
エソメプラゾールのナトリウム塩の製造に使用するための結晶の改変型の作成方法
【課題】 エソメプラゾールのナトリウム塩の結晶化のための溶液を調製すること。
【解決手段】
以下の工程:
i) エソメプラゾールのカリウム塩をトルエンに懸濁し、1モル当量の酸HAを水溶液として添加することによりpHを調整する;
ii) トルエン相にメタノールからなる追加の溶媒を加える;そして、
iii) 塩基Bのナトリウム塩の1モル当量を水溶液として加える、
を含むエソメプラゾールのナトリウム塩の結晶化用溶液を調製する方法。
【解決手段】
以下の工程:
i) エソメプラゾールのカリウム塩をトルエンに懸濁し、1モル当量の酸HAを水溶液として添加することによりpHを調整する;
ii) トルエン相にメタノールからなる追加の溶媒を加える;そして、
iii) 塩基Bのナトリウム塩の1モル当量を水溶液として加える、
を含むエソメプラゾールのナトリウム塩の結晶化用溶液を調製する方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明はエソメプラゾールのナトリウム塩の製造に使用するための結晶の改変型(crystal modifications)の新規な作成方法に関する。さらに本発明はまた、この新規な結晶
の改変型の胃腸疾患の治療のための使用、それらを含有する医薬組成物ならびに結晶の改変型それ自体、にも関する。
【背景技術】
【0002】
オメプラゾール、すなわち化合物5−メトキシ−2−[[(4−メトキシ−3,5−ジメチル−2−ピリジニル)メチル]スルフィニル]−1H−ベンゾイミダゾール、およびその治療上許容
される塩はEP 5129に記載されている。いくつかのオメプラゾールの特定のアルカリ塩がEP 124 495に記載されている。
【0003】
オメプラゾールはスルホキシドでありそしてキラル化合物であるが、この化合物では硫黄原子が立体異性中心である。したがって、オメプラゾールはその2つの単一のエナンチオマー、オメプラゾールのR−およびS−エナンチオマーのラセミ混合物であり、後者はエソメプラゾールという一般名を有している。エソメプラゾールは最近になって新世代のプロトンポンプ阻害剤として市場に送り出されており、その活性医薬成分はエソメプラゾールのマグネシウム塩である。エソメプラゾールはそれ以前の薬物療法に比べてGERDの治療において改善を示している。
【0004】
オメプラゾールのエナンチオマーの絶対立体配置は、非塩形態の(+)−エナンチオマーのN−アルキル化誘導体のX線研究から決定されている。この非塩形態の(+)−エナンチオマーおよび非塩形態の(−)−エナンチオマーは、それぞれRおよびS配置を有することがわかっており、そしてマグネシウム塩の(+)−エナンチオマーおよびマグネシウム塩の(−)−エナンチオマーは、またそれぞれRおよびS配置を有することがわかっている。それらのエナンチオマーのそれぞれの旋光度測定のための条件はWO 94/27988に記載してい
る。
【0005】
オメプラゾールの単一エナンチオマーの特定の塩およびその製造はWO 94/27988で開示
されている。それらの化合物は改良された薬物動態学的および代謝特性を有しており、それにより改善された、例えば個人間のばらつきの程度の小ささのような治療的プロフィルを提供するだろう。
【0006】
WO 96/02535は、オメプラゾールおよびその塩、例えばナトリウム塩の単一のエナンチ
オマーの製造方法を開示している。
【0007】
WO 98/54171は、オメプラゾール三水和物のS−エナンチオマーのマグネシウム塩の製
造方法を開示し、そこではS−オメプラゾールのカリウム塩が中間体として使用される。
【0008】
WO 00/44744は、メタノールを含まないS−オメプラゾールのカリウム塩を開示してい
る。
【0009】
WO 03/089408 (Sun Pharmaceutical Industries Limited)は、ナトリウム塩を含むエソメプラゾールのアルカリまたはアルカリ土類金属塩を開示している。
【発明の概要】
【発明が解決しようとする課題】
【0010】
驚くべきことに、エソメプラゾールのナトリウム塩の調製中に、いくつかの新規な結晶の改変型が形成されていることが見出された。これらの新規な中間体のいくつかは安定であるために、分離して特徴付けることが可能である。他のものは特徴付けるためにはあまりにも短命すぎ、さらに他のものは湿潤な状態では結晶性であるが、乾燥により種々の無晶形態へと転換し、その結果特徴付けることが困難である。乾燥工程の間、これらの改変型は、結晶含量のより少ないいくつかの追加の形態を経てゆくだろう。しかしながら全ての結晶の改変型は本発明の方法により得られる。
【課題を解決するための手段】
【0011】
本発明の方法は、エソメプラゾールのナトリウム塩を、良好な濾過特性(filtering properties)を有する結晶の改変型を用いて、その対応するカリウム塩から高収率および良好な品質で直接に製造することを可能にすることから有利なものである。さらなる利点は、高い再現性、安全性および、全工程、好ましくは酸化工程ならびにそれに続く工程および操作を通しての1つの主溶媒系の使用を含む良好なプロセス能力である。より好ましくは、本発明ではエソメプラゾールのカリウム塩を調製するために使用されたものと同じ溶媒系が使用される。
【発明の効果】
【0012】
本発明の方法は新規な結晶の改変型およびそれらの特性を最適に用いて、それによりエソメプラゾールのナトリウム塩をより効果的および効率的な方法で製造することを可能にする。
【図面の簡単な説明】
【0013】
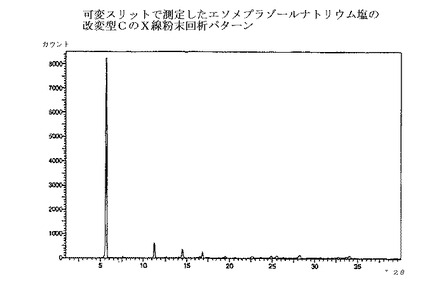
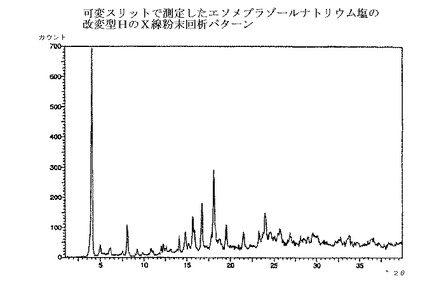
【図1】エソメプラゾールナトリウム塩の改変型CのX線粉末回析パターンである。
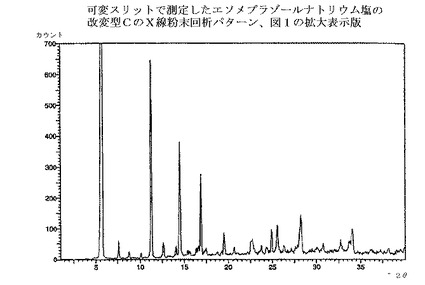
【図2】図1の拡大表示である。
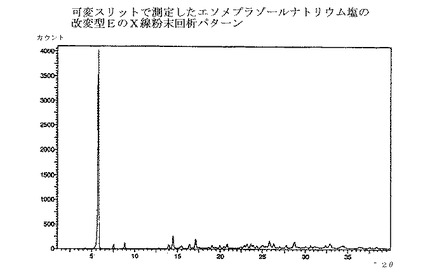
【図3】エソメプラゾールナトリウム塩の改変型EのX線粉末回析パターンである。
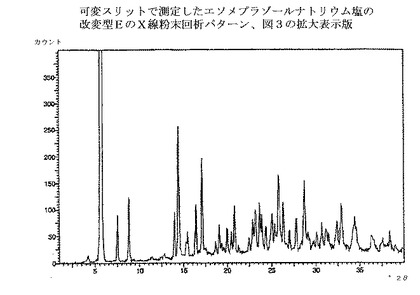
【図4】図3の拡大表示である。
【図5】エソメプラゾールナトリウム塩の改変型HのX線粉末回析パターンである。
【発明を実施するための形態】
【0014】
本発明の方法は本質的に以下の工程を含んでいる:
i)エソメプラゾールの中性形態を適切な溶媒S1中に溶解し;
ii)追加の適切な溶媒S2を加え;
iii)約1モル当量の適切な塩基Bのナトリウム塩を加え;
iv)エソメプラゾールのナトリウム塩を結晶化させそして形成した塩を分離する。
【0015】
