
エストロゲン受容体リガンドおよびその使用方法
本発明は、男性対象者において、黄体形成ホルモン(LH)の減少によって、またはLHレベルと無関係に、テストステロンレベルを低下させる方法、および進行した前立腺癌を治療、抑制、発生を低減、重症度を軽減、または阻害する方法、および進行した前立腺の緩和治療する方法に関する。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、男性対象者において、黄体形成ホルモン(LH)の減少による、またはLHレベルと無関係である、テストステロンレベルを低下させる方法、および進行した前立腺癌の治療、抑制、発生を低減、重症度を軽減、または阻害する方法、および進行した前立腺癌の緩和治療の方法に関する。
【背景技術】
【0002】
エストロゲンは、組織および骨の維持にとって重要であり、そのために使用される内因性ホルモンおよび合成ホルモンの群を指す。エストロゲンは、生殖器系の発達および維持に関与する細胞過程における内分泌調節剤である。生殖に関する生物現象、閉経後のほてりの防止、および閉経後の骨粗しょう症の防止におけるエストロゲンの役割は、十分に確立されている。エストラジオールは、主な内因性ヒトエストロゲンであり、女性および男性の両方に見られる。
【0003】
エストロゲンおよび抗エストロゲンの生物学的作用は、2つの異なる細胞内受容体である、エストロゲン受容体アルファ(ERα)およびエストロゲン受容体ベータ(ERβ)を介して現れる。内因性エストロゲンは、典型的には、両方の受容体サブタイプの強力な活性剤である。例えば、エストラジオールは、乳房組織、骨組織、心臓血管組織、および中枢神経系組織を含む多くの組織において、ERα作動薬としての機能を果たす。選択的エストロゲン受容体モジュレータは、一般に、異なる組織において異なる機能を有する。例えば、SERMは、乳房において、ERα拮抗薬であり得るが、子宮、骨、および心臓血管系において、部分的にERα作動薬であり得る。したがって、エストロゲン受容体リガンドとしての機能を果たす化合物は、種々の病状および疾患を治療する上で有用である。
【0004】
前立腺癌は、米国において、男性で最も頻繁に診断される非皮膚癌のうちの1つであり、今年は18万件以上の新規症例、および2万9千の死亡件数が予想される、2番目の一般的な癌による死亡原因である。進行した前立腺癌を罹患する患者は、典型的に、黄体形成ホルモン放出ホルモン(LHRH)作動薬、または両側精巣摘除のいずれかによる、アンドロゲン除去療法(ADT)を受ける。エストロゲンがテストステロンの芳香族化に由来し、そのレベルがADTによって欠損するため、アンドロゲン除去療法は、テストステロンを減少させるだけでなく、エストロゲンレベルもまた低下させる。アンドロゲン除去療法誘発エストロゲン欠損は、ほてり、女性化乳房および乳房痛、骨量の減少、骨質および強度の低下、骨粗しょう症および致命的な骨折、有害な脂質変化、ならびにより高度の心臓血管疾患および心筋梗塞、ならびに鬱および他の情緒変化を含む、著しい副作用をもたらす。ADTのエストロゲン欠損の副作用の多くは、ERαによって媒介されると考えられている。
【0005】
酢酸ロイプロリド(Lupron(R))は、天然に存在するゴナドトロピン放出ホルモン(GnRHまたはLH−RH)の合成非ペプチド類似体である。酢酸ロイプロリドは、最終的に、下垂体によるLH分泌を抑制するLH−RH超作動薬である。酢酸ロイプロリドは、ゴナドトロピン分泌の強力阻害剤としての機能を果たし、卵巣および精巣のステロイド産生の抑制をもたらす。ヒトでは、酢酸ロイプロリドの投与は、黄体形成ホルモン(LH)および卵胞刺激ホルモン(FSH)の血中レベルの初期増加をもたらし、性腺ステロイド(男性におけるテストステロンおよびジヒドロテストステロン、ならびに閉経前の女性におけるエストロンおよびエストラジオール)の一過的なレベルの増加につながる。しかしながら、酢酸ロイプロリドの継続投与は、LHおよびFSHのレベルの低下をもたらす。男性では、テストステロンは、去勢レベル(50ng/dL以下)まで減少する。閉経前の女性では、エストロゲンは、閉経後レベルまで減少する。テストステロンは、前立腺の癌細胞に対する公知の刺激剤である。したがって、テストステロン分泌の抑制およびテストステロン作用の阻害は、前立腺癌療法の必要な構成要素である。酢酸ロイプロリドは、前立腺癌を治療するための去勢レベルまでの血清テストステロンの減少および低減である、LH抑制のために使用することができる。
【0006】
LHRH作動薬の導入前に、エストロゲン、主に、ジエチルスチルベストロール(DES)を介して、下垂体内のエストロゲン活性を増加させることによって、去勢テストステロンレベルを達成した。DESは、去勢レベルまでテストステロンを抑制する上でLHRH作動薬と同等に有効であった。DESで治療された患者は、ほてりまたは骨量の減少を有さなかったが、LHRH作動薬を用いるADTよりも高い比率で女性化乳房を有した。残念ながら、DESおよびエストラジオール等の非常に強力な純粋なエストロゲンは、しばしば、重度の心臓血管および血栓塞栓性合併症の高いリスクを伴い、それらの臨床使用を制限している。
【0007】
本発明の化合物は、テストステロンレベルを去勢レベルまで抑制し、骨量の減少、ほてり、および/または女性化乳房をもたらすことなく、血栓事象のリスクの増大を防止しながら、前立腺癌を治療するために使用し得る。
【発明の概要】
【0008】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、本明細書において下記に説明する、治療的有効量の式I〜XIIの化合物を投与することを含む。
【0009】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、本明細書において下記に説明する、治療的有効量の式I〜XIIの化合物を投与することを含み、血清総テストステロンの低下は、血清黄体形成ホルモンレベルの減少によって生じる。
【0010】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、本明細書において下記に説明する、治療的有効量の式I〜XIIの化合物を投与することを含み、血清総テストステロンの低下は、血清黄体形成ホルモンレベルの減少と無関係である。
【0011】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、本明細書において下記に説明する、治療的有効量の式I〜XIIの化合物を投与することを含み、式I〜XIIの該化合物の投与は、アンドロゲン除去療法(ADT)に伴う副作用が生じることを防止、または治療し、該対象者は、前立腺癌を有する。
【0012】
一実施形態では、本発明は、対象者において、アンドロゲン除去療法のための方法を提供し、本明細書において下記に説明する、治療的有効量の式I〜XIIの化合物を投与することを含み、該対象者は、前立腺癌を有する。
【0013】
一実施形態では、本発明は、進行した前立腺癌を抑制、発生を低減、重症度を軽減、または阻害する方法を提供し、本明細書において下記に説明する、治療的有効量の式I〜XIIの化合物を投与することを含む。
【0014】
一実施形態では、本発明は、進行した前立腺癌の緩和治療の方法を提供し、本明細書において下記に説明する、治療的有効量の式I〜XIIの化合物を投与することを含む。
本発明に関する主題を、具体的に、明細書のまとめの部分に示し、明確に請求する。しかしながら、本発明は、構成および操作方法双方に関して、その目的、特徴、および利点とともに、以下の添付の図面をとともに読む際に、詳細な説明を参照することにより、最も良く理解され得る。
【図面の簡単な説明】
【0015】
【図1】化合物IVの30mg/kgの経口投与(0日目の第1の投与量)後の無損傷のオスのサルにおける血清テストステロン(太線)および総アンドロゲン(点線)レベルを図示する(実施例8を参照)。
【図2】化合物IV(0.3、1、10、30mg/kg)で処置された無損傷のラットにおけるテストステロンレベルを図示する。「I」は、無損傷の媒体対照に対するP<0.05を示す。BLOQ値を、0.08ng/mLの定量限界で図式的に表す(実施例9を参照)。
【図3】17β−HSD5酵素活性への化合物IVの阻害作用を図示する(実施例12を参照)。
【図4】DES、17β−エストラジオール(E2)、および化合物IVの存在下のヒト血小板のインビトロ凝集を図示する。多血小板血漿(PRP)を、0.3ユニットのトロンビンとの凝集を誘発する前に、媒体、E2、DES、または化合物IVで、30秒間、培養した。凝集を、5分間監視し、媒体対照に対する割合で表示した(実施例13を参照)。
【図5】化合物II〜XIIの調製のための一般合成スキーム(実施例1を参照)。
【図6】化合物IVの調製のための合成スキーム(実施例2を参照)。
【図7】化合物VIの調製のための合成スキーム(実施例3を参照)。
【図8】化合物IXおよびXの調製のための合成スキーム(実施例5を参照)。
【図9】3mg/kg、10mg/kg、および300mg/kgの用量で、化合物IVで処置された無損傷ラットにおける、24時間後、72時間後、および168時間後のテストステロンレベルを図示する(実施例9を参照)。
【図10A】0.3mg/kg、1mg/kg、3mg/kg、10mg/kg、および30mg/kgの用量の化合物IVを用いて処置された、無損傷ラットおよび精巣摘除された(ORX)ラットのLHレベルを図示する。「I」は、無損傷の媒体対照に対するP<0.05を示す。「O」は、ORX媒体対照に対するP<0.05を示す。BLOQ値を、定量限界0.08ng/mLで図式的に表す(実施例9を参照)。
【図10B】0.3mg/kg、1mg/kg、3mg/kg、10mg/kg、および30mg/kgの用量の化合物IVを用いて処置された、無損傷ラットおよび精巣摘除された(ORX)ラットのFSHレベルを図示する。「I」は、無損傷の媒体対照に対するP<0.05を示す。「O」は、ORX媒体対照に対するP<0.05を示す。BLOQ値を、定量限界0.08ng/mLで図式的に表す(実施例9を参照)。
【図10C】0.3mg/kg、1mg/kg、3mg/kg、10mg/kg、および30mg/kgの用量の化合物IVを用いて処置された、無損傷ラットおよび精巣摘除された(ORX)ラットのテストステロンレベルを図示する。「I」は、無損傷の媒体対照に対するP<0.05を示す。「O」は、ORX媒体対照に対するP<0.05を示す。BLOQ値を、定量限界0.08ng/mLで図式的に表す(実施例9を参照)。
【図10D】0.3mg/kg、1mg/kg、3mg/kg、10mg/kg、および30mg/kgの用量の化合物IVを用いて処置された、無損傷ラットおよび精巣摘除された(ORX)ラットの前立腺重量レベルを図示する。「I」は、無損傷の媒体対照に対するP<0.05を示す。「O」は、ORX媒体対照に対するP<0.05を示す。BLOQ値を、定量限界0.08ng/mLで図式的に表す(実施例9を参照)。
【図10E】0.3mg/kg、1mg/kg、3mg/kg、10mg/kg、および30mg/kgの用量の化合物IVを用いて処置された、無損傷ラットおよび精巣摘除された(ORX)ラットの精嚢重量レベルを図示する。「I」は、無損傷の媒体対照に対するP<0.05を示す。「O」は、ORX媒体対照に対するP<0.05を示す。BLOQ値を、定量限界0.08ng/mLで図式的に表す(実施例9を参照)。
【図10F】0.3mg/kg、1mg/kg、3mg/kg、10mg/kg、および30mg/kgの用量の化合物IVを用いて処置された、無損傷ラットおよび精巣摘除された(ORX)ラットの肛門挙筋重量を図示する。「I」は、無損傷の媒体対照に対するP<0.05を示す。「O」は、ORX媒体対照に対するP<0.05を示す。BLOQ値を、定量限界0.08ng/mLで図式的に表す(実施例9を参照)。
【図11】化合物IV(A)およびDES(B)を異なる用量で投与することによる、無損傷ラットおよびORXラットにおける前立腺の大きさを図示する(実施例15を参照)。
【図12A】DESと化合物IVとの差異を図示し、DESは、グルココルチコイド受容体(GR)と交差反応するが、化合物IVは交差反応しない(実施例15を参照)。
【図12B】DESと化合物IVとの差異を図示し、DESは、アンドロゲン受容体(AR)と交差反応する。それは、AR作用を軽度に刺激し、軽度に阻害する(すなわち、それは、部分的作動薬/拮抗薬である)が、化合物IVは、それらをしない(実施例15を参照)。
【図12C】DESと化合物IVとの差異を図示し、DESは、エストロゲン関連受容体(ERR)トランス活性化を無効にするが、化合物IVは無効にしない(実施例15を参照)。
【図13】モルヒネ禁断モデルにおける、5mg/kg、10mg/kg、15mg/kg、および30mg/kgの用量での、ほてりの軽減への化合物IVの作用を図示する。グループ当たりN=7匹の動物。17β−E2を、100%のDMSO中で5mg/kgで使用した(実施例14を参照)。
【図14】化合物IVを91日間投与することによる、サルの用量依存性の体重(kg)低下(100mg/kgで約20%)を図示する。女性化乳房または高エストロゲン性の兆候は認められなかった(実施例16を参照)。
【図15】陽性対照(LHRH作動薬)と比較した、化合物IVの連日経口投与後のサルにおける、用量依存性の血清テストステロンレベルの減少(ng/mL)を図示する。点線は、化学的に去勢された患者のテストステロンレベルを示し、太い点線は、外科的に去勢されたサルのテストステロンレベルを示す(実施例16を参照)。
【図16】ベースライン時に、および28日目に化合物IVを投与することによる、サルにおける、用量依存性の前立腺特異的抗原(PSA)レベル(ng/mL)を図示する。PSAレベルは、化合物IVの処置で顕著に減少した(実施例16)。
【図17】陽性対照(LHRH作動薬)と比較した、6週間目に化合物IVを投与することによる、サルにおける、経直腸超音波(TRUS)を使用した用量依存性前立腺体積を図示する(実施例16を参照)。
【図18】化合物IV(図18A)投与することによる、90日目の、用量依存性の臓器重量(前立腺、精嚢、および精巣)を、サルの対照との比率で図示する。化合物IV(図18B)の連日経口投与後のサルにおける13週間目の剖検での前立腺重量(実施例16を参照)。
【図19】化合物IV(100mg、300mg、600mg、および1000mg)を投与することによる、1〜11日間の期間のヒトにおける用量依存性の平均総テストステロンレベル(nmol/L)を図示する(実施例17を参照)。
【図20】化合物IV(100mg、300mg、600mg、および1000mg)を投与することによる、1〜10日間の期間のヒトにおける用量依存性の平均LHレベル(IU/L)を図示する。(実施例17を参照)。
【図21】化合物IV(100mg、300mg、600mg、および1000mg)を投与することによる、1〜10日間の期間のヒトにおける用量依存性の平均遊離テストステロンレベル(pg/mL)を図示する。(実施例17を参照)。
【図22】化合物IV(100mg、300mg、600mg、および1000mg)を投与することによる、1〜10日間の期間のヒトにおける用量依存性の平均PSAレベル(μg/L)を図示する。(実施例17を参照)。
【図23】化合物IVの投与の14日間の回復後の無損傷ラットにおける用量依存性血清テストステロンレベル(ng/mL)を図示する。「I」は、無損傷の媒体対照に対するP<0.05を示す。(実施例10を参照)。
【発明を実施するための形態】
【0016】
図解を簡潔および明確にするために、図面に示す要素は、必ずしも原寸に比例して描かれてはいないことを理解されたい。例えば、要素のうちのいくつかの寸法は、明確にするために、他の要素に対して誇張され得る。さらに、適切であると考えられる場合では、参照番号は、対応する要素または類似要素を指すために図面で反復され得る。
【0017】
以下の詳細な説明では、本発明についての完全な理解を提供するために、多くの具体的な詳細を説明する。しかしながら、当業者は、本発明がこれらの具体的な詳細なく実践し得ることを理解するであろう。他の例では、本発明を曖昧にしないよう、周知の方法、手順、および構成要素を詳細に説明していない。
【0018】
一実施形態では、本明細書に説明する化合物、および/またはそれを含む組成物は、男性対象者において、血清総テストステロンレベルを低下させるために使用され得る。
一実施形態では、本明細書に説明する化合物、および/またはそれを含む組成物は、男性対象者において、血清総テストステロンレベルを低下させるために使用され得、血清総テストステロンの低下は、血清黄体形成ホルモン(LH)レベルの減少により生じる。
【0019】
一実施形態では、本明細書に説明する化合物、および/またはそれを含む組成物は、男性対象者において、血清総テストステロンレベルを低下させるために使用され得、血清総テストステロンの低下は、血清黄体形成ホルモンレベルの減少と無関係である。
【0020】
一実施形態では、本発明は、男性対象者において血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、それは式I:
【0021】
【化1】
の構造で表され、式中、
Yは、C(O)またはCH2であり、
R1、R2は、独立して、水素、ハロゲン、ヒドロキシル、アルコキシ、シアノ、ニトロ、CF3、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、O−Alk−NR5R6、またはO−Alk−複素環であり、複素環は、3〜7員の置換または非置換複素環式環、任意に芳香族であり、
R3、R4は、独立して、水素、ハロゲン、ヒドロキシアルキル、ヒドロキシル、アルコキシ、シアノ、ニトロ、CF3、NHCOR、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、または保護ヒドロキシルであり、
Rは、アルキル、水素、ハロアルキル、ジハロアルキル、トリハロアルキル、CH2F、CHF2、CF3、CF2CF3、アリール、フェニル、ハロゲン、アルケニル、CN、NO2、またはOHであり、
R5およびR6は、独立して、水素、フェニル、1〜6個の炭素原子のアルキル基、3〜7員のシクロアルキル、3〜7員の複素環、5〜7員のアリールであるか、またはR5およびR6は、窒素原子とともに、3〜7員の環を形成し、
jおよびkは、独立して、1から4であり、
Alkは、1〜7個の炭素の直鎖アルキル、1〜7個の炭素の分岐鎖アルキル、または3〜8個の炭素の環状アルキルである。
【0022】
本明細書に説明する方法のさらなる実施形態では、式Iの化合物は、式IA:
【0023】
【化2】
によって表され、式中、R1、R2、R3、R4、j、およびkは、式Iについて定義するとおりである。
【0024】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、その化合物は式II:
【0025】
【化3】
で表される。
【0026】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、その化合物は式III:
【0027】
【化4】
で表される。
【0028】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、その化合物は式IV:
【0029】
【化5】
で表される。
【0030】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、その化合物は式V:
【0031】
【化6】
で表される。
【0032】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、その化合物は式VI:
【0033】
【化7】
で表される。
【0034】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、その化合物は式VII:
【0035】
【化8】
で表される。
【0036】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、その化合物は式VIII:
【0037】
【化9】
で表される。
【0038】
一実施形態では、本発明は、前立腺癌を有する男性対象者において、黄体形成ホルモン(LH)レベルの減少によって、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、その化合物は式IX:
【0039】
【化10】
で表される。
【0040】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、その化合物は式X:
【0041】
【化11】
で表される。
【0042】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、その化合物は式XI:
【0043】
【化12】
で表される。
【0044】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、その化合物は式XII:
【0045】
【化13】
で表される。
【0046】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の式IA、I〜XIIの化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含む。別の実施形態では、男性対象者は、前立腺癌を有する。別の実施形態では、血清総テストステロンは、約100ng/dL未満に低減される。別の実施形態では、血清総テストステロンは、約50ng/dL未満に低減される。別の実施形態では、血清総テストステロン濃度は、約25ng/dL未満に低減される。
【0047】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の式IA、I〜XIIの化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、血清総テストステロンの低下は、血清黄体形成ホルモン(LH)レベルの減少によって生じる。別の実施形態では、男性対象者は、前立腺癌を有する。別の実施形態では、血清総テストステロンは、約100ng/dL未満に低減される。別の実施形態では、血清総テストステロンは、約50ng/dL未満に低減される。別の実施形態では、血清総テストステロン濃度は、約25ng/dL未満に低減される。
【0048】
一実施形態では、本発明は、男性対象者において、遊離血清テストステロンレベルを低下させる方法を提供し、治療的有効量の式IA、I〜XIIの化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、遊離血清テストステロンの低下は、血清黄体形成ホルモン(LH)レベルの減少によって生じる。別の実施形態では、男性対象者は、前立腺癌を有する。
【0049】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の式IA、I〜XIIの化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、血清総テストステロンの低下は、血清黄体形成ホルモン(LH)レベルの減少と無関係である。別の実施形態では、男性対象者は、前立腺癌を有する。別の実施形態では、血清総テストステロンは、約100ng/dL未満に低減される。別の実施形態では、血清総テストステロンは、約50ng/dL未満に低減される。別の実施形態では、血清総テストステロン濃度は、約25ng/dL未満に低減される。
【0050】
一実施形態では、本発明は、男性対象者において、遊離血清テストステロンレベルを低下させる方法を提供し、治療的有効量の式IA、I〜XIIの化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、遊離血清テストステロンレベルの低下は、血清黄体形成ホルモン(LH)レベルの減少と無関係である。別の実施形態では、男性対象者は、前立腺癌を有する。
【0051】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベル、または遊離血清テストステロンレベルを低下させる方法を提供し、該男性対象者は、前立腺癌を有する。別の実施形態では、該対象者は、進行した前立腺癌を有する。
【0052】
一実施形態では、テストステロンの血清濃度の減少は、可逆的であり、本発明の化合物での治療後にベースラインレベルに戻る。
別の実施形態では、テストステロンの血清濃度は、図23および実施例10に従う化合物IVでの治療後に可逆的である。
【0053】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の式IA、I〜XIIの化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含む。別の実施形態では、血清総テストステロンは、約100ng/dL未満に低下される。別の実施形態では、血清総テストステロンは、約50ng/dL未満に低減される。別の実施形態では、血清総テストステロンは、約25ng/dL未満に低減される。別の実施形態では、血清総テストステロンは、約75ng/dL未満に低減される。別の実施形態では、血清総テストステロンは、約75ng/dL〜100ng/dLに低減される。別の実施形態では、血清総テストステロンは、約50ng/dL〜75ng/dLに低減される。別の実施形態では、血清総テストステロンは、約40ng/dL〜50ng/dLに低減される。別の実施形態では、血清総テストステロン濃度は、約25ng/dL〜50ng/dLに低減される。別の実施形態では、血清総テストステロンは、約40ng/dL〜60ng/dLに低減される。
【0054】
テストステロンは、「遊離」(つまり、生物学的に利用可能、および未結合)、または「総」(タンパク質結合したものの割合を含み、利用不可能)血清レベルとして測定することができる。40歳以上の前立腺癌を有さない男性は、250ng/dL(<8.7nmol/L)未満の総テストステロンレベル、または0.75ng/dL(<0.03nmol/L)未満の遊離テストステロンレベルを有し、低いテストステロンレベルを示す。
【0055】
一実施形態では、本発明は、前立腺癌を有する男性対象者において、黄体形成ホルモン(LH)レベルの減少と無関係に、またはLHレベルの減少によって、血清総テストステロンレベル、および/または遊離テストステロンレベルを低下させる方法を提供する。別の実施形態では、テストステロンレベルの変化は、治療前のレベルからの減少でなければならない。別の実施形態では、血清総テストステロンレベルは、100ng/dL未満に低減される。別の実施形態では、血清総テストステロンは、50ng/dL未満に低減される。別の実施形態では、血清総テストステロンは、25ng/dL未満に低減される。別の実施形態では、遊離テストステロンは、2ng/dL未満に低減される。別の実施形態では、遊離テストステロンは、約1ng/dL未満に低減される。別の実施形態では、遊離テストステロンは、約0.5ng/dL未満に低減される。別の実施形態では、遊離テストステロンは、約0.25ng/dL未満に低減される。
【0056】
遊離血清テストステロンレベルおよび血清総テストステロンレベルを決定する方法は、血液試験によって、一連の治療期間中にテストステロンレベルを監視することを含む。総テストステロンは、担体タンパク質(アルブミン、SHBG、トランスコルチン、トランスフェリン)に結合した循環テストステロンと、遊離/未結合ホルモンとの組み合わせである。総テストステロンレベルは、体内のホルモンを運搬する血中のタンパク質レベル、年齢、肥満、および一般に使用される試験方法に伴う干渉を含む、種々の要因によって影響される場合がある。
【0057】
遊離テストステロン(FT)を測定するために使用可能な方法は、類似体トレーサを使用して、複雑である(平衡透析および算出された遊離テストステロン)か、または単純であり得る(市販のFTキット「Coat−A−Count」)。別の実施形態では、総テストステロン血清レベル、および遊離テストステロン血清レベルの測定は、総テストステロンおよびSHBG(例えば、Irma−Count,DPC)の同時測定、次いで、算出された遊離テストステロン(CFT)によって達成することができる。別の実施形態では、総テストステロンおよび遊離テストステロンの測定は、当業者の知識に従う。
【0058】
一実施形態では、男性対象者において、血清総テストステロンレベル、または遊離血清テストステロンレベルを低下させる方法を提供し、治療的有効量の1つまたは複数の別の形態のADTと式IA、I〜XIIの化合物またはその異性体との組み合わせ、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含む。別の実施形態では、総テストステロン、または遊離血清テストステロンの低減は、血清黄体形成ホルモン(LH)レベルの減少によって生じる。別の実施形態では、総テストステロンレベル、または遊離血清テストステロンレベルは、血清黄体形成ホルモンレベルの減少と無関係である。
【0059】
本発明の方法は、別の形態のADTと本発明の化合物との組み合わせを投与することを含む。一実施形態では、別の形態のADTは、LHRH作動薬を含む。別の実施形態では、LHRH作動薬は、酢酸ロイプロリド(Lupron(R))(米国特許第5,480,656号、同第5,575,987号、同第5,631,020号、同第5,643,607号、同第5,716,640号、同第5,814,342号、同第6,036,976号はすべて、参照することにより、本明細書に組み込まれる)、または酢酸ゴセレリン(Zoladex(登録商標))(米国特許第7,118,552号、同第7,220,247、同第7,500,964号はすべて、参照することにより、本明細書に組み込まれる)を含む。一実施形態では、別の形態のADTは、LHRH拮抗薬を含む。別の実施形態では、LHRH拮抗薬は、デガレリクスを含む。一実施形態では、別の形態のADTは、抗アンドロゲンを含む。別の実施形態では、抗アンドロゲンは、ビカルタミド、フルタミド、フィナステリド、デュタステリド、MDV3100、ニルタミド、クロルマジノン、またはそれらの任意の組み合わせを含む。
【0060】
一実施形態では、本発明の方法は、治療的有効量の抗アンドロゲン、および本発明の化合物を投与することを含む。一実施形態では、本発明の方法は、治療的有効量のLHRH作動薬、および本発明の化合物を投与することを含む。一実施形態では、本発明の方法は、治療的有効量の抗アンドロゲン、LHRH作動薬、および本発明の化合物を投与することを含む。
【0061】
一実施形態では、本発明は、男性対象者において、アンドロゲン除去療法(ADT)を生成する目的で、前立腺癌を有する男性対象者において、黄体形成ホルモン(LH)レベルの減少により、または黄体形成ホルモンレベルの減少と無関係で、血清総テストステロンレベル、および/または遊離テストステロンレベルを低下させる方法を提供し、治療的有効量の式IA、I〜XIIの化合物を投与することを含む。別の実施形態では、化合物は、化合物IVである。
【0062】
別の実施形態では、本発明は、対象者において、アンドロゲン除去療法(ADT)のための方法を提供し、治療的有効量の式IA、I〜XIIの化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含む。別の実施形態では、該対象者は、前立腺癌を有する。別の実施形態では、化合物は、化合物IVである。
【0063】
別の実施形態では、ADTを、前立腺癌を治療するため、前立腺癌の進行を遅延するため、または前立腺癌の再発を防止する、および/または治療するために使用する。
一実施形態では、本発明は、前立腺癌を治療する、前立腺癌の進行を遅延する、前立腺癌の再発の防止する、および/または治療する方法を提供し、本発明の化合物を投与することを含む。別の実施形態では、LHRH類似体、可逆性抗アンドロゲン(ビカルタミドまたはフルタミド等)、抗エストロゲン、抗癌剤、5−アルファ還元酵素阻害剤、アロマターゼ阻害剤、プロゲスチン、選択的アンドロゲン受容体モジュレータ(SARMS)、または他の核ホルモン受容体を介して作用する薬剤と組み合わせて、本発明の化合物を投与する。
【0064】
一実施形態では、本発明は、前立腺癌を治療し、LHレベルを減少させることによるか、またはLHレベルの減少と無関係に、血清総テストステロン、および/または遊離血清テストステロンレベルを減少させる方法を提供し、式IA、I〜XIIの化合物を投与することを含む。別の実施形態では、化合物IVを投与する。
【0065】
アンドロゲン除去療法は、テストステロンを減少させるだけでなく、エストロゲンレベルもまた、テストステロンの芳香族化に由来するため、低減される。アンドロゲン除去療法により誘発されるエストロゲン欠乏は、ほてり、女性化乳房、および乳房痛、骨量の減少、骨質および強度の低下、骨粗しょう症、骨減少症、および致命的な骨折、有害な脂質変化、およびより高い心臓血管疾患、および心筋梗塞、性欲減弱、インポテンス、筋肉量の低下(サルコペニア)、疲労、認知機能障害、ならびに鬱および他の情緒変化を含む、著しい副作用をもたらす。
【0066】
他の実施形態では、本発明は、ADTに伴う任意の疾患、障害、または症状を治療する方法を提供する。他の実施形態では、本発明は、治療する方法を提供する。テストステロン枯渇に伴う任意の疾患、障害、または症状を治療する方法を提供する。各疾患、障害、または症状は、本発明の別個の実施形態を表す。
【0067】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の式IA、I〜XIIの化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、該式IA、I〜XIIの化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することは、アンドロゲン除去療法(ADT)に伴う副作用が発生することを防止し、抑制し、発症を低減し、阻害し、または治療し、該対象者は、前立腺癌を有する。別の実施形態では、総テストステロンレベル、または遊離血清テストステロンレベルの低減は、LHレベルを減少させることによるものであるか、またはLHレベルの減少と無関係である。
【0068】
一実施形態では、本発明の化合物を投与することは、従来のアンドロゲン除去療法(ADT)に伴う典型的な副作用の発生を抑制し、発症を低減し、阻害し、または治療する。別の実施形態では、対象者は、前立腺癌を有する。副作用のかかる防止および/または減少は、プラシーボまたは対照群と比較した、相対的なものである。一実施形態では、従来のアンドロゲン除去療法(ADT)に伴う典型的な副作用は、ほてり、女性化乳房、骨塩密度の低下、および骨折の増加を含む。別の実施形態では、本発明の化合物を投与することは、従来の形態のアンドロゲン除去療法(ADT)を使用すると認められるであろう、ほてりが発生することを防止する。別の実施形態では、本発明の化合物を投与することは、従来の形態のアンドロゲン除去療法(ADT)を使用すると認められるであろう、女性化乳房が発生することを防止する。別の実施形態では、本発明の化合物を投与することは、従来の形態のアンドロゲン除去療法(ADT)を使用すると認められるであろう、骨塩密度(BMD)の低下が発生することを防止する。別の実施形態では、本発明の化合物を投与することは、従来の形態のアンドロゲン除去療法(ADT)を使用すると認められるであろう、骨折の増加が発生することを防止する。別の実施形態では、骨折の増加は、病理学的骨折、非外傷性骨折、脊椎骨折、非脊椎骨折、新規形態学的骨折、臨床骨折、またはそれらの組み合わせである。
【0069】
一実施形態では、「従来のアンドロゲン除去療法」という用語は、外科医が精巣を除去する、精巣摘除(外科的去勢)を対象とする。別の実施形態では、「従来のアンドロゲン除去療法」という用語は、黄体形成ホルモン放出ホルモン(LHRH)類似体を投与することを対象とし、これらの薬剤は、精巣によって生成されるテストステロンの量を低下させる。米国で使用可能なLHRH類似体の例は、ロイプロリド(Lupron,Viadur(登録商標),Eligard(登録商標))、ゴセレリン(Zoladex(登録商標))、トリプトレリン(Trelstar)、およびヒストレリン(Vantas)を含む。別の実施形態では、「従来のアンドロゲン除去療法」という用語は、抗アンドロゲンを投与することを対象とし、抗アンドロゲンは、任意のアンドロゲンを使用する身体の能力を阻止する。精巣摘除後、またはLHRH類似体を用いた治療中であっても、少量のアンドロゲンは、依然として、副腎によって生成される。抗アンドロゲン薬剤の例は、フルタミド(Eulexin)、ビカルタミド(Casodex(登録商標))、およびニルタミド(Nilandron)を含む。別の実施形態では、「従来のアンドロゲン除去療法」という用語は、アバレリックス(Plenaxis(登録商標))等の黄体形成ホルモン放出ホルモン(LHRH)拮抗薬を投与することを対象とし、デガレリックス(Firmagon(登録商標))は、2008年にFDAによって、進行した前立腺癌を治療するために使用が認可された、新規LHRH拮抗薬である。別の実施形態では、「従来のアンドロゲン除去療法」という用語は、フィナステライド(Proscar(登録商標))およびデュタステリド(Avodart(登録商標))等の5α−還元酵素阻害剤を投与することを対象とし、5α−還元酵素阻害剤は、テストステロンを、より活性的なアンドロゲン、5α−ジヒドロテストステロン(DHT)に転換する身体の能力を阻止する。別の実施形態では、「従来のアンドロゲン除去療法」という用語は、ケトコナゾール(Nizoral(登録商標))等のテストステロン生合成の阻害剤を投与することを対象とする。別の実施形態では、「従来のアンドロゲン除去療法」という用語は、ジエチルスチルベストロール、または17β−エストラジオール等のエストロゲンを投与することを対象とする。
【0070】
一実施形態では、「ほてり」という用語は、上半身、または全身の急激な熱感、顔面および首筋の紅潮、胸部、背部、および腕に発現する赤色斑、多量の発汗、寒気等を指す。
一実施形態では、「女性化乳房」という用語は、乳房の腺構成要素の増殖から生じる、男性乳房の両性肥大を指し、疼痛を伴う場合、または伴わない場合がある。女性化乳房は、乳首から同心円状に伸びるゴム状または硬い腫瘤の存在によって、臨床的に定められている。擬似女性化乳房、または脂肪堆積(lipomastia)として公知の病状は、腺増殖のない脂肪沈着を特徴とする。女性化乳房は、通常、両側性であるが、片側性である場合もある。
【0071】
一実施形態では、本発明の方法は、テストステロンの減少によって、骨量の減少およびほてりをもたらさずに、前立腺癌または進行した前立腺癌を有する男性を治療することを対象とする。別の実施形態では、本発明の方法は、IA、I〜XIIの化合物を活用し、化合物は、前立腺癌のための現在のアンドロゲン枯渇療法(ADT)に一般である、骨量の減少およびほてり等の副作用をもたらさずに、前立腺癌の一次刺激であるテストステロンを減少させる可能性を有する。
【0072】
別の実施形態では、下記の表8(実施例11)は、化合物IVを投与することによる、骨量の減少をもたらさないテストステロンの減少を表す。
一実施形態では、本発明の方法は、式IA、I〜XIIの化合物を投与することによる、テストステロンレベルの減少を対象とし、それはさらに、進行した前立腺癌を治療する。一実施形態では、本発明の方法は、式IA、I〜XIIの化合物を投与することによる、テストステロンレベルの減少を対象とし、それはさらに、発症を抑制し、減少させ、重症度を軽減し、または進行した前立腺癌を阻害する。一実施形態では、本発明の方法は、式IA、I〜XIIの化合物を投与することによる、テストステロンレベルの減少を対象とし、それはさらに、進行した前立腺癌の緩和治療を提供する。
【0073】
一実施形態では、本発明の方法は、進行した前立腺癌を治療することを対象とする。一実施形態では、本発明の方法は、発症を抑制し、減少させ、重症度を軽減し、または進行した前立腺癌を阻害することを対象とする。一実施形態では、本発明の方法は、進行した前立腺癌の緩和治療を対象とする。別の実施形態では、本発明の方法は、IA、I〜XIIの化合物を活用する。別の実施形態では、本発明は、進行した前立腺癌を治療することを対象とする。別の実施形態では、本発明は、進行した前立腺癌を抑制することを対象とする。別の実施形態では、本発明は、進行した前立腺癌の発症を減少させることを対象とする。別の実施形態では、本発明は、進行した前立腺癌の重症度を軽減することを対象とする。別の実施形態では、本発明は、進行した前立腺癌を阻害することを対象とする。
【0074】
「進行した前立腺癌」という用語は、前立腺に由来し、精嚢、骨盤リンパ節、もしくは骨、または身体の他の部分を含むように、周辺組織等の前立腺を超えて広く転移している転移性癌を指す。前立腺癌病理学は、悪性度の増加の順に1〜5のグリーソン分類で分類される。別の実施形態では、前立腺癌による進行性疾患および/または死の有意なリスクを有する患者は、定義に含まれなければならず、IIBという低い疾患段階の前立腺被膜の外側に癌を有する任意の患者は、明らかに「進行した」疾患を有する。
【0075】
一実施形態では、本明細書に提供する方法および/または本明細書に提供する化合物の使用は、視床下部−下垂体−精巣軸(HPT軸)上のフィードバックを提供する上で有効である。フィードバックは、1つの臓器または組織内に生成された物質の、それ自体の活性に影響を及ぼす別の臓器または組織の活性を調節する能力を指す。一実施形態では、視床下部−下垂体−精巣軸(HPT軸)上のフィードバックは、LHレベルの減少をもたらす。一実施形態では、視床下部−下垂体−精巣軸(HPT軸)上のフィードバックは、血清総テストステロンレベルの減少をもたらす。一実施形態では、視床下部−下垂体−精巣軸(HPT軸)上のフィードバックは、遊離血清テストステロンレベルの減少をもたらす。一実施形態では、視床下部−下垂体−精巣軸(HPT軸)上のフィードバックは、アンドロゲンの血清レベル、組織レベル、または腫瘍レベルの減少をもたらす。
【0076】
視床下部−下垂体−精巣(HPT)軸は、視床下部、下垂体腺、および精巣内のホルモンレベルを調節する、内分泌生理学系を指す。LHRH(黄体形成ホルモン放出ホルモン)は、視床下部によって放出され、下垂体を刺激して、LHおよびFSH(ゴナドトロピン)を合成して分泌させる。次いで、LHおよびFSHは、精巣に作用し、テストステロンおよび精子産生を刺激する。次いで、テストステロンは、視床下部LHRH分泌に直接的な負のフィードバック作用、および下垂体LHおよびFSH産生に間接的負のフィードバックを有する。エストロゲン、アンドロゲン、および血清タンパク質(例えば、インヒビン)はまた、LHRH分泌、ならびにLHおよびFSHの分泌に負の影響を有する。
【0077】
下垂体腺は、体内のテストステロンのレベルを制御する、1つの腺である。
テストステロンレベルが低い時、下垂体腺は、黄体形成ホルモン(LH)を放出する。このホルモンは、
精巣を誘発して、より多くのテストステロンを生成させる。テストステロンのレベルは、思春期中に増大する。テストステロンのレベルは、約20歳〜40歳で最高であり、次いで、年配の男性では、徐々に少なくなる。女性は、男性と比較すると、体内にはるかに少量のテストステロンを有する。しかし、テストステロンは、男性および女性の体内にわたって重要な役割を担う。それは、脳、骨、筋肉量、脂肪分布、血管系、エネルギーレベル、生殖器組織、および性機能に影響を及ぼす。循環テストステロンの大半は、性ホルモン結合グロブリンと称されるタンパク質、またはアルブミンと称される別の血清タンパク質に結合している。結合しない(または「遊離」)テストステロンもまた、臨床的に判定することができる。
