
エゼチミブを合成する方法およびこのために有用な中間体
本発明は、特徴的なZ−異性体構造を共有する、エゼチミブ(EZT)の合成のための新規で有用な中間体を開示する。Z−5−(4−フルオロフェニル)−ペント−4−エン酸、およびさらなるZ−中間体を経る合成の進行に基づいて、全合成は、高収率で最終エゼチミブを得るために示される。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、エゼチミブ(以下、EZTと省略することがある)の製造の合成、およびこれを含有する医薬組成物の調製の方法、ならびにこのために有用な新規な中間体に関する。
【背景技術】
【0002】
(3R,4S)−1−(p−フルオロフェニル)−タイプ−((3S)−3−(p−フルオロフェニル)−3−ヒドロキシプロピル)−4−(p−ヒドロキシフェニル)−2−アゼチジノンとして化学的に定義されるエゼチミブ(EZT)は、有用な薬剤として、具体的にはコレステロール低下薬、抗高脂血症剤、とりわけ腸内コレステロール吸収阻害剤、抗脂血症薬、および抗代謝剤として知られている。
【0003】
文献US−A−5856473は、エゼチミブを調製するための様々な従来の方法について背景の項で論じており、この文献自体は、エゼチミブの新しい合成方法を提案している。1つの合成経路は、E−5−(4−フルオロフェニル)−ペント−4−エン酸から出発し、さらにE−中間体を経て進行する。同様の情報が、S.B.Rosenblumらによって、Tetrahedron 56、5735−5742頁(2000)に記載されている。E−5−(4−フルオロフェニル)−ペント−4−エン酸の合成についてのさらなる論文は、Journal of Organic Chemistry 31(1)、147−153頁(1966)、Chemical Communications 2006、643−645頁およびJournal of Organometallic Chemistry 692、2270−2281頁(2007)に示されている。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】米国特許第5856473号明細書
【非特許文献】
【0005】
【非特許文献1】S.B.Rosenblumら、Tetrahedron 56、5735−5742頁(2000)
【非特許文献2】Journal of Organic Chemistry 31(1)、147−153頁(1966)
【非特許文献3】Chemical Communications 2006、643−645頁
【非特許文献4】Journal of Organometallic Chemistry 692、2270−2281頁(2007)
【発明の概要】
【発明が解決しようとする課題】
【0006】
(発明の要旨)
本発明の目的は、具体的には、より高い総収率を得ることができる、エゼチミブ(EZT)の合成または製造のための改良された方法および新しい方法を提供することであった。
【課題を解決するための手段】
【0007】
第1の態様において、本発明は、特徴的なZ−異性体構造を共有する、エゼチミブ(EZT)の合成のための、新規で有用な中間体を提供する。本発明によれば、Z−5−(4−フルオロフェニル)−ペント−4−エン酸に基づく化合物で出発する場合、およびZ−中間体を経て合成を進行する場合に、最終エゼチミブの合成の総収率が著しく増加することが見出された。Z−前駆体およびZ−中間体を経た合成経路で出発し、続けていくと、それぞれのE−類似体を経るよりも驚くほど高い全収率が得られる。加えて、Z−ペント−4−エン酸は、論文で知られる方法によるE−ペント−4−エン酸より著しく高い収率で調製される。Z−前駆体およびZ−中間体における前述の構造特性は、本明細書において、それぞれ示された二重結合において、Z−異性体であるまたはZ−エン配置を有すると称されることがある。
【0008】
したがって、本発明の第1の態様において、本発明によるエゼチミブの改良された合成の代表的な鍵となる化合物として、以下のZ−中間体化合物が提供される。
(1)Z−エン配置を有する以下の式I
【0009】
【化1】

(式中、R1は、Hであり、またはそれぞれ非置換または置換の、アルキル、シクロアルキル、アルケニル、シクロアルケニル、アルキニル、アリール、アラルキル、アリールシクロアルキルである。)
を有する化合物。
(2)R1がHである、(1)に記載の化合物。
(3)Z−エン配置を有する以下の式II
【0010】
【化2】
(式中、R2は脱離基である。)
を有する化合物。
【0011】
本明細書で使用する、用語「脱離基」は、カルボニル基から化学的に開裂し、それによって環化ステップを実施する後続のステップを最終的に助け、エゼチミブ構造中に出現する特徴的なアゼチジン−2−オン構造部分を形成することができる、化学的部分である。脱離基は、式IIのカルボニル基に結合するN−原子を有することが好ましい。さらに、脱離基は、環構造を形成することが好ましく、および/またはその化学的部分にキラル原子を有することができる。
(4)R2が、式IIのカルボニル基とアミド基を形成するN−ヘテロ原子であるN−ヘテロ環基か、またはRおよびR’が同一または異なり、環を形成することができ、前で定義したN−ヘテロ環基に変換され得るNRR’基であり;
ここで、好ましくは、N−ヘテロ環基がアルキル化またはアリール化され、より好ましくはキラルS異性体中心によって定義される、(3)に記載の化合物。
(5)R2が以下の式III:
【0012】
【化3】
(式中、R3は置換基、好ましくはかさ高い置換基、例えばアリール、置換アリール、または分岐アルキルまたは少なくとも4個のC原子を有するアルキル、好ましくはフェニル、置換フェニル、iPr、BuまたはtBuを表す。)
によって定義される、(4)に記載の化合物。
(6)Z−エン配置を有する以下の式IV
【0013】
【化4】
(式中、R2は、(3)から(5)のいずれか1つで定義した通りであり、R4は、OH−保護基である。)
を有する化合物。
(7)Z−エン配置を有する以下の式V
【0014】
【化5】
(式中、R4は、OH−保護基である。)
を有する化合物。
【0015】
本発明の別の態様によれば、以下が提供される。
(8)4−フルオロ−ベンズアルデヒドおよび式Vlの化合物:
【0016】
【化6】
(式中、X−は、ホスホニウム塩の陰イオン、好ましくはハロゲン化物、酢酸、トリフルオロ酢酸またはスルホン酸、具体的にはハロゲン化物であり、R’’は、アルキル、置換アルキル、アリールまたは置換アリールであり、R1は上の(1)で定義した通りである。より好ましくは、R’’はフェニルである。)
をウィッティッヒ反応に供することを含む、5−(4−フルオロフェニル)ペント−4−エン酸またはこの酸誘導体を製造する方法。
(9)得られた5−(4−フルオロフェニル)ペント−4−エン酸またはこの酸誘導体の(E,Z)−異性体混合物が、それぞれE−異性体およびZ−異性体として単離される、(8)に記載の方法。
(10)単離された異性体が、以下の式I
【0017】
【化7】
(式中、R1は上の(1)で定義した通りである。)
を有するZ−異性体化合物である、(8)または(9)に記載の方法。
(11)エステル誘導体が、式Vlの化合物として使用され、ウィッティッヒ反応後、得られたエステル基を鹸化に供し、対応する5−(4−フルオロフェニル)−ペント−4−エン酸を生成する、(8)から(10)のいずれか1つに記載の方法。
【0018】
本発明の別の態様によれば、以下が提供される。
(12)4−フルオロ−ベンズアルデヒドの式VIIの化合物とのウィッティッヒ反応によって、式IIを有する化合物
【0019】
【化8】
(式中、X−は、ホスホニウム塩の陰イオン、好ましくはハロゲン化物、酢酸、トリフルオロ酢酸またはスルホン酸、具体的にはハロゲン化物であり、R’’は、アルキル、置換アルキル、アリールまたは置換アリールであり、R2は(3)から(5)のいずれか1つで定義した通りである。より好ましくは、R’’はフェニルである。)
を製造する方法。
【0020】
この実施形態において、所望のR2基を既に有する、適したホスホニウムイリド化合物を使用することによって、式Iの化合物を経る手順の必要が排除される。
【0021】
本発明のさらに別の態様によれば、以下が提供される。
(13)上の式IからVで定義した通りの任意の化合物を使用した、エゼチミブを合成する方法。これらの中間体化合物のいずれかからでも、当業者は、従来の合成方法を使用して、最終的にエゼチミブを得ることができる。
【0022】
本発明の特定の態様において、以下が提供される。
(14)a)Z−エン配置を有する式Vの化合物を酸化して、以下で示す式IX(式中、R4は上で定義した通りである。)のケトンを得るステップ;
b)ステップ(a)のケトン生成物を還元し、対応する(S)−ヒドロキシ異性体生成物を得るステップ、および
c)還元された(S)−ヒドロキシ異性体生成物をOH−脱保護反応に供して、エゼチミブを得るステップ
【0023】
【化9】
を含む、エゼチミブを合成する方法。
【0024】
本発明は、さらに以下を提供する。
(15)(13)または(14)に記載の方法に従ってエゼチミブを調製するステップ、および
こうして調製されたエゼチミブを、少なくとも1つの薬学的に許容される賦形剤と混和するステップ
を含む、エゼチミブを活性成分として含む医薬組成物を調製する方法。
(16)Z−エン配置を有する化合物Vを本質的に含まず、その脱保護された4−ヒドロキシフェニル類似体を本質的に含まないエゼチミブ。
(17)a)(3R,4S)−4−(4−(ベンジルオキシ)フェニル)−1−(4−フルオロフェニル)−3−((Z/E)−3−(4−フルオロフェニル)−アリル)アゼチジン−2−オンの3−Z−またはE−異性体のどちらかまたは両方を本質的に含まない、および/またはその脱保護された4−ヒドロキシフェニル類似体を本質的に含まないエゼチミブを提供するステップ、ならびに
b)こうして提供されたエゼチミブを、少なくとも1つの薬学的に許容される賦形剤と混和するステップ
を含む、エゼチミブを活性成分として含む医薬組成物を調製する方法。
(18)(17)に記載の方法によって得られる、活性成分としてのエゼチミブおよび少なくとも1つの薬学的に許容される賦形剤を含む医薬組成物。
【図面の簡単な説明】
【0025】
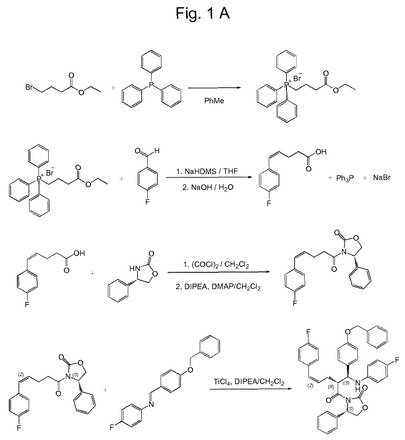
【図1A】本発明の実施形態による、例示的なZ−中間体を介するエゼチミブの全合成のための、好ましい合成経路を示す概略図である。
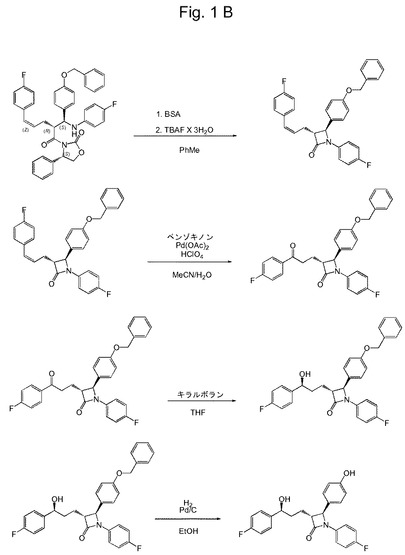
【図1B】本発明の実施形態による、例示的なZ−中間体を介するエゼチミブの全合成のための、好ましい合成経路を示す概略図である。
【発明を実施するための形態】
【0026】
本発明の特定の態様によれば、エゼチミブの全合成のための重要な中間体としての5−(4−フルオロフェニル)−ペント−4−エン酸およびこの誘導体は、ウィッティッヒ反応を介した非常に有効な合成手法によって高収率でもたらされる。この方法ステップにおいて、4−フルオロ−ベンズアルデヒドは、式VIの化合物と一緒にウィッティッヒ反応に供される。5−(4−フルオロフェニル)−ペント−4−エン酸の誘導体として、典型的に、酸性塩、酸エステルおよび酸アミド、好ましくは酸エステルが挙げられるが、これらだけには限らない。好ましい実施形態において、R1=水素である酸基が使用されるか、より好ましくは、酸基は、それぞれ非置換または置換の、アルキル、シクロアルキル、アルケニル、シクロアルケニル、アルキニル、アリール、アラルキルおよびアリールシクロアルキルから選択されるR1によって誘導体化される。
【0027】