本発明の1つの実施態様では、エソメプラゾールの中性形態は、対応するエソメプラゾールのカリウム塩から製造され、そしてそれに続く上記で定義された工程が、ほぼその直後に行なわれる。そうした場合、エソメプラゾールのカリウム塩は従来技術で記載されたどのような方法を用いて製造してもよく、そしてその後に溶媒S1に懸濁される。pHはその後エソメプラゾールをその中性形態で作られるように調整しなければならない。このpH調整は、約1モル当量の適切な酸HAを、好ましくは水溶液として加えることで行うことができる。そうした酸HAの例には、それに限定されないが、水溶性のカリウム塩を形成する全ての鉱酸、例えば塩酸、および酢酸が含まれる。その後水相は捨てられ、有機相は場合によっては水またはブラインで洗浄される。エソメプラゾールの中性形態は、こうしてほぼその直後に、上記で定義された工程ii)〜iv)において使用できるように準備される。
【0016】
本発明の他の実施態様において、エソメプラゾールの中性形態は、対応するエソメプラゾールのマグネシウム塩から製造され、そして上記で定義されたそれに続く工程がほぼそ
の直後に行なわれる。
【0017】
本発明の1つの実施態様において、溶媒S1はトルエンである。
本発明の1つの実施態様において、溶媒S2はメタノールである。
本発明の1つの実施態様において、溶媒S2はエタノールである。
本発明の1つの実施態様において、溶媒S2はイソプロピルアルコールである。
本発明の1つの実施態様において、塩基Bは水酸化物である。
本発明の1つの実施態様において、塩基Bは水溶液として与えられる。
本発明の1つの実施態様において、工程iv)の結晶化は結晶種付けによって開始される。
本発明の1つの実施態様において、上記の工程iii)は工程ii)の前に行われる。
【0018】
工程iv)で分離されたエソメプラゾールのナトリウム塩は、どのような溶媒S2が用いられたかに依存するだろう。その後、分離したエソメプラゾールのナトリウム塩は乾燥され、そして乾燥の過程の間にほとんどの溶媒S2は若干の追加の水と一緒に除去される。実施例にしたがって分離されたエソメプラゾールのナトリウム塩は湿潤状態での結晶であり、乾燥によって種々の無晶性形態へと変換される。乾燥の過程の間に、分離された結晶の改変型は、結晶含量がより少ないいくつかの追加の形態を経るだろう。
【0019】
本発明の方法で得られるエソメプラゾールのナトリウム塩は、全て慣用の乾燥方法で乾燥でき、場合によっては種々の無晶性形態へと変換される。乾燥方法は、エソメプラゾールのナトリウム塩の改変型C、EおよびHのX線回析パターンにおけるピークの位置と強度に少しばかり影響を与えるだろう。図1〜3の回析パターンを完全に再現するためには、実施例の方法に慎重に従うことが重要である。それから少しでも変更すればX線回析パターンにおけるピークの位置と強度は影響される。
【0020】
したがって本発明の結晶の改変型は、その良好な濾過特性により、中間体として最も有用である。しかしながら、それらは全て完全に乾燥させることができ、そしてそれを必要とする患者に用いるための医薬組成物へと製剤化することができる。
【0021】
メタノールが溶媒S2として使用された場合は、次に分離される結晶の改変型はエソメプラゾールのナトリウム塩の改変型Cである。
【0022】
エタノールが溶媒S2として使用された場合は、次に分離される結晶の改変型はエソメプラゾールのナトリウム塩の改変型Eである。
【0023】
イソプロピルアルコールが溶媒S2として使用された場合は、次に分離される結晶の改変型はエソメプラゾールのナトリウム塩の改変型Hである。
【0024】
疑義を避けるために、本願明細書において方法の工程または類似の行為が「本願で先に定義された」または「上記で定義された」と表記された場合は、その工程は最初に表されたそして最も広い定義であるだけでなくその工程の他の各および全ての定義を包含するものと理解されるべきである。
【0025】
本願明細書での「ほぼその直後」という表現は、続く工程または行為が、活性化合物の分解を避けられるような時間に行われることを意味すると理解されるべきである。したがってこの続く工程は、活性化合物の分解を避けるために相当の注意が為されれば、かなり後の時間になっても行うことができる。
【0026】
適切な溶媒S1は、それには限定されないが、トルエンを含む。
適切な溶媒S2は、それには限定されないが、メタノール、エタノールおよびイソプロピルアルコールを含む。
適切な塩基Bは、それに限定されないが、水酸化物、メトキシドおよびエトキシドを含み、好ましくは水溶液として加えられる。
【0027】
本発明の他の目的は、エソメプラゾールのナトリウム塩の新規で安定な結晶の改変型を提供することである。エソメプラゾールのナトリウム塩は1つ以上の結晶の改変型で存在できる。この結晶の改変型または形態は以下にエソメプラゾールのナトリウム塩の改変型C、E及びHと称される。表記C、E及びHは、それらの結晶の改変型が形成された時間の順に関連しており、それらの相対的な熱力学的安定性には関連していない。
【0028】
エソメプラゾールのナトリウム塩の改変型Cを提供することが本発明の1つの態様である。
【0029】
エソメプラゾールのナトリウム塩の改変型Cは、図1のような、X線粉末回析パターンを提供することによって特徴付けられ、実質的に下記のd値および強度を有する主要ピークを示す;
【表1】

【0030】
ブラッグ式(Bragg formula)から計算されたd値および強度で同定されたピークが、
エソメプラゾールのナトリウム塩の改変型Cの回析パターンから抽出されてきた。最も特徴的で、有意で、明瞭なおよび/または再現性のある、主要ピークのみを表に示したが、追加のピークも、慣用法を用いて、回析パターンから抽出できる。これらの主要ピークの、再現性のあるそして誤差範囲内での存在が、該結晶改変型の存在を確証するために十分な最も重要な証拠である。相対的な強度は信頼性が低く、数値の代わりに下記の定義が用いられた;
vs (非常に強い): >15% 相対的強度
s (強い): 7−15% 相対的強度
m (中程度): 3−7% 相対的強度
w (弱い): 1−3% 相対的強度
vw (非常に弱い): <1% 相対的強度
* この相対的強度は可変スリットを用いて測定した回析パターンから導かれた。
【0031】
エソメプラゾールのナトリウム塩の改変型Eを提供することが本発明のさらなる態様である。
【0032】
エソメプラゾールのナトリウム塩の改変型Eは、図3のような、X線粉末回析パターンを提供することによって特性付けられ、実質的に下記のd値および強度を有する主要ピークを示す;
【表2】
【0033】
ブラッグ式から計算されたd値および強度で同定されたピークが、エソメプラゾールのナトリウム塩の改変型Eの回析パターンから抽出されてきた。最も特徴的で、有意で、明瞭なおよび/または再現性のある、主要ピークのみを表に示したが、追加のピークも、慣用法を用いて、回析パターンから抽出できる。これらの主要ピークの、再現性のあるそして誤差範囲内での存在が、該結晶改変型の存在を確証するために十分な最も重要な証拠である。相対的な強度は信頼性が低く、数値の代わりに下記の定義が用いられた;
vs (非常に強い): >15% 相対的強度
s (強い): 8−15% 相対的強度
m (中程度): 5−8% 相対的強度
w (弱い): 1−5% 相対的強度
vw (非常に弱い): <1% 相対的強度
