
インテグリン結合小分子
本発明は、インテグリン結合小分子を含む組成物に関する。また、その小分子に対してインテグリンを結合する方法およびインテグリンに対して結合する小分子を同定する方法を開示する。
【発明の詳細な説明】
【発明の開示】
【0001】
関連出願
本出願は、2005年11月14日に提出された米国仮出願第60/736,780の優先権を主張するものであり、その内容は、その全体において参照することにより本明細書において組み込まれる。
【0002】
発明の分野
本発明は、一般的にインテグリン結合小分子に関する。更に詳しくは、本発明は、新規組成物およびこれらの組成物を種々の疾患の治療のために使用する方法を提供する。
【0003】
発明の背景
インテグリン、即ち、膜貫通接着受容体のファミリーは、細胞付着、遊走、分化および生存の主な媒介物質である1。構造的に、インテグリンは、各サブユニットとして大きな細胞外ドメイン、1つの膜貫通へリックス、および小さな細胞内ドメインで構成されるヘテロ二量体である2。これらの受容体は、α-及びβ-サブユニットからなり、これらは非共有結合的に示された組合せで結合する。現在までのところ、18 α-サブユニットおよび8 βサブユニットが確認されており、これらは結合し、選択的に少なくとも24のインテグリンを形成する。それらの接着性機能に加えて、インテグリンは、多様な情報伝達経路を介してメッセージを伝達し、並びに腫瘍細胞および活性化された内皮細胞の増殖およびアポトーシスに影響を与える3,4。細胞表面におけるインテグリンの独特の組合せは細胞が種々の細胞外リガンドを認識し、その後にそれに対して応答することを可能にする。インテグリンαvβ3はインテグリンファミリーの中でも突出したものである。それは、悪性腫瘍の病理生理学的において関連付けられており、腫瘍血管形成5のために必要であり、新血管における内皮細胞および攻撃性の高いヒト癌腫において発現が高い。インテグリン αvβ3は、種々の細胞外マトリックスタンパク質における腫瘍細胞の接着を媒介し、これらの細胞が浸潤中の遊走および血管外遊走を可能にする6,7。乳癌において、αvβ3は、このインテグリンが浸潤性腫瘍および遠位転移において上方制御されるように、転移性表現形を特徴づける8−10。従って、インテグリン αvβ3のアンタゴニストは、転移性および浸潤性の癌の治療のための新しいアプローチを提供すると期待される11,12。αvβ3アンタゴニストと従来の治療様式との組合せは、余分な毒性を伴わずに、転移性癌治療の有効性を増大可能であった。当該αvβ3受容体は、フィブリノーゲン、フィブロネクチン、オステオポンチン、スロンボスポンジンおよびビトロネクチンを含む種々の細胞外マトリックスタンパク質に対して、大部分は、当該Arg-Gly-Asp(RGD)トリペプチド配列との相互作用を介して結合する13、14。以前に、種々のペプチドミメチックス小分子αvβ3アンタゴニストが同定されており、その幾つかは、疾患モデル、例えば、骨粗鬆症および骨格転移性乳癌において活性である12、15−18。
【0004】
当該αvβ3 アンタゴニストは、マウス異種移植モデルおよび転移モデルを含む多くの動物モデルにおいて強力に血管形成を抑制する。mABsおよびサイクリックRGDペプチドによるαvβ3活性の阻害が、内皮のアポトーシスを誘導し、血管形成を阻害することが示されている19、20。当該αvβ3 アンタゴニストは、活性化された内皮細胞ばかりではなく、αvβ3陽性腫瘍細胞においてもアポトーシスを誘導することができ、腫瘍細胞における直接的な細胞毒性効果を結果として引き起こす21。αvβ3活性のアンタゴニストは、乳癌細胞異種移植およびメラノーマ異種移植における腫瘍増殖を結果的に減少する22,23。転移性癌のための臨床試験におけるシレンジタイド(Cilengitide)、サイクリックRGDペプチドが24、攻撃的な乳癌モデルにおいて試験されており、そこにおいて、シレンジタイドと放射免疫療法との併用が、何れかの薬剤での単一様式療法に比べて、更なる特性を伴わずに、著しく有効性が増強され、且つアポトーシスを増大しすることが示された25。これは、併用抗癌療法において、シレンジタイドおよび一般的なαvβ3アンタゴニストの実際の治療学的なポテンシャルを明確に示唆するものである。
【0005】
当該αvβ3受容体はまた、骨再吸収において中心的な役割を果たす。種々の試験は、αvβ3受容体が破骨細胞における最も豊富なインテグリンであることを示している26-29。αvβ3抗体、RGDペプチドおよびペプチドミメティックアンタゴニストは、著しい有害作用を伴わずにインビボにおける骨再吸収の阻害を示した30-34。これらの試験および初期の臨床的試験からの結果の基礎に、αvβ3アンタゴニストは骨粗鬆症の治療および予防のために大きな見込みを示すものである。
【0006】
発明の概要
本発明は、少なくとも一部分において、以下に記載の小分子化合物がインテグリンに結合し、それにより、種々の疾患および状態を診断および治療するために使用できるという予期しない発見に基づく。
【0007】
従って、1側面において、本発明は、化合物または薬学的若しくは薬用化粧品学的に許容されるその塩、その溶媒和物またはその水和物を含む組成物が特徴であり、ここで、当該化合物は、1のH結合供与体(HBD)、1のH結合受容体(HBA)、2の疎水性芳香族基(HAR1およびHAR2)および1の陰性イオン性基(NI)を含み、およびここで、当該化合物は化合物 A、BまたはCではない;
【化4】

【0008】
。
【0009】
当該組成物は、薬学的または薬用化粧品学的に許容された担体を更に含んでもよい。
【0010】
一つの態様において、当該HBD、HBA、HAR1、HAR2およびNIは、図8Aに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.81±1 Åでり、当該HBDとHBAの距離は7.52±1 Åであり、当該HBDとNIの距離は17.67±1 Åであり、当該HRA1とHBAの距離は7.52±1 Åであり、当該HRA1とHRA2の距離は9.41±1 Åであり、当該HBAとNIの距離は 10.20±1 Åであり、および当該HRA2とNIの距離は5.88±1 Åである。特に、当該HBDとHRA1の距離は2.81 Åであってよく、当該HBDとHBAとの距離は7.52 Åであってよく、当該HBDとNIの距離は17.67 Åであってよく、当該HRA1とHBAの距離は7.52 Åであってよく、当該HRA1とHRA2の距離は9.41 Åであってよく、当該HBAとNIの距離は10.20 Åであってよくおよび当該HRA2とNIの距離は5.88 Åであってよい。
【0011】
他の態様において、当該HBD、HBA、HAR1、HAR2,およびNIは、図8Bに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.80±1 Åであり、当該HBDとHBAの距離は7.34±1 Åであり、当該HBDとNIの距離は16.92±1 Åであり、当該HRA1とHBAの距離は5.20±1 Åであり、当該HRA1とNI の距離は14.34±1 Åであり、当該HBAとHRA2の距離は4.69±1 Åであり、当該HBAとNIの距離は9.85±1 Åであり、および当該HRA2とNI の距離は5.66±1 Åである。特に、当該HBDとHRA1の距離は2.80 Åでよく、当該HBDとHBAの距離は7.34 Åでよく、当該HBDとNI の距離は16.92 Åでよく、当該HRA1とHBAの距離は5.20 Åでよく、当該HRA1とNIの距離は14.34 Åでよく、当該HBAとHRA2の距離は4.69 Åでよく、当該HBAとNIの距離は9.85 Åでよく、および当該HRA2とNIの距離は5.66 Åでよい。
【0012】
もう一つの態様において、当該HBD、HBA、HAR1、HAR2およびNIは、図8Cに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.79±1 Åであり、当該HBDとHRA2の距離は11.94±1 Åであり、当該HBDとNI の距離は16.28±1 Åであり、当該HRA1とHBAの距離は5.20±1 Åであり、当該HRA1とNIの距離は14.64±1 Åであり、当該HBAとHRA2の距離は4.72±1 Åであり、および当該HRA2とNIの距離は5.63±1 Åである。特に、当該HBDとHRA1の距離は2.79 Åでよく、当該HBDとHRA2の距離は11.94 Åでよく、当該HBDとNIの距離は16.28 Åでよく、当該HRA1とHBAの距離は5.20 Åでよく、当該HRA1とNI の距離は14.64 Åでよく、当該HBAとHRA2の距離は4.72 Åでよく、および当該HRA2とNI の距離は5.63 Åでよい。
【0013】
例えば、本発明の化合物は、式 I、II、III,またはIV:
【化5】
【0014】
でよい。
【0015】
式I、IIおよびIIIにおいて、それぞれ、R1およびR3 は、脂肪族、芳香族またはヘテロ環基; R2 は置換基;およびX はN、OまたはSである。式 IVにおいて、それぞれ、R1およびR2 は水素またはハロゲン、ヒドロキシル、スルフヒドリル、アルコキシ、アリールオキシ、カルボキシル、ニトロ、シアノ、アミノ、アミドまたはスルホニル基または何れかの数の炭素原子を含む何れか他の有機官能基; R3 は水素、ヒドロキシル、アルコキシ、アリールオキシ、芳香族またはヘテロ環基または何れかの数の炭素原子の脂肪族鎖;それぞれ、R4およびR5 はヒドロキシルまたはアミノ基; X は C、N、OまたはSであり; m は炭素、窒素、酸素または硫黄原子を伴う 0 から 3 原子リンカー基;およびn は炭素、窒素、酸素または硫黄原子を伴う0 から 5 原子リンカー基である。
【0016】
特に、式I、IIおよびIIIにおいて、それぞれ、R1およびR3 はアルキル、置換されたアルキル、アルケニル、置換されたアルケニル、アルキニル、置換されたアルキニル、フェニル、置換されたフェニル、アリール、置換されたアリール、ヘテロアリールまたは置換されたヘテロアリール基であってよく; R2 は、水素またはハロゲン、ヒドロキシル、スルフヒドリル、アルコキシ、カルボキシル、ニトロ、シアノ、アミノ、アミドまたはスルホニル基または何れかの数の炭素原子を含む何れか他の有機官能基またはそれらの組合せであってよい。R1またはR3における、当該置換されたアルキル、アルケニル、アルキニル、フェニル、アリールまたはヘテロアリール基は、ハロ、ヒドロキシル、スルフヒドリル、アルコキシ、カルボキシル、カルボニル、ニトロ、シアノ、アミノ、アミド、スルホニル、または置換されたヘテロ環基により置換されたアルキル、アルケニル、アルキニル、フェニル、アリール、またはヘテロアリール基であってよい。式 IVにおいて、R4またはR5における当該アミノ基は、脂肪族、芳香族またはヘテロ環基で置換されてよい。本発明の化合物の例は、AV1-11、AV14-15、AV17-32およびAV34-38を含む。
【0017】
もう一つの側面において、本発明は、第一の化合物(即ち、上で記載された通りの本発明の化合物)、またはその薬学的若しくは薬用化粧品学的に許容される塩、溶媒和物または水和物と、1または1以上の第二の化合物とを含む、またはからなる組成物を特徴とし、ここで第二の化合物は治療剤、イメージング財またはその組合せである。
【0018】
例えば、当該第二の化合物は、血管増殖阻害剤(例えば、TNF-アルファ、アンジオスタチン、ベバシザン(Bevacizumab)、アレステン(Arresten)、カンスタチン(Canstatin)、コンブレタスタチン、エンドスタチン、NM-3、サリドマイド、スロンボスポンジン(Thrombospondin)、タムスタチン(Tumstatin)、2-メトキシエストラジオールおよびビタキシン(Vitaxin))、関節リウマチを治療するための治療剤、放射線治療剤、化学療法剤(例えば、シスプラチン、ドキソルビシン、ビンクリスチン、シクロホスファミド、トポテカン、パクリタキセル,および他の化学療法剤)、骨転移を治療するための治療剤骨転移を治療するための治療剤、骨粗鬆症を治療するための治療剤(例えば、ホセマックス(Fosemax)、アレジア(Aredia)、アクトネル(Actonel)、ジンドロネル(Dindronel)、コメタ(Cometa)、プレマリン(Premarin)、クリマラ(Climara)、エストラ(Estrae)、ビベラ(Vivelle)、エストラダーム(Estraderm)、プレモプロ(Prempro)、プラムフェーズ(Premphase)、フェマート(Femhrt)、プレフェスト(Prefest)、コンビパッチ(Combipatch)、エビスタ(Evista)、カルシトニン(Calcitonin)、ミアカルシン(Miacalcin)およびカルシトニン)、再狭窄を治療するための治療剤、血管イメージング剤(例えば、放射性核種)またはその組み合わせであってよい。
【0019】
幾つかの態様において、当該第一の化合物は、実施可能に少なくとも1の当該第二の化合物に実施可能に連結されている。
【0020】
本発明は、更に、当該発明は、インテグリンと本発明の化合物またはその薬学的若しくは薬用化粧品学的に許容される塩、溶媒和物または水和物とを接触させることを具備する、インテグリンを化合物に対して結合する方法を提供する。
【0021】
一つの態様において、当該インテグリンはαvβ3である。もう一つの態様において、当該インテグリンは細胞上に、例えば、癌細胞、良性腫瘍細胞、間質細胞、炎症に反応する、炎症を媒介する、または炎症を調節する細胞(例えば、白血球、マクロファージおよび樹状細胞)、免疫系の細胞(例えば、白血球、マクロファージおよび樹状細胞)、血管機能に関与する細胞(例えば、内皮細胞、内皮細胞に関与する細胞および平滑筋細胞)、骨芽細胞、破骨細胞またはウイルス(例えば、アデノウイルス(アデノビリダエ;Adenoviridae)、コックサッキーウイルス(Coxackievirus、Picornaviridae)、エコウイルス(Echovirus、Picornaviridae)、口蹄疫ウイルス(Picornaviridae)、ハンタウイルス(Buriyaviridae)、ヒトパレコウイルス(Picornaviridae)、ヒト免疫不全ウイルス 1(Retroviridae)およびロタウイルス(Reoviridae))に感染した細胞上にある。
【0022】
当該方法は、更に、当該細胞と1または1以上の治療剤、イメージング剤またはその組合せとを接触させることを具備する。当該治療剤およびイメージング剤は、上で記載した。当該インテグリンおよび当該細胞は、同時にまたは経時的に接触してよい。当該化合物は、少なくとも1の薬剤と混合されてもよく、または実施可能に連結されてもよい。
【0023】
また、本発明において、本発明は、対象においてインテグリンを化合物に対して結合する方法であって、それを必要とする対象において本発明の化合物またはその薬学的若しくは薬用化粧品学的に許容される塩、溶媒和物または水和物の有効量を投与することを具備する方法である。当該インテグリンはαvβ3であってよく、および当該対象は、動物またはヒトであってよい。
【0024】
一つの態様において、当該対象は、癌、例えば、悪性固形腫瘍の組織学的種類、例えば、肉腫または癌腫または高いレベルの血管形成および血管原性因子に関連する血液学的悪性度、例えば、急性または慢性白血病、多発性骨髄腫、骨髄増殖性疾患、非-ホジキンリンパ腫またはホジキン病、に罹患する、またはそれらの発展のリスクに面している。
【0025】
もう一つの態様において、当該対象は、良性腫瘍、例えば、良性腫瘍または高い血管新生に関連する新生物発生前の状態、尋常性疣贅、化膿性 肉芽腫、酒さ、ケロイド瘢痕,または脈絡叢に関連する眼部腫瘍または虹彩新血管新生に罹患している、またはそれらの発展のリスクに面している。
【0026】
更なるもう一つの態様において、当該対象は、炎症性疾患、例えば、炎症性腸疾患、例えば、クローン病または潰瘍性大腸炎;特発性炎症性ミオパチーなどの多発性筋炎(PM)、皮膚筋炎(DM)または増大した血管新生に関連する状態;他の眼部炎症性疾患;増大した炎症に関連する疾患または状態、例えば、歯肉炎または粘膜炎;ざ瘡;または特に慢性的な炎症状態に関連する胃腸管疾患、例えば、胃炎または十二指腸炎など、に罹患している、またはそれらの発展のリスクに面している。粘膜炎の例は、放射線または化学療法誘導粘膜炎である。
【0027】
更なるもう一つの態様において、当該対象は、免疫または自己免疫疾患、例えば、関節リウマチ; 乾癬性関節炎;乾癬;多発性硬化症;気管支喘息またはアレルギー;湿疹または皮膚炎;または他の皮膚または皮下組織の炎症性状態、例えば、紅斑性扁平皮膚症、斑不全角化、脂漏性皮膚炎、頭部脂漏症、他の脂漏性皮膚炎、アトピー性皮膚炎または関連する状態、紅斑、ベニエ痒疹、神経皮膚炎、接触性皮膚炎または他の湿疹、水疱性皮膚疾患、紅斑性状態、苔癬、掻痒症または関連する状態、疱疹状皮膚炎、疱疹状皮膚症、デューリング病、疱疹状皮膚炎、角層下膿疱症、スネッドン-ウィルキンソン病、若年性疱疹状皮膚炎、若年性天疱瘡、疱疹状膿痂疹、多巣性紅斑、紅斑虹彩、疱疹虹彩、ライル症候群、エリトマトーデス、他の特定の紅斑性状態、苔癬化またはハイド病に罹患している、またはそれらの発展の危険に面している。当該接触性皮膚炎および他の湿疹の例は、内部に取り込まれた物質のための皮膚炎、皮膚との接触における化学薬品、薬物および医薬品のための皮膚炎、日射のための皮膚炎、日焼け、ベルロック皮膚炎、光アレルギー反応皮膚炎、光毒性反応皮膚炎、多形性 光性発疹、播種性 浅在性 光線性 汗孔角化症(DSAP)、化粧品のための皮膚炎、赤外線のための皮膚炎、紫外線のための皮膚炎および電離放射線による治療のための皮膚炎を含む。当該他の特定の紅斑性状態は、リッター病、新生児剥脱性皮膚炎および紅斑間擦疹を含む。
【0028】
更なるもう一つの態様において、当該対象は、血管性疾患、例えば、子宮内膜症;機能性障害性の子宮性出血;子宮内膜過形成;骨髄線維症; 血管腫;動脈硬化症;急性または慢性移植拒絶反応または移植脈管障害;眼部新血管新生により特徴付けられる眼部疾患、例えば、加齢関連横斑性変性(AMD)、増殖性糖尿病網膜症(PDR)、未熟児網膜症(ROP)、ネオ血管緑内障、網膜芽細胞腫、水晶体後線維増殖症、ルベオーシス、ブドウ膜炎、横斑性変性または角膜移植新血管新生;不適当な血小板活性化、補充、凝集または血栓症により媒介される状態、例えば、冠動脈疾患または障害、心筋梗塞または心筋梗塞に続く障害、脳卒中、不安定狭心症、子癇前症、塞栓症、血小板関連虚血性疾患、経皮的冠動脈インターベンションに続く再狭窄、血栓性疾患、血栓症に続く再閉塞、深在性静脈血栓症(DVT)、肺塞栓症(PE)、一過性虚血性発作(transient ischemic attack;TIA)または血管閉塞が共通する基本をなす特徴である他の状態;心筋の血管新生;出血性関節;血管癒着;敗血症;成人呼吸困難症候群;末梢血管拡張;または創傷肉芽形成に罹患しており、またはそれらの発展するリスクに面している。当該血小板関連虚血性疾患の例は、肺虚血、冠血管虚血および脳虚血を含む。当該経皮的冠動脈インターベンションの例は、血管形成術、アテレクトミー、ステント設置およびバイパス手術を含む。当該血栓性疾患の例は、冠動脈血栓症、脳動脈血栓症、心臓内血栓症、末梢動脈血栓症、静脈血栓症および血栓症および外来性に対する露出または損傷された組織表面に関連する凝固障害を含む。
【0029】
更なるもう一つの態様において、当該対象は、または病理学的骨再吸収に主に関連する疾患、例えば、骨粗鬆症、骨のパジェット病または副甲状腺ホルモン(PTHrP)媒介高カルシウム血症に罹患している、またはそれらの発展のリスクに面している。
【0030】
更なるもう一つの態様において、当該対象は、ウイルス感染、例えば、an infection by アデノウイルス(Adenoviridae)、コックサッキーウイルス(Picornaviridae)、エコウイルス(Picornaviridae)、口蹄疫ウイルス(Picornaviridae)、ハンタウイルス(Buriyaviridae)、ヒトパレコウイルス(Picornaviridae)、ヒト免疫不全ウイルス 1(Retroviridae),またはロタウイルス(Reoviridae)による感染に罹患している、またはそれらの発展のリスクに面している。
【0031】
当該方法は、更に、当該対象に対して、有効量の1または1以上の治療剤、イメージング剤またはその組合せを投与することを具備してよい。当該治療剤およびイメージング剤は、上で記載した通りである。当該化合物および当該薬剤は、同時または経時的に投与されてよい。当該化合物は、少なくとも1の薬剤と混合されてもよく、実施可能に連結されてもよい。
【0032】
化合物、治療剤、イメージング剤またはその組合せは、非経口、皮内、皮下、経口、経皮、経粘膜,または直腸に投与されてよい。
【0033】
幾つかの態様において、当該対象は、血管形成方法(例えば、バルーン血管形成術;レーザー血管形成術;冠血管アテレクトミーまたは同様の技術;頚動脈動脈膜切除;血管グラフトの吻合;血栓形成の高リスクを有する外科手術、例えば、冠血管バイパス手術および補綴バルブまたは血管の挿入;アテレクトミー;ステント設置;慢性心血管装置の設置、例えば、インドルエリングカテーテル、補綴バルブまたは血管; 臓器移植;およびバイパス手術)、放射線治療または化学療法(例えば、有効量のシスプラチン、ドキソルビシン、ビンクリスチン、シクロホスファミド、トポテカン、パクリタキセルまたは他の化学療法剤を当該対象に投与することを含む化学療法)で処置されてもよい。
【0034】
本発明は、更に、ファルマコフォアの提示を具備するコンピュータ読み取り可能媒体であって、ここで、当該ファルマコフォアは1の H結合供与体(HBD)、1のH結合受容体(HBA)、2の疎水性芳香族基s(HAR1およびHAR2)および1の陰性イオン性基(NI)の特徴を含むコンピュータ読み取り可能媒体を提供する。
【0035】
一つの態様において、当該HBD、HBA、HAR1、HAR2およびNIは、図8Aに従う立体配置であり、ここで、当該HBDとHRA1の距離が2.81±1 Åであり、当該HBDとHBAの距離が7.52±1 Åであり、当該HBDとNIの距離が17.67±1 Åであり、当該HRA1とHBAの距離が7.52±1 Åであり、当該HRA1とHRA2の距離が9.41±1 Åであり、当該HBAとNIの距離が10.20±1 Åであり、および当該HRA2とNIの距離が5.88±1 Åである。特に、当該HBDとHRA1の距離は2.81 Åでよく、当該HBDとHBA の距離は7.52 Åでよく、当該HBDとNIの距離は17.67 Åでよく、当該HRA1とHBAの距離は7.52 Åでよく、当該HRA1とHRA2 の距離は9.41 Åでよく、当該HBAとNI の距離は10.20 Åでよく、および当該HRA2とNI の距離は5.88 Åでよい。
【0036】
もう一つの態様において、当該HBD、HBA、HAR1、HAR2およびNIは、図8Bに従う立体配置であり、ここで、当該HBDとHRA1の距離は 2.80±1 Åであり、当該HBDとHBAの距離は7.34±1 Åであり、当該HBDとNIの距離は16.92±1 Åであり、当該HRA1とHBAの距離は5.20±1 Åであり、当該HRA1とNI の距離は14.34±1 Åであり、当該HBAとHRA2の距離は4.69±1 Åであり、当該HBAとNI の距離は9.85±1 Åであり、および当該HRA2とNI の距離は5.66±1 Åである。特に、当該HBDとHRA1の距離は2.80 Åでよく、当該HBDとHBAの距離は7.34 Åでよく、当該HBDとNIの距離は16.92 Åでよく、当該HRA1とHBAの距離は5.20 Åでよく、当該HRA1とNIの距離は14.34 Åでよく、当該HBAとHRA2の距離は4.69 Åでよく、当該HBAとNIの距離は9.85 Åでよく、および当該HRA2とNIの距離は5.66 Åでよい。
【0037】
更なるもう一つの態様において、当該HBD、HBA、HAR1、HAR2およびNIは、図8Cに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.79±1 Åであり、当該HBDとHRA2の距離は11.94±1 Åであり、当該HBDとNIの距離は16.28±1 Åであり、当該HRA1と
HBAの距離は5.20±1 Åであり、当該HRA1とNIの距離は14.64±1 Åであり、当該HBAとHRA2の距離は4.72±1 Åであり、および当該HRA2とNIの距離は5.63±1 Åである。特に、当該HBDとHRA1の距離は2.79 Åでよく、当該HBDとHRA2の距離は11.94 Åでよく、当該HBDとNIの距離は16.28 Åでよく、当該HRA1とHBAの距離は5.20 Åでよく、当該HRA1とNI の距離は14.64 Åでよく、当該HBAとHRA2の距離は4.72 Åでよく、および当該HRA2とNIの距離は5.63 Åでよい。
【0038】
本発明のもう一つの特徴は、インテグリン結合小分子を同定する方法である。当該方法は、上述のファルマコフォアの三次元構造と化合物の三次元構造を比較すること、当該化合物が、当該ファルマコフォアの特徴と一致する場合に、当該化合物を選択することを具備する。
【0039】
以下の詳細な説明、添付の図面との組合せを参照することにより、上述および本発明の他の特徴およびそれらを得るおよび使用する方法がより明確になるであろうし、最も浴理解されるであろう。当該図面は、本発明の典型的な態様を示すものであり、その範囲を制限するためのものではない。
【0040】
発明の詳細な説明
インテグリン αvβ3は、腫瘍の進行、転移および破骨細胞骨再吸収の複数の面において関連している。多くの腫瘍は、αvβ3を高発現しており、この発現は、メラノーマ、神経膠腫、卵巣癌、前立腺癌、乳癌、並びに他の癌における腫瘍の進行に関連している。当該αvβ3受容体は、新規抗癌剤のための治療上の標的として評価されている。我々は、化学官能基を基礎にした共通の特徴のファルマコフォアモデル(ケミカル・ファンクション・ベース・コモン・フューチャー・ファルマコフォア・モデル)を使用して構造的に多用な小分子αvβ3アンタゴニストのシリーズを発見した。当該三次元(3D)ファルマコフォアモデルは、3つの最近報告されたαvβ3受容体アンタゴニストの訓練用セットを使用して作成した。αvβ3受容体の公知のアンタゴニストのデータベースを使用したバリデーションにおいて、高判別能を有する当該ファルマコフォアモデルが、新規の構造骨格および所望の化学的特徴を有する検索する化合物に対して3D照会として使用された。Hypo5を使用する約600,000化合物のデータベースの検索により832化合物を得た。構造的新規性、計算された物理化学的な性質およびサンプルの有用性を基礎にして、38化合物が選択されαvβ3受容体結合アッセイにおいてスクリーニングされた。αvβ3受容体特異的結合アッセイにおいて試験された29化合物のうち、4つの化合物がナノモーラーで結合する親和性を示した。活性物質(AV26)の1つにおける有限構造活性相関分析(limited structure-activity relationship analysis)は、ナノモーラーからサブナノモーラー結合親和性を有する2つの強力な非RGDミメティックアンタゴニストの発見に結果として至った。がん細胞株のパネルにおいて評価された全ての活性化合物は、AV26を除いて、低マイクロモーラー濃度で、細胞毒性を示さず、AV26は、乳癌細胞株において考慮すべき細胞毒性を示した。我々の新規小分子化合物は、αvβ3陽性転移性癌に対して選択的に送達するためにパクリタキセルなどの抗癌剤と結合することが可能である。
【0041】
組成物
従って、本発明は、化合物またはその薬学的または薬用化粧品学的に許容される塩、溶媒和物または水和物を含む組成物であって、ここで、当該化合物は、1のH結合供与体(HBD)、1のH結合受容体(HBA)、2の疎水性芳香族基(HAR1およびHAR2)および1の陰性イオン性基(NI)を含み、およびここで、当該化合物は化合物 A、B,またはCではない組成物を提供する。
【0042】
当該組成物は、更に、薬学的または薬用化粧品学的に許容される担体を含んでよい。「薬学的または薬用化粧品学的に許容される担体」は、薬学的または薬用化粧品学的投与に適合する溶媒、分散媒、コーティング、抗菌剤および抗真菌剤、等張剤および吸収遅延剤などを含む。
【0043】
一つの態様において、当該HBD、HBA、HAR1、HAR2,およびNIは、図8Aに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.81±1 Åであり、当該HBDとHBAの距離は7.52±1 Åであり、当該HBDとNIの距離は17.67±1 Åであり、当該HRA1とHBAの距離は7.52±1 Åであり、当該HRA1とHRA2の距離は9.41±1 Åであり、当該HBAとNIの距離は10.20±1 Åであり、および当該HRA2とNIの距離は5.88±1 Åである。特に、当該HBDとHRA1の距離は2.81 Åでよく、当該HBDとHBAの距離は7.52 Åでよく、当該HBDとNIの距離は17.67 Åでよく、当該HRA1とHBAの距離は7.52 Åでよく、当該HRA1とHRA2の距離は9.41 Åでよく、当該HBAとNIの距離は10.20 Åでよく、および当該HRA2とNIの距離は5.88 Åでよい。
【0044】
もう一つの態様において、当該HBD、HBA、HAR1、HAR2およびNIは、図8Bに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.80±1 Åであり、当該HBDとHBAの距離は7.34±1 Åであり、当該HBDとNIの距離は16.92±1 Åであり、当該HRA1とHBAの距離は5.20±1 Åであり、当該HRA1とNIの距離は14.34±1 Åであり、当該HBAとHRA2の距離は4.69±1 Åであり、当該HBAとNIの距離は9.85±1 Åであり、および当該HRA2とNIの距離は5.66±1 Åである。特に、当該HBDとHRA1の距離は2.80 Åでよく、当該HBDとHBAの距離は7.34 Åでよく、当該HBDとNIの距離は16.92 Åでよく、当該HRA1とHBA の距離は5.20 Åでよく、当該HRA1とNI の距離は14.34 Åでよく、当該HBAとHRA2の距離は4.69 Åでよく、当該HBAとNIの距離は9.85 Åでよく、および当該HRA2とNIの距離は5.66 Åでよい。
【0045】
更なるもう一つの態様において、当該HBD、HBA、HAR1、HAR2およびNIは、図8Cに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.79±1 Åであり、当該HBDとHRA2の距離は11.94±1 Åであり、当該HBDとNI の距離は16.28±1 Åであり、当該HRA1とHBAの距離は5.20±1 Åであり、当該HRA1とNIの距離は14.64±1 Åであり、当該HBAとHRA2の距離は4.72±1 Åであり、および当該HRA2とNIの距離は5.63±1 Åである。特に、当該HBDとHRA1の距離は2.79 Åでよく、当該HBDとHRA2の距離は11.94 Åでよく、当該HBDとNI の距離は16.28 Åでよく、当該HRA1とHBAの距離は5.20 Åでよく、当該HRA1とNI の距離は14.64 Åでよく、当該HBAとHRA2の距離は4.72 Åでよく、および当該HRA2とNIの距離は5.63 Åでよい。
【0046】
本発明の化合物は、式 I、II、IIIまたはIVの化合物であってよい。
【0047】
式I、IIおよびIIIにおいて、それぞれ、R1およびR3 は脂肪族、芳香族またはヘテロ環基; R2 は置換基;およびX は N、OまたはSである。幾つかの態様において、それぞれ、R1およびR3は、アルキル、置換されたアルキル、アルケニル、置換されたアルケニル、アルキニル、置換されたアルキニル、フェニル、置換されたフェニル、アリール、置換されたアリール、ヘテロアリールまたは置換されたヘテロアリール 基; R2 は水素またはハロゲン、ヒドロキシル、スルフヒドリル、アルコキシ、カルボキシル、ニトロ、シアノ、アミノ、アミドまたはスルホニル基または何れかの数の炭素原子を含む何れか他の有機官能基;またはその組み合わせである。他の態様において、R1またはR3における当該置換されたアルキル、アルケニル、アルキニル、フェニル、アリールまたはヘテロアリール基は、ハロ、ヒドロキシル、スルフヒドリル、アルコキシ、カルボキシル、カルボニル、ニトロ、シアノ、アミノ、アミド、スルホニルまたは置換されたヘテロ環基により置換されたアルキル、アルケニル、アルキニル、フェニル、アリール,またはヘテロアリール基である。
【0048】
式 IVにおいて、それぞれ、R1およびR2 は水素またはハロゲン、ヒドロキシル、スルフヒドリル、アルコキシ、アリールオキシ、カルボキシル、ニトロ、シアノ、アミノ、アミドまたはスルホニル基または何れかの数の炭素原子を含む何れか他の有機官能基; R3 は水素、ヒドロキシル、アルコキシ、アリールオキシ、芳香族またはヘテロ環基または何れかの数の炭素原子の脂肪族鎖;それぞれ、R4およびR5 はヒドロキシルまたはアミノ基; X はC、N、OまたはS; m は、炭素、窒素、酸素または硫黄原子を伴う 0 から 3 原子リンカー基であり;およびn は炭素、窒素、酸素または硫黄原子を伴う0 から 5 原子リンカー基である。幾つかの態様において、R4またはR5における当該アミノ基は、脂肪族、芳香族またはヘテロ環基で置換されている。
【0049】
本発明の化合物の例は、AV1-11、AV14-15、AV17-32およびAV34-38を含む。
【0050】
本発明の化合物は、当該技術分野において公知の方法を使用して化学合成により、または市販の供給源から得てもよい。本発明の組成物は、以下に記載する疾患の診断および治療のために使用される。
【0051】
幾つかの態様において、本発明の化合物は、1または1以上の治療剤、イメージング剤およびその組合せとの併用において使用されてよい。当該治療剤は、しかしながらこれらに限定するものではないが、何れかの小分子薬物、抗体、FCフラグメント、タンパク質ベースの薬物、オリゴヌクレオチド、siRNA、アプタマーまたはアンキリンリピートを含む。当該治療剤の詳細な例は、血管増殖阻害剤(例えば、TNF-アルファ、アンジオスタチン、ベバシザン、アレステン、カンスタチン、コンブレタスタチン、エンドスタチン、NM-3、サリドマイド、スロンボスポンジン、タムスタチン、2-メトキシエストラジオールおよびビタキシン)、関節リウマチを治療するための治療剤、放射線治療剤、化学療法剤(例えば、シスプラチン、ドキソルビシン、ビンクリスチン、シクロホスファミド、トポテカン、パクリタキセルおよび他の化学療法剤)、骨転移を治療するための治療剤、骨粗鬆症を治療するための治療剤(例えば、ホセマックス、アレジア、アクトネル、ジンドロネル、コメタ、プレマリン、クリマラ、エストラ、ビベラ、エストラダーム、プレモプロ、プレムフェーズ、フェマート、プレフェスト、コンビパッチ、エビスタ、カルシトニン、ミアカルシンおよびカルシトニン)および再狭窄を治療するための治療剤を含む。当該イメージング剤は、これらに限定するものではないが、放射性核種(すなわち、イメージング可能なガンマ線またはポジトロン放出を生成する放射性金属イオン)を含む。
【0052】
本発明の化合物は、少なくとも1の治療またはイメージング化合物と実施可能に連結し、複合体を形成してよい。「実施可能に連結される」により、本発明の化合物と治療剤またはイメージング剤が、当該化合物が細胞においてインテグリンに対して結合するように、且つ治療剤またはイメージング剤の機能が妨害されないように結合することを意味する。
【0053】
現在広く使用される大部分の癌化学療法は、これに限定するものではないが、癌細胞に殆ど選択性を有さないシスプラチン、ドキソルビシン、ビンクリスチン、シクロホスファミド、トポテカンおよびパクリタキセルを含む。同様の問題は、非癌的兆候、例えば、関節リウマチなどのために使用される多くの治療に伴って生じ、そこにおいて、副作用が、特に延長された治療過程で生じる。多くの場合、これは望まれない副作用、および正常組織に対する高い毒性を引き起こす。正常組織に対する毒性の結果として、多くの抗癌化学療法並びに非癌性薬物は、多くの場合、至適以下の用量で投与されるか、または当該治療が妨害されてしまい、その結果として効果が低減され、最終的に治療が失敗する。これは、薬物抵抗性を発生することにより伴う可能性がある。広く引用されたこの例は、アントラサイクリンによる用量を制限する心毒性である。問題の薬物の選択的毒性は、疾患に犯された組織に届く薬物の用量の増加することにより、または正常組織に届く用量を減少することの何れかにより、または、その両方によって増加する。
【0054】
薬物の標的細胞への送達は、薬物と、正常組織とは相対的に当該標的細胞において独自に発現または過剰発現されたかの何れかの抗原または受容体に結合する分子との関連により選択的に増大できる。これにより、薬物の当該標的細胞への特異的な送達が可能になる。例えば、αvβ3インテグリンは、腫瘍の新生血管および炎症疾患性のあいだ、並びに破骨細胞において過剰発現されていることが知られているので、本発明の複合体はこれらの細胞を選択的に標的にするために使用することが可能である。
【0055】
本発明の複合体は、当該技術分野において周知の何れかの方法を用いて構成されてよい。これらの方法の詳細な説明は、例えば、以下に見出すことが可能である:「Bioconjugate Techniques」 by G.T. Hermanson、Academic Press、San Diego、1996、これらの内容は、参照することにより、その全体において本明細書に組み込まれる。
【0056】
例えば、問題の他の分子(例えば、抗体、薬物、DNAおよびsiRNA分子など)に対する本発明の化合物の複合体は、一般的に入手可能な技術および公開されている技術を用いて化学的に架橋結合により最も容易に実施されてよい。架橋結合は、2またはそれ以上の分子を共有結合により化学的に結合する方法である。架橋結合剤は、タンパク質または他の分子における特異的な官能基(例えば、一級アミン、スルフヒドリルなど)に対する反応性末端を含む。反応のための標的であり得るタンパク質およびペプチドにおける幾つかの化学基の有効性のために、タンパク質およびペプチドは、容易に結合され、且つ別の架橋結合法を使用して試験されている。クロスリンカーは、また、一般的に核酸、薬物および固体表面を修飾するために使用される。それらはまた、抗体-酵素複合体、免疫毒素および他の標識されたタンパク質試薬を用意するために有用である。当該リガンド(例えば、本発明の化合物)との接触の前または後でクロスリンカーにより受容体を誘導体化することにより、受容体-リカ゛ント゛複合体の単離が可能である。放射ヨウ素標識クロスリンカーの使用は、オートラジオグラフィー検出により特定の受容体を同定することを可能にする。クロスリンカーは、それらの化学的反応性(即ち、特定の官能基に対する特異性)および応用を伴う反応の適合性を基礎に選択される。特異的な適用のために使用される最も好ましいクロスリンカーは、経験的に決定されるべきである。クロスリンカーは、以下の特徴を基に選択される:化学的特異性、スペーサーの腕の長さ、水溶解性および細胞膜透過性、同じ(ホモ二機能性)または異なる(ヘテロ二機能性)反応性基、自発的反応性基または光反応性基、および切断性(cleavability)。クロスリンカーは、少なくとも2の反応性基を含む。架橋のために標的とされ得る官能基は、一級アミン、スルフヒドリル、カルボニル、カルボ水和物およびカルボン酸を含む(表A)。カップリングもまた、光反応性フェニルアジドクロスリンカーを使用して非選択的である。
【表A】
多くの場合、異なるスペーサーの腕の長さが要求され、これは、立体効果が架橋のための潜在的な反応部位の間の距離を要求するためである。通常、短い(4〜8Å)スペーサーの腕を有するクロスリンカーが、第一におよび決定された架橋の程度で使用される。より長いスペーサーの腕を有するクロスリンカーは、次に最適化された架橋高率のために使用され得る。短いスペーサーの腕は、多くの場合、分子内架橋研究において使用され、分子間の架橋は、長いスペーサーの腕を含むクロスリンカーを好む。多くの適用において、タンパク質の天然の構造を維持する必要があり、そのために架橋は、最も多くの場合、穏やかなpHと緩衝液条件を使用して実施される。更に、反応のために最適なクロスリンカー対タンパク質のモル比が決定される。適用に依存して、複合の程度が重要な因子である。例えば、免疫原複合体を用意する場合、抗原の免疫原性を高めるために高い程度の複合が望まれる。しかしながら、抗体または酵素のための複合は、低度または中程度の複合が、タンパク質の生物学的な活性を保持するために至適であり得る。当該タンパク質表面における官能基の数もまた、考慮のために重要である。仮に多数の標的基がある場合、より低いクロスリンカー対タンパク質の比が使用される。制限された数の潜在的な標的のためには、より高いクロスリンカー対タンパク質の比が要求される。更に、多くの成分は、最小限に維持されるべきであり、それは2以上の成分からなる複合体は、分析することが困難であり、タンパク質サブユニットの空間的な配置における少ない情報しか提供されないためである。水溶解性および膜透過性もまた、カップリングのための最適なクロスリンカーを選択する上で重要な問題である。多くのクロスリンカーは、それらの疎水性のスペーサーの腕によって、水溶液における溶解度が制限される。これらのクロスリンカーは、一般的にDMFまたはDMSOに溶解され、次に生物系または架橋されるべき生体分子の溶液に添加される。疎水性のクロスリンカーは、細胞膜およびオルガネラ膜を通過することが可能であり、膜の外側表面、および膜と結合した空間内の両方における架橋に影響を及ぼす。多くの場合、生物系のための架橋手順に有機溶媒を導入することは都合が悪いか、または望ましくない。また、多くの場合、細胞またはオルガネラの内側を変化させることなく、細胞膜またはオルガネラ膜の外側表面だけにおける架橋に影響を及ぼすことが望ましく、そのような場合において、幾らかの水溶解、膜不透過のクロスリンカーが利用できる。幾つかのクロスリンカーは、ポリエチレングリコール(PEG)サブユニットから形成されるスペーサーの腕を含み、結果として大量の酸素原子と共にポリエチレンオキシド(PEO)を生じて水溶解性を与える。これらのクロスリンカーは、それらの名称に(PEO)nが示され、水可能性であり、且つ生物膜透過できない。それらは、架橋化された複合体に対してそれらの親水性のスペーサーを移行する更なる利点を提供し、従って、当該複合体の凝集および沈殿を減少する。他のクロスリンカーは、当該スペーサーの何れかの末端で荷電反応性基によって、それらの水溶解性および膜不透過性を含む。荷電反応記、例えば、スルホ-NHSエステルまたはイミドエステルなどは、当該架橋試薬に対して水溶解性を与えるが、当該反応性基は最終複合体の一部分ではないので、当該架橋された複合体には与えない。
【0057】
説明の目的のために、2つの例として、NVX-188(即ち、AV38)複合体を構成することを以下に示す。
【0058】
例1:カルボイル反応性化学を使用することによるNVX-188および類似体のカルボキシル基のカップリング
NVX-188基の幾つかは、種々のクロスリンカーのためのカップリングパートナーとして役立つカルボキシル基を含む。カルボジイミドは、一級アミンまたはヒドラジンに対してカルボキシルを結合し、結果としてアミドまたはヒドラゾン結合を形成する。カルボジイミドは、結合される分子の間にスペーサーが存在しない他の結合反応とは異なる。タンパク質のカルボキシ末端、並びにグルタミン酸およびアスパラギン酸側鎖もまた、標的とされ得る。この例において、EDC(製品#22980、22981、Pierce)はカルボン酸基と反応し、カルボキシル基を活性化し、活性O-アシルイソウレア中間体を形成し、当該反応混合物における当該アミノ基に対する結合を可能にする。EDC副産物は、可溶性尿素誘導体として、求核試薬による排除の後に放出される。当該O-アシルイソウレア中間体は水溶液中で不安定であり、N-ヒドロキシスクシンイミドを使用する中間体の安定性を増加することなく、2つの工程の複合体手順においてそれを無効にする。この中間体は、一級アミンと反応し、アミド誘導体を形成する。アミンとの反応ができないということは、結果として当該中間体の加水分解、当該カルボキシルの再生およびN-非置換ウレアの放出を生じる。当該架橋反応は、通常、pH4.5〜5の間で実施され、多くの適用のために数分しか必要としない。しかしながら、当該反応の終了は、pH4.5〜7.5で同様である。EDCの加水分解は、カップリング中競合的反応であり、温度、pHおよび緩衝液組成物に依存する。4-モルホリノエタンスルホン酸(MES、製品#28390、Pierce)は有効なカルボジイミド反応緩衝液である。リン酸緩衝液は、EDCの反応効率を低下するが、EDCの量を増加することで、低下した効率を代償できる。トリス、グリシンおよびアセテート緩衝液は、結合緩衝液として使用されない。NHS(製品 # 24500、Piercebiotechnology)またはその水可溶性類似体 スルホ-NHS(製品# 24510、Pierce)は、多くの場合、EDCカップリングプロトコールを含み、効率を改善する。EDCは、NHSをカルボキシルに対して結合し、結果として、分子におけるNHS活性化部位を生じる。形成された当該NHS-エステルと、当該カルボジイミドのO-アシルイソウレア中間体は、アミン反応性である;しかしながら、NHS-エステルは、僅かに酸性または中性近くのpH状態において非常に高い安定性を有する。水中で、NHS-エステルは、温度、pHおよび当該クロスリンカーの構造に依存して1〜数時間、または数日までもの半減期を有するが、O-アシルイソウレア中間体は、酸性または中性pH状態で秒単位で測定される半減期を有する。分子のEDC媒介カップリングは、多くの適用において、NHSまたはスルホ-NHSなしで良好に働き、これらは、一般的に必要とされるタンパク質濃度が非常に低い場合でない限り必要とされない。多量に過剰なEDCがNHSを伴わずに使用される場合には、しばしば、EDC/NHS系への変換の際に当該EDC量を減少させ、過剰な架橋と可能な沈殿を防止する必要がある。
【0059】
例2:非特異的化学(アリールアザイド)を使用するNVX-188および類似体のカップリング
光反応試薬は、紫外線または可視光に暴露された場合に、反応性となる化学的に不活性な試薬である。少しの例外はあるが、これらの試薬における当該光反応基は、アリールアザイドである。アリールアザイドがUV光に曝されたとき、それは、二重結合との更なる反応、C-HおよびN-H部位への挿入、または続く求核試薬(例えば、一級アミン)と反応するための環拡大を開始することが可能なニトレン基を形成する。当該サンプルに一級アミンが存在する場合、後者の反応経路が支配する。チオール含有還元剤(例えば、DTTまたは2-メルカプトエタノールなど)は、光反応前および光反応中における全ての工程の間に当該サンプル溶液中から避けられるべきである。これらの試薬は、アミンに対するアザイド官能基を減少し、光活性化を妨げるであろう。反応は、種々のアミン非含有緩衝液条件において実施され得る。ヘテロ二機能性光反応性クロスリンカーと作業する場合、緩衝液は、当該試薬の化学的反応性と適合可能に使用されるべきである。当該化学反応は、ホイルで覆われた反応容器で光を遮った中で実施される。当該光活性化は、当該反応容器上約1〜2インチ(約2.54〜5.08センチメートル)で明るいカメラフラッシュまたは紫外線ハンドホルダーランプを用いて実施される。明るいカメラフラッシュ右派、ニトロ-およびヒドロキシ置換アリールアザイドと共に良好に機能する。非置換のアリールアザイドは、紫外線光または多数のフラッシュを必要とするかもしれない。
【0060】
本発明の組成物は、その意図される投与経路に適合するように形成される。例えば、米国特許第 6,756,196を参照されたい。投与経路の例は、非経口、例えば、静脈内、皮内、皮下、経口(例えば、吸入)、経皮(局所的)、経粘膜および直腸投与を含む。非経口、皮内または皮下適用のために使用される溶液または懸濁液は、以下の成分を含んでよい:滅菌希釈剤、例えば、注射用の水、整理食塩溶液、特定のオイル、ポリエチレングリコール、グリセリン、プロピレングリコールまたは他の合成溶媒;抗菌剤、例えば、ベンジルアルコールまたはメチルパラベン;抗酸化剤、例えば、アスコルビン酸または亜硫酸ナトリウム;キレート剤、例えば、エチレンジアミンテトラ酢酸;緩衝液、例えば、アセテート、クエン酸塩、リン酸塩;張度の調整のための薬剤、例えば、塩化ナトリウムまたはデキストロース。pHは、酸または塩基、例えば、塩酸または水酸化ナトリウムなどで調整できる。
【0061】
非経口製剤は、ガラスまたはプラスチックで作られたアンプル、ディスポシリンジまたは多用量バイアルに入れることが可能である。
【0062】
注射に使用するために適切な組成物は、滅菌水溶液(水溶液中)または懸濁液および滅菌注射溶液または分散剤の即席製剤のための滅菌パウダーを含む。静脈投与のための適切な担体は、生理的食塩水、滅菌水、クレモフォア(Cremophor) EL(登録商標)(BASF
、Parsippany、NJ)またはリン酸緩衝化生理食塩水(PBS)を含む。全ての場合において、当該組成物は、滅菌されるべきであり、簡単にシリンジに存在可能な範囲の流体であるべきである。それは、製造および貯蔵の条件下で安定であるべきであり、細菌および真菌などの微生物の活性を混入作用に反して保存されるべきである。当該担体は、例えば、水、エタノール、ポリオール(例えば、グリセロール、プロピレングリコールおよび液体ポリエチレングリコールなど)、および適切なその混合物を含む溶媒または分散剤であってよい。その適切な流動性は、例えば、レシチンなどのコーティングなどの使用により、分散剤の場合においては、必要とされる粒子サイズの維持により、および界面活性剤の使用により維持される。微生物の作用の阻止は、種々の抗菌剤および抗真菌剤、例えば、パラベン、クロルブタノール、フェノール、アスコルビン酸、チメロサル(thimerosal)などにより達成される。多くの場合において、当該組成物において、等張剤、例えば、糖、ポリアルコール、例えば、マンニトール、ソルビトールまたは塩化ナトリウムが含まれることは好ましいであろう。注射用組成物の延長された吸収は、アルミニウムモノステアレートおよびゼラチンなどの吸収を遅延する薬剤を当該組成物に含ませることにより、凡そ達成され得る。
【0063】
滅菌注射溶液は、所望に応じて、1のまたは上で列記した成分の組合せと共に適切な溶媒に必要量で当該化合物を組み込み、続いてフィルター滅菌することにより製造できる。一般的に、分散剤は、基本的な分散媒および上で列記したものからの所望の他の成分を含む滅菌媒体に当該化合物を組み込むことにより製造される。滅菌注射溶液の製造のための滅菌パウダーの場合において、好ましい製造の方法は、予めフィルター滅菌されたその溶液から活性成分と更なる所望の成分を得る減圧乾燥および凍結乾燥である。
【0064】
経口組成物は、一般的に不活性な希釈液または可食担体を含む。経口治療投与の目的のために、当該化合物は、賦形剤と共に組み込まれ、錠剤、トローチまたはカプセル、例えば、ゼラチンカプセルなどの形態において使用される。経口組成物はまた、マウスウォッシュとして使用されるための流体担体を使用して製造されてもよい。薬学的または薬用化粧品学的に適合する結合剤および/またはアジュバント物質が当該組成物の一部分として含まれてもよい。当該錠剤、ピル、カプセル、トローチなどは、以下の成分または同様の天然の化合物の何れかを含んでもよい:結合剤、例えば、微小結晶性セルロース、トラガカントゴムまたはゼラチン;賦形剤、例えば、デンプンまたはラクトース、崩壊剤、例えば、アルギン酸、プリモゲル(Primogel)またはトウモロコシデンプン;潤滑剤、例えば、ステアリン酸マグネシウムまたはステロート(Sterotes);グリダント(glidant)、例えば、コロイド状シリコンジオキシド;甘味剤、例えば、ショ糖またはサッカリン;または香味剤、例えば、ペパーミント、メチルサリチル酸塩、またはオレンジ香料。
【0065】
吸入による投与のための当該組成物は、適切な噴霧剤、例えば、ガス、例えば、二酸化炭素を含むプレッシャーコンテナまたはディスペンサーまたはネブライザーからのエアロゾルスプレーの形態で送達される。
【0066】
全身投与もまた、経粘膜または経皮手段により行われてよい。経粘膜または経皮投与のために、透過されるべき障壁に対して適切な貫通刺激が製剤において使用される。そのような貫通刺激は、当該分野において一般的に公知であり、例えば、経粘膜投与、界面活性剤、胆汁塩、フシジン酸誘導体などが含まれる。経粘膜投与は、経鼻スプレーまたは坐剤の使用を介して達成されてよい。経粘膜投与のために、本発明の化合物が、当該技術分野において一般的に公知の軟膏(ointment)、軟膏(salves)、ゲルまたはクリームに製剤化される。
【0067】
本発明の組成物はまた、坐剤の形態(例えば、ココアバターおよび他のグリセリドなどを基剤する通常の坐剤)、または直腸送達のための維持浣腸の形態に製造されてよい。
【0068】
一つの態様において、当該組成物は、生体からの急速な消失に対して当該化合物を保護する担体と共に製造され、例えば、制御放出製剤、例えば、インプラントおよび微小被包性送達系などを含む形態に製造される。生分解性の生体適合性重合体、例えば、エチレンビニルアセテート、ポリアンヒドライド、ポリグリコール、コラーゲン、ポリオルトエステルおよびポリ乳酸などが使用されてよい。そのような製剤の製造方法は、当業者に明白であろう。当該材料も、Alza Corporation および Nova Pharmaceuticals,Incから商業的に得ることが可能である。リポソームの懸濁液(ウイルス性抗原に対するモノクローナル抗体を伴う感染細胞に対して標的するリポソームを含む)も、薬学的または薬用化粧品学的に許容される担体として使用されてよい。これらは、当業者に公知の方法に従って、例えば、米国特許第 4,522,811に従って製造される。
【0069】
投与が容易且つ均一な用量のために単位投与量での経口または非経口組成物が製剤化されることも有利である。ここで使用される「単位投与量形態」は、治療されるべき対象のための単位投与量として適切な物理的に分離した単位をいう:各単位は、必要とされる薬学的または薬用化粧品学的担体と共に所望の治療学的または薬用化粧品学的効果を生ずることが計算された活性化合物の予め決定された量を含む。
【0070】
本発明の組成物は、容器、パックまたはディスペンサーに含まれて、投与のための指示と共にパッケージされた製品を形成してもよい。他の活性化合物もまた、本組成物と組み合わされてもよい。
【0071】
組成物の使用
処理の方法
本発明の1つの目的は、インビトロまたはインビボにおいて化合物に対してインテグリンを結合し、それによりインテグリンの活性を調節するための方法を提供する。当該方法は、インテグリンを本発明の化合物またはその薬学的若しくは薬用化粧品学的に許容される塩、溶媒和物または水和物と接触することを具備する。
【0072】
インテグリンは、細胞接着およびシグナリング受容体を形成する糖タンパク質のファミリーである。少なくとも25のインテグリン受容体では、アルファおよびベータサブユニットのヘテロダイマーにより形成される各々が確認されている。αvβ3インテグリンは、インテグリンファミリーの突出したものである。
