
インビボ画像化剤として使用されるイサチン誘導体
本発明は、イサチン5−スルホンアミド誘導体、当該誘導体を含む薬剤組成物、これらの分子画像化剤としての使用、これらのアポトーシスの異常調節に関連する疾患若しくは障害の診断または治療のための使用、当該誘導体の合成方法、カスパーゼ活性およびアポトーシスの分子画像化方法、ならびにカスパーゼ活性に関する試験物質の治療効果の評価方法を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規なイサチン5−スルホンアミド誘導体、ならびにカスパーゼ活性およびカスパーゼ依存性アポトーシスの可視化および定量化、および治療用途における分子画像化剤(molecular imaging agent)としてのその使用を提供する。
【背景技術】
【0002】
アポトーシスまたはプログラム細胞死(PCD)は、最も普及している細胞死経路であり、非常に調節されたエネルギー保存メカニズムを介して進行する。健常な状態では、アポトーシスは、細胞の成長を制御し、細胞数を調節し、形態形成を容易にし、さらに有害なまたは異常な細胞を除去する際に重要な役割を果たす。このプロセスの異常調節は、癌や自己免疫疾患等の、アポトーシスの阻害に関連するもの、および神経変性疾患、血液病、AIDS、虚血及び同種移植の拒絶反応等の、過活動アポトーシスに関連するものなどの、多くの病状にかかわりがある。したがって、アポトーシスの可視化および定量化は、このようなアポトーシスに関連する病態生理学の診断に有用である。
【0003】
これらの疾患の治療のための処置は、このプロセスを刺激するまたは阻害することによって、均衡のとれたアポトーシスを回復させることを目的とする。したがって、細胞や組織におけるアポトーシスの非侵襲性の画像化(imaging)は、治療的介入に対する応答の初期評価にとって非常に有益であり、病理学的なプロセスの破壊への新しい知見を提供する。悪性の成長を症状が末期になる前に確実に制御するために癌治療の有効性を初期にモニターすることは特に関心が高い。
【0004】
細胞死を画像化するのに有用なプローブのうち、放射性標識されたアネキシンV(Annexin V)は最も注目を集めている1,2。しかしながら、アネキシンVは、細胞内のものではなく、細胞外表面のものしか検出せず、負に帯電したリン脂質にのみ結合するため、アポトーシスと壊死とを区別することができない。膜相互作用プローブに関しては、数多くのジ−ダンシルシステイン(di-dansyl cysteine)およびナフチル−エチル−フルオロアニリン(naphthyl-ethyl-fluoroalanine)誘導体がアポトーシスを画像化するために開発された3,4,5。しかしながら、このような膜相互作用プローブはまた、アポトーシス性細胞に対する特異性が低く、別の試験を必要とせずにはアポトーシスと壊死とを区別することができない6。より最近では、カスパーゼと呼ばれる酵素群に結合する特定の化合物の開発への興味が高まっている。
【0005】
カスパーゼは、アポトーシスの調節に中心的な役割を果たすシステインアスパルテートに特異的なプロテアーゼ(cysteine aspartate-specific proteases)群である。内因性のおよび外因性のシグナルネットワークは、「イニシエーター」カスパーゼ8(外因性)または9(内因性)を活性化し、さらに不活性なプロカスパーゼ3、6及び7を切断して活性のある「エクセキューショナー(executioner)」カスパーゼ3、6及び7にする。このエクセキューショナーカスパーゼは、最終的には細胞タンパク質の切断により細胞死をもたらし、これは非常に選択的にアスパラギン酸残基の右側に起こる。切断されるタンパク質としては、DNA修復酵素(例えば、PARP)、キーシグナルタンパク質(key signaling protein)(例えば、Akt、Ras)、核骨格タンパク質(nuclear skeletal protein)(例えば、アクチン、α−フォドリン、ラミン)および細胞サイクルレギュレーター(例えば、p27Kip1)がある。
【0006】
[131I]IZ−VAD−fmk等の、カスパーゼのペプチド系の不可逆的阻害剤を分子画像化剤として使用することは、選択性が中程度しかなく、細胞への取り込みがインビボでの画像化には不十分であるなど、細胞透過性が低いため、不十分であった。
【0007】
より最近では、イサチンとして知られている化学物質群が潜在的なカスパーゼ阻害剤として研究されてきた。イサチンの作用メカニズムは、活性化カスパーゼの酵素活性部位への共有結合を介したカスパーゼ3及び7との細胞内酵素−阻害剤複合体の形成がかかわっていると考えられる。イサチンのジカルボニル官能基がその作用メカニズムには必須である;この基が活性部位のシステイン残基と結合して、イサチンの求電子性のC−3カルボニル炭素および求核性のシステインチオレート官能基によるチオヘミケタルを形成する7。
【0008】
高スループットスクリーンについては、Lee et al.がカスパーゼ−3の阻害剤として非ペプチドイサチンN−(1−メチル)−5−ニトロイサチン(N-(1-methyl)-5-nitroisatin)を同定した。構造を最適化することによって、スルホンアミド(S)−1−ベンジル−5−(2−フェノキシメチル−ピロリジン−1−スルホニル)イサチン[(S)-1-benzyl-5-(2-phenoxymethyl-pyrrolidine-1-sulfonyl)isatin](2.5nM)8(本明細書中の化合物13を示す)が開発された。他のイサチンスルホンアミドがカスパーゼ3及び7の阻害剤として開発された9−12。Kopka13およびMach14は、それぞれ、18Fで標識された(S)−1−(4−(2−フルオロエトキシ)ベンジル)−5−[1−(2−フェノキシメチルピロリジニル)スルホニル]イサチン[18F-labeled (S)-1-(4-(2-fluoroethoxy)benzyl)-5-[1-(2-phenyoxymethylpyrrolidinyl)sulfonyl]isatin]をポジトロン放出型断層撮影(PET)の推定上のトレーサーとして開発し、Kopka13は、放射性標識された類似体である[125I](S)−1−(4−ヨードベンジル)−5−(2−フェノキシメチル−ピロリジン−1−スルホニル)イサチン)(本明細書中の化合物14を示す)の生物学的特性を調査した。WO 99/06367およびWO 01/22966には、多数のイサチン誘導体およびカスパーゼの阻害のためのこれらの使用が記載される。WO 2006/074799、US 2005/0250798およびGB 1,240,648には、数多くのイサチン5−スルホンアミド誘導体およびアポトーシスの画像化剤としてのこれらの使用が記載される。しかしながら、これらの既知の化合物は、分子安定性が低い、カスパーゼ−3との親和性が比較的低いおよび親油性が高いなどの、数多くの欠点がある。分子安定性が低いと、代謝が速いため、低い信号対ノイズ比でコントラストの像に劣る。また、カスパーゼ−3との親和性が低いと、画像のコントラストが劣る。親油性が高いと、系からの排泄(system elimination)が悪く、高分子への全体的な非特異的な結合が増加してしまう。
【発明の概要】
【0009】
したがって、分子安定性が向上し、カスパーゼ酵素に対する親和性が向上し、かつ親和性が低減した新規なイサチン誘導体を提供することが、本発明の目的である。
【0010】
本発明の第一の態様は、下記式Aを有する新規なイサチン5−スルホンアミド誘導体、またはこの塩、水和物もしくはプロドラッグを提供する:
【0011】
【化1】

【0012】
ただし、Rは、フェニル、3−フルオロフェニル、2,4−ジフルオロフェニル、3,5−ジフルオロフェニル、置換されてもよいテトラヒドロピラン、置換されてもよいジアジンおよび置換されてもよいトリアゾールであり;R’は、置換されてもよいフェニルまたは置換されてもよいトリアゾールであり;
この際、Rがフェニルである場合には、R’は、置換されてもよいトリアゾールである。
【0013】
好ましい実施形態では、Rは置換されてもよいトリアゾールを含み、R’は置換されてもよいフェニルを含む。他の好ましい実施形態では、Rは置換されてもよいフェニルを含み、R’は置換されてもよいトリアゾールを含む。
【0014】
好ましくは、前記置換されてもよいフェニル、置換されてもよいテトラヒドロピランおよび置換されてもよいジアジンは、必要であれば、1以上の電子求引基で置換される。好ましくは、前記電子求引基は、ハロゲン、ニトロ基およびカルボン酸基またはアルデヒドまたはケトン等の他のカルボニル含有官能基からなる群より選択される。より好ましくは、前記電子求引基はハロゲンである。好ましくは、前記電子求引基はフッ素である。
【0015】
好ましくは、置換されてもよいフェニルは2,4−ジフルオロフェニルである。
【0016】
一実施形態では、置換されてもよいトリアゾールは、必要であれば、置換されたアルキル基で置換される。好ましくは、前記置換されたアルキル基は、ハロゲンで置換されたアルキルである。より好ましくは、前記ハロゲンで置換されたアルキルはC1−4のフルオロアルキルである。好ましくは、前記C1−4のフルオロアルキルは、フルオロメチル、2−フルオロエチル、3−フルオロプロピルまたは4−フルオロブチルである。最も好ましくは、前記C1−4のフルオロアルキルは2−フルオロエチルである。
【0017】
他の実施形態では、置換されてもよいトリアゾールは、必要であれば、置換されたアルキル基で置換される。好ましくは、前記アルキルはメチルである。
【0018】
本発明はまた、これらの光学異性体およびジアステレオ異性体などの、本発明の化合物のすべての立体異性体を含むことは当業者に理解されるであろう。本発明の化合物は、特定の光学異性体の実質的な純粋な溶液の形態でまたはラセミ混合物として存在してもよい。好ましくは、本発明の化合物は、S光学異性体の実質的な純粋な溶液として、またはS光学異性体から実質的に構成される溶液として存在する。好ましくは、S光学異性体およびR光学異性体双方を含むラセミ混合物では、S光学異性体がラセミ混合物中本発明の化合物の少なくとも50%を構成する。より好ましくは、S光学異性体が、ラセミ混合物中本発明の化合物の少なくとも60%、少なくとも70%、少なくとも80%、少なくとも90%、少なくとも95%または少なくとも99%を構成する。
【0019】
本発明の特に好ましい化合物としては、RおよびR’が下記に規定される式Aの化合物がある:
【0020】
【表1−1】
【0021】
【表1−2】
【0022】
【表1−3】
【0023】
【表1−4】
【0024】
好ましくは、「塩」ということばは、酢酸、プロピオン酸、乳酸、酒石酸、コハク酸、フマル酸、マレイン酸、マロン酸、マンデル酸、リンゴ酸、フタル酸、塩酸、臭化水素酸、リン酸、硝酸、硫酸、メタンスルホン酸、ナルタレンスルホン酸、ベンゼンスルホン酸、トルエンスルホン酸、カンファースルホン酸などの有機及び無機酸、ならびに本発明の化合物が塩基部分を含む際に同様にして既知の許容できる酸由来の塩を包含する。塩はまた、本発明の化合物がカルボン酸若しくはフェノール酸部分を含む際には、有機及び無機塩基、好ましくはアルカリ金属塩、例えば、ナトリウム、リチウム若しくはカリウム、または塩基付加塩を形成できる同様の部分から形成されてもよい。
【0025】
本明細書中で使用される「水和物」ということばは、予め水と化学的に結合した本発明の化合物の形態を意味する。
【0026】
本明細書中で使用される「プロドラッグ」ということばは、代謝によってインビボで本発明の化合物に変換可能な化合物を意味する。
【0027】
本明細書中で使用される、「置換されてもよいフェニル」という表現は、必要であれば1以上の置換基が環の2、3、4、5、および6位の1以上の位置に配置されてなるフェニル基を意味する。
【0028】
本明細書中で使用される「テトラヒドロピラン」ということばは、5つの炭素原子及び1つの酸素原子を含む飽和の6員環から構成される有機化合物を意味する。
【0029】
本明細書中で使用される「トリアゾール」ということばは、2つの炭素原子及び3つの窒素原子を有する5員環を有する、分子式:C2HN3を有する化合物を意味する。これらの化合物としては、異性体 1,4−及び1,5で2置換された1,2,3 トリアゾールがある。
【0030】
本明細書中で使用される「ハロゲン」ということばは、臭素、塩素、フッ素およびヨウ素を意味する。
【0031】
本明細書中で使用される「アルキル」ということばは、脂肪族炭素化水素鎖を意味し、特記しない限り、以下に制限されないが、1、2、3、4、5または6個の炭素原子を有する直鎖または分岐鎖を包含する。アルキルとしては、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、sec−ブチル、イソブチル、tert−ブチルなどが挙げられる。
【0032】
本明細書中で使用される「置換されたアルキル」という表現は、ハロゲン、ヒドロキシル、チオール、アミノ、別のヘテロ原子、芳香族またはヘテロ芳香族基ならびにポリエチレングリコール等のポリエーテル、スクシニジル(succinidyl)、−NH−(CH2)n−NH−及びポリアミドなどのスペーサー基からなる群より選択される1以上の置換基をさらに有するアルキルを意味する。
【0033】
本明細書中で使用される「ジアジン」ということばは、分子式:C4H4N2を有する有機化合物群を意味し、各化合物は炭素原子の2個が窒素で置換されるベンゼン環を含む。これらの化合物としては、異性体 ピラジン(1,4−ジアジン)、ピリミジン(1,3−ジアジン)、およびピリダジン(1,2−ジアジン)がある。
【0034】
本発明の化合物は、活性化カスパーゼに特異的であり、活性化カスパーゼに対して高い親和性を有する。本発明の化合物は、カスパーゼ3及び7の強力かつ選択的な阻害剤である。特に本発明の化合物は、カスパーゼ−3及びカスパーゼ−7に対する高い親和性を有する。本発明の化合物は、アポトーシス中に活性形態で発現されるのみであり、ゆえに、カスパーゼ−3および/またはカスパーゼ−7の活性はカスパーゼに依存するアポトーシスの信頼性のあるインジケータである。
【0035】
本発明の化合物は、多くの既知のイサチン5−スルホンアミド誘導体と比べて親油性が低いため、全身で良好に除去でき、高分子への一般的な非特異的な結合が低減する。しかしながら、カスパーゼは細胞内プロテアーゼであるため、本発明の化合物は、非促進拡散によって細胞膜を通過するのに十分な親油性を有するため、細胞内外を自由に通過できる。このように、本化合物は、活性化カスパーゼ−3および/またはカスパーゼ−7を有する細胞中でのみ迅速に取り込みおよび保持を双方向に行うであろう。理想的な親油性(LogPとして表わされる)は、多くの他の要因に加えて、化合物が通過するのに必要な細胞タイプによって異なる。一実施形態では、親油性は1.0〜2.0の領域であることが好ましい。
【0036】
本発明の化合物は、代謝安定性が向上し、これにより化合物が代謝され、また、場合によっては排出される速度が減少する。代謝安定性が増すと、活性化カスパーゼと結合すると、アポトーシスを受ける細胞内に本発明の化合物が蓄積される。これにより、信号対ノイズ比の高いコントラストの良好な画像が得られる。これは、細胞及び組織内のカスパーゼ活性の正確な可視化および定量化を可能にするため、分子画像化剤では好ましい。
【0037】
好ましくは、本発明の化合物は、非処置腫瘍、心臓および脳組織では取り込みが低い。これにより、カスパーゼ活性の変化に関連する、哺乳動物組織におけるこれらの化合物の活性化カスパーゼへの結合の変化をモニターしやすくなる。
【0038】
本発明の第二の態様は、画像化部分(imaging moiety)で標識される、本発明の第一の態様の化合物を提供する。
【0039】
前記標識は、官能基内に画像化部分を有していても、または別の物質としての画像化部分が結合されていてもよい。
【0040】
前記画像化部分は、検出可能なシグナルを製造できる部分を有していてもよい。このような部分としては、蛍光ラベル及び放射性ラベルがある。
【0041】
蛍光ラベルはフルオロフォアが共有結合してなる。好ましいフルオロフォアとしては、フルオレセインイソチオシアネート、ローダミンの誘導体、クマリンならびにシアニン色素である、Alexa Fluors及びDyLight Fluorsがある。
【0042】
好ましくは、本化合物はラジオアイソトープで標識される。ラジオアイソトープは本発明の化合物の分子量を実質的に増加させず、臨床的な非侵襲性イメージングに使用できるため、画像化部分としてラジオアイソトープを使用することが好ましい。
【0043】
好ましくは、ラジオアイソトープはポジトロン放射体である。放射性同位体は、3H、14C、18F、11C、120I、123I、124I、125I、131I、94mTc、66Ga、68Ga、64Cu、61Cu、67Cu、75Br、76Br、94mTc、99mTc、201Tl、111In、86Yおよび89Zrからなる群より選択されうる。
【0044】
画像化部分をポジトロン放出型断層撮影(PET)を用いて画像化する場合には、放射性同位体は18F、11C、120I、124I、94mTc、66Ga、68Ga、64Cu、67Cu、86Y、75Brおよび76Brからなる群より選択されることが好ましい。
【0045】
画像化部分を単光子放出コンピュータ断層撮影(SPECT)を用いて画像化する場合には、放射性同位体は123I、99mTcおよび111Inからなる群より選択されることが好ましい。
【0046】
好ましくは、画像化部分は18Fまたは11Cである。
【0047】
好ましくは、化合物1〜12および29〜34のいずれかにおいて、Rおよび/またはR’内のフッ素原子の1以上が18Fである。
【0048】
例えば、化合物6〜12、および29〜36におけるように、RまたはR’での1,2,3−トリアゾール基の付加が、化合物が18Fまたは11Cで容易に標識できるため、さらに好ましい。
【0049】
本発明の特に好ましい実施形態は、下記式を有する。
【0050】
【化2】
【0051】
本発明の他の好ましい化合物は、下記一般式を有する。
【0052】
【化3】
【0053】
本発明の第三の態様は、本発明の第一の態様に係る化合物を、必要であれば一以上の製薬上許容できる担体、希釈剤または賦形剤と組み合わせてなる組成物を提供する。
【0054】
本発明の第四の態様は、本発明の第二の態様に係る化合物を、必要であれば一以上の製薬上許容できる担体、希釈剤または賦形剤と組み合わせてなる組成物を提供する。
【0055】
本組成物はまた、一以上の別の活性剤を含んでもよい。前記別の活性剤は、本発明の化合物によって生産されるシグナルを促進する物質を含んでもよい。好ましくは、前記活性剤は、マレイン酸ジエチル(DEM)、L−ブチオニン−(S,R)−スルホキシミン(BSO)及びこれらの誘導体などの、細胞グルタチオンレベルを枯渇するまたは調節する化合物を含む。
【0056】
本発明の第三または第四の態様の組成物は、既知の方法によって投与されうる。
【0057】
本組成物は、細胞に接近できる化合物を暴露する(exposing)、インキュベートする(incubating)、接触する(touching)、結合する(associating)または作製する(making)ことによって、細胞と接触させてもよい。
【0058】
本組成物は、経口(吸入によることを含む)、非経口、粘膜(例えば、口腔、舌下、鼻腔内)、直腸または経皮投与によって被検者に投与されてもよく、本組成物はそれに従って適用されてもよい。
【0059】
経口投与では、本組成物は、液体または固体として、例えば、溶液、シロップ、懸濁液またはエマルジョン、錠剤、カプセル及びロゼンジとして配合できる。
【0060】
液剤は、通常、適当な水性または非水性液状担体、例えば、水、エタノール、グリセリン、ポリエチレングリコールまたは油における、化合物またはその生理学上許容できる塩、水和物若しくはプロドラッグの懸濁液または溶液から構成されるであろう。液剤はまた、懸濁化剤、防腐剤、香料添加剤または着色剤を含んでもよい。
【0061】
錠剤形態の組成物は、固体製剤を調製するのに一般的に使用される適当な医薬担体を用いて調製できる。このような担体の例としては、ステアリン酸マグネシウム、デンプン、ラクトース、スクロース及び微結晶性セルロースが挙げられる。
【0062】
カプセル形態の組成物は、一般的なカプセル化方法を用いて調製できる。例えば、活性成分を含む粉末、顆粒またはペレットを標準的な担体を用いて調製した後、硬質ゼラチンカプセル中に充填してもよい。または、分散液または懸濁液を適当な医薬担体、例えば、水性ゴム、セルロース、ケイ酸塩または油を用いて調製した後、前記分散液または懸濁液を軟質ゼラチンカプセル中に充填してもよい。
【0063】
経口投与用組成物は、例えば、錠剤またはカプセルに配合物を外側コーティングすることによって、消化管を通過する際に活性成分が分解するのに対して保護するように設計されてもよい。
【0064】
鼻腔内または経口投与用組成物は、エアロゾル、液滴、ゲル及び粉末として簡便に配合されてもよい。エアロゾル製剤は、具体的には生理学上許容できる水性または非水性溶媒における活性物質の溶液または細かい懸濁液(fine suspension)を含み、一般的に密閉容器中で滅菌形態で1回量または複数回量で提示され、これは噴霧装置と共に用いられるカートリッジまたはレフィルの形態をとってもよい。または、密閉容器は、容器の内容物が排出されたら処分されるような用途である、単回投与鼻吸入器等の単回調剤装置(unitary dispensing device)または絞り弁を備えたエアロゾルディスペンサーであってもよい。投与形態がエアロゾルディスペンサーを含む場合には、製薬上許容できる推進剤を含むであろう。エアロゾル投与形態はまたポンプ−アトマイザーの形態をとってもよい。
【0065】
口腔または舌下投与に適する組成物としては、活性成分が糖及びアカシア、トラガカント、またはゼラチン及びグリセリン等の担体と共に配合される、錠剤、ロゼンジ及びトローチが挙げられる。
【0066】
直腸または膣内投与用の組成物は、簡便には坐剤(ココアバター等の公知の坐剤の基剤を含む)、ペッサリー、膣錠、泡剤または浣腸剤の形態を有する。
【0067】
経皮投与に適する組成物としては、軟膏、ゲル、パッチ及び粉体噴射剤等の噴射剤が挙げられる。
【0068】
簡便には、本組成物は、錠剤、カプセルまたはアンプル等の単回投与形態を有する。
【0069】
好ましくは、本発明の化合物または組成物は、非経口投与によって被検者に投与される。特に、本組成物は、静脈内、腹腔内、髄腔内、リンパ管内または筋肉内に投与されてもよい。
【0070】
具体的な非経口組成物は、適当なpH、等張性および安定性を有する滅菌した水性または非水性担体における化合物またはその生理学上許容できる塩の経口投与に許容できる溶液または懸濁液から構成される。当業者は、例えば、塩化ナトリウム注射液、リンガー注射液、乳酸加リンガー注射液等の等張ベヒクルを用いて適当な溶液を調製できる。防腐剤、安定化剤、緩衝剤、抗酸化剤および/または他の添加剤を必要であれば含んでもよい。
【0071】
本発明の第五の態様は、分子画像化剤(molecular imaging agent)として使用されるための本発明の第一若しくは第二の態様の化合物または本発明の第三若しくは第四の態様の薬剤組成物を提供する。
【0072】
好ましくは、前記分子画像化剤は、細胞及び組織におけるカスパーゼ活性の可視化および定量化を目的とする。好ましくは、前記分子画像化剤は、ヒトの細胞及び組織等の哺乳動物の細胞及び組織におけるカスパーゼ活性の可視化および定量化を目的とする。前記定量は、細胞または組織の放射線量、または取り込み、解離若しくは分配速度の分析を含んでもよい。
【0073】
活性カスパーゼと複合体を形成すると、細胞及び組織内への標識化合物の取り込みおよび蓄積を画像化して、前記細胞及び組織内のカスパーゼ活性のレベルを示す。したがって、本発明の第六の態様は、
a)細胞または組織を本発明の第二の態様の化合物または本発明の第四の態様の組成物と接触させ;および
b)カスパーゼ活性を検出する
段階を有する、カスパーゼ活性の分子画像化方法を提供する。
【0074】
好ましくは、カスパーゼ活性の検出段階は、検出装置の検出域内に被検者をおき、前記検出装置を用いて被検者内の前記化合物を検出する段階を有する。
【0075】
本方法は、インビトロで行われてもよい。細胞または組織の本発明の化合物または組成物との接触は、細胞または組織に接近できる化合物を暴露する(exposing)、インキュベートする(incubating)、接触する(touching)、結合する(associating)または作製する(making)ことを含んでもよい。
【0076】
本発明の化合物または組成物が放射性標識される際には、カスパーゼ活性を適当な放射線検出装置を用いてインビトロで検出できる。前記装置としては、Packard Topcount等の、ベータカウンター、またはPackard Cobra IITMガンマカウンター(Perkin Elmer, UK)等の、ガンマカウンター、またはラジオ−TLCスキャナー(radio-TLC scanner)が挙げられる。
【0077】
本発明の化合物または組成物が蛍光標識される場合には、前記カスパーゼ活性は、適当な蛍光読み取り装置を用いてインビトロで検出できる。このような装置としては、蛍光顕微鏡、蛍光光度計、またはPerkin Elmer Victor等の、蛍光板リーダーがある。
【0078】
または、本方法は、インビボで行われてもよい。本化合物または本組成物を、本発明の第三および第四の態様で記載される方法によって被検者に投与されてもよい。好ましくは、本化合物は非経口で投与される。
【0079】
本発明の化合物または組成物が放射性標識される際には、カスパーゼ活性は、放射線検出装置を用いて検出してもよい。前記放射線検出装置としては、ポジトロン放出断層撮影(PET)スキャナーまたは単光子放出コンピュータ断層撮影(SPECT)スキャナーが挙げられる。好ましくは、前記放射線検出装置はポジトロン放出断層撮影(PET)スキャナーである。前記PETスキャナーは、18F等のポジトロンを放出するラジオアイソトープによって間接的に放出されるガンマ線の対を検出し、組織内のラジオアイソトープ濃度の3Dイメージを作製できる。したがって、PETは、哺乳動物の組織内の本発明の放射性標識された化合物または組成物の位置を確認する3Dイメージを作製するのに使用できる。
【0080】
本発明の化合物または組成物が蛍光標識される場合には、カスパーゼ活性は、適当な蛍光読み取り装置を用いて検出されてもよい。前記蛍光読み取り装置としては、蛍光内視鏡がある。
【0081】
好ましくは、カスパーゼ活性は、カスパーゼ−3活性である。
【0082】
本発明はまた、カスパーゼ活性の可視化及び定量化のための薬剤または診断組成物の製造を目的とする本発明の第二の態様の化合物の使用を提供する。
【0083】
第六の態様の他の実施形態では、本発明の第一の態様に係る化合物または本発明の第三の態様に係る薬剤組成物は、段階(a)で投与されてもよい。次に、前記化合物を投与後に蛍光または放射性で標識してもよい。
【0084】
上述したように、カスパーゼ酵素はアポトーシスをもたらし、このようなカスパーゼ活性はアポトーシスの指標として使用できる。したがって、本発明の第七の態様は、哺乳動物の細胞または組織におけるカスパーゼに依存するアポトーシスのインビボにおける画像化のための本発明の第二の態様の化合物または本発明の第四の態様の薬剤組成物の使用を提供する。
【0085】
本方法は、
a)本発明の第二の態様の化合物または本発明の第四の態様の組成物を被検者に投与し;
b)被検者を検出装置の検出域に置き;および
c)被検者における化合物を前記検出装置で検出する、
ことを有する。
【0086】
好ましくは、前記化合物または組成物は注射によって被検者に投与される。
【0087】
画像化部分は、非侵襲で外部からまたは血管内放射線(intravascular radiation)検出器若しくは光検出器等の、インビボでの使用のために設計される検出器、または術中での使用のために設計される放射線検出器を用いることによって内部から検出されうる。好ましくは、画像化部分は非侵襲的に検出される。
【0088】
一実施形態では、前記化合物は蛍光標識され、第六の態様で上記で規定されるような蛍光読み取り装置を用いて蛍光標識された化合物から放出される蛍光を測定することによって検出される。
【0089】
第二の好ましい実施形態では、前記化合物は放射性標識され、第六の態様で上記で規定されるような放射線検出装置を用いて放射性標識された化合物から放出される放射線を測定することによって検出される。
【0090】
好ましくは、カスパーゼ活性は、カスパーゼ−3活性である。
【0091】
第七の態様の別の実施形態では、本発明の第一の態様に係る化合物または本発明の第三の態様に係る薬剤組成物を、段階(a)で投与してもよい。次に、前記化合物を投与後にインビボで蛍光または放射性で標識してもよい。
【0092】
上述したように、多数の病気の治療のための処置は、このプロセスを刺激または阻害することによって、正常で釣り合いのとれたアポトーシスを取り戻すことを目的とする。したがって、本発明の第八の態様は、哺乳動物の細胞または組織におけるカスパーゼ活性への試験物質の治療効果を評価するための、本発明の第二の態様の化合物または本発明の第四の態様の薬剤組成物の使用を提供する。
【0093】
本方法は、
a)哺乳動物の細胞または組織を本発明の第二の態様の化合物または本発明の第四の態様に係る組成物と接触させ;
b)前記哺乳動物の細胞または組織を検出装置の検出域に置き:
c)化合物を前記検出装置で検出し;さらに
d)段階a)、b)およびc)を繰り返す、
ことを有してもよい。
【0094】
一実施形態では、前記化合物は蛍光標識され、第六の態様で上記で規定されるような蛍光読み取り装置を用いて蛍光標識された化合物から放出される蛍光を測定することによって検出される。
【0095】
第二の好ましい実施形態では、前記化合物は放射性標識され、第六の態様で上記で規定されるような放射線検出装置を用いて放射性標識された化合物から放出される放射線を測定することによって検出される。
【0096】
第八の態様の別の実施形態では、本発明の第一の態様に係る化合物または本発明の第三の態様に係る薬剤組成物を段階(a)で投与してもよい。次に、前記化合物を投与後にインビボで蛍光または放射性で標識してもよい。
【0097】
好ましくは、本発明の方法は、インビボで行われてもよく、
a)本発明の第二の態様に係る化合物または本発明の第四の態様に係る組成物を被検者に投与し;
b)被検者を放射線検出装置の検出域に置き;
c)被検者における放射性標識された化合物から放出される放射線を前記放射線検出装置を用いて測定し;
d)段階a)、b)およびc)を繰り返す、
ことを有してもよい。
【0098】
第八の態様のインビトロおよびインビボ方法双方で、段階d)は、繰り返しが経時的にカスパーゼ活性の変化を追跡するのに有効である所定の間隔で行うことが好ましい。段階d)の間隔は、問題になっている被検者および試験物質に適切であるべきである。ヒトでは、適当な間隔は、以下に制限されないが、12〜24時間、48時間、1週間、2週間、3週間、4週間、5週間または6週間が挙げられる。
【0099】
各繰り返しで測定されるカスパーゼ活性を比較することによって、経時的な、カスパーゼ活性の程度及び位置の定量的な変化、およびゆえにカスパーゼに依存するアポトーシスの定量的なまたは半定量的な変化を評価できる。カスパーゼ活性の程度および位置の変化は、カスパーゼ活性の刺激または阻害によって、カスパーゼに依存するアポトーシスへの試験物質の治療効果を示しうる。本方法は、確立された薬を用いて治療のための処置に対する被検者の応答を評価するのに、および新規な薬の薬効の評価に使用できる。
【0100】
本発明の化合物または組成物は、試験物質の前に、後にまたはと同時に投与してもよい。好ましくは、段階(a)〜(c)は、試験物質の投与前に被検者で行われ、カスパーゼ活性の1回目の測定結果を提供する。次に、試験物質を投与し、段階(a)〜(c)を上記したような間隔をあけた後繰り返して、カスパーゼ活性の2回目の測定結果を提供する。必要であれば、試験物質をさらに投与する前または後に、段階(a)〜(c)をさらに繰り返して、カスパーゼ活性のさらなる測定結果を提供してもよい。
【0101】
好ましくは、前記化合物または組成物を注射によって被検者に投与する。
【0102】
試験物質は、以下に制限されないが、癌、自己免疫疾患、血液病、HIV、AIDS、虚血、心血管疾患、神経疾患および移植拒絶反応などの病気を治療するのに使用される薬(drug)または薬剤(agent)含んでもよい。特に試験物質は、癌の処置のための化学療法薬、放射線療法薬または免疫療法薬を含んでもよい。
【0103】
上述したように、段階(a)〜(c)を2回以上繰り返してもよい。好ましい一実施形態では、段階(a)の1回目の実施で被検者に投与される化合物は、11Cで放射性標識された本発明に係る化合物である。好ましくは、前記化合物は、化合物35および36からなる群より選択される。次に、段階(a)の2回目の実施で被検者に投与される化合物は、放射性標識された画像化剤である。前記放射性標識された画像化剤は、例えば、フルオロデオキシグルコース(FDG)、3’−デオキシ−3’−18F−フルオロチミジン(FLT)または本発明に係る化合物であってもよい。好ましい一実施形態では、前記画像化剤は18Fで標識される。本実施形態は、特に好ましい。炭素−11(11C)は20分という比較的短い半減期を有する。ゆえに、11Cで標識された化合物を使用することによって、2回目以降のスキャンの間のバックグランドノイズがほとんどないまたはない。これにより、より高い信号対ノイズ比、ならびにカスパーゼ活性およびカスパーゼに依存するアポトーシスのより正確な可視化及び定量化が可能になる。
【0104】
同様にして、本発明の第六および第七の態様に係る方法を比較的短い時間内で繰り返そうとする場合には、11C標識された化合物、さらには18F標識された化合物等の別の放射性標識された画像化剤を使用することによっても、2回目のスキャン中に存在するバックグランドノイズを低減して、信号対ノイズ比を高める。
【0105】
本発明の第九の態様は、カスパーゼ活性の阻害に使用される本発明の第一の態様に係る化合物または本発明の第三の態様に係る組成物を提供する。本方法は、有効量の本発明の第一の態様の化合物または本発明の第三の態様の組成物をこのような処置の必要のある被検者に投与することを有していてもよい。
【0106】
本態様はまた、カスパーゼ活性の阻害のための医薬を製造する際の本発明の第一の態様の化合物または本発明の第三の態様の組成物の使用を提供する。
【0107】
カスパーゼ酵素がアポトーシスをもたらすため、本発明の第九の態様はまた、アポトーシスの阻害に使用される本発明の第一の態様の化合物または本発明の第三の態様の組成物を提供する。
【0108】
カスパーゼ活性の阻害は、過剰のまたは不適切なアポトーシスによって引き起こされるまたは前記アポトーシスに関連する病気若しくは症状を処置するのに有効でありうる。このような病気または症状の例としては、アルツハイマー病等の神経変性疾患、血液病、肝細胞の変性、変形性関節症、AIDS、虚血および同種移植の拒絶反応が挙げられる。
【0109】
本発明の第一の態様の化合物または本発明の第三の態様の組成物は、一般的に、1日投与量レジメで被検者に投与されるであろう。例えば、1日投与量レジメは、約0.001〜約100mg/kg、好ましくは約0.001〜約10mg/kg被検者の体重を必要とするかもしれない。1日投与量は、より大きな哺乳動物では、好ましくは約1mg〜約1000mg、好ましくは1mg〜500mgであり、前記化合物は1日1〜4回投与されうる。または、本発明の化合物が安定である場合には、投与量は、1週間で、1、2または3回投与されてもよい。
【0110】
本発明の化合物および組成物の投与の投与量、レジメおよび形式は、病気または症状および処置される個体によって異なり、かかわりのある開業医の判断に従うであろうことは理解される。
【0111】
本発明の第十の態様は、病態生理を診断するのに使用される本発明の第二の態様の化合物または本発明の第四の態様の組成物を提供する。特に、本発明の第二の態様の化合物または本発明の第四の態様の組成物は、以下に制限されないが、慢性心不全、急性心筋梗塞、発作、神経変性疾患、自己免疫疾患、局所性血液病、局所性AIDS、虚血(心虚血を含む)、および移植拒絶反応などの、過剰なまたは不適切なアポトーシスに関連する病気および疾患を診断するのに使用できる。
【0112】
本方法は、
a)哺乳動物の細胞または組織を本発明の第二の態様の化合物または本発明の第四の態様に係る組成物と接触させ;
b)前記哺乳動物の細胞または組織を検出装置の検出域内に置き;
c)前記検出装置を用いて前記化合物を検出して、前記哺乳動物の細胞または組織内のカスパーゼ活性の定量的な測定結果を提供し、この際、カスパーゼ活性がアポトーシスの指標である、
ことを有しうる。
【0113】
一実施形態では、前記化合物は蛍光標識され、第六の態様で上記で規定されるような蛍光読み取り装置を用いて蛍光標識された化合物から放出される蛍光を測定することによって検出される。
【0114】
第二の好ましい実施形態では、前記化合物は放射性標識され、第六の態様で上記で規定されるような放射線検出装置を用いて放射性標識された化合物から放出される放射線を測定することによって検出される。
【0115】
次に、好ましくは、前記測定結果を標準値と比較することによって、病態生理を診断できる。
【0116】
第十の態様の別の実施形態では、本発明の第一の態様に係る化合物または本発明の第三の態様に係る薬剤組成物は段階(a)で投与されてもよい。次に、前記化合物は投与後に蛍光標識または放射性標識されてもよい。
【0117】
本発明の第十一の態様は、本発明の第一の化合物の合成方法を提供する。本発明の第一の化合物は、適当な方法で調製されうる。実施例1、3及び10に記載され、図1、2(i)、2(ii)、12、19、20及び21に示されるスキームには、本発明の第一の化合物が調製される方法が示される。
【0118】
図1は、化合物1〜5、10〜12および中間体19a〜h、20a〜h、21〜24が調製できる方法を示す。化合物29〜34は類似した方法で調製される。さらなる詳細を実施例1に示される。
【0119】
トリアゾール基を含む本発明の化合物はまた、図2(i)、図12、及び図19に示され、実施例1及び3に記載されるような、「クリック−ラベリング(click-labelling)」と称される方法によって製造できる。このような方法は、例えば、化合物11、35及び36を作製するのに使用できる。本発明の第一の態様の化合物の製造には、非標識の化合物は、もちろん、これらの実施例に開示される放射性標識された化合物の代わりに使用されるであろう。例えば、図2(i)に示され、実施例3に記載されるスキームでは、2−フルオロエチルアジド(化合物27)が2−[18F]フルオロエチルアジド(化合物[18F]27)の代わりに使用される。
【0120】
また、付加環化して置換トリアゾールを得ることを、N−1で官能化されたイサチンを末端アジド及び末端アルキンを有する補欠分子族と反応することによって、逆に行ってもよい(「逆クリック−ラベリング(click-labelling)」と称される方法における)。このような方法は、例えば、化合物6、7、8及び9を作製するのに使用できる。中間体18F標識された末端アルキンの合成方法をも含む、この方法の適切な例は、図2(ii)に示され、実施例1、及びMarik, J and Sutcliffe, J. L.26, Sirion, U et al27and Li, Z. et al28に記載され、これらは参考で本明細書中に引用される。
【0121】
本発明の第十二の態様は、本発明の第二の態様の化合物の合成方法を提供する。本発明の第二の態様の化合物は、適当な方法で調製されうる。実施例1及び3に記載され、図1、2(i)、2(ii)及び12に示されるスキームは、本発明の第二の態様の化合物を調製されうる方法を示し、この際、標識された中間体化合物を使用する。
【0122】
実施例1に記載され、図1に示されるスキームは、本発明の第二の態様の化合物を調製するのに使用でき、この際、1以上の前駆体分子または中間体化合物、例えば、化合物19a〜h、20a〜hおよび/または21〜25は標識されたRまたはR’基を有する。このような方法は、例えば、標識形態の化合物1〜5、10〜12および29〜34を製造するのに使用できる。
【0123】
図2(i)、図12、図19に示され、実施例1及び実施例3に記載されるスキームは、[18F]11、[11C]35おおび[11C]36等の、本発明の第二の態様の特定の化合物を調製する「クリック−ラベリング」方法を示す。
【0124】
図2(ii)に示され、実施例1に記載される「逆クリック−ラベリング」方法もまた、標識形態の化合物6、7、8及び9等の、本発明の第二の態様に係る特定の化合物を作製するのに使用できる。例えば、図2(ii)に示され、実施例1に記載されるスキームでは、3−[18F]フルオロプロプ−1−イン[3-[18F]fluoroprop-1-yne](化合物[18F]28)が3−フルオロプロプ−1−イン[3-fluoroprop-1-yne](化合物28)の代わりに使用される。
【0125】
しかしながら、実施例3に記載され、図19に示される「クリック−ラベリング」方法には、安定した不純物が生産され、イサチン誘導体産物の特定の活性を低減するという欠点がある。実施例10に記載され、図20及び21に示されるように、この欠点は、一般式38及び43(ただし、n=0、1、2、3、4、5または6(すなわち、n=0〜6)、x=離脱基、例えば、メシラート、トシレート、ノシレートまたは他のスルホネートエステル若しくはハライド)に示される化合物などの、保護された前駆体を用いて、イサチン誘導体、例えば、[18F]11を合成することによって、解消できる。
【0126】
【化4】
【0127】
特に好ましい保護されたアルキン前駆体は、(S)−1−{[1’−[1−(2−プロピニル)]−(1’2’−ジヒドロ−2’−オキソスピロ(1,3−ジオキサン−2,3’−[3H]インドール)−5’−スルホニル}−2−(2,4−ジフルオロフェノキシメチル)−ピロリジン[(S)-1-{[1’-[1-(2-Propynyl)]-(1’2’-dihydro-2’-oxospiro(1,3-dioxane-2,3’-[3H]indol)-5’-sulfonyl}-2-(2,4-difluor ophenoxymethyl)-pyrrolidine](化合物39)である。このような保護されたアルキン前駆体の使用によって、C−3位置での望ましくない副反応が防止される。
【0128】
【化5】
【0129】
これらの方法に加えて、本発明の第二の態様に係る化合物は、例えば、適当な錫前駆体のハロ−脱メタル化(halo-demetallation)、望ましい金属による、好ましくはイサチンに結合した適当なリガンドによるキレート化、適当なアリールまたはアルキル離脱基の存在下での[18F]フッ素による置換によって、放射性標識されうる。
【図面の簡単な説明】
【0130】
図面
【図1】図1は、ターゲット化合物の合成の概略図である。試薬および条件は下記のとおりである:(a)フェノール/フッ素置換フェノール/4−テトラヒドロピラン、NaH、DMF、80℃、17h;(b)4(3H)−ピリミドン、PPh3、DIAD、DCM、rt、48h;(c)臭化プロパルギル、KOH、DMF、rt、18h;(d)TFA、DCM、0℃、1h;(e)5−クロロスルホニルイサチン、TEA、THF/DCM、rt、19h;(f)4−フルオロベンジルブロミド、K2CO3、DMF、rt、2h;(g)臭化プロパルギル、K2CO3、DMF、rt、2h;(h)2−フルオロエチルアジド、CuSO4、L−アスコルビン酸、DMF、rt、2h。
【図2−1】図2(i)は、(S)−1−(2−プロピニル)−5−(2−(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物25)と[18F]フルオロエチルアジド(化合物27)との反応による化合物[18F]11の合成の概略図である。化合物26は、2−(トルエン−4−スルホニル)エチルアジドである。試薬は下記のとおりである:(a)[18F]KF,Kryptofix[2,2,2],アセトニトリル;(b)CuSO4、アスコルビン酸ナトリウム、リン酸緩衝液 pH6.0、化合物25。
【図2−2】図2(ii)は、(S)−1−(アジドメチル)−5−(2−(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン](化合物37)と(3−フルオロプロプ−1−イン)(化合物28)との反応による、化合物6 ((S)−1−[4−(2−フルオロエチル)−1H−[1,2,3]−トリアゾール−1−イル]メチル−5−(2−(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン)の合成の概略図である。試薬は下記のとおりである:(a)CuSO4、L−アスコルビン酸、DMF。
【図3−1】図3は、[18F]11を含む反応混合物の予備HPLCトレース(左)および分析ピーク分離(右)である。上図:放射能チャンネル;下図:254nmでのUVチャンネル。
【図3−2】図3は、[18F]11を含む反応混合物の予備HPLCトレース(左)および分析ピーク分離(右)である。上図:放射能チャンネル;下図:254nmでのUVチャンネル。
【図4】図4は、2、10、30及び60分後のRIF−1腫瘍を有するマウスにおける[18F]11の生体内分布を示す。データは、平均値±SEMである;n=3〜6匹マウス/時点。
