
エステル誘導体の製造方法
【課題】酵素反応を用いた、(メタ)アクリル酸誘導体と、水酸基を有してもよい脂肪族カルボン酸、又は多価カルボン酸とを反応させることを特徴とする高分子材料モノマーとして有用なアクリレート化合物の製造方法の提供。
【解決手段】特定の微生物から得られるリパーゼ又はリパーゼを細胞表層に提示したアーミング酵母を用いて、温和な条件下で、水酸基を有してもよい脂肪族カルボン酸、又は多価カルボン酸とヒドロキシアクリレートとのエステル化反応による、(メタ)アクリレートの製造方法の提供。
【解決手段】特定の微生物から得られるリパーゼ又はリパーゼを細胞表層に提示したアーミング酵母を用いて、温和な条件下で、水酸基を有してもよい脂肪族カルボン酸、又は多価カルボン酸とヒドロキシアクリレートとのエステル化反応による、(メタ)アクリレートの製造方法の提供。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、酵素反応を用いた、(メタ)アクリル酸誘導体と、水酸基を有してもよい脂肪族カルボン酸、又は多価カルボン酸とを反応させることを特徴とする高分子材料モノマーとして有用なアクリレート化合物の製造方法に関する。
【背景技術】
【0002】
2‐ヒドロキシエチル(メタ)アクリレート(HE(M)A)に代表されるヒドロキシル基を有する(メタ)アクリル酸誘導体は種々のアクリル樹脂の原料に用いられている。アクリル樹脂中にヒドロキシル基を導入することで、樹脂の水溶性付与やポリイソシアネート化合物との反応による架橋反応などが可能になり、塗料や粘/接着剤などの分野で広く用いられている。しかしながら、HEMAのようなアクリレートをアクリル樹脂に共重合した場合、アクリル樹脂の主鎖近傍にヒドロキシル基が存在するためポリイソシアネート化合物との硬化物物性が脆くなるといった問題があった。このような問題解決のためPEGなどの片末端に(メタ)アクリル基を導入したモノマーが用いられているが、吸水率の上昇などの問題があった。
【0003】
一方、再生可能資源として植物由来原料を用いた材料の開発が近年盛んに行われている。脂肪酸を原料に用いた疎水性の高い(メタ)アクリレート化合物およびその製造方法としては、以下の特許文献1、特許文献2、及び特許文献3の開示がある。
しかしながら、熱、酸、酸素等に不安定な原料を用いた場合には著しい着色や分解、ゲル化などの副反応により製造できないこともある等の問題点を有していた。
更には、ヒドロキシ脂肪酸とアクリル酸アルカノールを原料に用いたエステル化反応では、アクリル酸アルカノールおよびヒドロキシ脂肪酸のカルボキシル基間の交差反応に加え、それぞれのヒドロキシル基間の交差反応のため上記特許文献における手法では、更にヒドロキシル基の官能基保護/脱保護等の工程が必要となるなどの問題があった。
【0004】
【特許文献1】特開平2−110115号
【特許文献2】特開平5−229991号
【特許文献3】特許第3829101号
【発明の開示】
【発明が解決しようとする課題】
【0005】
本発明が解決しようとする課題は、酵素反応を用いた、(メタ)アクリル酸誘導体と、水酸基を有してもよい脂肪族カルボン酸、又は多価カルボン酸とを反応させることを特徴とする高分子材料モノマーとして有用なアクリレート化合物の製造方法の提供にある。
【課題を解決するための手段】
【0006】
本発明者らは、鋭意研究を重ねた結果、アスペルギルス(Aspergillus oryzae)属、アクロモバクター(Achromobacter)属、バシラス(Bacillus)属、カンジダ(Candida)属、クロモバクター(Chromobacter)属、フザリウム(Fusarium)属、フミコラ(Humicola)属、ハイフォザイマ(Hyphozyma)属、シュードモナス(Pseudomonas)属、リゾムーコル(Rhizomucor)属、リゾプス(Rhizopus)属、又はテルモマイセス(Thermomyces)属の微生物から得られるリパーゼ(A)又はリパーゼを細胞表層に提示したアーミング酵母(B)(以下リパーゼ・アーミング酵母(B)と記す。)を用いて、温和な条件下で、(メタ)アクリル酸誘導体と、水酸基を有してもよい炭素数6〜24の脂肪族カルボン酸とのエステル化反応により、高分子材料モノマーとして有用なアクリレート化合物の製造方法を見出し、本発明を完成させるに至った。
【0007】
即ち、本発明は、リパーゼ(A)又はリパーゼ・アーミング酵母(B)の存在下に、一般式(1)
【0008】
【化1】

【0009】
(式中、R1は水素原子又はメチル基、Xは酸素原子又は窒素原子、nは2〜4の整数を表す。)
で表される(メタ)アクリル酸誘導体と、一般式(2)
【0010】
【化2】
【0011】
(式中、R2は水酸基又はカルボキシ基を有してもよい炭素数5〜23のアルキル基又はアルケニル基を表す。)
で表されるカルボン酸とを反応させる一般式(3)
【0012】
【化3】
【0013】
(式中、R1は水素原子又はメチル基、Xは酸素原子又は窒素原子、nは2〜4の整数、R2は水酸基又はカルボキシ基を有してもよい炭素数5〜23のアルキル基又はアルケニル基を表す。)
で表されるアクリレート化合物の製造方法に関する。
【発明の効果】
【0014】
本発明によれば、(メタ)アクリル酸誘導体と、水酸基を有してもよい脂肪族カルボン酸、又は多価カルボン酸とを反応させることを特徴とする高分子材料モノマーとして有用なアクリレート化合物の製造方法を提供できる。
【発明を実施するための最良の形態】
【0015】
本発明は、リパーゼ(A)又はリパーゼ・アーミング酵母(B)の存在下に、一般式(1)
【0016】
【化4】
【0017】
(式中、R1は水素原子又はメチル基、Xは酸素原子又は窒素原子、nは2〜4の整数を表す。)
で表される(メタ)アクリル酸誘導体と、一般式(2)
【0018】
【化5】
【0019】
(式中、R2は水酸基又はカルボキシ基を有してもよい炭素数5〜23のアルキル基又はアルケニル基を表す。)
で表されるカルボン酸とを反応させる一般式(3)
【0020】
【化6】
【0021】
(式中、R1は水素原子又はメチル基、Xは酸素原子又は窒素原子、nは2〜4の整数、R2は水酸基又はカルボキシ基を有してもよい炭素数5〜23のアルキル基又はアルケニル基を表す。)
で表されるアクリレート化合物の製造方法を提供する。
【0022】
一般式(1)で表される化合物は、(メタ)アクリル酸と2価アルコール又はヒドロキシアミンとの脱水縮合反応によって、通常公知の方法により得ることができる。
式中、R1は水素原子であってもメチル基であってもよい。また、Xが酸素原子を表すときは、一般式(1)で表される化合物はエステル誘導体であり、Xが窒素原子を表すときは、一般式(1)で表される化合物はアミド誘導体であるが、本発明ではどちらであってもよい。nは2〜4の整数であれば特に制限なく用いることが可能であるが、特に好ましくは、n=2の場合である。
また、一般式(1)で表される化合物とのエステル反応に供される脂肪族カルボン酸は、一般式(2)
【0023】
【化7】
【0024】
(式中、R2は水酸基又はカルボキシ基を有してもよい炭素数5〜23のアルキル基又はアルケニル基を表す。)
で表されるカルボン酸であって、得られるエステル誘導体の用途によって、R2に水酸基又はカルボキシ基を有していても有していなくてもよい。
本発明に使用される水酸基を有してもよい炭素数6〜24のカルボン酸の例としては、例えば、カプロン酸、カプリル酸、カプリン酸、ウンデシレン酸、パルミチン酸、オレイン酸、ステアリン酸、リシノール酸、ヒドロキシステアリン酸等の脂肪族カルボン酸を挙げることができるが、これらに限定されない。
さらに、本発明に使用されるカルボキシ基を有する一般式(2)で表されるカルボン酸としては、多価カルボン酸として公知公用のものを好ましく用いることができ、具体的には、アジピン酸、セバシン酸、ドデカン2酸等の多価カルボン酸を挙げることができるが、これらに限定されない。
エステル化の反応においては、一般式(2)で表されるカルボン酸と一般式(1)で表される(メタ)アクリル酸誘導体を反応させてもよいが、一般式(2)で表されるカルボン酸のエステル誘導体を用いても好適にエステル化反応を行うことができる。ここで用いることのできるエステル基としては、例えば、メチルエステル、エチルエステル等の低級アルキルエステルを挙げることができるが、これらに限定されるものではなく、公知公用のエステル基を好ましく用いるこができる。
【0025】
本発明で使用される酵素は、アスペルギルス(Aspergillus oryzae)属、アクロモバクター(Achromobacter)属、バシラス(Bacillus)属、カンジダ(Candida)属、クロモバクター(Chromobacter)属、フザリウム(Fusarium)属、フミコラ(Humicola)属、ハイフォザイマ(Hyphozyma)属、シュードモナス(Pseudomonas)属、リゾムーコル(Rhizomucor)属、リゾプス(Rhizopus)属、又はテルモマイセス(Thermomyces)属の微生物から得られるものが好ましい。
より具体的に、本発明に用いられる好ましい市販の酵素を列挙すると、名糖産業株式会社のリパーゼQL、同社リパーゼPL、これらの固定化酵素であるリパーゼQLC、リパーゼQLG、リパーゼPLC、リパーゼPLGがあり、他にも、天野製薬社製リパーゼPS、ノボザイムズ社製リポザイムRM−IM、リポザイムTL−IM、ノボザイム435が挙げられる。これらの酵素は、混合して使用しても良く、また反応過程に伴い酵素を変えたり、又は異なった酵素を足し込んでも良い。
【0026】
反応槽中に添加する酵素量は反応後に酵素を取り除く工程上の手間と経済性の点から少ない方が好ましく、この観点から高活性の酵素が好ましい。具体的には1000U/g(脂肪酸エステルの加水分解による活性測定法)以上が好ましく、粉体で分散しにくい酵素の場合は5000U/g以上が好ましい。
酵素の形態は溶液状態、固体粉末、担体に固定化された形態を問わないが、生成物の精製時に不要な成分を含まない固体粉末もしくは担体に固定化されたものが好ましい。また、酵素の再利用を考慮すると担体に固定化されたものが、より好ましい。
酵素の回収は、未反応原料の回収と共に行うことが可能で、反応後の反応液を静置又は冷却後に沈降した酵素と未反応原料と共にデカンテーション、遠心分離又は濾別によって容易に分離できる。冷却は反応時の組成にも依存するが室温以下が好ましい。濾別された酵素及び未反応原料はこのまま、もしくは一度、加温及び減圧下に乾燥し再利用できる。
【0027】
また、本発明では、エステル反応に適したエステル合成活性の高いリパーゼを細胞表層に発現させた高活性アーミング酵母も好ましく用いることができる。
【0028】
本発明で用いられるリパーゼ・アーミング酵母(B)は、以下の特徴を有する。
1.リパーゼ・アーミング酵母(B)が、一倍体a細胞アーミング酵母と一倍体α細胞アーミング酵母を細胞融合したものであること、
2.リパーゼ・アーミング酵母(B)が、前記一倍体a細胞アーミング酵母及び一倍体α細胞アーミング酵母のいずれか一方あるいは両方の細胞表層へ提示されるタンパク質が、分泌シグナル配列、前記タンパク質の構造遺伝子配列、細胞表層局在タンパク質の一部をコードする配列及びGPIアンカリングドメインをコードする配列をこの順で有するDNAによって細胞表層に発現されるものであること、
3.リパーゼ・アーミング酵母(B)が、前記一倍体a細胞アーミング酵母及び一倍体α細胞アーミング酵母のいずれか一方あるいは両方の細胞表層へ提示されるタンパク質が、分泌シグナル配列、前記タンパク質の構造遺伝子配列、GPIアンカリングドメインをコードする配列をこの順で有するDNAによって細胞表層に発現されるものであること、
4.リパーゼ・アーミング酵母(B)が、前記一倍体a細胞アーミング酵母及び一倍体α細胞アーミング酵母のいずれか一方あるいは両方の細胞表層へ提示されるタンパク質が、分泌シグナル配列、前記タンパク質の構造遺伝子配列、糖鎖結合タンパク質ドメインをコードする配列をこの順で有するDNA、あるいは分泌シグナル配列、糖鎖結合タンパク質ドメインをコードする配列、前記細胞表層へ提示されるタンパク質の構造遺伝子配列をこの順で有するDNAによって細胞表層に発現されるものであること。
【0029】
本発明におけるリパーゼを細胞表層に発現させたアーミング酵母の作製は、公開特許公報(特開平11−290078、WO2002/085935)を基に行うことができる。
【0030】
(アーミング酵母)
本発明において、アーミング酵母とは細胞表層局在タンパク質と種々の機能性タンパク質(酵素・抗原・抗体・レポータータンパク質など)やペプチドを融合させ、細胞表層にディスプレイさせることにより、通常の酵母では有していない新しい機能を有する、あるいは元来有している機能を増強した酵母細胞のことをいう。また、細胞表層とは細胞膜・細胞壁ならびにその間の空間であるペリプラズムのことであり、これらの層を利用し上記の様な要件を満たす酵母細胞をアーミング酵母という。
【0031】
本発明におけるリパーゼに関しては次に説明する通りである。
【0032】
(分泌型酵素)
リパーゼとは、油脂から脂肪酸を遊離させ得る活性を有する酵素であり、その起源については特に限定されないが、通常、リゾプス・オリザエ(Rhizopus orizae、以下、リゾプス・オリザエと略記する)などのカビ由来のリパーゼや、キャンディダ・アンタルクチカ(Candida Antarctica、以下、キャンディダ・アンタルクチカと略記する)などの酵母由来のリパーゼが好適に用いられている。
【0033】
(細胞表層局在タンパク質)
本発明において、細胞表層局在タンパク質とは、酵母の細胞表層に固定され、細胞表層に存在するタンパク質をいう。例えば、凝集性タンパク質であるα―又はa−アグルチニン、FLOタンパク質、大腸菌の外膜タンパク質OmpAなどが挙げられる。一般に細胞表層局在タンパク質は、N末端側に分泌シグナル配列及びC末端側にGPIアンカー付着認識シグナル配列を有している。分泌シグナル配列を有する点では分泌性タンパク質と共通しているが、細胞表層局在タンパク質は、GPIアンカーを介して細胞膜に固定されて輸送される点が分泌性タンパク質と異なる。細胞表層局在タンパク質は細胞膜通過の際、GPIアンカー付着認識シグナル配列が選択的に切断され、新たに突出したC末端部分でGPIアンカーと結合して細胞膜に固定される。その後ホスファチジルイノシトール依存性ホスホリパーゼC(以下、PI−PLCと略記する)によりGPIアンカーの根元部分が切断される。次いで、細胞膜から切り離されたタンパク質は、細胞壁に組み込まれて細胞表層に固定され、細胞表層に局在する。
【0034】
(分泌シグナル配列)
本発明において、分泌シグナル配列とは、一般に細胞外に分泌されるタンパク質のN−末端に結合している、疎水性に富んだアミノ酸を多く含むアミノ酸配列であり、通常、分泌性タンパク質が細胞内から細胞膜を通して細胞外(ペリプラズムも含む)へ分泌される際に除去される。分泌シグナル配列であれば、どのような分泌シグナル配列でも用いることができ、その起源は限定されない。例えば、グルコアミラーゼの分泌シグナル配列、酵母のα―又はa―アグルチニンの分泌シグナル配列、リパーゼの分泌シグナル配列などが好適に用いられている。また、リパーゼの活性に影響を与えないのであれば、分泌シグナル配列の一部又は全部が酵素のN−末端側に残っても良い(特開平11−290078号公報、国際公開第2002/085935号パンフレット参照)。
【0035】
(GPIアンカー及びGPIアンカリングドメイン)
GPIアンカーとは、グリコシルホスファチジルイノシトール(GPI)と呼ばれる、エタノールアミンリン酸―6マンノースα1−2マンノースα1−6マンノースα1−4グルコサミンα1−6イノシトールリン脂質を基本構造とする糖脂質をいう。
【0036】
GPIアンカリングドメインは、通常、細胞表層局在タンパク質のC末端あるいはその近傍に位置する。例えば、α―アグルチニンのC末端から320アミノ酸の配列をコードする配列がこれに相当し、この配列には、GPIアンカーが細胞表層局在タンパク質と結合する際に認識される配列であるGPIアンカー認識付着シグナル配列の他に、4カ所の糖鎖結合部位がある。GPIアンカーの根元部分がPI−PLCにより切断された後、これらの糖鎖結合部位に結合した糖鎖と細胞壁を構成する多糖類とが共有結合することで、α―アグルチニンのC末端配列部分が細胞壁と結合し、α―アグルチニンは細胞表層に保持される。
【0037】
(糖鎖結合タンパク質ドメイン)
本発明において、糖鎖結合タンパク質ドメインとは、複数の糖鎖を有し、この糖鎖が、細胞壁中の糖鎖と相互作用又は絡み合うことによって、細胞表層に留まることのできるドメインをいう。例えば、レクチンなどの糖鎖結合部位や、α―アグルチニン、a―アグルチニン、FLOタンパク質などの凝集タンパク質の凝集機能ドメインなどが挙げられる。
(凝集機能ドメイン)
細胞表層局在タンパク質の凝集機能ドメインとはGPIアンカリングドメインよりもN末端側にあり、複数の糖鎖を有し、凝集に関与していると考えられているドメインをいう。
【0038】
本発明において、リパーゼを細胞表層に発現させるDNAは、以下の配列を有するDNAであることが好ましい。