* この相対的強度は可変スリットを用いて測定した回析パターンから導かれた。
【0034】
エソメプラゾールのナトリウム塩の改変型Hを提供することが本発明の1つの態様である。
【0035】
エソメプラゾールのナトリウム塩の改変型Hは、図5のように、X線粉末回析パターンを提供することによって特性付けられ、実質的に下記のd値および強度を有する主要ピー
クを示す;
【表3】
【0036】
ブラッグ式から計算されたd値および強度で同定されたピークが、エソメプラゾールの
ナトリウム塩の改変型Hの回析パターンから抽出されてきた。最も特徴的で、有意で、明瞭なおよび/または再現性のある、主要ピークのみを表に示したが、追加のピークも、慣用法を用いて、回析パターンから抽出できる。これらの主要ピークの、再現性のあるそして誤差範囲内での存在が、該結晶改変型の存在を確証するために十分な最も重要な証拠である。相対的な強度は信頼性が低く、数値の代わりに下記の定義が用いられた;
vs (非常に強い): >50% 相対的強度
s (強い): 27−50% 相対的強度
m (中程度): 11−27% 相対的強度
w (弱い): 3−11% 相対的強度
vw (非常に弱い): <3% 相対的強度
* この相対的強度は可変スリットを用いて測定した回析パターンから導かれた。
【0037】
複数の溶媒を含む適切な溶媒系からの本発明の結晶の改変型の結晶化は、溶媒蒸留、温度低下および/または逆溶媒(すなわち、結晶の改変型が難溶性である溶媒)による過飽和を達成することにより行うことができる。
【0038】
結晶化が無水物であるか溶媒和物であるかは、特定の条件下での各結晶の改変の反応速度論および平衡条件に関連する。したがって、当業者が理解するように、得られる結晶の改変型は、結晶化工程の反応速度論および熱力学の両方に依存している。特定の熱力学的条件下(溶媒系、温度、圧および本発明の化合物の濃度)では、1つの結晶の改変型が他のもの(または実際は他の全て)より安定だろう。しかしながら、相対的に低い熱力学的安定性を有する結晶の改変型は反応速度論的には好ましいだろう。したがって、さらに、反応速度論的要因、例えば時間、不純物のプロフィル、攪拌、シードの存在または不在等もまた、どの結晶の改変型が結晶化するかに影響を及ぼすだろう。
【0039】
特定の結晶の改変型が、実質的に他の結晶の改変型の不在下で製造できるようにするために、結晶化は、所望の結晶の改変型のシード結晶で種付けをすることにより行うことが
好ましい。このことは特に実施例に記載された特定の結晶の改変型のそれぞれについて適用される。
【0040】
本発明により得られるエソメプラゾールのナトリウム塩の改変型C、E及びHは、実質的にエソメプラゾールのナトリウム塩の他の結晶型および非結晶形態は含まない。「実質的にエソメプラゾールのナトリウム塩の他の結晶型および非結晶形態は含まない」という表現は、エソメプラゾールのナトリウム塩の所望の結晶型が、エソメプラゾールのナトリウム塩の他のどのような形態も15%未満、好ましくは10%未満、より好ましくは5%未満しか含んでいないことを意味すると理解されるべきである。
【0041】
本発明の結晶の改変型は、胃酸分泌阻害剤として有効であり、したがって抗潰瘍剤として有用である。より一般的な意味では、これらは、哺乳類、特にヒトにおける胃酸関連症状の予防および治療のために用いることができ、その症状には、例えば逆流性食道炎、胃炎、十二指腸炎、胃潰瘍および十二指腸潰瘍が含まれる。さらに、これらは胃酸抑制効果が望ましい他の胃腸疾患の治療、例えばNSAID治療を受けている患者において、非潰瘍性
消化不良の患者において、兆候性胃食道逆流性疾患を有する患者において、およびにガストリノーマを有する患者においても有効であると考えられる。これらはまた、集中治療状態にいる患者において、急性上部胃腸出血を有する患者において、手術の前後に胃酸の吸引を予防するために、ストレス性潰瘍化および喘息の予防および治療のために、そして睡眠の改善のためにも使用できるであろう。さらに、本発明の結晶の改変型は、乾癬の治療だけでなくヘリコバクター感染症および関連疾患の治療においても有用であろう。本発明の結晶の改変型はまた、ヒトを含む哺乳類の炎症症状の治療のためにも有用であろう。
【0042】
全ての適切な投与経路を、結晶の改変型の有効量を患者に提供するために用いることができる。例えば、経口または非経口製剤(i.v.等を含む)を用いることができる。投与剤形には、カプセル、錠剤、分散剤、懸濁剤、溶液等が含まれる。
【0043】
さらに、製薬的に受容できる担体、希釈剤または賦形剤および場合によっては他の活性医薬成分と組み合わせた、本発明の結晶の改変型を活性成分として含有する医薬組成物を提供する。他の治療成分を含有する組成物は上記の症状の治療において関心がある。本発明はまた、該症状で使用するための医薬の製造における結晶のこの改変型の使用、ならびに、胃液関連症状の治療方法であって、該症状を有する患者に医薬として有効量の結晶の改変型を投与することを含む方法も提供する。
【0044】
本発明の組成物は、経口、i.v.または非経口投与に適した組成物を含む。最も好ましい経路はi.v.経路である。この組成物は使いやすいように単位投与剤形で存在でき、そして製剤製薬学の技術分野で知られたどのような方法を用いても製造できる。
【0045】
本発明の実施において、最も適切な投与経路および治療投与量の大きさは、治療されるべき疾患の性質および重症度に依存するだろう。投与量、および投与頻度は、個々の患者の年齢、体重および応答性によって変えることもできる。ゾーリンガー・エリソン症候群を有する患者に関しては、平均の患者より高用量を必要とるような、特別の要請が求められるだろう。小児および肝疾患を有する患者は、一般に、ある程度平均より少ない用量から利益を受けるだろう。そうしたより高いおよびより低い投与量も本発明の範囲内である。そうした一日用量は5mgから300mgの間で変えることができる。
【0046】
一般に、本発明の化合物の適切な経口投与剤形は、単回用量または等しく分割した用量で投与される、一日総用量で5mgから300mgの用量範囲を包含する。好ましい用量範囲は10mgから80mgの範囲である。
【0047】
本発明の化合物は、WO 96/01623およびEP 0 247 983に記載された経口製剤のような従
来技術にしたがって、密接な(intimate)混合物中の活性成分として医薬用担体と組み合わせることができ、その開示は参照により全体で本願に含まれる。
【0048】
本発明の化合物および他の活性成分を含む組み合わせ製剤もまた使用することができる。そうした活性成分の例には、それに限定されないが、抗細菌化合物、非ステロイド抗炎症剤、制酸剤、アルギナートおよび運動促進剤(prokinetic agent)が含まれる。
【0049】
本発明の化合物は、適切な医薬製剤へと製剤化される前にさらに加工できる。例えば、結晶の改変型はより小さな粒子にすりつぶされまたは粉砕されるだろう。
【0050】
疑義を避けるために「治療」には症状の治療的処置だけでなく予防も含む。
【0051】
