
ウレアーゼ遺伝子を欠失又は不活性化させたロドコッカス属細菌
【課題】ニトリルヒドラターゼを安定的、効率的に発現させたロドコッカス属細菌および、低コストかつ高効率なアミド化合物の製造方法の提供。
【解決手段】ロドコッカス属細菌が有するウレアーゼ遺伝子を欠失または不活性化した遺伝子欠損ロドコッカス属細菌。
【解決手段】ロドコッカス属細菌が有するウレアーゼ遺伝子を欠失または不活性化した遺伝子欠損ロドコッカス属細菌。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ニトリルヒドラターゼ活性を有するロドコッカス属細菌におけるウレアーゼ遺伝子を欠失又は不活性化させたロドコッカス属細菌に関する。また、当該ロドコッカス属細菌を利用した、ニトリルヒドラターゼの製造方法及びアミド化合物の製造方法に関する。
【背景技術】
【0002】
近年、ニトリル基を水和しアミド基に変換するニトリル水和活性を有する酵素であるニトリルヒドラターゼが発見され、該酵素または該酵素を含有する微生物菌体等を用いてニトリル化合物より対応するアミド化合物を製造する方法が開示されている。この製造方法は、従来の化学合成法と比較し、ニトリル化合物から対応するアミド化合物への転化率及び選択率が高いことで知られている。
【0003】
ニトリルヒドラターゼを生産する微生物としては、例えば、コリネバクテリウム(Corynebacterium)属、シュードモナス(Pseudomonas)属、ロドコッカス(Rhodococcus)属、リゾビウム(Rhizobium)属、クレビシエラ(Klebsiella)属、シュードノカルディア(Pseudonocardia)属、ノカルディア(Nocardia)属等に属する細菌(微生物)を挙げることができる。中でも、ロドコッカス属細菌の一種であるロドコッカス・ロドクロウス(Rhodococcus rhodochrous)J1株(以下、J1株と称する)は、アクリルアミドの工業的生産に使用されており、有用性が実証されている。また、その菌株が産生するニトリルヒドラターゼをコードする遺伝子も明らかとなっている(特許文献1)。
【0004】
ロドコッカス・ロドクロウスは尿素を誘導剤としてニトリルヒドラターゼを産生し、同時に尿素を分解するウレアーゼ活性も持っていることが知られている(特許文献2、非特許文献1)。これらの微生物において、尿素を誘導剤としてニトリルヒドラーゼを発現させると、ウレアーゼにより尿素が分解されることから酵素発現が不安定になり、完全に発現を制御することは困難であった。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特許第3162091号公報
【特許文献2】特表2007−531535号公報
【非特許文献】
【0006】
【非特許文献1】Appl.Microbiol.Biotechnol(1991)34:783−788
【発明の概要】
【発明が解決しようとする課題】
【0007】
従って、本発明の主な目的は、ニトリルヒドラターゼを安定的、効率的に発現させたロドコッカス属細菌を提供し、低コストかつ高効率でアミド化合物を製造することにある。
【課題を解決するための手段】
【0008】
本発明は、上記状況を考慮してなされたもので、以下に示す、ウレアーゼ遺伝子が欠失又は不活性化されたロドコッカス属細菌、当該ロドコッカス属細菌を用いたニトリルヒドラターゼの製造方法、及び当該ロドコッカス属細菌を用いたアミド化合物の製造方法等を提供するものである。すなわち、本発明は以下の通りである。
【0009】
(1)ニトリルヒドラターゼ活性を有するロドコッカス属細菌において、ウレアーゼ遺伝子が欠失又は不活性化されたロドコッカス属細菌。
(2)ロドコッカス属細菌が、ロドコッカス ロドクロウス(Rhodococcus rhodochrous)である、(1)記載のロドコッカス属細菌。
(3)欠失または不活性化させるウレアーゼ遺伝子が、配列番号1に示される塩基配列を有するウレアーゼアルファサブユニットもしくはそのオーソログである、(1)又は(2)記載のロドコッカス属細菌。
(4)欠失または不活性化させるウレアーゼ遺伝子が、以下の(a)又は(b)に示されるいずれかの塩基配列を有するDNAからなる、(1)〜(3)のいずれか1項に記載のロドコッカス属細菌。
(a)配列番号1記載の塩基配列を含むDNA
(b)配列番号1記載の塩基配列と相同性が65%以上の塩基配列からなり、かつウレアーゼ活性を有するタンパク質をコードするDNA
(5)ロドコッカス属細菌が、ロドコッカス・ロドクロウス(Rhodococcus rhodochrous)J1株又はその変異株である、請求項1〜4記載のいずれか1項に記載のロドコッカス属細菌。
(6)(1)〜(5)のいずれか1項に記載のロドコッカス属細菌を培養し、得られる培養物からニトリルヒドラターゼを採取することを特徴とする、ニトリルヒドラターゼの製造方法。
(7)(1)〜(5)のいずれか1項に記載のロドコッカス属細菌を培養して得られる培養物又は当該培養物の処理物をニトリル化合物に接触させ、当該接触により生成されるアミド化合物を採取することを特徴とする、アミド化合物の製造方法。
【発明の効果】
【0010】
本発明によれば、尿素を誘導剤としてニトリルヒドラターゼを産生するロドコッカス属細菌において、安定的、効率的にニトリルヒドラターゼを発現させることができる。また、当該微生物を微生物触媒として用いて様々なニトリル化合物から低コストかつ高効率でアミド化合物を製造することもできる。
【図面の簡単な説明】
【0011】
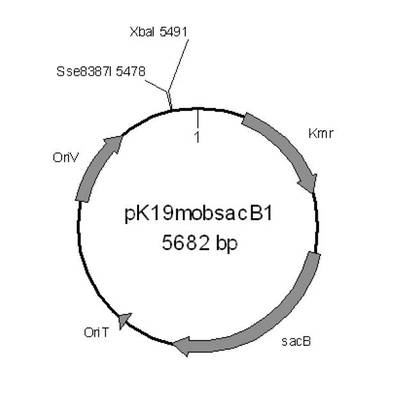
【図1】本実施例における、プラスミドpK19mobsacB1の構造を示す模式図である。
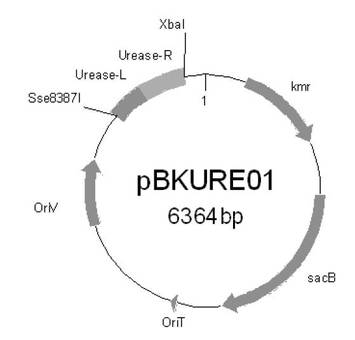
【図2】本実施例における、プラスミドpBKURE01の構造を示す模式図である。
【発明を実施するための最良の形態】
【0012】
以下、本発明を詳細に説明する。本発明の範囲はこれらの説明に拘束されることはなく、以下の例示以外についても、本発明の趣旨を損なわない範囲で適宜変更し実施することができる。なお、本明細書において引用された全ての刊行物、例えば先行技術文献、及び公開公報、特許公報その他の特許文献は、参照として本明細書に組み込まれる。
【0013】
1.ウレアーゼ遺伝子が改変されたロドコッカス属細菌
本発明に係るウレアーゼ遺伝子が改変(欠失又は不活性化(以下、「欠失等」と称する))されたロドコッカス属細菌(以下、「本発明のロドコッカス属細菌」と称する)は、前述した通り、ニトリルヒドラターゼ活性を有するロドコッカス属細菌におけるウレアーゼ遺伝子が欠失又は不活性化されたロドコッカス属細菌である。
【0014】
本発明のロドコッカス属細菌は、ニトリルヒドラターゼ活性を有するロドコッカス属細菌であって、ニトリルヒドラターゼおよび、ウレアーゼを産生し、それらの活性を示すものであればよく、特に限定はされない。
【0015】
例えば、ロドコッカス・ロドクロウス(Rhodococcus rhodochrous)及びロドコッカス・エリスロポリス(Rhodococcus erythropolis)、ロドコッカス・オパカス(Rhodococcus opacus),ロドコッカス・ジョスティ(Rhodococcus jostii)、ロドコッカス・エクイ(Rhodococcus equi)等が好ましく挙げられる。
【0016】
ロドコッカス・ロドクロウスとしては、例えば、ロドコッカス・ロドクロウス(Rhodococcus rhodochrous)J1(FERM BP−1478)、ロドコッカス・ロドクロウス(Rhodococcus rhodochrous)M8(SU1731814)(VKPMB−S926)、ロドコッカス・ロドクロウス(Rhodococcus rhodochrous)M33(VKM Ac−1515D)及びロドコッカス・ロドクロウス(Rhodococcus rhodochrous)NCIMB41164(国際公開第05/054456号)等が好ましく挙げられる。ロドコッカス エリスロポリス(Rhodococcus erythropolis)としては、例えばロドコッカス エリスロポリス(Rhodococcus erythropolis)PR4(NBRC 100887)、ロドコッカス エリスロポリス(Rhodococcus erythropolis)SK121等が好ましく挙げられる。ロドコッカス オパカス(Rhodococcus opacus)としては、例えばロドコッカス オパカス(Rhodococcus opacus)B4(NBRC 108011)が好ましく挙げられる。ロドコッカス ジョスティ(Rhodococcus jostii)としては、例えばロドコッカス ジョスティ(Rhodococcus jostii)RHA1が好ましく挙げられる。 ロドコッカス・エクイ(Rhodococcus equi)としては、例えば、ロドコッカス・エクイ(Rhodococcus equi)103S等が好ましく挙げられる。
【0017】
なお、上記NBRC株は独立行政法人製品評価技術基盤機構バイオテクノロジー本部生物遺伝資源部門 (NBRC) から、FERM株は、独立行政法人 産業技術総合研究所 特許生物寄託センターから、VKPM株はルシアン・ナショナル・コレクション・オブ・インダストリアル・マイクロオーガニズムから、VKM株はルシアン・コレクション・オブ・マイクロオーガニズムからそれぞれ入手可能である。
【0018】
ウレアーゼ(EC3.5.1.5)は、以下の反応式に示すように、尿素を二酸化炭素とアンモニアへ加水分解する反応を触媒する酵素である。
(NH2)2CO + H2O → CO2 + 2 NH3
ここで、「ウレアーゼ活性」とは、尿素をアンモニアと二酸化炭素に変換させる活性を意味する。本発明のロドコッカス属細菌のウレアーゼ活性の測定方法としては、本発明のロドコッカス属細菌を尿素に接触させ、生成するアンモニアの量の増加を定量することで、ウレアーゼ活性を測定することができる。あるいは、尿素の消費量を定量することによってもウレアーゼ活性を測定することができる。
【0019】
ニトリルヒドラターゼは、以下の反応式に示すように、ニトリル化合物のニトリル基に作用し、対応するアミド化合物に変換する水和反応を触媒する酵素である。
R−CN + H2O → R−CONH2
(式中、Rは、置換又は無置換のアルキル基、置換又は無置換のアルケニル基、置換又は無置換のシクロアルキル基、置換又は無置換のアリール基、あるいは置換若しくは無置換の飽和又は不飽和複素環基を表す。)
【0020】
ここで、「ニトリルヒドラターゼ活性」とは、ニトリル化合物のニトリル基に作用して対応するアミド化合物に変換させる活性を意味する。本発明のロドコッカス属細菌のニトリルヒドラターゼ活性の測定方法としては、例えば、ニトリル化合物の1種であるアクリロニトリルを用いる方法が挙げられる。この方法では、ニトリルヒドラターゼ活性の結果としてアクリロニトリルに対応するアミド化合物、すなわちアクリルアミドが得られる。従って、本発明のロドコッカス属細菌をアクリロニトリルに接触させ、生成するアクリルアミドの量の増加を定量することで、ニトリルヒドラターゼ活性を測定することができる。あるいは、アクリロニトリルの消費量を定量することによってもニトリルヒドラターゼ活性を測定することができる。
【0021】
本発明のロドコッカス属細菌は、本来有するウレアーゼ遺伝子が欠失又は不活性化されたものであるが、この欠失等される遺伝子(標的遺伝子)としては、対象微生物のゲノムDNAにコードされるウレアーゼ遺伝子の一部又は全部が欠失等されたもの、プロモーター配列の一部又は全部が欠失等されたもの、発現調節に関連する遺伝子の一部又は全部が欠失等されたものなどが挙げられる。これらの欠失等された領域は単独でもよいし複数を組み合わせたものでもよい。
【0022】
2.ウレアーゼ遺伝子が改変されたロドコッカス属細菌の製造
ロドコッカス属細菌のウレアーゼは3つのサブユニット(アルファ及びベータ及びガンマ)が組み合わさって構成されており、それぞれのサブユニットは隣り合う3つの遺伝子(ureC及びureB及びureA)にコードされている。加えて、ウレアーゼの活性部位への金属取り込みに寄与する数種類のウレアーゼアクセサリータンパク質が知られている。ウレアーゼ活性は、これらのいずれかを欠失することで失活させることが可能である。すなわち、ウレアーゼ活性失活株を取得するには、ureA又はureB又はureC又はウレアーゼアクセサリータンパク質遺伝子のいずれかを欠失又は不活性化させればよく、その組合せや欠損の順序は問わない。特にウレアーゼアルファサブユニットは活性部位をもつことが知られており、該当酵素サブユニットをコードするureCを欠失させることが好ましい。
【0023】
本発明に用いられるウレアーゼ遺伝子は、ロドコッカス・ロドクロウス(Rhodococcus rhodochrous)J1から単離されたウレアーゼ遺伝子ureCであり、配列番号1に示される塩基配列を含む(配列番号1:ORF)。
J1菌以外のロドコッカス属細菌については、上記J1菌のウレアーゼアルファサブユニットに対応するオーソログを欠失または不活性化させることができる。
ここで、「オーソログ」とは、異なる生物に存在する相同な機能を有するタンパクをコードする類縁遺伝子であって、ウレアーゼ遺伝子の場合であれば、そのオーソログとはJ1菌以外の生物においてウレアーゼ活性を有するタンパクをコードする類縁遺伝子である。
上記したウレアーゼ遺伝子のオーソログは、ウレアーゼ遺伝子と高い配列相同性を有する。それゆえ、上記配列番号1に塩基配列を有する遺伝子に加えて、これらの配列と約65%以上、好ましくは約70%以上、より好ましくは約80%以上、特に好ましくは約90%以上、さらに特に好ましくは約95%以上の相同性(同一性)を有する塩基配列を有するDNAも本発明に用いられるウレアーゼ遺伝子に含まれる。
【0024】
本発明の微生物を製造するための、ウレアーゼ遺伝子を欠失又は不活性化する方法は、特に限定されない。例えば、自然変異誘発法、紫外線照射や変異誘発剤(N‐メチル−N´−ニトロ−N−ニトロソグアニジン、メチルメタンスルホン酸等)を用いる突然変異誘発法、ゲノムDNAの相同領域を有するプラスミドを形質転換する方法を用いることができる。形質転換の方法としては、エレクトロポレーション法や接合伝達法が使用できる。
