
ウイルス感染症およびその他の内科疾患を治療するための化合物、組成物および方法
【課題】 本出願は、脂質含有抗ウイルス性化合物、および詳細には、抗ウイルス性脂質含有化合物の生物学的利用能を改善するための方法および組成物を提供する。
【解決手段】 ある実施形態では、抗ウイルス性脂質含有化合物、またはそれらの塩、エステルもしくはプロドラッグ、および例えばシトクロムP450酵素の阻害剤などの1つまたは複数の生物学的利用能エンハンサー化合物を含む、医薬上許容される組成物が提供される。
【解決手段】 ある実施形態では、抗ウイルス性脂質含有化合物、またはそれらの塩、エステルもしくはプロドラッグ、および例えばシトクロムP450酵素の阻害剤などの1つまたは複数の生物学的利用能エンハンサー化合物を含む、医薬上許容される組成物が提供される。
【発明の詳細な説明】
【技術分野】
【0001】
<関連出願の相互参照>
本出願は、その開示がこれにより全体として参照して組み込まれる、2005年4月8日に出願された米国出願番号第60/669,765号に対する優先権を主張する。
【0002】
<発明の分野>
本出願は、ウイルス感染症を治療するためのヌクレオシドもしくは非環式ヌクレオシドなどの脂質含有化合物の生物学的利用能、活性もしくは他の特性を増強するための方法を提供する。
【背景技術】
【0003】
薬物の生物学的利用能を改善する工程は、医学分野において確定されている目標である。薬理学においては、薬物がその治療目的のために十分な生物学的利用能を有することが重要である。経口組成物についての事象の順序には、様々な粘膜表面を通しての吸収、血流を介しての様々な組織への分布、肝臓およびその他の組織中での生体内変換、標的部位での作用、および尿もしくは胆汁中への薬物もしくは代謝産物の排出が含まれる。生物学的利用能は、循環器系に到達する前の薬物の消化管からの不良な吸収、肝初回通過作用、または分解によって減少させられることがある。
【0004】
プロドラッグは、身体内(インビボ)で活性化合物へ代謝されるように設計される。脂質プロドラッグは、消化管からの薬物の不良な吸収が限定因子である場合は、通常は経口による生物学的利用能を改善するように設計される。脂質プロドラッグは、それらの標的組織に対する薬物の選択性を改善することもできる。脂肪酸を含む脂質分子は、薬物とコンジュゲート化されることによって、そのコンジュゲートをコンジュゲート化されていない薬物より脂肪親和性にする。一般に、増加した脂肪親和性は、リンパ系内への薬物の腸取り込みを増強し、それにより脳内へのコンジュゲートの進入を増強し、さらに肝臓内でのコンジュゲートの初回通過代謝を回避するための機序として提案される。使用される脂質分子のタイプには、リン脂質および脂肪酸が含まれる。
【0005】
多数の薬物のリン脂質プロドラッグが開発されてきた。これらの化合物の一部は、親化合物に比して増強された活性もしくは生物学的利用能を有することが証明されている。
【0006】
薬物を含有する非ホスホネートに共有結合するアルキルグリセロールホスホネートの調製物および使用は、記載されている(米国特許第5,411,947号および米国特許出願第08/487,081号:それぞれ特許文献1及び2)。抗ウイルス性ヌクレオシド(米国特許第5,223,263号:特許文献3)またはホスホノ−カルボキシレート(米国特許第5,463,092号:特許文献4)に結合したアルキルグリセロールホスホネート残基を含むプロドラッグもまた記載されている。Hostetlerへの米国特許第6,716,825号(特許文献5)は、シドフォビルなどの抗ウイルス性化合物の所定のプロドラッグについて記載している。詳細には、抗ウイルス性化合物の所定の誘導体、および詳細にはシドフォビルのプロドラッグは、ウイルスの治療において親薬物より有効である。詳細には、1−O−ヘキサデシルオキシプロピル−シドフォビルは、詳細には痘瘡などのポックスウイルスの治療において、シドフォビルに比して増強された有効性を有する。
【0007】
薬物の分解は、肝臓もしくは腸内で発生し得る。消化管からのすべての血液は、全哺乳動物において身体のいずれかに進行する前に肝臓を通過する。その場所のために、経口投与された薬物の肝臓内形質転換は、小腸内での酵素活性の作用を上回ると考えられた薬物の生物学的利用能に実質的な「初回通過作用」を有する(Tam,Y.K.,「IndividualVariation inFirst−Pass Metabolism,」Clin.Pharmacokinetics1993,25,300−328:非特許文献1)。
【0008】
肝臓による活性薬物の排出は、2つの一般的経路、つまり薬物の生体内変換および胆汁中への薬物の排泄の一方もしくは両方によって発生する。生体内変換反応は、広範に定義された2つの期に分類される。第1生体内変換期はしばしば、肝臓内で多種多様かつ活性であり、多数の化学的に様々な薬物を変換させるシトクロムP450酵素によって触媒される反応を利用する。第2生体内変換期は、水溶性を増加させて腎臓を通しての排出を促進するために、グルタチオン、グルクロン酸もしくは硫酸塩などの親水基を加えることができる。
【0009】
肝細胞は、例えば門静脈(腸からの栄養分および薬物が豊富な血液)、肝動脈(心臓からの直接の酸素を豊富に含む血液)、肝静脈(駆出)、リンパ管(脂質およびリンパ球)、および胆管などの、多数のタイプの血液およびその他の流体輸送管と接触する。胆管は、上方の腸内に胆汁を排泄して消化を支援する胆嚢および総胆管内に合流する。胆汁は、疎水性薬物および薬物代謝産物を含む様々な排出生成物もまた含有する。
【0010】
一部の場合には、経口投与後の薬物の不良な生物学的利用能は、膜結合P糖タンパク質である多剤トランスポーターの活性の結果であり、これは細胞から生体異物を排出することによって薬物の細胞内蓄積を減少するために、エネルギー依存型輸送もしくは排出ポンプとして機能すると推測されてきた。P糖タンパク質排出ポンプは、所定の医薬化合物が小腸の粘膜細胞を横断すること、そしてこのために全身性循環中に吸収されることを防止すると考えられる。多数の既知である非細胞毒性薬理学的物質は、特にシクロスポリン、ベラパミル、タモキシフェン、キニジンおよびフェノチアジンを含むP糖タンパク質を阻害することが証明されている(Fisheret al.,Proc.Am.Soc.Clin.Oncol.,13:143,1994;Bartlettet al.,J.Clin.Onc.12:835−842,1994;Lumet al.,J.Clin.Onc.10:1635−42,1992:それぞれ非特許文献2〜4)。所定の抗癌薬の投与前、または同時の静脈内シクロスポリンの投与は、おそらく減少した身体クリアランスによってそれらの薬物のより高い血中濃度を生じさせ、実質的に低用量レベルで予想される毒性を示した。これらの観察所見は、シクロスポリンの併用投与はP−糖タンパク質のMDR作用を抑制し、治療薬のより大きな細胞内蓄積を可能にすることを示す傾向があった。P−糖タンパク質の臨床使用についての薬理学的意味に関する一般的考察は、Lumet al.,DrugResist.Clin.Onc.Hemat,9:319−336(1995)及びSchinkelet al.,Eur.J.Cancer,31A:1295−1298(1995)(それぞれ、非特許文献5及び6)に提供されている。
【0011】
薬物の分解、または生体内変換を最小限に抑えるために薬物とともに投与できる化合物は、バイオエンハンサーと呼ばれている。公開されたPCT特許出願WO第95/20980号(1995年8月10日に公開:特許文献6)では、Benetらは、シトクロムP4503A酵素の阻害剤またはP糖タンパク質−媒介性膜輸送の阻害剤を含む、バイオエンハンサーの使用を開示する。
【0012】
インビボでの薬物への代謝性もしくは他の望ましくない作用のために、身体内での脂質プロドラッグもしくは誘導体の有効性を最大化するという課題がある。これは、脂質を分解および合成するための身体の精巧かつ複雑な機序を前提にすると、脂質誘導体に関する特別な問題であった。
【0013】
そこで、治療薬への薬物代謝および相互作用の影響を最小限に抑えながら、改善された脂肪親和性化合物でウイルス感染症を治療するための方法および組成物に対する必要がある。
【0014】
詳細には、投与される治療薬に薬物の代謝が及ぼす影響を減少させる方法でオルソポックスウイルス感染症を治療する有効な方法に対する必要がある。
【0015】
さらに、抗ウイルス剤の生物学的利用能を改善するための方法に対する必要がある。
【先行技術文献】
【特許文献】
【0016】
【特許文献1】米国特許第5,411,947号
【特許文献2】および米国特許出願第08/487,081号
【特許文献3】米国特許第5,223,263号
【特許文献4】米国特許第5,463,092号
【特許文献5】米国特許第6,716,825号
【特許文献6】WO第95/20980号
【非特許文献】
【0017】
【非特許文献1】Tam,Y.K.,「Individual Variation in First−Pass Metabolism,」Clin.Pharmacokinetics1993,25,300−328
【非特許文献2】Fisher et al.,Proc.Am.Soc.Clin.Oncol.,13:143,1994
【非特許文献3】Bartlett et al.,J.Clin.Onc.12:835−842,1994
【非特許文献4】Lum et al.,J.Clin.Onc.10:1635−42,1992
【非特許文献5】Lum et al.,Drug Resist.Clin.Onc.Hemat,9:319−336(1995)
【非特許文献6】Schinkel et al.,Eur.J.Cancer,31A:1295−1298(1995)
【発明の概要】
【0018】
本出願は、プロドラッグ、またはその医薬上許容される塩もしくはエステル、詳細には脂質含有化合物、および特定実施形態では抗ウイルス性脂質含有ヌクレオシドの、生物学的利用能を改善するための方法および組成物を提供する。1つの実施形態では、脂質含有ヌクレオシドプロドラッグもしくは他の活性化合物は、脂質成分の代謝もしくは分解を防止もしくは最小限に抑えるバイオエンハンサーと組み合わせて投与される。本発明は、医薬物質の改善された生物学的利用能、血液中の医薬物質の増加した濃度、疾患および障害を治療するために必要とされる薬物の用量の減少、およびそれらの薬物に関連する副作用の減少を提供できる。所定の態様では、バイオエンハンサーは、シトクロムP450酵素の1つもしくはイミダゾールなどの、薬物生体内変換に関連する阻害剤もしくは基質である。1つの実施形態では、抗ウイルス性脂質含有化合物は、抗痘瘡薬などの抗オルソポックス薬である。他の実施形態では、抗ウイルス性化合物は、HIV、B型肝炎、C型肝炎もしくは他のウイルスに対して活性である。
【0019】
アルコキシルアルキルホスフェートエステルなどのシドフォビルの所定のプロドラッグが経口投与される場合は、肝臓および腸内でのP450酵素などの酵素はプロドラッグの生体内変換を誘発し、それによりその薬物の有効性を減少させることができる。任意の理論に限定されることなく、生体内変換は、例えば末端アルキル鎖のω酸化によって発生することがある。そのような生体内変換の不都合な影響を回避するために、抗ウイルス性化合物のプロドラッグ、詳細にはヌクレオシド、および詳細にはシドフォビルのプロドラッグの、生物学的利用能を改善するための方法が提供される。
【0020】
抗ウイルス性脂質含有化合物の生物学的利用能を増強できる様々なバイオエンハンサーを使用できる。化合物の投与後にインビボで発生できる化合物に脂質基の生体内変換を減少させるエンハンサーを使用できる。1つの実施形態では、生物学的利用能エンハンサーは、シトクロムP450酵素の1つ、および詳細には酵素のCYP3ファミリーなどの、薬物生体内変換に関連する酵素の阻害剤もしくは基質である。1つの実施形態では、エンハンサーは、例えば、ケトコナゾールもしくはトロレアンドマイシンなどのイミダゾール系(それらの一部は抗真菌活性を有する);エリスロマイシンなどのマクロライド系;ニフェジピンなどのカルシウムチャネルブロッカー;またはゲストデンなどのステロイド系である。任意選択的に、本化合物は、グレープフルーツ中で見いだされるナリンゲニンなどのシトクロムP4503A(CYP3A)の阻害剤である。
【0021】
1つの実施形態では、抗ウイルス性脂質含有化合物、またはそれらの塩、エステルもしくはプロドラッグ、および1つまたは複数の生物学的利用能増強化合物を含む、医薬上許容される組成物が提供される。本組成物は、オルソポックスウイルスなどのウイルスに感染した宿主の治療または予防のための有効量で、それを必要とする宿主へ投与できる。
【0022】
1つの実施形態では、ウイルス感染症、例えば、オルソポックス感染症を治療する方法であって、それを必要とする宿主へ有効量の抗ウイルス性脂質含有化合物、またはそれらの塩、エステルもしくはプロドラッグ、および1つまたは複数の生物学的利用能増強化合物を投与する工程を含む方法が提供される。本組成物は、任意選択的に医薬上許容される担体と組み合わせて、オルソポックスウイルスなどのウイルスに感染した宿主の治療または予防のための有効量で投与できる。本化合物もしくは組成物は、例えば経口もしくは非経口投与される。
【0023】
1つの実施形態では、ウイルス感染症、例えば、オルソポックスウイルス感染症を治療する方法であって、それを必要とする宿主へ有効量の脂質基を含有する抗ウイルス性ヌクレオシドのプロドラッグ、またはそれらの塩、エステルもしくはプロドラッグ、および1つまたは複数の生物学的利用能増強化合物を投与する工程を含み、1つの実施形態では生物学的利用能エンハンサーが脂質基の分解を減少させる物質である方法が提供される。本組成物は、任意選択的に医薬上許容される担体と組み合わせて、オルソポックスウイルスなどのウイルスに感染した宿主の治療または予防のための有効量で組み合わせて、または交互に投与できる。本化合物もしくは組成物は、例えば経口もしくは非経口投与される。
【0024】
1つの実施形態では、化合物が単独で投与された場合に比較して、抗ウイルス性脂質含有化合物の生物学的利用能を改善するために有効な量の生物学的利用能エンハンサーを含んでもよい、医薬組成物が提供される。また別の実施形態では、エンハンサーは、抗ウイルス性化合物がエンハンサーを含めずに投与された場合に比較して、抗ウイルス性化合物の生物学的利用能を改善するために有効な量で、抗ウイルス性脂質含有化合物と連続的に、または一緒に投与される。
【0025】
1つの実施形態では、抗ウイルス性化合物は、任意選択的に脂質に共有結合した、またはアルキルグリセロール、アルキルプロパンジオール、1−S−アルキルチオグリセロール、アルコキシアルカノールもしくはアルキルエタンジオールに結合した、シドフォビル、アデフォビル、環式シドフォビルもしくはテノフォビルである。エンハンサーは、例えば、ケトコナゾールもしくはトロレアンドマイシンなどのイミダゾール系抗真菌薬;エリスロマイシンなどのマクロライド系;ニフェジピンなどのカルシウムチャネルブロッカー;またはゲストデンなどのステロイド系である。任意選択的に、本化合物は、グレープフルーツ中で見いだされるナリンゲニンなどのシトクロムP4503A(CYP3A)の阻害剤である。
【0026】
1つの特定実施形態では、シドフォビル脂質プロドラッグおよび抗真菌薬などの生物学的利用能エンハンサーを含む組成物が提供されるが、この組成物はオルソポックス感染症などのウイルス感染症を治療するための有効量で投与できる。1つの実施形態では、ヌクレオシドプロドラッグは、シドフォビルのアルコキシアルカノールなどのシドフォビルのアルコキシアルキルエステルである。例えば、この化合物は、構造:
【化1】

を有してもよい。
【0027】
詳細には、本明細書に記載した組成物は、ウイルス、詳細にはオルソポックスウイルス、例えば大痘瘡および小痘瘡、ワクシニア、痘瘡、ウシ痘、ラクダ痘、マウス痘、ウサギ痘、およびサル痘などに感染した宿主を、予防もしくは治療するための方法において使用できる。
【図面の簡単な説明】
【0028】
【図1】ウサギ、Cynサル、Rhサル、ヒト、マウスおよびラットにおける、HDP−シドフォビルにおける経時的な減少を示した図である。
【図2】マウスにおけるHDP−シドフォビル、HDP−シドフォビルから遊離したシドフォビル、およびHDP−シドフォビルの不活性代謝産物である代謝産物M−8の血清中濃度を示した図である。
【図3】NZWウサギにおけるHDP−シドフォビル、HDP−シドフォビルから遊離したシドフォビル、およびHDP−シドフォビルの不活性代謝産物である代謝産物M−8の血清中濃度を示した図である。
【図4】サルにおけるHDP−シドフォビル、HDP−シドフォビルから遊離したシドフォビル、およびHDP−シドフォビルの不活性代謝産物である代謝産物M−8の血清中濃度を示した図である。
【図5】HDP−シドフォビルの酸化によるM−8の形成について考えられる機序を示した図である。
【図6】薬物投与後の様々なタイプの組織中のサル痘力価のウイルス量を示した図である。
【発明を実施するための形態】
【0029】
脂質含有プロドラッグの生物学的利用能を改善するための方法であって、プロドラッグが生物学的利用能エンハンサーと組み合わせて、または交互に投与される方法が提供される。さらに脂質含有プロドラッグおよび生物学的利用能エンハンサーを含む医薬上許容される組成物もまた提供される。1つの実施形態におけるプロドラッグは、脂質に結合したシドフォビルなどの抗ウイルス性脂質含有化合物である。
【0030】
1つの実施形態では、疾患もしくは障害を治療する方法であって、脂質含有ヌクレオシドもしくは他の活性化合物を、脂質成分の代謝もしくは分解を防止もしくは最小限に抑えるバイオエンハンサーと組み合わせて投与するステップを含む方法が提供される。所定の態様では、生物学的利用能エンハンサーは、例えば、薬物との酵素反応、例えばシトクロムP450酵素との反応のために発生し得る、抗ウイルス性化合物の生体内変換を減少させる化合物である。生物学的利用能エンハンサーは、例えば、抗真菌化合物または抗ウイルス性化合物の生体内変換と関係している酵素の活性を減少させるように作用する他の化合物である。抗ウイルス性脂質含有化合物は、例えば、天然痘(痘瘡)などのオルソポックス感染症を治療するための有効量で投与される。生物学的利用能エンハンサーは、例えば消化管もしくは肝臓内での酵素との反応のために発生し得る薬物の生体内変換を減少させるための有効量で、組成物中に存在する。
【0031】
さらに、オルソポックス感染症などのウイルス感染症を治療するための方法であって、有効量の生物学的利用能エンハンサーおよび抗ウイルス性脂質含有ヌクレオシドを投与する工程を含む方法もまた提供される。
【0032】
<プロドラッグ化合物>
本明細書に開示した方法および組成物においては、様々な化合物のプロドラッグを使用できる。詳細には、プロドラッグは、炭化水素鎖、例えば、C4−C30、またはC8−22炭化水素鎖を含むプロドラッグであってよい。本薬物は、様々な抗がん化合物もしくは抗ウイルス性化合物などの様々な薬物のいずれかであってよい。
【0033】
1つの実施形態では、本プロドラッグは、ホスホネートおよびホスフェートを含むヌクレオシドのプロドラッグである。特定の実施形態では、本プロドラッグは、抗オルソポックス剤などの抗ウイルス性脂質含有ヌクレオシドである。
【0034】
1つの実施形態におけるプロドラッグは、抗ウイルス性化合物のプロドラッグである。本プロドラッグは、例えば、アルキルグリセロール、アルキルプロパンジオール、1−S−アルキルチオグリセロール、アルコキシアルカノールもしくはアルキルエタンジオール、またC8−30アルキル、アルケニルもしくはアルキニルを含有する脂質などの脂質に共有結合した、シドフォビル、アデフォビル、環式シドフォビルもしくはテノフォビルである。本明細書で使用するように、化合物が「脂質に共有結合する」場合は、本化合物は、化合物と脂質基との間にリンカーを含んでもよい。脂質基は、例えば、C8−30アルキル、アルケニルもしくはアルキニルである。
【0035】
1つの実施形態では、抗ウイルス性プロドラッグは、例えば、脂質に共有結合したシドフォビルである。
【0036】
1つの実施形態では、抗ウイルス性プロドラッグは、構造:
【化2】
(式中、Rは、H;任意選択的に置換されたアルキル、例えば、C1−C30アルキル;アルケニル、例えば、C1−C30アルケニル;もしくはアルキニル、例えば、C1−C30アルキニル;アシル;モノホスフェートもしくはジホスフェート;アルキルグリセロール、アルキルプロパンジオール、1−S−アルキルチオグリセロール、アルコキシアルカノールもしくはアルキルエタンジオールである)を有する。1つの実施形態では、Rは、アルコキシアルカノールである。例えば、Rは、−(CH2)m−O−(CH2)n−CH3(式中、例えば、mは1〜5であり、nは1〜25である;またはmは2〜4であり、nは10〜25である)である。
【0037】
また別の実施形態では、抗ウイルス性プロドラッグ化合物は、以下の構造:
【化3】
を有する。
【0038】
1つの実施形態では、抗ウイルス性プロドラッグは、例えば、脂質基に共有結合したアデフォビルである。
【0039】
特定の実施形態では、抗ウイルス性プロドラッグは、以下の構造:
【化4】
(式中、Rは、H;任意選択的に置換されたアルキル、例えば、C1−C30アルキル;アルケニル、例えば、C1−C30アルケニル;アルキニル、例えば、C1−C30アルキニル;アシル;モノ−もしくはジ−ホスフェート;アルキルグリセロール、アルキルプロパンジオール、1−S−アルキルチオグリセロール、アルコキシアルカノールもしくはアルキルエタンジオールである)を有する。1つの実施形態では、Rは、アルコキシアルカノールである。例えば、Rは、−(CH2)m−O−(CH2)n−CH3(式中、例えば、mは1〜5であり、nは1〜25である;またはmは2〜4であり、nは10〜25である)である。
【0040】
1つの実施形態では、抗ウイルス性プロドラッグは、例えば、脂質に共有結合したテノフォビルである。
【0041】
特定の実施形態では、抗ウイルス性プロドラッグは、以下の構造:
【化5】
(式中、Rは、H;任意選択的に置換されたアルキル、例えば、C1−C30アルキル;アルケニル、例えば、C1−C30アルケニル;アルキニル、例えば、C1−C30アルキニル;アシル;モノホスフェートもしくはジホスフェート;アルキルグリセロール;アルキルプロパンジオール;1−S−アルキルチオグリセロール;アルコキシアルカノール;もしくはアルキルエタンジオールである)を有する。1つの実施形態では、Rは、アルコキシアルカノールである。Rは、例えば、−(CH2)m−O−(CH2)n−CH3(式中、例えば、mは1〜5であり、nは1〜25である;またはmは2〜4であり、nは10〜25である)である。
【0042】
1つの実施形態では、抗ウイルス性プロドラッグは、例えば、脂質に共有結合した環式シドフォビルである。
【0043】
また別の実施形態では、抗ウイルス性プロドラッグは、以下の式:
【化6】
(式中、Rは、H;任意選択的に置換されたアルキル、例えば、C1−C30アルキル;アルケニル、例えば、C1−C30アルケニル;アルキニル、例えば、C1−C30アルキニル;アシル;モノホスフェートもしくはジホスフェート;アルキルグリセロール、アルキルプロパンジオール、1−S−アルキルチオグリセロール、アルコキシアルカノールもしくはアルキルエタンジオールである)を有する。1つの実施形態では、Rは、アルコキシアルカノールである。Rは、例えば、−(CH2)m−O−(CH2)n−CH3(式中、例えば、mは1〜5であり、nは1〜25である;またはmは2〜4であり、nは10〜25である)である。
【0044】
また別の実施形態では、プロドラッグは、1−O−オクタデシルプロパンジオール−3−シドフォビル、1−O−オクタデシルエタンジオール−2−シドフォビル、1−O−ヘキサデシルプロパンジオール−3−環式シドフォビル、1−O−オクタデシルプロパンジオール−3−環式シドフォビル、1−O−オクタデシルエタンジオール−2−環式シドフォビル、1−O−ヘキサデシルプロパンジオール−3−アデフォビル、もしくは1−O−オクタデシル−sn−グリセロ−3−アデフォビルである。
【0045】
また別の実施形態では、プロドラッグは、ヘキサデシルオキシプロピルシドフォビル、オクタデシルオキシエチルシドフォビル、オレイルオキシプロピルシドフォビル、オクチルオキシプロピルシドフォビル、ドデシルオキシプロピルシドフォビル、オレイルオキシエチルシドフォビル、1−O−オクタデシル−2−O−ベンジル−グリセリルシドフォビル、テトラデシルオキシプロピルシドフォビル、エイコシルシドフォビル、ドコシルシドフォビル、ヘキサデシルシドフォビル、ヘキサデシルオキシプロピル環式シドフォビル、オクタデシルオキシエチル環式シドフォビル、オレイルオキシプロピル環式シドフォビル、オクチルオキシプロピル環式シドフォビル、ドデシルオキシプロピル環式シドフォビル、オレイルオキシエチル環式シドフォビル、1−O−オクタデシル−2−O−ベンジル−グリセリル環式シドフォビル、テトラデシルオキシプロピル環式シドフォビル、エイコシル環式シドフォビル、ドコシル環式シドフォビル、もしくはヘキサデシル環式シドフォビルである。
【0046】
その開示が本明細書に組み込まれる米国特許第6,716,825号に記載された化合物は、本明細書に提供した方法および組成物において使用できる。これらの化合物は、例えば米国特許第6,716,825号に記載された方法などの当分野において利用できる方法によって作製できる。
【0047】
抗ウイルス性化合物の広範囲の脂質誘導体、例えば抗ウイルス性ヌクレオシドは、本明細書に提供した方法および組成物において使用できる。1つの実施形態では、抗ウイルス性化合物はプロドラッグ形にあり、以下の構造:
【化7A】
【化7B】
(式中、W1、W2、およびW3は各々、独立して−O−、−S−、−SO−、−SO2、−O(C=O)−、−(C=O)O−、−NH(C=O)−、−(C=O)NH−もしくは−NH−である;および1つの実施形態では、各々は独立してO、S、もしくは−O(C=O)−である;
nは、0もしくは1である;mは、0もしくは1である;pは、0もしくは1である;
R1は、任意選択的に置換されたアルキル、アルケニルもしくはアルキニル、例えば、C1−30アルキル、アルケニル、もしくはアルキニルである;または1つの実施形態では、R1は任意選択的にC8−30アルキル、アルケニルもしくはアルキニルである、またはR1はC8−24アルキル、アルケニルもしくはアルキニル(例、C17、C18、C19、C20、C21、C22、C23、もしくはC24アルキル、アルケニルもしくはアルキニル)である;
R2およびR3は各々、独立して任意選択的に置換されたC1−25アルキル、アルケニルもしくはアルキニルである;または1つの実施形態では、任意選択的にR2およびR3は各々、独立してC1−5アルキル、アルケニルもしくはアルキニル(例、C1、C2、もしくはC3アルキル、アルケニルもしくはアルキニル;例えば、メチル、エチルもしくはプロピル)である;または別の実施形態では、CF3である;または別の実施形態では、アリール、例えば、ベンジルである;
tは、0もしくは1である;および
Dは、抗ウイルス薬、例えば抗ウイルス性環式もしくは非環式ヌクレオシドである)のうちの1つを有する。
【0048】
本明細書に開示した式V〜Xを含む式の1つの部分実施形態では、
式中、tは1である、および
【化8】
は、5’−O−リン酸ヌクレオシド、2’−O−リン酸ヌクレオシド、もしくは3’−O−リン酸ヌクレオシドを含むがそれらに限定されない、生物活性ホスフェート薬の残基である。
【0049】
本明細書に開示した、式V〜Xを含む式のまた別の部分実施形態では、
式中、tは0である、および
【化9】
は、シドフォビル、アデフォビル、テノフォビル、環式シドフォビル、HPMPA、PMEG、または生物活性ヌクレオシドもしくは非環式ヌクレオシドの任意の他のホスホン酸塩誘導体を含むがそれらに限定されない、生物活性ホスホン酸塩薬の残基である。
【0050】
また別の実施形態では、プロドラッグは、抗ウイルス性ヌクレオシドの1−O−アルキル−プロパンジオール−ホスフェートであるが、このとき抗ウイルス性ヌクレオシドは、(1)置換もしくは未置換プリンもしくはピリミジンを(a)リボース残基の非環式ヒドロキシル化フラグメント(例、ヒドロキシル化2−プロポキシメチルもしくはエトキシメチル)、(b)リボースもしくは2’−デオキシリボース、または(c)デオキシペントースのいずれかとともに有する。
【0051】
また別の実施形態では、プロドラッグは、抗ウイルス性ヌクレオシドの1−O−アルキル−エタンジオール−ホスフェートであるが、このとき抗ウイルス性ヌクレオシドは、(1)置換もしくは未置換プリンもしくはピリミジンを(a)リボース残基の非環式ヒドロキシル化フラグメント(例、ヒドロキシル化2−プロポキシメチルもしくはエトキシメチル)、(b)リボースもしくは2’−デオキシリボース、または(c)デオキシペントースのいずれかとともに有する。
【0052】
式V〜Xの1つの部分実施形態では、
W1、W2、およびW3は各々、独立して−O−、−S−、もしくは−O(CO)−である;
nは、0もしくは1である;mは、0もしくは1である;pは、0もしくは1である。
R1は、任意選択的に置換されたC18−24アルキルもしくはアルケニル(例、C18、C19、C20、C21、C22、C23、もしくはC24アルキル)である;