(1)分泌シグナル配列、リパーゼの構造遺伝子配列、細胞表層局在タンパク質の一部をコードする配列及びGPIアンカリングドメインをコードする配列をこの順で有するDNA
(2)分泌シグナル配列、リパーゼの構造遺伝子配列、糖鎖結合タンパク質ドメインをコードする配列をこの順で有するDNA
(3)分泌シグナル配列、糖鎖結合タンパク質ドメインをコードする配列、リパーゼの構造遺伝子配列をこの順で有するDNA
(4)分泌シグナル配列、リパーゼの構造遺伝子配列、GPIアンカリングドメインをコードする配列をこの順で有するDNA
上記(2)、(3)の場合は、糖鎖結合タンパク質ドメインが、少なくとも細胞表層局在タンパク質の凝集機能ドメインを含む部分であることが好ましい。
【0039】
また、上記(1)〜(4)の場合、リパーゼの構造遺伝子配列と、細胞表層局在タンパク質の一部をコードする配列もしくは糖鎖結合タンパク質ドメインをコードする配列又はGPIアンカリングドメインをコードする配列との間には、適当な長さのリンカー配列が挟みこまれてもよく、アミノ酸の種類は限定されないが、アミノ酸8個〜21個をコードするDNA配列が好ましい。
【0040】
前記(1)〜(4)のDNAは、従来公知の手法を用いて合成することができる。例えば、(1)のDNAの場合、分泌シグナル配列とリパーゼの構造遺伝子配列との結合は、部位特異的突然変異法を用いて行うことができ、正確な分泌シグナル配列の切断と高活性なリパーゼBの発現が可能である。さらに、このようにして得られた配列と、細胞表層局在タンパク質の一部をコードする配列及びGPIアンカー付着シグナル配列とを結合すればよい。結合は、適切な制限酵素、リンカー等を用いて行うことができる。
【0041】
また、係るDNAは、プラスミドの形態でも、宿主となるサッカロミセス属酵母の遺伝子に組み込まれた形態でも、いずれでもよい。
【0042】
アーミング酵母は、係るDNAをサッカロミセス属酵母に導入することにより得ることができる。
【0043】
このようにして得られるアーミング酵母は、従来のアーミング酵母に比べ、エステル反応を触媒する活性が高く、エステル反応により適したアーミング酵母である。更に、従来のアーミング酵母よりも耐熱性が高く、高温でエステル反応を触媒できるため、エステル合成反応のような吸熱反応を効率よく触媒することが可能である。
一般式(1)で表される(メタ)アクリレートと脂肪族カルボン酸の使用比率は、好ましくは等モル、又は一般式(1)で表される(メタ)アクリレートと脂肪族カルボン酸に対して1.1〜1.5倍モルの比率で使用してもよい。
反応に使用する酵素量は、一般式(3)で表される(メタ)アクリレートと2級水酸基を有する脂肪酸の合計質量に対して、0.5〜10質量%、好ましくは、1〜5質量%、より好ましくは、2〜4質量%である。
本発明の反応は、一般式(1)で表される(メタ)アクリレートを基質とすると共に反応溶媒として用いて、酵素反応を行なうことができるが、酵素活性に影響しない有機溶媒を添加して行うこともできる。このような反応溶媒は、用いないことが好ましいが、反応系の撹拌効率の向上や基質の溶解促進を目的として、必要に応じて反応系に添加しても良い。
【0044】
用いられる溶媒としては、例えば、水、緩衝水溶液、塩水溶液、メタノール、エタノール、プロパノール、イソプロパノール、ブタノール、ターシャリーブタノール、アミルアルコール、イソアミルアルコール、エチレングリコール、グリセリン、モノグリム、ジグリム、アセトニトリル、硝酸メチル、ジエチルエーテル、ジメチルスルホキシド(DMSO)、ジメチルホルムアミド(DMF)、ジエチルエーテル、ジイソプロピルエーテル、THF、ジオキサン、酢酸メチル、酢酸エチル、酢酸プロピル、酢酸イソプロピル、酢酸ブチル、酢酸イソブチル、ベンゼン、トルエン、キシレン、ジクロロメタン、クロロホルム、四塩化炭素、ジクロロエタン、トリクロロエタン、クロロベンゼン、ジクロロベンゼン、トリクロロベンゼン、ピリジン、ペンタン、ヘキサン、ヘプタン、オクタン、イソオクタン、シクロヘキサン等が好ましく、これら2種以上の混合物でもかまわない。
反応で副生する水は、反応系内を減圧にし、窒素等の不活性ガスを吹き込むことにより、減圧留去することが好ましい。
【0045】
酵素活性を考慮すると反応系の水分を制御することが好ましく、水分量は10ppmから10%(100000ppm)、酵素の活性のためには10ppmから1%(10000ppm)添加することが好ましく、反応の平衡を考慮すると10ppmから1000ppmとすることが好ましい。これらの水分は、添加する酵素に含ませても、反応系に別途に添加しても良く、反応途中で順次、少量の水分を添加しても良い。
反応温度は、基質の溶解性と反応速度を考慮すると高温が好ましいが、一方、酵素の熱による失活を考慮すると低温が好ましく、相反する条件を満たす必要があり、極めて重要である。本発明に使用するリパーゼでは、好ましい反応温度として、30〜60℃の温度を挙げることができ、更に好ましくは、35〜45℃の温度を挙げることができる。
反応圧力は沸点による温度制御等を目的とし可変であり、減圧、加圧、0.13から1013khPa(1mmHgから7600mmHg)の範囲で反応可能であり、通常1.3から202.6KPa(10mmHgから1520mmHg)の範囲で可能である。反応時間は、反応系により異なり、一概に規定されないが、反応効率と収量を考慮すると、一般に3時間から48時間が好ましく、更に好ましくは5時間から30時間であって、この程度の反応時間で留めて、未反応の原料を全て回収し、再反応に用いることが好ましい。
【0046】
本発明に用いる反応装置は特に限定されるものではないが、基質と酵素の分散衝突性を向上させる為に、攪拌装置を有する反応槽での反応が好ましい。即ち、反応槽から留出する水を凝縮器に通し、次いで凝集したこれらをモレキュラーシーブス等の充填剤を充填した副生成物除去槽に通して、水をモレキュラーシーブスで吸着除去する。
副生成物除去槽に充填する充填材としては、(メタ)アクリレートと溶媒を吸着しないものを選択することが好ましく、モレキュラーシーブス、シリカゲル、塩化カルシウム、塩化カリウム、塩化ナトリウム、酸化マグネシウム、硫化マグネシウム、炭酸カリウム、硫酸ナトリウム等が挙げられる。モレキュラーシーブスは、副生物の種類により適宜変える必要があるが、副生生物が水、メタノールならば3Aタイプ、これらもしくはエタノール等では4Aタイプが好ましい。副生成物除去槽は反応槽の1%から100%の体積があれば十分である。
【0047】
モレキュラーシーブは、その重量の30%程度の重量の水等を吸着することが可能であるが、反応効率等の観点から過剰量を充填することが好ましい。反応中又は反応後に充填材は再生する必要があり、再生方法としては洗浄、加熱、減圧の方法がある。一般には水、アルコールでの洗浄後に加熱乾燥及び減圧乾燥を行う。温度は100℃以上が好ましく、180℃以上が速い再生を望める。
副生成物除去槽の規模を小さくするには、反応中に充填剤の再生を行うことが効率的であり、例えば2つ以上の除去槽を並列に使用し、使用槽を切換えて連続運転することが好ましい。また副生成物除去槽はソクスレー抽出機のような構造を有していることも好ましい。また副生成物除去槽を持たずに反応槽に直接、これらの充填物を反応槽内部に入れて反応させ、反応後に反応物から除去することも可能である。
撹拌装置はスクリュー翼、ヘリカル翼、ファードラー翼、タービン翼、パドル翼等を使用でき、均質な撹拌が可能な用いることが好ましい。撹拌回転数は撹拌効率、撹拌動力に依存するもののため、撹拌翼、反応槽のスケール等に依存するが、基質が不均質な場合、沈殿が生じない範囲で低速度が好ましい。
【実施例】
【0048】
以下に本発明を実施例により具体的に説明する。
【0049】
(実施例1) カプリル酸とHEMAとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
カプリル酸 28.8g、0.200mol
2−ヒドロキシエチルメタクリレート 28.6g、0.220mol
メトキシハイドロキノン 9mg
水 1.7g
リポザイムRM−IM 1.7g(モノマー全量に対し3重量%)
を入れ、油浴にて45℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。4時間後、反応系内を減圧(3.5kPa)し、かつ乾燥空気を供給した。反応開始より24時間後の反応転化率は>97%(GCピーク)であった。反応系内の減圧および窒素の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。室温にて透明液体(純度93%(GCピーク比)、ハーゼン色数20)を得た。
【0050】
本発明において反応追跡および純度の決定はガスクロマトグラムにより行った。
<ガスクロマトグラム条件>
測定機器: 島津製 GC−2010
カラム: キャピラリーカラムTC−5(0.32mmI.D.×30m,df=0.25μm,GLサイエンス製)
インジェクション方法: スプリット法(30:1)
インジェクション温度: 300℃
検出方法:FID
検出器温度:320℃
カラム温度:100℃(2分間保持)→20℃/分→300℃(13分間保持)
また、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
<GC−MS条件>
測定機器: 日本電子製 JMS−K9(四重極型)
カラム: キャピラリーカラムDB−5ms(0.25mmI.D.×30m,df=0.25μm,アジレントテクノロジー製)
インジェクション方法: スプリット法(30:1)
インジェクション温度: 300℃
カラム温度:50℃(2分間保持)→15℃/分→300℃
イオン化方法:EI または CI(イソブタン)
【0051】
(実施例2)カプリル酸メチルとHEMAとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
カプリル酸メチル 34.8gl、0.220mol
2−ヒドロキシエチルメタクリレート 31.5g、0.242mol
メトキシハイドロキノン 7mg
リポザイムRM−IM 3.3g(モノマー全量に対し5重量%)
を入れ、油浴にて45℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。30分後、反応系内を減圧(2.5kPa)し、かつ乾燥空気を供給した。反応開始より8時間後、反応系内の減圧度を1.5kPa、ついで反応開始より24時間後に0.6kPaにまで高め反応を継続した。反応開始より24時間後の反応転化率は93%、36時間後の転化率97%であった。反応開始より72時間後、カプリル酸メチルのピークの消失を確認した後、反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。室温にて透明液体を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0052】
(比較例1)カプリル酸とHEMAとの反応(化学合成例)
デカンターおよび気体導入管を取り付けた三ツ口フラスコに、
カプリル酸 28.8g、0.200mol
2−ヒドロキシエチルメタクリレート 31.2g、0.240mol
メトキシハイドロキノン 0.1g
p−トルエンスルホン酸 1.5g
トルエン 240.0g
を入れ、油浴にて125〜130℃に加温した。攪拌はマグネチックスターラーを用い、また、乾燥空気をバブリングにより供給した。トルエンの還流とともに縮合水の生成が確認された。反応開始より6時間後、縮合水の生成が停止し、また、反応追跡結果より反応の進行が認められないため反応を停止した。このときの転化率は91%であった。反応混合物を5%炭酸ナトリウム水溶液によるアルカリ洗浄、その後、飽和食塩水および蒸留水による洗浄を行った。反応混合物を無水硫酸マグネシウムで乾燥後、トルエンを留去し、淡黄色液体(純度85%(GCピーク比)、ハーゼン色数150)を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0053】
(比較例2) カプリル酸とHEMAとの反応(選択性を示さないリパーゼを用いた場合の実験例)
酵素触媒としてリポザイムRM−IMの代わりにノボザイム435(Candida antarctica由来リパーゼBの固定化酵素)を用いた以外は実施例1と同様の反応を行った。
【0054】
GC分析による反応追跡の結果、反応の進行に伴い目的物のピーク(Rt:8.9分)以外のピークも複数種類生成した。反応開始より24時間後の反応転化率は>98%であった。反応系内の減圧および窒素の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。室温にて透明液体を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0055】
得られた化合物についてGC分析を行ったところ、目的化合物の純度は51%(GCピーク比)であった。
【0056】
(実施例1と比較例1および2の分析)
GC分析およびGC質量分析により反応生成物の分析を行い、生成物の同定及びGCピーク面積より反応選択率(純度)を算出した。
【0057】
【表1】
【0058】
(表中、EGDMAはエチレングリコールジメタクリレート、HO−EG−C8はエチレングリコールモノカプリレート、C8−EG−C8はエチレングリコールジカプリレートを示す。)
【0059】
実施例1および比較例1、2の結果より、実施例1では高純度、且つ、着色の無い目的物が得られた。
【0060】
(実施例3) カプロン酸とHEMAとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
カプロン酸 28.3、0.250mol
2−ヒドロキシエチルメタクリレート 35.8g、0.275mol
メトキシハイドロキノン 6mg
水 3.2g
リポザイムRM−IM 1.9g(モノマー全量に対し3重量%)
を入れ、油浴にて45℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。1.5時間後、反応系内を減圧(3.5kPa)し、かつ乾燥空気を供給した。反応開始より8.5時間後、反応系内の減圧度を5.0kPaにまで高め反応を継続した。反応開始より48時間後、反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。室温にて透明液体を得た。目的物の純度は90%(GCピーク比)であった。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0061】
(実施例4) パルミチン酸とHEMAとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
パルミチン酸38.5g、0.150mol
2−ヒドロキシエチルメタクリレート 21.5g、0.165mol
メトキシハイドロキノン 7mg
リポザイムRM−IM 1.8g(モノマー全量に対し3重量%)
を入れ、油浴にて60℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。反応開始時にはパルミチン酸は一部溶解した懸濁状態であった。15分後、反応系内を減圧(3.5kPa)し、かつ乾燥空気を供給した。反応の進行に伴い固体のパルミチン酸はすべて溶解した。反応開始より20時間後(反応転化率>98%)、反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。生成物の純度は85%(GCピーク比)であった。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0062】
(実施例5) オレイン酸とHEMAとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
オレイン酸 42.4g、0.150mol
2−ヒドロキシエチルメタクリレート 21.5g、0.165mol
メトキシハイドロキノン 12.7mg
リポザイムRM−IM 2.0g(モノマー全量に対し3重量%)
を入れ、油浴にて45℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。30分後、反応系内を減圧(3.5kPa)し、かつ乾燥空気を供給しながら縮合水を除去した。反応開始より2時間後、反応系内の減圧度を2.0kPaにまで高め反応を継続した。反応開始より18時間後の反応転化率は>96%であった。反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。室温にて淡黄色液体(ハーゼン色数<1)を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0063】
(比較例3) オレイン酸とHEMAとの反応(化学合成法)
デカンターおよび気体導入管を取り付けた四ツ口フラスコに。
オレイン酸 42.4g、0.150mol
2−ヒドロキシエチルメタクリレート 23.4g、0.180mol
メトキシハイドロキノン 100mg
トルエン 270.0g
p−トルエンスルホン酸一水和物 2.0g(モノマー全量に対し3重量%)