【図5−1】図5は、ラジオ−HPLCにより血漿中で評価された[18F]11のインビボでの代謝を示す。上左図:[18F]11標準;上右図:2分後の血漿;下左図:15分後の血漿;下右図:60分後の血漿。
【図5−2】図5は、ラジオ−HPLCにより血漿中で評価された[18F]11のインビボでの代謝を示す。上左図:[18F]11標準;上右図:2分後の血漿;下左図:15分後の血漿;下右図:60分後の血漿。
【図6−1】図6は、ラジオ−HPLCにより肝臓及び尿中で評価された[18F]11のインビボでの代謝を示す。上図:肝臓抽出物;下図:尿抽出物;左〜右図:それぞれ、2、15及び60分。
【図6−2】図6は、ラジオ−HPLCにより肝臓及び尿中で評価された[18F]11のインビボでの代謝を示す。上図:肝臓抽出物;下図:尿抽出物;左〜右図:それぞれ、2、15及び60分。
【図6−3】図6は、ラジオ−HPLCにより肝臓及び尿中で評価された[18F]11のインビボでの代謝を示す。上図:肝臓抽出物;下図:尿抽出物;左〜右図:それぞれ、2、15及び60分。
【図7】図7は、2、10、30及び60分後の[125I]標識された(S)−1−(4−ヨードベンジル)−5−(2−フェノキシメチル−ピロリジン−1−スルホニル)イサチン(本明細書中では、化合物[125I]14と称する)の生体内分布を示す。データは、平均値±SEMである;n=3匹マウス/時点。
【図8−1】図8は、HPLCによってマウス肝臓S9画分中で評価された既知の化合物 (S)−1−(4−ヨードベンジル)−5−(2−フェノキシメチル−ピロリジン−1−スルホニル)イサチン(化合物14)、(S)−1−(4−フルオロベンジル)−5−(2−フェノキシメチル−ピロリジン−1−スルホニル)イサチン(化合物15)および1−(4−フルオロベンジル)−5−(ピロリジン−1−スルホニル)イサチン(化合物16)のインビトロ代謝を示す。上図、左〜右図:0時での化合物14、15及び16;下図、左〜右図:60分間インキュベーション後の化合物14、15及び16。
【図8−2】図8は、HPLCによってマウス肝臓S9画分中で評価された既知の化合物 (S)−1−(4−ヨードベンジル)−5−(2−フェノキシメチル−ピロリジン−1−スルホニル)イサチン(化合物14)、(S)−1−(4−フルオロベンジル)−5−(2−フェノキシメチル−ピロリジン−1−スルホニル)イサチン(化合物15)および1−(4−フルオロベンジル)−5−(ピロリジン−1−スルホニル)イサチン(化合物16)のインビトロ代謝を示す。上図、左〜右図:0時での化合物14、15及び16;下図、左〜右図:60分間インキュベーション後の化合物14、15及び16。
【図8−3】図8は、HPLCによってマウス肝臓S9画分中で評価された既知の化合物 (S)−1−(4−ヨードベンジル)−5−(2−フェノキシメチル−ピロリジン−1−スルホニル)イサチン(化合物14)、(S)−1−(4−フルオロベンジル)−5−(2−フェノキシメチル−ピロリジン−1−スルホニル)イサチン(化合物15)および1−(4−フルオロベンジル)−5−(ピロリジン−1−スルホニル)イサチン(化合物16)のインビトロ代謝を示す。上図、左〜右図:0時での化合物14、15及び16;下図、左〜右図:60分間インキュベーション後の化合物14、15及び16。
【図9】図9は、A)10μg/mL CDDPまたは10μg/mL CHXで24時間処置したRIF−1細胞、B)50μg/mL CDDP(シスプラチン)で処置したPEO1/4細胞、C)100μM CDDPまたは100μM VP−16を、単独でまたは組み合わせて、処置したLNM35細胞、における60分後の[18F]11の細胞への取り込みを示す。カスパーゼ−3アッセイ(C右パネル)を、実施例で記載したようにして行った。*スチューデントt−テスト(Student’s t-test)、p<0.005。
【図10】図10は、CDDPで処置したマウスにおける[18F]11の分布の全身矢状イメージ(whole body sagittal image)を示す。イメージは、約100μCi [18F]11を静脈内注射してから30〜60分で合計した。
【図11】図11は、100mg/kg シクロホスファミドで処置された38C18異種移植片を有するマウスにおける[18F]11の分布の全身矢状イメージ(whole body sagittal image)を示す。左から右へのイメージは、約100μCi [18F]11を静脈内注射してから24時間に撮影された、軸性(axial)イメージ、冠性(coronal)イメージおよび矢状(sagittal)イメージである。
【図12】図12は、それぞれ、化合物35(上図)および化合物36(下図)の合成の概略図である。
【図13】図13は、ベヒクルまたはCDDPで処置されたRIF−1細胞における[18F]11の取り込みプロフィールを示す。データは、1mgの全細胞タンパク質当たりで平均された減水補正されたカウント/分として表わされる。データは、平均値±SEMであり、3連で行われた。
【図14】図14は、ベヒクル(50%DMSO)またはCDDP(10mg/kg 単回投与)で処置されたRIF−1腫瘍における[18F]11の取り込みを示す。放射性トレーサーを注射してから60分での[18F]由来の放射能レベルを分析し、血中の放射能レベルに対する割合として表わす。データは、平均値±SEMであり、n=8匹マウス/群である。
【図15】図15は、シクロホスファミド(CPA、100mg/kg)またはベヒクルで処置された腫瘍における[18F]の生体内分布を示し、この際、カウント密度を各19時点での興味のある各領域で平均して、興味のある領域に関する時間対放射能曲線(time versus radioactivity curve)(TAC)を得た。
【図16】図16は、シクロホスファミド(CPA、100mg/kg)またはベヒクル(Veh)で処置された腫瘍における、注射してから60分後(NUV60)の標準化取り込み値(Normalised Uptake Value)(NUV)、および0〜60分のNUVの積分として算出されるNUV曲線下の面積(Area Under the NUV curve)(AUC)のヒストグラムである。
【図17】図17は、ベヒクルまたはシクロホスファミドで処置された2匹の38C13異種移植片を有するマウスの代表的なOSEM3D再構築[18F]11 PETイメージである。丸は腫瘍を表わす。
【図18】図18は、カスパーゼ−3と同種のターゲットであるポリ(ADP−リボース)ポリメラーゼ(caspase-3 cognate target poly(ADP-ribose)polymerase)(PARP)に関する化合物11(「イサチン−7」として示す)および化合物14(「イサチン−4」として示す)の阻害活性を示す。
【図19】図19は、(S)−1−(2−プロピニル)−5−(2−(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物25)と[18F]フルオロエチルアジド(化合物27)との反応による化合物[18F]11の合成の第2の概略図である。
【図20】図20は、保護されたアルキン前駆体 (S)−1−{[1’−[1−(2−プロピニル)]−(1’2’−ジヒドロ−2’−オキソスピロ(1,3−ジオキサン−2,3’−[3H]インドール)−5’−スルホニル}−2−(2,4−ジフルオロフェノキシメチル)−ピロリジン(化合物39)および保護されたトリゾール (S)−1−{[1’−[1−(2−フルオロエチル)−1H−[1,2,3]−トリアゾール−4−イル]−(1’2’−ジヒドロ−2’−オキソスピロ(1,3−ジオキサン−2,3’−[3H]インドール)−5’−スルホニル}−2−(2,4−ジフルオロフェノキシメチル)−ピロリジン(化合物40)の合成に関する一般的な反応スキームである。
【図21】図21は、保護されたアルキン前駆体 (S)−1−{[1’−[1−(2−プロピニル)]−(1’2’−ジヒドロ−2’−オキソスピロ(1,3−ジオキサン−2,3’−[3H]インドール)−5’−スルホニル}−2−(2,4−ジフルオロフェノキシメチル)−ピロリジン(化合物39)からの[18F]11の製造に関する具体的な放射化学反応の概略図である。
【図22】図22は、脱保護前の標識混合物のHPLC分析を示す。赤:放射能チャンネル、青:254nmでのUVチャンネル。残りの[18F]フルオロエチルアジド:1.3分、標識されたイサチンアセタール(isatin acetale):6.8分。
【図23】図23は、[18F]11の予備HPLCを示す。上図:放射能チャンネル。11:45分のピークは[18F]11に相当する。下図:UVチャンネル、254nm。11:51分のシグナルは安定した不純物である。
【図24】図24は、配合された[18F]11の分析HPLCを示す。上図:254nmでのUVチャンネル、下図:3.3分に[18F]11を示す放射能シグナル。
【図25】図25は、N−(2−プロピニル)イサチン(化合物41)からの弱いイサチンカスパーゼ−3阻害剤であるN−[1−(2−フルオロエチル)−1H−[1,2,3]−トリアゾール−4−イル]イサチン(化合物42)の合成の概略図である。
【図26】図26は、処置によりアポトーシスが誘導された癌細胞における[18F]11及び[18F]42の結合性を示す。(左パネル)4−ヒドロキシペルオキシシクロホスファミド 4−HC(4−HC;1μg/mL;24h)で処置して、アポトーシスを誘導した38C13リンパ腫細胞における化学構造および[18F]11結合性。すべての処置およびコントロールサンプルについて、放射能データは、1mg全細胞タンパク質当たりの減衰補正されたカウントとして表わされる。(右パネル)38C13リンパ腫細胞における、低親和性プローブ、[18F]42、の結合に関する4−HCの効果。
【実施例】
【0131】
実施例1−化合物の合成
((S)−1−(4−フルオロベンジル)−5−(2−フェノキシメチル−ピロリジン−1−シルホニル)イサチン)をリード化合物として使用して、ターゲット化合物のライブラリーを作製した。左側のエーテル部分およびN−1位置で修飾をした。フッ素基を左側のフェニルエーテル基に導入して、この位置での複素環及びアルキンのトレランスを調べた。N−1位置での1,2,3トリアゾール基に対するトレランスもまた調べた。官能化ピロリジンを5−クロロスルホニルイサチンで縮合した後、炭酸カリウム/DMFを用いてイサチン窒素をアルキル化することによって図1に示されるように、ターゲット化合物を合成した。全ての必要な出発材料は市販されている、またはLee, D. et al 8, Chu, W. et al9及びKopka, K. et al13に記載されるのと同様にして製造する。
【0132】
4−ヒドロキシテトラヒドロピランおよび市販のフェノールをトシレート17と反応させることによって、良好な収率でピロリジン19a〜fを得た。4(3H)−ピリミドンを、Wipf et al.15に報告される方法の修飾を用いて(S)−1−(tert−ブトキシカルボニル)−2−ピロリジンメタノール(化合物18)と結合させ、69%の収率でピロリジン19gを得た。18を臭化プロパルギルでO−アルキル化することによって、67%の収率で相当するエーテル19hを得た。BOCで保護されたピロリジン19a〜hをトリフルオロ酢酸で脱保護した後、5−クロロスルホニルイサチンと結合させることによって、並から良好な収率でスルホンアミド20a〜hを得た。次に、塩基性条件下で20b〜hを4−フルオロベンジルブロミドで処理することによって、新規な化合物1〜5ならびに中間化合物22及び23を得、一方、20a及び20dを臭化プロパルギルで処理することによって、それぞれ、中間アルキン24及び25を得た。2−フルオロエチルアジドを各アルキン前駆体23、24及び25で銅触媒による付加環化を行うことによって、新規なトリアゾール10〜12を調製した。いささか驚くべきことに、イサチン足場は、硫酸銅及びアスコルビン酸の存在下で90℃で加熱すると分解し、これにより新規なトリアゾール10〜12の収率が悪くなった。この問題は、アルキン前駆体に対する硫酸銅濃度を5%から50%に上げ、周囲温度で反応を行い、さらに反応時間を1時間にまで短くすることによって一部解決し、これにより、トリアゾール10〜12を48〜57%の収率で得た。
【0133】
化学 試薬及び溶媒は、Sigma-Aldrich(Gillingham, United Kingdom)から購入し、さらに精製することなく使用した。水酸化カリウム及び炭酸カリウムは、5酸化リンで真空デシケーター内で保存した。特記しない限り、すべての反応はアルゴン下で行われた。石油エーテルは、40℃〜60℃で蒸留する画分を意味する。自動化フラッシュクロマトグラフィーは、RediSep 4gまたは12g 正常相シリカカートリッジ(流速 12mL/minまたは26mL/min)を用いて、CombiFlash Companion machine(Companion Presearch Ltd.)で行った。マニュアルフラッシュクロマトグラフィーは、Davisil中性シリカ(neutral silica)(60Å、60〜200ミクロン、Fisher Scientific, Loughborough, UK)を用いて行い、溶媒混合物は容積/容積として記載する。1H NMRスペクトルをBruker Avance 600 MHz NMR machineで得、スペクトルを残りの溶媒で参照する。結合定数(J)はヘルツ(Hz)で示す。GC−MSデータは、Agilent 6890Nシステムを用いて電子イオン化下で得た。質量スペクトルは、WatersMicromass LCT Premier machineで陽極電気スプレーイオン化(positive electrospray ionisation)形式で得た。融点は、Stuart Scientific SMP1融点測定装置で毛細管中で測定し、修正しない。薄層クロマトグラフィ(TLC)用の溶媒混合物は容積/容積で記載し、サンプルをアルミニウムで塗布された中性シリカ板(aluminium backed neutral silica plates)(厚み 0.2mm)(Fluka, Seelze, Germany)で発色させた。化合物1〜5、10〜16、20b〜h及び22〜25の純度の分析は分析HPLCで評価し;化合物19b〜hはGC−MSで分析した。すべての化合物の純度は95%超であった。[18F]フッ化物を、濃縮(enriched)[18O]H2Oターゲットの16.4MeVプロトン照射による18O(p,n)18F核反応を用いてサイクロトロン(GE PETrace)によって製造した。
【0134】
HPLC法 a)非放射性化合物の純度分析は、Phenomenex Luna 50×4.6mm(3μm)HPLCカラムを備えたLaura 3 software(Lablogic, Sheffield, UK)を有するAgilent 1100シリーズシステムで行い、この際、0.1M ギ酸アンモニウム及びメタノール/アセトニトリル(1.8:1 v/v)の移動相、グラジエント(50%有機相で1分;50(R)90%有機相で14分;90%有機で4分;90(R)50%有機相で4分)、流速 1mL/min及び波長254nmであった。
【0135】
b)予備ラジオ−HPLCを、Bioscan Flowcount FC-3400 PINダイオード検出器(Lablogic)及びlinear UV-200 detector(波長 254nm)を備えたBeckman System Goldで行った。Phenomenex Onyx C18 100×10mm HPLCカラムならびに水およびメタノール/アセトニトリル(1.8:1 v/v)の移動相、グラジエント 45(R)90%有機相で20分および流速 3mL/minであった。
【0136】
c)分析ラジオ−HPLCを、水およびメタノール/アセトニトリル(1.8:1 v/v)の移動相、グラジエント 60(R)90%有機相で20分、流速 1mL/minを用いて、Bioscan Flowcount FC3200 ヨウ化ナトリウム/PMT ガンマ線検出器(Lablogic)およびPhenomenex Luna 50×4.6mm(3μm)カラムを使用する以外は、上記と同様にして行った。
【0137】
既知の化合物の製造
下記化合物を確立された文献の方法に従って合成したところ、1H NMRスペクトルデータが報告された値と一致した:(S)−1−ベンジル−5−(2−フェノキシメチル−ピロリジン−1−スルホニル)イサチン(化合物13)、(S)−1−(4−ヨードベンジル)−5−(2−フェノキシメチル−ピロリジン−1−スルホニル)イサチン(化合物14)及び(S)−1−(4−フルオロベンジル)−5−(2−フェノキシメチル−ピロリジン−1−スルホニル)イサチン(化合物15)。
【0138】
1−(4−フルオロベンジル)−5−(ピロリジン−1−スルホニル)イサチン(化合物16) 乾燥DMF(8mL)における5−ピロリジン−1−スルホニルイサチン8(0.14g、0.5mmol)の氷冷した攪拌溶液に、水素化ナトリウム(40mg、1mmol)を添加した。30分後、4−フルオロベンジルブロミド(0.38g、2mmol)を添加し、混合物を室温まで加温した。19時間後、オレンジ色の溶液を10%NH4Cl水溶液(25mL)に注ぎ、DCMで抽出した(3×15mL)。真空中で濃縮後、残渣をジエチルエーテル(10mL)中にとり、水で洗浄し(3×10mL)、Na2SO4で乾燥した。クロマトグラフィー(ジエチルエーテル/ヘキサン)によって、標題化合物をオレンジ色のゴム状物として得た(83mg、43%)。HRMS (ESI) = 389.0988 (M + H)+. C19H18FN2O4Sに関する算出値 389.0971. 1H NMR (600 MHz, CDCl3) δ 8.05 (d, J = 1.5 Hz, 1H), 7.99 (dd, J = 8.4 Hz, J = 1.5 Hz, 1 H), 7.35-7.32 (m, 2 H), 7.09-7.06 (m, 2 H), 6.91 (d, J = 8.4 Hz, 1 H), 4.94 (s, 2 H), 3.25-3.23 (4 H, m), 1.84-1.79 (4 H, m). TLC (UV254) Rf= 0.63 (4:1 酢酸エチル/ヘキサン). HPLC tR = 6.83 min。
【0139】
新規な化合物およびその中間体の製造
(S)−tert−ブチル2−(4−フルオロフェノキシメチル)ピロリジン−1−カルボキシレート(化合物19b) 乾燥DMF(10mL)における4−フルオロフェノール(0.27g、2.4mmol)の攪拌溶液に、水素化ナトリウム(鉱油における60%w/w)(0.11g、2.8mmol)を添加した。30分後、乾燥DMF(5mL)における((S)−1−(tert−ブトキシカルボニル)ピロリジン−2−イル)トルエン−4−スルホネート8(化合物)15(0.71g、2.0mmol)を添加し、この混合物を80℃で17時間加熱した。反応物を室温にまで冷却し、1M NaOH(25mL)に注ぎ、DCMで抽出した(3×15mL)。有機画分を合わせたものを真空中で還元し、ジエチルエーテル(20mL)を添加した後、1M NaOH(1×20mL)、水(1×20mL)、さらにブライン(1×20mL)で洗浄し、Na2SO4で乾燥した。クロマトグラフィー(ヘキサン/酢酸エチル)によって、生成物を無色の油として得た(0.36g、61%)。HRMS (ESI) = 296.1654 (M + H)+. C16H23FNO3に関する算出値 296.1656. 1H NMR (600 MHz, CDCl3) δ 6.97-6.93 (m, 2 H), 6.88-6.84 (m, 2 H), 4.18-4.03 (m, 2 H), 3.94-3.73 (m, 1 H), 3.46-3.32 (m, 2 H), 2.07-1.81 (m, 4 H), 1.47 (s, 9 H). TLC (UV254) Rf = 0.51 (2:1 ヘキサン/酢酸エチル)。
【0140】
(S)−tert−ブチル 2−(3−フルオロフェノキシメチル)ピロリジン−1−カルボキシレート(化合物19c) 3−フルオロフェノールを使用する以外は、19bの方法に従って、19cを調製した。クロマトグラフィー(ヘキサン/酢酸エチル)によって、標題化合物を無色の油として得た(0.32g、54%)。HRMS (ESI) = 296.1657 (M + H)+. C16H23FNO3に関する算出値 296.1656 1H NMR (600 MHz, CDCl3) δ 7.20-7.15 (m, 1 H), 6.73-6.69 (m, 1 H), 6.65-6.59 (m, 2 H), 4.18-4.03 (m, 2 H), 3.97-3.74 (m, 1 H), 3.48-3.29 (m, 2 H), 2.05-1.79 (m, 4 H), 1.48 (s, 9 H). TLC (UV254) Rf = 0.40 (3:1 ヘキサン/酢酸エチル)。
【0141】
(S)−tert−ブチル 2−(2,4−ジフルオロフェノキシメチル)ピロリジン−1−カルボキシレート(化合物19d) 2,4−ジフルオロフェノールを使用する以外は、19bの方法に従って、19dを調製した。クロマトグラフィー(ヘキサン/酢酸エチル)によって、標題化合物を無色の油として得た(1.92g、58%)。HRMS (ESI) = 314.1560 (M + H)+. C16H22F2NO3に関する算出値 314.1562. 1H NMR (600 MHz, CDCl3) δ 7.08-6.76 (m, 3 H), 4.21-3.87 (m, 3 H), 3.47-3.32 (m, 2 H), 2.16-1.85 (m, 4 H), 1.48 (s, 9 H). TLC (UV254) Rf = 0.51 (2:1 ヘキサン/酢酸エチル)。
【0142】
(S)−tert−ブチル 2−(3,5−ジフルオロフェノキシメチル)ピロリジン−1−カルボキシレート(化合物19e) 3,5−ジフルオロフェノールを使用する以外は、19bの方法に従って、19eを調製した。クロマトグラフィー(ヘキサン/酢酸エチル)によって、標題化合物を無色の油として得た(0.31g、53%)。HRMS (ESI) = 314.1563 (M + H)+. C16H22F2NO3に関する算出値 314.1562. 1H NMR (600 MHz, CDCl3) δ 6.44-6.29 (m, 3 H), 4.11-3.98 (m, 2 H), 3.91-3.72 (m, 1 H), 3.42-3.24 (m, 2H), 1.99-1.79 (m, 4 H), 1.42 (s, 9 H). TLC (UV254) Rf= 0.56 (2:1 ヘキサン/酢酸エチル)。
【0143】
(S)−tert−ブチル 2−(4−テトラヒドロピラニルオキシメチル)ピロリジン−1−カルボキシレート(化合物19f) 4−ヒドロキシテトラヒドロピランを使用する以外は、19bの方法に従って、19fを調製した。クロマトグラフィー(酢酸エチル)によって、標題化合物を無色の油として得た(0.14g、25%)。HRMS (ESI) = 286.2012 (M + H)+. C15H28NO4に関する算出値 286.2013. 1H NMR (600 MHz, CDCl3) δ 3.97-3.83 (m, 2 H), 3.64-3.23 (m, 9 H), 1.97-1.72 (m, 5 H), 1.60-1.52 (m, 2 H), 1.46 (s, 9 H). TLC (I2) Rf = 0.62 (酢酸エチル)。
【0144】
(S)−tert−ブチル 2−(ピリミジン−4−イルオキシメチル)ピロリジン−1−カルボキシレート(化合物19g) 乾燥DCM(10mL)におけるN−tert−ブトキシカルボニル−L−ピロリノール(0.81g、4mmol)18の攪拌溶液に、トリフェニルホスフィン(5.24g、20mmol)、さらには4(3H)−ピリミドン(0.77g、8mmol)を加えた。この溶液を氷浴で冷却し、DIAD(3.24g、16mmol)を10分間滴下した。48時間後、GC−MSでは、18の完全な変換が示され、この反応混合物を水(30mL)に注ぎ、有機画分を集め、水相を別のDCM(2×20mL)で洗浄した。有機画分を合わせたものを1M NaOH(2×15mL)、さらにはブライン(1×15mL)で洗浄し、Na2SO4で乾燥した。大量の溶媒を除去することによって、オレンジ色のゴム状物として得、ヘキサン/ジエチルエーテル(1:1)を添加することによって、トリフェニルホスフィンオキシドの沈殿物が形成し、これを濾別した。クロマトグラフィー(酢酸エチル)によって、目的生成物を無色の油として得た(0.77g、69%)。HRMS (ESI) = 280.1655 (M + H)+. C14H22N3O3に関する算出値 280.1656. 1H NMR (600 MHz, CDCl3) δ 8.71 (s, 1 H), 8.39 (d, J= 6 Hz, 1 H), 6,69 (d, J = 6 Hz, 1 H), 4.45-3.91 (m, 3 H), 3.40-3.33 (m, 2 H), 2.00-1.82 (m, 4 H), 1.42 (s, 9 H). TLC (UV254) Rf= 0.49 (酢酸エチル)。
【0145】
(S)−tert−ブチル 2−(2−プロピニルオキシメチル)ピロリジン−1−カルボキシレート(化合物19h) 乾燥DMF(10mL)における18(0.40g、2mmol)の攪拌溶液に、水酸化カリウム(0.56g、10mmol)を添加した後、臭化プロパルギル(トルエンにおいて、80wt.%)(0.48g、4mmol)を5分かけて滴下した。18時間後、反応混合物を水(30mL)に注ぎ、DCMで洗浄した(3×15mL)。有機画分を合わせたものを真空中で濃縮し、残った液体をジエチルエーテル(15mL)にとり、水(2×10mL)、次にブライン(1×10mL)で洗浄し、Na2SO4で乾燥した。クロマトグラフィー(ヘキサン/酢酸エチル)によって、目的生成物を無色の油として得た(0.32g、67%)。HRMS (ESI) = 240.1597 (M + H)+. C13H22NO3に関する算出値 240.1594. 1H NMR (600 MHz, CDCl3) δ 4.13 (s, 2 H), 3.96-3.88 (m, 1 H), 3.64 (dd, J = 9 Hz, J = 3.6 Hz, 1 H), 3.49-3.22 (m, 3 H), 2.40 (s, 1 H), 1.94-1.78 (m, 4 H), 1.46 (s, 9 H). TLC (I2) Rf = 0.67 (1:1 ヘキサン/酢酸エチル)。
【0146】
(S)−5−(2−(4−フルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物20b) 氷浴中で冷却した乾燥DCM(4mL)における19b(0.15g、0.5mmol)の攪拌溶液に、TFA(0.6mL、10mmol)を添加した。1時間後、大量の溶媒を真空中で除去し、残った残渣を乾燥DCM(8mL)にとり、氷浴中で冷却した。次に、乾燥トリエチルアミン(1.5mL)、さらに乾燥THF(4mL)における5−クロロスルホニルイサチン8(0.16g、0.65mmol)を添加した後、溶液を攪拌した。19時間後、大量の溶媒を真空中で除去し、DCM(10mL)中に再溶解し、水(2×10mL)、次にブライン(1×10mL)で洗浄し、Na2SO4で乾燥した。クロマトグラフィー(ヘキサン/酢酸エチル)によって、目的生成物をオレンジ色の固体として得た(104mg、51%)。Mp: 205-207℃. HRMS (ESI) = 405.0941 (M + H)+. C19H18FN2O5Sに関する算出値 405.0920. 1H NMR (600 MHz, CDCl3) δ 8.10 (s,1 H), 8.08 (dd, J = 8.4 Hz, J = 1.8 Hz, 1 H), 8.00 (br, 1 H), 7.00 (d, J = 7.8 Hz, 1 H), 6.99-6.95 (m, 2 H), 6.83-6.81 (m, 2 H), 4.17 (dd, J= 9.6 Hz, J = 3.6 Hz, 1 H), 3.98-3.95 (m, 1 H), 3.91 (dd, J= 9 Hz, J = 7.8 Hz, 1 H), 3.54-3.50 (m, 1 H), 3.24-3.19 (m, 1 H), 2.10-1.99 (m, 2 H), 1.87-1.77 (m, 2 H). TLC (UV254) Rf= 0.27 (2:1 酢酸エチル/ヘキサン). HPLC tR = 7.83。
【0147】
(S)−5−(2−(3−フルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物20c) 19cを使用する以外は、20bの方法に従って、20cを調製して、オレンジ色の固体を得た(93mg、46%)。Mp: 201-203℃. HRMS (ESI) = 405.0933 (M + H)+. C19H18FN2O5Sに関する算出値 405.0920. 1H NMR (600 MHz, CDCl3) δ 8.14 (br, 1 H), 8.09-8.06 (m, 2 H), 7.21 (q, J = 7.2 Hz, 1 H), 7.03 (d, J = 7.8 Hz, 1 H), 6.67-6.62 (m, 2 H), 6.55 (dt, J = 10.8 Hz, J = 2.4 Hz, 1 H), 4.17 (dd, J = 9 Hz, J = 3 Hz, 1 H), 4.01-3.97 (m, 1 H), 3.94 (dd, J = 9 Hz, J = 7.2 Hz, 1 H), 3.54-3.50 (m, 1 H), 3.27-3.25 (m, 1 H), 2.08-1.96 (m, 2 H), 1.88-1.77 (m, 2 H). TLC (UV254) Rf = 0.36 (2:1 酢酸エチル/ヘキサン). HPLC tR = 8.27 min。
【0148】
(S)−5−(2−(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物20d) 19dを使用する以外は、20bの方法に従って、20dを調製して、オレンジ色の固体を得た(0.86g、34%)。Mp: 185-187℃. HRMS (ESI) = 423.0834 (M + H)+. C19H17F2N2O5Sに関する算出値 423.0826. 1H NMR (600 MHz, CDCl3) δ 8.08-8.06 (m, 2 H), 7.97 (br, 1 H), 7.03 (d, J = 9 Hz, 1 H), 6.97-6.92 (m, 1 H), 6.87-6.77 (m, 2 H), 4.21 (dd, J= 8.4 Hz, J = 2.4 Hz, 1 H), 4.03-3.97 (m, 2 H), 3.56-3.52 (m, 1 H), 3.23-3.17 (m, 1 H), 2.11-2.01 (m, 2 H), 1.88-1.75 (m, 2 H). TLC (UV254) Rf = 0.46 (2:1 酢酸エチル/ヘキサン). HPLC tR = 8.12 min。
【0149】
(S)−5−(2−(3,5−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物20e) 19eを使用する以外は、20bの方法に従って、20eを調製して、オレンジ色の固体を得た(112mg、53%)。Mp: 196-198℃. HRMS (ESI) = 423.0834 (M + H)+. C19H17F2N2O5Sに関する算出値 423.0826. 1H NMR (600 MHz, CDCl3) δ 8.10-8.06 (m, 2 H), 8.04 (br, 1 H), 7.05 (d, J = 8.4 Hz), 6.45-6.38 (m, 3 H), 4.18 (dd, J = 8.4 Hz, J = 2.4 Hz, 1 H), 3.99-3.92 (m, 2 H), 3.56-3.49 (m, 1 H), 3.24-3.17 (m, 1 H), 2.04-1.92 (m, 2 H), 1.86-1.77 (m, 2 H). TLC (UV254) Rf= 0.36 (2:1 酢酸エチル/ヘキサン). HPLC tR = 9.18 min。
【0150】
(S)−5−(2−(テトラヒドロ−2H−ピラン−4−イルオキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物20f) 19fを使用する以外は、20bの方法に従って、20fを調製して、オレンジ色のゴム状物を得た(63mg、32%)。HRMS (ESI) = 395.1282 (M + H)+. C18H23N2O6Sに関する算出値 395.1277. 1H NMR (600 MHz, CDCl3) δ 8.17 (br, 1 H), 8.10-8.08 (m, 2 H), 7.07 (d, J = 8.4 Hz, 1 H), 3.94-3.90 (m, 2 H), 3.77-3.73 (m, 1 H), 3.70 (dd, J = 9.6 Hz, J = 3 Hz, 1 H), 3.56-3.51 (septet, J = 4.2 Hz, 1 H), 3.48-3.44 (m, 3 H), 3.13-3.11 (m, 1 H), 2.05-1.87 (m, 4 H), 1.72-1.64 (m, 2 H), 1.60-1.52 (m, 2 H). TLC (UV254) Rf = 0.26 (4:1 酢酸エチル/ヘキサン). HPLC tR = 2.65 min。
【0151】
(S)−5−(2−(ピリミジン−4−イルオキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物20g) 19gを使用する以外は、20bの方法に従って、20gを調製した。クロマトグラフィー(酢酸エチル)によって、オレンジ色のゴム状物を得た(51mg、27%)。HRMS (ESI) = 389.025 (M + H)+. C17H17N4O5Sに関する算出値 389.020. 1H NMR (600 MHz, CDCl3) δ 8.79 (s, 1 H), 8.45 (d, J = 5.4 Hz, 1 H), 8.12 (d, J = 1.8 Hz, 1 H), 8.09 (dd, J = 8.4 Hz, J = 1.8 Hz, 1 H), 7.95 (br, 1 H), 7.03 (d, J = 8.4 Hz, 1 H), 6.72 (d, J = 5.4 Hz, 1 H), 4.57 (dd, J = 10.8 Hz, J = 4.8 Hz, 1 H), 4.39 (dd, J = 10.8 Hz, J=7.2 Hz, 1 H), 4.08-4.04 (m, 1 H), 3.53-3.46 (m, 1 H), 3.27-3.22 (m, 1 H), 2.01-1.93 (m, 2 H), 1.84-1.74 (m, 2 H). TLC (UV254) Rf= 0.49 (9:1 酢酸エチル/メタノール). HPLC tR = 1.90 min。
【0152】
(S)−5−(2−(2−プロピニルオキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物20h) 19hを使用する以外は、20bの方法に従って、20hを調製して、オレンジ色のゴム状物を得た(92mg、53%)。HRMS (ESI) = 349.0867 (M + H)+. C16H17N2O5Sに関する算出値 349.067. 1H NMR (600 MHz, CDCl3) δ 8.10-8.07 (m, 2 H), 7.84 (br, 1 H), 7.04 (d, J = 8.4 Hz, 1 H), 4.16 (s, 2 H), 3.83-3.79 (m, 1 H), 3.72 (dd, J= 9.6 Hz, J = 3.6 Hz, 1 H), 3.54 (dd, J = 9.6 Hz, J = 3.6 Hz, 1 H), 3.45-3.43 (m, 1 H), 3.19-3.15 (m, 1 H), 2.46 (t, J= 2.4 Hz, 1 H), 1.96-1.89 (m, 2 H), 1.74-1.67 (m, 2 H). TLC (UV254) Rf = 0.28 (2:1 酢酸エチル/ヘキサン). HPLC tR =2.93 min。
【0153】
(S)−1−(4−フルオロベンジル)−5−(2−(4−フルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物22) 乾燥DMF(3mL)における20b(40mg、0.1mmol)の攪拌溶液に、炭酸カリウム(21mg、0.15mmol)、さらには4−フルオロベンジルブロミド(76mg、0.4mmol)を添加した。2時間後、TLCにより、20bの完全な変換が示され、この溶液を10%NH4Cl水溶液(10mL)に注ぎ、DCMで抽出した(3×10mL)。有機画分を合わせたものを真空中で濃縮し、ジエチルエーテル(10mL)中にとり、水(2×10mL)、次にブライン(1×10mL)で洗浄し、Na2SO4で乾燥した。クロマトグラフィー(ヘキサン/酢酸エチル)によって、標題化合物をオレンジ色のゴム状物として得た(34mg、66%)。HRMS (ESI) = 513.1306 (M + H)+. C26H23F2N2O5Sに関する算出値 513.1296. 1H NMR (600 MHz, CDCl3) δ 8.04 (d, J = 1.8 Hz, 1 H), 7.97 (dd, J = 8.4 Hz, J = 1.8 Hz, 1 H), 7.33-7.30 (m, 2 H), 7.09-7.05 (m, 2 H), 6.96-6.92 (m, 2 H), 6.86 (d, J= 8.4 Hz, 1 H), 6.81-6.77 (m, 2 H), 4.92 (d, J = 15.6, 1 H), 4.91 (d, J= 15.6, 1 H), 4.14 (dd, J = 9.6 Hz, J = 3.6 Hz, 1 H), 3.95-3.92 (m, 1 H), 3.88 (dd, J = 9.6 Hz, J = 7.2 Hz, 1 H), 3.51-3.47 (m, 1 H), 3.20-3.15 (m, 1 H), 2.06-1.93 (m, 2 H), 1.83-1.73 (m, 2 H). TLC (UV254) Rf = 0.61 (2:1 酢酸エチル/ヘキサン). HPLC tR = 12.25 min。
【0154】
(S)−1−(4−フルオロベンジル)−5−(2−(3−フルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物1) 20cを使用する以外は、22の方法に従って、1を調製して、オレンジ色のゴム状物を得た(31mg、61%)。HRMS (ESI) = 513.1298 (M + H)+. C26H23F2N2O5Sに関する算出値 513.1296. 1H NMR (600 MHz, CDCl3) δ 8.02 (d, J = 1.8 Hz, 1 H), 7.98 (dd,J = 8.4 Hz, J = 1.8 Hz, 1 H), 7.33-7.29 (m, 2 H), 7.18 (m, 1 H), 7.09-7.04 (m, 2 H), 6.86 (d, J = 8.4 Hz, 1 H), 6.66-6.62 (m, 2 H), 6.53 (dt, J = 10.8, J = 2.4 Hz, 1 H), 4.88 (s, 2 H), 4.15 (dd, J = 9.6 Hz, J = 3.6 Hz, 1 H), 3.98-3.88 (m, 2 H), 3.51-3.47 (m, 1 H), 3.23-3.18 (m, 1 H), 2.08-1.97 (m, 2 H), 1.84-1.72 (m, 2 H). TLC (UV254) Rf= 0.64 (2:1 酢酸エチル/ヘキサン). HPLC tR = 12.57 min。
【0155】
(S)−1−(4−フルオロベンジル)−5−(2−(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物2) 20dを使用する以外は、22の方法に従って、2を調製して、オレンジ色のゴム状物を得た(32mg、60%)。HRMS (ESI) = 531.1204 (M + H)+. C26H22F3N2O5Sに関する算出値 531.1202. 1H NMR (600 MHz, CDCl3) δ 8.03 (d, J = 1.8 Hz, 1 H), 7.98 (dd, J = 8.4 Hz, J = 1.8 Hz, 1 H), 7.33-7.30 (m, 2 H), 7.09-7.06 (m, 2 H), 6.95-6.91 (m, 1 H), 6.88 (d, J = 8.4 Hz, 1 H), 6.81-6.76 (m, 2 H), 4.93 (d, J = 16.2 Hz, 1 H), 4.92 (d, J = 16.2 Hz, 1 H), 4.18 (dd, J = 9 Hz, J = 3 Hz, 1 H), 4.00-3.95 (m, 2 H), 3.51-3.49 (m, 1 H), 3.21-3.17 (m, 1 H), 2.09-1.98 (m, 2 H), 1.85-1.74 (m, 2 H). TLC (UV254) Rf= 0.67 (2:1 酢酸エチル/ヘキサン). HPLC tR = 12.50 min。
【0156】
(S)−1−(4−フルオロベンジル)−5−(2−(3,5−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物3) 20eを使用する以外は、22の方法に従って、3を調製して、オレンジ色のゴム状物を得た(31mg、58%)。HRMS (ESI) = 531.1213 (M + H)+. C26H22F3-N2O5Sに関する算出値 531.1202. 1H NMR (600 MHz, CDCl3) δ 8.04 (d, J = 1.8 Hz, 1 H), 7.99 (dd, J = 8.4 Hz, J = 1.8 Hz, 1 H), 7.34-7.29 (m, 2 H), 7.09-7.06 (m, 2 H), 6.90 (d, J= 8.4 Hz, 1 H), 6.44-6.38 (m, 3 H), 4.93 (s, 2 H), 4.18-4.15 (m, 1 H), 3.94-3.89 (m, 2 H), 3.51-3.48 (m, 1 H), 3.19-3.15 (m, 1 H), 2.03-1.93 (m, 2 H), 1.83-1.73 (m, 2 H). TLC (UV254) Rf = 0.64 (2:1 酢酸エチル/ヘキサン). HPLC tR = 13.00 min。
【0157】
(S)−1−(4−フルオロベンジル)−5−(2−(テトラヒドロ−2H−ピラン−4−イルオキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物4) 20fを使用する以外は、22の方法に従って、4を調製して、オレンジ色のゴム状物を得た(26mg、52%)。HRMS (ESI) = 503.1646 (M + H)+. C25H28FN2O6Sに関する算出値 503.1652. 1H NMR (600 MHz, CDCl3) δ 8.06 (d, J = 1.8 Hz, 1 H), 8.01 (dd, J = 8.4 Hz, J = 1.8 Hz, 1 H), 7.34-7.31 (m, 2 H), 7.10-7.06 (m, 2 H), 6.91 (d, J = 8.4 Hz, 1 H), 4.87 (s, 2 H), 3.93-3.88 (m, 2 H), 3.73-3.70 (m, 1 H), 3.67 (dd, J = 9 Hz, J = 3 Hz, 1 H), 3.55-3.49 (m, 1 H), 3.47-3.41 (m, 4 H), 3.10-3.05 (m, 1 H), 1.97-1.85 (m, 4 H), 1.70-1.63 (m, 2 H), 1.59-1.54 (m, 2 H). TLC (UV254) Rf = 0.55 (4:1 酢酸エチル/ヘキサン). HPLC tR = 8.58 min。
【0158】
(S)−1−(4−フルオロベンジル)−5−(2−(ピリミジン−4−イルオキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物5) 20gを使用する以外は、22の方法に従って、5を調製して、オレンジ色のゴム状物を得た(12mg、24%)。HRMS (ESI) = 497.1287 (M + H)+. C24H22FN4O5Sに関する算出値 497.1295. 1H NMR (600 MHz, CDCl3) δ 8.77 (s, 1 H), 8.43 (d, J = 5.4 Hz, 1 H), 8.08 (d, J = 1.8 Hz, 1 H), 8.01 (dd, J = 8.4 Hz, J = 1.8 Hz, 1 H), 7.34-7.32 (m, 2 H), 7.09-7.06 (m, 2 H), 6.89 (d, J = 8.4 Hz, 1 H), 6.70 (d, J = 5.4 Hz, 1 H), 4.90 (s, 2 H), 4.55 (dd, J = 10.8 Hz, J = 4.2 Hz, 1 H), 4.37 (dd, J = 10.8 Hz, J = 7.8 Hz, 1 H), 4.05-4.01 (m, 1 H), 3.50-3.46 (m, 1 H), 3.22-3.19 (m, 1 H), 1.98-1.87 (m, 2 H), 1.81-1.72 (m, 2 H). TLC (UV254) Rf = 0.32 (2:1 酢酸エチル). HPLC tR = 7.45 min。
