
インフルエンザワクチンを製造するためのプロセス
本発明は、予防目的、診断目的、免疫治療目的、治療目的のためのインフルエンザウイルスまたはインフルエンザウイルス抗原の生産のための、商用規模のプロセスに関する。本発明は、特に、インフルエンザワクチン生産のためのMadin−Darby Canine Kidney(MDCK)由来の細胞株、および細胞培養ベースのプロセスを提供し、そして特にインフルエンザA型およびインフルエンザB型を含むヒトワクチンを提供する。

【発明の詳細な説明】
【技術分野】
【0001】
本出願は、2004年5月20日に出願された米国特許仮出願第60/572,612号の利益を主張する。この出願は、本明細書中で参考として援用される。
【0002】
(発明の分野)
本発明は、一般に、予防目的、診断目的、免疫治療目的または治療目的のためのインフルエンザウイルスまたは抗原の製造のための商用規模のプロセスに関する。特に、本発明は、インフルエンザワクチン製造、そして特にインフルエンザA型およびインフルエンザB型を含むヒトワクチン製造のための、Madin−Darby Canine Kidney(MDCK)由来の細胞培養ベースのプロセスを提供する。
【背景技術】
【0003】
(発明の背景)
伝統的に、商用のインフルエンザワクチンは、胚含有の鶏卵においてワクチンウイルス種を増殖させることによって製造されている。このウイルスは、尿膜流体から収集され、そして処理されてワクチンを形成する。しかしながら、この手順は、大きな労力がかかり、そして卵当たりの収量が低いことを生じるという不利を有し、それらの要因は、流行期間の間に深刻な制限を提供する。従って、胚含有鶏卵の方法について、コスト、時間および収量の不利を克服する、インフルエンザワクチンの大規模製造のための必要性が存在する。
【0004】
上記の代替的な方法は、インフルエンザ粒子またはウイルスタンパク質を製造するための細胞培養の使用を含む。
【0005】
細胞培養を用いて製造されたインフルエンザワクチンは、卵において製造されたワクチンよりも安全であると考えられ、そして子供および成人により体験される、卵ベースのワクチンに対する過感受性を誘導しないはずである。このようなワクチンはまた、特に高齢者において、広範な種の野生株に対するよりよい保護を授けるはずである。なぜなら、ウイルス複製の忠実度は、卵におけるよりも細胞培養におけるほうがずっと大きいと考えられるからである(非特許文献1)。さらに、流行性および汎発性の場合において、卵の供給の制限に起因した、インフルエンザワクチンの現在可能である供給よりもより大きな供給が製造され得る。
【0006】
インフルエンザウイルスA型およびインフルエンザウイルスB型の伝播は、種々の組織培養系において実証されており、その培養系としては、細片化された(minced)ニワトリ胚、ヒト胎児肺およびヒト胎児腎臓、サル腎臓およびウシ胎仔腎臓が挙げられる。
【0007】
特に、イヌ腎臓(canine kidney)細胞は、インフルエンザウイルスの製造に関して有用であると示されているが、低収量であり、すなわち、ワクチン製造目的には不十分である。
【0008】
イヌ腎臓細胞は、そもそも、S.H.MadinおよびN.B.Darbyによって、外観上正常である、成体のメスCocker Spanielの腎臓より1958年に誘導された(非特許文献2)。このMDCK細胞株は、1964年にATCCにより(番号CCL34の下で)受託され、そしていくつかのウイルス(水疱性口内炎ウイルス、ワクシニアウイルス、コサッキーウイルス、レオウイルス、およびアデノウイルスが挙げられる)の複製を可能にするとして同定された。
【0009】
インフルエンザウイルスB型の連続伝播は、最初に、MDCK細胞株において、Greenによって1962年に実証された(非特許文献3)。伝播は、連続する6回の組織培養の継代の各々における細胞変性効果およびヘマグルチニン(HA)(インフルエンザウイルスの主要な糖タンパク質)の存在によって、そして組織培養流体における卵感染力価によって、明らかであった。しかしながら、これらのデータは小規模に限定され、そしてワクチン目的のためのウイルス粒子の大規模生産を達成する手段もウイルスタンパク質の大規模生産を達成する手段も提供しない。Gaushらは、MDCK細胞がまたインフルエンザA型の感染に感受性であることを1966年に実証した。しかしながら、この論文は、感染性のみを報告しており、そしてこの培地におけるウイルスの伝播の問題については取り組んでいなかった(非特許文献4)。
【0010】
1975年に、Tobitaらは初めて、トリプシンを含むオーバーレイ培地において、構築されたMDCK細胞株において、広範な種々のインフルエンザウイルスA型の増殖を記載した。これらウイルス伝播は、それらウイルスの以前の継代歴に関係なく十分に定義されるプラークを形成し、そしてHAポリペプチドを切断し、これによってインフルエンザウイルスの成熟を加速させることにトリプシンが寄与することを提案した。しかし、このトリプシンの使用がもたらす進歩にもかかわらず、寒天培地におけるウイルスの単離は、ウイルスの大量生産を達成する手段を提供しなかった。同参考文献において、またMDCK細胞が、患者の喉洗浄物(throat washing)からのインフルエンザウイルスA型の一次単離に関して首尾よく使用された。
【0011】
続いて、Reuvenyらは、バッチ培養において、セルロースベースのマイクロキャリア上でのMDCK細胞において、インフルエンザウイルスA型を増殖させた(非特許文献5)。得られたインフルエンザウイルスの力価は、胚含有卵において得られたものと同様にトリプシン含有培地中に存在した。
【0012】
特許文献1(Brownら)は、ウイルス培養の間にトリプシンのようなタンパク質加水分解酵素を培養物中に含むことによって、同じ液体培養物の逐次数の細胞中でのインフルエンザウイルスの複製のための方法を記載する。タンパク質分解酵素は、HAを機能性にさせ、そしてこれによって既往の液体培養技術の1段階増殖サイクルに勝るために必要とされる。このことは、液体細胞培養物からの潜在的な「商用」インフルエンザワクチン製造の最初の記述である。しかしながら、現在、溶液中のトリプシンの存在が、MDCK細胞の特定の割合をそれらの固体支持体から浮遊させるという不利を有することが認められる。結果的に、この特許されたプロセスの潜在的な有用性にもかかわらず、トリプシンを要求することが、インフルエンザワクチンの商用生産に対する深刻な制限である。従って、インフルエンザワクチンの生産のための細胞培養ベースの商用プロセスのための必要性がさらに存在する。このことは、以下の記述によって支持される:非特許文献6。
【0013】
また1995年に、World Health Organization(WHO)は、その分野の専門家たちが、インフルエンザウイルスの生産のための細胞培養ベースのプロセスに関する近年の進展を評価するために会合した、1995年の夏に開催された議論を要約する摘要を発表した(非特許文献7)。この文書は、迅速にスケールアップの可能な、細胞培養物系において大量のインフルエンザワクチンの迅速な生産を達成するためのさらなる研究を奨励する。
【0014】
研究目的に関して、細胞培養物中でのウイルスの増殖のため、MDCK細胞およびベロ(Vero)細胞が、いくつかのウイルスの良好な生産体であるとして最も頻繁に参照される。しかしながら、大規模生産目的のため、いくつかの要因(例えば、1つのウイルスまたは複数のウイルスに対する感受性、ウイルス力価の出来高(output)、足場依存性、腫瘍形成性、など)が、別の細胞株の選択以上に特定の細胞株の選択に影響を与える。
【0015】
MDCK細胞はいくつかのウイルス株に感受性であるが、結果的に乏しいウイルス力価により、大規模生産目的のためのこの細胞株の有用性が制限され得る。MDCK細胞株の特性は、いくつかの研究の課題であった。1970年に、Leightonら(非特許文献8)は、MDCK細胞が、コラーゲンでコーティングされたセルローススポンジ上での三次元的な組織培養の組織病理学的調製物において、乳頭状の腺癌の形態学的パターンを提示することを報告した。この細胞株の新生物性の特色(quality)は、11日齢または12日齢のニワトリ胚に注入されるた細胞懸濁物が、脳転移の多くの病巣を形成する場合により実証された。
【0016】
細胞株の腫瘍形成性の評価は、生物学的産物の製造における使用に対するその望ましさを評価するために重要である。米国食品医薬品局のCenter for Biologics Evaluation and Researchは、生物学的物品の製造のための基体(substrate)として細胞培養物を使用する場合に考慮する要点を発表した(非特許文献9を参照のこと)。このような要点の1つは、使用される細胞株の腫瘍形成性であり、そしてFDAは、インビボ腫瘍形成性試験のためのガイドラインを考案した。これらのガイドラインは、とりわけ、ヌードマウス(nu/nu)において、皮下経路または筋肉内経路により投与された細胞を試験することを指示する。
【特許文献1】米国特許第4,500,513号明細書
【非特許文献1】Katz,J.M.ら、J.Infect Diseases(1989)160:191
【非特許文献2】American Type Culture Collection(ATCC)、Catalogue of Cell Lines and Hybridoma(1992)第7版、p21
【非特許文献3】Green、Science(1962)138:42
【非特許文献4】Gaushら,Proc.Soc.Exp.Biol,and Med.(1966)122:931
【非特許文献5】Develop.Biol.Standard(1982)50:115
【非特許文献6】Kodihalliら、J.Virol.(1995)69(8):4888:「Embryonated chicken eggs are currently the only host in which sufficient quantities of virus can be cultivated economically and within the short time necessary to ensure a vaccine supply」
【非特許文献7】Bull.W.H.O.73(4):431,1995
【非特許文献8】LeightonらCancer(1970)26:1022
【非特許文献9】Points to Consider in the Characterization of Cell Lines Used to Produce Biologicals,Office of Biologics Research and Review,Center for Drugs and Biologics,FDA(USA),1993
【発明の開示】
【課題を解決するための手段】
【0017】
(発明の要旨)
従って、本発明は、インフルエンザウイルス粒子またはインフルエンザタンパク質の大規模生産のためのプロセスに関し、このプロセスは、以下の工程を包含する:
(a)上記インフルエンザウイルスを複製可能であるMDCK細胞株を増殖させる工程;
(b)このMDCK細胞株にインフルエンザウイルス株を感染させ、そしてインキュベートしてそのウイルスの複製の可能にする工程;および
(c)複製されたウイルスを収集し、ウイルス粒子またはそのタンパク質を精製する工程。
【0018】
本発明は、ウイルス感染に高感受性であるMDCK細胞株に由来し、そしてそれら由来の親細胞株よりも高いウイルス力価でインフルエンザウイルスを産生する細胞株を、さらに提供する。
【発明を実施するための最良の形態】
【0019】
(発明の詳細な説明)
1実施形態において、本発明のMDCK由来の細胞株は、インフルエンザウイルスの多段階複製を可能にする。別の実施形態において、本発明のMDCK細胞株は、足場依存性であり、かつ非腫瘍形成性である。
【0020】
本発明は、ウイルス感染に超感受性であるMDCKクローンの誘導体を提供する。「超感受性」との記載は、少なくとも1つのウイルスに高感受性であり、これによって親MDCK細胞株よりもより高力価のウイルス粒子を産生するMDCK由来の細胞株を示すために、使用される。この誘導化クローン(MDCK.5F1)は、番号CRL−12042の下で1996年2月8日にAmerican Type Culture Collection(ATCC)に受託された。1実施形態において、MDCK由来の本発明の細胞株は、FDAガイドラインに従って行われた試験において非腫瘍形成性であり、そしてこれによって予防目的、診断目的、免疫治療目的または治療目的のためのウイルスまたは抗原の調製における使用に適切であり得る。この細胞株は、混入する微生物の存在について試験されており、そしていずれも検出されていない。また、本発明に従って、ワクチン製造のためのインフルエンザウイルス粒子またはインフルエンザウイルスタンパク質の大規模生産のためのプロセスが提供される。
【0021】
また、本発明の1局面に従って、インフルエンザワクチンの製造のためのインフルエンザウイルス粒子またはインフルエンザウイルスタンパク質の生産のための、大規模で細胞培養によるマイクロキャリアベースの商用プロセスが提供される。
【0022】
本発明のプロセスは、インフルエンザウイルスの複数回の複製のためにMDCK細胞株を使用して実施される。1実施形態において、本発明のプロセスにおいて使用されるMDCK細胞株は、ATCC細胞株番号CRL−12042と同じ生物学的特性を有する。1実施形態において、本プロセスにおいて使用される細胞株は、ATCC番号CRL−12042の下で受託された、MDCK.5F1として本明細書中で称されるMDCK細胞株のクローンである。
【0023】
本発明のMDCK由来の細胞株は、ウイルス感染に高感受性である。この文脈において、ウイルス感染に「高感受性である」とは、その細胞株が、少なくとも1つのウイルス種に対して親細胞株によって産生される力価よりも高い力価を産生し得ることを意味する。
【0024】
1実施形態において、「高感受性」とは、親細胞株によって産生される力価よりも少なくとも約1.2倍の力価でウイルスを産生し得る細胞株として定義される。1実施形態において、より高感受性の細胞株は、3B5、5Fl、1D11、5H12、9C2、9D9、P79、9E9、7C1およびP123からなる群より選択されるクローンである。
【0025】
さらに1実施形態において、より高感受性とは、それらの同じウイルスに関して、親細胞株によって産生される力価の少なくとも2倍の力価で多数のウイルスを産生し得る細胞株として定義される。1実施形態において、このようなクローンは、3B5、5F1、5H12、9C2、7C1およびP123からなる群より選択される。
【0026】
1実施形態において、より高感受性とは、同じウイルスの2つの異なる株について、親のウイルス力価の少なくとも約2倍を産生し得るとして定義される。1実施形態において、このようなクローンは、RSウイルスおよびインフルエンザA型およびインフルエンザB型に関して親のウイルス力価の2倍を産生し得る。1実施形態において、このようなクローンは、5F1および5H12からなる群より選択される。
【0027】
1実施形態において、細胞株は、インフルエンザウイルス、RSウイルス、パポバウイルス、パラインフルエンザウイルス、水疱性口内炎ウイルス、ワクシニアウイルス、コクサッキーウイルス、レオウイルス、パルボウイルス、アデノウイルス、ポリオウイルス、麻疹ウイルス、狂犬病ウイルス、ヘルペスウイルスおよび他のウイルスからなる群より選択されるウイルスによって感染され得る。
