
エチレンジシステイン(EC)−薬物結合体
【課題】画像化用に組織を標的化するための新しい放射性標識の戦略を提供することによって、先行技術の欠点および他の欠点を克服すること。
【解決手段】本発明は、放射性標識した組織特異的リガンド、ならびにこの放射性標識したリガンドを作製するための方法および組織特異的疾患を画像化するためにこの放射性標識したリガンドを使用するための方法を提供する。本発明は、組織特異的疾患画像化のための組成物を提供する。本発明のこの画像化組成物は、一般に、エチレンジシステインおよび酸アーム(acid arm)の一方または両方上のエチレンジシステインに結合体化された組織特異的リガンドでキレート化された放射性核種標識を含む。このエチレンジシステインは、放射性核種標識を有するN2S2キレートを形成する。当然、このキレート化した化合物は、放射性核種とキレート化号物との間にイオン結合を含む。
【解決手段】本発明は、放射性標識した組織特異的リガンド、ならびにこの放射性標識したリガンドを作製するための方法および組織特異的疾患を画像化するためにこの放射性標識したリガンドを使用するための方法を提供する。本発明は、組織特異的疾患画像化のための組成物を提供する。本発明のこの画像化組成物は、一般に、エチレンジシステインおよび酸アーム(acid arm)の一方または両方上のエチレンジシステインに結合体化された組織特異的リガンドでキレート化された放射性核種標識を含む。このエチレンジシステインは、放射性核種標識を有するN2S2キレートを形成する。当然、このキレート化した化合物は、放射性核種とキレート化号物との間にイオン結合を含む。
【発明の詳細な説明】
【技術分野】
【0001】
(発明の背景)
政府は、本発明の権利を所有しない。
【0002】
(1.発明の分野)
本発明は、一般に、標識、放射画像化(radioimaging)および化学合成の分野に関する。より詳細には、本発明は、標的化リガンドを放射画像化するための戦略に関する。本発明は、さらに、腫瘍の画像化および組織特異的障害の画像化において、これらの放射性標識したリガンドを使用する方法に関する。
【背景技術】
【0003】
(2.関連技術の説明)
シンチグラフ腫瘍画像化の改善は、さらなる腫瘍特異的放射性医薬品の開発によって、広範囲にわたって決定される。より高い腫瘍特異性に起因して、放射性標識したリガンドおよび放射性標識した抗体は、腫瘍のシンチグラグ検出の新しい時代を開き、そして広範囲の前臨床試験の開発および評価を受けている(非特許文献1〜3(Mathiasら、1996、1997a、1997b))。放射性核種画像化様式(陽子射出放出断層撮影、PET;単一光子放射型コンピュータ断層撮影、SPECT)は、放射性核種標識された放射性トレーサーの位置および密度をマッピングする診断用断層画像化技術である。CTおよびMRIは、腫瘍の位置および範囲に関するかなりの解剖的な情報を提供するが、これらの画像化様式は、浮腫、放射線壊死、グレーディング(grading)または神経膠症由来の侵襲性損傷を十分に区別し得ない。PETおよびSPECTは、代謝活性を測定することによって、腫瘍を局在化および特徴付けするために使用され得る。
【0004】
新しい腫瘍低酸素剤の開発は、一次性損傷および転移性損傷を検出するため、ならびに放射線応答性および再発までの時間を予測するために、臨床的に所望されている。現代の画像化様式のどれも、低酸素症を正確に測定しない。なぜならば、この腫瘍低酸素症の診断は、病理学的試験を必要とするからである。処置される各腫瘍において、少なくとも低酸素症のベースラインを知ることなく、低酸素腫瘍のための治療の成果を予測することは、しばしば困難である。Eppendorfポーラログラフ酸素微小電極は、腫瘍中の酸素電圧を測定し得るが、この技術は、侵襲性であり、かつ熟練した操作者を必要とする。さらに、この技術は、アクセス可能な腫瘍(例えば、頭部および首(頚部))でのみ使用され得、そして複数の読み取りが必要とされる。従って、腫瘍低酸素症を測定するための正確かつ容易な方法は、患者選択のために有用である。しかし、正常な組織に対する腫瘍の取込み比は、使用される放射性医薬品に依存して変化する。従って、新しい放射性医薬品を臨床的な実施に導入する場合に、低酸素症の標準的なEppendorf金電極測定を用いて、正常組織に対する腫瘍の取込み比を補正することが合理的である。
【0005】
[18F]FMISOは、頭部および頚部の腫瘍、心筋梗塞、炎症および脳虚血を診断するために使用されている(Martinら、1992;Yehら、1994;Yetら、1996;Liuら、1994)。正常組織に対する腫瘍の取込み比は、腫瘍低酸素症を評価するためのベースラインとして使用された(Yehら、1996)。[18F]FMISOを使用する腫瘍低酸素症は明確に実証されたが、新しい画像化剤を臨床的な実施に導入することは、容易に利用可能であることおよび同位体の費用などのいくつかの他の因子に依存する。[18F]FDGを使用する腫瘍代謝性画像化は明確に実証されたが、分子画像化剤を臨床的な実施に導入することは、容易に利用可能であることおよび同位体の費用などのいくつかの他の因子に依存する。[18F]フルオロデオキシグルコース(FDG)は、腫瘍、心筋梗塞、および神経学的疾患を診断するために使用されている。さらに、PET放射性合成は、ポジトロン同位体の半減期が短いので、迅速でなければならない。18F化学もまた、複雑である。この18F化学は、異なる分子において再現不可能である。従って、種々の薬物に結合体化し得るキレート剤を開発することが、理想的である。好ましい同位体は、低い費用(18Fの$50/mCiに対して$0.21/mCi)および低いエネルギー(18Fの571Kevに対して140Kev)に起因して99mTcである。99mTcは、99Mo発生器から容易に得られる。望ましい物理学的特性およびかなり低い値段に起因して、99mTcは、放射性医薬品を標識するのに好ましい。
【0006】
いくつかの化合物は、窒素キレート剤および硫黄キレート剤を使用して、99mTcで標識されている(Blondeauら、1967;Davisonら、1980)。ビス−アミノエタンチオール四座配位子リガンド(これはまた、ジアミノジチオール化合物とも呼ばれる)は、2個のチオール硫酸原子および2個のアミン窒素原子に対するオキソテクネチウム基の効率的な結合に基づいて、非常に安定なTc(V)O錯体を形成することで公知である。99mTc−L,L−エチレンジシステイン(99mTc−EC)は、N2S2キレートの最近、かつ有用な例である。ECは、放射性化学的に高い純度および安定性を有する、99mTcで容易におよび効率的に標識され得、そして活性な管状輸送体によって腎臓を介して排出される(Surmaら、1994;Van Neromら、1990、1993;Verbruggenら、1990、1992)。他のECの適用は、PET用のガリウム−68(ポジトロン放出、t1/2=68分)およびガドリニウム、磁気共鳴画像法(MRI)用の鉄もしくはマンガンでキレート化される。99mTc−EC−ネオマイシンおよび99mTc−EC−デオキシグルコースが開発され、そして腫瘍の特徴付けにおけるそれらの潜在的な使用が評価された。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Mathiasら,J Nucl Med,37:1003−1008,1996
【非特許文献2】Mathiasら,J Nucl Med,(Supplement)38:87P,1997a
【非特許文献3】Mathiasら,J Nucl Med,(Supplement)38:133P,1997b
【発明の概要】
【発明が解決しようとする課題】
【0008】
画像化用に組織を標的化するための新しい放射性標識の戦略を提供することによって、先行技術のこれらの欠点および他の欠点を克服すること。
【課題を解決するための手段】
【0009】
(発明の要旨)
本発明は、画像化用に組織を標的化するための新しい放射性標識の戦略を提供することによって、先行技術のこれらの欠点および他の欠点を克服する。本発明は、放射性標識した組織特異的リガンド、ならびにこの放射性標識したリガンドを作製するための方法および組織特異的疾患を画像化するためにこの放射性標識したリガンドを使用するための方法を提供する。
本発明は、組織特異的疾患画像化のための組成物を提供する。本発明のこの画像化組成物は、一般に、エチレンジシステインおよび酸アーム(acid arm)の一方または両方上のエチレンジシステインに結合体化された組織特異的リガンドでキレート化された放射性核種標識を含む。このエチレンジシステインは、放射性核種標識を有するN2S2キレートを形成する。当然、このキレート化した化合物は、放射性核種とキレート化号物との間にイオン結合を含む。用語「EC−組織特異的リガンド結合体」、「EC−誘導体」および「EC−薬物結合体」は、標識されていないエチレンジシステイン−組織特異的リガンド化合物をいうために、本明細書中で交換可能に使用される。本明細書中で使用される場合、用語「結合体」は、共有結合した化合物をいう。
【0010】
エチレンジシステインは、ビス−アミノエタンチオール(BAT)四座配位子である(これはまた、ジアミノジチオール(DADT)化合物としても公知である)。このような化合物は、2個のチオール硫酸原子および2個のアミン窒素原子に対するオキソテクネチウム基の効率的な結合に基づいて、非常に安定なTc(V)O錯体を形成することで公知である。この99mTc標識化ジエチルエステル(99mTc−L,L−ECD)は、脳内物質(brain agent)として公知である。99mTc−L,L−エチレンジシステイン(99mTc−L,L−EC)は、最も極性のある代謝物であり、尿中に迅速かつ効率的に排出されることが発見された。従って、99mTc−L,L−ECは、腎機能剤として使用されてきた(Verbruggenら、1992)。
【0011】
組織特異的リガンドは、哺乳動物または患者の体内に導入された場合、組織の特定の型に特異的に結合する化合物である。本発明の組成物は、実質的に任意の公知の組織特異的化合物を含み得ることが、想定される。好ましくは、本発明と組み合わせて使用される組織特異的リガンドには、抗癌剤、DNAトポイソメラーゼインヒビター、抗代謝剤、腫瘍マーカー、ホレート(folate)レセプター標的化リガンド、腫瘍アポトーシス細胞標的化リガンド、腫瘍低酸素症標的化リガンド、DNAインターカレーター(intercalator)、レセプターマーカー、ペプチド、ヌクレオチド、器官特異的リガンド、抗菌剤(例えば、抗生物質もしくは抗真菌剤)、グルタメートペンタペプチド、またはグルコースを模倣する薬剤がある。このグルコースを模倣する薬剤はまた、「糖(sugar)」とも呼ばれ得る。
【0012】
好ましい抗癌剤としては、メトトレキセート、ドキソルビシン、タモキシフェン、パクリタキセル、トポテカン(topotecan)、LHRH、マイトマイシンC、エトポシド、トムデックス(tomudex)、ポドフィロトキシン、ミトザントロン、カプトセシン(captothecin)、コルヒチン、エンドスタチン(endostatin)、フルダラビンおよびゲムシタビンが挙げられる。好ましい腫瘍マーカーとしては、PSA、ER、PR、AFP、CA−125、CA−199、CEA、インターフェロン、BRCA1、シトキサン(cytoxan)、p53、VEGF、インテグリン、エンドスタチン、HER−2/neu、アンチセンスマーカーまたはモノクローナル抗体が挙げられる。任意の他の公知の腫瘍マーカーまたは任意のモノクローナル抗体が本発明と組み合わせた使用に効果的であることを想定する。好ましいホレートレセプター標的化リガンドとしては、ホレート、メトトレキセートおよびトムデクスが挙げられる。好ましい腫瘍アポトーシス細胞標的化リガンドまたは腫瘍低酸素症標的化リガンドとしては、アネキシンV、コルヒチン、ニトロイミダゾール、マイトマイシンまたはメトロニダゾルが挙げられる。好ましい抗菌剤としては、グラム陽性細菌およびグラム陰性細菌に対して、アンピシリン、アモキシリン、ペニシリン、セファロスポリン、クリダマイシン、ゲンタマイシン、カナマイシン、ネオマイシン、ピマリシン(natamycin)、ナフシリン、リファンピン、テトラサイクリン、バンコマイシン、ブレオマイシン、およびドキシサイクリン、ならびに真菌に対して、アンホテリシンB、アマンタジン、ナイスタチン、ケトコナゾール、ポリミキシン(polymycin)、アシクロビル、およびガンシクロビルが挙げられる。グルコースまたは糖を模倣する好ましい薬剤としては、ネオマイシン、カナマイシン、ゲンタマイシン、プロマイシン(paromycin)、アミカシン、トブラマイシン、ネチルマイシン、リボスタマイシン、シソマイシン、ミクロマイシン、リビドマイシン、ジベカシン(dibekacin)、イセパマイシン(isepamicin)、アストロマイシン、アミノグリコシド、グルコースまたはグルコサミンが挙げられる。
【0013】
特定の実施形態において、エチレンジシステインと組織特異的リガンドとの間にリンカーを含むことが必要である。リンカーは、代表的に、薬物の水溶液中の水溶性を増加させるため、および薬物の親和性の変化を最小にするために使用される。この組成物の水溶性を増加させる任意のリンカーは、実質的に、本発明と組み合わせて使用するために想定されるが、このリンカーは、一般に、ポリアミノ酸、水溶性ペプチド、または単一のアミノ酸のいずれかである。例えば、組織特異的リガンド上の官能基、または薬物が、エストラジオール、トポテカン、パクリタキセルまたはラロキシフェンエトポシドのような脂肪族OHまたはフェノールOHである場合、このリンカーは、ポリグルタミン酸(MW 約750〜約15,000)、ポリアスパラギン酸(MW 約2,000〜約15,000)、ブロモエチルアセテート、グルタミン酸、またはアスパラギン酸である得る。この薬物の官能基は、脂肪族NH2もしくは芳香族NH2またはペプチド(例えば、ドキソルビシン、マイトマイシン C、エンドスタチン、アネキシン V、LHRH、オクトレオチド,およびVIP)である場合、このリンカーは、ポリグルタミン酸(MW 約750〜約15,000)、ポリアスパラギン酸(MW 約2,000〜約15,000)、グルタミン酸またはアスパラギン酸であり得る。この薬物の官能基が、カルボン酸またはペプチド(例えば、メトトレキセートまたは葉酸)である場合、このリンカーは、エチレンジアミン、またはリジンであり得る。
【0014】
画像化のための好ましい放射性核種は、99mTcであるが、他の放射性核種が、特に治療剤としての使用のために、EC組織特異的リガンド結合体または本発明のEC薬物結合体にキレート化され得ることが想定される。例えば、他の有用な放射性核種には、188Re、186Re、153Sm、166Ho、90Y、89Sr、67Ga、68Ga、111In、153Gd、および59Feがある。これらの組成物は、体内の特定の損傷部(例えば、乳癌、卵巣癌、前立腺癌(例えば、l86/l88Re−EC−ホレートを使用する)ならびに頭部および頚部の癌(例えば、186/188Re−EC−ニトロイミダゾールを使用する))に治療放射性核種を送達するために使用される。
【0015】
本発明の特定の実施形態としては、99mTc−EC−アネキシン V、99mTc−EC−コルヒチン、99mTc−EC−ニトロイミダゾール、99mTc−EC−グルタメートペンタペプチド、99mTc−EC−メトロニダゾル、99mTc−EC−ホレート、99mTc−EC−メトトレキセート、99mTc−EC−トムデクス、99mTc−EC−ネオマイシン、99mTc−EC−カナマイシン、99mTc−EC−アミノグリコシド、(グルコサミン、EC−デオキシグルコース)、99mTc−EC−ゲンタマイシン、および99mTc−EC−トブラマイシンが挙げられる。
【0016】
本発明は、さらに、画像化または治療用途のために、放射性標識したエチレンジシステイン薬物結合体またはエチレンジシステイン薬物誘導体を合成するための方法を提供する。この方法は、組織特異的リガンドを得る工程、このリガンドをエチレンジシステイン(EC)と混合して、EC組織特異的リガンド誘導体を得る工程、ならびに放射性核種および還元剤と共にEC組織特異的リガンド誘導体を混合して、放射性核種標識されたEC組織特異的リガンド誘導体を得る工程を包含する。この放射性核種は、N2S2キレートを介してECにキレート化される。この組織特異的リガンドは、上記のように直接的またはリンカーを介してのいずれかで、ECの酸アームの一方または両方のに結合体化される。この還元剤は、好ましくは、ジチオナイト(dithionite)イオン、スズイオンまたは鉄(II)イオンである。
【0017】
本発明は、さらに、画像化、治療用途のため、または診断用途もしくは予後用途のために組織特異的リガンドを標識するための方法を提供する。この標識方法は、組織特異的リガンドを得る工程、エチレンジシステイン(EC)と共に組織特異的リガンドを混合して、EC−リガンド薬物結合体を得る工程、および還元剤の存在下で、薬物結合体を99mTcと反応させて、エチレンジシステインとこの99mTcとの間にN2S2キレートを形成する工程を包含する。
【0018】
この実施形態の目的のために、組織特異的リガンドは、上記または本明細書中で議論されるいずれかのリガンドであり得る。還元剤は、任意の公知の還元剤であり得るが、好ましくは、ジチオナイトイオン、スズイオンまたは鉄(II)イオンである。
【0019】
別の実施形態において、本発明は、哺乳動物の体内のある部位を画像化する方法を提供する。この画像化方法は、99mTc標識されたエチレンジシステイン組織特異的リガンド結合体を含む組成物の診断有効量を投与する工程、およびその部位に局在化された99mTcからの放射能シグナルを検出する工程を包含する。この検出する工程は、代表的に、哺乳動物の体内にこの組成物を導入した約10分〜約4時間後に実施される。より好ましくは、この検出する工程は、哺乳動物の体内にこの組成物を注入した約1時間後に実施される。
【0020】
特定の好ましい実施形態において、この部位は、感染、腫瘍、心臓、肺、脳、肝臓、脾臓、膵臓、腸または任意の他の器官である。腫瘍または感染は、哺乳動物の体内のどこかに局在化され得るが、一般に、胸部、卵巣、前立腺、子宮内膜、肺、脳、または肝臓である。この部位は、ホレート陽性癌またはエストロゲン陽性癌であり得る。
【0021】
本発明はまた、放射性薬学的調製物を調製するためのキットを提供する。このキットは、一般に、予め決められた量のエチレンジシステイン組織特異的リガンド結合体組成物およびこの結合体を99mTcで標識するために十分な量の還元剤を含む、密閉されたバックまたは他の任意の種の適切な容器を含む。特定の場合において、このエチレンジシステイン組織特異的リガンド結合体組成物はまた、このエチレンジシステインと組織特異的リガンドとの間にリンカーを含む。この組織特異的リガンドは、任意の特異的な組織型(例えば、本明細書中で議論される組織型)に特異的に結合する任意のリガンドであり得る。リンカーがこの組成物中に含まれる場合、このリンカーは、本明細書中で記載されるようなリンカーであり得る。
【0022】
このキット成分は、任意の適切な形態(例えば、液体形態、凍結形態または乾燥形態)であり得る。好ましい実施形態において、このキット成分は、凍結乾燥形態で提供される。このキットはまた、抗酸化剤および/または捕捉剤を含み得る。この抗酸化剤は、公知の任意の抗酸化剤であるが、好ましくは、ビタミンCであり得る。捕捉剤はまた、残りの放射性核種を結合するために存在し得る。最も市販されているキットは、捕捉剤としてグルコヘプトネートを含む。しかし、グルコへプトネートは、約10〜15%を残したまま、代表的なキット成分と完全には反応しない。この残りのグルコへプトネートは腫瘍に達し、そして画像化結果をゆがめる。従って、本発明は、より安価で、かつより完全に反応するように、捕捉剤としてEDTAを使用することが好ましい。
【0023】
本発明の別の局面は、特定の腫瘍の治療のために、候補化合物の潜在的な有用性を決定するための予後方法である。現在、ほとんどの腫瘍は、数ヶ月および数千ドルを費やすまで、この薬物が特定の腫瘍に対して実際に有用であるどうかのいずれの指標もなく、化学療法において「通常の選択された薬物」で処置される。本発明の画像化組成物は、標識化されたEC薬物結合体の形態で、特定の薬物を腫瘍部位に送達すること、次いで特定の薬物かどうかを決定するために数時間内で部位を画像化することに有用である。
【0024】
このことに関連して、本発明の予後方法は、哺乳動物の体内の腫瘍の部位を決定する工程、腫瘍特異的な癌化学療法薬物候補物に結合体化されるECにキレート化された放射性核種を含む画像化組成物を得る工程、この組成物を哺乳動物の体内に投与する工程、ならびにこの腫瘍に対するこの候補薬物の有効性を決定するためにこの部位を画像化する工程を包含する。代表的に、この画像化工程は、哺乳動物の体内に組成物を注入した約10分〜約4時間後以内で実施される。好ましくは、この画像化工程は、哺乳動物の体内に組成物を注入した約1時間後以内に実施される。
【0025】
予後組成物中のECに結合体化されたこの癌化学療法薬物候補物は、公知の癌化学療法薬物から選択され得る。このような薬物は、表2にある。癌の特定の型に特異的であることが公知の多くの抗癌剤が存在する。しかし、癌の特定の型に対する全ての抗癌剤が、あらゆる患者に有用とは限らない。従って、本発明は、処置に莫大な時間とお金を費やす前に、候補薬物の潜在的な有用性を決定する方法を初めて提供する。
【0026】
本発明のなお別の実施形態は、シンチグラフ画像化剤を調製するための試薬である。本発明の試薬は、組織特異的リガンドを含み、この組織特異的リガンドは、シンチグラフで検出可能な画像を提供するのに十分なインビボの標識部位に対する親和性を有し、99mTc結合部分に共有結合する。99mTc結合部分は、上記のように、この組織特異的リガンドに直接的に結合されるか、またはリンカーを介してリガンドに結合されるかのいずれかである。この99mTc結合部分は、好ましくは、+4酸化状態の99mTcとエチレンジシステイン(EC)との間のN2S2キレートである。この組織特異的リガンドは、上記のように直接的またはリンカーを介してのいずれかで、ECの酸アームの一方または両方に共有結合される。この組織特異的リガンドは、上記のようなリガンドのいずれかであり得る。
【発明の効果】
【0027】
本発明により、例えば、以下が提供される:
(項目1) 画像化のための組成物であって、以下:
放射線核種標識;
エチレンジシステイン;および
該エチレンジシステインに結合体化した組織特異的リガンド;を含み、ここで、該エチレンジシステインが、該放射線核種標識とN2S2キレートを形成する、組成物。
(項目2) 前記組織特異的リガンドが、前記エチレンジシステインの1つまたは両方の酸アームで該エチレンジシステインと結合体化し得る、項目1に記載の組成物。 (項目3) 前記放射線核種が、99mTc、188Re、186Re、183Sm、166Ho、90Y、89Sr、67Ga、68Ga、111In、183Gd、59Fe、225Ac、212Bi、211At、64Cuまたは62Cuである、項目1に記載の組成物。
(項目4) 前記放射線核種が99mTcである、項目3に記載の組成物。 (項目5) 前記組織特異的リガンドが、抗癌剤、DNAトポイソメラーゼインヒビター、代謝拮抗剤、腫瘍マーカー、ホレートレセプター標的化リガンド、腫瘍アポトーシス細胞標的化リガンド、腫瘍低酸素標的化リガンド、DNAインターカレーター、レセプターマーカー、ペプチド、ヌクレオチド、器官特異的リガンド、抗生物質、抗真菌剤、抗体、グルタミン酸ペンタペプチドまたはグルコースを模倣する因子である、項目1に記載の組成物。
(項目6) 前記組織特異的リガンドが、抗癌剤である、項目5に記載の組成物。 (項目7) 前記抗癌剤が、メトトレキセート、ドキソルビシン、タモキシフェン、パクリタキセル、トポテカン、LHRH、マイトマイシンC、エトポシドトムデックス、ポドフィロトキシン、ミトザントロン、カンプトセシン、コルヒチン、エンドスタチン、フルダラビン、ゲムシタビンおよびトムデックスからなる群より選択され得る、項目6に記載の組成物。
(項目8) 前記組織特異的リガンドが腫瘍マーカーである、項目5に記載の組成物。 (項目9) 前記腫瘍マーカーが、PSA、ER、PR、CA−125、CA−199、CEA AFP、インターフェロン、BRCA1、HER−2/neu、シトキサン、p53、エンドスタチンまたはモノクローナル抗体(例えば、アンチセンス)である、項目8に記載の組成物。
(項目10) 前記組織特異的リガンドが、ホレートレセプター標的化リガンドである、項目5に記載の組成物。
(項目11) 前記ホレートレセプター標的化リガンドが、ホレート、メトトレキセートまたはトムデックスである、項目10に記載の組成物。
(項目12) 99mTc−EC−ホレートとしてさらに規定される、項目11に記載の組成物。
(項目13) 99mTc−EC−メトトレキセートとしてさらに規定される、項目11に記載の組成物。
(項目14) 99mTc−EC−トムデックスとしてさらに規定される、項目11に記載の組成物。
(項目15) 前記組織特異的リガンドが、腫瘍アポトーシス細胞標的化リガンドまたは腫瘍低酸素標的化リガンドである、項目5に記載の組成物。
(項目16) 前記組織特異的リガンドが、アネキシンV、コルヒチン、ニトロイミダゾール、マイトマイシンまたはメトロニダゾルである、項目15に記載の組成物。 (項目17) 99mTc−EC−アネキシンVとしてさらに規定される、項目16に記載の組成物。
(項目18) 99mTc−EC−コルヒチンとしてさらに規定される、項目16に記載の組成物。
(項目19) 99mTc−EC−ニトロイミダゾールとしてさらに規定される、項目16に記載の組成物。
(項目20) 99mTC−EC−メトロニダゾールとしてさらに規定される、項目16に記載の組成物。
(項目21) 前記組織特異的リガンドが、グルタミン酸ペンタペプチド(分子量750〜15,000)である、項目5に記載の組成物。
(項目22) 99mTc−EC−グルタミン酸ペンタペプチドとしてさらに規定される、項目21に記載の組成物。
(項目23) 前記組織特異的リガンドが、グルコースを模倣する因子である、項目5に記載の組成物。
(項目24) グルコースを模倣する前記因子が、ネオマイシン、カナマイシン、ゲンタマインシン、パロマイシン、アミカシン、トブラマイシン、ネチルマイシン、リボスタマイシン、シソマイシン、ミクロマイシン、リビドマイシン、ジベカシン、イセパマイシン、アストロマイシン、またはアミノグルコシドである、項目23に記載の組成物。 (項目25) 99mTc−EC−ネオマイシンとしてさらに規定される、項目24に記載の組成物。
(項目26) 99mTc−EC−カナマイシンとしてさらに規定される、項目24に記載の組成物。
(項目27) 99mTc−EC−アミノグリコシドとしてさらに規定される、項目24に記載の組成物。
(項目28) 99mTc−EC−ゲンタマイシンとしてさらに規定される、項目24に記載の組成物。
(項目29) 99mTc−EC−トブラマイシンとしてさらに規定される、項目24に記載の組成物。
(項目30) ECを前記組織特異的リガンドに結合体化するリンカーをさらに含む。項目2に記載の組成物。
(項目31) 前記リンカーが、水溶性ペプチド、グルタミン酸、アスパラギン酸、ブロモ酢酸エチル、エチレンジアミンまたはリジンである、項目30に記載の組成物。 (項目32) 前記組織特異的リガンドが、エストラジオール、トポテカン、パクリタキセル、ラロキシフェン、エトポシド、ドキソルビシン、マイトマイシンC、エンドスタチン、アネキシンV、LHRH、オクトレオチド、VIP、メトトレキセートまたは葉酸である、項目31に記載の組成物。
(項目33) 画像化のための放射標識されたエチレンジシステイン誘導体を合成する方法であって、該方法は、以下の工程:
a)組織特異的リガンドを得る工程;
b)該リガンドをエチレンジシステイン(EC)と混合して、EC−組織特異的リガンド誘導体を得る工程;ならびに
c)該EC−組織特異的リガンド誘導体を、放射線核種および還元剤と混合して、放射線核種標識されたEC−組織特異的リガンド誘導体を得る工程であって、ここで、該ECが、該放射線核種とN2S2キレートを形成する、工程、を包含する、方法。
(項目34) 前記還元剤が、亜ジチオン酸イオン、第一スズイオン、または第一鉄イオンである、項目33に記載の方法。
(項目35) 画像化のために組織特異的リガンドを標識化する方法であって、該方法は、以下の工程:
a)組織特異的リガンドを得る工程;
b)該組織特異的リガンドをエチレンジシステイン(EC)と混合して、EC−リガンド薬物結合体を得る工程;ならびに
c)該薬物結合体を、還元剤の存在下で99mTcと反応させて、該エチレンジシステイン(リンカーありまたはリンカーなし)と該99mTcとの間にN2S2キレートを形成する工程、
を包含する、方法。
(項目36) 前記組織特異的リガンドが、抗癌剤、DNAトポイソメラーゼインヒビター、代謝拮抗剤、腫瘍マーカー、ホレートレセプター標的化リガンド、腫瘍アポトーシス細胞標的化リガンド、腫瘍低酸素標的化リガンド、DNAインターカレーター、レセプターマーカー、ペプチド、器官特異的リガンド、抗生物質、抗真菌剤、グルタミン酸ペンタペプチドまたはグルコースを模倣する因子である、項目35に記載の方法。 (項目37) 前記還元剤が、亜ジチオン酸イオン、第一スズイオン、または第一鉄イオンである、項目36に記載の方法。
(項目38) 哺乳動物身体内の部位を画像化する方法であって、99mTc標識されたエチレンジシステイン−組織特異的リガンド結合体を含む診断有効量の組成物を投与する工程、および該部位に局在する99mTcからの放射活性シグナルを検出する工程、を包含する、方法。
(項目39) 前記部位が腫瘍である、項目38に記載の方法。 (項目40) 前記部位が感染である、項目38に記載の方法。 (項目41) 前記部位が、乳癌、卵巣癌、前立腺癌、子宮内膜、心臓、肺、脳、肝臓、ホレート(+)癌、ER(+)癌、脾臓、膵臓、または腸である、項目38に記載の方法。
(項目42) 放射性薬学的調製物を調製するためのキットであって、該キットは、所定量のエチレンジシステイン−組織特異的リガンド結合体組成物および99mTcを用いて該結合体を標識するために十分量の還元剤を含むシールされた容器を備える、キット。 (項目43) 前記エチレンジシステイン−組織特異的リガンド結合体組成物が、該エチレンジシステインと該組織特異的リガンドとの間にさらにリンカーを含む、項目42に記載のキット。
(項目44) 前記組織特異的リガンドが、抗癌剤、DNAトポイソメラーゼインヒビター、代謝拮抗剤、腫瘍マーカー、ホレートレセプター標的化リガンド、 腫瘍アポトーシス細胞標的化リガンド、腫瘍低酸素標的化リガンド、DNAインターカレーター、レセプターマーカー、ペプチド、器官リガンド、抗生物質、抗真菌剤、グルタミン酸ペンタペプチドまたはグルコースを模倣する因子である、項目42に記載のキット。 (項目45) 前記組織特異的リガンドが、エストラジオール、トポテカン、パクリタキセル、ラロキシフェン、エトポシド、ドキソルビシン、マイトマイシンC、エンドスタチン、アネキシンV、LHRH、オクトレオチド、VIP、メトトレキセートまたは葉酸である、項目43に記載のキット。
(項目46) 前記リンカーが、水溶性ペプチド、グルタミン酸、ポリグルタミン酸、アスパラギン酸、ポリアスパラギン酸、ブロモ酢酸エチル、エチレンジアミンまたはリジンである、項目45に記載のキット。
(項目47) シンチグラフィー画像化剤を調製するための試薬であって、99mTc結合部位に共有結合した組織特異的リガンドを含む、試薬。
(項目48) 前記99mTc結合部分が、エチレンジシステインである、項目47に記載の試薬。
(項目49) 前記組織特異的リガンドが、抗癌剤、DNAトポイソメラーゼインヒビター、代謝拮抗剤、腫瘍マーカー、ホレートレセプター標的化リガンド、腫瘍アポトーシス細胞標的化リガンド、腫瘍低酸素標的化リガンド、DNAインターカレーター、レセプターマーカー、ペプチド、器官特異的リガンド、抗生物質、抗真菌剤、グルタミン酸ペンタペプチドまたはグルコースを模倣する因子である、項目48に記載の試薬。 (項目50) 前記組織特異的リガンドと前記99mTc結合部位との間にさらにリンカーを含む、項目48に記載の試薬。
