
エストロゲン受容体リガンドおよびその使用方法
本発明は、アンドロゲン遮断療法(ADT)により誘発されるほてりおよび重度のほてりを処置する方法に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、対象へのアンドロゲン遮断療法(ADT)により誘発されるほてりおよび重度のほてりを処置する方法に関する。
【背景技術】
【0002】
エストロゲンとは、組織および骨の維持にとって重要であり、かつ使用される内因性および合成ホルモンの群のことを言う。エストロゲンは、生殖器系の発達および維持に関与する細胞過程の内分泌制御因子である。生殖生物学におけるエストロゲンの役割、閉経後のほてり(一過性熱感)の予防、および閉経後骨粗鬆症の予防は確立されている。エストラジオールは主要な内因性ヒトエストロゲンであり、女性と男性両方で見つかっている。
【0003】
エストロゲンおよび抗エストロゲンの生理学的作用は、2つの異なる細胞内受容体、エストロゲンアルファ(ERα)およびエストロゲンベータ(ERβ)を通じた発現である。内因性エストロゲンは、両方の受容体亜型の典型的に強力な活性剤である。例えば、エストラジオールは、乳房、骨、心血管、および中枢神経系組織を含む多くの組織内でERαとして作用する。選択的エストロゲン受容体モジュレーターは、さまざまな組織で、一般に異なる作用をする。例えば、SERMは乳房ではERαアンタゴニストである場合があるが、子宮、骨および心血管系では部分ERαアゴニストである場合がある。そのため、エストロゲン受容体リガンドとしての機能を果たす化合物は、さまざまな病状および疾患の処置に有用である。
【0004】
前立腺癌は、米国の男性で最もよく診断される非皮膚癌の1つであり、癌による死亡の2番目に多い原因であり、今年には180,000件を超える新規症例とほぼ29,000人の死亡が見込まれている。進行前立腺癌の患者は、典型的には黄体形成ホルモン放出ホルモン(LHRH)アゴニストまたは両側精巣摘除術のいずれかによる、アンドロゲン遮断療法(ADT)を受ける。アンドロゲン遮断療法ではテストステロンを削減するだけでなく、エストロゲンがテストステロンの芳香族化から生じ、テストステロンの濃度はADTによって激減させられるため、エストロゲン濃度も低くなる。アンドロゲン遮断療法により誘発されるエストロゲン欠乏は、ほてり(hot flash)、女性化乳房および乳房痛、骨量減少、骨の質および強度の低下、骨粗鬆症および致命的な骨折、有害な脂質変化および高い心血管疾患および心筋梗塞、ならびに鬱病およびその他の情緒の変化を含む、有意な副作用を引き起こす。ADTのエストロゲン欠乏副作用の多くはERαが関与していると考えられる。
【0005】
酢酸ロイプロリド(Lupron(登録商標))は、天然ゴナドトロピン放出ホルモン(GnRHまたはLH−RH)の合成非ペプチド類似体である。酢酸ロイプロリドは、下垂体によるLH分泌を最終的に抑えるLH−RHスーパーアゴニストである。酢酸ロイプロリドは、ゴナドトロピン分泌の強力な阻害剤としての機能を果たし、卵巣および精巣ステロイド合成を抑制する。ヒトでは、酢酸ロイプロリドの投与によって、黄体形成ホルモン(LH)および卵胞刺激ホルモン(FSH)の血中濃度の初期増加を引き起こし、性腺ステロイド(男性でのテストステロンおよびジヒドロテストステロン、並びに閉経前女性でのエストロンおよびエストラジオール)の濃度を一時的に増加させる。しかし、酢酸ロイプロリドの継続投与、LHおよびFSHの濃度低下を招く。男性では、テストステロンが去勢レベル(50ng/dL未満)にまで減少する。閉経前女性では、エストロゲンが閉経後レベルにまで減少する。テストステロンは、前立腺の癌細胞に対する既知の刺激物質である。したがって、テストステロンの分泌を抑制することまたはテストステロンの作用を阻害することは、前立腺癌治療の必要な構成要素である。酢酸ロイプロリドは、前立腺癌を処置するための去勢レベルへの血清テストステロンの減少および低減であるLH抑制に使用できる。
【0006】
LHRHアゴニストを導入する前に、エストロゲン(主としてジエチルスチルベストロール(DES))を介して、下垂体でのエストロゲン活性を増加させることで、去勢テストステロンレベルが達成された。DESは、去勢濃度へのテストステロンの抑制においてLHRHアゴニストと同じように有効であった。DESによって処置された患者はほてりまたは骨量減少を示さなかったが、LHRHアゴニストによるADTよりも高い率で女性化乳房を示した。残念ながら、DESおよびエストラジオールのような極めて強力な純エストロゲンは、その臨床使用を制限してきた重篤な心血管および血栓塞栓合併症の高いリスクと関係がある場合が多い。
【0007】
本発明の化合物は、テストステロンレベルを去勢レベルに抑制する。このレベルは、血栓症のリスク増加を予防しながら、骨量減少、ほてり、および/または女性化乳房なしに、前立腺癌の処置に使用してもよいレベルである。
【発明の概要】
【課題を解決するための手段】
【0008】
別の実施形態では、本発明は、アンドロゲン遮断療法(ADT)により誘発されるほてりを処置する方法であって、対象に対して、式Iの化合物、またはその異性体、薬学的に許容される塩、N−酸化物、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与するステップを含む方法を提供する。
【0009】
【化1】

【0010】
式中、
Yは、C(O)またはCH2であり;
R1、R2は、互いに独立して、水素、ハロゲン、ヒドロキシル、アルコキシ、シアン、ニトロ、CF3、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、O−Alk−NR5R6、または、置換されたもしくは非置換の3〜7員のO−Alk−複素環(任意選択で芳香族である)であり;
R3、R4は、互いに独立して、水素、ハロゲン、ヒドロキシアルキル、ヒドロキシル、アルコキシ、シアン、ニトロ、CF3、NHCOR、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、または保護ヒドロキシルであり;
Rは、アルキル、水素、ハロアルキル、ジハロアルキル、トリハロアルキル、CH2F、CHF2、CF3、CF2CF3、アリール、フェニル、ハロゲン、アルケニル、CN、NO2、またはOHであり;
R5およびR6は、互いに独立して、水素、フェニル、炭素数1〜6のアルキル基、3〜7員のシクロアルキル、3〜7員の複素環、5〜7員のアリールであり;あるいは、R5およびR6は、窒素原子を含む3〜7員環を形成し;
jおよびkは、互いに独立して、1〜4であり;
Alkは、炭素数1〜7の直鎖アルキル、炭素数1〜7の分岐鎖アルキル、または炭素数3〜8の環状アルキルである。
【0011】
別の実施形態では、本発明は、アンドロゲン遮断療法(ADT)により誘発されるほてりを抑制もしくは防止、または該ほてりの発症率を低減する方法であって、対象に対して、式Iの合物、またはその異性体、薬学的に許容される塩、N−酸化物、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与するステップを含む方法を提供する。
【0012】
別の実施形態では、本発明は、アンドロゲン遮断療法(ADT)により誘発される重度のほてりを処置する方法であって、対象に対して、式Iの化合物、またはその異性体、薬学的に許容される塩、N−酸化物、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与するステップを含む方法を提供する。
【0013】
別の実施形態では、本発明は、アンドロゲン遮断療法(ADT)により誘発される重度のほてりを抑制もしくは防止、または該ほてりの発症率を低減する方法であって、対象に対して、式Iの化合物、またはその異性体、薬学的に許容される塩、N−酸化物、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与するステップを含む方法を提供する。
【図面の簡単な説明】
【0014】
発明と見なされる主題は具体的に指摘され、明細書の最終部分で明確に主張される。しかしながら、その目的、特徴、および利点とともに、構成および実施方法の両方に関して、本発明は、以下の添付図面とともに読むならば、以下の詳細な説明を参照することによって最もよく理解できる。
【0015】
【図1】化合物IVを30mg/kg連日経口投与(0日目に初回投与)した後の未去勢雄ザルの血清テストステロン(実線)および全アンドロゲン(点線)の濃度を示す(実施例8を参照)。
【図2】化合物IV(0.3、1、10、30mg/kg)で処置された未去勢ラットのテストステロン濃度を示す。Iは、P<0.05 対未去勢賦形剤対照を示す。BLOQ値は、0.08ng/mLの定量限界で図示されている(実施例9を参照)。
【図3】17β−HSD5酵素活性に対する化合物IVの阻害効果を示す(実施形12を参照)。
【図4】DES、17β−エストラジオール(E2)、および化合物IVの存在におけるヒト血小板のインビトロ凝集を示す。0.3単位のトロンビンで凝集を誘導する前の30秒間、多血小板血漿(PRP)を、賦形剤、E2、DES、または化合物IVとともにインキュベートした。凝集を5分間監視し、賦形剤対照の割合として表した(実施例13を参照)。
【図5】化合物II〜XIIの調製のための一般的な合成スキームである(実施例1を参照)。
【図6】化合物IVの調製のための合成スキームである(実施例2を参照)。
【図7】化合物VIの調製のための合成スキームである(実施例3を参照)。
【図8】化合物IXおよびXの調製のための合成スキームである(実施例5を参照)。
【図9】3mg/kg、10mg/kg、および300mg/kgの用量で化合物IVによる処置を受けた未去勢ラットの24時間後、72時間後、および168時間後のテストステロン濃度を示す(実施例9を参照)。
【図10A】化合物IVの0.3mg/kg、1mg/kg、3mg/kg、10mg/kg、30mg/kgの用量で処置を受けた未去勢および精巣摘徐術済みラットのLH濃度を示す。Iは、P<0.05対未去勢賦形剤対照を示す。Oは、P<0.05対ORX賦形剤対照を示す。BLOQ値は、0.08ng/mLの定量限界で図示されている(実施例9を参照)。
【図10B】化合物IVの0.3mg/kg、1mg/kg、3mg/kg、10mg/kg、30mg/kgの用量で処置を受けた未去勢および精巣摘徐術済みラットのFSH濃度を示す。Iは、P<0.05対未去勢賦形剤対照を示す。Oは、P<0.05対ORX賦形剤対照を示す。BLOQ値は、0.08ng/mLの定量限界で図示されている(実施例9を参照)。
【図10C】化合物IVの0.3mg/kg、1mg/kg、3mg/kg、10mg/kg、30mg/kgの用量で処置を受けた未去勢および精巣摘徐術済みラットのテストステロン濃度を示す。Iは、P<0.05対未去勢賦形剤対照を示す。Oは、P<0.05対ORX賦形剤対照を示す。BLOQ値は、0.08ng/mLの定量限界で図示されている(実施例9を参照)。
【図10D】化合物IVの0.3mg/kg、1mg/kg、3mg/kg、10mg/kg、30mg/kgの用量で処置を受けた未去勢および精巣摘徐術済みラットの前立腺重量レベルを示す。Iは、P<0.05対未去勢賦形剤対照を示す。Oは、P<0.05対ORX賦形剤対照を示す。BLOQ値は、0.08ng/mLの定量限界で図示されている(実施例9を参照)。
【図10E】化合物IVの0.3mg/kg、1mg/kg、3mg/kg、10mg/kg、30mg/kgの用量で処置を受けた未去勢および精巣摘徐術済みラットの精嚢重量レベルを示す。Iは、P<0.05対未去勢賦形剤対照を示す。Oは、P<0.05対ORX賦形剤対照を示す。BLOQ値は、0.08ng/mLの定量限界で図示されている(実施例9を参照)。
【図10F】化合物IVの0.3mg/kg、1mg/kg、3mg/kg、10mg/kg、30mg/kgの用量で処置を受けた未去勢および精巣摘徐術済みラットの肛門拳筋重量を示す。Iは、P<0.05対未去勢賦形剤対照を示す。Oは、P<0.05対ORX賦形剤対照を示す。BLOQ値は、0.08ng/mLの定量限界で図示されている(実施例9を参照)。
【図11】さまざまな用量で化合物IV(図11A)およびDES(図11B)を投与することによる未去勢およびORXラットの前立腺の大きさを示す(実施例15を参照)。
【図12A】DESと化合物IVとの間の違いを示し;DESはグルココルチコイド受容体(GR)と交差反応するが、化合物IVは反応しない(図12A);DESはアンドロゲン受容体(AR)と交差反応する。ARの活動を穏やかに刺激し、穏やかに阻害するが(すなわち、部分アゴニスト/アンタゴニストである)、化合物IVはしない(図12B);DESはエストロゲン関連受容体(ERR)トランス活性化を無効にするが、化合物IVはしない(図12C)(実施例15を参照)。
【図12B】DESと化合物IVとの間の違いを示し;DESはグルココルチコイド受容体(GR)と交差反応するが、化合物IVは反応しない(図12A);DESはアンドロゲン受容体(AR)と交差反応する。ARの活動を穏やかに刺激し、穏やかに阻害するが(すなわち、部分アゴニスト/アンタゴニストである)、化合物IVはしない(図12B);DESはエストロゲン関連受容体(ERR)トランス活性化を無効にするが、化合物IVはしない(図12C)(実施例15を参照)。
【図12C】DESと化合物IVとの間の違いを示し;DESはグルココルチコイド受容体(GR)と交差反応するが、化合物IVは反応しない(図12A);DESはアンドロゲン受容体(AR)と交差反応する。ARの活動を穏やかに刺激し、穏やかに阻害するが(すなわち、部分アゴニスト/アンタゴニストである)、化合物IVはしない(図12B);DESはエストロゲン関連受容体(ERR)トランス活性化を無効にするが、化合物IVはしない(図12C)(実施例15を参照)。
【図13】5mg/kg、10mg/kg、15mg/kg、および30mg/kgの用量によるモルヒネ禁断モデルのほてりの軽減に対する化合物IVの効果を示す。N=7匹/群。17β−E2は、100%DMSO溶液で5mg/kgを使用した(実施例14を参照)。
【図14】91日間、化合物IVを投与することによるサルの用量依的な体重(kg)減少(100mg/kgで最高20%)を示す。女性化乳房または高エストロゲン症の兆候は観察されなかった(実施例16を参照)。
【図15】化合物IVを連日経口投与した後のサルの用量依存的な血清テストステロン濃度の減少(ng/mL)を、陽性対照(LHRHアゴニスト)と比較して示す。点線は化学的に去勢された患者のテストステロン濃度を示し、太い破線は外科的に去勢されたサルのテストステロン濃度を示す(実施例16を参照)。
【図16】化合物IVを投与よることによる、ベースライン時と第28日のサルの用量依存的な前立腺特異的抗原(PSA)濃度(ng/mL)を示す。PSA濃度は、化合物IVの処置により有意に減少した(実施例16を参照)。
【図17】第6週に化合物IVを投与することによる、陽性対照(LHRHアゴニスト)と比較した、経直腸超音波(TRUS)を用いたサルの用量依存的な前立腺体積を示す(実施例16を参照)。
【図18】化合物IVの投与による、第90日の用量依存的な臓器重量(前立腺、精嚢、および睾丸)をサルの対照の割合として示す(図18A)。化合物IVを連日経口投与した後、サルの死体解剖による第13週時の前立腺の重量(図18B)(実施例16を参照)。
【図19】化合物IVの投与(100mg、300mg、600mg、および1000mg)による、第1日〜第11日の期間中のヒトの用量依存的な平均全テストステロン濃度(nmol/L)を示す(実施例17を参照)。
【図20】化合物IVの投与(100mg、300mg、600mg、おび1000mg)による、第1日〜第10日の期間中のヒトの用量依存的な平均LH濃度(IU/L)を示す(実施例17を参照)。
【図21】化合物IVの投与(100mg、300mg、600mg、および1000mg)による、第1日〜第10日の期間中のヒトの用量依存的な平均遊離テストステロン濃度(pg/mL)を示す(実施例17を参照)。
【図22】化合物IVの投与(100mg、300mg、600mg、および1000mg)による、第1日〜第10日の期間中のヒトの用量依存的な平均PSA濃度(μg/L)を示す(実施例17を参照)。
【図23】化合物IV投与の14日間の回復後の未去勢ラットの用量依存的な血清テストステロン濃度(ng/mL)を示す。Iは、P<0.05対未去勢対照を示す(実施例10を参照)。
【0016】
図の説明を簡単かつ明瞭にするために、図に示された要素が必ずしも一定の縮尺には描かれていないことは十分に理解されるであろう。例えば、要素の一部の寸法は、明瞭化のために多の要素と比較して誇張されている場合がある。さらに、適切と考える場合、参照数字が、対応する類似要素を示すために、複数の図の間で繰り返される場合がある。
【発明を実施するための形態】
【0017】
以下の詳細説明では、発明が十分に理解されるように、多数の具体的詳細が示される。しかし、これらの具体的詳細なしに本発明を実施できる場合があるということは、当業者によって理解される。他の例では、本発明を曖昧にしないように、公知の方法、手順、および構成要素は詳細に説明されていない。
【0018】
一実施形態では、本願明細書で説明されるような化合物および/またはそれを含む組成物を、男性対象へのアンドロゲン遮断療法(ADT)により誘発されるほてり(hot flash)または重度のほてりの処置に使用できる。一実施形態では、被検者は前立腺癌を患っている。別の実施形態では、被検者は進行前立腺癌を患っている。
【0019】
一実施形態では、本願明細書で説明されるような化合物および/またはそれを含む組成物を、男性対象へのアンドロゲン遮断療法(ADT)により誘発されるほてりまたは重度のほてりの発生率減少、抑制、重症度軽減、または阻害を行うために使用できる。一実施形態では、被検者は前立腺癌を患っている。別の実施形態では、被検者は進行前立腺癌を患っている。
【0020】
一実施形態では、本発明は、以下の式Iの構造で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象へのアンドロゲン遮断療法(ADT)により誘発されるほてりの処置、発生率減少、抑制、重症度軽減、または阻害を行う方法を提供する。
【0021】
【化1】
【0022】
式中、
Yは、C(O)またはCH2であり;
R1、R2は、互いに独立して、水素、ハロゲン、ヒドロキシル、アルコキシ、シアン、ニトロ、CF3、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、O−Alk−NR5R6、または、置換されたもしくは非置換の3〜7員のO−Alk−複素環(任意選択で芳香族である)であり;
R3、R4は、互いに独立して、水素、ハロゲン、ヒドロキシアルキル、ヒドロキシル、アルコキシ、シアン、ニトロ、CF3、NHCOR、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、または保護ヒドロキシルであり;
Rは、アルキル、水素、ハロアルキル、ジハロアルキル、トリハロアルキル、CH2F、CHF2、CF3、CF2CF3、アリール、フェニル、ハロゲン、アルケニル、CN、NO2、またはOHであり;
R5およびR6は、互いに独立して、水素、フェニル、炭素数1〜6のアルキル基、3〜7員のシクロアルキル、3〜7員の複素環、5〜7員のアリールであり;あるいは、R5およびR6は、窒素原子を含む3〜7員環を形成し;
jおよびkは、互いに独立して、1〜4であり;
Alkは、炭素数1〜7の直鎖アルキル、炭素数1〜7の分岐鎖アルキル、または炭素数3〜8の環状アルキルである。
【0023】
本願明細書に記載の方法の別の実施形態では、式Iの化合物が式IAで表される:
【0024】
【化14】
【0025】
式中、R1、R2、R3、R4、jおよびkは、式Iに定義された通りである。
【0026】
一実施形態では、本発明は、以下の式IIの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象へのアンドロゲン遮断療法(ADT)により誘発されるほてりまたは重度のほてりの処置、発生率減少、抑制、重症度軽減、または阻害を行う方法を提供する。
【0027】
【化2】
【0028】
一実施形態では、本発明は、以下の式IIIの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象へのアンドロゲン遮断療法(ADT)により誘発されるほてりまたは重度のほてりの処置、発生率減少、抑制、重症度軽減、または阻害を行う方法を提供する。
【0029】
【化3】
【0030】
一実施形態では、本発明は、以下の式IVの化合物で表される化合物、またはその異性体、薬学的生成物、薬学的に許容される塩、多形体、水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象へのアンドロゲン遮断療法(ADT)により誘発されるほてりまたは重度のほてりの処置、発生率減少、抑制、重症度軽減、または阻害を行う方法を提供する。
【0031】
【化4】
【0032】
一実施形態では、本発明は、以下の式Vの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象へのアンドロゲン遮断療法(ADT)により誘発されるほてりまたは重度のほてりの処置、発生率減少、抑制、重症度軽減、または阻害を行う方法を提供する。
【0033】
【化5】
【0034】
一実施形態では、本発明は、以下の式VIの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象へのアンドロゲン遮断療法(ADT)により誘発されるほてりまたは重度のほてりの処置、発生率減少、抑制、重症度軽減、または阻害を行う方法を提供する。
【0035】
【化6】
【0036】
一実施形態では、本発明は、以下の式VIIの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象へのアンドロゲン遮断療法(ADT)により誘発されるほてりまたは重度のほてりの処置、発生率減少、抑制、重症度軽減、または阻害を行う方法を提供する。
【0037】
【化7】
【0038】
一実施形態では、本発明は、以下の式VIIIの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象へのアンドロゲン遮断療法(ADT)により誘発されるほてりまたは重度のほてりの処置、発生率減少、抑制、重症度軽減、または阻害を行う方法を提供する。
【0039】
【化8】
【0040】
一実施形態では、本発明は、以下の式IXの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、前立腺癌を患う男性対象へのアンドロゲン遮断療法(ADT)により誘発されるほてりまたは重度のほてりの処置、発生率減少、抑制、重症度軽減、または阻害を行う方法を提供する。
【0041】
【化9】
【0042】
一実施形態では、本発明は、以下の式Xの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象へのアンドロゲン遮断療法(ADT)により誘発されるほてりまたは重度のほてりの処置、発生率減少、抑制、重症度軽減、または阻害を行う方法を提供する。
【0043】
【化10】
【0044】
一実施形態では、本発明は、以下の式XIの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象へのアンドロゲン遮断療法(ADT)により誘発されるほてりまたは重度のほてりの処置、発生率減少、抑制、重症度軽減、または阻害を行う方法を提供する。
【0045】
【化11】
【0046】
一実施形態では、本発明は、以下の式XIIの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象へのアンドロゲン遮断療法(ADT)により誘発されるほてりまたは重度のほてりの処置、発生率減少、抑制、重症度軽減、または阻害を行う方法を提供する。
【0047】
【化12】
【0048】
一実施形態では、本発明は、式IA、I〜XIIの化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象へのアンドロゲン遮断療法(ADT)により誘発されるほてりまたは重度のほてりの処置、発生率減少、抑制、重症度軽減、または阻害を行う方法を提供する。別の実施形態では、男性被検者は前立腺癌を患っている。別の実施形態では、男性被検者は進行前立腺癌を患っている。別の実施形態では、化合物は化合物IVである。
【0049】
用語「進行前立腺癌」は、前立腺に起源を持ち、かつ精嚢、骨盤リンパ節、骨、または体の他の部分を含む周辺組織など前立腺を超えて幅広く転移している転移性癌のことを言う。前立腺癌の病状は、悪性度が増加する順に1〜5のグリソン分類で格付けされる。別の実施形態では、進行性疾患および/または前立腺癌による死亡の重大な危険性がある患者を定義に含める必要があり、IIBほどの低い病期で前立腺被膜の外に癌がある患者には、明らかに「進行」疾患がある。
【0050】
一実施形態では、本発明は、式IA、I〜XIIの化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、また任意の組み合わせを治療有効量投与することを含む、男性対象のほてりまたは重度のほてりの処置、発生率減少、抑制、重症度軽減、または阻害を行う方法を提供する。別の実施形態では、化合物は化合物IVである。
【0051】
一実施形態では、用語「ほてり(hot flash)」は、対象の体温の急上昇などの体温の一過性撹乱のことを言う。別の実施形態では、撹乱は発汗を伴う。一実施形態では、用語「ほてり」は、体、顔、および首の上部またはすべての紅潮、乳房、背中、および腕に現れる赤色斑、激しい発汗、寒気による震え、またはそれらの任意の組み合わせの突発性熱感のことを言う。一実施形態では、ほてりはヒト対象が経験し、別の実施形態では男性対象が経験する。一実施形態では、ほてりはADTの結果発生したものである。別の実施形態では、ほてりはADTの結果発生したものではない。一実施形態では、ほてりは前立腺癌処置の副作用である。別の実施形態では、ほてりは前立腺癌処置の副作用ではない。
【0052】
用語「重度のほてり」は、多量の発汗を引き起こし、異常な高熱を発生させ、長く持続し、行っている活動を妨げるほてりのことを言う。一実施形態では、重度のほてりは睡眠障害を特徴とする(不眠症を特徴とすることがよくある)。
【0053】
一実施形態では、ほてりを処置する本発明の方法を、例えば、閉経、酢酸タモキシフェン処置、前立腺癌処置、アルコール脱水素酵素欠損症、またはカルチノイド症候群/褐色細胞腫によって起こるほてりの処置に使用できる。各種類のほてりは、本発明の個別の実施形態を表す。
【0054】
本願明細書に示した通り、例えば化合物IVなど治療有効量の式Iの化合物を投与することで、対象のほてりを軽減したことを結果が示している。
【0055】
一実施形態では、本願明細書で説明されるような化合物および/またはそれを含む組成物を、男性対象の全血清テストステロン濃度を下げるために使用できる。
【0056】
一実施形態では、本願明細書で説明されるような化合物および/またはそれを含む組成物を、男性対象の全血清テストステロン濃度を下げるために使用でき、全血清テストステロンの低下は黄体形成ホルモン(LH)濃度の低下によって生じる。
【0057】
一実施形態では、本願明細書で説明されるような化合物および/またはそれを含む組成物を、男性対象の全血清テストステロン濃度を下げるために使用でき、全血清テストステロンの低下が黄体形成ホルモン濃度の低下と無関係である。
【0058】
一実施形態では、本発明は、式Iの構造で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象の全血清テストステロン濃度を下げる方法を提供する。
【0059】
【化1】
【0060】
式中、
Yは、C(O)またはCH2であり;
R1、R2は、互いに独立して、水素、ハロゲン、ヒドロキシル、アルコキシ、シアン、ニトロ、CF3、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、O−Alk−NR5R6、または、置換されたもしくは非置換の3〜7員のO−Alk−複素環(任意選択で芳香族である)であり;
R3、R4は、互いに独立して、水素、ハロゲン、ヒドロキシアルキル、ヒドロキシル、アルコキシ、シアン、ニトロ、CF3、NHCOR、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、または保護ヒドロキシルであり;
Rは、アルキル、水素、ハロアルキル、ジハロアルキル、トリハロアルキル、CH2F、CHF2、CF3、CF2CF3、アリール、フェニル、ハロゲン、アルケニル、CN、NO2、またはOHであり;
R5およびR6は、互いに独立して、水素、フェニル、炭素数1〜6のアルキル基、3〜7員のシクロアルキル、3〜7員の複素環、5〜7員のアリールであり;あるいは、R5およびR6は、窒素原子を含む3〜7員環を形成し;
jおよびkは、互いに独立して、1〜4であり;
Alkは、炭素数1〜7の直鎖アルキル、炭素数1〜7の分岐鎖アルキル、または炭素数3〜8の環状アルキルである。
【0061】
本願明細書に記載の方法の追加実施形態では、式Iの化合物が以下の式IAで表される。
【0062】
【化14】
【0063】
式中、R1、R2、R3、R4、jおよびkは、式Iに定義された通りである。
【0064】
一実施形態では、本発明は、式IIの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象の全血清テストステロン濃度を下げる方法を提供する。
【0065】
【化2】
【0066】
一実施形態では、本発明は、式IIIの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象の全血清テストステロン濃度を下げる方法を提供する。
【0067】
【化3】
【0068】
一実施形態では、本発明は、式IVの化合物で表される化合物、またはその異性体、薬学的生成物、薬学的に許容される塩、多形体、水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象の全血清テストステロン濃度を下げる方法を提供する。
【0069】
【化4】
【0070】
一実施形態では、本発明は、式Vの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象の全血清テストステロン濃度を下げる方法を提供する。
【0071】
【化5】
【0072】
一実施形態では、本発明は、式VIの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象の全血清テストステロン濃度を下げる方法を提供する。
【0073】
【化6】
【0074】
一実施形態では、本発明は、式VIIの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象の全血清テストステロン濃度を下げる方法を提供する。
【0075】
【化7】
【0076】
一実施形態では、本発明は、式VIIIの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象の全血清テストステロン濃度を下げる方法を提供する。
【0077】
【化8】
【0078】
一実施形態では、本発明は、式IXの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象の黄体形成ホルモン(LH)濃度を下げることで全血清テストステロン濃度を下げる方法を提供する。
【0079】
【化9】
【0080】
一実施形態では、本発明は、式Xの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象の全血清テストステロン濃度を下げる方法を提供する。
【0081】
【化10】
【0082】
一実施形態では、本発明は、式XIの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象の全血清テストステロン濃度を下げる方法を提供する。
【0083】
【化11】
【0084】
一実施形態では、本発明は、式XIIの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象の全血清テストステロン濃度を下げる方法を提供する。
【0085】
【化12】
【0086】
一実施形態では、本発明は、式IA、I〜XIIの化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象の全血清テストステロン濃度を下げる方法を提供する。別の実施形態では、男性被検者は前立腺癌を患っている。別の実施形態では、全血清テストステロンを約100ng/dL未満に下げる。別の実施形態では、全血清テストステロンを約50ng/dL未満に下げる。別の実施形態では、全血清テストステロン濃度を約25ng/dL未満に下げる。
【0087】
一実施形態では、本発明は、式IA、I〜XIIの化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、全血清テストステロンの削減を血清黄体形成ホルモン(LH)濃度を下げることで行う、男性対象の全血清テストステロン濃度を下げる方法を提供する。別の実施形態では、男性被検者は前立腺癌を患っている。別の実施形態では、全血清テストステロンを約100ng/dL未満に下げる。別の実施形態では、全血清テストステロンを約50ng/dL未満に下げる。別の実施形態では、全血清テストステロン濃度を約25ng/dL未満に下げる。
【0088】
一実施形態では、本発明は、式IA、I〜XIIの化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、遊離血清テストステロンの削減を血清黄体形成ホルモン(LH)濃度を下げることで行う、男性対象の遊離血清テストステロン濃度を下げる方法を提供する。別の実施形態では、男性被検者は前立腺癌を患っている。
【0089】
一実施形態では、本発明は、式IA、I〜XIIの化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、全血清テストステロンの削減は血清黄体形成ホルモン(LH)濃度の削減と関係がない、男性対象の全血清テストステロン濃度を下げる方法を提供する。別の実施形態では、男性被検者は前立腺癌を患っている。別の実施形態では、全血清テストステロンを約100ng/dL未満に下げる。別の実施形態では、全血清テストステロンを約50ng/dL未満に下げる。別の実施形態では、全血清テストステロン濃度を約25ng/dL未満に下げる。
【0090】
一実施形態では、本発明は、式IA、I〜XIIの化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、遊離血清テストステロンの削減は血清黄体形成ホルモン(LH)濃度の削減と関係がない、男性対象の遊離血清テストステロン濃度を下げる方法を提供する。別の実施形態では、男性被検者は前立腺癌を患っている。
【0091】
一実施形態では、本発明は、男性対象の全血清テストステロンまたは遊離血清テストステロンの濃度を下げる方法を提供する。ここで、当該男性対象は前立腺癌を患っている。別の実施形態では、当該被検者は進行前立腺癌を患っている。
【0092】
一実施形態では、テストステロンの血清濃度の削減は可逆的であり、本発明の化合物による処置後には基準濃度に戻る。
【0093】
別の実施形態では、図23および実施例10に準じて、テストステロンの血清濃度は、化合物IVによる処置後に可逆的である。
【0094】
一実施形態では、本発明は、式IA、I〜XIIの化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象の全血清テストステロン濃度を下げる方法を提供する。別の実施形態では、全血清テストステロンを約100ng/dL未満に下げる。別の実施形態では、全血清テストステロンを約50ng/dL未満に下げる。別の実施形態では、全血清テストステロンを約25ng/dL未満に下げる。別の実施形態では、全血清テストステロンを約75ng/dL未満に下げる。別の実施形態では、全血清テストステロンを約75ng/dL〜100ng/dLに下げる。別の実施形態では、全血清テストステロンを約50ng/dL〜75ng/dLに下げる。別の実施形態では、全血清テストステロンを約40ng/dL〜50ng/dLに下げる。別の実施形態では、全血清テストステロン濃度を約25ng/dL〜50ng/dLに下げる。別の実施形態では、全血清テストステロンを約40ng/dL〜60ng/dLに下げる。
【0095】
テストステロンを「遊離」(すなわち、生物学的に利用可能かつ未結合)または「全」(タンパク結合し、利用不可な分を含む)血清濃度として測定できる。前立腺癌に罹ってなく、40歳を超える男性では、全テストステロン濃度が250ng/dL未満(<8.7nmol/L)に下がり、あるいは遊離テストステロン濃度が0.75ng/dL未満(<0.03nmol/L)に下がることを示す。
【0096】
一実施形態では、本発明は、前立腺癌を患う男性対象において、黄体形成ホルモン(LH)濃度を下げることと関係なく、あるいはLH濃度を下げることで、全血清および/または遊離テストステロン濃度を下げる方法を提供する。別の実施形態では、テストステロン濃度の変化は、処置前の濃度から下がる必要がある。別の実施形態では、全血清テストステロン濃度を100ng/dL未満に下げる。別の実施形態では、全血清テストステロンを50ng/dL未満に下げる。別の実施形態では、全血清テストステロンを25ng/dL未満に下げる。別の実施形態では、遊離テストステロン濃度を2ng/dL未満に下げる。別の実施形態では、遊離テストステロン濃度を1ng/dL未満に下げる。別の実施形態では、遊離テストステロン濃度を0.5ng/dL未満に下げる。別の実施形態では、遊離テストステロン濃度を0.25ng/dL未満に下げる。
【0097】
血液検査による処置期間中テストステロン濃度のモニタリングを含む、遊離血清テストステロン濃度および全血清テストステロン濃度を測定する方法。全テストステロンは、担体タンパク質(アルブミン、SHBG、トランスコルチン、トランスフェリン)に結合したテストステロンと遊離/未結合ホルモンとの計算の組み合わせである。全テストステロン濃度は、ホルモンを体内に運ぶ血中タンパク質濃度、年齢、肥満、およびよく用いられる検査方法と関係がある干渉を含むいくつかの要因の影響を受けることがある。
【0098】
遊離テストステロン(FT)の測定に利用可能な方法は、複雑(平衡透析および算出遊離テストステロン(CFT))に、またはアナログトレーサーを用いて簡素(市販FTキット「Coat−A−Count」)になり得る。別の実施形態では、全テストステロンおよび遊離テストステロンの濃度測定を、全テストステロンおよびSHBG(例えば、Irma−Count、DPC)の同時測定し、その後、算出遊離テストステロン(CFT)を導き出すことによって達成できる。別の実施形態では、全テストステロンおよび遊離テストステロンの測定は、当業者の知識に準じている。
【0099】
一実施形態では、本発明は、ADTの別の形体の1つ以上と、式IA、I〜XIIの化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせとの組み合わせを治療有効量投与することを含む、男性対象の全血清テストステロン濃度または遊離血清テストステロン濃度を下げる方法を提供する。別の実施形態では、全または遊離血清テストステロンの削減は、血清黄体形成ホルモン(LH)濃度を下げることで生じる。別の実施形態では、全または遊離血清テストステロン濃度の削減は、血清黄体形成ホルモン濃度を下げることと関係がない。
【0100】
本発明の方法は、ADTの他の形態と、本発明の化合物との組み合わせを投与することを含む。一実施形態では、ADTの他の形態として、LHRHアゴニストを含む。別の実施形態では、LHRH他動薬として、酢酸ロイプロリド(Lupron(登録商標))(参照することにより本書にすべて援用されるUS5,480,656;US5,575,987;5,631,020;5,643,607;5,716,640;5,814,342;6,036,976)、または酢酸ゴセレリン(Zoladex(登録商標))(参照することにより本書にすべて援用されるUS7,118,552;7,220,247;7,500,964)。一実施形態では、ADTの他の形態として、LHRHアンタゴニストを含む。別の実施形態では、LHRHアンタゴニストはデガレリクスを含む。一実施形態では、ADTの他の形態として、抗アンドロゲンを含む。別の実施形態では、抗アンドロゲンとして、ビカルタミド、フルタミド、フィナステライド、デュタステライド、MDV3100、ニルタミド、クロルマジノン、またはそれらの任意の組み合わせが挙げられる。
【0101】
一実施形態では、本発明の方法は、抗アンドロゲンと、本発明の化合物とを治療有効量投与することを含む。一実施形態では、本発明の方法は、LHRHアゴニストと、本発明の化合物とを治療有効量投与することを含む。一実施形態では、本発明の方法は、抗アンドロゲンと、LHRHアゴニストと、本発明の化合物とを治療有効量投与することを含む。
【0102】
一実施形態では、本発明は、式IA、I〜XIIの化合物を治療有効量投与することを含む、アンドロゲン遮断療法(ADT)を行うことを目的として、前立腺癌を患う男性対象において、黄体形成ホルモン(LH)濃度を下げることにより、あるいは黄体形成ホルモン濃度の下げることと関係なく、全血清テストステロン濃度および/または遊離テストステロン濃度を下げる方法を提供する。別の実施形態では、化合物は化合物IVである。
【0103】
別の実施形態では、本発明は、式IA、I〜XIIの化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、対象のアンドロゲン遮断療法(ADT)の方法を提供する。別の実施形態では、当該被検者は前立腺癌を患っている。別の実施形態では、化合物は化合物IVである。
【0104】
別の実施形態では、ADTは、進行前立腺癌を処置するために、前立腺癌の進行を遅らせるために、または前立腺癌の再発を予防および/または処置するために使用される。
【0105】
一実施形態では、本発明は、本発明の化合物を投与することを含む、前立腺癌を処置する、前立腺癌の進行を遅らせる、前立腺癌の再発を予防および/または処置する方法を提供する。別の実施形態では、LHRH類似体、可逆性抗アンドロゲン(ビカルタミドまたはフルタミドなど)、抗エストロゲン、抗癌剤、5−α還元酵素阻害薬、アロマターゼ阻害薬、プロゲスチン、選択的アンドロゲン受容体モジュレーター(SARMS)、または他の核ホルモン受容体を通じて作用する薬剤の併用による、本発明の化合物の投与。
【0106】
一実施形態では、本発明は、化合物IA、I〜XIIの化合物を投与することを含む、前立腺癌を処置する、並びにLH濃度を下げることで、あるいはLH濃度を下げることと関係なく、全血清テストステロンおよび/または遊離血清テストステロン濃度を下げる方法を提供する。別の実施形態では、化合物IVを投与する。
【0107】
アンドロゲン遮断療法ではテストステロンを削減するだけでなく、エストロゲンがテストステロンの芳香族化から生じるため、エストロゲン濃度も低くなる。アンドロゲン遮断療法により誘発されるエストロゲン欠乏は、ほてり、女性化乳房および乳房痛、骨量減少、骨の質および強度の低下、骨粗鬆症、骨減少症、および致命的な骨折、有害な脂質変化および高い心血管疾患および心筋梗塞、性欲減退、インポテンス、筋肉量の低下(サルコペニア)、疲労、認知機能傷害、および鬱病、およびその他の情緒の変化を含む重大な副作用を引き起こす。
【0108】
一実施形態では、本発明は、ADTに関連する疾患、障害、または症状を処置する方法を提供する。他の実施形態では、本発明は、テストステロンの欠乏に関連する疾患、障害、または症状を処置する方法を提供する。各疾病、障害または症状は、本発明の個別の実施形態を表す。
【0109】
一実施形態では、本発明は、式IA、I〜XIIの化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象の全血清テストステロン濃度を下げる方法を示し、ここで、前記の当該式IA、I〜XIIの化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを投与することで、アンドロゲン遮断療法(ADT)に関連する副作用の発症を予防、抑制、軽減、阻害、または処置を行い、当該対象は前立腺癌を患っている。別の実施形態では、全または遊離血清テストステロン濃度の削減は、LH濃度を下げることによる、またはLH濃度を下げることと関係がない。
【0110】
一実施形態では、本発明の化合物を投与することで、従来のアンドロゲン遮断療法(ADT)に関連する典型的な副作用の発症の抑制、軽減、阻害、または処置を行う。別の実施形態では、被検者は前立腺癌を患っている。前記副作用の予防および/または軽減は、プラセボ群または対照群と比べる。一実施形態では、従来のアンドロゲン遮断療法(ADT)に関連がある典型的な副作用としては、ほてり、女性化乳房、骨ミネラル濃度の減少、および骨折の増加が挙げられる。別の実施形態では、本発明の化合物を投与することで、従来型のアンドロゲン遮断療法(ADT)を用いて分かるように、ほてりの発症を予防する。別の実施形態では、本発明の化合物を投与することで、従来型のアンドロゲン遮断療法(ADT)を用いて分かるように、女性化乳房の発症を予防する。別の実施形態では、本発明の化合物を投与することで、従来型のアンドロゲン遮断療法(ADT)を用いて分かるように、骨ミネラル濃度(BMD)の減少の発症を予防する。別の実施形態では、本発明の化合物を投与することで、従来型のアンドロゲン遮断療法(ADT)を用いて分かるように、骨折の増加が生じるのを予防する。別の実施形態では、骨折の増加は、病的骨折、非外傷性骨折、脊椎骨折、非脊椎骨折、新たな形態学的骨折、臨床骨折、またはそれらの組み合わせである。