を入れ、油浴にて125〜130℃に加温した。攪拌はマグネチックスターラーを用い、また、乾燥空気をバブリングにより供給した。トルエンの還流とともに縮合水の生成が確認されたが、反応溶液は褐色に変化した。反応開始より4時間後、縮合水の生成が停止し、また、反応追跡結果より反応の進行が認められないため反応を停止した。このときの転化率は76%であった。反応混合物のアルカリ洗浄を試みたが乳化がひどく、水洗は困難であった。また、アルカリおよび水洗による反応混合物からの脱色はできず得られた化合物は褐色液体(ハーゼン色数>18)であった。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0064】
(実施例6) 10−ウンデセン酸とHEMAとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
10−ウンデセン酸 36.9g、0.200mol
2−ヒドロキシエチルメタクリレート 28.6g、0.220mol
メトキシハイドロキノン 7mg
水 1.3g
リポザイムRM−IM 2.0g(モノマー全量に対し3重量%)
を入れ、油浴にて45℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。4時間後、反応系内を減圧(6.0kPa)し、かつ乾燥空気を供給した。反応開始より7時間後、反応系内の減圧度を3.5kPaにまで高め反応を継続した。反応開始より28時間後(反応転化率97%)、反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。室温にて透明液体(純度90%)を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0065】
(実施例7) 脱水ヒマシ油脂肪酸DCO/FAとHEMAとの反応(酸素に不安定な化合物の合成例)
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
DCO/FA42.4g、0.150mol
(平均値より算出)
2−ヒドロキシエチルメタクリレート 21.5g、0.165mol
メトキシハイドロキノン 7mg
リポザイムRM−IM 1.9g(モノマー全量に対し3重量%)
を入れ、油浴にて45℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。15分後、反応系内を減圧(2.5kPa)し、かつ乾燥窒素を供給した。反応開始より2.5時間後、反応系内の減圧度を1.5kPaにまで高め反応を継続した。反応開始より20時間後、反応系内の減圧および窒素の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。室温にて淡黄色透明液体を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0066】
得られた反応生成物のGC/MS分析により、原料DCO/FA(混合物)の各ピークとHEMAとの選択的なエステル化反応によって生成した混合物であることが示された(図1(原料)および図2(生成物)にGCチャートを示す。)
【0067】
(比較例4) 脱水ヒマシ油脂肪酸DCO/FAとHEMAとの反応(酸素に不安定な化合物の化学合成例)
フラスコに
DCO/FA42.4g、0.150mol
2−ヒドロキシエチルメタクリレート 21.5g、0.165mol
メトキシハイドロキノン 100mg
トルエン 270.0g
を入れ。油浴にて100℃に加温した。攪拌はマグネチックスターラーを用い、また、乾燥空気をバブリングにより供給した。反応開始より2.5時間後、沈殿物が生じたため反応を停止した。
【0068】
実施例7および比較例4の結果より、酸素および熱等に不安定な脱水ヒマシ油脂肪酸などを原料に用いた反応にも適用可能である。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0069】
(実施例8) カプリン酸とHEAとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
カプリン酸 37.9g、0.220mol
2−ヒドロキシエチルアクリレート 26.8g、0.231mol
メトキシハイドロキノン 8mg
水 1.3g
リポザイムRM−IM 3.2g(モノマー全量に対し5重量%)
を入れ、油浴にて40℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。2.5時間後、反応系内を減圧(5.0kPa)し、かつ乾燥空気を供給した。反応開始より6時間後、反応系内の減圧度を一時停止した。反応開始より10時間後(転化率90%)、反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。室温にて透明液体(純度93%(GCピーク比))を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0070】
(実施例9) パルミチン酸とHEAとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
パルミチン酸153.9g、0.600mol
2−ヒドロキシエチルアクリレート 76.6g、0.630mol
メトキシハイドロキノン 31mg
リポザイムRM−IM 6.9g(モノマー全量に対し3重量%)
を入れ、油浴にて60℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。反応開始時にはパルミチン酸は一部溶解した懸濁状態であった。1時間後、反応系内を減圧(5.0kPa)し、かつ乾燥空気を供給した。反応の進行に伴い固体のパルミチン酸はすべて溶解した。反応開始より2時間後、反応系内の減圧度を3.5kPaにまで高め反応を継続した。反応開始より24時間後(反応転化率>98%)、反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0071】
少量の高融点副生成物を除去するために以下の精製を行った。
【0072】
反応混合物をメタノールに分散し不溶物を濾過により取り除いた。濾液を濃縮後に得られたオイル状液体を氷水に注ぎ分散した。オイル状液体は固化した。白色固体を濾別、乾燥を行い、室温にて白色結晶(純度95%(GCピーク比)、融点 27.9℃)を得た。
【0073】
(実施例10) ステアリン酸とHEAとの反応(溶剤使用)
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)を取り付けたディーンスタークトラップおよび気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
ステアリン酸128.0g、0.450mol
2−ヒドロキシエチルアクリレート 57.5g、0.495mol
メトキシハイドロキノン 38mg
リポザイムRM−IM 3.7g(モノマー全量に対し2重量%)
トルエン 100g
を入れ、油浴にて45℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。27.5時間後、反応系内を減圧(50kPa)し、かつ乾燥空気を供給した。反応開始より32時間後、反応温度を65℃にまで昇温し、トルエンを除去しながら反応を継続した。反応開始より48時間後、反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0074】
未反応原料を除去するために以下の精製を行った。
反応混合物をトルエンに溶解し、5%炭酸水素ナトリウム水溶液、飽和食塩水、蒸留水でそれぞれ2回洗浄を行った。無水硫酸マグネシウムで乾燥後、トルエンを減圧留去し、室温にて白色結晶(純度96%(GCピーク比)、融点:34.5℃)を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0075】
(実施例11) カプリン酸とHBMAとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
カプリン酸 25.8g、0.150mol
2−ヒドロキシブチルメタクリレート 24.9g、0.158mol
メトキシハイドロキノン 5mg
水 1g
リポザイムRM−IM 1.5g(モノマー全量に対し3重量%)
を入れ、油浴にて40℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。5時間後、反応系内を減圧(8.0kPa)し、かつ乾燥空気を供給した。反応開始より30時間後、減圧を停止しそのまま反応を続けた。反応開始より48時間後、再度、反応系内を減圧(5.0kPa)し、かつ乾燥空気を供給した。反応開始より58時間後、反応系内の減圧度を2.5kPaにまで高め反応を継続した。反応開始より74時間後、反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。室温にて透明液体を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0076】
(実施例12) カプリン酸とHEAAmとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
カプリン酸 43.1g、0.250mol
2−ヒドロキシエチルメタクリレート 30.2g、0.250mol
メトキシハイドロキノン 43mg
ノボザイム435 1.5g(モノマー全量に対し2重量%)
を入れ、油浴にて45℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。15分後、反応系内を減圧(3.5kPa)し、かつ乾燥空気を供給した。反応開始より4時間後、反応系内の減圧度を2.5kPaにまで高め反応を継続した。結晶析出のため反応開始より23時間後に60℃に昇温した。反応開始より24時間後、反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。室温にて白色固体(純度90%(GCピーク比)、融点:44.7℃)を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0077】
(実施例13) パルミチン酸とHEAAmとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
パルミチン酸153.9g、0.600mol
2−ヒドロキシエチルアクリルアミド 72.5g、0.630mol
メトキシハイドロキノン 154mg
ノボザイム435 4.5g(モノマー全量に対し2重量%)
を入れ、油浴にて65℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。15分後、反応系内を減圧(15.0kPa)し、かつ乾燥空気を供給した。反応開始より4時間後、結晶析出のため反応温度を70℃に昇温した。反応開始より8時間後、反応系全体が結晶化したためトルエン100mlを加え濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。得られた濾液に更にトルエン500mlを加え、室温に放冷し、析出した白色結晶(純度94%(GCピーク比)、融点 73.9℃)を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0078】
(実施例14) 10−ウンデセン酸とHEAAmとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
10−ウンデセン酸 46.1g、0.250mol
2−ヒドロキシエチルアクリルアミド 30.2g、0.263mol
メトキシハイドロキノン 46mg
ノボザイム435 0.8g(モノマー全量に対し1重量%)
を入れ、油浴にて45℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。15分後、反応系内を減圧(5kPa)し、かつ乾燥空気を供給した。反応開始より2時間後、反応系内の減圧度を2.5kPaにまで高め反応を継続した。反応開始より24時間後、反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。室温にて固化した。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0079】
(実施例15) アジピン酸ジビニルとHEMAとの反応(リポザイムTL−IMを用いた反応)
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
アジピン酸ジビニル 99.1g、0.501mol
2−ヒドロキシエチルメタクリレート 136.6g、1.050mol
メトキシハイドロキノン 50mg
リポザイムTL−IM 7.1g(モノマー全量に対し3重量%)
を入れ、油浴にて40℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。15分後、反応系内を減圧(30kPa)し、かつ乾燥空気を供給した。反応開始より4時間後、反応系内の減圧度を25kPa、ついで8.5時間後に10kPaにまで高め反応を継続した。反応開始より28時間後、反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。得られた濾液を飽和食塩水で洗浄後、室温にて淡黄色透明液体(純度92%(GCピーク比))を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0080】
(実施例16) アジピン酸ジビニルとHEMAとの反応(ノボザイム435を用いた反応)
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
アジピン酸ジビニル 19.8g、0.100mol
2−ヒドロキシエチルメタクリレート 27.3g、0.210mol
メトキシハイドロキノン 23mg
ノボザイム435 1.4g(モノマー全量に対し3重量%)
を入れ、油浴にて40℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。反応系内を減圧(10kPa)し、かつ乾燥空気を供給した。反応開始より2時間後、反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。室温にて淡黄色透明液体(純度86%(GCピーク比))を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0081】
(実施例17) セバシン酸とHEMAとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
セバシン酸 40.5g、0.200mol
2−ヒドロキシエチルメタクリレート 53.4g、0.410mol
メトキシハイドロキノン 8mg
水 1.9g
リポザイムRM−IM 4.7g(モノマー全量に対し5重量%)
を入れ、油浴にて55℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。反応開始時にはセバシン酸は一部溶解した懸濁状態であった。3時間後、反応系内を減圧(5.0kPa)し、かつ乾燥空気を供給した。反応の進行に伴い固体のセバシン酸はすべて溶解した。反応開始より24時間後、反応系内の減圧度を2.5kPaにまで高め反応を継続した。反応開始より30時間後、反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。室温にて透明液体(純度90%(GCピーク比))を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0082】
(実施例18) ドデカン二酸とHEMAとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
ドデカン二酸34.6g、0.150mol
2−ヒドロキシエチルメタクリレート 40.0g、0.308mol
メトキシハイドロキノン 7mg
水 1.5g
リポザイムRM−IM 3.7g(モノマー全量に対し5重量%)
を入れ、油浴にて50℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。反応開始時にはドデカン二酸は一部溶解した懸濁状態であった。3時間後、反応系内を減圧(5.0kPa)し、かつ乾燥空気を供給した。反応の進行に伴い固体のドデカン二酸はすべて溶解した。反応開始より25時間後、反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。室温にて透明液体(純度85%(GCピーク比))を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0083】
(実施例19) カプリル酸メチルとHEMAとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
カプリル酸 43.2g、0.300mol