次いで、得られた5−(4−フルオロフェニル)−ペント−4−エン酸またはこの誘導体のE−およびZ−異性体の混合物は、それぞれE−異性体およびZ−異性体として単離され得る。驚くべきことに、本発明による方法におけるウィッティッヒ反応は、高い総収率を達成することに大いに寄与するだけでなく、より重要なことには、大部分は所望のZ−異性体(すなわちZ/E−モル比がより高く、具体的には実質的に50%より高い)を生成し、これによって、エゼチミブの合成のための代表的な鍵となるZ−中間体を選択的に単離し、得ることが可能になる。注目すべきは、Z−およびE−異性体の混合物に対して90%を上回る収率で、Z−異性体の範囲が約80%の混合物(Z:Eの比=88:12)は、具体的には、本発明の実施形態においてウィッティッヒ反応を使用することによって達成され得る。
【0028】
望ましいZ−異性体は、Z−およびE−異性体混合物から容易に単離され、場合によって精製され、その結果、式I
【0029】
【化10】
の化合物を提供することができる。
【0030】
残基R1は、Hであり、または非置換もしくは置換アルキル、非置換もしくは置換シクロアルキル、非置換もしくは置換アルケニル、非置換もしくは置換シクロアルケニル、非置換もしくは置換アルキニル、非置換もしくは置換アリール、非置換もしくは置換アラルキル、または非置換もしくは置換アリールシクロアルキルである。前述のR1に対応する残基におけるアルキル、アルケニルもしくはアルキニル基またはこれらの環構造として、1から12個、好ましくは1から8個、より好ましくは1から6個の炭素原子を含有する有機基を挙げることができるが、これらだけには限らない。好ましい残基R1は、Hであり、または例えばメチル、エチル、プロピル、イソプロピル、n−ブチル、tert−ブチル、ペンチル、ヘキシルなどの1から6個の炭素原子を有する低級アルキルである。適したアリール基として、フェニル、ナフチル、ジフェニルなどを挙げることができるが、これらだけには限らない。適した置換基として、1から6個の炭素原子を有する低級アルキル、ハロゲン、アミン基などを挙げることができるが、これらだけには限らない。
【0031】
同様の意味が、他に記述がなければ、本明細書で使用するアルキル、置換アルキル、アリール、置換アリールおよび類似の用語の定義に適用される。
【0032】
好ましい実施形態によれば、ウィッティッヒ反応は、R1が上述の基から選択されるがHではない場合に、より効率的に進行し、対応する5−(4−フルオロフェニル)ペント−4−エン酸誘導体、とりわけエステル誘導体が得られ、その後すぐに、得られたエステル基を鹸化に供し、対応する5−(4−フルオロフェニル)−ペント−4−エン酸を生成することができる。鹸化は、エステル誘導体の単離無しに、ウィッティッヒ反応後にワンポットプロセスとして実施されることが好ましい。次いで、式Iの化合物(具体的には、式中、R1はHである。)は、式IIの化合物(式中、R2は上で示した脱離基を示す。)に変換することができる。好ましい実施形態によれば、R2は、N−ヘテロ原子が式IIにおけるカルボニル基とアミド基を形成する、N−複素環基である。エゼチミブを生成するためのさらなるエナンチオ選択的合成ステップを考慮すると、N−複素環基はキラル中心を有し、より好ましくはキラルS−異性体中心を有することが、より好ましい。したがって、より好ましいN−複素環基は、例えば、これらだけには限らないが、置換されたまたは置換されていない、メチル、エチル、プロピル、イソプロピル、n−ブチル、tert−ブチルなどの低級アルキルから選択されるアルキル基、またはフェニル、ナフチルなどのアリールによってアルキル化またはアリール化される。
【0033】
したがって、式IIを有する化合物は、所望のZ−形態で提供することができ、ここでR2は上で定義されている。R2がNRR’基の場合、RおよびR’は、同一または異なることができ、これらは、当業者に利用可能な知識に従って、前記N−複素環基に変換され得る環を形成するために選択することができる。R2は、下の式III(式中、アルキルおよびアリールは上で定義した通りである。)の構造部分によって定義されることが好ましい。
【0034】
【化11】
【0035】
この式IIIにおいて、R3は置換基、好ましくはかさ高い置換基、例えばアリール、置換アリールまたは少なくとも4個のC原子を有する分岐アルキルまたはアルキルを表す。R3は、好ましくはフェニル、置換フェニル、iPr、BuまたはtBuである。
【0036】
本発明の一実施形態による場合、式IIの化合物は、Z−5−(4−フルオロフェニル)−ペント−4−エン酸との反応から得られ、酸基は、特に、残基R2が式IIIによって定義される、後続の式IIの化合物の形成に対して活性化され得る。Z−5−(4−フルオロフェニル)−ペント−4−エン酸の酸基の活性化は、例として、例えば、ホスゲン、塩化オキサリル(COCl)2、塩化チオニルClSOCl、五塩化リンPCl5、臭化ベンゾイルなどのそれぞれとの反応によってなど、酸塩化物または酸臭化物などの酸ハロゲン化物の形成を介する、および対応する無水炭酸を介する、既知の方法によって実行することができる。
【0037】
上述のウィッティッヒ反応に対して、適切なトリフェニルホスホニウム塩を調製して、式VI
【0038】
【化12】
(式中、Xは、ハロゲン、アセテート、トリフルオロアセテートまたはスルホネートであってよく、より好ましくは、ClまたはBrなどのハロゲンであり、さらに、式中、R’’は、アルキル、置換アルキル、アリールまたは置換アリールであり、
R1は上の通り定義される。)
の化合物を得る。
【0039】
好ましい実施例において、R’’は、以下で示す例示的な式VIを採用するためのフェニルである(XおよびR1は、上の通り定義される。)。
【0040】
【化13】
【0041】
本発明による方法の変形において、式II
の化合物は、4−フルオロ−ベンズアルデヒドの、対応する式VII
【0042】
【化14】
(式中、Xは、ハロゲン、アセテート、トリフルオロアセテートまたはスルホネートであってよく、より好ましくは、ClまたはBrなどのハロゲンであり、さらに、式中、R’’は、アルキル、置換アルキル、アリールまたは置換アリールであり、R2は上の通り定義される。)
の化合物とのウィッティッヒ反応を実行することによって、直接得ることができる。
【0043】
好ましい実施形態において、R’’は、以下で示す例示的な式VIIを採用するためのフェニルである(XおよびR2は、上の通り定義される。)。
【0044】
【化15】
【0045】
式IIの望ましいZ−中間体化合物は、エゼチミブ合成に使用されるので、ウィッティッヒ反応から得られるZ−異性体形態は、異性体の混合物から単離されるものとする。
【0046】
エゼチミブの全合成経路の後続のステップにおいて、式IIの化合物、より好ましくは、式IIIによって示される通りの基R2を有する化合物は、以下で示す式VIII(式中、R4は、これらだけには限らないが、ベンジル(Bn)、炭酸ベンジル(Cbz)、ベンジルオキシメチルエーテル(BOM);シリル、より好ましくはC1−C8トリアルキルシリル、C1−C8ジアルキルアリールシリル、C1−C8アルキルジアリールシリル(ここで、アルキルは、同一または異なっていてもよく、好ましくは、アリールはフェニルであり、アルキルは1から4個のC原子を有する。)などを含めたOH−保護基である。)によって定義されるイミン化合物と反応し、
【0047】
【化16】
【0048】
【化17】
(式中、R2およびR4は、上で定義した通りである。)を有する化合物を生成する。
【0049】
次いで、化合物IVは、適切に、ビス−トリメチルシリルアセトアミド(BSA)などのシリル化剤での処理、およびテトラブチルアンモニウムフルオリド(TBAF)などのフッ化物イオン触媒による後続の処理によって環化され、その結果、式V:
【0050】
【化18】
(式中、R4は、上で定義した通りである。)を有する化合物を生成することができる。
【0051】
次いで、Z−異性体構造を有する式Vの化合物は、エゼチミブの合成におけるステップに有用な中間体を構成する。
【0052】
ここで、最初に、この化合物を酸化して、上で定義した通りの式IXのケトンを得て、次いで、例えばキラルボランとの反応によって、ケトンを還元して、所望のS−OH−異性体生成物を得て、単離し、最後に、還元されたS−OH−異性体生成物をOH−脱保護反応に供し、最終的にエゼチミブを得る。適したOH−脱保護反応として、通常、遷移金属のPt、Pd、Rhなどの金属触媒を、それぞれ単独または混和して、好ましくは、活性炭、アルミナ、シリカなどの担持物質と組み合わせて用いる接触水素化が挙げられる。最も好ましい触媒は、炭素担持パラジウムである。水素化反応に存在するべき水素源として、H2ガス、ギ酸アンモニウム/ギ酸などが、使用されてもよい。
【0053】
本発明によるエゼチミブを合成する方法は、Z−出発および中間体化合物を使用すると、対応するE−出発物質または中間体の使用と比較して、予想外に高い収率(エゼチミブの総収率と称される。)をもたらすことができるという点で特に有利である。このことは、全方法の設計(複合体混合物が、反応後に得られるTetrahedron 2000、56、5735−5742頁と比較して、純粋なZ−酸を生成する「クリーンな」ウィッティッヒ反応の適用)および、高純度での単離を可能にし、したがって、全収率を低下させる副反応の程度がより低い後続のステップのそれぞれにおいて変換されるZ−出発および中間体化合物の物性(溶解性、クロマトグラフィーの挙動)によるものである。この効果は、アルケンV酸化ステップおよび化合物IXのキラルボラン還元ステップにおいて、特に顕著になる。すなわち、Z−アルケンの酸化は、収率86%のケトンIXを生成するが、E−アルケン酸化は、US−A−5856473に記載されているように、収率70−80%でケトンを生成する。さらに、Z−中間体を経て誘導されるケトンのケトン還元ステップによる収率は、95%になるが、US−A−5856473に記載されているように、E−中間体から誘導されるケトンの収率は、わずか42%になる。したがって、エゼチミブを合成する方法のための中間体として上で述べた化合物の使用は、本発明の有益な側面である。上で述べた特別で好ましい反応の他に、最終的にエゼチミブを得るその他の従来の合成ステップまたは知られている変形(さらに今開示したZ−中間体化合物の使用を含めて)が、適用され得る。
【0054】
次いで、本発明による方法によって調製されるエゼチミブは(通常、所望の純度を得た後に)、少なくとも1つの薬学的に許容される賦形剤と混和され得る。エゼチミブを含む、薬学的に安全で安定な医薬組成物を得るために、エゼチミブが任意の医薬賦形剤と混和される前に、化合物Vを本質的に含まない、好ましくは全く含まないことを確認することが重要である。化合物Vの存在自体は、最終医薬品の薬理学的特徴および安定性、具体的には化学安定性にとって有害であると考えられ、さらに、化合物V中間体の後のエゼチミブ合成ステップで形成される可能性のある、さらなる有害な不純物を示すことがある。不純物を無くしてから医薬賦形剤と混和することの重要性は、特に固形剤形が予定されている場合、不純物が、活性なエゼチミブおよび賦形剤を一緒に最終組成物中に同時処理した後では、もはや取り除くことができないことにある。いかなる理論によっても制限されないが、化合物V中に存在するアリル基の二重結合は、前述の最終エゼチミブ合成ステップ(例えば、化合物の3位での(S)−ヒドロキシル基にエナンチオピュアな還元[上の項(14)のステップ(b)参照]、および/または最終OH−脱保護反応[上の項(14)のステップ(c)参照])またはその他の最終エゼチミブ合成ステップでの反応の妨害、および/または、特に、エゼチミブを含有する医薬組成物中の賦形剤との最終混和物に、深刻に関与する恐れがあると考えることができる。最終エゼチミブ生成物中に化合物Vを本質的に含まないこと、またはさらには存在しないことが、化合物Vが遊離体として使用されるステップ中、またはステップの後の適切な精製および単離方法によって、確実となるべきである。化合物Vが遊離体として使用された合成ステップ後に、化合物Vの全てを除去することはできないという観点から、エゼチミブを得るための最終脱保護ステップにおける別の深刻な不純物として最終的に形成された可能性のある化合物Vの脱保護された4−ヒドロキシフェニル類似体も、エゼチミブが医薬組成物の調製において医薬賦形剤と混和される前に確実に除去されることが、さらに妥当である。
