
ウイルス増殖用の非腫瘍形成性MDCK細胞株
【課題】血清の存在下又は非存在下で増殖可能な新規のMDCK由来の付着性の非腫瘍形成性細胞株を提供する。
【解決手段】本発明は、血清の存在下又は非存在下で増殖可能な新規のMDCK由来の付着性の非腫瘍形成性細胞株を提供する。本発明の細胞株は、ワクチン物質(例えば、ウイルス)の生産に有用である。より具体的に言えば、本発明の細胞株は一般にインフルエンザウイルス、特にca/tsインフルエンザウイルスの生産に有用である。本発明はさらに、MDCK細胞が非腫瘍形成性を維持するような、該細胞の適応及び培養の方法、並びに培地処方物を提供する。さらに、本発明は、本発明の新規細胞株でのワクチン物質(例えば、インフルエンザウイルス)の生産方法を提供する。
【解決手段】本発明は、血清の存在下又は非存在下で増殖可能な新規のMDCK由来の付着性の非腫瘍形成性細胞株を提供する。本発明の細胞株は、ワクチン物質(例えば、ウイルス)の生産に有用である。より具体的に言えば、本発明の細胞株は一般にインフルエンザウイルス、特にca/tsインフルエンザウイルスの生産に有用である。本発明はさらに、MDCK細胞が非腫瘍形成性を維持するような、該細胞の適応及び培養の方法、並びに培地処方物を提供する。さらに、本発明は、本発明の新規細胞株でのワクチン物質(例えば、インフルエンザウイルス)の生産方法を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
発明の分野
本発明は、ワクチン物質の生産に利用することのできる新規非腫瘍形成性MDCK細胞に関する。非腫瘍形成性MDCK細胞は無血清培地に適応することができる。本発明は、さらに非腫瘍形成性MDCK細胞の増殖用培地処方物及び培養方法、並びに本発明の細胞株の非腫瘍形成性質を維持する方法に関する。本発明は、さらに非腫瘍形成性MDCK細胞を用いた細胞培養でのインフルエンザウイルスの生産方法に関する。本発明はまた、記載された方法によって得ることができるウイルス(例えば、インフルエンザ)、及びこのタイプのウイルスを含む免疫原性組成物及び/又はその成分に関する。
【背景技術】
【0002】
発明の背景
ワクチン接種は、年1度のインフルエンザの流行によりもたらされる病気を予防する上で最も重要な公衆衛生措置である。ワクチンの効率的な利用は、大量のワクチン物質(例えば、ウイルス)を、安定かつ培養の容易な供給源からすばやく生産できることに依る。ワクチンの早急な開発とそれらを十分に利用できるようにすることは、ヒト及び動物の多くの病気に対抗する上で重要である。ワクチン生産の遅延やその量の不足は、病気の発生に対処する上で問題となり得る。例えば、最近の研究では世界的流行性(パンデミック)インフルエンザに対するワクチンの生産に長期の準備期間を要することについての心配が示唆されている。例えば、非特許文献1(Wood, J. M., 2001, Philos. Trans. R. Soc. Lond. B. Biol. Sci., 356:1953)を参照のこと。効率的なワクチン生産には、宿主系から高収量で生産されるワクチン物質の大量増殖が必要である。異なるワクチン物質から満足のいく収量を得るためには異なる増殖条件が必要となる。ワクチン物質は、孵化卵、初代組織培養細胞、又は株化細胞で生産することができる。しかし、これらの宿主系は、現在以下で詳述する多くの制限を受けている。
【0003】
孵化卵は、通常、鶏の飼育や卵の受精の管理を要する時間、労力及びコスト集約的方法の下でインフルエンザワクチンウイルスの生産に利用されている。さらに、卵中で作られるインフルエンザワクチンは、卵アレルギーをもつ人にとっては重度の即時性過敏反応を起こし得るため禁忌である。それ故、ワクチン業界が努力して、例えば細胞培養系でインフルエンザワクチンを生産するといった卵を利用しない代替の生産基盤を開発した。
【0004】
初代組織培養細胞の利用は、安定した初代細胞集団を開発し、維持することの難しさに直面し、それがネックとなっている。株化細胞がしばしば使用され、初代細胞の技術的制限を回避している。しかし、これらの細胞株の多くは、腫瘍原性であることが知られており、それ自体が安全性の問題を引き起こすので、ワクチン生産の使用に対して規制面でかなりの制約を受ける。事実、世界保健機関の適用ガイドラインには、ごくわずかの細胞株しかワクチン生産に許可されていないことが示されている。さらなる問題は、細胞培養の培地中の動物若しくはヒト由来の血清及び/又はタンパク質添加物の使用に起因している。例えば、多数の添加物間の質及び組成の変動、並びにマイコプラズマ、ウイルス、BSE剤、及びその他の伝染性物質による汚染の危険性が、よく知られている。一般に、血清、又はアルブミン、トランスフェリン、若しくはインスリンのような血清由来の物質は、培養物を汚染する可能性のある厄介な物質を含み得る。また生物学的な製品は、それらから生産されている。それゆえ、多数の研究グループが血清や血清由来の製品を必要としない効率的な宿主系及び培養条件の開発のために勤しんでいる。
【0005】
したがって、低コストで、安全性が高く、かつ安定した様態で、好ましくは無血清若しくは動物性タンパク質フリーの培養条件下で、ワクチン物質を生産するのに有用な非腫瘍形成性細胞株の樹立に対する要望があった。このような細胞系は、特にインフルエンザワクチン物質の生産に有用であると思われる。
【0006】
イヌ腎臓由来(Madin Darby Canine Kidney:MDCK)細胞は、元々インフルエンザウイルスの滴定に使用されていた(非特許文献2(Zambon M.著, in Textbook of Influenza, Nicholson編, Webster and Hay, ch 22, pg 291-313, Blackwell Science (1998)))。この細胞は、1958年にコッカースパニエルの正常なオスの腎臓から株化された。ATCC(American Type Culture Collection)リストのMDCK(CCL 34)株は、S.MadinとN.B.Darbyにより寄託されたが、その他にも多くのMDCK細胞の細胞系が利用できる。Leighton Jとその共同研究者らは、MDCK細胞の発癌の特性を実証した一連の論文を発表した(非特許文献3(Leightonら、1968, Science 163:472);非特許文献4(Leightonら、1970, Cancer 26:1022);及び非特許文献5(Leightonら、1971 Europ J. Cancer 8:281))。しかし、それらの研究に使用されたMDCK細胞の系統及び継代数は記載されていなかった。また、異なる系統と異なる継代由来のMDCK細胞が、染色体数及び構造に違いを示すことは既に知られていた(非特許文献6(Gaushら, 1966, Proc. Soc. Exp. Biol. Med., 122: 931))。このような違いは、腫瘍形成性の性質をもった細胞を生じ得る。
【0007】
ワクチン生産用細胞株の受容性に関して考慮すべき主要な点の一つは、それらの細胞の潜在的悪性度と関係がある。それゆえ、現在記載されている細胞株を利用したワクチン物質生産のためのMDCK細胞の使用は制限される。Gronerら(特許文献1(米国特許番号第6,656,720号))、及びMakizumiら(特許文献2(米国特許番号第6,825,036号))の両者は、無血清培地において浮遊状態での増殖に適応し、またインフルエンザウイルスの生産に利用できるMDCK細胞由来の細胞株の開示を主張している。しかし、足場を必要としないことと、正常動物細胞の腫瘍形成性細胞への形質転換との間に相関関係があることが報告されている(非特許文献7(Stilesら、1976, Cancer Res., 36:3300))。いくつかのグループ(非特許文献8(Kesslerら、1999、Cell Culture Dev Biol Stand, 98:13);非特許文献9(Mertenら、1999, Cell Culture Dev Biol Stand, 98:23);及び非特許文献10(Treeら、2001, Vaccine, 19:3444))は、インフルエンザウイルスの大量生産用にMDCK細胞を使用することを記載し、主張している。しかし、彼らは使用したMDCK細胞の潜在的な形質転換に対しては何ら取り組んでいない。
【0008】
本明細書中の参考文献の引用又は考察は、それらを本発明の先行技術と認めたものとは解釈しないものとする。さらに特許の引用は、その効力を認めたものとは解釈しないものとする。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】米国特許番号第6,656,720号
【特許文献2】米国特許番号第6,825,036号
【非特許文献】
【0010】
【非特許文献1】Wood, J. M., 2001, Philos. Trans. R. Soc. Lond. B. Biol. Sci., 356:1953
【非特許文献2】Zambon M.著, in Textbook of Influenza, Nicholson編, Webster and Hay, ch 22, pg 291-313, Blackwell Science (1998)
【非特許文献3】Leightonら、1968, Science 163:472
【非特許文献4】Leightonら、1970, Cancer 26:1022
【非特許文献5】Leightonら、1971 Europ J. Cancer 8:281
【非特許文献6】Gaushら, 1966, Proc. Soc. Exp. Biol. Med., 122: 931
【非特許文献7】Stilesら、1976, Cancer Res., 36:3300
【非特許文献8】Kesslerら、1999、Cell Culture Dev Biol Stand, 98:13
【非特許文献9】Mertenら、1999, Cell Culture Dev Biol Stand, 98:23
【非特許文献10】Treeら、2001, Vaccine, 19:3444
【発明の概要】
【課題を解決するための手段】
【0011】
発明の概要
本発明は、動物性タンパク質非含有(animal protein-free:APF)の製剤を含む血清含有又は無血清の培地処方物のいずれかで増殖するのに適応した非腫瘍形成性MDCK細胞を提供する。一の実施形態において、本発明の非腫瘍形成性MDCK細胞は付着性である。他の実施形態において、本発明の非腫瘍形成性MDCK細胞は、上皮形態を有している。さらに他の実施形態において、本発明の非腫瘍形成性MDCK細胞は付着性で、かつ上皮形態を有している。発癌性が一の実施形態において、アダルトヌードマウスモデルで実証された(例えば、Stilesら,1976, Cancer Res, 36:1353、及び後述の実施例2)。発癌性については、例えば、鶏胚への注入、及び/又は漿尿膜への局所適用による他のアッセイででもテストすることができる(Leightonら,1970, Cancer, 26:1024)。
【0012】
本発明のMDCK細胞内で増殖できるウイルスは、マイナス鎖RNAウイルスを含むがそれに限定されない。また、当該マイナス鎖RNAウイルスは、インフルエンザ、RSV、パラインフルエンザウイルス1、2及び3型、並びにヒトメタニューモウイルスを含むが、これらに限定されない。
【0013】
本発明は、さらに非腫瘍形成性MDCK細胞の誘導とその維持に有用な方法及び培地処方物を提供する。本発明のMDCK細胞は、例えばウイルス等のワクチン物質の生産に特に有用である。
【0014】
本発明のその他の態様は、本発明のいずれかのMDCK細胞を、ワクチン物質の生産を可能にする条件の下で適切な培地にて培養すること、及びその物質を一以上の宿主細胞から又は宿主細胞を増殖させる培地から分離することによるワクチン物質(例えばウイルス)の生産方法を含む。
【0015】
免疫原性組成物も、本発明の特徴である。例えば、上記のように生産されるワクチン物質、及び場合により、製薬上許容される賦形剤等の賦形剤又は一以上の製薬上許容される投薬成分、を含む免疫原性組成物が挙げられる。
【0016】
被験体に、一以上の上記免疫原性組成物を効果的な量で投与することによって被験体に免疫原性反応を生じさせる方法も本発明に含まれる。さらに、ウイルス感染に対して免疫原性反応を引き起こすのに効果的な量で、一以上の上記の免疫原性組成物を投与することで被験体にウイルス感染(例えば、インフルエンザウイルスの)の予防的又は治療的処置を施す方法も本発明の一部である。前記処置を受ける被験体は、哺乳動物(例えば、ヒト)を含み得る。さらに、前記方法は、本発明のMDCK細胞で生産された一以上のウイルス及び製薬上許容される賦形剤からなる組成物を、予防的又は治療的にウイルス感染を治療するのに効果的な量で被験体に投与することも含むことができる。
【0017】
本発明のこれら及びその他の目的並びに特徴は、以下の詳細な説明を添付の図面と合わせて読むことで、より一層明らかとなるであろう。
【図面の簡単な説明】
【0018】
【図1A】細胞中のインフルエンザ株の増殖。図1Aは、MDCK細胞とベロ細胞のクローン(27F9)における代表的なca/tsインフルエンザ株の感染の広がりを比較した蛍光焦点アッセイの結果を示す写真である。
【図1B】細胞中のインフルエンザ株の増殖。図1Bは、MDCK細胞におけるインフルエンザ株ca A/Vietnam/1203/2004 (H5N1)の増殖曲線である。タイターは、感染後48時間に〜8 log10 TCID50/mLでピークに達し、その後3〜4日間は安定したままであった。
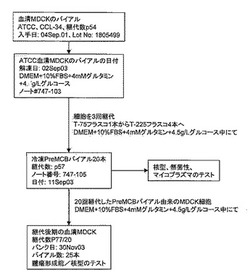
【図2】図2は、MDCK-S PreMCB(継代数57)の誘導に使用される方法を概説している。この方法については実施例2で詳述する。
【図3】図3は、MDCK-S細胞が上皮様形態を有することを示す写真である。写真は、接種後3日目に撮影された。
【図4】図4は、10% FBS DMEM培地におけるMDCK-S細胞の成長曲線である。細胞は約1日の誘導期後、指数関数的増殖へと続き、接種後4日目に静止期に入り、5日目に最大密度〜29×106細胞に達している。
【図5】図5は、10% FBS DMEM培地におけるMDCK-S細胞のグルコース消費及び乳酸産生のグラフである。誘導期中のそれらの速度は、グルコース及び乳酸についてそれぞれ2.93mM/日及び3.43mM/日の増加であり低かった。
【図6】図6は、10% FBS DMEM培地におけるMDCK-S細胞のグルタミン消費、並びにグルタミン酸及びアンモニア産生のグラフである。グルタミン消費速度は4日目までで0.49mM/日であり、アンモニア産生速度は、5日目までで0.32mM/日であった。グルタミン酸は本実験では蓄積しなかった。
【図7】図7は、低継代(P61/4)及び高継代(P81/24)の中期MDCK-S細胞100個における染色体数の分配のグラフである。高低の両継代細胞に関して78本の最頻値染色体数をもち、カウントされた染色体数は中期あたり70〜84本の範囲に及んでいた。
【図8】図8は、MDCK-T PreMCB(継代数64/5)の誘導に使用される方法を概説している。本方法については実施例3で詳述する。
【図9】図9は、MDCK-T細胞が上皮様形態を有することを示す写真である。写真は、接種後3日目に撮影された。
【図10】図10は、Taub培地におけるMDCK-T細胞の成長曲線である。細胞は誘導期を有さず、接種後4日目で静止期に入るまで指数関数的に増殖した。
【図11】図11は、Taub培地におけるMDCK-T細胞のグルコース消費及び乳酸産生のグラフである。対数期中、それらの速度はグルコース及び乳酸でそれぞれ1.78mM/日及び2.88mM/日であった。
【図12】図12は、Taub培地におけるMDCK-T細胞のグルタミン消費、並びにグルタミン酸及びアンモニア産生のグラフである。グルタミン消費速度は4日目までで0.36mM/日であり、アンモニア産生速度は7日目まで0.22mM/日で線形的に増加した。グルタミン酸は本実験では蓄積しなかった。
【図13】図13は、低継代(P61/4)及び高継代(P81/24)の中期MDCK-T細胞100個における染色体数の分布のグラフである。カウントされた染色体数は、低継代細胞では中期あたり52〜82本、また高継代細胞では54〜82本の範囲に及んでいた。
【図14】図14は、MDCK-T、MDCK-SF101細胞(継代数71/9)、及びMDCK-SF102細胞(継代数71/9)の各中期100個における染色体数の分布のグラフである。SF101及びSF102の両細胞とも78本の最頻値染色体数を有し、カウントされた染色体数は、SF101及びSF102でそれぞれ70〜82本及び60〜80本の範囲に及んでいた。
【図15】図15は、MDCK- SF103細胞が上皮様形態を有することを示す写真である。写真は、接種後3日目に撮影された。
【図16】図16は、MediV SFM103培地におけるMDCK-SF103細胞の成長曲線である。細胞は約1日の誘導期後、指数関数的増殖へと続き、接種後4日目に静止期に入り、4日目に最大密度〜17×106に達している。
【図17】図17は、MediV SFM103培地におけるMDCK-SF103細胞のグルコース消費、及び乳酸産生のグラフである。対数期中、グルコース消費及び乳酸産生は、お互いに酷似している。すなわち、グルコース濃度が減少したときに乳酸濃度が増加している。
【図18】図18は、MediV SFM103培地におけるMDCK-SF103細胞のグルタミン消費、並びにアンモニア及びグルタミン酸産生のグラフである。アンモニア産生速度は、7日目までほぼ線形的に増加した。グルタミン酸は、本実験では蓄積しなかった。
【図19】図19は、87継代の中期MDCK-SF103細胞100個における染色体数の分布のグラフである。SF103細胞は、78本の最頻値染色体数を有し、カウントされた染色体数は66〜80本の範囲に及んでいた。
【図20A】生産規模の増殖及び精製。図20Aは、数種のワクチンリアソータント株、すなわちB/Victoria/504/2000株(〜8 Log TCID50/mL)、A/Sydney/05/97株(〜7.85 Log TCID50/mL)、及びA/New Caledonia/20/99株(~8.2 Log TCID50/mL)について、Cytodexビーズ上で増殖させたMDCK-SF103細胞の250mLスピナーフラスコから得られた収量のグラフである。
【図20B】生産規模の増殖及び精製。図20Bは、ワクチン物質の工業規模生産に利用することができるようにスケールアップされた細胞培養方法の一つを概説している。
【発明を実施するための形態】
【0019】
発明の詳細な説明
本発明は、一部には、MDCK細胞を非腫瘍形成性の状態に維持する条件の下で培養できるという発見に基づく。本発明は、無血清培地処方物を包含する様々な細胞培養条件に適応し、また本明細書で「本発明の細胞」と呼ぶMDCK細胞株及びその他の細胞型を含む非腫瘍形成性細胞株を提供する。さらに、本発明は、本発明の細胞、及びその他の成分を含む細胞培養(cell culture)組成物を提供する。ここで言うその他の成分は、培地(例えば、本明細書で開示された培地)、培地成分、バッファ、化合物、その他の細胞型、ウイルス物質(例えば、ウイルスゲノム、ウイルス粒子)、及び異種タンパク質を含むが、これらに限定されない。本発明は、MDCK細胞を含む、一以上の特有の性質をもった非腫瘍形成性細胞の培養に有用な方法及び培地処方物も提供する。ここで言う特有の性質は、非腫瘍形成性であること(例えば、ヌードマウスで小結節を形成しないこと)、及び/又は付着性細胞として増殖すること、及び/又は上皮様形態を呈すること、及び/又は様々なウイルスの複製を支持することを含むが、これらに限定はされない。また、前記様々なウイルスは、オルソミクソウイルス(orthomyxovirus)、パラミクソウイルス(paramyxovirus)、ラブドウイルス(rhabdovirus)及びフラボウイルス(flavovirus)を含むが、これらに限られない。本発明の培養条件は、血清含有及び無血清培地処方物と共に動物性タンパク質非含有(APF)製剤を含む。さらに本発明は、MDCK細胞を含む非腫瘍形成性細胞でワクチン物質(例えば、インフルエンザウイルス)を生産する方法、非腫瘍形成性細胞からワクチン物質を調製すること、及び非腫瘍形成性細胞で生産されたワクチン物質を利用してインフルエンザ感染を予防する方法も提供する。本発明の細胞は、他の哺乳動物細胞株(例えば、Vero、PerC6、HEK-293、MRC-5及びWI-38細胞)中で効率的に複製されない低温適応性/温度感受性/弱毒性(ca/ts/att)インフルエンザ株(例えば、FluMist(登録商標)のウイルス株)の生産に特に有用である。
【0020】
<細胞特性>
本発明に係る細胞は、一の実施形態において脊椎動物細胞である。他の実施形態において、本発明の細胞は、例えばハムスター、ウシ、サル又はイヌ由来の哺乳動物細胞であり、特に腎臓細胞又はそれ由来の細胞株である。さらなる他の実施形態において、本発明の細胞は、(例えば、ATCC CCL-34 MDCK由来の)MDCK細胞であり、具体的には本明細書中で「本発明のMDCK細胞」と称され、また「本発明の細胞」の用語に包含される。具体的実施形態において、本発明の細胞はATCC CCL-34 MDCK由来である。本発明の細胞は、当業分野で周知の方法によるCCL-34 MDCK細胞由来の細胞であってもよい。例えば、まずCCL-34 MDCK細胞を血清含有培地(例えば、ダルベッコ改良イーグル培地(DMEM)+10%ウシ胎仔血清(FBS)+4mMグルタミン+4.5g/Lグルコース、又は本明細書に記載の他の培地)で限られた回数継代し、続いて個々の細胞をクローニングし、そのクローンを特徴付けてもよい。倍加時間、発癌性の特徴、及びウイルス生産を含むがこれらに限定されない生物学的及び生理学的に優れた特性をもつクローンを、マスター細胞バンク(master cell bank:MCB)生成用に選択する。一の態様において、本発明の細胞は、選択した培地(例えば、本明細書に記載したような無血清又はAPF培地)での増殖に適応している。このような適応は、個々の細胞のクローニング前、クローニング時、又はクローニング後に起こり得る。特定の実施形態において、本発明の細胞は、MediV SF101、MediV SF102、MediV SF103、MediV SF104、又はMediV SF105で増殖できるように適応している。これらの培地での増殖に適応した本発明の細胞を、本明細書では、それぞれ「MDCK-SF101、MDCK-SF102、MDCK-SF103、MDCK-SF104、そしてMDCK-SF105」細胞と呼び、また、まとめて「MDCK-SF細胞」と呼ぶ。他の実施形態において、本発明の細胞は、血清含有培地(例えばダルベッコ改良イーグル培地(DMEM)+10%ウシ胎仔血清(FBS)+4mMグルタミン+4.5g/Lグルコース)で増殖するように適応しており、そのような細胞を本明細書では「MDCK-S」細胞と呼ぶ。MDCK
-SF細胞及びMDCK-S細胞はまた、「本発明の細胞」及び「本発明のMDCK細胞」の用語に包含される。
【0021】
本発明の特定の実施形態において、細胞は株細胞である。本株細胞は、American Type Culture Collection(10801 University Boulevard, Manassas, Va. 20110-2209)に寄託され、ATCC寄託番号 PTA-6500 (2005年1月5日寄託)、PTA-6501 (2005年1月5日寄託)、PTA-6502(2005年1月5日寄託)、PTA-6503 (2005年1月5日寄託)が割り振られた株細胞であり、また本明細書ではそれぞれ「MDCK-S、MDCK-SF101、MDCK-SF102、及びMDCK-SF103」と呼び、まとめて「本発明のMDCK細胞」と呼ぶ株細胞を含むが、これらに限定はされない。これらの寄託物は、特許手続上の微生物の寄託の国際承認に関するブダペスト条約の条項に基づいて保存されるであろう。一の実施形態において、本発明のMDCK細胞は、ヒト用としてアメリカ食品医薬品局(the U.S. Food and Drug Administration)の承認に適したワクチン物質を調製する上で有用な細胞バンクを作製するのに利用される。
【0022】
株細胞MDCK-S、MDCK-SF101、MDCK-SF102、MDCK-SF103、MDCK-SF104、及びMDCK-SF105は、継代及び一以上の特有の性質に関する選択によって株細胞MDCK(CCL 34)から得られる。その性質は、血清含有培地、又は無血清若しくは動物性タンパク質非含有培地のいずれかで付着性細胞として増殖すること、上皮様形態を呈すること、非腫瘍形成性であること(例えば、ヌードマウスで小結節を形成しないこと)、及び/又は様々なウイルスの複製を支持することを含むが、これらに限定されない。また、前記ウイルスは、オルソミクソウイルス、パラミクソウイルス、ラブドウイルス及びフラボウイルスを含むが、これらに限定されない。
【0023】
一の実施形態において、本発明のMDCK細胞は非腫瘍形成性である。細胞が腫瘍形成性であるかどうかを決定する方法は、当該分野では周知である(例えば、Leightonらの1970, Cancer, 26:1024、及びStilesらの1976, Cancer Res, 36:1353を参照されたい)。アメリカ食品医薬品局で現在推奨のヌードマウスモデルを用いた方法を、以下の実施例2で詳述している。具体的な実施形態において、本発明のMDCK細胞はアダルトヌードマウスモデルで非腫瘍形成性である(前述のStilesらの文献、及び以下の実施例2を参照)。もう一つの具体的実施形態において、本発明のMDCK細胞は、鶏胚へ注射した際、及び/又は漿尿膜に局所的に適用した際に非腫瘍形成性である(前述のLeightonらの文献を参照)。さらなる他の実施形態において、本発明のMDCK細胞はアダルトヌードマウスモデルでは非腫瘍形成性であるが、鶏胚への注射、及び/又は漿尿膜に局所的に適用した場合は非腫瘍形成性ではない。さらに他の実施形態において、本発明のMDCK細胞は、アダルトヌードマウスモデルでも、また鶏胚への注射、及び/又は漿尿膜に局所的に適用した場合にも非腫瘍形成性である。さらなる他の実施形態において、本発明のMDCK細胞は、培地で少なくとも20継代後、少なくとも30継代後、少なくとも40継代後、少なくとも50継代後、少なくとも60継代後、少なくとも70継代後、少なくとも80継代後、少なくとも90継代後、又は少なくとも100継代後も非腫瘍形成性である。さらなる他の具体的実施形態において、培地は本明細書に記載した培地(例えばMedi SF103)である。
【0024】
腫瘍形成性は、当業者に多くの様々な方法で数値化することができる。一般的に利用される方法の一つは、「TD50」値を測定することである。この値は、テストした動物の50%に腫瘍を生じさせるのに必要な細胞数として定義される(例えば、Hill R. The TD50 assay for tumor cells. In: Potten C, Hendry J, editors. Cell clones. London: Churchill Livingstone; 1985. p. 223を参照されたい)。一の実施形態において、本発明のMDCK細胞は、約1010〜約101、約108〜約103、又は約107〜約104のTD50値を有する。具体的な実施形態において、本発明のMDCK細胞は約1010より大きい、約109より大きい、約108より大きい、約107より大きい、約106より大きい、約105より大きい、約104より大きい、約103より大きい、約102より大きい、又は約101より大きい、TD50値を有する。
【0025】
他の実施形態において本発明の非腫瘍形成性細胞は、血清含有培地、又は無血清若しくは動物性タンパク質非含有培地のいずれかで付着性細胞として増殖する。さらなる他の実施形態において、本発明の非腫瘍形成性細胞は上皮様形態を呈する。さらに他の実施形態において、本発明のMDCK細胞は、様々なウイルスの複製を支持する。ここで言うウイルスは、オルソミクソウイルス、パラミクソウイルス、ラブドウイルス及びフラボウイルスを含むが、これらに限定はされない。本発明のMDCK細胞は、一以上の特有の性質をどのような組合せで有していてもよいことが意図されている。ここでいう特有の性質は、非腫瘍形成性であること、付着性細胞として増殖すること、上皮様形態を有すること、及び様々なウイルスの複製を支持することを含むが、これらに限定はされない。
【0026】
本発明のMDCK細胞のあらゆる継代は、各株細胞の完全な系譜を利用できるように十分詳細に記録されていることが意図されている。あらゆる継代の記録は、ワクチン物質の調製のために本発明のMDCK細胞を使用することに対し、アメリカ食品医薬品局や世界中の他の取締機関の承認を円滑にすることができる。
【0027】
他の実施形態において、本発明のMDCK細胞は、微生物汚染(例えば、細菌、ウイルス、真菌の汚染)を含まない。細菌や真菌の汚染の有無を調べる方法は、当業分野で周知であり、通常民間の受託業者(例えば、BioReliance(登録商標);Rockville、MD)によって行われている。一般に承認されている微生物の無菌性及びマイコプラズマテストを、以下の実施例2で詳述している。テストされ得る微生物剤の具体例については、表6に列挙している。
【0028】
さらなる他の実施形態において、本発明のMDCK細胞は、ウイルスの複製を支持する。ここでいうウイルスは、オルソミクソウイルス(インフルエンザA型株及び/又はB型株を含む)、パラミクソウイルス(RSV A型及び/又はB型、ヒトメタニューモウイルス、並びにパラインフルエンザ1、2及び/又は3型を含む)、ラブドウイルス及びフラボウイルスを含むが、これらに限定はされない。具体的な実施例において、本発明のMDCK細胞は、低温適応性/温度感受性(ca/ts)インフルエンザウイルスの複製を支持する。ここでいうインフルエンザウイルスは、例えば、FluMist(登録商標) (Belsheら, 1998, N Engl J Med 338:1405;Nicholら, 1999, JAMA 282:137;Jacksonら, 1999, Vaccine, 17:1905)、及び/又はそれらのウイルスのバックボーンを含む、若しくは一以上の以下の特徴、すなわち低温適応性、弱毒性、及び温度感受性を有するインフルエンザウイルスのバックボーン(若しくは一以上のvRNA断片)を含むリアソータントウイルス等が該当する。ウイルス複製を支持する細胞の能力の指標の一つとして、感染した培養細胞から得られるウイルスの収量がある。ウイルスの収量は、当業者に既知の多くの方法によって測定することができる。例えば、ウイルスの収量は、感染性ウイルス粒子を測定する中間値(50%)組織培養感染量(TCID50)アッセイによりサンプル中に存在するウイルスの濃度を測定することで数値化することができる。TCID50値は、しばしばlog10 TCID50/mLとして報告されいている。一の実施形態において、本発明のMDCK細胞は、インフルエンザウイルス(例えばca/ts株)の複製を少なくとも6.0、少なくとも6.2、少なくとも6.4、少なくとも6.6、少なくとも6.8、少なくとも7.0、少なくとも7.2、少なくとも7.4、少なくとも7.6、少なくとも7.8、少なくとも8.0、少なくとも8.2、少なくとも8.4、少なくとも8.6、少なくとも8.8、少なくとも9.0、少なくとも9.2、少なくとも9.4、少なくとも9.6、又は少なくとも9.8のlog10 TCID50/mLまで支持する。他の実施形態において、本発明のMDCK細胞は、インフル
エンザウイルス(例えばca/ts株)の複製を少なくとも6.0、少なくとも6.2、少なくとも6.4、少なくとも6.6、少なくとも6.8、少なくとも7.0、少なくとも7.2、少なくとも7.4、少なくとも7.6、少なくとも7.8、少なくとも8.0、少なくとも8.2、少なくとも8.4、少なくとも8.6、少なくとも8.8、少なくとも9.0、少なくとも9.2、少なくとも9.4、少なくとも9.6、又は少なくとも9.8のlog10 TCID50/mLにまで支持する。
【0029】
本発明の細胞が一般に細胞培養組成物の一部であることは、当業者にはわかるであろう。細胞培養組成物の成分は、細胞や使用目的によって変化し得る。例えば、培養目的であれば、細胞培養組成物は、本発明の細胞とその細胞を増殖するための適切な培地を含み得る。したがって、本発明は、本発明の細胞及びその他の成分を含む細胞培養組成物を提供する。ここでいうその他の成分は、培地(例えば、本明細書に開示した培地)、培地成分、バッファ、化合物、他の細胞型、ウイルス物質(例えば、ウイルスゲノム、ウイルス粒子)、並びに異種タンパク質を含むが、これらに限定はされない。一の実施形態において、細胞培養組成物は、本発明の細胞、及び培地又はその成分から構成される。細胞培養組成物中に存在する培地は、無血清培地、血清含有培地及びAPF培地を含む。一の実施形態において、細胞組成物は、本明細書で開示する培地(例えば、MediV SF101、MediV SF102、MediV SF103、MediV SF104、若しくはMediV SF105)、又はそれらの成分を含む。
【0030】
<方法及び培地処方物>
本発明は、血清含有培地での非腫瘍形成性MDCK細胞の培養の方法及び培地処方物を提供する。本発明はまた、APF培地処方物を含む無血清培地での非腫瘍形成性MDCK細胞の適応方法、及びその後の培養方法も提供する。本発明のいくつかの態様において、前記培地はMDCK細胞が培養時に一以上の特性を維持するように処方されている。ここでいう一以上の特性は、非腫瘍形成性であること、付着性細胞として増殖すること、上皮様形態を有すること、及び様々なウイルスの複製を支持することを含むが、これらに限られない。本明細書で開示した培地処方物又はそれらの成分は、細胞培養組成物中に存在し得ることが意図されている。
【0031】
血清含有培地処方物は、当該分野で周知である。血清含有培地処方物は、ダルベッコ改良イーグル培地(DMEM)+ウシ胎仔血清(FBS)+グルタミン+グルコースを含むが、これに限定されない。一の実施形態において、FBSは、血清含有培地に約1%〜約20%、約5%〜約15%、又は約5%〜約10%の濃度で存在する。具体的な実施形態において、FBSは、血清含有培地に10%の濃度で存在する。他の実施形態において、グルタミンは、血清含有培地に約0.5mM〜約10mM、約1mM〜約10mM、又は約2mM〜約5mMの濃度で存在する。具体的な実施形態において、グルタミンは、血清含有培地に4mMの濃度で存在する。さらなる他の実施形態において、グルコースは、血清含有培地に約1g/L〜約10g/L、又は約2g/L〜約5g/L間の濃度で存在する。具体的な実施形態において、グルコースは、血清含有培地に4.5g/Lの濃度で存在する。さらなる他の実施形態において、血清含有培地処方物は、約1%〜約20%の濃度のFBS、約0.5mM〜約10mMの濃度のグルタミン、及び約1g/L〜約10g/Lの濃度のグルコースを含む。具体的な実施形態において、血清含有培地処方物は、ダルベッコ改良イーグル培地(DMEM)+10%ウシ胎仔血清(FBS)+4mMグルタミン+4.5g/Lグルコースからなる。DMEMは、例えばGibco/BRL社(Cat. No.11965-084)を含む多数の商業的供給源から容易に調達できる。FBSは、例えばJRH Biosciences社(Cat. No.12107-500M)を含む多数の商業的供給源から容易に調達できる。FBSは、動物細胞培地で最も一般的に用いられる補助添加剤であるが、新生の子牛、ウマ及びヒトを含むその他の血清源も日常的に使用され、本発明に包含される。
【0032】
一の実施形態において、本発明のMDCK-S血清適応非腫瘍形成性細胞は、血清を追加した化学的に規定された培地中でAmerican Type Culture Collection(ATCC CCL34)より入手したイヌ腎臓由来(MDCK)細胞を培養することによって、当該細胞から得られる。具体的な実施形態において、MDCK細胞(ATCC CCL34)を、血清を補充した化学的に規定された培地中で増やし、以下のようにしてMDCK-S株を作製する。すなわち、MDCK (ATCC CCL34)細胞を必要に応じてウシ胎仔血清(10% v/v)、4mM グルタミン、及び4.5 g/Lグルコースを補足したダルベッコ改良イーグル培地(DMEM)に継代し、MDCK-Sと呼ぶ冷凍プレマスター細胞バンク(PreMCB)を調製するのに十分な細胞を得る。他の具体的な実施形態において、細胞は下記の実施例2で詳述する工程を用いて培養される。特に、MDCK-S血清適合細胞を、PreMCBのバイアルから他へ20回以上継代すること、並びにアダルトヌードマウスモデルにおけるインビボでの腫瘍形成性、及び核型分析での核型をテストすることが意図されている。いくつかの実施形態において、増やしたMDCK-S細胞は、アダルトヌードマウスの皮下に注射しても小結節を生じず、また約60本〜88本以下の、約65本〜85本以下の、約65本〜80本以下の、又は約70本〜85本以下の染色体数の範囲で78本の最頻値染色体数を有している。一の実施形態において、MDCK-S細胞は、培地(例えば、本明細書に記載された培地)で、少なくとも20回の継代後、少なくとも30回の継代後、少なくとも40回の継代後、少なくとも50回の継代後、少なくとも60回の継代後、少なくとも70回の継代後、少なくとも80回の継代後、少なくとも90回の継代後、又は少なくとも100回の継代後でも非腫瘍形成性である。
【0033】
当業者であれば、組織培養を適用する上で血清又は動物抽出物の使用に欠点があることがわかるであろう(Lambert, K.J.ら, In: Animal Cell Biotechnology, Vol 1、Spier, R.E.ら編, Academic Pres New York, pp. 85-122 (1985))。例えば、これらの補助添加剤の化学組成は、同一生産業メーカーものであってもロット間で変わり得る。さらに、動物又はヒト起源の補助添加剤は、外来物質(例えば、マイコプラズマ、ウイルス、及びプリオン)で汚染されてもいるかもしれない。このような汚染された補助添加剤が細胞培地処方物に使用されれば、これらの作用物質によって培養細胞の健康状態が著しく損なわれる。さらに、外来作用物質で汚染された培養液中で作られた物質が細胞療法やその他の臨床応用で使用された場合、これらの物質は健康上のリスクを引き起こすかもしれない。最大の懸念は、動物の海綿状脳症やヒトのクロイツフェルト・ヤコブ症の原因となるプリオンの存在である。したがって、本発明はさらに無血清培地処方物を提供する。
【0034】
本発明の無血清培地処方物は、MediV SF101 (Taub’s培地+植物加水分解物)、MediV SF102 (Taub’s培地+脂質)、MediV SF103 (Taub’s培地+脂質+植物加水分解物)、MediV SF104 (Taub’s培地+脂質+植物加水分解物+成長因子)、及びMedi SF105(トランスフェリンをクエン酸鉄アンモニウム/トロポロン、又は硫酸鉄アンモニウム/トロポロンに置換した以外はMediV SF104と同じ)を含むが、これらの限定はされない。特に、Taub’s SF培地(TaubのSF培地;TaubとLivingston, 1981, Ann NY Acad Sci.,372:406)はホルモン、すなわち5μg/mLインスリン、5μg/mL トランスフェリン、25ng/mL プロスタグランジンE1、 50nMヒドロコルチゾン、5pMトリヨードチロニン、並びに10nM Na2SeO3、4.5g/Lグルコース、2.2g/L NaHCO3及び4mM L-グルタミンを追加したDMEM培地とHamの F12培地の50:50の混合物であることが意図されている。TaubのSF培地も、本明細書ではTaub’s SF培地、又は単に「Taub’s」と呼ぶ。
【0035】
植物加水分解物は、一以上の以下の植物、すなわち、トウモロコシ、綿実、エンドウ豆、大豆、麦芽、ジャガイモ、及び小麦由来の加水分解物を含む。ただし、これらに限定されない。植物加水分解物は、酵素的加水分解で作ることができ、通常、ペプチドの混合物、遊離アミノ酸、及び成長因子を含んでいる。植物加水分解物は、例えば、Marcor Development社、HyClone社、及びOrgano Technie社を含む多くの商業的供給源から容易に調達できる。酵母加水分解物も植物加水分解物の代わりに、又はそれと組み合わせて利用できることが意図されている。酵母加水分解物は、例えば、Sigma-Aldrich社、USB Corp社、Gibco/BRL社、その他を含む商業的供給源から容易に調達できる。
【0036】