(項目51) 腫瘍に対する候補薬物の有効性を決定する方法であって、該方法は、以下:
a)候補薬物を得る工程;
b)該候補薬物をエチレンジシステイン(EC)と結合体化して、EC−候補薬物結合体を生成する工程;
c)該候補薬物結合体を、99mTcを用いてキレート化して、99mTc−EC−候補薬物結合体を生成する工程;
d)該99mTc−EC−候補薬物結合体を、腫瘍を有する患者に導入する工程;および
e)該患者を画像化して、該腫瘍に対する該候補薬物の有効性を決定する工程、を包含する、方法。
【図面の簡単な説明】
【0028】
【図1】図1は、99mTc−EC−ホレートの合成スキームである。
【図2】図2は、99mTc−EC−MTX(メトトレキサート)の合成スキームである。
【図3】図3は、99mTc−EC−TDX(トムデクス(tomudex))の合成スキームである。
【図4】図4は、99mTc−ECおよび99mTc−EC−ホレートの生体分布研究である。
【図5】図5は、99mTc−ECホレートを用いた腫瘍/筋肉カウント比および腫瘍/血液カウント比についてのブロッキング研究である。
【図6】図6Aおよび図6Bは、99mTc−EC注射群と比較した、99mTc−ECホレート注射群における腫瘍のシンチグラフィ画像である。
【図7】図7は、EC−MN(メトロニダゾール)の合成スキームである。
【図8A】図8Aは、EC−NIMについての合成スキームを示す。
【図8B】図8Bは、EC−NIMについての構造の1H−NMR確認を示す。
【図9】図9は、99mTc−EC−MN、[18F]FMISOおよび[131I]IMISOについての生体分布研究(腫瘍/血液比)である。
【図10】図10は、99mTc−EC、[18F]FMISOおよび[131I]IMISOについての生体分布研究(腫瘍/筋肉比)である。
【図11】図11Aは、99mTc−EC−MN注射群の腫瘍のシンチグラフィ画像であり、図11Bは、99mTc−EC注射群の腫瘍のシンチグラフィ画像である。
【図12】図12は、99mTc−EC−MNでの注射の1時間後において実施したオートラジオグラムである。
【図13】図13は、イヌ血清サンプル中の99mTc−EC−MNの安定性を示す。
【図14A】図14Aは、ラットにおける99mTc−EC−MN 対 99mTc−ECの胸部腫瘍取り込みを示す。
【図14B】図14Bは、コントロールと比較した、パクリタキセルで処理したラットにおける99mTc−EC−MN 対 99mTc−ECの胸部腫瘍取り込みを示す。
【図15A】図15Aは、ラットにおける99mTc−EC−NIM 対 99mTc−ECの卵巣腫瘍取り込みを示す。パクリタキセルを用いて処理したラットにおける腫瘍取り込み(図15B)は、パクリタキセルで処理していないラットにおける腫瘍取り込み(図15A)よりも少ない。肉腫を有するラットにおける99mTc−EC−NIMの腫瘍取り込みもまた示す。図15Cは、パクリタキセルで処理した肉腫保有ラットにおける腫瘍取り込みを示し、一方、図15Dは、パクリタキセルで処理していないラットにおける腫瘍取り込みを示す。パクリタキセルで処理した後に、99mTc−EC−NIMの取り込みの減少したが存在した。
【図15B】図15Aは、ラットにおける99mTc−EC−NIM 対 99mTc−ECの卵巣腫瘍取り込みを示す。パクリタキセルを用いて処理したラットにおける腫瘍取り込み(図15B)は、パクリタキセルで処理していないラットにおける腫瘍取り込み(図15A)よりも少ない。肉腫を有するラットにおける99mTc−EC−NIMの腫瘍取り込みもまた示す。図15Cは、パクリタキセルで処理した肉腫保有ラットにおける腫瘍取り込みを示し、一方、図15Dは、パクリタキセルで処理していないラットにおける腫瘍取り込みを示す。パクリタキセルで処理した後に、99mTc−EC−NIMの取り込みの減少したが存在した。
【図15C】図15Aは、ラットにおける99mTc−EC−NIM 対 99mTc−ECの卵巣腫瘍取り込みを示す。パクリタキセルを用いて処理したラットにおける腫瘍取り込み(図15B)は、パクリタキセルで処理していないラットにおける腫瘍取り込み(図15A)よりも少ない。肉腫を有するラットにおける99mTc−EC−NIMの腫瘍取り込みもまた示す。図15Cは、パクリタキセルで処理した肉腫保有ラットにおける腫瘍取り込みを示し、一方、図15Dは、パクリタキセルで処理していないラットにおける腫瘍取り込みを示す。パクリタキセルで処理した後に、99mTc−EC−NIMの取り込みの減少したが存在した。
【図15D】図15Aは、ラットにおける99mTc−EC−NIM 対 99mTc−ECの卵巣腫瘍取り込みを示す。パクリタキセルを用いて処理したラットにおける腫瘍取り込み(図15B)は、パクリタキセルで処理していないラットにおける腫瘍取り込み(図15A)よりも少ない。肉腫を有するラットにおける99mTc−EC−NIMの腫瘍取り込みもまた示す。図15Cは、パクリタキセルで処理した肉腫保有ラットにおける腫瘍取り込みを示し、一方、図15Dは、パクリタキセルで処理していないラットにおける腫瘍取り込みを示す。パクリタキセルで処理した後に、99mTc−EC−NIMの取り込みの減少したが存在した。
【図16】図16は、EC−GAP(ペンタグルタメート)の合成スキームである。
【図17】図17は、99mTc−EC−GAP注射群における胸部腫瘍のシンチグラフィー画像である。
【図18】図18は、異なる時間間隔での、99mTc−EC−ANNEX V注射群における胸部腫瘍のシンチグラフィー画像である。
【図19A】図19Aおよび図19Bは、卵巣腫瘍保有群におけるパクリタキセル処理前(図19A)とパクリタキセル処理後(図19B)との間の、99mTc−EC−ANNEX Vの取り込み差異の比較である。
【図19B】図19Aおよび図19Bは、卵巣腫瘍保有群におけるパクリタキセル処理前(図19A)とパクリタキセル処理後(図19B)との間の、99mTc−EC−ANNEX Vの取り込み差異の比較である。
【図20A】図20Aおよび図20Bは、肉腫腫瘍保有群におけるパクリタキセル処理前(図20A)とパクリタキセル処理後(図20B)との間の、99mTc−EC−ANNEX Vの取り込み差異の比較である。
【図20B】図20Aおよび図20Bは、肉腫腫瘍保有群におけるパクリタキセル処理前(図20A)とパクリタキセル処理後(図20B)との間の、99mTc−EC−ANNEX Vの取り込み差異の比較である。
【図21】図21は、EC−COL(コルヒチン)の合成スキームである。
【図22】図22は、EC−COL合成において分解生成物が観察されなかったことを示す。
【図23】図23は、99mTc−EC−COLについての、時間関数としての腫瘍 対 筋肉比および腫瘍 対 血液比である。
【図24】図24は、99mTc−ECについての、時間関数としての腫瘍 対 筋肉比および腫瘍 対 血液比である。
【図25】図25は、99mTc−EC−COLを用いた胸部腫瘍保有ラットにおけるインビボ画像研究である。
【図26】図26は、99mTc−ECを用いた胸部腫瘍保有ラットにおけるインビボ画像研究である。
【図27】図27は、99mTc−EC−COL 対 99mTc−ECの、注射後のコンピューターで輪郭をとった目的の領域である。
【図28】図28は、発作に罹患した59歳の男性患者の99mTc−EC−MNを用いたSPECTである。画像は、注射1時間後に撮影した。
【図29】図29は、図28と同じ患者のMRI T1重み付け画像である。
【図30】図30は、発作1日後の73歳の男性患者の、99mTc−EC−MNを用いた注射の1時間後でのSPECTである。
【図31】図31は、発作12日後の図30で画像化した同じ73歳の男性患者の、99mTc−EC−MNを用いた注射の1時間後でのSPECTである。
【図32】図32は、発作1日後の図30で画像化した同じ73歳の男性発作患者のCTである。
【図33】図33は、発作12日後の図32で画像化した同じ73歳の男性発作患者のCTである。画像化にCTを用いた1日目と12日目との間に顕著な差がないことに注意する。
【図34】図34は、発作に罹患した72歳の男性患者の99mTc−EC−MNを用いた注射の1時間後でのSPECTである。
【図35】図35は、図34で画像化した同じ72歳の男性発作患者のCTである。CTがいかに病変サイズを誇張しているかに注意すること。
【図36】図36は、99mTc−EC−ネオマイシンの合成スキームである。
【図37A】99mTc−ECおよび99mTc−EC−ネオマイシン(100μCi/ラット、静脈内)の投与後の胸部腫瘍保有ラットのシンチグラフィー画像は、この腫瘍が注射後0.5〜4時間で良好に可視化され得ることを示す。
【図37B】図37Bは、乳癌患者の99mTc−EC−ネオマイシン(30mCi、静脈内)を用いたシンチマンモグラフィである。画像は、注射2時間後に撮影した。
【図38A】図38Aは、ECの1H−NMRである。
【図38B】図38Bは、ネオマイシンの1H−NMRである。
【図38C】図38Cは、EC−ネオマイシンの1H−NMRである。
【図39】図39Aおよび図39Bは、EC−ネオマイシンの質量分析である(M+1112.55)。
【図40A】図40Aは、ECのUV波長スキャンである。
【図40B】図40Bは、ネオマイシンのUV波長スキャンである。
【図40C】図40Cは、EC−ネオマイシンのUV波長スキャンである。
【図41】図41は、99mTc−EC−ネオマイシンのラジオ−TLC分析である。
【図42】図42は、99mTc−EC−ネオマイシンのHPLC分析である(放射能検出器)。
【図43】図43は、99mTc−EC−ネオマイシンのHPLC分析である(UV 254nm)。
【図44】図44は、18F−FDGのHPLC分析である(放射能検出器)。
【図45】図45は、18F−FDGのHPLC分析である(UV 254nm)。
【図46】図46は、肺癌細胞株における一連の99mTc−EC薬物結合体のインビトロ細胞取り込みアッセイである。99mTc−EC−ネオマイシンは、試験した薬剤において最も高い取り込みを示した。
【図47】図47は、99mTc−EC−ネオマイシンおよび18F−FDGの細胞(A549)取り込みに対するグルコースの影響を示す。
【図48A】図48Aは、薬物取り込み(%)として示され、99mTc−EC−ネオマイシンおよび18F−FDGの細胞(H1299)取り込みに対するグルコースの影響を示す。
【図48B】図48Bは、グルコース充填に伴なう変化(%)として示され、99mTc−EC−ネオマイシンおよび18F−FDGの細胞(H1299)取り込みに対するグルコースの影響を示す。
【図49】図49は、99mTc−EC−グルコサミンの合成スキームである。
【図50】図50は、グルコースのヘキソキナーゼアッセイである。
【図51】図51は、グルコサミンのヘキソキナーゼアッセイである。
【図52】図52は、EC−グルコサミンのヘキソキナーゼアッセイである。
【図53】図53は、EC−GAP−グルコサミンのヘキソキナーゼアッセイである。
【図54】図54は、99mTc−EC−GAP−グルコサミンの合成スキームである。
【図55A】図55A〜図55Cは、肺癌細胞株(A549)における99mTc−EC(図55A)、99mTc−EC−デオキシグルコース−GAP(図55B)、および18F−FDG(図55C)のインビトロ細胞取り込みアッセイである。99mTc−EC−DGは、18F−FDGと比較して類似の取り込みを示した。
【図55B】図55A〜図55Cは、肺癌細胞株(A549)における99mTc−EC(図55A)、99mTc−EC−デオキシグルコース−GAP(図55B)、および18F−FDG(図55C)のインビトロ細胞取り込みアッセイである。99mTc−EC−DGは、18F−FDGと比較して類似の取り込みを示した。
【図55C】図55A〜図55Cは、肺癌細胞株(A549)における99mTc−EC(図55A)、99mTc−EC−デオキシグルコース−GAP(図55B)、および18F−FDG(図55C)のインビトロ細胞取り込みアッセイである。99mTc−EC−DGは、18F−FDGと比較して類似の取り込みを示した。
【図56】図56は、胸部腫瘍保有ラットにおける99mTc−EC−GAPの腫瘍 対 組織カウント密度比である。
【図57】図57は、乳癌細胞株(13762)における注射2時間後の、グルコース充填を伴なう18FDGのインビトロ細胞取り込みである。
【図58】図58は、胸部腫瘍保有マウスにおける99mTc−EC−ネオマイシンのインビボ組織取り込みである。
【図59】図59は、99mTc−EC−デオキシグルコースの合成スキームである。
【図60】図60は、EC−デオキシグルコースの質量分析である。
【図61】図61は、EC−デオキシグルコース(EC−DG)の1H−NMRである。
【図62】図62は、グルコサミンの1H−NMRである。
【図63】図63は、99mTc−EC−DGのラジオ−TLC分析である。
【図64】図64は、99mTc−EC−デオキシグルコースおよび99mTc−ECのHPLC分析である(放射能検出器)。
【図65】図65は、99mTc−EC−デオキシグルコースおよび99mTc−ECのHPLC分析である(放射能検出器、混合)。
【図66】図66は、グルコースのヘキソキナーゼアッセイである。
【図67】図67は、FDGのヘキソキナーゼアッセイである。
【図68】図68は、EC−DGのヘキソキナーゼアッセイである。
【図69】図69は、肺癌細胞株(A549)における99mTc−EC−デオキシグルコース、99mTc−ECおよび18F−FDGのインビトロ細胞取り込みアッセイである。99mTc−EC−DGは、18F−FDGと比較して類似した取り込みを示した。
【図70】図70は、99mTc−EC−DGの胸部細胞(13762細胞株)取り込みに対するd−グルコースおよびl−グルコースの影響である。
【図71】図71は、18F−FDGの胸部細胞(13762細胞株)取り込みに対するd−グルコースおよびl−グルコースの影響である。
【図72】図72は、18F−FDGの肺細胞(A549細胞株)取り込みに対するd−グルコースおよびl−グルコースの影響である。
【図73】図73は、99mTc−EC−DGの肺細胞(A549細胞株)取り込みに対するd−グルコースおよびl−グルコースの影響である。
【図74】図74は、グルコサミンおよびEC−DG(1.2mmol/kg、静脈内)によって誘導されたインビボ血中グルコースレベルの影響である。
【図75】図75は、FDG(1.2mmol/kgおよび1.9mmol/kg、静脈内)およびインスリンによって誘導されたインビボ血中グルコースレベルの影響である。
【図76】図76は、胸部腫瘍保有ラットにおける99mTc−EC−デオキシグルコースの腫瘍 対 組織カウント密度比である。
【図77】図77は、胸部腫瘍保有ラットにおける99mTc−EC−デオキシグルコースのインビボ生体分布である。
【図78】図78は、肺腫瘍保有マウスにおける99mTc−EC−デオキシグルコースのインビボ組織取り込みである。
【図79】図79は、肺腫瘍保有マウスにおける99mTc−EC−ネオマイシンのインビボ組織取り込みである。
【図80】図80は、肺腫瘍保有マウスにおける18F−FDGのインビボ組織取り込みである。
【図81】99mTc−ECおよび99mTc−EC−デオキシグルコース(100μCi/ラット、静脈内)投与後の胸部腫瘍保有ラットの平面画像は、この腫瘍が、注射後0.5〜4時間で良好に可視化され得ることを示した。
【図82A】図82Aは、悪性神経膠星状細胞腫を有する患者のMRIである。
【図82B】図82Bは、悪性神経膠星状細胞腫を有する患者の99mTc−EC−DGを用いたSPECTである。
【図83A】図83Aは、出血性神経膠星状細胞腫を有する患者のMRIである。
【図83B】図83Bは、悪性神経膠星状細胞腫を有する患者の99mTc−EC−DGを用いたSPECTである。
【図84A】図84Aは、良性髄膜腫を有する患者のMRIである。
【図84B】良性髄膜腫を有する患者の99mTc−EC−DGを用いたSPECTは、焦点が強調された取り込みを示さなかった。
【図85A】図85Aは、肺におけるTBを有する患者のCTである。
【図85B】TBを有する患者の99mTc−EC−DGを用いたSPECTは、焦点が強調された取り込みを示さなかった。
【図86A】図86Aは、肺癌を有する患者のCTである。
【図86B】図86Bは、肺癌を有する患者の99mTc−EC−DGを用いた全身画像である。
【図86C】図86Cは、肺癌を有する患者の99mTc−EC−DGを用いたSPECTであり、腫瘍は、焦点が強調された取り込みを示した。
【発明を実施するための形態】
【0029】
(例示的な実施形態の説明)
核医学の分野において、特定の病理的状態は、局在化するか、またはそれらの程度が、少量の内部投与される放射性標識されたトレーサ化合物(放射性トレーサまたは放射性薬品と呼ばれる)の分布を検出することによって評価される。これらの放射性薬品を検出するための方法は、画像化法または放射性画像化法として一般的に公知である。
【0030】
放射性画像化において、放射性標識は、γ線放射放射性核種であり、そして放射性トレーサは、γ線検出カメラを使用して位置決めされる(このプロセスは、しばしば、γシンチグラフィと呼ばれる)。画像化部位は、検出可能である。なぜなら、放射性トレーサは、病理学的部位に局在化する(陽性コントラストと呼ばれる)か、あるいは、放射性トレーサがこのような病理学的部位に特異的に局在化しないように選択される(陰性コントラストと呼ばれる)かのいずれかであるからである。
【0031】
種々の放射性核種(67Ga、99mTc、111In、123I、125I、169Ybまたは186Reを含む)は、放射性画像化に有用であることが公知である。より良い画像化特性および低価格に起因して、可能な場合、123I、131I、67Gaおよび111In標識された化合物を、対応する99mTc標識化合物に置き換える試みがなされている。好ましい物理的特質および非常に低い価格(1mCi当たり0.21ドル)に起因して、99mTcは、放射性薬品を標識するのに好ましかった。DTPA薬物結合体が99mTcで効果的に標識され得ることが報告された(Mathiasら、1997)が、DTPA部分は、111Inほど安定には99mTcにキレート化しない(Goldsmith、1997)。
【0032】
ヒトにおける最適な放射線画像化のために、多くの因子が考慮されなければならない。検出の効率を最大にするために、100〜200keVの範囲でγエネルギーを放射する放射性核種が好ましい。患者に対する吸収される放射線量を最小にするために、放射性核種の物理的半減期は、画像化手順が可能である限り短くあるべきである。検査が任意の日にそしてその日の任意の時間で行われ得るために、臨床場所においていつでも利用可能な放射性核種の供給源を有することが有利である。99mTcは、140keVでγ線を放射し、6時間の物理的半減期を有し、そしてモリブデン−99/テクネチウム−99m発生器を使用して、現場で容易に利用可能であるので、好ましい放射性核種である。
【0033】
ビスアミノエタンチオール4座配位リガンド(ジアミノジチオール化合物とも呼ばれる)は、2つのチオール硫黄および2つの窒素原子に対するオキソテクネチウム基の効率的な結合に基づいて、非常に安定なTc(V)O−錯体を形成することが公知である(Davisonら、1980;1981;Verbruggenら、1992)。99mTc−L,L−エチレンジシステイン(99mTc−EC)は、最近成功したN2S2キレートの例である(Verbruggenら、1992;Van Neromら、1993;Surmaら、1994)。EC(新規な腎臓画像化剤)は、99mTcで容易に標識化され得、そして高い放射化学純度および安定性で効率的に標識され得、そして活性な管輸送によって腎臓を通って排出され得る(Verbruggenら、1992;Van Neromら、1993;Surmaら、1994;Verbruggenら、1990;Van Neromら、1990;Jamarら、1993)。ECの他の用途は、PETのためのガリウム−68(陽電子放射体、t1/2=68分)および磁気共鳴画像化(MRI)のためのガドリニウム、鉄またはマンガンとキレート化される。
【0034】
本発明は、画像化のために、リガンドを特定の組織型に標的化させるための標識剤として99mTc−ECを利用する。ECを組織標的リガンドに結合する利点は、組織標的リガンドの特異的結合特性が目的の領域上で放射性シグナルを濃縮することである。標識化戦略としての99mTc−ECの使用が、実質的に任意の型の化合物で効果的であり得ることが考慮されるが、いくつかの提案される好ましいリガンドは、例示の目的のために本明細書中において提供される。本発明の99mTc−EC薬物結合体が腫瘍だけでなく、他の組織特異的状態(例えば、感染、低酸素組織(発作)、心筋梗塞、アポトーシス細胞、アルツハイマー病および子宮内膜症)をも画像化するのに有用であり得る。
【0035】
放射標識タンパク質およびペプチドは、先行技術において報告されている(Egeら、米国特許第4,832,940号、Abramsら、1990;Bakkerら、1990;Goldsmithら、1995,1997;Olexaら、1982;Ranbyら、1988;Hadleyら、1988;Leesら、1989;Sobelら、1989;Stuttle、1990;Maraganoreら、1991;Rodwellら、1991;Tubisら、1968;Sandrehagen 1983)。しかし、99mTc−ECは、本発明の前に、任意のリガンド(ジエチルエステル以外)と組み合わせて使用されなかった(Kabasakal、2000)。ECのジエチルエステルは、脳血流剤として使用された(Kikukawaら、2000)。
【0036】
放射性画像化について最適ではあるが、99mTcの化学は、他の元素の化学ほど完全には研究されておらず、このため、99mTcでの放射標識の方法は、豊富ではない。99mTcは、通常、モリブデン−99/テクネチウム−99m発生器から、99mTcペルテクネテート(TcO4−;+7の酸化状態のテクネチウム)として通常得られる。しかし、ペルテクネテートは、他の化合物と十分結合しない。従って、化合物を放射標識するために、99mTcペルテクネテートは、別の形態に変換されなければならない。テクネチウムが水溶液において安定なイオンを形成しないので、分解を妨げ、99mTcの不溶性の二酸化テクネチウムへの変換も、ペルテクネテートへ戻る変換も生じることを妨げるのに十分な動力学的および熱力学的安定性を有する配位錯体の形態でこのような溶液中で保持されなければならない。
【0037】
放射標識化のために、99mTc錯体が、テクネチウムイオンの周りのドナー基のすべてが単一のキレートリガンドによって提供される(この場合、エチレンジシステイン)であるキレートとして形成されることが特に有利である。これによって、キレート化された99mTcが、エチレンジシステインとリガンドとの間で直接かまたは単一のリンカーを介してのいずれかで、組織特異的リガンドに共有結合され得る。
【0038】
テクネチウムは、多くの酸化状態(+1、+2、+4、+5、+6および+7)を有する。+1の酸化状態である場合、Tc MIBIと呼ばれる。Tc MIBIは、熱反応を用いて作製されなければならない(Seaboldら、1999)。本発明の目的のために、Tcが+4の酸化状態であることが重要である。この酸化状態は、ECを用いるN2S2キレートを形成するのに理想的である。従って、本発明の薬物結合体と放射性テクネチウムとの錯体の形成において、テクネチウム錯体(好ましくは、99mTcペルテクネテートの塩)は、還元剤の存在下で、本発明の薬物結合体と反応する。
【0039】
本発明における使用のための好ましい還元剤は、Tcをその+4の酸化状態に還元するための塩化スズ(SnCl2)の形態のスズイオンである。しかし、他の還元剤(例えば、ジチオネートイオンまたは鉄(II)イオン)が本発明と組み合わせて有用であり得る。還元剤が固相還元剤であり得ることも意図される。還元剤の量は、重要であり得る。なぜなら、コロイドの形成を避けることが必要であるからである。例えば、約100〜約300mCiのTcペルテクネテート当たり、約10〜約100μgのSnCl2を使用することが好ましい。最も好ましい量は、約200mCiのTcペルテクネテートおよび約2mlの生理食塩水当たり、約0.1mgのSnCl2である。これは、代表的には、5人の患者における使用に十分なTc−EC組織特異的リガンド結合体を作製する。
【0040】
エチレンジシステインの酸化を防ぐために、組成物中に酸化防止剤を含むこともしばしば重要である。本発明と組み合わせた使用に好ましい酸化防止剤は、ビタミンC(アスコルビン酸)である。しかし、他の酸化防止剤(例えば、トコフェロール、ピリドキシン、チアミンまたはルチン)もまた有用であり得ることが意図される。
【0041】
一般的に、本発明と組み合わせた使用のためのリガンドは、酸のアームのいずれかまたは両方でECに結合し得るアミノ基またはヒドロキシ基のいずれかを有する。アミノ基またはヒドロキシル基が利用可能でない場合(例えば、酸官能基)、所望のリガンドは、リンカー(例えば、エチレンジアミン、アミノプロパノール、ジエチレントリアミン、アスパラギン酸、ポリアスパラギン酸、グルタミン酸、ポリグルタミン酸、またはリジン)を加えることによって本発明の方法を使用して、なおECに結合し、そして99mTcで標識される。本発明における使用に意図されるリガンドとしては、限定しないが、新脈管形成/抗新脈管形成リガンド、DNAトポイソメラーゼインヒビター、解糖マーカー、抗代謝リガンド、アポトーシス/低酸素リガンド、DNAインターカレーター、レセプターマーカー、ペプチド、ヌクレオチド、抗菌剤(例えば、抗生物質または抗真菌剤)、器官特異的リガンドおよび糖または薬剤(グルコースを模倣する)が挙げられる。
【0042】
EC自体は、水溶性である。本発明のEC−薬物結合体もまた水溶性であることが必要である。本発明と組み合わせて使用されるリガンドの多くは、水溶性であるか、またはECと結合した場合に水溶性化合物を形成する。しかし、組織特異的リガンドが水溶性でない場合、リガンドの溶解性を増加するリンカーが使用され得る。リンカーは、脂肪族アルコールまたは芳香族アルコール、アミンまたはペプチドにあるいはカルボキシルおよびまたはペプチドに結合し得る。リンカーは、ポリアミノ酸(ペプチド)またはアミノ酸(例えば、グルタミン酸、アスパラギン酸またはリジン)のいずれかであり得る。表1は、特異的薬物官能基に対する所望のリンカーを示す。
【0043】
【表1】

例:
A.エストラジオール、トポテカン、パクリタキセル、ラロキシルフェン(raloxfen)エトポシド
B.ドキソルビシン、マイトマイシンC、エンドスタチン、アネキシンV.LHRH、オクトレオチド、VIP
C.メトトレキサート、葉酸
本発明のEC組織特異的リガンド薬物結合体が他の放射性核種にキレート化し得、放射性核種療法に使用され得ることも想定される。一般的に、実質的に任意のα,β−放射体、γ−放射体、またはβ,γ−放射体が、本発明とともに使用され得ることが考えられる。好ましいβ,γ−放射体としては、166Ho、188Re、186Re、153Sm、および89Srが挙げられる。好ましいβ−放射体としては、90Yおよび225Acが挙げられる。好ましいγ−放射体としては、67Ga、68Ga、64Cu、62Cuおよび111Inが挙げられる。好ましいα−放射体としては、211Atおよび212Biが挙げられる。常磁性物質(例えば、Gd、MnおよびFe)が、本発明と組み合わせた使用のためにECとキレート化され得ることも想定される。
【0044】
錯体およびこのような錯体を調製するための手段は、標識される本発明のEC−組織特異的リガンドの所定量および99mTcで結合体を標識するのに十分な量の還元剤を含む密封バイアルを備えるキット形態で便利に提供される。本発明に従う99mTc標識シンチグラフィ画像化剤は、EC−組織特異的結合体および還元剤を含むバイアルへの適切な量の99mTcまたは99mTc錯体の添加、および本明細書中、以下の実施例1に記載される条件下での反応によって調製され得る。キットはまた、例えば、浸透圧を調節するための薬学的に受容可能な塩、緩衝液、保存剤、酸化防止剤などのような従来の薬学的補助物質を含み得る。キットの成分は、液体形態、凍結形態または乾燥形態であり得る。好ましい実施形態において、キット成分は、凍結乾燥形態で提供される。
【0045】
本発明によって提供される放射性標識試薬または結合体は、適切な量の放射能を有して提供される。99mTc放射性錯体を形成する際に、1mL当たり約0.01ミリキュリー(mCi)〜約300mCiの濃度で放射能を含む溶液で放射性錯体を形成することが一般的に好ましい。
【0046】
本発明によって提供される99mTc標識シンチグラフィ画像化剤は、哺乳動物の身体における部位を視覚化するために使用され得る。本発明に従って、99mTc標識シンチグラフィ画像化剤は、単一の単位注射可能線量で投与される。当業者に公知の任意の通常のキャリア(例えば、滅菌生理食塩水溶液または血漿)は、本発明に従って、種々の器官、腫瘍などを診断的に画像化するための注射可能溶液を調製するための放射標識の後に、利用され得る。一般的に、投与される単位線量は、約0.01mCi〜約300mCi、好ましくは10mCi〜約200mCiの放射能を有する。単位投薬量で注入される溶液は、約0.01mL〜約10mLである。静脈内投与の後に、器官または腫瘍のインビボでの画像化は、所望である場合、放射標識試薬が、患者に導入された後の数時間でまたはそれより長くにおいて行われ得る。大部分の場合、十分な量の投与線量が、シンチフォトグラフをとることを可能にするように約0.1時間以内に画像化される領域に蓄積する。診断目的または予後判定目的のためのシンチグラフィ画像化の任意の従来の方法が、本発明に従って利用され得る。
【0047】
本発明の99mTc−EC標識戦略はまた、予後判定の目的のために使用され得る。ECが癌化学療法のために選択した公知の薬物(例えば、表2に列挙されるもの)に結合され得ることが想定される。次いで、これらのEC−薬物結合体は、99mTcで放射標識され得、そして腫瘍を有する患者に投与され得る。標識EC−薬物結合体は、腫瘍に特異的に結合する。画像化は、特定の患者の特定の腫瘍に対する癌化学療法薬物の効力を決定するために実行され得る。この方法において、医師は、続行するどの処置様式がもっとも有効か、どの化学療法薬物がもっとも有効かを迅速に決定し得る。これは、薬物を選択する工程および1回の化学療法を施す工程を包含する現在の方法に対して劇的な改善を表す。これは、薬物の効果が決定され得る前の、数か月の患者の時間および数千ドルに関係する。
【0048】
本発明によって提供される99mTc標識EC−組織特異的リガンド結合体および錯体は、水性生理食塩水媒体のような静脈内注射のための任意の従来の媒体で、または血漿媒体において静脈内で投与され得る。このような媒体はまた、例えば、浸透圧を調節するための薬学的に受容可能な塩、緩衝液、保存剤、酸化防止剤などのような従来の薬学的補助物質を含み得る。好ましい媒体には、ノーマルセーラインおよび血漿がある。
【0049】
特定の好ましい標的化戦略は、さらに詳細に以下に議論される。
【0050】
(腫瘍葉酸レセプター標的化)
放射標識リガンド(例えば、ペンテトレオチド(pentetreotide)および血管作用性腸ペプチド)は、細胞レセプターに結合し、これらのうちのいくつかは、腫瘍細胞において過剰発現される(BrittonおよびGranowska、1996;Krenningら、1995;Reubiら、1992;Goldsmithら、1995;Virgoliniら、1994)。これらのリガンドが免疫原性でなく、そして血漿から迅速に排除されるので、レセプター画像化は、抗体画像化と比較してより見込みがあるようである。
【0051】