【0055】
エゼチミブが対応するE−中間体を経て合成された場合、同じ見解が同様に適用される。本発明は、Z−中間体を介して進行することができるが、任意の医薬賦形剤と混和されるエゼチミブはまた、(3R,4S)−4−(4−(ベンジルオキシ)フェニル)−1−(4−フルオロフェニル)−3−(E)−3−(4−フルオロフェニル)−アリル)アゼチジン−2−オンの対応するE−異性体を含まないことが確実となる。したがって、E−異性体を本質的に含まないことおよびさらに全く存在しないことは、薬学的に安全で安定なエゼチミブ含有医薬組成物を可能にする本発明の別の利点である。
【0056】
本明細書で使用する、用語「化合物Vを本質的に含まない」(すなわちZ−異性体)、または同様に化合物VのE−異性体もしくはそれぞれの脱保護された4−ヒドロキシフェニル(ZまたはE)類似体を「本質的に含まない」とは、得られたEZT生成物の総重量に対して、その量が、例えば約0.2重量%より低い、好ましくは約0.1重量%未満、より好ましくは約0.05重量%未満の対照を意味するものとする。化合物V、これに対応するE−異性体、またはこれらのそれぞれの脱保護された4−ヒドロキシフェニル類似体の検出は、それぞれEZT生成物に対して、HPLCによって実行され得る。しかし、用語「を含まない」は、EZT合成がZ−またはE−中間体を経由したかどうかを示す、少なくとも微量の同定されたEZT不純物の存在を含めることができる。
【0057】
医薬組成物は、コレステロールの、高脂血症のおよび脂肪血症の状態、癌から選択される疾病のいずれか1つ、ならびに抗菌状態の予防および/または治療に使用することができる。好ましい使用は、高コレステロール血症である。適したエゼチミブの用量は、0.1mgから100mg、より適切には1mgから20mgの範囲とすることができる。薬学的に許容される賦形剤は、当業者に知られる適した担体および賦形剤から選択され得る。例えば、適した薬学的に許容される担体および/または賦形剤として、これらだけには限らないが、結合剤、充填剤、マトリックス形成剤、希釈剤、酸化防止剤、緩衝剤、消泡剤、保存剤、キレート化剤、粘度調整剤、香味料、着色剤、臭気剤、乳白剤、安定化剤、可溶化剤、可塑剤、潤滑剤およびこれらの混合物が挙げられる。適した具体的な例として、これらだけには限らないが、ラクトース、微結晶性セルロース、ヒドロキシプロピルセルロースなどのセルロース誘導体、ポリアクリレート、炭酸カルシウム、デンプン、コロイド状二酸化シリコーン、デンプングリコール酸ナトリウム、タルク、ステアリン酸マグネシウム、ポリビニルピロリドン、ポリエチレングリコールおよび医薬技術の分野で知られるその他の賦形剤からなる群のものが挙げられる。
【0058】
賦形剤はまた、所望の投与形態および剤形によって選択されてもよい。適した投与形態として、経口、非経口、注射、経皮、経鼻、経直腸、吸入が挙げられるが、これらだけには限らない。適した剤形として、錠剤、カプセル、顆粒、粉末、丸薬、懸濁剤、液剤、坐薬、徐放剤形が挙げられるが、これらだけには限らない。本発明による医薬調製物において、エゼチミブはまた、1つまたは複数の活性成分と組み合わせてもよい。
【0059】
本発明は、単に例示的な目的として提供される実施例を参照することで、下でより詳細に説明されるが、本発明は、決してこれに限定されない。
【0060】
好ましい反応ステップによるエゼチミブの全合成のための特に好ましい合成経路は、図1Aおよび1Bに示される。関連する反応ステップは、以下の実施例でより詳細に説明される。
【0061】
(実施例)
【実施例1】
【0062】
(4−エトキシ−4−オキソブチル)トリフェニルホスホニウムブロミド(1)の合成
【0063】
【化19】
【0064】
4−ブロモ酪酸エチル(51.0g、261mmol)およびトリフェニルホスフィン(67.2g、256mmol)の乾燥トルエン(250mL)中溶液を撹拌し、還流下およびアルゴン雰囲気下で4日間加熱した。反応混合物を室温に冷却後、形成された不溶性の白色沈殿物を濾別し、加熱したトルエン(100mL)およびヘキサン(80mL)で洗浄した。この生成物を粉砕し、減圧下で乾燥し、白色の粉末として表題化合物1(113.7g、97%)を得た。
【実施例2】
【0065】
(Z)−5−(4−フルオロフェニル)ペント−4−エン酸(3)の合成
【0066】
【化20】
【0067】
乾燥THF(30mL)中の粉末(4−エトキシ−4−オキソブチル)トリフェニルホスホニウムブロミド(1)(10.5g、23.7mmol)を、アルゴン雰囲気下で、撹拌しながら、18℃でナトリウムヘキサメチルジシラジド(12.5mL、25mmol;THF中2M溶液)で処理した。20分後、4−フルオロベンズアルデヒド(2.61g、21.0mmol)を18℃で加え、赤オレンジ色の混合物を速やかに脱色した。室温で4時間撹拌した後、反応混合物を氷浴で冷却し、水10mL中のNaOH(1.6g、40mmol)を加えた。得られた溶液を室温でさらに12時間撹拌した。水(100mL)およびエーテル(200mL)を加えた。相を分離させ、水相をエーテル(2x50mL)ですすぎ、酸性化し(4M HCl)、EtOAc(4x50mL)で抽出した。採集したEtOAc抽出物を無水Na2SO4で乾燥し、濃縮した。クーゲルロール蒸留によって、88%のcis異性体を含有する白色の結晶として、3.52g(90.6%)の純粋な5−(4−フルオロフェニル)ペント−4−エン酸(3)を得た。このcis異性体は、エステル誘導体または酸性塩誘導体の再結晶化、続いて酸性化、または粗酸の再結晶化後に、純粋な形態で得ることができる
化合物2(cis−エステル):
1H−NMR(300MHz,CDCl3):δ(ppm)=1.26(t,3H,J=7.1Hz,−CH3)、2.42−2.47(m,2H,CH2CH2CO)、2.60−2.68(m,2H,CH2CH2CO)、4.15(q,2H,J=7.1Hz,−CH2−CH3)、5.63(td,1H,J=7.2Hz,J=11.6Hz,CH=CHCH2)、6.44(d,1H,J=11.6Hz,PhCH=CH)、7.00−7.08(m,2H,Ph−H)、7.22−7.28(m,2H,Ph−H)ppm。
化合物3(cis−カルボン酸):
1H−NMR(300MHz,CDCl3):δ(ppm)=2.46−2.51(m,2H,CH2CH2CO);2.59−2.66(m,2H,CH2CH2CO)、5.64(td,1H,J=7.0Hz,J=11.6Hz,CH=CHCH2)、6.44(d,1H,J=11.6Hz,PhCH=CH)、7.00−7.08(m,2H,Ph−H)、7.22−7.27(m,2H,Ph−H)、9.99(bs,1H,−COOH)ppm。
【実施例3】
【0068】
(S,Z)−3−(5−(4−フルオロフェニル)ペント−4−エノイル)−4−フェニルオキサゾリジン−2−オン(4)の合成
【0069】
【化21】
【0070】
p−フルオロフェニルペンテン酸(3)(2.3g)のCH2Cl2(40mL)中溶液に、塩化オキサリル(1.81g)およびDMF(2滴)を、0℃でゆっくり加えた。この溶液を室温まで温めて、1時間還流させた。溶媒を減圧下で蒸発させ、過剰の塩化オキサリルをCH2Cl2(2x50mL)と共沸除去した。得られた酸塩化物をCH2Cl2(40mL)に再溶解し、この溶液を、(S)(+)−4−フェニル−2−オキサゾリドン(1.99g)、N,N−ジイソプロピルエチルアミン(DIPEA)(3.07g)およびN,N−ジメチルアミノピリジン(0.05g)のCH2Cl2(20mL)中の冷却した溶液に滴下した。得られた混合物を室温で2時間撹拌した。CH2Cl2(100mL)を加え、有機相を0.5MのHCl(2×40mL)、水(1×40mL)およびブライン(1×40mL)で洗浄した後、次いで乾燥させ、濃縮して、所望の生成物を結晶化した。生成物を酢酸エチル/ヘキサンから再結晶させ、濾過して採集した(3.7g、93%)。
1H−NMR:(300MHz,CDCl3):δ 2.55−2.70(m,2H,CH2CH2CO)、3.05−3.15(m,2H,CH2CH2CO)、4.29(dd,J=8.91Hz,J=3.69Hz,1H,CHCH2OCO)、4.69(t,J=8.82Hz,1H,CHCH2OCO)、5,43(dd,J=8.71Hz,J=3.66Hz,1H,CHCH2OCO)、5.57−5.66(m,1H,ArCH=CH)、6.40(d,J=11.56Hz,1H,ArCH=CH)、6.95−7.04(m,2H,ArH)、7.20−7.43(m,7H,ArH)ppm。
【実施例4】
【0071】
(S)−3−((R,Z)−2−((S)−(4−(ベンジルオキシ)フェニル)(4−フルオロフェニルアミノ)メチル)−5−(4−フルオロフェニル)ペント−4−エノイル)−4−フェニルオキサゾリジン−2−オン(6)の合成
【0072】
【化22】
【0073】
−20℃の(S,Z)−3−(5−(4−フルオロフェニル)ペント−4−エノイル)−4−フェニルオキサゾリジン−2−オン(4)(1.0g)のCH2Cl2(15mL)中溶液に、TiCl4のCH2Cl2(3.25mL)中1M溶液をゆっくり加え、−20℃で撹拌した。15分後、DIPEA(0.97mL)を加え、混合物を30分間、−20℃で撹拌した。(E)−N−(4−(ベンジルオキシ)ベンジリデン)−4−フルオロアニリン(5)(1.53g)をCH2Cl2(30mL)に溶解し、温度を−20℃より低く維持しながら溶液に加えた。1.5時間後、反応を、CH2Cl2(3mL)中の氷酢酸(1mL)で、−20℃でクエンチして、30分間撹拌した。反応混合物を1MのH2SO4(40mL)の水溶液に注ぎ、さらに30分混合した。その後、酢酸エチル150mLを加え、有機相をNaHCO3(3x40mL)の飽和溶液およびブライン(1×40mL)で洗浄し、次いでNa2SO4で乾燥して、濃縮した。生成物を酢酸エチル/ヘキサンから再結晶させ、濾過して採集した。結晶(1.15g;59%)を冷却したメタノールで洗浄した。
1H−NMR:(300MHz,CDCl3):δ 2.31−2.40(m,1H,=CHCH2CH)、2.65−2.75(m,1H,=CHCH2CH)、4.22(dd,J=8.76Hz,J=3.13Hz,1H,CHCH2OCO)、4.39(t,J=9.42Hz,1H,=CHCH2CH)4.36−4.60(m,1H,CHNH)、4.69(t,J=8.67Hz,1H,CHCH2OCO)、4.95(d,J=10.32Hz,1H,NH)、5.03(s,2H,CH2Ph)、5.43(dd,J=8.45Hz,J=3.11Hz,1H,CHCH2OCO)、5.51−5.60(m,1H,ArCH=CH)、6.24−6.42(m,1H,ArCH=CH)、6.69−6.81(m,2H,ArH)、6.84−6.96(m,4H,ArH)、7.08−7.18(m,10H,ArH)、7.38−7.50(m,7H,ArH)ppm。
MS(ES+)m/z(%):645(MH+,60)、534(100)。
【実施例5】
【0074】
(3R,4S)−4−(4−(ベンジルオキシ)フェニル)−1−(4−フルオロフェニル)−3−((Z)−3−(4−フルオロフェニル)−アリル)アゼチジン−2−オン(7)の合成