【0111】
一実施形態では、用語「従来のアンドロゲン遮断療法」は、外科医が睾丸を除去する睾丸摘出(去勢手術)を対象にする。別の実施形態では、用語「従来のアンドロゲン遮断療法」は、黄体形成ホルモン放出ホルモン(LHRH)類似体を対象にする。これらの薬剤は、睾丸で作られたテストステロンの量を下げる。米国で入手可能なLHRH類似体の例としては、ロイプロリド(Lupron、Viadur、Eligard)、ゴセレリン(Zoladex)、トリプトレリン(Trelstar)、およびヒストレリン(Vantas)が挙げられる。別の実施形態では、用語「従来のアンドロゲン遮断療法」は、抗アンドロゲン剤を投与することを対象とする。すなわち、抗アンドロゲンは、アンドロゲンを使用する体の能力を阻害する。睾丸摘出後でも、あるいはLHRH類似体による処置中でも、少量のアンドロゲンが副腎でまだ作られる。抗アンドロゲン剤の例としては、フルタミド(Eulexin)、ビカルタミド(Casodex)、およびニルタミド(Nilandron)が挙げられる。別の実施形態では、用語「従来のアンドロゲン遮断療法」は、アバレリクス(Plenaxis)などの黄体形成ホルモン放出ホルモン(LHRH)アンタゴニストを投与することを対象とする。デガレリクス(Firmasgon)は、2008年にFDAによって進行前立腺癌の処置用に認可された新規のLHRHアンタゴニストである。別の実施形態では、用語「従来のアンドロゲン遮断療法」は、フィナステライド(Proscar)およびデュタステライド(Avodart)などの5α−還元酵素阻害薬を投与することを対象にする。5α−還元酵素阻害薬は、テストステロンをより活性なアンドロゲン、5α−ジヒドロテストステロン(DHT)に変換する体の機能を阻害する。別の実施形態では、用語「従来のアンドロゲン遮断療法」は、ケトコナゾール(Nizoral)などのテストステロン生合成の阻害薬を投与することを対象にする。別の実施形態では、用語「従来のアンドロゲン遮断療法」は、ジエチルスチルベストロールまたは17βエストラジオールなどのエストロゲンを投与することを対象にする。
【0112】
一実施形態では、用語「女性化乳房」とは、腺構成要素の増殖によって生じる男性の乳房の良性肥大のことを言い、痛みと関係がある場合も、無い場合もある。女性化乳房は、ゴム状の存在、または乳頭から同心円状に存在する硬い腫瘤によって臨床的に定義される。偽性女性化乳房または脂肪腫乳房として公知の症状は、腺の増殖のない脂肪堆積を特徴とする。通常、女性化乳房は左右対称であるが、前記症状は片側だけの場合がある。
【0113】
一実施形態では、本発明の方法は、骨量の減少およびほてりも生じること無く、テストステロンの削減による前立腺癌または進行前立腺癌の男性対象の処置を対象とする。別の実施形態では、本発明の方法は、化合物IA、I〜XIIを使用し、当該化合物には、前立腺癌に対する現在のアンドロゲン遮断療法(ADT)でよく見られる骨量減少およびほてりなどの特定の副作用も生じることなく、前立腺癌の主要刺激剤であるテストステロンを削減する能力がある。
【0114】
別の実施形態では、後述の表8(実施例11)によって、化合物IVを投与することで骨量減少も生じることなく、テストステロンを削減することを示す。
【0115】
一実施形態では、本願明細書で示される方法および/または本願明細書で示される化合物を利用することは、視床下部・下垂体・精巣軸(HPT軸)に関するフィードバックの提供において効果的である。フィードバックとは、自らの活動に影響を及ぼす別の臓器または組織の活動を調整する、1つの臓器または組織で生成される物質の能力のことを言う。一実施形態では、視床下部・下垂体・精巣軸(HPT軸)に関するフィードバックによって、LH濃度を削減する。一実施形態では、視床下部・下垂体・精巣軸(HPT軸)に関するフィードバックによって、全血清テストステロン濃度を削減する。一実施形態では、視床下部・下垂体・精巣軸(HPT軸)に関するフィードバックによって、遊離血清テストステロン濃度を削減する。一実施形態では、視床下部・下垂体・精巣軸(HPT軸)に関するフィードバックによって、血清、組織、または腫瘍のアンドロゲン濃度を削減する。
【0116】
視床下部・下垂体・精巣(HPT)軸とは、視床下部、下垂体、および睾丸のホルモン濃度を調整する内分泌生理学系のことを言う。LHRH(黄体形成ホルモン放出ホルモン)は視床下部によって放出され、LHおよびFSH(ゴナドトロピン)を合成および分泌するように下垂体を刺激する。その後、LHおよびFSHは睾丸に作用し、テストステロンおよび精液の産生を刺激する。その後、テストステロンは、視床下部LHRH分泌に対して直接的な負のフィードバック効果、および下垂体LHおよびFSH産生に対して間接的な負のフィードバック効果を持つ。エストロゲン、アンドロゲン、および血清タンパク質(インヒビンなど)も、LHRH分泌およびLHおよびFSHの分泌に対して負のフィードバックを持つ。
【0117】
下垂体は、体内のテストステロン濃度を調節する1つの腺である。テストステロン濃度が低い場合、下垂体が黄体形成ホルモン(LH)を放出する。このホルモンは、より多くのテストステロンを作るように睾丸を誘導する。思春期の間、テストステロンの濃度は増加する。テストステロンの濃度は約20〜40歳で最高になり、その後、高齢男性では徐々に低くなる。女性は、男性と比較してずっと少ない量のテストステロンを体内に持つ。しかし、男性と女性の両方において、テストステロンは体の隅々で重要な役割を果たす。脳、骨、および筋肉の量、脂肪分布、血管系、エネルギー準位、生殖器組織、および性機能に影響を及ぼす。血中のテストステロンの大部分は、性ホルモン結合グロブリン(SHBG)と呼ばれるタンパク質に、またはアルブミンと呼ばれる別の血清タンパク質に結合している。結合していない(または「遊離」)テストステロンも臨床的に測定できる。
【0118】
別の実施形態では、血清黄体形成ホルモン濃度を下げることと関係がない全血清テストステロンまたは遊離血清テストステロンの濃度の削減は、性ホルモン結合グロブリン(SHBG)の増加による。別の実施形態では、血清黄体形成ホルモン濃度を下げることと関係がない遊離テストステロン濃度の削減は、性ホルモン結合グロブリン(SHBG)の増加による。別の実施形態では、血清黄体形成ホルモン(LH)濃度を下げることと関係がない全血清または遊離血清テストステロンの濃度の削減は、睾丸のライディッヒ細胞によるテストステロン産生または分泌の阻害による。別の実施形態では、血清黄体形成ホルモン(LH)濃度を下げることと関係がない全血清または遊離血清テストステロン濃度の低下は、副腎ステロイド産生の減少による。
【0119】
一実施形態では、本願明細書で説明されるような化合物および/またはそれを含む組成物を、黄体形成ホルモン(LH)濃度を下げるために使用できる。別の実施形態では、本発明の化合物および/または組成物を、内因性の性ホルモンを減らすために使用できる。
【0120】
ヒドロキシステロイド脱水素酵素(HSD)群の構成要素は、血中ステロイドの変換に関与する。17β−HSD5は、アンドロステンジオンをテストステロンに、エストロンをエストラジオールに変換する。さらに、プロスタグランジン合成にも関与する。一実施形態では、本発明の化合物は、HSD、特に17β−ヒドロキシステロイド脱水素酵素5(17β−HSD5)を阻害する。前記阻害は、末梢/性腺外テストステロン合成を防止することで、ADTにおいて有益な場合があり、前記合成によって、HPT軸制御を避け、全または遊離血清テストステロンの不完全な削減を生じる、あるいは細胞内テストステロン濃度の局所的な上昇を可能にし、そのいずれかがADTでは有害である可能性がある。
【0121】
LHRHアゴニスト治療、すなわち黄体形成ホルモン放出ホルモンアゴニスト(LHRH)またはその類似体の投与によった達成されるアンドロゲン遮断療法(ADT)によって、下垂体からのゴナドトロピン放出、および睾丸からのテストステロン産生(「発赤反応」と呼ばれる)の初期刺激をもたらし、続いてゴナドトロピン放出の減少、およびテストステロンとエストロゲン両方の濃度の減少をもたらす。LHRHアゴニスト療法によって生じる「発赤反応」は、アンドロゲン/テストステロン濃度の増加によって前立腺癌の処置に悪影響をもたらす。さらに、LHRH療法は、糖尿病と心血管疾患のリスク増加に関連があった(Smith(2008)Current Prostate Reports.6:149-154)。
【0122】
LHRH療法の発赤の影響を克服する努力において、抗アンドロゲン単剤療法(ビカルタミド、フルタミド、クロルマジノン)、LHRH/抗アンドロゲン併用療法、およびLHRHアンタゴニスト(デガレリクス)が提案されている(Suzuki et al., (2008) Int. J. Clin. Oncol. 13: 401-410; Sharifi, N. et al., (2005) JAMA. 294(2): 238-244)。抗アンドロゲン単剤療法では、対象のアンドロゲン濃度を下げない。ビカルタミド抗アンドロゲン単剤療法では、骨転移のある前立腺癌患者においてADTより効果が低いことが示された。さらに、ビカルタミド療法で見られる副作用としては、乳房の圧痛および乳房肥大(女性化乳房および乳房痛)が挙げられる(Suzuki et al., ibid)。抗アンドロゲン療法による付加的リスクとしては、肝臓アミノ基転移酵素の増加が挙げられる(Sharifi et al., ibid)。
【0123】
一実施形態では、本発明は、LH濃度を低下させ、その結果、「発赤」の影響を生じることなく、かつ従来のADT法を用いてテストステロン低下によって生じるエストロゲン不足な関連がある副作用を克服しながら、全血清テストステロンおよび/または遊離血清テストステロン濃度を低下させる。方法/対象化合物の使用によって、骨組織の維持(骨組織に対するアゴニスト作用)、トロンビン産生能および/またはほてりの低下、および/またはエストラジオールまたはジエチルスチルベストロールよりも乳房組織に対する少ない影響、または中立的影響を示す組織選択的エストロゲン活性を提供する。
【0124】
一実施形態では、化合物IVはアゴニスト作用を示すが、アンタゴニスト作用は示さず(実施例6および7)、そのため、化合物IVはゴナドトロピンおよびテストステロンの増加を生じないだろう。
【0125】
一実施形態では、化合物IVは、血清ホルモン、テストステロン、および全アンドロゲンを下げる堅牢な薬理反応を示すアゴニスト活性(実施例8〜11)を示す。
【0126】
一実施形態では、本願明細書で示される化合物および/または組成物を利用する本願明細書で示される方法は、従来の形態のADTを用いたLHの削減によって生じる骨吸収作用の削減または除去において効果的である。一実施形態では、本願明細書で示される方法および/または本願明細書で示される組成物の利用は、従来の形態のADTを用いたテストステロン濃度の削減によって生じる骨吸収作用の削減または除去において効果的である。一実施形態では、本願明細書で示される組成物を利用した本願明細書で示される方法は、LH濃度低下の結果としてのエストロゲンの削減によって生じる骨吸収作用の削減または除去において効果的である。一実施形態では、本願明細書で示される化合物および/または組成物を利用する本願明細書で示される方法は、従来の形態のADTを用いたLH濃度低下に関係がある骨吸収作用を予防する。一実施形態では、本願明細書で示される化合物および/または組成物を利用する本願明細書で示される方法は、従来の形態のADTを用いた内因性LH、テストステロン、および/またはエストラジオールの低下に関係がある骨吸収作用を予防する。一実施形態では、本願明細書で示される化合物および/または組成物を利用する本願明細書で示される方法は、LH濃度の低下を示しながら、骨ミネラル濃度(BMD)を増加させる。一実施形態では、本願明細書で示される化合物および/または組成物を利用する本願明細書で示される方法は、内因性LH、テストステロン、および/またはエストラジオール濃度の低下を示しながら、パーセント骨量を増加させる。
【0127】
一部の実施形態では、本発明は、本発明の化合物、またはその異性体、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを投与することで、血栓塞栓症を避ける、および/または軽減する方法を提供する。
【0128】
一実施形態では、本願明細書で示される化合物および/または組成物を利用する本願明細書で示される方法は、乳房組織に効果的である。一実施形態では、本願明細書で示される化合物および/または組成物を利用する本願明細書で示される方法は、従来のADTによって達成されるLH濃度の低下に関係がある女性化乳房を予防しながら、LH濃度を低下させる。
【0129】
一実施形態では、実施例13によって特別な毒性研究を開示し、ヒト血小板によるそのインビトロ研究から、化合物IVがDESよりもずっと低い前凝固性活性を示すことが分かった。そのため、化合物IV、ER選択的アゴニストは、DESよりも血栓症の低いリスクでDESによる前立腺癌処置の利点を実現する必要があり、骨量減少、ほてり、または有害な脂質状態の変化を生じることなくLHRHアゴニストまたはアンタゴニストの利点も実現する必要がある。
【0130】
ジエチルスチルベストロール(DES)療法単独またはADTとの併用によって、DESが前立腺癌患者の骨吸収を予防することを示した。DESの使用は、前立腺癌用の治療として推進されてきたが、血管形成および悪性腫瘍に対するDESの影響は、DES代謝物によって仲介されると考えられ、エストロゲン受容体を通じて作用するとは考えられていない。さらに、治療用に投与されるDESの用量レベルは、血管疾患、心血管疾患の罹患率、血栓毒性、女性化乳房、勃起障害、および性欲減退を含む多数の副作用を示す(Scherr and Pitts, ibid and Presti, J.C. Jr. (1996) JAMA. 275(15): 1153-6)。
【0131】
一実施形態では、本発明は、LHRHアゴニストまたはアンタゴニスト療法の単独または抗アンドロゲン剤またはDESとの併用の副作用を克服する。別の実施形態では、対象発明の方法によって、有害な骨関連の症状などの有害なエストロゲン遮断副作用なしに、および女性化乳房などの有害なエストロゲン刺激副作用なしにアンドロゲン遮断療法を提供する。別の実施形態では、本発明の方法は、LH濃度を低下させ、その結果、「発赤」の影響を生じることなく、LHの低下によって生じるエストロゲン不足に関連がある副作用を克服し、並びにDES療法で見られる一般的なエストロゲンアゴニストの増加に関連がある副作用を克服しながら、総血清テストステロン濃度および/または遊離血清テストステロン濃度を低下させる。方法/対象化合物の使用によって、組織選択的エストロゲン活性を提供し、その結果、骨組織の維持(骨組織に対するアゴニスト作用)、トロンビン産生能の低下、乳房組織に対する中立的影響を示す。
【0132】
視床下部濃度でのタモキシフェン、トレミフェン、およびラロキシフェンなどの従来の選択的エストロゲン受容体モジュレーター(SERMs)の抗エストロゲン効果によって、男性でゴナドトロピン濃度の増加、またはLH濃度の増加を生じ、その結果、テストステロン血清濃度の増加を生じる可能性がある(Tsouri et al., 2008, Fertility and Sterility doi: 10.1016)。その一方、本発明の方法は、式I〜XIIの化合物を投与することを含み、男性対象のLHを低下させる。
【0133】
式Iの化合物の追加実施形態
【0134】
本発明の方法の一実施形態では、式Iの化合物のYがC(O)である。別の実施形態では、YがCH2である。別の実施形態では、式IまたはIAの化合物のR1およびR2は、互いに独立して、O−Alk−NR5R6またはO−Alk−複素環である。別の実施形態では、本願明細書の前述のように前記のO−Alk−複素環、O−Alk−NR5R6、−Alk−複素環、およびAlk−NR5R6のAlkは、炭素数1〜7の直鎖アルキル、炭素数1〜7の分岐鎖アルキル、または炭素数3〜8の環状アルキルである。別の実施形態では、アルキルはエチレン(−CH2CH2−)である。別の実施形態では、Alkはメチレン(−CH2−)である。別の実施形態では、Alkはプロピレン(−CH2CH2CH2−)である。別の実施形態では、Alkは2−メチルプロピレン(−CH2CH(CH3)CH2−)である。
【0135】
本発明の方法の一実施形態では、式IまたはIAの化合物のR1がパラ位にある。本発明の方法の一実施形態では、式IまたはIAの化合物のR1とR2は異なる。本発明の方法の別の実施形態では、式IまたはIAの化合物のR1とR2は同じである。本発明の方法の別の実施形態では、式IまたはIAの化合物のR1は、
【0136】
【化13】
【0137】
である。方法の別の実施形態では、式IまたはIAの化合物のR1は、ヒドロキシルである。方法の別の実施形態では、式IまたはIAの化合物のR1は、ヒドロキシルである。方法の別の実施形態で、R1、およびR2は、互いに独立して、水素、ハロゲン、ヒドロキシル、アルコキシ、シアン、ニトロ、CF3、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、O−Alk−NR5R6、または複素環が3〜7員置換または未置換の複素環であるO−Alk−複素環である。方法の別の実施形態で、式IまたはIAの化合物のR1およびR2は、互いに独立して、ハロゲン、ヒドロキシル、アルコキシ、シアン、ニトロ、CF3、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、O−Alk−NR5R6、または、置換されたもしくは非置換の3〜7員のO−Alk−複素環(任意選択で芳香族である)。方法の別の実施形態では、式IまたはIAの化合物のR2は、ハロゲンである。方法の別の実施形態では、式IまたはIAの化合物のR2は、Fである。方法の別の実施形態では、式Iの化合物のR2は、Clである。方法の別の実施形態では、式IまたはIAの化合物のR2は、Brである。方法の別の実施形態では、式IまたはIAの化合物のR2は、Iである。方法の別の実施形態では、式IまたはIAの化合物のR2は、ヒドロキシルである。方法の別の実施形態で、R1および/またはR2はCF3である。別の実施形態で、R1および/またはR2はCH3である。別の実施形態で、R1および/またはR2はハロゲンである。別の実施形態で、R1および/またはR2はFである。別の実施形態で、R1および/またはR2はClである。別の実施形態で、R1および/またはR2はBrである。別の実施形態で、R1および/またはR2はIである。別の実施形態で、式Iの化合物のR2はパラ位にある。
【0138】
本発明の方法の一実施形態では、式IまたはIAの化合物のR3とR4は同じである。本発明の方法の別の実施形態では、式IまたはIAの化合物のR3とR4は異なる。方法の別の実施形態では、式IまたはIAの化合物のjおよびkは、互いに独立して、1である。方法の別の実施形態では、式IまたはIAの化合物のR3およびR4は、互いに独立して、ハロゲン、ハロアルキル、ヒドロキシル、またはアルキルである。方法の別の実施形態では、式IまたはIAの化合物のR3およびR4は、互いに独立して、Fである。方法の別の実施形態では、式IまたはIAの化合物のR3およびR4は、互いに独立して、Brである。方法の別の実施形態では、式IまたはIAの化合物のR3およびR4は、互いに独立して、Clである。別の実施形態では、R4はパラ位にある。別の実施形態では、R3はオルト位にある。別の実施形態では、R3はメタ位にある。別の実施形態で、R3および/またはR4はCF3である。別の実施形態で、R3および/またはR4はCH3である。
【0139】
本発明の方法の一実施形態では、式IまたはIAの化合物のR5とR6は、窒素原子を含む3〜7員環を形成する。別の実施形態では、環が飽和または不飽和環である。別の実施形態では、環が置換または非置換環である。本発明の方法の別の実施形態では、式IまたはIAの化合物のR5およびR6は、窒素原子を含むピペリジン環を形成する。方法の別の実施形態では、式IまたはIAの化合物のR5およびR6は、窒素原子を含むピラジン環を形成する。方法の別の実施形態では、式IまたはIAの化合物のR5およびR6は、窒素原子を含むピペラジン環を形成する。方法の別の実施形態では、式IまたはIAの化合物のR5およびR6は、窒素原子を含むモルホリン環を形成する。方法の別の実施形態では、式IまたはIAの化合物のR5およびR6は、窒素原子を含むピロール環を形成する。方法の別の実施形態では、式IまたはIAの化合物のR5およびR6は、ピロリジンを形成する。方法の別の実施形態では、式IまたはIAの化合物のR5およびR6は、窒素原子を含むピリジン環を形成する。別の実施形態では、環は、ハロゲン、アルキル、アルコキシ、ヒドロキシル、シアノ、ニトロ、アミノ、アミド、COOH、またはアルデヒドで置換される。
【0140】
本発明の方法の別の実施形態では、式IまたはIAの化合物のR1と、式IまたはIAの化合物のR2は、互いに独立して、O−Alk−複素環またはOCH2CH2−複素環である。別の実施形態では、用語「複素環」基とは、環の一部として炭素原子に加えて、硫黄、水素、窒素、またはそれらの任意の組み合わせを含む、一実施形態での環構造のことを言う。別の実施形態では、複素環は3〜12員環である。別の実施形態では、複素環は6員環である。別の実施形態では、複素環は5〜7員環である。別の実施形態では、複素環は4〜8員環である。別の実施形態では、複素環基は非置換であるか、またはハロゲン、ハロアルキル、ヒドロキシル、アルコキシ、カルボニル、アミド、アルキルアミド、ジアルキルアミド、シアノ、ニトロ、CO2H、アミノ、アルキルアミノ、カルボキシル、チオ、および/またはチオアルキルで置換されてもよい。別の実施形態では、複素環は、別の飽和または不飽和シクロアルキルまたは3〜8員複素環と融合されることがある。別の実施形態では、複素環が飽和環である。別の実施形態では、複素環が不飽和環である。別の実施形態では、複素環がピペリジンである。別の実施形態では、複素環がピリジンである。別の実施形態では、複素環がピペリジン、ピリジン、フラン、チオフェン、ピロール、ピロリジン、ピラジン、ピペラジン、またはピリミジンである。
【0141】
用語「シクロアルキル」は、炭素と水素の原子を含む非芳香族環、単環、または多環のことを言う。シクロアルキル基は、環がその存在によって芳香族環の状態にならない限りは、環に1つ以上の炭素−炭素二重結合を持ってもよい。シクロアルキル基の例としては、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシルおよびシクロヘプチルなどの炭素数3〜7のシクロアルキル基、飽和環式および二環式テルペン、シクロプロペニル、シクロブテニル、シクロペンテニル、シクロヘキセニル、およびシクロヘプテニルなど炭素数3〜7のシクロアルケニル基、不飽和環式および二環式テルペンが挙げられるが、これに限定されるものではない。シクロアルキル基は、非置換、または1つまたは2つの置換基で置換されてもよい。好適には、シクロアルキル基は単環または二環である。
【0142】
一実施形態では、用語「アルキル」とは、直鎖、分岐鎖、および環式アルキル基を含む飽和脂肪族炭化水素のことを言う。一実施形態では、アルキル基は1〜12個の炭素を持つ。別の実施形態では、アルキル基は1〜7個の炭素を持つ。別の実施形態では、アルキル基は1〜6個の炭素を持つ。別の実施形態では、アルキル基は1〜4個の炭素を持つ。別の実施形態では、環式アルキル基は3〜8個の炭素を持つ。別の実施形態では、環式アルキル基は3〜12個の炭素を持つ。別の実施形態では、分岐アルキルは、1〜5個の炭素のアルキル側鎖で置換されたアルキルである。別の実施形態では、分岐アルキルは、1〜5個の炭素のハロアルキル側鎖で置換されたアルキルである。アルキル基は非置換であるか、またはハロゲン、ハロアルキル、ヒドロキシル、アルコキシ、カルボニル、アミド、アルキルアミド、ジアルキルアミド、ニトロ、アミノ、アルキルアミノ、ジアルキルアミノ、カルボキシル、チオ、および/またはチオアルキルで置換されてもよい。
【0143】
別の実施形態では、「アルケニル」基とは、直鎖、分岐鎖、および1つ以上の二重結合を有する環式基を含む、不飽和炭化水素のことを言う。アルケニル基は、1つの二重結合、2つの二重結合、3つの二重結合などを有してもよい。別の実施形態では、アルケニル基は2〜12個の炭素を持つ。別の実施形態では、アルケニル基は2〜6個の炭素を持つ。別の実施形態では、アルケニル基は2〜4個の炭素を持つ。アルケニル基の例としては、エテニル、プロペニル、ブテニル、シクロヘキセニルなどである。アルケニル基は非置換であるか、またはハロゲン、ヒドロキシル、アルコキシカルボニル、アミド、アルキルアミド、ジアルキルアミド、ニトロ、アミノ、アルキルアミノ、ジアルキルアミノ、カルボキシル、チオ、および/またはチオアルキルで置換されてもよい。
【0144】
「アリール」基は、少なくとも1つの炭素環式芳香族基または複素環式芳香族基を有する芳香族基のことを言い、非置換であるか、またはハロゲン、ハロアルキル、ヒドロキシル、アルコキシカルボニル、アミド、アルキルアミド、ジアルキルアミド、ニトロ、アミノ、アルキルアミノ、ジアルキルアミノ、カルボキシル、あるいはチオ、またはチオアルキルから選ばれる1つ以上の基で置換されてもよい。アリール環の限定されない例としては、フェニル、ナフチル、ピラニル、ピロリル、ピラジニル、ピリミジニル、ピラゾリル、ピリジニル、フラニル、チオフェニル、チアゾリル、イミダゾリル、イソオキサゾリルなどである。一実施形態では、アリール基は4〜8員環である。別の実施形態では、アリール基は4〜12員環である。別の実施形態では、アリール基は6員環である。別の実施形態では、アリール基は5員環である。別の実施形態では、アリール基は2〜4個の縮合環系である。
【0145】
一実施形態では、「アルデヒド」基とは、ホルミル基で置換されたアルキルまたはアルケニルのことを言い、アルキルまたはアルケニルは本書の上で定義したとおりである。別の実施形態では、アルデヒド基は、ホルミル基で置換されたアリール基またはフェニル基であり、アリールは本書の上で定義したとおりである。アルデヒドの例としては、ホルミル、アセタール、プロパナール、ブタナール、ペンタナール、ベンズアルデヒドである。別の実施形態では、アルデヒド基はホルミル基である。
【0146】
別の実施形態では、「ハロアルキル」基は、上に定義したようなアルキル基のことを言い、このアルキル基は1つ以上のハロゲン原子、例えばF、Cl、Br、またはIで置換される。
【0147】
別の実施形態では、「ヒドロキシル」基はOH基のことを言う。本発明の化合物のR1、R2、またはR3がORである場合、RはOHではないことは、当業者には明らかである。
【0148】
一実施形態では、用語「ハロゲン」または「ハロ」は、F、Cl、Br、またはIなどのハロゲンのことを言う。
【0149】
別の実施形態では、語句「フェノール」は、ベンゼンのアルコール(OH)誘導体のことを言う。
【0150】
一部の実施形態では、保護ヒドロキシルへの言及には、ベンゼン環の酸素部分に結合した置換基の取り込みを含み、当該置換基は容易に取り除くことができる。一部の実施形態では、フェノール保護基が以下を含む場合がある。すなわち、メチルエーテル(メトキシ)、アルキルエーテル(アルコキシ)、ベンジルエーテル(Bn)、メトキシメチル(MOM)エーテル、ベンゾイルオキシメチル(BOM)エーテル、ベンジル、カルボベンゾキシ、メトキシエトキシメチル(MEM)エーテル、2−(トリメチルシリル)エトキシメチル(SEM)エーテル、メチルチオメチル(MTM)エーテル、フェニルチオメチル(PTM)エーテル、アジドメチルエーテル、シアノメチルエーテル、2,2−ジクロロ−1,1−ジフルオロエチルエーテル、2−クロロエチルエーテル、2−ブロモエチルエーテル、テトラヒドロピラニル(THP)エーテル、1−エトキシエチル(EE)エーテル、フェナシルエーテル、4−ブロモフェナシルエーテル、シクロプロピルメチルエーテル、アリルエーテル、プロパルギルエーテル、イソプロピルエーテル、シクロヘキシルエーテル、t−ブチルエーテル、2,6−ジメチルベンジルエーテル、4−メトキシベンジルエーテル、o−ニトロベンジルエーテル、2,6−ジクロロベンジルエーテル、3,4−ジクロロベンジルエーテル、4−(ジメチルアミノ)カルボニルベンジルエーテル、4−メチルスルフィニルベンジルエーテル、4−アントリルメチルエーテル、4−ピコリルエーテル、ヘプタフルオロ−p−トリル、テトラフルオロ−4−ピリジルエーテル、トリメチルシリル(TMS)エーテル、t−ブチルジメチルシリル(TBDMS)エーテル、t−ブチルジフェニルシリル(TBDPS)エーテル、トリイソプロピルシリル(TIPS)エーテル、ギ酸アリール、酢酸アリール、レブリン酸アリール、アリールピバレート、安息香酸アリール、アリール9−フルオレンカルボキシレート、アリールメチルカルボネート、1−アダマンチルカルボネート、t−ブチルカルボネート、4−メチルスルフィニルベンジルカルボネート、2,4−ジメチルペンタ−3−イルカルボネート、アリール2,2,2−トリクロロエチルカルボネート、アリールベンジルカルボネート、アリールカルバメート、ジメチルホスフィニルエステル(Dmp−OAr)、ジメチルホスフィノチオニルエステル(Mpt−OAr)、ジフェニルホスフィノチオニルエステル(Dpt−OAr)、メタンスルホン酸アリール、トルエンスルホン酸アリール、または2−ホルミルベンゼンスルホン酸アリール。
【0151】
一実施形態では、本発明の方法は、N,N−ビス(4−ヒドロキシフェニル)−4−プロピルベズアミド(II)、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを使用する。別の実施形態では、本発明の方法は、4,4´−(2,3−ジメチル−ベンジルアザンジイル)ジフェノール(III)、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを使用する。別の実施形態では、本発明の方法は、3−フルオロ−N−(4−フルオロフェニル)−4−ヒドロキシ−N−(4−ヒドロキシフェニル)ベズアミド(IV)、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを使用する。別の実施形態では、本発明の方法は、N,N−ビス(4−ヒドロキシフェニル)−2,3−ジメチルベズアミド(V)、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを使用する。別の実施形態では、本発明の方法は、N,N−ビス(4−ヒドロキシフェニル)−2−ナフチルズアミド(VI)、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを使用する。別の実施形態では、本発明の方法は、3−フルオロ−4−ヒドロキシ−N,N−ビス(4−ヒドロキシフェニル)−ベズアミド(VII)、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを使用する。別の実施形態では、本発明の方法は、4−((4−フルオロフェニル)(4−ヒドロキシベンジル)アミノ)フェノール(VIII)、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを使用する。別の実施形態では、本発明の方法は、4−フルオロ−N−(4−ヒドロキシ−フェニル)−N−[4−(2−ピペリジン−1−イル−エトキシ)−フェニル]−2−トリフルオロメチル−ベズアミド(IX)、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを使用する。別の実施形態では、本発明の方法は、IXの塩酸塩(IXのHCl塩)または塩酸4−フルオロ−N−(4−ヒドロキシ−フェニル)−N−[4−(2−ピペリジン−1−イル−エトキシ)−フェニル]−2−トリフルオロメチル−ベズアミド(X)、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを使用する。別の実施形態では、本発明の方法は、3−フルオロ−4−ヒドロキシ−N−(4−ヒドロキシフェニル)−N−フェニルベズアミド(XI)、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを使用する。別の実施形態では、本発明の方法は、3−フルオロ−N,N−ビス−(4−ヒドロキシ−フェニル)−2−メチル−ベズアミド(XII)、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを使用する。
【0152】
一実施形態では、本発明の方法は、化合物の「薬学的に許容される塩」を使用し、この塩は、本発明の化合物の酸または塩基との反応によって生成することができる。
【0153】
本発明の方法の化合物のアミンの好適な薬学的に許容される塩は、無機酸または有機酸から調製できる。一実施形態では、アミンの無機塩の例としては、重硫酸塩、ホウ酸塩、臭化物、塩化物、ヘミ硫酸塩、臭化水素酸塩、塩酸塩、2−ヒドロキシエチルスルホン酸塩(スルホン酸ヒドロキシエタン)、ヨウ素酸塩、ヨウ化物、イソチオン酸塩、硝酸塩、過硫酸塩、リン酸塩、硫酸塩、スルファミン酸塩、スルファニル酸塩、スルホン酸(スルホン酸アルキル、スルホン酸アリール、ハロゲン置換スルホン酸アルキル、ハロゲン置換スルホン酸アリール)、スルホン酸塩、およびチオシアン酸塩である。
【0154】
一実施形態では、アミンの有機塩の例は、有機酸の脂肪族、脂環式、芳香族、芳香脂肪族、複素環、カルボン酸、およびスルホン酸類から選択でき、それらの例は、酢酸塩、アルギニン、アスパラギン酸塩、アスコルビン酸塩、アジピン酸塩、アントラニル酸塩、アルギン酸塩、カルボン酸アルカン、置換カルボン酸アルカン、アルギン酸塩、ベンゼンスルホン酸塩、安息香酸エステル塩、重硫酸塩、酪酸塩、重炭酸塩、酒石酸水素塩、カルボン酸塩、クエン酸塩、樟脳酸塩、樟脳スルホン酸塩、シクロヘキシルスルファミン酸塩、シクロペンタンプロピオン酸塩、エデト酸カルシウム塩、カンシル酸塩、炭酸塩、クラブラン酸塩、桂皮酸塩、ジカルボン酸塩、ジグルコン酸塩、ドデシルスルホン酸塩、二塩酸塩、デカン酸塩、エナンタート、エタンスルホン酸塩、エデト酸塩、エジシル酸塩、エストレート、エシレート、フマル酸塩、ギ酸塩、フッ化物、ガラクツロン酸塩、グルコン酸塩、グルタミン酸塩、グリコール酸塩、グルカ酸、グルコヘプタン酸塩、グリセロリン酸塩、グルセプテート、グリコリルアルサニレート、グルタル酸塩、グルタミン酸塩、ヘプタン酸塩、ヘキサン酸塩、ヒドロキシマレイン酸塩、ヒドロキシカルボン酸、ヘキシルレゾルシン酸塩、ヒドロキシ安息香酸塩、ヒドロキシナフトエ酸塩、フッ化水素酸塩、乳酸塩、ラクトビオン酸塩、ラウリン酸塩、リンゴ酸塩、マレイン酸塩、メチレンビス(β−オキシナフトエート)、マロン酸塩、マンデル酸塩、メシル酸塩、メタンスルホン酸塩、臭化メチル、硝酸メチル、スルホン酸メチル、マレイン酸カリウム、ムチン酸塩、モノカルボン酸塩、硝酸塩、ナフタレンスルホン酸塩、2−ナフタレンスルホン酸塩、ニコチン酸塩、ナフシレート、N−メチルグルカミン、シュウ酸塩、オクタン酸塩、オレイン酸塩、パモ酸塩、酢酸フェニル、ピクリン酸塩、安息香酸フェニル、ピバレート、プロピオン酸塩、フタル酸塩、ペクチン酸塩、プロピオン酸フェニル、パルミチン酸塩、パントテン酸塩、ポリガラクツロン酸塩、ピルビン酸塩、クインネート、サリチル酸塩、コハク酸塩、ステアリン酸塩、スルファニル酸塩、塩基性酢酸塩、酒石酸塩、酢酸テオフィリン、p−トルエンスルホン酸塩(トシレート)、トリフルオロ酢酸塩、テレフタル酸塩、タンニン酸塩、テオクル酸塩、トリハロ酢酸塩、トリエチオダイド、トリカルボン酸塩、ウンデカン酸塩、および吉草酸塩である。
【0155】
一実施形態では、カルボン酸またはフェノールの無機塩の例は、アンモニウムと、リチウム、ナトリウム、カリウム、セシウムを含むアルカリ金属と;カルシウム、マグネシウム、アルミニウムを含むアルカリ土類金属と;亜鉛と、バリウムと、コリンと、第4級アンモニウムとから選択できる。
【0156】
別の実施形態では、カルボン酸またはフェノールの有機塩の例は、アルギニンと、脂肪族有機アミン、脂環式有機アミン、芳香族有機アミン、ベンザチン、t−ブチルアミン、ベネタミン(N−ベンジルフェネチルアミン)、ジシクロヘキシルアミン、ジメチルアミン、ジエタノールアミン、エタノールアミン、エチレンジアミン、ヒドラバミン、イミダゾール、リジン、メチルアミン、メグルミン、N−メチル−D−グルカミン、N,N´−ジベンジルエチレンジアミン、ニコチンアミド、有機アミン、オルニチン、ピリジン、ピコリル、ピペラジン、プロカイン、トリス(ヒドロキシメチル)メチルアミン、トリエチルアミン、トリエタノールアミン、トリメチルアミン、トロメタミン、および尿素を含む有機アミンとから選択できる。
【0157】
一実施形態では、塩は、塩が溶けない溶媒または媒体、または水などの溶媒の中で生成物の形態の遊離塩基または遊離酸を1つ以上の同等の適切な酸または塩基に反応させるなど、従来の手段で形成できる。これらの溶媒は、真空、または凍結乾燥、または既存の塩のイオンを別のイオンまたは好適なイオン交換樹脂に交換することで取り除かれる。
【0158】
一実施形態では、本発明の方法は、本発明の化合物の薬学的に許容される塩を使用する。一実施形態では、本発明の方法は、式IA、I〜XIIの化合物の薬学的に許容される塩を使用する。一実施形態では、本発明の方法は、本発明の式IA、I〜XIIの化合物のアミンの塩を使用する。一実施形態では、本発明の方法は、本発明の式IA、I〜XIIの化合物のフェノールの塩を使用する。
【0159】
一実施形態では、本発明の方法は、式IA、I〜XIIの遊離塩基、遊離酸、非荷電、または非複合化合物、および/またはその異性体、薬学的生成物、水和物、多形体またはそれらの任意の組み合わせを使用する。
【0160】
本発明の一部の実施形態では、本発明の化合物は、アミン結合によって一緒に保持される3つのフェニル基を含む。一実施形態では、本発明の化合物は非荷電構造である。別の実施形態では、本発明の化合物は遊離塩基構造である。別の実施形態では、本発明の化合物は遊離酸構造である。別の実施形態では、本発明の化合物は非複合構造である。別の実施形態では、本発明の化合物は非イオン化構造である。別の実施形態では、本発明の化合物は薬学的に許容される塩である。別の実施形態では、本発明の一部の化合物は塩酸(HCl)塩を含む。
【0161】
一実施形態では、本発明の方法は、式IA、I〜XIIの化合物の異性体を使用する。一実施形態では、本発明の方法は、式IA、I〜XIIの化合物の薬学的生成物を使用する。一実施形態では、本発明の方法は、式IA、I〜XIIの化合物の水和物を使用する。一実施形態では、本発明の方法は、式IA、I〜XIIの化合物の多形体を使用する。一実施形態では、本発明の方法は、式IA、I〜XIIの化合物の代謝物を使用する。別の実施形態では、本発明の方法は、本書に記載の通り、式IA、I〜XIIの化合物を含む組成物を使用し、あるいは、別の実施形態では、式IA、I〜XIIの化合物の異性体、代謝物、薬学的生成物、水和物、多形体の組み合わせを使用する。
【0162】
一実施形態では、用語「異性体」として、光学異性体および類似体、構造異性体および類似体、配座異性体および類似体などが挙げられるが、これに限定されるものではない。
【0163】
一実施形態では、用語「異性体」は、化合物の光学異性体を含むことになっている。一実施形態では、用語「異性体」は、化合物の立体異性体を含むことになっている。本発明の化合物は、シスおよびトランス異性化する可能性があるアミド結合を有する。当然のことながら、本発明は、光学活性、または立体異性体、またはその混合物を含み、用途に対してこれらを使用する場合には、本発明の範囲内で配慮すべきである。
【0164】
別の実施形態では、本発明はさらに化合物の水和物を含む。一実施形態では、用語「水和物」は、当該技術分野で公知のように、半水和物、一水和物、二水和物、三水和物、またはその他のことを言う。
【0165】
合成プロセス
【0166】
式IAの化合物は、例えば、ベンズアミドを生じるように塩基の存在で、置換ジフェニルアミンを安息香酸またはハロゲン化ベンゾイルと反応させることで、簡単に調製できる。一実施形態では、塩基はピリジンである。別の実施形態では、ハロゲン化ベンゾイルは塩化ベンゾイルである。別の実施形態では、ジフェニルアミンと安息香酸またはハロゲン化ベンゾイルとの反応中、ヒドロキシル置換基が保護される。別の実施形態では、ヒドロキシルに対する保護基は、任意に最終段階で取り除かれる。米国公開番号2009/00624231も参照するものとし、参照することにより本書に援用される。
【0167】
例えば、式IAの化合物:
【0168】
【化14】
【0169】
式中、R1、R2、R3、R4、jおよびkの調製は、
【0170】
【化15】
【0171】
と一緒に
【0172】
【化16】
【0173】
を反応させて、
【0174】
【化17】
【0175】
を得る。ジフェニルアミン(3)を
【0176】
【化18】
【0177】
と、塩基の存在で反応させて、以下のものを得る。
【0178】
【化19】
【0179】
式中、R1、R2、R3およびR4が、互いに独立して、OH、O−Alk−R5R6、O−Alk−複素環である場合、R1´、R2´、R3´、R4´は保護ヒドロキシル基である。ここで、保護基は、遊離ヒドロキシを得るために取り除かれる。あるいは必要に応じて、式IAの化合物を生じさせるために、続いてCl−Alk−複素環またはCl−Alk−NR5R6と反応させる。
【0180】
【化14】
【0181】
式中、R1、R2、R3、およびR4が、互いに独立して、OH、O−Alk−NR5R6、またはO−Alk−複素環と異なる場合、R1´、R2´、R3´およびR4´はそれぞれR1、R2、R3およびR4である。
【0182】
別の例として、式IAの化合物を調製するためのプロセス:
【0183】
【化14】
【0184】
式中、R1、R2、R3、およびR4は、上述の通りであり、該プロセスは、
【0185】
【化17】
【0186】
と
【0187】
【化18】
【0188】
とを、塩基の存在で反応させ、以下のものを得る。
【0189】
【化19】
【0190】
式中、R1、R2、R3およびR4が、互いに独立して、OH、O−Alk−R5R6、O−Alk−複素環である場合、R1´、R2´、R3´、R4´は保護ヒドロキシル基である。ここで、保護基は、遊離ヒドロキシを得るために取り除かれる。あるいは必要に応じて、式IAの化合物を生じさせるために、続いてCl−Alk−複素環またはCl−Alk−NR5R6と反応させる。
【0191】
【化14】
【0192】
式中、R1、R2、R3、およびR4が、互いに独立して、OH、O−Alk−NR5R6、またはO−Alk−複素環と異なる場合、R1´、R2´、R3´およびR4´はそれぞれR1、R2、R3およびR4である。
【0193】
一例では、化合物IIは実施例1、および図5にしたがって調製される。
【0194】
別の例では、化合物IIIは実施例1、および図5にしたがって調製される。
【0195】
さらなる例では、式IVの化合物:
【0196】
【化4】
【0197】
の調製は以下のようにしてもよい。すなわち、
【0198】
【化20】
【0199】
と
【0200】
【化21】
【0201】
とを、塩基の存在下で、反応させて、
【0202】
【化22】
【0203】
を得て、続いて化合物IVを生じさせるために、保護基の脱保護を行う。
【0204】
【化4】
【0205】
式中、PおよびP´は同じか、または異なる保護基である。一例では、化合物IVは実施例2、および図6にしたがって調製される。
【0206】
別の例では、化合物Vは実施例1、および図5にしたがって調製される。
【0207】
さらなる例では、化合物VIは実施例3、および図7にしたがって調製される。
【0208】
別の例では、化合物VIIは実施例1、および図5にしたがって調製される。
【0209】
別の例では、化合物VIIIは実施例4、および図5にしたがって調製される。
【0210】
別の例では、化合物IXは実施例5、および図8にしたがって調製される。
【0211】
別の例では、化合物X塩酸塩は実施例5、および図8にしたがって調製される。
【0212】
別の例では、化合物XIは実施例1、および図5にしたがって調製される。
【0213】
別の例では、化合物XIIは実施例1、および図5にしたがって調製される。
【0214】
例えば、好適なヒドロキシル保護基としては、メチルエーテル(メトキシ)、ベンジルエーテル(ベンジルオキシ)、メトキシメチル(MOM)エーテル、ベンゾイルオキシメチル(BOM)エーテル、ベンジル、カルボベンゾキシ、メトキシエトキシメチル(MEM)エーテル、2−(トリメチルシリル)エトキシメチル(SEM)エーテル、メチルチオメチル(MTM)エーテル、フェニルチオメチル(PTM)エーテル、アジドメチルエーテル、シアノメチルエーテル、2,2−ジクロロ−1,1−ジフルオロエチルエーテル、2−クロロエチルエーテル、2−ブロモエチルエーテル、テトラヒドロピラニル(THP)エーテル、1−エトキシエチル(EE)エーテル、フェナシルエーテル、4−ブロモフェナシルエーテル、シクロプロピルメチルエーテル、アリルエーテル、プロパルギルエーテル、イソプロピルエーテル、シクロヘキシルエーテル、t−ブチルエーテル、2,6−ジメチルベンジルエーテル、4−メトキシベンジルエーテル、o−ニトロベンジルエーテル、2,6−ジクロロベンジルエーテル、3,4−ジクロロベンジルエーテル、4−(ジメチルアミノ)カルボニルベンジルエーテル、4−メチルスルフィニルベンジルエーテル、4−アントリルメチルエーテル、4−ピコリルエーテル、ヘプタフルオロ−p−トリル、テトラフルオロ−4−ピリジルエーテル、トリメチルシリル(TMS)エーテル、t−ブチルジメチルシリル(TBDMS)エーテル、t−ブチルジフェニルシリル(TBDPS)エーテル、トリイソプロピルシリル(TIPS)エーテル、ギ酸アリール、酢酸アリール、レブリン酸アリール、アリールピバレート、安息香酸アリール、アリール9−フルオレンカルボキシレート、アリールメチルカルボネート、1−アダマンチルカルボネート、t−ブチルカルボネート、4−メチルスルフィニルベンジルカルボネート、2,4−ジメチルペンタ−3−イルカルボネート、アリール2,2,2−トリクロロエチルカルボネート、アリールベンジルカルボネート、アリールカルバメート、ジメチルホスフィニルエステル(Dmp−OAr)、ジメチルホスフィノチオニルエステル(Mpt−OAr)、ジフェニルホスフィノチオニルエステル(Dpt−OAr)、メタンスルホン酸アリール、トルエンスルホン酸アリール、または2−ホルミルベンゼンスルホン酸アリールが挙げられる。