培地補助添加剤に用いることのできる脂質は、動物及び植物由来の化学的に規定された脂質補助添加剤と合成的に誘導された脂質を含むが、これらに限定はされない。脂質補助添加剤中に存在し得る脂質には、コレステロール、飽和及び/又は不飽和脂肪酸(例えば、アラキドン酸、リノール酸、リノレン酸、ミリスチン酸、オレイン酸、パルミチン酸、及びステアリン酸)が含まれるが、これらに限られない。コレステロールは、100×ストックの脂質補助添加剤中に0.10mg/ml〜0.40mg/mlの濃度で存在し得る。脂肪酸は、100×ストックの脂質補助添加剤中に1μg/ml〜20μg/mlの濃度で存在し得る。培地処方物に適した脂質は、例えばHyClone社、Gibco/BRL社、及びSigma-Aldrich社を含む多くの商業的供給源から容易に調達できる。
【0037】
一の実施形態において、Taub’s培地には植物加水分解物が少なくとも0.5g/L、少なくとも1.0g/L、少なくとも1.5g/L、少なくとも2.0g/L、少なくとも2.5g/L、少なくとも3.0g/L、少なくとも5.0g/L、少なくとも10g/L、又は少なくとも20g/Lの最終濃度で追加されている。具体的な実施形態において、Taub’s培地には小麦の加水分解物が追加されている。他の具体的な実施形態において、Taub’s培地には小麦の加水分解物が2.5g/Lの最終濃度で追加されている。本発明は、本明細書でMediV SFM 101と呼ばれる無血清培地であって、小麦の加水分解物を最終濃度2.5g/Lで追加したTaub’s培地からなる無血清培地を提供する。
【0038】
他の実施形態において、Taub’s培地には脂質混合物が製造メーカーの推奨最終濃度の少なくとも50%、少なくとも60%、少なくとも70%、少なくとも80%、少なくとも90%、少なくとも100%、少なくとも125%、少なくとも150%、少なくとも200%、少なくとも300%の最終濃度で追加されている。具体的な実施形態において、Taub’s培地には化学的に規定された脂質混合物が追加されている。他の具体的な実施形態において、Taub’s培地には、化学的に規定された脂質混合物が製造メーカーの推奨最終濃度の100%の最終濃度で補充されている(例えば、製造メーカーから入手した100×ストックであれば、最終濃度1×で培地に補充される)。本発明は、本明細書でMediV SFM 102と呼ばれる無血清培地であって、化学的に規定された脂質混合物を製造メーカーの推奨最終濃度の100%の最終濃度で追加したTaub’s培地からなる無血清培地を提供する。
【0039】
さらなる他の実施形態において、Taub’s培地には植物加水分解物が少なくとも0.5g/L、少なくとも1.0g/L、少なくとも1.5g/L、少なくとも2.0g/L、少なくとも2.5g/L、少なくとも3.0g/L、少なくとも5.0g/L、少なくとも10g/L、又は少なくとも20g/Lの最終濃度で、かつ脂質混合物が製造メーカーの推奨濃度の少なくとも50%、少なくとも60%、少なくとも70%、少なくとも80%、少なくとも90%、少なくとも100%、少なくとも125%、少なくとも150%、少なくとも175%、又は少なくとも200%の最終濃度で、追加されている。具体的な実施形態において、Taub’s培地には小麦の加水分解物、及び化学的に規定された脂質混合物が追加されている。他の具体的な実施形態において、Taub’s培地には小麦の加水分解物が2.5g/Lの最終濃度で、かつ化学的に規定された脂質混合物が製造メーカーの推奨最終濃度の100%の最終濃度で追加されている。本発明は、本明細書でMediV SFM 103と呼ばれる無血清培地であって、小麦の加水分解物を最終濃度2.5g/Lで、かつ化学的に規定された脂質混合物を製造メーカーの推奨最終濃度の100%の最終濃度で追加したTaub’s培地からなる無血清培地を提供する。
【0040】
さらなる他の実施形態において、Taub’s培地には成長ホルモンが追加されている。使用することのできる成長ホルモンには、上皮成長因子(EGF)、インスリン成長因子(IGF)、形質転換増殖因子(TGF)、及び線維芽細胞増殖因子(FGF)が含まれるが、これらに限定はされない。具体的な実施形態において、成長ホルモンは上皮成長因子(EGF)である。一の実施形態において、Taub’s培地には、成長因子が約0.1〜約50.0ng/ml、約0.5〜約25.0ng/ml、約1.0〜約20ng/ml、約5.0〜約15.0ng/ml、又は約8〜約12ng/mlの最終濃度で添加されている。具体的な実施形態において、Taub’s培地にはEGFが約10ng/mlの最終濃度で追加されている。さらなる他の実施形態において、Taub’s培地には成長因子が約0.1〜約50.0ng/ml、約0.5〜約25.0ng/ml、約1.0〜約20ng/ml、約5.0〜約15.0ng/ml、又は約8〜約12.0ng/mlの最終濃度で、かつ植物加水分解物が少なくとも0.5g/L、少なくとも1.0g/L、少なくとも1.5g/L、少なくとも2.0g/L、少なくとも2.5g/L、少なくとも3.0g/L、少なくとも5.0g/L、少なくとも10g/L、又は少なくとも20g/Lの最終濃度で、かつ脂質混合物が製造メーカーの推奨最終濃度の少なくとも50%、少なくとも60%、少なくとも70%、少なくとも80%、少なくとも90%、少なくとも100%、少なくとも125%、少なくとも150%、少なくとも175%、又は少なくとも200%の最終濃度で、追加されている。他の具体的な実施形態において、Taub’s培地には小麦の加水分解物が2.5g/Lの最終濃度で、かつ化学的に規定された脂質混合物が製造メーカーの推奨最終濃度の100%の最終濃度で、かつEGFが約10ng/mlの最終濃度で、追加されている。本発明は、本明細書でMediV SFM 104と呼ばれる無血清培地であって、小麦の加水分解物を最終濃度2.5g/Lで、かつ化学的に規定された脂質混合物を製造メーカーの推奨最終濃度の100%の最終濃度で、かつEGFを約10ng/mlの最終濃度で追加したTaub’s培地からなる無血清培地を提供する。
【0041】
当業者であれば、動物性タンパク質フリーの培地処方物がワクチンの製造に使用するウイルスの生産に望ましいことも理解できるであろう。したがって、いくつかの実施形態において、本明細書で開示された無血清培地(例えば、MediV SF101、MediV SF102、MediV SF103、MediV SF104、及びMedi SF105)の1以上又は全ての動物由来の成分は、非動物性誘導体で置換される。例えば、市販の非動物源由来の組換えインスリン(例えば、Biological Industries社のCat. No. 01-818-1)は、動物源から得られたインスリンの代わりに利用することができる。同様に、鉄結合剤(例えば、米国特許第5,045,454号;同第5,118,513号;同第6,593,140号;及びPCT国際公開第WO 01/16294号を参照されたい)を動物源由来のトランスフェリンの代わりに利用することができる。一の実施形態において、本発明の無血清培地処方物は、トランスフェリンの代わりにトロポロン(2-ヒドロキシ-2,4,6-シクロヘプタトリエン-1)及び鉄源(例えばクエン酸鉄アンモニウム、硫酸鉄アンモニウム)を含む。例えば、トロポロン又はトロポロン誘導体は、培地中に存在する鉄に対して、約5対1〜約70対1、又は約10対1〜約70対1のモル比のような過剰モル濃度で存在し得る。したがって、培地中の鉄濃度は0.3μM前後であり、トロポロン又はその誘導体は、約1.5μMから約20μM、例えば約3μMから約20μMの濃度で使用することができる。前記鉄は、例えば、硫酸鉄、塩化第二鉄、硝酸鉄、特にクエン酸鉄アンモニウム等の培地中の鉄単塩又は鉄錯塩を使用することによって生じる二価又は三価の鉄イオンとして存在することができる。本発明は、本明細書でMediV SFM 105と呼ばれる無血清培地でえあって、小麦の加水分解物を最終濃度2.5g/Lで、かつ化学的に規定された脂質混合物を製造メーカーの推奨最終濃度の100%の最終濃度で、かつEGFを約10ng/mlの最終濃度で、かつクエン酸鉄アンモニウム:トロポロン又は硫酸鉄アンモニウム:トロポロンを10:1〜70:1の比率で追加したトランスフェリンを含まないTaub’s培地からなる無血清培地を提供する。
【0042】
一の実施形態において、MDCK-SF101、MDCK-SF102、MDCK-SF103、MDCK-SF104、及びMDCK-SF105無血清培地に適応した非腫瘍形成性細胞(本明細書では、まとめてMDCK-SFと呼ぶ)は、American Type Culture Collection(ATCC CCL34)株より入手したイヌ腎臓由来(MDCK)細胞を、少なくとも1回の継代については血清を加えた化学的に規定された培地中で、その後それらを例えば上記無血清培地等の無血清培地で継代することによって得られる。具体的な実施形態において、MDCK細胞(ATCC CCL34)を無血清培地に対して以下のように適応させて、MDCK-SF細胞株を作製する。すなわち、MDCK細胞(ATCC CCL34)をウシ胎仔血清(10% v/v)、4mMグルタミン及び4.5g/Lグルコースを追加したダルベッコ改良イーグル培地(DMEM)で少なくとも1回継代し、その後無血清培地で継代する。MDCK-SF細胞をその後必要に応じて無血清培地で継代し、冷凍プレマスター細胞バンク(PreMCB)を調製するのに十分な細胞を得る。いくつかの実施形態において、前記細胞を、血清含有培地(例えば、ウシ胎仔血清(10% v/v)、4mMグルタミン及び4.5g/Lグルコースを追加したダルベッコ改変イーグル培地(DMEM))で1〜5回、4〜10回、9〜20回、又は20回以上継代する。その後、無血清培地(例えば、MediV SF101、MediV SF102、MediV SF103、MediV SF104、及びMedi SF105)で継代する。特に、MDCK-SF無血清培地に適応した細胞をPreMCBのビンから他に20回以上継代すること、並びにアダルトヌードマウスモデルのインビボでの腫瘍原性、及び核型測定での核特性をテストすることが意図されている。いくつかの実施形態において、増やしたMDCK-SF細胞は、アダルトヌードマウスの皮下に導入しても小結節を生じず、及び/又は78本の最頻染色体数を有している。他の実施形態において、増やしたMDCK-SF細胞は、わずか約60本〜88本の、わずか約65本〜85本の、わずか65本〜85本の、わずか約65本〜80本の、又はわずか約70本〜85本の染色体数の範囲で、78本の最頻染色体数を有している。一の実施形態において、MDCK-SF細胞は、培地(例えば、本明細書に記載された培地)中で、少なくとも100回の継代後、少なくとも90回の継代後、少なくとも80回の継代後、少なくとも70回の継代後、少なくとも60回の継代後、少なくとも50回の継代後、少なくとも40回の継代後、少なくとも30回の継代後、又は少なくとも20回の継代後も非腫瘍形成性である。
【0043】
一の実施形態において、MDCK-SF細胞の誘導用に使用される無血清培地は、MediV SF101である。他の実施形態において、MDCK-SF細胞の誘導用に使用される無血清培地はMediV SF102である。さらに、他の実施形態において、MDCK-SF細胞の誘導用に使用される無血清培地はMediV SF103である。さらなる他の実施形態において、MDCK-SF細胞の誘導用に使用される無血清培地はMediV SF104である。他の実施形態において、MDCK-SF細胞の誘導用に使用される無血清培地はMediV SF105である。更なる他の実施形態において、MDCK-SF細胞の誘導用に使用される無血清培地はAPF培地である。本明細書に記載された培地を処方して動物性タンパク質を排除できることが意図されている。例えば、ウシのトランスフェリンを非動物源由来の組換えトランスフェリンと置換することができる。
【0044】
<培養条件>
本発明は、血清含有又は無血清培地処方物(上記)でのMDCK細胞(好ましくは非腫瘍形成性)、及びその他の動物細胞(腫瘍形成性又は非腫瘍形成性)の培養方法を提供する。特に、さらなる培養条件として、MDCK-S細胞及びMDCK-SF細胞を非腫瘍形成性状態で維持する役割を果たし得ることが意図されている。これらの培養条件は、付着表面の選択、細胞密度、温度、CO2濃度、培養方法、溶存酸素含有量、及びpHを含む。ただし、これらに限定はされない。
【0045】
特に、当業者が多数の方法の培養条件を適応させて、本発明のMDCK細胞の増殖を最適にしてもよいことが意図されている。このような適応はまた、ウイルス物質(例えば、ウイルス)の生産増加に繋がり得る。あるいは、当業者が培養条件を適応させて、細胞の増殖に関係なく本発明のMDCK細胞からワクチン物質の生産を最適にすることができる。これらの培養条件は、付着表面、細胞密度、温度、CO2濃度、培養方法、溶存酸素含有量、及びpHを含むが、これらの限定はされない。
【0046】
一の実施形態において、本発明のMDCK細胞は、付着性細胞としてそれらが付着する表面上で培養される。組織培養細胞が増殖可能な付着表面は、当業分野では周知である。付着表面は、表面修飾したポリスチレンプラスチック、タンパク質被覆表面(例えば、フィブロネクチン及び/又はコラーゲン被覆ガラス/プラスチック)と市販の多岐にわたるマイクロキャリア(例えば、Dormacell [Pfeifer & Lange社]等のDEAE-デキストランマイクロキャリアビーズ; Superbead [Flow Laboratories社];Hillex [SoloHill, Ann Arbor]等のスチレン共重合体トリメチルアミンビーズ)を含むが、これらの限定はされない。マイクロキャリアビーズは、小球(直径100〜200ミクロンの範囲)であって、細胞培養量につき付着性細胞の増殖のための大きな表面積領域を提供する。例えば、培地1リットルは、8000平方センチメートルを越える増殖表面を提供する2000万個以上のビーズを含むことができる。付着表面の選択は、本発明のMDCK細胞の培養に使用する方法によって決まり、また当業者が決めることができる。本発明による工程の過程で使用することのできる適切な培養容器は、当業者に既知の全ての容器である。例えば、スピナーボトル、ローラーボトル、培養槽、又は生物反応槽(バイオリアクター)等が該当する。例えばワクチン生産用のようにウイルスを量産するためには、細胞をバイオリアクターや培養槽で培養することが望ましい。生物反応槽は、1リットル未満から100リットルを越える量まで市販されている。例えば、Cyto3バイオリアクター(Osmonics社, Minnetonka, MN);NBSバイオリアクター(New Brunswick Scientific社,Edison, NJ);B.Braun Biotech International社の研究・業務用 バイオリアクター(B. Braun Biotech社,Melsungen, Germany)が挙げられる。
【0047】
一の実施形態において、本実施形態のMDCK細胞は、付着性細胞としてバッチ培養システムで培養される。さらに他の実施形態において、本発明のMDCK細胞は、付着性細胞として灌流培養システムで培養される。特に、本発明のMDCK細胞は、ワクチン物質(例えば、ウイルス)の生産に灌流システム(例えば、攪拌培養槽内で、例えば遠心、濾過、スピンフィルターといったような当業者に既知の細胞維持方式を用いて)で培養され得ることが意図されている。
【0048】
一の実施形態において、本発明のMDCK細胞は、少なくとも1%、少なくとも2%、少なくとも3%、少なくとも4%、少なくとも5%、少なくとも6%、少なくとも7%、少なくとも8%、少なくとも9%、少なくとも10%、又は少なくとも20%のCO2濃度で培養される。
【0049】
一の実施形態において、溶存酸素(dissolved oxygen:DO)濃度(pO2値)は、本発明のMDCK細胞の培養中で適宜調節される。その濃度範囲は、(空気飽和に基づいて)5%〜95%、又は10%〜60%である。具体的な実施形態において、溶解酸素(DO)濃度(pO2値)は、少なくとも10%、少なくとも20%、少なくとも30%、少なくとも50%又は少なくとも60%である。
【0050】
他の実施形態において、本発明のMDCK細胞の培養に用いる培地のpHは、培養中に調節される。その範囲は、pH 6.4〜pH 8.0又はpH 6.8〜pH 7.4である。具体的な実施形態において、本培地のpHは、少なくとも6.4、少なくとも6.6、少なくとも6.8、少なくとも7.0、少なくとも7.2、少なくとも7.4、少なくとも7.6、少なくとも7.8、又は少なくとも8.0である。
【0051】
さらなる実施形態において、本発明のMDCK細胞は、25℃〜39℃の温度で培養される。特に、培養温度は、所望の工程によって変えることができることを意図している。例えば、本発明のMDCK細胞は、細胞増殖のため37℃で増やし、その後ワクチン物質(例えば、ウイルス)生産のため、より低い温度(例えば、25℃〜30℃)で増やすことができる。他の実施形態において、前記細胞は、ワクチン物質の生産のために30℃未満、31℃未満、32℃未満、33℃未満、又は34℃未満で培養される。他の実施形態において、前記細胞は、ワクチン物質の生産のために30℃、31℃、32℃、33℃、又は34℃で培養される。
【0052】
ワクチン物質(例えば、ウイルス)を生産するため、特に本発明のMDCK細胞が培地を容易に交換できる(例えば、灌流システムで)ように培養されることを意図している。細胞を非常に高い密度、例えば1×106〜25×106細胞/mLまで培養することができる。
【0053】
培地のグルコース、グルタミン、乳酸含有量とpH及びpO2値、及び攪拌等の当業者に既知の他のパラメータを本発明のMDCK細胞の培養中に細胞密度、及び/又はウイルス生産量を最適化するために容易に操作することができる。
【0054】
<ワクチン物質(例えばウイルス)の生産>
本発明は、本発明のMDCK細胞を使用する細胞培養でのウイルス生産の方法(本明細書では以降「本発明の方法」と呼ぶ)を提供する。一の実施形態において、本方法は以下のステップ、すなわち
i) 培養培地中で本発明のMDCK細胞の増殖させるステップ、
ii) 細胞にウイルスを感染させるステップ、及び
iii) さらなる培養期の後、非腫瘍形成性細胞内で複製されたウイルスを単離するステップ、
を含む。
【0055】
一の実施形態において、本発明のMDCK細胞は、ステップ(i)で付着性細胞として増殖する。本発明のMDCK細胞は、前記方法の過程で上記培地を含むがそれに限定されないいずれの培地においても培養することができる。いくつかの実施形態において、本発明のMDCK細胞は、前記方法の過程において例えばMediV-SF101、MediV-SF102、MediV-SF103、MediV-SF104、MediV-SF105及びそれらのAPF処方物等の無血清培地で培養される。任意で、本発明のMDCK細胞を、前記方法の過程において血清含有培地(例えば、DMEM+10%FBS+4mMグルタミン+4.5g/Lグルコース)で培養することができる。例えば、温度、pH、pO2値、CO2濃度、及び細胞密度等のさらなる培養条件については、上記で詳細に述べている。当業者は、ウイルス生産のために本発明のMDCK細胞の増殖についての培養条件の組合せを確立することができる。
【0056】
ウイルスによる感染前の細胞増殖用温度は、一の形態において22℃〜40℃である。いくつかの実施形態において、ウイルスによる感染前の細胞増殖用温度は、39℃未満、38℃未満、37℃未満、36℃未満、35℃未満、34℃未満、33℃未満、32℃未満、30℃未満、28℃未満、26℃未満、又は24℃未満である。細胞増殖のための培養(ステップ(i))は、前記方法の一の実施形態において灌流システムで行われる。例えば攪拌培養槽内で、例えば遠心、濾過、スピンフィルター、マイクロキャリアといったような当業者に既知の細胞維持方式を用いて行われる。
【0057】
この場合、前記細胞を1〜20日間、又は3〜11日間増やす。培地の交換は、この過程で一日あたり0〜培養槽容量の約1〜5倍に増やして行われる。前記細胞は、この方法で例えば最大で少なくとも1×106〜25×106細胞/mLの高細胞密度まで増大される。灌流システムにおける培養中の灌流速度は、細胞カウント、培地中のグルコース、グルタミン又は乳酸含有量によって、そして当業者に既知の他のパラメータによって、調節することができる。さらに、本発明による方法のステップ(i)の細胞は、バッチ工程で培養される。
【0058】
本発明による方法の一の実施形態において、pH、pO2値、グルコース濃度、及びステップ(i)で使用される他の培地パラメータは、当業者に既知の方法を用いて上述のように培養中に調節される。
【0059】
他の実施形態において、ウイルスによる細胞の感染を約0.0001〜約10、約0.0005〜約5、又は約0.002〜約0.5のM.O.I.(multiplicity of infection:感染多重度)で行われる。さらに他の実施形態において、ウイルスによる細胞の感染は、0.0001〜10、0.0005〜5、又は0.002〜0.5のM.O.I.(multiplicity of infection:感染多重度)で行う。感染後、感染した培養細胞をさらに培養して、ウイルスを複製させる。具体的には最大細胞変性効果、又はウイルス抗原の最大量を検出することができるまで培養する。一の実施形態において、感染後に前記細胞を22℃〜40℃の温度で培養する。いくつかの実施形態において、ウイルスによる感染後、前記細胞を39℃未満、38℃未満、37℃未満、36℃未満、35℃未満、34℃未満、33℃未満、32℃未満、30℃未満、28℃未満、26℃未満、又は24℃未満の温度で培養する。他の実施形態において、感染後に前記細胞を33℃以下の温度で培養する。さらに他の実施形態において、感染後に前記細胞を31℃の温度で培養する。いくつかの実施形態において、前記細胞の培養を2〜10日間行う。培養は、灌流システムで、又は任意でバッチ方式で実施することができる。
【0060】
ウイルスによる感染後の細胞の培養(ステップ(iii))を、上記のようにpH及びpO2値を維持するように同様に行う。本方法のステップ(iii)による細胞培養又はウイルス複製の間、抗原収量を最適化するために、新たに調製された培地、培地濃縮物で、又はアミノ酸、ビタミン、脂質画分、リン酸等の既知構成成分で、細胞培地を置換することも可能である。前記細胞を、さらなる培地又は培地濃縮物の添加により数日にわたって徐々に希釈するか、さらなる灌流中にあるいは培地又は培地濃縮物でインキュベートすることができる。灌流速度は、この場合、細胞数、培地中のグルコース、グルタミン、乳酸若しくは乳酸デヒドロゲナーゼの含有量、又は当業者に既知の他のパラメータを用いて調節することができる。灌流システムとフィード−バッチ(fed-batch)方式の組合せが、さらに可能である。
【0061】
本発明方法の一の実施形態において、生産されたウイルスの回収と単離(ステップ(iii))は、ウイルスが適切な収量で生産されるのに十分な期間後に行われる。例えば、感染後2〜10日間、又は場合により3〜7日間等が挙げられる。本発明方法の一の実施形態において、生産されたウイルスの回収と単離(ステップ(iii))は、感染の2日後、3日後、4日後、5日後、6日後、7日後、8日後、9日後又は10日後に行われる。
【0062】
本発明のMDCK細胞で生産され得るウイルスは、動物ウイルス、すなわちオルソミクソウイルス科、パラミクソウイルス科、トガウイルス科、ヘルペスウイルス科、ラブドウイルス科、レトロウイルス科、レオウイルス科、フラビウイルス科、アデノウイルス科、ピコルナウイルス科、アレナウイルス科、及びポックスウイルス科の各科に含まれるウイルスを含むが、これらに限定されない。
【0063】
細胞培養でインフルエンザウイルスを生産する系も、近年になって開発された(例えば、Furminger著, in Textbook of Influenza,Nicholson編,Webster and Hay, pp.324-332, Blackwell Science (1998); Mertenら著. in Novel Strategies in The Design and Production of Vaccines,Cohen & Shafferman編,pp.141-151, Kluwer Academic (1996)を参照されたい)。これらの方法は、主として選択したウイルス株により適当な不死化宿主細胞が感染することを必要とする。これらの方法により鶏卵におけるワクチン生産に関連する多くの問題を排除できるが、インフルエンザの全ての病原性株が十分に増殖し、また従来の組織培養方法により生産することができるというわけではない。さらに、例えば弱毒化された生ワクチンの生産に適した弱毒性、温度感受性、及び低温適応性といった所望の性質をもつ多数の株を、従来方法を用いた組織培養では、特に工業規模でうまく増やすことができなかった。
【0064】
本発明は、いくつかの非腫瘍形成性MDCK細胞株を提供する。これらの株は、培養時に血清含有、又は無血清のいずれの培地中でも増殖するように適応しており、またインフルエンザを含むがそれに限定されないウイルスの複製を支持することができる。これらの細胞株は、ワクチン物質として利用するための細胞培養でウイルスを経済的に複製するのに適している。本発明のMDCK細胞は、従来の細胞株(下記実施例1を参照)を用いた場合は十分に増殖しない低温適応性、温度感受性(ca/ts)のインフルエンザ株(例えば、FluMist(登録商標)に含まれるインフルエンザ株)の生産に特に有効である。さらに、本発明のMDCK細胞は、ヒトでも病気の原因となり得るトリインフルエンザウイルス(例えば、「パンデミック(世界的流行)」株)のように孵化卵で増殖できないインフルエンザ株の生産に有効である。
【0065】
本発明のMDCK細胞内で本発明の方法によって生産することができるインフルエンザウイルスは、リアソータント(reassortant)ウイルスを含むが、それに限定はされない。当該ウイルスは、弱毒性、温度感受性、低温適応性(ca/ts/att)のマスター株の中で選択された赤血球凝集素ヘマグルチニン及び/又はノイラミニダーゼ抗原が合体したウイルスである。例えば、ウイルスは、一以上の、例えば温度感受性(ts)、低温適応性(ca)、又は弱毒性(att)を有するマスター株のバックボーン(又は一以上のvRNAセグメント)を含み得る(例えば、A/Ann Arbor/6/60、B/Ann Arbor/1/66、PR8、B/Leningrad/14/17/55、B/14/5/1、B/USSR/60/69、B/Leningrad/179/86、B/Leningrad/14/55、B/England/2608/76等)。卵又は細胞株のどちらかのリアソータントインフルエンザワクチン株の生産方法は、当該分野で既知である。また当該方法は、例えばKilbourne, E.D著. in Vaccines (2nd Edition), PlotkinとMortimer編, WB Saunders Co. (1988)、及び国際出願、国際公開第WO 05/062820号及び同第WO 03/091401号で開示された方法を含む。本発明のMDCK細胞内で本発明の工程により生産することができるその他のインフルエンザウイルスは、異種遺伝子産物を発現することができる組換えインフルエンザウイルスを含む。当該ウイルスについては、例えば、米国特許公開第2004/0241139号、及び同第2004/0253273号を参照されたい。
【0066】
一の実施形態において、前記細胞を上記のように増殖(成長)し(ステップ(i))、その後細胞をインフルエンザウイルスで感染させる、(ステップ(ii))。いくつかの実施形態において、前記感染を0.0001〜10、0.0005〜5、又は0.002〜0.5のM.O.I.(感染多重度)で行う。他の実施形態において、前記感染を約0.0001〜約10、約0.0005〜約5、又は約0.002〜約0.5のM.O.I.(感染多重度)で行う。任意でヘマグルチニン(HA0)前駆体タンパク質の切断、及びそれによる細胞上へのウイルスの吸着を引き起こすプロテアーゼが添加される。プロテアーゼの添加は、インフルエンザウイルスによる細胞の感染(ステップ(ii))の直前、同時、又は直後に本発明に従って行われる。前記添加を感染と同時に行う場合には、プロテアーゼを細胞培養液に直接添加して感染させることも、又は例えば濃縮物としてウイルスの接種と共に添加し感染させることも可能である。前記プロテアーゼは、本発明のいくつかの局面で、セリンプロテアーゼ、システインプロテアーゼ、又はアスパラギンプロテアーゼである。一の実施形態において、トリプシンが使用される。具体的な実施形態において、TPCK処理したトリプシンが使用される。
【0067】
一の実施形態において、トリプシンは細胞培養液に1〜5000mU/ml、5〜1000mU/ml、又は100〜500mU/mlの最終濃度で添加される。さらなる実施形態において、トリプシンは細胞培養液に1〜200μg/ml、5〜50μg/ml、又は5〜30μg/mlの最終濃度で添加される。
【0068】
本発明に係る方法のステップ(iii)で感染した細胞をさらに培養する間に、トリプシン再活性化を実施することができる。すなわち、バッチ工程の場合にはトリプシンの新たな添加によって、又は灌流システムの場合にはトリプシン溶液の連続的添加又は断続的添加によって行われる。
【0069】
感染後、感染した培養細胞をさらに培養して、ウイルスを複製させる。具体的には最大細胞変性効果、又はウイルス及び/又はウイルス抗原の最大量を検出することができるまで培養する。いくつかの実施形態において、細胞の培養を2〜10日間行う。培養は、灌流システムで、又は任意でバッチ工程で順番に行うことができる。さらなる実施形態において、細胞をインフルエンザウイルスでの感染後25℃〜36℃、又は29℃〜34℃の温度で培養する。33℃よりも低い温度での、具体的には上記で示した温度範囲内での感染細胞の培養は、例えばB型株のようないくつかのインフルエンザウイルスの高い生産収量をもたらす。それ故、35℃よりも低い温度での感染細胞の培養は、温度感受性、低温適応性(ts/ca)インフルエンザウイルスの生産が意図されている。ts/caウイルスが弱毒化(att)され得ることも意図されている。他の実施形態において、細胞を、ts/caウイルス株の生産のため30℃未満、31℃未満、32℃未満、33℃未満、又は34℃未満の温度で培養する。具体的な実施形態において、細胞をB型インフルエンザウイルス株の生産用に31℃の温度で培養する。
【0070】
インフルエンザウイルスで感染後の細胞培養(ステップ(iii))は、例えば上記のように順次行われる。
【0071】
本発明方法の一の実施形態において、生産されたインフルエンザの回収と単離(ステップ(iii))は、感染の2〜10日後、又は3〜7日後等のウイルスが適当な収量で生産されるのに十分な期間後に行われる。ウイルスは、通常、感染細胞が増殖した培地から回収される。通常はインフルエンザウイルスの濃縮前に粗製培地を清浄化する。一般的な方法は、濾過、限外濾過、硫酸バリウムへの吸着及び溶出、並びに遠心を含む。例を挙げると、感染培養物由来のクルードな培地を、まず例えば1000〜2000×gで、細胞片及びその他の大型粒状物質を除くのに十分な時間(例えば、10〜30分)遠心して清浄化することができる。あるいは、培地を0.8μmの酢酸セルロースフィルターに通して濾過して、無傷の細胞及びその他の大型粒状物質を除く。場合によりその後清浄化した培地の上清を、例えば15,000×gで約3〜5時間遠心し、インフルエンザウイルスを沈殿させる。STE(0.01MのTris-HCl;0.15MのNaCl;0.0001MのEDTA)又はpH7.4のリン酸バッファ(PBS)等の適当なバッファにウイルス沈殿物を再懸濁した後、ウイルスをショ糖(60%〜12%)又は酒石酸カリウム(50%〜10%)の密度勾配遠心で濃縮してもよい。連続的勾配か、又は段階的勾配(例えば12%〜60%間で12%ごと4段階)のショ糖勾配のいずれかが適している。勾配を、ウイルスを回収するため目に見えるバンドに凝集させる上で十分な速度及び時間で遠心分離で分離する。あるいは、大規模の工業用に適用するため、ウイルスを連続様式で稼動するゾーン遠心ローターを用いて密度勾配から溶出する。技術者が組織培養からインフルエンザウイルスを調製するためのさらに詳しい説明については、例えば、Furminger, Textbook of Influenza pp. 324-332 Nicholsonら編、Mertenら, in Novel Strategies in Design and Production of Vaccines pp.141-151 CohenとShafferman編、及び米国特許第5,690,937号で提供されている。もし所望であれば、回収したウイルスをショ糖-リン酸-グルタミン酸(SPG)等の安定剤の存在下で−80℃にて保存することができる。
【0072】
本発明方法のいくつかの実施形態において、ウイルスはBenzonase(登録商標)、又はその他の非特異的エンドヌクレアーゼで処理される。任意でBenzonase処理は、生産されたインフルエンザウイルスの回収と分離(ステップ(iii))の初期に行う。本工程のその他の実施形態では、Benzonase処理後に材料を清浄化する。清浄化に有用な方法は、ダイレクトフロー濾過(direct flow filtration:DFF)を含む。ただし、これに限られない。生産されたインフルエンザウイルスの回収と単離(ステップ(iii))に利用することができるさらなるステップは、タンジェンシャルフロー濾過(tangential flow filtration;TFF)、アフィニティークロマトグラフィーと、イオン交換クロマトグラフィー及び/又はヒドロキシアパタイトクロマトグラフィーを含むが、これに限られない。その他のステップについては下記実施例の項で例証している。
【0073】
<ワクチン組成物と使用方法>
本発明は、さらに本発明の方法で得ることのできるウイルス(例えばインフルエンザ)に関する。これらのウイルスを既知の方法で製剤化し、ヒトや動物に投与するワクチンを提供することができる。ウイルスは、完全ウイルス粒子(例えば、弱毒化した生ウイルス)として、又は不活性化/非組込み型ウイルス(例えば、ホルムアルデヒド、界面活性剤で処理したウイルス)として存在し得る。任意で、既知のウイルス成分(例えば、タンパク質)を当業者に既知の方法でウイルスから分離し、ワクチンの調製に使用することができる。
【0074】
完全ウイルス粒子(例えば、弱毒化した生ウイルス)の製剤は、最終製剤にする濾過によるバッファ交換、それに続く滅菌ステップと、さらなるステップを含むことができるが、それらに限られない。前記製剤に有用なバッファは、アルギニンなどの他のアミノ酸賦形剤を追加した200mMショ糖及びpH7.0〜7.2のリン酸又はヒスチジンバッファを含み得る。いくつかの実施形態において、ブタゼラチン等のタンパク質加水分解物安定化剤が添加される。いくつかの実施形態において、本発明の最終ウイルス溶液/ワクチンは、生きたウイルスを含み得る。当該ウイルスは、インフルエンザワクチン接種期(例えば、北半球では一般に大体9月から3月)の間、「現場で」保存(例えば、販売や市場で2〜8℃、4℃、5℃等で冷蔵される場合)のに十分な期間、液体状態で安定である。したがって、前記ウイルス/ワクチン組成物は、それらの効力が維持されること、又は保存期間以降は許容できる速さでその効力が失われることが望ましい。その他の実施形態において、前記溶液/ワクチンは、例えば、冷蔵庫内温度である約2℃〜約8℃において液体状態で安定である。例えば、冷蔵庫内で安定な弱毒化インフルエンザワクチンを処方するための方法と組成物は、2005年10月4日出願の国際出願第PCT/US2005/035614号に記載されている。国際公開第WO 05/014862号も参照されたい。任意で、液体製剤試料を乾燥加熱気体の気流下で微細液滴に噴霧化する噴霧乾燥法の急速乾燥工程を利用して、ワクチン製剤の保存時間を延長することができる。微細液滴の蒸発により、溶解溶質からなる乾燥粉末が生じる(例えば、米国特許公開第2004/0042972号を参照されたい)。ワクチン組成物用の不活性化/非組込み型ウイルス粒子の生成及び製剤化の方法は、当業分野では周知であり、40年間にわたって利用されている。
【0075】
一般にウイルスやウイルス組成物を予防的に適当な担体又は賦形剤と共に投与して、一以上のウイルス株に特異的な免疫反応を刺激することができる。通常前記担体又は賦形剤は、製薬上許容される担体又は賦形剤である。すなわち、滅菌水、生理食塩水、生理食塩バッファ、グルコース溶液、グリセロール溶液、エタノール、又はそれらの組合せ等が挙げられる。無菌性、pH、等張性、及び安定性が保証された前記溶液の調製は、当該分野の従来のプロトコルに従って達成される。通常、担体又は賦形剤は、アレルギーやその他の望ましくない効果を最小限に抑えるように、又は例えば皮下、筋肉内、鼻腔内等の個々の投与手段に合うように選択される。
【0076】
任意で、ウイルスの予防投与用の製剤又はそれらの成分はまた、インフルエンザ抗原に対する免疫反応を増強するための一以上のアジュバントを含む。適切なアジュバントには以下のものが含まれる。すなわち、サポニン、水酸化アルミニウムのようなミネラルゲル、リゾレシチン、界面活性物質、プルロニックポリオール、多価陰イオン、ペプチド、油又は炭化水素エマルジョン、カルメット−ゲラン桿菌(BCG)、コリネバクテリウム・パルヴム(Corynebacterium parvum)、並びに合成アジュバントQS-21及びMF59が挙げられる。
【0077】
一般に、ワクチン製剤は、一以上のインフルエンザウイルス株に対して特異的な免疫反応を刺激する上で十分な量で投与される。好ましくはウイルスの投与が防御免疫反応を誘導することである。一以上のウイルス株に対して防御免疫反応を誘導するための投与量及び方法は、当業者に既知である。例えば、不活性化したインフルエンザウイルスは、約1〜1000HID50(human infectious dose;ヒト感染用量)、すなわち1回の投与量あたり約105〜108pfu(plaque forming units;プラーク形成単位)の範囲で投与される。あるいは、約10〜50μg、例えば約15μgのHAをアジュバントなしで、アジュバントと共に投与される量よりも少量で投与する。一般に、投与量は、上記範囲で例えば年齢、健康状態、体重、性別、食事療法、投与時間、及びその他の臨床学的因子に基づいて調節され得る。予防用ワクチン製剤は、例えば針とシリンジを用いた皮下若しくは筋肉内注射、又は無針注射器具を用いて全身投与される。あるいは、ワクチン製剤は、鼻腔内への点滴、大粒子エアロゾル(約10ミクロンよりも大きい)、又は上気道内へのスプレーのいずれかによって投与される。上記送達経路のいずれかによって全身性の防御免疫反応が生じると同時に、鼻腔内投与は、インフルエンザウイルスの侵入部位である粘膜免疫を誘発するさらなる利益をもたらす。鼻腔内投与については、弱毒化した生ウイルスワクチンがよく選ばれる。例えば、弱毒化、低温適応性、及び/又は温度感受性の組換えインフルエンザウイルス若しくはリアソータントインフルエンザウイルスが挙げられる。1回の投与で防御免疫反応が刺激されることが好ましいが、同一の若しくは異なる経路で追加投与し、所望の予防効果を達成することができる。これらの方法は、いずれのウイルスにも適用することができる。ここで言うウイルスは、オルソミクソウイルス(A型及びB型インフルエンザ株を含む)、パラミクソウイルス(RSV、ヒトメタニューモウイルス、及びパラインフルエンザを含む)、ラブドウイルス及びフラボウイルスを含む。ただし、これらに限定はされない。
【0078】
<インフルエンザウイルス>
本明細書に記載の方法、工程及び組成物は、主としてワクチン用のインフルエンザウイルスの生産に関係している。インフルエンザウイルスは、セグメント化した一本鎖RNAゲノムを含む内部のリボ核タンパク質コアと、マトリックスタンパク質によって裏打ちされた外部のリポタンパク質エンベロープから構成されている。A型インフルエンザ及びB型インフルエンザは、それぞれ8セグメントの一本鎖のマイナス鎖センスRNAを含んでいる。A型インフルエンザゲノムは11種のポリペプチドをコードしている。第1〜3セグメントは、RNA依存性RNAポリメラーゼを構成する3つのポリペプチドをコードしている。第1セグメントは、ポリメラーゼ複合体タンパク質PB2をコードしている。残るポリメラーゼタンパク質PB1及びPAは、それぞれ第2セグメント及び第3セグメントにコードされている。さらに、いくつかのインフルエンザ株の第1セグメントは、PB1コード領域内の別の読み取り枠から生成される小タンパク質PB1-F2をコードしている。第4セグメントは、感染中の細胞への接着及び侵入に関与する表面糖タンパク質ヘマグルチニン(HA)をコードする。第5セグメントは、ウイルスRNAと結びつく主要な構造成分であるヌクレオカプシドヌクレオタンパク質(NP)ポリペプチドをコードしている。第6セグメントは、エンベロープ糖タンパク質ノイラミニダーゼ(NA)をコードしている。第7セグメントは、異なるスプライシングを受けたmRNAより翻訳される2種の基質タンパク質M1及びM2をコードしている。第8セグメントは、選択的スプライスシングを受けたmRNAから翻訳される2種の非構造タンパク質NS1及びNS2をコードしている。