【0028】
1実施形態において、ウイルスは、インフルエンザA型、インフルエンザB型およびインフルエンザC型;RSウイルス;パポバウイルス;水疱性口内炎ウイルス(インド株);コクサッキーウイルスB−5;レオウイルス2型およびレオウイルス3型;ならびにアデノウイルス4型およびアデノウイルス5型からなる群より選択される。
【0029】
1実施形態において、ウイルスは、インフルエンザA型、インフルエンザB型およびインフルエンザC型、ならびにRSウイルスからなる群より選択される。
【0030】
1実施形態において、ウイルスは、ヒトインフルエンザ株、ウマインフルエンザ株、ブタインフルエンザ株またはトリインフルエンザ株から選択される。
【0031】
1実施形態において、ウイルスは、ヒトインフルエンザA型、ヒトインフルエンザB型またはヒトインフルエンザC型から選択される。
【0032】
1実施形態において、本発明の細胞株は、ヒトインフルエンザウイルスA型またはヒトインフルエンザウイルスB型によって感染され得る。
【0033】
1実施形態において、本発明の細胞株は、ヒトインフルエンザA型およびヒトインフルエンザB型のの両方に感染され得る。
【0034】
1実施形態において、本発明の細胞株は、トリプシン、キモトリプシン、ペプシン、パンクレアチン、パパイン、プロナーゼおよびカルボキシペプチダーゼのようなタンパク質分解酵素の添加によってインフルエンザウイルスの多段階複製を可能にする。1実施形態において、このような細胞株は、トリプシンの添加によってインフルエンザウイルスの多段階複製を可能にする。
【0035】
あるいは、本発明の細胞株は、トリプシン、キモトリプシン、ペプシン、パンクレアチン、パパイン、プロナーゼおよびカルボキシペプチダーゼのようなタンパク質分解酵素の添加を必要とすることなく、インフルエンザウイルスの多段階複製を可能にする。1実施形態において、このような細胞株は、トリプシンの添加を必要とすることなくインフルエンザウイルスの多段階複製を可能にする。
【0036】
どのウイルスまたはウイルス株がタンパク質分解酵素の使用を必要とし得るかは、当業者に明らかである。
【0037】
本発明の細胞株を懸濁して培養し得るが、この細胞株を足場依存性様式で増殖させることが好ましい。1実施形態において、この細胞株はマイクロキャリアビーズ上で増殖し得、これによって高密度の細胞が細胞培養物中に得られることを可能にする。
【0038】
1実施形態において、本発明のMDCK由来の細胞株は、非腫瘍形成性である。1実施形態において、本発明の細胞株は、軟寒天中で最小限の効率(すなわち、1%未満の効率)で増殖する。1実施形態において、本発明の細胞株は、ヌードマウスにおいて、少なくとも3ヶ月間観察される場合、小結節を生じない。
【0039】
本発明は、バルク量でウイルス粒子またはウイルスタンパク質の産生のために、本発明に従う細胞株の使用をさらに企図する。
【0040】
このようなウイルス粒子またはタンパク質は、宿主におけるウイルス感染の予防のためのワクチンの製造において使用され得る。1実施形態において、この宿主は哺乳動物である。哺乳動物としては、例えば、ヒト種、ウマ種、ブタ種が挙げられる。別の実施形態において、この宿主はヒトである。さらなる実施形態において、この宿主はトリ種(例えば、ガチョウまたはニワトリ)である。
【0041】
本発明のプロセスは、足場依存性MDCK細胞が培養物中で増殖する能力にトリプシンの存在が影響を与えない限り、トリプシンの存在下で実施されてもトリプシンの非存在下で実施されてもよい。1実施形態において、本プロセスは、約4μg/ml以下の濃度のトリプシンの存在下で実施される。あるいは、本プロセスはトリプシンの非存在下で実施される。
【0042】
本明細書中に記載のプロセスは、RSウイルスおよび種々のインフルエンザウイルス(例えば、ヒトインフルエンザウイルス株、ウマインフルエンザウイルス株、ブタインフルエンザウイルス株およびトリインフルエンザウイルス株)の産生のために意図される。1実施形態において、本明細書中に記載されるプロセスは、ヒトインフルエンザA型、ヒトインフルエンザB型、またはヒトインフルエンザC型の産生のために意図される。1実施形態において、本明細書中に記載されるプロセスは、ヒトインフルエンザA型またはヒトインフルエンザB型の産生のために意図される。1実施形態において、本明細書中に記載されるプロセスは、ヒトインフルエンザA型またはヒトインフルエンザB型のいずれかのウイルス型に感染され得るMDCK細胞株を使用して、ヒトインフルエンザA型およびヒトインフルエンザB型の産生のために意図される。
【0043】
本発明のプロセスは、106個のMDCK細胞あたり4μg HAの範囲である収量を提供する。実施例9に記載されるように、本プロセスは106個のMDCK細胞当たり4μg HAより多くの量のウイルスタンパク質を生じ、より具体的には106個の細胞当たり9μg HAの範囲で生じる。
【0044】
「大規模プロセス」とは、多量のインフルエンザウイルスを製造するためのプロセスを意味する。このようなプロセスは、通常、培養フラスコとは対照的に、バイオリアクターにおいてなされる。このようなバイオリアクターは、(最終的な収量)/(必要とされる用量)に依存して変動性のサイズであり得る。例えば、このようなバイオリアクターは、約5リットルのサイズ(約4リットルの作業容量)であり得るか、または5000リットルまでであり得る。無論、大規模な細胞培養ベースのワクチン製造の当業者に明らかであるように、全ての反応物、培地、栄養分、細胞濃度、マイクロキャリア濃度、灌流速度などは、そのバイオリアクターのサイズに従って調節され得る。
【0045】
本発明のプロセスは、懸濁培養において実施され得るが、細胞濃度を上昇させる(そしてこれによってウイルス出来高を向上させる)ためにマイクロキャリア手段において実施されることが好ましい。例えば、デキストランポリマー(CytodexTM)として当該分野で通常公知の型のマイクロキャリアビーズが選択され得る。これらのマイクロキャリアは、約5g/L〜25g/Lの範囲の濃度で使用され得る。1実施形態において、このマイクロキャリア濃度は、約10g/L〜25g/Lの範囲にある。1実施形態において、このマイクロキャリア濃度は、約15g/L〜約20g/Lの範囲にある。
【0046】
灌流手段は、プロセス中の細胞増殖およびウイルス複製の両方を最大にするように導入される。灌流は、同時に培養培地中の潜在的に毒性の副産物の蓄積を避ける手段を提供しながら、栄養分の一定の供給を可能にする。灌流を介して、栄養分の型および質量は、プロセスの種々の段階の間に変動され得る。例えば、血清は、増殖期の間に細胞に導入され得るが、理想的には、細胞の集密期(confluency)においてそしてウイルスの導入前に排除されるべきである。その灌流速度は、適切な栄養供給を提供するために、細胞増殖の間に徐々に上昇され得る。灌流はウイルス複製の間に継続される。
【0047】
灌流速度は、プロセスの段階に依存して、1日当たりバイオリアクターの約0.5〜4倍容量の間で調節される。
【0048】
灌流手段が、バイオリアクターに追加され得る。この灌流手段は、そのバイオリアクター中に培地を連続的に導入するための流入口手段、およびそのバイオリアクターから消費された培地を連続的に除去するための2つの出口手段を備え(これによってそのバイオリアクター中でマイクロキャリアの懸濁を介して培地の連続的な流動を形成する)、そしてその出口手段と一体化したデカンターを備える。本明細書中に記載されそして本発明のプロセスにおいて使用されるデカンターは、上記に提案されるような灌流手段を用い、10g/Lの過剰なマイクロキャリア濃度を使用して、上方へと移動しそしてその出口手段を通って抜け出るかまたはその出口手段を詰まらせるかのどちらかのマイクロキャリアの傾向によって細胞増殖が制限されることが認められる場合の、5L(3.7Lの作業容量)のバイオリアクターに対して具体的に設計された。従って、より高いマイクロキャリア濃度を達成するため、以下の、デカンター内で乱流形成を最小にし、そして懸濁の上方への流速よりも速いマイクロキャリアの沈降速度を達成する特徴を有するデカンターが、構築された。
【0049】
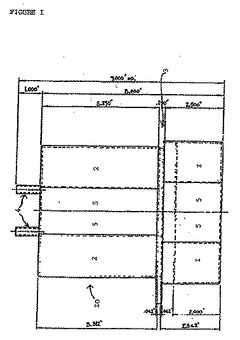
本発明の好ましい実施形態に従って、図1に示されるようなデカンター10は、下側チャンバ1を備え、この下側チャンバ1はより大きな上側チャンバ2と放射状プレート3によって接続される。2つの出口手段4は、このデカンターの頂部において上側チャンバ2に取り付けられる。この下側チャンバ1および上側チャンバ2は、半円筒形状であり、そして同軸上に向けられる中央の円形空洞5を取り囲み、この中央の円形空間5は、このバイオリアクターの中央回転シャフト、および本発明のプロセスをモニタリングするためにこのバイオリアクター中に導入される種々のプローブを収容するように意図される。中実の金属壁は、このデカンターを完全にする。
【0050】
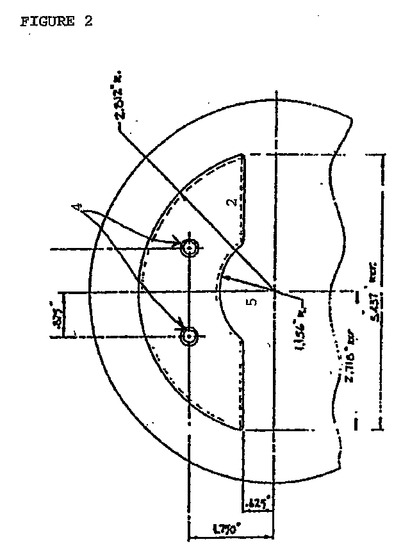
図2は、上記の構成要素2、4および5の上正面図を示す。
【0051】
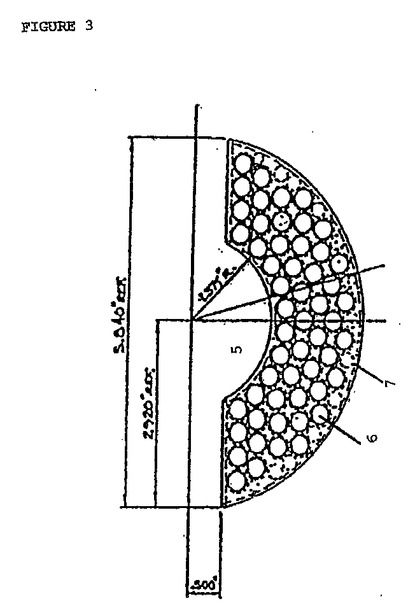
図3は、デカンターの下側チャンバ1の下正面図を示す。下側チャンバ1は、規則正しく分配した複数の長手軸チャネル6を有し、この長手軸チャネル6は、円周方向に向けられる長手軸方向の区画7によって分離され、この長手軸チャネルは、そのデカンターの上側チャンバ2および放射状プレート3を介して互いに連絡している。1実施形態において、長手軸チャネル6は同一であり、そして各々は円形の断面を有する。さらに、複数の長手軸チャネル6は、同軸上に向けられた中央の円形空洞5の周りに規則正しく分配され、その周囲の長手軸チャネル6は、円周方向に向いた長手軸方向の区画7によって互いから分離されている。
【0052】
灌流手段の出口手段は、感染の際に廃液レザバから収集レシピエントへと切り換えられる。連続的な収集は、複製が完了するまで(約4〜5日)生じる。
【0053】
本発明のプロセスは、10:1〜1:1010の間の感染多重度(M.O.I)でのウイルスによってMDCK細胞培養物を播種することによって実施される。1実施形態において、このプロセスは、1:10〜1:108の間の感染多重度(M.O.I.)で実施される。1実施形態において、このプロセスは、約1:104〜1:107のM.O.Iにおいて実施される。1実施形態において、1:105〜1:106のM.O.Iにおいて実施される。
【0054】
1実施形態において、本発明のプロセスにおけるMDCK細胞の増殖は、温度約33℃〜40℃において約7日〜10日間実施される。1実施形態において、このプロセスにおけるMDCK細胞の増殖は温度約36℃〜38℃において、約7日間実施される。
【0055】
1実施形態において、本プロセスにおけるMDCK細胞の増殖は、温度約37℃で約7日間実施される。1実施形態において、本発明のプロセスにおけるウイルス複製は、温度約30℃〜37℃において約4日〜6日間実施される。1実施形態において、このプロセスにおけるウイルス複製は、温度約32℃〜約34℃で約5日間実施される。1実施形態において、このプロセスにおけるウイルス複製は、温度約33℃で実施される。
【0056】
ウイルス粒子またはウイルスタンパク質は、以下の方法で精製される。ウイルス収集物の濾過の後に、ホルムアルデヒドを用いた濾過物の不活性化が続く。次いで、得られた不活性化ウイルス懸濁物は遠心分離され、そして富化されたウイルス画分は、ワクチン調製物における使用のために選択される。
【0057】
本発明のワクチンを投与する工程を包含する、哺乳動物におけるインフルエンザ感染の予防のための方法もまた、提供される。
【0058】
精製されたウイルス粒子またはタンパク質を使用してインフルエンザウイルス感染の検出のための診断キットを生産するプロセスもまた、提供される。
【0059】
哺乳動物におけるインフルエンザウイルス感染の検出および診断のためのキットを生産するための、単離されたバルクのウイルス粒子またはウイルスタンパク質の使用もまた、提供される。
【0060】
本発明者らのプロセスの使用の特定の例および選択されたヒトインフルエンザウイルスA型株およびヒトインフルエンザウイルスB型株の増幅は、以下に考察される。本発明の配置および構成要素において、本発明の本質および範囲から逸脱することもなく、その物質の利点の全てを損なうこともなく、種々の変更がなされ得ることは明らかであり、以下に記載される形態は、単なる好ましい実施形態または単なる例示的な実施形態である。
【実施例】
【0061】
実施例1は、クローンMDCK.5F1の誘導を記載する。実施例2は、MDCK.5F1細胞株の純度を検討する。実施例3は、トリプシン有りおよび無しでの、MDCK.5F1細胞株における、インフルエンザウイルスの増殖を示す。実施例4は、MDCK.5F1についての腫瘍形成能研究のまとめである。実施例5は、MDCK.5F1の細胞懸濁物による、胸腺欠損ヌードマウスの接種後の結果を議論する。実施例6は、プロセスの特定の実施形態の段階的な説明である。実施例7は、トリプシン有りでの親MDCK細胞とインフルエンザ株A/Shanghai/ll/87とを使用して実行されたアッセイの結果を記載する。実施例8は、トリプシン無しでのMDCK.5F1細胞とインフルエンザ株A/Shanghai/ll/87とを使用して実行されたアッセイの結果を記載する。実施例9は、トリプシン無しでのMDCK.5F1細胞とインフルエンザ株B/Harbin/7/94とを使用して実行されたアッセイの結果を記載する。
【0062】
(実施例1)
(クローンMDCK.5F1の誘導)
MDCK細胞No.CCL 34をAmerican Type Culture Collection、Rockville、Marylandから取得した。このストックを、3.4×106細胞を含む1mlアンプル中の凍結状態で受け取った。この細胞株は、54回継代したものだった。
【0063】
継代後、MDCK細胞を、64回目の継代で回収し、10%(v/v)ウシ胎児血清(FBS)を含み、1:1の比でのダルベッコ変法イーグル培地(DMEM)および培地199からなる栄養培地(DMEM−199)中に希釈した。次に、各ウェルが細胞を1個未満受容するように、希釈した細胞懸濁物を96ウェルプレートにアリコートし、溶液中の細胞の均一な分配とみなした。これらのプレートを、37℃でCO2インキュベーター中に置き、増殖について、これらのウェルをスコア付けするために、一週間の間隔において光学顕微鏡下で調べた。
【0064】
クローンにおいて求めた特徴を、以下から選択した:
(1)ウイルス感染に対する、親株よりも高い感受性(すなわち、クローンが、親株より高いウイルスの力価を生成する);
(2)1つより多いウイルスに対する、より高い感受性(一実施形態において、インフルエンザウイルスのいくつかの株に対する感受性);
(3)タンパク質分解酵素(例えば、トリプシン)の添加の必要なく、インフルエンザウイルスの多段階の複製を可能にする能力;および、必要に応じて、
(4)足場依存性(すなわち、培養中でのより高い細胞の濃度を得るため)。
表1は、トリプシンの添加なしでの、インフルエンザウイルスA型およびインフルエンザウイルスB型による感染に対する、いくつかのクローンの感受性を示す。
【0065】
【表1】