【0215】
本発明の方法は、化合物IA、I〜XIIの使用を含み、本発明の化合物の調製のためのプロセスは、塩基の存在でのジフェニルアミンと塩化ベンゾイルの反応を含む。例えば、好適な塩基としては、ピリジン、トリエチルアミン、K2CO3、Cs2CO3、Na2CO3、メチルアミン、イミダゾール、ベンズイミダゾール、ヒスチジン、トリブチルアミン、またはそれらの任意の組み合わせが挙げられる。一実施形態では、塩基はピリジンである。
【0216】
本発明の方法は、化合物IA、I〜XIIの使用を含み、本発明の化合物の調製のためのプロセスは、保護されたヒドロキシルの脱保護を含む。別の実施形態では、脱保護の条件は保護基に依存する。一部の実施形態では、脱保護段階は、Pd/Cの存在での水素化を含む。別の実施形態では、脱保護はBBr3との反応を含む。別の実施形態では、脱保護段階は酸との反応を含む。
【0217】
さらなる例では、化合物IA、I〜XIIは図5〜8、および実施例1〜5にしたがって調製される。
【0218】
医薬組成物
【0219】
一部の実施形態では、本発明は、必要な化合物を含む組成物を投与することを含む使用の方法を提供する。本願明細書で使用される場合、「医薬組成物」は「治療有効量」の有効成分、すなわち、薬学的に許容される担体または希釈剤とともに本発明の化合物を意味する。本願明細書で使用される場合、「治療有効量」とは、所定の条件および投薬計画に対して治療効果をもたらす量のことを言う。
【0220】
本願明細書で使用される場合、用語「投与」とは、対象を本発明の化合物に接触させることを意味する。本願明細書で使用される場合、投与はインビトロ、すなわち試験管内で、またはインビボ、すなわち生体、例えばヒトの細胞または組織で達成できる。一実施形態では、本発明には、男性対象に本発明の化合物を投与することを含む。
【0221】
他の実施形態では、本発明は、本願明細書で述べられる化合物の薬学的生成物を示す。他の実施形態では、用語「薬学的生成物」とは、例えば本書で述べたように、製薬学的用途(医薬組成物)に好適な組成物のことを言う。
【0222】
本発明の化合物は、単独で、または製剤の有効成分として投与できる。したがって、本発明は、例えば薬学的に許容される担体を1つ以上含む式Iの化合物の医薬組成物も含む。
【0223】
多数の標準的基準によって、本発明に従った化合物の投与に好適なさまざまな製剤を調製するための手順を説明することができる。例えば、有望な製剤および調製の例は、米国薬学会、製薬賦形剤ハンドブック(最新版);薬の剤形:錠剤(編集者、Lieberman、LachmanおよびSchwartz)最新版、Marcel Dekker,Inc.発行のほか、Remington's Pharmaceutical Science(編集者、Arthur Osol)、1553〜1593(最新版)に含まれている;
【0224】
投与方法および剤形は、所定の処置用途に望ましくおよび有効な化合物または組成物の治療量と密接な関係がある。
【0225】
好適な剤形としては、経口、直腸、舌下、粘膜、経鼻、眼、皮下、筋肉内、静脈内、経皮、脊椎、くも膜下腔内、関節内、動脈内、くも膜下、気管支、リンパ管、および子宮内の投与、並びに有効成分の全身送達のための他の剤形が挙げられるが、これに限定されるものではない。経口投与に好適な製剤が望ましい。
【0226】
上記薬の剤形を調製するために、従来の薬学的生成物配合法にしたがって、有効成分を医薬担体に混ぜることができる。担体は、投与のために望まれる調製の形態に応じて、幅広い種類の形態を取ることができる。
【0227】
経口剤形の組成物の調製において、通常の製剤媒体のいずれも採用できる。このため、例えば懸濁液、エリキシル剤、および溶液などの液体経口製剤の場合、好適な担体および添加剤としては、水、グリコール、オイル、アルコール、香料添加剤、保存料、着色剤などが挙げられる。例えば粉末、カプセル、および錠剤などの固形経口製剤の場合、好適な担体および添加剤としては、スターチ、砂糖、希釈剤、造粒剤、潤滑剤、バインダー、崩壊剤などが挙げられる。投与のしやすさにより、錠剤およびカプセルが最も有利な経口単位剤形である。必要に応じて、標準的な方法で錠剤に糖衣または腸溶コ―ティングを施すことができる。
【0228】
非経口製剤の場合、通常、担体は滅菌水を含むが、他の原料、例えば、溶解性の支援または保存のための原料を含む場合がある。注射剤も調製でき、その場合、適切な安定化剤が採用されることがある。
【0229】
一部の用途では、リポソームまたは他のカプセル材料媒体中での活性薬剤のカプセル化による、またはタンパク質、リポタンパク質、糖タンパク質、および多糖から選択されるものなどの好適な生体分子への活性薬剤の固定による、例えば共有結合、キレート化、または配位結合などによる「ベクター化(vectorized)」で活性薬剤を利用することに有利な場合がある。
【0230】
経口投与に好適な製剤を用いた本発明の処置方法は、それぞれ、例えば粉末または顆粒として所定量の有効成分を含むカプセル、カシェー、錠剤、またはトローチ剤などの個別の単位として提示されることがある。任意に、シロップ、エリキシル剤、エマルション、または水薬一服(draught)など、水性液体または非水性液体の懸濁液を採用できる。
【0231】
圧縮または成形、または必要に応じて1つ以上の副原料との湿式造粒法によって、錠剤を製造できる。圧縮錠剤は、例えば、必要に応じてバインダー、崩壊剤、潤滑剤、不活性希釈剤、または排出剤に混ぜられる粉末または顆粒などの自由流動形態の活性化合物とともに好適な機械で圧縮することで製造できる。粉末活性化合物と好適な担体の混合物を含む成形錠剤は、好適な機械による成型によって製造できる。
【0232】
シロップは、砂糖、例えば蔗糖の濃縮水溶液に活性化合物を添加することで製造でき、これには、副原料を添加することもできる。前記副原料には、砂糖の結晶化を遅らせるために香料添加剤、好適な保存料と、そして、多価アルコール、例えばグリセロールまたはソルビトールなどの他の原料の溶解性を高める薬剤とを含む場合がある。
【0233】
非経口投与に好適な製剤は、好適には受容者の血液と等張である活性化合物の無菌水性調合液(例えば、生理食塩水)を含んでもよい。前記製剤は、懸濁化剤および増粘剤およびリポソーム、または化合物が血液成分または1つ以上の臓器を標的にするように設計された他の微粒子系を含んでもよい。製剤は、単回投与または複数回投与剤形で調製してもよい。
【0234】
非経口投与は、全身送達の好適な剤形を含んでもよい。例えば、投与は、静脈内、動脈内、くも膜下腔内、筋肉内、皮下、筋肉内、腹内(例えば、腹腔内)などであってもよく、そして注入ポンプ(外部または埋め込み型)または望ましい投与法に適した他の好適な手段による影響を受ける場合がある。
【0235】
鼻および他の粘膜スプレー製剤(例えば、吸引可能剤形)は、保存料および等張剤とともに活性化合物の精製水溶液を含むことができる。前記製剤は、好適には、鼻または他の粘膜に適合するpHおよび等張状態に調整される。あるいは、ガス担体に懸濁された微粉固体粉末の剤形であってもよい。前記製剤は、好適な手段または方法、例えば、噴霧器、アトマイザー、定量吸入具などで送達してもよい。
【0236】
直腸投与のための製剤は、ココアバター、硬化油脂、または硬化脂肪カルボン酸などの好適な担体とともに座薬として提供してもよい。
【0237】
経皮製剤は、セルロース媒体、例えば、メチルセルロースまたはヒドロキシエチルセルロースなどのチキソトロピーまたはゼラチン状担体に活性薬剤を組み込むことで調製してもよく、得られる製剤は、着用者の皮膚との皮膚接触を確保するように適合された経皮投与装置に詰められることになる。
【0238】
前述の原料に加えて、本発明の製剤は、例えば、希釈剤、緩衝剤、香料添加剤、バインダー、崩壊剤、界面活性剤、増粘剤、潤滑剤、保存料(抗酸化剤を含む)などから選択される1つ以上の副原料をさらに含んでもよい。
【0239】
本発明の製剤は、即時放出、持続放出、遅発性放出、または当業者には公知の他の放出プロファイルを有することができる。
【0240】
一実施形態では、本発明は、式IA、I〜XIIの化合物を含む経口組成物を投与することを含む、前立腺癌を患う男性対象において、黄体形成ホルモン(LH)の削減によって、またはLHホルモンの削減と関係なく、a)全血清テストステロン濃度を下げる;b)遊離血清テストステロン濃度を下げる方法を提供する。追加の実施形態では、本発明の方法は、式II、式III、式IV、式V、式VI、式VII、式VIII、式IX、式X、式XI、または式XIIの化合物を含む経口組成物を使用する。
【0241】
一実施形態では、本発明は、式IA、I〜XIIの化合物を含む経口組成物を投与することを含む、前立腺癌を患う男性対象において、LH濃度の削減によって、またはLH濃度の削減と関係なく、前立腺癌を処置する方法を提供する。追加の実施形態では、本発明は、式II、式III、式IV、式V、式VI、式VII、式VIII、式IX、式X、式XI、または式XIIの化合物を含む経口組成物を投与することを含む、前立腺癌治を患う男性対象において、LH濃度を下げることで、またはLH濃度を下げることとは関係なく、前立腺癌を処置する方法を提供する。
【0242】
当然のことながら、本発明は、本願明細書で述べられるような化合物のいかなる実施形態も含み、一部の実施形態では、「本発明の化合物」と呼ばれる。
【0243】
一実施形態では、本発明の方法は、さまざまな用量での本発明の化合物の投与を含んでもよい。一実施形態では、本発明の化合物は、1〜1500/日の用量で投与される。追加の実施形態では、本発明の化合物は、1〜10mg/日、3〜26mg/日、3〜60mg/日、3〜16mg/日、3〜30mg/日、10〜26mg/日、15〜60mg/日、50〜100mg/日、50〜200mg/日、150〜300mg/日、20〜50mg/日、5〜50mg/日、200〜500mg/日、150〜500mg/日、200〜1000mg/日、300〜1500mg/日、または100〜1000mg/日の用量で投与される。
【0244】
一実施形態では、本発明の方法は、さまざまな用量での本発明の化合物の投与を含んでもよい。一実施形態では、本発明の化合物は、3mgの用量で投与される。追加の実施形態では、本発明の化合物は、10mg、30mg、50mg、100mg、200mg、300mg、450mg、500mg、600mg、900mg、1000mg、または1500mgの用量で投与される。
【0245】
一実施形態では、本発明の方法は、さまざまな用量での本発明の化合物の投与を含んでもよい。一実施形態では、本発明の化合物は、0.1mg/Kg/日の用量で投与される。追加の実施形態では、本発明の化合物は、0.2〜30mg/kg/日、または0.2mg/kg/日、0.3mg/kg/日、1mg/kg/日、3mg/kg/日、5mg/kg/日、10mg/kg/日、20mg/kg/日、または30mg/kg/日の用量で投与される。
【0246】
一実施形態では、本発明の方法は、式IA、I〜XIIの化合物を含む医薬組成物を使用するために提供される。追加の実施形態では、本発明の方法は、式II、式III、式IV、式V、式VI、式VII、式VIII、式IX、式X、式XI、または式XIIの化合物を含む医薬組成物を使用するために提供される。
【0247】
いくつかの実施形態では、医薬組成物は固体剤形である。別の実施形態では、医薬組成物は錠剤である。別の実施形態では、医薬組成物はカプセルである。別の実施形態では、医薬組成物は溶液である。別の実施形態では、医薬組成物は経皮貼布である。
【0248】
一実施形態では、本発明の化合物、またはこれを含む組成物を使用することには、当業者には公知の通り、対象に望まれる反応の阻害、抑制、増進、または刺激において有用性がある。別の実施形態では、組成物は追加有効成分をさらに含み、この成分の活性は、本発明の化合物が投与される特定の用途に有益である。
【0249】
動物、特にヒトへの投与の場合、個人に最も好適で、特定の個人の年齢、体重、遺伝、および/または反応によって変動する可能性がある、実際の用量および処置期間を医師が決定することが予想される。
【0250】
一部の実施形態では、本発明の組成物はいずれも、本願明細書で述べられるような剤形または実施形態で本発明の化合物を含むことになる。一部の実施形態では、本発明の組成物はいずれも、本願明細書で述べられるような剤形または実施形態で本発明の化合物からなる。一部の実施形態では、本発明の組成物はいずれも、本願明細書で述べられるような剤形または実施形態で、本発明の化合物から本質的になる。一部の実施形態では、用語「含む(comprise)」とは、本発明の化合物などの示された活性薬剤の含有のほか、製薬業界では公知のように、他の活性薬剤、および薬学的に許容される担体、賦形剤、皮膚軟化剤、安定化剤などの含有のことも言う。一部の実施形態では、用語「本質的になる(consisting essentially of)」とは、唯一の有効成分が示された有効成分であるが、安定化、保存などのための他の化合物を含むことができるが、示された有効成分の治療効果に製剤が直接関与することはない組成物のことを言う。一部の実施形態では、用語「本質的になる(consisting essentially of)」とは、有効成分の放出を促進する成分のことを言う場合がある。一部の実施形態では、用語「からなる(consisting of)」とは、有効成分および薬学的に許容される担体または賦形剤を含む組成物のことを言う。
【0251】
当然のことながら、本願明細書で述べられるような化合物の使用は、本願明細書で述べられるような疾患、障害、または症状で使用でき、本発明の実施形態を表す。一実施形態では、化合物は、遊離塩基、遊離酸、または非複合化合物である。
【0252】
以下の実施例は、本発明の望ましい実施形態をさらに十分に説明するために提供される。しかしながら、本発明の幅広い範囲を制限するものと解釈されることは全くない。
【実施例】
【0253】
実施例1:式II〜XIIの化合物の一般的な合成手順および中間体による合成
【0254】
有機溶媒、界面活性剤、および抗酸化剤など、これらは本願明細書で述べられる組成物で使用でき、典型的には商業用供給源から容易に入手できる。例えば、PEG−300、polysorbate 80、Captex(商標)200、Capmul(商標)MCM C8は、例えばDow Chemical Company(Midland, MI)、ICI Americas, Inc(Wilmington, DE)、またはAbitec Corporation(Janesville, WI)から購入できる。
【0255】
本願明細書で述べられるエストロゲン受容体リガンドは、当業者には公知のさまざまな方法で調製できる。例えば、本願明細書で述べられるエストロゲン受容体リガンドは、米国特許出願公開番号2009/0062341で述べられた合成方法で調製でき、その全体の各開示を参照することにより本書に組み込まれる。
【0256】
N,N−ビスアリールベンズアミド誘導体の一般的な合成
【0257】
ジアリールアニリンの一般的な合成(図5)アリールアミン(1.5当量)、ヨウ化アリール(1当量)、K2CO3(2当量)、CuI(0.1当量)、およびL−プロリン(0.2当量)の混合物を一緒に混ぜ、室温で無水DMSOに溶かした。次に、反応混合物を攪拌し、28時間90℃に加熱した。混合物を室温に冷却し、水で加水分解した。EtOAcを添加し、溶液を分離させた。EtOAc層を分離し、塩水で洗浄し、無水MgSO4によって乾燥させた。減圧下で溶媒を取り除いた。該当するジアリールアニリンを利用可能にするために、溶離液として5%EtOAc/ヘキサンを用いて、フラッシュカラムクロマトグラフィー(シリカゲル)で固体残留物を精製した。
【0258】
ビス−(4−メトキシフェニル)アミン(1a):淡黄色固体、収率73%、融点98.6〜99.0℃。1H NMR(CDCl3、300MHz)δ6.93〜6.81(m、8H)、5.37(s、br、1H)、3.78(s、6H)。MS m/z 228.4(M−H)+。
【0259】
N−(4−メトキシフェニル)−フェニルアミン(1b):淡黄色固体、収率70%、融点106.3〜106.5℃。1H NMR(CDCl3、300MHz)δ 7.24〜7.18(m、3H)、7.08〜7.06(m、2H)、6.92〜6.84(m、4H)、5.61(s、br、1H)、3.79(s、3H)。MS m/z 200.1(M+H)+。
【0260】
N−(4−フルオロフェニル)−N−4−メトキシフェニルアミン(1c):淡黄色固体、収率54%、融点60.6〜61.0℃。1H NMR(CDCl3、300MHz)δ7.01〜6.83(m、8H)、3.78(s、3H)。MS m/z 217(M)+。
【0261】
N−(4−ベンジルオキシフェニル)−N−4−メトキシフェニルアミン(1d):淡黄色固体、収率54%、融点108.0〜108.4℃。1H NMR(CDCl3、300MHz)δ 7.34〜7.08(m、5H)、6.90〜6.81(s、3H)、3.78(s、3H)。MS m/z 306(M+H)+。
【0262】
ベンズアミドの一般的な合成。アリールアミン(1当量)、塩化ベンゾイル(1.3当量)、およびピリジン(6当量)の混合物を一緒に混ぜ、室温で無水THFに溶かした。24時間、混合物を攪拌し、還流させた。反応液を室温に冷却し、2N HCl溶液を添加して加水分解した。溶液は、酢酸エチルで抽出した。有機層をNaHCO3の飽和水溶液で洗浄し、余分な酸を取り除き、無水MgSO4で乾燥させ、濾過し、減圧下で濃縮した。該当するベンズアミドを利用可能にするために、EtOAc/ヘキサン(3/7v/v)を用いて、フラッシュカラムクロマトグラフィーで残留物を精製した。
【0263】
3−フルオロ−N−(4−フルオロフェニル)−4−メトキシ−N−(4−メトキシフェニル)ベンズアミド(2a):黄色固体、融点54〜56℃。1H NMR(CDCl3、/TMS)δ 7.24〜7.11(m、4H)、7.05〜6.97(m、4H)、6.85〜6.78(m、3H)、3.86(s、3H)、3.79(s、3H)。MS (ESI) m/z 370.1 [M +H]+。
【0264】
4−フルオロ−N,N−ビス(4−メトキシフェニル)−2−(トリフルオロメチル)ベンズアミド(2b):無色油、収率84.2%。1H NMR(CDCl3、300MHz)δ 7.34〜7.26(m、4H)、7.09〜7.01(m、3H)、6.91(d、2H、J=8.7Hz)、6.87(d、2H、J=8.7Hz)、3.80(s、3H)、3.71(s、3H)。MS m/z 442.1(M+Na)+。
【0265】
4−メトキシ−N−(4−メトキシフェニル)−N−(4−フルオロフェニル)−ベンズアミド(2c):白色固体、収率97%、融点133.5〜134.5℃。 1H NMR(CDCl3、300MHz)δ 8.11〜6.66(m、15H)、3.74(s、3H)、3.73(s、3H)。MS m/z 384 (M+H)+。
【0266】
N−(4−メトキシフェニル)−N−(4−ベンジルオキシフェニル)−2−ナフチルアミド(1d):白色固体、収率58%、融点174.9〜175.5℃。1H NMR(CDCl3、300MHz)δ 8.04(s、1H)、7.77〜7.74(m、2H)、7.64〜7.61(m、1H)、7.51〜7.43(m、4H)、7.40〜7.31(m、4H)、7.13〜7.10(m、4H)、6.88〜6.78(m、4H)、4.99(s、2H)、3.74(s、3H)。MS m/z 460 (M+H)+。
【0267】
4−フルオロ−N,N−ビス(4−メトキシフェニル)−2−(トリフルオロメチル)ベンズアミド(2e):無色油、収率84.2%。1H NMR(CDCl3、300MHz)δ 7.34〜7.26(m、4H)、7.09〜7.01(m、3H)、6.91(d、2H、J=8.7Hz)、6.87(d、2H、J=8.7Hz)、3.80(s、3H)、3.71(s、3H)。MS m/z 442.1(M+Na)+。
【0268】
BBr3を用いたベンズアミド誘導体の脱メチル反応の一般的手順。メトキシベンズアミド化合物を、乾燥したCH2Cl2に溶かした。BBr3(1.0M CH2Cl2溶液)を0℃で滴加した。反応溶液を室温までゆっくりと温め、室温で一晩、攪拌したまま置いた。混合物を氷浴にて0℃に冷却し、水を添加して加水分解した。EtOAcを添加し、溶液を分離させた。有機層が分離した。水層をEtOAcで抽出した。有機層を塩水で洗浄し、無水MgSO4によって乾燥させた。減圧下で溶媒を取り除いた。該当するフェノール化合物を利用可能にするために、CH3OH/CH2Cl2(1/9 v/v)を用いて、フラッシュカラムクロマトグラフィーで残留物を精製した。
【0269】
4−フルオロ−N,N−ビス(4−ヒドロキシフェニル)−2−(トリフルオロメチル)ベンズアミド(3a):白色固体、収率92.5%。1H NMR(DMSO−d6、300MHz)δ 9.55(s、1H)、9.53(s、1H)、7.69〜7.58(m、2H)、7.46〜7.39(m、1H)、7.18(d、2H、J=8.7Hz)、6.93(d、4H、J=8.7Hz)、7.03(d、2H、J=8.4Hz)、6.78(d、2H、J=8.7Hz)、6.57(d、2H、J=8.7Hz)。MS m/z 392.1(M+H)+。
【0270】
以下の化合物は上述の通り合成され、表1にて特徴付けられ、要約されている。すなわち、N,N−ビス(4−ヒドロキシフェニル)−4−プロピルベンズアミド(II);3−フルオロ−N−(4−フルオロフェニル)−4−ヒドロキシ−N−(4−ヒドロキシフェニル)ベンズアミド(IV);N,N−ビス(4−ヒドロキシフェニル)−2,3−ジメチルベンズアミド(V);3−フルオロ−4−ヒドロキシ−N,N−ビス(4−ヒドロキシフェニル)−ベンズアミド(VII);3−フルオロ−4−ヒドロキシ−N−(4−ヒドロキシフェニル)−N−フェニルベンズアミド(XI);および3−フルオロ−N,N−ビス(4−ヒドロキシフェニル)−2−メチルベンズアミド(XII)。
【0271】
ベンジルオキシフェニル−ベンズアミドの脱ベンジル化の一般的手順。化合物を、250mL水素化ボトルに入ったEtOHに溶かした。溶液にPd/C粉末(5%モル)を添加した。反応容器を、水素ガス圧力20psiの下で水素化装置に取り付けた。出発物質が消えるまで、TLCで反応をモニタリングした。その後、減圧下で溶媒を取り除いた。所望の生成物を利用可能にするために、ヘキサン/EtOAc=3/2 v/vを用いて、フラッシュカラムクロマトグラフィーで残留物を精製した。
【0272】
以下の化合物は上述の通り合成され、表1にて特徴付けられ、要約されている。すなわち、N,N−ビス(4−ヒドロキシフェニル)−2−ナフチルアミド(VI)。
【0273】
脱保護ベンズアミドの還元の一般的手順。ベンズアミド化合物を、室温で20mLの無水THFに溶かした。アルゴン下で、室温にてシリンジを用いてH3B(SMe2)を添加した。6時間、反応溶液を攪拌し、加熱して還流させた。その後、0℃でMeOHを10mL添加して反応を抑えた。減圧下で溶媒を取り除いた。所望の生成物を利用可能にするために、フラッシュカラムクロマトグラフィー(シリカゲル、CH2Cl2/MeOH=9/1 v/v)に残留物を通した。
【0274】
以下の化合物は上述の通り合成され、表1にて特徴付けられ、要約されている。すなわち、4,4´−(2,3−ジメチルベンジルアザネジイル)ジフェノール(III);4−((4−フルオロフェニル)(4−ヒドロキシベンジル)アミノ)フェノール(VIII)。
【0275】
O−(2−ピペリジン−1−イルエトキシ)−ベンズアミドおよび類似体の一般的な合成。ベンズアミド類似体(1当量)を含むヒドロキシフェニルのアセトン溶液に、K2CO3(3当量)および塩酸N−クロロエチル−ピペリジン(1.2当量)を添加した。6時間、反応溶液を加熱して還流させた。溶液を蒸発乾固させた。水を添加して残留物を加水分解し、その後、酢酸エチルで抽出した。有機層を分離させ、無水MgSO4によって乾燥させた。減圧下で溶媒を取り除いた。所望の生成物を利用可能にするために、塩化メチレン/メタノール=9/1 v/vを用いて、フラッシュカラムクロマトグラフィーで残留物を精製した。化合物のメタノール溶液にHClのEt2O溶液を添加して、塩酸塩を調製した。
【0276】
以下の化合物は上述の通り合成され、表1にて特徴付けられ、要約されている。すなわち、4−フルオロ−N−(4−ヒドロキシフェニル)−N−(4−(2−(ピペリジン−1−イル)エトキシ)フェニル)−2−(トリフルオロメチル)ベンズアミド(IX);およびIXのHCl塩である4−フルオロ−N−(4−ヒドロキシフェニル)−N−(4−(2−(ピペリジン−1−イル)エトキシ)フェニル)−2−(トリフルオロメチル)ベンズアミド塩酸塩(X)。
【0277】
【表1A】
【0278】
【表1B】
【0279】
【表1C】
【0280】
実施例2:式IVの化合物の合成(図6)
【0281】
【化23】
【0282】
ステップ1:4−フルオロ−N−(4−メトキシフェニル)アニリン(1c)
【0283】
4−フルオロアニリン(78.63g、0.708mol)、4−ヨードアニソール(138.00g、0.590mol)、無水K2CO3(122.23g、0.884mol)、CuI(11.23g、58.96mol)、およびL−プロリン(13.58g、0.118mol)の混合物を、攪拌子、還流冷却器、およびアルゴン入口の付いた1Lの3つ口丸底フラスコで一緒に混ぜた。室温で無水DMSO(300mL)を添加した。反応混合物を攪拌し、アルゴン下で20時間90℃に加熱した。次に、混合物を室温に冷却し、水(300mL)で加水分解した。EtOAc(200mL)を添加し、溶液を分離した。EtOAc層を分けた。EtOAc100mLで水層を抽出した。EtOAc層に塩水(2x100mL)を添加し、洗浄し、無水MgSO4(50g)によって乾燥させた。減圧下で溶媒を取り除いた。収率77.8%、融点46〜48℃の黄色固体99.70gとして4−フルオロ−N−(4−メトキシフェニル)アニリン(1c)を利用可能にするために、茶色の油状残留物をフラッシュカラムクロマトグラフィー(シリカゲル、ヘキサン/EtOAc=9/1 v/v)で精製した。MS(ESI) m/z 218.1 [M + H] +、1H NMR(DMSO−d6、300MHz) δ 7.77(bs、1H)、7.03〜6.98(m、4H)、6.93〜6.82(m、4H)、3.70(s、3H)。
【0284】
【化24】
【0285】
ステップ2:3−フルオロ−N−(4−フルオロフェニル)−4−メトキシ−N−(4−メトキシフェニル)ベンズアミドの合成(2a)
【0286】
4−フルオロ−N−(4−メトキシフェニル)アニリン(1c)(90.78g、0.418mol)および塩酸3−フルオロ−4−メトキシベンゾイル(94.55g、0.501mol)を、攪拌子、還流冷却器、およびアルゴン入口の付いた1Lの3つ口丸底フラスコで無水THF(200mL)に一緒に混ぜ、溶かした。アルゴン下で、室温にてシリンジを用いて無水ピリジン(132.22g、1.672mol)を添加した。一晩、反応混合物を攪拌し、加熱して還流させた。次に、反応混合物を室温に冷却し、濾過してピリジン塩を取り除いた。溶液を濃縮し、THF溶剤を取り除いた。残留油を2N HCl溶液200mLで洗浄し、酢酸エチル(2x200mL)で抽出した。組み合わせた有機層をNa2HCO3の飽和水溶液(150mL)で洗浄し、余分な塩化ベンゾイルと酸とを取り除き、無水MgSO4(50g)で乾燥させ、濾過し、減圧下で濃縮し、油状物を得た。融点54〜56℃の黄色固体として該当する純粋なベンズアミド化合物を利用可能にするために、CH2Cl2/アセトン(50/1 v/v)で、シリカゲルを用いたフラッシュカラムクロマトグラフィーによって残留物を精製した。MS(ESI) m/z 370.1 [M +H]+、1H NMR(CDCl3/TMS) δ 7.24〜7.11(m、4H)、7.05〜6.97(m、4H)、6.85〜6.78(m、3H)、3.86(s、3H)、3.79(s、3H)。
【0287】
【化25】
【0288】
ステップ3:3−フルオロ−N−(4−フルオロフェニル)−4−ヒドロキシ−N−(4−ヒドロキシフェニル)ベンズアミド(IV)の合成
【0289】
化合物3−フルオロ−N−(4−フルオロフェニル)−4−メトキシ−N−(4−メトキシフェニル)ベンズアミド(2a)(138.0g、0.374mol)を、アルゴン下、室温でCH2Cl2(600mL)に溶かした。BBr3(374.75g、1.496mol)を、アルゴン下、0℃で、氷浴にて攪拌しながらシリンジを用いて滴加した。一晩、室温にて反応溶液を攪拌し続けた。次に、溶液を攪拌しながら氷水1Lに注いだ。2時間、室温にてスラリー混合物を攪拌した。白色沈殿物を濾過し、水(2x100mL)で洗浄し、真空下で乾燥させた。CH2Cl2層を分離し、無水MgSO4(50g)によって乾燥させ、濾過し、真空下で濃縮し、乾燥させた。収率81.6%、融点110〜112℃の白色結晶性固体を利用可能にするために、CH2Cl2溶液から得た白色沈殿物および残留物を組み合わせ、フラッシュカラムクロマトグラフィー(シリカゲル、CH2Cl2/アセトン/MeOH=90/7/3 v/v/v)で精製し、加熱したEtOAc/ヘキサン溶液から2回再結晶させた淡褐色固体を得た。MS(ESI) m/z 364.1 [M + Na]+、1H NMR(DMSO−d6) ? 10.14(bs、1H)、9.71(bs、1H)、7.25〜7.11(m, 5H)、7.05〜6.99(m、3H)、6.78(t、J=8.6 Hz、1H)、6.68(d、J=8.7 Hz、2H)。
【0290】
実施例3:式VIの化合物の合成(図7)
【0291】
4−(ベンジルオキシ)−N−(4−メトキシフェニル)アニリンの合成(1d)
【0292】
4−ベンジルオキシアニリン(16.6g、83.31mmol)、4−ヨードアニソール(15.0g、64.09mmol)、K2CO3(17.72g、128.18mmol)、CuI(1.22g、6.41mmol)、およびL−プロリン(1.48g、12.82mmol)の混合物を一緒に混ぜ、室温で無水DMSO(120mL)に溶かした。次に、反応混合物を攪拌し、48時間90℃に加熱した。混合物を室温に冷却し、水で加水分解した。EtOAcを添加し、溶液を分離させた。EtOAc層を分離し、塩水で洗浄し、無水MgSO4によって乾燥させた。減圧下で溶媒を取り除いた。収率9.8g、融点108.0〜108.4℃の黄色固体9.8gとして該当するジアリールアニリンを利用可能にするために、EtOAc/ヘキサン(1/9 v/v)で、フラッシュカラムクロマトグラフィー(シリカゲル)によって固体残留物を精製した。1H NMR(CDCl3、300 MHz) δ 7.34〜7.25(m、5H)、6.90〜6.81(m、8H)、5.02(s、2H)、3.78(s、3H)。MS m/z 306(M+H)+。
【0293】
N−(4−ベンジルオキシフェニル)−N−(4−メトキシフェニル)−2−ナフトアミドの合成(2d)
【0294】
1当量の4−(ベンジルオキシ)−N−(4−メトキシフェニル)アニリン(0.80g、2.62mmol)を、攪拌子および還流冷却器の付いた乾燥3口丸底フラスコで1.5当量の塩化2−ナフトイル(0.75g、3.93mmol)および4当量のピリジン(0.83g、10.48mmol)に混ぜた。混合物を無水THF(30mL)に溶かし、加熱して、20時間還流させた。反応溶液を室温に冷却し、濾過した。減圧下で溶媒を取り除いた。収率58%、融点174.9〜175.5℃の白色固体として該当する純粋なナフトアミド化合物を利用可能にするために、EtOAc/ヘキサン(3/7 v/v)で、シリカゲルを用いたフラッシュカラムクロマトグラフィーによって残留物を精製した。1H NMR(CDCl3、300MHz) δ 8.04(s、1H)、7.77〜7.74(m、2H)、7.64〜7.61(m、1H)、7.51〜7.43(m、4H)、7.40〜7.31(m、4H)、7.13〜7.10(m、4H)、6.88〜6.78(m、4H)、4.99(s、2H)、3.74(s、3H)。MS m/z 460 (M+H)+。
【0295】
N,N−ビス(4−ヒドロキシフェニル)−2−ナフチルアミド(VI)の合成
【0296】
化合物N−(4−ベンジルオキシフェニル)−N−(4−メトキシフェニル)−2−ナフトアミド(2d)(0.5g、1.09mmol)を、室温で乾燥したCH2Cl2(30mL)に溶かした。BBr3(1.0M CH2Cl2溶液3.26mL、3.26mmol)を、室温で攪拌しながら、シリンジを用いて滴加した。一晩、室温にて反応溶液を攪拌し続けた。混合物を氷浴にて0℃に冷却し、水を添加して加水分解した。EtOAcを添加し、溶液を分離させた。有機層が分離させた。水層をEtOAcで2回抽出した。有機層を組み合わせ、塩水で洗浄し、無水MgSO4によって乾燥させた。真空下で溶媒を取り除いた。収率70%、融点264.3〜265.2℃(分解温度)の白色固体として所望の純粋なフェノール化合物を利用可能にするために、CH3OH/CH2Cl2(1/9 v/v)で、シリカゲルを用いたフラッシュカラムクロマトグラフィーによって残留物を精製した。1H NMR(DMSO−d6、500MHz)δ 9.46(s、2H)、7.98 (s、1H)、7.85〜7.75(m、2H)、7.75〜7.73(m、2H)、7.54〜7.48(m、2H)、7.45〜7.43(m、1H)、7.05(s、4H)、6.66(s、4H)。MS m/z 356 (M+H)+。
【0297】
実施例4:式VIIIの化合物の合成
【0298】
【化26】
【0299】
4−((4−フルオロフェニル)(4−ヒドロキシベンジル)アミノ)フェノール(VIII)の合成
【0300】
化合物N−(4−フルオロフェニル)−4−ヒドロキシ−N−(ヒドロキシフェニル)ベンズアミド(0.30g、0.93mmol)を、室温で無水THF20mLに溶かした。アルゴン下で、室温にてシリンジを用いてH3B(SMe2)(2M THF溶液1.86mL、3.71mmol)を添加した。6時間、反応溶液を攪拌し、加熱して還流させた。その後、0℃でMeOHを10mL添加して反応を抑えた。減圧下で溶媒を取り除いた。フラッシュカラムクロマトグラフィー(シリカゲル、CH2Cl2/MeOH=9/1 v/v)に残留物を通し、黄色油状物(0.26g、収率92%)を得た。1H NMR(DMSO−d6、500MHz) δ 9.29(s、1H)、9.24(s、1H)、7.09(d、2H、J=8.3 Hz)、6.98(d、2H、J=9.0 Hz)、6.94〜6.91(m、2H)、6.73(d、2H、J=9.0 Hz)、6.68〜6.64(m、4H)、4.70(s、2H)。MS m/z 307.8 (M−H)−。
【0301】
実施例5:式IXおよびXの化合物の合成(図8)
【0302】
ジアリールアニリンの合成。アリールアミン(1.5当量)、ヨウ化アリール(1当量)、K2CO3(2当量)、CuI(0.1当量)、およびL−プロリン(0.2当量)の混合物を一緒に混ぜ、室温で無水DMSOに溶かした。次に、反応混合物を攪拌し、28時間90℃に加熱した。混合物を室温に冷却し、水で加水分解した。EtOAcを添加し、溶液を分離させた。EtOAc層を分離し、塩水で洗浄し、無水MgSO4によって乾燥させた。減圧下で溶媒を取り除いた。該当するジアリールアニリンを利用可能にするために、溶媒としてEtOAc/ヘキサン(3/7 v/v)を用いて、フラッシュカラムクロマトグラフィー(シリカゲル)で固体残留物を精製した。ビス−(4−メトキシフェニル)アミン(1a):淡黄色固体、収率73%。1H NMR(CDCl3、300MHz) δ 6.93〜6.81(m、8H)、5.37(s、br、1H)、3.78(s、6H)。MS m/z 228.4(M−H)+。
【0303】
4−フルオロ−N,N−ビス(4−メトキシフェニル)−2−(トリフルオロメチル)ベンズアミドの合成(2e)
【0304】
1当量のビス−(4−メトキシフェニル)アミン(1a)(0.73g、3.18mmol)を、攪拌子および還流冷却器を備えた乾燥3口丸底フラスコで1.2当量の塩化4−フルオロ−2−トリフルオロメチルベンゾイル(0.87g、3.82mmol)および6当量のピリジン(1.51g、19.08mmol)に混ぜた。混合物を無水THF(20mL)に溶かし、20時間、90℃に加熱した。反応溶液を室温に冷却し、濾過した。減圧下で溶媒を取り除いた。収率84.2%の無色油1.12gとして該当する純粋なベンズアミド化合物を利用可能にするために、EtOAc/ヘキサン(3/7 v/v)で、シリカゲルを用いたフラッシュカラムクロマトグラフィーによって残留物を精製した。1H NMR(CDCl3、300MHz)δ 7.34〜7.26(m、4H)、7.09〜7.01(m、3H)、6.91(d、2H、J=8.7Hz)、6.87(d、2H、J=8.7Hz)、3.80(s、3H)、3.71(s、3H)。MS m/z 442.1(M+Na)+。
【0305】
4−フルオロ−N,N−ビス(4−ヒドロキシフェニル)−2−(トリフルオロメチル)ベンズアミドの合成(3a)
【0306】
化合物4−フルオロ−N,N−ビス(4−メトキシフェニル)−2−(トリフルオロメチル)ベンズアミド(2e)(1.00g、2.38mmol)を、室温で乾燥したCH2Cl2(30mL)に溶かした。BBr3(1.0M CH2Cl2溶液10mL、10.0mmol)を、室温で攪拌しながら、シリンジを用いて滴加した。一晩、室温にて反応溶液を攪拌し続けた。混合物を氷浴にて0℃に冷却し、水を添加して加水分解した。EtOAcを添加し、溶液を分離させた。有機層が分離させた。水層をEtOAcで2回抽出した。有機層を組み合わせ、塩水で洗浄し、無水MgSO4によって乾燥させた。真空下で溶媒を取り除いた。収率92.5%の白色固体0.86gとして所望の純粋なフェノール化合物を利用可能にするために、CH3OH/CH2Cl2(1/9 v/v)で、シリカゲルを用いたフラッシュカラムクロマトグラフィーによって残留物を精製した。1H NMR(DMSO−d6、300MHz)δ 9.55(s、1H)、9.53(s、1H)、7.69〜7.58(m、2H)、7.46〜7.39(m、1H)、7.18(d、2H、J=8.7Hz)、6.93(d、4H、J=8.7Hz)、7.03(d、2H、J=8.4Hz)、6.78(d、2H、J=8.7Hz)、6.57(d、2H、J=8.7Hz)。MS m/z 392.1(M+H)+。
【0307】
4−フルオロ−N−(4−ヒドロキシフェニル)−N−[4−(2−ピペリジン−1−イル)−エトキシ]フェニル]−2−(トリフルオロメチル)ベンズアミド(IX)の合成
【0308】
4−フルオロ−N,N−ビス(4−ヒドロキシフェニル)−2−(トリフルオロメチル)ベンズアミド(3a)のアセトン溶液に、K2CO3(1.29g、9.36mmol)および塩酸N−クロロエチル−ピペリジン(0.34g、1.87mmol)を添加した。20時間、反応溶液を加熱して還流させた。溶液を蒸発乾固させた。収率57.7%の白色固体0.45gとして所望の化合物を利用可能にするために、フラッシュカラムクロマトグラフィー(シリカゲル;塩化メチレン/メタノール=9/1 v/v)で残留物を精製した。1H NMR(DMSO−d6、300MHz)δ 9.57(s、1H)、7.71〜7.68(m、2H)、7.47〜7.44(m、1H)、7.28(d、1H、J=9.0 Hz)、7.18(d、1H、J=8.7 Hz)、7.13(d、1H、J=8.7 Hz)、7.05(d、1H、J=8.4 Hz)、6.97(d、1H、J=9.0 Hz)、6.80〜6.76(m、2H)、6.57(d、1H、J=87. Hz)、4.06(t、1H、J=6.0 Hz)、3.93(t、1H、J=6.0 Hz)、2.66(t、1H、J=5.7 Hz)、2.55(t、1H、J=5.4 Hz)、2.44(s、2H)、2.36(s、2H)、1.49〜1.37(m、6H)。MS m/z 501.0 (M−H)−。
【0309】
化合物のメタノール溶液にHClのEt2O溶液を添加し、続いて溶媒を蒸発させて、塩酸塩(X)を調製した。
【0310】
実施例6:エストロゲン受容体結合親和性、アゴニスト、およびアンタゴニスト活性
【0311】
化合物のER結合親和性は、[2,4,6,7−3H(N)]−エストラジオール([3H]E2)、天然高親和性ERリガンド、および細菌発現GST融合ER−αまたはER−βリガンド結合ドメイン(LBD)タンパク質によるインビトロ競合放射性リガンド結合試験を用いて測定された。
【0312】
方法
【0313】
組み換え型ER−αまたはER−βを[3H]E2に結合させ、[3H]E2の平衡解離定数(Kd)を測定した。全特異的結合および非特異的結合を測定するために、[3H]E2の濃度を上げながら、高濃度の非標識E2がある場合とない場合で、4℃で18時間、タンパク質をインキュベートした。非特異的結合を減算し、非線形回帰を用いてE2(ERα:0.71nM;ERβ:1.13nM)のKdを測定した。さらに、ER−αおよびER−βLBDを飽和状態にするために必要な[3H]E2の濃度を4〜6nMと測定した。
【0314】
上述の条件を用いて、[3H]E2(5.7nM)およびER LBDとともに、濃度を上げた化合物(範囲:10−11〜10−6M)をインキュベートした。培養に続いて、Unifilter−96ハーベスターでGF/Bフィルターを用いてプレートを採取し、氷冷した緩衝液B(50mMトリス、pH7.2)で3回洗浄した。室温でフィルタープレートを乾燥させ、その後、MicroScint−Oカクテル35μLを各ウェルに添加し、フィルタープレートをTopSeal−Aで密閉した。3HのMicroscintカクテル(PerkinElmer)用設定を用いて、TopCount(登録商標)NXTマイクロプレートシンチレーションカウンターで、放射線を計測した。
【0315】
[3H]E2の非特異的結合(10−6M非標識E2で培養して決定)を減算し、検査化合物がない状態での特異的結合の割合として表して、各濃度の化合物での[3H]E2の特異的結合を決定した。50%(IC50)で、[3H]E2の特異的結合を減らした化合物の濃度を決定した。次に、化合物の平衡結合定数(Ki)を、Ki=KdXIC50/(Kd+L)で計算した。ここで、Kdは、[3H]E2の平衡解離定数であり(ER−α=0.71nM;ER−β=1.13nM)、Lは[3H]E2の濃度である(ER−α:5.7nM;ER−β:5.7nM)。
【0316】
結果
【0317】
結合試験から、リガンド結合ER−αおよびER−βは、3.75nMから1,000nM以上の範囲に及ぶさまざまな濃度であり、そして選択性はアイソフォーム選択的から非アイソフォーム選択的までに及ぶことが明らかになった。代表的な化合物からの結果を表2に記載する。
【0318】
【表2A】
【0319】
【表2B】
【0320】
化合物IVはERαおよびERβに結合する。化合物IVのER結合親和性は、[2,4,6,7−3H(N)]−エストラジオール([3H]E2)、天然高親和性ERリガンド、および細菌発現GST融合ERαまたはERβリガンド結合ドメイン(LBD)タンパク質によるインビトロ競合放射性リガンド結合試験を用いて測定した。この試験では、化合物IVのERαおよびERβ結合親和性(Ki値)はそれぞれ、21.7±1.7nM(n=3)および15.2±4.1nM(n=3)だった。ERに結合するとき、化合物IVは、組織選択的な方法での薬理反応に携わる標的遺伝子の発現または抑制につながる複雑な一連の分子事象を開始する。一過性形質転換試験では、化合物IVは、ERβと比べると、ERα媒介転写活性化を促進する立証済みの効力が大きいERαおよびERβアゴニストである。エストラジオールは、ERαに対する5.1倍以上の選択性を持ってERαおよびERβを活性化させるのに対して、化合物IVはERαに対して49.0倍の選択性を示す。したがって、化合物IVは、ERβよりもERαに対して、相対的トランス活性化能において相対的に9.7倍の選択性を有する。さらに、最高10μMの濃度の化合物では、エストラジオール(1nM)−促進転写活性化でアンタゴニスト作用は見られなかった。多くのステロイドリガンドは他の核ホルモン受容体と交差反応するが、化合物IVの作用はERαおよびERβに対して特異的である。転写活性化試験におけるアゴニストおよびアンタゴニストの両モードで、グリココルチコイド受容体(GR)、ミネラルコルチコイド受容体(MR)、プロゲステロン受容体(PR)、アンドロゲン受容体(AR)のラットアイソフォームと、およびファルネソイドX受容体(FXR)、肝臓X受容体(LXR)、ペルオキシソーム増殖剤応答性受容体(PPAR−αおよびPPAR−γ)、およびレチノイドX受容体(RXR−α)のヒトアイソフォームとに対する交差反応について、化合物IVをスクリーニングした。化合物IVは、これらの試験のいずれでもアゴニストまたはアンタゴニスト活性を示さず、化合物IVが核ホルモン受容体スパーファミリーメンバーと機能的に交差反応しないということを裏付けた。
【0321】
実施例7:選択化合物のトランス活性化
