
ウイルスレスキューのための改善された逆遺伝学
【要約書】
逆遺伝学によってウイルスをレスキューするための方法が提供され、ここで細胞は、トランスフェクション後に添加される。一実施形態において、本発明のウイルスを調製するための方法は、(i)宿主細胞の培養物をウイルスRNA分子をコードする少なくとも1個の発現構築物でトランスフェクトする工程;(ii)細胞を、(i)のトランスフェクトされた該宿主細胞に添加して、細胞の混合物を提供する工程;および(iii)ウイルスを生成するために、該細胞の混合物を培養する工程を包含する。
逆遺伝学によってウイルスをレスキューするための方法が提供され、ここで細胞は、トランスフェクション後に添加される。一実施形態において、本発明のウイルスを調製するための方法は、(i)宿主細胞の培養物をウイルスRNA分子をコードする少なくとも1個の発現構築物でトランスフェクトする工程;(ii)細胞を、(i)のトランスフェクトされた該宿主細胞に添加して、細胞の混合物を提供する工程;および(iii)ウイルスを生成するために、該細胞の混合物を培養する工程を包含する。
【発明の詳細な説明】
【技術分野】
【0001】
この出願は、2009年10月20日に出願された米国仮出願第61/279,487号(この完全な内容は、参考として本明細書に援用される)の利益を主張する。
【0002】
(技術分野)
本発明は、逆遺伝学の分野にある。さらに、種々のウイルスに対して防御するためのワクチンを製造することに関する。
【背景技術】
【0003】
(背景技術)
逆遺伝学は、細胞培養物においてRNAウイルスの組換え発現および操作を可能にする。逆遺伝学は、ウイルス学およびワクチン製造において強力なツールである。なぜなら、逆遺伝学は、組換えウイルス(リアソータントを含む)の迅速な生成および/もしくはそれらの変異を可能にするからである。上記方法は、宿主細胞を、ウイルスゲノムをコードする1個以上の発現構築物でトランスフェクトする工程および上記ウイルスを上記細胞から単離する工程を包含する。
【0004】
逆遺伝学によるウイルスレスキュー(virus rescue)の1つの欠点は、そのプロセスが非効率的であり、頻繁に、結果として得られるウイルス収量が十分ではないことである。従って、代替のより効率的な逆遺伝学法を提供することが当該分野には未だに必要である。
【発明の概要】
【課題を解決するための手段】
【0005】
(好ましい実施形態の要旨)
本発明者らは、ここで、逆遺伝学法の不十分な効率が、細胞を上記逆遺伝学構築物でトランスフェクトする工程およびその後、より多くの細胞を上記トランスフェクトした細胞に添加する工程によって、顕著に改善され得ることを驚くべきことに発見した。本発明者らは、観察された効率の増大が、上記細胞に対してトランスフェクションが有害であるという事実に起因すると推量する。従って、レスキューされる任意のウイルスは、疾患細胞において拡大するか、もしくは上記ウイルスが健康な細胞に移入されるまで待つことが必要である。このことは、生存可能なウイルスの喪失を生じる。上記トランスフェクトされた宿主細胞への細胞の添加は、上記ウイルス収量を顕著に増大させる健康な細胞基質上で、上記レスキューされたウイルスが繁殖することを可能にする。
【0006】
一実施形態において、本発明は、ウイルスを調製するための方法を提供し、上記方法は、(i)宿主細胞の培養物を、ウイルスRNA分子をコードする少なくとも1個の発現構築物でトランスフェクトする工程;(ii)細胞を、上記(i)のトランスフェクトされた宿主細胞に添加して、細胞の混合物を提供する工程;および(iii)ウイルスを生成するために、上記細胞の混合物を培養する工程を包含する。
【0007】
さらなる実施形態において、本発明は、ワクチン製造のためにウイルスを調製する方法を提供し、上記方法は、(i)宿主細胞の培養物を、ウイルスRNA分子をコードする少なくとも1個の発現構築物でトランスフェクトする工程;(ii)細胞を、上記(1)のトランスフェクトされた宿主細胞に添加して、細胞の混合物を提供する工程;(iii)ウイルスを生成するために、上記細胞の混合物を培養する工程;(iv)上記工程(iii)において得られたウイルスを精製する工程および必要に応じて(v)上記ウイルスをワクチンへと処方する工程を包含する。
【0008】
(逆遺伝学)
逆遺伝学は、広く種々のRNAウイルス(プラス鎖RNAウイルス[1、2]、マイナス鎖RNAウイルス[3、4]および二本鎖RNAウイルス[5]を含む)の生成のために使用され得る。従って、本発明は、組換えウイルスを生成するための方法を提供し、ここで上記ウイルスは、本発明の逆遺伝学法を使用して得られる。
【0009】
既知の逆遺伝学システムは、pol Iプロモーター、細菌RNAポリメラーゼプロモーター、バクテリオファージポリメラーゼプロモーターなどから、所望のウイルスRNA(vRNA)分子をコードするDNA分子を発現する工程を包含する。さらに、ウイルスが、感染性ウイルスを形成するために特定のタンパク質を必要とする場合、上記システムはまた、これらタンパク質を提供し、例えば、上記システムは、ウイルスタンパク質をコードするDNA分子をさらに含み、その結果、DNAの両方のタイプの発現が、完全な感染性ウイルスの構築をもたらす。
【0010】
逆遺伝学が、vRNAの発現のために使用される場合、互いに対する配列エレメントの正確な間隔が、複製を開始するための上記ポリメラーゼの中枢であることは、当業者に明らかである。従って、上記ウイルスRNAをコードするDNA分子が、上記pol Iプロモーターと終結配列との間に正確に配置されることは重要であるが、この配置は、逆遺伝学システムを取り扱う者の能力の十分範囲内である。
【0011】
一般に、逆遺伝学は、ウイルスの生活環の間にゲノムRNAの生成を必要とすることが公知の任意のウイルスの発現に適している。このようなウイルスとしては、プラス鎖RNAウイルスおよびマイナス鎖RNAウイルス(例えば、以下に記載されるもの)が挙げられるが、これらに限定されない。好ましくは、上記ウイルスは、オルソミクソウイルス(例えば、インフルエンザウイルス)である。本発明の方法は、さらに、非セグメント化ウイルスおよびセグメント化ウイルスにも適している。
【0012】
上記ウイルスがプラス鎖RNAウイルスである場合、上記ウイルスゲノムを含む発現構築物で細胞をトランスフェクトすることはしばしば十分である。例えば、ポリオウイルスゲノムを含むプラスミドのトランスフェクションは、感染性ポリオウイルスの回収をもたらした[1、2]。マイナス鎖RNAウイルスの逆遺伝学は、より大きな難題を呈した。なぜなら、上記アンチセンスウイルスRNAは、通常は、非感染性であり、その生活環を完了するのにRNAポリメラーゼを必要とするからである。従って、上記ウイルスポリメラーゼは、タンパク質としてか、もしくはインサイチュでのタンパク質発現のための遺伝子としてかのいずれかで、供給されなければならない。
【0013】
上記ウイルスが感染性のためにタンパク質を必要とする場合、二方向性発現構築物(bi−directional expression construct)を使用することは一般に好ましい。なぜなら、これは、上記宿主細胞が必要とする発現構築物の総数を減らすからである。従って、本発明の方法は、少なくとも1個の二方向性発現構築物を利用し得る。ここで遺伝子もしくはcDNAは、上流のpol IIプロモーターと下流の非内因性pol Iプロモーターとの間に位置する。上記pol IIプロモーターからの上記遺伝子もしくはcDNAの転写は、キャップされたプラス−センスウイルスmRNAを生成し、これはタンパク質へと翻訳され得る一方で、上記非内因性pol Iプロモーターからの転写は、マイナス−センスvRNAを生成する。上記二方向性発現構築物は、二方向性発現ベクターであり得る。
【0014】
組換えウイルスを生成するために、細胞は、ビリオンを構築するために必要なウイルスゲノムのすべてのセグメントを発現しなければならない。本発明の発現構築物へとクローニングされたDNAは、好ましくは、上記ウイルスRNAおよびタンパク質のすべてを提供する。しかし、上記RNAおよびタンパク質のうちのいくらかを提供するためにヘルパーウイルスを使用することも考えられ得るものの、ヘルパーウイルスを使用しないシステムが好ましい。上記ウイルスが非セグメント化ウイルスである場合、これは、1個より多くの発現構築物を使用して非セグメント化ウイルスのウイルスゲノムを発現することは、本発明の範囲内にあるとしても、通常は、本発明の方法において単一の発現構築物を利用することによって達成され得る。上記ウイルスがセグメント化ウイルスである場合、上記ウイルスゲノムは、通常は、本発明の方法において1個より多くの発現構築物を使用して発現される。しかし、単一の発現構築物の上で上記ウイルスゲノムのうちの1個より多くのセグメントもしくはさらにはすべてのセグメントを合わせることもまた、想定される。
【0015】
本発明の方法は、リアソータントウイルス株の生成に特に適している。その技術は、プラスミドのインビトロでの操作を使用して、ウイルスセグメントの組み合わせを生成し、上記ウイルスセグメントにおけるコード配列もしくは非コード配列の操作を容易にし、変異などを導入し得る。リアソータントウイルス株の生成のための上記発現系の使用は、好ましい。なぜなら、これは、ワクチンの迅速な生成が流行に対抗するために必要とされる状況において特に有益であるリアソータント種ウイルスを得るために必要とされる時間を顕著に減少させ得るからである。従って、本発明のこの局面の方法は、少なくとも2つの異なる野生型株に由来するかもしくはこれらから得られるウイルス遺伝子を発現する1個以上の発現構築物を使用することが好ましい。
【0016】
上記宿主細胞においてアクセサリタンパク質の発現をもたらす発現構築物もまた、使用され得る。例えば、非ウイルス性セリンプロテアーゼ(例えば、トリプシン)を逆遺伝学システムの一部として発現することもまた、有利であり得る。
【0017】
(発現構築物)
本発明における使用のために上記ウイルスRNA分子をコードする発現構築物は、ウイルスをレスキューするために当該分野で一般に使用される任意の発現構築物であり得る。
【0018】
上記使用される発現構築物は、一方向性発現構築物であってもよいし、二方向性発現構築物であってもよい。宿主細胞は1つより多い導入遺伝子を発現する場合(同じ発現構築物においてであろうと、異なる発現構築物においてであろうと)、一方向性および/もしくは二方向性の発現を使用することは可能である。
【0019】
二方向性発現構築物は、同じ構築物から異なる方向(すなわち、5’→3’および3’→5’の両方)において発現を駆動する少なくとも2個のプロモーターを含む。上記2個のプロモーターは、同じ二本鎖DNAの異なる鎖に作動可能に連結され得る。好ましくは、上記プロモーターのうちの一方は、pol Iプロモーターであり他方のプロモーターのうちの少なくとも1つは、pol IIプロモーターである。このことは、上記pol Iプロモーターが、キャップされていないcRNAを発現するために使用され得る一方で、上記pol IIプロモーターが、タンパク質へと後に翻訳され得るmRNAを転写するために使用され得、よって同じ構築物からRNAおよびタンパク質の同時の発現を可能にするので有用である。
【0020】
上記発現構築物(一方向性であろうと二方向性であろうと)は、代表的には、RNA転写終結配列を含む。上記終結配列は、内因性終結配列であってもよいし、上記宿主細胞に対して内因性ではない終結配列であってもよい。適切な終結配列は、当業者に明らかであり、RNAポリメラーゼI転写終結配列、RNAポリメラーゼII転写終結配列、およびリボザイムが挙げられるが、これらに限定されない。さらに、上記発現構築物は、mRNAについて、特に発現がpol IIプロモーターによって制御される遺伝子の末端において、1個以上のポリアデニル化シグナルを含み得る。
【0021】
上記発現構築物において使用される上記pol Iプロモーターおよびpol IIプロモーターは、上記発現構築物が発現される宿主細胞と分類学上同じ目にある生物に由来し得る。あるいは、上記プロモーターは、上記宿主細胞とは異なる分類学上の目にある生物に由来し得る。用語「目」とは、従来の分類学上の順位をいい、目の例は、霊長目、齧歯目、食肉目、有袋目、クジラ目などである。ヒトおよびチンパンジーは、分類学上同じ目(霊長目)にあるが、ヒトおよびイヌは、異なる目にある(霊長類 対 食肉目)。
【0022】
本発明の方法において、宿主細胞は、少なくとも2個、少なくとも3個、少なくとも4個、少なくとも5個、少なくとも6個、少なくとも7個、少なくとも8個、少なくとも9個、少なくとも10個、少なくとも11個、もしくは少なくとも12個の発現構築物でトランスフェクトされ得る。
【0023】
発現構築物は、ベクター(例えば、プラスミドもしくは他のエピソーム構築物)であり得る。このようなベクターは、代表的には、少なくとも1個の細菌複製起点および/もしくは真核生物複製起点を含む。さらに、上記ベクターは、原核生物細胞および/もしくは真核生物細胞における選択を可能にする選択マーカーを含み得る。このような選択マーカーの例は、抗生物質(例えば、アンピシリンもしくはカナマイシン)に対する耐性を付与する遺伝子である。上記ベクターは、DNA配列のクローニングを促進するために、1個以上の、マルチクローニング部位をさらに含み得る。
【0024】
代わりとして、発現構築物は、線形の発現構築物であり得る。このような線形の発現構築物は、代表的には、任意の増幅配列および/もしくは選択配列を含まない。しかし、このような増幅配列および/もしくは選択配列を含む線形の構築物はまた、本発明の範囲内である。インフルエンザウイルスの発現のために、このような線形の発現構築物を使用する方法の例は、参考文献6に記載される。
【0025】
本発明の発現構築物は、当該分野で公知の方法を使用して、生成され得る。このような方法は、例えば、参考文献7に記載された。上記発現構築物が線形の発現構築物である場合、単一の制限酵素部位を利用して上記宿主細胞に導入する前に、発現構築物を線形にすることは可能である。あるいは、少なくとも2個の制限酵素部位を使用して、上記発現構築物をベクターから切り出すことは可能である。さらに、核酸増幅技術を使用して(例えば、PCRによって)増幅することによって、線形の発現構築物を得ることもまた、可能である。
【0026】
上記発現宿主がイヌ細胞(例えば、MDCK細胞株)である場合、タンパク質コード領域は、例えば、野生型イヌ遺伝子に由来するかもしくはイヌウイルスに由来するプロモーターを使用して、そして/またはヒト細胞よりイヌ細胞に適したコドン使用頻度を有して、イヌの発現に最適化され得る。例えば、ヒト遺伝子がPheのコドンとしてわずかにUUCを好む(54%)のに対して、イヌ細胞では、優先度はより強い(59%)。同様に、ヒト細胞においてIleコドンに大きな優先度はないのに対して、イヌのコドンのうちの53%は、Ileに関してAUCを使用する。イヌウイルス(例えば、イヌパルボウイルス(ssDNAウイルス))はまた、コドン最適化についてガイダンスを提供し得る(例えば、イヌパルボウイルス配列におけるPheコドンのうちの95%は、UUUであり(イヌゲノムにおける41%に対して)、Ileコドンのうちの68%は、AUUであり(32%に対して)、Valコドンのうちの46%は、GUUであり(14%に対して)、Proコドンのうちの72%は、CCAであり(25%に対して)、Tyrコドンのうちの87%は、UAUであり(40%に対して)、Hisコドンのうちの87%は、CAUであり(39%に対して)、Glnコドンのうちの92%は、CAAであり(25%に対して)、Gluコドンのうちの81%は、GAAであり(40%に対して)、Cysコドンのうちの94%は、UGUであり(42%に対して)、Serコドンのうちの1%のみがUCUであり(24%に対して)、CCCは、Pheについては決して使用されず、UAGは、終止コドンとして決して使用されない。従って、タンパク質コード遺伝子は、性質がイヌ細胞における発現のためにすでに最適化され、発現を促進したより類似の遺伝子にされ得る。
【0027】
(トランスフェクション)
「トランスフェクション」とは、細胞へのDNAの導入をいう。上記発現構築物は、当業者に公知の任意の技術を使用して、宿主細胞へと導入され得る。例えば、それらは、エレクトロポレーション、DEAE−デキストラン、リン酸カルシウム沈殿、リポソーム、マイクロインジェクション、もしくは微粒子銃(microparticle−bombardment)を使用することによって、宿主細胞へと導入され得る。逆遺伝学は、一般に、リポソーム法を使用して、例えば、トランスフェクション試薬LipofectamineTMを使用して実施される。本発明の方法は、リポソームでのトランスフェクションに限定されないが、他のトランスフェクション法で等しく十分に機能すると予測される。
【0028】
上記細胞は、トランスフェクション後最大72時間までのいずれかのときに、上記トランスフェクトされた宿主細胞に添加され得る。例えば、上記細胞は、トランスフェクション直後、トランスフェクションの5分後、10分後、20分後、30分後、1時間後、2時間後、3時間後、6時間後、12時間後、24時間後、36時間後、48時間後、60時間後、もしくは72時間後に、添加され得る。一般に、トランスフェクション後に細胞が添加される最大時間は、上記トランスフェクトされた細胞が生存し得る最大時間と適合する。
【0029】
期間「トランスフェクション後」とは、いったん上記DNAが上記培養物内で宿主細胞に添加されたら始まる。上記DNAが導入された時点は、種々の因子(例えば、使用される細胞株、トランスフェクション法、もしくはトランスフェクションが行われる温度)とともに変動する。上記時間は、標準的な方法を使用して、当業者によって容易に決定され得る。例えば、いくつかの実験を並行して行い得、そこで上記細胞は、トランスフェクションが異なる時点で停止されることを除いて同一な条件下で、逆遺伝学構築物でトランスフェクトされる。上記DNAが上記宿主細胞に導入された場合、少なくとも1個のウイルスが、形成されたはずである。従って、当業者は、例えば、標準的なアッセイを使用してプラーク形成単位の数を決定することによって、ウイルスの存在についてアッセイし得る。少なくとも1プラーク形成単位(PFU)が検出可能である後の最初の時点は、上記宿主細胞へと上記DNAが導入された時点である。
【0030】
あるいは、細胞が上記トランスフェクトされた細胞に添加された後の期間は、上記トランスフェクションの始まりから決定され得る。トランスフェクションの開始は、上記ウイルス分子をコードする発現構築物が上記宿主細胞の培養物と接触させられた時点である。従って、上記細胞は、トランスフェクション開始の最大96時間までのいずれかのとき(例えば、12時間、24時間、36時間、48時間、60時間、72時間、84時間もしくは96時間)に、上記トランスフェクトされた細胞に添加され得る。「トランスフェクション後」の時点および「トランスフェクションの開始から」の時点は、異なる出発点に対して測定される一方で、上記細胞が上記感染した細胞に添加される絶対時間は、同じである。例えば、上記DNAが培養物中で上記宿主細胞に導入されるのに1時間かかり、上記細胞が、上記DNAが導入された30分後に添加された場合、上記細胞は、トランスフェクションの開始から90分もしくはトランスフェクションの30分後で添加されている。
【0031】
トランスフェクション後に添加される細胞の数は、一般に、トランスフェクションのための宿主細胞として使用される細胞の数、および同様にトランスフェクションのために使用される上記細胞培養容器のサイズに依存して変動する。本発明の方法において使用され得る細胞の数は、当業者によって容易に決定され得る。例えば、本発明の方法は、添加される細胞の数が異なることを除いて、いくつかの並行した実験において同一の条件下で行われ得る。最高のウイルス収量を与える細胞の数は、さらなる実験において使用され得る。例えば、約6×105細胞が、LipofectamineTMでのトランスフェクションのために出発培養物として使用される場合、約4×105細胞が添加され得る。
【0032】
トランスフェクション後に添加される上記細胞は、上記レスキューされるウイルスが複製するための基質を提供する。従って、上記細胞は、感染していない細胞であってもよく、これは、上記細胞が本発明の方法において使用される前に、上記特定の細胞が特定の時間(例えば、48時間)内でウイルスに感染もせず、ウイルスRNAをコードする発現構築物でトランスフェクトもされなかったことを意味する。細胞を不死化させるためにウイルス(例えば、エプスタイン−バー・ウイルス)に感染した細胞は、本発明における使用に適している。同様に、トランスフェクトされた細胞は、本発明の方法において使用され得るが、ただし、トランスフェクションは、上記細胞が本発明の方法において使用される直前に(すなわち、48時間以内に)は行われない。
【0033】
上記レスキューされたウイルスが、感染性のためにプロテアーゼの存在を必要とする場合、トランスフェクション後に上記細胞と一緒に上記プロテアーゼを添加することは有利である。例えば、上記インフルエンザウイルスは、感染のためにセリンプロテアーゼ(トリプシンのような)の活性を必要とする。従って、本発明の方法が、インフルエンザウイルスをレスキューするために使用される場合、上記セリンプロテアーゼは、トランスフェクション後に上記細胞と同時に添加され得る。あるいは、上記プロテアーゼは、上記細胞が上記トランスフェクトされた細胞に添加される前もしくは添加された後に、添加され得る。
【0034】
(細胞)
本発明は、上記目的のウイルスを生成し得る任意の真核生物細胞で実施され得る。本発明は、代表的には、細胞株を使用するが、例えば、初代細胞は、代替手段として使用され得る。上記細胞は、代表的には、哺乳動物のものである。適切な哺乳動物細胞としては、ハムスター、ウシ、霊長類(ヒトおよびサルを含む)およびイヌの細胞が挙げられるが、これらに限定されない。種々の細胞タイプが使用され得る(例えば、腎臓細胞、線維芽細胞、網膜細胞、肺細胞など)。適切なハムスター細胞の例は、名称BHK21もしくはHKCCを有する細胞株である。適切なサル細胞は、例えば、アフリカミドリザル細胞(例えば、Vero細胞株のような腎臓細胞である[8〜10]。適切なイヌ細胞は、例えば、CLDKおよびMDCK細胞株のような腎臓細胞である。
【0035】
さらなる適切な細胞としては、CHO;293T;BHK;MRC 5;PER.C6[11];FRhL2;WI−38などが挙げられるが、これらに限定されない。適切な細胞は、広く入手可能である(例えば、アメリカンタイプセルカルチャー(ATCC)コレクションから[12]、Coriell Cell Repositories[13]から、もしくはEuropean Collection of Cell Cultures(ECACC)から)。例えば、上記ATCCは、カタログ番号CCL 81、CCL 81.2、CRL 1586およびCRL−1587の下で種々の異なるVero細胞を供給し、カタログ番号CCL 34の下でMDCK細胞を供給する。PER.C6は、寄託番号96022940の下でECACCから入手可能である。MDCK細胞、Vero細胞およびPER.C6細胞は、ウイルスの生成のために一般に使用され、従って、これら細胞株の各々は、本発明の方法とともに使用するのに特に適している。
【0036】
本発明において使用するための好ましい細胞(特にインフルエンザウイルスを増殖させるため)は、メイディン・ダービー・イヌ腎臓に由来するMDCK細胞[14〜16]である。元々のMDCK細胞は、CCL 34としてATCCから入手可能である。これら細胞の派生物もしくは他のMDCK細胞の派生物が使用されることは、好ましい。MDCK細胞の派生物は、例えば、懸濁培養における増殖に適合されたMDCK細胞(DSM ACC 2219として寄託された「MDCK 33016」もしくは「33016−PF」;参考文献14もまた参照のこと)を開示する参考文献14において記載された。さらに、参考文献17は、無血清培養において懸濁物中で増殖するMDCK由来細胞(FERM BP−7449として寄託された「B−702」)を開示する。上記使用されるMDCK細胞株は、腫瘍形成性であり得る。非腫瘍形成性MDCK細胞を使用することもまた、想定される。例えば、参考文献18は、非腫瘍形成性MDCK細胞(「MDCK−S」(ATCC PTA−6500)、「MDCK−SF101」(ATCC PTA−6501)、「MDCK−SF102」(ATCC PTA−6502)および「MDCK−SF103」(ATCC PTA−6503)が挙げられる)を開示する。参考文献19は、感染に対して高い感受性を有するMDCK細胞(「MDCK.5F1」細胞(ATCC CRL 12042)が挙げられる)を開示する。
