘弾性試験は、動的粘弾性測定装置(商品名「DMS 6100」、セイコーインスツルメンツ社製)を用い、20Hz、5℃/分、−100〜400℃の昇温過程にて行い、貯蔵弾性率(E’)、損失正接(tanδ)を測定した。その結果を図3に示す。同グラフにおいて、左側の縦軸は貯蔵弾性率E’(Pa)、左側の縦軸は損失正接tanδを示し、横軸は温度(℃)を示す。
【0103】
図3に示した結果から、実施例1及び比較例2(溶液重合)のいずれの場合でも、イミド変性エラストマー(1)シートの、温度に対する動的粘弾性の挙動に差は無いことが示された。
【0104】
[イミド変性エラストマー(1)シートの明度評価]
実施例1で得られたイミド変性エラストマー(1)シートは、薄い黄色を呈しているのに対して、比較例2で得られたイミド変性エラストマー(1)シートは、こげ茶色を呈しており、目視での差異は明らかであった。このような明度の差異を客観的に評価するため、イミド変性エラストマー(1)シートの明度をJIS Z8729に準拠して測定し、結果を下記表1の「明度」の欄に示した。
【0105】
表1に示した結果から、実施例1で得られたイミド変性エラストマー(1)シートは、優れた明度を有することが客観的に示された。
【0106】
[イミド変性エラストマー(1)シートの耐溶剤性評価]
イミド変性エラストマー(1)シートを、室温(23℃)で96時間、N−メチル−2−ピロリドン(NMP)に浸漬し、イミド変性エラストマー(1)シートの浸漬前後の重量増加を計量した。より具体的には、イミド変性エラストマー(1)シートから幅2.0cm、長さ3.0cmの試験片を切り出し、この試験片を前記条件で溶剤に浸漬し、浸漬前後の重量を下記式(β)に当てはめて膨潤率(%)を算出し、耐溶剤性の指標とした。
膨潤率(%)=[(S2−S1)/S1]×100 ・・・(β)
S1:浸漬前の試験片の重量
S2:浸漬後の試験片の重量
【0107】
表1に示した結果から、実施例1で得られたイミド変性エラストマー(1)シートは、優れた耐溶剤性を有することが示された。
【0108】
[イミド変性エラストマー(1)シートの重量平均分子量評価]
イミド変性エラストマー(1)シートの重量平均分子量を測定し、結果を下記表1の「分子量(Mw)」の欄に示した。
【0109】
表1に示した結果から、実施例1で得られたイミド変性エラストマー(1)シートは、重量平均分子量が高いことが示された。
【0110】
【表1】
【産業上の利用可能性】
【0111】
本発明のイミド変性エラストマーの製造方法によれば、溶媒を使用せずに済むため製造時の生産効率が高くなり、溶媒を除去する必要も無いためコストが低減でき、さらに、着色が低減され、耐溶剤性が向上したイミド変性エラストマーが得られるため、特に、ガスケット等のゴム成形品の製造方法として有用である。
【技術分野】
【0001】
本発明は、イミド変性エラストマーの製造方法に関する。
【背景技術】
【0002】
柔軟な低い弾性率を有するゴム状物で、かつ高強度及び高耐熱性を有する新規なイミド変性エラストマーの製造方法として、ジイソシアネートとポリオールから得た分子両末端にイソシアネート基を有するウレタンプレポリマーを、ジアミン化合物でウレア結合により鎖延長し、さらにテトラカルボン酸二無水物との反応によりポリウレタンアミック酸(PUA)溶液を得て、これを遠心成形後、加熱処理により閉環反応(脱水縮合反応)させることにより、ウレア結合部にイミドユニットを導入したブロック共重合体であるイミド変性エラストマーを合成する製造方法が知られている(特許文献1)。
【0003】
【特許文献1】特開2008−101195号公報
【発明の概要】
【発明が解決しようとする課題】
【0004】
しかしながら、上記製造方法では、PUA溶液を得るために、あらかじめ重合したウレタンプレポリマーをN−メチル−2−ピロリドン(NMP)等の有機溶媒に溶解させる必要がある。この場合、ポリマー量の3〜5倍量の溶媒を使用するため、製造時の生産効率が低く、コストが増大し、環境への影響が懸念されるという問題があった。さらに、反応後に溶媒を除去する必要があるため、反応工程が長く、この点でもコストが増大するという問題があった。
【0005】
また、上記製造方法によって得られるイミド変性エラストマーは、着色の程度が強く、カラーリングをする用途には不向きであるという問題があった。さらに、上記製造方法によって得られるイミド変性エラストマーは、耐溶剤性についても充分ではないという問題もあった。
【0006】
上記製造方法では、PUA溶液を得て、これを遠心成形後、加熱処理するのに対して、溶媒を用いずに塊状重合させた場合には、反応が進まないため、得られるPUAは粘調体となる。そのため、PUAを長時間の熱処理(100℃程度で約12時間)によって粘度を上げて粘土状にしてから成形する必要があった。また、得られるイミド変性エラストマー中に溶媒に不溶な凝集物が残存して製品の品質を損ねるという問題があった。このように、単に溶媒を用いずに塊状重合させれば良いというものでもないのである。
【0007】
従って、本発明の目的は、着色が低減され、耐溶剤性が向上したイミド変性エラストマーを得ることができ、製造時の生産効率が高く、環境に配慮し、反応工程が短く、コストが低減された、イミド変性エラストマーの製造方法を提供することにある。
【課題を解決するための手段】
【0008】
そこで、本発明者らが、上記課題を解決するために鋭意検討した結果、テトラカルボン酸二無水物を添加する際に、テトラカルボン酸二無水物を含む微粒子の平均粒子径を10μm以下とすることにより、溶媒を用いずに塊状重合させても凝集物が生じず、しかも、得られるPUAの粘度が高く粘土状であるため、長時間の熱処理をせずに成形できることを見いだし、本発明を完成させた。
【0009】
すなわち、本発明は、
下記式(c)で表されるウレタンプレポリマー(c)と、下記式(d)で表されるジアミン化合物(d)と、下記式(f)で表されるテトラカルボン酸二無水物(f)とを、溶媒非存在下で加熱下に混合して重合させる第一の工程、及び
ジアミン化合物(d)及びテトラカルボン酸二無水物(f)の固体を粉砕して、平均粒子径が10μm以下の微粒子として、前記微粒子を含む混合物を生成する第二の工程、
を含むポリウレタンアミック酸合成工程、並びに
前記ポリウレタンアミック酸合成工程で得られたポリウレタンアミック酸を加熱処理することにより閉環反応を起こさせるポリウレタンアミック酸閉環工程を含み、
下記式(I)で表されるイミド変性エラストマー(I)を得ることを特徴とするイミド変性エラストマーの製造方法、を提供する。
【化1】
[式中、R1は、芳香族環又は脂肪族環を含む2価の有機基を示す。R2は、分子量100〜10,000の2価の有機基を示す。R3は、芳香族環、脂肪族環又は脂肪族鎖を含む2価の有機基を示す。R4は、4個以上の炭素原子を含む4価の有機基を示す。nは1〜100の整数を示す。]
【化2】
[式中、R1、R2、R3、R4、nは上記と同じ。mは5〜200の整数を示す。]
【0010】
前記ポリウレタンアミック酸合成工程において、第二の工程を第一の工程後に行い、且つ、
前記ポリウレタンアミック酸合成工程が、前記第二の工程の後に、前記微粒子を含む混合物をさらに加熱下に混合して重合させる第三の工程をさらに含むことが好ましい。
