
エステラーゼ活性を有するタンパク質
【課題】エステラーゼ活性を示す新規タンパク質およびその突然変異体、それらをコードする核酸配列、発現カセット、ベクターおよび組換え微生物を提供する。
【解決手段】Pseudomonas glumae(Burkholderia plantarii)LU 2023から出発して得られる前記タンパク質を生産する方法およびそれらの酵素的、特にエナンチオ選択的エステ加水分解または有機エステルのエステル交換に対する使用。
【解決手段】Pseudomonas glumae(Burkholderia plantarii)LU 2023から出発して得られる前記タンパク質を生産する方法およびそれらの酵素的、特にエナンチオ選択的エステ加水分解または有機エステルのエステル交換に対する使用。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、エステラーゼ活性を有する新規タンパク質、その機能的等価体および突然変異体、それらをコードする核酸配列、発現カセット、ベクターおよび組換え微生物;前記タンパク質を調製する方法;ならびに有機エステルの酵素性の、特に、エナンチオ選択的酵素性の、エステル加水分解またはエステル交換を行うためのそれらの使用に関する。
【背景技術】
【0002】
エステラーゼやリパーゼは光学活性有機化合物を合成する工業的方法で使用できる加水分解酵素であって、高い基質特異性を特徴とする。これらの酵素はセリンプロテアーゼと同様の作用機序により、アシル基を例えばカルボニル基のような求核基に転移したり、エステル結合を加水分解で切断できる。エステラーゼ、リパーゼおよびセリンプロテアーゼは触媒三つ組残基(triad)(すなわち、Ser、HisおよびAspのアミノ酸からなる配列モチーフ)を共有し、前記カルボニル基の炭素原子は活性Serによる求核攻撃を受け、他の2つのアミノ酸が参加して電荷分布を形成する。エステラーゼとリパーゼはまた、チオエーテルのチオ基または活性化アミンなどの他の求核基にアシル基を転移することもできる。
【0003】
リパーゼは長鎖グリセロールエステルを加水分解し、表面活性化により特徴づけられる、すなわち、活性部位が脂質基質の存在下でのみ利用可能になる。リパーゼは非水性有機溶媒中で安定しており、反応速度論的ラセミ体分割を行う多数の工業的方法に利用されており、すなわち、一つのエナンチオマーを他のエナンチオマーより実質的に速く変換する。このエナンチオマーはその後、物理化学的性質の相異によって反応溶液から得ることができる。
【0004】
Nakamura (非特許文献1)は、市販のリパーゼ(Amano AK、AHおよびPS;Amano Pharmaceuticals Co. Ltd.)を使って、疎水性溶媒中のエステル交換による1-アルキン-3-オールのラセミ体分割を記載している。この反応においては、エナンチオ選択性がアシル供与体の鎖長とともに増加し、また、立体的に大きな残基(クロロアセテート、ビニルベンゾエート)は反応に悪影響を及ぼす。Yang(非特許文献2)はCandida antarctica由来のリパーゼBを触媒として用いて、ビニルエステルとエステル交換することによる、光学活性酸のエナンチオ選択的製法を記載している。この場合、エチルエステルは明らかに低い反応速度と選択率をもたらす。Burkholderia plantarii(Pseudomonas plantariiまたはglumae)DSM 6535から単離されたリパーゼは、メトキシ酢酸エチルを使ってラセミ体アミンをエナンチオ選択的にアシル化するのに利用されている(非特許文献3)。
【0005】
エステラーゼはエステル結合の形成および分解(すなわち、正反応および逆反応)をエナンチオ選択的に触媒する。光学活性アルコールを得るには、エステル交換反応でビニルエステルを使用することが好ましい。何故ならば、このエステルのアルコール官能基はアルデヒドやケトンへの互変異性化のため変換後にはもはや利用されず、従って逆反応を回避できるからである。リパーゼとは対照的に、エステラーゼは表面活性化されることなく、比較的短い鎖長の有機化合物も変換する。さまざまな基質特異性を示すエステラーゼが種々の生物から単離されている。
【0006】
例えば、Pseudocardia thermophila FERM-BP-6275由来のエステラーゼは、光学活性クロマン酢酸エステルの加水分解に利用される(EP-A-0 892 044)。
【0007】
Bacillus acidocaldarius由来のエステラーゼは、低いエナンチオ選択性で、狭い基質範囲からのエステルを加水分解する(非特許文献4)。
【0008】
Aspergillus由来のアシラーゼ1は、無極性有機溶媒中でビニルエステルを用いてエステル交換することにより第二級アルコールを得るために用いられ、これは短い側鎖をもつ第二級アルコールを変換するのに好適である(非特許文献5)。Pseudomonas fluorescens DSM 50 106から、膜結合型のラクトン特異的エステラーゼがクローニングされており(非特許文献6)、また、大腸菌Q突然変異体からはアセチルエステラーゼがクローニングされている(非特許文献7)。しかし、これらの2種のエステラーゼのエナンチオ選択性および基質特異性は詳細に研究されていない。Rhodococcus sp. NCBM 11216は4種類のエステラーゼ、RR1〜RR4を発現するが、これらは異なる特異性をもっている。ナフトールと酸からのエステル合成の場合、RR1とRR2は炭素鎖の短い酸のほうを優先し、一方RR3とRR4は比較的長い炭素鎖と立体的に比較的大きい残基をもつ酸を特異的に変換する(非特許文献8)。
【0009】
しかし最近まで、短い炭素鎖をもつ光学活性アルコール、酸またはエステルのような小有機分子を調製するための、広い基質範囲を持ち、高いエナンチオ選択性を有しかつ工業プロセスに利用できるエステラーゼは入手できなかった。
【0010】
特許文献1は初めて、エステラーゼ活性を有する有用なタンパク質を記載し、このタンパク質を、広範囲の光学活性エステルをエナンチオ選択的に加水分解できるエステラーゼとして言及した。さらに具体的に、この特許は510個のアミノ酸を含むタンパク質(配列番号2)およびそのコード配列(配列番号1)を記載している。
【0011】
本発明の目的は、さらに、場合によっては活性が最適化された、上記特性の少なくとも1つを有するエステラーゼを提供することである。
【先行技術文献】
【特許文献】
【0012】
【特許文献1】WO-A-02/18560
【非特許文献】
【0013】
【非特許文献1】Nakamura, K. et al., Tetrahedron; Asymmetry 9, (1999), 4429-4439
【非特許文献2】Yang, H. et al., J. Org. Chem. 64, (1999), 1709-1712
【非特許文献3】Balkenhohl, F. et al., J. prakt. Chem. 339, (1997), 381-384
【非特許文献4】Manco, G. et al., Biochem. J. 332, (1998), 203-212
【非特許文献5】Faraldos, J. et al., Synlett 4, (1997), 367-370
【非特許文献6】Khalameyzer, V. et al., Appl. and Environ. Microbiol. 65(2), (1999), 477-482
【非特許文献7】Peist, R. et al., J. Bacteriol. 179, (1997), 7679-7686
【非特許文献8】Gudelj, M. et al., J. Mol. Cat. B, Enzymatic 5, (1998), 261-266
【発明の概要】
【0014】
前記目的は、驚くべきことに、エステラーゼ活性を有するタンパク質またはその機能的に等価なタンパク質であって、前記タンパク質が510個未満のアミノ酸の全長をもつポリペプチド鎖を有しかつ前記鎖が少なくとも1つの配列番号3に記載のアミノ酸部分配列を含む前記タンパク質を提供することにより達成できた。
【図面の簡単な説明】
【0015】
【図1】図1は、ブチノールIエステラーゼのアミノ酸部分配列とPseudomonas fluorescens由来のラクトン特異的エステラーゼの部分配列との配列アラインメントを示す。Query:本発明のクローンLU2898の部分配列。Sbjct:P. fluorescens 酵素の部分配列、(受託番号:O87637)。
【図2】図2は、LU2898に対するクローニングチャートを図解する。
【図3】図3は、LU2898由来のブチノールエステラーゼ遺伝子とERGOデータベースとの比較を示す。高相同性領域を黒色で示す。右側のテキストカラムはERGOからの注釈であり、対応するe値を示す;e値はチャンスに基づく配列相同性存在の確率である。ブチノールエステラーゼ遺伝子のPstI切断部位の位置を示す。比較のために、Ps. glumae、LU2023由来の非組み換え酵素のアミノ酸配列を示す。
【図4】図4は、長さ335個のアミノ酸の配列を有する本発明のエステラーゼ(クローンLU11147由来)および活性が増強されたそれ由来の突然変異体の酵素活性を説明する。
【発明を実施するための形態】
【0016】
好ましい実施形態
本発明の第一の主題は、エステラーゼ活性を有するタンパク質またはその機能的に等価なタンパク質であって、前記タンパク質が510個未満のアミノ酸の全長をもつポリペプチド鎖を有しかつ前記鎖が少なくとも1つの配列番号3に記載の部分配列を含む前記タンパク質に関するものであり、前記タンパク質は好ましくは、参照基質としてのエステル、酪酸ブタ-3-イン-2-イルの切断により特徴付ることができるエステラーゼ活性を持つ。
【0017】
本発明のタンパク質は特にさらに、少なくとも1つのさらなる配列番号4、5または6に記載のアミノ酸部分配列を含んでもよい。
【0018】
配列番号3、4、5または6に記載の前記アミノ酸部分配列は、次の通り規定される(それぞれ、アミノ酸の一文字記号で示し、それぞれの第1アミノ酸は特有のアミノ末端に対応する)
a)FIETLGLERPVLVGHSLGGAIALAVGLDYPER(配列番号3)、
b)IALIAPLTHTETEP(配列番号4)、
c)GGGMMGLRPEAFYAASSDLV(配列番号5)、および
d)AIDAIFAPEPV(配列番号6)
本発明のタンパク質は特に、全長450個未満、例えば300〜445個のアミノ酸(例えば350個未満のアミノ酸、特に300〜345個、特に約330〜340個、とりわけ335個のアミノ酸)を有するポリペプチド鎖を含む。
【0019】
特に強調しうるのは、配列番号8に記載のアミノ酸配列を含むタンパク質である。
【0020】
本発明はさらに、これら新規の末端切断されたエステラーゼの、活性が増強された突然変異体、および同様に調製されたWO-A-02/18560に開示されかつ約510個のアミノ酸およびその機能的等価体を含むエステラーゼの突然変異体にも関する。
【0021】
本発明は特に、配列番号2または8のアミノ酸配列領域12〜20、185〜195および258〜268のいずれかに少なくとも1つの機能性突然変異を有する、とりわけ、配列番号2または8のアミノ酸配列の16、190および263位のいずれかに少なくとも1つの機能性突然変異を有するエステラーゼ突然変異体に関する。
【0022】
かかる突然変異の例は、限定されるものでないが、単独でまたはいずれかの組合わせで存在する次のアミノ酸置換:Leu16Pro、Ile190Thr、Ile190ArgおよびIle263Valである。
【0023】
本発明のタンパク質はさらに、例えば約60kDa以下、例えば56kDa以下または55.5kDa以下の計算分子量を有するポリペプチド鎖である。例えば、510個以下のアミノ酸をもつ末端切断したブチノールエステラーゼは、例えば、約56kDa〜20kDa、例えば約55.5〜30kDaまたは55.5〜35kDaまたは55.5〜40kDaまたは40〜30kDa、または55.5〜45kDaまたは38〜34kDaまたは36.5〜35.5kDaの範囲の分子量を有しうる。
【0024】
配列番号2によるタンパク質の突然変異体は、好ましくは約60〜40kDa、または56〜50kDaまたは55.5〜54kDaの範囲の計算分子量を有する。
【0025】
配列番号8によるタンパク質の突然変異体は、好ましくは約38〜34kDa、37〜35kDaまたは36.5〜35.5kDaの範囲の計算分子量を有する。
【0026】
好ましいタンパク質は受託番号DSM13176(DSMZに1999年12月2日寄託した)をもつPseudomonas glumae(Burkholderia plantariiとも呼ぶ)LU2023、および、適宜、その後の突然変異から得ることができる。
【0027】
エステラーゼ活性を有する本発明のタンパク質、機能的等価体および突然変異体はさらに次の反応:
a)式I:
R1-COO-R2 (I)
[式中、R1は直鎖または分枝鎖の、任意に一置換または多置換されたC1〜C10-アルキル、C2〜C10-アルケニル、C2〜C10-アルキニルであり、そしてR2は直鎖または分枝鎖の、任意に一置換または多置換されたC1〜C10-アルキル、C2〜C10-アルケニル、C2〜C10-アルキニル、C7〜C15-アラルキル、または単環または多環の、任意に一置換または多置換された芳香族基であり、そしてR1および/またはR2は少なくとも1個の不斉炭素を含む]で表わされる光学活性エステルのエナンチオ選択的加水分解;および
b)式Iのエステルと式II:
R2-OH (II)
[式中、R2は上記の意味の1つを有し、任意に、少なくとも1個の不斉炭素を有する]で表わされる光学活性アルコールとのエナンチオ選択的エステル交換
の両反応のうちの少なくとも1つの反応を触媒する。
【0028】
好適なC1〜C10-アルキル基の例として挙げることができるのは、1〜10個の炭素をもつ直鎖または分枝鎖の基、例えばメチル、エチル、イソプロピルまたはn-プロピル、n-、イソ-、sec-またはtert-ブチル、n-ペンチルまたはイソペンチル;さらにn-ヘキシル、n-ヘプチル、n-オクチル、n-ノニル、n-デシル、およびまた、それらの単分枝または多分枝した鎖の類似体である。
【0029】
好適なC2〜C10-アルケニル基の例は、2〜10個の炭素をもつ前記アルキル基の単不飽和または多不飽和類似体であり、この類似体は好ましくは1つまたは2つの炭素-炭素二重結合を有し、この結合は炭素鎖のいずれの位置にあってもよい。
【0030】
好適なC2〜C10-アルキニル基の例は、2〜10個の炭素をもつ前記アルキル基の単不飽和または多不飽和類似体であり、この類似体は好ましくは1つまたは2つの炭素-炭素三重結合を有し、この結合は炭素鎖のいずれの位置にあってもよい。
【0031】
C7〜C15-アラルキルは好ましくはフェニル-C1〜C5-アルキルまたはナフチル-C1〜C5-アルキルである。
【0032】
単環または多環の、任意に一置換または多置換の芳香族基の例は、C1〜C5-アルキル、例えばメチル、エチル、イソプロピルもしくはn-プロピル、n-、イソ-、sec-またはtert-ブチル、n-ペンチルもしくはイソペンチル;ヒドロキシ、メルカプト、アミノ、ニトロまたはハロ、例えばF、Br、Clのなかから選択される1、2または3個の同一または異なる置換基により置換されたフェニルおよびナフチルである。
【0033】
式Iのエステルは、例えば、直鎖または分枝鎖の、任意に単不飽和または多不飽和、任意に置換されたC1〜C11-モノカルボン酸から誘導される。飽和酸の例として挙げることができるのは、例えば蟻酸、酢酸、プロピオン酸およびn-酪酸およびイソ酪酸、n-吉草酸およびイソ吉草酸、カプロン酸、ヘプタン酸、カプリル酸、ペラルゴン酸、カプリン酸、ウンデカン酸;単不飽和酸、例えばアクリル酸、クロトン酸;および二不飽和酸、例えばソルビン酸である。もし酸が二重結合を含めば、cis型とtrans型の両方が存在しうる。
【0034】
本発明はさらに、以上規定したタンパク質、機能的等価体または突然変異体をコードするポリヌクレオチド、および前記ポリヌクレオチドの機能的等価体、それに相補的なポリヌクレオチド、およびそれとハイブリダイズしうる核酸配列に関する。
【0035】
本発明は特に、配列番号7に記載の核酸配列中の少なくとも30個の連続ヌクレオチド残基のヌクレオチド配列を含むポリヌクレオチドに関する。
【0036】
本発明はさらに、配列番号8のアミノ酸263位に対応する領域のコドンがGTTおよびGTCのなかから選択されるポリヌクレオチドに関する。
【0037】
本発明はさらに、少なくとも1つの、以上に規定されかつ少なくとも1つの調節核酸配列と機能しうる形で連結されたポリヌクレオチドを含む発現カセットに関する。
【0038】
本発明はさらに、真核生物または原核生物宿主を形質転換するための、以上規定したポリヌクレオチドまたは前記発現カセットを含む組換えベクターに関する。
【0039】
本発明はさらに、以上規定したタンパク質を調製する方法であって、前記タンパク質を内因的に産生する微生物または以上規定したベクターを用いて形質転換した微生物を培養するステップ、および前記タンパク質を培養物から単離するステップを含んでなる前記方法に関する。
【0040】
これに関する好適な方法は、受託番号DSM13176をもつ微生物Pseudomonas glumae(Burkholderia plantarii)LU2023またはそれから誘導された微生物を用いる。
【0041】
本発明はまた、以上規定した方法により得られる、前記タンパク質およびその機能的等価体または突然変異体に関する。
【0042】
本発明はさらに、受託番号DSM13176をもつPseudomonas glumae(Burkholderia plantarii)LU2023およびその変異体および突然変異体に関する。
【0043】
本発明はさらに、以上規定したベクターを担持する微生物に関する。
【0044】
本発明はさらに、以上規定したタンパク質(機能的等価体および突然変異体を含む)を用いるエナンチオ選択的エステル加水分解法であって、
a)前記タンパク質を、式Iで表される光学活性エステルの立体異性体混合物と接触させるステップ;および
b)前記立体異性体のいずれかの立体選択的加水分解により生成する光学活性化合物および/または加水分解されないエステルエナンチオマーを反応媒体から取得するステップを含んでなる前記方法に関する。
【0045】
本発明はさらに、エナンチオ選択的エステル交換法であって、
a)式IIで表される光学活性アルコールの立体異性体混合物を、前記タンパク質(機能的等価体および突然変異体を含む)の存在下に、式Iで表されるエステルと接触させ、未反応のアルコール立体異性体を反応媒体から取得するステップ;または
b)式Iで表される光学活性エステルの立体異性体混合物を、前記タンパク質(機能的等価体および突然変異体を含む)の存在下に、式IIで表されるアルコールと接触させ、前記エステルに含まれる光学活性アルコールの立体異性体を反応媒体から取得するステップを含んでなる前記方法に関する。
【0046】
本方法の特別な変形によれば、エステル交換に用いる光学活性アルコールに対するアシル化剤はビニルエステルである。
【0047】
本発明は特に、用いる反応媒体が有機溶媒である前記方法に関する。
【0048】
2.一般用語の説明
「エステラーゼ」または「ブチノールエステラーゼ」または「ブチノールIエステラーゼ」は、本明細書に記載した酵素性変換の少なくとも1つ、特に少なくともカルボン酸、特に酪酸の参照ブチノールエステル、例えば酪酸ブタ-3イン-2イルなどのブチノール酪酸エステルの切断を触媒する酵素である。
【0049】
具体的に開示した配列から「誘導された」またはそれと「相同的な」配列、例えば、誘導されたアミノ酸配列または核酸配列は、本発明によれば、特に断らない限り、出発配列と少なくとも80%または少なくとも90%、特に91%、92%、93%、94%、95%、96%、97%、98%および99%、同一である配列を意味する。
【0050】
二つの核酸間の「同一性」は、それぞれの場合に、全核酸長にわたるヌクレオチドの同一性、特にInformax(USA)のソフトウエアであるVector NTI Suite 7.1を使い、クラスタルの方法 (Higgins DG, Sharp PM. Fast and sensitive multiple sequence alignments on a microcomputer. Comput Appl. Biosci. 1989 Apr;5(2):151-1)を用いて、次のパラメーター:
マルチプル・アラインメント・パラメーター:
ギャップ・オープニング・ペナルティ 10
ギャップ・エキステンション・ペナルティ 10
ギャップ分離ペナルティ範囲 8
ギャップ分離ペナルティ オフ
アラインメント遅れに対する%同一性 40
残基特異的ギャップ オフ
親水残基ギャップ オフ
トランジション重み付け(Transition weighing) 0
ペアワイズ・アラインメントパラメーター:
FASTアルゴリズム オン
K-tuple サイズ 1
ギャップペナルティ 3
ウインドウサイズ 5
最良対角数(Number of best diagonals) 5
を設定して比較することにより計算した同一性を意味する。
【0051】
3. 本発明のさらなる実施形態
3.1 本発明によるタンパク質
本発明は、具体的に開示したエステラーゼ活性を有するタンパク質および酵素に限定されるものでなく、その機能的等価体も包含する。
【0052】
本発明の目的にとって、具体的に開示した酵素の「機能的等価体」 または 類似体は、前記酵素と異なるポリペプチドで、さらに、例えば、加水分解活性などの所望の生物学的活性を有するポリペプチドである。
【0053】
「機能的等価体」とは例えば、使用したエステラーゼ活性アッセイにおける活性が、配列番号8に記載のアミノ酸配列を含む酵素の活性より、少なくとも1%、例えば少なくとも10%または20%、例えば少なくとも50%または75%または90%、高いかまたは低い酵素を意味する。さらに、機能的等価体は好ましくはpH 4〜10の間で安定であり、最適pHは有利なのはpH 5〜9、例えば6〜8であり、かつ最適温度は15℃〜80℃ または 20℃〜70℃の範囲にある。
【0054】
エステラーゼ活性は様々な公知のアッセイによって検出できる。限定されるものでないが、例えば、標準化した条件下(例えば20mM基質、10mMリン酸バッファー、pH7.4、T=20℃など)で、酪酸ブタ-3イン-2イルなどのブチノール酪酸エステルのような参照基質を用いるアッセイを挙げることができる。
【0055】
本発明によれば「機能的等価体」はまた、特に、以上記載したアミノ酸配列の少なくとも1つの配列位置において具体的に記載したものと異なるアミノ酸を有するが、それにも関らず、以上記載した生物学的活性の1つを有する「突然変異体」も意味する。「機能的等価体」は従って、1個以上のアミノ酸付加、置換、欠失および/または逆転によって得られる突然変異体を包含し、本発明の特性プロファイルを有する突然変異体を生じる限り、前記改変がいずれの配列位置で起こってもよい。機能的等価は特にまた、突然変異体と未改変のポリペプチドの間の反応性パターンが定性的に一致する場合、すなわち、例えば、同じ基質が異なる速度で変換される場合にも存在する。好適なアミノ酸置換の例を、次表に総括する。
【0056】

本発明による突然変異を行うのに好適な個々の配列領域の具体的な例は既に先に規定している、すなわち、配列番号2または8からのアミノ酸配列領域12〜20、185〜195および258〜268、特に配列番号2または8のアミノ酸配列の16、190および263位である。
【0057】
好適なアミノ酸置換の具体的な例はLeu16Pro、Ile190Thr、Ile190ArgおよびIle263Valである。本発明の教示を知れば、当業者はこれらのさらなる改変を容易に提供しうる。
【0058】
以上の意味での「機能的等価体」はまた、記載したポリペプチドの「前駆体」およびまた、前記ポリペプチドの「機能的誘導体」および「塩」でもある。
【0059】
この場合、「前駆体」は所望の生物学的活性を有するまたは有しないポリペプチドの天然または合成の前駆体である。
【0060】
表現「塩」は、本発明によるタンパク質分子のカルボキシル基の塩ならびにアミノ基の酸付加塩を意味する。カルボキシル基の塩は、公知の方法で生産することができ、無機塩、例えばナトリウム、カルシウム、アンモニウム、鉄および亜鉛塩、および有機塩基の塩、例えばアミン、例えばトリエタノールアミン、アルギニン、リシン、ピペリジンなどを含む。酸付加塩、例えば鉱酸との塩、例えば塩酸または硫酸および有機酸との塩、例えば酢酸およびシュウ酸も本発明に包含される。
【0061】
本発明によるポリペプチドの「機能的誘導体」はまた、機能的アミノ酸側鎖基でまたはこれらのN末端またはC末端で公知の技法を用いて作製することができる。かかる誘導体には、例えばカルボン酸基の脂肪族エステル、アンモニアとのまたは一級もしくは二級アミンとの反応により得られるカルボン酸基のアミド;アシル基との反応により得られる遊離アミノ基のN-アシル誘導体;またはアシル基との反応により生成する遊離ヒドロキシ基のO-アシル誘導体が含まれる。
【0062】
「機能的等価体」は勿論、他生物から得られるポリペプチドならびに天然に存在する変異体も包含する。例えば、相同配列の領域を配列比較によって確立できるし、また、等価酵素を本発明の具体的なパラメーターに基づいて確認できる可能性がある。
【0063】
同様に、「機能的等価体」は、例えば所望の生物学的機能を有する、本発明によるポリペプチドの断片、好ましくは、個々のドメインまたは配列モチーフも含む。
【0064】
「機能的等価体」はさらに融合タンパク質であって、上記のポリペプチド配列またはそれから誘導される機能的等価体の1つおよび機能的N-またはC-末端結合に(すなわち、融合タンパク質部分の相互機能を実質的に損なうことなく)少なくとも1つのさらなる機能的に異なる異種配列を有する融合タンパク質である。かかる異種配列の例は、限定するものではないが、例えばシグナルペプチド、ヒスチジンアンカーまたは酵素である。
【0065】
本発明に含まれる「機能的等価体」はまた、具体的に開示したタンパク質の相同体である。これらは、Pearson and Lipman, Proc. Natl. Acad, Sci. (USA) 85(8), 1988, 2444-2448のアルゴリズムに従って計算して、具体的に開示したアミノ酸配列の1つと少なくとも60%、好ましくは少なくとも75%、特に少なくとも85%、例えば90、91、92、93、94、95、96、97、98または99%の相同性を有する。本発明による相同性ポリペプチドのパーセント相同性は、特に、本明細書に具体的に記載したアミノ酸配列の1つの全長に基づくアミノ酸残基の同一性パーセントを意味する。
【0066】
タンパク質のグリコシル化されている可能性がある場合、本発明による「機能的等価体」は、脱グリコシル化型またはグリコシル化型で先に示したタンパク質、ならびにグリコシル化パターンを変えることにより得られる改変型を包含する。
【0067】
本発明によるタンパク質またはポリペプチドの相同体は、突然変異誘発により、例えば、タンパク質の点突然変異、伸長または末端切断により作製することができる。
【0068】
本発明によるタンパク質の相同体は、突然変異体、例えば末端切断突然変異体のコンビナトリアルライブラリーをスクリーニングすることにより同定できる。例えば、タンパク質変異体の多様性に富んだライブラリーは、合成オリゴヌクレオチドの混合物の酵素的連結による、核酸レベルでのコンビナトリアル突然変異誘発により作製することができる。縮重オリゴヌクレオチド配列から可能性のある相同体のライブラリーを作製するために使用できる方法は数多くある。縮重遺伝子配列の化学合成を自動DNA合成機で実施し、合成遺伝子を次いで好適な発現ベクターに連結することができる。遺伝子の縮重セットを使用すると、1つの混合物中に、可能性のあるタンパク質配列の所望セットをコードする全ての配列を提供することが可能となる。縮重オリゴヌクレオチドを合成する方法は当業者に公知である(例えば Narang, S.A. (1983) Tetrahedron 39:3;Itakura et al. (1984) Annu. Rev. Biochem. 53:323;Itakura et al., (1984) Science 198:1056;Ike et al. (1983) Nucleic Acids Res. 11:477)。
【0069】