葉酸および葉酸代謝拮抗薬(例えば、メトトレキサート)が、古典的な還元型葉酸キャリアシステムに加えて、高い親和性の葉酸レセプター(グリコシルホスファチジルイノシトール結合膜葉酸結合タンパク質)を介して、細胞に入る(Westerhofら、1991;Orrら、1995;HsuehおよびDolnick、1993)。葉酸レセプター(FR)は、多くの新生物細胞型(例えば、肺癌、乳房癌、卵巣癌、頚部癌、結腸直腸癌、鼻咽頭癌、腎腺癌、悪性黒色腫および上衣細胞腫)において過剰発現されるが、主に、いくつかの正常な分化組織(例えば、脈絡叢、胎盤、甲状腺および腎臓)において発現される(Orrら、1995;Weitmanら、1992a;Campbellら、1991;Weitmanら、1992b;Holmら、1994;Rossら、1994;Franklinら、1994;Weitmanら、1994)。FRは、葉酸結合タンパク質毒素、薬物/アンチセンスオリゴヌクレオチドおよびリポソームを、葉酸レセプターを過剰発現する腫瘍細胞に送達するために使用された(Ginobbiら、1997;LeamonおよびLow、1991;LeamonおよびLow、1992;Leamonら、1993;LeeおよびLow、1994)。さらに、抗T細胞レセプター抗体に結合される抗FR抗体を含む二特異的抗体は、T細胞をFR陽性腫瘍に標的化するために使用されており、現在、卵巣癌について臨床試験にある(Canevariら、1993;Bolhuisら、1992;Patrickら、1997;Coneyら、1994;Kranzら、1995)。同様に、この性質は、放射標識葉酸結合体(例えば、葉酸レセプター陽性腫瘍の画像化のための、67Gaデフェロキサミン−葉酸および111In−DTPA−葉酸)を開発するために鼓舞された(Mathiasら、1996;Wangら、1997;Wangら、1996;Mathiasら、1997b)。これらの薬剤を用いた限定されたインビトロおよびインビボ研究の結果は、葉酸レセプターが、腫瘍画像化のための潜在的な標的であり得ることを示唆する。本発明において、本発明者らは、一連の新規な葉酸レセプターリガンドを開発した。これらのリガンドは、99mTc−EC−葉酸、99mTc−EC−メトトレキセート(99mTc−EC−MTX)、99mTc−EC−トムデックス(tomudex)(99mTc−EC−TDX)である。
【0052】
(腫瘍低酸素標的化)
腫瘍細胞は、酸素の非存在化におけるよりも酸素の存在下において従来の照射により感受性である;腫瘍内の少ない割合の低酸素細胞でさえ、放射線に対する応答を制限し得る(Hall、1988;Bushら、1978;Grayら、1953)。低酸素放射線抵抗性は、多くの動物腫瘍において示されたが、ヒトにおいては少しの腫瘍型のみで示された(Dische、1991;Gatenbyら、1988;Nordsmarkら、1996)。ヒト腫瘍における低酸素の発生は、大部分において、組織学的知見および動物腫瘍研究から推論された。低酸素のインビボ実証は、酸素電極を用いた組織測定を必要とし、そしてこれらの技術の侵襲性は、それらの臨床的適用を限定している。
【0053】
ミソニダゾール(MISO)は、低酸素細胞増感剤であり、そして異なる放射性同位体(例えば、18F、123I、99mTc)でのMISOの標識は、PETまたは平面(planar)シンチグラフィによる十分に酸素化された活性腫瘍から、低酸素だか代謝的に活性な腫瘍を区別するのに有用であり得る。[18F]フルオロソニダゾール(FMISO)は、腫瘍低酸素を評価するためにPETとともに使用される。最近の研究は、PET([18F]FMISOによる細胞酸素含有量をモニターするその能力を有する)が、放射線に対する腫瘍応答を推測するための高い可能性を有することを示した(Kohら、1992;Valkら、1992;Martinら、1989;Raseyら、1989;Raseyetalら、1990;Yangら、1995)。PETは、視準なしで高い分解能を提供するが、臨床設定においてPET同位体を使用するコストは、ひどく高い。ヨウ素でのMISOの標識化が選択されたが、甲状腺組織における高い取り込みが観測された。従って、同位体があまり高価でなく、そして大部分の主用な医療設備において容易に入手可能である平面シンチグラフィのための化合物を開発することが望ましい。本発明において、発明者らは、99mTc−EC−2−ニトロイミダゾールおよび99mTc−EC−メトロニダゾールの合成を提供し、そして腫瘍低酸素マーカーとしてそれらの潜在的な使用を示す。
【0054】
(癌のペプチド画像化)
ペプチドおよびアミノ酸は、種々の型の腫瘍の画像化において首尾よく使用されてきた(Westerら、1999;CoenenおよびStocklin、1988;Radererら、1996;Lambertら、1990;Bakkerら、1990;StellaおよびMathew、1990;Butterfieldら、1998;Piperら、1983;Mochizukiら、DickinsonおよびHiltner、1981)。グルタミン酸ベースのペプチドは、癌処置のための薬物キャリアとして使用されてきた(StellaおよびMathew、1990;Butterfieldら、1998;Piperら、1983;Mochizukiら、1985;DickinsonおよびHiltner、1981)。ホレートのグルタメート部分が分解し、そしてインビボでポリグルタメートを形成することは公知である。次いで、このポリグルタメートは、ホレートに再結合されて、ホリルポリグルタメートを形成し、このホリルポリグルタメートは、グルコース代謝に関係する。グルタミン酸ペプチドを標識することは、腫瘍の悪性度を区別する際に有用であり得る。本発明において、本発明者らは、EC−グルタミン酸ペンタペプチドの合成を報告し、そして腫瘍の画像化におけるその潜在的な使用を評価する。
【0055】
(腫瘍アポトーシス細胞の画像化)
アポトーシスは、化学療法および放射線を用いる癌の処置の間に生じる(Lennonら、1991;Abramsら、1990;Blakenbergら、1998;Blakenbergら、1999;TaitおよびSmith、1991)。アネキシンVは、ホスホチジルセリンに結合することが知られており、このホスホチジルセリンは、腫瘍アポトーシス細胞によって過剰発現される(Blakenbergら、1999;TaitおよびSmith、1991)。アネキシンVによるアポトーシスの評価は、治療の効力(例えば、疾患の進行または後退)を評価するために有用である。本発明において、本発明者らは、99mTc−EC−アネキシンV(EC−ANNEX)を合成し、そして腫瘍画像化におけるその潜在的な使用を評価する。
【0056】
(腫瘍新脈管形成の画像化)
新脈管形成は、部分的に、腫瘍増殖および転移の発生の原因である。抗有糸分裂性化合物は、抗脈管形成性であり、そして抗癌薬物としてのそれらの潜在的な使用について知られている。これらの化合物は、細胞サイクルの分裂期の間の細胞分裂を阻害する。細胞機能の生化学的プロセス(例えば、細胞分裂、細胞運動性、分泌、線毛運動およびべん毛運動、細胞内輸送、ならびに細胞の形態の維持)の間に、微小管が関係する。抗有糸分裂性化合物は、高い親和性で、微小管タンパク質(チューブリン)に結合し、微小管アセンブリを乱し、そして増幅細胞の有糸分裂の停止を引き起こすことが知られている。従って、抗有糸分裂性化合物は、微小管インヒビターとして、または紡錘体毒として、みなされる(Lu、1995)。
【0057】
多くの型の抗有糸分裂性化合物は、チューブリンに結合することによって、微小管アセンブリ−ディスアセンブリを制御する(Lu、1995;Gohら、1998;Wangら、1998;Rowinskyら、1990;Imbert、1998)。コルヒチノイド(colchicinoid)のような化合物は、コルヒチン結合部位上のチューブリンと相互作用し、そしてチューブリンの構築を阻害する(Lu、1995;Gohら、1998;Wangら、1998)。コルヒチノイドの中で、コルヒチンは、急性痛風の予防を処置するために使用される有効な抗炎症性薬物である。コルヒチンはまた、慢性骨髄性白血病において使用され得る。コルヒチノイドは、特定の型の腫瘍増殖に対して強力であるが、臨床的な治療可能性は、治療的効果および毒性効果を分離することが不可能であるために、制限される(Lu、1995)。しかし、コルヒチンは、細胞機能を評価するための生化学的ツールとして有用である。本発明のおいて、本発明者らは、チューブンリン機能に対する生化学的プロセスの評価のために、99mTc−EC−コルヒチン(EC−COL)を開発した。
【0058】
(腫瘍アポトーシス細胞の画像化)
アポトーシスは、化学療法および放射線を用いる癌の処置の間に生じる。アネキシンVは、ホスホチジルセリンに結合することが知られており、このホスホチジルセリンは、腫瘍アポトーシス細胞によって過剰発現される。アネキシンVによるアポトーシスの評価は、治療の効力(例えば、疾患の進行または後退)を評価するために有用である。従って、99mTc−EC−アネキシンV(EC−ANNEX)を開発した。
【0059】
(腫瘍低酸素症の画像化)
放射線治療の前の、画像化様式による腫瘍低酸素症の評価により、放射線増感剤または生体還元薬剤(bioreductive drug)(例えば、チラパザミン(tirapazamine)、マイトマイシンC)での処置のための患者を選択する合理的な手段が提供される。このような患者の選択は、低酸素症性腫瘍を有する患者のより正確な処置を可能にする。さらに、腫瘍サプレッサ遺伝子(P53)は、多重薬物耐性に関係する。化学療法の前後の組織病理学により画像化の知見とP53の過剰発現とを相関付けることは、後の腫瘍処置応答において有用である。99mTc−EC−2−ニトロイミダゾールおよび99mTc−EC−メトロニダゾルを開発した。
【0060】
(腫瘍新脈管形成の画像化)
新脈管形成は、部分的に、腫瘍増殖および転移の発生の原因である。抗有糸分裂性化合物は、抗脈管形成性であり、そして抗癌薬物としてのそれらの潜在的な使用について知られている。これらの化合物は、細胞サイクルの分裂期の間の細胞分裂を阻害する。細胞機能の生化学的プロセス(例えば、細胞分裂、細胞運動性、分泌、線毛運動およびべん毛運動、細胞内輸送、ならびに細胞の形態の維持)の間に、微小管が関係する。抗有糸分裂性化合物は、高い親和性で、微小管タンパク質(チューブリン)に結合し、微小管アセンブリを乱し、そして増幅細胞の有糸分裂の停止を引き起こすことが知られている。従って、抗有糸分裂性化合物は、微小管インヒビターとして、または紡錘体毒として、見なされる。コルヒチン(これは、強力な抗脈管形成薬剤である)は、微小管重合および中期での細胞停止を阻害することが知られている。コルヒチン(COL)は、細胞機能を評価するための生化学的ツールとして有用であり得る。次いで、99mTc−EC−COLを開発した。
【0061】
(発作に起因する低酸素症の画像化)
腫瘍細胞は、多少低酸素症的であるが、それは、酸素プローブが、圧力を測定することを必要とする。低酸素症的な条件を模倣するために、本発明者らは、発作を経験した11人の患者を、99mTc−EC−メトロニダゾル(99mTc−EC−MN)を使用して、画像化した。メトロニダゾルは、腫瘍低酸素症マーカーである。発作の範囲の組織は、酸素の欠乏に起因して、低酸素症的となる。このSPECT画像を、99mTc−EC−MNの注射の1時間後と3時間後に行った。全てのこれらの画像化研究は、積極的に、病変を局在化させた。CTは、非常に十分にまたは正確に、病変を示さなかった。いくつかの場合におけるMRIおよびCTは、病変のサイズを誇張した。以下は、3人の患者から選択されたケースである。
【0062】
ケース1. 59歳の男性患者は、左脳幹神経節に発作を患った。SPECT
99mTc−EC−MNは、注入後1時間で病変を同定し(図28)、これは、MRI
T1負荷画像に対応する(図29)。
【0063】
ケース2. 73歳の男性患者は、左中大脳動脈(MCA)領域において、発作を患った。SPECT 99mTc−EC−MNを、注入後1時間で、1日目および12日目(図30および31)で得た。これらの病変は、12日目での有意な増加した取り込みを示した。CTは、病変における広範な脳出血を示した。1日目と12日目との間に、顕著な差異は、観察されなかった(図32および図33)。これらの発見は、組織生存度に起因して、患者の症状(無酸素症から低酸素症へ)を改善したことを示す。SPECT 99mTc−EC−MNにより、CT画像化よりも良好な機能的情報が提供される。
【0064】
ケース3. 72歳の男性患者が、右MCAおよびPCA領域における発作を患った。SPECT 99mTc−EC−MNは、注入後1時間での病変を同定した(図34)。CTは、病変のサイズを誇張した(図35)。
【0065】
(腫瘍解糖標的化)
放射性標識リガンド(例えば、多糖(ネオマイシン、カナマイシン、トブラマイシン)および単糖(例えば、グルコサミン))は、細胞グルコーストランスポータ(腫瘍細胞で過剰発現される)に結合し、続いて、リン酸化される(Rogersら、1968;Fanciulliら、1994;Popoviciら、1971;Jonesら、1973;Hermannら、2000)。多糖(ネオマイシン、カナマイシン、トブラマイシン)および単糖(グルコサミン)によって誘導されるグルコースレベルは、インスリンによって抑制され得る(Haradaら、1995;Mollerら、1991;Offieldら、1996;Shankarら、1998;Yoshinoら、1999;Villevalois−Camら、2000)。これらのリガンドは、免疫原性ではなく、そして細胞質から迅速に浄化されるので、代謝画像化は、抗体画像化と比較して、有望であるようである。
【0066】
以下の実施例は、本発明の好ましい実施形態を例示することが意図される。以下の実施例に開示される技術は、本発明の実施において十分に機能することが本発明者らによって発見された技術を表し、従って、その実施のための好ましい様式を構成すると考えられ得ることが、当業者によって理解されるべきである。しかし、当業者は、本開示の観点から、多くの改変が、開示される特定の実施形態においてなされ得、そしてなおも本発明の意図および範囲から逸脱することなく同様または類似の結果を得ることを、理解する。
【実施例】
【0067】
(実施例1:腫瘍ホレートレセプター標的化)
(ECの合成)
ECを、以前に記載された方法(RatnerおよびClarke、1937;Blondeauら、1967;各々は、本明細書中に参考として援用される)に従って、2工程合成で調製した。前駆体L−チアゾリジン−4−カルボン酸を、合成した(m.p.195℃、報告値196−197℃)。次いで、ECを、調製した(m.p.237℃、報告値251−253℃)。この構造を、1H−NMRおよび高速原子ボンバードメント質量分析法(FAB−MS)によって確認した。
【0068】
(メトトレキサートのアミノエチルアミドアナログ(MTX−NH2)の合成)
MIX(227ma、0.5mmol)を、1mlのHCl溶液(2N)に溶解した。pH値を、<3とした。この攪拌溶液に、2mlの水および4mlのN−エトキシカルボニル−2−エトキシ−1,2−ジヒドロキノリン(EEDQ、メタノール中6.609%、1mmol)を、室温で添加した。エチレンジアミン(EDA、0.6 ml、10mmol)をゆっくりと添加した。この反応混合物を、一晩攪拌し、そして溶媒を減圧下でエバポレートした。未処理の固体物質を、ジエチルエーテル(10ml)、アセトニトリル(10ml)および95%エチルアルコール(50ml)で洗浄して、未反応のEEDQおよびEDAを除去した。次いで、生成物は、凍結乾燥によって乾燥し、そしてさらに精製することなく使用した。この生成物を、210mg(84.7%)であり、黄色粉末であった。生成物の融点:195−198℃(分解、MIX);1H−NMR(D2O)δ 2.98−3.04(d,8H,−(CH2)2CONH(CH0)2NH2),4.16−4.71(m,6H,−CH2−プテリジニル,芳香族−NCH3,NH−CH−COOHグルタメート),6.63−6.64(d,2H,芳香族−CO),7.51−753(d,2H.芳香族−N),8.36(s,1H,プテリジニル)。FAB MS m/z C22H28,N10,O4(M)+についての計算値496.515,実測値496.835。
【0069】
(ホレートのアミノエチルアミドアナログ(ホレート−NH2)の合成)
葉酸二水和物(1g、2.0mmol)を、10mlの水の添加した。pH値を、HCl(2N)を使用して、2に調節した。この攪拌溶液に、N−エトキシカルボニル−2−エトキシ−1,2−ジヒドロキノリン(EEDQ、10mlメタノール中1g、4.0mmol)およびエチレンジアミン(EDA、1.3ml、18mmol)をゆっくりと添加した。この反応混合物を、室温で一晩攪拌した。溶媒を、減圧下でエバポレートした。生成物を、メタノール(50ml)中で沈澱させ、そしてさらに、アセトン(100ml)で洗浄して、未反応のEEDQおよびEDITを除去した。次いで、この生成物を、凍結乾燥し、そしてさらに精製することなく使用した。ニンヒドリン(メタノール中2%)スプレーは、陽性のアミノ基を示した。この生成物は、0.6g(収率60%)であり、黄色粉末であった。生成物の融点:250℃(分解)。1H−NMR(D2O)δ 1.97−2.27(m,2H,ホレートの−CH2グルタメート),3.05−3.40(d,6H,−CH2CONH(CH2)2NH2),4.27−4.84(m,3H,−CH2−プテリジニル,NH−CH−COOHグルタメート),6.68−6.70(d,2H,芳香族−CO)、7.60−7.62(d,2H,芳香族−N),8.44(s,1H,プテリジニル)。FAB MS m/z C21H25N9,O5(M)+のついての計算値:483,実測値483.21。
【0070】
(エチレンジシステイン−ホレート(EC−ホレート)の合成)
ECを溶解するために、NaOH(2N、0.1ml)を、水(1.5ml)中のEC(114ma、0.425mmol)の攪拌溶液に添加した。この無色の溶液に、スルホ−NHS(92.3mg、0.425mmol)およびEDC(81.5mg、0.425mmol)を添加した。ホレート−NH2(205mg、0.425mmol)を、次いで、添加した。この混合物を、室温で24時間攪拌した。この混合物を、500の分子カットオフを有するSpectra/POR分子多孔性膜(Spectrum Medical Industries Inc.,Houston,TX)を使用して、48時間透析した。透析の後、生成物を凍結乾燥した。この生成物は、116mg(収率35%)であった。m.p.195℃(分解);1H−NMR(D2O)δ 1.98−2.28(m,2H,ホレートの−CH2グルタメート),2.60−2.95(m,4HおよびECの−CH2−SH),3.24−3.34(m,10H,−CH2−CO,ホレートのエチレンジアミンおよびECのエチレンジアミン),4.27−4.77(m,5H,−CH−プテリジニル,ホレートのNH−CH−COOHグルタメートおよびECのNH−CH−COOH),6.60−6.62(d,2H,芳香族−CO),7.58−7.59(d,2H.芳香族−N),8.59(s,1H,プテリジニル)。C29H37N11S2O8Na2(8H2O)についての分析計算値,FAB MS m/z(M)+777.3(水なし)。C,37.79;H,5.75;N,16.72;S,6.95。実測値:m/z (M)+777.7(20),489.4(100)。C,37.40;H,5.42;N.15.43;S,7.58。
【0071】
(EC−ホレートおよびECの99mTcでの放射標識化)
99mTc−EC−ホレートの放射合成(radiosynthesis)を、必要とされる量の99mTc−過テクネチウム酸を、自家製キット(EC−ホレートの凍結乾燥残渣(3mg)、SnCl2(100μg)、Na2HPO4(13.5mg)、アスコルビン酸(0.5mg)およびNaEDTA(0.5mg)を含む)に添加することによって達成した。調製物の最終pHは、7.4であった。99mTc−ECをまた、ECの凍結乾燥残渣(3mg)、SnCl2(100μg)、Na2HPO4(13.5mg)、アスコルビン酸(0.5mg)およびpH10のNaEDTA(0.5mg)を含む自家製キットを使用することによって、得た。次いで、調製物のpHを7.4に調節した。放射化学純度を、各々、アセトン(系A)および酢酸アンモニウム(水中1M):メタノール(4:1)(系B)で溶出するTLC(ITLC SG,Gelman Sciences,Ann Arbor,MI)によって、決定した。radio−TLC (Bioscan,Washington,DC)分析から、放射化学純度は、両方の放射性医薬品について、>95%であった。Radio−TLCデータを表2に要約する。99mTc−EC−ホレートの合成を、図1に示す。
【0072】
(表2)
(癌化学療法のための選択された薬物)
以下の表は、USAおよびカナダにおける癌の治療のために使用される薬物およびそれらの主要な有害な影響を列挙する。列挙する選択された薬物は、Medical Letterコンサルタントの意見に基づいた。いくつかの薬物を、それらが、米国食品医薬品庁によって承認されていない指標について列挙する。抗癌薬物およびそれらの有害な影響については、以下の通りである。本発明の目的のために、これらの列挙は、例示を意味し、全てを網羅するものではないことを意味する。
【0073】
【表2】
* 化学療法は、中程度の活性のみを有する。
** 化学療法は、少ない活性のみを有する。
1 化学療法を伴うかまたは伴わないタモキシフェンは、一般に、閉経後エストロゲン−レセプター陽性の、モード陽性患者に対して推奨され、そしてタモキシフェンを伴うかまたは伴わない化学療法が、閉経前のモード陽性な患者に対して一般に推奨される。化学療法および/またはタモフェキシンを伴うアジュバント処置が、より大きな腫瘍または他の有害な予後指標を有するモード陰性患者に対して推奨される。
2 メゲストロールおよび他のホルモン剤は、タモキシフェンが作用しないいくらかの患者において有用であり得る。
3 高用量化学療法の後(Medical Letter,34:79,1982)。
4 直腸癌について、フルオロウラシル単独での処置の前後に、フルオロウラシル(fluoroutacil)および放射線を用いる手術後アジュバント処置。
5 手術切除、放射線治療、または両方と組合された場合にのみ、薬物が主要な活性を有する。
6 ビタミンAアナログである、ラクトラチノル(lactratinoln)(Acgutana)は、前腫瘍性病変(白斑症(leukoplakla))を制御し得、そして第二の原発性腫瘍の割合を減少し得る(SE Bannerら、J Natl Cancer Inst,88:140 1994)。
† 調査用途のためにUSAのみで利用可能。
7 高リスク患者(例えば、高カウント、細胞遺伝異常、成人)は、誘導、維持および「強化」(寛解の達成後のさらなる薬物の使用)のためのさらなる薬物を必要とし得る。さらなる薬物としては、シクロホスファミド(cyclophosphamida)、ミトキサトロンおよびチオグアニン(thloguanine)が挙げられる。イギリスにおける1つの大きな制御された試行の結果は、強化が、ALLの全ての子供の生存率を改善し得ることを示唆する(JM Chasselleら、Lancet,34B:143,Jan 21,1995)。
8 初めに悪い予後を有する患者または寛解後に再発した患者。
9 急性前骨髄球白血病を有する何人かの患者は、トラチノイン(tratinoin)に対する完全な応答を有した。このような処置は、主に発熱および呼吸困難により特徴付けられる有毒な症状を引き起こし得る(RP Warrell,Jrら、N Engl J Med.328:177,1993)。
10 同種異系HLA同一同胞骨髄移植は、慢性期のCMLの患者の40%〜70%、加速期のCMLの患者の18%〜28%、急性転化の患者の15%未満を治癒し得る。骨髄移植後の疾患を有さない生存率は、50歳より大きい年齢、診断から3年より長い疾患の持続時間、および1抗原不適合または適合無関係のドナーの骨髄の使用により悪影響を受けた。インターフェロンαはまた、慢性期のCMLを有する患者(この患者は完全な細胞遺伝的応答(約10%)を達成する)において治癒的であり得;これは、新しく慢性期CMLと診断された80歳を越える年齢の患者、および同種異系骨髄移植の候補ではない全ての患者のために選択される処置である。化学治療単独は待機的である。
11 これらの組み合わせのいずれかを用いて第2の慢性期が達成される場合、同種異系骨髄移植が考慮されるべきである。第2の慢性期における骨髄移植は、CMLの患者の30%〜35%に対して治癒的であり得る。
12 極限段階のホジキン病(1段階および2段階)は、放射線治療によって治癒可能である。播種性疾患(3b段階および4段階)は、化学治療を必要とする。いくつかの中間段階および選択された臨床的状況が、この両方から利益を受ける。
【0074】
【表2−2】
† 研究用途のためのみにUSAで利用可能。
‡ 用量制限効果は太字である。皮膚反応(時折、重篤)、色素過剰、および眼毒性は、実質的に全ての非ホルモン性抗癌剤で報告された。他の薬物との悪い相互作用については、Medical Letter Handbook of Adverse Drug
Interactions,1995を参照のこと。
1. 研究用途のためのみにUSAで利用可能。
2. メゲストロールおよび他のホルモン剤は、タモキシフェンで失敗した場合の患者において有効であり得る。
3. 高用量化学治療後(Medical Letter,34:78,1992)。
4. 直腸癌について、フルオロウラシルのみを用いる処置の前および後の、フルオロウラシル+放射線を用いる手術後のアジュバント処置。
5. 薬物は、外科的切除、放射線治療またはその両方と組み合わせた場合にのみ、主な活性を有する。
6. ビタミンAアナログのイソトレチノイン(Accutane)は、新生物発生前のイシオン(ision)(白斑症)を制御し、そしてラットの第1の一次腫瘍を減少させ得る(SE Sennerら、J Natl Cancer Inst.88:140,1994)。
7. 高リスク患者(例えば、高カウント、細胞遺伝異常、成人)は、誘導、維持および「強化」(寛解の達成後のさらなる薬物の使用)のためのさらなる薬物を必要とし得る。さらなる薬物としては、シクロホスファミド、ミトキサトロンおよびチオグアニンが挙げられる。イギリスにおける1つの大きな制御された試行の結果は、強化がALLの全ての子供の生存率を改善し得ることを示唆する(JM Chassellaら、Lancet,348:143,Jan 21,1998)。
8. 初めに悪い予後を有する患者または寛解後に再発した患者。
9. 急性前骨髄球白血病を有する何人かの患者は、トレチノインに対する完全な応答を有した。このような処置は、主に発熱および呼吸困難により特徴付けられる有毒な症状を引き起こし得る(RP Warrell,Jrら、N Engl J Med.329:177,1993)。
10. 同種異系HLA同一同胞骨髄移植は、慢性期のCMLの患者の40%〜70%、加速期のCMLの患者の15%〜25%、急性転化の患者の15%未満を治癒し得る。骨髄移植後の疾患を有さない生存率は、50歳より大きい年齢、診断から3年より長い疾患の持続時間、および1抗原不適合または適合無関係のドナーの骨髄の使用により悪影響を受けた。インターフェロンαはまた、慢性期のCMLを有する患者(この患者は完全な細胞遺伝的応答(約10%)を達成する)において治癒的であり得;これは、新しく慢性期CMLと診断された50歳を越える年齢の患者、および同種異系骨髄移植の候補ではない全ての患者のために選択される処置である。化学治療単独は待機的である。
【0075】
(99mTcでのCE−MTXおよびEC−TDXの放射標識)
EC−ホレートの合成について記載された同じ方法を使用して、EC−MTXおよびEC−TDXを調製した。標識手順は、EC−MTXおよびEC−TDXを使用したこと以外は、99mTc−EC−ホレートの調製について記載された手順と同じである。99mTc−EC−MTXおよび99mTc−EC−TDXの合成を、図2および図3に示す。
【0076】
(99mTc−EC−ホレート、99mTc−EC−MTXおよび99mTc−EC−TDXの安定性アッセイ)
99mTc−EC−ホレート、99mTc−EC−MTXおよび99mTc−EC−TDXの安定性を、血清サンプル中で試験した。簡単には、740KBqの1mgの99mTc−EC−ホレート、99mTc−EC−MTXおよび99mTc−EC−TDXを、イヌ血清(200μl)中で、37℃で4時間インキュベートした。この血清サンプルを、水中50%メタノールで希釈し、そして放射性TLCを、上記のように、0.5時間、2時間および4時間で繰り返した。
【0077】
(組織分布研究)
雌性Fischer 344ラット(150±25g)(Harlan Sprague−Dawley,Indianapolis,IN)に、0.1mlの乳癌細胞を、25ゲージ針を使用して、013762腫瘍細胞株懸濁液(106細胞/ラット、Fixcherラットに特異的な腫瘍細胞株)から後脚に皮下接種した。腫瘍の直径が約1cmに達した場合、移植後14〜17日間研究を実施した。各手順の前に、動物をケタミン(10〜15mg/ラット、腹腔内)で麻酔した。
【0078】
組織分布研究において、各動物に、370〜550KBqの99mTc−EC−ホレートまたは99mTc−ECを静脈内注射した(n=3/時点)。各リガンドの注入質量は、10μg/ラットであった。放射性医薬の投与後20分、1時間、2時間および4時間において、麻酔した動物を屠殺し、そして腫瘍および選択した組織を切除し、計量し、そしてγ線計数器(Packard Instruments,Downers Grove,IL)で放射能をカウントした。各サンプル中のトレーサーの体内分布を、組織の湿潤重量1gあたりの注射用量の割合(%ID/g)として算出した。元の注射液の希釈サンプルのカウントを、参照のために使用した。腫瘍/非標的組織の計数密度比を、対応する%ID/g値から算出した。スチューデントt検定を使用して、2つの群間の差違の有意性を評価した。
【0079】
別の研究において、ブロッキング研究を行って、レセプター媒介型プロセスを決定した。ブロッキング研究において、99mTc−EC−ホレートを、腫瘍保有マウスに、50および150μmol/kgの葉酸と同時投与(i.v.)した(n=3/群)。注射の1時間後に動物を殺し、そしてデータを収集した。
【0080】
(シンチグラフィー画像化およびオートラジオグラフィー研究)
シンチグラフィー画像を、低エネルギー平行穴型コリメーターを備えるγ線カメラ(Siemens Medical Systems,Inc.,Hoffman Estates,IL)を使用して、18.5MBqの99mTc標識放射性トレーサーの静脈内注射後0.5時間、2時間および4時間で得た。
【0081】
全身オートラジオグラフィーを、定量用画像分析器(Cyclone Storage
Phosphor System,Packard,Meridian,CI.)によって得た。37MBqの99mTc−EC−ホレートの静脈内注射後1時間で動物を殺し、そして死体をカルボキシメチルセルロース(4%)中に固定した。冷凍した死体をクリオスタット(LKB 2250クリオマイクロトーム)中におき、そして100μmの冠状切片に切断した。各切片を解凍し、そしてスライドに取り付けた。次いで、このスライドを汎用リン光ストレージスクリーン(MP,7001480)に接触して配置し、そして15時間99mTc標識に暴露した。このリン光スクリーンを赤色レーザー光で励起し、そして以前に吸収されたエネルギーに比例する得られる青色光を記録した。
【0082】
(結果)
(99mTc−EC−ホレートの化学および安定性)
単純であり、高速かつ高収率のホレートのアミノエチルアミドおよびECアナログ、MTXおよびTDXを開発した。これらのアナログの構造を、NMRおよび質量分光分析により確認した。EC−ホレートの99mTcでの放射性合成を、高い(>95%)放射化学的純度で達成した。99mTc−ECホレートは、イヌ血清サンプル中で、20分、1時間、2時間および4時間において安定であることが見出された。
【0083】
(99mTc−EC−ホレートの体内分布)