【0037】
上記トランスフェクションのために使用される細胞および上記トランスフェクション後に添加される細胞は、同じ細胞タイプもしくは異なる細胞タイプのものであり得る。例えば、上記発現構築物は、MDCK細胞へとトランスフェクトされ得、上記添加される細胞は、Vero細胞であり得、その逆もまた同様である。別の例は、上記発現構築物がMDCK細胞の1つの株へトランスフェクトされ、上記添加される細胞は異なるMDCK株のものである方法であり得る。このアプローチは、上記レスキューされるウイルスが、容易にトランスフェクトされ得ない細胞株において最良に増殖する場合に有利である。この局面において、上記ウイルスは、より容易にトランスフェクションすることが可能な細胞へトランスフェクトされ得るが、ウイルス複製によりよく適した細胞株においてその後増殖させられ得る。しかし、一般に、トランスフェクションおよびその後の細胞の添加の両方のために宿主細胞と同じ細胞株(例えば、MDCK 33016細胞)を使用することは好ましい。なぜなら、これは、例えば、競合する培養選択圧もしくは異なる細胞培養条件が、避けられ得るという利点を有するからである。トランスフェクションおよび/もしくはトランスフェクション後の添加のための宿主細胞として1種より多い細胞タイプの混合物を使用することもまた、可能である。
【0038】
好ましくは、上記細胞は、夾雑物の共通する供給源を回避するために、血清の非存在下で培養される。真核生物細胞培養物のための種々の無血清培地は、当業者に公知である(例えば、Iscove培地、ウルトラCHO培地(BioWhittaker)、EX−CELL(JRH Biosciences))。さらに、無タンパク質培地が使用され得る(例えば、PF−CHO(JRH Biosciences))。さもなければ、上記複製のための細胞はまた、習慣的な血清含有培地(例えば、0.5%〜10%のウシ胎仔血清を含む、MEM培地もしくはDMEM培地)中で培養され得る。
【0039】
上記宿主細胞および上記添加され得る細胞は、接着培養物中もしくは懸濁培養物中に存在し得る。例えば、上記トランスフェクションのために使用される細胞は、接着性の細胞であり得、上記トランスフェクション後に添加される細胞もまた、接着性の細胞であり得る。上記トランスフェクション後に添加される細胞が接着性の細胞である場合、上記細胞は、上記細胞が上記トランスフェクトされた細胞に添加される前にそれらが増殖させられる培養容器から除去される必要があることは明らかである。このことは、例えば、トリプシンのようなセリンプロテアーゼの助けを借りて達成され得る。トランスフェクションのために宿主細胞として懸濁細胞を、そしてトランスフェクション後の添加のために接着性細胞を使用することも可能であり、その逆もまた同様である。
【0040】
本発明の方法は、通常、使用される具体的なトランスフェクション法のために、当該分野で一般に使用されている温度で実施される。例えば、トランスフェクションのためにリポソームが使用される場合、上記トランスフェクション反応は、最初に、上記DNA/リポソーム複合体が最初に添加される(例えば、約30分間にわたって)場合に室温でインキュベートされ、続いて、約37℃で特定された期間(例えば、約24時間)にわたってインキュベートされる。本発明の方法は、当該分野で公知の任意のトランスフェクション法で実施され得、従って、トランスフェクションが行われるはずである温度は、当業者に公知である。あるいは、トランスフェクションの間の上記インキュベーション温度が変動することを除いて、同一な条件下で並行していくつかのトランスフェクション実験を行うことによって、適切なインキュベーション温度を見いだすこともまた、可能である。レスキューされるウイルスの数は、トランスフェクションの効率を示し、上記のように、生じるPFUの数に関してアッセイすることによって決定され得る。最大数のPFUを生じる温度は、トランスフェクションのために使用され得る。
【0041】
上記細胞が上記トランスフェクトされた細胞に添加される温度および得られた細胞の混合物がインキュベートされる温度は、トランスフェクションの間に使用される温度と比較して、同じであってもよいし、異なっていてもよい。上記温度は、上記トランスフェクション後の添加のために使用される細胞株に依存し得、本発明の方法を使用してレスキューされる上記ウイルスに伴っても変動し得る。個々の細胞株を増殖させるか、もしくは特定のウイルスをレスキューするための適切な温度は、当該分野で公知であるが、それ故、当業者は、適切な温度を容易に同定し得る。例えば、Vero細胞株、Per.C6細胞株およびMDCK細胞株のような哺乳動物細胞株は、通常、36℃〜38℃の間の温度、もしくは約37℃で増殖させられる。この温度はまた、たとえいくつかのウイルス(インフルエンザのような)が、約33℃の温度でレスキューされ得るとしても、多くのウイルスのレスキューのために選択される。なぜなら、これは、得られるウイルスのよりよい抗原性を生じ得るからである[20]。適切な温度はまた、上記トランスフェクション後のインキュベーション温度が変動することを除いて、同一条件下で並行していくつかのトランスフェクション実験を行うことによって同定され得る。最大数のPFUを生じる温度は、トランスフェクション後に使用され得る。PFUについてアッセイするために適した方法は、トランスフェクションの間に使用され得る温度を同定するためのアッセイに関して既に上記に記載されている。
【0042】
(ウイルス調製)
さらなる局面において、本発明は、ワクチン製造のためにウイルスを調製する方法を提供し、上記方法は、(i)宿主細胞の培養物を、ウイルスRNA分子をコードする少なくとも1個の発現構築物でトランスフェクトする工程;(ii)上記(i)のトランスフェクトされた宿主細胞に細胞を添加して、細胞の混合物を提供する工程;(iii)ウイルスを生成するために、上記細胞の混合物を培養する工程;および(iv)上記工程(iii)で得られたウイルスを精製する工程、ならびに必要に応じて、(v)上記ウイルスをワクチンへと処方する工程を包含する。
【0043】
細胞が、本発明のこの局面において培養宿主として使用される場合、細胞培養条件(例えば、温度、細胞密度、pH値など)が、使用される上記細胞株および上記ウイルスを仮定した広い範囲にわたって変動性であり、本願の要件に適合され得ることは公知である。従って、以下の情報は、ガイドラインを表すに過ぎない。
【0044】
上記のように、細胞は、好ましくは、無血清培地もしくは無タンパク質培地中で培養される。
【0045】
上記細胞の増殖は、当業者に公知の方法に従って行われ得る。例えば、上記細胞は、遠心分離もしくは濾過のような通常の補助的方法を使用する灌流システムにおいて培養され得る。さらに、上記細胞は、感染前に、流加システム(fed−batch system)で、本発明に従って増殖させられ得る。本発明の状況において、培養システムは、上記細胞が最初にバッチシステムで培養され、培地中の栄養分(もしくは栄養分の一部)の枯渇が、濃縮された栄養分の制御された供給によって補償される流加システムに関して言及される。感染前の細胞の増殖の間に上記培地のpH値を、pH6.6〜pH7.8の値に、および特に、pH7.2〜pH7.3の間の値に調節することは、有利であり得る。細胞の培養は、好ましくは、30℃〜40℃の間の温度で行われる。工程(iii)において、上記細胞は、好ましくは、30℃〜36℃の間の温度もしくは32℃〜34℃の間の温度、または約33℃で培養される。これは、本発明の方法がインフルエンザウイルスを生成するために使用される場合に特に好ましい。なぜなら、この温度範囲での感染細胞のインキュベーションが、ワクチンへと処方される場合に改善された効率を生じるウイルスの生成を生じることが示されたからである[20]。
【0046】
酸素分圧は、感染前の培養の間に、好ましくは、25%〜95%の間の値において、および特には、35%〜60%の間の値において調節され得る。本発明の状況において示される酸素分圧の値は、空気の飽和に基づく。細胞の感染は、バッチシステムにおいては、好ましくは、約8〜25×105細胞/mLの細胞密度で、もしくは灌流システムにおいては、好ましくは、約5〜20×106細胞/mLの細胞密度で行われる。上記細胞は、10−8〜10の間、好ましくは、0.0001〜0.5の間のウイルス用量(MOI値(「感染多重度」;感染時の細胞あたりのウイルス単位の数に対応する)で感染させられ得る。
【0047】
ウイルスは、接着培養もしくは懸濁物中で、細胞において増殖させられ得る。マイクロキャリア培養が使用され得る。マイクロキャリア培養は、接着培養とみなされる。上記細胞はまた、懸濁物中での増殖に適合され得る。
【0048】
本発明に従う方法はまた、ウイルスの採取および単離、またはウイルスによって生成されるタンパク質の採取および単離を含む。ウイルスもしくはタンパク質の単離の間に、上記細胞は、分離、濾過もしくは限外濾過のような標準的な方法によって培養培地から分離される。次いで、上記ウイルスもしくは上記タンパク質は、勾配遠心分離、濾過、沈殿、クロマトグラフィーなどの当業者に十分公知の方法に従って濃縮され、次いで、精製される。また、上記ウイルスが精製の間もしくは精製後に不活性化されることは、本発明によれば好ましい。ウイルス不活性化は、上記精製プロセス内のいずれかの時点で、例えば、β−プロピオラクトンもしくはホルムアルデヒドによって行われ得る。
【0049】
本発明に従って単離される上記ウイルスはまた、工程(iii)において卵において増殖させられ得る。ワクチンのためのインフルエンザウイルス増殖の現在標準的な方法は、胚含有SPF鶏卵を使用し、ウイルスは、卵内容物(尿膜腔液)から精製される。卵を介してウイルスを継代し、その後、細胞培養においてウイルスを増殖させることもまた、可能である。
【0050】
(ウイルス)
本発明の方法は、細胞において逆遺伝学によって発現され得る任意のウイルスで実施され得る。このようなウイルスは、セグメント化ウイルスもしくは非セグメント化ウイルスであり得る。さらに、上記ウイルスは、プラス鎖RNAウイルスもしくはマイナス鎖RNAウイルスであり得る。上記ウイルスはまた、二本鎖RNAウイルスであり得る。
【0051】
上記ウイルスがマイナス鎖RNAウイルスである場合、上記ウイルスは、パラミクソウイルス科、ニューモウイルス亜科(Pneumovirinae)、ラブドウイルス科、フィロウイルス科、ボルナウイルス科(Bornaviridae)、オルソミクソウイルス科、ブンヤウイルス科、もしくはアレナウイルス科からなる群より選択される科に由来し得る。さらに、上記ウイルスは、パラミクソウイルス、オルソミクソウイルス、レスピロウイルス、モルビリウイルス、ルブラウイルス、ヘニパウイルス、アブラウイルス、ニューモウイルス、メタニューモウイルス、ベシクロウイルス、リッサウイルス、エフェメロウイルス、サイトラブドウイルス、ヌクレオラブドウイルス、ノビラブドウイルス、マールブルグウイルス、エボラウイルス、ボルナウイルス、インフルエンザウイルスA、インフルエンザウイルスB、インフルエンザウイルスC、トゴトウイルス、イサウイルス、オルソブンヤウイルス、ハンタウイルス、ナイロウイルス、フレボウイルス、トスポウイルス、アレナウイルス、オフィオウイルス、テヌイウイルス、もしくはデルタウイルスからなる群より選択される属に由来するウイルスであり得る。具体的実施形態において、上記マイナス鎖RNAウイルスは、センダイウイルス、麻疹ウイルス、ムンプスウイルス、ヘンドラウイルス、ニューキャッスル病ウイルス、ヒトRSウイルス、トリニューモウイルス、水疱性口内炎インディアナウイルス(Vesicular stomatitis Indiana virus)、狂犬病ウイルス、ウシ流行熱ウイルス、レタス壊死性黄変病ウイルス(Lettuce necrotic yellows virus)、ジャガイモ黄萎病ウイルス(Potato yellow dwarf virus)、伝染性造血器壊死症ウイルス(Infectious hematopoietic necrosis virus)、ビクトリア湖マールブルグウイルス、ザイールエボラウイルス、ボルナ病ウイルス、インフルエンザウイルス、トゴトウイルス、伝染性サケ貧血ウイルス(Infectious salmon anemia virus)、ブニヤムウェラーウイルス(Bunyamwera virus)、ハンタンウイルス、ジュグベウイルス(Dugbe virus)、リフトバレー熱ウイルス、トマト黄化え疽ウイルス(Tomato spotted wilt virus)、リンパ球性脈絡髄膜炎ウイルス(Lymphocytic choriomeningitis virus)、カンキツソローシスウイルス(Citrus psorosis virus)、イネ縞葉枯ウイルス(Rice stripe virus)、およびデルタ型肝炎ウイルスからなる群より選択される。好ましい実施形態において、上記ウイルスは、インフルエンザウイルスである(以下を参照のこと)。
【0052】
上記ウイルスがプラス鎖RNAウイルスである場合、上記ウイルスは、アルテリウイルス科、コロナウイルス科、ピコルナウイルス科およびロニウイルス科からなる群より選択される科に由来し得る。さらに、上記ウイルスは、アルテリウイルス、コロナウイルス、エンテロウイルス、トロウイルス、オカウイルス(Okavirus)、ライノウイルス、ヘパトウイルス、カルジオウイルス、アフトウイルス、パレコウイルス、エルボウイルス、コブウイルスおよびテッショウウイルスからなる群より選択される属に由来するウイルスであり得る。具体的実施形態において、上記ウイルスは、重症急性呼吸器症候群(SARS)ウイルス、ポリオウイルス、ヒトエンテロウイルスA(HEV−A)、ヒトエンテロウイルスB(HEV−B)、ヒトエンテロウイルスC、ヒトエンテロウイルスD、A型肝炎ウイルス、ならびにヒトライノウイルスAおよびBからなる群より選択される。
【0053】
上記ウイルスが二本鎖RNAウイルスである場合、上記ウイルスは、ビルナウイルス科、シストウイルス科、ハイポウイルス科、パルティティウイルス科、レオウイルス科およびトティウイルス科からなる群より選択される科に由来し得る。さらに、上記ウイルスは、アクアビルナウイルス、トリビルナウイルス、エントモビルナウイルス、シストウイルス、パルティティウイルス、アルファクリプトウイルス、ベータクリプトウイルス、アクアレオウイルス、コルティウイルス、サポウイルス、フィジウイルス、イドノレオウイルス(Idnoreovirus)、マイコレオウイルス、オルビウイルス、オルトレオウイルス、オリザウイルス、フィトレオウイルス、ロタウイルスおよびセドルナウイルス(Seadornavirus)からなる群より選択される属に由来するウイルスであり得る。
【0054】
本発明とともに使用するのに好ましいウイルスは、ロタウイルスである。これらウイルスでの逆遺伝学は、上記ウイルスレスキューの不十分な効率に起因して現在は困難であり、従って、本発明の方法は、このウイルスのレスキューを容易にし得る。
【0055】
本発明はまた、迅速な変異を受け、組換えアプローチがウイルスのより迅速な単離を可能にし、次いで、適切なワクチンを得るためにさらに増殖させられ得るウイルスに特に適している。従って、別の好ましい実施形態において、上記ウイルスはインフルエンザである。
【0056】
(インフルエンザウイルス)
インフルエンザウイルスは、本発明の方法における使用に特に適している(特に、インフルエンザAウイルスおよびインフルエンザBウイルス)。なぜなら、このウイルスについての逆遺伝学は、既に十分に特徴付けられているからである。インフルエンザウイルスは、セグメント化されたマイナス鎖RNAウイルスである。インフルエンザAウイルスおよびインフルエンザBウイルスは、8個のセグメントを有するのに対して、インフルエンザCウイルスは、7個のセグメントを有する。上記ウイルスは、複製および転写を開始するために、少なくとも4個のウイルスタンパク質(PB1、PB2、PAおよびヌクレオプロテイン)を必要とする。
【0057】
インフルエンザAウイルスおよびインフルエンザBウイルスについての逆遺伝学は、上記4個の必要とされるタンパク質および8個すべてのゲノムセグメントを発現するために、12個のプラスミドで実施され得る。しかし、構築物の数を減らすために、複数のRNAポリメラーゼI転写カセット(ウイルスRNA合成のため)が、単一のプラスミドに含まれ得(例えば、1個、2個、3個、4個、5個、6個、7個もしくは全8個のインフルエンザvRNAセグメントをコードする配列)、RNAポリメラーゼIIプロモーターを有する複数のタンパク質コード領域(例えば、1個、2個、3個、4個、5個、6個、7個もしくは8個のインフルエンザmRNA転写物をコードする配列)が別のプラスミドに含まれ得る[21]。1個以上のインフルエンザvRNAセグメントをpol Iプロモーターの制御下で、および同じプラスミド上に1個以上のインフルエンザタンパク質コード領域を別のプロモーター(特に、pol IIプロモーター)の制御下で含むこともまた、可能である。上記のように、これは、好ましくは、二方向性プラスミドを使用して行われる。参考文献21の方法の好ましい局面は、(a)単一のプラスミド上のPB1、PB2およびPAのmRNAコード領域;および(b)単一のプラスミド上の全8個のvRNAコードセグメントを含む。一方のプラスミド上にノイラミニダーゼ(NA)およびヘマグルチニン(HA)のセグメント、およびもう一方のプラスミド上に6個のほかのセグメントを含むことは、特に好ましい。なぜなら、新たに出現するインフルエンザウイルス株は、通常、上記NAセグメントおよび/もしくはHAセグメントに変異を有するからである。従って、この実施形態において、上記HA配列およびNA配列を含むベクターのみが、置換される必要がある。
【0058】
インフルエンザAウイルスの好ましい発現系は、複数の異なる野生型株に由来するゲノムセグメントをコードする。上記系は、PR/8/34株(A/Puerto Rico/8/34)に由来する1個以上の(例えば、1個、2個、3個、4個、5個もしくは6個の)ゲノムセグメントをコードし得るが、通常、これ/これらは、上記PR/8/34 HAセグメントを含まず、通常、上記PR/8/34 NAセグメントを含まない。よって、上記系は、PR/8/34に由来するセグメントNP、M、NS、PA、PB1および/もしくはPB2(おそらく6個すべて)のうちの少なくとも1個をコードし得る。
【0059】
インフルエンザAウイルスの他の有用な発現系は、AA/6/60 インフルエンザウイルス(A/Ann Arbor/6/60)に由来する1個以上の(例えば、1個、2個、3個、4個、5個もしくは6個の)ゲノムセグメントをコードし得るが、通常は、これ/これらは、上記AA/6/60 HAセグメントを含まず、通常、上記AA/6/60 NAセグメントを含まない。よって上記系は、AA/6/60に由来するセグメントNP、M、NS、PA、PB1および/もしくはPB2(おそらく6個すべて)のうちの少なくとも1個をコードし得る。
【0060】
上記系は、A/California/4/09株に由来する1個以上のゲノムセグメント(例えば、上記HAセグメントおよび/もしくは上記NAセグメント)をコードし得る。従って、例えば、上記HA遺伝子セグメントは、配列番号2より配列番号1に密接に関連する(すなわち、同じアルゴリズムおよびパラメーターを使用して、配列番号1と比較した場合、配列番号2より高い程度の配列同一性を有する)H1ヘマグルチニンをコードし得る。配列番号1および配列番号2は、80%同一である。同様に、上記NA遺伝子は、配列番号4より配列番号3に密接に関連しているN1ノイラミニダーゼをコードし得る。配列番号3および4は、82%同一である。
【0061】
インフルエンザBウイルスの発現系は、複数の異なる野生型株に由来するゲノムセグメントをコードし得る。上記系は、AA/1/66 インフルエンザウイルス(B/Ann Arbor/1/66)に由来する1個以上の(例えば、1個、2個、3個、4個、5個もしくは6個の)ゲノムセグメントをコードし得るが、通常は、これ/これらは、上記AA/1/66 HAセグメントを含まず、通常は、上記AA/1/66 NAセグメントを含まない。従って、上記系は、AA/1/66に由来するセグメントNP、M、NS、PA、PB1および/もしくはPB2のうちの少なくとも1個をコードし得る。
【0062】
A/PR/8/34株、AA/6/60株、AA/1/66株、A/Chile/1/83株およびA/California/04/09株に由来するウイルスセグメントおよび配列は、広く利用可能である。それらの配列は、公のデータベースで利用可能である(例えば、GI:89779337、GI:89779334、GI:89779332、GI:89779320, GI:89779327、GI:89779325、GI:89779322、GI:89779329)。
【0063】
インフルエンザウイルスの逆遺伝学システムは、上記宿主細胞においてアクセサリタンパク質の発現をもたらす発現構築物を含み得る。例えば、非ウイルス性セリンプロテアーゼ(例えば、トリプシン)を発現することは有利であり得る。
【0064】
(ワクチン)
一局面において、本発明は、本発明の方法に従って生成されるウイルスを利用して、ワクチンを生成する。
【0065】
ワクチン(特に、インフルエンザウイルスのための)は、一般に、生のウイルスもしくは不活性化ウイルスのいずれかに基づく。不活性化ワクチンは、完全ビリオン、「スプリット」ビリオン、もしくは精製表面抗原に基づき得る。抗原はまた、ビロソームの形態で提示され得る。本発明は、これらタイプのワクチンのうちのいずれかを製造するために使用され得る。
【0066】
不活性化ウイルスが使用される場合、上記ワクチンは、完全ビリオン、スプリットビリオン、もしくは精製表面抗原(インフルエンザについては、ヘマグルチニンを含み、通常はまた、ノイラミニダーゼを含む)を含み得る。ウイルスを不活性化するための化学的手段としては、以下の薬剤のうちの1つ以上の有効量での処置が挙げられる:洗剤、ホルムアルデヒド、β−プロピオラクトン、メチレンブルー、ソラレン、カルボキシフラーレン(C60)、バイナリーエチルアミン、アセチルエチレンイミン、もしくはこれらの組み合わせ。ウイルス不活性化の非化学的方法は、当該分野で公知である(例えば、UV光もしくはγ線照射)。
【0067】
ビリオンは、ウイルス含有流体(例えば、尿膜腔液もしくは細胞培養上清)から、種々の方法によって採取され得る。例えば、精製プロセスは、上記ビリオンを破壊するために洗剤を含む線形スクロース勾配溶液を使用するゾーン遠心分離を含み得る。次いで、抗原は、ダイアフィルトレーションによって、必要に応じた希釈の後に精製され得る。
【0068】
スプリットビリオンは、精製されたビリオンを洗剤(例えば、エチルエーテル、ポリソルベート 80、デオキシコレート、トリ−N−ブチルホスフェート、Triton X−100、Triton N101、セチルトリメチルアンモニウムブロミド、Tergitol NP9など)で処理して、サブビリオン調製物を生成する(「Tween−エーテル」スプリットプロセスを含む)ことによって得られる。インフルエンザウイルスをスプリットするための方法は、例えば、当該分野で周知である(例えば、参考文献22〜27などを参照のこと)。上記ウイルスのスプリットは、代表的には、感染性であろうと非感染性であろうと、破壊濃度のスプリット薬剤で完全ウイルスを破壊するかもしくはフラグメント化することによって行われる。上記破壊は、上記ウイルスタンパク質の完全なもしくは部分的な可溶化を生じ、上記ウイルスの完全性を変化させる。好ましいスプリット薬剤は、非イオン性界面活性剤およびイオン性(例えば、カチオン性)界面活性剤(例えば、アルキルグリコシド、アルキルチオグリコシド、アシル糖、スルホベタイン、ベタイン、ポリオキシエチレンアルキルエーテル、N,N−ジアルキル−グルカミド、Hecameg、アルキルフェノキシ−ポリエトキシエタノール、NP9、4級アンモニウム化合物、サルコシル、CTAB(セチルトリメチルアンモニウムブロミド)、トリ−N−ブチルホスフェート、Cetavlon、ミリスチルトリメチルアンモニウム塩、リポフェクチン、リポフェクタミン、およびDOT−MA)、オクチル−もしくはノニルフェノキシポリオキシエタノール(例えば、Triton界面活性剤(例えば、Triton X−100もしくはTriton N101))、ポリオキシエチレンソルビタンエステル(Tween界面活性剤)、ポリオキシエチレンエーテル、ポリオキシエチレンエステル(polyoxyethlene ester)などである。1つの有用なスプリット手順は、デオキシコール酸ナトリウムおよびホルムアルデヒドの連続的効果を使用し、スプリットは、最初のビリオン精製の間に起こり得る(例えば、スクロース密度勾配溶液において)。よって、スプリットプロセスは、上記ビリオン含有材料の清澄化(非ビリオン材料を除去するために)、上記採取したビリオンの濃縮(例えば、吸着法(例えば、CaHPO4吸着)を使用して)、非ビリオン材料からの完全ビリオンの分離、密度勾配遠心分離工程においてスプリット薬剤を使用する(例えば、デオキシコール酸ナトリウムのようなスプリット薬剤を含むスクロース勾配を使用する)ビリオンのスプリット、次いで、望ましくない物質を除去するための濾過(例えば、限外濾過)を含み得る。スプリットビリオンは、有用には、リン酸ナトリウム緩衝化等張性塩化ナトリウム溶液中で再懸濁され得る。スプリットインフルエンザワクチンの例は、BEGRIVACTM、FLUARIXTM、FLUZONETMおよびFLUSHIELDTM製品である。