【0159】
(S)−1−(4−フルオロベンジル)−5−(2−(2−プロピニルオキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物23) 20hを使用する以外は、22の方法に従って、23を調製して、オレンジ色のゴム状物を得た。酢酸エチル/ヘキサンからの再結晶化によって、目的生成物をオレンジ色の針状物として得た(93mg、58%)。HRMS (ESI) = 457.1236 (M + H)+. C23H22FN2O5Sに関する算出値 457.1233. 1H NMR (600 MHz, CDCl3) δ 8.07 (d, J = 1.8 Hz, 1 H), 8.01 (dd, J = 8.4 Hz, J = 1.8 Hz, 1 H), 7.43-7.31 (m, 2 H), 7.09-7.04 (m, 2 H), 6.90 (d, J= 8.4 Hz, 1 H), 4.91 (s, 2 H), 4.13 (d, J= 2.4 Hz, 2 H), 3.81-3.77 (m, 1 H), 3.69 (dd, J = 9.6 Hz, J = 4.2 Hz, 1 H), 3.51 (dd, J = 9 Hz, J = 7.2 Hz, 1 H), 3.43-3.40 (m, 1 H), 3.16-3.12 (m, 1 H), 2.43 (t, J= 2.4 Hz, 1 H), 1.94-1.87 (m, 2 H), 1.75-1.66 (m, 2 H). TLC (UV254) Rf = 0.62 (2:1 酢酸エチル/ヘキサン). HPLC tR = 8.80 min。
【0160】
(S)−1−(2−プロピニル)−5−(2−フェノキシメチル−ピロリジン−1−スルホニル)イサチン(化合物24) 乾燥DMF(10mL)における(S)−5−(2−フェノキシメチル−ピロリジン−1−スルホニル)イサチン20a(0.39g、1mmol)の溶液に、炭酸カリウム(0.21g、1.5mmol)、さらには臭化プロパルギル(トルエンにおける80wt%)(0.14g、1.2mmol)を添加した。2時間後、TLCにより、20aの完全な変換が示され、この溶液を10%NH4Cl水溶液(20mL)に注ぎ、DCMで抽出した(3×10mL)。次に、有機画分を合わせたものを真空中で還元し、残渣をジエチルエーテル(10mL)中にとり、水(2×10mL)、次にブライン(2×10mL)、次にブライン(1×10mL)で洗浄し、Na2SO4で乾燥した。クロマトグラフィー(ヘキサン/酢酸エチル)によって、生成物をオレンジ色のゴム状物として得た。酢酸エチル/ヘキサンからの再結晶化によって、生成物をオレンジ色の固体として得た(0.28g、66%)。Mp: 115-117℃. HRMS (ESI) = 425.1185 (M + H)+. C22H21N2O5Sに関する算出値 425.1171. 1H NMR (600 MHz, CDCl3) δ 8.12 (dd, J = 8.4 Hz, J = 1.8 Hz, 1 H), 8.05 (d, J = 1.8 Hz, 1 H), 7.25-7.21 (m, 2 H), 7.16 (d, J = 8.4 Hz, 1 H), 6.96-6.93 (m, 1 H), 6.81 (d, J= 9 Hz, 2 H), 4.56 (dd, J = 18 Hz, J = 2.4 Hz, 1 H), 4.53 (dd, J = 18 Hz, J = 2.4 Hz, 1 H), 4.19 (dd, J = 9.6 Hz, J = 3 Hz, 1 H), 4.07-4.01 (m, 1 H), 3.97 (dd, J = 9.6 Hz, J = 7.2 Hz, 1 H), 3.55-3.51 (m, 1 H), 3.33-3.28 (m, 1 H), 2.36 (t, J=2.4 Hz, 1 H), 2.09-1.99 (m, 2 H), 1.89-1.75 (m, 2 H). TLC (UV254) Rf = 0.54 (2:1 酢酸エチル/ヘキサン). HPLC tR = 9.00 min。
【0161】
(S)−1−(2−プロピニル)−5−(2−(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物25) 20dを使用する以外は、24の方法に従って、25を調製して、オレンジ色の固体を得た(0.48g、70%)。Mp: 101-103℃. HRMS (ESI) = 461.0976 (M + H)+. C22H19F2N2O5Sに関する算出値 461.0983. 1H NMR (600 MHz, CDCl3) δ 8.13 (dd, J = 8.4 Hz, J = 1.8 Hz, 1 H), 8.07 (d, J = 1.8 Hz, 1 H), 7.24 (d, J = 8.4 Hz, 1 H), 6.96-6.92 (m, 1 H), 6.85-6.78 (m, 2 H), 4.59 (dd, J = 18 Hz, J = 2.4 Hz, 1 H), 4.57 (dd, J = 18 Hz, J = 2.4 Hz, 1 H), 4.24-4.20 (m, 1 H), 4.03-3.98 (m, 2 H), 3.55-3.52 (m, 1 H), 3.26-3.21 (m, 1 H), 2.37 (t, J= 2.4 Hz, 1 H), 2.12-2.01 (m, 2 H), 1.88-1.76 (m, 2 H). TLC (UV254) Rf = 0.63 (2:1 酢酸エチル/ヘキサン). HPLC tR = 9.60 min。
【0162】
(S)−1−((1−(2−フルオロエチル)−1H−[1,2,3]−トリアゾール−4−イル)メチル)−5−(2−フェノキシメチル−ピロリジン−1−スルホニル)イサチン(化合物10) 乾燥DMF(3mL)における24(138mg、0.3mmol)の攪拌溶液に、水(0.2mL)における硫酸銅(38mg、0.15mmol)、次に水(0.2mL)におけるアルコルビン酸(53mg、0.3mmol)、次に乾燥DMF(1.5mL)における2−フルオロエチルアジド(33mg、0.36mmol)を添加し、混合物をアルゴン下で攪拌し続けた。2時間後、TLCによって、反応が完了したことを示され、混合物を10%NH4Cl水溶液(12mL)に注ぎ、DCMで抽出し(3×10mL)、Na2SO4で乾燥した。クロマトグラフィー(ヘキサン/酢酸エチル)によって、オレンジ色の固体を得た(26mg、51%)。Mp: 165-167℃. HRMS (ESI) = 514.1557 (M + H)+. C24H25FN5O5Sに関する算出値 514.1560. 1H NMR (600 MHz, CDCl3) δ 8.07 (dd, J = 8.4 Hz, J = 1.8 Hz, 1 H), 8.01 (d, J = 1.8 Hz, 1 H), 7.76 (s, 1 H), 7.47 (d, J = 8.4 Hz, 1 H), 7.23-7.19 (m, 2 H), 6.92 (t, J = 7.8 Hz, 1 H), 6.82 (d, J = 7.8 Hz, 2 H), 5.03 (d, J = 15.6 Hz, 1 H), 5.02 (d, J = 15.6 Hz, 1 H), 4.79 (dt, J = 46.2 Hz, J = 4.8 Hz, 2 H), 4.66 (dt, J = 27 Hz, J = 4.8 Hz, 2 H), 4.17 (dd, J = 9.6 Hz, J = 3.6 Hz, 1 H), 4.00-3.97 (m, 1 H), 3.93 (dd, J = 9 Hz, J = 7.2 Hz, 1 H), 3.53-3.49 (m, 1 H), 3.27-3.24 (m, 1 H), 2.08-1.97 (m, 2 H), 1.86-1.75 (m, 2 H). TLC (UV254) Rf = 0.56 (酢酸エチル). HPLC tR = 7.93 min。
【0163】
(S)−1−((1−(2−フルオロエチル)−1H−[1,2,3]−トリアゾール−4−イル)メチル)−5−(2(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物11) 25を使用する以外は、10の方法に従って、11を調製して、オレンジ色のゴム状物を得た。酢酸エチル/ヘキサンからの再結晶化によって、オレンジ色の固体を得た(94mg,57%)。Mp: 130-131℃. HRMS (ESI) = 550.1381 (M + H)+. C24H23F3N5O5Sに関する算出値 550.1372. 1H NMR (600 MHz, CDCl3) δ 8.08 (dd, J = 8.4 Hz, J = 1.8 Hz, 1 H), 8.02 (d, J = 1.8 Hz, 1 H), 7.77 (s, 1 H), 7.52 (d, J= 8.4 Hz, 1 H), 6.95-6.91 (m, 1 H), 6.82-6.75 (m, 2 H), 5.05 (s, 2 H), 4.79 (dt, J = 46.2 Hz, J = 4.8 Hz, 2 H), 4.67 (dt, J = 26.4 Hz, J = 4.8 Hz, 2 H), 4.20 (dd, J = 9.6 Hz, J = 3 Hz, 1 H), 4.01-3.94 (m, 2 H), 3.53-3.50 (m, 1 H), 3.22-3.17 (m, 1 H), 2.10-1.98 (m, 2 H), 1.85-1.74 (m, 2 H). TLC (UV254) Rf = 0.47 (酢酸エチル). HPLC tR = 8.45 min。
【0164】
(S)−1−((1−(フルオロメチル)−1H−[1,2,3]−トリアゾール−4−イル)メチル)−5−(2(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物29)を、フルオロメチルアジドを2−フルオロエチルアジドの代わりに使用する以外は、化合物11の方法に従って調製した。
【0165】
(S)−1−((1−(3−フルオロプロピル)−1H−[1,2,3]−トリアゾール−4−イル)メチル)−5−(2(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物30)を、3−フルオロプロピルアジドを2−フルオロエチルアジドの代わりに使用する以外は、化合物11の方法に従って調製した。
【0166】
(S)−1−((1−(4−フルオロブチル)−1H−[1,2,3]−トリアゾール−4−イル)メチル)−5−(2(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物31)を、4−フルオロブチルアジドを2−フルオロエチルアジドの代わりに使用する以外は、化合物11の方法に従って調製した。
【0167】
(S)−1−(4−フルオロベンジル)−5−(2−(1−((2−フルオロエチル)−1H−[1,2,3]−トリアゾール−4−イル)メトキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物12) 23を使用する以外は、10の方法に従って、12を調製して、オレンジ色のゴム状物を得た(26mg,48%)。HRMS (ESI) = 546.1632 (M + H)+. C25H26F2N5O5Sに関する算出値 546.1623. 1H NMR (600 MHz, CDCl3) δ 8.03 (d, J =1.8 Hz, 1 H), 8.00 (dd, J = 8.4 Hz, J = 1.8 Hz, 1 H), 7.67 (s, 1 H), 7.35-7.33 (m, 2 H), 7.08-7.05 (m, 2 H), 6.90 (d, J = 8.4 Hz, 1 H), 4.95 (s, 2 H), 4.81 (dt, J= 46.8 Hz, J = 4.8 Hz, 2 H), 4.67 (dt. J = 26.4 Hz, J = 4.8 Hz, 2 H), 4.62 (d, J = 12 Hz, 1 H), 4.60 (d, J = 12 Hz, 1 H), 3.81-3.78 (m, 1 H), 3.65 (dd, J = 9.6 Hz, J = 4.2 Hz, 1 H), 3.51 (dd, J = 9.6 Hz, J = 7.2 Hz, 1 H), 3.40-3.37 (m, 1 H), 3.19-3.15 (m, 1 H), 1.92-1.87 (m, 2 H), 1.72-1.66 (m, 2 H). TLC (UV254) Rf = 0.35 (酢酸エチル). HPLC tR = 6.58 min。
【0168】
(S)−1−(4−フルオロベンジル)−5−(2−(1−((フルオロメチル)−1H−[1,2,3]−トリアゾール−4−イル)メトキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物32)を、フルオロメチルアジドを2−フルオロエチルアジドの代わりに使用する以外は、化合物12の方法に従って調製する。
【0169】
(S)−1−(4−フルオロベンジル)−5−(2−((1−(3−フルオロプロピル)−1H−[1,2,3]−トリアゾール−4−イル)メトキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物33)を、3−フルオロプロピルアジドを2−フルオロエチルアジドの代わりに使用する以外は、化合物12の方法に従って調製する。
【0170】
(S)−1−(4−フルオロベンジル)−5−(2−((1−(4−フルオロブチル)−1H−[1,2,3]−トリアゾール−4−イル)メトキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物34)を、4−フルオロブチルアジドを2−フルオロエチルアジドの代わりに使用する以外は、化合物12の方法に従って調製する。
【0171】
(S)−1−[4−(フルオロメチル)−1H−[1,2,3]−トリアゾール−1−イル]メチル−5−(2−(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン)(化合物6)を、(S)−1−(アジドメチル)−5−(2−(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン](化合物37)を3−フルオロプロプ−1−イン(化合物28)と反応させることによって、図2(ii)に示されるように調製する。試薬は下記のとおりである:(a)CuSO4、L−アスコルビン酸、DMF。[18F]6の調製では、3−[18F]フルオロプロプ−1−インを3−フルオロプロプ−1−インの代わりに使用する。
【0172】
(S)−1−[4−(2−フルオロエチル)−1H−[1,2,3]−トリアゾール−1−イル]メチル−5−(2−(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン)(化合物7)を、4−フルオロブチ−1−インを(3−フルオロプロプ−1−イン)の代わりに使用する以外は、化合物6の方法に従って調製する。[18F]7の調製では、4−[18F]フルオロブチ−1−インを3−フルオロプロプ−1−インの代わりに使用する。
【0173】
(S)−1−[4−(3−フルオロプロピル)−1H−[1,2,3]−トリアゾール−1−イル]メチル−5−(2−(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン)(化合物8)を、5−フルオロペンチ−1−インを(3−フルオロプロプ−1−イン)の代わりに使用する以外は、化合物6の方法に従って調製する。[18F]8の調製では、5−[18F]フルオロペンチ−1−インを3−フルオロプロプ−1−インの代わりに使用する。
【0174】
(S)−1−[4−(4−フルオロブチル)−1H−[1,2,3]−トリアゾール−1−イル]メチル−5−(2−(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン)(化合物9)を、6−フルオロヘキシ−1−インを(3−フルオロプロプ−1−イン)の代わりに使用する以外は、化合物6の方法に従って調製する。[18F]9の調製では、6−[18F]フルオロヘキシ−1−インを3−フルオロプロプ−1−インの代わりに使用する。
【0175】
化合物35は、Rが前記規定と同様である化合物の一群を有する。化合物36は、R’が前記規定と同様である化合物の一群を有する。これらの化合物は、図12(上行=化合物35、下行=化合物36)で概略的に表わされる方法を用いて製造できる。このストラテジーは、in situのまたは別の反応としての、[11C]ヨウ化メチルとアジド源との反応、次に、適当なアルキンを用いた付加環化を含む。この方法は、近年Schirrmacher, R. et al 25で報告される、より既知のクリックケミストリー(Click Chemistry)ストラテジーの修飾である。
【0176】
実施例2−ターゲット化合物の親油性およびカスパーゼに対する親和性の測定
カスパーゼに対するターゲット化合物の親和性 様々な活性化カスパーゼ1、3、6、7、及び8に関する、新規なフッ素化イサチン1〜5及び10〜12、ならびに既知のイサチン誘導体及び中間対化合物22及び23の親和性を、Kopka and co-workers.13に記載されるのと同様の蛍光インビトロカスパーゼ阻害アッセイによって測定した。組換えヒトカスパーゼの阻害は、蛍光性生成物、7−アミノ−4−メチルクマリン(7−AMC)の蓄積を測定することによって評価した。
【0177】
組換えヒトカスパーゼ−1、−3、−6、−7、及び−8ならびにこれらのペプチドに特異的な基質は、Biomol International, UKから購入した。非放射性イサチンによる組換えカスパーゼの阻害は、蛍光性生成物、7−アミノ−4−メチルクマリン(7−AMC)の蓄積を測定する蛍光アッセイを用いて評価した。全てのアッセイは、1ウェルあたり200μlの容積で96ウェルプレートで行った。アッセイは、各カスパーゼについて以下に記載されるような適当な反応緩衝液中で37℃で行った。カスパーゼ1では、緩衝液は、0.1% CHAPS、100mM NaCl、5mM 2−メルカプトエタノール、100mM HEPES(pH 7.4)、2mM EDTA、10% スクロース、及び10μMのペプチド基質 Ac−YVAD−AMCから構成された。カスパーゼ3について:20mM HEPES(pH 7.4)、10% スクロース、100mM NaCl、0.1% CHAPS、2m,mM EDTA、及び10μM Ac−DEVD−AMC。カスパーゼ6について:20mM HEPES(pH 7.4)、10% スクロース、100mM NaCl、0.1% CHAPS、2mM EDTA、及び10μM Ac−VEID−AMC。カスパーゼ7について:20mM HEPES(pH 7.4)、10% スクロース、5mM 2−メルカプトエタノール、100mM NaCl、0.1% CHAPS、2mM EDTA、10μM Ac−DEVD−AMC。カスパーゼ8について:20mM HEPES(pH 7.4)、10% スクロース、100mM NaCl、0.1% CHAPS、2mM EDTA、および10μM Ac−IETD−AMC。これらの緩衝液は、500、50、5μM;500、50、5nM;500、50、5pMの最終濃度でDMSO中に非放射性イサチンを含んだ;全てのウェルにおけるDMSOの最終濃度は全容積の5%であった。組換えカスパーゼを、1アッセイ当たり0.5単位で使用した(〜500pmol 変換基質/時間)。ペプチド基質以外の全ての試薬は、10分間予めインキュベートした。次に、ペプチド基質(最終濃度 10μM)を添加して、プレートをさら30分間インキュベートした;30分は、10、30、60または90分後に反応が経過した初期直線性研究後に選択された。30分の時点が選択された。各コントロールウェルは、酵素以外の全ての反応成分を含んだ。7−AMCの生産量を、それぞれ、355nm及び460nmの励起波長及び放出波長で蛍光マイクロプレートリーダー(Victor2; Perkin-Elmer Life sciences)で測定した。カスパーゼ活性を50%阻害するイサチンの濃度(EC50)を、GraphPad Prism(Version 4.0 for Windows, GraphPad Software, San Diego California USA)を用いて非直線性回帰分析によって評価した。全てのイサチンは2連で分析した;アッセイは1回繰り返した。既知の化合物13〜16を参考化合物として含ませた。結果を下記表1に要約する。
【0178】
データは、5pM〜500μMの範囲の9種の濃度で、それぞれ重複した、2回の操作の平均である;各酵素を50%阻害するのに必要な薬剤濃度(EC50)を、阻害プロフィールの非直線性回帰分析から得た。
【0179】
結果 本アッセイでは、既知のイサチン13の親和性は、それぞれ、カスパーゼ3及び7に対して59.9及び25.3nMであった。フェニルエーテルでのフッ素置換は良好に許容され、新規なイサチン1では、カスパーゼ3に対する親和性は、既知の非フッ素化フェニルエーテル14に比べて2〜3倍増加した。2フッ素化イサチン2及び3はより強力であり、それぞれ、カスパーゼ3に対して、12.4及び10.4nMであった。テトラヒドロピラン4では同様の親和性が見出された。既知のベンジル誘導体15に比して4の効力の5倍の増加は、環が十分飽和であることを考えれば、驚くべきことである。ピリミジニル誘導体5におけるようなフェニルエーテルのピリミジニル基による置換により、さらに効力が増加し、カスパーゼ3及び7に対する親和性が、それぞれ、5.5及び2.3nMであった。分子のいずれかの側に2−フルオロエチル−1,2,3−トリアゾールを導入することによって、効力が急増し、トリアゾール10及び12に関して測定されたカスパーゼ3の親和性が、それぞれ、16.7及び12.6nMであった。フルオロエチルトリアゾール12の効力は1〜3と同様であり、このことは基がかなりより小さな大きさでありかつ極性性がより高いことを考えると驚くべきことである。Lee8は、親水性ポケットとしてイサチン窒素周辺の結合ドメインを示し、ほとんどの基がこの位置にベンジル部分を有する。したがって、トリアゾール10が既知のベンジル誘導体15より4倍強力であることは非常に予想し得ないことであった。試験した化合物のうち、トリアゾール11は、圧倒的に最も強力であり、カスパーゼ3及び7に対して、それぞれ、0.5及び2.5nMの親和性を有していた。試験したすべての化合物は、カスパーゼ1、6及び8に対して低い基質であった(EC50>5000nM)。
【0180】
Log P計算 親油性をACD/Chemsketch labs softwareから算出した。
【0181】
結果 結果を下記表1および1aに示す。
【0182】
【表2−1】
【0183】
【表2−2】
【0184】
実施例3−[18F]11の合成
好ましくは、化合物[18F]11は、(S)−1−(2−プロピニル)−5−(2−(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物25)を[18F]フルオロエチルアジド(化合物27)15, 16でクリック−ラベリング(click-labelling)することによって、製造される。トリアゾール(S)−1−((1−(2−フルオロエチル)−1H−[1,2,3]−トリアゾール−4−イル)メチル)−5−(2(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物11)は、図2(i)及び図19に示されるように、2−[18F]フルオロエチルアジド(化合物27)をアルキン前駆体である(S)−1−(2−プロピニル)−5−(2−(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物25)で銅触媒による付加環化を行うことによって標識される。同様の方法を用いて、非放射性標識の化合物11を製造でき、この際、フルオロエチルアジドを2−[18F]フルオロエチルアジドの代わりに使用する。
【0185】
窒素雰囲気下で、アスコルビン酸ナトリウム(50μl、8.7mg、43.2μmol)の緩衝液(リン酸ナトリム緩衝液、pH 6.0、250mM)を、硫酸銅(II)の水溶液(50μl、1.7mg 5水和物、7.0μmol)を含むホイートンバイアル瓶(Wheaton vial)(1mL)に添加した。1分後、ジメチルホルムアミド(25μl)におけるアルキン25(3.0mg、6.5μmol)の溶液、さらにアセトニトリル(100μl)における蒸留[18F]FEA(185−740 MBq)を添加した。この混合物を室温で30分間放置して、水(15μl)を添加した後、予備ラジオ−HPLCにかけた。単離したHPLC物溶液を水(5mL)で希釈し、予めエタノール(5mL)及び水(10mL)で調整された(conditioned)SepPak C18−lightカートリッジ(Waters)にのせた。次に、このカートリッジを水(5mL)でフラッシュし、18F−9を小画分(0.1mL)づつエタノールで溶出した。この生成物画分を、エタノール含量が10〜15%(v/v)になるようにPBSで希釈した。
【0186】
結果 27をアルキン前駆体25と共役させようとする初期の試みにより、放射性生成物の複合混合物が形成した。放射化学的収率への触媒システム、温度及びpHの影響をさらに調べたところ、最も高い収率が室温で得られたものの、[18F]9は熱安定性に劣ることが分かった。リン酸ナトリウム緩衝液(pH 6.0、250mM)を反応混合物に添加することによって、放射化学的収率が劇的に向上したものの、調べた2つの触媒システム(銅粉末または硫酸銅/アスコルビン酸塩)では若干の相違が観察されたのみであった。最適条件は、アルキン前駆体25に対して若干過剰な硫酸銅の存在下で室温で30分の反応時間であることが分かった。これにより、65±6%(n=26、27から減衰補正)の単離放射化学的収率で、HPLCによる精製後の放射化学的純度が>99%のトリアゾール[18F]11が得られた。生成混合物に関する具体的なHPLCクロマトグラムを図3に示す。本方法は少量の放射能で手動で行ったので、合成終了時に低い比放射能(1.2GBq/μmol)しか達成しなかった。[18F]11の同一性を、非放射性参照化合物と一緒に溶出することによって確認した。精製[18F]11を、91±6%(n=26、減衰補正)の効率で固相抽出することによって配合した(formulate)。[18F]11の配合(formulation)を含む放射合成(radiosynthesis)は全体で3時間かかった。
【0187】
実施例4−[18F]11の親油性
Log P測定 Barthel et al 18の方法を用いてオクタノール−水分配係数を測定することによって、親油性を評価した。簡単にいうと、[18F]11(25μLエタノールにおける〜180μCi)を水を用いて最終容積が100μLになるように希釈して、ストック溶液を得た。ストック溶液のアリコート(10μL)を水(490μL)及びオクタン−1−オール(Aldrich無水グレード)(500μL)に加えた。次に、この溶液を10分間激しく振盪した後、遠心した(13201g、20℃、30分)。遠心後、水層及びオクタノール層の部分(200μL)を注意深く除去し、Cobra II Auto-Gamma counter (Packard Instruments, Meriden, CT, USA)に置いて分析し、比較した。放射能を含む水で放射能を含むオクタノールを割ることによって、オクタノール−水分配係数を得た。Log Pを測定したところ、上記表1の脚注cに示されるように、1.61であった。
【0188】
実施例5−[18F]11および[125I]14の組織分布および代謝安定性
血漿及び肝臓における新規な化合物[18F]11及び既知の化合物[125I]14の安定性を、マウスで投与した後経時的に測定した。[18F]11を腫瘍を有するマウスの静脈内に注射し、その組織分布を注射してから2、15及び60分後に所定の組織中で測定した。[125I]14を腫瘍を持たないマウスの静脈内に注射し、その組織分布を注射してから2〜60分の所定の時点で測定した。さらに、放射性トレーサーのインビボでの代謝安定性を血漿及び肝臓サンプルから、および[18F]11では、尿サンプル中で測定した。
【0189】
インビトロでのマウス肝臓S9代謝研究 肝臓S9代謝研究をAboagye et al19に記載されるのと同様にして行った。マウス肝臓を迅速に切除し、実験を通して4℃に保持した。組織を秤量し、Ultra-Thurraxホモジナイザー(IKA, Staufen, Germany)を用いて等容積の50mM Tris−150mM KCl−HCl緩衝液(pH 7.4)中で均質化した。S9画分を得るために、ホモジネートを30分間遠心(10,000×g)して、核、ミトコンドリア及び細胞残屑を除去した。S9画分のタンパク質濃度を市販のBCAタンパク質アッセイキット(Perbio Science, Cheshire, UK)によって測定した。S9画分を最長6週間−80℃で貯蔵した。所定の非標識イサチン14、15、16(10mM、10μL)を、1mLの全容積で0.1mM Tris−HCl緩衝液(pH 7.4)中で37℃で60分間、S9画分(33.26mg/ml、20μL)及び0.5mM ニコチンアミドアデニンジヌクレオチドリン酸(還元形態)と共にインキュベートした。コントロールインキュベーションサンプルは、イサチンを含まなかった。氷冷したアセトニトリル(2mL)を添加することによって、反応を止めた;次に、サンプルを即座にドライアイスに置いた後、抽出及びHPLC分析を行った(下記)。
【0190】
インビボでの生体内分布および代謝研究 放射線誘発線維肉腫(RIF−1)腫瘍細胞20を、5% CO2の加湿インキュベーターで37℃で、10%ウシ胎児血清(BioWhittaker Europe Ltd, Verviers, Belgium)、2mM L−グルタミン、100U/mL ペニシリン、100μg/mL ストレプトマイシン及び0.25μg/mL ファンギゾン(Gibco, UK)を加えたRPMI 1640培地(Invitrogen Ltd, Paisley, UK)中で維持した。すべての動物の作業は、英国の"Guidance on the Operation of Animals (Scientific Procedures) Act 1986" (HMSO, London, United Kingdom, 1990)に従って、また、実験新生組織形成における動物の愛護に関する政府の規則及びガイドライン(government regulations and guidelines on the welfare of animals in experimental neoplasia)21に完全にのっとって、認可を受けた研究者によって行われた。オスのC3H/hejマウス(Harlan, Bicester, Oxfordshire, UK)の背中に5×105個の細胞を皮下注射することによって腫瘍を確立した。腫瘍の成長を電子キャリパー(electronic callipers)を用いて2日毎にモニターし、腫瘍の容積を式:(π/6)×L×W×D(L=長さ、W=幅およびD=深さ)を用いて評価した。腫瘍が約100〜150mm3に到達したら、[18F]11の生体内分布の研究のために動物を選択した;[125I]14の生体内分布は腫瘍を持たないマウスで行った。
【0191】
リン酸緩衝液に溶解した放射能(〜0.37MBqの[125I]14または3.7MBqの[18F]11) 0.08〜0.13mLを、側静脈を介して、マウスの静脈内に注射した。注射してから所定の時点(2〜60分)で、通常の麻酔下(イソフルオラン(isofluorane)吸入)で心穿刺による放血によってマウスを剖検した。ヘパリン化血のアリコートを直ぐに遠心(2000gで5分)して、血漿を得た。組織に含まれる放射能をCobra II Auto-Gamma counter (Packard Instruments, Meriden, CT, USA)でのガンマ−カウンティングによって測定し、注射量/g 組織の割合(%)(%ID/g)として表わした。3匹のマウスのうちの最小値を各時点で使用した。すべての動物は未処置(treatment-naive)であった。
【0192】
インビボでの代謝研究もまたC3H/hejマウスで行った。腫瘍を持たないマウスの静脈内に0.37MBqの[125I]14または7.4MBqの[18F]11を注射し、血漿を上記と同様にして得た。血漿、肝臓及び尿サンプルを液体窒素中でスナップフリーズし(snap-frozen)、分析するまで、ドライアイス上の予め秤量された(pre-weighed)シンチレーション計測用チューブ中に保持した。
【0193】
HPLC分析 抽出直前に、サンプルを解凍し、氷上に置いた。抽出では、氷冷したアセトニトリル(1.5mL)を血漿(0.2mL)に添加した;アセトニトリルを含むS9インキュベーションサンプル(3mL)もまた分析した。各混合物を遠心(15493×g、4℃、3分)し、得られた上清をロータリーエバポレーターを用いて40℃で真空中で蒸発乾固した。肝臓サンプルは、遠心前にIKA Ultra-Turrax T-25ホモゲナイザーを用いて氷冷アセトニトリル(1.5mL)と共に均質化した。次に、残渣をHPLC移動相(1.2mL)に再懸濁し、Minisart親水性シリンジフィルター(Minisart hydrophilic syringe filter)(0.2μm)(Sartorius, Goettingen, Germany)で濾過した。尿サンプルは、HPLC移動相で希釈し、上記と同様にして濾過した。次に、サンプル(1mL)を、γ−RAM モデル3 ガンマ−検出器(γ-RAM Model 3 gamma-detector)及びLauraソフトウェア(IN/US Systems inc., Florida, USA)を備えたAgilent 1100シリーズHPLCシステム(Agilent Technologies, Stockport, UK)によるラジオ−HPLCによって分析した。
【0194】
固定相は、Waters μBondapak C18逆相カラム(300×7.8mm)で構成された。[125I]14の分析では、血漿及び肝臓サンプルを上記と同様にして処理し、2mL/分の流速で定組成で(in isocratic mode)流す水(0.1% TFA)/プロパン−1−オール(0.1% TFA)(35:65)の移動相を用いて分析した。インビトロでの代謝後の非標識の14、15及び16の分析では、水(0.1% TFA)/プロパン−1−オール(0.1% TFA)から構成される移動相を、11分で2→80%有機相、3分で80(R)5%有機相後、6分で5%有機相の勾配を3mL/分の流速で供給しながら、用いることによって、サンプルを分析した。[18F]9の分析では、0.1M ギ酸アンモニウム/1.8:1 メタノール:アセトニトリルから構成される移動相を、1分で50%有機相、14分で50(R)90%有機相、3分で90%有機相、2分で90(R)50%有機相、4分で50%有機相の勾配を3mL/分の流速で供給しながら、用いることによって、血漿、肝臓及び尿サンプルを分析した。さらに、HPLC注入液中に残る[18F]9−由来の放射能に対する抽出後の血漿及び肝臓ペレットに関連する[18F]9−由来の放射能の割合を評価するために、HPLC注入液の全容積を記録し、アリコート(0.1mL)を計測用に除去した。次に、0.1mLアリコート及びペレット中の放射能をガンマカウンティング(gamma counting)(Packard Instruments)によって分析した。
【0195】
結果 図4から、[18F]11は迅速に組織に分布し、また迅速に排出されたことが分かる。[18F]11由来の放射能の高い極在化が腎臓、尿及び肝臓で見られたことから、排出の腎臓及び肝臓経路双方の重要性が示唆される。しかしながら、これらの組織でさえ、放射能の迅速な排出が見られた。重要なことに、イメージングの観点から、未処置の腫瘍、心臓及び脳での[18F]11の取り込みは低かった;これにより、これらの組織でのカスパーゼ活性化に関連する結合性の増加の測定が容易になるはずである。骨への取り込みが低かったことから、放射性トレーサーの脱フッ素化は存在せず、ゆえにフルオロエチルアジド部分が安定であることが示唆される。ラジオ−HPLC分析から、[18F]11は血漿及び肝臓で単一の極性代謝ピークを作成する[125I]14(図5)より代謝分解に対して比較的より安定であることが示された。親化合物は60分で血漿中に依然として存在し、主要なピークであった。これに対して、代謝産物は、すべての研究した時点で肝臓サンプルでは主要なピークであった。尿の放射能は主として代謝産物から構成された。所定の時点での血漿及び肝臓での親放射性トレーサーの割合を、見かけの抽出効率と共に、表2に要約する。すばやく減少した後は、インビボでの代謝速度は15〜60分の間は横ばいであるように見えた。しかしながら、血漿及び肝臓からの抽出効率はおそらく非特異的結合により経時的に減少した。また、血漿抽出後の残渣ペレットでの放射能は時間に依存して増加したことから、血漿タンパク質への結合が増加したことが示唆される。
【0196】
【表3】
【0197】
図7から、[125I]14は主要な器官に迅速に分布し、また迅速に排出されたことが分かる。[125I]14は10分以内に血漿から完全に排出され、この時点で肝臓で検出される放射能の<15%(n=3)を提示した。注射してから30分以内に、親[125I]14は肝臓で十分代謝され、この際、放射能の大部分を構成する1つの極性のある代謝産物が検出された。これらの知見と一致して、我々は、調査したすべての組織で迅速なクリアランスが観察され、10分以降、注射された放射能の大部分は尿中に排出された。
【0198】
図8に示されるように、既知の非標識のイサチン14及び15の代謝プロフィールは同様であり、ほとんど完全な分解が60分インキュベーション後に観察された。
【0199】
実施例6−4種の癌細胞系での[18F]11の「取り込み
細胞系および腫瘍モデル Wolf C.R. et al.22 and Langdon S.P. et al23に記載されるのと同様にして、ヒト卵巣癌細胞系PEO1/4を確立し、特性を明らかにした。簡単にいうと、PEO1/4細胞は、再発粘液卵巣腺癌の患者のシスプラチン化学療法に臨床的耐性が発現する前(PEO1)及び後(PEO4)の、患者の悪性腹水由来であった。当初は、放射線誘発線維肉腫(RIF−1)腫瘍細胞系の特性をTwentyman P.R. et al.20によって明らかにした。LNM35腫瘍細胞は、Kozaki K. et al.24に記載されるように、ヒト肺大細胞癌NCI−H460細胞系のリンパ行性転移性の高いサブラインである。すべての細胞系を、定常的に、5% CO2の加湿インキュベーターで37℃で、10%ウシ胎児血清(BioWhittaker Europe Ltd, Verviers, Belgium)、2mM L−グルタミン、100U/mL ペニシリン、100μg/mL ストレプトマイシン及び0.25μg/mL ファンギゾン(Gibco, UK)を加えたRPMI 1640培地(Invitrogen Ltd, Paisley, UK)中で維持した。
【0200】
6〜8週齢のメスのnu/nu−BALB/cマウスまたはオスのC3H/hejマウス(Harlan, Bicester, Oxfordshire, UK)の背中に、PEO1/4(10×106細胞、with v/v matrigel, BD Biosciences, Oxford, UK)、RIF−1(5×105細胞)及びLNM35(1×106細胞)を皮下注射することによって、インビボでの腫瘍モデルを確立した。腫瘍の成長を電子キャリパー(electronic callipers)を用いて2日毎にモニターし、腫瘍の容積を式:(π/6)×L×W×D(L=長さ、W=幅およびD=深さ)を用いて評価した。すべての動物の作業は、英国の"Guidance on the Operation of Animals (Scientific Procedures) Act 1986" (HMSO, London, United Kingdom, 1990)に従って、また、実験新生組織形成における動物の愛護に関する政府の規則及びガイドライン(government regulations and guidelines on the welfare of animals in experimental neoplasia)に完全にのっとって、認可を受けた研究者によって行われた。
【0201】
インビトロでの[18F]11取り込みおよびカスパーゼ3活性化アッセイ 細胞を、実験の2または3日前に12ウェルプレートに3連で播種し、所定の濃度および時間に白金由来の抗腫瘍薬であるシスプラチン、5−FU、タンパク質合成阻害剤であるシクロヘキシミド(CHX)またはトポイソメラーゼ阻害剤エトポシド(VP−16)(Sigma, UK)または対応するベヒクルで処理した。実験の日に、約10μCi/ウェルの[18F]11を添加して、37℃で60分間細胞中に蓄積させた。細胞を集め、洗浄し、400μlのPBS中に再懸濁した。260μlの各サンプルを計測用チューブに移した直後、フッ素−18の放射能を、Packard Cobra II gamma counter (Perkin Elmer, UK)を用いて測定した。製造者(caspase-Glo 3/7 assay, Promega, UK)の指示に従って、40μlの各サンプルを用いて、カスパーゼ3活性化比色分析アッセイを行った。簡単にいうと、細胞を乳白色の96ウェルプレートに移し、50μlのカスパーゼ−Gloと共に30分〜3時間インキュベートし、カスパーゼ3の酵素活性をMultiskan Luminometer (Thermo Electron, UK)を用いて測定した。全てのサンプルで、BCAプロテインアッセイ(BCA Protein assay)(Pierce, UK)を行い、データを正規化して、CCPMAまたはRLU(Relative Light Unit)/mg タンパク質として表わす。
【0202】
インビボでの[18F]11生体内分布およびPET画像化 生体内分布の研究のために、腫瘍を有するマウスを10mg/kgのシスプラチンまたはベヒクルで24時間処置し、イソフルオラン(isofluorane)/O2/N2Oで麻酔し、所定の時間、約100μCiの[18F]11を側尾静脈(lateral tail vein)に静脈内注射した。様々な組織を取り除き、秤量し、即座にフッ素−18放射能計測するために計測用チューブに置いた。
【0203】
シスプラチン(10mg/kgで24時間)またはベヒクルで処置した腫瘍を有するマウスを、小動物専用のPETスキャナー(dedicated small animal PET scanner)(quad-HIDAC; Oxford Positron Systems, Weston-on-the-Green, United Kingdom)でスキャンした。麻酔をかけた動物をサーモスタット制御のベッドに置き、スキャナー内に腹臥位にした。マウスの直腸温度が約37℃になるように、ベッドを調節した。[18F]11(約100μCi)のボーラス注射液(bolus injection)を、尾静脈カニューレを介して静脈内(i.v.)投与し、スキャンを開始した。動的放射スキャン(dynamic emission scan)を60分にわたってリスト形式(list-mode format)で得た。次に、画像再構築のために、得られたデータを、0.5−mm 副鼻腔撮影図(sinogram bin)および19のタイムフレーム(0.5×0.5×0.5mm ボクセル;4×15、4×60、および11×300秒)に分け、これを2次元のハミングフィルター(two-dimensional Hamming filter)(カットオフ 0.6)を用いたフィルター逆投影によって行った。このようにして得られた画像データ−セットをSUN workstation (Ultra 10; SUN Microsystems, Santa Clara, CA)に移し、Analyze ソフトウェア(version 6.0; Biomedical Imaging Resource, Mayo Clinic, Rochester, MN)を用いて可視化した。注射してから0〜1分および注射してから30〜60分から構成される動的データの累積画像を、放射性トレーサーの取り込みの可視化のためにおよび関心のある領域(regions of interest)(ROI)を規定するために使用した。各19時点で各ROIについて、カウント密度を平均して、ROIに関する時間に対する放射能曲線(TAC)を得た。腫瘍のTACを各時点で心臓のものに正規化して、正規化した取り込み値(normalized uptake value)(NUV)を得た。筋肉の[18F]9データを、腫瘍データを正規化するための内部入力関数として使用した。注射してから60分でのNUV(NUV60)、0〜60分のNUVの積分として算出されたNUV曲線下の面積(AUC)、およびトレーサーの分数保持(fractional retention of tracer)(FRT)、2.5分での放射能に対する60分での放射能を、比較に使用した。FRTは、保持される腫瘍にデリバリーされる放射性トレーサーの割合を示すという点で有用で可変である。したがって、これは腫瘍の[18F]9の取り込みからデリバリーまでを正規化する。