R2およびR3は各々、独立して任意選択的に置換されたC1−5アルキル、アルケニル、アルキニルまたはCF3;(例、C1、C2、もしくはC3アルキル;例、メチルもしくはエチル)またはシクロアルキルである;
tは、0もしくは1である;および
Dは、抗ウイルス性環式もしくは非環式ヌクレオシドである。
【0053】
また別の部分実施形態では、抗ウイルス性プロドラッグは、以下の構造:
【化10】
(式中、R1は、任意選択的に置換されたC8−24アルキル、例えばC18−24アルキル(例、C18、C19、C20、C21、C22、C23、もしくはC24アルキル)である;
R2およびR3は、独立してC1−5アルキル、ハロアルキル、アルケニル、アルキニル、もしくはシクロアルキル、例えばメチル、エチルもしくはCF3である;
tは、0もしくは1である;および
Dは、抗ウイルス薬、例えば抗ウイルス性環式もしくは非環式ヌクレオシドである)のうちの1つである。
【0054】
例えば上記の式V〜XVIに示したような、脂質基に結合することのできる代表的な抗ウイルス性ヌクレオシドは、ddA、ddI、ddG、L−FMAU、DXG、DAPD、L−dA、L−dI、L−(d)T、L−dC、L−dG、FTC、5−FC、1−(2’−デオキシ−2’−フルオロ−1−β−D−アラビノフラノシル)−5−ヨードシトシン(FIAC)もしくは1(2’−デオキシ−2’−フルオロ−1−β−D−アラビノフラノシル)−5−ヨードウラシル(FIAU)などである。
【0055】
例えば式V〜XVIに例示したように、脂質基を含むために任意選択的に共有的に誘導体化して使用できるその他のヌクレオシド(例えばポックスウイルス感染症を治療する際に有用である)には、8−メチルアデノシン、2−アミノ−7−[(1,3−ジヒドロキシ−2−プロポキシ)メチル]プリン(S2242)、Ara−A、PME−N6−(シクロプロピル)DAP、ホスホノメトキシエチルデオキシジアミノプリン(PMEADADP);PME−N6−(ジメチル)DAP、PME−N6−(トリフルオロエチル)DAP、PMEA−N6−(2−プロペニル)DAP、アデノシン−N(l)−オキシドのアナログ、1−(ベンジルオキシ)アデノシンのアナログ、IMPデヒドロゲナーゼ阻害剤(例、リバビリン、EICAR、チアゾフリン、およびセレナゾール)、SAHヒドロラーゼ阻害剤(例、5’−ノルアリステロマイシン、ネプラノシンAおよびC、炭素環式3−デアザ−アデノシン、DHCeA、C3DHCeA、6’−β−フルオロ−アリステロマイシン、5’−ノルアリステロマイシンおよびそのエピマー、3−デアザ−5’−ノルアリステロマイシン、6’−C−メチルネプラノシン、6’−ホモネプラノシン、2−フルオロネプラノシン、6’−ヨードアセチルAdo、および3−デアザネプラノシン)、OMPデカルボキシラーゼ阻害剤(例、ピラゾフリンおよび5’−デオキシピラゾフリン)CTPシンテターゼ阻害剤(例、シクロペンテニルシトシンおよびカルボジン)、チミジレートシンターゼ阻害剤(例、5−置換2’−デオキシウリジン)、および3’−フルオロ−3’−デオキシアデノシンが含まれる。
【0056】
その他のヌクレオシドには、3’−アジド−2’,3’−ジデオキシピリミジンヌクレオシド、例えば、AZT、AZT−P−AZT、AZT−P−ddA、AZT−P−ddI、AzddClU、AzddMeC、AzddMeCN4−OH、AzddMeC N4Me、AZT−P−CyE−ddA、AzddEtU(CS−85)、AzddU(CS−87)、AzddC(CS−91)、AzddFC、AzddBrU、およびAzddIU;3’−ハロピリミジンジデオキシヌクレオシドを含むクラス、例えば、3’−FddClU、3’−FddU、3’−FddT、3’−FddBrU、および3’−FddEtU;2’,3’−ジデヒドロ−2’,3’−ジデオキシヌクレオシド(D4ヌクレオシド)を含むクラス、例えば、D4T、D4C、D4MeC、およびD4A;2’,3’−未置換ジデオキシピリミジンヌクレオシドを含むクラス、例えば、5−F−ddC、ddCおよびddT;2’,3’−未置換ジデオキシプリンヌクレオシドを含むクラス、例えば、ddA、ddDAPR(ジアミノプリン)、ddG、ddI、およびddMeA(N6メチル);および糖−置換ジデオキシプリンヌクレオシドを含むクラス、例えば、3−N3ddDAPR、3−N3ddG、3−FddDAPR、3−FddG、3−FddaraA、および3−FddA(式中、Meはメチルであり、Etはエチルであり、そしてCyEtはシアノエチルである)が含まれる。
【0057】
その他の代表的なヌクレオシドには、ジデヒドロピリミジン、ならびにカルボビル、炭素環式2’,3’−ジデヒドログアノシン;デオキシグアノシン(AZG)およびピリミジンの3’−アジド誘導体、デオキシウリジン;デオキシチミジンおよびデオキシグアノシンの3’−フルオロ誘導体;2’,6’−ジアミノプリン、2’,3’−デオキシリボシドおよびその3’−フルオロおよび3’−アジド誘導体;2−クロロ−デオキシアデノシン;ガンシクロビル、アシクロビル、環式ガンシクロビル、9−(2−ホスホニルメトキシエチル)グアニン(PMEG)、9−(2−ホスホニルメトキシエチル)アデニン(PMEA)、ペンシクロビル、シドフォビル、アデフォビル、環式シドフォビル、9−(3−ヒドロキシ−2−ホスホニルメトキシプロピル)アデニン(HPMPA)、cHPMPA、8−アザ−HPMPA、HPMPG、(S)−9−(3−ヒドロキシ−2−ホスホニルメトキシプロピル)−2,6−ジアミノプリン((S)−HPMPDAP)、(S)−6−(3−ヒドロキシ−2−ホスホニルメトキシプロピル)オキシ−2,4−ジアミノピリミジン((S)−HPMPO−DAPy)、およびテノフォビルが含まれる。
【0058】
例えば、それらの各々がこれにより全体として参照して本明細書に組み込まれる以下の特許に開示されている化合物などの多数のホスホン酸塩化合物は、それらの薬理学的活性を改善するため、またはそれらの経口吸収を増加させるために脂質含有化合物に誘導体化することができる:米国特許第3,468,935号(Etidronate)、米国特許第4,327,039号(Pamidronate)、米国特許第4,705,651号(Alendronate)、米国特許第4,870,063号(Bisphosphonicacid derivatives)、米国特許第4,927,814号(Diphosphonates)、米国特許第5,043,437号(Phosphonatesof azidodideoxynucleosides)、米国特許第5,047,533号(Acyclicpurine phosphonatenucleotide analogs)、米国特許第5,142,051号(N−Phosphonylmethoxyalkylderivatives ofpyrimidine andpurine bases)、米国特許第5,183,815号(Boneacting agents)、米国特許第5,196,409号(Bisphosphonates)、米国特許第5,247,085号(Antiviralpurine compounds)、米国特許第5,300,671号(Gem−diphosphonicacids)、米国特許第5,300,687号(Trifluoromethylbenzylphosphonates)、米国特許第5,312,954号(Bis−and tetrakis−phosphonates)、米国特許第5,395,826号(Guanidinealkyl−1,1−bisphosphonicacid derivatives)、米国特許第5,428,181号(Bisphosphonatederivatives)、米国特許第5,442,101号(Methylenebisphosphonicacid derivatives)、米国特許第5,532,226号(Trifluoromethybenzylphosphonates)、米国特許第5,656,745号(Nucleotideanalogs)、米国特許第5,672,697号(Nucleoside−5’−methylenephosphonates)、米国特許第5,717,095号(Nucleotideanalogs)、米国特許第5,760,013号(Thymidylateanalogs)、米国特許第5,798,340号(Nucleotideanalogs)、米国特許第5,840,716号(Phosphonatenucleotide compounds)、米国特許第5,856,314号(Thio−substituted、nitrogen−containing、heterocyclicphosphonate compounds)、米国特許第5,885,973号(olpadronate)、米国特許第5,886,179号(Nucleotideanalogs)、米国特許第5,877,166号(Enantiomericallypure 2−aminopurinephosphonate nucleotideanalogs)、米国特許第5,922,695号(Antiviral phosphonomethoxy nucleotideanalogs)、米国特許第5,922,696号(Ethylenic and allenic phosphonate derivatives of purines)、米国特許第5,977,089号(Antiviralphosphonomethoxy nucleotide analogs)、米国特許第6,043,230号(Antiviralphosphonomethoxy nucleotide analogs)、米国特許第6,069,249号(Antiviralphosphonomethoxy nucleotide analogs);ベルギー国特許第672205号(Clodronate);欧州特許第753523号(Amino−substitutedbisphosphonic acids);欧州特許出願第186405号(geminal diphosphonates)などが含まれる。さらに、それらの各々は参照することによって全体として本明細書に組み込まれる以下の刊行物:J.Med.Chem.,2002,45:1918−1929;J.Med.Chem.,2003,46:5064−5073;Antimicrob.AgentsChemotherapy,2002,46:2185−2193に列挙した化合物は、それらの薬理学的活性を改善するため、またはそれらの経口吸収を増加させるために誘導体化できる。
【0059】
ヌクレオシドは、それらの各々は参照することによって全体として本明細書に組み込まれる以下の特許:米国特許第5,614,548号;米国特許第5,512,671号;米国特許第5,770,584号、米国特許第5,962,437号;米国特許第6,030,960号;米国特許第6,670,341号;米国特許第5,223,263号;米国特許第5,817,638号;米国特許第6,252,060号;米国特許第6,448,392号;米国特許第5,411,947号;米国特許第5,744,592号;米国特許第5,484,809号;米国特許第5,827,831号;米国特許第5,696,277号;米国特許第6,002,029号;米国特許第5,780,617号;米国特許第5,194,654号;米国特許第5,463,092号;米国特許第5,744,461号;米国特許第5,484,911号;WO第91/09602号;WO第91/05558号;米国特許第4,444,766号;米国特許第5,869,468号;米国特許第5,84,228号;米国特許出願第2002/0082242号;米国特許出願第2004/0161398号;米国特許出願第2004/0259845号;WO第98/38202号;米国特許第5,696,277号;米国特許第6,002,029号;米国特許第5,744,592号;米国特許第5,827,831号;米国特許第5,817,638号;ならびに米国特許第6,252,060号および米国特許第5,756,711号に記載した様々な脂肪親和性基を用いて誘導体化することができ、本明細書に提供した組成物および方法において使用できる。
【0060】
以下の物質:鎮痛薬;麻酔薬;食欲抑制薬;抗アドレナリン作動薬;抗アレルギー薬;抗狭心症薬;抗不安薬;抗関節炎薬;抗喘息薬;抗アテローム硬化症薬;抗細菌薬;抗凝固薬;抗痙攣薬;抗うつ薬;抗糖尿病薬;下痢止め薬;抗利尿薬;抗エストロゲン薬;抗線維素溶解薬;抗真菌薬;抗緑内障薬;抗ヒスタミン薬;抗感染薬;抗炎症薬;抗角化薬;抗マラリア薬;抗微生物薬;抗片頭痛薬;抗有糸分裂薬;抗カビ薬、制吐薬、抗腫瘍薬、抗好中球減少薬、antiobessionalagent;抗寄生虫薬;抗パーキンソン薬;抗蠕動薬、抗ニューモシスチス属薬;抗増殖薬;肝障害療法薬;向精神病薬;セロトニン阻害剤;セロトニン受容体アンタゴニスト;ステロイド;刺激剤;抑制剤;甲状腺ホルモン;甲状腺阻害剤;甲状腺ミメティック;トランキライザー;筋萎縮性側索硬化症の治療薬;脳虚血の治療薬;ページェット病の治療薬;不安定性狭心症の治療薬;尿酸排泄薬;血管収縮薬;血管拡張薬;傷薬;または創傷治療薬のプロドラッグを含む、他の化合物のプロドラッグもまた使用できる。
【0061】
使用できる以下の抗癌薬のプロドラッグには、例えば:アシビシン;アクラルビシン;塩酸アコダゾール;アクロニン;アドゼレシン;アドリアマイシン;アルデスロイキン;アリトレチノイン;アロプリノールナトリウム;アルトレタミン;アンボマイシン;酢酸アメタントロン;アミノグルテチミド;アムサクリン;アナストロゾール;アノナセオス・アセトゲニン;アントラマイシン;アシミシン;アスパラギナーゼ;アスペルリン;アザシチジン;アゼテパ;アゾトマイシン;バチマスタット;ベンゾデパ;ベキサロテン;ビカルタミド;塩酸ビサントレン;ビスナフィドジメシレート;ビゼレシン;硫酸ブレオマイシン;ブレキナールナトリウム;ブロピリミン;ブラタシン;ブスルファン;カベルゴリン;カクチノマイシン;カルステロン;カラセミド;カルベチマー;カルボプラチン;カルムスチン;塩酸カルビシン;カルゼレシン;セデフィンゴール;セレコキシブ;クロラムブシル;シロレマイシン;シスプラチン;クラドリビン;クリスナトールメシレート;シクロホスファミド;シタラビン;ダカルバジン;DACA(N−[2−(ピメチル−アミノ)エチル]アクリジン−4−カルボキサミド);ダクチノマイシン;塩酸ダウノルビシン;ダウノマイシン;デシタビン;デニロイキンディフチトクス;デキソルマプラチン;デザグアニン;デザグアニンメシレート;ジアジコン;ドセタキセル;ドキソルビシン;塩酸ドキソルビシン;ドロロキシフェン;クエン酸ドロロキシフェン;プロピオン酸ドロモスタノロン;デュアゾマイシン;エダトレキセート;塩酸エフロルニチン;エルサミトルシン;エンロプラチン;エンプロメート;エピプロピジン;塩酸エピルビシン;エルブロゾール;塩酸エソルビシン;エストラムスチン;リン酸エストラムスチンナトリウム;エタニダゾール;エチオダイズド油I131;エトポシド;リン酸エトポシド;エトプリン;塩酸ファドロゾール;ファザラビン;フェンレチニド;フロクスウリジン;リン酸フルダラビン;フルオロウラシル;5−FdUMP;フルロシタビン;フォスキドン;フォストリエシンナトリウム;FK−317;FK−973;FR−66979;FR−900482;ゲムシタビン;塩酸ゲムシタビン;ゲムツズマブオゾガマイシン;GoldAu 198;酢酸ゴセレリン;グアナコーン;ヒドロキシウレア;塩酸イダルビシン;イフォスファミド;イルモフォシン;インターフェロンα−2a;インターフェロンα−2b;インターフェロンα−n1;インターフェロンα−n3;インターフェロンβ−Ia;インターフェロンγ−Ib;イプロプラチン;塩酸イリノテカン;酢酸ランレオチド;レトロゾール;酢酸ロイプロリド;塩酸リアロゾール;ロメトレキソールナトリウム;ロムスチン;塩酸ロソキサントロン;マソプロコール;メイタンシン;塩酸メクロルエタミン;酢酸メゲストロール;酢酸メレンゲストロール;メルファラン;メノガリル;メルカプトプリン;メトトレキセート;メトトレキセートナトリウム;メトキサレン;メトプリン;メツルデパ;ミチンドミド;マイトカルシン;マイトクロミン;マイトギリン;マイトマルシン;マイトマイシン;マイトマイシンC;ミトスペル;ミトタン;塩酸ミトキサントロン;ミコフェノール酸;ノコダゾール;ノガラマイシン;オプレルベキン;オルマプラチン;オキシスラン;パクリタキセル;パミドロン酸二ナトリウム;ペグアスパルガーゼ;ペリオマイシン;ペンタムスチン;硫酸ペプロマイシン;ペルフォスファミド;ピポブロマン;ピポスルファン;塩酸ピロキサントロン;プリカマイシン;プロメスタン;ポルフィマーナトリウム;ポルフィロマイシン;プレドニムスチン;塩酸プロカルバジン;ピューロマイシン;塩酸ピューロマイシン;ピラゾフリン;リボプリン;リツキシマブ;ログレチミド;ロリニアスタチン;サフィンゴール;塩酸サフィンゴール;サマリウム/レキシドロナム;セムスチン;7−ヒドロキシスタウロスポリン;シムトラゼン;スパルフォセートナトリウム;スパルソマイシン;塩酸スピロゲルマニウム;スピロムスチン;スピロプラチン;スクアモシン;スクアモタシン;ストレプトニグリン;ストレプトゾシン;塩化ストロンチウムSr89;スロフェヌル;タリソマイシン;タキサン;タキソイド;テコガランナトリウム;テガフール;塩酸テロキサントロン;テモポルフィン;テニポシド;テロキシロン;テストラクトン;チアミプリン;チオグアニン;チオテパ;チミタック;チアゾフリン;チラパラザミン;トミュデックス(Tomudex);TOP−53;塩酸トポテカン;クエン酸トレミフェン;トラスツズマブ;酢酸トレストロン;リン酸トリシリビン;トリメトレキセート;グルクロン酸トリメトレキセート;トリプトレリン;塩酸トブロゾール;ウラシルマスタード;ウレデパ;バルルビシン;バプレオチド;ベルテポルフィン;ビンブラスチン;硫酸ビンブラスチン;ビンクリスチン;硫酸ビンクリスチン;ビンデシン;硫酸ビンデシン;硫酸ビネピジン;硫酸ビングリシネート;硫酸ビンロイロシン;酒石酸ビノレルビン;硫酸ビンロシジン;硫酸ビンゾリジン;ボロゾール;ゼニプラチン;ジノスタチン;塩酸ゾルビシン;2−クロロデオキシアデノシン;2’−デオキシホルマイシン;9−アミノカンプトテシン;ラルチトレキセド;N−プロパルジル−5,8−ジデアザ葉酸;2−クロロ−2’−アラビノ−フルオロ−2’−デオキシアデノシン;2−クロロ−2’−デオキシアデノシン;アニソマイシン;トリコスタチンA;hPRL−G129R;CEP−751;リノミド;サルファマスタード;ナイトロジェンマスタード(メクロルエタミン);シクロホスファミド;メルファラン;クロラムブシル;イフォスファミド;ブスルファン;N−メチル−N−ニトロソウレア(MNU);N,N’−ビス(2−クロロエチル)−N−ニトロソウレア(BCNU);N−(2−クロロエチル)−N’−シクロヘキシル−N−ニトロソウレア(CCNU);N−(2−クロロエチル)−N’−(トランス−4−メチルシクロヘキシル−N−ニトロソウレア(MeCCNU);N−(2−クロロエチル)−N’−(ジエチル)エチルホスホネート−N−ニトロソウレア(フォテムスチン);ストレプトゾトシン;ジアカルバジン(DTIC);ミトゾロマイド;テモゾロマイド;チオテパ;マイトマイシンC;AZQ;アドゼレシン;シスプラチン;カルボプラチン;オルマプラチン;オキサリプラチン;C1−973;DWA2114R;JM216;JM335;ビス(白金);トミュデックス;アザシチジン;シタラビン;ゲムシタビン;6−メルカプトプリン;6−チオグアニン;ヒポキサンチン;テニポシド;9−アミノカンプトテシン;トポテカン;CPT−11;ドキソルビシン;ダウノマイシン;エピルビシン;ダルビシン;ミトキサントロン;ロソキサントロン;ダクチノマイシン(アクチノマイシンD);アムサクリン;ピラゾロアクリジン;オール−トランスレチノール;14−ヒドロキシ−レトロ−レチノール;オール−トランスレチノイン酸;N−(4−ヒドロキシフェニル)レチンアミド;13−シスレチノイン酸;3−メチルTTNEB;9−シスレチノイン酸;フルダラビン(2−F−ara−AMP);および2−クロロデオキシアデノシン(2−Cda)などの抗腫瘍薬のプロドラッグが含まれる。
【0062】
<エンハンサー>
様々な生物学的利用能向上剤は、例えば抗ウイルス性脂質含有ヌクレオシドなどの脂質含有プロドラッグの生物学的利用能を増強するための有効量で投与されてよい、または医薬組成物中に存在していてよい。生物学的利用能エンハンサーは、薬物の分解、または生体内変換を最小限に抑えるために使用される。1つの実施形態では、生物学的利用能エンハンサーは、脂質プロドラッグの脂質成分の代謝もしくは分解を防止する、または最小限に抑える。
【0063】
「薬物の生物学的利用能」は、全身性で経時的に利用できる薬物の総量を意味する。薬物の生物学的利用能は、腸内での薬物生体内変換を阻害することによって、および/または腸内での腸上皮を横断する薬物の正味輸送を減少させる活性輸送系を阻害することによって、および/または肝臓内での薬物生体内変換を減少させることによって増加させることができる。プロドラッグの薬物の生物学的利用能の増加を引き起こす化合物は、本明細書ではバイオエンハンサーもしくは生物学的利用能エンハンサーと呼ぶ。
【0064】
所定の化合物がバイオエンハンサーに必要とされる阻害もしくは結合特性を有するかどうかを決定する任意選択的のバイオアッセイは、使用できる化合物を同定するために使用できる。
【0065】
1つの態様では、生物学的利用能エンハンサーは、シトクロムP450酵素の1つなどの、薬物生体内変換に関連する酵素の阻害剤もしくは基質である。1つの実施形態では、エンハンサーは、例えば、ケトコナゾールもしくはトロレアンドマイシンなどのイミダゾール系抗真菌薬;エリスロマイシンなどのマクロライド系;ニフェジピンなどのカルシウムチャネルブロッカー;またはゲストデンなどのステロイド系である。任意選択的に、本化合物は、グレープフルーツ中で見いだされるナリンゲニンなどのシトクロムP4503A(CYP3A)の阻害剤である。
【0066】
薬物生体内変換に関係している酵素もしくは受容体の活性に影響を及ぼす任意選択的の物質を使用できる。1つの実施形態では、その活性を減少させることのできる酵素はシトクロムP450酵素であり、詳細にはCYP3ファミリーの酵素である。
【0067】
シトクロムP450は、ヘムタンパク質のスーパーファミリーである。それらは、混合官能オキシダーゼ系の末端オキシダーゼを表している。シトクロムP450遺伝子スーパーファミリーは、シトクロムP450の進化的関係に基づいて命名される、少なくとも207個の遺伝子から構成される。この命名システムのためには、シトクロムP450遺伝子の全部の配列順序が比較され、少なくとも40%の同一性を共有するシトクロムP450が1ファミリー(CYPの後にローマもしくはアラビア数字が続く、例えばCYP3)であると規定され、さらにそれらの推定アミノ酸配列によって少なくとも55%関連するそれらの形態から構成されるサブファミリー(大文字によって表示される、例えば、CYP3A)に分割される。最後に、シトクロムP450の各個別形態についての遺伝子は、アラビア数字(例、CYP3A4)が指定される。
【0068】
3つのシトクロムP450遺伝子ファミリー(CYP1、CYP2およびCYP3)が大多数の薬物代謝の原因となると思われる。少なくとも15種のシトクロムP450が、ヒト肝において様々な程度まで特性付けられている。3型のシトクロムP450をコードするCYP3遺伝子ファミリーは、おそらくヒト薬物代謝において最も重要なファミリーである。ヒト3Aサブファミリーにおいては少なくとも5つの形態のシトクロムP450が見いだされており、これらの形態は極めて多数の構造的に多様な薬物の代謝の原因となっている。肝臓はシトクロムP450の多数のアイソフォームを含有しており、広範囲の物質を生体内変換させることができる。腸の内腔を内張する腸細胞はまた、重要なシトクロムP450活性を有しており、この活性は、薬物代謝において最も重要なアイソフォームである単一ファミリーのアイソザイム3Aによって決定付けられる。そこで、1つの実施形態では、薬物有効性は、例えば肝臓および/または腸内腔内でのCYP3a薬物生体内変換を減少させることによって増加させられる。
【0069】
詳細には、シトクロムP450酵素の活性は阻害することができる、またはシトクロムP450酵素は薬物および環境化合物によって不活性化することができる。これには、同一シトクロムP450の基質間の競合的阻害、活性部位以外のシトクロムP450上の部位に結合する物質による阻害、および物質の代謝中に形成される反応中間体によるシトクロムP450の自殺的不活性化が含まれる。例えば、阻害剤は、CYP3A薬物生体内変換の競合的、非競合的、競合不能、混合もしくは非可逆性阻害剤として作用することによって機能できる。阻害剤は、共有結合またはイオン性もしくは極性誘引のいずれかによって保護される薬物に結合することによって作用することもできる。
【0070】
CYP3Aの活性は、CYP3A基質からの反応生成物のCYP3A触媒性産生である。CYP3Aについての基質は、天然型基質または表1に列挙した成分などの他の成分である可能性がある。さらに、表1に列挙したCYP3A阻害剤の一部は、表の中に表示したように基質として同定される。阻害を受けるCYP3Aの触媒活性には、dealkyase、オキシダーゼ、およびヒドロラーゼ活性が含まれる。CYP3Aの様々な触媒活性に加えて、ある範囲の分子量(例えば、Komoriet al.,J.Biochem.1988,104:912−16に示されるように、51kD〜54kD)を備える様々な形態のCYP3Aが存在する。表1に列挙した化合物、詳細には阻害剤は、本明細書に記載した方法および組成物中のエンハンサーとして使用できる。
【0071】
【表1】
【0072】
特定の実施形態では、エンハンサーは、例えば、パロキセチン、フルオキセチン、セルトレリン、フルボキサミン、ネファゾドン、ベンラファキシン、シメチジン、フルフェナジン、ハロペリドール、ペルフェナジン、チオリダジン、ジルチアゼム、メトロニダゾール、troleandomyan、ジスルフィラム、オトギリソウ、およびオメプラゾールなどのCYA酵素の阻害剤である。
【0073】
エンハンサーには、例えばシクロスポリン、ベラパミル、タモキシフェン、キニジンおよびフェノチアジンなどのP糖タンパク質を阻害する化合物もまた含まれる。
【0074】
代表的なエンハンサーには、抗ウイルス性プロテアーゼ阻害薬、例えばインジナビル、ネルフィナビル、リトナビル、サキナビル;ならびに抗真菌薬、例えばフルコナゾール、イトラコナゾール、ケトコナゾール、およびミコナゾールが含まれる。
【0075】
その他のエンハンサーには、例えばクラリスロマイシン、エリスロマイシン、ノルトリプチリン、リグノカイン、およびanriodaroneなどのマクロライド系が含まれる。
【0076】
その他のエンハンサーには、17−エチニル−置換ステロイド系、例えば、ゲストデン、エチニル−エストラジオール、メトキサレン、およびレボノルゲストレルが含まれる。
【0077】
その他のエンハンサーには、例えばクエルセチンおよびナリンゲニンなどのフラボン系、および例えばエチニルエストラジオール、およびプレドニゾロンなどの他の化合物が含まれる。
【0078】
1つの実施形態では、生物学的利用能エンハンサーは、P糖タンパク質(P−gp)−媒介性膜輸送の阻害剤である。
【0079】
また別の実施形態では、生物学的利用能エンハンサーは、シクロスポリンA、活性ブロッカーであるGF120918(エラクリダル)、LY335989(zosuquidar)、バルスポダール(PSC833)、ビリコダル(VX710)、またはR101933である。
【0080】
当分野において利用できる活性エンハンサーについての試験を使用すると、適切な化合物を選択することができる。例えば、酵素阻害を測定できる。1つの実施形態では、肝細胞もしくは腸細胞の培養細胞または肝臓もしくは腸のいずれか由来の新しく調製した細胞は、化合物がCYP3A阻害剤として作用する能力を決定するために使用できる。Watkinset al.,J.Clin.Invest.1985;80:1029−36の方法などの腸上皮細胞単離のための様々な方法を使用できる。Schmiedlin−Ren,P.etal.,Biochem.Pharmacol.1993;46:905−918に記載された培養細胞もまた使用できる。細胞中でのCYP3A代謝産物の産生は、CYP3A活性のミクロソームアッセイについての以下のセクションに記載したような高圧液体クロマトグラフィ(HPLC)法を用いて測定できる。
【0081】
肝細胞もしくは腸細胞由来のミクロソームは、CYP3Aアッセイのためにも使用できる。ミクロソームは、Kronbachet al.,Clin.Pharmacol.Ther1988;43:630−5で考察された従来型方法を用いて肝臓から調製できる。または、ミクロソームは、Watkinset al.,J.Clin.Invest.1987;80:1029−1037の方法を用いて単離された腸細胞から調製できる。腸上皮細胞由来のミクロソームは、Bonkovsky,H.L.etal.,Gastroenterology 1985;88:458−467に記載されたカルシウム沈降法を用いて調製することもできる。ミクロソームは、薬物と一緒にインキュベートし、代謝産物を時間の関数として監視できる。さらに、組織サンプル中のこれらの酵素のレベルは、ラジオイムノアッセイもしくはウエスタンブロットを用いて測定できる。
【0082】