X線粉末回析により特徴付けられる、医薬賦形剤のような、試料中の付加的な物質の存在は、当然、上記で特徴付けられた結晶の改変型のどれかの小さなピークのいくつかをマスクする可能性がある。もちろんこの事実のみをもって、結晶の改変型が試料中に存在しないということを証明することはできない。そうした環境下では、適切な注意が用いられなければならず、そしてX線粉末回析パターンの実質的に全ての主要ピークの存在が結晶の改変型の特徴付けるためには十分である。したがって、付加的な物質の存在なしで、本発明の結晶の改変型を解析することが好ましい。
【0052】
本発明は、以下の実施例で説明されるが、それには限定されない。
【0053】
〔実施例〕
一般的な方法
試料のX線粉末回析分析(XRPD)は、標準的な方法、例えば以下に記載された方法にしたがって行った: Giacovazzo, C.等(1995), Fundamentals of Crystallography, Oxford
University Press; Jenkins, R.およびSnyder, R. L. (1996), Introduction to X-Ray Powder Diffractometry, John Wiley & Sons, New York; Bunn, C. W. (1948), Chemical
Crystallography, Clarendon Press, London; またはKlug, H. P.およびAlexander, L. E. (1974), X-ray Diffraction Procedures, John Wiley and Sons, New York. X線回析分析はPhilips X'Pert MPDを用いて16分間で1から40°2θまで行った。 付加をする
ことが試料の調製に費やされる時間に影響しそしてX線回析パターンのピークの位置および強度に影響するために、試料は内部標準なしで分析した。測定したピーク値は、前の実験(−0.05°2θ)に基づいて調整した。その後でd値への計算を行った。
【0054】
XRPD距離値(distance value)は得られた小数点の最後の桁で±2の範囲で変化しうる。
【0055】
実施例1.1
エソメプラゾールのナトリウム塩の改変型Cの製造
エソメプラゾールのカリウム塩(11.89g)を水(50ml)に溶解し、そしてトルエン(80ml)を加えた。次いで、酢酸(5.89ml, 25% v/v)を加えてpHを約7に調整した。2つの相を10分間混合しそして分離させた。水相を取り除き残余の有機相を塩化ナトリウム水溶液(50ml, 10%)で洗浄した。相分離後にメタノール(4.24ml)をトルエン相に加え、次に1当量(eq)のNaOH(1.52ml, 水溶液、45%)を加えた。この溶液をエソメプラゾールのナトリウム塩25mgで結晶種付けした。攪拌しながら終夜結晶化を行わせ、この結晶を減圧濾過で濾別し、そして直ぐにトルエンで2回洗浄した(2×10ml)。得られたフィルターケーキを、分析前に短時間(例えば2〜5分間)空気中で乾燥させた。
【0056】
実施例1.2
エソメプラゾールのナトリウム塩の改変型Eの製造
エソメプラゾールのカリウム塩(11.89g)を水(50ml)に溶解し、そしてトルエン(80ml)を加えた。次いで、酢酸(5.89ml, 25% v/v)を加えてpHを約7に調整した。2つの相を10分間混合しそして分離させた。水相を取り除き残余の有機相を塩化ナトリウム水溶液(50ml, 10%)で洗浄した。相分離後にエタノール(11.1ml)をトルエン相に加え、次に1当量(eq)のNaOH(1.52ml, 水溶液、45%)を加えた。この溶液をエソメプラゾールのナトリウム塩55mgで結晶種付けした。攪拌しながら終夜結晶化を行わせ、この結晶を減圧濾過で濾別し、そして直ぐにトルエンで2回洗浄した(2×10ml)。得られたフィルターケーキを、分析前に短時間(例えば2〜5分間)空気中で乾燥させた。
【0057】
実施例1.3
エソメプラゾールのナトリウム塩の改変型Hの製造
エソメプラゾールのカリウム塩(11.89g)を水(50ml)に溶解し、そしてトルエン(80ml)を加えた。次いで、酢酸(5.89ml, 25% v/v)を加えてpHを約7に調整した。2つの相を10分間混合しそして分離させた。水相を取り除き残余の有機相を塩化ナトリウム水溶液(50ml, 10%)で洗浄した。相分離後に2−プロパノール(3.6ml)をトルエン相に加え、次に1当量(eq)のNaOH(1.52ml, 水溶液、45%)を加えた。この溶液をエソメプラゾールのナトリウム塩53mgで結晶種付けした。攪拌しながら終夜結晶化を行わせ、この結晶を減圧濾過で濾別し、そして直ぐにトルエンで2回洗浄した(2×10ml)。得られたフィルターケーキを、分析前に短時間(例えば2〜5分間)空気中で乾燥させた。
【技術分野】
【0001】
本発明はエソメプラゾールのナトリウム塩の製造に使用するための結晶の改変型(crystal modifications)の新規な作成方法に関する。さらに本発明はまた、この新規な結晶
の改変型の胃腸疾患の治療のための使用、それらを含有する医薬組成物ならびに結晶の改変型それ自体、にも関する。
【背景技術】
【0002】
オメプラゾール、すなわち化合物5−メトキシ−2−[[(4−メトキシ−3,5−ジメチル−2−ピリジニル)メチル]スルフィニル]−1H−ベンゾイミダゾール、およびその治療上許容
される塩はEP 5129に記載されている。いくつかのオメプラゾールの特定のアルカリ塩がEP 124 495に記載されている。
【0003】
オメプラゾールはスルホキシドでありそしてキラル化合物であるが、この化合物では硫黄原子が立体異性中心である。したがって、オメプラゾールはその2つの単一のエナンチオマー、オメプラゾールのR−およびS−エナンチオマーのラセミ混合物であり、後者はエソメプラゾールという一般名を有している。エソメプラゾールは最近になって新世代のプロトンポンプ阻害剤として市場に送り出されており、その活性医薬成分はエソメプラゾールのマグネシウム塩である。エソメプラゾールはそれ以前の薬物療法に比べてGERDの治療において改善を示している。
【0004】
オメプラゾールのエナンチオマーの絶対立体配置は、非塩形態の(+)−エナンチオマーのN−アルキル化誘導体のX線研究から決定されている。この非塩形態の(+)−エナンチオマーおよび非塩形態の(−)−エナンチオマーは、それぞれRおよびS配置を有することがわかっており、そしてマグネシウム塩の(+)−エナンチオマーおよびマグネシウム塩の(−)−エナンチオマーは、またそれぞれRおよびS配置を有することがわかっている。それらのエナンチオマーのそれぞれの旋光度測定のための条件はWO 94/27988に記載してい
る。
【0005】
オメプラゾールの単一エナンチオマーの特定の塩およびその製造はWO 94/27988で開示
されている。それらの化合物は改良された薬物動態学的および代謝特性を有しており、それにより改善された、例えば個人間のばらつきの程度の小ささのような治療的プロフィルを提供するだろう。
【0006】