【0011】
さらに、前記イミド変性エラストマーの製造方法は、下記の反応工程式(A)に示すように、下記式(a)で表されるジイソシアネート(a)と下記式(b)で表されるポリオール(b)とを混合することにより反応させて、下記式(c)で表されるウレタンプレポリマー(c)を得るウレタンプレポリマー合成工程(A)を含み、ウレタンプレポリマー合成工程(A)によって得られたウレタンプレポリマー(c)を前記第一の工程に用いることが好ましい。
【化3】
[式中、R1、R2、nは上記と同じ。]
【発明の効果】
【0012】
本発明のイミド変性エラストマーの製造方法によれば、溶媒を使用せずに済むため、環境に与える影響が少なく、製造時の生産効率が高くなり、コストが低減できる。また、溶媒を除去したり、成形前に長時間の熱処理をしたりする必要等も無く、反応工程の短縮化が図られるため、この点でもコストが低減できる。さらに、着色が低減され、耐溶剤性が向上したイミド変性エラストマーが得られる。
【図面の簡単な説明】
【0013】
【図1】本発明のイミド変性エラストマーの製造方法によって得られたイミド変性エラストマーのFT−IR試験の結果を示すIRスペクトルの解析結果である。
【図2】本発明のイミド変性エラストマーの製造方法によって得られたイミド変性エラストマーの引張り試験の結果を示すグラフである。
【図3】本発明のイミド変性エラストマーの製造方法によって得られたイミド変性エラストマーの動的粘弾性試験の結果を示すグラフである。
【図4】実施例1における、乳鉢で混合したモノマー微粒子を示す顕微鏡写真である。
【図5】比較例1における、モノマーを含む凝集物を示す顕微鏡写真である。
【発明を実施するための形態】
【0014】
以下に、ポリウレタンアミック酸合成工程及びポリウレタンアミック酸閉環工程の各工程を詳細に説明する。
【0015】
[1.ポリウレタンアミック酸合成工程の第一の工程]
第一の工程は、上記式(c)で表されるウレタンプレポリマー(c)と、上記式(d)で表されるジアミン化合物(d)と、上記式(f)で表されるテトラカルボン酸二無水物(f)とを、溶媒非存在下で加熱下に混合して重合させる工程であれば良く、特に限定されない。
【0016】
(1−1.混合方法)
混合方法としては、特に限定されないが、まず、ウレタンプレポリマー(c)とジアミン化合物(d)とを混合し、その後に、テトラカルボン酸二無水物(f)を添加して混合することが好ましい。
【0017】
前記混合の際には、ジアミン化合物(d)及びテトラカルボン酸二無水物(f)の固体を破砕しながら混合することが好ましい。破砕しながら混合する際には、テトラカルボン酸二無水物(f)の固体の平均粒子径を、例えば、30μm以下となるように破砕することが考えられるが、20μm以下となるように破砕することが好ましく、15μm以下となるように破砕することがより好ましい。
【0018】
混合の条件としては、特に限定されないが、混練機を用いて混合することが好ましい。前記混練機としては、高せん断ミキサーを用いることが好ましい。特に、まず、ウレタンプレポリマー(c)とジアミン化合物(d)とを混合し、その後に、テトラカルボン酸二無水物(f)を添加して混合する場合に、テトラカルボン酸二無水物(f)の添加後に、混練機を用いて混合することが好ましく、高せん断ミキサーを用いることがより好ましい。混練機を用いる場合の混合条件としては、例えば、5〜200rpm程度であり、好ましくは10〜100rpm程度である。
【0019】
混合時の温度等の条件としては、特に限定されないが、窒素ガス、アルゴンガス等の不活性ガス雰囲気下、25〜150℃、好ましくは50〜100℃において、1〜5時間程度攪拌させればよい。
【0020】
(1−2.混合比)
ウレタンプレポリマー(c)、ジアミン化合物(d)、及びテトラカルボン酸二無水物(f)の混合比は、特に限定されない。しかし、ウレタンプレポリマー(c)に対して、ジアミン化合物(d)を、NCO/NH2(モル比)が0.9〜1.1程度の割合で混合することが好ましく、0.95〜1.05程度の割合で混合することがより好ましい。また、ウレタンプレポリマー(c)に対して、テトラカルボン酸二無水物(f)を、混合比(モル)が、ジアミン化合物(d):テトラカルボン酸二無水物(f)=1:2〜1:2.02の範囲となる割合で混合するのが好ましい。これにより、確実にウレア結合部にイミドユニットを導入することができる。
【0021】
次に、ウレタンプレポリマー(c)、ジアミン化合物(d)、及びテトラカルボン酸二無水物(f)のそれぞれについて説明する。
【0022】
(1−3.ウレタンプレポリマー(c))
ウレタンプレポリマー(c)としては、式(c)で表されるウレタンプレポリマー(c)であれば、特に制限されないが、下記反応工程式(A)で示す反応により得られるウレタンプレポリマー(c)であることが好ましい。具体的には、式(a)で表されるジイソシアネート(a)と式(b)で表されるポリオール(b)とを混合することにより反応させて、式(c)で表されるウレタンプレポリマー(c)が得られる。
【0023】
[反応工程式(A)]
【化4】
[式中、R1、R2、nは、前記と同じである。]
【0024】
上記反応工程式(A)では、ジイソシアネート(a)とポリオール(b)から、分子両末端にイソシアネート基を有するウレタンプレポリマー(c)を得る。このウレタンプレポリマー(c)をエラストマー成分とした場合、ゴム状領域(室温付近)の弾性率が低くなり、よりエラスティックにすることができると共に、このウレタンプレポリマー(c)の重量平均分子量を制御することにより、主鎖に連続した2つのイミドユニットを、その分布を制御しつつ所望の割合で導入することが可能となる。
【0025】
ジイソシアネート(a)としては、特に限定されないが、例えば、2,4−トリレンジイソシアネート(TDI)、2,6−トリレンジイソシアネート(TDI)、4,4’−ジフェニルメタンジイソシアネート(MDI)、ポリメリックMDI(Cr.MDI)、ジアニシジンジイソシアネート(DADL)、ジフェニルエーテルジイソシアネート(PEDI)、ピトリレンジイソシアネート(TODI)、ナフタレンジイソシアネート(NDI)、ヘキサメチレンジイソシアネート(HMDI)、イソホロンジイソシアネート(IPDI)、リジンジイソシアネートメチルエステル(LDI)、メタキシリレンジイソシアネート(MXDI)、2,2,4−トリメチルヘキサメチレンジイソシアネート(TMDI)、2,4,4−トリメチルヘキサメチレンジイソシアネート(TMDI)、ダイマー酸ジイソシアネート(DDI)、イソプロピリデンビス(4−シクロヘキシルイソシアネート)(IPCI)、シクロヘキシルメタンジイソシアネート(水添MDI)、メチルシクロヘキサンジイソシアネート(水添TDI)、TDI2量体(TT)等が挙げられるが、減圧蒸留したものを用いるのが好ましい。これらは1種又は2種以上を混合して用いてもよい。
【0026】
ポリオール(b)としては、特に限定されないが、例えば、ポリプロピレングリコール(PPG)、ポリオキシテトラメチレングリコール(PTMG)、ポリマーポリオール等のポリエーテルポリオール;アジペート系ポリオール(縮合ポリエステルポリオール)、ポリカプロラクトン系ポリオール等のポリエステルポリオール;ポリカーボネートポリオール(PCD);ポリブタジエンポリオール;アクリルポリオール等が挙げられる。これらは1種又は2種以上を混合して用いてもよい。