体内分布研究により、20分〜4時間における腫瘍/血液計数密度比は、99mTc−EC−ホレートについて、徐々に増加し、一方、これらの値は、同じ時間における99mTc−ECについて減少することが示された(図4)。99mTC−EC−ホレートおよび99mTc−ECの%ID/g取込み値、腫瘍/血液比、および腫瘍/筋肉比を、それぞれ、表3および4に示した。
【0084】
(表3)
乳房腫瘍保有ラットにおける99mTc−EC−ホレートの体内分布
【0085】
【表3】
示される値は、3匹の動物のデーターの平均±標準偏差を表す。
【0086】
(シンチグラフィー画像化およびオートラジオグラフィー研究)
異なる時点において得られたシンチグラフィー画像は、99mTc−EC−葉酸を注射した群における腫瘍の可視化を示した。対照的に、99mTc−ECを注射した群においては、明らかな腫瘍取り込みが存在しなかった(図6)。両方の放射性トレーサーは、全ての画像における明らかな腎臓取込みを示した。99mTc−EC−葉酸の注射の1時間後に実施したオートラジオグラムは、腫瘍の活性を明らかに示した。
【0087】
(実施例2:腫瘍低酸素症標的化)
(2−(2−メチル−5−ニトロ−1Hイミダゾリル)エチルアミン(メトロニダゾルのアミノアナログMN−NH2)の合成)
メトロニダゾルのアミノアナログを、以前に記載された方法に従って合成した(Hayら、1994)。簡単に言えば、メトロニダゾルをメシル化アナログ(m.p.149〜150℃、報告値153〜154℃、TLC:酢酸エチル、Rf=0.45)に転換し、75%を得た。次いで、メシル化メトロニダゾルをアジ化ナトリウムと反応させて、アジドアナログ(TLC:酢酸エチル、Rf=0.52)を、収率80%で得た。このアジドアナログを、トリフェニルホスフィンによって還元し、そして所望のアミノアナログ(m.p.190〜192℃、報告値194〜195℃、TLC:酢酸エチル、Rf=0.15)を得た(60%)。ニンヒドリン(メタノール中2%)のスプレーは、MN−NH2のアミノ基の陽性を示した。構造を、1H−NMRおよび質量分析(FAB−MS)m/z 171(M+H,100)によって確認した。
【0088】
(エチレンジシステイン−メトロニダゾル(EC−MN)の合成)
水酸化ナトリウム(2N、0.2ml)を、水(5ml)中EC(134ma、0.50mmol)の攪拌溶液に添加した。この無色の溶液に、スルホ−NHS(217mg、1.0mmol)および1〜)C(192ma.1.0mmol)を添加した。次いで、MN−NH:二塩酸塩(340mg、2.0mmol)を添加した。この材料(mature)を室温で24時間攪拌した。この混合物を、500でのカットオフを有するSpectra/POR分子多孔質膜(Spectrum MedicalIndustries Inc.,Houston,TX)を使用して、48時間透析した。透析後、生成物を、凍結乾燥機(Labconco,Kansas City,MO)を使用してフリーズドライした。生成物の重量は、315mgであった(収率55%)。1H−NMR(D2O)δ2.93(s、6H、ニトロイミダゾール−CH3)、2.60〜2.95(m,4HおよびECの−CH2−SH)、3.30〜3.66(m、8H、ECのエチレンジアミンおよびニトロイミダゾール−CH2−CH2−NH2)、3.70〜3.99(t、2H、ECのNH−CH−CO)、5.05(t、4H、メトロニダゾル−CH2−CH2−NH2)、(s、2H、ニトロイミダゾールC=CH)。FAB MS m/z
572(M+,20)。EC−MNの合成スキームを、図7に示す。
【0089】
(3−(2−ニトロ−1H−イミダゾリル)プロピルアミン(ニトロイミダゾールのアミノアナログNIM−NH2)の合成)
2−ニトロイミダゾール(1g、8.34mmol)およびSc2CO3(2.9g,8.90mmol)を含むジメチルホルムアミド(DMF、50ml)中の攪拌溶液に、1,3−ジトシルプロパン(3.84g、9.99mmol)を添加した。この反応物を80℃で3時間加熱した。溶媒を減圧下でエバポレートし、そして残渣を酢酸エチル中に懸濁させた。固体を濾過し、溶媒を濃縮し、シリカゲルを充填したカラムに装填し、そして、ヘキサン:酢酸エチル(1:1)で溶出した。生成物である、3−トシルプロピル−(2−ニトロイミダゾール)を、m.p.108〜111℃で単離した(1.67g、収率57.5%)。1H−NMR(CDCl3)δ2.23(m、2H)、2.48(S、3H)、4.06(t、2H、J=5.7Hz)、4.52(t、2H、J=6.8Hz)、7.09(S、1H)、7.24(S、1H)、7.40(d、2H、J=8.2Hz)、7.77(d、2H、J=8.2Hz)。
【0090】
次いで、トシル化2−ニトロイミダゾール(1.33g、4.08mmol)を、DMF(10ml)中アジ化ナトリウム(Q29g、4.49mmol)と、100℃で3時間反応させた。冷却後、水(20ml)を添加し、そして生成物を酢酸エチル(3×20ml)から抽出した。溶媒を、MgSO4で乾燥し、そして乾固するまでエバポレートして、アジドアナログ(0.6g、75%、TLC:ヘキサン:酢酸エチル;1:1、Rf=0.42)を得た。1H−NMR(CDCl3)δ2.14(m、2H)、3.41(t、2H、J=6.2Hz)、4.54(t、2H、J=6.9Hz)、7.17(S、2H)。
【0091】
このアジドアナログ(0.57g、2.90mmol)を、テトラヒドロフラン(PHI;)中のトリフェニル(taphenyl)ホスフィン(1.14g、4.35mmol)によって、室温で4時間還元した。濃HCl(12ml)を添加し、そしてさらに5時間加熱した。生成物を、酢酸エチルと水との混合物から抽出した。この酢酸エチルをMgSO4で乾燥し、そして乾固するまでエバポレートして、アミン塩酸塩アナログを得た(360ma、60%)。ニンヒドリン(メタノール中2%)のスプレーは、NIM−NHのアミノ基の陽性を示した。1H−NMR(D2O)δ2.29(m、2H)、3.13(t、2H、J=7.8Hz)、3.60(br、2H)、4.35(t、2H、J=7.4Hz)、7.50(d、1H、J=2.1Hz)、7.63(d、1H、J=2.1Hz)。
【0092】
(エチレンジシステイン−ニトロイミダゾール(EC−NIM)の合成)
水酸化ナトリウム(2N、0.6ml)を、EC(134ma、0.50mmol)の水(2ml)中の攪拌溶液に添加した。この無色の溶液に、スルホ−NHS(260.6mg、1.2mmol)、EDC(230ma、1.2mmol)および水酸化ナトリウム(2N、1ml)を添加した。次いでNIM−NH2塩酸塩(206.6mg、1.0mmol)を添加した。この混合物を室温で24時間攪拌した。この混合物を、500でのカットオフを有するSpectra/POR分子多孔質膜(Spectrum Medical Industries Inc.,Houston,TX)を使用して、48時間透析した。透析後、生成物を、凍結乾燥機(Labconco,Kansas City,MO)を使用してフリーズドライした。生成物の重量は、594.8mgであった(収率98%)。EC−NIMの合成スキームを、図8Aに示す。構造を、1H−NMR(D2O)によって確認する(図8B)。
【0093】
(EC−MNおよびEC−NIMの、99mTcでの放射性同位元素標識)
99mTc−EC−MNおよび99mTc−EC−NIMの放射線合成を、必要量の過テクネチウム酸を、EC−MNまたはEC−NIMの凍結乾燥した残渣(3mg)、SnCl2(100μg)、Na2HPO4(13.5mg)、アスコルビン酸(0.5mg)およびNaEDTA(0.5mg)を含む自家製のキットに添加することによって、達成した。調製物の最終pHは、7.4であった。放射化学的純度を、それぞれアセトン(系A)および酢酸アンモニウム(水中1M):メタノール(4:1)(系B)で溶出するTLC(ITLAC SG、Gelman Sciences,Ann Arbor、MI)によって決定した。放射線TLC(Bioscan,Washington,DC)分析から、両方の放射線トレースに対して放射化学的純度は96%より高かった。
【0094】
([18F]FMISOおよび[131I]IMISOの合成)
[F]フッ素を、小容量の銀標的において富化された18O−水のプロトン照射を使用するサイクロトロンによって、生成した。トシルMISO(Hayら、1994)(20mg)を、アセトニトリル(1.5ml)に溶解し、クリプトフィックス−フッ化物錯体に添加した。加熱後、加水分解およびカラム精製により、25〜40%の収率(崩壊を補正した)の純粋な生成物を、60分のボンバードメントの終了(EOB)において、単離した。HPLCを、C−18 ODS−20Tカラム(4.6×25mm)(Waters Corp.,Milford,Mass)において、水/アセトニトリル(80/20)を用いて、1ml/分の流速を使用して実施した。キャリアが添加されていない生成物は、類似の条件下での非標識FMISOの保持時間(6.12分)に対応した。放射化学的純度は、99%より高かった。UV検出器(310nm)のもとで、他の不純物は存在しなかった。決定した[18F]FMISOの比活性は、既知の質量および放射能のサンプルのUVおよび放射能検出に基づいて、1Ci/μmolであった。
【0095】
[13I]IMISOを、同じ手順を使用して調製した(Cherifら、1994)。簡単に言えば、5mgのトシルMISOをアセトニトリル(1ml)に溶解し、そしてNa131I(0.1mlの1N NaOH中1mCi)(Dupont New England Nuclear,Boston.MA)を添加した。加熱および精製の後、生成物(収率60〜70%)を得た。放射線TLCは、溶出液としてクロロホルムメタノール(7:3)を使用して、最終生成物に対して0.01のRf値を示した。
【0096】
(99mTc−EC−MNおよび99mTc−EC−NIMの安定性アッセイ)
標識された99mTc−EC−MNおよび99mTc−EC−NIMの安定性を、血清サンプルにおいて試験した。簡単に言えば、740KBqの1mgの99mTc−EC−MNおよび99mTc−EC−NIMを、イヌ血清(200μl)中37℃で4時間インキュベートした。これらの血清サンプルを、水中50%メタノールで希釈し、そして放射線TLCを、上記のように、0.5時間、2時間および4時間において繰り返した。
【0097】
(99mTc−EC−MNの組織分布研究)
雌性Fischer344ラット(150±25g)(Harlan Sprague−Dawley,Indianapolis,IN)に、13762腫瘍細胞株懸濁液(106細胞/ラット、Fischerラットに対して特異的な腫瘍細胞株)由来の0.1mlの哺乳動物腫瘍細胞を、25ゲージの針を使用して後肢に皮下接種した。研究を、移植の14〜17日後に、腫瘍がおおよそ1cmの直径に達したときに実施した。ラットをケタミン(10〜15mg/ラット、腹腔内)で麻酔し、その後、それぞれの手順を行った。
【0098】
組織分布研究において、各動物に、370〜550KBqの99mTc−EC−MNまたは99mTc−EC(n=3/時点)を静脈内注射した。99mTc−EC−MNの注射した質量は、1匹のラットあたり10μgであった。放射線トレーサーの投与の0.5時間後、2時間後および4時間後に、これらのラットを屠殺し、そして選択した組織を切除し、秤量し、そして放射能を計数した。各サンプルにおけるトレーサーの生体分布を、組織含水重量1グラムあたりの注射用量の百分率(%ID/g)として、算出した。腫瘍/非標的組織の計数密度比を、対応する%ID/g値から算出した。このデータを、同じ動物モデルを使用して[18F]FMISOおよび[131I]IMISOと比較した。Studentのt検定を使用して、群間の差異の有意性を評価した。
【0099】
(シンチグラフィー画像化およびオートラジオグラフィー研究)
低エネルギーの平行穴コリメータを備えるγ線カメラ(Siemens Medical Systems,Inc.,Hoffman Estates,IL)を使用するシンチグラフィー画像を、18.5MBqの各放射線トレーサーの静脈内注射の0.5時間後、2時間後および4時間後に得た。
【0100】
全身のオートラジオグラムを、定量的画像分析器(Cyclone Storage Phosphor System,Packard,Meridian,CT)によって得た。37MBqの99mTc−EC−MNの静脈内注射に続いて、これらの動物を1時間で殺傷し、そしてその身体をカルボキシメチルセルロース(4%)に、以前に記載されたように固定した(Yangら、1995)。凍結させた身体を低温槽(LKB 2250クリオミクロトーム)に設置し、そして100μmの冠状切片に切断した。各切片を解凍し、そしてスライドに載せた。次いで、このスライドを多目的リン貯蔵スクリーン(MP、7001480)と接触させ、そして15時間露光した。
【0101】
99mTc−EC−NIMが化学療法に対する腫瘍応答をモニタリングし得るか否かを確認するために、腫瘍容量1.5cmを有するラットおよび卵巣腫瘍保有マウスの群を、パクリタキセル(40mg/kg/ラット、80mg/kg/マウス、静脈内)で、単一用量で処置した。画像を、パクリタキセル処置の4日後に撮像した。処置ありまたは処置なしでの、1グラムの腫瘍重量あたりの注射された用量の百分率を、決定した。
【0102】
(ポーラログラフィー酸素微小電極によるpO2測定)
腫瘍の低酸素症を確認するために、腫瘍内のpO2測定を、エッペンドルフコンピュータ化ヒストグラムシステムを使用して、実施した。2〜3の直線軌道の各々に沿った、20〜25のpO2測定を各腫瘍において0.4mmの間隔で実施した(合計40〜75の測定)。腫瘍pO測定を、3匹の腫瘍保有ラットにおいて実施した。オンラインコンピュータシステムを使用して、各軌道のポット測定を、この軌道に沿った測定点の位置に対して絶対的な値として表現し、そして2.5mmのクラス幅の0mmHgと100mmHgとの間のpO2ヒストグラムの相対頻度として、表した。
【0103】
(結果)
(99mTc−EC−MNおよび99mTc−EC−NIMの放射線合成および安定性)
EC−MNおよびEC−NIMの、99mTcでの放射線合成を、高い放射化学的純度(95%より高い)で達成し、放射化学的収率は100%であった。99mTc−EC−MNおよび99mTc−EC−NIM(図13)は、イヌ血清サンプルにおいて、0.5時間、2時間および4時間安定であることが見出された。分解生成物は、観察されなかった。MISOの放射性フッ素化および放射性ヨウ素化は、同じ前駆体を使用して、容易に達成された。両方の標識されたMISOアナログにおいて、放射化学的純度は、99%より高かった。
【0104】
(インビボ組織分布研究)
99mTc−EC−MNおよび99mTc−ECの、腫瘍保有ラットにおける組織分布を、表4および5に示す。イオン性99mTcに対する高い親和性に起因して、有意な一貫した甲状腺取り込みは存在せず、インビボでの99mTc−EC−MNの安定性を示唆する(表5)。
【0105】
【表4】
示される値は、3匹の動物からのデータの平均±標準偏差を表す。
【0106】
ブロッキング研究において、腫瘍/筋肉および腫瘍/血液の計数密度比は、葉酸の同時投与によって、有意に低下した(p<0.01)(図5)。
【0107】
【表5】
1.各ラットが、99mTc−EC−メトロニダゾル(10μCi、静脈内)を受容した。各値は、1グラムの重量あたりの注射用量(n=3)/時間間隔の百分率である。各データは、標準偏差を伴う3回の測定の平均を表す。
【0108】
生体分布研究は、0.54時間における腫瘍/血液および腫瘍/筋肉の計数密度比が、99mTc−EC−NM、[18F]FMISOおよび[131I]IMISOにおいては次第に増加し、一方でこれらの値は、同じ時間において、99mTc−ECについては変化しないことを示した(図9および図10)。[18F]FMISOは、注射後30分、2時間および4時間において、[131I]IMISOおよび99mTc−EC−MNの取り込み比と比較して、最も高い腫瘍対血液の取り込み比を示した。注射後2時間および4時間における99mTc−EC−MNおよび[131I]IMISOについての、腫瘍/血液および腫瘍/筋肉の比は、有意には異ならなかった(p<0.05)。
【0109】
(シンチグラフ画像化およびオートラジオグラフィー研究)
異なる時点において得られたシンチグラフィー画像は、99mTc−EC−MNおよび99mTc−EC−NIM群における腫瘍の可視化を示した。対照的に、99mTc−ECを注射した群においては、明らかな腫瘍の取り込みが存在しなかった(図11)。99mTc−EC−MNの注射の1時間後に実施したオートラジオグラムは、明らかに、腫瘍活性を示した(図12)。99mTc−EC−NMと比較して、99mTc−EC−NIMは、より高い腫瘍対バックグラウンドの比に起因して、より良好なシンチグラフィー画像を提供するようであった。乳房腫瘍保有ラットにおいて、腫瘍の取り込みは、99mTc−ECと比較して、99mTc−EC−NIM群において顕著に高かった(図14A)。1グラムの腫瘍の重量あたりの99mTc−EC−NIMの注射用量の百分率から得たれたデータは、コントロール群と比較した場合に、パクリタキセルで処置したラットにおいて、25%が取り込みを低下したことを示した(図14B)。
【0110】
卵巣癌保有マウスにおいて、パクリタキセルで処置したマウスにおける低下した腫瘍取り込みが存在した(図15Aおよび図15B)。類似の結果が、肉腫保有において観察された(図15Cおよび図15D)。従って、99mTc−EC−NIMは、パクリタキセル処置に対する腫瘍応答を評価するために、使用され得る。
【0111】
(ポーラログラフィー酸素微小電極によるpO2測定)
腫瘍の腫瘍内PO2測定は、腫瘍の酸素圧力が、正常筋肉の35±10mmHgと比較して、4.6±4.1mmHgの範囲であることを示した。このデータは、腫瘍が低酸素症であることを示す。
【0112】
(実施例3:癌のペプチド画像化)
(エチレンジシステイン−ペンタグルタミン酸(EC−GAP)の合成)
水酸化ナトリウム(1N,1ml)を、水(10ml)中のEC(200mg、0.75mmol)の撹拌溶液に添加した。この無色溶液に、スルホNHS(162mg、0.75mmol)およびEDC(143mg、0.75mmol)を添加した。次いでペンタグルタミン酸ナトリウム塩(M.W.750〜1500、Sigma Chemical Company)(500mg、0.67mmol)を添加した。混合物を24時間室温で撹拌した。混合物を、500のカットオフを有するSpectra/POR分子多孔膜(Spectrum Medical Industries Inc.,Houston,TX)を使用して48時間透析した。透析後、生成物を、凍結乾燥機(Labconco,Kansas City,MO)を使用して凍結乾燥した。塩形態の生成物は、0.95gの重さであった。EC−GAPの合成スキームは、図16に示す。
【0113】
(99mTc−EC−GAPの安定性アッセイ)
99mTcでのEC−GAPの放射性標識を、以前に記載されたのと同じ手順を使用して達成した。放射化学純度は、100%であった。99mTc−EC−GAPの安定性を、血清サンプル中で試験した。手短にいうと、740KBqの1mg99mTc−EC−GAPを、37℃で4時間、イヌ血清(200μl)中でインキュベートした。血清サンプルを、水中50%メタノールで希釈し、そして放射性TLCを、上記のように、0.5時間、2時間、および4時間で繰り返した。
【0114】
(シンチグラフィー画像化研究)
低エネルギーの、平行な穴のコリメータを備えたγカメラを使用して、シンチグラフィー画像を、18.5MBqの各放射性トレーサーの静脈内注射の0.5時間、2時間および4時間後に得た。
【0115】
(結果)
(99mTc−EC−GAPの安定性アッセイ)
99mTc−EC−GAPは、イヌ血清サンプル中で、0.5時間、2時間および4時間で安定であることが見出された。分解産物は、観察されなかった。
【0116】
(シンチグラフィー画像化研究)
異なる時点で得られたシンチグラフィー画像は、99mTc−EC−GAP群において腫瘍の可視化を示した。最適な取り込みは、投与後30分〜1時間である(図17)。
【0117】
(実施例4:腫瘍アポトーシス細胞の画像化)
(エチレンジシステイン−アネキシンV(EC−ANNEX)の合成)
炭酸水素ナトリウム(1N、1ml)を、EC(5mg、0.019mmol)の撹拌溶液に添加した。この無色溶液に、スルホNHS(4mg、0.019mmol)およびEDC(4mg、0.019mmol)を添加した。次いでアネキシンV(M.W.33kD、ヒト、Sigma Chemical Company)(0.3mg)を添加した。混合物を24時間室温で撹拌した。混合物を、10,000のカットオフを有するSpectra/POR分子多孔膜(Spectrum Medical Industries Inc.,Houston,TX)を使用して48時間透析した。透析後、生成物を、凍結乾燥機(Labconco,Kansas City,MO)を使用して凍結乾燥した。塩形態の生成物は、12mgの重さであった。
【0118】
(99mTc−EC−ANNEXの安定性アッセイ)
99mTcでのEC−ANNEXの放射性標識を、EC−GAPにおいて記載されたのと同じ手順を使用して達成した。放射化学純度は、100%であった。標識した99mTc−EC−ANNEXの安定性を、血清サンプル中で試験した。手短にいうと、740KBqの1mg99mTc−EC−ANNEXを、37℃で4時間、イヌ血清(200μl)中でインキュベートした。血清サンプルを、水中50%メタノールで希釈し、そして放射性TLCを、上記のように、0.5時間、2時間、および4時間で繰り返した。
【0119】
(シンチグラフィー画像化研究)
低エネルギーの、平行な穴のコリメータを備えたγカメラを使用して、シンチグラフィー画像を、18.5MBqの各放射性トレーサーの静脈内注射の0.5時間、2時間および4時間後に得た。使用した動物モデルは、乳房、卵巣および肉腫であった。乳房腫瘍および卵巣腫瘍を有する両方のラットは、高いアポトーシス性の細胞を過剰発現することが知られている。画像化研究を、腫瘍細胞接種後14日で行なった。腫瘍処置応答を確かめるために、画像化前のマウスを、パクリタキセル投与し(80mg/Kg、静脈内注射、14日目)、そして画像を18日目に取った。
【0120】
(結果)
(99mTc−EC−ANNEXの安定性アッセイ)
99mTc−EC−ANNEXは、イヌ血清サンプル中で、0.5時間、2時間および4時間で安定であることが見出された。分解産物は、観察されなかった。
【0121】
(シンチグラフィー画像化研究)
異なる時点で得られたシンチグラフィー画像は、99mTc−EC−ANNEX群において腫瘍の可視化を示した(図18〜20)。画像は、高度にアポトーシス性の細胞が、より多くの99mTc−EC−ANNEXの取り込みを有することを示した。高いアポトーシス(卵巣腫瘍保有)群(図19Aおよび図19B)および低アポトーシス(肉腫腫瘍保有)群(図20Aおよび図20B)におけるパクリタキセル処理前とパクリタキセル処理後との間に、腫瘍取り込みの顕著な差異は、存在しなかった。
【0122】
(実施例5:腫瘍新脈管形成の画像化)
((コルヒチンのアミノアナログ、COL−NH2)の合成)
コルヒチンの脱メチル化アミノアナログおよび脱メチル化ヒドロキシアナログを以前に記載された方法(Orrら、1995)に従って合成した。手短にいうと、コルヒチン(4g)を、25%硫酸を含む100mlの水中に溶解した。反応混合物を、100℃で5時間加熱した。混合物を、炭酸ナトリウムを用いて中和した。生成物をろ過し、そして、凍結乾燥機で乾燥し、2.4g(70%)の所望のアミノアナログ(融点153〜155℃、155〜157℃と報告されている)を生じた。ニンヒドリン(メタノール中2%)噴霧は、COL−NH2のアミノ基の陽性を示した。構造を、1H−NMRおよび質量分析法(FAB−MS)によって確かめた。
【0123】
【数1】
(エチレンジシステイン−コルヒチン(EC−COL)の合成)
水酸化ナトリウム(2N,0.2ml)を、水(5ml)中のEC(134mg、0.50mmol)の撹拌溶液に添加した。この無色溶液に、スルホNHS(217mg、1.0mmol)およびEDC(192mg、1.0mmol)を添加した。次いでCOL−NH2(340mg、2.0mmol)を添加した。混合物を24時間室温で撹拌した。混合物を、500のカットオフを有するSpectra/POR分子多孔膜(Spectrum Medical Industries Inc.,Houston,TX)を使用して48時間透析した。透析後、生成物を、凍結乾燥機(Labconco,Kansas City,MO)を使用して凍結乾燥した。塩形態の生成物は、315mgの重さであった(収率55%)。
【0124】
【数2】
EC−COLの合成スキームを、図21に示す。
【0125】
(EC−COLおよびECの99mTcを用いる放射性標識)
99mTc−EC−COLの放射性合成を、EC−COLの凍結乾燥残渣(5mg)、SnCl2(100μg)、Na2HPO4(13.5mg)、アスコルビン酸(0.5mg)およびNaEDTA(0.5mg)を含む自家製キットへ必要な量の99mTc−過テクネチウム酸を添加することによって達成した。調製物の最終pHは、7.4であった。99mTc−ECはまた、pH10で、ECの凍結乾燥残渣(5mg)、SnCl2(100μg)、Na2HPO4(13.5mg)、アスコルビン酸(0.5mg)およびNaEDTA(0.5mg)を含む自家製キットを使用することによって得た。次いで、調製物の最終pHを7.4に調整した。放射化学純度を、酢酸アンモニウム(水中1M):メタノール(4:1)を用いて溶出するTLC(ITLC SG,Gelman Sciences,Ann Arbor,MI)によって決定した。放射線−薄層クロマトグラフィー(TLC,Bioscan,Washington,DC)を使用して、両方の放射線トレーサについての放射化学純度を分析した。
【0126】
(99mTc−EC−COLの安定性アッセイ)
標識された99mTc−EC−COLの安定性を、血清サンプル中で試験した。手短にいうと、740KBqの5mg99mTc−EC−COLを、37℃で4時間、ウサギ(rabbinate)血清(500μl)中でインキュベートした。血清サンプルを、水中50%メタノールで希釈し、そして放射性TLCを、上記のように、0.5時間、2時間、および4時間で繰り返した。
【0127】
(組織分布研究)
雌性Fischer344ラット(150±25g)(Harlan Sprague−Dawley,Indianapolis,IN)を、25ゲージ針を使用して後肢に、13762腫瘍細胞株懸濁液(10細胞/ラット、Fischerラットに特異的な腫瘍細胞株)由来の0.1mlの乳房腫瘍細胞で皮下接種した。研究を、腫瘍が直径約1cmに達した、移植後14〜17日で行なった。ラットを、各手順の前に、ケタミン(10〜15mg/ラット、腹腔内)で麻酔した。
【0128】
組織分布研究において、各動物を、370〜550KBqの99mTc−EC−COLまたは99mTc−ECで静脈内注射した(n=3/時点)。99mTc−EC−COLの注射された質量は、1匹のラット当り10μgであった。放射線トレーサの投与0.5時間、2時間および4時間後、ラットを屠殺し、そして選択した組織を切り出し、秤量し、そして放射能についてカウントした。各サンプルにおけるトレーサーの体内分布を、1gの組織湿重量当りの注射された用量の割合として計算した(%ID/g)。腫瘍/非標的組織カウント密度比を、対応する%ID/g値から計算した。スチューデントt検定を使用して、群の間の差異の有意性を評価した。
【0129】
(シンチグラフィー画像化研究)
低エネルギーの、平行な穴のコリメータを備えたγカメラ(Siemens Medical Systems,Inc.,Hoffman Estates,IL)を使用して、シンチグラフィー画像を、300μCiの99mTc−EC−COLおよび99mTc−ECの静脈内注射の0.5時間、2時間および4時間後に得た。コンピューターで概略した目的の領域(ROI)を使用して、腫瘍取り込み対正常筋肉取り込みを定量する(1画素当りのカウント)。
【0130】
(結果)
(99mTc−EC−COLの放射性合成および安定性)
99mTcでのEC−COLの放射性合成を、高い(>95%)放射化学的純度で達成した(図21)。99mTc−EC−COLは、は、ウサギ血清サンプル中で、0.5時間、2時間および4時間で安定であることが見出された。分解産物は、観察されなかった(図22)。
【0131】
(インビボ体内分布)
乳房腫瘍保有ラットにおける99mTc−EC−COLおよび99mTc−ECのインビボ体内分布を、表4および6に示す。0.5時間、2時間および4時間における99mTc−EC−COLの腫瘍取り込み値(%ID/g)は、0.436±0.089、0.395±0.154および0.221±0.006であったが(表6)、99mTc−ECについての体内分布は、それぞれ0.342±0.163、0.115±0.002および0.097±0.005であった(表4)。時間の関数としての増加した腫瘍対血液比(0.52±0.12から0.72±0.07)および腫瘍対筋肉比(3.47±0.40から7.97±0.93)は、99mTc−EC−COL群において観察された(図23)。逆に、腫瘍対血液値および腫瘍対筋肉値は、同じ期間における99mTc−EC−COL群と比較した場合、99mTc−ECでの時間依存性の減少を示した(図24)。
【0132】
【表6】
*各ラットは、99mTc−EC−コルヒチン(10μCi、静脈内)を受けた。各値は、時間間隔あたり1グラム組織重量(n=3)当りの注射された用量のパーセントである。各データは、標準偏差を有する3つの測定値の平均値を示す。
【0133】
【表7】
(乳房腫瘍保有ラットにおける99mTc−EC−COLのγシンチグラフィー画像化)
投与後1時間での3匹の乳房腫瘍保有ラットにおけるインビトロ画像化研究は、腫瘍が99mTc−EC−COL群で十分に可視化され得ることを示したが(図25)、99mTc−EC群においてより少ない腫瘍取り込みが観察された(図26)。コンピューターで概略した目的の領域(ROI)は、99mTc−EC−COL群における腫瘍/バックグラウンド比が、99mTc−EC群よりも有意に高いことを示した(図27)。
【0134】
(腫瘍解糖標的化)
(実施例6:99mTc−EC−ネオマイシンの開発)
(ECの合成)
ECを、以前に記載された方法(RatnerおよびClarke,1937;Blondeauら、1967)に従って、2工程合成で調製した。前駆体(L−チアゾリジン−4−カルボン酸)を合成した(融点195°、196〜197°と報告されている)。次いでECを調製した(融点237°、251〜253°と報告されている)。構造を、1H−NMRおよび高速原子衝撃質量分析(FAB−MS)によって確認した。
【0135】
(エチレンジシステイン−ネオマイシン(EC−ネオマイシン)の合成)
水酸化ナトリウム(2N、0.2ml)を、水中(5ml)のEC(134mg、0.50mmol)の攪拌した溶液に添加した。この無色の溶液に、スルホ−NHS(217mg、1.0mmol)およびEDC(192mg、1.0mmol)を添加した。次いで、ネオマイシントリスルフェート塩(909mg、1.0mmol)を添加した。この混合物を、室温にて24時間攪拌した。この混合物を、500のカットオフ値を有するSpectra/POR分子多孔性膜(Spectrum Medical Industries Inc.,Houston,TX)を使用して48時間透析した。透析後、生成物を、凍結乾燥器(Labconco,Kansas City,MO)を使用して凍結乾燥した。生成物の重量は、720mg(収率83%)であった。EC−ネオマイシンの合成スキームを図36に示す。この構造を、1H−NMR(図38A〜B)、質量分析計(図39A〜B)および元素分析(Galbraith Laboratories,Inc,Knoxville,TN)によって確かめる。元素分析C39H75N10S4O19・15H2O(C、H、N、S)、計算値 C:33.77、H:7.58、N:10.11、S:9.23;実測値 C:32.44、H:5.90、N:10.47、S:10.58.EC−ネオマイシンのUV波長は、ECおよびネオマイシンと比較した場合に、270.5nmシフトした(図40A〜C)。