【0078】
別の実施形態では、血清黄体形成ホルモンレベルの減少と無関係に血清総テストステロンまたは遊離血清テストステロンレベルを低下させるのは、性ホルモン結合グロブリン(SHBG)の増加によるものである。別の実施形態では、血清黄体形成ホルモンレベルの減少と無関係な遊離テストステロンレベルを低下させるのは、性ホルモン結合グロブリン(SHBG)の増加によるものである。別の実施形態では、血清黄体形成ホルモン(LH)レベルの減少と無関係に血清総テストステロンレベルまたは遊離血清テストステロンレベルを低下させるのは、精巣内のライディッヒ細胞によるテストステロン産生または分泌の阻害によるものである。別の実施形態では、血清黄体形成ホルモン(LH)レベルの減少と無関係に血清総テストステロンレベルまたは遊離血清テストステロンレベルを低下させるのは、副腎ステロイド産生の減少によるものである。
【0079】
一実施形態では、本明細書に説明する化合物、および/またはそれらを含む組成物は、黄体形成ホルモン(LH)レベルの減少のために使用し得る。別の実施形態では、本発明の化合物および/または組成物を使用して、内因性性ホルモンを減少させ得る。
【0080】
ヒドロキシステロイド脱水素酵素(HSD)ファミリー成員は、循環ステロイドの変換に関与する。17β−HSD5は、アンドロステンジオンをテストステロンに、エストロンをエストラジオールに転換する。加えて、それはまた、プロスタグランジン合成に関与する。一実施形態では、本発明の化合物は、HSDを阻害し、具体的には、17β−ヒドロキシステロイド脱水素酵素5(17β−HSD5)阻害である。かかる阻害は、末梢/性腺外テストステロン合成を防止することによって、ADTにおいて有用であり得、それは、HPT軸制御を逃れ、総テストステロンまたは遊離血清テストステロンの不完全な減少をもたらし得るか、または局所的に上昇した細胞内テストステロンレベルを可能にし得、それらのいずれかは、ADTにおいて有害である可能性がある。
【0081】
LHRH作動薬療法、すなわち、黄体形成ホルモン放出ホルモン作動薬(LHRH)、またはその類似体の投与によって達成される、アンドロゲン除去療法(ADT)は、下垂体からのゴナドトロピン放出の初期刺激、および精巣からのテストステロン産生(「フレア反応」と称される)、およびそれに続くゴナドトロピン放出の低下、ならびにテストステロンレベル、およびエストロゲンレベルの両方の低下をもたらす。LHRH作動薬療法によって引き起こされた「フレア反応」は、アンドロゲン/テストステロンレベルの増加により、前立腺癌の治療へのマイナスの影響を有する。加えて、LHRH療法には、糖尿病および心臓血管疾患のリスクの増大が伴う(Smith(2008)Current Prostate Reports.6:149−154)。
【0082】
LHRH療法のフレア反応を克服するために、LHRH/抗アンドロゲン療法アプローチと組み合わせた抗アンドロゲン単独療法(ビカルタミド、フルタミド、クロルマジノン)、およびLHRH拮抗薬(デガレリクス)が示唆されている(Suzuki et al.,(2008)Int.J.Clin.Oncol.13:401−410、Sharifi,N.et al.,(2005)JAMA。294(2):238−244)。抗アンドロゲン単独療法は、対象者におけるアンドロゲンレベルを減少させない。ビカルタミド抗アンドロゲン単独療法は、骨転移を有する前立腺癌の患者において、ADTよりも有効性が少ないことが分かっている。加えて、ビカルタミド療法で認められた悪影響は、乳房圧痛および乳房肥大(女性化乳房および乳房痛)を含む(Suzuki et al.,同書)。抗アンドロゲン療法に伴う付加的なリスクは、肝トランスアミナーゼの増加を含む。
【0083】
一実施形態では、本発明は、「フレア」反応の産生なく、かつ、従来のADT方法を使用して、テストステロンの減少によって引き起こされる、エストロゲン欠損に伴う悪影響を克服しながら、LHレベルの減少、ならびにそれによる、血清総テストステロンレベルおよび/または遊離血清テストステロンの減少を提供する。対象化合物の方法/使用は、骨組織の維持(骨組織への作動薬の作用)、血栓の潜在性の低下、および/または、エストラジオールもしくはジエチルスチルベストロールよりも少ない、もしくは中立的なほてり、および/または乳房組織への作用を提供する、組織選択的エストロゲン活性を提供する。
【0084】
一実施形態では、化合物IVは、作動作用を示すが、拮抗作用は示さず(実施例6および7)、したがって、化合物IVは、ゴナドトロピンおよびテストステロンの増加を引き起こさない。
【0085】
一実施形態では、化合物IVは、血清ホルモン、テストステロン、および総アンドロゲンの減少に対する、強力な薬理学的反応を表す、作動薬活性(実施例8〜11)を示す。
一実施形態では、本明細書において提供する化合物および/または組成物を使用する、本明細書において提供する方法は、従来の形態のADTの使用によるLHの減少によって引き起こされる骨吸収作用を減少させ、または除去する上で有効である。一実施形態では、本明細書において提供する方法、および/または本明細書において提供する組成物の使用は、従来の形態のADTの使用によるテストステロンレベルの減少によって引き起こされる骨吸収作用を減少させ、または除去する上で有効である。一実施形態では、本明細書において提供する組成物を使用する、本明細書において提供する方法は、LHレベルの減少の結果、エストロゲンの減少によって引き起こされる骨吸収作用を減少させ、または除去する上で有効である。一実施形態では、本明細書において提供する化合物および/または組成物を使用する、本明細書において提供する方法は、従来の形態のADTの使用によるLHレベルの減少に伴う骨吸収作用を防止する。一実施形態では、本明細書において提供する化合物および/または組成物を使用する、本明細書において提供する方法は、従来の形態のADTの使用による内因性LH、テストステロン、および/またはエストラジオール減少に伴う骨量の減少を防止する。一実施形態では、本明細書において提供する化合物および/または組成物を使用する、本明細書において提供する方法は、LHレベルの減少を提供しながら、骨量密度(BMD)を増加させる。一実施形態では、本明細書において提供する化合物および/または組成物を使用する、本明細書において提供する方法は、内因性LH、テストステロン、および/またはエストラジオールレベルの低下を提供しながら、骨容量の割合を増加させる。
【0086】
いくつかの実施形態では、本発明は、本発明の化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することによって、血栓塞栓症を回避し、および/または減少させる方法を提供する。
【0087】
一実施形態では、本明細書において提供する化合物および/または組成物を使用する、本明細書において提供する方法は、乳房組織において有効である。一実施形態では、本明細書において提供する化合物および/または組成物を使用する、本明細書において提供する方法は、従来のADTによって達成されたLHレベルの減少に伴う女性化乳房を防止しながら、LHレベルの減少を提供する。
【0088】
一実施形態では、実施例13は、特定の毒性研究を開示し、ヒトの血小板を用いるインビボ研究は、化合物IVが、DESよりもさらに低い凝固促進活性を有したことを示した。したがって、ER選択的作動薬である化合物IVは、DESよりも少ない血栓事象のリスクを有するとともにDESの前立腺癌の利点をもたらし、また、骨量の減少、ほてり、または有害な脂質プロファイルを引き起こすことなく、LHRH作動薬または拮抗薬の利点をもたらすはずである。
【0089】
単独の、または他のADTと組み合わせたジエチルスチルベストロール(DES)療法は、DESが前立腺癌を有する患者において骨吸収を防止したことを示した。DESの使用は、前立腺癌の療法として推進されているが、血管新生および悪性腫瘍へのDESの作用は、DES代謝物によって媒介されると考えられ、エストロゲン受容体を介して機能することは考えられない。加えて、治療的使用のために投与されるDESの用量レベルは、血管疾患、心臓血管罹患率、血栓毒性、女性化乳房、勃起不全、および性欲減退を含む、数々の有害な副作用を示す(Scherr and Pitts,同書、およびPresti,J.C.Jr.(1996)JAMA.275(15):1153−6)。
【0090】
一実施形態では、本発明は、単独の、または抗アンドロゲンまたはDESと組み合わせた、LHRH作動薬または拮抗薬療法のマイナスの副作用を克服する。別の実施形態では、対象発明の方法は、有害な骨関連病状等の有害なエストロゲン欠損の副作用がなく、かつ女性化乳房等の有害なエストロゲン刺激副作用がない、アンドロゲン除去療法を提供する。別の実施形態では、本発明の方法は、LHレベルの減少を提供し、それによって、LHの減少によって引き起こされるエストロゲン欠損に伴う悪影響を克服し、かつDES療法で認められた、一般的エストロゲン作動薬の増加に伴う悪影響を克服しながら、「フレア」作用を産生せずに、総テストステロンレベル、および/または遊離血清テストステロンレベルの減少を提供する。対象化合物の方法/使用は、組織選択的エストロゲン活性を提供し、それによって、骨組織の維持(骨組織への作動薬作用)、血栓の潜在性の低下、および乳房組織への中立的作用を提供する。
【0091】
視床下部レベルのタモキシフェン、トレミフェン、およびラロキシフェン等の従来の選択的エストロゲン受容体モジュレータ(SERM)の抗エストロゲン作用は、男性において、ゴナドトロピンレベルの増加、またはLHレベルの増加をもたらし、それによって、潜在的に、テストステロン血清レベルの増加をもたらす(Tsouri et al.,2008,Fertility and Sterility doi:10.1016)。対照的に、式IA、I〜XIIの化合物を投与することを含む、本発明の方法は、男性対象者において、LHの減少を提供する。
式Iの化合物のためのさらなる実施形態:
本発明の方法の一実施形態では、式Iの化合物のYは、C(O)である。別の実施形態では、Yは、CH2である。別の実施形態では、式IまたはIAの化合物のR1およびR2は、独立して、O−Alk−NR5R6、またはO−Alk−複素環である。別の実施形態では、上記に説明する、該O−Alk−複素環、O−Alk−NR5R6、−Alk−複素環、およびAlk−NR5R6のAlkは、1〜7個の炭素の直鎖アルキル、1〜7個の炭素の分岐鎖アルキル、または3〜8個の炭素の環状アルキルである。別の実施形態では、アルキルは、エチレン(−CH2CH2−)である。別の実施形態では、Alkは、メチレン(−CH2−)である。別の実施形態では、Alkは、プロピレン(−CH2CH2CH2−)である。別の実施形態では、Alkは、2−メチルプロピレン(−CH2CH(CH3)CH2−)である。
【0092】
本発明の方法の一実施形態では、式IまたはIAの化合物のR1は、パラ位にある。本発明の方法の一実施形態では、式IまたはIAの化合物のR1およびR2は、異なる。本発明の方法の別の実施形態では、式IまたはIAの化合物のR1およびR2は、同一である。本発明の方法の別の実施形態
では、式IまたはIAの化合物のR1は、
【0093】
【化14】
である。方法の別の実施形態では、式IまたはIAの化合物のR1は、ヒドロキシルである。方法の別の実施形態では、式IまたはIAの化合物のR1は、アルコキシである。方法の別の実施形態では、R1およびR2は、独立して、水素、ハロゲン、ヒドロキシル、アルコキシ、シアノ、ニトロ、CF3、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、O−Alk−NR5R6、またはO−Alk−複素環であり、複素環は、3〜7員の置換または非置換複素環式環、任意に芳香族である。方法の別の実施形態では、式IまたはIAの化合物のR1およびR2は、独立して、水素、ハロゲン、ヒドロキシル、アルコキシ、シアノ、ニトロ、CF3、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、O−Alk−NR5R6、またはO−Alk−複素環であり、複素環は、3〜7員の置換または非置換複素環式環、任意に芳香族である。方法の別の実施形態では、式IまたはIAの化合物のR2は、ハロゲンである。方法の別の実施形態では、式IまたはIAの化合物のR2は、Fである。方法の別の実施形態では、式Iの化合物のR2は、Clである。方法の別の実施形態では、式IまたはIAの化合物のR2は、Brである。方法の別の実施形態では、式IまたはIAの化合物のR2は、Iである。方法の別の実施形態では、式IまたはIAの化合物のR2は、ヒドロキシルである。方法の別の実施形態では、R1および/またはR2は、CF3である。別の実施形態では、R1および/またはR2は、CH3である。別の実施形態では、R1および/またはR2は、ハロゲンである。別の実施形態では、R1および/またはR2は、Fである。別の実施形態では、R1および/またはR2は、Clである。別の実施形態では、R1および/またはR2は、Brである。別の実施形態では、R1および/またはR2は、Iである。別の実施形態では、式Iの化合物のR2は、パラ位にある。
【0094】
本発明の方法の一実施形態では、式IまたはIAの化合物のR3およびR4は、同一である。本発明の方法の別の実施形態では、式IまたはIAの化合物のR3およびR4は、異なる。方法の別の実施形態では、式IまたはIAの化合物のjおよびkは、独立して、1である。方法の別の実施形態では、式IまたはIAの化合物のR3およびR4は、独立して、ハロゲン、ハロアルキル、ヒドロキシル、またはアルキルである。方法の別の実施形態では、式IまたはIAの化合物のR3およびR4は、独立して、Fである。方法の別の実施形態では、式IまたはIAの化合物のR3およびR4は、独立して、Brである。方法の別の実施形態では、式IまたはIAの化合物のR3およびR4は、独立して、Clである。別の実施形態では、R4は、パラ位にある。別の実施形態では、R3は、オルト位にある。別の実施形態では、R3は、メタ位にある。別の実施形態では、R3および/またはR4は、CF3である。別の実施形態では、R3および/またはR4は、CH3である。
【0095】
本発明の方法の一実施形態では、式IまたはIAの化合物のR5およびR6は、その窒素原子とともに、3〜7員の環を形成する。別の実施形態では、環は、飽和または不飽和環である。別の実施形態では、環は、飽和または不飽和環である。本発明の方法の別の実施形態では、式IまたはIAの化合物のR5およびR6は、その窒素とともに、ピペラジン環を形成する。方法の別の実施形態では、式IまたはIAの化合物のR5およびR6は、その窒素とともに、ピラジン環を形成する。方法の別の実施形態では、式IまたはIAの化合物のR5およびR6は、その窒素とともに、ピペラジン環を形成する。方法の別の実施形態では、式IまたはIAの化合物のR5およびR6は、その窒素とともに、モルホリン環を形成する。方法の別の実施形態では、式IまたはIAの化合物のR5およびR6は、その窒素とともに、ピロール環を形成する。方法の別の実施形態では、式IまたはIAの化合物のR5およびR6は、ピロリジンを形成する。方法の別の実施形態では、式IまたはIAの化合物のR5およびR6は、窒素を有する、ピリジン環を形成する。別の実施形態では、環は、ハロゲン、アルキル、アルコキシ、アルキレン、ヒドロキシル、シアノ、ニトロ、アミノ、アミド、COOH、またはアルデヒドによって置換される。
【0096】
本発明の方法の別の実施形態では、式IまたはIAの化合物のR1、および式IまたはIAの化合物のR2は、独立して、O−Alk−複素環、またはOCH2CH2−複素環である。別の実施形態では、「複素環」基という用語は、一実施形態では、環の一部として、炭素原子に加えて、硫黄、酸素、窒素、またはそれらの任意の組み合わせを含む、環構造を指す。別の実施形態では、複素環は、3〜12員環である。別の実施形態では、複素環は、6員環である。別の実施形態では、複素環は、5〜7員環である。別の実施形態では、複素環は、4〜8員環である。別の実施形態では、複素環基は、置換されないか、またはハロゲン、ハロアルキル、ヒドロキシル、アルコキシ、カルボニル、アミド、アルキルアミド、ジアルキルアミド、シアノ、ニトロ、CO2H、アミノ、アルキルアミノ、ジアルキルアミノ、カルボキシル、チオ、および/またはチオアルキルによって置換され得る。別の実施形態では、複素環式環は、別の飽和または不飽和シクロアルキル、または3〜8員の複素環に縮合され得る。別の実施形態では、複素環式環は、飽和環である。別の実施形態では、複素環式環は、不飽和環である。別の実施形態では、複素環は、ピペリジンである。別の実施形態では、複素環は、ピリジンである。別の実施形態では、複素環は、ピペリジン、ピリジン、フラン、チオフェン、ピロール、ピロリジン、ピラジン、ピペラジン、またはピリミジンである。
【0097】
「シクロアルキル」という用語は、炭素原子および水素原子を含む、非芳香族環、単環式環、または多環式環を指す。シクロアルキル基は、環内に1つまたは複数の炭素−炭素二重結合を有することができるが、それらの存在によって芳香族にされない場合に限られる。シクロアルキル基の例には、(C3−C7)シクロアルキル基(シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、およびシクロへプチル、ならびに飽和単環式および二環式テルペン等)、および(C3−C7)シクロアルケニル基(シクロプロペニル、シクロブテニル、シクロペンテニル、シクロヘキセニル、およびシクロヘプテニル、ならびに不飽和単環式および二環式テルペン等)が挙げられるが、これらに限定されない。シクロアルキル基は、置換されないか、または1個または2個の置換基によって置換することができる。好ましくは、シクロアルキル基は、単環式または二環式環である。
【0098】
「アルキル」という用語は、一実施形態では、直鎖アルキル基、分岐鎖アルキル基、および環状アルキル基を含む、飽和脂肪族炭化水素を指す。一実施形態では、アルキル基は、1〜12個の炭素を有する。別の実施形態では、アルキル基は、1〜7個の炭素を有する。別の実施形態では、アルキル基は、1〜6個の炭素を有する。別の実施形態では、アルキル基は、1〜4個の炭素を有する。別の実施形態では、環状アルキル基は、3〜8個の炭素を有する。別の実施形態では、環状アルキル基は、3〜12個の炭素を有する。別の実施形態では、分岐鎖アルキルは、1〜5個の炭素のアルキル側鎖によって置換されたアルキルである。別の実施形態では、分岐鎖アルキルは、1〜5個の炭素のハロアルキル側鎖によって置換されたアルキルである。アルキル基は、非置換であり得るか、またはハロゲン、ハロアルキル、ヒドロキシル、アルコキシカルボニル、アミド、アルキルアミド、ジアルキルアミド、ニトロ、アミノ、アルキルアミノ、ジアルキルアミノ、カルボキシル、チオ、および/またはチオアルキルによって置換され得る。
【0099】
「アルケニル」基は、別の実施形態では、1つまたは複数の二重結合を有する、直鎖基、分岐鎖基、および環状基を含む、不飽和炭化水素を指す。アルケニル基は、1つの二重結合、2つの二重結合、3つの二重結合等を有し得る。別の実施形態では、アルケニル基は、2〜12個の炭素を有する。別の実施形態では、アルケニル基は、2〜6個の炭素を有する。別の実施形態では、アルケニル基は、2〜4個の炭素を有する。アルケニル基の例は、エテニル、プロペニル、ブテニル、シクロヘキセニル等である。アルケニル基は、非置換であり得るか、またはハロゲン、ヒドロキシ、アルコキシカルボニル、アミド、アルキルアミド、ジアルキルアミド、ニトロ、アミノ、アルキルアミノ、ジアルキルアミノ、カルボキシル、チオ、および/またはチオアルキルによって置換され得る。
【0100】
「アリール」基は、少なくとも1つの炭素環式芳香族基または複素環式芳香族基を有する、芳香族基を指し、それは、非置換であり得るか、またはハロゲン、ハロアルキル、ヒドロキシ、アルコキシカルボニル、アミド、アルキルアミド、ジアルキルアミド、ニトロ、アミノ、アルキルアミノ、ジアルキルアミノ、カルボキシ、またはチオもしくはチオアルキルから選択される、1つまたは複数の基によって置換され得る。アリール環の非制限的例は、フェニル、ナフチル、ピラニル、ピロリル、ピラジニル、ピリミジニル、ピラゾリル、ピリジニル、フラニル、チオフェニル、チアゾリル、イミダゾリル、イソオキサゾリル等である。一実施形態では、アリール基は、4〜8員環である。別の実施形態では、アリール基は、4〜12員環である。別の実施形態では、アリール基は、6員環である。別の実施形態では、アリール基は、5員環である。別の実施形態では、アリール基は、2〜4個の縮合環系である。
【0101】
「アルデヒド」基は、一実施形態では、ホルミル基によって置換された、アルキル、またはアルケニルを指し、アルキルまたはアルケニルは、上で定義したとおりである。別の実施形態では、アルデヒド基は、ホルミル基によって置換された、アリールまたはフェニル基であり、アリールは、上で定義したとおりである。アルデヒドの例は、ホルミル、アセタル、プロパナール、ブタナール、ペンタナール、ベンズアルデヒドである。別の実施形態では、アルデヒド基は、ホルミル基である。
【0102】
「ハロアルキル」基は、別の実施形態では、1個または複数個のハロゲン原子によって、例えば、F、Cl、Br、またはIによって置換された、上で定義されるアルキル基を指す。
【0103】
「ヒドロキシル」基は、別の実施形態では、OH基を指す。当業者は、本発明の化合物中のR1、R2、またはR3が、ORである場合、RはOHではないことを理解する。
一実施形態では、「ハロゲン」または「ハロ」という用語は、F、Cl、Br、またはI等のハロゲンを指す。
【0104】
別の実施形態では、「フェノール」という句は、ベンゼンのアルコール(OH)誘導体を指す。
保護ヒドロキシルへの言及は、いくつかの実施形態において、ベンゼン環の酸素部分に結合した置換基の組込みを含み、置換基は、容易に除去し得る。いくつかの実施形態では、フェノール保護基は、メチルエーテル(メトキシ)、アルキルエーテル(アルコキシ)、ベンジルエーテル(Bn)、メトキシメチル(MOM)エーテル、ベンゾイルオキシメチル(BOM)エーテル、ベンジル、カルボベンゾキシ、メトキシエトキシメチル(MEM)エーテル、2−(トリメチルシリル)エトキシメチル(SEM)エーテル、メチルチオメチル(MTM)エーテル、フェニルチオメチル(PTM)エーテル、アジドメチルエーテル、シアノメチルエーテル、2,2−ジクロロ−1,1−ジフルオロエチルエーテル、2−クロロエチルエーテル、2−ブロモエチルエーテル、テトラヒドロピラニル(THP)エーテル、1−エトキシエチル(EE)エーテル、フェナシルエーテル、4−ブロモフェナシルエーテル、シクロプロピルメチルエーテル、アリルエーテル、プロパルギルエーテル、イソプロピルエーテル、シクロヘキシルエーテル、t−ブチルエーテル、2,6−ジメチルベンジルエーテル、4−メトキシベンジルエーテル、o−ニトロベンジルエーテル、2,6−ジクロロベンジルエーテル、3,4−ジクロロベンジルエーテル、4−(ジメチルアミノ)カルボニルベンジルエーテル、4−メチルスルフィニルベンジルエーテル、4−アントリルメチルエーテル、4−ピコリルエーテル、ヘプタフルオロ−p−トリル、テトラフルオロ−4−ピリジルエーテル、トリメチルシリル(TMS)エーテル、t−ブチルジメチルシリル(TBDMS)エーテル、t−ブチルジフェニルシリル(TBDPS)エーテル、トリイソプロピルシリル(TIPS)エーテル、アリールギ酸、アリール酢酸、アリールレブリン酸、アリールピバル酸、アリール安息香酸、アリール9−フルオレンカルボン酸、炭酸アリールメチル、炭酸1−アダマンチル、炭酸t−ブチル、炭酸4−メチルスルフィニルベンジル、炭酸2,4−ジメチルペント−3−イル、炭酸アリール2,2,2−トリクロロエチル、炭酸アリールベンジル、炭酸アリール、ジメチルホスフィニルエステル(Dmp−OAr)、ジメチルホスフィノチオニルエステル(Mpt−OAr)、ジフェニルホスフィノチオニルエステル(Dpt−OAr)、アリールメタンスルホン酸、アリールトルエンスルホン酸、またはアリール2−ホルミルベンゼンスルホン酸を含み得る。
【0105】
一実施形態では、本発明の方法は、N,N−ビス(4−ヒドロキシフェニル)−4−プロピルベンズアミド(II)またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを活用する。別の実施形態では、本発明の方法は、4,4’−(2,3−ジメチル−ベンジルアザンイジル)ジフェノール(III)またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを活用する。別の実施形態では、本発明の方法は、3−フルオロ−N−(4−フルオロフェニル)−4−ヒドロキシ−N−(4−ヒドロキシフェニル)ベンズアミド(IV)またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを活用する。別の実施形態では、本発明の方法は、N,N−ビス(4−ヒドロキシフェニル)−2,3−ジメチルベンズアミド(V)またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを活用する。別の実施形態では、本発明の方法は、N,N−ビス(4−ヒドロキシフェニル)−2−ナフチルアミド(VI)またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを活用する。別の実施形態では、本発明の方法は、3−フルオロ−4−ヒドロキシ−N,N−ビス(4−ヒドロキシフェニル)−ベンズアミド(VII)またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを活用する。別の実施形態では、本発明の方法は、4−((4−フルオロフェニル)(4−ヒドロキシベンジル)アミノ)フェノール(VIII)またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを活用する。別の実施形態では、本発明の方法は、4−フルオロ−N−(4−ヒドロキシ−フェニル)−N−[4−(2ピペリジン−1−イル−エトキシ)−フェニル]−2−トリフルオロメチル−ベンズアミド(IX)またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを活用する。別の実施形態では、本発明の方法は、IXの塩酸塩(IXのHCl塩)もしくは4−フルオロ−N−(4−ヒドロキシ−フェニル)−N−[4−(2−ピペリジン−1−イル−エトキシ)−フェニル]−2−トリフルオロメチル−ベンズアミド塩酸塩(X)またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを活用する。別の実施形態では、本発明の方法は、3−フルオロ−4−ヒドロキシ−N−(4−ヒドロキシフェニル)−N−フェニルベンズアミド(XI)またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを活用する。別の実施形態では、本発明の方法は、3−フルオロ−N,N−ビス−(4−ヒドロキシ−フェニル)−2−メチル−ベンズアミド(XII)またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを活用する。
【0106】
一実施形態では、本発明の方法は、化合物の「薬学的に許容される塩」を活用し、それは、酸または塩基との本発明の化合物の反応によって産生され得る。
本発明の方法の化合物のアミンの好適な薬学的に許容される塩は、無機酸から、または有機酸から調製し得る。一実施形態では、アミンの無機塩の例は、重硫酸塩、ホウ酸塩、臭化物、塩化物、ヘミ硫酸塩、臭化水素酸塩、塩酸塩、2−ヒドロキシエチルスルホン酸塩(ヒドロキシエタンスルホン酸塩)、ヨウ素酸塩、ヨウ化物、イソチオナート、硝酸塩、過硫酸塩、リン酸塩、硫酸塩、スルファミン酸塩、スルファニル酸塩、スルホン酸(アルキルスルホン酸塩、アリールスルホン酸塩、ハロゲン置換アルキルスルホン酸塩、ハロゲン置換アリールスルホン酸塩)、スルホン酸塩、およびチオシアン酸塩である。
【0107】
一実施形態では、アミンの有機塩の例は、有機酸の脂肪族、脂環式、芳香族、アル脂肪族、複素環式、カルボン酸およびスルホン酸クラスから選択され得、それらの例は、酢酸塩、アルギニン塩、アスパラギン酸塩、アスコルビン酸塩、アジピン酸塩、アントラニル酸塩、アルギン酸塩、アルカンカルボン酸塩、置換アルカンカルボン酸塩、アルギン酸塩、ベンゼンスルホン酸塩、安息香酸塩、重硫酸塩、酪酸塩、重炭酸塩、酸性酒石酸塩、カルボン酸塩、クエン酸塩、樟脳酸塩、樟脳スルホン酸塩、シクロヘキシルスルファミン酸塩、シクロペンタンプロピオン酸塩、エデト酸カルシウム、カンシル酸塩、炭酸塩、クラブラン酸塩、ケイ皮酸塩、ジカルボン酸塩、ジグルコン酸塩、ドデシルスルホン酸塩、二塩酸塩、デカン酸塩、エナント酸塩、エタンスルホン酸塩、エデト酸塩、エジシル酸塩、エストラート、エシラート、フマル酸塩、ギ酸塩、フッ化物、ガラクツロン酸塩、グルコン酸塩、グルタミン酸塩、グリコール酸塩、グルコレート(glucorates)、グルコヘプタン酸塩、グリセロリン酸塩、グルセプチン酸塩、アルサニル酸グリコリル、グルタラート、グルタミン酸塩、ヘプタン酸塩、ヘキサン酸塩、ヒドロキシマレイン酸塩、ヒドロキシカルボキシル酸、ヘキシルレゾルシン酸塩、ヒドロキシ安息香酸塩、ヒドロキシナフトエ酸塩、フッ化水素酸塩、乳酸塩、ラクトビオン酸塩、ラウリン酸塩、リンゴ酸塩、マレイン酸塩、メチレンビス(ベータ−オキシナフトエ酸塩)、マロン酸塩、マンデル酸塩、メシル酸塩、メタンスルホン酸塩、メチル臭化物、メチル硝酸塩、メチルスルホン酸塩、一カリウムマレイン酸塩、ムコ酸塩、モノカルボン酸塩、硝酸塩、ナフタレンスルホン酸塩、2−ナフタレンスルホン酸塩、ニコチン酸塩、ナプシル酸塩、N−メチルグルカミン、シュウ酸塩、オクタン酸塩、オイレン酸塩、パモ酸塩、フェニル酢酸、ピクリン酸塩、フェニル安息香酸塩、ピバル酸塩、プロピオン酸塩、フタル酸塩、ペクチニン酸塩、フェニルプロピオン酸塩、パルミチン酸塩、パントテン酸塩、ポリガラクツロン酸塩、ピルビン酸塩、キナ酸塩、サリチル酸塩、コハク酸塩、ステアリン酸塩、スフラニル酸塩、塩基性酢酸塩、酒石酸塩、テオフィリン酢酸塩、p−トルエンスルホン酸(トシラート)、トリフルオロ酢酸、テレフタル酸塩、タンニン酸塩、テオクル酸塩、トリハロ酢酸塩、トリエチオダイド、トリカルボン酸塩、ウンデカン酸塩、および吉草酸塩である。
【0108】
一実施形態では、カルボン酸またはフェノールの無機塩の例は、アンモニウム、アルカリ金属(リチウム、ナトリウム、カリウム、セシウムを含む)、アルカリ土類金属(カルシウム、マグネシウム、アルミニウム)、亜鉛、バリウム、コリン、第4級アンモニウムから選択され得る。
【0109】
別の実施形態では、カルボン酸またはフェノールの有機塩の例は、アルギニン、有機アミン(脂肪族有機アミンを含む)、脂環式有機アミン、芳香族有機アミン、ベンザチン、t−ブチルアミン、ベネタミン(N−ベンジルフェネチルアミン)、ジシクロヘキシルアミン、ジメチルアミン、ジエタノールアミン、エタノールアミン、エチレンジアミン、ヒドラバミン、イミダゾール、リジン、メチルアミン、メグラミン、N−メチル−D−グルカミン、N,N’−ジベンジルエチレンジアミン、ニコチンアミド、有機アミン、オルニチン、ピリジン、ピコリ、ピペラジン、プロカイン、トリス(ヒドロキシメチル)メチルアミン、トリエチルアミン、トリエタノールアミン、トリメチルアミン、トロメタミン、および尿素から選択され得る。
【0110】
一実施形態では、これらの塩は従来の方法によって形成されてもよく、例えば生成物の遊離塩基または遊離酸形態を、塩が不溶性である溶媒または媒体中または水等の溶媒中で、適切な酸または塩基の1つまたは複数の等価物と反応させることによって形成され、その溶媒または媒体は、真空で、または凍結乾燥によって、あるいは別のイオンまたは好適なイオン交換樹脂のために既存の塩のイオンを交換することによって、除去される。
【0111】
一実施形態では、本発明の方法は、本発明の化合物の薬学的に許容される塩を活用する。一実施形態では、本発明の方法は、式IA、I〜XIIの化合物の薬学的に許容される塩を活用する。一実施形態では、本発明の方法は、本発明の式IA、I〜XIIの化合物のアミンの塩を活用する。一実施形態では、本発明の方法は、本発明の式IA、I〜XIIの化合物のフェノールの塩を活用する。
【0112】
一実施形態では、本発明の方法は、式IA、I〜XIIの遊離塩基、遊離酸、非荷電、または非錯体化合物、またはその異性体、薬学的生成物、水和物、多形体、またはそれらの組み合わせを活用する。
【0113】
本発明のいくつかの実施形態では、本発明の化合物は、アミド結合によって互いに結合した3個のフェニル基を含む。一実施形態では、本発明の化合物は、非荷電構造である。別の実施形態では、本発明の化合物は、遊離塩基構造である。別の実施形態では、本発明の化合物は、遊離酸構造である。別の実施形態では、本発明の化合物は、非錯体構造である。別の実施形態では、本発明の化合物は、非イオン化構造である。別の実施形態では、本発明の化合物は、薬学的に許容される塩である。別の実施形態では、本発明のいくつかの化合物は、塩酸(HCl)塩を含む。
【0114】
一実施形態では、本発明の方法は、式IA、I〜XIIの化合物の異性体を活用する。一実施形態では、本発明の方法は、式IA、I〜XIIの化合物の薬学的生成物を活用する。一実施形態では、本発明の方法は、式IA、I〜XIIの化合物の水和物を活用する。一実施形態では、本発明の方法は、式IA、I〜XIIの化合物の多形体を活用する。一実施形態では、本発明の方法は、式IA、I〜XIIの化合物の代謝物を活用する。別の実施形態では、本発明の方法は、本明細書に説明する、式IA、I〜XIIの化合物を含む組成物を活用し、または別の実施形態では、式IA、I〜XIIの化合物の異性体、代謝物、薬学的生成物、水和物、多形体の組み合わせを活用する。
【0115】
一実施形態では、「異性体」という用語は、光学的異性体および類似体、構造的異性体および類似体、立体構造的異性体および類似体、ならびに同等物を含むが、これらに限定されない。
【0116】
一実施形態では、「異性体」という用語は、化合物の光学的異性体を包含することを意味する。一実施形態では、「異性体」という用語は、化合物の立体異性体を包含することが意図される。本発明の化合物は、そのシスまたはトランス異性体化にあり得る、アミド結合を保持する。本発明は、任意の光学的活性形態、または立体異性体形態、またはそれらの混合物を包含し、これらの任意の用途のための使用は本発明の範囲内と見なされることを理解されたい。
【0117】
別の実施形態では、本発明はさらに、化合物の水和物を含む。一実施形態では、「水和物」という用語は、当該技術分野において公知のとおり、半水和物、一水和物、二水和物、三水和物、またはその他を指す。
合成プロセス
式IまたはIAの化合物は、例えば、塩基の存在下で、置換ジフェニルアミンを安息香酸またはハロゲン化ベンゾイルと反応させて、ベンズアミドを得ることによって、容易に調製し得る。一実施形態では、塩基はピリジンである。別の実施形態では、ハロゲン化ベンゾイルは塩化ベンゾイルである。別の実施形態では、ヒドロキシル置換基は、ジフェニルアミンと安息香酸またはハロゲン化ベンゾイルとの反応中に保護される。別の実施形態では、ヒドロキシルのための保護基は、任意に、最終工程において除去される。また、米国公報第2009/00624231号も参照されたく、それは、参照することにより、その全体として組み込まれる。
【0118】
例えば、式IA:
【0119】
【化15】
(式中、R1、R2、R3、およびR4、jおよびKは、上述のとおりである)
の化合物は、
【0120】
【化16】
を、
【0121】
【化17】
ととも反応させて、
【0122】
【化18】
を得る工程を含むプロセスによって調製され得、
ジフェニルアミン(3)を、塩基の存在下で、
【0123】
【化19】
と反応させて、
【0124】
【化20】
を得、
式中、R1、R2、R3、およびR4が、独立して、OH、O−Alk−R5R6、またはO−Alk−複素環である場合は、R1’、R2’、R3’、およびR4’は、保護されたヒドロキシル基であり、保護基は、除去されて、遊離ヒドロキシルを得るか、または任意に、その後にCl−Alk−複素環、またはCl−Alk−NR5R6と反応させて、式IA:
【0125】
【化21】
の化合物を得、
式中、R1、R2、R3、およびR4が、独立して、OH、O−Alk−NR5R6、またはO−Alk−複素環と異なる場合は、R1’、R2’、R3’、およびR4’はそれぞれ、R1、R2、R3、およびR4である。
【0126】
別の例として、式IA:
【0127】
【化22】
(式中、R1、R2、R3、およびR4は、上述のとおりである)、の化合物の調製のためのプロセスは、塩基の存在下で、
【0128】
【化23】
を、
【0129】
【化24】
と反応させて、
【0130】
【化25】
を得る工程を含み、
式中、R1、R2、R3、およびR4が、独立して、OH、O−Alk−R5R6、またはO−Alk−複素環である場合は、R1’、R2’、R3’、およびR4’は、保護されたヒドロキシル基であり、保護基は、除去されて、遊離ヒドロキシルを得るか、または任意に、その後にCl−Alk−複素環、またはCl−Allk−NR5R6と反応させて、式IA:
【0131】
【化26】
の化合物を得、
式中、R1、R2、R3、およびR4が、独立して、OH、O−Alk−NR5R6、またはO−Alk−複素環と異なる場合は、R1’、R2’、R3’、およびR4’はそれぞれ、R1、R2、R3、およびR4である。
【0132】
一例では、化合物IIは、実施例1および図5に従い調製される。
別の例では、化合物IIIは、実施例1および図5に従い調製される。
さらなる例では、式IV:
【0133】
【化27】
の化合物は、塩基の存在下で、
【0134】
【化28】
を、
【0135】
【化29】
と反応させて、
【0136】
【化30】
を得た後に、保護基の脱保護により、化合物IV:
【0137】
【化31】
を得ることによって、調製され得、
式中、PおよびP’は、同一、または異なる保護基である。一例では、化合物IVは、実施例2、および図6に従い調製される。
【0138】
別の例では、化合物Vは、実施例1、および図5に従い調製される。
さらなる例では、化合物VIは、実施例3、および図7に従い調製される。
別の例では、化合物VIIは、実施例1、および図5に従い調製される。
【0139】
別の例では、化合物VIIIは、実施例4、および図5に従い調製される。
別の例では、化合物IXは、実施例5、および図8に従い調製される。
別の例では、化合物X塩酸塩は、実施例5、および図8に従い調製される。
【0140】
別の例では、化合物XIは、実施例1、および図5に従い調製される。
別の例では、化合物XIIは、実施例1、および図5に従い調製される。
好適なヒドロキシル保護基は、例えば、メチルエーテル(メトキシ)、ベンジルエーテル(ベンジルオキシ)メトキシメチル(MOM)エーテル、ベンゾイルオキシメチル(BOM)エーテル、ベンジル、カルボベンゾオキシ、メトキシエトキシメチル(MEM)エーテル、2−(トリメチルシリル)エトキシメチル(SEM)エーテル、メチルチオメチル(MTM)エーテル、フェニルチオメチル(PTM)エーテル、アジドメチルエーテル、シアノメチルエーテル、2,2−ジクロロ−1,1−ジフルオロエチルエーテル、2−クロロエチルエーテル、2−ブロモエチルエーテル、テトラヒドロピラニル(THP)エーテル、1−エトキシエチル(EE)エーテル、フェナシルエーテル、4−ブロモフェナシルエーテル、シクロプロピルメチルエーテル、アリルエーテル、プロパルギルエーテル、イソプロピルエーテル、シクロヘキシルエーテル、t−ブチルエーテル、ベンジルエーテル、2,6−ジメチルベンジルエーテル、4−メトキシベンジルエーテル、o−ニトロベンジルエーテル、2,6−ジクロロベンジルエーテル、3,4−ジクロロベンジルエーテル、4−(ジメチルアミノ)カルボニルベンジルエーテル、4−メチルスルフィニルベンジルエーテル、4−アントリルメチルエーテル、4−ピコリルエーテル、ヘプタフルオロ−p−トリル、テトラフルオロ−4−ピリジルエーテル、トリメチルシリル(TMS)エーテル、t−ブチルジメチルシリル(TBDMS)エーテル、t−ブチルジフェニルシリル(TBDPS)エーテル、トリイソプロピルシリル(TIPS)エーテル、アリールギ酸、アリール酢酸、アリールレブリン酸、アリールピバル酸、アリール安息香酸、アリール9−フルオレンカルボン酸、アリールメチルカルボン酸、1−アダマンチルカルボン酸、t−ブチルカルボン酸、4−メチルスルフィニルベンジルカルボン酸、2,4−ジメチルペント−3−イルカルボン酸、アリール2,2,2−トリクロロエチルカルボン酸、アリールベンジルカルボン酸、アリールカルバミン酸、ジメチルホスフィニルエステル(Dmp−OAr)、ジメチルホスフィノチオニルエステル(Mpt−OAr)、ジフェニルホスフィノチオニルエステル(Dpt−OAr)、アリールメタンスルホン酸、アリールトルエンスルホン酸、またはアリール2−ホルミルベンゼンスルホン酸を含む。
【0141】
本発明の方法は、化合物IA、I〜XIIの使用を含み、本発明の化合物の調製のためのプロセスは、塩基の存在下で、塩化ベンゾイルとのジフェニルアミンの反応を含む。好適な塩基は、例えば、ピリジン、トリエチルアミン、K2CO3、Cs2CO3、Na2CO3、メチルアミン、イミダゾール、ベンズイミダゾール、ヒスチジン、トリブチルアミン、またはそれらの任意の組み合わせを含む。一実施形態では、塩基は、ピリジンである。
【0142】
本発明の方法は、化合物IA、I〜XIIの使用を含み、本発明の化合物の調製のためのプロセスは、保護されたヒドロキシルの脱保護を含む。別の実施形態では、脱保護条件は、保護基によって異なる。いくつかの実施形態では、脱保護工程は、Pd/Cの存在下での水素化を含む。別の実施形態では、脱保護は、BBr3との反応を含む。別の実施形態では、脱保護工程は、酸との反応を含む。
【0143】
さらなる実施形態では、化合物IA、I〜XIIは、図5〜8、および実施例1〜5に従い調製される。
薬学的組成物
いくつかの実施形態では、本発明は、説明した化合物を含有する組成物を投与する工程を含む使用方法を提供する。本明細書において使用されるとき、「薬学的組成物」は、薬学的に許容される担体または希釈剤を伴う「治療的有効量」の活性成分、すなわち、本発明の化合物を意味する。本明細書において使用されるとき、「治療的有効量」は、所与の病状および投与レジメンに対して治療効果を提供する量を指す。
【0144】
本明細書において使用されるとき、「投与する」という用語は、対象者を本発明の化合物と接触させることを指す。本明細書において使用されるとき、投与は、インビトロ(すなわち試験管内)、またはインビボ(すなわち有機体、例えばヒトの細胞もしくは組織内)で達成することができる。一実施形態では、本発明は、男性対象者に本発明の化合物を投与することを含む。
【0145】
本発明は、他の実施形態では、本明細書に説明する化合物の薬学的生成物を提供する。「薬学的生成物」という用語は、他の実施形態では、例えば、本明細書に説明する、薬学的使用に好適な組成物(薬学的組成物)を指す。
【0146】