【0025】
上記欠失等する方法については、本発明では以下の方法により行われるものが好ましい。
すなわち、ドナー微生物からニトリルヒドラターゼ活性を有するレシピエント微生物への接合伝達を利用した形質転換方法を用いることを含む、ウレアーゼ遺伝子の欠失又は不活性化方法であって、以下の工程(イ)〜(ニ)を含むことを特徴とする方法である。
(イ)レシピエント微生物として、前記接合伝達に供するドナー微生物が感受性を示す薬剤への耐性が強化された微生物を作製する工程;
(ロ)ドナー微生物として、下記(i)〜(v):
(i) レシピエント微生物中のウレアーゼ遺伝子(標的遺伝子)とその周辺の塩基配列とを含む塩基配列において当該ウレアーゼ遺伝子を欠失又は不活性化させた塩基配列領域、
(ii) 当該ドナー微生物において機能する接合伝達開始領域、
(iii) 当該ドナー微生物において機能する複製開始領域、
(iv) レシピエント微生物が感受性を示す薬剤に対する耐性遺伝子、及び
(v) レシピエント微生物に対する条件致死遺伝子
を含む、遺伝子改変用プラスミドを用いて形質転換された微生物を作製する工程;
(ハ)工程(ロ)で作製されたドナー微生物から工程(イ)で作製されたレシピエント微生物への接合伝達を行うことにより、当該レシピエント微生物の形質転換体を作製する工程;並びに
(ニ)工程(ハ)で作製された形質転換体を、前記条件致死遺伝子が機能し得る培養条件で培養する工程。
【0026】
上記欠失又は不活性化方法において用いるレシピエント微生物としては、ニトリルヒドラターゼ活性を有し、ウレアーゼ遺伝子を有する微生物であれば限定はされない。中でもロドコッカス属細菌が、前述した理由でより好ましい。ロドコッカス属細菌等の具体例についても、前述と同様のものが挙げられる。
一方、ドナー微生物としては、上記レシピエント微生物と接合伝達可能な微生物であれば限定はされない。例えば、大腸菌が好ましい。
【0027】
2.1. 工程(イ)
工程(イ)では、接合伝達に供するレシピエント微生物として、接合伝達に供するドナー微生物が感受性を示す薬剤への耐性を強化した微生物を作製する。「薬剤への耐性を強化する」とは、レシピエント微生物が薬剤耐性を有していない場合には、薬剤耐性を付与することをいい、レシピエント微生物が薬剤耐性の乏しい場合には、当該耐性をより強くすることをいう。
【0028】
接合伝達法を使用する場合、レシピエントとなる微生物には薬剤耐性マーカーが必要である。よって、薬剤耐性を有していない微生物、又は薬剤耐性の乏しい微生物を使用する場合、薬剤選択可能な程度の十分な薬剤耐性を有する株(レシピエント)の作製が必要となる。ここで、薬剤としては、接合伝達法を使用することを考慮し、接合伝達に供するドナー微生物(接合伝達時のドナー微生物)が感受性を示す薬剤が好ましい。当該薬剤としては、クロラムフェニコール、アンピシリン、カナマイシン、トリメトプリム、ゲンタマイシン、ナルジクス酸、カルベニシン、チオストレプトン、テトラサイクリン及びストレプトマイシン等が好ましく、クロラムフェニコール及びアンピシリンがより好ましい。
【0029】
上述のように薬剤耐性を強化したレシピエント微生物を作製する方法としては、特には限定されない。例えば、自然変異誘発法、紫外線照射や変異誘発剤を用いる突然変異誘発法、EZ−Tn5(Epicentre社製)のようなランダム変異導入ツール等を用いることにより、本来レシピエント微生物が持たない薬剤耐性遺伝子を人為的に当該微生物ゲノム上に導入する方法、あらかじめ遺伝子改変用プラスミドとは別の、抗生物質耐性獲得用のプラスミドを導入する方法等が好ましい。これらの中でも自然変異誘発法がより好ましい。
自然変異誘発法は、所望の薬剤を含有する培地中で対象とする微生物を継代培養等することにより、もともとは当該培地中で生育不可又は困難な微生物に自然変異を誘発させて、より高濃度の薬剤を含有する当該培地中でも生育し得る株を取得する方法である。どの程度まで薬剤耐性を強化するかは、使用するレシピエント微生物、ドナー微生物、選択する薬剤により異なるが、レシピエント微生物の生育が抑制されない、且つ、ドナー微生物の生育が阻害される薬剤濃度を選ぶことが好ましい。例えば、レシピエント微生物としてロドコッカス属細菌を、ドナー微生物として大腸菌を、選択用薬剤としてクロラムフェニコールを用いる場合、自然突然変異によりクロラムフェニコール1〜200mg/L、好ましくは10〜100mg/Lを含有する培地において生育可能なレシピエント微生物(クロラムフェニコール耐性強化株)を得ることが望ましい。
【0030】
2.2. 工程(ロ)
工程(ロ)では、接合伝達に供するドナー微生物として、所定の遺伝子改変用プラスミドを用いて形質転換された微生物を作製する。遺伝子改変用プラスミド、すなわちレシピエント微生物中のウレアーゼ遺伝子を改変するためのプラスミドDNAとしては、前述の(i)〜(v)の構成(遺伝子・塩基配列)を含むものを用いる。
ここで、前記(i)の塩基配列領域は、改変の対象とするレシピエント微生物中のウレアーゼ遺伝子と当該遺伝子の周辺の塩基配列とを含む塩基配列において、当該ウレアーゼ遺伝子を欠失又はさせた塩基配列領域である。当該塩基配列領域の作製は、レシピエント微生物のゲノムから、ウレアーゼ遺伝子と当該遺伝子の周辺の塩基配列とを含む塩基配列の単離(クローニング)及び遺伝子ライブラリー作製やPCR等の公知技術を用いて行うことができる。
なお、ウレアーゼ遺伝子の周辺の塩基配列としては、限定はされない。例えば、当該遺伝子に加え、発現調節に関連する遺伝子の上流及び下流の相同領域が両端からそれぞれ100〜3000bpの塩基配列を含む配列であることが好ましい。より好ましくは500〜2000bpの塩基配列を含む配列である。単離した塩基配列を用いて、前述(i)を作製する方法は特に限定されず、PCR法や制限酵素を用いた標的遺伝子部分の切除もしくは置換等の公知技術を用いて行うことができる。
【0031】
単離したウレアーゼ遺伝子を欠失又は不活性化する方法としては、限定はされず、如何なる方法を適用してもよい。例えば、SOE−PCR法や、ゲノムDNAの相同領域を有するプラスミドを形質転換する方法を用いることができる。さらに、形質転換の方法としては、エレクトロポレーション法や接合伝達法が使用できる。
上記欠失又は不活性化されたウレアーゼ遺伝子としては、レシピエント微生物のゲノムDNAにコードされる当該ウレアーゼ遺伝子の一部又は全部が欠失等されたもの、プロモーター配列の一部又は全部が欠失等されたもの、発現調節に関連する遺伝子の一部又は全部が欠失等されたものなどが挙げられる。これらの欠失等された領域は単独でもよいし複数を組み合わせたものでもよい。
【0032】
前記(ii)の接合伝達開始領域は、使用するドナー微生物中において接合伝達の開始点となる塩基配列を含む領域であれば、限定はされない。例えば、プラスミドRP4由来のoriTが好ましい。
【0033】
前記(iii)の複製開始領域は、使用するドナー微生物中において前記遺伝子改変用プラスミドの自己複製起点として機能し得る塩基配列を含む領域であれば、限定はされない。例えば、oriVが好ましい。
【0034】
前記(iv)の薬剤耐性遺伝子は、接合伝達に供するレシピエント微生物(例えばロドコッカス属細菌等)が感受性を示す薬剤に対する耐性遺伝子であれば、限定はされない。例えば、アンピシリン耐性遺伝子、カナマイシン耐性遺伝子、クロラムフェニコール耐性遺伝子、トリメトプリム耐性遺伝子、ゲンタマイシン耐性遺伝子、ナルジクス酸耐性遺伝子、カルベニシン耐性遺伝子、チオストレプトン耐性遺伝子等が好ましく挙げられる。レシピエント微生物がこれらの薬剤耐性を有していない場合、所望の薬剤耐性を有する変異株を作製し、使用することができる。
【0035】
前記(v)の条件致死遺伝子は、レシピエント微生物のゲノム上に導入された場合に、当該微生物を死に至らしめる作用を有し得る遺伝子であれば、限定はされない。例えば、sacB遺伝子が好ましい。sacB遺伝子は、当該遺伝子を保有し発現する微生物(例えばロドコッカス属細菌等)をスクロース含有培地で培養した場合に、スクロースを基質とし当該微生物に対して致死作用を有する有害物質を産生する酵素をコードする遺伝子である。
【0036】
接合伝達に用いる前記遺伝子改変用プラスミドは、如何なるベクターをベースとして構築されたものであってもよく、限定はされない。例えば、レシピエント微生物がロドコッカス属細菌の場合は、pK19mobベクター(Schaefer et al,Gene1,vol.45,p.69−73(1994))等を用いることが好ましい。
【0037】
前記遺伝子改変用プラスミドにおける(i)〜(v)の構成は、例えば、上流から順に、前記(i)の塩基配列領域、前記(iv)の耐性遺伝子、前記(v)の条件致死遺伝子、前記(ii)の接合伝達開始領域、前記(iii)の複製開始領域の順で配されていることが好ましい。前記(i)〜(v)の構成を含む遺伝子改変用プラスミドの構築は、公知の遺伝子組換え技術を用いて実施することができる。
工程(ロ)では、上述したような各構成を有する遺伝子改変用プラスミドをドナー微生物内に導入して形質転換された微生物、すなわち接合伝達に供するドナー微生物を作製する。その際、形質転換の方法としては、エレクトロポレーション法やカルシウム法(アルカリSDS法)等の、微生物の形質転換方法として公知の方法を用いることができる。
【0038】
2.3. 工程(ハ)
工程(ハ)では、工程(ロ)で作製されたドナー微生物から工程(イ)で作製されたレシピエント微生物への接合伝達を行う。通常は、ドナー微生物及びレシピエント微生物のそれぞれの細胞懸濁液を混合し、適当なプレート培地(LB培地等)上に均一に広げて、両微生物の接合を行わせる。
当該接合においては、ドナー微生物中の遺伝子改変用プラスミドがレシピエント微生物内に移動し、レシピエント微生物のゲノムと上記プラスミドとの相同配列で2重交叉が起こり当該ゲノム中のウレアーゼ遺伝子が欠失される。この接合により、レシピエント微生物の形質転換体が作製される。すなわち、当該形質転換体は、ドナー微生物中の遺伝子改変用プラスミドの一部が相同組換えによりレシピエント微生物のゲノム上に導入されたものである。
所望の形質転換体であるかどうかの確認は、レシピエント微生物自体の薬剤耐性、及び前記遺伝子改変用プラスミド由来の薬剤耐性を利用して行うことができる。具体的には、両薬剤を含む培地(例えば、カナマイシン及びクロラムフェニコール含有培地等)において上記接合後の微生物を培養することにより、所望の形質転換体を選択することができる。
【0039】
2.4. 工程(ニ)
工程(ニ)では、工程(ハ)で作製されたレシピエント微生物の形質転換体(形質転換微生物)を、前記遺伝子改変用プラスミド由来の条件致死遺伝子が機能し得る培養条件で培養(継代培養)する。条件致死遺伝子が機能し得る培養条件としては、限定はされないが、例えば条件致死遺伝子がsacB遺伝子の場合は、スクロース含有培地を用いた培養が好ましく挙げられる。
当該培養においては、上記条件致死遺伝子を有する形質転換微生物は生育困難であるため、継代培養により、自然誘発的に、当該微生物のゲノム上から相同組換えにより上記致死遺伝子を含む塩基配列領域が除かれた(脱落した)形質転換微生物を得ることができる。
ただし、当該得られた微生物の中には、レシピエント微生物中のウレアーゼ遺伝子が、当初の目的通り欠失又は不活性化しているものと、そうでないもの(上記脱落の際の相同組換えにより元のウレアーゼ遺伝子の機能が復活したもの)が含まれている。
よって、通常は、さらに別の培養条件でも培養したり、培養物を用いてウレアーゼ活性測定や各種タンパク質分析法を適用して分析することにより、所望の形質転換微生物を選択することがより好ましい。
【0040】
3.ニトリルヒドラターゼの製造方法
本発明のニトリルヒドラターゼの製造方法は、前述した本発明の微生物を培養し、得られる培養物から当該ニトリルヒドラターゼを採取することを特徴とする方法である。
ここで、当該製造方法に用いる本発明の微生物としては、上記ウレアーゼ遺伝子が改変された微生物に、ニトリルヒドラターゼ遺伝子を当該微生物の遺伝子に挿入(付加)させたものであってもよいし、ニトリルヒドラターゼ遺伝子を含む組換えベクターを導入することによりなされたものであってもよいし、その両方を兼ね備えてあるものであってもよい。
組換えベクターを用いる場合は、ニトリルヒドラターゼ遺伝子が、形質転換される本発明の微生物において発現可能なように、ベクターに組み込まれることが必要である。例えば、ニトリルヒドラターゼを産生し得る微生物のゲノム等からニトリルヒドラターゼ遺伝子を含むPCR断片を得て、得られた断片を発現ベクターに連結することで、ニトリルヒド
ラターゼ遺伝子を含む発現ベクターを得ることができる。
【0041】
発現ベクターとしては、例えば、プラスミドDNA、バクテリオファージDNA、レトロトランスポゾンDNA、人工染色体DNAなどが挙げられ、中でもプラスミドDNAが好ましい。異種ニトリルヒドラターゼを発現させるためのベクター(プラスミドDNA)としては、限定されないが、例えば、ロドコッカス属細菌を宿主とする場合は、pSJ034、pSJ041、pSJ042、pSJ043等が好ましい。pSJ034は、ロドコッカス属細菌においてニトリルヒドラターゼを発現するベクターであり、特開平10−337185号公報に示す方法でpSJ023より作製することができる。このpSJ023は、形質転換体 ATCC16274/pSJ023(FERM BP−6232)として、独立行政法人産業技術総合研究所 特許生物寄託センター(茨城県つくば市東1丁目1番地1中央第6)に平成9年3月4日付けで国際寄託されている。
当該発現ベクターには、ニトリルヒドラターゼ遺伝子のほか、プロモーター、ターミネーター、エンハンサー、スプライシングシグナル、ポリA付加シグナル、選択マーカー、リボソーム結合配列(SD配列)等を連結することができる。なお、選択マーカーとしては、例えば、アンピシリン耐性遺伝子、カナマイシン耐性遺伝子、ジヒドロ葉酸還元酵素遺伝子、ネオマイシン耐性遺伝子等が挙げられる。
