
エカランチドに関する配合物
【課題】室温にて安定しておりかつ薬学的配合物として有用な新規のエカランチド配合物の提供。
【解決手段】ヒスチジンおよびリン酸塩からなる群から選択される緩衝剤;スクロースおよびスクロースとマンニトールとの組み合わせからなる群から選択される増量剤/凍結保護物質;ならびにエカランチドを含むエカランチド配合物であって、該配合物が約6.0〜7.0のpHを有する、配合物が提供される。ヒスチジンおよびリン酸塩からなる群から選択される緩衝剤;スクロースおよびスクロースとマンニトールとの組み合わせからなる群から選択される増量剤/凍結保護物質;ならびにエカランチドを含む、凍結乾燥エカランチド配合物もまた提供される。
【解決手段】ヒスチジンおよびリン酸塩からなる群から選択される緩衝剤;スクロースおよびスクロースとマンニトールとの組み合わせからなる群から選択される増量剤/凍結保護物質;ならびにエカランチドを含むエカランチド配合物であって、該配合物が約6.0〜7.0のpHを有する、配合物が提供される。ヒスチジンおよびリン酸塩からなる群から選択される緩衝剤;スクロースおよびスクロースとマンニトールとの組み合わせからなる群から選択される増量剤/凍結保護物質;ならびにエカランチドを含む、凍結乾燥エカランチド配合物もまた提供される。
【発明の詳細な説明】
【背景技術】
【0001】
(背景)
エカランチド(ecallantide)は、クニッツドメインの一般構造を有する60アミノ酸のペプチドである。エカランチドは、血漿カリクレインの強力な阻害剤であることが明らかにされている。
【発明の概要】
【課題を解決するための手段】
【0002】
(発明の要旨)
本明細書には、室温にて安定しておりかつ薬学的配合物として有用な新規のエカランチド配合物が開示されている。
【0003】
本開示内容は、緩衝剤、緩衝剤/凍結保護物質、およびエカランチドを含めたエカランチド含有組成物(「エカランチド配合物」)を提供する。緩衝剤はpHを約6.0〜7.0に調節するヒスチジン緩衝剤またはリン酸緩衝剤であってもよく、増量剤はスクロースまたはスクロースとマンニトールの組み合わせであってもよい。場合により、増量剤/凍結保護物質はデキストラン40などのデキストランも含む。
【0004】
さらには、本明細書に開示される方法で製造される組成物も提供される。
【0005】
いくつかの実施形態において、緩衝剤はヒスチジンであり、これは10mMだけ存在する場合がある。いくつかの実施形態において、配合物のpHは約6.5である。
【0006】
いくつかの実施形態において、増量剤/凍結保護物質はスクロースであり、これは10%(w/v)で存在し得る。
【0007】
いくつかの実施形態において、エカランチドは、10mg/mL、20mg/mL、または30mg/mLで存在する。
【0008】
いくつかの実施形態において、本配合物は等張性を有する。
【0009】
本明細書に開示されるエカランチド配合物は凍結乾燥され得る。したがって、本開示内容では、緩衝剤、緩衝剤/凍結保護物質、およびエカランチドを含む、凍結乾燥エカランチド配合物を提供する。緩衝剤はpHを約6.0〜7.0に調節するヒスチジン緩衝剤またはリン酸緩衝剤である場合があり、増量剤はスクロースまたはスクロースとマンニトールの組み合わせであり得る。場合により、増量剤/凍結保護物質はデキストラン40などのデキストランも含む。
【0010】
凍結乾燥エカランチド配合物の成分は、約1:1〜約7.5:1もしくは約2:1〜約2.5:1(緩衝剤:エカランチド)、または約250:1〜約45:1もしくは約75:1〜約60:1(増量剤/凍結保護物質:エカランチド)、または約2.5:75:1〜約2:65:1、もしくは約7:208:1、約2.4:70:1、もしくは約1.4:41:1(緩衝剤:増量剤/凍結保護物質:エカランチド)などの種々のモル比で存在し得る。
【0011】
凍結乾燥エカランチド配合物の成分は、約1%〜約2%(w/w)の緩衝剤、約90%〜約60%の増量剤/凍結保護物質、および約9%〜約37%のエカランチドなど、種々の百分率(w/w)で存在し得る。
【0012】
本明細書ではまた、緩衝剤、増量剤/凍結保護物質、およびエカランチドの混合物を取得または産生し、その混合物を凍結乾燥させることによって、本明細書に開示される凍結乾燥エカランチド配合物を産生する方法も提供される。
【0013】
また、有効量の本開示内容のエカランチド配合物を血管性浮腫の被験体または血管性浮腫の疑いのある被験体に投与することにより、血管性浮腫(遺伝性血管性浮腫、アンギオテンシン変換酵素(ACE)阻害剤誘発性の血管性浮腫、後天性(例えば、C1エステラーゼ阻害剤の欠損)血管性浮腫、特発性慢性血管性浮腫、アレルギー性血管性浮腫、および非ステロイド系抗炎症薬(NSAID)誘発性の血管性浮腫)を治療する方法も提供される。
【0014】
また、本開示内容のエカランチド配合物を含むキットも提供される。こうしたキットは、本開示内容のエカランチド配合物を含む少なくとも1つの容器を含み、また血管性浮腫を治療するためのエカランチドの使用に関する指示書を含む場合もある。容器は、アンプル、バイアル、プレフィルドシリンジ、または自動注入装置(もしくは自動注入装置のカートリッジ)であり得る。
例えば、本願発明は以下の項目を提供する。
(項目1)
ヒスチジンおよびリン酸塩からなる群から選択される緩衝剤;
スクロースおよびスクロースとマンニトールとの組み合わせからなる群から選択される増量剤/凍結保護物質;ならびに
エカランチド
を含むエカランチド配合物であって、
該配合物が約6.0〜7.0のpHを有する、
配合物。
(項目2)
前記緩衝剤がヒスチジンである、項目1に記載の配合物。
(項目3)
前記増量剤/凍結保護物質がスクロースである、項目2に記載の配合物。
(項目4)
前記pHが約pH6.5である、項目3に記載の配合物。
(項目5)
前記緩衝剤および前記エカランチドが2:1〜2.5:1(緩衝剤:エカランチド)のモル比で存在する、項目4に記載の配合物。
(項目6)
前記増量剤/凍結保護物質および前記エカランチドが75:1〜60:1(増量剤/凍結保護物質:エカランチド)のモル比で存在する、項目5に記載の配合物。
(項目7)
前記緩衝剤、前記増量剤/凍結保護物質、および前記エカランチドが2.5:75:1〜2:65:1(緩衝剤:増量剤/凍結保護物質:エカランチド)のモル比で存在する、項目6に記載の配合物。
(項目8)
前記増量剤/凍結保護物質がスクロースである、項目1に記載の配合物。
(項目9)
前記pHが約pH6.5である、項目1に記載の配合物。
(項目10)
前記配合物が凍結乾燥されている、項目1に記載の配合物。
(項目11)
前記緩衝剤および前記エカランチドが1:1〜7.5:1(緩衝剤:エカランチド)のモル比で存在する、項目1に記載の配合物。
(項目12)
前記緩衝剤および前記エカランチドが2:1〜2.5:1(緩衝剤:エカランチド)のモル比で存在する、項目11に記載の配合物。
(項目13)
前記増量剤/凍結保護物質および前記エカランチドが300:1〜45:1(増量剤/凍結保護物質:エカランチド)のモル比で存在する、項目1に記載の配合物。
(項目14)
前記増量剤/凍結保護物質および前記エカランチドが75:1〜60:1(増量剤/凍結保護物質:エカランチド)のモル比で存在する、項目13に記載の配合物。
(項目15)
前記緩衝剤、前記増量剤/凍結保護物質、および前記エカランチドが2.5:75:1〜2:65:1(緩衝剤:増量剤/凍結保護物質:エカランチド)のモル比で存在する、項目1に記載の配合物。
(項目16)
ヒスチジンおよびリン酸塩からなる群から選択される緩衝剤;
スクロースおよびスクロースとマンニトールとの組み合わせからなる群から選択される増量剤/凍結保護物質;ならびに
エカランチド
から本質的になる、項目1に記載の配合物。
(項目17)
ヒスチジンおよびリン酸塩からなる群から選択される緩衝剤;
スクロースおよびスクロースとマンニトールとの組み合わせからなる群から選択される増量剤/凍結保護物質;ならびに
エカランチド
を含む、凍結乾燥エカランチド配合物。
(項目18)
前記緩衝剤がヒスチジンである、項目17に記載の配合物。
(項目19)
前記増量剤/凍結保護物質がスクロースである、項目18に記載の配合物。
(項目20)
再構成された配合物のpHが約pH6.5である、項目19に記載の配合物。
(項目21)
前記緩衝剤および前記エカランチドが2:1〜2.5:1(緩衝剤:エカランチド)のモル比で存在する、項目20に記載の配合物。
(項目22)
前記増量剤/凍結保護物質および前記エカランチドが75:1〜60:1(増量剤/凍結保護物質:エカランチド)のモル比で存在する、項目21に記載の配合物。
(項目23)
前記緩衝剤、前記増量剤/凍結保護物質、および前記エカランチドが2.5:75:1〜2:65:1(緩衝剤:増量剤/凍結保護物質:エカランチド)のモル比で存在する、項目22に記載の配合物。
(項目24)
前記増量剤/凍結保護物質がスクロースである、項目17に記載の配合物。
(項目25)
前記pHが約pH6.5である、項目17に記載の配合物。
(項目26)
前記配合物が凍結乾燥されている、項目17に記載の配合物。
(項目27)
前記緩衝剤および前記エカランチドが1:1〜7.5:1(緩衝剤:エカランチド)のモル比で存在する、項目17に記載の配合物。
(項目28)
前記緩衝剤および前記エカランチドが2:1〜2.5:1(緩衝剤:エカランチド)のモル比で存在する、項目27に記載の配合物。
(項目29)
前記増量剤/凍結保護物質および前記エカランチドが300:1〜45:1(増量剤/凍結保護物質:エカランチド)のモル比で存在する、項目17に記載の配合物。
(項目30)
前記増量剤/凍結保護物質および前記エカランチドが75:1〜60:1(増量剤/凍結保護物質:エカランチド)のモル比で存在する、項目29に記載の配合物。
(項目31)
前記緩衝剤、前記増量剤/凍結保護物質、および前記エカランチドが2.5:75:1〜2:65:1(緩衝剤:増量剤/凍結保護物質:エカランチド)のモル比で存在する、求項17に記載の配合物。
(項目32)
(a)ヒスチジンおよびリン酸塩からなる群から選択される緩衝剤と、スクロースおよびスクロースとマンニトールとの組み合わせからなる群から選択される増量剤/凍結保護物質と、エカランチドとの混合物を得ること;ならびに
(b)該混合物を凍結乾燥させること
というプロセスにより産生される、凍結乾燥エカランチド配合物。
(項目33)
(a)ヒスチジンおよびリン酸塩からなる群から選択される緩衝剤と、スクロースおよびスクロースとマンニトールとの組み合わせからなる群から選択される増量剤/凍結保護物質と、エカランチドとの混合物を得ること;ならびに
(b)該混合物を凍結乾燥させること
というプロセスにより産生される凍結乾燥エカランチド配合物を製造するための方法。
【図面の簡単な説明】
【0015】
【図1】プラスミドpPIC K503の図を示す。
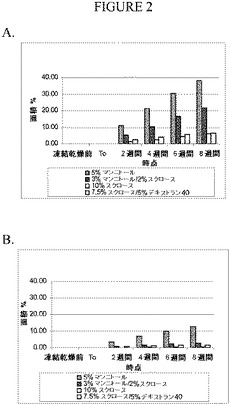
【図2】PBSで緩衝処理された配合物中のピログルタメートのレベル(図2A)または10mMのヒスチジンで緩衝処理された配合物中のピログルタメートのレベル(図2B)を測定したRP‐HPLCのデータのグラフを示す。
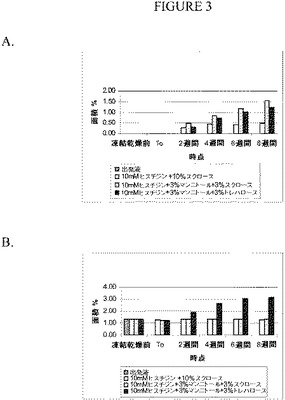
【図3】ピログルタメートのレベル(図3A)およびピーク4のレベル(図3B)を測定したRP‐HPLCのデータのグラフを示す。
【発明を実施するための形態】
【0016】
(発明の詳細な説明)
本明細書には、室温にて安定しておりかつ薬学的配合物として有用な新規のエカランチド配合物が開示される。
【0017】
本明細書で使用される、百分率、モル濃度またはモル比に関連して使用される「約」という言葉は、示される値の±10%の範囲を指す(例えば、「約10mM」は9mM〜11mMを意味する)。pHの値に関連して「約」が使用される場合、これは示される値の±0.2pH単位の範囲を指す(例えば、「約pH7.0」はpH6.8〜7.2を意味する)。
【0018】
エカランチド
クニッツドメインをベースにした多くのタンパク質が当該技術分野で既知である(例えば、米国特許第4,245,051号、第5,278,285号、第5,436,153号、第5,728,674号、第5,563,123号、第5,589,359号、第5,696,088号,第5,663,143号、第5,880,256号、第5,968,897号、第5,977,057号、第6,103,500号、第5,990,079号、第6,063,764号、第6,414,124号、第6,583,108号、第6,593,291号;および第6,914,135号など)。