【0079】
B型インフルエンザ8つのゲノムセグメントは、11種のタンパク質をコードしている。最も大きい3つの遺伝子はRNAポリメラーゼの成分、PB1、PB2及びPAをコードしている。第4セグメントは、HAタンパク質をコードしている。第5セグメントは、NPをコードしている。第6セグメントは、NAタンパク質とNBタンパク質をコードしている。NB及びNAの両タンパク質は、バイシストロニックmRNAの重複した読み取り枠から翻訳される。B型インフルエンザの第7セグメントも、二つのタンパク質をコードしている。すなわち、M1及びM2である。最も小さいセグメントは、全長RNAから翻訳されるNS1とスプライシングされたmRNA変異体から翻訳されるNS2の2つの産物をコードしている。
【0080】
リアソータントウイルスは、選択されたヘマグルチニン抗原、及びノイラミニダーゼ抗原をマスタードナーウイルス(MDV)とも呼ばれる認可されたマスター株の中で合体させて生産される。FluMist(登録商標)は、認可された低温適応性、弱毒性、温度感受性MDV株(例えばA/AnnArbor/6/60及びB/Ann Arbor/1/66)を使用している。卵をベースとした方法や最近の細胞培養方法を含む多くの方法は、リアソータントウイルスの生成に有用である(例えば、国際公開第WO 03/091401号;同第05/062820号、及び米国特許第6,544,785号;同第6,649,372号;同第6,951,754号を参照されたい)。本発明のMDCK細胞、培地及び工程は、インフルエンザウイルスの生産に有用であることが意図されている。ここでいうインフルエンザウイルスは、本明細書で開示するインフルエンザ株(例えば、A/AnnArbor/6/60及びB/AnnArbor/1/66)、並びにA/AnnArbor/6/60、B/AnnArbor/1/66、PR8の遺伝子を含むリアソータントウイルスを含むが、これらに限定はされない。さらに本発明のMDCK細胞、培地、及び方法は、一以上の以下の表現型、すなわち温度感受性、低温適応性、弱毒性を有するリアソータントウイルスを含むインフルエンザウイルスの生産に有用であることが意図されている。リアソータントウイルスは、古典的なリアソータント技術、例えば共感染法によって、又は任意でプラスミドレスキュー技術によって生成することができる(例えば、国際公開第WO 03/091401号;国際公開第WO 05/062820号、及び米国特許第6,544,785号;同第6,649,372号、同第6,951,754号を参照されたい。)
【実施例】
【0081】
以下の実施例を参照して本発明をこれから説明する。これらの実施例は、例示の目的のみのために提供されるのであって、本発明がこれらの実施例に限定されると解されるべきではない。むしろ本明細書で提供される教示の結果明らかとなる、ありとあらゆる変更を包含すると解されるべきである。
【0082】
実施例1
株細胞におけるca/tsインフルエンザ株の感染拡大の測定とMDCK細胞で生産されるインフルエンザの特徴解析
卵を利用しない代替生産プラットフォームの開発や動物又は昆虫の培養細胞系でのインフルエンザワクチンの生産にはワクチン業界の努力があった。明らかな利点として、スケールアップが簡単なことと、工程制御が増えたこと、及びいくつかのワクチンでアレルギー反応の原因となっていた卵タンパク質の排除が挙げられる。細胞培養に基づく系は、スケールアップが素早くできるので、卵の供給不足が予想されると共にワクチンの迅速な生産が必要とされるインフルエンザの世界的流行時にさらなる利点を提供する。最初の実験では、計7種の異なる株細胞を用いて行った。すなわち、2種の二倍体肺線維芽細胞株(MRC-5株とWI-38株)(データは示さず)、ヒト網膜芽細胞腫及びヒト腎臓細胞株(それぞれPER.C6株と293株;通常これらはいずれもアデノウイルス産物の生産用に構築された)(データは示さず)、胎児アカゲザル肺細胞株(FRhL2)(データは示さず)、アフリカミドリザル腎臓由来細胞株(Vero)、及びMarin-Darbyイヌ腎臓由来(MDCK)細胞株である。MDCK細胞は、低温適応性、温度感受性、弱毒性(ca/ts/att)のリアソータントインフルエンザウイルス株H1N1、H3N2、潜在的パンデミックワクチン株H5N1、そしてB型株の4種全てを商業的に妥当なタイター(>107 Log TCID50/mL)まで増やすことのできることを試験した唯一の細胞株である(図1、データは示さず)。MDCK細胞で増殖したウイルスの遺伝学的な及び抗原性の特徴を卵で増殖したウイルスのそれと比較した。ゲノム配列に有意な差異は見られなかった(データは示さず)。またHAIタイターで測定された抗原性は同程度であった(表1)。
【0083】
<蛍光焦点アッセイ>
MDCK細胞及びVero細胞を96ウェルブラックプレートで4日間増やした(DMEM+4 mMグルタミン+PEN/Strep)。各ウェルをca/ts B型ンフルエンザ株(B/HongKong/330/01、及びB/Yamanashi/166/98)で〜0.01のMOIにてDMEM+4mMグルタミン+60mU/mL TPCKトリプシン中で感染させた。ウイルスに感染したプレートを固定し、免疫染色した後、感染の広がりを以下のように測定した。ウイルスを含んだ培地を各プレートから除き、プレートを200μl/ウェルのDPBS(Ca2+/Mg2+非含有)で1回洗浄し、その後プレートを200μl/ウェルの冷やした4%(v/v)パラホルムアルデヒド/PBSで固定した。プレートを200μl/ウェルのDPBS(Ca2+/Mg2+非含有)で2回洗浄し、続いて細胞を一次抗体(0.1%サポニン+1%BSA/PBSで1:1000の比率で希釈したヒツジ抗Yamanashi B型、及びヒツジ抗HongKong B型)と共にインキュベートした。1時間のインキュベーションの後、一次抗体を除き、細胞をPBS中の0.1%Tween20で3回洗浄し、ウェルを二次抗体(0.1%サポニン+1%BSA/PBSを用いて1:100の比率で希釈したFITC標識したウサギ抗ヒツジ抗体)と共にインキュベートした。ウェルを毎日4日間、蛍光顕微鏡を用いて視覚化し、画像を毎日SPOTプログラムを用いて取り込んだ。
【0084】
<結果と考察>
蛍光焦点アッセイを用いて、MDCK細胞及びVero細胞でca/tsのB型インフルエンザ株の感染の広がりがあるか否かを評価した。また、50個のVeroクローン細胞間でウイルス感染の広がりに何らかの違いがあるかどうかも評価した。単分子層の蛍光は、MDCK細胞では4日間にわたって増加したが、Vero細胞では増加しなかったので(図1A参照)、Vero細胞はca/tsのB型株の生産に対して許容状態にないが、MDCK細胞は許容状態にあると結論付けられた。このデータは、B型株がMDCK細胞では7〜7.5log10TCID50生産されるが、Vero細胞ではわずか4〜4.5log10TCID50しか生産されないことを示した初期の実験データに類似している(データは示さず)。
【0085】
MDCK細胞を、潜在的なパンデミックワクチン株ca A/Vietnam/1203/2004株を含む多くのca/ts/attリアソータント株の複製を支持する能力についても試験した。MDCK細胞をca A/Vietnam/1203/2004を用いて低感染多重度で感染した。また、上清中のウイルスは感染後に様々な時間で定量化された。感染後48時間までにca A/Vietnam/1203/2004のタイターは、約8log10 TCID50/mLに達し、その後3〜4日間はその値で安定していた。図1B及び表2を参照されたい。
【0086】
A/H1N1型、A/H5N1型、A/H3N2型及びB型のca/ts/att株は、MDCK細胞内で比較的高いタイターで複製された。さらに、MDCK細胞中でこれらのca/ts/att株は、継代にて、そのゲノム配列を有意に変えなかった。3種のca/ts/att株、ca A/Sydney/05/97、ca A/Beijing/262/95、及びca B/Ann Arbor/1/94をMDCK細胞内で1回、又は2回継代し、全部で6つの内部遺伝子の完全なコード領域を配列決定し、開始時の配列と比較した。ヌクレオチドの変異は見られず(データは示さず)、本細胞を介した継代が、これらの株の遺伝子組成を変えないことが実証された。さらなる配列の特徴づけを培地組成、導入量(MOI)、インキュベーション温度、及び回収時間を含む生産工程を再現した条件下においてMDCK細胞で生産された異なるワクチン株で行った。予備データに基づき、MDCK産生ウイルスのゲノム配列には有意な変化がないことが期待された。
【0087】
ゲノムはMDCK細胞内では継代後も遺伝学的には安定であるので、卵又はMDCK細胞で生産されるワクチンの生物学的形質は見分けがつかないことが期待された。しかし培養細胞由来の主なウイルス産物は、卵由来の産物と比べていくつかの僅かな違い、特にHA及びNAを含むウイルスタンパク質の翻訳後修飾、又はウイルス膜中の脂質の組成に関して違いが見られた。これらはどちらもウイルス粒子の全体的な物理的特性を潜在的に変えることができる。培養細胞で生産されたワクチンと卵で生産されたワクチンの抗原性における予備的な前臨床データから、前記の重要なパラメータの違いを検出できないことが実証された。いくつかのワクチン株の卵のストックがMDCK細胞を介して継代され、両産物の抗原性を基準抗血清を用いてHAIタイターを測定することで実証した。表1で示すように、全てのHAIタイターは、お互いに2倍以内であった。これは、卵由来のワクチンと比べて、細胞内でのワクチンの複製がワクチンの抗原性を変えないことを示している。
【表1】

【0088】
実施例2
非腫瘍形成性血清MDCK細胞の誘導
MDCK細胞は、昔からインフルエンザウイルスのタイトレーションに使用されてきた(Zambon著,1988,in Textbook of Influenza,Nicholson編, Webster and Hay, ch24, pg 324-332, Blackwell Science)。それ故、ワクチン物質生産のためインフルエンザの増殖用に使用することができた。しかし、MDCK細胞は、伝統的にFBSを追加したイーグル最小必須培地(Eagle’s Minimal Essential Medium;EMEM)のような基本培地処方物で増殖されていた。そのような条件下で及び/又は長期間インキュベートするとMDCK細胞が腫瘍形成性となる可能性があることを示した報告が多数ある(例えば、Gaushら,Proc Soc Exp Biol Med,122:931;Leightonら,1968,Science,63:472;及びLeightonら,1970,Cancer,26:1022を参照されたい)。ゆえに、ワクチン物質の生産にMDCK細胞を使用することについては懸念があり、その他の細胞株の開発(例えば、PER.C6及びVERO)に努力が向けられた。残念なことに、インフルエンザ株の全てが他の哺乳動物細胞株中で十分に増殖するわけではない。特に弱毒化されたインフルエンザ生ワクチンであるFluMist(登録商標)を含む低温適応したインフルエンザウイルスは、MDCK細胞内でのみ適当なタイター(>107 TCID50/mL)まで増殖できる(上記実施例1を参照)。MDCK細胞の特徴解析を行った初期の報告では、MDCK細胞の早期の継代では腫瘍形成性が起き得ないことを示している(Gaushら,1966, Proc Soc Exp Biol Med.122:931)。非腫瘍形成性の状態にMDCK細胞を維持する培地及び継代プロトコルを確立することが本実験の目標であった。
【0089】
ATCC(CCL34)から入手したMDCK細胞を、Tフラスコ内で10%FBS、4mMグルタミン及び4.5g/Lグルコースを追加したDMEMを増殖培地に用いて増やした。前マスターMDCK細胞バンクを血清増殖MDCK細胞(MDCK-S細胞)で確立した。この血清増殖MDCK細胞は、民間の受託業者(例えば、BioReliance、Rockville、MD)によって行われる通常のテストを用いて細菌/真菌の汚染、及びマイコプラズマの汚染についてテストされた。前記細胞は、細菌/真菌の汚染物質の存在についてネガティブであることがわかった。MDCK-S細胞は、培養可能なマイコプラズマの存在についてもネガティブであることがわかった。前記バンク由来のMDCK-S細胞を核型解析でもテストした。その結果、その起源がイヌ科であることが判明し、また約70本〜84本の染色体数の範囲で78本の最頻値染色体数を有していた。その後、MDCK-S細胞をPreMCBの容器から起こして新たに20継代し、インビボでアダルトヌードマウスのモデルの核型と腫瘍形成についてテストした。核型テストでは、継代後期のMDCK-S細胞(p81/24)は継代初期のMDCK-S細胞と同じ染色体数最頻値(78本)及び染色体の範囲(70〜84)を示した。これは、継代を続けても細胞が変化しなかったことを示している。1×107のMDCK-S細胞をアダルトヌードマウスの皮下に注射しても、どんな小結節の形成も生じなかった。したがって、当該細胞を非腫瘍形成性とみなした。
【0090】
<材料と方法>
材料
MDCK細胞(ATCC, Cat. No:CCL-34);T-25、T-75、T-225フラスコ(Corning社、Cat No.: 430639, 430641, 431082);ダルベッコ改良イーグル培地(DMEM)粉末(Gibco社、Grand Island NY, Formulation No.:01-5052EF);ガンマ線照射済みウシ胎仔血清(JRH社、Lenexa KS, Cat. No.:12107-500M);L-グルタミン(JRH社、Lenexa KS, Cat. No.: 59202-100M);D-グルコース(Amresco社、Cat. No.:0188-1KG);Ca2+及びMg2+非含有のダルベッコリン酸バッファ(DPBS)(Gibco社、Grand Isand NY, Cat. No.:21600-069);0.05%トリプシン-EDTA(Gibco社、Grand Island NY, Cat. No.:25300);ジメチルスルホキシド(DMSO)(Sigma社、St. Louis MS, Cat. No.:D2650);0.4%(w/v)トリパンブルー染料/PBS溶液(Sigma社、St. Louis MS, Cat. No.:T8154);CO2インキュベータ(Forma Scientific社、Model No.:3110);YSIバイオアナライザ(YSI社、Model No.: 2700 select);VITROS化学システム(Ortho clinic社、Model: DT60 II);改良型ノイバウア血球測定器(Hausser Scientific社、Bright-Line 0.1mm deep/Reichert, Bright-Line 0.1mm deep)。
【0091】
組織培養フラスコでの血清MDCK(MDCK-S)細胞の継代培養
血清MDCK細胞のバイアルをATCCから入手した。細胞をT-75フラスコ内の10%(v/v)FBS、4.5g/Lグルコース、2.2g/L NaHCO3、及び4mM L-グルタミンを追加したDMEM培地で増やした。細胞を接種後3〜4日で継代するとともに、もし細胞を4日目に継代するのであれば接種後3日目に全培地交換を行った。細胞を以下に記載のようにTフラスコから回収した。
【0092】
使用済み増殖培地を除き、単分子層細胞をDPBS(カルシウム、マグネシウム無し)で2回洗浄した。37℃ウォーターバスで予熱したトリプシン‐EDTAを適当量(3mL/T-75、7.5mL/T-225)、各フラスコに加え、その後Tフラスコを37℃、5%CO2インキュベータ内で約15〜20分間インキュベートした。フラスコを5分ごとに確認し、細胞が剥離しているかどうかを調べた。またフラスコを数回軽く叩いて細胞の剥離を促した。細胞がTフラスコから完全に剥離したら、10%血清を含む等量の完全成長培地(3mL/T-75、7.5mL/T-225)を加えてトリプシンを不活性化した。細胞懸濁液を適当なサイズのピペットで吸引/排出によって数回上下させ、大きな細胞塊をバラバラにした。0.5ml細胞懸濁液サンプル二つを血球測定器でカウントした。二つのカウントの結果が相互に15%以内にない場合には細胞カウントを繰り返した。細胞を新鮮な保温した増殖培地(DMEM+10%FBS+4.5g/Lグルコース+4mMグルタミン)で新しいフラスコ内で0.05×106生細胞/mLに希釈した。そしてTフラスコに接種した(35mL/T75又は100mL/T-225)。その後、継代又は培地交換を行う前にそのフラスコを37±1℃、5% CO2の環境下で3日間培養した。
【0093】
MDCK-S細胞バンクの調製
MDCK-S細胞を上記のTフラスコ内でバンクに必要な細胞の総必要量を回収できるまで増やした(4×106細胞/バイアル×バイアル数)。MDCK-S細胞が対数増殖期にあるとき(接種後3日目)に記載したトリプシン処理によって当該細胞を回収した。個々のフラスコ由来のMDCK-S細胞の懸濁液をプールし、150〜250Gで7±1分間遠心して細胞を回収した。各チューブから上清を吸引して除き、沈殿した細胞を新しい完全成長培地(DMEM+10%FBS+4.5g/Lグルコース+4mMグルタミン)で再懸濁した。異なる遠心ボトル由来の細胞懸濁液をプールし、細胞懸濁液を適当なサイズのピペットで吸引/排出によって数回上下させ、大きな細胞塊をバラバラにした。総細胞数を測定し、4×106細胞/バイアルで冷凍できるバイアルの総数を決定した。
【0094】
その後、細胞懸濁液の量を新しい増殖培地を用いて上記の値に合わせた。等量の新たに調製した2×冷凍培地(DMEM+10%FBS+4mMグルタミン+4.5/Lグルコース+15%DMSO)を細胞懸濁液に加えた。細胞懸濁液を十分に混合して、1mLの細胞懸濁液を各冷凍バイアルに分注した。全てのバイアルをNalgene社の冷凍コンテナに移し、≦−60℃フリーザ内に置いた。冷凍バイアルを液体窒素保存タンクに移した。
【0095】
T-75フラスコでのMDCK-S細胞の成長曲線の作成
細胞を細胞成長曲線の実験前に少なくとも4回(解凍後)、成長培地にて継代した。MDCK-S細胞を、T-225フラスコ内で少なくとも総細胞数2.7×107個得るために増やした。フラスコを上記のトリプシン処理前にコンフルエントの80〜95%まで増やした。回収したMDCK-S細胞をプールし、細胞懸濁液をピペットで吸引/排出によって数回上下させ、大きな細胞塊をバラバラにした。2サンプル(0.5mL)を細胞カウント用に取り、細胞密度を測定した。2サンプルのカウントが、相互に15%以内にない場合には再度測定を行った。その後、総計2.7×107MDCK細胞を総量540mLの完全成長培地に希釈した(5.0×104細胞/mL)。それから、このMDCK-S細胞懸濁液を14本のT-75フラスコ(35mL/T-75フラスコ)に分注した。そのフラスコを37±1℃、5% CO2インキュベータに置いた。
【0096】
二つのTフラスコを、細胞カウント及び代謝解析のために毎日インキュベータから取り出した。細胞培養培地を2サンプル(約1.0mL)、各フラスコから代謝解析用に取り出した。一のサンプルを使用して、YSI及びVITROSアナライザーを用いてグルコース、乳酸、グルタミン、グルタミン酸、及びアンモニア濃度を測定した。他方のサンプルについては、後日アミノ酸解析を行うため−70℃で冷凍した。MDCK-S細胞を上記のトリプシン処理によって各フラスコから回収した。細胞密度を測定し、また細胞総数/Tフラスコも測定した。二つのカウントが、お互い15%以内にない場合には再度カウントを行った。存在数は、二つの異なる継代数(p63及びp65)のMDCK-S細胞で行った二つの独立した成長曲線解析の平均である。
【0097】
核型試験
核型試験は、フロリダ州メルボルンのApplied Genetics Laboratoriesで行われた。簡単に言えば、T-225フラスコで増殖したMDCK-S細胞をApplied Genetics Laboratories社に送った。細胞は上で記載した方法により維持され、また継代された。細胞が有糸分裂状態の細胞を十分に含んでいると思われるときは、その細胞を有糸分裂解析用に回収した。細胞をコルセミド(0.02μg/mL)で37℃にて150分間処理した。その後、細胞をトリプシン処理して回収し、200Gで5分間遠心した。上清を吸引にて除去し、予熱した低張溶液に再懸濁し、37℃で10分間インキュベートした。膨張した細胞を遠心で沈殿させ、それからCarnoy’s溶液(3:1 メタノール:氷酢酸)中で室温にて40分間インキュベートして固定した。細胞を再度遠心し、その後その細胞を少なくとも2回Carnoy’s固定剤で洗浄した。最後の遠心の後、細胞を1〜3mLの新しい固定剤で再懸濁し、乳白色の細胞懸濁液を生成した。最終細胞懸濁液の液滴をきれいなスライド上に落とし、風乾した。
【0098】
ライト染色液/リン酸バッファを前記スライドに加えて7〜10分間インキュベートして細胞を染色した。スライドを7〜10分後に水道水で洗浄し、その後風乾した。細胞を低倍率対物レンズ(10×)で走査し、細胞分裂の中期ステージにある細胞を検出した。また中期の細胞の染色体を高倍率油浸レンズ(100×)により解析した。100個の中期細胞を細胞発生異常及び染色体カウントについて解析した。1000個の細胞を走査し、倍数体頻度及び分裂指数(有糸分裂をしている細胞のパーセント)を測定した。
【0099】
MDCK-S PRE-MCBの無菌テスト(静菌性、静真菌性、及び4つの培地の滅菌性)
MDCK-S PRE-MCBの静菌活性、及び静真菌活性をBioreliance Inc.社(ロックビル,メリーランド)でテストした。本アッセイは、US 26及びUS21 CFR 610.12要件を満たして行われた。本アッセイは、0.1 mLのテストサンプルを含む適当な培地とコントロール生物のみを含む培地に接種したコントロール生物(バチルス・サブチルス(Bacillus subtilis)、カンジダ・アルビカンス(Candida albicans)、クロストリジウム・スポロゲネス(Clostridium sporogenes)、スタフィロコッカス・アウレウス(Staphylococcus aureus)、シュードモナス・エアルギノサ(Pseudomonas aeruginosa)、アスペルギルス・ニガー(Aspergillus niger))の増殖と違いがあるか否かをテストするものである。簡単に言えば、被験物質を3本のTSB(大豆カゼイン分解培地)チューブ、4本のTHIO(fluid thioglycollate medium:流動性チオグリコール酸培地)、2本のSAB(Sabourand Dextrose Agar:Sabourandデキストロースアガー)、及び1本のPYG(peptone yeast extract:ペプトン酵母エクストラクト)に接種した。その後、100cfu未満のコントロール生物を含む各コントロール生物を適当なタイプの培地に接種した。ポジティブコントロールは、TSB及びTHIOのバチルス・サブチルス、TSB及びSAB(20〜25℃及び30〜35℃)のカンジダ・アルビカンス、THIO及びPYGのクロストリジウム・スポロゲネス、シュードモナス・エアルギノサ、及びアスペルギルス・ニガーからなる。ネガティブコントロールは、滅菌したPBSである。培地を3〜5日間インキュベートし、生物の増殖を確認した。
【0100】
被験物質を、細菌及び真菌汚染の有無についても4種の培地無菌テストを用いてBioreliance社(ロックビル,メリーランド)で解析した。本アッセイは、USP 26, EP、及び21CFR610.12要件を満たすことが意図されている。簡単に言えば、被験物質を、2本のTSB(大豆カゼイン分解培地)チューブ、2本のTHIO(流動性チオグリコール酸培地)、3本のSAB(Sabourandデキストロースアガー)、及び2本のPYG(ペプトン酵母エクストラクト)に接種した。培地を適当な温度(SABスラントについては2つの温度でインキュベートした)でインキュベートし、チューブをテスト第3日目/第4日目又は第5日目、7日目又は8日目、及び14日目に確認することで、全てのチューブを14日間にわたって観察した。混濁が現れる被験物質を接種したいずれのチューブもプレートに広げ、そのプレート上でグラム染色を行った。ネガティブコントロールは滅菌したPBSである。
【0101】
マイコプラズマ/静マイコプラズマ性テスト
冷凍MDCK-S細胞のバイアル(MDCK preMCB lot No.747p105)をBioreliance社に送った。細胞は上記で説明したようにTフラスコ内で増やされて培養された。5×105cells/mLの濃度の細胞溶解物を調製し、−70℃で冷凍した。被験物質をマイコプラズマ・ニューモニエ(Mycoplasma pneumoniae)、マイコプラズマ・オラレ(Mycoplasma orale)、及びマイコプラズマ・ハイオライニス(Mycoplasma hyorhinis)の増殖を抑制する能力についてアガーブロス/プレートで、及び/又はVERO細胞内でテストした。
【0102】
アガー分離アッセイについては、被験物質をアガープレート又はブロスボトルでスパイク、又は非スパイクのいずれかをテストした。被験物質は、マイコプラズマ・ニューモニエ、及びマイコプラズマ・オラレでスパイクされ、10〜100cfu/0.2mL(アガーテスト用)及び10〜100cfu/10mL(準ブロスアッセイ用)の希釈を達成した。テストサンプルの一部はスパイクされなかった。4つの半固体ブロスボトルをスパイク(2ボトル)、又は非スパイク(2ボトル)の各10mlで接種した。スパイク/非スパイクの各1本のボトルを、有酸素的にか、又は嫌気的にかのいずれかで適当な温度でインキュベートした。10枚のA型アガープレートと10枚のB型アガープレートをそれぞれスパイクサンプル又は非スパイクサンプルで接種した。A型アガープレートとB型アガープレートの半分を有酸素的にか又は嫌気的にかのいずれかで適当な温度でインキュベートした。非接種マイコプラズマ半固体ブロスを非接種ネガティブコントロールとした。全ブロスボトルを21日間観察した。各ブロスボトル(非接種ネガティブコントロールを除く)を3日目、7日目、及び14日目にA型アガープレート又はB型アガープレートに継代し(それぞれ10プレート、0.2mL/プレート)、同一条件下で適切なボトルとしてインキュベートした。それらを1日1回、21日間観察した。
【0103】
増強VERO細胞培養アッセイについて、被験物質のスパイク、又は非スパイクをテストした。被験物質をM.オラレ及びM.ハイオライニスを用いて10〜100cfu/0.2mLの濃度でスパイクした。スパイクした被験物質、非スパイクした被験物質、ポジティブコントロール、及びネガティブコントロールをそれぞれVERO細胞培養液のT-75フラスコに接種した。3〜5日のインキュベーション後、各フラスコの細胞を掻き取り、直ちに凍結した。各フラスコの細胞溶解物1mLの10分の2をVERO細胞の入った6ウェルプレートの各ウェルに接種した。さらに、ポジティブ及びネガティブコントロールをVERO細胞の入った6ウェルプレートの適当なウェルに接種した。3〜5日後、細胞を固定し、DNA結合へキスト色素で染色し、マイコプラズマの存在を評価した。
【0104】
ヌードマウスにおけるMDCK-S細胞の腫瘍形成性テスト
無胸腺ヌードマウス(nu/nu)における腫瘍形成の評価は、BioReliance(登録商標)(ロックビル、MD)によって行われた。簡単に説明すると、30匹のメス無胸腺マウス(4週齢)の皮下に、0.2mL(1×107細胞/マウス)のポジティブコントロール(18Cl-10T細胞)、ネガティブコントロール(シリアンハムスター胚細胞;SHE細胞)、又はテスト細胞(血清MDCK細胞、747p105高継代)を注射した。前記動物を注射前に無作為に選択し、全マウスに22ゲージ針を用いて同日に注射した。全動物を就業日に毎日観察し、注射部位を84日の期間、1週間に2回、病変発生の確認のため触診した。それぞれの病変を測定し、病変サイズの増加が見らる間は動物を維持した。この病変サイズの増加が見られる期間は、最大で3ヶ月間であった。84日後に全マウスを絞めて解剖し、注射部位、肺、肩甲リンパ節、及び全病変部を組織病理学的方法で解析した。
【0105】
MDCK-Sにおける低温適応性インフルエンザ株の複製
T-75フラスコに5×104細胞/mL(DMEM 35mL+10%FBS+4mMグルタミン)を接種し、37℃、5%CO2維持されたインキュベータで3日間、増やした。接種後3日目にTフラスコあたりの全細胞をトリプシンEDTAを用いて回収することによって測定した。また、細胞のカウントは、トリパンブルー分染排除法で測定した。残りのTフラスコは、その後次のように感染させた。増殖培地を吸引して除去し、細胞を1フラスコあたり10mLのDPBS(Ca2+/Mg2+なし)で2回洗浄した。0.01の感染多重度(MOI)でそれぞれのTフラスコに感染させるウイルス量を以下の方程式により測定した。
【数1】
【0106】
MOIは、加えた1細胞あたりのウイルス粒子として定義される。
【0107】
その後、必要量のウイルスを各Tフラスコ中の35mLの後感染培地(DMEM+4mMグルタミン+60mu/mL TPCKトリプシン)に加えた。それから、そのTフラスコを33℃、5%CO2でインキュベートし、6日間毎日サンプルを取った。10×SPを各サンプルに安定剤として加え、そのサンプルを感染性をテストする前に<−70℃に保存した。
【0108】
各サンプル中に存在するウイルスの濃度を、感染ウイルスを測定する50%組織培養感染量(TCID50)アッセイによって測定した。簡単に説明すると、MDCK細胞を96ウェルマイクロタイタープレートにて単分子層でコンフルエントまで増やし、ca/tsインフルエンザウイルスサンプルの連続希釈物を加えた。MDCK細胞アッセイプレート内のサンプルは、通常10−4〜10−10の最終希釈物となった。カラム1〜5及び8〜12のウェルには、ウイルス希釈サンプルを入れた。また、カラム6〜7にはウイルス希釈物のみを入れ、細胞コントロールとしての役目をさせた。この構成により、プレートあたり2点のデータ(n=2)が得られた。MDCK細胞でのウイルスの複製は、細胞死及び培養液上清中への子孫ウイルスの遊離を生じた。子孫ウイルスは他の細胞に感染し、最終的に単分子層を破壊した。感染の結果生じる細胞変性効果(cytopathogenic effect;CPE)をCO2環境下33±1℃で6日間インキュベーションする間、進展させた。その後プレートをインキュベータから取り出し、ウェル内の培地を廃棄した。続いて100μlのMEM+4mMグルタミン+ペニシリン/ストレプトマイシン+MTTを各ウェルに加えた。プレートを37℃、5%CO2で6時間インキュベートし、CPEを示すウェル数を各ウェル内で形成される色(黄色/橙色はCPEウェルを意味し、濃淡のない紫色はCPEがないことを意味する)に基づいた目視検査によって測定した。それぞれ半分のプレートでCPEを示したウェル数を用いて、Reed-Muench法のKarber改変法に基づいたタイター(log10 TCID50/mL)を算出した。
【0109】
<結果と考察>
血清MDCK細胞の冷凍バイアル2本を、2本のT-75フラスコに分離して完全成長培地(DMEM+10% FBS+4mMグルタミン+4.5g/Lグルコース)中で解凍した。解凍による細胞生存率は、それぞれ97%と98%であった。細胞は、解凍後3日間でコンフルエントに達した。細胞形態は、上皮様で、ATCCより入手したストック株に似ていた(図3)。これらの細胞を5回継代し、血清増殖MDCK細胞(MDCK-S細胞)の前マスター細胞バンク(Pre-master cell bank;PreMCB)を樹立した。図2は、MDCK-Sの前マスター細胞バンク(PreMCB)の誘導に用いられる方法を概説している。
【0110】
10%FBS DMEM培地におけるMDCK-S細胞の成長曲線を図4に示す。この結果は、異なる継代数(P63及びP65)の細胞を用いた2つの実験の平均である。MDCK-S細胞は、約1日の誘導期(lag phase)を有している(接種時の総細胞数1.75×106細胞/T75フラスコ、及び1日目の総細胞数2.9×106細胞/T75)。誘導期とは、接種してからの細胞数が2倍に達しない時期をいう。グルコース消費/乳酸産生比は、初日ではほぼ0であった。これは、細胞が誘導期にあることを示している(図5)。その後細胞は、接種後4日目に静止期に入るまでの細胞増殖期間の間、指数関数的に増殖した。対数増殖期におけるMDCK-S細胞の倍化時間は、23.1時間であった。対数期の間、グルコース消費と乳酸産生は、乳酸濃度の増加とグルコース濃度の減少においてお互いに酷似していた(図5)。グルコース消費/乳酸産生比は、細胞成長曲線とよく対応していた(図4、5を比較されたい)。この速度は誘導期間の間は低く、1日目〜4日目の対数期の間はグルコースが2.93mM/日、及び乳酸が3.43 mM/日増加した。
【0111】
MDCK-S細胞は、接種後4日目に静止期に入った。そして、到達した最大細胞密度は、接種後5日目におけるおよそ29±0.99×106細胞であった(図4)。細胞数は、本実験では最大細胞密度に達した後も7日目まで一定に維持された。グルコース消費速度と乳酸産生速度は、静止期ではグルコースが0.33mM/日、及び乳酸が0.25mM/日に落ちた。7日間の培養後の約12mMのグルコースが培地中にまだ残っていた。4日目のグルコース消費量対乳酸産生の比率は1.2であった。
【0112】
MDCK-S細胞のグルタミン消費、並びにグルタミン酸及びアンモニアの両産生を図6に示す。グルタミン消費率とアンモニア産生率は、細胞成長曲線とよく対応していた(図4と図6を比較されたい)。MDCK-S細胞は、4日目までの対数増殖期間中、グルタミンを0.49mM/日の速度で消費し、同時にアンモニアを5日目まで0.32mM/日の速度で産生した。その後、細胞が静止期に入ると、グルタミン消費速度は0.24mM/日まで下がり、同時にアンモニア産生速度も0.11mM/日まで下がった。アンモニア産生対グルタミン消費の比率は、接種後4日目で0.7であった。グルタミン代謝から生じるグルタミン酸は、今回の7日間の細胞増殖実験では蓄積しなかった。
【0113】
MDCK-S細胞の核型を継代61/4、及び継代81/24でテストした。Gバンド染色体解析は細胞がイヌ科起源であることを示した。100個の中期細胞における染色体数の分布を図7に示す。染色体の総数は、低継代61/4については細胞中期あたり70本〜84本、また高継代81/24については70本〜84本に分布していた。両継代とも最頻値染色体数は78本であった。染色体の分布は、継代で変わらなかった。細胞の様相は、正常なイヌ腎臓細胞(Starkeら, 1972, Prog Immunobiol Stand., 5:178)から予想された通りであった。
【0114】
MDCK-SのpreMCBを細菌汚染、真菌汚染、又はマイコプラズマ汚染のいずれかの存在についてテストした。pre-MCBは無菌テスト(細菌及び真菌の汚染を確認するための直接接種法を用いた4培地無菌テスト)に合格し、マイコプラズマの存在(アガー培養可能、及び非アガー培養可能アッセイ)についてネガティブであることがわかった。被験物質は、静菌性/静真菌性テスト、及び静マイコプラズマ性テストのどちらもポジティブコントロールの増殖を阻害しないこともわかった。
【0115】
継代81/24のMDCK-S細胞(pre-MCB+20継代)をヌードマウス上に接種し、3ヶ月間腫瘍形成テストを行った。MDCK-S細胞を接種したいずれのマウスにおいても腫瘍が診断されなかったことから、MDCK-S細胞は腫瘍形成性でないことが実証された(表4)。
【0116】
MDCK-S細胞をテストし、低温適応性、温度感受性、弱毒性のリアソータントインフルエンザ株の複製を支持できることが判明した(表2)。
【表2】
【0117】
実施例3
Taub培地における無血清MDCK細胞の誘導
上記実施例2で詳述した結果は、MDCK細胞を上皮形態及び正常な核型、同時に低温適応性インフルエンザ株を複製する能力を維持した状態で培養することが可能なことを証明している。さらに、本発明者らは、上記実験で開発された条件下でMDCK細胞を培養すると、非腫瘍形成性のMDCK細胞を生じることを立証した。しかし、実施例2で使用した培地は、ウシ胎仔血清(FBS)を含んでいる。FBSは、複合成分混合物であり、ロット間の変動の問題が報告されている。また、ウシの牛海綿状脳症(BSE)に伴う現在継続中の問題が、安全面での不安をもたらしている。MDCK-S細胞株の非腫瘍形成性の性質と増殖特性を維持する無血清培地の開発は、治療及び予防接種用に生産される生物製剤の安全性を高める上で重要である。
【0118】
ATCC(CCL34)から入手したイヌ腎臓由来(MDCK)細胞を、Tフラスコ内で10%FBS、4mMグルタミン、及び4.5g/Lグルコースを追加したDMEMを増殖培地として用いて、5回継代して増やした。その後、細胞を無血清Taub培地(以下の処方を参照)に移した。Taub培地処方物中での増殖に適応した細胞をMDCK-Tとした。pre-MCBをMDCK-T細胞について樹立し(図8参照)、細菌/真菌汚染、及びマイコプラズマ汚染についてテストした。MDCK-T細胞の前マスター細胞バンクの細胞を核型アッセイでもテストし、イヌ起源であること、52本〜84本に及ぶ染色体数と共に78本の最頻値染色体数を有することがわかった。さらに、MDCK-T細胞をPreMCBのバイアルから起こして新たに少なくとも20回継代した。その後、核型及びインビボアダルトヌードマウスモデルでの腫瘍形成能についてテストした。しかしながら、MDCK-T細胞は、本モデルで腫瘍形成性であった。これは、公知のTaub培地がヒトワクチン物質を生産する上でMDCK細胞を安定して培養できないことを示している。
【0119】
<材料と方法>
材料
MDCK細胞(ATCC, Cat. No:CCL-34);T-25、T-75、T-225フラスコ(Corning社、Cat No.: 430639, 430641, 431082);ダルベッコ改良イーグル培地(DMEM)粉末(Gibco社、Grand Island NY, 製剤番号:01-5052EF);Ham F12栄養素混合物粉末(Gibco社、Grand Island NY, 製剤番号:21700-075);ガンマ線照射済みウシ胎仔血清(JRH社、Lenexa KS, Cat. No.:12107-500M);L-グルタミン(JRH社、Lenexa KS, Cat. No. 59202-100M);D-グルコース(Amresco社、Cat. No.:0188-1KG);Ca2+及びMg2+非含有のダルベッコリン酸バッファ(DPBS)(Gibco社、Grand Isand NY, Cat. No.:21600-069);インスリン粉末(Serological社、Cat. No.4506);トランスフェリン(APO型)(Gibco社、Grand Island NY, Cat. No.:11108-016);プロスタグランジンE1(Sigma社、St. Louis MS, Cat. No.:P7527);ヒドロコルチゾン(Mallinckrodt社、Cat. No.:8830(-05));トリヨードチロニン(Sigma社、St. Louis MS, Cat. No.:T5516);亜セレン酸ナトリウム(EMD社、Cat. No.:6607-31);0.05%トリプシン-EDTA(Gibco社、Grand Island NY, Cat. No.:25300);ライマメ・トリプシンインヒビター(Worthington社、Cat. No.:LS002829);ジメチルスルホキシド(DMSO)(Sigma社、St. Louis MS, Cat. No.:D2650);0.4%(w/v)トリパンブルー/PBS溶液(Sigma社、St. Louis MS, Cat. No.:T8154);改良型ノイバウェル血球測定器(Hausser Scientific社、Bright-Line 0.1mm deep/Reichert, Bright-Line 0.1mm deep);YSIバイオアナライザ(YSI社、Model No.: 2700 select);VITROS化学システム(Ortho clinic社、Model: DT60 II)。
【0120】
Taub's無血清培地(SFM)の処方
Taub's培地(TaubとLivingston, 1981, Ann NY Acad Sci.,372:406)は、4.5 g/Lグルコース及び4mMグルタミンを含むDMEM/Ham F12(1:1)からなる無血清培地処方物を基礎培地処方として、表3に示したようなホルモン/因子を添加したものである。
【表3】
【0121】