単離されたミクロソームは、CYP3A薬物生体内変換の阻害を決定するために使用できる。一般に、薬物はCYP3Aの基質である。阻害剤の添加は、CYP3Aが薬物代謝を触媒する能力を減少させる。本アッセイにおいて同定される阻害剤は、CYP3A機能の阻害剤であり、基質触媒を減少させる。代謝産物の産生は、高圧液体クロマトグラフィーシステム(HPLC)を用いて監視することができ、保持時間に基づいて同定することができる。CYP3A活性は、Wrighton,etal.,Mol.Pharmacol.1985;28:312−321 and Nash,T.,Biochem.J.1953;55:416−421におけるように、ホルムアルデヒドの産生としてエリスロマイシンデメチラーゼ活性を熱量測定法により測定することによってアッセイすることもできる。
【0083】
<治療方法>
以下では、ウイルス感染症などの障害を治療する、予防する、または改善する方法を提供する。本発明の実施においては、連続的もしくは併用して、有効量の、例えば抗ウイルス性化合物、詳細には抗ウイルス性脂質含有化合物のプロドラッグおよびエンハンサーが投与される。本化合物は、任意の所望の方法で、例えば、経口、経直腸、経鼻、局所(経口腔および舌下を含む)、経膣、または非経口(皮下、筋肉内、皮下、静脈内、皮内、眼内、気管内、槽内、腹腔内、および硬膜外)投与することができる。本化合物は、同一もしくは相違する投与経路によって組み合わせて、または交互に投与することができる。
【0084】
所定の実施形態では、治療できるウイルス感染症には、インフルエンザ;ウシウイルス性下痢ウイルス(BVDV)、古典的ブタ熱ウイルス(CSFV、ブタコレラウイルスとしても既知である)、およびヒツジのボーダー病ウイルス(BDV)などのペスチウイルス;デング出血熱ウイルス(DHFもしくはDENV)、黄熱病ウイルス(YFV)、西ナイル熱ウイルス(WNV)、ショック症候群および日本脳炎ウイルスのようなフラビウイルス;B型およびC型肝炎ウイルス;サイトメガロウイルス(CMV);水疱−帯状疱疹ウイルス、単純ヘルペスウイルス1および2型、ヒトヘルペスウイルス6型、エプスタイン−バーウイルス、ヘルペス6型(HHV−6)および8型(HHV−8)などによって誘発されるヘルペス感染症;帯状ヘルペスもしくは水疱などの水疱−帯状疱疹ウイルス感染症;感染性の単核球症/腺を含むがそれに限定されないエプスタイン−バーウイルス感染症;SIV、HIV−1およびHIV−2;エボラウイルス;アデノウイルスおよびパピローマウイルスを含むがそれらに限定されないレトロウイルス感染症が含まれる。
【0085】
特定のフラビウイルスには、さらに制限なく:アブセットアローブ(Absettarov)、アルフイ(Alfuy)、アポイ(Apoi)、アロア(Aroa)、バガザ(Bagaza)、バンジ(Banzi)、ボウボウ(Bouboui)、ブスクアラ(Bussuquara)、カシパコール(Cacipacore)、カーレー島(CareyIsland)、ダカーコウモリ(Dakar bat)、デング(Dengue)1型、デング2型、デング3型、デング4型、エッジヒル(Edge Hill)、エンテベコウモリ(Entebbe bat)、ガジェット・ガリー(GadgetsGully)、ハンザローバ(Hanzalova)、ハイパア(Hypr)、イレウス、イスラエルシチメンチョウ髄膜脳炎、日本脳炎、ジュグラ(Jugra)、ジュチアパ(Jutiapa)、カダム(Kadam)、カルシー(Karshi)、ケドーゴー(Kedougou)、ココベラ、Koutango、クムリンゲ(Kumlinge)、クンジン(Kunjin)、キャサヌール森林病(KyasanurForest disease)、Langat、跳躍病、Meaban、Modoc、モンタナミオチス白質脳炎、マレー渓谷脳炎、Naranjal、Negishi、Ntaya、オムスク出血熱、プノンペンコウモリ、ポワッサン(Powassan)、リオ・ブラボー(RioBravo)、ロシオ(Rocio)、ロイヤルファーム、ロシア春夏脳炎、サボヤ(Saboya)、セントルイス脳炎、SalVieja、サン・ペリータ(San Perlita)、Saumarez Reef、セピック(Sepik)、Sokuluk、スポンドウェニ(Spondweni)、ストラトフォード(Stratford)、テムブス(Tembusu)、Tyuleniy、ウガンダS、ウスツ(Usutu)、ヴェッセルスブロン(Wesselsbron)、西ナイル、ヤウンデ(Yaounde)、黄熱、およびジカ(Zika)が含まれる。
【0086】
さらにまた別の実施形態では、抗ウイルス性化合物およびエンハンサーは、例えば大痘瘡(variola major)および小痘瘡(variola minor)、ワクシニア、伝染性軟属腫、オルフ(感染性嚢胞)痘瘡、ウシ痘、ラクダ痘、マウス痘、ウサギ痘、およびサル痘などのオルソポックスウイルスの結果として生じるウイルス感染症を治療または予防するために、有効量で投与される。
【0087】
1つの実施形態では、オルソポックス感染症などを治療するための治療有効用量は、抗ウイルス薬の約0.1ng/mL〜約50〜100μg/mLの血清中濃度を生じさせるはずである。また別の実施形態では、医薬組成物は、1日当たり体重1kg当たり約0.001mg〜約2,000mgの化合物の用量を提供しなければならない。医薬製剤単位は、例えば、単位製剤当たり約0.01mg、0.1mgもしくは1mg〜約500mg、1,000mgもしくは2,000mgの、および1つの実施形態では約10mg〜約500mgの有効成分または必須成分の組み合わせを提供するために調製される。
【0088】
エンハンサーの量は、抗ウイルス薬の生物学的利用能を増強するために当分野において既知の方法を用いて選択できる。所望の応答を提供する任意の量を使用できる。用量は、非限定的実施例では、1日当たり体重1kg当たり約0.001mg〜約2,000mgの化合物,例えば0.01〜500mg/kg、または例えば、0.1〜10mg/kgの範囲に及んでよい。
【0089】
<併用療法>
本明細書に提供する化合物および組成物は、組み合わせて、およびまたは、他の有効成分と組み合わせて使用することもできる。所定の実施形態では、本化合物は、また別の治療薬と組み合わせて、または連続的に投与できる。そのような他の治療薬には、ウイルス感染症に関連する1つまたは複数の症状の治療、予防、または改善のために既知である治療薬が含まれる。本明細書に提供した化合物と上記に挙げた化合物の1つまたは複数、および任意の1つまたは複数のさらなる薬理学的に活性の物質との任意の適切な組み合わせは、本開示の範囲内に含まれると見なされることを理解されたい。また別の実施形態では、本明細書に提供した化合物は、1つまたは複数の追加の有効成分に先行して、または引き続いて投与される。1つの実施形態では、本明細書に開示した2つまたはそれ以上の抗ウイルス薬が連続して、または組み合わせて投与される。
【0090】
<医薬組成物>
本明細書に提供した化合物を投与するために適合する医薬担体には、特定投与様式のために適合することが当業者に既知である任意の担体が含まれる。本化合物は、組成物中の単独医薬有効成分として調製できる、または他の有効成分と結合できる。
【0091】
本明細書に開示した化合物を含む組成物は、経口、経直腸、経鼻、局所(経口腔および舌下を含む)、経膣、または非経口(皮下、筋肉内、皮下、静脈内、皮内、眼内、気管内、槽内、腹腔内、および硬膜外)投与のために適合する可能性がある。
【0092】
本組成物は、便宜的にも単位製剤形で提示することができ、従来型の製薬技術によって調製できる。そのような技術には、本明細書に提供した1つまたは複数の組成物と1つまたは複数の医薬担体もしくは賦形剤を結合させる工程が含まれる。
【0093】
本化合物は、経口投与用の液剤、懸濁剤、錠剤、分散性錠剤、ピル剤、カプセル剤、散剤、徐放性製剤もしくはエリキシル剤などの適切な医薬調製物、または非経口投与用の無菌液剤もしくは懸濁剤中、ならびに経皮的パッチ製剤およびドライパウダー吸入剤中に調製することができる。1つの実施形態では、上述した化合物は当分野において周知の技術および手順を用いて医薬組成物中に調製される(例えば、AnselIntroduction to Pharmaceutical Dosage Forms,Fourth Edition 1985,126を参照されたい)。
【0094】
組成物中では、有効濃度の1つまたは複数の化合物もしくはそれらの医薬上許容される誘導体は、1つまたは複数の適切な医薬担体と混合できる。本化合物は、調製する前に、対応する塩、エステル、エノールエーテルもしくはエステル、アセタール、ケタール、オルトエステル、ヘミアセタール、ヘミケタール、酸、塩基、溶媒和物、水和物もしくはプロドラッグとして誘導体化できる。組成物中の本化合物の濃度は、投与すると、標的疾患もしくは障害の1つまたは複数の症状を治療する、予防する、または改善する量を送達するために有効である。1つの実施形態では、本組成物は単位用量投与のために調製される。組成物を調製するためには、治療される状態が緩和される、予防される、または1つまたは複数の症状が改善されるように有効濃度で選択された担体中に、化合物の重量分画が溶解される、懸濁される、分散される、さもなければ混合される。
【0095】
経口投与のために適合する組成物は、例えば、各々が規定量の1つまたは複数の組成物を含有する錠剤、カプレット、ピル剤、糖衣錠カプセル、もしくはカシェ剤などであるがそれらに限定されない個別単位として;散剤もしくは顆粒剤として;水性液もしくは非水性液中の液剤もしくは懸濁剤として;水中油型液体エマルジョンもしくは油中水型エマルジョンとして、もしくはボーラスなどとして提示できる。
【0096】
液体の医薬上投与可能な組成物は、例えば、水、食塩液、デキストロース水溶液、グリセロール、グリコール、エタノールなどの担体中で、それにより液剤もしくは懸濁剤を形成するために、上記に規定した活性化合物と任意選択的に追加する医薬アジュバントとを溶解させる、分散させる、またはさもなければ混合する工程によって調製できる。所望であれば、投与すべき医薬組成物は、湿潤剤、乳化剤、可溶化剤、pH緩衝剤、保存料、フレーバー剤などの少量の非毒性補助物質、例えば酢酸塩、クエン酸ナトリウム、シクロデキストリン誘導体、ソルビタンモノラウレート、酢酸トリエタノールアミンナトリウム、オレイン酸トリエタノールアミンおよび他のそのような物質を、さらに含有してもよい。そのような製剤形を調製する方法は既知である、または当業者には明白であろう;例えば、Remington’sPharmaceutical Sciences,Mack PublishingCompany,Easton,Pa.,15th Edition,1975を参照されたい。
【0097】
口腔内に局所投与するために適合する本発明の組成物には、例えば、フレーバー基剤中の成分、通常はスクロースおよびアカシアもしくはトラガカントを有するロゼンジ剤;ゼラチンおよびグリセリン、またはスクロースおよびアカシアなどの不活性基剤中に本発明の1つまたは複数の組成物を有するトローチ剤;および適切な液体担体中に投与された本発明の1つまたは複数の組成物を有するマウスウォッシュが含まれる。
【0098】
錠剤、ピル剤、カプセル剤、トローチ剤などは、以下の成分のうちの1つまたは複数、または類似の性質の化合物を含有することができる:結合剤;潤滑剤;希釈剤;流動促進剤;崩壊剤;着色剤;甘味剤;フレーバー剤;湿潤化剤;催吐性コーティング;およびフィルムコーティング。結合剤の例には、微結晶セルロース、トラガカントゴム、ブドウ糖溶液、アカシア粘液、ゼラチン液、糖蜜、ポリビニルピロリジン、ポビドン、クロスポビドン、スクロースおよびデンプン糊が含まれる。潤滑剤には、タルク、デンプン、ステアリン酸マグネシウムもしくはカルシウム、セキショウシおよびステアリン酸が含まれる。希釈剤には、例えば、ラクトース、スクロース、デンプン、カオリン、塩、マンニトールおよびリン酸二カルシウムが含まれる。流動促進剤には、コロイド状二酸化ケイ素が含まれるが、それには限定されない。崩壊剤には、クロスカルメロースナトリウム、グリコール酸デンプンナトリウム、アルギン酸、コーンスターチ、ジャガイモデンプン、ベントナイト、メチルセルロース、寒天およびカルボキシメチルセルロースが含まれる。着色剤には、例えば任意の承認かつ認証された水溶性FDおよびC色素、それらの混合物;およびアルミナ水和物に懸濁させた非水溶性FDおよびC色素が含まれる。甘味料には、スクロース、ラクトース、マンニトール、およびサッカリンなどの人工甘味料、ならびに任意の数のスプレードライフレーバーが含まれる。フレーバー剤には、果物などの植物から抽出された天然フレーバー、およびペパーミントおよびサリチル酸メチルなどであるがそれらに限定されない、快い感覚を生成する化合物の合成混合物が含まれる。湿潤剤には、プロピレングリコールモノステアレート、ソルビタンモノオレエート、ジエチレングリコールモノラウレートおよびポリオキシエチレンlauralエーテルが含まれる。催吐性コーティングには、脂肪酸、脂肪、ロウ、シェラック、アンモニア処理シェラックおよび酢酸フタル酸セルロースが含まれる。フィルムコーティングには、ヒドロキシエチルセルロース、カルボキシメチルセルロースナトリウム、ポリエチレングリコール4000および酢酸フタル酸セルロースが含まれる。
【0099】
皮膚への局所投与のために適合する組成物は、医薬上許容される担体中で投与される1つまたは複数の組成物を有する、軟膏剤、クリーム剤、ジェル剤、およびペースト剤として提示できる。
【0100】
直腸投与のための組成物は、例えばカカオ脂もしくはサリチル酸塩を含む適切な基剤を備える坐剤として提示できる。
【0101】
経鼻投与のために適切な組成物は、担体が固体の場合は、例えば鼻から吸う方法で投与される20〜500ミクロンの範囲内の粒径を有する粗粉末を含む(すなわち、鼻の近くに保持された粉末の容器から鼻腔を通した迅速な吸入によって)。担体が液体(例えば、鼻腔用スプレーもしくは点鼻剤)である場合は、1つまたは複数の組成物は水性溶液もしくは油性溶液中に混合できる、そして鼻腔内へ吸入もしくはスプレーすることができる。
【0102】
膣投与のために適切な組成物は、1つまたは複数の組成物および適切な担体を含有するペッサリー、タンポン、クリーム剤、ジェル剤、ペースト剤、フォーム剤もしくはスプレー製剤として提示できる。
【0103】
非経口投与のために適切な組成物には、酸化防止剤、緩衝剤、静菌剤、および製剤を所定のレシピエントの血液と等張性にさせる溶質を含有してもよい、水性および非水性無菌注射液;ならびに懸濁化剤および増粘剤を含んでもよい水性および非水性無菌懸濁剤が含まれる。本組成物は、単位用量もしくは多用量容器、例えば密封アンプルおよびバイアルに入れて提示することができ、使用直前に例えば注射用水などの無菌液体担体の添加だけを必要とするフリーズドライ(凍結乾燥)状態で保存することができる。即時注射溶液および懸濁剤は、無菌散剤、顆粒剤、および上述した種類の錠剤から調製できる。
【0104】
腸内もしくは非経口投与のために適合する医薬有機もしくは無機固体もしくは液体担体媒体を使用して組成物を作製することができる。ゼラチン、ラクトース、デンプン、ステアリン酸マグネシウム、タルク、植物性および動物性脂肪および油、ガム、ポリアルキレングリコール、水、またはその他の既知の担体は、すべてが担体媒体として適合する可能性がある。
【0105】
組成物は、1つまたは複数の医薬上許容される担体媒体および/または賦形剤と組み合わせて有効成分として使用できる。本明細書で使用する「医薬上許容される担体」には、望ましい特定の製剤形に適合するように、任意およびすべての担体、溶媒、希釈剤、または他の液体ビヒクル、分散もしくは懸濁補助剤、界面活性剤(surfaceactive agent)、等張化剤、増粘剤もしくは乳化剤、保存料、固体結合剤、潤滑剤、アジュバント、ビヒクル、送達系、崩壊剤、吸収剤、保存料、界面活性剤(surfactant)、着色剤、香味剤、または甘味剤などが含まれる。
【0106】
さらに、本組成物は、治療用組成物を生成するために、医薬上許容される賦形剤、および任意選択的に、例えば生分解性ポリマーなどの徐放性マトリックスと結合することができる。「医薬上許容される賦形剤」には、非毒性の固体、半固体もしくは液体充填剤、希釈剤、任意のタイプの被包化材料もしくは処方補助剤が含まれる。
【0107】
しかし、組成物の総1日量は、正確な医学的判断の範囲内で担当医師によって決定されることが理解されるであろう。任意の特定宿主のための特異的な治療有効量レベルは、例えば、治療される障害および障害の重症度;使用される特定組成物の活性;使用される特定組成物、患者の年齢、体重、全身状態、性別および食事;投与時期;投与経路;使用される特定化合物の排出速度;治療期間;使用される特定組成物と組み合わせて、もしくは同時に使用される薬物;ならびに、医学分野において周知である同様の要素を含む、様々な要素に依存する。例えば、所望の治療作用を達成するため、および所望の作用が達成されるまで用量を徐々に増加させるために必要とされる用量より低いレベルで組成物の用量を開始することは、明確に当分野の技術の範囲内である。
【0108】
組成物は、好ましくは投与の容易さおよび用量の一様性のために単位製剤形で調製される。本明細書で使用する「単位製剤形」は、治療対象の宿主にとって適切な組成物の物理的に別個の単位である。各用量は、単独で、または選択された医薬担体媒体と結び付けてのいずれかで所望の治療作用を生成するように計算された組成物の量を含有していなければならない。
【0109】
代表的な単位製剤は、投与される成分の1日量もしくは単位、1日分割用量、または適切なその分画を含有する製剤である。用量は、体重、年齢、表面積、代謝、組織分布、吸収速度および排泄速度などの宿主の要素に依存する。本明細書に記載した状態すべてのための代表的な全身性用量は、単一1日量もしくは分割1日量として、1日当たり0.01mg/kg〜2,000mg/kg(体重)の範囲に及ぶ用量である。局所投与のための典型的用量は、活性化合物の重量で0.001〜100%の範囲に及ぶ用量である。
【0110】
治療有効量レベルは、上述した多数の要素に依存する。さらに、組成物の用量を相当に低いレベルで開始すること、そして所望の作用が達成されるまで用量を増加させることは、明確に当分野の技術の範囲内である。
【0111】
本明細書に開示した化合物を含む組成物は、酵素的もしくは酸に基づく加水分解または溶解によって分解性である、通常はポリマーである材料から製造できる徐放性マトリックスと一緒に使用できる。身体内に挿入されると、マトリックスは酵素および体液によって作用する。徐放性マトリックスは、例えば、リポソーム、ポリラクチド(ポリ乳酸)、ポリグリコリド(グリコール酸のポリマー)、ポリラクチドコ−グリコリド(乳酸とグリコール酸のコポリマー)、ポリアンヒドリド、ポリ(オルト)エステル、ポリペプチド、ヒアルロン酸、コラーゲン、硫酸コンドロイチン、カルボン酸、脂肪酸、リン脂質、多糖、核酸、ポリアミノ酸、例えばフェニルアラニン、チロシン、イソロイシンなどのアミノ酸、ポリヌクレオチド、ポリビニルプロピレン、ポリビニルピロリドンおよびシリコーンなどの生体適合性材料から選択される。好ましい生体分解性マトリックスは、ポリラクチド、ポリグリコリド、もしくはポリラクチドコ−グリコリド(乳酸とグリコール酸のコポリマー)のいずれか1つのマトリックスである。
【0112】
本化合物は、リポソームの形態で投与することもできる。当分野において既知のように、リポソームは一般に、リン脂質もしくは他の脂質物質に由来する。リポソームは、水性媒体中に分散する単層状もしくは多層状の水和した液晶によって形成される。リポソームを形成できる任意の非毒性の、生理学的に許容できる、そして代謝可能な脂質を使用できる。リポソームは、本発明の1つまたは複数の組成物に加えて、安定剤、保存料、賦形剤などを含有することができる。脂質の例は、天然および合成の両方のリン脂質およびホスファチジルコリン(レシチン)である。リポソームを形成する方法は、当分野において既知である。
【0113】
本化合物は、吸入によるなどの、適用のためのエーロゾルとして調製できる。呼吸管へ投与するためのこれらの製剤は、ネブライザー用のエーロゾルもしくは液剤の形態であってよい、または単独もしくはラクトースなどの不活性担体と組み合わせて吸入用の微粉末としてよい。そのような場合には、製剤の粒子は、1つの実施形態では50ミクロン未満の、1つの実施形態では10ミクロン未満の径を有する。
【0114】
本明細書に開示した化合物を含む組成物は、他の組成物と組み合わせて、および/または上述した状態を治療するための方法において使用できる。
【0115】
<用語の定義>
本明細書で使用する用語「アルキル」は、他に特に規定しない限り、飽和直鎖状、分枝状、もしくは環状の、例えばC1−30もしくはC1−22の1級、2級、もしくは3級炭化水素が含まれ、詳細には、メチル、エチル、プロピル、イソプロピル、シクロプロピル、ブチル、イソブチル、secブチル、t−ブチル、ペンチル、シクロペンチル、イソペンチル、ネオペンチル、ヘキシル、イソヘキシル、シクロヘキシル、シクロヘキシルメチル、ヘプチル、シクロヘプチル、オクチル、シクロ−オクチル、ドデシル、トリデシル、ペンタデシル、イコシル、ヘミコシル、およびデコシルが含まれる。アルキル基は、例えば、参照することによって本明細書に組み込まれるGreene,etal.,Protective Groups in Organic Synthesis,John Wileyand Sons,SecondEdition,1991に教示されるように、当業者には既知のように、必要に応じて保護されていない、または保護されているいずれかである、ハロゲン(フルオロ、クロロ、ブロモ、もしくはヨード)、ヒドロキシル、アミノ、アルキルアミノ、アリールアミノ、アルコキシ、アリールオキシ、ニトロ、シアノ、スルホン酸、硫酸塩、ホスホン酸、ホスフェート、もしくはホスホネートで、任意選択的に置換されてよい。
【0116】
本明細書で使用する用語「低級アルキル」には、他に特に規定しない限り、任意選択的に置換される、C1〜C4飽和直鎖状、分枝状、もしくは適切であれば環状(例えば、シクロプロピル)アルキル基が含まれる。
【0117】
炭素原子の範囲について言及される場合は必ず、それは独立して個別にその範囲の各数を含む。非限定的例として、用語「C1−C10アルキル」は、例えば、C1−C10アルキルが直鎖状、分枝状および適切な場合は環状のC1、C2、C3、C4、C5、C6、C7、C8、C9およびC10アルキル官能基を含むように、独立して、各数の基を含むと見なされる。
【0118】
本明細書で使用する用語「保護(された)」には、他に特に規定しない限り、それ以上の反応を防止する、または他の目的で酸素、窒素、もしくはリン原子などの原子に加えられている基を含む。広範囲の酸素および窒素保護基は、有機合成の当業者に既知である。
【0119】
本明細書で使用する用語「ハロ」には、詳細にはクロロ、ブロモ、ヨード、およびフルオロが含まれる。
【0120】
用語「アルケニル」には、少なくとも1つの二重結合を備える、例えば、C2−100、もしくはC2−22の直鎖状、分枝状、もしくは環状炭化水素が含まれる。例には、ビニル、アリル、およびメチル−ビニルが含まれるが、それらに限定されない。アルケニル基は、アルキル基について上述した方法と同一方法で任意選択的に置換されてよい。
【0121】
用語「アルキニル」には、例えば、少なくとも1つの三重結合を備える、例えば、C2−100、もしくはC2−22の直鎖状もしくは分枝状炭化水素が含まれる。アルキニル基は、アルキル基について上述した方法と同一方法で任意選択的に置換されてよい。
【0122】
用語「アルコキシ」には、構造−O−アルキル基の成分が含まれる。
【0123】
用語「アシル」には、式R’C(O)(式中、R’は直鎖状、分枝状、もしくは環状の置換もしくは未置換アルキルもしくはアリールである)基が含まれる。
【0124】
本明細書で使用する用語「アリール」には、6〜14個の範囲内の炭素原子を有する芳香族基が含まれ、「置換アリール」は上記に規定した1つまたは複数の置換基をさらに有するアリール基を意味する。
【0125】
本明細書で使用する「ヘテロアリール」には、環状構造の一部として1つまたは複数のヘテロ原子(例、N、O、Sなど)を含有し、3〜14個の範囲内の炭素原子を有する芳香族基が含まれ、「置換ヘテロアリール」は、上記に規定した1つまたは複数の置換基をさらに有するヘテロアリール基を意味する。
【0126】
本明細書で使用する用語「結合」もしくは「原子価結合」には、電子対からなる原子間の連鎖を含む。
【0127】
本明細書で使用する用語「宿主」は、他に特に規定しない限り、哺乳動物(例、ネコ、イヌ、ウマ、マウス、サルなど)、ヒト、または治療を必要とする他の生物が含まれる。宿主は、例えばヒト、または制限なく、マカク属のサル、ヒヒ、ならびにチンパンジー、ゴリラ、およびオランウータンを含む霊長類;ヒツジ、ヤギ、シカ、および畜牛、例えば乳牛、去勢牛(steer)、雄牛(bull)、および去勢牛(ox)などを含む反芻動物;ブタ(pig)を含むブタ(swine);ならびにニワトリ、シチメンチョウ、アヒル、もしくはガチョウを含む家禽類を含む動物である。
【0128】
本明細書で使用する用語「医薬上許容される塩」には、他に特に規定しない限り、正確な医学的判断の範囲内で、過度の毒性、刺激、アレルギー反応などを伴わずに宿主の組織と接触させて使用するために適合する、そして合理的なベネフィット/リスク比と釣り合っていてそれらの所定の使用のために有効である塩が含まれる。塩は、組成物の1つまたは複数の化合物の最終単離および精製中にinsitu(インサイチュ)で、または遊離塩基官能基と適切な有機酸とを反応させる工程によって個別に調製できる。医薬上許容されない酸および塩基もまた、本明細書では、例えば当該の化合物の合成および/または精製において使用される。そのような塩の非限定的例は、(a)無機塩(例えば、塩酸、臭化水素酸、硫酸、リン酸、硝酸など)を用いて形成された、および酢酸、シュウ酸、酒石酸、コハク酸、アスコルビン酸、安息香酸、タンニン酸などの有機塩を用いて形成された酸付加塩;(b)亜鉛、カルシウム、マグネシウム、アルミニウム、ナトリウム、カリウム、銅、ニッケルなどの金属カチオンを用いて形成された塩基付加塩;(c)(a)および(b)の組み合わせである。さらにアミン塩もまた「医薬上許容される塩」として含まれる。
【0129】
本明細書で使用する用語「医薬上許容されるエステル」には、他に特に規定しない限り、正確な医学的判断の範囲内で、過度の毒性、刺激、アレルギー反応などを伴わずに宿主の組織と接触させて使用するために適合する、そして合理的なベネフィット/リスク比と釣り合っていてそれらの所定の使用のために有効である、1つまたは複数の化合物のエステルが含まれる。
【0130】
用語「医薬上許容されるプロドラッグ」には、活性化合物を形成するために宿主において代謝される、例えば加水分解もしくは酸化される化合物が含まれる。プロドラッグの典型的な例には、活性化合物の官能基成分上に生物学的に不安定な保護基を有する化合物が含まれる。プロドラッグには、活性化合物を生成するために酸化する、還元する、アミノ化する、脱アミノ化する、ヒドロキシル化する、脱ヒドロキシル化する、加水分解する、脱水縮合する(dehydrolyzed)、アルキル化する、脱アルキル化する、アシル化する、脱アシル化する、リン酸化する、脱リン酸化することのできる化合物が含まれる。
【0131】
本明細書で使用する用語「エナンチオマーが豊富である」は、1つのエナンチオマーが過剰に存在する、好ましくは95%以上まで存在する、およびより好ましくは100%を含めて98%以上まで存在するエナンチオマーの混合物である化合物を意味する。
【0132】
用語「有効量」には、本明細書に提供した疾患もしくは障害の1つまたは複数の症状の予防、治療、または改善のために必要とされる量が含まれる。
【0133】
本明細書に開示した化合物はキラル中心を含有する可能性があることを理解されたい。そのようなキラル中心は、(R)もしくは(S)立体配置いずれかの中心であってよい、またはそれらの混合物であってよい。そこで、本明細書に提供した化合物は、エナンチオマー的に純粋、または立体異性体的もしくはジアステレオマー的混合物であってよい。本明細書の化合物の開示は、好ましくは本明細書に記載した有用な特性を有する、任意のラセミ体の、光学活性形、多形体形、もしくは立体異性体形、またはそれらの混合物を含むことは理解されているが、当分野においては、光学活性形を調製する方法、および本明細書に記載した標準試験を用いて、または当分野において既知である他の類似の試験を用いて活性を決定する方法は、周知である。本化合物の光学異性体を入手するために使用できる方法の例には、以下が含まれる:
i)結晶の物理的分離−それにより個別エナンチオマーの肉眼的結晶が手動で分離される技術。この技術は、個別エナンチオマーの結晶が存在する場合、すなわち材料が集合体であり、結晶が視覚的に識別される場合に使用できる;
ii)同時の結晶化−それにより個別エナンチオマーがラセミ化合物の溶液から個別に結晶化される技術であり、後者が固体状態において集合体である場合にのみ可能である;
iii)酵素的分割−それによりエナンチオマーと酵素の相違する反応速度により、ラセミ化合物を部分的もしくは完全分離する技術;
iv)非対称性酵素的合成−それにより合成の少なくとも1つの工程が、所望のエナンチオマーのエナンチオマー的に純粋もしくはエナンチオマーが豊富な合成前駆物質を入手するために酵素反応を使用する合成技術;