2−ヒドロキシエチルメタクリレート 42.9g、0.330mol
メトキシハイドロキノン 15mg
CALB−アーミング酵母 7.5g(モノマー全量に対し8.7重量%)
を入れ、油浴にて40℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。3時間後、反応系内を減圧(5kPa)し、かつ乾燥空気を供給した。反応開始より24時間後、反応系内の減圧度を1kPaにまで高め反応を継続した。反応開始より46時間後の反応転化率は96.6%であった。この時点で反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)によりCALB−アーミング酵母を濾別した。室温にて透明液体を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0084】
(実施例20)12−ヒドロキシステアリン酸とHEMAとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
12−ヒドロキシステアリン酸 150.2g、0.500mol
メトキシハイドロキノン 45mg
を入れ、油浴(90℃)にて12−ヒドロキシステアリン酸を融解した後、80℃まで放冷した。
2−ヒドロキシエチルメタクリレート 71.6g、0.550mol
リポザイムRM−IM 6.7g(モノマー全量に対し3重量%)
を加えて反応開始した。反応温度70℃まで徐々に放冷し、2時間後、反応系内を減圧(5.0kPa)かつ乾燥空気を供給した。反応開始より20時間後に減圧を3.5kPa、30時間後に2.0kPaにまで高め反応を継続した(反応転化率96%)。反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。室温にて白色ワックス状固体(収量187.3g、収率88%、純度92%(GCピーク比))を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0085】
以下に上記実施例で用いたアーミング酵母の調整法を記す。
(製造例1)<CALB表層発現プラスミドの作製>
(1−A.キャンディダ・アンタルクチカ由来リパーゼB遺伝子の取得)
次のようにしてキャンディダ・アンタルクチカ由来リパーゼB(CALB)遺伝子を取得した。
【0086】
すなわち、キャンディダ・アンタルクチカCBS6678株ゲノムをテンプレートとし、従来公知の方法でDNA合成装置にて合成した、配列番号1及び2に示す塩基配列からなるプライマーを用いてPCR増幅を行い、BglII及びXhoIで切断して、約1000bpの長さのBglII−XhoI断片(BglII−XhoI ProCALB CBS6678断片)を得た。このCALBは、Uppenbergらの文献(structure,2:293(1994))に記載され、もっとも広く知られているキャンディダ・アンタルクチカ LF058株由来リパーゼBのアミノ酸配列(CALB WT)と比較して7アミノ酸が変異している。すなわち25番目のアラニンがトレオニンに、28番目のセリンがトレオニンに、31番目のセリンがトレオニンに、46番目のグルタミンがグリシンに、89番目のアラニンがトレオニンに、97番目のアスパラギンがアルギニンに、286番目のバリンがイソロイシンにそれぞれ変異している、特徴的なCALBであった。
(1−B.CALB遺伝子導入プラスミドの取得)
CALB遺伝子中への変異の導入を効率よく行うため、上記1−Aで得られたCALB CBS6678遺伝子を、一度サイズの小さいプラスミドに導入した。
【0087】
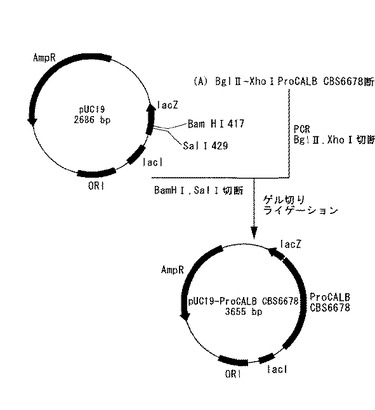
すなわち、プラスミドpUC19(TaKaRa BIO社製)をBamHI及びSalIで切断し、上記Aで得られたBglII−XhoI ProCALB CBS6678断片を挿入して、プラスミドpUC19−ProCALB CBS6678を得た。作製の模式図を図1に示す。
【0088】
(1−C.CALB1遺伝子ならびにCALB1導入プラスミドの取得)
上記1−Bで得られたプラスミドpUC19−ProCALB CBS6678をテンプレートとし、従来公知の方法でDNA合成装置にて合成した、配列番号3及び4に示す塩基配列からなるプライマーを用いてPCR増幅を行うことにより、CALB CBS6678中にポイントミューテーションを導入し、CALB CBS6678中のアミノ末端から25番目のトレオニンをアラニンに、28番目のトレオニンをセリンに、31番目のトレオニンをセリンにそれぞれ置換したCALB1遺伝子を含むプラスミドpUC19−ProCALB1を得た。
【0089】
(1−D.CALB2遺伝子ならびにCALB2導入プラスミドの取得)
上記1−Cで得られたプラスミドpUC19−ProCALB1をテンプレートとし、従来公知の方法でDNA合成装置にて合成した、配列番号5及び6に示す塩基配列からなるプライマーを用いてPCR増幅を行うことにより、CALB1中にポイントミューテーションを導入し、CALB1中のアミノ末端から46番目のグリシンをグルタミンに置換したCALB2遺伝子を含むプラスミドpUC19−ProCALB2を得た。
【0090】
(1−E.リンカー導入型CALB遺伝子の取得)
上記1−Dで得られた各CALB2導入プラスミドをテンプレートとし、従来公知の方法でDNA合成装置にて合成した、配列番号7及び8に示す塩基配列からなるプライマーを用いてPCR増幅を行い、次いでBglII及びXhoIで切断して、約1000bpの長さのBglII−XhoI FLAG−ProCALB2断片を得た。BglII−XhoI FLAG−ProCALB2断片は、FLAG tag、リパーゼのプロ配列、及びCALB2の成熟タンパク質配列を有している。ここで、プロ配列とは、リパーゼの立体構造を形成するために必要とされる配列のことであり、FLAG tagとは、リンカー配列のことであり、成熟タンパク質配列とは、実際にリパーゼの触媒反応機能を有する配列のことである。
【0091】
(1−F.CALB表層発現カセットの作製)
CALB表層発現カセットは、発現プロモーターの配列とターミネーターの配列、並びにこれらの間にFLO1誘導体であるShort型FLO1アンカー遺伝子と上記1−Eで得られたリンカー導入型CALB遺伝子を接続したものをそれぞれ有するDNA配列である。該CALB表層発現カセットを作製するために、以下の操作を行った、作製の模式図を図2に示す。
【0092】
すなわち、プラスミドpWIFS(T.Matsumotoら、Appl.Environ.Microbiol.,68:4517(2002))をBglIIとXhoIで切断し、上記1−Eで得られたBglII−XhoI FLAG−ProCALB2断片を挿入して、pWIFS−FLAG−ProCALB2を得た。そしてこれをBssHIIで切断し、約6300bpのCALB表層発現カセットを得た。
【0093】
(1−G.CALB表層発現プラスミドの作製)
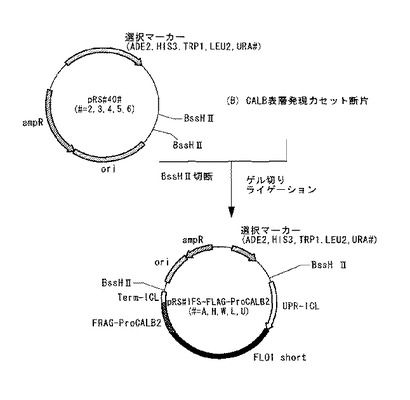
目的のDNAを有するプラスミドは、上記1−Fで得られたCALB表層発現カセットを、各種選択マーカーを有するプラスミドに導入することで得られる。作製の模式図を図3に示す。
【0094】
すなわち、プラスミドpRS402(ATCC87477)、pRS403(ATCC87514)、pRS404(ATCC87515)、pRS405(ATCC87516)、pRS402(ATCC87517)をそれぞれBssHIIで切断し、上記Fで得られたCALB表層発現カセットを挿入してpRSAIFS−FLAG−ProCALB2、pRSHIFS−FLAG−ProCALB2、pRSWIFS−FLAG−ProCALB2、pRSLIFS−FLAG−ProCALB2、pRSUIFS−FLAG−ProCALB2を得た。
【0095】
(製造例2)<ゲノム組み込み型二倍体CALB−アーミング酵母の調製>
(2−A.サッカロミセス属酵母への遺伝子の導入)
上記1−Gで得られたCALB表層発現プラスミドを、表1に示す制限酵素を用いて、それぞれ選択マーカー中に存在する制限酵素サイトで切断した。次いで、YEAST MAKERTM(Clontech Laboratories Inc. USA)を用いて、サッカロミセス・セレヴィシエ(Saccharomyces cerevisiae、以下、サッカロミセス・セレヴィシエと略記する)YPH499(MATa,ura3,lys2,ade2,his4,trp1,leu2)には、pRSUIFS−FLAG−ProCALB2、pRSLIFS−FLAG−ProCALB2、pRSHIFS−FLAG−ProCALB2、pRSAIFS−FLAG−ProCALB2をこの順で、サッカロミセス・セレヴィシエYPH500(MATalpha,ura3,lys2,ade2,his4,trp1,leu2)には、pRSUIFS−FLAG−ProCALB2、pRSLIFS−FLAG−ProCALB2、pRSWIFS−FLAG−ProCALB2、pRSAIFS−FLAG−ProCALB2をこの順で、それぞれのゲノム中に導入した。各形質転換の選択においては、アミノ酸並びに核酸(0.002%L−ヒスチジン、0.01%L−ロイシン、0.002%L−トリプトファン、0.002%L−リシン、0.002%アデニン、0.002%ウラシル)のうち、選択に必要なものを含むSD寒天培地(2%グルコース、0.67% Yeast Nitrogen Base without amino acids)を用いて培養した。生育した酵母を選択し、ゲノム組み込み型CALBアーミング酵母である、サッカロミセス・セレヴィシエYPH499−U、YPH499−UL、YPH499−ULH、YPH499−ULHA、並びにYPH500−U、YPH500−UL、YPH500−ULW、YPH500−ULWAアーミング酵母を得た
【0096】
表2 各プラスミドの選択マーカー中における切断に用いた制限酵素
【0097】
【表2】
【0098】
(2−B.一倍体サッカロミセス−CALBアーミング酵母の調製)
上記2−Aで創製したサッカロミセス・セレヴィシエYPH499−U、YPH499−UL、YPH499−ULH、YPH499−ULHA、並びにYPH500−U、YPH500−UL、YPH500−ULW、YPH500−ULWAアーミング酵母を、SDC培地(2%グルコース、0.67%Yeast Nitrogen Base without amino acids、2%カザミノ酸、appropriate amino acids and nucleic acids)に植菌し、30℃で6日間浸透培養した。次いで、遠心分離により培地と菌体に分離し、得られた菌体を一倍体サッカロミセス−CALBアーミング酵母とした。得られた一倍体サッカロミセス−CALBアーミング酵母の、酪酸p−ニトロフェニル(以下、PNPBと略記する)を基質とした30℃におけるリパーゼ活性を確認した。
【0099】
(2−C.二倍体サッカロミセス−CALBアーミング酵母の創製)
一倍体アーミング酵母YPH499−ULHA、YPH500−ULWAをYPD培地(2%グルコース、2%peptone、1%yeast extract)に植菌し、30℃で1時間培養後、遠心分離により培地と菌体に分離し、得られた菌体を混合してYPD培地に再懸濁し、30℃で一晩培養した。これを適切な菌体密度に希釈した後、SD+K寒天培地(2%グルコース、0.67%Yeast Nitrogen Base without amino acids、0.002%L−リシン)を用いて培養した。生育した酵母を選択し、ゲノム導入型二倍体CALB−アーミング酵母である、サッカロミセス・セレヴィシエYPH501−U2L2HWA2アーミング酵母を得た。
【0100】
(2−D.二倍体サッカロミセス−CALBアーミング酵母の調製)
上記2−Cで得られたサッカロミセス・セレヴィシエYPH501−U2L2HWA2アーミング酵母を、SDC+K培地(2%グルコース、0.67%Yeast Nitrogen Base without amino acids、2%カザミノ酸、0.002%L−リシン)に植菌し、30℃で6日間浸透培養した。次いで、遠心分離により培地と菌体に分離し、PNPBを基質とした菌体の30℃におけるリパーゼ活性(エステル分解活性)を確認した。
【0101】
エステル分解活性の測定には、PNPB法を用いた。培養液を遠心分離することで菌体を回収し、得られた菌体を蒸留水で2回洗浄した。次いで、菌体を20mMリン酸緩衝液に懸濁し、30℃で5分間プレインキュベートした。一方、基質にはPNPBを用いた。PNPBは少量のエタノールに溶解させた後、蒸留水で適当に希釈して基質液を調製した。プレインキュベートした菌体懸濁液へ基質液を加え、よく撹拌し、振盪させながら30℃で10分間反応させた。次いで、5%トリクロロ酢酸水溶液を添加し、反応を停止したのち、遠心分離し、上清を回収し、200mMリン酸緩衝液で希釈し、400nmの吸光度を測定した。そして、生成したp−ニトロフェノール(PNP)量を求め、エステル分解活性とした。活性を定義するにあたり、PNPが1分間に1μmol生成する酵素量を1Uと定義した。
【図面の簡単な説明】
【0102】
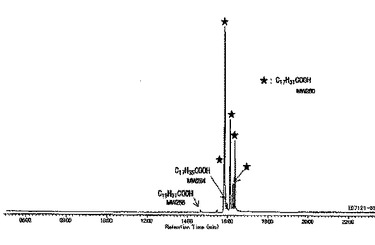
【図1】反応に用いた脱水ヒマシ油脂肪酸DCO/FAのGCチャートである。
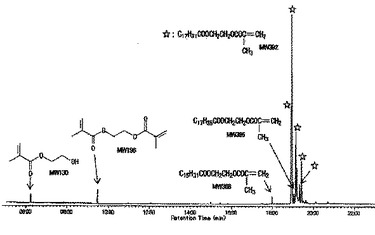
【図2】実施例7における生成物のGCチャートである。保持時間18〜20分に観測されるピークの分子量m/zは、いずれも原料(図1)のピークにHEMAが付加した分子量を示すことから、目的物であることが分かる。
【図3】製造例におけるプラスミドpUC19−ProCALB CBS6678作成の模式図である。
【図4】製造例におけるCALB表層発現カセット作成の模式図である。
【図5】製造例におけるCALB表層発現プラスミド作成の模式図である。
【技術分野】
【0001】
本発明は、酵素反応を用いた、(メタ)アクリル酸誘導体と、水酸基を有してもよい脂肪族カルボン酸、又は多価カルボン酸とを反応させることを特徴とする高分子材料モノマーとして有用なアクリレート化合物の製造方法に関する。
【背景技術】
【0002】
2‐ヒドロキシエチル(メタ)アクリレート(HE(M)A)に代表されるヒドロキシル基を有する(メタ)アクリル酸誘導体は種々のアクリル樹脂の原料に用いられている。アクリル樹脂中にヒドロキシル基を導入することで、樹脂の水溶性付与やポリイソシアネート化合物との反応による架橋反応などが可能になり、塗料や粘/接着剤などの分野で広く用いられている。しかしながら、HEMAのようなアクリレートをアクリル樹脂に共重合した場合、アクリル樹脂の主鎖近傍にヒドロキシル基が存在するためポリイソシアネート化合物との硬化物物性が脆くなるといった問題があった。このような問題解決のためPEGなどの片末端に(メタ)アクリル基を導入したモノマーが用いられているが、吸水率の上昇などの問題があった。
【0003】
一方、再生可能資源として植物由来原料を用いた材料の開発が近年盛んに行われている。脂肪酸を原料に用いた疎水性の高い(メタ)アクリレート化合物およびその製造方法としては、以下の特許文献1、特許文献2、及び特許文献3の開示がある。
しかしながら、熱、酸、酸素等に不安定な原料を用いた場合には著しい着色や分解、ゲル化などの副反応により製造できないこともある等の問題点を有していた。
更には、ヒドロキシ脂肪酸とアクリル酸アルカノールを原料に用いたエステル化反応では、アクリル酸アルカノールおよびヒドロキシ脂肪酸のカルボキシル基間の交差反応に加え、それぞれのヒドロキシル基間の交差反応のため上記特許文献における手法では、更にヒドロキシル基の官能基保護/脱保護等の工程が必要となるなどの問題があった。
【0004】
【特許文献1】特開平2−110115号
【特許文献2】特開平5−229991号
【特許文献3】特許第3829101号
【発明の開示】
【発明が解決しようとする課題】
【0005】
本発明が解決しようとする課題は、酵素反応を用いた、(メタ)アクリル酸誘導体と、水酸基を有してもよい脂肪族カルボン酸、又は多価カルボン酸とを反応させることを特徴とする高分子材料モノマーとして有用なアクリレート化合物の製造方法の提供にある。
【課題を解決するための手段】
【0006】
本発明者らは、鋭意研究を重ねた結果、アスペルギルス(Aspergillus oryzae)属、アクロモバクター(Achromobacter)属、バシラス(Bacillus)属、カンジダ(Candida)属、クロモバクター(Chromobacter)属、フザリウム(Fusarium)属、フミコラ(Humicola)属、ハイフォザイマ(Hyphozyma)属、シュードモナス(Pseudomonas)属、リゾムーコル(Rhizomucor)属、リゾプス(Rhizopus)属、又はテルモマイセス(Thermomyces)属の微生物から得られるリパーゼ(A)又はリパーゼを細胞表層に提示したアーミング酵母(B)(以下リパーゼ・アーミング酵母(B)と記す。)を用いて、温和な条件下で、(メタ)アクリル酸誘導体と、水酸基を有してもよい炭素数6〜24の脂肪族カルボン酸とのエステル化反応により、高分子材料モノマーとして有用なアクリレート化合物の製造方法を見出し、本発明を完成させるに至った。