【0322】
化合物がアゴニストか、アンタゴニストか、あるいは部分的かを確認するために、アゴニストおよびアンタゴニストモードでのトランス活性化試験を行った。
【0323】
方法
【0324】
ラットエストロゲン受容体(ER−αおよびER−β)を、ラット卵巣cDBAからpCR3.1プラスミドベクターバックボーンに複製した。いかなる変異も存在しないことを確認するために、シーケンシングを行った。HEK−293細胞を、Dulbeccoの最小必須培地(DMEM)+5%チャコールストリップウシ胎仔血清(csFBS)の24ウェルプレートのウェルあたり細胞100,000個で蒔いた。0.25μgのERE−LUC、0.02μgのCMV−LUC(レニラルシフェラーゼ)、および12.5ngのラットER−αまたは25ngのラットER−βに、Lipofectamine(Invitrogen、Carlsbad、CA)を用いて、細胞をトランスフェクトした。アンタゴニスト活性を確認するために、さまざまな濃度の化合物、または化合物の組み合わせ、エストラジオールでのトランスフェクションの24時間後に細胞を処理した。トランスフェクションの48時間後にルシフェラーゼ試験を行った。
【0325】
結果
【0326】
トランス活性システムでの本発明の化合物のスクリーニングによって、化合物は3つの分類、すなわちアゴニスト、アンタゴニスト、および部分的アゴニストのすべてに属することが明らかになった。アゴニストおよびアンタゴニストの例を表3に示す。トランス活性化の結果は、アイソフォーム選択性に関する結合結果に極めてよく一致した。
【0327】
表3は、本発明の一部の選択化合物に関するEC50およびIC50トランス活性化値を示す。
【0328】
【表3】
【0329】
実施例8:カニクイザルでのテストステロン抑制
【0330】
試験の間、USDA指針にしたがって、霊長類用餌と水を自由に摂れる状態で(経口投与前の絶食を除き)、2歳の生殖腺非去勢でオスのカニクイザル(n=2)を飼った。Tween 80/脱イオン水のマイクロエマルジョン賦形剤で、連続7日間、毎日1回、式IVの化合物30mg/kgを強制経口投与で与えた。第1日(基準値)、第3日、第4日、第5日、第6日、第7日の経口投与前に静脈穿刺によって血清検体を採取した。それぞれ、HPLC法を組み合わした場合と組み合わさない場合で、酵素免疫測定(EIA)法を用いてテストステロンおよび全アンドロゲンを定量した。式IVの化合物による6日間の処置後、テストステロンおよび全アンドロゲン(テストステロン/ジヒドロテストステロン)に関して、時間依存的な増加が明らかだった。式IVの化合物は、動物No.1および動物No.3において、それぞれ基準値に対して58%および64%にテストステロンの濃度を抑制した(図1の実線を参照;表4)。同様に、動物No.1およびNo.3の両方で、基準値と比較して56%に全アンドロゲン濃度を抑制した(図1の点線を参照;表4)。
【0331】
オスでの下垂体・精巣軸のエストロゲンフィードバックと一致するこれらの結果から、式IVの化合物の経口投与(30mg/kg)を繰り返した後、未去勢非ヒト霊長類(カニクイザル)での血清ホルモン(テストステロンおよび全アンドロゲン)の抑制に関する堅牢な薬理反応を実証する。
【0332】
【表4】
【0333】
実施例9:ラットのLHおよびテストステロンホルモン濃度の抑制
【0334】
未去勢および精巣摘出(ORX)雄ラットでのLH抑制に対する化合物IVの効果を評価するために、インビトロ用量依存的試験を行った。未去勢およびORX動物では、1日あたり10mg/kg以上の用量での化合物IVでは、それぞれの対照と比較した場合に、LH濃度を有意に抑制した。(FSH濃度において、同じパターンの抑制が見られた。)LH抑制によって、テストステロン濃度を、0.08ng/mLである定量限界未満(BLOQ)までしっかりと低下させ、前立腺、精嚢、および肛門拳筋はアンドロゲン依存臓器であるため、これらの重量を低下させた。未去勢動物では、去勢対照のレベルに対して精嚢および肛門拳筋の重量において、これらの対象臓器重量の用量依存的な減少が顕著だった。未去勢動物では前立腺重量が有意に減少したが、これらの値は去勢対照のレベルに達しなかった。結果を下記の表6にまとめる。
【0335】
材料および方法:
【0336】
体重約200gの雄Sprague−Dawleyラットを、餌(2016 Teklad Global 16%タンパク質齧歯類餌、Harlan、Madison、WI)および水が自由摂取できるようにして、12時間明暗サイクルで維持した。動物実験プロトコルは、テネシー大学動物実験委員会による審査および承認を受けた。
【0337】
適切な用量の製剤を調製するために、本試験の被験物質を秤量し、PEG 300(Acros Organics、NJ)で希釈した10%DMSO(Fisher)に溶かした。本試験の場合、60歳の雄Sprague−Dawleyラットを体重で無作為抽出し、12の処置群(n=動物5匹/群)のうちの1つに割り当てた。処置群は、表5に記載している。ケージあたり動物2〜3匹の群で、動物を飼った。対照群(未去勢および精巣摘出済み(ORX))には、毎日、賦形剤を投与した。未去勢群およびORX群の両方に、0.3、1、3、10、および30mg/kg/日の用量で、皮下注射(200μL)を用いて化合物IVを投与した。
【0338】
14日の投与計画の後、麻酔(ケタミン/キシラジン、87:13mg/kg)をかけて動物を屠殺し、体重を記録した。さらに、腹側前立腺、精嚢、および肛門拳筋を取り除き、外部から組織を洗浄し、個別に秤量した。臓器重量を体重で正規化し、未去勢対照に対する割合として表した。イソフルラン麻酔下で腹部大動脈から血液を採取し、凝固させた。遠心分離によって血清を分離し、血清ホルモン濃度の測定まで−80℃で保管した。メーカーの指示にしたがって、ラット下垂体ルミネックスアッセイ(Millipore、Billerica、MA)によって、血清黄体形成ホルモン(LH)および卵胞刺激ホルモン(FSH)の濃度を測定した。この試験の定量下限は、LHに対して3.2pg/mLで、FSHに対して32pg/mLだった。LLOQが0.08ng/mLのテストステロンEIA(Alpco Diagnostics、Salem、NH)でテストステロンを測定した。定量限界未満(BLOQ)の血清ホルモン値を群平均の解析から除いた。そのため、検体BLOQである群でのLHおよびTの報告値は、実測値よりも高い。この方法の解析では、LHおよびT抑制の最も控えめな推量を示した。フィッシャーの最小有意差試験を用いて、個別の用量群を未去勢およびORX賦形剤対照群と比較した。P値<0.05として、推測的に有意性を定義した。
【0339】
【表5】
【0340】
未去勢およびORXラットの黄体形成ホルモン濃度(表6)
【0341】
未去勢群およびORX賦形剤対照群のLH濃度(平均±SD)はそれぞれ、1.46±0.64および11.1±3.9ng/mLだった。化合物IVは、未去勢動物のLH濃度を用量依存的に下げ、3mg/kg以上の日用量で統計的有意な減少を実現した。0.3、1、3、10、および30mg/kg/日の投与後、未去勢化合物IV処置動物のLH濃度はそれぞれ、0.863±0.384、0.704±0.530、0.395±0.302、0.226±0.165、および0.236±0.176ng/mLだった。ORX雄動物のLH濃度も、化合物IV処置によって有意に低下した。ORX動物では、0.3、1、3、10、および30mg/kg/日の投与後、LH濃度はそれぞれ、15.4±2.9、13.5±2.2、6.5±5.6、0.425±0.135、および0.368±0.119ng/mLだった。結果は、図10Aに図示する。
【0342】
未去勢およびORXラットの卵胞刺激ホルモン濃度(表6)
【0343】
未去勢群およびORX賦形剤対照群の血清FSH濃度はそれぞれ、20.9±8.5および93.5±13.8ng/mLだった。未去勢動物では、化合物IVは、FSH濃度を用量依存的に下げ、10mg/kg/日以上の用量で有意な低下が見られた。0.3、1、3、10、および30mg/kg/日の投与後、未去勢化合物IV処置動物のFSH濃度はそれぞれ、17.3±6.4、15.7±7.3、18.4±7.7、9.2±4.0、および6.3±1.8ng/mLだった。ORX動物では、0.3、1、3、10、および30mg/kg/日の投与後、LH濃度はそれぞれ、115±17、114±22、65.2±31.9、27.6±8.2、および15.1±4.1ng/mLだった。結果は、図10Bに図示する。
【0344】
未去勢およびORXラットのテストステロン濃度
【0345】
未去勢賦形剤対照群の血清テストステロン濃度は2.4±1.1ng/mLだった。Tの定量下限は0.08ng/mLだった。0.08ng/mL未満の値は、定量限界未満(BLOQ)に指定した。未去勢動物では、式IVの化合物は、T濃度を用量依存的に下げ、3mg/kg/日以上の用量で有意な低下が見られた。そのため、0.3、1、3、10、および30mg/kg/日の投与後、式IVの化合物による処置を受けた未去勢動物のテストステロン濃度はそれぞれ、2.6±1.7、1.6±1.0、0.7±0.4ng/mLだった。ORX動物では、化合物IVによる処置を受けたすべての群および賦形剤処置群で、T濃度はBLOQだった。未去勢動物に対する結果は、図10C(および図2)に図示する(BLOQ値は、図示のために定量限界で表される)。
【0346】
図9にした通り、3mg/kg、10mg/kg、および300mg/kgの用量で化合物IVを投与することで、24時間後、72時間後、および168時間後の未去勢雄ラットの血清テストステロン濃度の迅速かつ強力な抑制が測定された。
【0347】
臓器重量(表6)
【0348】
Tの抑制を確認するために、前立腺、精嚢、および肛門拳筋の重量を測定した。前記臓器の重量(平均±SD)をそれぞれ、図10D、10E、および10Fに示す。化合物IVによる処置を受けた未去勢動物で、前立腺、精嚢、および肛門拳筋の重量の用量依存的低下が見られた。0.3、1、3、10、および30mg/kg/日の投与後、未去勢動物の前立腺重量はそれぞれ、84.0±19.2、75.2±20.7、68.2±8.1、45.1±20.0、および43.6±8.8だった。0.3、1、3、10、および30mg/kg/日の投与後、ORX動物の前立腺重量はそれぞれ、19.0±4.2、17.4±3.4、19.6±6.7、22.9±5.4、および20.6±2.1だった。0.3、1、3、10、および30mg/kg/日の投与後、未去勢動物の精嚢重量はそれぞれ、76.2±7.8、66.3±27.2、51.8±28.5、19.1±7.0、および17.9±3.3だった。0.3、1、3、10、および30mg/kg/日の投与後、ORX動物の精嚢重量はそれぞれ、12.2±1.3、16.6±5.4、16.5±4.8、13.3±1.9、および12.9±2.1だった。0.3、1、3、10、および30mg/kg/日の投与後、未去勢動物の肛門拳筋重量はそれぞれ、86.9±10.0、82.1±12.1、65.2±4.4、57.8±11.2、および58.1±4.7だった。0.3、1、3、10、および30mg/kg/日の投与後、ORX動物の肛門拳筋重量はそれぞれ、54.5±6.6、49.6±7.0、53.6±10.0、51.1±4.9、および49.2±4.2だった。LH抑制および臓器重量のデータを表6に要約する。
【0349】
【表6】
【0350】
実施例10:ラットおよびサルにおける化合物IVによる抑制後のテストステロン濃度の回復
【0351】
化合物IVによる化学的去勢の可逆性を研究した。
【0352】
材料および方法:
【0353】
体重約200gの雄Sprague−Dawleyラット35匹を、餌(2016 Teklad Global 16%タンパク質齧歯類餌、Harlan、Madison、WI)および水が自由摂取できるようにして、12時間明暗サイクルで維持した。動物実験プロトコルは、テネシー大学動物実験委員会による審査および承認を受けた。
【0354】
適切な用量の製剤を調製するために、本試験の被験物質を秤量し、PEG 300(100%)(Acros Organics、NJ)に溶かした。10の処置群(n=動物5匹/群)の1つに動物を不作為で割り当てた。処置群は、表7に記載している。ケージあたり動物2〜3匹の群で、動物を飼った。群1は、未去勢動物のテストステロン濃度を測定するために、試験の開始時(第1日)に犠牲にした。群2〜7は、3日間、強制経口投与(最大200uL)で毎日1、3、または30mg/kgの用量を摂取した。群2、3、および4は、最大のテストステロン抑制を測定するために第4日に犠牲にした。群5、6、および7は、14日間の薬物排出期間を設けて、回復させた。
【0355】
【表7】
【0356】
結果:
【0357】
未去勢ラットの血清テストステロン濃度は、ベースライン時に6.4±3.1ng/mL(平均±標準偏差)だった。3日間、3および30mg/kgの用量で化合物IVを投与することで、血清テストステロン濃度をそれぞれ1.47±0.26および1.62±0.49ng/mLに有意に低下させた。3日間、1mg/kgの化合物IVを摂取した動物では、有意な抑制は見られなかった。最も重要なことには、14日間の回復期間後に測定した場合、3日間、それぞれ1、3、または30mg/kgの化合物IVを投与された動物では、血清テストステロン濃度が3.3±1.92、3.00±1.06、および3.8±1.72だった。そして、図23に示した通り、未去勢ラットでの基準血清テストステロン濃度からの統計的に有意な違いは見られなかった。
【0358】
本研究によって、化合物IVが未去勢ラットの血清テストステロン濃度を素早く抑制することを示す以前の結果を確認する。3日間3mg/kg/日以上投与された用量群で血清テストステロン濃度の抑制を観察した。1mg/kgの用量群では、血清テストステロンに有意な低下は見られなかった。しかし、14日の回復期間内に、血清テストステロン濃度は未去勢対照の濃度に戻った。本研究から、ラットにおいて化合物IVによる薬理的去勢は可逆的であることが分かる。
【0359】
経口薬物動態研究とともに、未去勢雄ザルにおけるテストステロン濃度の抑制および回復に対する化合物IVの効果を評価した。3匹の処置未経験雄カニクイザル(2〜3歳)が、7日間連続で、強制経口投与によって、毎日30mg/kgで化合物IVの投与を受けた。血液献体を採取し、それぞれテストステロンおよび化合物IV定量測定のために、血清と血漿に分けた。結果からは、化合物IVを連日経口投与することで、3匹の雄ザルすべての血中アンドロゲン(主としてテストステロンおよびジヒドロテストステロン)濃度を、基準値と比較して最高47%に有意に低下させたことが分かる(それぞれ、基準値に対して、1591±72.5、997±104、および852±136ng/mL、処置の第2日および第6日[平均±SEM])。18日間の薬物を摂取しない回復期間の後、アンドロゲン濃度は通常に戻り、処置前の基準濃度との有意な違いは無かった(回復後、1757.7±369.5ng/mL)。
【0360】
実施例11:ラットのLHおよびテストステロンが低下するにもかかわらず骨量を維持する(表8)
【0361】
骨の処置に対する式IVの化合物の効果を研究した。式IVの化合物を経口投与することで、未去勢雄ラットでのLH抑制に関連した骨量減少を完全に予防した。LHの有意な低下は、1日あたり10mg/kg以上の用量で未去勢動物に式IVの化合物を投与することによって誘導された。1日あたり1mg/kgでは、式IVの化合物はLHを有意に下げなかったが、この用量で前立腺、精嚢、および肛門拳筋の重量を有意に減らすことが明らかになり、血中テストステロンの低下がこれらのアンドロゲン応答性臓器と生理学的に関係していることが分かった。しかし、1日あたり1mg/kgの式IVの化合物では、未去勢対照のレベルに骨梁量(大腿骨遠位)を維持した。1日あたり10および30mg/kgの用量で投与した場合、式IVの化合物は、未去勢対照の骨量以上に、骨梁の骨量を有意に増加させた。これらのデータから、化合物IVによって、未去勢ラットのLH濃度を下げる用量レベルで骨梁骨ミネラル濃度(BMD)および骨稜率を増やしたことが分かる。この研究から得られたデータを表8に記載する。
【0362】
【表8】
【0363】
実施例12:17βヒドロキシステロイド脱水素酵素5(17β−HSD5)酵素活性に対する効果
【0364】
HSD群の構成要素は、血中ステロイドの変換に関与する。17β−HSD5は、アンドロステンジオンをテストステロンに、エストロンをエストラジオールに変換する。さらに、プロスタグランジン合成にも関与する。ここで、17β−HSD5活性を阻害する本発明の一部の選択化合物の能力を実証した。
【0365】
方法
【0366】
pGEX 4t1ベクターでヒト17β−HSD5を複製し、精製タンパク質を調製した。適切な緩衝液中で、本発明の代表的な化合物、14Cアンドロステンジオン、NADPHとともに、精製タンパク質をインキュベートした。酢酸エチルを用いて合成したテストステロンを抽出し、乾燥させ、薄層クロマトグラフィー(TLC)プレートに置き、分析した。TLCをフォスフォイメージャーにさらし、テストステロン結合の強度を定量した。陽性対照(LHRHアゴニスト)としてインドメタシンを使用した。
【0367】
結果
【0368】
化合物IVを検査し、17β−HSD5酵素活性に対して部分的抑制効果があった。図3に示した通り、陽性対照(LHRHアゴニスト)、インドメタシンは、予想通りに、本酵素の強力な抑制を示した。
【0369】
実施例13:毒性研究
【0370】
ヒト血小板凝集試験を用いて、化合物IVとジエチルスチルベストール(DES、陽性対照)の血栓症の可能性を比較するために、研究を行った。男性が化合物IVの対照処置集団(LH抑制)であるため、研究では健常男性供血者からの血液を使用した。30秒間、エストラジオール(E2)、DES、化合物IV、または賦形剤のいずれかとともに、多血小板血漿を前保温し、その後、血小板凝集を誘導するためにトロンビン(0.3単位)を添加した。研究の結果から、DESとの前保温によって、トロンビン誘導血小板凝集が約10倍増えたことが分かる。しかし、化合物IVおよびエストラジオールは多血小板血漿の凝集を減らした。これらのデータから、DESと比較して、化合物IVはインビトロのヒト血小板の反応性を下げたことを実証し、化合物IVはDESよりも血栓塞栓症の可能性が低いことを示唆する(図4)。
【0371】
実施例14:ほてりに対する化合物IVの効果
【0372】
Simpkins等によって開発された(1983年)モルヒネ依存ラットモデル(MDモデル)を用いて、ほてりに対する化合物IVの効果を調査するために研究を行い、閉経によるほてりとの幾つかの類似性があることが分かった。ヒトの症状との類似性に加えて、本実験動物モデルは、尾皮膚温度(TST)を用いて血管運動症状を緩和できる化合物を特定する便利な高性能ツールにする短い応答時間を持つ。TSTプローブTA−40(Data Sciences International, MN)を尾の根元にテープで貼り、15分間、基準温度を得た。15分後、モルヒネの効果を無効にするために、動物をナロキソン(1mg/kgSQ)で処置した。一連の実験の間中、5秒間のサンプリング頻度で、ナロキソン処置前の1時間の尾皮膚温度(TST)を測定した。データ収集に続いて、各動物に対して60秒ごとに記録された温度の移動平均を計算し、さらに解析した。ナロキソン投与前の15分にわたって取得された平均温度として、基準温度を計算した。線形台形を用いて、基準値からすべてのナロキソン投与前値を減算することで、濃度曲線下面積(AUC)を計算した。
【0373】
化合物IVは、モルヒネ禁断モデル(図13)のほてりを弱め、10mgの化合物IVで最高の結果が得られた。17βE2は、100%DMSO溶液で5mg/kgを使用した。
【0374】
実施例15:ラットでの化合物IV対DES
【0375】
LHRHアゴニストを導入する前に、エストロゲン、主としてジエチルスチルベストロール(DES)を介して下垂体でのエストロゲン活性を増加させることで、去勢テストステロン濃度が達成された。DESは、去勢濃度へのテストステロンの抑制においてLHRHアゴニストとして同じように有効であった。DESによって処置された患者はほてりまたは骨量減少を示さなかったが、LHRHアゴニストによるADTよりも高い率で女性化乳房を示した。残念ながら、DESおよびエストラジオールのような極めて強力な純エストロゲンは、その臨床使用を制限してきた重篤な心血管および血栓塞栓合併症の高いリスクと関係がある場合が多い。DESによる静脈血栓塞栓合併症のリスト上昇は、他のホルモン受容体とのその交差反応性によることが、証明されていないが、仮定してきた。ヒト血小板を用いたインビトロ研究から、化合物IVがDESよりもかなり低い前凝固活性を示した。そのため、化合物IV、ER−α選択的アゴニストは、DESの前立腺癌に関する利点を実現でき、骨粗鬆症または有害な脂質状態の変化を生じることなくLHRHアゴニストの利点も実現できる。
【0376】
化合物IVは、ラットの前立腺サイズの削減でDESと同じくらい効果があり(図11A)、また、ORXラットの前立腺サイズの中程度の増加を示す(図11B)。
【0377】
DESと化合物IVとの差を図12A〜12Cに示す。ここで、化合物IVは交差反応しなかったが、DESはグルココルチコイド受容体(GR)(図12A)およびアンドロゲン受容体(AR)(図12B)と交差反応した。さらに、DESはエストロゲン関連受容体(ERR)トランス活性化を無効にした。一方で化合物IVは無効にしなかった。化合物IVは、図12Cに示した通り、3種類のERRアイソフォーム(ERR−α、ERR−β、およびERR−γ)のいずれとも交差反応しなかった。
【0378】
実施例16:サル毒性研究−90日
【0379】
モーリシャス育ちの集団飼育カニクイザルを入手した。13週の暫定期間で、雄カニクイザルにおける化合物IVおよび陽性対照(LHRHアゴニスト)の39週経口薬理および毒性評価として、プロスペクティブ研究を計画した。処置開始前に、合計39匹の性的に成熟した5〜8歳の雄ザルを5つの群に無作為に割り当てた。群としては次のものが挙げられる。すなわち、1)賦形剤、2)1mg/kgの化合物IV、3)10mg/kgの化合物IV、4)100mg/kgの化合物IV、および5)陽性対照(LHRHアゴニスト)。群1および5に対して賦形剤対照品(Tween 80/PRANG(商標))または群2、3、および4に対して化合物IVを含む賦形剤を用いて、39週間、1日1回のケージサイド投与によって薬剤を経口で送達した。化合物IVの用量レベルは、群2、3、および4に対して、それぞれ1、10、および100mg/kg/日だった。各動物に対して直近に入手した体重に基づく計算通りに、10mg/kgの用量で経口量を送達した。群5の動物は、39週の研究期間中、陽性対照(LHRHアゴニスト)(0.02mL定量)の1日1回の皮下注射も受けた。毎日、全体的な外観および臨床兆候を観察、記録した。研究プロトコルに示した通り、所定の評価および厳選した他の研究調査を行った。厳選したパラメータとしては、テストステロン、前立腺特異抗原(PSA)、および前立腺の容積および重量が挙げられるが、これに限定されるものではない。
【0380】
酵素免疫測定(EIA)法および化学発光免疫測定(LIA、ALPCO Diagnostics、Salem NH)それぞれを用いて、血清検体のテストステロンおよび全PSA濃度を定量した(標準手順にしたって)。ベースライン時(すなわち、処置開始前)と、第1、3、7、14、28、64、および90日にすべての動物(絶食状態で)からテストステロン評価用の血液検体を採取した。ベースライン時と第6週の間にすべての動物(絶食状態で)PSA測定用の血液検体を採取した。議論のために、テストステロンおよびPSA試験の定量限界未満(BLQ)の濃度を持つ検体の結果は、試験の定量下限(LLOQ)の1/2として計算され、「予想最終濃度」と考えられる。表9〜16のデータは、「予想最終濃度」(すなわち、検体BLQを持つ検体が試験のLLOQの1/2として含まれた)に加えて、「定量化可能濃度のみ」(BLQ値を除く)として示される。ベースライン時と第6週に経直腸超音波(TRUS)検査を用いて、麻酔下の生きた動物で前立腺容積を測定した。前立腺の幅と高さを記録した。幅*幅*高さ*π/6[ref]として前立腺容積を計算し、体重に正規化した。組織を脂肪のないように、そして異質組織を刈り込んだ後、剖検時に前立腺の湿重量を記録した。
【0381】
結果および議論:
【0382】
血清テストステロン濃度を図15および表9〜12に示す。ベースライン時、研究のすべてのサルのテストステロン濃度が、性的に成熟した大人の雄カニクイザルに対して通常範囲内だった。しかし、100mg/kg/日で化合物IVを摂取しているサル、および陽性対照(LHRHアゴニスト)によって処置されたサルでは、テストステロン濃度が有意に低下した。陽性対照(LHRHアゴニスト)群のテストステロン濃度は、それぞれ、第1日と第3日に47.4%と547%(p<0.01)の有意な初期増加(すなわち、フレア)とともに、二相性変化を示し、続いて、第7、14、28、64、および90日に、3.6%、67%、73%、83%、および85%の低下を示した(図15および表9〜12を参照)。最高の用量レベル(すなわち、100mg/kg/日)でも、化合物IVにより処置されたどの動物にも、同様のフレアは見られなかった。用量および処置期間は化合物IVの薬理作用にとって重要だった。ここで、100mg/kg/日の用量では、第3、7、14、28、および64日にそれぞれ、血清テストステロンを基準値に対して60%、51%、42%、79%、および92%抑制した(図15および表9および10を参照)。100mg/kg/日の化合物による処置の90日後、群4のサルの10匹中6匹のテストステロン濃度は、試験の定量限界未満の濃度に低下した(表11を参照)。群4のサルの平均血清テストステロン濃度は、それぞれの基準値と比較して96%低下した(「予想最終濃度」、すなわち、BLQ値を持つ6/10匹のサルのテストステロン濃度は、LLOQ濃度の50%として計算される。表10を参照)。第90日で、100mg/kg/日の化合物IVによって、血清テストステロンを陽性対照(LHRHアゴニスト)よりも有意に低い濃度に下げたことに留意することが重要である(p=0.013)。
【0383】
【表9】
【0384】
【表10】
【0385】
【表11】
【0386】
【表12】
【0387】
処置開始の4週間以内に化合物IVによって血清PSA濃度も有意に抑制された。陽性対照(LHRHアゴニスト)群ではPSA濃度が60%低下した一方で、4週間、10mg/kgおよび100mg/kgで化合物IVを摂取しているサルに対して、67%および87%(平均で)のPSA低下が分かった(図16および表13〜16)。
【0388】
【表13】
【0389】
【表14】
【0390】
【表15】
【0391】
【表16】
【0392】
研究を通じて定期的に、TRUSによって前立腺容積を測定した。6週間の処置後に得られる結果から、サルの前立腺に対する化合物IVおよび陽性対照(LHRHアゴニスト)の強力な効果を実証する。化合物IVは、それぞれ10mg/kgおよび100mg/kgで前立腺容積を25%および45%、有意に抑制した。一方で、陽性対照(LHRHアゴニスト)群では28%、前立腺を低下させた(図17ならびに表17および18)。
【0393】
【表17】
【0394】
【表18】
【0395】
剖検時の前立腺重量の評価によって、前立腺容積の化合物IVに関連した減少が確認された。13週間の処置後、化合物IVは、それぞれ10および100mg/kg/日を摂取している動物において、平均前立腺重量を24%および21%と有意に減少させた(図18および表19および20)。
【0396】
【表19】
【0397】
【表20】
【0398】
血小板凝集、プロトロンビン時間(PT)、または活性化部分トロンボプラスチン時間(APTT)に対する明らかな効果は見られなかった。
【0399】
実施例17:ヒトに対する化合物IV研究
【0400】
ヒトの男性に対する化合物IVの効果を確認するために、研究を行った。化合物IVの100、300、600、および1000mgの用量で、群あたり12人の対象を調べた。表21には、100、300、600、および1000mgの用量で化合物IVを投与することによる、男性のLH、血清PSA、遊離テストステロン、および全テストステロン濃度の平均変化を示す。第1日〜第11日の期間中のヒトでの用量依存的平均全テストステロン濃度(nmol/L)を測定した(図19)。それぞれ、600mgおよび1000mgの用量で、全テストステロン濃度は51.9%および47.9%低下した。
【0401】
第1日〜第10日の期間中のヒトでの用量依存的平均LH濃度(IU/L)を測定した(図20)。それぞれ、100mg、300mg、600mgおよび1000mgの用量で、LH濃度は20.7%、46.9%、27.6%および29.2%低下した。
【0402】
第1日〜第10日の期間中のヒトでの用量依存的遊離テストステロン濃度(pg/mL)を測定した(図21)。それぞれ、100mg、300mg、600mgおよび1000mgの用量で、遊離テストステロン濃度は17.0%、18.5%、72.7%、および53.2%低下した。
【0403】
第1日〜第10日の期間中のヒトでの用量依存的平均PSA濃度(μg/L)を測定した(図22)。それぞれ、100mg、300mg、600mgおよび1000mgの用量で、PSA濃度は9.2%、24.4%、27.5%、および29.9%低下した。10および30mgの用量に対しては、変化が無いことが分かった。
【0404】
【表21】
【0405】
実施例18:化合物IVのバイオアベイラビリティ
【0406】
ラット、イヌ、およびサルへの経口投与の後、化合物IVは迅速に吸収された。1回分を投与した剤形に応じて、ラットでの化合物IVの経口バイオアベイラビリティは、6%〜25%に及んだ。ポリエチレングリコール300(PEG300)を用いた製剤は、脱イオン水で希釈されたTween 80で調製されるマイクロエマルションよりも高い暴露を典型的に生じる。イヌでは、血書濃度−時間プロファイルの目視検査から、終末期の第2ピークでも分かるように、化合物IVが腸肝循環を経ることが分かった。重要なことには、イヌでは、雄のPEG300を30mg/kg経口投与した群の暴露は、LH抑制のラットモデルでの前立腺減少に対する最大作用を生じるために必要な暴露を超えた。サルでは、予備薬物動態学的研究によって、化合物IVの血漿濃度、および7日間の期間にわたる血漿テストステロンの抑制でも分かるように、この種の経口バイオアベイラビリティはイヌのものに近似する、または超えることが示された。総じて、これらのデータから、所望の薬理作用を生じるように、2つの非齧歯動物種で十分な経口暴露を達成できることを示している(AUCデータに基づく)。さらに、ラットおよびサルの内分泌データから、化合物IVの薬理作用は可逆的(すなわち、化合物IVによる処置が停止されると、テストステロンの血清濃度は基準値または通常濃度に戻る)であることを示している。
【0407】
実施例19:化合物IVの薬物動態
【0408】
インビトロ(マウス、ラット、イヌ、サルおよびヒト)およびインビボ(ラット)代謝研究からの予備データによって、動物およびヒトでは、化合物IVの結合、そのヒドロキシル化代謝産物、およびそのN−脱アルキル化代謝産物が、化合物IVの全般的な分解に寄与することを示している。定性のみだが、種間比較の結果から、非臨床種の全般的な代謝物プロファイルは、ヒト肝臓ミクロソームで作られるプロファイルに十分に反映することを示す。これらの結果に基づき、ラットおよびイヌはそれぞれ、薬理および毒性評価に適した齧歯および非齧歯種である。インビトロ研究から、化合物IVは、30μM以下の濃度では、関連かるCYP450アイソフォーム(CYP1A2、CYP2B6、またはCYP3A4)の減少を誘導せず、かつCYP1A2、CYP2C19、CYP2D6、またはCYP3A4/5を阻害しない。CYP2C9は、高濃度(Ki=8μM)のみで化合物IVによって阻害されるが、強力な薬物動態薬物・薬物相互作用がごくわずかにあると考えられる。
【0409】
実施例20:化合物IVの生物活性
【0410】
化合物IVは、hERGチャンネルに対してインビトロの阻害作用(IC50≧300μM)をわずかに及ぼす、または及ぼさない。化合物は、インビトロで分離したイヌプルキンエ線維において、10および100μMの濃度でAPD50およびAPD90を用量依存的に減らした。しかし、化合物IVは、あらゆる用量(最高300mg/kg)での遠隔計測器が付けられたイヌにおいて、血流動態または心臓機能(血圧、心拍数、心電図形態、またはQT間隔)に影響を及ぼさなかった。神経薬理または肺の効果は見られなかった。最高30mg/kgの化合物IVの単回経口投与では、腎臓機能に対して有意な効果は示されなかった。高用量研究(100mg/kg)では、尿排出量と、カリウムおよび塩化物の尿中排泄との増加のみが見られた。ラットにおいて30〜300mg/kgの用量で化合物IVを経口投与することによって蠕動の有意な増加を生じさせ、ラットにおいて30mg/kgの用量で化合物IVを経口投与することによって、胃腸の運動および胃液の酸度の有意な増加を生じさせた(おそらく、平滑筋に対する効果によるものではない)。
【0411】
化合物IVには変異原性はなく、インビトロでのヒト末梢血リンパ球中で最高200μMの濃度では構造的または数的染色体異常を誘導しなかった。化合物IVは、単回および反復経口投与(最高28日)の後、ラットおよびイヌで良好な耐容性を示した。腎臓、肝臓、心臓、および他の非対象関連臓器では、病変は見られなかった。最長28日間の雄および雌のイヌに対する化合物IVの投与に関連した深刻な身体的兆候、体重に関する効果、臨床病理的変化、眼科、心電図、または組織病理学的変化は無かった。
【0412】
本願明細書では、本発明のいくつかの特徴が説明、記述されているが、現在では多くの修正、代用、変更、および同等物を当業者が思い付くことになる。そのため、当然のことながら、追加請求項は、本発明の真意の範囲内でそのような修正および変更のすべてを対象にするためのものである。
【技術分野】
【0001】
本発明は、対象へのアンドロゲン遮断療法(ADT)により誘発されるほてりおよび重度のほてりを処置する方法に関する。
【背景技術】
【0002】
エストロゲンとは、組織および骨の維持にとって重要であり、かつ使用される内因性および合成ホルモンの群のことを言う。エストロゲンは、生殖器系の発達および維持に関与する細胞過程の内分泌制御因子である。生殖生物学におけるエストロゲンの役割、閉経後のほてり(一過性熱感)の予防、および閉経後骨粗鬆症の予防は確立されている。エストラジオールは主要な内因性ヒトエストロゲンであり、女性と男性両方で見つかっている。
【0003】
エストロゲンおよび抗エストロゲンの生理学的作用は、2つの異なる細胞内受容体、エストロゲンアルファ(ERα)およびエストロゲンベータ(ERβ)を通じた発現である。内因性エストロゲンは、両方の受容体亜型の典型的に強力な活性剤である。例えば、エストラジオールは、乳房、骨、心血管、および中枢神経系組織を含む多くの組織内でERαとして作用する。選択的エストロゲン受容体モジュレーターは、さまざまな組織で、一般に異なる作用をする。例えば、SERMは乳房ではERαアンタゴニストである場合があるが、子宮、骨および心血管系では部分ERαアゴニストである場合がある。そのため、エストロゲン受容体リガンドとしての機能を果たす化合物は、さまざまな病状および疾患の処置に有用である。
【0004】
前立腺癌は、米国の男性で最もよく診断される非皮膚癌の1つであり、癌による死亡の2番目に多い原因であり、今年には180,000件を超える新規症例とほぼ29,000人の死亡が見込まれている。進行前立腺癌の患者は、典型的には黄体形成ホルモン放出ホルモン(LHRH)アゴニストまたは両側精巣摘除術のいずれかによる、アンドロゲン遮断療法(ADT)を受ける。アンドロゲン遮断療法ではテストステロンを削減するだけでなく、エストロゲンがテストステロンの芳香族化から生じ、テストステロンの濃度はADTによって激減させられるため、エストロゲン濃度も低くなる。アンドロゲン遮断療法により誘発されるエストロゲン欠乏は、ほてり(hot flash)、女性化乳房および乳房痛、骨量減少、骨の質および強度の低下、骨粗鬆症および致命的な骨折、有害な脂質変化および高い心血管疾患および心筋梗塞、ならびに鬱病およびその他の情緒の変化を含む、有意な副作用を引き起こす。ADTのエストロゲン欠乏副作用の多くはERαが関与していると考えられる。
【0005】
酢酸ロイプロリド(Lupron(登録商標))は、天然ゴナドトロピン放出ホルモン(GnRHまたはLH−RH)の合成非ペプチド類似体である。酢酸ロイプロリドは、下垂体によるLH分泌を最終的に抑えるLH−RHスーパーアゴニストである。酢酸ロイプロリドは、ゴナドトロピン分泌の強力な阻害剤としての機能を果たし、卵巣および精巣ステロイド合成を抑制する。ヒトでは、酢酸ロイプロリドの投与によって、黄体形成ホルモン(LH)および卵胞刺激ホルモン(FSH)の血中濃度の初期増加を引き起こし、性腺ステロイド(男性でのテストステロンおよびジヒドロテストステロン、並びに閉経前女性でのエストロンおよびエストラジオール)の濃度を一時的に増加させる。しかし、酢酸ロイプロリドの継続投与、LHおよびFSHの濃度低下を招く。男性では、テストステロンが去勢レベル(50ng/dL未満)にまで減少する。閉経前女性では、エストロゲンが閉経後レベルにまで減少する。テストステロンは、前立腺の癌細胞に対する既知の刺激物質である。したがって、テストステロンの分泌を抑制することまたはテストステロンの作用を阻害することは、前立腺癌治療の必要な構成要素である。酢酸ロイプロリドは、前立腺癌を処置するための去勢レベルへの血清テストステロンの減少および低減であるLH抑制に使用できる。
【0006】
LHRHアゴニストを導入する前に、エストロゲン(主としてジエチルスチルベストロール(DES))を介して、下垂体でのエストロゲン活性を増加させることで、去勢テストステロンレベルが達成された。DESは、去勢濃度へのテストステロンの抑制においてLHRHアゴニストと同じように有効であった。DESによって処置された患者はほてりまたは骨量減少を示さなかったが、LHRHアゴニストによるADTよりも高い率で女性化乳房を示した。残念ながら、DESおよびエストラジオールのような極めて強力な純エストロゲンは、その臨床使用を制限してきた重篤な心血管および血栓塞栓合併症の高いリスクと関係がある場合が多い。
【0007】
本発明の化合物は、テストステロンレベルを去勢レベルに抑制する。このレベルは、血栓症のリスク増加を予防しながら、骨量減少、ほてり、および/または女性化乳房なしに、前立腺癌の処置に使用してもよいレベルである。
【発明の概要】
【課題を解決するための手段】
【0008】
別の実施形態では、本発明は、アンドロゲン遮断療法(ADT)により誘発されるほてりを処置する方法であって、対象に対して、式Iの化合物、またはその異性体、薬学的に許容される塩、N−酸化物、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与するステップを含む方法を提供する。
【0009】
【化1】
【0010】
式中、
Yは、C(O)またはCH2であり;
R1、R2は、互いに独立して、水素、ハロゲン、ヒドロキシル、アルコキシ、シアン、ニトロ、CF3、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、O−Alk−NR5R6、または、置換されたもしくは非置換の3〜7員のO−Alk−複素環(任意選択で芳香族である)であり;
R3、R4は、互いに独立して、水素、ハロゲン、ヒドロキシアルキル、ヒドロキシル、アルコキシ、シアン、ニトロ、CF3、NHCOR、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、または保護ヒドロキシルであり;
Rは、アルキル、水素、ハロアルキル、ジハロアルキル、トリハロアルキル、CH2F、CHF2、CF3、CF2CF3、アリール、フェニル、ハロゲン、アルケニル、CN、NO2、またはOHであり;
R5およびR6は、互いに独立して、水素、フェニル、炭素数1〜6のアルキル基、3〜7員のシクロアルキル、3〜7員の複素環、5〜7員のアリールであり;あるいは、R5およびR6は、窒素原子を含む3〜7員環を形成し;
jおよびkは、互いに独立して、1〜4であり;
Alkは、炭素数1〜7の直鎖アルキル、炭素数1〜7の分岐鎖アルキル、または炭素数3〜8の環状アルキルである。
【0011】
別の実施形態では、本発明は、アンドロゲン遮断療法(ADT)により誘発されるほてりを抑制もしくは防止、または該ほてりの発症率を低減する方法であって、対象に対して、式Iの合物、またはその異性体、薬学的に許容される塩、N−酸化物、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与するステップを含む方法を提供する。
【0012】
別の実施形態では、本発明は、アンドロゲン遮断療法(ADT)により誘発される重度のほてりを処置する方法であって、対象に対して、式Iの化合物、またはその異性体、薬学的に許容される塩、N−酸化物、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与するステップを含む方法を提供する。
【0013】
別の実施形態では、本発明は、アンドロゲン遮断療法(ADT)により誘発される重度のほてりを抑制もしくは防止、または該ほてりの発症率を低減する方法であって、対象に対して、式Iの化合物、またはその異性体、薬学的に許容される塩、N−酸化物、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与するステップを含む方法を提供する。
【図面の簡単な説明】
【0014】
発明と見なされる主題は具体的に指摘され、明細書の最終部分で明確に主張される。しかしながら、その目的、特徴、および利点とともに、構成および実施方法の両方に関して、本発明は、以下の添付図面とともに読むならば、以下の詳細な説明を参照することによって最もよく理解できる。
【0015】
【図1】化合物IVを30mg/kg連日経口投与(0日目に初回投与)した後の未去勢雄ザルの血清テストステロン(実線)および全アンドロゲン(点線)の濃度を示す(実施例8を参照)。
【図2】化合物IV(0.3、1、10、30mg/kg)で処置された未去勢ラットのテストステロン濃度を示す。Iは、P<0.05 対未去勢賦形剤対照を示す。BLOQ値は、0.08ng/mLの定量限界で図示されている(実施例9を参照)。
【図3】17β−HSD5酵素活性に対する化合物IVの阻害効果を示す(実施形12を参照)。
【図4】DES、17β−エストラジオール(E2)、および化合物IVの存在におけるヒト血小板のインビトロ凝集を示す。0.3単位のトロンビンで凝集を誘導する前の30秒間、多血小板血漿(PRP)を、賦形剤、E2、DES、または化合物IVとともにインキュベートした。凝集を5分間監視し、賦形剤対照の割合として表した(実施例13を参照)。
【図5】化合物II〜XIIの調製のための一般的な合成スキームである(実施例1を参照)。
【図6】化合物IVの調製のための合成スキームである(実施例2を参照)。
【図7】化合物VIの調製のための合成スキームである(実施例3を参照)。
【図8】化合物IXおよびXの調製のための合成スキームである(実施例5を参照)。
【図9】3mg/kg、10mg/kg、および300mg/kgの用量で化合物IVによる処置を受けた未去勢ラットの24時間後、72時間後、および168時間後のテストステロン濃度を示す(実施例9を参照)。
【図10A】化合物IVの0.3mg/kg、1mg/kg、3mg/kg、10mg/kg、30mg/kgの用量で処置を受けた未去勢および精巣摘徐術済みラットのLH濃度を示す。Iは、P<0.05対未去勢賦形剤対照を示す。Oは、P<0.05対ORX賦形剤対照を示す。BLOQ値は、0.08ng/mLの定量限界で図示されている(実施例9を参照)。
【図10B】化合物IVの0.3mg/kg、1mg/kg、3mg/kg、10mg/kg、30mg/kgの用量で処置を受けた未去勢および精巣摘徐術済みラットのFSH濃度を示す。Iは、P<0.05対未去勢賦形剤対照を示す。Oは、P<0.05対ORX賦形剤対照を示す。BLOQ値は、0.08ng/mLの定量限界で図示されている(実施例9を参照)。
【図10C】化合物IVの0.3mg/kg、1mg/kg、3mg/kg、10mg/kg、30mg/kgの用量で処置を受けた未去勢および精巣摘徐術済みラットのテストステロン濃度を示す。Iは、P<0.05対未去勢賦形剤対照を示す。Oは、P<0.05対ORX賦形剤対照を示す。BLOQ値は、0.08ng/mLの定量限界で図示されている(実施例9を参照)。
【図10D】化合物IVの0.3mg/kg、1mg/kg、3mg/kg、10mg/kg、30mg/kgの用量で処置を受けた未去勢および精巣摘徐術済みラットの前立腺重量レベルを示す。Iは、P<0.05対未去勢賦形剤対照を示す。Oは、P<0.05対ORX賦形剤対照を示す。BLOQ値は、0.08ng/mLの定量限界で図示されている(実施例9を参照)。
【図10E】化合物IVの0.3mg/kg、1mg/kg、3mg/kg、10mg/kg、30mg/kgの用量で処置を受けた未去勢および精巣摘徐術済みラットの精嚢重量レベルを示す。Iは、P<0.05対未去勢賦形剤対照を示す。Oは、P<0.05対ORX賦形剤対照を示す。BLOQ値は、0.08ng/mLの定量限界で図示されている(実施例9を参照)。
【図10F】化合物IVの0.3mg/kg、1mg/kg、3mg/kg、10mg/kg、30mg/kgの用量で処置を受けた未去勢および精巣摘徐術済みラットの肛門拳筋重量を示す。Iは、P<0.05対未去勢賦形剤対照を示す。Oは、P<0.05対ORX賦形剤対照を示す。BLOQ値は、0.08ng/mLの定量限界で図示されている(実施例9を参照)。