【0069】
本発明の方法はまた、生ワクチンを生成するために使用され得る。このようなワクチンは、通常は、ビリオン含有流体からビリオンを精製することによって調製される。例えば、上記流体は、遠心分離によって清澄にされ得、緩衝液(例えば、スクロース、リン酸カリウム、およびグルタミン酸一ナトリウムを含む)で安定化され得る。インフルエンザウイルスワクチンの種々の形態が、現在利用可能である(例えば、参考文献28の第17章および第18章を参照のこと)。生のウイルスは、MedImmuneのFLUMISTTM製品(三価の生のウイルス)を含む。
【0070】
精製されたインフルエンザウイルス表面抗原ワクチンは、表面抗原であるヘマグルチニン、および代表的には、同様にノイラミニダーゼを含む。精製形態でこれらタンパク質を調製するためのプロセスは、当該分野で周知である。上記FLUVIRINTM、AGRIPPALTMおよびINFLUVACTM製品は、インフルエンザサブユニットワクチンである。
【0071】
別の形態の不活性化抗原は、ビロソーム[29](核酸を含まないウイルス様リポソーム粒子)である。ビロソームは、洗剤でのウイルスの可溶化、続いて、ヌクレオキャプシドの除去およびウイルス糖タンパク質を含む膜の再構成によって、調製され得る。ビロソームを調製するための代替法は、ウイルス膜糖タンパク質を過剰量のリン脂質に添加して、それらの膜中においてリポソームにウイルスタンパク質を与えることを含む。
【0072】
上記ウイルスは、弱毒化され得る。上記ウイルスは、温度感受性であり得る。上記ウイルスは、低温適合(cold−adapted)され得る。これら3つの特徴は、生のウイルスを抗原として使用する場合に特に有用である。
【0073】
HAは、現在の不活性化インフルエンザワクチンにおける主要な免疫原であり、ワクチン用量は、HAレベルを参照することによって標準化されている(代表的には、SRIDによって測定される)。既存のワクチンは、代表的には、1株あたり約15μgのHAを含むが、より低用量が、使用され得る(例えば、小児に関して、もしくは汎流行的状況において、またはアジュバントを使用する場合)。分割用量(例えば、1/2(すなわち、7.5μg HA/株)、1/4および1/8)が使用されてきたことと同様に、より高い用量も使用されてきた(例えば、3×もしくは9×用量[30、31])。従って、ワクチンは、0.1〜150μgの間のHA/インフルエンザ株、好ましくは、0.1〜50μgの間(例えば、0.1〜20μg、0.1〜15μg、0.1〜10μg、0.1〜7.5μg、0.5〜5μgなど)を含み得る。特定の用量としては、1株あたり、例えば、約45、約30、約15、約10、約7.5、約5、約3.8、約3.75、約1.9、約1.5などが挙げられる。
【0074】
生ワクチンに関して、投与は、HA含有量よりむしろ、50%組織培養感染量(median tissue culture infectious dose)(TCID50)によって測定され、1株あたり106〜108の間の(好ましくは、106.5〜107.5の間の)TCID50が、代表的である。
【0075】
本発明とともに使用されるインフルエンザ株は、野生型ウイルスにおいて見いだされるような天然のHA、もしくは改変されたHAを有し得る。例えば、HAを改変して、トリ種においてウイルスの病原性を高くする決定因子(例えば、上記HA1/HA2切断部位の周りの超塩基性領域)を除去することは、公知である。逆遺伝学の使用は、このような改変を促進する。
【0076】
ワクチンにおける使用のためのインフルエンザウイルス株は、流行期ごとに変化する。汎流行期間の間には、ワクチンは、代表的には、2つのインフルエンザA株(H1N1およびH3N2)および1つのインフルエンザB株を含み、三価ワクチンが代表的である。本発明はまた、汎流行性ウイルス株(すなわち、上記ワクチンレシピエントおよび一般的なヒト集団が免疫学的に経験したことがない株(特にインフルエンザAウイルスの)(例えば、H2、H5、H7もしくはH9サブタイプ株))を使用し得、汎流行性株のインフルエンザワクチンは、1価であってもよいし、汎流行性株が補充された通常の三価ワクチンに基づいていてもよい。しかし、流行期および上記ワクチン中に含まれる抗原の性質に依存して、本発明は、HAサブタイプであるH1、H2、H3、H4、H5、H6、H7、H8、H9、H10、H11、H12、H13、H14、H15もしくはH16のうちの1つ以上に対して防御し得る。本発明は、インフルエンザAウイルス NAサブタイプN1、N2、N3、N4、N5、N6、N7、N8もしくはN9のうちの1つ以上に対して防御し得る。
【0077】
汎流行期間の株に対して免疫化するために適しているのに加えて、本発明の組成物は、汎流行性もしくは潜在的に汎流行性の株に対して免疫化するために特に有用である。汎流行のアウトブレイクを引き起こす可能性を与えるインフルエンザ株の特徴は、以下である:(a)現在広まっているヒト株のヘマグルチニンと比較して、新たなヘマグルチニンを含む(すなわち、10年間を超えてヒト集団において顕性でなかったもの(例えば、H2)、または上記ヒト集団において以前に全く認められなかったもの(例えば、一般に、トリ集団においてのみ見いだされてきたH5、H6もしくはH9)。その結果、上記ヒト集団は、上記株のヘマグルチニンに対して免疫学的に経験したことがない;(b)上記ヒト集団において水平伝播し得る;および(c)ヒトに対して病原性である。H5ヘマグルチニンタイプを有するウイルスは、汎流行性インフルエンザ(例えば、H5N1株)に対して免疫化するのに好ましい。他の考えられる株としては、H5N3、H9N2、H2N2、H7N1およびH7N7、ならびに任意の他の潜在的に出現する汎流行性株が挙げられる。本発明は、非ヒト動物集団からヒトへと拡がり得るかもしくは拡がってしまった潜在的汎流行性株(例えば、ブタ起源のH1N1インフルエンザ株)に対して防御するのに特に適している。次いで、本発明は、ヒトおよび非ヒト動物にワクチン接種するのに適している。
【0078】
抗原が上記組成物中に有用に含まれ得る他の株は、抗ウイルス治療に抵抗性である(例えば、オセルタミビル[32]および/もしくはザナミビルに抵抗性である)株である(耐性汎流行性株を含む[33])。
【0079】
本発明の組成物は、1つ以上の(例えば、1つ、2つ、3つ、4つもしくはこれ以上の)インフルエンザウイルス株(インフルエンザAウイルスおよび/もしくはインフルエンザBウイルスを含む)に由来する抗原を含み得る。ワクチンが1より多くのインフルエンザ株を含む場合、異なる株は、代表的には、別個に増殖させられ、上記ウイルスが採取された後に混合され、抗原が調製されてきた。従って、本発明のプロセスは、1より多くのインフルエンザ株に由来する抗原を混合する工程を包含し得る。三価ワクチンは、代表的である(2つのインフルエンザAウイルス株および1つのインフルエンザBウイルス株に由来する抗原を含む)。四価ワクチンもまた有用である[34](2つのインフルエンザAウイルス株および2つのインフルエンザBウイルス株に由来する抗原を含むか、または3つのインフルエンザAウイルス株および1つのインフルエンザBウイルス株に由来する抗原を含む)。
【0080】
(薬学的組成物)
本発明に従って製造されるワクチン組成物は、薬学的に受容可能である。それらは、通常、上記抗原に加えて、成分を含む。例えば、それらは、代表的には、1種以上の薬学的キャリアおよび/もしくは賦形剤を含む。以下に記載されるように、アジュバントもまた含まれ得る。このような成分の詳細な議論は、参考文献35で入手される。
【0081】
ワクチン組成物は、一般に、水性形態にある。しかし、いくつかのワクチンは、乾燥形態、例えば、注射可能な固体、またはパッチ上の、乾燥したかもしくは重合した調製物の形態にあり得る。
【0082】
ワクチン組成物は、チオメルサールもしくは2−フェノキシエタノールのような保存剤を含み得る。しかし、上記ワクチンは、実質的に水銀物質を実質的に含まないべきである(すなわち、5μg/ml未満)ことが好ましい(例えば、チオメルサールを含まない[26、36])。水銀を含まないワクチンは、より好ましい。α−トコフェロールスクシネートは、水銀化合物の代替として含まれ得る[26]。保存剤非含有ワクチンは、特に好ましい。
【0083】
張度を制御するために、組成物は、生理学的塩(例えば、ナトリウム塩)を含み得ることは好ましい。塩化ナトリウム(NaCl)が好ましく、塩化ナトリウムは、1〜20mg/mlの間で存在し得る。存在し得る他の塩としては、塩化カリウム、リン酸二水素カリウム、無水リン酸二ナトリウム(disodium phosphate dehydrate)、塩化マグネシウム、塩化カルシウムなどが挙げられる。
【0084】
ワクチン組成物は、一般に、200mOsm/kg〜400mOsm/kgの間(好ましくは、240〜360mOsm/kgの間)の重量オスモル濃度を有し、より好ましくは、290〜310mOsm/kgの範囲内に入る。ワクチン接種によって引き起こされる疼痛に対して影響を有さない重量オスモル濃度が以前に報告されている[37]が、この範囲において重量オスモル濃度を維持することは好ましい。
【0085】
ワクチン組成物は、1種以上の緩衝剤を含み得る。代表的な緩衝剤としては、以下が挙げられる:リン酸塩緩衝剤;Tris緩衝剤;ホウ酸塩緩衝剤;コハク酸塩緩衝剤;ヒスチジン緩衝剤(特に、水酸化アルミニウムアジュバントで);またはクエン酸塩緩衝剤。緩衝剤は、代表的には、5〜20mM範囲で含まれる。
【0086】
ワクチン組成物のpHは、一般に、一般に、5.0〜8.1の間、より代表的には、6.0〜8.0の間(例えば、6.5〜7.5の間、または7.0〜7.8の間)である。本発明のプロセスは、従って、バルクワクチンのpHを、パッケージ前に調節する工程を包含し得る。
【0087】
上記ワクチン組成物は、好ましくは、無菌である。上記ワクチン組成物は、好ましくは、非発熱性である(例えば、1用量あたり<1 EU(エンドトキシン単位、標準尺度)、好ましくは、<0.1 EU/用量を含む)。上記ワクチン組成物は、好ましくは、グルテン非含有である。
【0088】
本発明のワクチン組成物は、洗剤(例えば、ポリオキシエチレンソルビタンエステル界面活性剤(「Tweens」として公知)、オクトキシノール(例えば、オクトキシノール−9(Triton X−100)もしくはt−オクチルフェノキシポリエトキシエタノール)、セチルトリメチルアンモニウムブロミド(「CTAB」)、またはデオキシコール酸ナトリウム(特に、スプリットワクチンもしくは表面抗原ワクチンのために))を含み得る。上記洗剤は、微量においてのみ存在し得る。従って、上記ワクチンは、オクトキシノール−10およびポリソルベート80の各々について1mg/ml未満で含み得る。他の残りの微量の成分は、抗生物質(例えば、ネオマイシン、カナマイシン、ポリミキシンB)であり得る。
【0089】
ワクチン組成物は、1回の免疫化のための物質を含んでいてもよいし、複数回の免疫化のための物質を含んでいてもよい(すなわち、「複数用量」キット)。保存剤を含めることは、複数用量の準備において好ましい。複数用量組成物中に保存剤を含める代替として(もしくはそれに加えて)、上記組成物は、物質の取り出しのための無菌アダプタを有する容器中に含まれ得る。
【0090】
インフルエンザワクチンは、代表的には、約0.5mlの投与体積で投与されるが、半用量(すなわち、約0.25ml)が、小児に投与され得る。
【0091】
組成物およびキットは、好ましくは、2℃〜8℃の間で貯蔵される。それらは、凍結されるべきではない。それらは、理想的には、遮光して保持されるべきである。
【0092】
(宿主細胞DNA)
ウイルスが単離され、そして/または細胞株上で増殖させられた場合、最終ワクチン中に残っている細胞株DNAのいかなる腫瘍形成活性をも最小限にするために、上記DNAの量を最小限にすることは、標準的な実務である。
【0093】
従って、本発明に従って調製されるワクチン組成物は、好ましくは、1用量あたり10ng(好ましくは、1ng未満、より好ましくは、100pg未満)の残留宿主細胞DNAを含むが、微量の宿主細胞DNAが存在し得る。
【0094】
任意の残留宿主細胞DNAの平均長が500bp未満(例えば、400bp未満、300bp未満、200bp未満、100bp未満など)であることは、好ましい。
【0095】
夾雑するDNAは、標準的精製手順(例えば、クロマトグラフィーなど)を使用するワクチン調製の間に除去され得る。残留宿主細胞DNAの除去は、ヌクレアーゼ処理によって(例えば、DNaseを使用することによって)増強され得る。宿主細胞DNA夾雑物を減らすための便利な方法は、2工程処理(第1は、DNase(例えば、Benzonase)(これは、ウイルス増殖の間に使用され得る)を、次いで、カチオン性洗剤(例えば、CTAB)(これは、ビリオン破壊の間に使用され得る)使用する)を含む参考文献38および39において開示される。アルキル化剤(例えば、β−プロピオラクトン)での処理はまた、宿主細胞DNAを除去するために使用され得、有利なことには、ビリオンを不活性化するためにも使用され得る[40]。
【0096】
(アジュバント)
本発明の組成物は、有利には、アジュバントを含み得る。これは、上記組成物を受ける被験体において誘発される免疫応答(液性および/もしくは細胞性)を高めるように機能し得る。好ましいアジュバントは、水中油型エマルジョンを含む。種々のこのようなアジュバントは公知であり、それらは、代表的には、少なくとも1種の油および少なくとも1種の界面活性剤を含み、上記油および界面活性剤は、生体分解性(代謝可能)かつ生体適合性である。上記エマルジョン中の油滴は、一般に、直径5μm未満であり、理想的には、サブミクロンの直径を有し、これら小さなサイズは、マイクロフルイダイザー(microfluidiser)で達成されて、安定なエマルジョンを提供する。220nm未満のサイズを有する液滴は、濾過滅菌に供され得るので、好ましい。
【0097】
前記エマルジョンは、動物(例えば魚類)供給源または植物供給源からのものなどの油を含むことができる。植物油の供給源としては、堅果、種子および穀粒が挙げられる。ラッカセイ油、ダイズ油、ヤシ油およびオリーブ油が、最も一般的に利用できる堅果油の例である。例えばホホバ豆から得られる、ホホバ油を使用することができる。種子油としては、紅花油、綿実油、ヒマワリ種子油、ゴマ種子油およびこれらに類するものが挙げられる。穀粒の群の中で、トウモロコシ油が最も容易に入手できるが、他の穀物粒、例えばコムギ、オートムギ、ライムギ、イネ、テフ、ライコムギおよびこれらに類するものの油も使用することができる。グリセロールおよび1,2−プロパンジオールの6〜10炭素脂肪酸エステルは、種子油中に天然に存在しないが、堅果油および種子油から出発して適切な材料の加水分解、分離およびエステル化によって調製することができる。哺乳動物の乳からの脂肪および油は代謝性であり、従って、本発明の実施の際に使用することができる。動物供給源から純粋な油を得るために必要な分離、精製、鹸化および他の手段についての手順は、当該技術分野において周知である。殆どの魚類は、容易に回収できる代謝性の油を含有する。例えば、タラ肝油、サメ肝油、および鯨油、例えば鯨ろうが、ここで使用することができる魚油のいくつかの例である。多数の分岐鎖油が5炭素イソプレン単位で生化学的に合成されており、一般にテルペノイドと呼ばれる。サメ肝油は、スクアレン、2,6,10,15,19,23−ヘキサメチル−2,6,10,14,18,22−テトラコサヘキサン、として公知の、分岐した不飽和テルペノイドを含有し、これは、ここで特に好ましい。スクワラン、スクアレンの飽和類似体、も好ましい油である。スクアレンおよびスクワランを含む、魚油は、商業的供給源から容易に入手することができ、または当該技術分野において公知の方法によって得ることができる。別の好ましい油は、α―トコフェロール(下記参照)である。油の混合物を使用することができる。
【0098】
界面活性剤をそれらの「HLB」(親水親油バランス)によって分類することができる。本発明の好ましい界面活性剤は、少なくとも10、好ましくは少なくとも15、およびさらに好ましくは少なくとも16のHLBを有する。本発明は、ポリオキシエチレンソルビタンエステル界面活性剤(一般にTweenと呼ばれる)、特にポリソルベート20およびポリソルベート80;商品名DOWFAXTMで販売されている、エチレンオキシド(EO)、プロピレンオキシド(PO)および/またはブチレンオキシド(BO)のコポリマー、例えば、線状EO/POブロックコポリマー;反復エトキシ(オキシ−1,2−エタンジイル)基の数が様々であり得るオクトキシノール、オクトキシノール−9(Triton X−100、すなわちt−オクチルフェノキシポリエトキシエタノール)が特に興味深い;(オクチルフェノキシ)ポリエトキシエタノール(IGEPAL CA−630/NP−40);リン脂質、例えばホスファチジルコリン(レシチン);ノニルフェノールエトキシレート、例えばTergitolTMNPシリーズ;ラウリル、セチル、ステアリルおよびオレイルアルコールから誘導されたポリオキシエチレン脂肪エーテル(Brij界面活性剤として公知)、例えばトリエチレングリコールモノラウリルエーテル(Brij 30);ならびにソルビタンエステル(一般にSPANとして公知)、例えばソルビタントリオレエート(Span 85)およびソルビタンモノラウレートを含む(しかしこれらに限定されない)界面活性剤と共に用いることができる。非イオン性界面活性剤が好ましい。前記エマルジョンに含めるために好ましい界面活性剤は、Tween 80(ポリオキシエチレンソルビタンモノオレエート)、Span 85(ソルビタントリオレエート)、レシチンおよびTriton X−100である。
【0099】
界面活性剤の混合物、例えばTween 80/Span 85混合物、を使用することができる。ポリオキシエチレンソルビタンエステル、例えばポリオキシエチレンソルビタンモノオレエート(Tween 80)、およびオクトキシノール、例えばt−オクチルフェノキシポリエトキシエタノール(Triton X−100)、の組み合わせも適する。別の有用な組み合わせは、ラウレス9とポリオキシエチレンソルビタンエステルおよび/またはオクトキシノールを含む。
【0100】
界面活性剤の好ましい量(重量%)は、ポリオキシエチレンソルビタンエステル(例えば、Tween 80)0.01%から1%、特に約0.1%;オクチル−またはノニルフェノキシポリオキシエタノール(例えば、Triton X−100、またはTritonシリーズの他の洗剤)0.001%から0.1%、特に0.005%から0.02%;ポリオキシエチレンエーテル(例えば、ラウレス9)0.1%から20%、好ましくは0.1%から10%および特に0.1%から1%または約0.5%である。
【0101】
上記ワクチンがスプリットウイルスを含む場合、上記ワクチンは、水相中に遊離界面活性剤を含むことが好ましい。これは、上記遊離界面活性剤が、上記抗原に対して「スプリット効果」を発揮し得、それによって、他の状態で存在し得る任意の非スプリットビリオンおよび/もしくはビリオン凝集物を破壊するので、有利である。これは、スプリットウイルスワクチンの安全性を改善し得る[41]。
【0102】
好ましいエマルジョンは、<1μmの平均液滴サイズ(例えば、≦750nm、≦500nm、≦400nm、≦300nm、≦250nm、≦220nm、≦200nm、もしくはこれより小さい)を有し得る。これら液滴サイズは、便利なことには、微小流動化(microfluidisation)のような技術によって達成され得る。
【0103】
本発明で有用な特定の水中油型エマルジョンアジュバントとしては、以下が挙げられるが、これらに限定されない。
【0104】
・スクアレン、Tween 80、およびSpan85のサブミクロンエマルジョン。上記エマルジョンの容積での組成は、約5% スクアレン、約0.5% ポリソルベート80および約0.5% Span 85であり得る。重量では、これらの比は、4.3% スクアレン、0.5% ポリソルベート80および0.48% Span 85になる。このアジュバントは、引用文献45の第10章および引用文献46の第12章により詳細に記載されるように、「MF59」として公知である[42〜44]。上記MF59エマルジョンは、有利なことには、クエン酸イオン(例えば、10mM クエン酸ナトリウム緩衝液)を含む。
【0105】
・スクアレン、DL−α−トコフェロール、およびポリソルベート80(Tween 80)のエマルジョン。このエマルジョンは、リン酸緩衝生理食塩水を含み得る。これはまた、Span85(たとえば、1%)および/またはレシチンを含み得る。これらエマルジョンは、2〜10% スクアレン、2〜10% トコフェロールおよび0.3〜3% Tween 80を有し得、スクアレン:トコフェロールの重量比は、好ましくは、≦1である。なぜなら、これは、より安定なエマルジョンを提供するからである。スクアレンおよびTween 80は、約5:2の容積比もしくは約11:5の重量比で存在し得る。1種のこのようなエマルジョンは、Tween 80をPBS中に溶解して、2%溶液を与え、次いで、この溶液90mlと、(5gのDL−α−トコフェロールおよび5ml スクアレン)の混合物とを混合し、次いで、上記混合物を微小流動化する(microfluidise)ことによって、作製され得る。得られたエマルジョンは、例えば、100〜250nmの間、好ましくは、約180nmの平均直径を有するサブミクロン油滴を有し得る。上記エマルジョンはまた、3−de−O−アシル化モノホスホリルリピドA(3d−MPL)を含み得る。このタイプの別の有用なエマルジョンは、ヒトの用量あたり、0.5〜10mg スクアレン、0.5〜11mg トコフェロール、および0.1〜4mg ポリソルベート80を含み得る[47]。
【0106】
・スクアレン、トコフェロール、およびTriton洗剤(例えば、Triton X−100)のエマルジョン。上記エマルジョンはまた、3d−MPL(下記を参照のこと)を含み得る。上記エマルジョンは、リン酸緩衝液を含み得る。
【0107】
・ポリソルベート(例えば、ポリソルベート80)、Triton洗剤(例えば、Triton X−100)およびトコフェロール(例えば、α−トコフェロールスクシネート)を含むエマルジョン。上記エマルジョンは、約75:11:10(例えば、750μg/ml ポリソルベート80、110μg/ml Triton X−100および100μg/ml α−トコフェロールスクシネート)の質量比でこれら3種の成分を含み得、これら濃度は、抗原由来のこれら成分の何らかの寄与を含むはずである。上記エマルジョンはまた、スクアレンを含み得る。上記エマルジョンはまた、3d−MPL(下記を参照のこと)を含み得る。その水相は、リン酸緩衝液を含み得る。
【0108】
・スクアラン、ポリソルベート80およびポロキサマー401(「PluronicTM L121」)のエマルジョン。上記エマルジョンは、リン酸緩衝生理食塩水(pH7.4)中に処方され得る。このエマルジョンは、ムラミルジペプチドの有用な送達ビヒクルであり、「SAF−1」アジュバント中でトレオニル−MDPとともに使用されてきた[48](0.05〜1% Thr−MDP、5% スクアラン、2.5% Pluronic L121および0.2% ポリソルベート80)。このエマルジョンはまた、「AF」アジュバントでのように[49](5% スクアラン、1.25% Pluronic L121および0.2% ポリソルベート80)、上記Thr−MDPなしでも使用され得る。微小流動化が好ましい。