Taub's無血清培地は、継代時に新たに作られ、又は無血清(SF)DMEM/Ham F12培地+4mMグルタミン+4.5g/Lグルコース+10−8M亜セレン酸ナトリウムにホルモン補足剤のストック溶液を添加することによって再供給される。100mLのTaub's培地は、100μlのインスリンストック溶液(5mg/mL)、100μlのトランスフェリンストック溶液(5mg/mL)、100μlのトリヨードチロニン(T3)ストック溶液(5×10−9M)、5μlのヒドロコルチゾンストック溶液(10−3M)、及び50μlのプロスタグランジンE1ストック溶液(50μg/mL)を、基礎となるDMEM/Ham F12培地+4mMグルタミン+4.5g/Lグルコース+10−8M亜セレン酸ナトリウムに添加して作られる。全ストック溶液は、次のように調製した。
【0122】
インスリンストック溶液
5mg/mlのストック溶液は、適当量のインスリンを0.01NのHClに溶かして作製された。その溶液を0.2ミクロンの滅菌フィルターに通し、Nalgene社の冷凍バイアルに分注し、4℃で保存した。
【0123】
トランスフェリンストック溶液
5mg/mlのストック溶液は、適当量のトランスフェリンをMilliQ水に溶かして作製された。その溶液を滅菌フィルターに通し、その後Nalgene社の冷凍バイアルに分注し、<−20℃で保存した。
【0124】
トリヨードチロニン(T3)ストック溶液
ストック溶液は、適当量のT3を0.02NのNaOHに溶かし、10−4M溶液を得ることによって作製された。このストック溶液を0.02NのNaOHでさらに5×10−9Mストック溶液の濃度にまで希釈し、滅菌フィルターに通し、その後Nalgene社の冷凍バイアルに分注した後、<−20℃で保存した。
【0125】
ヒドロコルチゾンストック溶液
10−3Mストック溶液は、適当量のヒドロコルチゾンを100%のEtOHに溶かして作製された。これをNalgene社の冷凍バイアルに分注した。そのバイアルを4℃で3〜4ヶ月間保存した。
【0126】
プロスタグランジンE1ストック溶液
50μg/mLのストック溶液は、適当量のプロスタグランジンE1を100%のEtOHに溶かして作製された。これをNalgene社の冷凍バイアルに分注した後、<−20℃で保存した。
【0127】
亜セレン酸ナトリウム(Na2SeO3)ストック溶液
10−2Mのストック溶液は、適当量のセレン化ナトリウムをWFI水、又はMilliQ水に溶かして作製された。これをさらに水で10−5の最終濃度にまで希釈し、滅菌フィルターに通し、その後、4℃で保存した。
【0128】
MDCK-S細胞の無血清Taub's培地への適応
ATCC由来のMDCK細胞の冷凍バイアル(継代54)を、無血清Taub's培地に継代する前に4.5g/Lグルコース、2.2g/L NaHCO3、及び4mM L-グルタミンを含んだ10%FBS DMEM培地で5回継代して増やした(上記のようにした)。T-75フラスコで増やした血清MDCKをトリプシン処理で回収した。使用した増殖培地を除き、細胞単分子層をDPBS(カルシウム及びマグネシウムなし)で2回洗浄した。その後、DPBSを廃棄した。適当量の予熱したトリプシン-EDTAを加え(3mL/T-75)、Tフラスコを37℃、5%CO2インキュベータで約15分間インキュベートした。フラスコを手のひらで数回叩き、細胞を完全に剥離させた。等量のライマメ・トリプシンインヒビターを加え、トリプシンを中和した後、サンプルを二つ取り、細胞懸濁液の細胞濃度を測定した。それから1.75×106細胞を新しいT75フラスコのTaub's培地35mLに希釈した。そのフラスコを5%CO2、37±1℃に維持された細胞培養インキュベータ内に入れた。細胞を接種後3日継代培養するか、全培地交換を接種後3日目に行い、続く4日目に継代した。
【0129】
Taub's培地に適応したMDCK細胞の継代
使用した培地を除き、細胞単分子層をDPBS(カルシウム及びマグネシウムなし)で2回洗浄した。適当量の予熱したトリプシン-EDTAを加え(3mL/T-75、7.5mL/T-225)、そのTフラスコを37℃、5%CO2インキュベータで約15分間インキュベートした。フラスコを手のひらで数回叩き、細胞を完全に剥離させた。その後トリプシンを、等量のライマメ・トリプシンインヒビターを加えること(3mL/T-75、7.5mL/T-225)によって阻害した。細胞懸濁液を適切なサイズのピペットで上下に吸引、排出をすることによって均質にした。0.5mLの二つの細胞懸濁液サンプルを、細胞数をカウントするために取った。二つのカウント結果がお互いに15%以内にない場合は、細胞のカウントを繰り返した。カウント後、細胞を新しいフラスコの予熱した新しいTaub's培地で0.05×106生細胞/mLに希釈し、総量35mL/T75、又は100mL/T-225とした。その後、フラスコを37±1℃、5%CO2の環境下でインキュベートした。細胞を3日目に新しいTフラスコに継代する(下記のように)か、又は全培地交換を行い、接種後4日目に新しいTフラスコに継代した。
【0130】
Taub's培地に適応したMDCK細胞PreMCBバンクの調製
Taub's無血清に適応したMDCK細胞株(MDCK-T)の前マスター細胞バンクを、上記実施例2のように調製した。ただし、2×冷凍培地はTaub's培地+15%DMSOとした。
【0131】
Taub's培地に適応したMDCK(MDCK−T)細胞の特徴解析
MDCK-T pre-MCBの核型テスト、無菌テスト及びマイコプラズマテストを、実施例2に記載したように行った。ただし、血清含有完全培地の代わりにTaub's培地を使用した。さらに、T-75フラスコでのMDCK-T細胞の成長曲線の特徴、及びMDCK-T細胞での低温適応性インフルエンザ株の複製を、実施例2に記載したように行った。ただし、血清含有完全培地の代わりにTaub's培地を使用した。腫瘍形成能実験は、上記実施例2のようにBioRelianceによって継代88/29におけるMDCK-T(pre-MCB+20継代)で行われた。
【0132】
<結果と考察>
MDCK-T preMCB(継代64/5)細胞の冷凍バイアルを、T-75フラスコ内の無血清Taub's培地中で解凍した。細胞生存率は97%であった。また5.25×106細胞を解凍時に冷凍バイアルから回収した。細胞は解凍後3日でコンフルエントに達した。細胞形態は、親細胞であるMDCK-S細胞に似た上皮様細胞形態を示した(図9)。
【0133】
Taub's無血清(SF)培地でのMDCK-T細胞の成長曲線を図10に示す。この結果は、異なる継代数(P71/12及びP73/14)の細胞を用いた2つの実験の平均である。MDCK-T細胞は、接種後1日で細胞が倍化し、誘導期が見られなかった(1日目の総細胞数3.42×106細胞/T75フラスコ 対 0日目の総細胞数1.75×106細胞/T75フラスコ)。静止期に入る4日目まで、細胞は対数増殖期にあった。対数期における細胞の倍化時間は、20.4時間であった。対数期(0日目〜4日目)の間、細胞はグルコースとグルタミンを利用する(図11、及び12)と同時に、乳酸及びアンモニアを産生した。グルコース消費/乳酸産生比率は、細胞の成長曲線とよく対応していた(図10と11を比較されたい)。グルコース消費速度は0日目〜4日目の対数期の間で1.78mM/日であり、乳酸は2.88mM/日の速度で産生された。MDCK-T細胞は、7日間までの培養で培地中のグルコースを合計約10mM消費しただけだった。接種後4日目のグルコース消費量対乳酸産生量の比率は、1.2であった。グルコース消費率と乳酸産生率は、細胞が静止期に入った4日目以降に減速した。すなわち、グルコース消費は0.65mM/日であり、乳酸は0.46mM/日の速度で産生された。37±0.24×106の最大細胞密度には、接種後およそ4日目で達した。細胞密度は静止期の間も下がらず、7日目まで一定であった。
【0134】
グルタミン消費率とアンモニア産生率は、MDCK-T細胞の増殖、及びグルコース/乳酸のプロファイルに似ていた(図10、11、及び12を比較されたい)。MDCK-T細胞は、対数増殖期(0日目〜4日目)の間は0.36mM/日の速度でグルタミンを消費したが、静止期(4日目〜7日目)に入ると0.27mM/日に減速した。アンモニア産生は、7日目まで0.22mM/日の速度で直線的に増加した。アンモニア産生対グルタミン消費の比率は、接種後4日目で0.49であった。グルタミン酸消費は、7日間の全期間中、目に見える変化はなかった。
【0135】
MDCK-T細胞を、実施例2のようにca/tsインフルエンザ株の複製を支持する能力についてテストした。表2に示す結果は、MDCK-T細胞がca/tsインフルエンザ株の複製をMDCK-S細胞で見られた結果とほとんど同じレベルで支持できることを示している。
【0136】
MDCK-T細胞の核型を継代68/9及び継代88/29でテストした。Gバンド染色体解析は細胞がイヌ科起源であることを示した。100個の中期細胞における染色体数の分布を図13に示した。染色体の総数は、低継代68/9の細胞については中期あたり52本〜82本、また高継代81/24については54本〜84本の範囲にあった。これは、染色体の分布が継代によって変化しないことを示している。しかし、MDCK-S細胞(70本〜84本)と比べると、MDCK-T細胞(52本〜84本)では染色体数が広範囲に広がる結果が見られた
MDCK-T preMCBを細菌汚染、真菌汚染、又はマイコプラズマ汚染のいずれかの存在についてテストした。MDCK-T pre-MCBは無菌テスト(細菌及び真菌の汚染を確認するための直接接種法を用いた4培地無菌テスト)に合格し、マイコプラズマの存在(アガー培養可能、及び非アガー培養可能アッセイ)についてネガティブであることがわかった。被験物質は、静菌性/静真菌性テスト、及び静マイコプラズマ性テストのどちらもポジティブコントロールの増殖を阻害しないこともわかった。
【0137】
継代88/29のMDCK-T細胞(pre-MCB+20継代)をヌードマウス上に接種し、3ヶ月間腫瘍形成テストを行った。被験物質は、被験マウス10匹のうち6匹において注射部位で腺癌と診断された。これは無血清Taub's培地で増殖したMDCK細胞が、腫瘍形成性であることを示している。MDCK-S及びMDCK-T細胞についてのTP50で評価した腫瘍形成能、及び核型は、以下の表4にまとめている。
【0138】
実施例4
MediV無血清培地における無血清MDCK細胞の誘導
実施例3で詳述した結果は、無血清Taub's培地での増殖に適応したMDCK細胞(MDCK-T)が卓越した増殖特性を有し、またca/tsインフルエンザ株の複製を支持できるものの、腫瘍形成性であることを証明している。それゆえ、これらの結果は、MDCK細胞が文献で報告された標準的な無血清培地処方で容易に形質転換し得ることを示している。本発明にしたがって、いくつかのさらなる無血清培地処方物を開発し、MDCK-S細胞の非腫瘍形成性の性質を維持する能力をテストした。MDCK-S細胞を、新しい無血清製剤MediV SFM101、102及び103のそれぞれに適応させた。これらの無血清適応細胞株を、それぞれMDCK-SF101、-SF102及び-SF103とし、まとめて「MDCK-SF」と呼ぶ。PreMCBを、各MDCK-SF適応細胞株について作製した。MDCK-SF細胞株PreMCBを、細菌/真菌汚染、及びマイコプラズマ汚染についてテストした(最終結果待ち)。またMDCK-SF preMCBを核型解析でもテストした。その結果、MDCK-SF101及びMDCK-SF102細胞は、78本の最頻値染色体数を有し、染色体数はそれぞれ70本〜82本、及び60本〜80本に及んでいた。さらに、それぞれの無血清培地バンク由来の細胞をPreMCBのバイアルから起こして少なくとも新たに20回継代した。そして、MDCK-SF103を核型及びインビボアダルトヌードマウスモデルでの腫瘍形成能についてテストした。継代87のMDCK-SF103は、78本の最頻値染色体数を有し、染色体数は66本〜80本に及んでいたことから、非腫瘍形成性であるとみなした。
【0139】
<材料と方法>
材料
MDCK細胞(ATCC, Cat. No:CCL-34);T-25、T-75、T-225フラスコ(Corning社、Cat No.: 430639, 430641, 431082);ダルベッコ改良イーグル培地(DMEM)粉末(Gibco社、Grand Island NY, Formulation No.:01-5052EF);Ham F12栄養素混合物粉末(Gibco社、Grand Island NY, Formulation No.:21700-075);ガンマ線照射済みウシ胎仔血清(JRH社、Lenexa KS, Cat. No.:12107-500M);L-グルタミン(JRH社、Lenexa KS, Cat. No. 59202-100M);D-グルコース(Amresco社、Cat. No.:0188-1KG);Ca2+及びMg2+非含有のダルベッコリン酸バッファ(DPBS)(Gibco社、Grand Isand NY, Cat. No.:21600-069);インスリン粉末(Serological社、Cat. No.4506);トランスフェリン(APO型)(Gibco社、Grand Island NY, Cat. No.:11108-016);プロスタグランジンE1(Sigma社、St. Louis MS, Cat. No.:P7527);ヒドロコルチゾン(Mallinckrodt社、Cat. No.:8830(-05));トリヨードチロニン(Sigma社、St. Louis MS, Cat. No.:T5516);亜セレン酸ナトリウム(EMD社、Cat. No.:6607-31);0.05%トリプシン-EDTA(Gibco社、Grand Island NY, Cat. No.:25300);ライマメ・トリプシンインヒビター(Worthington社、Cat. No.:LS002829);ジメチルスルホキシド(DMSO)(Sigma社、St. Louis MS, Cat. No.:D2650);0.4%(w/v)トリパンブルー/PBS溶液(Sigma社、St. Louis MS, Cat. No.:T8154);改良型ノイバウェル血球測定器(Hausser Scientific社、Bright-Line 0.1mm deep/Reichert, Bright-Line 0.1mm deep);YSIバイオアナライザ(YSI社、Model No.: 2700 select);VITROS化学システム(Ortho clinic社、Model: DT60 II)。
【0140】
MediV無血清培地の処方(MediV SFM101、102及び103)
各MediV無血清培地処方物は、基礎培地としてTaub's培地(上記実施例2の方法の項参照)を用い、以下の補足剤を添加している。
【0141】
MediV SFM 101: Taub's+2.5g/L コムギペプトンE1(Organo Techine社、cat no.19559)。コムギペプトンE1については、水に溶かして250g/Lの滅菌済ストック溶液として保存する。
【0142】
MediV SFM 102: Taub's+最終濃度1×で添加される100×化学的に規定された脂質濃縮物(GIBCO BRL社、cat no.11905)。
【0143】
MediV SFM 103: Taub's+最終濃度1×の脂質濃縮物(GIBCO社)+2.5g/L コムギペプトンE1(Organo Techine社)。
【0144】
MediV SFM 104: Taub's+最終濃度1×の脂質濃縮物(GIBCO社)+2.5g/L コムギペプトンE1(Organo Techine社)+0.01μg/mL EGF(複数の販売元から)。
【0145】
MediV SFM 105: トランスフェリン非含有Taub's培地+最終濃度1×の脂質濃縮物(GIBCO社)+2.5g/L コムギペプトンE1(Organo Techine社)+0.01μg/mL EGF+10:1〜70:1の比率でクエン酸第二鉄アンモニウム:トロポロン、又は硫酸鉄アンモニウム:トロポロン。
【0146】
無血清MediV SFM培地処方物へのMDCK-S細胞の適応
ATCC由来のMDCK細胞の冷凍バイアルを、MediV SFM培地処方物(MediV SFM101、MediV SFM102、又はMediV SFM103)に継代する前に4.5g/Lグルコース、2.2g/L NaHCO3、及び4mM L-グルタミンを含んだ10%FBS DMEM培地で5回継代して増やした(上記のようにした)。T-75フラスコで増やした血清MDCKをトリプシン処理で回収した。使用した増殖培地を除き、細胞単分子層をDPBS(カルシウム及びマグネシウムなし)で2回洗浄した。その後、DPBSを廃棄した。適当量の予熱したトリプシン-EDTAを加え(3mL/T-75)、Tフラスコを37℃、5%CO2インキュベータで約15分間インキュベートした。フラスコを手のひらで数回叩き、細胞を完全に剥離させた。等量のライマメ・トリプシンインヒビターを加え、トリプシンを中和した後、サンプルを二つ取り、細胞懸濁液の細胞濃度を測定した。その後、1.75×106細胞を新しいT75フラスコ中の所望のMediV SFM培地35mLに希釈した。そのフラスコを5%CO2、37±1℃に維持された細胞培養インキュベータ内に入れた。細胞を接種後3日継代培養するか、全培地交換を接種後3日目に行い、続く4日目に継代した。細胞は、上記と同じ方法を用いたMediV SF104及びMediV SF105にも適応すると思われる。
【0147】
MediV SFM培地に適応したMDCK細胞の継代
使用した培地を除き、細胞単分子層をDPBS(カルシウム及びマグネシウムなし)で2回洗浄した。適当量の予熱したトリプシン-EDTAを加え(3mL/T-75、7.5mL/T-225)、Tフラスコを37℃、5%CO2インキュベータで約15分間インキュベートした。フラスコを手のひらで数回叩き、細胞を完全に剥離させた。その後、トリプシンを、等量のライマメ・トリプシンインヒビターを加えること(3mL/T-75、7.5mL/T-225)によって阻害した。細胞懸濁液を適切なサイズのピペットで上下に吸引、排出をすることによって均質にした。0.5mLの二つの細胞懸濁液サンプルを、細胞数カウント用に取った。二つのカウント結果がお互いに15%以内にない場合は、細胞のカウントを繰り返した。カウント後、細胞を新しいフラスコの予熱した適当な新しいMediV SFM培地処方物で0.05×106生細胞/mLに希釈し、総量35mL/T75、又は100mL/T-225とした。その後、フラスコを37±1℃、5%CO2の環境下でインキュベートした。細胞を3日目に新しいTフラスコに継代する(下記のように)か、又は全培地交換を行い、接種後4日目に新しいTフラスコに継代した。注記:MDCK-SF細胞については、いつもその細胞が適応したときと同じMediV SFM培地処方物で継代すること。
【0148】
MediV SFM培地に適応したMDCK細胞PreMCBバンクの調製
無血清適応MDCK細胞株の前マスター細胞バンクを、上記実施例1のように調製した。ただし、2×冷凍培地は適当なMediV SFM培地処方物+15%DMSOとした。
【0149】
MediV SFM培地に適応したMDCK(MDCK-SF)細胞の特徴解析
MDCK-SF pre-MCBの核型テスト、無菌テスト及びマイコプラズマテストを本明細書に記載の手順、例えば実施例2に記載の手順にしたがってテストした。ただし、血清含有完全培地の代わりに適当なMediV SFM培地処方物を使用した。さらに、T-75フラスコでのMDCK-SF細胞の成長曲線の特徴、及びMDCK-SF細胞での低温適応性インフルエンザ株の複製を、実施例2に記載したように実験した。ただし、血清含有完全培地の代わりに適当なMediV SFM培地処方物を使用した。さらに、腫瘍形成能実験が、さらなる継代(例えば、pre-MCB+20継代)の後に上記実施例2のような通常民間の受託業者(例えば、BioReliance)によってMDCK-SF細胞で行われ得る。
【0150】
<結果と考察>
MDCK-SF101細胞及びMDCK-SF102細胞の核型を継代71/9で、またMDCK-SF103細胞の核型を継代87でテストした。MDCK-T、MDCK-SF101、及びMDCK-SF102の100個の中期細胞における染色体数の分布を図14に、またMDCK-SF103のそれを図19に示す。MDCK-T細胞は、MDCK-SF101、MDCK-SF102又はMDCK-SF103の各細胞(それぞれ70本〜82本、60本〜80本、及び66本〜80本)と比べると、染色体数が広範囲に広がる結果(52本〜84本)を示すことがわかった。MDCK-SF101、MDCK-SF102及びMDCK-SF103の各細胞の染色体数の広がりは、非腫瘍形成性MDCK-S血清増殖細胞(70本〜84本)で見られたものに非常に近い。これは、MediV SF101、MediV SF102、及びMediV SF103培地処方物が、当該製剤で増殖したMDCK細胞の正常な染色体数をより適切に維持できることを示唆している。
【0151】
MediV SF103培地におけるMDCK-SF103細胞の典型的な予備成長曲線を図16に示す。MDCK-SF103細胞では約1日の誘導期が見られた。静止期に入る4日目まで、細胞は対数増殖期にあった。対数期(0日目〜4日目)の間、細胞はグルコースとグルタミンを利用(図17、及び18)すると同時に、乳酸及びアンモニアを産生した。グルコース消費/乳酸産生比率は、細胞の成長曲線とよく対応していた(図16と17を参照)。〜17×106の最大細胞密度には、接種後およそ4日目で達した。細胞密度は静止期の間も下がらず、7日目までほぼ一定であった。
【0152】
グルタミン消費率とアンモニア産生率は、MDCK- SF103細胞の増殖、及びグルコース/乳酸のプロファイルに似ていた(図18参照)。アンモニア産生は7日目まで線形的に増加したが、グルタミン酸濃度は、7日の期間中、目に見える変化はなかった。
【0153】
MDCK-SF103細胞を、以下の実施例7に記載するようないくつかのリアソータントインフルエンザ株の複製を支持する能力についてテストした。表20Aに示すその結果は、MDCK-SF103細胞がテストした各インフルエンザ株の複製を支持できることを示している。
【0154】
MDCK-SF103細胞をヌードマウス上に接種し、上記のように3ヶ月間腫瘍形成テストを行った。被験物質は、アダルトヌードマウスモデルで非腫瘍形成性であると見なされた(RioReliance 実験番号 AB09EU.001000.BSV)。
【表4】
【0155】
実施例5
培養におけるヒト上皮細胞の感染
ヒト起源の細胞内におけるMDCK及び卵で作られたワクチンの複製後の生化学的、生物学的及び構造的類似性を評価するため、ワクチンを正常なヒト気管支上皮細胞(human bronchial epithelial cells:NHBE)等の関連二倍体ヒト細胞で1回継代する。この継代は、ヒトの気道内における感染の一形態を模倣するのに役立ち、また子孫ウイルスとの比較を可能にする。この子孫ウイルスは、詰まるところ免疫反応の有効な誘導に関与する。ワクチンのヘマグルチニン活性(結合及び融合)並びにノイラミニダーゼ活性を、電子顕微鏡法、感染総粒子率、ウイルスゲノムの等価性を含む他の生化学的及び構造的実験と共にこれらの材料で測定した。概して、これらの比較は、効率的かつ安全な卵で作られたワクチンに対する細胞由来ワクチンの比較可能性を証明するのに役立つ。細菌及び真菌汚染の有無をテストする方法は当該分野では周知であり、通常民間の受託業者(例えば、BioReliance(登録商標);Rockville, MD)で行われる。実施され得る分析的研究の概要は、表5にまとめた。
【表5】
【0156】
実施例6
マスター細胞バンクの製造、テスト、及び特徴解析
マスター細胞バンク(MCB)の作製を始めるために、一以上の上記preMCB(実施例2〜4参照)由来の細胞を、生成する細胞が遺伝学的に一つの群に由来することを確実にするために限定希釈法により生物学的にクローン化した。その後、クローンを、倍化時間や関連する腫瘍形成性、そしてウイルス生産を含む様々な表現型特性についてスクリーニングした。最初の概念実証実験で、54個のMDCKクローンがFCSを含む培地で得られた。これらのクローンを継代し、それぞれを低感染多重度のca A/New Caledonia/20/99で感染させた。感染後数日して上清を取り出し、上清中のウイルス量をTCID50で測定した。少数のクローンは、クローンでない親細胞における生産を超える比較的高いタイターのウイルスを生産していた。生物学的及び生理学的に優れた特性を持つクローンをマスター細胞バンク(MCB)の樹立に使用した。
【0157】
MCBを広範囲にわたってテストし、外来の作用因子の痕跡がないことを確実にした。例えば、以下の表6で示すようなウイルス性因子に利用できる一以上のPCR及び/又は市販の抗体特異的テストを実施した。
【表6】
【0158】
実施例7
ワクチン物質の製法と製剤
近年認可されたポリオワクチンの生産に使用されるものに類似するスケールアップ性の高いマイクロキャリア技術の使用をMDCK細胞でのインフルエンザの生産に使用可能にした。デキストランでできた球状ビーズは、2〜10Lバイオリアクター内でMDCK細胞の増殖を支持するのに優れている。SFMV103培地中で増殖した親MDCK細胞は、Cytodex 1マイクロキャリアで、両スピナーフラスコのバッチモードで2×106核/mlの密度まで増殖することができることがわかった。また、MDCK細胞を最大10Lスケールのバイオリアクター内で1×106細胞/ml以上まで増やした(データ示さず)。最初のパイロットスケールでの稼動で、MDCK細胞が無血清法でワクチンインフルエンザ株を高タイターで生産できることが証明された。またそのタイターは、Tフラスコ内で血清増殖細胞を使用して得た生産性と同等か又はそれ以上であることがわかった。図20Aで示したように、250mLのスピナーフラスコ内のCytodexビーズ中で増殖したMDCK細胞は、高タイターのH1N1、H3N2及びB型ワクチン株を生産した。医療用製造については、インフルエンザウイルスをMDCK細胞内で20L〜150L規模で生産することができる。一方、商業規模生産の場合、2,500Lのバイオリアクターを利用することもできる。図20Bは、商業生産レベルまでスケールアップした細胞培養を利用できる一のプロセスを概説している。まず、使用する細胞バンクをT-75フラスコからT-225フラスコ、1Lスピナーフラスコ、20Lバイオリアクター、そして300Lバイオリアクターへと連続的に増やし、最終的には2500Lバイオリアクターにまで拡大する。最適細胞密度が得られたときに、培養液にマスターウイルス株を接種する。その後、ウイルスを培養液上清から一括回収する。
【0159】
細胞培養ベースのインフルエンザワクチンの精製工程は、卵ベースのインフルエンザワクチンの精製をモデルにしている(例えば、国際公開番号第05/014862号、及び2005年10月4日出願の国際出願番号第US05/035614号を参照されたい)。細胞からのウイルスワクチン物質の精製は、以下の工程のいずれか、又は全てを含み得る。すなわち、ホモジネーション、清浄化遠心分離、限外濾過、硫酸バリウムへの吸着及び溶出、タンジェンシャルフロー濾過(tangential flow filtration)、密度勾配超遠心、クロマトグラフィー、及び濾過滅菌が挙げられる。その他の精製ステップを含んでいてもよい。例を挙げると、まず、感染培養物由来のクルードな培地を例えば1000〜2000×gで細胞片及びその他の大型粒状物質を除くのに十分な時間(例えば、10〜30分)遠心して清澄化することができる。あるいは、培地を0.8μmの酢酸セルロースフィルターに通して濾過して、無傷の細胞及びその他の大型粒状物質を除く。その後、任意で清浄化した培地の上清を、例えば15,000×gで約3〜5時間遠心し、インフルエンザウイルスを沈殿させる。STE (0.01M Tris-HCl;0.15M NaCl;0.0001M EDTA)又はpH7.4のリン酸バッファ(PBS)等の適当なバッファにウイルス沈殿物を再懸濁した後、ウイルスをショ糖(60%〜12%)、又は酒石酸カリウム(50%〜10%)の密度勾配遠心で濃縮してもよい。連続的勾配か、又は例えば12%〜60%間で12%ごと4段階といった段階的勾配のショ糖勾配のいずれかが適している。勾配は、ウイルスを回収のため目に見えるバンドに凝集させる上で十分な速度、及び時間で遠心分離される。あるいは、市販の大規模なアプリケーションのほとんどは、ウイルスを密度勾配から連続モードで稼動して帯状遠心分離ロータを使用して溶出する。
【0160】
細胞からのウイルスワクチン物質の精製に含めることのできる特徴として、初期工程での非特異的エンドヌクレアーゼBenzonase(登録商標)の使用がある。細胞DNAの発癌性を評価する研究によればMDCK細胞DNAは発癌リスクがないが、Benzonase処理によってあらゆる可能性のあるリスク又は仮説的なリスクを実質的に排除することができる。一の精製工程において、Benzonase処理後、当該材料をダイレクトフロー濾過(DFF)によって清浄化した。DFFは、バルク材中のあらゆる残余無傷哺乳動物細胞をも除くことができる。その後、濾過したバルクを次の精製ステップ前にタンジェンシャルフロー濾過(TFF)で濃縮する。他のウイルスシステムにもよく使用されるアフィニティークロマトグラフィーとイオン交換クロマトグラフィー、及び/又はヒドロキシアパタイトを含む精製方法は、細胞培養ベースのインフルエンザワクチン生産に有用である。開発した方法で得られた高精製ウイルス物質は、その後、ワクチン物質の製造に利用される。例えば、弱毒化生ワクチン製造(例えばFluMist(登録商標))での使用に関しては、ウイルス物質を最終処方中に濾過によってバッファ交換し、その後滅菌ステップをすることができる。このような処方に有用なバッファは、アルギニン等のその他のアミノ酸賦形剤を添加した200mMショ糖、及びリン酸又はヒスチジンバッファ(pH7.0〜7.2)を含み得る。安定化が必要であれば、ブタゼラチン等のタンパク質加水分解物も添加することができる。ワクチン物質を長期保管時間、安定するように処方するのが理想的である。長期保管時間に利用することができる一の方法として噴霧乾燥法がある。これは液体製剤試料を乾燥加熱気体の気流下で微細液滴に噴霧化することによって、試料を急速乾燥させる方法である。微細液滴の蒸発によって溶解溶質からなる乾燥粉末が生じる(例えば、米国特許公開公報番号2004/0042972号を参照されたい)。噴霧乾燥法は、スケールアップ化が容易な点や従来の凍結乾燥法と比べると生産原価の点で有利である。さらに、ワクチン物質を製剤化し、当該分野で既知の方法を用いて冷蔵庫で安定な液体製剤として安定化させる。例えば、冷蔵庫で安定な弱毒性インフルエンザワクチンを処方するための方法と組成物は、2005年10月4日出願の国際出願第PCT/US2005/035614号に記載されている。
【0161】
製造過程において、特徴解析ステップは精製ステップの中に組み込まれて、その生産がモニターされる。利用可能な特徴解析ステップは、ウイルス感染性を測定するための単一の抗体結合を用いる蛍光焦点アッセイ法(FFA、例えば上記参照)及び蛍光染色法を含むが、これらに限定はされない。総タンパク質及びDNAの測定は、当業者に既知の多くの方法を用いて実施することができ、またこれらの測定は残存する初期不純物のパーセントを測定するのに使用される。調製物の特定の活性を、ワクチン量あたりのウイルス感染価(例えば、感染価/mg)を算出することで測定することができる。
【0162】
実施例8
前臨床動物モデル
フェレットは丈夫な動物モデルで、弱毒化したインフルエンザワクチン及びワクチン株の成分の弱毒性、並びに免疫原性を評価するのに使用される。MCBから作られた細胞由来のインフルエンザ株の性能を、卵で作られた同一株と比較する。コントロール実験におけるこれらの材料の直接的な比較は、これらのウイルス産物の比較の確かさを高レベルにすることができる。
【0163】
二つのワクチンのフェレットでの感染能力又は「(免疫)獲得」能力を評価するために、動物を軽く麻酔し、細胞又は卵で作られたウイルス調製物のどちらかを鼻腔内に接種した。接種後に鼻洗浄物を数時点で回収し、ウイルス量を利用可能ないくつかの方法のうちの一つで、その動物の上気道における動力学及びウイルスの複製の広がりを判断するために、評価する。実験を投与量の範囲で実施し、複数株及び異なる三つの混合物を包含して細胞培養増殖株対卵生産株の相対的感染力を標準化する。同様の研究がインフルエンザ株の免疫原性を評価するのにも使用される。免疫原性は、ウイルスが感染を起こす能力と本質的に関連する性質である。動物を飼育し、鼻洗浄物を接種後の様々な時点(週)で回収した。これらの試料を感染に対する血清抗体及び鼻のIgA応答を評価するのに使用した。これらのデータ、感染力、血清抗体、及び粘膜抗体応答の最高点は、細胞産生ワクチン対卵産生ワクチンの相対感染力の比較及び評価に利用することができる。最もあり得そうな結果は、細胞及び卵で作られたワクチン株が同様の感染力及び免疫原性を持っているということである。細胞由来のワクチンが卵由来の産物よりも感染力や免疫原性がより高いように見えるのであれば、低位側の投与量の可能性を評価するさらなる研究を行う。
【0164】
多数の免疫原性及び複製の研究がフェレットモデルで行われ、細胞培養由来ワクチンを単一ユニットのヒト投与量で評価する。ca/ts/att株による感染は、フェレットで通常急速かつ強い抗体反応を引き起こす。さらに、個々のca/ts/att株を定期的にテストする。当該株を鼻咽頭内で比較的高いタイターまで複製し、しかしこれらの動物の肺では検出されないレベルまで複製することで、弱毒性(att)表現型が発現することがわかる。これらの生物学的形質で増殖した細胞培養の影響も評価される。att表現型はこれらの株の遺伝学的構成と一体化した部分であるので、あらゆる相違が見られるというのは考えにくい。これらの株の増殖動力学と交差反応性をこれらの動物にヒト1回分の投与量で投与後に評価する。卵由来の材料から生産された弱毒性生ワクチンは、同一遺伝系統内の複数の株と交差反応する血清抗体を誘導する。そして、細胞由来のワクチンも同じ能力を持つであろうことが予想される。
【0165】
これらの比較可能性評価は、主要なウイルス産物の潜在的な生化学的及び/又は生物物理学的違いに重要な洞察を提供し、またヒトの細胞におけるウイルスの最初の継代又は動物実験によって測定されるca/ts/att株の性能の後成的な相違がもたらす影響を証明するはずである。現在までのところ配列情報に基づけば、MDCK細胞で生産されたことによって生じるca/ts/att株の免疫原性能力において予期された衝撃的な影響はない。
【0166】
フェレットは、インフルエンザ用に文献上十分証明されたモデル動物であり、弱毒表現型とca/ts/att株の免疫原性を評価するのに日常的に使用されている。通常、8〜10週齢の動物を使用して弱毒性を評価する。一般的な実験計画では1回のテスト又はコントロール群につきn=3〜5匹の動物が評価される。免疫原性実験では、8週〜6月齢の動物で評価し、通常1回の被験物質又はコントロール群につきn=3〜5匹の動物を必要とする。これらの匹数は、グループ間で統計的に有効な又は観察上重要な比較結果を得る上で十分な情報を提供する。ほとんどの実験中にインフルエンザ様の徴候に気づくかもしれないが、簡単ではないであろう。フェレットは食欲又は体重の減少、鼻汁又は眼漏等の病気の徴候を示さないからである。インフルエンザ様の疾患の徴候を観察することは、研究に必要な一部であり、鎮痛剤等の介入を行うべきでない。宿弊又は深刻な体重減少等の他の不快症状は、担当する獣医と話し合った後、その動物にとって適切な処置がなされるであろう。
【0167】
本発明は特定の実施形態に関して開示したが、本発明の他の実施形態や変形型が本発明の本質及び範囲を逸脱することなしに当業者により考え出され得ることは明らかである。添付の特許請求の範囲は、そのような全ての実施形態及び均等の変更を含むと解されることが意図されている。例えば、上記全ての技術及び装置を様々な組合せで使用することができる。本出願で引用された全ての刊行物、特許、特許出願、又はその他の資料は、各個の刊行物、特許、特許出願、又はその他の資料が、参照によって全ての目的に援用するために個別に示される場合と同程度に、参照によって全ての目的に援用される。さらに、2004年の12月23日出願の米国仮出願第60/638,166号、及び2005年1月5日出願の第60/641,139号を、この記載によって、本明細書に引用して全ての目的にその全体を援用する。
【技術分野】
【0001】
発明の分野
本発明は、ワクチン物質の生産に利用することのできる新規非腫瘍形成性MDCK細胞に関する。非腫瘍形成性MDCK細胞は無血清培地に適応することができる。本発明は、さらに非腫瘍形成性MDCK細胞の増殖用培地処方物及び培養方法、並びに本発明の細胞株の非腫瘍形成性質を維持する方法に関する。本発明は、さらに非腫瘍形成性MDCK細胞を用いた細胞培養でのインフルエンザウイルスの生産方法に関する。本発明はまた、記載された方法によって得ることができるウイルス(例えば、インフルエンザ)、及びこのタイプのウイルスを含む免疫原性組成物及び/又はその成分に関する。
【背景技術】
【0002】
発明の背景
ワクチン接種は、年1度のインフルエンザの流行によりもたらされる病気を予防する上で最も重要な公衆衛生措置である。ワクチンの効率的な利用は、大量のワクチン物質(例えば、ウイルス)を、安定かつ培養の容易な供給源からすばやく生産できることに依る。ワクチンの早急な開発とそれらを十分に利用できるようにすることは、ヒト及び動物の多くの病気に対抗する上で重要である。ワクチン生産の遅延やその量の不足は、病気の発生に対処する上で問題となり得る。例えば、最近の研究では世界的流行性(パンデミック)インフルエンザに対するワクチンの生産に長期の準備期間を要することについての心配が示唆されている。例えば、非特許文献1(Wood, J. M., 2001, Philos. Trans. R. Soc. Lond. B. Biol. Sci., 356:1953)を参照のこと。効率的なワクチン生産には、宿主系から高収量で生産されるワクチン物質の大量増殖が必要である。異なるワクチン物質から満足のいく収量を得るためには異なる増殖条件が必要となる。ワクチン物質は、孵化卵、初代組織培養細胞、又は株化細胞で生産することができる。しかし、これらの宿主系は、現在以下で詳述する多くの制限を受けている。
【0003】
孵化卵は、通常、鶏の飼育や卵の受精の管理を要する時間、労力及びコスト集約的方法の下でインフルエンザワクチンウイルスの生産に利用されている。さらに、卵中で作られるインフルエンザワクチンは、卵アレルギーをもつ人にとっては重度の即時性過敏反応を起こし得るため禁忌である。それ故、ワクチン業界が努力して、例えば細胞培養系でインフルエンザワクチンを生産するといった卵を利用しない代替の生産基盤を開発した。
【0004】
初代組織培養細胞の利用は、安定した初代細胞集団を開発し、維持することの難しさに直面し、それがネックとなっている。株化細胞がしばしば使用され、初代細胞の技術的制限を回避している。しかし、これらの細胞株の多くは、腫瘍原性であることが知られており、それ自体が安全性の問題を引き起こすので、ワクチン生産の使用に対して規制面でかなりの制約を受ける。事実、世界保健機関の適用ガイドラインには、ごくわずかの細胞株しかワクチン生産に許可されていないことが示されている。さらなる問題は、細胞培養の培地中の動物若しくはヒト由来の血清及び/又はタンパク質添加物の使用に起因している。例えば、多数の添加物間の質及び組成の変動、並びにマイコプラズマ、ウイルス、BSE剤、及びその他の伝染性物質による汚染の危険性が、よく知られている。一般に、血清、又はアルブミン、トランスフェリン、若しくはインスリンのような血清由来の物質は、培養物を汚染する可能性のある厄介な物質を含み得る。また生物学的な製品は、それらから生産されている。それゆえ、多数の研究グループが血清や血清由来の製品を必要としない効率的な宿主系及び培養条件の開発のために勤しんでいる。
【0005】
したがって、低コストで、安全性が高く、かつ安定した様態で、好ましくは無血清若しくは動物性タンパク質フリーの培養条件下で、ワクチン物質を生産するのに有用な非腫瘍形成性細胞株の樹立に対する要望があった。このような細胞系は、特にインフルエンザワクチン物質の生産に有用であると思われる。
【0006】