【0019】
エカランチドは、クニッツドメインの一般構造を有する60アミノ酸のペプチドである。エカランチドは、Glu Ala Met His Ser Phe Cys Ala
Phe Lys Ala Asp Asp Gly Pro Cys Arg Ala
Ala His Pro Arg Trp Phe Phe Asn Ile Phe
Thr Arg Gln Cys Glu Glu Phe Ile Tyr Gly
Gly Cys Glu Gly Asn Gln Asn Arg Phe Glu
Ser Leu Glu Glu Cys Lys Lys Met Cys Thr
Arg Asp(配列番号1)の配列を有する。エカランチドの分子量は7,054ダルトンである。エカランチドは、血漿カリクレインの非常に効果的な阻害剤であり、遺伝性血管性浮腫および虚血防止を含めたいくつかの適応症の治療剤として提案されてきた(Williams, et al., 2003, Transfus. Apher. Sci. 29(3):255−58;米国特許出願公開第2004/0038893号)。
【0020】
エカランチドは、いずれかの標準的なポリペプチド合成手順および機器を使用して合成され得る。例えば、エカランチドの段階的合成は、最初の(すなわち、カルボキシ末端)アミノ酸からアミノ(N)末端保護基を除去し、ポリペプチドの配列中の次のアミノ酸のカルボキシル末端をそれに結合させることにより、行われ得る。このアミノ酸はまた好適に保護される。入ってくるアミノ酸のカルボキシル基は、カルボジイミド、対称酸無水物、または「活性エステル」基(ヒドロキシベンゾトリアゾールまたはペンタフルオロフェニルエステルなど)を形成するなど、反応性基を形成することにより、結合するアミノ酸のN末端と反応するように活性化させることができる。有用な固相ペプチド合成方法には、a‐アミノ保護基としてtert−ブチルオキシカルボニルを利用するBoc法、および9‐フルオレニルメトキシカルボニルを利用してアミノ酸残基のa‐アミノを保護するFmoc法が含まれる。いずれの方法も当業者に周知である(教示内容全体が参考として本明細書で援用される、Stewart, J. and Young, J., Solid‐Phase Peptide Synthesis(W. H. Freeman Co.,San Francisco)1989);Merrifield, J., 1963. Am. Chem. Soc., 85:2149−2154; Bodanszky, M. and Bodanszky, A., The Practice of Peptide Synthesis(Springer‐Verlag, New York 1984))。
【0021】
あるいは、エカランチドは、いくつかの細胞および対応する発現ベクター(細菌発現ベクター、酵母発現ベクター、バキュロウイルス発現ベクター、哺乳類ウイルス発現ベクターを含むがこれらに限定されない)のいずれかを使用した組換え方法により産生され得る。エカランチドは、エカランチドをコードする配列を含む核酸分子を使用して遺伝子導入により産生される場合もあり、その場合、核酸分子は、当該技術分野で使用可能な遺伝子導入方法を使用して宿主動物のゲノムに組み込み、そのゲノムから発現させることができる。場合によっては、エカランチドのコード配列を発現ベクター中の別のコード配列と融合させ、宿主細胞内で容易に発現される融合ポリペプチドを形成させることが必要であるか、または有利であり得る。このような融合ポリペプチドを発現する宿主細胞はまた、融合ポリペプチドのプロセシングを行って、所望のアミノ酸配列(すなわち、エカランチド)のみを得るのが好ましい。その他いずれかの1つ以上のアミノ酸が、発現したエカランチドに結合したままである場合、このような1つ以上の付加的なアミノ酸は、本明細書に開示される配合物中でポリペプチドを使用できなくなるほどエカランチドの活性を減少させるべきではない。
【0022】
実施例に開示されるエカランチドの特定の産生方法では、酵母宿主細胞における組換え発現が利用される。酵母発現ベクターでは、同じ読み枠において、エカランチドのアミノ酸配列をコードする核酸配列が、サッカロマイセス・セレビシエのmatαプレプロリーダーペプチド配列をコードするヌクレオチド配列と結合することができるため、このベクターが操作可能な酵母プロモーターの制御下にある。次いで、得られた組換え酵母発現プラスミドは、標準的な方法によって適合性のある適切な酵母宿主の細胞に形質転換され、これらの細胞は組換え酵母発現ベクターから組換えタンパク質を発現させることができる。このような組換え発現ベクターによって形質転換される酵母宿主細胞は、好ましくは、融合タンパク質をプロセッシングして、本明細書に開示される方法および組成物において有用な活性エカランチドを提供することもできる。このような方法で組換えエカランチドを産生させるのに有用な酵母宿主細胞は、ピキア・パストリスである。
【0023】
薬学的配合物で使用するためのエカランチドは実質的に均質であるべきである。したがって、エカランチドは通常、(合成または組換え発現によって)産生された後に精製される。エカランチドの精製は、サイズ排除クロマトグラフィー、イオン交換(陰イオンおよび/または陽イオン交換)クロマトグラフィー、疎水性相互作用クロマトグラフィー、アフィニティークロマトグラフィー、および逆相クロマトグラフィー、またはそれらのいずれかの組み合わせを含めた、当該技術分野で既知の技法を使用して行われ得る。さらには、緩衝剤交換および/または濃縮技術も所望により使用され得る。
【0024】
本明細書の実施例に記載の通り、エカランチドは特定の条件下で不安定であり、貯蔵されると、高分子量(例えば、凝集生成物)劣化生成物および低分子量(例えば、断片化生成物)劣化生成物の両方、ならびに修飾生成物(例えば、アミノ末端ピログルタミン酸塩)を生じる。本明細書に開示される配合物では、エカランチドが実質的に安定され、凝集生成物、断片化生成物、または修飾生成物の生成が阻止されるかまたは減少する。
【0025】
エカランチドは、所期の用途(例えば、所期の用量)に応じて、種々のレベルで本配合物中に存在し得る。液体配合物の場合、エカランチドは、約5mg/mL(0.7mM)〜約50mg/mL(7mM)、または約7mg/mL(1mM)〜約40mg/mL(5.7mM)、または約10mg/mL(1.4mM)〜約30mg/mL(4.2mM)、または約30mg/mLの濃度で存在し得る。百分率(w/v)で表わした場合、エカランチドは、約0.5%〜約5%、または約0.7%〜約4%、または約1%〜約3%の濃度で存在し得る。凍結乾燥した配合物の場合、エカランチドは、約5%〜約45%(w/w)、または約7%〜約40%(w/w)または約9%〜約37%(w/w)だけ存在し得る。
【0026】
pHおよび緩衝剤
本明細書に開示される配合物は、緩衝剤でpHが制御される。実施例で記載の通り、エカランチドは約6.0〜約7.0のpH範囲で安定している。したがって、本明細書では、液体形態である場合(例えば、産生時または再構成時)に、pHが約6.0〜約7.0、例えば約6.0(例えば、pH5.8〜6.2)、約6.5(例えば、pH6.3〜6.7)、または約7.0(例えば、pH6.8〜7.2)である配合物が提供される。
【0027】
pH約6.0〜約7.0の範囲に調節するのに好適ないずれの緩衝剤も使用され得る。いくつかの実施形態において、緩衝剤はまた薬学的に許容される。好適な緩衝剤には、クエン酸塩、コハク酸塩、リンゴ酸塩、カコジル酸塩、(N−モルホリノ)エタンスルホン酸水和物(MES)、クエン酸塩、マレアート、ヒスチジン、リン酸塩、および炭酸塩がある。特定の実施形態において、緩衝剤はヒスチジンまたはリン酸塩である。特定の実施形態において、緩衝剤はヒスチジンである。
【0028】
緩衝剤は、予想される貯蔵条件および(凍結乾燥配合物の場合は)再構成条件において十分なpH制御を行える濃度で含まれる。液体形態の配合物の場合、緩衝剤は一般的に、約3mM〜約20mM、または約5mM〜約15mM、または約8mM〜約12mM、または約10mMで含まれる。百分率(w/v)として計算した場合、緩衝剤は、約0.045%〜約0.31%、または約0.08%〜約0.23%、または約0.12%〜約0.19%、または約0.15%の濃度で存在し得る。凍結乾燥配合物の場合、緩衝剤は一般的に、約0.25%〜約5%(w/w)、または約0.5%〜約2.5%(w/w)、または約1%〜約2%(w/w)で含まれる。
【0029】
増量剤/凍結保護物質
本明細書に開示される配合物は、増量剤/凍結保護物質を含む。本発明者等は、スクロースが、単独でまたはマンニトールと組み合わせて、エカランチド配合物の増量剤/凍結保護物質として有用であることを発見した。さらに、配合物はデキストラン(実施形態によってはデキストラン40)を含みうる。
【0030】
予想外にも、本発明者等は、安定させていると予想されていた通常使用される増量剤/凍結保護物質のトレハロースが、エカランチド配合物に含まれる場合には不安定にさせていることも見出した。したがって、トレハロースがエカランチド配合物を不安定にすることを本発明者等は見出したため、本明細書に開示される配合物は、実質的にまたは完全にトレハロースを含まない場合ができる。本明細書で使用される「実質的にトレハロースを含まない」とは、配合物において(液体形態の場合)トレハロースが1mM未満であるか、または(凍結乾燥形態の場合)トレハロースが1重量%未満であることを意味する。
【0031】
増量剤/凍結保護物質は、乾燥させたときに許容される凍結乾燥したケーキを産生して、少なくともエカランチドの凍結保護の指標を提供するのに十分な容積となる量だけ、本配合物に含まれる。液体配合物の場合、配合物の百分率で測定すると、増量剤/凍結保護物質は、約3%〜約15%(w/v)、または約4%〜約15%、または約5%〜約10%だけ存在する。液体配合物の場合、増量剤/凍結保護物質のモル濃度で測定すると、増量剤/凍結保護物質は、約200mM〜約350mM、または約250mM〜約300mMで存在する。増量剤/凍結保護物質がスクロースである実施形態において、増量剤/凍結保護物質は約292mMで存在し得る。凍結乾燥配合物の場合、増量剤は約95%〜約55%(w/w)、または約90%〜約60%(w/w)で存在する。
【0032】
配合物
本明細書に開示される配合物は、エカランチド、pH緩衝剤、および増量剤/凍結保護物質を含む。本配合物は薬学的配合物として使用することを意図していることから、特定の実施形態において、配合物は等張性を有する(例えば、250〜350mOsM、または約300mOsMの重量オスモル濃度を有する)。当業者であれば分かるように、成分の比率は、成分(特にエカランチド)の濃度により変動する(エカランチドは所期の投与量により変動し得る)。医薬品の用途では、本明細書に開示される配合物の成分は、米国薬局方(USP)または同様の等級のものであるか、あるいは医薬品の製造及び品質管理に関する基準(GMP)に従って産生されなければならない。
【0033】
液体形態の場合、配合物の成分の量は、モル濃度または百分率(w/v)濃度で容易に記載することができる。モル濃度で表わす場合、本配合物は、緩衝剤が約3mM〜約20mM、または約5mM〜約15mM、または約8mM〜約12mM、または約10mMである場合があり、増量剤/凍結保護物質が約200mM〜約350mM、または約250mM〜約300mM、または約292mMである場合があり、エカランチドが約1mM〜約5mM、または約1.4、2.8もしくは4.2mMである場合がある。百分率(w/v)濃度で表わす場合、本配合物は、緩衝剤が0.045%〜約0.31%、または約0.08%〜約0.23%、または約0.12%〜約0.19%、または約0.15%である場合があり、増量剤/凍結保護物質が3%〜約15%、または約4%〜約15%、または約5%〜約10%である場合があり、エカランチドが約0.5%〜約5%、または約0.7%〜約4%、または約1%〜約3%である場合がある。
【0034】