【0073】
インテグリンは、細胞死、増殖、遊走および組織リモデリングを含む細胞応答を調節する細胞外マトリックスからの指示に対して応答する。シグナリングは、FAKおよびSrcファミリーからのチロシンキナーゼの補充を介して調節される。これは、MAPキナーゼおよびPI3K伝達カスケードの活性を調節する。細胞外マトリックスに対するインテグリン媒介接着の欠如は内皮細胞におけるアポトーシスを引き起こす。多くのインテグリンは、多くの細胞外タンパク質において見出されているRGDトリペプチドモチーフ(Arg-Gly-Asp)に対して特異的に結合する。
【0074】
αvβ3の発現は、殆どの組織において低いが、リモデリングまたは増殖組織においては非常に高い。αvβ3は、細胞外マトリックスタンパク質、フィブロネクチン、フィブリノーゲン、オステオポンチン、スロンボスポンジンおよびビトロネクチンのRGDモチーフに対して結合する。それは、腫瘍の増殖および転移、骨再吸収、ある炎症性疾患および血管構造(血管形成)において重要な役割を演じている。αvβ3インテグリンの生物学的活性は、症状の範囲において、選択的αvβ3アンタゴニストを使用する治療学的および薬用化粧品学的介入のための基礎を提供する。これらの症状は、しかしながらこれらに限定するものではないが、癌、骨粗鬆症、関節リウマチおよび加齢関連横斑性変性を含む。
【0075】
従って、一つの態様において、本発明の化合物は、細胞、例えば、癌細胞、良性腫瘍細胞、間質細胞、炎症に反応する、炎症を媒介する、または炎症を調節する細胞(例えば、白血球、マクロファージおよび樹状細胞)、免疫系の細胞(例えば、白血球、マクロファージおよび樹状細胞)、血管機能に関与する細胞(例えば、内皮細胞、内皮細胞に関与する細胞および平滑筋細胞)、骨芽細胞、破骨細胞またはウイルスに感染した細胞(例えば、アデノウイルス(Adenoviridae)、コックサッキーウイルス(Picornaviridae)、エコウイルス(Picornaviridae)、口蹄疫ウイルス(Picornaviridae)、ハンタウイルス(Buriyaviridae)、ヒトパレコウイルス(Picornaviridae)、ヒト免疫不全ウイルス 1(Retroviridae)およびロタウイルス(Reoviridae))においてインテグリンと接触する。
【0076】
本発明は更に、それを必要とする対象を処置するための予防学的および治療学的の両方または薬用化粧品学的方法のために有効量の上記の化合物を提供する。
【0077】
ここで使用される「対象」は、ヒト、または以下を含む動物、全ての脊椎動物、例えば、哺乳類、例えば、霊長類(特に、高等霊長類(higher primates))、ヒツジ、イヌ、げっ歯類(例えば、マウスまたはラット)、モルモット、ヤギ、ブタ、ネコ、ウサギ、ウシ;および非哺乳類、例えば、ニワトリ、両生類、爬虫類などをいう。好ましい態様において、当該対象はヒトである。もう一つの態様において、当該対象は動物である。
【0078】
処置されるべき対象は、例えば、当該分野に公知の診断方法を使用して、疾患または以下に記載の状態に罹患しているか、またはそれらの進展のリスクに面しているかが同定されてもよい。当該対象は、対象またはヘルスケアのプロフェッショナルの判断により同定されてもよく、主観的に(例えば、見解)または客観的に(例えば、検査または診断方法により測定可)であってもよい。
【0079】
本明細書に使用される、用語「処置」は、当該疾患、疾患の症状または疾患に向かう素因を治療、治癒、緩和、軽減、変化、療法、寛解、改善するまたはそれらに影響する目的を伴って、治療剤または化粧剤の対象への適用または投与、または対象から単離された組織または細胞株への治療剤または化粧剤の適用または投与、(ここで、当該対象は、疾患、疾患の症状または疾患に向かう素因を有する)と定義される。
【0080】
「有効量」は、処置される対象において、本明細書において描写された通りの医学的または薬用化粧品学的所望の血管を生じることが可能である治療剤または化粧剤の量である。当該医学的または薬用化粧品学的所望の結果は、(即ち、幾つかの試験またはマーカーにより測定可能な)客観的または(即ち、対象が効果の兆候を示す、または効果を感じる)主観的なものであってよい。
【0081】
本発明の特性および治療学的または薬用化粧品学的有効性は、細胞培養物または実験動物において、例えば、LD50(集団の50%までの致死的用量)およびED50(集団の50%において治療学的または薬用化粧品学的に有効な用量)を決定するための標準的な薬学的または薬用化粧品学的手続により決定されてよい。毒性と治療学的または薬用化粧品学的有効性の間の当該用量比は、治療学的指標であり、LD50/ED50 比と表される。高い治療学的または薬用化粧品学的指標を示す化合物が好ましい。有毒な副作用を示す化合物が使用されてよい一方で、非感染細胞への可能な障害を最少化し、それにより副作用を低減するために、そのような化合物を患部組織の部位に対して向ける送達系を設計する配慮がなされるべきである。
【0082】
細胞培養アッセイおよび動物実験から得られたデータは、ヒトにおいて使用するための用量の範囲を決定することにおいて使用できる。当該化合物の投与量は、好ましくは殆ど毒性がないか、全く毒性のないED50を含む循環濃度の範囲内に位置する。当該投与量は、使用される投与量形態および利用される投与経路に依存するこの範囲内で変動してよい。本発明の方法において使用される幾つかの化合物については、当該治療学的または薬用化粧品学的有効用量は、細胞培養アッセイから初期に推定され得る。用量は、動物モデルにおいて、細胞培養において決定されたようなIC50(即ち、症状の最大半減阻害を達成する化合物の濃度)を含む循環血漿濃度範囲を達しするために処方されてよい。そのような情報は、ヒトにおいて有用な用量をより正確に決定するために使用され得る。血漿におけるレベル、例えば、高速液体クロマトグラフィにより測定されてよい。
【0083】
本発明の化合物の治療学的または薬用化粧品学的有効量(即ち、有効投与量)は、例えば、キログラム当たり約1 マイクログラムからキログラム当たり約500 ミリグラム、キログラム当たり約100 マイクログラムからキログラム当たり約5 ミリグラム、またはキログラム当たり約1 マイクログラムからキログラム当たり約50 マイクログラムの範囲であってよい。当該化合物は、例えば、約1から10週間の間、好ましくは2から8週間の間、より好ましくは約3から7週間の間、より更に好ましくは約4、5または6週間に亘り、1週当たり1回投与され得る。慢性疾患、例えば、関節炎または骨粗鬆症に罹患する対象に置いては、長期間に亘る治療、例えば、1日に1回または好ましくは1週間に1回などが必要であってよい。更に、化合物の適切な用量は、当該化合物の効力に依存することが理解される。これらの化合物の1または1以上が対象(例えば、動物またはヒト)に対して投与される場合、医師、獣医師または研究者が、例えば、最初は相対的に低用量で、引き続いて、適切な応答が得られるまで当該用量を増加して処方してよい。加えて、何れかの特定の対象のための当該具体的な用量レベルは、使用される具体的な化合物の活性、当該患者の年齢、体重、一般的な身体の状態、性別および食事制限、投与の時間、投与経路、排出率、何れかの薬物の併用、疾患または障害の重症度、以前の治療および他の疾患の存在を含む多様な因子に依存するであろうことは理解される。更に、当該化合物の治療学的または薬用化粧品学的有効量での対象の処置は、単一の処置を含み、または好ましくは一連の処置を含む。
【0084】
本発明の方法は、多数の適用を有する。当該適用の1つの例は、これらに限定するものではないが、全ての組織学的種類の悪性固形腫瘍、例えば、肉腫および癌腫および高レベルの血管形成および血管原性因子に関連する血液学的悪性物、例えば、急性および慢性白血病、多発性骨髄腫、骨髄増殖性疾患、非ホジキンリンパ腫およびホジキン病を含む癌の処置である。
【0085】
腫瘍の増殖&転移: αvβ3 インテグリンは、悪性メラノーマ、乳癌、卵巣癌および神経膠腫および大腸癌を含む多くの腫瘍により発現されている。これらの腫瘍における過剰発現は、浸潤および転移を含む悪性進行と関連する。癌治療のためのαvβ3インテグリンアンタゴニストの潜在的な役割についての実験的サポートは以下を含む:
αvβ3インテグリンアンタゴニストで治療されるマウスにおける、マウス大腸癌細胞の注入に続く肝転移の低下、
サイクリックRGDペンタペプチドに対する暴露に続く、インビトロでの脳腫瘍細胞におけるアポトーシスの誘導、
マウスにおけるメラノーマ異種移植の増殖のオリゴペプチドαvβ3インテグリンアンタゴニストによる阻害、
マウスにおける乳癌異種移植の腫瘍アポトーシスの増加および治療率に対する放射免疫療法およびシレンジタイド(サイクリックRGDペプチド)の相乗効果、
αvβ3インテグリンに対するモノクローナル抗体によるSCIDマウスにおけるヒト皮膚移植における乳癌腫の増殖阻害(および血管形成の減少)、
メラノーマ、神経膠腫、乳癌および卵巣癌を含む幾つかの腫瘍タイプにおける腫瘍進行とαvβ3インテグリンの発現の相関、
αvβ3インテグリンの変異体(常時活性型)変種と関連するMDA-MB 435乳癌細胞の転移可能性の増加、および
高レベルのαvβ3インテグリン発現に形質転換された腫瘍からの骨転移の発生率の増加、およびマウスにおけるMDA-MB 435乳癌異種移植のαvβ3インテグリンアンタゴニストによる治療での骨転移の抑制。
【0086】
αvβ3インテグリンは、腫瘍細胞浸潤および腫瘍細胞核酸において重要な役割を果たしていることから、αvβ3インテグリンアンタゴニスト、特に毒性プロフィールを伴わずに、細胞外マトリックスタンパク質に対する結合から浸潤している癌細胞をブロックすることが可能なもの、例えば、NVX-188、は、この適用のために特別に興味深く、また、併用治療戦略のために良好に適しているようである。
【0087】
血管新生: αvβ3インテグリンは、新血管新生の間に血管平滑筋細胞において発現する。血管発生におけるαvβ3インテグリンの中心的役割は、実験において証明されている。これらの実験において、(即ち、腫瘍における)新たな血管増殖は実質的にαvβ3インテグリンアンタゴニストによりブロックされた。癌における当該αvβ3インテグリンアンタゴニストの治療的利点は、直接的に腫瘍を殺すことに由来するものではなく、腫瘍血管新生を阻害することに由来するものである。例えば、動物モデルにおけるメラノーマ細胞による肺転移発生は、αvβ3インテグリンアンタゴニストにより防止できることが示されている。これは、当該メラノーマ細胞がαvβ3インテグリンを発現していないのだが生じた。従って、非細胞障害性のαvβ3ブロッカーは、癌治療において潜在的な血管形成阻害適用を有する。
【0088】
標的治療: 転移性癌細胞における重要な細胞表面受容体へのNVX-188の高特異性は、複合性(composite)の標的癌化学療法学の発達において利用され得る。これらの化合物は、標的細胞に高い親和性を有する特異的標的剤、例えば、NVX-188などに結合し、当該細胞を殺傷することが可能な細胞障害性の薬剤を伴う。当該2つの成分は、ナノ粒子表面をコートするNVX-188を有するナノ粒子として共有結合または処方され、特異的な取り込みを亢進する。それから当該細胞障害性薬剤が当該ナノ粒子に被包される。
【0089】
本発明の方法のもう一つの例示的適用は、良性腫瘍および新生物発生前の高い血管新生に関連する状態、尋常性疣贅、化膿性肉芽腫、酒さ、ケロイド瘢痕の治療である。脈絡叢または光彩新血管新生に関連する眼部疾患(腫瘍)もまた、本発明に従い治療され得る。
【0090】
加えて、本発明の方法は、炎症性疾患の治療のために使用され得る。そのような疾患の例は、これらに限定するものではないが、炎症性腸疾患、例えば、クローン病および潰瘍性大腸炎;多発性炎症性ミオパシー、例えば、多発性筋炎(PM)、皮膚筋炎(DM)および高い血管新生に関連する状態;および他の眼部炎症性疾患を含む。
【0091】
高い炎症と関連する疾患または状態、例えば、歯肉炎および粘膜炎(例えば、放射線および化学療法に誘導される粘膜炎)もまた、本発明の方法により治療されてよい。これらの適用において、本発明の化合物、例えば、NVX-188および類似体は、局所的に適用され、例えば、口腔粘膜炎などのためのマウスウォッシュ溶液であってよい。ざ瘡の治療のためには、本発明の化合物、例えば、NVX-188および類似体は単独、または抗細菌剤または他の抗起炎性療法と組み合わせて適用されてよい。特に慢性的な炎症状態に関連する胃腸管疾患、例えば、胃炎または十二指腸炎は、本発明の方法を使用して治療されてよく、本発明の化合物(例えば、NVX-188および類似体)の経口処方を適用することこにより最も効率的に治療されてよい。
【0092】
関節リウマチの中心的な特徴は、患部の関節における軟骨および骨の破壊を引き起こす慢性的な炎症である。この炎症は、サイトカイン系の活性化に起因する。これは、炎症細胞により、特に活性かれたマクロファージによる腫瘍壊死因子産生、IL-1およびIL-6を介して調節される。これらのマクロファージは、それらの表面においてαvβ3インテグリンを発現する。αvβ3阻害剤は、従って、関節リウマチ治療のために有用である。
【0093】
本発明の方法を使用して治療されるべき免疫および自己免疫疾患は、これらに限定うするものではないが、関節リウマチ、乾癬性関節炎、乾癬、多発性硬化症、気管支喘息およびアレルギーおよび皮膚の他の炎症性状態および皮下組織を含む。
【0094】
関節リウマチ(RA)は、関節における広範な骨および軟骨破壊に関連する衰弱性、全身性、自己免疫疾患である。αvβ3インテグリンが、RAの病因において重要な役割を果たしていることを見解を支持する実質的なデータがある。これらの役割には、活性化マクロファージ依存性炎症、破骨細胞の発達、骨再吸収および炎症性血管形成を含む。RAは、活性化された血管形成およびαvβ3 インテグリン脈管構造発現を伴う炎症性疾患である。
【0095】
ヒトにおいて関節リウマチを擬態した関節炎を誘導されたウサギおよびラットの実験モデルにおけるαvβ3阻害剤の動物実験は、アルスロジェニック(arthrogenic)変化の減少を示した。これらの変化は、ウサギにおける滑膜血管形成阻害、細胞浸潤の減少、パンヌス形成および軟骨侵食、およびラットにおける骨、軟骨および軟組織の有意な保護を含む。
【0096】
乾癬は、免疫媒介疾患であると主に考えられているにも拘わらず、また、過剰な血管形成により特徴付けられる。そのため、NVX-188および類似体などの本発明の化合物は、全身的のみならず、好ましく局所的にのみ、または乾癬の治療に現在使用される他の薬物と併用して使用されてもよい。これらの薬物は、例えば、皮膚および頭皮のための局所的なローション、軟膏、クリーム、ゲルおよびジャンプーとして添加されてもよい。FDAに承認された乾癬治療のための処方局所剤は、コルチコステロイド、レチノイド、カルシポトリエン(calcipotriene)およびコールタール製品を含む。
【0097】
多発性硬化症(MS)は、もう一つの自己免疫仮定により媒介される衰弱性疾患である。中枢神経系におけるMS病変は、αvβ3インテグリンを含むラミニン受容体発現における変化を示す。NVX-188などの発明の化合物は、αvβ3インテグリン受容体に結合する特異的なリガンドとして潜在的に有用である。
【0098】
本発明の方法を使用して値幼され得る皮膚および皮下組織の他の炎症性状態には、これらに限定するものではないが、紅斑性扁平皮膚症、アトピー性皮膚炎および関連状態、接触性皮膚炎および他の湿疹、内部に取り込まれた物質のための皮膚炎、水疱性皮膚疾患、紅斑性状態、苔癬および掻痒症および関連する状態が含まれる。
【0099】
本発明の化合物、例えば、NVX-188および類似体は、全身的にでも、更に好ましくは局所的のみでも、または現在使用されている他の薬剤との組合せにおいての何れにおいても使用されてよい。これらの薬剤は、例えば、皮膚および頭皮のための局所的ローション、軟膏、クリーム、ゲルおよびシャンプーとして添加されてもよい。治療されてよい状態の例は以下である: 紅斑性扁平皮膚症、斑不全角化、脂漏性皮膚炎、頭部脂漏症、他の脂漏性皮膚炎、紅斑性扁平皮膚症、アトピー性皮膚炎および関連状態、紅斑、アトピー性皮膚炎、ベニエ痒疹、湿疹:(アトピー性、曲性(flexural)、内因性(アレルギー性))、神経皮膚炎、接触性皮膚炎および皮膚炎を含む他の湿疹(接触性、職業性)、内部に取り込まれた物質のための皮膚炎、皮膚に接触した薬物性および医薬性、皮膚炎(アレルギー性)、例えば、皮膚に適用された何れかの薬物性または他の化学製品性、日射のための皮膚炎、日焼け、ベルロック皮膚炎、光アレルギー反応皮膚炎、光毒性反応皮膚炎、多形性 光性発疹、播種性浅在性光線性汗孔角化症(DSAP)、化粧品性皮膚炎、他の放射線、例えば、赤外線、紫外線などによる皮膚炎および電離放射線での治療による皮膚炎、水疱性皮膚疾患、疱疹状皮膚炎、疱疹状皮膚症、デューリング病、疱疹状皮膚炎、角層下膿疱症、スネッドン-ウィルキンソン病、若年性 疱疹状皮膚炎、若年性天疱瘡、疱疹状膿痂疹、多巣性紅斑、紅斑 虹彩、疱疹 虹彩、ライル症候群、エリトマトーデス、他の特定の紅斑性状態、例えば、リッター病、新生児剥脱性皮膚炎、紅斑間擦疹、苔癬、苔癬化およびハイド病、神経皮膚炎(限局性)(局所性)、他の特定の掻痒症および掻痒症。
【0100】
更に、本発明の方法は、血管性疾患を治療するために有用である。当該関連する血管性疾患は、これらに限定するものではないが、子宮内膜症、機能性障害性の子宮性出血、子宮内膜過形成、骨髄線維症、血管腫、動脈硬化症、急性および慢性移植拒絶反応および移植脈管障害、加齢関連 横斑性変性(AMD)を含む眼部 新血管新生を特徴とする眼部障害、増殖性糖尿病網膜症(PDR)、未熟児網膜症(ROP)、新血管緑内障、網膜芽細胞腫、水晶体後線維増殖症、ルベオーシス、ブドウ膜炎、横斑性変性および角膜移植新血管新生を含む。
【0101】
加齢関連横斑性変性(AMD)は、高齢者において共通する失明の原因である。最も重篤な形態は、網膜の患部領域における広範な新血管の形成を含む。この理由のために、抗血管形成性化合物が有効に投与され、血管の内殖を阻害し、視覚を保存している。αvβ3インテグリンアンタゴニストの血管形成阻害能は、本化合物がAMDの治療において価値があり得ることを示唆する。
【0102】
本発明の方法に従い治療され得る不適当な血小板活性化、補充、凝集または血栓症により媒介される状態の中には、冠動脈疾患または障害、心筋梗塞または心筋梗塞に続く障害、脳卒中、不安定狭心症、子癇前症、塞栓症、血小板関連虚血性疾患、例えば、肺虚血、冠血管虚血および脳虚血、血管形成術、アテレクトミー、ステント設置,およびバイパス手術を含む経皮的冠動脈インターベンションに続く再狭窄、血栓性疾患、例えば、冠動脈血栓症、脳動脈血栓症、心臓内血栓症、末梢動脈血栓症、静脈血栓症、血栓症および外来性に対する露出または損傷された組織表面に関連する凝固障害および血栓症に続く再閉塞、深在性静脈血栓症(DVT)、肺塞栓症(PE)、一過性虚血性発作(TIAs)および血管閉塞が共通する基本をなす特徴である他の状態がある。幾つかの態様において、本発明に従う方法は、血栓形成または再形成、進行型の冠動脈疾患または閉塞、再閉塞、血管の狭窄または再狭窄または脳卒中のハイリスクにある個体において使用される。他の態様において、本発明に従う方法は、血管形成方法、例えば、バルーン血管形成術、レーザー血管形成術、冠血管アテレクトミーまたは同様の技術、頚動脈動脈膜切除、血管グラフトの吻合、血栓形成の高リスクを有する外科手術(すなわち、冠血管バイパス手術、補綴バルブまたは血管の挿入など)、アテレクトミー、ステント設置、慢性心血管装置の設置、例えば、インドルエリングカテーテルまたは補綴バルブまたは血管、臓器移植またはバイパス手術と組み合わせて使用される。
【0103】
本発明の方法を使用して治療され得る他の疾患および状態は、心筋の血管新生、出血性関節s、血管癒着、敗血症、成人呼吸困難症候群、末梢血管拡張および創傷肉芽形成を含む。
【0104】
骨再吸収は、本発明の方法が適用され得る他の領域である。骨再吸収、例えば、骨粗鬆症において見られるように、骨マトリックスへの破骨細胞の接着に関連する。この重要な接着は、インテグリンにより媒介される。高レベルのαvβ3インテグリンの発現は、骨再吸収する破骨細胞において注目されている。αvβ3アンタゴニストによる破骨細胞接着の破損は、インビトロおよびインビボの両方における骨再吸収を阻害する。
【0105】
病理学的骨再吸収と主に関連する疾患は、これらに限定するものではないが、骨粗鬆症、骨のパジェット病および副甲状腺ホルモン(PTHrP)媒介高カルシウム血症を含む。
【0106】
αvβ3アンタゴニストは、骨粗鬆症の予防および治療のために評価されている。それらは、注目すべき有害作用を伴わずに骨減少の阻害を示している。この状態における長期治療のために必要なものは、何れの治療も非常に低毒性のプロフィールを有することが示される。非常に低い容量で投与できるNVX-188の高結合親和性およびその明らかに毒性を欠くということは、この化合物、同時に本発明の他の化合物をこの適用において魅力的にしている。
【0107】
感染した生物体における標的細胞との相互作用のための受容体としてαvβ3インテグリンを使用するウイルス感染は、本発明の方法により治療されてよい。例示的なウイルス感染は、これらの限定するものではないが、アデノウイルス(Adenoviridae)、コックサッキーウイルス(Picornaviridae)、エコウイルス(Picornaviridae)、口蹄疫ウイルス(Picornaviridae)、ハンタウイルス(Buriyaviridae)、ヒトパレコウイルス(Picornaviridae)、ヒト免疫不全ウイルス 1(Retroviridae)およびロタウイルス(Reoviridae)を含む。
【0108】
本発明の化合物は、単体で使用されてもよく、または他の薬剤、例えば、上述の治療剤およびイメージング剤と組み合わせて使用されてもよい。当該インテグリンおよび当該細胞は、同時にまたは経時的に当該化合物および混合されたまたは個々の投与量としての薬剤と接触されてもよい。当該化合物は、少なくとも1の上述の薬剤と混合されてもよく、または実施可能に結合されてもよい。化合物、治療剤、イメージング剤またはその組合せは、非経口、皮内、皮下、経口、経皮、経粘膜または直腸に投与されてよい。
【0109】
診断法
本発明の方法は、標的領域をイメージングする、例えば、癌のインテグリン受容体イメージングするために有用である。治療学的放射性医薬品は、治療学的に有用な放射性核種、例えば、β粒子、α粒子およびオージェまたはコスター-クロニッグ電子(Coster-Kronig electrons)などの電離放射線を放射する放射活性金属イオンから構成される医薬である。放射性核種に実施可能に結合される本発明の化合物は、インテグリンを発現している細胞に対して放射線核種をターゲティングすることを促進する。それから、イメージングは、患部領域、例えば、心または肢などの標準的な技術を使用して、実行することが可能であり、それにより疾患の存在を検出したり、または疾患の進行または治療学的処置の結果ををモニターしたりすることが可能である。
【0110】
スクリーニング法
本発明の1つの目的は、インテグリン結合化合物を同定する方法を提供することである。従って、本発明は、ファルマコフォアの提示を具備するコンピュータ読み取り可能媒体を提供し、ここで、当該ファルマコフォアは、1のH結合供与体(HBD)、1のH結合受容体(HBA)、2の疎水性芳香族基(HAR1およびHAR2)および1の陰性イオン性基(NI)の特徴を含む。
【0111】
一つの態様において、当該HBD、HBA、HAR1、HAR2およびNIは、図8Aに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.81±1 Åであり、当該HBDとHBAの距離は7.52±1 Åであり、当該HBDとNIの距離は17.67±1 Åであり、当該HRA1とHBAの距離は7.52±1 Åであり、当該HRA1と HRA2の距離は9.41±1 Åであり、当該HBAとNIの距離は10.20±1 Åであり、および当該HRA2とNIの距離は5.88±1 Åである。特に、当該HBDとHRA1の距離は2.81 Åでよく、当該HBDとHBAの距離は7.52 Åでよく、当該HBDとNIの距離は17.67 Åでよく、当該HRA1とHBA の距離は7.52 Åでよく、当該HRA1とHRA2の距離は9.41 Åでよく、当該HBAとNIの距離は10.20 Åでよく、および当該
HRA2とNI の距離は5.88 Åでよい。
【0112】
もう一つの態様において、当該HBD、HBA、HAR1、HAR2,およびNIは、図8Bに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.80±1 Åであり、当該HBDとHBAの距離は7.34±1 Åであり、当該HBDとNIの距離は16.92±1 Åであり、当該HRA1とHBAの距離は5.20±1 Åであり、当該HRA1とNIの距離は14.34±1 Åであり、当該HBAとHRA2の距離は4.69±1 Åであり、当該HBAとNI の距離は9.85±1 Åであり、および当該HRA2とNIの距離は5.66±1 Åである。特に、当該HBDとHRA1の距離は2.80 Åでよく、当該HBDとHBAの距離は7.34 Åでよく、当該HBDとNIの距離は16.92 Åでよく、当該HRA1とHBAの距離は5.20 Åでよく、当該HRA1とNIの距離は14.34 Åでよく、当該HBAとHRA2の距離は4.69 Åでよく、当該HBAとNIの距離は9.85 Åでよく、および当該HRA2とNIの距離は5.66 Åでよい。
【0113】
更なるもう一つの態様において、当該HBD、HBA、HAR1、HAR2およびNIは、図8Cに従う立体配置であり、ここで、当該HBDとHRA1の距離は 2.79±1 Åであり、当該HBDとHRA2の距離は11.94±1 Åであり、当該HBDとNIの距離は16.28±1 Åであり、当該HRA1とHBAの距離は5.20±1 Åであり、当該HRA1とNI の距離は14.64±1 Åであり、当該HBAとHRA2の距離は4.72±1 Åであり、および当該HRA2とNIの距離は5.63±1 Åである。特に、当該HBDとHRA1の距離は2.79 Åでよく、当該HBDとHRA2の距離は11.94 Åでよく、当該HBDとNIの距離は16.28 Åでよく、当該HRA1とHBAの距離は5.20 Åでよく、当該HRA1とNIの距離は14.64 Åでよく、当該HBAとHRA2の距離は4.72 Åでよく、および当該HRA2とNI の距離は5.63 Åでよい。
【0114】
本明細書に使用される、「コンピュータ読み取り可能媒体」は、コンピュータにより直接的に読まれるおよびアクセスされることが可能な何れかの媒体をいう。そのような媒体は、これらに限定するものではないが、磁気記憶媒体、例えば、フロッピー(登録商標)ディスク、ハードディスク記憶媒体、および磁気テープ;光学記憶媒体、例えば、CD-ROM;電気的記憶媒体、例えば、RAMおよびROM;およびこれらのカテゴリーのハイブリッド、例えば、磁気/光学記憶媒体などを含む。当業者は、当該分野において周知の何れかの方法を用いて、本発明のファルマコフォアの表示をそこにおいて記憶されたコンピュータ読み取り媒体を容易に作ることが可能である。
【0115】
コンピュータ読み取り可能形態において、本発明のファルマコフォアの表示を提供することにより、当業者は、多様な目的のためにファルマコフォアの情報にルーチンとしてアクセスすることが可能である。例えば、当業者は、コンピュータ読み取り可能形態中の本発明のファルマコフォアを、データ保管手段中に格納されている化合物情報と比較するために使用することが可能である。そのような手段は、当該ファルマコフォアの特徴とマッチする化合物を同定し、それにより、インテグリン結合分子の候補を出すために使用される。
【0116】
従って、本発明は、インテグリン結合小分子を同定する方法を提供する。当該方法は、化合物の三次元構造と本発明のファルマコフォアの三次元構造とを比較すること、およびもし当該化合物が当該ファルマコフォアの特徴と一致した場合に、当該化合物を選択することを具備する。
【0117】
続く例は、本発明の範囲を説明することが目的であるが、これを制限するものではない。このような例は、典型的に、使用され得るものの典型的なものでり、当業者にとって公知の他の手順は、変更可能に利用されてよい。実際、当業者は、過度の実験を行うことなしに、本明細書の教示を基に更なる態様を容易に思い描くことおよび作製することが可能である。
【0118】
例I
小分子インテグリンの発見
新規抗癌剤としてのαvβ3アンタゴニスト
実験部門
ファルマコフォア仮定の発生およびバリデーション。トレーニングセット化合物(A-C)の構造が、組み立てられ、カタリスト(Catalyst(Accelrys、Inc.))を使用して徹底的に最小化された40。各化合物の到達可能なコンフォメーションフレキシビリティが探索できた独特のコンフォメーションのセットが、カタリストのキャットコンフォモデル(Catconf module)を使用して生じた。当該カタリストにおいて実行された当該ポーリングアルゴリズム(poling algorithm)が使用され、コンフォメーションを生じた。当該ポーリングアルゴリズムは、高いコンフォメーションの変化を促進し、低いエネルギーのコンフォメーションスペースの広い適用範囲を保証する49−51。当該共通の特徴ファルマコフォア仮説が、カタリストのヒップホップアルゴリズム(HipHop algorithm)を使用して生じた。ヒップホップは、そのデフォルトセッティングを有する10のファルマコフォアを生じる。ヒップホップは、トレーニングセット分枝のコンフォメーションモデルの収集物および化学的特徴の選択を得て、当該トレーニングセット分子の共通する立体配置を同定する。化合物Aは、ファルマコフォア仮説発生例における主要な化合物とみなされる。当該トレーニングセット化合物の構造およびαvβ3活性部位特徴の化学的特徴を基本に、ファルマコフォア的特徴のセットが当該ファルマコフォア発生例の最初に選択される。公知のαvβ3アンタゴニストの検索可能なマルチコンフォーマーデータベースは、カタリストデータベースサーバーを使用して発生される。このデータベースは、当該ファルマコフォアモデルをバリデーションするために使用される。
【0119】
ドッキング試験。ドッキングは、ゴールド(GOLD)プログラム(Genetic Optimization for Ligand Docking)のバージョン1.2を使用して行った42。ゴールドは、遺伝子アルゴリズムを使用する自動化ドッキングプログラムであり、活性部位の部分的なフレキシビリティを有するリガンドコンフォメーションフレキシビリティを探索する52。当該アルゴリズムは、ブルックタンパク質データベース(the Brookhaven Protein DataBank)から抽出された300以上の複合体のデータセットにおいて試験される。ゴールドは、当該リガンドの実験的結合コンフォメーションを再現することにおいて70%よりも多くのケースにおいて成功した53。ゴールドは、ユーザーの定義した結合部位を必要とする。それは、定義された領域内の空洞を探索し、定義された領域において全ての溶媒到達可能な原子を活性部位原子とみなす。適切なプロトン化状態は、酸性および塩基性アミノ酸残基のために割り当てる。当該受容体において存在する全ての水分子は除去され、水素原子がインテグリンαvβ3受容体に対して与えられた。各分子の全てのコンフォマーは、αvβ3受容体活性部位にドッキングした。各実行の終点で、ゴールドは、適応度スコアおよび二乗平均平方根距離(RMSD)を基に全ての生じた結合コンフォメーションを分離し、ランク付けする。全てのドッキングの実行は、集合サイズ100、最大数100,000操作、変異およびクロスオーバー率95を伴う標準デフォルトセッティングを使用して実行された。ゴールドにおいて実行された当該適合度機能は、H-結合、複合体エネルギーおよびリガンド内部エネルギー期間(terms)をからなる。当該ドッキング試験は、24-CPU シリコン・グラフィックス・オニックス・ワーク(24-CPU Silicon Graphics Onyx workstation)において実行した。
【0120】
細胞培養。ヒト乳癌細胞(MCF-7、αvβ3-、過剰発現野生型p53、ER+; MDA-MB 468、p53 変異体、ER+;およびMDA-MB-435、αvβ3+、p53 変異体、ER-)および非小細胞肺癌細胞H1975をザ・アメリカン・タイプ・セル・カルチャー(the American Type Cell Culture(Rockville、MD Q4))から入手した。天然にシスプラチン(CDDP)に対して耐性である当該HEYヒト卵巣癌腫細胞系は、ドクター・デュビュー(Dr. Dubeau(University of Southern California Norris Cancer Center))から快く提供された。細胞は、単層培養物として、10%のウシ胎仔血清(Gemini-Bioproducts、WoodlおよびCA)および2mmol/L L-グルタミンを添加したRPMI 1640中で、37℃で、5%CO2の加湿した雰囲気において維持した。継代および計数のために当該フラスコから接着細胞を剥がすために、細胞をカルシウムまたはマグネシウム非含有のPBSで洗浄し、少量の0.25%のトリプシン-EDTA溶液(Sigma、St. Louis、MO)で5分〜10分間インキュベートし、培地で洗浄し、遠心した。全ての実験は、指数関数的な細胞増殖における細胞を使用して行った。
【0121】
薬物。全ての化合物の10mMストック溶液を、DMSO中で調製し、-20で保存した。更なる希釈は、PBS中で新鮮に作製した。
【0122】
受容体結合アッセイ。NCI-H1975細胞の表面における全ての化合物の結合親和性は、変更を加えた文献に記載の準じて、125I-標識エキスタチン(echistatin)を放射性リガンドとして使用し競合的結合実験において決定した41。即ち、NCI-H1975 細胞を収集し、2回PBSで洗浄し、結合緩衝液(20 mMトリス、pH 7.4、150 mM NaCl、2 mM CaCl2、1 mM MgCl2、1 mM MnCl2、0.1% BSA)中に再懸濁(2 x 106 cells/mL)した。96-ウェルマルチスクリーンDVプレート(フィルターポアサイズ:0.65μm、ミリポア、Billerica, MA)を、αvβ3アンタゴニストの大濃度の存在下で125I-エキスタチンインキュベートした。総インキュベート量は、200μLに調整した。当該細胞を3時間室温でインキュベートした後に、当該プレートをマルチスクリーン減圧マニフォールドを介して濾過し、2回冷結合緩衝液で洗浄した。親水性のPVDFフィルターを回収し、その放射能をNaI(TI)ガンマカウンター(Packard、Meriden、CT)で測定した。最も適合したIC50値をグラフィックパッド・プリズム(GraphPad Prism(GraphPad Software、Inc.、San Diego、CA))を使用した非線形回帰により当該データを適合させることにより計算した。実験は、トリプリケイトサンプルで実施した。
【0123】
細胞毒性試験。細胞毒性は、3-(4,5-ジメチルチアゾール-2-イル)-2,5-ジフェニルテトラゾリウムブロミド(MTT)アッセイで以前に記載した通りに評価した41、54。即ち、細胞を96ウェルマイクロタイタープレートに播種し、接着させる。細胞を次に、連続して72時間に亘り対応する薬物に暴露した。MTT 溶液(最終濃度0.5 mg/mL)を各ウェルに添加し、細胞を4時間37℃でインキュベートした。培地の除去後、DMSOを添加し、570nmで吸光度を測定した。全てのアッセイは、トリプリケイトで行った。次に、そのIC50 は各薬物について対数(薬物濃度)対細胞死滅パーセンテージとして測定した。
【0124】
結果および考察
インテグリンαvβ3アンタゴニストの設計。RGDペプチドを使用した以前の試験は、αvβ3は、RGDトリペプチド配列の重要なグアニジン(Arg)およびカルボン酸(Asp)基(ArgとAsp残基のCβ原子の間の距離)の間の短い全体的な分離を認識することを示唆した35、36。バックボーンコンフォメーションにおいて捩れを伴う環状RGDペプチドは、他のインテグリンよりもαvβ3の対してより選択性を示した24。このサイクリックRGDペプチド-αvβ3相互作用モデルは、多様な小分子ペプチドミメティックアンタゴニストの発見のための開始点を提供する。αvβ3の細胞外セグメントの結晶構造およびそのサイクリックRGDペプチドとの複合体は、以前に報告されている37、38。我々は、共通の特徴ファルマコフォアモデルを産生するための最近報告された小分子αvβ3アンタゴニストのセットを利用し、それらは次に、638の公知のαvβ3アンタゴニストのデータベースに対してバリデートした39。当該バリデーションしたファルマコフォアモデルを、新規構造骨格および所望の化学的特徴を有する分子を検索するために探索照会(search queries)として使用した。新規αvβ3アンタゴニストを同定し、設計するための我々の戦略は、図1に模式的に示す。
【0125】
共通の特徴ファルマコフォアモデルの発生。一般に、共通の特徴ファルマコフォアモデルの発生のためのトレーニングセットは、同様な活性プロフィールおよび活性部位結合メカニズムを有する化合物を含むべきであり、それによりこれらのトレーニング化合物がそれらの生物学的活性を招く比較できる特徴の3D配置を有する可能性を高める。カタリスト・ソフトウェア・パッケージにおける当該ヒップホップアルゴリズムは、共通する特徴ファルマコフォアモデルを引き出すために、30から49nM(図2)の範囲におけるαvβ3インテグリンに匹敵する結合親和性を有する最近報告された3つのαvβ3インテグリン(A、BおよびC)のアンタゴニストからなるトレーニングセットに対して適用される39、40。当該トレーニングセット化合物は、類似体をより集め、同様の結合コンフォメーションにおけるαvβ3受容体の活性部位における同様の部位に対する結合を予測する。当該ファルマコフォアの特徴は、(1)トレーニングセットアンタゴニストの構造および化学的特徴、(2)αvβ3受容体活性部位の構造、および(3)サイクリックRGDペプチド(PDB1L5G)と複合したαvβ3受容体の共結晶構造におけるサイクリックRGDペプチドと突出したαvβ3受容体活性残基との間で観察される重要な相互作用を基礎に選択される37。ファルマコフォアモデル発生実験において考慮される特徴は、H結合供与体(HBD)、H結合受容体(HBA)、環芳香族(HYR)、疎水性(HYA)および陰性イオン性基(negatively ionizable(NI))特徴である。ピップホップは、10 5の特徴とする(10 five-featured)ファルマコフォア仮定を生じた。これらの仮定がそれらのファルマコフォアの特徴において同様である一方で、種々の特徴の相対的な配向、位置およびベクター方向は異なる。カタリストプログラムにおいて入手可能な階層的完全連関法(hierarchical complete linkage method)を使用した10仮定のクラスター解析は、3つのクラスターを産生した。これらのクラスターのそれぞれからの代表的なモデル(ファルマコフォア仮定 Hypo1、Hypo5およびHypo9)を更なる解析およびバリデーションのために選択した。
【0126】
共通する特徴ファルマコフォアのバリデーション。内部のバリデーションとして、トレーニングセット化合物A-Cを、3つのファルマコフォア上にマッピングした。トレーニングセット化合物(表1)のマッピングされたコンフォメーションの適合度スコアおよび関連するエネルギーは、化合物AおよびBのためにはHypo1またはHypo9よりもHypo5の方が低いエネルギーを産生したが、化合物Cのためには相対的に高いエネルギーを産生した。図3に示すように、化合物AへのHypo5のマッピングは、この化合物の重要な化学的特徴と当該ファルマコフォアとの間の良好な一致を確認した。
【表1】
【0127】
不活性化合物からの強力なアンタゴニストの分離における、これらのファルマコフォアの判別可能な能力を評価するために、当該3つの代表的なファルマコフォアを3D照会として使用し、公知のαvβ3受容体アンタゴニストのデータベースを検索した。このデータベースは、αvβ3受容体に対する広い範囲の活性プロフィールを有する計638化合物を有する。当該638化合物のうち、αvβ3重要対阻害をIC50値≦20nMで示す303が、強力なαvβ3アンタゴニストとみなされた。検索結果を表2に要約する。147化合物を検索した他の2つのファルマコフォアよりも良好に実行されたHypo5の88(〜60%)化合物がαvβ3の強力なアンタゴニストであった。Hypo9はより活性な化合物を検索したが、強力とみなされた化合物の存在は、Hypo5よりも低かった(〜54%)。強力なアンタゴニストを選択する優れた能力を有するHypo5および3つのトレーニングセット化合物の2つに対するHypo5のマッチを基に、このファルマコフォアが次の工程のために選択された:新規構造骨格および所望の特徴を有する化合物の検索のための我々の構内のデータベースのサブセットを探索するための3D照会。
【表2】
【0128】
データベース検索および化合物選択。238,819化合物のNCI12000データベースのHypo5を使用する探索は、684ヒット得た。Hypo5により検索された684化合物のうち、分子量≦500を有する282化合物が、更なる物理化学的性質解析のために考えられ、残りは処分された。359,224化合物のケミカル・ダイバーシティ(ChemDiv、Inc、San Diego、CA)データベースの検索は148ヒットを生じた。物理化学的性質フィルターを使用し、インビトロスクリーニングの前に薬物様性質を有していない化合物を排除した41。ファルマコフォア適合値、計算物理化学的性質およびサンプル有効性を基に、8 化合物をNCI化合物レポジトリーから得て、21化合物のコレクションを、インビトロ評価のためにケミカル・ダイバーシティ(Chemical Diversity)から購入した。
【0129】
αvβ3 結合。 非小細胞肺癌 NCI-H1975 細胞の表面における全ての化合物の受容体結合親和性は、125I-標識エキスタチンを放射性リガンドとして使用する競合結合実験で測定し、結果を表3に示す。試験された29の化合物のうち、4つの化合物は、顕著なαvβ3結合親和性(AV3 = 52 nM、AV26 = 240 nM、AV27 = 18 nM、AV32 = 605 nM)を示した。2つの化合物、AV3およびAV27 は、トレーニング化合物A-Dと同様の範囲の結合親和性を示した(30 − 49 nM)39。バリデーション解析において得られた通り、Hypo5 は、ナノモーラーの結合親和性を有する構造的に多様な強力なアンタゴニストの検索において成功した。これらの化合物は、多様な構造骨格を有するアンタゴニストの新規セットを意味する。当該トレーニングセット化合物とは異なって、当該化合物は、親水性質の幾つかの官能基を有する。これは、αvβ3のRGDペプチド結合領域の高い静電気的性質を考慮する重要な特徴である。当該RGDペプチドは、αvβ3サイクリックRGDペプチド複合体結晶構造におけるαvβ3を有するその2つの負荷末端を介する強い静電気的相互作用を形成する。化合物AV3、AV27およびAV32は、RGDミメティックアンタゴニストの範疇に大まかに含まれ、これはそれらがαvβ3と同様の静電気的相互作用を確立することができる何れかの末端にカルボキシル基またはアミン/アミド基を有するからである。加えて、これらの化合物における幾つかの親水性官能基の存在は、αvβ3のRGD結合部位の高い静電気的領域との相互作用に助力する。これらの興味深い特徴が得られたので、我々は、AV27およびAV32の構造学的に近い更なる類似体を試験した。しかしながら、我々の構内データベースにおけるこれらの化合物のコア骨格を使用する下部構造検索照会では、近い類似体は得られない。これらの2つの化合物のコア骨格周辺の構造−活性研究は、αvβ3に向かうそれらの結合親和性を更に最適化するための代替オプションであってよい。独特な構造的特徴を有しているために、AV26は、αvβ3アンタゴニストの新規種類の非RGDミメティックを代表する。我々は、我々の結合アッセイにおいて化合物AV34からAV38を試験することにより、化合物AV26における制限された構造活性相関(SAR)分析を実施した(表3)。検索照会としてAC26のコア骨格を使用する下部構造探索は、359,224化合物の我々の構内データベースから幾つかの化合物を検索した。当該類似体AV34からAV38は、ケミカル・ダイバーシティ(Chemical Diversity(San Diego、CA))から購入し、αvβ3結合親和性をスクリーニングした。このSAR試験から、我々は2つの新規化合物を発見した:AV35 はIC50 値が24 nMであり、親化合物AV26よりも10倍高い親和性を示した。AV38 はIC50 値が0.03 nMであり、最も強力であり、親AV26よりも800倍高い親和性を示した。化合物 AV26、AV35およびAV38は、通常のRGDミミックαvβ3アンタゴニストから構造学的に逸脱しており、それらが新規な機序の相互作用を有するαvβ3の非RGDミミックアンタゴニストの新規種類を代表するものであることが示唆される。我々の知識に対して、これらの化合物は、これまでに記載された最も強力なαvβ3アンタゴニストの中に含まれる。併せて、我々の6つの化合物は、小分子αvβ3アンタゴニストを代表するものであり、研究は、抗癌剤としてのそれらのインビボでの有効性を示すために進行中である。また、研究は、共有結合を介してαvβ3の過剰発現癌に対する選択的送達細胞障害剤、例えば、パクリタキセルなどに対しても進行中である。
【表3】
【表S1】
癌細胞系のパネルにおける選択された化合物の細胞毒性。最初に、我々は、全ての化合物を、高および低αvβ3発現する2つの乳癌細胞系MDA-MB-435およびMCF7、並びに天然でシスプラチンに対する抵抗性のHEY卵巣癌細胞系において試験した。20μM(表4)での細胞増殖の顕著な阻害が見られた化合物は、続いて、5つの細胞系のパネルにおいて試験した(表5)。我々は、これらの細胞に対する当該化合物の幾つかについて、顕著な特異性を観察した。例えば、AV26は、MCF7、NIH3T3,およびCRL5908 細胞に比較して、MDA-MB-435細胞に対して60倍の選択性を示し、HEY細胞よりも40倍近くの選択性を示した。同様の経口が、しかし程度は小さいが、化合物AV34で観察された。一方、AV30は、MDA-MB-435、HEYおよびNIH3T3細胞においてはまさに同様のプロフィールが示されたが、MCF-7およびNCI-H1975細胞に対しては有意に活性が低かった。興味深いことに、AV26を除く当該新規アンタゴニストの何れか1項に記載のもが、注目すべき細胞毒性を示さず、これは、これらの新規のアンタゴニストは、治療非細胞毒性メカニズムを基礎とする抗癌治療として有用性を有していることを示す。αvβ3が癌細胞の遊走、接着およびアポトーシスを媒介するという事実を考慮すると、これらの新規の高親和性アンタゴニストと通常の細胞障害性薬物との組合せが、更なる毒性のない改良された治療学的な利点を示すものである。
【表4】
【表5】
【0130】
ドッキング試験。 我々の新規アゴニストの配向に結合することを確認するために、我々は、化合物A、AV3、AV26、AV27およびAV38を当該αvβ3 RGD 結合領域にゴールドを使用してドッキングさせた42。当該αvβ3RGD結合領域内側での化合物A、AV3、AV26、AV27およびAV38の当該予測される結合コンフォメーションは、図5a−eに示す。コールドは、各化合物について、幾つかの可能な結合コンフォメーションを生じ、それらの適応度スコアに従って、それらをランク付けした。最も有利なエネルギーを有する結合コンフォメーションが最高の結合配向であると考えられた。サイクリックRGDペプチドと複合したαvβ3受容体の結晶構造において、サイクリックRGDペプチドの1つのAspカルボキシル化酵素がαvβ3受容体のβ鎖におけるMIDAS(Metal Ion-Dependent Adhesion Site)でMn2+と相互作用する一方で、そのArg グアニジニウム基は、αvβ3受容体(PDB1L5G)β鎖のD218およびα鎖のD150と相互作用する。化合物 A、AV3、AV26およびAV27は、MIDASのMn2+を有するそれらのカルボキシ末端で同様に結合する相互作用を確立し、同様に、アミノ酸残基S121およびS123の側鎖のヒドロキシル基との幾つかのH-結合相互作用の形成を確立する。当該サイクリックRGDペプチドのArg側鎖の結合配向に対比して、化合物 A、AV3、AV27の当該アミン/アミド関連末端およびAV26のヒドロキシル関連フェニル基は、β鎖のD218から離れた空洞を占領するが、α鎖のD150には近い。この空洞は、αvβ3のβ鎖からのアミノ酸残基Y166、P170、D179、R214、N215およびR216およびα鎖のアミノ酸残基 K119、E121、D148、D150、Y178,およびR248により囲まれている。著しく、最も強力な非RGDミメティックアンタゴニストAV38は、逆向きの結合配向を採用し、αvβ3のサイクリックRGDペプチド結合領域において種々のアミノ酸残基と強い静電気的相互作用のセットを確立した。化合物 A、AV26,およびAV38と当該αvβ3RGDペプチド結合部位での種々のアミノ酸残基との間の観察された重要な相互作用の略図の提示は図6に示す。AV38の当該ジメトキシ関連フェニル基は、MIDASのMn2+および2つの、Mn2+に対応するメトキシ酸素原子の近くの領域を占める。3つのH結合相互作用が当該メトキシ酸素原子とアミノ酸残基S121およびS123のヒドロキシル基の間に観察された。当該カルボキシル化関連フェニル基は、αvβ3のβ鎖からのアミノ酸残基 Y116、P170、D179、R214およびR216およびα鎖からのD148、A149、D150およびY178により囲まれる領域を占めた。当該カルボキシル化酸素原子は、R216グアニジニウム基、A149の主鎖NHおよびY166のヒドロキシル基と、幾つかの強いH結合相互作用を形成した。最も強力なアンタゴニストAV38を除いて、一貫したパターンが、全てのアンタゴニストの予測された結合配向において観察された。同様の化学的性質を有するこれらのアンタゴニストの当該官能性基は、αvβ3RGDペプチド結合領域における同様の領域を占め、Mn2+と他のアミノ酸残基との同様な相互作用を形成した。これは、我々のドッキング試験により予測されたこれらのアンタゴニストの結合コンフォメーションの質を支持するものである。幾つかの矛盾が、以前に予測されたRGDミメティックペプチドミメティックおよび非ペプチド小分子アンタゴニストの結合配向において見出されている43−47。非ペプチド小分子アンタゴニストを使用しない以前に報告されたドッキング試験もまた、この試験において予測された配向に対して非常に類似する結合配向を予測した43,45。しかしながら、ドッキング試験は、αvβ3−RGD複合体結晶構造におけるサイクリックRGDの結合コンフォメーションに対して同様な結合配向を生成したサイクリックRGD類似体およびRGDミメティックにおいて行った46,47。我々の新規アンタゴニストの予測された結合配向とαvβ3ーRGD複合体結晶構造におけるサイクリックRGDペプチドの結合コンフォメーションとの間で観察された矛盾は、当該サイクリックRGDペプチドとこれらの新規アンタゴニストの間の有意な構造上の相違のためである。他方、観察された矛盾は、我々のドッキング手順におけるアーチファクトの結果かもしれない。しかしながら、我々の最も強力な非RGDミメティックアンタゴニストAV38により示された高結合親和性(親AV26よりも800倍高い)は、その予測される結合配向において見出される新規な結合相互作用により説明される。この予測される結合配向において、AV38は、幾つかの強いH結合相互作用を形成し、明らかな相補性が、その官能基の化学的性質とαvβ3におけるその結合部位の周辺の種々のアミノ酸残基の間で見いだされた。更なる構造研究は、この非RGDミメティックアンタゴニストおよびそのαvβ3との相互作用により及ぼされる高結合親和性の説明を保証するものである。
【0131】
最後に、ノックアウトマウスでの遺伝的結果と、全ての報告されているアンタゴニストによる薬理学的効果との間の分野における最近の矛盾を説明するために、我々は、リチャード・へネス(Richard Hynes)によるモデル(彼は、β3インテグリンを遺伝的にノックアウトしたマウスが増強された血管形成を示した理由を考察している)に言及する(図7)48。彼は、RGDペプチドは、実際に「アンタゴニスト」の代わりに「アゴニスト」と称されるべきであると主張している。仮に、当該文言アゴニストが使用された場合、当該遺伝子学的モデルは、薬理学的モデルに一致する。この明細書を通して、我々は、最も以前の報告において使用していたように文言「アンタゴニスト」を使用しているが、我々は、これらの化合物は実際にはアゴニストとして働く可能性に気付いている。どんなメカニズムであっても、我々の化合物は高度に強力であり、αvβ3陽性細胞に対して選択的に送達するための種々の化学療法剤に対して複合化するために非常に適切である。
【0132】
我々は、構造的に多様なインテグリンαvβ3アンタゴニストのシリーズを、小分子薬物様化合物のデータベースのファルマコフォアスクリーニングを介して同定してきた。共通の特徴3Dファルマコフォアモデルは、公知のインテグリン αvβ3アンタゴニストのセットを利用して発生させた。バリデーションされたファルマコフォアモデルは、当該ファルマコフォアモデルを生じるために使用したトレーニングセット化合物よりも高い効力を有する構造学的に新規な化合物をうまく検索した。更に、1つの強力なアンタゴニストにおける制限された構造活性相関解析は、非RGDミメティックαvβ3アンタゴニストとしてサブナノモーラーの効力を有する高い効力の化合物を発見した。受け入れられる構造骨格を有するこれらの小分子アンタゴニストは、強力な非RGDミメティックαvβ3アンタゴニストとして更に最適化するために役立つ手がかりを提供する。我々は、我々の最近の原稿に記載したとおり、当該3つの強力なアンタゴニストとパクリタキセルを複合化している41。これらの新規の薬剤のαvβ3陽性癌細胞への標的送達についての詳細な薬理学的性質を示す(Figure 7)。
【0133】
参考文献
1. Danen, E. H., Integrins: regulators of tissue function and cancer progression. Curr Pharm Des 2005, 11, (7), 881-91.
2. Humphries, M. J., Integrin structure. Biochem Soc Trans 2000, 28, (4), 311-39.
3. Martin, K. H.; Slack, J. K.; Boerner, S. A.; Martin, C. C.; Parsons, J. T., Integrin connections map: to infinity and beyond. Science 2002, 296, (5573), 1652-3.