イヌ腎臓由来(Madin Darby Canine Kidney:MDCK)細胞は、元々インフルエンザウイルスの滴定に使用されていた(非特許文献2(Zambon M.著, in Textbook of Influenza, Nicholson編, Webster and Hay, ch 22, pg 291-313, Blackwell Science (1998)))。この細胞は、1958年にコッカースパニエルの正常なオスの腎臓から株化された。ATCC(American Type Culture Collection)リストのMDCK(CCL 34)株は、S.MadinとN.B.Darbyにより寄託されたが、その他にも多くのMDCK細胞の細胞系が利用できる。Leighton Jとその共同研究者らは、MDCK細胞の発癌の特性を実証した一連の論文を発表した(非特許文献3(Leightonら、1968, Science 163:472);非特許文献4(Leightonら、1970, Cancer 26:1022);及び非特許文献5(Leightonら、1971 Europ J. Cancer 8:281))。しかし、それらの研究に使用されたMDCK細胞の系統及び継代数は記載されていなかった。また、異なる系統と異なる継代由来のMDCK細胞が、染色体数及び構造に違いを示すことは既に知られていた(非特許文献6(Gaushら, 1966, Proc. Soc. Exp. Biol. Med., 122: 931))。このような違いは、腫瘍形成性の性質をもった細胞を生じ得る。
【0007】
ワクチン生産用細胞株の受容性に関して考慮すべき主要な点の一つは、それらの細胞の潜在的悪性度と関係がある。それゆえ、現在記載されている細胞株を利用したワクチン物質生産のためのMDCK細胞の使用は制限される。Gronerら(特許文献1(米国特許番号第6,656,720号))、及びMakizumiら(特許文献2(米国特許番号第6,825,036号))の両者は、無血清培地において浮遊状態での増殖に適応し、またインフルエンザウイルスの生産に利用できるMDCK細胞由来の細胞株の開示を主張している。しかし、足場を必要としないことと、正常動物細胞の腫瘍形成性細胞への形質転換との間に相関関係があることが報告されている(非特許文献7(Stilesら、1976, Cancer Res., 36:3300))。いくつかのグループ(非特許文献8(Kesslerら、1999、Cell Culture Dev Biol Stand, 98:13);非特許文献9(Mertenら、1999, Cell Culture Dev Biol Stand, 98:23);及び非特許文献10(Treeら、2001, Vaccine, 19:3444))は、インフルエンザウイルスの大量生産用にMDCK細胞を使用することを記載し、主張している。しかし、彼らは使用したMDCK細胞の潜在的な形質転換に対しては何ら取り組んでいない。
【0008】
本明細書中の参考文献の引用又は考察は、それらを本発明の先行技術と認めたものとは解釈しないものとする。さらに特許の引用は、その効力を認めたものとは解釈しないものとする。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】米国特許番号第6,656,720号
【特許文献2】米国特許番号第6,825,036号
【非特許文献】
【0010】
【非特許文献1】Wood, J. M., 2001, Philos. Trans. R. Soc. Lond. B. Biol. Sci., 356:1953
【非特許文献2】Zambon M.著, in Textbook of Influenza, Nicholson編, Webster and Hay, ch 22, pg 291-313, Blackwell Science (1998)
【非特許文献3】Leightonら、1968, Science 163:472
【非特許文献4】Leightonら、1970, Cancer 26:1022
【非特許文献5】Leightonら、1971 Europ J. Cancer 8:281
【非特許文献6】Gaushら, 1966, Proc. Soc. Exp. Biol. Med., 122: 931
【非特許文献7】Stilesら、1976, Cancer Res., 36:3300
【非特許文献8】Kesslerら、1999、Cell Culture Dev Biol Stand, 98:13
【非特許文献9】Mertenら、1999, Cell Culture Dev Biol Stand, 98:23
【非特許文献10】Treeら、2001, Vaccine, 19:3444
【発明の概要】
【課題を解決するための手段】
【0011】
発明の概要
本発明は、動物性タンパク質非含有(animal protein-free:APF)の製剤を含む血清含有又は無血清の培地処方物のいずれかで増殖するのに適応した非腫瘍形成性MDCK細胞を提供する。一の実施形態において、本発明の非腫瘍形成性MDCK細胞は付着性である。他の実施形態において、本発明の非腫瘍形成性MDCK細胞は、上皮形態を有している。さらに他の実施形態において、本発明の非腫瘍形成性MDCK細胞は付着性で、かつ上皮形態を有している。発癌性が一の実施形態において、アダルトヌードマウスモデルで実証された(例えば、Stilesら,1976, Cancer Res, 36:1353、及び後述の実施例2)。発癌性については、例えば、鶏胚への注入、及び/又は漿尿膜への局所適用による他のアッセイででもテストすることができる(Leightonら,1970, Cancer, 26:1024)。
【0012】
本発明のMDCK細胞内で増殖できるウイルスは、マイナス鎖RNAウイルスを含むがそれに限定されない。また、当該マイナス鎖RNAウイルスは、インフルエンザ、RSV、パラインフルエンザウイルス1、2及び3型、並びにヒトメタニューモウイルスを含むが、これらに限定されない。
【0013】
本発明は、さらに非腫瘍形成性MDCK細胞の誘導とその維持に有用な方法及び培地処方物を提供する。本発明のMDCK細胞は、例えばウイルス等のワクチン物質の生産に特に有用である。
【0014】
本発明のその他の態様は、本発明のいずれかのMDCK細胞を、ワクチン物質の生産を可能にする条件の下で適切な培地にて培養すること、及びその物質を一以上の宿主細胞から又は宿主細胞を増殖させる培地から分離することによるワクチン物質(例えばウイルス)の生産方法を含む。
【0015】
免疫原性組成物も、本発明の特徴である。例えば、上記のように生産されるワクチン物質、及び場合により、製薬上許容される賦形剤等の賦形剤又は一以上の製薬上許容される投薬成分、を含む免疫原性組成物が挙げられる。
【0016】
被験体に、一以上の上記免疫原性組成物を効果的な量で投与することによって被験体に免疫原性反応を生じさせる方法も本発明に含まれる。さらに、ウイルス感染に対して免疫原性反応を引き起こすのに効果的な量で、一以上の上記の免疫原性組成物を投与することで被験体にウイルス感染(例えば、インフルエンザウイルスの)の予防的又は治療的処置を施す方法も本発明の一部である。前記処置を受ける被験体は、哺乳動物(例えば、ヒト)を含み得る。さらに、前記方法は、本発明のMDCK細胞で生産された一以上のウイルス及び製薬上許容される賦形剤からなる組成物を、予防的又は治療的にウイルス感染を治療するのに効果的な量で被験体に投与することも含むことができる。
【0017】
本発明のこれら及びその他の目的並びに特徴は、以下の詳細な説明を添付の図面と合わせて読むことで、より一層明らかとなるであろう。
【図面の簡単な説明】
【0018】
【図1A】細胞中のインフルエンザ株の増殖。図1Aは、MDCK細胞とベロ細胞のクローン(27F9)における代表的なca/tsインフルエンザ株の感染の広がりを比較した蛍光焦点アッセイの結果を示す写真である。
【図1B】細胞中のインフルエンザ株の増殖。図1Bは、MDCK細胞におけるインフルエンザ株ca A/Vietnam/1203/2004 (H5N1)の増殖曲線である。タイターは、感染後48時間に〜8 log10 TCID50/mLでピークに達し、その後3〜4日間は安定したままであった。
【図2】図2は、MDCK-S PreMCB(継代数57)の誘導に使用される方法を概説している。この方法については実施例2で詳述する。
【図3】図3は、MDCK-S細胞が上皮様形態を有することを示す写真である。写真は、接種後3日目に撮影された。
【図4】図4は、10% FBS DMEM培地におけるMDCK-S細胞の成長曲線である。細胞は約1日の誘導期後、指数関数的増殖へと続き、接種後4日目に静止期に入り、5日目に最大密度〜29×106細胞に達している。
【図5】図5は、10% FBS DMEM培地におけるMDCK-S細胞のグルコース消費及び乳酸産生のグラフである。誘導期中のそれらの速度は、グルコース及び乳酸についてそれぞれ2.93mM/日及び3.43mM/日の増加であり低かった。
【図6】図6は、10% FBS DMEM培地におけるMDCK-S細胞のグルタミン消費、並びにグルタミン酸及びアンモニア産生のグラフである。グルタミン消費速度は4日目までで0.49mM/日であり、アンモニア産生速度は、5日目までで0.32mM/日であった。グルタミン酸は本実験では蓄積しなかった。
【図7】図7は、低継代(P61/4)及び高継代(P81/24)の中期MDCK-S細胞100個における染色体数の分配のグラフである。高低の両継代細胞に関して78本の最頻値染色体数をもち、カウントされた染色体数は中期あたり70〜84本の範囲に及んでいた。
【図8】図8は、MDCK-T PreMCB(継代数64/5)の誘導に使用される方法を概説している。本方法については実施例3で詳述する。
【図9】図9は、MDCK-T細胞が上皮様形態を有することを示す写真である。写真は、接種後3日目に撮影された。
【図10】図10は、Taub培地におけるMDCK-T細胞の成長曲線である。細胞は誘導期を有さず、接種後4日目で静止期に入るまで指数関数的に増殖した。
【図11】図11は、Taub培地におけるMDCK-T細胞のグルコース消費及び乳酸産生のグラフである。対数期中、それらの速度はグルコース及び乳酸でそれぞれ1.78mM/日及び2.88mM/日であった。
【図12】図12は、Taub培地におけるMDCK-T細胞のグルタミン消費、並びにグルタミン酸及びアンモニア産生のグラフである。グルタミン消費速度は4日目までで0.36mM/日であり、アンモニア産生速度は7日目まで0.22mM/日で線形的に増加した。グルタミン酸は本実験では蓄積しなかった。
【図13】図13は、低継代(P61/4)及び高継代(P81/24)の中期MDCK-T細胞100個における染色体数の分布のグラフである。カウントされた染色体数は、低継代細胞では中期あたり52〜82本、また高継代細胞では54〜82本の範囲に及んでいた。
【図14】図14は、MDCK-T、MDCK-SF101細胞(継代数71/9)、及びMDCK-SF102細胞(継代数71/9)の各中期100個における染色体数の分布のグラフである。SF101及びSF102の両細胞とも78本の最頻値染色体数を有し、カウントされた染色体数は、SF101及びSF102でそれぞれ70〜82本及び60〜80本の範囲に及んでいた。
【図15】図15は、MDCK- SF103細胞が上皮様形態を有することを示す写真である。写真は、接種後3日目に撮影された。
【図16】図16は、MediV SFM103培地におけるMDCK-SF103細胞の成長曲線である。細胞は約1日の誘導期後、指数関数的増殖へと続き、接種後4日目に静止期に入り、4日目に最大密度〜17×106に達している。
【図17】図17は、MediV SFM103培地におけるMDCK-SF103細胞のグルコース消費、及び乳酸産生のグラフである。対数期中、グルコース消費及び乳酸産生は、お互いに酷似している。すなわち、グルコース濃度が減少したときに乳酸濃度が増加している。
【図18】図18は、MediV SFM103培地におけるMDCK-SF103細胞のグルタミン消費、並びにアンモニア及びグルタミン酸産生のグラフである。アンモニア産生速度は、7日目までほぼ線形的に増加した。グルタミン酸は、本実験では蓄積しなかった。
【図19】図19は、87継代の中期MDCK-SF103細胞100個における染色体数の分布のグラフである。SF103細胞は、78本の最頻値染色体数を有し、カウントされた染色体数は66〜80本の範囲に及んでいた。
【図20A】生産規模の増殖及び精製。図20Aは、数種のワクチンリアソータント株、すなわちB/Victoria/504/2000株(〜8 Log TCID50/mL)、A/Sydney/05/97株(〜7.85 Log TCID50/mL)、及びA/New Caledonia/20/99株(~8.2 Log TCID50/mL)について、Cytodexビーズ上で増殖させたMDCK-SF103細胞の250mLスピナーフラスコから得られた収量のグラフである。
【図20B】生産規模の増殖及び精製。図20Bは、ワクチン物質の工業規模生産に利用することができるようにスケールアップされた細胞培養方法の一つを概説している。
【発明を実施するための形態】
【0019】
発明の詳細な説明
本発明は、一部には、MDCK細胞を非腫瘍形成性の状態に維持する条件の下で培養できるという発見に基づく。本発明は、無血清培地処方物を包含する様々な細胞培養条件に適応し、また本明細書で「本発明の細胞」と呼ぶMDCK細胞株及びその他の細胞型を含む非腫瘍形成性細胞株を提供する。さらに、本発明は、本発明の細胞、及びその他の成分を含む細胞培養(cell culture)組成物を提供する。ここで言うその他の成分は、培地(例えば、本明細書で開示された培地)、培地成分、バッファ、化合物、その他の細胞型、ウイルス物質(例えば、ウイルスゲノム、ウイルス粒子)、及び異種タンパク質を含むが、これらに限定されない。本発明は、MDCK細胞を含む、一以上の特有の性質をもった非腫瘍形成性細胞の培養に有用な方法及び培地処方物も提供する。ここで言う特有の性質は、非腫瘍形成性であること(例えば、ヌードマウスで小結節を形成しないこと)、及び/又は付着性細胞として増殖すること、及び/又は上皮様形態を呈すること、及び/又は様々なウイルスの複製を支持することを含むが、これらに限定はされない。また、前記様々なウイルスは、オルソミクソウイルス(orthomyxovirus)、パラミクソウイルス(paramyxovirus)、ラブドウイルス(rhabdovirus)及びフラボウイルス(flavovirus)を含むが、これらに限られない。本発明の培養条件は、血清含有及び無血清培地処方物と共に動物性タンパク質非含有(APF)製剤を含む。さらに本発明は、MDCK細胞を含む非腫瘍形成性細胞でワクチン物質(例えば、インフルエンザウイルス)を生産する方法、非腫瘍形成性細胞からワクチン物質を調製すること、及び非腫瘍形成性細胞で生産されたワクチン物質を利用してインフルエンザ感染を予防する方法も提供する。本発明の細胞は、他の哺乳動物細胞株(例えば、Vero、PerC6、HEK-293、MRC-5及びWI-38細胞)中で効率的に複製されない低温適応性/温度感受性/弱毒性(ca/ts/att)インフルエンザ株(例えば、FluMist(登録商標)のウイルス株)の生産に特に有用である。
【0020】
<細胞特性>
本発明に係る細胞は、一の実施形態において脊椎動物細胞である。他の実施形態において、本発明の細胞は、例えばハムスター、ウシ、サル又はイヌ由来の哺乳動物細胞であり、特に腎臓細胞又はそれ由来の細胞株である。さらなる他の実施形態において、本発明の細胞は、(例えば、ATCC CCL-34 MDCK由来の)MDCK細胞であり、具体的には本明細書中で「本発明のMDCK細胞」と称され、また「本発明の細胞」の用語に包含される。具体的実施形態において、本発明の細胞はATCC CCL-34 MDCK由来である。本発明の細胞は、当業分野で周知の方法によるCCL-34 MDCK細胞由来の細胞であってもよい。例えば、まずCCL-34 MDCK細胞を血清含有培地(例えば、ダルベッコ改良イーグル培地(DMEM)+10%ウシ胎仔血清(FBS)+4mMグルタミン+4.5g/Lグルコース、又は本明細書に記載の他の培地)で限られた回数継代し、続いて個々の細胞をクローニングし、そのクローンを特徴付けてもよい。倍加時間、発癌性の特徴、及びウイルス生産を含むがこれらに限定されない生物学的及び生理学的に優れた特性をもつクローンを、マスター細胞バンク(master cell bank:MCB)生成用に選択する。一の態様において、本発明の細胞は、選択した培地(例えば、本明細書に記載したような無血清又はAPF培地)での増殖に適応している。このような適応は、個々の細胞のクローニング前、クローニング時、又はクローニング後に起こり得る。特定の実施形態において、本発明の細胞は、MediV SF101、MediV SF102、MediV SF103、MediV SF104、又はMediV SF105で増殖できるように適応している。これらの培地での増殖に適応した本発明の細胞を、本明細書では、それぞれ「MDCK-SF101、MDCK-SF102、MDCK-SF103、MDCK-SF104、そしてMDCK-SF105」細胞と呼び、また、まとめて「MDCK-SF細胞」と呼ぶ。他の実施形態において、本発明の細胞は、血清含有培地(例えばダルベッコ改良イーグル培地(DMEM)+10%ウシ胎仔血清(FBS)+4mMグルタミン+4.5g/Lグルコース)で増殖するように適応しており、そのような細胞を本明細書では「MDCK-S」細胞と呼ぶ。MDCK
-SF細胞及びMDCK-S細胞はまた、「本発明の細胞」及び「本発明のMDCK細胞」の用語に包含される。
【0021】
本発明の特定の実施形態において、細胞は株細胞である。本株細胞は、American Type Culture Collection(10801 University Boulevard, Manassas, Va. 20110-2209)に寄託され、ATCC寄託番号 PTA-6500 (2005年1月5日寄託)、PTA-6501 (2005年1月5日寄託)、PTA-6502(2005年1月5日寄託)、PTA-6503 (2005年1月5日寄託)が割り振られた株細胞であり、また本明細書ではそれぞれ「MDCK-S、MDCK-SF101、MDCK-SF102、及びMDCK-SF103」と呼び、まとめて「本発明のMDCK細胞」と呼ぶ株細胞を含むが、これらに限定はされない。これらの寄託物は、特許手続上の微生物の寄託の国際承認に関するブダペスト条約の条項に基づいて保存されるであろう。一の実施形態において、本発明のMDCK細胞は、ヒト用としてアメリカ食品医薬品局(the U.S. Food and Drug Administration)の承認に適したワクチン物質を調製する上で有用な細胞バンクを作製するのに利用される。
【0022】
株細胞MDCK-S、MDCK-SF101、MDCK-SF102、MDCK-SF103、MDCK-SF104、及びMDCK-SF105は、継代及び一以上の特有の性質に関する選択によって株細胞MDCK(CCL 34)から得られる。その性質は、血清含有培地、又は無血清若しくは動物性タンパク質非含有培地のいずれかで付着性細胞として増殖すること、上皮様形態を呈すること、非腫瘍形成性であること(例えば、ヌードマウスで小結節を形成しないこと)、及び/又は様々なウイルスの複製を支持することを含むが、これらに限定されない。また、前記ウイルスは、オルソミクソウイルス、パラミクソウイルス、ラブドウイルス及びフラボウイルスを含むが、これらに限定されない。
【0023】
一の実施形態において、本発明のMDCK細胞は非腫瘍形成性である。細胞が腫瘍形成性であるかどうかを決定する方法は、当該分野では周知である(例えば、Leightonらの1970, Cancer, 26:1024、及びStilesらの1976, Cancer Res, 36:1353を参照されたい)。アメリカ食品医薬品局で現在推奨のヌードマウスモデルを用いた方法を、以下の実施例2で詳述している。具体的な実施形態において、本発明のMDCK細胞はアダルトヌードマウスモデルで非腫瘍形成性である(前述のStilesらの文献、及び以下の実施例2を参照)。もう一つの具体的実施形態において、本発明のMDCK細胞は、鶏胚へ注射した際、及び/又は漿尿膜に局所的に適用した際に非腫瘍形成性である(前述のLeightonらの文献を参照)。さらなる他の実施形態において、本発明のMDCK細胞はアダルトヌードマウスモデルでは非腫瘍形成性であるが、鶏胚への注射、及び/又は漿尿膜に局所的に適用した場合は非腫瘍形成性ではない。さらに他の実施形態において、本発明のMDCK細胞は、アダルトヌードマウスモデルでも、また鶏胚への注射、及び/又は漿尿膜に局所的に適用した場合にも非腫瘍形成性である。さらなる他の実施形態において、本発明のMDCK細胞は、培地で少なくとも20継代後、少なくとも30継代後、少なくとも40継代後、少なくとも50継代後、少なくとも60継代後、少なくとも70継代後、少なくとも80継代後、少なくとも90継代後、又は少なくとも100継代後も非腫瘍形成性である。さらなる他の具体的実施形態において、培地は本明細書に記載した培地(例えばMedi SF103)である。
【0024】
腫瘍形成性は、当業者に多くの様々な方法で数値化することができる。一般的に利用される方法の一つは、「TD50」値を測定することである。この値は、テストした動物の50%に腫瘍を生じさせるのに必要な細胞数として定義される(例えば、Hill R. The TD50 assay for tumor cells. In: Potten C, Hendry J, editors. Cell clones. London: Churchill Livingstone; 1985. p. 223を参照されたい)。一の実施形態において、本発明のMDCK細胞は、約1010〜約101、約108〜約103、又は約107〜約104のTD50値を有する。具体的な実施形態において、本発明のMDCK細胞は約1010より大きい、約109より大きい、約108より大きい、約107より大きい、約106より大きい、約105より大きい、約104より大きい、約103より大きい、約102より大きい、又は約101より大きい、TD50値を有する。
【0025】
他の実施形態において本発明の非腫瘍形成性細胞は、血清含有培地、又は無血清若しくは動物性タンパク質非含有培地のいずれかで付着性細胞として増殖する。さらなる他の実施形態において、本発明の非腫瘍形成性細胞は上皮様形態を呈する。さらに他の実施形態において、本発明のMDCK細胞は、様々なウイルスの複製を支持する。ここで言うウイルスは、オルソミクソウイルス、パラミクソウイルス、ラブドウイルス及びフラボウイルスを含むが、これらに限定はされない。本発明のMDCK細胞は、一以上の特有の性質をどのような組合せで有していてもよいことが意図されている。ここでいう特有の性質は、非腫瘍形成性であること、付着性細胞として増殖すること、上皮様形態を有すること、及び様々なウイルスの複製を支持することを含むが、これらに限定はされない。
【0026】
本発明のMDCK細胞のあらゆる継代は、各株細胞の完全な系譜を利用できるように十分詳細に記録されていることが意図されている。あらゆる継代の記録は、ワクチン物質の調製のために本発明のMDCK細胞を使用することに対し、アメリカ食品医薬品局や世界中の他の取締機関の承認を円滑にすることができる。
【0027】
他の実施形態において、本発明のMDCK細胞は、微生物汚染(例えば、細菌、ウイルス、真菌の汚染)を含まない。細菌や真菌の汚染の有無を調べる方法は、当業分野で周知であり、通常民間の受託業者(例えば、BioReliance(登録商標);Rockville、MD)によって行われている。一般に承認されている微生物の無菌性及びマイコプラズマテストを、以下の実施例2で詳述している。テストされ得る微生物剤の具体例については、表6に列挙している。
【0028】
さらなる他の実施形態において、本発明のMDCK細胞は、ウイルスの複製を支持する。ここでいうウイルスは、オルソミクソウイルス(インフルエンザA型株及び/又はB型株を含む)、パラミクソウイルス(RSV A型及び/又はB型、ヒトメタニューモウイルス、並びにパラインフルエンザ1、2及び/又は3型を含む)、ラブドウイルス及びフラボウイルスを含むが、これらに限定はされない。具体的な実施例において、本発明のMDCK細胞は、低温適応性/温度感受性(ca/ts)インフルエンザウイルスの複製を支持する。ここでいうインフルエンザウイルスは、例えば、FluMist(登録商標) (Belsheら, 1998, N Engl J Med 338:1405;Nicholら, 1999, JAMA 282:137;Jacksonら, 1999, Vaccine, 17:1905)、及び/又はそれらのウイルスのバックボーンを含む、若しくは一以上の以下の特徴、すなわち低温適応性、弱毒性、及び温度感受性を有するインフルエンザウイルスのバックボーン(若しくは一以上のvRNA断片)を含むリアソータントウイルス等が該当する。ウイルス複製を支持する細胞の能力の指標の一つとして、感染した培養細胞から得られるウイルスの収量がある。ウイルスの収量は、当業者に既知の多くの方法によって測定することができる。例えば、ウイルスの収量は、感染性ウイルス粒子を測定する中間値(50%)組織培養感染量(TCID50)アッセイによりサンプル中に存在するウイルスの濃度を測定することで数値化することができる。TCID50値は、しばしばlog10 TCID50/mLとして報告されいている。一の実施形態において、本発明のMDCK細胞は、インフルエンザウイルス(例えばca/ts株)の複製を少なくとも6.0、少なくとも6.2、少なくとも6.4、少なくとも6.6、少なくとも6.8、少なくとも7.0、少なくとも7.2、少なくとも7.4、少なくとも7.6、少なくとも7.8、少なくとも8.0、少なくとも8.2、少なくとも8.4、少なくとも8.6、少なくとも8.8、少なくとも9.0、少なくとも9.2、少なくとも9.4、少なくとも9.6、又は少なくとも9.8のlog10 TCID50/mLまで支持する。他の実施形態において、本発明のMDCK細胞は、インフル
エンザウイルス(例えばca/ts株)の複製を少なくとも6.0、少なくとも6.2、少なくとも6.4、少なくとも6.6、少なくとも6.8、少なくとも7.0、少なくとも7.2、少なくとも7.4、少なくとも7.6、少なくとも7.8、少なくとも8.0、少なくとも8.2、少なくとも8.4、少なくとも8.6、少なくとも8.8、少なくとも9.0、少なくとも9.2、少なくとも9.4、少なくとも9.6、又は少なくとも9.8のlog10 TCID50/mLにまで支持する。
【0029】
本発明の細胞が一般に細胞培養組成物の一部であることは、当業者にはわかるであろう。細胞培養組成物の成分は、細胞や使用目的によって変化し得る。例えば、培養目的であれば、細胞培養組成物は、本発明の細胞とその細胞を増殖するための適切な培地を含み得る。したがって、本発明は、本発明の細胞及びその他の成分を含む細胞培養組成物を提供する。ここでいうその他の成分は、培地(例えば、本明細書に開示した培地)、培地成分、バッファ、化合物、他の細胞型、ウイルス物質(例えば、ウイルスゲノム、ウイルス粒子)、並びに異種タンパク質を含むが、これらに限定はされない。一の実施形態において、細胞培養組成物は、本発明の細胞、及び培地又はその成分から構成される。細胞培養組成物中に存在する培地は、無血清培地、血清含有培地及びAPF培地を含む。一の実施形態において、細胞組成物は、本明細書で開示する培地(例えば、MediV SF101、MediV SF102、MediV SF103、MediV SF104、若しくはMediV SF105)、又はそれらの成分を含む。
【0030】
<方法及び培地処方物>
本発明は、血清含有培地での非腫瘍形成性MDCK細胞の培養の方法及び培地処方物を提供する。本発明はまた、APF培地処方物を含む無血清培地での非腫瘍形成性MDCK細胞の適応方法、及びその後の培養方法も提供する。本発明のいくつかの態様において、前記培地はMDCK細胞が培養時に一以上の特性を維持するように処方されている。ここでいう一以上の特性は、非腫瘍形成性であること、付着性細胞として増殖すること、上皮様形態を有すること、及び様々なウイルスの複製を支持することを含むが、これらに限られない。本明細書で開示した培地処方物又はそれらの成分は、細胞培養組成物中に存在し得ることが意図されている。
【0031】
血清含有培地処方物は、当該分野で周知である。血清含有培地処方物は、ダルベッコ改良イーグル培地(DMEM)+ウシ胎仔血清(FBS)+グルタミン+グルコースを含むが、これに限定されない。一の実施形態において、FBSは、血清含有培地に約1%〜約20%、約5%〜約15%、又は約5%〜約10%の濃度で存在する。具体的な実施形態において、FBSは、血清含有培地に10%の濃度で存在する。他の実施形態において、グルタミンは、血清含有培地に約0.5mM〜約10mM、約1mM〜約10mM、又は約2mM〜約5mMの濃度で存在する。具体的な実施形態において、グルタミンは、血清含有培地に4mMの濃度で存在する。さらなる他の実施形態において、グルコースは、血清含有培地に約1g/L〜約10g/L、又は約2g/L〜約5g/L間の濃度で存在する。具体的な実施形態において、グルコースは、血清含有培地に4.5g/Lの濃度で存在する。さらなる他の実施形態において、血清含有培地処方物は、約1%〜約20%の濃度のFBS、約0.5mM〜約10mMの濃度のグルタミン、及び約1g/L〜約10g/Lの濃度のグルコースを含む。具体的な実施形態において、血清含有培地処方物は、ダルベッコ改良イーグル培地(DMEM)+10%ウシ胎仔血清(FBS)+4mMグルタミン+4.5g/Lグルコースからなる。DMEMは、例えばGibco/BRL社(Cat. No.11965-084)を含む多数の商業的供給源から容易に調達できる。FBSは、例えばJRH Biosciences社(Cat. No.12107-500M)を含む多数の商業的供給源から容易に調達できる。FBSは、動物細胞培地で最も一般的に用いられる補助添加剤であるが、新生の子牛、ウマ及びヒトを含むその他の血清源も日常的に使用され、本発明に包含される。
【0032】
一の実施形態において、本発明のMDCK-S血清適応非腫瘍形成性細胞は、血清を追加した化学的に規定された培地中でAmerican Type Culture Collection(ATCC CCL34)より入手したイヌ腎臓由来(MDCK)細胞を培養することによって、当該細胞から得られる。具体的な実施形態において、MDCK細胞(ATCC CCL34)を、血清を補充した化学的に規定された培地中で増やし、以下のようにしてMDCK-S株を作製する。すなわち、MDCK (ATCC CCL34)細胞を必要に応じてウシ胎仔血清(10% v/v)、4mM グルタミン、及び4.5 g/Lグルコースを補足したダルベッコ改良イーグル培地(DMEM)に継代し、MDCK-Sと呼ぶ冷凍プレマスター細胞バンク(PreMCB)を調製するのに十分な細胞を得る。他の具体的な実施形態において、細胞は下記の実施例2で詳述する工程を用いて培養される。特に、MDCK-S血清適合細胞を、PreMCBのバイアルから他へ20回以上継代すること、並びにアダルトヌードマウスモデルにおけるインビボでの腫瘍形成性、及び核型分析での核型をテストすることが意図されている。いくつかの実施形態において、増やしたMDCK-S細胞は、アダルトヌードマウスの皮下に注射しても小結節を生じず、また約60本〜88本以下の、約65本〜85本以下の、約65本〜80本以下の、又は約70本〜85本以下の染色体数の範囲で78本の最頻値染色体数を有している。一の実施形態において、MDCK-S細胞は、培地(例えば、本明細書に記載された培地)で、少なくとも20回の継代後、少なくとも30回の継代後、少なくとも40回の継代後、少なくとも50回の継代後、少なくとも60回の継代後、少なくとも70回の継代後、少なくとも80回の継代後、少なくとも90回の継代後、又は少なくとも100回の継代後でも非腫瘍形成性である。
【0033】
当業者であれば、組織培養を適用する上で血清又は動物抽出物の使用に欠点があることがわかるであろう(Lambert, K.J.ら, In: Animal Cell Biotechnology, Vol 1、Spier, R.E.ら編, Academic Pres New York, pp. 85-122 (1985))。例えば、これらの補助添加剤の化学組成は、同一生産業メーカーものであってもロット間で変わり得る。さらに、動物又はヒト起源の補助添加剤は、外来物質(例えば、マイコプラズマ、ウイルス、及びプリオン)で汚染されてもいるかもしれない。このような汚染された補助添加剤が細胞培地処方物に使用されれば、これらの作用物質によって培養細胞の健康状態が著しく損なわれる。さらに、外来作用物質で汚染された培養液中で作られた物質が細胞療法やその他の臨床応用で使用された場合、これらの物質は健康上のリスクを引き起こすかもしれない。最大の懸念は、動物の海綿状脳症やヒトのクロイツフェルト・ヤコブ症の原因となるプリオンの存在である。したがって、本発明はさらに無血清培地処方物を提供する。
【0034】
本発明の無血清培地処方物は、MediV SF101 (Taub’s培地+植物加水分解物)、MediV SF102 (Taub’s培地+脂質)、MediV SF103 (Taub’s培地+脂質+植物加水分解物)、MediV SF104 (Taub’s培地+脂質+植物加水分解物+成長因子)、及びMedi SF105(トランスフェリンをクエン酸鉄アンモニウム/トロポロン、又は硫酸鉄アンモニウム/トロポロンに置換した以外はMediV SF104と同じ)を含むが、これらの限定はされない。特に、Taub’s SF培地(TaubのSF培地;TaubとLivingston, 1981, Ann NY Acad Sci.,372:406)はホルモン、すなわち5μg/mLインスリン、5μg/mL トランスフェリン、25ng/mL プロスタグランジンE1、 50nMヒドロコルチゾン、5pMトリヨードチロニン、並びに10nM Na2SeO3、4.5g/Lグルコース、2.2g/L NaHCO3及び4mM L-グルタミンを追加したDMEM培地とHamの F12培地の50:50の混合物であることが意図されている。TaubのSF培地も、本明細書ではTaub’s SF培地、又は単に「Taub’s」と呼ぶ。
【0035】
植物加水分解物は、一以上の以下の植物、すなわち、トウモロコシ、綿実、エンドウ豆、大豆、麦芽、ジャガイモ、及び小麦由来の加水分解物を含む。ただし、これらに限定されない。植物加水分解物は、酵素的加水分解で作ることができ、通常、ペプチドの混合物、遊離アミノ酸、及び成長因子を含んでいる。植物加水分解物は、例えば、Marcor Development社、HyClone社、及びOrgano Technie社を含む多くの商業的供給源から容易に調達できる。酵母加水分解物も植物加水分解物の代わりに、又はそれと組み合わせて利用できることが意図されている。酵母加水分解物は、例えば、Sigma-Aldrich社、USB Corp社、Gibco/BRL社、その他を含む商業的供給源から容易に調達できる。
【0036】
培地補助添加剤に用いることのできる脂質は、動物及び植物由来の化学的に規定された脂質補助添加剤と合成的に誘導された脂質を含むが、これらに限定はされない。脂質補助添加剤中に存在し得る脂質には、コレステロール、飽和及び/又は不飽和脂肪酸(例えば、アラキドン酸、リノール酸、リノレン酸、ミリスチン酸、オレイン酸、パルミチン酸、及びステアリン酸)が含まれるが、これらに限られない。コレステロールは、100×ストックの脂質補助添加剤中に0.10mg/ml〜0.40mg/mlの濃度で存在し得る。脂肪酸は、100×ストックの脂質補助添加剤中に1μg/ml〜20μg/mlの濃度で存在し得る。培地処方物に適した脂質は、例えばHyClone社、Gibco/BRL社、及びSigma-Aldrich社を含む多くの商業的供給源から容易に調達できる。
【0037】
一の実施形態において、Taub’s培地には植物加水分解物が少なくとも0.5g/L、少なくとも1.0g/L、少なくとも1.5g/L、少なくとも2.0g/L、少なくとも2.5g/L、少なくとも3.0g/L、少なくとも5.0g/L、少なくとも10g/L、又は少なくとも20g/Lの最終濃度で追加されている。具体的な実施形態において、Taub’s培地には小麦の加水分解物が追加されている。他の具体的な実施形態において、Taub’s培地には小麦の加水分解物が2.5g/Lの最終濃度で追加されている。本発明は、本明細書でMediV SFM 101と呼ばれる無血清培地であって、小麦の加水分解物を最終濃度2.5g/Lで追加したTaub’s培地からなる無血清培地を提供する。
【0038】
他の実施形態において、Taub’s培地には脂質混合物が製造メーカーの推奨最終濃度の少なくとも50%、少なくとも60%、少なくとも70%、少なくとも80%、少なくとも90%、少なくとも100%、少なくとも125%、少なくとも150%、少なくとも200%、少なくとも300%の最終濃度で追加されている。具体的な実施形態において、Taub’s培地には化学的に規定された脂質混合物が追加されている。他の具体的な実施形態において、Taub’s培地には、化学的に規定された脂質混合物が製造メーカーの推奨最終濃度の100%の最終濃度で補充されている(例えば、製造メーカーから入手した100×ストックであれば、最終濃度1×で培地に補充される)。本発明は、本明細書でMediV SFM 102と呼ばれる無血清培地であって、化学的に規定された脂質混合物を製造メーカーの推奨最終濃度の100%の最終濃度で追加したTaub’s培地からなる無血清培地を提供する。
【0039】
さらなる他の実施形態において、Taub’s培地には植物加水分解物が少なくとも0.5g/L、少なくとも1.0g/L、少なくとも1.5g/L、少なくとも2.0g/L、少なくとも2.5g/L、少なくとも3.0g/L、少なくとも5.0g/L、少なくとも10g/L、又は少なくとも20g/Lの最終濃度で、かつ脂質混合物が製造メーカーの推奨濃度の少なくとも50%、少なくとも60%、少なくとも70%、少なくとも80%、少なくとも90%、少なくとも100%、少なくとも125%、少なくとも150%、少なくとも175%、又は少なくとも200%の最終濃度で、追加されている。具体的な実施形態において、Taub’s培地には小麦の加水分解物、及び化学的に規定された脂質混合物が追加されている。他の具体的な実施形態において、Taub’s培地には小麦の加水分解物が2.5g/Lの最終濃度で、かつ化学的に規定された脂質混合物が製造メーカーの推奨最終濃度の100%の最終濃度で追加されている。本発明は、本明細書でMediV SFM 103と呼ばれる無血清培地であって、小麦の加水分解物を最終濃度2.5g/Lで、かつ化学的に規定された脂質混合物を製造メーカーの推奨最終濃度の100%の最終濃度で追加したTaub’s培地からなる無血清培地を提供する。
【0040】
さらなる他の実施形態において、Taub’s培地には成長ホルモンが追加されている。使用することのできる成長ホルモンには、上皮成長因子(EGF)、インスリン成長因子(IGF)、形質転換増殖因子(TGF)、及び線維芽細胞増殖因子(FGF)が含まれるが、これらに限定はされない。具体的な実施形態において、成長ホルモンは上皮成長因子(EGF)である。一の実施形態において、Taub’s培地には、成長因子が約0.1〜約50.0ng/ml、約0.5〜約25.0ng/ml、約1.0〜約20ng/ml、約5.0〜約15.0ng/ml、又は約8〜約12ng/mlの最終濃度で添加されている。具体的な実施形態において、Taub’s培地にはEGFが約10ng/mlの最終濃度で追加されている。さらなる他の実施形態において、Taub’s培地には成長因子が約0.1〜約50.0ng/ml、約0.5〜約25.0ng/ml、約1.0〜約20ng/ml、約5.0〜約15.0ng/ml、又は約8〜約12.0ng/mlの最終濃度で、かつ植物加水分解物が少なくとも0.5g/L、少なくとも1.0g/L、少なくとも1.5g/L、少なくとも2.0g/L、少なくとも2.5g/L、少なくとも3.0g/L、少なくとも5.0g/L、少なくとも10g/L、又は少なくとも20g/Lの最終濃度で、かつ脂質混合物が製造メーカーの推奨最終濃度の少なくとも50%、少なくとも60%、少なくとも70%、少なくとも80%、少なくとも90%、少なくとも100%、少なくとも125%、少なくとも150%、少なくとも175%、又は少なくとも200%の最終濃度で、追加されている。他の具体的な実施形態において、Taub’s培地には小麦の加水分解物が2.5g/Lの最終濃度で、かつ化学的に規定された脂質混合物が製造メーカーの推奨最終濃度の100%の最終濃度で、かつEGFが約10ng/mlの最終濃度で、追加されている。本発明は、本明細書でMediV SFM 104と呼ばれる無血清培地であって、小麦の加水分解物を最終濃度2.5g/Lで、かつ化学的に規定された脂質混合物を製造メーカーの推奨最終濃度の100%の最終濃度で、かつEGFを約10ng/mlの最終濃度で追加したTaub’s培地からなる無血清培地を提供する。
【0041】