【図11】さまざまな用量で化合物IV(図11A)およびDES(図11B)を投与することによる未去勢およびORXラットの前立腺の大きさを示す(実施例15を参照)。
【図12A】DESと化合物IVとの間の違いを示し;DESはグルココルチコイド受容体(GR)と交差反応するが、化合物IVは反応しない(図12A);DESはアンドロゲン受容体(AR)と交差反応する。ARの活動を穏やかに刺激し、穏やかに阻害するが(すなわち、部分アゴニスト/アンタゴニストである)、化合物IVはしない(図12B);DESはエストロゲン関連受容体(ERR)トランス活性化を無効にするが、化合物IVはしない(図12C)(実施例15を参照)。
【図12B】DESと化合物IVとの間の違いを示し;DESはグルココルチコイド受容体(GR)と交差反応するが、化合物IVは反応しない(図12A);DESはアンドロゲン受容体(AR)と交差反応する。ARの活動を穏やかに刺激し、穏やかに阻害するが(すなわち、部分アゴニスト/アンタゴニストである)、化合物IVはしない(図12B);DESはエストロゲン関連受容体(ERR)トランス活性化を無効にするが、化合物IVはしない(図12C)(実施例15を参照)。
【図12C】DESと化合物IVとの間の違いを示し;DESはグルココルチコイド受容体(GR)と交差反応するが、化合物IVは反応しない(図12A);DESはアンドロゲン受容体(AR)と交差反応する。ARの活動を穏やかに刺激し、穏やかに阻害するが(すなわち、部分アゴニスト/アンタゴニストである)、化合物IVはしない(図12B);DESはエストロゲン関連受容体(ERR)トランス活性化を無効にするが、化合物IVはしない(図12C)(実施例15を参照)。
【図13】5mg/kg、10mg/kg、15mg/kg、および30mg/kgの用量によるモルヒネ禁断モデルのほてりの軽減に対する化合物IVの効果を示す。N=7匹/群。17β−E2は、100%DMSO溶液で5mg/kgを使用した(実施例14を参照)。
【図14】91日間、化合物IVを投与することによるサルの用量依的な体重(kg)減少(100mg/kgで最高20%)を示す。女性化乳房または高エストロゲン症の兆候は観察されなかった(実施例16を参照)。
【図15】化合物IVを連日経口投与した後のサルの用量依存的な血清テストステロン濃度の減少(ng/mL)を、陽性対照(LHRHアゴニスト)と比較して示す。点線は化学的に去勢された患者のテストステロン濃度を示し、太い破線は外科的に去勢されたサルのテストステロン濃度を示す(実施例16を参照)。
【図16】化合物IVを投与よることによる、ベースライン時と第28日のサルの用量依存的な前立腺特異的抗原(PSA)濃度(ng/mL)を示す。PSA濃度は、化合物IVの処置により有意に減少した(実施例16を参照)。
【図17】第6週に化合物IVを投与することによる、陽性対照(LHRHアゴニスト)と比較した、経直腸超音波(TRUS)を用いたサルの用量依存的な前立腺体積を示す(実施例16を参照)。
【図18】化合物IVの投与による、第90日の用量依存的な臓器重量(前立腺、精嚢、および睾丸)をサルの対照の割合として示す(図18A)。化合物IVを連日経口投与した後、サルの死体解剖による第13週時の前立腺の重量(図18B)(実施例16を参照)。
【図19】化合物IVの投与(100mg、300mg、600mg、および1000mg)による、第1日〜第11日の期間中のヒトの用量依存的な平均全テストステロン濃度(nmol/L)を示す(実施例17を参照)。
【図20】化合物IVの投与(100mg、300mg、600mg、おび1000mg)による、第1日〜第10日の期間中のヒトの用量依存的な平均LH濃度(IU/L)を示す(実施例17を参照)。
【図21】化合物IVの投与(100mg、300mg、600mg、および1000mg)による、第1日〜第10日の期間中のヒトの用量依存的な平均遊離テストステロン濃度(pg/mL)を示す(実施例17を参照)。
【図22】化合物IVの投与(100mg、300mg、600mg、および1000mg)による、第1日〜第10日の期間中のヒトの用量依存的な平均PSA濃度(μg/L)を示す(実施例17を参照)。
【図23】化合物IV投与の14日間の回復後の未去勢ラットの用量依存的な血清テストステロン濃度(ng/mL)を示す。Iは、P<0.05対未去勢対照を示す(実施例10を参照)。
【0016】
図の説明を簡単かつ明瞭にするために、図に示された要素が必ずしも一定の縮尺には描かれていないことは十分に理解されるであろう。例えば、要素の一部の寸法は、明瞭化のために多の要素と比較して誇張されている場合がある。さらに、適切と考える場合、参照数字が、対応する類似要素を示すために、複数の図の間で繰り返される場合がある。
【発明を実施するための形態】
【0017】
以下の詳細説明では、発明が十分に理解されるように、多数の具体的詳細が示される。しかし、これらの具体的詳細なしに本発明を実施できる場合があるということは、当業者によって理解される。他の例では、本発明を曖昧にしないように、公知の方法、手順、および構成要素は詳細に説明されていない。
【0018】
一実施形態では、本願明細書で説明されるような化合物および/またはそれを含む組成物を、男性対象へのアンドロゲン遮断療法(ADT)により誘発されるほてり(hot flash)または重度のほてりの処置に使用できる。一実施形態では、被検者は前立腺癌を患っている。別の実施形態では、被検者は進行前立腺癌を患っている。
【0019】
一実施形態では、本願明細書で説明されるような化合物および/またはそれを含む組成物を、男性対象へのアンドロゲン遮断療法(ADT)により誘発されるほてりまたは重度のほてりの発生率減少、抑制、重症度軽減、または阻害を行うために使用できる。一実施形態では、被検者は前立腺癌を患っている。別の実施形態では、被検者は進行前立腺癌を患っている。
【0020】
一実施形態では、本発明は、以下の式Iの構造で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象へのアンドロゲン遮断療法(ADT)により誘発されるほてりの処置、発生率減少、抑制、重症度軽減、または阻害を行う方法を提供する。
【0021】
【化1】
【0022】
式中、
Yは、C(O)またはCH2であり;
R1、R2は、互いに独立して、水素、ハロゲン、ヒドロキシル、アルコキシ、シアン、ニトロ、CF3、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、O−Alk−NR5R6、または、置換されたもしくは非置換の3〜7員のO−Alk−複素環(任意選択で芳香族である)であり;
R3、R4は、互いに独立して、水素、ハロゲン、ヒドロキシアルキル、ヒドロキシル、アルコキシ、シアン、ニトロ、CF3、NHCOR、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、または保護ヒドロキシルであり;
Rは、アルキル、水素、ハロアルキル、ジハロアルキル、トリハロアルキル、CH2F、CHF2、CF3、CF2CF3、アリール、フェニル、ハロゲン、アルケニル、CN、NO2、またはOHであり;
R5およびR6は、互いに独立して、水素、フェニル、炭素数1〜6のアルキル基、3〜7員のシクロアルキル、3〜7員の複素環、5〜7員のアリールであり;あるいは、R5およびR6は、窒素原子を含む3〜7員環を形成し;
jおよびkは、互いに独立して、1〜4であり;
Alkは、炭素数1〜7の直鎖アルキル、炭素数1〜7の分岐鎖アルキル、または炭素数3〜8の環状アルキルである。
【0023】
本願明細書に記載の方法の別の実施形態では、式Iの化合物が式IAで表される:
【0024】
【化14】
【0025】
式中、R1、R2、R3、R4、jおよびkは、式Iに定義された通りである。
【0026】
一実施形態では、本発明は、以下の式IIの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象へのアンドロゲン遮断療法(ADT)により誘発されるほてりまたは重度のほてりの処置、発生率減少、抑制、重症度軽減、または阻害を行う方法を提供する。
【0027】
【化2】
【0028】
一実施形態では、本発明は、以下の式IIIの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象へのアンドロゲン遮断療法(ADT)により誘発されるほてりまたは重度のほてりの処置、発生率減少、抑制、重症度軽減、または阻害を行う方法を提供する。
【0029】
【化3】
【0030】
一実施形態では、本発明は、以下の式IVの化合物で表される化合物、またはその異性体、薬学的生成物、薬学的に許容される塩、多形体、水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象へのアンドロゲン遮断療法(ADT)により誘発されるほてりまたは重度のほてりの処置、発生率減少、抑制、重症度軽減、または阻害を行う方法を提供する。
【0031】
【化4】
【0032】
一実施形態では、本発明は、以下の式Vの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象へのアンドロゲン遮断療法(ADT)により誘発されるほてりまたは重度のほてりの処置、発生率減少、抑制、重症度軽減、または阻害を行う方法を提供する。
【0033】
【化5】
【0034】
一実施形態では、本発明は、以下の式VIの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象へのアンドロゲン遮断療法(ADT)により誘発されるほてりまたは重度のほてりの処置、発生率減少、抑制、重症度軽減、または阻害を行う方法を提供する。
【0035】
【化6】
【0036】
一実施形態では、本発明は、以下の式VIIの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象へのアンドロゲン遮断療法(ADT)により誘発されるほてりまたは重度のほてりの処置、発生率減少、抑制、重症度軽減、または阻害を行う方法を提供する。
【0037】
【化7】
【0038】
一実施形態では、本発明は、以下の式VIIIの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象へのアンドロゲン遮断療法(ADT)により誘発されるほてりまたは重度のほてりの処置、発生率減少、抑制、重症度軽減、または阻害を行う方法を提供する。
【0039】
【化8】
【0040】
一実施形態では、本発明は、以下の式IXの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、前立腺癌を患う男性対象へのアンドロゲン遮断療法(ADT)により誘発されるほてりまたは重度のほてりの処置、発生率減少、抑制、重症度軽減、または阻害を行う方法を提供する。
【0041】
【化9】
【0042】
一実施形態では、本発明は、以下の式Xの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象へのアンドロゲン遮断療法(ADT)により誘発されるほてりまたは重度のほてりの処置、発生率減少、抑制、重症度軽減、または阻害を行う方法を提供する。
【0043】
【化10】
【0044】
一実施形態では、本発明は、以下の式XIの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象へのアンドロゲン遮断療法(ADT)により誘発されるほてりまたは重度のほてりの処置、発生率減少、抑制、重症度軽減、または阻害を行う方法を提供する。
【0045】
【化11】
【0046】
一実施形態では、本発明は、以下の式XIIの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象へのアンドロゲン遮断療法(ADT)により誘発されるほてりまたは重度のほてりの処置、発生率減少、抑制、重症度軽減、または阻害を行う方法を提供する。
【0047】
【化12】
【0048】
一実施形態では、本発明は、式IA、I〜XIIの化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象へのアンドロゲン遮断療法(ADT)により誘発されるほてりまたは重度のほてりの処置、発生率減少、抑制、重症度軽減、または阻害を行う方法を提供する。別の実施形態では、男性被検者は前立腺癌を患っている。別の実施形態では、男性被検者は進行前立腺癌を患っている。別の実施形態では、化合物は化合物IVである。
【0049】
用語「進行前立腺癌」は、前立腺に起源を持ち、かつ精嚢、骨盤リンパ節、骨、または体の他の部分を含む周辺組織など前立腺を超えて幅広く転移している転移性癌のことを言う。前立腺癌の病状は、悪性度が増加する順に1〜5のグリソン分類で格付けされる。別の実施形態では、進行性疾患および/または前立腺癌による死亡の重大な危険性がある患者を定義に含める必要があり、IIBほどの低い病期で前立腺被膜の外に癌がある患者には、明らかに「進行」疾患がある。
【0050】
一実施形態では、本発明は、式IA、I〜XIIの化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、また任意の組み合わせを治療有効量投与することを含む、男性対象のほてりまたは重度のほてりの処置、発生率減少、抑制、重症度軽減、または阻害を行う方法を提供する。別の実施形態では、化合物は化合物IVである。
【0051】
一実施形態では、用語「ほてり(hot flash)」は、対象の体温の急上昇などの体温の一過性撹乱のことを言う。別の実施形態では、撹乱は発汗を伴う。一実施形態では、用語「ほてり」は、体、顔、および首の上部またはすべての紅潮、乳房、背中、および腕に現れる赤色斑、激しい発汗、寒気による震え、またはそれらの任意の組み合わせの突発性熱感のことを言う。一実施形態では、ほてりはヒト対象が経験し、別の実施形態では男性対象が経験する。一実施形態では、ほてりはADTの結果発生したものである。別の実施形態では、ほてりはADTの結果発生したものではない。一実施形態では、ほてりは前立腺癌処置の副作用である。別の実施形態では、ほてりは前立腺癌処置の副作用ではない。
【0052】
用語「重度のほてり」は、多量の発汗を引き起こし、異常な高熱を発生させ、長く持続し、行っている活動を妨げるほてりのことを言う。一実施形態では、重度のほてりは睡眠障害を特徴とする(不眠症を特徴とすることがよくある)。
【0053】
一実施形態では、ほてりを処置する本発明の方法を、例えば、閉経、酢酸タモキシフェン処置、前立腺癌処置、アルコール脱水素酵素欠損症、またはカルチノイド症候群/褐色細胞腫によって起こるほてりの処置に使用できる。各種類のほてりは、本発明の個別の実施形態を表す。
【0054】
本願明細書に示した通り、例えば化合物IVなど治療有効量の式Iの化合物を投与することで、対象のほてりを軽減したことを結果が示している。
【0055】
一実施形態では、本願明細書で説明されるような化合物および/またはそれを含む組成物を、男性対象の全血清テストステロン濃度を下げるために使用できる。
【0056】
一実施形態では、本願明細書で説明されるような化合物および/またはそれを含む組成物を、男性対象の全血清テストステロン濃度を下げるために使用でき、全血清テストステロンの低下は黄体形成ホルモン(LH)濃度の低下によって生じる。
【0057】
一実施形態では、本願明細書で説明されるような化合物および/またはそれを含む組成物を、男性対象の全血清テストステロン濃度を下げるために使用でき、全血清テストステロンの低下が黄体形成ホルモン濃度の低下と無関係である。
【0058】
一実施形態では、本発明は、式Iの構造で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象の全血清テストステロン濃度を下げる方法を提供する。
【0059】
【化1】
【0060】
式中、
Yは、C(O)またはCH2であり;
R1、R2は、互いに独立して、水素、ハロゲン、ヒドロキシル、アルコキシ、シアン、ニトロ、CF3、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、O−Alk−NR5R6、または、置換されたもしくは非置換の3〜7員のO−Alk−複素環(任意選択で芳香族である)であり;
R3、R4は、互いに独立して、水素、ハロゲン、ヒドロキシアルキル、ヒドロキシル、アルコキシ、シアン、ニトロ、CF3、NHCOR、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、または保護ヒドロキシルであり;
Rは、アルキル、水素、ハロアルキル、ジハロアルキル、トリハロアルキル、CH2F、CHF2、CF3、CF2CF3、アリール、フェニル、ハロゲン、アルケニル、CN、NO2、またはOHであり;
R5およびR6は、互いに独立して、水素、フェニル、炭素数1〜6のアルキル基、3〜7員のシクロアルキル、3〜7員の複素環、5〜7員のアリールであり;あるいは、R5およびR6は、窒素原子を含む3〜7員環を形成し;
jおよびkは、互いに独立して、1〜4であり;
Alkは、炭素数1〜7の直鎖アルキル、炭素数1〜7の分岐鎖アルキル、または炭素数3〜8の環状アルキルである。
【0061】
本願明細書に記載の方法の追加実施形態では、式Iの化合物が以下の式IAで表される。
【0062】
【化14】
【0063】
式中、R1、R2、R3、R4、jおよびkは、式Iに定義された通りである。
【0064】
一実施形態では、本発明は、式IIの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象の全血清テストステロン濃度を下げる方法を提供する。
【0065】
【化2】
【0066】
一実施形態では、本発明は、式IIIの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象の全血清テストステロン濃度を下げる方法を提供する。
【0067】
【化3】
【0068】
一実施形態では、本発明は、式IVの化合物で表される化合物、またはその異性体、薬学的生成物、薬学的に許容される塩、多形体、水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象の全血清テストステロン濃度を下げる方法を提供する。
【0069】
【化4】
【0070】
一実施形態では、本発明は、式Vの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象の全血清テストステロン濃度を下げる方法を提供する。
【0071】
【化5】
【0072】
一実施形態では、本発明は、式VIの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象の全血清テストステロン濃度を下げる方法を提供する。
【0073】
【化6】
【0074】
一実施形態では、本発明は、式VIIの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象の全血清テストステロン濃度を下げる方法を提供する。
【0075】
【化7】
【0076】
一実施形態では、本発明は、式VIIIの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象の全血清テストステロン濃度を下げる方法を提供する。
【0077】
【化8】
【0078】
一実施形態では、本発明は、式IXの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象の黄体形成ホルモン(LH)濃度を下げることで全血清テストステロン濃度を下げる方法を提供する。
【0079】
【化9】
【0080】
一実施形態では、本発明は、式Xの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象の全血清テストステロン濃度を下げる方法を提供する。
【0081】
【化10】
【0082】
一実施形態では、本発明は、式XIの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象の全血清テストステロン濃度を下げる方法を提供する。
【0083】
【化11】
【0084】
一実施形態では、本発明は、式XIIの化合物で表される化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象の全血清テストステロン濃度を下げる方法を提供する。
【0085】
【化12】
【0086】
一実施形態では、本発明は、式IA、I〜XIIの化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象の全血清テストステロン濃度を下げる方法を提供する。別の実施形態では、男性被検者は前立腺癌を患っている。別の実施形態では、全血清テストステロンを約100ng/dL未満に下げる。別の実施形態では、全血清テストステロンを約50ng/dL未満に下げる。別の実施形態では、全血清テストステロン濃度を約25ng/dL未満に下げる。
【0087】
一実施形態では、本発明は、式IA、I〜XIIの化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、全血清テストステロンの削減を血清黄体形成ホルモン(LH)濃度を下げることで行う、男性対象の全血清テストステロン濃度を下げる方法を提供する。別の実施形態では、男性被検者は前立腺癌を患っている。別の実施形態では、全血清テストステロンを約100ng/dL未満に下げる。別の実施形態では、全血清テストステロンを約50ng/dL未満に下げる。別の実施形態では、全血清テストステロン濃度を約25ng/dL未満に下げる。
【0088】
一実施形態では、本発明は、式IA、I〜XIIの化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、遊離血清テストステロンの削減を血清黄体形成ホルモン(LH)濃度を下げることで行う、男性対象の遊離血清テストステロン濃度を下げる方法を提供する。別の実施形態では、男性被検者は前立腺癌を患っている。
【0089】
一実施形態では、本発明は、式IA、I〜XIIの化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、全血清テストステロンの削減は血清黄体形成ホルモン(LH)濃度の削減と関係がない、男性対象の全血清テストステロン濃度を下げる方法を提供する。別の実施形態では、男性被検者は前立腺癌を患っている。別の実施形態では、全血清テストステロンを約100ng/dL未満に下げる。別の実施形態では、全血清テストステロンを約50ng/dL未満に下げる。別の実施形態では、全血清テストステロン濃度を約25ng/dL未満に下げる。
【0090】
一実施形態では、本発明は、式IA、I〜XIIの化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、遊離血清テストステロンの削減は血清黄体形成ホルモン(LH)濃度の削減と関係がない、男性対象の遊離血清テストステロン濃度を下げる方法を提供する。別の実施形態では、男性被検者は前立腺癌を患っている。
【0091】
一実施形態では、本発明は、男性対象の全血清テストステロンまたは遊離血清テストステロンの濃度を下げる方法を提供する。ここで、当該男性対象は前立腺癌を患っている。別の実施形態では、当該被検者は進行前立腺癌を患っている。
【0092】
一実施形態では、テストステロンの血清濃度の削減は可逆的であり、本発明の化合物による処置後には基準濃度に戻る。
【0093】
別の実施形態では、図23および実施例10に準じて、テストステロンの血清濃度は、化合物IVによる処置後に可逆的である。
【0094】
一実施形態では、本発明は、式IA、I〜XIIの化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象の全血清テストステロン濃度を下げる方法を提供する。別の実施形態では、全血清テストステロンを約100ng/dL未満に下げる。別の実施形態では、全血清テストステロンを約50ng/dL未満に下げる。別の実施形態では、全血清テストステロンを約25ng/dL未満に下げる。別の実施形態では、全血清テストステロンを約75ng/dL未満に下げる。別の実施形態では、全血清テストステロンを約75ng/dL〜100ng/dLに下げる。別の実施形態では、全血清テストステロンを約50ng/dL〜75ng/dLに下げる。別の実施形態では、全血清テストステロンを約40ng/dL〜50ng/dLに下げる。別の実施形態では、全血清テストステロン濃度を約25ng/dL〜50ng/dLに下げる。別の実施形態では、全血清テストステロンを約40ng/dL〜60ng/dLに下げる。
【0095】
テストステロンを「遊離」(すなわち、生物学的に利用可能かつ未結合)または「全」(タンパク結合し、利用不可な分を含む)血清濃度として測定できる。前立腺癌に罹ってなく、40歳を超える男性では、全テストステロン濃度が250ng/dL未満(<8.7nmol/L)に下がり、あるいは遊離テストステロン濃度が0.75ng/dL未満(<0.03nmol/L)に下がることを示す。
【0096】
一実施形態では、本発明は、前立腺癌を患う男性対象において、黄体形成ホルモン(LH)濃度を下げることと関係なく、あるいはLH濃度を下げることで、全血清および/または遊離テストステロン濃度を下げる方法を提供する。別の実施形態では、テストステロン濃度の変化は、処置前の濃度から下がる必要がある。別の実施形態では、全血清テストステロン濃度を100ng/dL未満に下げる。別の実施形態では、全血清テストステロンを50ng/dL未満に下げる。別の実施形態では、全血清テストステロンを25ng/dL未満に下げる。別の実施形態では、遊離テストステロン濃度を2ng/dL未満に下げる。別の実施形態では、遊離テストステロン濃度を1ng/dL未満に下げる。別の実施形態では、遊離テストステロン濃度を0.5ng/dL未満に下げる。別の実施形態では、遊離テストステロン濃度を0.25ng/dL未満に下げる。
【0097】
血液検査による処置期間中テストステロン濃度のモニタリングを含む、遊離血清テストステロン濃度および全血清テストステロン濃度を測定する方法。全テストステロンは、担体タンパク質(アルブミン、SHBG、トランスコルチン、トランスフェリン)に結合したテストステロンと遊離/未結合ホルモンとの計算の組み合わせである。全テストステロン濃度は、ホルモンを体内に運ぶ血中タンパク質濃度、年齢、肥満、およびよく用いられる検査方法と関係がある干渉を含むいくつかの要因の影響を受けることがある。
【0098】
遊離テストステロン(FT)の測定に利用可能な方法は、複雑(平衡透析および算出遊離テストステロン(CFT))に、またはアナログトレーサーを用いて簡素(市販FTキット「Coat−A−Count」)になり得る。別の実施形態では、全テストステロンおよび遊離テストステロンの濃度測定を、全テストステロンおよびSHBG(例えば、Irma−Count、DPC)の同時測定し、その後、算出遊離テストステロン(CFT)を導き出すことによって達成できる。別の実施形態では、全テストステロンおよび遊離テストステロンの測定は、当業者の知識に準じている。
【0099】
一実施形態では、本発明は、ADTの別の形体の1つ以上と、式IA、I〜XIIの化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせとの組み合わせを治療有効量投与することを含む、男性対象の全血清テストステロン濃度または遊離血清テストステロン濃度を下げる方法を提供する。別の実施形態では、全または遊離血清テストステロンの削減は、血清黄体形成ホルモン(LH)濃度を下げることで生じる。別の実施形態では、全または遊離血清テストステロン濃度の削減は、血清黄体形成ホルモン濃度を下げることと関係がない。
【0100】
本発明の方法は、ADTの他の形態と、本発明の化合物との組み合わせを投与することを含む。一実施形態では、ADTの他の形態として、LHRHアゴニストを含む。別の実施形態では、LHRH他動薬として、酢酸ロイプロリド(Lupron(登録商標))(参照することにより本書にすべて援用されるUS5,480,656;US5,575,987;5,631,020;5,643,607;5,716,640;5,814,342;6,036,976)、または酢酸ゴセレリン(Zoladex(登録商標))(参照することにより本書にすべて援用されるUS7,118,552;7,220,247;7,500,964)。一実施形態では、ADTの他の形態として、LHRHアンタゴニストを含む。別の実施形態では、LHRHアンタゴニストはデガレリクスを含む。一実施形態では、ADTの他の形態として、抗アンドロゲンを含む。別の実施形態では、抗アンドロゲンとして、ビカルタミド、フルタミド、フィナステライド、デュタステライド、MDV3100、ニルタミド、クロルマジノン、またはそれらの任意の組み合わせが挙げられる。
【0101】
一実施形態では、本発明の方法は、抗アンドロゲンと、本発明の化合物とを治療有効量投与することを含む。一実施形態では、本発明の方法は、LHRHアゴニストと、本発明の化合物とを治療有効量投与することを含む。一実施形態では、本発明の方法は、抗アンドロゲンと、LHRHアゴニストと、本発明の化合物とを治療有効量投与することを含む。
【0102】
一実施形態では、本発明は、式IA、I〜XIIの化合物を治療有効量投与することを含む、アンドロゲン遮断療法(ADT)を行うことを目的として、前立腺癌を患う男性対象において、黄体形成ホルモン(LH)濃度を下げることにより、あるいは黄体形成ホルモン濃度の下げることと関係なく、全血清テストステロン濃度および/または遊離テストステロン濃度を下げる方法を提供する。別の実施形態では、化合物は化合物IVである。
【0103】
別の実施形態では、本発明は、式IA、I〜XIIの化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、対象のアンドロゲン遮断療法(ADT)の方法を提供する。別の実施形態では、当該被検者は前立腺癌を患っている。別の実施形態では、化合物は化合物IVである。
【0104】
別の実施形態では、ADTは、進行前立腺癌を処置するために、前立腺癌の進行を遅らせるために、または前立腺癌の再発を予防および/または処置するために使用される。
【0105】
一実施形態では、本発明は、本発明の化合物を投与することを含む、前立腺癌を処置する、前立腺癌の進行を遅らせる、前立腺癌の再発を予防および/または処置する方法を提供する。別の実施形態では、LHRH類似体、可逆性抗アンドロゲン(ビカルタミドまたはフルタミドなど)、抗エストロゲン、抗癌剤、5−α還元酵素阻害薬、アロマターゼ阻害薬、プロゲスチン、選択的アンドロゲン受容体モジュレーター(SARMS)、または他の核ホルモン受容体を通じて作用する薬剤の併用による、本発明の化合物の投与。
【0106】
一実施形態では、本発明は、化合物IA、I〜XIIの化合物を投与することを含む、前立腺癌を処置する、並びにLH濃度を下げることで、あるいはLH濃度を下げることと関係なく、全血清テストステロンおよび/または遊離血清テストステロン濃度を下げる方法を提供する。別の実施形態では、化合物IVを投与する。
【0107】
アンドロゲン遮断療法ではテストステロンを削減するだけでなく、エストロゲンがテストステロンの芳香族化から生じるため、エストロゲン濃度も低くなる。アンドロゲン遮断療法により誘発されるエストロゲン欠乏は、ほてり、女性化乳房および乳房痛、骨量減少、骨の質および強度の低下、骨粗鬆症、骨減少症、および致命的な骨折、有害な脂質変化および高い心血管疾患および心筋梗塞、性欲減退、インポテンス、筋肉量の低下(サルコペニア)、疲労、認知機能傷害、および鬱病、およびその他の情緒の変化を含む重大な副作用を引き起こす。
【0108】
一実施形態では、本発明は、ADTに関連する疾患、障害、または症状を処置する方法を提供する。他の実施形態では、本発明は、テストステロンの欠乏に関連する疾患、障害、または症状を処置する方法を提供する。各疾病、障害または症状は、本発明の個別の実施形態を表す。
【0109】
一実施形態では、本発明は、式IA、I〜XIIの化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与することを含む、男性対象の全血清テストステロン濃度を下げる方法を示し、ここで、前記の当該式IA、I〜XIIの化合物、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを投与することで、アンドロゲン遮断療法(ADT)に関連する副作用の発症を予防、抑制、軽減、阻害、または処置を行い、当該対象は前立腺癌を患っている。別の実施形態では、全または遊離血清テストステロン濃度の削減は、LH濃度を下げることによる、またはLH濃度を下げることと関係がない。
【0110】
一実施形態では、本発明の化合物を投与することで、従来のアンドロゲン遮断療法(ADT)に関連する典型的な副作用の発症の抑制、軽減、阻害、または処置を行う。別の実施形態では、被検者は前立腺癌を患っている。前記副作用の予防および/または軽減は、プラセボ群または対照群と比べる。一実施形態では、従来のアンドロゲン遮断療法(ADT)に関連がある典型的な副作用としては、ほてり、女性化乳房、骨ミネラル濃度の減少、および骨折の増加が挙げられる。別の実施形態では、本発明の化合物を投与することで、従来型のアンドロゲン遮断療法(ADT)を用いて分かるように、ほてりの発症を予防する。別の実施形態では、本発明の化合物を投与することで、従来型のアンドロゲン遮断療法(ADT)を用いて分かるように、女性化乳房の発症を予防する。別の実施形態では、本発明の化合物を投与することで、従来型のアンドロゲン遮断療法(ADT)を用いて分かるように、骨ミネラル濃度(BMD)の減少の発症を予防する。別の実施形態では、本発明の化合物を投与することで、従来型のアンドロゲン遮断療法(ADT)を用いて分かるように、骨折の増加が生じるのを予防する。別の実施形態では、骨折の増加は、病的骨折、非外傷性骨折、脊椎骨折、非脊椎骨折、新たな形態学的骨折、臨床骨折、またはそれらの組み合わせである。
【0111】
一実施形態では、用語「従来のアンドロゲン遮断療法」は、外科医が睾丸を除去する睾丸摘出(去勢手術)を対象にする。別の実施形態では、用語「従来のアンドロゲン遮断療法」は、黄体形成ホルモン放出ホルモン(LHRH)類似体を対象にする。これらの薬剤は、睾丸で作られたテストステロンの量を下げる。米国で入手可能なLHRH類似体の例としては、ロイプロリド(Lupron、Viadur、Eligard)、ゴセレリン(Zoladex)、トリプトレリン(Trelstar)、およびヒストレリン(Vantas)が挙げられる。別の実施形態では、用語「従来のアンドロゲン遮断療法」は、抗アンドロゲン剤を投与することを対象とする。すなわち、抗アンドロゲンは、アンドロゲンを使用する体の能力を阻害する。睾丸摘出後でも、あるいはLHRH類似体による処置中でも、少量のアンドロゲンが副腎でまだ作られる。抗アンドロゲン剤の例としては、フルタミド(Eulexin)、ビカルタミド(Casodex)、およびニルタミド(Nilandron)が挙げられる。別の実施形態では、用語「従来のアンドロゲン遮断療法」は、アバレリクス(Plenaxis)などの黄体形成ホルモン放出ホルモン(LHRH)アンタゴニストを投与することを対象とする。デガレリクス(Firmasgon)は、2008年にFDAによって進行前立腺癌の処置用に認可された新規のLHRHアンタゴニストである。別の実施形態では、用語「従来のアンドロゲン遮断療法」は、フィナステライド(Proscar)およびデュタステライド(Avodart)などの5α−還元酵素阻害薬を投与することを対象にする。5α−還元酵素阻害薬は、テストステロンをより活性なアンドロゲン、5α−ジヒドロテストステロン(DHT)に変換する体の機能を阻害する。別の実施形態では、用語「従来のアンドロゲン遮断療法」は、ケトコナゾール(Nizoral)などのテストステロン生合成の阻害薬を投与することを対象にする。別の実施形態では、用語「従来のアンドロゲン遮断療法」は、ジエチルスチルベストロールまたは17βエストラジオールなどのエストロゲンを投与することを対象にする。
【0112】
一実施形態では、用語「女性化乳房」とは、腺構成要素の増殖によって生じる男性の乳房の良性肥大のことを言い、痛みと関係がある場合も、無い場合もある。女性化乳房は、ゴム状の存在、または乳頭から同心円状に存在する硬い腫瘤によって臨床的に定義される。偽性女性化乳房または脂肪腫乳房として公知の症状は、腺の増殖のない脂肪堆積を特徴とする。通常、女性化乳房は左右対称であるが、前記症状は片側だけの場合がある。
【0113】
一実施形態では、本発明の方法は、骨量の減少およびほてりも生じること無く、テストステロンの削減による前立腺癌または進行前立腺癌の男性対象の処置を対象とする。別の実施形態では、本発明の方法は、化合物IA、I〜XIIを使用し、当該化合物には、前立腺癌に対する現在のアンドロゲン遮断療法(ADT)でよく見られる骨量減少およびほてりなどの特定の副作用も生じることなく、前立腺癌の主要刺激剤であるテストステロンを削減する能力がある。
【0114】
別の実施形態では、後述の表8(実施例11)によって、化合物IVを投与することで骨量減少も生じることなく、テストステロンを削減することを示す。
【0115】
一実施形態では、本願明細書で示される方法および/または本願明細書で示される化合物を利用することは、視床下部・下垂体・精巣軸(HPT軸)に関するフィードバックの提供において効果的である。フィードバックとは、自らの活動に影響を及ぼす別の臓器または組織の活動を調整する、1つの臓器または組織で生成される物質の能力のことを言う。一実施形態では、視床下部・下垂体・精巣軸(HPT軸)に関するフィードバックによって、LH濃度を削減する。一実施形態では、視床下部・下垂体・精巣軸(HPT軸)に関するフィードバックによって、全血清テストステロン濃度を削減する。一実施形態では、視床下部・下垂体・精巣軸(HPT軸)に関するフィードバックによって、遊離血清テストステロン濃度を削減する。一実施形態では、視床下部・下垂体・精巣軸(HPT軸)に関するフィードバックによって、血清、組織、または腫瘍のアンドロゲン濃度を削減する。
【0116】
視床下部・下垂体・精巣(HPT)軸とは、視床下部、下垂体、および睾丸のホルモン濃度を調整する内分泌生理学系のことを言う。LHRH(黄体形成ホルモン放出ホルモン)は視床下部によって放出され、LHおよびFSH(ゴナドトロピン)を合成および分泌するように下垂体を刺激する。その後、LHおよびFSHは睾丸に作用し、テストステロンおよび精液の産生を刺激する。その後、テストステロンは、視床下部LHRH分泌に対して直接的な負のフィードバック効果、および下垂体LHおよびFSH産生に対して間接的な負のフィードバック効果を持つ。エストロゲン、アンドロゲン、および血清タンパク質(インヒビンなど)も、LHRH分泌およびLHおよびFSHの分泌に対して負のフィードバックを持つ。
【0117】
下垂体は、体内のテストステロン濃度を調節する1つの腺である。テストステロン濃度が低い場合、下垂体が黄体形成ホルモン(LH)を放出する。このホルモンは、より多くのテストステロンを作るように睾丸を誘導する。思春期の間、テストステロンの濃度は増加する。テストステロンの濃度は約20〜40歳で最高になり、その後、高齢男性では徐々に低くなる。女性は、男性と比較してずっと少ない量のテストステロンを体内に持つ。しかし、男性と女性の両方において、テストステロンは体の隅々で重要な役割を果たす。脳、骨、および筋肉の量、脂肪分布、血管系、エネルギー準位、生殖器組織、および性機能に影響を及ぼす。血中のテストステロンの大部分は、性ホルモン結合グロブリン(SHBG)と呼ばれるタンパク質に、またはアルブミンと呼ばれる別の血清タンパク質に結合している。結合していない(または「遊離」)テストステロンも臨床的に測定できる。
【0118】
別の実施形態では、血清黄体形成ホルモン濃度を下げることと関係がない全血清テストステロンまたは遊離血清テストステロンの濃度の削減は、性ホルモン結合グロブリン(SHBG)の増加による。別の実施形態では、血清黄体形成ホルモン濃度を下げることと関係がない遊離テストステロン濃度の削減は、性ホルモン結合グロブリン(SHBG)の増加による。別の実施形態では、血清黄体形成ホルモン(LH)濃度を下げることと関係がない全血清または遊離血清テストステロンの濃度の削減は、睾丸のライディッヒ細胞によるテストステロン産生または分泌の阻害による。別の実施形態では、血清黄体形成ホルモン(LH)濃度を下げることと関係がない全血清または遊離血清テストステロン濃度の低下は、副腎ステロイド産生の減少による。
【0119】
一実施形態では、本願明細書で説明されるような化合物および/またはそれを含む組成物を、黄体形成ホルモン(LH)濃度を下げるために使用できる。別の実施形態では、本発明の化合物および/または組成物を、内因性の性ホルモンを減らすために使用できる。
【0120】
ヒドロキシステロイド脱水素酵素(HSD)群の構成要素は、血中ステロイドの変換に関与する。17β−HSD5は、アンドロステンジオンをテストステロンに、エストロンをエストラジオールに変換する。さらに、プロスタグランジン合成にも関与する。一実施形態では、本発明の化合物は、HSD、特に17β−ヒドロキシステロイド脱水素酵素5(17β−HSD5)を阻害する。前記阻害は、末梢/性腺外テストステロン合成を防止することで、ADTにおいて有益な場合があり、前記合成によって、HPT軸制御を避け、全または遊離血清テストステロンの不完全な削減を生じる、あるいは細胞内テストステロン濃度の局所的な上昇を可能にし、そのいずれかがADTでは有害である可能性がある。
【0121】
LHRHアゴニスト治療、すなわち黄体形成ホルモン放出ホルモンアゴニスト(LHRH)またはその類似体の投与によった達成されるアンドロゲン遮断療法(ADT)によって、下垂体からのゴナドトロピン放出、および睾丸からのテストステロン産生(「発赤反応」と呼ばれる)の初期刺激をもたらし、続いてゴナドトロピン放出の減少、およびテストステロンとエストロゲン両方の濃度の減少をもたらす。LHRHアゴニスト療法によって生じる「発赤反応」は、アンドロゲン/テストステロン濃度の増加によって前立腺癌の処置に悪影響をもたらす。さらに、LHRH療法は、糖尿病と心血管疾患のリスク増加に関連があった(Smith(2008)Current Prostate Reports.6:149-154)。
【0122】
LHRH療法の発赤の影響を克服する努力において、抗アンドロゲン単剤療法(ビカルタミド、フルタミド、クロルマジノン)、LHRH/抗アンドロゲン併用療法、およびLHRHアンタゴニスト(デガレリクス)が提案されている(Suzuki et al., (2008) Int. J. Clin. Oncol. 13: 401-410; Sharifi, N. et al., (2005) JAMA. 294(2): 238-244)。抗アンドロゲン単剤療法では、対象のアンドロゲン濃度を下げない。ビカルタミド抗アンドロゲン単剤療法では、骨転移のある前立腺癌患者においてADTより効果が低いことが示された。さらに、ビカルタミド療法で見られる副作用としては、乳房の圧痛および乳房肥大(女性化乳房および乳房痛)が挙げられる(Suzuki et al., ibid)。抗アンドロゲン療法による付加的リスクとしては、肝臓アミノ基転移酵素の増加が挙げられる(Sharifi et al., ibid)。
【0123】
一実施形態では、本発明は、LH濃度を低下させ、その結果、「発赤」の影響を生じることなく、かつ従来のADT法を用いてテストステロン低下によって生じるエストロゲン不足な関連がある副作用を克服しながら、全血清テストステロンおよび/または遊離血清テストステロン濃度を低下させる。方法/対象化合物の使用によって、骨組織の維持(骨組織に対するアゴニスト作用)、トロンビン産生能および/またはほてりの低下、および/またはエストラジオールまたはジエチルスチルベストロールよりも乳房組織に対する少ない影響、または中立的影響を示す組織選択的エストロゲン活性を提供する。
【0124】
一実施形態では、化合物IVはアゴニスト作用を示すが、アンタゴニスト作用は示さず(実施例6および7)、そのため、化合物IVはゴナドトロピンおよびテストステロンの増加を生じないだろう。
【0125】
一実施形態では、化合物IVは、血清ホルモン、テストステロン、および全アンドロゲンを下げる堅牢な薬理反応を示すアゴニスト活性(実施例8〜11)を示す。
【0126】
一実施形態では、本願明細書で示される化合物および/または組成物を利用する本願明細書で示される方法は、従来の形態のADTを用いたLHの削減によって生じる骨吸収作用の削減または除去において効果的である。一実施形態では、本願明細書で示される方法および/または本願明細書で示される組成物の利用は、従来の形態のADTを用いたテストステロン濃度の削減によって生じる骨吸収作用の削減または除去において効果的である。一実施形態では、本願明細書で示される組成物を利用した本願明細書で示される方法は、LH濃度低下の結果としてのエストロゲンの削減によって生じる骨吸収作用の削減または除去において効果的である。一実施形態では、本願明細書で示される化合物および/または組成物を利用する本願明細書で示される方法は、従来の形態のADTを用いたLH濃度低下に関係がある骨吸収作用を予防する。一実施形態では、本願明細書で示される化合物および/または組成物を利用する本願明細書で示される方法は、従来の形態のADTを用いた内因性LH、テストステロン、および/またはエストラジオールの低下に関係がある骨吸収作用を予防する。一実施形態では、本願明細書で示される化合物および/または組成物を利用する本願明細書で示される方法は、LH濃度の低下を示しながら、骨ミネラル濃度(BMD)を増加させる。一実施形態では、本願明細書で示される化合物および/または組成物を利用する本願明細書で示される方法は、内因性LH、テストステロン、および/またはエストラジオール濃度の低下を示しながら、パーセント骨量を増加させる。