【0042】
前述の通り、本発明においては、ニトリルヒドラターゼは、上記形質転換微生物を培養し、得られる培養物から採取することにより製造することができる。
本発明において、「培養物」とは、培養上清、培養菌体、又は菌体の破砕物のいずれをも意味するものである。形質転換微生物を培養する方法は、宿主の培養に用いられる通常の方法に従って行われる。目的のニトリルヒドラターゼは、上記培養物中に蓄積される。
【0043】
形質転換微生物を培養する培地は、宿主菌となる本発明の微生物が資化し得る炭素源、窒素源、無機塩類等を含有し、微生物の培養を効率的に行うことができる培地であれば、天然培地、合成培地のいずれを用いてもよい。炭素源としては、グルコース、フラクトース、スクロース、デンプン等の炭水化物、酢酸、プロピオン酸等の有機酸、エタノール、プロパノール等のアルコール類が挙げられる。窒素源としては、アンモニア、塩化アンモニウム、硫酸アンモニウム、酢酸アンモニウム、リン酸アンモニウム等の無機酸若しくは有機酸のアンモニウム塩又はその他の含窒素化合物のほか、ペプトン、肉エキス、コーンスティープリカー等が挙げられる。無機物としては、リン酸第一カリウム、リン酸第二カリウム、リン酸マグネシウム、硫酸マグネシウム、塩化ナトリウム、硫酸第一鉄、硫酸マンガン、硫酸銅若しくは炭酸カルシウム等が挙げられる。また、必要に応じ、培養中の発泡を防ぐために消泡剤を添加してもよい。さらに、培地にはニトリルヒドラターゼの補欠分子であるコバルトイオンや鉄イオンを添加し、酵素の誘導剤となるニトリル類やアミド類を添加してもよい。
【0044】
培養中、発現ベクターや目的遺伝子の脱落を防ぐために選択圧を掛けた状態で培養してもよい。すなわち、発現ベクター等における選択マーカーが薬剤耐性遺伝子である場合には相当する薬剤を培地に添加したり、選択マーカーが栄養要求性相補遺伝子である場合には相当する栄養因子を培地から除いたりしてもよい。また、選択マーカーが資化性付与遺伝子である場合には、相当する資化因子を必要に応じて唯一因子として添加することができる。例えば、アンピシリン耐性遺伝子を含むベクターで形質転換した微生物を培養する場合、培養中、必要に応じてアンピシリンを添加してもよい。
【0045】
プロモーターとして誘導性のプロモーターを用いた発現ベクターで形質転換した形質転換微生物を培養する場合は、必要に応じてインデューサーを培地に添加してもよい。例えば、イソプロピル−β−D−チオガラクトシド(IPTG)で誘導可能なプロモーターを有する
発現ベクターで形質転換した形質転換微生物を培養するときには、IPTG等を培地に添加することができる。また、インドール酢酸(IAA)で誘導可能なtrpプロモーターを用いた発現ベクターで形質転換した形質転換微生物を培養するときには、IAA等を培地に添加することができる。
【0046】
形質転換微生物の培養条件は、発現目的のニトリルヒドラターゼの生産性及び宿主の生育が妨げられない条件であれば、特段限定されるものではない。通常、10℃〜45℃、好ましくは10℃〜40℃の温度下で、5〜120時間、好ましくは5〜100時間程度行うことができる。pHの調整は、無機酸又は有機酸、アルカリ溶液等を用いて、適時調整を行うことが好ましい。
【0047】
培養方法としては、固体培養、静置培養、振盪培養、通気攪拌培養などが挙げられるが、振盪培養又は通気攪拌培養(ジャーファーメンター)により好気的条件下で培養することが好ましい。
【0048】
また、場合により、本培養に先立ち、少量の前培養を行うこともできる。例えば、ロドコッカス属細菌の形質転換微生物を培養する場合は、振盪培養又は通気攪拌培養などの好気的条件下、30〜40℃の温度下で行うことが好ましい。
上記培養条件で培養すると、高収率でニトリルヒドラターゼを上記培養物中、すなわち培養上清、培養菌体、又は菌体の破砕物の少なくともいずれかに蓄積することができる。これらニトリルヒドラターゼを含有する「培養物」は、後述するアミド化合物の製造方法に使用することができる。
【0049】
培養後、ニトリルヒドラターゼが菌体内に生産される場合には、菌体を破砕することにより、目的のニトリルヒドラターゼを採取することができる。菌体の破砕方法としては、フレンチプレス又はホモジナイザーによる高圧処理、超音波処理、ガラスビーズ等による磨砕処理、リゾチーム、セルラーゼ又はペクチナーゼ等を用いる酵素処理、凍結融解処理、低張液処理、ファージによる溶菌誘導処理等を利用することができる。
【0050】
破砕後、必要に応じて菌体の破砕残渣(細胞抽出液不溶性画分を含む)を除くことができる。残渣を除去する方法としては、例えば、遠心分離やろ過などが挙げられ、必要に応じて、凝集剤やろ過助剤等を使用して残渣除去効率を上げることもできる。残渣を除去した後に得られた上清は、細胞抽出液可溶性画分であり、粗精製したニトリルヒドラターゼ溶液とすることができる。
【0051】
また、ニトリルヒドラターゼが菌体内に生産される場合、菌体そのものを遠心分離、膜分離等で回収して、未破砕のまま使用することも可能である。
ニトリルヒドラターゼが菌体外に生産される場合には、培養液をそのまま使用するか、遠心分離やろ過等により菌体を除去する。その後、必要に応じて硫安沈澱による抽出等により前記培養物中から異種ニトリルヒドラターゼを採取し、さらに必要に応じて透析、各種クロマトグラフィー(ゲルろ過、イオン交換クロマトグラフィー、アフィニティクロマトグラフィー等)を用いて単離精製することもできる。
形質転換微生物を培養して得られたニトリルヒドラターゼの生産収率は、例えば、培養液あたり、菌体湿重量又は乾燥重量あたり、粗酵素液タンパク質あたりなどの単位で、SDS−PAGE(ポリアクリルアミドゲル電気泳動)やニトリルヒドラターゼ活性測定などにより確認することができるが、特段限定されるものではない。SDS−PAGEは当業者であれば公知の方法を用いて行うことができる。また、ニトリルヒドラターゼ活性は、上述した活性の値を適用することができる。
【0052】
4.アミド化合物の製造方法
上述のように製造されたニトリルヒドラターゼ(前述した培養物を含む)は、酵素触媒(菌体のまま利用する微生物触媒等を含む)として物質生産に利用することができる。例えば、ニトリル化合物に、ニトリルヒドラターゼを接触させることにより、当該接触によりニトリル化合物が変換されてアミド化合物を製造することができる。
【0053】
酵素触媒としては、前述のように適当な宿主内でニトリルヒドラターゼ遺伝子が発現するように遺伝子導入を行い、宿主を培養した後の培養物、又は当該培養物の処理物を利用することができる。処理物としては、例えば、培養後の菌体をアクリルアミド等のゲルで包含したもの、グルタルアルデヒドで処理したもの、アルミナ、シリカ、ゼオライト及び珪藻土等の無機担体に担持したもの等が挙げられる。
【0054】
基質として使用されるニトリル化合物は、酵素の基質特異性、酵素の基質に対する安定性等を考慮して選択される。ニトリル化合物としては、アクリロニトリルが好ましい。
反応方法、及び反応終了後のアミド化合物の採取方法は限定されず、基質及び酵素触媒の特性により適宜選択することができる。例えば、当該反応において、基質となるニトリル化合物の濃度は、0.1〜10%(W/V)が好ましく、5%(W/V)程度が特に好ましい。また、当該反応は、pH5〜10の緩衝液又は水中で行うことが好ましく、例えば、50mMリン酸緩衝液(pH7.0)中で行うことができる。反応後の酵素触媒は、その活性が失活しない限り、リサイクル使用することが好ましい。失活の防止やリサイクルを容易にすることに鑑み、酵素触媒は処理物の形態で使用されることが好ましい。
上記反応により生成したアクリルアミドは、ガスクロマトグラフィーなどの公知の方法を用いて定量することができる。また、アクリルアミドの産生量から前記酵素触媒のニトリルヒドラターゼ活性を換算することができる。
【0055】
以下に、実施例を挙げて本発明をより具体的に説明するが、本発明はこれらに限定されるものではない。
【実施例】
【0056】
[調製例1]
ウレアーゼ欠失用プラスミドの作製
(1)接合伝達プラスミド(pK19mobsacB1)の作製
pDNR−1r(clontech社製)中のsacB遺伝子を、NspV切断サイトを付加したプライマーSAC−01(配列番号2)及びSAC−02(配列番号3)を使用したPCRにより増幅し、約1.9kbのsacB遺伝子断片を得た。PCRは以下の反応条件で行った。
【0057】
プライマー:
SAC−01:5’−GGTTCGAATACCTGCCGTTCACTATTATTTAGTG−3’(配列番号2)
SAC−02:5’−GGTTCGAATCGGCATTTTCTTTTGCGTTTTTATTTG−3’(配列番号3)
【0058】
反応液組成:
滅菌水 22μl
PrimeSTAR(登録商標) Max Premix(2×) (タカラバイオ社製) 25μl
SAC−01(10μM)(配列番号2) 1μl
SAC−02(10μM)(配列番号3) 1μl
pDNR−1r(100ng)(clontech社製) 1μl
総量 50μl
温度サイクル:
98℃ 10秒、55℃ 15秒及び72℃ 20秒の反応を30サイクル
【0059】
sacB遺伝子断片を制限酵素NspV(タカラバイオ社製)で消化後、pK19mobのNspVサイトに接続し、sacB遺伝子が正方向に導入されたプラスミドpK19mobsacB1を構築した。pK19mob NspV切断断片とsacB遺伝子断片及びsacB遺伝子NspV切断断片の精製にはGFX PCR DNA band and Gel Band Purification kit(GE Healthcare社製)、両断片の接続にはDNA Ligation Kit <Mighty Mix>、プラスミドの抽出にはQIAprep miniprep kit(QIAGEN社製)を用いた。
【0060】
(2)J1株ゲノムDNAの調製
ロドコッカス・ロドクロウス(Rhodococcus rhodochrous)J1株を100mlのMYK培地(0.5%ポリペプトン、0.3%バクトイーストエキス、0.3%バクトモルトエキス、1% グルコース、0.2% K2HPO4、0.2% KH2PO4、pH 7.0)中、30℃にて72時間振盪培養した。
培養後、集菌し、集菌された菌体をSaline−EDTA溶液(0.1M EDTA、0.15M NaCl(pH8.0))4 mlに懸濁した。懸濁液にリゾチーム40 mgを加えて、37℃で1〜2時間振盪した後、−20℃で凍結した。
次に、10mlのTris−SDS液(1%SDS、0.1M NaCl、0.1M Tris−HCl(pH9.0))を穏やかに振盪しながら加え、さらにプロテイナーゼK(メルク社)を10μl(終濃度10mg/ml)加えて37℃で1時間振盪した。
次に、等量のTE(10mM Tris−HCl、1mM EDTA(pH8.0)) 飽和フェノールを加え、撹拌した後遠心した。遠心後、上層をとり2倍量のエタノールを加えた後、ガラス棒でDNAを巻きとり、90%、80%、70%のエタノールで順次フェノールを取り除いた。
次に、DNAを3mlのTE緩衝液に溶解させ、リボヌクレアーゼA溶液(100℃、15分間の加熱処理済)を10μg/mlになるよう加え、37℃で30分間振盪した。さらに、プロテイナーゼKを加え37℃で30分間振盪した後、等量のTE飽和フェノールを加えて遠心し、上層と下層に分離させた。
上層についてこの操作を2回繰り返した後、同量のクロロホルム(4%イソアミルアルコール含有)を加え、同様の抽出操作を繰り返した。その後、上層に2倍量のエタノールを加え、ガラス棒でDNAを巻きとり回収し、J1株ゲノムゲノムDNAを得た。
【0061】
(3)ウレアーゼ遺伝子(ureC)の増幅と発現プラスミドの構築
J1菌のウレアーゼ遺伝子ureCを含む配列を特定するために、GenBankに登録されているロドコッカス エリスロポリス(Rhodococcus erythropolis)PR4株の塩基配列(登録番号AP008957)を基に、以下示すプライマーおよび反応液組成を用いてPCRを行った。
プライマー:
ureC−01:5’−tgtacggcccgaccgctggcgaccag−3’(配列番号4)
ureC−02:5’−caccggctgcggggtcgggatagcagc−3’(配列番号5)
【0062】
反応液組成:
滅菌水 22μl
PrimeSTAR(登録商標) Max Premix(2×) (タカラバイオ社製) 25μl
ureC−01(10μM)(配列番号4) 1μl
ureC−02(10μM)(配列番号5) 1μl
J1株ゲノムDNA(100ng) 1μl
総量 50μl
温度サイクル:
98℃:10秒、55℃:15秒及び72℃:20秒の反応を30サイクル
【0063】
PCR終了後、反応液2μlを0.7%アガロースゲルにおける電気泳動に供し、約1.4kbpのPCR産物の検出を行った。PCR産物を確認した後、反応液をGFX PCR DNA band and GelBand Purification kit(アマシャムバイオサイエンス社製)で精製した。次に、精製したPCR産物をMighty Cloning Reagent Set(Blunt End)(タカラバイオ株式会社製)でpUC118にクローニングし、プライマーウォーキング法により、塩基配列を解析した。その結果、配列番号6に示す配列が得られた。ホモロジー検索(blastx)を実施した結果、表1の結果が得られ、ウレアーゼのアルファサブユニットと相同性が認められた。
【0064】
【表1】

【0065】
次に、J1菌のウレアーゼアルファサブユニットをコードするureC遺伝子の全長配列を特定するために以下の操作を行った。
BamHI、BsaI、BglII、NcoI、PvuI,SacI、SmaI、XhoIそれぞれで消化したJ1菌ゲノムDNAに対し、後述の方法で調製したUreCのプローブを用いてサザンハイブリダイゼーションを行ったところ、BglIIで消化した断片から、約3.5kbの単一シグナルが得られた。
【0066】
なお、ureCのプローブは以下のようにして調製した。