当業者であれば、動物性タンパク質フリーの培地処方物がワクチンの製造に使用するウイルスの生産に望ましいことも理解できるであろう。したがって、いくつかの実施形態において、本明細書で開示された無血清培地(例えば、MediV SF101、MediV SF102、MediV SF103、MediV SF104、及びMedi SF105)の1以上又は全ての動物由来の成分は、非動物性誘導体で置換される。例えば、市販の非動物源由来の組換えインスリン(例えば、Biological Industries社のCat. No. 01-818-1)は、動物源から得られたインスリンの代わりに利用することができる。同様に、鉄結合剤(例えば、米国特許第5,045,454号;同第5,118,513号;同第6,593,140号;及びPCT国際公開第WO 01/16294号を参照されたい)を動物源由来のトランスフェリンの代わりに利用することができる。一の実施形態において、本発明の無血清培地処方物は、トランスフェリンの代わりにトロポロン(2-ヒドロキシ-2,4,6-シクロヘプタトリエン-1)及び鉄源(例えばクエン酸鉄アンモニウム、硫酸鉄アンモニウム)を含む。例えば、トロポロン又はトロポロン誘導体は、培地中に存在する鉄に対して、約5対1〜約70対1、又は約10対1〜約70対1のモル比のような過剰モル濃度で存在し得る。したがって、培地中の鉄濃度は0.3μM前後であり、トロポロン又はその誘導体は、約1.5μMから約20μM、例えば約3μMから約20μMの濃度で使用することができる。前記鉄は、例えば、硫酸鉄、塩化第二鉄、硝酸鉄、特にクエン酸鉄アンモニウム等の培地中の鉄単塩又は鉄錯塩を使用することによって生じる二価又は三価の鉄イオンとして存在することができる。本発明は、本明細書でMediV SFM 105と呼ばれる無血清培地でえあって、小麦の加水分解物を最終濃度2.5g/Lで、かつ化学的に規定された脂質混合物を製造メーカーの推奨最終濃度の100%の最終濃度で、かつEGFを約10ng/mlの最終濃度で、かつクエン酸鉄アンモニウム:トロポロン又は硫酸鉄アンモニウム:トロポロンを10:1〜70:1の比率で追加したトランスフェリンを含まないTaub’s培地からなる無血清培地を提供する。
【0042】
一の実施形態において、MDCK-SF101、MDCK-SF102、MDCK-SF103、MDCK-SF104、及びMDCK-SF105無血清培地に適応した非腫瘍形成性細胞(本明細書では、まとめてMDCK-SFと呼ぶ)は、American Type Culture Collection(ATCC CCL34)株より入手したイヌ腎臓由来(MDCK)細胞を、少なくとも1回の継代については血清を加えた化学的に規定された培地中で、その後それらを例えば上記無血清培地等の無血清培地で継代することによって得られる。具体的な実施形態において、MDCK細胞(ATCC CCL34)を無血清培地に対して以下のように適応させて、MDCK-SF細胞株を作製する。すなわち、MDCK細胞(ATCC CCL34)をウシ胎仔血清(10% v/v)、4mMグルタミン及び4.5g/Lグルコースを追加したダルベッコ改良イーグル培地(DMEM)で少なくとも1回継代し、その後無血清培地で継代する。MDCK-SF細胞をその後必要に応じて無血清培地で継代し、冷凍プレマスター細胞バンク(PreMCB)を調製するのに十分な細胞を得る。いくつかの実施形態において、前記細胞を、血清含有培地(例えば、ウシ胎仔血清(10% v/v)、4mMグルタミン及び4.5g/Lグルコースを追加したダルベッコ改変イーグル培地(DMEM))で1〜5回、4〜10回、9〜20回、又は20回以上継代する。その後、無血清培地(例えば、MediV SF101、MediV SF102、MediV SF103、MediV SF104、及びMedi SF105)で継代する。特に、MDCK-SF無血清培地に適応した細胞をPreMCBのビンから他に20回以上継代すること、並びにアダルトヌードマウスモデルのインビボでの腫瘍原性、及び核型測定での核特性をテストすることが意図されている。いくつかの実施形態において、増やしたMDCK-SF細胞は、アダルトヌードマウスの皮下に導入しても小結節を生じず、及び/又は78本の最頻染色体数を有している。他の実施形態において、増やしたMDCK-SF細胞は、わずか約60本〜88本の、わずか約65本〜85本の、わずか65本〜85本の、わずか約65本〜80本の、又はわずか約70本〜85本の染色体数の範囲で、78本の最頻染色体数を有している。一の実施形態において、MDCK-SF細胞は、培地(例えば、本明細書に記載された培地)中で、少なくとも100回の継代後、少なくとも90回の継代後、少なくとも80回の継代後、少なくとも70回の継代後、少なくとも60回の継代後、少なくとも50回の継代後、少なくとも40回の継代後、少なくとも30回の継代後、又は少なくとも20回の継代後も非腫瘍形成性である。
【0043】
一の実施形態において、MDCK-SF細胞の誘導用に使用される無血清培地は、MediV SF101である。他の実施形態において、MDCK-SF細胞の誘導用に使用される無血清培地はMediV SF102である。さらに、他の実施形態において、MDCK-SF細胞の誘導用に使用される無血清培地はMediV SF103である。さらなる他の実施形態において、MDCK-SF細胞の誘導用に使用される無血清培地はMediV SF104である。他の実施形態において、MDCK-SF細胞の誘導用に使用される無血清培地はMediV SF105である。更なる他の実施形態において、MDCK-SF細胞の誘導用に使用される無血清培地はAPF培地である。本明細書に記載された培地を処方して動物性タンパク質を排除できることが意図されている。例えば、ウシのトランスフェリンを非動物源由来の組換えトランスフェリンと置換することができる。
【0044】
<培養条件>
本発明は、血清含有又は無血清培地処方物(上記)でのMDCK細胞(好ましくは非腫瘍形成性)、及びその他の動物細胞(腫瘍形成性又は非腫瘍形成性)の培養方法を提供する。特に、さらなる培養条件として、MDCK-S細胞及びMDCK-SF細胞を非腫瘍形成性状態で維持する役割を果たし得ることが意図されている。これらの培養条件は、付着表面の選択、細胞密度、温度、CO2濃度、培養方法、溶存酸素含有量、及びpHを含む。ただし、これらに限定はされない。
【0045】
特に、当業者が多数の方法の培養条件を適応させて、本発明のMDCK細胞の増殖を最適にしてもよいことが意図されている。このような適応はまた、ウイルス物質(例えば、ウイルス)の生産増加に繋がり得る。あるいは、当業者が培養条件を適応させて、細胞の増殖に関係なく本発明のMDCK細胞からワクチン物質の生産を最適にすることができる。これらの培養条件は、付着表面、細胞密度、温度、CO2濃度、培養方法、溶存酸素含有量、及びpHを含むが、これらの限定はされない。
【0046】
一の実施形態において、本発明のMDCK細胞は、付着性細胞としてそれらが付着する表面上で培養される。組織培養細胞が増殖可能な付着表面は、当業分野では周知である。付着表面は、表面修飾したポリスチレンプラスチック、タンパク質被覆表面(例えば、フィブロネクチン及び/又はコラーゲン被覆ガラス/プラスチック)と市販の多岐にわたるマイクロキャリア(例えば、Dormacell [Pfeifer & Lange社]等のDEAE-デキストランマイクロキャリアビーズ; Superbead [Flow Laboratories社];Hillex [SoloHill, Ann Arbor]等のスチレン共重合体トリメチルアミンビーズ)を含むが、これらの限定はされない。マイクロキャリアビーズは、小球(直径100〜200ミクロンの範囲)であって、細胞培養量につき付着性細胞の増殖のための大きな表面積領域を提供する。例えば、培地1リットルは、8000平方センチメートルを越える増殖表面を提供する2000万個以上のビーズを含むことができる。付着表面の選択は、本発明のMDCK細胞の培養に使用する方法によって決まり、また当業者が決めることができる。本発明による工程の過程で使用することのできる適切な培養容器は、当業者に既知の全ての容器である。例えば、スピナーボトル、ローラーボトル、培養槽、又は生物反応槽(バイオリアクター)等が該当する。例えばワクチン生産用のようにウイルスを量産するためには、細胞をバイオリアクターや培養槽で培養することが望ましい。生物反応槽は、1リットル未満から100リットルを越える量まで市販されている。例えば、Cyto3バイオリアクター(Osmonics社, Minnetonka, MN);NBSバイオリアクター(New Brunswick Scientific社,Edison, NJ);B.Braun Biotech International社の研究・業務用 バイオリアクター(B. Braun Biotech社,Melsungen, Germany)が挙げられる。
【0047】
一の実施形態において、本実施形態のMDCK細胞は、付着性細胞としてバッチ培養システムで培養される。さらに他の実施形態において、本発明のMDCK細胞は、付着性細胞として灌流培養システムで培養される。特に、本発明のMDCK細胞は、ワクチン物質(例えば、ウイルス)の生産に灌流システム(例えば、攪拌培養槽内で、例えば遠心、濾過、スピンフィルターといったような当業者に既知の細胞維持方式を用いて)で培養され得ることが意図されている。
【0048】
一の実施形態において、本発明のMDCK細胞は、少なくとも1%、少なくとも2%、少なくとも3%、少なくとも4%、少なくとも5%、少なくとも6%、少なくとも7%、少なくとも8%、少なくとも9%、少なくとも10%、又は少なくとも20%のCO2濃度で培養される。
【0049】
一の実施形態において、溶存酸素(dissolved oxygen:DO)濃度(pO2値)は、本発明のMDCK細胞の培養中で適宜調節される。その濃度範囲は、(空気飽和に基づいて)5%〜95%、又は10%〜60%である。具体的な実施形態において、溶解酸素(DO)濃度(pO2値)は、少なくとも10%、少なくとも20%、少なくとも30%、少なくとも50%又は少なくとも60%である。
【0050】
他の実施形態において、本発明のMDCK細胞の培養に用いる培地のpHは、培養中に調節される。その範囲は、pH 6.4〜pH 8.0又はpH 6.8〜pH 7.4である。具体的な実施形態において、本培地のpHは、少なくとも6.4、少なくとも6.6、少なくとも6.8、少なくとも7.0、少なくとも7.2、少なくとも7.4、少なくとも7.6、少なくとも7.8、又は少なくとも8.0である。
【0051】
さらなる実施形態において、本発明のMDCK細胞は、25℃〜39℃の温度で培養される。特に、培養温度は、所望の工程によって変えることができることを意図している。例えば、本発明のMDCK細胞は、細胞増殖のため37℃で増やし、その後ワクチン物質(例えば、ウイルス)生産のため、より低い温度(例えば、25℃〜30℃)で増やすことができる。他の実施形態において、前記細胞は、ワクチン物質の生産のために30℃未満、31℃未満、32℃未満、33℃未満、又は34℃未満で培養される。他の実施形態において、前記細胞は、ワクチン物質の生産のために30℃、31℃、32℃、33℃、又は34℃で培養される。
【0052】
ワクチン物質(例えば、ウイルス)を生産するため、特に本発明のMDCK細胞が培地を容易に交換できる(例えば、灌流システムで)ように培養されることを意図している。細胞を非常に高い密度、例えば1×106〜25×106細胞/mLまで培養することができる。
【0053】
培地のグルコース、グルタミン、乳酸含有量とpH及びpO2値、及び攪拌等の当業者に既知の他のパラメータを本発明のMDCK細胞の培養中に細胞密度、及び/又はウイルス生産量を最適化するために容易に操作することができる。
【0054】
<ワクチン物質(例えばウイルス)の生産>
本発明は、本発明のMDCK細胞を使用する細胞培養でのウイルス生産の方法(本明細書では以降「本発明の方法」と呼ぶ)を提供する。一の実施形態において、本方法は以下のステップ、すなわち
i) 培養培地中で本発明のMDCK細胞の増殖させるステップ、
ii) 細胞にウイルスを感染させるステップ、及び
iii) さらなる培養期の後、非腫瘍形成性細胞内で複製されたウイルスを単離するステップ、
を含む。
【0055】
一の実施形態において、本発明のMDCK細胞は、ステップ(i)で付着性細胞として増殖する。本発明のMDCK細胞は、前記方法の過程で上記培地を含むがそれに限定されないいずれの培地においても培養することができる。いくつかの実施形態において、本発明のMDCK細胞は、前記方法の過程において例えばMediV-SF101、MediV-SF102、MediV-SF103、MediV-SF104、MediV-SF105及びそれらのAPF処方物等の無血清培地で培養される。任意で、本発明のMDCK細胞を、前記方法の過程において血清含有培地(例えば、DMEM+10%FBS+4mMグルタミン+4.5g/Lグルコース)で培養することができる。例えば、温度、pH、pO2値、CO2濃度、及び細胞密度等のさらなる培養条件については、上記で詳細に述べている。当業者は、ウイルス生産のために本発明のMDCK細胞の増殖についての培養条件の組合せを確立することができる。
【0056】
ウイルスによる感染前の細胞増殖用温度は、一の形態において22℃〜40℃である。いくつかの実施形態において、ウイルスによる感染前の細胞増殖用温度は、39℃未満、38℃未満、37℃未満、36℃未満、35℃未満、34℃未満、33℃未満、32℃未満、30℃未満、28℃未満、26℃未満、又は24℃未満である。細胞増殖のための培養(ステップ(i))は、前記方法の一の実施形態において灌流システムで行われる。例えば攪拌培養槽内で、例えば遠心、濾過、スピンフィルター、マイクロキャリアといったような当業者に既知の細胞維持方式を用いて行われる。
【0057】
この場合、前記細胞を1〜20日間、又は3〜11日間増やす。培地の交換は、この過程で一日あたり0〜培養槽容量の約1〜5倍に増やして行われる。前記細胞は、この方法で例えば最大で少なくとも1×106〜25×106細胞/mLの高細胞密度まで増大される。灌流システムにおける培養中の灌流速度は、細胞カウント、培地中のグルコース、グルタミン又は乳酸含有量によって、そして当業者に既知の他のパラメータによって、調節することができる。さらに、本発明による方法のステップ(i)の細胞は、バッチ工程で培養される。
【0058】
本発明による方法の一の実施形態において、pH、pO2値、グルコース濃度、及びステップ(i)で使用される他の培地パラメータは、当業者に既知の方法を用いて上述のように培養中に調節される。
【0059】
他の実施形態において、ウイルスによる細胞の感染を約0.0001〜約10、約0.0005〜約5、又は約0.002〜約0.5のM.O.I.(multiplicity of infection:感染多重度)で行われる。さらに他の実施形態において、ウイルスによる細胞の感染は、0.0001〜10、0.0005〜5、又は0.002〜0.5のM.O.I.(multiplicity of infection:感染多重度)で行う。感染後、感染した培養細胞をさらに培養して、ウイルスを複製させる。具体的には最大細胞変性効果、又はウイルス抗原の最大量を検出することができるまで培養する。一の実施形態において、感染後に前記細胞を22℃〜40℃の温度で培養する。いくつかの実施形態において、ウイルスによる感染後、前記細胞を39℃未満、38℃未満、37℃未満、36℃未満、35℃未満、34℃未満、33℃未満、32℃未満、30℃未満、28℃未満、26℃未満、又は24℃未満の温度で培養する。他の実施形態において、感染後に前記細胞を33℃以下の温度で培養する。さらに他の実施形態において、感染後に前記細胞を31℃の温度で培養する。いくつかの実施形態において、前記細胞の培養を2〜10日間行う。培養は、灌流システムで、又は任意でバッチ方式で実施することができる。
【0060】
ウイルスによる感染後の細胞の培養(ステップ(iii))を、上記のようにpH及びpO2値を維持するように同様に行う。本方法のステップ(iii)による細胞培養又はウイルス複製の間、抗原収量を最適化するために、新たに調製された培地、培地濃縮物で、又はアミノ酸、ビタミン、脂質画分、リン酸等の既知構成成分で、細胞培地を置換することも可能である。前記細胞を、さらなる培地又は培地濃縮物の添加により数日にわたって徐々に希釈するか、さらなる灌流中にあるいは培地又は培地濃縮物でインキュベートすることができる。灌流速度は、この場合、細胞数、培地中のグルコース、グルタミン、乳酸若しくは乳酸デヒドロゲナーゼの含有量、又は当業者に既知の他のパラメータを用いて調節することができる。灌流システムとフィード−バッチ(fed-batch)方式の組合せが、さらに可能である。
【0061】
本発明方法の一の実施形態において、生産されたウイルスの回収と単離(ステップ(iii))は、ウイルスが適切な収量で生産されるのに十分な期間後に行われる。例えば、感染後2〜10日間、又は場合により3〜7日間等が挙げられる。本発明方法の一の実施形態において、生産されたウイルスの回収と単離(ステップ(iii))は、感染の2日後、3日後、4日後、5日後、6日後、7日後、8日後、9日後又は10日後に行われる。
【0062】
本発明のMDCK細胞で生産され得るウイルスは、動物ウイルス、すなわちオルソミクソウイルス科、パラミクソウイルス科、トガウイルス科、ヘルペスウイルス科、ラブドウイルス科、レトロウイルス科、レオウイルス科、フラビウイルス科、アデノウイルス科、ピコルナウイルス科、アレナウイルス科、及びポックスウイルス科の各科に含まれるウイルスを含むが、これらに限定されない。
【0063】
細胞培養でインフルエンザウイルスを生産する系も、近年になって開発された(例えば、Furminger著, in Textbook of Influenza,Nicholson編,Webster and Hay, pp.324-332, Blackwell Science (1998); Mertenら著. in Novel Strategies in The Design and Production of Vaccines,Cohen & Shafferman編,pp.141-151, Kluwer Academic (1996)を参照されたい)。これらの方法は、主として選択したウイルス株により適当な不死化宿主細胞が感染することを必要とする。これらの方法により鶏卵におけるワクチン生産に関連する多くの問題を排除できるが、インフルエンザの全ての病原性株が十分に増殖し、また従来の組織培養方法により生産することができるというわけではない。さらに、例えば弱毒化された生ワクチンの生産に適した弱毒性、温度感受性、及び低温適応性といった所望の性質をもつ多数の株を、従来方法を用いた組織培養では、特に工業規模でうまく増やすことができなかった。
【0064】
本発明は、いくつかの非腫瘍形成性MDCK細胞株を提供する。これらの株は、培養時に血清含有、又は無血清のいずれの培地中でも増殖するように適応しており、またインフルエンザを含むがそれに限定されないウイルスの複製を支持することができる。これらの細胞株は、ワクチン物質として利用するための細胞培養でウイルスを経済的に複製するのに適している。本発明のMDCK細胞は、従来の細胞株(下記実施例1を参照)を用いた場合は十分に増殖しない低温適応性、温度感受性(ca/ts)のインフルエンザ株(例えば、FluMist(登録商標)に含まれるインフルエンザ株)の生産に特に有効である。さらに、本発明のMDCK細胞は、ヒトでも病気の原因となり得るトリインフルエンザウイルス(例えば、「パンデミック(世界的流行)」株)のように孵化卵で増殖できないインフルエンザ株の生産に有効である。
【0065】
本発明のMDCK細胞内で本発明の方法によって生産することができるインフルエンザウイルスは、リアソータント(reassortant)ウイルスを含むが、それに限定はされない。当該ウイルスは、弱毒性、温度感受性、低温適応性(ca/ts/att)のマスター株の中で選択された赤血球凝集素ヘマグルチニン及び/又はノイラミニダーゼ抗原が合体したウイルスである。例えば、ウイルスは、一以上の、例えば温度感受性(ts)、低温適応性(ca)、又は弱毒性(att)を有するマスター株のバックボーン(又は一以上のvRNAセグメント)を含み得る(例えば、A/Ann Arbor/6/60、B/Ann Arbor/1/66、PR8、B/Leningrad/14/17/55、B/14/5/1、B/USSR/60/69、B/Leningrad/179/86、B/Leningrad/14/55、B/England/2608/76等)。卵又は細胞株のどちらかのリアソータントインフルエンザワクチン株の生産方法は、当該分野で既知である。また当該方法は、例えばKilbourne, E.D著. in Vaccines (2nd Edition), PlotkinとMortimer編, WB Saunders Co. (1988)、及び国際出願、国際公開第WO 05/062820号及び同第WO 03/091401号で開示された方法を含む。本発明のMDCK細胞内で本発明の工程により生産することができるその他のインフルエンザウイルスは、異種遺伝子産物を発現することができる組換えインフルエンザウイルスを含む。当該ウイルスについては、例えば、米国特許公開第2004/0241139号、及び同第2004/0253273号を参照されたい。
【0066】
一の実施形態において、前記細胞を上記のように増殖(成長)し(ステップ(i))、その後細胞をインフルエンザウイルスで感染させる、(ステップ(ii))。いくつかの実施形態において、前記感染を0.0001〜10、0.0005〜5、又は0.002〜0.5のM.O.I.(感染多重度)で行う。他の実施形態において、前記感染を約0.0001〜約10、約0.0005〜約5、又は約0.002〜約0.5のM.O.I.(感染多重度)で行う。任意でヘマグルチニン(HA0)前駆体タンパク質の切断、及びそれによる細胞上へのウイルスの吸着を引き起こすプロテアーゼが添加される。プロテアーゼの添加は、インフルエンザウイルスによる細胞の感染(ステップ(ii))の直前、同時、又は直後に本発明に従って行われる。前記添加を感染と同時に行う場合には、プロテアーゼを細胞培養液に直接添加して感染させることも、又は例えば濃縮物としてウイルスの接種と共に添加し感染させることも可能である。前記プロテアーゼは、本発明のいくつかの局面で、セリンプロテアーゼ、システインプロテアーゼ、又はアスパラギンプロテアーゼである。一の実施形態において、トリプシンが使用される。具体的な実施形態において、TPCK処理したトリプシンが使用される。
【0067】
一の実施形態において、トリプシンは細胞培養液に1〜5000mU/ml、5〜1000mU/ml、又は100〜500mU/mlの最終濃度で添加される。さらなる実施形態において、トリプシンは細胞培養液に1〜200μg/ml、5〜50μg/ml、又は5〜30μg/mlの最終濃度で添加される。
【0068】
本発明に係る方法のステップ(iii)で感染した細胞をさらに培養する間に、トリプシン再活性化を実施することができる。すなわち、バッチ工程の場合にはトリプシンの新たな添加によって、又は灌流システムの場合にはトリプシン溶液の連続的添加又は断続的添加によって行われる。
【0069】
感染後、感染した培養細胞をさらに培養して、ウイルスを複製させる。具体的には最大細胞変性効果、又はウイルス及び/又はウイルス抗原の最大量を検出することができるまで培養する。いくつかの実施形態において、細胞の培養を2〜10日間行う。培養は、灌流システムで、又は任意でバッチ工程で順番に行うことができる。さらなる実施形態において、細胞をインフルエンザウイルスでの感染後25℃〜36℃、又は29℃〜34℃の温度で培養する。33℃よりも低い温度での、具体的には上記で示した温度範囲内での感染細胞の培養は、例えばB型株のようないくつかのインフルエンザウイルスの高い生産収量をもたらす。それ故、35℃よりも低い温度での感染細胞の培養は、温度感受性、低温適応性(ts/ca)インフルエンザウイルスの生産が意図されている。ts/caウイルスが弱毒化(att)され得ることも意図されている。他の実施形態において、細胞を、ts/caウイルス株の生産のため30℃未満、31℃未満、32℃未満、33℃未満、又は34℃未満の温度で培養する。具体的な実施形態において、細胞をB型インフルエンザウイルス株の生産用に31℃の温度で培養する。
【0070】
インフルエンザウイルスで感染後の細胞培養(ステップ(iii))は、例えば上記のように順次行われる。
【0071】
本発明方法の一の実施形態において、生産されたインフルエンザの回収と単離(ステップ(iii))は、感染の2〜10日後、又は3〜7日後等のウイルスが適当な収量で生産されるのに十分な期間後に行われる。ウイルスは、通常、感染細胞が増殖した培地から回収される。通常はインフルエンザウイルスの濃縮前に粗製培地を清浄化する。一般的な方法は、濾過、限外濾過、硫酸バリウムへの吸着及び溶出、並びに遠心を含む。例を挙げると、感染培養物由来のクルードな培地を、まず例えば1000〜2000×gで、細胞片及びその他の大型粒状物質を除くのに十分な時間(例えば、10〜30分)遠心して清浄化することができる。あるいは、培地を0.8μmの酢酸セルロースフィルターに通して濾過して、無傷の細胞及びその他の大型粒状物質を除く。場合によりその後清浄化した培地の上清を、例えば15,000×gで約3〜5時間遠心し、インフルエンザウイルスを沈殿させる。STE(0.01MのTris-HCl;0.15MのNaCl;0.0001MのEDTA)又はpH7.4のリン酸バッファ(PBS)等の適当なバッファにウイルス沈殿物を再懸濁した後、ウイルスをショ糖(60%〜12%)又は酒石酸カリウム(50%〜10%)の密度勾配遠心で濃縮してもよい。連続的勾配か、又は段階的勾配(例えば12%〜60%間で12%ごと4段階)のショ糖勾配のいずれかが適している。勾配を、ウイルスを回収するため目に見えるバンドに凝集させる上で十分な速度及び時間で遠心分離で分離する。あるいは、大規模の工業用に適用するため、ウイルスを連続様式で稼動するゾーン遠心ローターを用いて密度勾配から溶出する。技術者が組織培養からインフルエンザウイルスを調製するためのさらに詳しい説明については、例えば、Furminger, Textbook of Influenza pp. 324-332 Nicholsonら編、Mertenら, in Novel Strategies in Design and Production of Vaccines pp.141-151 CohenとShafferman編、及び米国特許第5,690,937号で提供されている。もし所望であれば、回収したウイルスをショ糖-リン酸-グルタミン酸(SPG)等の安定剤の存在下で−80℃にて保存することができる。
【0072】
本発明方法のいくつかの実施形態において、ウイルスはBenzonase(登録商標)、又はその他の非特異的エンドヌクレアーゼで処理される。任意でBenzonase処理は、生産されたインフルエンザウイルスの回収と分離(ステップ(iii))の初期に行う。本工程のその他の実施形態では、Benzonase処理後に材料を清浄化する。清浄化に有用な方法は、ダイレクトフロー濾過(direct flow filtration:DFF)を含む。ただし、これに限られない。生産されたインフルエンザウイルスの回収と単離(ステップ(iii))に利用することができるさらなるステップは、タンジェンシャルフロー濾過(tangential flow filtration;TFF)、アフィニティークロマトグラフィーと、イオン交換クロマトグラフィー及び/又はヒドロキシアパタイトクロマトグラフィーを含むが、これに限られない。その他のステップについては下記実施例の項で例証している。
【0073】
<ワクチン組成物と使用方法>
本発明は、さらに本発明の方法で得ることのできるウイルス(例えばインフルエンザ)に関する。これらのウイルスを既知の方法で製剤化し、ヒトや動物に投与するワクチンを提供することができる。ウイルスは、完全ウイルス粒子(例えば、弱毒化した生ウイルス)として、又は不活性化/非組込み型ウイルス(例えば、ホルムアルデヒド、界面活性剤で処理したウイルス)として存在し得る。任意で、既知のウイルス成分(例えば、タンパク質)を当業者に既知の方法でウイルスから分離し、ワクチンの調製に使用することができる。
【0074】
完全ウイルス粒子(例えば、弱毒化した生ウイルス)の製剤は、最終製剤にする濾過によるバッファ交換、それに続く滅菌ステップと、さらなるステップを含むことができるが、それらに限られない。前記製剤に有用なバッファは、アルギニンなどの他のアミノ酸賦形剤を追加した200mMショ糖及びpH7.0〜7.2のリン酸又はヒスチジンバッファを含み得る。いくつかの実施形態において、ブタゼラチン等のタンパク質加水分解物安定化剤が添加される。いくつかの実施形態において、本発明の最終ウイルス溶液/ワクチンは、生きたウイルスを含み得る。当該ウイルスは、インフルエンザワクチン接種期(例えば、北半球では一般に大体9月から3月)の間、「現場で」保存(例えば、販売や市場で2〜8℃、4℃、5℃等で冷蔵される場合)のに十分な期間、液体状態で安定である。したがって、前記ウイルス/ワクチン組成物は、それらの効力が維持されること、又は保存期間以降は許容できる速さでその効力が失われることが望ましい。その他の実施形態において、前記溶液/ワクチンは、例えば、冷蔵庫内温度である約2℃〜約8℃において液体状態で安定である。例えば、冷蔵庫内で安定な弱毒化インフルエンザワクチンを処方するための方法と組成物は、2005年10月4日出願の国際出願第PCT/US2005/035614号に記載されている。国際公開第WO 05/014862号も参照されたい。任意で、液体製剤試料を乾燥加熱気体の気流下で微細液滴に噴霧化する噴霧乾燥法の急速乾燥工程を利用して、ワクチン製剤の保存時間を延長することができる。微細液滴の蒸発により、溶解溶質からなる乾燥粉末が生じる(例えば、米国特許公開第2004/0042972号を参照されたい)。ワクチン組成物用の不活性化/非組込み型ウイルス粒子の生成及び製剤化の方法は、当業分野では周知であり、40年間にわたって利用されている。
【0075】
一般にウイルスやウイルス組成物を予防的に適当な担体又は賦形剤と共に投与して、一以上のウイルス株に特異的な免疫反応を刺激することができる。通常前記担体又は賦形剤は、製薬上許容される担体又は賦形剤である。すなわち、滅菌水、生理食塩水、生理食塩バッファ、グルコース溶液、グリセロール溶液、エタノール、又はそれらの組合せ等が挙げられる。無菌性、pH、等張性、及び安定性が保証された前記溶液の調製は、当該分野の従来のプロトコルに従って達成される。通常、担体又は賦形剤は、アレルギーやその他の望ましくない効果を最小限に抑えるように、又は例えば皮下、筋肉内、鼻腔内等の個々の投与手段に合うように選択される。
【0076】
任意で、ウイルスの予防投与用の製剤又はそれらの成分はまた、インフルエンザ抗原に対する免疫反応を増強するための一以上のアジュバントを含む。適切なアジュバントには以下のものが含まれる。すなわち、サポニン、水酸化アルミニウムのようなミネラルゲル、リゾレシチン、界面活性物質、プルロニックポリオール、多価陰イオン、ペプチド、油又は炭化水素エマルジョン、カルメット−ゲラン桿菌(BCG)、コリネバクテリウム・パルヴム(Corynebacterium parvum)、並びに合成アジュバントQS-21及びMF59が挙げられる。
【0077】
一般に、ワクチン製剤は、一以上のインフルエンザウイルス株に対して特異的な免疫反応を刺激する上で十分な量で投与される。好ましくはウイルスの投与が防御免疫反応を誘導することである。一以上のウイルス株に対して防御免疫反応を誘導するための投与量及び方法は、当業者に既知である。例えば、不活性化したインフルエンザウイルスは、約1〜1000HID50(human infectious dose;ヒト感染用量)、すなわち1回の投与量あたり約105〜108pfu(plaque forming units;プラーク形成単位)の範囲で投与される。あるいは、約10〜50μg、例えば約15μgのHAをアジュバントなしで、アジュバントと共に投与される量よりも少量で投与する。一般に、投与量は、上記範囲で例えば年齢、健康状態、体重、性別、食事療法、投与時間、及びその他の臨床学的因子に基づいて調節され得る。予防用ワクチン製剤は、例えば針とシリンジを用いた皮下若しくは筋肉内注射、又は無針注射器具を用いて全身投与される。あるいは、ワクチン製剤は、鼻腔内への点滴、大粒子エアロゾル(約10ミクロンよりも大きい)、又は上気道内へのスプレーのいずれかによって投与される。上記送達経路のいずれかによって全身性の防御免疫反応が生じると同時に、鼻腔内投与は、インフルエンザウイルスの侵入部位である粘膜免疫を誘発するさらなる利益をもたらす。鼻腔内投与については、弱毒化した生ウイルスワクチンがよく選ばれる。例えば、弱毒化、低温適応性、及び/又は温度感受性の組換えインフルエンザウイルス若しくはリアソータントインフルエンザウイルスが挙げられる。1回の投与で防御免疫反応が刺激されることが好ましいが、同一の若しくは異なる経路で追加投与し、所望の予防効果を達成することができる。これらの方法は、いずれのウイルスにも適用することができる。ここで言うウイルスは、オルソミクソウイルス(A型及びB型インフルエンザ株を含む)、パラミクソウイルス(RSV、ヒトメタニューモウイルス、及びパラインフルエンザを含む)、ラブドウイルス及びフラボウイルスを含む。ただし、これらに限定はされない。
【0078】
<インフルエンザウイルス>
本明細書に記載の方法、工程及び組成物は、主としてワクチン用のインフルエンザウイルスの生産に関係している。インフルエンザウイルスは、セグメント化した一本鎖RNAゲノムを含む内部のリボ核タンパク質コアと、マトリックスタンパク質によって裏打ちされた外部のリポタンパク質エンベロープから構成されている。A型インフルエンザ及びB型インフルエンザは、それぞれ8セグメントの一本鎖のマイナス鎖センスRNAを含んでいる。A型インフルエンザゲノムは11種のポリペプチドをコードしている。第1〜3セグメントは、RNA依存性RNAポリメラーゼを構成する3つのポリペプチドをコードしている。第1セグメントは、ポリメラーゼ複合体タンパク質PB2をコードしている。残るポリメラーゼタンパク質PB1及びPAは、それぞれ第2セグメント及び第3セグメントにコードされている。さらに、いくつかのインフルエンザ株の第1セグメントは、PB1コード領域内の別の読み取り枠から生成される小タンパク質PB1-F2をコードしている。第4セグメントは、感染中の細胞への接着及び侵入に関与する表面糖タンパク質ヘマグルチニン(HA)をコードする。第5セグメントは、ウイルスRNAと結びつく主要な構造成分であるヌクレオカプシドヌクレオタンパク質(NP)ポリペプチドをコードしている。第6セグメントは、エンベロープ糖タンパク質ノイラミニダーゼ(NA)をコードしている。第7セグメントは、異なるスプライシングを受けたmRNAより翻訳される2種の基質タンパク質M1及びM2をコードしている。第8セグメントは、選択的スプライスシングを受けたmRNAから翻訳される2種の非構造タンパク質NS1及びNS2をコードしている。
【0079】
B型インフルエンザ8つのゲノムセグメントは、11種のタンパク質をコードしている。最も大きい3つの遺伝子はRNAポリメラーゼの成分、PB1、PB2及びPAをコードしている。第4セグメントは、HAタンパク質をコードしている。第5セグメントは、NPをコードしている。第6セグメントは、NAタンパク質とNBタンパク質をコードしている。NB及びNAの両タンパク質は、バイシストロニックmRNAの重複した読み取り枠から翻訳される。B型インフルエンザの第7セグメントも、二つのタンパク質をコードしている。すなわち、M1及びM2である。最も小さいセグメントは、全長RNAから翻訳されるNS1とスプライシングされたmRNA変異体から翻訳されるNS2の2つの産物をコードしている。
【0080】
リアソータントウイルスは、選択されたヘマグルチニン抗原、及びノイラミニダーゼ抗原をマスタードナーウイルス(MDV)とも呼ばれる認可されたマスター株の中で合体させて生産される。FluMist(登録商標)は、認可された低温適応性、弱毒性、温度感受性MDV株(例えばA/AnnArbor/6/60及びB/Ann Arbor/1/66)を使用している。卵をベースとした方法や最近の細胞培養方法を含む多くの方法は、リアソータントウイルスの生成に有用である(例えば、国際公開第WO 03/091401号;同第05/062820号、及び米国特許第6,544,785号;同第6,649,372号;同第6,951,754号を参照されたい)。本発明のMDCK細胞、培地及び工程は、インフルエンザウイルスの生産に有用であることが意図されている。ここでいうインフルエンザウイルスは、本明細書で開示するインフルエンザ株(例えば、A/AnnArbor/6/60及びB/AnnArbor/1/66)、並びにA/AnnArbor/6/60、B/AnnArbor/1/66、PR8の遺伝子を含むリアソータントウイルスを含むが、これらに限定はされない。さらに本発明のMDCK細胞、培地、及び方法は、一以上の以下の表現型、すなわち温度感受性、低温適応性、弱毒性を有するリアソータントウイルスを含むインフルエンザウイルスの生産に有用であることが意図されている。リアソータントウイルスは、古典的なリアソータント技術、例えば共感染法によって、又は任意でプラスミドレスキュー技術によって生成することができる(例えば、国際公開第WO 03/091401号;国際公開第WO 05/062820号、及び米国特許第6,544,785号;同第6,649,372号、同第6,951,754号を参照されたい。)
【実施例】
【0081】
以下の実施例を参照して本発明をこれから説明する。これらの実施例は、例示の目的のみのために提供されるのであって、本発明がこれらの実施例に限定されると解されるべきではない。むしろ本明細書で提供される教示の結果明らかとなる、ありとあらゆる変更を包含すると解されるべきである。
【0082】
実施例1
株細胞におけるca/tsインフルエンザ株の感染拡大の測定とMDCK細胞で生産されるインフルエンザの特徴解析
卵を利用しない代替生産プラットフォームの開発や動物又は昆虫の培養細胞系でのインフルエンザワクチンの生産にはワクチン業界の努力があった。明らかな利点として、スケールアップが簡単なことと、工程制御が増えたこと、及びいくつかのワクチンでアレルギー反応の原因となっていた卵タンパク質の排除が挙げられる。細胞培養に基づく系は、スケールアップが素早くできるので、卵の供給不足が予想されると共にワクチンの迅速な生産が必要とされるインフルエンザの世界的流行時にさらなる利点を提供する。最初の実験では、計7種の異なる株細胞を用いて行った。すなわち、2種の二倍体肺線維芽細胞株(MRC-5株とWI-38株)(データは示さず)、ヒト網膜芽細胞腫及びヒト腎臓細胞株(それぞれPER.C6株と293株;通常これらはいずれもアデノウイルス産物の生産用に構築された)(データは示さず)、胎児アカゲザル肺細胞株(FRhL2)(データは示さず)、アフリカミドリザル腎臓由来細胞株(Vero)、及びMarin-Darbyイヌ腎臓由来(MDCK)細胞株である。MDCK細胞は、低温適応性、温度感受性、弱毒性(ca/ts/att)のリアソータントインフルエンザウイルス株H1N1、H3N2、潜在的パンデミックワクチン株H5N1、そしてB型株の4種全てを商業的に妥当なタイター(>107 Log TCID50/mL)まで増やすことのできることを試験した唯一の細胞株である(図1、データは示さず)。MDCK細胞で増殖したウイルスの遺伝学的な及び抗原性の特徴を卵で増殖したウイルスのそれと比較した。ゲノム配列に有意な差異は見られなかった(データは示さず)。またHAIタイターで測定された抗原性は同程度であった(表1)。
【0083】
<蛍光焦点アッセイ>
MDCK細胞及びVero細胞を96ウェルブラックプレートで4日間増やした(DMEM+4 mMグルタミン+PEN/Strep)。各ウェルをca/ts B型ンフルエンザ株(B/HongKong/330/01、及びB/Yamanashi/166/98)で〜0.01のMOIにてDMEM+4mMグルタミン+60mU/mL TPCKトリプシン中で感染させた。ウイルスに感染したプレートを固定し、免疫染色した後、感染の広がりを以下のように測定した。ウイルスを含んだ培地を各プレートから除き、プレートを200μl/ウェルのDPBS(Ca2+/Mg2+非含有)で1回洗浄し、その後プレートを200μl/ウェルの冷やした4%(v/v)パラホルムアルデヒド/PBSで固定した。プレートを200μl/ウェルのDPBS(Ca2+/Mg2+非含有)で2回洗浄し、続いて細胞を一次抗体(0.1%サポニン+1%BSA/PBSで1:1000の比率で希釈したヒツジ抗Yamanashi B型、及びヒツジ抗HongKong B型)と共にインキュベートした。1時間のインキュベーションの後、一次抗体を除き、細胞をPBS中の0.1%Tween20で3回洗浄し、ウェルを二次抗体(0.1%サポニン+1%BSA/PBSを用いて1:100の比率で希釈したFITC標識したウサギ抗ヒツジ抗体)と共にインキュベートした。ウェルを毎日4日間、蛍光顕微鏡を用いて視覚化し、画像を毎日SPOTプログラムを用いて取り込んだ。