【0109】
・スクアレン、水性溶媒、ポリオキシエチレンアルキルエーテル親水性非イオン性界面活性剤(例えば、ポリオキシエチレン(12)セトステアリルエーテル)および疎水性非イオン性界面活性剤(例えば、ソルビタンエステルもしくはマンニドエステル(mannide ester)(例えば、ソルビタンモノオレエート(monoleate)もしくは「Span 80」))を含むエマルジョン。上記エマルジョンは、好ましくは、熱可逆性であり、そして/または上記油滴のうちの少なくとも90%(容積で)が、200nm未満のサイズを有する[50]。上記エマルジョンはまた、アルジトール;凍結保護剤(cryoprotective agent)(例えば、糖(例えば、ドデシルマルトシドおよび/もしくはスクロース));および/またはアルキルポリグリコシドのうちの1種以上を含み得る。上記エマルジョンは、TLR4アゴニストを含み得る[51]。このようなエマルジョンは、凍結乾燥され得る。
【0110】
・スクアレン、ポロキサマー105およびAbil−Careのエマルジョン[52]。アジュバント添加ワクチン中のこれら成分の最終濃度(重量)は、5% スクアレン、4% ポロキサマー105(プルロニックポリオール)および2% Abil−Care 85(ビス−PEG/PPG−16/16 PEG/PPG−16/16ジメチコン;カプリル/カプリントリグリセリド)である。
【0111】
・0.5〜50%の油、0.1〜10%のリン脂質、および0.05〜5%の非イオン性界面活性剤を有するエマルジョン。参考文献53に記載されるように、好ましいリン脂質成分は、ホスファチジルコリン、ホスファチジルエタノールアミン、ホスファチジルセリン、ホスファチジルイノシトール、ホスファチジルグリセロール、ホスファチジン酸、スフィンゴミエリンおよびカルジオリピンである。サブミクロン液滴サイズが有利である。
【0112】
・非代謝性の油(例えば、軽油(light mineral oil))および少なくとも1種の界面活性剤(例えば、レシチン、Tween 80もしくはSpan 80)のサブミクロン水中油型エマルジョン。添加剤が含まれ得る(例えば、QuilAサポニン、コレステロール、サポニン−親油性結合体(例えば、参考文献54に記載され、グルクロン酸のカルボキシル基を介して、脂肪族アミンを、デスアシルサポニン(desacylsaponin)に添加することにより生成されるGPI−0100)、ジメチルジオクタデシルアンモニウムブロミド(dimethyidioctadecylammonium bromide)および/もしくはN,N−ジオクタデシル−N,N−ビス(2−ヒドロキシエチル)プロパンジアミン)。
【0113】
・サポニン(例えば、QuilAもしくはQS21)およびステロール(例えば、コレステロール)が螺旋状ミセルとして会合されるエマルジョン[55]。
【0114】
・鉱油、非イオン性親油性エトキシ化脂肪アルコール、および非イオン性親水性界面活性剤(例えば、エトキシ化脂肪アルコールおよび/もしくはポリオキシエチレン−ポリオキシプロピレンブロックコポリマー)を含むエマルジョン[56]。
【0115】
・鉱油、非イオン性親水性エトキシ化脂肪アルコール、および非イオン性親油性界面活性剤(例えば、エトキシ化脂肪アルコールおよび/もしくはポリオキシエチレン−ポリオキシプロピレンブロックコポリマー)を含むエマルジョン[56]。
【0116】
いくつかの実施形態において、エマルジョンは、送達時に抗原と即座に混合され得、従って、上記アジュバントおよび抗原は、パッケージされたもしくは配布されたワクチン(使用時に最終処方物の準備ができている)では別個に保持され得る。他の実施形態において、エマルジョンは、製造の間に抗原と混合され、従って、上記組成物は、液体アジュバント添加形態(liquid adjuvanted form)においてパッケージされる。上記抗原は、一般に、水性形態にあり、その結果、上記ワクチンは、2種の液体を混合することによって最終的に調製される。混合するための上記2種の液体の容積比は、変動し得る(例えば、5:1〜1:5の間)が、一般に、約1:1である。成分濃度が、上記の特定のエマルジョンの説明において与えられる場合、これら濃度は、代表的には、非希釈組成物に関するものであり、したがって抗原溶液と混合した後の濃度は低下する。
【0117】
(ワクチン組成物のパッケージング)
本発明の組成物(もしくはキット成分)に適した容器としては、バイアル、シリンジ(例えば、使い捨てシリンジ)、鼻スプレーなどが挙げられる。これら容器は、滅菌であるべきである。
【0118】
組成物/成分がバイアル中に配置される場合、上記バイアルは、好ましくは、ガラス材料もしくはプラスチック材料から作製され得る。上記バイアルは、好ましくは、上記組成物が上記バイアルに添加される前に、滅菌される。ラテックス感受性患者に伴う問題を回避するために、バイアルは、好ましくは、ラテックス非含有ストッパーでシールされ、全てのパッケージング材料中にラテックスが存在しないことが好ましい。上記バイアルは、ワクチンの単一用量を含んでいてもよいし、1用量より多い用量(「複数用量」バイアル)、例えば、10用量を含んでいてもよい。好ましいバイアルは、無色ガラスから作製される。
【0119】
バイアルは、あらかじめ充填されたシリンジがキャップに挿入され得るように適合されたキャップ(例えば、ルアーロック)を有し得、上記シリンジの内容物は、上記バイアルへと排出され得(例えば、その中の凍結乾燥物質を再構成するために)、上記バイアルの内容物は、取り出されて上記シリンジへと戻され得る。上記バイアルから上記シリンジを取り出した後、次いで、ニードルが取り付けられ得、上記組成物が、患者へと投与され得る。上記キャップは、好ましくは、シールもしくはカバーの内部に配置され得る。その結果、上記シールもしくはカバーは、上記キャップに到達し得る前に除去されなければならない。バイアルは、特に、複数用量バイアルについては、その内容物の無菌的取り出しを可能にするキャップを有し得る。
【0120】
成分がシリンジにパッケージされる場合、上記シリンジは、これに取り付けられるニードルを有し得る。ニードルが取り付けられていない場合、別個のニードルが、組み立ておよび使用のために、上記シリンジと共に供給され得る。このようなニードルは、鞘に入れられ得る。安全ニードル(safety needle)が好ましい。1インチ 23ゲージ、1インチ 25ゲージおよび5/8インチ 25ゲージのニードルが代表的である。シリンジは、記録保持を容易にするために、ロット番号、インフルエンザ流行期、および内容物の使用期限がプリントされ得るピールオフラベルとともに提供され得る。上記シリンジにおけるプランジャーは、好ましくは、上記プランジャーが吸引の間に偶発的に除去されてしまわないように、ストッパーを有し得る。上記シリンジは、ラテックスゴムキャップおよび/もしくはプランジャーを有し得る。使い捨てシリンジは、単一用量のワクチンを含む。上記シリンジは、一般に、ニードルの取り付け前に、先端をシールするために先端キャップを有し、上記先端キャップは、好ましくは、ブチルゴムから作製され得る。上記シリンジおよびニードルが別個にパッケージされる場合、上記ニードルは、好ましくは、ブチルゴムシールドと適合させられる。好ましいシリンジは、商品名「Tip−Lok」TMの下で市販されるものである。
【0121】
容器は、例えば、小児への送達を容易にするために、半用量容積を示すために印が付けられ得る。例えば、0.5ml用量を含むシリンジは、0.25ml容積を示す印を有し得る。
【0122】
ガラス容器(例えば、シリンジもしくはバイアル)が使用される場合、ソーダ石灰ガラスよりむしろホウケイ酸ガラスから作製された容器を使用することが好ましい。
【0123】
キットもしくは組成物は、(例えば、同じボックスの中に)上記ワクチンの詳細(例えば、投与の説明書、上記ワクチン内の抗原の詳細など)を含むリーフレットとともにパッケージされ得る。上記説明書はまた、警告(例えば、ワクチン接種後のアナフィラキシー反応の場合に容易に利用可能なアドレナリン溶液を保持することなど)を含み得る。
【0124】
(処置方法、および上記ワクチンの投与)
本発明は、本発明に従って製造されるワクチンを提供する。これらワクチン組成物は、ヒト被験体もしくはヒト以外の動物被験体(例えば、ブタ)への投与に適しており、本発明は、上記被験体に本発明の組成物を投与する工程を包含する、被験体における免疫応答を惹起するための方法を提供する。本発明はまた、医薬として使用するための本発明の組成物を提供し、被験体における免疫応答を惹起するための医薬の製造のための、本発明の組成物の使用を提供する。
【0125】
これら方法および使用によって惹起される免疫応答は、一般に、抗体応答(好ましくは、防御的抗体応答)を含む。インフルエンザウイルスワクチン接種後の抗体応答、中和能力および防御を評価するための方法は、当該分野で周知である。ヒトでの研究から、ヒトインフルエンザウイルスのヘマグルチニンに対する抗体力価が、防御と相関する(約30〜40の血清サンプル赤血球凝集阻害力価が、同種のウイルスによる感染から約50%防御を与える)ことが示された[57]。抗体応答は、代表的には、赤血球凝集阻害によって、マイクロ中和によって、単一放射免疫拡散(single radial immunodiffusion)(SRID)によって、および/もしくは単一放射溶血(SRH)によって、測定される。これらアッセイ技術は、当該分野で周知である。
【0126】
本発明の組成物は、種々の方法において投与され得る。最も好ましい免疫化経路は、筋肉内注射(例えば、腕もしくは脚への)によるが、他の利用可能な経路としては、皮下注射、鼻内[58〜60]、経口[61]、皮内[62、63]、経皮的(transcutaneous)、経皮的(transdermal)[64]などが挙げられる。
【0127】
本発明に従って調製されるワクチンは、小児および成人の両方を処置するために使用され得る。インフルエンザワクチンは、6ヶ月齢からの小児および成人での免疫化における使用に関して現在推奨されている。従って、ヒト被験体は、1歳未満、1〜5歳、5〜15歳、15〜55歳、もしくは少なくとも55歳であってもよい。上記ワクチンを受けるのに好ましい被験体は、高齢(例えば、≧50歳、≧60歳、および好ましくは、≧65歳)、若年齢(例えば、≦5歳)、入院している被験体、ヘルスケアワーカー、軍隊および軍職員、妊婦、慢性疾患の、免疫不全の被験体、上記ワクチンを受ける7日前までに抗ウイルス化合物(例えば、オセルタミビルおよびザナミビル化合物;以下を参照のこと)を摂取している被験体、卵アレルギーがある人々、および外国に旅行する人々である。上記ワクチンは、これらの群に適しているのみではないが、一般に、集団においてより多く使用され得る。汎流行性株については、すべての年齢群への投与が好ましい。
【0128】
好ましい本発明の組成物は、効力についてCPMP基準のうちの1つ、2つもしくは3つを満たす。成人(18〜60歳)において、これら基準は、以下である:(1)≧70%血清保有率(seroprotection);(2)≧40%セロコンバージョン;および/もしくは(3)GMT増加≧2.5倍。高齢者(>60歳)において、これら基準は、以下である:(1)≧60%血清保有率;(2)≧30%セロコンバージョン;および/もしくは(3)GMT増加≧2倍。これら基準は、少なくとも50名の患者でのオープンラベル研究に基づく。
【0129】
処置は、単一用量スケジュールもしくは複数用量スケジュールによってであり得る。複数用量は、初回免疫化スケジュールおよび/もしくは追加免疫化スケジュールにおいて使用され得る。複数用量スケジュールにおいて上記種々の用量は、同じ経路もしくは異なる経路によって与えられ得る(例えば、非経口の初回免疫(prime)および粘膜の追加免疫(boost)、粘膜の初回免疫および非経口の追加免疫など)。1より多くの用量の投与(代表的には、2用量)は、免疫学的に経験したことがない患者(例えば、これまでインフルエンザワクチンを受けたことのない人に関して)において、または新たなHAサブタイプ(汎流行性アウトブレイクにおけるような)に対するワクチン接種のために、特に有用である。複数用量は、代表的には、少なくとも1週間空けて(例えば、約2週間、約3週間、約4週間、約6週間、約8週間、約10週間、約12週間、約16週間など)投与される。
【0130】
本発明によって生成されるワクチンは、他のワクチン(例えば、麻疹ワクチン、ムンプス・ワクチン、風疹ワクチン、MMRワクチン、水痘ワクチン、MMRVワクチン、ジフテリアワクチン、破傷風ワクチン、百日咳ワクチン、DTPワクチン、結合体化H.influenzaeタイプbワクチン、不活性化ポリオウイルスワクチン、B型肝炎ウイルスワクチン、髄膜炎菌結合型ワクチン(例えば、四価A−C−W135−Yワクチン)、RSウイルスワクチン、肺炎球菌結合型ワクチンなど)と実質的に同時(例えば、同じ医療相談の間に、またはヘルスケア専門家およびワクチン接種施設への訪問の間に)に患者に投与され得る。肺炎球菌ワクチンおよび/もしくは髄膜炎菌ワクチンと実施的に同時の投与は、高齢の患者において特に有用である。
【0131】
同様に、本発明のワクチンは、抗ウイルス化合物、および特に、インフルエンザウイルスに対して活性な抗ウイルス化合物(例えば、オセルタミビルおよび/もしくはザナミビル)と実質的に同時(例えば、同じ医療相談の間に、またはヘルスケア専門家への訪問の間に)に、患者に投与され得る。これら抗ウイルス剤としては、ノイラミニダーゼインヒビター(例えば、(3R,4R,5S)−4−アセチルアミノ−5−アミノ−3(1−エチルプロポキシ)−1−シクロヘキセン−1−カルボン酸もしくは5−(アセチルアミノ)−4−[(アミノイミノメチル)−アミノ]−2,6−アンヒドロ−3,4,5−トリデオキシ−D−グリセロ−D−ガラクトノン−2−エノン酸(そのエステル(例えば、そのエチルエステル)およびその塩(例えば、そのリン酸塩)を含む)が挙げられる。好ましい抗ウイルス剤は、(3R,4R,5S)−4−アセチルアミノ−5−アミノ−3(1−エチルプロポキシ)−1−シクロヘキセン−1−カルボン酸、エチルエステル、ホスフェート(1:1)(オセルタミビルホスフェート(TAMIFLUTM)としても公知)である。
【0132】
(一般)
用語「含む(comprising)」は、「含む(including)」および「からなる(consisting)」を含み、例えば、Xを「含む」組成物は、もっぱらXからなってもよいし、さらなる何かを含んでいてもよい(例えば、X+Y)。
【0133】
語句「実質的に」は、「完全に」を排除しない。例えば、Yを「実質的に含まない」組成物は、完全にYを含まなくてもよい。必要であれば、語句「実質的に」は、本発明の定義から省略され得る。
【0134】
数値xに関する用語「約」とは、任意であり、例えば、x±10%を意味する。
【0135】
別段示されなければ、2種以上の成分を混合する工程を包含するプロセスは、いかなる特定の混合する順序も必要としない。従って、成分は、任意の順序で混合され得る。3つの成分が存在する場合、2つの成分が、互いに合わされ得、次いで、その組み合わせが、第3の成分と合わせられ得るなど。
【0136】
動物(および特にウシ)の物質が、細胞培養において使用される場合、それら物質は、伝染性海綿状脳症(TSE)を含まない、特に、ウシ海綿状脳症(BSE)を含まない供給源から得られるべきである。全体として、動物由来物質が完全に存在しない状態で細胞を培養することが好ましい。
【0137】
化合物が、組成物の一部として身体に投与される場合、その化合物は、代わりに、適切なプロドラッグによって置換され得る。
【図面の簡単な説明】
【0138】
【図1】図1:本発明の実施形態の概略図;MDCK 33016懸濁細胞(CDM中で培養)を、6ウェルディッシュA(無血清培地中もしくはDMEM+5% FBS中);B:同時トランスフェクションMDCK 33016;C:洗浄2×,無血清DMEMに交換,+trypzean,+/−新鮮な非感染MDCK細胞,D:RGウイルス中に播種する。
【図2】図2:PR8−A/CAバックボーン(backbone)を使用するMDCK 33016細胞でのインフルエンザウイルスレスキューの効率;y軸は、感染力価(FFU/mL)を示す;白抜き四角は、PR8−A/CAおよび5% 血清を使用した結果を示し、斜線を入れた四角は、PR8−A/CAありおよび血清なしでの結果を示す;Aは、新鮮な非感染細胞を添加したことを示す;Bは、新鮮な非感染細胞を添加しなかったことを示す。
【図3】図3:A/WSN/33バックボーンを使用するMDCK 33016細胞でのインフルエンザウイルスレスキューの効率;y軸は、感染力価(FFU/mL)を示す;白抜き四角は、WSNおよび5% 血清を使用した結果を示し、斜線を入れた四角は、WSNありおよび血清なしでの結果を示す;Aは、新鮮な非感染細胞を添加したことを示す;Bは、新鮮な非感染細胞を添加しなかったことを示す。
【図4】図4:PR8−WSNバックボーンを使用するMDCK 33016細胞でのインフルエンザウイルスレスキューの効率。y軸は、FFU/mLを示す;白抜き四角は、PR8−WSNおよび5%血清を使用した結果を示し、斜線を入れた四角は、PR8−WSNありおよび血清なしでの結果を示す;Aは、新鮮な非感染細胞を添加したことを示す;Bは、新鮮な非感染細胞を添加しなかったことを示す。
【発明を実施するための形態】
【0139】
(本発明を実施するための態様)
(ウイルスレスキュー)
MDCK 33016細胞を、CDM培地中、温度37℃において懸濁して培養した。トランスフェクションの前日に、6×105細胞/ウェルを、2mL DMEM培地(5% FCSを含む)中、CELL−BINDTM 6ウェルディッシュの中に播種した。並行実験において、上記細胞を、FCSの非存在下で播種した。上記細胞を、37℃において一晩インキュベートした。
【0140】
翌日、上記播種した細胞を、製造業者のプロトコルを使用して、LipofectamineTM LTX Plusトランスフェクション試薬でトランスフェクトした。簡潔には、1μgの各プラスミド(PA、PB1、PB2、NP、NS、M、HA、NAと、TMPRSS2)を、500μlの無血清DMEM培地中に希釈した。10μlのPlus試薬を、上記希釈したDNAに直接添加した。上記混合物を、室温で5分間にわたってインキュベートし、その後、25μlのLipofectamineをそれに添加した。次いで、上記混合物を、室温で30分間にわたってインキュベートした。インキュベーション後、上記トランスフェクション試薬:DNA複合体を、滴下様式において上記細胞に添加した。上記細胞を、その後、37℃において24時間にわたってインキュベートした。
【0141】
インキュベーション後、培地を細胞から吸引し、1:2000希釈のTrypzeanTM(1mg/mL ストック溶液)を含む無血清DMEM培地中の4×105 MDCK 33016細胞の懸濁物を、各ウェルに添加した。並行した実験において、1:2000希釈のTrypzeanTM(1mg/mL ストック溶液)単独(すなわち、細胞の添加なし)を、上記トランスフェクトされた細胞に添加した。次いで、上記細胞を、37℃においてさらに48時間にわたって、または上記細胞が溶解するまでインキュベートし、その後、ウイルス力価を、病巣形成アッセイを使用して決定した。
【0142】
(病巣形成アッセイ(Focus−Forming Assay))
非感染MDCK細胞を、密度6.25×104細胞/ウェルにおいて、48ウェルプレートの中に、500μlの10% FCS含有DMEM中にプレートした。翌日、細胞に、容積100〜150μlにおいてウイルスを2時間にわたって37℃において感染させた。上記細胞に、上記ウイルスの種々の希釈物を感染させた。感染の2時間後、培地を吸引し、500μlの10% FCS含有DMEMを各ウェルに添加した。上記細胞を、翌日まで37℃においてインキュベートした。
【0143】
感染の24時間後、培地を吸引し、細胞をPBSで1回洗浄した。500μlの氷冷したPBS中80% アセトンを、各ウェルに添加し、続いて、4℃において30分間にわたってインキュベートした。アセトン混合物を吸引し、上記細胞を、PBST(PBS+0.1% Tween)で1回洗浄した。500μlのPBS中2% BSAを、各ウェルに添加し、続いて、室温(RT)において30分間にわたってインキュベートした。500μlの1:6000希釈の抗NPを、ブロッキング緩衝液中に添加し、続いて、室温において2時間にわたってインキュベートした。上記抗体溶液を吸引し、上記細胞をPBSTで2回洗浄した。2次抗体(ヤギ抗マウス)を、1:2000希釈において500μl ブロッキング緩衝液中で添加し、上記プレートを、室温において2時間にわたってインキュベートした。抗体溶液を吸引し、細胞をPBSTで3回洗浄した。500μlのKPL True Blueを各ウェルに添加し、10分間にわたってインキュベートした。True−Blueを吸引し、dH2Oで1回洗浄することによって、上記反応を停止した。上記水を吸引し、上記細胞を乾燥させた。
【0144】
(結果)
3種の異なるウイルスバックボーンでの上記病巣形成アッセイの結果を、図2〜4に示す。上記結果は、感染の24時間後に細胞の添加ありおよびなしでのウイルスレスキューの効率を示す。認められ得るように、すべての場合において、上記ウイルスレスキューの効率は、細胞を添加しなかった実験と比較して、トランスフェクションの24時間後に細胞を添加した実験において最大3倍まで増大した。この効率の増大は、試験したすべてのウイルスバックボーンで、ならびに血清含有条件下および無血清条件下の両方認められ得る。
【0145】
本発明は、例示によって記載されてきたに過ぎず、改変が行われ得る一方で、本発明の範囲および趣旨内にとどまっていることが理解される。
【0146】
【化1】

【0147】
【化2】
【0148】
【化3】
【技術分野】
【0001】
この出願は、2009年10月20日に出願された米国仮出願第61/279,487号(この完全な内容は、参考として本明細書に援用される)の利益を主張する。
【0002】
(技術分野)
本発明は、逆遺伝学の分野にある。さらに、種々のウイルスに対して防御するためのワクチンを製造することに関する。
【背景技術】
【0003】
(背景技術)
逆遺伝学は、細胞培養物においてRNAウイルスの組換え発現および操作を可能にする。逆遺伝学は、ウイルス学およびワクチン製造において強力なツールである。なぜなら、逆遺伝学は、組換えウイルス(リアソータントを含む)の迅速な生成および/もしくはそれらの変異を可能にするからである。上記方法は、宿主細胞を、ウイルスゲノムをコードする1個以上の発現構築物でトランスフェクトする工程および上記ウイルスを上記細胞から単離する工程を包含する。
【0004】
逆遺伝学によるウイルスレスキュー(virus rescue)の1つの欠点は、そのプロセスが非効率的であり、頻繁に、結果として得られるウイルス収量が十分ではないことである。従って、代替のより効率的な逆遺伝学法を提供することが当該分野には未だに必要である。
【発明の概要】
【課題を解決するための手段】
【0005】
(好ましい実施形態の要旨)
本発明者らは、ここで、逆遺伝学法の不十分な効率が、細胞を上記逆遺伝学構築物でトランスフェクトする工程およびその後、より多くの細胞を上記トランスフェクトした細胞に添加する工程によって、顕著に改善され得ることを驚くべきことに発見した。本発明者らは、観察された効率の増大が、上記細胞に対してトランスフェクションが有害であるという事実に起因すると推量する。従って、レスキューされる任意のウイルスは、疾患細胞において拡大するか、もしくは上記ウイルスが健康な細胞に移入されるまで待つことが必要である。このことは、生存可能なウイルスの喪失を生じる。上記トランスフェクトされた宿主細胞への細胞の添加は、上記ウイルス収量を顕著に増大させる健康な細胞基質上で、上記レスキューされたウイルスが繁殖することを可能にする。
【0006】
一実施形態において、本発明は、ウイルスを調製するための方法を提供し、上記方法は、(i)宿主細胞の培養物を、ウイルスRNA分子をコードする少なくとも1個の発現構築物でトランスフェクトする工程;(ii)細胞を、上記(i)のトランスフェクトされた宿主細胞に添加して、細胞の混合物を提供する工程;および(iii)ウイルスを生成するために、上記細胞の混合物を培養する工程を包含する。