4. Aplin, A. E.; Howe, A. K.; Juliano, R. L., Cell adhesion molecules, signal transduction and cell growth. Curr Opin Cell Biol 1999, 11, (6), 737-44.
5. Brooks, P. C.; Clark, R. A.; Cheresh, D. A., Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science 1994, 264, (5158), 569-71.
6. Hood, J. D.; Cheresh, D. A., Role of integrins in cell invasion and migration. Nat Rev Cancer 2002, 2, (2), 91-100.
7. Felding-Habermann, B., Integrin adhesion receptors in tumor metastasis. Clin Exp Metastasis 2003, 20, (3), 203-13.
8. Natali, P. G.; Hamby, C. V.; Felding-Habermann, B.; Liang, B.; Nicotra, M. R.; Di Filippo, F.; Giannarelli, D.; Temponi, M.; Ferrone, S., Clinical significance of alpha(v)beta3 integrin and intercellular adhesion molecule-1 expression in cutaneous malignant melanoma lesions. Cancer Res 1997, 57, (8), 1554-60.
9. Felding-Habermann, B.; O'Toole, T. E.; Smith, J. W.; Fransvea, E.; Ruggeri, Z. M.; Ginsberg, M. H.; Hughes, P. E.; Pampori, N.; Shattil, S. J.; Saven, A.; Mueller, B. M., Integrin activation controls metastasis in human breast cancer. Proc Natl Acad Sci U S A 2001, 98, (4), 1853-8.
10. Pecheur, I.; Peyruchaud, O.; Serre, C. M.; Guglielmi, J.; Voland, C.; Bourre, F.; Margue, C.; Cohen-Solal, M.; Buffet, A.; Kieffer, N.; Clezardin, P., Integrin alpha(v)beta3 expression confers on tumor cells a greater propensity to metastasize to bone. Faseb J 2002, 16, (10), 1266-8.
11. Kumar, C. C.; Armstrong, L.; Yin, Z.; Malkowski, M.; Maxwell, E.; Ling, H.; Yaremko, B.; Liu, M.; Varner, J.; Smith, E. M.; Neustadt, B.; Nechuta, T., Targeting integrins alpha v beta 3 and alpha v beta 5 for blocking tumor-induced angiogenesis. Adv Exp Med Biol 2000, 476, 169-80.
12. Shannon, K. E.; Keene, J. L.; Settle, S. L.; Duffin, T. D.; Nickols, M. A.; Westlin, M.; Schroeter, S.; Ruminski, P. G.; Griggs, D. W., Anti-metastatic properties of RGD-peptidomimetic agents S137 and S247. Clin Exp Metastasis 2004, 21, (2), 129-38.
13. Ruoslahti, E.; Pierschbacher, M. D., Arg-Gly-Asp: a versatile cell recognition signal. Cell 1986, 44, (4), 517-8.
14. Hynes, R. O., Integrins: versatility, modulation, and signaling in cell adhesion. Cell 1992, 69, (1), 11-25.
15. Kerr, J. S.; Wexler, R. S.; Mousa, S. A.; Robinson, C. S.; Wexler, E. J.; Mohamed, S.; Voss, M. E.; Devenny, J. J.; Czerniak, P. M.; Gudzelak, A., Jr.; Slee, A. M., Novel small molecule alpha v integrin antagonists: comparative anti-cancer efficacy with known angiogenesis inhibitors. Anticancer Res 1999, 19, (2A), 959-68.
16. Lark, M. W.; Stroup, G. B.; Hwang, S. M.; James, I. E.; Rieman, D. J.; Drake, F. H.; Bradbeer, J. N.; Mathur, A.; Erhard, K. F.; Newlander, K. A.; Ross, S. T.; Salyers, K. L.; Smith, B. R.; Miller, W. H.; Huffman, W. F.; Gowen, M., Design and characterization of orally active Arg-Gly-Asp peptidomimetic vitronectin receptor antagonist SB 265123 for prevention of bone loss in osteoporosis. J Pharmacol Exp Ther 1999, 291, (2), 612-7.
17. Reinmuth, N.; Liu, W.; Ahmad, S. A.; Fan, F.; Stoeltzing, O.; Parikh, A. A.; Bucana, C. D.; Gallick, G. E.; Nickols, M. A.; Westlin, W. F.; Ellis, L. M., Alphavbeta3 integrin antagonist S247 decreases colon cancer metastasis and angiogenesis and improves survival in mice. Cancer Res 2003, 63, (9), 2079-87.
18. Harms, J. F.; Welch, D. R.; Samant, R. S.; Shevde, L. A.; Miele, M. E.; Babu, G. R.; Goldberg, S. F.; Gilman, V. R.; Sosnowski, D. M.; Campo, D. A.; Gay, C. V.; Budgeon, L. R.; Mercer, R.; Jewell, J.; Mastro, A. M.; Donahue, H. J.; Erin, N.; Debies, M. T.; Meehan, W. J.; Jones, A. L.; Mbalaviele, G.; Nickols, A.; Christensen, N. D.; Melly, R.; Beck, L. N.; Kent, J.; Rader, R. K.; Kotyk, J. J.; Pagel, M. D.; Westlin, W. F.; Griggs, D. W., A small molecule antagonist of the alpha(v)beta3 integrin suppresses MDA-MB-435 skeletal metastasis. Clin Exp Metastasis 2004, 21, (2), 119-28.
19. Brooks, P. C.; Stromblad, S.; Klemke, R.; Visscher, D.; Sarkar, F. H.; Cheresh, D. A., Antiintegrin alpha v beta 3 blocks human breast cancer growth and angiogenesis in human skin. J Clin Invest 1995, 96, (4), 1815-22.
20. Brooks, P. C.; Montgomery, A. M.; Rosenfeld, M.; Reisfeld, R. A.; Hu, T.; Klier, G.; Cheresh, D. A., Integrin alpha v beta 3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell 1994, 79, (7), 1157-64.
21. Taga, T.; Suzuki, A.; Gonzalez-Gomez, I.; Gilles, F. H.; Stins, M.; Shimada, H.; Barsky, L.; Weinberg, K. I.; Laug, W. E., alpha v-Integrin antagonist EMD 121974 induces apoptosis in brain tumor cells growing on vitronectin and tenascin. Int J Cancer 2002, 98, (5), 690-7.
22. Mitjans, F.; Meyer, T.; Fittschen, C.; Goodman, S.; Jonczyk, A.; Marshall, J. F.; Reyes, G.; Piulats, J., In vivo therapy of malignant melanoma by means of antagonists of alphav integrins. Int J Cancer 2000, 87, (5), 716-23.
23. Allman, R.; Cowburn, P.; Mason, M., In vitro and in vivo effects of a cyclic peptide with affinity for the alpha(nu)beta3 integrin in human melanoma cells. Eur J Cancer 2000, 36, (3), 410-22.
24. Dechantsreiter, M. A.; Planker, E.; Matha, B.; Lohof, E.; Holzemann, G.; Jonczyk, A.; Goodman, S. L.; Kessler, H., N-Methylated cyclic RGD peptides as highly active and selective alpha(V)beta(3) integrin antagonists. J Med Chem 1999, 42, (16), 3033-40.
25. Burke, P. A.; DeNardo, S. J.; Miers, L. A.; Lamborn, K. R.; Matzku, S.; DeNardo, G. L., Cilengitide targeting of alpha(v)beta(3) integrin receptor synergizes with radioimmunotherapy to increase efficacy and apoptosis in breast cancer xenografts. Cancer Res 2002, 62, (15), 4263-72.
26. Rodan, S. B.; Rodan, G. A., Integrin function in osteoclasts. J Endocrinol 1997, 154 Suppl, S47-56.
27. Nakamura, I.; Pilkington, M. F.; Lakkakorpi, P. T.; Lipfert, L.; Sims, S. M.; Dixon, S. J.; Rodan, G. A.; Duong, L. T., Role of alpha(v)beta(3) integrin in osteoclast migration and formation of the sealing zone. J Cell Sci 1999, 112 ( Pt 22), 3985-93.
28. Duong, L. T.; Rodan, G. A., The role of integrins in osteoclast function. J Bone Miner Metab 1999, 17, (1), 1-6.
29. Teitelbaum, S. L., Bone resorption by osteoclasts. Science 2000, 289, (5484), 1504-8.
30. Fisher, J. E.; Caulfield, M. P.; Sato, M.; Quartuccio, H. A.; Gould, R. J.; Garsky, V. M.; Rodan, G. A.; Rosenblatt, M., Inhibition of osteoclastic bone resorption in vivo by echistatin, an "arginyl-glycyl-aspartyl" (RGD)-containing protein. Endocrinology 1993, 132, (3), 1411-3.
31. Engleman, V. W.; Nickols, G. A.; Ross, F. P.; Horton, M. A.; Griggs, D. W.; Settle, S. L.; Ruminski, P. G.; Teitelbaum, S. L., A peptidomimetic antagonist of the alpha(v)beta3 integrin inhibits bone resorption in vitro and prevents osteoporosis in vivo. J Clin Invest 1997, 99, (9), 2284-92.
32. Yamamoto, M.; Fisher, J. E.; Gentile, M.; Seedor, J. G.; Leu, C. T.; Rodan, S. B.; Rodan, G. A., The integrin ligand echistatin prevents bone loss in ovariectomized mice and rats. Endocrinology 1998, 139, (3), 1411-9.
33. Hutchinson, J. H.; Halczenko, W.; Brashear, K. M.; Breslin, M. J.; Coleman, P. J.; Duong le, T.; Fernandez-Metzler, C.; Gentile, M. A.; Fisher, J. E.; Hartman, G. D.; Huff, J. R.; Kimmel, D. B.; Leu, C. T.; Meissner, R. S.; Merkle, K.; Nagy, R.; Pennypacker, B.; Perkins, J. J.; Prueksaritanont, T.; Rodan, G. A.; Varga, S. L.; Wesolowski, G. A.; Zartman, A. E.; Rodan, S. B.; Duggan, M. E., Nonpeptide alphavbeta3 antagonists. 8. In vitro and in vivo evaluation of a potent alphavbeta3 antagonist for the prevention and treatment of osteoporosis. J Med Chem 2003, 46, (22), 4790-8.
34. Murphy, M. G.; Cerchio, K.; Stoch, S. A.; Gottesdiener, K.; Wu, M.; Recker, R., Effect of L-000845704, an alphaVbeta3 integrin antagonist, on markers of bone turnover and bone mineral density in postmenopausal osteoporotic women. J Clin Endocrinol Metab 2005, 90, (4), 2022-8.
35. Pfaff, M.; Tangemann, K.; Muller, B.; Gurrath, M.; Muller, G.; Kessler, H.; Timpl, R.; Engel, J., Selective recognition of cyclic RGD peptides of NMR defined conformation by alpha IIb beta 3, alpha V beta 3, and alpha 5 beta 1 integrins. J Biol Chem 1994, 269, (32), 20233-8.
36. Burgess, K.; Lim, D.; Mousa, S. A., Synthesis and solution conformation of cyclo[RGDRGD]: a cyclic peptide with selectivity for the alpha V beta 3 receptor. J Med Chem 1996, 39, (22), 4520-6.
37. Xiong, J. P.; Stehle, T.; Zhang, R.; Joachimiak, A.; Frech, M.; Goodman, S. L.; Arnaout, M. A., Crystal structure of the extracellular segment of integrin alpha Vbeta3 in complex with an Arg-Gly-Asp ligand. Science 2002, 296, (5565), 151-5.
38. Xiong, J. P.; Stehle, T.; Diefenbach, B.; Zhang, R.; Dunker, R.; Scott, D. L.; Joachimiak, A.; Goodman, S. L.; Arnaout, M. A., Crystal structure of the extracellular segment of integrin alpha V beta3. Science 2001, 294, (5541), 339-45.
39. Marugan, J. J.; Manthey, C.; Anaclerio, B.; Lafrance, L.; Lu, T.; Markotan, T.; Leonard, K. A.; Crysler, C.; Eisennagel, S.; Dasgupta, M.; Tomczuk, B., Design, synthesis, and biological evaluation of novel potent and selective alphavbeta3/alphavbeta5 integrin dual inhibitors with improved bioavailability. Selection of the molecular core. J Med Chem 2005, 48, (4), 926-34.
40. Catalyst 4.9, Accelrys Inc: San Diego, USA, 2004.
41. Chen, X.; Plasencia, C.; Hou, Y.; Neamati, N., Synthesis and biological evaluation of dimeric RGD peptide-paclitaxel conjugate as a model for integrin-targeted drug delivery. J Med Chem 2005, 48, (4), 1098-106.
42. GOLD 1.2, CCDC: Cambridge, UK., 2002.
43. Gottschalk, K. E.; Gunther, R.; Kessler, H., A three-state mechanism of integrin activation and signal transduction for integrin alpha(v)beta(3). Chembiochem 2002, 3, (5), 470-3.
44. Gottschalk, K. E.; Kessler, H., The structures of integrins and integrin-ligand complexes: implications for drug design and signal transduction. Angew Chem Int Ed Engl 2002, 41, (20), 3767-74.
45. Feuston, B. P.; Culberson, J. C.; Duggan, M. E.; Hartman, G. D.; Leu, C. T.; Rodan, S. B., Binding model for nonpeptide antagonists of alpha(v)beta(3) integrin. J Med Chem 2002, 45, (26), 5640-8.
46. Marinelli, L.; Lavecchia, A.; Gottschalk, K. E.; Novellino, E.; Kessler, H., Docking studies on alphavbeta3 integrin ligands: pharmacophore refinement and implications for drug design. J Med Chem 2003, 46, (21), 4393-404.
47. Moitessier, N.; Henry, C.; Maigret, B.; Chapleur, Y., Combining pharmacophore search, automated docking, and molecular dynamics simulations as a novel strategy for flexible docking. Proof of concept: docking of arginine-glycine-aspartic acid-like compounds into the alphavbeta3 binding site. J Med Chem 2004, 47, (17), 4178-87.
48. Hynes, R. O., A reevaluation of integrins as regulators of angiogenesis. Nat Med 2002, 8, (9), 918-21.
49. Smellie, A.; Kahn, S. D.; Teig, S. L., Analysis of Conformational Coverage 1. Validation and Estimation of Coverage. J Chem Inf Comput Sci 1995, 35, (2), 285-294.
50. Smellie, A.; Kahn, S. D.; Teig, S. L., Analysis of Conformational Coverage 2. Application of Conformational Models. J Chem Inf Comput Sci 1995, 35, (2), 295-304.
51. Smellie, A.; Teig, S. L.; Towbin, P., Poling - Promoting Conformational Variation. J Comput Chem 1995, 16, (2), 171-187.
52. Jones, G.; Willett, P.; Glen, R. C.; Leach, A. R.; Taylor, R., Development and validation of a genetic algorithm for flexible docking. J Mol Biol 1997, 267, (3), 727-48.
53. Nissink, J. W.; Murray, C.; Hartshorn, M.; Verdonk, M. L.; Cole, J. C.; Taylor, R., A new test set for validating predictions of protein-ligand interaction. Proteins 2002, 49, (4), 457-71.
54. Plasencia, C.; Dayam, R.; Wang, Q.; Pinski, J.; Burke, T. R., Jr.; Quinn, D. I.; Neamati, N., Discovery and preclinical evaluation of a novel class of small-molecule compounds in hormone-dependent and -independent cancer cell lines. Mol Cancer Ther 2005, 4, (7), 1105-13。
【0134】
例II
NVX-188(AV38)研究
NVX-188とαvβ3 インテグリンの立体的相互作用
NVX-188は、新規種類の非RGDミメティックのαvβ3アンタゴニストを代表するものである。αvβ3インテグリンとのNVX-188のドッキング立体配置は、ゴールドソフトウェアパッケージを使用してモデル化した。図9に示す通り、NVX-188とαvβ3インテグリンの当該最小エネルギードッキングコンフォメーションは、当該2つのサブユニットのジャンクション近くの活性結合部位で溝を占有していることを示す。
【0135】
図10における代替的な表現により、予測されるNVX-188(水色)のドッキングを、細胞外リガンドタンパク質(藤色)のRGDペプチドの予測される位置と比較して示す。赤色の球は、αvβ3インテグリンの活性結合領域において抱埋されるMn2+イオンを示す。このコンフォメーションにおけるNVX-188により、当該RGDトリペプチド結合部位が効率的にブロッキングされる。機能性アッセイは、αvβインテグリンに対するNVX−188の結合がどのようにインテグリンの生物学的機能に影響を与えるかを決定するために必要である。NVX-188が直接的には、αvβ3インテグリンとの立体相互作用においてRGDペプチドを擬態していないという観察は、NVX-188がアゴニスト(活性化剤)としてよりもアンタゴニスト(ブロッキング剤)として作用することを示唆する。それは、当該αvβ3受容体に関連するおよびそのRGD結合部位に関連する小分子の配向であり、新規および異なる種類としてαvβ3結合剤のNVX-188を特徴付けるものである。
【0136】
NVX-188は、αvβ3インテグリン受容体へのその結合親和性が著しく、ピコモーラー濃度で活性化される。NVX-188クラスの他の化合物は、NVX-188の開発中に同定され(上記を参照されたい)、実質的に低い親和性を示す。それらの親和性は、公知のαvβ3インテグリンアンタゴニストについての文献において報告された親和性を有する系においてより大きい。
【0137】
NVX-188の高親和性についての基礎は、図11から明らかである。図11は、NVX-188とαvβ3ヘテロダイマーにおけるアミノ酸の間に形成される水素結合を示す。NVX-188の高結合親和性(以下を参照されたい)は、当該小分子および当該タンパク質の間で形成された比較的多くの水素結合に起因する可能性がある。
【0138】
αvβ3 結合する親和性
NVX-188の受容体結合親和性をNCI-H1975細胞において125I-放射性標識エキスタチンを使用した競合結合実験を介して決定した。NVX-188の濃度の増加を、エキスタチンの固定した量に加えて行い、当該細胞表面におけるαvβ3インテグリンへの結合するエキスタチンを50%阻害するNVX-188の濃度を決定する(即ち、IC50)。
【0139】
当該αvβ3インテグリン/NVX-188結合親和性は、0.03nM(30pM)であると測定された。これは他の文献に報告されている一般的にナノモーラーレンジで結合を示すαvβ3アンタゴニストに比較して有用である。
【0140】
細胞毒性(細胞障害性)アッセイ
NVX-188 を、乳癌細胞系、高αvβ3インテグリン発現を有するMDA-MB-435および低αvβ3インテグリン発現を有するMCF7、卵巣癌細胞系、HEY(これは細胞増殖阻害証明のための試験に使用した)におけるMTTアッセにより細胞障害性について試験した。NVX-188 は、肺癌細胞系、NCI-H1975およびマウス繊維芽細胞、NIH3T3において更に評価した。NVX-188は、試験された最高濃度においても細胞障害性を示さなかった(20 uM)。
【表B】
例III
αvβ3 アンタゴニストの新規種類
関節リウマチの治療について
背景
関節リウマチにおけるαvβ3 インテグリンの主な役割。 インテグリン、膜貫通接着受容体のファミリーは、細胞の接着、遊走、分化および生存の主なメディエーターである1 。インテグリンは、ヘテロダイマータンパク質であり、それぞれ1つのαおよび1つのβサブユニットで構成され、これらは組合せにおいて非共有結合的に結合している。データに対して、少なくとも18の異なるα-と、8のβ-サブユニットが同定されており、少なくとも24の機能的に多様なインテグリンを形成する。当該インテグリン αvβ3はビトロネクチン受容体として公知であり、125kDaのαvサブユニットと105kDaのβ3サブユニットからなる。幾つかの異なる病態生理学的な過程、特に、関節リウマチおよび他の新血管新生に関連する疾患、炎症およびは骨細胞活性の増加などにおけるその主な役割のためにそれは集中的な探索の焦点である。それらの良好に記載された細胞接着についての機能に加えて、αvβ3インテグリンは、種々のシグナル伝達経路(MAP-キナーゼ、Akt-キナーゼ、VEGFR,およびその他)を調節し、それにより、破骨細胞、内皮細胞および腫瘍細胞の増殖およびアポトーシスに影響を及ぼす3-12 。フィブリノーゲン、ビトロネクチン、オステオポンチンおよびスロンボスポンジンを含む種々の細胞外マトリックスタンパク質(ECM)に対して結合する当該αvβ3受容体は、大部分は、Arg-Gly-Asp(RGD)トリペプチド配列との相互作用を介する13、14 。リガンド結合モチーフの配列を基に、多様なペプチドミメチック賞分子αvβ3アンタゴニストが種々の疾患モデルにおける活性を有して合成されている12、15-25 。当該αvβ3 受容体は、破骨細胞による骨再吸収において中心的な役割を果たしており、αvβ3アンタゴニストは、注目すべき有害作用を伴わずにインビボにおいて骨再吸収を阻害することが示されている26-34 。
【0141】
まとめると、インテグリン αvβ3 の役割は、破骨細胞形成および骨再吸収、マクロファージの遊走および活性化、および血管形成に関連して最も広く研究されている。αvβ3 は、活性化されたマクロファージおよび破骨細胞において過剰発現され、これは関節リウマチ患者における骨破壊部位で多数見られる。これらの患者において、成熟した破骨細胞数は、関節周辺および全身性骨損失の両方に関連して高い。動物関節炎モデルからのデータを含む実質的に膨大な数のエビデンスが、現在、存在し、決定的ではないが、αvβ3が活性化されたマクロファージ依存の炎症、破骨細胞の進展および遊走、骨再吸収、並びに炎症性形成において重要な役割を果たしているという知見を支持している。そのため、このエビデンスおよびリウマチ様疾患の進行した段階のための有効な治療学的選択の欠乏が、αvβ3を標的とする新規化合物の開発がこの疾患のために大いに理性的、選択的および潜在的な有効なアプローチであるということを強く支持している35。
【0142】
新しい種類のαvβ3 アンタゴニストの発見
予備データ。 我々は最近、化学的機能ベースの共通特徴ファルマコフォアモデルを使用して新規小分子αvβ3アンタゴニストのシリーズを発見した36。三次元ファルマコフォアモデルを3つの現在報告されているαvβ3受容体アンタゴニストのトレーニングセットを使用して生成した。当該αvβ3受容体の公知のアンタゴニストの化学的データベースを使用するバリデーションにおいて、高い識別能力を有するファルマコフォアモデルを新規構造骨格と所望の化学的特徴を有する化合物を検索するための三次元照会として使用した。600,000化合物を超える化学的データベースのコンピュータによる探索により、αvβ3アンタゴニストとして特有の構造学的特徴を有する800よりも多い化合物が得られた。構造学的新規性および計算された物理化学的および薬物様性質を基に、化合物のパネルを更なるインビトロスクリーニングのために選択した。αvβ3受容体特異的競合結合アッセイにおいて最終的に試験された29の化合物のうち、幾つかの化合物がナノモーラーからピコモーラーレンジの結合親和性を示した(例Iの表3)36。これらの化合物の1つ(AV38)が、新規にNVX-188と命名され、ピコモーラー濃度での高親和性結合特性のためにリード化合物として選択された。
【0143】
実験プラン
あらまし
一般的考察および目的。 上記の予備データからのエビデンスの通り、高親和性結合特性および優れた「薬物様性質」を示す6つの化合物が同定された。これらの化合物は、優れた特異的結合特性を有しているのみならず、それらは作用様式は、現存のαvβ3アンタゴニストとは異なるようである。NVX-188は、リード化合物として定義されると同時に、それにも拘らず、このグループの他の化合物はインビトロアッセイにおいてより都合のよい機能的性質を有することが可能である。リード化合物のグループについての更なる詳しい記載(例1の表3)は、最近我々のグループによりthe Journal of Medicinal 化学に公開された36。αvβ3機能に批判的に依存する細胞ベースのアッセイは、更なるこれらの化合物の特徴付けるために利用できる。これらのアッセイは、更なる前臨床および臨床開発にための最高の候補を効果的に選択するための基礎を提供する。インシリコ(in silico)で最適化され、結合パイロット試験において評価されたこれらのリード化合物は、例えば、続くインビトロおよびインビボモデル系を使用して試験されてよい:
αvβ3阻害剤の最も重要な標的組織のためのバリデーションモデルインビトロ試験
初代ヒト内皮細胞(HUVEC)
初代破骨細胞/骨芽細胞
αvβ3を発現する骨肉腫(SAOS-2)細胞株
ラットにおけるαvβ3薬物候補の薬物動態学的および毒物学的特徴における重要なデータを得ることに焦点を当てたインビトロ試験。
【0144】
材料および方法
新規αvβ3阻害薬物候補。 当該新規薬物候補(NVX-188および類似体)は、ChemDiv(San Diego、CA)により合成、およびChemDiv(San Diego、CA)から購入した。化合物は、DMSO中で1mM のストック溶液で-20℃で使用時まで所蔵した。
【0145】
試薬および抗体。 ウシゼラチン、ウシ血漿フィブロネクチン、ヒト結成ビトロネクチン、ラットコラーゲンI、BSA、マウスラミニン、およびポリ-L-リジンはシグマから得た(Sigma-Aldrich Co.)。抗ヒトアクチン抗体はシグマから得た。機能ブロッキングmAbs: FB12(抗-α1)、P1E6(抗-α2)、P1B5(抗-α3),およびLM609(抗αvβ3)は、Chemicon(Temecula、CA)から; mAbs Lia1/2(抗-β1)、mAb GI9(抗-α2)、Sam-1(抗-α5)およびGoH3(抗-α6)は、Beckman Coulter(Nyon、Switzerland)から得た。プロテアーゼ阻害剤混合物は、4-(2-アミノエチル)-ベンゼンスルホニルフルオライド、アプロチニン、ロイペプチン、べスタチン、ペプスタチン AおよびE-64を含み、これは、シグマから得た。エキスタチンはBachem California Inc.(Torrance、CA)から得た。
【0146】
細胞培養および処理。 当該ヒト骨肉腫細胞株SAOS-2をthe American Type Culture Collection(ATCC、Manassas、VA)から得て、および現在、我々の研究室において使用している。興味深いことに、それは最近、αvβ3インテグリン発現が、特異的に肺に対する遊走を当該細胞に可能にすることを増強することによりSAOS-2細胞の転移の可能性において役割を担っていることを示している37。骨肉腫細胞の転移の可能性およびビトロネクチンに対する細胞接着に関連するαvβ3 発現は、強力なαvβ3アンタゴニストであると良好に記載されているエキスタチンでの処理の後に減少した。αvβ3 インテグリン は、従って、骨肉腫のための新しい標的である可能性がある。αvβ3を発現している正常なヒト骨芽細胞は、Promocell(Heidelberg、Germany)から得た。PromoCellの骨芽細胞培養系は、正常なヒト骨芽細胞と、最適化された骨芽細胞成長用培地と継代用試薬と組み合わせて提供されるように開発された。正常ヒト内皮細胞(HUVEC)は、Promocellから入手し、製造者の示唆どおりに培養した。破骨細胞培養および破骨細胞分化アッセイは、以下により詳細に記載した。
【0147】
毛細血管形成アッセイ。 血管形成(管構築の範囲)における阻害剤の効果のインビトロアッセイのために、商業的に入手可能なアッセイキットをChemiconから入手し、使用した。このアッセイキットは、血管形成のシンプルなモデルを代表するものであり、そこにおいて、外来性シグナルによる管形成の誘導または阻害が簡単にモニター可能である。環形成の阻害または刺激をアッセイするために、内皮細胞懸濁液を異なる濃度の試験されるべき化合物と共にインキュベートし、次に、当該細胞に特殊分化されたマトリックスの上層に細胞を添加した。the Engelbreth Holm-Swarm(EHS)マウス腫瘍から調製された基底タンパク質の固体ゲルである当該マトリックスにおいて培養した場合、内皮細胞は整列し、中空管様の構造を形成した。管形成は、複数の工程、即ち、細胞接着、遊走、分化および成長を含む工程である。当該アッセイを、使用し、種々の内皮細胞、例えば、ヒト臍帯静脈細胞(HUVEC)およびウシ毛細血管内皮(BCE)細胞におけるαvβ3阻害剤の存在において管構築の範囲をモニターした。
【0148】
αvβ3シグナリングの影響。αvβ3シグナリングにおける阻害剤の効果を測定するために、細胞をトリプシン処理し、阻害剤含有または非含有であり、αvβ3結合タンパク質の存在または非存在の10%血清を含む培地に懸濁した。異なる時間(5-30分)の後に、総細胞性タンパク質を得て、アッセイを行った。限局性接着キナーゼ(FAK)の免疫沈降アッセイのために、FAK を300μgから1mtの総タンパク質から免疫沈降させた。最終的にウエスタンブロットをリン酸化トリプシンおよびFAKについて以下の通りに行った。関連するが、g別々の実験においては、ビトロネクチンコートフラスコで成長されたHUVECを10ng/mlのVEGF含有または非含有の10%FBS-含有培地中で1時間インキュベートした。次に、阻害剤を5から30分間添加し、タンパク質を細胞ライセートから採取する。ウエスタンブロットアッセイは、総およびリン酸化形態のVEGFRおよびAktについて行った。幾つかの方向は、マイトジェン活性化タンパクキナーゼ(MAPK)が、PI3キナーゼシグナルカスケードと同様に、インテグリン阻害剤により調節されることを示している。MAPKおよびAktシグナリング経路において研究されている幾つかの工程のための抗体は、幾つかの会社から入手した(例えば、Cell SignallingおよびUpstat)。種々のリン酸化(活性化)段階を示す異なる抗体はも、入手可能であり(Cell Signalling および/または Upstatから)、総ライセート並びに免疫組織化学的分析のために使用してよい。
【0149】
細胞接着アッセイおよびコーティング。 内皮細胞は、トリプシン消化により回収し、血清非含有M199培地に2 x 104 cells/well(96-ウェルプレート)または12または24-ウェルプレートには1-2 x 105 cells/well で播種した。ブロッキング抗インテグリンmAbsは、この工程で添加される(10 μg/ml)。37℃での1時間の後、やさしくPBSで洗浄し、4%パラホルムアルデヒド(Fluka Chemie、Buchs、Switzerland)で接着細胞を固定化し、0.5%のクリスタルバイオレットで染色した。各ウェルの吸光度を620nmでプレートリーダーで読む。結果は、トリプリケート判定の平均値±S.Dで示す。培養プレートは、実験の種類に依存して、PBS中のフィブロネクチン(3μg/ml)、ゼラチン(0.5%)、コラーゲンI(10μg/ml)、ラミニン(10μg/ml)またはビトロネクチン(1μg/ml)で1時間37℃でプレコートされた。次にそれらを、0.5%の熱不活性脂肪酸非含有ウシ血清アルブミンで1時間37℃でブロックし、リン酸緩衝化生理食塩水(PBS)で洗浄される。実験において、ECMタンパク質が世ゆえ記に添加された実験においては、細胞をトリプシン処理により回収し、PBSで洗浄し、血清非含有のM199培地に再懸濁し1から2時間37℃でインキュベートする。ECMタンパク質を次に、上記で示された濃度で添加し、細胞を更に30から60分間懸濁液中でインキュベートする。細胞接着における阻害剤の効果を測定するために、細胞は96ウェルプレートに阻害剤あり、またはなしで、播種される。60分後に、細胞を洗浄し、接着細胞をカウントする。
【0150】
処理細胞の増殖、アポトーシスおよびクローン原性細胞死。 HUVEC、骨肉腫細胞および骨芽細胞を、ECMタンパク質コート(細胞の種類に依存してビトロネクチン、フィブリノーゲン、オステオポンチン)フラスコに播種し、48時間インキュベートし、血清含有培地において最終的に60から80%のコンフルエントにする。阻害剤を、適切な濃度で同時に添加する。浮遊および接着細胞を8および24時間後に採取し、血清含有培地に再懸濁し、カウントする。アポトーシスは、ヘキスト染料(Hoechst dye)で染色することにより測定する。ECMPコートプレートについて、阻害剤は、播種の後3時間の細胞が良好に付着または広がったところ添加する。即ち、当該細胞の処理の後、浮遊細胞および付着細胞を採取し、FCSエンリッチの実験媒地中で1時間37℃でインキュベートし、次に、アネキシンV/ヨウ化プロピジウム(PI)染色をモレキュラー・プローブ・アンドからのアネキシンV−アレキシア・フルオア488とシグマからのPIを使用して行った。得られた蛍光をフローサイトメトリーにより測定する。アネキシンVおよびPIにより染色された細胞は非アポトーシス細胞(アネックスV-/PI-)、早期アポトーシス細胞(アネックスV+/PI-)、後期アポトーシス細胞(アネックスV-/PI-)、ネクローシス細胞(アネックスV-/PI+)と決定される。細胞カウントおよび生存度の測定は、トリパンブルー排除により実行される。
【0151】
SDS PAGEおよびウエスタンブロット。 指示処理に続き、細胞を4%SDS、20%のグリセロールおよび200mMのTRISを含有するLaemmli緩衝液(pH6.8)に溶解する。タンパク質濃度ををBCAアッセイ(シグマ)を使用して測定する。等量の総タンパク質をSDS-PAGEで分離し、次に、一次抗体に依存してPVDFまたはニトロセルロース膜にブロットする。膜は、PBS中の5%スキムミルクでブロックし、次に、目的の一次抗体とハイブリダイズする。膜をPBSで洗浄し、適切な二次抗体をハイブリダイズする。抗マウスおよび抗ラビットペルオキシダーゼまたはアルカリフォスファターゼ複合二次抗体を5%のスキムミルク含有のPBSで希釈した。PBSでの洗浄の後、膜上の免疫複合体を、増強された化学発光により検出する(Pierce、Rockford、IL、USA)。
【0152】
破骨細胞形成のエクスビボによる測定のための破骨細胞の調製。 破骨細胞は、マウス脾臓から、ナイロンメッシュ(70μm)を使用して5mlのPBS中で2つの脾臓をホモジナイズすることにより得る。Cellsを次に、15mlのチューブに移し、1200U/minで10分間延伸することにより回収する。PBSでの洗浄の後、30ng/mlのM-CSFを含有する15mlのアルファMIM(10%PBS)中に再懸濁し、37℃で o.n.で培養し、採取する。PBSでの洗浄の後、細胞を30mlの総容量中での15mlのフィコール上に置き、1420U/minで8℃で30分間遠心する。内相の細胞を回収し、PBSで2回洗浄し、再懸濁し、カウントする。適切な数の細胞を次に、αvβ3シグナリングおよび/または受容体結合のモジュレータの存在または非存在の48ウェルプレートに配置する。破骨細胞分化における阻害剤の影響を測定するために、マウス脾臓細胞を、一晩、配置し、非接着細胞は回収し、24ウェルプレートに(1ウェル当たり106 cells)でM-CSF(20 ng/ml)含有、誘導物質、例えば、RANKL(50 ng/ml)存在または非存在で、αvβ3インテグリン特異的阻害剤の存在または非存在下に配置する。破骨細胞分化の範囲は、白血球アシッドフォスファターゼキットを使用してTRAP染色により培養4日後に評価する。3以上の核を有する全てのTRAT陽性細胞を破骨細胞としてカウントされる。
【0153】
薬物動態学的および毒物学的試験。 要するに、ベーシックな抽出およびLC-MSまたはLC-MS/MS 分析方法を各化合物に使用するだろう。薬物動態学的試験は、化合物の経口および静脈内投与について行った。投与に続いて、経口経路により処置した各ラットについて血液を7時点(0.5、1、2、4、6、8および12 時間)、および9点(5 min、15 min、30 minおよび1、2、4、6、8および12 時間)で採取した。薬物動態学的なパラメータ(Cmax、Tmax、Vd,および生物学的利用能)は、個々のラットについて、WinNonlin(Pharsight Corp)を使用した非コンパートメント分析を使用して行った。更に、全体的な血算と、肝臓および腎臓血液パラメータを各動物について測定した。
【0154】
参考文献
1. Danen EH. Integrins: regulators of tissue function and cancer progression. Curr Pharm Des 2005, 11: 881-91.
2. Humphries MJ. Integrin structure. Biochem Soc Trans 2000, 28: 311-39.
3. Martin KH, Slack JK, Boerner SA, Martin CC, Parsons JT. Integrin connections map: to infinity and beyond. Science 2002, 296: 652-53.
4. Aplin AE, Howe AK, Juliano RL. Cell adhesion molecules, signal transduction and cell growth. Curr Opin Cell Biol 1999, 11: 737-44.
5. Brooks PC, Clark RA, Cheresh DA. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science 1994, 264: 569-71.
6. Hood JD, Cheresh DA. Role of integrins in cell invasion and migration. Nat Rev Cancer 2002, 2: 91-100.
7. Felding-Habermann B. Integrin adhesion receptors in tumor metastasis. Clin Exp Metastasis 2003, 20: 203-13.
8. Natali PG, Hamby CV, Felding-Habermann B, et al. Clinical significance of alpha(v)beta3 integrin and intercellular adhesion molecule-1 expression in cutaneous malignant melanoma lesions. Cancer Res 1997, 57: 1554-60.
9. Felding-Habermann B, O'Toole TE, Smith JW, et al. Integrin activation controls metastasis in human breast cancer. Proc Natl Acad Sci USA 2001, 98: 1853-58.
10. Pecheur I, Peyruchaud O, Serre CM, et al. Integrin alpha(v)beta3 expression confers on tumor cells a greater propensity to metastasize to bone. Faseb J 2002, 16: 1266-68.
11. Kumar CC, Armstrong L, Yin Z, et al. Targeting integrins alpha v beta 3 and alpha v beta 5 for blocking tumor-induced angiogenesis. Adv Exp Med Biol 2000, 476: 169-80.
12. Shannon KE, Keene JL, Settle SL, et al. Anti-metastatic properties of RGD peptidomimetic agents S137 and S247. Clin Exp Metastasis 2004, 21: 129-38.
13. Ruoslahti E, Pierschbacher MD. Arg-Gly-Asp: a versatile cell recognition signal. Cell 1986, 44: 517-18.
14. Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell 1992, 69: 11-25.
15. Kerr JS, Wexler RS, Mousa SA, et al. Novel small molecule alpha v integrin antagonists: comparative anti-cancer efficacy with known angiogenesis inhibitors. Anticancer Res 1999, 19: 959-68.
16. Lark MW, Stroup GB, Hwang SM, et al. Design and characterization of orally active Arg-Gly-Asp peptidomimetic vitronectin receptor antagonist SB 265123 for prevention of bone loss in osteoporosis. J Pharmacol Exp Ther 1999, 291: 612-17.
17. Reinmuth N, Liu W, Ahmad SA, et al. Alphavbeta3 integrin antagonist S247 decreases colon cancer metastasis and angiogenesis and improves survival in mice. Cancer Res 2003, 63: 2079-87.
18. Harms JF, Welch DR, Samant RS, et al. A small molecule antagonist of the alpha(v)beta3 integrin suppresses MDA-MB-435 skeletal metastasis. Clin Exp Metastasis 2004, 21: 119-28.
19. Brooks PC, Stromblad S, Klemke R, Visscher D, Sarkar FH, Cheresh DA. Antiintegrin alpha v beta 3 blocks human breast cancer growth and angiogenesis in human skin. J Clin Invest 1995, 96: 1815-22.
20. Brooks PC, Montgomery AM, Rosenfeld M, Reisfeld RA, Hu T, Klier G, Cheresh DA. Integrin alpha v beta 3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell 1994, 79: 1157-64.
21. Taga T, Suzuki A, Gonzalez-Gomez I, et al. alpha v-Integrin antagonist EMD 121974 induces apoptosis in brain tumor cells growing on vitronectin and tenascin. Int J Cancer 2002, 98: 690-97.
22. Mitjans F, Meyer T, Fittschen C, Goodman S, Jonczyk A, Marshall JF, Reyes G, Piulats J. In vivo therapy of malignant melanoma by means of antagonists of alphav integrins. Int J Cancer 2000, 87: 716-23.
23. Allman R, Cowburn P, Mason M. In vitro and in vivo effects of a cyclic peptide with affinity for the alpha (nu) beta3 integrin in human melanoma cells. Eur J Cancer 2000, 36: 410-22.
24. Dechantsreiter MA, Planker E, Matha B, Lohof E, Holzemann G, Jonczyk A, Goodman SL, Kessler H. N-Methylated cyclic RGD peptides as highly active and selective alpha(V)beta(3) integrin antagonists. J Med Chem 1999, 42: 3033-40.
25. Burke PA, DeNardo SJ, Miers LA, Lamborn KR, Matzku S, DeNardo GL. Cilengitide targeting of alpha(v)beta(3) integrin receptor synergizes with radioimmunotherapy to increase efficacy and apoptosis in breast cancer xenografts. Cancer Res 2002, 62: 4263-72.
26. Rodan SB, Rodan GA. Integrin function in osteoclasts. J Endocrinol 1997, 154 (Suppl.): 47-56.
27. Nakamura I, Pilkington MF, Lakkakorpi PT, Lipfert L, Sims SM, Dixon SJ, Rodan GA, Duong LT. Role of alpha(v)beta(3) integrin in osteoclast migration and formation of the sealing zone. J Cell Sci 1999, 112: 3985-93.
28. Duong LT, Rodan GA. The role of integrins in osteoclast function. J Bone Miner Metab 1999, 17: 1-6.
29. Teitelbaum SL. Bone resorption by osteoclasts. Science 2000, 289: 1504-8.
30. Fisher JE, Caulfield MP, Sato M, Quartuccio HA, Gould RJ, Garsky VM, Rodan, GA, Rosenblatt, M. Inhibition of osteoclastic bone resorption in vivo by echistatin, an "arginyl-glycyl-aspartyl" (RGD)-containing protein. Endocrinology 1993, 132: 1411- 13.
31. Engleman VW, Nickols GA, Ross FP, Horton MA, Griggs DW, Settle SL, Ruminski PG, Teitelbaum SL. A peptidomimetic antagonist of the alpha(v)beta3 integrin inhibits bone resorption in vitro and prevents osteoporosis in vivo. J Clin Invest 1997, 99: 2284-92.
32. Yamamoto M, Fisher JE, Gentile M, Seedor JG, Leu CT, Rodan SB, Rodan GA. The integrin ligand echistatin prevents bone loss in ovariectomized mice and rats. Endocrinology 1998, 139: 1411-19.
33. Hutchinson JH, Halczenko W, Brashear KM, et al. Nonpeptide alphavbeta3 antagonists. In vitro and in vivo evaluation of a potent alphavbeta3 antagonist for the prevention and treatment of osteoporosis. J Med Chem 2003, 46: 4790-98.
34. Murphy MG, Cerchio K, Stoch SA, Gottesdiener K, Wu M, Recker R. Effect of L-000845704, an alphaVbeta3 integrin antagonist, on markers of bone turnover and bone mineral density in postmenopausal osteoporotic women. J Clin Endocrinol Metab 2005, 90: 2022-28.
35. Wilder RL. Integrin alpha V beta 3 as a target for treatment of rheumatoid arthritis and related rheumatic diseases. Ann Rheum Dis 2002, 61: 96−99.
36. Dayam R, Aiello F, Deng J, Wu Y, Garofalo A, Chen X, Neamati N. Discovery of small molecule integrin alphavbeta3 antagonists as novel anticancer agents. J Med Chem. 2006, 49: 4526-34.