本発明の化合物は、単独で、または製剤の活性成分として投与することができる。したがって、本発明はまた、例えば、1つまたは複数の薬学的に許容される担体を含有する、式Iの化合物の薬学的組成物も含む。
【0147】
本発明に従う、化合物を投与するために好適な種々の製剤を調製するための処置を説明する、数々の標準的参考文献が利用可能である。潜在的製剤の例および調製は、例えば、Handbook of Pharmaceutical Excipients,American Pharmaceutical Association(最新版)、Pharmaceutical Dosage Forms:Tablets(Lieberman,Lachman and Schwartz,editors)最新版、(Marcel Dekker,Inc.ならびにRemington‘s Pharmaceutical Sciences(Arthur Osol,editor)から刊行、1553−1593(current edition)に含まれている。
【0148】
投与方法および剤形は、所与の治療用途に望ましく、かつ有効である化合物または組成物の治療量に密接に関連する。
好適な剤形は、経口、直腸、舌下、粘膜、鼻腔、眼、皮下、筋肉内、静脈内、経皮、脊髄、髄腔内、関節内、動脈内、くも膜下、気管支、リンパ管、および子宮内投与、ならびに活性成分全身送達のための他の剤形を含むが、これらに限定されない。経口投与に好適な製剤が望ましい。
【0149】
かかる薬学的剤形を調製するために、従来の薬学的配合技術に従い、活性成分を薬学的担体と混合し得る。担体は、投与に望ましい調製の形態によって多種多様な形態を成し得る。
【0150】
経口剤形に組成物を調製する際に、通常の薬学的媒体のうちのいずれをも使用し得る。したがって、例えば、懸濁液、エリキシル剤、および溶液等の液体経口調製剤では、好適な担体および添加剤は、水、グリコール、油、アルコール、香味剤、防腐剤、着色剤、および同等物を含む。例えば、粉末、カプセル、および錠剤等の固体経口調製剤では、好適な担体および添加剤は、スターチ、糖、希釈剤、造粒剤、滑沢剤、結合剤、崩壊剤を含む。錠剤およびカプセルは、それらの容易な投与のため、最良の有益な経口投与単位形態を示す。必要に応じて、錠剤は、標準的技術によって、糖衣化または腸溶化され得る。
【0151】
非経口製剤では、他の成分、例えば、溶解を補助するか、または保存のための成分が含まれ得るが、担体は、通常、無菌水を含む。注入可能な溶液はまた、調製され得、その場合、適切な安定剤が用いられ得る。
【0152】
いくつかの用途では、リポソームまたは他のカプセル媒体中の活性剤のカプセル化による、またはタンパク質、リポタンパク質、糖タンパク質、および多糖等の生体分子への活性剤の固着による、例えば、共有結合、キレート化、または関連配位等による、「ベクトル化」形態の活性剤を用いることが有益であり得る。
【0153】
経口投与に好適な製剤を使用する、本発明の治療方法は、カプセル、カシェー、錠剤、またはロゼンジ剤等の不連続単位で表され得、それぞれ、例えば、粉末または顆粒として、所定量の活性成分を含有する。任意に、シロップ、エリキシル剤、乳剤または注ぎ出し(draught)等の水性液または非水性液体中の懸濁液を使用し得る。
【0154】
錠剤は、任意に1つまたは複数の副成分を用いて、圧縮もしくは成形、または湿式造粒により作製し得る。圧縮された錠剤は、例えば、結合剤、崩壊剤、滑沢剤、不活性希釈剤、界面活性剤、または抜染剤と任意に混合される、粉末または顆粒等の自由流動性形態にある活性化合物を用いて、好適な機械で圧縮することによって調製され得る。粉末状活性化合物と好適な担体との混合物から成る、成形された錠剤は、好適な機械で成形することによって作製され得る。
【0155】
シロップは、糖、例えば、スクロースの濃縮水溶液に活性化合物を添加することによって作製され得、そこに副成分も添加し得る。かかる副成分は、香味剤、好適な防腐剤、糖の結晶化を遅延する薬剤、任意の他の成分の溶解度を増大する薬剤(ポリヒドロキシアルコール、例えば、グリセロールまたはソルビトール等)を含み得る。
【0156】
非経口投与に好適な製剤は、好ましくは、受容者の血液と等張性である、活性化合物の無菌水性調製剤(例えば、生理食塩溶液)を含み得る。かかる製剤は、懸濁剤および濃化剤、ならびにリポソーム、または、化合物を血液構成要素、もしくは1つまたは複数の臓器に標的化するように設計される他の微粒子系を含み得る。製剤は、単位容量、または多用量形態で提示され得る。
【0157】
非経口投与は、全身送達のための任意の好適な形態を含み得る。投与は、例えば、静脈内、動脈内、髄腔内、筋肉内、皮下、筋肉内、腹内(例えば、腹腔内)等であり得、輸液ポンプ(外部または埋め込み型)、または所望の投与モダリティに適切な、任意の他の好適な手段によって達成され得る。
【0158】
鼻腔および他の粘膜スプレー製剤(例えば、吸入型形態)は、防腐剤または等張剤を有する、活性化合物の精製された水溶液を含むことができる。かかる製剤は、好ましくは、鼻腔または他の粘膜に適合するpHおよび等張状態に調節される。代替的には、それらは、ガス担体中に懸濁された、微細固体粉末の形態である可能性がある。かかる製剤は、任意の好適な手段または方法によって、例えば、ネブライザー、アトマイザー、定量吸入器、または同等物によって送達され得る。
【0159】
直腸投与の製剤は、ココアバター、硬化油脂、または硬化油脂カルボン酸等の好適な担体を有する、座薬として提供され得る。
経皮製剤は、セルロース媒体、例えば、メチルセルロースまたはヒドロキシエチルセルロース等のチキソトロピーまたはゼラチン状担体中に活性剤を組み込むことによって調製し得、次いで、得られた製剤は、使用者の皮膚と皮膚接触して固着されるように適合された経皮デバイス内に充填される。
【0160】
先述の成分に加えて、本発明の製剤はさらに、希釈剤、緩衝剤、香味剤、結合剤、崩壊剤、界面活性剤、増粘剤、滑沢剤、防腐剤(抗酸化剤を含む)、および同等物から選択される、1つまたは複数の副成分を含み得る。
【0161】
本発明の製剤は、当業者に公知の即効放出、持続放出、遅延放出、または任意の他の放出プロファイルを有することができる。
一実施形態では、本発明は、a)血清総テストステロンレベルを低下させる方法、b)前立腺癌を有する男性対象者において、黄体形成ホルモン(LH)の減少によって、またはLHホルモンの減少と無関係に、遊離血清テストステロンレベルを低下させる方法を提供し、式IA、I〜XIIの化合物を含有する、経口組成物を投与することを含む。付加的な実施形態では、本発明の方法は、式II、式III、式IV、式V、式VI、式VII、式VIII、式IX、式X、式XI、または式XIIの化合物を含有する経口組成物を活用する。
【0162】
一実施形態では、本発明は、前立腺癌を有する男性対象者において、LHレベルを減少させることによるか、またはLHレベルの減少と無関係に、前立腺癌を治療する方法を提供し、式IA、I〜XIIの化合物を含有する経口組成物を投与することを含む。さらなる実施形態では、本発明の方法は、前立腺癌を有する男性対象者において、LHレベルを減少させることによるか、またはLHレベルの減少と無関係に、前立腺癌を治療する方法を提供し、式II、式III、式IV、式V、式VI、式VII、式VIII、式IX、式X、式XI、または式XIIの化合物を含有する経口組成物を投与することを含む。
【0163】
本発明は、いくつかの実施形態では、「本発明の化合物」と称される、本明細書に説明する化合物の任意の実施形態を包含することを理解されたい。
一実施形態では、本発明の方法は、種々の用量での本発明の化合物の投与を含む。一実施形態では、本発明の化合物は、1日当たり1〜1500mgの用量で投与される。付加的な実施形態では、本発明の化合物は、1日当たり1〜10mg、1日当たり3〜26mg、1日当たり3〜60mg、1日当たり3〜16mg、1日当たり3〜30mg、1日当たり10〜26mg、1日当たり15〜60mg、1日当たり50〜100mg、1日当たり50〜200mg、1日当たり150〜300mg、1日当たり20〜50mg、1日当たり5〜50mg、1日当たり200〜500mg、1日当たり150〜500mg、1日当たり200〜1000mg、1日当たり300〜1500mg、または1日当たり100〜1000mgの用量で投与される。
【0164】
一実施形態では、本発明の方法は、種々の用量での本発明の化合物の投与を含む。一実施形態では、本発明の化合物は、3mgの用量で投与される。付加的な実施形態では、本発明の化合物は、10mg、30mg、50mg、100mg、200mg、300mg、450mg、500mg、600mg、900mg、1000mg、または1500mgの用量で投与される。
【0165】
一実施形態では、本発明の方法は、種々の用量での本発明の化合物の投与を含む。一実施形態では、本発明の化合物は、0.1mg/kg/日の用量で投与される。付加的な実施形態では、本発明の化合物は、0.2〜30mg/kg/日、または0.2mg/kg/日、0.3mg/kg/日、1mg/kg/日、3mg/kg/日、5mg/kg/日、10mg/kg/日、20mg/kg/日、もしくは30mg/kg/日の用量で投与される。
【0166】
本発明の方法の一実施形態では、式IA、I〜XIIの化合物を含有する薬学的組成物の使用を提供する。付加的な実施形態では、本発明の方法は、式II、式III、式IV、式V、式VI、式VII、式VIII、式IX、式X、式XI、または式XIIの化合物を含有する薬学的組成物の使用のために提供される。
【0167】
ある実施形態では、薬学的組成物は、固体剤形である。別の実施形態では、薬学的組成物は、錠剤である。別の実施形態では、薬学的組成物は、カプセルである。別の実施形態では、薬学的組成物は、溶液である。別の実施形態では、薬学的組成物は、経皮パッチである。
【0168】
一実施形態では、本発明の化合物またはそれを含有する組成物の使用は、当業者に理解されるとおり、対象者において、所望の反応を阻害、抑制、強化、または刺激する上で有用性がある。別の実施形態では、組成物はさらに、付加的な活性成分を含有し得、その活性は、本発明の化合物が投与される特定の用途に有用である。
【0169】
哺乳類、および特にヒトへの投与のために、医師が、実際の用量および治療期間を決定することが予期されており、それは、個人に最も好適であり、特定の個人の年齢、体重、遺伝子、および/または反応によって異なる可能性がある。
【0170】
いくつかの実施形態では、本発明の組成物のうちのいずれかは、本明細書に説明する任意の形態または実施形態の本発明の化合物を含有する。いくつかの実施形態では、本発明の組成物のうちのいずれもが本明細書に説明する任意の形態または実施形態の本発明の化合物から成る。いくつかの実施形態では、本発明の組成物は、本明細書に説明する任意の形態または実施形態の本発明の化合物から本質的に成る。いくつかの実施形態では、「含有する」という用語は、薬学業界において公知のとおり、本発明の化合物等の指示した活性剤の含有、ならびに他の活性剤、および薬学的に許容される担体、賦形剤、皮膚軟化剤、安定剤等の含有を指す。いくつかの実施形態では、「から本質的に成る」という用語は、その活性成分のみが指示した活性成分である組成物を指すが、指示した活性成分の治療効果に直接関与しない、製剤を安定、保存する等のための他の化合物が含まれ得る。いくつかの実施形態では、「から本質的に成る」という用語は、活性成分の放出を促進する構成要素を指す。いくつかの実施形態では、「から成る」という用語は、活性成分および薬学的に許容される担体または賦形剤を含む組成物を指す。
【0171】
本明細書に説明する化合物のうちのいずれの、いかなる使用も、本明細書において説明する、任意の疾患、障害、または病状の治療に使用され得、本明細書の一実施形態を表すことを理解されたい。一実施形態では、化合物は、遊離塩基、遊離酸、非荷電または非錯体化合物である。
【0172】
本発明の好ましい実施形態をより完全に例証するために、以下の実施例を提示する。しかしながら、それらは、本発明の広い範囲を制限するものと一切解釈されてはならない。
実施例
実施例1
式II〜XIIの化合物および合成中間体のための一般的合成手順
有機溶媒、界面活性剤、および抗酸化剤等は、本明細書に説明する組成物に使用され得、典型的には、商業的供給源から容易に入手可能である。例えば、PEG−300、ポリソルベート80、Captex(商標)200、Capmul(商標)MCM C8は、例えば、Dow Chemical社(ミシガン州ミッドランド)、ICI Americas社(デラウェア州ウィルミントン)、またはAbitec Corporation(ウィスコンシン州ジェーンズビル)から購入可能である。
【0173】
本明細書に説明するエストロゲン受容体リガンドは、当業者に公知の多くの方法で調製され得る。例えば、本明細書に説明するエストロゲン受容体リガンドは、米国特許出願公開第2009/0062341号明細書において説明される合成方法によって調製され得、それぞれの開示は、参照することにより、その全体として組み込まれる。
N,N−ビスアリール ベンズアミド誘導体の一般的合成
ジアリールアニリンの一般的合成(図5)。アリールアミン(1.5当量)、アリールヨウ化物(1当量)、K2CO3(2当量)、CuI(0.1当量)、およびL−プロリン(0.2当量)の混合物をともに混合し、室温で、無水DMSO中で溶解した。次いで、反応混合物を28時間、90℃で攪拌および加熱した。混合物を室温まで冷却し、水で加水分解した。EtOAcを添加して、溶液を分割した。EtOAc層を分離し、ブラインで洗浄し、無水MgSO4上で乾燥させた。溶媒を減圧下で除去した。固体残渣を、溶離液として5%のEtOAc/ヘキサンを使用するフラッシュカラムクロマトグラフィー(シリカゲル)によって精製し、対応するジアリールアニリンを得た。
【0174】
ビス−(4−メトキシフェニル)アミン(1a):淡黄色固体、73%の収率。融点98.6〜99.0℃。1H NMR(CDCl3,300MHz)δ6.93−6.81(m,8H),5.37(s,br,1H),3.78(s,6H)。MS m/z 228.4(M−H)+
N−(4−メトキシフェニル)−フェニルアミン(1b):淡黄色固体、70%の収率。融点106.3〜106.5℃。1H NMR(CDCl3,300MHz)δ7.24−7.18(m,3H),7.08−7.06(m,2H),6.92−6.84(m,4H),5.61(s,br,1H),3.79(s,3H)。MS m/z 200.1(M+H)+。
【0175】
N−(4−フルオロフェニル)−N−4−メトキシフェニルアミン(1c):淡黄色固体、54%の収率。融点60.6〜61.0℃。1H NMR(CDCl3,300MHz)δ7.01−6.83(m,8H),3.78(s,3H)。MS m/z 217(M)+。
【0176】
N−(4−ベンジルオキシフェニル)−N−4−メトキシフェニルアミン(1d):淡黄色固体、54%の収率。融点108.0〜108.4℃。1H NMR(CDCl3,300MHz)δ7.34−7.08(m,5H),6.90−6.81(s,3H),3.78(s,3H)。MS m/z 306(M+H)+。
【0177】
ベンズアミドの一般的合成。アリールアリニン(1当量)、ベンゾイル塩化物(1.3当量)、およびピリジン(6当量)をともに混合し、室温で、無水THF中で溶解した。24時間、反応混合物を攪拌し、還流させた。反応溶液を室温まで冷却し、2N HCl溶液の添加により加水分解した。溶液を酢酸エチルで抽出した。有機層を、飽和NaHCO3水溶液で洗浄して、過剰な酸を除去し、無水MgSO4上で乾燥させ、ろ過し、減圧下で濃縮した。残渣を、EtOAc/ヘキサン(3/7v/v)を使用するフラッシュカラムクロマトグラフィーによって精製し、対応するベンズアミド化合物を得た。
【0178】
3−フルオロ−N−(4−フルオロフェニル)−4−メトキシ−N−(4−メトキシフェニル)ベンズアミド(2a):黄色固体、融点54〜56℃,1H NMR(CDCl3/TMS)δ7.24−7.11(m,4H),7.05−6.97(m,4H),6.85−6.78(m,3H),3.86(s,3H),3.79(s,3H)。MS(ESI)m/z 370.1[M+H]+
4−フルオロ−N,N−ビス(4−メトキシフェニル)−2−(トリフルオロメチル)ベンズアミド(2b):無色油、84.2%の収率。1H NMR(CDCl3,300MHz)δ7.34−7.26(m,4H),7.09−7.01(m,3H),6.91(d,2H,J=8.7Hz),6.87(d,2H,J=8.7Hz),3.80(s,3H),3.71(s,3H)。MS m/z 442.1(M+Na)+。
【0179】
4−メトキシ−N−(4−メトキシフェニル)−N−(4−フルオロフェニル)−ベンズアミド(2c)(2c):白色固体、97%の収率、融点133.5.0〜134.5℃。1H NMR(CDCl3,300MHz)δ8.11−6.66(m,15H),3.74(s,3H),3.73(s,3H)。MS m/z 384(M+H)+。
【0180】
N−(4−メトキシフェニル)−N−(4−ベンジルオキシフェニル)−2−ナフチルアミド(2d):白色固体、58%の収率。融点174.9〜175.5℃。1H NMR(CDCl3,300MHz)δ8.04(s,1H),7.77−7.74(m,2H),7.64−7.61(m,1H),7.51−7.43(m,4H),7.40−7.31(m,4H),7.13−7.10 m,4H),6.88−6.78(m,4H),4.99(s,2H),3.74(s,3H)。MS m/z 460(M+H)+。
【0181】
4−フルオロ−N,N−ビス(4−メトキシフェニル)−2−(トリフルオロメチル)ベンズアミド(2e):無色油、84.2%の収率。1H NMR(CDCl3,300MHz)δ7.34−7.26(m,4H),7.09−7.01(m,3H),6.91(d,2H,J=8.7Hz),6.87(d,2H,J=8.7Hz),3.80(s,3H),3.71(s,3H)。MS m/z 442.1(M+Na)+。
【0182】
BBr3を使用した、ベンズアミド誘導体のデメチル化のための一般手順。メトキシベンズアミド化合物を、乾燥CH2Cl2中で溶解した。BBr3(1.0MのCH2Cl2溶液)を、0℃で滴加した。反応溶液を、ゆっくりと室温まで温め、室温で一晩攪拌した。混合物を、氷浴中で0℃に冷却し、水を添加して加水分解した。EtOAcを添加し、溶液を分割した。有機層を分離し、水性層をEtOAcで抽出した。有機層をブラインで洗浄し、無水MgSO4上で乾燥させた。溶媒を減圧下で除去した。残渣を、CH3OH/CH2Cl2(1/9 v/v)を使用するフラッシュカラムクロマトグラフィーによって精製し、対応するフェノール化合物を得た。
【0183】
4−フルオロ−N,N−ビス(4−ヒドロキシフェニル)−2−(トリフルオロメチル)ベンズアミド(3a):白色固体、92.5%の収率。1H NMR(DMSO−d6,300MHz)δ9.55(s,1H),9.53(s,1H),7.69−7.58(m,2H),7.46−7.39(m,1H),7.18(d,2H,J=8.7Hz),6.93(d,4H,J=8.7Hz),7.03(d,2H,J=8.4Hz),6.78(d,2H,J=8.7Hz),6.57(d,2H,J=8.7Hz)。MS m/z 392.1(M+H)+。
【0184】
以下の化合物を上述のとおり合成し、表1に特徴付け、要約する:N,N−ビス(4−ヒドロキシフェニル)−4−プロピルベンズアミド(II)、3−フルオロ−N−(4−フルオロフェニル)−4−ヒドロキシ−N−(4−ヒドロキシフェニル)ベンズアミド(IV)、N,N−ビス(4−ヒドロキシフェニル)−2,3−ジメチルベンズアミド(V)、3−フルオロ−4−ヒドロキシ−N,N−ビス(4−ヒドロキシフェニル)−ベンズアミド(VII)、3−フルオロ−4−ヒドロキシ−N−(4−ヒドロキシフェニル)−N−フェニルベンズアミド(XI)、および3−フルオロ−N,N−ビス(4−ヒドロキシフェニル)−2−メチルベンズアミド(XII)。
【0185】
ベンジルオキシフェニル−ベンズアミドのデベンジル化の一般手順。化合物を、250mLの水素化ボトル内のEtOH中で溶解した。Pd/C粉末(5%mol)を溶液に添加した。反応容器を、138kPa(20psi)圧力水素ガス下で水素化装置に装着した。出発物質が消失するまで、反応をTLCによって監視した。次いで、溶媒を減圧下で除去した。残渣を、ヘキサン/EtOAc=3/2v/vを有するフラッシュカラムクロマトグラフィーによって精製し、所望の生成物を得た。
【0186】
以下の化合物を上述のとおり合成し、表1に特徴付け、要約する:N,N−ビス(4−ヒドロキシフェニル)−2−ナフチルアミド(VI)。
脱保護ベンズアミドの還元のための一般手順。ベンズアミド化合物を、室温で、20mLの無水THF中で溶解した。H3B(SMe2)を、アルゴン下で、室温で、注射器を介して添加した。反応溶液を攪拌し、加熱して6時間還流させた。次いで、0℃の10mLのMeOHを添加することにより、反応を停止した。溶媒を減圧下で除去した。残渣を、フラッシュカラムクロマトグラフィー(シリカゲル、CH2Cl2/MeOH=9/1v/v)に供し、所望の生成物を得た。
【0187】
以下の化合物を上述のとおり合成し、表1に特徴付け、要約する:4,4’−(2,3−ジメチルベンジルアザンイジル)ジフェノール(III)、4−((4−フルオロフェニル)(4−ヒドロキシベンジル)アミノ)フェノール(VIII)。
【0188】
O−(2−ピペリジン−1−イルエトキシ)−ベンズアミド、および類似体の一般的合成。アセトン中のベンズアミド類似体(1当量)を含有するヒドロキシフェニルの溶液に、K2CO3(3当量)およびN−クロロエチル−ピペリジン塩酸塩(1.2当量)を添加した。溶液を、6時間、加熱して還流させた。乾燥するまで溶液を蒸発させた。残渣を、水を添加することにより加水分解し、次いで、酢酸エチルで抽出した。有機層を分離し、無水MgSO4上で乾燥させた。溶媒を減圧下で除去した。残渣を、塩化メチレン/メタノール=9/1 v/vで、フラッシュクロマトグラフィーによって精製し、所望の化合物を得た。Et2O中のHClを化合物のメタノール溶液に添加した後、溶媒を蒸発させることにより、塩酸塩を調製した。
【0189】
以下の化合物を上述のとおり合成し、表1に特徴付け、要約する:4−フルオロ−N−(4−ヒドロキシフェニル)−N−(4−(2−ピペリジン−1−イル)エトキシ)フェニル)−2−(トリフルオロメチル)ベンズアミド(IX)、および4−フルオロ−N−(4−ヒドロキシフェニル)−N−(4−(2−(ピペリジン−1−イル)エトキシ)フェニル)−2−(トリフルオロメチル)ベンズアミド塩酸塩(X)(IXのHCl塩である)。
【0190】
【表1】
実施例2
式IVの化合物の合成(図6)。
【0191】
【化32】
4−フルオロアニリン(78.63g、0.708mol)、4−ヨードアニソール(138.00g、0.590mol)、無水K2CO3(122.23g、0.884mol)、CuI(11.23g、58.96mmol)、およびL−プロリン(13.58g、0.118mol)の混合物をともに、攪拌棒、還流冷却器、およびアルゴン導入口を装着した、乾燥した1Lの三つ口丸底フラスコ中で混合した。無水DMSO(300mL)を室温で添加した。反応混合物を、アルゴン下で、20時間、90℃で攪拌および加熱した。次いで、混合物を室温まで冷却し、水(300mL)で加水分解した。EtOAc(200mL)を添加して、溶液を分割した。EtOAc層を分離した。水性層を、100mLのEtOAcで抽出した。EtOAc層を混合し、ブライン(2×100mL)で洗浄し、無水MgSO4(50g)上で乾燥させた。溶媒を減圧下で除去した。褐色油の残渣を、フラッシュカラムクロマトグラフィー(シリカゲル、ヘキサン/EtOAc=9/1v/v)によって精製し、黄色固体生成物(99.70g、77.8%の収率)として、4−フルオロ−N−(4−メトキシフェニル)アニリン(1c)を得た。融点46〜48℃。MS(ESI)m/z 218.1[M+H]+,1H NMR(DMSO−d6,300MHz)δ7.77(bs,1H),7.03−6.98(m,4H),6.93−6.82(m,4H),3.70(s,3H)。
【0192】
【化33】
4−フルオロ−N−(4−メトキシフェニル)アニリン(1c)(90.78g、0.418mol)、および3−フルオロ−4−塩化メトキシベンゾイル(94.55g、0.501mol)を、攪拌棒、還流冷却器、およびアルゴン導入口を装着した、乾燥した1Lの三つ口丸底フラスコ内で、ともに混合し、無水THF(200mL)中で溶解した。無水ピリジン(132.22g、1.672mol)を、アルゴン下で、室温で、注射器を介して添加した。反応混合物を、攪拌し、加熱して一晩還流させた。次いで、反応混合物を室温まで冷却し、ろ過して、ピリジン塩を除去した。溶液を濃縮して、THF溶媒を除去した。残渣油を、200mLの2N HCl溶液で洗浄し、酢酸エチル(2x200mL)で抽出した。混合した有機層を、飽和Na2CO3水溶液(150mL)で洗浄して、過剰塩化ベンゾイルおよび酸を除去し、MgSO4(50g)上で乾燥させ、ろ過し、減圧下で濃縮して、油を得た。残渣を、CH2Cl2/アセトン(50/1v/v)を有する、シリカゲル使用するフラッシュカラムクロマトグラフィーによって精製し、黄色固体として純粋な対応するベンズアミド化合物を得た。融点54〜56℃。MS(ESI)m/z 370.1[M+H]+,1H NMR(CDCl3/TMS)δ7.24−7.11(m,4H),7.05−6.97(m,4H),6.85−6.78(m,3H),3.86(s,3H),3.79(s,3H)。
【0193】
【化34】
化合物3−フルオロ−N−(4−フルオロフェニル)−4−メトキシ−N−(4−メトキシフェニル)ベンズアミド(2a)(138.0g、0.374mol)を、アルゴン下で、室温で、無水CH2Cl2(600mL)中で溶解した。BBr3(374.75g、1.496mol)を、アルゴン下で、氷浴中で、0℃で、注射器を介して、攪拌しながら滴加した。反応溶液を、一晩、室温で攪拌した。次いで、溶液を、1Lの氷水に攪拌しながら注入した。スラリー混合物を、2時間、室温で攪拌した。白色沈殿物をろ過し、水(2×100mL)で洗浄し、真空下で乾燥させた。CH2Cl2層を分離し、無水MgSO4(50g)上で乾燥させ、ろ過し、減圧下で乾燥するまで濃縮した。CH2Cl2溶液からの白色沈殿物および残渣を混合し、フラッシュカラムクロマトグラフィー(シリカゲル、CH2Cl2/アセトン/MeOH=90/7/3v/v/v)によって精製して、淡黄褐色固体を得、それを、二度、熱いEtOAc/ヘキサン溶液から再結晶させて、白色結晶固体(104.0g、81.6%の収率)を得た。融点110〜112℃。MS(ESI)m/z 364.1[M+Na]+,1H NMR(DMSO−d6)δ10.14(bs,1H),9.71(bs,1H),7.25−7.11(m,5H),7.05−6.99(m,3H),6.78(t,J=8.6Hz,1H),6.68(d,J=8.7Hz,2H)。
実施例3
式VIの化合物の合成(図7)
4−(ベンジルオキシ)−N−(4−メトキシフェニル)アニリン(1d)の合成
4−ベンジルオキシアニリン(16.6g、83.31mmol)、4−ヨードアニソール(15.0g、64.09mmol)、K2CO3(17.72g、128.18mmol)、CuI(1.22g、6.41mmol)、およびL−プロリン(1.48g、12.82mmol)の混合物をともに、室温で混合し、無水DMSO(120mL)中で溶解した。次いで、反応混合物を、48時間、90℃で攪拌および加熱した。混合物を室温まで冷却し、水で加水分解した。EtOAcを添加して、溶液を分割した。EtOAc層を分離し、ブラインで洗浄し、無水MgSO4上で乾燥させた。溶媒を減圧下で除去した。固体残渣を、EtOAc/ヘキサン(1/9 v/v)を使用するフラッシュカラムクロマトグラフィー(シリカゲル)によって精製し、黄色固体(9.8g、50%の収率)として、対応するジアリールアニリンを得た。融点108.0〜108.4℃。1H NMR(CDCl3,300MHz)δ7.34−7.25(m,5H),6.90−6.81(m,8H),5.02(s,2H),3.78(s,3H)。MS m/z 306(M+H)+。
N−(4−ベンジルオキシフェニル)−N−(4−メトキシフェニル)−2−ナフトアミド(2d)の合成
1当量の4−(ベンジルオキシ)−N−(4−メトキシフェニル)アニリン(0.80g、2.62mmol)を、磁気攪拌棒および還流冷却器を装着した、乾燥した三つ口丸底フラスコ内で、1.5当量の2−塩化ナフトイル(0.75g、3.93mmol)、および4当量のピリジン(0.83g、10.48mmol)と混合した。混合物を、無水THF(30mL)中で溶解し、加熱して、20時間還流させた。反応溶液を室温まで冷却し、ろ過した。溶媒を減圧下で除去した。残渣を、EtOAc/ヘキサン(3/7 v/v)を用いる、シリカゲルを使用するフラッシュカラムクロマトグラフィーによって精製し、白色固体(0.70g、58%の収率)として、純粋な対応するナフトアミド化合物を得た。融点174.9〜175.5℃。1H NMR(CDCl3,300MHz)δ8.04(s,1H),7.77−7.74(m,2H),7.64−7.61(m,1H),7.51−7.43(m,4H),7.40−7.31(m,4H),7.13−7.10(m,4H),6.88−6.78(m,4H),4.99(s,2H),3.74(s,3H)。MS m/z 460(M+H)+。
N,N−ビス(4−ヒドロキシフェニル)−2−ナフチルアミド(VI)の合成。
【0194】
化合物N−(4−ベンジルオキシフェニル)−N−(4−メトキシフェニル)−2−ナフトアミド(2d)(0.50g、1.09mmol)を、室温で、無水CH2Cl2(30mL)中で溶解した。BBr3(3.26mLの1.0MのCH2Cl2溶液、3.26mmol)を、室温で、注射器を介して滴加した。反応溶液を、室温で一晩攪拌した。混合物を、氷浴内で、0℃まで冷却し、水を添加して加水分解した。EtOAcを添加して、溶液を分割した。有機層を分離し、水性層を、二度、EtOAcで抽出した。有機層を混合し、ブラインで洗浄し、無水MgSO4上で乾燥させた。溶媒を真空下で除去した。残渣を、CH3OH/CH2Cl2(1/9 v/v)を用いる、シリカゲルを使用するフラッシュカラムクロマトグラフィーによって精製して、白色固体(0.27g)、白色固体(70%の収率)として、純粋な所望のフェノール化合物を得た。融点264.3〜265.2℃(分解)。1H NMR(DMSO−d6,500MHz)δ9.46(s,2H),7.98(s,1H),7.85−7.75(m,2H),7.75−7.73(m,2H),7.54−7.48(m,2H),7.45−7.43(m,1H),7.05(s,4H),6.66(s,4H)。MS m/z 356(M+H)+。
実施例4
式VIIIの化合物の合成
【0195】
【化35】
化合物N−(4−フルオロフェニル)−4−ヒドロキシ−N−(ヒドロキシフェニル)ベンズアミド(0.30g、0.93mmol)を、室温で、20mLの無水THF中で溶解した。H3B(SMe2)(1.86mLの2MのTHF溶液、3.71mmol)を、アルゴン下で、室温で、注射器を介して添加した。反応溶液を、攪拌し、加熱して、6時間還流させた。次いで、0℃の10mLのMeOHを添加することによって、反応を停止した。溶媒を減圧下で除去した。残渣を、フラッシュカラムクロマトグラフィー(シリカゲル、CH2Cl2/MeOH=9/1 v/v)に供し、黄色油(0.26g、92%の収率)を得た。1H NMR(DMSO−d6,500MHz)δ9.29(s,1H),9.24(s,1H),7.09(d,2H,J=8.3Hz),6.98(d,2H,J=9.0Hz),6.94−6.91(m,2H),6.73(d,2H,J=9.0Hz),6.68−6.64(m,4H),4.70(s,2H)。MS m/z 307.8(M−H)。
実施例5
化合物IXおよびXの化合物の合成(図8)。
【0196】
ジアリールアニリンの合成。アリールアミン(1.5当量)、ヨードアリール(1当量)、K2CO3(2当量)、CuI(0.1当量)、およびL−プロリン(0.2当量)の混合物をともに、室温で混合し、無水DMSO中で溶解した。次いで、反応混合物を、28時間、90℃で攪拌および加熱した。混合物を室温まで冷却し、水で加水分解した。EtOAcを添加して、溶液を分割した。EtOAc層を分離し、ブラインで洗浄し、無水MgSO4上で乾燥させた。溶媒を減圧下で除去した。固体残渣を、溶媒としてEtOAc/ヘキサン(3/7 v/v)を使用するフラッシュカラムクロマトグラフィー(シリカゲル)によって精製して、対応するジアリールアニリンを得た。ビス−(4−メトキシフェニル)アミン(1a):淡黄色固体、73%の収率。1H NMR(CDCl3,300MHz)δ6.93−6.81(m,8H),5.37(s,br,1H),3.78(s,6H)。MS m/z 228.4(M−H)+。
4−フルオロ−N,N−ビス(4−メトキシフェニル)−2−(トリフルオロメチル)ベンズアミド(2e)の合成
1当量のビス−(4−メトキシフェニル)アミン(1a)(0.73g、3.18mmol)を、磁気攪拌棒および還流冷却器を装着した、乾燥した三つ口丸底フラスコ内で、1.2当量の4−フルオロ−2−塩化トリフルオロメチルベンゾイル(0.87g、3.82mmol)、および6当量のピリジン(1.51g、19.08mmol)と混合した。混合物を無水THF(20mL)中で溶解し、90℃で20時間加熱した。反応溶液を室温まで冷却し、ろ過した。溶媒を減圧下で除去した。残渣を、EtOAc/ヘキサン(3/7 v/v)を有する、シリカゲルを使用するフラッシュカラムクロマトグラフィーによって精製して、無色油(1.12g、84.2%の収率)として、純粋な対応するベンズアミド化合物を得た。1H NMR(CDCl3,300MHz)δ7.34−7.26(m,4H),7.09−7.01(m,3H),6.91(d,2H,J=8.7Hz),6.87(d,2H,J=8.7Hz),3.80(s,3H),3.71(s,3H)。MS m/z 442.1(M+Na)+。
4−フルオロ−N,N−ビス(4−ヒドロキシフェニル)−2−(トリフルオロメチル)ベンズアミド(3a)の合成。
【0197】
化合物4−フルオロ−N,N−ビス(4−メトキシフェニル)−2−(トリフルオロメチル)ベンズアミド(2e)(1.00g、2.38mmol)を、室温で、無水CH2Cl2(30mL)中で溶解した。BBr3(10mLの1.0MのCH2Cl2溶液、10.0mmol)を、室温で、注射器を介して滴加した。反応溶液を室温で一晩攪拌した。混合物を、氷浴内で0℃まで冷却し、水を添加して加水分解した。EtOAcを添加して、溶液を分割した。有機層を分離し、水性層を、二度、EtOAcで抽出した。有機層を混合し、ブラインで洗浄し、無水MgSO4上で乾燥させた。溶媒を真空下で除去した。残渣を、CH3OH/CH2Cl2(1/9 v/v)を有する、シリカゲルを使用するフラッシュカラムクロマトグラフィーによって精製して、白色固体(0.86g、92.5%の収率)として、純粋な所望のフェノール化合物を得た。1H NMR(DMSO−d6,300MHz)δ9.55(s,1H),9.53(s,1H),7.69−7.58(m,2H),7.46−7.39(m,1H),7.18(d,2H,J=8.7Hz),6.93(d,4H,J=8.7Hz),7.03(d,2H,J=8.4Hz),6.78(d,2H,J=8.7Hz),6.57(d,2H,J=8.7Hz)。MS m/z 392.1(M+H)+。
4−フルオロ−N−(4−ヒドロキシフェニル)−N−[4−(2−ピペリジン−1−イル)エトキシ]フェニル]−2−(トリフルオロメチル)ベンズアミド(IX)の合成
アセトン中の4−フルオロ−N,N−ビス(4−ヒドロキシフェニル)−2−(トリフルオロメチル)ベンズアミド(3a)(0.61g、1.56mmol)の溶液に、K2CO3(1.29g、9.36mmol)、およびN−クロロエチル−ピペリジン塩酸塩(0.34g、1.87mmol)を添加した。溶液を加熱して20時間還流させた。乾燥するまで溶液を蒸発させた。残渣を、フラッシュクロマトグラフィー(シリカゲル、塩化メチレン/メタノール=9/1 v/v)によって精製して、白色固体(0.45g、57.5%の収率)として、所望の化合物を得た。1H NMR(DMSO−d6,300MHz)δ9.57(s,1H),7.71−7.68(m,2H),7.47−7.44(m,1H),7.28(d,1H,J=9.0Hz),7.18(d,1H,J=8.7Hz),7.13(d,1H,J=8.7Hz),7.05(d,1H,J=8.4Hz),6.97(d,1H,J=9.0Hz),6.80−6.76(m,2H),6.57(d,1H,J=87。Hz),4.06(t,1H,J=6.0Hz),3.93(t,1H,J=6.0Hz),2.66(t,1H,J=5.7Hz),2.55(t,1H,J=5.4Hz),2.44(s,2H),2.36(s,2H),1.49−1.37(m,6H)。MS m/z 501.0(M−H)。
【0198】
Et2O中のHClを化合物のメタノール溶液に添加した後に、溶媒を蒸発させることによって塩酸塩(X)を調製した。
実施例6
エストロゲン受容体結合親和性、作動薬および拮抗薬活性
インビボ競合的ラジオリガンド結合アッセイを使用して、天然の高親和性ERリガンドである[2,4,6,7−3H(N)]−エストラジオール([3H]E2)、および細菌で発現したGST縮合ER−α、またはER−βリガンド結合ドメイン(LBD)タンパク質との化合物のET結合親和性を決定した。
方法
組み換えER−α、またはER−βを、[3H]E2と組み合わせて、[3H]E2の平衡解離定数(Kd)を決定した。全結合または非特異的結合を決定するために、タンパク質を、漸増濃度の[3H]E2とともに、高濃度の非標識E2の存在下または不在下で、18時間、4℃で培養した。非特異的結合を減算し、非線形回帰を使用して、E2のKd(ERα:0.71nM、ERβ:1.13nM)を判定した。加えて、ER−αおよびER−βLBDを飽和させるのに必要とされる[3H]E2の濃度は、4〜6nMであると決定した。
【0199】
漸増濃度の化合物(範囲:10−11〜10−6M)を、上述の条件を使用して、[3H]E2(5.7nM)、およびER LBDとともに培養した。培養後、プレートを、Unifilter−96 Harvester(PerkinElmer)上のGF/Bろ過で採取し、氷冷緩衝液B(50mMトリス、pH7.2)で、3度洗浄した。ろ過プレートを室温で乾燥させ、次いで、35μlのMicroscint−Oカクテルを各ウェルに添加し、ろ過プレートをTopSeal−Aで密封した。放射能を、Microscintカクテル(PerkinElmer)内の3Hの設定を使用して、TopCount(R)NXTマイクロプレートシンチレーション計測器内で計数した。
【0200】
化合物の各濃度における、[3H]E2の非特異的結合を減算することによって、[3H]E2の特異的結合を決定(10−6Mの非標識E2とともに培養することによって決定)し、それを、試験化合物の不在下での特異的結合に対する割合で示した。[3H]E2の特異的結合を50%減少させる化合物の濃度(IC50)を決定した。次いで、化合物の平衡結合定数(Ki)を、Ki=Kd×IC50/(Kd+L)により、算出した。式中、Kdは、[3H]E2(ER−α=0.71nM、ER−β=1.13nM)の平衡解離定数であり、Lは、[3H]E2(ER−α:5.7nM、ER−β:5.7nM)の濃度である。
結果
結合アッセイは、リガンドが、3.75nMから1000nM以上の範囲の種々の濃度、および化合物が、イソ型選択的から、非イソ型選択的である選択的範囲において、ER−α、およびER−βに結合したことを明らかにした。代表的化合物の結果を表2に記載する。
【0201】
【表2】
化合物IVは、ERαおよびERβに結合する。インビトロ競合的ラジオリガンド結合アッセイを使用して、[2,4,6,7−3H(N)]−エストラジオール([3H]E2)、天然の高親和性ERリガンド、および細菌で発現したGST縮合ERαまたはERβリガンド結合ドメイン(LBD)タンパク質との化合物IVのER結合親和性を決定した。このアッセイでは、化合物IVのERαおよびERβ結合親和性(Ki値)はそれぞれ、21.7±1.7nM(n=3)、および15.2±4.1nM(n=3)であった。ERへの結合の際、化合物IVは、組織選択的な方法で、薬理反応に関与する標的遺伝子の発現または抑制につながる、複雑な一連の分子事象を開始する。遷移トランスフェクションアッセイでは、化合物IVは、ERαおよびERβ作動薬であり、ERβ媒介転写活性化と比較して、ERα媒介転写活性化を刺激する効率がより高いことが示された。エストラジオールはERαおよびERβを活性化するが、ERαに対する選択性は5.1倍高い。一方、化合物IVは、ERαに対して、49.0倍の選択性を示す。したがって、相対的トランス活性化能(エストラジオール値に標準化)において、化合物IVは、ERβよりもERαに対して相対的に9.7倍の選択性を有する。さらに、拮抗薬作用は、最大10μMの濃度まで、化合物IVのエストラジオール(1nM)−刺激転写活性化において認められなかった。多くのステロイドリガンドは、他の核ホルモン受容体と交差反応するが、化合物IVの作用は、ERαおよびERβに対して特異的である。化合物IVを、転写活性化アッセイにおける作動薬および拮抗薬モードの両方で、グルココルチコイド受容体(GR)のラットイソ型、ミネラルコルチコイド受容体(MR)のラットイソ型、プロゲステロン受容体(PR)のラットイソ型、アンドロゲン受容体(AR)のラットイソ型、ならびにファルネソイドX受容体(FXR)のヒトイソ型、肝臓X受容体(LXR)、ペルオキシソーム活性受容体(PPAR−αおよびPPAR−γ)のヒトイソ型、およびレチノイドX受容体(RXR−α)のヒトイソ型に対する交差反応に対してスクリーニングした。化合物IVは、これらのアッセイのうちのいずれかにおいて、任意の作動薬活性または拮抗薬活性を表わさず、化合物IVが、これらの核ホルモン受容体スーパーファミリー成員と機能的に交差反応しないという結論を支持した。
実施例7
選択した化合物のトランス活性化
作動薬および拮抗薬モードでトランス活性化アッセイを実施して、化合物が、作動薬、拮抗薬、または、部分的であるか否かを特定した。
【0202】
方法
ラットエストロゲン受容体(ER−αおよびER−β)を、ラット卵巣cDNAから、pCR3.1プラスミドベクター骨格にクローン化した。配列決定を実施して、任意の突然変異の欠如を判定した。HEK−293細胞を、ダルベッコ最小必須培地(DMEM)+5%活性炭除去ウシ胎児血清(csFBS)において、プレーティングした。0.25μgのERE−LUC、0.02μgのCMV−LUC(ウミシイタケルシフェラーゼ)、および12.5ngのラットER−α、または25ngのラットER−βを有する、Lipofectamine(Invitrogen社、カリフォルニア州カールズバッド)を使用して、細胞をトランスフェクトした。種々の濃度の化合物、または化合物およびエストラジオールの組み合わせのトランスフェクト24時間後、細胞を処理して、拮抗薬活性を判定した。ルシフェラーゼアッセイを、トランスフェクト48時間後、実施した。
結果
トランス活性化システムにおける本発明の化合物のスクリーニングは、化合物が、3つのすべての分類、すなわち、作動薬、拮抗薬、および部分的作動薬に属したことを明らかにした。作動薬および拮抗薬の一例を表3に示す。トランス活性化結果は、イソ型選択性に対する結合結果と非常によく一致した。