従来の技術分野において、点突然変異または末端切断により作製されたコンビナトリアルライブラリーの遺伝子産物をスクリーニングする技法、および選択した特性をもつ遺伝子産物に対するcDNAライブラリーをスクリーニングする技法はいくつかが公知である。これらの技法を、本発明による相同体のコンビナトリアル突然変異誘発により作製した遺伝子ライブラリーの迅速スクリーニングに適合させることができる。大型の遺伝子ライブラリーをハイスループット分析によりスクリーニングするために最も広く用いられる技法は、遺伝子ライブラリーを複製可能な発現ベクターにクローニングするステップ;得られるベクターライブラリーで好適な細胞を形質転換するステップ;所望の活性の検出により、検出した産物の遺伝子をコードするベクターの単離が容易になる条件で、コンビナトリアル遺伝子を発現させるステップを含んでなる。ライブラリー中の機能的突然変異体の頻度を高める技法である帰納的集合突然変異誘発(REM)を、相同体を同定するためのスクリーニング試験と組み合わせて使用してもよい(Arkin and Yourvan (1992) PNAS 89:7811-7815;Delgrave et al. (1993) Protein Engineering 6(3):327-331)
本発明のエステラーゼの機能的等価体のさらなる例には、例えば、配列番号3、4、5または6から誘導された少なくとも1つの部分配列であって、1個以上のアミノ酸が具体的に示した部分配列と比較して置換、欠失、逆転または付加されていて、かつ天然のタンパク質のエステラーゼ活性と±90%または±50%以下、好ましくは±30%以下しか異ならないエステラーゼ活性を有する前記部分配列が含まれる。
【0070】
本発明によるエステラーゼは特にPseudomonas glumae LU2023、受託番号DSM 13176から得ることができる。さらに菌株変異体を、例えばPseudomonas glumae LU8093から出発して選択により、例えば最少培地プレートで、唯一の炭素源として酢酸エチルフェニルを用いて培養する方法により得ることができる。
【0071】
3.2 コード核酸配列
本発明はさらに、エステラーゼ活性をもつ酵素をコードする核酸配列に関する。配列番号7による配列を含む核酸配列;または配列番号8によるアミノ酸配列から誘導される核酸配列が好ましい。
【0072】
本明細書に記載の全ての核酸配列(1本鎖および2本鎖のDNAおよびRNA配列、例えばcDNAおよびmRNA)は、公知の方法でヌクレオチド・ビルディングブロックから化学合成により、例えば、二重へリックスの個々のオーバーラップする、相補的な核酸ビルディングブロックの断片縮合により作製することができる。オリゴヌクレオチドの化学合成は、例えば、公知の方法で、ホスホアミダイト法により行うことができる(Voet, Voet, 2nd edition, Wiley Press, New York, pages 896-897)。合成オリゴヌクレオチドの蓄積およびDNAポリメラーゼのクレノウ断片の方法によるギャップの充填およびライゲーション反応ならびに一般的クローニング技法が、Sambrook et al. (1989), Molecular Cloning: A laboratory manual, Cold Spring Harbor Laboratory Pressに記載されている。
【0073】
本発明はまた、上記ポリペプチドおよびそれらの機能的等価体のいずれかをコードする核酸配列(1本鎖および2本鎖のDNAおよびRNA配列、例えばcDNAおよびmRNA)にも関するものであり、これらの核酸配列は、例えば、人工のヌクレオチド類似体を用いて得ることができる。
【0074】
本発明は、本発明のポリペプチドもしくはタンパク質またはその生物学的に活性なセグメントをコードする単離された核酸分子と、例えば、本発明のコード核酸を同定または増幅するためのハイブリダイゼーションプローブまたはプライマーとして使用することができる核酸断片との両方に関する。
【0075】
本発明による核酸分子は、さらに、コード遺伝子領域の3'および/または5'末端から得られる非翻訳配列を含んでもよい。
【0076】
本発明はさらに、具体的に記載したヌクレオチド配列またはそのセグメントに相補的である核酸分子に関する。
【0077】
本発明のヌクレオチド配列によって、他の細胞タイプおよび生物に含まれる相同配列を同定および/またはクローニングするために有用なプローブおよびプライマーを作製することが可能となる。かかるプローブおよびプライマーは通常、「ストリンジェントな」条件(下記参照)下で、本発明の核酸配列のセンス鎖または対応するアンチセンス鎖の少なくとも約12個、好ましくは、少なくとも約25個、例えば、約40、50または75個の連続ヌクレオチドにハイブリダイズするヌクレオチド配列領域を含む。
【0078】
「単離された」核酸分子は、該核酸の天然源中に存在する他の核酸分子から分離されており、さらに、それが組換え法により産生されたのであれば、他の細胞性物質や培地を実質的に含まず、また、それが化学合成されたのであれば、化学的前駆物質や他の化学薬品を実質的に含まないものでありうる。
【0079】
本発明の核酸分子は、標準的な分子生物学の手法および本発明により提供される配列情報を用いることにより単離することができる。例えば、cDNAは好適なcDNAライブラリーから、ハイブリダイゼーションプローブとして具体的に開示した完全な配列またはそのセグメントの1つと標準的なハイブリダイゼーション技法とを用いて単離することができる(例えば、Sambrook, J., Fritsch, E. F. und Maniatis, T. Molecular Cloning: A Laboratory Manual. 2. Aufl., CoId Spring Harbor Laboratory, CoId Spring Harbor Laboratory Press, CoId Spring Harbor, NY, 1989に記載のように行う)。さらに、開示した配列またはそのセグメントの1つを含む核酸分子は、この配列に基づいて構築されたオリゴヌクレオチドプライマーを用いてポリメラーゼ連鎖反応を行うことにより単離することができる。このようにして増幅した核酸を好適なベクターにクローニングして、DNA配列解析により特徴づけることができる。本発明のオリゴヌクレオチドはまた、標準的な合成方法により、例えば自動DNA合成機を使って、作製することもできる。
【0080】
配列番号7またはその誘導体、相同体またはこれらの配列の部分などの、本発明による核酸配列は、例えばゲノムまたはcDNAライブラリーを介して、他の細菌から、例えば通常のハイブリダイゼーション技法またはPCR技法により単離することができる。これらのDNA配列は標準条件で本発明による配列とハイブリダイズする。
【0081】
「ハイブリダイズする」は、ポリヌクレオチドまたはオリゴヌクレオチドがほとんど相補的な配列と標準条件で結合できるが、非特異的結合が非相補的なパートナーの間ではこの条件で起こらないことを意味する。このためには、配列が90〜100%相補的でありうる。特異的にお互いに結合できる相補的配列の特性は、例えばノーザンブロットもしくはサザンブロットに、またはPCRもしくはRT-PCRのプライマー結合に利用される。
【0082】
短い保存領域のオリゴヌクレオチドはハイブリダイゼーション用に使うと有利である。しかし、本発明による核酸の、より長い断片または完全な配列をハイブリダイゼーション用に使うことも可能である。これらの標準条件は使用する核酸(オリゴヌクレオチド、より長い断片または完全な配列)に応じてまたはハイブリダイゼーションに使用する核酸の型(DNAもしくはRNA)に応じて変化する。例えば、DNA:DNA ハイブリッドに対する融点は、同じ長さのDNA:RNA ハイブリッドの融点よりほぼ10℃低い。
【0083】
例えば、特定の核酸に応じて、標準条件は42〜58℃の温度、0.1〜5×SSC(1×SSC=0.15M NaCl、15mMクエン酸ナトリウム、pH 7.2)の濃度のバッファ水溶液中、またはさらに50%ホルムアミドが存在する条件を意味し、例えば42℃、5×SSC、50%ホルムアミド中である。有利なDNA:DNAハイブリッドに対するハイブリダイゼーション条件は0.1×SSCおよび約20℃〜45℃の温度、好ましくは約30℃〜45℃の温度である。DNA:RNA ハイブリッドに対する有利なハイブリダイゼーション条件は0.1×SSCおよび約30℃〜55℃、好ましくは約45℃〜55℃の温度である。ハイブリダイゼーション用のこれらの記載温度は、長さほぼ100ヌクレオチドおよびG+C含量50%の核酸に対してホルムアミドの非存在のもとで計算した融点値の例である。DNAハイブリダイゼーションの実験条件は、関連する遺伝学教科書、例えば、Sambrook et al., "Molecular Cloning”, Cold Spring Harbor Laboratory, 1989に記載されており、当業者に公知である式を用いて、例えば、核酸の長さ、ハイブリッドの型またはG+C含量に応じて計算することができる。当業者はハイブリダイゼーションについてのさらなる情報を次の教科書から得ることができる:Ausubel et al. (eds), 1985, Current Protocols in Molecular Biology, John Wiley & Sons, New York;Hames and Higgins (eds), 1985, Nucleic Acids Hybridization: A Practical Approach, IRL Press at Oxford University Press, Oxford;Brown (ed), 1991, Essential Molecular Biology: A Practical Approach, IRL Press at Oxford University Press, Oxford。
【0084】
「ハイブリダイゼーション」は特にストリンジェントな条件のもとで行うことができる。かかるハイブリダイゼーション条件は、例えば、Sambrook, J., Fritsch, E.F., Maniatis, T., in: Molecular Cloning (A Laboratory Manual), 2nd edition, Cold Spring Harbor Laboratory Press, 1989, pages 9.31-9.57、またはCurrent Protocols in Molecular Biology, John Wiley & Sons, N.Y. (1989), 6.3.1-6.3.6に記載されている。
【0085】
「ストリンジェントな」ハイブリダイゼーション条件は、特に:42℃にて一晩、50%ホルムアミド、5×SSC(750mM NaCl、75mMクエン酸三ナトリウム)、50mMリン酸ナトリウム(pH7.6)、5×Denhardt溶液、10%デキストラン硫酸塩および20g/ml変性剪断サケ精子DNAからなる溶液中のインキュベーションと、次いで0.1×SSCによる65℃におけるフィルターの洗浄を意味する。
【0086】
本発明はまた、具体的に開示したまたは誘導可能な核酸配列の誘導体に関する。
【0087】
従って、本発明によるさらなる核酸配列を、例えば、配列番号7から誘導することができ、この核酸配列は単一のまたは複数のヌクレオチドの付加、置換、挿入または欠失により元のものとは異なるが、なお所望の特性のプロファイルをもつポリペプチドをコードすることができる。
【0088】
本発明はまた、「サイレントな」突然変異を含むかまたは具体的に記載した配列と比較して、特別な供給源または宿主生物のコドン利用、ならびに天然の変異体、例えばそのスプライシング変異体または対立遺伝子変異体によって改変されている核酸配列を包含する。
【0089】
本発明はまた、保存ヌクレオチド置換により得ることができる(すなわち、問題のアミノ酸を同じ電荷、サイズ、極性および/または溶解度のアミノ酸により置換した)配列に関する。
【0090】
本発明はまた、具体的に開示した核酸から配列多形により誘導された分子に関する。これらの遺伝的多形は天然変異によって一集団内の個体間で存在しうる。これらの自然変異は通常、遺伝子のヌクレオチド配列に1〜5%のばらつきを生じる。
【0091】
本発明による配列番号7をもつ核酸配列の誘導体とは、例えば、全配列領域にわたって、誘導されたアミノ酸のレベルで少なくとも60%相同性、好ましくは少なくとも80%相同性、非常に特に好ましくは少なくとも90%相同性(アミノ酸レベルの相同性については、先にポリペプチドについて記載した詳細な説明を参照されたい)を有する対立遺伝子変異体を意味する。相同性が配列の部分領域にわたってより高いと有利でありうる。
【0092】
さらに、誘導体はまた、本発明による核酸配列の、特に配列番号7の相同体、例えば、真菌もしくは細菌の相同体、末端切断配列、コードおよび非コードDNA配列の1本鎖DNAもしくはRNAであるとも解釈すべきである。例えば、配列番号7の相同体は、配列番号7に与えられた全DNA領域にわたって、DNAレベルで、少なくとも40%、好ましくは少なくとも60%、特に好ましくは少なくとも70%、非常に好ましくは少なくとも80%の相同性を有する。
【0093】
さらに、誘導体は、例えば、プロモーターとの融合体であると解釈すべきである。記載したヌクレオチド配列の上流にあるプロモーターを、少なくとも1つのヌクレオチド交換、少なくとも1つの挿入、逆転および/または欠失により改変し、しかし、プロモーターの機能または効力を損なわないようにすることができる。さらに、プロモーターの効力を、プロモーターの配列を変えることにより向上することができるし、または、プロモーターをより有効なプロモーター(異なる属の生物のものであってもよい)により完全に置き換えることもできる。
【0094】
3.3 本発明による構築物
本発明はさらに、調節核酸配列の遺伝子制御のもとで、本発明によるポリペプチドをコードする核酸配列を含む発現構築物;ならびにこれらの発現構築物の少なくとも1つを含むベクターに関する。
【0095】
「発現ユニット」は、本発明によれば、本明細書に規定したプロモーターを含むものであって、発現すべき核酸もしくは遺伝子との機能的連結に従って、この核酸もしくはこの遺伝子の発現、転写および翻訳を調節する、発現活性をもつ核酸を意味する。従って、この意味で「調節核酸配列」とも呼ばれる。プロモーターだけでなく、他の調節配列、例えばエンハンサーが存在してもよい。
【0096】
「発現カセット」または「発現構築物」は、本発明によれば、発現すべき核酸または発現すべき遺伝子と機能しうる形で結び付けた発現ユニットを意味する。単なる発現ユニットとは対照的に、発現カセットは従って、転写および翻訳を調節する核酸配列だけでなく、転写および翻訳の結果、タンパク質として発現すべき核酸配列も含むものである。
【0097】
用語「発現」または「過剰発現」は、本発明の関係では、対応するDNAによりコードされた微生物中の1以上の酵素の細胞内活性の産生または増加を記載する。これについては、例えば、生物に遺伝子を挿入し、存在する遺伝子を他の遺伝子と置き換え、遺伝子のコピー数を増加し、強いプロモーターを使用し、または高活性をもつ対応する酵素をコードする遺伝子を使用することが可能であり、また任意にこれらの対策を組合わせることもできる。
【0098】
好ましくは、本発明によるかかる構築物はそれぞれのコード配列の5'上流にプロモーター配列、および3'下流にターミネーター配列、ならびに任意にさらなる通常の調節配列を含むものであり、それぞれの調節配列はコード配列と機能しうる形で連結されている。
【0099】
「プロモーター」、「プロモーター活性をもつ核酸」または「プロモーター配列」は、本発明によれば、転写すべき核酸と機能しうる形で連結されて、この核酸の転写を調節する核酸を意味する。
【0100】
「機能的」または「機能しうる」連結は、この文脈では、例えば、プロモーター活性をもつ核酸、および転写すべき核酸配列、および任意にさらなる調節配列(例えば、核酸の転写を確実なものにする核酸配列)、および例えばターミネーターの1つの配列を、それぞれの調節配列が核酸配列の転写中にその機能を遂行できる方式で配置することを意味する。これは必ずしも化学的な意味での直接連結を必要としない。エンハンサー配列などの遺伝子調節配列はまた、例えば、より離れた位置からまたは他のDNA分子からですら、標的配列に作用することができる。転写すべき核酸配列をプロモーター配列の下流(すなわち3'末端)に置いて、2つの配列がお互いに共有結合している配置が好ましい。プロモーター配列と遺伝子組換えで発現される核酸配列の間の距離は200塩基対未満、または100塩基対未満、または50塩基対未満でありうる。
【0101】
プロモーターおよびターミネーターとは別に、挙げることができる他の調節配列の例は標的化配列、エンハンサー、ポリアデニル化シグナル、選択マーカー、増幅シグナル、複製起点などである。好適な調節配列は、例えばGoeddel, Gene Expression Technology: Methods in Enzymology 185, Academic Press, San Diego, CA (1990)に記載されている。
【0102】
本発明による核酸構築物は、特に、配列番号7の配列またはその誘導体および相同体、ならびに配列番号8から誘導しうる核酸配列を含むものであり、これは、遺伝子発現を制御する、例えば増加するための1以上の調節シグナルと機能しうる形でまたは機能的に連結されていることが有利である。
【0103】
これらの調節配列に加えて、これらの配列の自然調節がさらに現行の構造遺伝子の上流に存在することがあり、任意に遺伝的に改変して、自然調節がスイッチオフされて遺伝子の発現を増加させることができる。核酸構築物は、しかしまた、もっとシンプルな設計であってもよく、すなわち、さらなる調節シグナルをコード配列(例えば配列番号7またはその相同体)の上流に挿入せず、また、それを調節する自然のプロモーターを取り除かなくてもよい。代わりに、自然の調節配列を突然変異させて調節がもはや起こらないようにして、遺伝子発現を増加するようにしてもよい。
【0104】
好ましい核酸構築物はまた、有利な方法として、核酸配列の発現の増加を可能にするプロモーターと機能しうる形で連結された前記エンハンサー配列を1つ以上含む。他の調節配列またはターミネーターなどのさらなる有利な配列をまた、DNA配列の3'末端に挿入することもできる。本発明による核酸の1以上のコピーが構築物に含まれてもよい。構築物はまた、任意に構築物を選択するために、抗生物質耐性または栄養要求性を補完しうる遺伝子などの他のマーカーを含んでもよい。
【0105】
プロモーターに含まれる好適な調節配列の例は、グラム陰性菌で有利に使われるcos、tac、trp、tet、trp-tet、lpp、lac、lpp-lac、lacIq、T7、T5、T3、gal、trc、ara、rhaP(rhaPBAD)SP6、λ-PRまたはλ-PLプロモーターである。他の有利な調節配列は、例えば、グラム陽性菌プロモーターのamyおよびSPO2、酵母または真菌プロモーターのADC1、MFα、AC、P-60、CYC1、GAPDH、TEF、rp28、ADHである。人工プロモーターも調節に用いることができる。
【0106】
発現については、核酸構築物を宿主生物に、有利な方法としては、宿主での遺伝子の最適な発現を可能にするベクター、例えばプラスミドまたはファージ中に挿入する。ベクターは、プラスミドおよびファージだけでなく、当業者に公知の全ての他のベクター、すなわち、例えばウイルス(SV40、CMV、バキュロウイルスおよびアデノウイルスなど)、トランスポゾン、ISエレメント、ファスミド、コスミド、および直鎖または環状DNAも意味すると解釈すべきである。これらのベクターは宿主生物中で自律的に複製できるかまたは染色体を介して複製できる。これらのベクターは本発明のさらなる実施形態を示す。
【0107】
好適なプラスミドは、例えば、大腸菌ではpLG338、pACYC184、pBR322、pUC18、pUC19、pKC30、pRep4、pHS1、pKK223-3、pDHE19.2、pHS2、pPLc236、pMBL24、pLG200、pUR290、pIN-III113-B1、λgt11またはpBdCI;ストレプトマイセス菌ではpIJ101、pIJ364、pIJ702またはpIJ361;バシラス菌ではpUB110、pC194またはpBD214;コリネバクテリウム菌ではpSA77またはpAJ667;真菌ではpALS1、pIL2またはpBB116;酵母では2alphaM、pAG-1、YEp6、YEp13またはpEMBLYe23、あるいは植物ではpLGV23、pGHlac+、pBIN19、pAK2004またはpDH51である。以上記載したプラスミドは可能性のあるプラスミドから選択したわずかなものである。その他のプラスミドが当業者に周知であり、それらは例えば、書物のCloning Vectors (Eds. Pouwels P.H. ら Elsevier, Amsterdam-New York-Oxford, 1985, ISBN 0 444 904018)に見出しうる。
【0108】
さらなるベクターの実施形態においては、本発明による核酸構築物または本発明による核酸を含むベクターを、有利な方法としては直鎖DNAの形態で微生物に導入して、宿主生物のゲノム中に異種または相同的遺伝子組換えを介して組み込むことができる。この直鎖DNAはプラスミドなどの直線化ベクター、または本発明による核酸構築物、または核酸そのものであってもよい。
【0109】
生物において異種遺伝子を最適に発現させるためには、前記生物で使われる特定のコドン使用頻度に従って核酸配列を改変することが有利である。コドン使用頻度は、問題の生物の他の公知の遺伝子のコンピューター評価に基づいて容易に決定することができる。
【0110】
本発明による発現カセットの作製は、好適なプロモーターと好適なコードヌクレオチド配列およびターミネーターシグナルまたはポリアデニル化シグナルとの融合に基づく。このために使われる通常の組換えおよびクローニング技法は、例えば、T. Maniatis, E.F. Fritsch and J. Sambrook, Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY (1989)、ならびにT.J. Silhavy, M.L. Berman and L.W. Enquist, Experiments with Gene Fusions, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY (1984)、およびAusubel, F.M. et al., Current Protocols in Molecular Biology, Greene Publishing Assoc. and Wiley Interscience (1987)に記載されている。
【0111】
組換え核酸構築物または遺伝子構築物は、有利な方法として、好適な宿主生物における発現用の宿主特異的ベクターに挿入し、これにより遺伝子が宿主中で最適に発現されることを可能にする。ベクターは当業者に周知であり、例えば、"Cloning Vectors" (Pouwels P.H. et al., Publ. Elsevier, Amsterdam-New York-Oxford, 1985)に見出すことができる。
【0112】
3.4 本発明によって利用できる微生物
状況に応じて、用語「微生物」は出発微生物(野生型)または遺伝的に改変した組換え微生物またはそれらの両方を意味する。
【0113】
本発明によるベクターを用いることによって、例えば、少なくとも1つの本発明によるベクターにより形質転換されかつ本発明によるポリペプチドの産生に利用できる組換え微生物を作製することができる。有利な方法として、以上記載した本発明による組換え構築物を好適な宿主系に挿入して発現させる。好ましくは、特別な発現系における記載した核酸の発現を確保するために、当業者が手慣れた通常のクローニングおよびトランスフェクション法、例えば共沈降、プロトプラスト融合、エレクトロポレーション、レトロウイルスのトランスフェクションなどを用いる。好適な系は、例えばCurrent Protocols in Molecular Biology, F. Ausubel et al., Publ. Wiley Interscience, New York 1997、またはSambrook et al. Molecular Cloning: A Laboratory Manual. 2nd edition, Cold Spring Harbor Laboratory, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 1989に記載されている。
【0114】
原則として、全ての原核生物のまたは真核生物は本発明による核酸または核酸構築物用の組換え宿主生物とみなしうる。細菌、真菌または酵母などの微生物を宿主生物として用いることは有利である。グラム陽性菌もしくはグラム陰性菌、好ましくは腸内細菌科、シュードモナス科、リゾビウム科、ストレプトミセス科またはノカルジア科のファミリーの細菌、とりわけ好ましくはエシェリキア属、シュードモナス属、ストレプトマイセス属、ノカルジア属、ブルコルデリア属、サルモネラ属、アグロバクター属またはロドコッカス属の細菌を用いることが有利である。大腸菌の属および種はとりわけ特に好ましい。他の有利な細菌も次のグループ中に見出しうる:α-プロテオバクテリア、β-プロテオバクテリアまたはγ-プロテオバクテリア。
【0115】
本発明による宿主生物は、前記エステラーゼ活性をもつ酵素をコードする本発明に記載の核酸配列、核酸構築物またはベクターの少なくとも1つを含むことが好ましい。
【0116】
本発明の方法に利用する生物を、当業者の手慣れた方法で、宿主生物に応じて、生育または増殖する。通常、微生物を、一般に糖類の形態の炭素源、一般に窒素の有機源(酵母エキスなど)または塩(硫酸アンモニウムなど)の形態の窒素源、微量元素(鉄、マンガンおよびマグネシウム塩ならびに、適宜、ビタミン)を含む液培地中で、0℃〜100℃、好ましくは10℃〜60℃の温度で酸素曝気して増殖する。栄養液体培地のpHは増殖の間、固定した値、すなわち調節したまたは調節しない値に維持してもよい。増殖はバッチ式、半バッチ式または連続的に行ってもよい。栄養分は発酵の開始時に補給してもまたは、その後、半連続的または連続的に補給してもよい。ケトンは増殖中に直接加えてもよいし、または、有利な方法として、増殖後に加えてもよい。酵素は、実施例に記載の方法により生物から単離してもよいし、または粗抽出物として反応用に用いてもよい。
【0117】
3.5 エステラーゼの組換え生産
本発明はさらに、本発明によるポリペプチドまたはその機能的な生物学的活性断片を遺伝子組換え生産する方法であって、ポリペプチド産生微生物を培養し、適宜、ポリペプチドの発現を誘導し、そしてポリペプチドを培養物から単離することによる前記方法に関する。ポリペプチドはまた、所望であれば、この方法で工業的規模で生産することもできる。
【0118】
本発明によって作製する微生物は、連続的にまたはバッチ式(バッチ法、または1回供給バッチ法もしくは繰り返し供給バッチ法)で培養することができる。公知の培養方法の総括は、Chmiel(Bioprocesstechnik 1. Einfuehrung in die Bioverfahrenstechnik (Gustav Fischer Verlag, Stuttgart, 1991))の教科書またはStorhas(Bioreaktoren und periphere Einrichtungen (Vieweg Verlag, Braunschweig/Wiesbaden, 1994))の教科書に見出しうる。
【0119】
使用する培地は、適当な方式で、特定の菌株の要件を満たすものでなければならない。様々な微生物に対する培地の説明は、米国細菌学協会(the American Society)のハンドブック"Manual of Methods for General Bacteriology"(Washington D. C., USA, 1981)に記載されている。
【0120】
本発明に使用しうるこれらの培地は一般に1種以上の炭素源、窒素源、無機塩、ビタミンおよび/または微量元素を含む。
【0121】
好ましい炭素源は糖類、例えば単糖類、二糖類または多糖類である。非常に優れた炭素源は例えばグルコース、フルクトース、マンノース、ガラクトース、リボース、ソルボース、リブロース、ラクトース、マルトース、スクロース、ラフィノース、デンプンまたはセルロースである。糖類はまた、複雑な化合物、例えば糖蜜または糖精製から得られる他の副産物を介して培地に加えることもできる。様々な炭素源の混合物を加えることも有利でありうる。他の可能性のある炭素源は油類および脂肪類(ダイズ油、ヒマワリ油、ピーナッツ油およびヤシ油など)、脂肪酸類(パルミチン酸、ステアリン酸またはリノール酸など)、アルコール類(グリセロール、メタノールまたはエタノールなど)および有機酸類(酢酸または乳酸など)である。
【0122】