乾燥(例えば、凍結乾燥)形態の場合、成分の量は、百分率(w/w)またはモル比で極めて容易に記載される。百分率で表わす場合、本配合物は、緩衝剤が約0.25%〜約5%(w/w)、または約0.5%〜約2.5%(w/w)、または約1%〜約2%(w/w)である場合があり、増量剤/凍結保護物質が約95%〜約55%(w/w)、または約90%〜約60%(w/w)である場合があり、エカランチドが5%〜約45%(w/w)、または約7%〜約40%(w/w)、または約9%〜約37%(w/w)である場合がある。当業者であれば分かるように、緩衝剤、増量剤/凍結保護物質、およびエカランチドの百分率量の合計は、100%に満たない場合があるが、実際に一般的には100%に満たず、残りは残留溶媒である。モル比(緩衝剤:増量剤/凍結保護物質:エカランチド)で表わす場合、本配合物は、約7.5:208:1〜約1:45:1、または約2:100:1〜約2.5:75:1、または約7:208:1、または約2.4:70:1、または約1.4:41:1であり得る。
【0035】
1つの例示的な配合物は、(液体形態の場合)緩衝剤として約10mMのヒスチジンを、増量剤/凍結保護物質として約10%(w/v)のスクロースを、また約10mg/mLのエカランチドを含み、pHが6.5である。乾燥(凍結乾燥)形態の場合、本配合物は、緩衝剤が約1.4%(w/w)、増量剤/凍結保護物質が88.8%(w/w)、エカランチドが約8.9%(w/w)であり、モル比が約7:208:1(ヒスチジン:スクロース:エカランチド)である。
【0036】
別の例示的な配合物は、(液体形態の場合)緩衝剤として約10mMのヒスチジンを、増量剤/凍結保護物質として約10%(w/v)のスクロース、また約30mg/mLのエカランチドを含み、pHが6.5である。乾燥(凍結乾燥)形態の場合、本配合物は緩衝剤が約1.2%、増量剤/凍結保護物質が約75.4%、エカランチドが約22.6%であり、モル比が約2.4:70:1である。
【0037】
本明細書に開示される配合物は、所望の最終組成物を生じる従来の技法によって製造され得る。成分は、水に直接溶解して最終濃度にする場合もあれば、濃縮物として製造し、これらを混合・希釈されて最終生成物を得る場合もある。あるいは、緩衝剤交換方法が使用される場合もある。
【0038】
一般的に、エカランチドは、エカランチド産生の最終処理手順の結果、水溶液中に含まれることになる。次いで、本エカランチド溶液は、(例えば、ダイアフィルトレーションによって)緩衝剤の交換を行い、所望の配合物を得る場合もあれば、あるいは、緩衝剤の交換を実行できない場合(例えば、増量剤/凍結保護物質によって、緩衝剤を交換するにはあまりに粘性がある配合物が得られる場合)、エカランチドは緩衝剤の交換を行い(必要に応じて濃縮し)濃縮溶液を得て、それを残りの成分と混合して所望の配合物を産生する場合もある(例えば、ヒスチジンが10mM、pHが6.5、スクロースが10%、エカランチドが30mg/mLである所望の配合物の場合、エカランチド溶液は緩衝剤の交換を行い、必要に応じて濃縮して原液を製造する。この原液は、濃縮スクロース溶液と混合するか、あるいは乾燥スクロースと混合したとしても、10mMのヒスチジン、pH6.5、10%のスクロース、および30mg/mLのエカランチドの最終配合物が得られる)。
【0039】
凍結乾燥
凍結乾燥(lyophilization)(すなわち、凍結乾燥(freeze‐drying))は、液体組成物を凍らせてから、凍結液体(例えば、水)の昇華によって脱水するプロセスである。昇華は一次乾燥に適した温度で行われる。一次乾燥に適した温度とは、配合物の共融点または崩壊温度以下の温度に生成物を保持する温度である。
【0040】
凍結乾燥させる物質(例えば、エカランチド配合物)は、凍結乾燥装置に充填する前に凍結させる場合もあれば、液体形態で装置に充填し、装置内にある間に凍結させる場合もある。液体配合物の凍結は、単一の傾斜(例えば、所望の温度まで温度を連続して低下させる)として、または一連の手順/傾斜において、所望の温度まで下げる単一の手順を含めたいずれかの方法で行われ得る。凍結液体配合物の「所望の温度」は、物質が凍結するが一般的に物質の凝固点よりも低い、いずれかの温度である場合があり、約0℃〜約−50℃の範囲であり得る。所望の温度に達したら(または所望の温度に達した後の平衡期間に続いて)、部分真空が確立されるが、これは、約50〜約250mTorr、または約60〜約200mTorr、または約75〜約100mTorrの範囲であり得る。
【0041】
凍結乾燥装置内の温度は、凍結乾燥プロセスの間一定に維持される場合があり、より一般的には本プロセスの間に調整される(一般的には上昇させる)。例えば、凍結乾燥器は、真空にする前に平衡状態にして約−40℃または約−45℃にし、その後、凍結乾燥プロセスの一次乾燥段階が進行するにつれて、一連の手順または傾斜にて徐々に加温され得る。例えば、約−40℃で開始する凍結乾燥プロセスの場合、凍結乾燥器は、一次乾燥段階の最初の部分の間に一連の凝固点下の温度を経て(例えば、連続して約5℃または10℃ずつ増加させながら、あるいは約−40℃〜約−35℃へ、次いで約−25℃へ、次いで約−10℃へ、または約−40℃〜約−30℃へ、次いで約−15℃へ、または−40℃〜約−30℃へ、次いで約−25℃へ、または約−40℃〜約−25℃へといったように一連の不規則な手順で)上昇/増大させ得る。一次乾燥の後半の段階は、約0℃〜約10℃(例えば、約3℃、約5℃、約7℃、または約10℃)の温度など、同じ温度または高い温度で行われ得る。
【0042】
主に、試料を貯蔵する容器(例えば、ガラスバイアル)の正確な配合、サイズおよび種類、液体の容積、ならびに凍結乾燥の温度および圧力によって、乾燥に必要な時間が決定され、この時間は数時間から数日(例えば、40〜60時間)に及んでもよい。一次乾燥条件の例には、(1)75mTorrの真空レベル、一次乾燥段階の大半で約−25℃の温度の後に約5℃の期間、および約30〜35時間の一次乾燥時間、ならびに(2)75mTorrの真空レベル、一次乾燥段階での約−25℃の温度、および約15〜20時間の一次乾燥時間が含まれる。
【0043】
二次乾燥段階は、主に、使用する容器の種類およびサイズと正確な配合に応じて行われ得る。場合によっては、高温(例えば、約0℃〜約40℃、または約10℃〜約30℃、または約20℃または約30℃)での二次乾燥段階が使用される。しかし、場合によっては、二次乾燥手順が必要でない場合もある。二次乾燥に必要な時間および圧力は、好適な凍結乾燥したケーキを産生する時間および圧力となる。したがって、二次乾燥条件(およびいずれにせよ二次乾燥手順に必要なもの)は、温度および他のパラメータによって決まる。二次乾燥時間は、生成物中の所望の残留水分レベルによって決まり、一般的には少なくとも約5時間(例えば、約5〜約20時間(約8、約9、約10、約12、約15または約18時間など))かかる。圧力は、一次乾燥手順時に使用するものと同じであり得る。凍結乾燥条件は、配合およびバイアルサイズによって変更することができる。
【0044】
場合によっては、移送手順を回避するために、タンパク質を元に戻す容器中でタンパク質配合物を凍結乾燥させるのが望ましいことがある。この場合の容器は、例えば、3、5、10、20、50または100ccのバイアルであり得る。
【0045】
凍結乾燥(および必要に応じていずれかの移送手順)の後、凍結乾燥した配合物を一般的には容器内に入れて密封する。密封は、非弾性の蓋(例えば、全体がガラスでできたバイアルの端を溶かしてバイアルを密閉する)により、または弾性の蓋を取り付ける(例えば、容器の開口部を弾性の栓で塞いだ後、所定の位置にシール保持止め具を圧着して固定する)ことにより行うことができる。場合によっては、(例えば、減圧窒素環境で密閉することにより行われる場合のように)内容物を減圧下および/または減圧酸素下に置くような条件のもとで容器を密閉する。
【0046】
一般的な事柄として、凍結乾燥を行うと、含水量が約5%未満、例えば、約3%未満、または約2%未満の凍結乾燥配合物が生成される。
【0047】
再構成および投与
所望の段階で(一般的にはタンパク質を患者に投与するのが相応しい時に)、凍結乾燥配合物を希釈剤で再構成する場合がある。再構成に使用する希釈剤の容量は、再構成した配合物が所望のエカランチド濃度を有するようになる容量である。いくつかの実施形態においては、凍結乾燥配合物が再構成されて(例えば、適切な量の希釈剤を加えて)、エカランチドが10、20、30または40mg/mLである再構成配合物が得られ、また特定の実施形態においては、凍結乾燥配合物が再構成されて、エカランチドが30mg/mL、ヒスチジンが10mM、pHが6.5、スクロースが10%(w/v)である再構成配合物が得られる。
【0048】
希釈剤の例には、注射用滅菌水(WFI)、および注射用静菌性水(BWFI)が含まれるが、pH緩衝剤(例えば、リン酸塩緩衝食塩水)、滅菌食塩水、リンゲル液またはブドウ糖溶液などの他の希釈剤が使用される場合もある。
【0049】
希釈剤は場合により防腐剤を含む。有用な防腐剤には、ベンジルアルコールまたはフェノールアルコールなどの芳香族アルコールが含まれる。使用する防腐剤の量は、タンパク質との適合性に関する種々の防腐剤の濃度の評価および防腐剤の効力試験により決定される。例えば、防腐剤が芳香族アルコール(ベンジルアルコールなど)である場合、これは約0.1〜2.0%、約0.5〜1.5%、または約1.0〜1.2%の量だけ存在する場合がある。
【0050】
凍結乾燥配合物の再構成は一般的に、完全な水和が確実に行われるように室温(例えば、20℃〜25℃)にて行われるが、所望により他の温度が使用される場合もある。再構成に必要な時間は、配合物の正確な構成成分(例えば、希釈剤の種類、1つ以上の賦形剤およびエカランチドの量)により異なる。再構成は、手作業で(例えば、凍結乾燥配合物を含む容器に注入ポートを介して注入することにより、希釈剤を凍結乾燥配合物に手作業で加える)または自動的に(例えば、自動的に再構成するように構成されたBecton‐Dickinson BD(登録商標)のLiquid Dry Injectorなどの装置で希釈剤を凍結乾燥配合物に自動添加する)行う場合がある。
【0051】
配合物(液体配合物および再構成した凍結乾燥配合物)は、薬学的配合物(一般的には非経口投与用)として有用である。非経口投与には、静脈内(IV)、筋肉内(IM)、皮下(SC)、腹腔内(IP)、鼻腔内、および吸入の経路が含まれるが、これらに限定されない。IV、IM、SCおよびIP投与は、ボーラスまたは注入によって行われる場合があり、SCの場合には、徐放性埋込み式デバイス(ポンプ、徐放性配合物、および機械装置を含むがこれらに限定されない)で行われる場合もある。投与の用量、経路および方法は、治療する障害および患者の病歴により異なる。
【0052】
使用方法
本明細書ではまた、本明細書に開示される配合物を利用して、過剰なまたは調節不全の血漿カリクレイン活性に関連した障害を治療する方法も提供される。本明細書で使用される「治療」という用語は、治療する障害の症状を安定化、回復、改善または除去することを指す。過剰/調節不全の血漿カリクレイン活性に関連する臨床的障害はいくつかあり、これには、遺伝性血管性浮腫(I、IIおよびIII型の遺伝性血管性浮腫)、アンギオテンシン変換酵素(ACE)阻害剤誘発性の血管性浮腫、後天性(例えば、C1エステラーゼ阻害剤の欠損)血管性浮腫、特発性慢性血管性浮腫、アレルギー性血管性浮腫、および非ステロイド系抗炎症薬(NSAID)誘発性血管性浮腫が含まれる(本明細書では、遺伝性、ACE阻害剤誘発性、特発性慢性、アレルギー性、およびNSAID誘発性の血管性浮腫を「血管性浮腫」と総称する)。本開示内容のエカランチド配合物を投与することで、治療する血管性浮腫の少なくとも1つの症状(例えば、限局性浮腫)が安定化、回復、改善または除去される。
【0053】
したがって、本開示内容は、(1)本明細書に開示される有効量のエカランチド配合物を遺伝性血管性浮腫の被験体またはその疑いのある被験体に投与すことにより、遺伝性血管浮腫を治療する方法、(2)本明細書に開示される有効量のエカランチド配合物をACE阻害剤誘発性の血管性浮腫の被験体またはその疑いのある被験体に投与することにより、ACE阻害剤誘発性の血管性浮腫を治療する方法、(3)本明細書に開示される有効量のエカランチド配合物を後天性血管性浮腫の被験体またはその疑いのある被験体に投与することにより、後天性(例えば、C1エステラーゼ阻害剤の欠損)血管性浮腫を治療する方法、(4)本明細書に開示される有効量のエカランチド配合物を特発性慢性血管性浮腫の被験体またはその疑いのある被験体に投与することにより、特発性慢性血管性浮腫を治療する方法、(5)本明細書に開示される有効量のエカランチド配合物をアレルギー性血管性浮腫の被験体またはその疑いのある被験体に投与することにより、アレルギー性血管性浮腫を治療する方法、および(6)本明細書に開示される有効量のエカランチド配合物をNSAID誘発性血管性浮腫の被験体またはその疑いのある被験体に投与することにより、NSAID誘発性血管性浮腫を治療する方法を提供する。場合により、治療方法はさらに、投与に先立って凍結乾燥エカランチド配合物を再構成する手順も含む場合がある。