プライマーureC-01およびureC-02で調製したPCR産物(配列番号6)をGFX PCR DNA band and Gel Band Purification kit(GEヘルスケア バイオサイエンス社)を用いて精製した。精製したPCR産物に対してAlkPhos Direct Labeling kit(GEヘルスケア バイオサイエンス社)を用い、添付のマニュアルにしたがってラベリングを行い、ureCのプローブとした。
【0067】
続いて、J1菌ゲノムDNAを制限酵素BglIIで分解して0.7%アガロースゲル電気泳動で分離し、ゲルからGFX PCR DNA band and Gel Band Purification kit(GEヘルスケア バイオサイエンス社)を使用して約3.5kbの断片を回収した。得られた断片は、pBluescriptII SK(+)ベクター(Stratagene社製)にDNA ligation kit<Mighty mix>(タカラバイオ社製)を用いて連結した。反応条件は以下の通りである。
【0068】
反応液組成:
ligation mighty mix(タカラバイオ社製) 5μl
J1菌ゲノムDNA/BglII切断断片 4μl
pBluescriptII SK(+)/BglII切断断片 1μl
総量 10μl
反応:
16℃,1時間
【0069】
上記ライゲーション産物の全量を、大腸菌JM109株コンピテントセル200μlに加え、0℃で30分放置した。続いて、42℃で45秒間ヒートショックを与え、0℃で2分間冷却した。その後、SOC 培地(20 mMグルコース、2%バクトトリプトン、0.5%バクトイーストエキス、10mM NaCl,2.5mM KCl,1mM MgSO4,1mM MgCl2)を1ml添加し、37℃にて1時間振盪培養した。培養後の培養液を200μlずつ、LB AIXプレート(100μg/lアンピシリン、100μM IPTG,50μg/l X−galを含むLB寒天培地)に塗布し、37℃で一晩放置した。プレート上に生育した白色の組換コロニーを新しいLB AIXプレートに、プレート1枚に付き94個、プレート10枚分単離した。各プレートにはインサートを含まないpBluescriptII SK(+)で形質転換したJM109株を2コロニー/プレート植菌した。コロニー単離したプレートを37℃で一晩放置した後、Hybond−N+(GEヘルスケア バイオサイエンス社)膜にコロニーを写し取り、ureCのプローブを用いてコロニーハイブリダイゼーションを行った。
【0070】
検出されたコロニーを培養して得られた培養液を集菌後、QIAprep miniprep kit(QIAGEN社製)を用いて組換えプラスミドを回収した。キャピラリーDNAシーケンサーCEQ2000(ベックマン・コールター社製)を用いて、添付のマニュアルに従って、プラスミド中にクローニングされているゲノムDNA断片の塩基配列を解析した。その結果、配列番号7に示される塩基配列が得られた。配列番号7に示される塩基配列中に、配列番号1に示す約1.7kbpのオープンリーディングフレーム(ORF)を見出した。配列番号1の塩基配列のホモロジー検索(blastn)を実施した結果、表2の結果が得られ、ウレアーゼのアルファサブユニットと相同性が認められた。
【0071】
【表2】
【0072】
続いて、ウレアーゼ遺伝子(ureC)を含んだ配列を取得するため、以下に示す反応液組成及びプライマーを用いてPCRを行った。
プライマー:
ureC−03: 5’− ggCCTGCAGGGACAGTCGGCGACCAGGTACGCC −3’(配列番号8)
ureC−04: 5’− ACGGCGACAGCAGGTGCACGTGCGAGTCGAAG−3’(配列番号9)
【0073】
反応液組成:
滅菌水 22μl
PrimeSTAR(登録商標) Max Premix(2×) (タカラバイオ社製) 25μl
ureC−03(10μM)(配列番号8) 1μl
ureC−04(10μM)(配列番号9) 1μl
J1株ゲノムDNA(100ng) 1μl
総量 50μl
温度サイクル:
98℃:10秒、55℃:15秒及び72℃:20秒の反応を30サイクル
【0074】
同様に、下記プライマーureC−05(配列番号10)及びureC−06(配列番号11)を使用し、上記と同じ条件でPCRを実施した。
プライマー:
ureC−05: 5’− GTCGCATCGGTGAGGTCGTCACCCGCAC−3’(配列番号10)
ureC−06: 5’− ccTCTAGAGATCGACGCGTTCGGGTCGCCGAG−3’(配列番号11)
【0075】
PCR終了後、反応液2μlを0.7%アガロースゲルにおける電気泳動に供し、ureC−03とureC−04で約400 bp、ureC−05とureC−06で約300bpのPCR産物の検出を行った。PCR産物を確認した後、反応液をGFX PCR DNA band and GelBand Purification kit(アマシャムバイオサイエンス社製)で精製した。
【0076】
次に、上記で増幅した2つのPCR断片を連結するため、以下の条件でAssembly PCRを行った。
【0077】
反応液組成:
滅菌水 21μl
PrimeSTAR(登録商標) Max Premix(2×) (タカラバイオ社製) 25μl
ureC−03(10μM)(配列番号8) 1μl
ureC−06(10μM)(配列番号11) 1μl
ureC−03とureC−04の増幅産物(50ng) 1μl
ureC−05とureC−06の増幅産物(50ng) 1μl
総量 50μl
温度サイクル:
98℃:10秒、55℃:15秒及び72℃:40秒の反応を30サイクル
【0078】
PCR終了後、約1Kbの増幅産物をアガロース電気泳動で確認し、Mighty Cloning Reagent Set(Blunt End)(タカラバイオ株式会社製)でpUC118にクローニングした。
続いて、上記でクローニングした断片を制限酵素XbaI及びSse8387Iで切断し、約1kbの断片を回収し、接合伝達用のプラスミド(pK19mobsacB1)のXbaI−Sse8387I部位に連結し、プラスミドを作製した。得られたプラスミドはウレアーゼ遺伝子欠失プラスミド:pBKAMD01と名付けた。図1に、プラスミドpK19mobsacB1の構造を示す模式図を、図2に、プラスミドpBKURE01の構造を示す模式図を示した。
【0079】
[調製例2]
薬剤耐性を有するJ1株の作製
接合伝達に使用するドナーは、遺伝子欠失株のセレクションに薬剤耐性が必要である。そこで、種々の薬剤耐性株の取得を試み、アンピシリン耐性を有するJ1株の変異株を下記の方法で取得した。
2μg/mlのアンピシリンを含んだMYKプレート(0.5%ポリペプトン、0.3%バクトイーストエキス、0.3%マルツエキス、0.2% KH2PO4、0.2% K2HPO4、1.5%寒天)にJ1株をストリークし、コロニーが生育するまで30℃で保温した。約2週間後、アンピシリン耐性株が生育してきたので、再度2μg/mlのアンピシリンプレートにストリークして、30℃で保温した。
次に、2μg/mlのアンピシリンプレートから生育したコロニーを、5μg/mlのアンピシリンプレートにストリークし、耐性株が出現するまで30℃で保温した。以下、同様の操作を、アンピシリン濃度を10μg/ml→15μg/ml→50μg/ml→100μg/mlに高めながら繰り返し、100μg/mlのアンピシリン濃度で生育するアンピシリン耐性株(J1−Amp株)を得た。
【0080】
[実施例1]
接合伝達によるウレアーゼ遺伝子の欠失
(1)ドナーの調製
乾熱滅菌した試験管に大腸菌S17−1λpirのコンピテントセル20μlにプラスミドpBKURE01 1μlを加え、氷上で30分静置した。42℃で30秒ヒートショック後、SOC培地を180μl添加し、37℃で1時間振とう培養を行った。その後、50μg/mlカナマイシンを含んだLBプレートに塗布し、37℃で一晩静置した。
翌日、プレートに生育したコロニーをLB培地1mlで回収し、遠心分離により菌体を回収し、遠心上清を除去した。同様の操作をもう一度繰り返し、最後に0.5 mlのLB培地を添加し、菌体懸濁液を調製した。この菌体懸濁液をドナー溶液とした。
【0081】
(2)レシピエントの調整
J1−Amp株をMYKプレートにストリークし、30℃で2日生育させた。生育したコロニーは実施例1(1)と同様の方法で回収、洗浄し、レシピエントとなる菌体懸濁液を調製した。
【0082】
(3)接合伝達
実施例1(1)で調製したドナー溶液と、実施例1(2)で調製したレシピエント溶液を100μlずつ混合し、抗生物質を含まないMYKプレートに塗布し、30℃で一晩静置した。
翌日、生育したコロニーは1mlのLB培地で回収し、100μlずつカナマイシン濃度を10、30、50μg/mlとした選抜プレート(すべて100μg/mlアンピシリンを含む)に塗布した。
塗布したプレートは組み換え菌コロニーが出現するまで30℃で1週間保温した。
その結果、100μg/mlアンピシリン、10μg/mlカナマイシンを含んだプレートにのみ3個のコロニーが出現した。得られたコロニーの一つを#U1と命名し、以後の実験に使用した。
【0083】
(4)ウレアーゼ遺伝子の欠失
接合伝達により得られた組み換え菌#U1は、ゲノムのウレアーゼ遺伝子の領域に相同組換えによりプラスミドが挿入されているが、ウレアーゼ遺伝子を欠失するには2段階の相同組換えが必要である。そこで、次に、sacB遺伝子を利用した選抜を実施した。
10%ショ糖を含んだMYKプレートを作製し、#U1のコロニーを滅菌水に懸濁した液を適度に希釈して塗布し、30℃で静置した。生育したコロニーについて10個のコロニーからゲノムDNAを調製し、以下に示す反応液組成及びプライマーを用いてPCRを行って、得られた断片(PCR産物)のサイズを電気泳動で調べた。なお、プライマーureC−03及びureC−06は、調製例1で用いたものと同様である。
【0084】
反応液組成:
滅菌水 22μl
PrimeSTAR(登録商標) Max Premix(2×) (タカラバイオ社製) 25μl
ureC−03(10μM)(配列番号8) 1μl
ureC−06(10μM)(配列番号11) 1μl
ゲノムDNA(100ng) 1μl
総量 50μl
温度サイクル:
98℃:10秒、55℃:15秒及び72℃:20秒の反応を30サイクル
その結果、10個の内、5個のコロニーはウレアーゼ遺伝子が欠失していることが確認された。得られたウレアーゼ遺伝子欠失株を、それぞれUD1〜UD5株とした。
【0085】
[実施例2]
UD1〜UD5株の培養
UD1〜UD5株とJ1株5株(J1−1〜J1−5)を下記の培地で培養し、ウレアーゼ活性およびニトリルヒドラターゼ活性を確認した。本培養の培地は、ニトリルヒドラターゼの誘導剤である尿素とコバルト、を含むものを用いた。前培養は30℃で3日間、本培養は30℃で2日間、振とう培養した。
【0086】
[前培養培地組成](pH7.0)
グルコース 20g/L
味液 20g/L
ポリペプトン 5g/L
酵母エキス 3g/L
MgSO4・7H2O 1g/L
KH2PO4 1g/L
K2HPO4 1g/L
【0087】
[本培養液組成](pH7.0)
グルコース 15g/L
ポリペプトン 3g/L
酵母エキス 3g/L
MgSO4・7H2O 1g/L
KH2PO4 1g/L
K2HPO4 1g/L
エタノール 2g/L
CoCl2・6H2O 0.025g/L
チアミン 0.002g/L
ビタミンK 0.002g/L
尿素 15g/L
【0088】
菌体を含む培養液のウレアーゼ活性の測定は、以下の方法に従って行った。
まず、本培養後に得られた培養液を遠心分離して菌体を取り除き、上清を50mMリン酸緩衝液(pH7.0)で100倍希釈する。市販の尿素測定キット(F−キット 尿素/アンモニア;ロシュ・ダイアグノスティクス社製)を用いて希釈液に含まれる尿素濃度を測定し、ウレアーゼ活性を評価した。
培養液中の尿素濃度を測定した結果、培養開始時の尿素濃度15g/Lに対して、J1株の場合は平均で約3.5g/L残っていたが、ウレアーゼ遺伝子を欠失させたUD1〜UD5株ではほとんど減少しておらず平均14.5g/L残っていた。以上の結果よりウレアーゼ活性が欠失していることを確認した。
【配列表フリーテキスト】
【0089】
配列番号2:プライマーSAC−01
配列番号3:プライマーSAC−02
配列番号4:プライマーureC−01
配列番号5:プライマーureC−02
配列番号8:プライマーureC−03
配列番号9:プライマーureC−04
配列番号10:プライマーureC−05
配列番号11:プライマーureC−06
【技術分野】
【0001】
本発明は、ニトリルヒドラターゼ活性を有するロドコッカス属細菌におけるウレアーゼ遺伝子を欠失又は不活性化させたロドコッカス属細菌に関する。また、当該ロドコッカス属細菌を利用した、ニトリルヒドラターゼの製造方法及びアミド化合物の製造方法に関する。
【背景技術】
【0002】
近年、ニトリル基を水和しアミド基に変換するニトリル水和活性を有する酵素であるニトリルヒドラターゼが発見され、該酵素または該酵素を含有する微生物菌体等を用いてニトリル化合物より対応するアミド化合物を製造する方法が開示されている。この製造方法は、従来の化学合成法と比較し、ニトリル化合物から対応するアミド化合物への転化率及び選択率が高いことで知られている。
【0003】
ニトリルヒドラターゼを生産する微生物としては、例えば、コリネバクテリウム(Corynebacterium)属、シュードモナス(Pseudomonas)属、ロドコッカス(Rhodococcus)属、リゾビウム(Rhizobium)属、クレビシエラ(Klebsiella)属、シュードノカルディア(Pseudonocardia)属、ノカルディア(Nocardia)属等に属する細菌(微生物)を挙げることができる。中でも、ロドコッカス属細菌の一種であるロドコッカス・ロドクロウス(Rhodococcus rhodochrous)J1株(以下、J1株と称する)は、アクリルアミドの工業的生産に使用されており、有用性が実証されている。また、その菌株が産生するニトリルヒドラターゼをコードする遺伝子も明らかとなっている(特許文献1)。
【0004】