窒素源は通常、有機または無機窒素化合物またはこれらの化合物を含む物質である。窒素源の例としては、アンモニアガスまたはアンモニウム塩(硫酸アンモニウム、塩化アンモニウム、リン酸アンモニウム、炭酸アンモニウムまたは硝酸アンモニウム、硝酸塩など)、尿素、アミノ酸類または複雑な窒素源(トウモロコシ浸漬液、ダイズ粉、ダイズタンパク質、酵母エキス、肉エキスなど)などが挙げられる。窒素源は別々にまたは混合物として使用することができる。
【0123】
培地に存在してもよい無機塩化合物にはカルシウム、マグネシウム、ナトリウム、コバルト、モリブデン、カリウム、マンガン、亜鉛、銅および鉄の塩化物、リン酸塩または硫酸塩が含まれる。
【0124】
無機硫黄を含有する化合物(硫酸塩、亜硫酸塩、ジチオニット、テトラチオナート、チオ硫酸塩、スルフィドなど)だけでなく、有機硫黄化合物(メルカプタンおよびチオールなど)を硫黄源として使用することができる。
【0125】
リン酸、リン酸二水素カリウムまたはリン酸水素二カリウムまたは対応するナトリウム含有塩をリン源として使用することができる。
【0126】
溶液中の金属イオンを保持するためにキレート剤を培地に加えてもよい。とりわけ好適なキレート剤にはジヒドロフェノール(カテコールまたはプロトカテキュ酸など)、または有機酸(クエン酸など)が含まれる。
【0127】
本発明による発酵培地はまた、通常、他の成長因子(ビタミンなど)または成長促進物質(例えば、ビオチン、リボフラビン、チアミン、葉酸、ニコチン酸、パントテン酸およびピリドキシンなど)も含む。成長因子と塩はしばしば培地の複雑な成分(酵母エキス、糖蜜、トウモロコシ浸漬液など)に由来する。さらに好適な前駆体を培地に加えることができる。培地中の化合物の正確な組成は特定の実験に強く依存するものであり、それぞれの具体的な事例に対して個々に決定しなければならない。培地最適化に関する情報は、教科書"Applied Microbiol. Physiology, A Practical Approach"(Publ. P.M. Rhodes, P.F. Stanbury, IRL Press (1997) p. 53-73, ISBN 0 19 963577 3)に見出しうる。増殖培地はまた、商業的供給者(例えば、Standard 1(Merck)またはBHI(脳心臓注入液、DIFCO)など)から得ることもできる。
【0128】
全ての培地成分を、加熱(20分間、1.5bar、121℃にて)によりまたは滅菌濾過により無菌化する。成分を一緒にまたは、必要であれば、別々に無菌化してもよい。培地の全ての成分は増殖の出発時に存在してもよいし、または、任意に連続的にもしくはバッチ式供給により加えてもよい。
【0129】
培養の温度は通常、15℃〜45℃、好ましくは25℃〜40℃であり、実験中、一定に保持してもよいしまたは変化させてもよい。培地のpHは5〜8.5の範囲、好ましくはほぼ7.0にすべきである。増殖のためのpHは増殖中に、塩基化合物(水酸化ナトリウム、水酸化カリウム、アンモニアまたはアンモニア水など)または酸化合物(リン酸または硫酸など)を加えることにより制御することができる。消泡剤、脂肪酸ポリグリコールエステルを発泡を制御するために使用してもよい。プラスミドの安定性を維持するために、選択作用をもつ好適な物質、例えば抗生物質を培地に加えてもよい。酸素または酸素含有気体混合物、例えば大気を培地に供給して好気性条件を維持する。培養温度は通常20℃〜45℃である。培養は、所望の産物の最大量が得られるまで続ける。これは通常、10時間〜160時間以内に達成される。
【0130】
次に発酵ブロスをさらに処理する。要件に応じて、バイオマスを発酵ブロスから分離技法(例えば、遠心分離、濾過、デカントまたはこれらの方法の組合わせなど)により完全にまたは部分的に取り出してもよいしまたは発酵ブロス中に残してもよい。
【0131】
もしポリペプチドが培地中に分泌されなければ、細胞を破壊して、溶菌液からタンパク質を単離する公知の技法により産物を取得してもよい。細胞を、任意に、高周波超音波により、高圧により(例えば、フレンチプレス細胞破砕機で)、浸透圧溶解により、界面活性剤、溶解性酵素もしくは有機溶媒の作用により、ホモジナイザーを用いて、または掲げた方法のいくつかを組合わせることにより破壊することができる。
【0132】
ポリペプチドは公知のクロマトグラフィ法(モレキュラーシーブ・クロマトグラフィ(ゲル濾過)、Q-セファロース・クロマトグラフィ、イオン交換クロマトグラフィおよび疎水性クロマトグラフィなど)を用いて、および他の通常の方法(限界濾過、結晶化、塩析、透析および未変性ゲル電気泳動など)により精製することができる。好適な方法は例えば、Cooper, F.G., Biochemische Arbeitsmethoden, Verlag Walter de Gruyter, Berlin, New YorkまたはScopes, R., Protein Purification, Springer Verlag, New York, Heidelberg, Berlinに記載されている。
【0133】
組換えタンパク質を単離するには、例えば精製をより簡単にするために利用しうる改変したポリペプチドまたは融合タンパク質をコードする、規定したヌクレオチド配列だけcDNAの伸長したベクター系またはオリゴヌクレオチドを用いることが有利でありうる。この種類の好適な改変は、例えば、アンカーとして機能するいわゆる「タグ」、例えば、ヘキサ-ヒスチジンアンカーとして公知の改変、または抗原として抗体が認識しうるエピトープである(Harlow, E. and Lane, D., 1988, Antibodies: A Laboratory Manual. Cold Spring Harbor (N.Y.) Pressに記載されている)。これらのアンカーは、タンパク質と固体支持体、例えばクロマトグラフィカラムに充填する、例えばポリマーマトリックスとの接着を可能にするか、またはマイクロタイタープレート上でまたはいくつかの他の支持体上で利用することができる。
【0134】
同時に、これらのアンカーはまた、タンパク質の認識用にも使うことができる。タンパク質を認識するために、通常のマーカー、例えば、蛍光染料および酵素マーカー(基質との反応後、検出可能な反応生成物を生じる)、または放射性マーカーを、単独でもしくはタンパク質の誘導体化用のアンカーと組合わせて利用することもできる。
【0135】
3.6 本発明による、エステラーゼの用途
本発明はまた、エステラーゼを用いるエナンチオ選択的エステル加水分解法にも関し、この方法は、本発明のエステラーゼを、式Iで表される光学活性エステルの立体異性体混合物と接触させ、2種の立体異性体のいずれかの立体選択的加水分解により生成する光学活性化合物および/または加水分解されないエステルエナンチオマーを反応媒体から取得する。しかし、該エステラーゼは、光学的に活性でない式Iのエステルを加水分解することも可能である。
【0136】
本発明はさらに、エナンチオ選択的エステル交換法にも関し、この方法は、式IIで表される光学活性アルコールの立体異性体混合物を、本発明のエステラーゼの存在下で、式Iで表されるエステルと接触させ、未反応のアルコール立体異性体を反応媒体から取得するか、または、式Iで表される光学活性エステルの立体異性体混合物を、該エステラーゼの存在下で、式IIで表されるアルコールと接触させ、該エステルに含まれる光学活性アルコールの立体異性体を反応媒体から取得する。エステル交換法では、光学活性アルコールのアシル化剤としてビニルエステルを使用することが好ましい。このことは、ビニルエステルのアルコール官能基が変換後に互変異性化のため逆反応にもはや利用されないので、有利である。該エステラーゼはまた、エステルとアルコールのどちらも光学活性でないエステル交換法も触媒する。
【0137】
エステル加水分解の好ましい基質は、エタノール、プロパノール、ブタノールのエステル、そして特に好ましくは、ブチノールのカルボン酸(酢酸、プロピオン酸、酪酸、ペンタン酸、ヘキサン酸、ヘプタン酸、オクタン酸、乳酸、2-エチルヘキサン酸、3-メチル酪酸、メトキシ酢酸、2-メチルプロピオン酸、2-ブテン酸、3-クロロプロピオン酸、および2-メチルペンタン酸など)とのエステル(ブチノールエステル、1-メチルプロパ-2-イン-1-オールのエステル)である。特に好ましいのは、酪酸ブチニルとメチル酪酸ブチニルである。
【0138】
エステル交換に用いるのに好ましいアルコールはエタノール、プロパノールおよびブタノール、特に好ましいのはブチノールである。
【0139】
エステル交換に用いるのに好ましいエステルは、ビニルエステル、例えば、酢酸ビニル、プロピオン酸ビニルおよび酪酸ビニルである。
【0140】
以上の方法で用いる反応媒体は有機溶媒であり、例えば、アルカン類、エーテル類、トルエン、ジオキサン、メチルイソブチルケトン、メチルtert-ブチルエーテル(MTBE)などである。エステル加水分解においては、使用したバッファー溶液と有機溶媒(例えば、MTBEおよびヘプタンまたはトルエン)からなる混合溶液を使用してもよい。
【0141】
ラセミ化合物の分割(すなわち、エナンチオ選択性)および反応速度は、酸部分の大きさと疎水性により影響を受けることがある。
【0142】
本発明による反応は、好ましくは、室温にてpH 6〜9で、特に好ましくは、pH 7.0〜7.4で実施する。エステラーゼは、単離されたまたは精製された酵素の形態で、エステラーゼを発現する微生物の細胞として、前記微生物の培養上清、細胞溶解物または抽出物として、あるいは固定化されたエステラーゼとして使用される。反応生成物は当業者に公知の化学的または物理的分離方法により反応溶液から単離することができる。エステラーゼは膜濾過によって反応混合物から単離することができる。
【0143】
エステラーゼは、ポリアクリルアミド、アルギン酸またはカラギーナンを用いて固定化することが可能である。またエステラーゼを、公知の方法を用いて共有結合によりまたは吸着により好適な担体と結合することもできる。エステラーゼをケイソウ土への凍結乾燥により、または硫酸アンモニウム沈降により固定化することが好ましい。
【0144】
(実施例)
特に断らない限り、本発明の範囲内で行ったクローニングのステップ、例えば、制限酵素切断、アガロースゲル電気泳動、DNA断片の精製、核酸のニトロセルロースおよびナイロン膜への転移、DNA断片の連結、微生物の形質転換、微生物の培養、ファージの増殖、および組換えDNAの配列分析は、Sambrookら(1989、前記引用)に記載の通り行った。
【実施例1】
【0145】
エステラーゼを発現するPseudomonas glumae突然変異体の選択
このスクリーニングの開始点は、リパーゼ産生株であるPseudomonas (Burkholderia) glumae LU 8093であった。この菌株から得られたリパーゼにより、いくつかの興味深い反応を実施することができる(Balkenhohl, F. et al., J. prakt. Chem. 339, (1997), 381-384)。しかし、乳酸エステル、メチル酪酸エステルおよびフェニル酢酸エステルはこのリパーゼの基質ではなく、この菌株はこれらのエステルをどうしても加水分解することができない。
【0146】
しかし、加水分解産物は炭素源として利用可能である。それ故に、上記エステルを加水分解して、その加水分解産物を炭素源として利用して生育する能力があるLU 8093の突然変異体を探した。新規のエステラーゼ活性を有する突然変異体は、したがって、上記エステルで生育させることによってその正体を現すに違いない。
【0147】
選択条件:LU 8093を培地上で16時間培養し、遠心分離により回収した。細胞を食塩水で2回洗浄した。106個の細胞を、唯一の炭素源として0.5または1.0g/lのフェニル酢酸エチル(EPA)を含む最少培地プレートにまいた。ところが、最初は全く生育しなかった。やっと4〜6日後に、単一のコロニーが確認できた。その数はそれ以後さらに増えた。
【0148】
このようにして得られたエステラーゼ陽性突然変異体から、LU 2023突然変異株を選択した。驚いたことに、この新規エステラーゼの活性は比較的小さい有機分子の選択的加水分解にも適していた。例えば、ブチノールエステルを選択的に加水分解することがわかった。
【実施例2】
【0149】
Pseudomonas glumae LU 2023の発酵
エステラーゼを得るために、Pseudomonas glumae LU 2023を14Lのスケールで培養し、活性バイオマスを収穫した。
【0150】
実験室で、Pseudomonas glumae LU 2023を、M12ミネラル塩培地と1g/LのEPAを含む寒天プレート上にストリーク接種し、28℃で36〜48時間インキュベートした。その後、このプレートは4℃で4週間保存した。
【0151】
この菌株の発酵は、Infors xxy 14 l発酵槽で実施した。前培養のため、250mlの培地に2〜3の白金耳を用いて接種し、200rpm、28℃で24時間インキュベートした。主培養は下記の条件下で行った:
温度 28℃
空気供給量 7 L/分
攪拌 600rpm
発酵実施時間 約24時間
内蔵pHおよびpO2の測定
前培養および主培養用の培地
Springer酵母自己分解物質65% 15 g/l
硫酸マグネシウム×7水 1.6 g/l
塩化カルシウム×2水 0.02 g/l
リン酸二水素カリウム 3.5 g/l
リン酸水素二カリウム 3.5 g/l
リン酸水素二アンモニウム 5 g/l
Pluriol P2000消泡剤 6 ml
上記の成分を脱イオン水に溶解し、この溶液を25%濃度のアンモニア水を用いてpH6.5に調整した。5 ml/lの微量元素溶液および2 g/lグルコースを別々に滅菌濾過した。
【0152】
培地を滅菌して完成した後、0.5 g/lのフェニル酢酸エチルを発酵槽に導入した。Pluriol P2000を添加して発酵中の泡の発生を防止した。発酵槽内のpO2が85%以上に再度増加したとき発酵を停止させた。次いで、発酵槽の内容物を15℃以下、約9000〜10,000gで遠心分離し、透明な上清を廃棄した。細胞塊は-16℃で凍結させた。
【実施例3】
【0153】
Pseudomonas glumae LU 2023から得たエステラーゼの精製
Pseudomonas glumae LU 2023細胞(100 ml、湿重量:50 g)を4℃、3000rpmでガラスビーズミル(100mlのガラスビーズ、直径:0.5mm)で溶解した。遠心分離(10,000rpm、30分)して、ガラスビーズを洗浄した後、上清(300ml)を塩化マンガン沈降(pH7〜7.5;最終濃度:50mM)に付した。もう一回遠心分離した後、上清をpH8.0に調整し、EDTAを50mMの濃度で添加した。この容量をQ−Sepharose(300ml)クロマトグラフィーで精製した。サンプルをアプライした後、カラムを50mMトリス/HClで洗浄した。対象となる画分を集めて、限外濾過(100 kDa)により濃縮した。モレキュラーシーブ・クロマトグラフィー(直径:5cm、高さ:90cm;材料:S−300)にかけて非特異的エステラーゼからブチノール加水分解性エステラーゼを分離した。得られた活性画分は濁っており、再度濃縮した。このエステラーゼは明らかに膜と結合していた。次いで、膜画分をまずプロテアーゼ(トリプシン、重量比:1:50から1:100)で消化した。この操作により、SDSポリアクリルアミドゲル電気泳動において2,3のバンドを別にすれば、膜画分から全タンパク質が消失した。活性は保存されていた。前記バンドを未変性ゲル電気泳動(0.04%のSDS)で互いに分離し、活性アッセイにより未変性ゲル中のエステラーゼを同定した。前記エステラーゼをゲルから溶出した後に、変性SDSポリアクリルアミドゲル電気泳動にかけたところ、きれいな1本のバンドとして現れた。
【0154】
このようにして精製したタンパク質は、PVDF膜へのブロッティングにより移行させてから配列決定するか、または、トリプシンで切断した後にペプチドを逆相HPLCで分離してから配列決定した。このタンパク質のアミノ末端はブロックしたので、トリプシン消化ペプチドのみを得た。様々なアミノ酸配列がAcinetobacter iwoffiiおよびPseudomonas putida由来のムコン酸エステルシクロイソメラーゼ(muconate cycloisomerase)EC 5.5.1.1に対して、また、Pseudomonas fluorescens由来のラクトンエステラーゼに対して弱い相同性を示した。配列AIDAIFAPEGVをもつペプチドはペクチンエステラーゼ(EC 3.1.1.11)に対する相同性を示した。
【0155】
図1は、本発明のアミノ酸部分配列と、Pseudomonas fluorescens由来のラクトン特異的エステラーゼの部分配列との、配列アラインメントを示す。
【実施例4】
【0156】
エステラーゼの固定化
様々な方法を固定化のために用いた。
【0157】
1. エステラーゼを、ケイソウ土の存在下にアセトンで沈澱させることにより実質的に不活性化した。25mgのタンパク質を3.5gのケイソウ土(Merck)と混合し、1.4Lのアセトン(-20℃)を10分間添加した。その後、エステラーゼをローディングした担体をG3ガラス吸引フィルターにより分離し、フィルター残留物を冷アセトンで洗浄して、乾燥させた。
【0158】
2. エステラーゼは、Accurel(Akzo)と結合しない。
【0159】
3. エステラーゼ(2.3単位/g、EPAアッセイ)を、凍結乾燥によりケイソウ土に固定させることができた。このためには、酵素溶液をケイソウ土と混合し、−80℃で凍結させた。続いて、この固形物質を凍結乾燥により乾燥した。
【0160】
4. エステラーゼ(454ミリ単位/g、EPAアッセイ)を、硫酸アンモニウム沈澱により固定化した。そのためには、酵素をケイソウ土の存在下に80%飽和の硫酸アンモニウムで沈澱させた。
【実施例5】
【0161】
Pseudomonas glumae LU 2023由来のエステラーゼを用いるラセミ体分割
手順(標準手法)
100単位のエステラーゼを、リン酸バッファー(200ml、10mM、pH7.4)中で20mmolのブチノール酪酸エステル(酪酸1-メチルプロパ-2-イニル)と攪拌しながら反応させた。pHを連続測定して、水酸化ナトリウム溶液の添加により約pH 7.4に維持した。表1に示した時間にサンプルを採取し、メチルt-ブチルエーテル(MTBE)で2回抽出し、有機相をGC(Chiraldex GTA)により分析した。エステラーゼを膜濾過により反応混合物から分離した。
【0162】
その濃度が増加すると、好ましくない方のエステルエナンチオマーの変換が増加した。約45分後、これは反応混合物中のS-ブチノールのeeの低下を招いた。生成物のeeは、約30〜40分後に84%(83〜97.9%)で最大値に達した。基質のeeは90分間で99%以上に増加した。ee(エナンチオマー過剰率)は、好ましい方の変換エナンチオマーの量(%)マイナス好ましくない方の変換エナンチオマーの量(%)として規定される。ほとんどの場合、これは光学純度に一致する。pHの低下は30分まで直線状であったが、約100分以後、pHの変化は無視しうる程度であった。
【0163】
抽出後、水相中の残留エステラーゼ活性はまだ50%ほど残っていた。
【表1】
【0164】
表1は、エステラーゼを用いてブチノール酪酸エステルを変換したときの時間依存的エナンチオマー過剰率を示す。Cahn、Prelog及びIngoldによるR/S変換に従うと、RおよびS立体配置が1つのキラル分子の2つのエナンチオマーを規定する。変換率は反応混合物中の変換されたエステルの比率である。
【実施例6】
【0165】
エステラーゼ特異性の、エステルの酸部分の大きさおよび疎水性/電荷に対する依存性
標準手法
100単位のエステラーゼを、リン酸バッファー(200ml、10mM、pH7.4)中で20mmolのブチノールエステルと攪拌しながら反応させた。pHを連続的に測定し、連続滴定によりpH 7.0に維持した。サンプルを採取し、メチルt-ブチルエーテル(MTBE)で2回抽出し、有機相をGC(Chiraldex GTA)により分析した。
【0166】
結果
ラセミ体分割の品質と反応速度は、酸部分の大きさおよび疎水性に依存した。ブチノールエステラーゼの最良の基質は、ブチノールの酪酸エステルとブチノールのメチル酪酸エステルであった。リパーゼはこれらの基質には不活性である。このことは、n-デカン酸ブチニルのような長鎖エステルにも当てはまる。
【表2】
【0167】
表2は、エステラーゼを用いてエステルを変換するときのエナンチオマー過剰率が、変換されるエステルの酸部分に依存することを示す。
【実施例7】
【0168】
エステラーゼを用いる有機媒体中でのエステル交換
10mmolのrac-ブチノールと5mmolの酪酸ビニルを50mlのメチルt-ブチルエーテル(MTBE)に溶解し、ケイソウ土に担持された9単位のブチノールIエステラーゼ(3.3g)と混合し、この混合物を室温で24時間振とうした。濾過後、溶媒を除去し、生成物の混合物をGCにより特徴づけた。
【0169】
47%の変換率で、(R)-ブチノール(18% ee)および(S)-ブチノールの酪酸エステル(45% ee)が残存した。
【0170】
メチルイソブチルケトン中では、(R)-ブチノール(16% ee)および(S)-ブチノールの酪酸エステル(52% ee)が43%の変換率で得られた。
【0171】
表3は、エステラーゼを用いてエステルを変換したときのエナンチオマー過剰率が、変換されるエステルの酸部分に依存することを示す。
【表3】
【実施例8】
【0172】
菌株LU2023の遺伝子ライブラリーの調製およびLU2898のクローニング
a)染色体DNAの取得:
LU2023(前記実施例1および2を参照)を100mlのFP培地(Becton Dickinson GmbH)で28℃および180rpmにて24時間増殖した。細胞を遠心分離(2000rpm/5分間)によって収穫し、5mlの溶解バッファー1(0.41Mスクロース、0.01M MgSO4*7H2O、50ml/L M12培地(10×conc.)、10ml/L 10%KH2PO4 pH 6.7、2.5 mg/mlリゾチーム[使用の直前に加える])に再懸濁し、そして37℃にてほぼ4時間インキュベートした。遠心分離(2000rpm/20分間)の後、得られるプロトプラストを、5mlのリゾチームを含まない溶解バッファー1で洗浄(2000rpm/20分間)し、10mM TRIS-HCl pH8.0に再懸濁した。
【0173】
もう1回10mM TRIS-HCl pH 8.0を用いて洗浄(3000rpm/5分間)した後、ペレットを4mlのTEバッファー(10mM TRIS-HCl、1mM EDTA、pH 8)に再懸濁し、それを0.5mlのSDS(10%w/v)および0.5mlのNaCl(5M)と混合した。この後、100μlのプロテイナーゼK溶液(200μg/ml)を加え、37℃にて16時間インキュベートした。次いで、TEバッファーを加えて、この溶液を10mlの最終容量とした。この溶液をフェノールと1:1で混合した。遠心分離(4000rpm/5分間)の後、上相を取り出してフェノール、クロロホルムおよびイソアミルアルコール(12:12:1)の混合物と混合した。上相を取り出して1容量のクロロホルム・イソアミルアルコール(24:1)と混合した。上相が透明になるまでこの手順を繰り返した。
【0174】
2容量のエタノールおよび1/50容量のCH3COONa(3M)を-20℃で加えることにより(時間:ほぼ30分間)、DNAを上相から沈降させ、遠心分離(12000rpm、30分間、4℃)によりペレット化して、TEバッファーに再懸濁した。RNase(1mlの20μg/ml溶液)はRNAを37℃で1時間以内に加水分解する。その溶液を次いでTEバッファーに対して4℃で3回それぞれ1時間透析する。それぞれ0.4mlのアリコートのDNAに、エタノール(2容量+1/3容量のLiCl、2M)を加え、-20℃で30分間のインキュベーションにより沈降させる。遠心分離(15000rpm、30分間、4℃)でペレット化したDNAを次いで乾燥した。0.4mlアリコートのDNAを0.5ml TEバッファーに再懸濁した。
【0175】
b)DNAの制限酵素切断とクローニング
1μgの染色体DNAを120U PstIを用いて37℃にて180分間消化した。0.4μgのpUC19(例えば New England Biolabs)を160UのPstI(37℃にて1時間)により制限酵素切断し、次いでアルカリホスファターゼ(37℃にて1時間+不活性化(65℃にて15分間))により脱リン酸した。DNA断片をラピッドライゲーションキット(Roche)を用いて連結した(2時間、室温にて)。ライゲーション混合物を、製造業者の情報に従って形質転換コンピテント大腸菌(Stratagene)中に形質転換した。細胞をIPTG/X-Gal FPプレート(100μg/mlアンピシリンを含有するFP培地)にストリーク接種し、37℃にて16時間インキュベートした。白色のコロニーをアンピシリンを含有する新しいFPプレートにストリーク接種した。
【0176】
c)遺伝子ライブラリーのスクリーニング
コロニーを、無菌ベルベットクッションを用いてWhatman濾紙に移した。フィルターを2.5mlのアッセイ溶液(10mM TRIS*HCl、pH7.5、0.01%ブロモクレゾール・パープル、0.1%ラセミエステル[rac-乳酸メチル(MEE)またはrac-メチル酪酸エチルまたはrac-フェニル酢酸エチル])中に置いた。エステラーゼ活性をもつ細胞のコロニーは青色から黄色へ5分間以内に変色する。試験した2900個のコロニーのなかで、強い変色を生じるものが1つあった。この菌株をLU2898と名付けた。図2はLU2898のクローニング計画を表す模式図である。
【実施例9】
【0177】
LU2898の組換え体DNAの配列分析
LU2898から得たプラスミドは7.7kbインサートを有する。大腸菌LU2898のブチノールエステラーゼ遺伝子を保有するプラスミドを完全に配列決定した。DNAデータベースの最初の検索は、加水分解酵素と相同性を有するインサートをもつオープンリーディングフレーム(位置1394〜2923)を生じた。
【0178】
ERGOデータベース(Integrated Genomics, Inc. 1999-2004, Chicago, USA)をLU2898ブチノールエステラーゼのDNA配列を用いて検索した。ブチノールエステラーゼ遺伝子の領域1384〜2397bpだけが公知の加水分解酵素と相同的であることを見出した。
【0179】
領域1384〜2397bpは明らかに所望のブチノールエステラーゼのDNAである。他の良い徴候は、セリンカルボキシルエステラーゼに典型的でありかつLU2023ブチノールエステラーゼのアミノ酸位置127〜131に位置するGXSXGモチーフである。
【0180】
LU2898ブチノールエステラーゼ遺伝子とB. cepacia 加水分解酵素の間の良い一致はインサートのほぼ位置2400で終わる。さらにリーディングフレームの下流で、NADPレダクターゼの3'末端と非常に明確な相同性が存在する。
【0181】
これらの結果を添付した図3に示す。図3はERGO分析の総括である。LU2898ブチノールエステラーゼの相同性の急激な移行はPstI切断部位と一致する。PstI制限酵素の切断部位は、選んだクローニング計画で予想されなかった。プラスミドライブラリーを調製するために、この酵素を用いてLU2023染色体のDNAを完全に切断した。完全な加水分解の後、PstIに対する内部認識部位はもう存在しないはずである。LU2898プラスミド中のPstI切断部位の発見は、前記PstI制限部位により作製される色々な遺伝子断片がプラスミド構築中にお互いに接続され、その結果、連結されてpUC19のPstI切断部位になったという事実により説明することができ、この事実は、ブチノールエステラーゼに割りつけられたリーディングフレームがおそらくPs. glumae LU2023由来の実際の遺伝子の一部分だけを表すことを意味する。ブチノールエステラーゼ遺伝子はPstI部位で破壊され、次いでNADPレダクターゼ遺伝子の断片と接続されたのである。
【0182】
この仮説はタンパク質配列決定データにより支持される。Ps. glumae LU2023ブチノールエステラーゼを精製した。該タンパク質のアミノ酸配列をEdman分解により決定した。完全配列は、いくつかのN-およびC-末端アミノ酸を除くと公知である。それはLU2898のDNAデータから誘導したブチノールエステラーゼの最初の部分からのアミノ酸配列に対応する。41.3kDaのブチノールエステラーゼの実験で決定した分子量は、組換えLU2898ブチノールエステラーゼ遺伝子のDNA配列から誘導した値より著しく低い。DNA配列データによると、ブチノールエステラーゼは55kDaの分子量を持つはずである。
【0183】
データベース分析の助けで、ブチノールエステラーゼはその全長がクローニングされていなかったという確証が見出された。LU2898から得たプラスミドはブチノールエステラーゼ遺伝子の3'末端を含んでいない。