【0007】
さらなる実施形態において、本発明は、ワクチン製造のためにウイルスを調製する方法を提供し、上記方法は、(i)宿主細胞の培養物を、ウイルスRNA分子をコードする少なくとも1個の発現構築物でトランスフェクトする工程;(ii)細胞を、上記(1)のトランスフェクトされた宿主細胞に添加して、細胞の混合物を提供する工程;(iii)ウイルスを生成するために、上記細胞の混合物を培養する工程;(iv)上記工程(iii)において得られたウイルスを精製する工程および必要に応じて(v)上記ウイルスをワクチンへと処方する工程を包含する。
【0008】
(逆遺伝学)
逆遺伝学は、広く種々のRNAウイルス(プラス鎖RNAウイルス[1、2]、マイナス鎖RNAウイルス[3、4]および二本鎖RNAウイルス[5]を含む)の生成のために使用され得る。従って、本発明は、組換えウイルスを生成するための方法を提供し、ここで上記ウイルスは、本発明の逆遺伝学法を使用して得られる。
【0009】
既知の逆遺伝学システムは、pol Iプロモーター、細菌RNAポリメラーゼプロモーター、バクテリオファージポリメラーゼプロモーターなどから、所望のウイルスRNA(vRNA)分子をコードするDNA分子を発現する工程を包含する。さらに、ウイルスが、感染性ウイルスを形成するために特定のタンパク質を必要とする場合、上記システムはまた、これらタンパク質を提供し、例えば、上記システムは、ウイルスタンパク質をコードするDNA分子をさらに含み、その結果、DNAの両方のタイプの発現が、完全な感染性ウイルスの構築をもたらす。
【0010】
逆遺伝学が、vRNAの発現のために使用される場合、互いに対する配列エレメントの正確な間隔が、複製を開始するための上記ポリメラーゼの中枢であることは、当業者に明らかである。従って、上記ウイルスRNAをコードするDNA分子が、上記pol Iプロモーターと終結配列との間に正確に配置されることは重要であるが、この配置は、逆遺伝学システムを取り扱う者の能力の十分範囲内である。
【0011】
一般に、逆遺伝学は、ウイルスの生活環の間にゲノムRNAの生成を必要とすることが公知の任意のウイルスの発現に適している。このようなウイルスとしては、プラス鎖RNAウイルスおよびマイナス鎖RNAウイルス(例えば、以下に記載されるもの)が挙げられるが、これらに限定されない。好ましくは、上記ウイルスは、オルソミクソウイルス(例えば、インフルエンザウイルス)である。本発明の方法は、さらに、非セグメント化ウイルスおよびセグメント化ウイルスにも適している。
【0012】
上記ウイルスがプラス鎖RNAウイルスである場合、上記ウイルスゲノムを含む発現構築物で細胞をトランスフェクトすることはしばしば十分である。例えば、ポリオウイルスゲノムを含むプラスミドのトランスフェクションは、感染性ポリオウイルスの回収をもたらした[1、2]。マイナス鎖RNAウイルスの逆遺伝学は、より大きな難題を呈した。なぜなら、上記アンチセンスウイルスRNAは、通常は、非感染性であり、その生活環を完了するのにRNAポリメラーゼを必要とするからである。従って、上記ウイルスポリメラーゼは、タンパク質としてか、もしくはインサイチュでのタンパク質発現のための遺伝子としてかのいずれかで、供給されなければならない。
【0013】
上記ウイルスが感染性のためにタンパク質を必要とする場合、二方向性発現構築物(bi−directional expression construct)を使用することは一般に好ましい。なぜなら、これは、上記宿主細胞が必要とする発現構築物の総数を減らすからである。従って、本発明の方法は、少なくとも1個の二方向性発現構築物を利用し得る。ここで遺伝子もしくはcDNAは、上流のpol IIプロモーターと下流の非内因性pol Iプロモーターとの間に位置する。上記pol IIプロモーターからの上記遺伝子もしくはcDNAの転写は、キャップされたプラス−センスウイルスmRNAを生成し、これはタンパク質へと翻訳され得る一方で、上記非内因性pol Iプロモーターからの転写は、マイナス−センスvRNAを生成する。上記二方向性発現構築物は、二方向性発現ベクターであり得る。
【0014】
組換えウイルスを生成するために、細胞は、ビリオンを構築するために必要なウイルスゲノムのすべてのセグメントを発現しなければならない。本発明の発現構築物へとクローニングされたDNAは、好ましくは、上記ウイルスRNAおよびタンパク質のすべてを提供する。しかし、上記RNAおよびタンパク質のうちのいくらかを提供するためにヘルパーウイルスを使用することも考えられ得るものの、ヘルパーウイルスを使用しないシステムが好ましい。上記ウイルスが非セグメント化ウイルスである場合、これは、1個より多くの発現構築物を使用して非セグメント化ウイルスのウイルスゲノムを発現することは、本発明の範囲内にあるとしても、通常は、本発明の方法において単一の発現構築物を利用することによって達成され得る。上記ウイルスがセグメント化ウイルスである場合、上記ウイルスゲノムは、通常は、本発明の方法において1個より多くの発現構築物を使用して発現される。しかし、単一の発現構築物の上で上記ウイルスゲノムのうちの1個より多くのセグメントもしくはさらにはすべてのセグメントを合わせることもまた、想定される。
【0015】
本発明の方法は、リアソータントウイルス株の生成に特に適している。その技術は、プラスミドのインビトロでの操作を使用して、ウイルスセグメントの組み合わせを生成し、上記ウイルスセグメントにおけるコード配列もしくは非コード配列の操作を容易にし、変異などを導入し得る。リアソータントウイルス株の生成のための上記発現系の使用は、好ましい。なぜなら、これは、ワクチンの迅速な生成が流行に対抗するために必要とされる状況において特に有益であるリアソータント種ウイルスを得るために必要とされる時間を顕著に減少させ得るからである。従って、本発明のこの局面の方法は、少なくとも2つの異なる野生型株に由来するかもしくはこれらから得られるウイルス遺伝子を発現する1個以上の発現構築物を使用することが好ましい。
【0016】
上記宿主細胞においてアクセサリタンパク質の発現をもたらす発現構築物もまた、使用され得る。例えば、非ウイルス性セリンプロテアーゼ(例えば、トリプシン)を逆遺伝学システムの一部として発現することもまた、有利であり得る。
【0017】
(発現構築物)
本発明における使用のために上記ウイルスRNA分子をコードする発現構築物は、ウイルスをレスキューするために当該分野で一般に使用される任意の発現構築物であり得る。
【0018】
上記使用される発現構築物は、一方向性発現構築物であってもよいし、二方向性発現構築物であってもよい。宿主細胞は1つより多い導入遺伝子を発現する場合(同じ発現構築物においてであろうと、異なる発現構築物においてであろうと)、一方向性および/もしくは二方向性の発現を使用することは可能である。
【0019】
二方向性発現構築物は、同じ構築物から異なる方向(すなわち、5’→3’および3’→5’の両方)において発現を駆動する少なくとも2個のプロモーターを含む。上記2個のプロモーターは、同じ二本鎖DNAの異なる鎖に作動可能に連結され得る。好ましくは、上記プロモーターのうちの一方は、pol Iプロモーターであり他方のプロモーターのうちの少なくとも1つは、pol IIプロモーターである。このことは、上記pol Iプロモーターが、キャップされていないcRNAを発現するために使用され得る一方で、上記pol IIプロモーターが、タンパク質へと後に翻訳され得るmRNAを転写するために使用され得、よって同じ構築物からRNAおよびタンパク質の同時の発現を可能にするので有用である。
【0020】
上記発現構築物(一方向性であろうと二方向性であろうと)は、代表的には、RNA転写終結配列を含む。上記終結配列は、内因性終結配列であってもよいし、上記宿主細胞に対して内因性ではない終結配列であってもよい。適切な終結配列は、当業者に明らかであり、RNAポリメラーゼI転写終結配列、RNAポリメラーゼII転写終結配列、およびリボザイムが挙げられるが、これらに限定されない。さらに、上記発現構築物は、mRNAについて、特に発現がpol IIプロモーターによって制御される遺伝子の末端において、1個以上のポリアデニル化シグナルを含み得る。
【0021】
上記発現構築物において使用される上記pol Iプロモーターおよびpol IIプロモーターは、上記発現構築物が発現される宿主細胞と分類学上同じ目にある生物に由来し得る。あるいは、上記プロモーターは、上記宿主細胞とは異なる分類学上の目にある生物に由来し得る。用語「目」とは、従来の分類学上の順位をいい、目の例は、霊長目、齧歯目、食肉目、有袋目、クジラ目などである。ヒトおよびチンパンジーは、分類学上同じ目(霊長目)にあるが、ヒトおよびイヌは、異なる目にある(霊長類 対 食肉目)。
【0022】
本発明の方法において、宿主細胞は、少なくとも2個、少なくとも3個、少なくとも4個、少なくとも5個、少なくとも6個、少なくとも7個、少なくとも8個、少なくとも9個、少なくとも10個、少なくとも11個、もしくは少なくとも12個の発現構築物でトランスフェクトされ得る。
【0023】
発現構築物は、ベクター(例えば、プラスミドもしくは他のエピソーム構築物)であり得る。このようなベクターは、代表的には、少なくとも1個の細菌複製起点および/もしくは真核生物複製起点を含む。さらに、上記ベクターは、原核生物細胞および/もしくは真核生物細胞における選択を可能にする選択マーカーを含み得る。このような選択マーカーの例は、抗生物質(例えば、アンピシリンもしくはカナマイシン)に対する耐性を付与する遺伝子である。上記ベクターは、DNA配列のクローニングを促進するために、1個以上の、マルチクローニング部位をさらに含み得る。
【0024】
代わりとして、発現構築物は、線形の発現構築物であり得る。このような線形の発現構築物は、代表的には、任意の増幅配列および/もしくは選択配列を含まない。しかし、このような増幅配列および/もしくは選択配列を含む線形の構築物はまた、本発明の範囲内である。インフルエンザウイルスの発現のために、このような線形の発現構築物を使用する方法の例は、参考文献6に記載される。
【0025】
本発明の発現構築物は、当該分野で公知の方法を使用して、生成され得る。このような方法は、例えば、参考文献7に記載された。上記発現構築物が線形の発現構築物である場合、単一の制限酵素部位を利用して上記宿主細胞に導入する前に、発現構築物を線形にすることは可能である。あるいは、少なくとも2個の制限酵素部位を使用して、上記発現構築物をベクターから切り出すことは可能である。さらに、核酸増幅技術を使用して(例えば、PCRによって)増幅することによって、線形の発現構築物を得ることもまた、可能である。
【0026】
上記発現宿主がイヌ細胞(例えば、MDCK細胞株)である場合、タンパク質コード領域は、例えば、野生型イヌ遺伝子に由来するかもしくはイヌウイルスに由来するプロモーターを使用して、そして/またはヒト細胞よりイヌ細胞に適したコドン使用頻度を有して、イヌの発現に最適化され得る。例えば、ヒト遺伝子がPheのコドンとしてわずかにUUCを好む(54%)のに対して、イヌ細胞では、優先度はより強い(59%)。同様に、ヒト細胞においてIleコドンに大きな優先度はないのに対して、イヌのコドンのうちの53%は、Ileに関してAUCを使用する。イヌウイルス(例えば、イヌパルボウイルス(ssDNAウイルス))はまた、コドン最適化についてガイダンスを提供し得る(例えば、イヌパルボウイルス配列におけるPheコドンのうちの95%は、UUUであり(イヌゲノムにおける41%に対して)、Ileコドンのうちの68%は、AUUであり(32%に対して)、Valコドンのうちの46%は、GUUであり(14%に対して)、Proコドンのうちの72%は、CCAであり(25%に対して)、Tyrコドンのうちの87%は、UAUであり(40%に対して)、Hisコドンのうちの87%は、CAUであり(39%に対して)、Glnコドンのうちの92%は、CAAであり(25%に対して)、Gluコドンのうちの81%は、GAAであり(40%に対して)、Cysコドンのうちの94%は、UGUであり(42%に対して)、Serコドンのうちの1%のみがUCUであり(24%に対して)、CCCは、Pheについては決して使用されず、UAGは、終止コドンとして決して使用されない。従って、タンパク質コード遺伝子は、性質がイヌ細胞における発現のためにすでに最適化され、発現を促進したより類似の遺伝子にされ得る。
【0027】
(トランスフェクション)
「トランスフェクション」とは、細胞へのDNAの導入をいう。上記発現構築物は、当業者に公知の任意の技術を使用して、宿主細胞へと導入され得る。例えば、それらは、エレクトロポレーション、DEAE−デキストラン、リン酸カルシウム沈殿、リポソーム、マイクロインジェクション、もしくは微粒子銃(microparticle−bombardment)を使用することによって、宿主細胞へと導入され得る。逆遺伝学は、一般に、リポソーム法を使用して、例えば、トランスフェクション試薬LipofectamineTMを使用して実施される。本発明の方法は、リポソームでのトランスフェクションに限定されないが、他のトランスフェクション法で等しく十分に機能すると予測される。
【0028】
上記細胞は、トランスフェクション後最大72時間までのいずれかのときに、上記トランスフェクトされた宿主細胞に添加され得る。例えば、上記細胞は、トランスフェクション直後、トランスフェクションの5分後、10分後、20分後、30分後、1時間後、2時間後、3時間後、6時間後、12時間後、24時間後、36時間後、48時間後、60時間後、もしくは72時間後に、添加され得る。一般に、トランスフェクション後に細胞が添加される最大時間は、上記トランスフェクトされた細胞が生存し得る最大時間と適合する。
【0029】
期間「トランスフェクション後」とは、いったん上記DNAが上記培養物内で宿主細胞に添加されたら始まる。上記DNAが導入された時点は、種々の因子(例えば、使用される細胞株、トランスフェクション法、もしくはトランスフェクションが行われる温度)とともに変動する。上記時間は、標準的な方法を使用して、当業者によって容易に決定され得る。例えば、いくつかの実験を並行して行い得、そこで上記細胞は、トランスフェクションが異なる時点で停止されることを除いて同一な条件下で、逆遺伝学構築物でトランスフェクトされる。上記DNAが上記宿主細胞に導入された場合、少なくとも1個のウイルスが、形成されたはずである。従って、当業者は、例えば、標準的なアッセイを使用してプラーク形成単位の数を決定することによって、ウイルスの存在についてアッセイし得る。少なくとも1プラーク形成単位(PFU)が検出可能である後の最初の時点は、上記宿主細胞へと上記DNAが導入された時点である。
【0030】
あるいは、細胞が上記トランスフェクトされた細胞に添加された後の期間は、上記トランスフェクションの始まりから決定され得る。トランスフェクションの開始は、上記ウイルス分子をコードする発現構築物が上記宿主細胞の培養物と接触させられた時点である。従って、上記細胞は、トランスフェクション開始の最大96時間までのいずれかのとき(例えば、12時間、24時間、36時間、48時間、60時間、72時間、84時間もしくは96時間)に、上記トランスフェクトされた細胞に添加され得る。「トランスフェクション後」の時点および「トランスフェクションの開始から」の時点は、異なる出発点に対して測定される一方で、上記細胞が上記感染した細胞に添加される絶対時間は、同じである。例えば、上記DNAが培養物中で上記宿主細胞に導入されるのに1時間かかり、上記細胞が、上記DNAが導入された30分後に添加された場合、上記細胞は、トランスフェクションの開始から90分もしくはトランスフェクションの30分後で添加されている。
【0031】
トランスフェクション後に添加される細胞の数は、一般に、トランスフェクションのための宿主細胞として使用される細胞の数、および同様にトランスフェクションのために使用される上記細胞培養容器のサイズに依存して変動する。本発明の方法において使用され得る細胞の数は、当業者によって容易に決定され得る。例えば、本発明の方法は、添加される細胞の数が異なることを除いて、いくつかの並行した実験において同一の条件下で行われ得る。最高のウイルス収量を与える細胞の数は、さらなる実験において使用され得る。例えば、約6×105細胞が、LipofectamineTMでのトランスフェクションのために出発培養物として使用される場合、約4×105細胞が添加され得る。
【0032】
トランスフェクション後に添加される上記細胞は、上記レスキューされるウイルスが複製するための基質を提供する。従って、上記細胞は、感染していない細胞であってもよく、これは、上記細胞が本発明の方法において使用される前に、上記特定の細胞が特定の時間(例えば、48時間)内でウイルスに感染もせず、ウイルスRNAをコードする発現構築物でトランスフェクトもされなかったことを意味する。細胞を不死化させるためにウイルス(例えば、エプスタイン−バー・ウイルス)に感染した細胞は、本発明における使用に適している。同様に、トランスフェクトされた細胞は、本発明の方法において使用され得るが、ただし、トランスフェクションは、上記細胞が本発明の方法において使用される直前に(すなわち、48時間以内に)は行われない。
【0033】
上記レスキューされたウイルスが、感染性のためにプロテアーゼの存在を必要とする場合、トランスフェクション後に上記細胞と一緒に上記プロテアーゼを添加することは有利である。例えば、上記インフルエンザウイルスは、感染のためにセリンプロテアーゼ(トリプシンのような)の活性を必要とする。従って、本発明の方法が、インフルエンザウイルスをレスキューするために使用される場合、上記セリンプロテアーゼは、トランスフェクション後に上記細胞と同時に添加され得る。あるいは、上記プロテアーゼは、上記細胞が上記トランスフェクトされた細胞に添加される前もしくは添加された後に、添加され得る。
【0034】
(細胞)
本発明は、上記目的のウイルスを生成し得る任意の真核生物細胞で実施され得る。本発明は、代表的には、細胞株を使用するが、例えば、初代細胞は、代替手段として使用され得る。上記細胞は、代表的には、哺乳動物のものである。適切な哺乳動物細胞としては、ハムスター、ウシ、霊長類(ヒトおよびサルを含む)およびイヌの細胞が挙げられるが、これらに限定されない。種々の細胞タイプが使用され得る(例えば、腎臓細胞、線維芽細胞、網膜細胞、肺細胞など)。適切なハムスター細胞の例は、名称BHK21もしくはHKCCを有する細胞株である。適切なサル細胞は、例えば、アフリカミドリザル細胞(例えば、Vero細胞株のような腎臓細胞である[8〜10]。適切なイヌ細胞は、例えば、CLDKおよびMDCK細胞株のような腎臓細胞である。
【0035】
さらなる適切な細胞としては、CHO;293T;BHK;MRC 5;PER.C6[11];FRhL2;WI−38などが挙げられるが、これらに限定されない。適切な細胞は、広く入手可能である(例えば、アメリカンタイプセルカルチャー(ATCC)コレクションから[12]、Coriell Cell Repositories[13]から、もしくはEuropean Collection of Cell Cultures(ECACC)から)。例えば、上記ATCCは、カタログ番号CCL 81、CCL 81.2、CRL 1586およびCRL−1587の下で種々の異なるVero細胞を供給し、カタログ番号CCL 34の下でMDCK細胞を供給する。PER.C6は、寄託番号96022940の下でECACCから入手可能である。MDCK細胞、Vero細胞およびPER.C6細胞は、ウイルスの生成のために一般に使用され、従って、これら細胞株の各々は、本発明の方法とともに使用するのに特に適している。
【0036】
本発明において使用するための好ましい細胞(特にインフルエンザウイルスを増殖させるため)は、メイディン・ダービー・イヌ腎臓に由来するMDCK細胞[14〜16]である。元々のMDCK細胞は、CCL 34としてATCCから入手可能である。これら細胞の派生物もしくは他のMDCK細胞の派生物が使用されることは、好ましい。MDCK細胞の派生物は、例えば、懸濁培養における増殖に適合されたMDCK細胞(DSM ACC 2219として寄託された「MDCK 33016」もしくは「33016−PF」;参考文献14もまた参照のこと)を開示する参考文献14において記載された。さらに、参考文献17は、無血清培養において懸濁物中で増殖するMDCK由来細胞(FERM BP−7449として寄託された「B−702」)を開示する。上記使用されるMDCK細胞株は、腫瘍形成性であり得る。非腫瘍形成性MDCK細胞を使用することもまた、想定される。例えば、参考文献18は、非腫瘍形成性MDCK細胞(「MDCK−S」(ATCC PTA−6500)、「MDCK−SF101」(ATCC PTA−6501)、「MDCK−SF102」(ATCC PTA−6502)および「MDCK−SF103」(ATCC PTA−6503)が挙げられる)を開示する。参考文献19は、感染に対して高い感受性を有するMDCK細胞(「MDCK.5F1」細胞(ATCC CRL 12042)が挙げられる)を開示する。
【0037】
上記トランスフェクションのために使用される細胞および上記トランスフェクション後に添加される細胞は、同じ細胞タイプもしくは異なる細胞タイプのものであり得る。例えば、上記発現構築物は、MDCK細胞へとトランスフェクトされ得、上記添加される細胞は、Vero細胞であり得、その逆もまた同様である。別の例は、上記発現構築物がMDCK細胞の1つの株へトランスフェクトされ、上記添加される細胞は異なるMDCK株のものである方法であり得る。このアプローチは、上記レスキューされるウイルスが、容易にトランスフェクトされ得ない細胞株において最良に増殖する場合に有利である。この局面において、上記ウイルスは、より容易にトランスフェクションすることが可能な細胞へトランスフェクトされ得るが、ウイルス複製によりよく適した細胞株においてその後増殖させられ得る。しかし、一般に、トランスフェクションおよびその後の細胞の添加の両方のために宿主細胞と同じ細胞株(例えば、MDCK 33016細胞)を使用することは好ましい。なぜなら、これは、例えば、競合する培養選択圧もしくは異なる細胞培養条件が、避けられ得るという利点を有するからである。トランスフェクションおよび/もしくはトランスフェクション後の添加のための宿主細胞として1種より多い細胞タイプの混合物を使用することもまた、可能である。
【0038】
好ましくは、上記細胞は、夾雑物の共通する供給源を回避するために、血清の非存在下で培養される。真核生物細胞培養物のための種々の無血清培地は、当業者に公知である(例えば、Iscove培地、ウルトラCHO培地(BioWhittaker)、EX−CELL(JRH Biosciences))。さらに、無タンパク質培地が使用され得る(例えば、PF−CHO(JRH Biosciences))。さもなければ、上記複製のための細胞はまた、習慣的な血清含有培地(例えば、0.5%〜10%のウシ胎仔血清を含む、MEM培地もしくはDMEM培地)中で培養され得る。
【0039】
上記宿主細胞および上記添加され得る細胞は、接着培養物中もしくは懸濁培養物中に存在し得る。例えば、上記トランスフェクションのために使用される細胞は、接着性の細胞であり得、上記トランスフェクション後に添加される細胞もまた、接着性の細胞であり得る。上記トランスフェクション後に添加される細胞が接着性の細胞である場合、上記細胞は、上記細胞が上記トランスフェクトされた細胞に添加される前にそれらが増殖させられる培養容器から除去される必要があることは明らかである。このことは、例えば、トリプシンのようなセリンプロテアーゼの助けを借りて達成され得る。トランスフェクションのために宿主細胞として懸濁細胞を、そしてトランスフェクション後の添加のために接着性細胞を使用することも可能であり、その逆もまた同様である。
【0040】