v)非対称性化学合成−それにより所望のエナンチオマーが、キラル触媒もしくはキラル補助剤を用いて達成できる、生成物中において非対称性(すなわち、キラリティ)を生成する条件下でアキラル性前駆物質から合成される合成技術;
vi)ジアステレオマーの分離−それによりラセミ化合物が、個別エナンチオマーをジアステレオマーへ変換させるエナンチオマー的に純粋な試薬(キラル補助剤)と反応させられる技術。結果として生じるジアステレオマーは、次にクロマトグラフィによって、またはそれらの現在のより明確な構造的相違による結晶化によって分離され、後に所望のエナンチオマーを入手するためにキラル補助剤は除去される;
vii)一次および二次非対称性変換−それによりラセミ化合物からのジアステレオマーが釣り合って所望のエナンチオマーからジアステレオマーの溶液中で優勢を生じさせる、または所望のエナンチオマーからのジアステレオマーの優先的結晶化は、原理的には最終的に全材料が所望のエナンチオマーからの結晶ジアステレオマーへ変換されるように平衡を混乱させる技術。所望のエナンチオマーは、次にジアステレオマーから遊離させられる;
viii)速度論的分割−この技術は、反応速度的条件下でエナンチオマーとキラル、非ラセミ化合物試薬もしくは触媒との不同の反応速度による、ラセミ化合物(または、部分的に溶解した化合物のいっそうの分割)の部分もしくは完全分割の達成を意味する;
ix)非ラセミ前駆体からのエナンチオ特異的合成−それにより所望のエナンチオマーが非キラル出発物質から入手され、立体化学的完全性が合成の経過に渡って損なわれない、または極めてわずかしか損なわれない合成技術;
x)キラル液体クロマトグラフィ−それによりラセミ化合物のエナンチオマーが、固定相とのそれらの相違する相互作用によって液体移動相中で分離される技術。固定相は、キラル材料から作製されてよい、または移動相は相違する相互作用を誘発するために追加のキラル材料を含有してもよい;
xi)キラルガスクロマトグラフィ−それによりラセミ化合物が揮発化され、エナンチオマーが、固定非ラセミキラル吸着相を含有するカラムを用いて気体状移動相中でのそれらの相違する相互作用によって分離される技術;
xii)キラル溶媒を用いた抽出−それによりエナンチオマーが特定キラル溶媒中への1つのエナンチオマーの優先的溶解によって分離される技術;
xiii)キラル膜を横断する輸送−それによりラセミ化合物が薄膜障壁と接触させられる技術。障壁は、典型的には一方がラセミ化合物を含有する2つの混和性流体を分離し、濃度差もしくは圧力差などの駆動力が、薄膜障壁を横断する優先的輸送を引き起こす。分離は、ラセミ化合物の1つだけのエナンチオマーが通過することを可能にする膜の非ラセミ性のキラル性質の結果として発生する。
【0134】
<抗ウイルス性化合物の合成>
抗ウイルス性化合物、および詳細には抗ウイルス性脂質含有化合物は、その開示が参照することにより本明細書に組み込まれる米国特許第6,716,825号に記載されているように、当分野において利用できる方法を用いて合成できる。本明細書に提供する抗ウイルス性化合物および脂質含有プロドラッグは、一般にはスキームI〜IIに描出したような、様々な方法で調製できる。以下に記載する一般的ホスホン酸エステル化法は、単に具体的に示すためにのみ提供されており、決して限定するものと考えられてはならない。実際に、アルコールを用いたホスホン酸の直接縮合のために数種の方法が開発されている(例えば、R.C.Larock,ComprehensiveOrganic Transformations,VCH,NewYork,1989,p.966およびその中に言及された文献を参照されたい)。実施例に記載した化合物および中間体の単離および精製は、所望であれば、例えば、濾過、抽出、結晶化、フラッシュカラムクロマトグラフィ、薄層クロマトグラフィ、蒸留、またはこれらの方法の組み合わせなどの、任意の適切な分離もしくは精製方法によって実施できる。適切な分離および単離方法の特別の具体例は、以下の実施例に記載されている。その他の等価の分離および単離方法もまた、当然ながら使用できる。
【0135】
スキームIは、シドフォビル、環式シドフォビル、およびその他のホスホン酸塩のアルキルグリセロールもしくはアルキルプロパンジオールアナログの一般的合成法を例示する。2,3−イソプロピリデングリセロール(1)とジメチルホルムアミド中のNaHとの処理、次にアルキルメタンスルホネートとの反応は、アルキルエーテル(2)を産生させる。酢酸による処理、次にピリジン中の塩化トリチルとの反応によってイソプロピリデン基を除去すると、中間体(3)が産生する。中間体(3)のハロゲン化アルキルを用いたアルキル化は化合物(4)を生じさせる。80%酢酸水溶液を用いたトリチル基の除去によりO,O−ジアルキルグリセロール(5)が得られる。化合物(5)の臭素化、その後の環式シドフォビルもしくは他のホスホン酸塩含有ヌクレオチドのナトリウム塩との反応は、所望のホスホン酸塩付加物(7)を生じさせる。環状付加物の開環は、水酸化ナトリウム水溶液との反応によって遂行される。好ましいプロパンジオール種は、スキームIにおける化合物(5)に対して1−O−アルキルプロパン−3−オールを置換することによって合成できる。テノフォビルおよびアデフォビルアナログは、これらのヌクレオチドリン酸をスキームIの反応(f)においてcCDVと置換することによって合成できる。同様に、他のヌクレオチドホスホン酸塩もこの方法で形成できる。
【0136】
スキームI
【化11】
試薬:a)NaH、R1OSO2Me、DMF;b)80%酢酸水溶液;c)塩化トリチル、ピリジン;d)NaH、R2−−B4、DMF、e)CBr4、トリフェニルホスフィン、THF;f)環式シドフォビル(DCMC塩)、DMF;g)0.5NのNaOH
【0137】
スキームIIは、実施例として1−O−ヘキサデシルオキシプロピル−アデフォビルを用いたヌクレオチドホスホン酸塩を合成するための一般的方法を例示する。ヌクレオチドホスホン酸塩(5mmol)を無水ピリジン中に懸濁させ、アルコキシアルカノールもしくはアルキルグリセロール誘導体(6mmol)および1,3−ジシクロヘキシルカルボジイミド(DCC、10mmol)を加える。この混合液を還流するように加熱し、薄層クロマトグラフィによって監視して縮合反応が完了するまで強力に攪拌する。この混合液を次に冷却して濾過する。濾液を減圧下で濃縮し、残留物をシリカゲルに吸着させてフラッシュカラムクロマトグラフィ(およそ9:1のジクロロメタン/メタノールを用いた溶出)によって精製すると、対応するホスホン酸モノエステルが産生する。
【0138】
スキームII
【化12】
【0139】
本発明は、以下の非限定的な実施例から詳細に理解されるであろう。
【実施例1】
【0140】
(米国特許第6,716,825号に記載されているとおり)
アデフォビルヘキサデシルオキシプロピルおよび1−O−オクタデシル−sn−グリセリルエステルの合成
無水ピリジン中のアデフォビル(1.36g、5mmol)および3−ヘキサデシルオキシ−1−プロパノール(1.8g、6mmol)の混合液にDCC(2.06g、10mmol)を加えた。この混合液を還流するように加熱し、18時間攪拌し、その後に冷却して濾過した。濾液を減圧下で濃縮し、残留物をシリカゲルのショートカラムに装填した。9:1のジクロロメタン/メタノールを備えるカラムによる溶出は、白色粉末としてヘキサデシルオキシプロピル−アデフォビル(HDP−ADV)を産生した。
【0141】
無水ピリジン(30mL)中のアデフォビル(1.36g、5mmol)および1−O−オクタデシル−sn−グリセロール(2.08g、6mmol)の混合液にDCC(2.06g、10mmol)を加えた。この混合液を還流するように加熱し、一晩攪拌し、その後に冷却して濾過した。濾液を減圧下で濃縮し、残留物をシリカゲルのカラムに装填した。9:1のジクロロメタン/メタノールを備えるカラムによる溶出は、1−O−オクタデシル−sn−グリセリル−3−アデフォビルを産生した。
【実施例2】
【0142】
(米国特許第6,716,825号に記載されているとおり)
AZT−ホスホン酸ヘキサデシルオキシプロピルエステルの合成
AZT(3’−アジド−3’−5’−ジデオキシチミジン−5’−ホスホン酸)のホスホン酸塩アナログは、公表された方法:Hakimelahi,G.H.;Moosavi−Movahedi,A.A.;Sadeghi,M.M.;Tsay,S−C.;Hwu,J.R.Journalof MedicinalChemistry,1995 38,4648−4659を用いて合成した。
【0143】
AZTホスホン酸塩(1.65g、5mmol)を無水ピリジン(30mL)中に懸濁させ、次に3−ヘキサデシルオキシ−1−プロパノール(1.8g、6mmol)およびDCC(2.06g、10mmol)を加え、この混合液を還流するように加熱し、6時間攪拌し、次に冷却して濾過した。濾液を減圧下で濃縮し、残留物をシリカゲルのカラムに装填した。9:1のジクロロメタン/メタノールを備えるカラムによる溶出は、3’−アジド−3’−5’−ジデオキシチミジン−5’−ホスホン酸、ヘキサデシルオキシプロピルエステルを産生した。
【実施例3】
【0144】
(米国特許第6,716,825号に記載されているとおり)
環式シドフォビルのヘキサデシルオキシプロピルエステル、オクタデシルオキシプロピルエステル、オクタデシルオキシエチルエステルおよびヘキサデシルエステルの合成
N,N−DMF(25mL)中のシドフォビルの攪拌懸濁液(1.0g、3.17mmol)にN,N−ジシクロヘキシル−4−モルホリンカルボキサミジン(DCMC、1.0g、3.5mmol)を加えた。シドフォビルを溶解させるために、この混合液を一晩攪拌した。この透明溶液を次に添加漏斗に装填し、1,3−ジシクロヘキシルカルボジイミド(1.64g、7.9mmol)の攪拌した高温ピリジン溶液(25mL、60℃)へ緩徐に(30分間)加えた。この反応混合液を100℃で16時間攪拌し、次に室温へ冷却し、減圧下で溶媒を除去した。残留物をシリカゲルに吸着させ、勾配溶離(CH2Cl2+MeOH)を用いてフラッシュカラムクロマトグラフィによって精製した。最後にUV活性生成物を5:5:1のCH2Cl2/MeOH/H2Oにより溶出した。溶媒を蒸発させると860mgの白色固体が得られた。1Hおよび31PNMRスペクトルは、これが環式シドフォビルのDCMC塩であることを証明した(収率=44%)。
【0145】
無水DMF(35mL)中の環式シドフォビルの溶液(DCMC塩)(0.5g、0.8mmol)に1−ブロモ−3−ヘキサデシルオキシプロパン(1.45g、4mmol)を加え、この混合液を攪拌して80℃で6時間加熱した。この溶液を次にインバキュオで濃縮し、残留物をシリカゲルに吸着させ、勾配溶離(CH2Cl2+EtOH)を用いてフラッシュカラムクロマトグラフィによって精製した。アルキル化生成物は、90:10のCH2Cl2/EtOHで溶出した。純粋生成物を含有する分画を蒸発させると260mgのHDP−環式シドフォビルが産生した(収率55%)。
【0146】
無水DMF(35mL)中の環式シドフォビルの溶液(DCMC塩)(1.0g、3.7mmol)に1−ブロモ−3−オクタデシルオキシプロパン(2.82g、7.2mmol)を加え、この混合液を攪拌して85℃で5時間加熱した。この溶液を次にインバキュオで濃縮し、残留物をシリカゲルに吸着させ、勾配溶離(CH2Cl2+MeOH)を用いてフラッシュカラムクロマトグラフィによって精製した。アルキル化生成物は、9:1のCH2Cl2/MeOHで溶出した。純粋生成物を含有する分画を蒸発させると450mgのODP−環式シドフォビルが産生した。
【0147】
無水DMF(35mL)中のcCDVの溶液(DCMC塩)(1.0g、3.7mmol)に1−ブロモ−3−オクタデシルオキシエタン(3.0g、7.9mmol)を加え、この混合液を攪拌して80℃で4時間加熱した。この溶液を次にインバキュオで濃縮し、残留物をシリカゲルに吸着させ、勾配溶離(CH2Cl2+MeOH)を用いてフラッシュカラムクロマトグラフィによって精製した。アルキル化生成物は、9:1のCH2Cl2/MeOHで溶出した。純粋生成物を含有する分画を蒸発させると320mgのオクタデシルオキシエチル−cCDVが産生した。
【0148】
無水DMF(35mL)中の環式シドフォビルの溶液(DCMC塩)(0.5g、0.8mmol)に1−ブロモ−ヘキサデカン(1.2g、4mmol)を加え、この混合液を攪拌して80℃で6時間加熱した。この溶液を次にインバキュオで濃縮し、残留物をシリカゲルに吸着させ、勾配溶離(CH2Cl2+MeOH)を用いてフラッシュカラムクロマトグラフィによって精製した。アルキル化生成物は、9:1のCH2Cl2/MeOHで溶出した。純粋生成物を含有する分画を蒸発させると160mgのヘキサデシル−cCDVが産生した。
【実施例4】
【0149】
(米国特許第6,716,825号に記載されているとおり)
シドフォビルのヘキサデシルオキシプロピルエステル、オクタデシルオキシプロピルエステル、オクタデシルオキシエチルエステルおよびヘキサデシルエステルの合成
上記からのヘキサデシルオキシプロピル−環式CDVを0.5MのNaOH中に溶解させ、室温で1.5時間攪拌した。pHを約9に調整するために、50%酢酸水溶液を滴下した。沈降したHDP−CDVを濾過によって単離し、水ですすぎ洗いし、乾燥させ、次に再結晶化させると(3:1p−ジオキサン/水)HDP−CDVが得られた。同様に、オクタデシルオキシプロピルエステル、オクタデシルオキシエチルエステル、およびヘキサデシル−cCDVエステルを0.5MのNaOHにより加水分解して精製すると、対応するシドフォビルジエステルが得られた。
【実施例5】
【0150】
(米国特許第6,716,825号に記載されているとおり)
環式−ガンシクロビルホスホン酸ヘキサデシルオキシプロピルエステルの合成
ガンシクロビルの環式ホスホン酸塩アナログは、公表された方法を用いて調製した:(Reist,E.J.;Sturm,P.A.;Pong,R.Y.;Tanga,M.J.andSidwell,R.W.Synthesis of acyclonucleoside phosphonates for evaluation as antiviral agents,p.17−34.In J,C.Martin(ed.),Nucleotide Analoguesas AntiviralAgents,American Chemical Society,Washington,D.C.)。DMF中でDCMC塩へ変換させた後、cGCVホスホン酸塩は1−ブロモ−3−ヘキサデシルオキシプロパンにより処理し、この混合液を80℃で6時間加熱した。アルキル化生成物をフラッシュクロマトグラフィによって単離すると、HDP−環式−GCVホスホン酸塩が産生した。
【実施例6】
【0151】
(米国特許第6,716,825号に記載されているとおり)
ガンシクロビルホスホン酸ヘキサデシルオキシプロピルエステルの合成
上記からのHDP−環式GCVホスホン酸塩を0.5MのNaOH中に溶解させ、室温で攪拌してそれを非環式ジエステルに変換させた。この溶液を50%酢酸水溶液で中和すると生成物が沈降したので、これを3:1のp−ジオキサン/水中で再結晶化させた。
【実施例7】
【0152】
ヒトサイトメガロウイルス(HCMV)に対するホスホン酸塩ヌクレオチドアナログの抗ウイルス活性および選択性
HCMV抗ウイルスアッセイ:HCMV DNAについての抗ウイルスアッセイは、Dankner,W.M.,Scholl,D.,Stanat,S.C,Martin,M.,Souke,R.L.andSpector,S.A,J.Virol.Methods 21:293−298,1990によって報告されたDNAハイブリダイゼーションによって実施された。簡単に説明すれば、24ウェル培養皿内のサブコンフルエントなMRC−5細胞は、2%FBSおよび抗生物質を含有するイーグルの最小必須培地(E−MEM)中で様々な濃度の薬物を用いて24時間にわたり前処理した。培地を除去し、薬物を含有しないウェル中では5日間で3〜4+の細胞変性作用(CPE)を生じさせる希釈率でHCMV菌株を加えた。ウイルスを37℃で1’時間にわたり吸収させ、吸引させ、薬物希釈液と置換した。5日間のインキュベーション後、HCMVDNAはDiagnostic Hybrids社(オハイオ州アセンズ)製のCMV抗ウイルス感受性試験キットを用いて、核酸ハイブリダイゼーションによって3回ずつ定量した。培地を除去し、細胞は製造業者の取扱説明書にしたがって溶解させた。溶解液を吸収させた後、Hybriwix(商標)フィルタを60℃で一晩ハイブリダイズさせた。Hybriwix(商標)を73℃で30分間にわたり洗浄し、γ計数器で計数した。結果は、EC50(50%阻害濃度)として表示する。予備実験は、表2に示したように、シドフォビルおよびアデフォビルの1−O−ヘキサデシルプロパンジオール(HDP)誘導体上で実施した。
【表2】
【0153】
表2の結果が示すように、1−O−ヘキサデシルプロパンジオール−3−環式CDV(HDP−cCDV)は、CDVもしくは環式CDVより>900倍活性であった。より細胞毒性であるが、迅速に分割する細胞中でのHCMVに対する選択指数は、未誘導体化CDVについての1,900から>2,100に対して>59,000であった。
【0154】
インビトロでの試験化合物の細胞毒性:24ウェルプレート中のサブコンフルエントなヒト肺線維芽細胞(MRC−5、アメリカンタイプカルチャーコレクション、メリーランド州ロックビル)は、2%ウシ胎児血清および抗生物質を補給したE−MEM(GibcoBRL社、ニューヨーク州グランドアイランド)中に希釈した薬物で処理した。37℃での5日間のインキュベーション後、細胞単層を顕微鏡下で視覚的に検査し、細胞数の50%減少を誘発した薬物濃度を推定した。
【0155】
これらの実験から入手したデータは、表3に示されている。
【表3】
【実施例8】
【0156】
(米国特許第6,716,825号に記載されているとおり)
インビトロでの、HDP−cCDVがポックスウイルス複製に及ぼす作用
シドフォビル(CDV)、環式シドフォビル(cCDV)、および1−O−ヘキサデシルプロパンジオール−3−cCDV(HDP−cCDV)の活性は、細胞変性作用(CPE)における用量依存性減少を測定することによって、ワクシニアウイルスもしくはウシ痘ウイルスに感染したヒト包皮線維芽細胞中の抗ウイルス活性について試験した。ワクシニアおよびウシ痘ウイルスのEC50の予備試験値は、ヒト包皮線維芽(HFF)細胞中でのCPE減少アッセイによって決定した。このように入手したデータは、表4に示されている。
【表4】
【0157】
表4に示したように、HDP−cCDVはワクシニアウイルスに対して高度に活性であり、IC50値はcCDVおよびCDVについて各々0.97および1.8μMに対して0.11μMであった。ウシ痘感染細胞中では、HDP−cCDVは、極度に有効であり、IC50はcCDVおよびCDVについて各々0.72および2.1に対して<0.03μMであった。
【0158】
ポックスウイルスに対する抗ウイルス細胞変性作用(CPE)アッセイ:各薬物濃度では、ベロ細胞を含有する3つのウェルを1,000pfu/ウェルのオルソポックスウイルスで感染させ、他の3つのウェルは毒性決定のために未感染のままに残した。プレートを試験し、ウイルス感染後に染色すると、未処理細胞は4+CPEを示した。培地にニュートラルレッドを加え、CPEは540nmでのニュートラルレッド取込みによって評価した。50%阻害(EC50)および細胞毒性濃度(CC50)は、用量反応曲線のプロットから決定した。結果は表5に示されている。
【0159】
【表5】
【実施例9】
【0160】
(米国特許第6,716,825号に記載されているとおり)
1−O−ヘキサデシルプロパンジオール−3−アデフォビル(HDP−ADV)がインビボでHIV−1複製に及ぼす作用
本明細書に提供した化合物によるHIV−1複製の阻害における予備実験は、以下のとおりに実施した。薬物アッセイは、Larderet.al.,Antimicrobial Agents & Chemotherapy,34:436−441,1990によって以前に記載されたとおりに実施した。HIV−1LAI感染HT4−6C細胞は、指示したように薬物に曝露させ、37℃で3日間にわたりインキュベートした。細胞は、プラークを視認するためにクリスタルバイオレットで固定した。抗ウイルス活性は、薬物処理サンプル中で測定したコントロールプラーク(薬物なし)のパーセンテージとして評価した。EC50は、プラーク数を50%減少させるマイクロモル濃度である。アデフォビルの活性は、HIV−1感染HT4−6C細胞中でAZT(ジドブジン)および1−O−ヘキサデシルプロパンジオール−3−アデフォビル(HDP−ADV)と比較した。結果は表6に示されている。
【表6】
【0161】
アデフォビルは、中等度に活性であり、EC50は16μMであった。AZTは予想どおり高度に活性であったが(EC500.007μM)、HDP−ADVは3種の化合物中で最も活性であり、0.0001μMのEC50を備えており、アデフォビル自体より5対数を超えて活性であった。
【0162】
HIV−1抗ウイルスアッセイ:抗ウイルス性化合物がCD4を発現するHeLaHT4−6C細胞中でのHIV複製に及ぼす作用を、プラーク減少アッセイ(Larder,B.A.,Chesebro,B.andRichman,D.D.Antimirob.Agents Chemother.,34:436−441,1990)によって測定した。手短には、HT4−6C細胞の単層を、24ウェルマイクロ希釈プレート中の1ウェル当たり100〜300プラーク形成単位(PFU)のウイルスで感染させた。様々な濃度の薬物を培養培地である、上述した5%FBSおよび抗生物質を含有するダルベッコの修飾イーグル培地へ加えた。37℃で3日間後、単層はリン酸緩衝食塩液(PBS)中の10%ホルムアルデヒド溶液で固定し、0.25%のクリスタルバイオレットを用いて染色してウイルスプラークを視認した。抗ウイルス活性は、薬物処理サンプル中で測定したコントロールプラークのパーセンテージとして評価した。細胞毒性は、Hostetleret al.,AntiviralResearch,31:59−67,1996の方法によって評価した。結果は表7に示されている。
【0163】
【表7】
【実施例10】
【0164】
(米国特許第6,716,825号に記載されているとおり)
シドフォビルアナログがヘルペスウイルス複製に及ぼす作用
HSV−1抗ウイルスアッセイ:24ウェル培養皿中でのサブコンフルエントなMRC−5細胞は、培地を除去し、20〜24時間中に薬物無含有ウェル内で3〜4+CPEを生じさせる希釈率でHSV−1ウイルスを加えることによって接種した。これを37℃で1時間吸収させ、吸引し、2%FBSおよび抗生物質を含有するE−MEM中で様々な濃度の薬物と置換した。およそ24時間のインキュベーション後、HSVDNAはDiagnostic Hybrids社(オハイオ州アセンズ)製のHSV抗ウイルス感受性試験キットを用いて核酸ハイブリダイゼーションによって3回ずつ定量した。培地を除去し、細胞は製造業者の取扱説明書にしたがって溶解させた。溶解液を吸収させた後、Hybriwix(商標)フィルタを60℃で一晩ハイブリダイズさせた。Hybriwix(商標)を73℃で30分間にわたり洗浄し、γ計数器で計数した。細胞毒性は、実施例17に記載したように評価した。このようにして入手したEC50およびCC50値は、表8に示されている。
【0165】
【表8】
【実施例11】
【0166】
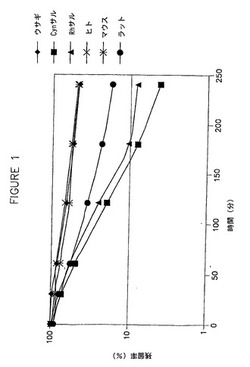
代謝安定性の評価
冷凍保存した一次肝細胞中のHDP−シドフォビル(1μM)の代謝安定性を、ウサギ、Cynサル、Rhサル、ヒト、マウスおよびラットにおいて試験した。結果は、HDP−シドフォビルの経時的な減少を示している図1に示されている。さらに、単回経口投与後のマウス、NZW系ウサギ、およびサルにおけるHDP−シドフォビル、HDP−シドフォビルから遊離されたシドフォビル、およびHDP−シドフォビルの不活性代謝産物である代謝産物M−8の血清中濃度を評価し、結果を図2、3および4に示した。図5は、HDP−シドフォビルの酸化によるM−8の形成について考えられる機序を示した図である。5mg/kgの[2−14C]HDP−シドフォビル(ヘキサデシルオキシプロピル−シドフォビル)の経口投与4時間後のマウスの定量的全身オートラジオグラフでは、この薬物は主として消化管、ならびに肺、心臓、肝臓、および腎臓を含む器官中に分布することが同定された。
【実施例12】
【0167】
サルにおける力価試験
16匹のカニクイザル(Cynomolgus Macaques)を各4匹の4つの治療群に分けて試験した。動物には、5×107PFU IVでサル痘ウイルスのZaire79菌株を接種した。動物には、図6にHD、LD、IDおよびプラセボとして示した、プラセボ、または相違する用量レベル(TD:35、30、20、0mg/kg)を投与した。図6は、薬物投与後の様々なタイプの組織中のサル痘力価のウイルス量を示す。
【技術分野】
【0001】
<関連出願の相互参照>
本出願は、その開示がこれにより全体として参照して組み込まれる、2005年4月8日に出願された米国出願番号第60/669,765号に対する優先権を主張する。
【0002】
<発明の分野>
本出願は、ウイルス感染症を治療するためのヌクレオシドもしくは非環式ヌクレオシドなどの脂質含有化合物の生物学的利用能、活性もしくは他の特性を増強するための方法を提供する。
【背景技術】
【0003】
薬物の生物学的利用能を改善する工程は、医学分野において確定されている目標である。薬理学においては、薬物がその治療目的のために十分な生物学的利用能を有することが重要である。経口組成物についての事象の順序には、様々な粘膜表面を通しての吸収、血流を介しての様々な組織への分布、肝臓およびその他の組織中での生体内変換、標的部位での作用、および尿もしくは胆汁中への薬物もしくは代謝産物の排出が含まれる。生物学的利用能は、循環器系に到達する前の薬物の消化管からの不良な吸収、肝初回通過作用、または分解によって減少させられることがある。
【0004】
プロドラッグは、身体内(インビボ)で活性化合物へ代謝されるように設計される。脂質プロドラッグは、消化管からの薬物の不良な吸収が限定因子である場合は、通常は経口による生物学的利用能を改善するように設計される。脂質プロドラッグは、それらの標的組織に対する薬物の選択性を改善することもできる。脂肪酸を含む脂質分子は、薬物とコンジュゲート化されることによって、そのコンジュゲートをコンジュゲート化されていない薬物より脂肪親和性にする。一般に、増加した脂肪親和性は、リンパ系内への薬物の腸取り込みを増強し、それにより脳内へのコンジュゲートの進入を増強し、さらに肝臓内でのコンジュゲートの初回通過代謝を回避するための機序として提案される。使用される脂質分子のタイプには、リン脂質および脂肪酸が含まれる。
【0005】
多数の薬物のリン脂質プロドラッグが開発されてきた。これらの化合物の一部は、親化合物に比して増強された活性もしくは生物学的利用能を有することが証明されている。
【0006】
薬物を含有する非ホスホネートに共有結合するアルキルグリセロールホスホネートの調製物および使用は、記載されている(米国特許第5,411,947号および米国特許出願第08/487,081号:それぞれ特許文献1及び2)。抗ウイルス性ヌクレオシド(米国特許第5,223,263号:特許文献3)またはホスホノ−カルボキシレート(米国特許第5,463,092号:特許文献4)に結合したアルキルグリセロールホスホネート残基を含むプロドラッグもまた記載されている。Hostetlerへの米国特許第6,716,825号(特許文献5)は、シドフォビルなどの抗ウイルス性化合物の所定のプロドラッグについて記載している。詳細には、抗ウイルス性化合物の所定の誘導体、および詳細にはシドフォビルのプロドラッグは、ウイルスの治療において親薬物より有効である。詳細には、1−O−ヘキサデシルオキシプロピル−シドフォビルは、詳細には痘瘡などのポックスウイルスの治療において、シドフォビルに比して増強された有効性を有する。
【0007】
薬物の分解は、肝臓もしくは腸内で発生し得る。消化管からのすべての血液は、全哺乳動物において身体のいずれかに進行する前に肝臓を通過する。その場所のために、経口投与された薬物の肝臓内形質転換は、小腸内での酵素活性の作用を上回ると考えられた薬物の生物学的利用能に実質的な「初回通過作用」を有する(Tam,Y.K.,「IndividualVariation inFirst−Pass Metabolism,」Clin.Pharmacokinetics1993,25,300−328:非特許文献1)。
【0008】
肝臓による活性薬物の排出は、2つの一般的経路、つまり薬物の生体内変換および胆汁中への薬物の排泄の一方もしくは両方によって発生する。生体内変換反応は、広範に定義された2つの期に分類される。第1生体内変換期はしばしば、肝臓内で多種多様かつ活性であり、多数の化学的に様々な薬物を変換させるシトクロムP450酵素によって触媒される反応を利用する。第2生体内変換期は、水溶性を増加させて腎臓を通しての排出を促進するために、グルタチオン、グルクロン酸もしくは硫酸塩などの親水基を加えることができる。
【0009】