37. Duan X, Jia SF, Zhou Z, Langley RR, Bolontrade MF, Kleinerman ES. Association of alphavbeta3 integrin expression with the metastatic potential and migratory and chemotactic ability of human osteosarcoma cells. Clin Exp Metastasis. 2004, 21: 747-53。
【図面の簡単な説明】
【0155】
【図1】図1は、新規αvβ3アンタゴニストのファルマコフォアにより導かれる設計および発見の模式図である。
【図2】図2は、トレーニングセット化合物A-Cの構造を示す。
【図3】図3。(a)共通特徴ファルマコフォアHypo5。(b)Hypo5 はトレーニングセット化合物Aの1つに位置づけられる。化合物Aの重要な化学的特徴は、Hypo5のファルマコフォアの特徴により位置付けられる。ファルマコフォア特徴は、マゼンタのH結合供与体(HBD)、緑色のH結合受容体(HBA)、茶色の疎水性芳香族(HAR1-HAR2)および青色の陰性イオン性機能(NI)として示される。特徴間の距離はÅで示される。
【図4】図4は、新規αvβ3 受容体 アンタゴニスト AV3(a)およびAV26(b)へのHypo5の位置づけを示す。Hypo5のファルマコフォア的特徴は、適度に当該アンタゴニストの重要な化学的特徴に位置づけされている。当該ファルマコフォアの特徴はマゼンタのH結合供与体、緑色のH結合受容体、茶色の疎水性芳香族および青色の陰性イオン化特徴として示される。
【図5】図5は、αvβ3RGDペプチド結合部位の内部におけるアンタゴニストA(a)、AV3(b)、AV26(c)、AV27(d)およびAV38(e)の 予測される二重結合コンフォメーションを示す。黄色および赤色部分は、αvβ3受容体のαおよびβ鎖を示す。突出した活性部位アミノ酸残基は、当該受容体表面におけるスティックモデルとして示した。当該緑色のスティックモデルは、サイクリックRGDペプチド(PDB1L5G)の結合配向を示す。当該活性部位Mn2+(MIDAS)は青緑色の球として示す。
【図6】図6は、アンタゴニストA(a)、AV26(b)およびAV38(c)とαvβ3RGDペプチド結合部位における突出したアミノ酸残基との間に得られた相互作用を示す模式図である。ダッシュの線はH結合相互作用を示す。H結合距離はÅで表す。
【図7】図7は、4つの内皮アポトーシスモデルを図示する。(a)古典的モデル; そこにおいてリガンドによるインテグリン結合が生存シグナルを提供するために必要である。阻害剤がリガンドの結合をブロックし、それにより、生存シグナルがブロックされる。(b)カスパーゼ活性化モデル;そこにおいて、RGDペプチドは直接にカスパーゼを活性化し、インテグリンの如何なる介入もなしにアポトーシスを引き起こす。(c)非ライゲーションモデルまたは「インテグリン-媒介細胞死」;そこにおいて結合していないインテグリンが、直接に結合し、カスパーゼ-8を活性化する。ECM リガンドはこれをブロックするが、それらがインテグリンを活性化することが知られて入るが、RGD ペプチドおよび同じインテグリンに対して結合する抗体はそのようなことはしない。(d)例えば、パクリタキセルなどの細胞毒性剤に対して一旦結合したαvβ3 アンタゴニストは、αvβ3陽性癌細胞に対して、および腫瘍新生血管におけるαvβ3陽性内皮細胞に対して効率的に送達される(Richard Hynesから改変された、参考文献 48)。
【図8】図8は、共通する特徴のファルマコフォア Hypos 1(A)、5(B)および9(C)を示す。当該ファルマコフォア特徴は、H結合供与体(HBD)はマゼンタで、H結合受容体(HBA)は緑色で、疎水性芳香族(HAR1-HAR2)は茶色で、および陰性イオン化特徴(NI)は青色で示す。当該特徴間の距離はÅで示す。
【図9】図 9は、NVX-188の予測される結合コンフォメーションを示す。
【図10】図10 は、αvβ3インテグリン上のNVX-188(水色)および当該RGDトリペプチド(藤色)の予測されるドッキングを示す。
【図11】図11は、当該αvβ3ヘテロダイマーにおけるアミノ酸に対するNVX-188(右側)およびRGD-ミメティック小分子(左側)の関係を示す。
【発明の開示】
【0001】
関連出願
本出願は、2005年11月14日に提出された米国仮出願第60/736,780の優先権を主張するものであり、その内容は、その全体において参照することにより本明細書において組み込まれる。
【0002】
発明の分野
本発明は、一般的にインテグリン結合小分子に関する。更に詳しくは、本発明は、新規組成物およびこれらの組成物を種々の疾患の治療のために使用する方法を提供する。
【0003】
発明の背景
インテグリン、即ち、膜貫通接着受容体のファミリーは、細胞付着、遊走、分化および生存の主な媒介物質である1。構造的に、インテグリンは、各サブユニットとして大きな細胞外ドメイン、1つの膜貫通へリックス、および小さな細胞内ドメインで構成されるヘテロ二量体である2。これらの受容体は、α-及びβ-サブユニットからなり、これらは非共有結合的に示された組合せで結合する。現在までのところ、18 α-サブユニットおよび8 βサブユニットが確認されており、これらは結合し、選択的に少なくとも24のインテグリンを形成する。それらの接着性機能に加えて、インテグリンは、多様な情報伝達経路を介してメッセージを伝達し、並びに腫瘍細胞および活性化された内皮細胞の増殖およびアポトーシスに影響を与える3,4。細胞表面におけるインテグリンの独特の組合せは細胞が種々の細胞外リガンドを認識し、その後にそれに対して応答することを可能にする。インテグリンαvβ3はインテグリンファミリーの中でも突出したものである。それは、悪性腫瘍の病理生理学的において関連付けられており、腫瘍血管形成5のために必要であり、新血管における内皮細胞および攻撃性の高いヒト癌腫において発現が高い。インテグリン αvβ3は、種々の細胞外マトリックスタンパク質における腫瘍細胞の接着を媒介し、これらの細胞が浸潤中の遊走および血管外遊走を可能にする6,7。乳癌において、αvβ3は、このインテグリンが浸潤性腫瘍および遠位転移において上方制御されるように、転移性表現形を特徴づける8−10。従って、インテグリン αvβ3のアンタゴニストは、転移性および浸潤性の癌の治療のための新しいアプローチを提供すると期待される11,12。αvβ3アンタゴニストと従来の治療様式との組合せは、余分な毒性を伴わずに、転移性癌治療の有効性を増大可能であった。当該αvβ3受容体は、フィブリノーゲン、フィブロネクチン、オステオポンチン、スロンボスポンジンおよびビトロネクチンを含む種々の細胞外マトリックスタンパク質に対して、大部分は、当該Arg-Gly-Asp(RGD)トリペプチド配列との相互作用を介して結合する13、14。以前に、種々のペプチドミメチックス小分子αvβ3アンタゴニストが同定されており、その幾つかは、疾患モデル、例えば、骨粗鬆症および骨格転移性乳癌において活性である12、15−18。
【0004】
当該αvβ3 アンタゴニストは、マウス異種移植モデルおよび転移モデルを含む多くの動物モデルにおいて強力に血管形成を抑制する。mABsおよびサイクリックRGDペプチドによるαvβ3活性の阻害が、内皮のアポトーシスを誘導し、血管形成を阻害することが示されている19、20。当該αvβ3 アンタゴニストは、活性化された内皮細胞ばかりではなく、αvβ3陽性腫瘍細胞においてもアポトーシスを誘導することができ、腫瘍細胞における直接的な細胞毒性効果を結果として引き起こす21。αvβ3活性のアンタゴニストは、乳癌細胞異種移植およびメラノーマ異種移植における腫瘍増殖を結果的に減少する22,23。転移性癌のための臨床試験におけるシレンジタイド(Cilengitide)、サイクリックRGDペプチドが24、攻撃的な乳癌モデルにおいて試験されており、そこにおいて、シレンジタイドと放射免疫療法との併用が、何れかの薬剤での単一様式療法に比べて、更なる特性を伴わずに、著しく有効性が増強され、且つアポトーシスを増大しすることが示された25。これは、併用抗癌療法において、シレンジタイドおよび一般的なαvβ3アンタゴニストの実際の治療学的なポテンシャルを明確に示唆するものである。
【0005】
当該αvβ3受容体はまた、骨再吸収において中心的な役割を果たす。種々の試験は、αvβ3受容体が破骨細胞における最も豊富なインテグリンであることを示している26-29。αvβ3抗体、RGDペプチドおよびペプチドミメティックアンタゴニストは、著しい有害作用を伴わずにインビボにおける骨再吸収の阻害を示した30-34。これらの試験および初期の臨床的試験からの結果の基礎に、αvβ3アンタゴニストは骨粗鬆症の治療および予防のために大きな見込みを示すものである。
【0006】
発明の概要
本発明は、少なくとも一部分において、以下に記載の小分子化合物がインテグリンに結合し、それにより、種々の疾患および状態を診断および治療するために使用できるという予期しない発見に基づく。
【0007】
従って、1側面において、本発明は、化合物または薬学的若しくは薬用化粧品学的に許容されるその塩、その溶媒和物またはその水和物を含む組成物が特徴であり、ここで、当該化合物は、1のH結合供与体(HBD)、1のH結合受容体(HBA)、2の疎水性芳香族基(HAR1およびHAR2)および1の陰性イオン性基(NI)を含み、およびここで、当該化合物は化合物 A、BまたはCではない;
【化4】
【0008】
。
【0009】
当該組成物は、薬学的または薬用化粧品学的に許容された担体を更に含んでもよい。
【0010】
一つの態様において、当該HBD、HBA、HAR1、HAR2およびNIは、図8Aに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.81±1 Åでり、当該HBDとHBAの距離は7.52±1 Åであり、当該HBDとNIの距離は17.67±1 Åであり、当該HRA1とHBAの距離は7.52±1 Åであり、当該HRA1とHRA2の距離は9.41±1 Åであり、当該HBAとNIの距離は 10.20±1 Åであり、および当該HRA2とNIの距離は5.88±1 Åである。特に、当該HBDとHRA1の距離は2.81 Åであってよく、当該HBDとHBAとの距離は7.52 Åであってよく、当該HBDとNIの距離は17.67 Åであってよく、当該HRA1とHBAの距離は7.52 Åであってよく、当該HRA1とHRA2の距離は9.41 Åであってよく、当該HBAとNIの距離は10.20 Åであってよくおよび当該HRA2とNIの距離は5.88 Åであってよい。
【0011】
他の態様において、当該HBD、HBA、HAR1、HAR2,およびNIは、図8Bに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.80±1 Åであり、当該HBDとHBAの距離は7.34±1 Åであり、当該HBDとNIの距離は16.92±1 Åであり、当該HRA1とHBAの距離は5.20±1 Åであり、当該HRA1とNI の距離は14.34±1 Åであり、当該HBAとHRA2の距離は4.69±1 Åであり、当該HBAとNIの距離は9.85±1 Åであり、および当該HRA2とNI の距離は5.66±1 Åである。特に、当該HBDとHRA1の距離は2.80 Åでよく、当該HBDとHBAの距離は7.34 Åでよく、当該HBDとNI の距離は16.92 Åでよく、当該HRA1とHBAの距離は5.20 Åでよく、当該HRA1とNIの距離は14.34 Åでよく、当該HBAとHRA2の距離は4.69 Åでよく、当該HBAとNIの距離は9.85 Åでよく、および当該HRA2とNIの距離は5.66 Åでよい。
【0012】
もう一つの態様において、当該HBD、HBA、HAR1、HAR2およびNIは、図8Cに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.79±1 Åであり、当該HBDとHRA2の距離は11.94±1 Åであり、当該HBDとNI の距離は16.28±1 Åであり、当該HRA1とHBAの距離は5.20±1 Åであり、当該HRA1とNIの距離は14.64±1 Åであり、当該HBAとHRA2の距離は4.72±1 Åであり、および当該HRA2とNIの距離は5.63±1 Åである。特に、当該HBDとHRA1の距離は2.79 Åでよく、当該HBDとHRA2の距離は11.94 Åでよく、当該HBDとNIの距離は16.28 Åでよく、当該HRA1とHBAの距離は5.20 Åでよく、当該HRA1とNI の距離は14.64 Åでよく、当該HBAとHRA2の距離は4.72 Åでよく、および当該HRA2とNI の距離は5.63 Åでよい。
【0013】
例えば、本発明の化合物は、式 I、II、III,またはIV:
【化5】
【0014】
でよい。
【0015】
式I、IIおよびIIIにおいて、それぞれ、R1およびR3 は、脂肪族、芳香族またはヘテロ環基; R2 は置換基;およびX はN、OまたはSである。式 IVにおいて、それぞれ、R1およびR2 は水素またはハロゲン、ヒドロキシル、スルフヒドリル、アルコキシ、アリールオキシ、カルボキシル、ニトロ、シアノ、アミノ、アミドまたはスルホニル基または何れかの数の炭素原子を含む何れか他の有機官能基; R3 は水素、ヒドロキシル、アルコキシ、アリールオキシ、芳香族またはヘテロ環基または何れかの数の炭素原子の脂肪族鎖;それぞれ、R4およびR5 はヒドロキシルまたはアミノ基; X は C、N、OまたはSであり; m は炭素、窒素、酸素または硫黄原子を伴う 0 から 3 原子リンカー基;およびn は炭素、窒素、酸素または硫黄原子を伴う0 から 5 原子リンカー基である。
【0016】
特に、式I、IIおよびIIIにおいて、それぞれ、R1およびR3 はアルキル、置換されたアルキル、アルケニル、置換されたアルケニル、アルキニル、置換されたアルキニル、フェニル、置換されたフェニル、アリール、置換されたアリール、ヘテロアリールまたは置換されたヘテロアリール基であってよく; R2 は、水素またはハロゲン、ヒドロキシル、スルフヒドリル、アルコキシ、カルボキシル、ニトロ、シアノ、アミノ、アミドまたはスルホニル基または何れかの数の炭素原子を含む何れか他の有機官能基またはそれらの組合せであってよい。R1またはR3における、当該置換されたアルキル、アルケニル、アルキニル、フェニル、アリールまたはヘテロアリール基は、ハロ、ヒドロキシル、スルフヒドリル、アルコキシ、カルボキシル、カルボニル、ニトロ、シアノ、アミノ、アミド、スルホニル、または置換されたヘテロ環基により置換されたアルキル、アルケニル、アルキニル、フェニル、アリール、またはヘテロアリール基であってよい。式 IVにおいて、R4またはR5における当該アミノ基は、脂肪族、芳香族またはヘテロ環基で置換されてよい。本発明の化合物の例は、AV1-11、AV14-15、AV17-32およびAV34-38を含む。
【0017】
もう一つの側面において、本発明は、第一の化合物(即ち、上で記載された通りの本発明の化合物)、またはその薬学的若しくは薬用化粧品学的に許容される塩、溶媒和物または水和物と、1または1以上の第二の化合物とを含む、またはからなる組成物を特徴とし、ここで第二の化合物は治療剤、イメージング財またはその組合せである。
【0018】
例えば、当該第二の化合物は、血管増殖阻害剤(例えば、TNF-アルファ、アンジオスタチン、ベバシザン(Bevacizumab)、アレステン(Arresten)、カンスタチン(Canstatin)、コンブレタスタチン、エンドスタチン、NM-3、サリドマイド、スロンボスポンジン(Thrombospondin)、タムスタチン(Tumstatin)、2-メトキシエストラジオールおよびビタキシン(Vitaxin))、関節リウマチを治療するための治療剤、放射線治療剤、化学療法剤(例えば、シスプラチン、ドキソルビシン、ビンクリスチン、シクロホスファミド、トポテカン、パクリタキセル,および他の化学療法剤)、骨転移を治療するための治療剤骨転移を治療するための治療剤、骨粗鬆症を治療するための治療剤(例えば、ホセマックス(Fosemax)、アレジア(Aredia)、アクトネル(Actonel)、ジンドロネル(Dindronel)、コメタ(Cometa)、プレマリン(Premarin)、クリマラ(Climara)、エストラ(Estrae)、ビベラ(Vivelle)、エストラダーム(Estraderm)、プレモプロ(Prempro)、プラムフェーズ(Premphase)、フェマート(Femhrt)、プレフェスト(Prefest)、コンビパッチ(Combipatch)、エビスタ(Evista)、カルシトニン(Calcitonin)、ミアカルシン(Miacalcin)およびカルシトニン)、再狭窄を治療するための治療剤、血管イメージング剤(例えば、放射性核種)またはその組み合わせであってよい。
【0019】
幾つかの態様において、当該第一の化合物は、実施可能に少なくとも1の当該第二の化合物に実施可能に連結されている。
【0020】
本発明は、更に、当該発明は、インテグリンと本発明の化合物またはその薬学的若しくは薬用化粧品学的に許容される塩、溶媒和物または水和物とを接触させることを具備する、インテグリンを化合物に対して結合する方法を提供する。
【0021】
一つの態様において、当該インテグリンはαvβ3である。もう一つの態様において、当該インテグリンは細胞上に、例えば、癌細胞、良性腫瘍細胞、間質細胞、炎症に反応する、炎症を媒介する、または炎症を調節する細胞(例えば、白血球、マクロファージおよび樹状細胞)、免疫系の細胞(例えば、白血球、マクロファージおよび樹状細胞)、血管機能に関与する細胞(例えば、内皮細胞、内皮細胞に関与する細胞および平滑筋細胞)、骨芽細胞、破骨細胞またはウイルス(例えば、アデノウイルス(アデノビリダエ;Adenoviridae)、コックサッキーウイルス(Coxackievirus、Picornaviridae)、エコウイルス(Echovirus、Picornaviridae)、口蹄疫ウイルス(Picornaviridae)、ハンタウイルス(Buriyaviridae)、ヒトパレコウイルス(Picornaviridae)、ヒト免疫不全ウイルス 1(Retroviridae)およびロタウイルス(Reoviridae))に感染した細胞上にある。
【0022】
当該方法は、更に、当該細胞と1または1以上の治療剤、イメージング剤またはその組合せとを接触させることを具備する。当該治療剤およびイメージング剤は、上で記載した。当該インテグリンおよび当該細胞は、同時にまたは経時的に接触してよい。当該化合物は、少なくとも1の薬剤と混合されてもよく、または実施可能に連結されてもよい。
【0023】
また、本発明において、本発明は、対象においてインテグリンを化合物に対して結合する方法であって、それを必要とする対象において本発明の化合物またはその薬学的若しくは薬用化粧品学的に許容される塩、溶媒和物または水和物の有効量を投与することを具備する方法である。当該インテグリンはαvβ3であってよく、および当該対象は、動物またはヒトであってよい。
【0024】
一つの態様において、当該対象は、癌、例えば、悪性固形腫瘍の組織学的種類、例えば、肉腫または癌腫または高いレベルの血管形成および血管原性因子に関連する血液学的悪性度、例えば、急性または慢性白血病、多発性骨髄腫、骨髄増殖性疾患、非-ホジキンリンパ腫またはホジキン病、に罹患する、またはそれらの発展のリスクに面している。
【0025】
もう一つの態様において、当該対象は、良性腫瘍、例えば、良性腫瘍または高い血管新生に関連する新生物発生前の状態、尋常性疣贅、化膿性 肉芽腫、酒さ、ケロイド瘢痕,または脈絡叢に関連する眼部腫瘍または虹彩新血管新生に罹患している、またはそれらの発展のリスクに面している。
【0026】
更なるもう一つの態様において、当該対象は、炎症性疾患、例えば、炎症性腸疾患、例えば、クローン病または潰瘍性大腸炎;特発性炎症性ミオパチーなどの多発性筋炎(PM)、皮膚筋炎(DM)または増大した血管新生に関連する状態;他の眼部炎症性疾患;増大した炎症に関連する疾患または状態、例えば、歯肉炎または粘膜炎;ざ瘡;または特に慢性的な炎症状態に関連する胃腸管疾患、例えば、胃炎または十二指腸炎など、に罹患している、またはそれらの発展のリスクに面している。粘膜炎の例は、放射線または化学療法誘導粘膜炎である。
【0027】
更なるもう一つの態様において、当該対象は、免疫または自己免疫疾患、例えば、関節リウマチ; 乾癬性関節炎;乾癬;多発性硬化症;気管支喘息またはアレルギー;湿疹または皮膚炎;または他の皮膚または皮下組織の炎症性状態、例えば、紅斑性扁平皮膚症、斑不全角化、脂漏性皮膚炎、頭部脂漏症、他の脂漏性皮膚炎、アトピー性皮膚炎または関連する状態、紅斑、ベニエ痒疹、神経皮膚炎、接触性皮膚炎または他の湿疹、水疱性皮膚疾患、紅斑性状態、苔癬、掻痒症または関連する状態、疱疹状皮膚炎、疱疹状皮膚症、デューリング病、疱疹状皮膚炎、角層下膿疱症、スネッドン-ウィルキンソン病、若年性疱疹状皮膚炎、若年性天疱瘡、疱疹状膿痂疹、多巣性紅斑、紅斑虹彩、疱疹虹彩、ライル症候群、エリトマトーデス、他の特定の紅斑性状態、苔癬化またはハイド病に罹患している、またはそれらの発展の危険に面している。当該接触性皮膚炎および他の湿疹の例は、内部に取り込まれた物質のための皮膚炎、皮膚との接触における化学薬品、薬物および医薬品のための皮膚炎、日射のための皮膚炎、日焼け、ベルロック皮膚炎、光アレルギー反応皮膚炎、光毒性反応皮膚炎、多形性 光性発疹、播種性 浅在性 光線性 汗孔角化症(DSAP)、化粧品のための皮膚炎、赤外線のための皮膚炎、紫外線のための皮膚炎および電離放射線による治療のための皮膚炎を含む。当該他の特定の紅斑性状態は、リッター病、新生児剥脱性皮膚炎および紅斑間擦疹を含む。
【0028】
更なるもう一つの態様において、当該対象は、血管性疾患、例えば、子宮内膜症;機能性障害性の子宮性出血;子宮内膜過形成;骨髄線維症; 血管腫;動脈硬化症;急性または慢性移植拒絶反応または移植脈管障害;眼部新血管新生により特徴付けられる眼部疾患、例えば、加齢関連横斑性変性(AMD)、増殖性糖尿病網膜症(PDR)、未熟児網膜症(ROP)、ネオ血管緑内障、網膜芽細胞腫、水晶体後線維増殖症、ルベオーシス、ブドウ膜炎、横斑性変性または角膜移植新血管新生;不適当な血小板活性化、補充、凝集または血栓症により媒介される状態、例えば、冠動脈疾患または障害、心筋梗塞または心筋梗塞に続く障害、脳卒中、不安定狭心症、子癇前症、塞栓症、血小板関連虚血性疾患、経皮的冠動脈インターベンションに続く再狭窄、血栓性疾患、血栓症に続く再閉塞、深在性静脈血栓症(DVT)、肺塞栓症(PE)、一過性虚血性発作(transient ischemic attack;TIA)または血管閉塞が共通する基本をなす特徴である他の状態;心筋の血管新生;出血性関節;血管癒着;敗血症;成人呼吸困難症候群;末梢血管拡張;または創傷肉芽形成に罹患しており、またはそれらの発展するリスクに面している。当該血小板関連虚血性疾患の例は、肺虚血、冠血管虚血および脳虚血を含む。当該経皮的冠動脈インターベンションの例は、血管形成術、アテレクトミー、ステント設置およびバイパス手術を含む。当該血栓性疾患の例は、冠動脈血栓症、脳動脈血栓症、心臓内血栓症、末梢動脈血栓症、静脈血栓症および血栓症および外来性に対する露出または損傷された組織表面に関連する凝固障害を含む。
【0029】
更なるもう一つの態様において、当該対象は、または病理学的骨再吸収に主に関連する疾患、例えば、骨粗鬆症、骨のパジェット病または副甲状腺ホルモン(PTHrP)媒介高カルシウム血症に罹患している、またはそれらの発展のリスクに面している。
【0030】
更なるもう一つの態様において、当該対象は、ウイルス感染、例えば、an infection by アデノウイルス(Adenoviridae)、コックサッキーウイルス(Picornaviridae)、エコウイルス(Picornaviridae)、口蹄疫ウイルス(Picornaviridae)、ハンタウイルス(Buriyaviridae)、ヒトパレコウイルス(Picornaviridae)、ヒト免疫不全ウイルス 1(Retroviridae),またはロタウイルス(Reoviridae)による感染に罹患している、またはそれらの発展のリスクに面している。
【0031】
当該方法は、更に、当該対象に対して、有効量の1または1以上の治療剤、イメージング剤またはその組合せを投与することを具備してよい。当該治療剤およびイメージング剤は、上で記載した通りである。当該化合物および当該薬剤は、同時または経時的に投与されてよい。当該化合物は、少なくとも1の薬剤と混合されてもよく、実施可能に連結されてもよい。
【0032】
化合物、治療剤、イメージング剤またはその組合せは、非経口、皮内、皮下、経口、経皮、経粘膜,または直腸に投与されてよい。
【0033】
幾つかの態様において、当該対象は、血管形成方法(例えば、バルーン血管形成術;レーザー血管形成術;冠血管アテレクトミーまたは同様の技術;頚動脈動脈膜切除;血管グラフトの吻合;血栓形成の高リスクを有する外科手術、例えば、冠血管バイパス手術および補綴バルブまたは血管の挿入;アテレクトミー;ステント設置;慢性心血管装置の設置、例えば、インドルエリングカテーテル、補綴バルブまたは血管; 臓器移植;およびバイパス手術)、放射線治療または化学療法(例えば、有効量のシスプラチン、ドキソルビシン、ビンクリスチン、シクロホスファミド、トポテカン、パクリタキセルまたは他の化学療法剤を当該対象に投与することを含む化学療法)で処置されてもよい。
【0034】
本発明は、更に、ファルマコフォアの提示を具備するコンピュータ読み取り可能媒体であって、ここで、当該ファルマコフォアは1の H結合供与体(HBD)、1のH結合受容体(HBA)、2の疎水性芳香族基s(HAR1およびHAR2)および1の陰性イオン性基(NI)の特徴を含むコンピュータ読み取り可能媒体を提供する。
【0035】
一つの態様において、当該HBD、HBA、HAR1、HAR2およびNIは、図8Aに従う立体配置であり、ここで、当該HBDとHRA1の距離が2.81±1 Åであり、当該HBDとHBAの距離が7.52±1 Åであり、当該HBDとNIの距離が17.67±1 Åであり、当該HRA1とHBAの距離が7.52±1 Åであり、当該HRA1とHRA2の距離が9.41±1 Åであり、当該HBAとNIの距離が10.20±1 Åであり、および当該HRA2とNIの距離が5.88±1 Åである。特に、当該HBDとHRA1の距離は2.81 Åでよく、当該HBDとHBA の距離は7.52 Åでよく、当該HBDとNIの距離は17.67 Åでよく、当該HRA1とHBAの距離は7.52 Åでよく、当該HRA1とHRA2 の距離は9.41 Åでよく、当該HBAとNI の距離は10.20 Åでよく、および当該HRA2とNI の距離は5.88 Åでよい。
【0036】
もう一つの態様において、当該HBD、HBA、HAR1、HAR2およびNIは、図8Bに従う立体配置であり、ここで、当該HBDとHRA1の距離は 2.80±1 Åであり、当該HBDとHBAの距離は7.34±1 Åであり、当該HBDとNIの距離は16.92±1 Åであり、当該HRA1とHBAの距離は5.20±1 Åであり、当該HRA1とNI の距離は14.34±1 Åであり、当該HBAとHRA2の距離は4.69±1 Åであり、当該HBAとNI の距離は9.85±1 Åであり、および当該HRA2とNI の距離は5.66±1 Åである。特に、当該HBDとHRA1の距離は2.80 Åでよく、当該HBDとHBAの距離は7.34 Åでよく、当該HBDとNIの距離は16.92 Åでよく、当該HRA1とHBAの距離は5.20 Åでよく、当該HRA1とNIの距離は14.34 Åでよく、当該HBAとHRA2の距離は4.69 Åでよく、当該HBAとNIの距離は9.85 Åでよく、および当該HRA2とNIの距離は5.66 Åでよい。
【0037】
更なるもう一つの態様において、当該HBD、HBA、HAR1、HAR2およびNIは、図8Cに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.79±1 Åであり、当該HBDとHRA2の距離は11.94±1 Åであり、当該HBDとNIの距離は16.28±1 Åであり、当該HRA1と
HBAの距離は5.20±1 Åであり、当該HRA1とNIの距離は14.64±1 Åであり、当該HBAとHRA2の距離は4.72±1 Åであり、および当該HRA2とNIの距離は5.63±1 Åである。特に、当該HBDとHRA1の距離は2.79 Åでよく、当該HBDとHRA2の距離は11.94 Åでよく、当該HBDとNIの距離は16.28 Åでよく、当該HRA1とHBAの距離は5.20 Åでよく、当該HRA1とNI の距離は14.64 Åでよく、当該HBAとHRA2の距離は4.72 Åでよく、および当該HRA2とNIの距離は5.63 Åでよい。
【0038】
本発明のもう一つの特徴は、インテグリン結合小分子を同定する方法である。当該方法は、上述のファルマコフォアの三次元構造と化合物の三次元構造を比較すること、当該化合物が、当該ファルマコフォアの特徴と一致する場合に、当該化合物を選択することを具備する。
【0039】
以下の詳細な説明、添付の図面との組合せを参照することにより、上述および本発明の他の特徴およびそれらを得るおよび使用する方法がより明確になるであろうし、最も浴理解されるであろう。当該図面は、本発明の典型的な態様を示すものであり、その範囲を制限するためのものではない。
【0040】
発明の詳細な説明
インテグリン αvβ3は、腫瘍の進行、転移および破骨細胞骨再吸収の複数の面において関連している。多くの腫瘍は、αvβ3を高発現しており、この発現は、メラノーマ、神経膠腫、卵巣癌、前立腺癌、乳癌、並びに他の癌における腫瘍の進行に関連している。当該αvβ3受容体は、新規抗癌剤のための治療上の標的として評価されている。我々は、化学官能基を基礎にした共通の特徴のファルマコフォアモデル(ケミカル・ファンクション・ベース・コモン・フューチャー・ファルマコフォア・モデル)を使用して構造的に多用な小分子αvβ3アンタゴニストのシリーズを発見した。当該三次元(3D)ファルマコフォアモデルは、3つの最近報告されたαvβ3受容体アンタゴニストの訓練用セットを使用して作成した。αvβ3受容体の公知のアンタゴニストのデータベースを使用したバリデーションにおいて、高判別能を有する当該ファルマコフォアモデルが、新規の構造骨格および所望の化学的特徴を有する検索する化合物に対して3D照会として使用された。Hypo5を使用する約600,000化合物のデータベースの検索により832化合物を得た。構造的新規性、計算された物理化学的な性質およびサンプルの有用性を基礎にして、38化合物が選択されαvβ3受容体結合アッセイにおいてスクリーニングされた。αvβ3受容体特異的結合アッセイにおいて試験された29化合物のうち、4つの化合物がナノモーラーで結合する親和性を示した。活性物質(AV26)の1つにおける有限構造活性相関分析(limited structure-activity relationship analysis)は、ナノモーラーからサブナノモーラー結合親和性を有する2つの強力な非RGDミメティックアンタゴニストの発見に結果として至った。がん細胞株のパネルにおいて評価された全ての活性化合物は、AV26を除いて、低マイクロモーラー濃度で、細胞毒性を示さず、AV26は、乳癌細胞株において考慮すべき細胞毒性を示した。我々の新規小分子化合物は、αvβ3陽性転移性癌に対して選択的に送達するためにパクリタキセルなどの抗癌剤と結合することが可能である。
【0041】
組成物
従って、本発明は、化合物またはその薬学的または薬用化粧品学的に許容される塩、溶媒和物または水和物を含む組成物であって、ここで、当該化合物は、1のH結合供与体(HBD)、1のH結合受容体(HBA)、2の疎水性芳香族基(HAR1およびHAR2)および1の陰性イオン性基(NI)を含み、およびここで、当該化合物は化合物 A、B,またはCではない組成物を提供する。
【0042】
当該組成物は、更に、薬学的または薬用化粧品学的に許容される担体を含んでよい。「薬学的または薬用化粧品学的に許容される担体」は、薬学的または薬用化粧品学的投与に適合する溶媒、分散媒、コーティング、抗菌剤および抗真菌剤、等張剤および吸収遅延剤などを含む。
【0043】
一つの態様において、当該HBD、HBA、HAR1、HAR2,およびNIは、図8Aに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.81±1 Åであり、当該HBDとHBAの距離は7.52±1 Åであり、当該HBDとNIの距離は17.67±1 Åであり、当該HRA1とHBAの距離は7.52±1 Åであり、当該HRA1とHRA2の距離は9.41±1 Åであり、当該HBAとNIの距離は10.20±1 Åであり、および当該HRA2とNIの距離は5.88±1 Åである。特に、当該HBDとHRA1の距離は2.81 Åでよく、当該HBDとHBAの距離は7.52 Åでよく、当該HBDとNIの距離は17.67 Åでよく、当該HRA1とHBAの距離は7.52 Åでよく、当該HRA1とHRA2の距離は9.41 Åでよく、当該HBAとNIの距離は10.20 Åでよく、および当該HRA2とNIの距離は5.88 Åでよい。
【0044】
もう一つの態様において、当該HBD、HBA、HAR1、HAR2およびNIは、図8Bに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.80±1 Åであり、当該HBDとHBAの距離は7.34±1 Åであり、当該HBDとNIの距離は16.92±1 Åであり、当該HRA1とHBAの距離は5.20±1 Åであり、当該HRA1とNIの距離は14.34±1 Åであり、当該HBAとHRA2の距離は4.69±1 Åであり、当該HBAとNIの距離は9.85±1 Åであり、および当該HRA2とNIの距離は5.66±1 Åである。特に、当該HBDとHRA1の距離は2.80 Åでよく、当該HBDとHBAの距離は7.34 Åでよく、当該HBDとNIの距離は16.92 Åでよく、当該HRA1とHBA の距離は5.20 Åでよく、当該HRA1とNI の距離は14.34 Åでよく、当該HBAとHRA2の距離は4.69 Åでよく、当該HBAとNIの距離は9.85 Åでよく、および当該HRA2とNIの距離は5.66 Åでよい。
【0045】
更なるもう一つの態様において、当該HBD、HBA、HAR1、HAR2およびNIは、図8Cに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.79±1 Åであり、当該HBDとHRA2の距離は11.94±1 Åであり、当該HBDとNI の距離は16.28±1 Åであり、当該HRA1とHBAの距離は5.20±1 Åであり、当該HRA1とNIの距離は14.64±1 Åであり、当該HBAとHRA2の距離は4.72±1 Åであり、および当該HRA2とNIの距離は5.63±1 Åである。特に、当該HBDとHRA1の距離は2.79 Åでよく、当該HBDとHRA2の距離は11.94 Åでよく、当該HBDとNI の距離は16.28 Åでよく、当該HRA1とHBAの距離は5.20 Åでよく、当該HRA1とNI の距離は14.64 Åでよく、当該HBAとHRA2の距離は4.72 Åでよく、および当該HRA2とNIの距離は5.63 Åでよい。
【0046】
本発明の化合物は、式 I、II、IIIまたはIVの化合物であってよい。
【0047】
式I、IIおよびIIIにおいて、それぞれ、R1およびR3 は脂肪族、芳香族またはヘテロ環基; R2 は置換基;およびX は N、OまたはSである。幾つかの態様において、それぞれ、R1およびR3は、アルキル、置換されたアルキル、アルケニル、置換されたアルケニル、アルキニル、置換されたアルキニル、フェニル、置換されたフェニル、アリール、置換されたアリール、ヘテロアリールまたは置換されたヘテロアリール 基; R2 は水素またはハロゲン、ヒドロキシル、スルフヒドリル、アルコキシ、カルボキシル、ニトロ、シアノ、アミノ、アミドまたはスルホニル基または何れかの数の炭素原子を含む何れか他の有機官能基;またはその組み合わせである。他の態様において、R1またはR3における当該置換されたアルキル、アルケニル、アルキニル、フェニル、アリールまたはヘテロアリール基は、ハロ、ヒドロキシル、スルフヒドリル、アルコキシ、カルボキシル、カルボニル、ニトロ、シアノ、アミノ、アミド、スルホニルまたは置換されたヘテロ環基により置換されたアルキル、アルケニル、アルキニル、フェニル、アリール,またはヘテロアリール基である。
【0048】
式 IVにおいて、それぞれ、R1およびR2 は水素またはハロゲン、ヒドロキシル、スルフヒドリル、アルコキシ、アリールオキシ、カルボキシル、ニトロ、シアノ、アミノ、アミドまたはスルホニル基または何れかの数の炭素原子を含む何れか他の有機官能基; R3 は水素、ヒドロキシル、アルコキシ、アリールオキシ、芳香族またはヘテロ環基または何れかの数の炭素原子の脂肪族鎖;それぞれ、R4およびR5 はヒドロキシルまたはアミノ基; X はC、N、OまたはS; m は、炭素、窒素、酸素または硫黄原子を伴う 0 から 3 原子リンカー基であり;およびn は炭素、窒素、酸素または硫黄原子を伴う0 から 5 原子リンカー基である。幾つかの態様において、R4またはR5における当該アミノ基は、脂肪族、芳香族またはヘテロ環基で置換されている。
【0049】
本発明の化合物の例は、AV1-11、AV14-15、AV17-32およびAV34-38を含む。
【0050】
本発明の化合物は、当該技術分野において公知の方法を使用して化学合成により、または市販の供給源から得てもよい。本発明の組成物は、以下に記載する疾患の診断および治療のために使用される。
【0051】
幾つかの態様において、本発明の化合物は、1または1以上の治療剤、イメージング剤およびその組合せとの併用において使用されてよい。当該治療剤は、しかしながらこれらに限定するものではないが、何れかの小分子薬物、抗体、FCフラグメント、タンパク質ベースの薬物、オリゴヌクレオチド、siRNA、アプタマーまたはアンキリンリピートを含む。当該治療剤の詳細な例は、血管増殖阻害剤(例えば、TNF-アルファ、アンジオスタチン、ベバシザン、アレステン、カンスタチン、コンブレタスタチン、エンドスタチン、NM-3、サリドマイド、スロンボスポンジン、タムスタチン、2-メトキシエストラジオールおよびビタキシン)、関節リウマチを治療するための治療剤、放射線治療剤、化学療法剤(例えば、シスプラチン、ドキソルビシン、ビンクリスチン、シクロホスファミド、トポテカン、パクリタキセルおよび他の化学療法剤)、骨転移を治療するための治療剤、骨粗鬆症を治療するための治療剤(例えば、ホセマックス、アレジア、アクトネル、ジンドロネル、コメタ、プレマリン、クリマラ、エストラ、ビベラ、エストラダーム、プレモプロ、プレムフェーズ、フェマート、プレフェスト、コンビパッチ、エビスタ、カルシトニン、ミアカルシンおよびカルシトニン)および再狭窄を治療するための治療剤を含む。当該イメージング剤は、これらに限定するものではないが、放射性核種(すなわち、イメージング可能なガンマ線またはポジトロン放出を生成する放射性金属イオン)を含む。
【0052】
本発明の化合物は、少なくとも1の治療またはイメージング化合物と実施可能に連結し、複合体を形成してよい。「実施可能に連結される」により、本発明の化合物と治療剤またはイメージング剤が、当該化合物が細胞においてインテグリンに対して結合するように、且つ治療剤またはイメージング剤の機能が妨害されないように結合することを意味する。
【0053】
現在広く使用される大部分の癌化学療法は、これに限定するものではないが、癌細胞に殆ど選択性を有さないシスプラチン、ドキソルビシン、ビンクリスチン、シクロホスファミド、トポテカンおよびパクリタキセルを含む。同様の問題は、非癌的兆候、例えば、関節リウマチなどのために使用される多くの治療に伴って生じ、そこにおいて、副作用が、特に延長された治療過程で生じる。多くの場合、これは望まれない副作用、および正常組織に対する高い毒性を引き起こす。正常組織に対する毒性の結果として、多くの抗癌化学療法並びに非癌性薬物は、多くの場合、至適以下の用量で投与されるか、または当該治療が妨害されてしまい、その結果として効果が低減され、最終的に治療が失敗する。これは、薬物抵抗性を発生することにより伴う可能性がある。広く引用されたこの例は、アントラサイクリンによる用量を制限する心毒性である。問題の薬物の選択的毒性は、疾患に犯された組織に届く薬物の用量の増加することにより、または正常組織に届く用量を減少することの何れかにより、または、その両方によって増加する。
【0054】
薬物の標的細胞への送達は、薬物と、正常組織とは相対的に当該標的細胞において独自に発現または過剰発現されたかの何れかの抗原または受容体に結合する分子との関連により選択的に増大できる。これにより、薬物の当該標的細胞への特異的な送達が可能になる。例えば、αvβ3インテグリンは、腫瘍の新生血管および炎症疾患性のあいだ、並びに破骨細胞において過剰発現されていることが知られているので、本発明の複合体はこれらの細胞を選択的に標的にするために使用することが可能である。
【0055】
本発明の複合体は、当該技術分野において周知の何れかの方法を用いて構成されてよい。これらの方法の詳細な説明は、例えば、以下に見出すことが可能である:「Bioconjugate Techniques」 by G.T. Hermanson、Academic Press、San Diego、1996、これらの内容は、参照することにより、その全体において本明細書に組み込まれる。
【0056】
例えば、問題の他の分子(例えば、抗体、薬物、DNAおよびsiRNA分子など)に対する本発明の化合物の複合体は、一般的に入手可能な技術および公開されている技術を用いて化学的に架橋結合により最も容易に実施されてよい。架橋結合は、2またはそれ以上の分子を共有結合により化学的に結合する方法である。架橋結合剤は、タンパク質または他の分子における特異的な官能基(例えば、一級アミン、スルフヒドリルなど)に対する反応性末端を含む。反応のための標的であり得るタンパク質およびペプチドにおける幾つかの化学基の有効性のために、タンパク質およびペプチドは、容易に結合され、且つ別の架橋結合法を使用して試験されている。クロスリンカーは、また、一般的に核酸、薬物および固体表面を修飾するために使用される。それらはまた、抗体-酵素複合体、免疫毒素および他の標識されたタンパク質試薬を用意するために有用である。当該リガンド(例えば、本発明の化合物)との接触の前または後でクロスリンカーにより受容体を誘導体化することにより、受容体-リカ゛ント゛複合体の単離が可能である。放射ヨウ素標識クロスリンカーの使用は、オートラジオグラフィー検出により特定の受容体を同定することを可能にする。クロスリンカーは、それらの化学的反応性(即ち、特定の官能基に対する特異性)および応用を伴う反応の適合性を基礎に選択される。特異的な適用のために使用される最も好ましいクロスリンカーは、経験的に決定されるべきである。クロスリンカーは、以下の特徴を基に選択される:化学的特異性、スペーサーの腕の長さ、水溶解性および細胞膜透過性、同じ(ホモ二機能性)または異なる(ヘテロ二機能性)反応性基、自発的反応性基または光反応性基、および切断性(cleavability)。クロスリンカーは、少なくとも2の反応性基を含む。架橋のために標的とされ得る官能基は、一級アミン、スルフヒドリル、カルボニル、カルボ水和物およびカルボン酸を含む(表A)。カップリングもまた、光反応性フェニルアジドクロスリンカーを使用して非選択的である。
【表A】
多くの場合、異なるスペーサーの腕の長さが要求され、これは、立体効果が架橋のための潜在的な反応部位の間の距離を要求するためである。通常、短い(4〜8Å)スペーサーの腕を有するクロスリンカーが、第一におよび決定された架橋の程度で使用される。より長いスペーサーの腕を有するクロスリンカーは、次に最適化された架橋高率のために使用され得る。短いスペーサーの腕は、多くの場合、分子内架橋研究において使用され、分子間の架橋は、長いスペーサーの腕を含むクロスリンカーを好む。多くの適用において、タンパク質の天然の構造を維持する必要があり、そのために架橋は、最も多くの場合、穏やかなpHと緩衝液条件を使用して実施される。更に、反応のために最適なクロスリンカー対タンパク質のモル比が決定される。適用に依存して、複合の程度が重要な因子である。例えば、免疫原複合体を用意する場合、抗原の免疫原性を高めるために高い程度の複合が望まれる。しかしながら、抗体または酵素のための複合は、低度または中程度の複合が、タンパク質の生物学的な活性を保持するために至適であり得る。当該タンパク質表面における官能基の数もまた、考慮のために重要である。仮に多数の標的基がある場合、より低いクロスリンカー対タンパク質の比が使用される。制限された数の潜在的な標的のためには、より高いクロスリンカー対タンパク質の比が要求される。更に、多くの成分は、最小限に維持されるべきであり、それは2以上の成分からなる複合体は、分析することが困難であり、タンパク質サブユニットの空間的な配置における少ない情報しか提供されないためである。水溶解性および膜透過性もまた、カップリングのための最適なクロスリンカーを選択する上で重要な問題である。多くのクロスリンカーは、それらの疎水性のスペーサーの腕によって、水溶液における溶解度が制限される。これらのクロスリンカーは、一般的にDMFまたはDMSOに溶解され、次に生物系または架橋されるべき生体分子の溶液に添加される。疎水性のクロスリンカーは、細胞膜およびオルガネラ膜を通過することが可能であり、膜の外側表面、および膜と結合した空間内の両方における架橋に影響を及ぼす。多くの場合、生物系のための架橋手順に有機溶媒を導入することは都合が悪いか、または望ましくない。また、多くの場合、細胞またはオルガネラの内側を変化させることなく、細胞膜またはオルガネラ膜の外側表面だけにおける架橋に影響を及ぼすことが望ましく、そのような場合において、幾らかの水溶解、膜不透過のクロスリンカーが利用できる。幾つかのクロスリンカーは、ポリエチレングリコール(PEG)サブユニットから形成されるスペーサーの腕を含み、結果として大量の酸素原子と共にポリエチレンオキシド(PEO)を生じて水溶解性を与える。これらのクロスリンカーは、それらの名称に(PEO)nが示され、水可能性であり、且つ生物膜透過できない。それらは、架橋化された複合体に対してそれらの親水性のスペーサーを移行する更なる利点を提供し、従って、当該複合体の凝集および沈殿を減少する。他のクロスリンカーは、当該スペーサーの何れかの末端で荷電反応性基によって、それらの水溶解性および膜不透過性を含む。荷電反応記、例えば、スルホ-NHSエステルまたはイミドエステルなどは、当該架橋試薬に対して水溶解性を与えるが、当該反応性基は最終複合体の一部分ではないので、当該架橋された複合体には与えない。
【0057】
説明の目的のために、2つの例として、NVX-188(即ち、AV38)複合体を構成することを以下に示す。
【0058】
例1:カルボイル反応性化学を使用することによるNVX-188および類似体のカルボキシル基のカップリング
NVX-188基の幾つかは、種々のクロスリンカーのためのカップリングパートナーとして役立つカルボキシル基を含む。カルボジイミドは、一級アミンまたはヒドラジンに対してカルボキシルを結合し、結果としてアミドまたはヒドラゾン結合を形成する。カルボジイミドは、結合される分子の間にスペーサーが存在しない他の結合反応とは異なる。タンパク質のカルボキシ末端、並びにグルタミン酸およびアスパラギン酸側鎖もまた、標的とされ得る。この例において、EDC(製品#22980、22981、Pierce)はカルボン酸基と反応し、カルボキシル基を活性化し、活性O-アシルイソウレア中間体を形成し、当該反応混合物における当該アミノ基に対する結合を可能にする。EDC副産物は、可溶性尿素誘導体として、求核試薬による排除の後に放出される。当該O-アシルイソウレア中間体は水溶液中で不安定であり、N-ヒドロキシスクシンイミドを使用する中間体の安定性を増加することなく、2つの工程の複合体手順においてそれを無効にする。この中間体は、一級アミンと反応し、アミド誘導体を形成する。アミンとの反応ができないということは、結果として当該中間体の加水分解、当該カルボキシルの再生およびN-非置換ウレアの放出を生じる。当該架橋反応は、通常、pH4.5〜5の間で実施され、多くの適用のために数分しか必要としない。しかしながら、当該反応の終了は、pH4.5〜7.5で同様である。EDCの加水分解は、カップリング中競合的反応であり、温度、pHおよび緩衝液組成物に依存する。4-モルホリノエタンスルホン酸(MES、製品#28390、Pierce)は有効なカルボジイミド反応緩衝液である。リン酸緩衝液は、EDCの反応効率を低下するが、EDCの量を増加することで、低下した効率を代償できる。トリス、グリシンおよびアセテート緩衝液は、結合緩衝液として使用されない。NHS(製品 # 24500、Piercebiotechnology)またはその水可溶性類似体 スルホ-NHS(製品# 24510、Pierce)は、多くの場合、EDCカップリングプロトコールを含み、効率を改善する。EDCは、NHSをカルボキシルに対して結合し、結果として、分子におけるNHS活性化部位を生じる。形成された当該NHS-エステルと、当該カルボジイミドのO-アシルイソウレア中間体は、アミン反応性である;しかしながら、NHS-エステルは、僅かに酸性または中性近くのpH状態において非常に高い安定性を有する。水中で、NHS-エステルは、温度、pHおよび当該クロスリンカーの構造に依存して1〜数時間、または数日までもの半減期を有するが、O-アシルイソウレア中間体は、酸性または中性pH状態で秒単位で測定される半減期を有する。分子のEDC媒介カップリングは、多くの適用において、NHSまたはスルホ-NHSなしで良好に働き、これらは、一般的に必要とされるタンパク質濃度が非常に低い場合でない限り必要とされない。多量に過剰なEDCがNHSを伴わずに使用される場合には、しばしば、EDC/NHS系への変換の際に当該EDC量を減少させ、過剰な架橋と可能な沈殿を防止する必要がある。
【0059】
例2:非特異的化学(アリールアザイド)を使用するNVX-188および類似体のカップリング
光反応試薬は、紫外線または可視光に暴露された場合に、反応性となる化学的に不活性な試薬である。少しの例外はあるが、これらの試薬における当該光反応基は、アリールアザイドである。アリールアザイドがUV光に曝されたとき、それは、二重結合との更なる反応、C-HおよびN-H部位への挿入、または続く求核試薬(例えば、一級アミン)と反応するための環拡大を開始することが可能なニトレン基を形成する。当該サンプルに一級アミンが存在する場合、後者の反応経路が支配する。チオール含有還元剤(例えば、DTTまたは2-メルカプトエタノールなど)は、光反応前および光反応中における全ての工程の間に当該サンプル溶液中から避けられるべきである。これらの試薬は、アミンに対するアザイド官能基を減少し、光活性化を妨げるであろう。反応は、種々のアミン非含有緩衝液条件において実施され得る。ヘテロ二機能性光反応性クロスリンカーと作業する場合、緩衝液は、当該試薬の化学的反応性と適合可能に使用されるべきである。当該化学反応は、ホイルで覆われた反応容器で光を遮った中で実施される。当該光活性化は、当該反応容器上約1〜2インチ(約2.54〜5.08センチメートル)で明るいカメラフラッシュまたは紫外線ハンドホルダーランプを用いて実施される。明るいカメラフラッシュ右派、ニトロ-およびヒドロキシ置換アリールアザイドと共に良好に機能する。非置換のアリールアザイドは、紫外線光または多数のフラッシュを必要とするかもしれない。
【0060】
本発明の組成物は、その意図される投与経路に適合するように形成される。例えば、米国特許第 6,756,196を参照されたい。投与経路の例は、非経口、例えば、静脈内、皮内、皮下、経口(例えば、吸入)、経皮(局所的)、経粘膜および直腸投与を含む。非経口、皮内または皮下適用のために使用される溶液または懸濁液は、以下の成分を含んでよい:滅菌希釈剤、例えば、注射用の水、整理食塩溶液、特定のオイル、ポリエチレングリコール、グリセリン、プロピレングリコールまたは他の合成溶媒;抗菌剤、例えば、ベンジルアルコールまたはメチルパラベン;抗酸化剤、例えば、アスコルビン酸または亜硫酸ナトリウム;キレート剤、例えば、エチレンジアミンテトラ酢酸;緩衝液、例えば、アセテート、クエン酸塩、リン酸塩;張度の調整のための薬剤、例えば、塩化ナトリウムまたはデキストロース。pHは、酸または塩基、例えば、塩酸または水酸化ナトリウムなどで調整できる。
【0061】
非経口製剤は、ガラスまたはプラスチックで作られたアンプル、ディスポシリンジまたは多用量バイアルに入れることが可能である。
【0062】
注射に使用するために適切な組成物は、滅菌水溶液(水溶液中)または懸濁液および滅菌注射溶液または分散剤の即席製剤のための滅菌パウダーを含む。静脈投与のための適切な担体は、生理的食塩水、滅菌水、クレモフォア(Cremophor) EL(登録商標)(BASF
、Parsippany、NJ)またはリン酸緩衝化生理食塩水(PBS)を含む。全ての場合において、当該組成物は、滅菌されるべきであり、簡単にシリンジに存在可能な範囲の流体であるべきである。それは、製造および貯蔵の条件下で安定であるべきであり、細菌および真菌などの微生物の活性を混入作用に反して保存されるべきである。当該担体は、例えば、水、エタノール、ポリオール(例えば、グリセロール、プロピレングリコールおよび液体ポリエチレングリコールなど)、および適切なその混合物を含む溶媒または分散剤であってよい。その適切な流動性は、例えば、レシチンなどのコーティングなどの使用により、分散剤の場合においては、必要とされる粒子サイズの維持により、および界面活性剤の使用により維持される。微生物の作用の阻止は、種々の抗菌剤および抗真菌剤、例えば、パラベン、クロルブタノール、フェノール、アスコルビン酸、チメロサル(thimerosal)などにより達成される。多くの場合において、当該組成物において、等張剤、例えば、糖、ポリアルコール、例えば、マンニトール、ソルビトールまたは塩化ナトリウムが含まれることは好ましいであろう。注射用組成物の延長された吸収は、アルミニウムモノステアレートおよびゼラチンなどの吸収を遅延する薬剤を当該組成物に含ませることにより、凡そ達成され得る。
【0063】
滅菌注射溶液は、所望に応じて、1のまたは上で列記した成分の組合せと共に適切な溶媒に必要量で当該化合物を組み込み、続いてフィルター滅菌することにより製造できる。一般的に、分散剤は、基本的な分散媒および上で列記したものからの所望の他の成分を含む滅菌媒体に当該化合物を組み込むことにより製造される。滅菌注射溶液の製造のための滅菌パウダーの場合において、好ましい製造の方法は、予めフィルター滅菌されたその溶液から活性成分と更なる所望の成分を得る減圧乾燥および凍結乾燥である。
【0064】
経口組成物は、一般的に不活性な希釈液または可食担体を含む。経口治療投与の目的のために、当該化合物は、賦形剤と共に組み込まれ、錠剤、トローチまたはカプセル、例えば、ゼラチンカプセルなどの形態において使用される。経口組成物はまた、マウスウォッシュとして使用されるための流体担体を使用して製造されてもよい。薬学的または薬用化粧品学的に適合する結合剤および/またはアジュバント物質が当該組成物の一部分として含まれてもよい。当該錠剤、ピル、カプセル、トローチなどは、以下の成分または同様の天然の化合物の何れかを含んでもよい:結合剤、例えば、微小結晶性セルロース、トラガカントゴムまたはゼラチン;賦形剤、例えば、デンプンまたはラクトース、崩壊剤、例えば、アルギン酸、プリモゲル(Primogel)またはトウモロコシデンプン;潤滑剤、例えば、ステアリン酸マグネシウムまたはステロート(Sterotes);グリダント(glidant)、例えば、コロイド状シリコンジオキシド;甘味剤、例えば、ショ糖またはサッカリン;または香味剤、例えば、ペパーミント、メチルサリチル酸塩、またはオレンジ香料。
【0065】
吸入による投与のための当該組成物は、適切な噴霧剤、例えば、ガス、例えば、二酸化炭素を含むプレッシャーコンテナまたはディスペンサーまたはネブライザーからのエアロゾルスプレーの形態で送達される。
【0066】
全身投与もまた、経粘膜または経皮手段により行われてよい。経粘膜または経皮投与のために、透過されるべき障壁に対して適切な貫通刺激が製剤において使用される。そのような貫通刺激は、当該分野において一般的に公知であり、例えば、経粘膜投与、界面活性剤、胆汁塩、フシジン酸誘導体などが含まれる。経粘膜投与は、経鼻スプレーまたは坐剤の使用を介して達成されてよい。経粘膜投与のために、本発明の化合物が、当該技術分野において一般的に公知の軟膏(ointment)、軟膏(salves)、ゲルまたはクリームに製剤化される。