本発明の方法は、通常、使用される具体的なトランスフェクション法のために、当該分野で一般に使用されている温度で実施される。例えば、トランスフェクションのためにリポソームが使用される場合、上記トランスフェクション反応は、最初に、上記DNA/リポソーム複合体が最初に添加される(例えば、約30分間にわたって)場合に室温でインキュベートされ、続いて、約37℃で特定された期間(例えば、約24時間)にわたってインキュベートされる。本発明の方法は、当該分野で公知の任意のトランスフェクション法で実施され得、従って、トランスフェクションが行われるはずである温度は、当業者に公知である。あるいは、トランスフェクションの間の上記インキュベーション温度が変動することを除いて、同一な条件下で並行していくつかのトランスフェクション実験を行うことによって、適切なインキュベーション温度を見いだすこともまた、可能である。レスキューされるウイルスの数は、トランスフェクションの効率を示し、上記のように、生じるPFUの数に関してアッセイすることによって決定され得る。最大数のPFUを生じる温度は、トランスフェクションのために使用され得る。
【0041】
上記細胞が上記トランスフェクトされた細胞に添加される温度および得られた細胞の混合物がインキュベートされる温度は、トランスフェクションの間に使用される温度と比較して、同じであってもよいし、異なっていてもよい。上記温度は、上記トランスフェクション後の添加のために使用される細胞株に依存し得、本発明の方法を使用してレスキューされる上記ウイルスに伴っても変動し得る。個々の細胞株を増殖させるか、もしくは特定のウイルスをレスキューするための適切な温度は、当該分野で公知であるが、それ故、当業者は、適切な温度を容易に同定し得る。例えば、Vero細胞株、Per.C6細胞株およびMDCK細胞株のような哺乳動物細胞株は、通常、36℃〜38℃の間の温度、もしくは約37℃で増殖させられる。この温度はまた、たとえいくつかのウイルス(インフルエンザのような)が、約33℃の温度でレスキューされ得るとしても、多くのウイルスのレスキューのために選択される。なぜなら、これは、得られるウイルスのよりよい抗原性を生じ得るからである[20]。適切な温度はまた、上記トランスフェクション後のインキュベーション温度が変動することを除いて、同一条件下で並行していくつかのトランスフェクション実験を行うことによって同定され得る。最大数のPFUを生じる温度は、トランスフェクション後に使用され得る。PFUについてアッセイするために適した方法は、トランスフェクションの間に使用され得る温度を同定するためのアッセイに関して既に上記に記載されている。
【0042】
(ウイルス調製)
さらなる局面において、本発明は、ワクチン製造のためにウイルスを調製する方法を提供し、上記方法は、(i)宿主細胞の培養物を、ウイルスRNA分子をコードする少なくとも1個の発現構築物でトランスフェクトする工程;(ii)上記(i)のトランスフェクトされた宿主細胞に細胞を添加して、細胞の混合物を提供する工程;(iii)ウイルスを生成するために、上記細胞の混合物を培養する工程;および(iv)上記工程(iii)で得られたウイルスを精製する工程、ならびに必要に応じて、(v)上記ウイルスをワクチンへと処方する工程を包含する。
【0043】
細胞が、本発明のこの局面において培養宿主として使用される場合、細胞培養条件(例えば、温度、細胞密度、pH値など)が、使用される上記細胞株および上記ウイルスを仮定した広い範囲にわたって変動性であり、本願の要件に適合され得ることは公知である。従って、以下の情報は、ガイドラインを表すに過ぎない。
【0044】
上記のように、細胞は、好ましくは、無血清培地もしくは無タンパク質培地中で培養される。
【0045】
上記細胞の増殖は、当業者に公知の方法に従って行われ得る。例えば、上記細胞は、遠心分離もしくは濾過のような通常の補助的方法を使用する灌流システムにおいて培養され得る。さらに、上記細胞は、感染前に、流加システム(fed−batch system)で、本発明に従って増殖させられ得る。本発明の状況において、培養システムは、上記細胞が最初にバッチシステムで培養され、培地中の栄養分(もしくは栄養分の一部)の枯渇が、濃縮された栄養分の制御された供給によって補償される流加システムに関して言及される。感染前の細胞の増殖の間に上記培地のpH値を、pH6.6〜pH7.8の値に、および特に、pH7.2〜pH7.3の間の値に調節することは、有利であり得る。細胞の培養は、好ましくは、30℃〜40℃の間の温度で行われる。工程(iii)において、上記細胞は、好ましくは、30℃〜36℃の間の温度もしくは32℃〜34℃の間の温度、または約33℃で培養される。これは、本発明の方法がインフルエンザウイルスを生成するために使用される場合に特に好ましい。なぜなら、この温度範囲での感染細胞のインキュベーションが、ワクチンへと処方される場合に改善された効率を生じるウイルスの生成を生じることが示されたからである[20]。
【0046】
酸素分圧は、感染前の培養の間に、好ましくは、25%〜95%の間の値において、および特には、35%〜60%の間の値において調節され得る。本発明の状況において示される酸素分圧の値は、空気の飽和に基づく。細胞の感染は、バッチシステムにおいては、好ましくは、約8〜25×105細胞/mLの細胞密度で、もしくは灌流システムにおいては、好ましくは、約5〜20×106細胞/mLの細胞密度で行われる。上記細胞は、10−8〜10の間、好ましくは、0.0001〜0.5の間のウイルス用量(MOI値(「感染多重度」;感染時の細胞あたりのウイルス単位の数に対応する)で感染させられ得る。
【0047】
ウイルスは、接着培養もしくは懸濁物中で、細胞において増殖させられ得る。マイクロキャリア培養が使用され得る。マイクロキャリア培養は、接着培養とみなされる。上記細胞はまた、懸濁物中での増殖に適合され得る。
【0048】
本発明に従う方法はまた、ウイルスの採取および単離、またはウイルスによって生成されるタンパク質の採取および単離を含む。ウイルスもしくはタンパク質の単離の間に、上記細胞は、分離、濾過もしくは限外濾過のような標準的な方法によって培養培地から分離される。次いで、上記ウイルスもしくは上記タンパク質は、勾配遠心分離、濾過、沈殿、クロマトグラフィーなどの当業者に十分公知の方法に従って濃縮され、次いで、精製される。また、上記ウイルスが精製の間もしくは精製後に不活性化されることは、本発明によれば好ましい。ウイルス不活性化は、上記精製プロセス内のいずれかの時点で、例えば、β−プロピオラクトンもしくはホルムアルデヒドによって行われ得る。
【0049】
本発明に従って単離される上記ウイルスはまた、工程(iii)において卵において増殖させられ得る。ワクチンのためのインフルエンザウイルス増殖の現在標準的な方法は、胚含有SPF鶏卵を使用し、ウイルスは、卵内容物(尿膜腔液)から精製される。卵を介してウイルスを継代し、その後、細胞培養においてウイルスを増殖させることもまた、可能である。
【0050】
(ウイルス)
本発明の方法は、細胞において逆遺伝学によって発現され得る任意のウイルスで実施され得る。このようなウイルスは、セグメント化ウイルスもしくは非セグメント化ウイルスであり得る。さらに、上記ウイルスは、プラス鎖RNAウイルスもしくはマイナス鎖RNAウイルスであり得る。上記ウイルスはまた、二本鎖RNAウイルスであり得る。
【0051】
上記ウイルスがマイナス鎖RNAウイルスである場合、上記ウイルスは、パラミクソウイルス科、ニューモウイルス亜科(Pneumovirinae)、ラブドウイルス科、フィロウイルス科、ボルナウイルス科(Bornaviridae)、オルソミクソウイルス科、ブンヤウイルス科、もしくはアレナウイルス科からなる群より選択される科に由来し得る。さらに、上記ウイルスは、パラミクソウイルス、オルソミクソウイルス、レスピロウイルス、モルビリウイルス、ルブラウイルス、ヘニパウイルス、アブラウイルス、ニューモウイルス、メタニューモウイルス、ベシクロウイルス、リッサウイルス、エフェメロウイルス、サイトラブドウイルス、ヌクレオラブドウイルス、ノビラブドウイルス、マールブルグウイルス、エボラウイルス、ボルナウイルス、インフルエンザウイルスA、インフルエンザウイルスB、インフルエンザウイルスC、トゴトウイルス、イサウイルス、オルソブンヤウイルス、ハンタウイルス、ナイロウイルス、フレボウイルス、トスポウイルス、アレナウイルス、オフィオウイルス、テヌイウイルス、もしくはデルタウイルスからなる群より選択される属に由来するウイルスであり得る。具体的実施形態において、上記マイナス鎖RNAウイルスは、センダイウイルス、麻疹ウイルス、ムンプスウイルス、ヘンドラウイルス、ニューキャッスル病ウイルス、ヒトRSウイルス、トリニューモウイルス、水疱性口内炎インディアナウイルス(Vesicular stomatitis Indiana virus)、狂犬病ウイルス、ウシ流行熱ウイルス、レタス壊死性黄変病ウイルス(Lettuce necrotic yellows virus)、ジャガイモ黄萎病ウイルス(Potato yellow dwarf virus)、伝染性造血器壊死症ウイルス(Infectious hematopoietic necrosis virus)、ビクトリア湖マールブルグウイルス、ザイールエボラウイルス、ボルナ病ウイルス、インフルエンザウイルス、トゴトウイルス、伝染性サケ貧血ウイルス(Infectious salmon anemia virus)、ブニヤムウェラーウイルス(Bunyamwera virus)、ハンタンウイルス、ジュグベウイルス(Dugbe virus)、リフトバレー熱ウイルス、トマト黄化え疽ウイルス(Tomato spotted wilt virus)、リンパ球性脈絡髄膜炎ウイルス(Lymphocytic choriomeningitis virus)、カンキツソローシスウイルス(Citrus psorosis virus)、イネ縞葉枯ウイルス(Rice stripe virus)、およびデルタ型肝炎ウイルスからなる群より選択される。好ましい実施形態において、上記ウイルスは、インフルエンザウイルスである(以下を参照のこと)。
【0052】
上記ウイルスがプラス鎖RNAウイルスである場合、上記ウイルスは、アルテリウイルス科、コロナウイルス科、ピコルナウイルス科およびロニウイルス科からなる群より選択される科に由来し得る。さらに、上記ウイルスは、アルテリウイルス、コロナウイルス、エンテロウイルス、トロウイルス、オカウイルス(Okavirus)、ライノウイルス、ヘパトウイルス、カルジオウイルス、アフトウイルス、パレコウイルス、エルボウイルス、コブウイルスおよびテッショウウイルスからなる群より選択される属に由来するウイルスであり得る。具体的実施形態において、上記ウイルスは、重症急性呼吸器症候群(SARS)ウイルス、ポリオウイルス、ヒトエンテロウイルスA(HEV−A)、ヒトエンテロウイルスB(HEV−B)、ヒトエンテロウイルスC、ヒトエンテロウイルスD、A型肝炎ウイルス、ならびにヒトライノウイルスAおよびBからなる群より選択される。
【0053】
上記ウイルスが二本鎖RNAウイルスである場合、上記ウイルスは、ビルナウイルス科、シストウイルス科、ハイポウイルス科、パルティティウイルス科、レオウイルス科およびトティウイルス科からなる群より選択される科に由来し得る。さらに、上記ウイルスは、アクアビルナウイルス、トリビルナウイルス、エントモビルナウイルス、シストウイルス、パルティティウイルス、アルファクリプトウイルス、ベータクリプトウイルス、アクアレオウイルス、コルティウイルス、サポウイルス、フィジウイルス、イドノレオウイルス(Idnoreovirus)、マイコレオウイルス、オルビウイルス、オルトレオウイルス、オリザウイルス、フィトレオウイルス、ロタウイルスおよびセドルナウイルス(Seadornavirus)からなる群より選択される属に由来するウイルスであり得る。
【0054】
本発明とともに使用するのに好ましいウイルスは、ロタウイルスである。これらウイルスでの逆遺伝学は、上記ウイルスレスキューの不十分な効率に起因して現在は困難であり、従って、本発明の方法は、このウイルスのレスキューを容易にし得る。
【0055】
本発明はまた、迅速な変異を受け、組換えアプローチがウイルスのより迅速な単離を可能にし、次いで、適切なワクチンを得るためにさらに増殖させられ得るウイルスに特に適している。従って、別の好ましい実施形態において、上記ウイルスはインフルエンザである。
【0056】
(インフルエンザウイルス)
インフルエンザウイルスは、本発明の方法における使用に特に適している(特に、インフルエンザAウイルスおよびインフルエンザBウイルス)。なぜなら、このウイルスについての逆遺伝学は、既に十分に特徴付けられているからである。インフルエンザウイルスは、セグメント化されたマイナス鎖RNAウイルスである。インフルエンザAウイルスおよびインフルエンザBウイルスは、8個のセグメントを有するのに対して、インフルエンザCウイルスは、7個のセグメントを有する。上記ウイルスは、複製および転写を開始するために、少なくとも4個のウイルスタンパク質(PB1、PB2、PAおよびヌクレオプロテイン)を必要とする。
【0057】
インフルエンザAウイルスおよびインフルエンザBウイルスについての逆遺伝学は、上記4個の必要とされるタンパク質および8個すべてのゲノムセグメントを発現するために、12個のプラスミドで実施され得る。しかし、構築物の数を減らすために、複数のRNAポリメラーゼI転写カセット(ウイルスRNA合成のため)が、単一のプラスミドに含まれ得(例えば、1個、2個、3個、4個、5個、6個、7個もしくは全8個のインフルエンザvRNAセグメントをコードする配列)、RNAポリメラーゼIIプロモーターを有する複数のタンパク質コード領域(例えば、1個、2個、3個、4個、5個、6個、7個もしくは8個のインフルエンザmRNA転写物をコードする配列)が別のプラスミドに含まれ得る[21]。1個以上のインフルエンザvRNAセグメントをpol Iプロモーターの制御下で、および同じプラスミド上に1個以上のインフルエンザタンパク質コード領域を別のプロモーター(特に、pol IIプロモーター)の制御下で含むこともまた、可能である。上記のように、これは、好ましくは、二方向性プラスミドを使用して行われる。参考文献21の方法の好ましい局面は、(a)単一のプラスミド上のPB1、PB2およびPAのmRNAコード領域;および(b)単一のプラスミド上の全8個のvRNAコードセグメントを含む。一方のプラスミド上にノイラミニダーゼ(NA)およびヘマグルチニン(HA)のセグメント、およびもう一方のプラスミド上に6個のほかのセグメントを含むことは、特に好ましい。なぜなら、新たに出現するインフルエンザウイルス株は、通常、上記NAセグメントおよび/もしくはHAセグメントに変異を有するからである。従って、この実施形態において、上記HA配列およびNA配列を含むベクターのみが、置換される必要がある。
【0058】
インフルエンザAウイルスの好ましい発現系は、複数の異なる野生型株に由来するゲノムセグメントをコードする。上記系は、PR/8/34株(A/Puerto Rico/8/34)に由来する1個以上の(例えば、1個、2個、3個、4個、5個もしくは6個の)ゲノムセグメントをコードし得るが、通常、これ/これらは、上記PR/8/34 HAセグメントを含まず、通常、上記PR/8/34 NAセグメントを含まない。よって、上記系は、PR/8/34に由来するセグメントNP、M、NS、PA、PB1および/もしくはPB2(おそらく6個すべて)のうちの少なくとも1個をコードし得る。
【0059】
インフルエンザAウイルスの他の有用な発現系は、AA/6/60 インフルエンザウイルス(A/Ann Arbor/6/60)に由来する1個以上の(例えば、1個、2個、3個、4個、5個もしくは6個の)ゲノムセグメントをコードし得るが、通常は、これ/これらは、上記AA/6/60 HAセグメントを含まず、通常、上記AA/6/60 NAセグメントを含まない。よって上記系は、AA/6/60に由来するセグメントNP、M、NS、PA、PB1および/もしくはPB2(おそらく6個すべて)のうちの少なくとも1個をコードし得る。
【0060】
上記系は、A/California/4/09株に由来する1個以上のゲノムセグメント(例えば、上記HAセグメントおよび/もしくは上記NAセグメント)をコードし得る。従って、例えば、上記HA遺伝子セグメントは、配列番号2より配列番号1に密接に関連する(すなわち、同じアルゴリズムおよびパラメーターを使用して、配列番号1と比較した場合、配列番号2より高い程度の配列同一性を有する)H1ヘマグルチニンをコードし得る。配列番号1および配列番号2は、80%同一である。同様に、上記NA遺伝子は、配列番号4より配列番号3に密接に関連しているN1ノイラミニダーゼをコードし得る。配列番号3および4は、82%同一である。
【0061】
インフルエンザBウイルスの発現系は、複数の異なる野生型株に由来するゲノムセグメントをコードし得る。上記系は、AA/1/66 インフルエンザウイルス(B/Ann Arbor/1/66)に由来する1個以上の(例えば、1個、2個、3個、4個、5個もしくは6個の)ゲノムセグメントをコードし得るが、通常は、これ/これらは、上記AA/1/66 HAセグメントを含まず、通常は、上記AA/1/66 NAセグメントを含まない。従って、上記系は、AA/1/66に由来するセグメントNP、M、NS、PA、PB1および/もしくはPB2のうちの少なくとも1個をコードし得る。
【0062】
A/PR/8/34株、AA/6/60株、AA/1/66株、A/Chile/1/83株およびA/California/04/09株に由来するウイルスセグメントおよび配列は、広く利用可能である。それらの配列は、公のデータベースで利用可能である(例えば、GI:89779337、GI:89779334、GI:89779332、GI:89779320, GI:89779327、GI:89779325、GI:89779322、GI:89779329)。
【0063】
インフルエンザウイルスの逆遺伝学システムは、上記宿主細胞においてアクセサリタンパク質の発現をもたらす発現構築物を含み得る。例えば、非ウイルス性セリンプロテアーゼ(例えば、トリプシン)を発現することは有利であり得る。
【0064】
(ワクチン)
一局面において、本発明は、本発明の方法に従って生成されるウイルスを利用して、ワクチンを生成する。
【0065】
ワクチン(特に、インフルエンザウイルスのための)は、一般に、生のウイルスもしくは不活性化ウイルスのいずれかに基づく。不活性化ワクチンは、完全ビリオン、「スプリット」ビリオン、もしくは精製表面抗原に基づき得る。抗原はまた、ビロソームの形態で提示され得る。本発明は、これらタイプのワクチンのうちのいずれかを製造するために使用され得る。
【0066】
不活性化ウイルスが使用される場合、上記ワクチンは、完全ビリオン、スプリットビリオン、もしくは精製表面抗原(インフルエンザについては、ヘマグルチニンを含み、通常はまた、ノイラミニダーゼを含む)を含み得る。ウイルスを不活性化するための化学的手段としては、以下の薬剤のうちの1つ以上の有効量での処置が挙げられる:洗剤、ホルムアルデヒド、β−プロピオラクトン、メチレンブルー、ソラレン、カルボキシフラーレン(C60)、バイナリーエチルアミン、アセチルエチレンイミン、もしくはこれらの組み合わせ。ウイルス不活性化の非化学的方法は、当該分野で公知である(例えば、UV光もしくはγ線照射)。
【0067】
ビリオンは、ウイルス含有流体(例えば、尿膜腔液もしくは細胞培養上清)から、種々の方法によって採取され得る。例えば、精製プロセスは、上記ビリオンを破壊するために洗剤を含む線形スクロース勾配溶液を使用するゾーン遠心分離を含み得る。次いで、抗原は、ダイアフィルトレーションによって、必要に応じた希釈の後に精製され得る。
【0068】
スプリットビリオンは、精製されたビリオンを洗剤(例えば、エチルエーテル、ポリソルベート 80、デオキシコレート、トリ−N−ブチルホスフェート、Triton X−100、Triton N101、セチルトリメチルアンモニウムブロミド、Tergitol NP9など)で処理して、サブビリオン調製物を生成する(「Tween−エーテル」スプリットプロセスを含む)ことによって得られる。インフルエンザウイルスをスプリットするための方法は、例えば、当該分野で周知である(例えば、参考文献22〜27などを参照のこと)。上記ウイルスのスプリットは、代表的には、感染性であろうと非感染性であろうと、破壊濃度のスプリット薬剤で完全ウイルスを破壊するかもしくはフラグメント化することによって行われる。上記破壊は、上記ウイルスタンパク質の完全なもしくは部分的な可溶化を生じ、上記ウイルスの完全性を変化させる。好ましいスプリット薬剤は、非イオン性界面活性剤およびイオン性(例えば、カチオン性)界面活性剤(例えば、アルキルグリコシド、アルキルチオグリコシド、アシル糖、スルホベタイン、ベタイン、ポリオキシエチレンアルキルエーテル、N,N−ジアルキル−グルカミド、Hecameg、アルキルフェノキシ−ポリエトキシエタノール、NP9、4級アンモニウム化合物、サルコシル、CTAB(セチルトリメチルアンモニウムブロミド)、トリ−N−ブチルホスフェート、Cetavlon、ミリスチルトリメチルアンモニウム塩、リポフェクチン、リポフェクタミン、およびDOT−MA)、オクチル−もしくはノニルフェノキシポリオキシエタノール(例えば、Triton界面活性剤(例えば、Triton X−100もしくはTriton N101))、ポリオキシエチレンソルビタンエステル(Tween界面活性剤)、ポリオキシエチレンエーテル、ポリオキシエチレンエステル(polyoxyethlene ester)などである。1つの有用なスプリット手順は、デオキシコール酸ナトリウムおよびホルムアルデヒドの連続的効果を使用し、スプリットは、最初のビリオン精製の間に起こり得る(例えば、スクロース密度勾配溶液において)。よって、スプリットプロセスは、上記ビリオン含有材料の清澄化(非ビリオン材料を除去するために)、上記採取したビリオンの濃縮(例えば、吸着法(例えば、CaHPO4吸着)を使用して)、非ビリオン材料からの完全ビリオンの分離、密度勾配遠心分離工程においてスプリット薬剤を使用する(例えば、デオキシコール酸ナトリウムのようなスプリット薬剤を含むスクロース勾配を使用する)ビリオンのスプリット、次いで、望ましくない物質を除去するための濾過(例えば、限外濾過)を含み得る。スプリットビリオンは、有用には、リン酸ナトリウム緩衝化等張性塩化ナトリウム溶液中で再懸濁され得る。スプリットインフルエンザワクチンの例は、BEGRIVACTM、FLUARIXTM、FLUZONETMおよびFLUSHIELDTM製品である。
【0069】
本発明の方法はまた、生ワクチンを生成するために使用され得る。このようなワクチンは、通常は、ビリオン含有流体からビリオンを精製することによって調製される。例えば、上記流体は、遠心分離によって清澄にされ得、緩衝液(例えば、スクロース、リン酸カリウム、およびグルタミン酸一ナトリウムを含む)で安定化され得る。インフルエンザウイルスワクチンの種々の形態が、現在利用可能である(例えば、参考文献28の第17章および第18章を参照のこと)。生のウイルスは、MedImmuneのFLUMISTTM製品(三価の生のウイルス)を含む。
【0070】
精製されたインフルエンザウイルス表面抗原ワクチンは、表面抗原であるヘマグルチニン、および代表的には、同様にノイラミニダーゼを含む。精製形態でこれらタンパク質を調製するためのプロセスは、当該分野で周知である。上記FLUVIRINTM、AGRIPPALTMおよびINFLUVACTM製品は、インフルエンザサブユニットワクチンである。
【0071】
別の形態の不活性化抗原は、ビロソーム[29](核酸を含まないウイルス様リポソーム粒子)である。ビロソームは、洗剤でのウイルスの可溶化、続いて、ヌクレオキャプシドの除去およびウイルス糖タンパク質を含む膜の再構成によって、調製され得る。ビロソームを調製するための代替法は、ウイルス膜糖タンパク質を過剰量のリン脂質に添加して、それらの膜中においてリポソームにウイルスタンパク質を与えることを含む。
【0072】
上記ウイルスは、弱毒化され得る。上記ウイルスは、温度感受性であり得る。上記ウイルスは、低温適合(cold−adapted)され得る。これら3つの特徴は、生のウイルスを抗原として使用する場合に特に有用である。
【0073】
HAは、現在の不活性化インフルエンザワクチンにおける主要な免疫原であり、ワクチン用量は、HAレベルを参照することによって標準化されている(代表的には、SRIDによって測定される)。既存のワクチンは、代表的には、1株あたり約15μgのHAを含むが、より低用量が、使用され得る(例えば、小児に関して、もしくは汎流行的状況において、またはアジュバントを使用する場合)。分割用量(例えば、1/2(すなわち、7.5μg HA/株)、1/4および1/8)が使用されてきたことと同様に、より高い用量も使用されてきた(例えば、3×もしくは9×用量[30、31])。従って、ワクチンは、0.1〜150μgの間のHA/インフルエンザ株、好ましくは、0.1〜50μgの間(例えば、0.1〜20μg、0.1〜15μg、0.1〜10μg、0.1〜7.5μg、0.5〜5μgなど)を含み得る。特定の用量としては、1株あたり、例えば、約45、約30、約15、約10、約7.5、約5、約3.8、約3.75、約1.9、約1.5などが挙げられる。