【0075】
【化23】
【0076】
6(0.484g、0.75mmol)の乾燥トルエン(5.0mL)中懸濁液を、アルゴンで脱酸素化し、15分後、BSA(1.50mmoL、0.37ml)を加えた。室温で30分撹拌した後、TBAF×3H2O(5.0mol%、0.0375mmol、11.8mg)を加え、4時間撹拌させたままにした。次いで、氷酢酸0.027mlおよびMeOH 4.0mlを加えた。5分後、反応混合物を減圧化で濃縮し、EtOAc(50ml)を加えた。有機相を、5%の重炭酸ナトリウム溶液(25ml)、水(25ml)およびブライン(25ml)で洗浄し、硫酸ナトリウムで乾燥し、濾過して、減圧下で濃縮した。粗生成物を、溶出液としてEtOAc/ヘキサン(1/4)を用いるフラッシュクロマトグラフィーによって精製し、0.256g(71%)の化合物7を得た。
1H−NMR(300MHz,CDCl3):δ(ppm)=2.74−2.95(m,1H,CH2)、3.13−3.19(m,1H,CHCO)、4.52(d,J=2.3Hz,1H,CHN)、5.07(s,2H,OCH2Ph)、5.63−5.71(m,1H,PhCHCH)、6.52(d,J=11.5Hz,1H,PhCHCH)、6.86−7.04(m,6H,Ph−H)、7.15−7.24(m,6H,Ph−H)、7.30−7.41(m,5H,Ph−H)。
【実施例6】
【0077】
(3R、4S)−4−(4−(ベンジルオキシ)フェニル)−1−(4−フルオロフェニル)−3−(3−(4−フルオロフェニル)−3−オキソプロピル)アゼチジン−2−オン(8)の合成
【0078】
【化24】
【0079】
Pd(OAc)2(3.0mol%、0.042mmol、9.43mg)、ベンゾキノン(2.1mmol、227mg)および過塩素酸(70%水溶液、0.15M、0.100ml)をアセトニトリル(3.5mL)に溶解し、少なくとも20分間、アルゴンでパージして脱酸素化した。次いで、同様にアルゴンで脱酸素化した水(0.7ml)を加えた。反応混合物を、さらに5分間、アルゴン雰囲気下で激しく撹拌し、同様にあらかじめアルゴンで30分間パージして脱酸素化した7(1.40mmol)のアセトニトリル(3.5ml)中溶液を加えた。4時間後、過塩素酸(70%水溶液、0.100ml)を、再度加えた。得られた溶液を48時間撹拌し、次いでEtOAc(50ml)で希釈した。水相を別の30mLのEtOAcで洗浄した。有機相を合わせて、水(2×40m)およびブライン(40ml)で洗浄し、硫酸ナトリウムで乾燥し、濾過して、減圧下で濃縮した。粗生成物を、EtOAc/ヘキサン(1/4)で溶出するフラッシュクロマトグラフィーによって精製し、化合物8(0.600g)を収率86%で得た。
1H−NMR(300MHz,CDCl3):δ(ppm)=2.21−2.47(m,2H,CH2CH)、3.10−3.21(m,2H,CH2CO)、3.24−3.35(m,1H,CHCO)、4.68(d,J=2,3Hz,1H,CHN)、5.05(s,2H,OCH2Ph)、6.90−6.99(m,4H,Ph−H)、7.09−7.17(m,2H,Ph−H)、7.22−7.27(m,4H,Ph−H)、7.33−7.44(m,5H,Ph−H)、7.96−8.02(m,2H,Ph−H)。
【実施例7】
【0080】
(3R,4S)−4−(4−(ベンジルオキシ)フェニル)−1−(4−フルオロフェニル)−3−((S)−3−(4−フルオロフェニル)−3−ヒドロキシプロピル)アゼチジン−2−オン(9)の合成
【0081】
【化25】
【0082】
(R)−テトラヒドロ−1−メチル−3,3−ジフェニル−1H,3H−ピロロ[1,2−c]−[1,3,2]オキサザボロール(96mg;0.346mmol)および(3R,4S)−4−(4−(ベンジルオキシ)フェニル)−1−(4−フルオロフェニル)−3−(3−(4−フルオロフェニル)−3−オキソプロピル)アゼチジン−2−オン(8)(760mg;1.528mmol)を、アルゴン雰囲気下で乾燥THF(2mL)に溶解した。溶液を−22℃に冷却し、5分間撹拌した後、ボロヒドリド−ジメチルスルフィド複合体(THF中2M溶液;0.864mL;1.728mmol)を、2時間かけて滴下した。−22℃で合計5時間撹拌した後、反応をメタノール(3mL)の添加によってクエンチした。EtOAc(30mL)および1MのHCl(15mL)を加え、相を分離させた。水相をEtOAc(20mL)で2度抽出した。合わせたEtOAc抽出物を無水Na2SO4で乾燥し、濃縮して泡(0.900g)を安定させた。得られた粗生成物を、移動相としてEtOAc/ヘキサン=1/3を用いるフラッシュクロマトグラフィーによって精製し、表題化合物9(0.731g、95.8%)を得た。
1H−NMR(300MHz,CDCl3):δ(ppm)=1.84−2.05(m,4H;CH2CH2);3.07−3.17(m,1H,CHCO);4.59(d,J=2.4Hz;1H,CHN);4.71−4.76(m,1H,CHOH);5.07(s,2H,OCH2Ph);6.91−7.06(m,6H,Ph−H);7.22−7.46(m,11H,Ph−H)ppm
【実施例8】
【0083】
エゼチミブ(10)の合成
【0084】
【化26】
【0085】
(3R,4S)−4−(4−(ベンジルオキシ)フェニル)−1−(4−フルオロフェニル)−3−((S)−3−(4−フルオロフェニル)−3−ヒドロキシプロピル)アゼチジン−2−オン(9)を、エタノールに溶解し、5%Pd/C(0.5mol%)を、得られた溶液に加えた。得られた懸濁液を、出発物質9が消費されるまで、高圧下でのH2ガス雰囲気下で撹拌した。触媒を濾過して除去した。得られた溶液を減圧下で濃縮し、再結晶化して、表題化合物10を得た。
【0086】
結果:
個々のステップにおける収率に関する、およびエゼチミブの全合成の全収率に関するデータを、Z−中間体に基づく本発明による結果およびE−中間体に基づく従来技術による結果をそれぞれ別々に、下記の表1および2に示す。
【0087】
【表1】
【0088】
【表2】
【0089】
【表3】
US 5,856,473で開示されたE−誘導体を用いるステップの全収率:16.8%−19.2%。
手作業でUS 5,856,473のE−誘導体を用いる方法の全収率:1.5%
本発明によって得られたZ−誘導体を用いる同様のステップの全収率:31.8%
Tetrahedron 56(2000)5735−5742およびUS 5,856,473を合わせた全収率:9.4%−10.7%
Tetrahedron 56(2000)5735−5742およびUS 5,856,473を手作業で合わせた全収率:0.07%−0.25%
Z−誘導体を用いる本発明によって得られた全収率:24.7%
【技術分野】
【0001】
本発明は、エゼチミブ(以下、EZTと省略することがある)の製造の合成、およびこれを含有する医薬組成物の調製の方法、ならびにこのために有用な新規な中間体に関する。
【背景技術】
【0002】
(3R,4S)−1−(p−フルオロフェニル)−タイプ−((3S)−3−(p−フルオロフェニル)−3−ヒドロキシプロピル)−4−(p−ヒドロキシフェニル)−2−アゼチジノンとして化学的に定義されるエゼチミブ(EZT)は、有用な薬剤として、具体的にはコレステロール低下薬、抗高脂血症剤、とりわけ腸内コレステロール吸収阻害剤、抗脂血症薬、および抗代謝剤として知られている。
【0003】
文献US−A−5856473は、エゼチミブを調製するための様々な従来の方法について背景の項で論じており、この文献自体は、エゼチミブの新しい合成方法を提案している。1つの合成経路は、E−5−(4−フルオロフェニル)−ペント−4−エン酸から出発し、さらにE−中間体を経て進行する。同様の情報が、S.B.Rosenblumらによって、Tetrahedron 56、5735−5742頁(2000)に記載されている。E−5−(4−フルオロフェニル)−ペント−4−エン酸の合成についてのさらなる論文は、Journal of Organic Chemistry 31(1)、147−153頁(1966)、Chemical Communications 2006、643−645頁およびJournal of Organometallic Chemistry 692、2270−2281頁(2007)に示されている。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】米国特許第5856473号明細書
【非特許文献】
【0005】
【非特許文献1】S.B.Rosenblumら、Tetrahedron 56、5735−5742頁(2000)
【非特許文献2】Journal of Organic Chemistry 31(1)、147−153頁(1966)
【非特許文献3】Chemical Communications 2006、643−645頁
【非特許文献4】Journal of Organometallic Chemistry 692、2270−2281頁(2007)
【発明の概要】
【発明が解決しようとする課題】
【0006】
(発明の要旨)
本発明の目的は、具体的には、より高い総収率を得ることができる、エゼチミブ(EZT)の合成または製造のための改良された方法および新しい方法を提供することであった。
【課題を解決するための手段】
【0007】
第1の態様において、本発明は、特徴的なZ−異性体構造を共有する、エゼチミブ(EZT)の合成のための、新規で有用な中間体を提供する。本発明によれば、Z−5−(4−フルオロフェニル)−ペント−4−エン酸に基づく化合物で出発する場合、およびZ−中間体を経て合成を進行する場合に、最終エゼチミブの合成の総収率が著しく増加することが見出された。Z−前駆体およびZ−中間体を経た合成経路で出発し、続けていくと、それぞれのE−類似体を経るよりも驚くほど高い全収率が得られる。加えて、Z−ペント−4−エン酸は、論文で知られる方法によるE−ペント−4−エン酸より著しく高い収率で調製される。Z−前駆体およびZ−中間体における前述の構造特性は、本明細書において、それぞれ示された二重結合において、Z−異性体であるまたはZ−エン配置を有すると称されることがある。
【0008】
したがって、本発明の第1の態様において、本発明によるエゼチミブの改良された合成の代表的な鍵となる化合物として、以下のZ−中間体化合物が提供される。
(1)Z−エン配置を有する以下の式I
【0009】
【化1】
(式中、R1は、Hであり、またはそれぞれ非置換または置換の、アルキル、シクロアルキル、アルケニル、シクロアルケニル、アルキニル、アリール、アラルキル、アリールシクロアルキルである。)
を有する化合物。
(2)R1がHである、(1)に記載の化合物。
(3)Z−エン配置を有する以下の式II
【0010】
【化2】
(式中、R2は脱離基である。)
を有する化合物。
【0011】
本明細書で使用する、用語「脱離基」は、カルボニル基から化学的に開裂し、それによって環化ステップを実施する後続のステップを最終的に助け、エゼチミブ構造中に出現する特徴的なアゼチジン−2−オン構造部分を形成することができる、化学的部分である。脱離基は、式IIのカルボニル基に結合するN−原子を有することが好ましい。さらに、脱離基は、環構造を形成することが好ましく、および/またはその化学的部分にキラル原子を有することができる。
(4)R2が、式IIのカルボニル基とアミド基を形成するN−ヘテロ原子であるN−ヘテロ環基か、またはRおよびR’が同一または異なり、環を形成することができ、前で定義したN−ヘテロ環基に変換され得るNRR’基であり;
ここで、好ましくは、N−ヘテロ環基がアルキル化またはアリール化され、より好ましくはキラルS異性体中心によって定義される、(3)に記載の化合物。
(5)R2が以下の式III:
【0012】