「高感受性」を、TCID50により評価した場合に、少なくとも親細胞株の約1.2倍の感受性として定義した。クローン3B5、5F1、1D11、5H12、9C2、9D9、P79、9E9、7C1およびP123を、感受性が高いとして同定した。クローン3B5、5F1、1D11、5H12、9C2、9D9、7C1およびP123を、親株の少なくとも2倍の感受性であるとして同定した。
クローン5F1および5H12を、感受性が最も高い2クローンとして選択し、そして5F1を、MDCK.5F1として本明細書中で称される細胞株を樹立するために選択した。
【0066】
細胞の世代数を、クローニングの時点において0として定義し、続く各々の細胞培養物における細胞の数え上げにより計算した。最初、マルチウェルプレート培養で培養物を継代し、最終的にプラスチック製フラスコに移した。
さらに、実験を、親MDCK細胞株およびMDCK.5F1クローンの、RSウイルスによる感染に対する感受性を測定するために、このウイルスを用いて実行した。ウイルス力価は、MDCK細胞株は、RSウイルスの宿主Hep2細胞株よりも感染に対し、より非感受性であったが、MDCK.5F1クローンは、このウイルスによる感染に対し、親MDCK細胞株よりも約10倍感受性が高かったことを示した。
【0067】
(実施例2)
(MDCK.5F1のクローン性の決定)
選択された任意の特定のクローンのクローン性を決定するパラメータは、クローンが選択されたマルチウェルプレート上のパーセンテージ増殖である。選択された培養物が実際にクローンである可能性(すなわち、P(1))は、このパーセンテージから決定される(CollerおよびColler、Methods in Enzymology、vol.121、pp.412〜417(Academic Press、1986)を参照のこと)。
【0068】
5%のウェルが、このクローニングにおいて増殖を示したと仮定すると、細胞株MDCK.5F1が単一の細胞に由来する可能性は、≧97.5%である。
【0069】
299アンプルの親細胞バンク(Master Cell Bank)(MCB)および283アンプルの製造者の作業用細胞バンク(Working Cell Bank)(WCB)を、MDCK.5F1細胞株から調製した。これらのバンクを、Good Manufacturing Practiceの方針上のカナダ国の指針にしたがって調製し、真菌因子、酵母因子、マイコプラズマ因子、細菌因子およびウイルス因子の形態での汚染について評価した。いかなる種類の汚染も見出さなかった。
【0070】
持続可能かつ生存可能な培養物を、調製した細胞バンクから取得し、細胞株を、WCBよりも50回集団倍化したものについての産物の産生の安定性、形態学、腫瘍形成能およびイソ酵素の特徴について試験した。これらの結果は、これらの特徴について安定である細胞株を示した。
【0071】
(実施例3)
(トリプシンの添加有りおよびトリプシンの添加無しでの、MDCK.5F1クローンにおける、インフルエンザウイルスの増殖を示す実験)
トリプシンの存在下および非存在下において、クローンMDCK.5F1の培養物中で、インフルエンザウイルスが複製する能力を決定するために、実験を実行した。インフルエンザ株A/Johannesburg、インフルエンザ株A/Texasおよびインフルエンザ株B/Harbinを使用した3つの実験の結果を表2に示す。
【0072】
【表2】
これらの結果は、血球凝集(HA)力価の値により示されるように、トリプシン有りおよび無しで、インフルエンザが、細胞培養物中で複製される場合に実質的な違いが何もないことを明らかにする。
【0073】
(実施例4)
(インビトロ腫瘍形成能研究のまとめ)
Fureszら(Develop.Biol.Stand.vol.70、pp.233〜243、S.Kargel編、Basel、1989)により記載される方法にしたがって、10%(v/v)FBSを含むDMEM−199培地中の0.6%寒天の4mlを、直径35mmのウェルを6つ備える組織培養皿内に固化させた。固化後、これらのウェルを、42℃に維持された、0.3% W/Vの非ゲル状の寒天と60,000細胞/mlの細胞濃度とを含む3mlの培地で重層した。プレートを、5% CO2、37℃でインキュベートした。光学顕微鏡観察を、3日目、7日目、10日目および14日目に行った。コロニーは、軟寒天中に球状の群を形成する、4個以上の細胞からなった。パーセンテージ有効性を、プレートした細胞の総数で割った、計数された細胞コロニーの数の比により決定した。
【0074】
【表3】
表3の結果は、MDCK.5F1細胞株が、軟寒天において最低の有効性で増殖し、これが動物において非腫瘍形成性であり得ることの示唆を示す。この特性は、18回継代後も維持され、この表現型が安定であることを示した。
【0075】
非腫瘍形成能のこの所見をさらに支持するために、発明者らは、胸腺欠損ヌードマウスにおいて、MDCK.5F1細胞株の腫瘍形成の可能性を試験した。
【0076】
(実施例5)
(MDCK.5F1クローンの細胞懸濁物の皮下接種後の、胸腺欠損ヌード(nu/nu)マウスにおける腫瘍形成の評価)
胸腺欠損ヌード(nu/nu)マウスは、外来物質に対する細胞媒介性の応答を確立できず、それゆえ同種異系の腫瘍細胞株および異種遺伝子型の腫瘍細胞株の増殖を補助する。このことは、接種物がインビボで新生物を形成する能力の評価を可能にする。
【0077】
6週齢のヌードマウスを、約1×107細胞の試験物質(MDCK.5F1)により皮下接種し、84日間臨床的に置き、そして検死した。ポジティブコントロールの細胞およびネガティブコントロールの細胞で接種したヌードマウスを、同様に処置した。接種部位(皮膚)、肺、肩甲骨のリンパ節および肉眼的病変を、加工、切り出し、染色し、そして顕微鏡で調べた。この実験のさらなる詳細は、以下に示される。
【0078】
(試験物質およびコントロール物質の接種)
各ケージ内の全てのマウスを、同様に処置した。
【0079】
各マウスを、以下に記載の0.2mlの適切な接種物により、肩甲骨の間に皮下接種した。22ゲージ針を、接種のために使用し、全てのマウスを、同じ日に接種した。
【0080】
群1および群2:試験物質、MDCK.5F1(5×107細胞/mlの濃度で)。
【0081】
群3および群4:ポジティブコントロール(5×107細胞/mlの濃度の18C1−10T細胞)。
【0082】
群5および群6:ネガティブコントロール(1×107細胞/mlの濃度のSHE細胞)。
【0083】
全ての動物を、毎作業日(working day)観察し、接種部位を、84日目までの期間、週に二回触診した。
【0084】
(結果)
(臨床所見)
全てのポジティブコントロールのマウスは、接種部位において、少なくとも一つの長さが1cmより大きい大型の塊を有したので、これら全てを、接種後14日目に、屠殺および検死した。
【0085】
全てのネガティブコントロールのマウスを、接種後84日目に、屠殺および検死した。
【0086】
10匹の試験物質(5F1)マウスのうち9匹を、接種後84日目に、屠殺および検死した。進行していた接種部位の病変が退行し始めたので、1匹の5F1接種されたマウスを、接種後33日目に、屠殺および検死した。この病変は、後に嚢胞であることが明らかになった。
【0087】
(触診)
ポジティブコントロール物質により接種した10匹のヌードマウスは、接種後14日目までに、少なくとも一つの長さが1cmより大きい触知可能な病変を有した。
【0088】
10匹のネガティブコントロール物質を接種したヌードマウスの接種部位において、小型の非進行性の病変が触知可能であった。これらの病変は、最初、接種後4日目に発覚し、観察期間の間、10匹のネガティブコントロールのマウスのうち8匹において残存した。触診の結果を、表4にまとめる。
【0089】
10匹の5F1マウス全てが、接種後4日目までに病変を有した。10匹の5F1マウスのうちの9匹において、これらの病変は、小さく、進行しなかった。接種後56日目までに、試験物質のマウスのうち8匹において、触知可能な病変は存在しなかった。1匹の5F1を接種したマウスは、25日目と28日目との間に、大きさが顕著に進行し、そして接種後32日目までに、大きさが顕著に減少した接種部位の病変を有した。この病変を、顕微鏡検査により、嚢胞として同定した(表6を参照のこと)。病変を示す他のマウスは、局在化した炎症を有した。
【0090】
【表4】
(肉眼的な検死所見)
処置に関連する肉眼的な検死所見を、表5にまとめる。
【0091】
【表5】
接種部位における塊を、全てのポジティブコントロールの動物において見出した。10匹の5F1マウスのうち3匹、および10匹のネガティブコントロールのマウスのうちの6匹において、接種部位に、塊または小結節を見出した。
【0092】
(顕微鏡の所見)
目的の病変を、表6にまとめる。
【0093】
【表6】
新形成(線維肉腫)を、全てのポジティブコントロールのマウス中の接種部位において診断した。線維肉腫は、織り交ぜられたパターンを伴う、可変の密度の束で配置される紡錘細胞(spindloid cell)からなった。コラーゲンの沈着は、最小限であった。隣接する組織は新生物により圧迫されるが、ほとんど新生物により浸潤されなかった。
【0094】
どのネガティブコントロールのマウスにおいても、新生物は診断されなかったが、骨様の増殖の病巣を、ネガティブコントロール物質のマウスのうちの2匹中の接種部位において確認した。これは、ネガティブコントロールとして、SHE細胞の分化および増殖の選択を表すと考えられる。
【0095】
どの5F1マウスにおいても、新生物は診断されなかったが、1匹のマウスにおいて嚢胞を確認し、他の1匹の5F1マウスにおいて病巣の亜急性の炎症を確認した。
【0096】
(結論)
10匹全てのポジティブコントロールのマウスの接種部位において、線維肉腫を診断した。
【0097】
どのネガティブコントロールにおいても、どの試験物質のマウスにおいても、新生物は存在しなかった。
【0098】
上記研究の条件下で、試験物質MDCK.5F1は、腫瘍形成性であるとはみなされない。
【0099】
(実施例6)
(一般的な手順)
(a)細胞の増殖)
バイオリアクターに播種する前に、MDCK.5F1(ATCC番号CRL−12042)細胞を、細胞の増殖のために、数回継代した。約5〜10×106細胞を、37℃の水浴で解凍し、栄養培地を含むポリスチレン製の細胞培養フラスコに移し、そして37℃でインキュベートした。3〜4日後、細胞をこのフラスコから乖離し、5個の他のフラスコに播種するために使用した。次に、この5個のフラスコを、同様な様式で、20個のフラスコに播種するために使用した。細胞増殖のために使用した栄養培地は、脱イオン水で調製された1:1の比でのダルベッコ変法イーグル培地および培地199であり、4.5g/Lのグルコース、0.58g/Lのグルタミンおよび1g/Lの炭酸水素ナトリウムを含んだ(DMEM−199)。この栄養培地に、10%のγ線照射されたウシ胎児血清(I−FBS)を補充した。細胞の継代のために、フラスコ内の細胞を乖離するために使用した溶液は、マグネシウムおよびカルシウムを含まないリン酸緩衝化生理食塩水(PBS)により調製した、0.02%エチレンジアミン四酢酸(EDTA)を含む0.25%トリプシン溶液であった。
【0100】
20個のフラスコ中の細胞を、3〜4日間増殖させ、トリプシン処理し、収集し、そして3〜5g/Lのマイクロキャリアビーズを含む1000mlの攪拌フラスコ内の3つのマイクロキャリア細胞培養に播種するために使用した。攪拌フラスコを、約50rpmに攪拌を維持しながら、37℃でインキュベートした。細胞の増殖を、5〜7日間続け、その後、細胞を、マイクロキャリアからトリプシン処理し、5Lバイオリアクター(New Brunswick of Edison、N.J.によるCelliGenTM)に播種するために使用した。
【0101】
マイクロキャリアからの細胞のトリプシン処理を、以下の方法で行った。マイクロキャリア細胞培養を、PBSおよび0.02%EDTAの溶液で2回洗浄した。二回目の細胞の洗浄後、約200mlのトリプシン溶液を、フラスコに注ぎ、攪拌しながら37℃で約20分間置いた。光学顕微鏡により決定される場合、細胞の乖離が完全になった後、細胞を、2%I−FBSを含むDMEM−199を使用して、遊離のマイクロキャリアから回収し、それからペレット化し、10%I−FBSを含むDMEM−199中に再懸濁した。細胞の計数を実行し、適切な数(約1010個)の細胞をバイオリアクターに播種するために使用した。
【0102】
細胞培養のために使用された球状のビーズまたはマイクロキャリアは、Pharmacia(スウェーデン)により製造され、Cytodex 1の商品名の下に頒布される。Cytodex 1マイクロキャリアの濃度は、1.03(g/ml、0.9%NaCl中で)であり、その大きさは、131μmと220μmとの間、180μmの平均値で変動した。細胞増殖のためのおおよその表面積は、マイクロキャリア(乾燥重量)1gあたり4,500cm2であり、マイクロキャリア1gは、約6.8×106個のマイクロキャリアを含んだ。
【0103】
(b)バイオリアクター内での細胞の播種および増殖)
15〜25g/Lの濃度のCytodex 1 マイクロキャリアを、5Lバイオリアクター(3.7L作業用容量(working volume))に導入した。バイオリアクターの播種を、以下のように実行した。先に調製した株(上記を参照のこと)から取得した約4×109〜1×1010個の細胞を、管状のガラス製のボトル内に配置した。20g/Lのマイクロキャリアの無菌溶液からの、55.5〜92.5gのマイクロキャリア(培養内の望ましい濃度に依存する)を、DMEM−199で二回すすぎ、そしてガラス製のボトル内の細胞に添加した。次に、このボトルを、10%I−FBSを含むDMEM−199で満たし、3.7Lの最終容量にした。それから、このボトルの内容物(細胞、DMEM−199中のマイクロキャリア)を、バイオリアクター容器に注ぎ、中央シャフトを約20rpmで回転させた。この容器を満たすにつれて、攪拌を50rpmに上げ、温度を37℃に調整し、溶存酸素量を5〜50%の間の空気の飽和度に維持した。培養物のpHもまた、6.8〜7.4で維持した。マイクロキャリア細胞培養の灌流を、2.5%I−FBSと0.5g/L硫酸マグネシウムとを含むDMEM−199を使用して、一日あたり0.5容量で1日目に開始した。細胞の増殖を、おおよそ7〜10日間続け、灌流の流速を一日あたり2容量まで徐々に増加させた。
【0104】
(c)ウイルス感染)
ウイルスをマイクロキャリア細胞培養に添加する前に、感染の日に、灌流を一日あたり4容量(血清を含まない)まで増加させ、温度を33℃まで下げ、酸素の分圧を15%の空気の飽和度に制御した。細胞のコンフルーエンシーおよびウイルス感染の直前(同じ日)において、培養培地を、血清を含まない同一の培養培地により置き換え、灌流速度を、7時間、一日あたり4容量まで増加させた。このことは、感染前に、培養物の血清含量を最小限のレベルまで下げることを確実にした。この期間後、灌流を停止し、ヒトインフルエンザウイルスA型またはヒトインフルエンザウイルスB型をマイクロキャリア細胞培養に導入した。1:10〜1:108の範囲のM.O.I.を取得するために、バイオリアクターへの導入の前に、ウイルスを、栄養培地(6.5gグルコース/Lを含むDMEM−199)により標準的に希釈した。次の日、細胞変性効果が完全になるまで、灌流を一日あたり2容量で維持した。MDCK細胞の完全な破壊を、代表的に5日以内に観察した。それから、インフルエンザウイルス懸濁物を含む溶出物を、収集し、当該分野において周知であるように、ワクチンを生成するために処理した。
【0105】
(d)不活性化および精製)