【0074】
生ワクチンに関して、投与は、HA含有量よりむしろ、50%組織培養感染量(median tissue culture infectious dose)(TCID50)によって測定され、1株あたり106〜108の間の(好ましくは、106.5〜107.5の間の)TCID50が、代表的である。
【0075】
本発明とともに使用されるインフルエンザ株は、野生型ウイルスにおいて見いだされるような天然のHA、もしくは改変されたHAを有し得る。例えば、HAを改変して、トリ種においてウイルスの病原性を高くする決定因子(例えば、上記HA1/HA2切断部位の周りの超塩基性領域)を除去することは、公知である。逆遺伝学の使用は、このような改変を促進する。
【0076】
ワクチンにおける使用のためのインフルエンザウイルス株は、流行期ごとに変化する。汎流行期間の間には、ワクチンは、代表的には、2つのインフルエンザA株(H1N1およびH3N2)および1つのインフルエンザB株を含み、三価ワクチンが代表的である。本発明はまた、汎流行性ウイルス株(すなわち、上記ワクチンレシピエントおよび一般的なヒト集団が免疫学的に経験したことがない株(特にインフルエンザAウイルスの)(例えば、H2、H5、H7もしくはH9サブタイプ株))を使用し得、汎流行性株のインフルエンザワクチンは、1価であってもよいし、汎流行性株が補充された通常の三価ワクチンに基づいていてもよい。しかし、流行期および上記ワクチン中に含まれる抗原の性質に依存して、本発明は、HAサブタイプであるH1、H2、H3、H4、H5、H6、H7、H8、H9、H10、H11、H12、H13、H14、H15もしくはH16のうちの1つ以上に対して防御し得る。本発明は、インフルエンザAウイルス NAサブタイプN1、N2、N3、N4、N5、N6、N7、N8もしくはN9のうちの1つ以上に対して防御し得る。
【0077】
汎流行期間の株に対して免疫化するために適しているのに加えて、本発明の組成物は、汎流行性もしくは潜在的に汎流行性の株に対して免疫化するために特に有用である。汎流行のアウトブレイクを引き起こす可能性を与えるインフルエンザ株の特徴は、以下である:(a)現在広まっているヒト株のヘマグルチニンと比較して、新たなヘマグルチニンを含む(すなわち、10年間を超えてヒト集団において顕性でなかったもの(例えば、H2)、または上記ヒト集団において以前に全く認められなかったもの(例えば、一般に、トリ集団においてのみ見いだされてきたH5、H6もしくはH9)。その結果、上記ヒト集団は、上記株のヘマグルチニンに対して免疫学的に経験したことがない;(b)上記ヒト集団において水平伝播し得る;および(c)ヒトに対して病原性である。H5ヘマグルチニンタイプを有するウイルスは、汎流行性インフルエンザ(例えば、H5N1株)に対して免疫化するのに好ましい。他の考えられる株としては、H5N3、H9N2、H2N2、H7N1およびH7N7、ならびに任意の他の潜在的に出現する汎流行性株が挙げられる。本発明は、非ヒト動物集団からヒトへと拡がり得るかもしくは拡がってしまった潜在的汎流行性株(例えば、ブタ起源のH1N1インフルエンザ株)に対して防御するのに特に適している。次いで、本発明は、ヒトおよび非ヒト動物にワクチン接種するのに適している。
【0078】
抗原が上記組成物中に有用に含まれ得る他の株は、抗ウイルス治療に抵抗性である(例えば、オセルタミビル[32]および/もしくはザナミビルに抵抗性である)株である(耐性汎流行性株を含む[33])。
【0079】
本発明の組成物は、1つ以上の(例えば、1つ、2つ、3つ、4つもしくはこれ以上の)インフルエンザウイルス株(インフルエンザAウイルスおよび/もしくはインフルエンザBウイルスを含む)に由来する抗原を含み得る。ワクチンが1より多くのインフルエンザ株を含む場合、異なる株は、代表的には、別個に増殖させられ、上記ウイルスが採取された後に混合され、抗原が調製されてきた。従って、本発明のプロセスは、1より多くのインフルエンザ株に由来する抗原を混合する工程を包含し得る。三価ワクチンは、代表的である(2つのインフルエンザAウイルス株および1つのインフルエンザBウイルス株に由来する抗原を含む)。四価ワクチンもまた有用である[34](2つのインフルエンザAウイルス株および2つのインフルエンザBウイルス株に由来する抗原を含むか、または3つのインフルエンザAウイルス株および1つのインフルエンザBウイルス株に由来する抗原を含む)。
【0080】
(薬学的組成物)
本発明に従って製造されるワクチン組成物は、薬学的に受容可能である。それらは、通常、上記抗原に加えて、成分を含む。例えば、それらは、代表的には、1種以上の薬学的キャリアおよび/もしくは賦形剤を含む。以下に記載されるように、アジュバントもまた含まれ得る。このような成分の詳細な議論は、参考文献35で入手される。
【0081】
ワクチン組成物は、一般に、水性形態にある。しかし、いくつかのワクチンは、乾燥形態、例えば、注射可能な固体、またはパッチ上の、乾燥したかもしくは重合した調製物の形態にあり得る。
【0082】
ワクチン組成物は、チオメルサールもしくは2−フェノキシエタノールのような保存剤を含み得る。しかし、上記ワクチンは、実質的に水銀物質を実質的に含まないべきである(すなわち、5μg/ml未満)ことが好ましい(例えば、チオメルサールを含まない[26、36])。水銀を含まないワクチンは、より好ましい。α−トコフェロールスクシネートは、水銀化合物の代替として含まれ得る[26]。保存剤非含有ワクチンは、特に好ましい。
【0083】
張度を制御するために、組成物は、生理学的塩(例えば、ナトリウム塩)を含み得ることは好ましい。塩化ナトリウム(NaCl)が好ましく、塩化ナトリウムは、1〜20mg/mlの間で存在し得る。存在し得る他の塩としては、塩化カリウム、リン酸二水素カリウム、無水リン酸二ナトリウム(disodium phosphate dehydrate)、塩化マグネシウム、塩化カルシウムなどが挙げられる。
【0084】
ワクチン組成物は、一般に、200mOsm/kg〜400mOsm/kgの間(好ましくは、240〜360mOsm/kgの間)の重量オスモル濃度を有し、より好ましくは、290〜310mOsm/kgの範囲内に入る。ワクチン接種によって引き起こされる疼痛に対して影響を有さない重量オスモル濃度が以前に報告されている[37]が、この範囲において重量オスモル濃度を維持することは好ましい。
【0085】
ワクチン組成物は、1種以上の緩衝剤を含み得る。代表的な緩衝剤としては、以下が挙げられる:リン酸塩緩衝剤;Tris緩衝剤;ホウ酸塩緩衝剤;コハク酸塩緩衝剤;ヒスチジン緩衝剤(特に、水酸化アルミニウムアジュバントで);またはクエン酸塩緩衝剤。緩衝剤は、代表的には、5〜20mM範囲で含まれる。
【0086】
ワクチン組成物のpHは、一般に、一般に、5.0〜8.1の間、より代表的には、6.0〜8.0の間(例えば、6.5〜7.5の間、または7.0〜7.8の間)である。本発明のプロセスは、従って、バルクワクチンのpHを、パッケージ前に調節する工程を包含し得る。
【0087】
上記ワクチン組成物は、好ましくは、無菌である。上記ワクチン組成物は、好ましくは、非発熱性である(例えば、1用量あたり<1 EU(エンドトキシン単位、標準尺度)、好ましくは、<0.1 EU/用量を含む)。上記ワクチン組成物は、好ましくは、グルテン非含有である。
【0088】
本発明のワクチン組成物は、洗剤(例えば、ポリオキシエチレンソルビタンエステル界面活性剤(「Tweens」として公知)、オクトキシノール(例えば、オクトキシノール−9(Triton X−100)もしくはt−オクチルフェノキシポリエトキシエタノール)、セチルトリメチルアンモニウムブロミド(「CTAB」)、またはデオキシコール酸ナトリウム(特に、スプリットワクチンもしくは表面抗原ワクチンのために))を含み得る。上記洗剤は、微量においてのみ存在し得る。従って、上記ワクチンは、オクトキシノール−10およびポリソルベート80の各々について1mg/ml未満で含み得る。他の残りの微量の成分は、抗生物質(例えば、ネオマイシン、カナマイシン、ポリミキシンB)であり得る。
【0089】
ワクチン組成物は、1回の免疫化のための物質を含んでいてもよいし、複数回の免疫化のための物質を含んでいてもよい(すなわち、「複数用量」キット)。保存剤を含めることは、複数用量の準備において好ましい。複数用量組成物中に保存剤を含める代替として(もしくはそれに加えて)、上記組成物は、物質の取り出しのための無菌アダプタを有する容器中に含まれ得る。
【0090】
インフルエンザワクチンは、代表的には、約0.5mlの投与体積で投与されるが、半用量(すなわち、約0.25ml)が、小児に投与され得る。
【0091】
組成物およびキットは、好ましくは、2℃〜8℃の間で貯蔵される。それらは、凍結されるべきではない。それらは、理想的には、遮光して保持されるべきである。
【0092】
(宿主細胞DNA)
ウイルスが単離され、そして/または細胞株上で増殖させられた場合、最終ワクチン中に残っている細胞株DNAのいかなる腫瘍形成活性をも最小限にするために、上記DNAの量を最小限にすることは、標準的な実務である。
【0093】
従って、本発明に従って調製されるワクチン組成物は、好ましくは、1用量あたり10ng(好ましくは、1ng未満、より好ましくは、100pg未満)の残留宿主細胞DNAを含むが、微量の宿主細胞DNAが存在し得る。
【0094】
任意の残留宿主細胞DNAの平均長が500bp未満(例えば、400bp未満、300bp未満、200bp未満、100bp未満など)であることは、好ましい。
【0095】
夾雑するDNAは、標準的精製手順(例えば、クロマトグラフィーなど)を使用するワクチン調製の間に除去され得る。残留宿主細胞DNAの除去は、ヌクレアーゼ処理によって(例えば、DNaseを使用することによって)増強され得る。宿主細胞DNA夾雑物を減らすための便利な方法は、2工程処理(第1は、DNase(例えば、Benzonase)(これは、ウイルス増殖の間に使用され得る)を、次いで、カチオン性洗剤(例えば、CTAB)(これは、ビリオン破壊の間に使用され得る)使用する)を含む参考文献38および39において開示される。アルキル化剤(例えば、β−プロピオラクトン)での処理はまた、宿主細胞DNAを除去するために使用され得、有利なことには、ビリオンを不活性化するためにも使用され得る[40]。
【0096】
(アジュバント)
本発明の組成物は、有利には、アジュバントを含み得る。これは、上記組成物を受ける被験体において誘発される免疫応答(液性および/もしくは細胞性)を高めるように機能し得る。好ましいアジュバントは、水中油型エマルジョンを含む。種々のこのようなアジュバントは公知であり、それらは、代表的には、少なくとも1種の油および少なくとも1種の界面活性剤を含み、上記油および界面活性剤は、生体分解性(代謝可能)かつ生体適合性である。上記エマルジョン中の油滴は、一般に、直径5μm未満であり、理想的には、サブミクロンの直径を有し、これら小さなサイズは、マイクロフルイダイザー(microfluidiser)で達成されて、安定なエマルジョンを提供する。220nm未満のサイズを有する液滴は、濾過滅菌に供され得るので、好ましい。
【0097】
前記エマルジョンは、動物(例えば魚類)供給源または植物供給源からのものなどの油を含むことができる。植物油の供給源としては、堅果、種子および穀粒が挙げられる。ラッカセイ油、ダイズ油、ヤシ油およびオリーブ油が、最も一般的に利用できる堅果油の例である。例えばホホバ豆から得られる、ホホバ油を使用することができる。種子油としては、紅花油、綿実油、ヒマワリ種子油、ゴマ種子油およびこれらに類するものが挙げられる。穀粒の群の中で、トウモロコシ油が最も容易に入手できるが、他の穀物粒、例えばコムギ、オートムギ、ライムギ、イネ、テフ、ライコムギおよびこれらに類するものの油も使用することができる。グリセロールおよび1,2−プロパンジオールの6〜10炭素脂肪酸エステルは、種子油中に天然に存在しないが、堅果油および種子油から出発して適切な材料の加水分解、分離およびエステル化によって調製することができる。哺乳動物の乳からの脂肪および油は代謝性であり、従って、本発明の実施の際に使用することができる。動物供給源から純粋な油を得るために必要な分離、精製、鹸化および他の手段についての手順は、当該技術分野において周知である。殆どの魚類は、容易に回収できる代謝性の油を含有する。例えば、タラ肝油、サメ肝油、および鯨油、例えば鯨ろうが、ここで使用することができる魚油のいくつかの例である。多数の分岐鎖油が5炭素イソプレン単位で生化学的に合成されており、一般にテルペノイドと呼ばれる。サメ肝油は、スクアレン、2,6,10,15,19,23−ヘキサメチル−2,6,10,14,18,22−テトラコサヘキサン、として公知の、分岐した不飽和テルペノイドを含有し、これは、ここで特に好ましい。スクワラン、スクアレンの飽和類似体、も好ましい油である。スクアレンおよびスクワランを含む、魚油は、商業的供給源から容易に入手することができ、または当該技術分野において公知の方法によって得ることができる。別の好ましい油は、α―トコフェロール(下記参照)である。油の混合物を使用することができる。
【0098】
界面活性剤をそれらの「HLB」(親水親油バランス)によって分類することができる。本発明の好ましい界面活性剤は、少なくとも10、好ましくは少なくとも15、およびさらに好ましくは少なくとも16のHLBを有する。本発明は、ポリオキシエチレンソルビタンエステル界面活性剤(一般にTweenと呼ばれる)、特にポリソルベート20およびポリソルベート80;商品名DOWFAXTMで販売されている、エチレンオキシド(EO)、プロピレンオキシド(PO)および/またはブチレンオキシド(BO)のコポリマー、例えば、線状EO/POブロックコポリマー;反復エトキシ(オキシ−1,2−エタンジイル)基の数が様々であり得るオクトキシノール、オクトキシノール−9(Triton X−100、すなわちt−オクチルフェノキシポリエトキシエタノール)が特に興味深い;(オクチルフェノキシ)ポリエトキシエタノール(IGEPAL CA−630/NP−40);リン脂質、例えばホスファチジルコリン(レシチン);ノニルフェノールエトキシレート、例えばTergitolTMNPシリーズ;ラウリル、セチル、ステアリルおよびオレイルアルコールから誘導されたポリオキシエチレン脂肪エーテル(Brij界面活性剤として公知)、例えばトリエチレングリコールモノラウリルエーテル(Brij 30);ならびにソルビタンエステル(一般にSPANとして公知)、例えばソルビタントリオレエート(Span 85)およびソルビタンモノラウレートを含む(しかしこれらに限定されない)界面活性剤と共に用いることができる。非イオン性界面活性剤が好ましい。前記エマルジョンに含めるために好ましい界面活性剤は、Tween 80(ポリオキシエチレンソルビタンモノオレエート)、Span 85(ソルビタントリオレエート)、レシチンおよびTriton X−100である。
【0099】
界面活性剤の混合物、例えばTween 80/Span 85混合物、を使用することができる。ポリオキシエチレンソルビタンエステル、例えばポリオキシエチレンソルビタンモノオレエート(Tween 80)、およびオクトキシノール、例えばt−オクチルフェノキシポリエトキシエタノール(Triton X−100)、の組み合わせも適する。別の有用な組み合わせは、ラウレス9とポリオキシエチレンソルビタンエステルおよび/またはオクトキシノールを含む。
【0100】
界面活性剤の好ましい量(重量%)は、ポリオキシエチレンソルビタンエステル(例えば、Tween 80)0.01%から1%、特に約0.1%;オクチル−またはノニルフェノキシポリオキシエタノール(例えば、Triton X−100、またはTritonシリーズの他の洗剤)0.001%から0.1%、特に0.005%から0.02%;ポリオキシエチレンエーテル(例えば、ラウレス9)0.1%から20%、好ましくは0.1%から10%および特に0.1%から1%または約0.5%である。
【0101】
上記ワクチンがスプリットウイルスを含む場合、上記ワクチンは、水相中に遊離界面活性剤を含むことが好ましい。これは、上記遊離界面活性剤が、上記抗原に対して「スプリット効果」を発揮し得、それによって、他の状態で存在し得る任意の非スプリットビリオンおよび/もしくはビリオン凝集物を破壊するので、有利である。これは、スプリットウイルスワクチンの安全性を改善し得る[41]。
【0102】
好ましいエマルジョンは、<1μmの平均液滴サイズ(例えば、≦750nm、≦500nm、≦400nm、≦300nm、≦250nm、≦220nm、≦200nm、もしくはこれより小さい)を有し得る。これら液滴サイズは、便利なことには、微小流動化(microfluidisation)のような技術によって達成され得る。
【0103】
本発明で有用な特定の水中油型エマルジョンアジュバントとしては、以下が挙げられるが、これらに限定されない。
【0104】
・スクアレン、Tween 80、およびSpan85のサブミクロンエマルジョン。上記エマルジョンの容積での組成は、約5% スクアレン、約0.5% ポリソルベート80および約0.5% Span 85であり得る。重量では、これらの比は、4.3% スクアレン、0.5% ポリソルベート80および0.48% Span 85になる。このアジュバントは、引用文献45の第10章および引用文献46の第12章により詳細に記載されるように、「MF59」として公知である[42〜44]。上記MF59エマルジョンは、有利なことには、クエン酸イオン(例えば、10mM クエン酸ナトリウム緩衝液)を含む。
【0105】
・スクアレン、DL−α−トコフェロール、およびポリソルベート80(Tween 80)のエマルジョン。このエマルジョンは、リン酸緩衝生理食塩水を含み得る。これはまた、Span85(たとえば、1%)および/またはレシチンを含み得る。これらエマルジョンは、2〜10% スクアレン、2〜10% トコフェロールおよび0.3〜3% Tween 80を有し得、スクアレン:トコフェロールの重量比は、好ましくは、≦1である。なぜなら、これは、より安定なエマルジョンを提供するからである。スクアレンおよびTween 80は、約5:2の容積比もしくは約11:5の重量比で存在し得る。1種のこのようなエマルジョンは、Tween 80をPBS中に溶解して、2%溶液を与え、次いで、この溶液90mlと、(5gのDL−α−トコフェロールおよび5ml スクアレン)の混合物とを混合し、次いで、上記混合物を微小流動化する(microfluidise)ことによって、作製され得る。得られたエマルジョンは、例えば、100〜250nmの間、好ましくは、約180nmの平均直径を有するサブミクロン油滴を有し得る。上記エマルジョンはまた、3−de−O−アシル化モノホスホリルリピドA(3d−MPL)を含み得る。このタイプの別の有用なエマルジョンは、ヒトの用量あたり、0.5〜10mg スクアレン、0.5〜11mg トコフェロール、および0.1〜4mg ポリソルベート80を含み得る[47]。
【0106】
・スクアレン、トコフェロール、およびTriton洗剤(例えば、Triton X−100)のエマルジョン。上記エマルジョンはまた、3d−MPL(下記を参照のこと)を含み得る。上記エマルジョンは、リン酸緩衝液を含み得る。
【0107】
・ポリソルベート(例えば、ポリソルベート80)、Triton洗剤(例えば、Triton X−100)およびトコフェロール(例えば、α−トコフェロールスクシネート)を含むエマルジョン。上記エマルジョンは、約75:11:10(例えば、750μg/ml ポリソルベート80、110μg/ml Triton X−100および100μg/ml α−トコフェロールスクシネート)の質量比でこれら3種の成分を含み得、これら濃度は、抗原由来のこれら成分の何らかの寄与を含むはずである。上記エマルジョンはまた、スクアレンを含み得る。上記エマルジョンはまた、3d−MPL(下記を参照のこと)を含み得る。その水相は、リン酸緩衝液を含み得る。
【0108】
・スクアラン、ポリソルベート80およびポロキサマー401(「PluronicTM L121」)のエマルジョン。上記エマルジョンは、リン酸緩衝生理食塩水(pH7.4)中に処方され得る。このエマルジョンは、ムラミルジペプチドの有用な送達ビヒクルであり、「SAF−1」アジュバント中でトレオニル−MDPとともに使用されてきた[48](0.05〜1% Thr−MDP、5% スクアラン、2.5% Pluronic L121および0.2% ポリソルベート80)。このエマルジョンはまた、「AF」アジュバントでのように[49](5% スクアラン、1.25% Pluronic L121および0.2% ポリソルベート80)、上記Thr−MDPなしでも使用され得る。微小流動化が好ましい。
【0109】
・スクアレン、水性溶媒、ポリオキシエチレンアルキルエーテル親水性非イオン性界面活性剤(例えば、ポリオキシエチレン(12)セトステアリルエーテル)および疎水性非イオン性界面活性剤(例えば、ソルビタンエステルもしくはマンニドエステル(mannide ester)(例えば、ソルビタンモノオレエート(monoleate)もしくは「Span 80」))を含むエマルジョン。上記エマルジョンは、好ましくは、熱可逆性であり、そして/または上記油滴のうちの少なくとも90%(容積で)が、200nm未満のサイズを有する[50]。上記エマルジョンはまた、アルジトール;凍結保護剤(cryoprotective agent)(例えば、糖(例えば、ドデシルマルトシドおよび/もしくはスクロース));および/またはアルキルポリグリコシドのうちの1種以上を含み得る。上記エマルジョンは、TLR4アゴニストを含み得る[51]。このようなエマルジョンは、凍結乾燥され得る。
【0110】
・スクアレン、ポロキサマー105およびAbil−Careのエマルジョン[52]。アジュバント添加ワクチン中のこれら成分の最終濃度(重量)は、5% スクアレン、4% ポロキサマー105(プルロニックポリオール)および2% Abil−Care 85(ビス−PEG/PPG−16/16 PEG/PPG−16/16ジメチコン;カプリル/カプリントリグリセリド)である。
【0111】
・0.5〜50%の油、0.1〜10%のリン脂質、および0.05〜5%の非イオン性界面活性剤を有するエマルジョン。参考文献53に記載されるように、好ましいリン脂質成分は、ホスファチジルコリン、ホスファチジルエタノールアミン、ホスファチジルセリン、ホスファチジルイノシトール、ホスファチジルグリセロール、ホスファチジン酸、スフィンゴミエリンおよびカルジオリピンである。サブミクロン液滴サイズが有利である。
【0112】
・非代謝性の油(例えば、軽油(light mineral oil))および少なくとも1種の界面活性剤(例えば、レシチン、Tween 80もしくはSpan 80)のサブミクロン水中油型エマルジョン。添加剤が含まれ得る(例えば、QuilAサポニン、コレステロール、サポニン−親油性結合体(例えば、参考文献54に記載され、グルクロン酸のカルボキシル基を介して、脂肪族アミンを、デスアシルサポニン(desacylsaponin)に添加することにより生成されるGPI−0100)、ジメチルジオクタデシルアンモニウムブロミド(dimethyidioctadecylammonium bromide)および/もしくはN,N−ジオクタデシル−N,N−ビス(2−ヒドロキシエチル)プロパンジアミン)。
【0113】
・サポニン(例えば、QuilAもしくはQS21)およびステロール(例えば、コレステロール)が螺旋状ミセルとして会合されるエマルジョン[55]。
【0114】
・鉱油、非イオン性親油性エトキシ化脂肪アルコール、および非イオン性親水性界面活性剤(例えば、エトキシ化脂肪アルコールおよび/もしくはポリオキシエチレン−ポリオキシプロピレンブロックコポリマー)を含むエマルジョン[56]。
【0115】