【0204】
結果 図9に示されるように、予想とおりシスプラチン耐性卵巣癌細胞(PEO4)以外の、全ての薬剤処置細胞は、コントロール(ベヒクル処置)に比して[18F]9の取り込みの有意な増加を示す。さらに、薬剤処置LNM35での[18F]9の取り込みの増加は、細胞内の活性カスパーゼ3量と相関し、これからCDDP処置とVP−16処置との拮抗作用が明らかになった。
【0205】
表3は、CDDPで処置したPEO1異種移植片を有するマウスの腫瘍における[18F]9の取り込みの有意な増加を示すが、この際、CDDP耐性PEO4腫瘍はコントロールと処置との間に取り込みの相違は示していない。PEO1腫瘍を有するマウスの脾臓及び筋肉以外の、他の組織における[18F]9の取り込みは、コントロールと処置との間に有意な差はない。
【0206】
【表4】
【0207】
PEO1、PEO4及びRIF−1異種移植片を有するこれらのCDDP処置マウスにおけるPET画像(図10)から、腫瘍における[18F]11の有意な取り込みが明らかになった。
【0208】
下記文書はすべて参考で本明細書中に引用される。
【0209】
実施例7−アポトーシス細胞への[18F]11の取り込み
RIF−1細胞を、ベヒクル(0.1% DMSO)またはcis−ジアミンジクロロ白金(II)(cis-diamminedichloro-platinum(II))(シスプラチン)(CDDP)(100μM)で、48時間処置した。次に、これらの細胞を[18F]11と共に1時間インキュベートし、洗浄し、放射能を分析した。図13に示されるように、細胞アッセイにより、アポトーシス細胞への[18F]11の取り込みが約1.5倍増加したことが示された。データは、1mg 全細胞タンパク質当たりで平均化した減衰補正したカウント/分として表わす。データは、平均値±SEMであり、3連で行った。
【0210】
RIF−1腫瘍をベヒクル(50% DMSO)またはCDDP(10mg/kg 単回投与)で処置した。放射性トレーサーを注入してから60分での[18F]由来の放射能レベルを分析し、血中レベルに対する割合として表わす。図14に示されるように、RIF−1腫瘍を有するマウスにおけるインビボでの取り込み研究でも、ベヒクル処置したマウスに比べて処置マウスでは[18F]11の取り込みが約1.5倍増加した。データは、平均値±SEMであり、1群当たりn=8匹マウスである。
【0211】
実施例8−アポトーシス画像化トレーサーとしての[18F]11の機能性
6〜8週齢のオスのC3H/hejマウス(Harlan, Bicester, Oxfordshire, UK)の背中に38C13 マウスリンパ腫細胞(5000細胞)を皮下注射することによって、腫瘍アポトーシスのインビボ実験モデルを確立した。異種移植片が約100mm3に到達したら、マウスを、シクロホスファミド(CPA、100mg/kg)またはベヒクルで24時間処置した後、小動物専用のPETスキャナー(dedicated small animal PET scanner)(Siemens Inveon PET module)でスキャンした。麻酔をかけた動物をサーモスタット制御のベッドに置き、スキャナー内に腹臥位にした。[18F]11(約100μCi)のボーラス注射液(bolus injection)を、尾静脈カニューレを介して静脈内(i.v.)投与し、スキャンを開始した。
【0212】
動的放射スキャン(dynamic emission scan)を60分にわたってリスト形式(list-mode format)で得た。次に、画像再構築のために、得られたデータを、0.5−mm 副鼻腔撮影図(sinogram bin)および19のタイムフレームに分け、これをフィルター逆投影によって行った。このようにして得られた画像データ−セットをSiemens Inveon Research Workplace softwareを用いて可視化した。動的データの累積画像を、放射性トレーサーの取り込みの可視化のためにおよび関心のある領域(regions of interest)(ROI)を規定するために使用した。各19時点で各ROIについて、カウント密度を平均して、ROIに関する時間に対する放射能曲線(TAC)を得た(図15参照)。
【0213】
腫瘍のTACを各時点で全身のものに正規化して、正規化した取り込み値(normalized uptake value)(NUV)を得た。注射してから60分でのNUV(NUV60)、0〜60分のNUVの積分として算出されたNUV曲線下の面積(AUC)を比較に使用した(図16参照)。
【0214】
スキャン後、様々な組織を取り除き、秤量し、即座にフッ素−18放射能計測するために計測用チューブに置いて、[18F]11の生体内分布を評価した(下記表4参照)。
【0215】
ベヒクルまたはシクロホスファミドで処置した2匹の38C13異種移植片を有するマウスの代表的なOSEM3D 再構築[18F]11 PET画像を図17に示し、この際、丸は腫瘍を示す。
【0216】
表4の生体内分布の結果から、ベヒクルに比して、CPAで処置したマウスの腫瘍では、[18F]11の取り込みが有意に2倍増加したことが示され、これは、PETイメージングパラメーター、腫瘍のTAC、NUV60及びAUCから得られた半定量データによって確認された(図15及び16を参照)。したがって、図15〜17及び表4に示される生体内分布およびPETスキャンデータから、[18F]11によりインビボでの腫瘍のアポトーシスを検出できる可能性があることが示され、これにより、初めて、腫瘍のためのカスパーゼ−3に特異的なPET画像化剤の使用が記載される。
【0217】
【表5】
【0218】
実施例9−化合物11の細胞活性
化合物11の細胞活性を酵素アッセイを用いて評価し、化合物14の細胞活性と比較した。
【0219】
マウス放射線誘発線維肉腫(RIF−1)細胞を、カスパーゼ阻害剤である、z−VAD−fmk(100μM)、化合物11及び化合物14で15分間処理した後、48時間、シスプラチン(100μM)でアポトーシスを誘導した。Δ−PARP免疫ブロットバンドは、カスパーゼ 3の切断により得られるPARPの内因性の89kDaの大きさの断片に相当する。α−チューブリンをローディングコントロールとして使用した。
【0220】
図18に示されるように、RIF−1細胞を、化合物14ではなく、化合物11と予めインキュベーションすると、シスプラチンで誘導されるアポトーシスによるカスパーゼ−3と同種のターゲットPARPの誘導が妨害されたことから、化合物11によるカスパーゼ−3の細胞不活性化が示唆される。
【0221】
薬剤により誘導されるアポトーシスの条件下で化合物11の結合性をさらに確立するために、我々は、実施例6で上記されるのと同様にしておよび図9に示されるように、LNM35、RIF−1及びPEO1/4癌細胞における放射線標識された[18F]11の取り込みを評価した。予想とおり、シスプラチン耐性卵巣癌細胞(PEO4)以外の、全ての薬剤で処理された細胞は、コントロールに比べて[18F]11の取り込みが有意に増加した。さらに、薬剤処理したLNM35細胞における[18F]11の取り込みの増加は、カスパーゼ−Gloアッセイを用いた細胞活性カスパーゼ−3の量と相関した。
【0222】
実施例10−保護前駆体を用いた[18F]11の合成
イサチン放射性リガンド[18F]11は、図2(i)、図19に及び実施例3に記載されるのと同様の2段階法を用いて合成できる。この放射性合成(radiosynthesis)の欠点には、安定した不純物の存在があり、これにより配合される生成物の比放射能が減少する(1〜4Ci/μmol)。より根本的には、安定した不純物は、かなりの量(5〜10μg/mL)で存在し、この量は臨床的進行には多すぎる値である。毒性試験を行うためにこの不純物の特性を明らかにしようと試みたが結論は得られず、安定した不純物が、「冷たい」フッ素−19トリアゾール化合物でなく、イサチン由来であることが確認されたにとどまった。
【0223】
HPLC分析によって、不純物が放射性合成中の副反応の結果であり、アルキン前駆体中に存在する物質ではないことが確認された。イサチン分子構造の試験から、化学反応性が高い2つの部位、上記クリック化学付加環化(click chemistry cycloaddition)で使用される末端アルキン基およびカスパーゼ−3活性部位での結合に必要なC−3カルボニル位置のみが示された。C−3カルボニル位置に関する保護基ストラテジーを調べ、特に、下記一般式38及び43で示される(この際、n=0、1、2、3、4、5または6であり、x=離脱基、例えば、メシレート、トシレート、ノシレート(nosylate)、または他のスルホネートエステルまたはハライド)ような、アセタールジオキソランとしてのC−3カルボニル位置の保護を調べた:
【0224】
【化6】
【0225】
保護アルキン、(S)−1−{[1’−[1−(2−プロピニル)]−(1’2’−ジヒドロ−2’−オキソスピロ(1,3−ジオキサン−2,3’−[3H]インドール)−5’−スルホニル}−2−(2,4−ジフルオロフェノキシメチル)−ピロリジン(化合物39)、及び保護トリアゾール(化合物40)を図20に要約されるようにして合成した。
【0226】
一般的な実験の詳細は、本実施例の最初におよびSmith et al29に既に記載されている。全ての反応は、アルゴン雰囲気下で行われた。
【0227】
(S)−1−{[1’−[1−(2−プロピニル)]−(1’2’−ジヒドロ−2’−オキソスピロ(1,3−ジオキサン−2,3’−[3H]インドール)−5’−スルホニル}−2−(2,4−ジフルオロフェノキシメチル)−ピロリジン(化合物39)を、下記方法に従って製造した。無水トルエン(6mL)における化合物25(92mg、0.2mmol)の溶液に、1,3−プロパンジオール(0.3mL)及び4−トルエンスルホン酸(10mg、0.005mmol)を添加した。次に、形成した水を共沸蒸留によって除去しながら、この溶液を24時間還流した。さらに、この反応混合物を、室温まで冷却して、バルク溶媒を減圧下で除去した。次に、サンプルをDCM(10mL)に再溶解し、飽和Na2CO3(1×10mL)、水(1×10mL)及びブライン(1×10mL)で洗浄した後、Na2SO4で乾燥した。カラムクロマトグラフィー(2:1 酢酸エチル/ヘキサン)によって、目的とする生成物を第一の画分として得た(無色の油、46mg、44%)。HRMS (ESI) = 519.1393 (M + H)+. C25H25F2N2O6Sに関する算出値 519.1401. 1H NMR (400 MHz, CDCl3) δ 7.90 (d, J = 1.8 Hz, 1 H), 7.87 (dd, J = 8.2 Hz, 1.8 Hz, 1 H), 7.09 (d, J = 8.2 Hz, 1 H), 7.04-6.98 (m, 1 H), 6.88-6.78 (m, 1 H), 4.93 (t, J = 12.1 Hz, 2 H), 4.45 (d, J = 2.4 Hz, 2 H), 4.32-4.27 (m, 1 H), 3.99-3.93 (m, 4 H), 3.56-3.49 (m, 1 H), 3.14-3.08 (m, 1 H), 2.43-2.37 (m, 1 H), 2.30 (t, J = 2.4 Hz, 1 H), 2.09-1.93 (m, 2 H), 1.78-1.64 (m, 3 H)。
【0228】
(S)−1−{[1’−[1−(2−フルオロエチル)−1H−[1,2,3]−トリアゾール−4−イル]−(1’2’−ジヒドロ−2’−オキソスピロ(1,3−ジオキサン−2,3’−[3H]インドール)−5’−スルホニル}−2−(2,4−ジフルオロフェノキシメチル)−ピロリジン(化合物40)を、下記方法に従って製造した。無水トルエン(4mL)における(S)−1−((1−(2−フルオロエチル)−1H−[1,2,3]−トリアゾール−4−イル)メチル)−5−(2(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン)(化合物11)(12mg、0.02mmol)の溶液に、1,3−プロパンジオール(0.15mL)及び4−トルエンスルホン酸(1mg、0.005mmol)を添加した。次に、形成した水を共沸蒸留によって除去しながら、この溶液を24時間還流した。さらに、この反応混合物を、室温まで冷却して、バルク溶媒を減圧下で除去した。次に、サンプルをDCM(10mL)に再溶解し、飽和Na2CO3(1×10mL)、水(1×10mL)及びブライン(1×10mL)で洗浄した後、Na2SO4で乾燥した。カラムクロマトグラフィー(4:1 酢酸エチル/ヘキサン)によって、目的とする生成物を第一の画分として得た(無色の油、7mg、58%)。HRMS (ESI) = 519.1393 (M + H)+; C25H25F2N2O6S 519.1401に関する算出値. 1H NMR (400 MHz, CDCl3) δ.7.84 (s, 1 H), 7.81 (d, J = 1.6 Hz, 1 H), 7.67 (s, 1 H), 7.43 (dd, J = 8.4 Hz, 1.6 Hz, 1 H), 7.04-6.98 (m, 1 H), 6.88-6.79 (m, 1 H), 4.96 (s, 1 H), 4.92 (t, J = 2.8 Hz, 2 H), 4.79 (dt, J = 46.6 Hz, 4.8 Hz, 2 H), 4.64 (dt, J = 27 Hz, 4.8 Hz, 2 H), 4.29 (dd, J = 8.4 Hz, 2.6 Hz, 1 H), 4.00-3.89 (m, 4 H), 3.53-3.48 (m, 1 H), 3.10-3.07 (m, 1 H), 2.43-2.37 (m, 1 H), 2.05-1.92 (m, 2 H), 1.75-1.65 (m, 3 H)。
【0229】
次に、[18F]11は、1以上のこれらの保護前駆体を用いて合成できる。保護アルキン前駆体39を用いた具体的な放射化学反応を図21に示す。このような保護アルキン前駆体を使用することによって、C−3位置での望ましく副反応を防止する。
【0230】
硫酸銅(II)・6水和物(0.51mg、2.06μmol、25μL)の水溶液に、窒素下で、リン酸ナトリウム緩衝液(pH6.0、250mM、25μL)におけるアスコルビン酸ナトリウム(2.53mg,12.79μmol)、DMF(25μl)におけるアルキンアセタール前駆体(1.0mg、1.93μmol)(化合物39)、及びアセトニトリル(100μL)における[18F]−フルオロエチルアジド(1.27mCi)(化合物27)を添加した。この反応混合物を80℃で30分間加熱した(図22参照)。塩酸(6N、100μL)を添加した後、攪拌混合物をマイクロ波空洞(2s、50W、設定温度 80℃)を用いて加熱した。HPLC移動相(50μL、35% MeCNを用いた50% MeOH含む水)を添加した後、混合物を予備HPLCによって精製した(図23参照)。[18F]11を24%の減衰補正した放射化学的収率(出発[18F]−フルオロエチルアジドに対する)で単離し、PBS/10%EtOHでC18−SepPak SPSを用いて配合した。[18F]11と共に溶出する安定した不純物のレベルは0.84μg/mLであった。比放射能は0.05Ci/μmol(2GBq/μmol)であった(図24参照)。
【0231】
類似の保護先駆体を用いて本発明の他の化合物を合成できることはいうまでもない。
【0232】
実施例11−アポトーシス細胞への[18F]11及び[18F]42の取り込みの比較
本プロジェクトのさらなる態様は、弱いイサチンカスパーゼ−3阻害剤であるN−[1−(2−フルオロエチル)−1H−[1,2,3]−トリアゾール−4−イル]イサチン(化合物42)の分析を包含する。この化合物を、図25に示されるようにして、N−(2−プロピニル)イサチン(化合物41)を用いて合成した。化合物[18F]42は、コアのイサチンフレームワークを有するため、この化合物は公知のイサチン結合形式で活性化カスパーゼ−3に結合でき、また、放射性標識トリアゾール機能性がありうる。しかしながら、[18F]42は、カスパーゼ−3に対する高い親和性及び選択性を付与するピロリジンスルホンアミド機能性がない。[18F]42を、[18F]11と直接比較するために、細胞取り込みアッセイで調べた。この目的は、取り込みが細胞透過性の増加の結果、アポトーシス細胞の特性、あるいはカスパーゼ−3結合の結果であるのかを決定する目的で、化合物の取り込みプロフィールを調べることである。[18F]11とは異なり、薬剤処置後に[18F]42結合性の増加はなかった(図26参照)ことから、[18F]11結合はカスパーゼ3活性化によるものであるという知見が支持された。
【0233】
N−(2−プロピニル)イサチン(化合物41)を、Smith et al29に記載されるのと同様にして合成した(アルキン12及び13)。1H NMR (600 MHz, CDCl3) δ 7.65-7.61 (m, 2 H), 7.19-7.12 (m, 2 H), 4.61 (s, 2 H), 2.30 (s, 1 H)。
【0234】
N−[1−(2−フルオロエチル)−1H−[1,2,3]−トリアゾール−4−イル]イサチン(化合物42)を、Smith et al29に記載されるのと同様にして合成した(トリアゾール14及び15)。1H NMR (600 MHz, CDCl3) δ 7.76 (s, 1 H), 7.63-7.57 (m, 2 H), 7.34 (d, J = 8.4 Hz, 1 H), 7.13 (t, J = 3.6 Hz, 1 H), 5.03 (s, 2 H), 4.81 (dt, J = 46.8 Hz, 4.8 Hz, 2 H), 4.68 (dt, J = 27 Hz, 4.8 Hz, 2 H)。
【0235】
注意(N.B.) 番号の間違いにより、化合物15及び21は、実際同じ化合物である。
【0236】
参考文献
1. Lahorte et al. (2004) European Journal of Nuclear Medicine and Molecular Imaging. 31. 887 - 919.
2. Boersma et al. (2005) Journal of Nuclear Medicine. 46, 2035 - 2050.
3. Aloya, R. et al. (2006) アポトーシス. 11, 2089 - 2101.
4. Damianovich, M. et al. (2006) European Journal of Nuclear Medicine and Molecular Imaging.. 33, 281 - 291.
5. WO 2005/067388
6. Neuss, M. et al (2001). Cardiovascular Drugs and Therapy. 15, 507-523.
7. Lee, D. et al (2000) Journal of Biological Chemistry. 275, 16007 - 16014.
8. Lee, D. et al. (2001) Journal of Medicinal Chemistry.44, 2015-2026
9. Chu, W. et al (2005) Journal of Medicinal Chemistry. 48. 7637 - 7647
10. Chu, W. et al (2007) Journal of Medicinal Chemistry. 50. 3751 - 3755.
11. WO 2005/053752
12. Faust, A. et al (2007) Quarterly Journal of Nuclear Medicine and Molecular Imaging. 51. 67 - 71
13. Kopka, K. et al (2006) Journal of Medicinal Chemistry. 49, 6704 - 6715.
14. Zhou, D. et al (2006) Bioorganic and Medicinal Chemistry Letters. 16, 5041 - 5046.
15. WO 2006/067376
16. Glaser et al (2007) Bioconjugate Chemistry. 18. 989 - 993
17. Wipf, P. et al (2001) Tetrahedron. 57, 283 - 296.
18. Barthel, H. et al (2004) British Journal of Cancer. 90, 2232 - 2242.
19. Aboagye, E.O. et al, (1997) Biochemical Pharmacology. 54, 1217-1224.
20. Twentyman, P.R. et al.(1980) J Natl. Cancer Inst. 64, 595-604
21. Workman, P., et al (1998) British Journal of Cancer. 77, 1-10.
22. Wolf, C.R. et al. (1987) Int. J. Cancer. 39, 695-702
23. Langdon, S.P. et al (1988) Cancer Research. 48, 6166-6172
24. Kozaki, K et al (2000) Cancer Research. 60, 2535-40
25. Schirrmacher, R. et al (2008) Tet. Lett. 49, 4824 - 4827.
26. Marik, J. and Sutcliffe, J. L. (2006) Click for PET: Rapid preparation of [18F]fluoropeptides using CuIcatalyzed 1,3-dipolar cycloaddition. Tet. Lett. 47, 6681-6684
27. Sirion, U. et al, (2007) An efficient F-18 labeling method for PET study: Huisgen 1,3-dipolar cycloaddition of bioactive substances and F-18-labeled compounds. Tet. Lett., 48, 3953-3957
28. Li, Z, et al. (2007) Click Chemistry for 18F-Labeling of RGD Peptides and microPET Imaging of Tumor Integrin αvβ3 expression. Bioconjugate Chem., 18, 1987-1994.
29. Smith G. et al, (2008) Design, Synthesis and Biological Characterization of a Caspase 3/7 Selective Isatin Labeled with 2-[18F]fluoroethylazide. Journal of Medicinal Chemistry. 51. 8057-8067
【技術分野】
【0001】
本発明は、新規なイサチン5−スルホンアミド誘導体、ならびにカスパーゼ活性およびカスパーゼ依存性アポトーシスの可視化および定量化、および治療用途における分子画像化剤(molecular imaging agent)としてのその使用を提供する。
【背景技術】
【0002】
アポトーシスまたはプログラム細胞死(PCD)は、最も普及している細胞死経路であり、非常に調節されたエネルギー保存メカニズムを介して進行する。健常な状態では、アポトーシスは、細胞の成長を制御し、細胞数を調節し、形態形成を容易にし、さらに有害なまたは異常な細胞を除去する際に重要な役割を果たす。このプロセスの異常調節は、癌や自己免疫疾患等の、アポトーシスの阻害に関連するもの、および神経変性疾患、血液病、AIDS、虚血及び同種移植の拒絶反応等の、過活動アポトーシスに関連するものなどの、多くの病状にかかわりがある。したがって、アポトーシスの可視化および定量化は、このようなアポトーシスに関連する病態生理学の診断に有用である。
【0003】
これらの疾患の治療のための処置は、このプロセスを刺激するまたは阻害することによって、均衡のとれたアポトーシスを回復させることを目的とする。したがって、細胞や組織におけるアポトーシスの非侵襲性の画像化(imaging)は、治療的介入に対する応答の初期評価にとって非常に有益であり、病理学的なプロセスの破壊への新しい知見を提供する。悪性の成長を症状が末期になる前に確実に制御するために癌治療の有効性を初期にモニターすることは特に関心が高い。
【0004】
細胞死を画像化するのに有用なプローブのうち、放射性標識されたアネキシンV(Annexin V)は最も注目を集めている1,2。しかしながら、アネキシンVは、細胞内のものではなく、細胞外表面のものしか検出せず、負に帯電したリン脂質にのみ結合するため、アポトーシスと壊死とを区別することができない。膜相互作用プローブに関しては、数多くのジ−ダンシルシステイン(di-dansyl cysteine)およびナフチル−エチル−フルオロアニリン(naphthyl-ethyl-fluoroalanine)誘導体がアポトーシスを画像化するために開発された3,4,5。しかしながら、このような膜相互作用プローブはまた、アポトーシス性細胞に対する特異性が低く、別の試験を必要とせずにはアポトーシスと壊死とを区別することができない6。より最近では、カスパーゼと呼ばれる酵素群に結合する特定の化合物の開発への興味が高まっている。
【0005】
カスパーゼは、アポトーシスの調節に中心的な役割を果たすシステインアスパルテートに特異的なプロテアーゼ(cysteine aspartate-specific proteases)群である。内因性のおよび外因性のシグナルネットワークは、「イニシエーター」カスパーゼ8(外因性)または9(内因性)を活性化し、さらに不活性なプロカスパーゼ3、6及び7を切断して活性のある「エクセキューショナー(executioner)」カスパーゼ3、6及び7にする。このエクセキューショナーカスパーゼは、最終的には細胞タンパク質の切断により細胞死をもたらし、これは非常に選択的にアスパラギン酸残基の右側に起こる。切断されるタンパク質としては、DNA修復酵素(例えば、PARP)、キーシグナルタンパク質(key signaling protein)(例えば、Akt、Ras)、核骨格タンパク質(nuclear skeletal protein)(例えば、アクチン、α−フォドリン、ラミン)および細胞サイクルレギュレーター(例えば、p27Kip1)がある。
【0006】
[131I]IZ−VAD−fmk等の、カスパーゼのペプチド系の不可逆的阻害剤を分子画像化剤として使用することは、選択性が中程度しかなく、細胞への取り込みがインビボでの画像化には不十分であるなど、細胞透過性が低いため、不十分であった。
【0007】
より最近では、イサチンとして知られている化学物質群が潜在的なカスパーゼ阻害剤として研究されてきた。イサチンの作用メカニズムは、活性化カスパーゼの酵素活性部位への共有結合を介したカスパーゼ3及び7との細胞内酵素−阻害剤複合体の形成がかかわっていると考えられる。イサチンのジカルボニル官能基がその作用メカニズムには必須である;この基が活性部位のシステイン残基と結合して、イサチンの求電子性のC−3カルボニル炭素および求核性のシステインチオレート官能基によるチオヘミケタルを形成する7。
【0008】
高スループットスクリーンについては、Lee et al.がカスパーゼ−3の阻害剤として非ペプチドイサチンN−(1−メチル)−5−ニトロイサチン(N-(1-methyl)-5-nitroisatin)を同定した。構造を最適化することによって、スルホンアミド(S)−1−ベンジル−5−(2−フェノキシメチル−ピロリジン−1−スルホニル)イサチン[(S)-1-benzyl-5-(2-phenoxymethyl-pyrrolidine-1-sulfonyl)isatin](2.5nM)8(本明細書中の化合物13を示す)が開発された。他のイサチンスルホンアミドがカスパーゼ3及び7の阻害剤として開発された9−12。Kopka13およびMach14は、それぞれ、18Fで標識された(S)−1−(4−(2−フルオロエトキシ)ベンジル)−5−[1−(2−フェノキシメチルピロリジニル)スルホニル]イサチン[18F-labeled (S)-1-(4-(2-fluoroethoxy)benzyl)-5-[1-(2-phenyoxymethylpyrrolidinyl)sulfonyl]isatin]をポジトロン放出型断層撮影(PET)の推定上のトレーサーとして開発し、Kopka13は、放射性標識された類似体である[125I](S)−1−(4−ヨードベンジル)−5−(2−フェノキシメチル−ピロリジン−1−スルホニル)イサチン)(本明細書中の化合物14を示す)の生物学的特性を調査した。WO 99/06367およびWO 01/22966には、多数のイサチン誘導体およびカスパーゼの阻害のためのこれらの使用が記載される。WO 2006/074799、US 2005/0250798およびGB 1,240,648には、数多くのイサチン5−スルホンアミド誘導体およびアポトーシスの画像化剤としてのこれらの使用が記載される。しかしながら、これらの既知の化合物は、分子安定性が低い、カスパーゼ−3との親和性が比較的低いおよび親油性が高いなどの、数多くの欠点がある。分子安定性が低いと、代謝が速いため、低い信号対ノイズ比でコントラストの像に劣る。また、カスパーゼ−3との親和性が低いと、画像のコントラストが劣る。親油性が高いと、系からの排泄(system elimination)が悪く、高分子への全体的な非特異的な結合が増加してしまう。
【発明の概要】
【0009】
したがって、分子安定性が向上し、カスパーゼ酵素に対する親和性が向上し、かつ親和性が低減した新規なイサチン誘導体を提供することが、本発明の目的である。
【0010】
本発明の第一の態様は、下記式Aを有する新規なイサチン5−スルホンアミド誘導体、またはこの塩、水和物もしくはプロドラッグを提供する:
【0011】
【化1】
【0012】
ただし、Rは、フェニル、3−フルオロフェニル、2,4−ジフルオロフェニル、3,5−ジフルオロフェニル、置換されてもよいテトラヒドロピラン、置換されてもよいジアジンおよび置換されてもよいトリアゾールであり;R’は、置換されてもよいフェニルまたは置換されてもよいトリアゾールであり;
この際、Rがフェニルである場合には、R’は、置換されてもよいトリアゾールである。
【0013】
好ましい実施形態では、Rは置換されてもよいトリアゾールを含み、R’は置換されてもよいフェニルを含む。他の好ましい実施形態では、Rは置換されてもよいフェニルを含み、R’は置換されてもよいトリアゾールを含む。
【0014】
好ましくは、前記置換されてもよいフェニル、置換されてもよいテトラヒドロピランおよび置換されてもよいジアジンは、必要であれば、1以上の電子求引基で置換される。好ましくは、前記電子求引基は、ハロゲン、ニトロ基およびカルボン酸基またはアルデヒドまたはケトン等の他のカルボニル含有官能基からなる群より選択される。より好ましくは、前記電子求引基はハロゲンである。好ましくは、前記電子求引基はフッ素である。
【0015】
好ましくは、置換されてもよいフェニルは2,4−ジフルオロフェニルである。
【0016】
一実施形態では、置換されてもよいトリアゾールは、必要であれば、置換されたアルキル基で置換される。好ましくは、前記置換されたアルキル基は、ハロゲンで置換されたアルキルである。より好ましくは、前記ハロゲンで置換されたアルキルはC1−4のフルオロアルキルである。好ましくは、前記C1−4のフルオロアルキルは、フルオロメチル、2−フルオロエチル、3−フルオロプロピルまたは4−フルオロブチルである。最も好ましくは、前記C1−4のフルオロアルキルは2−フルオロエチルである。
【0017】
他の実施形態では、置換されてもよいトリアゾールは、必要であれば、置換されたアルキル基で置換される。好ましくは、前記アルキルはメチルである。
【0018】
本発明はまた、これらの光学異性体およびジアステレオ異性体などの、本発明の化合物のすべての立体異性体を含むことは当業者に理解されるであろう。本発明の化合物は、特定の光学異性体の実質的な純粋な溶液の形態でまたはラセミ混合物として存在してもよい。好ましくは、本発明の化合物は、S光学異性体の実質的な純粋な溶液として、またはS光学異性体から実質的に構成される溶液として存在する。好ましくは、S光学異性体およびR光学異性体双方を含むラセミ混合物では、S光学異性体がラセミ混合物中本発明の化合物の少なくとも50%を構成する。より好ましくは、S光学異性体が、ラセミ混合物中本発明の化合物の少なくとも60%、少なくとも70%、少なくとも80%、少なくとも90%、少なくとも95%または少なくとも99%を構成する。
【0019】
本発明の特に好ましい化合物としては、RおよびR’が下記に規定される式Aの化合物がある:
【0020】
【表1−1】
【0021】
【表1−2】
【0022】
【表1−3】
【0023】
【表1−4】
【0024】
好ましくは、「塩」ということばは、酢酸、プロピオン酸、乳酸、酒石酸、コハク酸、フマル酸、マレイン酸、マロン酸、マンデル酸、リンゴ酸、フタル酸、塩酸、臭化水素酸、リン酸、硝酸、硫酸、メタンスルホン酸、ナルタレンスルホン酸、ベンゼンスルホン酸、トルエンスルホン酸、カンファースルホン酸などの有機及び無機酸、ならびに本発明の化合物が塩基部分を含む際に同様にして既知の許容できる酸由来の塩を包含する。塩はまた、本発明の化合物がカルボン酸若しくはフェノール酸部分を含む際には、有機及び無機塩基、好ましくはアルカリ金属塩、例えば、ナトリウム、リチウム若しくはカリウム、または塩基付加塩を形成できる同様の部分から形成されてもよい。
【0025】
本明細書中で使用される「水和物」ということばは、予め水と化学的に結合した本発明の化合物の形態を意味する。
【0026】
本明細書中で使用される「プロドラッグ」ということばは、代謝によってインビボで本発明の化合物に変換可能な化合物を意味する。
【0027】
本明細書中で使用される、「置換されてもよいフェニル」という表現は、必要であれば1以上の置換基が環の2、3、4、5、および6位の1以上の位置に配置されてなるフェニル基を意味する。
【0028】
本明細書中で使用される「テトラヒドロピラン」ということばは、5つの炭素原子及び1つの酸素原子を含む飽和の6員環から構成される有機化合物を意味する。
【0029】
本明細書中で使用される「トリアゾール」ということばは、2つの炭素原子及び3つの窒素原子を有する5員環を有する、分子式:C2HN3を有する化合物を意味する。これらの化合物としては、異性体 1,4−及び1,5で2置換された1,2,3 トリアゾールがある。
【0030】
本明細書中で使用される「ハロゲン」ということばは、臭素、塩素、フッ素およびヨウ素を意味する。
【0031】
本明細書中で使用される「アルキル」ということばは、脂肪族炭素化水素鎖を意味し、特記しない限り、以下に制限されないが、1、2、3、4、5または6個の炭素原子を有する直鎖または分岐鎖を包含する。アルキルとしては、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、sec−ブチル、イソブチル、tert−ブチルなどが挙げられる。
【0032】
本明細書中で使用される「置換されたアルキル」という表現は、ハロゲン、ヒドロキシル、チオール、アミノ、別のヘテロ原子、芳香族またはヘテロ芳香族基ならびにポリエチレングリコール等のポリエーテル、スクシニジル(succinidyl)、−NH−(CH2)n−NH−及びポリアミドなどのスペーサー基からなる群より選択される1以上の置換基をさらに有するアルキルを意味する。
【0033】
本明細書中で使用される「ジアジン」ということばは、分子式:C4H4N2を有する有機化合物群を意味し、各化合物は炭素原子の2個が窒素で置換されるベンゼン環を含む。これらの化合物としては、異性体 ピラジン(1,4−ジアジン)、ピリミジン(1,3−ジアジン)、およびピリダジン(1,2−ジアジン)がある。
【0034】
本発明の化合物は、活性化カスパーゼに特異的であり、活性化カスパーゼに対して高い親和性を有する。本発明の化合物は、カスパーゼ3及び7の強力かつ選択的な阻害剤である。特に本発明の化合物は、カスパーゼ−3及びカスパーゼ−7に対する高い親和性を有する。本発明の化合物は、アポトーシス中に活性形態で発現されるのみであり、ゆえに、カスパーゼ−3および/またはカスパーゼ−7の活性はカスパーゼに依存するアポトーシスの信頼性のあるインジケータである。
【0035】
本発明の化合物は、多くの既知のイサチン5−スルホンアミド誘導体と比べて親油性が低いため、全身で良好に除去でき、高分子への一般的な非特異的な結合が低減する。しかしながら、カスパーゼは細胞内プロテアーゼであるため、本発明の化合物は、非促進拡散によって細胞膜を通過するのに十分な親油性を有するため、細胞内外を自由に通過できる。このように、本化合物は、活性化カスパーゼ−3および/またはカスパーゼ−7を有する細胞中でのみ迅速に取り込みおよび保持を双方向に行うであろう。理想的な親油性(LogPとして表わされる)は、多くの他の要因に加えて、化合物が通過するのに必要な細胞タイプによって異なる。一実施形態では、親油性は1.0〜2.0の領域であることが好ましい。
【0036】
本発明の化合物は、代謝安定性が向上し、これにより化合物が代謝され、また、場合によっては排出される速度が減少する。代謝安定性が増すと、活性化カスパーゼと結合すると、アポトーシスを受ける細胞内に本発明の化合物が蓄積される。これにより、信号対ノイズ比の高いコントラストの良好な画像が得られる。これは、細胞及び組織内のカスパーゼ活性の正確な可視化および定量化を可能にするため、分子画像化剤では好ましい。
【0037】
好ましくは、本発明の化合物は、非処置腫瘍、心臓および脳組織では取り込みが低い。これにより、カスパーゼ活性の変化に関連する、哺乳動物組織におけるこれらの化合物の活性化カスパーゼへの結合の変化をモニターしやすくなる。
【0038】
本発明の第二の態様は、画像化部分(imaging moiety)で標識される、本発明の第一の態様の化合物を提供する。
【0039】
前記標識は、官能基内に画像化部分を有していても、または別の物質としての画像化部分が結合されていてもよい。
【0040】
前記画像化部分は、検出可能なシグナルを製造できる部分を有していてもよい。このような部分としては、蛍光ラベル及び放射性ラベルがある。
【0041】
蛍光ラベルはフルオロフォアが共有結合してなる。好ましいフルオロフォアとしては、フルオレセインイソチオシアネート、ローダミンの誘導体、クマリンならびにシアニン色素である、Alexa Fluors及びDyLight Fluorsがある。
【0042】
好ましくは、本化合物はラジオアイソトープで標識される。ラジオアイソトープは本発明の化合物の分子量を実質的に増加させず、臨床的な非侵襲性イメージングに使用できるため、画像化部分としてラジオアイソトープを使用することが好ましい。
【0043】
好ましくは、ラジオアイソトープはポジトロン放射体である。放射性同位体は、3H、14C、18F、11C、120I、123I、124I、125I、131I、94mTc、66Ga、68Ga、64Cu、61Cu、67Cu、75Br、76Br、94mTc、99mTc、201Tl、111In、86Yおよび89Zrからなる群より選択されうる。
【0044】
画像化部分をポジトロン放出型断層撮影(PET)を用いて画像化する場合には、放射性同位体は18F、11C、120I、124I、94mTc、66Ga、68Ga、64Cu、67Cu、86Y、75Brおよび76Brからなる群より選択されることが好ましい。
【0045】
画像化部分を単光子放出コンピュータ断層撮影(SPECT)を用いて画像化する場合には、放射性同位体は123I、99mTcおよび111Inからなる群より選択されることが好ましい。
【0046】
好ましくは、画像化部分は18Fまたは11Cである。
【0047】
好ましくは、化合物1〜12および29〜34のいずれかにおいて、Rおよび/またはR’内のフッ素原子の1以上が18Fである。
【0048】
例えば、化合物6〜12、および29〜36におけるように、RまたはR’での1,2,3−トリアゾール基の付加が、化合物が18Fまたは11Cで容易に標識できるため、さらに好ましい。
【0049】
本発明の特に好ましい実施形態は、下記式を有する。
【0050】
【化2】
【0051】
本発明の他の好ましい化合物は、下記一般式を有する。
【0052】
【化3】
【0053】
本発明の第三の態様は、本発明の第一の態様に係る化合物を、必要であれば一以上の製薬上許容できる担体、希釈剤または賦形剤と組み合わせてなる組成物を提供する。
【0054】
本発明の第四の態様は、本発明の第二の態様に係る化合物を、必要であれば一以上の製薬上許容できる担体、希釈剤または賦形剤と組み合わせてなる組成物を提供する。
【0055】
本組成物はまた、一以上の別の活性剤を含んでもよい。前記別の活性剤は、本発明の化合物によって生産されるシグナルを促進する物質を含んでもよい。好ましくは、前記活性剤は、マレイン酸ジエチル(DEM)、L−ブチオニン−(S,R)−スルホキシミン(BSO)及びこれらの誘導体などの、細胞グルタチオンレベルを枯渇するまたは調節する化合物を含む。
【0056】
本発明の第三または第四の態様の組成物は、既知の方法によって投与されうる。
【0057】
本組成物は、細胞に接近できる化合物を暴露する(exposing)、インキュベートする(incubating)、接触する(touching)、結合する(associating)または作製する(making)ことによって、細胞と接触させてもよい。
【0058】
本組成物は、経口(吸入によることを含む)、非経口、粘膜(例えば、口腔、舌下、鼻腔内)、直腸または経皮投与によって被検者に投与されてもよく、本組成物はそれに従って適用されてもよい。
【0059】
経口投与では、本組成物は、液体または固体として、例えば、溶液、シロップ、懸濁液またはエマルジョン、錠剤、カプセル及びロゼンジとして配合できる。
【0060】
液剤は、通常、適当な水性または非水性液状担体、例えば、水、エタノール、グリセリン、ポリエチレングリコールまたは油における、化合物またはその生理学上許容できる塩、水和物若しくはプロドラッグの懸濁液または溶液から構成されるであろう。液剤はまた、懸濁化剤、防腐剤、香料添加剤または着色剤を含んでもよい。
【0061】
錠剤形態の組成物は、固体製剤を調製するのに一般的に使用される適当な医薬担体を用いて調製できる。このような担体の例としては、ステアリン酸マグネシウム、デンプン、ラクトース、スクロース及び微結晶性セルロースが挙げられる。
【0062】
カプセル形態の組成物は、一般的なカプセル化方法を用いて調製できる。例えば、活性成分を含む粉末、顆粒またはペレットを標準的な担体を用いて調製した後、硬質ゼラチンカプセル中に充填してもよい。または、分散液または懸濁液を適当な医薬担体、例えば、水性ゴム、セルロース、ケイ酸塩または油を用いて調製した後、前記分散液または懸濁液を軟質ゼラチンカプセル中に充填してもよい。
【0063】
経口投与用組成物は、例えば、錠剤またはカプセルに配合物を外側コーティングすることによって、消化管を通過する際に活性成分が分解するのに対して保護するように設計されてもよい。
【0064】
鼻腔内または経口投与用組成物は、エアロゾル、液滴、ゲル及び粉末として簡便に配合されてもよい。エアロゾル製剤は、具体的には生理学上許容できる水性または非水性溶媒における活性物質の溶液または細かい懸濁液(fine suspension)を含み、一般的に密閉容器中で滅菌形態で1回量または複数回量で提示され、これは噴霧装置と共に用いられるカートリッジまたはレフィルの形態をとってもよい。または、密閉容器は、容器の内容物が排出されたら処分されるような用途である、単回投与鼻吸入器等の単回調剤装置(unitary dispensing device)または絞り弁を備えたエアロゾルディスペンサーであってもよい。投与形態がエアロゾルディスペンサーを含む場合には、製薬上許容できる推進剤を含むであろう。エアロゾル投与形態はまたポンプ−アトマイザーの形態をとってもよい。
【0065】
口腔または舌下投与に適する組成物としては、活性成分が糖及びアカシア、トラガカント、またはゼラチン及びグリセリン等の担体と共に配合される、錠剤、ロゼンジ及びトローチが挙げられる。
【0066】
直腸または膣内投与用の組成物は、簡便には坐剤(ココアバター等の公知の坐剤の基剤を含む)、ペッサリー、膣錠、泡剤または浣腸剤の形態を有する。
【0067】
経皮投与に適する組成物としては、軟膏、ゲル、パッチ及び粉体噴射剤等の噴射剤が挙げられる。
【0068】
簡便には、本組成物は、錠剤、カプセルまたはアンプル等の単回投与形態を有する。
【0069】
好ましくは、本発明の化合物または組成物は、非経口投与によって被検者に投与される。特に、本組成物は、静脈内、腹腔内、髄腔内、リンパ管内または筋肉内に投与されてもよい。
【0070】
具体的な非経口組成物は、適当なpH、等張性および安定性を有する滅菌した水性または非水性担体における化合物またはその生理学上許容できる塩の経口投与に許容できる溶液または懸濁液から構成される。当業者は、例えば、塩化ナトリウム注射液、リンガー注射液、乳酸加リンガー注射液等の等張ベヒクルを用いて適当な溶液を調製できる。防腐剤、安定化剤、緩衝剤、抗酸化剤および/または他の添加剤を必要であれば含んでもよい。