【0027】
ポリオール(b)は、70〜90℃、1〜5mmHg、30分〜30時間程度の条件で減圧乾燥したものを用いるのが好ましい。また、ポリオール(b)の重量平均分子量は100〜10,000、好ましくは300〜5,000である。前記重量平均分子量は、ポリオール(b)をゲルパーミエーションクロマトグラフィー(GPC)で測定し、得られた測定値をポリスチレン換算した値である。
【0028】
反応は、上記で例示したジイソシアネート(a)とポリオール(b)とを所定の割合で混合した後、窒素ガス、アルゴンガス等の不活性ガス雰囲気下、室温〜100℃で1時間〜5時間程度反応させればよい。ジイソシアネート(a)とポリオール(b)との混合比(モル)は、ジイソシアネート(a):ポリオール(b)=1.01:1〜2:1の範囲にするのが好ましい。これにより、得られるウレタンプレポリマー(c)の重量平均分子量を、下記で説明する所定の値にすることができる。
【0029】
すなわち、得られるウレタンプレポリマー(c)の重量平均分子量は、300〜50,000、好ましくは500〜45,000であるのがよい。この範囲内でウレタンプレポリマー(c)の重量平均分子量を制御して、イミドユニットを所望の割合で導入すると、柔軟な低い弾性率を有するゴム状物で、かつ高強度及び高耐熱性を有するイミド変性エラストマー(I)を得ることができる。
【0030】
より具体的には、ウレタンプレポリマー(c)の重量平均分子量を上記所定の範囲にすると、イミド変性エラストマー(I)のイミド分率(イミド成分含有率)を5〜45重量%、好ましくは5〜40重量%にすることができる。該イミド分率は、イミド変性エラストマー中のイミド成分の割合を意味している。該イミド分率が前記所定の範囲にあると、主鎖に導入される連続した2つのイミドユニットの分布及び割合が最適化され、その結果、イミド変性エラストマー(I)が、柔軟な低い弾性率を有するゴム状物で、かつ高強度及び高耐熱性を有するようになる。これに対し、前記イミド分率が5重量%より低いと、強度や耐熱性が低下するおそれがあり、45重量%を超えると、柔軟性が低下するおそれがある。前記イミド分率は、原料、すなわちジイソシアネート(a)、ポリオール(b)、後述するジアミン化合物(d)及びテトラカルボン酸二無水物(f)の仕込み量から算出される値であり、より具体的には、下記式(α)から算出される値である。
イミド分率(%)=[(Wa'+Wc+Wd)/Wtotal]×100・・・(α)
Wtotal:Wa+Wb+Wc+Wd
Wa:ジイソシアネート仕込量(モル)×ジイソシアネート式量
Wb:ポリオール仕込量(モル)×ポリオール式量
Wc:ジアミン化合物仕込量(モル)×ジアミン化合物式量
Wd:テトラカルボン酸二無水物仕込量(モル)×テトラカルボン酸二無水物式量
Wa':[ジイソシアネート仕込量(モル)−ポリオール仕込量(モル)]×ジイソシアネート式量
【0031】
また、ウレタンプレポリマー(c)の重量平均分子量が、前記範囲内において小さいほど、ハードなイミド変性エラストマー(I)を得ることができる。これに対し、前記分子量が300より小さいと、イミド変性エラストマー(I)がハードになりすぎ、柔軟性が低下するおそれがある。また、50,000より大きいと、イミド変性エラストマー(I)がソフトになりすぎ、強度や耐熱性が低下するおそれがあるので好ましくない。前記重量平均分子量は、ウレタンプレポリマー(c)をGPCで測定し、得られた測定値をポリスチレン換算した値である。
【0032】
(1−4.ジアミン化合物(d))
ジアミン化合物(d)としては、特に限定されないが、例えば、1,4−ジアミノベンゼン(別名:p−フェニレンジアミン、略称:PPD)、1,3−ジアミノベンゼン(別名:m−フェニレンジアミン、略称:MPD)、2,4−ジアミノトルエン(別名:2,4−トルエンジアミン、略称:2、4−TDA)、4,4’−ジアミノジフェニルメタン(別名:4,4’−メチレンジアニリン、略称:MDA)、4,4’−ジアミノジフェニルエーテル(別名:4,4’−オキシジアニリン、略称:ODA、DPE)、3,4’−ジアミノジフェニルエーテル(別名:3,4’−オキシジアニリン、略称:3,4’−DPE)、3,3’−ジメチル−4,4’−ジアミノビフェニル(別名:o−トリジン、略称:TB)、2,2’−ジメチル−4,4’−ジアミノビフェニル(別名:m−トリジン、略称:m−TB)、2,2’−ビス(トリフルオロメチル)−4,4’−ジアミノビフェニル(略称:TFMB)、3,7−ジアミノ−ジメチルジベンゾチオフェン−5,5−ジオキシド(別名:o−トリジンスルホン、略称:TSN)、4,4’−ジアミノベンゾフェノン、3,3’−ジアミノベンゾフェノン、4,4’−ビス(4−アミノフェニル)スルフィド(別名:4,4’−チオジアニリン、略称:ASD)、4,4’−ジアミノジフェニルスルホン(別名:4,4’−スルホニルジアニリン、略称:ASN)、4,4’−ジアミノベンズアニリド(略称:DABA)、1,n−ビス(4−アミノフェノキシ)アルカン(n=3,4,5、略称:DAnMG)、1,3−ビス(4−アミノフェノキシ)−2,2−ジメチルプロパン(略称:DANPG)、1,2−ビス[2−(4−アミノフェノキシ)エトキシ]エタン(略称:DA3EG)、9,9−ビス(4−アミノフェニル)フルオレン(略称:FDA)、5(6)−アミノ−1−(4−アミノメチル)−1,3,3−トリメチルインダン、1,4−ビス(4−アミノフェノキシ)ベンゼン(略称:TPE−Q)、1,3−ビス(4−アミノフェノキシ)ベンゼン(別名:レゾルシンオキシジアニリン、略称:TPE−R)、1,3−ビス(3−アミノフェノキシ)ベンゼン(略称:APB)、4,4’−ビス(4−アミノフェノキシ)ビフェニル(略称:BAPB)、4,4’−ビス(3−アミノフェノキシ)ビフェニル、2,2−ビス(4−アミノフェノキシフェニル)プロパン(略称:BAPP)、ビス[4−(4−アミノフェノキシ)フェニル]スルホン(略称:BAPS)、ビス[4−(3−アミノフェノキシ)フェニル]スルホン(略称:BAPS−M)、2,2−ビス[4−(4−アミノフェノキシ)フェニル]ヘキサフルオロプロパン(略称:HFBAPP)、3,3’−ジカルボキシ−4,4’−ジアミノジフェニルメタン(略称:MBAA)、4,6−ジヒドロキシ−1,3−フェニレンジアミン(別名:4,6−ジアミノレゾルシン)、3,3’−ジヒドロキシ−4,4’−ジアミノビフェニル(別名:3,3’−ジヒドロキシベンジジン、略称:HAB)、3,3’,4,4’−テトラアミノビフェニル(別名:3,3’−ジアミノベンジジン、略称:TAB)等の炭素数6〜27の芳香族ジアミン化合物;1,6−ヘキサメチレンジアミン(HMDA)、1,8−オクタメチレンジアミン(OMDA)、1,9−ノナメチレンジアミン、1,12−ドデカメチレンジアミン(DMDA)、1−アミノ−3−アミノメチル−3,5,5−トリメチルシクロヘキサン(別名:イソホロンジアミン)、4,4’−ジシクロヘキシルメタンジアミン、シクロヘキサンジアミン等の炭素数6〜24の脂肪族又は脂環式ジアミン化合物;1,3−ビス(3−アミノプロピル)−1,1,3,3−テトラメチルジシロキサン等のシリコーン系ジアミン化合物等が挙げられ、これらは1種又は2種以上を混合して用いてもよい。特に、1,6−ヘキサメチレンジアミン(HMDA)を用いると、強度に優れるイミド変性エラストマー(I)を得ることができる。1,4−ビス(4−アミノフェノキシ)ベンゼン(TPE−Q)、1,3−ビス(4−アミノフェノキシ)ベンゼン(TPE−R)を用いると、耐溶剤性に優れるイミド変性エラストマー(I)を得ることができる。