【実施例10】
【0184】
末端切断された335AAブチノールエステラーゼ突然変異体を含むクローンLU11147の発見
次のPCRプライマーを設計するためにLU2898由来のプラスミドの配列データを利用した。
【0185】
HindIII
Breu0811: GGCGAGAAGCTTAGAAATCATGATCGTCCA (配列番号20)
XbaI
Breu0812: GGATCCTCTAGAGTCTCACTGCAGCGCGCC (配列番号21)
これらのオリゴヌクレオチドを用いてPCRを使い、以上記載した相同性をエステラーゼに対して有するLU2898由来のDNAを増幅した。
【0186】
PCR条件:
10*Taqポリメラーゼバッファー 5μl
プライマーBreu0811 125ng
プライマーBreu0812 125ng
dNTP(10mM) 2μl
LU11147由来のプラスミドDNA 50ng
DMSO 2.5μl、および
H20 50μ
Taq-DNAポリメラーゼ(Roche Diagnostics)0.5Uを加えてホットスタート。
【0187】
PCR温度プログラム:
5分間、95℃,
45秒、55℃,
3分間、72℃、(30サイクル)
30秒、95℃、
45秒、55℃,
10分間、72℃,
∞、4℃
HindIIIとXbaIによる制限酵素切断の後、PCR産物を、適当に調製したpUC19(例えばNew England Biolabs)中にライゲートした。
【0188】
この方法で調製したプラスミドベクターを次に大腸菌XL1ブルーを形質転換するのに用いた。ライゲーション混合物を形質転換コンピテント大腸菌(Stratagene)中に、製造業者の情報に従って形質転換した。細胞をIPTG/X-Gal FPプレート(100μg/mlアンピシリンを含有するFP培地)上にストリーク接種し、37℃にて16時間インキュベートした。プラスミドDNAを形質転換体から調製し(Birnboim et al.、 Nucleic Acids Research、1979、7、1513)、HindIIIとXbaIによる制御制限を用いたPCR産物の挿入の成功をチェックした。
【実施例11】
【0189】
末端切断された335AAブチノールエステラーゼの発現
次いでエステラーゼ遺伝子をベクターpDHE1650(WO-A-2004/050877に記載)中にNdeIとHindIII切断部位を用いてクローニングした。
【0190】
この目的で、エステラーゼ遺伝子を最初に、次のフォワードおよびリバースプライマー:
Breu1499 TATACATATGATCGTCCAACTGATCGCCATCGTG、
Breu1500 ATTTAAGCTTTTACTGCAGCGCGCCGGCCTGCGTGACCTC
を用いて増幅し、次いでNdeIとHindIIIを用いて制限酵素切断を行った。インサートを、同じ制限酵素(NdeIとHindIII)により予め消化しておいたpDHE1650ベクター中にライゲートした。同じ方法で、pDHE1650中に存在する場所ホルダー遺伝子を本発明のエステラーゼ遺伝子と置き換えた。2つの形質転換体を単離し、プラスミドを公知の方法でDNA沈降により抽出した。前記プラスミドをpDHE1650-Est2およびpDHE1615-Est4と名付けた。
【0191】
タンパク質発現については、エステラーゼ遺伝子を保持するプラスミドを大腸菌TG1(DSMZ No.6056)にTSS法を用いて形質転換した[Chung CT, Niemela SL and Miller RH,1989, "One-step preparation of competent Escherichia coli: Transformation and Storage of Bacterial Cells in the Same Solution" Proc. Natl. Acad. Sci. U.S.A. 86: 2172-2175]。単一コロニーを単離し、5mlのLB/Amp/Tet/Spec/Cm培地(=Sambrook, J., Fritsch, E.F. and Maniatis, T. Molecular cloning: A Laboratory Manual. 2nd Ed., Cold Spring Harbor Laboratory, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 1989によるLB培地+100μg/mlアンピシリン、10μg/mlテトラサイクリン,100μg/mlスペクチノマイシンおよび20μg/mlクロラムフェニコール)で37℃にて250rpmで一晩増殖した。10ml/l微量元素溶液を添加した100mlのLB/Amp/Tet/Spec/Cm培地に500μlの一晩培養物を接種し、37℃および200rpmにて増殖した(微量元素溶液は次の構成である:FeSO4*H2O 200mg/l、ZnSO4*7H2O 10mg/l、MnCl2*4H2O 3mg/l、H3BO3 30mg/l、CoCl2*6H2O 20mg/l、CuCl2*6H2O 1mg/l、NiCl2*6H2O 2mg/l、Na2MoO4*2H2O 3mg/l、Titriplex III 500mg/l)。タンパク質発現を1mM L-ラムノースを用いて1回誘導し、OD600は0.5〜0.6の値に達した。温度を30℃に低下したが、回転速度は200rpmに保った。培養物を一晩培養した。次いで、細胞を遠心分離し(15分間、4℃、3220g)、細胞ペレットを-20℃にて一晩保存した。
【0192】
細胞を、細胞ペレット(50mlのTB培養物)をリン酸ナトリウムバッファー(25mM、イオン強度154mM、pH7.5)に再懸濁することにより破壊した。細胞をホモジナイザーで破壊した(1500 bar、2回パス)。細胞断片を遠心分離(30分間、4℃、20817g)により取り出した。上清をタンパク質200 Plus Labchipにアプライし、Agilent 2100バイオアナライザーを用いてタンパク質を分析した。取り出した細胞断片を8M尿素に再懸濁し、次いで遠心分離(30分間、4℃、20817g)により処理して未溶解粒子を取り除いた。透明な上清(膜画分)をタンパク質200 Plus-Labchipにアプライし、Agilent 2100バイオアナライザーを用いてタンパク質を分析した。
【実施例12】
【0193】
ブチノールエステラーゼ(335AA)突然変異体の調製
a)材料
使用した全ての化学品は分析用に好適であるかまたはより高品質であって、Sigma-Aldrich Chemie, Taufkirchen, Germany, Applichem(Darmstadt, Germany)およびCarl Roth(Karlsruhe、Germany)から購入したものである。
【0194】
サーモサイクル(Mastercycler gradient、Eppendorf、Hamburg、Germany)および薄層PCRチューブ(Multi-Ultra tubes;0.2ml;CarlRoth)を全てのPCR実験に用いた。PCR容量はいずれの場合も50μlであり;より大きい容量は複数の50μl PCR混合物を用いて調製した。使用したDNAの量はNanoDrop光度計(NanoDrop Technologies、Wilmington、DE)を用いて数値化した。
【0195】
b)ブチノールエステラーゼ遺伝子のpASK-IBA7ベクター中へのクローニング:
pASK-IBA7ベクター(IBAGmbH、Goettingen、Germany)(配列番号19、N-末端Strep-タグによるタンパク質発現用のプラスミド)を次のプライマー:
フォワードプライマー
5'-TTTTTGCCCTCGTTATCTAGATTT-3'(配列番号9)
リバースプライマー
5'-CCGGAATTCCGGTATCTAACTAAGCTTGACCTG-3'(配列番号10)
を用いて増幅した。
【0196】
50μl PCR混合物(95℃ 3分間;1サイクル//95℃ 30秒、56.2℃ 45秒、72℃ 7分間;30サイクル//72℃ 10分間;1サイクル)は、20pmolのフォワードプライマー、20pmolのリバースプライマー、0.2mMの各dNTP(Roche Diagnostics GmbH, Mannheim, Germany)、150ngのpASK-IBA7プラスミドおよび2.5UのPfuDNAポリメラーゼ(Fermentas GmbH, St. Leon-Rot、Germany)を含んだ。生成物をQIAquickゲル抽出キット(Qiagen、Hilden、Germany)を用いてゲルにより精製し、以上の条件と同一の条件下で再び増幅した。ゲル精製(QIAquickゲル抽出キット、Qiagen)の後、2μgのDNA産物を40 UのEcoRI(New England Biolabs GmbH、Frankfurt、Germany)により100μl反応混合物中で37℃にて4時間消化した。消化した産物を、QIAquick PCR精製キット(Qiagen)を用いてPCR精製した。
【0197】
ブチノールエステラーゼ遺伝子を次のプライマー:
フォワードプライマー
5’-[Phos]GACCATGATTACGCCAAGCTTGC-3’(配列番号11)
リバースプライマー
5’-CCGGAATTCCGGTCACTGCAGCGCGCCGGCCTG-3’(配列番号12).
を用いて増幅した。
【0198】
50μl PCR混合物(95℃2分間;1サイクル//95℃30秒、59℃45秒、72℃3分間;30サイクル//72℃ 10分間;1サイクル)は、20pmolのフォワードプライマー、20pmolのリバースプライマー、0.2mMの各dNTP(Roche Diagnostics GmbH, Mannheim, Germany)、150ngのプラスミドLU11147(ブチノールエステラーゼ遺伝子を保有するpUC19ベクター)、0.02%DMSOおよび2.5UのPfuDNAポリメラーゼ(Fermentas)を含んだ。ゲル精製(QIAquickゲル抽出キット、Qiagen)の後、2μgのDNA産物を40UのEcoRI(New England Biolabs GmbH、Frankfurt、Germany)により100μl反応混合物中で37℃にて4時間消化した。消化した産物を、QIAquick PCR精製キット(Qiagen)を用いてPCR精製した。
【0199】
ブチノールエステラーゼ遺伝子をベクター中に平滑末端とEcoRI切断部位を用いてライゲートした。反応混合物(20μl)は、20ngのベクター、120ngのインサートおよび2UのT4DNAリガーゼ(Roche Diagnostics GmbH)を含んだ。反応混合物を最初に室温で1時間インキュベートし、次いで一晩冷蔵庫内でインキュベートした。得られるプラスミドをpASK-IBA7エステラーゼと名付けた。
【0200】
c)第1のブチノールエステラーゼ突然変異体ライブラリーの調製:
ブチノールエステラーゼ遺伝子を最初にエラープローンPCR(95℃ 2分間;1サイクル//95℃ 30秒、59℃ 45秒、72℃ 3分間;40サイクル//72℃ 10分間)を用いて増幅した。次のプライマー:
フォワード プライマー:
5’-CGACAAAAATCTAGATAACGAGGGCAA-3’ (配列番号13)
リバース プライマー:
5’-TTCACTTCACAGGTCAAGCTTAGTTAG-3’ (配列番号14).
を用いた。
【0201】
反応混合物(50μl)は、20pmolのフォワードプライマー、20pmolのリバースプライマー、0.2mMの各dNTP(Roche Diagnostics GmbH)、100ngのプラスミドpASK-IBA7エステラーゼ、0.02% DMSO、0.1mM MnCl2および2.5UのTaq DNAポリメラーゼ(Qiagen)を含んだ。ゲル精製(QIAquickゲル抽出キット、Qiagen)の後、4μgのDNA産物を40UのEcoRI(New England Biolabs GmbH、Frankfurt、Germany)と30UのXbaI(New England Biolabs GmbH)により二重消化した。産物をPCR精製(QIAquick PCR-精製 キット; Qiagen)した後、EcoRIとXbaIで予め消化したpASK-IBA7 プラスミド中にライゲートした。突然変異体ライブラリーを大腸菌XL2ブルー(Stratagene、Amsterdam)中にTSS法[Chung CT, Niemela SL and Miller RH,1989. "One-step preparation of competent Escherichia coli: Transformation and Storage of Bacterial Cells in the Same Solution" Proc. Natl. Acad. Sci. U.S.A. 86: 2172-2175]を用いて形質転換した。
【0202】
d)ブチノールエステラーゼ突然変異体ライブラリーの発現:
LBampプレート(=Sambrook, J., Fritsch, E.F. and Maniatis, T. Molecular Cloning: A Laboratory Manual. 2nd Ed., Cold Spring Harbor Laboratory, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 1989によるLB培地+10g/l寒天および100μg/mアンピシリン)上のコロニーを、つまようじを用いて拾い、150μlのLBamp培地を含有する96ウエルのマイクロタイタープレートへ移した。マイクロプレート振とう機(Multitron II; Infors GmbH、Einsbach、Germany;37℃、900rpmおよび70%湿度)で培養した後、ほぼ5μlの各培養物をDuetzシステム (Kuehner, Birsfelden, Switzerland) を用いて、550μlのTB培地と100μg ml アンピシリン (Becton Dickinson GmbH)を含有する2mlウエルのプレートへ移した。クローンを、Multitron II振とう機(Infors GmbH;30℃および500rpmおよび70%湿度)で8時間培養した。タンパク質発現を、2.4μg/ml無水テトラサイクリン(IBA GmbH)を含む50μl TBを各ウエルに加えることにより誘導した。クローンをさらに6時間、Multitron II振とう機(Infors GmbH; 30℃および500 rpm)で8時間培養した。
【0203】
e)pH指示薬アッセイ (96-ウエルのフォーマット):
各ウエルからの50μl TB培養物を96-ウエルアッセイプレートに、Multimek 96-チャネル自動ピペッター(Beckman Coulter、Krefeld、Germany)を用いて移した。アッセイプレートの各ウエルにMultimek 96(Beckman Coulter)を用いて、100μlの基質溶液(25mM NaH2PO4 バッファー、pH 7.5、NaClにより調節してイオン強度154mM、0.05% Triton X-100、7.5μg/mlフルオレセイン-ナトリウムおよび285mM酪酸ブタ-3-イン-2-イルを含む)を加えることにより加水分解反応を開始した。使用前に5分間、基質溶液を激しく攪拌した。フルオレッセンス・シグナル(吸光485nm、発光520nm)の低下の反応速度を、Tecan Safire (Tecan GmbH、 Crailsheim、Germany)を用いて10分間観察した。活性を初期スロープから決定した。930クローンのスクリーニング後、クローン8H1が改良された突然変異体であると同定した。
【0204】
f)第2のブチノールエステラーゼ突然変異体ライブラリー:
第2のブチノールエステラーゼ突然変異体ライブラリーを、8H1を親遺伝子として用いたことを除くと、以上の記載と正確に同じ条件下で作製した。ライブラリーを以上のように発現し、スクリーニングした。1116クローンのスクリーニング後、クローン8C9が改良された突然変異体であると同定した。
【0205】
g)アミノ酸位置190および263における飽和突然変異誘発:
アミノ酸位置190および263の飽和突然変異誘発を、次の改変QuikChangeプロトコル(Stratagene)を用いて実施した。アミノ酸位置190に対しては次の突然変異誘発プライマー:
5’-GGCATCCCGATCATGNNNCTGCAAAGCCGCAAG-3’ (配列番号15)
5’-CTTGCGGCTTTGCAGNNNCATGATCGGGATGCC-3’ (配列番号16)
を用いた。アミノ酸位置263に対しては次の突然変異誘発プライマー:
5’-GGGCGCCAGGACGCGNNNCTCGATTTCCACAAG-3’ (配列番号17)
5’-CTTGTGGAAATCGAGNNNCGCGTCCTGGCGCCC-3’ (配列番号18)
を用いた。
【0206】
50μl PCR混合物(95℃30秒;1サイクル//95℃30秒、55℃1分、68℃4分45秒;18サイクル)は、20pmolの各プライマー、0.2mMの各dNTP(Roche Diagnostics GmbH)、50 ngのプラスミド8C9(出発DNA)および2.5 UのPfu Turbo DNAポリメラーゼ(Stratagene)の混合物を含んだ。PCRの後、メチル化および半メチル化親DNAを、40UのDpnI(New England Biolabs GmbH)により37℃にて2〜3 時間消化した。産物をPCR精製し(QIAquick PCR-精製 キット; Qiagen)、次いで、TSS 方法 [Chung CT、loc. cit]を用いて大腸菌 XL2ブルー(Stratagene)中に形質転換した。
【0207】
2つのライブラリーを先に記載したのと同じ方法で発現してスクリーニングした。それぞれのライブラリーについて186クローンをスクリーニングした後、2つのクローンを改良された突然変異体として同定した。これらをEst190-1B2およびEst263-2D6と名付けた。
【0208】
h)振とうフラスコでのタンパク質発現
一晩培養物を5 mlのLBamp 培地中でMultitron-II振とう機(Infors GmbH; 37℃、250 rpm)を用いて新しく形質転換したコロニーを拾うことにより増殖した。100mlのTBamp培地に800μlの前記一晩培養物を接種し、Multitron-II振とう機(Infors GmbH;37℃、200rpm)中で培養した。0.5〜0.6のOD600値に達すると、タンパク質発現を25μlの無水テトラサイクリン(DMF中の0.2 mg/mlのストック溶液)を加えることにより誘導した。培養物をさらに12時間増殖した(Multitron-II振とう機、Infors GmbH; 30℃、200 rpm)。細胞を遠心分離(3220 gおよび4℃にて30 分間)により取り除き、湿細胞塊を測定した。細胞ペレットを-20℃にて保存して、さらなる特徴付けを行った。
【0209】
i)pHスタットを用いるタンパク質特徴付け:
ブチノールエステラーゼの加水分解活性を、pHスタット(716 DMS Titrino、Deutsche Metrohm GmbH & Co.、Filderstadt、Germany)を用いて測定した。
【0210】
細胞ペレットを最初に、0.1g湿細胞塊/mlの細胞濃度を得るのに好適な容量のリン酸ナトリウムバッファー(25mMリン酸二水素ナトリウムバッファー、pH 7.5、NaClにより調節してイオン強度154mM)に再懸濁した。リン酸ナトリウムバッファー(25mMリン酸二水素ナトリウムバッファー、pH7.5、NaClにより調節してイオン強度154mM)、0.05% TritonX-100および350mM酪酸ブタ-3-イン-2-イルを含む20 mlの基質溶液を調製した。基質溶液を使用前に5分間激しく攪拌した。
【0211】
加水分解反応の開始前に、基質溶液(20 ml)のpHを1N NaOHによりpH 7.5に調節した。0.01gの湿細胞塊を加えることにより、反応を開始した。7.5のpHを、バイオ変換中、0.1N NaOHを用いる滴定により維持した。加水分解活性はNaOHの滴定速度と相関があった(単位時間当たり加えた容量)。
【0212】
結果を図4に示す。次表に掲げた酵素と突然変異体を試験した。アミノ酸配列と核酸配列中の特別な突然変異に関する情報をこの表から同様に見出すことができる。
【表4】
【受託番号】
【0213】
DSM13176
【技術分野】
【0001】
本発明は、エステラーゼ活性を有する新規タンパク質、その機能的等価体および突然変異体、それらをコードする核酸配列、発現カセット、ベクターおよび組換え微生物;前記タンパク質を調製する方法;ならびに有機エステルの酵素性の、特に、エナンチオ選択的酵素性の、エステル加水分解またはエステル交換を行うためのそれらの使用に関する。
【背景技術】
【0002】
エステラーゼやリパーゼは光学活性有機化合物を合成する工業的方法で使用できる加水分解酵素であって、高い基質特異性を特徴とする。これらの酵素はセリンプロテアーゼと同様の作用機序により、アシル基を例えばカルボニル基のような求核基に転移したり、エステル結合を加水分解で切断できる。エステラーゼ、リパーゼおよびセリンプロテアーゼは触媒三つ組残基(triad)(すなわち、Ser、HisおよびAspのアミノ酸からなる配列モチーフ)を共有し、前記カルボニル基の炭素原子は活性Serによる求核攻撃を受け、他の2つのアミノ酸が参加して電荷分布を形成する。エステラーゼとリパーゼはまた、チオエーテルのチオ基または活性化アミンなどの他の求核基にアシル基を転移することもできる。
【0003】
リパーゼは長鎖グリセロールエステルを加水分解し、表面活性化により特徴づけられる、すなわち、活性部位が脂質基質の存在下でのみ利用可能になる。リパーゼは非水性有機溶媒中で安定しており、反応速度論的ラセミ体分割を行う多数の工業的方法に利用されており、すなわち、一つのエナンチオマーを他のエナンチオマーより実質的に速く変換する。このエナンチオマーはその後、物理化学的性質の相異によって反応溶液から得ることができる。
【0004】
Nakamura (非特許文献1)は、市販のリパーゼ(Amano AK、AHおよびPS;Amano Pharmaceuticals Co. Ltd.)を使って、疎水性溶媒中のエステル交換による1-アルキン-3-オールのラセミ体分割を記載している。この反応においては、エナンチオ選択性がアシル供与体の鎖長とともに増加し、また、立体的に大きな残基(クロロアセテート、ビニルベンゾエート)は反応に悪影響を及ぼす。Yang(非特許文献2)はCandida antarctica由来のリパーゼBを触媒として用いて、ビニルエステルとエステル交換することによる、光学活性酸のエナンチオ選択的製法を記載している。この場合、エチルエステルは明らかに低い反応速度と選択率をもたらす。Burkholderia plantarii(Pseudomonas plantariiまたはglumae)DSM 6535から単離されたリパーゼは、メトキシ酢酸エチルを使ってラセミ体アミンをエナンチオ選択的にアシル化するのに利用されている(非特許文献3)。
【0005】
エステラーゼはエステル結合の形成および分解(すなわち、正反応および逆反応)をエナンチオ選択的に触媒する。光学活性アルコールを得るには、エステル交換反応でビニルエステルを使用することが好ましい。何故ならば、このエステルのアルコール官能基はアルデヒドやケトンへの互変異性化のため変換後にはもはや利用されず、従って逆反応を回避できるからである。リパーゼとは対照的に、エステラーゼは表面活性化されることなく、比較的短い鎖長の有機化合物も変換する。さまざまな基質特異性を示すエステラーゼが種々の生物から単離されている。
【0006】
例えば、Pseudocardia thermophila FERM-BP-6275由来のエステラーゼは、光学活性クロマン酢酸エステルの加水分解に利用される(EP-A-0 892 044)。
【0007】
Bacillus acidocaldarius由来のエステラーゼは、低いエナンチオ選択性で、狭い基質範囲からのエステルを加水分解する(非特許文献4)。
【0008】
Aspergillus由来のアシラーゼ1は、無極性有機溶媒中でビニルエステルを用いてエステル交換することにより第二級アルコールを得るために用いられ、これは短い側鎖をもつ第二級アルコールを変換するのに好適である(非特許文献5)。Pseudomonas fluorescens DSM 50 106から、膜結合型のラクトン特異的エステラーゼがクローニングされており(非特許文献6)、また、大腸菌Q突然変異体からはアセチルエステラーゼがクローニングされている(非特許文献7)。しかし、これらの2種のエステラーゼのエナンチオ選択性および基質特異性は詳細に研究されていない。Rhodococcus sp. NCBM 11216は4種類のエステラーゼ、RR1〜RR4を発現するが、これらは異なる特異性をもっている。ナフトールと酸からのエステル合成の場合、RR1とRR2は炭素鎖の短い酸のほうを優先し、一方RR3とRR4は比較的長い炭素鎖と立体的に比較的大きい残基をもつ酸を特異的に変換する(非特許文献8)。
【0009】
しかし最近まで、短い炭素鎖をもつ光学活性アルコール、酸またはエステルのような小有機分子を調製するための、広い基質範囲を持ち、高いエナンチオ選択性を有しかつ工業プロセスに利用できるエステラーゼは入手できなかった。
【0010】
特許文献1は初めて、エステラーゼ活性を有する有用なタンパク質を記載し、このタンパク質を、広範囲の光学活性エステルをエナンチオ選択的に加水分解できるエステラーゼとして言及した。さらに具体的に、この特許は510個のアミノ酸を含むタンパク質(配列番号2)およびそのコード配列(配列番号1)を記載している。
【0011】
本発明の目的は、さらに、場合によっては活性が最適化された、上記特性の少なくとも1つを有するエステラーゼを提供することである。
【先行技術文献】
【特許文献】
【0012】
【特許文献1】WO-A-02/18560
【非特許文献】
【0013】
【非特許文献1】Nakamura, K. et al., Tetrahedron; Asymmetry 9, (1999), 4429-4439
【非特許文献2】Yang, H. et al., J. Org. Chem. 64, (1999), 1709-1712
【非特許文献3】Balkenhohl, F. et al., J. prakt. Chem. 339, (1997), 381-384
【非特許文献4】Manco, G. et al., Biochem. J. 332, (1998), 203-212
【非特許文献5】Faraldos, J. et al., Synlett 4, (1997), 367-370
【非特許文献6】Khalameyzer, V. et al., Appl. and Environ. Microbiol. 65(2), (1999), 477-482
【非特許文献7】Peist, R. et al., J. Bacteriol. 179, (1997), 7679-7686
【非特許文献8】Gudelj, M. et al., J. Mol. Cat. B, Enzymatic 5, (1998), 261-266
【発明の概要】
【0014】
前記目的は、驚くべきことに、エステラーゼ活性を有するタンパク質またはその機能的に等価なタンパク質であって、前記タンパク質が510個未満のアミノ酸の全長をもつポリペプチド鎖を有しかつ前記鎖が少なくとも1つの配列番号3に記載のアミノ酸部分配列を含む前記タンパク質を提供することにより達成できた。
【図面の簡単な説明】
【0015】
【図1】図1は、ブチノールIエステラーゼのアミノ酸部分配列とPseudomonas fluorescens由来のラクトン特異的エステラーゼの部分配列との配列アラインメントを示す。Query:本発明のクローンLU2898の部分配列。Sbjct:P. fluorescens 酵素の部分配列、(受託番号:O87637)。
【図2】図2は、LU2898に対するクローニングチャートを図解する。
【図3】図3は、LU2898由来のブチノールエステラーゼ遺伝子とERGOデータベースとの比較を示す。高相同性領域を黒色で示す。右側のテキストカラムはERGOからの注釈であり、対応するe値を示す;e値はチャンスに基づく配列相同性存在の確率である。ブチノールエステラーゼ遺伝子のPstI切断部位の位置を示す。比較のために、Ps. glumae、LU2023由来の非組み換え酵素のアミノ酸配列を示す。
【図4】図4は、長さ335個のアミノ酸の配列を有する本発明のエステラーゼ(クローンLU11147由来)および活性が増強されたそれ由来の突然変異体の酵素活性を説明する。
【発明を実施するための形態】
【0016】
好ましい実施形態