【0071】
本発明の第五の態様は、分子画像化剤(molecular imaging agent)として使用されるための本発明の第一若しくは第二の態様の化合物または本発明の第三若しくは第四の態様の薬剤組成物を提供する。
【0072】
好ましくは、前記分子画像化剤は、細胞及び組織におけるカスパーゼ活性の可視化および定量化を目的とする。好ましくは、前記分子画像化剤は、ヒトの細胞及び組織等の哺乳動物の細胞及び組織におけるカスパーゼ活性の可視化および定量化を目的とする。前記定量は、細胞または組織の放射線量、または取り込み、解離若しくは分配速度の分析を含んでもよい。
【0073】
活性カスパーゼと複合体を形成すると、細胞及び組織内への標識化合物の取り込みおよび蓄積を画像化して、前記細胞及び組織内のカスパーゼ活性のレベルを示す。したがって、本発明の第六の態様は、
a)細胞または組織を本発明の第二の態様の化合物または本発明の第四の態様の組成物と接触させ;および
b)カスパーゼ活性を検出する
段階を有する、カスパーゼ活性の分子画像化方法を提供する。
【0074】
好ましくは、カスパーゼ活性の検出段階は、検出装置の検出域内に被検者をおき、前記検出装置を用いて被検者内の前記化合物を検出する段階を有する。
【0075】
本方法は、インビトロで行われてもよい。細胞または組織の本発明の化合物または組成物との接触は、細胞または組織に接近できる化合物を暴露する(exposing)、インキュベートする(incubating)、接触する(touching)、結合する(associating)または作製する(making)ことを含んでもよい。
【0076】
本発明の化合物または組成物が放射性標識される際には、カスパーゼ活性を適当な放射線検出装置を用いてインビトロで検出できる。前記装置としては、Packard Topcount等の、ベータカウンター、またはPackard Cobra IITMガンマカウンター(Perkin Elmer, UK)等の、ガンマカウンター、またはラジオ−TLCスキャナー(radio-TLC scanner)が挙げられる。
【0077】
本発明の化合物または組成物が蛍光標識される場合には、前記カスパーゼ活性は、適当な蛍光読み取り装置を用いてインビトロで検出できる。このような装置としては、蛍光顕微鏡、蛍光光度計、またはPerkin Elmer Victor等の、蛍光板リーダーがある。
【0078】
または、本方法は、インビボで行われてもよい。本化合物または本組成物を、本発明の第三および第四の態様で記載される方法によって被検者に投与されてもよい。好ましくは、本化合物は非経口で投与される。
【0079】
本発明の化合物または組成物が放射性標識される際には、カスパーゼ活性は、放射線検出装置を用いて検出してもよい。前記放射線検出装置としては、ポジトロン放出断層撮影(PET)スキャナーまたは単光子放出コンピュータ断層撮影(SPECT)スキャナーが挙げられる。好ましくは、前記放射線検出装置はポジトロン放出断層撮影(PET)スキャナーである。前記PETスキャナーは、18F等のポジトロンを放出するラジオアイソトープによって間接的に放出されるガンマ線の対を検出し、組織内のラジオアイソトープ濃度の3Dイメージを作製できる。したがって、PETは、哺乳動物の組織内の本発明の放射性標識された化合物または組成物の位置を確認する3Dイメージを作製するのに使用できる。
【0080】
本発明の化合物または組成物が蛍光標識される場合には、カスパーゼ活性は、適当な蛍光読み取り装置を用いて検出されてもよい。前記蛍光読み取り装置としては、蛍光内視鏡がある。
【0081】
好ましくは、カスパーゼ活性は、カスパーゼ−3活性である。
【0082】
本発明はまた、カスパーゼ活性の可視化及び定量化のための薬剤または診断組成物の製造を目的とする本発明の第二の態様の化合物の使用を提供する。
【0083】
第六の態様の他の実施形態では、本発明の第一の態様に係る化合物または本発明の第三の態様に係る薬剤組成物は、段階(a)で投与されてもよい。次に、前記化合物を投与後に蛍光または放射性で標識してもよい。
【0084】
上述したように、カスパーゼ酵素はアポトーシスをもたらし、このようなカスパーゼ活性はアポトーシスの指標として使用できる。したがって、本発明の第七の態様は、哺乳動物の細胞または組織におけるカスパーゼに依存するアポトーシスのインビボにおける画像化のための本発明の第二の態様の化合物または本発明の第四の態様の薬剤組成物の使用を提供する。
【0085】
本方法は、
a)本発明の第二の態様の化合物または本発明の第四の態様の組成物を被検者に投与し;
b)被検者を検出装置の検出域に置き;および
c)被検者における化合物を前記検出装置で検出する、
ことを有する。
【0086】
好ましくは、前記化合物または組成物は注射によって被検者に投与される。
【0087】
画像化部分は、非侵襲で外部からまたは血管内放射線(intravascular radiation)検出器若しくは光検出器等の、インビボでの使用のために設計される検出器、または術中での使用のために設計される放射線検出器を用いることによって内部から検出されうる。好ましくは、画像化部分は非侵襲的に検出される。
【0088】
一実施形態では、前記化合物は蛍光標識され、第六の態様で上記で規定されるような蛍光読み取り装置を用いて蛍光標識された化合物から放出される蛍光を測定することによって検出される。
【0089】
第二の好ましい実施形態では、前記化合物は放射性標識され、第六の態様で上記で規定されるような放射線検出装置を用いて放射性標識された化合物から放出される放射線を測定することによって検出される。
【0090】
好ましくは、カスパーゼ活性は、カスパーゼ−3活性である。
【0091】
第七の態様の別の実施形態では、本発明の第一の態様に係る化合物または本発明の第三の態様に係る薬剤組成物を、段階(a)で投与してもよい。次に、前記化合物を投与後にインビボで蛍光または放射性で標識してもよい。
【0092】
上述したように、多数の病気の治療のための処置は、このプロセスを刺激または阻害することによって、正常で釣り合いのとれたアポトーシスを取り戻すことを目的とする。したがって、本発明の第八の態様は、哺乳動物の細胞または組織におけるカスパーゼ活性への試験物質の治療効果を評価するための、本発明の第二の態様の化合物または本発明の第四の態様の薬剤組成物の使用を提供する。
【0093】
本方法は、
a)哺乳動物の細胞または組織を本発明の第二の態様の化合物または本発明の第四の態様に係る組成物と接触させ;
b)前記哺乳動物の細胞または組織を検出装置の検出域に置き:
c)化合物を前記検出装置で検出し;さらに
d)段階a)、b)およびc)を繰り返す、
ことを有してもよい。
【0094】
一実施形態では、前記化合物は蛍光標識され、第六の態様で上記で規定されるような蛍光読み取り装置を用いて蛍光標識された化合物から放出される蛍光を測定することによって検出される。
【0095】
第二の好ましい実施形態では、前記化合物は放射性標識され、第六の態様で上記で規定されるような放射線検出装置を用いて放射性標識された化合物から放出される放射線を測定することによって検出される。
【0096】
第八の態様の別の実施形態では、本発明の第一の態様に係る化合物または本発明の第三の態様に係る薬剤組成物を段階(a)で投与してもよい。次に、前記化合物を投与後にインビボで蛍光または放射性で標識してもよい。
【0097】
好ましくは、本発明の方法は、インビボで行われてもよく、
a)本発明の第二の態様に係る化合物または本発明の第四の態様に係る組成物を被検者に投与し;
b)被検者を放射線検出装置の検出域に置き;
c)被検者における放射性標識された化合物から放出される放射線を前記放射線検出装置を用いて測定し;
d)段階a)、b)およびc)を繰り返す、
ことを有してもよい。
【0098】
第八の態様のインビトロおよびインビボ方法双方で、段階d)は、繰り返しが経時的にカスパーゼ活性の変化を追跡するのに有効である所定の間隔で行うことが好ましい。段階d)の間隔は、問題になっている被検者および試験物質に適切であるべきである。ヒトでは、適当な間隔は、以下に制限されないが、12〜24時間、48時間、1週間、2週間、3週間、4週間、5週間または6週間が挙げられる。
【0099】
各繰り返しで測定されるカスパーゼ活性を比較することによって、経時的な、カスパーゼ活性の程度及び位置の定量的な変化、およびゆえにカスパーゼに依存するアポトーシスの定量的なまたは半定量的な変化を評価できる。カスパーゼ活性の程度および位置の変化は、カスパーゼ活性の刺激または阻害によって、カスパーゼに依存するアポトーシスへの試験物質の治療効果を示しうる。本方法は、確立された薬を用いて治療のための処置に対する被検者の応答を評価するのに、および新規な薬の薬効の評価に使用できる。
【0100】
本発明の化合物または組成物は、試験物質の前に、後にまたはと同時に投与してもよい。好ましくは、段階(a)〜(c)は、試験物質の投与前に被検者で行われ、カスパーゼ活性の1回目の測定結果を提供する。次に、試験物質を投与し、段階(a)〜(c)を上記したような間隔をあけた後繰り返して、カスパーゼ活性の2回目の測定結果を提供する。必要であれば、試験物質をさらに投与する前または後に、段階(a)〜(c)をさらに繰り返して、カスパーゼ活性のさらなる測定結果を提供してもよい。
【0101】
好ましくは、前記化合物または組成物を注射によって被検者に投与する。
【0102】
試験物質は、以下に制限されないが、癌、自己免疫疾患、血液病、HIV、AIDS、虚血、心血管疾患、神経疾患および移植拒絶反応などの病気を治療するのに使用される薬(drug)または薬剤(agent)含んでもよい。特に試験物質は、癌の処置のための化学療法薬、放射線療法薬または免疫療法薬を含んでもよい。
【0103】
上述したように、段階(a)〜(c)を2回以上繰り返してもよい。好ましい一実施形態では、段階(a)の1回目の実施で被検者に投与される化合物は、11Cで放射性標識された本発明に係る化合物である。好ましくは、前記化合物は、化合物35および36からなる群より選択される。次に、段階(a)の2回目の実施で被検者に投与される化合物は、放射性標識された画像化剤である。前記放射性標識された画像化剤は、例えば、フルオロデオキシグルコース(FDG)、3’−デオキシ−3’−18F−フルオロチミジン(FLT)または本発明に係る化合物であってもよい。好ましい一実施形態では、前記画像化剤は18Fで標識される。本実施形態は、特に好ましい。炭素−11(11C)は20分という比較的短い半減期を有する。ゆえに、11Cで標識された化合物を使用することによって、2回目以降のスキャンの間のバックグランドノイズがほとんどないまたはない。これにより、より高い信号対ノイズ比、ならびにカスパーゼ活性およびカスパーゼに依存するアポトーシスのより正確な可視化及び定量化が可能になる。
【0104】
同様にして、本発明の第六および第七の態様に係る方法を比較的短い時間内で繰り返そうとする場合には、11C標識された化合物、さらには18F標識された化合物等の別の放射性標識された画像化剤を使用することによっても、2回目のスキャン中に存在するバックグランドノイズを低減して、信号対ノイズ比を高める。
【0105】
本発明の第九の態様は、カスパーゼ活性の阻害に使用される本発明の第一の態様に係る化合物または本発明の第三の態様に係る組成物を提供する。本方法は、有効量の本発明の第一の態様の化合物または本発明の第三の態様の組成物をこのような処置の必要のある被検者に投与することを有していてもよい。
【0106】
本態様はまた、カスパーゼ活性の阻害のための医薬を製造する際の本発明の第一の態様の化合物または本発明の第三の態様の組成物の使用を提供する。
【0107】
カスパーゼ酵素がアポトーシスをもたらすため、本発明の第九の態様はまた、アポトーシスの阻害に使用される本発明の第一の態様の化合物または本発明の第三の態様の組成物を提供する。
【0108】
カスパーゼ活性の阻害は、過剰のまたは不適切なアポトーシスによって引き起こされるまたは前記アポトーシスに関連する病気若しくは症状を処置するのに有効でありうる。このような病気または症状の例としては、アルツハイマー病等の神経変性疾患、血液病、肝細胞の変性、変形性関節症、AIDS、虚血および同種移植の拒絶反応が挙げられる。
【0109】
本発明の第一の態様の化合物または本発明の第三の態様の組成物は、一般的に、1日投与量レジメで被検者に投与されるであろう。例えば、1日投与量レジメは、約0.001〜約100mg/kg、好ましくは約0.001〜約10mg/kg被検者の体重を必要とするかもしれない。1日投与量は、より大きな哺乳動物では、好ましくは約1mg〜約1000mg、好ましくは1mg〜500mgであり、前記化合物は1日1〜4回投与されうる。または、本発明の化合物が安定である場合には、投与量は、1週間で、1、2または3回投与されてもよい。
【0110】
本発明の化合物および組成物の投与の投与量、レジメおよび形式は、病気または症状および処置される個体によって異なり、かかわりのある開業医の判断に従うであろうことは理解される。
【0111】
本発明の第十の態様は、病態生理を診断するのに使用される本発明の第二の態様の化合物または本発明の第四の態様の組成物を提供する。特に、本発明の第二の態様の化合物または本発明の第四の態様の組成物は、以下に制限されないが、慢性心不全、急性心筋梗塞、発作、神経変性疾患、自己免疫疾患、局所性血液病、局所性AIDS、虚血(心虚血を含む)、および移植拒絶反応などの、過剰なまたは不適切なアポトーシスに関連する病気および疾患を診断するのに使用できる。
【0112】
本方法は、
a)哺乳動物の細胞または組織を本発明の第二の態様の化合物または本発明の第四の態様に係る組成物と接触させ;
b)前記哺乳動物の細胞または組織を検出装置の検出域内に置き;
c)前記検出装置を用いて前記化合物を検出して、前記哺乳動物の細胞または組織内のカスパーゼ活性の定量的な測定結果を提供し、この際、カスパーゼ活性がアポトーシスの指標である、
ことを有しうる。
【0113】
一実施形態では、前記化合物は蛍光標識され、第六の態様で上記で規定されるような蛍光読み取り装置を用いて蛍光標識された化合物から放出される蛍光を測定することによって検出される。
【0114】
第二の好ましい実施形態では、前記化合物は放射性標識され、第六の態様で上記で規定されるような放射線検出装置を用いて放射性標識された化合物から放出される放射線を測定することによって検出される。
【0115】
次に、好ましくは、前記測定結果を標準値と比較することによって、病態生理を診断できる。
【0116】
第十の態様の別の実施形態では、本発明の第一の態様に係る化合物または本発明の第三の態様に係る薬剤組成物は段階(a)で投与されてもよい。次に、前記化合物は投与後に蛍光標識または放射性標識されてもよい。
【0117】
本発明の第十一の態様は、本発明の第一の化合物の合成方法を提供する。本発明の第一の化合物は、適当な方法で調製されうる。実施例1、3及び10に記載され、図1、2(i)、2(ii)、12、19、20及び21に示されるスキームには、本発明の第一の化合物が調製される方法が示される。
【0118】
図1は、化合物1〜5、10〜12および中間体19a〜h、20a〜h、21〜24が調製できる方法を示す。化合物29〜34は類似した方法で調製される。さらなる詳細を実施例1に示される。
【0119】
トリアゾール基を含む本発明の化合物はまた、図2(i)、図12、及び図19に示され、実施例1及び3に記載されるような、「クリック−ラベリング(click-labelling)」と称される方法によって製造できる。このような方法は、例えば、化合物11、35及び36を作製するのに使用できる。本発明の第一の態様の化合物の製造には、非標識の化合物は、もちろん、これらの実施例に開示される放射性標識された化合物の代わりに使用されるであろう。例えば、図2(i)に示され、実施例3に記載されるスキームでは、2−フルオロエチルアジド(化合物27)が2−[18F]フルオロエチルアジド(化合物[18F]27)の代わりに使用される。
【0120】
また、付加環化して置換トリアゾールを得ることを、N−1で官能化されたイサチンを末端アジド及び末端アルキンを有する補欠分子族と反応することによって、逆に行ってもよい(「逆クリック−ラベリング(click-labelling)」と称される方法における)。このような方法は、例えば、化合物6、7、8及び9を作製するのに使用できる。中間体18F標識された末端アルキンの合成方法をも含む、この方法の適切な例は、図2(ii)に示され、実施例1、及びMarik, J and Sutcliffe, J. L.26, Sirion, U et al27and Li, Z. et al28に記載され、これらは参考で本明細書中に引用される。
【0121】
本発明の第十二の態様は、本発明の第二の態様の化合物の合成方法を提供する。本発明の第二の態様の化合物は、適当な方法で調製されうる。実施例1及び3に記載され、図1、2(i)、2(ii)及び12に示されるスキームは、本発明の第二の態様の化合物を調製されうる方法を示し、この際、標識された中間体化合物を使用する。
【0122】
実施例1に記載され、図1に示されるスキームは、本発明の第二の態様の化合物を調製するのに使用でき、この際、1以上の前駆体分子または中間体化合物、例えば、化合物19a〜h、20a〜hおよび/または21〜25は標識されたRまたはR’基を有する。このような方法は、例えば、標識形態の化合物1〜5、10〜12および29〜34を製造するのに使用できる。
【0123】
図2(i)、図12、図19に示され、実施例1及び実施例3に記載されるスキームは、[18F]11、[11C]35おおび[11C]36等の、本発明の第二の態様の特定の化合物を調製する「クリック−ラベリング」方法を示す。
【0124】
図2(ii)に示され、実施例1に記載される「逆クリック−ラベリング」方法もまた、標識形態の化合物6、7、8及び9等の、本発明の第二の態様に係る特定の化合物を作製するのに使用できる。例えば、図2(ii)に示され、実施例1に記載されるスキームでは、3−[18F]フルオロプロプ−1−イン[3-[18F]fluoroprop-1-yne](化合物[18F]28)が3−フルオロプロプ−1−イン[3-fluoroprop-1-yne](化合物28)の代わりに使用される。
【0125】
しかしながら、実施例3に記載され、図19に示される「クリック−ラベリング」方法には、安定した不純物が生産され、イサチン誘導体産物の特定の活性を低減するという欠点がある。実施例10に記載され、図20及び21に示されるように、この欠点は、一般式38及び43(ただし、n=0、1、2、3、4、5または6(すなわち、n=0〜6)、x=離脱基、例えば、メシラート、トシレート、ノシレートまたは他のスルホネートエステル若しくはハライド)に示される化合物などの、保護された前駆体を用いて、イサチン誘導体、例えば、[18F]11を合成することによって、解消できる。
【0126】
【化4】
【0127】
特に好ましい保護されたアルキン前駆体は、(S)−1−{[1’−[1−(2−プロピニル)]−(1’2’−ジヒドロ−2’−オキソスピロ(1,3−ジオキサン−2,3’−[3H]インドール)−5’−スルホニル}−2−(2,4−ジフルオロフェノキシメチル)−ピロリジン[(S)-1-{[1’-[1-(2-Propynyl)]-(1’2’-dihydro-2’-oxospiro(1,3-dioxane-2,3’-[3H]indol)-5’-sulfonyl}-2-(2,4-difluor ophenoxymethyl)-pyrrolidine](化合物39)である。このような保護されたアルキン前駆体の使用によって、C−3位置での望ましくない副反応が防止される。
【0128】
【化5】
【0129】
これらの方法に加えて、本発明の第二の態様に係る化合物は、例えば、適当な錫前駆体のハロ−脱メタル化(halo-demetallation)、望ましい金属による、好ましくはイサチンに結合した適当なリガンドによるキレート化、適当なアリールまたはアルキル離脱基の存在下での[18F]フッ素による置換によって、放射性標識されうる。
【図面の簡単な説明】
【0130】
図面
【図1】図1は、ターゲット化合物の合成の概略図である。試薬および条件は下記のとおりである:(a)フェノール/フッ素置換フェノール/4−テトラヒドロピラン、NaH、DMF、80℃、17h;(b)4(3H)−ピリミドン、PPh3、DIAD、DCM、rt、48h;(c)臭化プロパルギル、KOH、DMF、rt、18h;(d)TFA、DCM、0℃、1h;(e)5−クロロスルホニルイサチン、TEA、THF/DCM、rt、19h;(f)4−フルオロベンジルブロミド、K2CO3、DMF、rt、2h;(g)臭化プロパルギル、K2CO3、DMF、rt、2h;(h)2−フルオロエチルアジド、CuSO4、L−アスコルビン酸、DMF、rt、2h。
【図2−1】図2(i)は、(S)−1−(2−プロピニル)−5−(2−(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物25)と[18F]フルオロエチルアジド(化合物27)との反応による化合物[18F]11の合成の概略図である。化合物26は、2−(トルエン−4−スルホニル)エチルアジドである。試薬は下記のとおりである:(a)[18F]KF,Kryptofix[2,2,2],アセトニトリル;(b)CuSO4、アスコルビン酸ナトリウム、リン酸緩衝液 pH6.0、化合物25。
【図2−2】図2(ii)は、(S)−1−(アジドメチル)−5−(2−(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン](化合物37)と(3−フルオロプロプ−1−イン)(化合物28)との反応による、化合物6 ((S)−1−[4−(2−フルオロエチル)−1H−[1,2,3]−トリアゾール−1−イル]メチル−5−(2−(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン)の合成の概略図である。試薬は下記のとおりである:(a)CuSO4、L−アスコルビン酸、DMF。
【図3−1】図3は、[18F]11を含む反応混合物の予備HPLCトレース(左)および分析ピーク分離(右)である。上図:放射能チャンネル;下図:254nmでのUVチャンネル。
【図3−2】図3は、[18F]11を含む反応混合物の予備HPLCトレース(左)および分析ピーク分離(右)である。上図:放射能チャンネル;下図:254nmでのUVチャンネル。
【図4】図4は、2、10、30及び60分後のRIF−1腫瘍を有するマウスにおける[18F]11の生体内分布を示す。データは、平均値±SEMである;n=3〜6匹マウス/時点。
【図5−1】図5は、ラジオ−HPLCにより血漿中で評価された[18F]11のインビボでの代謝を示す。上左図:[18F]11標準;上右図:2分後の血漿;下左図:15分後の血漿;下右図:60分後の血漿。
【図5−2】図5は、ラジオ−HPLCにより血漿中で評価された[18F]11のインビボでの代謝を示す。上左図:[18F]11標準;上右図:2分後の血漿;下左図:15分後の血漿;下右図:60分後の血漿。
【図6−1】図6は、ラジオ−HPLCにより肝臓及び尿中で評価された[18F]11のインビボでの代謝を示す。上図:肝臓抽出物;下図:尿抽出物;左〜右図:それぞれ、2、15及び60分。
【図6−2】図6は、ラジオ−HPLCにより肝臓及び尿中で評価された[18F]11のインビボでの代謝を示す。上図:肝臓抽出物;下図:尿抽出物;左〜右図:それぞれ、2、15及び60分。
【図6−3】図6は、ラジオ−HPLCにより肝臓及び尿中で評価された[18F]11のインビボでの代謝を示す。上図:肝臓抽出物;下図:尿抽出物;左〜右図:それぞれ、2、15及び60分。
【図7】図7は、2、10、30及び60分後の[125I]標識された(S)−1−(4−ヨードベンジル)−5−(2−フェノキシメチル−ピロリジン−1−スルホニル)イサチン(本明細書中では、化合物[125I]14と称する)の生体内分布を示す。データは、平均値±SEMである;n=3匹マウス/時点。
【図8−1】図8は、HPLCによってマウス肝臓S9画分中で評価された既知の化合物 (S)−1−(4−ヨードベンジル)−5−(2−フェノキシメチル−ピロリジン−1−スルホニル)イサチン(化合物14)、(S)−1−(4−フルオロベンジル)−5−(2−フェノキシメチル−ピロリジン−1−スルホニル)イサチン(化合物15)および1−(4−フルオロベンジル)−5−(ピロリジン−1−スルホニル)イサチン(化合物16)のインビトロ代謝を示す。上図、左〜右図:0時での化合物14、15及び16;下図、左〜右図:60分間インキュベーション後の化合物14、15及び16。
【図8−2】図8は、HPLCによってマウス肝臓S9画分中で評価された既知の化合物 (S)−1−(4−ヨードベンジル)−5−(2−フェノキシメチル−ピロリジン−1−スルホニル)イサチン(化合物14)、(S)−1−(4−フルオロベンジル)−5−(2−フェノキシメチル−ピロリジン−1−スルホニル)イサチン(化合物15)および1−(4−フルオロベンジル)−5−(ピロリジン−1−スルホニル)イサチン(化合物16)のインビトロ代謝を示す。上図、左〜右図:0時での化合物14、15及び16;下図、左〜右図:60分間インキュベーション後の化合物14、15及び16。
【図8−3】図8は、HPLCによってマウス肝臓S9画分中で評価された既知の化合物 (S)−1−(4−ヨードベンジル)−5−(2−フェノキシメチル−ピロリジン−1−スルホニル)イサチン(化合物14)、(S)−1−(4−フルオロベンジル)−5−(2−フェノキシメチル−ピロリジン−1−スルホニル)イサチン(化合物15)および1−(4−フルオロベンジル)−5−(ピロリジン−1−スルホニル)イサチン(化合物16)のインビトロ代謝を示す。上図、左〜右図:0時での化合物14、15及び16;下図、左〜右図:60分間インキュベーション後の化合物14、15及び16。
【図9】図9は、A)10μg/mL CDDPまたは10μg/mL CHXで24時間処置したRIF−1細胞、B)50μg/mL CDDP(シスプラチン)で処置したPEO1/4細胞、C)100μM CDDPまたは100μM VP−16を、単独でまたは組み合わせて、処置したLNM35細胞、における60分後の[18F]11の細胞への取り込みを示す。カスパーゼ−3アッセイ(C右パネル)を、実施例で記載したようにして行った。*スチューデントt−テスト(Student’s t-test)、p<0.005。
【図10】図10は、CDDPで処置したマウスにおける[18F]11の分布の全身矢状イメージ(whole body sagittal image)を示す。イメージは、約100μCi [18F]11を静脈内注射してから30〜60分で合計した。
【図11】図11は、100mg/kg シクロホスファミドで処置された38C18異種移植片を有するマウスにおける[18F]11の分布の全身矢状イメージ(whole body sagittal image)を示す。左から右へのイメージは、約100μCi [18F]11を静脈内注射してから24時間に撮影された、軸性(axial)イメージ、冠性(coronal)イメージおよび矢状(sagittal)イメージである。
【図12】図12は、それぞれ、化合物35(上図)および化合物36(下図)の合成の概略図である。
【図13】図13は、ベヒクルまたはCDDPで処置されたRIF−1細胞における[18F]11の取り込みプロフィールを示す。データは、1mgの全細胞タンパク質当たりで平均された減水補正されたカウント/分として表わされる。データは、平均値±SEMであり、3連で行われた。
【図14】図14は、ベヒクル(50%DMSO)またはCDDP(10mg/kg 単回投与)で処置されたRIF−1腫瘍における[18F]11の取り込みを示す。放射性トレーサーを注射してから60分での[18F]由来の放射能レベルを分析し、血中の放射能レベルに対する割合として表わす。データは、平均値±SEMであり、n=8匹マウス/群である。
【図15】図15は、シクロホスファミド(CPA、100mg/kg)またはベヒクルで処置された腫瘍における[18F]の生体内分布を示し、この際、カウント密度を各19時点での興味のある各領域で平均して、興味のある領域に関する時間対放射能曲線(time versus radioactivity curve)(TAC)を得た。
【図16】図16は、シクロホスファミド(CPA、100mg/kg)またはベヒクル(Veh)で処置された腫瘍における、注射してから60分後(NUV60)の標準化取り込み値(Normalised Uptake Value)(NUV)、および0〜60分のNUVの積分として算出されるNUV曲線下の面積(Area Under the NUV curve)(AUC)のヒストグラムである。
【図17】図17は、ベヒクルまたはシクロホスファミドで処置された2匹の38C13異種移植片を有するマウスの代表的なOSEM3D再構築[18F]11 PETイメージである。丸は腫瘍を表わす。
【図18】図18は、カスパーゼ−3と同種のターゲットであるポリ(ADP−リボース)ポリメラーゼ(caspase-3 cognate target poly(ADP-ribose)polymerase)(PARP)に関する化合物11(「イサチン−7」として示す)および化合物14(「イサチン−4」として示す)の阻害活性を示す。
【図19】図19は、(S)−1−(2−プロピニル)−5−(2−(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物25)と[18F]フルオロエチルアジド(化合物27)との反応による化合物[18F]11の合成の第2の概略図である。
【図20】図20は、保護されたアルキン前駆体 (S)−1−{[1’−[1−(2−プロピニル)]−(1’2’−ジヒドロ−2’−オキソスピロ(1,3−ジオキサン−2,3’−[3H]インドール)−5’−スルホニル}−2−(2,4−ジフルオロフェノキシメチル)−ピロリジン(化合物39)および保護されたトリゾール (S)−1−{[1’−[1−(2−フルオロエチル)−1H−[1,2,3]−トリアゾール−4−イル]−(1’2’−ジヒドロ−2’−オキソスピロ(1,3−ジオキサン−2,3’−[3H]インドール)−5’−スルホニル}−2−(2,4−ジフルオロフェノキシメチル)−ピロリジン(化合物40)の合成に関する一般的な反応スキームである。
【図21】図21は、保護されたアルキン前駆体 (S)−1−{[1’−[1−(2−プロピニル)]−(1’2’−ジヒドロ−2’−オキソスピロ(1,3−ジオキサン−2,3’−[3H]インドール)−5’−スルホニル}−2−(2,4−ジフルオロフェノキシメチル)−ピロリジン(化合物39)からの[18F]11の製造に関する具体的な放射化学反応の概略図である。
【図22】図22は、脱保護前の標識混合物のHPLC分析を示す。赤:放射能チャンネル、青:254nmでのUVチャンネル。残りの[18F]フルオロエチルアジド:1.3分、標識されたイサチンアセタール(isatin acetale):6.8分。
【図23】図23は、[18F]11の予備HPLCを示す。上図:放射能チャンネル。11:45分のピークは[18F]11に相当する。下図:UVチャンネル、254nm。11:51分のシグナルは安定した不純物である。
【図24】図24は、配合された[18F]11の分析HPLCを示す。上図:254nmでのUVチャンネル、下図:3.3分に[18F]11を示す放射能シグナル。
【図25】図25は、N−(2−プロピニル)イサチン(化合物41)からの弱いイサチンカスパーゼ−3阻害剤であるN−[1−(2−フルオロエチル)−1H−[1,2,3]−トリアゾール−4−イル]イサチン(化合物42)の合成の概略図である。
【図26】図26は、処置によりアポトーシスが誘導された癌細胞における[18F]11及び[18F]42の結合性を示す。(左パネル)4−ヒドロキシペルオキシシクロホスファミド 4−HC(4−HC;1μg/mL;24h)で処置して、アポトーシスを誘導した38C13リンパ腫細胞における化学構造および[18F]11結合性。すべての処置およびコントロールサンプルについて、放射能データは、1mg全細胞タンパク質当たりの減衰補正されたカウントとして表わされる。(右パネル)38C13リンパ腫細胞における、低親和性プローブ、[18F]42、の結合に関する4−HCの効果。
【実施例】
【0131】
実施例1−化合物の合成
((S)−1−(4−フルオロベンジル)−5−(2−フェノキシメチル−ピロリジン−1−シルホニル)イサチン)をリード化合物として使用して、ターゲット化合物のライブラリーを作製した。左側のエーテル部分およびN−1位置で修飾をした。フッ素基を左側のフェニルエーテル基に導入して、この位置での複素環及びアルキンのトレランスを調べた。N−1位置での1,2,3トリアゾール基に対するトレランスもまた調べた。官能化ピロリジンを5−クロロスルホニルイサチンで縮合した後、炭酸カリウム/DMFを用いてイサチン窒素をアルキル化することによって図1に示されるように、ターゲット化合物を合成した。全ての必要な出発材料は市販されている、またはLee, D. et al 8, Chu, W. et al9及びKopka, K. et al13に記載されるのと同様にして製造する。
【0132】
4−ヒドロキシテトラヒドロピランおよび市販のフェノールをトシレート17と反応させることによって、良好な収率でピロリジン19a〜fを得た。4(3H)−ピリミドンを、Wipf et al.15に報告される方法の修飾を用いて(S)−1−(tert−ブトキシカルボニル)−2−ピロリジンメタノール(化合物18)と結合させ、69%の収率でピロリジン19gを得た。18を臭化プロパルギルでO−アルキル化することによって、67%の収率で相当するエーテル19hを得た。BOCで保護されたピロリジン19a〜hをトリフルオロ酢酸で脱保護した後、5−クロロスルホニルイサチンと結合させることによって、並から良好な収率でスルホンアミド20a〜hを得た。次に、塩基性条件下で20b〜hを4−フルオロベンジルブロミドで処理することによって、新規な化合物1〜5ならびに中間化合物22及び23を得、一方、20a及び20dを臭化プロパルギルで処理することによって、それぞれ、中間アルキン24及び25を得た。2−フルオロエチルアジドを各アルキン前駆体23、24及び25で銅触媒による付加環化を行うことによって、新規なトリアゾール10〜12を調製した。いささか驚くべきことに、イサチン足場は、硫酸銅及びアスコルビン酸の存在下で90℃で加熱すると分解し、これにより新規なトリアゾール10〜12の収率が悪くなった。この問題は、アルキン前駆体に対する硫酸銅濃度を5%から50%に上げ、周囲温度で反応を行い、さらに反応時間を1時間にまで短くすることによって一部解決し、これにより、トリアゾール10〜12を48〜57%の収率で得た。
【0133】
化学 試薬及び溶媒は、Sigma-Aldrich(Gillingham, United Kingdom)から購入し、さらに精製することなく使用した。水酸化カリウム及び炭酸カリウムは、5酸化リンで真空デシケーター内で保存した。特記しない限り、すべての反応はアルゴン下で行われた。石油エーテルは、40℃〜60℃で蒸留する画分を意味する。自動化フラッシュクロマトグラフィーは、RediSep 4gまたは12g 正常相シリカカートリッジ(流速 12mL/minまたは26mL/min)を用いて、CombiFlash Companion machine(Companion Presearch Ltd.)で行った。マニュアルフラッシュクロマトグラフィーは、Davisil中性シリカ(neutral silica)(60Å、60〜200ミクロン、Fisher Scientific, Loughborough, UK)を用いて行い、溶媒混合物は容積/容積として記載する。1H NMRスペクトルをBruker Avance 600 MHz NMR machineで得、スペクトルを残りの溶媒で参照する。結合定数(J)はヘルツ(Hz)で示す。GC−MSデータは、Agilent 6890Nシステムを用いて電子イオン化下で得た。質量スペクトルは、WatersMicromass LCT Premier machineで陽極電気スプレーイオン化(positive electrospray ionisation)形式で得た。融点は、Stuart Scientific SMP1融点測定装置で毛細管中で測定し、修正しない。薄層クロマトグラフィ(TLC)用の溶媒混合物は容積/容積で記載し、サンプルをアルミニウムで塗布された中性シリカ板(aluminium backed neutral silica plates)(厚み 0.2mm)(Fluka, Seelze, Germany)で発色させた。化合物1〜5、10〜16、20b〜h及び22〜25の純度の分析は分析HPLCで評価し;化合物19b〜hはGC−MSで分析した。すべての化合物の純度は95%超であった。[18F]フッ化物を、濃縮(enriched)[18O]H2Oターゲットの16.4MeVプロトン照射による18O(p,n)18F核反応を用いてサイクロトロン(GE PETrace)によって製造した。
【0134】
HPLC法 a)非放射性化合物の純度分析は、Phenomenex Luna 50×4.6mm(3μm)HPLCカラムを備えたLaura 3 software(Lablogic, Sheffield, UK)を有するAgilent 1100シリーズシステムで行い、この際、0.1M ギ酸アンモニウム及びメタノール/アセトニトリル(1.8:1 v/v)の移動相、グラジエント(50%有機相で1分;50(R)90%有機相で14分;90%有機で4分;90(R)50%有機相で4分)、流速 1mL/min及び波長254nmであった。
【0135】
b)予備ラジオ−HPLCを、Bioscan Flowcount FC-3400 PINダイオード検出器(Lablogic)及びlinear UV-200 detector(波長 254nm)を備えたBeckman System Goldで行った。Phenomenex Onyx C18 100×10mm HPLCカラムならびに水およびメタノール/アセトニトリル(1.8:1 v/v)の移動相、グラジエント 45(R)90%有機相で20分および流速 3mL/minであった。
【0136】
c)分析ラジオ−HPLCを、水およびメタノール/アセトニトリル(1.8:1 v/v)の移動相、グラジエント 60(R)90%有機相で20分、流速 1mL/minを用いて、Bioscan Flowcount FC3200 ヨウ化ナトリウム/PMT ガンマ線検出器(Lablogic)およびPhenomenex Luna 50×4.6mm(3μm)カラムを使用する以外は、上記と同様にして行った。
【0137】
既知の化合物の製造
下記化合物を確立された文献の方法に従って合成したところ、1H NMRスペクトルデータが報告された値と一致した:(S)−1−ベンジル−5−(2−フェノキシメチル−ピロリジン−1−スルホニル)イサチン(化合物13)、(S)−1−(4−ヨードベンジル)−5−(2−フェノキシメチル−ピロリジン−1−スルホニル)イサチン(化合物14)及び(S)−1−(4−フルオロベンジル)−5−(2−フェノキシメチル−ピロリジン−1−スルホニル)イサチン(化合物15)。
【0138】
1−(4−フルオロベンジル)−5−(ピロリジン−1−スルホニル)イサチン(化合物16) 乾燥DMF(8mL)における5−ピロリジン−1−スルホニルイサチン8(0.14g、0.5mmol)の氷冷した攪拌溶液に、水素化ナトリウム(40mg、1mmol)を添加した。30分後、4−フルオロベンジルブロミド(0.38g、2mmol)を添加し、混合物を室温まで加温した。19時間後、オレンジ色の溶液を10%NH4Cl水溶液(25mL)に注ぎ、DCMで抽出した(3×15mL)。真空中で濃縮後、残渣をジエチルエーテル(10mL)中にとり、水で洗浄し(3×10mL)、Na2SO4で乾燥した。クロマトグラフィー(ジエチルエーテル/ヘキサン)によって、標題化合物をオレンジ色のゴム状物として得た(83mg、43%)。HRMS (ESI) = 389.0988 (M + H)+. C19H18FN2O4Sに関する算出値 389.0971. 1H NMR (600 MHz, CDCl3) δ 8.05 (d, J = 1.5 Hz, 1H), 7.99 (dd, J = 8.4 Hz, J = 1.5 Hz, 1 H), 7.35-7.32 (m, 2 H), 7.09-7.06 (m, 2 H), 6.91 (d, J = 8.4 Hz, 1 H), 4.94 (s, 2 H), 3.25-3.23 (4 H, m), 1.84-1.79 (4 H, m). TLC (UV254) Rf= 0.63 (4:1 酢酸エチル/ヘキサン). HPLC tR = 6.83 min。
【0139】
新規な化合物およびその中間体の製造
(S)−tert−ブチル2−(4−フルオロフェノキシメチル)ピロリジン−1−カルボキシレート(化合物19b) 乾燥DMF(10mL)における4−フルオロフェノール(0.27g、2.4mmol)の攪拌溶液に、水素化ナトリウム(鉱油における60%w/w)(0.11g、2.8mmol)を添加した。30分後、乾燥DMF(5mL)における((S)−1−(tert−ブトキシカルボニル)ピロリジン−2−イル)トルエン−4−スルホネート8(化合物)15(0.71g、2.0mmol)を添加し、この混合物を80℃で17時間加熱した。反応物を室温にまで冷却し、1M NaOH(25mL)に注ぎ、DCMで抽出した(3×15mL)。有機画分を合わせたものを真空中で還元し、ジエチルエーテル(20mL)を添加した後、1M NaOH(1×20mL)、水(1×20mL)、さらにブライン(1×20mL)で洗浄し、Na2SO4で乾燥した。クロマトグラフィー(ヘキサン/酢酸エチル)によって、生成物を無色の油として得た(0.36g、61%)。HRMS (ESI) = 296.1654 (M + H)+. C16H23FNO3に関する算出値 296.1656. 1H NMR (600 MHz, CDCl3) δ 6.97-6.93 (m, 2 H), 6.88-6.84 (m, 2 H), 4.18-4.03 (m, 2 H), 3.94-3.73 (m, 1 H), 3.46-3.32 (m, 2 H), 2.07-1.81 (m, 4 H), 1.47 (s, 9 H). TLC (UV254) Rf = 0.51 (2:1 ヘキサン/酢酸エチル)。
【0140】
(S)−tert−ブチル 2−(3−フルオロフェノキシメチル)ピロリジン−1−カルボキシレート(化合物19c) 3−フルオロフェノールを使用する以外は、19bの方法に従って、19cを調製した。クロマトグラフィー(ヘキサン/酢酸エチル)によって、標題化合物を無色の油として得た(0.32g、54%)。HRMS (ESI) = 296.1657 (M + H)+. C16H23FNO3に関する算出値 296.1656 1H NMR (600 MHz, CDCl3) δ 7.20-7.15 (m, 1 H), 6.73-6.69 (m, 1 H), 6.65-6.59 (m, 2 H), 4.18-4.03 (m, 2 H), 3.97-3.74 (m, 1 H), 3.48-3.29 (m, 2 H), 2.05-1.79 (m, 4 H), 1.48 (s, 9 H). TLC (UV254) Rf = 0.40 (3:1 ヘキサン/酢酸エチル)。
【0141】
(S)−tert−ブチル 2−(2,4−ジフルオロフェノキシメチル)ピロリジン−1−カルボキシレート(化合物19d) 2,4−ジフルオロフェノールを使用する以外は、19bの方法に従って、19dを調製した。クロマトグラフィー(ヘキサン/酢酸エチル)によって、標題化合物を無色の油として得た(1.92g、58%)。HRMS (ESI) = 314.1560 (M + H)+. C16H22F2NO3に関する算出値 314.1562. 1H NMR (600 MHz, CDCl3) δ 7.08-6.76 (m, 3 H), 4.21-3.87 (m, 3 H), 3.47-3.32 (m, 2 H), 2.16-1.85 (m, 4 H), 1.48 (s, 9 H). TLC (UV254) Rf = 0.51 (2:1 ヘキサン/酢酸エチル)。
【0142】
(S)−tert−ブチル 2−(3,5−ジフルオロフェノキシメチル)ピロリジン−1−カルボキシレート(化合物19e) 3,5−ジフルオロフェノールを使用する以外は、19bの方法に従って、19eを調製した。クロマトグラフィー(ヘキサン/酢酸エチル)によって、標題化合物を無色の油として得た(0.31g、53%)。HRMS (ESI) = 314.1563 (M + H)+. C16H22F2NO3に関する算出値 314.1562. 1H NMR (600 MHz, CDCl3) δ 6.44-6.29 (m, 3 H), 4.11-3.98 (m, 2 H), 3.91-3.72 (m, 1 H), 3.42-3.24 (m, 2H), 1.99-1.79 (m, 4 H), 1.42 (s, 9 H). TLC (UV254) Rf= 0.56 (2:1 ヘキサン/酢酸エチル)。
【0143】