一価のインフルエンザウイルスの精製を、以下の様式により実行した。バイオリアクターから集めたウイルス収集物(大体15〜30Lの間)を、大型の細胞残渣を除去するために、まず1.2μmフィルター(Sartorius Sartopure GF(登録商標)、長さ10インチ、0.6m2)を通して、清澄化した。次に、インフルエンザウイルスを含む、清澄化した懸濁物を、16時間の、0.125%(V/V)ホルムアルデヒド(終濃度)の添加により不活性化した。それから、不活性化したウイルス懸濁物を、イオン交換、DNAase処理およびゲル濾過により精製した。
【0106】
濃縮されたウイルス画分を選択し、それは、ワクチン調製において使用されるべき上等な材料と表した。次に、これらの画分をプールし、現在、国際機関により推奨される、1用量あたり、1株あたり15μgのHAの終濃度を与えるように希釈した。
【0107】
(e)ワクチン調製)
ウイルスタンパク質を、ワクチンの各用量に対して15μgになるように希釈した。チメロサール(0.01%)を、保存および安定化それぞれのために添加し、ワクチンを完成させた。
【0108】
標準的な三価ワクチンのために、3つの流行性の(circulating)株の各々の一価の用量が混合され得、上記の保存剤および安定化剤を添加され得る。
【0109】
他の公知の保存剤(例えば、アミノメチルプロパノール、ソルビン酸およびポリアミノプロピルビグアナイド(polyaminopropyl biguanid)、硝酸フェニル水銀、ホウ酸フェニル水銀、ホルムアルデヒドを含む2−フェノキシエタノール、フェノール、塩化ベンゼトニウムおよび2−フェノキシエタノール)は、ワクチン調製のために使用され得る。これらの保存剤の濃度は、産業基準に受容可能である必要がある。
【0110】
(実施例7)
(トリプシンの添加有りでの、株A/Shanghai/11/87による親細胞株の感染)
親株由来のMDCK細胞を、CelliGenTMバイオリアクターにより増殖させた。バイオリアクターの作業用容量は3.7Lであり、マイクロキャリアの濃度は25g/Lであり、そして攪拌を50rpmに設定した。7日目に、培養物を、A/Shanghai/11/87と称されるヒトインフルエンザウイルスにより感染させた。M.O.I.は1:133,000であり、2.5μg/mlトリプシンを、ウイルス複製を増大させるために添加した。表7は、このアッセイに関するデータを示し、表8は、これらの結果をまとめる。ワクチンの収量は、18Lの総収集容量に基づいて、l5μg HAの一価の用量9,828、ならびに、7.38μg HA/mlおよび9μg HA/mlの一元放射拡散(SRD)アッセイ値であった(表8を参照のこと)。
【0111】
【表7】
感染の時点での細胞の総数は、以下の方法により計算される:9.34×106個のMDCK細胞/ml×3700mlは、34,558×106個の細胞に等しい。
【0112】
【表8】
HAの総量の計算は、以下の方法により決定される:9,828用量×l5μg/用量=147,420μg 総HA。34558×106個の細胞で割った場合=
106個のMDCK細胞あたり、4.26μg HA。
【0113】
(実施例8)
(トリプシン無しでの、株A/Shanghai/11/87によるMDCK.5F1クローンの感染)
クローンMDCK.5F1(ATCC番号CRL−12042)に由来する細胞を、25g/Lのマイクロキャリア濃度を含むCelliGenTMバイオリアクターにより増殖させた。バイオリアクターの作業用容量は3.7Lであり、そして攪拌を50〜55rpmに設定した。7日目に、培養物を、A/Shanghai/11/87と称されるヒトインフルエンザウイルスにより感染させた。ウイルスの増殖を増大させるためのトリプシンを、バイオリアクターに添加しなかった。M.O.I.は1:133,000であり、アッセイは9.2μg HA/mlのSRD値を伴う32Lを生成し、一価の用量19,626をもたらした。表9は、このアッセイについてのデータをまとめ、表10は、これらの結果を列挙する。
【0114】
【表9】
感染の時点での細胞の総数は、以下の方法により計算される:16.8×106個のMDCK細胞/ml×3700mlは、62,160×106個の細胞に等しい。
【0115】
【表10】
HAの総量の計算は、以下の方法により決定される:19,626用量×l5μg/用量=294,390μg 総HA。62,160×106個の細胞で割った場合=
106個のMDCK細胞あたり、4.73μg HA。
【0116】
(実施例9)
(トリプシン無しでの、株B/Harbin/7/94によるMDCK.5F1クローンの感染)
クローンMDCK.5F1に由来する細胞を、実施例3と同様だが、15g/Lのマイクロキャリア濃度を用いて増殖させた。8日目に、培養物を、B−Harbin/7/94と称されるヒトインフルエンザウイルスにより感染させた。M.O.I.は1:10,000であった。ウイルス複製を促進させるためのトリプシンを添加しなかった。このアッセイからのデータおよび結果は、表11および12に再現される。収量は、35Lの収集物に基づいて、38,150用量、および16.35μg HA/mlのSRD値であった。
【0117】
【表11】
感染の時点での細胞の総数は、以下の方法により計算される:16.7×106個のMDCK細胞/ml×3700mlは、61,790×106個の細胞に等しい。
【0118】
【表12】
HAの総量の計算は、以下の方法により決定される:38,150用量×l5μg/用量=572,250μg 総HA。61,790×106個の細胞で割った場合=
106個のMDCK細胞あたり、9.26μg HA。
【0119】
もう一度繰り返すと、本発明のプロセスは、トリプシンの存在を必要とする先行技術のプロセスにより与えられる収量と同じか、またはそれより多い収量を与えることを示す。
【図面の簡単な説明】
【0120】
【図1】図1は、本発明の灌流手段において使用されるデカンターの前断面図である。
【図2】図2は、上記デカンターの上正面図である。
【図3】図3は、上記デカンターの下正面図である。
【技術分野】
【0001】
本出願は、2004年5月20日に出願された米国特許仮出願第60/572,612号の利益を主張する。この出願は、本明細書中で参考として援用される。
【0002】
(発明の分野)
本発明は、一般に、予防目的、診断目的、免疫治療目的または治療目的のためのインフルエンザウイルスまたは抗原の製造のための商用規模のプロセスに関する。特に、本発明は、インフルエンザワクチン製造、そして特にインフルエンザA型およびインフルエンザB型を含むヒトワクチン製造のための、Madin−Darby Canine Kidney(MDCK)由来の細胞培養ベースのプロセスを提供する。
【背景技術】
【0003】
(発明の背景)
伝統的に、商用のインフルエンザワクチンは、胚含有の鶏卵においてワクチンウイルス種を増殖させることによって製造されている。このウイルスは、尿膜流体から収集され、そして処理されてワクチンを形成する。しかしながら、この手順は、大きな労力がかかり、そして卵当たりの収量が低いことを生じるという不利を有し、それらの要因は、流行期間の間に深刻な制限を提供する。従って、胚含有鶏卵の方法について、コスト、時間および収量の不利を克服する、インフルエンザワクチンの大規模製造のための必要性が存在する。
【0004】
上記の代替的な方法は、インフルエンザ粒子またはウイルスタンパク質を製造するための細胞培養の使用を含む。
【0005】
細胞培養を用いて製造されたインフルエンザワクチンは、卵において製造されたワクチンよりも安全であると考えられ、そして子供および成人により体験される、卵ベースのワクチンに対する過感受性を誘導しないはずである。このようなワクチンはまた、特に高齢者において、広範な種の野生株に対するよりよい保護を授けるはずである。なぜなら、ウイルス複製の忠実度は、卵におけるよりも細胞培養におけるほうがずっと大きいと考えられるからである(非特許文献1)。さらに、流行性および汎発性の場合において、卵の供給の制限に起因した、インフルエンザワクチンの現在可能である供給よりもより大きな供給が製造され得る。
【0006】
インフルエンザウイルスA型およびインフルエンザウイルスB型の伝播は、種々の組織培養系において実証されており、その培養系としては、細片化された(minced)ニワトリ胚、ヒト胎児肺およびヒト胎児腎臓、サル腎臓およびウシ胎仔腎臓が挙げられる。
【0007】
特に、イヌ腎臓(canine kidney)細胞は、インフルエンザウイルスの製造に関して有用であると示されているが、低収量であり、すなわち、ワクチン製造目的には不十分である。
【0008】
イヌ腎臓細胞は、そもそも、S.H.MadinおよびN.B.Darbyによって、外観上正常である、成体のメスCocker Spanielの腎臓より1958年に誘導された(非特許文献2)。このMDCK細胞株は、1964年にATCCにより(番号CCL34の下で)受託され、そしていくつかのウイルス(水疱性口内炎ウイルス、ワクシニアウイルス、コサッキーウイルス、レオウイルス、およびアデノウイルスが挙げられる)の複製を可能にするとして同定された。
【0009】
インフルエンザウイルスB型の連続伝播は、最初に、MDCK細胞株において、Greenによって1962年に実証された(非特許文献3)。伝播は、連続する6回の組織培養の継代の各々における細胞変性効果およびヘマグルチニン(HA)(インフルエンザウイルスの主要な糖タンパク質)の存在によって、そして組織培養流体における卵感染力価によって、明らかであった。しかしながら、これらのデータは小規模に限定され、そしてワクチン目的のためのウイルス粒子の大規模生産を達成する手段もウイルスタンパク質の大規模生産を達成する手段も提供しない。Gaushらは、MDCK細胞がまたインフルエンザA型の感染に感受性であることを1966年に実証した。しかしながら、この論文は、感染性のみを報告しており、そしてこの培地におけるウイルスの伝播の問題については取り組んでいなかった(非特許文献4)。
【0010】
1975年に、Tobitaらは初めて、トリプシンを含むオーバーレイ培地において、構築されたMDCK細胞株において、広範な種々のインフルエンザウイルスA型の増殖を記載した。これらウイルス伝播は、それらウイルスの以前の継代歴に関係なく十分に定義されるプラークを形成し、そしてHAポリペプチドを切断し、これによってインフルエンザウイルスの成熟を加速させることにトリプシンが寄与することを提案した。しかし、このトリプシンの使用がもたらす進歩にもかかわらず、寒天培地におけるウイルスの単離は、ウイルスの大量生産を達成する手段を提供しなかった。同参考文献において、またMDCK細胞が、患者の喉洗浄物(throat washing)からのインフルエンザウイルスA型の一次単離に関して首尾よく使用された。
【0011】
続いて、Reuvenyらは、バッチ培養において、セルロースベースのマイクロキャリア上でのMDCK細胞において、インフルエンザウイルスA型を増殖させた(非特許文献5)。得られたインフルエンザウイルスの力価は、胚含有卵において得られたものと同様にトリプシン含有培地中に存在した。
【0012】
特許文献1(Brownら)は、ウイルス培養の間にトリプシンのようなタンパク質加水分解酵素を培養物中に含むことによって、同じ液体培養物の逐次数の細胞中でのインフルエンザウイルスの複製のための方法を記載する。タンパク質分解酵素は、HAを機能性にさせ、そしてこれによって既往の液体培養技術の1段階増殖サイクルに勝るために必要とされる。このことは、液体細胞培養物からの潜在的な「商用」インフルエンザワクチン製造の最初の記述である。しかしながら、現在、溶液中のトリプシンの存在が、MDCK細胞の特定の割合をそれらの固体支持体から浮遊させるという不利を有することが認められる。結果的に、この特許されたプロセスの潜在的な有用性にもかかわらず、トリプシンを要求することが、インフルエンザワクチンの商用生産に対する深刻な制限である。従って、インフルエンザワクチンの生産のための細胞培養ベースの商用プロセスのための必要性がさらに存在する。このことは、以下の記述によって支持される:非特許文献6。
【0013】
また1995年に、World Health Organization(WHO)は、その分野の専門家たちが、インフルエンザウイルスの生産のための細胞培養ベースのプロセスに関する近年の進展を評価するために会合した、1995年の夏に開催された議論を要約する摘要を発表した(非特許文献7)。この文書は、迅速にスケールアップの可能な、細胞培養物系において大量のインフルエンザワクチンの迅速な生産を達成するためのさらなる研究を奨励する。
【0014】
研究目的に関して、細胞培養物中でのウイルスの増殖のため、MDCK細胞およびベロ(Vero)細胞が、いくつかのウイルスの良好な生産体であるとして最も頻繁に参照される。しかしながら、大規模生産目的のため、いくつかの要因(例えば、1つのウイルスまたは複数のウイルスに対する感受性、ウイルス力価の出来高(output)、足場依存性、腫瘍形成性、など)が、別の細胞株の選択以上に特定の細胞株の選択に影響を与える。
【0015】
MDCK細胞はいくつかのウイルス株に感受性であるが、結果的に乏しいウイルス力価により、大規模生産目的のためのこの細胞株の有用性が制限され得る。MDCK細胞株の特性は、いくつかの研究の課題であった。1970年に、Leightonら(非特許文献8)は、MDCK細胞が、コラーゲンでコーティングされたセルローススポンジ上での三次元的な組織培養の組織病理学的調製物において、乳頭状の腺癌の形態学的パターンを提示することを報告した。この細胞株の新生物性の特色(quality)は、11日齢または12日齢のニワトリ胚に注入されるた細胞懸濁物が、脳転移の多くの病巣を形成する場合により実証された。
【0016】
細胞株の腫瘍形成性の評価は、生物学的産物の製造における使用に対するその望ましさを評価するために重要である。米国食品医薬品局のCenter for Biologics Evaluation and Researchは、生物学的物品の製造のための基体(substrate)として細胞培養物を使用する場合に考慮する要点を発表した(非特許文献9を参照のこと)。このような要点の1つは、使用される細胞株の腫瘍形成性であり、そしてFDAは、インビボ腫瘍形成性試験のためのガイドラインを考案した。これらのガイドラインは、とりわけ、ヌードマウス(nu/nu)において、皮下経路または筋肉内経路により投与された細胞を試験することを指示する。
【特許文献1】米国特許第4,500,513号明細書
【非特許文献1】Katz,J.M.ら、J.Infect Diseases(1989)160:191
【非特許文献2】American Type Culture Collection(ATCC)、Catalogue of Cell Lines and Hybridoma(1992)第7版、p21
【非特許文献3】Green、Science(1962)138:42
【非特許文献4】Gaushら,Proc.Soc.Exp.Biol,and Med.(1966)122:931
【非特許文献5】Develop.Biol.Standard(1982)50:115
【非特許文献6】Kodihalliら、J.Virol.(1995)69(8):4888:「Embryonated chicken eggs are currently the only host in which sufficient quantities of virus can be cultivated economically and within the short time necessary to ensure a vaccine supply」