【化3】
(式中、R3は置換基、好ましくはかさ高い置換基、例えばアリール、置換アリール、または分岐アルキルまたは少なくとも4個のC原子を有するアルキル、好ましくはフェニル、置換フェニル、iPr、BuまたはtBuを表す。)
によって定義される、(4)に記載の化合物。
(6)Z−エン配置を有する以下の式IV
【0013】
【化4】
(式中、R2は、(3)から(5)のいずれか1つで定義した通りであり、R4は、OH−保護基である。)
を有する化合物。
(7)Z−エン配置を有する以下の式V
【0014】
【化5】
(式中、R4は、OH−保護基である。)
を有する化合物。
【0015】
本発明の別の態様によれば、以下が提供される。
(8)4−フルオロ−ベンズアルデヒドおよび式Vlの化合物:
【0016】
【化6】
(式中、X−は、ホスホニウム塩の陰イオン、好ましくはハロゲン化物、酢酸、トリフルオロ酢酸またはスルホン酸、具体的にはハロゲン化物であり、R’’は、アルキル、置換アルキル、アリールまたは置換アリールであり、R1は上の(1)で定義した通りである。より好ましくは、R’’はフェニルである。)
をウィッティッヒ反応に供することを含む、5−(4−フルオロフェニル)ペント−4−エン酸またはこの酸誘導体を製造する方法。
(9)得られた5−(4−フルオロフェニル)ペント−4−エン酸またはこの酸誘導体の(E,Z)−異性体混合物が、それぞれE−異性体およびZ−異性体として単離される、(8)に記載の方法。
(10)単離された異性体が、以下の式I
【0017】
【化7】
(式中、R1は上の(1)で定義した通りである。)
を有するZ−異性体化合物である、(8)または(9)に記載の方法。
(11)エステル誘導体が、式Vlの化合物として使用され、ウィッティッヒ反応後、得られたエステル基を鹸化に供し、対応する5−(4−フルオロフェニル)−ペント−4−エン酸を生成する、(8)から(10)のいずれか1つに記載の方法。
【0018】
本発明の別の態様によれば、以下が提供される。
(12)4−フルオロ−ベンズアルデヒドの式VIIの化合物とのウィッティッヒ反応によって、式IIを有する化合物
【0019】
【化8】
(式中、X−は、ホスホニウム塩の陰イオン、好ましくはハロゲン化物、酢酸、トリフルオロ酢酸またはスルホン酸、具体的にはハロゲン化物であり、R’’は、アルキル、置換アルキル、アリールまたは置換アリールであり、R2は(3)から(5)のいずれか1つで定義した通りである。より好ましくは、R’’はフェニルである。)
を製造する方法。
【0020】
この実施形態において、所望のR2基を既に有する、適したホスホニウムイリド化合物を使用することによって、式Iの化合物を経る手順の必要が排除される。
【0021】
本発明のさらに別の態様によれば、以下が提供される。
(13)上の式IからVで定義した通りの任意の化合物を使用した、エゼチミブを合成する方法。これらの中間体化合物のいずれかからでも、当業者は、従来の合成方法を使用して、最終的にエゼチミブを得ることができる。
【0022】
本発明の特定の態様において、以下が提供される。
(14)a)Z−エン配置を有する式Vの化合物を酸化して、以下で示す式IX(式中、R4は上で定義した通りである。)のケトンを得るステップ;
b)ステップ(a)のケトン生成物を還元し、対応する(S)−ヒドロキシ異性体生成物を得るステップ、および
c)還元された(S)−ヒドロキシ異性体生成物をOH−脱保護反応に供して、エゼチミブを得るステップ
【0023】
【化9】
を含む、エゼチミブを合成する方法。
【0024】
本発明は、さらに以下を提供する。
(15)(13)または(14)に記載の方法に従ってエゼチミブを調製するステップ、および
こうして調製されたエゼチミブを、少なくとも1つの薬学的に許容される賦形剤と混和するステップ
を含む、エゼチミブを活性成分として含む医薬組成物を調製する方法。
(16)Z−エン配置を有する化合物Vを本質的に含まず、その脱保護された4−ヒドロキシフェニル類似体を本質的に含まないエゼチミブ。
(17)a)(3R,4S)−4−(4−(ベンジルオキシ)フェニル)−1−(4−フルオロフェニル)−3−((Z/E)−3−(4−フルオロフェニル)−アリル)アゼチジン−2−オンの3−Z−またはE−異性体のどちらかまたは両方を本質的に含まない、および/またはその脱保護された4−ヒドロキシフェニル類似体を本質的に含まないエゼチミブを提供するステップ、ならびに
b)こうして提供されたエゼチミブを、少なくとも1つの薬学的に許容される賦形剤と混和するステップ
を含む、エゼチミブを活性成分として含む医薬組成物を調製する方法。
(18)(17)に記載の方法によって得られる、活性成分としてのエゼチミブおよび少なくとも1つの薬学的に許容される賦形剤を含む医薬組成物。
【図面の簡単な説明】
【0025】
【図1A】本発明の実施形態による、例示的なZ−中間体を介するエゼチミブの全合成のための、好ましい合成経路を示す概略図である。
【図1B】本発明の実施形態による、例示的なZ−中間体を介するエゼチミブの全合成のための、好ましい合成経路を示す概略図である。
【発明を実施するための形態】
【0026】
本発明の特定の態様によれば、エゼチミブの全合成のための重要な中間体としての5−(4−フルオロフェニル)−ペント−4−エン酸およびこの誘導体は、ウィッティッヒ反応を介した非常に有効な合成手法によって高収率でもたらされる。この方法ステップにおいて、4−フルオロ−ベンズアルデヒドは、式VIの化合物と一緒にウィッティッヒ反応に供される。5−(4−フルオロフェニル)−ペント−4−エン酸の誘導体として、典型的に、酸性塩、酸エステルおよび酸アミド、好ましくは酸エステルが挙げられるが、これらだけには限らない。好ましい実施形態において、R1=水素である酸基が使用されるか、より好ましくは、酸基は、それぞれ非置換または置換の、アルキル、シクロアルキル、アルケニル、シクロアルケニル、アルキニル、アリール、アラルキルおよびアリールシクロアルキルから選択されるR1によって誘導体化される。
【0027】
次いで、得られた5−(4−フルオロフェニル)−ペント−4−エン酸またはこの誘導体のE−およびZ−異性体の混合物は、それぞれE−異性体およびZ−異性体として単離され得る。驚くべきことに、本発明による方法におけるウィッティッヒ反応は、高い総収率を達成することに大いに寄与するだけでなく、より重要なことには、大部分は所望のZ−異性体(すなわちZ/E−モル比がより高く、具体的には実質的に50%より高い)を生成し、これによって、エゼチミブの合成のための代表的な鍵となるZ−中間体を選択的に単離し、得ることが可能になる。注目すべきは、Z−およびE−異性体の混合物に対して90%を上回る収率で、Z−異性体の範囲が約80%の混合物(Z:Eの比=88:12)は、具体的には、本発明の実施形態においてウィッティッヒ反応を使用することによって達成され得る。
【0028】
望ましいZ−異性体は、Z−およびE−異性体混合物から容易に単離され、場合によって精製され、その結果、式I
【0029】
【化10】
の化合物を提供することができる。
【0030】
残基R1は、Hであり、または非置換もしくは置換アルキル、非置換もしくは置換シクロアルキル、非置換もしくは置換アルケニル、非置換もしくは置換シクロアルケニル、非置換もしくは置換アルキニル、非置換もしくは置換アリール、非置換もしくは置換アラルキル、または非置換もしくは置換アリールシクロアルキルである。前述のR1に対応する残基におけるアルキル、アルケニルもしくはアルキニル基またはこれらの環構造として、1から12個、好ましくは1から8個、より好ましくは1から6個の炭素原子を含有する有機基を挙げることができるが、これらだけには限らない。好ましい残基R1は、Hであり、または例えばメチル、エチル、プロピル、イソプロピル、n−ブチル、tert−ブチル、ペンチル、ヘキシルなどの1から6個の炭素原子を有する低級アルキルである。適したアリール基として、フェニル、ナフチル、ジフェニルなどを挙げることができるが、これらだけには限らない。適した置換基として、1から6個の炭素原子を有する低級アルキル、ハロゲン、アミン基などを挙げることができるが、これらだけには限らない。
【0031】
同様の意味が、他に記述がなければ、本明細書で使用するアルキル、置換アルキル、アリール、置換アリールおよび類似の用語の定義に適用される。
【0032】
好ましい実施形態によれば、ウィッティッヒ反応は、R1が上述の基から選択されるがHではない場合に、より効率的に進行し、対応する5−(4−フルオロフェニル)ペント−4−エン酸誘導体、とりわけエステル誘導体が得られ、その後すぐに、得られたエステル基を鹸化に供し、対応する5−(4−フルオロフェニル)−ペント−4−エン酸を生成することができる。鹸化は、エステル誘導体の単離無しに、ウィッティッヒ反応後にワンポットプロセスとして実施されることが好ましい。次いで、式Iの化合物(具体的には、式中、R1はHである。)は、式IIの化合物(式中、R2は上で示した脱離基を示す。)に変換することができる。好ましい実施形態によれば、R2は、N−ヘテロ原子が式IIにおけるカルボニル基とアミド基を形成する、N−複素環基である。エゼチミブを生成するためのさらなるエナンチオ選択的合成ステップを考慮すると、N−複素環基はキラル中心を有し、より好ましくはキラルS−異性体中心を有することが、より好ましい。したがって、より好ましいN−複素環基は、例えば、これらだけには限らないが、置換されたまたは置換されていない、メチル、エチル、プロピル、イソプロピル、n−ブチル、tert−ブチルなどの低級アルキルから選択されるアルキル基、またはフェニル、ナフチルなどのアリールによってアルキル化またはアリール化される。
【0033】
したがって、式IIを有する化合物は、所望のZ−形態で提供することができ、ここでR2は上で定義されている。R2がNRR’基の場合、RおよびR’は、同一または異なることができ、これらは、当業者に利用可能な知識に従って、前記N−複素環基に変換され得る環を形成するために選択することができる。R2は、下の式III(式中、アルキルおよびアリールは上で定義した通りである。)の構造部分によって定義されることが好ましい。
【0034】
【化11】
【0035】
この式IIIにおいて、R3は置換基、好ましくはかさ高い置換基、例えばアリール、置換アリールまたは少なくとも4個のC原子を有する分岐アルキルまたはアルキルを表す。R3は、好ましくはフェニル、置換フェニル、iPr、BuまたはtBuである。
【0036】
本発明の一実施形態による場合、式IIの化合物は、Z−5−(4−フルオロフェニル)−ペント−4−エン酸との反応から得られ、酸基は、特に、残基R2が式IIIによって定義される、後続の式IIの化合物の形成に対して活性化され得る。Z−5−(4−フルオロフェニル)−ペント−4−エン酸の酸基の活性化は、例として、例えば、ホスゲン、塩化オキサリル(COCl)2、塩化チオニルClSOCl、五塩化リンPCl5、臭化ベンゾイルなどのそれぞれとの反応によってなど、酸塩化物または酸臭化物などの酸ハロゲン化物の形成を介する、および対応する無水炭酸を介する、既知の方法によって実行することができる。
【0037】
上述のウィッティッヒ反応に対して、適切なトリフェニルホスホニウム塩を調製して、式VI
【0038】
【化12】
(式中、Xは、ハロゲン、アセテート、トリフルオロアセテートまたはスルホネートであってよく、より好ましくは、ClまたはBrなどのハロゲンであり、さらに、式中、R’’は、アルキル、置換アルキル、アリールまたは置換アリールであり、
R1は上の通り定義される。)
の化合物を得る。
【0039】
好ましい実施例において、R’’は、以下で示す例示的な式VIを採用するためのフェニルである(XおよびR1は、上の通り定義される。)。
【0040】
【化13】
【0041】