【0007】
即ち、本発明は、リパーゼ(A)又はリパーゼ・アーミング酵母(B)の存在下に、一般式(1)
【0008】
【化1】
【0009】
(式中、R1は水素原子又はメチル基、Xは酸素原子又は窒素原子、nは2〜4の整数を表す。)
で表される(メタ)アクリル酸誘導体と、一般式(2)
【0010】
【化2】
【0011】
(式中、R2は水酸基又はカルボキシ基を有してもよい炭素数5〜23のアルキル基又はアルケニル基を表す。)
で表されるカルボン酸とを反応させる一般式(3)
【0012】
【化3】
【0013】
(式中、R1は水素原子又はメチル基、Xは酸素原子又は窒素原子、nは2〜4の整数、R2は水酸基又はカルボキシ基を有してもよい炭素数5〜23のアルキル基又はアルケニル基を表す。)
で表されるアクリレート化合物の製造方法に関する。
【発明の効果】
【0014】
本発明によれば、(メタ)アクリル酸誘導体と、水酸基を有してもよい脂肪族カルボン酸、又は多価カルボン酸とを反応させることを特徴とする高分子材料モノマーとして有用なアクリレート化合物の製造方法を提供できる。
【発明を実施するための最良の形態】
【0015】
本発明は、リパーゼ(A)又はリパーゼ・アーミング酵母(B)の存在下に、一般式(1)
【0016】
【化4】
【0017】
(式中、R1は水素原子又はメチル基、Xは酸素原子又は窒素原子、nは2〜4の整数を表す。)
で表される(メタ)アクリル酸誘導体と、一般式(2)
【0018】
【化5】
【0019】
(式中、R2は水酸基又はカルボキシ基を有してもよい炭素数5〜23のアルキル基又はアルケニル基を表す。)
で表されるカルボン酸とを反応させる一般式(3)
【0020】
【化6】
【0021】
(式中、R1は水素原子又はメチル基、Xは酸素原子又は窒素原子、nは2〜4の整数、R2は水酸基又はカルボキシ基を有してもよい炭素数5〜23のアルキル基又はアルケニル基を表す。)
で表されるアクリレート化合物の製造方法を提供する。
【0022】
一般式(1)で表される化合物は、(メタ)アクリル酸と2価アルコール又はヒドロキシアミンとの脱水縮合反応によって、通常公知の方法により得ることができる。
式中、R1は水素原子であってもメチル基であってもよい。また、Xが酸素原子を表すときは、一般式(1)で表される化合物はエステル誘導体であり、Xが窒素原子を表すときは、一般式(1)で表される化合物はアミド誘導体であるが、本発明ではどちらであってもよい。nは2〜4の整数であれば特に制限なく用いることが可能であるが、特に好ましくは、n=2の場合である。
また、一般式(1)で表される化合物とのエステル反応に供される脂肪族カルボン酸は、一般式(2)
【0023】
【化7】
【0024】
(式中、R2は水酸基又はカルボキシ基を有してもよい炭素数5〜23のアルキル基又はアルケニル基を表す。)
で表されるカルボン酸であって、得られるエステル誘導体の用途によって、R2に水酸基又はカルボキシ基を有していても有していなくてもよい。
本発明に使用される水酸基を有してもよい炭素数6〜24のカルボン酸の例としては、例えば、カプロン酸、カプリル酸、カプリン酸、ウンデシレン酸、パルミチン酸、オレイン酸、ステアリン酸、リシノール酸、ヒドロキシステアリン酸等の脂肪族カルボン酸を挙げることができるが、これらに限定されない。
さらに、本発明に使用されるカルボキシ基を有する一般式(2)で表されるカルボン酸としては、多価カルボン酸として公知公用のものを好ましく用いることができ、具体的には、アジピン酸、セバシン酸、ドデカン2酸等の多価カルボン酸を挙げることができるが、これらに限定されない。
エステル化の反応においては、一般式(2)で表されるカルボン酸と一般式(1)で表される(メタ)アクリル酸誘導体を反応させてもよいが、一般式(2)で表されるカルボン酸のエステル誘導体を用いても好適にエステル化反応を行うことができる。ここで用いることのできるエステル基としては、例えば、メチルエステル、エチルエステル等の低級アルキルエステルを挙げることができるが、これらに限定されるものではなく、公知公用のエステル基を好ましく用いるこができる。
【0025】
本発明で使用される酵素は、アスペルギルス(Aspergillus oryzae)属、アクロモバクター(Achromobacter)属、バシラス(Bacillus)属、カンジダ(Candida)属、クロモバクター(Chromobacter)属、フザリウム(Fusarium)属、フミコラ(Humicola)属、ハイフォザイマ(Hyphozyma)属、シュードモナス(Pseudomonas)属、リゾムーコル(Rhizomucor)属、リゾプス(Rhizopus)属、又はテルモマイセス(Thermomyces)属の微生物から得られるものが好ましい。
より具体的に、本発明に用いられる好ましい市販の酵素を列挙すると、名糖産業株式会社のリパーゼQL、同社リパーゼPL、これらの固定化酵素であるリパーゼQLC、リパーゼQLG、リパーゼPLC、リパーゼPLGがあり、他にも、天野製薬社製リパーゼPS、ノボザイムズ社製リポザイムRM−IM、リポザイムTL−IM、ノボザイム435が挙げられる。これらの酵素は、混合して使用しても良く、また反応過程に伴い酵素を変えたり、又は異なった酵素を足し込んでも良い。
【0026】
反応槽中に添加する酵素量は反応後に酵素を取り除く工程上の手間と経済性の点から少ない方が好ましく、この観点から高活性の酵素が好ましい。具体的には1000U/g(脂肪酸エステルの加水分解による活性測定法)以上が好ましく、粉体で分散しにくい酵素の場合は5000U/g以上が好ましい。
酵素の形態は溶液状態、固体粉末、担体に固定化された形態を問わないが、生成物の精製時に不要な成分を含まない固体粉末もしくは担体に固定化されたものが好ましい。また、酵素の再利用を考慮すると担体に固定化されたものが、より好ましい。
酵素の回収は、未反応原料の回収と共に行うことが可能で、反応後の反応液を静置又は冷却後に沈降した酵素と未反応原料と共にデカンテーション、遠心分離又は濾別によって容易に分離できる。冷却は反応時の組成にも依存するが室温以下が好ましい。濾別された酵素及び未反応原料はこのまま、もしくは一度、加温及び減圧下に乾燥し再利用できる。
【0027】
また、本発明では、エステル反応に適したエステル合成活性の高いリパーゼを細胞表層に発現させた高活性アーミング酵母も好ましく用いることができる。
【0028】
本発明で用いられるリパーゼ・アーミング酵母(B)は、以下の特徴を有する。
1.リパーゼ・アーミング酵母(B)が、一倍体a細胞アーミング酵母と一倍体α細胞アーミング酵母を細胞融合したものであること、
2.リパーゼ・アーミング酵母(B)が、前記一倍体a細胞アーミング酵母及び一倍体α細胞アーミング酵母のいずれか一方あるいは両方の細胞表層へ提示されるタンパク質が、分泌シグナル配列、前記タンパク質の構造遺伝子配列、細胞表層局在タンパク質の一部をコードする配列及びGPIアンカリングドメインをコードする配列をこの順で有するDNAによって細胞表層に発現されるものであること、
3.リパーゼ・アーミング酵母(B)が、前記一倍体a細胞アーミング酵母及び一倍体α細胞アーミング酵母のいずれか一方あるいは両方の細胞表層へ提示されるタンパク質が、分泌シグナル配列、前記タンパク質の構造遺伝子配列、GPIアンカリングドメインをコードする配列をこの順で有するDNAによって細胞表層に発現されるものであること、
4.リパーゼ・アーミング酵母(B)が、前記一倍体a細胞アーミング酵母及び一倍体α細胞アーミング酵母のいずれか一方あるいは両方の細胞表層へ提示されるタンパク質が、分泌シグナル配列、前記タンパク質の構造遺伝子配列、糖鎖結合タンパク質ドメインをコードする配列をこの順で有するDNA、あるいは分泌シグナル配列、糖鎖結合タンパク質ドメインをコードする配列、前記細胞表層へ提示されるタンパク質の構造遺伝子配列をこの順で有するDNAによって細胞表層に発現されるものであること。
【0029】
本発明におけるリパーゼを細胞表層に発現させたアーミング酵母の作製は、公開特許公報(特開平11−290078、WO2002/085935)を基に行うことができる。
【0030】
(アーミング酵母)
本発明において、アーミング酵母とは細胞表層局在タンパク質と種々の機能性タンパク質(酵素・抗原・抗体・レポータータンパク質など)やペプチドを融合させ、細胞表層にディスプレイさせることにより、通常の酵母では有していない新しい機能を有する、あるいは元来有している機能を増強した酵母細胞のことをいう。また、細胞表層とは細胞膜・細胞壁ならびにその間の空間であるペリプラズムのことであり、これらの層を利用し上記の様な要件を満たす酵母細胞をアーミング酵母という。
【0031】
本発明におけるリパーゼに関しては次に説明する通りである。
【0032】
(分泌型酵素)
リパーゼとは、油脂から脂肪酸を遊離させ得る活性を有する酵素であり、その起源については特に限定されないが、通常、リゾプス・オリザエ(Rhizopus orizae、以下、リゾプス・オリザエと略記する)などのカビ由来のリパーゼや、キャンディダ・アンタルクチカ(Candida Antarctica、以下、キャンディダ・アンタルクチカと略記する)などの酵母由来のリパーゼが好適に用いられている。
【0033】
(細胞表層局在タンパク質)
本発明において、細胞表層局在タンパク質とは、酵母の細胞表層に固定され、細胞表層に存在するタンパク質をいう。例えば、凝集性タンパク質であるα―又はa−アグルチニン、FLOタンパク質、大腸菌の外膜タンパク質OmpAなどが挙げられる。一般に細胞表層局在タンパク質は、N末端側に分泌シグナル配列及びC末端側にGPIアンカー付着認識シグナル配列を有している。分泌シグナル配列を有する点では分泌性タンパク質と共通しているが、細胞表層局在タンパク質は、GPIアンカーを介して細胞膜に固定されて輸送される点が分泌性タンパク質と異なる。細胞表層局在タンパク質は細胞膜通過の際、GPIアンカー付着認識シグナル配列が選択的に切断され、新たに突出したC末端部分でGPIアンカーと結合して細胞膜に固定される。その後ホスファチジルイノシトール依存性ホスホリパーゼC(以下、PI−PLCと略記する)によりGPIアンカーの根元部分が切断される。次いで、細胞膜から切り離されたタンパク質は、細胞壁に組み込まれて細胞表層に固定され、細胞表層に局在する。
【0034】
(分泌シグナル配列)
本発明において、分泌シグナル配列とは、一般に細胞外に分泌されるタンパク質のN−末端に結合している、疎水性に富んだアミノ酸を多く含むアミノ酸配列であり、通常、分泌性タンパク質が細胞内から細胞膜を通して細胞外(ペリプラズムも含む)へ分泌される際に除去される。分泌シグナル配列であれば、どのような分泌シグナル配列でも用いることができ、その起源は限定されない。例えば、グルコアミラーゼの分泌シグナル配列、酵母のα―又はa―アグルチニンの分泌シグナル配列、リパーゼの分泌シグナル配列などが好適に用いられている。また、リパーゼの活性に影響を与えないのであれば、分泌シグナル配列の一部又は全部が酵素のN−末端側に残っても良い(特開平11−290078号公報、国際公開第2002/085935号パンフレット参照)。
【0035】
(GPIアンカー及びGPIアンカリングドメイン)
GPIアンカーとは、グリコシルホスファチジルイノシトール(GPI)と呼ばれる、エタノールアミンリン酸―6マンノースα1−2マンノースα1−6マンノースα1−4グルコサミンα1−6イノシトールリン脂質を基本構造とする糖脂質をいう。
【0036】
GPIアンカリングドメインは、通常、細胞表層局在タンパク質のC末端あるいはその近傍に位置する。例えば、α―アグルチニンのC末端から320アミノ酸の配列をコードする配列がこれに相当し、この配列には、GPIアンカーが細胞表層局在タンパク質と結合する際に認識される配列であるGPIアンカー認識付着シグナル配列の他に、4カ所の糖鎖結合部位がある。GPIアンカーの根元部分がPI−PLCにより切断された後、これらの糖鎖結合部位に結合した糖鎖と細胞壁を構成する多糖類とが共有結合することで、α―アグルチニンのC末端配列部分が細胞壁と結合し、α―アグルチニンは細胞表層に保持される。
【0037】
(糖鎖結合タンパク質ドメイン)
本発明において、糖鎖結合タンパク質ドメインとは、複数の糖鎖を有し、この糖鎖が、細胞壁中の糖鎖と相互作用又は絡み合うことによって、細胞表層に留まることのできるドメインをいう。例えば、レクチンなどの糖鎖結合部位や、α―アグルチニン、a―アグルチニン、FLOタンパク質などの凝集タンパク質の凝集機能ドメインなどが挙げられる。
(凝集機能ドメイン)
細胞表層局在タンパク質の凝集機能ドメインとはGPIアンカリングドメインよりもN末端側にあり、複数の糖鎖を有し、凝集に関与していると考えられているドメインをいう。
【0038】
本発明において、リパーゼを細胞表層に発現させるDNAは、以下の配列を有するDNAであることが好ましい。
(1)分泌シグナル配列、リパーゼの構造遺伝子配列、細胞表層局在タンパク質の一部をコードする配列及びGPIアンカリングドメインをコードする配列をこの順で有するDNA
(2)分泌シグナル配列、リパーゼの構造遺伝子配列、糖鎖結合タンパク質ドメインをコードする配列をこの順で有するDNA
(3)分泌シグナル配列、糖鎖結合タンパク質ドメインをコードする配列、リパーゼの構造遺伝子配列をこの順で有するDNA
(4)分泌シグナル配列、リパーゼの構造遺伝子配列、GPIアンカリングドメインをコードする配列をこの順で有するDNA
上記(2)、(3)の場合は、糖鎖結合タンパク質ドメインが、少なくとも細胞表層局在タンパク質の凝集機能ドメインを含む部分であることが好ましい。
【0039】
また、上記(1)〜(4)の場合、リパーゼの構造遺伝子配列と、細胞表層局在タンパク質の一部をコードする配列もしくは糖鎖結合タンパク質ドメインをコードする配列又はGPIアンカリングドメインをコードする配列との間には、適当な長さのリンカー配列が挟みこまれてもよく、アミノ酸の種類は限定されないが、アミノ酸8個〜21個をコードするDNA配列が好ましい。
【0040】
前記(1)〜(4)のDNAは、従来公知の手法を用いて合成することができる。例えば、(1)のDNAの場合、分泌シグナル配列とリパーゼの構造遺伝子配列との結合は、部位特異的突然変異法を用いて行うことができ、正確な分泌シグナル配列の切断と高活性なリパーゼBの発現が可能である。さらに、このようにして得られた配列と、細胞表層局在タンパク質の一部をコードする配列及びGPIアンカー付着シグナル配列とを結合すればよい。結合は、適切な制限酵素、リンカー等を用いて行うことができる。
【0041】
また、係るDNAは、プラスミドの形態でも、宿主となるサッカロミセス属酵母の遺伝子に組み込まれた形態でも、いずれでもよい。
【0042】
アーミング酵母は、係るDNAをサッカロミセス属酵母に導入することにより得ることができる。
【0043】
このようにして得られるアーミング酵母は、従来のアーミング酵母に比べ、エステル反応を触媒する活性が高く、エステル反応により適したアーミング酵母である。更に、従来のアーミング酵母よりも耐熱性が高く、高温でエステル反応を触媒できるため、エステル合成反応のような吸熱反応を効率よく触媒することが可能である。
一般式(1)で表される(メタ)アクリレートと脂肪族カルボン酸の使用比率は、好ましくは等モル、又は一般式(1)で表される(メタ)アクリレートと脂肪族カルボン酸に対して1.1〜1.5倍モルの比率で使用してもよい。
反応に使用する酵素量は、一般式(3)で表される(メタ)アクリレートと2級水酸基を有する脂肪酸の合計質量に対して、0.5〜10質量%、好ましくは、1〜5質量%、より好ましくは、2〜4質量%である。
本発明の反応は、一般式(1)で表される(メタ)アクリレートを基質とすると共に反応溶媒として用いて、酵素反応を行なうことができるが、酵素活性に影響しない有機溶媒を添加して行うこともできる。このような反応溶媒は、用いないことが好ましいが、反応系の撹拌効率の向上や基質の溶解促進を目的として、必要に応じて反応系に添加しても良い。
【0044】
用いられる溶媒としては、例えば、水、緩衝水溶液、塩水溶液、メタノール、エタノール、プロパノール、イソプロパノール、ブタノール、ターシャリーブタノール、アミルアルコール、イソアミルアルコール、エチレングリコール、グリセリン、モノグリム、ジグリム、アセトニトリル、硝酸メチル、ジエチルエーテル、ジメチルスルホキシド(DMSO)、ジメチルホルムアミド(DMF)、ジエチルエーテル、ジイソプロピルエーテル、THF、ジオキサン、酢酸メチル、酢酸エチル、酢酸プロピル、酢酸イソプロピル、酢酸ブチル、酢酸イソブチル、ベンゼン、トルエン、キシレン、ジクロロメタン、クロロホルム、四塩化炭素、ジクロロエタン、トリクロロエタン、クロロベンゼン、ジクロロベンゼン、トリクロロベンゼン、ピリジン、ペンタン、ヘキサン、ヘプタン、オクタン、イソオクタン、シクロヘキサン等が好ましく、これら2種以上の混合物でもかまわない。
反応で副生する水は、反応系内を減圧にし、窒素等の不活性ガスを吹き込むことにより、減圧留去することが好ましい。
【0045】