【0084】
<結果と考察>
蛍光焦点アッセイを用いて、MDCK細胞及びVero細胞でca/tsのB型インフルエンザ株の感染の広がりがあるか否かを評価した。また、50個のVeroクローン細胞間でウイルス感染の広がりに何らかの違いがあるかどうかも評価した。単分子層の蛍光は、MDCK細胞では4日間にわたって増加したが、Vero細胞では増加しなかったので(図1A参照)、Vero細胞はca/tsのB型株の生産に対して許容状態にないが、MDCK細胞は許容状態にあると結論付けられた。このデータは、B型株がMDCK細胞では7〜7.5log10TCID50生産されるが、Vero細胞ではわずか4〜4.5log10TCID50しか生産されないことを示した初期の実験データに類似している(データは示さず)。
【0085】
MDCK細胞を、潜在的なパンデミックワクチン株ca A/Vietnam/1203/2004株を含む多くのca/ts/attリアソータント株の複製を支持する能力についても試験した。MDCK細胞をca A/Vietnam/1203/2004を用いて低感染多重度で感染した。また、上清中のウイルスは感染後に様々な時間で定量化された。感染後48時間までにca A/Vietnam/1203/2004のタイターは、約8log10 TCID50/mLに達し、その後3〜4日間はその値で安定していた。図1B及び表2を参照されたい。
【0086】
A/H1N1型、A/H5N1型、A/H3N2型及びB型のca/ts/att株は、MDCK細胞内で比較的高いタイターで複製された。さらに、MDCK細胞中でこれらのca/ts/att株は、継代にて、そのゲノム配列を有意に変えなかった。3種のca/ts/att株、ca A/Sydney/05/97、ca A/Beijing/262/95、及びca B/Ann Arbor/1/94をMDCK細胞内で1回、又は2回継代し、全部で6つの内部遺伝子の完全なコード領域を配列決定し、開始時の配列と比較した。ヌクレオチドの変異は見られず(データは示さず)、本細胞を介した継代が、これらの株の遺伝子組成を変えないことが実証された。さらなる配列の特徴づけを培地組成、導入量(MOI)、インキュベーション温度、及び回収時間を含む生産工程を再現した条件下においてMDCK細胞で生産された異なるワクチン株で行った。予備データに基づき、MDCK産生ウイルスのゲノム配列には有意な変化がないことが期待された。
【0087】
ゲノムはMDCK細胞内では継代後も遺伝学的には安定であるので、卵又はMDCK細胞で生産されるワクチンの生物学的形質は見分けがつかないことが期待された。しかし培養細胞由来の主なウイルス産物は、卵由来の産物と比べていくつかの僅かな違い、特にHA及びNAを含むウイルスタンパク質の翻訳後修飾、又はウイルス膜中の脂質の組成に関して違いが見られた。これらはどちらもウイルス粒子の全体的な物理的特性を潜在的に変えることができる。培養細胞で生産されたワクチンと卵で生産されたワクチンの抗原性における予備的な前臨床データから、前記の重要なパラメータの違いを検出できないことが実証された。いくつかのワクチン株の卵のストックがMDCK細胞を介して継代され、両産物の抗原性を基準抗血清を用いてHAIタイターを測定することで実証した。表1で示すように、全てのHAIタイターは、お互いに2倍以内であった。これは、卵由来のワクチンと比べて、細胞内でのワクチンの複製がワクチンの抗原性を変えないことを示している。
【表1】
【0088】
実施例2
非腫瘍形成性血清MDCK細胞の誘導
MDCK細胞は、昔からインフルエンザウイルスのタイトレーションに使用されてきた(Zambon著,1988,in Textbook of Influenza,Nicholson編, Webster and Hay, ch24, pg 324-332, Blackwell Science)。それ故、ワクチン物質生産のためインフルエンザの増殖用に使用することができた。しかし、MDCK細胞は、伝統的にFBSを追加したイーグル最小必須培地(Eagle’s Minimal Essential Medium;EMEM)のような基本培地処方物で増殖されていた。そのような条件下で及び/又は長期間インキュベートするとMDCK細胞が腫瘍形成性となる可能性があることを示した報告が多数ある(例えば、Gaushら,Proc Soc Exp Biol Med,122:931;Leightonら,1968,Science,63:472;及びLeightonら,1970,Cancer,26:1022を参照されたい)。ゆえに、ワクチン物質の生産にMDCK細胞を使用することについては懸念があり、その他の細胞株の開発(例えば、PER.C6及びVERO)に努力が向けられた。残念なことに、インフルエンザ株の全てが他の哺乳動物細胞株中で十分に増殖するわけではない。特に弱毒化されたインフルエンザ生ワクチンであるFluMist(登録商標)を含む低温適応したインフルエンザウイルスは、MDCK細胞内でのみ適当なタイター(>107 TCID50/mL)まで増殖できる(上記実施例1を参照)。MDCK細胞の特徴解析を行った初期の報告では、MDCK細胞の早期の継代では腫瘍形成性が起き得ないことを示している(Gaushら,1966, Proc Soc Exp Biol Med.122:931)。非腫瘍形成性の状態にMDCK細胞を維持する培地及び継代プロトコルを確立することが本実験の目標であった。
【0089】
ATCC(CCL34)から入手したMDCK細胞を、Tフラスコ内で10%FBS、4mMグルタミン及び4.5g/Lグルコースを追加したDMEMを増殖培地に用いて増やした。前マスターMDCK細胞バンクを血清増殖MDCK細胞(MDCK-S細胞)で確立した。この血清増殖MDCK細胞は、民間の受託業者(例えば、BioReliance、Rockville、MD)によって行われる通常のテストを用いて細菌/真菌の汚染、及びマイコプラズマの汚染についてテストされた。前記細胞は、細菌/真菌の汚染物質の存在についてネガティブであることがわかった。MDCK-S細胞は、培養可能なマイコプラズマの存在についてもネガティブであることがわかった。前記バンク由来のMDCK-S細胞を核型解析でもテストした。その結果、その起源がイヌ科であることが判明し、また約70本〜84本の染色体数の範囲で78本の最頻値染色体数を有していた。その後、MDCK-S細胞をPreMCBの容器から起こして新たに20継代し、インビボでアダルトヌードマウスのモデルの核型と腫瘍形成についてテストした。核型テストでは、継代後期のMDCK-S細胞(p81/24)は継代初期のMDCK-S細胞と同じ染色体数最頻値(78本)及び染色体の範囲(70〜84)を示した。これは、継代を続けても細胞が変化しなかったことを示している。1×107のMDCK-S細胞をアダルトヌードマウスの皮下に注射しても、どんな小結節の形成も生じなかった。したがって、当該細胞を非腫瘍形成性とみなした。
【0090】
<材料と方法>
材料
MDCK細胞(ATCC, Cat. No:CCL-34);T-25、T-75、T-225フラスコ(Corning社、Cat No.: 430639, 430641, 431082);ダルベッコ改良イーグル培地(DMEM)粉末(Gibco社、Grand Island NY, Formulation No.:01-5052EF);ガンマ線照射済みウシ胎仔血清(JRH社、Lenexa KS, Cat. No.:12107-500M);L-グルタミン(JRH社、Lenexa KS, Cat. No.: 59202-100M);D-グルコース(Amresco社、Cat. No.:0188-1KG);Ca2+及びMg2+非含有のダルベッコリン酸バッファ(DPBS)(Gibco社、Grand Isand NY, Cat. No.:21600-069);0.05%トリプシン-EDTA(Gibco社、Grand Island NY, Cat. No.:25300);ジメチルスルホキシド(DMSO)(Sigma社、St. Louis MS, Cat. No.:D2650);0.4%(w/v)トリパンブルー染料/PBS溶液(Sigma社、St. Louis MS, Cat. No.:T8154);CO2インキュベータ(Forma Scientific社、Model No.:3110);YSIバイオアナライザ(YSI社、Model No.: 2700 select);VITROS化学システム(Ortho clinic社、Model: DT60 II);改良型ノイバウア血球測定器(Hausser Scientific社、Bright-Line 0.1mm deep/Reichert, Bright-Line 0.1mm deep)。
【0091】
組織培養フラスコでの血清MDCK(MDCK-S)細胞の継代培養
血清MDCK細胞のバイアルをATCCから入手した。細胞をT-75フラスコ内の10%(v/v)FBS、4.5g/Lグルコース、2.2g/L NaHCO3、及び4mM L-グルタミンを追加したDMEM培地で増やした。細胞を接種後3〜4日で継代するとともに、もし細胞を4日目に継代するのであれば接種後3日目に全培地交換を行った。細胞を以下に記載のようにTフラスコから回収した。
【0092】
使用済み増殖培地を除き、単分子層細胞をDPBS(カルシウム、マグネシウム無し)で2回洗浄した。37℃ウォーターバスで予熱したトリプシン‐EDTAを適当量(3mL/T-75、7.5mL/T-225)、各フラスコに加え、その後Tフラスコを37℃、5%CO2インキュベータ内で約15〜20分間インキュベートした。フラスコを5分ごとに確認し、細胞が剥離しているかどうかを調べた。またフラスコを数回軽く叩いて細胞の剥離を促した。細胞がTフラスコから完全に剥離したら、10%血清を含む等量の完全成長培地(3mL/T-75、7.5mL/T-225)を加えてトリプシンを不活性化した。細胞懸濁液を適当なサイズのピペットで吸引/排出によって数回上下させ、大きな細胞塊をバラバラにした。0.5ml細胞懸濁液サンプル二つを血球測定器でカウントした。二つのカウントの結果が相互に15%以内にない場合には細胞カウントを繰り返した。細胞を新鮮な保温した増殖培地(DMEM+10%FBS+4.5g/Lグルコース+4mMグルタミン)で新しいフラスコ内で0.05×106生細胞/mLに希釈した。そしてTフラスコに接種した(35mL/T75又は100mL/T-225)。その後、継代又は培地交換を行う前にそのフラスコを37±1℃、5% CO2の環境下で3日間培養した。
【0093】
MDCK-S細胞バンクの調製
MDCK-S細胞を上記のTフラスコ内でバンクに必要な細胞の総必要量を回収できるまで増やした(4×106細胞/バイアル×バイアル数)。MDCK-S細胞が対数増殖期にあるとき(接種後3日目)に記載したトリプシン処理によって当該細胞を回収した。個々のフラスコ由来のMDCK-S細胞の懸濁液をプールし、150〜250Gで7±1分間遠心して細胞を回収した。各チューブから上清を吸引して除き、沈殿した細胞を新しい完全成長培地(DMEM+10%FBS+4.5g/Lグルコース+4mMグルタミン)で再懸濁した。異なる遠心ボトル由来の細胞懸濁液をプールし、細胞懸濁液を適当なサイズのピペットで吸引/排出によって数回上下させ、大きな細胞塊をバラバラにした。総細胞数を測定し、4×106細胞/バイアルで冷凍できるバイアルの総数を決定した。
【0094】
その後、細胞懸濁液の量を新しい増殖培地を用いて上記の値に合わせた。等量の新たに調製した2×冷凍培地(DMEM+10%FBS+4mMグルタミン+4.5/Lグルコース+15%DMSO)を細胞懸濁液に加えた。細胞懸濁液を十分に混合して、1mLの細胞懸濁液を各冷凍バイアルに分注した。全てのバイアルをNalgene社の冷凍コンテナに移し、≦−60℃フリーザ内に置いた。冷凍バイアルを液体窒素保存タンクに移した。
【0095】
T-75フラスコでのMDCK-S細胞の成長曲線の作成
細胞を細胞成長曲線の実験前に少なくとも4回(解凍後)、成長培地にて継代した。MDCK-S細胞を、T-225フラスコ内で少なくとも総細胞数2.7×107個得るために増やした。フラスコを上記のトリプシン処理前にコンフルエントの80〜95%まで増やした。回収したMDCK-S細胞をプールし、細胞懸濁液をピペットで吸引/排出によって数回上下させ、大きな細胞塊をバラバラにした。2サンプル(0.5mL)を細胞カウント用に取り、細胞密度を測定した。2サンプルのカウントが、相互に15%以内にない場合には再度測定を行った。その後、総計2.7×107MDCK細胞を総量540mLの完全成長培地に希釈した(5.0×104細胞/mL)。それから、このMDCK-S細胞懸濁液を14本のT-75フラスコ(35mL/T-75フラスコ)に分注した。そのフラスコを37±1℃、5% CO2インキュベータに置いた。
【0096】
二つのTフラスコを、細胞カウント及び代謝解析のために毎日インキュベータから取り出した。細胞培養培地を2サンプル(約1.0mL)、各フラスコから代謝解析用に取り出した。一のサンプルを使用して、YSI及びVITROSアナライザーを用いてグルコース、乳酸、グルタミン、グルタミン酸、及びアンモニア濃度を測定した。他方のサンプルについては、後日アミノ酸解析を行うため−70℃で冷凍した。MDCK-S細胞を上記のトリプシン処理によって各フラスコから回収した。細胞密度を測定し、また細胞総数/Tフラスコも測定した。二つのカウントが、お互い15%以内にない場合には再度カウントを行った。存在数は、二つの異なる継代数(p63及びp65)のMDCK-S細胞で行った二つの独立した成長曲線解析の平均である。
【0097】
核型試験
核型試験は、フロリダ州メルボルンのApplied Genetics Laboratoriesで行われた。簡単に言えば、T-225フラスコで増殖したMDCK-S細胞をApplied Genetics Laboratories社に送った。細胞は上で記載した方法により維持され、また継代された。細胞が有糸分裂状態の細胞を十分に含んでいると思われるときは、その細胞を有糸分裂解析用に回収した。細胞をコルセミド(0.02μg/mL)で37℃にて150分間処理した。その後、細胞をトリプシン処理して回収し、200Gで5分間遠心した。上清を吸引にて除去し、予熱した低張溶液に再懸濁し、37℃で10分間インキュベートした。膨張した細胞を遠心で沈殿させ、それからCarnoy’s溶液(3:1 メタノール:氷酢酸)中で室温にて40分間インキュベートして固定した。細胞を再度遠心し、その後その細胞を少なくとも2回Carnoy’s固定剤で洗浄した。最後の遠心の後、細胞を1〜3mLの新しい固定剤で再懸濁し、乳白色の細胞懸濁液を生成した。最終細胞懸濁液の液滴をきれいなスライド上に落とし、風乾した。
【0098】
ライト染色液/リン酸バッファを前記スライドに加えて7〜10分間インキュベートして細胞を染色した。スライドを7〜10分後に水道水で洗浄し、その後風乾した。細胞を低倍率対物レンズ(10×)で走査し、細胞分裂の中期ステージにある細胞を検出した。また中期の細胞の染色体を高倍率油浸レンズ(100×)により解析した。100個の中期細胞を細胞発生異常及び染色体カウントについて解析した。1000個の細胞を走査し、倍数体頻度及び分裂指数(有糸分裂をしている細胞のパーセント)を測定した。
【0099】
MDCK-S PRE-MCBの無菌テスト(静菌性、静真菌性、及び4つの培地の滅菌性)
MDCK-S PRE-MCBの静菌活性、及び静真菌活性をBioreliance Inc.社(ロックビル,メリーランド)でテストした。本アッセイは、US 26及びUS21 CFR 610.12要件を満たして行われた。本アッセイは、0.1 mLのテストサンプルを含む適当な培地とコントロール生物のみを含む培地に接種したコントロール生物(バチルス・サブチルス(Bacillus subtilis)、カンジダ・アルビカンス(Candida albicans)、クロストリジウム・スポロゲネス(Clostridium sporogenes)、スタフィロコッカス・アウレウス(Staphylococcus aureus)、シュードモナス・エアルギノサ(Pseudomonas aeruginosa)、アスペルギルス・ニガー(Aspergillus niger))の増殖と違いがあるか否かをテストするものである。簡単に言えば、被験物質を3本のTSB(大豆カゼイン分解培地)チューブ、4本のTHIO(fluid thioglycollate medium:流動性チオグリコール酸培地)、2本のSAB(Sabourand Dextrose Agar:Sabourandデキストロースアガー)、及び1本のPYG(peptone yeast extract:ペプトン酵母エクストラクト)に接種した。その後、100cfu未満のコントロール生物を含む各コントロール生物を適当なタイプの培地に接種した。ポジティブコントロールは、TSB及びTHIOのバチルス・サブチルス、TSB及びSAB(20〜25℃及び30〜35℃)のカンジダ・アルビカンス、THIO及びPYGのクロストリジウム・スポロゲネス、シュードモナス・エアルギノサ、及びアスペルギルス・ニガーからなる。ネガティブコントロールは、滅菌したPBSである。培地を3〜5日間インキュベートし、生物の増殖を確認した。
【0100】
被験物質を、細菌及び真菌汚染の有無についても4種の培地無菌テストを用いてBioreliance社(ロックビル,メリーランド)で解析した。本アッセイは、USP 26, EP、及び21CFR610.12要件を満たすことが意図されている。簡単に言えば、被験物質を、2本のTSB(大豆カゼイン分解培地)チューブ、2本のTHIO(流動性チオグリコール酸培地)、3本のSAB(Sabourandデキストロースアガー)、及び2本のPYG(ペプトン酵母エクストラクト)に接種した。培地を適当な温度(SABスラントについては2つの温度でインキュベートした)でインキュベートし、チューブをテスト第3日目/第4日目又は第5日目、7日目又は8日目、及び14日目に確認することで、全てのチューブを14日間にわたって観察した。混濁が現れる被験物質を接種したいずれのチューブもプレートに広げ、そのプレート上でグラム染色を行った。ネガティブコントロールは滅菌したPBSである。
【0101】
マイコプラズマ/静マイコプラズマ性テスト
冷凍MDCK-S細胞のバイアル(MDCK preMCB lot No.747p105)をBioreliance社に送った。細胞は上記で説明したようにTフラスコ内で増やされて培養された。5×105cells/mLの濃度の細胞溶解物を調製し、−70℃で冷凍した。被験物質をマイコプラズマ・ニューモニエ(Mycoplasma pneumoniae)、マイコプラズマ・オラレ(Mycoplasma orale)、及びマイコプラズマ・ハイオライニス(Mycoplasma hyorhinis)の増殖を抑制する能力についてアガーブロス/プレートで、及び/又はVERO細胞内でテストした。
【0102】
アガー分離アッセイについては、被験物質をアガープレート又はブロスボトルでスパイク、又は非スパイクのいずれかをテストした。被験物質は、マイコプラズマ・ニューモニエ、及びマイコプラズマ・オラレでスパイクされ、10〜100cfu/0.2mL(アガーテスト用)及び10〜100cfu/10mL(準ブロスアッセイ用)の希釈を達成した。テストサンプルの一部はスパイクされなかった。4つの半固体ブロスボトルをスパイク(2ボトル)、又は非スパイク(2ボトル)の各10mlで接種した。スパイク/非スパイクの各1本のボトルを、有酸素的にか、又は嫌気的にかのいずれかで適当な温度でインキュベートした。10枚のA型アガープレートと10枚のB型アガープレートをそれぞれスパイクサンプル又は非スパイクサンプルで接種した。A型アガープレートとB型アガープレートの半分を有酸素的にか又は嫌気的にかのいずれかで適当な温度でインキュベートした。非接種マイコプラズマ半固体ブロスを非接種ネガティブコントロールとした。全ブロスボトルを21日間観察した。各ブロスボトル(非接種ネガティブコントロールを除く)を3日目、7日目、及び14日目にA型アガープレート又はB型アガープレートに継代し(それぞれ10プレート、0.2mL/プレート)、同一条件下で適切なボトルとしてインキュベートした。それらを1日1回、21日間観察した。
【0103】
増強VERO細胞培養アッセイについて、被験物質のスパイク、又は非スパイクをテストした。被験物質をM.オラレ及びM.ハイオライニスを用いて10〜100cfu/0.2mLの濃度でスパイクした。スパイクした被験物質、非スパイクした被験物質、ポジティブコントロール、及びネガティブコントロールをそれぞれVERO細胞培養液のT-75フラスコに接種した。3〜5日のインキュベーション後、各フラスコの細胞を掻き取り、直ちに凍結した。各フラスコの細胞溶解物1mLの10分の2をVERO細胞の入った6ウェルプレートの各ウェルに接種した。さらに、ポジティブ及びネガティブコントロールをVERO細胞の入った6ウェルプレートの適当なウェルに接種した。3〜5日後、細胞を固定し、DNA結合へキスト色素で染色し、マイコプラズマの存在を評価した。
【0104】
ヌードマウスにおけるMDCK-S細胞の腫瘍形成性テスト
無胸腺ヌードマウス(nu/nu)における腫瘍形成の評価は、BioReliance(登録商標)(ロックビル、MD)によって行われた。簡単に説明すると、30匹のメス無胸腺マウス(4週齢)の皮下に、0.2mL(1×107細胞/マウス)のポジティブコントロール(18Cl-10T細胞)、ネガティブコントロール(シリアンハムスター胚細胞;SHE細胞)、又はテスト細胞(血清MDCK細胞、747p105高継代)を注射した。前記動物を注射前に無作為に選択し、全マウスに22ゲージ針を用いて同日に注射した。全動物を就業日に毎日観察し、注射部位を84日の期間、1週間に2回、病変発生の確認のため触診した。それぞれの病変を測定し、病変サイズの増加が見らる間は動物を維持した。この病変サイズの増加が見られる期間は、最大で3ヶ月間であった。84日後に全マウスを絞めて解剖し、注射部位、肺、肩甲リンパ節、及び全病変部を組織病理学的方法で解析した。
【0105】
MDCK-Sにおける低温適応性インフルエンザ株の複製
T-75フラスコに5×104細胞/mL(DMEM 35mL+10%FBS+4mMグルタミン)を接種し、37℃、5%CO2維持されたインキュベータで3日間、増やした。接種後3日目にTフラスコあたりの全細胞をトリプシンEDTAを用いて回収することによって測定した。また、細胞のカウントは、トリパンブルー分染排除法で測定した。残りのTフラスコは、その後次のように感染させた。増殖培地を吸引して除去し、細胞を1フラスコあたり10mLのDPBS(Ca2+/Mg2+なし)で2回洗浄した。0.01の感染多重度(MOI)でそれぞれのTフラスコに感染させるウイルス量を以下の方程式により測定した。
【数1】
【0106】
MOIは、加えた1細胞あたりのウイルス粒子として定義される。
【0107】
その後、必要量のウイルスを各Tフラスコ中の35mLの後感染培地(DMEM+4mMグルタミン+60mu/mL TPCKトリプシン)に加えた。それから、そのTフラスコを33℃、5%CO2でインキュベートし、6日間毎日サンプルを取った。10×SPを各サンプルに安定剤として加え、そのサンプルを感染性をテストする前に<−70℃に保存した。
【0108】
各サンプル中に存在するウイルスの濃度を、感染ウイルスを測定する50%組織培養感染量(TCID50)アッセイによって測定した。簡単に説明すると、MDCK細胞を96ウェルマイクロタイタープレートにて単分子層でコンフルエントまで増やし、ca/tsインフルエンザウイルスサンプルの連続希釈物を加えた。MDCK細胞アッセイプレート内のサンプルは、通常10−4〜10−10の最終希釈物となった。カラム1〜5及び8〜12のウェルには、ウイルス希釈サンプルを入れた。また、カラム6〜7にはウイルス希釈物のみを入れ、細胞コントロールとしての役目をさせた。この構成により、プレートあたり2点のデータ(n=2)が得られた。MDCK細胞でのウイルスの複製は、細胞死及び培養液上清中への子孫ウイルスの遊離を生じた。子孫ウイルスは他の細胞に感染し、最終的に単分子層を破壊した。感染の結果生じる細胞変性効果(cytopathogenic effect;CPE)をCO2環境下33±1℃で6日間インキュベーションする間、進展させた。その後プレートをインキュベータから取り出し、ウェル内の培地を廃棄した。続いて100μlのMEM+4mMグルタミン+ペニシリン/ストレプトマイシン+MTTを各ウェルに加えた。プレートを37℃、5%CO2で6時間インキュベートし、CPEを示すウェル数を各ウェル内で形成される色(黄色/橙色はCPEウェルを意味し、濃淡のない紫色はCPEがないことを意味する)に基づいた目視検査によって測定した。それぞれ半分のプレートでCPEを示したウェル数を用いて、Reed-Muench法のKarber改変法に基づいたタイター(log10 TCID50/mL)を算出した。
【0109】
<結果と考察>
血清MDCK細胞の冷凍バイアル2本を、2本のT-75フラスコに分離して完全成長培地(DMEM+10% FBS+4mMグルタミン+4.5g/Lグルコース)中で解凍した。解凍による細胞生存率は、それぞれ97%と98%であった。細胞は、解凍後3日間でコンフルエントに達した。細胞形態は、上皮様で、ATCCより入手したストック株に似ていた(図3)。これらの細胞を5回継代し、血清増殖MDCK細胞(MDCK-S細胞)の前マスター細胞バンク(Pre-master cell bank;PreMCB)を樹立した。図2は、MDCK-Sの前マスター細胞バンク(PreMCB)の誘導に用いられる方法を概説している。
【0110】
10%FBS DMEM培地におけるMDCK-S細胞の成長曲線を図4に示す。この結果は、異なる継代数(P63及びP65)の細胞を用いた2つの実験の平均である。MDCK-S細胞は、約1日の誘導期(lag phase)を有している(接種時の総細胞数1.75×106細胞/T75フラスコ、及び1日目の総細胞数2.9×106細胞/T75)。誘導期とは、接種してからの細胞数が2倍に達しない時期をいう。グルコース消費/乳酸産生比は、初日ではほぼ0であった。これは、細胞が誘導期にあることを示している(図5)。その後細胞は、接種後4日目に静止期に入るまでの細胞増殖期間の間、指数関数的に増殖した。対数増殖期におけるMDCK-S細胞の倍化時間は、23.1時間であった。対数期の間、グルコース消費と乳酸産生は、乳酸濃度の増加とグルコース濃度の減少においてお互いに酷似していた(図5)。グルコース消費/乳酸産生比は、細胞成長曲線とよく対応していた(図4、5を比較されたい)。この速度は誘導期間の間は低く、1日目〜4日目の対数期の間はグルコースが2.93mM/日、及び乳酸が3.43 mM/日増加した。
【0111】
MDCK-S細胞は、接種後4日目に静止期に入った。そして、到達した最大細胞密度は、接種後5日目におけるおよそ29±0.99×106細胞であった(図4)。細胞数は、本実験では最大細胞密度に達した後も7日目まで一定に維持された。グルコース消費速度と乳酸産生速度は、静止期ではグルコースが0.33mM/日、及び乳酸が0.25mM/日に落ちた。7日間の培養後の約12mMのグルコースが培地中にまだ残っていた。4日目のグルコース消費量対乳酸産生の比率は1.2であった。
【0112】
MDCK-S細胞のグルタミン消費、並びにグルタミン酸及びアンモニアの両産生を図6に示す。グルタミン消費率とアンモニア産生率は、細胞成長曲線とよく対応していた(図4と図6を比較されたい)。MDCK-S細胞は、4日目までの対数増殖期間中、グルタミンを0.49mM/日の速度で消費し、同時にアンモニアを5日目まで0.32mM/日の速度で産生した。その後、細胞が静止期に入ると、グルタミン消費速度は0.24mM/日まで下がり、同時にアンモニア産生速度も0.11mM/日まで下がった。アンモニア産生対グルタミン消費の比率は、接種後4日目で0.7であった。グルタミン代謝から生じるグルタミン酸は、今回の7日間の細胞増殖実験では蓄積しなかった。
【0113】
MDCK-S細胞の核型を継代61/4、及び継代81/24でテストした。Gバンド染色体解析は細胞がイヌ科起源であることを示した。100個の中期細胞における染色体数の分布を図7に示す。染色体の総数は、低継代61/4については細胞中期あたり70本〜84本、また高継代81/24については70本〜84本に分布していた。両継代とも最頻値染色体数は78本であった。染色体の分布は、継代で変わらなかった。細胞の様相は、正常なイヌ腎臓細胞(Starkeら, 1972, Prog Immunobiol Stand., 5:178)から予想された通りであった。
【0114】
MDCK-SのpreMCBを細菌汚染、真菌汚染、又はマイコプラズマ汚染のいずれかの存在についてテストした。pre-MCBは無菌テスト(細菌及び真菌の汚染を確認するための直接接種法を用いた4培地無菌テスト)に合格し、マイコプラズマの存在(アガー培養可能、及び非アガー培養可能アッセイ)についてネガティブであることがわかった。被験物質は、静菌性/静真菌性テスト、及び静マイコプラズマ性テストのどちらもポジティブコントロールの増殖を阻害しないこともわかった。
【0115】
継代81/24のMDCK-S細胞(pre-MCB+20継代)をヌードマウス上に接種し、3ヶ月間腫瘍形成テストを行った。MDCK-S細胞を接種したいずれのマウスにおいても腫瘍が診断されなかったことから、MDCK-S細胞は腫瘍形成性でないことが実証された(表4)。
【0116】
MDCK-S細胞をテストし、低温適応性、温度感受性、弱毒性のリアソータントインフルエンザ株の複製を支持できることが判明した(表2)。
【表2】
【0117】
実施例3
Taub培地における無血清MDCK細胞の誘導
上記実施例2で詳述した結果は、MDCK細胞を上皮形態及び正常な核型、同時に低温適応性インフルエンザ株を複製する能力を維持した状態で培養することが可能なことを証明している。さらに、本発明者らは、上記実験で開発された条件下でMDCK細胞を培養すると、非腫瘍形成性のMDCK細胞を生じることを立証した。しかし、実施例2で使用した培地は、ウシ胎仔血清(FBS)を含んでいる。FBSは、複合成分混合物であり、ロット間の変動の問題が報告されている。また、ウシの牛海綿状脳症(BSE)に伴う現在継続中の問題が、安全面での不安をもたらしている。MDCK-S細胞株の非腫瘍形成性の性質と増殖特性を維持する無血清培地の開発は、治療及び予防接種用に生産される生物製剤の安全性を高める上で重要である。
【0118】
ATCC(CCL34)から入手したイヌ腎臓由来(MDCK)細胞を、Tフラスコ内で10%FBS、4mMグルタミン、及び4.5g/Lグルコースを追加したDMEMを増殖培地として用いて、5回継代して増やした。その後、細胞を無血清Taub培地(以下の処方を参照)に移した。Taub培地処方物中での増殖に適応した細胞をMDCK-Tとした。pre-MCBをMDCK-T細胞について樹立し(図8参照)、細菌/真菌汚染、及びマイコプラズマ汚染についてテストした。MDCK-T細胞の前マスター細胞バンクの細胞を核型アッセイでもテストし、イヌ起源であること、52本〜84本に及ぶ染色体数と共に78本の最頻値染色体数を有することがわかった。さらに、MDCK-T細胞をPreMCBのバイアルから起こして新たに少なくとも20回継代した。その後、核型及びインビボアダルトヌードマウスモデルでの腫瘍形成能についてテストした。しかしながら、MDCK-T細胞は、本モデルで腫瘍形成性であった。これは、公知のTaub培地がヒトワクチン物質を生産する上でMDCK細胞を安定して培養できないことを示している。
【0119】
<材料と方法>
材料
MDCK細胞(ATCC, Cat. No:CCL-34);T-25、T-75、T-225フラスコ(Corning社、Cat No.: 430639, 430641, 431082);ダルベッコ改良イーグル培地(DMEM)粉末(Gibco社、Grand Island NY, 製剤番号:01-5052EF);Ham F12栄養素混合物粉末(Gibco社、Grand Island NY, 製剤番号:21700-075);ガンマ線照射済みウシ胎仔血清(JRH社、Lenexa KS, Cat. No.:12107-500M);L-グルタミン(JRH社、Lenexa KS, Cat. No. 59202-100M);D-グルコース(Amresco社、Cat. No.:0188-1KG);Ca2+及びMg2+非含有のダルベッコリン酸バッファ(DPBS)(Gibco社、Grand Isand NY, Cat. No.:21600-069);インスリン粉末(Serological社、Cat. No.4506);トランスフェリン(APO型)(Gibco社、Grand Island NY, Cat. No.:11108-016);プロスタグランジンE1(Sigma社、St. Louis MS, Cat. No.:P7527);ヒドロコルチゾン(Mallinckrodt社、Cat. No.:8830(-05));トリヨードチロニン(Sigma社、St. Louis MS, Cat. No.:T5516);亜セレン酸ナトリウム(EMD社、Cat. No.:6607-31);0.05%トリプシン-EDTA(Gibco社、Grand Island NY, Cat. No.:25300);ライマメ・トリプシンインヒビター(Worthington社、Cat. No.:LS002829);ジメチルスルホキシド(DMSO)(Sigma社、St. Louis MS, Cat. No.:D2650);0.4%(w/v)トリパンブルー/PBS溶液(Sigma社、St. Louis MS, Cat. No.:T8154);改良型ノイバウェル血球測定器(Hausser Scientific社、Bright-Line 0.1mm deep/Reichert, Bright-Line 0.1mm deep);YSIバイオアナライザ(YSI社、Model No.: 2700 select);VITROS化学システム(Ortho clinic社、Model: DT60 II)。
【0120】
Taub's無血清培地(SFM)の処方
Taub's培地(TaubとLivingston, 1981, Ann NY Acad Sci.,372:406)は、4.5 g/Lグルコース及び4mMグルタミンを含むDMEM/Ham F12(1:1)からなる無血清培地処方物を基礎培地処方として、表3に示したようなホルモン/因子を添加したものである。
【表3】
【0121】
Taub's無血清培地は、継代時に新たに作られ、又は無血清(SF)DMEM/Ham F12培地+4mMグルタミン+4.5g/Lグルコース+10−8M亜セレン酸ナトリウムにホルモン補足剤のストック溶液を添加することによって再供給される。100mLのTaub's培地は、100μlのインスリンストック溶液(5mg/mL)、100μlのトランスフェリンストック溶液(5mg/mL)、100μlのトリヨードチロニン(T3)ストック溶液(5×10−9M)、5μlのヒドロコルチゾンストック溶液(10−3M)、及び50μlのプロスタグランジンE1ストック溶液(50μg/mL)を、基礎となるDMEM/Ham F12培地+4mMグルタミン+4.5g/Lグルコース+10−8M亜セレン酸ナトリウムに添加して作られる。全ストック溶液は、次のように調製した。
【0122】
インスリンストック溶液
5mg/mlのストック溶液は、適当量のインスリンを0.01NのHClに溶かして作製された。その溶液を0.2ミクロンの滅菌フィルターに通し、Nalgene社の冷凍バイアルに分注し、4℃で保存した。
【0123】
トランスフェリンストック溶液
5mg/mlのストック溶液は、適当量のトランスフェリンをMilliQ水に溶かして作製された。その溶液を滅菌フィルターに通し、その後Nalgene社の冷凍バイアルに分注し、<−20℃で保存した。
【0124】
トリヨードチロニン(T3)ストック溶液
ストック溶液は、適当量のT3を0.02NのNaOHに溶かし、10−4M溶液を得ることによって作製された。このストック溶液を0.02NのNaOHでさらに5×10−9Mストック溶液の濃度にまで希釈し、滅菌フィルターに通し、その後Nalgene社の冷凍バイアルに分注した後、<−20℃で保存した。
【0125】
ヒドロコルチゾンストック溶液
10−3Mストック溶液は、適当量のヒドロコルチゾンを100%のEtOHに溶かして作製された。これをNalgene社の冷凍バイアルに分注した。そのバイアルを4℃で3〜4ヶ月間保存した。
【0126】
プロスタグランジンE1ストック溶液
50μg/mLのストック溶液は、適当量のプロスタグランジンE1を100%のEtOHに溶かして作製された。これをNalgene社の冷凍バイアルに分注した後、<−20℃で保存した。
【0127】
亜セレン酸ナトリウム(Na2SeO3)ストック溶液
10−2Mのストック溶液は、適当量のセレン化ナトリウムをWFI水、又はMilliQ水に溶かして作製された。これをさらに水で10−5の最終濃度にまで希釈し、滅菌フィルターに通し、その後、4℃で保存した。
【0128】
MDCK-S細胞の無血清Taub's培地への適応
ATCC由来のMDCK細胞の冷凍バイアル(継代54)を、無血清Taub's培地に継代する前に4.5g/Lグルコース、2.2g/L NaHCO3、及び4mM L-グルタミンを含んだ10%FBS DMEM培地で5回継代して増やした(上記のようにした)。T-75フラスコで増やした血清MDCKをトリプシン処理で回収した。使用した増殖培地を除き、細胞単分子層をDPBS(カルシウム及びマグネシウムなし)で2回洗浄した。その後、DPBSを廃棄した。適当量の予熱したトリプシン-EDTAを加え(3mL/T-75)、Tフラスコを37℃、5%CO2インキュベータで約15分間インキュベートした。フラスコを手のひらで数回叩き、細胞を完全に剥離させた。等量のライマメ・トリプシンインヒビターを加え、トリプシンを中和した後、サンプルを二つ取り、細胞懸濁液の細胞濃度を測定した。それから1.75×106細胞を新しいT75フラスコのTaub's培地35mLに希釈した。そのフラスコを5%CO2、37±1℃に維持された細胞培養インキュベータ内に入れた。細胞を接種後3日継代培養するか、全培地交換を接種後3日目に行い、続く4日目に継代した。
【0129】
Taub's培地に適応したMDCK細胞の継代
使用した培地を除き、細胞単分子層をDPBS(カルシウム及びマグネシウムなし)で2回洗浄した。適当量の予熱したトリプシン-EDTAを加え(3mL/T-75、7.5mL/T-225)、そのTフラスコを37℃、5%CO2インキュベータで約15分間インキュベートした。フラスコを手のひらで数回叩き、細胞を完全に剥離させた。その後トリプシンを、等量のライマメ・トリプシンインヒビターを加えること(3mL/T-75、7.5mL/T-225)によって阻害した。細胞懸濁液を適切なサイズのピペットで上下に吸引、排出をすることによって均質にした。0.5mLの二つの細胞懸濁液サンプルを、細胞数をカウントするために取った。二つのカウント結果がお互いに15%以内にない場合は、細胞のカウントを繰り返した。カウント後、細胞を新しいフラスコの予熱した新しいTaub's培地で0.05×106生細胞/mLに希釈し、総量35mL/T75、又は100mL/T-225とした。その後、フラスコを37±1℃、5%CO2の環境下でインキュベートした。細胞を3日目に新しいTフラスコに継代する(下記のように)か、又は全培地交換を行い、接種後4日目に新しいTフラスコに継代した。
【0130】
Taub's培地に適応したMDCK細胞PreMCBバンクの調製
Taub's無血清に適応したMDCK細胞株(MDCK-T)の前マスター細胞バンクを、上記実施例2のように調製した。ただし、2×冷凍培地はTaub's培地+15%DMSOとした。
【0131】
Taub's培地に適応したMDCK(MDCK−T)細胞の特徴解析
MDCK-T pre-MCBの核型テスト、無菌テスト及びマイコプラズマテストを、実施例2に記載したように行った。ただし、血清含有完全培地の代わりにTaub's培地を使用した。さらに、T-75フラスコでのMDCK-T細胞の成長曲線の特徴、及びMDCK-T細胞での低温適応性インフルエンザ株の複製を、実施例2に記載したように行った。ただし、血清含有完全培地の代わりにTaub's培地を使用した。腫瘍形成能実験は、上記実施例2のようにBioRelianceによって継代88/29におけるMDCK-T(pre-MCB+20継代)で行われた。
【0132】
<結果と考察>
MDCK-T preMCB(継代64/5)細胞の冷凍バイアルを、T-75フラスコ内の無血清Taub's培地中で解凍した。細胞生存率は97%であった。また5.25×106細胞を解凍時に冷凍バイアルから回収した。細胞は解凍後3日でコンフルエントに達した。細胞形態は、親細胞であるMDCK-S細胞に似た上皮様細胞形態を示した(図9)。