【0033】
(1−5.テトラカルボン酸二無水物(f))
テトラカルボン酸二無水物(f)としては、特に限定されないが、例えば、無水ピロメリット酸(PMDA)、オキシジフタル酸二無水物(ODPA)、ベンゾフェノン−3,4,3’,4’−テトラカルボン酸二無水物(BTDA)、ビフェニル−3,4,3’,4’−テトラカルボン酸二無水物(BPDA)、ジフェニルスルホン−3,4,3’,4’−テトラカルボン酸二無水物(DSDA)、4,4’−(2,2−ヘキサフルオロイソプロピリデン)ジフタル酸二無水物(6FDA)、m(p)−ターフェニル−3,4,3’,4’−テトラカルボン酸二無水物、シクロブタン−1,2,3,4−テトラカルボン酸二無水物、1−カルボキシメチル−2,3,5−シクロペンタントリカルボン酸−2,6:3,5−二無水物等が挙げられる。なかでも、耐熱性、溶剤性、加工性の観点から、無水ピロメリット酸(PMDA)、オキシジフタル酸二無水物(ODPA)、ベンゾフェノン−3,4,3’,4’−テトラカルボン酸二無水物(BTDA)が好ましい。これらは1種又は2種以上を混合して用いてもよい。
【0034】
[2.ポリウレタンアミック酸合成工程の第二の工程]
第二の工程は、ジアミン化合物(d)及びテトラカルボン酸二無水物(f)の固体を粉砕して、平均粒子径が10μm以下の微粒子として、前記微粒子を含む混合物を生成する工程であれば、特に制限されないが、微粒子の平均粒子径を10μm以下とすることがより好ましく、5μm以下とすることがさらに好ましい。
【0035】
第二の工程は、第一の工程の前若しくは後、又は第一の工程とともに行ってもよいが、第一の工程後に行うことが好ましい。第二の工程を、第一の工程後に行い、さらに第三の工程を行った場合、反応物を混合することがより容易になるという利点がある。
【0036】
本願発明によるイミド変性エラストマーの製造方法は、第二の工程を含むことにより、溶媒を用いずに反応を行った場合にイミド変性エラストマー(I)中に見られる溶媒不溶の凝集物が低減されるため、製品の品質を損ねることがなくなる。
【0037】
ジアミン化合物(d)及びテトラカルボン酸二無水物(f)の固体を粉砕して、平均粒子径が10μm以下である微粒子とする方法としては、特に限定されない。具体的には、例えば、乳鉢、三本ロール等を用いることが考えられ、乳鉢を用いることがより好ましい。
【0038】
[3.ポリウレタンアミック酸合成工程の第三の工程]
第三の工程は、前記第二の工程の後に、前記微粒子を含む混合物をさらに加熱下に混合して重合させる工程であれば良く、特に限定されない。また、ポリウレタンアミック酸合成工程は、必ずしも第三の工程を含む必要は無い。
【0039】
上述のように、第二の工程を、第一の工程後に行い、さらに第三の工程を行った場合、反応物を混合することがより容易になるという利点がある。また、第二の工程でジアミン化合物(d)及びテトラカルボン酸二無水物(f)の固体が粉砕され、重合反応に供されることによって、未反応のジアミン化合物(d)及びテトラカルボン酸二無水物(f)が大幅に減少するという利点もある。
【0040】
混合方法としては、特に限定されないが、混練機を用いて混合することが好ましい。前記混練機としては、密閉式混練機を用いることが好ましく、小型密閉式混練機を用いることがより好ましい。また、1軸押出し機や2軸押出し機を用いて連続混合することも好ましい。また、前記混練機としては、加温機能を有する混練機であることが好ましい。また、混練機を用いる場合の混合条件としては、例えば、10〜200rpm程度であり、好ましくは20〜100rpm程度である。
【0041】
混合時の温度としては、特に限定されないが、25〜200℃、好ましくは50〜150℃において攪拌させればよい。混合の時間としては、特に限定されないが、トルクが上昇し極大となった後、低下して安定となった時点で、混合を停止することが好ましい。
【0042】
[4.ポリウレタンアミック酸閉環工程]
ポリウレタンアミック酸閉環工程は、前記ポリウレタンアミック酸合成工程で得られたポリウレタンアミック酸(PUA)を加熱処理することにより閉環反応を起こさせる工程であれば良く、特に限定されない。
【0043】
PUAを加熱処理に供することにより、閉環反応(脱水縮合反応)が起こり、前記式(I)で表されるイミド変性エラストマー(I)が得られる。
【0044】
前記加熱処理における加熱条件としては、特に限定されないが、例えば、減圧条件下であり、例えば150〜450℃、好ましくは150〜250℃であり、例えば1時間〜5時間程度である。これらの条件下では、PUAがより熱分解しにくいという点で好ましい。
【0045】
前記ポリウレタンアミック酸合成工程で得られたポリウレタンアミック酸(PUA)を成形した後に、成形したポリウレタンアミック酸(PUA)について前記加熱処理を行っても良いし、前記ポリウレタンアミック酸合成工程で得られたポリウレタンアミック酸(PUA)を成形する前に、前記加熱処理を行っても良い。前記成形の方法としては、特に限定されないが、例えば、1軸押出し機や2軸押出し機での連続混合後に押出し成型する方法、熱盤プレスのようなプレス成形、又は、オープンロール練りのようなロール練りであることが好ましい。
【0046】
前記成形によりPUAをどのような形状とするかについては、特に限定されないが、例えば、シート状、フィルム状又は板状に成形することができ、シート状に成形することが
好ましい。
【0047】
PUAを成形して、シート状、フィルム状又は板状とした場合に得られるイミド変性エラストマー(I)(又はPUI)の厚みとしては、例えば、0.1〜10mm程度が挙げられる。
【0048】
[5.本願発明の製造方法における化学反応]
本願発明の製造方法における化学反応においては、下記反応工程式(D)で示すように、下記式(c)で表されるウレタンプレポリマー(c)が、まずジアミン化合物(d)と反応して、下記式(e)で表されるポリウレタン−ウレア化合物(e)が得られ、その後に、ポリウレタン−ウレア化合物(e)がテトラカルボン酸二無水物(f)と反応することにより、下記式(I)で表されるイミド変性エラストマー(I)が得られると考えられる。
【0049】
【化5】
[式中、R1、R2、R3、R4、n、mは、前記と同じである。]
【0050】
以下、本願発明における化学反応について、ウレタンプレポリマー(c)とジアミン化合物(d)が反応してポリウレタン−ウレア化合物(e)が生じるまでの反応と、ポリウレタン−ウレア化合物(e)とテトラカルボン酸二無水物(f)が反応してイミド変性エラストマー(I)が生じるまでの反応とに分けて説明する。
【0051】
(5−1.ポリウレタン−ウレア化合物(e)が生じるまでの反応)