【0136】
(99mTcでのEC−MNおよびEC−ネオマイシンの放射標識)
99mTc−ECおよび99mTc−EC−ネオマイシンの放射合成(radiosynthesis)を、ECまたはEC−ネオマイシンの凍結乾燥化残渣(10mg)、SnCl2(100μg)、Na2HPO4(13.5mg)およびアスコルビン酸(0.5mg)を含む手製のキットに、必要量の99mTc−パラテクネテート(pertechnetate)を添加することによって達成した。次いで、0.1ml水中のNaEDTA(0.5mg)を添加した。調製物の最終pHは、7.4であった。放射化学的純度を、酢酸アンモニウム(水中1M):メタノール(4:1)で溶出されるTLC(ITLC SG,Gelman Sciences,Ann Arbor,MI)によって決定した。放射TLC(Bioscan,Washington,DC)分析(図41)およびHPLC分析(図42〜45)から、放射化学的純度は、両方の放射性トレーサーについて95%を超えた。
【0137】
(99mTc−ECおよび99mTc−EC−ネオマイシンの安定性アッセイ)
標識した99mTc−ECおよび99mTc−EC−ネオマイシンの安定性を、イヌ血清サンプル中で試験した。手短に言うと、740KBqの1mgの99mTc−ECおよび99mTc−EC−ネオマイシンを、イヌ血清中(200μl)で37℃にて4時間インキュベートした。この血清サンプルを、水中の50%メタノールで希釈し、そして放射−TLCを、上記のように0.5時間、2時間および4時間で繰り返した。
【0138】
(99mTc−EC−ネオマイシンの組織分布研究)
雌性Fishcer 344ラット(150±25g)(Harlan Sprague−Dawley,Indianapolis,IN)に、25ゲージ針を用いて、13762腫瘍細胞株懸濁液由来の0.1mlの乳房腫瘍細胞(106細胞/ラット、Fischerラットに特異的な腫瘍細胞株)を後脚に皮下接種した。腫瘍が約1cm直径に到達する移植後14〜17日に、研究を行った。ラットを、各手順の前にケタミン(10〜15mg/ラット、腹腔内)で麻酔した。
【0139】
組織分布研究において、各動物に、10〜20μCiの99mTc−ECまたは99mTc−EC−ネオマイシン(n=3/時点)を静脈内注射した。99mTc−EC−ネオマイシンの注射した量は、200μg/ラットであった。これらの放射トレーサーの投与後0.5時間、2時間および4時間で、ラットを屠殺し、そして選択した組織を切り出し、重さを量り、そして放射活性を計数した。各サンプル中のトレーサーの体内分布を、1g組織湿重量あたりの注射用量のパーセンテージ(%ID/g)として計算した。腫瘍/非標的組織の計数密度比を、対応する%ID/g値から計算した。99mTc−EC(表4)および遊離テクネチウム(表9)と比較した場合、腫瘍対組織比は、99mTc−EC−ネオマイシン群において時間の関数として増加した(表8)。
【0140】
(シンチグラフィー画像化研究)
低エネルギーの平行穴コリメーターを備えたγカメラ(Simens Medical
Systems,Inc.,Hoffman Estates,IL)を使用して、シンチグラフィー画像を、100μCiの各放射トレーサーの静脈内注射後0.5時間、2時間および4時間で得た。99mTc−ECと比較して、腫瘍中の高い取り込みが観察された(図37A)。予備的な臨床画像化研究を、乳癌を有する患者において行った。腫瘍は、99mTc−EC−ネオマイシンの投与後2時間で十分に可視化された(図37B)。
【0141】
(表8)
(乳房腫瘍保有ラットにおける99mTc−EC−ネオマイシンの体内分布)
【0142】
【表8】
示された値は、3匹の動物からのデータの平均±標準偏差を表す。
【0143】
(表9)
(乳房腫瘍保有ラットにおける99mTcパーテクネテートの体内分布)
【0144】
【表9】
示された値は、3匹の動物からのデータの平均±標準偏差を表す。
【0145】
(99mTc−EC−薬物結合体のインビトロ細胞取り込み)
99mTc−EC−薬物結合体の細胞取り込みを評価するために、80,000個の細胞(A549肺癌細胞株)を含む各ウェルに、2μCiの99mTc−EC−ネオマイシンおよび18F−FDGを添加した。インキュベーション後0.5〜4時間で、細胞を、リン酸緩衝化生理食塩水で3回洗浄し、次いで、トリプシンによって細胞を死なせた。次いで、細胞を、γカウンターによって計数した。99mTc−EC−ネオマイシンは、ヒト肺癌細胞株中で試験した薬剤中で最も高い取り込みを示した(図46)。
【0146】
(99mTc−EC−ネオマイシンおよび18F−FDGの細胞取り込みに対するグルコースの効果)
ネオマイシンは、グルコース吸収に影響を及ぼすことが知られている(Rogersら、1968;Fanciulliら、1994)。先の実験は、99mTc−EC−ネオマイシンが、ヒト肺癌細胞株(A549)中で18F−FDGよりも高い取り込みを有することを示した。99mTc−EC−ネオマイシンの取り込みがグルコース関連機構を介して媒介されるか否かを決定するために、グルコース(0.1mg〜2.0mg)を、50,000個の(乳房)細胞または80,000個の(肺)細胞のいずれかを含有する各ウェルに、2μCiの99mTc−EC−ネオマイシンおよび18F−FDGと共に添加した。インキュベーション後、細胞を、リン酸緩衝化生理食塩水で3回洗浄し、次いで、トリプシンによって細胞を死なせた。次いで、細胞を、γカウンターによって計数した。
【0147】
0.1〜2.0mg/ウェルの濃度でグルコースを添加することによって、2つの肺癌細胞株および1つの乳房細胞株における99mTc−EC−ネオマイシンの取り込みの減少が観察された。類似の結果が、18F−FDG群において観察された。99mTc−EC(コントロール)は、取り込みを全く示さなかった。これらの知見は、99mTc−EC−ネオマイシンの細胞取り込みが、グルコース関連機構を介して媒介され得ることを示唆する(図47、48Aおよび48B)。
【0148】
(実施例7:99mTc−EC−デオキシグルコースでの腫瘍の代謝画像化)
(EC−デオキシグルコース(EC−DC)の合成)
水酸化ナトリウム(1N、1ml)を、水中(5ml)のEC(110mg、0.41mmol)の攪拌した溶液に添加した。この無色の溶液に、スルホ−NHS(241.6mg、1.12mmol)およびEDC(218.8mg、1.15mmol)を添加した。次いで、D−グルコサミン塩酸塩(356.8mg、1.65mmol)を添加した。この混合物を、室温にて24時間攪拌した。この混合物を、500のカットオフ値を有するSpectra/POR分子多孔性膜(Spectrum Medical Industries Inc.,Houston,TX)を使用して48時間透析した。透析後、生成物を、凍結乾燥器(Labconco,Kansas City,MO)を使用して凍結乾燥した。塩形態の生成物の重量は、568.8mgであった。この合成スキームを図59に示す。この構造を、質量分析計(図60)およびプロトンNMR(図61および62)で確かめた。99mTc−EC−DCの放射化学的純度は、放射−TLC(図63)およびHPLC(図64および65)分析によって決定した場合100%であった。
【0149】
(ヘキソキナーゼアッセイ)
EC−DGがグルコースリン酸化を模倣するか否かを決定するために、ヘキソキナーゼアッセイを行った。既製のキット(Sigma Chemical Company)を使用して、EC−DG、グルコサミンおよびグルコース(標準)を、UV波長340nmでアッセイした。グルコース、EC−DGおよびグルコサミンは、陽性ヘキソキナーゼアッセイを示した(図66〜68)。
【0150】
(インビトロ細胞取り込みアッセイ)
インビトロ細胞取り込みアッセイを、ヒト肺癌細胞株(A549)を使用することによって行った。2μCiの99mTc−EC−DGおよび18F−FDGを、各80,000個の細胞を含有するウェルに添加した。インキュベーション後0.5〜4時間で、細胞を、リン酸緩衝化生理食塩水で3回洗浄し、次いで、トリプシンによって細胞を死なせた。次いで、細胞を、γカウンターによって計数した。99mTc−EC−DGの取り込みは、FDGに匹敵した(図69)。
【0151】
(99mTc−EC−デオキシグルコースおよび18F−FDGの細胞取り込みに対するd−グルコースおよびl−グルコースの効果)
99mTc−EC−デオキシグルコースの取り込みがd−グルコース関連機構を介して媒介されるか否かを評価するために、d−グルコースおよびl−グルコース(1mgおよび2.0mg)を、乳癌細胞または肺癌細胞(50,000個/0.5ml/ウェル)のいずれかを含有する各ウェルに、2μCiの99mTc−EC−デオキシグルコースおよび18F−FDGと共に添加した。2時間のインキュベーション後、細胞を、リン酸緩衝化生理食塩水で3回洗浄し、次いで、トリプシンによって細胞を死なせた。細胞を、γカウンターによって計数した。
【0152】
1〜2.0mg/ウェルの濃度でグルコースを添加することによって、乳癌細胞および肺癌細胞における99mTc−EC−デオキシグルコースおよび18F−FDGの取り込みの減少が観察された。しかし、l−グルコースによる両薬剤に対する影響は存在しなかった(図70〜73)。これらの知見は、99mTc−EC−デオキシグルコースの細胞取り込みが、d−グルコース機構を介して媒介されることを示唆する。
【0153】
(正常ラットにおける血中グルコースレベルに対するEC−デオキシグルコース負荷の効果)
先の実験は、99mTc−EC−デオキシグルコースの細胞取り込みが、FDGに類似することを示した。例えば、ヘキソキナーゼアッセイ(グルコースリン酸化)は、陽性であった。99mTc−EC−デオキシグルコースの取り込みは、d−グルコース機構を介して媒介される。この研究は、血中グルコースレベルがFDGまたはEC−デオキシグルコースのいずれかによって誘導され得、そしてインスリンによって抑制され得るか否かを決定することである。
【0154】
正常な健常Fischer 344ラット(重量145〜155g)を、実験前に一晩絶食させた。調製した塩酸グルコサミン、FDGおよびEC−デオキシグルコースの濃度は、60%および164%(mg/ml)であった。血中グルコースレベル(mg/dl)を、グルコースメーター(Glucometer DEX、Bayer Corporation,Elkhart,IN)によって測定した。この研究の前に、血中グルコースレベルのベースラインを得た。各ラット(n=3/群)に、1.2mmol/kgのグルコサミン、FDGおよびEC−デオキシグルコースを投与した。別の実験において、1群のラットに、EC−デオキシグルコースおよびFDGを投与した。インスリン(5ユニット)を、30分後に投与した。血液サンプルを、尾静脈から30分毎に投与後6時間まで収集した。
【0155】
血中グルコースレベルは、グルコサミン、FDGおよびEC−デオキシグルコースのボーラス静脈内投与によって誘導された。この血中グルコースレベルの増加は、EC−デオキシグルコースまたはFDGおよびインスリンの投与によって抑制され得た(図74および75)。
【0156】
(99mTc−EC−DGの組織分布研究)
乳房腫瘍保有動物モデルについて、雌性Fishcer 344ラット(150±25g)(Harlan Sprague−Dawley,Indianapolis,IN)に、25ゲージ針を用いて、13762腫瘍細胞株懸濁液由来の0.1mlの乳房腫瘍細胞(106細胞/ラット、Fischerラットに特異的な腫瘍細胞株)を後脚に皮下接種した。腫瘍が約1cm直径に到達する移植後14〜17日に、研究を行った。ラットを、各手順の前にケタミン(10〜15mg/ラット、腹腔内)で麻酔した。
【0157】
肺腫瘍保有動物モデルについて、各胸腺欠損ヌードマウス(20〜25g)に、25ゲージ針を用いて、A549腫瘍細胞株懸濁液由来の0.1mlのヒト肺腫瘍細胞(106細胞/マウス)を後脚に皮下接種した。腫瘍が約0.6cm直径に到達する移植後17〜21日に、研究を行った。
【0158】
組織分布研究において、各動物に、10〜20μCi(1匹のラットあたり)または1〜2μCi(1匹のマウスあたり)の99mTc−ECまたは99mTc−EC−DG(n=3/時点)を静脈内注射した。99mTc−EC−DGの注射した量は、1mg/ラットであった。これらの放射トレーサーの投与後0.5時間、2時間および4時間で、これらの齧歯動物を屠殺し、そして選択した組織を切り出し、重さを量り、そして放射活性を計数した。各サンプル中のトレーサーの体内分布を、1g組織湿重量あたりの注射用量のパーセンテージ(%ID/g)として計算した。腫瘍/非標的組織の計数密度比を、対応する%ID/g値から計算した。99mTc−EC(表4)および遊離テクネチウム(表9)と比較した場合、腫瘍対組織比は、99mTc−EC−DG群において、時間の関数として増加した(図76〜80)。
【0159】
(シンチグラフィー画像化研究)
低エネルギーの平行穴コリメーターを備えたγカメラを使用して、シンチグラフィー画像を、100μCiの放射トレーサーの静脈内注射後0.5時間、2時間および4時間で得た。使用した動物モデルは、乳房腫瘍保有ラットであった。99mTc−EC(コントロール群)と比較した場合、腫瘍は十分に可視化され得た(図81)。予備的な臨床画像化研究を、5人の患者(3人の脳腫瘍および2人肺疾患患者)において行った。画像を、投与後1〜2時間で得た。99mTc−EC−DGは、良性腫瘍を悪性腫瘍に対して区別し得た。例えば、悪性星状細胞腫は、高い取り込みを示した(図82A,82B、83Aおよび83B)。良性髄膜腫は、悪性髄膜腫と比較して乏しい取り込みを示した(図84AおよびB)。乏しい取り込みは、TBを有する患者において観察されたが(図85Aおよび図85B)、高い取り込みが、肺腫瘍において観察された(図86A,図86B、および図86C)。
【0160】
本明細書中に開示されそして特許請求される全ての組成物および/または方法は、本開示を考慮して過度の実験を伴うことなく作製および実行される。本発明の組成物および方法は、好ましい実施形態について記載されたが、これらの組成物および/または方法に対して、および本明細書中に記載される方法の工程または工程の順序において、本発明の概念、精神および範囲を逸脱することなく改変が適用され得ることは、当業者に明らかである。より詳細には、化学的および生理学的の両方で関連する特定の薬剤が、本明細書中に記載される薬剤と置換され得、同じかまたは類似の結果が達成されることが、明らかである。当業者に明らかな全てのこのような類似の置換物および改変物は、添付の特許請求の範囲に規定されるように、本発明の精神、範囲および概念内にあるとみなされる。
【0161】
(参考文献)
以下の参考文献は、これらが本明細書中に記載されるものに対して補助的な、例示的手順または他の詳細を提供する程度まで、本明細書中に参考として詳細に援用される。
【0162】
【表10】
以下の図面は、本明細書の一部を形成し、そして本発明の特定の局面をさらに示すために含まれる。本発明は、本明細書中に示される特定の実施形態の詳細な説明と組み合わせて、これらの図面のうちの1つ以上を参照することによって良好に理解され得る。
【技術分野】
【0001】
(発明の背景)
政府は、本発明の権利を所有しない。
【0002】
(1.発明の分野)
本発明は、一般に、標識、放射画像化(radioimaging)および化学合成の分野に関する。より詳細には、本発明は、標的化リガンドを放射画像化するための戦略に関する。本発明は、さらに、腫瘍の画像化および組織特異的障害の画像化において、これらの放射性標識したリガンドを使用する方法に関する。
【背景技術】
【0003】
(2.関連技術の説明)
シンチグラフ腫瘍画像化の改善は、さらなる腫瘍特異的放射性医薬品の開発によって、広範囲にわたって決定される。より高い腫瘍特異性に起因して、放射性標識したリガンドおよび放射性標識した抗体は、腫瘍のシンチグラグ検出の新しい時代を開き、そして広範囲の前臨床試験の開発および評価を受けている(非特許文献1〜3(Mathiasら、1996、1997a、1997b))。放射性核種画像化様式(陽子射出放出断層撮影、PET;単一光子放射型コンピュータ断層撮影、SPECT)は、放射性核種標識された放射性トレーサーの位置および密度をマッピングする診断用断層画像化技術である。CTおよびMRIは、腫瘍の位置および範囲に関するかなりの解剖的な情報を提供するが、これらの画像化様式は、浮腫、放射線壊死、グレーディング(grading)または神経膠症由来の侵襲性損傷を十分に区別し得ない。PETおよびSPECTは、代謝活性を測定することによって、腫瘍を局在化および特徴付けするために使用され得る。
【0004】
新しい腫瘍低酸素剤の開発は、一次性損傷および転移性損傷を検出するため、ならびに放射線応答性および再発までの時間を予測するために、臨床的に所望されている。現代の画像化様式のどれも、低酸素症を正確に測定しない。なぜならば、この腫瘍低酸素症の診断は、病理学的試験を必要とするからである。処置される各腫瘍において、少なくとも低酸素症のベースラインを知ることなく、低酸素腫瘍のための治療の成果を予測することは、しばしば困難である。Eppendorfポーラログラフ酸素微小電極は、腫瘍中の酸素電圧を測定し得るが、この技術は、侵襲性であり、かつ熟練した操作者を必要とする。さらに、この技術は、アクセス可能な腫瘍(例えば、頭部および首(頚部))でのみ使用され得、そして複数の読み取りが必要とされる。従って、腫瘍低酸素症を測定するための正確かつ容易な方法は、患者選択のために有用である。しかし、正常な組織に対する腫瘍の取込み比は、使用される放射性医薬品に依存して変化する。従って、新しい放射性医薬品を臨床的な実施に導入する場合に、低酸素症の標準的なEppendorf金電極測定を用いて、正常組織に対する腫瘍の取込み比を補正することが合理的である。
【0005】
[18F]FMISOは、頭部および頚部の腫瘍、心筋梗塞、炎症および脳虚血を診断するために使用されている(Martinら、1992;Yehら、1994;Yetら、1996;Liuら、1994)。正常組織に対する腫瘍の取込み比は、腫瘍低酸素症を評価するためのベースラインとして使用された(Yehら、1996)。[18F]FMISOを使用する腫瘍低酸素症は明確に実証されたが、新しい画像化剤を臨床的な実施に導入することは、容易に利用可能であることおよび同位体の費用などのいくつかの他の因子に依存する。[18F]FDGを使用する腫瘍代謝性画像化は明確に実証されたが、分子画像化剤を臨床的な実施に導入することは、容易に利用可能であることおよび同位体の費用などのいくつかの他の因子に依存する。[18F]フルオロデオキシグルコース(FDG)は、腫瘍、心筋梗塞、および神経学的疾患を診断するために使用されている。さらに、PET放射性合成は、ポジトロン同位体の半減期が短いので、迅速でなければならない。18F化学もまた、複雑である。この18F化学は、異なる分子において再現不可能である。従って、種々の薬物に結合体化し得るキレート剤を開発することが、理想的である。好ましい同位体は、低い費用(18Fの$50/mCiに対して$0.21/mCi)および低いエネルギー(18Fの571Kevに対して140Kev)に起因して99mTcである。99mTcは、99Mo発生器から容易に得られる。望ましい物理学的特性およびかなり低い値段に起因して、99mTcは、放射性医薬品を標識するのに好ましい。
【0006】
いくつかの化合物は、窒素キレート剤および硫黄キレート剤を使用して、99mTcで標識されている(Blondeauら、1967;Davisonら、1980)。ビス−アミノエタンチオール四座配位子リガンド(これはまた、ジアミノジチオール化合物とも呼ばれる)は、2個のチオール硫酸原子および2個のアミン窒素原子に対するオキソテクネチウム基の効率的な結合に基づいて、非常に安定なTc(V)O錯体を形成することで公知である。99mTc−L,L−エチレンジシステイン(99mTc−EC)は、N2S2キレートの最近、かつ有用な例である。ECは、放射性化学的に高い純度および安定性を有する、99mTcで容易におよび効率的に標識され得、そして活性な管状輸送体によって腎臓を介して排出される(Surmaら、1994;Van Neromら、1990、1993;Verbruggenら、1990、1992)。他のECの適用は、PET用のガリウム−68(ポジトロン放出、t1/2=68分)およびガドリニウム、磁気共鳴画像法(MRI)用の鉄もしくはマンガンでキレート化される。99mTc−EC−ネオマイシンおよび99mTc−EC−デオキシグルコースが開発され、そして腫瘍の特徴付けにおけるそれらの潜在的な使用が評価された。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Mathiasら,J Nucl Med,37:1003−1008,1996
【非特許文献2】Mathiasら,J Nucl Med,(Supplement)38:87P,1997a
【非特許文献3】Mathiasら,J Nucl Med,(Supplement)38:133P,1997b
【発明の概要】
【発明が解決しようとする課題】
【0008】
画像化用に組織を標的化するための新しい放射性標識の戦略を提供することによって、先行技術のこれらの欠点および他の欠点を克服すること。
【課題を解決するための手段】
【0009】
(発明の要旨)
本発明は、画像化用に組織を標的化するための新しい放射性標識の戦略を提供することによって、先行技術のこれらの欠点および他の欠点を克服する。本発明は、放射性標識した組織特異的リガンド、ならびにこの放射性標識したリガンドを作製するための方法および組織特異的疾患を画像化するためにこの放射性標識したリガンドを使用するための方法を提供する。
本発明は、組織特異的疾患画像化のための組成物を提供する。本発明のこの画像化組成物は、一般に、エチレンジシステインおよび酸アーム(acid arm)の一方または両方上のエチレンジシステインに結合体化された組織特異的リガンドでキレート化された放射性核種標識を含む。このエチレンジシステインは、放射性核種標識を有するN2S2キレートを形成する。当然、このキレート化した化合物は、放射性核種とキレート化号物との間にイオン結合を含む。用語「EC−組織特異的リガンド結合体」、「EC−誘導体」および「EC−薬物結合体」は、標識されていないエチレンジシステイン−組織特異的リガンド化合物をいうために、本明細書中で交換可能に使用される。本明細書中で使用される場合、用語「結合体」は、共有結合した化合物をいう。
【0010】
エチレンジシステインは、ビス−アミノエタンチオール(BAT)四座配位子である(これはまた、ジアミノジチオール(DADT)化合物としても公知である)。このような化合物は、2個のチオール硫酸原子および2個のアミン窒素原子に対するオキソテクネチウム基の効率的な結合に基づいて、非常に安定なTc(V)O錯体を形成することで公知である。この99mTc標識化ジエチルエステル(99mTc−L,L−ECD)は、脳内物質(brain agent)として公知である。99mTc−L,L−エチレンジシステイン(99mTc−L,L−EC)は、最も極性のある代謝物であり、尿中に迅速かつ効率的に排出されることが発見された。従って、99mTc−L,L−ECは、腎機能剤として使用されてきた(Verbruggenら、1992)。
【0011】
組織特異的リガンドは、哺乳動物または患者の体内に導入された場合、組織の特定の型に特異的に結合する化合物である。本発明の組成物は、実質的に任意の公知の組織特異的化合物を含み得ることが、想定される。好ましくは、本発明と組み合わせて使用される組織特異的リガンドには、抗癌剤、DNAトポイソメラーゼインヒビター、抗代謝剤、腫瘍マーカー、ホレート(folate)レセプター標的化リガンド、腫瘍アポトーシス細胞標的化リガンド、腫瘍低酸素症標的化リガンド、DNAインターカレーター(intercalator)、レセプターマーカー、ペプチド、ヌクレオチド、器官特異的リガンド、抗菌剤(例えば、抗生物質もしくは抗真菌剤)、グルタメートペンタペプチド、またはグルコースを模倣する薬剤がある。このグルコースを模倣する薬剤はまた、「糖(sugar)」とも呼ばれ得る。
【0012】
好ましい抗癌剤としては、メトトレキセート、ドキソルビシン、タモキシフェン、パクリタキセル、トポテカン(topotecan)、LHRH、マイトマイシンC、エトポシド、トムデックス(tomudex)、ポドフィロトキシン、ミトザントロン、カプトセシン(captothecin)、コルヒチン、エンドスタチン(endostatin)、フルダラビンおよびゲムシタビンが挙げられる。好ましい腫瘍マーカーとしては、PSA、ER、PR、AFP、CA−125、CA−199、CEA、インターフェロン、BRCA1、シトキサン(cytoxan)、p53、VEGF、インテグリン、エンドスタチン、HER−2/neu、アンチセンスマーカーまたはモノクローナル抗体が挙げられる。任意の他の公知の腫瘍マーカーまたは任意のモノクローナル抗体が本発明と組み合わせた使用に効果的であることを想定する。好ましいホレートレセプター標的化リガンドとしては、ホレート、メトトレキセートおよびトムデクスが挙げられる。好ましい腫瘍アポトーシス細胞標的化リガンドまたは腫瘍低酸素症標的化リガンドとしては、アネキシンV、コルヒチン、ニトロイミダゾール、マイトマイシンまたはメトロニダゾルが挙げられる。好ましい抗菌剤としては、グラム陽性細菌およびグラム陰性細菌に対して、アンピシリン、アモキシリン、ペニシリン、セファロスポリン、クリダマイシン、ゲンタマイシン、カナマイシン、ネオマイシン、ピマリシン(natamycin)、ナフシリン、リファンピン、テトラサイクリン、バンコマイシン、ブレオマイシン、およびドキシサイクリン、ならびに真菌に対して、アンホテリシンB、アマンタジン、ナイスタチン、ケトコナゾール、ポリミキシン(polymycin)、アシクロビル、およびガンシクロビルが挙げられる。グルコースまたは糖を模倣する好ましい薬剤としては、ネオマイシン、カナマイシン、ゲンタマイシン、プロマイシン(paromycin)、アミカシン、トブラマイシン、ネチルマイシン、リボスタマイシン、シソマイシン、ミクロマイシン、リビドマイシン、ジベカシン(dibekacin)、イセパマイシン(isepamicin)、アストロマイシン、アミノグリコシド、グルコースまたはグルコサミンが挙げられる。
【0013】
特定の実施形態において、エチレンジシステインと組織特異的リガンドとの間にリンカーを含むことが必要である。リンカーは、代表的に、薬物の水溶液中の水溶性を増加させるため、および薬物の親和性の変化を最小にするために使用される。この組成物の水溶性を増加させる任意のリンカーは、実質的に、本発明と組み合わせて使用するために想定されるが、このリンカーは、一般に、ポリアミノ酸、水溶性ペプチド、または単一のアミノ酸のいずれかである。例えば、組織特異的リガンド上の官能基、または薬物が、エストラジオール、トポテカン、パクリタキセルまたはラロキシフェンエトポシドのような脂肪族OHまたはフェノールOHである場合、このリンカーは、ポリグルタミン酸(MW 約750〜約15,000)、ポリアスパラギン酸(MW 約2,000〜約15,000)、ブロモエチルアセテート、グルタミン酸、またはアスパラギン酸である得る。この薬物の官能基は、脂肪族NH2もしくは芳香族NH2またはペプチド(例えば、ドキソルビシン、マイトマイシン C、エンドスタチン、アネキシン V、LHRH、オクトレオチド,およびVIP)である場合、このリンカーは、ポリグルタミン酸(MW 約750〜約15,000)、ポリアスパラギン酸(MW 約2,000〜約15,000)、グルタミン酸またはアスパラギン酸であり得る。この薬物の官能基が、カルボン酸またはペプチド(例えば、メトトレキセートまたは葉酸)である場合、このリンカーは、エチレンジアミン、またはリジンであり得る。
【0014】
画像化のための好ましい放射性核種は、99mTcであるが、他の放射性核種が、特に治療剤としての使用のために、EC組織特異的リガンド結合体または本発明のEC薬物結合体にキレート化され得ることが想定される。例えば、他の有用な放射性核種には、188Re、186Re、153Sm、166Ho、90Y、89Sr、67Ga、68Ga、111In、153Gd、および59Feがある。これらの組成物は、体内の特定の損傷部(例えば、乳癌、卵巣癌、前立腺癌(例えば、l86/l88Re−EC−ホレートを使用する)ならびに頭部および頚部の癌(例えば、186/188Re−EC−ニトロイミダゾールを使用する))に治療放射性核種を送達するために使用される。
【0015】
本発明の特定の実施形態としては、99mTc−EC−アネキシン V、99mTc−EC−コルヒチン、99mTc−EC−ニトロイミダゾール、99mTc−EC−グルタメートペンタペプチド、99mTc−EC−メトロニダゾル、99mTc−EC−ホレート、99mTc−EC−メトトレキセート、99mTc−EC−トムデクス、99mTc−EC−ネオマイシン、99mTc−EC−カナマイシン、99mTc−EC−アミノグリコシド、(グルコサミン、EC−デオキシグルコース)、99mTc−EC−ゲンタマイシン、および99mTc−EC−トブラマイシンが挙げられる。
【0016】
本発明は、さらに、画像化または治療用途のために、放射性標識したエチレンジシステイン薬物結合体またはエチレンジシステイン薬物誘導体を合成するための方法を提供する。この方法は、組織特異的リガンドを得る工程、このリガンドをエチレンジシステイン(EC)と混合して、EC組織特異的リガンド誘導体を得る工程、ならびに放射性核種および還元剤と共にEC組織特異的リガンド誘導体を混合して、放射性核種標識されたEC組織特異的リガンド誘導体を得る工程を包含する。この放射性核種は、N2S2キレートを介してECにキレート化される。この組織特異的リガンドは、上記のように直接的またはリンカーを介してのいずれかで、ECの酸アームの一方または両方のに結合体化される。この還元剤は、好ましくは、ジチオナイト(dithionite)イオン、スズイオンまたは鉄(II)イオンである。
【0017】
本発明は、さらに、画像化、治療用途のため、または診断用途もしくは予後用途のために組織特異的リガンドを標識するための方法を提供する。この標識方法は、組織特異的リガンドを得る工程、エチレンジシステイン(EC)と共に組織特異的リガンドを混合して、EC−リガンド薬物結合体を得る工程、および還元剤の存在下で、薬物結合体を99mTcと反応させて、エチレンジシステインとこの99mTcとの間にN2S2キレートを形成する工程を包含する。
【0018】
この実施形態の目的のために、組織特異的リガンドは、上記または本明細書中で議論されるいずれかのリガンドであり得る。還元剤は、任意の公知の還元剤であり得るが、好ましくは、ジチオナイトイオン、スズイオンまたは鉄(II)イオンである。
【0019】
別の実施形態において、本発明は、哺乳動物の体内のある部位を画像化する方法を提供する。この画像化方法は、99mTc標識されたエチレンジシステイン組織特異的リガンド結合体を含む組成物の診断有効量を投与する工程、およびその部位に局在化された99mTcからの放射能シグナルを検出する工程を包含する。この検出する工程は、代表的に、哺乳動物の体内にこの組成物を導入した約10分〜約4時間後に実施される。より好ましくは、この検出する工程は、哺乳動物の体内にこの組成物を注入した約1時間後に実施される。
【0020】
特定の好ましい実施形態において、この部位は、感染、腫瘍、心臓、肺、脳、肝臓、脾臓、膵臓、腸または任意の他の器官である。腫瘍または感染は、哺乳動物の体内のどこかに局在化され得るが、一般に、胸部、卵巣、前立腺、子宮内膜、肺、脳、または肝臓である。この部位は、ホレート陽性癌またはエストロゲン陽性癌であり得る。
【0021】