【0067】
本発明の組成物はまた、坐剤の形態(例えば、ココアバターおよび他のグリセリドなどを基剤する通常の坐剤)、または直腸送達のための維持浣腸の形態に製造されてよい。
【0068】
一つの態様において、当該組成物は、生体からの急速な消失に対して当該化合物を保護する担体と共に製造され、例えば、制御放出製剤、例えば、インプラントおよび微小被包性送達系などを含む形態に製造される。生分解性の生体適合性重合体、例えば、エチレンビニルアセテート、ポリアンヒドライド、ポリグリコール、コラーゲン、ポリオルトエステルおよびポリ乳酸などが使用されてよい。そのような製剤の製造方法は、当業者に明白であろう。当該材料も、Alza Corporation および Nova Pharmaceuticals,Incから商業的に得ることが可能である。リポソームの懸濁液(ウイルス性抗原に対するモノクローナル抗体を伴う感染細胞に対して標的するリポソームを含む)も、薬学的または薬用化粧品学的に許容される担体として使用されてよい。これらは、当業者に公知の方法に従って、例えば、米国特許第 4,522,811に従って製造される。
【0069】
投与が容易且つ均一な用量のために単位投与量での経口または非経口組成物が製剤化されることも有利である。ここで使用される「単位投与量形態」は、治療されるべき対象のための単位投与量として適切な物理的に分離した単位をいう:各単位は、必要とされる薬学的または薬用化粧品学的担体と共に所望の治療学的または薬用化粧品学的効果を生ずることが計算された活性化合物の予め決定された量を含む。
【0070】
本発明の組成物は、容器、パックまたはディスペンサーに含まれて、投与のための指示と共にパッケージされた製品を形成してもよい。他の活性化合物もまた、本組成物と組み合わされてもよい。
【0071】
組成物の使用
処理の方法
本発明の1つの目的は、インビトロまたはインビボにおいて化合物に対してインテグリンを結合し、それによりインテグリンの活性を調節するための方法を提供する。当該方法は、インテグリンを本発明の化合物またはその薬学的若しくは薬用化粧品学的に許容される塩、溶媒和物または水和物と接触することを具備する。
【0072】
インテグリンは、細胞接着およびシグナリング受容体を形成する糖タンパク質のファミリーである。少なくとも25のインテグリン受容体では、アルファおよびベータサブユニットのヘテロダイマーにより形成される各々が確認されている。αvβ3インテグリンは、インテグリンファミリーの突出したものである。
【0073】
インテグリンは、細胞死、増殖、遊走および組織リモデリングを含む細胞応答を調節する細胞外マトリックスからの指示に対して応答する。シグナリングは、FAKおよびSrcファミリーからのチロシンキナーゼの補充を介して調節される。これは、MAPキナーゼおよびPI3K伝達カスケードの活性を調節する。細胞外マトリックスに対するインテグリン媒介接着の欠如は内皮細胞におけるアポトーシスを引き起こす。多くのインテグリンは、多くの細胞外タンパク質において見出されているRGDトリペプチドモチーフ(Arg-Gly-Asp)に対して特異的に結合する。
【0074】
αvβ3の発現は、殆どの組織において低いが、リモデリングまたは増殖組織においては非常に高い。αvβ3は、細胞外マトリックスタンパク質、フィブロネクチン、フィブリノーゲン、オステオポンチン、スロンボスポンジンおよびビトロネクチンのRGDモチーフに対して結合する。それは、腫瘍の増殖および転移、骨再吸収、ある炎症性疾患および血管構造(血管形成)において重要な役割を演じている。αvβ3インテグリンの生物学的活性は、症状の範囲において、選択的αvβ3アンタゴニストを使用する治療学的および薬用化粧品学的介入のための基礎を提供する。これらの症状は、しかしながらこれらに限定するものではないが、癌、骨粗鬆症、関節リウマチおよび加齢関連横斑性変性を含む。
【0075】
従って、一つの態様において、本発明の化合物は、細胞、例えば、癌細胞、良性腫瘍細胞、間質細胞、炎症に反応する、炎症を媒介する、または炎症を調節する細胞(例えば、白血球、マクロファージおよび樹状細胞)、免疫系の細胞(例えば、白血球、マクロファージおよび樹状細胞)、血管機能に関与する細胞(例えば、内皮細胞、内皮細胞に関与する細胞および平滑筋細胞)、骨芽細胞、破骨細胞またはウイルスに感染した細胞(例えば、アデノウイルス(Adenoviridae)、コックサッキーウイルス(Picornaviridae)、エコウイルス(Picornaviridae)、口蹄疫ウイルス(Picornaviridae)、ハンタウイルス(Buriyaviridae)、ヒトパレコウイルス(Picornaviridae)、ヒト免疫不全ウイルス 1(Retroviridae)およびロタウイルス(Reoviridae))においてインテグリンと接触する。
【0076】
本発明は更に、それを必要とする対象を処置するための予防学的および治療学的の両方または薬用化粧品学的方法のために有効量の上記の化合物を提供する。
【0077】
ここで使用される「対象」は、ヒト、または以下を含む動物、全ての脊椎動物、例えば、哺乳類、例えば、霊長類(特に、高等霊長類(higher primates))、ヒツジ、イヌ、げっ歯類(例えば、マウスまたはラット)、モルモット、ヤギ、ブタ、ネコ、ウサギ、ウシ;および非哺乳類、例えば、ニワトリ、両生類、爬虫類などをいう。好ましい態様において、当該対象はヒトである。もう一つの態様において、当該対象は動物である。
【0078】
処置されるべき対象は、例えば、当該分野に公知の診断方法を使用して、疾患または以下に記載の状態に罹患しているか、またはそれらの進展のリスクに面しているかが同定されてもよい。当該対象は、対象またはヘルスケアのプロフェッショナルの判断により同定されてもよく、主観的に(例えば、見解)または客観的に(例えば、検査または診断方法により測定可)であってもよい。
【0079】
本明細書に使用される、用語「処置」は、当該疾患、疾患の症状または疾患に向かう素因を治療、治癒、緩和、軽減、変化、療法、寛解、改善するまたはそれらに影響する目的を伴って、治療剤または化粧剤の対象への適用または投与、または対象から単離された組織または細胞株への治療剤または化粧剤の適用または投与、(ここで、当該対象は、疾患、疾患の症状または疾患に向かう素因を有する)と定義される。
【0080】
「有効量」は、処置される対象において、本明細書において描写された通りの医学的または薬用化粧品学的所望の血管を生じることが可能である治療剤または化粧剤の量である。当該医学的または薬用化粧品学的所望の結果は、(即ち、幾つかの試験またはマーカーにより測定可能な)客観的または(即ち、対象が効果の兆候を示す、または効果を感じる)主観的なものであってよい。
【0081】
本発明の特性および治療学的または薬用化粧品学的有効性は、細胞培養物または実験動物において、例えば、LD50(集団の50%までの致死的用量)およびED50(集団の50%において治療学的または薬用化粧品学的に有効な用量)を決定するための標準的な薬学的または薬用化粧品学的手続により決定されてよい。毒性と治療学的または薬用化粧品学的有効性の間の当該用量比は、治療学的指標であり、LD50/ED50 比と表される。高い治療学的または薬用化粧品学的指標を示す化合物が好ましい。有毒な副作用を示す化合物が使用されてよい一方で、非感染細胞への可能な障害を最少化し、それにより副作用を低減するために、そのような化合物を患部組織の部位に対して向ける送達系を設計する配慮がなされるべきである。
【0082】
細胞培養アッセイおよび動物実験から得られたデータは、ヒトにおいて使用するための用量の範囲を決定することにおいて使用できる。当該化合物の投与量は、好ましくは殆ど毒性がないか、全く毒性のないED50を含む循環濃度の範囲内に位置する。当該投与量は、使用される投与量形態および利用される投与経路に依存するこの範囲内で変動してよい。本発明の方法において使用される幾つかの化合物については、当該治療学的または薬用化粧品学的有効用量は、細胞培養アッセイから初期に推定され得る。用量は、動物モデルにおいて、細胞培養において決定されたようなIC50(即ち、症状の最大半減阻害を達成する化合物の濃度)を含む循環血漿濃度範囲を達しするために処方されてよい。そのような情報は、ヒトにおいて有用な用量をより正確に決定するために使用され得る。血漿におけるレベル、例えば、高速液体クロマトグラフィにより測定されてよい。
【0083】
本発明の化合物の治療学的または薬用化粧品学的有効量(即ち、有効投与量)は、例えば、キログラム当たり約1 マイクログラムからキログラム当たり約500 ミリグラム、キログラム当たり約100 マイクログラムからキログラム当たり約5 ミリグラム、またはキログラム当たり約1 マイクログラムからキログラム当たり約50 マイクログラムの範囲であってよい。当該化合物は、例えば、約1から10週間の間、好ましくは2から8週間の間、より好ましくは約3から7週間の間、より更に好ましくは約4、5または6週間に亘り、1週当たり1回投与され得る。慢性疾患、例えば、関節炎または骨粗鬆症に罹患する対象に置いては、長期間に亘る治療、例えば、1日に1回または好ましくは1週間に1回などが必要であってよい。更に、化合物の適切な用量は、当該化合物の効力に依存することが理解される。これらの化合物の1または1以上が対象(例えば、動物またはヒト)に対して投与される場合、医師、獣医師または研究者が、例えば、最初は相対的に低用量で、引き続いて、適切な応答が得られるまで当該用量を増加して処方してよい。加えて、何れかの特定の対象のための当該具体的な用量レベルは、使用される具体的な化合物の活性、当該患者の年齢、体重、一般的な身体の状態、性別および食事制限、投与の時間、投与経路、排出率、何れかの薬物の併用、疾患または障害の重症度、以前の治療および他の疾患の存在を含む多様な因子に依存するであろうことは理解される。更に、当該化合物の治療学的または薬用化粧品学的有効量での対象の処置は、単一の処置を含み、または好ましくは一連の処置を含む。
【0084】
本発明の方法は、多数の適用を有する。当該適用の1つの例は、これらに限定するものではないが、全ての組織学的種類の悪性固形腫瘍、例えば、肉腫および癌腫および高レベルの血管形成および血管原性因子に関連する血液学的悪性物、例えば、急性および慢性白血病、多発性骨髄腫、骨髄増殖性疾患、非ホジキンリンパ腫およびホジキン病を含む癌の処置である。
【0085】
腫瘍の増殖&転移: αvβ3 インテグリンは、悪性メラノーマ、乳癌、卵巣癌および神経膠腫および大腸癌を含む多くの腫瘍により発現されている。これらの腫瘍における過剰発現は、浸潤および転移を含む悪性進行と関連する。癌治療のためのαvβ3インテグリンアンタゴニストの潜在的な役割についての実験的サポートは以下を含む:
αvβ3インテグリンアンタゴニストで治療されるマウスにおける、マウス大腸癌細胞の注入に続く肝転移の低下、
サイクリックRGDペンタペプチドに対する暴露に続く、インビトロでの脳腫瘍細胞におけるアポトーシスの誘導、
マウスにおけるメラノーマ異種移植の増殖のオリゴペプチドαvβ3インテグリンアンタゴニストによる阻害、
マウスにおける乳癌異種移植の腫瘍アポトーシスの増加および治療率に対する放射免疫療法およびシレンジタイド(サイクリックRGDペプチド)の相乗効果、
αvβ3インテグリンに対するモノクローナル抗体によるSCIDマウスにおけるヒト皮膚移植における乳癌腫の増殖阻害(および血管形成の減少)、
メラノーマ、神経膠腫、乳癌および卵巣癌を含む幾つかの腫瘍タイプにおける腫瘍進行とαvβ3インテグリンの発現の相関、
αvβ3インテグリンの変異体(常時活性型)変種と関連するMDA-MB 435乳癌細胞の転移可能性の増加、および
高レベルのαvβ3インテグリン発現に形質転換された腫瘍からの骨転移の発生率の増加、およびマウスにおけるMDA-MB 435乳癌異種移植のαvβ3インテグリンアンタゴニストによる治療での骨転移の抑制。
【0086】
αvβ3インテグリンは、腫瘍細胞浸潤および腫瘍細胞核酸において重要な役割を果たしていることから、αvβ3インテグリンアンタゴニスト、特に毒性プロフィールを伴わずに、細胞外マトリックスタンパク質に対する結合から浸潤している癌細胞をブロックすることが可能なもの、例えば、NVX-188、は、この適用のために特別に興味深く、また、併用治療戦略のために良好に適しているようである。
【0087】
血管新生: αvβ3インテグリンは、新血管新生の間に血管平滑筋細胞において発現する。血管発生におけるαvβ3インテグリンの中心的役割は、実験において証明されている。これらの実験において、(即ち、腫瘍における)新たな血管増殖は実質的にαvβ3インテグリンアンタゴニストによりブロックされた。癌における当該αvβ3インテグリンアンタゴニストの治療的利点は、直接的に腫瘍を殺すことに由来するものではなく、腫瘍血管新生を阻害することに由来するものである。例えば、動物モデルにおけるメラノーマ細胞による肺転移発生は、αvβ3インテグリンアンタゴニストにより防止できることが示されている。これは、当該メラノーマ細胞がαvβ3インテグリンを発現していないのだが生じた。従って、非細胞障害性のαvβ3ブロッカーは、癌治療において潜在的な血管形成阻害適用を有する。
【0088】
標的治療: 転移性癌細胞における重要な細胞表面受容体へのNVX-188の高特異性は、複合性(composite)の標的癌化学療法学の発達において利用され得る。これらの化合物は、標的細胞に高い親和性を有する特異的標的剤、例えば、NVX-188などに結合し、当該細胞を殺傷することが可能な細胞障害性の薬剤を伴う。当該2つの成分は、ナノ粒子表面をコートするNVX-188を有するナノ粒子として共有結合または処方され、特異的な取り込みを亢進する。それから当該細胞障害性薬剤が当該ナノ粒子に被包される。
【0089】
本発明の方法のもう一つの例示的適用は、良性腫瘍および新生物発生前の高い血管新生に関連する状態、尋常性疣贅、化膿性肉芽腫、酒さ、ケロイド瘢痕の治療である。脈絡叢または光彩新血管新生に関連する眼部疾患(腫瘍)もまた、本発明に従い治療され得る。
【0090】
加えて、本発明の方法は、炎症性疾患の治療のために使用され得る。そのような疾患の例は、これらに限定するものではないが、炎症性腸疾患、例えば、クローン病および潰瘍性大腸炎;多発性炎症性ミオパシー、例えば、多発性筋炎(PM)、皮膚筋炎(DM)および高い血管新生に関連する状態;および他の眼部炎症性疾患を含む。
【0091】
高い炎症と関連する疾患または状態、例えば、歯肉炎および粘膜炎(例えば、放射線および化学療法に誘導される粘膜炎)もまた、本発明の方法により治療されてよい。これらの適用において、本発明の化合物、例えば、NVX-188および類似体は、局所的に適用され、例えば、口腔粘膜炎などのためのマウスウォッシュ溶液であってよい。ざ瘡の治療のためには、本発明の化合物、例えば、NVX-188および類似体は単独、または抗細菌剤または他の抗起炎性療法と組み合わせて適用されてよい。特に慢性的な炎症状態に関連する胃腸管疾患、例えば、胃炎または十二指腸炎は、本発明の方法を使用して治療されてよく、本発明の化合物(例えば、NVX-188および類似体)の経口処方を適用することこにより最も効率的に治療されてよい。
【0092】
関節リウマチの中心的な特徴は、患部の関節における軟骨および骨の破壊を引き起こす慢性的な炎症である。この炎症は、サイトカイン系の活性化に起因する。これは、炎症細胞により、特に活性かれたマクロファージによる腫瘍壊死因子産生、IL-1およびIL-6を介して調節される。これらのマクロファージは、それらの表面においてαvβ3インテグリンを発現する。αvβ3阻害剤は、従って、関節リウマチ治療のために有用である。
【0093】
本発明の方法を使用して治療されるべき免疫および自己免疫疾患は、これらに限定うするものではないが、関節リウマチ、乾癬性関節炎、乾癬、多発性硬化症、気管支喘息およびアレルギーおよび皮膚の他の炎症性状態および皮下組織を含む。
【0094】
関節リウマチ(RA)は、関節における広範な骨および軟骨破壊に関連する衰弱性、全身性、自己免疫疾患である。αvβ3インテグリンが、RAの病因において重要な役割を果たしていることを見解を支持する実質的なデータがある。これらの役割には、活性化マクロファージ依存性炎症、破骨細胞の発達、骨再吸収および炎症性血管形成を含む。RAは、活性化された血管形成およびαvβ3 インテグリン脈管構造発現を伴う炎症性疾患である。
【0095】
ヒトにおいて関節リウマチを擬態した関節炎を誘導されたウサギおよびラットの実験モデルにおけるαvβ3阻害剤の動物実験は、アルスロジェニック(arthrogenic)変化の減少を示した。これらの変化は、ウサギにおける滑膜血管形成阻害、細胞浸潤の減少、パンヌス形成および軟骨侵食、およびラットにおける骨、軟骨および軟組織の有意な保護を含む。
【0096】
乾癬は、免疫媒介疾患であると主に考えられているにも拘わらず、また、過剰な血管形成により特徴付けられる。そのため、NVX-188および類似体などの本発明の化合物は、全身的のみならず、好ましく局所的にのみ、または乾癬の治療に現在使用される他の薬物と併用して使用されてもよい。これらの薬物は、例えば、皮膚および頭皮のための局所的なローション、軟膏、クリーム、ゲルおよびジャンプーとして添加されてもよい。FDAに承認された乾癬治療のための処方局所剤は、コルチコステロイド、レチノイド、カルシポトリエン(calcipotriene)およびコールタール製品を含む。
【0097】
多発性硬化症(MS)は、もう一つの自己免疫仮定により媒介される衰弱性疾患である。中枢神経系におけるMS病変は、αvβ3インテグリンを含むラミニン受容体発現における変化を示す。NVX-188などの発明の化合物は、αvβ3インテグリン受容体に結合する特異的なリガンドとして潜在的に有用である。
【0098】
本発明の方法を使用して値幼され得る皮膚および皮下組織の他の炎症性状態には、これらに限定するものではないが、紅斑性扁平皮膚症、アトピー性皮膚炎および関連状態、接触性皮膚炎および他の湿疹、内部に取り込まれた物質のための皮膚炎、水疱性皮膚疾患、紅斑性状態、苔癬および掻痒症および関連する状態が含まれる。
【0099】
本発明の化合物、例えば、NVX-188および類似体は、全身的にでも、更に好ましくは局所的のみでも、または現在使用されている他の薬剤との組合せにおいての何れにおいても使用されてよい。これらの薬剤は、例えば、皮膚および頭皮のための局所的ローション、軟膏、クリーム、ゲルおよびシャンプーとして添加されてもよい。治療されてよい状態の例は以下である: 紅斑性扁平皮膚症、斑不全角化、脂漏性皮膚炎、頭部脂漏症、他の脂漏性皮膚炎、紅斑性扁平皮膚症、アトピー性皮膚炎および関連状態、紅斑、アトピー性皮膚炎、ベニエ痒疹、湿疹:(アトピー性、曲性(flexural)、内因性(アレルギー性))、神経皮膚炎、接触性皮膚炎および皮膚炎を含む他の湿疹(接触性、職業性)、内部に取り込まれた物質のための皮膚炎、皮膚に接触した薬物性および医薬性、皮膚炎(アレルギー性)、例えば、皮膚に適用された何れかの薬物性または他の化学製品性、日射のための皮膚炎、日焼け、ベルロック皮膚炎、光アレルギー反応皮膚炎、光毒性反応皮膚炎、多形性 光性発疹、播種性浅在性光線性汗孔角化症(DSAP)、化粧品性皮膚炎、他の放射線、例えば、赤外線、紫外線などによる皮膚炎および電離放射線での治療による皮膚炎、水疱性皮膚疾患、疱疹状皮膚炎、疱疹状皮膚症、デューリング病、疱疹状皮膚炎、角層下膿疱症、スネッドン-ウィルキンソン病、若年性 疱疹状皮膚炎、若年性天疱瘡、疱疹状膿痂疹、多巣性紅斑、紅斑 虹彩、疱疹 虹彩、ライル症候群、エリトマトーデス、他の特定の紅斑性状態、例えば、リッター病、新生児剥脱性皮膚炎、紅斑間擦疹、苔癬、苔癬化およびハイド病、神経皮膚炎(限局性)(局所性)、他の特定の掻痒症および掻痒症。
【0100】
更に、本発明の方法は、血管性疾患を治療するために有用である。当該関連する血管性疾患は、これらに限定するものではないが、子宮内膜症、機能性障害性の子宮性出血、子宮内膜過形成、骨髄線維症、血管腫、動脈硬化症、急性および慢性移植拒絶反応および移植脈管障害、加齢関連 横斑性変性(AMD)を含む眼部 新血管新生を特徴とする眼部障害、増殖性糖尿病網膜症(PDR)、未熟児網膜症(ROP)、新血管緑内障、網膜芽細胞腫、水晶体後線維増殖症、ルベオーシス、ブドウ膜炎、横斑性変性および角膜移植新血管新生を含む。
【0101】
加齢関連横斑性変性(AMD)は、高齢者において共通する失明の原因である。最も重篤な形態は、網膜の患部領域における広範な新血管の形成を含む。この理由のために、抗血管形成性化合物が有効に投与され、血管の内殖を阻害し、視覚を保存している。αvβ3インテグリンアンタゴニストの血管形成阻害能は、本化合物がAMDの治療において価値があり得ることを示唆する。
【0102】
本発明の方法に従い治療され得る不適当な血小板活性化、補充、凝集または血栓症により媒介される状態の中には、冠動脈疾患または障害、心筋梗塞または心筋梗塞に続く障害、脳卒中、不安定狭心症、子癇前症、塞栓症、血小板関連虚血性疾患、例えば、肺虚血、冠血管虚血および脳虚血、血管形成術、アテレクトミー、ステント設置,およびバイパス手術を含む経皮的冠動脈インターベンションに続く再狭窄、血栓性疾患、例えば、冠動脈血栓症、脳動脈血栓症、心臓内血栓症、末梢動脈血栓症、静脈血栓症、血栓症および外来性に対する露出または損傷された組織表面に関連する凝固障害および血栓症に続く再閉塞、深在性静脈血栓症(DVT)、肺塞栓症(PE)、一過性虚血性発作(TIAs)および血管閉塞が共通する基本をなす特徴である他の状態がある。幾つかの態様において、本発明に従う方法は、血栓形成または再形成、進行型の冠動脈疾患または閉塞、再閉塞、血管の狭窄または再狭窄または脳卒中のハイリスクにある個体において使用される。他の態様において、本発明に従う方法は、血管形成方法、例えば、バルーン血管形成術、レーザー血管形成術、冠血管アテレクトミーまたは同様の技術、頚動脈動脈膜切除、血管グラフトの吻合、血栓形成の高リスクを有する外科手術(すなわち、冠血管バイパス手術、補綴バルブまたは血管の挿入など)、アテレクトミー、ステント設置、慢性心血管装置の設置、例えば、インドルエリングカテーテルまたは補綴バルブまたは血管、臓器移植またはバイパス手術と組み合わせて使用される。
【0103】
本発明の方法を使用して治療され得る他の疾患および状態は、心筋の血管新生、出血性関節s、血管癒着、敗血症、成人呼吸困難症候群、末梢血管拡張および創傷肉芽形成を含む。
【0104】
骨再吸収は、本発明の方法が適用され得る他の領域である。骨再吸収、例えば、骨粗鬆症において見られるように、骨マトリックスへの破骨細胞の接着に関連する。この重要な接着は、インテグリンにより媒介される。高レベルのαvβ3インテグリンの発現は、骨再吸収する破骨細胞において注目されている。αvβ3アンタゴニストによる破骨細胞接着の破損は、インビトロおよびインビボの両方における骨再吸収を阻害する。
【0105】
病理学的骨再吸収と主に関連する疾患は、これらに限定するものではないが、骨粗鬆症、骨のパジェット病および副甲状腺ホルモン(PTHrP)媒介高カルシウム血症を含む。
【0106】
αvβ3アンタゴニストは、骨粗鬆症の予防および治療のために評価されている。それらは、注目すべき有害作用を伴わずに骨減少の阻害を示している。この状態における長期治療のために必要なものは、何れの治療も非常に低毒性のプロフィールを有することが示される。非常に低い容量で投与できるNVX-188の高結合親和性およびその明らかに毒性を欠くということは、この化合物、同時に本発明の他の化合物をこの適用において魅力的にしている。
【0107】
感染した生物体における標的細胞との相互作用のための受容体としてαvβ3インテグリンを使用するウイルス感染は、本発明の方法により治療されてよい。例示的なウイルス感染は、これらの限定するものではないが、アデノウイルス(Adenoviridae)、コックサッキーウイルス(Picornaviridae)、エコウイルス(Picornaviridae)、口蹄疫ウイルス(Picornaviridae)、ハンタウイルス(Buriyaviridae)、ヒトパレコウイルス(Picornaviridae)、ヒト免疫不全ウイルス 1(Retroviridae)およびロタウイルス(Reoviridae)を含む。
【0108】
本発明の化合物は、単体で使用されてもよく、または他の薬剤、例えば、上述の治療剤およびイメージング剤と組み合わせて使用されてもよい。当該インテグリンおよび当該細胞は、同時にまたは経時的に当該化合物および混合されたまたは個々の投与量としての薬剤と接触されてもよい。当該化合物は、少なくとも1の上述の薬剤と混合されてもよく、または実施可能に結合されてもよい。化合物、治療剤、イメージング剤またはその組合せは、非経口、皮内、皮下、経口、経皮、経粘膜または直腸に投与されてよい。
【0109】
診断法
本発明の方法は、標的領域をイメージングする、例えば、癌のインテグリン受容体イメージングするために有用である。治療学的放射性医薬品は、治療学的に有用な放射性核種、例えば、β粒子、α粒子およびオージェまたはコスター-クロニッグ電子(Coster-Kronig electrons)などの電離放射線を放射する放射活性金属イオンから構成される医薬である。放射性核種に実施可能に結合される本発明の化合物は、インテグリンを発現している細胞に対して放射線核種をターゲティングすることを促進する。それから、イメージングは、患部領域、例えば、心または肢などの標準的な技術を使用して、実行することが可能であり、それにより疾患の存在を検出したり、または疾患の進行または治療学的処置の結果ををモニターしたりすることが可能である。
【0110】
スクリーニング法
本発明の1つの目的は、インテグリン結合化合物を同定する方法を提供することである。従って、本発明は、ファルマコフォアの提示を具備するコンピュータ読み取り可能媒体を提供し、ここで、当該ファルマコフォアは、1のH結合供与体(HBD)、1のH結合受容体(HBA)、2の疎水性芳香族基(HAR1およびHAR2)および1の陰性イオン性基(NI)の特徴を含む。
【0111】
一つの態様において、当該HBD、HBA、HAR1、HAR2およびNIは、図8Aに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.81±1 Åであり、当該HBDとHBAの距離は7.52±1 Åであり、当該HBDとNIの距離は17.67±1 Åであり、当該HRA1とHBAの距離は7.52±1 Åであり、当該HRA1と HRA2の距離は9.41±1 Åであり、当該HBAとNIの距離は10.20±1 Åであり、および当該HRA2とNIの距離は5.88±1 Åである。特に、当該HBDとHRA1の距離は2.81 Åでよく、当該HBDとHBAの距離は7.52 Åでよく、当該HBDとNIの距離は17.67 Åでよく、当該HRA1とHBA の距離は7.52 Åでよく、当該HRA1とHRA2の距離は9.41 Åでよく、当該HBAとNIの距離は10.20 Åでよく、および当該
HRA2とNI の距離は5.88 Åでよい。
【0112】
もう一つの態様において、当該HBD、HBA、HAR1、HAR2,およびNIは、図8Bに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.80±1 Åであり、当該HBDとHBAの距離は7.34±1 Åであり、当該HBDとNIの距離は16.92±1 Åであり、当該HRA1とHBAの距離は5.20±1 Åであり、当該HRA1とNIの距離は14.34±1 Åであり、当該HBAとHRA2の距離は4.69±1 Åであり、当該HBAとNI の距離は9.85±1 Åであり、および当該HRA2とNIの距離は5.66±1 Åである。特に、当該HBDとHRA1の距離は2.80 Åでよく、当該HBDとHBAの距離は7.34 Åでよく、当該HBDとNIの距離は16.92 Åでよく、当該HRA1とHBAの距離は5.20 Åでよく、当該HRA1とNIの距離は14.34 Åでよく、当該HBAとHRA2の距離は4.69 Åでよく、当該HBAとNIの距離は9.85 Åでよく、および当該HRA2とNIの距離は5.66 Åでよい。
【0113】
更なるもう一つの態様において、当該HBD、HBA、HAR1、HAR2およびNIは、図8Cに従う立体配置であり、ここで、当該HBDとHRA1の距離は 2.79±1 Åであり、当該HBDとHRA2の距離は11.94±1 Åであり、当該HBDとNIの距離は16.28±1 Åであり、当該HRA1とHBAの距離は5.20±1 Åであり、当該HRA1とNI の距離は14.64±1 Åであり、当該HBAとHRA2の距離は4.72±1 Åであり、および当該HRA2とNIの距離は5.63±1 Åである。特に、当該HBDとHRA1の距離は2.79 Åでよく、当該HBDとHRA2の距離は11.94 Åでよく、当該HBDとNIの距離は16.28 Åでよく、当該HRA1とHBAの距離は5.20 Åでよく、当該HRA1とNIの距離は14.64 Åでよく、当該HBAとHRA2の距離は4.72 Åでよく、および当該HRA2とNI の距離は5.63 Åでよい。
【0114】
本明細書に使用される、「コンピュータ読み取り可能媒体」は、コンピュータにより直接的に読まれるおよびアクセスされることが可能な何れかの媒体をいう。そのような媒体は、これらに限定するものではないが、磁気記憶媒体、例えば、フロッピー(登録商標)ディスク、ハードディスク記憶媒体、および磁気テープ;光学記憶媒体、例えば、CD-ROM;電気的記憶媒体、例えば、RAMおよびROM;およびこれらのカテゴリーのハイブリッド、例えば、磁気/光学記憶媒体などを含む。当業者は、当該分野において周知の何れかの方法を用いて、本発明のファルマコフォアの表示をそこにおいて記憶されたコンピュータ読み取り媒体を容易に作ることが可能である。
【0115】
コンピュータ読み取り可能形態において、本発明のファルマコフォアの表示を提供することにより、当業者は、多様な目的のためにファルマコフォアの情報にルーチンとしてアクセスすることが可能である。例えば、当業者は、コンピュータ読み取り可能形態中の本発明のファルマコフォアを、データ保管手段中に格納されている化合物情報と比較するために使用することが可能である。そのような手段は、当該ファルマコフォアの特徴とマッチする化合物を同定し、それにより、インテグリン結合分子の候補を出すために使用される。
【0116】
従って、本発明は、インテグリン結合小分子を同定する方法を提供する。当該方法は、化合物の三次元構造と本発明のファルマコフォアの三次元構造とを比較すること、およびもし当該化合物が当該ファルマコフォアの特徴と一致した場合に、当該化合物を選択することを具備する。
【0117】
続く例は、本発明の範囲を説明することが目的であるが、これを制限するものではない。このような例は、典型的に、使用され得るものの典型的なものでり、当業者にとって公知の他の手順は、変更可能に利用されてよい。実際、当業者は、過度の実験を行うことなしに、本明細書の教示を基に更なる態様を容易に思い描くことおよび作製することが可能である。
【0118】
例I
小分子インテグリンの発見
新規抗癌剤としてのαvβ3アンタゴニスト
実験部門
ファルマコフォア仮定の発生およびバリデーション。トレーニングセット化合物(A-C)の構造が、組み立てられ、カタリスト(Catalyst(Accelrys、Inc.))を使用して徹底的に最小化された40。各化合物の到達可能なコンフォメーションフレキシビリティが探索できた独特のコンフォメーションのセットが、カタリストのキャットコンフォモデル(Catconf module)を使用して生じた。当該カタリストにおいて実行された当該ポーリングアルゴリズム(poling algorithm)が使用され、コンフォメーションを生じた。当該ポーリングアルゴリズムは、高いコンフォメーションの変化を促進し、低いエネルギーのコンフォメーションスペースの広い適用範囲を保証する49−51。当該共通の特徴ファルマコフォア仮説が、カタリストのヒップホップアルゴリズム(HipHop algorithm)を使用して生じた。ヒップホップは、そのデフォルトセッティングを有する10のファルマコフォアを生じる。ヒップホップは、トレーニングセット分枝のコンフォメーションモデルの収集物および化学的特徴の選択を得て、当該トレーニングセット分子の共通する立体配置を同定する。化合物Aは、ファルマコフォア仮説発生例における主要な化合物とみなされる。当該トレーニングセット化合物の構造およびαvβ3活性部位特徴の化学的特徴を基本に、ファルマコフォア的特徴のセットが当該ファルマコフォア発生例の最初に選択される。公知のαvβ3アンタゴニストの検索可能なマルチコンフォーマーデータベースは、カタリストデータベースサーバーを使用して発生される。このデータベースは、当該ファルマコフォアモデルをバリデーションするために使用される。
【0119】
ドッキング試験。ドッキングは、ゴールド(GOLD)プログラム(Genetic Optimization for Ligand Docking)のバージョン1.2を使用して行った42。ゴールドは、遺伝子アルゴリズムを使用する自動化ドッキングプログラムであり、活性部位の部分的なフレキシビリティを有するリガンドコンフォメーションフレキシビリティを探索する52。当該アルゴリズムは、ブルックタンパク質データベース(the Brookhaven Protein DataBank)から抽出された300以上の複合体のデータセットにおいて試験される。ゴールドは、当該リガンドの実験的結合コンフォメーションを再現することにおいて70%よりも多くのケースにおいて成功した53。ゴールドは、ユーザーの定義した結合部位を必要とする。それは、定義された領域内の空洞を探索し、定義された領域において全ての溶媒到達可能な原子を活性部位原子とみなす。適切なプロトン化状態は、酸性および塩基性アミノ酸残基のために割り当てる。当該受容体において存在する全ての水分子は除去され、水素原子がインテグリンαvβ3受容体に対して与えられた。各分子の全てのコンフォマーは、αvβ3受容体活性部位にドッキングした。各実行の終点で、ゴールドは、適応度スコアおよび二乗平均平方根距離(RMSD)を基に全ての生じた結合コンフォメーションを分離し、ランク付けする。全てのドッキングの実行は、集合サイズ100、最大数100,000操作、変異およびクロスオーバー率95を伴う標準デフォルトセッティングを使用して実行された。ゴールドにおいて実行された当該適合度機能は、H-結合、複合体エネルギーおよびリガンド内部エネルギー期間(terms)をからなる。当該ドッキング試験は、24-CPU シリコン・グラフィックス・オニックス・ワーク(24-CPU Silicon Graphics Onyx workstation)において実行した。
【0120】
細胞培養。ヒト乳癌細胞(MCF-7、αvβ3-、過剰発現野生型p53、ER+; MDA-MB 468、p53 変異体、ER+;およびMDA-MB-435、αvβ3+、p53 変異体、ER-)および非小細胞肺癌細胞H1975をザ・アメリカン・タイプ・セル・カルチャー(the American Type Cell Culture(Rockville、MD Q4))から入手した。天然にシスプラチン(CDDP)に対して耐性である当該HEYヒト卵巣癌腫細胞系は、ドクター・デュビュー(Dr. Dubeau(University of Southern California Norris Cancer Center))から快く提供された。細胞は、単層培養物として、10%のウシ胎仔血清(Gemini-Bioproducts、WoodlおよびCA)および2mmol/L L-グルタミンを添加したRPMI 1640中で、37℃で、5%CO2の加湿した雰囲気において維持した。継代および計数のために当該フラスコから接着細胞を剥がすために、細胞をカルシウムまたはマグネシウム非含有のPBSで洗浄し、少量の0.25%のトリプシン-EDTA溶液(Sigma、St. Louis、MO)で5分〜10分間インキュベートし、培地で洗浄し、遠心した。全ての実験は、指数関数的な細胞増殖における細胞を使用して行った。
【0121】
薬物。全ての化合物の10mMストック溶液を、DMSO中で調製し、-20で保存した。更なる希釈は、PBS中で新鮮に作製した。
【0122】
受容体結合アッセイ。NCI-H1975細胞の表面における全ての化合物の結合親和性は、変更を加えた文献に記載の準じて、125I-標識エキスタチン(echistatin)を放射性リガンドとして使用し競合的結合実験において決定した41。即ち、NCI-H1975 細胞を収集し、2回PBSで洗浄し、結合緩衝液(20 mMトリス、pH 7.4、150 mM NaCl、2 mM CaCl2、1 mM MgCl2、1 mM MnCl2、0.1% BSA)中に再懸濁(2 x 106 cells/mL)した。96-ウェルマルチスクリーンDVプレート(フィルターポアサイズ:0.65μm、ミリポア、Billerica, MA)を、αvβ3アンタゴニストの大濃度の存在下で125I-エキスタチンインキュベートした。総インキュベート量は、200μLに調整した。当該細胞を3時間室温でインキュベートした後に、当該プレートをマルチスクリーン減圧マニフォールドを介して濾過し、2回冷結合緩衝液で洗浄した。親水性のPVDFフィルターを回収し、その放射能をNaI(TI)ガンマカウンター(Packard、Meriden、CT)で測定した。最も適合したIC50値をグラフィックパッド・プリズム(GraphPad Prism(GraphPad Software、Inc.、San Diego、CA))を使用した非線形回帰により当該データを適合させることにより計算した。実験は、トリプリケイトサンプルで実施した。
【0123】
細胞毒性試験。細胞毒性は、3-(4,5-ジメチルチアゾール-2-イル)-2,5-ジフェニルテトラゾリウムブロミド(MTT)アッセイで以前に記載した通りに評価した41、54。即ち、細胞を96ウェルマイクロタイタープレートに播種し、接着させる。細胞を次に、連続して72時間に亘り対応する薬物に暴露した。MTT 溶液(最終濃度0.5 mg/mL)を各ウェルに添加し、細胞を4時間37℃でインキュベートした。培地の除去後、DMSOを添加し、570nmで吸光度を測定した。全てのアッセイは、トリプリケイトで行った。次に、そのIC50 は各薬物について対数(薬物濃度)対細胞死滅パーセンテージとして測定した。
【0124】
結果および考察
インテグリンαvβ3アンタゴニストの設計。RGDペプチドを使用した以前の試験は、αvβ3は、RGDトリペプチド配列の重要なグアニジン(Arg)およびカルボン酸(Asp)基(ArgとAsp残基のCβ原子の間の距離)の間の短い全体的な分離を認識することを示唆した35、36。バックボーンコンフォメーションにおいて捩れを伴う環状RGDペプチドは、他のインテグリンよりもαvβ3の対してより選択性を示した24。このサイクリックRGDペプチド-αvβ3相互作用モデルは、多様な小分子ペプチドミメティックアンタゴニストの発見のための開始点を提供する。αvβ3の細胞外セグメントの結晶構造およびそのサイクリックRGDペプチドとの複合体は、以前に報告されている37、38。我々は、共通の特徴ファルマコフォアモデルを産生するための最近報告された小分子αvβ3アンタゴニストのセットを利用し、それらは次に、638の公知のαvβ3アンタゴニストのデータベースに対してバリデートした39。当該バリデーションしたファルマコフォアモデルを、新規構造骨格および所望の化学的特徴を有する分子を検索するために探索照会(search queries)として使用した。新規αvβ3アンタゴニストを同定し、設計するための我々の戦略は、図1に模式的に示す。
【0125】
共通の特徴ファルマコフォアモデルの発生。一般に、共通の特徴ファルマコフォアモデルの発生のためのトレーニングセットは、同様な活性プロフィールおよび活性部位結合メカニズムを有する化合物を含むべきであり、それによりこれらのトレーニング化合物がそれらの生物学的活性を招く比較できる特徴の3D配置を有する可能性を高める。カタリスト・ソフトウェア・パッケージにおける当該ヒップホップアルゴリズムは、共通する特徴ファルマコフォアモデルを引き出すために、30から49nM(図2)の範囲におけるαvβ3インテグリンに匹敵する結合親和性を有する最近報告された3つのαvβ3インテグリン(A、BおよびC)のアンタゴニストからなるトレーニングセットに対して適用される39、40。当該トレーニングセット化合物は、類似体をより集め、同様の結合コンフォメーションにおけるαvβ3受容体の活性部位における同様の部位に対する結合を予測する。当該ファルマコフォアの特徴は、(1)トレーニングセットアンタゴニストの構造および化学的特徴、(2)αvβ3受容体活性部位の構造、および(3)サイクリックRGDペプチド(PDB1L5G)と複合したαvβ3受容体の共結晶構造におけるサイクリックRGDペプチドと突出したαvβ3受容体活性残基との間で観察される重要な相互作用を基礎に選択される37。ファルマコフォアモデル発生実験において考慮される特徴は、H結合供与体(HBD)、H結合受容体(HBA)、環芳香族(HYR)、疎水性(HYA)および陰性イオン性基(negatively ionizable(NI))特徴である。ピップホップは、10 5の特徴とする(10 five-featured)ファルマコフォア仮定を生じた。これらの仮定がそれらのファルマコフォアの特徴において同様である一方で、種々の特徴の相対的な配向、位置およびベクター方向は異なる。カタリストプログラムにおいて入手可能な階層的完全連関法(hierarchical complete linkage method)を使用した10仮定のクラスター解析は、3つのクラスターを産生した。これらのクラスターのそれぞれからの代表的なモデル(ファルマコフォア仮定 Hypo1、Hypo5およびHypo9)を更なる解析およびバリデーションのために選択した。
【0126】
共通する特徴ファルマコフォアのバリデーション。内部のバリデーションとして、トレーニングセット化合物A-Cを、3つのファルマコフォア上にマッピングした。トレーニングセット化合物(表1)のマッピングされたコンフォメーションの適合度スコアおよび関連するエネルギーは、化合物AおよびBのためにはHypo1またはHypo9よりもHypo5の方が低いエネルギーを産生したが、化合物Cのためには相対的に高いエネルギーを産生した。図3に示すように、化合物AへのHypo5のマッピングは、この化合物の重要な化学的特徴と当該ファルマコフォアとの間の良好な一致を確認した。
【表1】
【0127】
不活性化合物からの強力なアンタゴニストの分離における、これらのファルマコフォアの判別可能な能力を評価するために、当該3つの代表的なファルマコフォアを3D照会として使用し、公知のαvβ3受容体アンタゴニストのデータベースを検索した。このデータベースは、αvβ3受容体に対する広い範囲の活性プロフィールを有する計638化合物を有する。当該638化合物のうち、αvβ3重要対阻害をIC50値≦20nMで示す303が、強力なαvβ3アンタゴニストとみなされた。検索結果を表2に要約する。147化合物を検索した他の2つのファルマコフォアよりも良好に実行されたHypo5の88(〜60%)化合物がαvβ3の強力なアンタゴニストであった。Hypo9はより活性な化合物を検索したが、強力とみなされた化合物の存在は、Hypo5よりも低かった(〜54%)。強力なアンタゴニストを選択する優れた能力を有するHypo5および3つのトレーニングセット化合物の2つに対するHypo5のマッチを基に、このファルマコフォアが次の工程のために選択された:新規構造骨格および所望の特徴を有する化合物の検索のための我々の構内のデータベースのサブセットを探索するための3D照会。
【表2】
【0128】
データベース検索および化合物選択。238,819化合物のNCI12000データベースのHypo5を使用する探索は、684ヒット得た。Hypo5により検索された684化合物のうち、分子量≦500を有する282化合物が、更なる物理化学的性質解析のために考えられ、残りは処分された。359,224化合物のケミカル・ダイバーシティ(ChemDiv、Inc、San Diego、CA)データベースの検索は148ヒットを生じた。物理化学的性質フィルターを使用し、インビトロスクリーニングの前に薬物様性質を有していない化合物を排除した41。ファルマコフォア適合値、計算物理化学的性質およびサンプル有効性を基に、8 化合物をNCI化合物レポジトリーから得て、21化合物のコレクションを、インビトロ評価のためにケミカル・ダイバーシティ(Chemical Diversity)から購入した。
【0129】
αvβ3 結合。 非小細胞肺癌 NCI-H1975 細胞の表面における全ての化合物の受容体結合親和性は、125I-標識エキスタチンを放射性リガンドとして使用する競合結合実験で測定し、結果を表3に示す。試験された29の化合物のうち、4つの化合物は、顕著なαvβ3結合親和性(AV3 = 52 nM、AV26 = 240 nM、AV27 = 18 nM、AV32 = 605 nM)を示した。2つの化合物、AV3およびAV27 は、トレーニング化合物A-Dと同様の範囲の結合親和性を示した(30 − 49 nM)39。バリデーション解析において得られた通り、Hypo5 は、ナノモーラーの結合親和性を有する構造的に多様な強力なアンタゴニストの検索において成功した。これらの化合物は、多様な構造骨格を有するアンタゴニストの新規セットを意味する。当該トレーニングセット化合物とは異なって、当該化合物は、親水性質の幾つかの官能基を有する。これは、αvβ3のRGDペプチド結合領域の高い静電気的性質を考慮する重要な特徴である。当該RGDペプチドは、αvβ3サイクリックRGDペプチド複合体結晶構造におけるαvβ3を有するその2つの負荷末端を介する強い静電気的相互作用を形成する。化合物AV3、AV27およびAV32は、RGDミメティックアンタゴニストの範疇に大まかに含まれ、これはそれらがαvβ3と同様の静電気的相互作用を確立することができる何れかの末端にカルボキシル基またはアミン/アミド基を有するからである。加えて、これらの化合物における幾つかの親水性官能基の存在は、αvβ3のRGD結合部位の高い静電気的領域との相互作用に助力する。これらの興味深い特徴が得られたので、我々は、AV27およびAV32の構造学的に近い更なる類似体を試験した。しかしながら、我々の構内データベースにおけるこれらの化合物のコア骨格を使用する下部構造検索照会では、近い類似体は得られない。これらの2つの化合物のコア骨格周辺の構造−活性研究は、αvβ3に向かうそれらの結合親和性を更に最適化するための代替オプションであってよい。独特な構造的特徴を有しているために、AV26は、αvβ3アンタゴニストの新規種類の非RGDミメティックを代表する。我々は、我々の結合アッセイにおいて化合物AV34からAV38を試験することにより、化合物AV26における制限された構造活性相関(SAR)分析を実施した(表3)。検索照会としてAC26のコア骨格を使用する下部構造探索は、359,224化合物の我々の構内データベースから幾つかの化合物を検索した。当該類似体AV34からAV38は、ケミカル・ダイバーシティ(Chemical Diversity(San Diego、CA))から購入し、αvβ3結合親和性をスクリーニングした。このSAR試験から、我々は2つの新規化合物を発見した:AV35 はIC50 値が24 nMであり、親化合物AV26よりも10倍高い親和性を示した。AV38 はIC50 値が0.03 nMであり、最も強力であり、親AV26よりも800倍高い親和性を示した。化合物 AV26、AV35およびAV38は、通常のRGDミミックαvβ3アンタゴニストから構造学的に逸脱しており、それらが新規な機序の相互作用を有するαvβ3の非RGDミミックアンタゴニストの新規種類を代表するものであることが示唆される。我々の知識に対して、これらの化合物は、これまでに記載された最も強力なαvβ3アンタゴニストの中に含まれる。併せて、我々の6つの化合物は、小分子αvβ3アンタゴニストを代表するものであり、研究は、抗癌剤としてのそれらのインビボでの有効性を示すために進行中である。また、研究は、共有結合を介してαvβ3の過剰発現癌に対する選択的送達細胞障害剤、例えば、パクリタキセルなどに対しても進行中である。
【表3】
【表S1】
癌細胞系のパネルにおける選択された化合物の細胞毒性。最初に、我々は、全ての化合物を、高および低αvβ3発現する2つの乳癌細胞系MDA-MB-435およびMCF7、並びに天然でシスプラチンに対する抵抗性のHEY卵巣癌細胞系において試験した。20μM(表4)での細胞増殖の顕著な阻害が見られた化合物は、続いて、5つの細胞系のパネルにおいて試験した(表5)。我々は、これらの細胞に対する当該化合物の幾つかについて、顕著な特異性を観察した。例えば、AV26は、MCF7、NIH3T3,およびCRL5908 細胞に比較して、MDA-MB-435細胞に対して60倍の選択性を示し、HEY細胞よりも40倍近くの選択性を示した。同様の経口が、しかし程度は小さいが、化合物AV34で観察された。一方、AV30は、MDA-MB-435、HEYおよびNIH3T3細胞においてはまさに同様のプロフィールが示されたが、MCF-7およびNCI-H1975細胞に対しては有意に活性が低かった。興味深いことに、AV26を除く当該新規アンタゴニストの何れか1項に記載のもが、注目すべき細胞毒性を示さず、これは、これらの新規のアンタゴニストは、治療非細胞毒性メカニズムを基礎とする抗癌治療として有用性を有していることを示す。αvβ3が癌細胞の遊走、接着およびアポトーシスを媒介するという事実を考慮すると、これらの新規の高親和性アンタゴニストと通常の細胞障害性薬物との組合せが、更なる毒性のない改良された治療学的な利点を示すものである。
【表4】
【表5】
【0130】
ドッキング試験。 我々の新規アゴニストの配向に結合することを確認するために、我々は、化合物A、AV3、AV26、AV27およびAV38を当該αvβ3 RGD 結合領域にゴールドを使用してドッキングさせた42。当該αvβ3RGD結合領域内側での化合物A、AV3、AV26、AV27およびAV38の当該予測される結合コンフォメーションは、図5a−eに示す。コールドは、各化合物について、幾つかの可能な結合コンフォメーションを生じ、それらの適応度スコアに従って、それらをランク付けした。最も有利なエネルギーを有する結合コンフォメーションが最高の結合配向であると考えられた。サイクリックRGDペプチドと複合したαvβ3受容体の結晶構造において、サイクリックRGDペプチドの1つのAspカルボキシル化酵素がαvβ3受容体のβ鎖におけるMIDAS(Metal Ion-Dependent Adhesion Site)でMn2+と相互作用する一方で、そのArg グアニジニウム基は、αvβ3受容体(PDB1L5G)β鎖のD218およびα鎖のD150と相互作用する。化合物 A、AV3、AV26およびAV27は、MIDASのMn2+を有するそれらのカルボキシ末端で同様に結合する相互作用を確立し、同様に、アミノ酸残基S121およびS123の側鎖のヒドロキシル基との幾つかのH-結合相互作用の形成を確立する。当該サイクリックRGDペプチドのArg側鎖の結合配向に対比して、化合物 A、AV3、AV27の当該アミン/アミド関連末端およびAV26のヒドロキシル関連フェニル基は、β鎖のD218から離れた空洞を占領するが、α鎖のD150には近い。この空洞は、αvβ3のβ鎖からのアミノ酸残基Y166、P170、D179、R214、N215およびR216およびα鎖のアミノ酸残基 K119、E121、D148、D150、Y178,およびR248により囲まれている。著しく、最も強力な非RGDミメティックアンタゴニストAV38は、逆向きの結合配向を採用し、αvβ3のサイクリックRGDペプチド結合領域において種々のアミノ酸残基と強い静電気的相互作用のセットを確立した。化合物 A、AV26,およびAV38と当該αvβ3RGDペプチド結合部位での種々のアミノ酸残基との間の観察された重要な相互作用の略図の提示は図6に示す。AV38の当該ジメトキシ関連フェニル基は、MIDASのMn2+および2つの、Mn2+に対応するメトキシ酸素原子の近くの領域を占める。3つのH結合相互作用が当該メトキシ酸素原子とアミノ酸残基S121およびS123のヒドロキシル基の間に観察された。当該カルボキシル化関連フェニル基は、αvβ3のβ鎖からのアミノ酸残基 Y116、P170、D179、R214およびR216およびα鎖からのD148、A149、D150およびY178により囲まれる領域を占めた。当該カルボキシル化酸素原子は、R216グアニジニウム基、A149の主鎖NHおよびY166のヒドロキシル基と、幾つかの強いH結合相互作用を形成した。最も強力なアンタゴニストAV38を除いて、一貫したパターンが、全てのアンタゴニストの予測された結合配向において観察された。同様の化学的性質を有するこれらのアンタゴニストの当該官能性基は、αvβ3RGDペプチド結合領域における同様の領域を占め、Mn2+と他のアミノ酸残基との同様な相互作用を形成した。これは、我々のドッキング試験により予測されたこれらのアンタゴニストの結合コンフォメーションの質を支持するものである。幾つかの矛盾が、以前に予測されたRGDミメティックペプチドミメティックおよび非ペプチド小分子アンタゴニストの結合配向において見出されている43−47。非ペプチド小分子アンタゴニストを使用しない以前に報告されたドッキング試験もまた、この試験において予測された配向に対して非常に類似する結合配向を予測した43,45。しかしながら、ドッキング試験は、αvβ3−RGD複合体結晶構造におけるサイクリックRGDの結合コンフォメーションに対して同様な結合配向を生成したサイクリックRGD類似体およびRGDミメティックにおいて行った46,47。我々の新規アンタゴニストの予測された結合配向とαvβ3ーRGD複合体結晶構造におけるサイクリックRGDペプチドの結合コンフォメーションとの間で観察された矛盾は、当該サイクリックRGDペプチドとこれらの新規アンタゴニストの間の有意な構造上の相違のためである。他方、観察された矛盾は、我々のドッキング手順におけるアーチファクトの結果かもしれない。しかしながら、我々の最も強力な非RGDミメティックアンタゴニストAV38により示された高結合親和性(親AV26よりも800倍高い)は、その予測される結合配向において見出される新規な結合相互作用により説明される。この予測される結合配向において、AV38は、幾つかの強いH結合相互作用を形成し、明らかな相補性が、その官能基の化学的性質とαvβ3におけるその結合部位の周辺の種々のアミノ酸残基の間で見いだされた。更なる構造研究は、この非RGDミメティックアンタゴニストおよびそのαvβ3との相互作用により及ぼされる高結合親和性の説明を保証するものである。
【0131】
最後に、ノックアウトマウスでの遺伝的結果と、全ての報告されているアンタゴニストによる薬理学的効果との間の分野における最近の矛盾を説明するために、我々は、リチャード・へネス(Richard Hynes)によるモデル(彼は、β3インテグリンを遺伝的にノックアウトしたマウスが増強された血管形成を示した理由を考察している)に言及する(図7)48。彼は、RGDペプチドは、実際に「アンタゴニスト」の代わりに「アゴニスト」と称されるべきであると主張している。仮に、当該文言アゴニストが使用された場合、当該遺伝子学的モデルは、薬理学的モデルに一致する。この明細書を通して、我々は、最も以前の報告において使用していたように文言「アンタゴニスト」を使用しているが、我々は、これらの化合物は実際にはアゴニストとして働く可能性に気付いている。どんなメカニズムであっても、我々の化合物は高度に強力であり、αvβ3陽性細胞に対して選択的に送達するための種々の化学療法剤に対して複合化するために非常に適切である。
【0132】
我々は、構造的に多様なインテグリンαvβ3アンタゴニストのシリーズを、小分子薬物様化合物のデータベースのファルマコフォアスクリーニングを介して同定してきた。共通の特徴3Dファルマコフォアモデルは、公知のインテグリン αvβ3アンタゴニストのセットを利用して発生させた。バリデーションされたファルマコフォアモデルは、当該ファルマコフォアモデルを生じるために使用したトレーニングセット化合物よりも高い効力を有する構造学的に新規な化合物をうまく検索した。更に、1つの強力なアンタゴニストにおける制限された構造活性相関解析は、非RGDミメティックαvβ3アンタゴニストとしてサブナノモーラーの効力を有する高い効力の化合物を発見した。受け入れられる構造骨格を有するこれらの小分子アンタゴニストは、強力な非RGDミメティックαvβ3アンタゴニストとして更に最適化するために役立つ手がかりを提供する。我々は、我々の最近の原稿に記載したとおり、当該3つの強力なアンタゴニストとパクリタキセルを複合化している41。これらの新規の薬剤のαvβ3陽性癌細胞への標的送達についての詳細な薬理学的性質を示す(Figure 7)。