本発明の第一の主題は、エステラーゼ活性を有するタンパク質またはその機能的に等価なタンパク質であって、前記タンパク質が510個未満のアミノ酸の全長をもつポリペプチド鎖を有しかつ前記鎖が少なくとも1つの配列番号3に記載の部分配列を含む前記タンパク質に関するものであり、前記タンパク質は好ましくは、参照基質としてのエステル、酪酸ブタ-3-イン-2-イルの切断により特徴付ることができるエステラーゼ活性を持つ。
【0017】
本発明のタンパク質は特にさらに、少なくとも1つのさらなる配列番号4、5または6に記載のアミノ酸部分配列を含んでもよい。
【0018】
配列番号3、4、5または6に記載の前記アミノ酸部分配列は、次の通り規定される(それぞれ、アミノ酸の一文字記号で示し、それぞれの第1アミノ酸は特有のアミノ末端に対応する)
a)FIETLGLERPVLVGHSLGGAIALAVGLDYPER(配列番号3)、
b)IALIAPLTHTETEP(配列番号4)、
c)GGGMMGLRPEAFYAASSDLV(配列番号5)、および
d)AIDAIFAPEPV(配列番号6)
本発明のタンパク質は特に、全長450個未満、例えば300〜445個のアミノ酸(例えば350個未満のアミノ酸、特に300〜345個、特に約330〜340個、とりわけ335個のアミノ酸)を有するポリペプチド鎖を含む。
【0019】
特に強調しうるのは、配列番号8に記載のアミノ酸配列を含むタンパク質である。
【0020】
本発明はさらに、これら新規の末端切断されたエステラーゼの、活性が増強された突然変異体、および同様に調製されたWO-A-02/18560に開示されかつ約510個のアミノ酸およびその機能的等価体を含むエステラーゼの突然変異体にも関する。
【0021】
本発明は特に、配列番号2または8のアミノ酸配列領域12〜20、185〜195および258〜268のいずれかに少なくとも1つの機能性突然変異を有する、とりわけ、配列番号2または8のアミノ酸配列の16、190および263位のいずれかに少なくとも1つの機能性突然変異を有するエステラーゼ突然変異体に関する。
【0022】
かかる突然変異の例は、限定されるものでないが、単独でまたはいずれかの組合わせで存在する次のアミノ酸置換:Leu16Pro、Ile190Thr、Ile190ArgおよびIle263Valである。
【0023】
本発明のタンパク質はさらに、例えば約60kDa以下、例えば56kDa以下または55.5kDa以下の計算分子量を有するポリペプチド鎖である。例えば、510個以下のアミノ酸をもつ末端切断したブチノールエステラーゼは、例えば、約56kDa〜20kDa、例えば約55.5〜30kDaまたは55.5〜35kDaまたは55.5〜40kDaまたは40〜30kDa、または55.5〜45kDaまたは38〜34kDaまたは36.5〜35.5kDaの範囲の分子量を有しうる。
【0024】
配列番号2によるタンパク質の突然変異体は、好ましくは約60〜40kDa、または56〜50kDaまたは55.5〜54kDaの範囲の計算分子量を有する。
【0025】
配列番号8によるタンパク質の突然変異体は、好ましくは約38〜34kDa、37〜35kDaまたは36.5〜35.5kDaの範囲の計算分子量を有する。
【0026】
好ましいタンパク質は受託番号DSM13176(DSMZに1999年12月2日寄託した)をもつPseudomonas glumae(Burkholderia plantariiとも呼ぶ)LU2023、および、適宜、その後の突然変異から得ることができる。
【0027】
エステラーゼ活性を有する本発明のタンパク質、機能的等価体および突然変異体はさらに次の反応:
a)式I:
R1-COO-R2 (I)
[式中、R1は直鎖または分枝鎖の、任意に一置換または多置換されたC1〜C10-アルキル、C2〜C10-アルケニル、C2〜C10-アルキニルであり、そしてR2は直鎖または分枝鎖の、任意に一置換または多置換されたC1〜C10-アルキル、C2〜C10-アルケニル、C2〜C10-アルキニル、C7〜C15-アラルキル、または単環または多環の、任意に一置換または多置換された芳香族基であり、そしてR1および/またはR2は少なくとも1個の不斉炭素を含む]で表わされる光学活性エステルのエナンチオ選択的加水分解;および
b)式Iのエステルと式II:
R2-OH (II)
[式中、R2は上記の意味の1つを有し、任意に、少なくとも1個の不斉炭素を有する]で表わされる光学活性アルコールとのエナンチオ選択的エステル交換
の両反応のうちの少なくとも1つの反応を触媒する。
【0028】
好適なC1〜C10-アルキル基の例として挙げることができるのは、1〜10個の炭素をもつ直鎖または分枝鎖の基、例えばメチル、エチル、イソプロピルまたはn-プロピル、n-、イソ-、sec-またはtert-ブチル、n-ペンチルまたはイソペンチル;さらにn-ヘキシル、n-ヘプチル、n-オクチル、n-ノニル、n-デシル、およびまた、それらの単分枝または多分枝した鎖の類似体である。
【0029】
好適なC2〜C10-アルケニル基の例は、2〜10個の炭素をもつ前記アルキル基の単不飽和または多不飽和類似体であり、この類似体は好ましくは1つまたは2つの炭素-炭素二重結合を有し、この結合は炭素鎖のいずれの位置にあってもよい。
【0030】
好適なC2〜C10-アルキニル基の例は、2〜10個の炭素をもつ前記アルキル基の単不飽和または多不飽和類似体であり、この類似体は好ましくは1つまたは2つの炭素-炭素三重結合を有し、この結合は炭素鎖のいずれの位置にあってもよい。
【0031】
C7〜C15-アラルキルは好ましくはフェニル-C1〜C5-アルキルまたはナフチル-C1〜C5-アルキルである。
【0032】
単環または多環の、任意に一置換または多置換の芳香族基の例は、C1〜C5-アルキル、例えばメチル、エチル、イソプロピルもしくはn-プロピル、n-、イソ-、sec-またはtert-ブチル、n-ペンチルもしくはイソペンチル;ヒドロキシ、メルカプト、アミノ、ニトロまたはハロ、例えばF、Br、Clのなかから選択される1、2または3個の同一または異なる置換基により置換されたフェニルおよびナフチルである。
【0033】
式Iのエステルは、例えば、直鎖または分枝鎖の、任意に単不飽和または多不飽和、任意に置換されたC1〜C11-モノカルボン酸から誘導される。飽和酸の例として挙げることができるのは、例えば蟻酸、酢酸、プロピオン酸およびn-酪酸およびイソ酪酸、n-吉草酸およびイソ吉草酸、カプロン酸、ヘプタン酸、カプリル酸、ペラルゴン酸、カプリン酸、ウンデカン酸;単不飽和酸、例えばアクリル酸、クロトン酸;および二不飽和酸、例えばソルビン酸である。もし酸が二重結合を含めば、cis型とtrans型の両方が存在しうる。
【0034】
本発明はさらに、以上規定したタンパク質、機能的等価体または突然変異体をコードするポリヌクレオチド、および前記ポリヌクレオチドの機能的等価体、それに相補的なポリヌクレオチド、およびそれとハイブリダイズしうる核酸配列に関する。
【0035】
本発明は特に、配列番号7に記載の核酸配列中の少なくとも30個の連続ヌクレオチド残基のヌクレオチド配列を含むポリヌクレオチドに関する。
【0036】
本発明はさらに、配列番号8のアミノ酸263位に対応する領域のコドンがGTTおよびGTCのなかから選択されるポリヌクレオチドに関する。
【0037】
本発明はさらに、少なくとも1つの、以上に規定されかつ少なくとも1つの調節核酸配列と機能しうる形で連結されたポリヌクレオチドを含む発現カセットに関する。
【0038】
本発明はさらに、真核生物または原核生物宿主を形質転換するための、以上規定したポリヌクレオチドまたは前記発現カセットを含む組換えベクターに関する。
【0039】
本発明はさらに、以上規定したタンパク質を調製する方法であって、前記タンパク質を内因的に産生する微生物または以上規定したベクターを用いて形質転換した微生物を培養するステップ、および前記タンパク質を培養物から単離するステップを含んでなる前記方法に関する。
【0040】
これに関する好適な方法は、受託番号DSM13176をもつ微生物Pseudomonas glumae(Burkholderia plantarii)LU2023またはそれから誘導された微生物を用いる。
【0041】
本発明はまた、以上規定した方法により得られる、前記タンパク質およびその機能的等価体または突然変異体に関する。
【0042】
本発明はさらに、受託番号DSM13176をもつPseudomonas glumae(Burkholderia plantarii)LU2023およびその変異体および突然変異体に関する。
【0043】
本発明はさらに、以上規定したベクターを担持する微生物に関する。
【0044】
本発明はさらに、以上規定したタンパク質(機能的等価体および突然変異体を含む)を用いるエナンチオ選択的エステル加水分解法であって、
a)前記タンパク質を、式Iで表される光学活性エステルの立体異性体混合物と接触させるステップ;および
b)前記立体異性体のいずれかの立体選択的加水分解により生成する光学活性化合物および/または加水分解されないエステルエナンチオマーを反応媒体から取得するステップを含んでなる前記方法に関する。
【0045】
本発明はさらに、エナンチオ選択的エステル交換法であって、
a)式IIで表される光学活性アルコールの立体異性体混合物を、前記タンパク質(機能的等価体および突然変異体を含む)の存在下に、式Iで表されるエステルと接触させ、未反応のアルコール立体異性体を反応媒体から取得するステップ;または
b)式Iで表される光学活性エステルの立体異性体混合物を、前記タンパク質(機能的等価体および突然変異体を含む)の存在下に、式IIで表されるアルコールと接触させ、前記エステルに含まれる光学活性アルコールの立体異性体を反応媒体から取得するステップを含んでなる前記方法に関する。
【0046】
本方法の特別な変形によれば、エステル交換に用いる光学活性アルコールに対するアシル化剤はビニルエステルである。
【0047】
本発明は特に、用いる反応媒体が有機溶媒である前記方法に関する。
【0048】
2.一般用語の説明
「エステラーゼ」または「ブチノールエステラーゼ」または「ブチノールIエステラーゼ」は、本明細書に記載した酵素性変換の少なくとも1つ、特に少なくともカルボン酸、特に酪酸の参照ブチノールエステル、例えば酪酸ブタ-3イン-2イルなどのブチノール酪酸エステルの切断を触媒する酵素である。
【0049】
具体的に開示した配列から「誘導された」またはそれと「相同的な」配列、例えば、誘導されたアミノ酸配列または核酸配列は、本発明によれば、特に断らない限り、出発配列と少なくとも80%または少なくとも90%、特に91%、92%、93%、94%、95%、96%、97%、98%および99%、同一である配列を意味する。
【0050】
二つの核酸間の「同一性」は、それぞれの場合に、全核酸長にわたるヌクレオチドの同一性、特にInformax(USA)のソフトウエアであるVector NTI Suite 7.1を使い、クラスタルの方法 (Higgins DG, Sharp PM. Fast and sensitive multiple sequence alignments on a microcomputer. Comput Appl. Biosci. 1989 Apr;5(2):151-1)を用いて、次のパラメーター:
マルチプル・アラインメント・パラメーター:
ギャップ・オープニング・ペナルティ 10
ギャップ・エキステンション・ペナルティ 10
ギャップ分離ペナルティ範囲 8
ギャップ分離ペナルティ オフ
アラインメント遅れに対する%同一性 40
残基特異的ギャップ オフ
親水残基ギャップ オフ
トランジション重み付け(Transition weighing) 0
ペアワイズ・アラインメントパラメーター:
FASTアルゴリズム オン
K-tuple サイズ 1
ギャップペナルティ 3
ウインドウサイズ 5
最良対角数(Number of best diagonals) 5
を設定して比較することにより計算した同一性を意味する。
【0051】
3. 本発明のさらなる実施形態
3.1 本発明によるタンパク質
本発明は、具体的に開示したエステラーゼ活性を有するタンパク質および酵素に限定されるものでなく、その機能的等価体も包含する。
【0052】
本発明の目的にとって、具体的に開示した酵素の「機能的等価体」 または 類似体は、前記酵素と異なるポリペプチドで、さらに、例えば、加水分解活性などの所望の生物学的活性を有するポリペプチドである。
【0053】
「機能的等価体」とは例えば、使用したエステラーゼ活性アッセイにおける活性が、配列番号8に記載のアミノ酸配列を含む酵素の活性より、少なくとも1%、例えば少なくとも10%または20%、例えば少なくとも50%または75%または90%、高いかまたは低い酵素を意味する。さらに、機能的等価体は好ましくはpH 4〜10の間で安定であり、最適pHは有利なのはpH 5〜9、例えば6〜8であり、かつ最適温度は15℃〜80℃ または 20℃〜70℃の範囲にある。
【0054】
エステラーゼ活性は様々な公知のアッセイによって検出できる。限定されるものでないが、例えば、標準化した条件下(例えば20mM基質、10mMリン酸バッファー、pH7.4、T=20℃など)で、酪酸ブタ-3イン-2イルなどのブチノール酪酸エステルのような参照基質を用いるアッセイを挙げることができる。
【0055】
本発明によれば「機能的等価体」はまた、特に、以上記載したアミノ酸配列の少なくとも1つの配列位置において具体的に記載したものと異なるアミノ酸を有するが、それにも関らず、以上記載した生物学的活性の1つを有する「突然変異体」も意味する。「機能的等価体」は従って、1個以上のアミノ酸付加、置換、欠失および/または逆転によって得られる突然変異体を包含し、本発明の特性プロファイルを有する突然変異体を生じる限り、前記改変がいずれの配列位置で起こってもよい。機能的等価は特にまた、突然変異体と未改変のポリペプチドの間の反応性パターンが定性的に一致する場合、すなわち、例えば、同じ基質が異なる速度で変換される場合にも存在する。好適なアミノ酸置換の例を、次表に総括する。
【0056】
本発明による突然変異を行うのに好適な個々の配列領域の具体的な例は既に先に規定している、すなわち、配列番号2または8からのアミノ酸配列領域12〜20、185〜195および258〜268、特に配列番号2または8のアミノ酸配列の16、190および263位である。
【0057】
好適なアミノ酸置換の具体的な例はLeu16Pro、Ile190Thr、Ile190ArgおよびIle263Valである。本発明の教示を知れば、当業者はこれらのさらなる改変を容易に提供しうる。
【0058】
以上の意味での「機能的等価体」はまた、記載したポリペプチドの「前駆体」およびまた、前記ポリペプチドの「機能的誘導体」および「塩」でもある。
【0059】
この場合、「前駆体」は所望の生物学的活性を有するまたは有しないポリペプチドの天然または合成の前駆体である。
【0060】
表現「塩」は、本発明によるタンパク質分子のカルボキシル基の塩ならびにアミノ基の酸付加塩を意味する。カルボキシル基の塩は、公知の方法で生産することができ、無機塩、例えばナトリウム、カルシウム、アンモニウム、鉄および亜鉛塩、および有機塩基の塩、例えばアミン、例えばトリエタノールアミン、アルギニン、リシン、ピペリジンなどを含む。酸付加塩、例えば鉱酸との塩、例えば塩酸または硫酸および有機酸との塩、例えば酢酸およびシュウ酸も本発明に包含される。
【0061】
本発明によるポリペプチドの「機能的誘導体」はまた、機能的アミノ酸側鎖基でまたはこれらのN末端またはC末端で公知の技法を用いて作製することができる。かかる誘導体には、例えばカルボン酸基の脂肪族エステル、アンモニアとのまたは一級もしくは二級アミンとの反応により得られるカルボン酸基のアミド;アシル基との反応により得られる遊離アミノ基のN-アシル誘導体;またはアシル基との反応により生成する遊離ヒドロキシ基のO-アシル誘導体が含まれる。
【0062】
「機能的等価体」は勿論、他生物から得られるポリペプチドならびに天然に存在する変異体も包含する。例えば、相同配列の領域を配列比較によって確立できるし、また、等価酵素を本発明の具体的なパラメーターに基づいて確認できる可能性がある。
【0063】
同様に、「機能的等価体」は、例えば所望の生物学的機能を有する、本発明によるポリペプチドの断片、好ましくは、個々のドメインまたは配列モチーフも含む。
【0064】
「機能的等価体」はさらに融合タンパク質であって、上記のポリペプチド配列またはそれから誘導される機能的等価体の1つおよび機能的N-またはC-末端結合に(すなわち、融合タンパク質部分の相互機能を実質的に損なうことなく)少なくとも1つのさらなる機能的に異なる異種配列を有する融合タンパク質である。かかる異種配列の例は、限定するものではないが、例えばシグナルペプチド、ヒスチジンアンカーまたは酵素である。
【0065】
本発明に含まれる「機能的等価体」はまた、具体的に開示したタンパク質の相同体である。これらは、Pearson and Lipman, Proc. Natl. Acad, Sci. (USA) 85(8), 1988, 2444-2448のアルゴリズムに従って計算して、具体的に開示したアミノ酸配列の1つと少なくとも60%、好ましくは少なくとも75%、特に少なくとも85%、例えば90、91、92、93、94、95、96、97、98または99%の相同性を有する。本発明による相同性ポリペプチドのパーセント相同性は、特に、本明細書に具体的に記載したアミノ酸配列の1つの全長に基づくアミノ酸残基の同一性パーセントを意味する。
【0066】
タンパク質のグリコシル化されている可能性がある場合、本発明による「機能的等価体」は、脱グリコシル化型またはグリコシル化型で先に示したタンパク質、ならびにグリコシル化パターンを変えることにより得られる改変型を包含する。
【0067】
本発明によるタンパク質またはポリペプチドの相同体は、突然変異誘発により、例えば、タンパク質の点突然変異、伸長または末端切断により作製することができる。
【0068】
本発明によるタンパク質の相同体は、突然変異体、例えば末端切断突然変異体のコンビナトリアルライブラリーをスクリーニングすることにより同定できる。例えば、タンパク質変異体の多様性に富んだライブラリーは、合成オリゴヌクレオチドの混合物の酵素的連結による、核酸レベルでのコンビナトリアル突然変異誘発により作製することができる。縮重オリゴヌクレオチド配列から可能性のある相同体のライブラリーを作製するために使用できる方法は数多くある。縮重遺伝子配列の化学合成を自動DNA合成機で実施し、合成遺伝子を次いで好適な発現ベクターに連結することができる。遺伝子の縮重セットを使用すると、1つの混合物中に、可能性のあるタンパク質配列の所望セットをコードする全ての配列を提供することが可能となる。縮重オリゴヌクレオチドを合成する方法は当業者に公知である(例えば Narang, S.A. (1983) Tetrahedron 39:3;Itakura et al. (1984) Annu. Rev. Biochem. 53:323;Itakura et al., (1984) Science 198:1056;Ike et al. (1983) Nucleic Acids Res. 11:477)。
【0069】
従来の技術分野において、点突然変異または末端切断により作製されたコンビナトリアルライブラリーの遺伝子産物をスクリーニングする技法、および選択した特性をもつ遺伝子産物に対するcDNAライブラリーをスクリーニングする技法はいくつかが公知である。これらの技法を、本発明による相同体のコンビナトリアル突然変異誘発により作製した遺伝子ライブラリーの迅速スクリーニングに適合させることができる。大型の遺伝子ライブラリーをハイスループット分析によりスクリーニングするために最も広く用いられる技法は、遺伝子ライブラリーを複製可能な発現ベクターにクローニングするステップ;得られるベクターライブラリーで好適な細胞を形質転換するステップ;所望の活性の検出により、検出した産物の遺伝子をコードするベクターの単離が容易になる条件で、コンビナトリアル遺伝子を発現させるステップを含んでなる。ライブラリー中の機能的突然変異体の頻度を高める技法である帰納的集合突然変異誘発(REM)を、相同体を同定するためのスクリーニング試験と組み合わせて使用してもよい(Arkin and Yourvan (1992) PNAS 89:7811-7815;Delgrave et al. (1993) Protein Engineering 6(3):327-331)
本発明のエステラーゼの機能的等価体のさらなる例には、例えば、配列番号3、4、5または6から誘導された少なくとも1つの部分配列であって、1個以上のアミノ酸が具体的に示した部分配列と比較して置換、欠失、逆転または付加されていて、かつ天然のタンパク質のエステラーゼ活性と±90%または±50%以下、好ましくは±30%以下しか異ならないエステラーゼ活性を有する前記部分配列が含まれる。
【0070】
本発明によるエステラーゼは特にPseudomonas glumae LU2023、受託番号DSM 13176から得ることができる。さらに菌株変異体を、例えばPseudomonas glumae LU8093から出発して選択により、例えば最少培地プレートで、唯一の炭素源として酢酸エチルフェニルを用いて培養する方法により得ることができる。
【0071】
3.2 コード核酸配列
本発明はさらに、エステラーゼ活性をもつ酵素をコードする核酸配列に関する。配列番号7による配列を含む核酸配列;または配列番号8によるアミノ酸配列から誘導される核酸配列が好ましい。
【0072】
本明細書に記載の全ての核酸配列(1本鎖および2本鎖のDNAおよびRNA配列、例えばcDNAおよびmRNA)は、公知の方法でヌクレオチド・ビルディングブロックから化学合成により、例えば、二重へリックスの個々のオーバーラップする、相補的な核酸ビルディングブロックの断片縮合により作製することができる。オリゴヌクレオチドの化学合成は、例えば、公知の方法で、ホスホアミダイト法により行うことができる(Voet, Voet, 2nd edition, Wiley Press, New York, pages 896-897)。合成オリゴヌクレオチドの蓄積およびDNAポリメラーゼのクレノウ断片の方法によるギャップの充填およびライゲーション反応ならびに一般的クローニング技法が、Sambrook et al. (1989), Molecular Cloning: A laboratory manual, Cold Spring Harbor Laboratory Pressに記載されている。
【0073】
本発明はまた、上記ポリペプチドおよびそれらの機能的等価体のいずれかをコードする核酸配列(1本鎖および2本鎖のDNAおよびRNA配列、例えばcDNAおよびmRNA)にも関するものであり、これらの核酸配列は、例えば、人工のヌクレオチド類似体を用いて得ることができる。
【0074】
本発明は、本発明のポリペプチドもしくはタンパク質またはその生物学的に活性なセグメントをコードする単離された核酸分子と、例えば、本発明のコード核酸を同定または増幅するためのハイブリダイゼーションプローブまたはプライマーとして使用することができる核酸断片との両方に関する。
【0075】
本発明による核酸分子は、さらに、コード遺伝子領域の3'および/または5'末端から得られる非翻訳配列を含んでもよい。
【0076】
本発明はさらに、具体的に記載したヌクレオチド配列またはそのセグメントに相補的である核酸分子に関する。
【0077】
本発明のヌクレオチド配列によって、他の細胞タイプおよび生物に含まれる相同配列を同定および/またはクローニングするために有用なプローブおよびプライマーを作製することが可能となる。かかるプローブおよびプライマーは通常、「ストリンジェントな」条件(下記参照)下で、本発明の核酸配列のセンス鎖または対応するアンチセンス鎖の少なくとも約12個、好ましくは、少なくとも約25個、例えば、約40、50または75個の連続ヌクレオチドにハイブリダイズするヌクレオチド配列領域を含む。
【0078】
「単離された」核酸分子は、該核酸の天然源中に存在する他の核酸分子から分離されており、さらに、それが組換え法により産生されたのであれば、他の細胞性物質や培地を実質的に含まず、また、それが化学合成されたのであれば、化学的前駆物質や他の化学薬品を実質的に含まないものでありうる。
【0079】
本発明の核酸分子は、標準的な分子生物学の手法および本発明により提供される配列情報を用いることにより単離することができる。例えば、cDNAは好適なcDNAライブラリーから、ハイブリダイゼーションプローブとして具体的に開示した完全な配列またはそのセグメントの1つと標準的なハイブリダイゼーション技法とを用いて単離することができる(例えば、Sambrook, J., Fritsch, E. F. und Maniatis, T. Molecular Cloning: A Laboratory Manual. 2. Aufl., CoId Spring Harbor Laboratory, CoId Spring Harbor Laboratory Press, CoId Spring Harbor, NY, 1989に記載のように行う)。さらに、開示した配列またはそのセグメントの1つを含む核酸分子は、この配列に基づいて構築されたオリゴヌクレオチドプライマーを用いてポリメラーゼ連鎖反応を行うことにより単離することができる。このようにして増幅した核酸を好適なベクターにクローニングして、DNA配列解析により特徴づけることができる。本発明のオリゴヌクレオチドはまた、標準的な合成方法により、例えば自動DNA合成機を使って、作製することもできる。
【0080】
配列番号7またはその誘導体、相同体またはこれらの配列の部分などの、本発明による核酸配列は、例えばゲノムまたはcDNAライブラリーを介して、他の細菌から、例えば通常のハイブリダイゼーション技法またはPCR技法により単離することができる。これらのDNA配列は標準条件で本発明による配列とハイブリダイズする。
【0081】
「ハイブリダイズする」は、ポリヌクレオチドまたはオリゴヌクレオチドがほとんど相補的な配列と標準条件で結合できるが、非特異的結合が非相補的なパートナーの間ではこの条件で起こらないことを意味する。このためには、配列が90〜100%相補的でありうる。特異的にお互いに結合できる相補的配列の特性は、例えばノーザンブロットもしくはサザンブロットに、またはPCRもしくはRT-PCRのプライマー結合に利用される。
【0082】
短い保存領域のオリゴヌクレオチドはハイブリダイゼーション用に使うと有利である。しかし、本発明による核酸の、より長い断片または完全な配列をハイブリダイゼーション用に使うことも可能である。これらの標準条件は使用する核酸(オリゴヌクレオチド、より長い断片または完全な配列)に応じてまたはハイブリダイゼーションに使用する核酸の型(DNAもしくはRNA)に応じて変化する。例えば、DNA:DNA ハイブリッドに対する融点は、同じ長さのDNA:RNA ハイブリッドの融点よりほぼ10℃低い。
【0083】
例えば、特定の核酸に応じて、標準条件は42〜58℃の温度、0.1〜5×SSC(1×SSC=0.15M NaCl、15mMクエン酸ナトリウム、pH 7.2)の濃度のバッファ水溶液中、またはさらに50%ホルムアミドが存在する条件を意味し、例えば42℃、5×SSC、50%ホルムアミド中である。有利なDNA:DNAハイブリッドに対するハイブリダイゼーション条件は0.1×SSCおよび約20℃〜45℃の温度、好ましくは約30℃〜45℃の温度である。DNA:RNA ハイブリッドに対する有利なハイブリダイゼーション条件は0.1×SSCおよび約30℃〜55℃、好ましくは約45℃〜55℃の温度である。ハイブリダイゼーション用のこれらの記載温度は、長さほぼ100ヌクレオチドおよびG+C含量50%の核酸に対してホルムアミドの非存在のもとで計算した融点値の例である。DNAハイブリダイゼーションの実験条件は、関連する遺伝学教科書、例えば、Sambrook et al., "Molecular Cloning”, Cold Spring Harbor Laboratory, 1989に記載されており、当業者に公知である式を用いて、例えば、核酸の長さ、ハイブリッドの型またはG+C含量に応じて計算することができる。当業者はハイブリダイゼーションについてのさらなる情報を次の教科書から得ることができる:Ausubel et al. (eds), 1985, Current Protocols in Molecular Biology, John Wiley & Sons, New York;Hames and Higgins (eds), 1985, Nucleic Acids Hybridization: A Practical Approach, IRL Press at Oxford University Press, Oxford;Brown (ed), 1991, Essential Molecular Biology: A Practical Approach, IRL Press at Oxford University Press, Oxford。