【0203】
表3は、いくつかの選択した本発明の化合物のEC50およびIC50トランス活性化値を提供する。
【0204】
【表3】
実施例8
カニクイザルにおけるテストステロン抑制
2歳の生殖腺無損傷のオスのカニクイザル(n=2)を、霊長類の餌および水への自由なアクセス(経口投与前の絶食を除く)で、USDAガイドラインに従い、研究の間収容した。動物は、連続7日間、Tween80/脱イオン水のマイクロエマルション媒体で、30mg/kgの式IVの化合物の1日1回の強制経口投与用量が与えられた。血清サンプルを、1日目(ベースライン)、3日目、4日目、5日目、6日目、および7日目に、経口用量投与前に静脈穿刺によって採取した。HPLC方法と組み合わせた、またはそれを用いない酵素免疫アッセイ(EIA)方法のそれぞれを使用して、テストステロンおよび総アンドロゲンを定量化した。6日間の式IVの化合物での処理後、テストステロンおよび総アンドロゲン(テストステロン/ジヒドロテストステロン)では、時間依存性の減少が明らかであった。式IVの化合物は、ベースライン値に対して、テストステロンのレベルを、動物#1および動物#3ではそれぞれ、58%および64%抑制した(図1の太線、表4を参照)。同様に、総アンドロゲンレベルは、ベースライン値と比較して、動物#1および#3の両方において、56%抑制された。
【0205】
男性の下垂体−精巣軸エストロゲンフィードバックと一致して、これらの結果は、式IVの化合物の反復経口用量(30mg/kg)の後、無損傷の非ヒト霊長類(カニクイザル)における血清ホルモン(テストステロンおよび総アンドロゲン)の抑制に対して、強い薬理反応を表す。
【0206】
【表4】
実施例9
ラットにおける、LHおよびテストステロンホルモンレベルの抑制
インビボ用量反応研究を実施して、無損傷および精巣摘除された(ORX)オスのラットにおける、LH抑制への化合物IVの作用を評価した。無損傷およびORX動物において、1日当たり≧10mg/kgの用量で、化合物は、対応する対照と比較した時、LHレベルを有意に抑制した(FSHレベルにおいて、同一の抑制パターンが認められた)。LH抑制は、0.08ng/mLである、定量下限以下(BLOQ)まで、テストステロンレベルの確実な減少をもたらし、また、前立腺、精嚢、肛門挙筋はアンドロゲン依存性が高い臓器であるため、これらの重量の減少をもたらした。無損傷の動物では、これらの標的臓器の重量における用量依存性の減少は、去勢された対照のレベルに対する精嚢および肛門挙筋重量で顕著であった。前立腺重量は、無損傷の動物において有意に減少したが、これらの値は、去勢された対照のレベルに到達しなかった。結果を下記の表6に要約する。
材料および方法:
約200gの重量のオスのスプラーグドーリーラットを、不断給の餌(2016Teklad Global 16%タンパク質げっ歯類食餌、Harlan社、ウィスコンシン州マディソン)および水で、12時間の明暗サイクルで維持した。動物プロトコルが再検討され、テネシー州の動物管理使用委員会により承認された。
【0207】
本研究のための試験物品を計量し、PEG300(Acros Organics社、ニュージャージー州)で希釈した10%のDMSO(Fisher)中で溶解して、適切な用量の製剤を調製した。本研究のために、60匹のオスのスプラーグドーリーラットを、体重によって無作為化し、12の治療グループのうちの1つに割り当てた(n=5匹の動物/グループ)。処置グループを表5に記載する。動物を、ケージ当たり2〜3匹の動物のグループに収容した。対照グループ(無損傷および精巣摘除された(ORX))に、連日、媒体を投与した。化合物IVを、0.3、1、3、10、および30mg/kg/日の用量で、無損傷およびORXグループに皮下注射(200μL)を介して投与した。
【0208】
14日間の投薬レジメン後、動物を麻酔(ケタミン/キシラジン、87:13mg/kg)化で安楽死させ、体重を記録した。加えて、腹側前立腺、精嚢、および肛門挙筋を除去し、外部組織を除去し、個々に計量した。臓器重量を、体重に対して標準化し、無損傷対照の割合として示した。イソフルラン麻酔下で、腹部大動脈から血液を採取し、凝血させた。血清ホルモンレベルの判定前に、血清を遠心分離によって分離し、−80℃で保管した。血清黄体形成ホルモン(LH)および卵胞刺激ホルモン(FSH)濃度を、製造者の指示に従い、ラット下垂体ルミネックスアッセイ(Millipore社、マサチューセッツ州ビレリカ)によって決定した。このアッセイの定量下限は、LHでは、3.2pg/mL、FSHでは、32pg/mLであった。テストステロンを、0.08ng/mLのLLOQを用いる、テストステロンEIA(Alpco Dianostics社、ニューハンプシャー州セーレム)によって測定した。定量下限以下の血清ホルモン値を、グループ平均の分析から除外した。したがって、サンプルBLOQを有するグループにおけるLHおよびTの報告された値は、実際の値よりも高い。この分析方法は、LHおよびT抑制の最も控えめな推定を提供した。フィッシャーの最小有意差検定を使用して、個々の用量グループを、無損傷およびORX媒体対照グループと比較した。有意性を、P値<0.05として事前に定義した。
【表5】
無損傷およびORXラットにおける黄体形成ホルモンレベル(表6)
無損傷およびORX媒体対照グループにおけるLHレベル(平均±標準偏差)はそれぞれ、1.46±0.64、および11.1±3.9ng/mLであった。化合物IVは、無損傷動物において、LHレベルを用量依存的に低下し、1日用量≧3mg/kgで、統計的に有意な減少に達した。無損傷の化合物IVで処置された動物におけるLHレベルは、0.3、1、3、10、および30mg/kg/日の用量後にそれぞれ、0.863±0.384、0.704±0.530、0.395±0.302、0.226±0.165、および0.236±0.176ng/mLであった。ORXのオスにおけるLHレベルもまた、化合物IV治療によって有意に減少した。ORX動物では、LHレベルは、0.3、1、3、10、および30mg/kg/日の用量後にそれぞれ、15.4±2.9、13.5±2.2、6.5±5.6、0.4.425±0.135、および0.368±0.119ng/mLであった。結果を、図10Aに図式的に表す。
無損傷およびORXラットにおける卵胞刺激ホルモンレベル(表6)
無損傷およびORX媒体対照グループにおける血清FSHレベルは、それぞれ、20.9±8.5、および93.5±13.8ng/mLであった。無損傷動物では、化合物IVは、≧10mg/kg/日の用量で認められた有意な減少で、FSHレベルを用量依存的に減少させた。無損傷の化合物IVで処置された動物におけるFSHレベルは、0.3、1、3、10、および30mg/kg/日の用量後にそれぞれ、17.3±6.4、15.7±7.3、18.4±7.7、9.2±4.0.および6.3±1.8ng/mLであった。ORX動物では、LHレベルは、0.3、1、3、10、および30mg/kg/日の用量後にそれぞれ、115±17、114±22、65.2±31.9、27.6±8.2、および15.1±4.1ng/mLであった。結果を、図10Bに図式的に表す。
無損傷およびORXラットにおけるテストステロンレベル
無損傷媒体対照グループにおける血清テストステロンレベルは、2.4±1.1ng/mLであった。Tの定量下限は、0.08ng/mLであった。0.08ng/mL未満の値を、定量下限以下(BLOQ)として指定した。無損傷動物では、式IVの化合物は、1日当たり≧3mg/kgの用量で認められた有意な減少で、Tレベルを用量依存的に低下させた。式IVの化合物で処置された無損傷動物におけるテストステロンレベルは、1日当たり0.3、1、3、10、および30mg/kgの用量後にそれぞれ、2.6±1.7、1.6±1.0、0.7±0.4、BLOQ、およびBLOQng/mLであった。ORX動物では、Tレベルは、化合物IVで治療されたすべてのグループ、および媒体で処置されたグループでは、BLOQであった。無損傷動物では、結果を図10C(および図2)に図式的に表す(BLOQ値を、図式目的のために、定量限界で表す)。
【0209】
図9に表すとおり、無損傷のオスのラットにおいて、3mg/kg、10mg/kg、および300mg/kgの用量で化合物IVを投与することにより、24時間後、72時間後、168時間後に、血清テストステロンの迅速かつ強力な抑制が測定された。
臓器重量(表6)
前立腺、精嚢、および肛門挙筋重量を測定して、Tの抑制を確認した。臓器重量(平均±標準偏差)をそれぞれ、図10D、10E、および10Fに表す。前立腺、精嚢、肛門挙筋重量における用量依存性の低下が、化合物IVで処置された無損傷動物において認められた。無損傷動物における前立腺重量は、0.3、1、3、10、および30mg/kg/日の用量後にそれぞれ、84.0±19.2、75.2±20.7、68.2±8.2、45.1±20.0、および43.6±8.8であった。ORX動物における前立腺重量は、0.3、1、3、10、および30mg/kg/日の用量後にそれぞれ、19.0±4.2、17.4±3.4、19.6±6.7、22.9±5.4、および20.6±2.1であった。無損傷動物における精嚢重量は、0.3、1、3、10、および30mg/kg/日の用量後にそれぞれ、76.2±7.8、66.3±27.2、51.8±28.5、19.1±7.0、および17.9±3.3であった。ORX動物における精嚢重量は、0.3、1、3、10、および30mg/kg/日の用量後にそれぞれ、12.2±1.3、16.6±5.4、16.5±4.8、13.3±1.9、および12.9±2.1であった。無損傷動物における肛門挙筋重量は、0.3、1、3、10、および30mg/kg/日の用量後にそれぞれ、86.9±10.0、82.1±12.1、65.2±4.4、57.8±11.2、および58.1±4.7であった。ORX動物における肛門挙筋重量は、0.3、1、3、10、および30mg/kg/日の用量後にそれぞれ、54.5±6.6、49.6±7.0、53.6±10.0、51.1±4.9、および49.2±4.2であった。
LH抑制および臓器重量データを、表6に要約する。
【0210】
【表6】
実施例10
ラットおよびサルにおける化合物IVによる抑制後のテストステロンレベルの回復
化合物IVとの化学的去勢の可逆性を研究した。
材料および方法
約200gの重量の35匹のオスのスプラーグドーリーラットを、不断給の餌(2016 Teklad Global 16%タンパク質げっ歯類食餌、Harlan社、ウィスコンシン州マディソン)および水で、12時間の明暗サイクルで維持した。動物プロトコルは再検討され、テネシー州の動物管理使用委員会により承認された。
【0211】
本研究のための試験物品を計量し、PEG300(100%)(Acros Organics社、ニュージャージー州)中で溶解して、適切な用量の製剤を調製した。動物を、10の処置グループのうちの1つに無作為に割り当てた(n=5匹の動物/グループ)。処置グループを表7に記載する。動物を、ケージ当たり2〜3匹の動物のグループに収容した。無損傷動物におけるベースラインの決定のために、グループ1を、研究の開始(1日目)に安楽死させた。グループ2〜7は、3日間、強制経口投与(〜200μL)を介して、1、3、または30mg/kgの1日用量を受けた。グループ2、3、および4を、4日目に安楽死させ、最大テストステロン抑制を測定した。グループ5、6、および7を、薬物を与えない休薬期間で14日間、回復させた。
【表7】
結果:
無損傷ラットにおける血清テストステロンレベルは、ベースラインにおいて、6.4±3.1ng/mL(平均±標準偏差)であった。3日間、3、および30mg/kgの用量で投与された化合物IVは、血清テストステロンレベルをそれぞれ、1.47±0.26、および1.62±0.49まで有意に抑制した。有意な抑制は、3日間、1mg/kgの化合物IVを受けた動物において認められなかった。最も重要なことには、14日間の回復期間後に測定された時、血清テストステロンレベルは、3日間、1、3、または30mg/kgの化合物IVを受けた動物においてそれぞれ、3.3±1.92、3.00±1.06、および3.8±1.72であり、図23に図示するとおり、無損傷ラットにおけるベースライン血清テストステロン濃度とは統計的に有意に異ならなかった。
【0212】
本研究は、化合物IVが、無損傷のオスのラットにおける血清テストステロンレベルを迅速に抑制することを示す前回の結果を裏付けている。我々は、3日間、≧3mg/kg/日を受けた用量グループにおいて、血清テストステロンレベル抑制を認めた。血清テストステロンの有意な減少は、1mg/kgの用量グループで認められなかった。しかしながら、14日間の回復以内で、血清テストステロンレベルは、無損傷対照のレベルまで戻った。本研究は、化合物IVによる薬理学的去勢は、ラットにおいて可逆性であることを示す。
【0213】
無損傷のオスのサルにおけるテストステロンレベルの抑制および回復への化合物IVの作用を、経口薬物動態研究と併せて評価した。これらの治療に使われていない3匹のオスのカニクイザル(2〜3歳)に、連続7日間、強制経口投与により、30mg/kgで化合物IVを投与した。血液サンプルを採取し、テストステロンおよび化合物IVの定量的測定のために、それぞれ、血清および血漿に分割した。結果は、化合物IVの1日経口用量が、ベースラインレベル(治療のベースライン、2日目、および6日目[平均±SEM]では、それぞれ、1591±72.5、997±104、および852±136ng/mLのレベル)と比較して、3匹のサルすべてにおいて、循環アンドロゲン(主に、テストステロンおよびジヒドロテストステロン)レベルを最大47%ほど有意に低下させたことを示す。18日間の薬物フリーの回復期間後、アンドロゲンレベルは、通常に戻り、治療前のベースラインレベル(回復後、1757.7±369.5ng/mL)とは、有意に異ならなかった。
実施例11
ラットにおけるLHおよびテストステロンの減少に反する骨の保存(表8)
骨への処置に対する式IVの化合物の作用を研究した。経口的に投与された、式IVの化合物は、無損傷のオスのラットにおいて、LH抑制に伴う骨量の減少を完全に防止した。LHの有意な減少は、無損傷動物において、1日当たり≧10mg/kgの用量レベルで、式IVの化合物によって誘発された。1日当たり1mg/kgでは、式IVの化合物はLHレベルを有意に減少させなかったものの、前立腺、精嚢、および肛門挙筋における有意な減少は、この用量で明らかであり、循環テストステロンの低下が、生理学的に、これらのアンドロゲン反応臓器に関連することを示す。しかしながら、1日当たり1mg/kgの式IVの化合物は、骨梁量(大腿遠位において測定)を無損傷対照のレベルに維持した。1日当たり10、および30mg/kgの用量で投与された時、式IVの化合物は、無損傷対照の大腿遠位よりも大腿遠位の骨量を有意に増大させた。これらのデータは、化合物IVが、無損傷ラットにおいてLHレベルを低減する用量レベルで、骨梁塩濃度(BMD)、および骨量の割合を増大させたことを示す。本研究からのデータを表8に示す。_Ref216166047_Toc217378480
【0214】
【表8】
実施例12
17βヒドロキシステロイド脱水素酵素5(17β−HSD5)酵素活性への作用
HSDファミリー成員は、循環ステロイドの転換に関与する。17β−HSD5は、アンドロステンジオンをテストステロンに、エストロンをエストラジオールに転換する。加えて、それはまた、プロスタグランジン合成にも関与する。ここで、いくつかの選択した本発明の化合物の17β−HSD5活性を阻害する能力を示す。
方法
ヒト17β−HSD5を、pGEX4t1ベクターにクローン化し、精製タンパク質を調製した。精製タンパク質を、適切な緩衝液中の代表的な本発明の化合物、14Cアンドロステンジオン、およびNADPHとともに培養した。合成テストステロンを、酢酸エチルを使用して抽出し、空気乾燥させ、薄層クロマトグラフィー(TLC)プレート上にスポットし、展開した。TLCをホスフォイメージャーに暴露し、テストステロンバンドの強度を定量化した。インドメトシンを陽性対照(LHRH作動薬)として使用した。
結果
化合物IVを試験し、それは、17β−HSD5酵素活性への部分的阻害作用を有した。陽性対照(LHRH作動薬)であるインドメトシンは、図3に示すとおり、予想通り、この酵素の強力な阻害を呈した。
実施例13
毒性研究
インビトロヒト血小板凝集アッセイを使用して、化合物IVおよびジエチルスチルベストロール(DES、陽性対照)の血栓の可能性を比較するために、研究を実施した。男性が化合物IV(LH抑制)の対象とする治療群であるため、健康な男性ドナーからの血液を研究に使用した。多血小板血漿を、30秒間、エストラジオール(E2)、DES、化合物IV、または媒体のいずれかとともに、事前に培養し、次いで、トロンビン(0.3単位)を添加して、血小板凝集を誘発した。研究結果は、DESを用いる事前培養が、トロンビン誘発血小板凝集を約10倍ほど増大させたことを示す。しかしながら、化合物IVおよびエストラジオールは、多血小板血漿において凝集を減少した。これらのデータは、化合物IVが、DESと比較して、インビトロでのヒト血小板の反応性を低減させたことを示し、化合物IVが、DESよりも低い血栓塞栓症の可能性を有し得ることを示唆する(図4)。
実施例14
ほてりへの化合物IVの作用
Simpkinsら(1983)によって開発され、閉経後のほてりとの種々の類似点を有することが示されている、モルヒネ依存ラットモデル(MDモデル)を使用して研究を実施し、ほてりへの化合物IVの作用を調査した。ヒトの状態との類似点に加えて、この実験動物モデルは、短い応答時間を有しているため、血管運動症状を緩和することのできる化合物を、尾部皮膚温度(TST)を使用して特定できる有用なハイスループットスクリーニングのツールである。TSTプローブTA−40(Data Sciences International社、ミネソタ州)を、尾基底にテープで貼りつけ、15分間、ベースライン温度を取得した。15分後、動物を、ナロキソン(1mg/kg、SQ)で処理して、モルヒネの作用を減弱させた。尾部皮膚温度(TST)を、ナロキソン処理後に、1時間、一連の実験にわたり、5秒のサンプリング度数で測定した。データ取得の後、各動物で60秒毎に記録された、温度の移動平均を算出し、さらに分析した。ベースライン温度を、ナロキソン投与前に、15分間にわたって取得された平均温度として計算した。線形台形法を使用して、ナロキソン投与後のすべての値をベースラインから減算することによって、湾曲(AUC)の下の面積を算出した。
【0215】
化合物IVは、モルヒネ禁断モデル(図13を参照)におけるほてりを軽減し、10mgの化合物IVで最良結果となった。17βE2を、100%のDMSO中で、5mg/kgで使用した。
実施例15
ラットにおける化合物IV対DES
LHRH作動薬の導入前に、去勢テストステロンレベルを、エストロゲン、主に、ジエチルスチルベストロール(DES)を介して、下垂体内のエストロゲン活性を増大させることによって達成した。DESは、テストステロンを去勢レベルに抑制する上でLHRH作動薬と同等に有効であった。DESで治療された患者は、ほてりまたは骨量の減少を有さなかったが、LHRH作動薬を用いたADTよりも高い比率で女性化乳房を有した。残念ながら、DESおよびエストラジオール等の非常に強力な純粋エストロゲンは、しばしば、それらの臨床使用を制限する、深刻な心臓血管および血栓塞栓性合併症の高リスクを伴う。DESでの静脈血栓塞栓性合併症のリスクの増加は、他のホルモン受容体とのその交差反応性によるものであると仮定されているが、証明されてはいない。ヒト血小板を用いるインビトロ研究は、化合物IVが、DESよりもさらに低い前凝固化成を有したことを明らかにした。したがって、ER−α選択的作動薬である化合物IVは、骨粗しょう症または有害な脂質プロファイルを引き起こすことなく、DESの前立腺癌の利点をもたらし得、LHRH作動薬の利点ももたらし得る。
【0216】
化合物IVは、ラットにおける前立腺の寸法を減少させ(図11A)、ORXラットの前立腺の寸法の中等度の増大を発現させる(図11B)上で、DESと同様に有効である。
【0217】
DESと化合物IVとの間の差異を、図12A〜12Cに提示する。DESは、グルココルチコイド受容体(GR)(図12A)、およびアンドロゲン受容体(AR)(図12B)と交差反応したが、化合物IVは、交差反応しなかった。加えて、DESは、エストロゲン関連受容体(ERR)トランス活性化を拮抗したが、化合物IVは、拮抗しなかった。化合物IVは、図12Cに図示するとおり、3つのERRイソ型(ERR−α、ERR−β、およびERR−γ)との交差反応に失敗した。
実施例16
サルの毒性研究−90日間
モーリシャス生息のコロニー繁殖のマカク属カニクイザルを入手した。前向き研究を、13週間の中間期間を有する、オスのカニクイザルにおける、化合物IVおよび陽性対照(LHRH作動薬)の39週間の経口薬理学および毒物学評価として、設計した。合計39匹の性成熟したオスのサル(5〜8歳)を、治療開始前に、5つのグループに無作為に割り当てた。グループには、1)媒体対照、2)1mg/kgの化合物IV、3)10mg/kgの化合物IV、4)100mg/kgの化合物IV、および5)陽性対照(LHRH作動薬)が含まれた。薬物を、グループ1および5では、媒体対照物品(Tween 80/PRANG(商標))を、グループ2、3、および4では、媒体中の化合物IVを用いて、39週間、1日1回、ケージ横投与によって、経口送達した。化合物IVの用量レベルは、グループ2、3、および4ではそれぞれ、1、10、100mg/kg/日であった。経口用量を、各動物の最新の利用可能な体重に基づき算出したとおり、10mL/kg用量体積で送達した(図14)。グループ5の動物はまた、39週間の研究期間の間、1日1回、陽性対照(LHRH作動薬)(0.02mLの定容量)の皮下注射を受けた。連日、一般的外見および臨床兆候を観察し、記録した。定期的評価および他の選択研究調査を、研究プロトコルに示すとおり、実施した。選択パラメータは、テストステロン、前立腺特異的抗原(PSA)、および前立腺体積および重量を含むが、これらに限定されない。
【0218】
テストステロンおよび全PSAレベルを、酵素免疫アッセイ(EIA)方法および化学発光免疫アッセイ(LIA,ALPCO Diagnostics社、ニューハンプシャー州セーレム)をそれぞれ使用して、(標準的処置に従い)血清サンプル中で定量化した。テストステロン評価の血液サンプルを、ベースライン(すなわち、治療開始前)、および1日目、3日目、7日目、14日目、28日目、64日目、および90日目に、すべての動物(絶食状態)から採取した。PSA決定のための血液試料を、ベースラインおよび6週目中に、すべての動物(絶食状態)から採取した。考察目的のために、テストステロンおよびPSAアッセイで、定量下限以下(BLQ)の濃度を有するサンプルの結果を、アッセイの定量下限(LLOQ)の半分として算出し、「推定最終濃度」と見なす。表9〜16のデータを、「推定最終濃度」(すなわち、アッセイのLLOQの半分として含まれた、BLQ結果を有するサンプル)に加えて、「定量化可能濃度のみ」(すなわち、BLQ値を除く)として提示する。ベースライン、および6週目に、経直腸超音波(TRUS)処置を使用して、前立腺体積を麻酔下の生きた動物で測定した。前立腺の幅および高さを記録した。前立腺体積を、幅×幅×高さ×パイ/6として算出し、体重に対して標準化した。前立腺の湿潤重量を、脂肪のない組織、および外部組織をトリミングした後に、剖検で記録した。
結果および考察:
血清テストステロンレベルを、図15および表9〜12に提示する。ベースラインにおいて、研究のすべてのサルのテストステロンレベルは、性成熟したオスのカニクイザルでは、正常範囲であった。しかしながら、テストステロンレベルは、100mg/kg/日で、化合物IVを受けたサル、および陽性対照(LHRH作動薬)で処置されたサルにおいて、有意に減少した。陽性対照(LHRH作動薬)グループにおけるテストステロンレベルは、二相性変化を示し、1日目、および3日目にそれぞれ、47.4%および547%(p<0.01)の初期の有意な増加(すなわち、炎症)の後、7日目、14日目、28日目、64日目、および90日目に、3.6%、67%、73%、83%、および85%の減少を有した(図15、および表9〜12を参照)。同様の炎症は、最高の用量レベル(すなわち、100mg/kg/日)でさえも、化合物IVで処置されたいかなる動物においても認められなかった。用量および治療期間は、化合物IVの薬理活性に重要であり、100mg/kg/日の用量は、ベースライン値に対して、血清テストステロンを、3日目、7日目、14日目、28日目、および64日目にそれぞれ、60%、51%、42%、79%、および92%ほど抑制した(図15、および表9および10を参照)。100mg/kg/日の化合物IVでの治療の90日後に、10匹のグループ4のサルのうちの6匹のテストステロンレベルは、アッセイの定量下限以下の濃度まで減少した(表11を参照)。グループ4のサルの平均血清テストステロンレベルは、相対的ベースライン値と比較して、96%ほど減少した(「推定最終濃度」、すなわち、BLQ値を有する、6/10匹のサルのテストステロンレベルを、LLOQ濃度の50%として算出した表10を参照)。90日目までに、100mg/kg/日で、化合物IVは、血清テストステロンを、陽性対照(LHRH作動薬)よりも有意に低いレベルまで減少させたこと(p=0.013)に留意することが重要である。
【0219】
【表9】
【0220】
【表10】
【0221】
【表11】
【0222】
【表12】
血清PSAレベルもまた、治療開始の4週間以内に、化合物IVによって有意に抑制された。69%および87%(平均)のPSA減少は、4週間、10mg/kg、および100mg/kgで、化合物IVを受けたサルでは顕著であり、PSAレベルは、陽性対照(LHRH作動薬)グループにおいて、60%ほど減少した(図16、および表13〜16)。
【0223】
【表13】
【0224】
【表14】
【0225】
【表15】
【0226】
【表16】
前立腺体積を、研究期間にわたり、定期的に、TRUSによって測定した。治療の6週間後に取得された結果は、サルの前立腺への化合物IVおよび陽性対照(LHRH作動薬)の強力な作用を示す。化合物IVは、前立腺体積を、10mg/kg、および100mg/kg用量レベルでそれぞれ、25%および45%、有意に抑制し、前立腺体積は、陽性対照(LHRH作動薬)グループにおいて、28%減少した(図17、ならびに表17および18)。
【0227】
【表17】
【0228】
【表18】
前立腺体積における化合物IV関連の減少は、剖検において、前立腺重量の評価により裏付けられた。治療の13週間後に、化合物IVは、10および100mg/kg/日の動物において、平均前立腺重量をそれぞれ、24%および21%、有意に減少させた(図18B、ならびに表19および20)。
【0229】
【表19】
【0230】
【表20】
血小板凝集、プロトロンビン時間(PT)、または活性化部分的トロンボプラスチン時間(APTT)への明らかな作用は、認められなかった。
実施例17
ヒトへの化合物IVの研究
男性への化合物IVの作用を判定するための研究を実施した。コホート毎に12名の対象者を、100、300、600、および1000mgの化合物IVの用量で調査した。表21は、100、300、600、および1000mgの用量で化合物IVを投与することによる、男性における、LH、血清PSA、遊離テストステロン、および総テストステロンレベルの平均変化を表す。ヒトにおける用量依存性平均総テストステロンレベル(nmol/L)を、1〜11日間の期間で測定した(図19)。総テストステロンレベルは、600mgおよび1000mgの用量でそれぞれ、51.9%、47.9%低下した。
【0231】
ヒトにおける用量依存性平均LHレベル(IU/L)を、1〜10日間の期間で測定した(図20)。LHレベルは、100mg、300mg、600mg、および1000mgの用量でそれぞれ、20.7%、46.9%、27.6%、および29.2%低下した。
【0232】
ヒトにおける用量依存性平均遊離テストステロンレベル(pg/mL)を、1〜10日間の期間で測定した(図21)。遊離テストステロンレベルは、100mg、300mg、600mg、および1000mgの用量でそれぞれ、17.0%、18.5%、72.7%、および53.2%低下した。
【0233】
ヒトにおける用量依存性平均PSAレベル(μg/L)を、1〜10日間の期間で測定した(図22)。PSAレベルは、100mg、300mg、600mg、および1000mgの用量でそれぞれ、9.2%、24.4%、27.5%、および29.9%低下した。10および30mgの用量では、変化を示さなかった。
【0234】
【表21】
実施例18
化合物IVの生物学的利用率
化合物IVは、ラット、イヌ、サルへの経口投与後に迅速に吸収された。ラットにおける、化合物IVの経口生物学的利用率は、用量が投与される製剤によって、6%〜25%の範囲であった。ポリエチレングリコール300(PEG300)を使用する製剤は、一般に、脱イオン水中で希釈されたTween80中で調製されたマイクロエマルションよりも高い暴露を生じた。イヌでは、血漿濃度−時間プロファイルの目視検査は、最終相の第2のピークで分かるとおり、化合物IVが、腸肝再循環を受けることを示唆した。重要なことには、イヌでは、オスの30mg/kgのPEG300の経口用量グループにおける暴露は、LH抑制のラットモデルにおける前立腺減少への最大作用を産生するために必要な暴露を超過した。サルでは、予備の薬物動態研究は、7日間の期間にわたる、化合物IVの血漿濃度および血清テストステロンの抑制によって明らかなとおり、この種における経口生物学的利用率は、イヌの経口生物学的利用率に近似するか、または超過することを示唆した。総じて、これらのデータは、十分な経口暴露を、2つの非げっ歯類の動物種において達成して、所望の薬理作用を産生することができることを示唆する(AUCデータに基づく)。さらに、ラットおよびサルにおける内分泌データは、化合物IVの薬理作用が可逆性である(すなわち、化合部IVによる治療を停止した時に、テストステロンの血清濃度が、ベースラインまたは正常レベルに戻る)ことを示唆する。
実施例19
化合物IVの薬物動態
インビトロ(マウス、ラット、イヌ、サル、およびヒト)、およびインビボ(ラット)代謝研究からの予備データは、化合物IV、そのヒドロキシル化された代謝物、およびそのN−ジアルキル化された代謝物の共役が、動物およびヒトにおいて、化合物IVの全体の体内動態に寄与することを示唆する。種間比較の結果は、定性的のみであるが、非臨床種の全代謝物プロファイルが、ヒト肝ミクロソーム内に生成されたプロファイルを的確に反映することを示す。これらの結果に基づき、ラットおよびイヌはそれぞれ、薬理評価および毒性評価において、適切なげっ歯種および非げっ歯種である。インビトロ研究は、化合物IVが、関連CYP450イソ型(CYP1A2、CYP2B6、またはCYP3A4)を誘導せず、<30μMの濃度ではCYP1A2、CYP2C9、CYP2D6、またはCYP3A4/5を阻害しないことを示す。CYP2C9は、化合物IVによって阻害されるが、高濃度(Ki=8μM)のみにおいてであり、薬物動態的薬物−薬物相互作用の可能性は、高くないと見なされる。
実施例20
化合物IVの生物活性
化合物IVは、hERGチャネルへのインビトロ阻害作用(IC50≧300μM)をほとんど、または全く発揮しない。化合物は、インビトロの単離したイヌのプルキンエ線維中で、10および100μMの濃度で、APD50およびAPD90を、化合物用量依存的に減少させた。しかしながら、化合物IVは、遠隔計測されたイヌにおいて、いかなる用量(最大300mg/kg)でも、血行動態または心臓機能(血圧、心拍数、心電図形態、またはQT間隔)に影響を及ぼさなかった。神経薬理学的作用または経肺作用は、認められなかった。最大の30mg/kgまでの化合物IVの単一経口用量では、腎機能への有意な作用を示さなかった。尿排出量および尿中のカリウムおよび塩素の排出量の増大のみが、試験した最大用量(100mg/kg)で認められた。ラットにおいて、30〜300mg/kgの用量での化合物IVの経口投与は、蠕動の有意な増加をもたらし、ラットにおいて、30mg/kgの用量での化合物IVの経口投与は、消化管運動および胃内酸度の有意な増加をもたらした(恐らく、平滑筋への影響によるものではない)。
【0235】
化合物IVは、変異原性ではなく、インビトロのヒト末梢血リンパ球において、最大200μMまでの濃度で、構造的または数的染色体異常を誘発しなかった。化合物IVは、単一および反復経口投与(最大28日間)後に、ラットおよびイヌによる耐容性が良好であった。腎臓、肝臓、心臓、および他の非標的関連臓器において、病理学的変化は、認められなかった。最大28日間のオスまたはメスのイヌへの化合物IVの経口投与に伴う深刻な物理的兆候、体重効果、臨床的病理学変化、眼科的変化、心電図変化、または組織病理学的変化は、存在しなかった。
【0236】
本発明のある特徴を、本明細書に例証し、説明してきたが、当業者には、多くの修正、変更、および等価物が思い当たるであろう。したがって、添付の特許請求の範囲は、本発明の真の精神に当てはまる、すべてのかかる修正および変更を網羅することを意図していることを理解されたい。
【技術分野】
【0001】
本発明は、男性対象者において、黄体形成ホルモン(LH)の減少による、またはLHレベルと無関係である、テストステロンレベルを低下させる方法、および進行した前立腺癌の治療、抑制、発生を低減、重症度を軽減、または阻害する方法、および進行した前立腺癌の緩和治療の方法に関する。
【背景技術】
【0002】
エストロゲンは、組織および骨の維持にとって重要であり、そのために使用される内因性ホルモンおよび合成ホルモンの群を指す。エストロゲンは、生殖器系の発達および維持に関与する細胞過程における内分泌調節剤である。生殖に関する生物現象、閉経後のほてりの防止、および閉経後の骨粗しょう症の防止におけるエストロゲンの役割は、十分に確立されている。エストラジオールは、主な内因性ヒトエストロゲンであり、女性および男性の両方に見られる。
【0003】
エストロゲンおよび抗エストロゲンの生物学的作用は、2つの異なる細胞内受容体である、エストロゲン受容体アルファ(ERα)およびエストロゲン受容体ベータ(ERβ)を介して現れる。内因性エストロゲンは、典型的には、両方の受容体サブタイプの強力な活性剤である。例えば、エストラジオールは、乳房組織、骨組織、心臓血管組織、および中枢神経系組織を含む多くの組織において、ERα作動薬としての機能を果たす。選択的エストロゲン受容体モジュレータは、一般に、異なる組織において異なる機能を有する。例えば、SERMは、乳房において、ERα拮抗薬であり得るが、子宮、骨、および心臓血管系において、部分的にERα作動薬であり得る。したがって、エストロゲン受容体リガンドとしての機能を果たす化合物は、種々の病状および疾患を治療する上で有用である。
【0004】
前立腺癌は、米国において、男性で最も頻繁に診断される非皮膚癌のうちの1つであり、今年は18万件以上の新規症例、および2万9千の死亡件数が予想される、2番目の一般的な癌による死亡原因である。進行した前立腺癌を罹患する患者は、典型的に、黄体形成ホルモン放出ホルモン(LHRH)作動薬、または両側精巣摘除のいずれかによる、アンドロゲン除去療法(ADT)を受ける。エストロゲンがテストステロンの芳香族化に由来し、そのレベルがADTによって欠損するため、アンドロゲン除去療法は、テストステロンを減少させるだけでなく、エストロゲンレベルもまた低下させる。アンドロゲン除去療法誘発エストロゲン欠損は、ほてり、女性化乳房および乳房痛、骨量の減少、骨質および強度の低下、骨粗しょう症および致命的な骨折、有害な脂質変化、ならびにより高度の心臓血管疾患および心筋梗塞、ならびに鬱および他の情緒変化を含む、著しい副作用をもたらす。ADTのエストロゲン欠損の副作用の多くは、ERαによって媒介されると考えられている。
【0005】
酢酸ロイプロリド(Lupron(R))は、天然に存在するゴナドトロピン放出ホルモン(GnRHまたはLH−RH)の合成非ペプチド類似体である。酢酸ロイプロリドは、最終的に、下垂体によるLH分泌を抑制するLH−RH超作動薬である。酢酸ロイプロリドは、ゴナドトロピン分泌の強力阻害剤としての機能を果たし、卵巣および精巣のステロイド産生の抑制をもたらす。ヒトでは、酢酸ロイプロリドの投与は、黄体形成ホルモン(LH)および卵胞刺激ホルモン(FSH)の血中レベルの初期増加をもたらし、性腺ステロイド(男性におけるテストステロンおよびジヒドロテストステロン、ならびに閉経前の女性におけるエストロンおよびエストラジオール)の一過的なレベルの増加につながる。しかしながら、酢酸ロイプロリドの継続投与は、LHおよびFSHのレベルの低下をもたらす。男性では、テストステロンは、去勢レベル(50ng/dL以下)まで減少する。閉経前の女性では、エストロゲンは、閉経後レベルまで減少する。テストステロンは、前立腺の癌細胞に対する公知の刺激剤である。したがって、テストステロン分泌の抑制およびテストステロン作用の阻害は、前立腺癌療法の必要な構成要素である。酢酸ロイプロリドは、前立腺癌を治療するための去勢レベルまでの血清テストステロンの減少および低減である、LH抑制のために使用することができる。
【0006】
LHRH作動薬の導入前に、エストロゲン、主に、ジエチルスチルベストロール(DES)を介して、下垂体内のエストロゲン活性を増加させることによって、去勢テストステロンレベルを達成した。DESは、去勢レベルまでテストステロンを抑制する上でLHRH作動薬と同等に有効であった。DESで治療された患者は、ほてりまたは骨量の減少を有さなかったが、LHRH作動薬を用いるADTよりも高い比率で女性化乳房を有した。残念ながら、DESおよびエストラジオール等の非常に強力な純粋なエストロゲンは、しばしば、重度の心臓血管および血栓塞栓性合併症の高いリスクを伴い、それらの臨床使用を制限している。
【0007】
本発明の化合物は、テストステロンレベルを去勢レベルまで抑制し、骨量の減少、ほてり、および/または女性化乳房をもたらすことなく、血栓事象のリスクの増大を防止しながら、前立腺癌を治療するために使用し得る。
【発明の概要】
【0008】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、本明細書において下記に説明する、治療的有効量の式I〜XIIの化合物を投与することを含む。
【0009】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、本明細書において下記に説明する、治療的有効量の式I〜XIIの化合物を投与することを含み、血清総テストステロンの低下は、血清黄体形成ホルモンレベルの減少によって生じる。
【0010】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、本明細書において下記に説明する、治療的有効量の式I〜XIIの化合物を投与することを含み、血清総テストステロンの低下は、血清黄体形成ホルモンレベルの減少と無関係である。
【0011】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、本明細書において下記に説明する、治療的有効量の式I〜XIIの化合物を投与することを含み、式I〜XIIの該化合物の投与は、アンドロゲン除去療法(ADT)に伴う副作用が生じることを防止、または治療し、該対象者は、前立腺癌を有する。
【0012】
一実施形態では、本発明は、対象者において、アンドロゲン除去療法のための方法を提供し、本明細書において下記に説明する、治療的有効量の式I〜XIIの化合物を投与することを含み、該対象者は、前立腺癌を有する。
【0013】
一実施形態では、本発明は、進行した前立腺癌を抑制、発生を低減、重症度を軽減、または阻害する方法を提供し、本明細書において下記に説明する、治療的有効量の式I〜XIIの化合物を投与することを含む。
【0014】
一実施形態では、本発明は、進行した前立腺癌の緩和治療の方法を提供し、本明細書において下記に説明する、治療的有効量の式I〜XIIの化合物を投与することを含む。
本発明に関する主題を、具体的に、明細書のまとめの部分に示し、明確に請求する。しかしながら、本発明は、構成および操作方法双方に関して、その目的、特徴、および利点とともに、以下の添付の図面をとともに読む際に、詳細な説明を参照することにより、最も良く理解され得る。
【図面の簡単な説明】
【0015】
【図1】化合物IVの30mg/kgの経口投与(0日目の第1の投与量)後の無損傷のオスのサルにおける血清テストステロン(太線)および総アンドロゲン(点線)レベルを図示する(実施例8を参照)。
【図2】化合物IV(0.3、1、10、30mg/kg)で処置された無損傷のラットにおけるテストステロンレベルを図示する。「I」は、無損傷の媒体対照に対するP<0.05を示す。BLOQ値を、0.08ng/mLの定量限界で図式的に表す(実施例9を参照)。
【図3】17β−HSD5酵素活性への化合物IVの阻害作用を図示する(実施例12を参照)。
【図4】DES、17β−エストラジオール(E2)、および化合物IVの存在下のヒト血小板のインビトロ凝集を図示する。多血小板血漿(PRP)を、0.3ユニットのトロンビンとの凝集を誘発する前に、媒体、E2、DES、または化合物IVで、30秒間、培養した。凝集を、5分間監視し、媒体対照に対する割合で表示した(実施例13を参照)。
【図5】化合物II〜XIIの調製のための一般合成スキーム(実施例1を参照)。
【図6】化合物IVの調製のための合成スキーム(実施例2を参照)。
【図7】化合物VIの調製のための合成スキーム(実施例3を参照)。
【図8】化合物IXおよびXの調製のための合成スキーム(実施例5を参照)。
【図9】3mg/kg、10mg/kg、および300mg/kgの用量で、化合物IVで処置された無損傷ラットにおける、24時間後、72時間後、および168時間後のテストステロンレベルを図示する(実施例9を参照)。
【図10A】0.3mg/kg、1mg/kg、3mg/kg、10mg/kg、および30mg/kgの用量の化合物IVを用いて処置された、無損傷ラットおよび精巣摘除された(ORX)ラットのLHレベルを図示する。「I」は、無損傷の媒体対照に対するP<0.05を示す。「O」は、ORX媒体対照に対するP<0.05を示す。BLOQ値を、定量限界0.08ng/mLで図式的に表す(実施例9を参照)。
【図10B】0.3mg/kg、1mg/kg、3mg/kg、10mg/kg、および30mg/kgの用量の化合物IVを用いて処置された、無損傷ラットおよび精巣摘除された(ORX)ラットのFSHレベルを図示する。「I」は、無損傷の媒体対照に対するP<0.05を示す。「O」は、ORX媒体対照に対するP<0.05を示す。BLOQ値を、定量限界0.08ng/mLで図式的に表す(実施例9を参照)。
【図10C】0.3mg/kg、1mg/kg、3mg/kg、10mg/kg、および30mg/kgの用量の化合物IVを用いて処置された、無損傷ラットおよび精巣摘除された(ORX)ラットのテストステロンレベルを図示する。「I」は、無損傷の媒体対照に対するP<0.05を示す。「O」は、ORX媒体対照に対するP<0.05を示す。BLOQ値を、定量限界0.08ng/mLで図式的に表す(実施例9を参照)。