【0054】
有効量となるエカランチド配合物の量は、患者の病歴および疾患の重症度(あるいは疾患の悪化または急性発作)により異なる場合がある。いくつかの実施形態において、エカランチド配合物の有効量は、30mgのエカランチドを含む量である。
【0055】
本方法によれば、エカランチド配合物はいずれかの非経口経路で投与される場合がある。特定の実施形態において、エカランチド配合物は皮下のボーラス注射で投与される。
【0056】
エカランチド配合物は、被験体以外の人(例えば、医療専門家)によって被験体に投与される場合もあれば、被験体が自己投与する場合もある。注射器、輸液ポンプ、静脈内または皮下カテーテル、および自動注入装置を含め、選択された投与方式に適合するいずれのデバイスも使用される場合もある。
【0057】
キット
さらには、本明細書に開示される配合物を含むキットも提供される。本明細書に開示されるキットは、本開示内容の配合物を含む1つ以上のパッケージを含み、また配合物の使用に関する指示書(例えば、血管性浮腫の治療に関するもの)もさらに含む場合がある。キットに付属する指示書は、一般的には書面のものであるが、電子的なものである場合もあり(またワールドワイドウェブ上の1つ以上のサイトへのリンクを含む場合もある)、一般的には血管性浮腫の治療に関する投与量、服薬スケジュール、および投与経路についての情報を含む。エカランチド配合物のパッケージは、単位用量、大量包装(例えば、複数用量パッケージ)または副次的単位用量である場合がある。
【0058】
エカランチド配合物のパッケージは、所期の用途に適したいずれのパッケージングである場合もある。液体配合物の場合、適切なパッケージには、弾性栓付きアンプル、非弾性蓋付きアンプル(例えば、密封ガラスアンプル)、プレフィルドシリンジ、および自動注入装置(Bioject IJECT(登録商標)ニードルレス注射器またはDIAPEN(登録商標)注射器など)、ならびにオートインジェクターのカートリッジが含まれるが、これらに限定されない。凍結乾燥配合物の場合、適切なパッケージには、弾性栓付きアンプル、自己投与用デバイス(例えば、再構成および注入を自動的に行うBD(登録商標)Liquid Dry Injector)、およびプレフィルドシリンジが含まれるが、これらに限定されない。
【0059】
以下の実施例は、本開示内容を例示するためのものであるが、これを限定するものではない。
【実施例】
【0060】
(実施例1:エカランチドの産生)
エカランチドは、酵母(P.パストリス)での組換え発現によって産生された。S.セレビシエプレプロ‐matαのシグナル配列とエカランチドとの融合をコードする配列を、(アンピシリン耐性遺伝子およびHIS4を保有する)pHIL‐D2由来プラスミドのAOXl領域にクローン化して、pPIC‐K503を生成した。
【0061】
His4−表現型を有するP.パストリス菌株GS115のスフェロプラストを、(SaCI部位で)線状化されたpPIC-K503で形質転換し、宿主の5’AOX1座へ
プラスミドDNAの相同組換えを行った。プラスミドは宿主細胞のAOX1座へ入り、宿主細胞をHis4+表現型に変換し、AOX1座によって制御されるエカランチド発現カセットを生成した。
【0062】
組換え菌株は、外因性ヒスチジンの非存在下でメタノールを唯一の炭素源として増殖させることにより選択した。選択したコロニーをクローン化し、発現の調査を実施して多量のエカランチドを培地に分泌するクローンを同定した。高発現クローンを使用して、作業用細胞バンクを生成した。
【0063】
無菌接種物ブロス(酵母窒素塩基、リン酸カリウム、およびグリセロール、pH=5)を含むフラスコに作業用細胞バンクの細胞を接種することにより、接種物の培養を確立した。接種培養物を30℃でおよそ20時間にわたって保温した。
【0064】
この接種培養物を使用して、種発酵培養物の接種を行った。種発酵培養物は、合成培地(正リン酸、硫酸カルシウム、硫酸カリウム、硫酸マグネシウム、水酸化カリウム、グリセロール、d‐ビオチン、金属塩(硫酸、硫酸銅、ヨウ化ナトリウム、硫酸マンガン、モリブデン酸ナトリウム、ホウ酸、塩化コバルト、塩化亜鉛、および硫酸鉄)、消泡剤溶液、および水酸化アンモニウム)中で培養し、これを30℃にて発酵槽中で、OD600が28〜56になるまで行った。
【0065】
次いで種発酵培養物を使用して、産生用発酵培養物の接種を行った。種発酵培養物を、発酵槽中のあらかじめ加温した産生用発酵培地(正リン酸、グリセロール、硫酸カルシウム、硫酸カリウム、硫酸マグネシウム、水酸化カリウム、金属塩(硫酸、硫酸銅、ヨウ化ナトリウム、硫酸マンガン、モリブデン酸ナトリウム、ホウ酸、塩化コバルト、塩化亜鉛、および硫酸鉄)、消泡剤溶液、および水酸化アンモニウム)、d‐ビオチン、消泡剤溶液、および水酸化アンモニウム)に添加し、培地中の最初のグリセロールが使い果たされるまでグリセロールのバッチ期で増殖させた。次いで、グリセロールを培地に添加するグリセロールのバッチ供給期に培養を移行して、産生菌株をさらに増殖させた。最後に、グリセロールとメタノールの供給に切り替えて、培養をおよそ83時間の混合供給期に移行させた。
【0066】
発酵段階はすべて撹拌および通気(必要に応じて酸素を添加)しながら行った。
【0067】
発酵槽の内容物を冷却し、精製水で希釈した。初期の精製手順では、拡張床クロマトグラフィー(EBC)を利用して、希釈発酵槽ブロスからエカランチドを捕捉し、発酵から酵母を除去した。希釈した発酵槽の培養物を、下降流方式で拡張床カラム(STREAMLINE(商標)SP樹脂)に注加し、上昇流方式で洗浄し、沈降させてから、下降流方式で洗浄および溶出を行った。
【0068】
さらなる精製を、結合/洗浄/溶出の構成で操作される一連のカラムクロマトグラフィーの手順で実施した。EBC溶出液を陽イオン交換(CEX)樹脂(Bio‐Rad MACRO‐PREP(登録商標))High S)上に注加し、それを洗浄し溶出させた。CEX溶出液を硫酸アンモニウムが1.1Mとなるように調整し、次いで疎水性相互作用クロマトグラフィー(HIC)樹脂上に注加し、それを洗浄し溶出させた。HIC溶出液は、1kDa MWCO再生セルロース膜(UFDF)を使用した限外濾過/ダイアフィルトレーションによって緩衝剤を交換し、次いで陰イオン交換(AEX)クロマトグラフィー樹脂(BioSepra Q HYPERD(登録商標))上に注加し、それを洗浄してから溶出させた。AEX溶出液は、UFDFによって緩衝剤を交換してPBS(pH7.0)にし、0.22μm膜で無菌的に濾過し、無菌PETGビンに無菌分注し、−20℃で貯蔵した。
【0069】
(実施例2:pHおよび緩衝剤の選択)
エカランチドの安定性を種々の緩衝剤中でpH6.0、6.5および7.0にて調べた。エカランチド(等張性リン酸塩緩衝食塩水中に10mg/mL、pH7.0)を透析により緩衝剤交換して、(a)10mMのコハク酸塩、pH6.0、150mMのNaCl、(b)10mMのヒスチジン、pH6.0、150mMのNaCl、(c)10mMのヒスチジン、pH6.5、150mMのNaCl、(d)リン酸塩緩衝食塩水(PBS、4.3mMのリン酸ナトリウム、1.5mMのリン酸カリウム、137mMのNaCl)、pH6.5、または(e)10mMのヒスチジン、pH7.0、150mMのNaClにした。
【0070】
それぞれの配合物の試料を滅菌濾過して個々の管に入れ、4℃または30℃で6週間貯蔵した。試料を1、2、3.5、5および6週間の時点でHPLCサイズ排除クロマトグラフィー(SEC)により分析して、凝集体の形成および断片化を検出し、さらに逆相(RP)HPLCによってピログルタメートの形成を検出した。
【0071】
SEC‐HPLCの結果では、pHが上昇すると、高分子量の種類のもの(凝集体)が増加することが示された。逆に言えば、pHが上昇すると、低分子量の種類のもの(断片)が減少した。
【0072】
RP‐HPLC分析では、pH7.0の試料で非常に多くのピログルタメートが生成されることが示された。pH6.0および6.5の試料のうち、pH6.5のPBS試料では、ピログルタメートのレベルがやや高かった。
【0073】
(実施例3:低用量のエカランチドの場合の増量剤/凍結保護物質の選択)
増量剤/凍結保護物質の種々の構成を利用して、凍結乾燥配合物中のエカランチドの安定性を調べた。エカランチド(PBS中に10mg/mL、pH7.0)を、透析によって緩衝剤交換して、10mMのヒスチジンで緩衝化されるか(pH6.5)またはPBSで緩衝化され(pH6.5)、さらに(a)5%のマンニトール、(b)3%のマンニトール/3%のスクロース、(c)10%のスクロース、または(d)7.5%のスクロース/5%のデキストラン40を増量剤/凍結保護物質として含む配合物にした。
【0074】
それぞれの配合物の試料を滅菌濾過して、ガラスバイアルに入れ、凍結させ、凍結乾燥させた後、4℃または40℃にて8週間貯蔵した。試料を水で再構成してから、2週間、4週間、6週間および8週間の時点でSEC‐HPLCおよびRP‐HPLCにより分析した。
【0075】
40℃に維持した試料のピログルタメート(RP-HPLC)のデータを図2に示す(図2AはPBS緩衝剤であり、図2Bはヒスチジン緩衝剤である)。マンニトールは一般的に安定させる増量剤/凍結保護物質であると見なされているが、マンニトールは、図2に示すように、エカランチド配合物中では不安定にさせている。唯一の増量剤/凍結保護物質としてマンニトールを含有する配合物は、他の配合物よりもピログルタメートのレベルがかなり高かった。また、スクロースとマンニトールの混合物を含有する配合物の安定性は、マンニトールのみを有する配合物よりも優れていたが、これらの配合物もやはりスクロースの配合物およびスクロース/デキストランの配合物よりもピログルタメートのレベルが高かった。凝集生成物および断片化生成物に関するSEC‐HPLCのデータは類似していた。
【0076】
(実施例4:用量を増加させたエカランチドの場合の増量剤/凍結保護物質の選択)
増量剤/凍結保護物質の種々の構成を利用して、凍結乾燥配合物中のエカランチドの安定性を調べた。エカランチド(PBS中に20mg/mL、pH7.0)を、透析によって緩衝剤交換して、10mMのヒスチジンで緩衝化され(pH6.5)、さらに(a)10%のスクロース、(b)3%のマンニトール/3%のスクロース、または(c)3%のマンニトール/3%のトレハロースを増量剤/凍結保護物質として含む配合物にした。
【0077】
それぞれの配合物の試料を滅菌濾過してガラスバイアルに入れ、凍結させ、凍結乾燥器で−40℃まで凍結させることによって凍結乾燥させた後、75mTorrにて−40℃で30分間、−25℃で23時間、5℃で10時間一次乾燥させ、次いで75mTorrにて30℃で9時間二次乾燥させた。凍結乾燥試料を4℃または40℃で8週間貯蔵した。試料を2週間(40°の試料のみ)、4週間、6週間および8週間の時点でSEC‐HPLCおよびRP‐HPLCにより分析した。
【0078】
RP‐HPLCおよびSEC‐HPLCの分析では、10%のスクロースを増量剤/凍結保護物質として含む試料は、スクロース/マンニトール配合物およびマンニトール/トレハロース配合物よりも劣化がかなり少ないことが示された。図3に示すように、マンニトール含有配合物の場合、かなりの量のピログルタメート(図3A)および「ピーク4」の不純物があった(図3B:「ピーク4」は、この方法では分解できない、酸化およびグリコシル化されたエカランチドの混合物であると考えられる)。
【0079】
上述したまたは添付の特許請求の範囲で定義された本発明から逸脱することなく、記載した実施態様および特徴の置換、改変および変更が追加で行われる場合があることが、当業者により理解されるであろう。
【0080】
本明細書に引用した刊行物は全体が参考として本明細書で援用される。