【0084】
「ハイブリダイゼーション」は特にストリンジェントな条件のもとで行うことができる。かかるハイブリダイゼーション条件は、例えば、Sambrook, J., Fritsch, E.F., Maniatis, T., in: Molecular Cloning (A Laboratory Manual), 2nd edition, Cold Spring Harbor Laboratory Press, 1989, pages 9.31-9.57、またはCurrent Protocols in Molecular Biology, John Wiley & Sons, N.Y. (1989), 6.3.1-6.3.6に記載されている。
【0085】
「ストリンジェントな」ハイブリダイゼーション条件は、特に:42℃にて一晩、50%ホルムアミド、5×SSC(750mM NaCl、75mMクエン酸三ナトリウム)、50mMリン酸ナトリウム(pH7.6)、5×Denhardt溶液、10%デキストラン硫酸塩および20g/ml変性剪断サケ精子DNAからなる溶液中のインキュベーションと、次いで0.1×SSCによる65℃におけるフィルターの洗浄を意味する。
【0086】
本発明はまた、具体的に開示したまたは誘導可能な核酸配列の誘導体に関する。
【0087】
従って、本発明によるさらなる核酸配列を、例えば、配列番号7から誘導することができ、この核酸配列は単一のまたは複数のヌクレオチドの付加、置換、挿入または欠失により元のものとは異なるが、なお所望の特性のプロファイルをもつポリペプチドをコードすることができる。
【0088】
本発明はまた、「サイレントな」突然変異を含むかまたは具体的に記載した配列と比較して、特別な供給源または宿主生物のコドン利用、ならびに天然の変異体、例えばそのスプライシング変異体または対立遺伝子変異体によって改変されている核酸配列を包含する。
【0089】
本発明はまた、保存ヌクレオチド置換により得ることができる(すなわち、問題のアミノ酸を同じ電荷、サイズ、極性および/または溶解度のアミノ酸により置換した)配列に関する。
【0090】
本発明はまた、具体的に開示した核酸から配列多形により誘導された分子に関する。これらの遺伝的多形は天然変異によって一集団内の個体間で存在しうる。これらの自然変異は通常、遺伝子のヌクレオチド配列に1〜5%のばらつきを生じる。
【0091】
本発明による配列番号7をもつ核酸配列の誘導体とは、例えば、全配列領域にわたって、誘導されたアミノ酸のレベルで少なくとも60%相同性、好ましくは少なくとも80%相同性、非常に特に好ましくは少なくとも90%相同性(アミノ酸レベルの相同性については、先にポリペプチドについて記載した詳細な説明を参照されたい)を有する対立遺伝子変異体を意味する。相同性が配列の部分領域にわたってより高いと有利でありうる。
【0092】
さらに、誘導体はまた、本発明による核酸配列の、特に配列番号7の相同体、例えば、真菌もしくは細菌の相同体、末端切断配列、コードおよび非コードDNA配列の1本鎖DNAもしくはRNAであるとも解釈すべきである。例えば、配列番号7の相同体は、配列番号7に与えられた全DNA領域にわたって、DNAレベルで、少なくとも40%、好ましくは少なくとも60%、特に好ましくは少なくとも70%、非常に好ましくは少なくとも80%の相同性を有する。
【0093】
さらに、誘導体は、例えば、プロモーターとの融合体であると解釈すべきである。記載したヌクレオチド配列の上流にあるプロモーターを、少なくとも1つのヌクレオチド交換、少なくとも1つの挿入、逆転および/または欠失により改変し、しかし、プロモーターの機能または効力を損なわないようにすることができる。さらに、プロモーターの効力を、プロモーターの配列を変えることにより向上することができるし、または、プロモーターをより有効なプロモーター(異なる属の生物のものであってもよい)により完全に置き換えることもできる。
【0094】
3.3 本発明による構築物
本発明はさらに、調節核酸配列の遺伝子制御のもとで、本発明によるポリペプチドをコードする核酸配列を含む発現構築物;ならびにこれらの発現構築物の少なくとも1つを含むベクターに関する。
【0095】
「発現ユニット」は、本発明によれば、本明細書に規定したプロモーターを含むものであって、発現すべき核酸もしくは遺伝子との機能的連結に従って、この核酸もしくはこの遺伝子の発現、転写および翻訳を調節する、発現活性をもつ核酸を意味する。従って、この意味で「調節核酸配列」とも呼ばれる。プロモーターだけでなく、他の調節配列、例えばエンハンサーが存在してもよい。
【0096】
「発現カセット」または「発現構築物」は、本発明によれば、発現すべき核酸または発現すべき遺伝子と機能しうる形で結び付けた発現ユニットを意味する。単なる発現ユニットとは対照的に、発現カセットは従って、転写および翻訳を調節する核酸配列だけでなく、転写および翻訳の結果、タンパク質として発現すべき核酸配列も含むものである。
【0097】
用語「発現」または「過剰発現」は、本発明の関係では、対応するDNAによりコードされた微生物中の1以上の酵素の細胞内活性の産生または増加を記載する。これについては、例えば、生物に遺伝子を挿入し、存在する遺伝子を他の遺伝子と置き換え、遺伝子のコピー数を増加し、強いプロモーターを使用し、または高活性をもつ対応する酵素をコードする遺伝子を使用することが可能であり、また任意にこれらの対策を組合わせることもできる。
【0098】
好ましくは、本発明によるかかる構築物はそれぞれのコード配列の5'上流にプロモーター配列、および3'下流にターミネーター配列、ならびに任意にさらなる通常の調節配列を含むものであり、それぞれの調節配列はコード配列と機能しうる形で連結されている。
【0099】
「プロモーター」、「プロモーター活性をもつ核酸」または「プロモーター配列」は、本発明によれば、転写すべき核酸と機能しうる形で連結されて、この核酸の転写を調節する核酸を意味する。
【0100】
「機能的」または「機能しうる」連結は、この文脈では、例えば、プロモーター活性をもつ核酸、および転写すべき核酸配列、および任意にさらなる調節配列(例えば、核酸の転写を確実なものにする核酸配列)、および例えばターミネーターの1つの配列を、それぞれの調節配列が核酸配列の転写中にその機能を遂行できる方式で配置することを意味する。これは必ずしも化学的な意味での直接連結を必要としない。エンハンサー配列などの遺伝子調節配列はまた、例えば、より離れた位置からまたは他のDNA分子からですら、標的配列に作用することができる。転写すべき核酸配列をプロモーター配列の下流(すなわち3'末端)に置いて、2つの配列がお互いに共有結合している配置が好ましい。プロモーター配列と遺伝子組換えで発現される核酸配列の間の距離は200塩基対未満、または100塩基対未満、または50塩基対未満でありうる。
【0101】
プロモーターおよびターミネーターとは別に、挙げることができる他の調節配列の例は標的化配列、エンハンサー、ポリアデニル化シグナル、選択マーカー、増幅シグナル、複製起点などである。好適な調節配列は、例えばGoeddel, Gene Expression Technology: Methods in Enzymology 185, Academic Press, San Diego, CA (1990)に記載されている。
【0102】
本発明による核酸構築物は、特に、配列番号7の配列またはその誘導体および相同体、ならびに配列番号8から誘導しうる核酸配列を含むものであり、これは、遺伝子発現を制御する、例えば増加するための1以上の調節シグナルと機能しうる形でまたは機能的に連結されていることが有利である。
【0103】
これらの調節配列に加えて、これらの配列の自然調節がさらに現行の構造遺伝子の上流に存在することがあり、任意に遺伝的に改変して、自然調節がスイッチオフされて遺伝子の発現を増加させることができる。核酸構築物は、しかしまた、もっとシンプルな設計であってもよく、すなわち、さらなる調節シグナルをコード配列(例えば配列番号7またはその相同体)の上流に挿入せず、また、それを調節する自然のプロモーターを取り除かなくてもよい。代わりに、自然の調節配列を突然変異させて調節がもはや起こらないようにして、遺伝子発現を増加するようにしてもよい。
【0104】
好ましい核酸構築物はまた、有利な方法として、核酸配列の発現の増加を可能にするプロモーターと機能しうる形で連結された前記エンハンサー配列を1つ以上含む。他の調節配列またはターミネーターなどのさらなる有利な配列をまた、DNA配列の3'末端に挿入することもできる。本発明による核酸の1以上のコピーが構築物に含まれてもよい。構築物はまた、任意に構築物を選択するために、抗生物質耐性または栄養要求性を補完しうる遺伝子などの他のマーカーを含んでもよい。
【0105】
プロモーターに含まれる好適な調節配列の例は、グラム陰性菌で有利に使われるcos、tac、trp、tet、trp-tet、lpp、lac、lpp-lac、lacIq、T7、T5、T3、gal、trc、ara、rhaP(rhaPBAD)SP6、λ-PRまたはλ-PLプロモーターである。他の有利な調節配列は、例えば、グラム陽性菌プロモーターのamyおよびSPO2、酵母または真菌プロモーターのADC1、MFα、AC、P-60、CYC1、GAPDH、TEF、rp28、ADHである。人工プロモーターも調節に用いることができる。
【0106】
発現については、核酸構築物を宿主生物に、有利な方法としては、宿主での遺伝子の最適な発現を可能にするベクター、例えばプラスミドまたはファージ中に挿入する。ベクターは、プラスミドおよびファージだけでなく、当業者に公知の全ての他のベクター、すなわち、例えばウイルス(SV40、CMV、バキュロウイルスおよびアデノウイルスなど)、トランスポゾン、ISエレメント、ファスミド、コスミド、および直鎖または環状DNAも意味すると解釈すべきである。これらのベクターは宿主生物中で自律的に複製できるかまたは染色体を介して複製できる。これらのベクターは本発明のさらなる実施形態を示す。
【0107】
好適なプラスミドは、例えば、大腸菌ではpLG338、pACYC184、pBR322、pUC18、pUC19、pKC30、pRep4、pHS1、pKK223-3、pDHE19.2、pHS2、pPLc236、pMBL24、pLG200、pUR290、pIN-III113-B1、λgt11またはpBdCI;ストレプトマイセス菌ではpIJ101、pIJ364、pIJ702またはpIJ361;バシラス菌ではpUB110、pC194またはpBD214;コリネバクテリウム菌ではpSA77またはpAJ667;真菌ではpALS1、pIL2またはpBB116;酵母では2alphaM、pAG-1、YEp6、YEp13またはpEMBLYe23、あるいは植物ではpLGV23、pGHlac+、pBIN19、pAK2004またはpDH51である。以上記載したプラスミドは可能性のあるプラスミドから選択したわずかなものである。その他のプラスミドが当業者に周知であり、それらは例えば、書物のCloning Vectors (Eds. Pouwels P.H. ら Elsevier, Amsterdam-New York-Oxford, 1985, ISBN 0 444 904018)に見出しうる。
【0108】
さらなるベクターの実施形態においては、本発明による核酸構築物または本発明による核酸を含むベクターを、有利な方法としては直鎖DNAの形態で微生物に導入して、宿主生物のゲノム中に異種または相同的遺伝子組換えを介して組み込むことができる。この直鎖DNAはプラスミドなどの直線化ベクター、または本発明による核酸構築物、または核酸そのものであってもよい。
【0109】
生物において異種遺伝子を最適に発現させるためには、前記生物で使われる特定のコドン使用頻度に従って核酸配列を改変することが有利である。コドン使用頻度は、問題の生物の他の公知の遺伝子のコンピューター評価に基づいて容易に決定することができる。
【0110】
本発明による発現カセットの作製は、好適なプロモーターと好適なコードヌクレオチド配列およびターミネーターシグナルまたはポリアデニル化シグナルとの融合に基づく。このために使われる通常の組換えおよびクローニング技法は、例えば、T. Maniatis, E.F. Fritsch and J. Sambrook, Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY (1989)、ならびにT.J. Silhavy, M.L. Berman and L.W. Enquist, Experiments with Gene Fusions, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY (1984)、およびAusubel, F.M. et al., Current Protocols in Molecular Biology, Greene Publishing Assoc. and Wiley Interscience (1987)に記載されている。
【0111】
組換え核酸構築物または遺伝子構築物は、有利な方法として、好適な宿主生物における発現用の宿主特異的ベクターに挿入し、これにより遺伝子が宿主中で最適に発現されることを可能にする。ベクターは当業者に周知であり、例えば、"Cloning Vectors" (Pouwels P.H. et al., Publ. Elsevier, Amsterdam-New York-Oxford, 1985)に見出すことができる。
【0112】
3.4 本発明によって利用できる微生物
状況に応じて、用語「微生物」は出発微生物(野生型)または遺伝的に改変した組換え微生物またはそれらの両方を意味する。
【0113】
本発明によるベクターを用いることによって、例えば、少なくとも1つの本発明によるベクターにより形質転換されかつ本発明によるポリペプチドの産生に利用できる組換え微生物を作製することができる。有利な方法として、以上記載した本発明による組換え構築物を好適な宿主系に挿入して発現させる。好ましくは、特別な発現系における記載した核酸の発現を確保するために、当業者が手慣れた通常のクローニングおよびトランスフェクション法、例えば共沈降、プロトプラスト融合、エレクトロポレーション、レトロウイルスのトランスフェクションなどを用いる。好適な系は、例えばCurrent Protocols in Molecular Biology, F. Ausubel et al., Publ. Wiley Interscience, New York 1997、またはSambrook et al. Molecular Cloning: A Laboratory Manual. 2nd edition, Cold Spring Harbor Laboratory, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 1989に記載されている。
【0114】
原則として、全ての原核生物のまたは真核生物は本発明による核酸または核酸構築物用の組換え宿主生物とみなしうる。細菌、真菌または酵母などの微生物を宿主生物として用いることは有利である。グラム陽性菌もしくはグラム陰性菌、好ましくは腸内細菌科、シュードモナス科、リゾビウム科、ストレプトミセス科またはノカルジア科のファミリーの細菌、とりわけ好ましくはエシェリキア属、シュードモナス属、ストレプトマイセス属、ノカルジア属、ブルコルデリア属、サルモネラ属、アグロバクター属またはロドコッカス属の細菌を用いることが有利である。大腸菌の属および種はとりわけ特に好ましい。他の有利な細菌も次のグループ中に見出しうる:α-プロテオバクテリア、β-プロテオバクテリアまたはγ-プロテオバクテリア。
【0115】
本発明による宿主生物は、前記エステラーゼ活性をもつ酵素をコードする本発明に記載の核酸配列、核酸構築物またはベクターの少なくとも1つを含むことが好ましい。
【0116】
本発明の方法に利用する生物を、当業者の手慣れた方法で、宿主生物に応じて、生育または増殖する。通常、微生物を、一般に糖類の形態の炭素源、一般に窒素の有機源(酵母エキスなど)または塩(硫酸アンモニウムなど)の形態の窒素源、微量元素(鉄、マンガンおよびマグネシウム塩ならびに、適宜、ビタミン)を含む液培地中で、0℃〜100℃、好ましくは10℃〜60℃の温度で酸素曝気して増殖する。栄養液体培地のpHは増殖の間、固定した値、すなわち調節したまたは調節しない値に維持してもよい。増殖はバッチ式、半バッチ式または連続的に行ってもよい。栄養分は発酵の開始時に補給してもまたは、その後、半連続的または連続的に補給してもよい。ケトンは増殖中に直接加えてもよいし、または、有利な方法として、増殖後に加えてもよい。酵素は、実施例に記載の方法により生物から単離してもよいし、または粗抽出物として反応用に用いてもよい。
【0117】
3.5 エステラーゼの組換え生産
本発明はさらに、本発明によるポリペプチドまたはその機能的な生物学的活性断片を遺伝子組換え生産する方法であって、ポリペプチド産生微生物を培養し、適宜、ポリペプチドの発現を誘導し、そしてポリペプチドを培養物から単離することによる前記方法に関する。ポリペプチドはまた、所望であれば、この方法で工業的規模で生産することもできる。
【0118】
本発明によって作製する微生物は、連続的にまたはバッチ式(バッチ法、または1回供給バッチ法もしくは繰り返し供給バッチ法)で培養することができる。公知の培養方法の総括は、Chmiel(Bioprocesstechnik 1. Einfuehrung in die Bioverfahrenstechnik (Gustav Fischer Verlag, Stuttgart, 1991))の教科書またはStorhas(Bioreaktoren und periphere Einrichtungen (Vieweg Verlag, Braunschweig/Wiesbaden, 1994))の教科書に見出しうる。
【0119】
使用する培地は、適当な方式で、特定の菌株の要件を満たすものでなければならない。様々な微生物に対する培地の説明は、米国細菌学協会(the American Society)のハンドブック"Manual of Methods for General Bacteriology"(Washington D. C., USA, 1981)に記載されている。
【0120】
本発明に使用しうるこれらの培地は一般に1種以上の炭素源、窒素源、無機塩、ビタミンおよび/または微量元素を含む。
【0121】
好ましい炭素源は糖類、例えば単糖類、二糖類または多糖類である。非常に優れた炭素源は例えばグルコース、フルクトース、マンノース、ガラクトース、リボース、ソルボース、リブロース、ラクトース、マルトース、スクロース、ラフィノース、デンプンまたはセルロースである。糖類はまた、複雑な化合物、例えば糖蜜または糖精製から得られる他の副産物を介して培地に加えることもできる。様々な炭素源の混合物を加えることも有利でありうる。他の可能性のある炭素源は油類および脂肪類(ダイズ油、ヒマワリ油、ピーナッツ油およびヤシ油など)、脂肪酸類(パルミチン酸、ステアリン酸またはリノール酸など)、アルコール類(グリセロール、メタノールまたはエタノールなど)および有機酸類(酢酸または乳酸など)である。
【0122】
窒素源は通常、有機または無機窒素化合物またはこれらの化合物を含む物質である。窒素源の例としては、アンモニアガスまたはアンモニウム塩(硫酸アンモニウム、塩化アンモニウム、リン酸アンモニウム、炭酸アンモニウムまたは硝酸アンモニウム、硝酸塩など)、尿素、アミノ酸類または複雑な窒素源(トウモロコシ浸漬液、ダイズ粉、ダイズタンパク質、酵母エキス、肉エキスなど)などが挙げられる。窒素源は別々にまたは混合物として使用することができる。
【0123】
培地に存在してもよい無機塩化合物にはカルシウム、マグネシウム、ナトリウム、コバルト、モリブデン、カリウム、マンガン、亜鉛、銅および鉄の塩化物、リン酸塩または硫酸塩が含まれる。
【0124】
無機硫黄を含有する化合物(硫酸塩、亜硫酸塩、ジチオニット、テトラチオナート、チオ硫酸塩、スルフィドなど)だけでなく、有機硫黄化合物(メルカプタンおよびチオールなど)を硫黄源として使用することができる。
【0125】
リン酸、リン酸二水素カリウムまたはリン酸水素二カリウムまたは対応するナトリウム含有塩をリン源として使用することができる。
【0126】
溶液中の金属イオンを保持するためにキレート剤を培地に加えてもよい。とりわけ好適なキレート剤にはジヒドロフェノール(カテコールまたはプロトカテキュ酸など)、または有機酸(クエン酸など)が含まれる。
【0127】
本発明による発酵培地はまた、通常、他の成長因子(ビタミンなど)または成長促進物質(例えば、ビオチン、リボフラビン、チアミン、葉酸、ニコチン酸、パントテン酸およびピリドキシンなど)も含む。成長因子と塩はしばしば培地の複雑な成分(酵母エキス、糖蜜、トウモロコシ浸漬液など)に由来する。さらに好適な前駆体を培地に加えることができる。培地中の化合物の正確な組成は特定の実験に強く依存するものであり、それぞれの具体的な事例に対して個々に決定しなければならない。培地最適化に関する情報は、教科書"Applied Microbiol. Physiology, A Practical Approach"(Publ. P.M. Rhodes, P.F. Stanbury, IRL Press (1997) p. 53-73, ISBN 0 19 963577 3)に見出しうる。増殖培地はまた、商業的供給者(例えば、Standard 1(Merck)またはBHI(脳心臓注入液、DIFCO)など)から得ることもできる。
【0128】
全ての培地成分を、加熱(20分間、1.5bar、121℃にて)によりまたは滅菌濾過により無菌化する。成分を一緒にまたは、必要であれば、別々に無菌化してもよい。培地の全ての成分は増殖の出発時に存在してもよいし、または、任意に連続的にもしくはバッチ式供給により加えてもよい。
【0129】
培養の温度は通常、15℃〜45℃、好ましくは25℃〜40℃であり、実験中、一定に保持してもよいしまたは変化させてもよい。培地のpHは5〜8.5の範囲、好ましくはほぼ7.0にすべきである。増殖のためのpHは増殖中に、塩基化合物(水酸化ナトリウム、水酸化カリウム、アンモニアまたはアンモニア水など)または酸化合物(リン酸または硫酸など)を加えることにより制御することができる。消泡剤、脂肪酸ポリグリコールエステルを発泡を制御するために使用してもよい。プラスミドの安定性を維持するために、選択作用をもつ好適な物質、例えば抗生物質を培地に加えてもよい。酸素または酸素含有気体混合物、例えば大気を培地に供給して好気性条件を維持する。培養温度は通常20℃〜45℃である。培養は、所望の産物の最大量が得られるまで続ける。これは通常、10時間〜160時間以内に達成される。
【0130】
次に発酵ブロスをさらに処理する。要件に応じて、バイオマスを発酵ブロスから分離技法(例えば、遠心分離、濾過、デカントまたはこれらの方法の組合わせなど)により完全にまたは部分的に取り出してもよいしまたは発酵ブロス中に残してもよい。
【0131】
もしポリペプチドが培地中に分泌されなければ、細胞を破壊して、溶菌液からタンパク質を単離する公知の技法により産物を取得してもよい。細胞を、任意に、高周波超音波により、高圧により(例えば、フレンチプレス細胞破砕機で)、浸透圧溶解により、界面活性剤、溶解性酵素もしくは有機溶媒の作用により、ホモジナイザーを用いて、または掲げた方法のいくつかを組合わせることにより破壊することができる。
【0132】
ポリペプチドは公知のクロマトグラフィ法(モレキュラーシーブ・クロマトグラフィ(ゲル濾過)、Q-セファロース・クロマトグラフィ、イオン交換クロマトグラフィおよび疎水性クロマトグラフィなど)を用いて、および他の通常の方法(限界濾過、結晶化、塩析、透析および未変性ゲル電気泳動など)により精製することができる。好適な方法は例えば、Cooper, F.G., Biochemische Arbeitsmethoden, Verlag Walter de Gruyter, Berlin, New YorkまたはScopes, R., Protein Purification, Springer Verlag, New York, Heidelberg, Berlinに記載されている。
【0133】
組換えタンパク質を単離するには、例えば精製をより簡単にするために利用しうる改変したポリペプチドまたは融合タンパク質をコードする、規定したヌクレオチド配列だけcDNAの伸長したベクター系またはオリゴヌクレオチドを用いることが有利でありうる。この種類の好適な改変は、例えば、アンカーとして機能するいわゆる「タグ」、例えば、ヘキサ-ヒスチジンアンカーとして公知の改変、または抗原として抗体が認識しうるエピトープである(Harlow, E. and Lane, D., 1988, Antibodies: A Laboratory Manual. Cold Spring Harbor (N.Y.) Pressに記載されている)。これらのアンカーは、タンパク質と固体支持体、例えばクロマトグラフィカラムに充填する、例えばポリマーマトリックスとの接着を可能にするか、またはマイクロタイタープレート上でまたはいくつかの他の支持体上で利用することができる。
【0134】
同時に、これらのアンカーはまた、タンパク質の認識用にも使うことができる。タンパク質を認識するために、通常のマーカー、例えば、蛍光染料および酵素マーカー(基質との反応後、検出可能な反応生成物を生じる)、または放射性マーカーを、単独でもしくはタンパク質の誘導体化用のアンカーと組合わせて利用することもできる。
【0135】
3.6 本発明による、エステラーゼの用途
本発明はまた、エステラーゼを用いるエナンチオ選択的エステル加水分解法にも関し、この方法は、本発明のエステラーゼを、式Iで表される光学活性エステルの立体異性体混合物と接触させ、2種の立体異性体のいずれかの立体選択的加水分解により生成する光学活性化合物および/または加水分解されないエステルエナンチオマーを反応媒体から取得する。しかし、該エステラーゼは、光学的に活性でない式Iのエステルを加水分解することも可能である。
【0136】
本発明はさらに、エナンチオ選択的エステル交換法にも関し、この方法は、式IIで表される光学活性アルコールの立体異性体混合物を、本発明のエステラーゼの存在下で、式Iで表されるエステルと接触させ、未反応のアルコール立体異性体を反応媒体から取得するか、または、式Iで表される光学活性エステルの立体異性体混合物を、該エステラーゼの存在下で、式IIで表されるアルコールと接触させ、該エステルに含まれる光学活性アルコールの立体異性体を反応媒体から取得する。エステル交換法では、光学活性アルコールのアシル化剤としてビニルエステルを使用することが好ましい。このことは、ビニルエステルのアルコール官能基が変換後に互変異性化のため逆反応にもはや利用されないので、有利である。該エステラーゼはまた、エステルとアルコールのどちらも光学活性でないエステル交換法も触媒する。
【0137】
エステル加水分解の好ましい基質は、エタノール、プロパノール、ブタノールのエステル、そして特に好ましくは、ブチノールのカルボン酸(酢酸、プロピオン酸、酪酸、ペンタン酸、ヘキサン酸、ヘプタン酸、オクタン酸、乳酸、2-エチルヘキサン酸、3-メチル酪酸、メトキシ酢酸、2-メチルプロピオン酸、2-ブテン酸、3-クロロプロピオン酸、および2-メチルペンタン酸など)とのエステル(ブチノールエステル、1-メチルプロパ-2-イン-1-オールのエステル)である。特に好ましいのは、酪酸ブチニルとメチル酪酸ブチニルである。
【0138】
エステル交換に用いるのに好ましいアルコールはエタノール、プロパノールおよびブタノール、特に好ましいのはブチノールである。
【0139】
エステル交換に用いるのに好ましいエステルは、ビニルエステル、例えば、酢酸ビニル、プロピオン酸ビニルおよび酪酸ビニルである。
【0140】
以上の方法で用いる反応媒体は有機溶媒であり、例えば、アルカン類、エーテル類、トルエン、ジオキサン、メチルイソブチルケトン、メチルtert-ブチルエーテル(MTBE)などである。エステル加水分解においては、使用したバッファー溶液と有機溶媒(例えば、MTBEおよびヘプタンまたはトルエン)からなる混合溶液を使用してもよい。
【0141】
ラセミ化合物の分割(すなわち、エナンチオ選択性)および反応速度は、酸部分の大きさと疎水性により影響を受けることがある。
【0142】