WO 96/02535は、オメプラゾールおよびその塩、例えばナトリウム塩の単一のエナンチ
オマーの製造方法を開示している。
【0007】
WO 98/54171は、オメプラゾール三水和物のS−エナンチオマーのマグネシウム塩の製
造方法を開示し、そこではS−オメプラゾールのカリウム塩が中間体として使用される。
【0008】
WO 00/44744は、メタノールを含まないS−オメプラゾールのカリウム塩を開示してい
る。
【0009】
WO 03/089408 (Sun Pharmaceutical Industries Limited)は、ナトリウム塩を含むエソメプラゾールのアルカリまたはアルカリ土類金属塩を開示している。
【発明の概要】
【発明が解決しようとする課題】
【0010】
驚くべきことに、エソメプラゾールのナトリウム塩の調製中に、いくつかの新規な結晶の改変型が形成されていることが見出された。これらの新規な中間体のいくつかは安定であるために、分離して特徴付けることが可能である。他のものは特徴付けるためにはあまりにも短命すぎ、さらに他のものは湿潤な状態では結晶性であるが、乾燥により種々の無晶形態へと転換し、その結果特徴付けることが困難である。乾燥工程の間、これらの改変型は、結晶含量のより少ないいくつかの追加の形態を経てゆくだろう。しかしながら全ての結晶の改変型は本発明の方法により得られる。
【課題を解決するための手段】
【0011】
本発明の方法は、エソメプラゾールのナトリウム塩を、良好な濾過特性(filtering properties)を有する結晶の改変型を用いて、その対応するカリウム塩から高収率および良好な品質で直接に製造することを可能にすることから有利なものである。さらなる利点は、高い再現性、安全性および、全工程、好ましくは酸化工程ならびにそれに続く工程および操作を通しての1つの主溶媒系の使用を含む良好なプロセス能力である。より好ましくは、本発明ではエソメプラゾールのカリウム塩を調製するために使用されたものと同じ溶媒系が使用される。
【発明の効果】
【0012】
本発明の方法は新規な結晶の改変型およびそれらの特性を最適に用いて、それによりエソメプラゾールのナトリウム塩をより効果的および効率的な方法で製造することを可能にする。
【図面の簡単な説明】
【0013】
【図1】エソメプラゾールナトリウム塩の改変型CのX線粉末回析パターンである。
【図2】図1の拡大表示である。
【図3】エソメプラゾールナトリウム塩の改変型EのX線粉末回析パターンである。
【図4】図3の拡大表示である。
【図5】エソメプラゾールナトリウム塩の改変型HのX線粉末回析パターンである。
【発明を実施するための形態】
【0014】
本発明の方法は本質的に以下の工程を含んでいる:
i)エソメプラゾールの中性形態を適切な溶媒S1中に溶解し;
ii)追加の適切な溶媒S2を加え;
iii)約1モル当量の適切な塩基Bのナトリウム塩を加え;
iv)エソメプラゾールのナトリウム塩を結晶化させそして形成した塩を分離する。
【0015】
本発明の1つの実施態様では、エソメプラゾールの中性形態は、対応するエソメプラゾールのカリウム塩から製造され、そしてそれに続く上記で定義された工程が、ほぼその直後に行なわれる。そうした場合、エソメプラゾールのカリウム塩は従来技術で記載されたどのような方法を用いて製造してもよく、そしてその後に溶媒S1に懸濁される。pHはその後エソメプラゾールをその中性形態で作られるように調整しなければならない。このpH調整は、約1モル当量の適切な酸HAを、好ましくは水溶液として加えることで行うことができる。そうした酸HAの例には、それに限定されないが、水溶性のカリウム塩を形成する全ての鉱酸、例えば塩酸、および酢酸が含まれる。その後水相は捨てられ、有機相は場合によっては水またはブラインで洗浄される。エソメプラゾールの中性形態は、こうしてほぼその直後に、上記で定義された工程ii)〜iv)において使用できるように準備される。
【0016】
本発明の他の実施態様において、エソメプラゾールの中性形態は、対応するエソメプラゾールのマグネシウム塩から製造され、そして上記で定義されたそれに続く工程がほぼそ
の直後に行なわれる。
【0017】
本発明の1つの実施態様において、溶媒S1はトルエンである。
本発明の1つの実施態様において、溶媒S2はメタノールである。
本発明の1つの実施態様において、溶媒S2はエタノールである。
本発明の1つの実施態様において、溶媒S2はイソプロピルアルコールである。
本発明の1つの実施態様において、塩基Bは水酸化物である。
本発明の1つの実施態様において、塩基Bは水溶液として与えられる。
本発明の1つの実施態様において、工程iv)の結晶化は結晶種付けによって開始される。
本発明の1つの実施態様において、上記の工程iii)は工程ii)の前に行われる。
【0018】
工程iv)で分離されたエソメプラゾールのナトリウム塩は、どのような溶媒S2が用いられたかに依存するだろう。その後、分離したエソメプラゾールのナトリウム塩は乾燥され、そして乾燥の過程の間にほとんどの溶媒S2は若干の追加の水と一緒に除去される。実施例にしたがって分離されたエソメプラゾールのナトリウム塩は湿潤状態での結晶であり、乾燥によって種々の無晶性形態へと変換される。乾燥の過程の間に、分離された結晶の改変型は、結晶含量がより少ないいくつかの追加の形態を経るだろう。
【0019】
本発明の方法で得られるエソメプラゾールのナトリウム塩は、全て慣用の乾燥方法で乾燥でき、場合によっては種々の無晶性形態へと変換される。乾燥方法は、エソメプラゾールのナトリウム塩の改変型C、EおよびHのX線回析パターンにおけるピークの位置と強度に少しばかり影響を与えるだろう。図1〜3の回析パターンを完全に再現するためには、実施例の方法に慎重に従うことが重要である。それから少しでも変更すればX線回析パターンにおけるピークの位置と強度は影響される。
【0020】
したがって本発明の結晶の改変型は、その良好な濾過特性により、中間体として最も有用である。しかしながら、それらは全て完全に乾燥させることができ、そしてそれを必要とする患者に用いるための医薬組成物へと製剤化することができる。
【0021】
メタノールが溶媒S2として使用された場合は、次に分離される結晶の改変型はエソメプラゾールのナトリウム塩の改変型Cである。
【0022】
エタノールが溶媒S2として使用された場合は、次に分離される結晶の改変型はエソメプラゾールのナトリウム塩の改変型Eである。
【0023】
イソプロピルアルコールが溶媒S2として使用された場合は、次に分離される結晶の改変型はエソメプラゾールのナトリウム塩の改変型Hである。
【0024】
疑義を避けるために、本願明細書において方法の工程または類似の行為が「本願で先に定義された」または「上記で定義された」と表記された場合は、その工程は最初に表されたそして最も広い定義であるだけでなくその工程の他の各および全ての定義を包含するものと理解されるべきである。
【0025】
本願明細書での「ほぼその直後」という表現は、続く工程または行為が、活性化合物の分解を避けられるような時間に行われることを意味すると理解されるべきである。したがってこの続く工程は、活性化合物の分解を避けるために相当の注意が為されれば、かなり後の時間になっても行うことができる。