本発明による方法の変形において、式II
の化合物は、4−フルオロ−ベンズアルデヒドの、対応する式VII
【0042】
【化14】
(式中、Xは、ハロゲン、アセテート、トリフルオロアセテートまたはスルホネートであってよく、より好ましくは、ClまたはBrなどのハロゲンであり、さらに、式中、R’’は、アルキル、置換アルキル、アリールまたは置換アリールであり、R2は上の通り定義される。)
の化合物とのウィッティッヒ反応を実行することによって、直接得ることができる。
【0043】
好ましい実施形態において、R’’は、以下で示す例示的な式VIIを採用するためのフェニルである(XおよびR2は、上の通り定義される。)。
【0044】
【化15】
【0045】
式IIの望ましいZ−中間体化合物は、エゼチミブ合成に使用されるので、ウィッティッヒ反応から得られるZ−異性体形態は、異性体の混合物から単離されるものとする。
【0046】
エゼチミブの全合成経路の後続のステップにおいて、式IIの化合物、より好ましくは、式IIIによって示される通りの基R2を有する化合物は、以下で示す式VIII(式中、R4は、これらだけには限らないが、ベンジル(Bn)、炭酸ベンジル(Cbz)、ベンジルオキシメチルエーテル(BOM);シリル、より好ましくはC1−C8トリアルキルシリル、C1−C8ジアルキルアリールシリル、C1−C8アルキルジアリールシリル(ここで、アルキルは、同一または異なっていてもよく、好ましくは、アリールはフェニルであり、アルキルは1から4個のC原子を有する。)などを含めたOH−保護基である。)によって定義されるイミン化合物と反応し、
【0047】
【化16】
【0048】
【化17】
(式中、R2およびR4は、上で定義した通りである。)を有する化合物を生成する。
【0049】
次いで、化合物IVは、適切に、ビス−トリメチルシリルアセトアミド(BSA)などのシリル化剤での処理、およびテトラブチルアンモニウムフルオリド(TBAF)などのフッ化物イオン触媒による後続の処理によって環化され、その結果、式V:
【0050】
【化18】
(式中、R4は、上で定義した通りである。)を有する化合物を生成することができる。
【0051】
次いで、Z−異性体構造を有する式Vの化合物は、エゼチミブの合成におけるステップに有用な中間体を構成する。
【0052】
ここで、最初に、この化合物を酸化して、上で定義した通りの式IXのケトンを得て、次いで、例えばキラルボランとの反応によって、ケトンを還元して、所望のS−OH−異性体生成物を得て、単離し、最後に、還元されたS−OH−異性体生成物をOH−脱保護反応に供し、最終的にエゼチミブを得る。適したOH−脱保護反応として、通常、遷移金属のPt、Pd、Rhなどの金属触媒を、それぞれ単独または混和して、好ましくは、活性炭、アルミナ、シリカなどの担持物質と組み合わせて用いる接触水素化が挙げられる。最も好ましい触媒は、炭素担持パラジウムである。水素化反応に存在するべき水素源として、H2ガス、ギ酸アンモニウム/ギ酸などが、使用されてもよい。
【0053】
本発明によるエゼチミブを合成する方法は、Z−出発および中間体化合物を使用すると、対応するE−出発物質または中間体の使用と比較して、予想外に高い収率(エゼチミブの総収率と称される。)をもたらすことができるという点で特に有利である。このことは、全方法の設計(複合体混合物が、反応後に得られるTetrahedron 2000、56、5735−5742頁と比較して、純粋なZ−酸を生成する「クリーンな」ウィッティッヒ反応の適用)および、高純度での単離を可能にし、したがって、全収率を低下させる副反応の程度がより低い後続のステップのそれぞれにおいて変換されるZ−出発および中間体化合物の物性(溶解性、クロマトグラフィーの挙動)によるものである。この効果は、アルケンV酸化ステップおよび化合物IXのキラルボラン還元ステップにおいて、特に顕著になる。すなわち、Z−アルケンの酸化は、収率86%のケトンIXを生成するが、E−アルケン酸化は、US−A−5856473に記載されているように、収率70−80%でケトンを生成する。さらに、Z−中間体を経て誘導されるケトンのケトン還元ステップによる収率は、95%になるが、US−A−5856473に記載されているように、E−中間体から誘導されるケトンの収率は、わずか42%になる。したがって、エゼチミブを合成する方法のための中間体として上で述べた化合物の使用は、本発明の有益な側面である。上で述べた特別で好ましい反応の他に、最終的にエゼチミブを得るその他の従来の合成ステップまたは知られている変形(さらに今開示したZ−中間体化合物の使用を含めて)が、適用され得る。
【0054】
次いで、本発明による方法によって調製されるエゼチミブは(通常、所望の純度を得た後に)、少なくとも1つの薬学的に許容される賦形剤と混和され得る。エゼチミブを含む、薬学的に安全で安定な医薬組成物を得るために、エゼチミブが任意の医薬賦形剤と混和される前に、化合物Vを本質的に含まない、好ましくは全く含まないことを確認することが重要である。化合物Vの存在自体は、最終医薬品の薬理学的特徴および安定性、具体的には化学安定性にとって有害であると考えられ、さらに、化合物V中間体の後のエゼチミブ合成ステップで形成される可能性のある、さらなる有害な不純物を示すことがある。不純物を無くしてから医薬賦形剤と混和することの重要性は、特に固形剤形が予定されている場合、不純物が、活性なエゼチミブおよび賦形剤を一緒に最終組成物中に同時処理した後では、もはや取り除くことができないことにある。いかなる理論によっても制限されないが、化合物V中に存在するアリル基の二重結合は、前述の最終エゼチミブ合成ステップ(例えば、化合物の3位での(S)−ヒドロキシル基にエナンチオピュアな還元[上の項(14)のステップ(b)参照]、および/または最終OH−脱保護反応[上の項(14)のステップ(c)参照])またはその他の最終エゼチミブ合成ステップでの反応の妨害、および/または、特に、エゼチミブを含有する医薬組成物中の賦形剤との最終混和物に、深刻に関与する恐れがあると考えることができる。最終エゼチミブ生成物中に化合物Vを本質的に含まないこと、またはさらには存在しないことが、化合物Vが遊離体として使用されるステップ中、またはステップの後の適切な精製および単離方法によって、確実となるべきである。化合物Vが遊離体として使用された合成ステップ後に、化合物Vの全てを除去することはできないという観点から、エゼチミブを得るための最終脱保護ステップにおける別の深刻な不純物として最終的に形成された可能性のある化合物Vの脱保護された4−ヒドロキシフェニル類似体も、エゼチミブが医薬組成物の調製において医薬賦形剤と混和される前に確実に除去されることが、さらに妥当である。
【0055】
エゼチミブが対応するE−中間体を経て合成された場合、同じ見解が同様に適用される。本発明は、Z−中間体を介して進行することができるが、任意の医薬賦形剤と混和されるエゼチミブはまた、(3R,4S)−4−(4−(ベンジルオキシ)フェニル)−1−(4−フルオロフェニル)−3−(E)−3−(4−フルオロフェニル)−アリル)アゼチジン−2−オンの対応するE−異性体を含まないことが確実となる。したがって、E−異性体を本質的に含まないことおよびさらに全く存在しないことは、薬学的に安全で安定なエゼチミブ含有医薬組成物を可能にする本発明の別の利点である。
【0056】
本明細書で使用する、用語「化合物Vを本質的に含まない」(すなわちZ−異性体)、または同様に化合物VのE−異性体もしくはそれぞれの脱保護された4−ヒドロキシフェニル(ZまたはE)類似体を「本質的に含まない」とは、得られたEZT生成物の総重量に対して、その量が、例えば約0.2重量%より低い、好ましくは約0.1重量%未満、より好ましくは約0.05重量%未満の対照を意味するものとする。化合物V、これに対応するE−異性体、またはこれらのそれぞれの脱保護された4−ヒドロキシフェニル類似体の検出は、それぞれEZT生成物に対して、HPLCによって実行され得る。しかし、用語「を含まない」は、EZT合成がZ−またはE−中間体を経由したかどうかを示す、少なくとも微量の同定されたEZT不純物の存在を含めることができる。
【0057】
医薬組成物は、コレステロールの、高脂血症のおよび脂肪血症の状態、癌から選択される疾病のいずれか1つ、ならびに抗菌状態の予防および/または治療に使用することができる。好ましい使用は、高コレステロール血症である。適したエゼチミブの用量は、0.1mgから100mg、より適切には1mgから20mgの範囲とすることができる。薬学的に許容される賦形剤は、当業者に知られる適した担体および賦形剤から選択され得る。例えば、適した薬学的に許容される担体および/または賦形剤として、これらだけには限らないが、結合剤、充填剤、マトリックス形成剤、希釈剤、酸化防止剤、緩衝剤、消泡剤、保存剤、キレート化剤、粘度調整剤、香味料、着色剤、臭気剤、乳白剤、安定化剤、可溶化剤、可塑剤、潤滑剤およびこれらの混合物が挙げられる。適した具体的な例として、これらだけには限らないが、ラクトース、微結晶性セルロース、ヒドロキシプロピルセルロースなどのセルロース誘導体、ポリアクリレート、炭酸カルシウム、デンプン、コロイド状二酸化シリコーン、デンプングリコール酸ナトリウム、タルク、ステアリン酸マグネシウム、ポリビニルピロリドン、ポリエチレングリコールおよび医薬技術の分野で知られるその他の賦形剤からなる群のものが挙げられる。
【0058】
賦形剤はまた、所望の投与形態および剤形によって選択されてもよい。適した投与形態として、経口、非経口、注射、経皮、経鼻、経直腸、吸入が挙げられるが、これらだけには限らない。適した剤形として、錠剤、カプセル、顆粒、粉末、丸薬、懸濁剤、液剤、坐薬、徐放剤形が挙げられるが、これらだけには限らない。本発明による医薬調製物において、エゼチミブはまた、1つまたは複数の活性成分と組み合わせてもよい。
【0059】
本発明は、単に例示的な目的として提供される実施例を参照することで、下でより詳細に説明されるが、本発明は、決してこれに限定されない。
【0060】
好ましい反応ステップによるエゼチミブの全合成のための特に好ましい合成経路は、図1Aおよび1Bに示される。関連する反応ステップは、以下の実施例でより詳細に説明される。
【0061】
(実施例)
【実施例1】
【0062】
(4−エトキシ−4−オキソブチル)トリフェニルホスホニウムブロミド(1)の合成
【0063】
【化19】
【0064】
4−ブロモ酪酸エチル(51.0g、261mmol)およびトリフェニルホスフィン(67.2g、256mmol)の乾燥トルエン(250mL)中溶液を撹拌し、還流下およびアルゴン雰囲気下で4日間加熱した。反応混合物を室温に冷却後、形成された不溶性の白色沈殿物を濾別し、加熱したトルエン(100mL)およびヘキサン(80mL)で洗浄した。この生成物を粉砕し、減圧下で乾燥し、白色の粉末として表題化合物1(113.7g、97%)を得た。
【実施例2】
【0065】
(Z)−5−(4−フルオロフェニル)ペント−4−エン酸(3)の合成
【0066】
【化20】
【0067】
乾燥THF(30mL)中の粉末(4−エトキシ−4−オキソブチル)トリフェニルホスホニウムブロミド(1)(10.5g、23.7mmol)を、アルゴン雰囲気下で、撹拌しながら、18℃でナトリウムヘキサメチルジシラジド(12.5mL、25mmol;THF中2M溶液)で処理した。20分後、4−フルオロベンズアルデヒド(2.61g、21.0mmol)を18℃で加え、赤オレンジ色の混合物を速やかに脱色した。室温で4時間撹拌した後、反応混合物を氷浴で冷却し、水10mL中のNaOH(1.6g、40mmol)を加えた。得られた溶液を室温でさらに12時間撹拌した。水(100mL)およびエーテル(200mL)を加えた。相を分離させ、水相をエーテル(2x50mL)ですすぎ、酸性化し(4M HCl)、EtOAc(4x50mL)で抽出した。採集したEtOAc抽出物を無水Na2SO4で乾燥し、濃縮した。クーゲルロール蒸留によって、88%のcis異性体を含有する白色の結晶として、3.52g(90.6%)の純粋な5−(4−フルオロフェニル)ペント−4−エン酸(3)を得た。このcis異性体は、エステル誘導体または酸性塩誘導体の再結晶化、続いて酸性化、または粗酸の再結晶化後に、純粋な形態で得ることができる