【非特許文献7】Bull.W.H.O.73(4):431,1995
【非特許文献8】LeightonらCancer(1970)26:1022
【非特許文献9】Points to Consider in the Characterization of Cell Lines Used to Produce Biologicals,Office of Biologics Research and Review,Center for Drugs and Biologics,FDA(USA),1993
【発明の開示】
【課題を解決するための手段】
【0017】
(発明の要旨)
従って、本発明は、インフルエンザウイルス粒子またはインフルエンザタンパク質の大規模生産のためのプロセスに関し、このプロセスは、以下の工程を包含する:
(a)上記インフルエンザウイルスを複製可能であるMDCK細胞株を増殖させる工程;
(b)このMDCK細胞株にインフルエンザウイルス株を感染させ、そしてインキュベートしてそのウイルスの複製の可能にする工程;および
(c)複製されたウイルスを収集し、ウイルス粒子またはそのタンパク質を精製する工程。
【0018】
本発明は、ウイルス感染に高感受性であるMDCK細胞株に由来し、そしてそれら由来の親細胞株よりも高いウイルス力価でインフルエンザウイルスを産生する細胞株を、さらに提供する。
【発明を実施するための最良の形態】
【0019】
(発明の詳細な説明)
1実施形態において、本発明のMDCK由来の細胞株は、インフルエンザウイルスの多段階複製を可能にする。別の実施形態において、本発明のMDCK細胞株は、足場依存性であり、かつ非腫瘍形成性である。
【0020】
本発明は、ウイルス感染に超感受性であるMDCKクローンの誘導体を提供する。「超感受性」との記載は、少なくとも1つのウイルスに高感受性であり、これによって親MDCK細胞株よりもより高力価のウイルス粒子を産生するMDCK由来の細胞株を示すために、使用される。この誘導化クローン(MDCK.5F1)は、番号CRL−12042の下で1996年2月8日にAmerican Type Culture Collection(ATCC)に受託された。1実施形態において、MDCK由来の本発明の細胞株は、FDAガイドラインに従って行われた試験において非腫瘍形成性であり、そしてこれによって予防目的、診断目的、免疫治療目的または治療目的のためのウイルスまたは抗原の調製における使用に適切であり得る。この細胞株は、混入する微生物の存在について試験されており、そしていずれも検出されていない。また、本発明に従って、ワクチン製造のためのインフルエンザウイルス粒子またはインフルエンザウイルスタンパク質の大規模生産のためのプロセスが提供される。
【0021】
また、本発明の1局面に従って、インフルエンザワクチンの製造のためのインフルエンザウイルス粒子またはインフルエンザウイルスタンパク質の生産のための、大規模で細胞培養によるマイクロキャリアベースの商用プロセスが提供される。
【0022】
本発明のプロセスは、インフルエンザウイルスの複数回の複製のためにMDCK細胞株を使用して実施される。1実施形態において、本発明のプロセスにおいて使用されるMDCK細胞株は、ATCC細胞株番号CRL−12042と同じ生物学的特性を有する。1実施形態において、本プロセスにおいて使用される細胞株は、ATCC番号CRL−12042の下で受託された、MDCK.5F1として本明細書中で称されるMDCK細胞株のクローンである。
【0023】
本発明のMDCK由来の細胞株は、ウイルス感染に高感受性である。この文脈において、ウイルス感染に「高感受性である」とは、その細胞株が、少なくとも1つのウイルス種に対して親細胞株によって産生される力価よりも高い力価を産生し得ることを意味する。
【0024】
1実施形態において、「高感受性」とは、親細胞株によって産生される力価よりも少なくとも約1.2倍の力価でウイルスを産生し得る細胞株として定義される。1実施形態において、より高感受性の細胞株は、3B5、5Fl、1D11、5H12、9C2、9D9、P79、9E9、7C1およびP123からなる群より選択されるクローンである。
【0025】
さらに1実施形態において、より高感受性とは、それらの同じウイルスに関して、親細胞株によって産生される力価の少なくとも2倍の力価で多数のウイルスを産生し得る細胞株として定義される。1実施形態において、このようなクローンは、3B5、5F1、5H12、9C2、7C1およびP123からなる群より選択される。
【0026】
1実施形態において、より高感受性とは、同じウイルスの2つの異なる株について、親のウイルス力価の少なくとも約2倍を産生し得るとして定義される。1実施形態において、このようなクローンは、RSウイルスおよびインフルエンザA型およびインフルエンザB型に関して親のウイルス力価の2倍を産生し得る。1実施形態において、このようなクローンは、5F1および5H12からなる群より選択される。
【0027】
1実施形態において、細胞株は、インフルエンザウイルス、RSウイルス、パポバウイルス、パラインフルエンザウイルス、水疱性口内炎ウイルス、ワクシニアウイルス、コクサッキーウイルス、レオウイルス、パルボウイルス、アデノウイルス、ポリオウイルス、麻疹ウイルス、狂犬病ウイルス、ヘルペスウイルスおよび他のウイルスからなる群より選択されるウイルスによって感染され得る。
【0028】
1実施形態において、ウイルスは、インフルエンザA型、インフルエンザB型およびインフルエンザC型;RSウイルス;パポバウイルス;水疱性口内炎ウイルス(インド株);コクサッキーウイルスB−5;レオウイルス2型およびレオウイルス3型;ならびにアデノウイルス4型およびアデノウイルス5型からなる群より選択される。
【0029】
1実施形態において、ウイルスは、インフルエンザA型、インフルエンザB型およびインフルエンザC型、ならびにRSウイルスからなる群より選択される。
【0030】
1実施形態において、ウイルスは、ヒトインフルエンザ株、ウマインフルエンザ株、ブタインフルエンザ株またはトリインフルエンザ株から選択される。
【0031】
1実施形態において、ウイルスは、ヒトインフルエンザA型、ヒトインフルエンザB型またはヒトインフルエンザC型から選択される。
【0032】
1実施形態において、本発明の細胞株は、ヒトインフルエンザウイルスA型またはヒトインフルエンザウイルスB型によって感染され得る。
【0033】
1実施形態において、本発明の細胞株は、ヒトインフルエンザA型およびヒトインフルエンザB型のの両方に感染され得る。
【0034】
1実施形態において、本発明の細胞株は、トリプシン、キモトリプシン、ペプシン、パンクレアチン、パパイン、プロナーゼおよびカルボキシペプチダーゼのようなタンパク質分解酵素の添加によってインフルエンザウイルスの多段階複製を可能にする。1実施形態において、このような細胞株は、トリプシンの添加によってインフルエンザウイルスの多段階複製を可能にする。
【0035】
あるいは、本発明の細胞株は、トリプシン、キモトリプシン、ペプシン、パンクレアチン、パパイン、プロナーゼおよびカルボキシペプチダーゼのようなタンパク質分解酵素の添加を必要とすることなく、インフルエンザウイルスの多段階複製を可能にする。1実施形態において、このような細胞株は、トリプシンの添加を必要とすることなくインフルエンザウイルスの多段階複製を可能にする。
【0036】
どのウイルスまたはウイルス株がタンパク質分解酵素の使用を必要とし得るかは、当業者に明らかである。
【0037】
本発明の細胞株を懸濁して培養し得るが、この細胞株を足場依存性様式で増殖させることが好ましい。1実施形態において、この細胞株はマイクロキャリアビーズ上で増殖し得、これによって高密度の細胞が細胞培養物中に得られることを可能にする。
【0038】
1実施形態において、本発明のMDCK由来の細胞株は、非腫瘍形成性である。1実施形態において、本発明の細胞株は、軟寒天中で最小限の効率(すなわち、1%未満の効率)で増殖する。1実施形態において、本発明の細胞株は、ヌードマウスにおいて、少なくとも3ヶ月間観察される場合、小結節を生じない。
【0039】
本発明は、バルク量でウイルス粒子またはウイルスタンパク質の産生のために、本発明に従う細胞株の使用をさらに企図する。
【0040】
このようなウイルス粒子またはタンパク質は、宿主におけるウイルス感染の予防のためのワクチンの製造において使用され得る。1実施形態において、この宿主は哺乳動物である。哺乳動物としては、例えば、ヒト種、ウマ種、ブタ種が挙げられる。別の実施形態において、この宿主はヒトである。さらなる実施形態において、この宿主はトリ種(例えば、ガチョウまたはニワトリ)である。
【0041】
本発明のプロセスは、足場依存性MDCK細胞が培養物中で増殖する能力にトリプシンの存在が影響を与えない限り、トリプシンの存在下で実施されてもトリプシンの非存在下で実施されてもよい。1実施形態において、本プロセスは、約4μg/ml以下の濃度のトリプシンの存在下で実施される。あるいは、本プロセスはトリプシンの非存在下で実施される。
【0042】
本明細書中に記載のプロセスは、RSウイルスおよび種々のインフルエンザウイルス(例えば、ヒトインフルエンザウイルス株、ウマインフルエンザウイルス株、ブタインフルエンザウイルス株およびトリインフルエンザウイルス株)の産生のために意図される。1実施形態において、本明細書中に記載されるプロセスは、ヒトインフルエンザA型、ヒトインフルエンザB型、またはヒトインフルエンザC型の産生のために意図される。1実施形態において、本明細書中に記載されるプロセスは、ヒトインフルエンザA型またはヒトインフルエンザB型の産生のために意図される。1実施形態において、本明細書中に記載されるプロセスは、ヒトインフルエンザA型またはヒトインフルエンザB型のいずれかのウイルス型に感染され得るMDCK細胞株を使用して、ヒトインフルエンザA型およびヒトインフルエンザB型の産生のために意図される。
【0043】
本発明のプロセスは、106個のMDCK細胞あたり4μg HAの範囲である収量を提供する。実施例9に記載されるように、本プロセスは106個のMDCK細胞当たり4μg HAより多くの量のウイルスタンパク質を生じ、より具体的には106個の細胞当たり9μg HAの範囲で生じる。
【0044】
「大規模プロセス」とは、多量のインフルエンザウイルスを製造するためのプロセスを意味する。このようなプロセスは、通常、培養フラスコとは対照的に、バイオリアクターにおいてなされる。このようなバイオリアクターは、(最終的な収量)/(必要とされる用量)に依存して変動性のサイズであり得る。例えば、このようなバイオリアクターは、約5リットルのサイズ(約4リットルの作業容量)であり得るか、または5000リットルまでであり得る。無論、大規模な細胞培養ベースのワクチン製造の当業者に明らかであるように、全ての反応物、培地、栄養分、細胞濃度、マイクロキャリア濃度、灌流速度などは、そのバイオリアクターのサイズに従って調節され得る。
【0045】
本発明のプロセスは、懸濁培養において実施され得るが、細胞濃度を上昇させる(そしてこれによってウイルス出来高を向上させる)ためにマイクロキャリア手段において実施されることが好ましい。例えば、デキストランポリマー(CytodexTM)として当該分野で通常公知の型のマイクロキャリアビーズが選択され得る。これらのマイクロキャリアは、約5g/L〜25g/Lの範囲の濃度で使用され得る。1実施形態において、このマイクロキャリア濃度は、約10g/L〜25g/Lの範囲にある。1実施形態において、このマイクロキャリア濃度は、約15g/L〜約20g/Lの範囲にある。
【0046】
灌流手段は、プロセス中の細胞増殖およびウイルス複製の両方を最大にするように導入される。灌流は、同時に培養培地中の潜在的に毒性の副産物の蓄積を避ける手段を提供しながら、栄養分の一定の供給を可能にする。灌流を介して、栄養分の型および質量は、プロセスの種々の段階の間に変動され得る。例えば、血清は、増殖期の間に細胞に導入され得るが、理想的には、細胞の集密期(confluency)においてそしてウイルスの導入前に排除されるべきである。その灌流速度は、適切な栄養供給を提供するために、細胞増殖の間に徐々に上昇され得る。灌流はウイルス複製の間に継続される。
【0047】
灌流速度は、プロセスの段階に依存して、1日当たりバイオリアクターの約0.5〜4倍容量の間で調節される。
【0048】
灌流手段が、バイオリアクターに追加され得る。この灌流手段は、そのバイオリアクター中に培地を連続的に導入するための流入口手段、およびそのバイオリアクターから消費された培地を連続的に除去するための2つの出口手段を備え(これによってそのバイオリアクター中でマイクロキャリアの懸濁を介して培地の連続的な流動を形成する)、そしてその出口手段と一体化したデカンターを備える。本明細書中に記載されそして本発明のプロセスにおいて使用されるデカンターは、上記に提案されるような灌流手段を用い、10g/Lの過剰なマイクロキャリア濃度を使用して、上方へと移動しそしてその出口手段を通って抜け出るかまたはその出口手段を詰まらせるかのどちらかのマイクロキャリアの傾向によって細胞増殖が制限されることが認められる場合の、5L(3.7Lの作業容量)のバイオリアクターに対して具体的に設計された。従って、より高いマイクロキャリア濃度を達成するため、以下の、デカンター内で乱流形成を最小にし、そして懸濁の上方への流速よりも速いマイクロキャリアの沈降速度を達成する特徴を有するデカンターが、構築された。
【0049】
本発明の好ましい実施形態に従って、図1に示されるようなデカンター10は、下側チャンバ1を備え、この下側チャンバ1はより大きな上側チャンバ2と放射状プレート3によって接続される。2つの出口手段4は、このデカンターの頂部において上側チャンバ2に取り付けられる。この下側チャンバ1および上側チャンバ2は、半円筒形状であり、そして同軸上に向けられる中央の円形空洞5を取り囲み、この中央の円形空間5は、このバイオリアクターの中央回転シャフト、および本発明のプロセスをモニタリングするためにこのバイオリアクター中に導入される種々のプローブを収容するように意図される。中実の金属壁は、このデカンターを完全にする。
【0050】
図2は、上記の構成要素2、4および5の上正面図を示す。
【0051】
図3は、デカンターの下側チャンバ1の下正面図を示す。下側チャンバ1は、規則正しく分配した複数の長手軸チャネル6を有し、この長手軸チャネル6は、円周方向に向けられる長手軸方向の区画7によって分離され、この長手軸チャネルは、そのデカンターの上側チャンバ2および放射状プレート3を介して互いに連絡している。1実施形態において、長手軸チャネル6は同一であり、そして各々は円形の断面を有する。さらに、複数の長手軸チャネル6は、同軸上に向けられた中央の円形空洞5の周りに規則正しく分配され、その周囲の長手軸チャネル6は、円周方向に向いた長手軸方向の区画7によって互いから分離されている。
【0052】
灌流手段の出口手段は、感染の際に廃液レザバから収集レシピエントへと切り換えられる。連続的な収集は、複製が完了するまで(約4〜5日)生じる。
【0053】