【背景技術】
【0001】
(背景)
エカランチド(ecallantide)は、クニッツドメインの一般構造を有する60アミノ酸のペプチドである。エカランチドは、血漿カリクレインの強力な阻害剤であることが明らかにされている。
【発明の概要】
【課題を解決するための手段】
【0002】
(発明の要旨)
本明細書には、室温にて安定しておりかつ薬学的配合物として有用な新規のエカランチド配合物が開示されている。
【0003】
本開示内容は、緩衝剤、緩衝剤/凍結保護物質、およびエカランチドを含めたエカランチド含有組成物(「エカランチド配合物」)を提供する。緩衝剤はpHを約6.0〜7.0に調節するヒスチジン緩衝剤またはリン酸緩衝剤であってもよく、増量剤はスクロースまたはスクロースとマンニトールの組み合わせであってもよい。場合により、増量剤/凍結保護物質はデキストラン40などのデキストランも含む。
【0004】
さらには、本明細書に開示される方法で製造される組成物も提供される。
【0005】
いくつかの実施形態において、緩衝剤はヒスチジンであり、これは10mMだけ存在する場合がある。いくつかの実施形態において、配合物のpHは約6.5である。
【0006】
いくつかの実施形態において、増量剤/凍結保護物質はスクロースであり、これは10%(w/v)で存在し得る。
【0007】
いくつかの実施形態において、エカランチドは、10mg/mL、20mg/mL、または30mg/mLで存在する。
【0008】
いくつかの実施形態において、本配合物は等張性を有する。
【0009】
本明細書に開示されるエカランチド配合物は凍結乾燥され得る。したがって、本開示内容では、緩衝剤、緩衝剤/凍結保護物質、およびエカランチドを含む、凍結乾燥エカランチド配合物を提供する。緩衝剤はpHを約6.0〜7.0に調節するヒスチジン緩衝剤またはリン酸緩衝剤である場合があり、増量剤はスクロースまたはスクロースとマンニトールの組み合わせであり得る。場合により、増量剤/凍結保護物質はデキストラン40などのデキストランも含む。
【0010】
凍結乾燥エカランチド配合物の成分は、約1:1〜約7.5:1もしくは約2:1〜約2.5:1(緩衝剤:エカランチド)、または約250:1〜約45:1もしくは約75:1〜約60:1(増量剤/凍結保護物質:エカランチド)、または約2.5:75:1〜約2:65:1、もしくは約7:208:1、約2.4:70:1、もしくは約1.4:41:1(緩衝剤:増量剤/凍結保護物質:エカランチド)などの種々のモル比で存在し得る。
【0011】
凍結乾燥エカランチド配合物の成分は、約1%〜約2%(w/w)の緩衝剤、約90%〜約60%の増量剤/凍結保護物質、および約9%〜約37%のエカランチドなど、種々の百分率(w/w)で存在し得る。
【0012】
本明細書ではまた、緩衝剤、増量剤/凍結保護物質、およびエカランチドの混合物を取得または産生し、その混合物を凍結乾燥させることによって、本明細書に開示される凍結乾燥エカランチド配合物を産生する方法も提供される。
【0013】
また、有効量の本開示内容のエカランチド配合物を血管性浮腫の被験体または血管性浮腫の疑いのある被験体に投与することにより、血管性浮腫(遺伝性血管性浮腫、アンギオテンシン変換酵素(ACE)阻害剤誘発性の血管性浮腫、後天性(例えば、C1エステラーゼ阻害剤の欠損)血管性浮腫、特発性慢性血管性浮腫、アレルギー性血管性浮腫、および非ステロイド系抗炎症薬(NSAID)誘発性の血管性浮腫)を治療する方法も提供される。
【0014】
また、本開示内容のエカランチド配合物を含むキットも提供される。こうしたキットは、本開示内容のエカランチド配合物を含む少なくとも1つの容器を含み、また血管性浮腫を治療するためのエカランチドの使用に関する指示書を含む場合もある。容器は、アンプル、バイアル、プレフィルドシリンジ、または自動注入装置(もしくは自動注入装置のカートリッジ)であり得る。
例えば、本願発明は以下の項目を提供する。
(項目1)
ヒスチジンおよびリン酸塩からなる群から選択される緩衝剤;
スクロースおよびスクロースとマンニトールとの組み合わせからなる群から選択される増量剤/凍結保護物質;ならびに
エカランチド
を含むエカランチド配合物であって、
該配合物が約6.0〜7.0のpHを有する、
配合物。
(項目2)
前記緩衝剤がヒスチジンである、項目1に記載の配合物。
(項目3)
前記増量剤/凍結保護物質がスクロースである、項目2に記載の配合物。
(項目4)
前記pHが約pH6.5である、項目3に記載の配合物。
(項目5)
前記緩衝剤および前記エカランチドが2:1〜2.5:1(緩衝剤:エカランチド)のモル比で存在する、項目4に記載の配合物。
(項目6)
前記増量剤/凍結保護物質および前記エカランチドが75:1〜60:1(増量剤/凍結保護物質:エカランチド)のモル比で存在する、項目5に記載の配合物。
(項目7)
前記緩衝剤、前記増量剤/凍結保護物質、および前記エカランチドが2.5:75:1〜2:65:1(緩衝剤:増量剤/凍結保護物質:エカランチド)のモル比で存在する、項目6に記載の配合物。
(項目8)
前記増量剤/凍結保護物質がスクロースである、項目1に記載の配合物。
(項目9)
前記pHが約pH6.5である、項目1に記載の配合物。
(項目10)
前記配合物が凍結乾燥されている、項目1に記載の配合物。
(項目11)
前記緩衝剤および前記エカランチドが1:1〜7.5:1(緩衝剤:エカランチド)のモル比で存在する、項目1に記載の配合物。
(項目12)
前記緩衝剤および前記エカランチドが2:1〜2.5:1(緩衝剤:エカランチド)のモル比で存在する、項目11に記載の配合物。
(項目13)
前記増量剤/凍結保護物質および前記エカランチドが300:1〜45:1(増量剤/凍結保護物質:エカランチド)のモル比で存在する、項目1に記載の配合物。
(項目14)
前記増量剤/凍結保護物質および前記エカランチドが75:1〜60:1(増量剤/凍結保護物質:エカランチド)のモル比で存在する、項目13に記載の配合物。
(項目15)
前記緩衝剤、前記増量剤/凍結保護物質、および前記エカランチドが2.5:75:1〜2:65:1(緩衝剤:増量剤/凍結保護物質:エカランチド)のモル比で存在する、項目1に記載の配合物。
(項目16)
ヒスチジンおよびリン酸塩からなる群から選択される緩衝剤;
スクロースおよびスクロースとマンニトールとの組み合わせからなる群から選択される増量剤/凍結保護物質;ならびに
エカランチド
から本質的になる、項目1に記載の配合物。
(項目17)
ヒスチジンおよびリン酸塩からなる群から選択される緩衝剤;
スクロースおよびスクロースとマンニトールとの組み合わせからなる群から選択される増量剤/凍結保護物質;ならびに
エカランチド
を含む、凍結乾燥エカランチド配合物。
(項目18)
前記緩衝剤がヒスチジンである、項目17に記載の配合物。
(項目19)
前記増量剤/凍結保護物質がスクロースである、項目18に記載の配合物。
(項目20)
再構成された配合物のpHが約pH6.5である、項目19に記載の配合物。
(項目21)
前記緩衝剤および前記エカランチドが2:1〜2.5:1(緩衝剤:エカランチド)のモル比で存在する、項目20に記載の配合物。
(項目22)
前記増量剤/凍結保護物質および前記エカランチドが75:1〜60:1(増量剤/凍結保護物質:エカランチド)のモル比で存在する、項目21に記載の配合物。
(項目23)
前記緩衝剤、前記増量剤/凍結保護物質、および前記エカランチドが2.5:75:1〜2:65:1(緩衝剤:増量剤/凍結保護物質:エカランチド)のモル比で存在する、項目22に記載の配合物。
(項目24)
前記増量剤/凍結保護物質がスクロースである、項目17に記載の配合物。
(項目25)
前記pHが約pH6.5である、項目17に記載の配合物。
(項目26)
前記配合物が凍結乾燥されている、項目17に記載の配合物。
(項目27)
前記緩衝剤および前記エカランチドが1:1〜7.5:1(緩衝剤:エカランチド)のモル比で存在する、項目17に記載の配合物。
(項目28)
前記緩衝剤および前記エカランチドが2:1〜2.5:1(緩衝剤:エカランチド)のモル比で存在する、項目27に記載の配合物。
(項目29)
前記増量剤/凍結保護物質および前記エカランチドが300:1〜45:1(増量剤/凍結保護物質:エカランチド)のモル比で存在する、項目17に記載の配合物。
(項目30)
前記増量剤/凍結保護物質および前記エカランチドが75:1〜60:1(増量剤/凍結保護物質:エカランチド)のモル比で存在する、項目29に記載の配合物。
(項目31)
前記緩衝剤、前記増量剤/凍結保護物質、および前記エカランチドが2.5:75:1〜2:65:1(緩衝剤:増量剤/凍結保護物質:エカランチド)のモル比で存在する、求項17に記載の配合物。
(項目32)
(a)ヒスチジンおよびリン酸塩からなる群から選択される緩衝剤と、スクロースおよびスクロースとマンニトールとの組み合わせからなる群から選択される増量剤/凍結保護物質と、エカランチドとの混合物を得ること;ならびに
(b)該混合物を凍結乾燥させること
というプロセスにより産生される、凍結乾燥エカランチド配合物。
(項目33)
(a)ヒスチジンおよびリン酸塩からなる群から選択される緩衝剤と、スクロースおよびスクロースとマンニトールとの組み合わせからなる群から選択される増量剤/凍結保護物質と、エカランチドとの混合物を得ること;ならびに
(b)該混合物を凍結乾燥させること
というプロセスにより産生される凍結乾燥エカランチド配合物を製造するための方法。
【図面の簡単な説明】
【0015】
【図1】プラスミドpPIC K503の図を示す。
【図2】PBSで緩衝処理された配合物中のピログルタメートのレベル(図2A)または10mMのヒスチジンで緩衝処理された配合物中のピログルタメートのレベル(図2B)を測定したRP‐HPLCのデータのグラフを示す。
【図3】ピログルタメートのレベル(図3A)およびピーク4のレベル(図3B)を測定したRP‐HPLCのデータのグラフを示す。
【発明を実施するための形態】
【0016】
(発明の詳細な説明)
本明細書には、室温にて安定しておりかつ薬学的配合物として有用な新規のエカランチド配合物が開示される。
【0017】
本明細書で使用される、百分率、モル濃度またはモル比に関連して使用される「約」という言葉は、示される値の±10%の範囲を指す(例えば、「約10mM」は9mM〜11mMを意味する)。pHの値に関連して「約」が使用される場合、これは示される値の±0.2pH単位の範囲を指す(例えば、「約pH7.0」はpH6.8〜7.2を意味する)。
【0018】
エカランチド
クニッツドメインをベースにした多くのタンパク質が当該技術分野で既知である(例えば、米国特許第4,245,051号、第5,278,285号、第5,436,153号、第5,728,674号、第5,563,123号、第5,589,359号、第5,696,088号,第5,663,143号、第5,880,256号、第5,968,897号、第5,977,057号、第6,103,500号、第5,990,079号、第6,063,764号、第6,414,124号、第6,583,108号、第6,593,291号;および第6,914,135号など)。
【0019】
エカランチドは、クニッツドメインの一般構造を有する60アミノ酸のペプチドである。エカランチドは、Glu Ala Met His Ser Phe Cys Ala
Phe Lys Ala Asp Asp Gly Pro Cys Arg Ala
Ala His Pro Arg Trp Phe Phe Asn Ile Phe
Thr Arg Gln Cys Glu Glu Phe Ile Tyr Gly
Gly Cys Glu Gly Asn Gln Asn Arg Phe Glu
Ser Leu Glu Glu Cys Lys Lys Met Cys Thr
Arg Asp(配列番号1)の配列を有する。エカランチドの分子量は7,054ダルトンである。エカランチドは、血漿カリクレインの非常に効果的な阻害剤であり、遺伝性血管性浮腫および虚血防止を含めたいくつかの適応症の治療剤として提案されてきた(Williams, et al., 2003, Transfus. Apher. Sci. 29(3):255−58;米国特許出願公開第2004/0038893号)。
【0020】