ロドコッカス・ロドクロウスは尿素を誘導剤としてニトリルヒドラターゼを産生し、同時に尿素を分解するウレアーゼ活性も持っていることが知られている(特許文献2、非特許文献1)。これらの微生物において、尿素を誘導剤としてニトリルヒドラーゼを発現させると、ウレアーゼにより尿素が分解されることから酵素発現が不安定になり、完全に発現を制御することは困難であった。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特許第3162091号公報
【特許文献2】特表2007−531535号公報
【非特許文献】
【0006】
【非特許文献1】Appl.Microbiol.Biotechnol(1991)34:783−788
【発明の概要】
【発明が解決しようとする課題】
【0007】
従って、本発明の主な目的は、ニトリルヒドラターゼを安定的、効率的に発現させたロドコッカス属細菌を提供し、低コストかつ高効率でアミド化合物を製造することにある。
【課題を解決するための手段】
【0008】
本発明は、上記状況を考慮してなされたもので、以下に示す、ウレアーゼ遺伝子が欠失又は不活性化されたロドコッカス属細菌、当該ロドコッカス属細菌を用いたニトリルヒドラターゼの製造方法、及び当該ロドコッカス属細菌を用いたアミド化合物の製造方法等を提供するものである。すなわち、本発明は以下の通りである。
【0009】
(1)ニトリルヒドラターゼ活性を有するロドコッカス属細菌において、ウレアーゼ遺伝子が欠失又は不活性化されたロドコッカス属細菌。
(2)ロドコッカス属細菌が、ロドコッカス ロドクロウス(Rhodococcus rhodochrous)である、(1)記載のロドコッカス属細菌。
(3)欠失または不活性化させるウレアーゼ遺伝子が、配列番号1に示される塩基配列を有するウレアーゼアルファサブユニットもしくはそのオーソログである、(1)又は(2)記載のロドコッカス属細菌。
(4)欠失または不活性化させるウレアーゼ遺伝子が、以下の(a)又は(b)に示されるいずれかの塩基配列を有するDNAからなる、(1)〜(3)のいずれか1項に記載のロドコッカス属細菌。
(a)配列番号1記載の塩基配列を含むDNA
(b)配列番号1記載の塩基配列と相同性が65%以上の塩基配列からなり、かつウレアーゼ活性を有するタンパク質をコードするDNA
(5)ロドコッカス属細菌が、ロドコッカス・ロドクロウス(Rhodococcus rhodochrous)J1株又はその変異株である、請求項1〜4記載のいずれか1項に記載のロドコッカス属細菌。
(6)(1)〜(5)のいずれか1項に記載のロドコッカス属細菌を培養し、得られる培養物からニトリルヒドラターゼを採取することを特徴とする、ニトリルヒドラターゼの製造方法。
(7)(1)〜(5)のいずれか1項に記載のロドコッカス属細菌を培養して得られる培養物又は当該培養物の処理物をニトリル化合物に接触させ、当該接触により生成されるアミド化合物を採取することを特徴とする、アミド化合物の製造方法。
【発明の効果】
【0010】
本発明によれば、尿素を誘導剤としてニトリルヒドラターゼを産生するロドコッカス属細菌において、安定的、効率的にニトリルヒドラターゼを発現させることができる。また、当該微生物を微生物触媒として用いて様々なニトリル化合物から低コストかつ高効率でアミド化合物を製造することもできる。
【図面の簡単な説明】
【0011】
【図1】本実施例における、プラスミドpK19mobsacB1の構造を示す模式図である。
【図2】本実施例における、プラスミドpBKURE01の構造を示す模式図である。
【発明を実施するための最良の形態】
【0012】
以下、本発明を詳細に説明する。本発明の範囲はこれらの説明に拘束されることはなく、以下の例示以外についても、本発明の趣旨を損なわない範囲で適宜変更し実施することができる。なお、本明細書において引用された全ての刊行物、例えば先行技術文献、及び公開公報、特許公報その他の特許文献は、参照として本明細書に組み込まれる。
【0013】
1.ウレアーゼ遺伝子が改変されたロドコッカス属細菌
本発明に係るウレアーゼ遺伝子が改変(欠失又は不活性化(以下、「欠失等」と称する))されたロドコッカス属細菌(以下、「本発明のロドコッカス属細菌」と称する)は、前述した通り、ニトリルヒドラターゼ活性を有するロドコッカス属細菌におけるウレアーゼ遺伝子が欠失又は不活性化されたロドコッカス属細菌である。
【0014】
本発明のロドコッカス属細菌は、ニトリルヒドラターゼ活性を有するロドコッカス属細菌であって、ニトリルヒドラターゼおよび、ウレアーゼを産生し、それらの活性を示すものであればよく、特に限定はされない。
【0015】
例えば、ロドコッカス・ロドクロウス(Rhodococcus rhodochrous)及びロドコッカス・エリスロポリス(Rhodococcus erythropolis)、ロドコッカス・オパカス(Rhodococcus opacus),ロドコッカス・ジョスティ(Rhodococcus jostii)、ロドコッカス・エクイ(Rhodococcus equi)等が好ましく挙げられる。
【0016】
ロドコッカス・ロドクロウスとしては、例えば、ロドコッカス・ロドクロウス(Rhodococcus rhodochrous)J1(FERM BP−1478)、ロドコッカス・ロドクロウス(Rhodococcus rhodochrous)M8(SU1731814)(VKPMB−S926)、ロドコッカス・ロドクロウス(Rhodococcus rhodochrous)M33(VKM Ac−1515D)及びロドコッカス・ロドクロウス(Rhodococcus rhodochrous)NCIMB41164(国際公開第05/054456号)等が好ましく挙げられる。ロドコッカス エリスロポリス(Rhodococcus erythropolis)としては、例えばロドコッカス エリスロポリス(Rhodococcus erythropolis)PR4(NBRC 100887)、ロドコッカス エリスロポリス(Rhodococcus erythropolis)SK121等が好ましく挙げられる。ロドコッカス オパカス(Rhodococcus opacus)としては、例えばロドコッカス オパカス(Rhodococcus opacus)B4(NBRC 108011)が好ましく挙げられる。ロドコッカス ジョスティ(Rhodococcus jostii)としては、例えばロドコッカス ジョスティ(Rhodococcus jostii)RHA1が好ましく挙げられる。 ロドコッカス・エクイ(Rhodococcus equi)としては、例えば、ロドコッカス・エクイ(Rhodococcus equi)103S等が好ましく挙げられる。
【0017】
なお、上記NBRC株は独立行政法人製品評価技術基盤機構バイオテクノロジー本部生物遺伝資源部門 (NBRC) から、FERM株は、独立行政法人 産業技術総合研究所 特許生物寄託センターから、VKPM株はルシアン・ナショナル・コレクション・オブ・インダストリアル・マイクロオーガニズムから、VKM株はルシアン・コレクション・オブ・マイクロオーガニズムからそれぞれ入手可能である。
【0018】
ウレアーゼ(EC3.5.1.5)は、以下の反応式に示すように、尿素を二酸化炭素とアンモニアへ加水分解する反応を触媒する酵素である。
(NH2)2CO + H2O → CO2 + 2 NH3
ここで、「ウレアーゼ活性」とは、尿素をアンモニアと二酸化炭素に変換させる活性を意味する。本発明のロドコッカス属細菌のウレアーゼ活性の測定方法としては、本発明のロドコッカス属細菌を尿素に接触させ、生成するアンモニアの量の増加を定量することで、ウレアーゼ活性を測定することができる。あるいは、尿素の消費量を定量することによってもウレアーゼ活性を測定することができる。
【0019】
ニトリルヒドラターゼは、以下の反応式に示すように、ニトリル化合物のニトリル基に作用し、対応するアミド化合物に変換する水和反応を触媒する酵素である。
R−CN + H2O → R−CONH2
(式中、Rは、置換又は無置換のアルキル基、置換又は無置換のアルケニル基、置換又は無置換のシクロアルキル基、置換又は無置換のアリール基、あるいは置換若しくは無置換の飽和又は不飽和複素環基を表す。)
【0020】
ここで、「ニトリルヒドラターゼ活性」とは、ニトリル化合物のニトリル基に作用して対応するアミド化合物に変換させる活性を意味する。本発明のロドコッカス属細菌のニトリルヒドラターゼ活性の測定方法としては、例えば、ニトリル化合物の1種であるアクリロニトリルを用いる方法が挙げられる。この方法では、ニトリルヒドラターゼ活性の結果としてアクリロニトリルに対応するアミド化合物、すなわちアクリルアミドが得られる。従って、本発明のロドコッカス属細菌をアクリロニトリルに接触させ、生成するアクリルアミドの量の増加を定量することで、ニトリルヒドラターゼ活性を測定することができる。あるいは、アクリロニトリルの消費量を定量することによってもニトリルヒドラターゼ活性を測定することができる。
【0021】
本発明のロドコッカス属細菌は、本来有するウレアーゼ遺伝子が欠失又は不活性化されたものであるが、この欠失等される遺伝子(標的遺伝子)としては、対象微生物のゲノムDNAにコードされるウレアーゼ遺伝子の一部又は全部が欠失等されたもの、プロモーター配列の一部又は全部が欠失等されたもの、発現調節に関連する遺伝子の一部又は全部が欠失等されたものなどが挙げられる。これらの欠失等された領域は単独でもよいし複数を組み合わせたものでもよい。
【0022】
2.ウレアーゼ遺伝子が改変されたロドコッカス属細菌の製造
ロドコッカス属細菌のウレアーゼは3つのサブユニット(アルファ及びベータ及びガンマ)が組み合わさって構成されており、それぞれのサブユニットは隣り合う3つの遺伝子(ureC及びureB及びureA)にコードされている。加えて、ウレアーゼの活性部位への金属取り込みに寄与する数種類のウレアーゼアクセサリータンパク質が知られている。ウレアーゼ活性は、これらのいずれかを欠失することで失活させることが可能である。すなわち、ウレアーゼ活性失活株を取得するには、ureA又はureB又はureC又はウレアーゼアクセサリータンパク質遺伝子のいずれかを欠失又は不活性化させればよく、その組合せや欠損の順序は問わない。特にウレアーゼアルファサブユニットは活性部位をもつことが知られており、該当酵素サブユニットをコードするureCを欠失させることが好ましい。
【0023】
本発明に用いられるウレアーゼ遺伝子は、ロドコッカス・ロドクロウス(Rhodococcus rhodochrous)J1から単離されたウレアーゼ遺伝子ureCであり、配列番号1に示される塩基配列を含む(配列番号1:ORF)。
J1菌以外のロドコッカス属細菌については、上記J1菌のウレアーゼアルファサブユニットに対応するオーソログを欠失または不活性化させることができる。
ここで、「オーソログ」とは、異なる生物に存在する相同な機能を有するタンパクをコードする類縁遺伝子であって、ウレアーゼ遺伝子の場合であれば、そのオーソログとはJ1菌以外の生物においてウレアーゼ活性を有するタンパクをコードする類縁遺伝子である。
上記したウレアーゼ遺伝子のオーソログは、ウレアーゼ遺伝子と高い配列相同性を有する。それゆえ、上記配列番号1に塩基配列を有する遺伝子に加えて、これらの配列と約65%以上、好ましくは約70%以上、より好ましくは約80%以上、特に好ましくは約90%以上、さらに特に好ましくは約95%以上の相同性(同一性)を有する塩基配列を有するDNAも本発明に用いられるウレアーゼ遺伝子に含まれる。
【0024】
本発明の微生物を製造するための、ウレアーゼ遺伝子を欠失又は不活性化する方法は、特に限定されない。例えば、自然変異誘発法、紫外線照射や変異誘発剤(N‐メチル−N´−ニトロ−N−ニトロソグアニジン、メチルメタンスルホン酸等)を用いる突然変異誘発法、ゲノムDNAの相同領域を有するプラスミドを形質転換する方法を用いることができる。形質転換の方法としては、エレクトロポレーション法や接合伝達法が使用できる。
【0025】
上記欠失等する方法については、本発明では以下の方法により行われるものが好ましい。
すなわち、ドナー微生物からニトリルヒドラターゼ活性を有するレシピエント微生物への接合伝達を利用した形質転換方法を用いることを含む、ウレアーゼ遺伝子の欠失又は不活性化方法であって、以下の工程(イ)〜(ニ)を含むことを特徴とする方法である。
(イ)レシピエント微生物として、前記接合伝達に供するドナー微生物が感受性を示す薬剤への耐性が強化された微生物を作製する工程;
(ロ)ドナー微生物として、下記(i)〜(v):
(i) レシピエント微生物中のウレアーゼ遺伝子(標的遺伝子)とその周辺の塩基配列とを含む塩基配列において当該ウレアーゼ遺伝子を欠失又は不活性化させた塩基配列領域、
(ii) 当該ドナー微生物において機能する接合伝達開始領域、
(iii) 当該ドナー微生物において機能する複製開始領域、
(iv) レシピエント微生物が感受性を示す薬剤に対する耐性遺伝子、及び
(v) レシピエント微生物に対する条件致死遺伝子
を含む、遺伝子改変用プラスミドを用いて形質転換された微生物を作製する工程;
(ハ)工程(ロ)で作製されたドナー微生物から工程(イ)で作製されたレシピエント微生物への接合伝達を行うことにより、当該レシピエント微生物の形質転換体を作製する工程;並びに
(ニ)工程(ハ)で作製された形質転換体を、前記条件致死遺伝子が機能し得る培養条件で培養する工程。
【0026】
上記欠失又は不活性化方法において用いるレシピエント微生物としては、ニトリルヒドラターゼ活性を有し、ウレアーゼ遺伝子を有する微生物であれば限定はされない。中でもロドコッカス属細菌が、前述した理由でより好ましい。ロドコッカス属細菌等の具体例についても、前述と同様のものが挙げられる。
一方、ドナー微生物としては、上記レシピエント微生物と接合伝達可能な微生物であれば限定はされない。例えば、大腸菌が好ましい。
【0027】
2.1. 工程(イ)
工程(イ)では、接合伝達に供するレシピエント微生物として、接合伝達に供するドナー微生物が感受性を示す薬剤への耐性を強化した微生物を作製する。「薬剤への耐性を強化する」とは、レシピエント微生物が薬剤耐性を有していない場合には、薬剤耐性を付与することをいい、レシピエント微生物が薬剤耐性の乏しい場合には、当該耐性をより強くすることをいう。