【図10D】0.3mg/kg、1mg/kg、3mg/kg、10mg/kg、および30mg/kgの用量の化合物IVを用いて処置された、無損傷ラットおよび精巣摘除された(ORX)ラットの前立腺重量レベルを図示する。「I」は、無損傷の媒体対照に対するP<0.05を示す。「O」は、ORX媒体対照に対するP<0.05を示す。BLOQ値を、定量限界0.08ng/mLで図式的に表す(実施例9を参照)。
【図10E】0.3mg/kg、1mg/kg、3mg/kg、10mg/kg、および30mg/kgの用量の化合物IVを用いて処置された、無損傷ラットおよび精巣摘除された(ORX)ラットの精嚢重量レベルを図示する。「I」は、無損傷の媒体対照に対するP<0.05を示す。「O」は、ORX媒体対照に対するP<0.05を示す。BLOQ値を、定量限界0.08ng/mLで図式的に表す(実施例9を参照)。
【図10F】0.3mg/kg、1mg/kg、3mg/kg、10mg/kg、および30mg/kgの用量の化合物IVを用いて処置された、無損傷ラットおよび精巣摘除された(ORX)ラットの肛門挙筋重量を図示する。「I」は、無損傷の媒体対照に対するP<0.05を示す。「O」は、ORX媒体対照に対するP<0.05を示す。BLOQ値を、定量限界0.08ng/mLで図式的に表す(実施例9を参照)。
【図11】化合物IV(A)およびDES(B)を異なる用量で投与することによる、無損傷ラットおよびORXラットにおける前立腺の大きさを図示する(実施例15を参照)。
【図12A】DESと化合物IVとの差異を図示し、DESは、グルココルチコイド受容体(GR)と交差反応するが、化合物IVは交差反応しない(実施例15を参照)。
【図12B】DESと化合物IVとの差異を図示し、DESは、アンドロゲン受容体(AR)と交差反応する。それは、AR作用を軽度に刺激し、軽度に阻害する(すなわち、それは、部分的作動薬/拮抗薬である)が、化合物IVは、それらをしない(実施例15を参照)。
【図12C】DESと化合物IVとの差異を図示し、DESは、エストロゲン関連受容体(ERR)トランス活性化を無効にするが、化合物IVは無効にしない(実施例15を参照)。
【図13】モルヒネ禁断モデルにおける、5mg/kg、10mg/kg、15mg/kg、および30mg/kgの用量での、ほてりの軽減への化合物IVの作用を図示する。グループ当たりN=7匹の動物。17β−E2を、100%のDMSO中で5mg/kgで使用した(実施例14を参照)。
【図14】化合物IVを91日間投与することによる、サルの用量依存性の体重(kg)低下(100mg/kgで約20%)を図示する。女性化乳房または高エストロゲン性の兆候は認められなかった(実施例16を参照)。
【図15】陽性対照(LHRH作動薬)と比較した、化合物IVの連日経口投与後のサルにおける、用量依存性の血清テストステロンレベルの減少(ng/mL)を図示する。点線は、化学的に去勢された患者のテストステロンレベルを示し、太い点線は、外科的に去勢されたサルのテストステロンレベルを示す(実施例16を参照)。
【図16】ベースライン時に、および28日目に化合物IVを投与することによる、サルにおける、用量依存性の前立腺特異的抗原(PSA)レベル(ng/mL)を図示する。PSAレベルは、化合物IVの処置で顕著に減少した(実施例16)。
【図17】陽性対照(LHRH作動薬)と比較した、6週間目に化合物IVを投与することによる、サルにおける、経直腸超音波(TRUS)を使用した用量依存性前立腺体積を図示する(実施例16を参照)。
【図18】化合物IV(図18A)投与することによる、90日目の、用量依存性の臓器重量(前立腺、精嚢、および精巣)を、サルの対照との比率で図示する。化合物IV(図18B)の連日経口投与後のサルにおける13週間目の剖検での前立腺重量(実施例16を参照)。
【図19】化合物IV(100mg、300mg、600mg、および1000mg)を投与することによる、1〜11日間の期間のヒトにおける用量依存性の平均総テストステロンレベル(nmol/L)を図示する(実施例17を参照)。
【図20】化合物IV(100mg、300mg、600mg、および1000mg)を投与することによる、1〜10日間の期間のヒトにおける用量依存性の平均LHレベル(IU/L)を図示する。(実施例17を参照)。
【図21】化合物IV(100mg、300mg、600mg、および1000mg)を投与することによる、1〜10日間の期間のヒトにおける用量依存性の平均遊離テストステロンレベル(pg/mL)を図示する。(実施例17を参照)。
【図22】化合物IV(100mg、300mg、600mg、および1000mg)を投与することによる、1〜10日間の期間のヒトにおける用量依存性の平均PSAレベル(μg/L)を図示する。(実施例17を参照)。
【図23】化合物IVの投与の14日間の回復後の無損傷ラットにおける用量依存性血清テストステロンレベル(ng/mL)を図示する。「I」は、無損傷の媒体対照に対するP<0.05を示す。(実施例10を参照)。
【発明を実施するための形態】
【0016】
図解を簡潔および明確にするために、図面に示す要素は、必ずしも原寸に比例して描かれてはいないことを理解されたい。例えば、要素のうちのいくつかの寸法は、明確にするために、他の要素に対して誇張され得る。さらに、適切であると考えられる場合では、参照番号は、対応する要素または類似要素を指すために図面で反復され得る。
【0017】
以下の詳細な説明では、本発明についての完全な理解を提供するために、多くの具体的な詳細を説明する。しかしながら、当業者は、本発明がこれらの具体的な詳細なく実践し得ることを理解するであろう。他の例では、本発明を曖昧にしないよう、周知の方法、手順、および構成要素を詳細に説明していない。
【0018】
一実施形態では、本明細書に説明する化合物、および/またはそれを含む組成物は、男性対象者において、血清総テストステロンレベルを低下させるために使用され得る。
一実施形態では、本明細書に説明する化合物、および/またはそれを含む組成物は、男性対象者において、血清総テストステロンレベルを低下させるために使用され得、血清総テストステロンの低下は、血清黄体形成ホルモン(LH)レベルの減少により生じる。
【0019】
一実施形態では、本明細書に説明する化合物、および/またはそれを含む組成物は、男性対象者において、血清総テストステロンレベルを低下させるために使用され得、血清総テストステロンの低下は、血清黄体形成ホルモンレベルの減少と無関係である。
【0020】
一実施形態では、本発明は、男性対象者において血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、それは式I:
【0021】
【化1】
の構造で表され、式中、
Yは、C(O)またはCH2であり、
R1、R2は、独立して、水素、ハロゲン、ヒドロキシル、アルコキシ、シアノ、ニトロ、CF3、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、O−Alk−NR5R6、またはO−Alk−複素環であり、複素環は、3〜7員の置換または非置換複素環式環、任意に芳香族であり、
R3、R4は、独立して、水素、ハロゲン、ヒドロキシアルキル、ヒドロキシル、アルコキシ、シアノ、ニトロ、CF3、NHCOR、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、または保護ヒドロキシルであり、
Rは、アルキル、水素、ハロアルキル、ジハロアルキル、トリハロアルキル、CH2F、CHF2、CF3、CF2CF3、アリール、フェニル、ハロゲン、アルケニル、CN、NO2、またはOHであり、
R5およびR6は、独立して、水素、フェニル、1〜6個の炭素原子のアルキル基、3〜7員のシクロアルキル、3〜7員の複素環、5〜7員のアリールであるか、またはR5およびR6は、窒素原子とともに、3〜7員の環を形成し、
jおよびkは、独立して、1から4であり、
Alkは、1〜7個の炭素の直鎖アルキル、1〜7個の炭素の分岐鎖アルキル、または3〜8個の炭素の環状アルキルである。
【0022】
本明細書に説明する方法のさらなる実施形態では、式Iの化合物は、式IA:
【0023】
【化2】
によって表され、式中、R1、R2、R3、R4、j、およびkは、式Iについて定義するとおりである。
【0024】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、その化合物は式II:
【0025】
【化3】
で表される。
【0026】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、その化合物は式III:
【0027】
【化4】
で表される。
【0028】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、その化合物は式IV:
【0029】
【化5】
で表される。
【0030】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、その化合物は式V:
【0031】
【化6】
で表される。
【0032】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、その化合物は式VI:
【0033】
【化7】
で表される。
【0034】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、その化合物は式VII:
【0035】
【化8】
で表される。
【0036】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、その化合物は式VIII:
【0037】
【化9】
で表される。
【0038】
一実施形態では、本発明は、前立腺癌を有する男性対象者において、黄体形成ホルモン(LH)レベルの減少によって、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、その化合物は式IX:
【0039】
【化10】
で表される。
【0040】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、その化合物は式X:
【0041】
【化11】
で表される。
【0042】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、その化合物は式XI:
【0043】
【化12】
で表される。
【0044】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、その化合物は式XII:
【0045】
【化13】
で表される。
【0046】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の式IA、I〜XIIの化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含む。別の実施形態では、男性対象者は、前立腺癌を有する。別の実施形態では、血清総テストステロンは、約100ng/dL未満に低減される。別の実施形態では、血清総テストステロンは、約50ng/dL未満に低減される。別の実施形態では、血清総テストステロン濃度は、約25ng/dL未満に低減される。
【0047】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の式IA、I〜XIIの化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、血清総テストステロンの低下は、血清黄体形成ホルモン(LH)レベルの減少によって生じる。別の実施形態では、男性対象者は、前立腺癌を有する。別の実施形態では、血清総テストステロンは、約100ng/dL未満に低減される。別の実施形態では、血清総テストステロンは、約50ng/dL未満に低減される。別の実施形態では、血清総テストステロン濃度は、約25ng/dL未満に低減される。
【0048】
一実施形態では、本発明は、男性対象者において、遊離血清テストステロンレベルを低下させる方法を提供し、治療的有効量の式IA、I〜XIIの化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、遊離血清テストステロンの低下は、血清黄体形成ホルモン(LH)レベルの減少によって生じる。別の実施形態では、男性対象者は、前立腺癌を有する。
【0049】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の式IA、I〜XIIの化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、血清総テストステロンの低下は、血清黄体形成ホルモン(LH)レベルの減少と無関係である。別の実施形態では、男性対象者は、前立腺癌を有する。別の実施形態では、血清総テストステロンは、約100ng/dL未満に低減される。別の実施形態では、血清総テストステロンは、約50ng/dL未満に低減される。別の実施形態では、血清総テストステロン濃度は、約25ng/dL未満に低減される。
【0050】
一実施形態では、本発明は、男性対象者において、遊離血清テストステロンレベルを低下させる方法を提供し、治療的有効量の式IA、I〜XIIの化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、遊離血清テストステロンレベルの低下は、血清黄体形成ホルモン(LH)レベルの減少と無関係である。別の実施形態では、男性対象者は、前立腺癌を有する。
【0051】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベル、または遊離血清テストステロンレベルを低下させる方法を提供し、該男性対象者は、前立腺癌を有する。別の実施形態では、該対象者は、進行した前立腺癌を有する。
【0052】
一実施形態では、テストステロンの血清濃度の減少は、可逆的であり、本発明の化合物での治療後にベースラインレベルに戻る。
別の実施形態では、テストステロンの血清濃度は、図23および実施例10に従う化合物IVでの治療後に可逆的である。
【0053】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の式IA、I〜XIIの化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含む。別の実施形態では、血清総テストステロンは、約100ng/dL未満に低下される。別の実施形態では、血清総テストステロンは、約50ng/dL未満に低減される。別の実施形態では、血清総テストステロンは、約25ng/dL未満に低減される。別の実施形態では、血清総テストステロンは、約75ng/dL未満に低減される。別の実施形態では、血清総テストステロンは、約75ng/dL〜100ng/dLに低減される。別の実施形態では、血清総テストステロンは、約50ng/dL〜75ng/dLに低減される。別の実施形態では、血清総テストステロンは、約40ng/dL〜50ng/dLに低減される。別の実施形態では、血清総テストステロン濃度は、約25ng/dL〜50ng/dLに低減される。別の実施形態では、血清総テストステロンは、約40ng/dL〜60ng/dLに低減される。
【0054】
テストステロンは、「遊離」(つまり、生物学的に利用可能、および未結合)、または「総」(タンパク質結合したものの割合を含み、利用不可能)血清レベルとして測定することができる。40歳以上の前立腺癌を有さない男性は、250ng/dL(<8.7nmol/L)未満の総テストステロンレベル、または0.75ng/dL(<0.03nmol/L)未満の遊離テストステロンレベルを有し、低いテストステロンレベルを示す。
【0055】
一実施形態では、本発明は、前立腺癌を有する男性対象者において、黄体形成ホルモン(LH)レベルの減少と無関係に、またはLHレベルの減少によって、血清総テストステロンレベル、および/または遊離テストステロンレベルを低下させる方法を提供する。別の実施形態では、テストステロンレベルの変化は、治療前のレベルからの減少でなければならない。別の実施形態では、血清総テストステロンレベルは、100ng/dL未満に低減される。別の実施形態では、血清総テストステロンは、50ng/dL未満に低減される。別の実施形態では、血清総テストステロンは、25ng/dL未満に低減される。別の実施形態では、遊離テストステロンは、2ng/dL未満に低減される。別の実施形態では、遊離テストステロンは、約1ng/dL未満に低減される。別の実施形態では、遊離テストステロンは、約0.5ng/dL未満に低減される。別の実施形態では、遊離テストステロンは、約0.25ng/dL未満に低減される。
【0056】
遊離血清テストステロンレベルおよび血清総テストステロンレベルを決定する方法は、血液試験によって、一連の治療期間中にテストステロンレベルを監視することを含む。総テストステロンは、担体タンパク質(アルブミン、SHBG、トランスコルチン、トランスフェリン)に結合した循環テストステロンと、遊離/未結合ホルモンとの組み合わせである。総テストステロンレベルは、体内のホルモンを運搬する血中のタンパク質レベル、年齢、肥満、および一般に使用される試験方法に伴う干渉を含む、種々の要因によって影響される場合がある。
【0057】
遊離テストステロン(FT)を測定するために使用可能な方法は、類似体トレーサを使用して、複雑である(平衡透析および算出された遊離テストステロン)か、または単純であり得る(市販のFTキット「Coat−A−Count」)。別の実施形態では、総テストステロン血清レベル、および遊離テストステロン血清レベルの測定は、総テストステロンおよびSHBG(例えば、Irma−Count,DPC)の同時測定、次いで、算出された遊離テストステロン(CFT)によって達成することができる。別の実施形態では、総テストステロンおよび遊離テストステロンの測定は、当業者の知識に従う。
【0058】
一実施形態では、男性対象者において、血清総テストステロンレベル、または遊離血清テストステロンレベルを低下させる方法を提供し、治療的有効量の1つまたは複数の別の形態のADTと式IA、I〜XIIの化合物またはその異性体との組み合わせ、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含む。別の実施形態では、総テストステロン、または遊離血清テストステロンの低減は、血清黄体形成ホルモン(LH)レベルの減少によって生じる。別の実施形態では、総テストステロンレベル、または遊離血清テストステロンレベルは、血清黄体形成ホルモンレベルの減少と無関係である。
【0059】
本発明の方法は、別の形態のADTと本発明の化合物との組み合わせを投与することを含む。一実施形態では、別の形態のADTは、LHRH作動薬を含む。別の実施形態では、LHRH作動薬は、酢酸ロイプロリド(Lupron(R))(米国特許第5,480,656号、同第5,575,987号、同第5,631,020号、同第5,643,607号、同第5,716,640号、同第5,814,342号、同第6,036,976号はすべて、参照することにより、本明細書に組み込まれる)、または酢酸ゴセレリン(Zoladex(登録商標))(米国特許第7,118,552号、同第7,220,247、同第7,500,964号はすべて、参照することにより、本明細書に組み込まれる)を含む。一実施形態では、別の形態のADTは、LHRH拮抗薬を含む。別の実施形態では、LHRH拮抗薬は、デガレリクスを含む。一実施形態では、別の形態のADTは、抗アンドロゲンを含む。別の実施形態では、抗アンドロゲンは、ビカルタミド、フルタミド、フィナステリド、デュタステリド、MDV3100、ニルタミド、クロルマジノン、またはそれらの任意の組み合わせを含む。
【0060】
一実施形態では、本発明の方法は、治療的有効量の抗アンドロゲン、および本発明の化合物を投与することを含む。一実施形態では、本発明の方法は、治療的有効量のLHRH作動薬、および本発明の化合物を投与することを含む。一実施形態では、本発明の方法は、治療的有効量の抗アンドロゲン、LHRH作動薬、および本発明の化合物を投与することを含む。
【0061】
一実施形態では、本発明は、男性対象者において、アンドロゲン除去療法(ADT)を生成する目的で、前立腺癌を有する男性対象者において、黄体形成ホルモン(LH)レベルの減少により、または黄体形成ホルモンレベルの減少と無関係で、血清総テストステロンレベル、および/または遊離テストステロンレベルを低下させる方法を提供し、治療的有効量の式IA、I〜XIIの化合物を投与することを含む。別の実施形態では、化合物は、化合物IVである。
【0062】
別の実施形態では、本発明は、対象者において、アンドロゲン除去療法(ADT)のための方法を提供し、治療的有効量の式IA、I〜XIIの化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含む。別の実施形態では、該対象者は、前立腺癌を有する。別の実施形態では、化合物は、化合物IVである。
【0063】
別の実施形態では、ADTを、前立腺癌を治療するため、前立腺癌の進行を遅延するため、または前立腺癌の再発を防止する、および/または治療するために使用する。
一実施形態では、本発明は、前立腺癌を治療する、前立腺癌の進行を遅延する、前立腺癌の再発の防止する、および/または治療する方法を提供し、本発明の化合物を投与することを含む。別の実施形態では、LHRH類似体、可逆性抗アンドロゲン(ビカルタミドまたはフルタミド等)、抗エストロゲン、抗癌剤、5−アルファ還元酵素阻害剤、アロマターゼ阻害剤、プロゲスチン、選択的アンドロゲン受容体モジュレータ(SARMS)、または他の核ホルモン受容体を介して作用する薬剤と組み合わせて、本発明の化合物を投与する。
【0064】
一実施形態では、本発明は、前立腺癌を治療し、LHレベルを減少させることによるか、またはLHレベルの減少と無関係に、血清総テストステロン、および/または遊離血清テストステロンレベルを減少させる方法を提供し、式IA、I〜XIIの化合物を投与することを含む。別の実施形態では、化合物IVを投与する。
【0065】
アンドロゲン除去療法は、テストステロンを減少させるだけでなく、エストロゲンレベルもまた、テストステロンの芳香族化に由来するため、低減される。アンドロゲン除去療法により誘発されるエストロゲン欠乏は、ほてり、女性化乳房、および乳房痛、骨量の減少、骨質および強度の低下、骨粗しょう症、骨減少症、および致命的な骨折、有害な脂質変化、およびより高い心臓血管疾患、および心筋梗塞、性欲減弱、インポテンス、筋肉量の低下(サルコペニア)、疲労、認知機能障害、ならびに鬱および他の情緒変化を含む、著しい副作用をもたらす。
【0066】
他の実施形態では、本発明は、ADTに伴う任意の疾患、障害、または症状を治療する方法を提供する。他の実施形態では、本発明は、治療する方法を提供する。テストステロン枯渇に伴う任意の疾患、障害、または症状を治療する方法を提供する。各疾患、障害、または症状は、本発明の別個の実施形態を表す。
【0067】
一実施形態では、本発明は、男性対象者において、血清総テストステロンレベルを低下させる方法を提供し、治療的有効量の式IA、I〜XIIの化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することを含み、該式IA、I〜XIIの化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することは、アンドロゲン除去療法(ADT)に伴う副作用が発生することを防止し、抑制し、発症を低減し、阻害し、または治療し、該対象者は、前立腺癌を有する。別の実施形態では、総テストステロンレベル、または遊離血清テストステロンレベルの低減は、LHレベルを減少させることによるものであるか、またはLHレベルの減少と無関係である。
【0068】
一実施形態では、本発明の化合物を投与することは、従来のアンドロゲン除去療法(ADT)に伴う典型的な副作用の発生を抑制し、発症を低減し、阻害し、または治療する。別の実施形態では、対象者は、前立腺癌を有する。副作用のかかる防止および/または減少は、プラシーボまたは対照群と比較した、相対的なものである。一実施形態では、従来のアンドロゲン除去療法(ADT)に伴う典型的な副作用は、ほてり、女性化乳房、骨塩密度の低下、および骨折の増加を含む。別の実施形態では、本発明の化合物を投与することは、従来の形態のアンドロゲン除去療法(ADT)を使用すると認められるであろう、ほてりが発生することを防止する。別の実施形態では、本発明の化合物を投与することは、従来の形態のアンドロゲン除去療法(ADT)を使用すると認められるであろう、女性化乳房が発生することを防止する。別の実施形態では、本発明の化合物を投与することは、従来の形態のアンドロゲン除去療法(ADT)を使用すると認められるであろう、骨塩密度(BMD)の低下が発生することを防止する。別の実施形態では、本発明の化合物を投与することは、従来の形態のアンドロゲン除去療法(ADT)を使用すると認められるであろう、骨折の増加が発生することを防止する。別の実施形態では、骨折の増加は、病理学的骨折、非外傷性骨折、脊椎骨折、非脊椎骨折、新規形態学的骨折、臨床骨折、またはそれらの組み合わせである。
【0069】
一実施形態では、「従来のアンドロゲン除去療法」という用語は、外科医が精巣を除去する、精巣摘除(外科的去勢)を対象とする。別の実施形態では、「従来のアンドロゲン除去療法」という用語は、黄体形成ホルモン放出ホルモン(LHRH)類似体を投与することを対象とし、これらの薬剤は、精巣によって生成されるテストステロンの量を低下させる。米国で使用可能なLHRH類似体の例は、ロイプロリド(Lupron,Viadur(登録商標),Eligard(登録商標))、ゴセレリン(Zoladex(登録商標))、トリプトレリン(Trelstar)、およびヒストレリン(Vantas)を含む。別の実施形態では、「従来のアンドロゲン除去療法」という用語は、抗アンドロゲンを投与することを対象とし、抗アンドロゲンは、任意のアンドロゲンを使用する身体の能力を阻止する。精巣摘除後、またはLHRH類似体を用いた治療中であっても、少量のアンドロゲンは、依然として、副腎によって生成される。抗アンドロゲン薬剤の例は、フルタミド(Eulexin)、ビカルタミド(Casodex(登録商標))、およびニルタミド(Nilandron)を含む。別の実施形態では、「従来のアンドロゲン除去療法」という用語は、アバレリックス(Plenaxis(登録商標))等の黄体形成ホルモン放出ホルモン(LHRH)拮抗薬を投与することを対象とし、デガレリックス(Firmagon(登録商標))は、2008年にFDAによって、進行した前立腺癌を治療するために使用が認可された、新規LHRH拮抗薬である。別の実施形態では、「従来のアンドロゲン除去療法」という用語は、フィナステライド(Proscar(登録商標))およびデュタステリド(Avodart(登録商標))等の5α−還元酵素阻害剤を投与することを対象とし、5α−還元酵素阻害剤は、テストステロンを、より活性的なアンドロゲン、5α−ジヒドロテストステロン(DHT)に転換する身体の能力を阻止する。別の実施形態では、「従来のアンドロゲン除去療法」という用語は、ケトコナゾール(Nizoral(登録商標))等のテストステロン生合成の阻害剤を投与することを対象とする。別の実施形態では、「従来のアンドロゲン除去療法」という用語は、ジエチルスチルベストロール、または17β−エストラジオール等のエストロゲンを投与することを対象とする。
【0070】
一実施形態では、「ほてり」という用語は、上半身、または全身の急激な熱感、顔面および首筋の紅潮、胸部、背部、および腕に発現する赤色斑、多量の発汗、寒気等を指す。
一実施形態では、「女性化乳房」という用語は、乳房の腺構成要素の増殖から生じる、男性乳房の両性肥大を指し、疼痛を伴う場合、または伴わない場合がある。女性化乳房は、乳首から同心円状に伸びるゴム状または硬い腫瘤の存在によって、臨床的に定められている。擬似女性化乳房、または脂肪堆積(lipomastia)として公知の病状は、腺増殖のない脂肪沈着を特徴とする。女性化乳房は、通常、両側性であるが、片側性である場合もある。
【0071】
一実施形態では、本発明の方法は、テストステロンの減少によって、骨量の減少およびほてりをもたらさずに、前立腺癌または進行した前立腺癌を有する男性を治療することを対象とする。別の実施形態では、本発明の方法は、IA、I〜XIIの化合物を活用し、化合物は、前立腺癌のための現在のアンドロゲン枯渇療法(ADT)に一般である、骨量の減少およびほてり等の副作用をもたらさずに、前立腺癌の一次刺激であるテストステロンを減少させる可能性を有する。
【0072】
別の実施形態では、下記の表8(実施例11)は、化合物IVを投与することによる、骨量の減少をもたらさないテストステロンの減少を表す。
一実施形態では、本発明の方法は、式IA、I〜XIIの化合物を投与することによる、テストステロンレベルの減少を対象とし、それはさらに、進行した前立腺癌を治療する。一実施形態では、本発明の方法は、式IA、I〜XIIの化合物を投与することによる、テストステロンレベルの減少を対象とし、それはさらに、発症を抑制し、減少させ、重症度を軽減し、または進行した前立腺癌を阻害する。一実施形態では、本発明の方法は、式IA、I〜XIIの化合物を投与することによる、テストステロンレベルの減少を対象とし、それはさらに、進行した前立腺癌の緩和治療を提供する。
【0073】
一実施形態では、本発明の方法は、進行した前立腺癌を治療することを対象とする。一実施形態では、本発明の方法は、発症を抑制し、減少させ、重症度を軽減し、または進行した前立腺癌を阻害することを対象とする。一実施形態では、本発明の方法は、進行した前立腺癌の緩和治療を対象とする。別の実施形態では、本発明の方法は、IA、I〜XIIの化合物を活用する。別の実施形態では、本発明は、進行した前立腺癌を治療することを対象とする。別の実施形態では、本発明は、進行した前立腺癌を抑制することを対象とする。別の実施形態では、本発明は、進行した前立腺癌の発症を減少させることを対象とする。別の実施形態では、本発明は、進行した前立腺癌の重症度を軽減することを対象とする。別の実施形態では、本発明は、進行した前立腺癌を阻害することを対象とする。
【0074】
「進行した前立腺癌」という用語は、前立腺に由来し、精嚢、骨盤リンパ節、もしくは骨、または身体の他の部分を含むように、周辺組織等の前立腺を超えて広く転移している転移性癌を指す。前立腺癌病理学は、悪性度の増加の順に1〜5のグリーソン分類で分類される。別の実施形態では、前立腺癌による進行性疾患および/または死の有意なリスクを有する患者は、定義に含まれなければならず、IIBという低い疾患段階の前立腺被膜の外側に癌を有する任意の患者は、明らかに「進行した」疾患を有する。
【0075】
一実施形態では、本明細書に提供する方法および/または本明細書に提供する化合物の使用は、視床下部−下垂体−精巣軸(HPT軸)上のフィードバックを提供する上で有効である。フィードバックは、1つの臓器または組織内に生成された物質の、それ自体の活性に影響を及ぼす別の臓器または組織の活性を調節する能力を指す。一実施形態では、視床下部−下垂体−精巣軸(HPT軸)上のフィードバックは、LHレベルの減少をもたらす。一実施形態では、視床下部−下垂体−精巣軸(HPT軸)上のフィードバックは、血清総テストステロンレベルの減少をもたらす。一実施形態では、視床下部−下垂体−精巣軸(HPT軸)上のフィードバックは、遊離血清テストステロンレベルの減少をもたらす。一実施形態では、視床下部−下垂体−精巣軸(HPT軸)上のフィードバックは、アンドロゲンの血清レベル、組織レベル、または腫瘍レベルの減少をもたらす。
【0076】
視床下部−下垂体−精巣(HPT)軸は、視床下部、下垂体腺、および精巣内のホルモンレベルを調節する、内分泌生理学系を指す。LHRH(黄体形成ホルモン放出ホルモン)は、視床下部によって放出され、下垂体を刺激して、LHおよびFSH(ゴナドトロピン)を合成して分泌させる。次いで、LHおよびFSHは、精巣に作用し、テストステロンおよび精子産生を刺激する。次いで、テストステロンは、視床下部LHRH分泌に直接的な負のフィードバック作用、および下垂体LHおよびFSH産生に間接的負のフィードバックを有する。エストロゲン、アンドロゲン、および血清タンパク質(例えば、インヒビン)はまた、LHRH分泌、ならびにLHおよびFSHの分泌に負の影響を有する。
【0077】
下垂体腺は、体内のテストステロンのレベルを制御する、1つの腺である。
テストステロンレベルが低い時、下垂体腺は、黄体形成ホルモン(LH)を放出する。このホルモンは、
精巣を誘発して、より多くのテストステロンを生成させる。テストステロンのレベルは、思春期中に増大する。テストステロンのレベルは、約20歳〜40歳で最高であり、次いで、年配の男性では、徐々に少なくなる。女性は、男性と比較すると、体内にはるかに少量のテストステロンを有する。しかし、テストステロンは、男性および女性の体内にわたって重要な役割を担う。それは、脳、骨、筋肉量、脂肪分布、血管系、エネルギーレベル、生殖器組織、および性機能に影響を及ぼす。循環テストステロンの大半は、性ホルモン結合グロブリンと称されるタンパク質、またはアルブミンと称される別の血清タンパク質に結合している。結合しない(または「遊離」)テストステロンもまた、臨床的に判定することができる。
【0078】
別の実施形態では、血清黄体形成ホルモンレベルの減少と無関係に血清総テストステロンまたは遊離血清テストステロンレベルを低下させるのは、性ホルモン結合グロブリン(SHBG)の増加によるものである。別の実施形態では、血清黄体形成ホルモンレベルの減少と無関係な遊離テストステロンレベルを低下させるのは、性ホルモン結合グロブリン(SHBG)の増加によるものである。別の実施形態では、血清黄体形成ホルモン(LH)レベルの減少と無関係に血清総テストステロンレベルまたは遊離血清テストステロンレベルを低下させるのは、精巣内のライディッヒ細胞によるテストステロン産生または分泌の阻害によるものである。別の実施形態では、血清黄体形成ホルモン(LH)レベルの減少と無関係に血清総テストステロンレベルまたは遊離血清テストステロンレベルを低下させるのは、副腎ステロイド産生の減少によるものである。
【0079】
一実施形態では、本明細書に説明する化合物、および/またはそれらを含む組成物は、黄体形成ホルモン(LH)レベルの減少のために使用し得る。別の実施形態では、本発明の化合物および/または組成物を使用して、内因性性ホルモンを減少させ得る。
【0080】
ヒドロキシステロイド脱水素酵素(HSD)ファミリー成員は、循環ステロイドの変換に関与する。17β−HSD5は、アンドロステンジオンをテストステロンに、エストロンをエストラジオールに転換する。加えて、それはまた、プロスタグランジン合成に関与する。一実施形態では、本発明の化合物は、HSDを阻害し、具体的には、17β−ヒドロキシステロイド脱水素酵素5(17β−HSD5)阻害である。かかる阻害は、末梢/性腺外テストステロン合成を防止することによって、ADTにおいて有用であり得、それは、HPT軸制御を逃れ、総テストステロンまたは遊離血清テストステロンの不完全な減少をもたらし得るか、または局所的に上昇した細胞内テストステロンレベルを可能にし得、それらのいずれかは、ADTにおいて有害である可能性がある。
【0081】
LHRH作動薬療法、すなわち、黄体形成ホルモン放出ホルモン作動薬(LHRH)、またはその類似体の投与によって達成される、アンドロゲン除去療法(ADT)は、下垂体からのゴナドトロピン放出の初期刺激、および精巣からのテストステロン産生(「フレア反応」と称される)、およびそれに続くゴナドトロピン放出の低下、ならびにテストステロンレベル、およびエストロゲンレベルの両方の低下をもたらす。LHRH作動薬療法によって引き起こされた「フレア反応」は、アンドロゲン/テストステロンレベルの増加により、前立腺癌の治療へのマイナスの影響を有する。加えて、LHRH療法には、糖尿病および心臓血管疾患のリスクの増大が伴う(Smith(2008)Current Prostate Reports.6:149−154)。
【0082】
LHRH療法のフレア反応を克服するために、LHRH/抗アンドロゲン療法アプローチと組み合わせた抗アンドロゲン単独療法(ビカルタミド、フルタミド、クロルマジノン)、およびLHRH拮抗薬(デガレリクス)が示唆されている(Suzuki et al.,(2008)Int.J.Clin.Oncol.13:401−410、Sharifi,N.et al.,(2005)JAMA。294(2):238−244)。抗アンドロゲン単独療法は、対象者におけるアンドロゲンレベルを減少させない。ビカルタミド抗アンドロゲン単独療法は、骨転移を有する前立腺癌の患者において、ADTよりも有効性が少ないことが分かっている。加えて、ビカルタミド療法で認められた悪影響は、乳房圧痛および乳房肥大(女性化乳房および乳房痛)を含む(Suzuki et al.,同書)。抗アンドロゲン療法に伴う付加的なリスクは、肝トランスアミナーゼの増加を含む。
【0083】
一実施形態では、本発明は、「フレア」反応の産生なく、かつ、従来のADT方法を使用して、テストステロンの減少によって引き起こされる、エストロゲン欠損に伴う悪影響を克服しながら、LHレベルの減少、ならびにそれによる、血清総テストステロンレベルおよび/または遊離血清テストステロンの減少を提供する。対象化合物の方法/使用は、骨組織の維持(骨組織への作動薬の作用)、血栓の潜在性の低下、および/または、エストラジオールもしくはジエチルスチルベストロールよりも少ない、もしくは中立的なほてり、および/または乳房組織への作用を提供する、組織選択的エストロゲン活性を提供する。
【0084】
一実施形態では、化合物IVは、作動作用を示すが、拮抗作用は示さず(実施例6および7)、したがって、化合物IVは、ゴナドトロピンおよびテストステロンの増加を引き起こさない。
【0085】
一実施形態では、化合物IVは、血清ホルモン、テストステロン、および総アンドロゲンの減少に対する、強力な薬理学的反応を表す、作動薬活性(実施例8〜11)を示す。
一実施形態では、本明細書において提供する化合物および/または組成物を使用する、本明細書において提供する方法は、従来の形態のADTの使用によるLHの減少によって引き起こされる骨吸収作用を減少させ、または除去する上で有効である。一実施形態では、本明細書において提供する方法、および/または本明細書において提供する組成物の使用は、従来の形態のADTの使用によるテストステロンレベルの減少によって引き起こされる骨吸収作用を減少させ、または除去する上で有効である。一実施形態では、本明細書において提供する組成物を使用する、本明細書において提供する方法は、LHレベルの減少の結果、エストロゲンの減少によって引き起こされる骨吸収作用を減少させ、または除去する上で有効である。一実施形態では、本明細書において提供する化合物および/または組成物を使用する、本明細書において提供する方法は、従来の形態のADTの使用によるLHレベルの減少に伴う骨吸収作用を防止する。一実施形態では、本明細書において提供する化合物および/または組成物を使用する、本明細書において提供する方法は、従来の形態のADTの使用による内因性LH、テストステロン、および/またはエストラジオール減少に伴う骨量の減少を防止する。一実施形態では、本明細書において提供する化合物および/または組成物を使用する、本明細書において提供する方法は、LHレベルの減少を提供しながら、骨量密度(BMD)を増加させる。一実施形態では、本明細書において提供する化合物および/または組成物を使用する、本明細書において提供する方法は、内因性LH、テストステロン、および/またはエストラジオールレベルの低下を提供しながら、骨容量の割合を増加させる。
【0086】
いくつかの実施形態では、本発明は、本発明の化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することによって、血栓塞栓症を回避し、および/または減少させる方法を提供する。
【0087】
一実施形態では、本明細書において提供する化合物および/または組成物を使用する、本明細書において提供する方法は、乳房組織において有効である。一実施形態では、本明細書において提供する化合物および/または組成物を使用する、本明細書において提供する方法は、従来のADTによって達成されたLHレベルの減少に伴う女性化乳房を防止しながら、LHレベルの減少を提供する。
【0088】
一実施形態では、実施例13は、特定の毒性研究を開示し、ヒトの血小板を用いるインビボ研究は、化合物IVが、DESよりもさらに低い凝固促進活性を有したことを示した。したがって、ER選択的作動薬である化合物IVは、DESよりも少ない血栓事象のリスクを有するとともにDESの前立腺癌の利点をもたらし、また、骨量の減少、ほてり、または有害な脂質プロファイルを引き起こすことなく、LHRH作動薬または拮抗薬の利点をもたらすはずである。
【0089】
単独の、または他のADTと組み合わせたジエチルスチルベストロール(DES)療法は、DESが前立腺癌を有する患者において骨吸収を防止したことを示した。DESの使用は、前立腺癌の療法として推進されているが、血管新生および悪性腫瘍へのDESの作用は、DES代謝物によって媒介されると考えられ、エストロゲン受容体を介して機能することは考えられない。加えて、治療的使用のために投与されるDESの用量レベルは、血管疾患、心臓血管罹患率、血栓毒性、女性化乳房、勃起不全、および性欲減退を含む、数々の有害な副作用を示す(Scherr and Pitts,同書、およびPresti,J.C.Jr.(1996)JAMA.275(15):1153−6)。
【0090】
一実施形態では、本発明は、単独の、または抗アンドロゲンまたはDESと組み合わせた、LHRH作動薬または拮抗薬療法のマイナスの副作用を克服する。別の実施形態では、対象発明の方法は、有害な骨関連病状等の有害なエストロゲン欠損の副作用がなく、かつ女性化乳房等の有害なエストロゲン刺激副作用がない、アンドロゲン除去療法を提供する。別の実施形態では、本発明の方法は、LHレベルの減少を提供し、それによって、LHの減少によって引き起こされるエストロゲン欠損に伴う悪影響を克服し、かつDES療法で認められた、一般的エストロゲン作動薬の増加に伴う悪影響を克服しながら、「フレア」作用を産生せずに、総テストステロンレベル、および/または遊離血清テストステロンレベルの減少を提供する。対象化合物の方法/使用は、組織選択的エストロゲン活性を提供し、それによって、骨組織の維持(骨組織への作動薬作用)、血栓の潜在性の低下、および乳房組織への中立的作用を提供する。
【0091】
視床下部レベルのタモキシフェン、トレミフェン、およびラロキシフェン等の従来の選択的エストロゲン受容体モジュレータ(SERM)の抗エストロゲン作用は、男性において、ゴナドトロピンレベルの増加、またはLHレベルの増加をもたらし、それによって、潜在的に、テストステロン血清レベルの増加をもたらす(Tsouri et al.,2008,Fertility and Sterility doi:10.1016)。対照的に、式IA、I〜XIIの化合物を投与することを含む、本発明の方法は、男性対象者において、LHの減少を提供する。
式Iの化合物のためのさらなる実施形態:
本発明の方法の一実施形態では、式Iの化合物のYは、C(O)である。別の実施形態では、Yは、CH2である。別の実施形態では、式IまたはIAの化合物のR1およびR2は、独立して、O−Alk−NR5R6、またはO−Alk−複素環である。別の実施形態では、上記に説明する、該O−Alk−複素環、O−Alk−NR5R6、−Alk−複素環、およびAlk−NR5R6のAlkは、1〜7個の炭素の直鎖アルキル、1〜7個の炭素の分岐鎖アルキル、または3〜8個の炭素の環状アルキルである。別の実施形態では、アルキルは、エチレン(−CH2CH2−)である。別の実施形態では、Alkは、メチレン(−CH2−)である。別の実施形態では、Alkは、プロピレン(−CH2CH2CH2−)である。別の実施形態では、Alkは、2−メチルプロピレン(−CH2CH(CH3)CH2−)である。