肝細胞は、例えば門静脈(腸からの栄養分および薬物が豊富な血液)、肝動脈(心臓からの直接の酸素を豊富に含む血液)、肝静脈(駆出)、リンパ管(脂質およびリンパ球)、および胆管などの、多数のタイプの血液およびその他の流体輸送管と接触する。胆管は、上方の腸内に胆汁を排泄して消化を支援する胆嚢および総胆管内に合流する。胆汁は、疎水性薬物および薬物代謝産物を含む様々な排出生成物もまた含有する。
【0010】
一部の場合には、経口投与後の薬物の不良な生物学的利用能は、膜結合P糖タンパク質である多剤トランスポーターの活性の結果であり、これは細胞から生体異物を排出することによって薬物の細胞内蓄積を減少するために、エネルギー依存型輸送もしくは排出ポンプとして機能すると推測されてきた。P糖タンパク質排出ポンプは、所定の医薬化合物が小腸の粘膜細胞を横断すること、そしてこのために全身性循環中に吸収されることを防止すると考えられる。多数の既知である非細胞毒性薬理学的物質は、特にシクロスポリン、ベラパミル、タモキシフェン、キニジンおよびフェノチアジンを含むP糖タンパク質を阻害することが証明されている(Fisheret al.,Proc.Am.Soc.Clin.Oncol.,13:143,1994;Bartlettet al.,J.Clin.Onc.12:835−842,1994;Lumet al.,J.Clin.Onc.10:1635−42,1992:それぞれ非特許文献2〜4)。所定の抗癌薬の投与前、または同時の静脈内シクロスポリンの投与は、おそらく減少した身体クリアランスによってそれらの薬物のより高い血中濃度を生じさせ、実質的に低用量レベルで予想される毒性を示した。これらの観察所見は、シクロスポリンの併用投与はP−糖タンパク質のMDR作用を抑制し、治療薬のより大きな細胞内蓄積を可能にすることを示す傾向があった。P−糖タンパク質の臨床使用についての薬理学的意味に関する一般的考察は、Lumet al.,DrugResist.Clin.Onc.Hemat,9:319−336(1995)及びSchinkelet al.,Eur.J.Cancer,31A:1295−1298(1995)(それぞれ、非特許文献5及び6)に提供されている。
【0011】
薬物の分解、または生体内変換を最小限に抑えるために薬物とともに投与できる化合物は、バイオエンハンサーと呼ばれている。公開されたPCT特許出願WO第95/20980号(1995年8月10日に公開:特許文献6)では、Benetらは、シトクロムP4503A酵素の阻害剤またはP糖タンパク質−媒介性膜輸送の阻害剤を含む、バイオエンハンサーの使用を開示する。
【0012】
インビボでの薬物への代謝性もしくは他の望ましくない作用のために、身体内での脂質プロドラッグもしくは誘導体の有効性を最大化するという課題がある。これは、脂質を分解および合成するための身体の精巧かつ複雑な機序を前提にすると、脂質誘導体に関する特別な問題であった。
【0013】
そこで、治療薬への薬物代謝および相互作用の影響を最小限に抑えながら、改善された脂肪親和性化合物でウイルス感染症を治療するための方法および組成物に対する必要がある。
【0014】
詳細には、投与される治療薬に薬物の代謝が及ぼす影響を減少させる方法でオルソポックスウイルス感染症を治療する有効な方法に対する必要がある。
【0015】
さらに、抗ウイルス剤の生物学的利用能を改善するための方法に対する必要がある。
【先行技術文献】
【特許文献】
【0016】
【特許文献1】米国特許第5,411,947号
【特許文献2】および米国特許出願第08/487,081号
【特許文献3】米国特許第5,223,263号
【特許文献4】米国特許第5,463,092号
【特許文献5】米国特許第6,716,825号
【特許文献6】WO第95/20980号
【非特許文献】
【0017】
【非特許文献1】Tam,Y.K.,「Individual Variation in First−Pass Metabolism,」Clin.Pharmacokinetics1993,25,300−328
【非特許文献2】Fisher et al.,Proc.Am.Soc.Clin.Oncol.,13:143,1994
【非特許文献3】Bartlett et al.,J.Clin.Onc.12:835−842,1994
【非特許文献4】Lum et al.,J.Clin.Onc.10:1635−42,1992
【非特許文献5】Lum et al.,Drug Resist.Clin.Onc.Hemat,9:319−336(1995)
【非特許文献6】Schinkel et al.,Eur.J.Cancer,31A:1295−1298(1995)
【発明の概要】
【0018】
本出願は、プロドラッグ、またはその医薬上許容される塩もしくはエステル、詳細には脂質含有化合物、および特定実施形態では抗ウイルス性脂質含有ヌクレオシドの、生物学的利用能を改善するための方法および組成物を提供する。1つの実施形態では、脂質含有ヌクレオシドプロドラッグもしくは他の活性化合物は、脂質成分の代謝もしくは分解を防止もしくは最小限に抑えるバイオエンハンサーと組み合わせて投与される。本発明は、医薬物質の改善された生物学的利用能、血液中の医薬物質の増加した濃度、疾患および障害を治療するために必要とされる薬物の用量の減少、およびそれらの薬物に関連する副作用の減少を提供できる。所定の態様では、バイオエンハンサーは、シトクロムP450酵素の1つもしくはイミダゾールなどの、薬物生体内変換に関連する阻害剤もしくは基質である。1つの実施形態では、抗ウイルス性脂質含有化合物は、抗痘瘡薬などの抗オルソポックス薬である。他の実施形態では、抗ウイルス性化合物は、HIV、B型肝炎、C型肝炎もしくは他のウイルスに対して活性である。
【0019】
アルコキシルアルキルホスフェートエステルなどのシドフォビルの所定のプロドラッグが経口投与される場合は、肝臓および腸内でのP450酵素などの酵素はプロドラッグの生体内変換を誘発し、それによりその薬物の有効性を減少させることができる。任意の理論に限定されることなく、生体内変換は、例えば末端アルキル鎖のω酸化によって発生することがある。そのような生体内変換の不都合な影響を回避するために、抗ウイルス性化合物のプロドラッグ、詳細にはヌクレオシド、および詳細にはシドフォビルのプロドラッグの、生物学的利用能を改善するための方法が提供される。
【0020】
抗ウイルス性脂質含有化合物の生物学的利用能を増強できる様々なバイオエンハンサーを使用できる。化合物の投与後にインビボで発生できる化合物に脂質基の生体内変換を減少させるエンハンサーを使用できる。1つの実施形態では、生物学的利用能エンハンサーは、シトクロムP450酵素の1つ、および詳細には酵素のCYP3ファミリーなどの、薬物生体内変換に関連する酵素の阻害剤もしくは基質である。1つの実施形態では、エンハンサーは、例えば、ケトコナゾールもしくはトロレアンドマイシンなどのイミダゾール系(それらの一部は抗真菌活性を有する);エリスロマイシンなどのマクロライド系;ニフェジピンなどのカルシウムチャネルブロッカー;またはゲストデンなどのステロイド系である。任意選択的に、本化合物は、グレープフルーツ中で見いだされるナリンゲニンなどのシトクロムP4503A(CYP3A)の阻害剤である。
【0021】
1つの実施形態では、抗ウイルス性脂質含有化合物、またはそれらの塩、エステルもしくはプロドラッグ、および1つまたは複数の生物学的利用能増強化合物を含む、医薬上許容される組成物が提供される。本組成物は、オルソポックスウイルスなどのウイルスに感染した宿主の治療または予防のための有効量で、それを必要とする宿主へ投与できる。
【0022】
1つの実施形態では、ウイルス感染症、例えば、オルソポックス感染症を治療する方法であって、それを必要とする宿主へ有効量の抗ウイルス性脂質含有化合物、またはそれらの塩、エステルもしくはプロドラッグ、および1つまたは複数の生物学的利用能増強化合物を投与する工程を含む方法が提供される。本組成物は、任意選択的に医薬上許容される担体と組み合わせて、オルソポックスウイルスなどのウイルスに感染した宿主の治療または予防のための有効量で投与できる。本化合物もしくは組成物は、例えば経口もしくは非経口投与される。
【0023】
1つの実施形態では、ウイルス感染症、例えば、オルソポックスウイルス感染症を治療する方法であって、それを必要とする宿主へ有効量の脂質基を含有する抗ウイルス性ヌクレオシドのプロドラッグ、またはそれらの塩、エステルもしくはプロドラッグ、および1つまたは複数の生物学的利用能増強化合物を投与する工程を含み、1つの実施形態では生物学的利用能エンハンサーが脂質基の分解を減少させる物質である方法が提供される。本組成物は、任意選択的に医薬上許容される担体と組み合わせて、オルソポックスウイルスなどのウイルスに感染した宿主の治療または予防のための有効量で組み合わせて、または交互に投与できる。本化合物もしくは組成物は、例えば経口もしくは非経口投与される。
【0024】
1つの実施形態では、化合物が単独で投与された場合に比較して、抗ウイルス性脂質含有化合物の生物学的利用能を改善するために有効な量の生物学的利用能エンハンサーを含んでもよい、医薬組成物が提供される。また別の実施形態では、エンハンサーは、抗ウイルス性化合物がエンハンサーを含めずに投与された場合に比較して、抗ウイルス性化合物の生物学的利用能を改善するために有効な量で、抗ウイルス性脂質含有化合物と連続的に、または一緒に投与される。
【0025】
1つの実施形態では、抗ウイルス性化合物は、任意選択的に脂質に共有結合した、またはアルキルグリセロール、アルキルプロパンジオール、1−S−アルキルチオグリセロール、アルコキシアルカノールもしくはアルキルエタンジオールに結合した、シドフォビル、アデフォビル、環式シドフォビルもしくはテノフォビルである。エンハンサーは、例えば、ケトコナゾールもしくはトロレアンドマイシンなどのイミダゾール系抗真菌薬;エリスロマイシンなどのマクロライド系;ニフェジピンなどのカルシウムチャネルブロッカー;またはゲストデンなどのステロイド系である。任意選択的に、本化合物は、グレープフルーツ中で見いだされるナリンゲニンなどのシトクロムP4503A(CYP3A)の阻害剤である。
【0026】
1つの特定実施形態では、シドフォビル脂質プロドラッグおよび抗真菌薬などの生物学的利用能エンハンサーを含む組成物が提供されるが、この組成物はオルソポックス感染症などのウイルス感染症を治療するための有効量で投与できる。1つの実施形態では、ヌクレオシドプロドラッグは、シドフォビルのアルコキシアルカノールなどのシドフォビルのアルコキシアルキルエステルである。例えば、この化合物は、構造:
【化1】
を有してもよい。
【0027】
詳細には、本明細書に記載した組成物は、ウイルス、詳細にはオルソポックスウイルス、例えば大痘瘡および小痘瘡、ワクシニア、痘瘡、ウシ痘、ラクダ痘、マウス痘、ウサギ痘、およびサル痘などに感染した宿主を、予防もしくは治療するための方法において使用できる。
【図面の簡単な説明】
【0028】
【図1】ウサギ、Cynサル、Rhサル、ヒト、マウスおよびラットにおける、HDP−シドフォビルにおける経時的な減少を示した図である。
【図2】マウスにおけるHDP−シドフォビル、HDP−シドフォビルから遊離したシドフォビル、およびHDP−シドフォビルの不活性代謝産物である代謝産物M−8の血清中濃度を示した図である。
【図3】NZWウサギにおけるHDP−シドフォビル、HDP−シドフォビルから遊離したシドフォビル、およびHDP−シドフォビルの不活性代謝産物である代謝産物M−8の血清中濃度を示した図である。
【図4】サルにおけるHDP−シドフォビル、HDP−シドフォビルから遊離したシドフォビル、およびHDP−シドフォビルの不活性代謝産物である代謝産物M−8の血清中濃度を示した図である。
【図5】HDP−シドフォビルの酸化によるM−8の形成について考えられる機序を示した図である。
【図6】薬物投与後の様々なタイプの組織中のサル痘力価のウイルス量を示した図である。
【発明を実施するための形態】
【0029】
脂質含有プロドラッグの生物学的利用能を改善するための方法であって、プロドラッグが生物学的利用能エンハンサーと組み合わせて、または交互に投与される方法が提供される。さらに脂質含有プロドラッグおよび生物学的利用能エンハンサーを含む医薬上許容される組成物もまた提供される。1つの実施形態におけるプロドラッグは、脂質に結合したシドフォビルなどの抗ウイルス性脂質含有化合物である。
【0030】
1つの実施形態では、疾患もしくは障害を治療する方法であって、脂質含有ヌクレオシドもしくは他の活性化合物を、脂質成分の代謝もしくは分解を防止もしくは最小限に抑えるバイオエンハンサーと組み合わせて投与するステップを含む方法が提供される。所定の態様では、生物学的利用能エンハンサーは、例えば、薬物との酵素反応、例えばシトクロムP450酵素との反応のために発生し得る、抗ウイルス性化合物の生体内変換を減少させる化合物である。生物学的利用能エンハンサーは、例えば、抗真菌化合物または抗ウイルス性化合物の生体内変換と関係している酵素の活性を減少させるように作用する他の化合物である。抗ウイルス性脂質含有化合物は、例えば、天然痘(痘瘡)などのオルソポックス感染症を治療するための有効量で投与される。生物学的利用能エンハンサーは、例えば消化管もしくは肝臓内での酵素との反応のために発生し得る薬物の生体内変換を減少させるための有効量で、組成物中に存在する。
【0031】
さらに、オルソポックス感染症などのウイルス感染症を治療するための方法であって、有効量の生物学的利用能エンハンサーおよび抗ウイルス性脂質含有ヌクレオシドを投与する工程を含む方法もまた提供される。
【0032】
<プロドラッグ化合物>
本明細書に開示した方法および組成物においては、様々な化合物のプロドラッグを使用できる。詳細には、プロドラッグは、炭化水素鎖、例えば、C4−C30、またはC8−22炭化水素鎖を含むプロドラッグであってよい。本薬物は、様々な抗がん化合物もしくは抗ウイルス性化合物などの様々な薬物のいずれかであってよい。
【0033】
1つの実施形態では、本プロドラッグは、ホスホネートおよびホスフェートを含むヌクレオシドのプロドラッグである。特定の実施形態では、本プロドラッグは、抗オルソポックス剤などの抗ウイルス性脂質含有ヌクレオシドである。
【0034】
1つの実施形態におけるプロドラッグは、抗ウイルス性化合物のプロドラッグである。本プロドラッグは、例えば、アルキルグリセロール、アルキルプロパンジオール、1−S−アルキルチオグリセロール、アルコキシアルカノールもしくはアルキルエタンジオール、またC8−30アルキル、アルケニルもしくはアルキニルを含有する脂質などの脂質に共有結合した、シドフォビル、アデフォビル、環式シドフォビルもしくはテノフォビルである。本明細書で使用するように、化合物が「脂質に共有結合する」場合は、本化合物は、化合物と脂質基との間にリンカーを含んでもよい。脂質基は、例えば、C8−30アルキル、アルケニルもしくはアルキニルである。
【0035】
1つの実施形態では、抗ウイルス性プロドラッグは、例えば、脂質に共有結合したシドフォビルである。
【0036】
1つの実施形態では、抗ウイルス性プロドラッグは、構造:
【化2】
(式中、Rは、H;任意選択的に置換されたアルキル、例えば、C1−C30アルキル;アルケニル、例えば、C1−C30アルケニル;もしくはアルキニル、例えば、C1−C30アルキニル;アシル;モノホスフェートもしくはジホスフェート;アルキルグリセロール、アルキルプロパンジオール、1−S−アルキルチオグリセロール、アルコキシアルカノールもしくはアルキルエタンジオールである)を有する。1つの実施形態では、Rは、アルコキシアルカノールである。例えば、Rは、−(CH2)m−O−(CH2)n−CH3(式中、例えば、mは1〜5であり、nは1〜25である;またはmは2〜4であり、nは10〜25である)である。
【0037】
また別の実施形態では、抗ウイルス性プロドラッグ化合物は、以下の構造:
【化3】
を有する。
【0038】
1つの実施形態では、抗ウイルス性プロドラッグは、例えば、脂質基に共有結合したアデフォビルである。
【0039】
特定の実施形態では、抗ウイルス性プロドラッグは、以下の構造:
【化4】
(式中、Rは、H;任意選択的に置換されたアルキル、例えば、C1−C30アルキル;アルケニル、例えば、C1−C30アルケニル;アルキニル、例えば、C1−C30アルキニル;アシル;モノ−もしくはジ−ホスフェート;アルキルグリセロール、アルキルプロパンジオール、1−S−アルキルチオグリセロール、アルコキシアルカノールもしくはアルキルエタンジオールである)を有する。1つの実施形態では、Rは、アルコキシアルカノールである。例えば、Rは、−(CH2)m−O−(CH2)n−CH3(式中、例えば、mは1〜5であり、nは1〜25である;またはmは2〜4であり、nは10〜25である)である。
【0040】
1つの実施形態では、抗ウイルス性プロドラッグは、例えば、脂質に共有結合したテノフォビルである。
【0041】
特定の実施形態では、抗ウイルス性プロドラッグは、以下の構造:
【化5】
(式中、Rは、H;任意選択的に置換されたアルキル、例えば、C1−C30アルキル;アルケニル、例えば、C1−C30アルケニル;アルキニル、例えば、C1−C30アルキニル;アシル;モノホスフェートもしくはジホスフェート;アルキルグリセロール;アルキルプロパンジオール;1−S−アルキルチオグリセロール;アルコキシアルカノール;もしくはアルキルエタンジオールである)を有する。1つの実施形態では、Rは、アルコキシアルカノールである。Rは、例えば、−(CH2)m−O−(CH2)n−CH3(式中、例えば、mは1〜5であり、nは1〜25である;またはmは2〜4であり、nは10〜25である)である。
【0042】
1つの実施形態では、抗ウイルス性プロドラッグは、例えば、脂質に共有結合した環式シドフォビルである。
【0043】
また別の実施形態では、抗ウイルス性プロドラッグは、以下の式:
【化6】
(式中、Rは、H;任意選択的に置換されたアルキル、例えば、C1−C30アルキル;アルケニル、例えば、C1−C30アルケニル;アルキニル、例えば、C1−C30アルキニル;アシル;モノホスフェートもしくはジホスフェート;アルキルグリセロール、アルキルプロパンジオール、1−S−アルキルチオグリセロール、アルコキシアルカノールもしくはアルキルエタンジオールである)を有する。1つの実施形態では、Rは、アルコキシアルカノールである。Rは、例えば、−(CH2)m−O−(CH2)n−CH3(式中、例えば、mは1〜5であり、nは1〜25である;またはmは2〜4であり、nは10〜25である)である。
【0044】
また別の実施形態では、プロドラッグは、1−O−オクタデシルプロパンジオール−3−シドフォビル、1−O−オクタデシルエタンジオール−2−シドフォビル、1−O−ヘキサデシルプロパンジオール−3−環式シドフォビル、1−O−オクタデシルプロパンジオール−3−環式シドフォビル、1−O−オクタデシルエタンジオール−2−環式シドフォビル、1−O−ヘキサデシルプロパンジオール−3−アデフォビル、もしくは1−O−オクタデシル−sn−グリセロ−3−アデフォビルである。
【0045】
また別の実施形態では、プロドラッグは、ヘキサデシルオキシプロピルシドフォビル、オクタデシルオキシエチルシドフォビル、オレイルオキシプロピルシドフォビル、オクチルオキシプロピルシドフォビル、ドデシルオキシプロピルシドフォビル、オレイルオキシエチルシドフォビル、1−O−オクタデシル−2−O−ベンジル−グリセリルシドフォビル、テトラデシルオキシプロピルシドフォビル、エイコシルシドフォビル、ドコシルシドフォビル、ヘキサデシルシドフォビル、ヘキサデシルオキシプロピル環式シドフォビル、オクタデシルオキシエチル環式シドフォビル、オレイルオキシプロピル環式シドフォビル、オクチルオキシプロピル環式シドフォビル、ドデシルオキシプロピル環式シドフォビル、オレイルオキシエチル環式シドフォビル、1−O−オクタデシル−2−O−ベンジル−グリセリル環式シドフォビル、テトラデシルオキシプロピル環式シドフォビル、エイコシル環式シドフォビル、ドコシル環式シドフォビル、もしくはヘキサデシル環式シドフォビルである。
【0046】
その開示が本明細書に組み込まれる米国特許第6,716,825号に記載された化合物は、本明細書に提供した方法および組成物において使用できる。これらの化合物は、例えば米国特許第6,716,825号に記載された方法などの当分野において利用できる方法によって作製できる。
【0047】
抗ウイルス性化合物の広範囲の脂質誘導体、例えば抗ウイルス性ヌクレオシドは、本明細書に提供した方法および組成物において使用できる。1つの実施形態では、抗ウイルス性化合物はプロドラッグ形にあり、以下の構造:
【化7A】
【化7B】
(式中、W1、W2、およびW3は各々、独立して−O−、−S−、−SO−、−SO2、−O(C=O)−、−(C=O)O−、−NH(C=O)−、−(C=O)NH−もしくは−NH−である;および1つの実施形態では、各々は独立してO、S、もしくは−O(C=O)−である;
nは、0もしくは1である;mは、0もしくは1である;pは、0もしくは1である;
R1は、任意選択的に置換されたアルキル、アルケニルもしくはアルキニル、例えば、C1−30アルキル、アルケニル、もしくはアルキニルである;または1つの実施形態では、R1は任意選択的にC8−30アルキル、アルケニルもしくはアルキニルである、またはR1はC8−24アルキル、アルケニルもしくはアルキニル(例、C17、C18、C19、C20、C21、C22、C23、もしくはC24アルキル、アルケニルもしくはアルキニル)である;
R2およびR3は各々、独立して任意選択的に置換されたC1−25アルキル、アルケニルもしくはアルキニルである;または1つの実施形態では、任意選択的にR2およびR3は各々、独立してC1−5アルキル、アルケニルもしくはアルキニル(例、C1、C2、もしくはC3アルキル、アルケニルもしくはアルキニル;例えば、メチル、エチルもしくはプロピル)である;または別の実施形態では、CF3である;または別の実施形態では、アリール、例えば、ベンジルである;
tは、0もしくは1である;および
Dは、抗ウイルス薬、例えば抗ウイルス性環式もしくは非環式ヌクレオシドである)のうちの1つを有する。
【0048】
本明細書に開示した式V〜Xを含む式の1つの部分実施形態では、
式中、tは1である、および
【化8】
は、5’−O−リン酸ヌクレオシド、2’−O−リン酸ヌクレオシド、もしくは3’−O−リン酸ヌクレオシドを含むがそれらに限定されない、生物活性ホスフェート薬の残基である。
【0049】
本明細書に開示した、式V〜Xを含む式のまた別の部分実施形態では、
式中、tは0である、および
【化9】
は、シドフォビル、アデフォビル、テノフォビル、環式シドフォビル、HPMPA、PMEG、または生物活性ヌクレオシドもしくは非環式ヌクレオシドの任意の他のホスホン酸塩誘導体を含むがそれらに限定されない、生物活性ホスホン酸塩薬の残基である。
【0050】
また別の実施形態では、プロドラッグは、抗ウイルス性ヌクレオシドの1−O−アルキル−プロパンジオール−ホスフェートであるが、このとき抗ウイルス性ヌクレオシドは、(1)置換もしくは未置換プリンもしくはピリミジンを(a)リボース残基の非環式ヒドロキシル化フラグメント(例、ヒドロキシル化2−プロポキシメチルもしくはエトキシメチル)、(b)リボースもしくは2’−デオキシリボース、または(c)デオキシペントースのいずれかとともに有する。
【0051】
また別の実施形態では、プロドラッグは、抗ウイルス性ヌクレオシドの1−O−アルキル−エタンジオール−ホスフェートであるが、このとき抗ウイルス性ヌクレオシドは、(1)置換もしくは未置換プリンもしくはピリミジンを(a)リボース残基の非環式ヒドロキシル化フラグメント(例、ヒドロキシル化2−プロポキシメチルもしくはエトキシメチル)、(b)リボースもしくは2’−デオキシリボース、または(c)デオキシペントースのいずれかとともに有する。
【0052】
式V〜Xの1つの部分実施形態では、
W1、W2、およびW3は各々、独立して−O−、−S−、もしくは−O(CO)−である;
nは、0もしくは1である;mは、0もしくは1である;pは、0もしくは1である。
R1は、任意選択的に置換されたC18−24アルキルもしくはアルケニル(例、C18、C19、C20、C21、C22、C23、もしくはC24アルキル)である;
R2およびR3は各々、独立して任意選択的に置換されたC1−5アルキル、アルケニル、アルキニルまたはCF3;(例、C1、C2、もしくはC3アルキル;例、メチルもしくはエチル)またはシクロアルキルである;
tは、0もしくは1である;および
Dは、抗ウイルス性環式もしくは非環式ヌクレオシドである。
【0053】
また別の部分実施形態では、抗ウイルス性プロドラッグは、以下の構造:
【化10】
(式中、R1は、任意選択的に置換されたC8−24アルキル、例えばC18−24アルキル(例、C18、C19、C20、C21、C22、C23、もしくはC24アルキル)である;
R2およびR3は、独立してC1−5アルキル、ハロアルキル、アルケニル、アルキニル、もしくはシクロアルキル、例えばメチル、エチルもしくはCF3である;
tは、0もしくは1である;および
Dは、抗ウイルス薬、例えば抗ウイルス性環式もしくは非環式ヌクレオシドである)のうちの1つである。
【0054】
例えば上記の式V〜XVIに示したような、脂質基に結合することのできる代表的な抗ウイルス性ヌクレオシドは、ddA、ddI、ddG、L−FMAU、DXG、DAPD、L−dA、L−dI、L−(d)T、L−dC、L−dG、FTC、5−FC、1−(2’−デオキシ−2’−フルオロ−1−β−D−アラビノフラノシル)−5−ヨードシトシン(FIAC)もしくは1(2’−デオキシ−2’−フルオロ−1−β−D−アラビノフラノシル)−5−ヨードウラシル(FIAU)などである。
【0055】
例えば式V〜XVIに例示したように、脂質基を含むために任意選択的に共有的に誘導体化して使用できるその他のヌクレオシド(例えばポックスウイルス感染症を治療する際に有用である)には、8−メチルアデノシン、2−アミノ−7−[(1,3−ジヒドロキシ−2−プロポキシ)メチル]プリン(S2242)、Ara−A、PME−N6−(シクロプロピル)DAP、ホスホノメトキシエチルデオキシジアミノプリン(PMEADADP);PME−N6−(ジメチル)DAP、PME−N6−(トリフルオロエチル)DAP、PMEA−N6−(2−プロペニル)DAP、アデノシン−N(l)−オキシドのアナログ、1−(ベンジルオキシ)アデノシンのアナログ、IMPデヒドロゲナーゼ阻害剤(例、リバビリン、EICAR、チアゾフリン、およびセレナゾール)、SAHヒドロラーゼ阻害剤(例、5’−ノルアリステロマイシン、ネプラノシンAおよびC、炭素環式3−デアザ−アデノシン、DHCeA、C3DHCeA、6’−β−フルオロ−アリステロマイシン、5’−ノルアリステロマイシンおよびそのエピマー、3−デアザ−5’−ノルアリステロマイシン、6’−C−メチルネプラノシン、6’−ホモネプラノシン、2−フルオロネプラノシン、6’−ヨードアセチルAdo、および3−デアザネプラノシン)、OMPデカルボキシラーゼ阻害剤(例、ピラゾフリンおよび5’−デオキシピラゾフリン)CTPシンテターゼ阻害剤(例、シクロペンテニルシトシンおよびカルボジン)、チミジレートシンターゼ阻害剤(例、5−置換2’−デオキシウリジン)、および3’−フルオロ−3’−デオキシアデノシンが含まれる。
【0056】
その他のヌクレオシドには、3’−アジド−2’,3’−ジデオキシピリミジンヌクレオシド、例えば、AZT、AZT−P−AZT、AZT−P−ddA、AZT−P−ddI、AzddClU、AzddMeC、AzddMeCN4−OH、AzddMeC N4Me、AZT−P−CyE−ddA、AzddEtU(CS−85)、AzddU(CS−87)、AzddC(CS−91)、AzddFC、AzddBrU、およびAzddIU;3’−ハロピリミジンジデオキシヌクレオシドを含むクラス、例えば、3’−FddClU、3’−FddU、3’−FddT、3’−FddBrU、および3’−FddEtU;2’,3’−ジデヒドロ−2’,3’−ジデオキシヌクレオシド(D4ヌクレオシド)を含むクラス、例えば、D4T、D4C、D4MeC、およびD4A;2’,3’−未置換ジデオキシピリミジンヌクレオシドを含むクラス、例えば、5−F−ddC、ddCおよびddT;2’,3’−未置換ジデオキシプリンヌクレオシドを含むクラス、例えば、ddA、ddDAPR(ジアミノプリン)、ddG、ddI、およびddMeA(N6メチル);および糖−置換ジデオキシプリンヌクレオシドを含むクラス、例えば、3−N3ddDAPR、3−N3ddG、3−FddDAPR、3−FddG、3−FddaraA、および3−FddA(式中、Meはメチルであり、Etはエチルであり、そしてCyEtはシアノエチルである)が含まれる。
【0057】
その他の代表的なヌクレオシドには、ジデヒドロピリミジン、ならびにカルボビル、炭素環式2’,3’−ジデヒドログアノシン;デオキシグアノシン(AZG)およびピリミジンの3’−アジド誘導体、デオキシウリジン;デオキシチミジンおよびデオキシグアノシンの3’−フルオロ誘導体;2’,6’−ジアミノプリン、2’,3’−デオキシリボシドおよびその3’−フルオロおよび3’−アジド誘導体;2−クロロ−デオキシアデノシン;ガンシクロビル、アシクロビル、環式ガンシクロビル、9−(2−ホスホニルメトキシエチル)グアニン(PMEG)、9−(2−ホスホニルメトキシエチル)アデニン(PMEA)、ペンシクロビル、シドフォビル、アデフォビル、環式シドフォビル、9−(3−ヒドロキシ−2−ホスホニルメトキシプロピル)アデニン(HPMPA)、cHPMPA、8−アザ−HPMPA、HPMPG、(S)−9−(3−ヒドロキシ−2−ホスホニルメトキシプロピル)−2,6−ジアミノプリン((S)−HPMPDAP)、(S)−6−(3−ヒドロキシ−2−ホスホニルメトキシプロピル)オキシ−2,4−ジアミノピリミジン((S)−HPMPO−DAPy)、およびテノフォビルが含まれる。