本発明による反応は、好ましくは、室温にてpH 6〜9で、特に好ましくは、pH 7.0〜7.4で実施する。エステラーゼは、単離されたまたは精製された酵素の形態で、エステラーゼを発現する微生物の細胞として、前記微生物の培養上清、細胞溶解物または抽出物として、あるいは固定化されたエステラーゼとして使用される。反応生成物は当業者に公知の化学的または物理的分離方法により反応溶液から単離することができる。エステラーゼは膜濾過によって反応混合物から単離することができる。
【0143】
エステラーゼは、ポリアクリルアミド、アルギン酸またはカラギーナンを用いて固定化することが可能である。またエステラーゼを、公知の方法を用いて共有結合によりまたは吸着により好適な担体と結合することもできる。エステラーゼをケイソウ土への凍結乾燥により、または硫酸アンモニウム沈降により固定化することが好ましい。
【0144】
(実施例)
特に断らない限り、本発明の範囲内で行ったクローニングのステップ、例えば、制限酵素切断、アガロースゲル電気泳動、DNA断片の精製、核酸のニトロセルロースおよびナイロン膜への転移、DNA断片の連結、微生物の形質転換、微生物の培養、ファージの増殖、および組換えDNAの配列分析は、Sambrookら(1989、前記引用)に記載の通り行った。
【実施例1】
【0145】
エステラーゼを発現するPseudomonas glumae突然変異体の選択
このスクリーニングの開始点は、リパーゼ産生株であるPseudomonas (Burkholderia) glumae LU 8093であった。この菌株から得られたリパーゼにより、いくつかの興味深い反応を実施することができる(Balkenhohl, F. et al., J. prakt. Chem. 339, (1997), 381-384)。しかし、乳酸エステル、メチル酪酸エステルおよびフェニル酢酸エステルはこのリパーゼの基質ではなく、この菌株はこれらのエステルをどうしても加水分解することができない。
【0146】
しかし、加水分解産物は炭素源として利用可能である。それ故に、上記エステルを加水分解して、その加水分解産物を炭素源として利用して生育する能力があるLU 8093の突然変異体を探した。新規のエステラーゼ活性を有する突然変異体は、したがって、上記エステルで生育させることによってその正体を現すに違いない。
【0147】
選択条件:LU 8093を培地上で16時間培養し、遠心分離により回収した。細胞を食塩水で2回洗浄した。106個の細胞を、唯一の炭素源として0.5または1.0g/lのフェニル酢酸エチル(EPA)を含む最少培地プレートにまいた。ところが、最初は全く生育しなかった。やっと4〜6日後に、単一のコロニーが確認できた。その数はそれ以後さらに増えた。
【0148】
このようにして得られたエステラーゼ陽性突然変異体から、LU 2023突然変異株を選択した。驚いたことに、この新規エステラーゼの活性は比較的小さい有機分子の選択的加水分解にも適していた。例えば、ブチノールエステルを選択的に加水分解することがわかった。
【実施例2】
【0149】
Pseudomonas glumae LU 2023の発酵
エステラーゼを得るために、Pseudomonas glumae LU 2023を14Lのスケールで培養し、活性バイオマスを収穫した。
【0150】
実験室で、Pseudomonas glumae LU 2023を、M12ミネラル塩培地と1g/LのEPAを含む寒天プレート上にストリーク接種し、28℃で36〜48時間インキュベートした。その後、このプレートは4℃で4週間保存した。
【0151】
この菌株の発酵は、Infors xxy 14 l発酵槽で実施した。前培養のため、250mlの培地に2〜3の白金耳を用いて接種し、200rpm、28℃で24時間インキュベートした。主培養は下記の条件下で行った:
温度 28℃
空気供給量 7 L/分
攪拌 600rpm
発酵実施時間 約24時間
内蔵pHおよびpO2の測定
前培養および主培養用の培地
Springer酵母自己分解物質65% 15 g/l
硫酸マグネシウム×7水 1.6 g/l
塩化カルシウム×2水 0.02 g/l
リン酸二水素カリウム 3.5 g/l
リン酸水素二カリウム 3.5 g/l
リン酸水素二アンモニウム 5 g/l
Pluriol P2000消泡剤 6 ml
上記の成分を脱イオン水に溶解し、この溶液を25%濃度のアンモニア水を用いてpH6.5に調整した。5 ml/lの微量元素溶液および2 g/lグルコースを別々に滅菌濾過した。
【0152】
培地を滅菌して完成した後、0.5 g/lのフェニル酢酸エチルを発酵槽に導入した。Pluriol P2000を添加して発酵中の泡の発生を防止した。発酵槽内のpO2が85%以上に再度増加したとき発酵を停止させた。次いで、発酵槽の内容物を15℃以下、約9000〜10,000gで遠心分離し、透明な上清を廃棄した。細胞塊は-16℃で凍結させた。
【実施例3】
【0153】
Pseudomonas glumae LU 2023から得たエステラーゼの精製
Pseudomonas glumae LU 2023細胞(100 ml、湿重量:50 g)を4℃、3000rpmでガラスビーズミル(100mlのガラスビーズ、直径:0.5mm)で溶解した。遠心分離(10,000rpm、30分)して、ガラスビーズを洗浄した後、上清(300ml)を塩化マンガン沈降(pH7〜7.5;最終濃度:50mM)に付した。もう一回遠心分離した後、上清をpH8.0に調整し、EDTAを50mMの濃度で添加した。この容量をQ−Sepharose(300ml)クロマトグラフィーで精製した。サンプルをアプライした後、カラムを50mMトリス/HClで洗浄した。対象となる画分を集めて、限外濾過(100 kDa)により濃縮した。モレキュラーシーブ・クロマトグラフィー(直径:5cm、高さ:90cm;材料:S−300)にかけて非特異的エステラーゼからブチノール加水分解性エステラーゼを分離した。得られた活性画分は濁っており、再度濃縮した。このエステラーゼは明らかに膜と結合していた。次いで、膜画分をまずプロテアーゼ(トリプシン、重量比:1:50から1:100)で消化した。この操作により、SDSポリアクリルアミドゲル電気泳動において2,3のバンドを別にすれば、膜画分から全タンパク質が消失した。活性は保存されていた。前記バンドを未変性ゲル電気泳動(0.04%のSDS)で互いに分離し、活性アッセイにより未変性ゲル中のエステラーゼを同定した。前記エステラーゼをゲルから溶出した後に、変性SDSポリアクリルアミドゲル電気泳動にかけたところ、きれいな1本のバンドとして現れた。
【0154】
このようにして精製したタンパク質は、PVDF膜へのブロッティングにより移行させてから配列決定するか、または、トリプシンで切断した後にペプチドを逆相HPLCで分離してから配列決定した。このタンパク質のアミノ末端はブロックしたので、トリプシン消化ペプチドのみを得た。様々なアミノ酸配列がAcinetobacter iwoffiiおよびPseudomonas putida由来のムコン酸エステルシクロイソメラーゼ(muconate cycloisomerase)EC 5.5.1.1に対して、また、Pseudomonas fluorescens由来のラクトンエステラーゼに対して弱い相同性を示した。配列AIDAIFAPEGVをもつペプチドはペクチンエステラーゼ(EC 3.1.1.11)に対する相同性を示した。
【0155】
図1は、本発明のアミノ酸部分配列と、Pseudomonas fluorescens由来のラクトン特異的エステラーゼの部分配列との、配列アラインメントを示す。
【実施例4】
【0156】
エステラーゼの固定化
様々な方法を固定化のために用いた。
【0157】
1. エステラーゼを、ケイソウ土の存在下にアセトンで沈澱させることにより実質的に不活性化した。25mgのタンパク質を3.5gのケイソウ土(Merck)と混合し、1.4Lのアセトン(-20℃)を10分間添加した。その後、エステラーゼをローディングした担体をG3ガラス吸引フィルターにより分離し、フィルター残留物を冷アセトンで洗浄して、乾燥させた。
【0158】
2. エステラーゼは、Accurel(Akzo)と結合しない。
【0159】
3. エステラーゼ(2.3単位/g、EPAアッセイ)を、凍結乾燥によりケイソウ土に固定させることができた。このためには、酵素溶液をケイソウ土と混合し、−80℃で凍結させた。続いて、この固形物質を凍結乾燥により乾燥した。
【0160】
4. エステラーゼ(454ミリ単位/g、EPAアッセイ)を、硫酸アンモニウム沈澱により固定化した。そのためには、酵素をケイソウ土の存在下に80%飽和の硫酸アンモニウムで沈澱させた。
【実施例5】
【0161】
Pseudomonas glumae LU 2023由来のエステラーゼを用いるラセミ体分割
手順(標準手法)
100単位のエステラーゼを、リン酸バッファー(200ml、10mM、pH7.4)中で20mmolのブチノール酪酸エステル(酪酸1-メチルプロパ-2-イニル)と攪拌しながら反応させた。pHを連続測定して、水酸化ナトリウム溶液の添加により約pH 7.4に維持した。表1に示した時間にサンプルを採取し、メチルt-ブチルエーテル(MTBE)で2回抽出し、有機相をGC(Chiraldex GTA)により分析した。エステラーゼを膜濾過により反応混合物から分離した。
【0162】
その濃度が増加すると、好ましくない方のエステルエナンチオマーの変換が増加した。約45分後、これは反応混合物中のS-ブチノールのeeの低下を招いた。生成物のeeは、約30〜40分後に84%(83〜97.9%)で最大値に達した。基質のeeは90分間で99%以上に増加した。ee(エナンチオマー過剰率)は、好ましい方の変換エナンチオマーの量(%)マイナス好ましくない方の変換エナンチオマーの量(%)として規定される。ほとんどの場合、これは光学純度に一致する。pHの低下は30分まで直線状であったが、約100分以後、pHの変化は無視しうる程度であった。
【0163】
抽出後、水相中の残留エステラーゼ活性はまだ50%ほど残っていた。
【表1】
【0164】
表1は、エステラーゼを用いてブチノール酪酸エステルを変換したときの時間依存的エナンチオマー過剰率を示す。Cahn、Prelog及びIngoldによるR/S変換に従うと、RおよびS立体配置が1つのキラル分子の2つのエナンチオマーを規定する。変換率は反応混合物中の変換されたエステルの比率である。
【実施例6】
【0165】
エステラーゼ特異性の、エステルの酸部分の大きさおよび疎水性/電荷に対する依存性
標準手法
100単位のエステラーゼを、リン酸バッファー(200ml、10mM、pH7.4)中で20mmolのブチノールエステルと攪拌しながら反応させた。pHを連続的に測定し、連続滴定によりpH 7.0に維持した。サンプルを採取し、メチルt-ブチルエーテル(MTBE)で2回抽出し、有機相をGC(Chiraldex GTA)により分析した。
【0166】
結果
ラセミ体分割の品質と反応速度は、酸部分の大きさおよび疎水性に依存した。ブチノールエステラーゼの最良の基質は、ブチノールの酪酸エステルとブチノールのメチル酪酸エステルであった。リパーゼはこれらの基質には不活性である。このことは、n-デカン酸ブチニルのような長鎖エステルにも当てはまる。
【表2】
【0167】
表2は、エステラーゼを用いてエステルを変換するときのエナンチオマー過剰率が、変換されるエステルの酸部分に依存することを示す。
【実施例7】
【0168】
エステラーゼを用いる有機媒体中でのエステル交換
10mmolのrac-ブチノールと5mmolの酪酸ビニルを50mlのメチルt-ブチルエーテル(MTBE)に溶解し、ケイソウ土に担持された9単位のブチノールIエステラーゼ(3.3g)と混合し、この混合物を室温で24時間振とうした。濾過後、溶媒を除去し、生成物の混合物をGCにより特徴づけた。
【0169】
47%の変換率で、(R)-ブチノール(18% ee)および(S)-ブチノールの酪酸エステル(45% ee)が残存した。
【0170】
メチルイソブチルケトン中では、(R)-ブチノール(16% ee)および(S)-ブチノールの酪酸エステル(52% ee)が43%の変換率で得られた。
【0171】
表3は、エステラーゼを用いてエステルを変換したときのエナンチオマー過剰率が、変換されるエステルの酸部分に依存することを示す。
【表3】
【実施例8】
【0172】
菌株LU2023の遺伝子ライブラリーの調製およびLU2898のクローニング
a)染色体DNAの取得:
LU2023(前記実施例1および2を参照)を100mlのFP培地(Becton Dickinson GmbH)で28℃および180rpmにて24時間増殖した。細胞を遠心分離(2000rpm/5分間)によって収穫し、5mlの溶解バッファー1(0.41Mスクロース、0.01M MgSO4*7H2O、50ml/L M12培地(10×conc.)、10ml/L 10%KH2PO4 pH 6.7、2.5 mg/mlリゾチーム[使用の直前に加える])に再懸濁し、そして37℃にてほぼ4時間インキュベートした。遠心分離(2000rpm/20分間)の後、得られるプロトプラストを、5mlのリゾチームを含まない溶解バッファー1で洗浄(2000rpm/20分間)し、10mM TRIS-HCl pH8.0に再懸濁した。
【0173】
もう1回10mM TRIS-HCl pH 8.0を用いて洗浄(3000rpm/5分間)した後、ペレットを4mlのTEバッファー(10mM TRIS-HCl、1mM EDTA、pH 8)に再懸濁し、それを0.5mlのSDS(10%w/v)および0.5mlのNaCl(5M)と混合した。この後、100μlのプロテイナーゼK溶液(200μg/ml)を加え、37℃にて16時間インキュベートした。次いで、TEバッファーを加えて、この溶液を10mlの最終容量とした。この溶液をフェノールと1:1で混合した。遠心分離(4000rpm/5分間)の後、上相を取り出してフェノール、クロロホルムおよびイソアミルアルコール(12:12:1)の混合物と混合した。上相を取り出して1容量のクロロホルム・イソアミルアルコール(24:1)と混合した。上相が透明になるまでこの手順を繰り返した。
【0174】
2容量のエタノールおよび1/50容量のCH3COONa(3M)を-20℃で加えることにより(時間:ほぼ30分間)、DNAを上相から沈降させ、遠心分離(12000rpm、30分間、4℃)によりペレット化して、TEバッファーに再懸濁した。RNase(1mlの20μg/ml溶液)はRNAを37℃で1時間以内に加水分解する。その溶液を次いでTEバッファーに対して4℃で3回それぞれ1時間透析する。それぞれ0.4mlのアリコートのDNAに、エタノール(2容量+1/3容量のLiCl、2M)を加え、-20℃で30分間のインキュベーションにより沈降させる。遠心分離(15000rpm、30分間、4℃)でペレット化したDNAを次いで乾燥した。0.4mlアリコートのDNAを0.5ml TEバッファーに再懸濁した。
【0175】
b)DNAの制限酵素切断とクローニング
1μgの染色体DNAを120U PstIを用いて37℃にて180分間消化した。0.4μgのpUC19(例えば New England Biolabs)を160UのPstI(37℃にて1時間)により制限酵素切断し、次いでアルカリホスファターゼ(37℃にて1時間+不活性化(65℃にて15分間))により脱リン酸した。DNA断片をラピッドライゲーションキット(Roche)を用いて連結した(2時間、室温にて)。ライゲーション混合物を、製造業者の情報に従って形質転換コンピテント大腸菌(Stratagene)中に形質転換した。細胞をIPTG/X-Gal FPプレート(100μg/mlアンピシリンを含有するFP培地)にストリーク接種し、37℃にて16時間インキュベートした。白色のコロニーをアンピシリンを含有する新しいFPプレートにストリーク接種した。
【0176】
c)遺伝子ライブラリーのスクリーニング
コロニーを、無菌ベルベットクッションを用いてWhatman濾紙に移した。フィルターを2.5mlのアッセイ溶液(10mM TRIS*HCl、pH7.5、0.01%ブロモクレゾール・パープル、0.1%ラセミエステル[rac-乳酸メチル(MEE)またはrac-メチル酪酸エチルまたはrac-フェニル酢酸エチル])中に置いた。エステラーゼ活性をもつ細胞のコロニーは青色から黄色へ5分間以内に変色する。試験した2900個のコロニーのなかで、強い変色を生じるものが1つあった。この菌株をLU2898と名付けた。図2はLU2898のクローニング計画を表す模式図である。
【実施例9】
【0177】
LU2898の組換え体DNAの配列分析
LU2898から得たプラスミドは7.7kbインサートを有する。大腸菌LU2898のブチノールエステラーゼ遺伝子を保有するプラスミドを完全に配列決定した。DNAデータベースの最初の検索は、加水分解酵素と相同性を有するインサートをもつオープンリーディングフレーム(位置1394〜2923)を生じた。
【0178】
ERGOデータベース(Integrated Genomics, Inc. 1999-2004, Chicago, USA)をLU2898ブチノールエステラーゼのDNA配列を用いて検索した。ブチノールエステラーゼ遺伝子の領域1384〜2397bpだけが公知の加水分解酵素と相同的であることを見出した。
【0179】
領域1384〜2397bpは明らかに所望のブチノールエステラーゼのDNAである。他の良い徴候は、セリンカルボキシルエステラーゼに典型的でありかつLU2023ブチノールエステラーゼのアミノ酸位置127〜131に位置するGXSXGモチーフである。
【0180】
LU2898ブチノールエステラーゼ遺伝子とB. cepacia 加水分解酵素の間の良い一致はインサートのほぼ位置2400で終わる。さらにリーディングフレームの下流で、NADPレダクターゼの3'末端と非常に明確な相同性が存在する。
【0181】
これらの結果を添付した図3に示す。図3はERGO分析の総括である。LU2898ブチノールエステラーゼの相同性の急激な移行はPstI切断部位と一致する。PstI制限酵素の切断部位は、選んだクローニング計画で予想されなかった。プラスミドライブラリーを調製するために、この酵素を用いてLU2023染色体のDNAを完全に切断した。完全な加水分解の後、PstIに対する内部認識部位はもう存在しないはずである。LU2898プラスミド中のPstI切断部位の発見は、前記PstI制限部位により作製される色々な遺伝子断片がプラスミド構築中にお互いに接続され、その結果、連結されてpUC19のPstI切断部位になったという事実により説明することができ、この事実は、ブチノールエステラーゼに割りつけられたリーディングフレームがおそらくPs. glumae LU2023由来の実際の遺伝子の一部分だけを表すことを意味する。ブチノールエステラーゼ遺伝子はPstI部位で破壊され、次いでNADPレダクターゼ遺伝子の断片と接続されたのである。
【0182】
この仮説はタンパク質配列決定データにより支持される。Ps. glumae LU2023ブチノールエステラーゼを精製した。該タンパク質のアミノ酸配列をEdman分解により決定した。完全配列は、いくつかのN-およびC-末端アミノ酸を除くと公知である。それはLU2898のDNAデータから誘導したブチノールエステラーゼの最初の部分からのアミノ酸配列に対応する。41.3kDaのブチノールエステラーゼの実験で決定した分子量は、組換えLU2898ブチノールエステラーゼ遺伝子のDNA配列から誘導した値より著しく低い。DNA配列データによると、ブチノールエステラーゼは55kDaの分子量を持つはずである。
【0183】
データベース分析の助けで、ブチノールエステラーゼはその全長がクローニングされていなかったという確証が見出された。LU2898から得たプラスミドはブチノールエステラーゼ遺伝子の3'末端を含んでいない。
【実施例10】
【0184】
末端切断された335AAブチノールエステラーゼ突然変異体を含むクローンLU11147の発見
次のPCRプライマーを設計するためにLU2898由来のプラスミドの配列データを利用した。
【0185】
HindIII
Breu0811: GGCGAGAAGCTTAGAAATCATGATCGTCCA (配列番号20)
XbaI
Breu0812: GGATCCTCTAGAGTCTCACTGCAGCGCGCC (配列番号21)
これらのオリゴヌクレオチドを用いてPCRを使い、以上記載した相同性をエステラーゼに対して有するLU2898由来のDNAを増幅した。
【0186】
PCR条件:
10*Taqポリメラーゼバッファー 5μl
プライマーBreu0811 125ng
プライマーBreu0812 125ng
dNTP(10mM) 2μl
LU11147由来のプラスミドDNA 50ng
DMSO 2.5μl、および
H20 50μ
Taq-DNAポリメラーゼ(Roche Diagnostics)0.5Uを加えてホットスタート。
【0187】
PCR温度プログラム:
5分間、95℃,
45秒、55℃,
3分間、72℃、(30サイクル)
30秒、95℃、
45秒、55℃,
10分間、72℃,
∞、4℃
HindIIIとXbaIによる制限酵素切断の後、PCR産物を、適当に調製したpUC19(例えばNew England Biolabs)中にライゲートした。
【0188】
この方法で調製したプラスミドベクターを次に大腸菌XL1ブルーを形質転換するのに用いた。ライゲーション混合物を形質転換コンピテント大腸菌(Stratagene)中に、製造業者の情報に従って形質転換した。細胞をIPTG/X-Gal FPプレート(100μg/mlアンピシリンを含有するFP培地)上にストリーク接種し、37℃にて16時間インキュベートした。プラスミドDNAを形質転換体から調製し(Birnboim et al.、 Nucleic Acids Research、1979、7、1513)、HindIIIとXbaIによる制御制限を用いたPCR産物の挿入の成功をチェックした。
【実施例11】
【0189】
末端切断された335AAブチノールエステラーゼの発現
次いでエステラーゼ遺伝子をベクターpDHE1650(WO-A-2004/050877に記載)中にNdeIとHindIII切断部位を用いてクローニングした。
【0190】
この目的で、エステラーゼ遺伝子を最初に、次のフォワードおよびリバースプライマー:
Breu1499 TATACATATGATCGTCCAACTGATCGCCATCGTG、
Breu1500 ATTTAAGCTTTTACTGCAGCGCGCCGGCCTGCGTGACCTC
を用いて増幅し、次いでNdeIとHindIIIを用いて制限酵素切断を行った。インサートを、同じ制限酵素(NdeIとHindIII)により予め消化しておいたpDHE1650ベクター中にライゲートした。同じ方法で、pDHE1650中に存在する場所ホルダー遺伝子を本発明のエステラーゼ遺伝子と置き換えた。2つの形質転換体を単離し、プラスミドを公知の方法でDNA沈降により抽出した。前記プラスミドをpDHE1650-Est2およびpDHE1615-Est4と名付けた。
【0191】
タンパク質発現については、エステラーゼ遺伝子を保持するプラスミドを大腸菌TG1(DSMZ No.6056)にTSS法を用いて形質転換した[Chung CT, Niemela SL and Miller RH,1989, "One-step preparation of competent Escherichia coli: Transformation and Storage of Bacterial Cells in the Same Solution" Proc. Natl. Acad. Sci. U.S.A. 86: 2172-2175]。単一コロニーを単離し、5mlのLB/Amp/Tet/Spec/Cm培地(=Sambrook, J., Fritsch, E.F. and Maniatis, T. Molecular cloning: A Laboratory Manual. 2nd Ed., Cold Spring Harbor Laboratory, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 1989によるLB培地+100μg/mlアンピシリン、10μg/mlテトラサイクリン,100μg/mlスペクチノマイシンおよび20μg/mlクロラムフェニコール)で37℃にて250rpmで一晩増殖した。10ml/l微量元素溶液を添加した100mlのLB/Amp/Tet/Spec/Cm培地に500μlの一晩培養物を接種し、37℃および200rpmにて増殖した(微量元素溶液は次の構成である:FeSO4*H2O 200mg/l、ZnSO4*7H2O 10mg/l、MnCl2*4H2O 3mg/l、H3BO3 30mg/l、CoCl2*6H2O 20mg/l、CuCl2*6H2O 1mg/l、NiCl2*6H2O 2mg/l、Na2MoO4*2H2O 3mg/l、Titriplex III 500mg/l)。タンパク質発現を1mM L-ラムノースを用いて1回誘導し、OD600は0.5〜0.6の値に達した。温度を30℃に低下したが、回転速度は200rpmに保った。培養物を一晩培養した。次いで、細胞を遠心分離し(15分間、4℃、3220g)、細胞ペレットを-20℃にて一晩保存した。
【0192】
細胞を、細胞ペレット(50mlのTB培養物)をリン酸ナトリウムバッファー(25mM、イオン強度154mM、pH7.5)に再懸濁することにより破壊した。細胞をホモジナイザーで破壊した(1500 bar、2回パス)。細胞断片を遠心分離(30分間、4℃、20817g)により取り出した。上清をタンパク質200 Plus Labchipにアプライし、Agilent 2100バイオアナライザーを用いてタンパク質を分析した。取り出した細胞断片を8M尿素に再懸濁し、次いで遠心分離(30分間、4℃、20817g)により処理して未溶解粒子を取り除いた。透明な上清(膜画分)をタンパク質200 Plus-Labchipにアプライし、Agilent 2100バイオアナライザーを用いてタンパク質を分析した。
【実施例12】
【0193】
ブチノールエステラーゼ(335AA)突然変異体の調製
a)材料
使用した全ての化学品は分析用に好適であるかまたはより高品質であって、Sigma-Aldrich Chemie, Taufkirchen, Germany, Applichem(Darmstadt, Germany)およびCarl Roth(Karlsruhe、Germany)から購入したものである。
【0194】
サーモサイクル(Mastercycler gradient、Eppendorf、Hamburg、Germany)および薄層PCRチューブ(Multi-Ultra tubes;0.2ml;CarlRoth)を全てのPCR実験に用いた。PCR容量はいずれの場合も50μlであり;より大きい容量は複数の50μl PCR混合物を用いて調製した。使用したDNAの量はNanoDrop光度計(NanoDrop Technologies、Wilmington、DE)を用いて数値化した。
【0195】
b)ブチノールエステラーゼ遺伝子のpASK-IBA7ベクター中へのクローニング:
pASK-IBA7ベクター(IBAGmbH、Goettingen、Germany)(配列番号19、N-末端Strep-タグによるタンパク質発現用のプラスミド)を次のプライマー:
フォワードプライマー
5'-TTTTTGCCCTCGTTATCTAGATTT-3'(配列番号9)
リバースプライマー
5'-CCGGAATTCCGGTATCTAACTAAGCTTGACCTG-3'(配列番号10)
を用いて増幅した。
【0196】
50μl PCR混合物(95℃ 3分間;1サイクル//95℃ 30秒、56.2℃ 45秒、72℃ 7分間;30サイクル//72℃ 10分間;1サイクル)は、20pmolのフォワードプライマー、20pmolのリバースプライマー、0.2mMの各dNTP(Roche Diagnostics GmbH, Mannheim, Germany)、150ngのpASK-IBA7プラスミドおよび2.5UのPfuDNAポリメラーゼ(Fermentas GmbH, St. Leon-Rot、Germany)を含んだ。生成物をQIAquickゲル抽出キット(Qiagen、Hilden、Germany)を用いてゲルにより精製し、以上の条件と同一の条件下で再び増幅した。ゲル精製(QIAquickゲル抽出キット、Qiagen)の後、2μgのDNA産物を40 UのEcoRI(New England Biolabs GmbH、Frankfurt、Germany)により100μl反応混合物中で37℃にて4時間消化した。消化した産物を、QIAquick PCR精製キット(Qiagen)を用いてPCR精製した。
【0197】
ブチノールエステラーゼ遺伝子を次のプライマー:
フォワードプライマー
5’-[Phos]GACCATGATTACGCCAAGCTTGC-3’(配列番号11)
リバースプライマー
5’-CCGGAATTCCGGTCACTGCAGCGCGCCGGCCTG-3’(配列番号12).