【0092】
本発明の方法の一実施形態では、式IまたはIAの化合物のR1は、パラ位にある。本発明の方法の一実施形態では、式IまたはIAの化合物のR1およびR2は、異なる。本発明の方法の別の実施形態では、式IまたはIAの化合物のR1およびR2は、同一である。本発明の方法の別の実施形態
では、式IまたはIAの化合物のR1は、
【0093】
【化14】
である。方法の別の実施形態では、式IまたはIAの化合物のR1は、ヒドロキシルである。方法の別の実施形態では、式IまたはIAの化合物のR1は、アルコキシである。方法の別の実施形態では、R1およびR2は、独立して、水素、ハロゲン、ヒドロキシル、アルコキシ、シアノ、ニトロ、CF3、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、O−Alk−NR5R6、またはO−Alk−複素環であり、複素環は、3〜7員の置換または非置換複素環式環、任意に芳香族である。方法の別の実施形態では、式IまたはIAの化合物のR1およびR2は、独立して、水素、ハロゲン、ヒドロキシル、アルコキシ、シアノ、ニトロ、CF3、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、O−Alk−NR5R6、またはO−Alk−複素環であり、複素環は、3〜7員の置換または非置換複素環式環、任意に芳香族である。方法の別の実施形態では、式IまたはIAの化合物のR2は、ハロゲンである。方法の別の実施形態では、式IまたはIAの化合物のR2は、Fである。方法の別の実施形態では、式Iの化合物のR2は、Clである。方法の別の実施形態では、式IまたはIAの化合物のR2は、Brである。方法の別の実施形態では、式IまたはIAの化合物のR2は、Iである。方法の別の実施形態では、式IまたはIAの化合物のR2は、ヒドロキシルである。方法の別の実施形態では、R1および/またはR2は、CF3である。別の実施形態では、R1および/またはR2は、CH3である。別の実施形態では、R1および/またはR2は、ハロゲンである。別の実施形態では、R1および/またはR2は、Fである。別の実施形態では、R1および/またはR2は、Clである。別の実施形態では、R1および/またはR2は、Brである。別の実施形態では、R1および/またはR2は、Iである。別の実施形態では、式Iの化合物のR2は、パラ位にある。
【0094】
本発明の方法の一実施形態では、式IまたはIAの化合物のR3およびR4は、同一である。本発明の方法の別の実施形態では、式IまたはIAの化合物のR3およびR4は、異なる。方法の別の実施形態では、式IまたはIAの化合物のjおよびkは、独立して、1である。方法の別の実施形態では、式IまたはIAの化合物のR3およびR4は、独立して、ハロゲン、ハロアルキル、ヒドロキシル、またはアルキルである。方法の別の実施形態では、式IまたはIAの化合物のR3およびR4は、独立して、Fである。方法の別の実施形態では、式IまたはIAの化合物のR3およびR4は、独立して、Brである。方法の別の実施形態では、式IまたはIAの化合物のR3およびR4は、独立して、Clである。別の実施形態では、R4は、パラ位にある。別の実施形態では、R3は、オルト位にある。別の実施形態では、R3は、メタ位にある。別の実施形態では、R3および/またはR4は、CF3である。別の実施形態では、R3および/またはR4は、CH3である。
【0095】
本発明の方法の一実施形態では、式IまたはIAの化合物のR5およびR6は、その窒素原子とともに、3〜7員の環を形成する。別の実施形態では、環は、飽和または不飽和環である。別の実施形態では、環は、飽和または不飽和環である。本発明の方法の別の実施形態では、式IまたはIAの化合物のR5およびR6は、その窒素とともに、ピペラジン環を形成する。方法の別の実施形態では、式IまたはIAの化合物のR5およびR6は、その窒素とともに、ピラジン環を形成する。方法の別の実施形態では、式IまたはIAの化合物のR5およびR6は、その窒素とともに、ピペラジン環を形成する。方法の別の実施形態では、式IまたはIAの化合物のR5およびR6は、その窒素とともに、モルホリン環を形成する。方法の別の実施形態では、式IまたはIAの化合物のR5およびR6は、その窒素とともに、ピロール環を形成する。方法の別の実施形態では、式IまたはIAの化合物のR5およびR6は、ピロリジンを形成する。方法の別の実施形態では、式IまたはIAの化合物のR5およびR6は、窒素を有する、ピリジン環を形成する。別の実施形態では、環は、ハロゲン、アルキル、アルコキシ、アルキレン、ヒドロキシル、シアノ、ニトロ、アミノ、アミド、COOH、またはアルデヒドによって置換される。
【0096】
本発明の方法の別の実施形態では、式IまたはIAの化合物のR1、および式IまたはIAの化合物のR2は、独立して、O−Alk−複素環、またはOCH2CH2−複素環である。別の実施形態では、「複素環」基という用語は、一実施形態では、環の一部として、炭素原子に加えて、硫黄、酸素、窒素、またはそれらの任意の組み合わせを含む、環構造を指す。別の実施形態では、複素環は、3〜12員環である。別の実施形態では、複素環は、6員環である。別の実施形態では、複素環は、5〜7員環である。別の実施形態では、複素環は、4〜8員環である。別の実施形態では、複素環基は、置換されないか、またはハロゲン、ハロアルキル、ヒドロキシル、アルコキシ、カルボニル、アミド、アルキルアミド、ジアルキルアミド、シアノ、ニトロ、CO2H、アミノ、アルキルアミノ、ジアルキルアミノ、カルボキシル、チオ、および/またはチオアルキルによって置換され得る。別の実施形態では、複素環式環は、別の飽和または不飽和シクロアルキル、または3〜8員の複素環に縮合され得る。別の実施形態では、複素環式環は、飽和環である。別の実施形態では、複素環式環は、不飽和環である。別の実施形態では、複素環は、ピペリジンである。別の実施形態では、複素環は、ピリジンである。別の実施形態では、複素環は、ピペリジン、ピリジン、フラン、チオフェン、ピロール、ピロリジン、ピラジン、ピペラジン、またはピリミジンである。
【0097】
「シクロアルキル」という用語は、炭素原子および水素原子を含む、非芳香族環、単環式環、または多環式環を指す。シクロアルキル基は、環内に1つまたは複数の炭素−炭素二重結合を有することができるが、それらの存在によって芳香族にされない場合に限られる。シクロアルキル基の例には、(C3−C7)シクロアルキル基(シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、およびシクロへプチル、ならびに飽和単環式および二環式テルペン等)、および(C3−C7)シクロアルケニル基(シクロプロペニル、シクロブテニル、シクロペンテニル、シクロヘキセニル、およびシクロヘプテニル、ならびに不飽和単環式および二環式テルペン等)が挙げられるが、これらに限定されない。シクロアルキル基は、置換されないか、または1個または2個の置換基によって置換することができる。好ましくは、シクロアルキル基は、単環式または二環式環である。
【0098】
「アルキル」という用語は、一実施形態では、直鎖アルキル基、分岐鎖アルキル基、および環状アルキル基を含む、飽和脂肪族炭化水素を指す。一実施形態では、アルキル基は、1〜12個の炭素を有する。別の実施形態では、アルキル基は、1〜7個の炭素を有する。別の実施形態では、アルキル基は、1〜6個の炭素を有する。別の実施形態では、アルキル基は、1〜4個の炭素を有する。別の実施形態では、環状アルキル基は、3〜8個の炭素を有する。別の実施形態では、環状アルキル基は、3〜12個の炭素を有する。別の実施形態では、分岐鎖アルキルは、1〜5個の炭素のアルキル側鎖によって置換されたアルキルである。別の実施形態では、分岐鎖アルキルは、1〜5個の炭素のハロアルキル側鎖によって置換されたアルキルである。アルキル基は、非置換であり得るか、またはハロゲン、ハロアルキル、ヒドロキシル、アルコキシカルボニル、アミド、アルキルアミド、ジアルキルアミド、ニトロ、アミノ、アルキルアミノ、ジアルキルアミノ、カルボキシル、チオ、および/またはチオアルキルによって置換され得る。
【0099】
「アルケニル」基は、別の実施形態では、1つまたは複数の二重結合を有する、直鎖基、分岐鎖基、および環状基を含む、不飽和炭化水素を指す。アルケニル基は、1つの二重結合、2つの二重結合、3つの二重結合等を有し得る。別の実施形態では、アルケニル基は、2〜12個の炭素を有する。別の実施形態では、アルケニル基は、2〜6個の炭素を有する。別の実施形態では、アルケニル基は、2〜4個の炭素を有する。アルケニル基の例は、エテニル、プロペニル、ブテニル、シクロヘキセニル等である。アルケニル基は、非置換であり得るか、またはハロゲン、ヒドロキシ、アルコキシカルボニル、アミド、アルキルアミド、ジアルキルアミド、ニトロ、アミノ、アルキルアミノ、ジアルキルアミノ、カルボキシル、チオ、および/またはチオアルキルによって置換され得る。
【0100】
「アリール」基は、少なくとも1つの炭素環式芳香族基または複素環式芳香族基を有する、芳香族基を指し、それは、非置換であり得るか、またはハロゲン、ハロアルキル、ヒドロキシ、アルコキシカルボニル、アミド、アルキルアミド、ジアルキルアミド、ニトロ、アミノ、アルキルアミノ、ジアルキルアミノ、カルボキシ、またはチオもしくはチオアルキルから選択される、1つまたは複数の基によって置換され得る。アリール環の非制限的例は、フェニル、ナフチル、ピラニル、ピロリル、ピラジニル、ピリミジニル、ピラゾリル、ピリジニル、フラニル、チオフェニル、チアゾリル、イミダゾリル、イソオキサゾリル等である。一実施形態では、アリール基は、4〜8員環である。別の実施形態では、アリール基は、4〜12員環である。別の実施形態では、アリール基は、6員環である。別の実施形態では、アリール基は、5員環である。別の実施形態では、アリール基は、2〜4個の縮合環系である。
【0101】
「アルデヒド」基は、一実施形態では、ホルミル基によって置換された、アルキル、またはアルケニルを指し、アルキルまたはアルケニルは、上で定義したとおりである。別の実施形態では、アルデヒド基は、ホルミル基によって置換された、アリールまたはフェニル基であり、アリールは、上で定義したとおりである。アルデヒドの例は、ホルミル、アセタル、プロパナール、ブタナール、ペンタナール、ベンズアルデヒドである。別の実施形態では、アルデヒド基は、ホルミル基である。
【0102】
「ハロアルキル」基は、別の実施形態では、1個または複数個のハロゲン原子によって、例えば、F、Cl、Br、またはIによって置換された、上で定義されるアルキル基を指す。
【0103】
「ヒドロキシル」基は、別の実施形態では、OH基を指す。当業者は、本発明の化合物中のR1、R2、またはR3が、ORである場合、RはOHではないことを理解する。
一実施形態では、「ハロゲン」または「ハロ」という用語は、F、Cl、Br、またはI等のハロゲンを指す。
【0104】
別の実施形態では、「フェノール」という句は、ベンゼンのアルコール(OH)誘導体を指す。
保護ヒドロキシルへの言及は、いくつかの実施形態において、ベンゼン環の酸素部分に結合した置換基の組込みを含み、置換基は、容易に除去し得る。いくつかの実施形態では、フェノール保護基は、メチルエーテル(メトキシ)、アルキルエーテル(アルコキシ)、ベンジルエーテル(Bn)、メトキシメチル(MOM)エーテル、ベンゾイルオキシメチル(BOM)エーテル、ベンジル、カルボベンゾキシ、メトキシエトキシメチル(MEM)エーテル、2−(トリメチルシリル)エトキシメチル(SEM)エーテル、メチルチオメチル(MTM)エーテル、フェニルチオメチル(PTM)エーテル、アジドメチルエーテル、シアノメチルエーテル、2,2−ジクロロ−1,1−ジフルオロエチルエーテル、2−クロロエチルエーテル、2−ブロモエチルエーテル、テトラヒドロピラニル(THP)エーテル、1−エトキシエチル(EE)エーテル、フェナシルエーテル、4−ブロモフェナシルエーテル、シクロプロピルメチルエーテル、アリルエーテル、プロパルギルエーテル、イソプロピルエーテル、シクロヘキシルエーテル、t−ブチルエーテル、2,6−ジメチルベンジルエーテル、4−メトキシベンジルエーテル、o−ニトロベンジルエーテル、2,6−ジクロロベンジルエーテル、3,4−ジクロロベンジルエーテル、4−(ジメチルアミノ)カルボニルベンジルエーテル、4−メチルスルフィニルベンジルエーテル、4−アントリルメチルエーテル、4−ピコリルエーテル、ヘプタフルオロ−p−トリル、テトラフルオロ−4−ピリジルエーテル、トリメチルシリル(TMS)エーテル、t−ブチルジメチルシリル(TBDMS)エーテル、t−ブチルジフェニルシリル(TBDPS)エーテル、トリイソプロピルシリル(TIPS)エーテル、アリールギ酸、アリール酢酸、アリールレブリン酸、アリールピバル酸、アリール安息香酸、アリール9−フルオレンカルボン酸、炭酸アリールメチル、炭酸1−アダマンチル、炭酸t−ブチル、炭酸4−メチルスルフィニルベンジル、炭酸2,4−ジメチルペント−3−イル、炭酸アリール2,2,2−トリクロロエチル、炭酸アリールベンジル、炭酸アリール、ジメチルホスフィニルエステル(Dmp−OAr)、ジメチルホスフィノチオニルエステル(Mpt−OAr)、ジフェニルホスフィノチオニルエステル(Dpt−OAr)、アリールメタンスルホン酸、アリールトルエンスルホン酸、またはアリール2−ホルミルベンゼンスルホン酸を含み得る。
【0105】
一実施形態では、本発明の方法は、N,N−ビス(4−ヒドロキシフェニル)−4−プロピルベンズアミド(II)またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを活用する。別の実施形態では、本発明の方法は、4,4’−(2,3−ジメチル−ベンジルアザンイジル)ジフェノール(III)またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを活用する。別の実施形態では、本発明の方法は、3−フルオロ−N−(4−フルオロフェニル)−4−ヒドロキシ−N−(4−ヒドロキシフェニル)ベンズアミド(IV)またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを活用する。別の実施形態では、本発明の方法は、N,N−ビス(4−ヒドロキシフェニル)−2,3−ジメチルベンズアミド(V)またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを活用する。別の実施形態では、本発明の方法は、N,N−ビス(4−ヒドロキシフェニル)−2−ナフチルアミド(VI)またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを活用する。別の実施形態では、本発明の方法は、3−フルオロ−4−ヒドロキシ−N,N−ビス(4−ヒドロキシフェニル)−ベンズアミド(VII)またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを活用する。別の実施形態では、本発明の方法は、4−((4−フルオロフェニル)(4−ヒドロキシベンジル)アミノ)フェノール(VIII)またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを活用する。別の実施形態では、本発明の方法は、4−フルオロ−N−(4−ヒドロキシ−フェニル)−N−[4−(2ピペリジン−1−イル−エトキシ)−フェニル]−2−トリフルオロメチル−ベンズアミド(IX)またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを活用する。別の実施形態では、本発明の方法は、IXの塩酸塩(IXのHCl塩)もしくは4−フルオロ−N−(4−ヒドロキシ−フェニル)−N−[4−(2−ピペリジン−1−イル−エトキシ)−フェニル]−2−トリフルオロメチル−ベンズアミド塩酸塩(X)またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを活用する。別の実施形態では、本発明の方法は、3−フルオロ−4−ヒドロキシ−N−(4−ヒドロキシフェニル)−N−フェニルベンズアミド(XI)またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを活用する。別の実施形態では、本発明の方法は、3−フルオロ−N,N−ビス−(4−ヒドロキシ−フェニル)−2−メチル−ベンズアミド(XII)またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを活用する。
【0106】
一実施形態では、本発明の方法は、化合物の「薬学的に許容される塩」を活用し、それは、酸または塩基との本発明の化合物の反応によって産生され得る。
本発明の方法の化合物のアミンの好適な薬学的に許容される塩は、無機酸から、または有機酸から調製し得る。一実施形態では、アミンの無機塩の例は、重硫酸塩、ホウ酸塩、臭化物、塩化物、ヘミ硫酸塩、臭化水素酸塩、塩酸塩、2−ヒドロキシエチルスルホン酸塩(ヒドロキシエタンスルホン酸塩)、ヨウ素酸塩、ヨウ化物、イソチオナート、硝酸塩、過硫酸塩、リン酸塩、硫酸塩、スルファミン酸塩、スルファニル酸塩、スルホン酸(アルキルスルホン酸塩、アリールスルホン酸塩、ハロゲン置換アルキルスルホン酸塩、ハロゲン置換アリールスルホン酸塩)、スルホン酸塩、およびチオシアン酸塩である。
【0107】
一実施形態では、アミンの有機塩の例は、有機酸の脂肪族、脂環式、芳香族、アル脂肪族、複素環式、カルボン酸およびスルホン酸クラスから選択され得、それらの例は、酢酸塩、アルギニン塩、アスパラギン酸塩、アスコルビン酸塩、アジピン酸塩、アントラニル酸塩、アルギン酸塩、アルカンカルボン酸塩、置換アルカンカルボン酸塩、アルギン酸塩、ベンゼンスルホン酸塩、安息香酸塩、重硫酸塩、酪酸塩、重炭酸塩、酸性酒石酸塩、カルボン酸塩、クエン酸塩、樟脳酸塩、樟脳スルホン酸塩、シクロヘキシルスルファミン酸塩、シクロペンタンプロピオン酸塩、エデト酸カルシウム、カンシル酸塩、炭酸塩、クラブラン酸塩、ケイ皮酸塩、ジカルボン酸塩、ジグルコン酸塩、ドデシルスルホン酸塩、二塩酸塩、デカン酸塩、エナント酸塩、エタンスルホン酸塩、エデト酸塩、エジシル酸塩、エストラート、エシラート、フマル酸塩、ギ酸塩、フッ化物、ガラクツロン酸塩、グルコン酸塩、グルタミン酸塩、グリコール酸塩、グルコレート(glucorates)、グルコヘプタン酸塩、グリセロリン酸塩、グルセプチン酸塩、アルサニル酸グリコリル、グルタラート、グルタミン酸塩、ヘプタン酸塩、ヘキサン酸塩、ヒドロキシマレイン酸塩、ヒドロキシカルボキシル酸、ヘキシルレゾルシン酸塩、ヒドロキシ安息香酸塩、ヒドロキシナフトエ酸塩、フッ化水素酸塩、乳酸塩、ラクトビオン酸塩、ラウリン酸塩、リンゴ酸塩、マレイン酸塩、メチレンビス(ベータ−オキシナフトエ酸塩)、マロン酸塩、マンデル酸塩、メシル酸塩、メタンスルホン酸塩、メチル臭化物、メチル硝酸塩、メチルスルホン酸塩、一カリウムマレイン酸塩、ムコ酸塩、モノカルボン酸塩、硝酸塩、ナフタレンスルホン酸塩、2−ナフタレンスルホン酸塩、ニコチン酸塩、ナプシル酸塩、N−メチルグルカミン、シュウ酸塩、オクタン酸塩、オイレン酸塩、パモ酸塩、フェニル酢酸、ピクリン酸塩、フェニル安息香酸塩、ピバル酸塩、プロピオン酸塩、フタル酸塩、ペクチニン酸塩、フェニルプロピオン酸塩、パルミチン酸塩、パントテン酸塩、ポリガラクツロン酸塩、ピルビン酸塩、キナ酸塩、サリチル酸塩、コハク酸塩、ステアリン酸塩、スフラニル酸塩、塩基性酢酸塩、酒石酸塩、テオフィリン酢酸塩、p−トルエンスルホン酸(トシラート)、トリフルオロ酢酸、テレフタル酸塩、タンニン酸塩、テオクル酸塩、トリハロ酢酸塩、トリエチオダイド、トリカルボン酸塩、ウンデカン酸塩、および吉草酸塩である。
【0108】
一実施形態では、カルボン酸またはフェノールの無機塩の例は、アンモニウム、アルカリ金属(リチウム、ナトリウム、カリウム、セシウムを含む)、アルカリ土類金属(カルシウム、マグネシウム、アルミニウム)、亜鉛、バリウム、コリン、第4級アンモニウムから選択され得る。
【0109】
別の実施形態では、カルボン酸またはフェノールの有機塩の例は、アルギニン、有機アミン(脂肪族有機アミンを含む)、脂環式有機アミン、芳香族有機アミン、ベンザチン、t−ブチルアミン、ベネタミン(N−ベンジルフェネチルアミン)、ジシクロヘキシルアミン、ジメチルアミン、ジエタノールアミン、エタノールアミン、エチレンジアミン、ヒドラバミン、イミダゾール、リジン、メチルアミン、メグラミン、N−メチル−D−グルカミン、N,N’−ジベンジルエチレンジアミン、ニコチンアミド、有機アミン、オルニチン、ピリジン、ピコリ、ピペラジン、プロカイン、トリス(ヒドロキシメチル)メチルアミン、トリエチルアミン、トリエタノールアミン、トリメチルアミン、トロメタミン、および尿素から選択され得る。
【0110】
一実施形態では、これらの塩は従来の方法によって形成されてもよく、例えば生成物の遊離塩基または遊離酸形態を、塩が不溶性である溶媒または媒体中または水等の溶媒中で、適切な酸または塩基の1つまたは複数の等価物と反応させることによって形成され、その溶媒または媒体は、真空で、または凍結乾燥によって、あるいは別のイオンまたは好適なイオン交換樹脂のために既存の塩のイオンを交換することによって、除去される。
【0111】
一実施形態では、本発明の方法は、本発明の化合物の薬学的に許容される塩を活用する。一実施形態では、本発明の方法は、式IA、I〜XIIの化合物の薬学的に許容される塩を活用する。一実施形態では、本発明の方法は、本発明の式IA、I〜XIIの化合物のアミンの塩を活用する。一実施形態では、本発明の方法は、本発明の式IA、I〜XIIの化合物のフェノールの塩を活用する。
【0112】
一実施形態では、本発明の方法は、式IA、I〜XIIの遊離塩基、遊離酸、非荷電、または非錯体化合物、またはその異性体、薬学的生成物、水和物、多形体、またはそれらの組み合わせを活用する。
【0113】
本発明のいくつかの実施形態では、本発明の化合物は、アミド結合によって互いに結合した3個のフェニル基を含む。一実施形態では、本発明の化合物は、非荷電構造である。別の実施形態では、本発明の化合物は、遊離塩基構造である。別の実施形態では、本発明の化合物は、遊離酸構造である。別の実施形態では、本発明の化合物は、非錯体構造である。別の実施形態では、本発明の化合物は、非イオン化構造である。別の実施形態では、本発明の化合物は、薬学的に許容される塩である。別の実施形態では、本発明のいくつかの化合物は、塩酸(HCl)塩を含む。
【0114】
一実施形態では、本発明の方法は、式IA、I〜XIIの化合物の異性体を活用する。一実施形態では、本発明の方法は、式IA、I〜XIIの化合物の薬学的生成物を活用する。一実施形態では、本発明の方法は、式IA、I〜XIIの化合物の水和物を活用する。一実施形態では、本発明の方法は、式IA、I〜XIIの化合物の多形体を活用する。一実施形態では、本発明の方法は、式IA、I〜XIIの化合物の代謝物を活用する。別の実施形態では、本発明の方法は、本明細書に説明する、式IA、I〜XIIの化合物を含む組成物を活用し、または別の実施形態では、式IA、I〜XIIの化合物の異性体、代謝物、薬学的生成物、水和物、多形体の組み合わせを活用する。
【0115】
一実施形態では、「異性体」という用語は、光学的異性体および類似体、構造的異性体および類似体、立体構造的異性体および類似体、ならびに同等物を含むが、これらに限定されない。
【0116】
一実施形態では、「異性体」という用語は、化合物の光学的異性体を包含することを意味する。一実施形態では、「異性体」という用語は、化合物の立体異性体を包含することが意図される。本発明の化合物は、そのシスまたはトランス異性体化にあり得る、アミド結合を保持する。本発明は、任意の光学的活性形態、または立体異性体形態、またはそれらの混合物を包含し、これらの任意の用途のための使用は本発明の範囲内と見なされることを理解されたい。
【0117】
別の実施形態では、本発明はさらに、化合物の水和物を含む。一実施形態では、「水和物」という用語は、当該技術分野において公知のとおり、半水和物、一水和物、二水和物、三水和物、またはその他を指す。
合成プロセス
式IまたはIAの化合物は、例えば、塩基の存在下で、置換ジフェニルアミンを安息香酸またはハロゲン化ベンゾイルと反応させて、ベンズアミドを得ることによって、容易に調製し得る。一実施形態では、塩基はピリジンである。別の実施形態では、ハロゲン化ベンゾイルは塩化ベンゾイルである。別の実施形態では、ヒドロキシル置換基は、ジフェニルアミンと安息香酸またはハロゲン化ベンゾイルとの反応中に保護される。別の実施形態では、ヒドロキシルのための保護基は、任意に、最終工程において除去される。また、米国公報第2009/00624231号も参照されたく、それは、参照することにより、その全体として組み込まれる。
【0118】
例えば、式IA:
【0119】
【化15】
(式中、R1、R2、R3、およびR4、jおよびKは、上述のとおりである)
の化合物は、
【0120】
【化16】
を、
【0121】
【化17】
ととも反応させて、
【0122】
【化18】
を得る工程を含むプロセスによって調製され得、
ジフェニルアミン(3)を、塩基の存在下で、
【0123】
【化19】
と反応させて、
【0124】
【化20】
を得、
式中、R1、R2、R3、およびR4が、独立して、OH、O−Alk−R5R6、またはO−Alk−複素環である場合は、R1’、R2’、R3’、およびR4’は、保護されたヒドロキシル基であり、保護基は、除去されて、遊離ヒドロキシルを得るか、または任意に、その後にCl−Alk−複素環、またはCl−Alk−NR5R6と反応させて、式IA:
【0125】
【化21】
の化合物を得、
式中、R1、R2、R3、およびR4が、独立して、OH、O−Alk−NR5R6、またはO−Alk−複素環と異なる場合は、R1’、R2’、R3’、およびR4’はそれぞれ、R1、R2、R3、およびR4である。
【0126】
別の例として、式IA:
【0127】
【化22】
(式中、R1、R2、R3、およびR4は、上述のとおりである)、の化合物の調製のためのプロセスは、塩基の存在下で、
【0128】
【化23】
を、
【0129】
【化24】
と反応させて、
【0130】
【化25】
を得る工程を含み、
式中、R1、R2、R3、およびR4が、独立して、OH、O−Alk−R5R6、またはO−Alk−複素環である場合は、R1’、R2’、R3’、およびR4’は、保護されたヒドロキシル基であり、保護基は、除去されて、遊離ヒドロキシルを得るか、または任意に、その後にCl−Alk−複素環、またはCl−Allk−NR5R6と反応させて、式IA:
【0131】
【化26】
の化合物を得、
式中、R1、R2、R3、およびR4が、独立して、OH、O−Alk−NR5R6、またはO−Alk−複素環と異なる場合は、R1’、R2’、R3’、およびR4’はそれぞれ、R1、R2、R3、およびR4である。
【0132】
一例では、化合物IIは、実施例1および図5に従い調製される。
別の例では、化合物IIIは、実施例1および図5に従い調製される。
さらなる例では、式IV:
【0133】
【化27】
の化合物は、塩基の存在下で、
【0134】
【化28】
を、
【0135】
【化29】
と反応させて、
【0136】
【化30】
を得た後に、保護基の脱保護により、化合物IV:
【0137】
【化31】
を得ることによって、調製され得、
式中、PおよびP’は、同一、または異なる保護基である。一例では、化合物IVは、実施例2、および図6に従い調製される。
【0138】
別の例では、化合物Vは、実施例1、および図5に従い調製される。
さらなる例では、化合物VIは、実施例3、および図7に従い調製される。
別の例では、化合物VIIは、実施例1、および図5に従い調製される。
【0139】
別の例では、化合物VIIIは、実施例4、および図5に従い調製される。
別の例では、化合物IXは、実施例5、および図8に従い調製される。
別の例では、化合物X塩酸塩は、実施例5、および図8に従い調製される。
【0140】
別の例では、化合物XIは、実施例1、および図5に従い調製される。
別の例では、化合物XIIは、実施例1、および図5に従い調製される。
好適なヒドロキシル保護基は、例えば、メチルエーテル(メトキシ)、ベンジルエーテル(ベンジルオキシ)メトキシメチル(MOM)エーテル、ベンゾイルオキシメチル(BOM)エーテル、ベンジル、カルボベンゾオキシ、メトキシエトキシメチル(MEM)エーテル、2−(トリメチルシリル)エトキシメチル(SEM)エーテル、メチルチオメチル(MTM)エーテル、フェニルチオメチル(PTM)エーテル、アジドメチルエーテル、シアノメチルエーテル、2,2−ジクロロ−1,1−ジフルオロエチルエーテル、2−クロロエチルエーテル、2−ブロモエチルエーテル、テトラヒドロピラニル(THP)エーテル、1−エトキシエチル(EE)エーテル、フェナシルエーテル、4−ブロモフェナシルエーテル、シクロプロピルメチルエーテル、アリルエーテル、プロパルギルエーテル、イソプロピルエーテル、シクロヘキシルエーテル、t−ブチルエーテル、ベンジルエーテル、2,6−ジメチルベンジルエーテル、4−メトキシベンジルエーテル、o−ニトロベンジルエーテル、2,6−ジクロロベンジルエーテル、3,4−ジクロロベンジルエーテル、4−(ジメチルアミノ)カルボニルベンジルエーテル、4−メチルスルフィニルベンジルエーテル、4−アントリルメチルエーテル、4−ピコリルエーテル、ヘプタフルオロ−p−トリル、テトラフルオロ−4−ピリジルエーテル、トリメチルシリル(TMS)エーテル、t−ブチルジメチルシリル(TBDMS)エーテル、t−ブチルジフェニルシリル(TBDPS)エーテル、トリイソプロピルシリル(TIPS)エーテル、アリールギ酸、アリール酢酸、アリールレブリン酸、アリールピバル酸、アリール安息香酸、アリール9−フルオレンカルボン酸、アリールメチルカルボン酸、1−アダマンチルカルボン酸、t−ブチルカルボン酸、4−メチルスルフィニルベンジルカルボン酸、2,4−ジメチルペント−3−イルカルボン酸、アリール2,2,2−トリクロロエチルカルボン酸、アリールベンジルカルボン酸、アリールカルバミン酸、ジメチルホスフィニルエステル(Dmp−OAr)、ジメチルホスフィノチオニルエステル(Mpt−OAr)、ジフェニルホスフィノチオニルエステル(Dpt−OAr)、アリールメタンスルホン酸、アリールトルエンスルホン酸、またはアリール2−ホルミルベンゼンスルホン酸を含む。
【0141】
本発明の方法は、化合物IA、I〜XIIの使用を含み、本発明の化合物の調製のためのプロセスは、塩基の存在下で、塩化ベンゾイルとのジフェニルアミンの反応を含む。好適な塩基は、例えば、ピリジン、トリエチルアミン、K2CO3、Cs2CO3、Na2CO3、メチルアミン、イミダゾール、ベンズイミダゾール、ヒスチジン、トリブチルアミン、またはそれらの任意の組み合わせを含む。一実施形態では、塩基は、ピリジンである。
【0142】
本発明の方法は、化合物IA、I〜XIIの使用を含み、本発明の化合物の調製のためのプロセスは、保護されたヒドロキシルの脱保護を含む。別の実施形態では、脱保護条件は、保護基によって異なる。いくつかの実施形態では、脱保護工程は、Pd/Cの存在下での水素化を含む。別の実施形態では、脱保護は、BBr3との反応を含む。別の実施形態では、脱保護工程は、酸との反応を含む。
【0143】
さらなる実施形態では、化合物IA、I〜XIIは、図5〜8、および実施例1〜5に従い調製される。
薬学的組成物
いくつかの実施形態では、本発明は、説明した化合物を含有する組成物を投与する工程を含む使用方法を提供する。本明細書において使用されるとき、「薬学的組成物」は、薬学的に許容される担体または希釈剤を伴う「治療的有効量」の活性成分、すなわち、本発明の化合物を意味する。本明細書において使用されるとき、「治療的有効量」は、所与の病状および投与レジメンに対して治療効果を提供する量を指す。
【0144】
本明細書において使用されるとき、「投与する」という用語は、対象者を本発明の化合物と接触させることを指す。本明細書において使用されるとき、投与は、インビトロ(すなわち試験管内)、またはインビボ(すなわち有機体、例えばヒトの細胞もしくは組織内)で達成することができる。一実施形態では、本発明は、男性対象者に本発明の化合物を投与することを含む。
【0145】
本発明は、他の実施形態では、本明細書に説明する化合物の薬学的生成物を提供する。「薬学的生成物」という用語は、他の実施形態では、例えば、本明細書に説明する、薬学的使用に好適な組成物(薬学的組成物)を指す。
【0146】
本発明の化合物は、単独で、または製剤の活性成分として投与することができる。したがって、本発明はまた、例えば、1つまたは複数の薬学的に許容される担体を含有する、式Iの化合物の薬学的組成物も含む。
【0147】
本発明に従う、化合物を投与するために好適な種々の製剤を調製するための処置を説明する、数々の標準的参考文献が利用可能である。潜在的製剤の例および調製は、例えば、Handbook of Pharmaceutical Excipients,American Pharmaceutical Association(最新版)、Pharmaceutical Dosage Forms:Tablets(Lieberman,Lachman and Schwartz,editors)最新版、(Marcel Dekker,Inc.ならびにRemington‘s Pharmaceutical Sciences(Arthur Osol,editor)から刊行、1553−1593(current edition)に含まれている。
【0148】
投与方法および剤形は、所与の治療用途に望ましく、かつ有効である化合物または組成物の治療量に密接に関連する。
好適な剤形は、経口、直腸、舌下、粘膜、鼻腔、眼、皮下、筋肉内、静脈内、経皮、脊髄、髄腔内、関節内、動脈内、くも膜下、気管支、リンパ管、および子宮内投与、ならびに活性成分全身送達のための他の剤形を含むが、これらに限定されない。経口投与に好適な製剤が望ましい。
【0149】
かかる薬学的剤形を調製するために、従来の薬学的配合技術に従い、活性成分を薬学的担体と混合し得る。担体は、投与に望ましい調製の形態によって多種多様な形態を成し得る。
【0150】
経口剤形に組成物を調製する際に、通常の薬学的媒体のうちのいずれをも使用し得る。したがって、例えば、懸濁液、エリキシル剤、および溶液等の液体経口調製剤では、好適な担体および添加剤は、水、グリコール、油、アルコール、香味剤、防腐剤、着色剤、および同等物を含む。例えば、粉末、カプセル、および錠剤等の固体経口調製剤では、好適な担体および添加剤は、スターチ、糖、希釈剤、造粒剤、滑沢剤、結合剤、崩壊剤を含む。錠剤およびカプセルは、それらの容易な投与のため、最良の有益な経口投与単位形態を示す。必要に応じて、錠剤は、標準的技術によって、糖衣化または腸溶化され得る。
【0151】
非経口製剤では、他の成分、例えば、溶解を補助するか、または保存のための成分が含まれ得るが、担体は、通常、無菌水を含む。注入可能な溶液はまた、調製され得、その場合、適切な安定剤が用いられ得る。
【0152】
いくつかの用途では、リポソームまたは他のカプセル媒体中の活性剤のカプセル化による、またはタンパク質、リポタンパク質、糖タンパク質、および多糖等の生体分子への活性剤の固着による、例えば、共有結合、キレート化、または関連配位等による、「ベクトル化」形態の活性剤を用いることが有益であり得る。
【0153】
経口投与に好適な製剤を使用する、本発明の治療方法は、カプセル、カシェー、錠剤、またはロゼンジ剤等の不連続単位で表され得、それぞれ、例えば、粉末または顆粒として、所定量の活性成分を含有する。任意に、シロップ、エリキシル剤、乳剤または注ぎ出し(draught)等の水性液または非水性液体中の懸濁液を使用し得る。
【0154】
錠剤は、任意に1つまたは複数の副成分を用いて、圧縮もしくは成形、または湿式造粒により作製し得る。圧縮された錠剤は、例えば、結合剤、崩壊剤、滑沢剤、不活性希釈剤、界面活性剤、または抜染剤と任意に混合される、粉末または顆粒等の自由流動性形態にある活性化合物を用いて、好適な機械で圧縮することによって調製され得る。粉末状活性化合物と好適な担体との混合物から成る、成形された錠剤は、好適な機械で成形することによって作製され得る。
【0155】
シロップは、糖、例えば、スクロースの濃縮水溶液に活性化合物を添加することによって作製され得、そこに副成分も添加し得る。かかる副成分は、香味剤、好適な防腐剤、糖の結晶化を遅延する薬剤、任意の他の成分の溶解度を増大する薬剤(ポリヒドロキシアルコール、例えば、グリセロールまたはソルビトール等)を含み得る。
【0156】
非経口投与に好適な製剤は、好ましくは、受容者の血液と等張性である、活性化合物の無菌水性調製剤(例えば、生理食塩溶液)を含み得る。かかる製剤は、懸濁剤および濃化剤、ならびにリポソーム、または、化合物を血液構成要素、もしくは1つまたは複数の臓器に標的化するように設計される他の微粒子系を含み得る。製剤は、単位容量、または多用量形態で提示され得る。
【0157】
非経口投与は、全身送達のための任意の好適な形態を含み得る。投与は、例えば、静脈内、動脈内、髄腔内、筋肉内、皮下、筋肉内、腹内(例えば、腹腔内)等であり得、輸液ポンプ(外部または埋め込み型)、または所望の投与モダリティに適切な、任意の他の好適な手段によって達成され得る。
【0158】
鼻腔および他の粘膜スプレー製剤(例えば、吸入型形態)は、防腐剤または等張剤を有する、活性化合物の精製された水溶液を含むことができる。かかる製剤は、好ましくは、鼻腔または他の粘膜に適合するpHおよび等張状態に調節される。代替的には、それらは、ガス担体中に懸濁された、微細固体粉末の形態である可能性がある。かかる製剤は、任意の好適な手段または方法によって、例えば、ネブライザー、アトマイザー、定量吸入器、または同等物によって送達され得る。
【0159】
直腸投与の製剤は、ココアバター、硬化油脂、または硬化油脂カルボン酸等の好適な担体を有する、座薬として提供され得る。
経皮製剤は、セルロース媒体、例えば、メチルセルロースまたはヒドロキシエチルセルロース等のチキソトロピーまたはゼラチン状担体中に活性剤を組み込むことによって調製し得、次いで、得られた製剤は、使用者の皮膚と皮膚接触して固着されるように適合された経皮デバイス内に充填される。
【0160】
先述の成分に加えて、本発明の製剤はさらに、希釈剤、緩衝剤、香味剤、結合剤、崩壊剤、界面活性剤、増粘剤、滑沢剤、防腐剤(抗酸化剤を含む)、および同等物から選択される、1つまたは複数の副成分を含み得る。
【0161】
本発明の製剤は、当業者に公知の即効放出、持続放出、遅延放出、または任意の他の放出プロファイルを有することができる。
一実施形態では、本発明は、a)血清総テストステロンレベルを低下させる方法、b)前立腺癌を有する男性対象者において、黄体形成ホルモン(LH)の減少によって、またはLHホルモンの減少と無関係に、遊離血清テストステロンレベルを低下させる方法を提供し、式IA、I〜XIIの化合物を含有する、経口組成物を投与することを含む。付加的な実施形態では、本発明の方法は、式II、式III、式IV、式V、式VI、式VII、式VIII、式IX、式X、式XI、または式XIIの化合物を含有する経口組成物を活用する。
【0162】
一実施形態では、本発明は、前立腺癌を有する男性対象者において、LHレベルを減少させることによるか、またはLHレベルの減少と無関係に、前立腺癌を治療する方法を提供し、式IA、I〜XIIの化合物を含有する経口組成物を投与することを含む。さらなる実施形態では、本発明の方法は、前立腺癌を有する男性対象者において、LHレベルを減少させることによるか、またはLHレベルの減少と無関係に、前立腺癌を治療する方法を提供し、式II、式III、式IV、式V、式VI、式VII、式VIII、式IX、式X、式XI、または式XIIの化合物を含有する経口組成物を投与することを含む。
【0163】
本発明は、いくつかの実施形態では、「本発明の化合物」と称される、本明細書に説明する化合物の任意の実施形態を包含することを理解されたい。
一実施形態では、本発明の方法は、種々の用量での本発明の化合物の投与を含む。一実施形態では、本発明の化合物は、1日当たり1〜1500mgの用量で投与される。付加的な実施形態では、本発明の化合物は、1日当たり1〜10mg、1日当たり3〜26mg、1日当たり3〜60mg、1日当たり3〜16mg、1日当たり3〜30mg、1日当たり10〜26mg、1日当たり15〜60mg、1日当たり50〜100mg、1日当たり50〜200mg、1日当たり150〜300mg、1日当たり20〜50mg、1日当たり5〜50mg、1日当たり200〜500mg、1日当たり150〜500mg、1日当たり200〜1000mg、1日当たり300〜1500mg、または1日当たり100〜1000mgの用量で投与される。
【0164】
一実施形態では、本発明の方法は、種々の用量での本発明の化合物の投与を含む。一実施形態では、本発明の化合物は、3mgの用量で投与される。付加的な実施形態では、本発明の化合物は、10mg、30mg、50mg、100mg、200mg、300mg、450mg、500mg、600mg、900mg、1000mg、または1500mgの用量で投与される。
【0165】
一実施形態では、本発明の方法は、種々の用量での本発明の化合物の投与を含む。一実施形態では、本発明の化合物は、0.1mg/kg/日の用量で投与される。付加的な実施形態では、本発明の化合物は、0.2〜30mg/kg/日、または0.2mg/kg/日、0.3mg/kg/日、1mg/kg/日、3mg/kg/日、5mg/kg/日、10mg/kg/日、20mg/kg/日、もしくは30mg/kg/日の用量で投与される。
【0166】
本発明の方法の一実施形態では、式IA、I〜XIIの化合物を含有する薬学的組成物の使用を提供する。付加的な実施形態では、本発明の方法は、式II、式III、式IV、式V、式VI、式VII、式VIII、式IX、式X、式XI、または式XIIの化合物を含有する薬学的組成物の使用のために提供される。
【0167】
ある実施形態では、薬学的組成物は、固体剤形である。別の実施形態では、薬学的組成物は、錠剤である。別の実施形態では、薬学的組成物は、カプセルである。別の実施形態では、薬学的組成物は、溶液である。別の実施形態では、薬学的組成物は、経皮パッチである。
【0168】
一実施形態では、本発明の化合物またはそれを含有する組成物の使用は、当業者に理解されるとおり、対象者において、所望の反応を阻害、抑制、強化、または刺激する上で有用性がある。別の実施形態では、組成物はさらに、付加的な活性成分を含有し得、その活性は、本発明の化合物が投与される特定の用途に有用である。
【0169】
哺乳類、および特にヒトへの投与のために、医師が、実際の用量および治療期間を決定することが予期されており、それは、個人に最も好適であり、特定の個人の年齢、体重、遺伝子、および/または反応によって異なる可能性がある。
【0170】