ポリウレタン−ウレア化合物(e)が生じるまでの反応としては、下記反応工程式(B)で示すように、ウレタンプレポリマー(c)をジアミン化合物(d)でウレア結合により鎖延長してポリウレタン−ウレア化合物(e)を得る。
【0052】
[反応工程式(B)]
【化6】
[式中、R1、R2、R3、n、mは、前記と同じである。]
【0053】
上記反応は、主に第一の工程中に、溶媒非存在下で起こっているものと推測される。
【0054】
(5−2.イミド変性エラストマー(I)が生じるまでの反応)
イミド変性エラストマー(I)が生じるまでの反応としては、下記反応工程式(C)で示すように、ポリウレタン−ウレア化合物(e)と、テトラカルボン酸二無水物(f)とが反応して、下記式(I)で表されるイミド変性エラストマー(I)が生じる。すなわち、テトラカルボン酸二無水物(f)により、ウレア結合部にイミドユニットを導入してブロック共重合体であるイミド変性エラストマー(I)が得られる。下記式(I)で表されるイミド変性エラストマー(I)のポリマー鎖の左側の末端は、通常、NCO基であり、右側の末端は、通常、NCO基、NH2基、(CO)2O基のいずれかである。
【0055】
[反応工程式(C)]
【化7】
[式中、R1、R2、R3、R4、n、mは、前記と同じである。]
【0056】
反応工程式(C)で示す反応は、ポリウレタン−ウレア化合物(e)とテトラカルボン酸二無水物(f)とのイミド化反応である。
【0057】
前記イミド化反応としては、まず、ポリウレタン−ウレア化合物(e)と、上記で例示したテトラカルボン酸二無水物(f)とが反応して、下記式(g)で表されるポリウレタンアミック酸(PUA)が得られる。上記反応は、主にポリウレタンアミック酸合成工程中に、溶媒非存在下で起こっているものと推測される。
【0058】
【化8】
【0059】
前記イミド化反応後のPUAは、粘土状であり、既に充分な粘度を有しているので、長時間の熱処理をすることなく、そのまま成形することができる。上述したように、単に溶媒を用いずに塊状重合させた従来の場合には、得られるPUAは粘調体であるため、PUAを長時間熱処理して粘土状にしてから成形する必要があった。そのため、本発明の方法によれば、反応工程が短くなり、この点でもコストが低減されることになる。上記反応後のPUAが既に充分な粘度を有している理由については、初期段階での均一混合により、反応の効率が向上したためであると推定される。
【0060】
(5−3.イミド変性エラストマー(I))
本発明のイミド変性エラストマーは、前記一般式(I)で表される。この式中において前記R1としては、例えば、2価の脂肪族炭化水素基、2価の芳香族炭化水素基、並びに、エーテル結合(−O−)、エステル結合(−C(=O)O−)、チオエーテル結合(−S−)、アミド結合(−C(=O)−NH2)、カルボニル基(−C(=O)−)、スルフィニル基(−SO−)、又はスルホニル基(−SO2−)を介して、又は介することなく、これらが2以上結合した炭化水素基等が挙げられる。
【0061】
前記2価の脂肪族炭化水素基、及び前記2価の芳香族炭化水素基は、置換基を有していても良い。前記置換基としては、例えば、アルキル基、アラルキル基、アリール基、アルコキシ基、水酸基、アミノ基、カルボキシル基、ハロゲン原子等が挙げられる。前記アルキル基としては、例えば、メチル基、エチル基、プロピル基等の炭素数1〜6のアルキル基が挙げられる。前記アラルキル基としては、例えば、ベンジル基、フェニルエチル基、フェニルプロピル基等の炭素数6〜12のアラルキル基が挙げられる。前記アリール基としては、例えば、フェニル基、トリル基、キシリル基、ビフェニリル基、ナフチル基、アントリル基、フェナントリル基等の炭素数6〜20のアリール基が挙げられる。前記アルコキシ基としては、例えば、メトキシ基、エトキシ基、プロポキシ基、ブトキシ基等の炭素数1〜6のアルコキシ基が挙げられる。前記ハロゲン原子としては、フッ素原子、塩素原子、臭素原子、ヨウ素原子等が挙げられる。
【0062】
前記2価の脂肪族炭化水素基、及び前記2価の芳香族炭化水素基は、ヘテロ原子を有していても良い。前記ヘテロ原子としては、例えば、窒素原子、酸素原子、イオウ原子等が挙げられる。
【0063】
前記2価の脂肪族炭化水素基としては、例えば、アルキレン基、アルケニレン基、アルキニレン基、アラルキレン基等が挙げられ、脂肪族環を有するものが好ましい。前記2価の脂肪族炭化水素基としては、炭素数1〜30(特に、炭素数2〜20)のものが好ましい。
【0064】
前記2価の芳香族炭化水素基としては、例えば、フェニレン基、ビフェニレン基、ナフチレン基等が挙げられる。前記2価の芳香族炭化水素基としては、炭素数1〜30(特に、炭素数6〜20)のものが好ましい。
【0065】
また、前記R1としては、上述した反応工程式(A)に従ってウレタンプレポリマー(c)を合成する際に用いるジイソシアネート(a)の残基等が挙げられる。中でも、ジイソシアネート(a)のうち、4,4’−ジフェニルメタンジイソシアネート(MDI)又はトリレンジイソシアネートの残基等が好ましい。
【0066】
前記R2は、分子量100〜10,000、好ましくは300〜5,000の2価の有機基を示すものである。該有機基としては、例えば、飽和又は不飽和の直鎖状又は分岐鎖状の多価の脂肪族炭化水素基、多価の脂環式炭化水素基、多価の芳香族炭化水素基からなる群から選択される化学基、又はこれらが2以上結合した炭化水素基等が挙げられる。前記炭化水素基は、ヘテロ原子及び/又は置換基を有していてもよく、ヘテロ原子、置換基として具体的に挙げられるものは、R1の場合と同様である。
【0067】
飽和又は不飽和の直鎖状又は分岐鎖状の多価の脂肪族炭化水素基としては、例えば、アルキレン基、アルケニレン基、アルカントリイル基、アルケントリイル基等が挙げられる。多価の脂環式炭化水素基としては、例えば、1,2−シクロペンチレン基、1,3−シクロペンチレン基、1,2−シクロヘキシレン基、1,3−シクロヘキシレン基、1,4−シクロヘキシレン基等のシクロアルキレン基;シクロヘキシリデン基等のシクロアルキリデン基;ノルボルナン−2,3−ジイル基、ノルボルナン−2,5−ジイル基、ノルボルナン−2,6−ジイル基等の多価の橋かけ環式炭化水素基等が挙げられる。多価の芳香族炭化水素基としては、例えば、1,2−フェニレン基、1,3−フェニレン基、1,4−フェニレン基等のフェニレン基等のアリレン基等が挙げられる。
【0068】
また、前記R2としては、上述した反応工程式(A)に従ってウレタンプレポリマー(c)を合成する際に用いるポリオール(b)の残基等が挙げられる。中でも、ポリオール(b)の残基のうち、ポリオキシテトラメチレングリコール(PTMG)又はポリプロピレングリコール(PPG)の残基等が好ましい。
【0069】
前記R3は、芳香族環、脂肪族環又は脂肪族鎖を含む2価の有機基を示すものである。前記有機基としては、例えば、2価の脂肪族炭化水素基、2価の芳香族炭化水素基、並びに、エーテル結合(−O−)、エステル結合(−C(=O)O−)、チオエーテル結合(−S−)、アミド結合(−C(=O)−NH2)、カルボニル基(−C(=O)−)、スルフィニル基(−SO−)、又はスルホニル基(−SO2−)を介して、又は介することなく、これらが2以上結合した炭化水素基等が挙げられる。前記炭化水素基は、ヘテロ原子及び/又は置換基を有していてもよく、ヘテロ原子、置換基として具体的に挙げられるものは、R1の場合と同様である。