【0127】
一部の実施形態では、本発明は、本発明の化合物、またはその異性体、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを投与することで、血栓塞栓症を避ける、および/または軽減する方法を提供する。
【0128】
一実施形態では、本願明細書で示される化合物および/または組成物を利用する本願明細書で示される方法は、乳房組織に効果的である。一実施形態では、本願明細書で示される化合物および/または組成物を利用する本願明細書で示される方法は、従来のADTによって達成されるLH濃度の低下に関係がある女性化乳房を予防しながら、LH濃度を低下させる。
【0129】
一実施形態では、実施例13によって特別な毒性研究を開示し、ヒト血小板によるそのインビトロ研究から、化合物IVがDESよりもずっと低い前凝固性活性を示すことが分かった。そのため、化合物IV、ER選択的アゴニストは、DESよりも血栓症の低いリスクでDESによる前立腺癌処置の利点を実現する必要があり、骨量減少、ほてり、または有害な脂質状態の変化を生じることなくLHRHアゴニストまたはアンタゴニストの利点も実現する必要がある。
【0130】
ジエチルスチルベストロール(DES)療法単独またはADTとの併用によって、DESが前立腺癌患者の骨吸収を予防することを示した。DESの使用は、前立腺癌用の治療として推進されてきたが、血管形成および悪性腫瘍に対するDESの影響は、DES代謝物によって仲介されると考えられ、エストロゲン受容体を通じて作用するとは考えられていない。さらに、治療用に投与されるDESの用量レベルは、血管疾患、心血管疾患の罹患率、血栓毒性、女性化乳房、勃起障害、および性欲減退を含む多数の副作用を示す(Scherr and Pitts, ibid and Presti, J.C. Jr. (1996) JAMA. 275(15): 1153-6)。
【0131】
一実施形態では、本発明は、LHRHアゴニストまたはアンタゴニスト療法の単独または抗アンドロゲン剤またはDESとの併用の副作用を克服する。別の実施形態では、対象発明の方法によって、有害な骨関連の症状などの有害なエストロゲン遮断副作用なしに、および女性化乳房などの有害なエストロゲン刺激副作用なしにアンドロゲン遮断療法を提供する。別の実施形態では、本発明の方法は、LH濃度を低下させ、その結果、「発赤」の影響を生じることなく、LHの低下によって生じるエストロゲン不足に関連がある副作用を克服し、並びにDES療法で見られる一般的なエストロゲンアゴニストの増加に関連がある副作用を克服しながら、総血清テストステロン濃度および/または遊離血清テストステロン濃度を低下させる。方法/対象化合物の使用によって、組織選択的エストロゲン活性を提供し、その結果、骨組織の維持(骨組織に対するアゴニスト作用)、トロンビン産生能の低下、乳房組織に対する中立的影響を示す。
【0132】
視床下部濃度でのタモキシフェン、トレミフェン、およびラロキシフェンなどの従来の選択的エストロゲン受容体モジュレーター(SERMs)の抗エストロゲン効果によって、男性でゴナドトロピン濃度の増加、またはLH濃度の増加を生じ、その結果、テストステロン血清濃度の増加を生じる可能性がある(Tsouri et al., 2008, Fertility and Sterility doi: 10.1016)。その一方、本発明の方法は、式I〜XIIの化合物を投与することを含み、男性対象のLHを低下させる。
【0133】
式Iの化合物の追加実施形態
【0134】
本発明の方法の一実施形態では、式Iの化合物のYがC(O)である。別の実施形態では、YがCH2である。別の実施形態では、式IまたはIAの化合物のR1およびR2は、互いに独立して、O−Alk−NR5R6またはO−Alk−複素環である。別の実施形態では、本願明細書の前述のように前記のO−Alk−複素環、O−Alk−NR5R6、−Alk−複素環、およびAlk−NR5R6のAlkは、炭素数1〜7の直鎖アルキル、炭素数1〜7の分岐鎖アルキル、または炭素数3〜8の環状アルキルである。別の実施形態では、アルキルはエチレン(−CH2CH2−)である。別の実施形態では、Alkはメチレン(−CH2−)である。別の実施形態では、Alkはプロピレン(−CH2CH2CH2−)である。別の実施形態では、Alkは2−メチルプロピレン(−CH2CH(CH3)CH2−)である。
【0135】
本発明の方法の一実施形態では、式IまたはIAの化合物のR1がパラ位にある。本発明の方法の一実施形態では、式IまたはIAの化合物のR1とR2は異なる。本発明の方法の別の実施形態では、式IまたはIAの化合物のR1とR2は同じである。本発明の方法の別の実施形態では、式IまたはIAの化合物のR1は、
【0136】
【化13】
【0137】
である。方法の別の実施形態では、式IまたはIAの化合物のR1は、ヒドロキシルである。方法の別の実施形態では、式IまたはIAの化合物のR1は、ヒドロキシルである。方法の別の実施形態で、R1、およびR2は、互いに独立して、水素、ハロゲン、ヒドロキシル、アルコキシ、シアン、ニトロ、CF3、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、O−Alk−NR5R6、または複素環が3〜7員置換または未置換の複素環であるO−Alk−複素環である。方法の別の実施形態で、式IまたはIAの化合物のR1およびR2は、互いに独立して、ハロゲン、ヒドロキシル、アルコキシ、シアン、ニトロ、CF3、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、O−Alk−NR5R6、または、置換されたもしくは非置換の3〜7員のO−Alk−複素環(任意選択で芳香族である)。方法の別の実施形態では、式IまたはIAの化合物のR2は、ハロゲンである。方法の別の実施形態では、式IまたはIAの化合物のR2は、Fである。方法の別の実施形態では、式Iの化合物のR2は、Clである。方法の別の実施形態では、式IまたはIAの化合物のR2は、Brである。方法の別の実施形態では、式IまたはIAの化合物のR2は、Iである。方法の別の実施形態では、式IまたはIAの化合物のR2は、ヒドロキシルである。方法の別の実施形態で、R1および/またはR2はCF3である。別の実施形態で、R1および/またはR2はCH3である。別の実施形態で、R1および/またはR2はハロゲンである。別の実施形態で、R1および/またはR2はFである。別の実施形態で、R1および/またはR2はClである。別の実施形態で、R1および/またはR2はBrである。別の実施形態で、R1および/またはR2はIである。別の実施形態で、式Iの化合物のR2はパラ位にある。
【0138】
本発明の方法の一実施形態では、式IまたはIAの化合物のR3とR4は同じである。本発明の方法の別の実施形態では、式IまたはIAの化合物のR3とR4は異なる。方法の別の実施形態では、式IまたはIAの化合物のjおよびkは、互いに独立して、1である。方法の別の実施形態では、式IまたはIAの化合物のR3およびR4は、互いに独立して、ハロゲン、ハロアルキル、ヒドロキシル、またはアルキルである。方法の別の実施形態では、式IまたはIAの化合物のR3およびR4は、互いに独立して、Fである。方法の別の実施形態では、式IまたはIAの化合物のR3およびR4は、互いに独立して、Brである。方法の別の実施形態では、式IまたはIAの化合物のR3およびR4は、互いに独立して、Clである。別の実施形態では、R4はパラ位にある。別の実施形態では、R3はオルト位にある。別の実施形態では、R3はメタ位にある。別の実施形態で、R3および/またはR4はCF3である。別の実施形態で、R3および/またはR4はCH3である。
【0139】
本発明の方法の一実施形態では、式IまたはIAの化合物のR5とR6は、窒素原子を含む3〜7員環を形成する。別の実施形態では、環が飽和または不飽和環である。別の実施形態では、環が置換または非置換環である。本発明の方法の別の実施形態では、式IまたはIAの化合物のR5およびR6は、窒素原子を含むピペリジン環を形成する。方法の別の実施形態では、式IまたはIAの化合物のR5およびR6は、窒素原子を含むピラジン環を形成する。方法の別の実施形態では、式IまたはIAの化合物のR5およびR6は、窒素原子を含むピペラジン環を形成する。方法の別の実施形態では、式IまたはIAの化合物のR5およびR6は、窒素原子を含むモルホリン環を形成する。方法の別の実施形態では、式IまたはIAの化合物のR5およびR6は、窒素原子を含むピロール環を形成する。方法の別の実施形態では、式IまたはIAの化合物のR5およびR6は、ピロリジンを形成する。方法の別の実施形態では、式IまたはIAの化合物のR5およびR6は、窒素原子を含むピリジン環を形成する。別の実施形態では、環は、ハロゲン、アルキル、アルコキシ、ヒドロキシル、シアノ、ニトロ、アミノ、アミド、COOH、またはアルデヒドで置換される。
【0140】
本発明の方法の別の実施形態では、式IまたはIAの化合物のR1と、式IまたはIAの化合物のR2は、互いに独立して、O−Alk−複素環またはOCH2CH2−複素環である。別の実施形態では、用語「複素環」基とは、環の一部として炭素原子に加えて、硫黄、水素、窒素、またはそれらの任意の組み合わせを含む、一実施形態での環構造のことを言う。別の実施形態では、複素環は3〜12員環である。別の実施形態では、複素環は6員環である。別の実施形態では、複素環は5〜7員環である。別の実施形態では、複素環は4〜8員環である。別の実施形態では、複素環基は非置換であるか、またはハロゲン、ハロアルキル、ヒドロキシル、アルコキシ、カルボニル、アミド、アルキルアミド、ジアルキルアミド、シアノ、ニトロ、CO2H、アミノ、アルキルアミノ、カルボキシル、チオ、および/またはチオアルキルで置換されてもよい。別の実施形態では、複素環は、別の飽和または不飽和シクロアルキルまたは3〜8員複素環と融合されることがある。別の実施形態では、複素環が飽和環である。別の実施形態では、複素環が不飽和環である。別の実施形態では、複素環がピペリジンである。別の実施形態では、複素環がピリジンである。別の実施形態では、複素環がピペリジン、ピリジン、フラン、チオフェン、ピロール、ピロリジン、ピラジン、ピペラジン、またはピリミジンである。
【0141】
用語「シクロアルキル」は、炭素と水素の原子を含む非芳香族環、単環、または多環のことを言う。シクロアルキル基は、環がその存在によって芳香族環の状態にならない限りは、環に1つ以上の炭素−炭素二重結合を持ってもよい。シクロアルキル基の例としては、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシルおよびシクロヘプチルなどの炭素数3〜7のシクロアルキル基、飽和環式および二環式テルペン、シクロプロペニル、シクロブテニル、シクロペンテニル、シクロヘキセニル、およびシクロヘプテニルなど炭素数3〜7のシクロアルケニル基、不飽和環式および二環式テルペンが挙げられるが、これに限定されるものではない。シクロアルキル基は、非置換、または1つまたは2つの置換基で置換されてもよい。好適には、シクロアルキル基は単環または二環である。
【0142】
一実施形態では、用語「アルキル」とは、直鎖、分岐鎖、および環式アルキル基を含む飽和脂肪族炭化水素のことを言う。一実施形態では、アルキル基は1〜12個の炭素を持つ。別の実施形態では、アルキル基は1〜7個の炭素を持つ。別の実施形態では、アルキル基は1〜6個の炭素を持つ。別の実施形態では、アルキル基は1〜4個の炭素を持つ。別の実施形態では、環式アルキル基は3〜8個の炭素を持つ。別の実施形態では、環式アルキル基は3〜12個の炭素を持つ。別の実施形態では、分岐アルキルは、1〜5個の炭素のアルキル側鎖で置換されたアルキルである。別の実施形態では、分岐アルキルは、1〜5個の炭素のハロアルキル側鎖で置換されたアルキルである。アルキル基は非置換であるか、またはハロゲン、ハロアルキル、ヒドロキシル、アルコキシ、カルボニル、アミド、アルキルアミド、ジアルキルアミド、ニトロ、アミノ、アルキルアミノ、ジアルキルアミノ、カルボキシル、チオ、および/またはチオアルキルで置換されてもよい。
【0143】
別の実施形態では、「アルケニル」基とは、直鎖、分岐鎖、および1つ以上の二重結合を有する環式基を含む、不飽和炭化水素のことを言う。アルケニル基は、1つの二重結合、2つの二重結合、3つの二重結合などを有してもよい。別の実施形態では、アルケニル基は2〜12個の炭素を持つ。別の実施形態では、アルケニル基は2〜6個の炭素を持つ。別の実施形態では、アルケニル基は2〜4個の炭素を持つ。アルケニル基の例としては、エテニル、プロペニル、ブテニル、シクロヘキセニルなどである。アルケニル基は非置換であるか、またはハロゲン、ヒドロキシル、アルコキシカルボニル、アミド、アルキルアミド、ジアルキルアミド、ニトロ、アミノ、アルキルアミノ、ジアルキルアミノ、カルボキシル、チオ、および/またはチオアルキルで置換されてもよい。
【0144】
「アリール」基は、少なくとも1つの炭素環式芳香族基または複素環式芳香族基を有する芳香族基のことを言い、非置換であるか、またはハロゲン、ハロアルキル、ヒドロキシル、アルコキシカルボニル、アミド、アルキルアミド、ジアルキルアミド、ニトロ、アミノ、アルキルアミノ、ジアルキルアミノ、カルボキシル、あるいはチオ、またはチオアルキルから選ばれる1つ以上の基で置換されてもよい。アリール環の限定されない例としては、フェニル、ナフチル、ピラニル、ピロリル、ピラジニル、ピリミジニル、ピラゾリル、ピリジニル、フラニル、チオフェニル、チアゾリル、イミダゾリル、イソオキサゾリルなどである。一実施形態では、アリール基は4〜8員環である。別の実施形態では、アリール基は4〜12員環である。別の実施形態では、アリール基は6員環である。別の実施形態では、アリール基は5員環である。別の実施形態では、アリール基は2〜4個の縮合環系である。
【0145】
一実施形態では、「アルデヒド」基とは、ホルミル基で置換されたアルキルまたはアルケニルのことを言い、アルキルまたはアルケニルは本書の上で定義したとおりである。別の実施形態では、アルデヒド基は、ホルミル基で置換されたアリール基またはフェニル基であり、アリールは本書の上で定義したとおりである。アルデヒドの例としては、ホルミル、アセタール、プロパナール、ブタナール、ペンタナール、ベンズアルデヒドである。別の実施形態では、アルデヒド基はホルミル基である。
【0146】
別の実施形態では、「ハロアルキル」基は、上に定義したようなアルキル基のことを言い、このアルキル基は1つ以上のハロゲン原子、例えばF、Cl、Br、またはIで置換される。
【0147】
別の実施形態では、「ヒドロキシル」基はOH基のことを言う。本発明の化合物のR1、R2、またはR3がORである場合、RはOHではないことは、当業者には明らかである。
【0148】
一実施形態では、用語「ハロゲン」または「ハロ」は、F、Cl、Br、またはIなどのハロゲンのことを言う。
【0149】
別の実施形態では、語句「フェノール」は、ベンゼンのアルコール(OH)誘導体のことを言う。
【0150】
一部の実施形態では、保護ヒドロキシルへの言及には、ベンゼン環の酸素部分に結合した置換基の取り込みを含み、当該置換基は容易に取り除くことができる。一部の実施形態では、フェノール保護基が以下を含む場合がある。すなわち、メチルエーテル(メトキシ)、アルキルエーテル(アルコキシ)、ベンジルエーテル(Bn)、メトキシメチル(MOM)エーテル、ベンゾイルオキシメチル(BOM)エーテル、ベンジル、カルボベンゾキシ、メトキシエトキシメチル(MEM)エーテル、2−(トリメチルシリル)エトキシメチル(SEM)エーテル、メチルチオメチル(MTM)エーテル、フェニルチオメチル(PTM)エーテル、アジドメチルエーテル、シアノメチルエーテル、2,2−ジクロロ−1,1−ジフルオロエチルエーテル、2−クロロエチルエーテル、2−ブロモエチルエーテル、テトラヒドロピラニル(THP)エーテル、1−エトキシエチル(EE)エーテル、フェナシルエーテル、4−ブロモフェナシルエーテル、シクロプロピルメチルエーテル、アリルエーテル、プロパルギルエーテル、イソプロピルエーテル、シクロヘキシルエーテル、t−ブチルエーテル、2,6−ジメチルベンジルエーテル、4−メトキシベンジルエーテル、o−ニトロベンジルエーテル、2,6−ジクロロベンジルエーテル、3,4−ジクロロベンジルエーテル、4−(ジメチルアミノ)カルボニルベンジルエーテル、4−メチルスルフィニルベンジルエーテル、4−アントリルメチルエーテル、4−ピコリルエーテル、ヘプタフルオロ−p−トリル、テトラフルオロ−4−ピリジルエーテル、トリメチルシリル(TMS)エーテル、t−ブチルジメチルシリル(TBDMS)エーテル、t−ブチルジフェニルシリル(TBDPS)エーテル、トリイソプロピルシリル(TIPS)エーテル、ギ酸アリール、酢酸アリール、レブリン酸アリール、アリールピバレート、安息香酸アリール、アリール9−フルオレンカルボキシレート、アリールメチルカルボネート、1−アダマンチルカルボネート、t−ブチルカルボネート、4−メチルスルフィニルベンジルカルボネート、2,4−ジメチルペンタ−3−イルカルボネート、アリール2,2,2−トリクロロエチルカルボネート、アリールベンジルカルボネート、アリールカルバメート、ジメチルホスフィニルエステル(Dmp−OAr)、ジメチルホスフィノチオニルエステル(Mpt−OAr)、ジフェニルホスフィノチオニルエステル(Dpt−OAr)、メタンスルホン酸アリール、トルエンスルホン酸アリール、または2−ホルミルベンゼンスルホン酸アリール。
【0151】
一実施形態では、本発明の方法は、N,N−ビス(4−ヒドロキシフェニル)−4−プロピルベズアミド(II)、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを使用する。別の実施形態では、本発明の方法は、4,4´−(2,3−ジメチル−ベンジルアザンジイル)ジフェノール(III)、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを使用する。別の実施形態では、本発明の方法は、3−フルオロ−N−(4−フルオロフェニル)−4−ヒドロキシ−N−(4−ヒドロキシフェニル)ベズアミド(IV)、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを使用する。別の実施形態では、本発明の方法は、N,N−ビス(4−ヒドロキシフェニル)−2,3−ジメチルベズアミド(V)、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを使用する。別の実施形態では、本発明の方法は、N,N−ビス(4−ヒドロキシフェニル)−2−ナフチルズアミド(VI)、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを使用する。別の実施形態では、本発明の方法は、3−フルオロ−4−ヒドロキシ−N,N−ビス(4−ヒドロキシフェニル)−ベズアミド(VII)、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを使用する。別の実施形態では、本発明の方法は、4−((4−フルオロフェニル)(4−ヒドロキシベンジル)アミノ)フェノール(VIII)、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを使用する。別の実施形態では、本発明の方法は、4−フルオロ−N−(4−ヒドロキシ−フェニル)−N−[4−(2−ピペリジン−1−イル−エトキシ)−フェニル]−2−トリフルオロメチル−ベズアミド(IX)、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを使用する。別の実施形態では、本発明の方法は、IXの塩酸塩(IXのHCl塩)または塩酸4−フルオロ−N−(4−ヒドロキシ−フェニル)−N−[4−(2−ピペリジン−1−イル−エトキシ)−フェニル]−2−トリフルオロメチル−ベズアミド(X)、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを使用する。別の実施形態では、本発明の方法は、3−フルオロ−4−ヒドロキシ−N−(4−ヒドロキシフェニル)−N−フェニルベズアミド(XI)、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを使用する。別の実施形態では、本発明の方法は、3−フルオロ−N,N−ビス−(4−ヒドロキシ−フェニル)−2−メチル−ベズアミド(XII)、またはその異性体、薬学的に許容される塩、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを使用する。
【0152】
一実施形態では、本発明の方法は、化合物の「薬学的に許容される塩」を使用し、この塩は、本発明の化合物の酸または塩基との反応によって生成することができる。
【0153】
本発明の方法の化合物のアミンの好適な薬学的に許容される塩は、無機酸または有機酸から調製できる。一実施形態では、アミンの無機塩の例としては、重硫酸塩、ホウ酸塩、臭化物、塩化物、ヘミ硫酸塩、臭化水素酸塩、塩酸塩、2−ヒドロキシエチルスルホン酸塩(スルホン酸ヒドロキシエタン)、ヨウ素酸塩、ヨウ化物、イソチオン酸塩、硝酸塩、過硫酸塩、リン酸塩、硫酸塩、スルファミン酸塩、スルファニル酸塩、スルホン酸(スルホン酸アルキル、スルホン酸アリール、ハロゲン置換スルホン酸アルキル、ハロゲン置換スルホン酸アリール)、スルホン酸塩、およびチオシアン酸塩である。
【0154】
一実施形態では、アミンの有機塩の例は、有機酸の脂肪族、脂環式、芳香族、芳香脂肪族、複素環、カルボン酸、およびスルホン酸類から選択でき、それらの例は、酢酸塩、アルギニン、アスパラギン酸塩、アスコルビン酸塩、アジピン酸塩、アントラニル酸塩、アルギン酸塩、カルボン酸アルカン、置換カルボン酸アルカン、アルギン酸塩、ベンゼンスルホン酸塩、安息香酸エステル塩、重硫酸塩、酪酸塩、重炭酸塩、酒石酸水素塩、カルボン酸塩、クエン酸塩、樟脳酸塩、樟脳スルホン酸塩、シクロヘキシルスルファミン酸塩、シクロペンタンプロピオン酸塩、エデト酸カルシウム塩、カンシル酸塩、炭酸塩、クラブラン酸塩、桂皮酸塩、ジカルボン酸塩、ジグルコン酸塩、ドデシルスルホン酸塩、二塩酸塩、デカン酸塩、エナンタート、エタンスルホン酸塩、エデト酸塩、エジシル酸塩、エストレート、エシレート、フマル酸塩、ギ酸塩、フッ化物、ガラクツロン酸塩、グルコン酸塩、グルタミン酸塩、グリコール酸塩、グルカ酸、グルコヘプタン酸塩、グリセロリン酸塩、グルセプテート、グリコリルアルサニレート、グルタル酸塩、グルタミン酸塩、ヘプタン酸塩、ヘキサン酸塩、ヒドロキシマレイン酸塩、ヒドロキシカルボン酸、ヘキシルレゾルシン酸塩、ヒドロキシ安息香酸塩、ヒドロキシナフトエ酸塩、フッ化水素酸塩、乳酸塩、ラクトビオン酸塩、ラウリン酸塩、リンゴ酸塩、マレイン酸塩、メチレンビス(β−オキシナフトエート)、マロン酸塩、マンデル酸塩、メシル酸塩、メタンスルホン酸塩、臭化メチル、硝酸メチル、スルホン酸メチル、マレイン酸カリウム、ムチン酸塩、モノカルボン酸塩、硝酸塩、ナフタレンスルホン酸塩、2−ナフタレンスルホン酸塩、ニコチン酸塩、ナフシレート、N−メチルグルカミン、シュウ酸塩、オクタン酸塩、オレイン酸塩、パモ酸塩、酢酸フェニル、ピクリン酸塩、安息香酸フェニル、ピバレート、プロピオン酸塩、フタル酸塩、ペクチン酸塩、プロピオン酸フェニル、パルミチン酸塩、パントテン酸塩、ポリガラクツロン酸塩、ピルビン酸塩、クインネート、サリチル酸塩、コハク酸塩、ステアリン酸塩、スルファニル酸塩、塩基性酢酸塩、酒石酸塩、酢酸テオフィリン、p−トルエンスルホン酸塩(トシレート)、トリフルオロ酢酸塩、テレフタル酸塩、タンニン酸塩、テオクル酸塩、トリハロ酢酸塩、トリエチオダイド、トリカルボン酸塩、ウンデカン酸塩、および吉草酸塩である。
【0155】
一実施形態では、カルボン酸またはフェノールの無機塩の例は、アンモニウムと、リチウム、ナトリウム、カリウム、セシウムを含むアルカリ金属と;カルシウム、マグネシウム、アルミニウムを含むアルカリ土類金属と;亜鉛と、バリウムと、コリンと、第4級アンモニウムとから選択できる。
【0156】
別の実施形態では、カルボン酸またはフェノールの有機塩の例は、アルギニンと、脂肪族有機アミン、脂環式有機アミン、芳香族有機アミン、ベンザチン、t−ブチルアミン、ベネタミン(N−ベンジルフェネチルアミン)、ジシクロヘキシルアミン、ジメチルアミン、ジエタノールアミン、エタノールアミン、エチレンジアミン、ヒドラバミン、イミダゾール、リジン、メチルアミン、メグルミン、N−メチル−D−グルカミン、N,N´−ジベンジルエチレンジアミン、ニコチンアミド、有機アミン、オルニチン、ピリジン、ピコリル、ピペラジン、プロカイン、トリス(ヒドロキシメチル)メチルアミン、トリエチルアミン、トリエタノールアミン、トリメチルアミン、トロメタミン、および尿素を含む有機アミンとから選択できる。
【0157】
一実施形態では、塩は、塩が溶けない溶媒または媒体、または水などの溶媒の中で生成物の形態の遊離塩基または遊離酸を1つ以上の同等の適切な酸または塩基に反応させるなど、従来の手段で形成できる。これらの溶媒は、真空、または凍結乾燥、または既存の塩のイオンを別のイオンまたは好適なイオン交換樹脂に交換することで取り除かれる。
【0158】
一実施形態では、本発明の方法は、本発明の化合物の薬学的に許容される塩を使用する。一実施形態では、本発明の方法は、式IA、I〜XIIの化合物の薬学的に許容される塩を使用する。一実施形態では、本発明の方法は、本発明の式IA、I〜XIIの化合物のアミンの塩を使用する。一実施形態では、本発明の方法は、本発明の式IA、I〜XIIの化合物のフェノールの塩を使用する。
【0159】
一実施形態では、本発明の方法は、式IA、I〜XIIの遊離塩基、遊離酸、非荷電、または非複合化合物、および/またはその異性体、薬学的生成物、水和物、多形体またはそれらの任意の組み合わせを使用する。
【0160】
本発明の一部の実施形態では、本発明の化合物は、アミン結合によって一緒に保持される3つのフェニル基を含む。一実施形態では、本発明の化合物は非荷電構造である。別の実施形態では、本発明の化合物は遊離塩基構造である。別の実施形態では、本発明の化合物は遊離酸構造である。別の実施形態では、本発明の化合物は非複合構造である。別の実施形態では、本発明の化合物は非イオン化構造である。別の実施形態では、本発明の化合物は薬学的に許容される塩である。別の実施形態では、本発明の一部の化合物は塩酸(HCl)塩を含む。
【0161】
一実施形態では、本発明の方法は、式IA、I〜XIIの化合物の異性体を使用する。一実施形態では、本発明の方法は、式IA、I〜XIIの化合物の薬学的生成物を使用する。一実施形態では、本発明の方法は、式IA、I〜XIIの化合物の水和物を使用する。一実施形態では、本発明の方法は、式IA、I〜XIIの化合物の多形体を使用する。一実施形態では、本発明の方法は、式IA、I〜XIIの化合物の代謝物を使用する。別の実施形態では、本発明の方法は、本書に記載の通り、式IA、I〜XIIの化合物を含む組成物を使用し、あるいは、別の実施形態では、式IA、I〜XIIの化合物の異性体、代謝物、薬学的生成物、水和物、多形体の組み合わせを使用する。
【0162】
一実施形態では、用語「異性体」として、光学異性体および類似体、構造異性体および類似体、配座異性体および類似体などが挙げられるが、これに限定されるものではない。
【0163】
一実施形態では、用語「異性体」は、化合物の光学異性体を含むことになっている。一実施形態では、用語「異性体」は、化合物の立体異性体を含むことになっている。本発明の化合物は、シスおよびトランス異性化する可能性があるアミド結合を有する。当然のことながら、本発明は、光学活性、または立体異性体、またはその混合物を含み、用途に対してこれらを使用する場合には、本発明の範囲内で配慮すべきである。
【0164】
別の実施形態では、本発明はさらに化合物の水和物を含む。一実施形態では、用語「水和物」は、当該技術分野で公知のように、半水和物、一水和物、二水和物、三水和物、またはその他のことを言う。
【0165】
合成プロセス
【0166】
式IAの化合物は、例えば、ベンズアミドを生じるように塩基の存在で、置換ジフェニルアミンを安息香酸またはハロゲン化ベンゾイルと反応させることで、簡単に調製できる。一実施形態では、塩基はピリジンである。別の実施形態では、ハロゲン化ベンゾイルは塩化ベンゾイルである。別の実施形態では、ジフェニルアミンと安息香酸またはハロゲン化ベンゾイルとの反応中、ヒドロキシル置換基が保護される。別の実施形態では、ヒドロキシルに対する保護基は、任意に最終段階で取り除かれる。米国公開番号2009/00624231も参照するものとし、参照することにより本書に援用される。
【0167】
例えば、式IAの化合物:
【0168】
【化14】
【0169】
式中、R1、R2、R3、R4、jおよびkの調製は、
【0170】
【化15】
【0171】
と一緒に
【0172】
【化16】
【0173】
を反応させて、
【0174】
【化17】
【0175】
を得る。ジフェニルアミン(3)を
【0176】
【化18】
【0177】
と、塩基の存在で反応させて、以下のものを得る。
【0178】
【化19】
【0179】
式中、R1、R2、R3およびR4が、互いに独立して、OH、O−Alk−R5R6、O−Alk−複素環である場合、R1´、R2´、R3´、R4´は保護ヒドロキシル基である。ここで、保護基は、遊離ヒドロキシを得るために取り除かれる。あるいは必要に応じて、式IAの化合物を生じさせるために、続いてCl−Alk−複素環またはCl−Alk−NR5R6と反応させる。
【0180】
【化14】
【0181】
式中、R1、R2、R3、およびR4が、互いに独立して、OH、O−Alk−NR5R6、またはO−Alk−複素環と異なる場合、R1´、R2´、R3´およびR4´はそれぞれR1、R2、R3およびR4である。
【0182】
別の例として、式IAの化合物を調製するためのプロセス:
【0183】
【化14】
【0184】
式中、R1、R2、R3、およびR4は、上述の通りであり、該プロセスは、
【0185】
【化17】
【0186】
と
【0187】
【化18】
【0188】
とを、塩基の存在で反応させ、以下のものを得る。
【0189】
【化19】
【0190】
式中、R1、R2、R3およびR4が、互いに独立して、OH、O−Alk−R5R6、O−Alk−複素環である場合、R1´、R2´、R3´、R4´は保護ヒドロキシル基である。ここで、保護基は、遊離ヒドロキシを得るために取り除かれる。あるいは必要に応じて、式IAの化合物を生じさせるために、続いてCl−Alk−複素環またはCl−Alk−NR5R6と反応させる。
【0191】
【化14】
【0192】
式中、R1、R2、R3、およびR4が、互いに独立して、OH、O−Alk−NR5R6、またはO−Alk−複素環と異なる場合、R1´、R2´、R3´およびR4´はそれぞれR1、R2、R3およびR4である。
【0193】
一例では、化合物IIは実施例1、および図5にしたがって調製される。
【0194】
別の例では、化合物IIIは実施例1、および図5にしたがって調製される。
【0195】
さらなる例では、式IVの化合物:
【0196】
【化4】
【0197】
の調製は以下のようにしてもよい。すなわち、
【0198】
【化20】
【0199】
と
【0200】
【化21】
【0201】
とを、塩基の存在下で、反応させて、
【0202】
【化22】
【0203】
を得て、続いて化合物IVを生じさせるために、保護基の脱保護を行う。
【0204】
【化4】
【0205】
式中、PおよびP´は同じか、または異なる保護基である。一例では、化合物IVは実施例2、および図6にしたがって調製される。
【0206】
別の例では、化合物Vは実施例1、および図5にしたがって調製される。
【0207】
さらなる例では、化合物VIは実施例3、および図7にしたがって調製される。
【0208】
別の例では、化合物VIIは実施例1、および図5にしたがって調製される。
【0209】
別の例では、化合物VIIIは実施例4、および図5にしたがって調製される。
【0210】
別の例では、化合物IXは実施例5、および図8にしたがって調製される。
【0211】
別の例では、化合物X塩酸塩は実施例5、および図8にしたがって調製される。
【0212】
別の例では、化合物XIは実施例1、および図5にしたがって調製される。
【0213】
別の例では、化合物XIIは実施例1、および図5にしたがって調製される。
【0214】
例えば、好適なヒドロキシル保護基としては、メチルエーテル(メトキシ)、ベンジルエーテル(ベンジルオキシ)、メトキシメチル(MOM)エーテル、ベンゾイルオキシメチル(BOM)エーテル、ベンジル、カルボベンゾキシ、メトキシエトキシメチル(MEM)エーテル、2−(トリメチルシリル)エトキシメチル(SEM)エーテル、メチルチオメチル(MTM)エーテル、フェニルチオメチル(PTM)エーテル、アジドメチルエーテル、シアノメチルエーテル、2,2−ジクロロ−1,1−ジフルオロエチルエーテル、2−クロロエチルエーテル、2−ブロモエチルエーテル、テトラヒドロピラニル(THP)エーテル、1−エトキシエチル(EE)エーテル、フェナシルエーテル、4−ブロモフェナシルエーテル、シクロプロピルメチルエーテル、アリルエーテル、プロパルギルエーテル、イソプロピルエーテル、シクロヘキシルエーテル、t−ブチルエーテル、2,6−ジメチルベンジルエーテル、4−メトキシベンジルエーテル、o−ニトロベンジルエーテル、2,6−ジクロロベンジルエーテル、3,4−ジクロロベンジルエーテル、4−(ジメチルアミノ)カルボニルベンジルエーテル、4−メチルスルフィニルベンジルエーテル、4−アントリルメチルエーテル、4−ピコリルエーテル、ヘプタフルオロ−p−トリル、テトラフルオロ−4−ピリジルエーテル、トリメチルシリル(TMS)エーテル、t−ブチルジメチルシリル(TBDMS)エーテル、t−ブチルジフェニルシリル(TBDPS)エーテル、トリイソプロピルシリル(TIPS)エーテル、ギ酸アリール、酢酸アリール、レブリン酸アリール、アリールピバレート、安息香酸アリール、アリール9−フルオレンカルボキシレート、アリールメチルカルボネート、1−アダマンチルカルボネート、t−ブチルカルボネート、4−メチルスルフィニルベンジルカルボネート、2,4−ジメチルペンタ−3−イルカルボネート、アリール2,2,2−トリクロロエチルカルボネート、アリールベンジルカルボネート、アリールカルバメート、ジメチルホスフィニルエステル(Dmp−OAr)、ジメチルホスフィノチオニルエステル(Mpt−OAr)、ジフェニルホスフィノチオニルエステル(Dpt−OAr)、メタンスルホン酸アリール、トルエンスルホン酸アリール、または2−ホルミルベンゼンスルホン酸アリールが挙げられる。
【0215】
本発明の方法は、化合物IA、I〜XIIの使用を含み、本発明の化合物の調製のためのプロセスは、塩基の存在でのジフェニルアミンと塩化ベンゾイルの反応を含む。例えば、好適な塩基としては、ピリジン、トリエチルアミン、K2CO3、Cs2CO3、Na2CO3、メチルアミン、イミダゾール、ベンズイミダゾール、ヒスチジン、トリブチルアミン、またはそれらの任意の組み合わせが挙げられる。一実施形態では、塩基はピリジンである。
【0216】
本発明の方法は、化合物IA、I〜XIIの使用を含み、本発明の化合物の調製のためのプロセスは、保護されたヒドロキシルの脱保護を含む。別の実施形態では、脱保護の条件は保護基に依存する。一部の実施形態では、脱保護段階は、Pd/Cの存在での水素化を含む。別の実施形態では、脱保護はBBr3との反応を含む。別の実施形態では、脱保護段階は酸との反応を含む。
【0217】
さらなる例では、化合物IA、I〜XIIは図5〜8、および実施例1〜5にしたがって調製される。
【0218】
医薬組成物
【0219】
一部の実施形態では、本発明は、必要な化合物を含む組成物を投与することを含む使用の方法を提供する。本願明細書で使用される場合、「医薬組成物」は「治療有効量」の有効成分、すなわち、薬学的に許容される担体または希釈剤とともに本発明の化合物を意味する。本願明細書で使用される場合、「治療有効量」とは、所定の条件および投薬計画に対して治療効果をもたらす量のことを言う。
【0220】
本願明細書で使用される場合、用語「投与」とは、対象を本発明の化合物に接触させることを意味する。本願明細書で使用される場合、投与はインビトロ、すなわち試験管内で、またはインビボ、すなわち生体、例えばヒトの細胞または組織で達成できる。一実施形態では、本発明には、男性対象に本発明の化合物を投与することを含む。
【0221】
他の実施形態では、本発明は、本願明細書で述べられる化合物の薬学的生成物を示す。他の実施形態では、用語「薬学的生成物」とは、例えば本書で述べたように、製薬学的用途(医薬組成物)に好適な組成物のことを言う。
【0222】
本発明の化合物は、単独で、または製剤の有効成分として投与できる。したがって、本発明は、例えば薬学的に許容される担体を1つ以上含む式Iの化合物の医薬組成物も含む。
【0223】
多数の標準的基準によって、本発明に従った化合物の投与に好適なさまざまな製剤を調製するための手順を説明することができる。例えば、有望な製剤および調製の例は、米国薬学会、製薬賦形剤ハンドブック(最新版);薬の剤形:錠剤(編集者、Lieberman、LachmanおよびSchwartz)最新版、Marcel Dekker,Inc.発行のほか、Remington's Pharmaceutical Science(編集者、Arthur Osol)、1553〜1593(最新版)に含まれている;
【0224】
投与方法および剤形は、所定の処置用途に望ましくおよび有効な化合物または組成物の治療量と密接な関係がある。
【0225】
好適な剤形としては、経口、直腸、舌下、粘膜、経鼻、眼、皮下、筋肉内、静脈内、経皮、脊椎、くも膜下腔内、関節内、動脈内、くも膜下、気管支、リンパ管、および子宮内の投与、並びに有効成分の全身送達のための他の剤形が挙げられるが、これに限定されるものではない。経口投与に好適な製剤が望ましい。
【0226】
上記薬の剤形を調製するために、従来の薬学的生成物配合法にしたがって、有効成分を医薬担体に混ぜることができる。担体は、投与のために望まれる調製の形態に応じて、幅広い種類の形態を取ることができる。
【0227】
経口剤形の組成物の調製において、通常の製剤媒体のいずれも採用できる。このため、例えば懸濁液、エリキシル剤、および溶液などの液体経口製剤の場合、好適な担体および添加剤としては、水、グリコール、オイル、アルコール、香料添加剤、保存料、着色剤などが挙げられる。例えば粉末、カプセル、および錠剤などの固形経口製剤の場合、好適な担体および添加剤としては、スターチ、砂糖、希釈剤、造粒剤、潤滑剤、バインダー、崩壊剤などが挙げられる。投与のしやすさにより、錠剤およびカプセルが最も有利な経口単位剤形である。必要に応じて、標準的な方法で錠剤に糖衣または腸溶コ―ティングを施すことができる。
【0228】
非経口製剤の場合、通常、担体は滅菌水を含むが、他の原料、例えば、溶解性の支援または保存のための原料を含む場合がある。注射剤も調製でき、その場合、適切な安定化剤が採用されることがある。
【0229】
一部の用途では、リポソームまたは他のカプセル材料媒体中での活性薬剤のカプセル化による、またはタンパク質、リポタンパク質、糖タンパク質、および多糖から選択されるものなどの好適な生体分子への活性薬剤の固定による、例えば共有結合、キレート化、または配位結合などによる「ベクター化(vectorized)」で活性薬剤を利用することに有利な場合がある。
【0230】
経口投与に好適な製剤を用いた本発明の処置方法は、それぞれ、例えば粉末または顆粒として所定量の有効成分を含むカプセル、カシェー、錠剤、またはトローチ剤などの個別の単位として提示されることがある。任意に、シロップ、エリキシル剤、エマルション、または水薬一服(draught)など、水性液体または非水性液体の懸濁液を採用できる。
【0231】
圧縮または成形、または必要に応じて1つ以上の副原料との湿式造粒法によって、錠剤を製造できる。圧縮錠剤は、例えば、必要に応じてバインダー、崩壊剤、潤滑剤、不活性希釈剤、または排出剤に混ぜられる粉末または顆粒などの自由流動形態の活性化合物とともに好適な機械で圧縮することで製造できる。粉末活性化合物と好適な担体の混合物を含む成形錠剤は、好適な機械による成型によって製造できる。
【0232】
シロップは、砂糖、例えば蔗糖の濃縮水溶液に活性化合物を添加することで製造でき、これには、副原料を添加することもできる。前記副原料には、砂糖の結晶化を遅らせるために香料添加剤、好適な保存料と、そして、多価アルコール、例えばグリセロールまたはソルビトールなどの他の原料の溶解性を高める薬剤とを含む場合がある。
【0233】
非経口投与に好適な製剤は、好適には受容者の血液と等張である活性化合物の無菌水性調合液(例えば、生理食塩水)を含んでもよい。前記製剤は、懸濁化剤および増粘剤およびリポソーム、または化合物が血液成分または1つ以上の臓器を標的にするように設計された他の微粒子系を含んでもよい。製剤は、単回投与または複数回投与剤形で調製してもよい。
【0234】
非経口投与は、全身送達の好適な剤形を含んでもよい。例えば、投与は、静脈内、動脈内、くも膜下腔内、筋肉内、皮下、筋肉内、腹内(例えば、腹腔内)などであってもよく、そして注入ポンプ(外部または埋め込み型)または望ましい投与法に適した他の好適な手段による影響を受ける場合がある。
【0235】
鼻および他の粘膜スプレー製剤(例えば、吸引可能剤形)は、保存料および等張剤とともに活性化合物の精製水溶液を含むことができる。前記製剤は、好適には、鼻または他の粘膜に適合するpHおよび等張状態に調整される。あるいは、ガス担体に懸濁された微粉固体粉末の剤形であってもよい。前記製剤は、好適な手段または方法、例えば、噴霧器、アトマイザー、定量吸入具などで送達してもよい。
【0236】
直腸投与のための製剤は、ココアバター、硬化油脂、または硬化脂肪カルボン酸などの好適な担体とともに座薬として提供してもよい。
【0237】
経皮製剤は、セルロース媒体、例えば、メチルセルロースまたはヒドロキシエチルセルロースなどのチキソトロピーまたはゼラチン状担体に活性薬剤を組み込むことで調製してもよく、得られる製剤は、着用者の皮膚との皮膚接触を確保するように適合された経皮投与装置に詰められることになる。
【0238】
前述の原料に加えて、本発明の製剤は、例えば、希釈剤、緩衝剤、香料添加剤、バインダー、崩壊剤、界面活性剤、増粘剤、潤滑剤、保存料(抗酸化剤を含む)などから選択される1つ以上の副原料をさらに含んでもよい。
【0239】
本発明の製剤は、即時放出、持続放出、遅発性放出、または当業者には公知の他の放出プロファイルを有することができる。
【0240】
一実施形態では、本発明は、式IA、I〜XIIの化合物を含む経口組成物を投与することを含む、前立腺癌を患う男性対象において、黄体形成ホルモン(LH)の削減によって、またはLHホルモンの削減と関係なく、a)全血清テストステロン濃度を下げる;b)遊離血清テストステロン濃度を下げる方法を提供する。追加の実施形態では、本発明の方法は、式II、式III、式IV、式V、式VI、式VII、式VIII、式IX、式X、式XI、または式XIIの化合物を含む経口組成物を使用する。
【0241】
一実施形態では、本発明は、式IA、I〜XIIの化合物を含む経口組成物を投与することを含む、前立腺癌を患う男性対象において、LH濃度の削減によって、またはLH濃度の削減と関係なく、前立腺癌を処置する方法を提供する。追加の実施形態では、本発明は、式II、式III、式IV、式V、式VI、式VII、式VIII、式IX、式X、式XI、または式XIIの化合物を含む経口組成物を投与することを含む、前立腺癌治を患う男性対象において、LH濃度を下げることで、またはLH濃度を下げることとは関係なく、前立腺癌を処置する方法を提供する。
【0242】
当然のことながら、本発明は、本願明細書で述べられるような化合物のいかなる実施形態も含み、一部の実施形態では、「本発明の化合物」と呼ばれる。
【0243】
一実施形態では、本発明の方法は、さまざまな用量での本発明の化合物の投与を含んでもよい。一実施形態では、本発明の化合物は、1〜1500/日の用量で投与される。追加の実施形態では、本発明の化合物は、1〜10mg/日、3〜26mg/日、3〜60mg/日、3〜16mg/日、3〜30mg/日、10〜26mg/日、15〜60mg/日、50〜100mg/日、50〜200mg/日、150〜300mg/日、20〜50mg/日、5〜50mg/日、200〜500mg/日、150〜500mg/日、200〜1000mg/日、300〜1500mg/日、または100〜1000mg/日の用量で投与される。
【0244】
一実施形態では、本発明の方法は、さまざまな用量での本発明の化合物の投与を含んでもよい。一実施形態では、本発明の化合物は、3mgの用量で投与される。追加の実施形態では、本発明の化合物は、10mg、30mg、50mg、100mg、200mg、300mg、450mg、500mg、600mg、900mg、1000mg、または1500mgの用量で投与される。
【0245】
一実施形態では、本発明の方法は、さまざまな用量での本発明の化合物の投与を含んでもよい。一実施形態では、本発明の化合物は、0.1mg/Kg/日の用量で投与される。追加の実施形態では、本発明の化合物は、0.2〜30mg/kg/日、または0.2mg/kg/日、0.3mg/kg/日、1mg/kg/日、3mg/kg/日、5mg/kg/日、10mg/kg/日、20mg/kg/日、または30mg/kg/日の用量で投与される。
【0246】
一実施形態では、本発明の方法は、式IA、I〜XIIの化合物を含む医薬組成物を使用するために提供される。追加の実施形態では、本発明の方法は、式II、式III、式IV、式V、式VI、式VII、式VIII、式IX、式X、式XI、または式XIIの化合物を含む医薬組成物を使用するために提供される。