本発明はまた、放射性薬学的調製物を調製するためのキットを提供する。このキットは、一般に、予め決められた量のエチレンジシステイン組織特異的リガンド結合体組成物およびこの結合体を99mTcで標識するために十分な量の還元剤を含む、密閉されたバックまたは他の任意の種の適切な容器を含む。特定の場合において、このエチレンジシステイン組織特異的リガンド結合体組成物はまた、このエチレンジシステインと組織特異的リガンドとの間にリンカーを含む。この組織特異的リガンドは、任意の特異的な組織型(例えば、本明細書中で議論される組織型)に特異的に結合する任意のリガンドであり得る。リンカーがこの組成物中に含まれる場合、このリンカーは、本明細書中で記載されるようなリンカーであり得る。
【0022】
このキット成分は、任意の適切な形態(例えば、液体形態、凍結形態または乾燥形態)であり得る。好ましい実施形態において、このキット成分は、凍結乾燥形態で提供される。このキットはまた、抗酸化剤および/または捕捉剤を含み得る。この抗酸化剤は、公知の任意の抗酸化剤であるが、好ましくは、ビタミンCであり得る。捕捉剤はまた、残りの放射性核種を結合するために存在し得る。最も市販されているキットは、捕捉剤としてグルコヘプトネートを含む。しかし、グルコへプトネートは、約10〜15%を残したまま、代表的なキット成分と完全には反応しない。この残りのグルコへプトネートは腫瘍に達し、そして画像化結果をゆがめる。従って、本発明は、より安価で、かつより完全に反応するように、捕捉剤としてEDTAを使用することが好ましい。
【0023】
本発明の別の局面は、特定の腫瘍の治療のために、候補化合物の潜在的な有用性を決定するための予後方法である。現在、ほとんどの腫瘍は、数ヶ月および数千ドルを費やすまで、この薬物が特定の腫瘍に対して実際に有用であるどうかのいずれの指標もなく、化学療法において「通常の選択された薬物」で処置される。本発明の画像化組成物は、標識化されたEC薬物結合体の形態で、特定の薬物を腫瘍部位に送達すること、次いで特定の薬物かどうかを決定するために数時間内で部位を画像化することに有用である。
【0024】
このことに関連して、本発明の予後方法は、哺乳動物の体内の腫瘍の部位を決定する工程、腫瘍特異的な癌化学療法薬物候補物に結合体化されるECにキレート化された放射性核種を含む画像化組成物を得る工程、この組成物を哺乳動物の体内に投与する工程、ならびにこの腫瘍に対するこの候補薬物の有効性を決定するためにこの部位を画像化する工程を包含する。代表的に、この画像化工程は、哺乳動物の体内に組成物を注入した約10分〜約4時間後以内で実施される。好ましくは、この画像化工程は、哺乳動物の体内に組成物を注入した約1時間後以内に実施される。
【0025】
予後組成物中のECに結合体化されたこの癌化学療法薬物候補物は、公知の癌化学療法薬物から選択され得る。このような薬物は、表2にある。癌の特定の型に特異的であることが公知の多くの抗癌剤が存在する。しかし、癌の特定の型に対する全ての抗癌剤が、あらゆる患者に有用とは限らない。従って、本発明は、処置に莫大な時間とお金を費やす前に、候補薬物の潜在的な有用性を決定する方法を初めて提供する。
【0026】
本発明のなお別の実施形態は、シンチグラフ画像化剤を調製するための試薬である。本発明の試薬は、組織特異的リガンドを含み、この組織特異的リガンドは、シンチグラフで検出可能な画像を提供するのに十分なインビボの標識部位に対する親和性を有し、99mTc結合部分に共有結合する。99mTc結合部分は、上記のように、この組織特異的リガンドに直接的に結合されるか、またはリンカーを介してリガンドに結合されるかのいずれかである。この99mTc結合部分は、好ましくは、+4酸化状態の99mTcとエチレンジシステイン(EC)との間のN2S2キレートである。この組織特異的リガンドは、上記のように直接的またはリンカーを介してのいずれかで、ECの酸アームの一方または両方に共有結合される。この組織特異的リガンドは、上記のようなリガンドのいずれかであり得る。
【発明の効果】
【0027】
本発明により、例えば、以下が提供される:
(項目1) 画像化のための組成物であって、以下:
放射線核種標識;
エチレンジシステイン;および
該エチレンジシステインに結合体化した組織特異的リガンド;を含み、ここで、該エチレンジシステインが、該放射線核種標識とN2S2キレートを形成する、組成物。
(項目2) 前記組織特異的リガンドが、前記エチレンジシステインの1つまたは両方の酸アームで該エチレンジシステインと結合体化し得る、項目1に記載の組成物。 (項目3) 前記放射線核種が、99mTc、188Re、186Re、183Sm、166Ho、90Y、89Sr、67Ga、68Ga、111In、183Gd、59Fe、225Ac、212Bi、211At、64Cuまたは62Cuである、項目1に記載の組成物。
(項目4) 前記放射線核種が99mTcである、項目3に記載の組成物。 (項目5) 前記組織特異的リガンドが、抗癌剤、DNAトポイソメラーゼインヒビター、代謝拮抗剤、腫瘍マーカー、ホレートレセプター標的化リガンド、腫瘍アポトーシス細胞標的化リガンド、腫瘍低酸素標的化リガンド、DNAインターカレーター、レセプターマーカー、ペプチド、ヌクレオチド、器官特異的リガンド、抗生物質、抗真菌剤、抗体、グルタミン酸ペンタペプチドまたはグルコースを模倣する因子である、項目1に記載の組成物。
(項目6) 前記組織特異的リガンドが、抗癌剤である、項目5に記載の組成物。 (項目7) 前記抗癌剤が、メトトレキセート、ドキソルビシン、タモキシフェン、パクリタキセル、トポテカン、LHRH、マイトマイシンC、エトポシドトムデックス、ポドフィロトキシン、ミトザントロン、カンプトセシン、コルヒチン、エンドスタチン、フルダラビン、ゲムシタビンおよびトムデックスからなる群より選択され得る、項目6に記載の組成物。
(項目8) 前記組織特異的リガンドが腫瘍マーカーである、項目5に記載の組成物。 (項目9) 前記腫瘍マーカーが、PSA、ER、PR、CA−125、CA−199、CEA AFP、インターフェロン、BRCA1、HER−2/neu、シトキサン、p53、エンドスタチンまたはモノクローナル抗体(例えば、アンチセンス)である、項目8に記載の組成物。
(項目10) 前記組織特異的リガンドが、ホレートレセプター標的化リガンドである、項目5に記載の組成物。
(項目11) 前記ホレートレセプター標的化リガンドが、ホレート、メトトレキセートまたはトムデックスである、項目10に記載の組成物。
(項目12) 99mTc−EC−ホレートとしてさらに規定される、項目11に記載の組成物。
(項目13) 99mTc−EC−メトトレキセートとしてさらに規定される、項目11に記載の組成物。
(項目14) 99mTc−EC−トムデックスとしてさらに規定される、項目11に記載の組成物。
(項目15) 前記組織特異的リガンドが、腫瘍アポトーシス細胞標的化リガンドまたは腫瘍低酸素標的化リガンドである、項目5に記載の組成物。
(項目16) 前記組織特異的リガンドが、アネキシンV、コルヒチン、ニトロイミダゾール、マイトマイシンまたはメトロニダゾルである、項目15に記載の組成物。 (項目17) 99mTc−EC−アネキシンVとしてさらに規定される、項目16に記載の組成物。
(項目18) 99mTc−EC−コルヒチンとしてさらに規定される、項目16に記載の組成物。
(項目19) 99mTc−EC−ニトロイミダゾールとしてさらに規定される、項目16に記載の組成物。
(項目20) 99mTC−EC−メトロニダゾールとしてさらに規定される、項目16に記載の組成物。
(項目21) 前記組織特異的リガンドが、グルタミン酸ペンタペプチド(分子量750〜15,000)である、項目5に記載の組成物。
(項目22) 99mTc−EC−グルタミン酸ペンタペプチドとしてさらに規定される、項目21に記載の組成物。
(項目23) 前記組織特異的リガンドが、グルコースを模倣する因子である、項目5に記載の組成物。
(項目24) グルコースを模倣する前記因子が、ネオマイシン、カナマイシン、ゲンタマインシン、パロマイシン、アミカシン、トブラマイシン、ネチルマイシン、リボスタマイシン、シソマイシン、ミクロマイシン、リビドマイシン、ジベカシン、イセパマイシン、アストロマイシン、またはアミノグルコシドである、項目23に記載の組成物。 (項目25) 99mTc−EC−ネオマイシンとしてさらに規定される、項目24に記載の組成物。
(項目26) 99mTc−EC−カナマイシンとしてさらに規定される、項目24に記載の組成物。
(項目27) 99mTc−EC−アミノグリコシドとしてさらに規定される、項目24に記載の組成物。
(項目28) 99mTc−EC−ゲンタマイシンとしてさらに規定される、項目24に記載の組成物。
(項目29) 99mTc−EC−トブラマイシンとしてさらに規定される、項目24に記載の組成物。
(項目30) ECを前記組織特異的リガンドに結合体化するリンカーをさらに含む。項目2に記載の組成物。
(項目31) 前記リンカーが、水溶性ペプチド、グルタミン酸、アスパラギン酸、ブロモ酢酸エチル、エチレンジアミンまたはリジンである、項目30に記載の組成物。 (項目32) 前記組織特異的リガンドが、エストラジオール、トポテカン、パクリタキセル、ラロキシフェン、エトポシド、ドキソルビシン、マイトマイシンC、エンドスタチン、アネキシンV、LHRH、オクトレオチド、VIP、メトトレキセートまたは葉酸である、項目31に記載の組成物。
(項目33) 画像化のための放射標識されたエチレンジシステイン誘導体を合成する方法であって、該方法は、以下の工程:
a)組織特異的リガンドを得る工程;
b)該リガンドをエチレンジシステイン(EC)と混合して、EC−組織特異的リガンド誘導体を得る工程;ならびに
c)該EC−組織特異的リガンド誘導体を、放射線核種および還元剤と混合して、放射線核種標識されたEC−組織特異的リガンド誘導体を得る工程であって、ここで、該ECが、該放射線核種とN2S2キレートを形成する、工程、を包含する、方法。
(項目34) 前記還元剤が、亜ジチオン酸イオン、第一スズイオン、または第一鉄イオンである、項目33に記載の方法。
(項目35) 画像化のために組織特異的リガンドを標識化する方法であって、該方法は、以下の工程:
a)組織特異的リガンドを得る工程;
b)該組織特異的リガンドをエチレンジシステイン(EC)と混合して、EC−リガンド薬物結合体を得る工程;ならびに
c)該薬物結合体を、還元剤の存在下で99mTcと反応させて、該エチレンジシステイン(リンカーありまたはリンカーなし)と該99mTcとの間にN2S2キレートを形成する工程、
を包含する、方法。
(項目36) 前記組織特異的リガンドが、抗癌剤、DNAトポイソメラーゼインヒビター、代謝拮抗剤、腫瘍マーカー、ホレートレセプター標的化リガンド、腫瘍アポトーシス細胞標的化リガンド、腫瘍低酸素標的化リガンド、DNAインターカレーター、レセプターマーカー、ペプチド、器官特異的リガンド、抗生物質、抗真菌剤、グルタミン酸ペンタペプチドまたはグルコースを模倣する因子である、項目35に記載の方法。 (項目37) 前記還元剤が、亜ジチオン酸イオン、第一スズイオン、または第一鉄イオンである、項目36に記載の方法。
(項目38) 哺乳動物身体内の部位を画像化する方法であって、99mTc標識されたエチレンジシステイン−組織特異的リガンド結合体を含む診断有効量の組成物を投与する工程、および該部位に局在する99mTcからの放射活性シグナルを検出する工程、を包含する、方法。
(項目39) 前記部位が腫瘍である、項目38に記載の方法。 (項目40) 前記部位が感染である、項目38に記載の方法。 (項目41) 前記部位が、乳癌、卵巣癌、前立腺癌、子宮内膜、心臓、肺、脳、肝臓、ホレート(+)癌、ER(+)癌、脾臓、膵臓、または腸である、項目38に記載の方法。
(項目42) 放射性薬学的調製物を調製するためのキットであって、該キットは、所定量のエチレンジシステイン−組織特異的リガンド結合体組成物および99mTcを用いて該結合体を標識するために十分量の還元剤を含むシールされた容器を備える、キット。 (項目43) 前記エチレンジシステイン−組織特異的リガンド結合体組成物が、該エチレンジシステインと該組織特異的リガンドとの間にさらにリンカーを含む、項目42に記載のキット。
(項目44) 前記組織特異的リガンドが、抗癌剤、DNAトポイソメラーゼインヒビター、代謝拮抗剤、腫瘍マーカー、ホレートレセプター標的化リガンド、 腫瘍アポトーシス細胞標的化リガンド、腫瘍低酸素標的化リガンド、DNAインターカレーター、レセプターマーカー、ペプチド、器官リガンド、抗生物質、抗真菌剤、グルタミン酸ペンタペプチドまたはグルコースを模倣する因子である、項目42に記載のキット。 (項目45) 前記組織特異的リガンドが、エストラジオール、トポテカン、パクリタキセル、ラロキシフェン、エトポシド、ドキソルビシン、マイトマイシンC、エンドスタチン、アネキシンV、LHRH、オクトレオチド、VIP、メトトレキセートまたは葉酸である、項目43に記載のキット。
(項目46) 前記リンカーが、水溶性ペプチド、グルタミン酸、ポリグルタミン酸、アスパラギン酸、ポリアスパラギン酸、ブロモ酢酸エチル、エチレンジアミンまたはリジンである、項目45に記載のキット。
(項目47) シンチグラフィー画像化剤を調製するための試薬であって、99mTc結合部位に共有結合した組織特異的リガンドを含む、試薬。
(項目48) 前記99mTc結合部分が、エチレンジシステインである、項目47に記載の試薬。
(項目49) 前記組織特異的リガンドが、抗癌剤、DNAトポイソメラーゼインヒビター、代謝拮抗剤、腫瘍マーカー、ホレートレセプター標的化リガンド、腫瘍アポトーシス細胞標的化リガンド、腫瘍低酸素標的化リガンド、DNAインターカレーター、レセプターマーカー、ペプチド、器官特異的リガンド、抗生物質、抗真菌剤、グルタミン酸ペンタペプチドまたはグルコースを模倣する因子である、項目48に記載の試薬。 (項目50) 前記組織特異的リガンドと前記99mTc結合部位との間にさらにリンカーを含む、項目48に記載の試薬。
(項目51) 腫瘍に対する候補薬物の有効性を決定する方法であって、該方法は、以下:
a)候補薬物を得る工程;
b)該候補薬物をエチレンジシステイン(EC)と結合体化して、EC−候補薬物結合体を生成する工程;
c)該候補薬物結合体を、99mTcを用いてキレート化して、99mTc−EC−候補薬物結合体を生成する工程;
d)該99mTc−EC−候補薬物結合体を、腫瘍を有する患者に導入する工程;および
e)該患者を画像化して、該腫瘍に対する該候補薬物の有効性を決定する工程、を包含する、方法。
【図面の簡単な説明】
【0028】
【図1】図1は、99mTc−EC−ホレートの合成スキームである。
【図2】図2は、99mTc−EC−MTX(メトトレキサート)の合成スキームである。
【図3】図3は、99mTc−EC−TDX(トムデクス(tomudex))の合成スキームである。
【図4】図4は、99mTc−ECおよび99mTc−EC−ホレートの生体分布研究である。
【図5】図5は、99mTc−ECホレートを用いた腫瘍/筋肉カウント比および腫瘍/血液カウント比についてのブロッキング研究である。
【図6】図6Aおよび図6Bは、99mTc−EC注射群と比較した、99mTc−ECホレート注射群における腫瘍のシンチグラフィ画像である。
【図7】図7は、EC−MN(メトロニダゾール)の合成スキームである。
【図8A】図8Aは、EC−NIMについての合成スキームを示す。
【図8B】図8Bは、EC−NIMについての構造の1H−NMR確認を示す。
【図9】図9は、99mTc−EC−MN、[18F]FMISOおよび[131I]IMISOについての生体分布研究(腫瘍/血液比)である。
【図10】図10は、99mTc−EC、[18F]FMISOおよび[131I]IMISOについての生体分布研究(腫瘍/筋肉比)である。
【図11】図11Aは、99mTc−EC−MN注射群の腫瘍のシンチグラフィ画像であり、図11Bは、99mTc−EC注射群の腫瘍のシンチグラフィ画像である。
【図12】図12は、99mTc−EC−MNでの注射の1時間後において実施したオートラジオグラムである。
【図13】図13は、イヌ血清サンプル中の99mTc−EC−MNの安定性を示す。
【図14A】図14Aは、ラットにおける99mTc−EC−MN 対 99mTc−ECの胸部腫瘍取り込みを示す。
【図14B】図14Bは、コントロールと比較した、パクリタキセルで処理したラットにおける99mTc−EC−MN 対 99mTc−ECの胸部腫瘍取り込みを示す。
【図15A】図15Aは、ラットにおける99mTc−EC−NIM 対 99mTc−ECの卵巣腫瘍取り込みを示す。パクリタキセルを用いて処理したラットにおける腫瘍取り込み(図15B)は、パクリタキセルで処理していないラットにおける腫瘍取り込み(図15A)よりも少ない。肉腫を有するラットにおける99mTc−EC−NIMの腫瘍取り込みもまた示す。図15Cは、パクリタキセルで処理した肉腫保有ラットにおける腫瘍取り込みを示し、一方、図15Dは、パクリタキセルで処理していないラットにおける腫瘍取り込みを示す。パクリタキセルで処理した後に、99mTc−EC−NIMの取り込みの減少したが存在した。
【図15B】図15Aは、ラットにおける99mTc−EC−NIM 対 99mTc−ECの卵巣腫瘍取り込みを示す。パクリタキセルを用いて処理したラットにおける腫瘍取り込み(図15B)は、パクリタキセルで処理していないラットにおける腫瘍取り込み(図15A)よりも少ない。肉腫を有するラットにおける99mTc−EC−NIMの腫瘍取り込みもまた示す。図15Cは、パクリタキセルで処理した肉腫保有ラットにおける腫瘍取り込みを示し、一方、図15Dは、パクリタキセルで処理していないラットにおける腫瘍取り込みを示す。パクリタキセルで処理した後に、99mTc−EC−NIMの取り込みの減少したが存在した。
【図15C】図15Aは、ラットにおける99mTc−EC−NIM 対 99mTc−ECの卵巣腫瘍取り込みを示す。パクリタキセルを用いて処理したラットにおける腫瘍取り込み(図15B)は、パクリタキセルで処理していないラットにおける腫瘍取り込み(図15A)よりも少ない。肉腫を有するラットにおける99mTc−EC−NIMの腫瘍取り込みもまた示す。図15Cは、パクリタキセルで処理した肉腫保有ラットにおける腫瘍取り込みを示し、一方、図15Dは、パクリタキセルで処理していないラットにおける腫瘍取り込みを示す。パクリタキセルで処理した後に、99mTc−EC−NIMの取り込みの減少したが存在した。
【図15D】図15Aは、ラットにおける99mTc−EC−NIM 対 99mTc−ECの卵巣腫瘍取り込みを示す。パクリタキセルを用いて処理したラットにおける腫瘍取り込み(図15B)は、パクリタキセルで処理していないラットにおける腫瘍取り込み(図15A)よりも少ない。肉腫を有するラットにおける99mTc−EC−NIMの腫瘍取り込みもまた示す。図15Cは、パクリタキセルで処理した肉腫保有ラットにおける腫瘍取り込みを示し、一方、図15Dは、パクリタキセルで処理していないラットにおける腫瘍取り込みを示す。パクリタキセルで処理した後に、99mTc−EC−NIMの取り込みの減少したが存在した。
【図16】図16は、EC−GAP(ペンタグルタメート)の合成スキームである。
【図17】図17は、99mTc−EC−GAP注射群における胸部腫瘍のシンチグラフィー画像である。
【図18】図18は、異なる時間間隔での、99mTc−EC−ANNEX V注射群における胸部腫瘍のシンチグラフィー画像である。
【図19A】図19Aおよび図19Bは、卵巣腫瘍保有群におけるパクリタキセル処理前(図19A)とパクリタキセル処理後(図19B)との間の、99mTc−EC−ANNEX Vの取り込み差異の比較である。
【図19B】図19Aおよび図19Bは、卵巣腫瘍保有群におけるパクリタキセル処理前(図19A)とパクリタキセル処理後(図19B)との間の、99mTc−EC−ANNEX Vの取り込み差異の比較である。
【図20A】図20Aおよび図20Bは、肉腫腫瘍保有群におけるパクリタキセル処理前(図20A)とパクリタキセル処理後(図20B)との間の、99mTc−EC−ANNEX Vの取り込み差異の比較である。
【図20B】図20Aおよび図20Bは、肉腫腫瘍保有群におけるパクリタキセル処理前(図20A)とパクリタキセル処理後(図20B)との間の、99mTc−EC−ANNEX Vの取り込み差異の比較である。
【図21】図21は、EC−COL(コルヒチン)の合成スキームである。
【図22】図22は、EC−COL合成において分解生成物が観察されなかったことを示す。
【図23】図23は、99mTc−EC−COLについての、時間関数としての腫瘍 対 筋肉比および腫瘍 対 血液比である。
【図24】図24は、99mTc−ECについての、時間関数としての腫瘍 対 筋肉比および腫瘍 対 血液比である。
【図25】図25は、99mTc−EC−COLを用いた胸部腫瘍保有ラットにおけるインビボ画像研究である。
【図26】図26は、99mTc−ECを用いた胸部腫瘍保有ラットにおけるインビボ画像研究である。
【図27】図27は、99mTc−EC−COL 対 99mTc−ECの、注射後のコンピューターで輪郭をとった目的の領域である。
【図28】図28は、発作に罹患した59歳の男性患者の99mTc−EC−MNを用いたSPECTである。画像は、注射1時間後に撮影した。
【図29】図29は、図28と同じ患者のMRI T1重み付け画像である。
【図30】図30は、発作1日後の73歳の男性患者の、99mTc−EC−MNを用いた注射の1時間後でのSPECTである。
【図31】図31は、発作12日後の図30で画像化した同じ73歳の男性患者の、99mTc−EC−MNを用いた注射の1時間後でのSPECTである。
【図32】図32は、発作1日後の図30で画像化した同じ73歳の男性発作患者のCTである。
【図33】図33は、発作12日後の図32で画像化した同じ73歳の男性発作患者のCTである。画像化にCTを用いた1日目と12日目との間に顕著な差がないことに注意する。
【図34】図34は、発作に罹患した72歳の男性患者の99mTc−EC−MNを用いた注射の1時間後でのSPECTである。
【図35】図35は、図34で画像化した同じ72歳の男性発作患者のCTである。CTがいかに病変サイズを誇張しているかに注意すること。
【図36】図36は、99mTc−EC−ネオマイシンの合成スキームである。
【図37A】99mTc−ECおよび99mTc−EC−ネオマイシン(100μCi/ラット、静脈内)の投与後の胸部腫瘍保有ラットのシンチグラフィー画像は、この腫瘍が注射後0.5〜4時間で良好に可視化され得ることを示す。
【図37B】図37Bは、乳癌患者の99mTc−EC−ネオマイシン(30mCi、静脈内)を用いたシンチマンモグラフィである。画像は、注射2時間後に撮影した。
【図38A】図38Aは、ECの1H−NMRである。
【図38B】図38Bは、ネオマイシンの1H−NMRである。
【図38C】図38Cは、EC−ネオマイシンの1H−NMRである。
【図39】図39Aおよび図39Bは、EC−ネオマイシンの質量分析である(M+1112.55)。
【図40A】図40Aは、ECのUV波長スキャンである。
【図40B】図40Bは、ネオマイシンのUV波長スキャンである。
【図40C】図40Cは、EC−ネオマイシンのUV波長スキャンである。
【図41】図41は、99mTc−EC−ネオマイシンのラジオ−TLC分析である。
【図42】図42は、99mTc−EC−ネオマイシンのHPLC分析である(放射能検出器)。
【図43】図43は、99mTc−EC−ネオマイシンのHPLC分析である(UV 254nm)。
【図44】図44は、18F−FDGのHPLC分析である(放射能検出器)。
【図45】図45は、18F−FDGのHPLC分析である(UV 254nm)。
【図46】図46は、肺癌細胞株における一連の99mTc−EC薬物結合体のインビトロ細胞取り込みアッセイである。99mTc−EC−ネオマイシンは、試験した薬剤において最も高い取り込みを示した。
【図47】図47は、99mTc−EC−ネオマイシンおよび18F−FDGの細胞(A549)取り込みに対するグルコースの影響を示す。
【図48A】図48Aは、薬物取り込み(%)として示され、99mTc−EC−ネオマイシンおよび18F−FDGの細胞(H1299)取り込みに対するグルコースの影響を示す。
【図48B】図48Bは、グルコース充填に伴なう変化(%)として示され、99mTc−EC−ネオマイシンおよび18F−FDGの細胞(H1299)取り込みに対するグルコースの影響を示す。
【図49】図49は、99mTc−EC−グルコサミンの合成スキームである。
【図50】図50は、グルコースのヘキソキナーゼアッセイである。
【図51】図51は、グルコサミンのヘキソキナーゼアッセイである。
【図52】図52は、EC−グルコサミンのヘキソキナーゼアッセイである。
【図53】図53は、EC−GAP−グルコサミンのヘキソキナーゼアッセイである。
【図54】図54は、99mTc−EC−GAP−グルコサミンの合成スキームである。
【図55A】図55A〜図55Cは、肺癌細胞株(A549)における99mTc−EC(図55A)、99mTc−EC−デオキシグルコース−GAP(図55B)、および18F−FDG(図55C)のインビトロ細胞取り込みアッセイである。99mTc−EC−DGは、18F−FDGと比較して類似の取り込みを示した。
【図55B】図55A〜図55Cは、肺癌細胞株(A549)における99mTc−EC(図55A)、99mTc−EC−デオキシグルコース−GAP(図55B)、および18F−FDG(図55C)のインビトロ細胞取り込みアッセイである。99mTc−EC−DGは、18F−FDGと比較して類似の取り込みを示した。
【図55C】図55A〜図55Cは、肺癌細胞株(A549)における99mTc−EC(図55A)、99mTc−EC−デオキシグルコース−GAP(図55B)、および18F−FDG(図55C)のインビトロ細胞取り込みアッセイである。99mTc−EC−DGは、18F−FDGと比較して類似の取り込みを示した。
【図56】図56は、胸部腫瘍保有ラットにおける99mTc−EC−GAPの腫瘍 対 組織カウント密度比である。
【図57】図57は、乳癌細胞株(13762)における注射2時間後の、グルコース充填を伴なう18FDGのインビトロ細胞取り込みである。
【図58】図58は、胸部腫瘍保有マウスにおける99mTc−EC−ネオマイシンのインビボ組織取り込みである。
【図59】図59は、99mTc−EC−デオキシグルコースの合成スキームである。
【図60】図60は、EC−デオキシグルコースの質量分析である。
【図61】図61は、EC−デオキシグルコース(EC−DG)の1H−NMRである。
【図62】図62は、グルコサミンの1H−NMRである。
【図63】図63は、99mTc−EC−DGのラジオ−TLC分析である。
【図64】図64は、99mTc−EC−デオキシグルコースおよび99mTc−ECのHPLC分析である(放射能検出器)。
【図65】図65は、99mTc−EC−デオキシグルコースおよび99mTc−ECのHPLC分析である(放射能検出器、混合)。
【図66】図66は、グルコースのヘキソキナーゼアッセイである。
【図67】図67は、FDGのヘキソキナーゼアッセイである。
【図68】図68は、EC−DGのヘキソキナーゼアッセイである。
【図69】図69は、肺癌細胞株(A549)における99mTc−EC−デオキシグルコース、99mTc−ECおよび18F−FDGのインビトロ細胞取り込みアッセイである。99mTc−EC−DGは、18F−FDGと比較して類似した取り込みを示した。
【図70】図70は、99mTc−EC−DGの胸部細胞(13762細胞株)取り込みに対するd−グルコースおよびl−グルコースの影響である。
【図71】図71は、18F−FDGの胸部細胞(13762細胞株)取り込みに対するd−グルコースおよびl−グルコースの影響である。
【図72】図72は、18F−FDGの肺細胞(A549細胞株)取り込みに対するd−グルコースおよびl−グルコースの影響である。
【図73】図73は、99mTc−EC−DGの肺細胞(A549細胞株)取り込みに対するd−グルコースおよびl−グルコースの影響である。
【図74】図74は、グルコサミンおよびEC−DG(1.2mmol/kg、静脈内)によって誘導されたインビボ血中グルコースレベルの影響である。
【図75】図75は、FDG(1.2mmol/kgおよび1.9mmol/kg、静脈内)およびインスリンによって誘導されたインビボ血中グルコースレベルの影響である。
【図76】図76は、胸部腫瘍保有ラットにおける99mTc−EC−デオキシグルコースの腫瘍 対 組織カウント密度比である。
【図77】図77は、胸部腫瘍保有ラットにおける99mTc−EC−デオキシグルコースのインビボ生体分布である。
【図78】図78は、肺腫瘍保有マウスにおける99mTc−EC−デオキシグルコースのインビボ組織取り込みである。
【図79】図79は、肺腫瘍保有マウスにおける99mTc−EC−ネオマイシンのインビボ組織取り込みである。
【図80】図80は、肺腫瘍保有マウスにおける18F−FDGのインビボ組織取り込みである。
【図81】99mTc−ECおよび99mTc−EC−デオキシグルコース(100μCi/ラット、静脈内)投与後の胸部腫瘍保有ラットの平面画像は、この腫瘍が、注射後0.5〜4時間で良好に可視化され得ることを示した。
【図82A】図82Aは、悪性神経膠星状細胞腫を有する患者のMRIである。
【図82B】図82Bは、悪性神経膠星状細胞腫を有する患者の99mTc−EC−DGを用いたSPECTである。
【図83A】図83Aは、出血性神経膠星状細胞腫を有する患者のMRIである。
【図83B】図83Bは、悪性神経膠星状細胞腫を有する患者の99mTc−EC−DGを用いたSPECTである。
【図84A】図84Aは、良性髄膜腫を有する患者のMRIである。
【図84B】良性髄膜腫を有する患者の99mTc−EC−DGを用いたSPECTは、焦点が強調された取り込みを示さなかった。
【図85A】図85Aは、肺におけるTBを有する患者のCTである。
【図85B】TBを有する患者の99mTc−EC−DGを用いたSPECTは、焦点が強調された取り込みを示さなかった。
【図86A】図86Aは、肺癌を有する患者のCTである。
【図86B】図86Bは、肺癌を有する患者の99mTc−EC−DGを用いた全身画像である。
【図86C】図86Cは、肺癌を有する患者の99mTc−EC−DGを用いたSPECTであり、腫瘍は、焦点が強調された取り込みを示した。
【発明を実施するための形態】
【0029】
(例示的な実施形態の説明)
核医学の分野において、特定の病理的状態は、局在化するか、またはそれらの程度が、少量の内部投与される放射性標識されたトレーサ化合物(放射性トレーサまたは放射性薬品と呼ばれる)の分布を検出することによって評価される。これらの放射性薬品を検出するための方法は、画像化法または放射性画像化法として一般的に公知である。
【0030】
放射性画像化において、放射性標識は、γ線放射放射性核種であり、そして放射性トレーサは、γ線検出カメラを使用して位置決めされる(このプロセスは、しばしば、γシンチグラフィと呼ばれる)。画像化部位は、検出可能である。なぜなら、放射性トレーサは、病理学的部位に局在化する(陽性コントラストと呼ばれる)か、あるいは、放射性トレーサがこのような病理学的部位に特異的に局在化しないように選択される(陰性コントラストと呼ばれる)かのいずれかであるからである。
【0031】
種々の放射性核種(67Ga、99mTc、111In、123I、125I、169Ybまたは186Reを含む)は、放射性画像化に有用であることが公知である。より良い画像化特性および低価格に起因して、可能な場合、123I、131I、67Gaおよび111In標識された化合物を、対応する99mTc標識化合物に置き換える試みがなされている。好ましい物理的特質および非常に低い価格(1mCi当たり0.21ドル)に起因して、99mTcは、放射性薬品を標識するのに好ましかった。DTPA薬物結合体が99mTcで効果的に標識され得ることが報告された(Mathiasら、1997)が、DTPA部分は、111Inほど安定には99mTcにキレート化しない(Goldsmith、1997)。
【0032】