エカランチドは、いずれかの標準的なポリペプチド合成手順および機器を使用して合成され得る。例えば、エカランチドの段階的合成は、最初の(すなわち、カルボキシ末端)アミノ酸からアミノ(N)末端保護基を除去し、ポリペプチドの配列中の次のアミノ酸のカルボキシル末端をそれに結合させることにより、行われ得る。このアミノ酸はまた好適に保護される。入ってくるアミノ酸のカルボキシル基は、カルボジイミド、対称酸無水物、または「活性エステル」基(ヒドロキシベンゾトリアゾールまたはペンタフルオロフェニルエステルなど)を形成するなど、反応性基を形成することにより、結合するアミノ酸のN末端と反応するように活性化させることができる。有用な固相ペプチド合成方法には、a‐アミノ保護基としてtert−ブチルオキシカルボニルを利用するBoc法、および9‐フルオレニルメトキシカルボニルを利用してアミノ酸残基のa‐アミノを保護するFmoc法が含まれる。いずれの方法も当業者に周知である(教示内容全体が参考として本明細書で援用される、Stewart, J. and Young, J., Solid‐Phase Peptide Synthesis(W. H. Freeman Co.,San Francisco)1989);Merrifield, J., 1963. Am. Chem. Soc., 85:2149−2154; Bodanszky, M. and Bodanszky, A., The Practice of Peptide Synthesis(Springer‐Verlag, New York 1984))。
【0021】
あるいは、エカランチドは、いくつかの細胞および対応する発現ベクター(細菌発現ベクター、酵母発現ベクター、バキュロウイルス発現ベクター、哺乳類ウイルス発現ベクターを含むがこれらに限定されない)のいずれかを使用した組換え方法により産生され得る。エカランチドは、エカランチドをコードする配列を含む核酸分子を使用して遺伝子導入により産生される場合もあり、その場合、核酸分子は、当該技術分野で使用可能な遺伝子導入方法を使用して宿主動物のゲノムに組み込み、そのゲノムから発現させることができる。場合によっては、エカランチドのコード配列を発現ベクター中の別のコード配列と融合させ、宿主細胞内で容易に発現される融合ポリペプチドを形成させることが必要であるか、または有利であり得る。このような融合ポリペプチドを発現する宿主細胞はまた、融合ポリペプチドのプロセシングを行って、所望のアミノ酸配列(すなわち、エカランチド)のみを得るのが好ましい。その他いずれかの1つ以上のアミノ酸が、発現したエカランチドに結合したままである場合、このような1つ以上の付加的なアミノ酸は、本明細書に開示される配合物中でポリペプチドを使用できなくなるほどエカランチドの活性を減少させるべきではない。
【0022】
実施例に開示されるエカランチドの特定の産生方法では、酵母宿主細胞における組換え発現が利用される。酵母発現ベクターでは、同じ読み枠において、エカランチドのアミノ酸配列をコードする核酸配列が、サッカロマイセス・セレビシエのmatαプレプロリーダーペプチド配列をコードするヌクレオチド配列と結合することができるため、このベクターが操作可能な酵母プロモーターの制御下にある。次いで、得られた組換え酵母発現プラスミドは、標準的な方法によって適合性のある適切な酵母宿主の細胞に形質転換され、これらの細胞は組換え酵母発現ベクターから組換えタンパク質を発現させることができる。このような組換え発現ベクターによって形質転換される酵母宿主細胞は、好ましくは、融合タンパク質をプロセッシングして、本明細書に開示される方法および組成物において有用な活性エカランチドを提供することもできる。このような方法で組換えエカランチドを産生させるのに有用な酵母宿主細胞は、ピキア・パストリスである。
【0023】
薬学的配合物で使用するためのエカランチドは実質的に均質であるべきである。したがって、エカランチドは通常、(合成または組換え発現によって)産生された後に精製される。エカランチドの精製は、サイズ排除クロマトグラフィー、イオン交換(陰イオンおよび/または陽イオン交換)クロマトグラフィー、疎水性相互作用クロマトグラフィー、アフィニティークロマトグラフィー、および逆相クロマトグラフィー、またはそれらのいずれかの組み合わせを含めた、当該技術分野で既知の技法を使用して行われ得る。さらには、緩衝剤交換および/または濃縮技術も所望により使用され得る。
【0024】
本明細書の実施例に記載の通り、エカランチドは特定の条件下で不安定であり、貯蔵されると、高分子量(例えば、凝集生成物)劣化生成物および低分子量(例えば、断片化生成物)劣化生成物の両方、ならびに修飾生成物(例えば、アミノ末端ピログルタミン酸塩)を生じる。本明細書に開示される配合物では、エカランチドが実質的に安定され、凝集生成物、断片化生成物、または修飾生成物の生成が阻止されるかまたは減少する。
【0025】
エカランチドは、所期の用途(例えば、所期の用量)に応じて、種々のレベルで本配合物中に存在し得る。液体配合物の場合、エカランチドは、約5mg/mL(0.7mM)〜約50mg/mL(7mM)、または約7mg/mL(1mM)〜約40mg/mL(5.7mM)、または約10mg/mL(1.4mM)〜約30mg/mL(4.2mM)、または約30mg/mLの濃度で存在し得る。百分率(w/v)で表わした場合、エカランチドは、約0.5%〜約5%、または約0.7%〜約4%、または約1%〜約3%の濃度で存在し得る。凍結乾燥した配合物の場合、エカランチドは、約5%〜約45%(w/w)、または約7%〜約40%(w/w)または約9%〜約37%(w/w)だけ存在し得る。
【0026】
pHおよび緩衝剤
本明細書に開示される配合物は、緩衝剤でpHが制御される。実施例で記載の通り、エカランチドは約6.0〜約7.0のpH範囲で安定している。したがって、本明細書では、液体形態である場合(例えば、産生時または再構成時)に、pHが約6.0〜約7.0、例えば約6.0(例えば、pH5.8〜6.2)、約6.5(例えば、pH6.3〜6.7)、または約7.0(例えば、pH6.8〜7.2)である配合物が提供される。
【0027】
pH約6.0〜約7.0の範囲に調節するのに好適ないずれの緩衝剤も使用され得る。いくつかの実施形態において、緩衝剤はまた薬学的に許容される。好適な緩衝剤には、クエン酸塩、コハク酸塩、リンゴ酸塩、カコジル酸塩、(N−モルホリノ)エタンスルホン酸水和物(MES)、クエン酸塩、マレアート、ヒスチジン、リン酸塩、および炭酸塩がある。特定の実施形態において、緩衝剤はヒスチジンまたはリン酸塩である。特定の実施形態において、緩衝剤はヒスチジンである。
【0028】
緩衝剤は、予想される貯蔵条件および(凍結乾燥配合物の場合は)再構成条件において十分なpH制御を行える濃度で含まれる。液体形態の配合物の場合、緩衝剤は一般的に、約3mM〜約20mM、または約5mM〜約15mM、または約8mM〜約12mM、または約10mMで含まれる。百分率(w/v)として計算した場合、緩衝剤は、約0.045%〜約0.31%、または約0.08%〜約0.23%、または約0.12%〜約0.19%、または約0.15%の濃度で存在し得る。凍結乾燥配合物の場合、緩衝剤は一般的に、約0.25%〜約5%(w/w)、または約0.5%〜約2.5%(w/w)、または約1%〜約2%(w/w)で含まれる。
【0029】
増量剤/凍結保護物質
本明細書に開示される配合物は、増量剤/凍結保護物質を含む。本発明者等は、スクロースが、単独でまたはマンニトールと組み合わせて、エカランチド配合物の増量剤/凍結保護物質として有用であることを発見した。さらに、配合物はデキストラン(実施形態によってはデキストラン40)を含みうる。
【0030】
予想外にも、本発明者等は、安定させていると予想されていた通常使用される増量剤/凍結保護物質のトレハロースが、エカランチド配合物に含まれる場合には不安定にさせていることも見出した。したがって、トレハロースがエカランチド配合物を不安定にすることを本発明者等は見出したため、本明細書に開示される配合物は、実質的にまたは完全にトレハロースを含まない場合ができる。本明細書で使用される「実質的にトレハロースを含まない」とは、配合物において(液体形態の場合)トレハロースが1mM未満であるか、または(凍結乾燥形態の場合)トレハロースが1重量%未満であることを意味する。
【0031】
増量剤/凍結保護物質は、乾燥させたときに許容される凍結乾燥したケーキを産生して、少なくともエカランチドの凍結保護の指標を提供するのに十分な容積となる量だけ、本配合物に含まれる。液体配合物の場合、配合物の百分率で測定すると、増量剤/凍結保護物質は、約3%〜約15%(w/v)、または約4%〜約15%、または約5%〜約10%だけ存在する。液体配合物の場合、増量剤/凍結保護物質のモル濃度で測定すると、増量剤/凍結保護物質は、約200mM〜約350mM、または約250mM〜約300mMで存在する。増量剤/凍結保護物質がスクロースである実施形態において、増量剤/凍結保護物質は約292mMで存在し得る。凍結乾燥配合物の場合、増量剤は約95%〜約55%(w/w)、または約90%〜約60%(w/w)で存在する。
【0032】
配合物
本明細書に開示される配合物は、エカランチド、pH緩衝剤、および増量剤/凍結保護物質を含む。本配合物は薬学的配合物として使用することを意図していることから、特定の実施形態において、配合物は等張性を有する(例えば、250〜350mOsM、または約300mOsMの重量オスモル濃度を有する)。当業者であれば分かるように、成分の比率は、成分(特にエカランチド)の濃度により変動する(エカランチドは所期の投与量により変動し得る)。医薬品の用途では、本明細書に開示される配合物の成分は、米国薬局方(USP)または同様の等級のものであるか、あるいは医薬品の製造及び品質管理に関する基準(GMP)に従って産生されなければならない。
【0033】
液体形態の場合、配合物の成分の量は、モル濃度または百分率(w/v)濃度で容易に記載することができる。モル濃度で表わす場合、本配合物は、緩衝剤が約3mM〜約20mM、または約5mM〜約15mM、または約8mM〜約12mM、または約10mMである場合があり、増量剤/凍結保護物質が約200mM〜約350mM、または約250mM〜約300mM、または約292mMである場合があり、エカランチドが約1mM〜約5mM、または約1.4、2.8もしくは4.2mMである場合がある。百分率(w/v)濃度で表わす場合、本配合物は、緩衝剤が0.045%〜約0.31%、または約0.08%〜約0.23%、または約0.12%〜約0.19%、または約0.15%である場合があり、増量剤/凍結保護物質が3%〜約15%、または約4%〜約15%、または約5%〜約10%である場合があり、エカランチドが約0.5%〜約5%、または約0.7%〜約4%、または約1%〜約3%である場合がある。
【0034】
乾燥(例えば、凍結乾燥)形態の場合、成分の量は、百分率(w/w)またはモル比で極めて容易に記載される。百分率で表わす場合、本配合物は、緩衝剤が約0.25%〜約5%(w/w)、または約0.5%〜約2.5%(w/w)、または約1%〜約2%(w/w)である場合があり、増量剤/凍結保護物質が約95%〜約55%(w/w)、または約90%〜約60%(w/w)である場合があり、エカランチドが5%〜約45%(w/w)、または約7%〜約40%(w/w)、または約9%〜約37%(w/w)である場合がある。当業者であれば分かるように、緩衝剤、増量剤/凍結保護物質、およびエカランチドの百分率量の合計は、100%に満たない場合があるが、実際に一般的には100%に満たず、残りは残留溶媒である。モル比(緩衝剤:増量剤/凍結保護物質:エカランチド)で表わす場合、本配合物は、約7.5:208:1〜約1:45:1、または約2:100:1〜約2.5:75:1、または約7:208:1、または約2.4:70:1、または約1.4:41:1であり得る。
【0035】
1つの例示的な配合物は、(液体形態の場合)緩衝剤として約10mMのヒスチジンを、増量剤/凍結保護物質として約10%(w/v)のスクロースを、また約10mg/mLのエカランチドを含み、pHが6.5である。乾燥(凍結乾燥)形態の場合、本配合物は、緩衝剤が約1.4%(w/w)、増量剤/凍結保護物質が88.8%(w/w)、エカランチドが約8.9%(w/w)であり、モル比が約7:208:1(ヒスチジン:スクロース:エカランチド)である。
【0036】