【0028】
接合伝達法を使用する場合、レシピエントとなる微生物には薬剤耐性マーカーが必要である。よって、薬剤耐性を有していない微生物、又は薬剤耐性の乏しい微生物を使用する場合、薬剤選択可能な程度の十分な薬剤耐性を有する株(レシピエント)の作製が必要となる。ここで、薬剤としては、接合伝達法を使用することを考慮し、接合伝達に供するドナー微生物(接合伝達時のドナー微生物)が感受性を示す薬剤が好ましい。当該薬剤としては、クロラムフェニコール、アンピシリン、カナマイシン、トリメトプリム、ゲンタマイシン、ナルジクス酸、カルベニシン、チオストレプトン、テトラサイクリン及びストレプトマイシン等が好ましく、クロラムフェニコール及びアンピシリンがより好ましい。
【0029】
上述のように薬剤耐性を強化したレシピエント微生物を作製する方法としては、特には限定されない。例えば、自然変異誘発法、紫外線照射や変異誘発剤を用いる突然変異誘発法、EZ−Tn5(Epicentre社製)のようなランダム変異導入ツール等を用いることにより、本来レシピエント微生物が持たない薬剤耐性遺伝子を人為的に当該微生物ゲノム上に導入する方法、あらかじめ遺伝子改変用プラスミドとは別の、抗生物質耐性獲得用のプラスミドを導入する方法等が好ましい。これらの中でも自然変異誘発法がより好ましい。
自然変異誘発法は、所望の薬剤を含有する培地中で対象とする微生物を継代培養等することにより、もともとは当該培地中で生育不可又は困難な微生物に自然変異を誘発させて、より高濃度の薬剤を含有する当該培地中でも生育し得る株を取得する方法である。どの程度まで薬剤耐性を強化するかは、使用するレシピエント微生物、ドナー微生物、選択する薬剤により異なるが、レシピエント微生物の生育が抑制されない、且つ、ドナー微生物の生育が阻害される薬剤濃度を選ぶことが好ましい。例えば、レシピエント微生物としてロドコッカス属細菌を、ドナー微生物として大腸菌を、選択用薬剤としてクロラムフェニコールを用いる場合、自然突然変異によりクロラムフェニコール1〜200mg/L、好ましくは10〜100mg/Lを含有する培地において生育可能なレシピエント微生物(クロラムフェニコール耐性強化株)を得ることが望ましい。
【0030】
2.2. 工程(ロ)
工程(ロ)では、接合伝達に供するドナー微生物として、所定の遺伝子改変用プラスミドを用いて形質転換された微生物を作製する。遺伝子改変用プラスミド、すなわちレシピエント微生物中のウレアーゼ遺伝子を改変するためのプラスミドDNAとしては、前述の(i)〜(v)の構成(遺伝子・塩基配列)を含むものを用いる。
ここで、前記(i)の塩基配列領域は、改変の対象とするレシピエント微生物中のウレアーゼ遺伝子と当該遺伝子の周辺の塩基配列とを含む塩基配列において、当該ウレアーゼ遺伝子を欠失又はさせた塩基配列領域である。当該塩基配列領域の作製は、レシピエント微生物のゲノムから、ウレアーゼ遺伝子と当該遺伝子の周辺の塩基配列とを含む塩基配列の単離(クローニング)及び遺伝子ライブラリー作製やPCR等の公知技術を用いて行うことができる。
なお、ウレアーゼ遺伝子の周辺の塩基配列としては、限定はされない。例えば、当該遺伝子に加え、発現調節に関連する遺伝子の上流及び下流の相同領域が両端からそれぞれ100〜3000bpの塩基配列を含む配列であることが好ましい。より好ましくは500〜2000bpの塩基配列を含む配列である。単離した塩基配列を用いて、前述(i)を作製する方法は特に限定されず、PCR法や制限酵素を用いた標的遺伝子部分の切除もしくは置換等の公知技術を用いて行うことができる。
【0031】
単離したウレアーゼ遺伝子を欠失又は不活性化する方法としては、限定はされず、如何なる方法を適用してもよい。例えば、SOE−PCR法や、ゲノムDNAの相同領域を有するプラスミドを形質転換する方法を用いることができる。さらに、形質転換の方法としては、エレクトロポレーション法や接合伝達法が使用できる。
上記欠失又は不活性化されたウレアーゼ遺伝子としては、レシピエント微生物のゲノムDNAにコードされる当該ウレアーゼ遺伝子の一部又は全部が欠失等されたもの、プロモーター配列の一部又は全部が欠失等されたもの、発現調節に関連する遺伝子の一部又は全部が欠失等されたものなどが挙げられる。これらの欠失等された領域は単独でもよいし複数を組み合わせたものでもよい。
【0032】
前記(ii)の接合伝達開始領域は、使用するドナー微生物中において接合伝達の開始点となる塩基配列を含む領域であれば、限定はされない。例えば、プラスミドRP4由来のoriTが好ましい。
【0033】
前記(iii)の複製開始領域は、使用するドナー微生物中において前記遺伝子改変用プラスミドの自己複製起点として機能し得る塩基配列を含む領域であれば、限定はされない。例えば、oriVが好ましい。
【0034】
前記(iv)の薬剤耐性遺伝子は、接合伝達に供するレシピエント微生物(例えばロドコッカス属細菌等)が感受性を示す薬剤に対する耐性遺伝子であれば、限定はされない。例えば、アンピシリン耐性遺伝子、カナマイシン耐性遺伝子、クロラムフェニコール耐性遺伝子、トリメトプリム耐性遺伝子、ゲンタマイシン耐性遺伝子、ナルジクス酸耐性遺伝子、カルベニシン耐性遺伝子、チオストレプトン耐性遺伝子等が好ましく挙げられる。レシピエント微生物がこれらの薬剤耐性を有していない場合、所望の薬剤耐性を有する変異株を作製し、使用することができる。
【0035】
前記(v)の条件致死遺伝子は、レシピエント微生物のゲノム上に導入された場合に、当該微生物を死に至らしめる作用を有し得る遺伝子であれば、限定はされない。例えば、sacB遺伝子が好ましい。sacB遺伝子は、当該遺伝子を保有し発現する微生物(例えばロドコッカス属細菌等)をスクロース含有培地で培養した場合に、スクロースを基質とし当該微生物に対して致死作用を有する有害物質を産生する酵素をコードする遺伝子である。
【0036】
接合伝達に用いる前記遺伝子改変用プラスミドは、如何なるベクターをベースとして構築されたものであってもよく、限定はされない。例えば、レシピエント微生物がロドコッカス属細菌の場合は、pK19mobベクター(Schaefer et al,Gene1,vol.45,p.69−73(1994))等を用いることが好ましい。
【0037】
前記遺伝子改変用プラスミドにおける(i)〜(v)の構成は、例えば、上流から順に、前記(i)の塩基配列領域、前記(iv)の耐性遺伝子、前記(v)の条件致死遺伝子、前記(ii)の接合伝達開始領域、前記(iii)の複製開始領域の順で配されていることが好ましい。前記(i)〜(v)の構成を含む遺伝子改変用プラスミドの構築は、公知の遺伝子組換え技術を用いて実施することができる。
工程(ロ)では、上述したような各構成を有する遺伝子改変用プラスミドをドナー微生物内に導入して形質転換された微生物、すなわち接合伝達に供するドナー微生物を作製する。その際、形質転換の方法としては、エレクトロポレーション法やカルシウム法(アルカリSDS法)等の、微生物の形質転換方法として公知の方法を用いることができる。
【0038】
2.3. 工程(ハ)
工程(ハ)では、工程(ロ)で作製されたドナー微生物から工程(イ)で作製されたレシピエント微生物への接合伝達を行う。通常は、ドナー微生物及びレシピエント微生物のそれぞれの細胞懸濁液を混合し、適当なプレート培地(LB培地等)上に均一に広げて、両微生物の接合を行わせる。
当該接合においては、ドナー微生物中の遺伝子改変用プラスミドがレシピエント微生物内に移動し、レシピエント微生物のゲノムと上記プラスミドとの相同配列で2重交叉が起こり当該ゲノム中のウレアーゼ遺伝子が欠失される。この接合により、レシピエント微生物の形質転換体が作製される。すなわち、当該形質転換体は、ドナー微生物中の遺伝子改変用プラスミドの一部が相同組換えによりレシピエント微生物のゲノム上に導入されたものである。
所望の形質転換体であるかどうかの確認は、レシピエント微生物自体の薬剤耐性、及び前記遺伝子改変用プラスミド由来の薬剤耐性を利用して行うことができる。具体的には、両薬剤を含む培地(例えば、カナマイシン及びクロラムフェニコール含有培地等)において上記接合後の微生物を培養することにより、所望の形質転換体を選択することができる。
【0039】
2.4. 工程(ニ)
工程(ニ)では、工程(ハ)で作製されたレシピエント微生物の形質転換体(形質転換微生物)を、前記遺伝子改変用プラスミド由来の条件致死遺伝子が機能し得る培養条件で培養(継代培養)する。条件致死遺伝子が機能し得る培養条件としては、限定はされないが、例えば条件致死遺伝子がsacB遺伝子の場合は、スクロース含有培地を用いた培養が好ましく挙げられる。
当該培養においては、上記条件致死遺伝子を有する形質転換微生物は生育困難であるため、継代培養により、自然誘発的に、当該微生物のゲノム上から相同組換えにより上記致死遺伝子を含む塩基配列領域が除かれた(脱落した)形質転換微生物を得ることができる。
ただし、当該得られた微生物の中には、レシピエント微生物中のウレアーゼ遺伝子が、当初の目的通り欠失又は不活性化しているものと、そうでないもの(上記脱落の際の相同組換えにより元のウレアーゼ遺伝子の機能が復活したもの)が含まれている。
よって、通常は、さらに別の培養条件でも培養したり、培養物を用いてウレアーゼ活性測定や各種タンパク質分析法を適用して分析することにより、所望の形質転換微生物を選択することがより好ましい。
【0040】
3.ニトリルヒドラターゼの製造方法
本発明のニトリルヒドラターゼの製造方法は、前述した本発明の微生物を培養し、得られる培養物から当該ニトリルヒドラターゼを採取することを特徴とする方法である。
ここで、当該製造方法に用いる本発明の微生物としては、上記ウレアーゼ遺伝子が改変された微生物に、ニトリルヒドラターゼ遺伝子を当該微生物の遺伝子に挿入(付加)させたものであってもよいし、ニトリルヒドラターゼ遺伝子を含む組換えベクターを導入することによりなされたものであってもよいし、その両方を兼ね備えてあるものであってもよい。
組換えベクターを用いる場合は、ニトリルヒドラターゼ遺伝子が、形質転換される本発明の微生物において発現可能なように、ベクターに組み込まれることが必要である。例えば、ニトリルヒドラターゼを産生し得る微生物のゲノム等からニトリルヒドラターゼ遺伝子を含むPCR断片を得て、得られた断片を発現ベクターに連結することで、ニトリルヒド
ラターゼ遺伝子を含む発現ベクターを得ることができる。
【0041】
発現ベクターとしては、例えば、プラスミドDNA、バクテリオファージDNA、レトロトランスポゾンDNA、人工染色体DNAなどが挙げられ、中でもプラスミドDNAが好ましい。異種ニトリルヒドラターゼを発現させるためのベクター(プラスミドDNA)としては、限定されないが、例えば、ロドコッカス属細菌を宿主とする場合は、pSJ034、pSJ041、pSJ042、pSJ043等が好ましい。pSJ034は、ロドコッカス属細菌においてニトリルヒドラターゼを発現するベクターであり、特開平10−337185号公報に示す方法でpSJ023より作製することができる。このpSJ023は、形質転換体 ATCC16274/pSJ023(FERM BP−6232)として、独立行政法人産業技術総合研究所 特許生物寄託センター(茨城県つくば市東1丁目1番地1中央第6)に平成9年3月4日付けで国際寄託されている。
当該発現ベクターには、ニトリルヒドラターゼ遺伝子のほか、プロモーター、ターミネーター、エンハンサー、スプライシングシグナル、ポリA付加シグナル、選択マーカー、リボソーム結合配列(SD配列)等を連結することができる。なお、選択マーカーとしては、例えば、アンピシリン耐性遺伝子、カナマイシン耐性遺伝子、ジヒドロ葉酸還元酵素遺伝子、ネオマイシン耐性遺伝子等が挙げられる。
【0042】
前述の通り、本発明においては、ニトリルヒドラターゼは、上記形質転換微生物を培養し、得られる培養物から採取することにより製造することができる。
本発明において、「培養物」とは、培養上清、培養菌体、又は菌体の破砕物のいずれをも意味するものである。形質転換微生物を培養する方法は、宿主の培養に用いられる通常の方法に従って行われる。目的のニトリルヒドラターゼは、上記培養物中に蓄積される。
【0043】
形質転換微生物を培養する培地は、宿主菌となる本発明の微生物が資化し得る炭素源、窒素源、無機塩類等を含有し、微生物の培養を効率的に行うことができる培地であれば、天然培地、合成培地のいずれを用いてもよい。炭素源としては、グルコース、フラクトース、スクロース、デンプン等の炭水化物、酢酸、プロピオン酸等の有機酸、エタノール、プロパノール等のアルコール類が挙げられる。窒素源としては、アンモニア、塩化アンモニウム、硫酸アンモニウム、酢酸アンモニウム、リン酸アンモニウム等の無機酸若しくは有機酸のアンモニウム塩又はその他の含窒素化合物のほか、ペプトン、肉エキス、コーンスティープリカー等が挙げられる。無機物としては、リン酸第一カリウム、リン酸第二カリウム、リン酸マグネシウム、硫酸マグネシウム、塩化ナトリウム、硫酸第一鉄、硫酸マンガン、硫酸銅若しくは炭酸カルシウム等が挙げられる。また、必要に応じ、培養中の発泡を防ぐために消泡剤を添加してもよい。さらに、培地にはニトリルヒドラターゼの補欠分子であるコバルトイオンや鉄イオンを添加し、酵素の誘導剤となるニトリル類やアミド類を添加してもよい。
【0044】
培養中、発現ベクターや目的遺伝子の脱落を防ぐために選択圧を掛けた状態で培養してもよい。すなわち、発現ベクター等における選択マーカーが薬剤耐性遺伝子である場合には相当する薬剤を培地に添加したり、選択マーカーが栄養要求性相補遺伝子である場合には相当する栄養因子を培地から除いたりしてもよい。また、選択マーカーが資化性付与遺伝子である場合には、相当する資化因子を必要に応じて唯一因子として添加することができる。例えば、アンピシリン耐性遺伝子を含むベクターで形質転換した微生物を培養する場合、培養中、必要に応じてアンピシリンを添加してもよい。
【0045】
プロモーターとして誘導性のプロモーターを用いた発現ベクターで形質転換した形質転換微生物を培養する場合は、必要に応じてインデューサーを培地に添加してもよい。例えば、イソプロピル−β−D−チオガラクトシド(IPTG)で誘導可能なプロモーターを有する