化合物2(cis−エステル):
1H−NMR(300MHz,CDCl3):δ(ppm)=1.26(t,3H,J=7.1Hz,−CH3)、2.42−2.47(m,2H,CH2CH2CO)、2.60−2.68(m,2H,CH2CH2CO)、4.15(q,2H,J=7.1Hz,−CH2−CH3)、5.63(td,1H,J=7.2Hz,J=11.6Hz,CH=CHCH2)、6.44(d,1H,J=11.6Hz,PhCH=CH)、7.00−7.08(m,2H,Ph−H)、7.22−7.28(m,2H,Ph−H)ppm。
化合物3(cis−カルボン酸):
1H−NMR(300MHz,CDCl3):δ(ppm)=2.46−2.51(m,2H,CH2CH2CO);2.59−2.66(m,2H,CH2CH2CO)、5.64(td,1H,J=7.0Hz,J=11.6Hz,CH=CHCH2)、6.44(d,1H,J=11.6Hz,PhCH=CH)、7.00−7.08(m,2H,Ph−H)、7.22−7.27(m,2H,Ph−H)、9.99(bs,1H,−COOH)ppm。
【実施例3】
【0068】
(S,Z)−3−(5−(4−フルオロフェニル)ペント−4−エノイル)−4−フェニルオキサゾリジン−2−オン(4)の合成
【0069】
【化21】
【0070】
p−フルオロフェニルペンテン酸(3)(2.3g)のCH2Cl2(40mL)中溶液に、塩化オキサリル(1.81g)およびDMF(2滴)を、0℃でゆっくり加えた。この溶液を室温まで温めて、1時間還流させた。溶媒を減圧下で蒸発させ、過剰の塩化オキサリルをCH2Cl2(2x50mL)と共沸除去した。得られた酸塩化物をCH2Cl2(40mL)に再溶解し、この溶液を、(S)(+)−4−フェニル−2−オキサゾリドン(1.99g)、N,N−ジイソプロピルエチルアミン(DIPEA)(3.07g)およびN,N−ジメチルアミノピリジン(0.05g)のCH2Cl2(20mL)中の冷却した溶液に滴下した。得られた混合物を室温で2時間撹拌した。CH2Cl2(100mL)を加え、有機相を0.5MのHCl(2×40mL)、水(1×40mL)およびブライン(1×40mL)で洗浄した後、次いで乾燥させ、濃縮して、所望の生成物を結晶化した。生成物を酢酸エチル/ヘキサンから再結晶させ、濾過して採集した(3.7g、93%)。
1H−NMR:(300MHz,CDCl3):δ 2.55−2.70(m,2H,CH2CH2CO)、3.05−3.15(m,2H,CH2CH2CO)、4.29(dd,J=8.91Hz,J=3.69Hz,1H,CHCH2OCO)、4.69(t,J=8.82Hz,1H,CHCH2OCO)、5,43(dd,J=8.71Hz,J=3.66Hz,1H,CHCH2OCO)、5.57−5.66(m,1H,ArCH=CH)、6.40(d,J=11.56Hz,1H,ArCH=CH)、6.95−7.04(m,2H,ArH)、7.20−7.43(m,7H,ArH)ppm。
【実施例4】
【0071】
(S)−3−((R,Z)−2−((S)−(4−(ベンジルオキシ)フェニル)(4−フルオロフェニルアミノ)メチル)−5−(4−フルオロフェニル)ペント−4−エノイル)−4−フェニルオキサゾリジン−2−オン(6)の合成
【0072】
【化22】
【0073】
−20℃の(S,Z)−3−(5−(4−フルオロフェニル)ペント−4−エノイル)−4−フェニルオキサゾリジン−2−オン(4)(1.0g)のCH2Cl2(15mL)中溶液に、TiCl4のCH2Cl2(3.25mL)中1M溶液をゆっくり加え、−20℃で撹拌した。15分後、DIPEA(0.97mL)を加え、混合物を30分間、−20℃で撹拌した。(E)−N−(4−(ベンジルオキシ)ベンジリデン)−4−フルオロアニリン(5)(1.53g)をCH2Cl2(30mL)に溶解し、温度を−20℃より低く維持しながら溶液に加えた。1.5時間後、反応を、CH2Cl2(3mL)中の氷酢酸(1mL)で、−20℃でクエンチして、30分間撹拌した。反応混合物を1MのH2SO4(40mL)の水溶液に注ぎ、さらに30分混合した。その後、酢酸エチル150mLを加え、有機相をNaHCO3(3x40mL)の飽和溶液およびブライン(1×40mL)で洗浄し、次いでNa2SO4で乾燥して、濃縮した。生成物を酢酸エチル/ヘキサンから再結晶させ、濾過して採集した。結晶(1.15g;59%)を冷却したメタノールで洗浄した。
1H−NMR:(300MHz,CDCl3):δ 2.31−2.40(m,1H,=CHCH2CH)、2.65−2.75(m,1H,=CHCH2CH)、4.22(dd,J=8.76Hz,J=3.13Hz,1H,CHCH2OCO)、4.39(t,J=9.42Hz,1H,=CHCH2CH)4.36−4.60(m,1H,CHNH)、4.69(t,J=8.67Hz,1H,CHCH2OCO)、4.95(d,J=10.32Hz,1H,NH)、5.03(s,2H,CH2Ph)、5.43(dd,J=8.45Hz,J=3.11Hz,1H,CHCH2OCO)、5.51−5.60(m,1H,ArCH=CH)、6.24−6.42(m,1H,ArCH=CH)、6.69−6.81(m,2H,ArH)、6.84−6.96(m,4H,ArH)、7.08−7.18(m,10H,ArH)、7.38−7.50(m,7H,ArH)ppm。
MS(ES+)m/z(%):645(MH+,60)、534(100)。
【実施例5】
【0074】
(3R,4S)−4−(4−(ベンジルオキシ)フェニル)−1−(4−フルオロフェニル)−3−((Z)−3−(4−フルオロフェニル)−アリル)アゼチジン−2−オン(7)の合成
【0075】
【化23】
【0076】
6(0.484g、0.75mmol)の乾燥トルエン(5.0mL)中懸濁液を、アルゴンで脱酸素化し、15分後、BSA(1.50mmoL、0.37ml)を加えた。室温で30分撹拌した後、TBAF×3H2O(5.0mol%、0.0375mmol、11.8mg)を加え、4時間撹拌させたままにした。次いで、氷酢酸0.027mlおよびMeOH 4.0mlを加えた。5分後、反応混合物を減圧化で濃縮し、EtOAc(50ml)を加えた。有機相を、5%の重炭酸ナトリウム溶液(25ml)、水(25ml)およびブライン(25ml)で洗浄し、硫酸ナトリウムで乾燥し、濾過して、減圧下で濃縮した。粗生成物を、溶出液としてEtOAc/ヘキサン(1/4)を用いるフラッシュクロマトグラフィーによって精製し、0.256g(71%)の化合物7を得た。
1H−NMR(300MHz,CDCl3):δ(ppm)=2.74−2.95(m,1H,CH2)、3.13−3.19(m,1H,CHCO)、4.52(d,J=2.3Hz,1H,CHN)、5.07(s,2H,OCH2Ph)、5.63−5.71(m,1H,PhCHCH)、6.52(d,J=11.5Hz,1H,PhCHCH)、6.86−7.04(m,6H,Ph−H)、7.15−7.24(m,6H,Ph−H)、7.30−7.41(m,5H,Ph−H)。
【実施例6】
【0077】
(3R、4S)−4−(4−(ベンジルオキシ)フェニル)−1−(4−フルオロフェニル)−3−(3−(4−フルオロフェニル)−3−オキソプロピル)アゼチジン−2−オン(8)の合成
【0078】
【化24】
【0079】
Pd(OAc)2(3.0mol%、0.042mmol、9.43mg)、ベンゾキノン(2.1mmol、227mg)および過塩素酸(70%水溶液、0.15M、0.100ml)をアセトニトリル(3.5mL)に溶解し、少なくとも20分間、アルゴンでパージして脱酸素化した。次いで、同様にアルゴンで脱酸素化した水(0.7ml)を加えた。反応混合物を、さらに5分間、アルゴン雰囲気下で激しく撹拌し、同様にあらかじめアルゴンで30分間パージして脱酸素化した7(1.40mmol)のアセトニトリル(3.5ml)中溶液を加えた。4時間後、過塩素酸(70%水溶液、0.100ml)を、再度加えた。得られた溶液を48時間撹拌し、次いでEtOAc(50ml)で希釈した。水相を別の30mLのEtOAcで洗浄した。有機相を合わせて、水(2×40m)およびブライン(40ml)で洗浄し、硫酸ナトリウムで乾燥し、濾過して、減圧下で濃縮した。粗生成物を、EtOAc/ヘキサン(1/4)で溶出するフラッシュクロマトグラフィーによって精製し、化合物8(0.600g)を収率86%で得た。
1H−NMR(300MHz,CDCl3):δ(ppm)=2.21−2.47(m,2H,CH2CH)、3.10−3.21(m,2H,CH2CO)、3.24−3.35(m,1H,CHCO)、4.68(d,J=2,3Hz,1H,CHN)、5.05(s,2H,OCH2Ph)、6.90−6.99(m,4H,Ph−H)、7.09−7.17(m,2H,Ph−H)、7.22−7.27(m,4H,Ph−H)、7.33−7.44(m,5H,Ph−H)、7.96−8.02(m,2H,Ph−H)。
【実施例7】
【0080】
(3R,4S)−4−(4−(ベンジルオキシ)フェニル)−1−(4−フルオロフェニル)−3−((S)−3−(4−フルオロフェニル)−3−ヒドロキシプロピル)アゼチジン−2−オン(9)の合成
【0081】
【化25】
【0082】
(R)−テトラヒドロ−1−メチル−3,3−ジフェニル−1H,3H−ピロロ[1,2−c]−[1,3,2]オキサザボロール(96mg;0.346mmol)および(3R,4S)−4−(4−(ベンジルオキシ)フェニル)−1−(4−フルオロフェニル)−3−(3−(4−フルオロフェニル)−3−オキソプロピル)アゼチジン−2−オン(8)(760mg;1.528mmol)を、アルゴン雰囲気下で乾燥THF(2mL)に溶解した。溶液を−22℃に冷却し、5分間撹拌した後、ボロヒドリド−ジメチルスルフィド複合体(THF中2M溶液;0.864mL;1.728mmol)を、2時間かけて滴下した。−22℃で合計5時間撹拌した後、反応をメタノール(3mL)の添加によってクエンチした。EtOAc(30mL)および1MのHCl(15mL)を加え、相を分離させた。水相をEtOAc(20mL)で2度抽出した。合わせたEtOAc抽出物を無水Na2SO4で乾燥し、濃縮して泡(0.900g)を安定させた。得られた粗生成物を、移動相としてEtOAc/ヘキサン=1/3を用いるフラッシュクロマトグラフィーによって精製し、表題化合物9(0.731g、95.8%)を得た。
1H−NMR(300MHz,CDCl3):δ(ppm)=1.84−2.05(m,4H;CH2CH2);3.07−3.17(m,1H,CHCO);4.59(d,J=2.4Hz;1H,CHN);4.71−4.76(m,1H,CHOH);5.07(s,2H,OCH2Ph);6.91−7.06(m,6H,Ph−H);7.22−7.46(m,11H,Ph−H)ppm
【実施例8】
【0083】
エゼチミブ(10)の合成
【0084】
【化26】
【0085】
(3R,4S)−4−(4−(ベンジルオキシ)フェニル)−1−(4−フルオロフェニル)−3−((S)−3−(4−フルオロフェニル)−3−ヒドロキシプロピル)アゼチジン−2−オン(9)を、エタノールに溶解し、5%Pd/C(0.5mol%)を、得られた溶液に加えた。得られた懸濁液を、出発物質9が消費されるまで、高圧下でのH2ガス雰囲気下で撹拌した。触媒を濾過して除去した。得られた溶液を減圧下で濃縮し、再結晶化して、表題化合物10を得た。