酵素活性を考慮すると反応系の水分を制御することが好ましく、水分量は10ppmから10%(100000ppm)、酵素の活性のためには10ppmから1%(10000ppm)添加することが好ましく、反応の平衡を考慮すると10ppmから1000ppmとすることが好ましい。これらの水分は、添加する酵素に含ませても、反応系に別途に添加しても良く、反応途中で順次、少量の水分を添加しても良い。
反応温度は、基質の溶解性と反応速度を考慮すると高温が好ましいが、一方、酵素の熱による失活を考慮すると低温が好ましく、相反する条件を満たす必要があり、極めて重要である。本発明に使用するリパーゼでは、好ましい反応温度として、30〜60℃の温度を挙げることができ、更に好ましくは、35〜45℃の温度を挙げることができる。
反応圧力は沸点による温度制御等を目的とし可変であり、減圧、加圧、0.13から1013khPa(1mmHgから7600mmHg)の範囲で反応可能であり、通常1.3から202.6KPa(10mmHgから1520mmHg)の範囲で可能である。反応時間は、反応系により異なり、一概に規定されないが、反応効率と収量を考慮すると、一般に3時間から48時間が好ましく、更に好ましくは5時間から30時間であって、この程度の反応時間で留めて、未反応の原料を全て回収し、再反応に用いることが好ましい。
【0046】
本発明に用いる反応装置は特に限定されるものではないが、基質と酵素の分散衝突性を向上させる為に、攪拌装置を有する反応槽での反応が好ましい。即ち、反応槽から留出する水を凝縮器に通し、次いで凝集したこれらをモレキュラーシーブス等の充填剤を充填した副生成物除去槽に通して、水をモレキュラーシーブスで吸着除去する。
副生成物除去槽に充填する充填材としては、(メタ)アクリレートと溶媒を吸着しないものを選択することが好ましく、モレキュラーシーブス、シリカゲル、塩化カルシウム、塩化カリウム、塩化ナトリウム、酸化マグネシウム、硫化マグネシウム、炭酸カリウム、硫酸ナトリウム等が挙げられる。モレキュラーシーブスは、副生物の種類により適宜変える必要があるが、副生生物が水、メタノールならば3Aタイプ、これらもしくはエタノール等では4Aタイプが好ましい。副生成物除去槽は反応槽の1%から100%の体積があれば十分である。
【0047】
モレキュラーシーブは、その重量の30%程度の重量の水等を吸着することが可能であるが、反応効率等の観点から過剰量を充填することが好ましい。反応中又は反応後に充填材は再生する必要があり、再生方法としては洗浄、加熱、減圧の方法がある。一般には水、アルコールでの洗浄後に加熱乾燥及び減圧乾燥を行う。温度は100℃以上が好ましく、180℃以上が速い再生を望める。
副生成物除去槽の規模を小さくするには、反応中に充填剤の再生を行うことが効率的であり、例えば2つ以上の除去槽を並列に使用し、使用槽を切換えて連続運転することが好ましい。また副生成物除去槽はソクスレー抽出機のような構造を有していることも好ましい。また副生成物除去槽を持たずに反応槽に直接、これらの充填物を反応槽内部に入れて反応させ、反応後に反応物から除去することも可能である。
撹拌装置はスクリュー翼、ヘリカル翼、ファードラー翼、タービン翼、パドル翼等を使用でき、均質な撹拌が可能な用いることが好ましい。撹拌回転数は撹拌効率、撹拌動力に依存するもののため、撹拌翼、反応槽のスケール等に依存するが、基質が不均質な場合、沈殿が生じない範囲で低速度が好ましい。
【実施例】
【0048】
以下に本発明を実施例により具体的に説明する。
【0049】
(実施例1) カプリル酸とHEMAとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
カプリル酸 28.8g、0.200mol
2−ヒドロキシエチルメタクリレート 28.6g、0.220mol
メトキシハイドロキノン 9mg
水 1.7g
リポザイムRM−IM 1.7g(モノマー全量に対し3重量%)
を入れ、油浴にて45℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。4時間後、反応系内を減圧(3.5kPa)し、かつ乾燥空気を供給した。反応開始より24時間後の反応転化率は>97%(GCピーク)であった。反応系内の減圧および窒素の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。室温にて透明液体(純度93%(GCピーク比)、ハーゼン色数20)を得た。
【0050】
本発明において反応追跡および純度の決定はガスクロマトグラムにより行った。
<ガスクロマトグラム条件>
測定機器: 島津製 GC−2010
カラム: キャピラリーカラムTC−5(0.32mmI.D.×30m,df=0.25μm,GLサイエンス製)
インジェクション方法: スプリット法(30:1)
インジェクション温度: 300℃
検出方法:FID
検出器温度:320℃
カラム温度:100℃(2分間保持)→20℃/分→300℃(13分間保持)
また、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
<GC−MS条件>
測定機器: 日本電子製 JMS−K9(四重極型)
カラム: キャピラリーカラムDB−5ms(0.25mmI.D.×30m,df=0.25μm,アジレントテクノロジー製)
インジェクション方法: スプリット法(30:1)
インジェクション温度: 300℃
カラム温度:50℃(2分間保持)→15℃/分→300℃
イオン化方法:EI または CI(イソブタン)
【0051】
(実施例2)カプリル酸メチルとHEMAとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
カプリル酸メチル 34.8gl、0.220mol
2−ヒドロキシエチルメタクリレート 31.5g、0.242mol
メトキシハイドロキノン 7mg
リポザイムRM−IM 3.3g(モノマー全量に対し5重量%)
を入れ、油浴にて45℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。30分後、反応系内を減圧(2.5kPa)し、かつ乾燥空気を供給した。反応開始より8時間後、反応系内の減圧度を1.5kPa、ついで反応開始より24時間後に0.6kPaにまで高め反応を継続した。反応開始より24時間後の反応転化率は93%、36時間後の転化率97%であった。反応開始より72時間後、カプリル酸メチルのピークの消失を確認した後、反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。室温にて透明液体を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0052】
(比較例1)カプリル酸とHEMAとの反応(化学合成例)
デカンターおよび気体導入管を取り付けた三ツ口フラスコに、
カプリル酸 28.8g、0.200mol
2−ヒドロキシエチルメタクリレート 31.2g、0.240mol
メトキシハイドロキノン 0.1g
p−トルエンスルホン酸 1.5g
トルエン 240.0g
を入れ、油浴にて125〜130℃に加温した。攪拌はマグネチックスターラーを用い、また、乾燥空気をバブリングにより供給した。トルエンの還流とともに縮合水の生成が確認された。反応開始より6時間後、縮合水の生成が停止し、また、反応追跡結果より反応の進行が認められないため反応を停止した。このときの転化率は91%であった。反応混合物を5%炭酸ナトリウム水溶液によるアルカリ洗浄、その後、飽和食塩水および蒸留水による洗浄を行った。反応混合物を無水硫酸マグネシウムで乾燥後、トルエンを留去し、淡黄色液体(純度85%(GCピーク比)、ハーゼン色数150)を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0053】
(比較例2) カプリル酸とHEMAとの反応(選択性を示さないリパーゼを用いた場合の実験例)
酵素触媒としてリポザイムRM−IMの代わりにノボザイム435(Candida antarctica由来リパーゼBの固定化酵素)を用いた以外は実施例1と同様の反応を行った。
【0054】
GC分析による反応追跡の結果、反応の進行に伴い目的物のピーク(Rt:8.9分)以外のピークも複数種類生成した。反応開始より24時間後の反応転化率は>98%であった。反応系内の減圧および窒素の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。室温にて透明液体を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0055】
得られた化合物についてGC分析を行ったところ、目的化合物の純度は51%(GCピーク比)であった。
【0056】
(実施例1と比較例1および2の分析)
GC分析およびGC質量分析により反応生成物の分析を行い、生成物の同定及びGCピーク面積より反応選択率(純度)を算出した。
【0057】
【表1】
【0058】
(表中、EGDMAはエチレングリコールジメタクリレート、HO−EG−C8はエチレングリコールモノカプリレート、C8−EG−C8はエチレングリコールジカプリレートを示す。)
【0059】
実施例1および比較例1、2の結果より、実施例1では高純度、且つ、着色の無い目的物が得られた。
【0060】
(実施例3) カプロン酸とHEMAとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
カプロン酸 28.3、0.250mol
2−ヒドロキシエチルメタクリレート 35.8g、0.275mol
メトキシハイドロキノン 6mg
水 3.2g
リポザイムRM−IM 1.9g(モノマー全量に対し3重量%)
を入れ、油浴にて45℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。1.5時間後、反応系内を減圧(3.5kPa)し、かつ乾燥空気を供給した。反応開始より8.5時間後、反応系内の減圧度を5.0kPaにまで高め反応を継続した。反応開始より48時間後、反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。室温にて透明液体を得た。目的物の純度は90%(GCピーク比)であった。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0061】
(実施例4) パルミチン酸とHEMAとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
パルミチン酸38.5g、0.150mol
2−ヒドロキシエチルメタクリレート 21.5g、0.165mol
メトキシハイドロキノン 7mg
リポザイムRM−IM 1.8g(モノマー全量に対し3重量%)
を入れ、油浴にて60℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。反応開始時にはパルミチン酸は一部溶解した懸濁状態であった。15分後、反応系内を減圧(3.5kPa)し、かつ乾燥空気を供給した。反応の進行に伴い固体のパルミチン酸はすべて溶解した。反応開始より20時間後(反応転化率>98%)、反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。生成物の純度は85%(GCピーク比)であった。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0062】
(実施例5) オレイン酸とHEMAとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
オレイン酸 42.4g、0.150mol
2−ヒドロキシエチルメタクリレート 21.5g、0.165mol
メトキシハイドロキノン 12.7mg
リポザイムRM−IM 2.0g(モノマー全量に対し3重量%)
を入れ、油浴にて45℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。30分後、反応系内を減圧(3.5kPa)し、かつ乾燥空気を供給しながら縮合水を除去した。反応開始より2時間後、反応系内の減圧度を2.0kPaにまで高め反応を継続した。反応開始より18時間後の反応転化率は>96%であった。反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。室温にて淡黄色液体(ハーゼン色数<1)を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0063】
(比較例3) オレイン酸とHEMAとの反応(化学合成法)
デカンターおよび気体導入管を取り付けた四ツ口フラスコに。
オレイン酸 42.4g、0.150mol
2−ヒドロキシエチルメタクリレート 23.4g、0.180mol
メトキシハイドロキノン 100mg
トルエン 270.0g
p−トルエンスルホン酸一水和物 2.0g(モノマー全量に対し3重量%)
を入れ、油浴にて125〜130℃に加温した。攪拌はマグネチックスターラーを用い、また、乾燥空気をバブリングにより供給した。トルエンの還流とともに縮合水の生成が確認されたが、反応溶液は褐色に変化した。反応開始より4時間後、縮合水の生成が停止し、また、反応追跡結果より反応の進行が認められないため反応を停止した。このときの転化率は76%であった。反応混合物のアルカリ洗浄を試みたが乳化がひどく、水洗は困難であった。また、アルカリおよび水洗による反応混合物からの脱色はできず得られた化合物は褐色液体(ハーゼン色数>18)であった。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0064】
(実施例6) 10−ウンデセン酸とHEMAとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
10−ウンデセン酸 36.9g、0.200mol
2−ヒドロキシエチルメタクリレート 28.6g、0.220mol
メトキシハイドロキノン 7mg
水 1.3g
リポザイムRM−IM 2.0g(モノマー全量に対し3重量%)
を入れ、油浴にて45℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。4時間後、反応系内を減圧(6.0kPa)し、かつ乾燥空気を供給した。反応開始より7時間後、反応系内の減圧度を3.5kPaにまで高め反応を継続した。反応開始より28時間後(反応転化率97%)、反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。室温にて透明液体(純度90%)を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0065】
(実施例7) 脱水ヒマシ油脂肪酸DCO/FAとHEMAとの反応(酸素に不安定な化合物の合成例)
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
DCO/FA42.4g、0.150mol
(平均値より算出)
2−ヒドロキシエチルメタクリレート 21.5g、0.165mol
メトキシハイドロキノン 7mg
リポザイムRM−IM 1.9g(モノマー全量に対し3重量%)
を入れ、油浴にて45℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。15分後、反応系内を減圧(2.5kPa)し、かつ乾燥窒素を供給した。反応開始より2.5時間後、反応系内の減圧度を1.5kPaにまで高め反応を継続した。反応開始より20時間後、反応系内の減圧および窒素の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。室温にて淡黄色透明液体を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0066】
得られた反応生成物のGC/MS分析により、原料DCO/FA(混合物)の各ピークとHEMAとの選択的なエステル化反応によって生成した混合物であることが示された(図1(原料)および図2(生成物)にGCチャートを示す。)
【0067】
(比較例4) 脱水ヒマシ油脂肪酸DCO/FAとHEMAとの反応(酸素に不安定な化合物の化学合成例)
フラスコに
DCO/FA42.4g、0.150mol
2−ヒドロキシエチルメタクリレート 21.5g、0.165mol
メトキシハイドロキノン 100mg
トルエン 270.0g
を入れ。油浴にて100℃に加温した。攪拌はマグネチックスターラーを用い、また、乾燥空気をバブリングにより供給した。反応開始より2.5時間後、沈殿物が生じたため反応を停止した。
【0068】
実施例7および比較例4の結果より、酸素および熱等に不安定な脱水ヒマシ油脂肪酸などを原料に用いた反応にも適用可能である。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0069】
(実施例8) カプリン酸とHEAとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
カプリン酸 37.9g、0.220mol
2−ヒドロキシエチルアクリレート 26.8g、0.231mol