別の例示的な配合物は、(液体形態の場合)緩衝剤として約10mMのヒスチジンを、増量剤/凍結保護物質として約10%(w/v)のスクロース、また約30mg/mLのエカランチドを含み、pHが6.5である。乾燥(凍結乾燥)形態の場合、本配合物は緩衝剤が約1.2%、増量剤/凍結保護物質が約75.4%、エカランチドが約22.6%であり、モル比が約2.4:70:1である。
【0037】
本明細書に開示される配合物は、所望の最終組成物を生じる従来の技法によって製造され得る。成分は、水に直接溶解して最終濃度にする場合もあれば、濃縮物として製造し、これらを混合・希釈されて最終生成物を得る場合もある。あるいは、緩衝剤交換方法が使用される場合もある。
【0038】
一般的に、エカランチドは、エカランチド産生の最終処理手順の結果、水溶液中に含まれることになる。次いで、本エカランチド溶液は、(例えば、ダイアフィルトレーションによって)緩衝剤の交換を行い、所望の配合物を得る場合もあれば、あるいは、緩衝剤の交換を実行できない場合(例えば、増量剤/凍結保護物質によって、緩衝剤を交換するにはあまりに粘性がある配合物が得られる場合)、エカランチドは緩衝剤の交換を行い(必要に応じて濃縮し)濃縮溶液を得て、それを残りの成分と混合して所望の配合物を産生する場合もある(例えば、ヒスチジンが10mM、pHが6.5、スクロースが10%、エカランチドが30mg/mLである所望の配合物の場合、エカランチド溶液は緩衝剤の交換を行い、必要に応じて濃縮して原液を製造する。この原液は、濃縮スクロース溶液と混合するか、あるいは乾燥スクロースと混合したとしても、10mMのヒスチジン、pH6.5、10%のスクロース、および30mg/mLのエカランチドの最終配合物が得られる)。
【0039】
凍結乾燥
凍結乾燥(lyophilization)(すなわち、凍結乾燥(freeze‐drying))は、液体組成物を凍らせてから、凍結液体(例えば、水)の昇華によって脱水するプロセスである。昇華は一次乾燥に適した温度で行われる。一次乾燥に適した温度とは、配合物の共融点または崩壊温度以下の温度に生成物を保持する温度である。
【0040】
凍結乾燥させる物質(例えば、エカランチド配合物)は、凍結乾燥装置に充填する前に凍結させる場合もあれば、液体形態で装置に充填し、装置内にある間に凍結させる場合もある。液体配合物の凍結は、単一の傾斜(例えば、所望の温度まで温度を連続して低下させる)として、または一連の手順/傾斜において、所望の温度まで下げる単一の手順を含めたいずれかの方法で行われ得る。凍結液体配合物の「所望の温度」は、物質が凍結するが一般的に物質の凝固点よりも低い、いずれかの温度である場合があり、約0℃〜約−50℃の範囲であり得る。所望の温度に達したら(または所望の温度に達した後の平衡期間に続いて)、部分真空が確立されるが、これは、約50〜約250mTorr、または約60〜約200mTorr、または約75〜約100mTorrの範囲であり得る。
【0041】
凍結乾燥装置内の温度は、凍結乾燥プロセスの間一定に維持される場合があり、より一般的には本プロセスの間に調整される(一般的には上昇させる)。例えば、凍結乾燥器は、真空にする前に平衡状態にして約−40℃または約−45℃にし、その後、凍結乾燥プロセスの一次乾燥段階が進行するにつれて、一連の手順または傾斜にて徐々に加温され得る。例えば、約−40℃で開始する凍結乾燥プロセスの場合、凍結乾燥器は、一次乾燥段階の最初の部分の間に一連の凝固点下の温度を経て(例えば、連続して約5℃または10℃ずつ増加させながら、あるいは約−40℃〜約−35℃へ、次いで約−25℃へ、次いで約−10℃へ、または約−40℃〜約−30℃へ、次いで約−15℃へ、または−40℃〜約−30℃へ、次いで約−25℃へ、または約−40℃〜約−25℃へといったように一連の不規則な手順で)上昇/増大させ得る。一次乾燥の後半の段階は、約0℃〜約10℃(例えば、約3℃、約5℃、約7℃、または約10℃)の温度など、同じ温度または高い温度で行われ得る。
【0042】
主に、試料を貯蔵する容器(例えば、ガラスバイアル)の正確な配合、サイズおよび種類、液体の容積、ならびに凍結乾燥の温度および圧力によって、乾燥に必要な時間が決定され、この時間は数時間から数日(例えば、40〜60時間)に及んでもよい。一次乾燥条件の例には、(1)75mTorrの真空レベル、一次乾燥段階の大半で約−25℃の温度の後に約5℃の期間、および約30〜35時間の一次乾燥時間、ならびに(2)75mTorrの真空レベル、一次乾燥段階での約−25℃の温度、および約15〜20時間の一次乾燥時間が含まれる。
【0043】
二次乾燥段階は、主に、使用する容器の種類およびサイズと正確な配合に応じて行われ得る。場合によっては、高温(例えば、約0℃〜約40℃、または約10℃〜約30℃、または約20℃または約30℃)での二次乾燥段階が使用される。しかし、場合によっては、二次乾燥手順が必要でない場合もある。二次乾燥に必要な時間および圧力は、好適な凍結乾燥したケーキを産生する時間および圧力となる。したがって、二次乾燥条件(およびいずれにせよ二次乾燥手順に必要なもの)は、温度および他のパラメータによって決まる。二次乾燥時間は、生成物中の所望の残留水分レベルによって決まり、一般的には少なくとも約5時間(例えば、約5〜約20時間(約8、約9、約10、約12、約15または約18時間など))かかる。圧力は、一次乾燥手順時に使用するものと同じであり得る。凍結乾燥条件は、配合およびバイアルサイズによって変更することができる。
【0044】
場合によっては、移送手順を回避するために、タンパク質を元に戻す容器中でタンパク質配合物を凍結乾燥させるのが望ましいことがある。この場合の容器は、例えば、3、5、10、20、50または100ccのバイアルであり得る。
【0045】
凍結乾燥(および必要に応じていずれかの移送手順)の後、凍結乾燥した配合物を一般的には容器内に入れて密封する。密封は、非弾性の蓋(例えば、全体がガラスでできたバイアルの端を溶かしてバイアルを密閉する)により、または弾性の蓋を取り付ける(例えば、容器の開口部を弾性の栓で塞いだ後、所定の位置にシール保持止め具を圧着して固定する)ことにより行うことができる。場合によっては、(例えば、減圧窒素環境で密閉することにより行われる場合のように)内容物を減圧下および/または減圧酸素下に置くような条件のもとで容器を密閉する。
【0046】
一般的な事柄として、凍結乾燥を行うと、含水量が約5%未満、例えば、約3%未満、または約2%未満の凍結乾燥配合物が生成される。
【0047】
再構成および投与
所望の段階で(一般的にはタンパク質を患者に投与するのが相応しい時に)、凍結乾燥配合物を希釈剤で再構成する場合がある。再構成に使用する希釈剤の容量は、再構成した配合物が所望のエカランチド濃度を有するようになる容量である。いくつかの実施形態においては、凍結乾燥配合物が再構成されて(例えば、適切な量の希釈剤を加えて)、エカランチドが10、20、30または40mg/mLである再構成配合物が得られ、また特定の実施形態においては、凍結乾燥配合物が再構成されて、エカランチドが30mg/mL、ヒスチジンが10mM、pHが6.5、スクロースが10%(w/v)である再構成配合物が得られる。
【0048】
希釈剤の例には、注射用滅菌水(WFI)、および注射用静菌性水(BWFI)が含まれるが、pH緩衝剤(例えば、リン酸塩緩衝食塩水)、滅菌食塩水、リンゲル液またはブドウ糖溶液などの他の希釈剤が使用される場合もある。
【0049】
希釈剤は場合により防腐剤を含む。有用な防腐剤には、ベンジルアルコールまたはフェノールアルコールなどの芳香族アルコールが含まれる。使用する防腐剤の量は、タンパク質との適合性に関する種々の防腐剤の濃度の評価および防腐剤の効力試験により決定される。例えば、防腐剤が芳香族アルコール(ベンジルアルコールなど)である場合、これは約0.1〜2.0%、約0.5〜1.5%、または約1.0〜1.2%の量だけ存在する場合がある。
【0050】
凍結乾燥配合物の再構成は一般的に、完全な水和が確実に行われるように室温(例えば、20℃〜25℃)にて行われるが、所望により他の温度が使用される場合もある。再構成に必要な時間は、配合物の正確な構成成分(例えば、希釈剤の種類、1つ以上の賦形剤およびエカランチドの量)により異なる。再構成は、手作業で(例えば、凍結乾燥配合物を含む容器に注入ポートを介して注入することにより、希釈剤を凍結乾燥配合物に手作業で加える)または自動的に(例えば、自動的に再構成するように構成されたBecton‐Dickinson BD(登録商標)のLiquid Dry Injectorなどの装置で希釈剤を凍結乾燥配合物に自動添加する)行う場合がある。
【0051】
配合物(液体配合物および再構成した凍結乾燥配合物)は、薬学的配合物(一般的には非経口投与用)として有用である。非経口投与には、静脈内(IV)、筋肉内(IM)、皮下(SC)、腹腔内(IP)、鼻腔内、および吸入の経路が含まれるが、これらに限定されない。IV、IM、SCおよびIP投与は、ボーラスまたは注入によって行われる場合があり、SCの場合には、徐放性埋込み式デバイス(ポンプ、徐放性配合物、および機械装置を含むがこれらに限定されない)で行われる場合もある。投与の用量、経路および方法は、治療する障害および患者の病歴により異なる。
【0052】
使用方法
本明細書ではまた、本明細書に開示される配合物を利用して、過剰なまたは調節不全の血漿カリクレイン活性に関連した障害を治療する方法も提供される。本明細書で使用される「治療」という用語は、治療する障害の症状を安定化、回復、改善または除去することを指す。過剰/調節不全の血漿カリクレイン活性に関連する臨床的障害はいくつかあり、これには、遺伝性血管性浮腫(I、IIおよびIII型の遺伝性血管性浮腫)、アンギオテンシン変換酵素(ACE)阻害剤誘発性の血管性浮腫、後天性(例えば、C1エステラーゼ阻害剤の欠損)血管性浮腫、特発性慢性血管性浮腫、アレルギー性血管性浮腫、および非ステロイド系抗炎症薬(NSAID)誘発性血管性浮腫が含まれる(本明細書では、遺伝性、ACE阻害剤誘発性、特発性慢性、アレルギー性、およびNSAID誘発性の血管性浮腫を「血管性浮腫」と総称する)。本開示内容のエカランチド配合物を投与することで、治療する血管性浮腫の少なくとも1つの症状(例えば、限局性浮腫)が安定化、回復、改善または除去される。
【0053】
したがって、本開示内容は、(1)本明細書に開示される有効量のエカランチド配合物を遺伝性血管性浮腫の被験体またはその疑いのある被験体に投与すことにより、遺伝性血管浮腫を治療する方法、(2)本明細書に開示される有効量のエカランチド配合物をACE阻害剤誘発性の血管性浮腫の被験体またはその疑いのある被験体に投与することにより、ACE阻害剤誘発性の血管性浮腫を治療する方法、(3)本明細書に開示される有効量のエカランチド配合物を後天性血管性浮腫の被験体またはその疑いのある被験体に投与することにより、後天性(例えば、C1エステラーゼ阻害剤の欠損)血管性浮腫を治療する方法、(4)本明細書に開示される有効量のエカランチド配合物を特発性慢性血管性浮腫の被験体またはその疑いのある被験体に投与することにより、特発性慢性血管性浮腫を治療する方法、(5)本明細書に開示される有効量のエカランチド配合物をアレルギー性血管性浮腫の被験体またはその疑いのある被験体に投与することにより、アレルギー性血管性浮腫を治療する方法、および(6)本明細書に開示される有効量のエカランチド配合物をNSAID誘発性血管性浮腫の被験体またはその疑いのある被験体に投与することにより、NSAID誘発性血管性浮腫を治療する方法を提供する。場合により、治療方法はさらに、投与に先立って凍結乾燥エカランチド配合物を再構成する手順も含む場合がある。
【0054】
有効量となるエカランチド配合物の量は、患者の病歴および疾患の重症度(あるいは疾患の悪化または急性発作)により異なる場合がある。いくつかの実施形態において、エカランチド配合物の有効量は、30mgのエカランチドを含む量である。
【0055】
本方法によれば、エカランチド配合物はいずれかの非経口経路で投与される場合がある。特定の実施形態において、エカランチド配合物は皮下のボーラス注射で投与される。
【0056】
エカランチド配合物は、被験体以外の人(例えば、医療専門家)によって被験体に投与される場合もあれば、被験体が自己投与する場合もある。注射器、輸液ポンプ、静脈内または皮下カテーテル、および自動注入装置を含め、選択された投与方式に適合するいずれのデバイスも使用される場合もある。
【0057】
キット