【0086】
結果:
個々のステップにおける収率に関する、およびエゼチミブの全合成の全収率に関するデータを、Z−中間体に基づく本発明による結果およびE−中間体に基づく従来技術による結果をそれぞれ別々に、下記の表1および2に示す。
【0087】
【表1】
【0088】
【表2】
【0089】
【表3】
US 5,856,473で開示されたE−誘導体を用いるステップの全収率:16.8%−19.2%。
手作業でUS 5,856,473のE−誘導体を用いる方法の全収率:1.5%
本発明によって得られたZ−誘導体を用いる同様のステップの全収率:31.8%
Tetrahedron 56(2000)5735−5742およびUS 5,856,473を合わせた全収率:9.4%−10.7%
Tetrahedron 56(2000)5735−5742およびUS 5,856,473を手作業で合わせた全収率:0.07%−0.25%
Z−誘導体を用いる本発明によって得られた全収率:24.7%
【特許請求の範囲】
【請求項1】
Z−エン配置を有する以下の式I
【化1】
(式中、R1は、Hであり、またはアルキル、シクロアルキル、アルケニル、シクロアルケニル、アルキニル、アリール、アラルキルおよびアリールシクロアルキルから選択される、それぞれ非置換または置換の有機基である。)
を有する化合物。
【請求項2】
R1がHである、請求項1に記載の化合物。
【請求項3】
Z−エン配置を有する以下の式II
【化2】
(式中、R2は脱離基である。)
を有する化合物。
【請求項4】
R2が、式IIのカルボニル基とアミド基を形成するN−ヘテロ原子であるN−ヘテロ環基か、またはRおよびR’が同一または異なり、環を形成することができ、前記N−ヘテロ環基に変換され得るNRR’基であり、好ましくは、式中、R2が以下の式III:
【化3】
(式中、R3は、置換基、好ましくはアリール、置換アリール、分岐アルキルおよび少なくとも4個のC原子を有するアルキルから選択される、特にフェニル、置換フェニル、iPr、BuおよびtBuから選択される置換基を表す。)
によって定義される、請求項3に記載の化合物。
【請求項5】
Z−エン配置を有する以下の式IV
【化4】
(式中、R2は、請求項3で定義した通りであり、R4は、OH−保護基である。)
を有する化合物。
【請求項6】
Z−エン配置を有する以下の式V
【化5】
(式中、R4は、OH−保護基である。)
を有する化合物。
【請求項7】
4−フルオロ−ベンズアルデヒドおよび式Vlの化合物:
【化6】
(式中、R’’は、アルキル、置換アルキル、アリールまたは置換アリール、好ましくはフェニルであり、X−は、ホスホニウム塩の陰イオン、好ましくはハロゲン化物、酢酸、トリフルオロ酢酸またはスルホン酸、具体的にはハロゲン化物であり、R1は、請求項1で定義した通りである。)
をウィッティッヒ反応に供することを含む、5−(4−フルオロフェニル)ペント−4−エン酸またはこの酸誘導体を製造する方法。
【請求項8】
得られた5−(4−フルオロフェニル)ペント−4−エン酸またはこの酸誘導体の(E,Z)異性体が、それぞれE−異性体およびZ−異性体として単離される、請求項7に記載の方法。
【請求項9】
単離された異性体が、以下の式I
【化7】
(式中、R1は、請求項1で定義した通りである。)
を有するZ−異性体化合物である、請求項7または8に記載の方法。
【請求項10】
エステル誘導体が、式Vlの化合物として使用され、ウィッティッヒ反応後、得られたエステル基を鹸化に供して、対応する5−(4−フルオロフェニル)−ペント−4−エン酸を生成する、請求項7から9のいずれか一項に記載の方法。
【請求項11】
4−フルオロ−ベンズアルデヒドの式VIIの化合物とのウィッティッヒ反応によって、請求項3に記載の式IIの化合物
【化8】
(式中、X−は、ホスホニウム塩の陰イオン、好ましくはハロゲン化物、酢酸、トリフルオロ酢酸またはスルホン酸、具体的にはハロゲン化物であり、R’’は、アルキル、置換アルキル、アリールまたは置換アリール、好ましくはフェニルであり、R2は、請求項3または4で定義した通りである。)
を製造する方法。
【請求項12】
エゼチミブの合成のための中間体としての、請求項1から6のいずれか一項に記載の化合物を使用する使用。
【請求項13】
a)請求項6に記載の式Vの化合物を酸化して、以下で示す式IX(式中、R4は上で定義した通りである。)のケトンを得るステップ;
b)ステップ(a)のケトン生成物を還元して、(S)−OH異性体生成物を得るステップ、および
c)還元された(S)−OH生成物をOH−脱保護反応に供して、エゼチミブを得るステップ
【化9】
を含む、エゼチミブを合成する方法。
【請求項14】
a)請求項12または請求項13に記載の方法に従ってエゼチミブを調製するステップ、および
b)こうして調製されたエゼチミブを、少なくとも1つの薬学的に許容される賦形剤と混和するステップ
を含む、エゼチミブを活性成分として含む医薬組成物を調製する方法。
【請求項15】
化合物Vを本質的に含まず、その脱保護された4−ヒドロキシフェニル類似体を本質的に含まないエゼチミブ。
【請求項16】
a)(3R,4S)−4−(4−(ベンジルオキシ)フェニル)−1−(4−フルオロフェニル)−3−((Z/E)−3−(4−フルオロフェニル)−アリル)アゼチジン−2−オンの3−Z−またはE−異性体のどちらか一方または両方を本質的に含まない、またはその脱保護された4−ヒドロキシフェニル類似体を本質的に含まないエゼチミブを提供するステップ、および
b)こうして提供されたエゼチミブを、少なくとも1つの薬学的に許容される賦形剤と混和するステップ
を含む、エゼチミブを活性成分として含む医薬組成物を調製する方法。
【請求項17】
請求項16に記載の方法によって得られる、活性成分としてのエゼチミブおよび少なくとも1つの薬学的に許容される賦形剤を含む医薬組成物。
【請求項1】
Z−エン配置を有する以下の式I
【化1】
(式中、R1は、Hであり、またはアルキル、シクロアルキル、アルケニル、シクロアルケニル、アルキニル、アリール、アラルキルおよびアリールシクロアルキルから選択される、それぞれ非置換または置換の有機基である。)
を有する化合物。
【請求項2】
R1がHである、請求項1に記載の化合物。
【請求項3】
Z−エン配置を有する以下の式II
【化2】
(式中、R2は脱離基である。)
を有する化合物。
【請求項4】
R2が、式IIのカルボニル基とアミド基を形成するN−ヘテロ原子であるN−ヘテロ環基か、またはRおよびR’が同一または異なり、環を形成することができ、前記N−ヘテロ環基に変換され得るNRR’基であり、好ましくは、式中、R2が以下の式III:
【化3】
(式中、R3は、置換基、好ましくはアリール、置換アリール、分岐アルキルおよび少なくとも4個のC原子を有するアルキルから選択される、特にフェニル、置換フェニル、iPr、BuおよびtBuから選択される置換基を表す。)
によって定義される、請求項3に記載の化合物。
【請求項5】
Z−エン配置を有する以下の式IV
【化4】
(式中、R2は、請求項3で定義した通りであり、R4は、OH−保護基である。)
を有する化合物。
【請求項6】
Z−エン配置を有する以下の式V
【化5】
(式中、R4は、OH−保護基である。)
を有する化合物。
【請求項7】
4−フルオロ−ベンズアルデヒドおよび式Vlの化合物:
【化6】
(式中、R’’は、アルキル、置換アルキル、アリールまたは置換アリール、好ましくはフェニルであり、X−は、ホスホニウム塩の陰イオン、好ましくはハロゲン化物、酢酸、トリフルオロ酢酸またはスルホン酸、具体的にはハロゲン化物であり、R1は、請求項1で定義した通りである。)
をウィッティッヒ反応に供することを含む、5−(4−フルオロフェニル)ペント−4−エン酸またはこの酸誘導体を製造する方法。
【請求項8】
得られた5−(4−フルオロフェニル)ペント−4−エン酸またはこの酸誘導体の(E,Z)異性体が、それぞれE−異性体およびZ−異性体として単離される、請求項7に記載の方法。
【請求項9】
単離された異性体が、以下の式I
【化7】
(式中、R1は、請求項1で定義した通りである。)
を有するZ−異性体化合物である、請求項7または8に記載の方法。
【請求項10】
エステル誘導体が、式Vlの化合物として使用され、ウィッティッヒ反応後、得られたエステル基を鹸化に供して、対応する5−(4−フルオロフェニル)−ペント−4−エン酸を生成する、請求項7から9のいずれか一項に記載の方法。
【請求項11】
4−フルオロ−ベンズアルデヒドの式VIIの化合物とのウィッティッヒ反応によって、請求項3に記載の式IIの化合物
【化8】
(式中、X−は、ホスホニウム塩の陰イオン、好ましくはハロゲン化物、酢酸、トリフルオロ酢酸またはスルホン酸、具体的にはハロゲン化物であり、R’’は、アルキル、置換アルキル、アリールまたは置換アリール、好ましくはフェニルであり、R2は、請求項3または4で定義した通りである。)
を製造する方法。
【請求項12】
エゼチミブの合成のための中間体としての、請求項1から6のいずれか一項に記載の化合物を使用する使用。
【請求項13】
a)請求項6に記載の式Vの化合物を酸化して、以下で示す式IX(式中、R4は上で定義した通りである。)のケトンを得るステップ;
b)ステップ(a)のケトン生成物を還元して、(S)−OH異性体生成物を得るステップ、および
c)還元された(S)−OH生成物をOH−脱保護反応に供して、エゼチミブを得るステップ
【化9】
を含む、エゼチミブを合成する方法。
【請求項14】
a)請求項12または請求項13に記載の方法に従ってエゼチミブを調製するステップ、および
b)こうして調製されたエゼチミブを、少なくとも1つの薬学的に許容される賦形剤と混和するステップ
を含む、エゼチミブを活性成分として含む医薬組成物を調製する方法。
【請求項15】
化合物Vを本質的に含まず、その脱保護された4−ヒドロキシフェニル類似体を本質的に含まないエゼチミブ。
【請求項16】
a)(3R,4S)−4−(4−(ベンジルオキシ)フェニル)−1−(4−フルオロフェニル)−3−((Z/E)−3−(4−フルオロフェニル)−アリル)アゼチジン−2−オンの3−Z−またはE−異性体のどちらか一方または両方を本質的に含まない、またはその脱保護された4−ヒドロキシフェニル類似体を本質的に含まないエゼチミブを提供するステップ、および
b)こうして提供されたエゼチミブを、少なくとも1つの薬学的に許容される賦形剤と混和するステップ
を含む、エゼチミブを活性成分として含む医薬組成物を調製する方法。
【請求項17】
請求項16に記載の方法によって得られる、活性成分としてのエゼチミブおよび少なくとも1つの薬学的に許容される賦形剤を含む医薬組成物。
【図1A】

【図1B】
【図1B】
【公表番号】特表2011−529473(P2011−529473A)
【公表日】平成23年12月8日(2011.12.8)
【国際特許分類】
【出願番号】特願2011−520504(P2011−520504)
【出願日】平成21年7月29日(2009.7.29)
【国際出願番号】PCT/EP2009/059813
【国際公開番号】WO2010/012775
【国際公開日】平成22年2月4日(2010.2.4)
【出願人】(504359293)レツク・フアーマシユーテイカルズ・デー・デー (60)
【Fターム(参考)】
【公表日】平成23年12月8日(2011.12.8)
【国際特許分類】
【出願日】平成21年7月29日(2009.7.29)
【国際出願番号】PCT/EP2009/059813
【国際公開番号】WO2010/012775
【国際公開日】平成22年2月4日(2010.2.4)
【出願人】(504359293)レツク・フアーマシユーテイカルズ・デー・デー (60)
【Fターム(参考)】
[ Back to top ]