ヒトにおける最適な放射線画像化のために、多くの因子が考慮されなければならない。検出の効率を最大にするために、100〜200keVの範囲でγエネルギーを放射する放射性核種が好ましい。患者に対する吸収される放射線量を最小にするために、放射性核種の物理的半減期は、画像化手順が可能である限り短くあるべきである。検査が任意の日にそしてその日の任意の時間で行われ得るために、臨床場所においていつでも利用可能な放射性核種の供給源を有することが有利である。99mTcは、140keVでγ線を放射し、6時間の物理的半減期を有し、そしてモリブデン−99/テクネチウム−99m発生器を使用して、現場で容易に利用可能であるので、好ましい放射性核種である。
【0033】
ビスアミノエタンチオール4座配位リガンド(ジアミノジチオール化合物とも呼ばれる)は、2つのチオール硫黄および2つの窒素原子に対するオキソテクネチウム基の効率的な結合に基づいて、非常に安定なTc(V)O−錯体を形成することが公知である(Davisonら、1980;1981;Verbruggenら、1992)。99mTc−L,L−エチレンジシステイン(99mTc−EC)は、最近成功したN2S2キレートの例である(Verbruggenら、1992;Van Neromら、1993;Surmaら、1994)。EC(新規な腎臓画像化剤)は、99mTcで容易に標識化され得、そして高い放射化学純度および安定性で効率的に標識され得、そして活性な管輸送によって腎臓を通って排出され得る(Verbruggenら、1992;Van Neromら、1993;Surmaら、1994;Verbruggenら、1990;Van Neromら、1990;Jamarら、1993)。ECの他の用途は、PETのためのガリウム−68(陽電子放射体、t1/2=68分)および磁気共鳴画像化(MRI)のためのガドリニウム、鉄またはマンガンとキレート化される。
【0034】
本発明は、画像化のために、リガンドを特定の組織型に標的化させるための標識剤として99mTc−ECを利用する。ECを組織標的リガンドに結合する利点は、組織標的リガンドの特異的結合特性が目的の領域上で放射性シグナルを濃縮することである。標識化戦略としての99mTc−ECの使用が、実質的に任意の型の化合物で効果的であり得ることが考慮されるが、いくつかの提案される好ましいリガンドは、例示の目的のために本明細書中において提供される。本発明の99mTc−EC薬物結合体が腫瘍だけでなく、他の組織特異的状態(例えば、感染、低酸素組織(発作)、心筋梗塞、アポトーシス細胞、アルツハイマー病および子宮内膜症)をも画像化するのに有用であり得る。
【0035】
放射標識タンパク質およびペプチドは、先行技術において報告されている(Egeら、米国特許第4,832,940号、Abramsら、1990;Bakkerら、1990;Goldsmithら、1995,1997;Olexaら、1982;Ranbyら、1988;Hadleyら、1988;Leesら、1989;Sobelら、1989;Stuttle、1990;Maraganoreら、1991;Rodwellら、1991;Tubisら、1968;Sandrehagen 1983)。しかし、99mTc−ECは、本発明の前に、任意のリガンド(ジエチルエステル以外)と組み合わせて使用されなかった(Kabasakal、2000)。ECのジエチルエステルは、脳血流剤として使用された(Kikukawaら、2000)。
【0036】
放射性画像化について最適ではあるが、99mTcの化学は、他の元素の化学ほど完全には研究されておらず、このため、99mTcでの放射標識の方法は、豊富ではない。99mTcは、通常、モリブデン−99/テクネチウム−99m発生器から、99mTcペルテクネテート(TcO4−;+7の酸化状態のテクネチウム)として通常得られる。しかし、ペルテクネテートは、他の化合物と十分結合しない。従って、化合物を放射標識するために、99mTcペルテクネテートは、別の形態に変換されなければならない。テクネチウムが水溶液において安定なイオンを形成しないので、分解を妨げ、99mTcの不溶性の二酸化テクネチウムへの変換も、ペルテクネテートへ戻る変換も生じることを妨げるのに十分な動力学的および熱力学的安定性を有する配位錯体の形態でこのような溶液中で保持されなければならない。
【0037】
放射標識化のために、99mTc錯体が、テクネチウムイオンの周りのドナー基のすべてが単一のキレートリガンドによって提供される(この場合、エチレンジシステイン)であるキレートとして形成されることが特に有利である。これによって、キレート化された99mTcが、エチレンジシステインとリガンドとの間で直接かまたは単一のリンカーを介してのいずれかで、組織特異的リガンドに共有結合され得る。
【0038】
テクネチウムは、多くの酸化状態(+1、+2、+4、+5、+6および+7)を有する。+1の酸化状態である場合、Tc MIBIと呼ばれる。Tc MIBIは、熱反応を用いて作製されなければならない(Seaboldら、1999)。本発明の目的のために、Tcが+4の酸化状態であることが重要である。この酸化状態は、ECを用いるN2S2キレートを形成するのに理想的である。従って、本発明の薬物結合体と放射性テクネチウムとの錯体の形成において、テクネチウム錯体(好ましくは、99mTcペルテクネテートの塩)は、還元剤の存在下で、本発明の薬物結合体と反応する。
【0039】
本発明における使用のための好ましい還元剤は、Tcをその+4の酸化状態に還元するための塩化スズ(SnCl2)の形態のスズイオンである。しかし、他の還元剤(例えば、ジチオネートイオンまたは鉄(II)イオン)が本発明と組み合わせて有用であり得る。還元剤が固相還元剤であり得ることも意図される。還元剤の量は、重要であり得る。なぜなら、コロイドの形成を避けることが必要であるからである。例えば、約100〜約300mCiのTcペルテクネテート当たり、約10〜約100μgのSnCl2を使用することが好ましい。最も好ましい量は、約200mCiのTcペルテクネテートおよび約2mlの生理食塩水当たり、約0.1mgのSnCl2である。これは、代表的には、5人の患者における使用に十分なTc−EC組織特異的リガンド結合体を作製する。
【0040】
エチレンジシステインの酸化を防ぐために、組成物中に酸化防止剤を含むこともしばしば重要である。本発明と組み合わせた使用に好ましい酸化防止剤は、ビタミンC(アスコルビン酸)である。しかし、他の酸化防止剤(例えば、トコフェロール、ピリドキシン、チアミンまたはルチン)もまた有用であり得ることが意図される。
【0041】
一般的に、本発明と組み合わせた使用のためのリガンドは、酸のアームのいずれかまたは両方でECに結合し得るアミノ基またはヒドロキシ基のいずれかを有する。アミノ基またはヒドロキシル基が利用可能でない場合(例えば、酸官能基)、所望のリガンドは、リンカー(例えば、エチレンジアミン、アミノプロパノール、ジエチレントリアミン、アスパラギン酸、ポリアスパラギン酸、グルタミン酸、ポリグルタミン酸、またはリジン)を加えることによって本発明の方法を使用して、なおECに結合し、そして99mTcで標識される。本発明における使用に意図されるリガンドとしては、限定しないが、新脈管形成/抗新脈管形成リガンド、DNAトポイソメラーゼインヒビター、解糖マーカー、抗代謝リガンド、アポトーシス/低酸素リガンド、DNAインターカレーター、レセプターマーカー、ペプチド、ヌクレオチド、抗菌剤(例えば、抗生物質または抗真菌剤)、器官特異的リガンドおよび糖または薬剤(グルコースを模倣する)が挙げられる。
【0042】
EC自体は、水溶性である。本発明のEC−薬物結合体もまた水溶性であることが必要である。本発明と組み合わせて使用されるリガンドの多くは、水溶性であるか、またはECと結合した場合に水溶性化合物を形成する。しかし、組織特異的リガンドが水溶性でない場合、リガンドの溶解性を増加するリンカーが使用され得る。リンカーは、脂肪族アルコールまたは芳香族アルコール、アミンまたはペプチドにあるいはカルボキシルおよびまたはペプチドに結合し得る。リンカーは、ポリアミノ酸(ペプチド)またはアミノ酸(例えば、グルタミン酸、アスパラギン酸またはリジン)のいずれかであり得る。表1は、特異的薬物官能基に対する所望のリンカーを示す。
【0043】
【表1】
例:
A.エストラジオール、トポテカン、パクリタキセル、ラロキシルフェン(raloxfen)エトポシド
B.ドキソルビシン、マイトマイシンC、エンドスタチン、アネキシンV.LHRH、オクトレオチド、VIP
C.メトトレキサート、葉酸
本発明のEC組織特異的リガンド薬物結合体が他の放射性核種にキレート化し得、放射性核種療法に使用され得ることも想定される。一般的に、実質的に任意のα,β−放射体、γ−放射体、またはβ,γ−放射体が、本発明とともに使用され得ることが考えられる。好ましいβ,γ−放射体としては、166Ho、188Re、186Re、153Sm、および89Srが挙げられる。好ましいβ−放射体としては、90Yおよび225Acが挙げられる。好ましいγ−放射体としては、67Ga、68Ga、64Cu、62Cuおよび111Inが挙げられる。好ましいα−放射体としては、211Atおよび212Biが挙げられる。常磁性物質(例えば、Gd、MnおよびFe)が、本発明と組み合わせた使用のためにECとキレート化され得ることも想定される。
【0044】
錯体およびこのような錯体を調製するための手段は、標識される本発明のEC−組織特異的リガンドの所定量および99mTcで結合体を標識するのに十分な量の還元剤を含む密封バイアルを備えるキット形態で便利に提供される。本発明に従う99mTc標識シンチグラフィ画像化剤は、EC−組織特異的結合体および還元剤を含むバイアルへの適切な量の99mTcまたは99mTc錯体の添加、および本明細書中、以下の実施例1に記載される条件下での反応によって調製され得る。キットはまた、例えば、浸透圧を調節するための薬学的に受容可能な塩、緩衝液、保存剤、酸化防止剤などのような従来の薬学的補助物質を含み得る。キットの成分は、液体形態、凍結形態または乾燥形態であり得る。好ましい実施形態において、キット成分は、凍結乾燥形態で提供される。
【0045】
本発明によって提供される放射性標識試薬または結合体は、適切な量の放射能を有して提供される。99mTc放射性錯体を形成する際に、1mL当たり約0.01ミリキュリー(mCi)〜約300mCiの濃度で放射能を含む溶液で放射性錯体を形成することが一般的に好ましい。
【0046】
本発明によって提供される99mTc標識シンチグラフィ画像化剤は、哺乳動物の身体における部位を視覚化するために使用され得る。本発明に従って、99mTc標識シンチグラフィ画像化剤は、単一の単位注射可能線量で投与される。当業者に公知の任意の通常のキャリア(例えば、滅菌生理食塩水溶液または血漿)は、本発明に従って、種々の器官、腫瘍などを診断的に画像化するための注射可能溶液を調製するための放射標識の後に、利用され得る。一般的に、投与される単位線量は、約0.01mCi〜約300mCi、好ましくは10mCi〜約200mCiの放射能を有する。単位投薬量で注入される溶液は、約0.01mL〜約10mLである。静脈内投与の後に、器官または腫瘍のインビボでの画像化は、所望である場合、放射標識試薬が、患者に導入された後の数時間でまたはそれより長くにおいて行われ得る。大部分の場合、十分な量の投与線量が、シンチフォトグラフをとることを可能にするように約0.1時間以内に画像化される領域に蓄積する。診断目的または予後判定目的のためのシンチグラフィ画像化の任意の従来の方法が、本発明に従って利用され得る。
【0047】
本発明の99mTc−EC標識戦略はまた、予後判定の目的のために使用され得る。ECが癌化学療法のために選択した公知の薬物(例えば、表2に列挙されるもの)に結合され得ることが想定される。次いで、これらのEC−薬物結合体は、99mTcで放射標識され得、そして腫瘍を有する患者に投与され得る。標識EC−薬物結合体は、腫瘍に特異的に結合する。画像化は、特定の患者の特定の腫瘍に対する癌化学療法薬物の効力を決定するために実行され得る。この方法において、医師は、続行するどの処置様式がもっとも有効か、どの化学療法薬物がもっとも有効かを迅速に決定し得る。これは、薬物を選択する工程および1回の化学療法を施す工程を包含する現在の方法に対して劇的な改善を表す。これは、薬物の効果が決定され得る前の、数か月の患者の時間および数千ドルに関係する。
【0048】
本発明によって提供される99mTc標識EC−組織特異的リガンド結合体および錯体は、水性生理食塩水媒体のような静脈内注射のための任意の従来の媒体で、または血漿媒体において静脈内で投与され得る。このような媒体はまた、例えば、浸透圧を調節するための薬学的に受容可能な塩、緩衝液、保存剤、酸化防止剤などのような従来の薬学的補助物質を含み得る。好ましい媒体には、ノーマルセーラインおよび血漿がある。
【0049】
特定の好ましい標的化戦略は、さらに詳細に以下に議論される。
【0050】
(腫瘍葉酸レセプター標的化)
放射標識リガンド(例えば、ペンテトレオチド(pentetreotide)および血管作用性腸ペプチド)は、細胞レセプターに結合し、これらのうちのいくつかは、腫瘍細胞において過剰発現される(BrittonおよびGranowska、1996;Krenningら、1995;Reubiら、1992;Goldsmithら、1995;Virgoliniら、1994)。これらのリガンドが免疫原性でなく、そして血漿から迅速に排除されるので、レセプター画像化は、抗体画像化と比較してより見込みがあるようである。
【0051】
葉酸および葉酸代謝拮抗薬(例えば、メトトレキサート)が、古典的な還元型葉酸キャリアシステムに加えて、高い親和性の葉酸レセプター(グリコシルホスファチジルイノシトール結合膜葉酸結合タンパク質)を介して、細胞に入る(Westerhofら、1991;Orrら、1995;HsuehおよびDolnick、1993)。葉酸レセプター(FR)は、多くの新生物細胞型(例えば、肺癌、乳房癌、卵巣癌、頚部癌、結腸直腸癌、鼻咽頭癌、腎腺癌、悪性黒色腫および上衣細胞腫)において過剰発現されるが、主に、いくつかの正常な分化組織(例えば、脈絡叢、胎盤、甲状腺および腎臓)において発現される(Orrら、1995;Weitmanら、1992a;Campbellら、1991;Weitmanら、1992b;Holmら、1994;Rossら、1994;Franklinら、1994;Weitmanら、1994)。FRは、葉酸結合タンパク質毒素、薬物/アンチセンスオリゴヌクレオチドおよびリポソームを、葉酸レセプターを過剰発現する腫瘍細胞に送達するために使用された(Ginobbiら、1997;LeamonおよびLow、1991;LeamonおよびLow、1992;Leamonら、1993;LeeおよびLow、1994)。さらに、抗T細胞レセプター抗体に結合される抗FR抗体を含む二特異的抗体は、T細胞をFR陽性腫瘍に標的化するために使用されており、現在、卵巣癌について臨床試験にある(Canevariら、1993;Bolhuisら、1992;Patrickら、1997;Coneyら、1994;Kranzら、1995)。同様に、この性質は、放射標識葉酸結合体(例えば、葉酸レセプター陽性腫瘍の画像化のための、67Gaデフェロキサミン−葉酸および111In−DTPA−葉酸)を開発するために鼓舞された(Mathiasら、1996;Wangら、1997;Wangら、1996;Mathiasら、1997b)。これらの薬剤を用いた限定されたインビトロおよびインビボ研究の結果は、葉酸レセプターが、腫瘍画像化のための潜在的な標的であり得ることを示唆する。本発明において、本発明者らは、一連の新規な葉酸レセプターリガンドを開発した。これらのリガンドは、99mTc−EC−葉酸、99mTc−EC−メトトレキセート(99mTc−EC−MTX)、99mTc−EC−トムデックス(tomudex)(99mTc−EC−TDX)である。
【0052】
(腫瘍低酸素標的化)
腫瘍細胞は、酸素の非存在化におけるよりも酸素の存在下において従来の照射により感受性である;腫瘍内の少ない割合の低酸素細胞でさえ、放射線に対する応答を制限し得る(Hall、1988;Bushら、1978;Grayら、1953)。低酸素放射線抵抗性は、多くの動物腫瘍において示されたが、ヒトにおいては少しの腫瘍型のみで示された(Dische、1991;Gatenbyら、1988;Nordsmarkら、1996)。ヒト腫瘍における低酸素の発生は、大部分において、組織学的知見および動物腫瘍研究から推論された。低酸素のインビボ実証は、酸素電極を用いた組織測定を必要とし、そしてこれらの技術の侵襲性は、それらの臨床的適用を限定している。
【0053】
ミソニダゾール(MISO)は、低酸素細胞増感剤であり、そして異なる放射性同位体(例えば、18F、123I、99mTc)でのMISOの標識は、PETまたは平面(planar)シンチグラフィによる十分に酸素化された活性腫瘍から、低酸素だか代謝的に活性な腫瘍を区別するのに有用であり得る。[18F]フルオロソニダゾール(FMISO)は、腫瘍低酸素を評価するためにPETとともに使用される。最近の研究は、PET([18F]FMISOによる細胞酸素含有量をモニターするその能力を有する)が、放射線に対する腫瘍応答を推測するための高い可能性を有することを示した(Kohら、1992;Valkら、1992;Martinら、1989;Raseyら、1989;Raseyetalら、1990;Yangら、1995)。PETは、視準なしで高い分解能を提供するが、臨床設定においてPET同位体を使用するコストは、ひどく高い。ヨウ素でのMISOの標識化が選択されたが、甲状腺組織における高い取り込みが観測された。従って、同位体があまり高価でなく、そして大部分の主用な医療設備において容易に入手可能である平面シンチグラフィのための化合物を開発することが望ましい。本発明において、発明者らは、99mTc−EC−2−ニトロイミダゾールおよび99mTc−EC−メトロニダゾールの合成を提供し、そして腫瘍低酸素マーカーとしてそれらの潜在的な使用を示す。
【0054】
(癌のペプチド画像化)
ペプチドおよびアミノ酸は、種々の型の腫瘍の画像化において首尾よく使用されてきた(Westerら、1999;CoenenおよびStocklin、1988;Radererら、1996;Lambertら、1990;Bakkerら、1990;StellaおよびMathew、1990;Butterfieldら、1998;Piperら、1983;Mochizukiら、DickinsonおよびHiltner、1981)。グルタミン酸ベースのペプチドは、癌処置のための薬物キャリアとして使用されてきた(StellaおよびMathew、1990;Butterfieldら、1998;Piperら、1983;Mochizukiら、1985;DickinsonおよびHiltner、1981)。ホレートのグルタメート部分が分解し、そしてインビボでポリグルタメートを形成することは公知である。次いで、このポリグルタメートは、ホレートに再結合されて、ホリルポリグルタメートを形成し、このホリルポリグルタメートは、グルコース代謝に関係する。グルタミン酸ペプチドを標識することは、腫瘍の悪性度を区別する際に有用であり得る。本発明において、本発明者らは、EC−グルタミン酸ペンタペプチドの合成を報告し、そして腫瘍の画像化におけるその潜在的な使用を評価する。
【0055】
(腫瘍アポトーシス細胞の画像化)
アポトーシスは、化学療法および放射線を用いる癌の処置の間に生じる(Lennonら、1991;Abramsら、1990;Blakenbergら、1998;Blakenbergら、1999;TaitおよびSmith、1991)。アネキシンVは、ホスホチジルセリンに結合することが知られており、このホスホチジルセリンは、腫瘍アポトーシス細胞によって過剰発現される(Blakenbergら、1999;TaitおよびSmith、1991)。アネキシンVによるアポトーシスの評価は、治療の効力(例えば、疾患の進行または後退)を評価するために有用である。本発明において、本発明者らは、99mTc−EC−アネキシンV(EC−ANNEX)を合成し、そして腫瘍画像化におけるその潜在的な使用を評価する。
【0056】
(腫瘍新脈管形成の画像化)
新脈管形成は、部分的に、腫瘍増殖および転移の発生の原因である。抗有糸分裂性化合物は、抗脈管形成性であり、そして抗癌薬物としてのそれらの潜在的な使用について知られている。これらの化合物は、細胞サイクルの分裂期の間の細胞分裂を阻害する。細胞機能の生化学的プロセス(例えば、細胞分裂、細胞運動性、分泌、線毛運動およびべん毛運動、細胞内輸送、ならびに細胞の形態の維持)の間に、微小管が関係する。抗有糸分裂性化合物は、高い親和性で、微小管タンパク質(チューブリン)に結合し、微小管アセンブリを乱し、そして増幅細胞の有糸分裂の停止を引き起こすことが知られている。従って、抗有糸分裂性化合物は、微小管インヒビターとして、または紡錘体毒として、みなされる(Lu、1995)。
【0057】
多くの型の抗有糸分裂性化合物は、チューブリンに結合することによって、微小管アセンブリ−ディスアセンブリを制御する(Lu、1995;Gohら、1998;Wangら、1998;Rowinskyら、1990;Imbert、1998)。コルヒチノイド(colchicinoid)のような化合物は、コルヒチン結合部位上のチューブリンと相互作用し、そしてチューブリンの構築を阻害する(Lu、1995;Gohら、1998;Wangら、1998)。コルヒチノイドの中で、コルヒチンは、急性痛風の予防を処置するために使用される有効な抗炎症性薬物である。コルヒチンはまた、慢性骨髄性白血病において使用され得る。コルヒチノイドは、特定の型の腫瘍増殖に対して強力であるが、臨床的な治療可能性は、治療的効果および毒性効果を分離することが不可能であるために、制限される(Lu、1995)。しかし、コルヒチンは、細胞機能を評価するための生化学的ツールとして有用である。本発明のおいて、本発明者らは、チューブンリン機能に対する生化学的プロセスの評価のために、99mTc−EC−コルヒチン(EC−COL)を開発した。
【0058】
(腫瘍アポトーシス細胞の画像化)
アポトーシスは、化学療法および放射線を用いる癌の処置の間に生じる。アネキシンVは、ホスホチジルセリンに結合することが知られており、このホスホチジルセリンは、腫瘍アポトーシス細胞によって過剰発現される。アネキシンVによるアポトーシスの評価は、治療の効力(例えば、疾患の進行または後退)を評価するために有用である。従って、99mTc−EC−アネキシンV(EC−ANNEX)を開発した。
【0059】
(腫瘍低酸素症の画像化)
放射線治療の前の、画像化様式による腫瘍低酸素症の評価により、放射線増感剤または生体還元薬剤(bioreductive drug)(例えば、チラパザミン(tirapazamine)、マイトマイシンC)での処置のための患者を選択する合理的な手段が提供される。このような患者の選択は、低酸素症性腫瘍を有する患者のより正確な処置を可能にする。さらに、腫瘍サプレッサ遺伝子(P53)は、多重薬物耐性に関係する。化学療法の前後の組織病理学により画像化の知見とP53の過剰発現とを相関付けることは、後の腫瘍処置応答において有用である。99mTc−EC−2−ニトロイミダゾールおよび99mTc−EC−メトロニダゾルを開発した。
【0060】
(腫瘍新脈管形成の画像化)
新脈管形成は、部分的に、腫瘍増殖および転移の発生の原因である。抗有糸分裂性化合物は、抗脈管形成性であり、そして抗癌薬物としてのそれらの潜在的な使用について知られている。これらの化合物は、細胞サイクルの分裂期の間の細胞分裂を阻害する。細胞機能の生化学的プロセス(例えば、細胞分裂、細胞運動性、分泌、線毛運動およびべん毛運動、細胞内輸送、ならびに細胞の形態の維持)の間に、微小管が関係する。抗有糸分裂性化合物は、高い親和性で、微小管タンパク質(チューブリン)に結合し、微小管アセンブリを乱し、そして増幅細胞の有糸分裂の停止を引き起こすことが知られている。従って、抗有糸分裂性化合物は、微小管インヒビターとして、または紡錘体毒として、見なされる。コルヒチン(これは、強力な抗脈管形成薬剤である)は、微小管重合および中期での細胞停止を阻害することが知られている。コルヒチン(COL)は、細胞機能を評価するための生化学的ツールとして有用であり得る。次いで、99mTc−EC−COLを開発した。
【0061】
(発作に起因する低酸素症の画像化)
腫瘍細胞は、多少低酸素症的であるが、それは、酸素プローブが、圧力を測定することを必要とする。低酸素症的な条件を模倣するために、本発明者らは、発作を経験した11人の患者を、99mTc−EC−メトロニダゾル(99mTc−EC−MN)を使用して、画像化した。メトロニダゾルは、腫瘍低酸素症マーカーである。発作の範囲の組織は、酸素の欠乏に起因して、低酸素症的となる。このSPECT画像を、99mTc−EC−MNの注射の1時間後と3時間後に行った。全てのこれらの画像化研究は、積極的に、病変を局在化させた。CTは、非常に十分にまたは正確に、病変を示さなかった。いくつかの場合におけるMRIおよびCTは、病変のサイズを誇張した。以下は、3人の患者から選択されたケースである。
【0062】
ケース1. 59歳の男性患者は、左脳幹神経節に発作を患った。SPECT
99mTc−EC−MNは、注入後1時間で病変を同定し(図28)、これは、MRI
T1負荷画像に対応する(図29)。
【0063】
ケース2. 73歳の男性患者は、左中大脳動脈(MCA)領域において、発作を患った。SPECT 99mTc−EC−MNを、注入後1時間で、1日目および12日目(図30および31)で得た。これらの病変は、12日目での有意な増加した取り込みを示した。CTは、病変における広範な脳出血を示した。1日目と12日目との間に、顕著な差異は、観察されなかった(図32および図33)。これらの発見は、組織生存度に起因して、患者の症状(無酸素症から低酸素症へ)を改善したことを示す。SPECT 99mTc−EC−MNにより、CT画像化よりも良好な機能的情報が提供される。
【0064】
ケース3. 72歳の男性患者が、右MCAおよびPCA領域における発作を患った。SPECT 99mTc−EC−MNは、注入後1時間での病変を同定した(図34)。CTは、病変のサイズを誇張した(図35)。
【0065】
(腫瘍解糖標的化)
放射性標識リガンド(例えば、多糖(ネオマイシン、カナマイシン、トブラマイシン)および単糖(例えば、グルコサミン))は、細胞グルコーストランスポータ(腫瘍細胞で過剰発現される)に結合し、続いて、リン酸化される(Rogersら、1968;Fanciulliら、1994;Popoviciら、1971;Jonesら、1973;Hermannら、2000)。多糖(ネオマイシン、カナマイシン、トブラマイシン)および単糖(グルコサミン)によって誘導されるグルコースレベルは、インスリンによって抑制され得る(Haradaら、1995;Mollerら、1991;Offieldら、1996;Shankarら、1998;Yoshinoら、1999;Villevalois−Camら、2000)。これらのリガンドは、免疫原性ではなく、そして細胞質から迅速に浄化されるので、代謝画像化は、抗体画像化と比較して、有望であるようである。
【0066】
以下の実施例は、本発明の好ましい実施形態を例示することが意図される。以下の実施例に開示される技術は、本発明の実施において十分に機能することが本発明者らによって発見された技術を表し、従って、その実施のための好ましい様式を構成すると考えられ得ることが、当業者によって理解されるべきである。しかし、当業者は、本開示の観点から、多くの改変が、開示される特定の実施形態においてなされ得、そしてなおも本発明の意図および範囲から逸脱することなく同様または類似の結果を得ることを、理解する。
【実施例】
【0067】
(実施例1:腫瘍ホレートレセプター標的化)
(ECの合成)
ECを、以前に記載された方法(RatnerおよびClarke、1937;Blondeauら、1967;各々は、本明細書中に参考として援用される)に従って、2工程合成で調製した。前駆体L−チアゾリジン−4−カルボン酸を、合成した(m.p.195℃、報告値196−197℃)。次いで、ECを、調製した(m.p.237℃、報告値251−253℃)。この構造を、1H−NMRおよび高速原子ボンバードメント質量分析法(FAB−MS)によって確認した。
【0068】
(メトトレキサートのアミノエチルアミドアナログ(MTX−NH2)の合成)
MIX(227ma、0.5mmol)を、1mlのHCl溶液(2N)に溶解した。pH値を、<3とした。この攪拌溶液に、2mlの水および4mlのN−エトキシカルボニル−2−エトキシ−1,2−ジヒドロキノリン(EEDQ、メタノール中6.609%、1mmol)を、室温で添加した。エチレンジアミン(EDA、0.6 ml、10mmol)をゆっくりと添加した。この反応混合物を、一晩攪拌し、そして溶媒を減圧下でエバポレートした。未処理の固体物質を、ジエチルエーテル(10ml)、アセトニトリル(10ml)および95%エチルアルコール(50ml)で洗浄して、未反応のEEDQおよびEDAを除去した。次いで、生成物は、凍結乾燥によって乾燥し、そしてさらに精製することなく使用した。この生成物を、210mg(84.7%)であり、黄色粉末であった。生成物の融点:195−198℃(分解、MIX);1H−NMR(D2O)δ 2.98−3.04(d,8H,−(CH2)2CONH(CH0)2NH2),4.16−4.71(m,6H,−CH2−プテリジニル,芳香族−NCH3,NH−CH−COOHグルタメート),6.63−6.64(d,2H,芳香族−CO),7.51−753(d,2H.芳香族−N),8.36(s,1H,プテリジニル)。FAB MS m/z C22H28,N10,O4(M)+についての計算値496.515,実測値496.835。
【0069】
(ホレートのアミノエチルアミドアナログ(ホレート−NH2)の合成)
葉酸二水和物(1g、2.0mmol)を、10mlの水の添加した。pH値を、HCl(2N)を使用して、2に調節した。この攪拌溶液に、N−エトキシカルボニル−2−エトキシ−1,2−ジヒドロキノリン(EEDQ、10mlメタノール中1g、4.0mmol)およびエチレンジアミン(EDA、1.3ml、18mmol)をゆっくりと添加した。この反応混合物を、室温で一晩攪拌した。溶媒を、減圧下でエバポレートした。生成物を、メタノール(50ml)中で沈澱させ、そしてさらに、アセトン(100ml)で洗浄して、未反応のEEDQおよびEDITを除去した。次いで、この生成物を、凍結乾燥し、そしてさらに精製することなく使用した。ニンヒドリン(メタノール中2%)スプレーは、陽性のアミノ基を示した。この生成物は、0.6g(収率60%)であり、黄色粉末であった。生成物の融点:250℃(分解)。1H−NMR(D2O)δ 1.97−2.27(m,2H,ホレートの−CH2グルタメート),3.05−3.40(d,6H,−CH2CONH(CH2)2NH2),4.27−4.84(m,3H,−CH2−プテリジニル,NH−CH−COOHグルタメート),6.68−6.70(d,2H,芳香族−CO)、7.60−7.62(d,2H,芳香族−N),8.44(s,1H,プテリジニル)。FAB MS m/z C21H25N9,O5(M)+のついての計算値:483,実測値483.21。
【0070】
(エチレンジシステイン−ホレート(EC−ホレート)の合成)
ECを溶解するために、NaOH(2N、0.1ml)を、水(1.5ml)中のEC(114ma、0.425mmol)の攪拌溶液に添加した。この無色の溶液に、スルホ−NHS(92.3mg、0.425mmol)およびEDC(81.5mg、0.425mmol)を添加した。ホレート−NH2(205mg、0.425mmol)を、次いで、添加した。この混合物を、室温で24時間攪拌した。この混合物を、500の分子カットオフを有するSpectra/POR分子多孔性膜(Spectrum Medical Industries Inc.,Houston,TX)を使用して、48時間透析した。透析の後、生成物を凍結乾燥した。この生成物は、116mg(収率35%)であった。m.p.195℃(分解);1H−NMR(D2O)δ 1.98−2.28(m,2H,ホレートの−CH2グルタメート),2.60−2.95(m,4HおよびECの−CH2−SH),3.24−3.34(m,10H,−CH2−CO,ホレートのエチレンジアミンおよびECのエチレンジアミン),4.27−4.77(m,5H,−CH−プテリジニル,ホレートのNH−CH−COOHグルタメートおよびECのNH−CH−COOH),6.60−6.62(d,2H,芳香族−CO),7.58−7.59(d,2H.芳香族−N),8.59(s,1H,プテリジニル)。C29H37N11S2O8Na2(8H2O)についての分析計算値,FAB MS m/z(M)+777.3(水なし)。C,37.79;H,5.75;N,16.72;S,6.95。実測値:m/z (M)+777.7(20),489.4(100)。C,37.40;H,5.42;N.15.43;S,7.58。