(S)−tert−ブチル 2−(4−テトラヒドロピラニルオキシメチル)ピロリジン−1−カルボキシレート(化合物19f) 4−ヒドロキシテトラヒドロピランを使用する以外は、19bの方法に従って、19fを調製した。クロマトグラフィー(酢酸エチル)によって、標題化合物を無色の油として得た(0.14g、25%)。HRMS (ESI) = 286.2012 (M + H)+. C15H28NO4に関する算出値 286.2013. 1H NMR (600 MHz, CDCl3) δ 3.97-3.83 (m, 2 H), 3.64-3.23 (m, 9 H), 1.97-1.72 (m, 5 H), 1.60-1.52 (m, 2 H), 1.46 (s, 9 H). TLC (I2) Rf = 0.62 (酢酸エチル)。
【0144】
(S)−tert−ブチル 2−(ピリミジン−4−イルオキシメチル)ピロリジン−1−カルボキシレート(化合物19g) 乾燥DCM(10mL)におけるN−tert−ブトキシカルボニル−L−ピロリノール(0.81g、4mmol)18の攪拌溶液に、トリフェニルホスフィン(5.24g、20mmol)、さらには4(3H)−ピリミドン(0.77g、8mmol)を加えた。この溶液を氷浴で冷却し、DIAD(3.24g、16mmol)を10分間滴下した。48時間後、GC−MSでは、18の完全な変換が示され、この反応混合物を水(30mL)に注ぎ、有機画分を集め、水相を別のDCM(2×20mL)で洗浄した。有機画分を合わせたものを1M NaOH(2×15mL)、さらにはブライン(1×15mL)で洗浄し、Na2SO4で乾燥した。大量の溶媒を除去することによって、オレンジ色のゴム状物として得、ヘキサン/ジエチルエーテル(1:1)を添加することによって、トリフェニルホスフィンオキシドの沈殿物が形成し、これを濾別した。クロマトグラフィー(酢酸エチル)によって、目的生成物を無色の油として得た(0.77g、69%)。HRMS (ESI) = 280.1655 (M + H)+. C14H22N3O3に関する算出値 280.1656. 1H NMR (600 MHz, CDCl3) δ 8.71 (s, 1 H), 8.39 (d, J= 6 Hz, 1 H), 6,69 (d, J = 6 Hz, 1 H), 4.45-3.91 (m, 3 H), 3.40-3.33 (m, 2 H), 2.00-1.82 (m, 4 H), 1.42 (s, 9 H). TLC (UV254) Rf= 0.49 (酢酸エチル)。
【0145】
(S)−tert−ブチル 2−(2−プロピニルオキシメチル)ピロリジン−1−カルボキシレート(化合物19h) 乾燥DMF(10mL)における18(0.40g、2mmol)の攪拌溶液に、水酸化カリウム(0.56g、10mmol)を添加した後、臭化プロパルギル(トルエンにおいて、80wt.%)(0.48g、4mmol)を5分かけて滴下した。18時間後、反応混合物を水(30mL)に注ぎ、DCMで洗浄した(3×15mL)。有機画分を合わせたものを真空中で濃縮し、残った液体をジエチルエーテル(15mL)にとり、水(2×10mL)、次にブライン(1×10mL)で洗浄し、Na2SO4で乾燥した。クロマトグラフィー(ヘキサン/酢酸エチル)によって、目的生成物を無色の油として得た(0.32g、67%)。HRMS (ESI) = 240.1597 (M + H)+. C13H22NO3に関する算出値 240.1594. 1H NMR (600 MHz, CDCl3) δ 4.13 (s, 2 H), 3.96-3.88 (m, 1 H), 3.64 (dd, J = 9 Hz, J = 3.6 Hz, 1 H), 3.49-3.22 (m, 3 H), 2.40 (s, 1 H), 1.94-1.78 (m, 4 H), 1.46 (s, 9 H). TLC (I2) Rf = 0.67 (1:1 ヘキサン/酢酸エチル)。
【0146】
(S)−5−(2−(4−フルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物20b) 氷浴中で冷却した乾燥DCM(4mL)における19b(0.15g、0.5mmol)の攪拌溶液に、TFA(0.6mL、10mmol)を添加した。1時間後、大量の溶媒を真空中で除去し、残った残渣を乾燥DCM(8mL)にとり、氷浴中で冷却した。次に、乾燥トリエチルアミン(1.5mL)、さらに乾燥THF(4mL)における5−クロロスルホニルイサチン8(0.16g、0.65mmol)を添加した後、溶液を攪拌した。19時間後、大量の溶媒を真空中で除去し、DCM(10mL)中に再溶解し、水(2×10mL)、次にブライン(1×10mL)で洗浄し、Na2SO4で乾燥した。クロマトグラフィー(ヘキサン/酢酸エチル)によって、目的生成物をオレンジ色の固体として得た(104mg、51%)。Mp: 205-207℃. HRMS (ESI) = 405.0941 (M + H)+. C19H18FN2O5Sに関する算出値 405.0920. 1H NMR (600 MHz, CDCl3) δ 8.10 (s,1 H), 8.08 (dd, J = 8.4 Hz, J = 1.8 Hz, 1 H), 8.00 (br, 1 H), 7.00 (d, J = 7.8 Hz, 1 H), 6.99-6.95 (m, 2 H), 6.83-6.81 (m, 2 H), 4.17 (dd, J= 9.6 Hz, J = 3.6 Hz, 1 H), 3.98-3.95 (m, 1 H), 3.91 (dd, J= 9 Hz, J = 7.8 Hz, 1 H), 3.54-3.50 (m, 1 H), 3.24-3.19 (m, 1 H), 2.10-1.99 (m, 2 H), 1.87-1.77 (m, 2 H). TLC (UV254) Rf= 0.27 (2:1 酢酸エチル/ヘキサン). HPLC tR = 7.83。
【0147】
(S)−5−(2−(3−フルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物20c) 19cを使用する以外は、20bの方法に従って、20cを調製して、オレンジ色の固体を得た(93mg、46%)。Mp: 201-203℃. HRMS (ESI) = 405.0933 (M + H)+. C19H18FN2O5Sに関する算出値 405.0920. 1H NMR (600 MHz, CDCl3) δ 8.14 (br, 1 H), 8.09-8.06 (m, 2 H), 7.21 (q, J = 7.2 Hz, 1 H), 7.03 (d, J = 7.8 Hz, 1 H), 6.67-6.62 (m, 2 H), 6.55 (dt, J = 10.8 Hz, J = 2.4 Hz, 1 H), 4.17 (dd, J = 9 Hz, J = 3 Hz, 1 H), 4.01-3.97 (m, 1 H), 3.94 (dd, J = 9 Hz, J = 7.2 Hz, 1 H), 3.54-3.50 (m, 1 H), 3.27-3.25 (m, 1 H), 2.08-1.96 (m, 2 H), 1.88-1.77 (m, 2 H). TLC (UV254) Rf = 0.36 (2:1 酢酸エチル/ヘキサン). HPLC tR = 8.27 min。
【0148】
(S)−5−(2−(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物20d) 19dを使用する以外は、20bの方法に従って、20dを調製して、オレンジ色の固体を得た(0.86g、34%)。Mp: 185-187℃. HRMS (ESI) = 423.0834 (M + H)+. C19H17F2N2O5Sに関する算出値 423.0826. 1H NMR (600 MHz, CDCl3) δ 8.08-8.06 (m, 2 H), 7.97 (br, 1 H), 7.03 (d, J = 9 Hz, 1 H), 6.97-6.92 (m, 1 H), 6.87-6.77 (m, 2 H), 4.21 (dd, J= 8.4 Hz, J = 2.4 Hz, 1 H), 4.03-3.97 (m, 2 H), 3.56-3.52 (m, 1 H), 3.23-3.17 (m, 1 H), 2.11-2.01 (m, 2 H), 1.88-1.75 (m, 2 H). TLC (UV254) Rf = 0.46 (2:1 酢酸エチル/ヘキサン). HPLC tR = 8.12 min。
【0149】
(S)−5−(2−(3,5−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物20e) 19eを使用する以外は、20bの方法に従って、20eを調製して、オレンジ色の固体を得た(112mg、53%)。Mp: 196-198℃. HRMS (ESI) = 423.0834 (M + H)+. C19H17F2N2O5Sに関する算出値 423.0826. 1H NMR (600 MHz, CDCl3) δ 8.10-8.06 (m, 2 H), 8.04 (br, 1 H), 7.05 (d, J = 8.4 Hz), 6.45-6.38 (m, 3 H), 4.18 (dd, J = 8.4 Hz, J = 2.4 Hz, 1 H), 3.99-3.92 (m, 2 H), 3.56-3.49 (m, 1 H), 3.24-3.17 (m, 1 H), 2.04-1.92 (m, 2 H), 1.86-1.77 (m, 2 H). TLC (UV254) Rf= 0.36 (2:1 酢酸エチル/ヘキサン). HPLC tR = 9.18 min。
【0150】
(S)−5−(2−(テトラヒドロ−2H−ピラン−4−イルオキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物20f) 19fを使用する以外は、20bの方法に従って、20fを調製して、オレンジ色のゴム状物を得た(63mg、32%)。HRMS (ESI) = 395.1282 (M + H)+. C18H23N2O6Sに関する算出値 395.1277. 1H NMR (600 MHz, CDCl3) δ 8.17 (br, 1 H), 8.10-8.08 (m, 2 H), 7.07 (d, J = 8.4 Hz, 1 H), 3.94-3.90 (m, 2 H), 3.77-3.73 (m, 1 H), 3.70 (dd, J = 9.6 Hz, J = 3 Hz, 1 H), 3.56-3.51 (septet, J = 4.2 Hz, 1 H), 3.48-3.44 (m, 3 H), 3.13-3.11 (m, 1 H), 2.05-1.87 (m, 4 H), 1.72-1.64 (m, 2 H), 1.60-1.52 (m, 2 H). TLC (UV254) Rf = 0.26 (4:1 酢酸エチル/ヘキサン). HPLC tR = 2.65 min。
【0151】
(S)−5−(2−(ピリミジン−4−イルオキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物20g) 19gを使用する以外は、20bの方法に従って、20gを調製した。クロマトグラフィー(酢酸エチル)によって、オレンジ色のゴム状物を得た(51mg、27%)。HRMS (ESI) = 389.025 (M + H)+. C17H17N4O5Sに関する算出値 389.020. 1H NMR (600 MHz, CDCl3) δ 8.79 (s, 1 H), 8.45 (d, J = 5.4 Hz, 1 H), 8.12 (d, J = 1.8 Hz, 1 H), 8.09 (dd, J = 8.4 Hz, J = 1.8 Hz, 1 H), 7.95 (br, 1 H), 7.03 (d, J = 8.4 Hz, 1 H), 6.72 (d, J = 5.4 Hz, 1 H), 4.57 (dd, J = 10.8 Hz, J = 4.8 Hz, 1 H), 4.39 (dd, J = 10.8 Hz, J=7.2 Hz, 1 H), 4.08-4.04 (m, 1 H), 3.53-3.46 (m, 1 H), 3.27-3.22 (m, 1 H), 2.01-1.93 (m, 2 H), 1.84-1.74 (m, 2 H). TLC (UV254) Rf= 0.49 (9:1 酢酸エチル/メタノール). HPLC tR = 1.90 min。
【0152】
(S)−5−(2−(2−プロピニルオキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物20h) 19hを使用する以外は、20bの方法に従って、20hを調製して、オレンジ色のゴム状物を得た(92mg、53%)。HRMS (ESI) = 349.0867 (M + H)+. C16H17N2O5Sに関する算出値 349.067. 1H NMR (600 MHz, CDCl3) δ 8.10-8.07 (m, 2 H), 7.84 (br, 1 H), 7.04 (d, J = 8.4 Hz, 1 H), 4.16 (s, 2 H), 3.83-3.79 (m, 1 H), 3.72 (dd, J= 9.6 Hz, J = 3.6 Hz, 1 H), 3.54 (dd, J = 9.6 Hz, J = 3.6 Hz, 1 H), 3.45-3.43 (m, 1 H), 3.19-3.15 (m, 1 H), 2.46 (t, J= 2.4 Hz, 1 H), 1.96-1.89 (m, 2 H), 1.74-1.67 (m, 2 H). TLC (UV254) Rf = 0.28 (2:1 酢酸エチル/ヘキサン). HPLC tR =2.93 min。
【0153】
(S)−1−(4−フルオロベンジル)−5−(2−(4−フルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物22) 乾燥DMF(3mL)における20b(40mg、0.1mmol)の攪拌溶液に、炭酸カリウム(21mg、0.15mmol)、さらには4−フルオロベンジルブロミド(76mg、0.4mmol)を添加した。2時間後、TLCにより、20bの完全な変換が示され、この溶液を10%NH4Cl水溶液(10mL)に注ぎ、DCMで抽出した(3×10mL)。有機画分を合わせたものを真空中で濃縮し、ジエチルエーテル(10mL)中にとり、水(2×10mL)、次にブライン(1×10mL)で洗浄し、Na2SO4で乾燥した。クロマトグラフィー(ヘキサン/酢酸エチル)によって、標題化合物をオレンジ色のゴム状物として得た(34mg、66%)。HRMS (ESI) = 513.1306 (M + H)+. C26H23F2N2O5Sに関する算出値 513.1296. 1H NMR (600 MHz, CDCl3) δ 8.04 (d, J = 1.8 Hz, 1 H), 7.97 (dd, J = 8.4 Hz, J = 1.8 Hz, 1 H), 7.33-7.30 (m, 2 H), 7.09-7.05 (m, 2 H), 6.96-6.92 (m, 2 H), 6.86 (d, J= 8.4 Hz, 1 H), 6.81-6.77 (m, 2 H), 4.92 (d, J = 15.6, 1 H), 4.91 (d, J= 15.6, 1 H), 4.14 (dd, J = 9.6 Hz, J = 3.6 Hz, 1 H), 3.95-3.92 (m, 1 H), 3.88 (dd, J = 9.6 Hz, J = 7.2 Hz, 1 H), 3.51-3.47 (m, 1 H), 3.20-3.15 (m, 1 H), 2.06-1.93 (m, 2 H), 1.83-1.73 (m, 2 H). TLC (UV254) Rf = 0.61 (2:1 酢酸エチル/ヘキサン). HPLC tR = 12.25 min。
【0154】
(S)−1−(4−フルオロベンジル)−5−(2−(3−フルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物1) 20cを使用する以外は、22の方法に従って、1を調製して、オレンジ色のゴム状物を得た(31mg、61%)。HRMS (ESI) = 513.1298 (M + H)+. C26H23F2N2O5Sに関する算出値 513.1296. 1H NMR (600 MHz, CDCl3) δ 8.02 (d, J = 1.8 Hz, 1 H), 7.98 (dd,J = 8.4 Hz, J = 1.8 Hz, 1 H), 7.33-7.29 (m, 2 H), 7.18 (m, 1 H), 7.09-7.04 (m, 2 H), 6.86 (d, J = 8.4 Hz, 1 H), 6.66-6.62 (m, 2 H), 6.53 (dt, J = 10.8, J = 2.4 Hz, 1 H), 4.88 (s, 2 H), 4.15 (dd, J = 9.6 Hz, J = 3.6 Hz, 1 H), 3.98-3.88 (m, 2 H), 3.51-3.47 (m, 1 H), 3.23-3.18 (m, 1 H), 2.08-1.97 (m, 2 H), 1.84-1.72 (m, 2 H). TLC (UV254) Rf= 0.64 (2:1 酢酸エチル/ヘキサン). HPLC tR = 12.57 min。
【0155】
(S)−1−(4−フルオロベンジル)−5−(2−(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物2) 20dを使用する以外は、22の方法に従って、2を調製して、オレンジ色のゴム状物を得た(32mg、60%)。HRMS (ESI) = 531.1204 (M + H)+. C26H22F3N2O5Sに関する算出値 531.1202. 1H NMR (600 MHz, CDCl3) δ 8.03 (d, J = 1.8 Hz, 1 H), 7.98 (dd, J = 8.4 Hz, J = 1.8 Hz, 1 H), 7.33-7.30 (m, 2 H), 7.09-7.06 (m, 2 H), 6.95-6.91 (m, 1 H), 6.88 (d, J = 8.4 Hz, 1 H), 6.81-6.76 (m, 2 H), 4.93 (d, J = 16.2 Hz, 1 H), 4.92 (d, J = 16.2 Hz, 1 H), 4.18 (dd, J = 9 Hz, J = 3 Hz, 1 H), 4.00-3.95 (m, 2 H), 3.51-3.49 (m, 1 H), 3.21-3.17 (m, 1 H), 2.09-1.98 (m, 2 H), 1.85-1.74 (m, 2 H). TLC (UV254) Rf= 0.67 (2:1 酢酸エチル/ヘキサン). HPLC tR = 12.50 min。
【0156】
(S)−1−(4−フルオロベンジル)−5−(2−(3,5−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物3) 20eを使用する以外は、22の方法に従って、3を調製して、オレンジ色のゴム状物を得た(31mg、58%)。HRMS (ESI) = 531.1213 (M + H)+. C26H22F3-N2O5Sに関する算出値 531.1202. 1H NMR (600 MHz, CDCl3) δ 8.04 (d, J = 1.8 Hz, 1 H), 7.99 (dd, J = 8.4 Hz, J = 1.8 Hz, 1 H), 7.34-7.29 (m, 2 H), 7.09-7.06 (m, 2 H), 6.90 (d, J= 8.4 Hz, 1 H), 6.44-6.38 (m, 3 H), 4.93 (s, 2 H), 4.18-4.15 (m, 1 H), 3.94-3.89 (m, 2 H), 3.51-3.48 (m, 1 H), 3.19-3.15 (m, 1 H), 2.03-1.93 (m, 2 H), 1.83-1.73 (m, 2 H). TLC (UV254) Rf = 0.64 (2:1 酢酸エチル/ヘキサン). HPLC tR = 13.00 min。
【0157】
(S)−1−(4−フルオロベンジル)−5−(2−(テトラヒドロ−2H−ピラン−4−イルオキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物4) 20fを使用する以外は、22の方法に従って、4を調製して、オレンジ色のゴム状物を得た(26mg、52%)。HRMS (ESI) = 503.1646 (M + H)+. C25H28FN2O6Sに関する算出値 503.1652. 1H NMR (600 MHz, CDCl3) δ 8.06 (d, J = 1.8 Hz, 1 H), 8.01 (dd, J = 8.4 Hz, J = 1.8 Hz, 1 H), 7.34-7.31 (m, 2 H), 7.10-7.06 (m, 2 H), 6.91 (d, J = 8.4 Hz, 1 H), 4.87 (s, 2 H), 3.93-3.88 (m, 2 H), 3.73-3.70 (m, 1 H), 3.67 (dd, J = 9 Hz, J = 3 Hz, 1 H), 3.55-3.49 (m, 1 H), 3.47-3.41 (m, 4 H), 3.10-3.05 (m, 1 H), 1.97-1.85 (m, 4 H), 1.70-1.63 (m, 2 H), 1.59-1.54 (m, 2 H). TLC (UV254) Rf = 0.55 (4:1 酢酸エチル/ヘキサン). HPLC tR = 8.58 min。
【0158】
(S)−1−(4−フルオロベンジル)−5−(2−(ピリミジン−4−イルオキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物5) 20gを使用する以外は、22の方法に従って、5を調製して、オレンジ色のゴム状物を得た(12mg、24%)。HRMS (ESI) = 497.1287 (M + H)+. C24H22FN4O5Sに関する算出値 497.1295. 1H NMR (600 MHz, CDCl3) δ 8.77 (s, 1 H), 8.43 (d, J = 5.4 Hz, 1 H), 8.08 (d, J = 1.8 Hz, 1 H), 8.01 (dd, J = 8.4 Hz, J = 1.8 Hz, 1 H), 7.34-7.32 (m, 2 H), 7.09-7.06 (m, 2 H), 6.89 (d, J = 8.4 Hz, 1 H), 6.70 (d, J = 5.4 Hz, 1 H), 4.90 (s, 2 H), 4.55 (dd, J = 10.8 Hz, J = 4.2 Hz, 1 H), 4.37 (dd, J = 10.8 Hz, J = 7.8 Hz, 1 H), 4.05-4.01 (m, 1 H), 3.50-3.46 (m, 1 H), 3.22-3.19 (m, 1 H), 1.98-1.87 (m, 2 H), 1.81-1.72 (m, 2 H). TLC (UV254) Rf = 0.32 (2:1 酢酸エチル). HPLC tR = 7.45 min。
【0159】
(S)−1−(4−フルオロベンジル)−5−(2−(2−プロピニルオキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物23) 20hを使用する以外は、22の方法に従って、23を調製して、オレンジ色のゴム状物を得た。酢酸エチル/ヘキサンからの再結晶化によって、目的生成物をオレンジ色の針状物として得た(93mg、58%)。HRMS (ESI) = 457.1236 (M + H)+. C23H22FN2O5Sに関する算出値 457.1233. 1H NMR (600 MHz, CDCl3) δ 8.07 (d, J = 1.8 Hz, 1 H), 8.01 (dd, J = 8.4 Hz, J = 1.8 Hz, 1 H), 7.43-7.31 (m, 2 H), 7.09-7.04 (m, 2 H), 6.90 (d, J= 8.4 Hz, 1 H), 4.91 (s, 2 H), 4.13 (d, J= 2.4 Hz, 2 H), 3.81-3.77 (m, 1 H), 3.69 (dd, J = 9.6 Hz, J = 4.2 Hz, 1 H), 3.51 (dd, J = 9 Hz, J = 7.2 Hz, 1 H), 3.43-3.40 (m, 1 H), 3.16-3.12 (m, 1 H), 2.43 (t, J= 2.4 Hz, 1 H), 1.94-1.87 (m, 2 H), 1.75-1.66 (m, 2 H). TLC (UV254) Rf = 0.62 (2:1 酢酸エチル/ヘキサン). HPLC tR = 8.80 min。
【0160】
(S)−1−(2−プロピニル)−5−(2−フェノキシメチル−ピロリジン−1−スルホニル)イサチン(化合物24) 乾燥DMF(10mL)における(S)−5−(2−フェノキシメチル−ピロリジン−1−スルホニル)イサチン20a(0.39g、1mmol)の溶液に、炭酸カリウム(0.21g、1.5mmol)、さらには臭化プロパルギル(トルエンにおける80wt%)(0.14g、1.2mmol)を添加した。2時間後、TLCにより、20aの完全な変換が示され、この溶液を10%NH4Cl水溶液(20mL)に注ぎ、DCMで抽出した(3×10mL)。次に、有機画分を合わせたものを真空中で還元し、残渣をジエチルエーテル(10mL)中にとり、水(2×10mL)、次にブライン(2×10mL)、次にブライン(1×10mL)で洗浄し、Na2SO4で乾燥した。クロマトグラフィー(ヘキサン/酢酸エチル)によって、生成物をオレンジ色のゴム状物として得た。酢酸エチル/ヘキサンからの再結晶化によって、生成物をオレンジ色の固体として得た(0.28g、66%)。Mp: 115-117℃. HRMS (ESI) = 425.1185 (M + H)+. C22H21N2O5Sに関する算出値 425.1171. 1H NMR (600 MHz, CDCl3) δ 8.12 (dd, J = 8.4 Hz, J = 1.8 Hz, 1 H), 8.05 (d, J = 1.8 Hz, 1 H), 7.25-7.21 (m, 2 H), 7.16 (d, J = 8.4 Hz, 1 H), 6.96-6.93 (m, 1 H), 6.81 (d, J= 9 Hz, 2 H), 4.56 (dd, J = 18 Hz, J = 2.4 Hz, 1 H), 4.53 (dd, J = 18 Hz, J = 2.4 Hz, 1 H), 4.19 (dd, J = 9.6 Hz, J = 3 Hz, 1 H), 4.07-4.01 (m, 1 H), 3.97 (dd, J = 9.6 Hz, J = 7.2 Hz, 1 H), 3.55-3.51 (m, 1 H), 3.33-3.28 (m, 1 H), 2.36 (t, J=2.4 Hz, 1 H), 2.09-1.99 (m, 2 H), 1.89-1.75 (m, 2 H). TLC (UV254) Rf = 0.54 (2:1 酢酸エチル/ヘキサン). HPLC tR = 9.00 min。
【0161】
(S)−1−(2−プロピニル)−5−(2−(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物25) 20dを使用する以外は、24の方法に従って、25を調製して、オレンジ色の固体を得た(0.48g、70%)。Mp: 101-103℃. HRMS (ESI) = 461.0976 (M + H)+. C22H19F2N2O5Sに関する算出値 461.0983. 1H NMR (600 MHz, CDCl3) δ 8.13 (dd, J = 8.4 Hz, J = 1.8 Hz, 1 H), 8.07 (d, J = 1.8 Hz, 1 H), 7.24 (d, J = 8.4 Hz, 1 H), 6.96-6.92 (m, 1 H), 6.85-6.78 (m, 2 H), 4.59 (dd, J = 18 Hz, J = 2.4 Hz, 1 H), 4.57 (dd, J = 18 Hz, J = 2.4 Hz, 1 H), 4.24-4.20 (m, 1 H), 4.03-3.98 (m, 2 H), 3.55-3.52 (m, 1 H), 3.26-3.21 (m, 1 H), 2.37 (t, J= 2.4 Hz, 1 H), 2.12-2.01 (m, 2 H), 1.88-1.76 (m, 2 H). TLC (UV254) Rf = 0.63 (2:1 酢酸エチル/ヘキサン). HPLC tR = 9.60 min。
【0162】
(S)−1−((1−(2−フルオロエチル)−1H−[1,2,3]−トリアゾール−4−イル)メチル)−5−(2−フェノキシメチル−ピロリジン−1−スルホニル)イサチン(化合物10) 乾燥DMF(3mL)における24(138mg、0.3mmol)の攪拌溶液に、水(0.2mL)における硫酸銅(38mg、0.15mmol)、次に水(0.2mL)におけるアルコルビン酸(53mg、0.3mmol)、次に乾燥DMF(1.5mL)における2−フルオロエチルアジド(33mg、0.36mmol)を添加し、混合物をアルゴン下で攪拌し続けた。2時間後、TLCによって、反応が完了したことを示され、混合物を10%NH4Cl水溶液(12mL)に注ぎ、DCMで抽出し(3×10mL)、Na2SO4で乾燥した。クロマトグラフィー(ヘキサン/酢酸エチル)によって、オレンジ色の固体を得た(26mg、51%)。Mp: 165-167℃. HRMS (ESI) = 514.1557 (M + H)+. C24H25FN5O5Sに関する算出値 514.1560. 1H NMR (600 MHz, CDCl3) δ 8.07 (dd, J = 8.4 Hz, J = 1.8 Hz, 1 H), 8.01 (d, J = 1.8 Hz, 1 H), 7.76 (s, 1 H), 7.47 (d, J = 8.4 Hz, 1 H), 7.23-7.19 (m, 2 H), 6.92 (t, J = 7.8 Hz, 1 H), 6.82 (d, J = 7.8 Hz, 2 H), 5.03 (d, J = 15.6 Hz, 1 H), 5.02 (d, J = 15.6 Hz, 1 H), 4.79 (dt, J = 46.2 Hz, J = 4.8 Hz, 2 H), 4.66 (dt, J = 27 Hz, J = 4.8 Hz, 2 H), 4.17 (dd, J = 9.6 Hz, J = 3.6 Hz, 1 H), 4.00-3.97 (m, 1 H), 3.93 (dd, J = 9 Hz, J = 7.2 Hz, 1 H), 3.53-3.49 (m, 1 H), 3.27-3.24 (m, 1 H), 2.08-1.97 (m, 2 H), 1.86-1.75 (m, 2 H). TLC (UV254) Rf = 0.56 (酢酸エチル). HPLC tR = 7.93 min。
【0163】
(S)−1−((1−(2−フルオロエチル)−1H−[1,2,3]−トリアゾール−4−イル)メチル)−5−(2(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物11) 25を使用する以外は、10の方法に従って、11を調製して、オレンジ色のゴム状物を得た。酢酸エチル/ヘキサンからの再結晶化によって、オレンジ色の固体を得た(94mg,57%)。Mp: 130-131℃. HRMS (ESI) = 550.1381 (M + H)+. C24H23F3N5O5Sに関する算出値 550.1372. 1H NMR (600 MHz, CDCl3) δ 8.08 (dd, J = 8.4 Hz, J = 1.8 Hz, 1 H), 8.02 (d, J = 1.8 Hz, 1 H), 7.77 (s, 1 H), 7.52 (d, J= 8.4 Hz, 1 H), 6.95-6.91 (m, 1 H), 6.82-6.75 (m, 2 H), 5.05 (s, 2 H), 4.79 (dt, J = 46.2 Hz, J = 4.8 Hz, 2 H), 4.67 (dt, J = 26.4 Hz, J = 4.8 Hz, 2 H), 4.20 (dd, J = 9.6 Hz, J = 3 Hz, 1 H), 4.01-3.94 (m, 2 H), 3.53-3.50 (m, 1 H), 3.22-3.17 (m, 1 H), 2.10-1.98 (m, 2 H), 1.85-1.74 (m, 2 H). TLC (UV254) Rf = 0.47 (酢酸エチル). HPLC tR = 8.45 min。
【0164】
(S)−1−((1−(フルオロメチル)−1H−[1,2,3]−トリアゾール−4−イル)メチル)−5−(2(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物29)を、フルオロメチルアジドを2−フルオロエチルアジドの代わりに使用する以外は、化合物11の方法に従って調製した。
【0165】
(S)−1−((1−(3−フルオロプロピル)−1H−[1,2,3]−トリアゾール−4−イル)メチル)−5−(2(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物30)を、3−フルオロプロピルアジドを2−フルオロエチルアジドの代わりに使用する以外は、化合物11の方法に従って調製した。
【0166】
(S)−1−((1−(4−フルオロブチル)−1H−[1,2,3]−トリアゾール−4−イル)メチル)−5−(2(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物31)を、4−フルオロブチルアジドを2−フルオロエチルアジドの代わりに使用する以外は、化合物11の方法に従って調製した。
【0167】
(S)−1−(4−フルオロベンジル)−5−(2−(1−((2−フルオロエチル)−1H−[1,2,3]−トリアゾール−4−イル)メトキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物12) 23を使用する以外は、10の方法に従って、12を調製して、オレンジ色のゴム状物を得た(26mg,48%)。HRMS (ESI) = 546.1632 (M + H)+. C25H26F2N5O5Sに関する算出値 546.1623. 1H NMR (600 MHz, CDCl3) δ 8.03 (d, J =1.8 Hz, 1 H), 8.00 (dd, J = 8.4 Hz, J = 1.8 Hz, 1 H), 7.67 (s, 1 H), 7.35-7.33 (m, 2 H), 7.08-7.05 (m, 2 H), 6.90 (d, J = 8.4 Hz, 1 H), 4.95 (s, 2 H), 4.81 (dt, J= 46.8 Hz, J = 4.8 Hz, 2 H), 4.67 (dt. J = 26.4 Hz, J = 4.8 Hz, 2 H), 4.62 (d, J = 12 Hz, 1 H), 4.60 (d, J = 12 Hz, 1 H), 3.81-3.78 (m, 1 H), 3.65 (dd, J = 9.6 Hz, J = 4.2 Hz, 1 H), 3.51 (dd, J = 9.6 Hz, J = 7.2 Hz, 1 H), 3.40-3.37 (m, 1 H), 3.19-3.15 (m, 1 H), 1.92-1.87 (m, 2 H), 1.72-1.66 (m, 2 H). TLC (UV254) Rf = 0.35 (酢酸エチル). HPLC tR = 6.58 min。
【0168】
(S)−1−(4−フルオロベンジル)−5−(2−(1−((フルオロメチル)−1H−[1,2,3]−トリアゾール−4−イル)メトキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物32)を、フルオロメチルアジドを2−フルオロエチルアジドの代わりに使用する以外は、化合物12の方法に従って調製する。
【0169】
(S)−1−(4−フルオロベンジル)−5−(2−((1−(3−フルオロプロピル)−1H−[1,2,3]−トリアゾール−4−イル)メトキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物33)を、3−フルオロプロピルアジドを2−フルオロエチルアジドの代わりに使用する以外は、化合物12の方法に従って調製する。
【0170】
(S)−1−(4−フルオロベンジル)−5−(2−((1−(4−フルオロブチル)−1H−[1,2,3]−トリアゾール−4−イル)メトキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物34)を、4−フルオロブチルアジドを2−フルオロエチルアジドの代わりに使用する以外は、化合物12の方法に従って調製する。
【0171】
(S)−1−[4−(フルオロメチル)−1H−[1,2,3]−トリアゾール−1−イル]メチル−5−(2−(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン)(化合物6)を、(S)−1−(アジドメチル)−5−(2−(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン](化合物37)を3−フルオロプロプ−1−イン(化合物28)と反応させることによって、図2(ii)に示されるように調製する。試薬は下記のとおりである:(a)CuSO4、L−アスコルビン酸、DMF。[18F]6の調製では、3−[18F]フルオロプロプ−1−インを3−フルオロプロプ−1−インの代わりに使用する。
【0172】
(S)−1−[4−(2−フルオロエチル)−1H−[1,2,3]−トリアゾール−1−イル]メチル−5−(2−(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン)(化合物7)を、4−フルオロブチ−1−インを(3−フルオロプロプ−1−イン)の代わりに使用する以外は、化合物6の方法に従って調製する。[18F]7の調製では、4−[18F]フルオロブチ−1−インを3−フルオロプロプ−1−インの代わりに使用する。
【0173】
(S)−1−[4−(3−フルオロプロピル)−1H−[1,2,3]−トリアゾール−1−イル]メチル−5−(2−(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン)(化合物8)を、5−フルオロペンチ−1−インを(3−フルオロプロプ−1−イン)の代わりに使用する以外は、化合物6の方法に従って調製する。[18F]8の調製では、5−[18F]フルオロペンチ−1−インを3−フルオロプロプ−1−インの代わりに使用する。
【0174】
(S)−1−[4−(4−フルオロブチル)−1H−[1,2,3]−トリアゾール−1−イル]メチル−5−(2−(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン)(化合物9)を、6−フルオロヘキシ−1−インを(3−フルオロプロプ−1−イン)の代わりに使用する以外は、化合物6の方法に従って調製する。[18F]9の調製では、6−[18F]フルオロヘキシ−1−インを3−フルオロプロプ−1−インの代わりに使用する。
【0175】
化合物35は、Rが前記規定と同様である化合物の一群を有する。化合物36は、R’が前記規定と同様である化合物の一群を有する。これらの化合物は、図12(上行=化合物35、下行=化合物36)で概略的に表わされる方法を用いて製造できる。このストラテジーは、in situのまたは別の反応としての、[11C]ヨウ化メチルとアジド源との反応、次に、適当なアルキンを用いた付加環化を含む。この方法は、近年Schirrmacher, R. et al 25で報告される、より既知のクリックケミストリー(Click Chemistry)ストラテジーの修飾である。
【0176】
実施例2−ターゲット化合物の親油性およびカスパーゼに対する親和性の測定
カスパーゼに対するターゲット化合物の親和性 様々な活性化カスパーゼ1、3、6、7、及び8に関する、新規なフッ素化イサチン1〜5及び10〜12、ならびに既知のイサチン誘導体及び中間対化合物22及び23の親和性を、Kopka and co-workers.13に記載されるのと同様の蛍光インビトロカスパーゼ阻害アッセイによって測定した。組換えヒトカスパーゼの阻害は、蛍光性生成物、7−アミノ−4−メチルクマリン(7−AMC)の蓄積を測定することによって評価した。
【0177】
組換えヒトカスパーゼ−1、−3、−6、−7、及び−8ならびにこれらのペプチドに特異的な基質は、Biomol International, UKから購入した。非放射性イサチンによる組換えカスパーゼの阻害は、蛍光性生成物、7−アミノ−4−メチルクマリン(7−AMC)の蓄積を測定する蛍光アッセイを用いて評価した。全てのアッセイは、1ウェルあたり200μlの容積で96ウェルプレートで行った。アッセイは、各カスパーゼについて以下に記載されるような適当な反応緩衝液中で37℃で行った。カスパーゼ1では、緩衝液は、0.1% CHAPS、100mM NaCl、5mM 2−メルカプトエタノール、100mM HEPES(pH 7.4)、2mM EDTA、10% スクロース、及び10μMのペプチド基質 Ac−YVAD−AMCから構成された。カスパーゼ3について:20mM HEPES(pH 7.4)、10% スクロース、100mM NaCl、0.1% CHAPS、2m,mM EDTA、及び10μM Ac−DEVD−AMC。カスパーゼ6について:20mM HEPES(pH 7.4)、10% スクロース、100mM NaCl、0.1% CHAPS、2mM EDTA、及び10μM Ac−VEID−AMC。カスパーゼ7について:20mM HEPES(pH 7.4)、10% スクロース、5mM 2−メルカプトエタノール、100mM NaCl、0.1% CHAPS、2mM EDTA、10μM Ac−DEVD−AMC。カスパーゼ8について:20mM HEPES(pH 7.4)、10% スクロース、100mM NaCl、0.1% CHAPS、2mM EDTA、および10μM Ac−IETD−AMC。これらの緩衝液は、500、50、5μM;500、50、5nM;500、50、5pMの最終濃度でDMSO中に非放射性イサチンを含んだ;全てのウェルにおけるDMSOの最終濃度は全容積の5%であった。組換えカスパーゼを、1アッセイ当たり0.5単位で使用した(〜500pmol 変換基質/時間)。ペプチド基質以外の全ての試薬は、10分間予めインキュベートした。次に、ペプチド基質(最終濃度 10μM)を添加して、プレートをさら30分間インキュベートした;30分は、10、30、60または90分後に反応が経過した初期直線性研究後に選択された。30分の時点が選択された。各コントロールウェルは、酵素以外の全ての反応成分を含んだ。7−AMCの生産量を、それぞれ、355nm及び460nmの励起波長及び放出波長で蛍光マイクロプレートリーダー(Victor2; Perkin-Elmer Life sciences)で測定した。カスパーゼ活性を50%阻害するイサチンの濃度(EC50)を、GraphPad Prism(Version 4.0 for Windows, GraphPad Software, San Diego California USA)を用いて非直線性回帰分析によって評価した。全てのイサチンは2連で分析した;アッセイは1回繰り返した。既知の化合物13〜16を参考化合物として含ませた。結果を下記表1に要約する。
【0178】
データは、5pM〜500μMの範囲の9種の濃度で、それぞれ重複した、2回の操作の平均である;各酵素を50%阻害するのに必要な薬剤濃度(EC50)を、阻害プロフィールの非直線性回帰分析から得た。
【0179】
結果 本アッセイでは、既知のイサチン13の親和性は、それぞれ、カスパーゼ3及び7に対して59.9及び25.3nMであった。フェニルエーテルでのフッ素置換は良好に許容され、新規なイサチン1では、カスパーゼ3に対する親和性は、既知の非フッ素化フェニルエーテル14に比べて2〜3倍増加した。2フッ素化イサチン2及び3はより強力であり、それぞれ、カスパーゼ3に対して、12.4及び10.4nMであった。テトラヒドロピラン4では同様の親和性が見出された。既知のベンジル誘導体15に比して4の効力の5倍の増加は、環が十分飽和であることを考えれば、驚くべきことである。ピリミジニル誘導体5におけるようなフェニルエーテルのピリミジニル基による置換により、さらに効力が増加し、カスパーゼ3及び7に対する親和性が、それぞれ、5.5及び2.3nMであった。分子のいずれかの側に2−フルオロエチル−1,2,3−トリアゾールを導入することによって、効力が急増し、トリアゾール10及び12に関して測定されたカスパーゼ3の親和性が、それぞれ、16.7及び12.6nMであった。フルオロエチルトリアゾール12の効力は1〜3と同様であり、このことは基がかなりより小さな大きさでありかつ極性性がより高いことを考えると驚くべきことである。Lee8は、親水性ポケットとしてイサチン窒素周辺の結合ドメインを示し、ほとんどの基がこの位置にベンジル部分を有する。したがって、トリアゾール10が既知のベンジル誘導体15より4倍強力であることは非常に予想し得ないことであった。試験した化合物のうち、トリアゾール11は、圧倒的に最も強力であり、カスパーゼ3及び7に対して、それぞれ、0.5及び2.5nMの親和性を有していた。試験したすべての化合物は、カスパーゼ1、6及び8に対して低い基質であった(EC50>5000nM)。