いくつかの実施形態では、本発明の組成物のうちのいずれかは、本明細書に説明する任意の形態または実施形態の本発明の化合物を含有する。いくつかの実施形態では、本発明の組成物のうちのいずれもが本明細書に説明する任意の形態または実施形態の本発明の化合物から成る。いくつかの実施形態では、本発明の組成物は、本明細書に説明する任意の形態または実施形態の本発明の化合物から本質的に成る。いくつかの実施形態では、「含有する」という用語は、薬学業界において公知のとおり、本発明の化合物等の指示した活性剤の含有、ならびに他の活性剤、および薬学的に許容される担体、賦形剤、皮膚軟化剤、安定剤等の含有を指す。いくつかの実施形態では、「から本質的に成る」という用語は、その活性成分のみが指示した活性成分である組成物を指すが、指示した活性成分の治療効果に直接関与しない、製剤を安定、保存する等のための他の化合物が含まれ得る。いくつかの実施形態では、「から本質的に成る」という用語は、活性成分の放出を促進する構成要素を指す。いくつかの実施形態では、「から成る」という用語は、活性成分および薬学的に許容される担体または賦形剤を含む組成物を指す。
【0171】
本明細書に説明する化合物のうちのいずれの、いかなる使用も、本明細書において説明する、任意の疾患、障害、または病状の治療に使用され得、本明細書の一実施形態を表すことを理解されたい。一実施形態では、化合物は、遊離塩基、遊離酸、非荷電または非錯体化合物である。
【0172】
本発明の好ましい実施形態をより完全に例証するために、以下の実施例を提示する。しかしながら、それらは、本発明の広い範囲を制限するものと一切解釈されてはならない。
実施例
実施例1
式II〜XIIの化合物および合成中間体のための一般的合成手順
有機溶媒、界面活性剤、および抗酸化剤等は、本明細書に説明する組成物に使用され得、典型的には、商業的供給源から容易に入手可能である。例えば、PEG−300、ポリソルベート80、Captex(商標)200、Capmul(商標)MCM C8は、例えば、Dow Chemical社(ミシガン州ミッドランド)、ICI Americas社(デラウェア州ウィルミントン)、またはAbitec Corporation(ウィスコンシン州ジェーンズビル)から購入可能である。
【0173】
本明細書に説明するエストロゲン受容体リガンドは、当業者に公知の多くの方法で調製され得る。例えば、本明細書に説明するエストロゲン受容体リガンドは、米国特許出願公開第2009/0062341号明細書において説明される合成方法によって調製され得、それぞれの開示は、参照することにより、その全体として組み込まれる。
N,N−ビスアリール ベンズアミド誘導体の一般的合成
ジアリールアニリンの一般的合成(図5)。アリールアミン(1.5当量)、アリールヨウ化物(1当量)、K2CO3(2当量)、CuI(0.1当量)、およびL−プロリン(0.2当量)の混合物をともに混合し、室温で、無水DMSO中で溶解した。次いで、反応混合物を28時間、90℃で攪拌および加熱した。混合物を室温まで冷却し、水で加水分解した。EtOAcを添加して、溶液を分割した。EtOAc層を分離し、ブラインで洗浄し、無水MgSO4上で乾燥させた。溶媒を減圧下で除去した。固体残渣を、溶離液として5%のEtOAc/ヘキサンを使用するフラッシュカラムクロマトグラフィー(シリカゲル)によって精製し、対応するジアリールアニリンを得た。
【0174】
ビス−(4−メトキシフェニル)アミン(1a):淡黄色固体、73%の収率。融点98.6〜99.0℃。1H NMR(CDCl3,300MHz)δ6.93−6.81(m,8H),5.37(s,br,1H),3.78(s,6H)。MS m/z 228.4(M−H)+
N−(4−メトキシフェニル)−フェニルアミン(1b):淡黄色固体、70%の収率。融点106.3〜106.5℃。1H NMR(CDCl3,300MHz)δ7.24−7.18(m,3H),7.08−7.06(m,2H),6.92−6.84(m,4H),5.61(s,br,1H),3.79(s,3H)。MS m/z 200.1(M+H)+。
【0175】
N−(4−フルオロフェニル)−N−4−メトキシフェニルアミン(1c):淡黄色固体、54%の収率。融点60.6〜61.0℃。1H NMR(CDCl3,300MHz)δ7.01−6.83(m,8H),3.78(s,3H)。MS m/z 217(M)+。
【0176】
N−(4−ベンジルオキシフェニル)−N−4−メトキシフェニルアミン(1d):淡黄色固体、54%の収率。融点108.0〜108.4℃。1H NMR(CDCl3,300MHz)δ7.34−7.08(m,5H),6.90−6.81(s,3H),3.78(s,3H)。MS m/z 306(M+H)+。
【0177】
ベンズアミドの一般的合成。アリールアリニン(1当量)、ベンゾイル塩化物(1.3当量)、およびピリジン(6当量)をともに混合し、室温で、無水THF中で溶解した。24時間、反応混合物を攪拌し、還流させた。反応溶液を室温まで冷却し、2N HCl溶液の添加により加水分解した。溶液を酢酸エチルで抽出した。有機層を、飽和NaHCO3水溶液で洗浄して、過剰な酸を除去し、無水MgSO4上で乾燥させ、ろ過し、減圧下で濃縮した。残渣を、EtOAc/ヘキサン(3/7v/v)を使用するフラッシュカラムクロマトグラフィーによって精製し、対応するベンズアミド化合物を得た。
【0178】
3−フルオロ−N−(4−フルオロフェニル)−4−メトキシ−N−(4−メトキシフェニル)ベンズアミド(2a):黄色固体、融点54〜56℃,1H NMR(CDCl3/TMS)δ7.24−7.11(m,4H),7.05−6.97(m,4H),6.85−6.78(m,3H),3.86(s,3H),3.79(s,3H)。MS(ESI)m/z 370.1[M+H]+
4−フルオロ−N,N−ビス(4−メトキシフェニル)−2−(トリフルオロメチル)ベンズアミド(2b):無色油、84.2%の収率。1H NMR(CDCl3,300MHz)δ7.34−7.26(m,4H),7.09−7.01(m,3H),6.91(d,2H,J=8.7Hz),6.87(d,2H,J=8.7Hz),3.80(s,3H),3.71(s,3H)。MS m/z 442.1(M+Na)+。
【0179】
4−メトキシ−N−(4−メトキシフェニル)−N−(4−フルオロフェニル)−ベンズアミド(2c)(2c):白色固体、97%の収率、融点133.5.0〜134.5℃。1H NMR(CDCl3,300MHz)δ8.11−6.66(m,15H),3.74(s,3H),3.73(s,3H)。MS m/z 384(M+H)+。
【0180】
N−(4−メトキシフェニル)−N−(4−ベンジルオキシフェニル)−2−ナフチルアミド(2d):白色固体、58%の収率。融点174.9〜175.5℃。1H NMR(CDCl3,300MHz)δ8.04(s,1H),7.77−7.74(m,2H),7.64−7.61(m,1H),7.51−7.43(m,4H),7.40−7.31(m,4H),7.13−7.10 m,4H),6.88−6.78(m,4H),4.99(s,2H),3.74(s,3H)。MS m/z 460(M+H)+。
【0181】
4−フルオロ−N,N−ビス(4−メトキシフェニル)−2−(トリフルオロメチル)ベンズアミド(2e):無色油、84.2%の収率。1H NMR(CDCl3,300MHz)δ7.34−7.26(m,4H),7.09−7.01(m,3H),6.91(d,2H,J=8.7Hz),6.87(d,2H,J=8.7Hz),3.80(s,3H),3.71(s,3H)。MS m/z 442.1(M+Na)+。
【0182】
BBr3を使用した、ベンズアミド誘導体のデメチル化のための一般手順。メトキシベンズアミド化合物を、乾燥CH2Cl2中で溶解した。BBr3(1.0MのCH2Cl2溶液)を、0℃で滴加した。反応溶液を、ゆっくりと室温まで温め、室温で一晩攪拌した。混合物を、氷浴中で0℃に冷却し、水を添加して加水分解した。EtOAcを添加し、溶液を分割した。有機層を分離し、水性層をEtOAcで抽出した。有機層をブラインで洗浄し、無水MgSO4上で乾燥させた。溶媒を減圧下で除去した。残渣を、CH3OH/CH2Cl2(1/9 v/v)を使用するフラッシュカラムクロマトグラフィーによって精製し、対応するフェノール化合物を得た。
【0183】
4−フルオロ−N,N−ビス(4−ヒドロキシフェニル)−2−(トリフルオロメチル)ベンズアミド(3a):白色固体、92.5%の収率。1H NMR(DMSO−d6,300MHz)δ9.55(s,1H),9.53(s,1H),7.69−7.58(m,2H),7.46−7.39(m,1H),7.18(d,2H,J=8.7Hz),6.93(d,4H,J=8.7Hz),7.03(d,2H,J=8.4Hz),6.78(d,2H,J=8.7Hz),6.57(d,2H,J=8.7Hz)。MS m/z 392.1(M+H)+。
【0184】
以下の化合物を上述のとおり合成し、表1に特徴付け、要約する:N,N−ビス(4−ヒドロキシフェニル)−4−プロピルベンズアミド(II)、3−フルオロ−N−(4−フルオロフェニル)−4−ヒドロキシ−N−(4−ヒドロキシフェニル)ベンズアミド(IV)、N,N−ビス(4−ヒドロキシフェニル)−2,3−ジメチルベンズアミド(V)、3−フルオロ−4−ヒドロキシ−N,N−ビス(4−ヒドロキシフェニル)−ベンズアミド(VII)、3−フルオロ−4−ヒドロキシ−N−(4−ヒドロキシフェニル)−N−フェニルベンズアミド(XI)、および3−フルオロ−N,N−ビス(4−ヒドロキシフェニル)−2−メチルベンズアミド(XII)。
【0185】
ベンジルオキシフェニル−ベンズアミドのデベンジル化の一般手順。化合物を、250mLの水素化ボトル内のEtOH中で溶解した。Pd/C粉末(5%mol)を溶液に添加した。反応容器を、138kPa(20psi)圧力水素ガス下で水素化装置に装着した。出発物質が消失するまで、反応をTLCによって監視した。次いで、溶媒を減圧下で除去した。残渣を、ヘキサン/EtOAc=3/2v/vを有するフラッシュカラムクロマトグラフィーによって精製し、所望の生成物を得た。
【0186】
以下の化合物を上述のとおり合成し、表1に特徴付け、要約する:N,N−ビス(4−ヒドロキシフェニル)−2−ナフチルアミド(VI)。
脱保護ベンズアミドの還元のための一般手順。ベンズアミド化合物を、室温で、20mLの無水THF中で溶解した。H3B(SMe2)を、アルゴン下で、室温で、注射器を介して添加した。反応溶液を攪拌し、加熱して6時間還流させた。次いで、0℃の10mLのMeOHを添加することにより、反応を停止した。溶媒を減圧下で除去した。残渣を、フラッシュカラムクロマトグラフィー(シリカゲル、CH2Cl2/MeOH=9/1v/v)に供し、所望の生成物を得た。
【0187】
以下の化合物を上述のとおり合成し、表1に特徴付け、要約する:4,4’−(2,3−ジメチルベンジルアザンイジル)ジフェノール(III)、4−((4−フルオロフェニル)(4−ヒドロキシベンジル)アミノ)フェノール(VIII)。
【0188】
O−(2−ピペリジン−1−イルエトキシ)−ベンズアミド、および類似体の一般的合成。アセトン中のベンズアミド類似体(1当量)を含有するヒドロキシフェニルの溶液に、K2CO3(3当量)およびN−クロロエチル−ピペリジン塩酸塩(1.2当量)を添加した。溶液を、6時間、加熱して還流させた。乾燥するまで溶液を蒸発させた。残渣を、水を添加することにより加水分解し、次いで、酢酸エチルで抽出した。有機層を分離し、無水MgSO4上で乾燥させた。溶媒を減圧下で除去した。残渣を、塩化メチレン/メタノール=9/1 v/vで、フラッシュクロマトグラフィーによって精製し、所望の化合物を得た。Et2O中のHClを化合物のメタノール溶液に添加した後、溶媒を蒸発させることにより、塩酸塩を調製した。
【0189】
以下の化合物を上述のとおり合成し、表1に特徴付け、要約する:4−フルオロ−N−(4−ヒドロキシフェニル)−N−(4−(2−ピペリジン−1−イル)エトキシ)フェニル)−2−(トリフルオロメチル)ベンズアミド(IX)、および4−フルオロ−N−(4−ヒドロキシフェニル)−N−(4−(2−(ピペリジン−1−イル)エトキシ)フェニル)−2−(トリフルオロメチル)ベンズアミド塩酸塩(X)(IXのHCl塩である)。
【0190】
【表1】
実施例2
式IVの化合物の合成(図6)。
【0191】
【化32】
4−フルオロアニリン(78.63g、0.708mol)、4−ヨードアニソール(138.00g、0.590mol)、無水K2CO3(122.23g、0.884mol)、CuI(11.23g、58.96mmol)、およびL−プロリン(13.58g、0.118mol)の混合物をともに、攪拌棒、還流冷却器、およびアルゴン導入口を装着した、乾燥した1Lの三つ口丸底フラスコ中で混合した。無水DMSO(300mL)を室温で添加した。反応混合物を、アルゴン下で、20時間、90℃で攪拌および加熱した。次いで、混合物を室温まで冷却し、水(300mL)で加水分解した。EtOAc(200mL)を添加して、溶液を分割した。EtOAc層を分離した。水性層を、100mLのEtOAcで抽出した。EtOAc層を混合し、ブライン(2×100mL)で洗浄し、無水MgSO4(50g)上で乾燥させた。溶媒を減圧下で除去した。褐色油の残渣を、フラッシュカラムクロマトグラフィー(シリカゲル、ヘキサン/EtOAc=9/1v/v)によって精製し、黄色固体生成物(99.70g、77.8%の収率)として、4−フルオロ−N−(4−メトキシフェニル)アニリン(1c)を得た。融点46〜48℃。MS(ESI)m/z 218.1[M+H]+,1H NMR(DMSO−d6,300MHz)δ7.77(bs,1H),7.03−6.98(m,4H),6.93−6.82(m,4H),3.70(s,3H)。
【0192】
【化33】
4−フルオロ−N−(4−メトキシフェニル)アニリン(1c)(90.78g、0.418mol)、および3−フルオロ−4−塩化メトキシベンゾイル(94.55g、0.501mol)を、攪拌棒、還流冷却器、およびアルゴン導入口を装着した、乾燥した1Lの三つ口丸底フラスコ内で、ともに混合し、無水THF(200mL)中で溶解した。無水ピリジン(132.22g、1.672mol)を、アルゴン下で、室温で、注射器を介して添加した。反応混合物を、攪拌し、加熱して一晩還流させた。次いで、反応混合物を室温まで冷却し、ろ過して、ピリジン塩を除去した。溶液を濃縮して、THF溶媒を除去した。残渣油を、200mLの2N HCl溶液で洗浄し、酢酸エチル(2x200mL)で抽出した。混合した有機層を、飽和Na2CO3水溶液(150mL)で洗浄して、過剰塩化ベンゾイルおよび酸を除去し、MgSO4(50g)上で乾燥させ、ろ過し、減圧下で濃縮して、油を得た。残渣を、CH2Cl2/アセトン(50/1v/v)を有する、シリカゲル使用するフラッシュカラムクロマトグラフィーによって精製し、黄色固体として純粋な対応するベンズアミド化合物を得た。融点54〜56℃。MS(ESI)m/z 370.1[M+H]+,1H NMR(CDCl3/TMS)δ7.24−7.11(m,4H),7.05−6.97(m,4H),6.85−6.78(m,3H),3.86(s,3H),3.79(s,3H)。
【0193】
【化34】
化合物3−フルオロ−N−(4−フルオロフェニル)−4−メトキシ−N−(4−メトキシフェニル)ベンズアミド(2a)(138.0g、0.374mol)を、アルゴン下で、室温で、無水CH2Cl2(600mL)中で溶解した。BBr3(374.75g、1.496mol)を、アルゴン下で、氷浴中で、0℃で、注射器を介して、攪拌しながら滴加した。反応溶液を、一晩、室温で攪拌した。次いで、溶液を、1Lの氷水に攪拌しながら注入した。スラリー混合物を、2時間、室温で攪拌した。白色沈殿物をろ過し、水(2×100mL)で洗浄し、真空下で乾燥させた。CH2Cl2層を分離し、無水MgSO4(50g)上で乾燥させ、ろ過し、減圧下で乾燥するまで濃縮した。CH2Cl2溶液からの白色沈殿物および残渣を混合し、フラッシュカラムクロマトグラフィー(シリカゲル、CH2Cl2/アセトン/MeOH=90/7/3v/v/v)によって精製して、淡黄褐色固体を得、それを、二度、熱いEtOAc/ヘキサン溶液から再結晶させて、白色結晶固体(104.0g、81.6%の収率)を得た。融点110〜112℃。MS(ESI)m/z 364.1[M+Na]+,1H NMR(DMSO−d6)δ10.14(bs,1H),9.71(bs,1H),7.25−7.11(m,5H),7.05−6.99(m,3H),6.78(t,J=8.6Hz,1H),6.68(d,J=8.7Hz,2H)。
実施例3
式VIの化合物の合成(図7)
4−(ベンジルオキシ)−N−(4−メトキシフェニル)アニリン(1d)の合成
4−ベンジルオキシアニリン(16.6g、83.31mmol)、4−ヨードアニソール(15.0g、64.09mmol)、K2CO3(17.72g、128.18mmol)、CuI(1.22g、6.41mmol)、およびL−プロリン(1.48g、12.82mmol)の混合物をともに、室温で混合し、無水DMSO(120mL)中で溶解した。次いで、反応混合物を、48時間、90℃で攪拌および加熱した。混合物を室温まで冷却し、水で加水分解した。EtOAcを添加して、溶液を分割した。EtOAc層を分離し、ブラインで洗浄し、無水MgSO4上で乾燥させた。溶媒を減圧下で除去した。固体残渣を、EtOAc/ヘキサン(1/9 v/v)を使用するフラッシュカラムクロマトグラフィー(シリカゲル)によって精製し、黄色固体(9.8g、50%の収率)として、対応するジアリールアニリンを得た。融点108.0〜108.4℃。1H NMR(CDCl3,300MHz)δ7.34−7.25(m,5H),6.90−6.81(m,8H),5.02(s,2H),3.78(s,3H)。MS m/z 306(M+H)+。
N−(4−ベンジルオキシフェニル)−N−(4−メトキシフェニル)−2−ナフトアミド(2d)の合成
1当量の4−(ベンジルオキシ)−N−(4−メトキシフェニル)アニリン(0.80g、2.62mmol)を、磁気攪拌棒および還流冷却器を装着した、乾燥した三つ口丸底フラスコ内で、1.5当量の2−塩化ナフトイル(0.75g、3.93mmol)、および4当量のピリジン(0.83g、10.48mmol)と混合した。混合物を、無水THF(30mL)中で溶解し、加熱して、20時間還流させた。反応溶液を室温まで冷却し、ろ過した。溶媒を減圧下で除去した。残渣を、EtOAc/ヘキサン(3/7 v/v)を用いる、シリカゲルを使用するフラッシュカラムクロマトグラフィーによって精製し、白色固体(0.70g、58%の収率)として、純粋な対応するナフトアミド化合物を得た。融点174.9〜175.5℃。1H NMR(CDCl3,300MHz)δ8.04(s,1H),7.77−7.74(m,2H),7.64−7.61(m,1H),7.51−7.43(m,4H),7.40−7.31(m,4H),7.13−7.10(m,4H),6.88−6.78(m,4H),4.99(s,2H),3.74(s,3H)。MS m/z 460(M+H)+。
N,N−ビス(4−ヒドロキシフェニル)−2−ナフチルアミド(VI)の合成。
【0194】
化合物N−(4−ベンジルオキシフェニル)−N−(4−メトキシフェニル)−2−ナフトアミド(2d)(0.50g、1.09mmol)を、室温で、無水CH2Cl2(30mL)中で溶解した。BBr3(3.26mLの1.0MのCH2Cl2溶液、3.26mmol)を、室温で、注射器を介して滴加した。反応溶液を、室温で一晩攪拌した。混合物を、氷浴内で、0℃まで冷却し、水を添加して加水分解した。EtOAcを添加して、溶液を分割した。有機層を分離し、水性層を、二度、EtOAcで抽出した。有機層を混合し、ブラインで洗浄し、無水MgSO4上で乾燥させた。溶媒を真空下で除去した。残渣を、CH3OH/CH2Cl2(1/9 v/v)を用いる、シリカゲルを使用するフラッシュカラムクロマトグラフィーによって精製して、白色固体(0.27g)、白色固体(70%の収率)として、純粋な所望のフェノール化合物を得た。融点264.3〜265.2℃(分解)。1H NMR(DMSO−d6,500MHz)δ9.46(s,2H),7.98(s,1H),7.85−7.75(m,2H),7.75−7.73(m,2H),7.54−7.48(m,2H),7.45−7.43(m,1H),7.05(s,4H),6.66(s,4H)。MS m/z 356(M+H)+。
実施例4
式VIIIの化合物の合成
【0195】
【化35】
化合物N−(4−フルオロフェニル)−4−ヒドロキシ−N−(ヒドロキシフェニル)ベンズアミド(0.30g、0.93mmol)を、室温で、20mLの無水THF中で溶解した。H3B(SMe2)(1.86mLの2MのTHF溶液、3.71mmol)を、アルゴン下で、室温で、注射器を介して添加した。反応溶液を、攪拌し、加熱して、6時間還流させた。次いで、0℃の10mLのMeOHを添加することによって、反応を停止した。溶媒を減圧下で除去した。残渣を、フラッシュカラムクロマトグラフィー(シリカゲル、CH2Cl2/MeOH=9/1 v/v)に供し、黄色油(0.26g、92%の収率)を得た。1H NMR(DMSO−d6,500MHz)δ9.29(s,1H),9.24(s,1H),7.09(d,2H,J=8.3Hz),6.98(d,2H,J=9.0Hz),6.94−6.91(m,2H),6.73(d,2H,J=9.0Hz),6.68−6.64(m,4H),4.70(s,2H)。MS m/z 307.8(M−H)。
実施例5
化合物IXおよびXの化合物の合成(図8)。
【0196】
ジアリールアニリンの合成。アリールアミン(1.5当量)、ヨードアリール(1当量)、K2CO3(2当量)、CuI(0.1当量)、およびL−プロリン(0.2当量)の混合物をともに、室温で混合し、無水DMSO中で溶解した。次いで、反応混合物を、28時間、90℃で攪拌および加熱した。混合物を室温まで冷却し、水で加水分解した。EtOAcを添加して、溶液を分割した。EtOAc層を分離し、ブラインで洗浄し、無水MgSO4上で乾燥させた。溶媒を減圧下で除去した。固体残渣を、溶媒としてEtOAc/ヘキサン(3/7 v/v)を使用するフラッシュカラムクロマトグラフィー(シリカゲル)によって精製して、対応するジアリールアニリンを得た。ビス−(4−メトキシフェニル)アミン(1a):淡黄色固体、73%の収率。1H NMR(CDCl3,300MHz)δ6.93−6.81(m,8H),5.37(s,br,1H),3.78(s,6H)。MS m/z 228.4(M−H)+。
4−フルオロ−N,N−ビス(4−メトキシフェニル)−2−(トリフルオロメチル)ベンズアミド(2e)の合成
1当量のビス−(4−メトキシフェニル)アミン(1a)(0.73g、3.18mmol)を、磁気攪拌棒および還流冷却器を装着した、乾燥した三つ口丸底フラスコ内で、1.2当量の4−フルオロ−2−塩化トリフルオロメチルベンゾイル(0.87g、3.82mmol)、および6当量のピリジン(1.51g、19.08mmol)と混合した。混合物を無水THF(20mL)中で溶解し、90℃で20時間加熱した。反応溶液を室温まで冷却し、ろ過した。溶媒を減圧下で除去した。残渣を、EtOAc/ヘキサン(3/7 v/v)を有する、シリカゲルを使用するフラッシュカラムクロマトグラフィーによって精製して、無色油(1.12g、84.2%の収率)として、純粋な対応するベンズアミド化合物を得た。1H NMR(CDCl3,300MHz)δ7.34−7.26(m,4H),7.09−7.01(m,3H),6.91(d,2H,J=8.7Hz),6.87(d,2H,J=8.7Hz),3.80(s,3H),3.71(s,3H)。MS m/z 442.1(M+Na)+。
4−フルオロ−N,N−ビス(4−ヒドロキシフェニル)−2−(トリフルオロメチル)ベンズアミド(3a)の合成。
【0197】
化合物4−フルオロ−N,N−ビス(4−メトキシフェニル)−2−(トリフルオロメチル)ベンズアミド(2e)(1.00g、2.38mmol)を、室温で、無水CH2Cl2(30mL)中で溶解した。BBr3(10mLの1.0MのCH2Cl2溶液、10.0mmol)を、室温で、注射器を介して滴加した。反応溶液を室温で一晩攪拌した。混合物を、氷浴内で0℃まで冷却し、水を添加して加水分解した。EtOAcを添加して、溶液を分割した。有機層を分離し、水性層を、二度、EtOAcで抽出した。有機層を混合し、ブラインで洗浄し、無水MgSO4上で乾燥させた。溶媒を真空下で除去した。残渣を、CH3OH/CH2Cl2(1/9 v/v)を有する、シリカゲルを使用するフラッシュカラムクロマトグラフィーによって精製して、白色固体(0.86g、92.5%の収率)として、純粋な所望のフェノール化合物を得た。1H NMR(DMSO−d6,300MHz)δ9.55(s,1H),9.53(s,1H),7.69−7.58(m,2H),7.46−7.39(m,1H),7.18(d,2H,J=8.7Hz),6.93(d,4H,J=8.7Hz),7.03(d,2H,J=8.4Hz),6.78(d,2H,J=8.7Hz),6.57(d,2H,J=8.7Hz)。MS m/z 392.1(M+H)+。
4−フルオロ−N−(4−ヒドロキシフェニル)−N−[4−(2−ピペリジン−1−イル)エトキシ]フェニル]−2−(トリフルオロメチル)ベンズアミド(IX)の合成
アセトン中の4−フルオロ−N,N−ビス(4−ヒドロキシフェニル)−2−(トリフルオロメチル)ベンズアミド(3a)(0.61g、1.56mmol)の溶液に、K2CO3(1.29g、9.36mmol)、およびN−クロロエチル−ピペリジン塩酸塩(0.34g、1.87mmol)を添加した。溶液を加熱して20時間還流させた。乾燥するまで溶液を蒸発させた。残渣を、フラッシュクロマトグラフィー(シリカゲル、塩化メチレン/メタノール=9/1 v/v)によって精製して、白色固体(0.45g、57.5%の収率)として、所望の化合物を得た。1H NMR(DMSO−d6,300MHz)δ9.57(s,1H),7.71−7.68(m,2H),7.47−7.44(m,1H),7.28(d,1H,J=9.0Hz),7.18(d,1H,J=8.7Hz),7.13(d,1H,J=8.7Hz),7.05(d,1H,J=8.4Hz),6.97(d,1H,J=9.0Hz),6.80−6.76(m,2H),6.57(d,1H,J=87。Hz),4.06(t,1H,J=6.0Hz),3.93(t,1H,J=6.0Hz),2.66(t,1H,J=5.7Hz),2.55(t,1H,J=5.4Hz),2.44(s,2H),2.36(s,2H),1.49−1.37(m,6H)。MS m/z 501.0(M−H)。
【0198】
Et2O中のHClを化合物のメタノール溶液に添加した後に、溶媒を蒸発させることによって塩酸塩(X)を調製した。
実施例6
エストロゲン受容体結合親和性、作動薬および拮抗薬活性
インビボ競合的ラジオリガンド結合アッセイを使用して、天然の高親和性ERリガンドである[2,4,6,7−3H(N)]−エストラジオール([3H]E2)、および細菌で発現したGST縮合ER−α、またはER−βリガンド結合ドメイン(LBD)タンパク質との化合物のET結合親和性を決定した。
方法
組み換えER−α、またはER−βを、[3H]E2と組み合わせて、[3H]E2の平衡解離定数(Kd)を決定した。全結合または非特異的結合を決定するために、タンパク質を、漸増濃度の[3H]E2とともに、高濃度の非標識E2の存在下または不在下で、18時間、4℃で培養した。非特異的結合を減算し、非線形回帰を使用して、E2のKd(ERα:0.71nM、ERβ:1.13nM)を判定した。加えて、ER−αおよびER−βLBDを飽和させるのに必要とされる[3H]E2の濃度は、4〜6nMであると決定した。
【0199】
漸増濃度の化合物(範囲:10−11〜10−6M)を、上述の条件を使用して、[3H]E2(5.7nM)、およびER LBDとともに培養した。培養後、プレートを、Unifilter−96 Harvester(PerkinElmer)上のGF/Bろ過で採取し、氷冷緩衝液B(50mMトリス、pH7.2)で、3度洗浄した。ろ過プレートを室温で乾燥させ、次いで、35μlのMicroscint−Oカクテルを各ウェルに添加し、ろ過プレートをTopSeal−Aで密封した。放射能を、Microscintカクテル(PerkinElmer)内の3Hの設定を使用して、TopCount(R)NXTマイクロプレートシンチレーション計測器内で計数した。
【0200】
化合物の各濃度における、[3H]E2の非特異的結合を減算することによって、[3H]E2の特異的結合を決定(10−6Mの非標識E2とともに培養することによって決定)し、それを、試験化合物の不在下での特異的結合に対する割合で示した。[3H]E2の特異的結合を50%減少させる化合物の濃度(IC50)を決定した。次いで、化合物の平衡結合定数(Ki)を、Ki=Kd×IC50/(Kd+L)により、算出した。式中、Kdは、[3H]E2(ER−α=0.71nM、ER−β=1.13nM)の平衡解離定数であり、Lは、[3H]E2(ER−α:5.7nM、ER−β:5.7nM)の濃度である。
結果
結合アッセイは、リガンドが、3.75nMから1000nM以上の範囲の種々の濃度、および化合物が、イソ型選択的から、非イソ型選択的である選択的範囲において、ER−α、およびER−βに結合したことを明らかにした。代表的化合物の結果を表2に記載する。
【0201】
【表2】
化合物IVは、ERαおよびERβに結合する。インビトロ競合的ラジオリガンド結合アッセイを使用して、[2,4,6,7−3H(N)]−エストラジオール([3H]E2)、天然の高親和性ERリガンド、および細菌で発現したGST縮合ERαまたはERβリガンド結合ドメイン(LBD)タンパク質との化合物IVのER結合親和性を決定した。このアッセイでは、化合物IVのERαおよびERβ結合親和性(Ki値)はそれぞれ、21.7±1.7nM(n=3)、および15.2±4.1nM(n=3)であった。ERへの結合の際、化合物IVは、組織選択的な方法で、薬理反応に関与する標的遺伝子の発現または抑制につながる、複雑な一連の分子事象を開始する。遷移トランスフェクションアッセイでは、化合物IVは、ERαおよびERβ作動薬であり、ERβ媒介転写活性化と比較して、ERα媒介転写活性化を刺激する効率がより高いことが示された。エストラジオールはERαおよびERβを活性化するが、ERαに対する選択性は5.1倍高い。一方、化合物IVは、ERαに対して、49.0倍の選択性を示す。したがって、相対的トランス活性化能(エストラジオール値に標準化)において、化合物IVは、ERβよりもERαに対して相対的に9.7倍の選択性を有する。さらに、拮抗薬作用は、最大10μMの濃度まで、化合物IVのエストラジオール(1nM)−刺激転写活性化において認められなかった。多くのステロイドリガンドは、他の核ホルモン受容体と交差反応するが、化合物IVの作用は、ERαおよびERβに対して特異的である。化合物IVを、転写活性化アッセイにおける作動薬および拮抗薬モードの両方で、グルココルチコイド受容体(GR)のラットイソ型、ミネラルコルチコイド受容体(MR)のラットイソ型、プロゲステロン受容体(PR)のラットイソ型、アンドロゲン受容体(AR)のラットイソ型、ならびにファルネソイドX受容体(FXR)のヒトイソ型、肝臓X受容体(LXR)、ペルオキシソーム活性受容体(PPAR−αおよびPPAR−γ)のヒトイソ型、およびレチノイドX受容体(RXR−α)のヒトイソ型に対する交差反応に対してスクリーニングした。化合物IVは、これらのアッセイのうちのいずれかにおいて、任意の作動薬活性または拮抗薬活性を表わさず、化合物IVが、これらの核ホルモン受容体スーパーファミリー成員と機能的に交差反応しないという結論を支持した。
実施例7
選択した化合物のトランス活性化
作動薬および拮抗薬モードでトランス活性化アッセイを実施して、化合物が、作動薬、拮抗薬、または、部分的であるか否かを特定した。
【0202】
方法
ラットエストロゲン受容体(ER−αおよびER−β)を、ラット卵巣cDNAから、pCR3.1プラスミドベクター骨格にクローン化した。配列決定を実施して、任意の突然変異の欠如を判定した。HEK−293細胞を、ダルベッコ最小必須培地(DMEM)+5%活性炭除去ウシ胎児血清(csFBS)において、プレーティングした。0.25μgのERE−LUC、0.02μgのCMV−LUC(ウミシイタケルシフェラーゼ)、および12.5ngのラットER−α、または25ngのラットER−βを有する、Lipofectamine(Invitrogen社、カリフォルニア州カールズバッド)を使用して、細胞をトランスフェクトした。種々の濃度の化合物、または化合物およびエストラジオールの組み合わせのトランスフェクト24時間後、細胞を処理して、拮抗薬活性を判定した。ルシフェラーゼアッセイを、トランスフェクト48時間後、実施した。
結果
トランス活性化システムにおける本発明の化合物のスクリーニングは、化合物が、3つのすべての分類、すなわち、作動薬、拮抗薬、および部分的作動薬に属したことを明らかにした。作動薬および拮抗薬の一例を表3に示す。トランス活性化結果は、イソ型選択性に対する結合結果と非常によく一致した。
【0203】
表3は、いくつかの選択した本発明の化合物のEC50およびIC50トランス活性化値を提供する。
【0204】
【表3】
実施例8
カニクイザルにおけるテストステロン抑制
2歳の生殖腺無損傷のオスのカニクイザル(n=2)を、霊長類の餌および水への自由なアクセス(経口投与前の絶食を除く)で、USDAガイドラインに従い、研究の間収容した。動物は、連続7日間、Tween80/脱イオン水のマイクロエマルション媒体で、30mg/kgの式IVの化合物の1日1回の強制経口投与用量が与えられた。血清サンプルを、1日目(ベースライン)、3日目、4日目、5日目、6日目、および7日目に、経口用量投与前に静脈穿刺によって採取した。HPLC方法と組み合わせた、またはそれを用いない酵素免疫アッセイ(EIA)方法のそれぞれを使用して、テストステロンおよび総アンドロゲンを定量化した。6日間の式IVの化合物での処理後、テストステロンおよび総アンドロゲン(テストステロン/ジヒドロテストステロン)では、時間依存性の減少が明らかであった。式IVの化合物は、ベースライン値に対して、テストステロンのレベルを、動物#1および動物#3ではそれぞれ、58%および64%抑制した(図1の太線、表4を参照)。同様に、総アンドロゲンレベルは、ベースライン値と比較して、動物#1および#3の両方において、56%抑制された。
【0205】
男性の下垂体−精巣軸エストロゲンフィードバックと一致して、これらの結果は、式IVの化合物の反復経口用量(30mg/kg)の後、無損傷の非ヒト霊長類(カニクイザル)における血清ホルモン(テストステロンおよび総アンドロゲン)の抑制に対して、強い薬理反応を表す。
【0206】
【表4】
実施例9
ラットにおける、LHおよびテストステロンホルモンレベルの抑制
インビボ用量反応研究を実施して、無損傷および精巣摘除された(ORX)オスのラットにおける、LH抑制への化合物IVの作用を評価した。無損傷およびORX動物において、1日当たり≧10mg/kgの用量で、化合物は、対応する対照と比較した時、LHレベルを有意に抑制した(FSHレベルにおいて、同一の抑制パターンが認められた)。LH抑制は、0.08ng/mLである、定量下限以下(BLOQ)まで、テストステロンレベルの確実な減少をもたらし、また、前立腺、精嚢、肛門挙筋はアンドロゲン依存性が高い臓器であるため、これらの重量の減少をもたらした。無損傷の動物では、これらの標的臓器の重量における用量依存性の減少は、去勢された対照のレベルに対する精嚢および肛門挙筋重量で顕著であった。前立腺重量は、無損傷の動物において有意に減少したが、これらの値は、去勢された対照のレベルに到達しなかった。結果を下記の表6に要約する。
材料および方法:
約200gの重量のオスのスプラーグドーリーラットを、不断給の餌(2016Teklad Global 16%タンパク質げっ歯類食餌、Harlan社、ウィスコンシン州マディソン)および水で、12時間の明暗サイクルで維持した。動物プロトコルが再検討され、テネシー州の動物管理使用委員会により承認された。
【0207】
本研究のための試験物品を計量し、PEG300(Acros Organics社、ニュージャージー州)で希釈した10%のDMSO(Fisher)中で溶解して、適切な用量の製剤を調製した。本研究のために、60匹のオスのスプラーグドーリーラットを、体重によって無作為化し、12の治療グループのうちの1つに割り当てた(n=5匹の動物/グループ)。処置グループを表5に記載する。動物を、ケージ当たり2〜3匹の動物のグループに収容した。対照グループ(無損傷および精巣摘除された(ORX))に、連日、媒体を投与した。化合物IVを、0.3、1、3、10、および30mg/kg/日の用量で、無損傷およびORXグループに皮下注射(200μL)を介して投与した。
【0208】
14日間の投薬レジメン後、動物を麻酔(ケタミン/キシラジン、87:13mg/kg)化で安楽死させ、体重を記録した。加えて、腹側前立腺、精嚢、および肛門挙筋を除去し、外部組織を除去し、個々に計量した。臓器重量を、体重に対して標準化し、無損傷対照の割合として示した。イソフルラン麻酔下で、腹部大動脈から血液を採取し、凝血させた。血清ホルモンレベルの判定前に、血清を遠心分離によって分離し、−80℃で保管した。血清黄体形成ホルモン(LH)および卵胞刺激ホルモン(FSH)濃度を、製造者の指示に従い、ラット下垂体ルミネックスアッセイ(Millipore社、マサチューセッツ州ビレリカ)によって決定した。このアッセイの定量下限は、LHでは、3.2pg/mL、FSHでは、32pg/mLであった。テストステロンを、0.08ng/mLのLLOQを用いる、テストステロンEIA(Alpco Dianostics社、ニューハンプシャー州セーレム)によって測定した。定量下限以下の血清ホルモン値を、グループ平均の分析から除外した。したがって、サンプルBLOQを有するグループにおけるLHおよびTの報告された値は、実際の値よりも高い。この分析方法は、LHおよびT抑制の最も控えめな推定を提供した。フィッシャーの最小有意差検定を使用して、個々の用量グループを、無損傷およびORX媒体対照グループと比較した。有意性を、P値<0.05として事前に定義した。
【表5】
無損傷およびORXラットにおける黄体形成ホルモンレベル(表6)
無損傷およびORX媒体対照グループにおけるLHレベル(平均±標準偏差)はそれぞれ、1.46±0.64、および11.1±3.9ng/mLであった。化合物IVは、無損傷動物において、LHレベルを用量依存的に低下し、1日用量≧3mg/kgで、統計的に有意な減少に達した。無損傷の化合物IVで処置された動物におけるLHレベルは、0.3、1、3、10、および30mg/kg/日の用量後にそれぞれ、0.863±0.384、0.704±0.530、0.395±0.302、0.226±0.165、および0.236±0.176ng/mLであった。ORXのオスにおけるLHレベルもまた、化合物IV治療によって有意に減少した。ORX動物では、LHレベルは、0.3、1、3、10、および30mg/kg/日の用量後にそれぞれ、15.4±2.9、13.5±2.2、6.5±5.6、0.4.425±0.135、および0.368±0.119ng/mLであった。結果を、図10Aに図式的に表す。
無損傷およびORXラットにおける卵胞刺激ホルモンレベル(表6)
無損傷およびORX媒体対照グループにおける血清FSHレベルは、それぞれ、20.9±8.5、および93.5±13.8ng/mLであった。無損傷動物では、化合物IVは、≧10mg/kg/日の用量で認められた有意な減少で、FSHレベルを用量依存的に減少させた。無損傷の化合物IVで処置された動物におけるFSHレベルは、0.3、1、3、10、および30mg/kg/日の用量後にそれぞれ、17.3±6.4、15.7±7.3、18.4±7.7、9.2±4.0.および6.3±1.8ng/mLであった。ORX動物では、LHレベルは、0.3、1、3、10、および30mg/kg/日の用量後にそれぞれ、115±17、114±22、65.2±31.9、27.6±8.2、および15.1±4.1ng/mLであった。結果を、図10Bに図式的に表す。
無損傷およびORXラットにおけるテストステロンレベル
無損傷媒体対照グループにおける血清テストステロンレベルは、2.4±1.1ng/mLであった。Tの定量下限は、0.08ng/mLであった。0.08ng/mL未満の値を、定量下限以下(BLOQ)として指定した。無損傷動物では、式IVの化合物は、1日当たり≧3mg/kgの用量で認められた有意な減少で、Tレベルを用量依存的に低下させた。式IVの化合物で処置された無損傷動物におけるテストステロンレベルは、1日当たり0.3、1、3、10、および30mg/kgの用量後にそれぞれ、2.6±1.7、1.6±1.0、0.7±0.4、BLOQ、およびBLOQng/mLであった。ORX動物では、Tレベルは、化合物IVで治療されたすべてのグループ、および媒体で処置されたグループでは、BLOQであった。無損傷動物では、結果を図10C(および図2)に図式的に表す(BLOQ値を、図式目的のために、定量限界で表す)。
【0209】
図9に表すとおり、無損傷のオスのラットにおいて、3mg/kg、10mg/kg、および300mg/kgの用量で化合物IVを投与することにより、24時間後、72時間後、168時間後に、血清テストステロンの迅速かつ強力な抑制が測定された。
臓器重量(表6)
前立腺、精嚢、および肛門挙筋重量を測定して、Tの抑制を確認した。臓器重量(平均±標準偏差)をそれぞれ、図10D、10E、および10Fに表す。前立腺、精嚢、肛門挙筋重量における用量依存性の低下が、化合物IVで処置された無損傷動物において認められた。無損傷動物における前立腺重量は、0.3、1、3、10、および30mg/kg/日の用量後にそれぞれ、84.0±19.2、75.2±20.7、68.2±8.2、45.1±20.0、および43.6±8.8であった。ORX動物における前立腺重量は、0.3、1、3、10、および30mg/kg/日の用量後にそれぞれ、19.0±4.2、17.4±3.4、19.6±6.7、22.9±5.4、および20.6±2.1であった。無損傷動物における精嚢重量は、0.3、1、3、10、および30mg/kg/日の用量後にそれぞれ、76.2±7.8、66.3±27.2、51.8±28.5、19.1±7.0、および17.9±3.3であった。ORX動物における精嚢重量は、0.3、1、3、10、および30mg/kg/日の用量後にそれぞれ、12.2±1.3、16.6±5.4、16.5±4.8、13.3±1.9、および12.9±2.1であった。無損傷動物における肛門挙筋重量は、0.3、1、3、10、および30mg/kg/日の用量後にそれぞれ、86.9±10.0、82.1±12.1、65.2±4.4、57.8±11.2、および58.1±4.7であった。ORX動物における肛門挙筋重量は、0.3、1、3、10、および30mg/kg/日の用量後にそれぞれ、54.5±6.6、49.6±7.0、53.6±10.0、51.1±4.9、および49.2±4.2であった。
LH抑制および臓器重量データを、表6に要約する。
【0210】
【表6】
実施例10
ラットおよびサルにおける化合物IVによる抑制後のテストステロンレベルの回復
化合物IVとの化学的去勢の可逆性を研究した。
材料および方法
約200gの重量の35匹のオスのスプラーグドーリーラットを、不断給の餌(2016 Teklad Global 16%タンパク質げっ歯類食餌、Harlan社、ウィスコンシン州マディソン)および水で、12時間の明暗サイクルで維持した。動物プロトコルは再検討され、テネシー州の動物管理使用委員会により承認された。
【0211】