【0133】
参考文献
1. Danen, E. H., Integrins: regulators of tissue function and cancer progression. Curr Pharm Des 2005, 11, (7), 881-91.
2. Humphries, M. J., Integrin structure. Biochem Soc Trans 2000, 28, (4), 311-39.
3. Martin, K. H.; Slack, J. K.; Boerner, S. A.; Martin, C. C.; Parsons, J. T., Integrin connections map: to infinity and beyond. Science 2002, 296, (5573), 1652-3.
4. Aplin, A. E.; Howe, A. K.; Juliano, R. L., Cell adhesion molecules, signal transduction and cell growth. Curr Opin Cell Biol 1999, 11, (6), 737-44.
5. Brooks, P. C.; Clark, R. A.; Cheresh, D. A., Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science 1994, 264, (5158), 569-71.
6. Hood, J. D.; Cheresh, D. A., Role of integrins in cell invasion and migration. Nat Rev Cancer 2002, 2, (2), 91-100.
7. Felding-Habermann, B., Integrin adhesion receptors in tumor metastasis. Clin Exp Metastasis 2003, 20, (3), 203-13.
8. Natali, P. G.; Hamby, C. V.; Felding-Habermann, B.; Liang, B.; Nicotra, M. R.; Di Filippo, F.; Giannarelli, D.; Temponi, M.; Ferrone, S., Clinical significance of alpha(v)beta3 integrin and intercellular adhesion molecule-1 expression in cutaneous malignant melanoma lesions. Cancer Res 1997, 57, (8), 1554-60.
9. Felding-Habermann, B.; O'Toole, T. E.; Smith, J. W.; Fransvea, E.; Ruggeri, Z. M.; Ginsberg, M. H.; Hughes, P. E.; Pampori, N.; Shattil, S. J.; Saven, A.; Mueller, B. M., Integrin activation controls metastasis in human breast cancer. Proc Natl Acad Sci U S A 2001, 98, (4), 1853-8.
10. Pecheur, I.; Peyruchaud, O.; Serre, C. M.; Guglielmi, J.; Voland, C.; Bourre, F.; Margue, C.; Cohen-Solal, M.; Buffet, A.; Kieffer, N.; Clezardin, P., Integrin alpha(v)beta3 expression confers on tumor cells a greater propensity to metastasize to bone. Faseb J 2002, 16, (10), 1266-8.
11. Kumar, C. C.; Armstrong, L.; Yin, Z.; Malkowski, M.; Maxwell, E.; Ling, H.; Yaremko, B.; Liu, M.; Varner, J.; Smith, E. M.; Neustadt, B.; Nechuta, T., Targeting integrins alpha v beta 3 and alpha v beta 5 for blocking tumor-induced angiogenesis. Adv Exp Med Biol 2000, 476, 169-80.
12. Shannon, K. E.; Keene, J. L.; Settle, S. L.; Duffin, T. D.; Nickols, M. A.; Westlin, M.; Schroeter, S.; Ruminski, P. G.; Griggs, D. W., Anti-metastatic properties of RGD-peptidomimetic agents S137 and S247. Clin Exp Metastasis 2004, 21, (2), 129-38.
13. Ruoslahti, E.; Pierschbacher, M. D., Arg-Gly-Asp: a versatile cell recognition signal. Cell 1986, 44, (4), 517-8.
14. Hynes, R. O., Integrins: versatility, modulation, and signaling in cell adhesion. Cell 1992, 69, (1), 11-25.
15. Kerr, J. S.; Wexler, R. S.; Mousa, S. A.; Robinson, C. S.; Wexler, E. J.; Mohamed, S.; Voss, M. E.; Devenny, J. J.; Czerniak, P. M.; Gudzelak, A., Jr.; Slee, A. M., Novel small molecule alpha v integrin antagonists: comparative anti-cancer efficacy with known angiogenesis inhibitors. Anticancer Res 1999, 19, (2A), 959-68.
16. Lark, M. W.; Stroup, G. B.; Hwang, S. M.; James, I. E.; Rieman, D. J.; Drake, F. H.; Bradbeer, J. N.; Mathur, A.; Erhard, K. F.; Newlander, K. A.; Ross, S. T.; Salyers, K. L.; Smith, B. R.; Miller, W. H.; Huffman, W. F.; Gowen, M., Design and characterization of orally active Arg-Gly-Asp peptidomimetic vitronectin receptor antagonist SB 265123 for prevention of bone loss in osteoporosis. J Pharmacol Exp Ther 1999, 291, (2), 612-7.
17. Reinmuth, N.; Liu, W.; Ahmad, S. A.; Fan, F.; Stoeltzing, O.; Parikh, A. A.; Bucana, C. D.; Gallick, G. E.; Nickols, M. A.; Westlin, W. F.; Ellis, L. M., Alphavbeta3 integrin antagonist S247 decreases colon cancer metastasis and angiogenesis and improves survival in mice. Cancer Res 2003, 63, (9), 2079-87.
18. Harms, J. F.; Welch, D. R.; Samant, R. S.; Shevde, L. A.; Miele, M. E.; Babu, G. R.; Goldberg, S. F.; Gilman, V. R.; Sosnowski, D. M.; Campo, D. A.; Gay, C. V.; Budgeon, L. R.; Mercer, R.; Jewell, J.; Mastro, A. M.; Donahue, H. J.; Erin, N.; Debies, M. T.; Meehan, W. J.; Jones, A. L.; Mbalaviele, G.; Nickols, A.; Christensen, N. D.; Melly, R.; Beck, L. N.; Kent, J.; Rader, R. K.; Kotyk, J. J.; Pagel, M. D.; Westlin, W. F.; Griggs, D. W., A small molecule antagonist of the alpha(v)beta3 integrin suppresses MDA-MB-435 skeletal metastasis. Clin Exp Metastasis 2004, 21, (2), 119-28.
19. Brooks, P. C.; Stromblad, S.; Klemke, R.; Visscher, D.; Sarkar, F. H.; Cheresh, D. A., Antiintegrin alpha v beta 3 blocks human breast cancer growth and angiogenesis in human skin. J Clin Invest 1995, 96, (4), 1815-22.
20. Brooks, P. C.; Montgomery, A. M.; Rosenfeld, M.; Reisfeld, R. A.; Hu, T.; Klier, G.; Cheresh, D. A., Integrin alpha v beta 3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell 1994, 79, (7), 1157-64.
21. Taga, T.; Suzuki, A.; Gonzalez-Gomez, I.; Gilles, F. H.; Stins, M.; Shimada, H.; Barsky, L.; Weinberg, K. I.; Laug, W. E., alpha v-Integrin antagonist EMD 121974 induces apoptosis in brain tumor cells growing on vitronectin and tenascin. Int J Cancer 2002, 98, (5), 690-7.
22. Mitjans, F.; Meyer, T.; Fittschen, C.; Goodman, S.; Jonczyk, A.; Marshall, J. F.; Reyes, G.; Piulats, J., In vivo therapy of malignant melanoma by means of antagonists of alphav integrins. Int J Cancer 2000, 87, (5), 716-23.
23. Allman, R.; Cowburn, P.; Mason, M., In vitro and in vivo effects of a cyclic peptide with affinity for the alpha(nu)beta3 integrin in human melanoma cells. Eur J Cancer 2000, 36, (3), 410-22.
24. Dechantsreiter, M. A.; Planker, E.; Matha, B.; Lohof, E.; Holzemann, G.; Jonczyk, A.; Goodman, S. L.; Kessler, H., N-Methylated cyclic RGD peptides as highly active and selective alpha(V)beta(3) integrin antagonists. J Med Chem 1999, 42, (16), 3033-40.
25. Burke, P. A.; DeNardo, S. J.; Miers, L. A.; Lamborn, K. R.; Matzku, S.; DeNardo, G. L., Cilengitide targeting of alpha(v)beta(3) integrin receptor synergizes with radioimmunotherapy to increase efficacy and apoptosis in breast cancer xenografts. Cancer Res 2002, 62, (15), 4263-72.
26. Rodan, S. B.; Rodan, G. A., Integrin function in osteoclasts. J Endocrinol 1997, 154 Suppl, S47-56.
27. Nakamura, I.; Pilkington, M. F.; Lakkakorpi, P. T.; Lipfert, L.; Sims, S. M.; Dixon, S. J.; Rodan, G. A.; Duong, L. T., Role of alpha(v)beta(3) integrin in osteoclast migration and formation of the sealing zone. J Cell Sci 1999, 112 ( Pt 22), 3985-93.
28. Duong, L. T.; Rodan, G. A., The role of integrins in osteoclast function. J Bone Miner Metab 1999, 17, (1), 1-6.
29. Teitelbaum, S. L., Bone resorption by osteoclasts. Science 2000, 289, (5484), 1504-8.
30. Fisher, J. E.; Caulfield, M. P.; Sato, M.; Quartuccio, H. A.; Gould, R. J.; Garsky, V. M.; Rodan, G. A.; Rosenblatt, M., Inhibition of osteoclastic bone resorption in vivo by echistatin, an "arginyl-glycyl-aspartyl" (RGD)-containing protein. Endocrinology 1993, 132, (3), 1411-3.
31. Engleman, V. W.; Nickols, G. A.; Ross, F. P.; Horton, M. A.; Griggs, D. W.; Settle, S. L.; Ruminski, P. G.; Teitelbaum, S. L., A peptidomimetic antagonist of the alpha(v)beta3 integrin inhibits bone resorption in vitro and prevents osteoporosis in vivo. J Clin Invest 1997, 99, (9), 2284-92.
32. Yamamoto, M.; Fisher, J. E.; Gentile, M.; Seedor, J. G.; Leu, C. T.; Rodan, S. B.; Rodan, G. A., The integrin ligand echistatin prevents bone loss in ovariectomized mice and rats. Endocrinology 1998, 139, (3), 1411-9.
33. Hutchinson, J. H.; Halczenko, W.; Brashear, K. M.; Breslin, M. J.; Coleman, P. J.; Duong le, T.; Fernandez-Metzler, C.; Gentile, M. A.; Fisher, J. E.; Hartman, G. D.; Huff, J. R.; Kimmel, D. B.; Leu, C. T.; Meissner, R. S.; Merkle, K.; Nagy, R.; Pennypacker, B.; Perkins, J. J.; Prueksaritanont, T.; Rodan, G. A.; Varga, S. L.; Wesolowski, G. A.; Zartman, A. E.; Rodan, S. B.; Duggan, M. E., Nonpeptide alphavbeta3 antagonists. 8. In vitro and in vivo evaluation of a potent alphavbeta3 antagonist for the prevention and treatment of osteoporosis. J Med Chem 2003, 46, (22), 4790-8.
34. Murphy, M. G.; Cerchio, K.; Stoch, S. A.; Gottesdiener, K.; Wu, M.; Recker, R., Effect of L-000845704, an alphaVbeta3 integrin antagonist, on markers of bone turnover and bone mineral density in postmenopausal osteoporotic women. J Clin Endocrinol Metab 2005, 90, (4), 2022-8.
35. Pfaff, M.; Tangemann, K.; Muller, B.; Gurrath, M.; Muller, G.; Kessler, H.; Timpl, R.; Engel, J., Selective recognition of cyclic RGD peptides of NMR defined conformation by alpha IIb beta 3, alpha V beta 3, and alpha 5 beta 1 integrins. J Biol Chem 1994, 269, (32), 20233-8.
36. Burgess, K.; Lim, D.; Mousa, S. A., Synthesis and solution conformation of cyclo[RGDRGD]: a cyclic peptide with selectivity for the alpha V beta 3 receptor. J Med Chem 1996, 39, (22), 4520-6.
37. Xiong, J. P.; Stehle, T.; Zhang, R.; Joachimiak, A.; Frech, M.; Goodman, S. L.; Arnaout, M. A., Crystal structure of the extracellular segment of integrin alpha Vbeta3 in complex with an Arg-Gly-Asp ligand. Science 2002, 296, (5565), 151-5.
38. Xiong, J. P.; Stehle, T.; Diefenbach, B.; Zhang, R.; Dunker, R.; Scott, D. L.; Joachimiak, A.; Goodman, S. L.; Arnaout, M. A., Crystal structure of the extracellular segment of integrin alpha V beta3. Science 2001, 294, (5541), 339-45.
39. Marugan, J. J.; Manthey, C.; Anaclerio, B.; Lafrance, L.; Lu, T.; Markotan, T.; Leonard, K. A.; Crysler, C.; Eisennagel, S.; Dasgupta, M.; Tomczuk, B., Design, synthesis, and biological evaluation of novel potent and selective alphavbeta3/alphavbeta5 integrin dual inhibitors with improved bioavailability. Selection of the molecular core. J Med Chem 2005, 48, (4), 926-34.
40. Catalyst 4.9, Accelrys Inc: San Diego, USA, 2004.
41. Chen, X.; Plasencia, C.; Hou, Y.; Neamati, N., Synthesis and biological evaluation of dimeric RGD peptide-paclitaxel conjugate as a model for integrin-targeted drug delivery. J Med Chem 2005, 48, (4), 1098-106.
42. GOLD 1.2, CCDC: Cambridge, UK., 2002.
43. Gottschalk, K. E.; Gunther, R.; Kessler, H., A three-state mechanism of integrin activation and signal transduction for integrin alpha(v)beta(3). Chembiochem 2002, 3, (5), 470-3.
44. Gottschalk, K. E.; Kessler, H., The structures of integrins and integrin-ligand complexes: implications for drug design and signal transduction. Angew Chem Int Ed Engl 2002, 41, (20), 3767-74.
45. Feuston, B. P.; Culberson, J. C.; Duggan, M. E.; Hartman, G. D.; Leu, C. T.; Rodan, S. B., Binding model for nonpeptide antagonists of alpha(v)beta(3) integrin. J Med Chem 2002, 45, (26), 5640-8.
46. Marinelli, L.; Lavecchia, A.; Gottschalk, K. E.; Novellino, E.; Kessler, H., Docking studies on alphavbeta3 integrin ligands: pharmacophore refinement and implications for drug design. J Med Chem 2003, 46, (21), 4393-404.
47. Moitessier, N.; Henry, C.; Maigret, B.; Chapleur, Y., Combining pharmacophore search, automated docking, and molecular dynamics simulations as a novel strategy for flexible docking. Proof of concept: docking of arginine-glycine-aspartic acid-like compounds into the alphavbeta3 binding site. J Med Chem 2004, 47, (17), 4178-87.
48. Hynes, R. O., A reevaluation of integrins as regulators of angiogenesis. Nat Med 2002, 8, (9), 918-21.
49. Smellie, A.; Kahn, S. D.; Teig, S. L., Analysis of Conformational Coverage 1. Validation and Estimation of Coverage. J Chem Inf Comput Sci 1995, 35, (2), 285-294.
50. Smellie, A.; Kahn, S. D.; Teig, S. L., Analysis of Conformational Coverage 2. Application of Conformational Models. J Chem Inf Comput Sci 1995, 35, (2), 295-304.
51. Smellie, A.; Teig, S. L.; Towbin, P., Poling - Promoting Conformational Variation. J Comput Chem 1995, 16, (2), 171-187.
52. Jones, G.; Willett, P.; Glen, R. C.; Leach, A. R.; Taylor, R., Development and validation of a genetic algorithm for flexible docking. J Mol Biol 1997, 267, (3), 727-48.
53. Nissink, J. W.; Murray, C.; Hartshorn, M.; Verdonk, M. L.; Cole, J. C.; Taylor, R., A new test set for validating predictions of protein-ligand interaction. Proteins 2002, 49, (4), 457-71.
54. Plasencia, C.; Dayam, R.; Wang, Q.; Pinski, J.; Burke, T. R., Jr.; Quinn, D. I.; Neamati, N., Discovery and preclinical evaluation of a novel class of small-molecule compounds in hormone-dependent and -independent cancer cell lines. Mol Cancer Ther 2005, 4, (7), 1105-13。
【0134】
例II
NVX-188(AV38)研究
NVX-188とαvβ3 インテグリンの立体的相互作用
NVX-188は、新規種類の非RGDミメティックのαvβ3アンタゴニストを代表するものである。αvβ3インテグリンとのNVX-188のドッキング立体配置は、ゴールドソフトウェアパッケージを使用してモデル化した。図9に示す通り、NVX-188とαvβ3インテグリンの当該最小エネルギードッキングコンフォメーションは、当該2つのサブユニットのジャンクション近くの活性結合部位で溝を占有していることを示す。
【0135】
図10における代替的な表現により、予測されるNVX-188(水色)のドッキングを、細胞外リガンドタンパク質(藤色)のRGDペプチドの予測される位置と比較して示す。赤色の球は、αvβ3インテグリンの活性結合領域において抱埋されるMn2+イオンを示す。このコンフォメーションにおけるNVX-188により、当該RGDトリペプチド結合部位が効率的にブロッキングされる。機能性アッセイは、αvβインテグリンに対するNVX−188の結合がどのようにインテグリンの生物学的機能に影響を与えるかを決定するために必要である。NVX-188が直接的には、αvβ3インテグリンとの立体相互作用においてRGDペプチドを擬態していないという観察は、NVX-188がアゴニスト(活性化剤)としてよりもアンタゴニスト(ブロッキング剤)として作用することを示唆する。それは、当該αvβ3受容体に関連するおよびそのRGD結合部位に関連する小分子の配向であり、新規および異なる種類としてαvβ3結合剤のNVX-188を特徴付けるものである。
【0136】
NVX-188は、αvβ3インテグリン受容体へのその結合親和性が著しく、ピコモーラー濃度で活性化される。NVX-188クラスの他の化合物は、NVX-188の開発中に同定され(上記を参照されたい)、実質的に低い親和性を示す。それらの親和性は、公知のαvβ3インテグリンアンタゴニストについての文献において報告された親和性を有する系においてより大きい。
【0137】
NVX-188の高親和性についての基礎は、図11から明らかである。図11は、NVX-188とαvβ3ヘテロダイマーにおけるアミノ酸の間に形成される水素結合を示す。NVX-188の高結合親和性(以下を参照されたい)は、当該小分子および当該タンパク質の間で形成された比較的多くの水素結合に起因する可能性がある。
【0138】
αvβ3 結合する親和性
NVX-188の受容体結合親和性をNCI-H1975細胞において125I-放射性標識エキスタチンを使用した競合結合実験を介して決定した。NVX-188の濃度の増加を、エキスタチンの固定した量に加えて行い、当該細胞表面におけるαvβ3インテグリンへの結合するエキスタチンを50%阻害するNVX-188の濃度を決定する(即ち、IC50)。
【0139】
当該αvβ3インテグリン/NVX-188結合親和性は、0.03nM(30pM)であると測定された。これは他の文献に報告されている一般的にナノモーラーレンジで結合を示すαvβ3アンタゴニストに比較して有用である。
【0140】
細胞毒性(細胞障害性)アッセイ
NVX-188 を、乳癌細胞系、高αvβ3インテグリン発現を有するMDA-MB-435および低αvβ3インテグリン発現を有するMCF7、卵巣癌細胞系、HEY(これは細胞増殖阻害証明のための試験に使用した)におけるMTTアッセにより細胞障害性について試験した。NVX-188 は、肺癌細胞系、NCI-H1975およびマウス繊維芽細胞、NIH3T3において更に評価した。NVX-188は、試験された最高濃度においても細胞障害性を示さなかった(20 uM)。
【表B】
例III
αvβ3 アンタゴニストの新規種類
関節リウマチの治療について
背景
関節リウマチにおけるαvβ3 インテグリンの主な役割。 インテグリン、膜貫通接着受容体のファミリーは、細胞の接着、遊走、分化および生存の主なメディエーターである1 。インテグリンは、ヘテロダイマータンパク質であり、それぞれ1つのαおよび1つのβサブユニットで構成され、これらは組合せにおいて非共有結合的に結合している。データに対して、少なくとも18の異なるα-と、8のβ-サブユニットが同定されており、少なくとも24の機能的に多様なインテグリンを形成する。当該インテグリン αvβ3はビトロネクチン受容体として公知であり、125kDaのαvサブユニットと105kDaのβ3サブユニットからなる。幾つかの異なる病態生理学的な過程、特に、関節リウマチおよび他の新血管新生に関連する疾患、炎症およびは骨細胞活性の増加などにおけるその主な役割のためにそれは集中的な探索の焦点である。それらの良好に記載された細胞接着についての機能に加えて、αvβ3インテグリンは、種々のシグナル伝達経路(MAP-キナーゼ、Akt-キナーゼ、VEGFR,およびその他)を調節し、それにより、破骨細胞、内皮細胞および腫瘍細胞の増殖およびアポトーシスに影響を及ぼす3-12 。フィブリノーゲン、ビトロネクチン、オステオポンチンおよびスロンボスポンジンを含む種々の細胞外マトリックスタンパク質(ECM)に対して結合する当該αvβ3受容体は、大部分は、Arg-Gly-Asp(RGD)トリペプチド配列との相互作用を介する13、14 。リガンド結合モチーフの配列を基に、多様なペプチドミメチック賞分子αvβ3アンタゴニストが種々の疾患モデルにおける活性を有して合成されている12、15-25 。当該αvβ3 受容体は、破骨細胞による骨再吸収において中心的な役割を果たしており、αvβ3アンタゴニストは、注目すべき有害作用を伴わずにインビボにおいて骨再吸収を阻害することが示されている26-34 。
【0141】
まとめると、インテグリン αvβ3 の役割は、破骨細胞形成および骨再吸収、マクロファージの遊走および活性化、および血管形成に関連して最も広く研究されている。αvβ3 は、活性化されたマクロファージおよび破骨細胞において過剰発現され、これは関節リウマチ患者における骨破壊部位で多数見られる。これらの患者において、成熟した破骨細胞数は、関節周辺および全身性骨損失の両方に関連して高い。動物関節炎モデルからのデータを含む実質的に膨大な数のエビデンスが、現在、存在し、決定的ではないが、αvβ3が活性化されたマクロファージ依存の炎症、破骨細胞の進展および遊走、骨再吸収、並びに炎症性形成において重要な役割を果たしているという知見を支持している。そのため、このエビデンスおよびリウマチ様疾患の進行した段階のための有効な治療学的選択の欠乏が、αvβ3を標的とする新規化合物の開発がこの疾患のために大いに理性的、選択的および潜在的な有効なアプローチであるということを強く支持している35。
【0142】
新しい種類のαvβ3 アンタゴニストの発見
予備データ。 我々は最近、化学的機能ベースの共通特徴ファルマコフォアモデルを使用して新規小分子αvβ3アンタゴニストのシリーズを発見した36。三次元ファルマコフォアモデルを3つの現在報告されているαvβ3受容体アンタゴニストのトレーニングセットを使用して生成した。当該αvβ3受容体の公知のアンタゴニストの化学的データベースを使用するバリデーションにおいて、高い識別能力を有するファルマコフォアモデルを新規構造骨格と所望の化学的特徴を有する化合物を検索するための三次元照会として使用した。600,000化合物を超える化学的データベースのコンピュータによる探索により、αvβ3アンタゴニストとして特有の構造学的特徴を有する800よりも多い化合物が得られた。構造学的新規性および計算された物理化学的および薬物様性質を基に、化合物のパネルを更なるインビトロスクリーニングのために選択した。αvβ3受容体特異的競合結合アッセイにおいて最終的に試験された29の化合物のうち、幾つかの化合物がナノモーラーからピコモーラーレンジの結合親和性を示した(例Iの表3)36。これらの化合物の1つ(AV38)が、新規にNVX-188と命名され、ピコモーラー濃度での高親和性結合特性のためにリード化合物として選択された。
【0143】
実験プラン
あらまし
一般的考察および目的。 上記の予備データからのエビデンスの通り、高親和性結合特性および優れた「薬物様性質」を示す6つの化合物が同定された。これらの化合物は、優れた特異的結合特性を有しているのみならず、それらは作用様式は、現存のαvβ3アンタゴニストとは異なるようである。NVX-188は、リード化合物として定義されると同時に、それにも拘らず、このグループの他の化合物はインビトロアッセイにおいてより都合のよい機能的性質を有することが可能である。リード化合物のグループについての更なる詳しい記載(例1の表3)は、最近我々のグループによりthe Journal of Medicinal 化学に公開された36。αvβ3機能に批判的に依存する細胞ベースのアッセイは、更なるこれらの化合物の特徴付けるために利用できる。これらのアッセイは、更なる前臨床および臨床開発にための最高の候補を効果的に選択するための基礎を提供する。インシリコ(in silico)で最適化され、結合パイロット試験において評価されたこれらのリード化合物は、例えば、続くインビトロおよびインビボモデル系を使用して試験されてよい:
αvβ3阻害剤の最も重要な標的組織のためのバリデーションモデルインビトロ試験
初代ヒト内皮細胞(HUVEC)
初代破骨細胞/骨芽細胞
αvβ3を発現する骨肉腫(SAOS-2)細胞株
ラットにおけるαvβ3薬物候補の薬物動態学的および毒物学的特徴における重要なデータを得ることに焦点を当てたインビトロ試験。
【0144】
材料および方法
新規αvβ3阻害薬物候補。 当該新規薬物候補(NVX-188および類似体)は、ChemDiv(San Diego、CA)により合成、およびChemDiv(San Diego、CA)から購入した。化合物は、DMSO中で1mM のストック溶液で-20℃で使用時まで所蔵した。
【0145】
試薬および抗体。 ウシゼラチン、ウシ血漿フィブロネクチン、ヒト結成ビトロネクチン、ラットコラーゲンI、BSA、マウスラミニン、およびポリ-L-リジンはシグマから得た(Sigma-Aldrich Co.)。抗ヒトアクチン抗体はシグマから得た。機能ブロッキングmAbs: FB12(抗-α1)、P1E6(抗-α2)、P1B5(抗-α3),およびLM609(抗αvβ3)は、Chemicon(Temecula、CA)から; mAbs Lia1/2(抗-β1)、mAb GI9(抗-α2)、Sam-1(抗-α5)およびGoH3(抗-α6)は、Beckman Coulter(Nyon、Switzerland)から得た。プロテアーゼ阻害剤混合物は、4-(2-アミノエチル)-ベンゼンスルホニルフルオライド、アプロチニン、ロイペプチン、べスタチン、ペプスタチン AおよびE-64を含み、これは、シグマから得た。エキスタチンはBachem California Inc.(Torrance、CA)から得た。
【0146】
細胞培養および処理。 当該ヒト骨肉腫細胞株SAOS-2をthe American Type Culture Collection(ATCC、Manassas、VA)から得て、および現在、我々の研究室において使用している。興味深いことに、それは最近、αvβ3インテグリン発現が、特異的に肺に対する遊走を当該細胞に可能にすることを増強することによりSAOS-2細胞の転移の可能性において役割を担っていることを示している37。骨肉腫細胞の転移の可能性およびビトロネクチンに対する細胞接着に関連するαvβ3 発現は、強力なαvβ3アンタゴニストであると良好に記載されているエキスタチンでの処理の後に減少した。αvβ3 インテグリン は、従って、骨肉腫のための新しい標的である可能性がある。αvβ3を発現している正常なヒト骨芽細胞は、Promocell(Heidelberg、Germany)から得た。PromoCellの骨芽細胞培養系は、正常なヒト骨芽細胞と、最適化された骨芽細胞成長用培地と継代用試薬と組み合わせて提供されるように開発された。正常ヒト内皮細胞(HUVEC)は、Promocellから入手し、製造者の示唆どおりに培養した。破骨細胞培養および破骨細胞分化アッセイは、以下により詳細に記載した。
【0147】
毛細血管形成アッセイ。 血管形成(管構築の範囲)における阻害剤の効果のインビトロアッセイのために、商業的に入手可能なアッセイキットをChemiconから入手し、使用した。このアッセイキットは、血管形成のシンプルなモデルを代表するものであり、そこにおいて、外来性シグナルによる管形成の誘導または阻害が簡単にモニター可能である。環形成の阻害または刺激をアッセイするために、内皮細胞懸濁液を異なる濃度の試験されるべき化合物と共にインキュベートし、次に、当該細胞に特殊分化されたマトリックスの上層に細胞を添加した。the Engelbreth Holm-Swarm(EHS)マウス腫瘍から調製された基底タンパク質の固体ゲルである当該マトリックスにおいて培養した場合、内皮細胞は整列し、中空管様の構造を形成した。管形成は、複数の工程、即ち、細胞接着、遊走、分化および成長を含む工程である。当該アッセイを、使用し、種々の内皮細胞、例えば、ヒト臍帯静脈細胞(HUVEC)およびウシ毛細血管内皮(BCE)細胞におけるαvβ3阻害剤の存在において管構築の範囲をモニターした。
【0148】
αvβ3シグナリングの影響。αvβ3シグナリングにおける阻害剤の効果を測定するために、細胞をトリプシン処理し、阻害剤含有または非含有であり、αvβ3結合タンパク質の存在または非存在の10%血清を含む培地に懸濁した。異なる時間(5-30分)の後に、総細胞性タンパク質を得て、アッセイを行った。限局性接着キナーゼ(FAK)の免疫沈降アッセイのために、FAK を300μgから1mtの総タンパク質から免疫沈降させた。最終的にウエスタンブロットをリン酸化トリプシンおよびFAKについて以下の通りに行った。関連するが、g別々の実験においては、ビトロネクチンコートフラスコで成長されたHUVECを10ng/mlのVEGF含有または非含有の10%FBS-含有培地中で1時間インキュベートした。次に、阻害剤を5から30分間添加し、タンパク質を細胞ライセートから採取する。ウエスタンブロットアッセイは、総およびリン酸化形態のVEGFRおよびAktについて行った。幾つかの方向は、マイトジェン活性化タンパクキナーゼ(MAPK)が、PI3キナーゼシグナルカスケードと同様に、インテグリン阻害剤により調節されることを示している。MAPKおよびAktシグナリング経路において研究されている幾つかの工程のための抗体は、幾つかの会社から入手した(例えば、Cell SignallingおよびUpstat)。種々のリン酸化(活性化)段階を示す異なる抗体はも、入手可能であり(Cell Signalling および/または Upstatから)、総ライセート並びに免疫組織化学的分析のために使用してよい。
【0149】
細胞接着アッセイおよびコーティング。 内皮細胞は、トリプシン消化により回収し、血清非含有M199培地に2 x 104 cells/well(96-ウェルプレート)または12または24-ウェルプレートには1-2 x 105 cells/well で播種した。ブロッキング抗インテグリンmAbsは、この工程で添加される(10 μg/ml)。37℃での1時間の後、やさしくPBSで洗浄し、4%パラホルムアルデヒド(Fluka Chemie、Buchs、Switzerland)で接着細胞を固定化し、0.5%のクリスタルバイオレットで染色した。各ウェルの吸光度を620nmでプレートリーダーで読む。結果は、トリプリケート判定の平均値±S.Dで示す。培養プレートは、実験の種類に依存して、PBS中のフィブロネクチン(3μg/ml)、ゼラチン(0.5%)、コラーゲンI(10μg/ml)、ラミニン(10μg/ml)またはビトロネクチン(1μg/ml)で1時間37℃でプレコートされた。次にそれらを、0.5%の熱不活性脂肪酸非含有ウシ血清アルブミンで1時間37℃でブロックし、リン酸緩衝化生理食塩水(PBS)で洗浄される。実験において、ECMタンパク質が世ゆえ記に添加された実験においては、細胞をトリプシン処理により回収し、PBSで洗浄し、血清非含有のM199培地に再懸濁し1から2時間37℃でインキュベートする。ECMタンパク質を次に、上記で示された濃度で添加し、細胞を更に30から60分間懸濁液中でインキュベートする。細胞接着における阻害剤の効果を測定するために、細胞は96ウェルプレートに阻害剤あり、またはなしで、播種される。60分後に、細胞を洗浄し、接着細胞をカウントする。
【0150】
処理細胞の増殖、アポトーシスおよびクローン原性細胞死。 HUVEC、骨肉腫細胞および骨芽細胞を、ECMタンパク質コート(細胞の種類に依存してビトロネクチン、フィブリノーゲン、オステオポンチン)フラスコに播種し、48時間インキュベートし、血清含有培地において最終的に60から80%のコンフルエントにする。阻害剤を、適切な濃度で同時に添加する。浮遊および接着細胞を8および24時間後に採取し、血清含有培地に再懸濁し、カウントする。アポトーシスは、ヘキスト染料(Hoechst dye)で染色することにより測定する。ECMPコートプレートについて、阻害剤は、播種の後3時間の細胞が良好に付着または広がったところ添加する。即ち、当該細胞の処理の後、浮遊細胞および付着細胞を採取し、FCSエンリッチの実験媒地中で1時間37℃でインキュベートし、次に、アネキシンV/ヨウ化プロピジウム(PI)染色をモレキュラー・プローブ・アンドからのアネキシンV−アレキシア・フルオア488とシグマからのPIを使用して行った。得られた蛍光をフローサイトメトリーにより測定する。アネキシンVおよびPIにより染色された細胞は非アポトーシス細胞(アネックスV-/PI-)、早期アポトーシス細胞(アネックスV+/PI-)、後期アポトーシス細胞(アネックスV-/PI-)、ネクローシス細胞(アネックスV-/PI+)と決定される。細胞カウントおよび生存度の測定は、トリパンブルー排除により実行される。
【0151】
SDS PAGEおよびウエスタンブロット。 指示処理に続き、細胞を4%SDS、20%のグリセロールおよび200mMのTRISを含有するLaemmli緩衝液(pH6.8)に溶解する。タンパク質濃度ををBCAアッセイ(シグマ)を使用して測定する。等量の総タンパク質をSDS-PAGEで分離し、次に、一次抗体に依存してPVDFまたはニトロセルロース膜にブロットする。膜は、PBS中の5%スキムミルクでブロックし、次に、目的の一次抗体とハイブリダイズする。膜をPBSで洗浄し、適切な二次抗体をハイブリダイズする。抗マウスおよび抗ラビットペルオキシダーゼまたはアルカリフォスファターゼ複合二次抗体を5%のスキムミルク含有のPBSで希釈した。PBSでの洗浄の後、膜上の免疫複合体を、増強された化学発光により検出する(Pierce、Rockford、IL、USA)。
【0152】
破骨細胞形成のエクスビボによる測定のための破骨細胞の調製。 破骨細胞は、マウス脾臓から、ナイロンメッシュ(70μm)を使用して5mlのPBS中で2つの脾臓をホモジナイズすることにより得る。Cellsを次に、15mlのチューブに移し、1200U/minで10分間延伸することにより回収する。PBSでの洗浄の後、30ng/mlのM-CSFを含有する15mlのアルファMIM(10%PBS)中に再懸濁し、37℃で o.n.で培養し、採取する。PBSでの洗浄の後、細胞を30mlの総容量中での15mlのフィコール上に置き、1420U/minで8℃で30分間遠心する。内相の細胞を回収し、PBSで2回洗浄し、再懸濁し、カウントする。適切な数の細胞を次に、αvβ3シグナリングおよび/または受容体結合のモジュレータの存在または非存在の48ウェルプレートに配置する。破骨細胞分化における阻害剤の影響を測定するために、マウス脾臓細胞を、一晩、配置し、非接着細胞は回収し、24ウェルプレートに(1ウェル当たり106 cells)でM-CSF(20 ng/ml)含有、誘導物質、例えば、RANKL(50 ng/ml)存在または非存在で、αvβ3インテグリン特異的阻害剤の存在または非存在下に配置する。破骨細胞分化の範囲は、白血球アシッドフォスファターゼキットを使用してTRAP染色により培養4日後に評価する。3以上の核を有する全てのTRAT陽性細胞を破骨細胞としてカウントされる。
【0153】
薬物動態学的および毒物学的試験。 要するに、ベーシックな抽出およびLC-MSまたはLC-MS/MS 分析方法を各化合物に使用するだろう。薬物動態学的試験は、化合物の経口および静脈内投与について行った。投与に続いて、経口経路により処置した各ラットについて血液を7時点(0.5、1、2、4、6、8および12 時間)、および9点(5 min、15 min、30 minおよび1、2、4、6、8および12 時間)で採取した。薬物動態学的なパラメータ(Cmax、Tmax、Vd,および生物学的利用能)は、個々のラットについて、WinNonlin(Pharsight Corp)を使用した非コンパートメント分析を使用して行った。更に、全体的な血算と、肝臓および腎臓血液パラメータを各動物について測定した。
【0154】
参考文献
1. Danen EH. Integrins: regulators of tissue function and cancer progression. Curr Pharm Des 2005, 11: 881-91.
2. Humphries MJ. Integrin structure. Biochem Soc Trans 2000, 28: 311-39.
3. Martin KH, Slack JK, Boerner SA, Martin CC, Parsons JT. Integrin connections map: to infinity and beyond. Science 2002, 296: 652-53.
4. Aplin AE, Howe AK, Juliano RL. Cell adhesion molecules, signal transduction and cell growth. Curr Opin Cell Biol 1999, 11: 737-44.
5. Brooks PC, Clark RA, Cheresh DA. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science 1994, 264: 569-71.
6. Hood JD, Cheresh DA. Role of integrins in cell invasion and migration. Nat Rev Cancer 2002, 2: 91-100.
7. Felding-Habermann B. Integrin adhesion receptors in tumor metastasis. Clin Exp Metastasis 2003, 20: 203-13.
8. Natali PG, Hamby CV, Felding-Habermann B, et al. Clinical significance of alpha(v)beta3 integrin and intercellular adhesion molecule-1 expression in cutaneous malignant melanoma lesions. Cancer Res 1997, 57: 1554-60.
9. Felding-Habermann B, O'Toole TE, Smith JW, et al. Integrin activation controls metastasis in human breast cancer. Proc Natl Acad Sci USA 2001, 98: 1853-58.
10. Pecheur I, Peyruchaud O, Serre CM, et al. Integrin alpha(v)beta3 expression confers on tumor cells a greater propensity to metastasize to bone. Faseb J 2002, 16: 1266-68.
11. Kumar CC, Armstrong L, Yin Z, et al. Targeting integrins alpha v beta 3 and alpha v beta 5 for blocking tumor-induced angiogenesis. Adv Exp Med Biol 2000, 476: 169-80.
12. Shannon KE, Keene JL, Settle SL, et al. Anti-metastatic properties of RGD peptidomimetic agents S137 and S247. Clin Exp Metastasis 2004, 21: 129-38.
13. Ruoslahti E, Pierschbacher MD. Arg-Gly-Asp: a versatile cell recognition signal. Cell 1986, 44: 517-18.
14. Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell 1992, 69: 11-25.
15. Kerr JS, Wexler RS, Mousa SA, et al. Novel small molecule alpha v integrin antagonists: comparative anti-cancer efficacy with known angiogenesis inhibitors. Anticancer Res 1999, 19: 959-68.
16. Lark MW, Stroup GB, Hwang SM, et al. Design and characterization of orally active Arg-Gly-Asp peptidomimetic vitronectin receptor antagonist SB 265123 for prevention of bone loss in osteoporosis. J Pharmacol Exp Ther 1999, 291: 612-17.
17. Reinmuth N, Liu W, Ahmad SA, et al. Alphavbeta3 integrin antagonist S247 decreases colon cancer metastasis and angiogenesis and improves survival in mice. Cancer Res 2003, 63: 2079-87.
18. Harms JF, Welch DR, Samant RS, et al. A small molecule antagonist of the alpha(v)beta3 integrin suppresses MDA-MB-435 skeletal metastasis. Clin Exp Metastasis 2004, 21: 119-28.
19. Brooks PC, Stromblad S, Klemke R, Visscher D, Sarkar FH, Cheresh DA. Antiintegrin alpha v beta 3 blocks human breast cancer growth and angiogenesis in human skin. J Clin Invest 1995, 96: 1815-22.
20. Brooks PC, Montgomery AM, Rosenfeld M, Reisfeld RA, Hu T, Klier G, Cheresh DA. Integrin alpha v beta 3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell 1994, 79: 1157-64.
21. Taga T, Suzuki A, Gonzalez-Gomez I, et al. alpha v-Integrin antagonist EMD 121974 induces apoptosis in brain tumor cells growing on vitronectin and tenascin. Int J Cancer 2002, 98: 690-97.
22. Mitjans F, Meyer T, Fittschen C, Goodman S, Jonczyk A, Marshall JF, Reyes G, Piulats J. In vivo therapy of malignant melanoma by means of antagonists of alphav integrins. Int J Cancer 2000, 87: 716-23.
23. Allman R, Cowburn P, Mason M. In vitro and in vivo effects of a cyclic peptide with affinity for the alpha (nu) beta3 integrin in human melanoma cells. Eur J Cancer 2000, 36: 410-22.
24. Dechantsreiter MA, Planker E, Matha B, Lohof E, Holzemann G, Jonczyk A, Goodman SL, Kessler H. N-Methylated cyclic RGD peptides as highly active and selective alpha(V)beta(3) integrin antagonists. J Med Chem 1999, 42: 3033-40.
25. Burke PA, DeNardo SJ, Miers LA, Lamborn KR, Matzku S, DeNardo GL. Cilengitide targeting of alpha(v)beta(3) integrin receptor synergizes with radioimmunotherapy to increase efficacy and apoptosis in breast cancer xenografts. Cancer Res 2002, 62: 4263-72.
26. Rodan SB, Rodan GA. Integrin function in osteoclasts. J Endocrinol 1997, 154 (Suppl.): 47-56.
27. Nakamura I, Pilkington MF, Lakkakorpi PT, Lipfert L, Sims SM, Dixon SJ, Rodan GA, Duong LT. Role of alpha(v)beta(3) integrin in osteoclast migration and formation of the sealing zone. J Cell Sci 1999, 112: 3985-93.
28. Duong LT, Rodan GA. The role of integrins in osteoclast function. J Bone Miner Metab 1999, 17: 1-6.
29. Teitelbaum SL. Bone resorption by osteoclasts. Science 2000, 289: 1504-8.
30. Fisher JE, Caulfield MP, Sato M, Quartuccio HA, Gould RJ, Garsky VM, Rodan, GA, Rosenblatt, M. Inhibition of osteoclastic bone resorption in vivo by echistatin, an "arginyl-glycyl-aspartyl" (RGD)-containing protein. Endocrinology 1993, 132: 1411- 13.