【0180】
Log P計算 親油性をACD/Chemsketch labs softwareから算出した。
【0181】
結果 結果を下記表1および1aに示す。
【0182】
【表2−1】
【0183】
【表2−2】
【0184】
実施例3−[18F]11の合成
好ましくは、化合物[18F]11は、(S)−1−(2−プロピニル)−5−(2−(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物25)を[18F]フルオロエチルアジド(化合物27)15, 16でクリック−ラベリング(click-labelling)することによって、製造される。トリアゾール(S)−1−((1−(2−フルオロエチル)−1H−[1,2,3]−トリアゾール−4−イル)メチル)−5−(2(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物11)は、図2(i)及び図19に示されるように、2−[18F]フルオロエチルアジド(化合物27)をアルキン前駆体である(S)−1−(2−プロピニル)−5−(2−(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン(化合物25)で銅触媒による付加環化を行うことによって標識される。同様の方法を用いて、非放射性標識の化合物11を製造でき、この際、フルオロエチルアジドを2−[18F]フルオロエチルアジドの代わりに使用する。
【0185】
窒素雰囲気下で、アスコルビン酸ナトリウム(50μl、8.7mg、43.2μmol)の緩衝液(リン酸ナトリム緩衝液、pH 6.0、250mM)を、硫酸銅(II)の水溶液(50μl、1.7mg 5水和物、7.0μmol)を含むホイートンバイアル瓶(Wheaton vial)(1mL)に添加した。1分後、ジメチルホルムアミド(25μl)におけるアルキン25(3.0mg、6.5μmol)の溶液、さらにアセトニトリル(100μl)における蒸留[18F]FEA(185−740 MBq)を添加した。この混合物を室温で30分間放置して、水(15μl)を添加した後、予備ラジオ−HPLCにかけた。単離したHPLC物溶液を水(5mL)で希釈し、予めエタノール(5mL)及び水(10mL)で調整された(conditioned)SepPak C18−lightカートリッジ(Waters)にのせた。次に、このカートリッジを水(5mL)でフラッシュし、18F−9を小画分(0.1mL)づつエタノールで溶出した。この生成物画分を、エタノール含量が10〜15%(v/v)になるようにPBSで希釈した。
【0186】
結果 27をアルキン前駆体25と共役させようとする初期の試みにより、放射性生成物の複合混合物が形成した。放射化学的収率への触媒システム、温度及びpHの影響をさらに調べたところ、最も高い収率が室温で得られたものの、[18F]9は熱安定性に劣ることが分かった。リン酸ナトリウム緩衝液(pH 6.0、250mM)を反応混合物に添加することによって、放射化学的収率が劇的に向上したものの、調べた2つの触媒システム(銅粉末または硫酸銅/アスコルビン酸塩)では若干の相違が観察されたのみであった。最適条件は、アルキン前駆体25に対して若干過剰な硫酸銅の存在下で室温で30分の反応時間であることが分かった。これにより、65±6%(n=26、27から減衰補正)の単離放射化学的収率で、HPLCによる精製後の放射化学的純度が>99%のトリアゾール[18F]11が得られた。生成混合物に関する具体的なHPLCクロマトグラムを図3に示す。本方法は少量の放射能で手動で行ったので、合成終了時に低い比放射能(1.2GBq/μmol)しか達成しなかった。[18F]11の同一性を、非放射性参照化合物と一緒に溶出することによって確認した。精製[18F]11を、91±6%(n=26、減衰補正)の効率で固相抽出することによって配合した(formulate)。[18F]11の配合(formulation)を含む放射合成(radiosynthesis)は全体で3時間かかった。
【0187】
実施例4−[18F]11の親油性
Log P測定 Barthel et al 18の方法を用いてオクタノール−水分配係数を測定することによって、親油性を評価した。簡単にいうと、[18F]11(25μLエタノールにおける〜180μCi)を水を用いて最終容積が100μLになるように希釈して、ストック溶液を得た。ストック溶液のアリコート(10μL)を水(490μL)及びオクタン−1−オール(Aldrich無水グレード)(500μL)に加えた。次に、この溶液を10分間激しく振盪した後、遠心した(13201g、20℃、30分)。遠心後、水層及びオクタノール層の部分(200μL)を注意深く除去し、Cobra II Auto-Gamma counter (Packard Instruments, Meriden, CT, USA)に置いて分析し、比較した。放射能を含む水で放射能を含むオクタノールを割ることによって、オクタノール−水分配係数を得た。Log Pを測定したところ、上記表1の脚注cに示されるように、1.61であった。
【0188】
実施例5−[18F]11および[125I]14の組織分布および代謝安定性
血漿及び肝臓における新規な化合物[18F]11及び既知の化合物[125I]14の安定性を、マウスで投与した後経時的に測定した。[18F]11を腫瘍を有するマウスの静脈内に注射し、その組織分布を注射してから2、15及び60分後に所定の組織中で測定した。[125I]14を腫瘍を持たないマウスの静脈内に注射し、その組織分布を注射してから2〜60分の所定の時点で測定した。さらに、放射性トレーサーのインビボでの代謝安定性を血漿及び肝臓サンプルから、および[18F]11では、尿サンプル中で測定した。
【0189】
インビトロでのマウス肝臓S9代謝研究 肝臓S9代謝研究をAboagye et al19に記載されるのと同様にして行った。マウス肝臓を迅速に切除し、実験を通して4℃に保持した。組織を秤量し、Ultra-Thurraxホモジナイザー(IKA, Staufen, Germany)を用いて等容積の50mM Tris−150mM KCl−HCl緩衝液(pH 7.4)中で均質化した。S9画分を得るために、ホモジネートを30分間遠心(10,000×g)して、核、ミトコンドリア及び細胞残屑を除去した。S9画分のタンパク質濃度を市販のBCAタンパク質アッセイキット(Perbio Science, Cheshire, UK)によって測定した。S9画分を最長6週間−80℃で貯蔵した。所定の非標識イサチン14、15、16(10mM、10μL)を、1mLの全容積で0.1mM Tris−HCl緩衝液(pH 7.4)中で37℃で60分間、S9画分(33.26mg/ml、20μL)及び0.5mM ニコチンアミドアデニンジヌクレオチドリン酸(還元形態)と共にインキュベートした。コントロールインキュベーションサンプルは、イサチンを含まなかった。氷冷したアセトニトリル(2mL)を添加することによって、反応を止めた;次に、サンプルを即座にドライアイスに置いた後、抽出及びHPLC分析を行った(下記)。
【0190】
インビボでの生体内分布および代謝研究 放射線誘発線維肉腫(RIF−1)腫瘍細胞20を、5% CO2の加湿インキュベーターで37℃で、10%ウシ胎児血清(BioWhittaker Europe Ltd, Verviers, Belgium)、2mM L−グルタミン、100U/mL ペニシリン、100μg/mL ストレプトマイシン及び0.25μg/mL ファンギゾン(Gibco, UK)を加えたRPMI 1640培地(Invitrogen Ltd, Paisley, UK)中で維持した。すべての動物の作業は、英国の"Guidance on the Operation of Animals (Scientific Procedures) Act 1986" (HMSO, London, United Kingdom, 1990)に従って、また、実験新生組織形成における動物の愛護に関する政府の規則及びガイドライン(government regulations and guidelines on the welfare of animals in experimental neoplasia)21に完全にのっとって、認可を受けた研究者によって行われた。オスのC3H/hejマウス(Harlan, Bicester, Oxfordshire, UK)の背中に5×105個の細胞を皮下注射することによって腫瘍を確立した。腫瘍の成長を電子キャリパー(electronic callipers)を用いて2日毎にモニターし、腫瘍の容積を式:(π/6)×L×W×D(L=長さ、W=幅およびD=深さ)を用いて評価した。腫瘍が約100〜150mm3に到達したら、[18F]11の生体内分布の研究のために動物を選択した;[125I]14の生体内分布は腫瘍を持たないマウスで行った。
【0191】
リン酸緩衝液に溶解した放射能(〜0.37MBqの[125I]14または3.7MBqの[18F]11) 0.08〜0.13mLを、側静脈を介して、マウスの静脈内に注射した。注射してから所定の時点(2〜60分)で、通常の麻酔下(イソフルオラン(isofluorane)吸入)で心穿刺による放血によってマウスを剖検した。ヘパリン化血のアリコートを直ぐに遠心(2000gで5分)して、血漿を得た。組織に含まれる放射能をCobra II Auto-Gamma counter (Packard Instruments, Meriden, CT, USA)でのガンマ−カウンティングによって測定し、注射量/g 組織の割合(%)(%ID/g)として表わした。3匹のマウスのうちの最小値を各時点で使用した。すべての動物は未処置(treatment-naive)であった。
【0192】
インビボでの代謝研究もまたC3H/hejマウスで行った。腫瘍を持たないマウスの静脈内に0.37MBqの[125I]14または7.4MBqの[18F]11を注射し、血漿を上記と同様にして得た。血漿、肝臓及び尿サンプルを液体窒素中でスナップフリーズし(snap-frozen)、分析するまで、ドライアイス上の予め秤量された(pre-weighed)シンチレーション計測用チューブ中に保持した。
【0193】
HPLC分析 抽出直前に、サンプルを解凍し、氷上に置いた。抽出では、氷冷したアセトニトリル(1.5mL)を血漿(0.2mL)に添加した;アセトニトリルを含むS9インキュベーションサンプル(3mL)もまた分析した。各混合物を遠心(15493×g、4℃、3分)し、得られた上清をロータリーエバポレーターを用いて40℃で真空中で蒸発乾固した。肝臓サンプルは、遠心前にIKA Ultra-Turrax T-25ホモゲナイザーを用いて氷冷アセトニトリル(1.5mL)と共に均質化した。次に、残渣をHPLC移動相(1.2mL)に再懸濁し、Minisart親水性シリンジフィルター(Minisart hydrophilic syringe filter)(0.2μm)(Sartorius, Goettingen, Germany)で濾過した。尿サンプルは、HPLC移動相で希釈し、上記と同様にして濾過した。次に、サンプル(1mL)を、γ−RAM モデル3 ガンマ−検出器(γ-RAM Model 3 gamma-detector)及びLauraソフトウェア(IN/US Systems inc., Florida, USA)を備えたAgilent 1100シリーズHPLCシステム(Agilent Technologies, Stockport, UK)によるラジオ−HPLCによって分析した。
【0194】
固定相は、Waters μBondapak C18逆相カラム(300×7.8mm)で構成された。[125I]14の分析では、血漿及び肝臓サンプルを上記と同様にして処理し、2mL/分の流速で定組成で(in isocratic mode)流す水(0.1% TFA)/プロパン−1−オール(0.1% TFA)(35:65)の移動相を用いて分析した。インビトロでの代謝後の非標識の14、15及び16の分析では、水(0.1% TFA)/プロパン−1−オール(0.1% TFA)から構成される移動相を、11分で2→80%有機相、3分で80(R)5%有機相後、6分で5%有機相の勾配を3mL/分の流速で供給しながら、用いることによって、サンプルを分析した。[18F]9の分析では、0.1M ギ酸アンモニウム/1.8:1 メタノール:アセトニトリルから構成される移動相を、1分で50%有機相、14分で50(R)90%有機相、3分で90%有機相、2分で90(R)50%有機相、4分で50%有機相の勾配を3mL/分の流速で供給しながら、用いることによって、血漿、肝臓及び尿サンプルを分析した。さらに、HPLC注入液中に残る[18F]9−由来の放射能に対する抽出後の血漿及び肝臓ペレットに関連する[18F]9−由来の放射能の割合を評価するために、HPLC注入液の全容積を記録し、アリコート(0.1mL)を計測用に除去した。次に、0.1mLアリコート及びペレット中の放射能をガンマカウンティング(gamma counting)(Packard Instruments)によって分析した。
【0195】
結果 図4から、[18F]11は迅速に組織に分布し、また迅速に排出されたことが分かる。[18F]11由来の放射能の高い極在化が腎臓、尿及び肝臓で見られたことから、排出の腎臓及び肝臓経路双方の重要性が示唆される。しかしながら、これらの組織でさえ、放射能の迅速な排出が見られた。重要なことに、イメージングの観点から、未処置の腫瘍、心臓及び脳での[18F]11の取り込みは低かった;これにより、これらの組織でのカスパーゼ活性化に関連する結合性の増加の測定が容易になるはずである。骨への取り込みが低かったことから、放射性トレーサーの脱フッ素化は存在せず、ゆえにフルオロエチルアジド部分が安定であることが示唆される。ラジオ−HPLC分析から、[18F]11は血漿及び肝臓で単一の極性代謝ピークを作成する[125I]14(図5)より代謝分解に対して比較的より安定であることが示された。親化合物は60分で血漿中に依然として存在し、主要なピークであった。これに対して、代謝産物は、すべての研究した時点で肝臓サンプルでは主要なピークであった。尿の放射能は主として代謝産物から構成された。所定の時点での血漿及び肝臓での親放射性トレーサーの割合を、見かけの抽出効率と共に、表2に要約する。すばやく減少した後は、インビボでの代謝速度は15〜60分の間は横ばいであるように見えた。しかしながら、血漿及び肝臓からの抽出効率はおそらく非特異的結合により経時的に減少した。また、血漿抽出後の残渣ペレットでの放射能は時間に依存して増加したことから、血漿タンパク質への結合が増加したことが示唆される。
【0196】
【表3】
【0197】
図7から、[125I]14は主要な器官に迅速に分布し、また迅速に排出されたことが分かる。[125I]14は10分以内に血漿から完全に排出され、この時点で肝臓で検出される放射能の<15%(n=3)を提示した。注射してから30分以内に、親[125I]14は肝臓で十分代謝され、この際、放射能の大部分を構成する1つの極性のある代謝産物が検出された。これらの知見と一致して、我々は、調査したすべての組織で迅速なクリアランスが観察され、10分以降、注射された放射能の大部分は尿中に排出された。
【0198】
図8に示されるように、既知の非標識のイサチン14及び15の代謝プロフィールは同様であり、ほとんど完全な分解が60分インキュベーション後に観察された。
【0199】
実施例6−4種の癌細胞系での[18F]11の「取り込み
細胞系および腫瘍モデル Wolf C.R. et al.22 and Langdon S.P. et al23に記載されるのと同様にして、ヒト卵巣癌細胞系PEO1/4を確立し、特性を明らかにした。簡単にいうと、PEO1/4細胞は、再発粘液卵巣腺癌の患者のシスプラチン化学療法に臨床的耐性が発現する前(PEO1)及び後(PEO4)の、患者の悪性腹水由来であった。当初は、放射線誘発線維肉腫(RIF−1)腫瘍細胞系の特性をTwentyman P.R. et al.20によって明らかにした。LNM35腫瘍細胞は、Kozaki K. et al.24に記載されるように、ヒト肺大細胞癌NCI−H460細胞系のリンパ行性転移性の高いサブラインである。すべての細胞系を、定常的に、5% CO2の加湿インキュベーターで37℃で、10%ウシ胎児血清(BioWhittaker Europe Ltd, Verviers, Belgium)、2mM L−グルタミン、100U/mL ペニシリン、100μg/mL ストレプトマイシン及び0.25μg/mL ファンギゾン(Gibco, UK)を加えたRPMI 1640培地(Invitrogen Ltd, Paisley, UK)中で維持した。
【0200】
6〜8週齢のメスのnu/nu−BALB/cマウスまたはオスのC3H/hejマウス(Harlan, Bicester, Oxfordshire, UK)の背中に、PEO1/4(10×106細胞、with v/v matrigel, BD Biosciences, Oxford, UK)、RIF−1(5×105細胞)及びLNM35(1×106細胞)を皮下注射することによって、インビボでの腫瘍モデルを確立した。腫瘍の成長を電子キャリパー(electronic callipers)を用いて2日毎にモニターし、腫瘍の容積を式:(π/6)×L×W×D(L=長さ、W=幅およびD=深さ)を用いて評価した。すべての動物の作業は、英国の"Guidance on the Operation of Animals (Scientific Procedures) Act 1986" (HMSO, London, United Kingdom, 1990)に従って、また、実験新生組織形成における動物の愛護に関する政府の規則及びガイドライン(government regulations and guidelines on the welfare of animals in experimental neoplasia)に完全にのっとって、認可を受けた研究者によって行われた。
【0201】
インビトロでの[18F]11取り込みおよびカスパーゼ3活性化アッセイ 細胞を、実験の2または3日前に12ウェルプレートに3連で播種し、所定の濃度および時間に白金由来の抗腫瘍薬であるシスプラチン、5−FU、タンパク質合成阻害剤であるシクロヘキシミド(CHX)またはトポイソメラーゼ阻害剤エトポシド(VP−16)(Sigma, UK)または対応するベヒクルで処理した。実験の日に、約10μCi/ウェルの[18F]11を添加して、37℃で60分間細胞中に蓄積させた。細胞を集め、洗浄し、400μlのPBS中に再懸濁した。260μlの各サンプルを計測用チューブに移した直後、フッ素−18の放射能を、Packard Cobra II gamma counter (Perkin Elmer, UK)を用いて測定した。製造者(caspase-Glo 3/7 assay, Promega, UK)の指示に従って、40μlの各サンプルを用いて、カスパーゼ3活性化比色分析アッセイを行った。簡単にいうと、細胞を乳白色の96ウェルプレートに移し、50μlのカスパーゼ−Gloと共に30分〜3時間インキュベートし、カスパーゼ3の酵素活性をMultiskan Luminometer (Thermo Electron, UK)を用いて測定した。全てのサンプルで、BCAプロテインアッセイ(BCA Protein assay)(Pierce, UK)を行い、データを正規化して、CCPMAまたはRLU(Relative Light Unit)/mg タンパク質として表わす。
【0202】
インビボでの[18F]11生体内分布およびPET画像化 生体内分布の研究のために、腫瘍を有するマウスを10mg/kgのシスプラチンまたはベヒクルで24時間処置し、イソフルオラン(isofluorane)/O2/N2Oで麻酔し、所定の時間、約100μCiの[18F]11を側尾静脈(lateral tail vein)に静脈内注射した。様々な組織を取り除き、秤量し、即座にフッ素−18放射能計測するために計測用チューブに置いた。
【0203】
シスプラチン(10mg/kgで24時間)またはベヒクルで処置した腫瘍を有するマウスを、小動物専用のPETスキャナー(dedicated small animal PET scanner)(quad-HIDAC; Oxford Positron Systems, Weston-on-the-Green, United Kingdom)でスキャンした。麻酔をかけた動物をサーモスタット制御のベッドに置き、スキャナー内に腹臥位にした。マウスの直腸温度が約37℃になるように、ベッドを調節した。[18F]11(約100μCi)のボーラス注射液(bolus injection)を、尾静脈カニューレを介して静脈内(i.v.)投与し、スキャンを開始した。動的放射スキャン(dynamic emission scan)を60分にわたってリスト形式(list-mode format)で得た。次に、画像再構築のために、得られたデータを、0.5−mm 副鼻腔撮影図(sinogram bin)および19のタイムフレーム(0.5×0.5×0.5mm ボクセル;4×15、4×60、および11×300秒)に分け、これを2次元のハミングフィルター(two-dimensional Hamming filter)(カットオフ 0.6)を用いたフィルター逆投影によって行った。このようにして得られた画像データ−セットをSUN workstation (Ultra 10; SUN Microsystems, Santa Clara, CA)に移し、Analyze ソフトウェア(version 6.0; Biomedical Imaging Resource, Mayo Clinic, Rochester, MN)を用いて可視化した。注射してから0〜1分および注射してから30〜60分から構成される動的データの累積画像を、放射性トレーサーの取り込みの可視化のためにおよび関心のある領域(regions of interest)(ROI)を規定するために使用した。各19時点で各ROIについて、カウント密度を平均して、ROIに関する時間に対する放射能曲線(TAC)を得た。腫瘍のTACを各時点で心臓のものに正規化して、正規化した取り込み値(normalized uptake value)(NUV)を得た。筋肉の[18F]9データを、腫瘍データを正規化するための内部入力関数として使用した。注射してから60分でのNUV(NUV60)、0〜60分のNUVの積分として算出されたNUV曲線下の面積(AUC)、およびトレーサーの分数保持(fractional retention of tracer)(FRT)、2.5分での放射能に対する60分での放射能を、比較に使用した。FRTは、保持される腫瘍にデリバリーされる放射性トレーサーの割合を示すという点で有用で可変である。したがって、これは腫瘍の[18F]9の取り込みからデリバリーまでを正規化する。
【0204】
結果 図9に示されるように、予想とおりシスプラチン耐性卵巣癌細胞(PEO4)以外の、全ての薬剤処置細胞は、コントロール(ベヒクル処置)に比して[18F]9の取り込みの有意な増加を示す。さらに、薬剤処置LNM35での[18F]9の取り込みの増加は、細胞内の活性カスパーゼ3量と相関し、これからCDDP処置とVP−16処置との拮抗作用が明らかになった。
【0205】
表3は、CDDPで処置したPEO1異種移植片を有するマウスの腫瘍における[18F]9の取り込みの有意な増加を示すが、この際、CDDP耐性PEO4腫瘍はコントロールと処置との間に取り込みの相違は示していない。PEO1腫瘍を有するマウスの脾臓及び筋肉以外の、他の組織における[18F]9の取り込みは、コントロールと処置との間に有意な差はない。
【0206】
【表4】
【0207】
PEO1、PEO4及びRIF−1異種移植片を有するこれらのCDDP処置マウスにおけるPET画像(図10)から、腫瘍における[18F]11の有意な取り込みが明らかになった。
【0208】
下記文書はすべて参考で本明細書中に引用される。
【0209】
実施例7−アポトーシス細胞への[18F]11の取り込み
RIF−1細胞を、ベヒクル(0.1% DMSO)またはcis−ジアミンジクロロ白金(II)(cis-diamminedichloro-platinum(II))(シスプラチン)(CDDP)(100μM)で、48時間処置した。次に、これらの細胞を[18F]11と共に1時間インキュベートし、洗浄し、放射能を分析した。図13に示されるように、細胞アッセイにより、アポトーシス細胞への[18F]11の取り込みが約1.5倍増加したことが示された。データは、1mg 全細胞タンパク質当たりで平均化した減衰補正したカウント/分として表わす。データは、平均値±SEMであり、3連で行った。
【0210】
RIF−1腫瘍をベヒクル(50% DMSO)またはCDDP(10mg/kg 単回投与)で処置した。放射性トレーサーを注入してから60分での[18F]由来の放射能レベルを分析し、血中レベルに対する割合として表わす。図14に示されるように、RIF−1腫瘍を有するマウスにおけるインビボでの取り込み研究でも、ベヒクル処置したマウスに比べて処置マウスでは[18F]11の取り込みが約1.5倍増加した。データは、平均値±SEMであり、1群当たりn=8匹マウスである。
【0211】
実施例8−アポトーシス画像化トレーサーとしての[18F]11の機能性
6〜8週齢のオスのC3H/hejマウス(Harlan, Bicester, Oxfordshire, UK)の背中に38C13 マウスリンパ腫細胞(5000細胞)を皮下注射することによって、腫瘍アポトーシスのインビボ実験モデルを確立した。異種移植片が約100mm3に到達したら、マウスを、シクロホスファミド(CPA、100mg/kg)またはベヒクルで24時間処置した後、小動物専用のPETスキャナー(dedicated small animal PET scanner)(Siemens Inveon PET module)でスキャンした。麻酔をかけた動物をサーモスタット制御のベッドに置き、スキャナー内に腹臥位にした。[18F]11(約100μCi)のボーラス注射液(bolus injection)を、尾静脈カニューレを介して静脈内(i.v.)投与し、スキャンを開始した。
【0212】
動的放射スキャン(dynamic emission scan)を60分にわたってリスト形式(list-mode format)で得た。次に、画像再構築のために、得られたデータを、0.5−mm 副鼻腔撮影図(sinogram bin)および19のタイムフレームに分け、これをフィルター逆投影によって行った。このようにして得られた画像データ−セットをSiemens Inveon Research Workplace softwareを用いて可視化した。動的データの累積画像を、放射性トレーサーの取り込みの可視化のためにおよび関心のある領域(regions of interest)(ROI)を規定するために使用した。各19時点で各ROIについて、カウント密度を平均して、ROIに関する時間に対する放射能曲線(TAC)を得た(図15参照)。
【0213】
腫瘍のTACを各時点で全身のものに正規化して、正規化した取り込み値(normalized uptake value)(NUV)を得た。注射してから60分でのNUV(NUV60)、0〜60分のNUVの積分として算出されたNUV曲線下の面積(AUC)を比較に使用した(図16参照)。
【0214】
スキャン後、様々な組織を取り除き、秤量し、即座にフッ素−18放射能計測するために計測用チューブに置いて、[18F]11の生体内分布を評価した(下記表4参照)。
【0215】
ベヒクルまたはシクロホスファミドで処置した2匹の38C13異種移植片を有するマウスの代表的なOSEM3D 再構築[18F]11 PET画像を図17に示し、この際、丸は腫瘍を示す。
【0216】
表4の生体内分布の結果から、ベヒクルに比して、CPAで処置したマウスの腫瘍では、[18F]11の取り込みが有意に2倍増加したことが示され、これは、PETイメージングパラメーター、腫瘍のTAC、NUV60及びAUCから得られた半定量データによって確認された(図15及び16を参照)。したがって、図15〜17及び表4に示される生体内分布およびPETスキャンデータから、[18F]11によりインビボでの腫瘍のアポトーシスを検出できる可能性があることが示され、これにより、初めて、腫瘍のためのカスパーゼ−3に特異的なPET画像化剤の使用が記載される。
【0217】
【表5】
【0218】
実施例9−化合物11の細胞活性
化合物11の細胞活性を酵素アッセイを用いて評価し、化合物14の細胞活性と比較した。
【0219】
マウス放射線誘発線維肉腫(RIF−1)細胞を、カスパーゼ阻害剤である、z−VAD−fmk(100μM)、化合物11及び化合物14で15分間処理した後、48時間、シスプラチン(100μM)でアポトーシスを誘導した。Δ−PARP免疫ブロットバンドは、カスパーゼ 3の切断により得られるPARPの内因性の89kDaの大きさの断片に相当する。α−チューブリンをローディングコントロールとして使用した。
【0220】
図18に示されるように、RIF−1細胞を、化合物14ではなく、化合物11と予めインキュベーションすると、シスプラチンで誘導されるアポトーシスによるカスパーゼ−3と同種のターゲットPARPの誘導が妨害されたことから、化合物11によるカスパーゼ−3の細胞不活性化が示唆される。
【0221】
薬剤により誘導されるアポトーシスの条件下で化合物11の結合性をさらに確立するために、我々は、実施例6で上記されるのと同様にしておよび図9に示されるように、LNM35、RIF−1及びPEO1/4癌細胞における放射線標識された[18F]11の取り込みを評価した。予想とおり、シスプラチン耐性卵巣癌細胞(PEO4)以外の、全ての薬剤で処理された細胞は、コントロールに比べて[18F]11の取り込みが有意に増加した。さらに、薬剤処理したLNM35細胞における[18F]11の取り込みの増加は、カスパーゼ−Gloアッセイを用いた細胞活性カスパーゼ−3の量と相関した。
【0222】
実施例10−保護前駆体を用いた[18F]11の合成
イサチン放射性リガンド[18F]11は、図2(i)、図19に及び実施例3に記載されるのと同様の2段階法を用いて合成できる。この放射性合成(radiosynthesis)の欠点には、安定した不純物の存在があり、これにより配合される生成物の比放射能が減少する(1〜4Ci/μmol)。より根本的には、安定した不純物は、かなりの量(5〜10μg/mL)で存在し、この量は臨床的進行には多すぎる値である。毒性試験を行うためにこの不純物の特性を明らかにしようと試みたが結論は得られず、安定した不純物が、「冷たい」フッ素−19トリアゾール化合物でなく、イサチン由来であることが確認されたにとどまった。
【0223】
HPLC分析によって、不純物が放射性合成中の副反応の結果であり、アルキン前駆体中に存在する物質ではないことが確認された。イサチン分子構造の試験から、化学反応性が高い2つの部位、上記クリック化学付加環化(click chemistry cycloaddition)で使用される末端アルキン基およびカスパーゼ−3活性部位での結合に必要なC−3カルボニル位置のみが示された。C−3カルボニル位置に関する保護基ストラテジーを調べ、特に、下記一般式38及び43で示される(この際、n=0、1、2、3、4、5または6であり、x=離脱基、例えば、メシレート、トシレート、ノシレート(nosylate)、または他のスルホネートエステルまたはハライド)ような、アセタールジオキソランとしてのC−3カルボニル位置の保護を調べた:
【0224】
【化6】
【0225】
保護アルキン、(S)−1−{[1’−[1−(2−プロピニル)]−(1’2’−ジヒドロ−2’−オキソスピロ(1,3−ジオキサン−2,3’−[3H]インドール)−5’−スルホニル}−2−(2,4−ジフルオロフェノキシメチル)−ピロリジン(化合物39)、及び保護トリアゾール(化合物40)を図20に要約されるようにして合成した。
【0226】
一般的な実験の詳細は、本実施例の最初におよびSmith et al29に既に記載されている。全ての反応は、アルゴン雰囲気下で行われた。
【0227】
(S)−1−{[1’−[1−(2−プロピニル)]−(1’2’−ジヒドロ−2’−オキソスピロ(1,3−ジオキサン−2,3’−[3H]インドール)−5’−スルホニル}−2−(2,4−ジフルオロフェノキシメチル)−ピロリジン(化合物39)を、下記方法に従って製造した。無水トルエン(6mL)における化合物25(92mg、0.2mmol)の溶液に、1,3−プロパンジオール(0.3mL)及び4−トルエンスルホン酸(10mg、0.005mmol)を添加した。次に、形成した水を共沸蒸留によって除去しながら、この溶液を24時間還流した。さらに、この反応混合物を、室温まで冷却して、バルク溶媒を減圧下で除去した。次に、サンプルをDCM(10mL)に再溶解し、飽和Na2CO3(1×10mL)、水(1×10mL)及びブライン(1×10mL)で洗浄した後、Na2SO4で乾燥した。カラムクロマトグラフィー(2:1 酢酸エチル/ヘキサン)によって、目的とする生成物を第一の画分として得た(無色の油、46mg、44%)。HRMS (ESI) = 519.1393 (M + H)+. C25H25F2N2O6Sに関する算出値 519.1401. 1H NMR (400 MHz, CDCl3) δ 7.90 (d, J = 1.8 Hz, 1 H), 7.87 (dd, J = 8.2 Hz, 1.8 Hz, 1 H), 7.09 (d, J = 8.2 Hz, 1 H), 7.04-6.98 (m, 1 H), 6.88-6.78 (m, 1 H), 4.93 (t, J = 12.1 Hz, 2 H), 4.45 (d, J = 2.4 Hz, 2 H), 4.32-4.27 (m, 1 H), 3.99-3.93 (m, 4 H), 3.56-3.49 (m, 1 H), 3.14-3.08 (m, 1 H), 2.43-2.37 (m, 1 H), 2.30 (t, J = 2.4 Hz, 1 H), 2.09-1.93 (m, 2 H), 1.78-1.64 (m, 3 H)。
【0228】
(S)−1−{[1’−[1−(2−フルオロエチル)−1H−[1,2,3]−トリアゾール−4−イル]−(1’2’−ジヒドロ−2’−オキソスピロ(1,3−ジオキサン−2,3’−[3H]インドール)−5’−スルホニル}−2−(2,4−ジフルオロフェノキシメチル)−ピロリジン(化合物40)を、下記方法に従って製造した。無水トルエン(4mL)における(S)−1−((1−(2−フルオロエチル)−1H−[1,2,3]−トリアゾール−4−イル)メチル)−5−(2(2,4−ジフルオロフェノキシメチル)−ピロリジン−1−スルホニル)イサチン)(化合物11)(12mg、0.02mmol)の溶液に、1,3−プロパンジオール(0.15mL)及び4−トルエンスルホン酸(1mg、0.005mmol)を添加した。次に、形成した水を共沸蒸留によって除去しながら、この溶液を24時間還流した。さらに、この反応混合物を、室温まで冷却して、バルク溶媒を減圧下で除去した。次に、サンプルをDCM(10mL)に再溶解し、飽和Na2CO3(1×10mL)、水(1×10mL)及びブライン(1×10mL)で洗浄した後、Na2SO4で乾燥した。カラムクロマトグラフィー(4:1 酢酸エチル/ヘキサン)によって、目的とする生成物を第一の画分として得た(無色の油、7mg、58%)。HRMS (ESI) = 519.1393 (M + H)+; C25H25F2N2O6S 519.1401に関する算出値. 1H NMR (400 MHz, CDCl3) δ.7.84 (s, 1 H), 7.81 (d, J = 1.6 Hz, 1 H), 7.67 (s, 1 H), 7.43 (dd, J = 8.4 Hz, 1.6 Hz, 1 H), 7.04-6.98 (m, 1 H), 6.88-6.79 (m, 1 H), 4.96 (s, 1 H), 4.92 (t, J = 2.8 Hz, 2 H), 4.79 (dt, J = 46.6 Hz, 4.8 Hz, 2 H), 4.64 (dt, J = 27 Hz, 4.8 Hz, 2 H), 4.29 (dd, J = 8.4 Hz, 2.6 Hz, 1 H), 4.00-3.89 (m, 4 H), 3.53-3.48 (m, 1 H), 3.10-3.07 (m, 1 H), 2.43-2.37 (m, 1 H), 2.05-1.92 (m, 2 H), 1.75-1.65 (m, 3 H)。
【0229】
次に、[18F]11は、1以上のこれらの保護前駆体を用いて合成できる。保護アルキン前駆体39を用いた具体的な放射化学反応を図21に示す。このような保護アルキン前駆体を使用することによって、C−3位置での望ましく副反応を防止する。
【0230】
硫酸銅(II)・6水和物(0.51mg、2.06μmol、25μL)の水溶液に、窒素下で、リン酸ナトリウム緩衝液(pH6.0、250mM、25μL)におけるアスコルビン酸ナトリウム(2.53mg,12.79μmol)、DMF(25μl)におけるアルキンアセタール前駆体(1.0mg、1.93μmol)(化合物39)、及びアセトニトリル(100μL)における[18F]−フルオロエチルアジド(1.27mCi)(化合物27)を添加した。この反応混合物を80℃で30分間加熱した(図22参照)。塩酸(6N、100μL)を添加した後、攪拌混合物をマイクロ波空洞(2s、50W、設定温度 80℃)を用いて加熱した。HPLC移動相(50μL、35% MeCNを用いた50% MeOH含む水)を添加した後、混合物を予備HPLCによって精製した(図23参照)。[18F]11を24%の減衰補正した放射化学的収率(出発[18F]−フルオロエチルアジドに対する)で単離し、PBS/10%EtOHでC18−SepPak SPSを用いて配合した。[18F]11と共に溶出する安定した不純物のレベルは0.84μg/mLであった。比放射能は0.05Ci/μmol(2GBq/μmol)であった(図24参照)。
【0231】
類似の保護先駆体を用いて本発明の他の化合物を合成できることはいうまでもない。
【0232】
実施例11−アポトーシス細胞への[18F]11及び[18F]42の取り込みの比較
本プロジェクトのさらなる態様は、弱いイサチンカスパーゼ−3阻害剤であるN−[1−(2−フルオロエチル)−1H−[1,2,3]−トリアゾール−4−イル]イサチン(化合物42)の分析を包含する。この化合物を、図25に示されるようにして、N−(2−プロピニル)イサチン(化合物41)を用いて合成した。化合物[18F]42は、コアのイサチンフレームワークを有するため、この化合物は公知のイサチン結合形式で活性化カスパーゼ−3に結合でき、また、放射性標識トリアゾール機能性がありうる。しかしながら、[18F]42は、カスパーゼ−3に対する高い親和性及び選択性を付与するピロリジンスルホンアミド機能性がない。[18F]42を、[18F]11と直接比較するために、細胞取り込みアッセイで調べた。この目的は、取り込みが細胞透過性の増加の結果、アポトーシス細胞の特性、あるいはカスパーゼ−3結合の結果であるのかを決定する目的で、化合物の取り込みプロフィールを調べることである。[18F]11とは異なり、薬剤処置後に[18F]42結合性の増加はなかった(図26参照)ことから、[18F]11結合はカスパーゼ3活性化によるものであるという知見が支持された。
【0233】
N−(2−プロピニル)イサチン(化合物41)を、Smith et al29に記載されるのと同様にして合成した(アルキン12及び13)。1H NMR (600 MHz, CDCl3) δ 7.65-7.61 (m, 2 H), 7.19-7.12 (m, 2 H), 4.61 (s, 2 H), 2.30 (s, 1 H)。
【0234】
N−[1−(2−フルオロエチル)−1H−[1,2,3]−トリアゾール−4−イル]イサチン(化合物42)を、Smith et al29に記載されるのと同様にして合成した(トリアゾール14及び15)。1H NMR (600 MHz, CDCl3) δ 7.76 (s, 1 H), 7.63-7.57 (m, 2 H), 7.34 (d, J = 8.4 Hz, 1 H), 7.13 (t, J = 3.6 Hz, 1 H), 5.03 (s, 2 H), 4.81 (dt, J = 46.8 Hz, 4.8 Hz, 2 H), 4.68 (dt, J = 27 Hz, 4.8 Hz, 2 H)。
【0235】
注意(N.B.) 番号の間違いにより、化合物15及び21は、実際同じ化合物である。
【0236】
参考文献
1. Lahorte et al. (2004) European Journal of Nuclear Medicine and Molecular Imaging. 31. 887 - 919.
2. Boersma et al. (2005) Journal of Nuclear Medicine. 46, 2035 - 2050.
3. Aloya, R. et al. (2006) アポトーシス. 11, 2089 - 2101.
4. Damianovich, M. et al. (2006) European Journal of Nuclear Medicine and Molecular Imaging.. 33, 281 - 291.
5. WO 2005/067388
6. Neuss, M. et al (2001). Cardiovascular Drugs and Therapy. 15, 507-523.
7. Lee, D. et al (2000) Journal of Biological Chemistry. 275, 16007 - 16014.
8. Lee, D. et al. (2001) Journal of Medicinal Chemistry.44, 2015-2026
9. Chu, W. et al (2005) Journal of Medicinal Chemistry. 48. 7637 - 7647
10. Chu, W. et al (2007) Journal of Medicinal Chemistry. 50. 3751 - 3755.
11. WO 2005/053752
12. Faust, A. et al (2007) Quarterly Journal of Nuclear Medicine and Molecular Imaging. 51. 67 - 71
13. Kopka, K. et al (2006) Journal of Medicinal Chemistry. 49, 6704 - 6715.
14. Zhou, D. et al (2006) Bioorganic and Medicinal Chemistry Letters. 16, 5041 - 5046.
15. WO 2006/067376
16. Glaser et al (2007) Bioconjugate Chemistry. 18. 989 - 993
17. Wipf, P. et al (2001) Tetrahedron. 57, 283 - 296.
18. Barthel, H. et al (2004) British Journal of Cancer. 90, 2232 - 2242.
19. Aboagye, E.O. et al, (1997) Biochemical Pharmacology. 54, 1217-1224.
20. Twentyman, P.R. et al.(1980) J Natl. Cancer Inst. 64, 595-604
21. Workman, P., et al (1998) British Journal of Cancer. 77, 1-10.
22. Wolf, C.R. et al. (1987) Int. J. Cancer. 39, 695-702
23. Langdon, S.P. et al (1988) Cancer Research. 48, 6166-6172
24. Kozaki, K et al (2000) Cancer Research. 60, 2535-40
25. Schirrmacher, R. et al (2008) Tet. Lett. 49, 4824 - 4827.
26. Marik, J. and Sutcliffe, J. L. (2006) Click for PET: Rapid preparation of [18F]fluoropeptides using CuIcatalyzed 1,3-dipolar cycloaddition. Tet. Lett. 47, 6681-6684
27. Sirion, U. et al, (2007) An efficient F-18 labeling method for PET study: Huisgen 1,3-dipolar cycloaddition of bioactive substances and F-18-labeled compounds. Tet. Lett., 48, 3953-3957
28. Li, Z, et al. (2007) Click Chemistry for 18F-Labeling of RGD Peptides and microPET Imaging of Tumor Integrin αvβ3 expression. Bioconjugate Chem., 18, 1987-1994.