・鉱油、非イオン性親水性エトキシ化脂肪アルコール、および非イオン性親油性界面活性剤(例えば、エトキシ化脂肪アルコールおよび/もしくはポリオキシエチレン−ポリオキシプロピレンブロックコポリマー)を含むエマルジョン[56]。
【0116】
いくつかの実施形態において、エマルジョンは、送達時に抗原と即座に混合され得、従って、上記アジュバントおよび抗原は、パッケージされたもしくは配布されたワクチン(使用時に最終処方物の準備ができている)では別個に保持され得る。他の実施形態において、エマルジョンは、製造の間に抗原と混合され、従って、上記組成物は、液体アジュバント添加形態(liquid adjuvanted form)においてパッケージされる。上記抗原は、一般に、水性形態にあり、その結果、上記ワクチンは、2種の液体を混合することによって最終的に調製される。混合するための上記2種の液体の容積比は、変動し得る(例えば、5:1〜1:5の間)が、一般に、約1:1である。成分濃度が、上記の特定のエマルジョンの説明において与えられる場合、これら濃度は、代表的には、非希釈組成物に関するものであり、したがって抗原溶液と混合した後の濃度は低下する。
【0117】
(ワクチン組成物のパッケージング)
本発明の組成物(もしくはキット成分)に適した容器としては、バイアル、シリンジ(例えば、使い捨てシリンジ)、鼻スプレーなどが挙げられる。これら容器は、滅菌であるべきである。
【0118】
組成物/成分がバイアル中に配置される場合、上記バイアルは、好ましくは、ガラス材料もしくはプラスチック材料から作製され得る。上記バイアルは、好ましくは、上記組成物が上記バイアルに添加される前に、滅菌される。ラテックス感受性患者に伴う問題を回避するために、バイアルは、好ましくは、ラテックス非含有ストッパーでシールされ、全てのパッケージング材料中にラテックスが存在しないことが好ましい。上記バイアルは、ワクチンの単一用量を含んでいてもよいし、1用量より多い用量(「複数用量」バイアル)、例えば、10用量を含んでいてもよい。好ましいバイアルは、無色ガラスから作製される。
【0119】
バイアルは、あらかじめ充填されたシリンジがキャップに挿入され得るように適合されたキャップ(例えば、ルアーロック)を有し得、上記シリンジの内容物は、上記バイアルへと排出され得(例えば、その中の凍結乾燥物質を再構成するために)、上記バイアルの内容物は、取り出されて上記シリンジへと戻され得る。上記バイアルから上記シリンジを取り出した後、次いで、ニードルが取り付けられ得、上記組成物が、患者へと投与され得る。上記キャップは、好ましくは、シールもしくはカバーの内部に配置され得る。その結果、上記シールもしくはカバーは、上記キャップに到達し得る前に除去されなければならない。バイアルは、特に、複数用量バイアルについては、その内容物の無菌的取り出しを可能にするキャップを有し得る。
【0120】
成分がシリンジにパッケージされる場合、上記シリンジは、これに取り付けられるニードルを有し得る。ニードルが取り付けられていない場合、別個のニードルが、組み立ておよび使用のために、上記シリンジと共に供給され得る。このようなニードルは、鞘に入れられ得る。安全ニードル(safety needle)が好ましい。1インチ 23ゲージ、1インチ 25ゲージおよび5/8インチ 25ゲージのニードルが代表的である。シリンジは、記録保持を容易にするために、ロット番号、インフルエンザ流行期、および内容物の使用期限がプリントされ得るピールオフラベルとともに提供され得る。上記シリンジにおけるプランジャーは、好ましくは、上記プランジャーが吸引の間に偶発的に除去されてしまわないように、ストッパーを有し得る。上記シリンジは、ラテックスゴムキャップおよび/もしくはプランジャーを有し得る。使い捨てシリンジは、単一用量のワクチンを含む。上記シリンジは、一般に、ニードルの取り付け前に、先端をシールするために先端キャップを有し、上記先端キャップは、好ましくは、ブチルゴムから作製され得る。上記シリンジおよびニードルが別個にパッケージされる場合、上記ニードルは、好ましくは、ブチルゴムシールドと適合させられる。好ましいシリンジは、商品名「Tip−Lok」TMの下で市販されるものである。
【0121】
容器は、例えば、小児への送達を容易にするために、半用量容積を示すために印が付けられ得る。例えば、0.5ml用量を含むシリンジは、0.25ml容積を示す印を有し得る。
【0122】
ガラス容器(例えば、シリンジもしくはバイアル)が使用される場合、ソーダ石灰ガラスよりむしろホウケイ酸ガラスから作製された容器を使用することが好ましい。
【0123】
キットもしくは組成物は、(例えば、同じボックスの中に)上記ワクチンの詳細(例えば、投与の説明書、上記ワクチン内の抗原の詳細など)を含むリーフレットとともにパッケージされ得る。上記説明書はまた、警告(例えば、ワクチン接種後のアナフィラキシー反応の場合に容易に利用可能なアドレナリン溶液を保持することなど)を含み得る。
【0124】
(処置方法、および上記ワクチンの投与)
本発明は、本発明に従って製造されるワクチンを提供する。これらワクチン組成物は、ヒト被験体もしくはヒト以外の動物被験体(例えば、ブタ)への投与に適しており、本発明は、上記被験体に本発明の組成物を投与する工程を包含する、被験体における免疫応答を惹起するための方法を提供する。本発明はまた、医薬として使用するための本発明の組成物を提供し、被験体における免疫応答を惹起するための医薬の製造のための、本発明の組成物の使用を提供する。
【0125】
これら方法および使用によって惹起される免疫応答は、一般に、抗体応答(好ましくは、防御的抗体応答)を含む。インフルエンザウイルスワクチン接種後の抗体応答、中和能力および防御を評価するための方法は、当該分野で周知である。ヒトでの研究から、ヒトインフルエンザウイルスのヘマグルチニンに対する抗体力価が、防御と相関する(約30〜40の血清サンプル赤血球凝集阻害力価が、同種のウイルスによる感染から約50%防御を与える)ことが示された[57]。抗体応答は、代表的には、赤血球凝集阻害によって、マイクロ中和によって、単一放射免疫拡散(single radial immunodiffusion)(SRID)によって、および/もしくは単一放射溶血(SRH)によって、測定される。これらアッセイ技術は、当該分野で周知である。
【0126】
本発明の組成物は、種々の方法において投与され得る。最も好ましい免疫化経路は、筋肉内注射(例えば、腕もしくは脚への)によるが、他の利用可能な経路としては、皮下注射、鼻内[58〜60]、経口[61]、皮内[62、63]、経皮的(transcutaneous)、経皮的(transdermal)[64]などが挙げられる。
【0127】
本発明に従って調製されるワクチンは、小児および成人の両方を処置するために使用され得る。インフルエンザワクチンは、6ヶ月齢からの小児および成人での免疫化における使用に関して現在推奨されている。従って、ヒト被験体は、1歳未満、1〜5歳、5〜15歳、15〜55歳、もしくは少なくとも55歳であってもよい。上記ワクチンを受けるのに好ましい被験体は、高齢(例えば、≧50歳、≧60歳、および好ましくは、≧65歳)、若年齢(例えば、≦5歳)、入院している被験体、ヘルスケアワーカー、軍隊および軍職員、妊婦、慢性疾患の、免疫不全の被験体、上記ワクチンを受ける7日前までに抗ウイルス化合物(例えば、オセルタミビルおよびザナミビル化合物;以下を参照のこと)を摂取している被験体、卵アレルギーがある人々、および外国に旅行する人々である。上記ワクチンは、これらの群に適しているのみではないが、一般に、集団においてより多く使用され得る。汎流行性株については、すべての年齢群への投与が好ましい。
【0128】
好ましい本発明の組成物は、効力についてCPMP基準のうちの1つ、2つもしくは3つを満たす。成人(18〜60歳)において、これら基準は、以下である:(1)≧70%血清保有率(seroprotection);(2)≧40%セロコンバージョン;および/もしくは(3)GMT増加≧2.5倍。高齢者(>60歳)において、これら基準は、以下である:(1)≧60%血清保有率;(2)≧30%セロコンバージョン;および/もしくは(3)GMT増加≧2倍。これら基準は、少なくとも50名の患者でのオープンラベル研究に基づく。
【0129】
処置は、単一用量スケジュールもしくは複数用量スケジュールによってであり得る。複数用量は、初回免疫化スケジュールおよび/もしくは追加免疫化スケジュールにおいて使用され得る。複数用量スケジュールにおいて上記種々の用量は、同じ経路もしくは異なる経路によって与えられ得る(例えば、非経口の初回免疫(prime)および粘膜の追加免疫(boost)、粘膜の初回免疫および非経口の追加免疫など)。1より多くの用量の投与(代表的には、2用量)は、免疫学的に経験したことがない患者(例えば、これまでインフルエンザワクチンを受けたことのない人に関して)において、または新たなHAサブタイプ(汎流行性アウトブレイクにおけるような)に対するワクチン接種のために、特に有用である。複数用量は、代表的には、少なくとも1週間空けて(例えば、約2週間、約3週間、約4週間、約6週間、約8週間、約10週間、約12週間、約16週間など)投与される。
【0130】
本発明によって生成されるワクチンは、他のワクチン(例えば、麻疹ワクチン、ムンプス・ワクチン、風疹ワクチン、MMRワクチン、水痘ワクチン、MMRVワクチン、ジフテリアワクチン、破傷風ワクチン、百日咳ワクチン、DTPワクチン、結合体化H.influenzaeタイプbワクチン、不活性化ポリオウイルスワクチン、B型肝炎ウイルスワクチン、髄膜炎菌結合型ワクチン(例えば、四価A−C−W135−Yワクチン)、RSウイルスワクチン、肺炎球菌結合型ワクチンなど)と実質的に同時(例えば、同じ医療相談の間に、またはヘルスケア専門家およびワクチン接種施設への訪問の間に)に患者に投与され得る。肺炎球菌ワクチンおよび/もしくは髄膜炎菌ワクチンと実施的に同時の投与は、高齢の患者において特に有用である。
【0131】
同様に、本発明のワクチンは、抗ウイルス化合物、および特に、インフルエンザウイルスに対して活性な抗ウイルス化合物(例えば、オセルタミビルおよび/もしくはザナミビル)と実質的に同時(例えば、同じ医療相談の間に、またはヘルスケア専門家への訪問の間に)に、患者に投与され得る。これら抗ウイルス剤としては、ノイラミニダーゼインヒビター(例えば、(3R,4R,5S)−4−アセチルアミノ−5−アミノ−3(1−エチルプロポキシ)−1−シクロヘキセン−1−カルボン酸もしくは5−(アセチルアミノ)−4−[(アミノイミノメチル)−アミノ]−2,6−アンヒドロ−3,4,5−トリデオキシ−D−グリセロ−D−ガラクトノン−2−エノン酸(そのエステル(例えば、そのエチルエステル)およびその塩(例えば、そのリン酸塩)を含む)が挙げられる。好ましい抗ウイルス剤は、(3R,4R,5S)−4−アセチルアミノ−5−アミノ−3(1−エチルプロポキシ)−1−シクロヘキセン−1−カルボン酸、エチルエステル、ホスフェート(1:1)(オセルタミビルホスフェート(TAMIFLUTM)としても公知)である。
【0132】
(一般)
用語「含む(comprising)」は、「含む(including)」および「からなる(consisting)」を含み、例えば、Xを「含む」組成物は、もっぱらXからなってもよいし、さらなる何かを含んでいてもよい(例えば、X+Y)。
【0133】
語句「実質的に」は、「完全に」を排除しない。例えば、Yを「実質的に含まない」組成物は、完全にYを含まなくてもよい。必要であれば、語句「実質的に」は、本発明の定義から省略され得る。
【0134】
数値xに関する用語「約」とは、任意であり、例えば、x±10%を意味する。
【0135】
別段示されなければ、2種以上の成分を混合する工程を包含するプロセスは、いかなる特定の混合する順序も必要としない。従って、成分は、任意の順序で混合され得る。3つの成分が存在する場合、2つの成分が、互いに合わされ得、次いで、その組み合わせが、第3の成分と合わせられ得るなど。
【0136】
動物(および特にウシ)の物質が、細胞培養において使用される場合、それら物質は、伝染性海綿状脳症(TSE)を含まない、特に、ウシ海綿状脳症(BSE)を含まない供給源から得られるべきである。全体として、動物由来物質が完全に存在しない状態で細胞を培養することが好ましい。
【0137】
化合物が、組成物の一部として身体に投与される場合、その化合物は、代わりに、適切なプロドラッグによって置換され得る。
【図面の簡単な説明】
【0138】
【図1】図1:本発明の実施形態の概略図;MDCK 33016懸濁細胞(CDM中で培養)を、6ウェルディッシュA(無血清培地中もしくはDMEM+5% FBS中);B:同時トランスフェクションMDCK 33016;C:洗浄2×,無血清DMEMに交換,+trypzean,+/−新鮮な非感染MDCK細胞,D:RGウイルス中に播種する。
【図2】図2:PR8−A/CAバックボーン(backbone)を使用するMDCK 33016細胞でのインフルエンザウイルスレスキューの効率;y軸は、感染力価(FFU/mL)を示す;白抜き四角は、PR8−A/CAおよび5% 血清を使用した結果を示し、斜線を入れた四角は、PR8−A/CAありおよび血清なしでの結果を示す;Aは、新鮮な非感染細胞を添加したことを示す;Bは、新鮮な非感染細胞を添加しなかったことを示す。
【図3】図3:A/WSN/33バックボーンを使用するMDCK 33016細胞でのインフルエンザウイルスレスキューの効率;y軸は、感染力価(FFU/mL)を示す;白抜き四角は、WSNおよび5% 血清を使用した結果を示し、斜線を入れた四角は、WSNありおよび血清なしでの結果を示す;Aは、新鮮な非感染細胞を添加したことを示す;Bは、新鮮な非感染細胞を添加しなかったことを示す。
【図4】図4:PR8−WSNバックボーンを使用するMDCK 33016細胞でのインフルエンザウイルスレスキューの効率。y軸は、FFU/mLを示す;白抜き四角は、PR8−WSNおよび5%血清を使用した結果を示し、斜線を入れた四角は、PR8−WSNありおよび血清なしでの結果を示す;Aは、新鮮な非感染細胞を添加したことを示す;Bは、新鮮な非感染細胞を添加しなかったことを示す。
【発明を実施するための形態】
【0139】
(本発明を実施するための態様)
(ウイルスレスキュー)
MDCK 33016細胞を、CDM培地中、温度37℃において懸濁して培養した。トランスフェクションの前日に、6×105細胞/ウェルを、2mL DMEM培地(5% FCSを含む)中、CELL−BINDTM 6ウェルディッシュの中に播種した。並行実験において、上記細胞を、FCSの非存在下で播種した。上記細胞を、37℃において一晩インキュベートした。
【0140】
翌日、上記播種した細胞を、製造業者のプロトコルを使用して、LipofectamineTM LTX Plusトランスフェクション試薬でトランスフェクトした。簡潔には、1μgの各プラスミド(PA、PB1、PB2、NP、NS、M、HA、NAと、TMPRSS2)を、500μlの無血清DMEM培地中に希釈した。10μlのPlus試薬を、上記希釈したDNAに直接添加した。上記混合物を、室温で5分間にわたってインキュベートし、その後、25μlのLipofectamineをそれに添加した。次いで、上記混合物を、室温で30分間にわたってインキュベートした。インキュベーション後、上記トランスフェクション試薬:DNA複合体を、滴下様式において上記細胞に添加した。上記細胞を、その後、37℃において24時間にわたってインキュベートした。
【0141】
インキュベーション後、培地を細胞から吸引し、1:2000希釈のTrypzeanTM(1mg/mL ストック溶液)を含む無血清DMEM培地中の4×105 MDCK 33016細胞の懸濁物を、各ウェルに添加した。並行した実験において、1:2000希釈のTrypzeanTM(1mg/mL ストック溶液)単独(すなわち、細胞の添加なし)を、上記トランスフェクトされた細胞に添加した。次いで、上記細胞を、37℃においてさらに48時間にわたって、または上記細胞が溶解するまでインキュベートし、その後、ウイルス力価を、病巣形成アッセイを使用して決定した。
【0142】
(病巣形成アッセイ(Focus−Forming Assay))
非感染MDCK細胞を、密度6.25×104細胞/ウェルにおいて、48ウェルプレートの中に、500μlの10% FCS含有DMEM中にプレートした。翌日、細胞に、容積100〜150μlにおいてウイルスを2時間にわたって37℃において感染させた。上記細胞に、上記ウイルスの種々の希釈物を感染させた。感染の2時間後、培地を吸引し、500μlの10% FCS含有DMEMを各ウェルに添加した。上記細胞を、翌日まで37℃においてインキュベートした。
【0143】
感染の24時間後、培地を吸引し、細胞をPBSで1回洗浄した。500μlの氷冷したPBS中80% アセトンを、各ウェルに添加し、続いて、4℃において30分間にわたってインキュベートした。アセトン混合物を吸引し、上記細胞を、PBST(PBS+0.1% Tween)で1回洗浄した。500μlのPBS中2% BSAを、各ウェルに添加し、続いて、室温(RT)において30分間にわたってインキュベートした。500μlの1:6000希釈の抗NPを、ブロッキング緩衝液中に添加し、続いて、室温において2時間にわたってインキュベートした。上記抗体溶液を吸引し、上記細胞をPBSTで2回洗浄した。2次抗体(ヤギ抗マウス)を、1:2000希釈において500μl ブロッキング緩衝液中で添加し、上記プレートを、室温において2時間にわたってインキュベートした。抗体溶液を吸引し、細胞をPBSTで3回洗浄した。500μlのKPL True Blueを各ウェルに添加し、10分間にわたってインキュベートした。True−Blueを吸引し、dH2Oで1回洗浄することによって、上記反応を停止した。上記水を吸引し、上記細胞を乾燥させた。
【0144】
(結果)
3種の異なるウイルスバックボーンでの上記病巣形成アッセイの結果を、図2〜4に示す。上記結果は、感染の24時間後に細胞の添加ありおよびなしでのウイルスレスキューの効率を示す。認められ得るように、すべての場合において、上記ウイルスレスキューの効率は、細胞を添加しなかった実験と比較して、トランスフェクションの24時間後に細胞を添加した実験において最大3倍まで増大した。この効率の増大は、試験したすべてのウイルスバックボーンで、ならびに血清含有条件下および無血清条件下の両方認められ得る。
【0145】
本発明は、例示によって記載されてきたに過ぎず、改変が行われ得る一方で、本発明の範囲および趣旨内にとどまっていることが理解される。
【0146】
【化1】
【0147】
【化2】
【0148】
【化3】
【特許請求の範囲】
【請求項1】
ウイルスを調製するための方法であって、該方法は、(i)宿主細胞の培養物をウイルスRNA分子をコードする少なくとも1個の発現構築物でトランスフェクトする工程;(ii)細胞を、(i)のトランスフェクトされた該宿主細胞に添加して、細胞の混合物を提供する工程;および(iii)ウイルスを生成するために、該細胞の混合物を培養する工程を包含する、方法。
【請求項2】
ワクチン製造のためのウイルスを調製する方法であって、該方法は、(i)宿主細胞の培養物をウイルスRNA分子をコードする少なくとも1個の発現構築物でトランスフェクトする工程;(ii)細胞を、(i)のトランスフェクトされた該宿主細胞に添加して、細胞の混合物を提供する工程;(iii)ウイルスを生成するために、該細胞の混合物を培養する工程;および(iv)工程(iii)において得られた該ウイルスを精製する工程を包含する、方法。
【請求項3】
工程(iii)において精製された前記ウイルスをワクチンへと処方する工程をさらに包含する、請求項2に記載の方法。
【請求項4】
前記宿主細胞は、VERO細胞、PER.C6細胞もしくはMDCK細胞である、前述の請求項のいずれか1項に記載の方法。
【請求項5】
前記MDCK細胞は、細胞株MDCK 33016(DSM ACC2219)である、請求項4に記載の方法。
【請求項6】
前記細胞は、懸濁物中で培養される、請求項1〜5のいずれか1項に記載の方法。
【請求項7】
前記細胞は、接着させて培養される、請求項1〜5のいずれか1項に記載の方法。
【請求項8】
前記ウイルスは、セグメント化ウイルスである、前述の請求項のいずれか1項に記載の方法。
【請求項9】
前記ウイルスは、マイナス鎖RNAウイルスである、請求項1〜8のいずれか1項に記載の方法。
【請求項10】
前記ウイルスは、インフルエンザウイルスである、請求項9に記載の方法。
【請求項11】
前記ウイルスは、二本鎖RNAウイルスである、請求項1〜8のいずれか1項に記載の方法。
【請求項12】
前記ウイルスは、ロタウイルスである、請求項11に記載の方法。
【請求項13】
前記ウイルスは、非セグメント化ウイルスである、請求項1〜9または請求項11のいずれか1項に記載の方法。
【請求項1】
ウイルスを調製するための方法であって、該方法は、(i)宿主細胞の培養物をウイルスRNA分子をコードする少なくとも1個の発現構築物でトランスフェクトする工程;(ii)細胞を、(i)のトランスフェクトされた該宿主細胞に添加して、細胞の混合物を提供する工程;および(iii)ウイルスを生成するために、該細胞の混合物を培養する工程を包含する、方法。
【請求項2】
ワクチン製造のためのウイルスを調製する方法であって、該方法は、(i)宿主細胞の培養物をウイルスRNA分子をコードする少なくとも1個の発現構築物でトランスフェクトする工程;(ii)細胞を、(i)のトランスフェクトされた該宿主細胞に添加して、細胞の混合物を提供する工程;(iii)ウイルスを生成するために、該細胞の混合物を培養する工程;および(iv)工程(iii)において得られた該ウイルスを精製する工程を包含する、方法。
【請求項3】
工程(iii)において精製された前記ウイルスをワクチンへと処方する工程をさらに包含する、請求項2に記載の方法。
【請求項4】
前記宿主細胞は、VERO細胞、PER.C6細胞もしくはMDCK細胞である、前述の請求項のいずれか1項に記載の方法。
【請求項5】
前記MDCK細胞は、細胞株MDCK 33016(DSM ACC2219)である、請求項4に記載の方法。
【請求項6】
前記細胞は、懸濁物中で培養される、請求項1〜5のいずれか1項に記載の方法。
【請求項7】
前記細胞は、接着させて培養される、請求項1〜5のいずれか1項に記載の方法。
【請求項8】
前記ウイルスは、セグメント化ウイルスである、前述の請求項のいずれか1項に記載の方法。
【請求項9】
前記ウイルスは、マイナス鎖RNAウイルスである、請求項1〜8のいずれか1項に記載の方法。
【請求項10】
前記ウイルスは、インフルエンザウイルスである、請求項9に記載の方法。
【請求項11】
前記ウイルスは、二本鎖RNAウイルスである、請求項1〜8のいずれか1項に記載の方法。
【請求項12】
前記ウイルスは、ロタウイルスである、請求項11に記載の方法。
【請求項13】
前記ウイルスは、非セグメント化ウイルスである、請求項1〜9または請求項11のいずれか1項に記載の方法。
【図1】

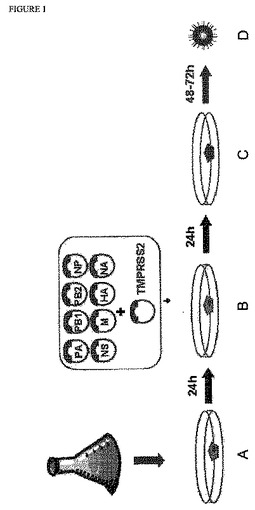
【図2】
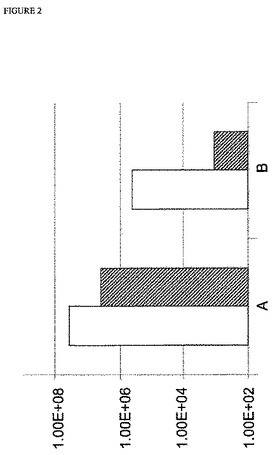
【図3】
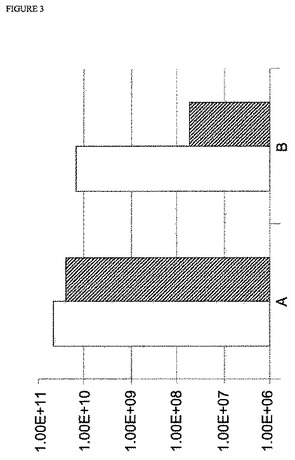
【図4】
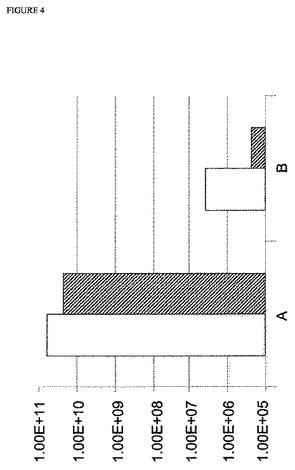
【図2】
【図3】
【図4】
【公表番号】特表2013−507952(P2013−507952A)
【公表日】平成25年3月7日(2013.3.7)
【国際特許分類】
【出願番号】特願2012−534814(P2012−534814)
【出願日】平成22年10月20日(2010.10.20)
【国際出願番号】PCT/IB2010/054752
【国際公開番号】WO2011/048560
【国際公開日】平成23年4月28日(2011.4.28)
【出願人】(504389991)ノバルティス アーゲー (806)
【Fターム(参考)】
【公表日】平成25年3月7日(2013.3.7)
【国際特許分類】
【出願日】平成22年10月20日(2010.10.20)
【国際出願番号】PCT/IB2010/054752
【国際公開番号】WO2011/048560
【国際公開日】平成23年4月28日(2011.4.28)
【出願人】(504389991)ノバルティス アーゲー (806)
【Fターム(参考)】
[ Back to top ]