本発明のプロセスは、10:1〜1:1010の間の感染多重度(M.O.I)でのウイルスによってMDCK細胞培養物を播種することによって実施される。1実施形態において、このプロセスは、1:10〜1:108の間の感染多重度(M.O.I.)で実施される。1実施形態において、このプロセスは、約1:104〜1:107のM.O.Iにおいて実施される。1実施形態において、1:105〜1:106のM.O.Iにおいて実施される。
【0054】
1実施形態において、本発明のプロセスにおけるMDCK細胞の増殖は、温度約33℃〜40℃において約7日〜10日間実施される。1実施形態において、このプロセスにおけるMDCK細胞の増殖は温度約36℃〜38℃において、約7日間実施される。
【0055】
1実施形態において、本プロセスにおけるMDCK細胞の増殖は、温度約37℃で約7日間実施される。1実施形態において、本発明のプロセスにおけるウイルス複製は、温度約30℃〜37℃において約4日〜6日間実施される。1実施形態において、このプロセスにおけるウイルス複製は、温度約32℃〜約34℃で約5日間実施される。1実施形態において、このプロセスにおけるウイルス複製は、温度約33℃で実施される。
【0056】
ウイルス粒子またはウイルスタンパク質は、以下の方法で精製される。ウイルス収集物の濾過の後に、ホルムアルデヒドを用いた濾過物の不活性化が続く。次いで、得られた不活性化ウイルス懸濁物は遠心分離され、そして富化されたウイルス画分は、ワクチン調製物における使用のために選択される。
【0057】
本発明のワクチンを投与する工程を包含する、哺乳動物におけるインフルエンザ感染の予防のための方法もまた、提供される。
【0058】
精製されたウイルス粒子またはタンパク質を使用してインフルエンザウイルス感染の検出のための診断キットを生産するプロセスもまた、提供される。
【0059】
哺乳動物におけるインフルエンザウイルス感染の検出および診断のためのキットを生産するための、単離されたバルクのウイルス粒子またはウイルスタンパク質の使用もまた、提供される。
【0060】
本発明者らのプロセスの使用の特定の例および選択されたヒトインフルエンザウイルスA型株およびヒトインフルエンザウイルスB型株の増幅は、以下に考察される。本発明の配置および構成要素において、本発明の本質および範囲から逸脱することもなく、その物質の利点の全てを損なうこともなく、種々の変更がなされ得ることは明らかであり、以下に記載される形態は、単なる好ましい実施形態または単なる例示的な実施形態である。
【実施例】
【0061】
実施例1は、クローンMDCK.5F1の誘導を記載する。実施例2は、MDCK.5F1細胞株の純度を検討する。実施例3は、トリプシン有りおよび無しでの、MDCK.5F1細胞株における、インフルエンザウイルスの増殖を示す。実施例4は、MDCK.5F1についての腫瘍形成能研究のまとめである。実施例5は、MDCK.5F1の細胞懸濁物による、胸腺欠損ヌードマウスの接種後の結果を議論する。実施例6は、プロセスの特定の実施形態の段階的な説明である。実施例7は、トリプシン有りでの親MDCK細胞とインフルエンザ株A/Shanghai/ll/87とを使用して実行されたアッセイの結果を記載する。実施例8は、トリプシン無しでのMDCK.5F1細胞とインフルエンザ株A/Shanghai/ll/87とを使用して実行されたアッセイの結果を記載する。実施例9は、トリプシン無しでのMDCK.5F1細胞とインフルエンザ株B/Harbin/7/94とを使用して実行されたアッセイの結果を記載する。
【0062】
(実施例1)
(クローンMDCK.5F1の誘導)
MDCK細胞No.CCL 34をAmerican Type Culture Collection、Rockville、Marylandから取得した。このストックを、3.4×106細胞を含む1mlアンプル中の凍結状態で受け取った。この細胞株は、54回継代したものだった。
【0063】
継代後、MDCK細胞を、64回目の継代で回収し、10%(v/v)ウシ胎児血清(FBS)を含み、1:1の比でのダルベッコ変法イーグル培地(DMEM)および培地199からなる栄養培地(DMEM−199)中に希釈した。次に、各ウェルが細胞を1個未満受容するように、希釈した細胞懸濁物を96ウェルプレートにアリコートし、溶液中の細胞の均一な分配とみなした。これらのプレートを、37℃でCO2インキュベーター中に置き、増殖について、これらのウェルをスコア付けするために、一週間の間隔において光学顕微鏡下で調べた。
【0064】
クローンにおいて求めた特徴を、以下から選択した:
(1)ウイルス感染に対する、親株よりも高い感受性(すなわち、クローンが、親株より高いウイルスの力価を生成する);
(2)1つより多いウイルスに対する、より高い感受性(一実施形態において、インフルエンザウイルスのいくつかの株に対する感受性);
(3)タンパク質分解酵素(例えば、トリプシン)の添加の必要なく、インフルエンザウイルスの多段階の複製を可能にする能力;および、必要に応じて、
(4)足場依存性(すなわち、培養中でのより高い細胞の濃度を得るため)。
表1は、トリプシンの添加なしでの、インフルエンザウイルスA型およびインフルエンザウイルスB型による感染に対する、いくつかのクローンの感受性を示す。
【0065】
【表1】
「高感受性」を、TCID50により評価した場合に、少なくとも親細胞株の約1.2倍の感受性として定義した。クローン3B5、5F1、1D11、5H12、9C2、9D9、P79、9E9、7C1およびP123を、感受性が高いとして同定した。クローン3B5、5F1、1D11、5H12、9C2、9D9、7C1およびP123を、親株の少なくとも2倍の感受性であるとして同定した。
クローン5F1および5H12を、感受性が最も高い2クローンとして選択し、そして5F1を、MDCK.5F1として本明細書中で称される細胞株を樹立するために選択した。
【0066】
細胞の世代数を、クローニングの時点において0として定義し、続く各々の細胞培養物における細胞の数え上げにより計算した。最初、マルチウェルプレート培養で培養物を継代し、最終的にプラスチック製フラスコに移した。
さらに、実験を、親MDCK細胞株およびMDCK.5F1クローンの、RSウイルスによる感染に対する感受性を測定するために、このウイルスを用いて実行した。ウイルス力価は、MDCK細胞株は、RSウイルスの宿主Hep2細胞株よりも感染に対し、より非感受性であったが、MDCK.5F1クローンは、このウイルスによる感染に対し、親MDCK細胞株よりも約10倍感受性が高かったことを示した。
【0067】
(実施例2)
(MDCK.5F1のクローン性の決定)
選択された任意の特定のクローンのクローン性を決定するパラメータは、クローンが選択されたマルチウェルプレート上のパーセンテージ増殖である。選択された培養物が実際にクローンである可能性(すなわち、P(1))は、このパーセンテージから決定される(CollerおよびColler、Methods in Enzymology、vol.121、pp.412〜417(Academic Press、1986)を参照のこと)。
【0068】
5%のウェルが、このクローニングにおいて増殖を示したと仮定すると、細胞株MDCK.5F1が単一の細胞に由来する可能性は、≧97.5%である。
【0069】
299アンプルの親細胞バンク(Master Cell Bank)(MCB)および283アンプルの製造者の作業用細胞バンク(Working Cell Bank)(WCB)を、MDCK.5F1細胞株から調製した。これらのバンクを、Good Manufacturing Practiceの方針上のカナダ国の指針にしたがって調製し、真菌因子、酵母因子、マイコプラズマ因子、細菌因子およびウイルス因子の形態での汚染について評価した。いかなる種類の汚染も見出さなかった。
【0070】
持続可能かつ生存可能な培養物を、調製した細胞バンクから取得し、細胞株を、WCBよりも50回集団倍化したものについての産物の産生の安定性、形態学、腫瘍形成能およびイソ酵素の特徴について試験した。これらの結果は、これらの特徴について安定である細胞株を示した。
【0071】
(実施例3)
(トリプシンの添加有りおよびトリプシンの添加無しでの、MDCK.5F1クローンにおける、インフルエンザウイルスの増殖を示す実験)
トリプシンの存在下および非存在下において、クローンMDCK.5F1の培養物中で、インフルエンザウイルスが複製する能力を決定するために、実験を実行した。インフルエンザ株A/Johannesburg、インフルエンザ株A/Texasおよびインフルエンザ株B/Harbinを使用した3つの実験の結果を表2に示す。
【0072】
【表2】
これらの結果は、血球凝集(HA)力価の値により示されるように、トリプシン有りおよび無しで、インフルエンザが、細胞培養物中で複製される場合に実質的な違いが何もないことを明らかにする。
【0073】
(実施例4)
(インビトロ腫瘍形成能研究のまとめ)
Fureszら(Develop.Biol.Stand.vol.70、pp.233〜243、S.Kargel編、Basel、1989)により記載される方法にしたがって、10%(v/v)FBSを含むDMEM−199培地中の0.6%寒天の4mlを、直径35mmのウェルを6つ備える組織培養皿内に固化させた。固化後、これらのウェルを、42℃に維持された、0.3% W/Vの非ゲル状の寒天と60,000細胞/mlの細胞濃度とを含む3mlの培地で重層した。プレートを、5% CO2、37℃でインキュベートした。光学顕微鏡観察を、3日目、7日目、10日目および14日目に行った。コロニーは、軟寒天中に球状の群を形成する、4個以上の細胞からなった。パーセンテージ有効性を、プレートした細胞の総数で割った、計数された細胞コロニーの数の比により決定した。
【0074】
【表3】
表3の結果は、MDCK.5F1細胞株が、軟寒天において最低の有効性で増殖し、これが動物において非腫瘍形成性であり得ることの示唆を示す。この特性は、18回継代後も維持され、この表現型が安定であることを示した。
【0075】
非腫瘍形成能のこの所見をさらに支持するために、発明者らは、胸腺欠損ヌードマウスにおいて、MDCK.5F1細胞株の腫瘍形成の可能性を試験した。
【0076】
(実施例5)
(MDCK.5F1クローンの細胞懸濁物の皮下接種後の、胸腺欠損ヌード(nu/nu)マウスにおける腫瘍形成の評価)
胸腺欠損ヌード(nu/nu)マウスは、外来物質に対する細胞媒介性の応答を確立できず、それゆえ同種異系の腫瘍細胞株および異種遺伝子型の腫瘍細胞株の増殖を補助する。このことは、接種物がインビボで新生物を形成する能力の評価を可能にする。
【0077】
6週齢のヌードマウスを、約1×107細胞の試験物質(MDCK.5F1)により皮下接種し、84日間臨床的に置き、そして検死した。ポジティブコントロールの細胞およびネガティブコントロールの細胞で接種したヌードマウスを、同様に処置した。接種部位(皮膚)、肺、肩甲骨のリンパ節および肉眼的病変を、加工、切り出し、染色し、そして顕微鏡で調べた。この実験のさらなる詳細は、以下に示される。
【0078】
(試験物質およびコントロール物質の接種)
各ケージ内の全てのマウスを、同様に処置した。
【0079】
各マウスを、以下に記載の0.2mlの適切な接種物により、肩甲骨の間に皮下接種した。22ゲージ針を、接種のために使用し、全てのマウスを、同じ日に接種した。
【0080】
群1および群2:試験物質、MDCK.5F1(5×107細胞/mlの濃度で)。
【0081】
群3および群4:ポジティブコントロール(5×107細胞/mlの濃度の18C1−10T細胞)。
【0082】
群5および群6:ネガティブコントロール(1×107細胞/mlの濃度のSHE細胞)。
【0083】
全ての動物を、毎作業日(working day)観察し、接種部位を、84日目までの期間、週に二回触診した。
【0084】
(結果)
(臨床所見)
全てのポジティブコントロールのマウスは、接種部位において、少なくとも一つの長さが1cmより大きい大型の塊を有したので、これら全てを、接種後14日目に、屠殺および検死した。
【0085】
全てのネガティブコントロールのマウスを、接種後84日目に、屠殺および検死した。
【0086】
10匹の試験物質(5F1)マウスのうち9匹を、接種後84日目に、屠殺および検死した。進行していた接種部位の病変が退行し始めたので、1匹の5F1接種されたマウスを、接種後33日目に、屠殺および検死した。この病変は、後に嚢胞であることが明らかになった。
【0087】
(触診)
ポジティブコントロール物質により接種した10匹のヌードマウスは、接種後14日目までに、少なくとも一つの長さが1cmより大きい触知可能な病変を有した。
【0088】
10匹のネガティブコントロール物質を接種したヌードマウスの接種部位において、小型の非進行性の病変が触知可能であった。これらの病変は、最初、接種後4日目に発覚し、観察期間の間、10匹のネガティブコントロールのマウスのうち8匹において残存した。触診の結果を、表4にまとめる。
【0089】
10匹の5F1マウス全てが、接種後4日目までに病変を有した。10匹の5F1マウスのうちの9匹において、これらの病変は、小さく、進行しなかった。接種後56日目までに、試験物質のマウスのうち8匹において、触知可能な病変は存在しなかった。1匹の5F1を接種したマウスは、25日目と28日目との間に、大きさが顕著に進行し、そして接種後32日目までに、大きさが顕著に減少した接種部位の病変を有した。この病変を、顕微鏡検査により、嚢胞として同定した(表6を参照のこと)。病変を示す他のマウスは、局在化した炎症を有した。
【0090】
【表4】
(肉眼的な検死所見)
処置に関連する肉眼的な検死所見を、表5にまとめる。
【0091】
【表5】
接種部位における塊を、全てのポジティブコントロールの動物において見出した。10匹の5F1マウスのうち3匹、および10匹のネガティブコントロールのマウスのうちの6匹において、接種部位に、塊または小結節を見出した。
【0092】
(顕微鏡の所見)
目的の病変を、表6にまとめる。
【0093】
【表6】
新形成(線維肉腫)を、全てのポジティブコントロールのマウス中の接種部位において診断した。線維肉腫は、織り交ぜられたパターンを伴う、可変の密度の束で配置される紡錘細胞(spindloid cell)からなった。コラーゲンの沈着は、最小限であった。隣接する組織は新生物により圧迫されるが、ほとんど新生物により浸潤されなかった。
【0094】
どのネガティブコントロールのマウスにおいても、新生物は診断されなかったが、骨様の増殖の病巣を、ネガティブコントロール物質のマウスのうちの2匹中の接種部位において確認した。これは、ネガティブコントロールとして、SHE細胞の分化および増殖の選択を表すと考えられる。
【0095】
どの5F1マウスにおいても、新生物は診断されなかったが、1匹のマウスにおいて嚢胞を確認し、他の1匹の5F1マウスにおいて病巣の亜急性の炎症を確認した。
【0096】
(結論)
10匹全てのポジティブコントロールのマウスの接種部位において、線維肉腫を診断した。
【0097】
どのネガティブコントロールにおいても、どの試験物質のマウスにおいても、新生物は存在しなかった。
【0098】
上記研究の条件下で、試験物質MDCK.5F1は、腫瘍形成性であるとはみなされない。
【0099】
(実施例6)
(一般的な手順)
(a)細胞の増殖)