【0026】
適切な溶媒S1は、それには限定されないが、トルエンを含む。
適切な溶媒S2は、それには限定されないが、メタノール、エタノールおよびイソプロピルアルコールを含む。
適切な塩基Bは、それに限定されないが、水酸化物、メトキシドおよびエトキシドを含み、好ましくは水溶液として加えられる。
【0027】
本発明の他の目的は、エソメプラゾールのナトリウム塩の新規で安定な結晶の改変型を提供することである。エソメプラゾールのナトリウム塩は1つ以上の結晶の改変型で存在できる。この結晶の改変型または形態は以下にエソメプラゾールのナトリウム塩の改変型C、E及びHと称される。表記C、E及びHは、それらの結晶の改変型が形成された時間の順に関連しており、それらの相対的な熱力学的安定性には関連していない。
【0028】
エソメプラゾールのナトリウム塩の改変型Cを提供することが本発明の1つの態様である。
【0029】
エソメプラゾールのナトリウム塩の改変型Cは、図1のような、X線粉末回析パターンを提供することによって特徴付けられ、実質的に下記のd値および強度を有する主要ピークを示す;
【表1】
【0030】
ブラッグ式(Bragg formula)から計算されたd値および強度で同定されたピークが、
エソメプラゾールのナトリウム塩の改変型Cの回析パターンから抽出されてきた。最も特徴的で、有意で、明瞭なおよび/または再現性のある、主要ピークのみを表に示したが、追加のピークも、慣用法を用いて、回析パターンから抽出できる。これらの主要ピークの、再現性のあるそして誤差範囲内での存在が、該結晶改変型の存在を確証するために十分な最も重要な証拠である。相対的な強度は信頼性が低く、数値の代わりに下記の定義が用いられた;
vs (非常に強い): >15% 相対的強度
s (強い): 7−15% 相対的強度
m (中程度): 3−7% 相対的強度
w (弱い): 1−3% 相対的強度
vw (非常に弱い): <1% 相対的強度
* この相対的強度は可変スリットを用いて測定した回析パターンから導かれた。
【0031】
エソメプラゾールのナトリウム塩の改変型Eを提供することが本発明のさらなる態様である。
【0032】
エソメプラゾールのナトリウム塩の改変型Eは、図3のような、X線粉末回析パターンを提供することによって特性付けられ、実質的に下記のd値および強度を有する主要ピークを示す;
【表2】
【0033】
ブラッグ式から計算されたd値および強度で同定されたピークが、エソメプラゾールのナトリウム塩の改変型Eの回析パターンから抽出されてきた。最も特徴的で、有意で、明瞭なおよび/または再現性のある、主要ピークのみを表に示したが、追加のピークも、慣用法を用いて、回析パターンから抽出できる。これらの主要ピークの、再現性のあるそして誤差範囲内での存在が、該結晶改変型の存在を確証するために十分な最も重要な証拠である。相対的な強度は信頼性が低く、数値の代わりに下記の定義が用いられた;
vs (非常に強い): >15% 相対的強度
s (強い): 8−15% 相対的強度
m (中程度): 5−8% 相対的強度
w (弱い): 1−5% 相対的強度
vw (非常に弱い): <1% 相対的強度
* この相対的強度は可変スリットを用いて測定した回析パターンから導かれた。
【0034】
エソメプラゾールのナトリウム塩の改変型Hを提供することが本発明の1つの態様である。
【0035】
エソメプラゾールのナトリウム塩の改変型Hは、図5のように、X線粉末回析パターンを提供することによって特性付けられ、実質的に下記のd値および強度を有する主要ピー
クを示す;
【表3】
【0036】
ブラッグ式から計算されたd値および強度で同定されたピークが、エソメプラゾールの
ナトリウム塩の改変型Hの回析パターンから抽出されてきた。最も特徴的で、有意で、明瞭なおよび/または再現性のある、主要ピークのみを表に示したが、追加のピークも、慣用法を用いて、回析パターンから抽出できる。これらの主要ピークの、再現性のあるそして誤差範囲内での存在が、該結晶改変型の存在を確証するために十分な最も重要な証拠である。相対的な強度は信頼性が低く、数値の代わりに下記の定義が用いられた;
vs (非常に強い): >50% 相対的強度
s (強い): 27−50% 相対的強度
m (中程度): 11−27% 相対的強度
w (弱い): 3−11% 相対的強度
vw (非常に弱い): <3% 相対的強度
* この相対的強度は可変スリットを用いて測定した回析パターンから導かれた。
【0037】
複数の溶媒を含む適切な溶媒系からの本発明の結晶の改変型の結晶化は、溶媒蒸留、温度低下および/または逆溶媒(すなわち、結晶の改変型が難溶性である溶媒)による過飽和を達成することにより行うことができる。
【0038】
結晶化が無水物であるか溶媒和物であるかは、特定の条件下での各結晶の改変の反応速度論および平衡条件に関連する。したがって、当業者が理解するように、得られる結晶の改変型は、結晶化工程の反応速度論および熱力学の両方に依存している。特定の熱力学的条件下(溶媒系、温度、圧および本発明の化合物の濃度)では、1つの結晶の改変型が他のもの(または実際は他の全て)より安定だろう。しかしながら、相対的に低い熱力学的安定性を有する結晶の改変型は反応速度論的には好ましいだろう。したがって、さらに、反応速度論的要因、例えば時間、不純物のプロフィル、攪拌、シードの存在または不在等もまた、どの結晶の改変型が結晶化するかに影響を及ぼすだろう。
【0039】
特定の結晶の改変型が、実質的に他の結晶の改変型の不在下で製造できるようにするために、結晶化は、所望の結晶の改変型のシード結晶で種付けをすることにより行うことが
好ましい。このことは特に実施例に記載された特定の結晶の改変型のそれぞれについて適用される。
【0040】
本発明により得られるエソメプラゾールのナトリウム塩の改変型C、E及びHは、実質的にエソメプラゾールのナトリウム塩の他の結晶型および非結晶形態は含まない。「実質的にエソメプラゾールのナトリウム塩の他の結晶型および非結晶形態は含まない」という表現は、エソメプラゾールのナトリウム塩の所望の結晶型が、エソメプラゾールのナトリウム塩の他のどのような形態も15%未満、好ましくは10%未満、より好ましくは5%未満しか含んでいないことを意味すると理解されるべきである。
【0041】
本発明の結晶の改変型は、胃酸分泌阻害剤として有効であり、したがって抗潰瘍剤として有用である。より一般的な意味では、これらは、哺乳類、特にヒトにおける胃酸関連症状の予防および治療のために用いることができ、その症状には、例えば逆流性食道炎、胃炎、十二指腸炎、胃潰瘍および十二指腸潰瘍が含まれる。さらに、これらは胃酸抑制効果が望ましい他の胃腸疾患の治療、例えばNSAID治療を受けている患者において、非潰瘍性