【0058】
例えば、それらの各々がこれにより全体として参照して本明細書に組み込まれる以下の特許に開示されている化合物などの多数のホスホン酸塩化合物は、それらの薬理学的活性を改善するため、またはそれらの経口吸収を増加させるために脂質含有化合物に誘導体化することができる:米国特許第3,468,935号(Etidronate)、米国特許第4,327,039号(Pamidronate)、米国特許第4,705,651号(Alendronate)、米国特許第4,870,063号(Bisphosphonicacid derivatives)、米国特許第4,927,814号(Diphosphonates)、米国特許第5,043,437号(Phosphonatesof azidodideoxynucleosides)、米国特許第5,047,533号(Acyclicpurine phosphonatenucleotide analogs)、米国特許第5,142,051号(N−Phosphonylmethoxyalkylderivatives ofpyrimidine andpurine bases)、米国特許第5,183,815号(Boneacting agents)、米国特許第5,196,409号(Bisphosphonates)、米国特許第5,247,085号(Antiviralpurine compounds)、米国特許第5,300,671号(Gem−diphosphonicacids)、米国特許第5,300,687号(Trifluoromethylbenzylphosphonates)、米国特許第5,312,954号(Bis−and tetrakis−phosphonates)、米国特許第5,395,826号(Guanidinealkyl−1,1−bisphosphonicacid derivatives)、米国特許第5,428,181号(Bisphosphonatederivatives)、米国特許第5,442,101号(Methylenebisphosphonicacid derivatives)、米国特許第5,532,226号(Trifluoromethybenzylphosphonates)、米国特許第5,656,745号(Nucleotideanalogs)、米国特許第5,672,697号(Nucleoside−5’−methylenephosphonates)、米国特許第5,717,095号(Nucleotideanalogs)、米国特許第5,760,013号(Thymidylateanalogs)、米国特許第5,798,340号(Nucleotideanalogs)、米国特許第5,840,716号(Phosphonatenucleotide compounds)、米国特許第5,856,314号(Thio−substituted、nitrogen−containing、heterocyclicphosphonate compounds)、米国特許第5,885,973号(olpadronate)、米国特許第5,886,179号(Nucleotideanalogs)、米国特許第5,877,166号(Enantiomericallypure 2−aminopurinephosphonate nucleotideanalogs)、米国特許第5,922,695号(Antiviral phosphonomethoxy nucleotideanalogs)、米国特許第5,922,696号(Ethylenic and allenic phosphonate derivatives of purines)、米国特許第5,977,089号(Antiviralphosphonomethoxy nucleotide analogs)、米国特許第6,043,230号(Antiviralphosphonomethoxy nucleotide analogs)、米国特許第6,069,249号(Antiviralphosphonomethoxy nucleotide analogs);ベルギー国特許第672205号(Clodronate);欧州特許第753523号(Amino−substitutedbisphosphonic acids);欧州特許出願第186405号(geminal diphosphonates)などが含まれる。さらに、それらの各々は参照することによって全体として本明細書に組み込まれる以下の刊行物:J.Med.Chem.,2002,45:1918−1929;J.Med.Chem.,2003,46:5064−5073;Antimicrob.AgentsChemotherapy,2002,46:2185−2193に列挙した化合物は、それらの薬理学的活性を改善するため、またはそれらの経口吸収を増加させるために誘導体化できる。
【0059】
ヌクレオシドは、それらの各々は参照することによって全体として本明細書に組み込まれる以下の特許:米国特許第5,614,548号;米国特許第5,512,671号;米国特許第5,770,584号、米国特許第5,962,437号;米国特許第6,030,960号;米国特許第6,670,341号;米国特許第5,223,263号;米国特許第5,817,638号;米国特許第6,252,060号;米国特許第6,448,392号;米国特許第5,411,947号;米国特許第5,744,592号;米国特許第5,484,809号;米国特許第5,827,831号;米国特許第5,696,277号;米国特許第6,002,029号;米国特許第5,780,617号;米国特許第5,194,654号;米国特許第5,463,092号;米国特許第5,744,461号;米国特許第5,484,911号;WO第91/09602号;WO第91/05558号;米国特許第4,444,766号;米国特許第5,869,468号;米国特許第5,84,228号;米国特許出願第2002/0082242号;米国特許出願第2004/0161398号;米国特許出願第2004/0259845号;WO第98/38202号;米国特許第5,696,277号;米国特許第6,002,029号;米国特許第5,744,592号;米国特許第5,827,831号;米国特許第5,817,638号;ならびに米国特許第6,252,060号および米国特許第5,756,711号に記載した様々な脂肪親和性基を用いて誘導体化することができ、本明細書に提供した組成物および方法において使用できる。
【0060】
以下の物質:鎮痛薬;麻酔薬;食欲抑制薬;抗アドレナリン作動薬;抗アレルギー薬;抗狭心症薬;抗不安薬;抗関節炎薬;抗喘息薬;抗アテローム硬化症薬;抗細菌薬;抗凝固薬;抗痙攣薬;抗うつ薬;抗糖尿病薬;下痢止め薬;抗利尿薬;抗エストロゲン薬;抗線維素溶解薬;抗真菌薬;抗緑内障薬;抗ヒスタミン薬;抗感染薬;抗炎症薬;抗角化薬;抗マラリア薬;抗微生物薬;抗片頭痛薬;抗有糸分裂薬;抗カビ薬、制吐薬、抗腫瘍薬、抗好中球減少薬、antiobessionalagent;抗寄生虫薬;抗パーキンソン薬;抗蠕動薬、抗ニューモシスチス属薬;抗増殖薬;肝障害療法薬;向精神病薬;セロトニン阻害剤;セロトニン受容体アンタゴニスト;ステロイド;刺激剤;抑制剤;甲状腺ホルモン;甲状腺阻害剤;甲状腺ミメティック;トランキライザー;筋萎縮性側索硬化症の治療薬;脳虚血の治療薬;ページェット病の治療薬;不安定性狭心症の治療薬;尿酸排泄薬;血管収縮薬;血管拡張薬;傷薬;または創傷治療薬のプロドラッグを含む、他の化合物のプロドラッグもまた使用できる。
【0061】
使用できる以下の抗癌薬のプロドラッグには、例えば:アシビシン;アクラルビシン;塩酸アコダゾール;アクロニン;アドゼレシン;アドリアマイシン;アルデスロイキン;アリトレチノイン;アロプリノールナトリウム;アルトレタミン;アンボマイシン;酢酸アメタントロン;アミノグルテチミド;アムサクリン;アナストロゾール;アノナセオス・アセトゲニン;アントラマイシン;アシミシン;アスパラギナーゼ;アスペルリン;アザシチジン;アゼテパ;アゾトマイシン;バチマスタット;ベンゾデパ;ベキサロテン;ビカルタミド;塩酸ビサントレン;ビスナフィドジメシレート;ビゼレシン;硫酸ブレオマイシン;ブレキナールナトリウム;ブロピリミン;ブラタシン;ブスルファン;カベルゴリン;カクチノマイシン;カルステロン;カラセミド;カルベチマー;カルボプラチン;カルムスチン;塩酸カルビシン;カルゼレシン;セデフィンゴール;セレコキシブ;クロラムブシル;シロレマイシン;シスプラチン;クラドリビン;クリスナトールメシレート;シクロホスファミド;シタラビン;ダカルバジン;DACA(N−[2−(ピメチル−アミノ)エチル]アクリジン−4−カルボキサミド);ダクチノマイシン;塩酸ダウノルビシン;ダウノマイシン;デシタビン;デニロイキンディフチトクス;デキソルマプラチン;デザグアニン;デザグアニンメシレート;ジアジコン;ドセタキセル;ドキソルビシン;塩酸ドキソルビシン;ドロロキシフェン;クエン酸ドロロキシフェン;プロピオン酸ドロモスタノロン;デュアゾマイシン;エダトレキセート;塩酸エフロルニチン;エルサミトルシン;エンロプラチン;エンプロメート;エピプロピジン;塩酸エピルビシン;エルブロゾール;塩酸エソルビシン;エストラムスチン;リン酸エストラムスチンナトリウム;エタニダゾール;エチオダイズド油I131;エトポシド;リン酸エトポシド;エトプリン;塩酸ファドロゾール;ファザラビン;フェンレチニド;フロクスウリジン;リン酸フルダラビン;フルオロウラシル;5−FdUMP;フルロシタビン;フォスキドン;フォストリエシンナトリウム;FK−317;FK−973;FR−66979;FR−900482;ゲムシタビン;塩酸ゲムシタビン;ゲムツズマブオゾガマイシン;GoldAu 198;酢酸ゴセレリン;グアナコーン;ヒドロキシウレア;塩酸イダルビシン;イフォスファミド;イルモフォシン;インターフェロンα−2a;インターフェロンα−2b;インターフェロンα−n1;インターフェロンα−n3;インターフェロンβ−Ia;インターフェロンγ−Ib;イプロプラチン;塩酸イリノテカン;酢酸ランレオチド;レトロゾール;酢酸ロイプロリド;塩酸リアロゾール;ロメトレキソールナトリウム;ロムスチン;塩酸ロソキサントロン;マソプロコール;メイタンシン;塩酸メクロルエタミン;酢酸メゲストロール;酢酸メレンゲストロール;メルファラン;メノガリル;メルカプトプリン;メトトレキセート;メトトレキセートナトリウム;メトキサレン;メトプリン;メツルデパ;ミチンドミド;マイトカルシン;マイトクロミン;マイトギリン;マイトマルシン;マイトマイシン;マイトマイシンC;ミトスペル;ミトタン;塩酸ミトキサントロン;ミコフェノール酸;ノコダゾール;ノガラマイシン;オプレルベキン;オルマプラチン;オキシスラン;パクリタキセル;パミドロン酸二ナトリウム;ペグアスパルガーゼ;ペリオマイシン;ペンタムスチン;硫酸ペプロマイシン;ペルフォスファミド;ピポブロマン;ピポスルファン;塩酸ピロキサントロン;プリカマイシン;プロメスタン;ポルフィマーナトリウム;ポルフィロマイシン;プレドニムスチン;塩酸プロカルバジン;ピューロマイシン;塩酸ピューロマイシン;ピラゾフリン;リボプリン;リツキシマブ;ログレチミド;ロリニアスタチン;サフィンゴール;塩酸サフィンゴール;サマリウム/レキシドロナム;セムスチン;7−ヒドロキシスタウロスポリン;シムトラゼン;スパルフォセートナトリウム;スパルソマイシン;塩酸スピロゲルマニウム;スピロムスチン;スピロプラチン;スクアモシン;スクアモタシン;ストレプトニグリン;ストレプトゾシン;塩化ストロンチウムSr89;スロフェヌル;タリソマイシン;タキサン;タキソイド;テコガランナトリウム;テガフール;塩酸テロキサントロン;テモポルフィン;テニポシド;テロキシロン;テストラクトン;チアミプリン;チオグアニン;チオテパ;チミタック;チアゾフリン;チラパラザミン;トミュデックス(Tomudex);TOP−53;塩酸トポテカン;クエン酸トレミフェン;トラスツズマブ;酢酸トレストロン;リン酸トリシリビン;トリメトレキセート;グルクロン酸トリメトレキセート;トリプトレリン;塩酸トブロゾール;ウラシルマスタード;ウレデパ;バルルビシン;バプレオチド;ベルテポルフィン;ビンブラスチン;硫酸ビンブラスチン;ビンクリスチン;硫酸ビンクリスチン;ビンデシン;硫酸ビンデシン;硫酸ビネピジン;硫酸ビングリシネート;硫酸ビンロイロシン;酒石酸ビノレルビン;硫酸ビンロシジン;硫酸ビンゾリジン;ボロゾール;ゼニプラチン;ジノスタチン;塩酸ゾルビシン;2−クロロデオキシアデノシン;2’−デオキシホルマイシン;9−アミノカンプトテシン;ラルチトレキセド;N−プロパルジル−5,8−ジデアザ葉酸;2−クロロ−2’−アラビノ−フルオロ−2’−デオキシアデノシン;2−クロロ−2’−デオキシアデノシン;アニソマイシン;トリコスタチンA;hPRL−G129R;CEP−751;リノミド;サルファマスタード;ナイトロジェンマスタード(メクロルエタミン);シクロホスファミド;メルファラン;クロラムブシル;イフォスファミド;ブスルファン;N−メチル−N−ニトロソウレア(MNU);N,N’−ビス(2−クロロエチル)−N−ニトロソウレア(BCNU);N−(2−クロロエチル)−N’−シクロヘキシル−N−ニトロソウレア(CCNU);N−(2−クロロエチル)−N’−(トランス−4−メチルシクロヘキシル−N−ニトロソウレア(MeCCNU);N−(2−クロロエチル)−N’−(ジエチル)エチルホスホネート−N−ニトロソウレア(フォテムスチン);ストレプトゾトシン;ジアカルバジン(DTIC);ミトゾロマイド;テモゾロマイド;チオテパ;マイトマイシンC;AZQ;アドゼレシン;シスプラチン;カルボプラチン;オルマプラチン;オキサリプラチン;C1−973;DWA2114R;JM216;JM335;ビス(白金);トミュデックス;アザシチジン;シタラビン;ゲムシタビン;6−メルカプトプリン;6−チオグアニン;ヒポキサンチン;テニポシド;9−アミノカンプトテシン;トポテカン;CPT−11;ドキソルビシン;ダウノマイシン;エピルビシン;ダルビシン;ミトキサントロン;ロソキサントロン;ダクチノマイシン(アクチノマイシンD);アムサクリン;ピラゾロアクリジン;オール−トランスレチノール;14−ヒドロキシ−レトロ−レチノール;オール−トランスレチノイン酸;N−(4−ヒドロキシフェニル)レチンアミド;13−シスレチノイン酸;3−メチルTTNEB;9−シスレチノイン酸;フルダラビン(2−F−ara−AMP);および2−クロロデオキシアデノシン(2−Cda)などの抗腫瘍薬のプロドラッグが含まれる。
【0062】
<エンハンサー>
様々な生物学的利用能向上剤は、例えば抗ウイルス性脂質含有ヌクレオシドなどの脂質含有プロドラッグの生物学的利用能を増強するための有効量で投与されてよい、または医薬組成物中に存在していてよい。生物学的利用能エンハンサーは、薬物の分解、または生体内変換を最小限に抑えるために使用される。1つの実施形態では、生物学的利用能エンハンサーは、脂質プロドラッグの脂質成分の代謝もしくは分解を防止する、または最小限に抑える。
【0063】
「薬物の生物学的利用能」は、全身性で経時的に利用できる薬物の総量を意味する。薬物の生物学的利用能は、腸内での薬物生体内変換を阻害することによって、および/または腸内での腸上皮を横断する薬物の正味輸送を減少させる活性輸送系を阻害することによって、および/または肝臓内での薬物生体内変換を減少させることによって増加させることができる。プロドラッグの薬物の生物学的利用能の増加を引き起こす化合物は、本明細書ではバイオエンハンサーもしくは生物学的利用能エンハンサーと呼ぶ。
【0064】
所定の化合物がバイオエンハンサーに必要とされる阻害もしくは結合特性を有するかどうかを決定する任意選択的のバイオアッセイは、使用できる化合物を同定するために使用できる。
【0065】
1つの態様では、生物学的利用能エンハンサーは、シトクロムP450酵素の1つなどの、薬物生体内変換に関連する酵素の阻害剤もしくは基質である。1つの実施形態では、エンハンサーは、例えば、ケトコナゾールもしくはトロレアンドマイシンなどのイミダゾール系抗真菌薬;エリスロマイシンなどのマクロライド系;ニフェジピンなどのカルシウムチャネルブロッカー;またはゲストデンなどのステロイド系である。任意選択的に、本化合物は、グレープフルーツ中で見いだされるナリンゲニンなどのシトクロムP4503A(CYP3A)の阻害剤である。
【0066】
薬物生体内変換に関係している酵素もしくは受容体の活性に影響を及ぼす任意選択的の物質を使用できる。1つの実施形態では、その活性を減少させることのできる酵素はシトクロムP450酵素であり、詳細にはCYP3ファミリーの酵素である。
【0067】
シトクロムP450は、ヘムタンパク質のスーパーファミリーである。それらは、混合官能オキシダーゼ系の末端オキシダーゼを表している。シトクロムP450遺伝子スーパーファミリーは、シトクロムP450の進化的関係に基づいて命名される、少なくとも207個の遺伝子から構成される。この命名システムのためには、シトクロムP450遺伝子の全部の配列順序が比較され、少なくとも40%の同一性を共有するシトクロムP450が1ファミリー(CYPの後にローマもしくはアラビア数字が続く、例えばCYP3)であると規定され、さらにそれらの推定アミノ酸配列によって少なくとも55%関連するそれらの形態から構成されるサブファミリー(大文字によって表示される、例えば、CYP3A)に分割される。最後に、シトクロムP450の各個別形態についての遺伝子は、アラビア数字(例、CYP3A4)が指定される。
【0068】
3つのシトクロムP450遺伝子ファミリー(CYP1、CYP2およびCYP3)が大多数の薬物代謝の原因となると思われる。少なくとも15種のシトクロムP450が、ヒト肝において様々な程度まで特性付けられている。3型のシトクロムP450をコードするCYP3遺伝子ファミリーは、おそらくヒト薬物代謝において最も重要なファミリーである。ヒト3Aサブファミリーにおいては少なくとも5つの形態のシトクロムP450が見いだされており、これらの形態は極めて多数の構造的に多様な薬物の代謝の原因となっている。肝臓はシトクロムP450の多数のアイソフォームを含有しており、広範囲の物質を生体内変換させることができる。腸の内腔を内張する腸細胞はまた、重要なシトクロムP450活性を有しており、この活性は、薬物代謝において最も重要なアイソフォームである単一ファミリーのアイソザイム3Aによって決定付けられる。そこで、1つの実施形態では、薬物有効性は、例えば肝臓および/または腸内腔内でのCYP3a薬物生体内変換を減少させることによって増加させられる。
【0069】
詳細には、シトクロムP450酵素の活性は阻害することができる、またはシトクロムP450酵素は薬物および環境化合物によって不活性化することができる。これには、同一シトクロムP450の基質間の競合的阻害、活性部位以外のシトクロムP450上の部位に結合する物質による阻害、および物質の代謝中に形成される反応中間体によるシトクロムP450の自殺的不活性化が含まれる。例えば、阻害剤は、CYP3A薬物生体内変換の競合的、非競合的、競合不能、混合もしくは非可逆性阻害剤として作用することによって機能できる。阻害剤は、共有結合またはイオン性もしくは極性誘引のいずれかによって保護される薬物に結合することによって作用することもできる。
【0070】
CYP3Aの活性は、CYP3A基質からの反応生成物のCYP3A触媒性産生である。CYP3Aについての基質は、天然型基質または表1に列挙した成分などの他の成分である可能性がある。さらに、表1に列挙したCYP3A阻害剤の一部は、表の中に表示したように基質として同定される。阻害を受けるCYP3Aの触媒活性には、dealkyase、オキシダーゼ、およびヒドロラーゼ活性が含まれる。CYP3Aの様々な触媒活性に加えて、ある範囲の分子量(例えば、Komoriet al.,J.Biochem.1988,104:912−16に示されるように、51kD〜54kD)を備える様々な形態のCYP3Aが存在する。表1に列挙した化合物、詳細には阻害剤は、本明細書に記載した方法および組成物中のエンハンサーとして使用できる。
【0071】
【表1】
【0072】
特定の実施形態では、エンハンサーは、例えば、パロキセチン、フルオキセチン、セルトレリン、フルボキサミン、ネファゾドン、ベンラファキシン、シメチジン、フルフェナジン、ハロペリドール、ペルフェナジン、チオリダジン、ジルチアゼム、メトロニダゾール、troleandomyan、ジスルフィラム、オトギリソウ、およびオメプラゾールなどのCYA酵素の阻害剤である。
【0073】
エンハンサーには、例えばシクロスポリン、ベラパミル、タモキシフェン、キニジンおよびフェノチアジンなどのP糖タンパク質を阻害する化合物もまた含まれる。
【0074】
代表的なエンハンサーには、抗ウイルス性プロテアーゼ阻害薬、例えばインジナビル、ネルフィナビル、リトナビル、サキナビル;ならびに抗真菌薬、例えばフルコナゾール、イトラコナゾール、ケトコナゾール、およびミコナゾールが含まれる。
【0075】
その他のエンハンサーには、例えばクラリスロマイシン、エリスロマイシン、ノルトリプチリン、リグノカイン、およびanriodaroneなどのマクロライド系が含まれる。
【0076】
その他のエンハンサーには、17−エチニル−置換ステロイド系、例えば、ゲストデン、エチニル−エストラジオール、メトキサレン、およびレボノルゲストレルが含まれる。
【0077】
その他のエンハンサーには、例えばクエルセチンおよびナリンゲニンなどのフラボン系、および例えばエチニルエストラジオール、およびプレドニゾロンなどの他の化合物が含まれる。
【0078】
1つの実施形態では、生物学的利用能エンハンサーは、P糖タンパク質(P−gp)−媒介性膜輸送の阻害剤である。
【0079】
また別の実施形態では、生物学的利用能エンハンサーは、シクロスポリンA、活性ブロッカーであるGF120918(エラクリダル)、LY335989(zosuquidar)、バルスポダール(PSC833)、ビリコダル(VX710)、またはR101933である。
【0080】
当分野において利用できる活性エンハンサーについての試験を使用すると、適切な化合物を選択することができる。例えば、酵素阻害を測定できる。1つの実施形態では、肝細胞もしくは腸細胞の培養細胞または肝臓もしくは腸のいずれか由来の新しく調製した細胞は、化合物がCYP3A阻害剤として作用する能力を決定するために使用できる。Watkinset al.,J.Clin.Invest.1985;80:1029−36の方法などの腸上皮細胞単離のための様々な方法を使用できる。Schmiedlin−Ren,P.etal.,Biochem.Pharmacol.1993;46:905−918に記載された培養細胞もまた使用できる。細胞中でのCYP3A代謝産物の産生は、CYP3A活性のミクロソームアッセイについての以下のセクションに記載したような高圧液体クロマトグラフィ(HPLC)法を用いて測定できる。
【0081】
肝細胞もしくは腸細胞由来のミクロソームは、CYP3Aアッセイのためにも使用できる。ミクロソームは、Kronbachet al.,Clin.Pharmacol.Ther1988;43:630−5で考察された従来型方法を用いて肝臓から調製できる。または、ミクロソームは、Watkinset al.,J.Clin.Invest.1987;80:1029−1037の方法を用いて単離された腸細胞から調製できる。腸上皮細胞由来のミクロソームは、Bonkovsky,H.L.etal.,Gastroenterology 1985;88:458−467に記載されたカルシウム沈降法を用いて調製することもできる。ミクロソームは、薬物と一緒にインキュベートし、代謝産物を時間の関数として監視できる。さらに、組織サンプル中のこれらの酵素のレベルは、ラジオイムノアッセイもしくはウエスタンブロットを用いて測定できる。
【0082】
単離されたミクロソームは、CYP3A薬物生体内変換の阻害を決定するために使用できる。一般に、薬物はCYP3Aの基質である。阻害剤の添加は、CYP3Aが薬物代謝を触媒する能力を減少させる。本アッセイにおいて同定される阻害剤は、CYP3A機能の阻害剤であり、基質触媒を減少させる。代謝産物の産生は、高圧液体クロマトグラフィーシステム(HPLC)を用いて監視することができ、保持時間に基づいて同定することができる。CYP3A活性は、Wrighton,etal.,Mol.Pharmacol.1985;28:312−321 and Nash,T.,Biochem.J.1953;55:416−421におけるように、ホルムアルデヒドの産生としてエリスロマイシンデメチラーゼ活性を熱量測定法により測定することによってアッセイすることもできる。
【0083】
<治療方法>
以下では、ウイルス感染症などの障害を治療する、予防する、または改善する方法を提供する。本発明の実施においては、連続的もしくは併用して、有効量の、例えば抗ウイルス性化合物、詳細には抗ウイルス性脂質含有化合物のプロドラッグおよびエンハンサーが投与される。本化合物は、任意の所望の方法で、例えば、経口、経直腸、経鼻、局所(経口腔および舌下を含む)、経膣、または非経口(皮下、筋肉内、皮下、静脈内、皮内、眼内、気管内、槽内、腹腔内、および硬膜外)投与することができる。本化合物は、同一もしくは相違する投与経路によって組み合わせて、または交互に投与することができる。
【0084】
所定の実施形態では、治療できるウイルス感染症には、インフルエンザ;ウシウイルス性下痢ウイルス(BVDV)、古典的ブタ熱ウイルス(CSFV、ブタコレラウイルスとしても既知である)、およびヒツジのボーダー病ウイルス(BDV)などのペスチウイルス;デング出血熱ウイルス(DHFもしくはDENV)、黄熱病ウイルス(YFV)、西ナイル熱ウイルス(WNV)、ショック症候群および日本脳炎ウイルスのようなフラビウイルス;B型およびC型肝炎ウイルス;サイトメガロウイルス(CMV);水疱−帯状疱疹ウイルス、単純ヘルペスウイルス1および2型、ヒトヘルペスウイルス6型、エプスタイン−バーウイルス、ヘルペス6型(HHV−6)および8型(HHV−8)などによって誘発されるヘルペス感染症;帯状ヘルペスもしくは水疱などの水疱−帯状疱疹ウイルス感染症;感染性の単核球症/腺を含むがそれに限定されないエプスタイン−バーウイルス感染症;SIV、HIV−1およびHIV−2;エボラウイルス;アデノウイルスおよびパピローマウイルスを含むがそれらに限定されないレトロウイルス感染症が含まれる。
【0085】
特定のフラビウイルスには、さらに制限なく:アブセットアローブ(Absettarov)、アルフイ(Alfuy)、アポイ(Apoi)、アロア(Aroa)、バガザ(Bagaza)、バンジ(Banzi)、ボウボウ(Bouboui)、ブスクアラ(Bussuquara)、カシパコール(Cacipacore)、カーレー島(CareyIsland)、ダカーコウモリ(Dakar bat)、デング(Dengue)1型、デング2型、デング3型、デング4型、エッジヒル(Edge Hill)、エンテベコウモリ(Entebbe bat)、ガジェット・ガリー(GadgetsGully)、ハンザローバ(Hanzalova)、ハイパア(Hypr)、イレウス、イスラエルシチメンチョウ髄膜脳炎、日本脳炎、ジュグラ(Jugra)、ジュチアパ(Jutiapa)、カダム(Kadam)、カルシー(Karshi)、ケドーゴー(Kedougou)、ココベラ、Koutango、クムリンゲ(Kumlinge)、クンジン(Kunjin)、キャサヌール森林病(KyasanurForest disease)、Langat、跳躍病、Meaban、Modoc、モンタナミオチス白質脳炎、マレー渓谷脳炎、Naranjal、Negishi、Ntaya、オムスク出血熱、プノンペンコウモリ、ポワッサン(Powassan)、リオ・ブラボー(RioBravo)、ロシオ(Rocio)、ロイヤルファーム、ロシア春夏脳炎、サボヤ(Saboya)、セントルイス脳炎、SalVieja、サン・ペリータ(San Perlita)、Saumarez Reef、セピック(Sepik)、Sokuluk、スポンドウェニ(Spondweni)、ストラトフォード(Stratford)、テムブス(Tembusu)、Tyuleniy、ウガンダS、ウスツ(Usutu)、ヴェッセルスブロン(Wesselsbron)、西ナイル、ヤウンデ(Yaounde)、黄熱、およびジカ(Zika)が含まれる。
【0086】
さらにまた別の実施形態では、抗ウイルス性化合物およびエンハンサーは、例えば大痘瘡(variola major)および小痘瘡(variola minor)、ワクシニア、伝染性軟属腫、オルフ(感染性嚢胞)痘瘡、ウシ痘、ラクダ痘、マウス痘、ウサギ痘、およびサル痘などのオルソポックスウイルスの結果として生じるウイルス感染症を治療または予防するために、有効量で投与される。
【0087】