【0247】
いくつかの実施形態では、医薬組成物は固体剤形である。別の実施形態では、医薬組成物は錠剤である。別の実施形態では、医薬組成物はカプセルである。別の実施形態では、医薬組成物は溶液である。別の実施形態では、医薬組成物は経皮貼布である。
【0248】
一実施形態では、本発明の化合物、またはこれを含む組成物を使用することには、当業者には公知の通り、対象に望まれる反応の阻害、抑制、増進、または刺激において有用性がある。別の実施形態では、組成物は追加有効成分をさらに含み、この成分の活性は、本発明の化合物が投与される特定の用途に有益である。
【0249】
動物、特にヒトへの投与の場合、個人に最も好適で、特定の個人の年齢、体重、遺伝、および/または反応によって変動する可能性がある、実際の用量および処置期間を医師が決定することが予想される。
【0250】
一部の実施形態では、本発明の組成物はいずれも、本願明細書で述べられるような剤形または実施形態で本発明の化合物を含むことになる。一部の実施形態では、本発明の組成物はいずれも、本願明細書で述べられるような剤形または実施形態で本発明の化合物からなる。一部の実施形態では、本発明の組成物はいずれも、本願明細書で述べられるような剤形または実施形態で、本発明の化合物から本質的になる。一部の実施形態では、用語「含む(comprise)」とは、本発明の化合物などの示された活性薬剤の含有のほか、製薬業界では公知のように、他の活性薬剤、および薬学的に許容される担体、賦形剤、皮膚軟化剤、安定化剤などの含有のことも言う。一部の実施形態では、用語「本質的になる(consisting essentially of)」とは、唯一の有効成分が示された有効成分であるが、安定化、保存などのための他の化合物を含むことができるが、示された有効成分の治療効果に製剤が直接関与することはない組成物のことを言う。一部の実施形態では、用語「本質的になる(consisting essentially of)」とは、有効成分の放出を促進する成分のことを言う場合がある。一部の実施形態では、用語「からなる(consisting of)」とは、有効成分および薬学的に許容される担体または賦形剤を含む組成物のことを言う。
【0251】
当然のことながら、本願明細書で述べられるような化合物の使用は、本願明細書で述べられるような疾患、障害、または症状で使用でき、本発明の実施形態を表す。一実施形態では、化合物は、遊離塩基、遊離酸、または非複合化合物である。
【0252】
以下の実施例は、本発明の望ましい実施形態をさらに十分に説明するために提供される。しかしながら、本発明の幅広い範囲を制限するものと解釈されることは全くない。
【実施例】
【0253】
実施例1:式II〜XIIの化合物の一般的な合成手順および中間体による合成
【0254】
有機溶媒、界面活性剤、および抗酸化剤など、これらは本願明細書で述べられる組成物で使用でき、典型的には商業用供給源から容易に入手できる。例えば、PEG−300、polysorbate 80、Captex(商標)200、Capmul(商標)MCM C8は、例えばDow Chemical Company(Midland, MI)、ICI Americas, Inc(Wilmington, DE)、またはAbitec Corporation(Janesville, WI)から購入できる。
【0255】
本願明細書で述べられるエストロゲン受容体リガンドは、当業者には公知のさまざまな方法で調製できる。例えば、本願明細書で述べられるエストロゲン受容体リガンドは、米国特許出願公開番号2009/0062341で述べられた合成方法で調製でき、その全体の各開示を参照することにより本書に組み込まれる。
【0256】
N,N−ビスアリールベンズアミド誘導体の一般的な合成
【0257】
ジアリールアニリンの一般的な合成(図5)アリールアミン(1.5当量)、ヨウ化アリール(1当量)、K2CO3(2当量)、CuI(0.1当量)、およびL−プロリン(0.2当量)の混合物を一緒に混ぜ、室温で無水DMSOに溶かした。次に、反応混合物を攪拌し、28時間90℃に加熱した。混合物を室温に冷却し、水で加水分解した。EtOAcを添加し、溶液を分離させた。EtOAc層を分離し、塩水で洗浄し、無水MgSO4によって乾燥させた。減圧下で溶媒を取り除いた。該当するジアリールアニリンを利用可能にするために、溶離液として5%EtOAc/ヘキサンを用いて、フラッシュカラムクロマトグラフィー(シリカゲル)で固体残留物を精製した。
【0258】
ビス−(4−メトキシフェニル)アミン(1a):淡黄色固体、収率73%、融点98.6〜99.0℃。1H NMR(CDCl3、300MHz)δ6.93〜6.81(m、8H)、5.37(s、br、1H)、3.78(s、6H)。MS m/z 228.4(M−H)+。
【0259】
N−(4−メトキシフェニル)−フェニルアミン(1b):淡黄色固体、収率70%、融点106.3〜106.5℃。1H NMR(CDCl3、300MHz)δ 7.24〜7.18(m、3H)、7.08〜7.06(m、2H)、6.92〜6.84(m、4H)、5.61(s、br、1H)、3.79(s、3H)。MS m/z 200.1(M+H)+。
【0260】
N−(4−フルオロフェニル)−N−4−メトキシフェニルアミン(1c):淡黄色固体、収率54%、融点60.6〜61.0℃。1H NMR(CDCl3、300MHz)δ7.01〜6.83(m、8H)、3.78(s、3H)。MS m/z 217(M)+。
【0261】
N−(4−ベンジルオキシフェニル)−N−4−メトキシフェニルアミン(1d):淡黄色固体、収率54%、融点108.0〜108.4℃。1H NMR(CDCl3、300MHz)δ 7.34〜7.08(m、5H)、6.90〜6.81(s、3H)、3.78(s、3H)。MS m/z 306(M+H)+。
【0262】
ベンズアミドの一般的な合成。アリールアミン(1当量)、塩化ベンゾイル(1.3当量)、およびピリジン(6当量)の混合物を一緒に混ぜ、室温で無水THFに溶かした。24時間、混合物を攪拌し、還流させた。反応液を室温に冷却し、2N HCl溶液を添加して加水分解した。溶液は、酢酸エチルで抽出した。有機層をNaHCO3の飽和水溶液で洗浄し、余分な酸を取り除き、無水MgSO4で乾燥させ、濾過し、減圧下で濃縮した。該当するベンズアミドを利用可能にするために、EtOAc/ヘキサン(3/7v/v)を用いて、フラッシュカラムクロマトグラフィーで残留物を精製した。
【0263】
3−フルオロ−N−(4−フルオロフェニル)−4−メトキシ−N−(4−メトキシフェニル)ベンズアミド(2a):黄色固体、融点54〜56℃。1H NMR(CDCl3、/TMS)δ 7.24〜7.11(m、4H)、7.05〜6.97(m、4H)、6.85〜6.78(m、3H)、3.86(s、3H)、3.79(s、3H)。MS (ESI) m/z 370.1 [M +H]+。
【0264】
4−フルオロ−N,N−ビス(4−メトキシフェニル)−2−(トリフルオロメチル)ベンズアミド(2b):無色油、収率84.2%。1H NMR(CDCl3、300MHz)δ 7.34〜7.26(m、4H)、7.09〜7.01(m、3H)、6.91(d、2H、J=8.7Hz)、6.87(d、2H、J=8.7Hz)、3.80(s、3H)、3.71(s、3H)。MS m/z 442.1(M+Na)+。
【0265】
4−メトキシ−N−(4−メトキシフェニル)−N−(4−フルオロフェニル)−ベンズアミド(2c):白色固体、収率97%、融点133.5〜134.5℃。 1H NMR(CDCl3、300MHz)δ 8.11〜6.66(m、15H)、3.74(s、3H)、3.73(s、3H)。MS m/z 384 (M+H)+。
【0266】
N−(4−メトキシフェニル)−N−(4−ベンジルオキシフェニル)−2−ナフチルアミド(1d):白色固体、収率58%、融点174.9〜175.5℃。1H NMR(CDCl3、300MHz)δ 8.04(s、1H)、7.77〜7.74(m、2H)、7.64〜7.61(m、1H)、7.51〜7.43(m、4H)、7.40〜7.31(m、4H)、7.13〜7.10(m、4H)、6.88〜6.78(m、4H)、4.99(s、2H)、3.74(s、3H)。MS m/z 460 (M+H)+。
【0267】
4−フルオロ−N,N−ビス(4−メトキシフェニル)−2−(トリフルオロメチル)ベンズアミド(2e):無色油、収率84.2%。1H NMR(CDCl3、300MHz)δ 7.34〜7.26(m、4H)、7.09〜7.01(m、3H)、6.91(d、2H、J=8.7Hz)、6.87(d、2H、J=8.7Hz)、3.80(s、3H)、3.71(s、3H)。MS m/z 442.1(M+Na)+。
【0268】
BBr3を用いたベンズアミド誘導体の脱メチル反応の一般的手順。メトキシベンズアミド化合物を、乾燥したCH2Cl2に溶かした。BBr3(1.0M CH2Cl2溶液)を0℃で滴加した。反応溶液を室温までゆっくりと温め、室温で一晩、攪拌したまま置いた。混合物を氷浴にて0℃に冷却し、水を添加して加水分解した。EtOAcを添加し、溶液を分離させた。有機層が分離した。水層をEtOAcで抽出した。有機層を塩水で洗浄し、無水MgSO4によって乾燥させた。減圧下で溶媒を取り除いた。該当するフェノール化合物を利用可能にするために、CH3OH/CH2Cl2(1/9 v/v)を用いて、フラッシュカラムクロマトグラフィーで残留物を精製した。
【0269】
4−フルオロ−N,N−ビス(4−ヒドロキシフェニル)−2−(トリフルオロメチル)ベンズアミド(3a):白色固体、収率92.5%。1H NMR(DMSO−d6、300MHz)δ 9.55(s、1H)、9.53(s、1H)、7.69〜7.58(m、2H)、7.46〜7.39(m、1H)、7.18(d、2H、J=8.7Hz)、6.93(d、4H、J=8.7Hz)、7.03(d、2H、J=8.4Hz)、6.78(d、2H、J=8.7Hz)、6.57(d、2H、J=8.7Hz)。MS m/z 392.1(M+H)+。
【0270】
以下の化合物は上述の通り合成され、表1にて特徴付けられ、要約されている。すなわち、N,N−ビス(4−ヒドロキシフェニル)−4−プロピルベンズアミド(II);3−フルオロ−N−(4−フルオロフェニル)−4−ヒドロキシ−N−(4−ヒドロキシフェニル)ベンズアミド(IV);N,N−ビス(4−ヒドロキシフェニル)−2,3−ジメチルベンズアミド(V);3−フルオロ−4−ヒドロキシ−N,N−ビス(4−ヒドロキシフェニル)−ベンズアミド(VII);3−フルオロ−4−ヒドロキシ−N−(4−ヒドロキシフェニル)−N−フェニルベンズアミド(XI);および3−フルオロ−N,N−ビス(4−ヒドロキシフェニル)−2−メチルベンズアミド(XII)。
【0271】
ベンジルオキシフェニル−ベンズアミドの脱ベンジル化の一般的手順。化合物を、250mL水素化ボトルに入ったEtOHに溶かした。溶液にPd/C粉末(5%モル)を添加した。反応容器を、水素ガス圧力20psiの下で水素化装置に取り付けた。出発物質が消えるまで、TLCで反応をモニタリングした。その後、減圧下で溶媒を取り除いた。所望の生成物を利用可能にするために、ヘキサン/EtOAc=3/2 v/vを用いて、フラッシュカラムクロマトグラフィーで残留物を精製した。
【0272】
以下の化合物は上述の通り合成され、表1にて特徴付けられ、要約されている。すなわち、N,N−ビス(4−ヒドロキシフェニル)−2−ナフチルアミド(VI)。
【0273】
脱保護ベンズアミドの還元の一般的手順。ベンズアミド化合物を、室温で20mLの無水THFに溶かした。アルゴン下で、室温にてシリンジを用いてH3B(SMe2)を添加した。6時間、反応溶液を攪拌し、加熱して還流させた。その後、0℃でMeOHを10mL添加して反応を抑えた。減圧下で溶媒を取り除いた。所望の生成物を利用可能にするために、フラッシュカラムクロマトグラフィー(シリカゲル、CH2Cl2/MeOH=9/1 v/v)に残留物を通した。
【0274】
以下の化合物は上述の通り合成され、表1にて特徴付けられ、要約されている。すなわち、4,4´−(2,3−ジメチルベンジルアザネジイル)ジフェノール(III);4−((4−フルオロフェニル)(4−ヒドロキシベンジル)アミノ)フェノール(VIII)。
【0275】
O−(2−ピペリジン−1−イルエトキシ)−ベンズアミドおよび類似体の一般的な合成。ベンズアミド類似体(1当量)を含むヒドロキシフェニルのアセトン溶液に、K2CO3(3当量)および塩酸N−クロロエチル−ピペリジン(1.2当量)を添加した。6時間、反応溶液を加熱して還流させた。溶液を蒸発乾固させた。水を添加して残留物を加水分解し、その後、酢酸エチルで抽出した。有機層を分離させ、無水MgSO4によって乾燥させた。減圧下で溶媒を取り除いた。所望の生成物を利用可能にするために、塩化メチレン/メタノール=9/1 v/vを用いて、フラッシュカラムクロマトグラフィーで残留物を精製した。化合物のメタノール溶液にHClのEt2O溶液を添加して、塩酸塩を調製した。
【0276】
以下の化合物は上述の通り合成され、表1にて特徴付けられ、要約されている。すなわち、4−フルオロ−N−(4−ヒドロキシフェニル)−N−(4−(2−(ピペリジン−1−イル)エトキシ)フェニル)−2−(トリフルオロメチル)ベンズアミド(IX);およびIXのHCl塩である4−フルオロ−N−(4−ヒドロキシフェニル)−N−(4−(2−(ピペリジン−1−イル)エトキシ)フェニル)−2−(トリフルオロメチル)ベンズアミド塩酸塩(X)。
【0277】
【表1A】
【0278】
【表1B】
【0279】
【表1C】
【0280】
実施例2:式IVの化合物の合成(図6)
【0281】
【化23】
【0282】
ステップ1:4−フルオロ−N−(4−メトキシフェニル)アニリン(1c)
【0283】
4−フルオロアニリン(78.63g、0.708mol)、4−ヨードアニソール(138.00g、0.590mol)、無水K2CO3(122.23g、0.884mol)、CuI(11.23g、58.96mol)、およびL−プロリン(13.58g、0.118mol)の混合物を、攪拌子、還流冷却器、およびアルゴン入口の付いた1Lの3つ口丸底フラスコで一緒に混ぜた。室温で無水DMSO(300mL)を添加した。反応混合物を攪拌し、アルゴン下で20時間90℃に加熱した。次に、混合物を室温に冷却し、水(300mL)で加水分解した。EtOAc(200mL)を添加し、溶液を分離した。EtOAc層を分けた。EtOAc100mLで水層を抽出した。EtOAc層に塩水(2x100mL)を添加し、洗浄し、無水MgSO4(50g)によって乾燥させた。減圧下で溶媒を取り除いた。収率77.8%、融点46〜48℃の黄色固体99.70gとして4−フルオロ−N−(4−メトキシフェニル)アニリン(1c)を利用可能にするために、茶色の油状残留物をフラッシュカラムクロマトグラフィー(シリカゲル、ヘキサン/EtOAc=9/1 v/v)で精製した。MS(ESI) m/z 218.1 [M + H] +、1H NMR(DMSO−d6、300MHz) δ 7.77(bs、1H)、7.03〜6.98(m、4H)、6.93〜6.82(m、4H)、3.70(s、3H)。
【0284】
【化24】
【0285】
ステップ2:3−フルオロ−N−(4−フルオロフェニル)−4−メトキシ−N−(4−メトキシフェニル)ベンズアミドの合成(2a)
【0286】
4−フルオロ−N−(4−メトキシフェニル)アニリン(1c)(90.78g、0.418mol)および塩酸3−フルオロ−4−メトキシベンゾイル(94.55g、0.501mol)を、攪拌子、還流冷却器、およびアルゴン入口の付いた1Lの3つ口丸底フラスコで無水THF(200mL)に一緒に混ぜ、溶かした。アルゴン下で、室温にてシリンジを用いて無水ピリジン(132.22g、1.672mol)を添加した。一晩、反応混合物を攪拌し、加熱して還流させた。次に、反応混合物を室温に冷却し、濾過してピリジン塩を取り除いた。溶液を濃縮し、THF溶剤を取り除いた。残留油を2N HCl溶液200mLで洗浄し、酢酸エチル(2x200mL)で抽出した。組み合わせた有機層をNa2HCO3の飽和水溶液(150mL)で洗浄し、余分な塩化ベンゾイルと酸とを取り除き、無水MgSO4(50g)で乾燥させ、濾過し、減圧下で濃縮し、油状物を得た。融点54〜56℃の黄色固体として該当する純粋なベンズアミド化合物を利用可能にするために、CH2Cl2/アセトン(50/1 v/v)で、シリカゲルを用いたフラッシュカラムクロマトグラフィーによって残留物を精製した。MS(ESI) m/z 370.1 [M +H]+、1H NMR(CDCl3/TMS) δ 7.24〜7.11(m、4H)、7.05〜6.97(m、4H)、6.85〜6.78(m、3H)、3.86(s、3H)、3.79(s、3H)。
【0287】
【化25】
【0288】
ステップ3:3−フルオロ−N−(4−フルオロフェニル)−4−ヒドロキシ−N−(4−ヒドロキシフェニル)ベンズアミド(IV)の合成
【0289】
化合物3−フルオロ−N−(4−フルオロフェニル)−4−メトキシ−N−(4−メトキシフェニル)ベンズアミド(2a)(138.0g、0.374mol)を、アルゴン下、室温でCH2Cl2(600mL)に溶かした。BBr3(374.75g、1.496mol)を、アルゴン下、0℃で、氷浴にて攪拌しながらシリンジを用いて滴加した。一晩、室温にて反応溶液を攪拌し続けた。次に、溶液を攪拌しながら氷水1Lに注いだ。2時間、室温にてスラリー混合物を攪拌した。白色沈殿物を濾過し、水(2x100mL)で洗浄し、真空下で乾燥させた。CH2Cl2層を分離し、無水MgSO4(50g)によって乾燥させ、濾過し、真空下で濃縮し、乾燥させた。収率81.6%、融点110〜112℃の白色結晶性固体を利用可能にするために、CH2Cl2溶液から得た白色沈殿物および残留物を組み合わせ、フラッシュカラムクロマトグラフィー(シリカゲル、CH2Cl2/アセトン/MeOH=90/7/3 v/v/v)で精製し、加熱したEtOAc/ヘキサン溶液から2回再結晶させた淡褐色固体を得た。MS(ESI) m/z 364.1 [M + Na]+、1H NMR(DMSO−d6) ? 10.14(bs、1H)、9.71(bs、1H)、7.25〜7.11(m, 5H)、7.05〜6.99(m、3H)、6.78(t、J=8.6 Hz、1H)、6.68(d、J=8.7 Hz、2H)。
【0290】
実施例3:式VIの化合物の合成(図7)
【0291】
4−(ベンジルオキシ)−N−(4−メトキシフェニル)アニリンの合成(1d)
【0292】
4−ベンジルオキシアニリン(16.6g、83.31mmol)、4−ヨードアニソール(15.0g、64.09mmol)、K2CO3(17.72g、128.18mmol)、CuI(1.22g、6.41mmol)、およびL−プロリン(1.48g、12.82mmol)の混合物を一緒に混ぜ、室温で無水DMSO(120mL)に溶かした。次に、反応混合物を攪拌し、48時間90℃に加熱した。混合物を室温に冷却し、水で加水分解した。EtOAcを添加し、溶液を分離させた。EtOAc層を分離し、塩水で洗浄し、無水MgSO4によって乾燥させた。減圧下で溶媒を取り除いた。収率9.8g、融点108.0〜108.4℃の黄色固体9.8gとして該当するジアリールアニリンを利用可能にするために、EtOAc/ヘキサン(1/9 v/v)で、フラッシュカラムクロマトグラフィー(シリカゲル)によって固体残留物を精製した。1H NMR(CDCl3、300 MHz) δ 7.34〜7.25(m、5H)、6.90〜6.81(m、8H)、5.02(s、2H)、3.78(s、3H)。MS m/z 306(M+H)+。
【0293】
N−(4−ベンジルオキシフェニル)−N−(4−メトキシフェニル)−2−ナフトアミドの合成(2d)
【0294】
1当量の4−(ベンジルオキシ)−N−(4−メトキシフェニル)アニリン(0.80g、2.62mmol)を、攪拌子および還流冷却器の付いた乾燥3口丸底フラスコで1.5当量の塩化2−ナフトイル(0.75g、3.93mmol)および4当量のピリジン(0.83g、10.48mmol)に混ぜた。混合物を無水THF(30mL)に溶かし、加熱して、20時間還流させた。反応溶液を室温に冷却し、濾過した。減圧下で溶媒を取り除いた。収率58%、融点174.9〜175.5℃の白色固体として該当する純粋なナフトアミド化合物を利用可能にするために、EtOAc/ヘキサン(3/7 v/v)で、シリカゲルを用いたフラッシュカラムクロマトグラフィーによって残留物を精製した。1H NMR(CDCl3、300MHz) δ 8.04(s、1H)、7.77〜7.74(m、2H)、7.64〜7.61(m、1H)、7.51〜7.43(m、4H)、7.40〜7.31(m、4H)、7.13〜7.10(m、4H)、6.88〜6.78(m、4H)、4.99(s、2H)、3.74(s、3H)。MS m/z 460 (M+H)+。
【0295】
N,N−ビス(4−ヒドロキシフェニル)−2−ナフチルアミド(VI)の合成
【0296】
化合物N−(4−ベンジルオキシフェニル)−N−(4−メトキシフェニル)−2−ナフトアミド(2d)(0.5g、1.09mmol)を、室温で乾燥したCH2Cl2(30mL)に溶かした。BBr3(1.0M CH2Cl2溶液3.26mL、3.26mmol)を、室温で攪拌しながら、シリンジを用いて滴加した。一晩、室温にて反応溶液を攪拌し続けた。混合物を氷浴にて0℃に冷却し、水を添加して加水分解した。EtOAcを添加し、溶液を分離させた。有機層が分離させた。水層をEtOAcで2回抽出した。有機層を組み合わせ、塩水で洗浄し、無水MgSO4によって乾燥させた。真空下で溶媒を取り除いた。収率70%、融点264.3〜265.2℃(分解温度)の白色固体として所望の純粋なフェノール化合物を利用可能にするために、CH3OH/CH2Cl2(1/9 v/v)で、シリカゲルを用いたフラッシュカラムクロマトグラフィーによって残留物を精製した。1H NMR(DMSO−d6、500MHz)δ 9.46(s、2H)、7.98 (s、1H)、7.85〜7.75(m、2H)、7.75〜7.73(m、2H)、7.54〜7.48(m、2H)、7.45〜7.43(m、1H)、7.05(s、4H)、6.66(s、4H)。MS m/z 356 (M+H)+。
【0297】
実施例4:式VIIIの化合物の合成
【0298】
【化26】
【0299】
4−((4−フルオロフェニル)(4−ヒドロキシベンジル)アミノ)フェノール(VIII)の合成
【0300】
化合物N−(4−フルオロフェニル)−4−ヒドロキシ−N−(ヒドロキシフェニル)ベンズアミド(0.30g、0.93mmol)を、室温で無水THF20mLに溶かした。アルゴン下で、室温にてシリンジを用いてH3B(SMe2)(2M THF溶液1.86mL、3.71mmol)を添加した。6時間、反応溶液を攪拌し、加熱して還流させた。その後、0℃でMeOHを10mL添加して反応を抑えた。減圧下で溶媒を取り除いた。フラッシュカラムクロマトグラフィー(シリカゲル、CH2Cl2/MeOH=9/1 v/v)に残留物を通し、黄色油状物(0.26g、収率92%)を得た。1H NMR(DMSO−d6、500MHz) δ 9.29(s、1H)、9.24(s、1H)、7.09(d、2H、J=8.3 Hz)、6.98(d、2H、J=9.0 Hz)、6.94〜6.91(m、2H)、6.73(d、2H、J=9.0 Hz)、6.68〜6.64(m、4H)、4.70(s、2H)。MS m/z 307.8 (M−H)−。
【0301】
実施例5:式IXおよびXの化合物の合成(図8)
【0302】
ジアリールアニリンの合成。アリールアミン(1.5当量)、ヨウ化アリール(1当量)、K2CO3(2当量)、CuI(0.1当量)、およびL−プロリン(0.2当量)の混合物を一緒に混ぜ、室温で無水DMSOに溶かした。次に、反応混合物を攪拌し、28時間90℃に加熱した。混合物を室温に冷却し、水で加水分解した。EtOAcを添加し、溶液を分離させた。EtOAc層を分離し、塩水で洗浄し、無水MgSO4によって乾燥させた。減圧下で溶媒を取り除いた。該当するジアリールアニリンを利用可能にするために、溶媒としてEtOAc/ヘキサン(3/7 v/v)を用いて、フラッシュカラムクロマトグラフィー(シリカゲル)で固体残留物を精製した。ビス−(4−メトキシフェニル)アミン(1a):淡黄色固体、収率73%。1H NMR(CDCl3、300MHz) δ 6.93〜6.81(m、8H)、5.37(s、br、1H)、3.78(s、6H)。MS m/z 228.4(M−H)+。
【0303】
4−フルオロ−N,N−ビス(4−メトキシフェニル)−2−(トリフルオロメチル)ベンズアミドの合成(2e)
【0304】
1当量のビス−(4−メトキシフェニル)アミン(1a)(0.73g、3.18mmol)を、攪拌子および還流冷却器を備えた乾燥3口丸底フラスコで1.2当量の塩化4−フルオロ−2−トリフルオロメチルベンゾイル(0.87g、3.82mmol)および6当量のピリジン(1.51g、19.08mmol)に混ぜた。混合物を無水THF(20mL)に溶かし、20時間、90℃に加熱した。反応溶液を室温に冷却し、濾過した。減圧下で溶媒を取り除いた。収率84.2%の無色油1.12gとして該当する純粋なベンズアミド化合物を利用可能にするために、EtOAc/ヘキサン(3/7 v/v)で、シリカゲルを用いたフラッシュカラムクロマトグラフィーによって残留物を精製した。1H NMR(CDCl3、300MHz)δ 7.34〜7.26(m、4H)、7.09〜7.01(m、3H)、6.91(d、2H、J=8.7Hz)、6.87(d、2H、J=8.7Hz)、3.80(s、3H)、3.71(s、3H)。MS m/z 442.1(M+Na)+。
【0305】
4−フルオロ−N,N−ビス(4−ヒドロキシフェニル)−2−(トリフルオロメチル)ベンズアミドの合成(3a)
【0306】
化合物4−フルオロ−N,N−ビス(4−メトキシフェニル)−2−(トリフルオロメチル)ベンズアミド(2e)(1.00g、2.38mmol)を、室温で乾燥したCH2Cl2(30mL)に溶かした。BBr3(1.0M CH2Cl2溶液10mL、10.0mmol)を、室温で攪拌しながら、シリンジを用いて滴加した。一晩、室温にて反応溶液を攪拌し続けた。混合物を氷浴にて0℃に冷却し、水を添加して加水分解した。EtOAcを添加し、溶液を分離させた。有機層が分離させた。水層をEtOAcで2回抽出した。有機層を組み合わせ、塩水で洗浄し、無水MgSO4によって乾燥させた。真空下で溶媒を取り除いた。収率92.5%の白色固体0.86gとして所望の純粋なフェノール化合物を利用可能にするために、CH3OH/CH2Cl2(1/9 v/v)で、シリカゲルを用いたフラッシュカラムクロマトグラフィーによって残留物を精製した。1H NMR(DMSO−d6、300MHz)δ 9.55(s、1H)、9.53(s、1H)、7.69〜7.58(m、2H)、7.46〜7.39(m、1H)、7.18(d、2H、J=8.7Hz)、6.93(d、4H、J=8.7Hz)、7.03(d、2H、J=8.4Hz)、6.78(d、2H、J=8.7Hz)、6.57(d、2H、J=8.7Hz)。MS m/z 392.1(M+H)+。
【0307】
4−フルオロ−N−(4−ヒドロキシフェニル)−N−[4−(2−ピペリジン−1−イル)−エトキシ]フェニル]−2−(トリフルオロメチル)ベンズアミド(IX)の合成
【0308】
4−フルオロ−N,N−ビス(4−ヒドロキシフェニル)−2−(トリフルオロメチル)ベンズアミド(3a)のアセトン溶液に、K2CO3(1.29g、9.36mmol)および塩酸N−クロロエチル−ピペリジン(0.34g、1.87mmol)を添加した。20時間、反応溶液を加熱して還流させた。溶液を蒸発乾固させた。収率57.7%の白色固体0.45gとして所望の化合物を利用可能にするために、フラッシュカラムクロマトグラフィー(シリカゲル;塩化メチレン/メタノール=9/1 v/v)で残留物を精製した。1H NMR(DMSO−d6、300MHz)δ 9.57(s、1H)、7.71〜7.68(m、2H)、7.47〜7.44(m、1H)、7.28(d、1H、J=9.0 Hz)、7.18(d、1H、J=8.7 Hz)、7.13(d、1H、J=8.7 Hz)、7.05(d、1H、J=8.4 Hz)、6.97(d、1H、J=9.0 Hz)、6.80〜6.76(m、2H)、6.57(d、1H、J=87. Hz)、4.06(t、1H、J=6.0 Hz)、3.93(t、1H、J=6.0 Hz)、2.66(t、1H、J=5.7 Hz)、2.55(t、1H、J=5.4 Hz)、2.44(s、2H)、2.36(s、2H)、1.49〜1.37(m、6H)。MS m/z 501.0 (M−H)−。
【0309】
化合物のメタノール溶液にHClのEt2O溶液を添加し、続いて溶媒を蒸発させて、塩酸塩(X)を調製した。
【0310】
実施例6:エストロゲン受容体結合親和性、アゴニスト、およびアンタゴニスト活性
【0311】
化合物のER結合親和性は、[2,4,6,7−3H(N)]−エストラジオール([3H]E2)、天然高親和性ERリガンド、および細菌発現GST融合ER−αまたはER−βリガンド結合ドメイン(LBD)タンパク質によるインビトロ競合放射性リガンド結合試験を用いて測定された。
【0312】
方法
【0313】
組み換え型ER−αまたはER−βを[3H]E2に結合させ、[3H]E2の平衡解離定数(Kd)を測定した。全特異的結合および非特異的結合を測定するために、[3H]E2の濃度を上げながら、高濃度の非標識E2がある場合とない場合で、4℃で18時間、タンパク質をインキュベートした。非特異的結合を減算し、非線形回帰を用いてE2(ERα:0.71nM;ERβ:1.13nM)のKdを測定した。さらに、ER−αおよびER−βLBDを飽和状態にするために必要な[3H]E2の濃度を4〜6nMと測定した。
【0314】
上述の条件を用いて、[3H]E2(5.7nM)およびER LBDとともに、濃度を上げた化合物(範囲:10−11〜10−6M)をインキュベートした。培養に続いて、Unifilter−96ハーベスターでGF/Bフィルターを用いてプレートを採取し、氷冷した緩衝液B(50mMトリス、pH7.2)で3回洗浄した。室温でフィルタープレートを乾燥させ、その後、MicroScint−Oカクテル35μLを各ウェルに添加し、フィルタープレートをTopSeal−Aで密閉した。3HのMicroscintカクテル(PerkinElmer)用設定を用いて、TopCount(登録商標)NXTマイクロプレートシンチレーションカウンターで、放射線を計測した。
【0315】
[3H]E2の非特異的結合(10−6M非標識E2で培養して決定)を減算し、検査化合物がない状態での特異的結合の割合として表して、各濃度の化合物での[3H]E2の特異的結合を決定した。50%(IC50)で、[3H]E2の特異的結合を減らした化合物の濃度を決定した。次に、化合物の平衡結合定数(Ki)を、Ki=KdXIC50/(Kd+L)で計算した。ここで、Kdは、[3H]E2の平衡解離定数であり(ER−α=0.71nM;ER−β=1.13nM)、Lは[3H]E2の濃度である(ER−α:5.7nM;ER−β:5.7nM)。
【0316】
結果
【0317】
結合試験から、リガンド結合ER−αおよびER−βは、3.75nMから1,000nM以上の範囲に及ぶさまざまな濃度であり、そして選択性はアイソフォーム選択的から非アイソフォーム選択的までに及ぶことが明らかになった。代表的な化合物からの結果を表2に記載する。
【0318】
【表2A】
【0319】
【表2B】
【0320】
化合物IVはERαおよびERβに結合する。化合物IVのER結合親和性は、[2,4,6,7−3H(N)]−エストラジオール([3H]E2)、天然高親和性ERリガンド、および細菌発現GST融合ERαまたはERβリガンド結合ドメイン(LBD)タンパク質によるインビトロ競合放射性リガンド結合試験を用いて測定した。この試験では、化合物IVのERαおよびERβ結合親和性(Ki値)はそれぞれ、21.7±1.7nM(n=3)および15.2±4.1nM(n=3)だった。ERに結合するとき、化合物IVは、組織選択的な方法での薬理反応に携わる標的遺伝子の発現または抑制につながる複雑な一連の分子事象を開始する。一過性形質転換試験では、化合物IVは、ERβと比べると、ERα媒介転写活性化を促進する立証済みの効力が大きいERαおよびERβアゴニストである。エストラジオールは、ERαに対する5.1倍以上の選択性を持ってERαおよびERβを活性化させるのに対して、化合物IVはERαに対して49.0倍の選択性を示す。したがって、化合物IVは、ERβよりもERαに対して、相対的トランス活性化能において相対的に9.7倍の選択性を有する。さらに、最高10μMの濃度の化合物では、エストラジオール(1nM)−促進転写活性化でアンタゴニスト作用は見られなかった。多くのステロイドリガンドは他の核ホルモン受容体と交差反応するが、化合物IVの作用はERαおよびERβに対して特異的である。転写活性化試験におけるアゴニストおよびアンタゴニストの両モードで、グリココルチコイド受容体(GR)、ミネラルコルチコイド受容体(MR)、プロゲステロン受容体(PR)、アンドロゲン受容体(AR)のラットアイソフォームと、およびファルネソイドX受容体(FXR)、肝臓X受容体(LXR)、ペルオキシソーム増殖剤応答性受容体(PPAR−αおよびPPAR−γ)、およびレチノイドX受容体(RXR−α)のヒトアイソフォームとに対する交差反応について、化合物IVをスクリーニングした。化合物IVは、これらの試験のいずれでもアゴニストまたはアンタゴニスト活性を示さず、化合物IVが核ホルモン受容体スパーファミリーメンバーと機能的に交差反応しないということを裏付けた。
【0321】
実施例7:選択化合物のトランス活性化
【0322】
化合物がアゴニストか、アンタゴニストか、あるいは部分的かを確認するために、アゴニストおよびアンタゴニストモードでのトランス活性化試験を行った。
【0323】
方法
【0324】
ラットエストロゲン受容体(ER−αおよびER−β)を、ラット卵巣cDBAからpCR3.1プラスミドベクターバックボーンに複製した。いかなる変異も存在しないことを確認するために、シーケンシングを行った。HEK−293細胞を、Dulbeccoの最小必須培地(DMEM)+5%チャコールストリップウシ胎仔血清(csFBS)の24ウェルプレートのウェルあたり細胞100,000個で蒔いた。0.25μgのERE−LUC、0.02μgのCMV−LUC(レニラルシフェラーゼ)、および12.5ngのラットER−αまたは25ngのラットER−βに、Lipofectamine(Invitrogen、Carlsbad、CA)を用いて、細胞をトランスフェクトした。アンタゴニスト活性を確認するために、さまざまな濃度の化合物、または化合物の組み合わせ、エストラジオールでのトランスフェクションの24時間後に細胞を処理した。トランスフェクションの48時間後にルシフェラーゼ試験を行った。
【0325】
結果
【0326】
トランス活性システムでの本発明の化合物のスクリーニングによって、化合物は3つの分類、すなわちアゴニスト、アンタゴニスト、および部分的アゴニストのすべてに属することが明らかになった。アゴニストおよびアンタゴニストの例を表3に示す。トランス活性化の結果は、アイソフォーム選択性に関する結合結果に極めてよく一致した。
【0327】
表3は、本発明の一部の選択化合物に関するEC50およびIC50トランス活性化値を示す。
【0328】
【表3】
【0329】
実施例8:カニクイザルでのテストステロン抑制
【0330】
試験の間、USDA指針にしたがって、霊長類用餌と水を自由に摂れる状態で(経口投与前の絶食を除き)、2歳の生殖腺非去勢でオスのカニクイザル(n=2)を飼った。Tween 80/脱イオン水のマイクロエマルジョン賦形剤で、連続7日間、毎日1回、式IVの化合物30mg/kgを強制経口投与で与えた。第1日(基準値)、第3日、第4日、第5日、第6日、第7日の経口投与前に静脈穿刺によって血清検体を採取した。それぞれ、HPLC法を組み合わした場合と組み合わさない場合で、酵素免疫測定(EIA)法を用いてテストステロンおよび全アンドロゲンを定量した。式IVの化合物による6日間の処置後、テストステロンおよび全アンドロゲン(テストステロン/ジヒドロテストステロン)に関して、時間依存的な増加が明らかだった。式IVの化合物は、動物No.1および動物No.3において、それぞれ基準値に対して58%および64%にテストステロンの濃度を抑制した(図1の実線を参照;表4)。同様に、動物No.1およびNo.3の両方で、基準値と比較して56%に全アンドロゲン濃度を抑制した(図1の点線を参照;表4)。
【0331】
オスでの下垂体・精巣軸のエストロゲンフィードバックと一致するこれらの結果から、式IVの化合物の経口投与(30mg/kg)を繰り返した後、未去勢非ヒト霊長類(カニクイザル)での血清ホルモン(テストステロンおよび全アンドロゲン)の抑制に関する堅牢な薬理反応を実証する。
【0332】
【表4】
【0333】
実施例9:ラットのLHおよびテストステロンホルモン濃度の抑制
【0334】
未去勢および精巣摘出(ORX)雄ラットでのLH抑制に対する化合物IVの効果を評価するために、インビトロ用量依存的試験を行った。未去勢およびORX動物では、1日あたり10mg/kg以上の用量での化合物IVでは、それぞれの対照と比較した場合に、LH濃度を有意に抑制した。(FSH濃度において、同じパターンの抑制が見られた。)LH抑制によって、テストステロン濃度を、0.08ng/mLである定量限界未満(BLOQ)までしっかりと低下させ、前立腺、精嚢、および肛門拳筋はアンドロゲン依存臓器であるため、これらの重量を低下させた。未去勢動物では、去勢対照のレベルに対して精嚢および肛門拳筋の重量において、これらの対象臓器重量の用量依存的な減少が顕著だった。未去勢動物では前立腺重量が有意に減少したが、これらの値は去勢対照のレベルに達しなかった。結果を下記の表6にまとめる。
【0335】
材料および方法:
【0336】
体重約200gの雄Sprague−Dawleyラットを、餌(2016 Teklad Global 16%タンパク質齧歯類餌、Harlan、Madison、WI)および水が自由摂取できるようにして、12時間明暗サイクルで維持した。動物実験プロトコルは、テネシー大学動物実験委員会による審査および承認を受けた。
【0337】
適切な用量の製剤を調製するために、本試験の被験物質を秤量し、PEG 300(Acros Organics、NJ)で希釈した10%DMSO(Fisher)に溶かした。本試験の場合、60歳の雄Sprague−Dawleyラットを体重で無作為抽出し、12の処置群(n=動物5匹/群)のうちの1つに割り当てた。処置群は、表5に記載している。ケージあたり動物2〜3匹の群で、動物を飼った。対照群(未去勢および精巣摘出済み(ORX))には、毎日、賦形剤を投与した。未去勢群およびORX群の両方に、0.3、1、3、10、および30mg/kg/日の用量で、皮下注射(200μL)を用いて化合物IVを投与した。
【0338】
14日の投与計画の後、麻酔(ケタミン/キシラジン、87:13mg/kg)をかけて動物を屠殺し、体重を記録した。さらに、腹側前立腺、精嚢、および肛門拳筋を取り除き、外部から組織を洗浄し、個別に秤量した。臓器重量を体重で正規化し、未去勢対照に対する割合として表した。イソフルラン麻酔下で腹部大動脈から血液を採取し、凝固させた。遠心分離によって血清を分離し、血清ホルモン濃度の測定まで−80℃で保管した。メーカーの指示にしたがって、ラット下垂体ルミネックスアッセイ(Millipore、Billerica、MA)によって、血清黄体形成ホルモン(LH)および卵胞刺激ホルモン(FSH)の濃度を測定した。この試験の定量下限は、LHに対して3.2pg/mLで、FSHに対して32pg/mLだった。LLOQが0.08ng/mLのテストステロンEIA(Alpco Diagnostics、Salem、NH)でテストステロンを測定した。定量限界未満(BLOQ)の血清ホルモン値を群平均の解析から除いた。そのため、検体BLOQである群でのLHおよびTの報告値は、実測値よりも高い。この方法の解析では、LHおよびT抑制の最も控えめな推量を示した。フィッシャーの最小有意差試験を用いて、個別の用量群を未去勢およびORX賦形剤対照群と比較した。P値<0.05として、推測的に有意性を定義した。
【0339】
【表5】
【0340】
未去勢およびORXラットの黄体形成ホルモン濃度(表6)
【0341】
未去勢群およびORX賦形剤対照群のLH濃度(平均±SD)はそれぞれ、1.46±0.64および11.1±3.9ng/mLだった。化合物IVは、未去勢動物のLH濃度を用量依存的に下げ、3mg/kg以上の日用量で統計的有意な減少を実現した。0.3、1、3、10、および30mg/kg/日の投与後、未去勢化合物IV処置動物のLH濃度はそれぞれ、0.863±0.384、0.704±0.530、0.395±0.302、0.226±0.165、および0.236±0.176ng/mLだった。ORX雄動物のLH濃度も、化合物IV処置によって有意に低下した。ORX動物では、0.3、1、3、10、および30mg/kg/日の投与後、LH濃度はそれぞれ、15.4±2.9、13.5±2.2、6.5±5.6、0.425±0.135、および0.368±0.119ng/mLだった。結果は、図10Aに図示する。
【0342】
未去勢およびORXラットの卵胞刺激ホルモン濃度(表6)
【0343】
未去勢群およびORX賦形剤対照群の血清FSH濃度はそれぞれ、20.9±8.5および93.5±13.8ng/mLだった。未去勢動物では、化合物IVは、FSH濃度を用量依存的に下げ、10mg/kg/日以上の用量で有意な低下が見られた。0.3、1、3、10、および30mg/kg/日の投与後、未去勢化合物IV処置動物のFSH濃度はそれぞれ、17.3±6.4、15.7±7.3、18.4±7.7、9.2±4.0、および6.3±1.8ng/mLだった。ORX動物では、0.3、1、3、10、および30mg/kg/日の投与後、LH濃度はそれぞれ、115±17、114±22、65.2±31.9、27.6±8.2、および15.1±4.1ng/mLだった。結果は、図10Bに図示する。
【0344】
未去勢およびORXラットのテストステロン濃度
【0345】
未去勢賦形剤対照群の血清テストステロン濃度は2.4±1.1ng/mLだった。Tの定量下限は0.08ng/mLだった。0.08ng/mL未満の値は、定量限界未満(BLOQ)に指定した。未去勢動物では、式IVの化合物は、T濃度を用量依存的に下げ、3mg/kg/日以上の用量で有意な低下が見られた。そのため、0.3、1、3、10、および30mg/kg/日の投与後、式IVの化合物による処置を受けた未去勢動物のテストステロン濃度はそれぞれ、2.6±1.7、1.6±1.0、0.7±0.4ng/mLだった。ORX動物では、化合物IVによる処置を受けたすべての群および賦形剤処置群で、T濃度はBLOQだった。未去勢動物に対する結果は、図10C(および図2)に図示する(BLOQ値は、図示のために定量限界で表される)。
【0346】
図9にした通り、3mg/kg、10mg/kg、および300mg/kgの用量で化合物IVを投与することで、24時間後、72時間後、および168時間後の未去勢雄ラットの血清テストステロン濃度の迅速かつ強力な抑制が測定された。
【0347】
臓器重量(表6)
【0348】
Tの抑制を確認するために、前立腺、精嚢、および肛門拳筋の重量を測定した。前記臓器の重量(平均±SD)をそれぞれ、図10D、10E、および10Fに示す。化合物IVによる処置を受けた未去勢動物で、前立腺、精嚢、および肛門拳筋の重量の用量依存的低下が見られた。0.3、1、3、10、および30mg/kg/日の投与後、未去勢動物の前立腺重量はそれぞれ、84.0±19.2、75.2±20.7、68.2±8.1、45.1±20.0、および43.6±8.8だった。0.3、1、3、10、および30mg/kg/日の投与後、ORX動物の前立腺重量はそれぞれ、19.0±4.2、17.4±3.4、19.6±6.7、22.9±5.4、および20.6±2.1だった。0.3、1、3、10、および30mg/kg/日の投与後、未去勢動物の精嚢重量はそれぞれ、76.2±7.8、66.3±27.2、51.8±28.5、19.1±7.0、および17.9±3.3だった。0.3、1、3、10、および30mg/kg/日の投与後、ORX動物の精嚢重量はそれぞれ、12.2±1.3、16.6±5.4、16.5±4.8、13.3±1.9、および12.9±2.1だった。0.3、1、3、10、および30mg/kg/日の投与後、未去勢動物の肛門拳筋重量はそれぞれ、86.9±10.0、82.1±12.1、65.2±4.4、57.8±11.2、および58.1±4.7だった。0.3、1、3、10、および30mg/kg/日の投与後、ORX動物の肛門拳筋重量はそれぞれ、54.5±6.6、49.6±7.0、53.6±10.0、51.1±4.9、および49.2±4.2だった。LH抑制および臓器重量のデータを表6に要約する。
【0349】
【表6】
【0350】
実施例10:ラットおよびサルにおける化合物IVによる抑制後のテストステロン濃度の回復
【0351】
化合物IVによる化学的去勢の可逆性を研究した。
【0352】
材料および方法:
【0353】
体重約200gの雄Sprague−Dawleyラット35匹を、餌(2016 Teklad Global 16%タンパク質齧歯類餌、Harlan、Madison、WI)および水が自由摂取できるようにして、12時間明暗サイクルで維持した。動物実験プロトコルは、テネシー大学動物実験委員会による審査および承認を受けた。
【0354】