31. Engleman VW, Nickols GA, Ross FP, Horton MA, Griggs DW, Settle SL, Ruminski PG, Teitelbaum SL. A peptidomimetic antagonist of the alpha(v)beta3 integrin inhibits bone resorption in vitro and prevents osteoporosis in vivo. J Clin Invest 1997, 99: 2284-92.
32. Yamamoto M, Fisher JE, Gentile M, Seedor JG, Leu CT, Rodan SB, Rodan GA. The integrin ligand echistatin prevents bone loss in ovariectomized mice and rats. Endocrinology 1998, 139: 1411-19.
33. Hutchinson JH, Halczenko W, Brashear KM, et al. Nonpeptide alphavbeta3 antagonists. In vitro and in vivo evaluation of a potent alphavbeta3 antagonist for the prevention and treatment of osteoporosis. J Med Chem 2003, 46: 4790-98.
34. Murphy MG, Cerchio K, Stoch SA, Gottesdiener K, Wu M, Recker R. Effect of L-000845704, an alphaVbeta3 integrin antagonist, on markers of bone turnover and bone mineral density in postmenopausal osteoporotic women. J Clin Endocrinol Metab 2005, 90: 2022-28.
35. Wilder RL. Integrin alpha V beta 3 as a target for treatment of rheumatoid arthritis and related rheumatic diseases. Ann Rheum Dis 2002, 61: 96−99.
36. Dayam R, Aiello F, Deng J, Wu Y, Garofalo A, Chen X, Neamati N. Discovery of small molecule integrin alphavbeta3 antagonists as novel anticancer agents. J Med Chem. 2006, 49: 4526-34.
37. Duan X, Jia SF, Zhou Z, Langley RR, Bolontrade MF, Kleinerman ES. Association of alphavbeta3 integrin expression with the metastatic potential and migratory and chemotactic ability of human osteosarcoma cells. Clin Exp Metastasis. 2004, 21: 747-53。
【図面の簡単な説明】
【0155】
【図1】図1は、新規αvβ3アンタゴニストのファルマコフォアにより導かれる設計および発見の模式図である。
【図2】図2は、トレーニングセット化合物A-Cの構造を示す。
【図3】図3。(a)共通特徴ファルマコフォアHypo5。(b)Hypo5 はトレーニングセット化合物Aの1つに位置づけられる。化合物Aの重要な化学的特徴は、Hypo5のファルマコフォアの特徴により位置付けられる。ファルマコフォア特徴は、マゼンタのH結合供与体(HBD)、緑色のH結合受容体(HBA)、茶色の疎水性芳香族(HAR1-HAR2)および青色の陰性イオン性機能(NI)として示される。特徴間の距離はÅで示される。
【図4】図4は、新規αvβ3 受容体 アンタゴニスト AV3(a)およびAV26(b)へのHypo5の位置づけを示す。Hypo5のファルマコフォア的特徴は、適度に当該アンタゴニストの重要な化学的特徴に位置づけされている。当該ファルマコフォアの特徴はマゼンタのH結合供与体、緑色のH結合受容体、茶色の疎水性芳香族および青色の陰性イオン化特徴として示される。
【図5】図5は、αvβ3RGDペプチド結合部位の内部におけるアンタゴニストA(a)、AV3(b)、AV26(c)、AV27(d)およびAV38(e)の 予測される二重結合コンフォメーションを示す。黄色および赤色部分は、αvβ3受容体のαおよびβ鎖を示す。突出した活性部位アミノ酸残基は、当該受容体表面におけるスティックモデルとして示した。当該緑色のスティックモデルは、サイクリックRGDペプチド(PDB1L5G)の結合配向を示す。当該活性部位Mn2+(MIDAS)は青緑色の球として示す。
【図6】図6は、アンタゴニストA(a)、AV26(b)およびAV38(c)とαvβ3RGDペプチド結合部位における突出したアミノ酸残基との間に得られた相互作用を示す模式図である。ダッシュの線はH結合相互作用を示す。H結合距離はÅで表す。
【図7】図7は、4つの内皮アポトーシスモデルを図示する。(a)古典的モデル; そこにおいてリガンドによるインテグリン結合が生存シグナルを提供するために必要である。阻害剤がリガンドの結合をブロックし、それにより、生存シグナルがブロックされる。(b)カスパーゼ活性化モデル;そこにおいて、RGDペプチドは直接にカスパーゼを活性化し、インテグリンの如何なる介入もなしにアポトーシスを引き起こす。(c)非ライゲーションモデルまたは「インテグリン-媒介細胞死」;そこにおいて結合していないインテグリンが、直接に結合し、カスパーゼ-8を活性化する。ECM リガンドはこれをブロックするが、それらがインテグリンを活性化することが知られて入るが、RGD ペプチドおよび同じインテグリンに対して結合する抗体はそのようなことはしない。(d)例えば、パクリタキセルなどの細胞毒性剤に対して一旦結合したαvβ3 アンタゴニストは、αvβ3陽性癌細胞に対して、および腫瘍新生血管におけるαvβ3陽性内皮細胞に対して効率的に送達される(Richard Hynesから改変された、参考文献 48)。
【図8】図8は、共通する特徴のファルマコフォア Hypos 1(A)、5(B)および9(C)を示す。当該ファルマコフォア特徴は、H結合供与体(HBD)はマゼンタで、H結合受容体(HBA)は緑色で、疎水性芳香族(HAR1-HAR2)は茶色で、および陰性イオン化特徴(NI)は青色で示す。当該特徴間の距離はÅで示す。
【図9】図 9は、NVX-188の予測される結合コンフォメーションを示す。
【図10】図10 は、αvβ3インテグリン上のNVX-188(水色)および当該RGDトリペプチド(藤色)の予測されるドッキングを示す。
【図11】図11は、当該αvβ3ヘテロダイマーにおけるアミノ酸に対するNVX-188(右側)およびRGD-ミメティック小分子(左側)の関係を示す。
【特許請求の範囲】
【請求項1】
化合物、またはその薬学的若しくは薬用化粧品学的に許容される塩、溶媒和物若しくは水和物を含む組成物であって、当該化合物が1のH結合供与体(HBD)、1のH結合受容体(HBA)、2の疎水性芳香族基(HAR1およびHAR2)、および1の陰性イオン性基(NI)を含み、
ここで、
当該HBD、HBA、HAR1、HAR2および NIが、図8Aに従う立体配置であり、当該HBDとHRA1の距離が2.81±1Åであり、当該HBDとHBAの距離が7.52±1Åであり、当該HBDとNIの距離が17.67±1Åであり、当該HRA1とHBAの距離が7.52±1Åであり、当該HRA1とHRA2の距離が9.41±1Åであり、当該HBAとNIの距離が10.20±1Åであり、当該HRA2とNIの距離が5.88±1Åであるか、
当該HBD、HBA、HAR1、HAR2およびNIは図8Bに従う立体配置であり、当該HBDとHRA1の距離が2.80±1Åであり、当該HBDとHBAの距離が7.31±1Åであり、当該HBDとNIの距離が16.92±1Åであり、当該HRA1とHBAの距離が5.20±1Åであり、当該HRA1とNIの距離が14.34±1Åであり、当該HBAとHRA2の距離が4.69±1であり、当該HBAとNIの距離が9.85±1Åであり、当該HRA2とNIの距離が5.66±1Åであるか、または
当該HBD、HBA、HAR1、HAR2およびNIは図8Cの立体配置であり、当該HBDとHRA1の距離が2.79±1Åであり、当該HBDとHRA2の距離が11.94±1Åであり、当該HBDとNIの距離が16.28±1Åであり、当該HRA1とHBAの距離が5.20±Åであり、当該HRA1とNIの距離が14.64±1Åであり、当該HBAとHRA2の距離が4.72±1Åであり、当該HRA2とNIの距離が5.63±1Åであり、
および当該化合物は、化合物A、BまたはC
【化1】
ではない
組成物。
【請求項2】
請求項1の組成物であって、
ここで、
当該 HBD、HBA、HAR1、HAR2およびNIは、図8Aに従う立体配置であり、当該HBDとHRA1の距離は 2.81 Åであり、当該 HBDとHBAの距離は 7.52 Åであり、当該HBDとNIの距離は17.67 Åであり、当該HRA1とHBAの距離は 7.52 Åであり、当該HRA1とHRA2 の距離は9.41 Åであり、当該HBAとNIの距離は10.20 Åであり、および当該HRA2と NI の距離は5.88 Åであるか、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Bに従う立体配置であり、図8Bに従う立体配置であり、ここで、当該 HBDとthe HRA1 の距離は2.80 Åであり、当該 HBDとthe HBA の距離は7.34 Åであり、当該HBDとNI の距離は16.92 Åであり、当該 HRA1とHBAの距離は 5.20 Åであり、当該HRA1と NI の距離は14.34 Åであり、当該HBAとHRA2 の距離は4.69 Åであり、当該HBAとNI の距離は9.85 Åであり、および当該HRA2とNI の距離は5.66 Åであるか、または
当該 HBD、HBA、HAR1、HAR2およびNIは、図8Cに従う立体配置であり、ここで、当該HBDと HRA1 の距離は2.79 Åであり、当該HBDと HRA2 の距離は11.94 Åであり、当該HBDと NI の距離は 16.28 Åであり、当該HRA1と HBA の距離は5.20 Åであり、当該HRA1とNI の距離は14.64 Åであり、当該HBAと HRA2 の距離は4.72 Åでありおよび当該HRA2とNI の距離は5.63 Åである
組成物。
【請求項3】
請求項1の化合物であって、ここで、当該化合物は、式 I、II、IIIまたはIVの化合物であって、
【化2】
ここで、式 I、IIおよびIIIにおいて
それぞれ、R1およびR3 は、脂肪族、芳香族またはヘテロ環基であり、
R2 は置換基であり、および
X は N、OまたはSであり、並びに
【化3】
ここで、式 IVにおいて、
それぞれ、R1およびR2 は水素またはハロゲン、ヒドロキシル、スルフヒドリル、アルコキシ、アリールオキシ、カルボキシル、ニトロ、シアノ、アミノ、アミドまたはスルホニル基、または何れかの数の炭素原子を含む何れか他の有機官能基であり、
R3 は、水素、ヒドロキシル、アルコキシ、アリールオキシ、芳香族またはヘテロ環基または何れかの数の炭素原子の脂肪族鎖であり、
それぞれ、R4およびR5 はヒドロキシルまたはアミノ基であり、
X は C、N、O,またはSであり,
m は、炭素、窒素、酸素または硫黄原子を伴う0 〜 3 原子リンカー基であり、および
n は、炭素、窒素、酸素または硫黄原子を伴う0 〜 5 原子リンカー基である
化合物。
【請求項4】
請求項3の組成物生物であって、
ここで、式 I、IIおよびIIIにおいて、
それぞれR1およびR3 は、アルキル、置換されたアルキル、アルケニル、置換されたアルケニル、アルキニル、置換されたアルキニル、フェニル、置換されたフェニル、アリール、置換されたアリール、ヘテロアリールまたは置換されたヘテロアリール基であり、
R2 は、水素またはハロゲン、ヒドロキシル、スルフヒドリル、アルコキシ、カルボキシル、ニトロ、シアノ、アミノ、アミドまたはスルホニル基または何れかの数の炭素原子を含む何れか他の有機官能基、または
その組み合わせであり、並びに
ここで、式 IVにおいて、R4またはR5 における当該アミノ基は、脂肪族、芳香族またはヘテロ環基で置換されている
組成物。
【請求項5】
請求項4の組成物であって、ここで、式 I、IIおよびIIIにおいて、R1またはR3における当該置換されたアルキル、アルケニル、アルキニル、フェニル、アリールまたはヘテロアリール基は、ハロ、ヒドロキシル、スルフヒドリル、アルコキシ、カルボキシル、カルボニル、ニトロ、シアノ、アミノ、アミド、スルホニルまたは置換されたヘテロ環で置換されたアルキル、アルケニル、アルキニル、フェニル、アリールまたはヘテロアリール基である組成物。
【請求項6】
請求項3の組成物であって、ここで、当該化合物がAV1-11、AV14-15、AV17-32,およびAV34-38からなる群より選択される組成物。
【請求項7】
請求項1の組成物であって、更に薬学的にまたは薬用化粧品学的に許容される担体を含む組成物。
【請求項8】
第一の化合物または、その薬学的または薬用化粧品学的に許容される塩、溶媒和物または水和物、および1または1以上の第二の化合物を含む組成物であって、ここで、
第一の化合物は、1の H結合供与体(HBD)、1の H結合受容体(HBA)、2の疎水性芳香族基s(HAR1およびHAR2),および1の陰性イオン性基(NI)を含み、ここで、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Aに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.81±1 Åであり、当該HBDとHBAの距離は 7.52±1Åであり、当該 HBDとNIの距離は17.67±1 Åであり、当該HRA1とHBA の距離は、7.52±1 Åであり、当該HRA1とHRA2の距離は9.41±1 Åであり、当該HBAとthe NI の距離は10.20±1 Åであり、および当該HRA2とNI の距離は5.88±1 Åであるか、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Bに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.80±1 Åであり、当該HBDとthe HBA の距離は7.34±1 Åであり、当該HBDとNIの距離は16.92±1 Åであり、当該HRA1とthe HBA の距離は5.20±1 Åであり、当該HRA1とNI の距離は14.34±1 Åであり、当該HBAとthe HRA2 の距離は4.69±1 Åであり、当該HBAとNI の距離は 9.85±1 Åであり、および当該HRA2と NIの距離は5.66±1 Åであるか、または
当該 HBD、HBA、HAR1、HAR2およびNIは、図8Cに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.79±1 Åであり、当該HBDと HRA2の距離は11.94±1 Åであり、当該HBDと NI の距離は16.28±1 Åであり、当該HRA1と HBA の距離は 5.20±1 Åであり、当該HRA1とNI の距離は14.64±1 Åであり、当該HBAとthe HRA2 の距離は 4.72±1Åであり、および当該HRA2と NI の距離は5.63±1 Åであり、
並びに、ここで、第一の化合物は化合物 A、BまたはCではなく、並びに
第二の化合物は、治療剤、イメージング剤またはその組み合わせである
組成物。
【請求項9】
請求項8の組成物であって、ここで、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Aに従う立体配置であり、ここで、 当該HBDとHRA1の距離は 2.81 Åであり、当該HBDと HBAの距離は7.52 Åであり、当該HBDとNI の距離は17.67 Åであり、当該HRA1とHBAの距離は 7.52 Åであり、当該HRA1とHRA2の距離は9.41 Åであり、当該HBAと NIの距離は10.20 Åであり、および当該HRA2とNI の距離は5.88 Åであるか、
当該 HBD、HBA、HAR1、HAR2およびNIは、図8Bに従う立体配置であり、ここで、 当該HBDとHRA1の距離は2.80 Åであり、当該HBDと HBAの距離は7.34 Åであり、当該HBDと NI の距離は16.92 Åであり、当該HRA1と HBA の距離は5.20 Åであり、当該HRA1とNI の距離は14.34 Åであり、当該HBAと HRA2 の距離は 4.69 Åであり、当該HBAとNI の距離は 9.85 Åであり、および当該HRA2とNI の距離は5.66 Åであるか、または、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Cに従う立体配置であり、ここで、 当該HBDとHRA1の距離は2.79 Åであり、当該HBDと HRA2の距離は11.94 Åであり、当該HBDとNI の距離は16.28 Åであり、当該HRA1と HBA の距離は5.20 Åであり、当該HRA1とNI の距離は14.64 Åであり、当該HBAとHRA2の距離は 4.72 Åであり、および当該HRA2とNI の距離は5.63 Åである
組成物。
【請求項10】
請求項8の組成物であって、ここで、第一の化合物は式 I、II、IIIまたはIVであり、
ここで、式 I、IIおよびIIIにおいて、
それぞれ、R1およびR3 は脂肪族、芳香族またはヘテロ環基であり、
R2 は置換基であり、および
X は N、OまたはSであり、および
ここで、式 IVにおいて、
それぞれ、R1およびR2 は水素またはハロゲン、ヒドロキシル、スルフヒドリル、アルコキシ、アリールオキシ、カルボキシル、ニトロ、シアノ、アミノ、アミドまたはスルホニル基または何れかの数の炭素原子を含む何れか他の有機官能基であり,
R3は、水素、ヒドロキシル、アルコキシ、アリールオキシ、芳香族またはヘテロ環基、または何れかの数の炭素原子の脂肪族鎖であり、
それぞれ、R4およびR5 はヒドロキシルまたはアミノ基であり、
X は C、N、OまたはSであり、
m は、炭素、窒素、酸素または硫黄原子を伴う 0 から 3 原子リンカー基であり、および
n は、炭素、窒素、酸素または硫黄原子を伴う 0 から 5 原子リンカー 基である
組成物。
【請求項11】
請求項10の組成物であって、
ここで、式 I、IIおよびIIIにおいて、
それぞれ、R1およびR3 はアルキル、置換されたアルキル、アルケニル、置換されたアルケニル、アルキニル、置換されたアルキニル、フェニル、置換されたフェニル、アリール、置換されたアリール、ヘテロアリールまたは置換されたヘテロアリール基であり、
R2 は、水素またはハロゲン、ヒドロキシル、スルフヒドリル、アルコキシ、カルボキシル、ニトロ、シアノ、アミノ、アミドまたはスルホニル基、または何れかの数の炭素原子を含む何れか他の有機官能基、またはその組み合わせであり、および
ここで、式 IVにおいて、R4またはR5 における当該アミノ基は脂肪族、芳香族,またはヘテロ環基で置換されている
組成物。
【請求項12】
請求項11の組成物であって、ここで、式s I、IIおよびIIIにおいて、R1またはR3における当該置換されたアルキル、アルケニル、アルキニル、フェニル、アリールまたはヘテロアリール基は、ハロ、ヒドロキシル、スルフヒドリル、アルコキシ、カルボキシル、カルボニル、ニトロ、シアノ、アミノ、アミド、スルホニルまたは置換されたヘテロ環基で置換されたアルキル、アルケニル、アルキニル、フェニル、アリールまたはヘテロアリール基である組成物。
【請求項13】
請求項10の組成物であって、ここで、当該第一の化合物がAV1-11、AV14-15、AV17-32,およびAV34-38からなる群より選択される組成物。
【請求項14】
請求項8の組成物であって、ここで、当該第二の化合物は、
血管増殖阻害剤,
関節リウマチを治療するための治療剤、
放射線治療剤、
化学療法剤、
骨転移の治療のための治療剤、
骨粗鬆症の治療のための治療剤、
再狭窄の治療のための治療剤、
血管イメージング剤、または
その組み合わせ
である組成物。
【請求項15】
請求項14の組成物であって、ここで、
当該血管増殖阻害剤が、TNF-アルファ、アンジオスタチン、ベバシザン、アレステン、カンスタチン、コンブレタスタチン、エンドスタチン、NM-3、サリドマイド、スロンボスポンジン、タムスタチン、2-メトキシエストラジオール,およびビタキシンからなる群より選択され、
当該化学療法剤が、シスプラチン、ドキソルビシン、ビンクリスチン、シクロホスファミド、トポテカン、パクリタキセルおよび他の化学療法剤からなる群より選択され、
骨粗鬆症の治療のための治療剤は、ホセマックス、アレジア、アクトネル、ジンドロネル、コメタ、プレマリン、クリマラ、エストラ、ビベラ、エストラダーム、プレモプロ、プレムフェーズ、フェマート、プレフェスト、コンビパッチ、エビスタ、カルシトニン、ミアカルシン,およびカルシトニンからなる群より選択され、および
当該血管イメージング剤は、放射線核種である
組成物。
【請求項16】
請求項8の組成物であって、ここで、当該組成物は第一の化合物および第二の化合物からなる組成物。
【請求項17】
請求項8の組成物であって、ここで、第一の化合物が実施可能に少なくとも1の第二の化合物に連結されている組成物。
【請求項18】
インテグリンを化合物に対して結合する方法であって、
インテグリンを化合物、またはその薬学的、または薬用化粧品学的に許容される塩、溶媒和物または水和物と接触すること、
ここで、当該化合物は、1のH結合供与体(HBD)、1のH結合受容体(HBA)、2の疎水性芳香族基(HAR1およびHAR2)および1の陰性イオン性基(NI)を含み、ここで、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Aに従う立体配置であり、ここで、当該HBDと HRA1の距離は2.81±1 Åであり、当該HBDとHBA の距離は7.52±1 Åであり、当該HBDと NI の距離は17.67±1 Åであり、当該HRA1とHBAの距離は7.52±1 Åであり、当該HRA1とHRA2 の距離は 9.41±1 Åであり、当該HBAとNIの距離は10.20±1 Åであり、および当該HRA2とNI の距離は5.88±1 Åであるか、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Bに従う立体配置であり、ここで、当該HBDと HRA1の距離は2.80±1 Åであり、当該HBDとHBAの距離は7.34±1 Åであり、当該HBDとNI の距離は16.92±1 Åであり、当該HRA1とHBAの距離は 5.20±1 Åであり、当該HRA1とNI の距離は14.34±1 Åであり、当該HBAとHRA2の距離は4.69±1 Åであり、当該HBAとNI の距離は9.85±1 Åであり、および当該 HRA2とNIの距離は 5.66±1 Åであるか、または、
当該 HBD、HBA、HAR1、HAR2およびNIは、図8Cに従う立体配置であり、ここで、当該HBDと HRA1 の距離は2.79±1 Åであり、当該HBDと HRA2 の距離は11.94±1 Åであり、当該HBDと NI の距離は16.28±1 Åであり、当該HRA1と HBAの距離は 5.20±1 Åであり、当該HRA1と NI の距離は14.64±1 Åであり、当該HBAとHRA2 の距離は4.72±1 Åであり、および当該HRA2と NI の距離は5.63±1 Åであり、
並びに、ここで、当該化合物は化合物 A、BまたはCではない、
を具備する方法。
【請求項19】
請求項18の方法であって、ここで、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Aに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.81 Åであり、当該HBDと HBA の距離は7.52 Åであり、当該HBDと NI の距離は17.67 Åであり、当該HRA1とHBA の距離は7.52 Åであり、当該HRA1と HRA2 の距離は9.41 Åであり、当該HBAとNI の距離は10.20 Åであり、および当該HRA2とNI の距離は5.88 Åであるか、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Bに従う立体配置であり、ここで、当該HBDとHRA1 の距離は2.80 Åであり、当該HBDと HBAの距離は7.34 Åであり、当該HBDと NI の距離は16.92 Åであり、当該HRA1と HBA の距離は5.20 Åであり、当該HRA1と NIの距離は14.34 Åであり、当該HBAと HRA2 の距離は4.69 Åであり、当該HBAとNI の距離は9.85 Åであり、および当該HRA2とNIの距離は5.66 Åであるか、または、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Cに従う立体配置であり、ここで、当該HBDとHRA1 の距離は2.79 Åであり、当該HBDとHRA2 の距離は11.94 Åであり、当該HBDとNI の距離は16.28 Åであり、当該HRA1と HBA の距離は5.20 Åであり、当該HRA1とNI の距離は14.64 Åであり、当該HBAとHRA2の距離は4.72 Åであり、および当該HRA2とNI の距離は5.63 Åである
方法。
【請求項20】
請求項18の方法であって、ここで、当該化合物が、式 I、II、IIIまたはIVの化合物であり、
ここで、式 I、IIおよびIIIにおいて、
それぞれ、R1およびR3 は脂肪族、芳香族またはヘテロ環基であり、
R2 は置換基であり、および
X は N、OまたはSであり、および
ここで、式 IVにおいて、
それぞれ、R1およびR2 は、水素またはハロゲン、ヒドロキシル、スルフヒドリル、アルコキシ、アリールオキシ、カルボキシル、ニトロ、シアノ、アミノ、アミドまたはスルホニル基または何れかの数の炭素原子を含む何れか他の有機官能基であり、
R3 は、水素、ヒドロキシル、アルコキシ、アリールオキシ、芳香族またはヘテロ環基、または何れかの数の炭素原子の脂肪族鎖であり、
それぞれR4およびR5 はヒドロキシルまたはアミノ基であり、
X は C、N、OまたはSであり、
m は、炭素、窒素、酸素または硫黄原子を伴う 0 から 3 原子リンカー基であり、および
n は、炭素、窒素、酸素または硫黄原子を伴う 0 から 5 原子リンカー基である
方法。
【請求項21】
請求項20の方法であって、
ここで、式I、IIおよびIIIにおいて、
それぞれ、R1およびR3 は、アルキル、置換されたアルキル、アルケニル、置換されたアルケニル、アルキニル、置換されたアルキニル、フェニル、置換されたフェニル、アリール、置換されたアリール、ヘテロアリールまたは置換されたヘテロアリール 基であり、
R2 は、水素またはハロゲン、ヒドロキシル、スルフヒドリル、アルコキシ、カルボキシル、ニトロ、シアノ、アミノ、アミドまたはスルホニル基または何れかの数の炭素原子を含む何れか他の有機官能基、または
その組み合わせ、および
ここで、式 IVにおいて、R4またはR5 における当該アミノ基が脂肪族、芳香族またはヘテロ環基で置換された
方法。
【請求項22】
請求項21の方法であって、ここで、式 I、IIおよびIIIにおいて、R1またはR3における当該置換されたアルキル、アルケニル、アルキニル、フェニル、アリールまたはヘテロアリール基は、ハロ、ヒドロキシル、スルフヒドリル、アルコキシ、カルボキシル、カルボニル、ニトロ、シアノ、アミノ、アミド、スルホニルまたは置換されたヘテロ環基によって置換されたアルキル、アルケニル、アルキニル、フェニル、アリールまたはヘテロアリール基である
方法。
【請求項23】
請求項20の方法であって、ここで、当該化合物がAV1-11、AV14-15、AV17-32,およびAV34-38からなる群より選択される方法。
【請求項24】
請求項18の方法であって、ここで、当該インテグリンがαvβ3である方法。
【請求項25】
請求項18の方法であって、ここで、当該インテグリンが細胞上にある方法。
【請求項26】
請求項25の方法であって、ここで、当該細胞が癌細胞、良性腫瘍細胞、間質細胞、炎症に反応する、炎症を媒介する、または炎症を調節する細胞、免疫系の細胞、血管機能に関与する細胞、骨芽細胞、破骨細胞またはウイルスに感染した細胞.
【請求項27】
請求項26の方法であって、ここで、
当該炎症に対して反応する、炎症を媒介する、若しくは炎症を調節する細胞、または免疫系の細胞が、白血球、マクロファージおよび樹状細胞からなる群より選択され、
当該血管機能に関連する細胞が、内皮細胞、内皮細胞に関与する細胞および平滑筋細胞からなる群より選択され、および、
当該ウイルスが、アデノウイルス(Adenoviridae)、コックサッキーウイルス(Picornaviridae)、エコウイルス(Picornaviridae)、口蹄疫ウイルス(Picornaviridae)、ハンタウイルス(Buriyaviridae)、ヒトパレコウイルス(Picornaviridae)、ヒト免疫不全ウイルス 1(Retroviridae),およびロタウイルス(Reoviridae)からなる群より選択される
方法。
【請求項28】
請求項25の方法であって、更に、当該細胞を1または1以上の治療剤、イメージング剤またはその組み合わせと接触させることを具備する方法。
【請求項29】
請求項28の方法であって、前記薬剤が、
血管増殖阻害剤、
関節リウマチを治療するための治療薬、
放射線治療剤、
化学療法剤、
骨転移を治療するための治療剤、
骨粗鬆症を治療するための治療剤、
再狭窄を治療するための治療剤、
血管イメージング剤、または
その組み合わせである
方法。
【請求項30】
請求項29の方法であって、ここで、
当該血管増殖阻害剤が、TNF-アルファ、アンジオスタチン、ベバシザン、アレステン、カンスタチン、コンブレタスタチン、エンドスタチン、NM-3、サリドマイド、スロンボスポンジン、タムスタチン、2-メトキシエストラジオール,およびビタキシンからなる群より選択され、
当該化学療法剤が、シスプラチン、ドキソルビシン、ビンクリスチン、シクロホスファミド、トポテカン、パクリタキセル,および他の化学療法剤からなる群より選択され、
当該骨粗鬆症を治療するための治療剤が、ホセマックス、アレジア、アクトネル、ジンドロネル、コメタ、プレマリン、クリマラ、エストラ、ビベラ、エストラダーム、プレモプロ、プレムフェーズ、フェマート、プレフェスト、コンビパッチ、エビスタ、カルシトニン、ミアカルシン,およびカルシトニンからなる群より選択され、および
当該血管イメージング剤が、放射性核種である
方法。
【請求項31】
請求項28の方法であって、ここで、当該インテグリンおよび当該細胞が、同時にまたは経時的に接触される方法。
【請求項32】
請求項28の方法であって、ここで、当該化合物が、少なくとも1の薬剤と混合される方法。
【請求項33】
請求項28の方法であって、ここで、 当該化合物が、少なくとも1の薬物に対して実施可能に連結される方法。
【請求項34】
対象においてインテグリンを化合物に対して結合する方法であって、
それを必要とする対象に対して、有効量の化合物、またはその薬学的若しくは薬用化粧品学的に許容される塩、溶媒和物または水和物を投与すること、
ここで、当該化合物は、1のH結合供与体(HBD)、1のH結合受容体(HBA)、2の疎水性芳香族基(HAR1およびHAR2)および1の陰性イオン性基(NI)を含み、ここで、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Aに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.81±1 Åであり、当該HBDとHBA の距離が7.52±1 Åであり、当該HBDとNI の距離は17.67±1 Åであり、当該HRA1と HBAの距離が7.52±1 Åであり、当該HRA1とthe HRA2の距離が9.41±1 Åであり、当該HBAと NI の距離が10.20±1 Åであり、および当該HRA2とNI の距離が5.88±1 Åであるか、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Bに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.80±1 Åであり、当該HBDとHBA の距離は7.34±1 Åであり、当該HBDとNI の距離は16.92±1 Åであり、当該HRA1とHBAの距離は5.20±1 Åであり、当該HRA1とNI の距離は14.34±1 Åであり、当該HBAとHRA2 の距離は4.69±1 Åであり、当該HBAとNI の距離は9.85±1 Åであり、および当該HRA2とNI の距離は5.66±1 Åであるか、または
the HBD、HBA、HAR1、HAR2,およびNIは、図8Cに従う立体配置であり、ここで、当該HBDと HRA1の距離は 2.79±1 Åであり、当該HBDと HRA2 の距離は11.94±1 Åであり、当該HBDと NI の距離は16.28±1 Åであり、当該HRA1とHBAの距離は5.20±1 Åであり、当該HRA1とNIの距離は14.64±1 Åであり、当該HBAとHRA2の距離は 4.72±1 Åであり、および当該HRA2とNI の距離は5.63±1 Åであり、
およびここで、当該化合物は化合物 A、BまたはCではない、
方法。
【請求項35】
請求項34の方法であって、ここで、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Aに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.81 Åであり、当該HBDとHBAの距離は7.52 Åであり、当該HBDとNI の距離は17.67 Åであり、当該HRA1とHBAの距離は 7.52 Åであり、当該HRA1とHRA2の距離は9.41 Åであり、当該HBAとNI の距離は 10.20 Åであり、および当該HRA2とNI の距離は5.88 Åであるか、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Bに従う立体配置であり、ここで、当該HBDとHRA1 の距離は2.80 Åであり、当該HBDと HBAの距離は7.34 Åであり、当該HBDとNI の距離は16.92 Åであり、当該HRA1とHBAの距離は 5.20 Åであり、当該HRA1とNIの距離は14.34 Åであり、当該HBAとHRA2 の距離は4.69 Åであり、当該HBAとNI の距離は9.85 Åであり、および当該HRA2とNI の距離は5.66 Åであるか、または、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Cに従う立体配置であり、ここで、当該HBDとHRA1 の距離は2.79 Åであり、当該HBDとHRA2 の距離は11.94 Åであり、当該HBDとNI の距離は16.28 Åであり、当該HRA1とHBAの距離は5.20 Åであり、当該HRA1とNI の距離は14.64 Åであり、当該HBAとHRA2の距離は4.72 Åであり、および当該HRA2とNI の距離は5.63 Åである
方法。
【請求項36】
請求項34の方法であって、ここで、当該化合物は、式 I、II、IIIまたはIVの化合物であって,
ここで、式s I、IIおよびIIIにおいて、
それぞれ、R1およびR3 は脂肪族、芳香族またはヘテロ環基であり、
R2 は置換基であり、および
X はN、OまたはSであり、および
ここで、式 IVにおいて、
それぞれ、R1およびR2 は水素またはハロゲン、ヒドロキシル、スルフヒドリル、アルコキシ、アリールオキシ、カルボキシル、ニトロ、シアノ、アミノ、アミドまたはスルホニル基または何れかの数の炭素原子を含む何れか他の有機官能基であり、
R3 は水素、ヒドロキシル、アルコキシ、アリールオキシ、芳香族またはヘテロ環基または何れかの数の炭素原子の脂肪族鎖であり、
それぞれ、R4およびR5 はヒドロキシルまたはアミノ基であり、
X は C、N、OまたはSであり、
m は 炭素、窒素、酸素または硫黄原子を伴う0 から 3 の原子リンカー基 with 炭素、窒素、酸素,または硫黄 原子であり、
n は、炭素、窒素、酸素または硫黄原子を伴う0 から 5 の原子リンカー基である
方法。
【請求項37】
請求項36の方法であって、
ここで、式 I、IIおよびIIIにおいて、
それぞれ、R1およびR3 は、アルキル、置換されたアルキル、アルケニル、置換されたアルケニル、アルキニル、置換されたアルキニル、フェニル、置換されたフェニル、アリール、置換された アリール、ヘテロアリールまたは置換されたヘテロアリール 基であり、
R2 は、水素またはハロゲン、ヒドロキシル、スルフヒドリル、アルコキシ、カルボキシル、ニトロ、シアノ、アミノ、アミドまたはスルホニル 基または何れかの数の炭素原子を含む何れか他の有機官能基であり、または
その組み合わせ、および
ここで、式 IVにおいて、R4またはR5における当該アミノ基は、脂肪族、芳香族またはヘテロ環基に置換されている方法。
【請求項38】
請求項37の方法であって、ここで、式 I、IIおよびIIIにおいて、R1またはR3における当該置換されたアルキル、アルケニル、アルキニル、フェニル、アリールまたはヘテロアリール基は、ハロ、ヒドロキシル、スルフヒドリル、アルコキシ、カルボキシル、カルボニル、ニトロ、シアノ、アミノ、アミド、スルホニル,または置換された ヘテロ環 基により置換されたアルキル、アルケニル、アルキニル、フェニル、アリールまたはヘテロアリール基である方法。
【請求項39】
請求項36の方法であって、ここで、当該化合物が、AV1-11、AV14-15、AV17-32,およびAV34-38からなる群より選択される方法。
【請求項40】
請求項34の方法であって、ここで、当該インテグリンはαvβ3である方法。
【請求項41】
請求項34の方法であって、ここで、当該対象が動物またはヒトである方法。
【請求項42】
請求項34の方法であって、ここで、当該対象が、癌、良性腫瘍、炎症性疾患、免疫または自己免疫疾患、血管性疾患、病理学的骨再吸収に主に関連する疾患またはウイルス感染に感染する、またはそれらの発展のリスクに曝されている方法。
【請求項43】
請求項42の方法であって、ここで、
当該癌が、悪性固形腫瘍、並びに高レベルの血管形成および血管原性因子と関連する血液学的悪性物の組織学的種類からなる群より選択され、
当該良性腫瘍が、高い血管新生(高い血管新生)に関連する良性腫瘍および新生物発生前状態、尋常性疣贅、化膿性 肉芽腫、酒さ、ケロイド瘢痕、脈絡叢または虹彩新血管新生に関連する眼部腫瘍からなる群より選択され、
当該炎症性疾患は、炎症性腸疾患、多発性炎症性ミオパシー、他の眼部炎症性疾患、上昇した炎症と関連ある疾患または状態、ざ瘡および特に慢性的な炎症状態に関連する胃腸管疾患からなる群より選択され、
当該免疫または自己免疫疾患は、関節リウマチ、乾癬性関節炎、乾癬、多発性硬化症、気管支喘息およびアレルギー、湿疹および皮膚炎並びに他の皮膚および皮下組織の他の炎症性状態からなる群より選択され、
当該血管性疾患は、子宮内膜症、機能性障害性の子宮性出血、子宮内膜過形成、骨髄線維症、血管腫、動脈硬化症、急性および慢性 移植 拒絶反応および移植 脈管障害、眼部新血管新生により特徴付けられる眼部障害、不適当な血小板活性化、補充、凝集または血栓症により媒介される状態、心筋の血管新生、出血性関節、血管癒着、敗血症、成人呼吸困難症候群、末梢血管拡張および創傷肉芽形成からなる群より選択され、
当該病理学的骨再吸収に主に関連する疾患が、骨粗鬆症、骨のパジェット病,および副甲状腺ホルモン(PTHrP)媒介高カルシウム血症からなる群より選択され、並びに、
当該ウイルス感染が、アデノウイルス(Adenoviridae)、コックサッキーウイルス(Picornaviridae)、エコウイルス(Picornaviridae)、口蹄疫ウイルス(Picornaviridae)、ハンタウイルス(Buriyaviridae)、ヒトパレコウイルス(Picornaviridae)、ヒト免疫不全ウイルス 1(Retroviridae),およびロタウイルス(Reoviridae)からなる群より選択されるウイルスによる感染である
方法。
【請求項44】
請求項43の方法であって、ここで、
当該悪性固形腫瘍の組織学的種類は、肉腫および癌腫からなる群より選択され、
当該高レベルの血管形成および血管原性因子に関連する血液学的悪性物は、急性および慢性白血病、多発性骨髄腫、骨髄増殖性疾患、非ホジキンリンパ腫およびホジキン病からなる群より選択され、
当該炎症性腸疾患は、クローン病および潰瘍性大腸炎からなる群より選択され、
当該多発性炎症性ミオパシーは、多発性筋炎(PM)、皮膚筋炎(DM),および高い血管新生に関連する状態からなる群より選択され、
増大した炎症に関連する疾患または状態は、歯肉炎および粘膜炎からなる群より選択され、
当該特に慢性的な炎症状態に関連する胃腸管疾患は、胃炎および十二指腸炎からなる群より選択され、
当該皮膚および皮下組織の他の炎症性状態は、紅斑性扁平皮膚症、斑不全角化、脂漏性皮膚炎、頭部脂漏症、他の脂漏性皮膚炎、アトピー性皮膚炎および関連状態、紅斑、ベニエ痒疹、神経皮膚炎、接触性皮膚炎および他の湿疹、水疱性皮膚疾患、紅斑性状態、苔癬、掻痒症および関連する状態、疱疹状皮膚炎、疱疹状皮膚症、デューリング病、疱疹状皮膚炎、角層下膿疱症、スネッドン-ウィルキンソン病、若年性疱疹状皮膚炎、若年性天疱瘡、疱疹状膿痂疹、多巣性紅斑、紅斑虹彩、疱疹虹彩、ライル症候群、エリトマトーデス、他の特定の紅斑性状態、苔癬化およびハイド病からなる群より選択される
当該眼部新血管新生により特徴付けられる眼部障害は、加齢関連横斑性変性(AMD)、増殖性糖尿病網膜症(PDR)、未熟児網膜症(ROP)、ネオ血管 緑内障、網膜芽細胞腫、水晶体後線維増殖症、ルベオーシス、ブドウ膜炎、横斑性変性および角膜移植新血管新生からなる群より選択され、並びに、
当該不適当な血小板活性化、補充、凝集または血栓症により媒介される状態は、冠動脈疾患または障害、心筋梗塞または心筋梗塞に続く障害、脳卒中、不安定狭心症、子癇前症、塞栓症、血小板関連虚血性疾患、経皮的冠動脈インターベンションに続く再狭窄、血栓性疾患、血栓症に続く再閉塞、深在性静脈血栓症(DVT)、肺塞栓症(PE)、一過性虚血性発作(TIAs)および血管閉塞が共通する基本をなす特徴である他の状態からなる群より選択される
方法。
【請求項45】
請求項44の方法であって、ここで、
当該粘膜炎は、放射線または化学療法誘導粘膜炎であり、
当該接触性皮膚炎および他の湿疹は、内部に取り込まれた物質のための皮膚炎、皮膚との接触における化学薬品、薬物および医薬品のための皮膚炎、日射のための皮膚炎、日焼け、ベルロック皮膚炎、光アレルギー反応皮膚炎、光毒性反応皮膚炎、多形性 光性発疹、播種性浅在性光線性汗孔角化症(DSAP)、化粧品のための皮膚炎、赤外線のための皮膚炎、紫外線のための皮膚炎および電離放射線による治療のための皮膚炎からなる群より選択され、
当該他の特定の紅斑性状態は、リッター病、新生児剥脱性皮膚炎および紅斑間擦疹からなる群より選択され、
当該血小板関連虚血性疾患は、肺虚血、冠血管虚血,および脳虚血からなる群より選択され、
当該経皮的冠動脈インターベンションは、血管形成術、アテレクトミー、ステント設置およびバイパス手術からなる群より選択され、並びに、
当該血栓性疾患は、冠動脈血栓症、脳動脈血栓症、心臓内血栓症、末梢動脈血栓症、静脈血栓症,および血栓症および外来性に対する露出または損傷された組織表面に関連する凝固障害からなる群より選択される
方法。
【請求項46】
請求項34の方法であって、ここで、当該化合物が非経口、皮内、皮下、経口、経皮、経粘膜または直腸に投与される方法。
【請求項47】
請求項34の方法であって、更に、当該対象に、有効量の1または1以上の治療剤、イメージング剤またはその組み合わせを投与することを具備する方法。
【請求項48】
請求項47の方法であって、ここで、当該薬剤が、
血管増殖阻害剤、
関節リウマチを治療するための治療剤、
骨転移を治療するための治療剤、
骨粗鬆症を治療するための治療剤、
再狭窄を治療するための治療剤、
血管イメージング剤、または
その組み合わせである
方法。
【請求項49】
請求項48の方法であって、ここで、
当該血管増殖阻害剤が、TNF-アルファ、アンジオスタチン、ベバシザン、アレステン、カンスタチン、コンブレタスタチン、エンドスタチン、NM-3、サリドマイド、スロンボスポンジン、タムスタチン、2-メトキシエストラジオールおよびビタキシンからなる群より選択され、
当該骨粗鬆症を治療するための治療剤が、ホセマックス、アレジア、アクトネル、ジンドロネル、コメタ、プレマリン、クリマラ、エストラ、ビベラ、エストラダーム、プレモプロ、プレムフェーズ、フェマート、プレフェスト、コンビパッチ、エビスタ、カルシトニン、ミアカルシン,およびカルシトニンからなる群より選択され、および
当該イメージング剤が、放射性核種である、
方法。
【請求項50】
請求項47の方法であって、ここで、当該化合物および当該薬剤が同時にまたは経時的に投与される方法。
【請求項51】
請求項47の方法であって、ここで、当該化合物が少なくとも1の当該薬剤が混合される方法。
【請求項52】
請求項47の方法であって、ここで、当該化合物は、少なくとも1の当該薬剤に対して実施可能に連結される方法。
【請求項53】
請求項47の方法であって、ここで、当該化合物または当該薬剤が非経口、皮内、皮下、経口、経皮的、経粘膜的または直腸に投与される方法。
【請求項54】
請求項34の方法であって、ここで、当該対象が血管形成処置、放射線治療または化学療法で治療される方法。
【請求項55】
請求項54の方法であって、ここで、
当該血管形成方法は、バルーン血管形成術、レーザー血管形成術、冠血管アテレクトミーまたは同様の技術、頚動脈動脈膜切除、血管グラフトの吻合、血栓形成の高リスクを有する外科手術、アテレクトミー、ステント設置、慢性心血管装置の設置、臓器移植およびバイパス手術からなる群より選択され、および
当該対象に対する有効量の薬剤の投与を含む化学療法は、シスプラチン、ドキソルビシン、ビンクリスチン、シクロホスファミド、トポテカン、パクリタキセル,および他の化学療法剤からなる群より選択される
方法。
【請求項56】
請求項55の方法であって、ここで、血栓形成の高リスクにある手術が、冠血管バイパス手術および補綴バルブまたは血管の挿入からなる群より選択され、並びに、
当該慢性心血管装置が、インドルエリングカテーテル、補綴バルブまたは血管である
方法。
【請求項57】
ファルマコフォアの表示を具備するコンピュータ読み取り可能媒体 であって、ここで、当該ファルマコフォアは、1のH結合供与体(HBD)、1のH結合受容体(HBA)、2の疎水性芳香族基(HAR1およびHAR2)および1の陰性イオン性基(NI)の特徴を含み、および、ここで、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Aに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.81±1 Åであり、当該HBDと HBAの距離は7.52±1 Åであり、当該HBDとNI の距離は17.67±1 Åであり、当該HRA1とHBAの距離は7.52±1 Åであり、当該HRA1とHRA2の距離は9.41±1 Åであり、当該HBAとNIの距離は10.20±1 Åであり、および当該HRA2とNIの距離は5.88±1 Åであるか、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Bに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.80±1 Åであり、当該HBDとHBAの距離は 7.34±1 Åであり、当該HBDとNI の距離は16.92±1 Åであり、当該HRA1とHBAの距離は5.20±1 Åであり、当該HRA1とNI の距離は14.34±1 Åであり、当該HBAとHRA2の距離は4.69±1 Åであり、当該HBAとNIの距離は 9.85±1 Åであり、および当該HRA2とNI の距離は5.66±1 Åであるか、または、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Cに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.79±1 Åであり、当該HBDとHRA2の距離は11.94±1 Åであり、当該HBDとNI の距離は16.28±1 Åであり、当該HRA1とHBA の距離は5.20±1 Åであり、当該HRA1とNI の距離は14.64±1 Åであり、当該HBAとHRA2 の距離は4.72±1 Åであり、当該HRA2とNI の距離は5.63±1 Åである
媒体。
【請求項58】
請求項57のコンピュータ読み取り可能媒体であって、ここで、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Aに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.81 Åであり、当該HBDとHBAの距離は7.52 Åであり、当該HBDとNIの距離は17.67 Åであり、当該HRA1とHBAの距離は7.52 Åであり、当該HRA1とHRA2の距離は9.41 Åであり、当該HBAとNIの距離は10.20 Åであり、および当該HRA2とNIの距離は5.88 Åであるか
当該HBD、HBA、HAR1、HAR2,およびNIは、図8Bに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.80 Åであり、当該HBDとHBAの距離は7.34 Åであり、当該HBDとNIの距離は16.92 Åであり、当該HRA1とHBAの距離は5.20 Åであり、当該HRA1とNIの距離は14.34 Åであり、当該HBAとHRA2の距離は4.69 Åであり、当該HBAとNIの距離は9.85 Åであり、および当該HRA2とNI の距離は5.66 Åであるか、または
当該HBD、HBA、HAR1、HAR2,およびNIは、図8Cに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.79 Åであり、当該HBDとHRA2の距離は11.94 Åであり、当該HBDとNI の距離は16.28 Åであり、当該HRA1とHBAの距離は5.20 Åであり、当該HRA1とNIの距離は14.64 Åであり、当該HBAとHRA2の距離は4.72 Åであり、および当該HRA2とNIの距離は5.63 Åである、
媒体。
【請求項59】
以下を具備するインテグリン結合小分子を同定する方法:
化合物の三次元構造とファルマコフォアの三次元構造とを比較すること、ここで、当該ファルマコフォアは、 includes features of 1のH結合供与体(HBD)、1のH結合受容体(HBA)、2の疎水性芳香族基(HAR1およびHAR2)および1の陰性イオン性基(NI)の特徴を含み、ここで、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Aに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.81±1 Åであり、当該HBDとHBAの距離は7.52±1 Åであり、当該HBDとNIの距離は17.67±1 Åであり、当該HRA1とHBAの距離は7.52±1 Åであり、当該HRA1とHRA2の距離は9.41±1 Åであり、当該HBAとNIの距離は10.20±1 Åであり、および当該HRA2とNIの距離は5.88±1 Åであるか、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Bに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.80±1 Åであり、当該HBDとHBAの距離は7.34±1 Åであり、当該HBDとNIの距離は16.92±1 Åであり、当該HRA1とHBAの距離は5.20±1 Åであり、当該HRA1とNIの距離は14.34±1 Åであり、当該HBAとHRA2の距離は4.69±1 Åであり、当該HBAとNI の距離は9.85±1 Åであり、および当該HRA2とNIの距離は5.66±1 Åであるか、または