発現ベクターで形質転換した形質転換微生物を培養するときには、IPTG等を培地に添加することができる。また、インドール酢酸(IAA)で誘導可能なtrpプロモーターを用いた発現ベクターで形質転換した形質転換微生物を培養するときには、IAA等を培地に添加することができる。
【0046】
形質転換微生物の培養条件は、発現目的のニトリルヒドラターゼの生産性及び宿主の生育が妨げられない条件であれば、特段限定されるものではない。通常、10℃〜45℃、好ましくは10℃〜40℃の温度下で、5〜120時間、好ましくは5〜100時間程度行うことができる。pHの調整は、無機酸又は有機酸、アルカリ溶液等を用いて、適時調整を行うことが好ましい。
【0047】
培養方法としては、固体培養、静置培養、振盪培養、通気攪拌培養などが挙げられるが、振盪培養又は通気攪拌培養(ジャーファーメンター)により好気的条件下で培養することが好ましい。
【0048】
また、場合により、本培養に先立ち、少量の前培養を行うこともできる。例えば、ロドコッカス属細菌の形質転換微生物を培養する場合は、振盪培養又は通気攪拌培養などの好気的条件下、30〜40℃の温度下で行うことが好ましい。
上記培養条件で培養すると、高収率でニトリルヒドラターゼを上記培養物中、すなわち培養上清、培養菌体、又は菌体の破砕物の少なくともいずれかに蓄積することができる。これらニトリルヒドラターゼを含有する「培養物」は、後述するアミド化合物の製造方法に使用することができる。
【0049】
培養後、ニトリルヒドラターゼが菌体内に生産される場合には、菌体を破砕することにより、目的のニトリルヒドラターゼを採取することができる。菌体の破砕方法としては、フレンチプレス又はホモジナイザーによる高圧処理、超音波処理、ガラスビーズ等による磨砕処理、リゾチーム、セルラーゼ又はペクチナーゼ等を用いる酵素処理、凍結融解処理、低張液処理、ファージによる溶菌誘導処理等を利用することができる。
【0050】
破砕後、必要に応じて菌体の破砕残渣(細胞抽出液不溶性画分を含む)を除くことができる。残渣を除去する方法としては、例えば、遠心分離やろ過などが挙げられ、必要に応じて、凝集剤やろ過助剤等を使用して残渣除去効率を上げることもできる。残渣を除去した後に得られた上清は、細胞抽出液可溶性画分であり、粗精製したニトリルヒドラターゼ溶液とすることができる。
【0051】
また、ニトリルヒドラターゼが菌体内に生産される場合、菌体そのものを遠心分離、膜分離等で回収して、未破砕のまま使用することも可能である。
ニトリルヒドラターゼが菌体外に生産される場合には、培養液をそのまま使用するか、遠心分離やろ過等により菌体を除去する。その後、必要に応じて硫安沈澱による抽出等により前記培養物中から異種ニトリルヒドラターゼを採取し、さらに必要に応じて透析、各種クロマトグラフィー(ゲルろ過、イオン交換クロマトグラフィー、アフィニティクロマトグラフィー等)を用いて単離精製することもできる。
形質転換微生物を培養して得られたニトリルヒドラターゼの生産収率は、例えば、培養液あたり、菌体湿重量又は乾燥重量あたり、粗酵素液タンパク質あたりなどの単位で、SDS−PAGE(ポリアクリルアミドゲル電気泳動)やニトリルヒドラターゼ活性測定などにより確認することができるが、特段限定されるものではない。SDS−PAGEは当業者であれば公知の方法を用いて行うことができる。また、ニトリルヒドラターゼ活性は、上述した活性の値を適用することができる。
【0052】
4.アミド化合物の製造方法
上述のように製造されたニトリルヒドラターゼ(前述した培養物を含む)は、酵素触媒(菌体のまま利用する微生物触媒等を含む)として物質生産に利用することができる。例えば、ニトリル化合物に、ニトリルヒドラターゼを接触させることにより、当該接触によりニトリル化合物が変換されてアミド化合物を製造することができる。
【0053】
酵素触媒としては、前述のように適当な宿主内でニトリルヒドラターゼ遺伝子が発現するように遺伝子導入を行い、宿主を培養した後の培養物、又は当該培養物の処理物を利用することができる。処理物としては、例えば、培養後の菌体をアクリルアミド等のゲルで包含したもの、グルタルアルデヒドで処理したもの、アルミナ、シリカ、ゼオライト及び珪藻土等の無機担体に担持したもの等が挙げられる。
【0054】
基質として使用されるニトリル化合物は、酵素の基質特異性、酵素の基質に対する安定性等を考慮して選択される。ニトリル化合物としては、アクリロニトリルが好ましい。
反応方法、及び反応終了後のアミド化合物の採取方法は限定されず、基質及び酵素触媒の特性により適宜選択することができる。例えば、当該反応において、基質となるニトリル化合物の濃度は、0.1〜10%(W/V)が好ましく、5%(W/V)程度が特に好ましい。また、当該反応は、pH5〜10の緩衝液又は水中で行うことが好ましく、例えば、50mMリン酸緩衝液(pH7.0)中で行うことができる。反応後の酵素触媒は、その活性が失活しない限り、リサイクル使用することが好ましい。失活の防止やリサイクルを容易にすることに鑑み、酵素触媒は処理物の形態で使用されることが好ましい。
上記反応により生成したアクリルアミドは、ガスクロマトグラフィーなどの公知の方法を用いて定量することができる。また、アクリルアミドの産生量から前記酵素触媒のニトリルヒドラターゼ活性を換算することができる。
【0055】
以下に、実施例を挙げて本発明をより具体的に説明するが、本発明はこれらに限定されるものではない。
【実施例】
【0056】
[調製例1]
ウレアーゼ欠失用プラスミドの作製
(1)接合伝達プラスミド(pK19mobsacB1)の作製
pDNR−1r(clontech社製)中のsacB遺伝子を、NspV切断サイトを付加したプライマーSAC−01(配列番号2)及びSAC−02(配列番号3)を使用したPCRにより増幅し、約1.9kbのsacB遺伝子断片を得た。PCRは以下の反応条件で行った。
【0057】
プライマー:
SAC−01:5’−GGTTCGAATACCTGCCGTTCACTATTATTTAGTG−3’(配列番号2)
SAC−02:5’−GGTTCGAATCGGCATTTTCTTTTGCGTTTTTATTTG−3’(配列番号3)
【0058】
反応液組成:
滅菌水 22μl
PrimeSTAR(登録商標) Max Premix(2×) (タカラバイオ社製) 25μl
SAC−01(10μM)(配列番号2) 1μl
SAC−02(10μM)(配列番号3) 1μl
pDNR−1r(100ng)(clontech社製) 1μl
総量 50μl
温度サイクル:
98℃ 10秒、55℃ 15秒及び72℃ 20秒の反応を30サイクル
【0059】
sacB遺伝子断片を制限酵素NspV(タカラバイオ社製)で消化後、pK19mobのNspVサイトに接続し、sacB遺伝子が正方向に導入されたプラスミドpK19mobsacB1を構築した。pK19mob NspV切断断片とsacB遺伝子断片及びsacB遺伝子NspV切断断片の精製にはGFX PCR DNA band and Gel Band Purification kit(GE Healthcare社製)、両断片の接続にはDNA Ligation Kit <Mighty Mix>、プラスミドの抽出にはQIAprep miniprep kit(QIAGEN社製)を用いた。
【0060】
(2)J1株ゲノムDNAの調製
ロドコッカス・ロドクロウス(Rhodococcus rhodochrous)J1株を100mlのMYK培地(0.5%ポリペプトン、0.3%バクトイーストエキス、0.3%バクトモルトエキス、1% グルコース、0.2% K2HPO4、0.2% KH2PO4、pH 7.0)中、30℃にて72時間振盪培養した。
培養後、集菌し、集菌された菌体をSaline−EDTA溶液(0.1M EDTA、0.15M NaCl(pH8.0))4 mlに懸濁した。懸濁液にリゾチーム40 mgを加えて、37℃で1〜2時間振盪した後、−20℃で凍結した。
次に、10mlのTris−SDS液(1%SDS、0.1M NaCl、0.1M Tris−HCl(pH9.0))を穏やかに振盪しながら加え、さらにプロテイナーゼK(メルク社)を10μl(終濃度10mg/ml)加えて37℃で1時間振盪した。
次に、等量のTE(10mM Tris−HCl、1mM EDTA(pH8.0)) 飽和フェノールを加え、撹拌した後遠心した。遠心後、上層をとり2倍量のエタノールを加えた後、ガラス棒でDNAを巻きとり、90%、80%、70%のエタノールで順次フェノールを取り除いた。
次に、DNAを3mlのTE緩衝液に溶解させ、リボヌクレアーゼA溶液(100℃、15分間の加熱処理済)を10μg/mlになるよう加え、37℃で30分間振盪した。さらに、プロテイナーゼKを加え37℃で30分間振盪した後、等量のTE飽和フェノールを加えて遠心し、上層と下層に分離させた。
上層についてこの操作を2回繰り返した後、同量のクロロホルム(4%イソアミルアルコール含有)を加え、同様の抽出操作を繰り返した。その後、上層に2倍量のエタノールを加え、ガラス棒でDNAを巻きとり回収し、J1株ゲノムゲノムDNAを得た。
【0061】
(3)ウレアーゼ遺伝子(ureC)の増幅と発現プラスミドの構築
J1菌のウレアーゼ遺伝子ureCを含む配列を特定するために、GenBankに登録されているロドコッカス エリスロポリス(Rhodococcus erythropolis)PR4株の塩基配列(登録番号AP008957)を基に、以下示すプライマーおよび反応液組成を用いてPCRを行った。
プライマー:
ureC−01:5’−tgtacggcccgaccgctggcgaccag−3’(配列番号4)
ureC−02:5’−caccggctgcggggtcgggatagcagc−3’(配列番号5)
【0062】
反応液組成:
滅菌水 22μl
PrimeSTAR(登録商標) Max Premix(2×) (タカラバイオ社製) 25μl
ureC−01(10μM)(配列番号4) 1μl
ureC−02(10μM)(配列番号5) 1μl
J1株ゲノムDNA(100ng) 1μl
総量 50μl
温度サイクル:
98℃:10秒、55℃:15秒及び72℃:20秒の反応を30サイクル
【0063】
PCR終了後、反応液2μlを0.7%アガロースゲルにおける電気泳動に供し、約1.4kbpのPCR産物の検出を行った。PCR産物を確認した後、反応液をGFX PCR DNA band and GelBand Purification kit(アマシャムバイオサイエンス社製)で精製した。次に、精製したPCR産物をMighty Cloning Reagent Set(Blunt End)(タカラバイオ株式会社製)でpUC118にクローニングし、プライマーウォーキング法により、塩基配列を解析した。その結果、配列番号6に示す配列が得られた。ホモロジー検索(blastx)を実施した結果、表1の結果が得られ、ウレアーゼのアルファサブユニットと相同性が認められた。
【0064】
【表1】
【0065】
次に、J1菌のウレアーゼアルファサブユニットをコードするureC遺伝子の全長配列を特定するために以下の操作を行った。
BamHI、BsaI、BglII、NcoI、PvuI,SacI、SmaI、XhoIそれぞれで消化したJ1菌ゲノムDNAに対し、後述の方法で調製したUreCのプローブを用いてサザンハイブリダイゼーションを行ったところ、BglIIで消化した断片から、約3.5kbの単一シグナルが得られた。
【0066】
なお、ureCのプローブは以下のようにして調製した。
プライマーureC-01およびureC-02で調製したPCR産物(配列番号6)をGFX PCR DNA band and Gel Band Purification kit(GEヘルスケア バイオサイエンス社)を用いて精製した。精製したPCR産物に対してAlkPhos Direct Labeling kit(GEヘルスケア バイオサイエンス社)を用い、添付のマニュアルにしたがってラベリングを行い、ureCのプローブとした。
【0067】
続いて、J1菌ゲノムDNAを制限酵素BglIIで分解して0.7%アガロースゲル電気泳動で分離し、ゲルからGFX PCR DNA band and Gel Band Purification kit(GEヘルスケア バイオサイエンス社)を使用して約3.5kbの断片を回収した。得られた断片は、pBluescriptII SK(+)ベクター(Stratagene社製)にDNA ligation kit<Mighty mix>(タカラバイオ社製)を用いて連結した。反応条件は以下の通りである。
【0068】
反応液組成:
ligation mighty mix(タカラバイオ社製) 5μl
J1菌ゲノムDNA/BglII切断断片 4μl
pBluescriptII SK(+)/BglII切断断片 1μl
総量 10μl
反応:
16℃,1時間
【0069】
上記ライゲーション産物の全量を、大腸菌JM109株コンピテントセル200μlに加え、0℃で30分放置した。続いて、42℃で45秒間ヒートショックを与え、0℃で2分間冷却した。その後、SOC 培地(20 mMグルコース、2%バクトトリプトン、0.5%バクトイーストエキス、10mM NaCl,2.5mM KCl,1mM MgSO4,1mM MgCl2)を1ml添加し、37℃にて1時間振盪培養した。培養後の培養液を200μlずつ、LB AIXプレート(100μg/lアンピシリン、100μM IPTG,50μg/l X−galを含むLB寒天培地)に塗布し、37℃で一晩放置した。プレート上に生育した白色の組換コロニーを新しいLB AIXプレートに、プレート1枚に付き94個、プレート10枚分単離した。各プレートにはインサートを含まないpBluescriptII SK(+)で形質転換したJM109株を2コロニー/プレート植菌した。コロニー単離したプレートを37℃で一晩放置した後、Hybond−N+(GEヘルスケア バイオサイエンス社)膜にコロニーを写し取り、ureCのプローブを用いてコロニーハイブリダイゼーションを行った。
【0070】