バイオリアクターに播種する前に、MDCK.5F1(ATCC番号CRL−12042)細胞を、細胞の増殖のために、数回継代した。約5〜10×106細胞を、37℃の水浴で解凍し、栄養培地を含むポリスチレン製の細胞培養フラスコに移し、そして37℃でインキュベートした。3〜4日後、細胞をこのフラスコから乖離し、5個の他のフラスコに播種するために使用した。次に、この5個のフラスコを、同様な様式で、20個のフラスコに播種するために使用した。細胞増殖のために使用した栄養培地は、脱イオン水で調製された1:1の比でのダルベッコ変法イーグル培地および培地199であり、4.5g/Lのグルコース、0.58g/Lのグルタミンおよび1g/Lの炭酸水素ナトリウムを含んだ(DMEM−199)。この栄養培地に、10%のγ線照射されたウシ胎児血清(I−FBS)を補充した。細胞の継代のために、フラスコ内の細胞を乖離するために使用した溶液は、マグネシウムおよびカルシウムを含まないリン酸緩衝化生理食塩水(PBS)により調製した、0.02%エチレンジアミン四酢酸(EDTA)を含む0.25%トリプシン溶液であった。
【0100】
20個のフラスコ中の細胞を、3〜4日間増殖させ、トリプシン処理し、収集し、そして3〜5g/Lのマイクロキャリアビーズを含む1000mlの攪拌フラスコ内の3つのマイクロキャリア細胞培養に播種するために使用した。攪拌フラスコを、約50rpmに攪拌を維持しながら、37℃でインキュベートした。細胞の増殖を、5〜7日間続け、その後、細胞を、マイクロキャリアからトリプシン処理し、5Lバイオリアクター(New Brunswick of Edison、N.J.によるCelliGenTM)に播種するために使用した。
【0101】
マイクロキャリアからの細胞のトリプシン処理を、以下の方法で行った。マイクロキャリア細胞培養を、PBSおよび0.02%EDTAの溶液で2回洗浄した。二回目の細胞の洗浄後、約200mlのトリプシン溶液を、フラスコに注ぎ、攪拌しながら37℃で約20分間置いた。光学顕微鏡により決定される場合、細胞の乖離が完全になった後、細胞を、2%I−FBSを含むDMEM−199を使用して、遊離のマイクロキャリアから回収し、それからペレット化し、10%I−FBSを含むDMEM−199中に再懸濁した。細胞の計数を実行し、適切な数(約1010個)の細胞をバイオリアクターに播種するために使用した。
【0102】
細胞培養のために使用された球状のビーズまたはマイクロキャリアは、Pharmacia(スウェーデン)により製造され、Cytodex 1の商品名の下に頒布される。Cytodex 1マイクロキャリアの濃度は、1.03(g/ml、0.9%NaCl中で)であり、その大きさは、131μmと220μmとの間、180μmの平均値で変動した。細胞増殖のためのおおよその表面積は、マイクロキャリア(乾燥重量)1gあたり4,500cm2であり、マイクロキャリア1gは、約6.8×106個のマイクロキャリアを含んだ。
【0103】
(b)バイオリアクター内での細胞の播種および増殖)
15〜25g/Lの濃度のCytodex 1 マイクロキャリアを、5Lバイオリアクター(3.7L作業用容量(working volume))に導入した。バイオリアクターの播種を、以下のように実行した。先に調製した株(上記を参照のこと)から取得した約4×109〜1×1010個の細胞を、管状のガラス製のボトル内に配置した。20g/Lのマイクロキャリアの無菌溶液からの、55.5〜92.5gのマイクロキャリア(培養内の望ましい濃度に依存する)を、DMEM−199で二回すすぎ、そしてガラス製のボトル内の細胞に添加した。次に、このボトルを、10%I−FBSを含むDMEM−199で満たし、3.7Lの最終容量にした。それから、このボトルの内容物(細胞、DMEM−199中のマイクロキャリア)を、バイオリアクター容器に注ぎ、中央シャフトを約20rpmで回転させた。この容器を満たすにつれて、攪拌を50rpmに上げ、温度を37℃に調整し、溶存酸素量を5〜50%の間の空気の飽和度に維持した。培養物のpHもまた、6.8〜7.4で維持した。マイクロキャリア細胞培養の灌流を、2.5%I−FBSと0.5g/L硫酸マグネシウムとを含むDMEM−199を使用して、一日あたり0.5容量で1日目に開始した。細胞の増殖を、おおよそ7〜10日間続け、灌流の流速を一日あたり2容量まで徐々に増加させた。
【0104】
(c)ウイルス感染)
ウイルスをマイクロキャリア細胞培養に添加する前に、感染の日に、灌流を一日あたり4容量(血清を含まない)まで増加させ、温度を33℃まで下げ、酸素の分圧を15%の空気の飽和度に制御した。細胞のコンフルーエンシーおよびウイルス感染の直前(同じ日)において、培養培地を、血清を含まない同一の培養培地により置き換え、灌流速度を、7時間、一日あたり4容量まで増加させた。このことは、感染前に、培養物の血清含量を最小限のレベルまで下げることを確実にした。この期間後、灌流を停止し、ヒトインフルエンザウイルスA型またはヒトインフルエンザウイルスB型をマイクロキャリア細胞培養に導入した。1:10〜1:108の範囲のM.O.I.を取得するために、バイオリアクターへの導入の前に、ウイルスを、栄養培地(6.5gグルコース/Lを含むDMEM−199)により標準的に希釈した。次の日、細胞変性効果が完全になるまで、灌流を一日あたり2容量で維持した。MDCK細胞の完全な破壊を、代表的に5日以内に観察した。それから、インフルエンザウイルス懸濁物を含む溶出物を、収集し、当該分野において周知であるように、ワクチンを生成するために処理した。
【0105】
(d)不活性化および精製)
一価のインフルエンザウイルスの精製を、以下の様式により実行した。バイオリアクターから集めたウイルス収集物(大体15〜30Lの間)を、大型の細胞残渣を除去するために、まず1.2μmフィルター(Sartorius Sartopure GF(登録商標)、長さ10インチ、0.6m2)を通して、清澄化した。次に、インフルエンザウイルスを含む、清澄化した懸濁物を、16時間の、0.125%(V/V)ホルムアルデヒド(終濃度)の添加により不活性化した。それから、不活性化したウイルス懸濁物を、イオン交換、DNAase処理およびゲル濾過により精製した。
【0106】
濃縮されたウイルス画分を選択し、それは、ワクチン調製において使用されるべき上等な材料と表した。次に、これらの画分をプールし、現在、国際機関により推奨される、1用量あたり、1株あたり15μgのHAの終濃度を与えるように希釈した。
【0107】
(e)ワクチン調製)
ウイルスタンパク質を、ワクチンの各用量に対して15μgになるように希釈した。チメロサール(0.01%)を、保存および安定化それぞれのために添加し、ワクチンを完成させた。
【0108】
標準的な三価ワクチンのために、3つの流行性の(circulating)株の各々の一価の用量が混合され得、上記の保存剤および安定化剤を添加され得る。
【0109】
他の公知の保存剤(例えば、アミノメチルプロパノール、ソルビン酸およびポリアミノプロピルビグアナイド(polyaminopropyl biguanid)、硝酸フェニル水銀、ホウ酸フェニル水銀、ホルムアルデヒドを含む2−フェノキシエタノール、フェノール、塩化ベンゼトニウムおよび2−フェノキシエタノール)は、ワクチン調製のために使用され得る。これらの保存剤の濃度は、産業基準に受容可能である必要がある。
【0110】
(実施例7)
(トリプシンの添加有りでの、株A/Shanghai/11/87による親細胞株の感染)
親株由来のMDCK細胞を、CelliGenTMバイオリアクターにより増殖させた。バイオリアクターの作業用容量は3.7Lであり、マイクロキャリアの濃度は25g/Lであり、そして攪拌を50rpmに設定した。7日目に、培養物を、A/Shanghai/11/87と称されるヒトインフルエンザウイルスにより感染させた。M.O.I.は1:133,000であり、2.5μg/mlトリプシンを、ウイルス複製を増大させるために添加した。表7は、このアッセイに関するデータを示し、表8は、これらの結果をまとめる。ワクチンの収量は、18Lの総収集容量に基づいて、l5μg HAの一価の用量9,828、ならびに、7.38μg HA/mlおよび9μg HA/mlの一元放射拡散(SRD)アッセイ値であった(表8を参照のこと)。
【0111】
【表7】
感染の時点での細胞の総数は、以下の方法により計算される:9.34×106個のMDCK細胞/ml×3700mlは、34,558×106個の細胞に等しい。
【0112】
【表8】
HAの総量の計算は、以下の方法により決定される:9,828用量×l5μg/用量=147,420μg 総HA。34558×106個の細胞で割った場合=
106個のMDCK細胞あたり、4.26μg HA。
【0113】
(実施例8)
(トリプシン無しでの、株A/Shanghai/11/87によるMDCK.5F1クローンの感染)
クローンMDCK.5F1(ATCC番号CRL−12042)に由来する細胞を、25g/Lのマイクロキャリア濃度を含むCelliGenTMバイオリアクターにより増殖させた。バイオリアクターの作業用容量は3.7Lであり、そして攪拌を50〜55rpmに設定した。7日目に、培養物を、A/Shanghai/11/87と称されるヒトインフルエンザウイルスにより感染させた。ウイルスの増殖を増大させるためのトリプシンを、バイオリアクターに添加しなかった。M.O.I.は1:133,000であり、アッセイは9.2μg HA/mlのSRD値を伴う32Lを生成し、一価の用量19,626をもたらした。表9は、このアッセイについてのデータをまとめ、表10は、これらの結果を列挙する。
【0114】
【表9】
感染の時点での細胞の総数は、以下の方法により計算される:16.8×106個のMDCK細胞/ml×3700mlは、62,160×106個の細胞に等しい。
【0115】
【表10】
HAの総量の計算は、以下の方法により決定される:19,626用量×l5μg/用量=294,390μg 総HA。62,160×106個の細胞で割った場合=
106個のMDCK細胞あたり、4.73μg HA。
【0116】
(実施例9)
(トリプシン無しでの、株B/Harbin/7/94によるMDCK.5F1クローンの感染)
クローンMDCK.5F1に由来する細胞を、実施例3と同様だが、15g/Lのマイクロキャリア濃度を用いて増殖させた。8日目に、培養物を、B−Harbin/7/94と称されるヒトインフルエンザウイルスにより感染させた。M.O.I.は1:10,000であった。ウイルス複製を促進させるためのトリプシンを添加しなかった。このアッセイからのデータおよび結果は、表11および12に再現される。収量は、35Lの収集物に基づいて、38,150用量、および16.35μg HA/mlのSRD値であった。
【0117】
【表11】
感染の時点での細胞の総数は、以下の方法により計算される:16.7×106個のMDCK細胞/ml×3700mlは、61,790×106個の細胞に等しい。
【0118】
【表12】
HAの総量の計算は、以下の方法により決定される:38,150用量×l5μg/用量=572,250μg 総HA。61,790×106個の細胞で割った場合=
106個のMDCK細胞あたり、9.26μg HA。
【0119】
もう一度繰り返すと、本発明のプロセスは、トリプシンの存在を必要とする先行技術のプロセスにより与えられる収量と同じか、またはそれより多い収量を与えることを示す。
【図面の簡単な説明】
【0120】
【図1】図1は、本発明の灌流手段において使用されるデカンターの前断面図である。
【図2】図2は、上記デカンターの上正面図である。
【図3】図3は、上記デカンターの下正面図である。
【特許請求の範囲】
【請求項1】
Madin−Darby Canine Kidney(MDCK)由来の細胞株であって、該MDCK由来細胞株の親のMDCK細胞株よりもウイルス感染に対してより高い感受性を有することで特徴付けられる、細胞株。
【請求項2】
前記より高い感受性が、前記親細胞株において産生されるウイルス力価の少なくとも約1.2倍として定義される、請求項1に記載の細胞株。
【請求項3】
請求項2に記載の細胞株であって、ここで前記ウイルスが:インフルエンザウイルス、RSウイルス、パポバウイルス、パラインフルエンザウイルス、水疱性口内炎ウイルス、ワクシニアウイルス、コクサッキーウイルス、レオウイルス、パルボウイルス、アデノウイルス、ポリオウイルス、麻疹ウイルス、狂犬病ウイルス、およびヘルペスウイルスからなる群より選択される、細胞株。
【請求項4】
前記ウイルスがインフルエンザウイルスまたはRSウイルスである、請求項3に記載の細胞株。
【請求項5】
前記インフルエンザウイルスがヒトインフルエンザウイルス株、ウマインフルエンザウイルス株、ブタインフルエンザウイルス株またはトリインフルエンザウイルス株を含む、請求項4に記載の細胞株。
【請求項6】
前記インフルエンザウイルスがヒトインフルエンザA型、ヒトインフルエンザB型、またはヒトインフルエンザC型から選択される、請求項5に記載の細胞株。
【請求項7】
前記インフルエンザウイルスがA型またはB型より選択される、請求項6に記載の細胞株。
【請求項8】
前記細胞株が、インフルエンザA型およびインフルエンザB型の両方に高感受性である、請求項7に記載の細胞株。
【請求項9】
非腫瘍形成性であることで特徴付けられる、MDCK由来の細胞株。
【請求項10】
非腫瘍形成性であることで特徴付けられる、請求項1〜8のいずれか1項に記載の細胞株。
【請求項11】
請求項10に記載の細胞株であって、ここで前記非腫瘍形成性が、ヌードマウスにおいて、約3ヵ月後の観察での触知可能な小結節が存在しないこととして定義される、細胞株。
【請求項12】
ATCC CRL−12042の生物学的特性を有する、細胞株。
【請求項13】
ATCC CRL−12042として規定される、細胞株。
【請求項14】
足場依存性であるとしてさらに特徴付けられる、請求項1〜13のいずれか1項に記載の細胞株。
【請求項15】
予防目的、診断目的、免疫治療目的または治療目的のためのウイルス粒子またはウイルスタンパク質の製造のための、請求項1〜14のいずれか1項に記載の細胞株の使用。
【請求項16】
前記ウイルス粒子またはウイルスタンパク質が、ウイルス感染の予防のために使用されるワクチンの製造のために使用される、請求項15に記載の使用。
【請求項17】
前記ウイルス感染がインフルエンザウイルスによって引き起こされる、請求項16に記載の使用。
【請求項18】
前記インフルエンザウイルスがヒトインフルエンザウイルス株、ウマインフルエンザウイルス株、ブタインフルエンザウイルス株またはトリインフルエンザウイルス株を含む、請求項17に記載の使用。
【請求項19】
前記インフルエンザウイルスがヒトインフルエンザウイルスA型、ヒトインフルエンザウイルスB型、またはヒトインフルエンザウイルスC型から選択される、請求項18に記載の使用。
【請求項20】
前記ウイルスがヒトインフルエンザA型またはヒトインフルエンザB型である、請求項19に記載の使用。
【請求項21】
インフルエンザウイルス感染の予防のためのワクチンの製造のために、ウイルス粒子またはウイルスタンパク質の産生が使用されるための、ATCC CRL−12042として規定される細胞株の、使用。
【請求項22】
前記ウイルス感染がヒトインフルエンザウイルスA型またはヒトインフルエンザウイルスB型によって引き起こされる、請求項21に記載の使用。
【請求項23】
前記ウイルス感染がヒトインフルエンザウイルスA型およびヒトインフルエンザウイルスB型の両方によって引き起こされる、請求項21に記載の使用。
【請求項24】
インフルエンザウイルス粒子またはインフルエンザウイルスタンパク質の産生のためのプロセスであって、以下:
a)該インフルエンザウイルスを複製可能であるMadin−Darby Canine Kidney(MDCK)細胞株を増殖させる工程;