1つの実施形態では、オルソポックス感染症などを治療するための治療有効用量は、抗ウイルス薬の約0.1ng/mL〜約50〜100μg/mLの血清中濃度を生じさせるはずである。また別の実施形態では、医薬組成物は、1日当たり体重1kg当たり約0.001mg〜約2,000mgの化合物の用量を提供しなければならない。医薬製剤単位は、例えば、単位製剤当たり約0.01mg、0.1mgもしくは1mg〜約500mg、1,000mgもしくは2,000mgの、および1つの実施形態では約10mg〜約500mgの有効成分または必須成分の組み合わせを提供するために調製される。
【0088】
エンハンサーの量は、抗ウイルス薬の生物学的利用能を増強するために当分野において既知の方法を用いて選択できる。所望の応答を提供する任意の量を使用できる。用量は、非限定的実施例では、1日当たり体重1kg当たり約0.001mg〜約2,000mgの化合物,例えば0.01〜500mg/kg、または例えば、0.1〜10mg/kgの範囲に及んでよい。
【0089】
<併用療法>
本明細書に提供する化合物および組成物は、組み合わせて、およびまたは、他の有効成分と組み合わせて使用することもできる。所定の実施形態では、本化合物は、また別の治療薬と組み合わせて、または連続的に投与できる。そのような他の治療薬には、ウイルス感染症に関連する1つまたは複数の症状の治療、予防、または改善のために既知である治療薬が含まれる。本明細書に提供した化合物と上記に挙げた化合物の1つまたは複数、および任意の1つまたは複数のさらなる薬理学的に活性の物質との任意の適切な組み合わせは、本開示の範囲内に含まれると見なされることを理解されたい。また別の実施形態では、本明細書に提供した化合物は、1つまたは複数の追加の有効成分に先行して、または引き続いて投与される。1つの実施形態では、本明細書に開示した2つまたはそれ以上の抗ウイルス薬が連続して、または組み合わせて投与される。
【0090】
<医薬組成物>
本明細書に提供した化合物を投与するために適合する医薬担体には、特定投与様式のために適合することが当業者に既知である任意の担体が含まれる。本化合物は、組成物中の単独医薬有効成分として調製できる、または他の有効成分と結合できる。
【0091】
本明細書に開示した化合物を含む組成物は、経口、経直腸、経鼻、局所(経口腔および舌下を含む)、経膣、または非経口(皮下、筋肉内、皮下、静脈内、皮内、眼内、気管内、槽内、腹腔内、および硬膜外)投与のために適合する可能性がある。
【0092】
本組成物は、便宜的にも単位製剤形で提示することができ、従来型の製薬技術によって調製できる。そのような技術には、本明細書に提供した1つまたは複数の組成物と1つまたは複数の医薬担体もしくは賦形剤を結合させる工程が含まれる。
【0093】
本化合物は、経口投与用の液剤、懸濁剤、錠剤、分散性錠剤、ピル剤、カプセル剤、散剤、徐放性製剤もしくはエリキシル剤などの適切な医薬調製物、または非経口投与用の無菌液剤もしくは懸濁剤中、ならびに経皮的パッチ製剤およびドライパウダー吸入剤中に調製することができる。1つの実施形態では、上述した化合物は当分野において周知の技術および手順を用いて医薬組成物中に調製される(例えば、AnselIntroduction to Pharmaceutical Dosage Forms,Fourth Edition 1985,126を参照されたい)。
【0094】
組成物中では、有効濃度の1つまたは複数の化合物もしくはそれらの医薬上許容される誘導体は、1つまたは複数の適切な医薬担体と混合できる。本化合物は、調製する前に、対応する塩、エステル、エノールエーテルもしくはエステル、アセタール、ケタール、オルトエステル、ヘミアセタール、ヘミケタール、酸、塩基、溶媒和物、水和物もしくはプロドラッグとして誘導体化できる。組成物中の本化合物の濃度は、投与すると、標的疾患もしくは障害の1つまたは複数の症状を治療する、予防する、または改善する量を送達するために有効である。1つの実施形態では、本組成物は単位用量投与のために調製される。組成物を調製するためには、治療される状態が緩和される、予防される、または1つまたは複数の症状が改善されるように有効濃度で選択された担体中に、化合物の重量分画が溶解される、懸濁される、分散される、さもなければ混合される。
【0095】
経口投与のために適合する組成物は、例えば、各々が規定量の1つまたは複数の組成物を含有する錠剤、カプレット、ピル剤、糖衣錠カプセル、もしくはカシェ剤などであるがそれらに限定されない個別単位として;散剤もしくは顆粒剤として;水性液もしくは非水性液中の液剤もしくは懸濁剤として;水中油型液体エマルジョンもしくは油中水型エマルジョンとして、もしくはボーラスなどとして提示できる。
【0096】
液体の医薬上投与可能な組成物は、例えば、水、食塩液、デキストロース水溶液、グリセロール、グリコール、エタノールなどの担体中で、それにより液剤もしくは懸濁剤を形成するために、上記に規定した活性化合物と任意選択的に追加する医薬アジュバントとを溶解させる、分散させる、またはさもなければ混合する工程によって調製できる。所望であれば、投与すべき医薬組成物は、湿潤剤、乳化剤、可溶化剤、pH緩衝剤、保存料、フレーバー剤などの少量の非毒性補助物質、例えば酢酸塩、クエン酸ナトリウム、シクロデキストリン誘導体、ソルビタンモノラウレート、酢酸トリエタノールアミンナトリウム、オレイン酸トリエタノールアミンおよび他のそのような物質を、さらに含有してもよい。そのような製剤形を調製する方法は既知である、または当業者には明白であろう;例えば、Remington’sPharmaceutical Sciences,Mack PublishingCompany,Easton,Pa.,15th Edition,1975を参照されたい。
【0097】
口腔内に局所投与するために適合する本発明の組成物には、例えば、フレーバー基剤中の成分、通常はスクロースおよびアカシアもしくはトラガカントを有するロゼンジ剤;ゼラチンおよびグリセリン、またはスクロースおよびアカシアなどの不活性基剤中に本発明の1つまたは複数の組成物を有するトローチ剤;および適切な液体担体中に投与された本発明の1つまたは複数の組成物を有するマウスウォッシュが含まれる。
【0098】
錠剤、ピル剤、カプセル剤、トローチ剤などは、以下の成分のうちの1つまたは複数、または類似の性質の化合物を含有することができる:結合剤;潤滑剤;希釈剤;流動促進剤;崩壊剤;着色剤;甘味剤;フレーバー剤;湿潤化剤;催吐性コーティング;およびフィルムコーティング。結合剤の例には、微結晶セルロース、トラガカントゴム、ブドウ糖溶液、アカシア粘液、ゼラチン液、糖蜜、ポリビニルピロリジン、ポビドン、クロスポビドン、スクロースおよびデンプン糊が含まれる。潤滑剤には、タルク、デンプン、ステアリン酸マグネシウムもしくはカルシウム、セキショウシおよびステアリン酸が含まれる。希釈剤には、例えば、ラクトース、スクロース、デンプン、カオリン、塩、マンニトールおよびリン酸二カルシウムが含まれる。流動促進剤には、コロイド状二酸化ケイ素が含まれるが、それには限定されない。崩壊剤には、クロスカルメロースナトリウム、グリコール酸デンプンナトリウム、アルギン酸、コーンスターチ、ジャガイモデンプン、ベントナイト、メチルセルロース、寒天およびカルボキシメチルセルロースが含まれる。着色剤には、例えば任意の承認かつ認証された水溶性FDおよびC色素、それらの混合物;およびアルミナ水和物に懸濁させた非水溶性FDおよびC色素が含まれる。甘味料には、スクロース、ラクトース、マンニトール、およびサッカリンなどの人工甘味料、ならびに任意の数のスプレードライフレーバーが含まれる。フレーバー剤には、果物などの植物から抽出された天然フレーバー、およびペパーミントおよびサリチル酸メチルなどであるがそれらに限定されない、快い感覚を生成する化合物の合成混合物が含まれる。湿潤剤には、プロピレングリコールモノステアレート、ソルビタンモノオレエート、ジエチレングリコールモノラウレートおよびポリオキシエチレンlauralエーテルが含まれる。催吐性コーティングには、脂肪酸、脂肪、ロウ、シェラック、アンモニア処理シェラックおよび酢酸フタル酸セルロースが含まれる。フィルムコーティングには、ヒドロキシエチルセルロース、カルボキシメチルセルロースナトリウム、ポリエチレングリコール4000および酢酸フタル酸セルロースが含まれる。
【0099】
皮膚への局所投与のために適合する組成物は、医薬上許容される担体中で投与される1つまたは複数の組成物を有する、軟膏剤、クリーム剤、ジェル剤、およびペースト剤として提示できる。
【0100】
直腸投与のための組成物は、例えばカカオ脂もしくはサリチル酸塩を含む適切な基剤を備える坐剤として提示できる。
【0101】
経鼻投与のために適切な組成物は、担体が固体の場合は、例えば鼻から吸う方法で投与される20〜500ミクロンの範囲内の粒径を有する粗粉末を含む(すなわち、鼻の近くに保持された粉末の容器から鼻腔を通した迅速な吸入によって)。担体が液体(例えば、鼻腔用スプレーもしくは点鼻剤)である場合は、1つまたは複数の組成物は水性溶液もしくは油性溶液中に混合できる、そして鼻腔内へ吸入もしくはスプレーすることができる。
【0102】
膣投与のために適切な組成物は、1つまたは複数の組成物および適切な担体を含有するペッサリー、タンポン、クリーム剤、ジェル剤、ペースト剤、フォーム剤もしくはスプレー製剤として提示できる。
【0103】
非経口投与のために適切な組成物には、酸化防止剤、緩衝剤、静菌剤、および製剤を所定のレシピエントの血液と等張性にさせる溶質を含有してもよい、水性および非水性無菌注射液;ならびに懸濁化剤および増粘剤を含んでもよい水性および非水性無菌懸濁剤が含まれる。本組成物は、単位用量もしくは多用量容器、例えば密封アンプルおよびバイアルに入れて提示することができ、使用直前に例えば注射用水などの無菌液体担体の添加だけを必要とするフリーズドライ(凍結乾燥)状態で保存することができる。即時注射溶液および懸濁剤は、無菌散剤、顆粒剤、および上述した種類の錠剤から調製できる。
【0104】
腸内もしくは非経口投与のために適合する医薬有機もしくは無機固体もしくは液体担体媒体を使用して組成物を作製することができる。ゼラチン、ラクトース、デンプン、ステアリン酸マグネシウム、タルク、植物性および動物性脂肪および油、ガム、ポリアルキレングリコール、水、またはその他の既知の担体は、すべてが担体媒体として適合する可能性がある。
【0105】
組成物は、1つまたは複数の医薬上許容される担体媒体および/または賦形剤と組み合わせて有効成分として使用できる。本明細書で使用する「医薬上許容される担体」には、望ましい特定の製剤形に適合するように、任意およびすべての担体、溶媒、希釈剤、または他の液体ビヒクル、分散もしくは懸濁補助剤、界面活性剤(surfaceactive agent)、等張化剤、増粘剤もしくは乳化剤、保存料、固体結合剤、潤滑剤、アジュバント、ビヒクル、送達系、崩壊剤、吸収剤、保存料、界面活性剤(surfactant)、着色剤、香味剤、または甘味剤などが含まれる。
【0106】
さらに、本組成物は、治療用組成物を生成するために、医薬上許容される賦形剤、および任意選択的に、例えば生分解性ポリマーなどの徐放性マトリックスと結合することができる。「医薬上許容される賦形剤」には、非毒性の固体、半固体もしくは液体充填剤、希釈剤、任意のタイプの被包化材料もしくは処方補助剤が含まれる。
【0107】
しかし、組成物の総1日量は、正確な医学的判断の範囲内で担当医師によって決定されることが理解されるであろう。任意の特定宿主のための特異的な治療有効量レベルは、例えば、治療される障害および障害の重症度;使用される特定組成物の活性;使用される特定組成物、患者の年齢、体重、全身状態、性別および食事;投与時期;投与経路;使用される特定化合物の排出速度;治療期間;使用される特定組成物と組み合わせて、もしくは同時に使用される薬物;ならびに、医学分野において周知である同様の要素を含む、様々な要素に依存する。例えば、所望の治療作用を達成するため、および所望の作用が達成されるまで用量を徐々に増加させるために必要とされる用量より低いレベルで組成物の用量を開始することは、明確に当分野の技術の範囲内である。
【0108】
組成物は、好ましくは投与の容易さおよび用量の一様性のために単位製剤形で調製される。本明細書で使用する「単位製剤形」は、治療対象の宿主にとって適切な組成物の物理的に別個の単位である。各用量は、単独で、または選択された医薬担体媒体と結び付けてのいずれかで所望の治療作用を生成するように計算された組成物の量を含有していなければならない。
【0109】
代表的な単位製剤は、投与される成分の1日量もしくは単位、1日分割用量、または適切なその分画を含有する製剤である。用量は、体重、年齢、表面積、代謝、組織分布、吸収速度および排泄速度などの宿主の要素に依存する。本明細書に記載した状態すべてのための代表的な全身性用量は、単一1日量もしくは分割1日量として、1日当たり0.01mg/kg〜2,000mg/kg(体重)の範囲に及ぶ用量である。局所投与のための典型的用量は、活性化合物の重量で0.001〜100%の範囲に及ぶ用量である。
【0110】
治療有効量レベルは、上述した多数の要素に依存する。さらに、組成物の用量を相当に低いレベルで開始すること、そして所望の作用が達成されるまで用量を増加させることは、明確に当分野の技術の範囲内である。
【0111】
本明細書に開示した化合物を含む組成物は、酵素的もしくは酸に基づく加水分解または溶解によって分解性である、通常はポリマーである材料から製造できる徐放性マトリックスと一緒に使用できる。身体内に挿入されると、マトリックスは酵素および体液によって作用する。徐放性マトリックスは、例えば、リポソーム、ポリラクチド(ポリ乳酸)、ポリグリコリド(グリコール酸のポリマー)、ポリラクチドコ−グリコリド(乳酸とグリコール酸のコポリマー)、ポリアンヒドリド、ポリ(オルト)エステル、ポリペプチド、ヒアルロン酸、コラーゲン、硫酸コンドロイチン、カルボン酸、脂肪酸、リン脂質、多糖、核酸、ポリアミノ酸、例えばフェニルアラニン、チロシン、イソロイシンなどのアミノ酸、ポリヌクレオチド、ポリビニルプロピレン、ポリビニルピロリドンおよびシリコーンなどの生体適合性材料から選択される。好ましい生体分解性マトリックスは、ポリラクチド、ポリグリコリド、もしくはポリラクチドコ−グリコリド(乳酸とグリコール酸のコポリマー)のいずれか1つのマトリックスである。
【0112】
本化合物は、リポソームの形態で投与することもできる。当分野において既知のように、リポソームは一般に、リン脂質もしくは他の脂質物質に由来する。リポソームは、水性媒体中に分散する単層状もしくは多層状の水和した液晶によって形成される。リポソームを形成できる任意の非毒性の、生理学的に許容できる、そして代謝可能な脂質を使用できる。リポソームは、本発明の1つまたは複数の組成物に加えて、安定剤、保存料、賦形剤などを含有することができる。脂質の例は、天然および合成の両方のリン脂質およびホスファチジルコリン(レシチン)である。リポソームを形成する方法は、当分野において既知である。
【0113】
本化合物は、吸入によるなどの、適用のためのエーロゾルとして調製できる。呼吸管へ投与するためのこれらの製剤は、ネブライザー用のエーロゾルもしくは液剤の形態であってよい、または単独もしくはラクトースなどの不活性担体と組み合わせて吸入用の微粉末としてよい。そのような場合には、製剤の粒子は、1つの実施形態では50ミクロン未満の、1つの実施形態では10ミクロン未満の径を有する。
【0114】
本明細書に開示した化合物を含む組成物は、他の組成物と組み合わせて、および/または上述した状態を治療するための方法において使用できる。
【0115】
<用語の定義>
本明細書で使用する用語「アルキル」は、他に特に規定しない限り、飽和直鎖状、分枝状、もしくは環状の、例えばC1−30もしくはC1−22の1級、2級、もしくは3級炭化水素が含まれ、詳細には、メチル、エチル、プロピル、イソプロピル、シクロプロピル、ブチル、イソブチル、secブチル、t−ブチル、ペンチル、シクロペンチル、イソペンチル、ネオペンチル、ヘキシル、イソヘキシル、シクロヘキシル、シクロヘキシルメチル、ヘプチル、シクロヘプチル、オクチル、シクロ−オクチル、ドデシル、トリデシル、ペンタデシル、イコシル、ヘミコシル、およびデコシルが含まれる。アルキル基は、例えば、参照することによって本明細書に組み込まれるGreene,etal.,Protective Groups in Organic Synthesis,John Wileyand Sons,SecondEdition,1991に教示されるように、当業者には既知のように、必要に応じて保護されていない、または保護されているいずれかである、ハロゲン(フルオロ、クロロ、ブロモ、もしくはヨード)、ヒドロキシル、アミノ、アルキルアミノ、アリールアミノ、アルコキシ、アリールオキシ、ニトロ、シアノ、スルホン酸、硫酸塩、ホスホン酸、ホスフェート、もしくはホスホネートで、任意選択的に置換されてよい。
【0116】
本明細書で使用する用語「低級アルキル」には、他に特に規定しない限り、任意選択的に置換される、C1〜C4飽和直鎖状、分枝状、もしくは適切であれば環状(例えば、シクロプロピル)アルキル基が含まれる。
【0117】
炭素原子の範囲について言及される場合は必ず、それは独立して個別にその範囲の各数を含む。非限定的例として、用語「C1−C10アルキル」は、例えば、C1−C10アルキルが直鎖状、分枝状および適切な場合は環状のC1、C2、C3、C4、C5、C6、C7、C8、C9およびC10アルキル官能基を含むように、独立して、各数の基を含むと見なされる。
【0118】
本明細書で使用する用語「保護(された)」には、他に特に規定しない限り、それ以上の反応を防止する、または他の目的で酸素、窒素、もしくはリン原子などの原子に加えられている基を含む。広範囲の酸素および窒素保護基は、有機合成の当業者に既知である。
【0119】
本明細書で使用する用語「ハロ」には、詳細にはクロロ、ブロモ、ヨード、およびフルオロが含まれる。
【0120】
用語「アルケニル」には、少なくとも1つの二重結合を備える、例えば、C2−100、もしくはC2−22の直鎖状、分枝状、もしくは環状炭化水素が含まれる。例には、ビニル、アリル、およびメチル−ビニルが含まれるが、それらに限定されない。アルケニル基は、アルキル基について上述した方法と同一方法で任意選択的に置換されてよい。
【0121】
用語「アルキニル」には、例えば、少なくとも1つの三重結合を備える、例えば、C2−100、もしくはC2−22の直鎖状もしくは分枝状炭化水素が含まれる。アルキニル基は、アルキル基について上述した方法と同一方法で任意選択的に置換されてよい。
【0122】
用語「アルコキシ」には、構造−O−アルキル基の成分が含まれる。
【0123】
用語「アシル」には、式R’C(O)(式中、R’は直鎖状、分枝状、もしくは環状の置換もしくは未置換アルキルもしくはアリールである)基が含まれる。
【0124】
本明細書で使用する用語「アリール」には、6〜14個の範囲内の炭素原子を有する芳香族基が含まれ、「置換アリール」は上記に規定した1つまたは複数の置換基をさらに有するアリール基を意味する。
【0125】
本明細書で使用する「ヘテロアリール」には、環状構造の一部として1つまたは複数のヘテロ原子(例、N、O、Sなど)を含有し、3〜14個の範囲内の炭素原子を有する芳香族基が含まれ、「置換ヘテロアリール」は、上記に規定した1つまたは複数の置換基をさらに有するヘテロアリール基を意味する。
【0126】
本明細書で使用する用語「結合」もしくは「原子価結合」には、電子対からなる原子間の連鎖を含む。
【0127】
本明細書で使用する用語「宿主」は、他に特に規定しない限り、哺乳動物(例、ネコ、イヌ、ウマ、マウス、サルなど)、ヒト、または治療を必要とする他の生物が含まれる。宿主は、例えばヒト、または制限なく、マカク属のサル、ヒヒ、ならびにチンパンジー、ゴリラ、およびオランウータンを含む霊長類;ヒツジ、ヤギ、シカ、および畜牛、例えば乳牛、去勢牛(steer)、雄牛(bull)、および去勢牛(ox)などを含む反芻動物;ブタ(pig)を含むブタ(swine);ならびにニワトリ、シチメンチョウ、アヒル、もしくはガチョウを含む家禽類を含む動物である。
【0128】
本明細書で使用する用語「医薬上許容される塩」には、他に特に規定しない限り、正確な医学的判断の範囲内で、過度の毒性、刺激、アレルギー反応などを伴わずに宿主の組織と接触させて使用するために適合する、そして合理的なベネフィット/リスク比と釣り合っていてそれらの所定の使用のために有効である塩が含まれる。塩は、組成物の1つまたは複数の化合物の最終単離および精製中にinsitu(インサイチュ)で、または遊離塩基官能基と適切な有機酸とを反応させる工程によって個別に調製できる。医薬上許容されない酸および塩基もまた、本明細書では、例えば当該の化合物の合成および/または精製において使用される。そのような塩の非限定的例は、(a)無機塩(例えば、塩酸、臭化水素酸、硫酸、リン酸、硝酸など)を用いて形成された、および酢酸、シュウ酸、酒石酸、コハク酸、アスコルビン酸、安息香酸、タンニン酸などの有機塩を用いて形成された酸付加塩;(b)亜鉛、カルシウム、マグネシウム、アルミニウム、ナトリウム、カリウム、銅、ニッケルなどの金属カチオンを用いて形成された塩基付加塩;(c)(a)および(b)の組み合わせである。さらにアミン塩もまた「医薬上許容される塩」として含まれる。
【0129】
本明細書で使用する用語「医薬上許容されるエステル」には、他に特に規定しない限り、正確な医学的判断の範囲内で、過度の毒性、刺激、アレルギー反応などを伴わずに宿主の組織と接触させて使用するために適合する、そして合理的なベネフィット/リスク比と釣り合っていてそれらの所定の使用のために有効である、1つまたは複数の化合物のエステルが含まれる。
【0130】
用語「医薬上許容されるプロドラッグ」には、活性化合物を形成するために宿主において代謝される、例えば加水分解もしくは酸化される化合物が含まれる。プロドラッグの典型的な例には、活性化合物の官能基成分上に生物学的に不安定な保護基を有する化合物が含まれる。プロドラッグには、活性化合物を生成するために酸化する、還元する、アミノ化する、脱アミノ化する、ヒドロキシル化する、脱ヒドロキシル化する、加水分解する、脱水縮合する(dehydrolyzed)、アルキル化する、脱アルキル化する、アシル化する、脱アシル化する、リン酸化する、脱リン酸化することのできる化合物が含まれる。
【0131】
本明細書で使用する用語「エナンチオマーが豊富である」は、1つのエナンチオマーが過剰に存在する、好ましくは95%以上まで存在する、およびより好ましくは100%を含めて98%以上まで存在するエナンチオマーの混合物である化合物を意味する。
【0132】
用語「有効量」には、本明細書に提供した疾患もしくは障害の1つまたは複数の症状の予防、治療、または改善のために必要とされる量が含まれる。
【0133】
本明細書に開示した化合物はキラル中心を含有する可能性があることを理解されたい。そのようなキラル中心は、(R)もしくは(S)立体配置いずれかの中心であってよい、またはそれらの混合物であってよい。そこで、本明細書に提供した化合物は、エナンチオマー的に純粋、または立体異性体的もしくはジアステレオマー的混合物であってよい。本明細書の化合物の開示は、好ましくは本明細書に記載した有用な特性を有する、任意のラセミ体の、光学活性形、多形体形、もしくは立体異性体形、またはそれらの混合物を含むことは理解されているが、当分野においては、光学活性形を調製する方法、および本明細書に記載した標準試験を用いて、または当分野において既知である他の類似の試験を用いて活性を決定する方法は、周知である。本化合物の光学異性体を入手するために使用できる方法の例には、以下が含まれる:
i)結晶の物理的分離−それにより個別エナンチオマーの肉眼的結晶が手動で分離される技術。この技術は、個別エナンチオマーの結晶が存在する場合、すなわち材料が集合体であり、結晶が視覚的に識別される場合に使用できる;
ii)同時の結晶化−それにより個別エナンチオマーがラセミ化合物の溶液から個別に結晶化される技術であり、後者が固体状態において集合体である場合にのみ可能である;
iii)酵素的分割−それによりエナンチオマーと酵素の相違する反応速度により、ラセミ化合物を部分的もしくは完全分離する技術;
iv)非対称性酵素的合成−それにより合成の少なくとも1つの工程が、所望のエナンチオマーのエナンチオマー的に純粋もしくはエナンチオマーが豊富な合成前駆物質を入手するために酵素反応を使用する合成技術;
v)非対称性化学合成−それにより所望のエナンチオマーが、キラル触媒もしくはキラル補助剤を用いて達成できる、生成物中において非対称性(すなわち、キラリティ)を生成する条件下でアキラル性前駆物質から合成される合成技術;
vi)ジアステレオマーの分離−それによりラセミ化合物が、個別エナンチオマーをジアステレオマーへ変換させるエナンチオマー的に純粋な試薬(キラル補助剤)と反応させられる技術。結果として生じるジアステレオマーは、次にクロマトグラフィによって、またはそれらの現在のより明確な構造的相違による結晶化によって分離され、後に所望のエナンチオマーを入手するためにキラル補助剤は除去される;
vii)一次および二次非対称性変換−それによりラセミ化合物からのジアステレオマーが釣り合って所望のエナンチオマーからジアステレオマーの溶液中で優勢を生じさせる、または所望のエナンチオマーからのジアステレオマーの優先的結晶化は、原理的には最終的に全材料が所望のエナンチオマーからの結晶ジアステレオマーへ変換されるように平衡を混乱させる技術。所望のエナンチオマーは、次にジアステレオマーから遊離させられる;
viii)速度論的分割−この技術は、反応速度的条件下でエナンチオマーとキラル、非ラセミ化合物試薬もしくは触媒との不同の反応速度による、ラセミ化合物(または、部分的に溶解した化合物のいっそうの分割)の部分もしくは完全分割の達成を意味する;
ix)非ラセミ前駆体からのエナンチオ特異的合成−それにより所望のエナンチオマーが非キラル出発物質から入手され、立体化学的完全性が合成の経過に渡って損なわれない、または極めてわずかしか損なわれない合成技術;
x)キラル液体クロマトグラフィ−それによりラセミ化合物のエナンチオマーが、固定相とのそれらの相違する相互作用によって液体移動相中で分離される技術。固定相は、キラル材料から作製されてよい、または移動相は相違する相互作用を誘発するために追加のキラル材料を含有してもよい;
xi)キラルガスクロマトグラフィ−それによりラセミ化合物が揮発化され、エナンチオマーが、固定非ラセミキラル吸着相を含有するカラムを用いて気体状移動相中でのそれらの相違する相互作用によって分離される技術;
xii)キラル溶媒を用いた抽出−それによりエナンチオマーが特定キラル溶媒中への1つのエナンチオマーの優先的溶解によって分離される技術;
xiii)キラル膜を横断する輸送−それによりラセミ化合物が薄膜障壁と接触させられる技術。障壁は、典型的には一方がラセミ化合物を含有する2つの混和性流体を分離し、濃度差もしくは圧力差などの駆動力が、薄膜障壁を横断する優先的輸送を引き起こす。分離は、ラセミ化合物の1つだけのエナンチオマーが通過することを可能にする膜の非ラセミ性のキラル性質の結果として発生する。
【0134】
<抗ウイルス性化合物の合成>
抗ウイルス性化合物、および詳細には抗ウイルス性脂質含有化合物は、その開示が参照することにより本明細書に組み込まれる米国特許第6,716,825号に記載されているように、当分野において利用できる方法を用いて合成できる。本明細書に提供する抗ウイルス性化合物および脂質含有プロドラッグは、一般にはスキームI〜IIに描出したような、様々な方法で調製できる。以下に記載する一般的ホスホン酸エステル化法は、単に具体的に示すためにのみ提供されており、決して限定するものと考えられてはならない。実際に、アルコールを用いたホスホン酸の直接縮合のために数種の方法が開発されている(例えば、R.C.Larock,ComprehensiveOrganic Transformations,VCH,NewYork,1989,p.966およびその中に言及された文献を参照されたい)。実施例に記載した化合物および中間体の単離および精製は、所望であれば、例えば、濾過、抽出、結晶化、フラッシュカラムクロマトグラフィ、薄層クロマトグラフィ、蒸留、またはこれらの方法の組み合わせなどの、任意の適切な分離もしくは精製方法によって実施できる。適切な分離および単離方法の特別の具体例は、以下の実施例に記載されている。その他の等価の分離および単離方法もまた、当然ながら使用できる。
【0135】
スキームIは、シドフォビル、環式シドフォビル、およびその他のホスホン酸塩のアルキルグリセロールもしくはアルキルプロパンジオールアナログの一般的合成法を例示する。2,3−イソプロピリデングリセロール(1)とジメチルホルムアミド中のNaHとの処理、次にアルキルメタンスルホネートとの反応は、アルキルエーテル(2)を産生させる。酢酸による処理、次にピリジン中の塩化トリチルとの反応によってイソプロピリデン基を除去すると、中間体(3)が産生する。中間体(3)のハロゲン化アルキルを用いたアルキル化は化合物(4)を生じさせる。80%酢酸水溶液を用いたトリチル基の除去によりO,O−ジアルキルグリセロール(5)が得られる。化合物(5)の臭素化、その後の環式シドフォビルもしくは他のホスホン酸塩含有ヌクレオチドのナトリウム塩との反応は、所望のホスホン酸塩付加物(7)を生じさせる。環状付加物の開環は、水酸化ナトリウム水溶液との反応によって遂行される。好ましいプロパンジオール種は、スキームIにおける化合物(5)に対して1−O−アルキルプロパン−3−オールを置換することによって合成できる。テノフォビルおよびアデフォビルアナログは、これらのヌクレオチドリン酸をスキームIの反応(f)においてcCDVと置換することによって合成できる。同様に、他のヌクレオチドホスホン酸塩もこの方法で形成できる。