消化不良の患者において、兆候性胃食道逆流性疾患を有する患者において、およびにガストリノーマを有する患者においても有効であると考えられる。これらはまた、集中治療状態にいる患者において、急性上部胃腸出血を有する患者において、手術の前後に胃酸の吸引を予防するために、ストレス性潰瘍化および喘息の予防および治療のために、そして睡眠の改善のためにも使用できるであろう。さらに、本発明の結晶の改変型は、乾癬の治療だけでなくヘリコバクター感染症および関連疾患の治療においても有用であろう。本発明の結晶の改変型はまた、ヒトを含む哺乳類の炎症症状の治療のためにも有用であろう。
【0042】
全ての適切な投与経路を、結晶の改変型の有効量を患者に提供するために用いることができる。例えば、経口または非経口製剤(i.v.等を含む)を用いることができる。投与剤形には、カプセル、錠剤、分散剤、懸濁剤、溶液等が含まれる。
【0043】
さらに、製薬的に受容できる担体、希釈剤または賦形剤および場合によっては他の活性医薬成分と組み合わせた、本発明の結晶の改変型を活性成分として含有する医薬組成物を提供する。他の治療成分を含有する組成物は上記の症状の治療において関心がある。本発明はまた、該症状で使用するための医薬の製造における結晶のこの改変型の使用、ならびに、胃液関連症状の治療方法であって、該症状を有する患者に医薬として有効量の結晶の改変型を投与することを含む方法も提供する。
【0044】
本発明の組成物は、経口、i.v.または非経口投与に適した組成物を含む。最も好ましい経路はi.v.経路である。この組成物は使いやすいように単位投与剤形で存在でき、そして製剤製薬学の技術分野で知られたどのような方法を用いても製造できる。
【0045】
本発明の実施において、最も適切な投与経路および治療投与量の大きさは、治療されるべき疾患の性質および重症度に依存するだろう。投与量、および投与頻度は、個々の患者の年齢、体重および応答性によって変えることもできる。ゾーリンガー・エリソン症候群を有する患者に関しては、平均の患者より高用量を必要とるような、特別の要請が求められるだろう。小児および肝疾患を有する患者は、一般に、ある程度平均より少ない用量から利益を受けるだろう。そうしたより高いおよびより低い投与量も本発明の範囲内である。そうした一日用量は5mgから300mgの間で変えることができる。
【0046】
一般に、本発明の化合物の適切な経口投与剤形は、単回用量または等しく分割した用量で投与される、一日総用量で5mgから300mgの用量範囲を包含する。好ましい用量範囲は10mgから80mgの範囲である。
【0047】
本発明の化合物は、WO 96/01623およびEP 0 247 983に記載された経口製剤のような従
来技術にしたがって、密接な(intimate)混合物中の活性成分として医薬用担体と組み合わせることができ、その開示は参照により全体で本願に含まれる。
【0048】
本発明の化合物および他の活性成分を含む組み合わせ製剤もまた使用することができる。そうした活性成分の例には、それに限定されないが、抗細菌化合物、非ステロイド抗炎症剤、制酸剤、アルギナートおよび運動促進剤(prokinetic agent)が含まれる。
【0049】
本発明の化合物は、適切な医薬製剤へと製剤化される前にさらに加工できる。例えば、結晶の改変型はより小さな粒子にすりつぶされまたは粉砕されるだろう。
【0050】
疑義を避けるために「治療」には症状の治療的処置だけでなく予防も含む。
【0051】
X線粉末回析により特徴付けられる、医薬賦形剤のような、試料中の付加的な物質の存在は、当然、上記で特徴付けられた結晶の改変型のどれかの小さなピークのいくつかをマスクする可能性がある。もちろんこの事実のみをもって、結晶の改変型が試料中に存在しないということを証明することはできない。そうした環境下では、適切な注意が用いられなければならず、そしてX線粉末回析パターンの実質的に全ての主要ピークの存在が結晶の改変型の特徴付けるためには十分である。したがって、付加的な物質の存在なしで、本発明の結晶の改変型を解析することが好ましい。
【0052】
本発明は、以下の実施例で説明されるが、それには限定されない。
【0053】
〔実施例〕
一般的な方法
試料のX線粉末回析分析(XRPD)は、標準的な方法、例えば以下に記載された方法にしたがって行った: Giacovazzo, C.等(1995), Fundamentals of Crystallography, Oxford
University Press; Jenkins, R.およびSnyder, R. L. (1996), Introduction to X-Ray Powder Diffractometry, John Wiley & Sons, New York; Bunn, C. W. (1948), Chemical
Crystallography, Clarendon Press, London; またはKlug, H. P.およびAlexander, L. E. (1974), X-ray Diffraction Procedures, John Wiley and Sons, New York. X線回析分析はPhilips X'Pert MPDを用いて16分間で1から40°2θまで行った。 付加をする
ことが試料の調製に費やされる時間に影響しそしてX線回析パターンのピークの位置および強度に影響するために、試料は内部標準なしで分析した。測定したピーク値は、前の実験(−0.05°2θ)に基づいて調整した。その後でd値への計算を行った。
【0054】
XRPD距離値(distance value)は得られた小数点の最後の桁で±2の範囲で変化しうる。
【0055】
実施例1.1
エソメプラゾールのナトリウム塩の改変型Cの製造
エソメプラゾールのカリウム塩(11.89g)を水(50ml)に溶解し、そしてトルエン(80ml)を加えた。次いで、酢酸(5.89ml, 25% v/v)を加えてpHを約7に調整した。2つの相を10分間混合しそして分離させた。水相を取り除き残余の有機相を塩化ナトリウム水溶液(50ml, 10%)で洗浄した。相分離後にメタノール(4.24ml)をトルエン相に加え、次に1当量(eq)のNaOH(1.52ml, 水溶液、45%)を加えた。この溶液をエソメプラゾールのナトリウム塩25mgで結晶種付けした。攪拌しながら終夜結晶化を行わせ、この結晶を減圧濾過で濾別し、そして直ぐにトルエンで2回洗浄した(2×10ml)。得られたフィルターケーキを、分析前に短時間(例えば2〜5分間)空気中で乾燥させた。
【0056】
実施例1.2
エソメプラゾールのナトリウム塩の改変型Eの製造
エソメプラゾールのカリウム塩(11.89g)を水(50ml)に溶解し、そしてトルエン(80ml)を加えた。次いで、酢酸(5.89ml, 25% v/v)を加えてpHを約7に調整した。2つの相を10分間混合しそして分離させた。水相を取り除き残余の有機相を塩化ナトリウム水溶液(50ml, 10%)で洗浄した。相分離後にエタノール(11.1ml)をトルエン相に加え、次に1当量(eq)のNaOH(1.52ml, 水溶液、45%)を加えた。この溶液をエソメプラゾールのナトリウム塩55mgで結晶種付けした。攪拌しながら終夜結晶化を行わせ、この結晶を減圧濾過で濾別し、そして直ぐにトルエンで2回洗浄した(2×10ml)。得られたフィルターケーキを、分析前に短時間(例えば2〜5分間)空気中で乾燥させた。