29. Smith G. et al, (2008) Design, Synthesis and Biological Characterization of a Caspase 3/7 Selective Isatin Labeled with 2-[18F]fluoroethylazide. Journal of Medicinal Chemistry. 51. 8057-8067
【特許請求の範囲】
【請求項1】
下記式Aを有する化合物、またはこの塩、水和物もしくはプロドラッグ:
【化1】
ただし、Rは、フェニル、3−フルオロフェニル、2,4−ジフルオロフェニル、3,5−ジフルオロフェニル、置換されてもよいテトラヒドロピラン、置換されてもよいジアジンまたは置換されてもよいトリアゾールであり;
R’は、置換されてもよいフェニルまたは置換されてもよいトリアゾールであり;
この際、Rがフェニルである場合には、R’は、置換されてもよいトリアゾールである。
【請求項2】
前記置換されてもよいフェニル、置換されてもよいテトラヒドロピランおよび/または置換されてもよいジアジンは、必要であれば、1以上の電子求引基で置換される、請求項1に記載の化合物。
【請求項3】
前記電子求引基は、フッ素である、請求項2に記載の化合物。
【請求項4】
前記置換されてもよいトリアゾールは、必要であれば、置換されたアルキル基で置換される、請求項1〜3のいずれか1項に記載の化合物。
【請求項5】
前記置換されたアルキル基はC1−4のフルオロアルキルである、請求項4に記載の化合物。
【請求項6】
前記C1−4のフルオロアルキルは2−フルオロエチルである、請求項5に記載の化合物。
【請求項7】
Rは、3−フルオロフェニル、2,4−ジフルオロフェニルおよび3,5−ジフルオロフェニルを含む群より選択され、R’は、2−フルオロエチルで置換されたトリアゾールである、請求項1〜6のいずれか1項に記載の化合物。
【請求項8】
前記RおよびR’は下記に定義される、請求項1に記載の化合物。
【表1−1】
【表1−2】
【表1−3】
【請求項9】
さらに画像化部分(imaging moiety)を有する、請求項1〜8のいずれか1項に記載の化合物。
【請求項10】
前記画像化部分(imaging moiety)は放射性標識である、請求項9に記載の化合物。
【請求項11】
前記放射性標識は18Fである、請求項10に記載の化合物。
【請求項12】
下記式[18F]11を有する、請求項11に記載の化合物。
【化2】
【請求項13】
下記式35を有する、請求項10に記載の化合物。
【化3】
【請求項14】
下記式36を有する、請求項10に記載の化合物。
【化4】
【請求項15】
請求項1〜8のいずれか1項に記載の化合物および必要であれば、1以上の別の活性成分、製薬上許容できる賦形剤、担体または希釈剤を含む、薬剤組成物。
【請求項16】
請求項9〜14のいずれか1項に記載の化合物および必要であれば、1以上の別の活性成分、製薬上許容できる賦形剤、担体または希釈剤を含む、薬剤組成物。
【請求項17】
前記化合物が実質的にS鏡像異性体から構成される、請求項15または16に記載の薬剤組成物。
【請求項18】
分子画像化剤として使用されるための、請求項9〜14のいずれか1項に記載の化合物または請求項16に記載の薬剤組成物。
【請求項19】
前記分子画像化剤がカスパーゼ活性の可視化および定量化を目的とする、請求項18に記載の化合物または薬剤組成物。
【請求項20】
a)細胞または組織を請求項9〜14のいずれか1項に記載の化合物または請求項16に記載の薬剤組成物と接触させ;
b)被検者を検出装置の検出域に置き;さらに
c)被検者における前記化合物を前記検出装置で検出する、
ことを有する、細胞または組織におけるカスパーゼ活性の分子画像化(molecular imaging)方法。
【請求項21】
a)被検者に請求項9〜14のいずれか1項に記載の化合物または請求項16に記載の薬剤組成物を投与し;
b)被検者を検出装置の検出域に置き;さらに
c)被検者における前記化合物を前記検出装置で検出する、
ことを有する、被検者におけるアポトーシスのインビボでの画像化方法。
【請求項22】
前記化合物は非侵襲的に被検者で検出される、請求項21に記載のインビボでの方法。
【請求項23】
a)哺乳動物の細胞または組織を請求項9〜14のいずれか1項に記載の化合物または請求項16に記載の薬剤組成物と接触させ;
b)前記哺乳動物の細胞または組織を検出装置の検出域に置き;
c)前記被検者における前記化合物を前記検出装置で検出し;さらに
d)段階a)、b)およびc)を繰り返す、
ことを有する、哺乳動物の細胞または組織におけるカスパーゼ活性への試験物質の治療効果の評価方法。
【請求項24】
前記試験物質は、心血管疾患、神経疾患およびHIVを治療する薬剤を含む群から選択される、請求項23に記載の方法。
【請求項25】
前記化合物は放射性標識され、前記検出装置は放射線検出装置であり、この際、前記放射性標識された化合物は放射線検出装置を用いて前記放射性標識された化合物から放出される放射線を測定することによって検出される、請求項23または24に記載の方法。
【請求項26】
前記化合物または薬剤組成物は、前記試験物質の前に、後にまたはと同時に前記哺乳動物の細胞または組織と接触させる、請求項23〜25のいずれか1項に記載の方法。
【請求項27】
段階(a)〜(c)は所定の間隔で繰り返され、この際、前記繰り返しは経時的なカスパーゼ活性の変化を追跡するのに効果的である、請求項23〜26のいずれか1項に記載の方法。
【請求項28】
1回目の段階(a)〜(c)を前記試験物質の投与前に被検者で行って、カスパーゼ活性の1回目の測定結果を得、次に試験物質を投与し、2回目の段階(a)〜(c)を所定の間隔で前記試験物質を投与した後に前記被検者で行って、カスパーゼ活性の2回目の測定結果を得る、請求項27に記載の方法。
【請求項29】
1回目の段階(a)で投与される前記化合物は請求項13または14に記載の化合物である、請求項28に記載の方法。
【請求項30】
前記カスパーゼ活性はカスパーゼ−3活性である、請求項19〜29のいずれか1項に記載の方法。
【請求項31】
薬剤に使用されるための、請求項1〜8のいずれか1項に記載の化合物または請求項15に記載の組成物。
【請求項32】
カスパーゼ活性の阻害に使用される、請求項31に記載の化合物または組成物。
【請求項33】
神経変性疾患、血液病、肝細胞の変性、変形性関節炎、AIDS、虚血、同種移植の拒絶反応を含む群より選択される疾患または症状の治療に使用される、請求項32に記載の化合物または組成物。
【請求項34】
a)哺乳動物の細胞または組織を請求項9〜14のいずれか1項に記載の化合物または請求項16に記載の薬剤組成物と接触させ;
b)前記哺乳動物の細胞または組織を検出装置の検出域に置き;さらに
c)前記化合物を前記検出装置で検出して、前記哺乳動物の細胞または組織内のカスパーゼ活性の定量的な測定結果を得、この際、カスパーゼ活性はアポトーシスのレベルの指標である、
ことを有する、過剰なまたは不適切なアポトーシスによって引き起こされるまたは前記アポトーシスと関連のある疾患または障害の診断方法。
【請求項35】
前記疾患または障害は、慢性心不全、急性心筋梗塞、発作、神経変性疾患、自己免疫疾患、限局性血液病(focal haematologic disease)、限局性AIDS、心虚血を含む虚血、および移植片拒絶反応からなる群より選択される、請求項34に記載の方法。
【請求項36】
請求項1〜14のいずれか1項に記載の化合物の合成方法。
【請求項37】
下記式38(ただし、n=0〜6)を有する化合物。
【化5】
【請求項38】
下記式39を有する化合物。
【化6】
【請求項39】
下記式43(ただし、n=0〜6およびx=脱離基)を有する化合物。
【化7】
【請求項40】
請求項12に記載の化合物の製造における請求項37、38または39に記載の化合物の使用。
【請求項1】
下記式Aを有する化合物、またはこの塩、水和物もしくはプロドラッグ:
【化1】
ただし、Rは、フェニル、3−フルオロフェニル、2,4−ジフルオロフェニル、3,5−ジフルオロフェニル、置換されてもよいテトラヒドロピラン、置換されてもよいジアジンまたは置換されてもよいトリアゾールであり;
R’は、置換されてもよいフェニルまたは置換されてもよいトリアゾールであり;
この際、Rがフェニルである場合には、R’は、置換されてもよいトリアゾールである。
【請求項2】
前記置換されてもよいフェニル、置換されてもよいテトラヒドロピランおよび/または置換されてもよいジアジンは、必要であれば、1以上の電子求引基で置換される、請求項1に記載の化合物。
【請求項3】
前記電子求引基は、フッ素である、請求項2に記載の化合物。
【請求項4】
前記置換されてもよいトリアゾールは、必要であれば、置換されたアルキル基で置換される、請求項1〜3のいずれか1項に記載の化合物。
【請求項5】
前記置換されたアルキル基はC1−4のフルオロアルキルである、請求項4に記載の化合物。
【請求項6】
前記C1−4のフルオロアルキルは2−フルオロエチルである、請求項5に記載の化合物。
【請求項7】
Rは、3−フルオロフェニル、2,4−ジフルオロフェニルおよび3,5−ジフルオロフェニルを含む群より選択され、R’は、2−フルオロエチルで置換されたトリアゾールである、請求項1〜6のいずれか1項に記載の化合物。
【請求項8】
前記RおよびR’は下記に定義される、請求項1に記載の化合物。
【表1−1】
【表1−2】
【表1−3】
【請求項9】
さらに画像化部分(imaging moiety)を有する、請求項1〜8のいずれか1項に記載の化合物。
【請求項10】
前記画像化部分(imaging moiety)は放射性標識である、請求項9に記載の化合物。
【請求項11】
前記放射性標識は18Fである、請求項10に記載の化合物。
【請求項12】
下記式[18F]11を有する、請求項11に記載の化合物。
【化2】
【請求項13】
下記式35を有する、請求項10に記載の化合物。
【化3】
【請求項14】
下記式36を有する、請求項10に記載の化合物。
【化4】
【請求項15】
請求項1〜8のいずれか1項に記載の化合物および必要であれば、1以上の別の活性成分、製薬上許容できる賦形剤、担体または希釈剤を含む、薬剤組成物。
【請求項16】
請求項9〜14のいずれか1項に記載の化合物および必要であれば、1以上の別の活性成分、製薬上許容できる賦形剤、担体または希釈剤を含む、薬剤組成物。
【請求項17】
前記化合物が実質的にS鏡像異性体から構成される、請求項15または16に記載の薬剤組成物。
【請求項18】
分子画像化剤として使用されるための、請求項9〜14のいずれか1項に記載の化合物または請求項16に記載の薬剤組成物。
【請求項19】
前記分子画像化剤がカスパーゼ活性の可視化および定量化を目的とする、請求項18に記載の化合物または薬剤組成物。
【請求項20】
a)細胞または組織を請求項9〜14のいずれか1項に記載の化合物または請求項16に記載の薬剤組成物と接触させ;
b)被検者を検出装置の検出域に置き;さらに
c)被検者における前記化合物を前記検出装置で検出する、
ことを有する、細胞または組織におけるカスパーゼ活性の分子画像化(molecular imaging)方法。
【請求項21】
a)被検者に請求項9〜14のいずれか1項に記載の化合物または請求項16に記載の薬剤組成物を投与し;
b)被検者を検出装置の検出域に置き;さらに
c)被検者における前記化合物を前記検出装置で検出する、
ことを有する、被検者におけるアポトーシスのインビボでの画像化方法。
【請求項22】
前記化合物は非侵襲的に被検者で検出される、請求項21に記載のインビボでの方法。
【請求項23】
a)哺乳動物の細胞または組織を請求項9〜14のいずれか1項に記載の化合物または請求項16に記載の薬剤組成物と接触させ;
b)前記哺乳動物の細胞または組織を検出装置の検出域に置き;
c)前記被検者における前記化合物を前記検出装置で検出し;さらに
d)段階a)、b)およびc)を繰り返す、
ことを有する、哺乳動物の細胞または組織におけるカスパーゼ活性への試験物質の治療効果の評価方法。
【請求項24】
前記試験物質は、心血管疾患、神経疾患およびHIVを治療する薬剤を含む群から選択される、請求項23に記載の方法。
【請求項25】
前記化合物は放射性標識され、前記検出装置は放射線検出装置であり、この際、前記放射性標識された化合物は放射線検出装置を用いて前記放射性標識された化合物から放出される放射線を測定することによって検出される、請求項23または24に記載の方法。
【請求項26】
前記化合物または薬剤組成物は、前記試験物質の前に、後にまたはと同時に前記哺乳動物の細胞または組織と接触させる、請求項23〜25のいずれか1項に記載の方法。
【請求項27】
段階(a)〜(c)は所定の間隔で繰り返され、この際、前記繰り返しは経時的なカスパーゼ活性の変化を追跡するのに効果的である、請求項23〜26のいずれか1項に記載の方法。
【請求項28】
1回目の段階(a)〜(c)を前記試験物質の投与前に被検者で行って、カスパーゼ活性の1回目の測定結果を得、次に試験物質を投与し、2回目の段階(a)〜(c)を所定の間隔で前記試験物質を投与した後に前記被検者で行って、カスパーゼ活性の2回目の測定結果を得る、請求項27に記載の方法。
【請求項29】
1回目の段階(a)で投与される前記化合物は請求項13または14に記載の化合物である、請求項28に記載の方法。
【請求項30】
前記カスパーゼ活性はカスパーゼ−3活性である、請求項19〜29のいずれか1項に記載の方法。
【請求項31】
薬剤に使用されるための、請求項1〜8のいずれか1項に記載の化合物または請求項15に記載の組成物。
【請求項32】
カスパーゼ活性の阻害に使用される、請求項31に記載の化合物または組成物。
【請求項33】
神経変性疾患、血液病、肝細胞の変性、変形性関節炎、AIDS、虚血、同種移植の拒絶反応を含む群より選択される疾患または症状の治療に使用される、請求項32に記載の化合物または組成物。
【請求項34】
a)哺乳動物の細胞または組織を請求項9〜14のいずれか1項に記載の化合物または請求項16に記載の薬剤組成物と接触させ;
b)前記哺乳動物の細胞または組織を検出装置の検出域に置き;さらに
c)前記化合物を前記検出装置で検出して、前記哺乳動物の細胞または組織内のカスパーゼ活性の定量的な測定結果を得、この際、カスパーゼ活性はアポトーシスのレベルの指標である、
ことを有する、過剰なまたは不適切なアポトーシスによって引き起こされるまたは前記アポトーシスと関連のある疾患または障害の診断方法。
【請求項35】
前記疾患または障害は、慢性心不全、急性心筋梗塞、発作、神経変性疾患、自己免疫疾患、限局性血液病(focal haematologic disease)、限局性AIDS、心虚血を含む虚血、および移植片拒絶反応からなる群より選択される、請求項34に記載の方法。
【請求項36】
請求項1〜14のいずれか1項に記載の化合物の合成方法。
【請求項37】
下記式38(ただし、n=0〜6)を有する化合物。
【化5】
【請求項38】
下記式39を有する化合物。
【化6】
【請求項39】
下記式43(ただし、n=0〜6およびx=脱離基)を有する化合物。
【化7】
【請求項40】
請求項12に記載の化合物の製造における請求項37、38または39に記載の化合物の使用。
【図1】

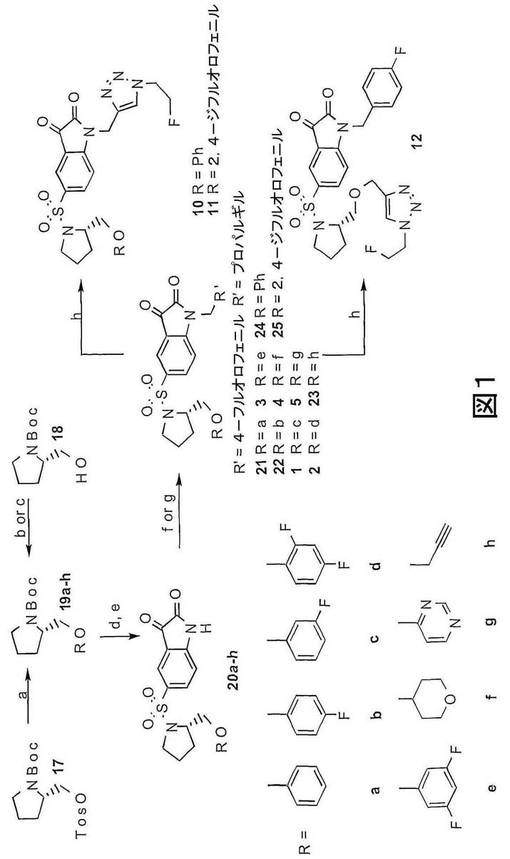
【図2−1】
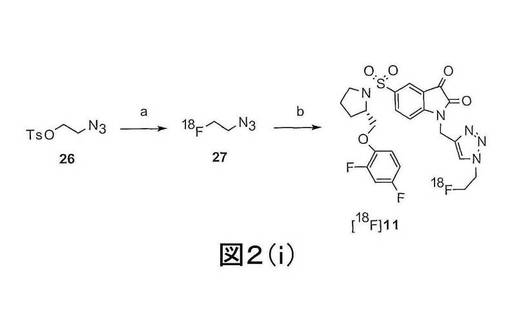
【図2−2】
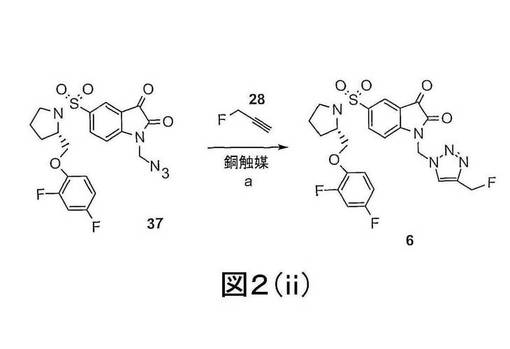
【図3−1】
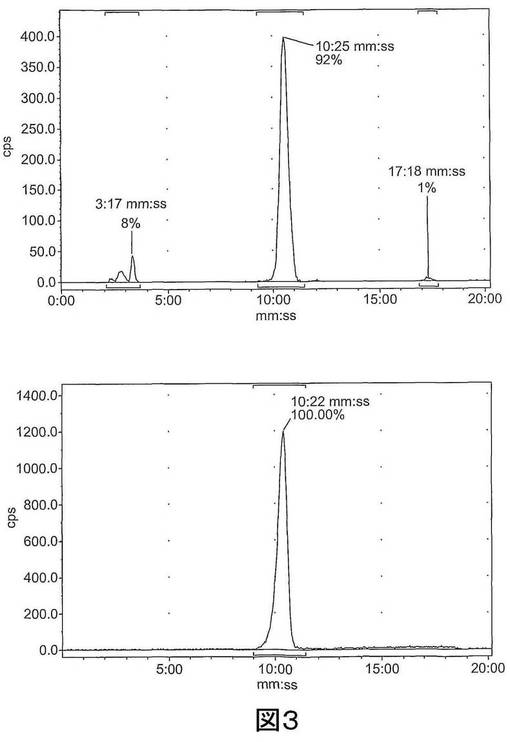
【図3−2】
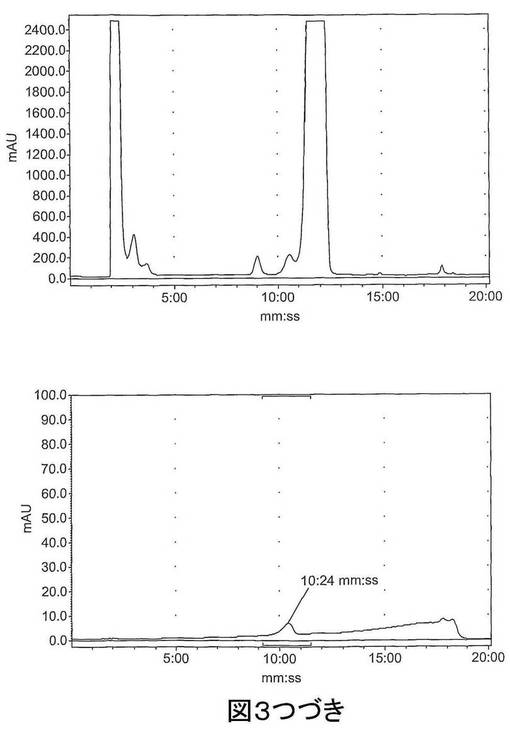
【図4】
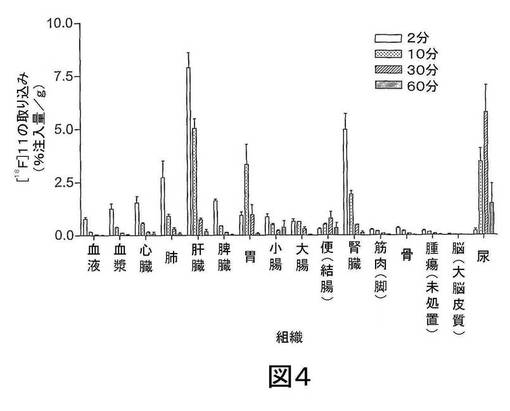
【図5−1】
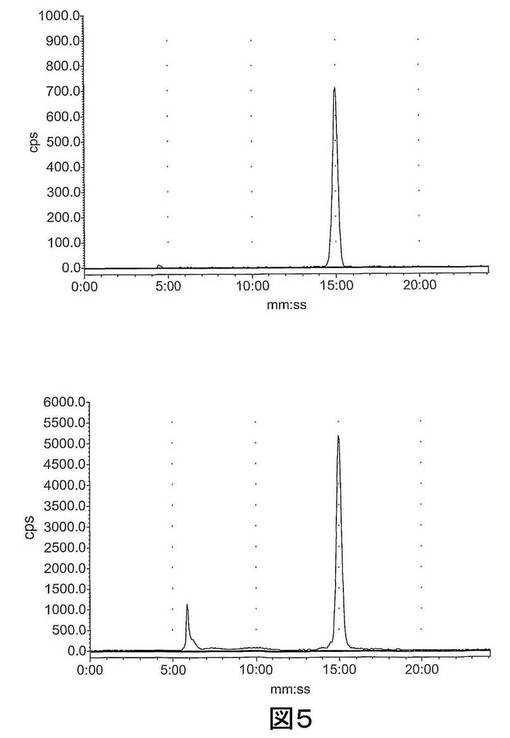
【図5−2】
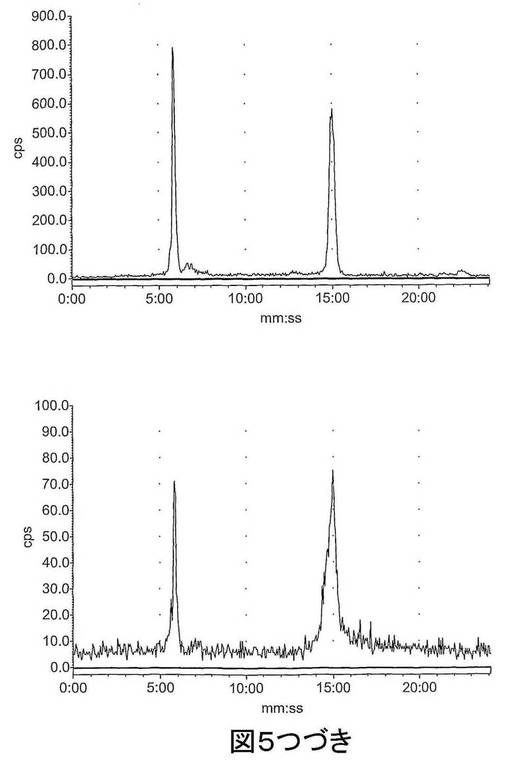
【図6−1】
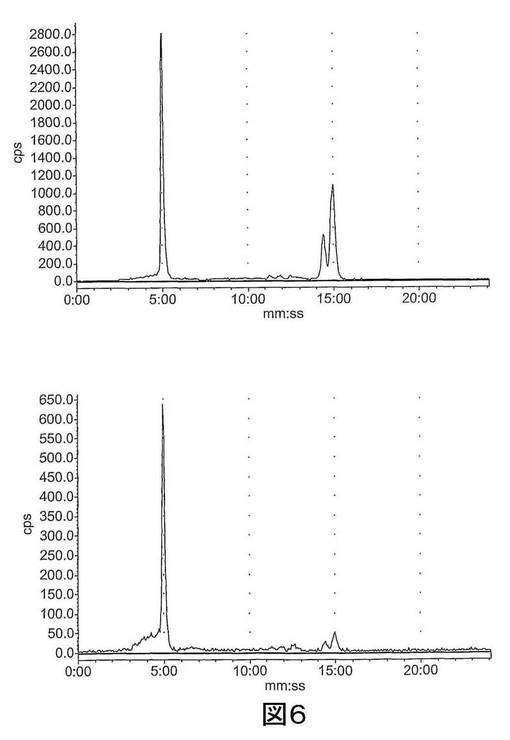
【図6−2】
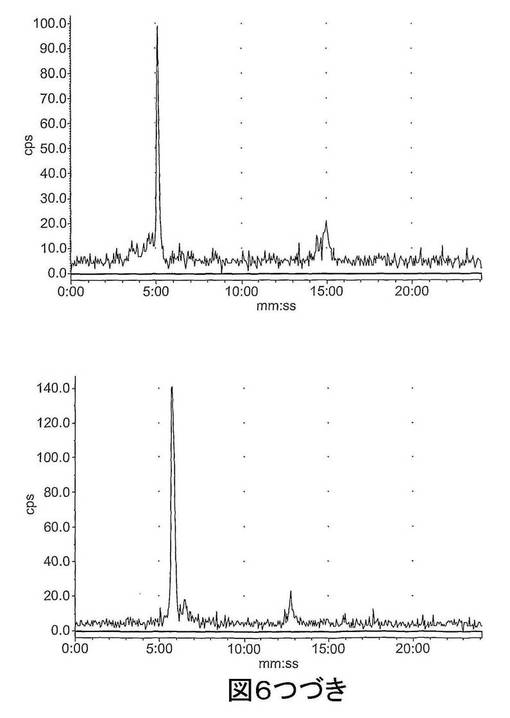
【図6−3】
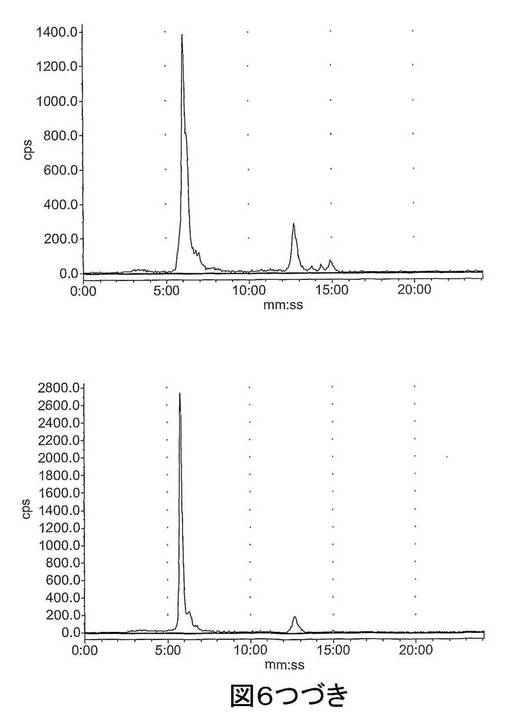
【図7】
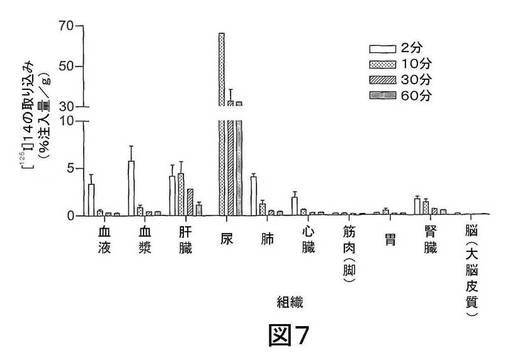
【図8−1】
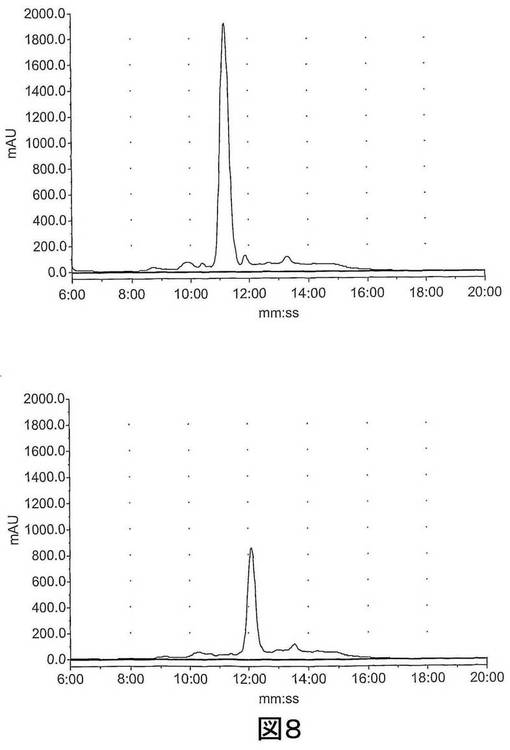
【図8−2】
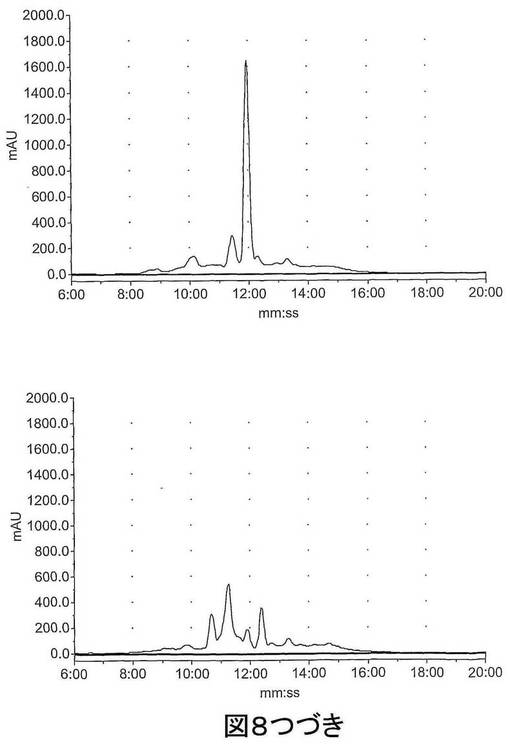
【図8−3】
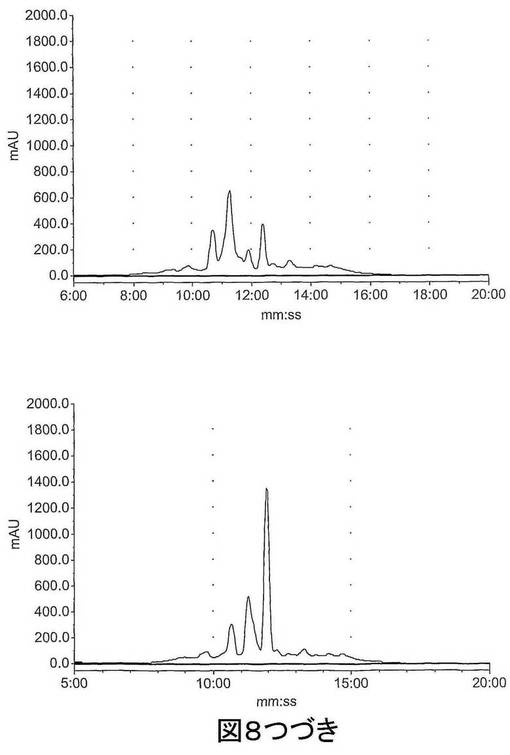
【図9】
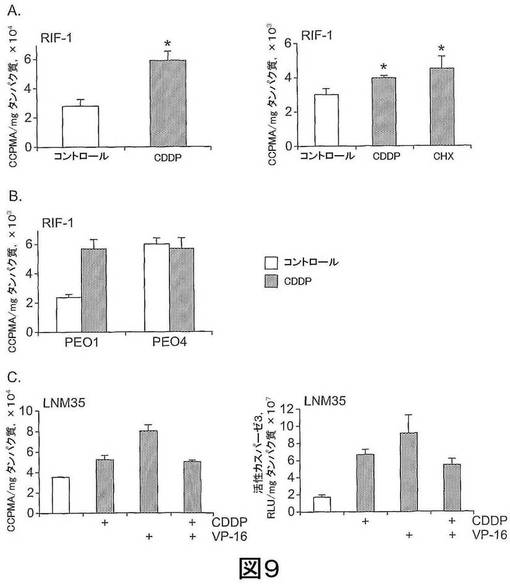
【図10】
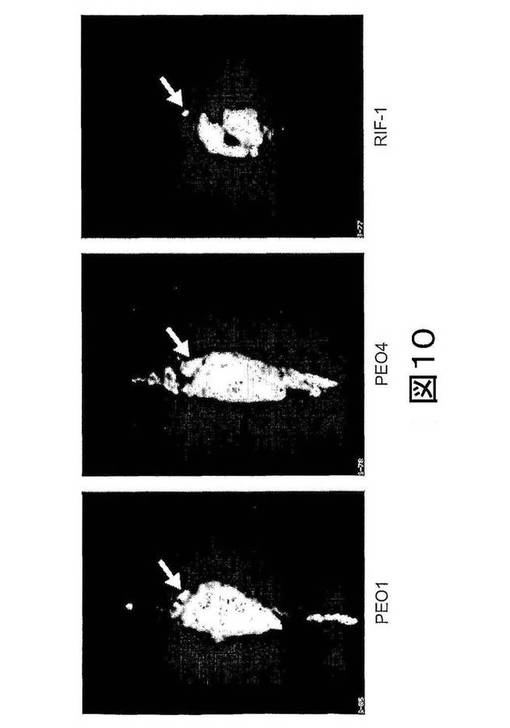
【図11】
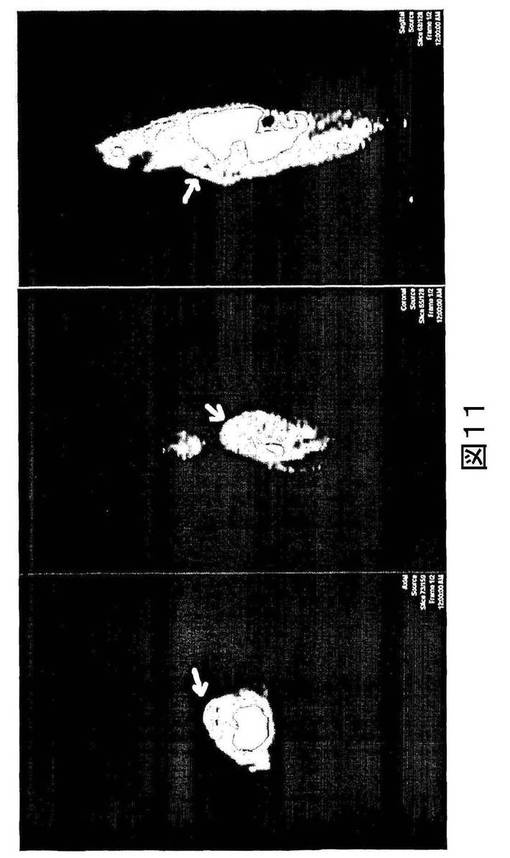
【図12】
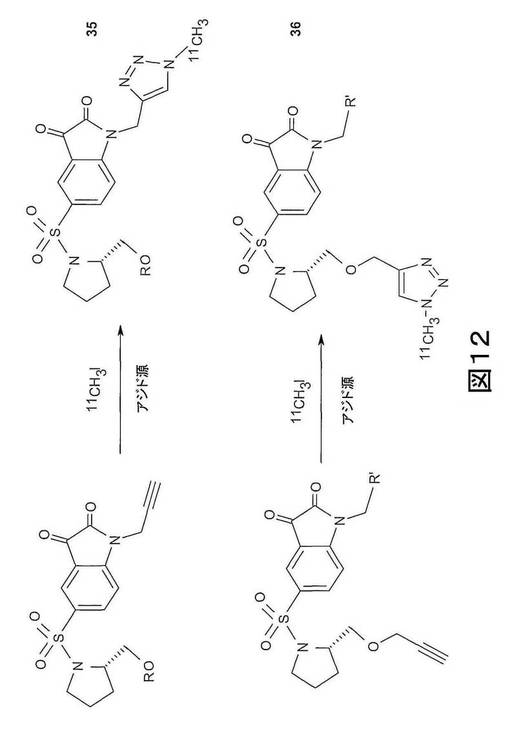
【図13】
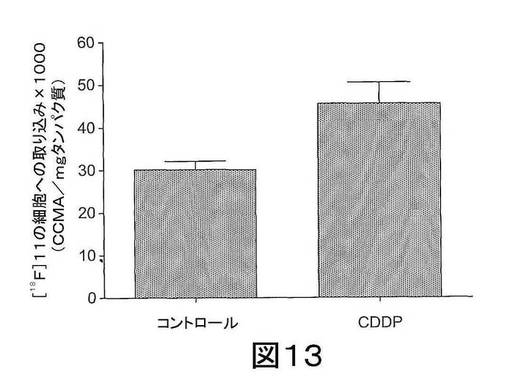
【図14】
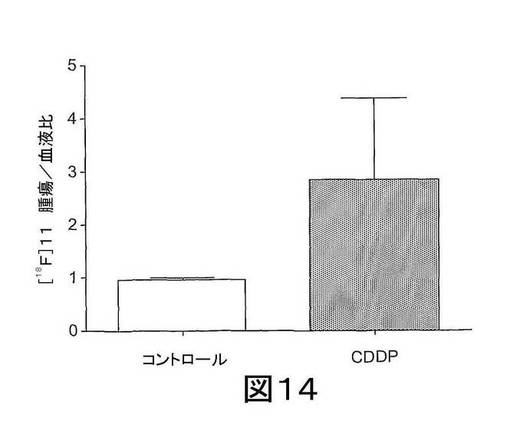
【図15】
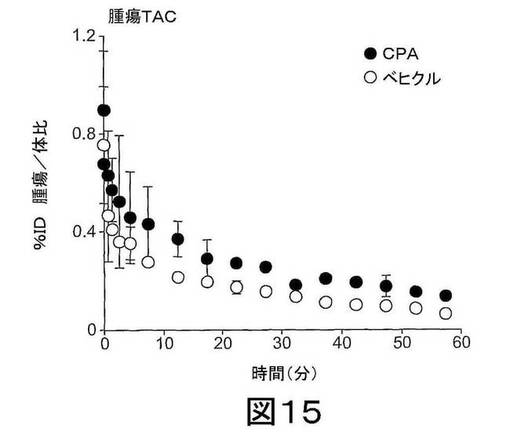
【図16】
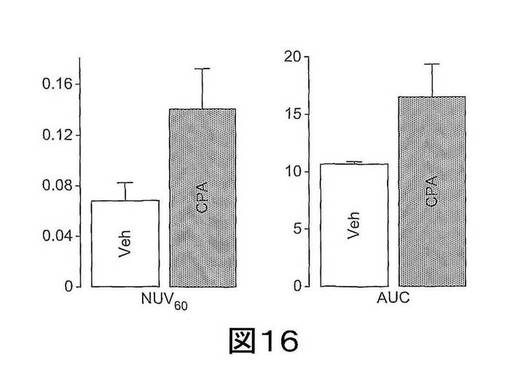
【図17】
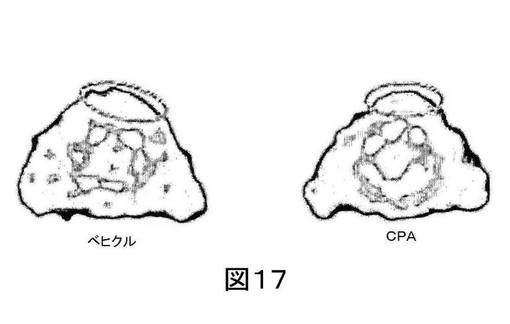
【図18】

【図19】
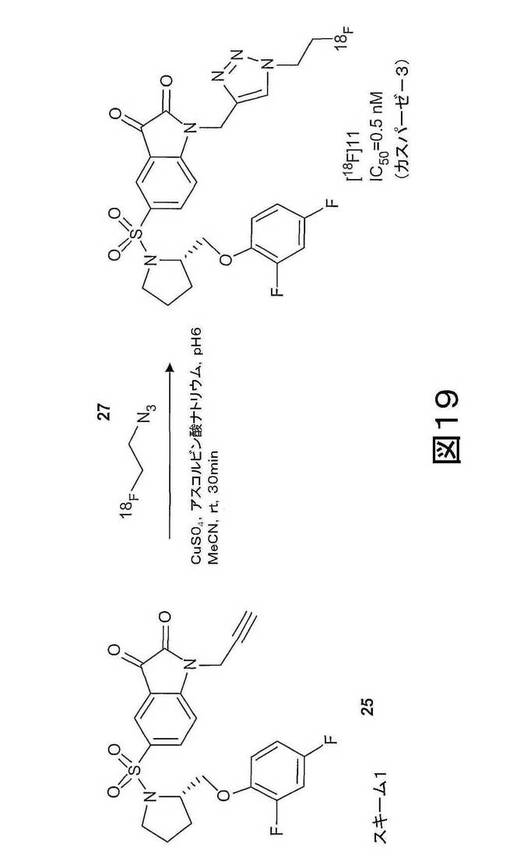
【図20】
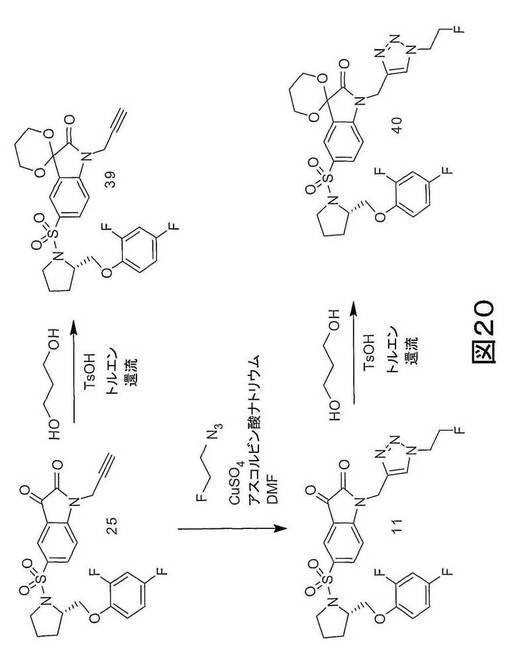
【図21】
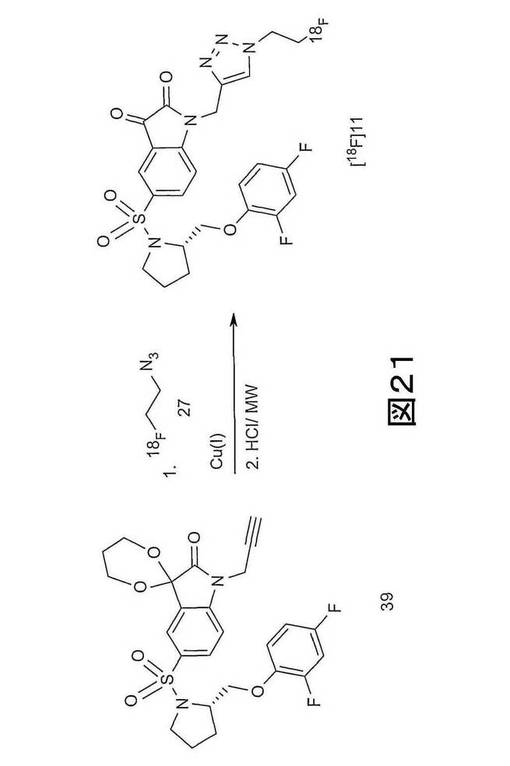
【図22】
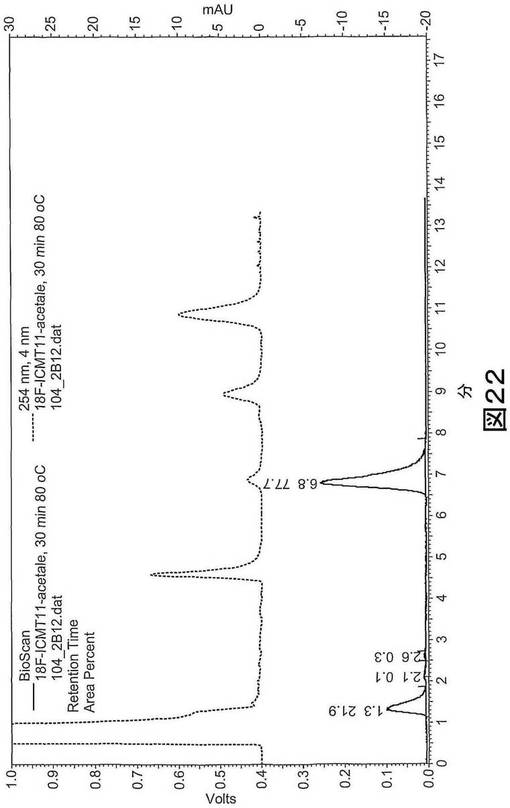
【図23】
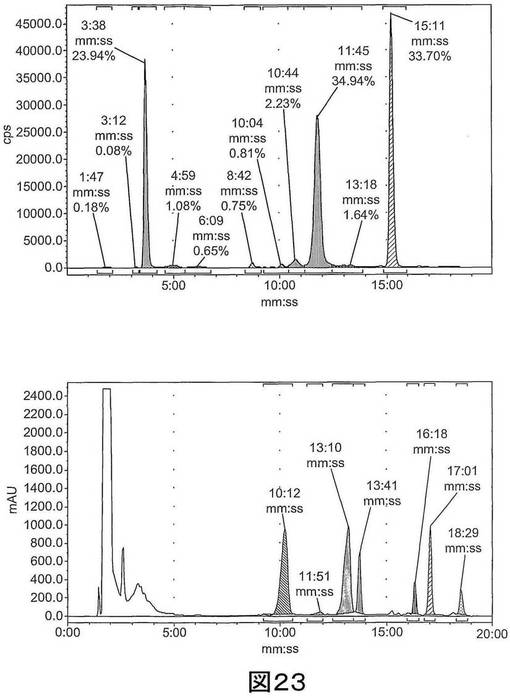
【図24】
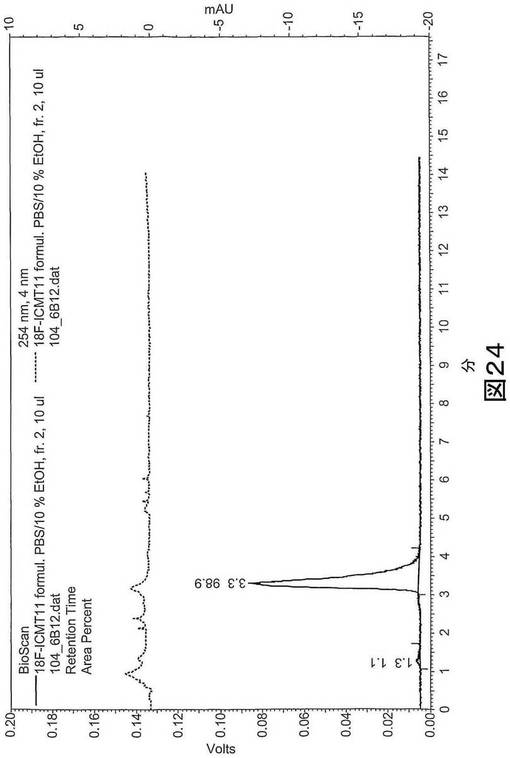
【図25】
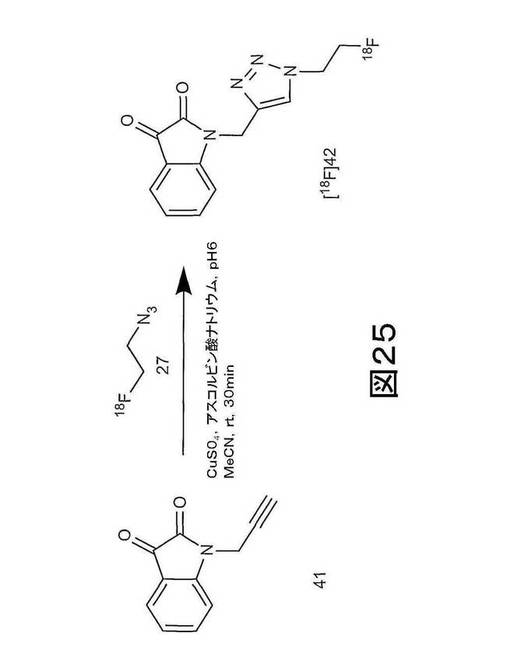
【図26】
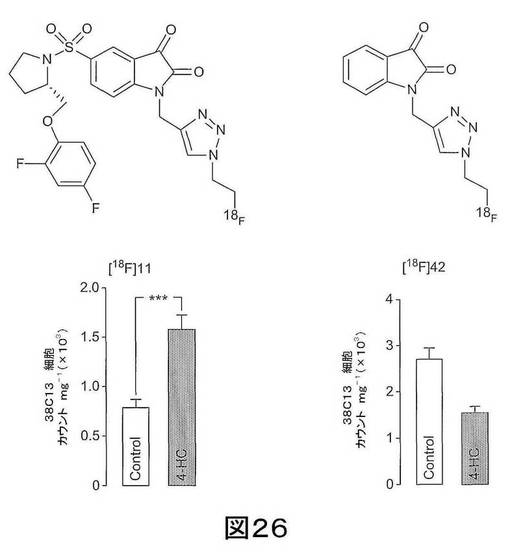
【図2−1】
【図2−2】
【図3−1】
【図3−2】
【図4】
【図5−1】
【図5−2】
【図6−1】
【図6−2】
【図6−3】
【図7】
【図8−1】
【図8−2】
【図8−3】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【公表番号】特表2012−502014(P2012−502014A)
【公表日】平成24年1月26日(2012.1.26)
【国際特許分類】
【出願番号】特願2011−525613(P2011−525613)
【出願日】平成21年9月4日(2009.9.4)
【国際出願番号】PCT/GB2009/002132
【国際公開番号】WO2010/026388
【国際公開日】平成22年3月11日(2010.3.11)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.WINDOWS
【出願人】(500165809)インペリアル・イノベイションズ・リミテッド (32)
【出願人】(504000591)ハマースミス・イメイネット・リミテッド (26)
【氏名又は名称原語表記】Hammersmith Imanet Ltd
【Fターム(参考)】
【公表日】平成24年1月26日(2012.1.26)
【国際特許分類】
【出願日】平成21年9月4日(2009.9.4)
【国際出願番号】PCT/GB2009/002132
【国際公開番号】WO2010/026388
【国際公開日】平成22年3月11日(2010.3.11)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.WINDOWS
【出願人】(500165809)インペリアル・イノベイションズ・リミテッド (32)
【出願人】(504000591)ハマースミス・イメイネット・リミテッド (26)
【氏名又は名称原語表記】Hammersmith Imanet Ltd
【Fターム(参考)】
[ Back to top ]