を用いて増幅した。
【0198】
50μl PCR混合物(95℃2分間;1サイクル//95℃30秒、59℃45秒、72℃3分間;30サイクル//72℃ 10分間;1サイクル)は、20pmolのフォワードプライマー、20pmolのリバースプライマー、0.2mMの各dNTP(Roche Diagnostics GmbH, Mannheim, Germany)、150ngのプラスミドLU11147(ブチノールエステラーゼ遺伝子を保有するpUC19ベクター)、0.02%DMSOおよび2.5UのPfuDNAポリメラーゼ(Fermentas)を含んだ。ゲル精製(QIAquickゲル抽出キット、Qiagen)の後、2μgのDNA産物を40UのEcoRI(New England Biolabs GmbH、Frankfurt、Germany)により100μl反応混合物中で37℃にて4時間消化した。消化した産物を、QIAquick PCR精製キット(Qiagen)を用いてPCR精製した。
【0199】
ブチノールエステラーゼ遺伝子をベクター中に平滑末端とEcoRI切断部位を用いてライゲートした。反応混合物(20μl)は、20ngのベクター、120ngのインサートおよび2UのT4DNAリガーゼ(Roche Diagnostics GmbH)を含んだ。反応混合物を最初に室温で1時間インキュベートし、次いで一晩冷蔵庫内でインキュベートした。得られるプラスミドをpASK-IBA7エステラーゼと名付けた。
【0200】
c)第1のブチノールエステラーゼ突然変異体ライブラリーの調製:
ブチノールエステラーゼ遺伝子を最初にエラープローンPCR(95℃ 2分間;1サイクル//95℃ 30秒、59℃ 45秒、72℃ 3分間;40サイクル//72℃ 10分間)を用いて増幅した。次のプライマー:
フォワード プライマー:
5’-CGACAAAAATCTAGATAACGAGGGCAA-3’ (配列番号13)
リバース プライマー:
5’-TTCACTTCACAGGTCAAGCTTAGTTAG-3’ (配列番号14).
を用いた。
【0201】
反応混合物(50μl)は、20pmolのフォワードプライマー、20pmolのリバースプライマー、0.2mMの各dNTP(Roche Diagnostics GmbH)、100ngのプラスミドpASK-IBA7エステラーゼ、0.02% DMSO、0.1mM MnCl2および2.5UのTaq DNAポリメラーゼ(Qiagen)を含んだ。ゲル精製(QIAquickゲル抽出キット、Qiagen)の後、4μgのDNA産物を40UのEcoRI(New England Biolabs GmbH、Frankfurt、Germany)と30UのXbaI(New England Biolabs GmbH)により二重消化した。産物をPCR精製(QIAquick PCR-精製 キット; Qiagen)した後、EcoRIとXbaIで予め消化したpASK-IBA7 プラスミド中にライゲートした。突然変異体ライブラリーを大腸菌XL2ブルー(Stratagene、Amsterdam)中にTSS法[Chung CT, Niemela SL and Miller RH,1989. "One-step preparation of competent Escherichia coli: Transformation and Storage of Bacterial Cells in the Same Solution" Proc. Natl. Acad. Sci. U.S.A. 86: 2172-2175]を用いて形質転換した。
【0202】
d)ブチノールエステラーゼ突然変異体ライブラリーの発現:
LBampプレート(=Sambrook, J., Fritsch, E.F. and Maniatis, T. Molecular Cloning: A Laboratory Manual. 2nd Ed., Cold Spring Harbor Laboratory, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 1989によるLB培地+10g/l寒天および100μg/mアンピシリン)上のコロニーを、つまようじを用いて拾い、150μlのLBamp培地を含有する96ウエルのマイクロタイタープレートへ移した。マイクロプレート振とう機(Multitron II; Infors GmbH、Einsbach、Germany;37℃、900rpmおよび70%湿度)で培養した後、ほぼ5μlの各培養物をDuetzシステム (Kuehner, Birsfelden, Switzerland) を用いて、550μlのTB培地と100μg ml アンピシリン (Becton Dickinson GmbH)を含有する2mlウエルのプレートへ移した。クローンを、Multitron II振とう機(Infors GmbH;30℃および500rpmおよび70%湿度)で8時間培養した。タンパク質発現を、2.4μg/ml無水テトラサイクリン(IBA GmbH)を含む50μl TBを各ウエルに加えることにより誘導した。クローンをさらに6時間、Multitron II振とう機(Infors GmbH; 30℃および500 rpm)で8時間培養した。
【0203】
e)pH指示薬アッセイ (96-ウエルのフォーマット):
各ウエルからの50μl TB培養物を96-ウエルアッセイプレートに、Multimek 96-チャネル自動ピペッター(Beckman Coulter、Krefeld、Germany)を用いて移した。アッセイプレートの各ウエルにMultimek 96(Beckman Coulter)を用いて、100μlの基質溶液(25mM NaH2PO4 バッファー、pH 7.5、NaClにより調節してイオン強度154mM、0.05% Triton X-100、7.5μg/mlフルオレセイン-ナトリウムおよび285mM酪酸ブタ-3-イン-2-イルを含む)を加えることにより加水分解反応を開始した。使用前に5分間、基質溶液を激しく攪拌した。フルオレッセンス・シグナル(吸光485nm、発光520nm)の低下の反応速度を、Tecan Safire (Tecan GmbH、 Crailsheim、Germany)を用いて10分間観察した。活性を初期スロープから決定した。930クローンのスクリーニング後、クローン8H1が改良された突然変異体であると同定した。
【0204】
f)第2のブチノールエステラーゼ突然変異体ライブラリー:
第2のブチノールエステラーゼ突然変異体ライブラリーを、8H1を親遺伝子として用いたことを除くと、以上の記載と正確に同じ条件下で作製した。ライブラリーを以上のように発現し、スクリーニングした。1116クローンのスクリーニング後、クローン8C9が改良された突然変異体であると同定した。
【0205】
g)アミノ酸位置190および263における飽和突然変異誘発:
アミノ酸位置190および263の飽和突然変異誘発を、次の改変QuikChangeプロトコル(Stratagene)を用いて実施した。アミノ酸位置190に対しては次の突然変異誘発プライマー:
5’-GGCATCCCGATCATGNNNCTGCAAAGCCGCAAG-3’ (配列番号15)
5’-CTTGCGGCTTTGCAGNNNCATGATCGGGATGCC-3’ (配列番号16)
を用いた。アミノ酸位置263に対しては次の突然変異誘発プライマー:
5’-GGGCGCCAGGACGCGNNNCTCGATTTCCACAAG-3’ (配列番号17)
5’-CTTGTGGAAATCGAGNNNCGCGTCCTGGCGCCC-3’ (配列番号18)
を用いた。
【0206】
50μl PCR混合物(95℃30秒;1サイクル//95℃30秒、55℃1分、68℃4分45秒;18サイクル)は、20pmolの各プライマー、0.2mMの各dNTP(Roche Diagnostics GmbH)、50 ngのプラスミド8C9(出発DNA)および2.5 UのPfu Turbo DNAポリメラーゼ(Stratagene)の混合物を含んだ。PCRの後、メチル化および半メチル化親DNAを、40UのDpnI(New England Biolabs GmbH)により37℃にて2〜3 時間消化した。産物をPCR精製し(QIAquick PCR-精製 キット; Qiagen)、次いで、TSS 方法 [Chung CT、loc. cit]を用いて大腸菌 XL2ブルー(Stratagene)中に形質転換した。
【0207】
2つのライブラリーを先に記載したのと同じ方法で発現してスクリーニングした。それぞれのライブラリーについて186クローンをスクリーニングした後、2つのクローンを改良された突然変異体として同定した。これらをEst190-1B2およびEst263-2D6と名付けた。
【0208】
h)振とうフラスコでのタンパク質発現
一晩培養物を5 mlのLBamp 培地中でMultitron-II振とう機(Infors GmbH; 37℃、250 rpm)を用いて新しく形質転換したコロニーを拾うことにより増殖した。100mlのTBamp培地に800μlの前記一晩培養物を接種し、Multitron-II振とう機(Infors GmbH;37℃、200rpm)中で培養した。0.5〜0.6のOD600値に達すると、タンパク質発現を25μlの無水テトラサイクリン(DMF中の0.2 mg/mlのストック溶液)を加えることにより誘導した。培養物をさらに12時間増殖した(Multitron-II振とう機、Infors GmbH; 30℃、200 rpm)。細胞を遠心分離(3220 gおよび4℃にて30 分間)により取り除き、湿細胞塊を測定した。細胞ペレットを-20℃にて保存して、さらなる特徴付けを行った。
【0209】
i)pHスタットを用いるタンパク質特徴付け:
ブチノールエステラーゼの加水分解活性を、pHスタット(716 DMS Titrino、Deutsche Metrohm GmbH & Co.、Filderstadt、Germany)を用いて測定した。
【0210】
細胞ペレットを最初に、0.1g湿細胞塊/mlの細胞濃度を得るのに好適な容量のリン酸ナトリウムバッファー(25mMリン酸二水素ナトリウムバッファー、pH 7.5、NaClにより調節してイオン強度154mM)に再懸濁した。リン酸ナトリウムバッファー(25mMリン酸二水素ナトリウムバッファー、pH7.5、NaClにより調節してイオン強度154mM)、0.05% TritonX-100および350mM酪酸ブタ-3-イン-2-イルを含む20 mlの基質溶液を調製した。基質溶液を使用前に5分間激しく攪拌した。
【0211】
加水分解反応の開始前に、基質溶液(20 ml)のpHを1N NaOHによりpH 7.5に調節した。0.01gの湿細胞塊を加えることにより、反応を開始した。7.5のpHを、バイオ変換中、0.1N NaOHを用いる滴定により維持した。加水分解活性はNaOHの滴定速度と相関があった(単位時間当たり加えた容量)。
【0212】
結果を図4に示す。次表に掲げた酵素と突然変異体を試験した。アミノ酸配列と核酸配列中の特別な突然変異に関する情報をこの表から同様に見出すことができる。
【表4】
【受託番号】
【0213】
DSM13176
【特許請求の範囲】
【請求項1】
エステラーゼ活性を有するタンパク質またはその機能的に等価なタンパク質であって、前記タンパク質が510個未満のアミノ酸の全長をもつポリペプチド鎖を有しかつ前記鎖が少なくとも1つの配列番号3に記載のアミノ酸部分配列を含む前記タンパク質。
【請求項2】
エステル、酪酸ブタ-3-イン-2-イルの切断により特徴付けることができるエステラーゼ活性をもつ、請求項1に記載のタンパク質。
【請求項3】
少なくとも1つのさらなる、配列番号 4、5または6に記載のアミノ酸部分配列をさらに含む、請求項1または2に記載のタンパク質。
【請求項4】
全長が450個未満または350個未満のアミノ酸のポリペプチド鎖を含む、請求項1〜3のいずれか1項に記載のタンパク質。
【請求項5】
全長が約335個のアミノ酸のポリペプチド鎖を含む、請求項1〜4のいずれか1項に記載のタンパク質。
【請求項6】
配列番号8に記載のアミノ酸配列を含む、請求項5に記載のタンパク質。
【請求項7】
配列番号2または8のアミノ酸配列領域12〜20、185〜195および258〜268のいずれかに少なくとも1つの機能的突然変異を有する、エステラーゼ突然変異体。
【請求項8】
配列番号2または8のアミノ酸配列の16、190および263位のいずれかに少なくとも1つの機能的突然変異を含む、請求項7に記載の突然変異体。
【請求項9】
次のアミノ酸置換:Leu16Pro、Ile190Thr、Ile190ArgおよびIle263Valの少なくとも1つを含む、請求項8に記載のタンパク質。
【請求項10】
約60kDa以下の計算分子量を有するポリペプチド鎖を含む、請求項1〜9のいずれか1項に記載のタンパク質。
【請求項11】
受託番号DSM 13176をもつPseudomonas glumae (Burkholderia plantarii) LU 2023から出発して得られる、請求項1〜10のいずれか1項に記載のタンパク質。
【請求項12】
次の反応:
a)式I:
R1-COO-R2 (I)
[式中、R1は直鎖または分枝鎖の、任意に一置換または多置換されたC1〜C10-アルキル、C2〜C10-アルケニル、C2〜C10-アルキニルであり、そしてR2は直鎖または分枝鎖の、任意に一置換または多置換されたC1〜C10-アルキル、C2〜C10-アルケニル、C2〜C10-アルキニル、C7〜C15-アラルキル、または単環または多環の、任意に一置換または多置換された芳香族基であり、R1および/またはR2は少なくとも1個の不斉炭素を含む]
で表わされる光学活性エステルのエナンチオ選択的加水分解;および
b)式Iのエステルと式II:
R2-OH (II)
[式中、R2は前記の意味の1つを有し、任意に、少なくとも1個の不斉炭素を有する]
で表わされる光学活性アルコールとのエナンチオ選択的エステル交換
の両反応のうちの少なくとも1つの反応を触媒する、請求項1〜11のいずれか1項に記載のタンパク質。
【請求項13】
請求項1〜12のいずれか1項に記載のタンパク質およびまたその機能的等価体をコードするポリヌクレオチド、それらと相補的なポリヌクレオチド、ならびにそれらとハイブリダイズしうる核酸配列。
【請求項14】
配列番号7に記載の核酸配列中の少なくとも30個の連続ヌクレオチド残基のヌクレオチド配列を含む、請求項13に記載のポリヌクレオチド。
【請求項15】
配列番号8のアミノ酸の263位に対応する領域のコドンがGTTおよびGTCのなかから選択される、請求項14に記載のポリヌクレオチド。
【請求項16】
少なくとも1つの調節核酸配列と機能しうる形で連結された請求項13〜15のいずれか1項に記載の少なくとも1つのポリヌクレオチドを含む発現カセット。
【請求項17】
真核生物のまたは原核生物の宿主を形質転換するための組換えベクターであって、請求項13〜15のいずれか1項に記載のポリヌクレオチドまたは請求項16に記載の発現カセットを含む前記組換えベクター。
【請求項18】
前記タンパク質を内因的に産生する微生物または請求項17に記載のベクターを用いて形質転換した微生物を培養するステップ、および前記タンパク質を該培養物から単離するステップを含んでなる、請求項1〜12のいずれか1項に記載のタンパク質を調製する方法。
【請求項19】
微生物が受託番号DSM13176をもつPseudomonas glumae(Burkholderia plantarii)LU2023である、請求項18に記載の方法。
【請求項20】
請求項18および19のいずれか1項に記載の方法により得ることができるタンパク質。
【請求項21】
受託番号DSM13176をもつ微生物Pseudomonas glumae(Burkholderia plantarii)LU2023ならびにその変異体および突然変異体。
【請求項22】
請求項17に記載のベクターを保有する微生物。
【請求項23】
請求項1〜12のいずれか1項に記載のタンパク質を用いるエナンチオ選択的エステル加水分解法であって、
a)前記タンパク質を、式Iで表される光学活性エステルの立体異性体混合物と接触させるステップ;および
b)前記立体異性体のいずれかの立体選択的加水分解により生成する光学活性化合物および/または加水分解されないエステルエナンチオマーを反応媒体から取得するステップを含んでなる前記方法。
【請求項24】
エナンチオ選択的エステル交換法であって、
a)式IIで表される光学活性アルコールの立体異性体混合物を、請求項1〜12のいずれか1項に記載のタンパク質の存在下に、式Iで表されるエステルと接触させ、未反応のアルコール立体異性体を反応媒体から取得するステップ;または
b)式Iで表される光学活性エステルの立体異性体混合物を、請求項1〜12のいずれか1項に記載のタンパク質の存在下に、式IIで表されるアルコールと接触させ、前記エステルに含まれる光学活性アルコールの立体異性体を反応媒体から取得するステップを含んでなる前記方法。
【請求項25】
エステル交換のために、ビニルエステルを光学活性アルコールに対するアシル化剤として用いるステップを含んでなる、請求項24に記載の方法。
【請求項26】
有機溶媒を反応媒体として用いることを含んでなる、請求項23〜25のいずれか1項に記載の方法。
【請求項1】
エステラーゼ活性を有するタンパク質またはその機能的に等価なタンパク質であって、前記タンパク質が510個未満のアミノ酸の全長をもつポリペプチド鎖を有しかつ前記鎖が少なくとも1つの配列番号3に記載のアミノ酸部分配列を含む前記タンパク質。
【請求項2】
エステル、酪酸ブタ-3-イン-2-イルの切断により特徴付けることができるエステラーゼ活性をもつ、請求項1に記載のタンパク質。
【請求項3】
少なくとも1つのさらなる、配列番号 4、5または6に記載のアミノ酸部分配列をさらに含む、請求項1または2に記載のタンパク質。
【請求項4】
全長が450個未満または350個未満のアミノ酸のポリペプチド鎖を含む、請求項1〜3のいずれか1項に記載のタンパク質。
【請求項5】
全長が約335個のアミノ酸のポリペプチド鎖を含む、請求項1〜4のいずれか1項に記載のタンパク質。
【請求項6】
配列番号8に記載のアミノ酸配列を含む、請求項5に記載のタンパク質。
【請求項7】
配列番号2または8のアミノ酸配列領域12〜20、185〜195および258〜268のいずれかに少なくとも1つの機能的突然変異を有する、エステラーゼ突然変異体。
【請求項8】
配列番号2または8のアミノ酸配列の16、190および263位のいずれかに少なくとも1つの機能的突然変異を含む、請求項7に記載の突然変異体。
【請求項9】
次のアミノ酸置換:Leu16Pro、Ile190Thr、Ile190ArgおよびIle263Valの少なくとも1つを含む、請求項8に記載のタンパク質。
【請求項10】
約60kDa以下の計算分子量を有するポリペプチド鎖を含む、請求項1〜9のいずれか1項に記載のタンパク質。
【請求項11】
受託番号DSM 13176をもつPseudomonas glumae (Burkholderia plantarii) LU 2023から出発して得られる、請求項1〜10のいずれか1項に記載のタンパク質。
【請求項12】
次の反応:
a)式I:
R1-COO-R2 (I)
[式中、R1は直鎖または分枝鎖の、任意に一置換または多置換されたC1〜C10-アルキル、C2〜C10-アルケニル、C2〜C10-アルキニルであり、そしてR2は直鎖または分枝鎖の、任意に一置換または多置換されたC1〜C10-アルキル、C2〜C10-アルケニル、C2〜C10-アルキニル、C7〜C15-アラルキル、または単環または多環の、任意に一置換または多置換された芳香族基であり、R1および/またはR2は少なくとも1個の不斉炭素を含む]
で表わされる光学活性エステルのエナンチオ選択的加水分解;および
b)式Iのエステルと式II:
R2-OH (II)
[式中、R2は前記の意味の1つを有し、任意に、少なくとも1個の不斉炭素を有する]
で表わされる光学活性アルコールとのエナンチオ選択的エステル交換
の両反応のうちの少なくとも1つの反応を触媒する、請求項1〜11のいずれか1項に記載のタンパク質。
【請求項13】
請求項1〜12のいずれか1項に記載のタンパク質およびまたその機能的等価体をコードするポリヌクレオチド、それらと相補的なポリヌクレオチド、ならびにそれらとハイブリダイズしうる核酸配列。
【請求項14】
配列番号7に記載の核酸配列中の少なくとも30個の連続ヌクレオチド残基のヌクレオチド配列を含む、請求項13に記載のポリヌクレオチド。
【請求項15】
配列番号8のアミノ酸の263位に対応する領域のコドンがGTTおよびGTCのなかから選択される、請求項14に記載のポリヌクレオチド。
【請求項16】
少なくとも1つの調節核酸配列と機能しうる形で連結された請求項13〜15のいずれか1項に記載の少なくとも1つのポリヌクレオチドを含む発現カセット。
【請求項17】
真核生物のまたは原核生物の宿主を形質転換するための組換えベクターであって、請求項13〜15のいずれか1項に記載のポリヌクレオチドまたは請求項16に記載の発現カセットを含む前記組換えベクター。
【請求項18】
前記タンパク質を内因的に産生する微生物または請求項17に記載のベクターを用いて形質転換した微生物を培養するステップ、および前記タンパク質を該培養物から単離するステップを含んでなる、請求項1〜12のいずれか1項に記載のタンパク質を調製する方法。
【請求項19】
微生物が受託番号DSM13176をもつPseudomonas glumae(Burkholderia plantarii)LU2023である、請求項18に記載の方法。
【請求項20】
請求項18および19のいずれか1項に記載の方法により得ることができるタンパク質。
【請求項21】
受託番号DSM13176をもつ微生物Pseudomonas glumae(Burkholderia plantarii)LU2023ならびにその変異体および突然変異体。
【請求項22】
請求項17に記載のベクターを保有する微生物。
【請求項23】
請求項1〜12のいずれか1項に記載のタンパク質を用いるエナンチオ選択的エステル加水分解法であって、
a)前記タンパク質を、式Iで表される光学活性エステルの立体異性体混合物と接触させるステップ;および
b)前記立体異性体のいずれかの立体選択的加水分解により生成する光学活性化合物および/または加水分解されないエステルエナンチオマーを反応媒体から取得するステップを含んでなる前記方法。
【請求項24】
エナンチオ選択的エステル交換法であって、
a)式IIで表される光学活性アルコールの立体異性体混合物を、請求項1〜12のいずれか1項に記載のタンパク質の存在下に、式Iで表されるエステルと接触させ、未反応のアルコール立体異性体を反応媒体から取得するステップ;または
b)式Iで表される光学活性エステルの立体異性体混合物を、請求項1〜12のいずれか1項に記載のタンパク質の存在下に、式IIで表されるアルコールと接触させ、前記エステルに含まれる光学活性アルコールの立体異性体を反応媒体から取得するステップを含んでなる前記方法。
【請求項25】
エステル交換のために、ビニルエステルを光学活性アルコールに対するアシル化剤として用いるステップを含んでなる、請求項24に記載の方法。
【請求項26】
有機溶媒を反応媒体として用いることを含んでなる、請求項23〜25のいずれか1項に記載の方法。
【図1】


【図2】
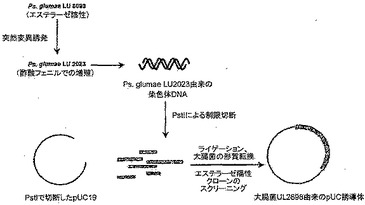
【図3】
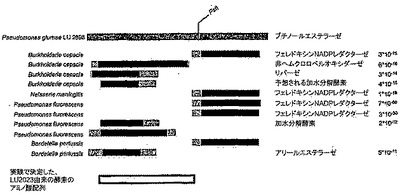
【図4】
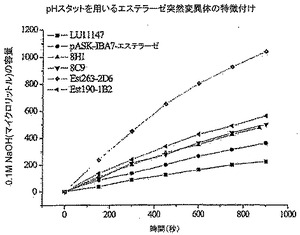
【図2】
【図3】
【図4】
【公開番号】特開2012−228257(P2012−228257A)
【公開日】平成24年11月22日(2012.11.22)
【国際特許分類】
【出願番号】特願2012−151968(P2012−151968)
【出願日】平成24年7月6日(2012.7.6)
【分割の表示】特願2009−517169(P2009−517169)の分割
【原出願日】平成19年6月26日(2007.6.26)
【出願人】(508020155)ビーエーエスエフ ソシエタス・ヨーロピア (2,842)
【氏名又は名称原語表記】BASF SE
【住所又は居所原語表記】D−67056 Ludwigshafen, Germany
【Fターム(参考)】
【公開日】平成24年11月22日(2012.11.22)
【国際特許分類】
【出願日】平成24年7月6日(2012.7.6)
【分割の表示】特願2009−517169(P2009−517169)の分割
【原出願日】平成19年6月26日(2007.6.26)
【出願人】(508020155)ビーエーエスエフ ソシエタス・ヨーロピア (2,842)
【氏名又は名称原語表記】BASF SE
【住所又は居所原語表記】D−67056 Ludwigshafen, Germany
【Fターム(参考)】
[ Back to top ]