当該HBD、HBA、HAR1、HAR2およびNIは、図8Cに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.79±1 Åであり、当該HBDとHRA2の距離は11.94±1 Åであり、当該HBDとNIの距離は16.28±1 Åであり、当該HRA1とHBAの距離は5.20±1 Åであり、当該HRA1とNIの距離は14.64±1 Åであり、当該HBAとHRA2の距離は4.72±1 Åであり、および当該HRA2とNIの距離は5.63±1 Åであり; 並びに
当該化合物がファルマコフォアの特徴と一致する場合には、当該化合物を選択すること。
【請求項60】
請求項59の方法であって、ここで、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Aに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.81 Åであり、当該HBDとHBA の距離は7.52 Åであり、当該HBDとNIの距離は17.67 Åであり、当該HRA1とHBA の距離は7.52 Åであり、当該HRA1とHRA2の距離は9.41 Åであり、当該HBAとNI の距離は10.20 Åであり、および当該HRA2とNIの距離は5.88Åであるか、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Bに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.80 Åであり、当該HBDとHBAの距離は7.34 Åであり、当該HBDとNIの距離は16.92 Åであり、当該HRA1とHBAの距離は5.20 Åであり、当該HRA1とNIの距離は14.34 Åであり、当該HBAとHRA2の距離は4.69 Åであり、当該HBAとNIの距離は9.85 Åであり、および当該HRA2とNIの距離は5.66 Åであるか、または、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Cに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.79 Åであり、当該HBDとHRA2の距離は11.94 Åであり、当該HBDとNIの距離は16.28 Åであり、当該HRA1とHBAの距離は5.20 Åであり、当該HRA1とNIの距離は14.64 Åであり、当該HBAとHRA2の距離は4.72 Åであり、および当該HRA2とNI の距離は5.63 Åである
方法。
【請求項1】
化合物、またはその薬学的若しくは薬用化粧品学的に許容される塩、溶媒和物若しくは水和物を含む組成物であって、当該化合物が1のH結合供与体(HBD)、1のH結合受容体(HBA)、2の疎水性芳香族基(HAR1およびHAR2)、および1の陰性イオン性基(NI)を含み、
ここで、
当該HBD、HBA、HAR1、HAR2および NIが、図8Aに従う立体配置であり、当該HBDとHRA1の距離が2.81±1Åであり、当該HBDとHBAの距離が7.52±1Åであり、当該HBDとNIの距離が17.67±1Åであり、当該HRA1とHBAの距離が7.52±1Åであり、当該HRA1とHRA2の距離が9.41±1Åであり、当該HBAとNIの距離が10.20±1Åであり、当該HRA2とNIの距離が5.88±1Åであるか、
当該HBD、HBA、HAR1、HAR2およびNIは図8Bに従う立体配置であり、当該HBDとHRA1の距離が2.80±1Åであり、当該HBDとHBAの距離が7.31±1Åであり、当該HBDとNIの距離が16.92±1Åであり、当該HRA1とHBAの距離が5.20±1Åであり、当該HRA1とNIの距離が14.34±1Åであり、当該HBAとHRA2の距離が4.69±1であり、当該HBAとNIの距離が9.85±1Åであり、当該HRA2とNIの距離が5.66±1Åであるか、または
当該HBD、HBA、HAR1、HAR2およびNIは図8Cの立体配置であり、当該HBDとHRA1の距離が2.79±1Åであり、当該HBDとHRA2の距離が11.94±1Åであり、当該HBDとNIの距離が16.28±1Åであり、当該HRA1とHBAの距離が5.20±Åであり、当該HRA1とNIの距離が14.64±1Åであり、当該HBAとHRA2の距離が4.72±1Åであり、当該HRA2とNIの距離が5.63±1Åであり、
および当該化合物は、化合物A、BまたはC
【化1】
ではない
組成物。
【請求項2】
請求項1の組成物であって、
ここで、
当該 HBD、HBA、HAR1、HAR2およびNIは、図8Aに従う立体配置であり、当該HBDとHRA1の距離は 2.81 Åであり、当該 HBDとHBAの距離は 7.52 Åであり、当該HBDとNIの距離は17.67 Åであり、当該HRA1とHBAの距離は 7.52 Åであり、当該HRA1とHRA2 の距離は9.41 Åであり、当該HBAとNIの距離は10.20 Åであり、および当該HRA2と NI の距離は5.88 Åであるか、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Bに従う立体配置であり、図8Bに従う立体配置であり、ここで、当該 HBDとthe HRA1 の距離は2.80 Åであり、当該 HBDとthe HBA の距離は7.34 Åであり、当該HBDとNI の距離は16.92 Åであり、当該 HRA1とHBAの距離は 5.20 Åであり、当該HRA1と NI の距離は14.34 Åであり、当該HBAとHRA2 の距離は4.69 Åであり、当該HBAとNI の距離は9.85 Åであり、および当該HRA2とNI の距離は5.66 Åであるか、または
当該 HBD、HBA、HAR1、HAR2およびNIは、図8Cに従う立体配置であり、ここで、当該HBDと HRA1 の距離は2.79 Åであり、当該HBDと HRA2 の距離は11.94 Åであり、当該HBDと NI の距離は 16.28 Åであり、当該HRA1と HBA の距離は5.20 Åであり、当該HRA1とNI の距離は14.64 Åであり、当該HBAと HRA2 の距離は4.72 Åでありおよび当該HRA2とNI の距離は5.63 Åである
組成物。
【請求項3】
請求項1の化合物であって、ここで、当該化合物は、式 I、II、IIIまたはIVの化合物であって、
【化2】
ここで、式 I、IIおよびIIIにおいて
それぞれ、R1およびR3 は、脂肪族、芳香族またはヘテロ環基であり、
R2 は置換基であり、および
X は N、OまたはSであり、並びに
【化3】
ここで、式 IVにおいて、
それぞれ、R1およびR2 は水素またはハロゲン、ヒドロキシル、スルフヒドリル、アルコキシ、アリールオキシ、カルボキシル、ニトロ、シアノ、アミノ、アミドまたはスルホニル基、または何れかの数の炭素原子を含む何れか他の有機官能基であり、
R3 は、水素、ヒドロキシル、アルコキシ、アリールオキシ、芳香族またはヘテロ環基または何れかの数の炭素原子の脂肪族鎖であり、
それぞれ、R4およびR5 はヒドロキシルまたはアミノ基であり、
X は C、N、O,またはSであり,
m は、炭素、窒素、酸素または硫黄原子を伴う0 〜 3 原子リンカー基であり、および
n は、炭素、窒素、酸素または硫黄原子を伴う0 〜 5 原子リンカー基である
化合物。
【請求項4】
請求項3の組成物生物であって、
ここで、式 I、IIおよびIIIにおいて、
それぞれR1およびR3 は、アルキル、置換されたアルキル、アルケニル、置換されたアルケニル、アルキニル、置換されたアルキニル、フェニル、置換されたフェニル、アリール、置換されたアリール、ヘテロアリールまたは置換されたヘテロアリール基であり、
R2 は、水素またはハロゲン、ヒドロキシル、スルフヒドリル、アルコキシ、カルボキシル、ニトロ、シアノ、アミノ、アミドまたはスルホニル基または何れかの数の炭素原子を含む何れか他の有機官能基、または
その組み合わせであり、並びに
ここで、式 IVにおいて、R4またはR5 における当該アミノ基は、脂肪族、芳香族またはヘテロ環基で置換されている
組成物。
【請求項5】
請求項4の組成物であって、ここで、式 I、IIおよびIIIにおいて、R1またはR3における当該置換されたアルキル、アルケニル、アルキニル、フェニル、アリールまたはヘテロアリール基は、ハロ、ヒドロキシル、スルフヒドリル、アルコキシ、カルボキシル、カルボニル、ニトロ、シアノ、アミノ、アミド、スルホニルまたは置換されたヘテロ環で置換されたアルキル、アルケニル、アルキニル、フェニル、アリールまたはヘテロアリール基である組成物。
【請求項6】
請求項3の組成物であって、ここで、当該化合物がAV1-11、AV14-15、AV17-32,およびAV34-38からなる群より選択される組成物。
【請求項7】
請求項1の組成物であって、更に薬学的にまたは薬用化粧品学的に許容される担体を含む組成物。
【請求項8】
第一の化合物または、その薬学的または薬用化粧品学的に許容される塩、溶媒和物または水和物、および1または1以上の第二の化合物を含む組成物であって、ここで、
第一の化合物は、1の H結合供与体(HBD)、1の H結合受容体(HBA)、2の疎水性芳香族基s(HAR1およびHAR2),および1の陰性イオン性基(NI)を含み、ここで、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Aに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.81±1 Åであり、当該HBDとHBAの距離は 7.52±1Åであり、当該 HBDとNIの距離は17.67±1 Åであり、当該HRA1とHBA の距離は、7.52±1 Åであり、当該HRA1とHRA2の距離は9.41±1 Åであり、当該HBAとthe NI の距離は10.20±1 Åであり、および当該HRA2とNI の距離は5.88±1 Åであるか、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Bに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.80±1 Åであり、当該HBDとthe HBA の距離は7.34±1 Åであり、当該HBDとNIの距離は16.92±1 Åであり、当該HRA1とthe HBA の距離は5.20±1 Åであり、当該HRA1とNI の距離は14.34±1 Åであり、当該HBAとthe HRA2 の距離は4.69±1 Åであり、当該HBAとNI の距離は 9.85±1 Åであり、および当該HRA2と NIの距離は5.66±1 Åであるか、または
当該 HBD、HBA、HAR1、HAR2およびNIは、図8Cに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.79±1 Åであり、当該HBDと HRA2の距離は11.94±1 Åであり、当該HBDと NI の距離は16.28±1 Åであり、当該HRA1と HBA の距離は 5.20±1 Åであり、当該HRA1とNI の距離は14.64±1 Åであり、当該HBAとthe HRA2 の距離は 4.72±1Åであり、および当該HRA2と NI の距離は5.63±1 Åであり、
並びに、ここで、第一の化合物は化合物 A、BまたはCではなく、並びに
第二の化合物は、治療剤、イメージング剤またはその組み合わせである
組成物。
【請求項9】
請求項8の組成物であって、ここで、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Aに従う立体配置であり、ここで、 当該HBDとHRA1の距離は 2.81 Åであり、当該HBDと HBAの距離は7.52 Åであり、当該HBDとNI の距離は17.67 Åであり、当該HRA1とHBAの距離は 7.52 Åであり、当該HRA1とHRA2の距離は9.41 Åであり、当該HBAと NIの距離は10.20 Åであり、および当該HRA2とNI の距離は5.88 Åであるか、
当該 HBD、HBA、HAR1、HAR2およびNIは、図8Bに従う立体配置であり、ここで、 当該HBDとHRA1の距離は2.80 Åであり、当該HBDと HBAの距離は7.34 Åであり、当該HBDと NI の距離は16.92 Åであり、当該HRA1と HBA の距離は5.20 Åであり、当該HRA1とNI の距離は14.34 Åであり、当該HBAと HRA2 の距離は 4.69 Åであり、当該HBAとNI の距離は 9.85 Åであり、および当該HRA2とNI の距離は5.66 Åであるか、または、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Cに従う立体配置であり、ここで、 当該HBDとHRA1の距離は2.79 Åであり、当該HBDと HRA2の距離は11.94 Åであり、当該HBDとNI の距離は16.28 Åであり、当該HRA1と HBA の距離は5.20 Åであり、当該HRA1とNI の距離は14.64 Åであり、当該HBAとHRA2の距離は 4.72 Åであり、および当該HRA2とNI の距離は5.63 Åである
組成物。
【請求項10】
請求項8の組成物であって、ここで、第一の化合物は式 I、II、IIIまたはIVであり、
ここで、式 I、IIおよびIIIにおいて、
それぞれ、R1およびR3 は脂肪族、芳香族またはヘテロ環基であり、
R2 は置換基であり、および
X は N、OまたはSであり、および
ここで、式 IVにおいて、
それぞれ、R1およびR2 は水素またはハロゲン、ヒドロキシル、スルフヒドリル、アルコキシ、アリールオキシ、カルボキシル、ニトロ、シアノ、アミノ、アミドまたはスルホニル基または何れかの数の炭素原子を含む何れか他の有機官能基であり,
R3は、水素、ヒドロキシル、アルコキシ、アリールオキシ、芳香族またはヘテロ環基、または何れかの数の炭素原子の脂肪族鎖であり、
それぞれ、R4およびR5 はヒドロキシルまたはアミノ基であり、
X は C、N、OまたはSであり、
m は、炭素、窒素、酸素または硫黄原子を伴う 0 から 3 原子リンカー基であり、および
n は、炭素、窒素、酸素または硫黄原子を伴う 0 から 5 原子リンカー 基である
組成物。
【請求項11】
請求項10の組成物であって、
ここで、式 I、IIおよびIIIにおいて、
それぞれ、R1およびR3 はアルキル、置換されたアルキル、アルケニル、置換されたアルケニル、アルキニル、置換されたアルキニル、フェニル、置換されたフェニル、アリール、置換されたアリール、ヘテロアリールまたは置換されたヘテロアリール基であり、
R2 は、水素またはハロゲン、ヒドロキシル、スルフヒドリル、アルコキシ、カルボキシル、ニトロ、シアノ、アミノ、アミドまたはスルホニル基、または何れかの数の炭素原子を含む何れか他の有機官能基、またはその組み合わせであり、および
ここで、式 IVにおいて、R4またはR5 における当該アミノ基は脂肪族、芳香族,またはヘテロ環基で置換されている
組成物。
【請求項12】
請求項11の組成物であって、ここで、式s I、IIおよびIIIにおいて、R1またはR3における当該置換されたアルキル、アルケニル、アルキニル、フェニル、アリールまたはヘテロアリール基は、ハロ、ヒドロキシル、スルフヒドリル、アルコキシ、カルボキシル、カルボニル、ニトロ、シアノ、アミノ、アミド、スルホニルまたは置換されたヘテロ環基で置換されたアルキル、アルケニル、アルキニル、フェニル、アリールまたはヘテロアリール基である組成物。
【請求項13】
請求項10の組成物であって、ここで、当該第一の化合物がAV1-11、AV14-15、AV17-32,およびAV34-38からなる群より選択される組成物。
【請求項14】
請求項8の組成物であって、ここで、当該第二の化合物は、
血管増殖阻害剤,
関節リウマチを治療するための治療剤、
放射線治療剤、
化学療法剤、
骨転移の治療のための治療剤、
骨粗鬆症の治療のための治療剤、
再狭窄の治療のための治療剤、
血管イメージング剤、または
その組み合わせ
である組成物。
【請求項15】
請求項14の組成物であって、ここで、
当該血管増殖阻害剤が、TNF-アルファ、アンジオスタチン、ベバシザン、アレステン、カンスタチン、コンブレタスタチン、エンドスタチン、NM-3、サリドマイド、スロンボスポンジン、タムスタチン、2-メトキシエストラジオール,およびビタキシンからなる群より選択され、
当該化学療法剤が、シスプラチン、ドキソルビシン、ビンクリスチン、シクロホスファミド、トポテカン、パクリタキセルおよび他の化学療法剤からなる群より選択され、
骨粗鬆症の治療のための治療剤は、ホセマックス、アレジア、アクトネル、ジンドロネル、コメタ、プレマリン、クリマラ、エストラ、ビベラ、エストラダーム、プレモプロ、プレムフェーズ、フェマート、プレフェスト、コンビパッチ、エビスタ、カルシトニン、ミアカルシン,およびカルシトニンからなる群より選択され、および
当該血管イメージング剤は、放射線核種である
組成物。
【請求項16】
請求項8の組成物であって、ここで、当該組成物は第一の化合物および第二の化合物からなる組成物。
【請求項17】
請求項8の組成物であって、ここで、第一の化合物が実施可能に少なくとも1の第二の化合物に連結されている組成物。
【請求項18】
インテグリンを化合物に対して結合する方法であって、
インテグリンを化合物、またはその薬学的、または薬用化粧品学的に許容される塩、溶媒和物または水和物と接触すること、
ここで、当該化合物は、1のH結合供与体(HBD)、1のH結合受容体(HBA)、2の疎水性芳香族基(HAR1およびHAR2)および1の陰性イオン性基(NI)を含み、ここで、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Aに従う立体配置であり、ここで、当該HBDと HRA1の距離は2.81±1 Åであり、当該HBDとHBA の距離は7.52±1 Åであり、当該HBDと NI の距離は17.67±1 Åであり、当該HRA1とHBAの距離は7.52±1 Åであり、当該HRA1とHRA2 の距離は 9.41±1 Åであり、当該HBAとNIの距離は10.20±1 Åであり、および当該HRA2とNI の距離は5.88±1 Åであるか、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Bに従う立体配置であり、ここで、当該HBDと HRA1の距離は2.80±1 Åであり、当該HBDとHBAの距離は7.34±1 Åであり、当該HBDとNI の距離は16.92±1 Åであり、当該HRA1とHBAの距離は 5.20±1 Åであり、当該HRA1とNI の距離は14.34±1 Åであり、当該HBAとHRA2の距離は4.69±1 Åであり、当該HBAとNI の距離は9.85±1 Åであり、および当該 HRA2とNIの距離は 5.66±1 Åであるか、または、
当該 HBD、HBA、HAR1、HAR2およびNIは、図8Cに従う立体配置であり、ここで、当該HBDと HRA1 の距離は2.79±1 Åであり、当該HBDと HRA2 の距離は11.94±1 Åであり、当該HBDと NI の距離は16.28±1 Åであり、当該HRA1と HBAの距離は 5.20±1 Åであり、当該HRA1と NI の距離は14.64±1 Åであり、当該HBAとHRA2 の距離は4.72±1 Åであり、および当該HRA2と NI の距離は5.63±1 Åであり、
並びに、ここで、当該化合物は化合物 A、BまたはCではない、
を具備する方法。
【請求項19】
請求項18の方法であって、ここで、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Aに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.81 Åであり、当該HBDと HBA の距離は7.52 Åであり、当該HBDと NI の距離は17.67 Åであり、当該HRA1とHBA の距離は7.52 Åであり、当該HRA1と HRA2 の距離は9.41 Åであり、当該HBAとNI の距離は10.20 Åであり、および当該HRA2とNI の距離は5.88 Åであるか、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Bに従う立体配置であり、ここで、当該HBDとHRA1 の距離は2.80 Åであり、当該HBDと HBAの距離は7.34 Åであり、当該HBDと NI の距離は16.92 Åであり、当該HRA1と HBA の距離は5.20 Åであり、当該HRA1と NIの距離は14.34 Åであり、当該HBAと HRA2 の距離は4.69 Åであり、当該HBAとNI の距離は9.85 Åであり、および当該HRA2とNIの距離は5.66 Åであるか、または、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Cに従う立体配置であり、ここで、当該HBDとHRA1 の距離は2.79 Åであり、当該HBDとHRA2 の距離は11.94 Åであり、当該HBDとNI の距離は16.28 Åであり、当該HRA1と HBA の距離は5.20 Åであり、当該HRA1とNI の距離は14.64 Åであり、当該HBAとHRA2の距離は4.72 Åであり、および当該HRA2とNI の距離は5.63 Åである
方法。
【請求項20】
請求項18の方法であって、ここで、当該化合物が、式 I、II、IIIまたはIVの化合物であり、
ここで、式 I、IIおよびIIIにおいて、
それぞれ、R1およびR3 は脂肪族、芳香族またはヘテロ環基であり、
R2 は置換基であり、および
X は N、OまたはSであり、および
ここで、式 IVにおいて、
それぞれ、R1およびR2 は、水素またはハロゲン、ヒドロキシル、スルフヒドリル、アルコキシ、アリールオキシ、カルボキシル、ニトロ、シアノ、アミノ、アミドまたはスルホニル基または何れかの数の炭素原子を含む何れか他の有機官能基であり、
R3 は、水素、ヒドロキシル、アルコキシ、アリールオキシ、芳香族またはヘテロ環基、または何れかの数の炭素原子の脂肪族鎖であり、
それぞれR4およびR5 はヒドロキシルまたはアミノ基であり、
X は C、N、OまたはSであり、
m は、炭素、窒素、酸素または硫黄原子を伴う 0 から 3 原子リンカー基であり、および
n は、炭素、窒素、酸素または硫黄原子を伴う 0 から 5 原子リンカー基である
方法。
【請求項21】
請求項20の方法であって、
ここで、式I、IIおよびIIIにおいて、
それぞれ、R1およびR3 は、アルキル、置換されたアルキル、アルケニル、置換されたアルケニル、アルキニル、置換されたアルキニル、フェニル、置換されたフェニル、アリール、置換されたアリール、ヘテロアリールまたは置換されたヘテロアリール 基であり、
R2 は、水素またはハロゲン、ヒドロキシル、スルフヒドリル、アルコキシ、カルボキシル、ニトロ、シアノ、アミノ、アミドまたはスルホニル基または何れかの数の炭素原子を含む何れか他の有機官能基、または
その組み合わせ、および
ここで、式 IVにおいて、R4またはR5 における当該アミノ基が脂肪族、芳香族またはヘテロ環基で置換された
方法。
【請求項22】
請求項21の方法であって、ここで、式 I、IIおよびIIIにおいて、R1またはR3における当該置換されたアルキル、アルケニル、アルキニル、フェニル、アリールまたはヘテロアリール基は、ハロ、ヒドロキシル、スルフヒドリル、アルコキシ、カルボキシル、カルボニル、ニトロ、シアノ、アミノ、アミド、スルホニルまたは置換されたヘテロ環基によって置換されたアルキル、アルケニル、アルキニル、フェニル、アリールまたはヘテロアリール基である
方法。
【請求項23】
請求項20の方法であって、ここで、当該化合物がAV1-11、AV14-15、AV17-32,およびAV34-38からなる群より選択される方法。
【請求項24】
請求項18の方法であって、ここで、当該インテグリンがαvβ3である方法。
【請求項25】
請求項18の方法であって、ここで、当該インテグリンが細胞上にある方法。
【請求項26】
請求項25の方法であって、ここで、当該細胞が癌細胞、良性腫瘍細胞、間質細胞、炎症に反応する、炎症を媒介する、または炎症を調節する細胞、免疫系の細胞、血管機能に関与する細胞、骨芽細胞、破骨細胞またはウイルスに感染した細胞.
【請求項27】
請求項26の方法であって、ここで、
当該炎症に対して反応する、炎症を媒介する、若しくは炎症を調節する細胞、または免疫系の細胞が、白血球、マクロファージおよび樹状細胞からなる群より選択され、
当該血管機能に関連する細胞が、内皮細胞、内皮細胞に関与する細胞および平滑筋細胞からなる群より選択され、および、
当該ウイルスが、アデノウイルス(Adenoviridae)、コックサッキーウイルス(Picornaviridae)、エコウイルス(Picornaviridae)、口蹄疫ウイルス(Picornaviridae)、ハンタウイルス(Buriyaviridae)、ヒトパレコウイルス(Picornaviridae)、ヒト免疫不全ウイルス 1(Retroviridae),およびロタウイルス(Reoviridae)からなる群より選択される
方法。
【請求項28】
請求項25の方法であって、更に、当該細胞を1または1以上の治療剤、イメージング剤またはその組み合わせと接触させることを具備する方法。
【請求項29】
請求項28の方法であって、前記薬剤が、
血管増殖阻害剤、
関節リウマチを治療するための治療薬、
放射線治療剤、
化学療法剤、
骨転移を治療するための治療剤、
骨粗鬆症を治療するための治療剤、
再狭窄を治療するための治療剤、
血管イメージング剤、または
その組み合わせである
方法。
【請求項30】
請求項29の方法であって、ここで、
当該血管増殖阻害剤が、TNF-アルファ、アンジオスタチン、ベバシザン、アレステン、カンスタチン、コンブレタスタチン、エンドスタチン、NM-3、サリドマイド、スロンボスポンジン、タムスタチン、2-メトキシエストラジオール,およびビタキシンからなる群より選択され、
当該化学療法剤が、シスプラチン、ドキソルビシン、ビンクリスチン、シクロホスファミド、トポテカン、パクリタキセル,および他の化学療法剤からなる群より選択され、
当該骨粗鬆症を治療するための治療剤が、ホセマックス、アレジア、アクトネル、ジンドロネル、コメタ、プレマリン、クリマラ、エストラ、ビベラ、エストラダーム、プレモプロ、プレムフェーズ、フェマート、プレフェスト、コンビパッチ、エビスタ、カルシトニン、ミアカルシン,およびカルシトニンからなる群より選択され、および
当該血管イメージング剤が、放射性核種である
方法。
【請求項31】
請求項28の方法であって、ここで、当該インテグリンおよび当該細胞が、同時にまたは経時的に接触される方法。
【請求項32】
請求項28の方法であって、ここで、当該化合物が、少なくとも1の薬剤と混合される方法。
【請求項33】
請求項28の方法であって、ここで、 当該化合物が、少なくとも1の薬物に対して実施可能に連結される方法。
【請求項34】
対象においてインテグリンを化合物に対して結合する方法であって、
それを必要とする対象に対して、有効量の化合物、またはその薬学的若しくは薬用化粧品学的に許容される塩、溶媒和物または水和物を投与すること、
ここで、当該化合物は、1のH結合供与体(HBD)、1のH結合受容体(HBA)、2の疎水性芳香族基(HAR1およびHAR2)および1の陰性イオン性基(NI)を含み、ここで、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Aに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.81±1 Åであり、当該HBDとHBA の距離が7.52±1 Åであり、当該HBDとNI の距離は17.67±1 Åであり、当該HRA1と HBAの距離が7.52±1 Åであり、当該HRA1とthe HRA2の距離が9.41±1 Åであり、当該HBAと NI の距離が10.20±1 Åであり、および当該HRA2とNI の距離が5.88±1 Åであるか、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Bに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.80±1 Åであり、当該HBDとHBA の距離は7.34±1 Åであり、当該HBDとNI の距離は16.92±1 Åであり、当該HRA1とHBAの距離は5.20±1 Åであり、当該HRA1とNI の距離は14.34±1 Åであり、当該HBAとHRA2 の距離は4.69±1 Åであり、当該HBAとNI の距離は9.85±1 Åであり、および当該HRA2とNI の距離は5.66±1 Åであるか、または
the HBD、HBA、HAR1、HAR2,およびNIは、図8Cに従う立体配置であり、ここで、当該HBDと HRA1の距離は 2.79±1 Åであり、当該HBDと HRA2 の距離は11.94±1 Åであり、当該HBDと NI の距離は16.28±1 Åであり、当該HRA1とHBAの距離は5.20±1 Åであり、当該HRA1とNIの距離は14.64±1 Åであり、当該HBAとHRA2の距離は 4.72±1 Åであり、および当該HRA2とNI の距離は5.63±1 Åであり、
およびここで、当該化合物は化合物 A、BまたはCではない、
方法。
【請求項35】
請求項34の方法であって、ここで、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Aに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.81 Åであり、当該HBDとHBAの距離は7.52 Åであり、当該HBDとNI の距離は17.67 Åであり、当該HRA1とHBAの距離は 7.52 Åであり、当該HRA1とHRA2の距離は9.41 Åであり、当該HBAとNI の距離は 10.20 Åであり、および当該HRA2とNI の距離は5.88 Åであるか、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Bに従う立体配置であり、ここで、当該HBDとHRA1 の距離は2.80 Åであり、当該HBDと HBAの距離は7.34 Åであり、当該HBDとNI の距離は16.92 Åであり、当該HRA1とHBAの距離は 5.20 Åであり、当該HRA1とNIの距離は14.34 Åであり、当該HBAとHRA2 の距離は4.69 Åであり、当該HBAとNI の距離は9.85 Åであり、および当該HRA2とNI の距離は5.66 Åであるか、または、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Cに従う立体配置であり、ここで、当該HBDとHRA1 の距離は2.79 Åであり、当該HBDとHRA2 の距離は11.94 Åであり、当該HBDとNI の距離は16.28 Åであり、当該HRA1とHBAの距離は5.20 Åであり、当該HRA1とNI の距離は14.64 Åであり、当該HBAとHRA2の距離は4.72 Åであり、および当該HRA2とNI の距離は5.63 Åである
方法。
【請求項36】
請求項34の方法であって、ここで、当該化合物は、式 I、II、IIIまたはIVの化合物であって,
ここで、式s I、IIおよびIIIにおいて、
それぞれ、R1およびR3 は脂肪族、芳香族またはヘテロ環基であり、
R2 は置換基であり、および
X はN、OまたはSであり、および
ここで、式 IVにおいて、
それぞれ、R1およびR2 は水素またはハロゲン、ヒドロキシル、スルフヒドリル、アルコキシ、アリールオキシ、カルボキシル、ニトロ、シアノ、アミノ、アミドまたはスルホニル基または何れかの数の炭素原子を含む何れか他の有機官能基であり、
R3 は水素、ヒドロキシル、アルコキシ、アリールオキシ、芳香族またはヘテロ環基または何れかの数の炭素原子の脂肪族鎖であり、
それぞれ、R4およびR5 はヒドロキシルまたはアミノ基であり、
X は C、N、OまたはSであり、
m は 炭素、窒素、酸素または硫黄原子を伴う0 から 3 の原子リンカー基 with 炭素、窒素、酸素,または硫黄 原子であり、
n は、炭素、窒素、酸素または硫黄原子を伴う0 から 5 の原子リンカー基である
方法。
【請求項37】
請求項36の方法であって、
ここで、式 I、IIおよびIIIにおいて、
それぞれ、R1およびR3 は、アルキル、置換されたアルキル、アルケニル、置換されたアルケニル、アルキニル、置換されたアルキニル、フェニル、置換されたフェニル、アリール、置換された アリール、ヘテロアリールまたは置換されたヘテロアリール 基であり、
R2 は、水素またはハロゲン、ヒドロキシル、スルフヒドリル、アルコキシ、カルボキシル、ニトロ、シアノ、アミノ、アミドまたはスルホニル 基または何れかの数の炭素原子を含む何れか他の有機官能基であり、または
その組み合わせ、および
ここで、式 IVにおいて、R4またはR5における当該アミノ基は、脂肪族、芳香族またはヘテロ環基に置換されている方法。
【請求項38】
請求項37の方法であって、ここで、式 I、IIおよびIIIにおいて、R1またはR3における当該置換されたアルキル、アルケニル、アルキニル、フェニル、アリールまたはヘテロアリール基は、ハロ、ヒドロキシル、スルフヒドリル、アルコキシ、カルボキシル、カルボニル、ニトロ、シアノ、アミノ、アミド、スルホニル,または置換された ヘテロ環 基により置換されたアルキル、アルケニル、アルキニル、フェニル、アリールまたはヘテロアリール基である方法。
【請求項39】
請求項36の方法であって、ここで、当該化合物が、AV1-11、AV14-15、AV17-32,およびAV34-38からなる群より選択される方法。
【請求項40】
請求項34の方法であって、ここで、当該インテグリンはαvβ3である方法。
【請求項41】
請求項34の方法であって、ここで、当該対象が動物またはヒトである方法。
【請求項42】
請求項34の方法であって、ここで、当該対象が、癌、良性腫瘍、炎症性疾患、免疫または自己免疫疾患、血管性疾患、病理学的骨再吸収に主に関連する疾患またはウイルス感染に感染する、またはそれらの発展のリスクに曝されている方法。
【請求項43】
請求項42の方法であって、ここで、
当該癌が、悪性固形腫瘍、並びに高レベルの血管形成および血管原性因子と関連する血液学的悪性物の組織学的種類からなる群より選択され、
当該良性腫瘍が、高い血管新生(高い血管新生)に関連する良性腫瘍および新生物発生前状態、尋常性疣贅、化膿性 肉芽腫、酒さ、ケロイド瘢痕、脈絡叢または虹彩新血管新生に関連する眼部腫瘍からなる群より選択され、
当該炎症性疾患は、炎症性腸疾患、多発性炎症性ミオパシー、他の眼部炎症性疾患、上昇した炎症と関連ある疾患または状態、ざ瘡および特に慢性的な炎症状態に関連する胃腸管疾患からなる群より選択され、
当該免疫または自己免疫疾患は、関節リウマチ、乾癬性関節炎、乾癬、多発性硬化症、気管支喘息およびアレルギー、湿疹および皮膚炎並びに他の皮膚および皮下組織の他の炎症性状態からなる群より選択され、
当該血管性疾患は、子宮内膜症、機能性障害性の子宮性出血、子宮内膜過形成、骨髄線維症、血管腫、動脈硬化症、急性および慢性 移植 拒絶反応および移植 脈管障害、眼部新血管新生により特徴付けられる眼部障害、不適当な血小板活性化、補充、凝集または血栓症により媒介される状態、心筋の血管新生、出血性関節、血管癒着、敗血症、成人呼吸困難症候群、末梢血管拡張および創傷肉芽形成からなる群より選択され、
当該病理学的骨再吸収に主に関連する疾患が、骨粗鬆症、骨のパジェット病,および副甲状腺ホルモン(PTHrP)媒介高カルシウム血症からなる群より選択され、並びに、
当該ウイルス感染が、アデノウイルス(Adenoviridae)、コックサッキーウイルス(Picornaviridae)、エコウイルス(Picornaviridae)、口蹄疫ウイルス(Picornaviridae)、ハンタウイルス(Buriyaviridae)、ヒトパレコウイルス(Picornaviridae)、ヒト免疫不全ウイルス 1(Retroviridae),およびロタウイルス(Reoviridae)からなる群より選択されるウイルスによる感染である
方法。
【請求項44】
請求項43の方法であって、ここで、
当該悪性固形腫瘍の組織学的種類は、肉腫および癌腫からなる群より選択され、
当該高レベルの血管形成および血管原性因子に関連する血液学的悪性物は、急性および慢性白血病、多発性骨髄腫、骨髄増殖性疾患、非ホジキンリンパ腫およびホジキン病からなる群より選択され、
当該炎症性腸疾患は、クローン病および潰瘍性大腸炎からなる群より選択され、
当該多発性炎症性ミオパシーは、多発性筋炎(PM)、皮膚筋炎(DM),および高い血管新生に関連する状態からなる群より選択され、
増大した炎症に関連する疾患または状態は、歯肉炎および粘膜炎からなる群より選択され、
当該特に慢性的な炎症状態に関連する胃腸管疾患は、胃炎および十二指腸炎からなる群より選択され、
当該皮膚および皮下組織の他の炎症性状態は、紅斑性扁平皮膚症、斑不全角化、脂漏性皮膚炎、頭部脂漏症、他の脂漏性皮膚炎、アトピー性皮膚炎および関連状態、紅斑、ベニエ痒疹、神経皮膚炎、接触性皮膚炎および他の湿疹、水疱性皮膚疾患、紅斑性状態、苔癬、掻痒症および関連する状態、疱疹状皮膚炎、疱疹状皮膚症、デューリング病、疱疹状皮膚炎、角層下膿疱症、スネッドン-ウィルキンソン病、若年性疱疹状皮膚炎、若年性天疱瘡、疱疹状膿痂疹、多巣性紅斑、紅斑虹彩、疱疹虹彩、ライル症候群、エリトマトーデス、他の特定の紅斑性状態、苔癬化およびハイド病からなる群より選択される
当該眼部新血管新生により特徴付けられる眼部障害は、加齢関連横斑性変性(AMD)、増殖性糖尿病網膜症(PDR)、未熟児網膜症(ROP)、ネオ血管 緑内障、網膜芽細胞腫、水晶体後線維増殖症、ルベオーシス、ブドウ膜炎、横斑性変性および角膜移植新血管新生からなる群より選択され、並びに、
当該不適当な血小板活性化、補充、凝集または血栓症により媒介される状態は、冠動脈疾患または障害、心筋梗塞または心筋梗塞に続く障害、脳卒中、不安定狭心症、子癇前症、塞栓症、血小板関連虚血性疾患、経皮的冠動脈インターベンションに続く再狭窄、血栓性疾患、血栓症に続く再閉塞、深在性静脈血栓症(DVT)、肺塞栓症(PE)、一過性虚血性発作(TIAs)および血管閉塞が共通する基本をなす特徴である他の状態からなる群より選択される
方法。
【請求項45】
請求項44の方法であって、ここで、
当該粘膜炎は、放射線または化学療法誘導粘膜炎であり、
当該接触性皮膚炎および他の湿疹は、内部に取り込まれた物質のための皮膚炎、皮膚との接触における化学薬品、薬物および医薬品のための皮膚炎、日射のための皮膚炎、日焼け、ベルロック皮膚炎、光アレルギー反応皮膚炎、光毒性反応皮膚炎、多形性 光性発疹、播種性浅在性光線性汗孔角化症(DSAP)、化粧品のための皮膚炎、赤外線のための皮膚炎、紫外線のための皮膚炎および電離放射線による治療のための皮膚炎からなる群より選択され、
当該他の特定の紅斑性状態は、リッター病、新生児剥脱性皮膚炎および紅斑間擦疹からなる群より選択され、
当該血小板関連虚血性疾患は、肺虚血、冠血管虚血,および脳虚血からなる群より選択され、
当該経皮的冠動脈インターベンションは、血管形成術、アテレクトミー、ステント設置およびバイパス手術からなる群より選択され、並びに、
当該血栓性疾患は、冠動脈血栓症、脳動脈血栓症、心臓内血栓症、末梢動脈血栓症、静脈血栓症,および血栓症および外来性に対する露出または損傷された組織表面に関連する凝固障害からなる群より選択される
方法。
【請求項46】
請求項34の方法であって、ここで、当該化合物が非経口、皮内、皮下、経口、経皮、経粘膜または直腸に投与される方法。
【請求項47】
請求項34の方法であって、更に、当該対象に、有効量の1または1以上の治療剤、イメージング剤またはその組み合わせを投与することを具備する方法。
【請求項48】
請求項47の方法であって、ここで、当該薬剤が、
血管増殖阻害剤、
関節リウマチを治療するための治療剤、
骨転移を治療するための治療剤、
骨粗鬆症を治療するための治療剤、
再狭窄を治療するための治療剤、
血管イメージング剤、または
その組み合わせである
方法。
【請求項49】
請求項48の方法であって、ここで、
当該血管増殖阻害剤が、TNF-アルファ、アンジオスタチン、ベバシザン、アレステン、カンスタチン、コンブレタスタチン、エンドスタチン、NM-3、サリドマイド、スロンボスポンジン、タムスタチン、2-メトキシエストラジオールおよびビタキシンからなる群より選択され、
当該骨粗鬆症を治療するための治療剤が、ホセマックス、アレジア、アクトネル、ジンドロネル、コメタ、プレマリン、クリマラ、エストラ、ビベラ、エストラダーム、プレモプロ、プレムフェーズ、フェマート、プレフェスト、コンビパッチ、エビスタ、カルシトニン、ミアカルシン,およびカルシトニンからなる群より選択され、および
当該イメージング剤が、放射性核種である、
方法。
【請求項50】
請求項47の方法であって、ここで、当該化合物および当該薬剤が同時にまたは経時的に投与される方法。
【請求項51】
請求項47の方法であって、ここで、当該化合物が少なくとも1の当該薬剤が混合される方法。
【請求項52】
請求項47の方法であって、ここで、当該化合物は、少なくとも1の当該薬剤に対して実施可能に連結される方法。
【請求項53】
請求項47の方法であって、ここで、当該化合物または当該薬剤が非経口、皮内、皮下、経口、経皮的、経粘膜的または直腸に投与される方法。
【請求項54】
請求項34の方法であって、ここで、当該対象が血管形成処置、放射線治療または化学療法で治療される方法。
【請求項55】
請求項54の方法であって、ここで、
当該血管形成方法は、バルーン血管形成術、レーザー血管形成術、冠血管アテレクトミーまたは同様の技術、頚動脈動脈膜切除、血管グラフトの吻合、血栓形成の高リスクを有する外科手術、アテレクトミー、ステント設置、慢性心血管装置の設置、臓器移植およびバイパス手術からなる群より選択され、および
当該対象に対する有効量の薬剤の投与を含む化学療法は、シスプラチン、ドキソルビシン、ビンクリスチン、シクロホスファミド、トポテカン、パクリタキセル,および他の化学療法剤からなる群より選択される
方法。
【請求項56】
請求項55の方法であって、ここで、血栓形成の高リスクにある手術が、冠血管バイパス手術および補綴バルブまたは血管の挿入からなる群より選択され、並びに、
当該慢性心血管装置が、インドルエリングカテーテル、補綴バルブまたは血管である
方法。
【請求項57】
ファルマコフォアの表示を具備するコンピュータ読み取り可能媒体 であって、ここで、当該ファルマコフォアは、1のH結合供与体(HBD)、1のH結合受容体(HBA)、2の疎水性芳香族基(HAR1およびHAR2)および1の陰性イオン性基(NI)の特徴を含み、および、ここで、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Aに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.81±1 Åであり、当該HBDと HBAの距離は7.52±1 Åであり、当該HBDとNI の距離は17.67±1 Åであり、当該HRA1とHBAの距離は7.52±1 Åであり、当該HRA1とHRA2の距離は9.41±1 Åであり、当該HBAとNIの距離は10.20±1 Åであり、および当該HRA2とNIの距離は5.88±1 Åであるか、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Bに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.80±1 Åであり、当該HBDとHBAの距離は 7.34±1 Åであり、当該HBDとNI の距離は16.92±1 Åであり、当該HRA1とHBAの距離は5.20±1 Åであり、当該HRA1とNI の距離は14.34±1 Åであり、当該HBAとHRA2の距離は4.69±1 Åであり、当該HBAとNIの距離は 9.85±1 Åであり、および当該HRA2とNI の距離は5.66±1 Åであるか、または、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Cに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.79±1 Åであり、当該HBDとHRA2の距離は11.94±1 Åであり、当該HBDとNI の距離は16.28±1 Åであり、当該HRA1とHBA の距離は5.20±1 Åであり、当該HRA1とNI の距離は14.64±1 Åであり、当該HBAとHRA2 の距離は4.72±1 Åであり、当該HRA2とNI の距離は5.63±1 Åである
媒体。
【請求項58】
請求項57のコンピュータ読み取り可能媒体であって、ここで、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Aに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.81 Åであり、当該HBDとHBAの距離は7.52 Åであり、当該HBDとNIの距離は17.67 Åであり、当該HRA1とHBAの距離は7.52 Åであり、当該HRA1とHRA2の距離は9.41 Åであり、当該HBAとNIの距離は10.20 Åであり、および当該HRA2とNIの距離は5.88 Åであるか
当該HBD、HBA、HAR1、HAR2,およびNIは、図8Bに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.80 Åであり、当該HBDとHBAの距離は7.34 Åであり、当該HBDとNIの距離は16.92 Åであり、当該HRA1とHBAの距離は5.20 Åであり、当該HRA1とNIの距離は14.34 Åであり、当該HBAとHRA2の距離は4.69 Åであり、当該HBAとNIの距離は9.85 Åであり、および当該HRA2とNI の距離は5.66 Åであるか、または
当該HBD、HBA、HAR1、HAR2,およびNIは、図8Cに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.79 Åであり、当該HBDとHRA2の距離は11.94 Åであり、当該HBDとNI の距離は16.28 Åであり、当該HRA1とHBAの距離は5.20 Åであり、当該HRA1とNIの距離は14.64 Åであり、当該HBAとHRA2の距離は4.72 Åであり、および当該HRA2とNIの距離は5.63 Åである、
媒体。
【請求項59】
以下を具備するインテグリン結合小分子を同定する方法:
化合物の三次元構造とファルマコフォアの三次元構造とを比較すること、ここで、当該ファルマコフォアは、 includes features of 1のH結合供与体(HBD)、1のH結合受容体(HBA)、2の疎水性芳香族基(HAR1およびHAR2)および1の陰性イオン性基(NI)の特徴を含み、ここで、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Aに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.81±1 Åであり、当該HBDとHBAの距離は7.52±1 Åであり、当該HBDとNIの距離は17.67±1 Åであり、当該HRA1とHBAの距離は7.52±1 Åであり、当該HRA1とHRA2の距離は9.41±1 Åであり、当該HBAとNIの距離は10.20±1 Åであり、および当該HRA2とNIの距離は5.88±1 Åであるか、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Bに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.80±1 Åであり、当該HBDとHBAの距離は7.34±1 Åであり、当該HBDとNIの距離は16.92±1 Åであり、当該HRA1とHBAの距離は5.20±1 Åであり、当該HRA1とNIの距離は14.34±1 Åであり、当該HBAとHRA2の距離は4.69±1 Åであり、当該HBAとNI の距離は9.85±1 Åであり、および当該HRA2とNIの距離は5.66±1 Åであるか、または
当該HBD、HBA、HAR1、HAR2およびNIは、図8Cに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.79±1 Åであり、当該HBDとHRA2の距離は11.94±1 Åであり、当該HBDとNIの距離は16.28±1 Åであり、当該HRA1とHBAの距離は5.20±1 Åであり、当該HRA1とNIの距離は14.64±1 Åであり、当該HBAとHRA2の距離は4.72±1 Åであり、および当該HRA2とNIの距離は5.63±1 Åであり; 並びに
当該化合物がファルマコフォアの特徴と一致する場合には、当該化合物を選択すること。
【請求項60】
請求項59の方法であって、ここで、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Aに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.81 Åであり、当該HBDとHBA の距離は7.52 Åであり、当該HBDとNIの距離は17.67 Åであり、当該HRA1とHBA の距離は7.52 Åであり、当該HRA1とHRA2の距離は9.41 Åであり、当該HBAとNI の距離は10.20 Åであり、および当該HRA2とNIの距離は5.88Åであるか、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Bに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.80 Åであり、当該HBDとHBAの距離は7.34 Åであり、当該HBDとNIの距離は16.92 Åであり、当該HRA1とHBAの距離は5.20 Åであり、当該HRA1とNIの距離は14.34 Åであり、当該HBAとHRA2の距離は4.69 Åであり、当該HBAとNIの距離は9.85 Åであり、および当該HRA2とNIの距離は5.66 Åであるか、または、
当該HBD、HBA、HAR1、HAR2およびNIは、図8Cに従う立体配置であり、ここで、当該HBDとHRA1の距離は2.79 Åであり、当該HBDとHRA2の距離は11.94 Åであり、当該HBDとNIの距離は16.28 Åであり、当該HRA1とHBAの距離は5.20 Åであり、当該HRA1とNIの距離は14.64 Åであり、当該HBAとHRA2の距離は4.72 Åであり、および当該HRA2とNI の距離は5.63 Åである
方法。
【図1】

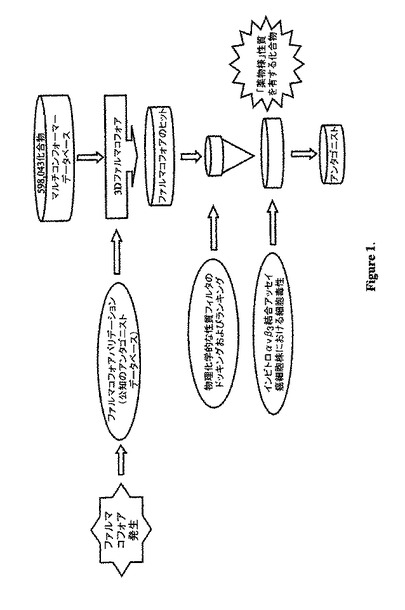
【図2】
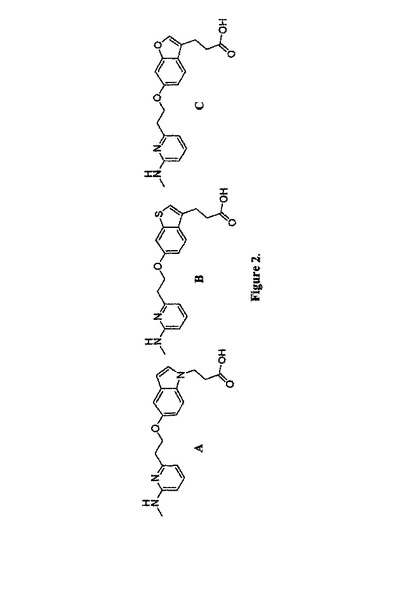
【図3】
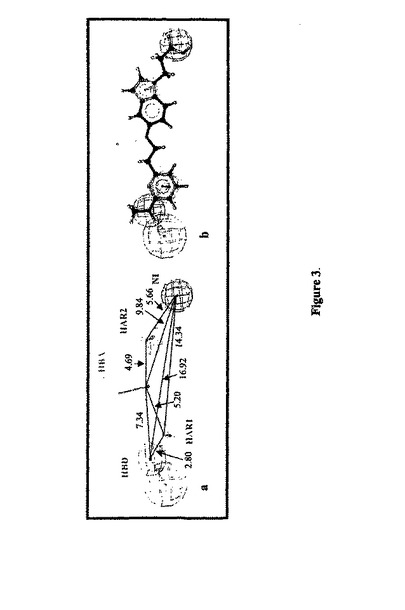
【図4】
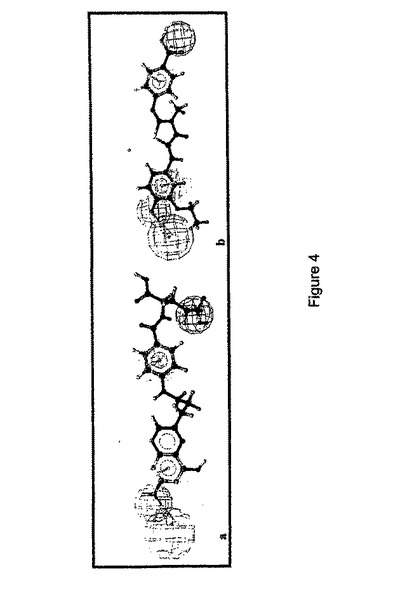
【図5】
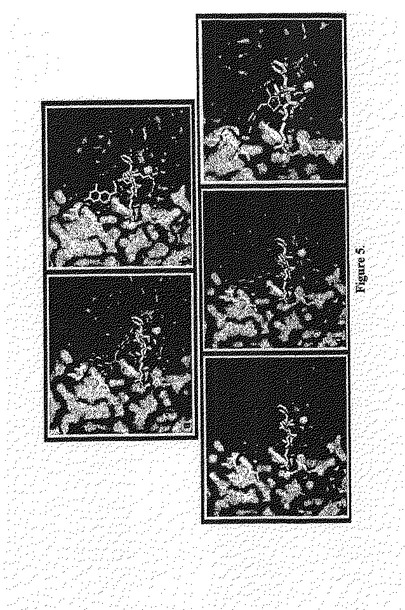
【図6】
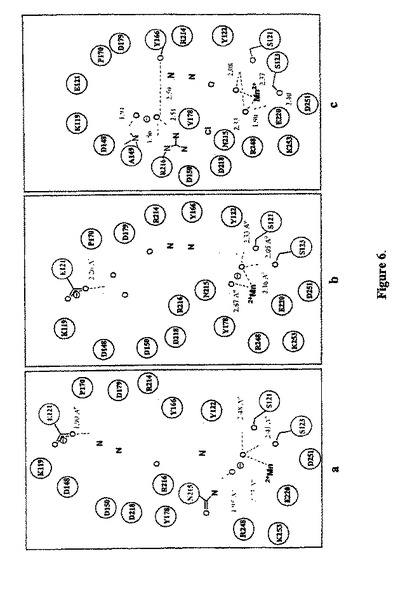
【図7】
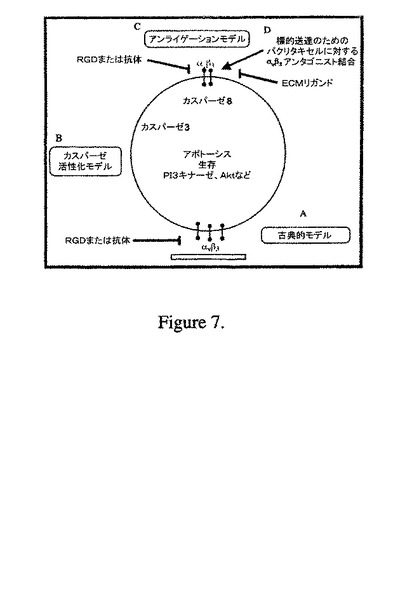
【図8】
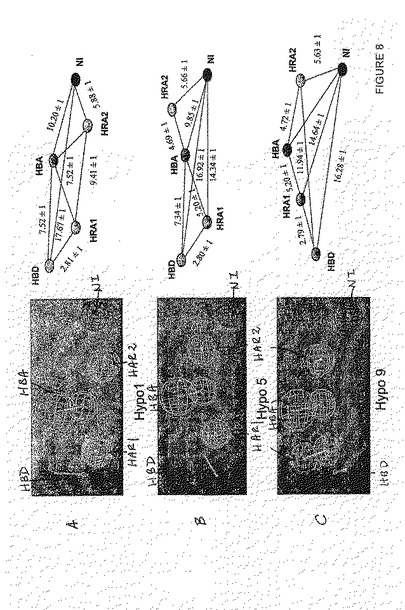
【図9】

【図10】

【図11】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【公表番号】特表2009−515980(P2009−515980A)
【公表日】平成21年4月16日(2009.4.16)
【国際特許分類】
【出願番号】特願2008−541300(P2008−541300)
【出願日】平成18年11月14日(2006.11.14)
【国際出願番号】PCT/US2006/044305
【国際公開番号】WO2007/059195
【国際公開日】平成19年5月24日(2007.5.24)
【出願人】(301040556)ユニヴァーシティー オブ サザン カリフォルニア (15)
【Fターム(参考)】
【公表日】平成21年4月16日(2009.4.16)
【国際特許分類】
【出願日】平成18年11月14日(2006.11.14)
【国際出願番号】PCT/US2006/044305
【国際公開番号】WO2007/059195
【国際公開日】平成19年5月24日(2007.5.24)
【出願人】(301040556)ユニヴァーシティー オブ サザン カリフォルニア (15)
【Fターム(参考)】
[ Back to top ]