適切な用量の製剤を調製するために、本試験の被験物質を秤量し、PEG 300(100%)(Acros Organics、NJ)に溶かした。10の処置群(n=動物5匹/群)の1つに動物を不作為で割り当てた。処置群は、表7に記載している。ケージあたり動物2〜3匹の群で、動物を飼った。群1は、未去勢動物のテストステロン濃度を測定するために、試験の開始時(第1日)に犠牲にした。群2〜7は、3日間、強制経口投与(最大200uL)で毎日1、3、または30mg/kgの用量を摂取した。群2、3、および4は、最大のテストステロン抑制を測定するために第4日に犠牲にした。群5、6、および7は、14日間の薬物排出期間を設けて、回復させた。
【0355】
【表7】
【0356】
結果:
【0357】
未去勢ラットの血清テストステロン濃度は、ベースライン時に6.4±3.1ng/mL(平均±標準偏差)だった。3日間、3および30mg/kgの用量で化合物IVを投与することで、血清テストステロン濃度をそれぞれ1.47±0.26および1.62±0.49ng/mLに有意に低下させた。3日間、1mg/kgの化合物IVを摂取した動物では、有意な抑制は見られなかった。最も重要なことには、14日間の回復期間後に測定した場合、3日間、それぞれ1、3、または30mg/kgの化合物IVを投与された動物では、血清テストステロン濃度が3.3±1.92、3.00±1.06、および3.8±1.72だった。そして、図23に示した通り、未去勢ラットでの基準血清テストステロン濃度からの統計的に有意な違いは見られなかった。
【0358】
本研究によって、化合物IVが未去勢ラットの血清テストステロン濃度を素早く抑制することを示す以前の結果を確認する。3日間3mg/kg/日以上投与された用量群で血清テストステロン濃度の抑制を観察した。1mg/kgの用量群では、血清テストステロンに有意な低下は見られなかった。しかし、14日の回復期間内に、血清テストステロン濃度は未去勢対照の濃度に戻った。本研究から、ラットにおいて化合物IVによる薬理的去勢は可逆的であることが分かる。
【0359】
経口薬物動態研究とともに、未去勢雄ザルにおけるテストステロン濃度の抑制および回復に対する化合物IVの効果を評価した。3匹の処置未経験雄カニクイザル(2〜3歳)が、7日間連続で、強制経口投与によって、毎日30mg/kgで化合物IVの投与を受けた。血液献体を採取し、それぞれテストステロンおよび化合物IV定量測定のために、血清と血漿に分けた。結果からは、化合物IVを連日経口投与することで、3匹の雄ザルすべての血中アンドロゲン(主としてテストステロンおよびジヒドロテストステロン)濃度を、基準値と比較して最高47%に有意に低下させたことが分かる(それぞれ、基準値に対して、1591±72.5、997±104、および852±136ng/mL、処置の第2日および第6日[平均±SEM])。18日間の薬物を摂取しない回復期間の後、アンドロゲン濃度は通常に戻り、処置前の基準濃度との有意な違いは無かった(回復後、1757.7±369.5ng/mL)。
【0360】
実施例11:ラットのLHおよびテストステロンが低下するにもかかわらず骨量を維持する(表8)
【0361】
骨の処置に対する式IVの化合物の効果を研究した。式IVの化合物を経口投与することで、未去勢雄ラットでのLH抑制に関連した骨量減少を完全に予防した。LHの有意な低下は、1日あたり10mg/kg以上の用量で未去勢動物に式IVの化合物を投与することによって誘導された。1日あたり1mg/kgでは、式IVの化合物はLHを有意に下げなかったが、この用量で前立腺、精嚢、および肛門拳筋の重量を有意に減らすことが明らかになり、血中テストステロンの低下がこれらのアンドロゲン応答性臓器と生理学的に関係していることが分かった。しかし、1日あたり1mg/kgの式IVの化合物では、未去勢対照のレベルに骨梁量(大腿骨遠位)を維持した。1日あたり10および30mg/kgの用量で投与した場合、式IVの化合物は、未去勢対照の骨量以上に、骨梁の骨量を有意に増加させた。これらのデータから、化合物IVによって、未去勢ラットのLH濃度を下げる用量レベルで骨梁骨ミネラル濃度(BMD)および骨稜率を増やしたことが分かる。この研究から得られたデータを表8に記載する。
【0362】
【表8】
【0363】
実施例12:17βヒドロキシステロイド脱水素酵素5(17β−HSD5)酵素活性に対する効果
【0364】
HSD群の構成要素は、血中ステロイドの変換に関与する。17β−HSD5は、アンドロステンジオンをテストステロンに、エストロンをエストラジオールに変換する。さらに、プロスタグランジン合成にも関与する。ここで、17β−HSD5活性を阻害する本発明の一部の選択化合物の能力を実証した。
【0365】
方法
【0366】
pGEX 4t1ベクターでヒト17β−HSD5を複製し、精製タンパク質を調製した。適切な緩衝液中で、本発明の代表的な化合物、14Cアンドロステンジオン、NADPHとともに、精製タンパク質をインキュベートした。酢酸エチルを用いて合成したテストステロンを抽出し、乾燥させ、薄層クロマトグラフィー(TLC)プレートに置き、分析した。TLCをフォスフォイメージャーにさらし、テストステロン結合の強度を定量した。陽性対照(LHRHアゴニスト)としてインドメタシンを使用した。
【0367】
結果
【0368】
化合物IVを検査し、17β−HSD5酵素活性に対して部分的抑制効果があった。図3に示した通り、陽性対照(LHRHアゴニスト)、インドメタシンは、予想通りに、本酵素の強力な抑制を示した。
【0369】
実施例13:毒性研究
【0370】
ヒト血小板凝集試験を用いて、化合物IVとジエチルスチルベストール(DES、陽性対照)の血栓症の可能性を比較するために、研究を行った。男性が化合物IVの対照処置集団(LH抑制)であるため、研究では健常男性供血者からの血液を使用した。30秒間、エストラジオール(E2)、DES、化合物IV、または賦形剤のいずれかとともに、多血小板血漿を前保温し、その後、血小板凝集を誘導するためにトロンビン(0.3単位)を添加した。研究の結果から、DESとの前保温によって、トロンビン誘導血小板凝集が約10倍増えたことが分かる。しかし、化合物IVおよびエストラジオールは多血小板血漿の凝集を減らした。これらのデータから、DESと比較して、化合物IVはインビトロのヒト血小板の反応性を下げたことを実証し、化合物IVはDESよりも血栓塞栓症の可能性が低いことを示唆する(図4)。
【0371】
実施例14:ほてりに対する化合物IVの効果
【0372】
Simpkins等によって開発された(1983年)モルヒネ依存ラットモデル(MDモデル)を用いて、ほてりに対する化合物IVの効果を調査するために研究を行い、閉経によるほてりとの幾つかの類似性があることが分かった。ヒトの症状との類似性に加えて、本実験動物モデルは、尾皮膚温度(TST)を用いて血管運動症状を緩和できる化合物を特定する便利な高性能ツールにする短い応答時間を持つ。TSTプローブTA−40(Data Sciences International, MN)を尾の根元にテープで貼り、15分間、基準温度を得た。15分後、モルヒネの効果を無効にするために、動物をナロキソン(1mg/kgSQ)で処置した。一連の実験の間中、5秒間のサンプリング頻度で、ナロキソン処置前の1時間の尾皮膚温度(TST)を測定した。データ収集に続いて、各動物に対して60秒ごとに記録された温度の移動平均を計算し、さらに解析した。ナロキソン投与前の15分にわたって取得された平均温度として、基準温度を計算した。線形台形を用いて、基準値からすべてのナロキソン投与前値を減算することで、濃度曲線下面積(AUC)を計算した。
【0373】
化合物IVは、モルヒネ禁断モデル(図13)のほてりを弱め、10mgの化合物IVで最高の結果が得られた。17βE2は、100%DMSO溶液で5mg/kgを使用した。
【0374】
実施例15:ラットでの化合物IV対DES
【0375】
LHRHアゴニストを導入する前に、エストロゲン、主としてジエチルスチルベストロール(DES)を介して下垂体でのエストロゲン活性を増加させることで、去勢テストステロン濃度が達成された。DESは、去勢濃度へのテストステロンの抑制においてLHRHアゴニストとして同じように有効であった。DESによって処置された患者はほてりまたは骨量減少を示さなかったが、LHRHアゴニストによるADTよりも高い率で女性化乳房を示した。残念ながら、DESおよびエストラジオールのような極めて強力な純エストロゲンは、その臨床使用を制限してきた重篤な心血管および血栓塞栓合併症の高いリスクと関係がある場合が多い。DESによる静脈血栓塞栓合併症のリスト上昇は、他のホルモン受容体とのその交差反応性によることが、証明されていないが、仮定してきた。ヒト血小板を用いたインビトロ研究から、化合物IVがDESよりもかなり低い前凝固活性を示した。そのため、化合物IV、ER−α選択的アゴニストは、DESの前立腺癌に関する利点を実現でき、骨粗鬆症または有害な脂質状態の変化を生じることなくLHRHアゴニストの利点も実現できる。
【0376】
化合物IVは、ラットの前立腺サイズの削減でDESと同じくらい効果があり(図11A)、また、ORXラットの前立腺サイズの中程度の増加を示す(図11B)。
【0377】
DESと化合物IVとの差を図12A〜12Cに示す。ここで、化合物IVは交差反応しなかったが、DESはグルココルチコイド受容体(GR)(図12A)およびアンドロゲン受容体(AR)(図12B)と交差反応した。さらに、DESはエストロゲン関連受容体(ERR)トランス活性化を無効にした。一方で化合物IVは無効にしなかった。化合物IVは、図12Cに示した通り、3種類のERRアイソフォーム(ERR−α、ERR−β、およびERR−γ)のいずれとも交差反応しなかった。
【0378】
実施例16:サル毒性研究−90日
【0379】
モーリシャス育ちの集団飼育カニクイザルを入手した。13週の暫定期間で、雄カニクイザルにおける化合物IVおよび陽性対照(LHRHアゴニスト)の39週経口薬理および毒性評価として、プロスペクティブ研究を計画した。処置開始前に、合計39匹の性的に成熟した5〜8歳の雄ザルを5つの群に無作為に割り当てた。群としては次のものが挙げられる。すなわち、1)賦形剤、2)1mg/kgの化合物IV、3)10mg/kgの化合物IV、4)100mg/kgの化合物IV、および5)陽性対照(LHRHアゴニスト)。群1および5に対して賦形剤対照品(Tween 80/PRANG(商標))または群2、3、および4に対して化合物IVを含む賦形剤を用いて、39週間、1日1回のケージサイド投与によって薬剤を経口で送達した。化合物IVの用量レベルは、群2、3、および4に対して、それぞれ1、10、および100mg/kg/日だった。各動物に対して直近に入手した体重に基づく計算通りに、10mg/kgの用量で経口量を送達した。群5の動物は、39週の研究期間中、陽性対照(LHRHアゴニスト)(0.02mL定量)の1日1回の皮下注射も受けた。毎日、全体的な外観および臨床兆候を観察、記録した。研究プロトコルに示した通り、所定の評価および厳選した他の研究調査を行った。厳選したパラメータとしては、テストステロン、前立腺特異抗原(PSA)、および前立腺の容積および重量が挙げられるが、これに限定されるものではない。
【0380】
酵素免疫測定(EIA)法および化学発光免疫測定(LIA、ALPCO Diagnostics、Salem NH)それぞれを用いて、血清検体のテストステロンおよび全PSA濃度を定量した(標準手順にしたって)。ベースライン時(すなわち、処置開始前)と、第1、3、7、14、28、64、および90日にすべての動物(絶食状態で)からテストステロン評価用の血液検体を採取した。ベースライン時と第6週の間にすべての動物(絶食状態で)PSA測定用の血液検体を採取した。議論のために、テストステロンおよびPSA試験の定量限界未満(BLQ)の濃度を持つ検体の結果は、試験の定量下限(LLOQ)の1/2として計算され、「予想最終濃度」と考えられる。表9〜16のデータは、「予想最終濃度」(すなわち、検体BLQを持つ検体が試験のLLOQの1/2として含まれた)に加えて、「定量化可能濃度のみ」(BLQ値を除く)として示される。ベースライン時と第6週に経直腸超音波(TRUS)検査を用いて、麻酔下の生きた動物で前立腺容積を測定した。前立腺の幅と高さを記録した。幅*幅*高さ*π/6[ref]として前立腺容積を計算し、体重に正規化した。組織を脂肪のないように、そして異質組織を刈り込んだ後、剖検時に前立腺の湿重量を記録した。
【0381】
結果および議論:
【0382】
血清テストステロン濃度を図15および表9〜12に示す。ベースライン時、研究のすべてのサルのテストステロン濃度が、性的に成熟した大人の雄カニクイザルに対して通常範囲内だった。しかし、100mg/kg/日で化合物IVを摂取しているサル、および陽性対照(LHRHアゴニスト)によって処置されたサルでは、テストステロン濃度が有意に低下した。陽性対照(LHRHアゴニスト)群のテストステロン濃度は、それぞれ、第1日と第3日に47.4%と547%(p<0.01)の有意な初期増加(すなわち、フレア)とともに、二相性変化を示し、続いて、第7、14、28、64、および90日に、3.6%、67%、73%、83%、および85%の低下を示した(図15および表9〜12を参照)。最高の用量レベル(すなわち、100mg/kg/日)でも、化合物IVにより処置されたどの動物にも、同様のフレアは見られなかった。用量および処置期間は化合物IVの薬理作用にとって重要だった。ここで、100mg/kg/日の用量では、第3、7、14、28、および64日にそれぞれ、血清テストステロンを基準値に対して60%、51%、42%、79%、および92%抑制した(図15および表9および10を参照)。100mg/kg/日の化合物による処置の90日後、群4のサルの10匹中6匹のテストステロン濃度は、試験の定量限界未満の濃度に低下した(表11を参照)。群4のサルの平均血清テストステロン濃度は、それぞれの基準値と比較して96%低下した(「予想最終濃度」、すなわち、BLQ値を持つ6/10匹のサルのテストステロン濃度は、LLOQ濃度の50%として計算される。表10を参照)。第90日で、100mg/kg/日の化合物IVによって、血清テストステロンを陽性対照(LHRHアゴニスト)よりも有意に低い濃度に下げたことに留意することが重要である(p=0.013)。
【0383】
【表9】
【0384】
【表10】
【0385】
【表11】
【0386】
【表12】
【0387】
処置開始の4週間以内に化合物IVによって血清PSA濃度も有意に抑制された。陽性対照(LHRHアゴニスト)群ではPSA濃度が60%低下した一方で、4週間、10mg/kgおよび100mg/kgで化合物IVを摂取しているサルに対して、67%および87%(平均で)のPSA低下が分かった(図16および表13〜16)。
【0388】
【表13】
【0389】
【表14】
【0390】
【表15】
【0391】
【表16】
【0392】
研究を通じて定期的に、TRUSによって前立腺容積を測定した。6週間の処置後に得られる結果から、サルの前立腺に対する化合物IVおよび陽性対照(LHRHアゴニスト)の強力な効果を実証する。化合物IVは、それぞれ10mg/kgおよび100mg/kgで前立腺容積を25%および45%、有意に抑制した。一方で、陽性対照(LHRHアゴニスト)群では28%、前立腺を低下させた(図17ならびに表17および18)。
【0393】
【表17】
【0394】
【表18】
【0395】
剖検時の前立腺重量の評価によって、前立腺容積の化合物IVに関連した減少が確認された。13週間の処置後、化合物IVは、それぞれ10および100mg/kg/日を摂取している動物において、平均前立腺重量を24%および21%と有意に減少させた(図18および表19および20)。
【0396】
【表19】
【0397】
【表20】
【0398】
血小板凝集、プロトロンビン時間(PT)、または活性化部分トロンボプラスチン時間(APTT)に対する明らかな効果は見られなかった。
【0399】
実施例17:ヒトに対する化合物IV研究
【0400】
ヒトの男性に対する化合物IVの効果を確認するために、研究を行った。化合物IVの100、300、600、および1000mgの用量で、群あたり12人の対象を調べた。表21には、100、300、600、および1000mgの用量で化合物IVを投与することによる、男性のLH、血清PSA、遊離テストステロン、および全テストステロン濃度の平均変化を示す。第1日〜第11日の期間中のヒトでの用量依存的平均全テストステロン濃度(nmol/L)を測定した(図19)。それぞれ、600mgおよび1000mgの用量で、全テストステロン濃度は51.9%および47.9%低下した。
【0401】
第1日〜第10日の期間中のヒトでの用量依存的平均LH濃度(IU/L)を測定した(図20)。それぞれ、100mg、300mg、600mgおよび1000mgの用量で、LH濃度は20.7%、46.9%、27.6%および29.2%低下した。
【0402】
第1日〜第10日の期間中のヒトでの用量依存的遊離テストステロン濃度(pg/mL)を測定した(図21)。それぞれ、100mg、300mg、600mgおよび1000mgの用量で、遊離テストステロン濃度は17.0%、18.5%、72.7%、および53.2%低下した。
【0403】
第1日〜第10日の期間中のヒトでの用量依存的平均PSA濃度(μg/L)を測定した(図22)。それぞれ、100mg、300mg、600mgおよび1000mgの用量で、PSA濃度は9.2%、24.4%、27.5%、および29.9%低下した。10および30mgの用量に対しては、変化が無いことが分かった。
【0404】
【表21】
【0405】
実施例18:化合物IVのバイオアベイラビリティ
【0406】
ラット、イヌ、およびサルへの経口投与の後、化合物IVは迅速に吸収された。1回分を投与した剤形に応じて、ラットでの化合物IVの経口バイオアベイラビリティは、6%〜25%に及んだ。ポリエチレングリコール300(PEG300)を用いた製剤は、脱イオン水で希釈されたTween 80で調製されるマイクロエマルションよりも高い暴露を典型的に生じる。イヌでは、血書濃度−時間プロファイルの目視検査から、終末期の第2ピークでも分かるように、化合物IVが腸肝循環を経ることが分かった。重要なことには、イヌでは、雄のPEG300を30mg/kg経口投与した群の暴露は、LH抑制のラットモデルでの前立腺減少に対する最大作用を生じるために必要な暴露を超えた。サルでは、予備薬物動態学的研究によって、化合物IVの血漿濃度、および7日間の期間にわたる血漿テストステロンの抑制でも分かるように、この種の経口バイオアベイラビリティはイヌのものに近似する、または超えることが示された。総じて、これらのデータから、所望の薬理作用を生じるように、2つの非齧歯動物種で十分な経口暴露を達成できることを示している(AUCデータに基づく)。さらに、ラットおよびサルの内分泌データから、化合物IVの薬理作用は可逆的(すなわち、化合物IVによる処置が停止されると、テストステロンの血清濃度は基準値または通常濃度に戻る)であることを示している。
【0407】
実施例19:化合物IVの薬物動態
【0408】
インビトロ(マウス、ラット、イヌ、サルおよびヒト)およびインビボ(ラット)代謝研究からの予備データによって、動物およびヒトでは、化合物IVの結合、そのヒドロキシル化代謝産物、およびそのN−脱アルキル化代謝産物が、化合物IVの全般的な分解に寄与することを示している。定性のみだが、種間比較の結果から、非臨床種の全般的な代謝物プロファイルは、ヒト肝臓ミクロソームで作られるプロファイルに十分に反映することを示す。これらの結果に基づき、ラットおよびイヌはそれぞれ、薬理および毒性評価に適した齧歯および非齧歯種である。インビトロ研究から、化合物IVは、30μM以下の濃度では、関連かるCYP450アイソフォーム(CYP1A2、CYP2B6、またはCYP3A4)の減少を誘導せず、かつCYP1A2、CYP2C19、CYP2D6、またはCYP3A4/5を阻害しない。CYP2C9は、高濃度(Ki=8μM)のみで化合物IVによって阻害されるが、強力な薬物動態薬物・薬物相互作用がごくわずかにあると考えられる。
【0409】
実施例20:化合物IVの生物活性
【0410】
化合物IVは、hERGチャンネルに対してインビトロの阻害作用(IC50≧300μM)をわずかに及ぼす、または及ぼさない。化合物は、インビトロで分離したイヌプルキンエ線維において、10および100μMの濃度でAPD50およびAPD90を用量依存的に減らした。しかし、化合物IVは、あらゆる用量(最高300mg/kg)での遠隔計測器が付けられたイヌにおいて、血流動態または心臓機能(血圧、心拍数、心電図形態、またはQT間隔)に影響を及ぼさなかった。神経薬理または肺の効果は見られなかった。最高30mg/kgの化合物IVの単回経口投与では、腎臓機能に対して有意な効果は示されなかった。高用量研究(100mg/kg)では、尿排出量と、カリウムおよび塩化物の尿中排泄との増加のみが見られた。ラットにおいて30〜300mg/kgの用量で化合物IVを経口投与することによって蠕動の有意な増加を生じさせ、ラットにおいて30mg/kgの用量で化合物IVを経口投与することによって、胃腸の運動および胃液の酸度の有意な増加を生じさせた(おそらく、平滑筋に対する効果によるものではない)。
【0411】
化合物IVには変異原性はなく、インビトロでのヒト末梢血リンパ球中で最高200μMの濃度では構造的または数的染色体異常を誘導しなかった。化合物IVは、単回および反復経口投与(最高28日)の後、ラットおよびイヌで良好な耐容性を示した。腎臓、肝臓、心臓、および他の非対象関連臓器では、病変は見られなかった。最長28日間の雄および雌のイヌに対する化合物IVの投与に関連した深刻な身体的兆候、体重に関する効果、臨床病理的変化、眼科、心電図、または組織病理学的変化は無かった。
【0412】
本願明細書では、本発明のいくつかの特徴が説明、記述されているが、現在では多くの修正、代用、変更、および同等物を当業者が思い付くことになる。そのため、当然のことながら、追加請求項は、本発明の真意の範囲内でそのような修正および変更のすべてを対象にするためのものである。
【特許請求の範囲】
【請求項1】
アンドロゲン遮断療法(ADT)により誘発されるほてりを処置する方法であって、
対象に対して、式Iの化合物、またはその異性体、薬学的に許容される塩、N−酸化物、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与するステップを含むことを特徴とする方法。
【化1】
式中、
Yは、C(O)またはCH2であり;
R1、R2は、互いに独立して、水素、ハロゲン、ヒドロキシル、アルコキシ、シアン、ニトロ、CF3、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、O−Alk−NR5R6、または、置換されたもしくは非置換の3〜7員のO−Alk−複素環(任意選択で芳香族である)であり;
R3、R4は、互いに独立して、水素、ハロゲン、ヒドロキシアルキル、ヒドロキシル、アルコキシ、シアン、ニトロ、CF3、NHCOR、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、または保護ヒドロキシルであり;
Rは、アルキル、水素、ハロアルキル、ジハロアルキル、トリハロアルキル、CH2F、CHF2、CF3、CF2CF3、アリール、フェニル、ハロゲン、アルケニル、CN、NO2、またはOHであり;
R5およびR6は、互いに独立して、水素、フェニル、炭素数1〜6のアルキル基、3〜7員のシクロアルキル、3〜7員の複素環、5〜7員のアリールであり;あるいは、R5およびR6は、窒素原子を含む3〜7員環を形成し;
jおよびkは、互いに独立して、1〜4であり;
Alkは、炭素数1〜7の直鎖アルキル、炭素数1〜7の分岐鎖アルキル、または炭素数3〜8の環状アルキルである。
【請求項2】
請求項1の方法であって、
前記式Iの化合物が以下から選択されることを特徴とする方法。
【化2】
【化3】
【化4】
【化5】
【化6】
【化7】
【化8】
【化9】
【化10】
【化11】
【化12】
【請求項3】
請求項1の方法であって、
前記対象がヒトであることを特徴とする方法。
【請求項4】
請求項3の方法であって、
前記ヒトが男性であることを特徴とする方法。
【請求項5】
請求項1の方法であって、
前記対象が前立腺癌を患っていることを特徴とする方法。
【請求項6】
請求項5の方法であって、
前記前立腺癌が進行前立腺癌であることを特徴とする方法。
【請求項7】
アンドロゲン遮断療法(ADT)により誘発されるほてりを抑制もしくは防止、または該ほてりの発症率を低減する方法であって、
アンドロゲン遮断療法(ADT)により誘発されるほてりを抑制もしくは防止、または該ほてりの発症率を低減する方法であって、
対象に対して、式Iの化合物、またはその異性体、薬学的に許容される塩、N−酸化物、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与するステップを含むことを特徴とする方法。
【化1】
式中、
Yは、C(O)またはCH2であり;
R1、R2は、互いに独立して、水素、ハロゲン、ヒドロキシル、アルコキシ、シアン、ニトロ、CF3、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、O−Alk−NR5R6、または、置換されたもしくは非置換の3〜7員のO−Alk−複素環(任意選択で芳香族である)であり;
R3、R4は、互いに独立して、水素、ハロゲン、ヒドロキシアルキル、ヒドロキシル、アルコキシ、シアン、ニトロ、CF3、NHCOR、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、または保護ヒドロキシルであり;
Rは、アルキル、水素、ハロアルキル、ジハロアルキル、トリハロアルキル、CH2F、CHF2、CF3、CF2CF3、アリール、フェニル、ハロゲン、アルケニル、CN、NO2、またはOHであり;
R5およびR6は、互いに独立して、水素、フェニル、炭素数1〜6のアルキル基、3〜7員のシクロアルキル、3〜7員の複素環、5〜7員のアリールであり;あるいは、R5およびR6は、窒素原子を含む3〜7員環を形成し;
jおよびkは、互いに独立して、1〜4であり;
Alkは、炭素数1〜7の直鎖アルキル、炭素数1〜7の分岐鎖アルキル、または炭素数3〜8の環状アルキルである。
【請求項8】
請求項7の方法であって、
前記式Iの化合物が以下から選択されることを特徴とする方法。
【化2】
【化3】
【化4】
【化5】
【化6】
【化7】
【化8】
【化9】
【化10】
【化11】
【化12】
【請求項9】
請求項7の方法であって、
前記対象がヒトであることを特徴とする方法。
【請求項10】
請求項9の方法であって、
前記ヒトが男性であることを特徴とする方法。
【請求項11】
請求項7の方法であって、
前記対象が前立腺癌を患っていることを特徴とする方法。
【請求項12】
請求項11の方法であって、
前記前立腺癌が進行前立腺癌であることを特徴とする方法。
【請求項13】
アンドロゲン遮断療法(ADT)により誘発される重度のほてりを処置する方法であって、
対象に対して、式Iの化合物、またはその異性体、薬学的に許容される塩、N−酸化物、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与するステップを含むことを特徴とする方法。
【化1】
式中、
Yは、C(O)またはCH2であり;
R1、R2は、互いに独立して、水素、ハロゲン、ヒドロキシル、アルコキシ、シアン、ニトロ、CF2、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、O−Alk−NR5R6、または、置換されたもしくは非置換の3〜7員のO−Alk−複素環(任意選択で芳香族である)であり;
R3、R4は、互いに独立して、水素、ハロゲン、ヒドロキシアルキル、ヒドロキシル、アルコキシ、シアン、ニトロ、CF3、NHCOR、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、または保護ヒドロキシルであり;
Rは、アルキル、水素、ハロアルキル、ジハロアルキル、トリハロアルキル、CH2F、CHF2、CF3、CF2CF3、アリール、フェニル、ハロゲン、アルケニル、CN、NO2、またはOHであり;
R5およびR6は、互いに独立して、水素、フェニル、炭素数1〜6のアルキル基、3〜7員のシクロアルキル、3〜7員の複素環、5〜7員のアリールであり;あるいは、R5およびR6は、窒素原子を含む3〜7員環を形成し;
jおよびkは、互いに独立して、1〜4であり;
Alkは、炭素数1〜7の直鎖アルキル、炭素数1〜7の分岐鎖アルキル、または炭素数3〜8の環状アルキルである。
【請求項14】
請求項13の方法であって、
前記式Iの化合物が以下から選択されることを特徴とする方法。
【化2】
【化3】
【化4】
【化5】
【化6】
【化7】
【化8】
【化9】
【化10】
【化11】
【化12】
【請求項15】
請求項13の方法であって、
前記対象がヒトであることを特徴とする方法。
【請求項16】
請求項15の方法であって、
前記ヒトが男性であることを特徴とする方法。
【請求項17】
請求項13の方法であって、
前記対象が前立腺癌を患っていることを特徴とする方法。
【請求項18】
請求項17の方法であって、
前記前立腺癌が進行前立腺癌であることを特徴とする方法。
【請求項19】
アンドロゲン遮断療法(ADT)により誘発される重度のほてりを抑制もしくは防止、または該ほてりの発症率を低減する方法であって、
対象に対して、式Iの化合物、またはその異性体、薬学的に許容される塩、N−酸化物、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与するステップを含むことを特徴とする方法。
【化1】
式中、
Yは、C(O)またはCH2であり;
R1、R2は、互いに独立して、水素、ハロゲン、ヒドロキシル、アルコキシ、シアン、ニトロ、CF2、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、O−Alk−NR5R6、または、置換されたもしくは非置換の3〜7員のO−Alk−複素環(任意選択で芳香族である)であり;
R3、R4は、互いに独立して、水素、ハロゲン、ヒドロキシアルキル、ヒドロキシル、アルコキシ、シアン、ニトロ、CF3、NHCOR、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、または保護ヒドロキシルであり;
Rは、アルキル、水素、ハロアルキル、ジハロアルキル、トリハロアルキル、CH2F、CHF2、CF3、CF2CF3、アリール、フェニル、ハロゲン、アルケニル、CN、NO2、またはOHであり;
R5およびR6は、互いに独立して、水素、フェニル、炭素数1〜6のアルキル基、3〜7員のシクロアルキル、3〜7員の複素環、5〜7員のアリールであり;あるいは、R5およびR6は、窒素原子を含む3〜7員環を形成し;
jおよびkは、互いに独立して、1〜4であり;
Alkは、炭素数1〜7の直鎖アルキル、炭素数1〜7の分岐鎖アルキル、または炭素数3〜8の環状アルキルである。
【請求項20】
請求項19の方法であって、
前記式Iの化合物が以下から選択されることを特徴とする方法。
【化2】
【化3】
【化4】
【化5】
【化6】
【化7】
【化8】
【化9】
【化10】
【化11】
【化12】
【請求項21】
請求項19の方法であって、
前記対象がヒトであることを特徴とする方法。
【請求項22】
請求項21の方法であって、
前記ヒトが男性であることを特徴とする方法。
【請求項23】
請求項19の方法であって、
前記対象が前立腺癌を患っていることを特徴とする方法。
【請求項24】
請求項23の方法であって、
前記前立腺癌が進行前立腺癌であることを特徴とする方法。
【請求項1】
アンドロゲン遮断療法(ADT)により誘発されるほてりを処置する方法であって、
対象に対して、式Iの化合物、またはその異性体、薬学的に許容される塩、N−酸化物、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与するステップを含むことを特徴とする方法。
【化1】
式中、
Yは、C(O)またはCH2であり;
R1、R2は、互いに独立して、水素、ハロゲン、ヒドロキシル、アルコキシ、シアン、ニトロ、CF3、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、O−Alk−NR5R6、または、置換されたもしくは非置換の3〜7員のO−Alk−複素環(任意選択で芳香族である)であり;
R3、R4は、互いに独立して、水素、ハロゲン、ヒドロキシアルキル、ヒドロキシル、アルコキシ、シアン、ニトロ、CF3、NHCOR、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、または保護ヒドロキシルであり;
Rは、アルキル、水素、ハロアルキル、ジハロアルキル、トリハロアルキル、CH2F、CHF2、CF3、CF2CF3、アリール、フェニル、ハロゲン、アルケニル、CN、NO2、またはOHであり;
R5およびR6は、互いに独立して、水素、フェニル、炭素数1〜6のアルキル基、3〜7員のシクロアルキル、3〜7員の複素環、5〜7員のアリールであり;あるいは、R5およびR6は、窒素原子を含む3〜7員環を形成し;
jおよびkは、互いに独立して、1〜4であり;
Alkは、炭素数1〜7の直鎖アルキル、炭素数1〜7の分岐鎖アルキル、または炭素数3〜8の環状アルキルである。
【請求項2】
請求項1の方法であって、
前記式Iの化合物が以下から選択されることを特徴とする方法。
【化2】
【化3】
【化4】
【化5】
【化6】
【化7】
【化8】
【化9】
【化10】
【化11】
【化12】
【請求項3】
請求項1の方法であって、
前記対象がヒトであることを特徴とする方法。
【請求項4】
請求項3の方法であって、
前記ヒトが男性であることを特徴とする方法。
【請求項5】
請求項1の方法であって、
前記対象が前立腺癌を患っていることを特徴とする方法。
【請求項6】
請求項5の方法であって、
前記前立腺癌が進行前立腺癌であることを特徴とする方法。
【請求項7】
アンドロゲン遮断療法(ADT)により誘発されるほてりを抑制もしくは防止、または該ほてりの発症率を低減する方法であって、
アンドロゲン遮断療法(ADT)により誘発されるほてりを抑制もしくは防止、または該ほてりの発症率を低減する方法であって、
対象に対して、式Iの化合物、またはその異性体、薬学的に許容される塩、N−酸化物、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与するステップを含むことを特徴とする方法。
【化1】
式中、
Yは、C(O)またはCH2であり;
R1、R2は、互いに独立して、水素、ハロゲン、ヒドロキシル、アルコキシ、シアン、ニトロ、CF3、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、O−Alk−NR5R6、または、置換されたもしくは非置換の3〜7員のO−Alk−複素環(任意選択で芳香族である)であり;
R3、R4は、互いに独立して、水素、ハロゲン、ヒドロキシアルキル、ヒドロキシル、アルコキシ、シアン、ニトロ、CF3、NHCOR、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、または保護ヒドロキシルであり;
Rは、アルキル、水素、ハロアルキル、ジハロアルキル、トリハロアルキル、CH2F、CHF2、CF3、CF2CF3、アリール、フェニル、ハロゲン、アルケニル、CN、NO2、またはOHであり;
R5およびR6は、互いに独立して、水素、フェニル、炭素数1〜6のアルキル基、3〜7員のシクロアルキル、3〜7員の複素環、5〜7員のアリールであり;あるいは、R5およびR6は、窒素原子を含む3〜7員環を形成し;
jおよびkは、互いに独立して、1〜4であり;
Alkは、炭素数1〜7の直鎖アルキル、炭素数1〜7の分岐鎖アルキル、または炭素数3〜8の環状アルキルである。
【請求項8】
請求項7の方法であって、
前記式Iの化合物が以下から選択されることを特徴とする方法。
【化2】
【化3】
【化4】
【化5】
【化6】
【化7】
【化8】
【化9】
【化10】
【化11】
【化12】
【請求項9】
請求項7の方法であって、
前記対象がヒトであることを特徴とする方法。
【請求項10】
請求項9の方法であって、
前記ヒトが男性であることを特徴とする方法。
【請求項11】
請求項7の方法であって、
前記対象が前立腺癌を患っていることを特徴とする方法。
【請求項12】
請求項11の方法であって、
前記前立腺癌が進行前立腺癌であることを特徴とする方法。
【請求項13】
アンドロゲン遮断療法(ADT)により誘発される重度のほてりを処置する方法であって、
対象に対して、式Iの化合物、またはその異性体、薬学的に許容される塩、N−酸化物、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与するステップを含むことを特徴とする方法。
【化1】
式中、
Yは、C(O)またはCH2であり;
R1、R2は、互いに独立して、水素、ハロゲン、ヒドロキシル、アルコキシ、シアン、ニトロ、CF2、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、O−Alk−NR5R6、または、置換されたもしくは非置換の3〜7員のO−Alk−複素環(任意選択で芳香族である)であり;
R3、R4は、互いに独立して、水素、ハロゲン、ヒドロキシアルキル、ヒドロキシル、アルコキシ、シアン、ニトロ、CF3、NHCOR、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、または保護ヒドロキシルであり;
Rは、アルキル、水素、ハロアルキル、ジハロアルキル、トリハロアルキル、CH2F、CHF2、CF3、CF2CF3、アリール、フェニル、ハロゲン、アルケニル、CN、NO2、またはOHであり;
R5およびR6は、互いに独立して、水素、フェニル、炭素数1〜6のアルキル基、3〜7員のシクロアルキル、3〜7員の複素環、5〜7員のアリールであり;あるいは、R5およびR6は、窒素原子を含む3〜7員環を形成し;
jおよびkは、互いに独立して、1〜4であり;
Alkは、炭素数1〜7の直鎖アルキル、炭素数1〜7の分岐鎖アルキル、または炭素数3〜8の環状アルキルである。
【請求項14】
請求項13の方法であって、
前記式Iの化合物が以下から選択されることを特徴とする方法。
【化2】
【化3】
【化4】
【化5】
【化6】
【化7】
【化8】
【化9】
【化10】
【化11】
【化12】
【請求項15】
請求項13の方法であって、
前記対象がヒトであることを特徴とする方法。
【請求項16】
請求項15の方法であって、
前記ヒトが男性であることを特徴とする方法。
【請求項17】
請求項13の方法であって、
前記対象が前立腺癌を患っていることを特徴とする方法。
【請求項18】
請求項17の方法であって、
前記前立腺癌が進行前立腺癌であることを特徴とする方法。
【請求項19】
アンドロゲン遮断療法(ADT)により誘発される重度のほてりを抑制もしくは防止、または該ほてりの発症率を低減する方法であって、
対象に対して、式Iの化合物、またはその異性体、薬学的に許容される塩、N−酸化物、薬学的生成物、多形体もしくは水和物、あるいはそれらの任意の組み合わせを治療有効量投与するステップを含むことを特徴とする方法。
【化1】
式中、
Yは、C(O)またはCH2であり;
R1、R2は、互いに独立して、水素、ハロゲン、ヒドロキシル、アルコキシ、シアン、ニトロ、CF2、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、O−Alk−NR5R6、または、置換されたもしくは非置換の3〜7員のO−Alk−複素環(任意選択で芳香族である)であり;
R3、R4は、互いに独立して、水素、ハロゲン、ヒドロキシアルキル、ヒドロキシル、アルコキシ、シアン、ニトロ、CF3、NHCOR、N(R)2、スルホンアミド、SO2R、アルキル、ハロアルキル、アリール、または保護ヒドロキシルであり;
Rは、アルキル、水素、ハロアルキル、ジハロアルキル、トリハロアルキル、CH2F、CHF2、CF3、CF2CF3、アリール、フェニル、ハロゲン、アルケニル、CN、NO2、またはOHであり;
R5およびR6は、互いに独立して、水素、フェニル、炭素数1〜6のアルキル基、3〜7員のシクロアルキル、3〜7員の複素環、5〜7員のアリールであり;あるいは、R5およびR6は、窒素原子を含む3〜7員環を形成し;
jおよびkは、互いに独立して、1〜4であり;
Alkは、炭素数1〜7の直鎖アルキル、炭素数1〜7の分岐鎖アルキル、または炭素数3〜8の環状アルキルである。
【請求項20】
請求項19の方法であって、
前記式Iの化合物が以下から選択されることを特徴とする方法。
【化2】
【化3】
【化4】
【化5】
【化6】
【化7】
【化8】
【化9】
【化10】
【化11】
【化12】
【請求項21】
請求項19の方法であって、
前記対象がヒトであることを特徴とする方法。
【請求項22】
請求項21の方法であって、
前記ヒトが男性であることを特徴とする方法。
【請求項23】
請求項19の方法であって、
前記対象が前立腺癌を患っていることを特徴とする方法。
【請求項24】
請求項23の方法であって、
前記前立腺癌が進行前立腺癌であることを特徴とする方法。
【図1】

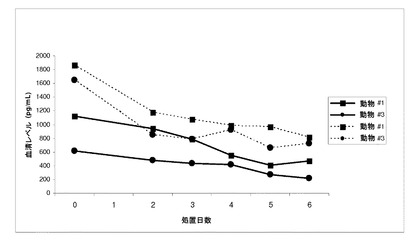
【図2】
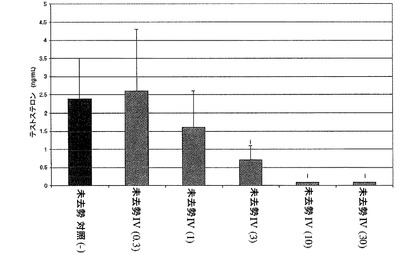
【図3】
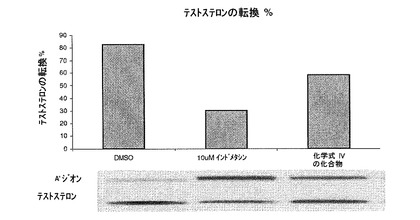
【図4】
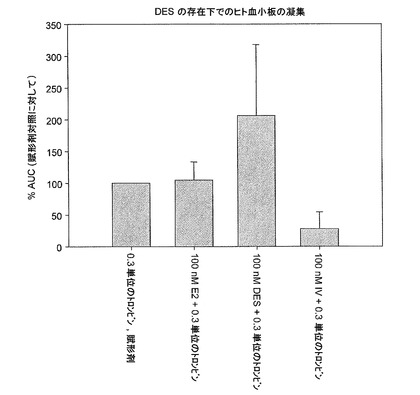
【図5】
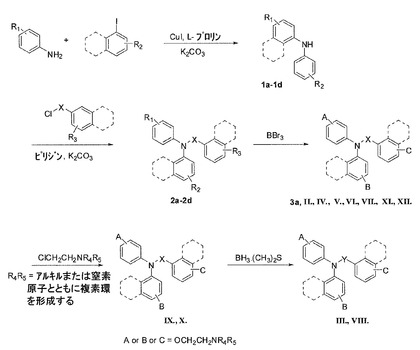
【図6】
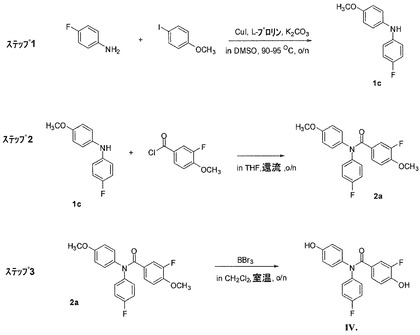
【図7】
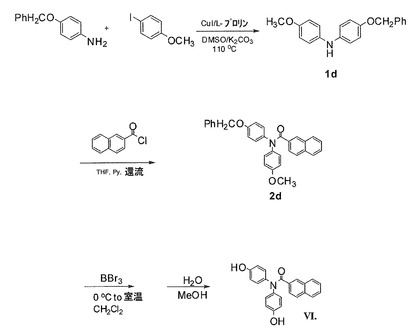
【図8】
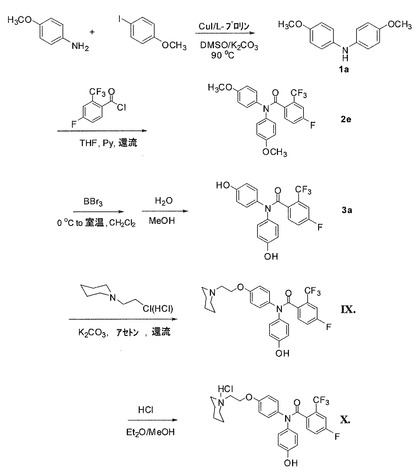
【図9】
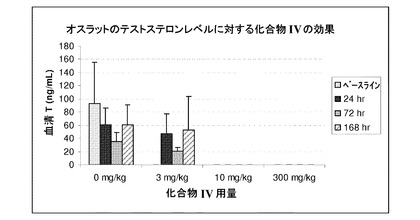
【図10A】
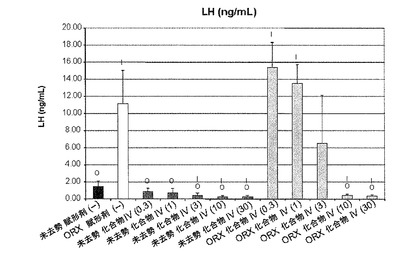
【図10B】
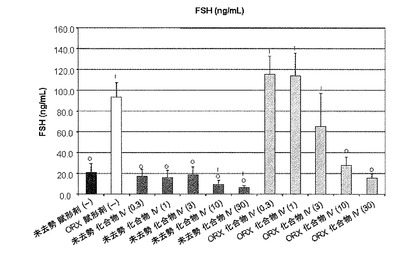
【図10C】
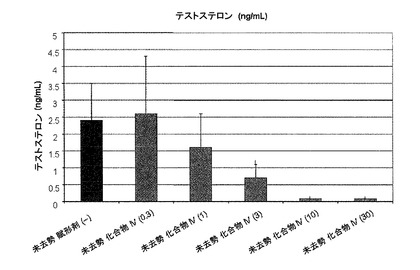
【図10D】
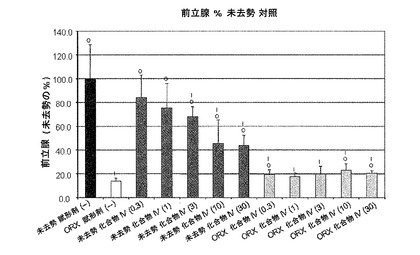
【図10E】
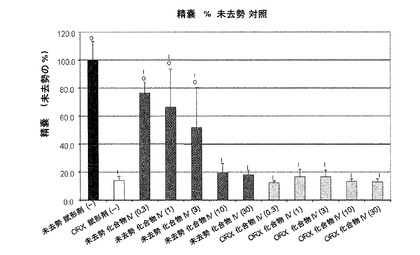
【図10F】
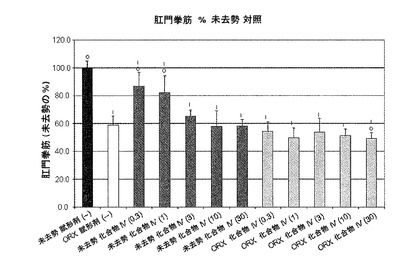
【図11】
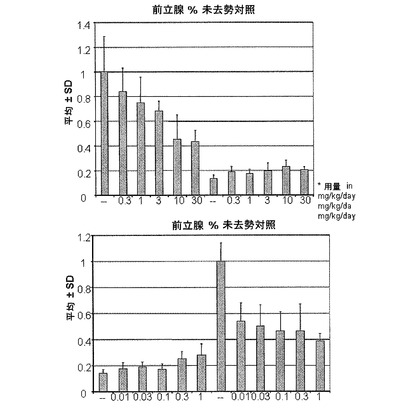
【図12A】
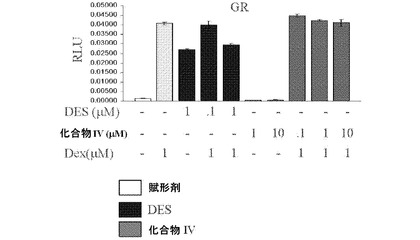
【図12B】
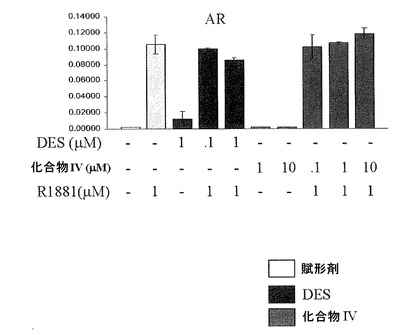
【図12C】
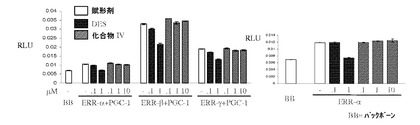
【図13】
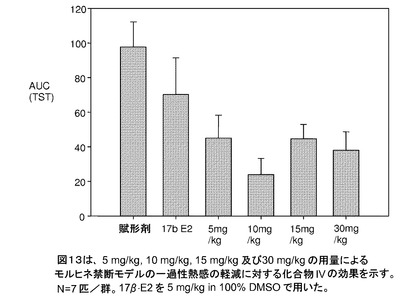
【図14】
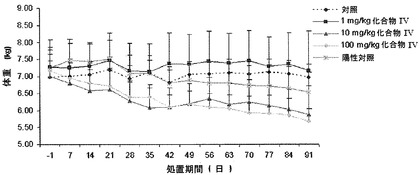
【図15】
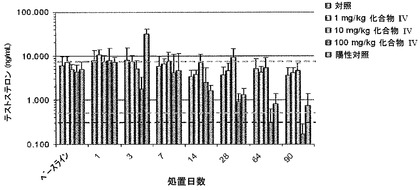
【図16】
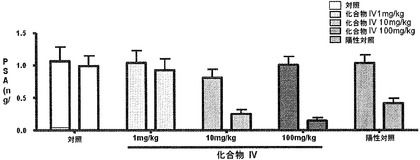
【図17】
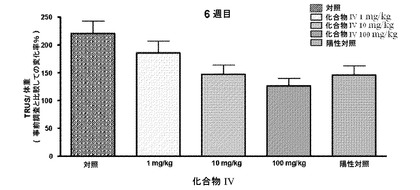
【図18】
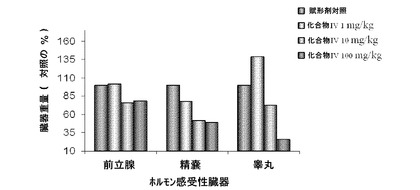
【図19】
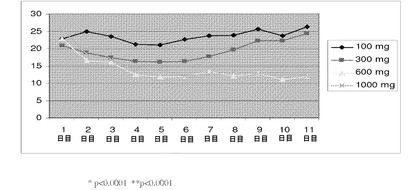
【図20】
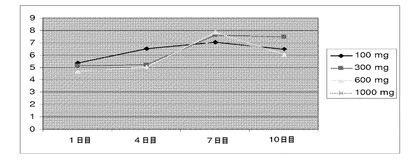
【図21】

【図22】
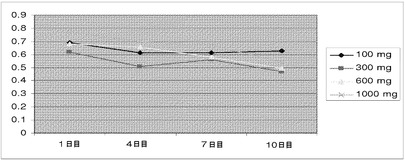
【図23】
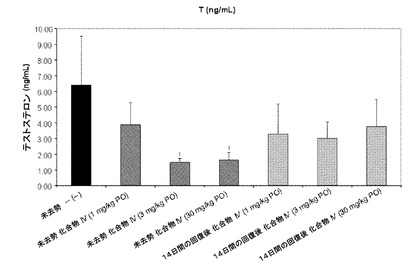
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10A】
【図10B】
【図10C】
【図10D】
【図10E】
【図10F】
【図11】
【図12A】
【図12B】
【図12C】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【公表番号】特表2013−520445(P2013−520445A)
【公表日】平成25年6月6日(2013.6.6)
【国際特許分類】
【出願番号】特願2012−554092(P2012−554092)
【出願日】平成23年2月22日(2011.2.22)
【国際出願番号】PCT/US2011/025724
【国際公開番号】WO2011/106317
【国際公開日】平成23年9月1日(2011.9.1)
【出願人】(504206621)ジーティーエックス・インコーポレイテッド (20)
【Fターム(参考)】
【公表日】平成25年6月6日(2013.6.6)
【国際特許分類】
【出願日】平成23年2月22日(2011.2.22)
【国際出願番号】PCT/US2011/025724
【国際公開番号】WO2011/106317
【国際公開日】平成23年9月1日(2011.9.1)
【出願人】(504206621)ジーティーエックス・インコーポレイテッド (20)
【Fターム(参考)】
[ Back to top ]