【0133】
Taub's無血清(SF)培地でのMDCK-T細胞の成長曲線を図10に示す。この結果は、異なる継代数(P71/12及びP73/14)の細胞を用いた2つの実験の平均である。MDCK-T細胞は、接種後1日で細胞が倍化し、誘導期が見られなかった(1日目の総細胞数3.42×106細胞/T75フラスコ 対 0日目の総細胞数1.75×106細胞/T75フラスコ)。静止期に入る4日目まで、細胞は対数増殖期にあった。対数期における細胞の倍化時間は、20.4時間であった。対数期(0日目〜4日目)の間、細胞はグルコースとグルタミンを利用する(図11、及び12)と同時に、乳酸及びアンモニアを産生した。グルコース消費/乳酸産生比率は、細胞の成長曲線とよく対応していた(図10と11を比較されたい)。グルコース消費速度は0日目〜4日目の対数期の間で1.78mM/日であり、乳酸は2.88mM/日の速度で産生された。MDCK-T細胞は、7日間までの培養で培地中のグルコースを合計約10mM消費しただけだった。接種後4日目のグルコース消費量対乳酸産生量の比率は、1.2であった。グルコース消費率と乳酸産生率は、細胞が静止期に入った4日目以降に減速した。すなわち、グルコース消費は0.65mM/日であり、乳酸は0.46mM/日の速度で産生された。37±0.24×106の最大細胞密度には、接種後およそ4日目で達した。細胞密度は静止期の間も下がらず、7日目まで一定であった。
【0134】
グルタミン消費率とアンモニア産生率は、MDCK-T細胞の増殖、及びグルコース/乳酸のプロファイルに似ていた(図10、11、及び12を比較されたい)。MDCK-T細胞は、対数増殖期(0日目〜4日目)の間は0.36mM/日の速度でグルタミンを消費したが、静止期(4日目〜7日目)に入ると0.27mM/日に減速した。アンモニア産生は、7日目まで0.22mM/日の速度で直線的に増加した。アンモニア産生対グルタミン消費の比率は、接種後4日目で0.49であった。グルタミン酸消費は、7日間の全期間中、目に見える変化はなかった。
【0135】
MDCK-T細胞を、実施例2のようにca/tsインフルエンザ株の複製を支持する能力についてテストした。表2に示す結果は、MDCK-T細胞がca/tsインフルエンザ株の複製をMDCK-S細胞で見られた結果とほとんど同じレベルで支持できることを示している。
【0136】
MDCK-T細胞の核型を継代68/9及び継代88/29でテストした。Gバンド染色体解析は細胞がイヌ科起源であることを示した。100個の中期細胞における染色体数の分布を図13に示した。染色体の総数は、低継代68/9の細胞については中期あたり52本〜82本、また高継代81/24については54本〜84本の範囲にあった。これは、染色体の分布が継代によって変化しないことを示している。しかし、MDCK-S細胞(70本〜84本)と比べると、MDCK-T細胞(52本〜84本)では染色体数が広範囲に広がる結果が見られた
MDCK-T preMCBを細菌汚染、真菌汚染、又はマイコプラズマ汚染のいずれかの存在についてテストした。MDCK-T pre-MCBは無菌テスト(細菌及び真菌の汚染を確認するための直接接種法を用いた4培地無菌テスト)に合格し、マイコプラズマの存在(アガー培養可能、及び非アガー培養可能アッセイ)についてネガティブであることがわかった。被験物質は、静菌性/静真菌性テスト、及び静マイコプラズマ性テストのどちらもポジティブコントロールの増殖を阻害しないこともわかった。
【0137】
継代88/29のMDCK-T細胞(pre-MCB+20継代)をヌードマウス上に接種し、3ヶ月間腫瘍形成テストを行った。被験物質は、被験マウス10匹のうち6匹において注射部位で腺癌と診断された。これは無血清Taub's培地で増殖したMDCK細胞が、腫瘍形成性であることを示している。MDCK-S及びMDCK-T細胞についてのTP50で評価した腫瘍形成能、及び核型は、以下の表4にまとめている。
【0138】
実施例4
MediV無血清培地における無血清MDCK細胞の誘導
実施例3で詳述した結果は、無血清Taub's培地での増殖に適応したMDCK細胞(MDCK-T)が卓越した増殖特性を有し、またca/tsインフルエンザ株の複製を支持できるものの、腫瘍形成性であることを証明している。それゆえ、これらの結果は、MDCK細胞が文献で報告された標準的な無血清培地処方で容易に形質転換し得ることを示している。本発明にしたがって、いくつかのさらなる無血清培地処方物を開発し、MDCK-S細胞の非腫瘍形成性の性質を維持する能力をテストした。MDCK-S細胞を、新しい無血清製剤MediV SFM101、102及び103のそれぞれに適応させた。これらの無血清適応細胞株を、それぞれMDCK-SF101、-SF102及び-SF103とし、まとめて「MDCK-SF」と呼ぶ。PreMCBを、各MDCK-SF適応細胞株について作製した。MDCK-SF細胞株PreMCBを、細菌/真菌汚染、及びマイコプラズマ汚染についてテストした(最終結果待ち)。またMDCK-SF preMCBを核型解析でもテストした。その結果、MDCK-SF101及びMDCK-SF102細胞は、78本の最頻値染色体数を有し、染色体数はそれぞれ70本〜82本、及び60本〜80本に及んでいた。さらに、それぞれの無血清培地バンク由来の細胞をPreMCBのバイアルから起こして少なくとも新たに20回継代した。そして、MDCK-SF103を核型及びインビボアダルトヌードマウスモデルでの腫瘍形成能についてテストした。継代87のMDCK-SF103は、78本の最頻値染色体数を有し、染色体数は66本〜80本に及んでいたことから、非腫瘍形成性であるとみなした。
【0139】
<材料と方法>
材料
MDCK細胞(ATCC, Cat. No:CCL-34);T-25、T-75、T-225フラスコ(Corning社、Cat No.: 430639, 430641, 431082);ダルベッコ改良イーグル培地(DMEM)粉末(Gibco社、Grand Island NY, Formulation No.:01-5052EF);Ham F12栄養素混合物粉末(Gibco社、Grand Island NY, Formulation No.:21700-075);ガンマ線照射済みウシ胎仔血清(JRH社、Lenexa KS, Cat. No.:12107-500M);L-グルタミン(JRH社、Lenexa KS, Cat. No. 59202-100M);D-グルコース(Amresco社、Cat. No.:0188-1KG);Ca2+及びMg2+非含有のダルベッコリン酸バッファ(DPBS)(Gibco社、Grand Isand NY, Cat. No.:21600-069);インスリン粉末(Serological社、Cat. No.4506);トランスフェリン(APO型)(Gibco社、Grand Island NY, Cat. No.:11108-016);プロスタグランジンE1(Sigma社、St. Louis MS, Cat. No.:P7527);ヒドロコルチゾン(Mallinckrodt社、Cat. No.:8830(-05));トリヨードチロニン(Sigma社、St. Louis MS, Cat. No.:T5516);亜セレン酸ナトリウム(EMD社、Cat. No.:6607-31);0.05%トリプシン-EDTA(Gibco社、Grand Island NY, Cat. No.:25300);ライマメ・トリプシンインヒビター(Worthington社、Cat. No.:LS002829);ジメチルスルホキシド(DMSO)(Sigma社、St. Louis MS, Cat. No.:D2650);0.4%(w/v)トリパンブルー/PBS溶液(Sigma社、St. Louis MS, Cat. No.:T8154);改良型ノイバウェル血球測定器(Hausser Scientific社、Bright-Line 0.1mm deep/Reichert, Bright-Line 0.1mm deep);YSIバイオアナライザ(YSI社、Model No.: 2700 select);VITROS化学システム(Ortho clinic社、Model: DT60 II)。
【0140】
MediV無血清培地の処方(MediV SFM101、102及び103)
各MediV無血清培地処方物は、基礎培地としてTaub's培地(上記実施例2の方法の項参照)を用い、以下の補足剤を添加している。
【0141】
MediV SFM 101: Taub's+2.5g/L コムギペプトンE1(Organo Techine社、cat no.19559)。コムギペプトンE1については、水に溶かして250g/Lの滅菌済ストック溶液として保存する。
【0142】
MediV SFM 102: Taub's+最終濃度1×で添加される100×化学的に規定された脂質濃縮物(GIBCO BRL社、cat no.11905)。
【0143】
MediV SFM 103: Taub's+最終濃度1×の脂質濃縮物(GIBCO社)+2.5g/L コムギペプトンE1(Organo Techine社)。
【0144】
MediV SFM 104: Taub's+最終濃度1×の脂質濃縮物(GIBCO社)+2.5g/L コムギペプトンE1(Organo Techine社)+0.01μg/mL EGF(複数の販売元から)。
【0145】
MediV SFM 105: トランスフェリン非含有Taub's培地+最終濃度1×の脂質濃縮物(GIBCO社)+2.5g/L コムギペプトンE1(Organo Techine社)+0.01μg/mL EGF+10:1〜70:1の比率でクエン酸第二鉄アンモニウム:トロポロン、又は硫酸鉄アンモニウム:トロポロン。
【0146】
無血清MediV SFM培地処方物へのMDCK-S細胞の適応
ATCC由来のMDCK細胞の冷凍バイアルを、MediV SFM培地処方物(MediV SFM101、MediV SFM102、又はMediV SFM103)に継代する前に4.5g/Lグルコース、2.2g/L NaHCO3、及び4mM L-グルタミンを含んだ10%FBS DMEM培地で5回継代して増やした(上記のようにした)。T-75フラスコで増やした血清MDCKをトリプシン処理で回収した。使用した増殖培地を除き、細胞単分子層をDPBS(カルシウム及びマグネシウムなし)で2回洗浄した。その後、DPBSを廃棄した。適当量の予熱したトリプシン-EDTAを加え(3mL/T-75)、Tフラスコを37℃、5%CO2インキュベータで約15分間インキュベートした。フラスコを手のひらで数回叩き、細胞を完全に剥離させた。等量のライマメ・トリプシンインヒビターを加え、トリプシンを中和した後、サンプルを二つ取り、細胞懸濁液の細胞濃度を測定した。その後、1.75×106細胞を新しいT75フラスコ中の所望のMediV SFM培地35mLに希釈した。そのフラスコを5%CO2、37±1℃に維持された細胞培養インキュベータ内に入れた。細胞を接種後3日継代培養するか、全培地交換を接種後3日目に行い、続く4日目に継代した。細胞は、上記と同じ方法を用いたMediV SF104及びMediV SF105にも適応すると思われる。
【0147】
MediV SFM培地に適応したMDCK細胞の継代
使用した培地を除き、細胞単分子層をDPBS(カルシウム及びマグネシウムなし)で2回洗浄した。適当量の予熱したトリプシン-EDTAを加え(3mL/T-75、7.5mL/T-225)、Tフラスコを37℃、5%CO2インキュベータで約15分間インキュベートした。フラスコを手のひらで数回叩き、細胞を完全に剥離させた。その後、トリプシンを、等量のライマメ・トリプシンインヒビターを加えること(3mL/T-75、7.5mL/T-225)によって阻害した。細胞懸濁液を適切なサイズのピペットで上下に吸引、排出をすることによって均質にした。0.5mLの二つの細胞懸濁液サンプルを、細胞数カウント用に取った。二つのカウント結果がお互いに15%以内にない場合は、細胞のカウントを繰り返した。カウント後、細胞を新しいフラスコの予熱した適当な新しいMediV SFM培地処方物で0.05×106生細胞/mLに希釈し、総量35mL/T75、又は100mL/T-225とした。その後、フラスコを37±1℃、5%CO2の環境下でインキュベートした。細胞を3日目に新しいTフラスコに継代する(下記のように)か、又は全培地交換を行い、接種後4日目に新しいTフラスコに継代した。注記:MDCK-SF細胞については、いつもその細胞が適応したときと同じMediV SFM培地処方物で継代すること。
【0148】
MediV SFM培地に適応したMDCK細胞PreMCBバンクの調製
無血清適応MDCK細胞株の前マスター細胞バンクを、上記実施例1のように調製した。ただし、2×冷凍培地は適当なMediV SFM培地処方物+15%DMSOとした。
【0149】
MediV SFM培地に適応したMDCK(MDCK-SF)細胞の特徴解析
MDCK-SF pre-MCBの核型テスト、無菌テスト及びマイコプラズマテストを本明細書に記載の手順、例えば実施例2に記載の手順にしたがってテストした。ただし、血清含有完全培地の代わりに適当なMediV SFM培地処方物を使用した。さらに、T-75フラスコでのMDCK-SF細胞の成長曲線の特徴、及びMDCK-SF細胞での低温適応性インフルエンザ株の複製を、実施例2に記載したように実験した。ただし、血清含有完全培地の代わりに適当なMediV SFM培地処方物を使用した。さらに、腫瘍形成能実験が、さらなる継代(例えば、pre-MCB+20継代)の後に上記実施例2のような通常民間の受託業者(例えば、BioReliance)によってMDCK-SF細胞で行われ得る。
【0150】
<結果と考察>
MDCK-SF101細胞及びMDCK-SF102細胞の核型を継代71/9で、またMDCK-SF103細胞の核型を継代87でテストした。MDCK-T、MDCK-SF101、及びMDCK-SF102の100個の中期細胞における染色体数の分布を図14に、またMDCK-SF103のそれを図19に示す。MDCK-T細胞は、MDCK-SF101、MDCK-SF102又はMDCK-SF103の各細胞(それぞれ70本〜82本、60本〜80本、及び66本〜80本)と比べると、染色体数が広範囲に広がる結果(52本〜84本)を示すことがわかった。MDCK-SF101、MDCK-SF102及びMDCK-SF103の各細胞の染色体数の広がりは、非腫瘍形成性MDCK-S血清増殖細胞(70本〜84本)で見られたものに非常に近い。これは、MediV SF101、MediV SF102、及びMediV SF103培地処方物が、当該製剤で増殖したMDCK細胞の正常な染色体数をより適切に維持できることを示唆している。
【0151】
MediV SF103培地におけるMDCK-SF103細胞の典型的な予備成長曲線を図16に示す。MDCK-SF103細胞では約1日の誘導期が見られた。静止期に入る4日目まで、細胞は対数増殖期にあった。対数期(0日目〜4日目)の間、細胞はグルコースとグルタミンを利用(図17、及び18)すると同時に、乳酸及びアンモニアを産生した。グルコース消費/乳酸産生比率は、細胞の成長曲線とよく対応していた(図16と17を参照)。〜17×106の最大細胞密度には、接種後およそ4日目で達した。細胞密度は静止期の間も下がらず、7日目までほぼ一定であった。
【0152】
グルタミン消費率とアンモニア産生率は、MDCK- SF103細胞の増殖、及びグルコース/乳酸のプロファイルに似ていた(図18参照)。アンモニア産生は7日目まで線形的に増加したが、グルタミン酸濃度は、7日の期間中、目に見える変化はなかった。
【0153】
MDCK-SF103細胞を、以下の実施例7に記載するようないくつかのリアソータントインフルエンザ株の複製を支持する能力についてテストした。表20Aに示すその結果は、MDCK-SF103細胞がテストした各インフルエンザ株の複製を支持できることを示している。
【0154】
MDCK-SF103細胞をヌードマウス上に接種し、上記のように3ヶ月間腫瘍形成テストを行った。被験物質は、アダルトヌードマウスモデルで非腫瘍形成性であると見なされた(RioReliance 実験番号 AB09EU.001000.BSV)。
【表4】
【0155】
実施例5
培養におけるヒト上皮細胞の感染
ヒト起源の細胞内におけるMDCK及び卵で作られたワクチンの複製後の生化学的、生物学的及び構造的類似性を評価するため、ワクチンを正常なヒト気管支上皮細胞(human bronchial epithelial cells:NHBE)等の関連二倍体ヒト細胞で1回継代する。この継代は、ヒトの気道内における感染の一形態を模倣するのに役立ち、また子孫ウイルスとの比較を可能にする。この子孫ウイルスは、詰まるところ免疫反応の有効な誘導に関与する。ワクチンのヘマグルチニン活性(結合及び融合)並びにノイラミニダーゼ活性を、電子顕微鏡法、感染総粒子率、ウイルスゲノムの等価性を含む他の生化学的及び構造的実験と共にこれらの材料で測定した。概して、これらの比較は、効率的かつ安全な卵で作られたワクチンに対する細胞由来ワクチンの比較可能性を証明するのに役立つ。細菌及び真菌汚染の有無をテストする方法は当該分野では周知であり、通常民間の受託業者(例えば、BioReliance(登録商標);Rockville, MD)で行われる。実施され得る分析的研究の概要は、表5にまとめた。
【表5】
【0156】
実施例6
マスター細胞バンクの製造、テスト、及び特徴解析
マスター細胞バンク(MCB)の作製を始めるために、一以上の上記preMCB(実施例2〜4参照)由来の細胞を、生成する細胞が遺伝学的に一つの群に由来することを確実にするために限定希釈法により生物学的にクローン化した。その後、クローンを、倍化時間や関連する腫瘍形成性、そしてウイルス生産を含む様々な表現型特性についてスクリーニングした。最初の概念実証実験で、54個のMDCKクローンがFCSを含む培地で得られた。これらのクローンを継代し、それぞれを低感染多重度のca A/New Caledonia/20/99で感染させた。感染後数日して上清を取り出し、上清中のウイルス量をTCID50で測定した。少数のクローンは、クローンでない親細胞における生産を超える比較的高いタイターのウイルスを生産していた。生物学的及び生理学的に優れた特性を持つクローンをマスター細胞バンク(MCB)の樹立に使用した。
【0157】
MCBを広範囲にわたってテストし、外来の作用因子の痕跡がないことを確実にした。例えば、以下の表6で示すようなウイルス性因子に利用できる一以上のPCR及び/又は市販の抗体特異的テストを実施した。
【表6】
【0158】
実施例7
ワクチン物質の製法と製剤
近年認可されたポリオワクチンの生産に使用されるものに類似するスケールアップ性の高いマイクロキャリア技術の使用をMDCK細胞でのインフルエンザの生産に使用可能にした。デキストランでできた球状ビーズは、2〜10Lバイオリアクター内でMDCK細胞の増殖を支持するのに優れている。SFMV103培地中で増殖した親MDCK細胞は、Cytodex 1マイクロキャリアで、両スピナーフラスコのバッチモードで2×106核/mlの密度まで増殖することができることがわかった。また、MDCK細胞を最大10Lスケールのバイオリアクター内で1×106細胞/ml以上まで増やした(データ示さず)。最初のパイロットスケールでの稼動で、MDCK細胞が無血清法でワクチンインフルエンザ株を高タイターで生産できることが証明された。またそのタイターは、Tフラスコ内で血清増殖細胞を使用して得た生産性と同等か又はそれ以上であることがわかった。図20Aで示したように、250mLのスピナーフラスコ内のCytodexビーズ中で増殖したMDCK細胞は、高タイターのH1N1、H3N2及びB型ワクチン株を生産した。医療用製造については、インフルエンザウイルスをMDCK細胞内で20L〜150L規模で生産することができる。一方、商業規模生産の場合、2,500Lのバイオリアクターを利用することもできる。図20Bは、商業生産レベルまでスケールアップした細胞培養を利用できる一のプロセスを概説している。まず、使用する細胞バンクをT-75フラスコからT-225フラスコ、1Lスピナーフラスコ、20Lバイオリアクター、そして300Lバイオリアクターへと連続的に増やし、最終的には2500Lバイオリアクターにまで拡大する。最適細胞密度が得られたときに、培養液にマスターウイルス株を接種する。その後、ウイルスを培養液上清から一括回収する。
【0159】
細胞培養ベースのインフルエンザワクチンの精製工程は、卵ベースのインフルエンザワクチンの精製をモデルにしている(例えば、国際公開番号第05/014862号、及び2005年10月4日出願の国際出願番号第US05/035614号を参照されたい)。細胞からのウイルスワクチン物質の精製は、以下の工程のいずれか、又は全てを含み得る。すなわち、ホモジネーション、清浄化遠心分離、限外濾過、硫酸バリウムへの吸着及び溶出、タンジェンシャルフロー濾過(tangential flow filtration)、密度勾配超遠心、クロマトグラフィー、及び濾過滅菌が挙げられる。その他の精製ステップを含んでいてもよい。例を挙げると、まず、感染培養物由来のクルードな培地を例えば1000〜2000×gで細胞片及びその他の大型粒状物質を除くのに十分な時間(例えば、10〜30分)遠心して清澄化することができる。あるいは、培地を0.8μmの酢酸セルロースフィルターに通して濾過して、無傷の細胞及びその他の大型粒状物質を除く。その後、任意で清浄化した培地の上清を、例えば15,000×gで約3〜5時間遠心し、インフルエンザウイルスを沈殿させる。STE (0.01M Tris-HCl;0.15M NaCl;0.0001M EDTA)又はpH7.4のリン酸バッファ(PBS)等の適当なバッファにウイルス沈殿物を再懸濁した後、ウイルスをショ糖(60%〜12%)、又は酒石酸カリウム(50%〜10%)の密度勾配遠心で濃縮してもよい。連続的勾配か、又は例えば12%〜60%間で12%ごと4段階といった段階的勾配のショ糖勾配のいずれかが適している。勾配は、ウイルスを回収のため目に見えるバンドに凝集させる上で十分な速度、及び時間で遠心分離される。あるいは、市販の大規模なアプリケーションのほとんどは、ウイルスを密度勾配から連続モードで稼動して帯状遠心分離ロータを使用して溶出する。
【0160】
細胞からのウイルスワクチン物質の精製に含めることのできる特徴として、初期工程での非特異的エンドヌクレアーゼBenzonase(登録商標)の使用がある。細胞DNAの発癌性を評価する研究によればMDCK細胞DNAは発癌リスクがないが、Benzonase処理によってあらゆる可能性のあるリスク又は仮説的なリスクを実質的に排除することができる。一の精製工程において、Benzonase処理後、当該材料をダイレクトフロー濾過(DFF)によって清浄化した。DFFは、バルク材中のあらゆる残余無傷哺乳動物細胞をも除くことができる。その後、濾過したバルクを次の精製ステップ前にタンジェンシャルフロー濾過(TFF)で濃縮する。他のウイルスシステムにもよく使用されるアフィニティークロマトグラフィーとイオン交換クロマトグラフィー、及び/又はヒドロキシアパタイトを含む精製方法は、細胞培養ベースのインフルエンザワクチン生産に有用である。開発した方法で得られた高精製ウイルス物質は、その後、ワクチン物質の製造に利用される。例えば、弱毒化生ワクチン製造(例えばFluMist(登録商標))での使用に関しては、ウイルス物質を最終処方中に濾過によってバッファ交換し、その後滅菌ステップをすることができる。このような処方に有用なバッファは、アルギニン等のその他のアミノ酸賦形剤を添加した200mMショ糖、及びリン酸又はヒスチジンバッファ(pH7.0〜7.2)を含み得る。安定化が必要であれば、ブタゼラチン等のタンパク質加水分解物も添加することができる。ワクチン物質を長期保管時間、安定するように処方するのが理想的である。長期保管時間に利用することができる一の方法として噴霧乾燥法がある。これは液体製剤試料を乾燥加熱気体の気流下で微細液滴に噴霧化することによって、試料を急速乾燥させる方法である。微細液滴の蒸発によって溶解溶質からなる乾燥粉末が生じる(例えば、米国特許公開公報番号2004/0042972号を参照されたい)。噴霧乾燥法は、スケールアップ化が容易な点や従来の凍結乾燥法と比べると生産原価の点で有利である。さらに、ワクチン物質を製剤化し、当該分野で既知の方法を用いて冷蔵庫で安定な液体製剤として安定化させる。例えば、冷蔵庫で安定な弱毒性インフルエンザワクチンを処方するための方法と組成物は、2005年10月4日出願の国際出願第PCT/US2005/035614号に記載されている。
【0161】
製造過程において、特徴解析ステップは精製ステップの中に組み込まれて、その生産がモニターされる。利用可能な特徴解析ステップは、ウイルス感染性を測定するための単一の抗体結合を用いる蛍光焦点アッセイ法(FFA、例えば上記参照)及び蛍光染色法を含むが、これらに限定はされない。総タンパク質及びDNAの測定は、当業者に既知の多くの方法を用いて実施することができ、またこれらの測定は残存する初期不純物のパーセントを測定するのに使用される。調製物の特定の活性を、ワクチン量あたりのウイルス感染価(例えば、感染価/mg)を算出することで測定することができる。
【0162】
実施例8
前臨床動物モデル
フェレットは丈夫な動物モデルで、弱毒化したインフルエンザワクチン及びワクチン株の成分の弱毒性、並びに免疫原性を評価するのに使用される。MCBから作られた細胞由来のインフルエンザ株の性能を、卵で作られた同一株と比較する。コントロール実験におけるこれらの材料の直接的な比較は、これらのウイルス産物の比較の確かさを高レベルにすることができる。
【0163】
二つのワクチンのフェレットでの感染能力又は「(免疫)獲得」能力を評価するために、動物を軽く麻酔し、細胞又は卵で作られたウイルス調製物のどちらかを鼻腔内に接種した。接種後に鼻洗浄物を数時点で回収し、ウイルス量を利用可能ないくつかの方法のうちの一つで、その動物の上気道における動力学及びウイルスの複製の広がりを判断するために、評価する。実験を投与量の範囲で実施し、複数株及び異なる三つの混合物を包含して細胞培養増殖株対卵生産株の相対的感染力を標準化する。同様の研究がインフルエンザ株の免疫原性を評価するのにも使用される。免疫原性は、ウイルスが感染を起こす能力と本質的に関連する性質である。動物を飼育し、鼻洗浄物を接種後の様々な時点(週)で回収した。これらの試料を感染に対する血清抗体及び鼻のIgA応答を評価するのに使用した。これらのデータ、感染力、血清抗体、及び粘膜抗体応答の最高点は、細胞産生ワクチン対卵産生ワクチンの相対感染力の比較及び評価に利用することができる。最もあり得そうな結果は、細胞及び卵で作られたワクチン株が同様の感染力及び免疫原性を持っているということである。細胞由来のワクチンが卵由来の産物よりも感染力や免疫原性がより高いように見えるのであれば、低位側の投与量の可能性を評価するさらなる研究を行う。
【0164】
多数の免疫原性及び複製の研究がフェレットモデルで行われ、細胞培養由来ワクチンを単一ユニットのヒト投与量で評価する。ca/ts/att株による感染は、フェレットで通常急速かつ強い抗体反応を引き起こす。さらに、個々のca/ts/att株を定期的にテストする。当該株を鼻咽頭内で比較的高いタイターまで複製し、しかしこれらの動物の肺では検出されないレベルまで複製することで、弱毒性(att)表現型が発現することがわかる。これらの生物学的形質で増殖した細胞培養の影響も評価される。att表現型はこれらの株の遺伝学的構成と一体化した部分であるので、あらゆる相違が見られるというのは考えにくい。これらの株の増殖動力学と交差反応性をこれらの動物にヒト1回分の投与量で投与後に評価する。卵由来の材料から生産された弱毒性生ワクチンは、同一遺伝系統内の複数の株と交差反応する血清抗体を誘導する。そして、細胞由来のワクチンも同じ能力を持つであろうことが予想される。
【0165】
これらの比較可能性評価は、主要なウイルス産物の潜在的な生化学的及び/又は生物物理学的違いに重要な洞察を提供し、またヒトの細胞におけるウイルスの最初の継代又は動物実験によって測定されるca/ts/att株の性能の後成的な相違がもたらす影響を証明するはずである。現在までのところ配列情報に基づけば、MDCK細胞で生産されたことによって生じるca/ts/att株の免疫原性能力において予期された衝撃的な影響はない。
【0166】
フェレットは、インフルエンザ用に文献上十分証明されたモデル動物であり、弱毒表現型とca/ts/att株の免疫原性を評価するのに日常的に使用されている。通常、8〜10週齢の動物を使用して弱毒性を評価する。一般的な実験計画では1回のテスト又はコントロール群につきn=3〜5匹の動物が評価される。免疫原性実験では、8週〜6月齢の動物で評価し、通常1回の被験物質又はコントロール群につきn=3〜5匹の動物を必要とする。これらの匹数は、グループ間で統計的に有効な又は観察上重要な比較結果を得る上で十分な情報を提供する。ほとんどの実験中にインフルエンザ様の徴候に気づくかもしれないが、簡単ではないであろう。フェレットは食欲又は体重の減少、鼻汁又は眼漏等の病気の徴候を示さないからである。インフルエンザ様の疾患の徴候を観察することは、研究に必要な一部であり、鎮痛剤等の介入を行うべきでない。宿弊又は深刻な体重減少等の他の不快症状は、担当する獣医と話し合った後、その動物にとって適切な処置がなされるであろう。
【0167】
本発明は特定の実施形態に関して開示したが、本発明の他の実施形態や変形型が本発明の本質及び範囲を逸脱することなしに当業者により考え出され得ることは明らかである。添付の特許請求の範囲は、そのような全ての実施形態及び均等の変更を含むと解されることが意図されている。例えば、上記全ての技術及び装置を様々な組合せで使用することができる。本出願で引用された全ての刊行物、特許、特許出願、又はその他の資料は、各個の刊行物、特許、特許出願、又はその他の資料が、参照によって全ての目的に援用するために個別に示される場合と同程度に、参照によって全ての目的に援用される。さらに、2004年の12月23日出願の米国仮出願第60/638,166号、及び2005年1月5日出願の第60/641,139号を、この記載によって、本明細書に引用して全ての目的にその全体を援用する。
【特許請求の範囲】
【請求項1】
細胞株MDCK(ATCC CCL34)の誘導体である非腫瘍形成性MDCK細胞を含む細胞培養組成物。
【請求項2】
組成物は血清を含まない、請求項1に記載の細胞培養組成物。
【請求項3】
前記非腫瘍形成性MDCK細胞は付着性である、請求項1に記載の細胞培養組成物。
【請求項4】
前記非腫瘍形成性MDCK細胞は細胞株MDCK-S(ATCC PTA-6500)由来である、請求項1〜3のいずれか1項に記載の細胞培養組成物。
【請求項5】
前記非腫瘍形成性MDCK細胞は無血清培地で増殖するように適応した、請求項1に記載の細胞培養組成物。
【請求項6】
前記非腫瘍形成性MDCK細胞はMDCK-SF101 (ATCC PTA-6501)、MDCK-SF102 (ATCC PTA-6502)、及びMDCK-SF103 (PTA-6503)よりなる群から選択される細胞株由来である、請求項5に記載の細胞培養組成物。
【請求項7】
インフルエンザウイルスの生産方法であって、
a.請求項1又は6に記載の細胞培養組成物中のMDCK細胞にインフルエンザウイルスを感染させること、
b.前記細胞培養組成物をインキュベートすること、及び
c.前記細胞培養組成物からインフルエンザウイルスを単離すること
を含む前記方法。
【請求項8】
MDCK細胞が付着性である、請求項7に記載の方法。
【請求項9】
請求項7記載の方法に従って生産されたインフルエンザウイルス。
【請求項10】
製薬上許容される担体又は希釈剤中に請求項9記載のインフルエンザウイルスのポリペプチドを含む免疫原性組成物。
【請求項11】
請求項10記載の免疫原性組成物を動物に投与することを含む、動物におけるインフルエンザ感染を予防する方法。
【請求項12】
請求項8記載の方法に従って生産されたインフルエンザウイルス。
【請求項13】
MDCK-S (ATCC PTA-6500)、MDCK-SF101 (ATCC PTA-6501)、MDCK-SF102 (ATCC PTA-6502)、及びMDCK-SF103 (PTA-6503)よりなる群から選択される非腫瘍形成性MDCK細胞株。
【請求項14】
血清含有培地で培養することができ、かつインフルエンザウイルスによる感染が可能な付着性の非腫瘍形成性MDCK細胞株を調製する方法であって、
(a)MDCK(ATCC CCL34)細胞を規定培地及び血清中に適応させて増殖させるステップ、
(b)増殖条件を維持するステップ、及び
(c)細胞バンクを樹立するステップ
を含む前記方法。
【請求項15】
請求項14記載の方法によって調製された細胞株。
【請求項16】
無血清培地中で培養することが可能であり、かつインフルエンザウイルスによる感染が可能である付着性の非腫瘍形成性MDCK細胞株を調製する方法であって、
(a)MDCK(ATCC CCL34)細胞を適応させて、脂質を含むTaubのSF培地;コムギ加水分解物を含むTaubのSF培地;脂質及びコムギ加水分解物を含むTaubのSF培地;脂質、コムギ加水分解物及びEGFを含むTaubのSF培地;並びに脂質、コムギ加水分解物、EGF、トロポロンを含むがトランスフェリンを欠くTaubのSF培地よりなる群から選択される無血清培地で増殖させるステップ、
(b)増殖条件を維持するステップ、及び
(c)細胞バンクを樹立するステップ
を含む前記方法。
【請求項17】
請求項16記載の方法によって調製された細胞株。
【請求項18】
MediV SF101、MediV SF102、MediV SF103、MediV SF104、及びMediV SF105よりなる群から選択される培地処方物。
【請求項19】
請求項18記載の培地で前記動物細胞を培養することを含む、動物細胞の非腫瘍形成性の性質を維持する方法。
【請求項1】
細胞株MDCK(ATCC CCL34)の誘導体である非腫瘍形成性MDCK細胞を含む細胞培養組成物。
【請求項2】
組成物は血清を含まない、請求項1に記載の細胞培養組成物。
【請求項3】
前記非腫瘍形成性MDCK細胞は付着性である、請求項1に記載の細胞培養組成物。
【請求項4】
前記非腫瘍形成性MDCK細胞は細胞株MDCK-S(ATCC PTA-6500)由来である、請求項1〜3のいずれか1項に記載の細胞培養組成物。
【請求項5】
前記非腫瘍形成性MDCK細胞は無血清培地で増殖するように適応した、請求項1に記載の細胞培養組成物。
【請求項6】
前記非腫瘍形成性MDCK細胞はMDCK-SF101 (ATCC PTA-6501)、MDCK-SF102 (ATCC PTA-6502)、及びMDCK-SF103 (PTA-6503)よりなる群から選択される細胞株由来である、請求項5に記載の細胞培養組成物。
【請求項7】
インフルエンザウイルスの生産方法であって、
a.請求項1又は6に記載の細胞培養組成物中のMDCK細胞にインフルエンザウイルスを感染させること、
b.前記細胞培養組成物をインキュベートすること、及び
c.前記細胞培養組成物からインフルエンザウイルスを単離すること
を含む前記方法。
【請求項8】
MDCK細胞が付着性である、請求項7に記載の方法。
【請求項9】
請求項7記載の方法に従って生産されたインフルエンザウイルス。
【請求項10】
製薬上許容される担体又は希釈剤中に請求項9記載のインフルエンザウイルスのポリペプチドを含む免疫原性組成物。
【請求項11】
請求項10記載の免疫原性組成物を動物に投与することを含む、動物におけるインフルエンザ感染を予防する方法。
【請求項12】
請求項8記載の方法に従って生産されたインフルエンザウイルス。
【請求項13】
MDCK-S (ATCC PTA-6500)、MDCK-SF101 (ATCC PTA-6501)、MDCK-SF102 (ATCC PTA-6502)、及びMDCK-SF103 (PTA-6503)よりなる群から選択される非腫瘍形成性MDCK細胞株。
【請求項14】
血清含有培地で培養することができ、かつインフルエンザウイルスによる感染が可能な付着性の非腫瘍形成性MDCK細胞株を調製する方法であって、
(a)MDCK(ATCC CCL34)細胞を規定培地及び血清中に適応させて増殖させるステップ、
(b)増殖条件を維持するステップ、及び
(c)細胞バンクを樹立するステップ
を含む前記方法。
【請求項15】
請求項14記載の方法によって調製された細胞株。
【請求項16】
無血清培地中で培養することが可能であり、かつインフルエンザウイルスによる感染が可能である付着性の非腫瘍形成性MDCK細胞株を調製する方法であって、
(a)MDCK(ATCC CCL34)細胞を適応させて、脂質を含むTaubのSF培地;コムギ加水分解物を含むTaubのSF培地;脂質及びコムギ加水分解物を含むTaubのSF培地;脂質、コムギ加水分解物及びEGFを含むTaubのSF培地;並びに脂質、コムギ加水分解物、EGF、トロポロンを含むがトランスフェリンを欠くTaubのSF培地よりなる群から選択される無血清培地で増殖させるステップ、
(b)増殖条件を維持するステップ、及び
(c)細胞バンクを樹立するステップ
を含む前記方法。
【請求項17】
請求項16記載の方法によって調製された細胞株。
【請求項18】
MediV SF101、MediV SF102、MediV SF103、MediV SF104、及びMediV SF105よりなる群から選択される培地処方物。
【請求項19】
請求項18記載の培地で前記動物細胞を培養することを含む、動物細胞の非腫瘍形成性の性質を維持する方法。
【図1A】

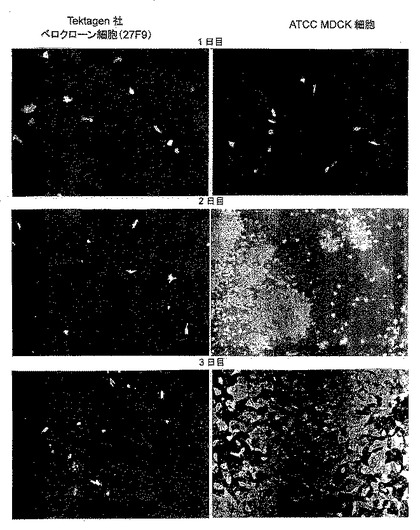
【図1B】
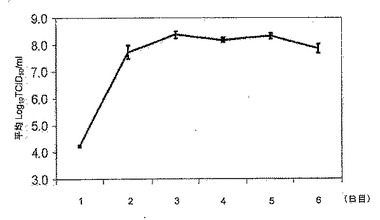
【図2】
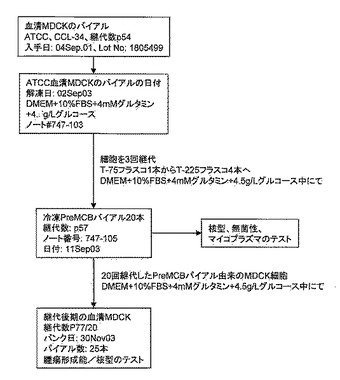
【図3】
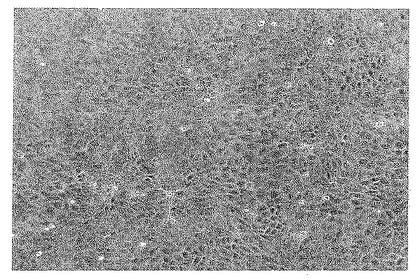
【図4】
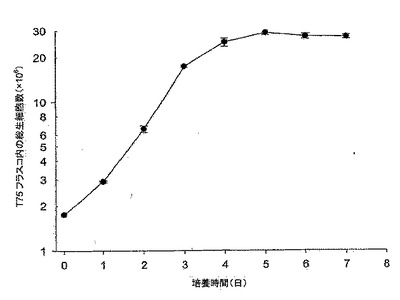
【図5】
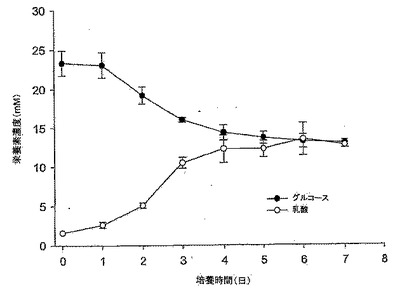
【図6】
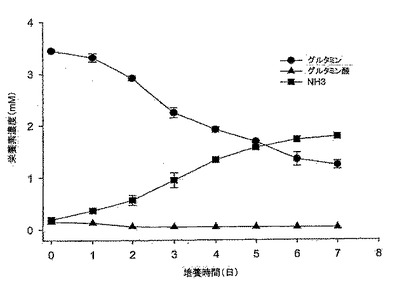
【図7】
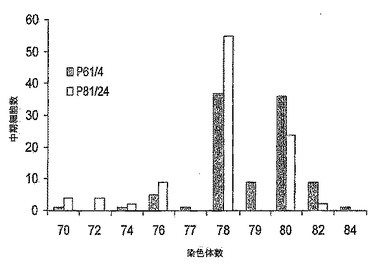
【図8】
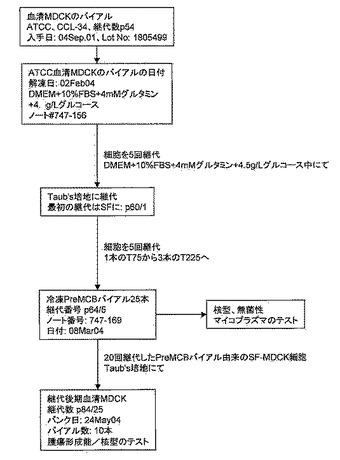
【図9】
【図10】
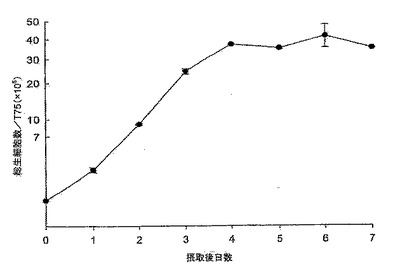
【図11】
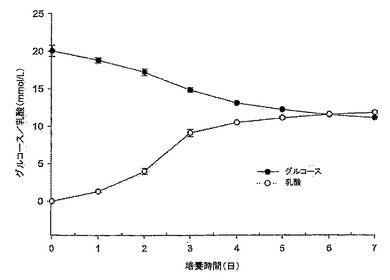
【図12】
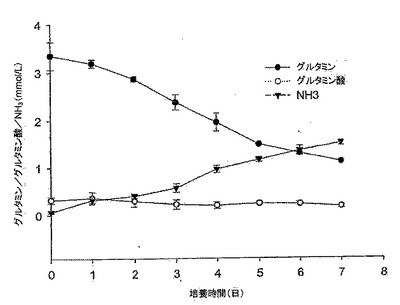
【図13】
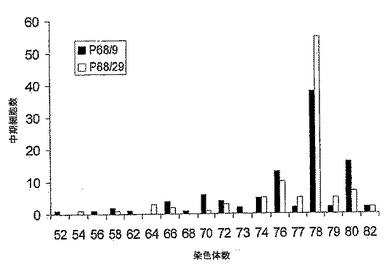
【図14】
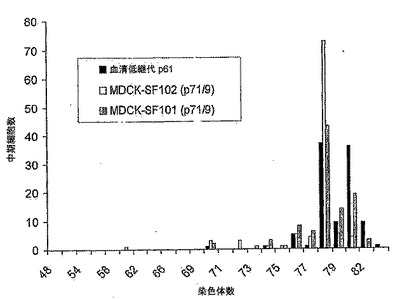
【図15】

【図16】
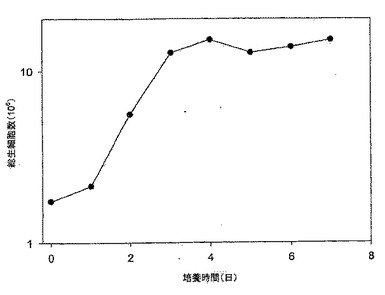
【図17】
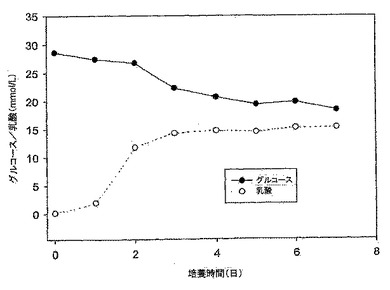
【図18】
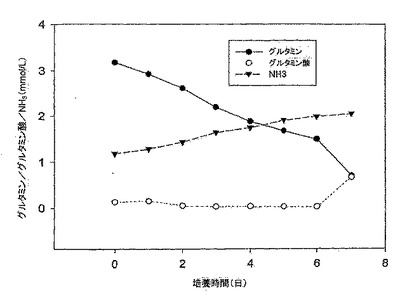
【図19】
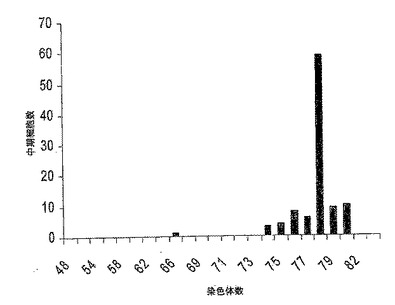
【図20A】
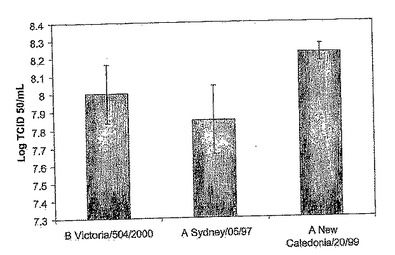
【図20B】
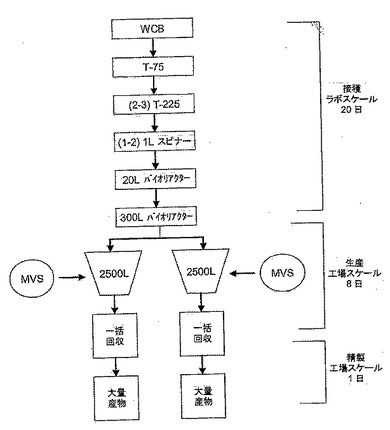
【図1B】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20A】
【図20B】
【公開番号】特開2012−210225(P2012−210225A)
【公開日】平成24年11月1日(2012.11.1)
【国際特許分類】
【出願番号】特願2012−163682(P2012−163682)
【出願日】平成24年7月24日(2012.7.24)
【分割の表示】特願2007−548322(P2007−548322)の分割
【原出願日】平成17年12月16日(2005.12.16)
【出願人】(504333972)メディミューン,エルエルシー (108)
【Fターム(参考)】
【公開日】平成24年11月1日(2012.11.1)
【国際特許分類】
【出願日】平成24年7月24日(2012.7.24)
【分割の表示】特願2007−548322(P2007−548322)の分割
【原出願日】平成17年12月16日(2005.12.16)
【出願人】(504333972)メディミューン,エルエルシー (108)
【Fターム(参考)】
[ Back to top ]