【0070】
前記2価の脂肪族炭化水素基としては、例えば、アルキレン基、アルケニレン基、アルキニレン基、アラルキレン基等が挙げられる。前記2価の脂肪族炭化水素基としては、炭素数1のものも含み、炭素数1〜30のものが好ましいが、炭素数6〜24であることがより好ましい。1,6−ヘキサメチレン基であると、強度に優れるイミド変性エラストマー(I)を得ることができ、特に好ましい。
【0071】
前記2価の芳香族炭化水素基としては、例えば、フェニレン基、ビフェニレン基、ナフチレン基等が挙げられる。前記2価の芳香族炭化水素基としては、炭素数1のものも含み、炭素数1〜30のものが好ましいが、炭素数6〜27であることがより好ましい。
【0072】
前記これらが2以上結合した炭化水素基としては、例えば、メチルフェニレン基、メチルビフェニレン基、ジフェニルエーテル基等が挙げられる。
【0073】
前記R3としては、例えば、上述した反応工程式(B)に従ってポリウレタン−ウレア化合物(e)を合成する際に用いるジアミン化合物(d)の残基等が挙げられる。該残基としては、具体的には、例えば、ジアミン化合物(d)の残基のうち、4,4’−ジアミノジフェニルメタン、1,4−ビス(4−アミノフェノキシ)ベンゼン(TPE−Q)、1,3−ビス(4−アミノフェノキシ)ベンゼン(TPE−R)の残基等が、耐溶剤性に優れるイミド変性エラストマー(I)を得ることができる点で、特に好ましい。
【0074】
前記R4は、4個以上の炭素原子を含む4価の有機基を示すものであり、例えば、4価の脂肪族炭化水素基、及び4価の芳香族炭化水素基等の炭化水素基が挙げられ、4価の芳香族炭化水素基が好ましい。前記炭化水素基は、ヘテロ原子及び/又は置換基を有していてもよく、ヘテロ原子、置換基として具体的に挙げられるものは、R1の場合と同様である。
【0075】
前記R4としては、例えば、上述した反応工程式(C)に従ってイミド変性エラストマー(I)を合成する際に用いるテトラカルボン酸二無水物(f)の残基等が挙げられる。該残基としては、具体的には、例えば、無水ピロメリット酸(PMDA)、オキシジフタル酸二無水物(ODPA)、ベンゾフェノン−3,4,3’,4’−テトラカルボン酸二無水物(BTDA)の残基等が好ましい。
【0076】
前記一般式(I)で表されるイミド変性エラストマー(以下、イミド変性エラストマー(I)とも言う。)の具体例としては、下記式(1)で表されるイミド変性エラストマー等が挙げられる。
【0077】
【化9】
[式中、n、mは、前記と同じである。xは5〜100の整数を示す。]
【0078】
本願発明の製造方法によって得られるイミド変性エラストマー(I)は、ジイソシアネートとポリオールから得た分子両末端にイソシアネート基を有するウレタンプレポリマーをジアミン化合物でウレア結合により鎖延長し、テトラカルボン酸二無水物でウレア結合部にイミドユニットを導入したブロック共重合体であるのが好ましい。
【0079】
本願発明の製造方法によって得られるイミド変性エラストマー(I)は、柔軟な低い弾性率を有するゴム状物で、高強度及び高耐熱性を有するだけでなく、重量平均分子量が高く、明度が高く、耐溶剤性にも優れる。
【0080】
具体的には、本願発明の製造方法によって得られるイミド変性エラストマー(I)の50℃での貯蔵弾性率E’は、5×106〜1×108Paであるのが好ましい。該貯蔵弾性率は、後述するように、動的粘弾性測定装置を用いて測定して得られる値である。このような柔軟な低い弾性率を有するイミド変性エラストマー(I)は、ガラス転移温度(Tg)が、通常−30〜−60℃であり、ゴム状弾性領域の温度範囲が広いものになる。イミド変性エラストマー(I)が、柔軟な低い弾性率を有するゴム状物で、かつ高強度及び高耐熱性を有するようになる理由としては、以下の理由が推察される。すなわち、上記で説明した通り、本発明のイミド変性エラストマー(I)は、主鎖に連続した2つのイミドユニットを、その分布を制御しつつ所望の割合(イミド分率)で導入することができるので、このイミドユニットからなるハードセグメントの凝集が均一かつ強固なものになる。このため、イミド変性エラストマー(I)は、より均一かつ強固なミクロ相分離構造を形成し、ガラス転移温度が低くなることで、ゴム状弾性領域の温度範囲が広くなる。その結果、ポリウレタンをエラストマー成分として含有し、柔軟な低い弾性率を有するゴム状物としても、高強度及び高耐熱性を有するようになる。
【0081】
本願発明の製造方法によって得られるイミド変性エラストマー(I)の重量平均分子量は10,000〜1,000,000、好ましくは50,000〜500,000、より好ましくは80,000〜100,000であるのがよい。これに対し、前記重量平均分子量が10,000より小さいと、耐溶剤性、強度、耐熱性が低下するおそれがあり、1,000,000より大きいと、成形性が低下するおそれがあるので好ましくない。前記重量平均分子量は、ポリウレタンアミック酸合成工程後の重合物をNMPに溶解して、GPC測定することにより測定した値である。
【0082】
上記重量平均分子量が、従来の溶媒を用いて溶液重合を行って得られるイミド変性エラストマー(I)よりも高くなる理由としては、以下の理由が推察される。すなわち、ウレタンプレポリマー(c)をジアミン化合物(d)でウレア結合により鎖延長させる工程後、ウレア結合が切断されることがあり、その切断されたウレア結合が再結合する頻度は、化合物の濃度によって決まると考えられる。この点、溶液重合の場合、比較的濃度が低いため、切断されたウレア結合が再結合する確率は低い。これに対して、本願発明の場合は溶媒を用いないため、濃度が高く、ウレア結合が再結合する確率が高くなり、その結果高い重量平均分子量が保持されると推定される。
【0083】
本願発明の製造方法によって得られるイミド変性エラストマー(I)の明度は70〜95、好ましくは80〜95である。これに対し、前記明度が70より小さいと、カラーリングがしにくくなり用途が限定されるおそれがあるので好ましくない。前記明度は、JIS Z8729に準拠して測定した値である。
【0084】
本願発明の製造方法によって得られるイミド変性エラストマー(I)の耐溶剤性は、従来の溶媒を用いて溶液重合を行って得られるイミド変性エラストマー(I)よりも優れている。前記耐溶剤性は、後述するように、イミド変性エラストマー(1)シートを、所定の溶媒に浸漬し、浸漬前後の重量増加を計量して膨潤率(%)を算出することにより評価される。
【0085】
本発明のイミド変性エラストマーの製造方法によれば、柔軟な低い弾性率を有するゴム状物で、かつ高強度、高耐熱性及び熱可塑性を有すると共に、重量平均分子量が高く、明度が高くカラーリングが容易であり、溶剤に対しても安定なイミド変性エラストマーを製造することができる。前記イミド変性エラストマーは、例えば、ガスケット、シート、ベルト、フィルム、チューブ、ホース、ロールギア、パッキング材、防音材、防振材、ブーツ、被覆材、パーベーパレーション用の分離膜、光学非線形材料、弾性繊維、圧電素子、アクチュエーター、その他の各種自動車部品、工業機械部品、スポーツ用品等に使用することができる。また、反応で得られるイミド変性エラストマー(I)を、プレス成型、ロール練り等に供して、シート状、フィルム状、又は板状に成形することができるため、特にガスケット、シート、ベルト等に有用であるが、これらの用途に限定されるものではない。