【0136】
スキームI
【化11】
試薬:a)NaH、R1OSO2Me、DMF;b)80%酢酸水溶液;c)塩化トリチル、ピリジン;d)NaH、R2−−B4、DMF、e)CBr4、トリフェニルホスフィン、THF;f)環式シドフォビル(DCMC塩)、DMF;g)0.5NのNaOH
【0137】
スキームIIは、実施例として1−O−ヘキサデシルオキシプロピル−アデフォビルを用いたヌクレオチドホスホン酸塩を合成するための一般的方法を例示する。ヌクレオチドホスホン酸塩(5mmol)を無水ピリジン中に懸濁させ、アルコキシアルカノールもしくはアルキルグリセロール誘導体(6mmol)および1,3−ジシクロヘキシルカルボジイミド(DCC、10mmol)を加える。この混合液を還流するように加熱し、薄層クロマトグラフィによって監視して縮合反応が完了するまで強力に攪拌する。この混合液を次に冷却して濾過する。濾液を減圧下で濃縮し、残留物をシリカゲルに吸着させてフラッシュカラムクロマトグラフィ(およそ9:1のジクロロメタン/メタノールを用いた溶出)によって精製すると、対応するホスホン酸モノエステルが産生する。
【0138】
スキームII
【化12】
【0139】
本発明は、以下の非限定的な実施例から詳細に理解されるであろう。
【実施例1】
【0140】
(米国特許第6,716,825号に記載されているとおり)
アデフォビルヘキサデシルオキシプロピルおよび1−O−オクタデシル−sn−グリセリルエステルの合成
無水ピリジン中のアデフォビル(1.36g、5mmol)および3−ヘキサデシルオキシ−1−プロパノール(1.8g、6mmol)の混合液にDCC(2.06g、10mmol)を加えた。この混合液を還流するように加熱し、18時間攪拌し、その後に冷却して濾過した。濾液を減圧下で濃縮し、残留物をシリカゲルのショートカラムに装填した。9:1のジクロロメタン/メタノールを備えるカラムによる溶出は、白色粉末としてヘキサデシルオキシプロピル−アデフォビル(HDP−ADV)を産生した。
【0141】
無水ピリジン(30mL)中のアデフォビル(1.36g、5mmol)および1−O−オクタデシル−sn−グリセロール(2.08g、6mmol)の混合液にDCC(2.06g、10mmol)を加えた。この混合液を還流するように加熱し、一晩攪拌し、その後に冷却して濾過した。濾液を減圧下で濃縮し、残留物をシリカゲルのカラムに装填した。9:1のジクロロメタン/メタノールを備えるカラムによる溶出は、1−O−オクタデシル−sn−グリセリル−3−アデフォビルを産生した。
【実施例2】
【0142】
(米国特許第6,716,825号に記載されているとおり)
AZT−ホスホン酸ヘキサデシルオキシプロピルエステルの合成
AZT(3’−アジド−3’−5’−ジデオキシチミジン−5’−ホスホン酸)のホスホン酸塩アナログは、公表された方法:Hakimelahi,G.H.;Moosavi−Movahedi,A.A.;Sadeghi,M.M.;Tsay,S−C.;Hwu,J.R.Journalof MedicinalChemistry,1995 38,4648−4659を用いて合成した。
【0143】
AZTホスホン酸塩(1.65g、5mmol)を無水ピリジン(30mL)中に懸濁させ、次に3−ヘキサデシルオキシ−1−プロパノール(1.8g、6mmol)およびDCC(2.06g、10mmol)を加え、この混合液を還流するように加熱し、6時間攪拌し、次に冷却して濾過した。濾液を減圧下で濃縮し、残留物をシリカゲルのカラムに装填した。9:1のジクロロメタン/メタノールを備えるカラムによる溶出は、3’−アジド−3’−5’−ジデオキシチミジン−5’−ホスホン酸、ヘキサデシルオキシプロピルエステルを産生した。
【実施例3】
【0144】
(米国特許第6,716,825号に記載されているとおり)
環式シドフォビルのヘキサデシルオキシプロピルエステル、オクタデシルオキシプロピルエステル、オクタデシルオキシエチルエステルおよびヘキサデシルエステルの合成
N,N−DMF(25mL)中のシドフォビルの攪拌懸濁液(1.0g、3.17mmol)にN,N−ジシクロヘキシル−4−モルホリンカルボキサミジン(DCMC、1.0g、3.5mmol)を加えた。シドフォビルを溶解させるために、この混合液を一晩攪拌した。この透明溶液を次に添加漏斗に装填し、1,3−ジシクロヘキシルカルボジイミド(1.64g、7.9mmol)の攪拌した高温ピリジン溶液(25mL、60℃)へ緩徐に(30分間)加えた。この反応混合液を100℃で16時間攪拌し、次に室温へ冷却し、減圧下で溶媒を除去した。残留物をシリカゲルに吸着させ、勾配溶離(CH2Cl2+MeOH)を用いてフラッシュカラムクロマトグラフィによって精製した。最後にUV活性生成物を5:5:1のCH2Cl2/MeOH/H2Oにより溶出した。溶媒を蒸発させると860mgの白色固体が得られた。1Hおよび31PNMRスペクトルは、これが環式シドフォビルのDCMC塩であることを証明した(収率=44%)。
【0145】
無水DMF(35mL)中の環式シドフォビルの溶液(DCMC塩)(0.5g、0.8mmol)に1−ブロモ−3−ヘキサデシルオキシプロパン(1.45g、4mmol)を加え、この混合液を攪拌して80℃で6時間加熱した。この溶液を次にインバキュオで濃縮し、残留物をシリカゲルに吸着させ、勾配溶離(CH2Cl2+EtOH)を用いてフラッシュカラムクロマトグラフィによって精製した。アルキル化生成物は、90:10のCH2Cl2/EtOHで溶出した。純粋生成物を含有する分画を蒸発させると260mgのHDP−環式シドフォビルが産生した(収率55%)。
【0146】
無水DMF(35mL)中の環式シドフォビルの溶液(DCMC塩)(1.0g、3.7mmol)に1−ブロモ−3−オクタデシルオキシプロパン(2.82g、7.2mmol)を加え、この混合液を攪拌して85℃で5時間加熱した。この溶液を次にインバキュオで濃縮し、残留物をシリカゲルに吸着させ、勾配溶離(CH2Cl2+MeOH)を用いてフラッシュカラムクロマトグラフィによって精製した。アルキル化生成物は、9:1のCH2Cl2/MeOHで溶出した。純粋生成物を含有する分画を蒸発させると450mgのODP−環式シドフォビルが産生した。
【0147】
無水DMF(35mL)中のcCDVの溶液(DCMC塩)(1.0g、3.7mmol)に1−ブロモ−3−オクタデシルオキシエタン(3.0g、7.9mmol)を加え、この混合液を攪拌して80℃で4時間加熱した。この溶液を次にインバキュオで濃縮し、残留物をシリカゲルに吸着させ、勾配溶離(CH2Cl2+MeOH)を用いてフラッシュカラムクロマトグラフィによって精製した。アルキル化生成物は、9:1のCH2Cl2/MeOHで溶出した。純粋生成物を含有する分画を蒸発させると320mgのオクタデシルオキシエチル−cCDVが産生した。
【0148】
無水DMF(35mL)中の環式シドフォビルの溶液(DCMC塩)(0.5g、0.8mmol)に1−ブロモ−ヘキサデカン(1.2g、4mmol)を加え、この混合液を攪拌して80℃で6時間加熱した。この溶液を次にインバキュオで濃縮し、残留物をシリカゲルに吸着させ、勾配溶離(CH2Cl2+MeOH)を用いてフラッシュカラムクロマトグラフィによって精製した。アルキル化生成物は、9:1のCH2Cl2/MeOHで溶出した。純粋生成物を含有する分画を蒸発させると160mgのヘキサデシル−cCDVが産生した。
【実施例4】
【0149】
(米国特許第6,716,825号に記載されているとおり)
シドフォビルのヘキサデシルオキシプロピルエステル、オクタデシルオキシプロピルエステル、オクタデシルオキシエチルエステルおよびヘキサデシルエステルの合成
上記からのヘキサデシルオキシプロピル−環式CDVを0.5MのNaOH中に溶解させ、室温で1.5時間攪拌した。pHを約9に調整するために、50%酢酸水溶液を滴下した。沈降したHDP−CDVを濾過によって単離し、水ですすぎ洗いし、乾燥させ、次に再結晶化させると(3:1p−ジオキサン/水)HDP−CDVが得られた。同様に、オクタデシルオキシプロピルエステル、オクタデシルオキシエチルエステル、およびヘキサデシル−cCDVエステルを0.5MのNaOHにより加水分解して精製すると、対応するシドフォビルジエステルが得られた。
【実施例5】
【0150】
(米国特許第6,716,825号に記載されているとおり)
環式−ガンシクロビルホスホン酸ヘキサデシルオキシプロピルエステルの合成
ガンシクロビルの環式ホスホン酸塩アナログは、公表された方法を用いて調製した:(Reist,E.J.;Sturm,P.A.;Pong,R.Y.;Tanga,M.J.andSidwell,R.W.Synthesis of acyclonucleoside phosphonates for evaluation as antiviral agents,p.17−34.In J,C.Martin(ed.),Nucleotide Analoguesas AntiviralAgents,American Chemical Society,Washington,D.C.)。DMF中でDCMC塩へ変換させた後、cGCVホスホン酸塩は1−ブロモ−3−ヘキサデシルオキシプロパンにより処理し、この混合液を80℃で6時間加熱した。アルキル化生成物をフラッシュクロマトグラフィによって単離すると、HDP−環式−GCVホスホン酸塩が産生した。
【実施例6】
【0151】
(米国特許第6,716,825号に記載されているとおり)
ガンシクロビルホスホン酸ヘキサデシルオキシプロピルエステルの合成
上記からのHDP−環式GCVホスホン酸塩を0.5MのNaOH中に溶解させ、室温で攪拌してそれを非環式ジエステルに変換させた。この溶液を50%酢酸水溶液で中和すると生成物が沈降したので、これを3:1のp−ジオキサン/水中で再結晶化させた。
【実施例7】
【0152】
ヒトサイトメガロウイルス(HCMV)に対するホスホン酸塩ヌクレオチドアナログの抗ウイルス活性および選択性
HCMV抗ウイルスアッセイ:HCMV DNAについての抗ウイルスアッセイは、Dankner,W.M.,Scholl,D.,Stanat,S.C,Martin,M.,Souke,R.L.andSpector,S.A,J.Virol.Methods 21:293−298,1990によって報告されたDNAハイブリダイゼーションによって実施された。簡単に説明すれば、24ウェル培養皿内のサブコンフルエントなMRC−5細胞は、2%FBSおよび抗生物質を含有するイーグルの最小必須培地(E−MEM)中で様々な濃度の薬物を用いて24時間にわたり前処理した。培地を除去し、薬物を含有しないウェル中では5日間で3〜4+の細胞変性作用(CPE)を生じさせる希釈率でHCMV菌株を加えた。ウイルスを37℃で1’時間にわたり吸収させ、吸引させ、薬物希釈液と置換した。5日間のインキュベーション後、HCMVDNAはDiagnostic Hybrids社(オハイオ州アセンズ)製のCMV抗ウイルス感受性試験キットを用いて、核酸ハイブリダイゼーションによって3回ずつ定量した。培地を除去し、細胞は製造業者の取扱説明書にしたがって溶解させた。溶解液を吸収させた後、Hybriwix(商標)フィルタを60℃で一晩ハイブリダイズさせた。Hybriwix(商標)を73℃で30分間にわたり洗浄し、γ計数器で計数した。結果は、EC50(50%阻害濃度)として表示する。予備実験は、表2に示したように、シドフォビルおよびアデフォビルの1−O−ヘキサデシルプロパンジオール(HDP)誘導体上で実施した。
【表2】
【0153】
表2の結果が示すように、1−O−ヘキサデシルプロパンジオール−3−環式CDV(HDP−cCDV)は、CDVもしくは環式CDVより>900倍活性であった。より細胞毒性であるが、迅速に分割する細胞中でのHCMVに対する選択指数は、未誘導体化CDVについての1,900から>2,100に対して>59,000であった。
【0154】
インビトロでの試験化合物の細胞毒性:24ウェルプレート中のサブコンフルエントなヒト肺線維芽細胞(MRC−5、アメリカンタイプカルチャーコレクション、メリーランド州ロックビル)は、2%ウシ胎児血清および抗生物質を補給したE−MEM(GibcoBRL社、ニューヨーク州グランドアイランド)中に希釈した薬物で処理した。37℃での5日間のインキュベーション後、細胞単層を顕微鏡下で視覚的に検査し、細胞数の50%減少を誘発した薬物濃度を推定した。
【0155】
これらの実験から入手したデータは、表3に示されている。
【表3】
【実施例8】
【0156】
(米国特許第6,716,825号に記載されているとおり)
インビトロでの、HDP−cCDVがポックスウイルス複製に及ぼす作用
シドフォビル(CDV)、環式シドフォビル(cCDV)、および1−O−ヘキサデシルプロパンジオール−3−cCDV(HDP−cCDV)の活性は、細胞変性作用(CPE)における用量依存性減少を測定することによって、ワクシニアウイルスもしくはウシ痘ウイルスに感染したヒト包皮線維芽細胞中の抗ウイルス活性について試験した。ワクシニアおよびウシ痘ウイルスのEC50の予備試験値は、ヒト包皮線維芽(HFF)細胞中でのCPE減少アッセイによって決定した。このように入手したデータは、表4に示されている。
【表4】
【0157】
表4に示したように、HDP−cCDVはワクシニアウイルスに対して高度に活性であり、IC50値はcCDVおよびCDVについて各々0.97および1.8μMに対して0.11μMであった。ウシ痘感染細胞中では、HDP−cCDVは、極度に有効であり、IC50はcCDVおよびCDVについて各々0.72および2.1に対して<0.03μMであった。
【0158】
ポックスウイルスに対する抗ウイルス細胞変性作用(CPE)アッセイ:各薬物濃度では、ベロ細胞を含有する3つのウェルを1,000pfu/ウェルのオルソポックスウイルスで感染させ、他の3つのウェルは毒性決定のために未感染のままに残した。プレートを試験し、ウイルス感染後に染色すると、未処理細胞は4+CPEを示した。培地にニュートラルレッドを加え、CPEは540nmでのニュートラルレッド取込みによって評価した。50%阻害(EC50)および細胞毒性濃度(CC50)は、用量反応曲線のプロットから決定した。結果は表5に示されている。
【0159】
【表5】
【実施例9】
【0160】
(米国特許第6,716,825号に記載されているとおり)
1−O−ヘキサデシルプロパンジオール−3−アデフォビル(HDP−ADV)がインビボでHIV−1複製に及ぼす作用
本明細書に提供した化合物によるHIV−1複製の阻害における予備実験は、以下のとおりに実施した。薬物アッセイは、Larderet.al.,Antimicrobial Agents & Chemotherapy,34:436−441,1990によって以前に記載されたとおりに実施した。HIV−1LAI感染HT4−6C細胞は、指示したように薬物に曝露させ、37℃で3日間にわたりインキュベートした。細胞は、プラークを視認するためにクリスタルバイオレットで固定した。抗ウイルス活性は、薬物処理サンプル中で測定したコントロールプラーク(薬物なし)のパーセンテージとして評価した。EC50は、プラーク数を50%減少させるマイクロモル濃度である。アデフォビルの活性は、HIV−1感染HT4−6C細胞中でAZT(ジドブジン)および1−O−ヘキサデシルプロパンジオール−3−アデフォビル(HDP−ADV)と比較した。結果は表6に示されている。
【表6】
【0161】
アデフォビルは、中等度に活性であり、EC50は16μMであった。AZTは予想どおり高度に活性であったが(EC500.007μM)、HDP−ADVは3種の化合物中で最も活性であり、0.0001μMのEC50を備えており、アデフォビル自体より5対数を超えて活性であった。
【0162】
HIV−1抗ウイルスアッセイ:抗ウイルス性化合物がCD4を発現するHeLaHT4−6C細胞中でのHIV複製に及ぼす作用を、プラーク減少アッセイ(Larder,B.A.,Chesebro,B.andRichman,D.D.Antimirob.Agents Chemother.,34:436−441,1990)によって測定した。手短には、HT4−6C細胞の単層を、24ウェルマイクロ希釈プレート中の1ウェル当たり100〜300プラーク形成単位(PFU)のウイルスで感染させた。様々な濃度の薬物を培養培地である、上述した5%FBSおよび抗生物質を含有するダルベッコの修飾イーグル培地へ加えた。37℃で3日間後、単層はリン酸緩衝食塩液(PBS)中の10%ホルムアルデヒド溶液で固定し、0.25%のクリスタルバイオレットを用いて染色してウイルスプラークを視認した。抗ウイルス活性は、薬物処理サンプル中で測定したコントロールプラークのパーセンテージとして評価した。細胞毒性は、Hostetleret al.,AntiviralResearch,31:59−67,1996の方法によって評価した。結果は表7に示されている。
【0163】
【表7】
【実施例10】
【0164】
(米国特許第6,716,825号に記載されているとおり)
シドフォビルアナログがヘルペスウイルス複製に及ぼす作用
HSV−1抗ウイルスアッセイ:24ウェル培養皿中でのサブコンフルエントなMRC−5細胞は、培地を除去し、20〜24時間中に薬物無含有ウェル内で3〜4+CPEを生じさせる希釈率でHSV−1ウイルスを加えることによって接種した。これを37℃で1時間吸収させ、吸引し、2%FBSおよび抗生物質を含有するE−MEM中で様々な濃度の薬物と置換した。およそ24時間のインキュベーション後、HSVDNAはDiagnostic Hybrids社(オハイオ州アセンズ)製のHSV抗ウイルス感受性試験キットを用いて核酸ハイブリダイゼーションによって3回ずつ定量した。培地を除去し、細胞は製造業者の取扱説明書にしたがって溶解させた。溶解液を吸収させた後、Hybriwix(商標)フィルタを60℃で一晩ハイブリダイズさせた。Hybriwix(商標)を73℃で30分間にわたり洗浄し、γ計数器で計数した。細胞毒性は、実施例17に記載したように評価した。このようにして入手したEC50およびCC50値は、表8に示されている。
【0165】
【表8】
【実施例11】
【0166】
代謝安定性の評価
冷凍保存した一次肝細胞中のHDP−シドフォビル(1μM)の代謝安定性を、ウサギ、Cynサル、Rhサル、ヒト、マウスおよびラットにおいて試験した。結果は、HDP−シドフォビルの経時的な減少を示している図1に示されている。さらに、単回経口投与後のマウス、NZW系ウサギ、およびサルにおけるHDP−シドフォビル、HDP−シドフォビルから遊離されたシドフォビル、およびHDP−シドフォビルの不活性代謝産物である代謝産物M−8の血清中濃度を評価し、結果を図2、3および4に示した。図5は、HDP−シドフォビルの酸化によるM−8の形成について考えられる機序を示した図である。5mg/kgの[2−14C]HDP−シドフォビル(ヘキサデシルオキシプロピル−シドフォビル)の経口投与4時間後のマウスの定量的全身オートラジオグラフでは、この薬物は主として消化管、ならびに肺、心臓、肝臓、および腎臓を含む器官中に分布することが同定された。
【実施例12】
【0167】
サルにおける力価試験
16匹のカニクイザル(Cynomolgus Macaques)を各4匹の4つの治療群に分けて試験した。動物には、5×107PFU IVでサル痘ウイルスのZaire79菌株を接種した。動物には、図6にHD、LD、IDおよびプラセボとして示した、プラセボ、または相違する用量レベル(TD:35、30、20、0mg/kg)を投与した。図6は、薬物投与後の様々なタイプの組織中のサル痘力価のウイルス量を示す。
【特許請求の範囲】
【請求項1】
ウイルス感染症を治療する方法であって、抗ウイルス性化合物、またはその塩、エステルもしくはプロドラッグの脂質含有プロドラッグを、ウイルス感染症を治療するための有効量で、治療を必要とする宿主に生物学的利用能エンハンサーと組み合わせて、または交互に投与するステップを含む方法。
【請求項2】
前記抗ウイルス性化合物はヌクレオシドである、請求項1に記載の方法。
【請求項3】
前記抗ウイルス性化合物は抗オルソポックス薬である、請求項1に記載の方法。
【請求項4】
前記抗ウイルス性化合物は、HIV、B型肝炎、またはC型肝炎に対して活性である、請求項1に記載の方法。
【請求項5】
前記生物学的利用能エンハンサーは、シトクロムP450酵素の阻害剤もしくは基質、イミダゾール;マクロライド;カルシウムチャネルブロッカー;またはステロイドである、請求項1に記載の方法。
【請求項6】
前記生物学的利用能エンハンサーは、シトクロムP450 3A(CYP3A)またはP糖タンパク質媒介性膜輸送の阻害剤である、請求項1に記載の方法。
【請求項7】
抗ウイルス性化合物、またはその塩、エステルもしくはプロドラッグの前記脂質含有プロドラッグ、および前記生物学的利用能エンハンサーは、医薬上許容される担体中で組み合わせて、または交互に投与される、請求項1に記載の方法。
【請求項8】
抗ウイルス性化合物、またはその塩、エステルもしくはプロドラッグの前記脂質含有プロドラッグ、および前記生物学的利用能エンハンサーは、経口、局所または非経口投与から選択された同一もしくは相違する経路によって投与される、請求項7に記載の方法。
【請求項9】
前記抗ウイルス性化合物は、シドフォビルもしくは環式シドフォビルである、請求項1に記載の方法。
【請求項10】
前記抗ウイルス性化合物は、アルキルプロパンジオール、1−S−アルキルチオグリセロール、アルコキシアルカノールもしくはアルキルエタンジオールに任意選択的に共有結合した、シドフォビル、アデフォビル、環式シドフォビルまたはテノフォビルである、請求項1に記載の方法。
【請求項11】
前記抗ウイルス性化合物は、構造:
【化1】
を有する、請求項9に記載の方法。
【請求項12】
前記ウイルス感染症は、大痘瘡(variola major)、小痘瘡(variola minor)、ワクシニア、天然痘、ウシ痘、ラクダ痘、マウス痘、ウサギ痘、またはサル痘である、請求項1に記載の方法。
【請求項13】
ウイルス感染症を治療するための薬剤の製造における、有効量の抗ウイルス性化合物、またはその塩、エステルもしくはプロドラッグの脂質含有プロドラッグの、生物学的利用能エンハンサーと組み合わせた、または交互の使用。
【請求項14】
前記抗ウイルス性化合物はヌクレオシドである、請求項13に記載の使用。
【請求項15】
前記抗ウイルス性化合物は抗オルソポックス薬である、請求項13に記載の使用。
【請求項16】
前記抗ウイルス性化合物は、HIV、B型肝炎、またはC型肝炎に対して活性である、請求項13に記載の使用。
【請求項17】
前記生物学的利用能エンハンサーは、シトクロムP450酵素の阻害剤もしくは基質;イミダゾール;マクロライド;カルシウムチャネルブロッカー;またはステロイドである、請求項13に記載の使用。
【請求項18】
前記生物学的利用能エンハンサーは、シトクロムP450 3A(CYP3A)またはP糖タンパク質媒介性膜輸送の阻害剤である、請求項13に記載の使用。
【請求項19】
前記抗ウイルス性化合物は、シドフォビルもしくは環式シドフォビルである、請求項13に記載の使用。
【請求項20】
前記抗ウイルス性化合物は、任意選択的にアルキルプロパンジオール、1−S−アルキルチオグリセロール、アルコキシアルカノールもしくはアルキルエタンジオールに共有結合した、シドフォビル、アデフォビル、環式シドフォビルもしくはテノフォビルである、請求項13に記載の使用。
【請求項21】
前記抗ウイルス性化合物は、構造:
【化2】
を有する、請求項19に記載の使用。
【請求項22】
前記ウイルス感染症は、大痘瘡、小痘瘡、ワクシニア、痘瘡、ウシ痘、ラクダ痘、マウス痘、ウサギ痘、またはサル痘である、請求項13に記載の使用。
【請求項1】
ウイルス感染症を治療する方法であって、抗ウイルス性化合物、またはその塩、エステルもしくはプロドラッグの脂質含有プロドラッグを、ウイルス感染症を治療するための有効量で、治療を必要とする宿主に生物学的利用能エンハンサーと組み合わせて、または交互に投与するステップを含む方法。
【請求項2】
前記抗ウイルス性化合物はヌクレオシドである、請求項1に記載の方法。
【請求項3】
前記抗ウイルス性化合物は抗オルソポックス薬である、請求項1に記載の方法。
【請求項4】
前記抗ウイルス性化合物は、HIV、B型肝炎、またはC型肝炎に対して活性である、請求項1に記載の方法。
【請求項5】
前記生物学的利用能エンハンサーは、シトクロムP450酵素の阻害剤もしくは基質、イミダゾール;マクロライド;カルシウムチャネルブロッカー;またはステロイドである、請求項1に記載の方法。
【請求項6】
前記生物学的利用能エンハンサーは、シトクロムP450 3A(CYP3A)またはP糖タンパク質媒介性膜輸送の阻害剤である、請求項1に記載の方法。
【請求項7】
抗ウイルス性化合物、またはその塩、エステルもしくはプロドラッグの前記脂質含有プロドラッグ、および前記生物学的利用能エンハンサーは、医薬上許容される担体中で組み合わせて、または交互に投与される、請求項1に記載の方法。
【請求項8】
抗ウイルス性化合物、またはその塩、エステルもしくはプロドラッグの前記脂質含有プロドラッグ、および前記生物学的利用能エンハンサーは、経口、局所または非経口投与から選択された同一もしくは相違する経路によって投与される、請求項7に記載の方法。
【請求項9】
前記抗ウイルス性化合物は、シドフォビルもしくは環式シドフォビルである、請求項1に記載の方法。
【請求項10】
前記抗ウイルス性化合物は、アルキルプロパンジオール、1−S−アルキルチオグリセロール、アルコキシアルカノールもしくはアルキルエタンジオールに任意選択的に共有結合した、シドフォビル、アデフォビル、環式シドフォビルまたはテノフォビルである、請求項1に記載の方法。
【請求項11】
前記抗ウイルス性化合物は、構造:
【化1】
を有する、請求項9に記載の方法。
【請求項12】
前記ウイルス感染症は、大痘瘡(variola major)、小痘瘡(variola minor)、ワクシニア、天然痘、ウシ痘、ラクダ痘、マウス痘、ウサギ痘、またはサル痘である、請求項1に記載の方法。
【請求項13】
ウイルス感染症を治療するための薬剤の製造における、有効量の抗ウイルス性化合物、またはその塩、エステルもしくはプロドラッグの脂質含有プロドラッグの、生物学的利用能エンハンサーと組み合わせた、または交互の使用。
【請求項14】
前記抗ウイルス性化合物はヌクレオシドである、請求項13に記載の使用。
【請求項15】
前記抗ウイルス性化合物は抗オルソポックス薬である、請求項13に記載の使用。
【請求項16】
前記抗ウイルス性化合物は、HIV、B型肝炎、またはC型肝炎に対して活性である、請求項13に記載の使用。
【請求項17】
前記生物学的利用能エンハンサーは、シトクロムP450酵素の阻害剤もしくは基質;イミダゾール;マクロライド;カルシウムチャネルブロッカー;またはステロイドである、請求項13に記載の使用。
【請求項18】
前記生物学的利用能エンハンサーは、シトクロムP450 3A(CYP3A)またはP糖タンパク質媒介性膜輸送の阻害剤である、請求項13に記載の使用。
【請求項19】
前記抗ウイルス性化合物は、シドフォビルもしくは環式シドフォビルである、請求項13に記載の使用。
【請求項20】
前記抗ウイルス性化合物は、任意選択的にアルキルプロパンジオール、1−S−アルキルチオグリセロール、アルコキシアルカノールもしくはアルキルエタンジオールに共有結合した、シドフォビル、アデフォビル、環式シドフォビルもしくはテノフォビルである、請求項13に記載の使用。
【請求項21】
前記抗ウイルス性化合物は、構造:
【化2】
を有する、請求項19に記載の使用。
【請求項22】
前記ウイルス感染症は、大痘瘡、小痘瘡、ワクシニア、痘瘡、ウシ痘、ラクダ痘、マウス痘、ウサギ痘、またはサル痘である、請求項13に記載の使用。
【図1】

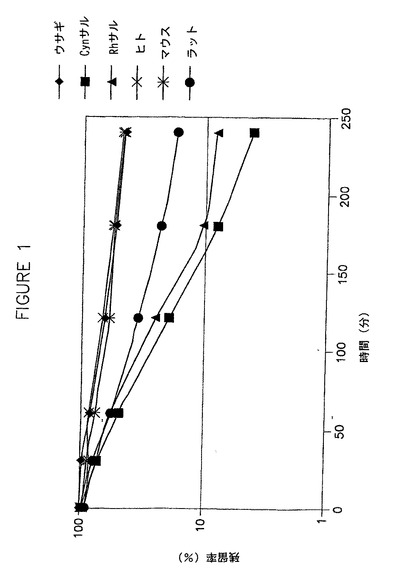
【図2】

【図3】

【図4】

【図5】
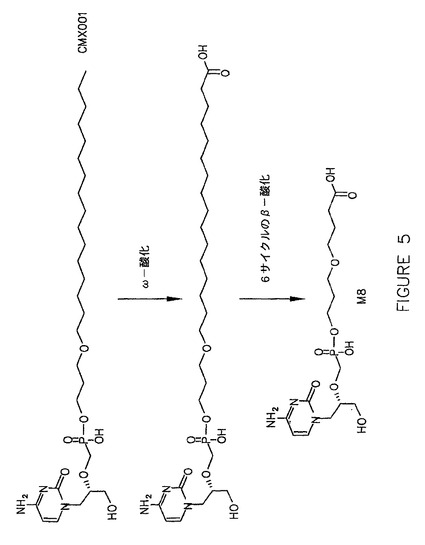
【図6】

【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2011−225615(P2011−225615A)
【公開日】平成23年11月10日(2011.11.10)
【国際特許分類】
【出願番号】特願2011−170912(P2011−170912)
【出願日】平成23年8月4日(2011.8.4)
【分割の表示】特願2008−505632(P2008−505632)の分割
【原出願日】平成18年4月10日(2006.4.10)
【出願人】(507334738)キメリクス,インコーポレイテッド (3)
【Fターム(参考)】
【公開日】平成23年11月10日(2011.11.10)
【国際特許分類】
【出願日】平成23年8月4日(2011.8.4)
【分割の表示】特願2008−505632(P2008−505632)の分割
【原出願日】平成18年4月10日(2006.4.10)
【出願人】(507334738)キメリクス,インコーポレイテッド (3)
【Fターム(参考)】
[ Back to top ]