メトキシハイドロキノン 8mg
水 1.3g
リポザイムRM−IM 3.2g(モノマー全量に対し5重量%)
を入れ、油浴にて40℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。2.5時間後、反応系内を減圧(5.0kPa)し、かつ乾燥空気を供給した。反応開始より6時間後、反応系内の減圧度を一時停止した。反応開始より10時間後(転化率90%)、反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。室温にて透明液体(純度93%(GCピーク比))を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0070】
(実施例9) パルミチン酸とHEAとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
パルミチン酸153.9g、0.600mol
2−ヒドロキシエチルアクリレート 76.6g、0.630mol
メトキシハイドロキノン 31mg
リポザイムRM−IM 6.9g(モノマー全量に対し3重量%)
を入れ、油浴にて60℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。反応開始時にはパルミチン酸は一部溶解した懸濁状態であった。1時間後、反応系内を減圧(5.0kPa)し、かつ乾燥空気を供給した。反応の進行に伴い固体のパルミチン酸はすべて溶解した。反応開始より2時間後、反応系内の減圧度を3.5kPaにまで高め反応を継続した。反応開始より24時間後(反応転化率>98%)、反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0071】
少量の高融点副生成物を除去するために以下の精製を行った。
【0072】
反応混合物をメタノールに分散し不溶物を濾過により取り除いた。濾液を濃縮後に得られたオイル状液体を氷水に注ぎ分散した。オイル状液体は固化した。白色固体を濾別、乾燥を行い、室温にて白色結晶(純度95%(GCピーク比)、融点 27.9℃)を得た。
【0073】
(実施例10) ステアリン酸とHEAとの反応(溶剤使用)
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)を取り付けたディーンスタークトラップおよび気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
ステアリン酸128.0g、0.450mol
2−ヒドロキシエチルアクリレート 57.5g、0.495mol
メトキシハイドロキノン 38mg
リポザイムRM−IM 3.7g(モノマー全量に対し2重量%)
トルエン 100g
を入れ、油浴にて45℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。27.5時間後、反応系内を減圧(50kPa)し、かつ乾燥空気を供給した。反応開始より32時間後、反応温度を65℃にまで昇温し、トルエンを除去しながら反応を継続した。反応開始より48時間後、反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0074】
未反応原料を除去するために以下の精製を行った。
反応混合物をトルエンに溶解し、5%炭酸水素ナトリウム水溶液、飽和食塩水、蒸留水でそれぞれ2回洗浄を行った。無水硫酸マグネシウムで乾燥後、トルエンを減圧留去し、室温にて白色結晶(純度96%(GCピーク比)、融点:34.5℃)を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0075】
(実施例11) カプリン酸とHBMAとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
カプリン酸 25.8g、0.150mol
2−ヒドロキシブチルメタクリレート 24.9g、0.158mol
メトキシハイドロキノン 5mg
水 1g
リポザイムRM−IM 1.5g(モノマー全量に対し3重量%)
を入れ、油浴にて40℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。5時間後、反応系内を減圧(8.0kPa)し、かつ乾燥空気を供給した。反応開始より30時間後、減圧を停止しそのまま反応を続けた。反応開始より48時間後、再度、反応系内を減圧(5.0kPa)し、かつ乾燥空気を供給した。反応開始より58時間後、反応系内の減圧度を2.5kPaにまで高め反応を継続した。反応開始より74時間後、反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。室温にて透明液体を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0076】
(実施例12) カプリン酸とHEAAmとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
カプリン酸 43.1g、0.250mol
2−ヒドロキシエチルメタクリレート 30.2g、0.250mol
メトキシハイドロキノン 43mg
ノボザイム435 1.5g(モノマー全量に対し2重量%)
を入れ、油浴にて45℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。15分後、反応系内を減圧(3.5kPa)し、かつ乾燥空気を供給した。反応開始より4時間後、反応系内の減圧度を2.5kPaにまで高め反応を継続した。結晶析出のため反応開始より23時間後に60℃に昇温した。反応開始より24時間後、反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。室温にて白色固体(純度90%(GCピーク比)、融点:44.7℃)を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0077】
(実施例13) パルミチン酸とHEAAmとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
パルミチン酸153.9g、0.600mol
2−ヒドロキシエチルアクリルアミド 72.5g、0.630mol
メトキシハイドロキノン 154mg
ノボザイム435 4.5g(モノマー全量に対し2重量%)
を入れ、油浴にて65℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。15分後、反応系内を減圧(15.0kPa)し、かつ乾燥空気を供給した。反応開始より4時間後、結晶析出のため反応温度を70℃に昇温した。反応開始より8時間後、反応系全体が結晶化したためトルエン100mlを加え濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。得られた濾液に更にトルエン500mlを加え、室温に放冷し、析出した白色結晶(純度94%(GCピーク比)、融点 73.9℃)を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0078】
(実施例14) 10−ウンデセン酸とHEAAmとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
10−ウンデセン酸 46.1g、0.250mol
2−ヒドロキシエチルアクリルアミド 30.2g、0.263mol
メトキシハイドロキノン 46mg
ノボザイム435 0.8g(モノマー全量に対し1重量%)
を入れ、油浴にて45℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。15分後、反応系内を減圧(5kPa)し、かつ乾燥空気を供給した。反応開始より2時間後、反応系内の減圧度を2.5kPaにまで高め反応を継続した。反応開始より24時間後、反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。室温にて固化した。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0079】
(実施例15) アジピン酸ジビニルとHEMAとの反応(リポザイムTL−IMを用いた反応)
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
アジピン酸ジビニル 99.1g、0.501mol
2−ヒドロキシエチルメタクリレート 136.6g、1.050mol
メトキシハイドロキノン 50mg
リポザイムTL−IM 7.1g(モノマー全量に対し3重量%)
を入れ、油浴にて40℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。15分後、反応系内を減圧(30kPa)し、かつ乾燥空気を供給した。反応開始より4時間後、反応系内の減圧度を25kPa、ついで8.5時間後に10kPaにまで高め反応を継続した。反応開始より28時間後、反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。得られた濾液を飽和食塩水で洗浄後、室温にて淡黄色透明液体(純度92%(GCピーク比))を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0080】
(実施例16) アジピン酸ジビニルとHEMAとの反応(ノボザイム435を用いた反応)
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
アジピン酸ジビニル 19.8g、0.100mol
2−ヒドロキシエチルメタクリレート 27.3g、0.210mol
メトキシハイドロキノン 23mg
ノボザイム435 1.4g(モノマー全量に対し3重量%)
を入れ、油浴にて40℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。反応系内を減圧(10kPa)し、かつ乾燥空気を供給した。反応開始より2時間後、反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。室温にて淡黄色透明液体(純度86%(GCピーク比))を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0081】
(実施例17) セバシン酸とHEMAとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
セバシン酸 40.5g、0.200mol
2−ヒドロキシエチルメタクリレート 53.4g、0.410mol
メトキシハイドロキノン 8mg
水 1.9g
リポザイムRM−IM 4.7g(モノマー全量に対し5重量%)
を入れ、油浴にて55℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。反応開始時にはセバシン酸は一部溶解した懸濁状態であった。3時間後、反応系内を減圧(5.0kPa)し、かつ乾燥空気を供給した。反応の進行に伴い固体のセバシン酸はすべて溶解した。反応開始より24時間後、反応系内の減圧度を2.5kPaにまで高め反応を継続した。反応開始より30時間後、反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。室温にて透明液体(純度90%(GCピーク比))を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0082】
(実施例18) ドデカン二酸とHEMAとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
ドデカン二酸34.6g、0.150mol
2−ヒドロキシエチルメタクリレート 40.0g、0.308mol
メトキシハイドロキノン 7mg
水 1.5g
リポザイムRM−IM 3.7g(モノマー全量に対し5重量%)
を入れ、油浴にて50℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。反応開始時にはドデカン二酸は一部溶解した懸濁状態であった。3時間後、反応系内を減圧(5.0kPa)し、かつ乾燥空気を供給した。反応の進行に伴い固体のドデカン二酸はすべて溶解した。反応開始より25時間後、反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。室温にて透明液体(純度85%(GCピーク比))を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0083】
(実施例19) カプリル酸メチルとHEMAとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
カプリル酸 43.2g、0.300mol
2−ヒドロキシエチルメタクリレート 42.9g、0.330mol
メトキシハイドロキノン 15mg
CALB−アーミング酵母 7.5g(モノマー全量に対し8.7重量%)
を入れ、油浴にて40℃に加温した。反応は攪拌羽根を取り付けた機械式攪拌器を用い攪拌を行った。3時間後、反応系内を減圧(5kPa)し、かつ乾燥空気を供給した。反応開始より24時間後、反応系内の減圧度を1kPaにまで高め反応を継続した。反応開始より46時間後の反応転化率は96.6%であった。この時点で反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)によりCALB−アーミング酵母を濾別した。室温にて透明液体を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0084】
(実施例20)12−ヒドロキシステアリン酸とHEMAとの反応
三方コック(一方は、冷却トラップを介して減圧ポンプと、他方は減圧ゲージと結合している)および気体導入管(ガス流量制御コントローラーに結合している)を取り付けた三ツ口フラスコに、
12−ヒドロキシステアリン酸 150.2g、0.500mol
メトキシハイドロキノン 45mg
を入れ、油浴(90℃)にて12−ヒドロキシステアリン酸を融解した後、80℃まで放冷した。
2−ヒドロキシエチルメタクリレート 71.6g、0.550mol
リポザイムRM−IM 6.7g(モノマー全量に対し3重量%)
を加えて反応開始した。反応温度70℃まで徐々に放冷し、2時間後、反応系内を減圧(5.0kPa)かつ乾燥空気を供給した。反応開始より20時間後に減圧を3.5kPa、30時間後に2.0kPaにまで高め反応を継続した(反応転化率96%)。反応系内の減圧および空気の吹き込みを停止し、濾過(アドバンテック製生産用濾紙No.28−3を使用)により固定化酵素を濾別した。室温にて白色ワックス状固体(収量187.3g、収率88%、純度92%(GCピーク比))を得た。
実施例1と同様にして反応追跡および純度の決定はガスクロマトグラムにより行い、混合物の化合物同定は、質量分析(GC−MS)によって確認した。
【0085】
以下に上記実施例で用いたアーミング酵母の調整法を記す。
(製造例1)<CALB表層発現プラスミドの作製>
(1−A.キャンディダ・アンタルクチカ由来リパーゼB遺伝子の取得)
次のようにしてキャンディダ・アンタルクチカ由来リパーゼB(CALB)遺伝子を取得した。
【0086】
すなわち、キャンディダ・アンタルクチカCBS6678株ゲノムをテンプレートとし、従来公知の方法でDNA合成装置にて合成した、配列番号1及び2に示す塩基配列からなるプライマーを用いてPCR増幅を行い、BglII及びXhoIで切断して、約1000bpの長さのBglII−XhoI断片(BglII−XhoI ProCALB CBS6678断片)を得た。このCALBは、Uppenbergらの文献(structure,2:293(1994))に記載され、もっとも広く知られているキャンディダ・アンタルクチカ LF058株由来リパーゼBのアミノ酸配列(CALB WT)と比較して7アミノ酸が変異している。すなわち25番目のアラニンがトレオニンに、28番目のセリンがトレオニンに、31番目のセリンがトレオニンに、46番目のグルタミンがグリシンに、89番目のアラニンがトレオニンに、97番目のアスパラギンがアルギニンに、286番目のバリンがイソロイシンにそれぞれ変異している、特徴的なCALBであった。
(1−B.CALB遺伝子導入プラスミドの取得)
CALB遺伝子中への変異の導入を効率よく行うため、上記1−Aで得られたCALB CBS6678遺伝子を、一度サイズの小さいプラスミドに導入した。
【0087】
すなわち、プラスミドpUC19(TaKaRa BIO社製)をBamHI及びSalIで切断し、上記Aで得られたBglII−XhoI ProCALB CBS6678断片を挿入して、プラスミドpUC19−ProCALB CBS6678を得た。作製の模式図を図1に示す。
【0088】
(1−C.CALB1遺伝子ならびにCALB1導入プラスミドの取得)