【実施例】
【0086】
以下、合成例及び実施例を挙げて本発明のイミド変性エラストマーを詳細に説明するが、本発明は以下の合成例及び実施例のみに限定されるものではない。
【0087】
[合成例]
(ウレタンプレポリマー(j)の合成)
ウレタンプレポリマー(j)を下記反応工程式に基づいて合成した。具体的には、4,4’−ジフェニルメタンジイソシアネート(MDI)(h)[日本ポリウレタン工業社製]を減圧蒸留した。また、ポリオキシテトラメチレングリコール(PTMG)(i)[保土谷化学社製の商品名「PTMG1000」、重量平均分子量:1,000]を80℃、2〜3mmHg、24時間の条件で減圧乾燥した。
【0088】
【化10】
[式中、n、xは、前記と同じである。]
【0089】
次に、PTMG(i)69.54gを、500mlセパラブルフラスコに加え、真空条件下、80℃で1時間脱気した後、さらに、上記MDI(h)30.46gを加え、窒素雰囲気下、80℃で2時間半、180rpmで攪拌して、分子両末端にイソシアネート基を有するウレタンプレポリマー(j)を得た。
【0090】
[実施例1]
上記で得たウレタンプレポリマー(j)から、イミド変性エラストマー(1)を下記反応工程式に基づいて合成した。具体的には、ウレタンプレポリマー(j)100gを高せん断ミキサー(商品名「T.K.ハイビスミックス」、プライミクス社製)に投入し、80℃で撹絆しながら、4,4’−ジアミノジフェニルメタン(MDA)(k)を10.34g投入し、さらに1時間撹絆した。続いて、無水ピロメリット酸(PMDA)(m)を22.75g投入し、さらに1時間撹絆した。生成物を取り出し、モノマー微粒子が目視で確認できない程度(平均粒子径が約5μm)に分散するまで、乳鉢で混合した。混合後、生成物を取り出し、小型密閉式混練機(商品名「ラボプラストミル」、東洋精機製作所社製)に投入し、100℃、50rpmにて混合し、トルクが上昇し極大後、低下して安定となった時点で、混合を停止して、生成物を取り出した。得た生成物を金型に仕込み、180℃で熱盤プレスした後、200℃オーブン中で熱処理を2時間行って、厚さ0.5mmのシート状のイミド変性エラストマー(1)を得た。
【0091】
なお、モノマー微粒子を乳鉢で混合した結果、平均粒子径が約5μmになったことは、顕微鏡(商品名「デジタルマイクロスコープVHX−900」、キーエンス社製)を用いて確認した。その写真を図4に示す。
【0092】
【化11】
[式中、n、m、xは上記と同じ。]
【0093】
[比較例1]
乳鉢での混合を行わなかった結果、モノマーを含む凝集物の平均粒子径が約300μmであった以外は、実施例1と同様の実験を行い、厚さ0.5mmのシート状のイミド変性エラストマー(1)を得た。
【0094】
モノマーを含む凝集物の平均粒子径が約300μmであったことは、顕微鏡(商品名「デジタルマイクロスコープVHX−900」、キーエンス社製)を用いて確認した。その写真を図5に示す。
【0095】
[比較例2]
以下のように溶液重合により反応を行い、同様に厚さ0.5mmのシート状のイミド変性エラストマー(1)を得た。
【0096】
まず、下記反応工程式のように、実施例1と同様の方法で得たウレタンプレポリマー(j)と4,4’−ジアミノジフェニルメタン(MDA)(k)とを反応させて、下記式(l)で表されるポリウレタン−ウレア化合物(l)を得た。具体的には、まず、ウレタンプレポリマー(j)100gを脱水処理したN−メチル−2−ピロリドン(NMP)200mlに溶解させたものと、4,4’−ジアミノジフェニルメタン(MDA)(k)10.34gを脱水処理したNMP50mlに溶解させたものとを、500mlの四つ口セパラブルフラスコにそれぞれ加え、アルゴン雰囲気下、室温(23℃)で24時間攪拌して、ポリウレタン−ウレア化合物(l)の溶液を得た。
【化12】
[式中、n、xは上記と同じ。m’は2〜100の整数を示す。]
【0097】
次に、下記反応工程式のように、上記で得たポリウレタン−ウレア化合物(l)と無水ピロメリット酸(PMDA)(m)とを反応させて、イミド変性エラストマー(1)のシートを得た。具体的には、まず、上記で得たポリウレタン−ウレア化合物(l)の溶液中に、無水ピロメリット酸(PMDA)(m)22.75gを加え、アルゴンガス雰囲気下、150℃で2時間攪拌して、ポリウレタンアミック酸(PUA)溶液を得た。ついで、該PUA溶液を遠心成形機に流し込み、150℃で1,000rpm、1時間遠心成形してPUAシートを得た。このPUAシートを減圧デシケータ内で200℃、2時間加熱処理(脱水縮合反応)して、厚さ0.5mmのシート状のイミド変性エラストマー(1)を得た。
【化13】
[式中、n、m、xは上記と同じ。]
【0098】
[FT−IR試験]
得られたイミド変性エラストマー(1)について、IRスペクトルを測定した。IRスペクトルの測定には、分光計(商品名「FTS−3000」、VALIAN社製)にてATR治具(PIKE社製)を用いた。また、IRスペクトルの測定は、サンプル押し付けによるATR法測定で、積算回数32回で行った。その結果を図1に示す。図1のグラフの横軸は波長数(cm-1)を示し、縦軸は吸光度(Abs.)を示す。
【0099】
図1に示した結果から、実施例1及び比較例2のいずれの場合でも、1780cm-1、1720cm-1及び1380cm-1にイミド環に由来する吸収が観察され、これらのイミド変性エラストマー(1)シートは同一の化学的構造を有することが示された。
【0100】
[引張り試験]
得られたイミド変性エラストマー(1)シートを3号ダンベルで打ち抜き、標線間20mm、引張り速度500mm/分の条件で、JIS K6251に準拠し、オートグラフ(商品名「AGS−G」、島津製作所社製)を用いて、応力、伸びをそれぞれ測定した。その結果を図2に示す。図2のグラフにおいて、縦軸は応力(MPa)、横軸は伸び(%)を示す。
【0101】
図2に示した結果から、実施例1及び比較例2(溶液重合)のいずれの場合でも、イミド変性エラストマー(1)シートの応力や柔軟性には差は無いことが示された。
【0102】
[動的粘弾性試験]
得られたイミド変性エラストマー(1)シートについて、動的粘弾性試験を行った。動的粘弾性試験は、動的粘弾性測定装置(商品名「DMS 6100」、セイコーインスツルメンツ社製)を用い、20Hz、5℃/分、−100〜400℃の昇温過程にて行い、貯蔵弾性率(E’)、損失正接(tanδ)を測定した。その結果を図3に示す。同グラフにおいて、左側の縦軸は貯蔵弾性率E’(Pa)、左側の縦軸は損失正接tanδを示し、横軸は温度(℃)を示す。
【0103】
図3に示した結果から、実施例1及び比較例2(溶液重合)のいずれの場合でも、イミド変性エラストマー(1)シートの、温度に対する動的粘弾性の挙動に差は無いことが示された。
【0104】
[イミド変性エラストマー(1)シートの明度評価]