【0057】
実施例1.3
エソメプラゾールのナトリウム塩の改変型Hの製造
エソメプラゾールのカリウム塩(11.89g)を水(50ml)に溶解し、そしてトルエン(80ml)を加えた。次いで、酢酸(5.89ml, 25% v/v)を加えてpHを約7に調整した。2つの相を10分間混合しそして分離させた。水相を取り除き残余の有機相を塩化ナトリウム水溶液(50ml, 10%)で洗浄した。相分離後に2−プロパノール(3.6ml)をトルエン相に加え、次に1当量(eq)のNaOH(1.52ml, 水溶液、45%)を加えた。この溶液をエソメプラゾールのナトリウム塩53mgで結晶種付けした。攪拌しながら終夜結晶化を行わせ、この結晶を減圧濾過で濾別し、そして直ぐにトルエンで2回洗浄した(2×10ml)。得られたフィルターケーキを、分析前に短時間(例えば2〜5分間)空気中で乾燥させた。
【特許請求の範囲】
【請求項1】
以下の工程:
i) エソメプラゾールの中性形態をトルエンに溶解する;
ii) メタノール、エタノールおよびイソプロピルアルコールから成る群から選択された追加の適切な溶媒を加える;
iii) 適切な塩基Bのナトリウム塩の約1モル当量を加える;
iv) エソメプラゾールのナトリウム塩を結晶化させそして形成された塩を分離する
を本質的に含むエソメプラゾールのナトリウム塩の製造方法。
【請求項2】
Bが水酸化物である、請求項1の方法。
【請求項3】
Bが水溶液として加えられる、請求項1の方法。
【請求項4】
以下の工程:
i) エソメプラゾールの中性形態をトルエンに溶解する;
ii) メタノール、エタノールおよびイソプロピルアルコールから成る群から選択された追加の適切な溶媒を加える;
iii) 適切な塩基Bのナトリウム塩の約1モル当量を加える;
iv) エソメプラゾールのナトリウム塩を結晶化させそして形成された塩を分離する
を本質的に含む方法によって得られるエソメプラゾールのナトリウム塩。
【請求項5】
Bが水酸化物である、請求項4の方法。
【請求項6】
Bが水溶液として加えられる、請求項4の方法。
【請求項7】
15.7、7.9、6.1、5.3、4.56、3.59、3.49、3.17Åのd値のピークを有するX線粉末回析パターンにより特徴付けられる、および/または本質的に図1で表わされる、エソメプラゾールのナトリウム塩の改変型C。
【請求項8】
15.5、11.8、10.1、6.4、6.2、5.4、5.2、4.28、3.46、3.40、3.12Åのd値のピークを有するX線粉末回析パターンにより特徴付けられる、および/または本質的に図2で表わされる、エソメプラゾールのナトリウム塩の改変型E。
【請求項9】
22.0、18.1、11.1、6.3、5.7、5.3、4.92、4.56、3.73Åのd値のピークを有するX線粉末回析パターンにより特徴付けられる、および/または本質的に図3で表わされる、エソメプラゾールのナトリウム塩の改変型H。
【請求項10】
請求項4〜9のいずれかに記載されたエソメプラゾールのナトリウム塩と、少なくとも1つの製薬的に受容できる賦形剤とを混合して含む医薬組成物。
【請求項11】
請求項4〜9のいずれかに記載されたエソメプラゾールのナトリウム塩の治療有効量を、それを必要としている患者に投与することを含む治療方法。
【請求項1】
以下の工程:
i) エソメプラゾールの中性形態をトルエンに溶解する;
ii) メタノール、エタノールおよびイソプロピルアルコールから成る群から選択された追加の適切な溶媒を加える;
iii) 適切な塩基Bのナトリウム塩の約1モル当量を加える;
iv) エソメプラゾールのナトリウム塩を結晶化させそして形成された塩を分離する
を本質的に含むエソメプラゾールのナトリウム塩の製造方法。
【請求項2】
Bが水酸化物である、請求項1の方法。
【請求項3】
Bが水溶液として加えられる、請求項1の方法。
【請求項4】
以下の工程:
i) エソメプラゾールの中性形態をトルエンに溶解する;
ii) メタノール、エタノールおよびイソプロピルアルコールから成る群から選択された追加の適切な溶媒を加える;
iii) 適切な塩基Bのナトリウム塩の約1モル当量を加える;
iv) エソメプラゾールのナトリウム塩を結晶化させそして形成された塩を分離する
を本質的に含む方法によって得られるエソメプラゾールのナトリウム塩。
【請求項5】
Bが水酸化物である、請求項4の方法。
【請求項6】
Bが水溶液として加えられる、請求項4の方法。
【請求項7】
15.7、7.9、6.1、5.3、4.56、3.59、3.49、3.17Åのd値のピークを有するX線粉末回析パターンにより特徴付けられる、および/または本質的に図1で表わされる、エソメプラゾールのナトリウム塩の改変型C。
【請求項8】
15.5、11.8、10.1、6.4、6.2、5.4、5.2、4.28、3.46、3.40、3.12Åのd値のピークを有するX線粉末回析パターンにより特徴付けられる、および/または本質的に図2で表わされる、エソメプラゾールのナトリウム塩の改変型E。
【請求項9】
22.0、18.1、11.1、6.3、5.7、5.3、4.92、4.56、3.73Åのd値のピークを有するX線粉末回析パターンにより特徴付けられる、および/または本質的に図3で表わされる、エソメプラゾールのナトリウム塩の改変型H。
【請求項10】
請求項4〜9のいずれかに記載されたエソメプラゾールのナトリウム塩と、少なくとも1つの製薬的に受容できる賦形剤とを混合して含む医薬組成物。
【請求項11】
請求項4〜9のいずれかに記載されたエソメプラゾールのナトリウム塩の治療有効量を、それを必要としている患者に投与することを含む治療方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図2】
【図3】
【図4】
【図5】
【公開番号】特開2012−246316(P2012−246316A)
【公開日】平成24年12月13日(2012.12.13)
【国際特許分類】
【出願番号】特願2012−203798(P2012−203798)
【出願日】平成24年9月18日(2012.9.18)
【分割の表示】特願2007−518003(P2007−518003)の分割
【原出願日】平成17年6月20日(2005.6.20)
【出願人】(391008951)アストラゼネカ・アクチエボラーグ (625)
【氏名又は名称原語表記】ASTRAZENECA AKTIEBOLAG
【Fターム(参考)】
【公開日】平成24年12月13日(2012.12.13)
【国際特許分類】
【出願日】平成24年9月18日(2012.9.18)
【分割の表示】特願2007−518003(P2007−518003)の分割
【原出願日】平成17年6月20日(2005.6.20)
【出願人】(391008951)アストラゼネカ・アクチエボラーグ (625)
【氏名又は名称原語表記】ASTRAZENECA AKTIEBOLAG
【Fターム(参考)】
[ Back to top ]