さらには、本明細書に開示される配合物を含むキットも提供される。本明細書に開示されるキットは、本開示内容の配合物を含む1つ以上のパッケージを含み、また配合物の使用に関する指示書(例えば、血管性浮腫の治療に関するもの)もさらに含む場合がある。キットに付属する指示書は、一般的には書面のものであるが、電子的なものである場合もあり(またワールドワイドウェブ上の1つ以上のサイトへのリンクを含む場合もある)、一般的には血管性浮腫の治療に関する投与量、服薬スケジュール、および投与経路についての情報を含む。エカランチド配合物のパッケージは、単位用量、大量包装(例えば、複数用量パッケージ)または副次的単位用量である場合がある。
【0058】
エカランチド配合物のパッケージは、所期の用途に適したいずれのパッケージングである場合もある。液体配合物の場合、適切なパッケージには、弾性栓付きアンプル、非弾性蓋付きアンプル(例えば、密封ガラスアンプル)、プレフィルドシリンジ、および自動注入装置(Bioject IJECT(登録商標)ニードルレス注射器またはDIAPEN(登録商標)注射器など)、ならびにオートインジェクターのカートリッジが含まれるが、これらに限定されない。凍結乾燥配合物の場合、適切なパッケージには、弾性栓付きアンプル、自己投与用デバイス(例えば、再構成および注入を自動的に行うBD(登録商標)Liquid Dry Injector)、およびプレフィルドシリンジが含まれるが、これらに限定されない。
【0059】
以下の実施例は、本開示内容を例示するためのものであるが、これを限定するものではない。
【実施例】
【0060】
(実施例1:エカランチドの産生)
エカランチドは、酵母(P.パストリス)での組換え発現によって産生された。S.セレビシエプレプロ‐matαのシグナル配列とエカランチドとの融合をコードする配列を、(アンピシリン耐性遺伝子およびHIS4を保有する)pHIL‐D2由来プラスミドのAOXl領域にクローン化して、pPIC‐K503を生成した。
【0061】
His4−表現型を有するP.パストリス菌株GS115のスフェロプラストを、(SaCI部位で)線状化されたpPIC-K503で形質転換し、宿主の5’AOX1座へ
プラスミドDNAの相同組換えを行った。プラスミドは宿主細胞のAOX1座へ入り、宿主細胞をHis4+表現型に変換し、AOX1座によって制御されるエカランチド発現カセットを生成した。
【0062】
組換え菌株は、外因性ヒスチジンの非存在下でメタノールを唯一の炭素源として増殖させることにより選択した。選択したコロニーをクローン化し、発現の調査を実施して多量のエカランチドを培地に分泌するクローンを同定した。高発現クローンを使用して、作業用細胞バンクを生成した。
【0063】
無菌接種物ブロス(酵母窒素塩基、リン酸カリウム、およびグリセロール、pH=5)を含むフラスコに作業用細胞バンクの細胞を接種することにより、接種物の培養を確立した。接種培養物を30℃でおよそ20時間にわたって保温した。
【0064】
この接種培養物を使用して、種発酵培養物の接種を行った。種発酵培養物は、合成培地(正リン酸、硫酸カルシウム、硫酸カリウム、硫酸マグネシウム、水酸化カリウム、グリセロール、d‐ビオチン、金属塩(硫酸、硫酸銅、ヨウ化ナトリウム、硫酸マンガン、モリブデン酸ナトリウム、ホウ酸、塩化コバルト、塩化亜鉛、および硫酸鉄)、消泡剤溶液、および水酸化アンモニウム)中で培養し、これを30℃にて発酵槽中で、OD600が28〜56になるまで行った。
【0065】
次いで種発酵培養物を使用して、産生用発酵培養物の接種を行った。種発酵培養物を、発酵槽中のあらかじめ加温した産生用発酵培地(正リン酸、グリセロール、硫酸カルシウム、硫酸カリウム、硫酸マグネシウム、水酸化カリウム、金属塩(硫酸、硫酸銅、ヨウ化ナトリウム、硫酸マンガン、モリブデン酸ナトリウム、ホウ酸、塩化コバルト、塩化亜鉛、および硫酸鉄)、消泡剤溶液、および水酸化アンモニウム)、d‐ビオチン、消泡剤溶液、および水酸化アンモニウム)に添加し、培地中の最初のグリセロールが使い果たされるまでグリセロールのバッチ期で増殖させた。次いで、グリセロールを培地に添加するグリセロールのバッチ供給期に培養を移行して、産生菌株をさらに増殖させた。最後に、グリセロールとメタノールの供給に切り替えて、培養をおよそ83時間の混合供給期に移行させた。
【0066】
発酵段階はすべて撹拌および通気(必要に応じて酸素を添加)しながら行った。
【0067】
発酵槽の内容物を冷却し、精製水で希釈した。初期の精製手順では、拡張床クロマトグラフィー(EBC)を利用して、希釈発酵槽ブロスからエカランチドを捕捉し、発酵から酵母を除去した。希釈した発酵槽の培養物を、下降流方式で拡張床カラム(STREAMLINE(商標)SP樹脂)に注加し、上昇流方式で洗浄し、沈降させてから、下降流方式で洗浄および溶出を行った。
【0068】
さらなる精製を、結合/洗浄/溶出の構成で操作される一連のカラムクロマトグラフィーの手順で実施した。EBC溶出液を陽イオン交換(CEX)樹脂(Bio‐Rad MACRO‐PREP(登録商標))High S)上に注加し、それを洗浄し溶出させた。CEX溶出液を硫酸アンモニウムが1.1Mとなるように調整し、次いで疎水性相互作用クロマトグラフィー(HIC)樹脂上に注加し、それを洗浄し溶出させた。HIC溶出液は、1kDa MWCO再生セルロース膜(UFDF)を使用した限外濾過/ダイアフィルトレーションによって緩衝剤を交換し、次いで陰イオン交換(AEX)クロマトグラフィー樹脂(BioSepra Q HYPERD(登録商標))上に注加し、それを洗浄してから溶出させた。AEX溶出液は、UFDFによって緩衝剤を交換してPBS(pH7.0)にし、0.22μm膜で無菌的に濾過し、無菌PETGビンに無菌分注し、−20℃で貯蔵した。
【0069】
(実施例2:pHおよび緩衝剤の選択)
エカランチドの安定性を種々の緩衝剤中でpH6.0、6.5および7.0にて調べた。エカランチド(等張性リン酸塩緩衝食塩水中に10mg/mL、pH7.0)を透析により緩衝剤交換して、(a)10mMのコハク酸塩、pH6.0、150mMのNaCl、(b)10mMのヒスチジン、pH6.0、150mMのNaCl、(c)10mMのヒスチジン、pH6.5、150mMのNaCl、(d)リン酸塩緩衝食塩水(PBS、4.3mMのリン酸ナトリウム、1.5mMのリン酸カリウム、137mMのNaCl)、pH6.5、または(e)10mMのヒスチジン、pH7.0、150mMのNaClにした。
【0070】
それぞれの配合物の試料を滅菌濾過して個々の管に入れ、4℃または30℃で6週間貯蔵した。試料を1、2、3.5、5および6週間の時点でHPLCサイズ排除クロマトグラフィー(SEC)により分析して、凝集体の形成および断片化を検出し、さらに逆相(RP)HPLCによってピログルタメートの形成を検出した。
【0071】
SEC‐HPLCの結果では、pHが上昇すると、高分子量の種類のもの(凝集体)が増加することが示された。逆に言えば、pHが上昇すると、低分子量の種類のもの(断片)が減少した。
【0072】
RP‐HPLC分析では、pH7.0の試料で非常に多くのピログルタメートが生成されることが示された。pH6.0および6.5の試料のうち、pH6.5のPBS試料では、ピログルタメートのレベルがやや高かった。
【0073】
(実施例3:低用量のエカランチドの場合の増量剤/凍結保護物質の選択)
増量剤/凍結保護物質の種々の構成を利用して、凍結乾燥配合物中のエカランチドの安定性を調べた。エカランチド(PBS中に10mg/mL、pH7.0)を、透析によって緩衝剤交換して、10mMのヒスチジンで緩衝化されるか(pH6.5)またはPBSで緩衝化され(pH6.5)、さらに(a)5%のマンニトール、(b)3%のマンニトール/3%のスクロース、(c)10%のスクロース、または(d)7.5%のスクロース/5%のデキストラン40を増量剤/凍結保護物質として含む配合物にした。
【0074】
それぞれの配合物の試料を滅菌濾過して、ガラスバイアルに入れ、凍結させ、凍結乾燥させた後、4℃または40℃にて8週間貯蔵した。試料を水で再構成してから、2週間、4週間、6週間および8週間の時点でSEC‐HPLCおよびRP‐HPLCにより分析した。
【0075】
40℃に維持した試料のピログルタメート(RP-HPLC)のデータを図2に示す(図2AはPBS緩衝剤であり、図2Bはヒスチジン緩衝剤である)。マンニトールは一般的に安定させる増量剤/凍結保護物質であると見なされているが、マンニトールは、図2に示すように、エカランチド配合物中では不安定にさせている。唯一の増量剤/凍結保護物質としてマンニトールを含有する配合物は、他の配合物よりもピログルタメートのレベルがかなり高かった。また、スクロースとマンニトールの混合物を含有する配合物の安定性は、マンニトールのみを有する配合物よりも優れていたが、これらの配合物もやはりスクロースの配合物およびスクロース/デキストランの配合物よりもピログルタメートのレベルが高かった。凝集生成物および断片化生成物に関するSEC‐HPLCのデータは類似していた。
【0076】
(実施例4:用量を増加させたエカランチドの場合の増量剤/凍結保護物質の選択)
増量剤/凍結保護物質の種々の構成を利用して、凍結乾燥配合物中のエカランチドの安定性を調べた。エカランチド(PBS中に20mg/mL、pH7.0)を、透析によって緩衝剤交換して、10mMのヒスチジンで緩衝化され(pH6.5)、さらに(a)10%のスクロース、(b)3%のマンニトール/3%のスクロース、または(c)3%のマンニトール/3%のトレハロースを増量剤/凍結保護物質として含む配合物にした。
【0077】
それぞれの配合物の試料を滅菌濾過してガラスバイアルに入れ、凍結させ、凍結乾燥器で−40℃まで凍結させることによって凍結乾燥させた後、75mTorrにて−40℃で30分間、−25℃で23時間、5℃で10時間一次乾燥させ、次いで75mTorrにて30℃で9時間二次乾燥させた。凍結乾燥試料を4℃または40℃で8週間貯蔵した。試料を2週間(40°の試料のみ)、4週間、6週間および8週間の時点でSEC‐HPLCおよびRP‐HPLCにより分析した。
【0078】
RP‐HPLCおよびSEC‐HPLCの分析では、10%のスクロースを増量剤/凍結保護物質として含む試料は、スクロース/マンニトール配合物およびマンニトール/トレハロース配合物よりも劣化がかなり少ないことが示された。図3に示すように、マンニトール含有配合物の場合、かなりの量のピログルタメート(図3A)および「ピーク4」の不純物があった(図3B:「ピーク4」は、この方法では分解できない、酸化およびグリコシル化されたエカランチドの混合物であると考えられる)。
【0079】
上述したまたは添付の特許請求の範囲で定義された本発明から逸脱することなく、記載した実施態様および特徴の置換、改変および変更が追加で行われる場合があることが、当業者により理解されるであろう。
【0080】
本明細書に引用した刊行物は全体が参考として本明細書で援用される。
【特許請求の範囲】
【請求項1】
本願明細書に記載された発明。
【請求項1】
本願明細書に記載された発明。
【図1】


【図2】
【図3】
【図2】
【図3】
【公開番号】特開2012−207047(P2012−207047A)
【公開日】平成24年10月25日(2012.10.25)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−165974(P2012−165974)
【出願日】平成24年7月26日(2012.7.26)
【分割の表示】特願2008−558556(P2008−558556)の分割
【原出願日】平成19年3月9日(2007.3.9)
【出願人】(506065987)ダイアックス コーポレーション (26)
【出願人】(500034653)ジェンザイム・コーポレーション (37)
【Fターム(参考)】
【公開日】平成24年10月25日(2012.10.25)
【国際特許分類】
【出願番号】特願2012−165974(P2012−165974)
【出願日】平成24年7月26日(2012.7.26)
【分割の表示】特願2008−558556(P2008−558556)の分割
【原出願日】平成19年3月9日(2007.3.9)
【出願人】(506065987)ダイアックス コーポレーション (26)
【出願人】(500034653)ジェンザイム・コーポレーション (37)
【Fターム(参考)】
[ Back to top ]