実施例1で得られたイミド変性エラストマー(1)シートは、薄い黄色を呈しているのに対して、比較例2で得られたイミド変性エラストマー(1)シートは、こげ茶色を呈しており、目視での差異は明らかであった。このような明度の差異を客観的に評価するため、イミド変性エラストマー(1)シートの明度をJIS Z8729に準拠して測定し、結果を下記表1の「明度」の欄に示した。
【0105】
表1に示した結果から、実施例1で得られたイミド変性エラストマー(1)シートは、優れた明度を有することが客観的に示された。
【0106】
[イミド変性エラストマー(1)シートの耐溶剤性評価]
イミド変性エラストマー(1)シートを、室温(23℃)で96時間、N−メチル−2−ピロリドン(NMP)に浸漬し、イミド変性エラストマー(1)シートの浸漬前後の重量増加を計量した。より具体的には、イミド変性エラストマー(1)シートから幅2.0cm、長さ3.0cmの試験片を切り出し、この試験片を前記条件で溶剤に浸漬し、浸漬前後の重量を下記式(β)に当てはめて膨潤率(%)を算出し、耐溶剤性の指標とした。
膨潤率(%)=[(S2−S1)/S1]×100 ・・・(β)
S1:浸漬前の試験片の重量
S2:浸漬後の試験片の重量
【0107】
表1に示した結果から、実施例1で得られたイミド変性エラストマー(1)シートは、優れた耐溶剤性を有することが示された。
【0108】
[イミド変性エラストマー(1)シートの重量平均分子量評価]
イミド変性エラストマー(1)シートの重量平均分子量を測定し、結果を下記表1の「分子量(Mw)」の欄に示した。
【0109】
表1に示した結果から、実施例1で得られたイミド変性エラストマー(1)シートは、重量平均分子量が高いことが示された。
【0110】
【表1】
【産業上の利用可能性】
【0111】
本発明のイミド変性エラストマーの製造方法によれば、溶媒を使用せずに済むため製造時の生産効率が高くなり、溶媒を除去する必要も無いためコストが低減でき、さらに、着色が低減され、耐溶剤性が向上したイミド変性エラストマーが得られるため、特に、ガスケット等のゴム成形品の製造方法として有用である。
【特許請求の範囲】
【請求項1】
下記式(c)で表されるウレタンプレポリマー(c)と、下記式(d)で表されるジアミン化合物(d)と、下記式(f)で表されるテトラカルボン酸二無水物(f)とを、溶媒非存在下で加熱下に混合して重合させる第一の工程、及び
ジアミン化合物(d)及びテトラカルボン酸二無水物(f)の固体を粉砕して、平均粒子径が10μm以下の微粒子として、前記微粒子を含む混合物を生成する第二の工程、
を含むポリウレタンアミック酸合成工程、並びに
前記ポリウレタンアミック酸合成工程で得られたポリウレタンアミック酸を加熱処理することにより閉環反応を起こさせるポリウレタンアミック酸閉環工程を含み、
下記式(I)で表されるイミド変性エラストマー(I)を得ることを特徴とするイミド変性エラストマーの製造方法。
【化1】
[式中、R1は、芳香族環又は脂肪族環を含む2価の有機基を示す。R2は、分子量100〜10,000の2価の有機基を示す。R3は、芳香族環、脂肪族環又は脂肪族鎖を含む2価の有機基を示す。R4は、4個以上の炭素原子を含む4価の有機基を示す。nは1〜100の整数を示す。]
【化2】
[式中、R1、R2、R3、R4、nは上記と同じ。mは5〜200の整数を示す。]
【請求項2】
前記ポリウレタンアミック酸合成工程において、第二の工程を第一の工程後に行い、且つ、
前記ポリウレタンアミック酸合成工程が、前記第二の工程の後に、前記微粒子を含む混合物をさらに加熱下に混合して重合させる第三の工程をさらに含む、請求項1記載のイミド変性エラストマーの製造方法。
【請求項3】
さらに、下記の反応工程式(A)に従い、下記式(a)で表されるジイソシアネート(a)と下記式(b)で表されるポリオール(b)とを混合することにより反応させて、下記式(c)で表されるウレタンプレポリマー(c)を得るウレタンプレポリマー合成工程(A)を含み、
ウレタンプレポリマー合成工程(A)によって得られたウレタンプレポリマー(c)を前記第一の工程に用いることを特徴とする請求項1又は2記載のイミド変性エラストマーの製造方法。
【化3】
[式中、R1は、芳香族環又は脂肪族環を含む2価の有機基を示す。R2は、分子量100〜10,000の2価の有機基を示す。nは1〜100の整数を示す。]
【請求項1】
下記式(c)で表されるウレタンプレポリマー(c)と、下記式(d)で表されるジアミン化合物(d)と、下記式(f)で表されるテトラカルボン酸二無水物(f)とを、溶媒非存在下で加熱下に混合して重合させる第一の工程、及び
ジアミン化合物(d)及びテトラカルボン酸二無水物(f)の固体を粉砕して、平均粒子径が10μm以下の微粒子として、前記微粒子を含む混合物を生成する第二の工程、
を含むポリウレタンアミック酸合成工程、並びに
前記ポリウレタンアミック酸合成工程で得られたポリウレタンアミック酸を加熱処理することにより閉環反応を起こさせるポリウレタンアミック酸閉環工程を含み、
下記式(I)で表されるイミド変性エラストマー(I)を得ることを特徴とするイミド変性エラストマーの製造方法。
【化1】
[式中、R1は、芳香族環又は脂肪族環を含む2価の有機基を示す。R2は、分子量100〜10,000の2価の有機基を示す。R3は、芳香族環、脂肪族環又は脂肪族鎖を含む2価の有機基を示す。R4は、4個以上の炭素原子を含む4価の有機基を示す。nは1〜100の整数を示す。]
【化2】
[式中、R1、R2、R3、R4、nは上記と同じ。mは5〜200の整数を示す。]
【請求項2】
前記ポリウレタンアミック酸合成工程において、第二の工程を第一の工程後に行い、且つ、
前記ポリウレタンアミック酸合成工程が、前記第二の工程の後に、前記微粒子を含む混合物をさらに加熱下に混合して重合させる第三の工程をさらに含む、請求項1記載のイミド変性エラストマーの製造方法。
【請求項3】
さらに、下記の反応工程式(A)に従い、下記式(a)で表されるジイソシアネート(a)と下記式(b)で表されるポリオール(b)とを混合することにより反応させて、下記式(c)で表されるウレタンプレポリマー(c)を得るウレタンプレポリマー合成工程(A)を含み、
ウレタンプレポリマー合成工程(A)によって得られたウレタンプレポリマー(c)を前記第一の工程に用いることを特徴とする請求項1又は2記載のイミド変性エラストマーの製造方法。
【化3】
[式中、R1は、芳香族環又は脂肪族環を含む2価の有機基を示す。R2は、分子量100〜10,000の2価の有機基を示す。nは1〜100の整数を示す。]
【図1】

【図2】
【図3】
【図4】

【図5】

【図2】
【図3】
【図4】
【図5】
【公開番号】特開2013−57019(P2013−57019A)
【公開日】平成25年3月28日(2013.3.28)
【国際特許分類】
【出願番号】特願2011−196556(P2011−196556)
【出願日】平成23年9月8日(2011.9.8)
【出願人】(303062093)
【出願人】(000111085)ニッタ株式会社 (588)
【Fターム(参考)】
【公開日】平成25年3月28日(2013.3.28)
【国際特許分類】
【出願日】平成23年9月8日(2011.9.8)
【出願人】(303062093)
【出願人】(000111085)ニッタ株式会社 (588)
【Fターム(参考)】
[ Back to top ]