検出されたコロニーを培養して得られた培養液を集菌後、QIAprep miniprep kit(QIAGEN社製)を用いて組換えプラスミドを回収した。キャピラリーDNAシーケンサーCEQ2000(ベックマン・コールター社製)を用いて、添付のマニュアルに従って、プラスミド中にクローニングされているゲノムDNA断片の塩基配列を解析した。その結果、配列番号7に示される塩基配列が得られた。配列番号7に示される塩基配列中に、配列番号1に示す約1.7kbpのオープンリーディングフレーム(ORF)を見出した。配列番号1の塩基配列のホモロジー検索(blastn)を実施した結果、表2の結果が得られ、ウレアーゼのアルファサブユニットと相同性が認められた。
【0071】
【表2】
【0072】
続いて、ウレアーゼ遺伝子(ureC)を含んだ配列を取得するため、以下に示す反応液組成及びプライマーを用いてPCRを行った。
プライマー:
ureC−03: 5’− ggCCTGCAGGGACAGTCGGCGACCAGGTACGCC −3’(配列番号8)
ureC−04: 5’− ACGGCGACAGCAGGTGCACGTGCGAGTCGAAG−3’(配列番号9)
【0073】
反応液組成:
滅菌水 22μl
PrimeSTAR(登録商標) Max Premix(2×) (タカラバイオ社製) 25μl
ureC−03(10μM)(配列番号8) 1μl
ureC−04(10μM)(配列番号9) 1μl
J1株ゲノムDNA(100ng) 1μl
総量 50μl
温度サイクル:
98℃:10秒、55℃:15秒及び72℃:20秒の反応を30サイクル
【0074】
同様に、下記プライマーureC−05(配列番号10)及びureC−06(配列番号11)を使用し、上記と同じ条件でPCRを実施した。
プライマー:
ureC−05: 5’− GTCGCATCGGTGAGGTCGTCACCCGCAC−3’(配列番号10)
ureC−06: 5’− ccTCTAGAGATCGACGCGTTCGGGTCGCCGAG−3’(配列番号11)
【0075】
PCR終了後、反応液2μlを0.7%アガロースゲルにおける電気泳動に供し、ureC−03とureC−04で約400 bp、ureC−05とureC−06で約300bpのPCR産物の検出を行った。PCR産物を確認した後、反応液をGFX PCR DNA band and GelBand Purification kit(アマシャムバイオサイエンス社製)で精製した。
【0076】
次に、上記で増幅した2つのPCR断片を連結するため、以下の条件でAssembly PCRを行った。
【0077】
反応液組成:
滅菌水 21μl
PrimeSTAR(登録商標) Max Premix(2×) (タカラバイオ社製) 25μl
ureC−03(10μM)(配列番号8) 1μl
ureC−06(10μM)(配列番号11) 1μl
ureC−03とureC−04の増幅産物(50ng) 1μl
ureC−05とureC−06の増幅産物(50ng) 1μl
総量 50μl
温度サイクル:
98℃:10秒、55℃:15秒及び72℃:40秒の反応を30サイクル
【0078】
PCR終了後、約1Kbの増幅産物をアガロース電気泳動で確認し、Mighty Cloning Reagent Set(Blunt End)(タカラバイオ株式会社製)でpUC118にクローニングした。
続いて、上記でクローニングした断片を制限酵素XbaI及びSse8387Iで切断し、約1kbの断片を回収し、接合伝達用のプラスミド(pK19mobsacB1)のXbaI−Sse8387I部位に連結し、プラスミドを作製した。得られたプラスミドはウレアーゼ遺伝子欠失プラスミド:pBKAMD01と名付けた。図1に、プラスミドpK19mobsacB1の構造を示す模式図を、図2に、プラスミドpBKURE01の構造を示す模式図を示した。
【0079】
[調製例2]
薬剤耐性を有するJ1株の作製
接合伝達に使用するドナーは、遺伝子欠失株のセレクションに薬剤耐性が必要である。そこで、種々の薬剤耐性株の取得を試み、アンピシリン耐性を有するJ1株の変異株を下記の方法で取得した。
2μg/mlのアンピシリンを含んだMYKプレート(0.5%ポリペプトン、0.3%バクトイーストエキス、0.3%マルツエキス、0.2% KH2PO4、0.2% K2HPO4、1.5%寒天)にJ1株をストリークし、コロニーが生育するまで30℃で保温した。約2週間後、アンピシリン耐性株が生育してきたので、再度2μg/mlのアンピシリンプレートにストリークして、30℃で保温した。
次に、2μg/mlのアンピシリンプレートから生育したコロニーを、5μg/mlのアンピシリンプレートにストリークし、耐性株が出現するまで30℃で保温した。以下、同様の操作を、アンピシリン濃度を10μg/ml→15μg/ml→50μg/ml→100μg/mlに高めながら繰り返し、100μg/mlのアンピシリン濃度で生育するアンピシリン耐性株(J1−Amp株)を得た。
【0080】
[実施例1]
接合伝達によるウレアーゼ遺伝子の欠失
(1)ドナーの調製
乾熱滅菌した試験管に大腸菌S17−1λpirのコンピテントセル20μlにプラスミドpBKURE01 1μlを加え、氷上で30分静置した。42℃で30秒ヒートショック後、SOC培地を180μl添加し、37℃で1時間振とう培養を行った。その後、50μg/mlカナマイシンを含んだLBプレートに塗布し、37℃で一晩静置した。
翌日、プレートに生育したコロニーをLB培地1mlで回収し、遠心分離により菌体を回収し、遠心上清を除去した。同様の操作をもう一度繰り返し、最後に0.5 mlのLB培地を添加し、菌体懸濁液を調製した。この菌体懸濁液をドナー溶液とした。
【0081】
(2)レシピエントの調整
J1−Amp株をMYKプレートにストリークし、30℃で2日生育させた。生育したコロニーは実施例1(1)と同様の方法で回収、洗浄し、レシピエントとなる菌体懸濁液を調製した。
【0082】
(3)接合伝達
実施例1(1)で調製したドナー溶液と、実施例1(2)で調製したレシピエント溶液を100μlずつ混合し、抗生物質を含まないMYKプレートに塗布し、30℃で一晩静置した。
翌日、生育したコロニーは1mlのLB培地で回収し、100μlずつカナマイシン濃度を10、30、50μg/mlとした選抜プレート(すべて100μg/mlアンピシリンを含む)に塗布した。
塗布したプレートは組み換え菌コロニーが出現するまで30℃で1週間保温した。
その結果、100μg/mlアンピシリン、10μg/mlカナマイシンを含んだプレートにのみ3個のコロニーが出現した。得られたコロニーの一つを#U1と命名し、以後の実験に使用した。
【0083】
(4)ウレアーゼ遺伝子の欠失
接合伝達により得られた組み換え菌#U1は、ゲノムのウレアーゼ遺伝子の領域に相同組換えによりプラスミドが挿入されているが、ウレアーゼ遺伝子を欠失するには2段階の相同組換えが必要である。そこで、次に、sacB遺伝子を利用した選抜を実施した。
10%ショ糖を含んだMYKプレートを作製し、#U1のコロニーを滅菌水に懸濁した液を適度に希釈して塗布し、30℃で静置した。生育したコロニーについて10個のコロニーからゲノムDNAを調製し、以下に示す反応液組成及びプライマーを用いてPCRを行って、得られた断片(PCR産物)のサイズを電気泳動で調べた。なお、プライマーureC−03及びureC−06は、調製例1で用いたものと同様である。
【0084】
反応液組成:
滅菌水 22μl
PrimeSTAR(登録商標) Max Premix(2×) (タカラバイオ社製) 25μl
ureC−03(10μM)(配列番号8) 1μl
ureC−06(10μM)(配列番号11) 1μl
ゲノムDNA(100ng) 1μl
総量 50μl
温度サイクル:
98℃:10秒、55℃:15秒及び72℃:20秒の反応を30サイクル
その結果、10個の内、5個のコロニーはウレアーゼ遺伝子が欠失していることが確認された。得られたウレアーゼ遺伝子欠失株を、それぞれUD1〜UD5株とした。
【0085】
[実施例2]
UD1〜UD5株の培養
UD1〜UD5株とJ1株5株(J1−1〜J1−5)を下記の培地で培養し、ウレアーゼ活性およびニトリルヒドラターゼ活性を確認した。本培養の培地は、ニトリルヒドラターゼの誘導剤である尿素とコバルト、を含むものを用いた。前培養は30℃で3日間、本培養は30℃で2日間、振とう培養した。
【0086】
[前培養培地組成](pH7.0)
グルコース 20g/L
味液 20g/L
ポリペプトン 5g/L
酵母エキス 3g/L
MgSO4・7H2O 1g/L
KH2PO4 1g/L
K2HPO4 1g/L
【0087】
[本培養液組成](pH7.0)
グルコース 15g/L
ポリペプトン 3g/L
酵母エキス 3g/L
MgSO4・7H2O 1g/L
KH2PO4 1g/L
K2HPO4 1g/L
エタノール 2g/L
CoCl2・6H2O 0.025g/L
チアミン 0.002g/L
ビタミンK 0.002g/L
尿素 15g/L
【0088】
菌体を含む培養液のウレアーゼ活性の測定は、以下の方法に従って行った。
まず、本培養後に得られた培養液を遠心分離して菌体を取り除き、上清を50mMリン酸緩衝液(pH7.0)で100倍希釈する。市販の尿素測定キット(F−キット 尿素/アンモニア;ロシュ・ダイアグノスティクス社製)を用いて希釈液に含まれる尿素濃度を測定し、ウレアーゼ活性を評価した。
培養液中の尿素濃度を測定した結果、培養開始時の尿素濃度15g/Lに対して、J1株の場合は平均で約3.5g/L残っていたが、ウレアーゼ遺伝子を欠失させたUD1〜UD5株ではほとんど減少しておらず平均14.5g/L残っていた。以上の結果よりウレアーゼ活性が欠失していることを確認した。
【配列表フリーテキスト】
【0089】
配列番号2:プライマーSAC−01
配列番号3:プライマーSAC−02
配列番号4:プライマーureC−01
配列番号5:プライマーureC−02
配列番号8:プライマーureC−03
配列番号9:プライマーureC−04
配列番号10:プライマーureC−05
配列番号11:プライマーureC−06
【特許請求の範囲】
【請求項1】
ニトリルヒドラターゼ活性を有するロドコッカス属細菌において、ウレアーゼ遺伝子が欠失又は不活性化されたロドコッカス属細菌。
【請求項2】
ロドコッカス属細菌が、ロドコッカス ロドクロウス(Rhodococcus rhodochrous)である、請求項1記載のロドコッカス属細菌。
【請求項3】
欠失または不活性化させるウレアーゼ遺伝子が、配列番号1に示される塩基配列を有するウレアーゼアルファサブユニットもしくはそのオーソログである、請求項1又は2記載のロドコッカス属細菌。
【請求項4】
欠失または不活性化させるウレアーゼ遺伝子が、以下の(a)又は(b)に示されるいずれかの塩基配列を有するDNAからなる、請求項1〜3記載のいずれか1項に記載のロドコッカス属細菌。
(a)配列番号1記載の塩基配列を含むDNA
(b)配列番号1記載の塩基配列と相同性が65%以上の塩基配列からなり、かつウレアーゼ活性を有するタンパク質をコードするDNA
【請求項5】
ロドコッカス属細菌が、ロドコッカス・ロドクロウス(Rhodococcus rhodochrous)J1株又はその変異株である、請求項1〜4記載のいずれか1項に記載のロドコッカス属細菌。
【請求項6】
請求項1〜5のいずれか1項に記載のロドコッカス属細菌を培養し、得られる培養物からニトリルヒドラターゼを採取することを特徴とする、ニトリルヒドラターゼの製造方法。
【請求項7】
請求項1〜5のいずれか1項に記載のロドコッカス属細菌を培養して得られる培養物又は当該培養物の処理物をニトリル化合物に接触させ、当該接触により生成されるアミド化合物を採取することを特徴とする、アミド化合物の製造方法。
【請求項1】
ニトリルヒドラターゼ活性を有するロドコッカス属細菌において、ウレアーゼ遺伝子が欠失又は不活性化されたロドコッカス属細菌。
【請求項2】
ロドコッカス属細菌が、ロドコッカス ロドクロウス(Rhodococcus rhodochrous)である、請求項1記載のロドコッカス属細菌。
【請求項3】
欠失または不活性化させるウレアーゼ遺伝子が、配列番号1に示される塩基配列を有するウレアーゼアルファサブユニットもしくはそのオーソログである、請求項1又は2記載のロドコッカス属細菌。
【請求項4】
欠失または不活性化させるウレアーゼ遺伝子が、以下の(a)又は(b)に示されるいずれかの塩基配列を有するDNAからなる、請求項1〜3記載のいずれか1項に記載のロドコッカス属細菌。
(a)配列番号1記載の塩基配列を含むDNA
(b)配列番号1記載の塩基配列と相同性が65%以上の塩基配列からなり、かつウレアーゼ活性を有するタンパク質をコードするDNA
【請求項5】
ロドコッカス属細菌が、ロドコッカス・ロドクロウス(Rhodococcus rhodochrous)J1株又はその変異株である、請求項1〜4記載のいずれか1項に記載のロドコッカス属細菌。
【請求項6】
請求項1〜5のいずれか1項に記載のロドコッカス属細菌を培養し、得られる培養物からニトリルヒドラターゼを採取することを特徴とする、ニトリルヒドラターゼの製造方法。
【請求項7】
請求項1〜5のいずれか1項に記載のロドコッカス属細菌を培養して得られる培養物又は当該培養物の処理物をニトリル化合物に接触させ、当該接触により生成されるアミド化合物を採取することを特徴とする、アミド化合物の製造方法。
【図1】

【図2】
【図2】
【公開番号】特開2013−66416(P2013−66416A)
【公開日】平成25年4月18日(2013.4.18)
【国際特許分類】
【出願番号】特願2011−206881(P2011−206881)
【出願日】平成23年9月22日(2011.9.22)
【出願人】(000006035)三菱レイヨン株式会社 (2,875)
【Fターム(参考)】
【公開日】平成25年4月18日(2013.4.18)
【国際特許分類】
【出願日】平成23年9月22日(2011.9.22)
【出願人】(000006035)三菱レイヨン株式会社 (2,875)
【Fターム(参考)】
[ Back to top ]