本研究のための試験物品を計量し、PEG300(100%)(Acros Organics社、ニュージャージー州)中で溶解して、適切な用量の製剤を調製した。動物を、10の処置グループのうちの1つに無作為に割り当てた(n=5匹の動物/グループ)。処置グループを表7に記載する。動物を、ケージ当たり2〜3匹の動物のグループに収容した。無損傷動物におけるベースラインの決定のために、グループ1を、研究の開始(1日目)に安楽死させた。グループ2〜7は、3日間、強制経口投与(〜200μL)を介して、1、3、または30mg/kgの1日用量を受けた。グループ2、3、および4を、4日目に安楽死させ、最大テストステロン抑制を測定した。グループ5、6、および7を、薬物を与えない休薬期間で14日間、回復させた。
【表7】
結果:
無損傷ラットにおける血清テストステロンレベルは、ベースラインにおいて、6.4±3.1ng/mL(平均±標準偏差)であった。3日間、3、および30mg/kgの用量で投与された化合物IVは、血清テストステロンレベルをそれぞれ、1.47±0.26、および1.62±0.49まで有意に抑制した。有意な抑制は、3日間、1mg/kgの化合物IVを受けた動物において認められなかった。最も重要なことには、14日間の回復期間後に測定された時、血清テストステロンレベルは、3日間、1、3、または30mg/kgの化合物IVを受けた動物においてそれぞれ、3.3±1.92、3.00±1.06、および3.8±1.72であり、図23に図示するとおり、無損傷ラットにおけるベースライン血清テストステロン濃度とは統計的に有意に異ならなかった。
【0212】
本研究は、化合物IVが、無損傷のオスのラットにおける血清テストステロンレベルを迅速に抑制することを示す前回の結果を裏付けている。我々は、3日間、≧3mg/kg/日を受けた用量グループにおいて、血清テストステロンレベル抑制を認めた。血清テストステロンの有意な減少は、1mg/kgの用量グループで認められなかった。しかしながら、14日間の回復以内で、血清テストステロンレベルは、無損傷対照のレベルまで戻った。本研究は、化合物IVによる薬理学的去勢は、ラットにおいて可逆性であることを示す。
【0213】
無損傷のオスのサルにおけるテストステロンレベルの抑制および回復への化合物IVの作用を、経口薬物動態研究と併せて評価した。これらの治療に使われていない3匹のオスのカニクイザル(2〜3歳)に、連続7日間、強制経口投与により、30mg/kgで化合物IVを投与した。血液サンプルを採取し、テストステロンおよび化合物IVの定量的測定のために、それぞれ、血清および血漿に分割した。結果は、化合物IVの1日経口用量が、ベースラインレベル(治療のベースライン、2日目、および6日目[平均±SEM]では、それぞれ、1591±72.5、997±104、および852±136ng/mLのレベル)と比較して、3匹のサルすべてにおいて、循環アンドロゲン(主に、テストステロンおよびジヒドロテストステロン)レベルを最大47%ほど有意に低下させたことを示す。18日間の薬物フリーの回復期間後、アンドロゲンレベルは、通常に戻り、治療前のベースラインレベル(回復後、1757.7±369.5ng/mL)とは、有意に異ならなかった。
実施例11
ラットにおけるLHおよびテストステロンの減少に反する骨の保存(表8)
骨への処置に対する式IVの化合物の作用を研究した。経口的に投与された、式IVの化合物は、無損傷のオスのラットにおいて、LH抑制に伴う骨量の減少を完全に防止した。LHの有意な減少は、無損傷動物において、1日当たり≧10mg/kgの用量レベルで、式IVの化合物によって誘発された。1日当たり1mg/kgでは、式IVの化合物はLHレベルを有意に減少させなかったものの、前立腺、精嚢、および肛門挙筋における有意な減少は、この用量で明らかであり、循環テストステロンの低下が、生理学的に、これらのアンドロゲン反応臓器に関連することを示す。しかしながら、1日当たり1mg/kgの式IVの化合物は、骨梁量(大腿遠位において測定)を無損傷対照のレベルに維持した。1日当たり10、および30mg/kgの用量で投与された時、式IVの化合物は、無損傷対照の大腿遠位よりも大腿遠位の骨量を有意に増大させた。これらのデータは、化合物IVが、無損傷ラットにおいてLHレベルを低減する用量レベルで、骨梁塩濃度(BMD)、および骨量の割合を増大させたことを示す。本研究からのデータを表8に示す。_Ref216166047_Toc217378480
【0214】
【表8】
実施例12
17βヒドロキシステロイド脱水素酵素5(17β−HSD5)酵素活性への作用
HSDファミリー成員は、循環ステロイドの転換に関与する。17β−HSD5は、アンドロステンジオンをテストステロンに、エストロンをエストラジオールに転換する。加えて、それはまた、プロスタグランジン合成にも関与する。ここで、いくつかの選択した本発明の化合物の17β−HSD5活性を阻害する能力を示す。
方法
ヒト17β−HSD5を、pGEX4t1ベクターにクローン化し、精製タンパク質を調製した。精製タンパク質を、適切な緩衝液中の代表的な本発明の化合物、14Cアンドロステンジオン、およびNADPHとともに培養した。合成テストステロンを、酢酸エチルを使用して抽出し、空気乾燥させ、薄層クロマトグラフィー(TLC)プレート上にスポットし、展開した。TLCをホスフォイメージャーに暴露し、テストステロンバンドの強度を定量化した。インドメトシンを陽性対照(LHRH作動薬)として使用した。
結果
化合物IVを試験し、それは、17β−HSD5酵素活性への部分的阻害作用を有した。陽性対照(LHRH作動薬)であるインドメトシンは、図3に示すとおり、予想通り、この酵素の強力な阻害を呈した。
実施例13
毒性研究
インビトロヒト血小板凝集アッセイを使用して、化合物IVおよびジエチルスチルベストロール(DES、陽性対照)の血栓の可能性を比較するために、研究を実施した。男性が化合物IV(LH抑制)の対象とする治療群であるため、健康な男性ドナーからの血液を研究に使用した。多血小板血漿を、30秒間、エストラジオール(E2)、DES、化合物IV、または媒体のいずれかとともに、事前に培養し、次いで、トロンビン(0.3単位)を添加して、血小板凝集を誘発した。研究結果は、DESを用いる事前培養が、トロンビン誘発血小板凝集を約10倍ほど増大させたことを示す。しかしながら、化合物IVおよびエストラジオールは、多血小板血漿において凝集を減少した。これらのデータは、化合物IVが、DESと比較して、インビトロでのヒト血小板の反応性を低減させたことを示し、化合物IVが、DESよりも低い血栓塞栓症の可能性を有し得ることを示唆する(図4)。
実施例14
ほてりへの化合物IVの作用
Simpkinsら(1983)によって開発され、閉経後のほてりとの種々の類似点を有することが示されている、モルヒネ依存ラットモデル(MDモデル)を使用して研究を実施し、ほてりへの化合物IVの作用を調査した。ヒトの状態との類似点に加えて、この実験動物モデルは、短い応答時間を有しているため、血管運動症状を緩和することのできる化合物を、尾部皮膚温度(TST)を使用して特定できる有用なハイスループットスクリーニングのツールである。TSTプローブTA−40(Data Sciences International社、ミネソタ州)を、尾基底にテープで貼りつけ、15分間、ベースライン温度を取得した。15分後、動物を、ナロキソン(1mg/kg、SQ)で処理して、モルヒネの作用を減弱させた。尾部皮膚温度(TST)を、ナロキソン処理後に、1時間、一連の実験にわたり、5秒のサンプリング度数で測定した。データ取得の後、各動物で60秒毎に記録された、温度の移動平均を算出し、さらに分析した。ベースライン温度を、ナロキソン投与前に、15分間にわたって取得された平均温度として計算した。線形台形法を使用して、ナロキソン投与後のすべての値をベースラインから減算することによって、湾曲(AUC)の下の面積を算出した。
【0215】
化合物IVは、モルヒネ禁断モデル(図13を参照)におけるほてりを軽減し、10mgの化合物IVで最良結果となった。17βE2を、100%のDMSO中で、5mg/kgで使用した。
実施例15
ラットにおける化合物IV対DES
LHRH作動薬の導入前に、去勢テストステロンレベルを、エストロゲン、主に、ジエチルスチルベストロール(DES)を介して、下垂体内のエストロゲン活性を増大させることによって達成した。DESは、テストステロンを去勢レベルに抑制する上でLHRH作動薬と同等に有効であった。DESで治療された患者は、ほてりまたは骨量の減少を有さなかったが、LHRH作動薬を用いたADTよりも高い比率で女性化乳房を有した。残念ながら、DESおよびエストラジオール等の非常に強力な純粋エストロゲンは、しばしば、それらの臨床使用を制限する、深刻な心臓血管および血栓塞栓性合併症の高リスクを伴う。DESでの静脈血栓塞栓性合併症のリスクの増加は、他のホルモン受容体とのその交差反応性によるものであると仮定されているが、証明されてはいない。ヒト血小板を用いるインビトロ研究は、化合物IVが、DESよりもさらに低い前凝固化成を有したことを明らかにした。したがって、ER−α選択的作動薬である化合物IVは、骨粗しょう症または有害な脂質プロファイルを引き起こすことなく、DESの前立腺癌の利点をもたらし得、LHRH作動薬の利点ももたらし得る。
【0216】
化合物IVは、ラットにおける前立腺の寸法を減少させ(図11A)、ORXラットの前立腺の寸法の中等度の増大を発現させる(図11B)上で、DESと同様に有効である。
【0217】
DESと化合物IVとの間の差異を、図12A〜12Cに提示する。DESは、グルココルチコイド受容体(GR)(図12A)、およびアンドロゲン受容体(AR)(図12B)と交差反応したが、化合物IVは、交差反応しなかった。加えて、DESは、エストロゲン関連受容体(ERR)トランス活性化を拮抗したが、化合物IVは、拮抗しなかった。化合物IVは、図12Cに図示するとおり、3つのERRイソ型(ERR−α、ERR−β、およびERR−γ)との交差反応に失敗した。
実施例16
サルの毒性研究−90日間
モーリシャス生息のコロニー繁殖のマカク属カニクイザルを入手した。前向き研究を、13週間の中間期間を有する、オスのカニクイザルにおける、化合物IVおよび陽性対照(LHRH作動薬)の39週間の経口薬理学および毒物学評価として、設計した。合計39匹の性成熟したオスのサル(5〜8歳)を、治療開始前に、5つのグループに無作為に割り当てた。グループには、1)媒体対照、2)1mg/kgの化合物IV、3)10mg/kgの化合物IV、4)100mg/kgの化合物IV、および5)陽性対照(LHRH作動薬)が含まれた。薬物を、グループ1および5では、媒体対照物品(Tween 80/PRANG(商標))を、グループ2、3、および4では、媒体中の化合物IVを用いて、39週間、1日1回、ケージ横投与によって、経口送達した。化合物IVの用量レベルは、グループ2、3、および4ではそれぞれ、1、10、100mg/kg/日であった。経口用量を、各動物の最新の利用可能な体重に基づき算出したとおり、10mL/kg用量体積で送達した(図14)。グループ5の動物はまた、39週間の研究期間の間、1日1回、陽性対照(LHRH作動薬)(0.02mLの定容量)の皮下注射を受けた。連日、一般的外見および臨床兆候を観察し、記録した。定期的評価および他の選択研究調査を、研究プロトコルに示すとおり、実施した。選択パラメータは、テストステロン、前立腺特異的抗原(PSA)、および前立腺体積および重量を含むが、これらに限定されない。
【0218】
テストステロンおよび全PSAレベルを、酵素免疫アッセイ(EIA)方法および化学発光免疫アッセイ(LIA,ALPCO Diagnostics社、ニューハンプシャー州セーレム)をそれぞれ使用して、(標準的処置に従い)血清サンプル中で定量化した。テストステロン評価の血液サンプルを、ベースライン(すなわち、治療開始前)、および1日目、3日目、7日目、14日目、28日目、64日目、および90日目に、すべての動物(絶食状態)から採取した。PSA決定のための血液試料を、ベースラインおよび6週目中に、すべての動物(絶食状態)から採取した。考察目的のために、テストステロンおよびPSAアッセイで、定量下限以下(BLQ)の濃度を有するサンプルの結果を、アッセイの定量下限(LLOQ)の半分として算出し、「推定最終濃度」と見なす。表9〜16のデータを、「推定最終濃度」(すなわち、アッセイのLLOQの半分として含まれた、BLQ結果を有するサンプル)に加えて、「定量化可能濃度のみ」(すなわち、BLQ値を除く)として提示する。ベースライン、および6週目に、経直腸超音波(TRUS)処置を使用して、前立腺体積を麻酔下の生きた動物で測定した。前立腺の幅および高さを記録した。前立腺体積を、幅×幅×高さ×パイ/6として算出し、体重に対して標準化した。前立腺の湿潤重量を、脂肪のない組織、および外部組織をトリミングした後に、剖検で記録した。
結果および考察:
血清テストステロンレベルを、図15および表9〜12に提示する。ベースラインにおいて、研究のすべてのサルのテストステロンレベルは、性成熟したオスのカニクイザルでは、正常範囲であった。しかしながら、テストステロンレベルは、100mg/kg/日で、化合物IVを受けたサル、および陽性対照(LHRH作動薬)で処置されたサルにおいて、有意に減少した。陽性対照(LHRH作動薬)グループにおけるテストステロンレベルは、二相性変化を示し、1日目、および3日目にそれぞれ、47.4%および547%(p<0.01)の初期の有意な増加(すなわち、炎症)の後、7日目、14日目、28日目、64日目、および90日目に、3.6%、67%、73%、83%、および85%の減少を有した(図15、および表9〜12を参照)。同様の炎症は、最高の用量レベル(すなわち、100mg/kg/日)でさえも、化合物IVで処置されたいかなる動物においても認められなかった。用量および治療期間は、化合物IVの薬理活性に重要であり、100mg/kg/日の用量は、ベースライン値に対して、血清テストステロンを、3日目、7日目、14日目、28日目、および64日目にそれぞれ、60%、51%、42%、79%、および92%ほど抑制した(図15、および表9および10を参照)。100mg/kg/日の化合物IVでの治療の90日後に、10匹のグループ4のサルのうちの6匹のテストステロンレベルは、アッセイの定量下限以下の濃度まで減少した(表11を参照)。グループ4のサルの平均血清テストステロンレベルは、相対的ベースライン値と比較して、96%ほど減少した(「推定最終濃度」、すなわち、BLQ値を有する、6/10匹のサルのテストステロンレベルを、LLOQ濃度の50%として算出した表10を参照)。90日目までに、100mg/kg/日で、化合物IVは、血清テストステロンを、陽性対照(LHRH作動薬)よりも有意に低いレベルまで減少させたこと(p=0.013)に留意することが重要である。
【0219】
【表9】
【0220】
【表10】
【0221】
【表11】
【0222】
【表12】
血清PSAレベルもまた、治療開始の4週間以内に、化合物IVによって有意に抑制された。69%および87%(平均)のPSA減少は、4週間、10mg/kg、および100mg/kgで、化合物IVを受けたサルでは顕著であり、PSAレベルは、陽性対照(LHRH作動薬)グループにおいて、60%ほど減少した(図16、および表13〜16)。
【0223】
【表13】
【0224】
【表14】
【0225】
【表15】
【0226】
【表16】
前立腺体積を、研究期間にわたり、定期的に、TRUSによって測定した。治療の6週間後に取得された結果は、サルの前立腺への化合物IVおよび陽性対照(LHRH作動薬)の強力な作用を示す。化合物IVは、前立腺体積を、10mg/kg、および100mg/kg用量レベルでそれぞれ、25%および45%、有意に抑制し、前立腺体積は、陽性対照(LHRH作動薬)グループにおいて、28%減少した(図17、ならびに表17および18)。
【0227】
【表17】
【0228】
【表18】
前立腺体積における化合物IV関連の減少は、剖検において、前立腺重量の評価により裏付けられた。治療の13週間後に、化合物IVは、10および100mg/kg/日の動物において、平均前立腺重量をそれぞれ、24%および21%、有意に減少させた(図18B、ならびに表19および20)。
【0229】
【表19】
【0230】
【表20】
血小板凝集、プロトロンビン時間(PT)、または活性化部分的トロンボプラスチン時間(APTT)への明らかな作用は、認められなかった。
実施例17
ヒトへの化合物IVの研究
男性への化合物IVの作用を判定するための研究を実施した。コホート毎に12名の対象者を、100、300、600、および1000mgの化合物IVの用量で調査した。表21は、100、300、600、および1000mgの用量で化合物IVを投与することによる、男性における、LH、血清PSA、遊離テストステロン、および総テストステロンレベルの平均変化を表す。ヒトにおける用量依存性平均総テストステロンレベル(nmol/L)を、1〜11日間の期間で測定した(図19)。総テストステロンレベルは、600mgおよび1000mgの用量でそれぞれ、51.9%、47.9%低下した。
【0231】
ヒトにおける用量依存性平均LHレベル(IU/L)を、1〜10日間の期間で測定した(図20)。LHレベルは、100mg、300mg、600mg、および1000mgの用量でそれぞれ、20.7%、46.9%、27.6%、および29.2%低下した。
【0232】
ヒトにおける用量依存性平均遊離テストステロンレベル(pg/mL)を、1〜10日間の期間で測定した(図21)。遊離テストステロンレベルは、100mg、300mg、600mg、および1000mgの用量でそれぞれ、17.0%、18.5%、72.7%、および53.2%低下した。
【0233】
ヒトにおける用量依存性平均PSAレベル(μg/L)を、1〜10日間の期間で測定した(図22)。PSAレベルは、100mg、300mg、600mg、および1000mgの用量でそれぞれ、9.2%、24.4%、27.5%、および29.9%低下した。10および30mgの用量では、変化を示さなかった。
【0234】
【表21】
実施例18
化合物IVの生物学的利用率
化合物IVは、ラット、イヌ、サルへの経口投与後に迅速に吸収された。ラットにおける、化合物IVの経口生物学的利用率は、用量が投与される製剤によって、6%〜25%の範囲であった。ポリエチレングリコール300(PEG300)を使用する製剤は、一般に、脱イオン水中で希釈されたTween80中で調製されたマイクロエマルションよりも高い暴露を生じた。イヌでは、血漿濃度−時間プロファイルの目視検査は、最終相の第2のピークで分かるとおり、化合物IVが、腸肝再循環を受けることを示唆した。重要なことには、イヌでは、オスの30mg/kgのPEG300の経口用量グループにおける暴露は、LH抑制のラットモデルにおける前立腺減少への最大作用を産生するために必要な暴露を超過した。サルでは、予備の薬物動態研究は、7日間の期間にわたる、化合物IVの血漿濃度および血清テストステロンの抑制によって明らかなとおり、この種における経口生物学的利用率は、イヌの経口生物学的利用率に近似するか、または超過することを示唆した。総じて、これらのデータは、十分な経口暴露を、2つの非げっ歯類の動物種において達成して、所望の薬理作用を産生することができることを示唆する(AUCデータに基づく)。さらに、ラットおよびサルにおける内分泌データは、化合物IVの薬理作用が可逆性である(すなわち、化合部IVによる治療を停止した時に、テストステロンの血清濃度が、ベースラインまたは正常レベルに戻る)ことを示唆する。
実施例19
化合物IVの薬物動態
インビトロ(マウス、ラット、イヌ、サル、およびヒト)、およびインビボ(ラット)代謝研究からの予備データは、化合物IV、そのヒドロキシル化された代謝物、およびそのN−ジアルキル化された代謝物の共役が、動物およびヒトにおいて、化合物IVの全体の体内動態に寄与することを示唆する。種間比較の結果は、定性的のみであるが、非臨床種の全代謝物プロファイルが、ヒト肝ミクロソーム内に生成されたプロファイルを的確に反映することを示す。これらの結果に基づき、ラットおよびイヌはそれぞれ、薬理評価および毒性評価において、適切なげっ歯種および非げっ歯種である。インビトロ研究は、化合物IVが、関連CYP450イソ型(CYP1A2、CYP2B6、またはCYP3A4)を誘導せず、<30μMの濃度ではCYP1A2、CYP2C9、CYP2D6、またはCYP3A4/5を阻害しないことを示す。CYP2C9は、化合物IVによって阻害されるが、高濃度(Ki=8μM)のみにおいてであり、薬物動態的薬物−薬物相互作用の可能性は、高くないと見なされる。
実施例20
化合物IVの生物活性
化合物IVは、hERGチャネルへのインビトロ阻害作用(IC50≧300μM)をほとんど、または全く発揮しない。化合物は、インビトロの単離したイヌのプルキンエ線維中で、10および100μMの濃度で、APD50およびAPD90を、化合物用量依存的に減少させた。しかしながら、化合物IVは、遠隔計測されたイヌにおいて、いかなる用量(最大300mg/kg)でも、血行動態または心臓機能(血圧、心拍数、心電図形態、またはQT間隔)に影響を及ぼさなかった。神経薬理学的作用または経肺作用は、認められなかった。最大の30mg/kgまでの化合物IVの単一経口用量では、腎機能への有意な作用を示さなかった。尿排出量および尿中のカリウムおよび塩素の排出量の増大のみが、試験した最大用量(100mg/kg)で認められた。ラットにおいて、30〜300mg/kgの用量での化合物IVの経口投与は、蠕動の有意な増加をもたらし、ラットにおいて、30mg/kgの用量での化合物IVの経口投与は、消化管運動および胃内酸度の有意な増加をもたらした(恐らく、平滑筋への影響によるものではない)。
【0235】
化合物IVは、変異原性ではなく、インビトロのヒト末梢血リンパ球において、最大200μMまでの濃度で、構造的または数的染色体異常を誘発しなかった。化合物IVは、単一および反復経口投与(最大28日間)後に、ラットおよびイヌによる耐容性が良好であった。腎臓、肝臓、心臓、および他の非標的関連臓器において、病理学的変化は、認められなかった。最大28日間のオスまたはメスのイヌへの化合物IVの経口投与に伴う深刻な物理的兆候、体重効果、臨床的病理学変化、眼科的変化、心電図変化、または組織病理学的変化は、存在しなかった。
【0236】
本発明のある特徴を、本明細書に例証し、説明してきたが、当業者には、多くの修正、変更、および等価物が思い当たるであろう。したがって、添付の特許請求の範囲は、本発明の真の精神に当てはまる、すべてのかかる修正および変更を網羅することを意図していることを理解されたい。
【特許請求の範囲】
【請求項1】
男性対象者において、血清総テストステロンレベルを低下させる方法であって、治療的有効量の式Iの化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することから本質的になり、
【化1】
式中、
Yは、C(O)またはCH2であり、
R1、R2は、独立して、水素、ハロゲン、ヒドロキシル、アルコキシ、シアノ、ニトロ、CF3、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、O−Alk−NR5R6、またはO−Alk−複素環であり、前記複素環は、3〜7員の置換または非置換複素環式環、任意に芳香族であり、
R3、R4は、独立して、水素、ハロゲン、ヒドロキシアルキル、ヒドロキシル、アルコキシ、シアノ、ニトロ、CF3、NHCOR、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、または保護ヒドロキシルであり、
Rは、アルキル、水素、ハロアルキル、ジハロアルキル、トリハロアルキル、CH2F、CHF2、CF3、CF2CF3、アリール、フェニル、ハロゲン、アルケニル、CN、NO2、またはOHであり、
R5およびR6は、独立して、水素、フェニル、1〜6個の炭素原子のアルキル基、3〜7員のシクロアルキル、3〜7員の複素環、5〜7員のアリールであるか、またはR5およびR6は、その窒素原子とともに、3〜7員の環を形成し、
jおよびkは、独立して、1〜4であり、
Alkは、1〜7個の炭素の直鎖アルキル、1〜7個の炭素の分岐鎖アルキル、または3〜8個の炭素の環状アルキルである、方法。
【請求項2】
式Iの前記化合物は、
【化2】
から選択される、請求項1に記載の方法。
【請求項3】
血清総テストステロンの低下は、血清黄体形成ホルモンレベルの減少により生じる、請求項1に記載の方法。
【請求項4】
血清総テストステロンの低下は、血清黄体形成ホルモンレベルの減少により生じる、請求項2に記載の方法。
【請求項5】
血清総テストステロンの低下は、血清黄体形成ホルモンレベルの減少と無関係である、請求項1に記載の方法。
【請求項6】
血清総テストステロンの低下は、SHBGの増加、精巣内のライディッヒ細胞によるテストステロン産生もしくは分泌の阻害、または副腎ステロイド産生の減少に起因する、請求項5に記載の方法。
【請求項7】
前記血清総テストステロンは、遊離テストステロンと、結合テストステロンとを含み、血清黄体形成ホルモンレベルの減少と無関係な前記遊離テストステロンレベルの低下は、SHBGの増加に起因する、請求項6に記載の方法。
【請求項8】
血清総テストステロンの低下は、血清黄体形成ホルモンレベルの減少と無関係である、請求項2に記載の方法。
【請求項9】
血清総テストステロンの低下は、SHBGの増加、精巣内のライディッヒ細胞によるテストステロン産生もしくは分泌の阻害、または副腎ステロイド産生の減少に起因する、請求項8に記載の方法。
【請求項10】
前記血清総テストステロンは、遊離テストステロンと、結合テストステロンとを含み、血清黄体形成ホルモンレベルの減少と無関係な前記遊離テストステロンレベルの低下は、SHBGの増加に起因する、請求項9に記載の方法。
【請求項11】
前記男性対象者は、前立腺癌を有する、請求項1に記載の方法。
【請求項12】
前記男性対象者は、前立腺癌を有する、請求項2に記載の方法。
【請求項13】
前記男性対象者は、前立腺癌を有する、請求項3に記載の方法。
【請求項14】
前記男性対象者は、前立腺癌を有する、請求項4に記載の方法。
【請求項15】
前記男性対象者は、前立腺癌を有する、請求項5に記載の方法。
【請求項16】
前記男性対象者は、前立腺癌を有する、請求項6に記載の方法。
【請求項17】
前記血清総テストステロンは、約100ng/dL未満に低減される、請求項1に記載の方法。
【請求項18】
前記血清総テストステロンは、約50ng/dL未満に低減される、請求項17に記載の方法。
【請求項19】
前記血清総テストステロンは、約25ng/dL未満に低減される、請求項17に記載の方法。
【請求項20】
前記血清総テストステロンは、約100ng/dL未満に低減される、請求項2に記載の方法。
【請求項21】
前記血清総テストステロンは、約50ng/dL未満に低減される、請求項20に記載の方法。
【請求項22】
前記血清総テストステロンは、約25ng/dL未満に低減される、請求項20に記載の方法。
【請求項23】
前記血清総テストステロンは、約100ng/dL未満に低減される、請求項3に記載の方法。
【請求項24】
前記血清総テストステロンは、約50ng/dL未満に低減される、請求項23に記載の方法。
【請求項25】
前記血清総テストステロンは、約25ng/dL未満に低減される、請求項23に記載の方法。
【請求項26】
前記血清総テストステロンは、約100ng/dL未満に低減される、請求項4に記載の方法。
【請求項27】
前記血清総テストステロンは、約50ng/dL未満に低減される、請求項26に記載の方法。
【請求項28】
前記血清総テストステロンは、約25ng/dL未満に低減される、請求項26に記載の方法。
【請求項29】
前記血清総テストステロンは、約100ng/dL未満に低減される、請求項5に記載の方法。
【請求項30】
前記血清総テストステロンは、約50ng/dL未満に低減される、請求項29に記載の方法。
【請求項31】
前記血清総テストステロンは、約25ng/dL未満に低減される、請求項29に記載の方法。
【請求項32】
前記血清総テストステロンは、約100ng/dL未満に低減される、請求項6に記載の方法。
【請求項33】
前記血清総テストステロンは、約50ng/dL未満に低減される、請求項32に記載の方法。
【請求項34】
前記血清総テストステロンは、約25ng/dL未満に低減される、請求項32に記載の方法。
【請求項35】
前記化合物の投与は、アンドロゲン除去療法(ADT)に伴う副作用が生じることを抑制し、発症を低減し、重症度を軽減し、阻害し、または治療するとともに、前記対象者は前立腺癌を有する、請求項1に記載の方法。
【請求項36】
血清総テストステロンの低下は、血清黄体形成ホルモンレベルの減少によって生じる、請求項35に記載の方法。
【請求項37】
血清総テストステロンの低下は、血清黄体形成ホルモンレベルの減少に無関係である、請求項35に記載の方法。
【請求項38】
ADTに伴う副作用は、ほてり、女性化乳房、骨塩密度の低下、および骨折の増加からなる群から選択される、請求項35に記載の方法。
【請求項39】
前記典型的な副作用は、ほてりである、請求項38に記載の方法。
【請求項40】
前記典型的な副作用は、女性化乳房である、請求項38に記載の方法。
【請求項41】
前記副作用は、骨塩密度の低下である、請求項38に記載の方法。
【請求項42】
前記骨折の増加は、病理学的骨折、非外傷性骨折、脊椎骨折、非脊椎骨折、新規形態学的骨折、臨床的骨折、またはそれらの組み合わせである、請求項38に記載の方法。
【請求項43】
前記化合物の投与は、アンドロゲン除去療法(ADT)に伴う副作用が生じることを防止または治療するとともに、前記対象者は前立腺癌を有する、請求項2に記載の方法。
【請求項44】
血清総テストステロンの低下は、血清黄体形成ホルモンレベルの減少により生じる、請求項43に記載の方法。
【請求項45】
血清総テストステロンの低下は、血清黄体形成ホルモンレベルの減少と無関係である、請求項43に記載の方法。
【請求項46】
ADTに伴う前記副作用は、ほてり、女性化乳房、骨塩密度の低下、および骨折の増加からなる群から選択される、請求項43に記載の方法。
【請求項47】
前記典型的な副作用は、ほてりである、請求項46に記載の方法。
【請求項48】
前記典型的な副作用は、女性化乳房である、請求項46に記載の方法。
【請求項49】
前記典型的な副作用は、骨塩密度の低下である、請求項46に記載の方法。
【請求項50】
前記方法はさらに、進行した前立腺癌を治療する、請求項1に記載の方法。
【請求項51】
前記方法はさらに、進行した前立腺癌を抑制し、発症を低減し、重症度を軽減し、または阻害する、請求項1に記載の方法。
【請求項52】
前記方法はさらに、進行した前立腺癌の緩和治療を提供する、請求項1に記載の方法。
【請求項1】
男性対象者において、血清総テストステロンレベルを低下させる方法であって、治療的有効量の式Iの化合物またはその異性体、薬学的に許容される塩、薬学的生成物、多形体、水和物、またはそれらの任意の組み合わせを投与することから本質的になり、
【化1】
式中、
Yは、C(O)またはCH2であり、
R1、R2は、独立して、水素、ハロゲン、ヒドロキシル、アルコキシ、シアノ、ニトロ、CF3、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、O−Alk−NR5R6、またはO−Alk−複素環であり、前記複素環は、3〜7員の置換または非置換複素環式環、任意に芳香族であり、
R3、R4は、独立して、水素、ハロゲン、ヒドロキシアルキル、ヒドロキシル、アルコキシ、シアノ、ニトロ、CF3、NHCOR、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、または保護ヒドロキシルであり、
Rは、アルキル、水素、ハロアルキル、ジハロアルキル、トリハロアルキル、CH2F、CHF2、CF3、CF2CF3、アリール、フェニル、ハロゲン、アルケニル、CN、NO2、またはOHであり、
R5およびR6は、独立して、水素、フェニル、1〜6個の炭素原子のアルキル基、3〜7員のシクロアルキル、3〜7員の複素環、5〜7員のアリールであるか、またはR5およびR6は、その窒素原子とともに、3〜7員の環を形成し、
jおよびkは、独立して、1〜4であり、
Alkは、1〜7個の炭素の直鎖アルキル、1〜7個の炭素の分岐鎖アルキル、または3〜8個の炭素の環状アルキルである、方法。
【請求項2】
式Iの前記化合物は、
【化2】
から選択される、請求項1に記載の方法。
【請求項3】
血清総テストステロンの低下は、血清黄体形成ホルモンレベルの減少により生じる、請求項1に記載の方法。
【請求項4】
血清総テストステロンの低下は、血清黄体形成ホルモンレベルの減少により生じる、請求項2に記載の方法。
【請求項5】
血清総テストステロンの低下は、血清黄体形成ホルモンレベルの減少と無関係である、請求項1に記載の方法。
【請求項6】
血清総テストステロンの低下は、SHBGの増加、精巣内のライディッヒ細胞によるテストステロン産生もしくは分泌の阻害、または副腎ステロイド産生の減少に起因する、請求項5に記載の方法。
【請求項7】
前記血清総テストステロンは、遊離テストステロンと、結合テストステロンとを含み、血清黄体形成ホルモンレベルの減少と無関係な前記遊離テストステロンレベルの低下は、SHBGの増加に起因する、請求項6に記載の方法。
【請求項8】
血清総テストステロンの低下は、血清黄体形成ホルモンレベルの減少と無関係である、請求項2に記載の方法。
【請求項9】
血清総テストステロンの低下は、SHBGの増加、精巣内のライディッヒ細胞によるテストステロン産生もしくは分泌の阻害、または副腎ステロイド産生の減少に起因する、請求項8に記載の方法。
【請求項10】
前記血清総テストステロンは、遊離テストステロンと、結合テストステロンとを含み、血清黄体形成ホルモンレベルの減少と無関係な前記遊離テストステロンレベルの低下は、SHBGの増加に起因する、請求項9に記載の方法。
【請求項11】
前記男性対象者は、前立腺癌を有する、請求項1に記載の方法。
【請求項12】
前記男性対象者は、前立腺癌を有する、請求項2に記載の方法。
【請求項13】
前記男性対象者は、前立腺癌を有する、請求項3に記載の方法。
【請求項14】
前記男性対象者は、前立腺癌を有する、請求項4に記載の方法。
【請求項15】
前記男性対象者は、前立腺癌を有する、請求項5に記載の方法。
【請求項16】
前記男性対象者は、前立腺癌を有する、請求項6に記載の方法。
【請求項17】
前記血清総テストステロンは、約100ng/dL未満に低減される、請求項1に記載の方法。
【請求項18】
前記血清総テストステロンは、約50ng/dL未満に低減される、請求項17に記載の方法。
【請求項19】
前記血清総テストステロンは、約25ng/dL未満に低減される、請求項17に記載の方法。
【請求項20】
前記血清総テストステロンは、約100ng/dL未満に低減される、請求項2に記載の方法。
【請求項21】
前記血清総テストステロンは、約50ng/dL未満に低減される、請求項20に記載の方法。
【請求項22】
前記血清総テストステロンは、約25ng/dL未満に低減される、請求項20に記載の方法。
【請求項23】
前記血清総テストステロンは、約100ng/dL未満に低減される、請求項3に記載の方法。
【請求項24】
前記血清総テストステロンは、約50ng/dL未満に低減される、請求項23に記載の方法。
【請求項25】
前記血清総テストステロンは、約25ng/dL未満に低減される、請求項23に記載の方法。
【請求項26】
前記血清総テストステロンは、約100ng/dL未満に低減される、請求項4に記載の方法。
【請求項27】
前記血清総テストステロンは、約50ng/dL未満に低減される、請求項26に記載の方法。
【請求項28】
前記血清総テストステロンは、約25ng/dL未満に低減される、請求項26に記載の方法。
【請求項29】
前記血清総テストステロンは、約100ng/dL未満に低減される、請求項5に記載の方法。
【請求項30】
前記血清総テストステロンは、約50ng/dL未満に低減される、請求項29に記載の方法。
【請求項31】
前記血清総テストステロンは、約25ng/dL未満に低減される、請求項29に記載の方法。
【請求項32】
前記血清総テストステロンは、約100ng/dL未満に低減される、請求項6に記載の方法。
【請求項33】
前記血清総テストステロンは、約50ng/dL未満に低減される、請求項32に記載の方法。
【請求項34】
前記血清総テストステロンは、約25ng/dL未満に低減される、請求項32に記載の方法。
【請求項35】
前記化合物の投与は、アンドロゲン除去療法(ADT)に伴う副作用が生じることを抑制し、発症を低減し、重症度を軽減し、阻害し、または治療するとともに、前記対象者は前立腺癌を有する、請求項1に記載の方法。
【請求項36】
血清総テストステロンの低下は、血清黄体形成ホルモンレベルの減少によって生じる、請求項35に記載の方法。
【請求項37】
血清総テストステロンの低下は、血清黄体形成ホルモンレベルの減少に無関係である、請求項35に記載の方法。
【請求項38】
ADTに伴う副作用は、ほてり、女性化乳房、骨塩密度の低下、および骨折の増加からなる群から選択される、請求項35に記載の方法。
【請求項39】
前記典型的な副作用は、ほてりである、請求項38に記載の方法。
【請求項40】
前記典型的な副作用は、女性化乳房である、請求項38に記載の方法。
【請求項41】
前記副作用は、骨塩密度の低下である、請求項38に記載の方法。
【請求項42】
前記骨折の増加は、病理学的骨折、非外傷性骨折、脊椎骨折、非脊椎骨折、新規形態学的骨折、臨床的骨折、またはそれらの組み合わせである、請求項38に記載の方法。
【請求項43】
前記化合物の投与は、アンドロゲン除去療法(ADT)に伴う副作用が生じることを防止または治療するとともに、前記対象者は前立腺癌を有する、請求項2に記載の方法。
【請求項44】
血清総テストステロンの低下は、血清黄体形成ホルモンレベルの減少により生じる、請求項43に記載の方法。
【請求項45】
血清総テストステロンの低下は、血清黄体形成ホルモンレベルの減少と無関係である、請求項43に記載の方法。
【請求項46】
ADTに伴う前記副作用は、ほてり、女性化乳房、骨塩密度の低下、および骨折の増加からなる群から選択される、請求項43に記載の方法。
【請求項47】
前記典型的な副作用は、ほてりである、請求項46に記載の方法。
【請求項48】
前記典型的な副作用は、女性化乳房である、請求項46に記載の方法。
【請求項49】
前記典型的な副作用は、骨塩密度の低下である、請求項46に記載の方法。
【請求項50】
前記方法はさらに、進行した前立腺癌を治療する、請求項1に記載の方法。
【請求項51】
前記方法はさらに、進行した前立腺癌を抑制し、発症を低減し、重症度を軽減し、または阻害する、請求項1に記載の方法。
【請求項52】
前記方法はさらに、進行した前立腺癌の緩和治療を提供する、請求項1に記載の方法。
【図4】

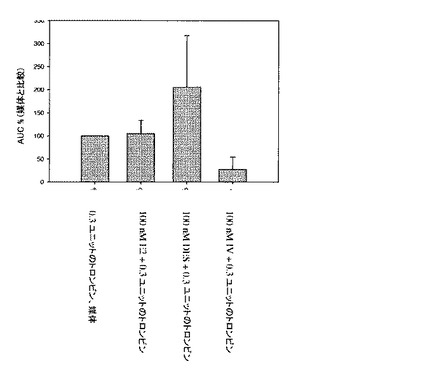
【図5】
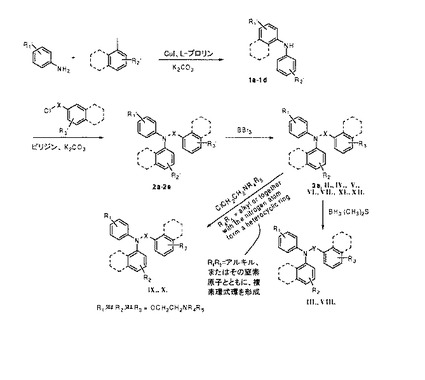
【図6】
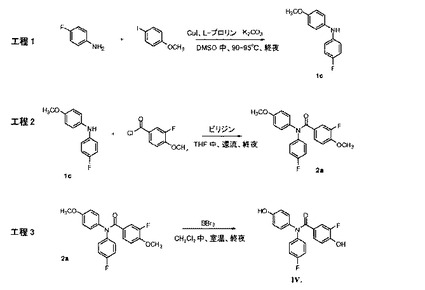
【図7】
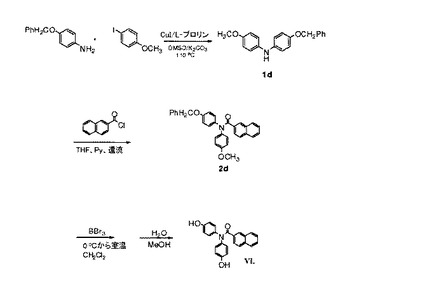
【図8】
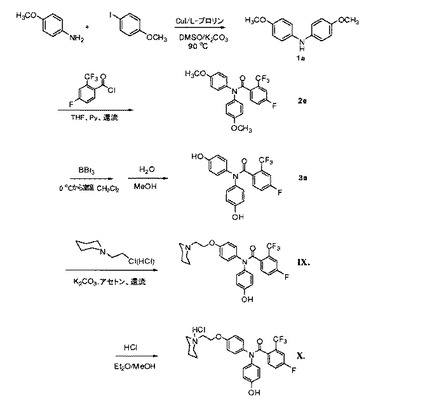
【図12A】
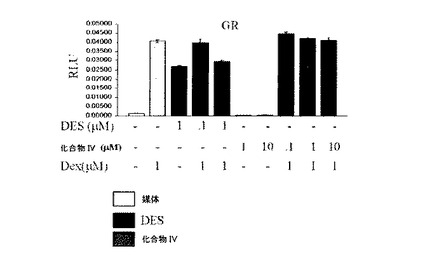
【図13】
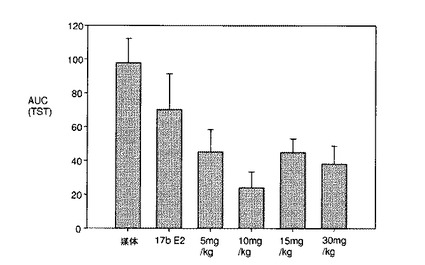
【図1】
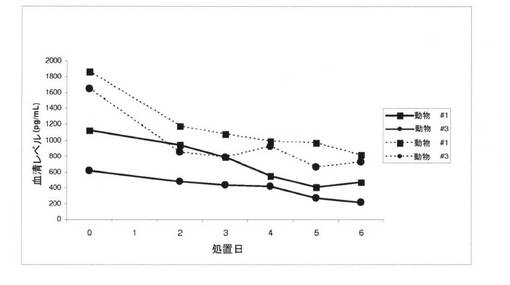
【図2】
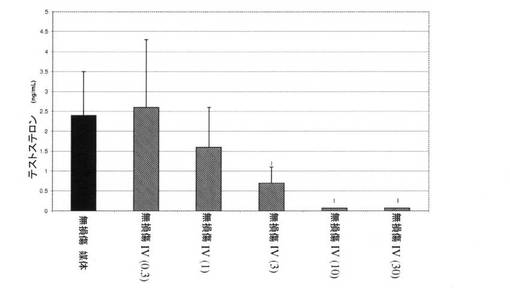
【図3】
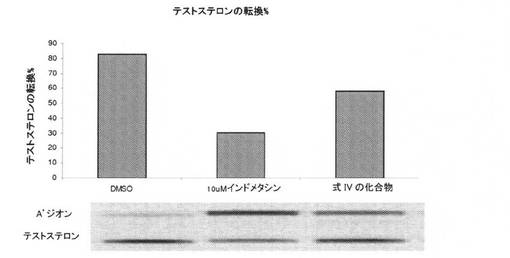
【図9】
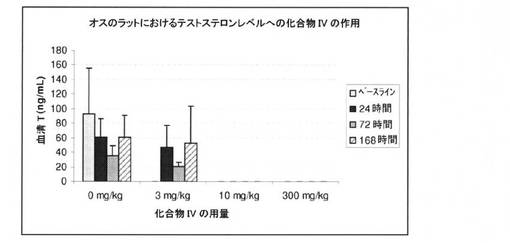
【図10A】
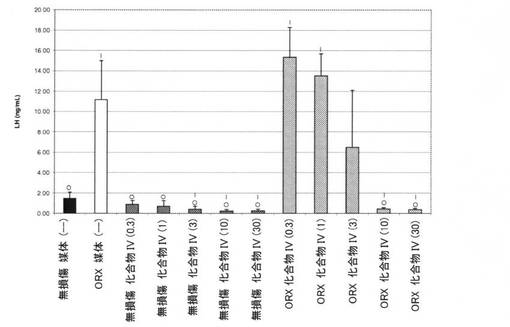
【図10B】
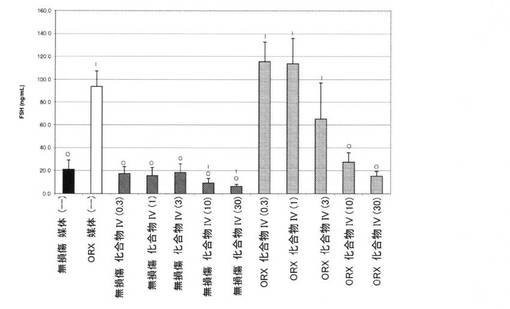
【図10C】
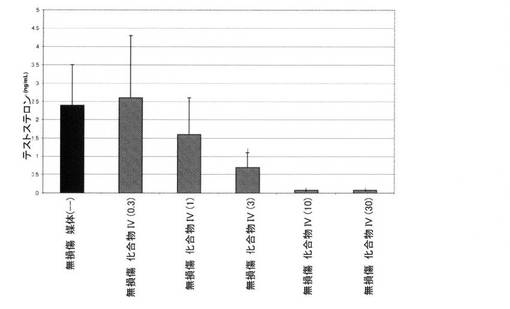
【図10D】
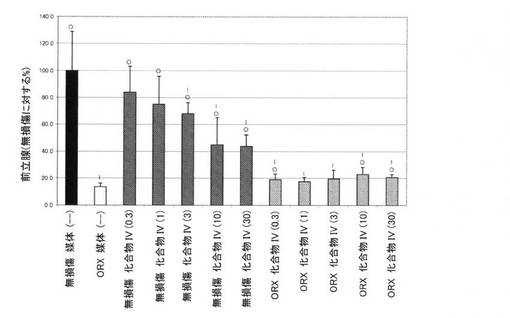
【図10E】
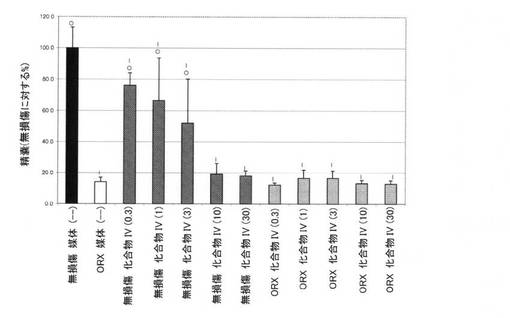
【図10F】
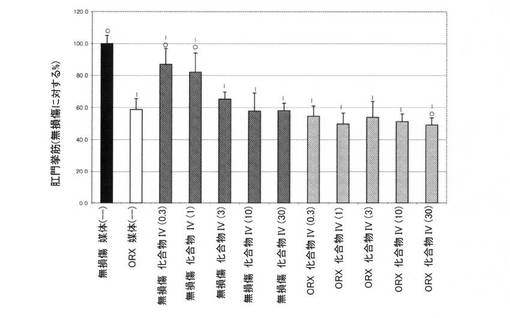
【図11】
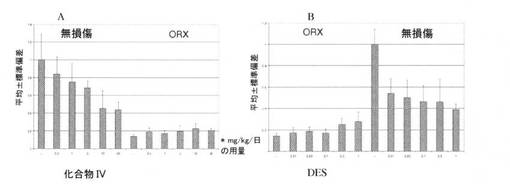
【図12B】
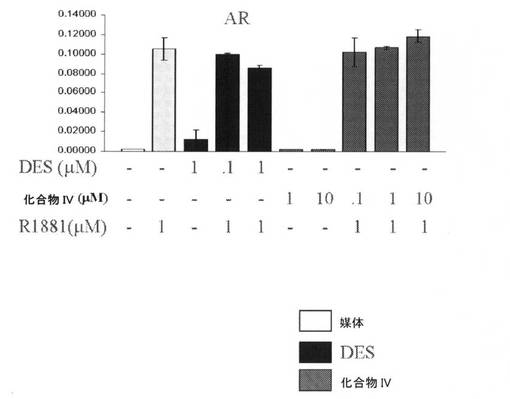
【図12C】
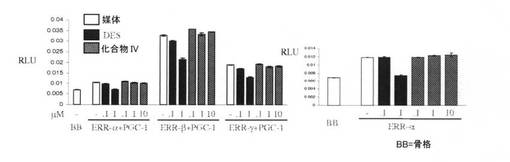
【図14】
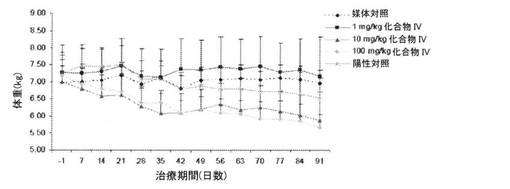
【図15】
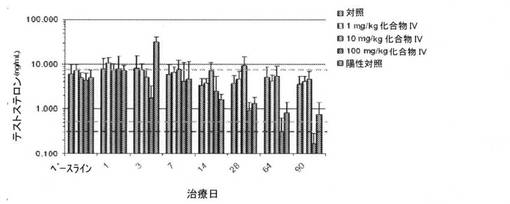
【図16】
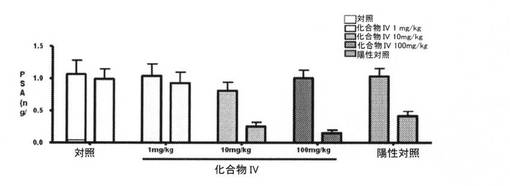
【図17】
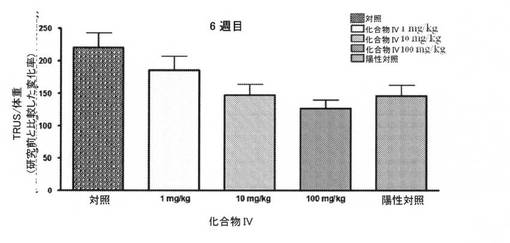
【図18】
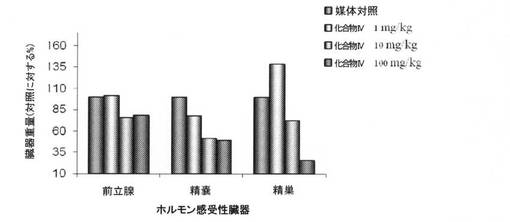
【図19】
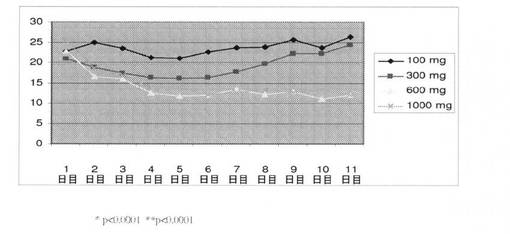
【図20】
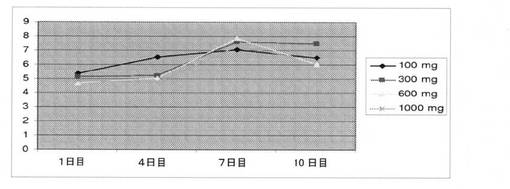
【図21】
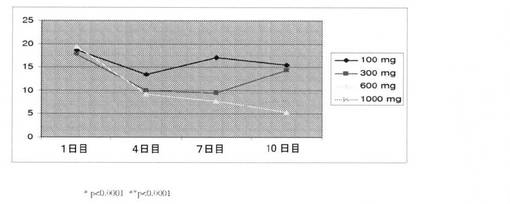
【図22】
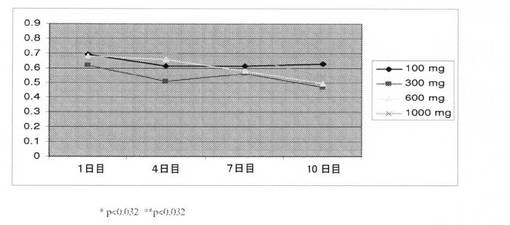
【図23】
【図5】
【図6】
【図7】
【図8】
【図12A】
【図13】
【図1】
【図2】
【図3】
【図9】
【図10A】
【図10B】
【図10C】
【図10D】
【図10E】
【図10F】
【図11】
【図12B】
【図12C】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【公表番号】特表2012−518654(P2012−518654A)
【公表日】平成24年8月16日(2012.8.16)
【国際特許分類】
【出願番号】特願2011−551292(P2011−551292)
【出願日】平成22年2月23日(2010.2.23)
【国際出願番号】PCT/US2010/025032
【国際公開番号】WO2010/096801
【国際公開日】平成22年8月26日(2010.8.26)
【出願人】(504206621)ジーティーエックス・インコーポレイテッド (20)
【Fターム(参考)】
【公表日】平成24年8月16日(2012.8.16)
【国際特許分類】
【出願日】平成22年2月23日(2010.2.23)
【国際出願番号】PCT/US2010/025032
【国際公開番号】WO2010/096801
【国際公開日】平成22年8月26日(2010.8.26)
【出願人】(504206621)ジーティーエックス・インコーポレイテッド (20)
【Fターム(参考)】
[ Back to top ]