上記1−Bで得られたプラスミドpUC19−ProCALB CBS6678をテンプレートとし、従来公知の方法でDNA合成装置にて合成した、配列番号3及び4に示す塩基配列からなるプライマーを用いてPCR増幅を行うことにより、CALB CBS6678中にポイントミューテーションを導入し、CALB CBS6678中のアミノ末端から25番目のトレオニンをアラニンに、28番目のトレオニンをセリンに、31番目のトレオニンをセリンにそれぞれ置換したCALB1遺伝子を含むプラスミドpUC19−ProCALB1を得た。
【0089】
(1−D.CALB2遺伝子ならびにCALB2導入プラスミドの取得)
上記1−Cで得られたプラスミドpUC19−ProCALB1をテンプレートとし、従来公知の方法でDNA合成装置にて合成した、配列番号5及び6に示す塩基配列からなるプライマーを用いてPCR増幅を行うことにより、CALB1中にポイントミューテーションを導入し、CALB1中のアミノ末端から46番目のグリシンをグルタミンに置換したCALB2遺伝子を含むプラスミドpUC19−ProCALB2を得た。
【0090】
(1−E.リンカー導入型CALB遺伝子の取得)
上記1−Dで得られた各CALB2導入プラスミドをテンプレートとし、従来公知の方法でDNA合成装置にて合成した、配列番号7及び8に示す塩基配列からなるプライマーを用いてPCR増幅を行い、次いでBglII及びXhoIで切断して、約1000bpの長さのBglII−XhoI FLAG−ProCALB2断片を得た。BglII−XhoI FLAG−ProCALB2断片は、FLAG tag、リパーゼのプロ配列、及びCALB2の成熟タンパク質配列を有している。ここで、プロ配列とは、リパーゼの立体構造を形成するために必要とされる配列のことであり、FLAG tagとは、リンカー配列のことであり、成熟タンパク質配列とは、実際にリパーゼの触媒反応機能を有する配列のことである。
【0091】
(1−F.CALB表層発現カセットの作製)
CALB表層発現カセットは、発現プロモーターの配列とターミネーターの配列、並びにこれらの間にFLO1誘導体であるShort型FLO1アンカー遺伝子と上記1−Eで得られたリンカー導入型CALB遺伝子を接続したものをそれぞれ有するDNA配列である。該CALB表層発現カセットを作製するために、以下の操作を行った、作製の模式図を図2に示す。
【0092】
すなわち、プラスミドpWIFS(T.Matsumotoら、Appl.Environ.Microbiol.,68:4517(2002))をBglIIとXhoIで切断し、上記1−Eで得られたBglII−XhoI FLAG−ProCALB2断片を挿入して、pWIFS−FLAG−ProCALB2を得た。そしてこれをBssHIIで切断し、約6300bpのCALB表層発現カセットを得た。
【0093】
(1−G.CALB表層発現プラスミドの作製)
目的のDNAを有するプラスミドは、上記1−Fで得られたCALB表層発現カセットを、各種選択マーカーを有するプラスミドに導入することで得られる。作製の模式図を図3に示す。
【0094】
すなわち、プラスミドpRS402(ATCC87477)、pRS403(ATCC87514)、pRS404(ATCC87515)、pRS405(ATCC87516)、pRS402(ATCC87517)をそれぞれBssHIIで切断し、上記Fで得られたCALB表層発現カセットを挿入してpRSAIFS−FLAG−ProCALB2、pRSHIFS−FLAG−ProCALB2、pRSWIFS−FLAG−ProCALB2、pRSLIFS−FLAG−ProCALB2、pRSUIFS−FLAG−ProCALB2を得た。
【0095】
(製造例2)<ゲノム組み込み型二倍体CALB−アーミング酵母の調製>
(2−A.サッカロミセス属酵母への遺伝子の導入)
上記1−Gで得られたCALB表層発現プラスミドを、表1に示す制限酵素を用いて、それぞれ選択マーカー中に存在する制限酵素サイトで切断した。次いで、YEAST MAKERTM(Clontech Laboratories Inc. USA)を用いて、サッカロミセス・セレヴィシエ(Saccharomyces cerevisiae、以下、サッカロミセス・セレヴィシエと略記する)YPH499(MATa,ura3,lys2,ade2,his4,trp1,leu2)には、pRSUIFS−FLAG−ProCALB2、pRSLIFS−FLAG−ProCALB2、pRSHIFS−FLAG−ProCALB2、pRSAIFS−FLAG−ProCALB2をこの順で、サッカロミセス・セレヴィシエYPH500(MATalpha,ura3,lys2,ade2,his4,trp1,leu2)には、pRSUIFS−FLAG−ProCALB2、pRSLIFS−FLAG−ProCALB2、pRSWIFS−FLAG−ProCALB2、pRSAIFS−FLAG−ProCALB2をこの順で、それぞれのゲノム中に導入した。各形質転換の選択においては、アミノ酸並びに核酸(0.002%L−ヒスチジン、0.01%L−ロイシン、0.002%L−トリプトファン、0.002%L−リシン、0.002%アデニン、0.002%ウラシル)のうち、選択に必要なものを含むSD寒天培地(2%グルコース、0.67% Yeast Nitrogen Base without amino acids)を用いて培養した。生育した酵母を選択し、ゲノム組み込み型CALBアーミング酵母である、サッカロミセス・セレヴィシエYPH499−U、YPH499−UL、YPH499−ULH、YPH499−ULHA、並びにYPH500−U、YPH500−UL、YPH500−ULW、YPH500−ULWAアーミング酵母を得た
【0096】
表2 各プラスミドの選択マーカー中における切断に用いた制限酵素
【0097】
【表2】
【0098】
(2−B.一倍体サッカロミセス−CALBアーミング酵母の調製)
上記2−Aで創製したサッカロミセス・セレヴィシエYPH499−U、YPH499−UL、YPH499−ULH、YPH499−ULHA、並びにYPH500−U、YPH500−UL、YPH500−ULW、YPH500−ULWAアーミング酵母を、SDC培地(2%グルコース、0.67%Yeast Nitrogen Base without amino acids、2%カザミノ酸、appropriate amino acids and nucleic acids)に植菌し、30℃で6日間浸透培養した。次いで、遠心分離により培地と菌体に分離し、得られた菌体を一倍体サッカロミセス−CALBアーミング酵母とした。得られた一倍体サッカロミセス−CALBアーミング酵母の、酪酸p−ニトロフェニル(以下、PNPBと略記する)を基質とした30℃におけるリパーゼ活性を確認した。
【0099】
(2−C.二倍体サッカロミセス−CALBアーミング酵母の創製)
一倍体アーミング酵母YPH499−ULHA、YPH500−ULWAをYPD培地(2%グルコース、2%peptone、1%yeast extract)に植菌し、30℃で1時間培養後、遠心分離により培地と菌体に分離し、得られた菌体を混合してYPD培地に再懸濁し、30℃で一晩培養した。これを適切な菌体密度に希釈した後、SD+K寒天培地(2%グルコース、0.67%Yeast Nitrogen Base without amino acids、0.002%L−リシン)を用いて培養した。生育した酵母を選択し、ゲノム導入型二倍体CALB−アーミング酵母である、サッカロミセス・セレヴィシエYPH501−U2L2HWA2アーミング酵母を得た。
【0100】
(2−D.二倍体サッカロミセス−CALBアーミング酵母の調製)
上記2−Cで得られたサッカロミセス・セレヴィシエYPH501−U2L2HWA2アーミング酵母を、SDC+K培地(2%グルコース、0.67%Yeast Nitrogen Base without amino acids、2%カザミノ酸、0.002%L−リシン)に植菌し、30℃で6日間浸透培養した。次いで、遠心分離により培地と菌体に分離し、PNPBを基質とした菌体の30℃におけるリパーゼ活性(エステル分解活性)を確認した。
【0101】
エステル分解活性の測定には、PNPB法を用いた。培養液を遠心分離することで菌体を回収し、得られた菌体を蒸留水で2回洗浄した。次いで、菌体を20mMリン酸緩衝液に懸濁し、30℃で5分間プレインキュベートした。一方、基質にはPNPBを用いた。PNPBは少量のエタノールに溶解させた後、蒸留水で適当に希釈して基質液を調製した。プレインキュベートした菌体懸濁液へ基質液を加え、よく撹拌し、振盪させながら30℃で10分間反応させた。次いで、5%トリクロロ酢酸水溶液を添加し、反応を停止したのち、遠心分離し、上清を回収し、200mMリン酸緩衝液で希釈し、400nmの吸光度を測定した。そして、生成したp−ニトロフェノール(PNP)量を求め、エステル分解活性とした。活性を定義するにあたり、PNPが1分間に1μmol生成する酵素量を1Uと定義した。
【図面の簡単な説明】
【0102】
【図1】反応に用いた脱水ヒマシ油脂肪酸DCO/FAのGCチャートである。
【図2】実施例7における生成物のGCチャートである。保持時間18〜20分に観測されるピークの分子量m/zは、いずれも原料(図1)のピークにHEMAが付加した分子量を示すことから、目的物であることが分かる。
【図3】製造例におけるプラスミドpUC19−ProCALB CBS6678作成の模式図である。
【図4】製造例におけるCALB表層発現カセット作成の模式図である。
【図5】製造例におけるCALB表層発現プラスミド作成の模式図である。
【特許請求の範囲】
【請求項1】
リパーゼ(A)又はリパーゼを細胞表層に提示したアーミング酵母(B)の存在下に、一般式(1)
【化1】
(式中、R1は水素原子又はメチル基、Xは酸素原子又は窒素原子、nは2〜4の整数を表す。)
で表される(メタ)アクリル酸誘導体と、一般式(2)
【化2】
(式中、R2は水酸基またはカルボキシ基を有してもよい炭素数5〜23のアルキル基又はアルケニル基を表す。)
で表されるカルボン酸またはそのエステル誘導体とを反応させる一般式(3)
【化3】
(式中、R1は水素原子又はメチル基、Xは酸素原子又は窒素原子、nは2〜4の整数、R2は水酸基またはカルボキシ基を有してもよい炭素数5〜23のアルキル基又はアルケニル基を表す。)
で表されるアクリレート化合物の製造方法。
【請求項2】
前記リパーゼ(A)が、アスペルギルス(Aspergillus oryzae)属、アクロモバクター(Achromobacter)属、バシラス(Bacillus)属、カンジダ(Candida)属、クロモバクター(Chromobacter)属、フザリウム(Fusarium)属、フミコラ(Humicola)属、ハイフォザイマ(Hyphozyma)属、シュードモナス(Pseudomonas)属、リゾムーコル(Rhizomucor)属、リゾプス(Rhizopus)属、又はテルモマイセス(Thermomyces)属の微生物から得られるものである請求項1に記載のアクリレート化合物の製造方法。
【請求項3】
前記リパーゼを細胞表層に提示したアーミング酵母(B)が二倍体アーミング酵母であって、一倍体a細胞アーミング酵母と一倍体α細胞アーミング酵母を細胞融合することを特徴とするものである請求項1に記載のアクリレート化合物の製造方法。
【請求項4】
前記リパーゼを細胞表層に提示したアーミング酵母(B)が、前記一倍体a細胞アーミング酵母及び一倍体α細胞アーミング酵母のいずれか一方あるいは両方の細胞表層へ提示されるタンパク質が、分泌シグナル配列、前記タンパク質の構造遺伝子配列、細胞表層局在タンパク質の一部をコードする配列及びGPIアンカリングドメインをコードする配列をこの順で有するDNAによって細胞表層に発現されるものである請求項3に記載のアクリレート化合物の製造方法。
【請求項5】
前記リパーゼを細胞表層に提示したアーミング酵母(B)が、前記一倍体a細胞アーミング酵母及び一倍体α細胞アーミング酵母のいずれか一方あるいは両方の細胞表層へ提示されるタンパク質が、分泌シグナル配列、前記タンパク質の構造遺伝子配列、GPIアンカリングドメインをコードする配列をこの順で有するDNAによって細胞表層に発現されるものである請求項3に記載のアクリレート化合物の製造方法。
【請求項6】
前記リパーゼを細胞表層に提示したアーミング酵母(B)が、前記一倍体a細胞アーミング酵母及び一倍体α細胞アーミング酵母のいずれか一方あるいは両方の細胞表層へ提示されるタンパク質が、分泌シグナル配列、前記タンパク質の構造遺伝子配列、糖鎖結合タンパク質ドメインをコードする配列をこの順で有するDNA、あるいは分泌シグナル配列、糖鎖結合タンパク質ドメインをコードする配列、前記細胞表層へ提示されるタンパク質の構造遺伝子配列をこの順で有するDNAによって細胞表層に発現されるものである請求項3に記載のアクリレート化合物の製造方法。
【請求項7】
前記一倍体a細胞アーミング酵母及び一倍体α細胞アーミング酵母が、サッカロミセス(Saccharomyces)属酵母である請求項3〜6のいずれか一項に記載のアクリレート化合物の製造方法。
【請求項1】
リパーゼ(A)又はリパーゼを細胞表層に提示したアーミング酵母(B)の存在下に、一般式(1)
【化1】
(式中、R1は水素原子又はメチル基、Xは酸素原子又は窒素原子、nは2〜4の整数を表す。)
で表される(メタ)アクリル酸誘導体と、一般式(2)
【化2】
(式中、R2は水酸基またはカルボキシ基を有してもよい炭素数5〜23のアルキル基又はアルケニル基を表す。)
で表されるカルボン酸またはそのエステル誘導体とを反応させる一般式(3)
【化3】
(式中、R1は水素原子又はメチル基、Xは酸素原子又は窒素原子、nは2〜4の整数、R2は水酸基またはカルボキシ基を有してもよい炭素数5〜23のアルキル基又はアルケニル基を表す。)
で表されるアクリレート化合物の製造方法。
【請求項2】
前記リパーゼ(A)が、アスペルギルス(Aspergillus oryzae)属、アクロモバクター(Achromobacter)属、バシラス(Bacillus)属、カンジダ(Candida)属、クロモバクター(Chromobacter)属、フザリウム(Fusarium)属、フミコラ(Humicola)属、ハイフォザイマ(Hyphozyma)属、シュードモナス(Pseudomonas)属、リゾムーコル(Rhizomucor)属、リゾプス(Rhizopus)属、又はテルモマイセス(Thermomyces)属の微生物から得られるものである請求項1に記載のアクリレート化合物の製造方法。
【請求項3】
前記リパーゼを細胞表層に提示したアーミング酵母(B)が二倍体アーミング酵母であって、一倍体a細胞アーミング酵母と一倍体α細胞アーミング酵母を細胞融合することを特徴とするものである請求項1に記載のアクリレート化合物の製造方法。
【請求項4】
前記リパーゼを細胞表層に提示したアーミング酵母(B)が、前記一倍体a細胞アーミング酵母及び一倍体α細胞アーミング酵母のいずれか一方あるいは両方の細胞表層へ提示されるタンパク質が、分泌シグナル配列、前記タンパク質の構造遺伝子配列、細胞表層局在タンパク質の一部をコードする配列及びGPIアンカリングドメインをコードする配列をこの順で有するDNAによって細胞表層に発現されるものである請求項3に記載のアクリレート化合物の製造方法。
【請求項5】
前記リパーゼを細胞表層に提示したアーミング酵母(B)が、前記一倍体a細胞アーミング酵母及び一倍体α細胞アーミング酵母のいずれか一方あるいは両方の細胞表層へ提示されるタンパク質が、分泌シグナル配列、前記タンパク質の構造遺伝子配列、GPIアンカリングドメインをコードする配列をこの順で有するDNAによって細胞表層に発現されるものである請求項3に記載のアクリレート化合物の製造方法。
【請求項6】
前記リパーゼを細胞表層に提示したアーミング酵母(B)が、前記一倍体a細胞アーミング酵母及び一倍体α細胞アーミング酵母のいずれか一方あるいは両方の細胞表層へ提示されるタンパク質が、分泌シグナル配列、前記タンパク質の構造遺伝子配列、糖鎖結合タンパク質ドメインをコードする配列をこの順で有するDNA、あるいは分泌シグナル配列、糖鎖結合タンパク質ドメインをコードする配列、前記細胞表層へ提示されるタンパク質の構造遺伝子配列をこの順で有するDNAによって細胞表層に発現されるものである請求項3に記載のアクリレート化合物の製造方法。
【請求項7】
前記一倍体a細胞アーミング酵母及び一倍体α細胞アーミング酵母が、サッカロミセス(Saccharomyces)属酵母である請求項3〜6のいずれか一項に記載のアクリレート化合物の製造方法。
【図1】

【図2】
【図3】
【図4】

【図5】
【図2】
【図3】
【図4】
【図5】
【公開番号】特開2009−254315(P2009−254315A)
【公開日】平成21年11月5日(2009.11.5)
【国際特許分類】
【出願番号】特願2008−110092(P2008−110092)
【出願日】平成20年4月21日(2008.4.21)
【出願人】(000002886)DIC株式会社 (2,597)
【Fターム(参考)】
【公開日】平成21年11月5日(2009.11.5)
【国際特許分類】
【出願日】平成20年4月21日(2008.4.21)
【出願人】(000002886)DIC株式会社 (2,597)
【Fターム(参考)】
[ Back to top ]