b)該細胞株にインフルエンザウイルス株を感染させる工程;および
c)インキュベートして該ウイルスの複製を可能にする工程;および該複製されたウイルスを収集する工程
を包含する、プロセス。
【請求項25】
ウイルス粒子または該ウイルス粒子由来のタンパク質を精製する工程をさらに包含する、請求項24に記載のプロセス。
【請求項26】
前記MDCK細胞株がATCC番号CRL−12042の生物学的特性を有するとして規定される、請求項24に記載のプロセス。
【請求項27】
前記MDCK細胞株がATCC番号CRL−12042として規定される、請求項24に記載のプロセス。
【請求項28】
前記インフルエンザウイルスがヒトインフルエンザウイルス株、ウマインフルエンザウイルス株、ブタインフルエンザウイルス株またはトリインフルエンザウイルス株を含む、請求項24に記載のプロセス。
【請求項29】
前記ウイルスがヒトインフルエンザウイルスA型、ヒトインフルエンザウイルスB型、およびヒトインフルエンザウイルスC型からなる群より選択される、請求項24に記載のプロセス。
【請求項30】
前記プロセスがバイオリアクターにおいて実施される、請求項24に記載のプロセス。
【請求項31】
前記バイオリアクターが約5リットルのサイズである、請求項30に記載のプロセス。
【請求項32】
前記バイオリアクターが約5リットル〜5000リットルのサイズである、請求項30に記載のプロセス。
【請求項33】
請求項24に記載のプロセスであって、ここで、工程a)において、前記細胞がマイクロキャリア手段を含む培地中で増殖され、そして
工程b)において、前記インキュベーションが灌流手段によって実施される、プロセス。
【請求項34】
前記灌流手段が、前記収集においてマイクロキャリアの流出を避けるためにデカンターを備える、請求項33に記載のプロセス。
【請求項35】
前記マイクロキャリア手段が、約5g/L〜25g/Lの濃度でマイクロキャリアビーズを含む、請求項33に記載のプロセス。
【請求項36】
前記マイクロキャリアビーズの濃度が約10g/L〜25g/Lの範囲にある、請求項35に記載のプロセス。
【請求項37】
前記マイクロキャリアビーズの濃度が約15g/L〜20g/Lの範囲にある、請求項35に記載のプロセス。
【請求項38】
前記灌流が、1日当たりバイオリアクターの約0.5〜4.0容量の速度で実施される、請求項34に記載のプロセス。
【請求項39】
請求項24に記載のプロセスであって、ここで工程b)において、前記インフルエンザウイルスが約10:1〜1:1010の感染多重度で播種される、プロセス。
【請求項40】
前記インフルエンザウイルスが、約1:10〜1:108の感染多重度で播種される、請求項24に記載のプロセス。
【請求項41】
前記インフルエンザウイルスが、約1:104〜1:107の感染多重度で播種される、請求項24に記載のプロセス。
【請求項42】
前記インフルエンザウイルスが、約1:105〜1:106の感染多重度で播種される、請求項24に記載のプロセス。
【請求項43】
請求項24に記載のプロセスであって、ここで工程a)において、前記MDCK細胞株が約7日間増殖される、プロセス。
【請求項44】
前記MDCK細胞株が、約33℃〜40℃の範囲の温度で増殖される、請求項43に記載のプロセス。
【請求項45】
前記MDCK細胞株が、約36℃〜38℃の範囲の温度で増殖される、請求項43に記載のプロセス。
【請求項46】
請求項24に記載のプロセスであって、ここで工程b)において、前記インフルエンザウイルスが約30℃〜37℃の温度で複製される、プロセス。
【請求項47】
前記インフルエンザウイルスが約32℃〜34℃の温度で複製される、請求項43に記載のプロセス。
【請求項48】
前記インフルエンザウイルスが約33℃の温度で複製される、請求項43に記載のプロセス。
【請求項49】
前記精製されたウイルス粒子またはウイルスタンパク質を使用して、インフルエンザウイルス感染を予防するためのワクチンを製造する、請求項24に記載のプロセス。
【請求項50】
請求項49のプロセスによって作製される、哺乳動物においてインフルエンザウイルス感染を予防するためのワクチン。
【請求項51】
前記哺乳動物がヒトである、請求項50に記載のワクチン。
【請求項52】
請求項50または51に記載のワクチンを投与する工程を包含する、哺乳動物においてインフルエンザ感染を予防するための、方法。
【請求項53】
請求項24に従って単離された前記ウイルス粒子またはウイルスタンパク質を含み、保存剤と混合された、哺乳動物においてインフルエンザウイルス感染を予防するためのワクチン。
【請求項54】
ワクチンを製造するための、請求項24に記載のプロセスの、使用。
【請求項55】
請求項24に記載のプロセスであって、ここで前記精製されたウイルス粒子またはウイルスタンパク質を使用して、インフルエンザウイルス感染の検出のための診断キットを製造する、プロセス。
【請求項56】
請求項24に従って単離されたバルクのウイルス粒子またはウイルスタンパク質の、哺乳動物におけるインフルエンザウイルス感染の検出および診断のための、キットの製造のための使用。
【請求項1】
Madin−Darby Canine Kidney(MDCK)由来の細胞株であって、該MDCK由来細胞株の親のMDCK細胞株よりもウイルス感染に対してより高い感受性を有することで特徴付けられる、細胞株。
【請求項2】
前記より高い感受性が、前記親細胞株において産生されるウイルス力価の少なくとも約1.2倍として定義される、請求項1に記載の細胞株。
【請求項3】
請求項2に記載の細胞株であって、ここで前記ウイルスが:インフルエンザウイルス、RSウイルス、パポバウイルス、パラインフルエンザウイルス、水疱性口内炎ウイルス、ワクシニアウイルス、コクサッキーウイルス、レオウイルス、パルボウイルス、アデノウイルス、ポリオウイルス、麻疹ウイルス、狂犬病ウイルス、およびヘルペスウイルスからなる群より選択される、細胞株。
【請求項4】
前記ウイルスがインフルエンザウイルスまたはRSウイルスである、請求項3に記載の細胞株。
【請求項5】
前記インフルエンザウイルスがヒトインフルエンザウイルス株、ウマインフルエンザウイルス株、ブタインフルエンザウイルス株またはトリインフルエンザウイルス株を含む、請求項4に記載の細胞株。
【請求項6】
前記インフルエンザウイルスがヒトインフルエンザA型、ヒトインフルエンザB型、またはヒトインフルエンザC型から選択される、請求項5に記載の細胞株。
【請求項7】
前記インフルエンザウイルスがA型またはB型より選択される、請求項6に記載の細胞株。
【請求項8】
前記細胞株が、インフルエンザA型およびインフルエンザB型の両方に高感受性である、請求項7に記載の細胞株。
【請求項9】
非腫瘍形成性であることで特徴付けられる、MDCK由来の細胞株。
【請求項10】
非腫瘍形成性であることで特徴付けられる、請求項1〜8のいずれか1項に記載の細胞株。
【請求項11】
請求項10に記載の細胞株であって、ここで前記非腫瘍形成性が、ヌードマウスにおいて、約3ヵ月後の観察での触知可能な小結節が存在しないこととして定義される、細胞株。
【請求項12】
ATCC CRL−12042の生物学的特性を有する、細胞株。
【請求項13】
ATCC CRL−12042として規定される、細胞株。
【請求項14】
足場依存性であるとしてさらに特徴付けられる、請求項1〜13のいずれか1項に記載の細胞株。
【請求項15】
予防目的、診断目的、免疫治療目的または治療目的のためのウイルス粒子またはウイルスタンパク質の製造のための、請求項1〜14のいずれか1項に記載の細胞株の使用。
【請求項16】
前記ウイルス粒子またはウイルスタンパク質が、ウイルス感染の予防のために使用されるワクチンの製造のために使用される、請求項15に記載の使用。
【請求項17】
前記ウイルス感染がインフルエンザウイルスによって引き起こされる、請求項16に記載の使用。
【請求項18】
前記インフルエンザウイルスがヒトインフルエンザウイルス株、ウマインフルエンザウイルス株、ブタインフルエンザウイルス株またはトリインフルエンザウイルス株を含む、請求項17に記載の使用。
【請求項19】
前記インフルエンザウイルスがヒトインフルエンザウイルスA型、ヒトインフルエンザウイルスB型、またはヒトインフルエンザウイルスC型から選択される、請求項18に記載の使用。
【請求項20】
前記ウイルスがヒトインフルエンザA型またはヒトインフルエンザB型である、請求項19に記載の使用。
【請求項21】
インフルエンザウイルス感染の予防のためのワクチンの製造のために、ウイルス粒子またはウイルスタンパク質の産生が使用されるための、ATCC CRL−12042として規定される細胞株の、使用。
【請求項22】
前記ウイルス感染がヒトインフルエンザウイルスA型またはヒトインフルエンザウイルスB型によって引き起こされる、請求項21に記載の使用。
【請求項23】
前記ウイルス感染がヒトインフルエンザウイルスA型およびヒトインフルエンザウイルスB型の両方によって引き起こされる、請求項21に記載の使用。
【請求項24】
インフルエンザウイルス粒子またはインフルエンザウイルスタンパク質の産生のためのプロセスであって、以下:
a)該インフルエンザウイルスを複製可能であるMadin−Darby Canine Kidney(MDCK)細胞株を増殖させる工程;
b)該細胞株にインフルエンザウイルス株を感染させる工程;および
c)インキュベートして該ウイルスの複製を可能にする工程;および該複製されたウイルスを収集する工程
を包含する、プロセス。
【請求項25】
ウイルス粒子または該ウイルス粒子由来のタンパク質を精製する工程をさらに包含する、請求項24に記載のプロセス。
【請求項26】
前記MDCK細胞株がATCC番号CRL−12042の生物学的特性を有するとして規定される、請求項24に記載のプロセス。
【請求項27】
前記MDCK細胞株がATCC番号CRL−12042として規定される、請求項24に記載のプロセス。
【請求項28】
前記インフルエンザウイルスがヒトインフルエンザウイルス株、ウマインフルエンザウイルス株、ブタインフルエンザウイルス株またはトリインフルエンザウイルス株を含む、請求項24に記載のプロセス。
【請求項29】
前記ウイルスがヒトインフルエンザウイルスA型、ヒトインフルエンザウイルスB型、およびヒトインフルエンザウイルスC型からなる群より選択される、請求項24に記載のプロセス。
【請求項30】
前記プロセスがバイオリアクターにおいて実施される、請求項24に記載のプロセス。
【請求項31】
前記バイオリアクターが約5リットルのサイズである、請求項30に記載のプロセス。
【請求項32】
前記バイオリアクターが約5リットル〜5000リットルのサイズである、請求項30に記載のプロセス。
【請求項33】
請求項24に記載のプロセスであって、ここで、工程a)において、前記細胞がマイクロキャリア手段を含む培地中で増殖され、そして
工程b)において、前記インキュベーションが灌流手段によって実施される、プロセス。
【請求項34】
前記灌流手段が、前記収集においてマイクロキャリアの流出を避けるためにデカンターを備える、請求項33に記載のプロセス。
【請求項35】
前記マイクロキャリア手段が、約5g/L〜25g/Lの濃度でマイクロキャリアビーズを含む、請求項33に記載のプロセス。
【請求項36】
前記マイクロキャリアビーズの濃度が約10g/L〜25g/Lの範囲にある、請求項35に記載のプロセス。
【請求項37】
前記マイクロキャリアビーズの濃度が約15g/L〜20g/Lの範囲にある、請求項35に記載のプロセス。
【請求項38】
前記灌流が、1日当たりバイオリアクターの約0.5〜4.0容量の速度で実施される、請求項34に記載のプロセス。
【請求項39】
請求項24に記載のプロセスであって、ここで工程b)において、前記インフルエンザウイルスが約10:1〜1:1010の感染多重度で播種される、プロセス。
【請求項40】
前記インフルエンザウイルスが、約1:10〜1:108の感染多重度で播種される、請求項24に記載のプロセス。
【請求項41】
前記インフルエンザウイルスが、約1:104〜1:107の感染多重度で播種される、請求項24に記載のプロセス。
【請求項42】
前記インフルエンザウイルスが、約1:105〜1:106の感染多重度で播種される、請求項24に記載のプロセス。
【請求項43】
請求項24に記載のプロセスであって、ここで工程a)において、前記MDCK細胞株が約7日間増殖される、プロセス。
【請求項44】
前記MDCK細胞株が、約33℃〜40℃の範囲の温度で増殖される、請求項43に記載のプロセス。
【請求項45】
前記MDCK細胞株が、約36℃〜38℃の範囲の温度で増殖される、請求項43に記載のプロセス。
【請求項46】
請求項24に記載のプロセスであって、ここで工程b)において、前記インフルエンザウイルスが約30℃〜37℃の温度で複製される、プロセス。
【請求項47】
前記インフルエンザウイルスが約32℃〜34℃の温度で複製される、請求項43に記載のプロセス。
【請求項48】
前記インフルエンザウイルスが約33℃の温度で複製される、請求項43に記載のプロセス。
【請求項49】
前記精製されたウイルス粒子またはウイルスタンパク質を使用して、インフルエンザウイルス感染を予防するためのワクチンを製造する、請求項24に記載のプロセス。
【請求項50】
請求項49のプロセスによって作製される、哺乳動物においてインフルエンザウイルス感染を予防するためのワクチン。
【請求項51】
前記哺乳動物がヒトである、請求項50に記載のワクチン。
【請求項52】
請求項50または51に記載のワクチンを投与する工程を包含する、哺乳動物においてインフルエンザ感染を予防するための、方法。
【請求項53】
請求項24に従って単離された前記ウイルス粒子またはウイルスタンパク質を含み、保存剤と混合された、哺乳動物においてインフルエンザウイルス感染を予防するためのワクチン。
【請求項54】
ワクチンを製造するための、請求項24に記載のプロセスの、使用。
【請求項55】
請求項24に記載のプロセスであって、ここで前記精製されたウイルス粒子またはウイルスタンパク質を使用して、インフルエンザウイルス感染の検出のための診断キットを製造する、プロセス。
【請求項56】
請求項24に従って単離されたバルクのウイルス粒子またはウイルスタンパク質の、哺乳動物におけるインフルエンザウイルス感染の検出および診断のための、キットの製造のための使用。
【図1】

【図2】
【図3】
【図2】
【図3】
【公表番号】特表2007−537760(P2007−537760A)
【公表日】平成19年12月27日(2007.12.27)
【国際特許分類】
【出願番号】特願2007−527447(P2007−527447)
【出願日】平成17年5月20日(2005.5.20)
【国際出願番号】PCT/US2005/017606
【国際公開番号】WO2005/113758
【国際公開日】平成17年12月1日(2005.12.1)
【出願人】(506383766)アイディー バイオメディカル コーポレイション (1)
【Fターム(参考)】
【公表日】平成19年12月27日(2007.12.27)
【国際特許分類】
【出願日】平成17年5月20日(2005.5.20)
【国際出願番号】PCT/US2005/017606
【国際公開番号】WO2005/113758
【国際公開日】平成17年12月1日(2005.12.1)
【出願人】(506383766)アイディー バイオメディカル コーポレイション (1)
【Fターム(参考)】
[ Back to top ]