【0071】
(EC−ホレートおよびECの99mTcでの放射標識化)
99mTc−EC−ホレートの放射合成(radiosynthesis)を、必要とされる量の99mTc−過テクネチウム酸を、自家製キット(EC−ホレートの凍結乾燥残渣(3mg)、SnCl2(100μg)、Na2HPO4(13.5mg)、アスコルビン酸(0.5mg)およびNaEDTA(0.5mg)を含む)に添加することによって達成した。調製物の最終pHは、7.4であった。99mTc−ECをまた、ECの凍結乾燥残渣(3mg)、SnCl2(100μg)、Na2HPO4(13.5mg)、アスコルビン酸(0.5mg)およびpH10のNaEDTA(0.5mg)を含む自家製キットを使用することによって、得た。次いで、調製物のpHを7.4に調節した。放射化学純度を、各々、アセトン(系A)および酢酸アンモニウム(水中1M):メタノール(4:1)(系B)で溶出するTLC(ITLC SG,Gelman Sciences,Ann Arbor,MI)によって、決定した。radio−TLC (Bioscan,Washington,DC)分析から、放射化学純度は、両方の放射性医薬品について、>95%であった。Radio−TLCデータを表2に要約する。99mTc−EC−ホレートの合成を、図1に示す。
【0072】
(表2)
(癌化学療法のための選択された薬物)
以下の表は、USAおよびカナダにおける癌の治療のために使用される薬物およびそれらの主要な有害な影響を列挙する。列挙する選択された薬物は、Medical Letterコンサルタントの意見に基づいた。いくつかの薬物を、それらが、米国食品医薬品庁によって承認されていない指標について列挙する。抗癌薬物およびそれらの有害な影響については、以下の通りである。本発明の目的のために、これらの列挙は、例示を意味し、全てを網羅するものではないことを意味する。
【0073】
【表2】
* 化学療法は、中程度の活性のみを有する。
** 化学療法は、少ない活性のみを有する。
1 化学療法を伴うかまたは伴わないタモキシフェンは、一般に、閉経後エストロゲン−レセプター陽性の、モード陽性患者に対して推奨され、そしてタモキシフェンを伴うかまたは伴わない化学療法が、閉経前のモード陽性な患者に対して一般に推奨される。化学療法および/またはタモフェキシンを伴うアジュバント処置が、より大きな腫瘍または他の有害な予後指標を有するモード陰性患者に対して推奨される。
2 メゲストロールおよび他のホルモン剤は、タモキシフェンが作用しないいくらかの患者において有用であり得る。
3 高用量化学療法の後(Medical Letter,34:79,1982)。
4 直腸癌について、フルオロウラシル単独での処置の前後に、フルオロウラシル(fluoroutacil)および放射線を用いる手術後アジュバント処置。
5 手術切除、放射線治療、または両方と組合された場合にのみ、薬物が主要な活性を有する。
6 ビタミンAアナログである、ラクトラチノル(lactratinoln)(Acgutana)は、前腫瘍性病変(白斑症(leukoplakla))を制御し得、そして第二の原発性腫瘍の割合を減少し得る(SE Bannerら、J Natl Cancer Inst,88:140 1994)。
† 調査用途のためにUSAのみで利用可能。
7 高リスク患者(例えば、高カウント、細胞遺伝異常、成人)は、誘導、維持および「強化」(寛解の達成後のさらなる薬物の使用)のためのさらなる薬物を必要とし得る。さらなる薬物としては、シクロホスファミド(cyclophosphamida)、ミトキサトロンおよびチオグアニン(thloguanine)が挙げられる。イギリスにおける1つの大きな制御された試行の結果は、強化が、ALLの全ての子供の生存率を改善し得ることを示唆する(JM Chasselleら、Lancet,34B:143,Jan 21,1995)。
8 初めに悪い予後を有する患者または寛解後に再発した患者。
9 急性前骨髄球白血病を有する何人かの患者は、トラチノイン(tratinoin)に対する完全な応答を有した。このような処置は、主に発熱および呼吸困難により特徴付けられる有毒な症状を引き起こし得る(RP Warrell,Jrら、N Engl J Med.328:177,1993)。
10 同種異系HLA同一同胞骨髄移植は、慢性期のCMLの患者の40%〜70%、加速期のCMLの患者の18%〜28%、急性転化の患者の15%未満を治癒し得る。骨髄移植後の疾患を有さない生存率は、50歳より大きい年齢、診断から3年より長い疾患の持続時間、および1抗原不適合または適合無関係のドナーの骨髄の使用により悪影響を受けた。インターフェロンαはまた、慢性期のCMLを有する患者(この患者は完全な細胞遺伝的応答(約10%)を達成する)において治癒的であり得;これは、新しく慢性期CMLと診断された80歳を越える年齢の患者、および同種異系骨髄移植の候補ではない全ての患者のために選択される処置である。化学治療単独は待機的である。
11 これらの組み合わせのいずれかを用いて第2の慢性期が達成される場合、同種異系骨髄移植が考慮されるべきである。第2の慢性期における骨髄移植は、CMLの患者の30%〜35%に対して治癒的であり得る。
12 極限段階のホジキン病(1段階および2段階)は、放射線治療によって治癒可能である。播種性疾患(3b段階および4段階)は、化学治療を必要とする。いくつかの中間段階および選択された臨床的状況が、この両方から利益を受ける。
【0074】
【表2−2】
† 研究用途のためのみにUSAで利用可能。
‡ 用量制限効果は太字である。皮膚反応(時折、重篤)、色素過剰、および眼毒性は、実質的に全ての非ホルモン性抗癌剤で報告された。他の薬物との悪い相互作用については、Medical Letter Handbook of Adverse Drug
Interactions,1995を参照のこと。
1. 研究用途のためのみにUSAで利用可能。
2. メゲストロールおよび他のホルモン剤は、タモキシフェンで失敗した場合の患者において有効であり得る。
3. 高用量化学治療後(Medical Letter,34:78,1992)。
4. 直腸癌について、フルオロウラシルのみを用いる処置の前および後の、フルオロウラシル+放射線を用いる手術後のアジュバント処置。
5. 薬物は、外科的切除、放射線治療またはその両方と組み合わせた場合にのみ、主な活性を有する。
6. ビタミンAアナログのイソトレチノイン(Accutane)は、新生物発生前のイシオン(ision)(白斑症)を制御し、そしてラットの第1の一次腫瘍を減少させ得る(SE Sennerら、J Natl Cancer Inst.88:140,1994)。
7. 高リスク患者(例えば、高カウント、細胞遺伝異常、成人)は、誘導、維持および「強化」(寛解の達成後のさらなる薬物の使用)のためのさらなる薬物を必要とし得る。さらなる薬物としては、シクロホスファミド、ミトキサトロンおよびチオグアニンが挙げられる。イギリスにおける1つの大きな制御された試行の結果は、強化がALLの全ての子供の生存率を改善し得ることを示唆する(JM Chassellaら、Lancet,348:143,Jan 21,1998)。
8. 初めに悪い予後を有する患者または寛解後に再発した患者。
9. 急性前骨髄球白血病を有する何人かの患者は、トレチノインに対する完全な応答を有した。このような処置は、主に発熱および呼吸困難により特徴付けられる有毒な症状を引き起こし得る(RP Warrell,Jrら、N Engl J Med.329:177,1993)。
10. 同種異系HLA同一同胞骨髄移植は、慢性期のCMLの患者の40%〜70%、加速期のCMLの患者の15%〜25%、急性転化の患者の15%未満を治癒し得る。骨髄移植後の疾患を有さない生存率は、50歳より大きい年齢、診断から3年より長い疾患の持続時間、および1抗原不適合または適合無関係のドナーの骨髄の使用により悪影響を受けた。インターフェロンαはまた、慢性期のCMLを有する患者(この患者は完全な細胞遺伝的応答(約10%)を達成する)において治癒的であり得;これは、新しく慢性期CMLと診断された50歳を越える年齢の患者、および同種異系骨髄移植の候補ではない全ての患者のために選択される処置である。化学治療単独は待機的である。
【0075】
(99mTcでのCE−MTXおよびEC−TDXの放射標識)
EC−ホレートの合成について記載された同じ方法を使用して、EC−MTXおよびEC−TDXを調製した。標識手順は、EC−MTXおよびEC−TDXを使用したこと以外は、99mTc−EC−ホレートの調製について記載された手順と同じである。99mTc−EC−MTXおよび99mTc−EC−TDXの合成を、図2および図3に示す。
【0076】
(99mTc−EC−ホレート、99mTc−EC−MTXおよび99mTc−EC−TDXの安定性アッセイ)
99mTc−EC−ホレート、99mTc−EC−MTXおよび99mTc−EC−TDXの安定性を、血清サンプル中で試験した。簡単には、740KBqの1mgの99mTc−EC−ホレート、99mTc−EC−MTXおよび99mTc−EC−TDXを、イヌ血清(200μl)中で、37℃で4時間インキュベートした。この血清サンプルを、水中50%メタノールで希釈し、そして放射性TLCを、上記のように、0.5時間、2時間および4時間で繰り返した。
【0077】
(組織分布研究)
雌性Fischer 344ラット(150±25g)(Harlan Sprague−Dawley,Indianapolis,IN)に、0.1mlの乳癌細胞を、25ゲージ針を使用して、013762腫瘍細胞株懸濁液(106細胞/ラット、Fixcherラットに特異的な腫瘍細胞株)から後脚に皮下接種した。腫瘍の直径が約1cmに達した場合、移植後14〜17日間研究を実施した。各手順の前に、動物をケタミン(10〜15mg/ラット、腹腔内)で麻酔した。
【0078】
組織分布研究において、各動物に、370〜550KBqの99mTc−EC−ホレートまたは99mTc−ECを静脈内注射した(n=3/時点)。各リガンドの注入質量は、10μg/ラットであった。放射性医薬の投与後20分、1時間、2時間および4時間において、麻酔した動物を屠殺し、そして腫瘍および選択した組織を切除し、計量し、そしてγ線計数器(Packard Instruments,Downers Grove,IL)で放射能をカウントした。各サンプル中のトレーサーの体内分布を、組織の湿潤重量1gあたりの注射用量の割合(%ID/g)として算出した。元の注射液の希釈サンプルのカウントを、参照のために使用した。腫瘍/非標的組織の計数密度比を、対応する%ID/g値から算出した。スチューデントt検定を使用して、2つの群間の差違の有意性を評価した。
【0079】
別の研究において、ブロッキング研究を行って、レセプター媒介型プロセスを決定した。ブロッキング研究において、99mTc−EC−ホレートを、腫瘍保有マウスに、50および150μmol/kgの葉酸と同時投与(i.v.)した(n=3/群)。注射の1時間後に動物を殺し、そしてデータを収集した。
【0080】
(シンチグラフィー画像化およびオートラジオグラフィー研究)
シンチグラフィー画像を、低エネルギー平行穴型コリメーターを備えるγ線カメラ(Siemens Medical Systems,Inc.,Hoffman Estates,IL)を使用して、18.5MBqの99mTc標識放射性トレーサーの静脈内注射後0.5時間、2時間および4時間で得た。
【0081】
全身オートラジオグラフィーを、定量用画像分析器(Cyclone Storage
Phosphor System,Packard,Meridian,CI.)によって得た。37MBqの99mTc−EC−ホレートの静脈内注射後1時間で動物を殺し、そして死体をカルボキシメチルセルロース(4%)中に固定した。冷凍した死体をクリオスタット(LKB 2250クリオマイクロトーム)中におき、そして100μmの冠状切片に切断した。各切片を解凍し、そしてスライドに取り付けた。次いで、このスライドを汎用リン光ストレージスクリーン(MP,7001480)に接触して配置し、そして15時間99mTc標識に暴露した。このリン光スクリーンを赤色レーザー光で励起し、そして以前に吸収されたエネルギーに比例する得られる青色光を記録した。
【0082】
(結果)
(99mTc−EC−ホレートの化学および安定性)
単純であり、高速かつ高収率のホレートのアミノエチルアミドおよびECアナログ、MTXおよびTDXを開発した。これらのアナログの構造を、NMRおよび質量分光分析により確認した。EC−ホレートの99mTcでの放射性合成を、高い(>95%)放射化学的純度で達成した。99mTc−ECホレートは、イヌ血清サンプル中で、20分、1時間、2時間および4時間において安定であることが見出された。
【0083】
(99mTc−EC−ホレートの体内分布)
体内分布研究により、20分〜4時間における腫瘍/血液計数密度比は、99mTc−EC−ホレートについて、徐々に増加し、一方、これらの値は、同じ時間における99mTc−ECについて減少することが示された(図4)。99mTC−EC−ホレートおよび99mTc−ECの%ID/g取込み値、腫瘍/血液比、および腫瘍/筋肉比を、それぞれ、表3および4に示した。
【0084】
(表3)
乳房腫瘍保有ラットにおける99mTc−EC−ホレートの体内分布
【0085】
【表3】
示される値は、3匹の動物のデーターの平均±標準偏差を表す。
【0086】
(シンチグラフィー画像化およびオートラジオグラフィー研究)
異なる時点において得られたシンチグラフィー画像は、99mTc−EC−葉酸を注射した群における腫瘍の可視化を示した。対照的に、99mTc−ECを注射した群においては、明らかな腫瘍取り込みが存在しなかった(図6)。両方の放射性トレーサーは、全ての画像における明らかな腎臓取込みを示した。99mTc−EC−葉酸の注射の1時間後に実施したオートラジオグラムは、腫瘍の活性を明らかに示した。
【0087】
(実施例2:腫瘍低酸素症標的化)
(2−(2−メチル−5−ニトロ−1Hイミダゾリル)エチルアミン(メトロニダゾルのアミノアナログMN−NH2)の合成)
メトロニダゾルのアミノアナログを、以前に記載された方法に従って合成した(Hayら、1994)。簡単に言えば、メトロニダゾルをメシル化アナログ(m.p.149〜150℃、報告値153〜154℃、TLC:酢酸エチル、Rf=0.45)に転換し、75%を得た。次いで、メシル化メトロニダゾルをアジ化ナトリウムと反応させて、アジドアナログ(TLC:酢酸エチル、Rf=0.52)を、収率80%で得た。このアジドアナログを、トリフェニルホスフィンによって還元し、そして所望のアミノアナログ(m.p.190〜192℃、報告値194〜195℃、TLC:酢酸エチル、Rf=0.15)を得た(60%)。ニンヒドリン(メタノール中2%)のスプレーは、MN−NH2のアミノ基の陽性を示した。構造を、1H−NMRおよび質量分析(FAB−MS)m/z 171(M+H,100)によって確認した。
【0088】
(エチレンジシステイン−メトロニダゾル(EC−MN)の合成)
水酸化ナトリウム(2N、0.2ml)を、水(5ml)中EC(134ma、0.50mmol)の攪拌溶液に添加した。この無色の溶液に、スルホ−NHS(217mg、1.0mmol)および1〜)C(192ma.1.0mmol)を添加した。次いで、MN−NH:二塩酸塩(340mg、2.0mmol)を添加した。この材料(mature)を室温で24時間攪拌した。この混合物を、500でのカットオフを有するSpectra/POR分子多孔質膜(Spectrum MedicalIndustries Inc.,Houston,TX)を使用して、48時間透析した。透析後、生成物を、凍結乾燥機(Labconco,Kansas City,MO)を使用してフリーズドライした。生成物の重量は、315mgであった(収率55%)。1H−NMR(D2O)δ2.93(s、6H、ニトロイミダゾール−CH3)、2.60〜2.95(m,4HおよびECの−CH2−SH)、3.30〜3.66(m、8H、ECのエチレンジアミンおよびニトロイミダゾール−CH2−CH2−NH2)、3.70〜3.99(t、2H、ECのNH−CH−CO)、5.05(t、4H、メトロニダゾル−CH2−CH2−NH2)、(s、2H、ニトロイミダゾールC=CH)。FAB MS m/z
572(M+,20)。EC−MNの合成スキームを、図7に示す。
【0089】
(3−(2−ニトロ−1H−イミダゾリル)プロピルアミン(ニトロイミダゾールのアミノアナログNIM−NH2)の合成)
2−ニトロイミダゾール(1g、8.34mmol)およびSc2CO3(2.9g,8.90mmol)を含むジメチルホルムアミド(DMF、50ml)中の攪拌溶液に、1,3−ジトシルプロパン(3.84g、9.99mmol)を添加した。この反応物を80℃で3時間加熱した。溶媒を減圧下でエバポレートし、そして残渣を酢酸エチル中に懸濁させた。固体を濾過し、溶媒を濃縮し、シリカゲルを充填したカラムに装填し、そして、ヘキサン:酢酸エチル(1:1)で溶出した。生成物である、3−トシルプロピル−(2−ニトロイミダゾール)を、m.p.108〜111℃で単離した(1.67g、収率57.5%)。1H−NMR(CDCl3)δ2.23(m、2H)、2.48(S、3H)、4.06(t、2H、J=5.7Hz)、4.52(t、2H、J=6.8Hz)、7.09(S、1H)、7.24(S、1H)、7.40(d、2H、J=8.2Hz)、7.77(d、2H、J=8.2Hz)。
【0090】
次いで、トシル化2−ニトロイミダゾール(1.33g、4.08mmol)を、DMF(10ml)中アジ化ナトリウム(Q29g、4.49mmol)と、100℃で3時間反応させた。冷却後、水(20ml)を添加し、そして生成物を酢酸エチル(3×20ml)から抽出した。溶媒を、MgSO4で乾燥し、そして乾固するまでエバポレートして、アジドアナログ(0.6g、75%、TLC:ヘキサン:酢酸エチル;1:1、Rf=0.42)を得た。1H−NMR(CDCl3)δ2.14(m、2H)、3.41(t、2H、J=6.2Hz)、4.54(t、2H、J=6.9Hz)、7.17(S、2H)。
【0091】
このアジドアナログ(0.57g、2.90mmol)を、テトラヒドロフラン(PHI;)中のトリフェニル(taphenyl)ホスフィン(1.14g、4.35mmol)によって、室温で4時間還元した。濃HCl(12ml)を添加し、そしてさらに5時間加熱した。生成物を、酢酸エチルと水との混合物から抽出した。この酢酸エチルをMgSO4で乾燥し、そして乾固するまでエバポレートして、アミン塩酸塩アナログを得た(360ma、60%)。ニンヒドリン(メタノール中2%)のスプレーは、NIM−NHのアミノ基の陽性を示した。1H−NMR(D2O)δ2.29(m、2H)、3.13(t、2H、J=7.8Hz)、3.60(br、2H)、4.35(t、2H、J=7.4Hz)、7.50(d、1H、J=2.1Hz)、7.63(d、1H、J=2.1Hz)。
【0092】
(エチレンジシステイン−ニトロイミダゾール(EC−NIM)の合成)
水酸化ナトリウム(2N、0.6ml)を、EC(134ma、0.50mmol)の水(2ml)中の攪拌溶液に添加した。この無色の溶液に、スルホ−NHS(260.6mg、1.2mmol)、EDC(230ma、1.2mmol)および水酸化ナトリウム(2N、1ml)を添加した。次いでNIM−NH2塩酸塩(206.6mg、1.0mmol)を添加した。この混合物を室温で24時間攪拌した。この混合物を、500でのカットオフを有するSpectra/POR分子多孔質膜(Spectrum Medical Industries Inc.,Houston,TX)を使用して、48時間透析した。透析後、生成物を、凍結乾燥機(Labconco,Kansas City,MO)を使用してフリーズドライした。生成物の重量は、594.8mgであった(収率98%)。EC−NIMの合成スキームを、図8Aに示す。構造を、1H−NMR(D2O)によって確認する(図8B)。
【0093】
(EC−MNおよびEC−NIMの、99mTcでの放射性同位元素標識)
99mTc−EC−MNおよび99mTc−EC−NIMの放射線合成を、必要量の過テクネチウム酸を、EC−MNまたはEC−NIMの凍結乾燥した残渣(3mg)、SnCl2(100μg)、Na2HPO4(13.5mg)、アスコルビン酸(0.5mg)およびNaEDTA(0.5mg)を含む自家製のキットに添加することによって、達成した。調製物の最終pHは、7.4であった。放射化学的純度を、それぞれアセトン(系A)および酢酸アンモニウム(水中1M):メタノール(4:1)(系B)で溶出するTLC(ITLAC SG、Gelman Sciences,Ann Arbor、MI)によって決定した。放射線TLC(Bioscan,Washington,DC)分析から、両方の放射線トレースに対して放射化学的純度は96%より高かった。
【0094】
([18F]FMISOおよび[131I]IMISOの合成)
[F]フッ素を、小容量の銀標的において富化された18O−水のプロトン照射を使用するサイクロトロンによって、生成した。トシルMISO(Hayら、1994)(20mg)を、アセトニトリル(1.5ml)に溶解し、クリプトフィックス−フッ化物錯体に添加した。加熱後、加水分解およびカラム精製により、25〜40%の収率(崩壊を補正した)の純粋な生成物を、60分のボンバードメントの終了(EOB)において、単離した。HPLCを、C−18 ODS−20Tカラム(4.6×25mm)(Waters Corp.,Milford,Mass)において、水/アセトニトリル(80/20)を用いて、1ml/分の流速を使用して実施した。キャリアが添加されていない生成物は、類似の条件下での非標識FMISOの保持時間(6.12分)に対応した。放射化学的純度は、99%より高かった。UV検出器(310nm)のもとで、他の不純物は存在しなかった。決定した[18F]FMISOの比活性は、既知の質量および放射能のサンプルのUVおよび放射能検出に基づいて、1Ci/μmolであった。
【0095】
[13I]IMISOを、同じ手順を使用して調製した(Cherifら、1994)。簡単に言えば、5mgのトシルMISOをアセトニトリル(1ml)に溶解し、そしてNa131I(0.1mlの1N NaOH中1mCi)(Dupont New England Nuclear,Boston.MA)を添加した。加熱および精製の後、生成物(収率60〜70%)を得た。放射線TLCは、溶出液としてクロロホルムメタノール(7:3)を使用して、最終生成物に対して0.01のRf値を示した。
【0096】
(99mTc−EC−MNおよび99mTc−EC−NIMの安定性アッセイ)
標識された99mTc−EC−MNおよび99mTc−EC−NIMの安定性を、血清サンプルにおいて試験した。簡単に言えば、740KBqの1mgの99mTc−EC−MNおよび99mTc−EC−NIMを、イヌ血清(200μl)中37℃で4時間インキュベートした。これらの血清サンプルを、水中50%メタノールで希釈し、そして放射線TLCを、上記のように、0.5時間、2時間および4時間において繰り返した。
【0097】
(99mTc−EC−MNの組織分布研究)
雌性Fischer344ラット(150±25g)(Harlan Sprague−Dawley,Indianapolis,IN)に、13762腫瘍細胞株懸濁液(106細胞/ラット、Fischerラットに対して特異的な腫瘍細胞株)由来の0.1mlの哺乳動物腫瘍細胞を、25ゲージの針を使用して後肢に皮下接種した。研究を、移植の14〜17日後に、腫瘍がおおよそ1cmの直径に達したときに実施した。ラットをケタミン(10〜15mg/ラット、腹腔内)で麻酔し、その後、それぞれの手順を行った。
【0098】
組織分布研究において、各動物に、370〜550KBqの99mTc−EC−MNまたは99mTc−EC(n=3/時点)を静脈内注射した。99mTc−EC−MNの注射した質量は、1匹のラットあたり10μgであった。放射線トレーサーの投与の0.5時間後、2時間後および4時間後に、これらのラットを屠殺し、そして選択した組織を切除し、秤量し、そして放射能を計数した。各サンプルにおけるトレーサーの生体分布を、組織含水重量1グラムあたりの注射用量の百分率(%ID/g)として、算出した。腫瘍/非標的組織の計数密度比を、対応する%ID/g値から算出した。このデータを、同じ動物モデルを使用して[18F]FMISOおよび[131I]IMISOと比較した。Studentのt検定を使用して、群間の差異の有意性を評価した。
【0099】
(シンチグラフィー画像化およびオートラジオグラフィー研究)
低エネルギーの平行穴コリメータを備えるγ線カメラ(Siemens Medical Systems,Inc.,Hoffman Estates,IL)を使用するシンチグラフィー画像を、18.5MBqの各放射線トレーサーの静脈内注射の0.5時間後、2時間後および4時間後に得た。
【0100】
全身のオートラジオグラムを、定量的画像分析器(Cyclone Storage Phosphor System,Packard,Meridian,CT)によって得た。37MBqの99mTc−EC−MNの静脈内注射に続いて、これらの動物を1時間で殺傷し、そしてその身体をカルボキシメチルセルロース(4%)に、以前に記載されたように固定した(Yangら、1995)。凍結させた身体を低温槽(LKB 2250クリオミクロトーム)に設置し、そして100μmの冠状切片に切断した。各切片を解凍し、そしてスライドに載せた。次いで、このスライドを多目的リン貯蔵スクリーン(MP、7001480)と接触させ、そして15時間露光した。
【0101】
99mTc−EC−NIMが化学療法に対する腫瘍応答をモニタリングし得るか否かを確認するために、腫瘍容量1.5cmを有するラットおよび卵巣腫瘍保有マウスの群を、パクリタキセル(40mg/kg/ラット、80mg/kg/マウス、静脈内)で、単一用量で処置した。画像を、パクリタキセル処置の4日後に撮像した。処置ありまたは処置なしでの、1グラムの腫瘍重量あたりの注射された用量の百分率を、決定した。
【0102】
(ポーラログラフィー酸素微小電極によるpO2測定)
腫瘍の低酸素症を確認するために、腫瘍内のpO2測定を、エッペンドルフコンピュータ化ヒストグラムシステムを使用して、実施した。2〜3の直線軌道の各々に沿った、20〜25のpO2測定を各腫瘍において0.4mmの間隔で実施した(合計40〜75の測定)。腫瘍pO測定を、3匹の腫瘍保有ラットにおいて実施した。オンラインコンピュータシステムを使用して、各軌道のポット測定を、この軌道に沿った測定点の位置に対して絶対的な値として表現し、そして2.5mmのクラス幅の0mmHgと100mmHgとの間のpO2ヒストグラムの相対頻度として、表した。
【0103】
(結果)
(99mTc−EC−MNおよび99mTc−EC−NIMの放射線合成および安定性)
EC−MNおよびEC−NIMの、99mTcでの放射線合成を、高い放射化学的純度(95%より高い)で達成し、放射化学的収率は100%であった。99mTc−EC−MNおよび99mTc−EC−NIM(図13)は、イヌ血清サンプルにおいて、0.5時間、2時間および4時間安定であることが見出された。分解生成物は、観察されなかった。MISOの放射性フッ素化および放射性ヨウ素化は、同じ前駆体を使用して、容易に達成された。両方の標識されたMISOアナログにおいて、放射化学的純度は、99%より高かった。
【0104】
(インビボ組織分布研究)
99mTc−EC−MNおよび99mTc−ECの、腫瘍保有ラットにおける組織分布を、表4および5に示す。イオン性99mTcに対する高い親和性に起因して、有意な一貫した甲状腺取り込みは存在せず、インビボでの99mTc−EC−MNの安定性を示唆する(表5)。
【0105】
【表4】
示される値は、3匹の動物からのデータの平均±標準偏差を表す。
【0106】
ブロッキング研究において、腫瘍/筋肉および腫瘍/血液の計数密度比は、葉酸の同時投与によって、有意に低下した(p<0.01)(図5)。
【0107】
【表5】
1.各ラットが、99mTc−EC−メトロニダゾル(10μCi、静脈内)を受容した。各値は、1グラムの重量あたりの注射用量(n=3)/時間間隔の百分率である。各データは、標準偏差を伴う3回の測定の平均を表す。
【0108】
生体分布研究は、0.54時間における腫瘍/血液および腫瘍/筋肉の計数密度比が、99mTc−EC−NM、[18F]FMISOおよび[131I]IMISOにおいては次第に増加し、一方でこれらの値は、同じ時間において、99mTc−ECについては変化しないことを示した(図9および図10)。[18F]FMISOは、注射後30分、2時間および4時間において、[131I]IMISOおよび99mTc−EC−MNの取り込み比と比較して、最も高い腫瘍対血液の取り込み比を示した。注射後2時間および4時間における99mTc−EC−MNおよび[131I]IMISOについての、腫瘍/血液および腫瘍/筋肉の比は、有意には異ならなかった(p<0.05)。
【0109】
(シンチグラフ画像化およびオートラジオグラフィー研究)
異なる時点において得られたシンチグラフィー画像は、99mTc−EC−MNおよび99mTc−EC−NIM群における腫瘍の可視化を示した。対照的に、99mTc−ECを注射した群においては、明らかな腫瘍の取り込みが存在しなかった(図11)。99mTc−EC−MNの注射の1時間後に実施したオートラジオグラムは、明らかに、腫瘍活性を示した(図12)。99mTc−EC−NMと比較して、99mTc−EC−NIMは、より高い腫瘍対バックグラウンドの比に起因して、より良好なシンチグラフィー画像を提供するようであった。乳房腫瘍保有ラットにおいて、腫瘍の取り込みは、99mTc−ECと比較して、99mTc−EC−NIM群において顕著に高かった(図14A)。1グラムの腫瘍の重量あたりの99mTc−EC−NIMの注射用量の百分率から得たれたデータは、コントロール群と比較した場合に、パクリタキセルで処置したラットにおいて、25%が取り込みを低下したことを示した(図14B)。
【0110】
卵巣癌保有マウスにおいて、パクリタキセルで処置したマウスにおける低下した腫瘍取り込みが存在した(図15Aおよび図15B)。類似の結果が、肉腫保有において観察された(図15Cおよび図15D)。従って、99mTc−EC−NIMは、パクリタキセル処置に対する腫瘍応答を評価するために、使用され得る。
【0111】
(ポーラログラフィー酸素微小電極によるpO2測定)
腫瘍の腫瘍内PO2測定は、腫瘍の酸素圧力が、正常筋肉の35±10mmHgと比較して、4.6±4.1mmHgの範囲であることを示した。このデータは、腫瘍が低酸素症であることを示す。
【0112】
(実施例3:癌のペプチド画像化)
(エチレンジシステイン−ペンタグルタミン酸(EC−GAP)の合成)
水酸化ナトリウム(1N,1ml)を、水(10ml)中のEC(200mg、0.75mmol)の撹拌溶液に添加した。この無色溶液に、スルホNHS(162mg、0.75mmol)およびEDC(143mg、0.75mmol)を添加した。次いでペンタグルタミン酸ナトリウム塩(M.W.750〜1500、Sigma Chemical Company)(500mg、0.67mmol)を添加した。混合物を24時間室温で撹拌した。混合物を、500のカットオフを有するSpectra/POR分子多孔膜(Spectrum Medical Industries Inc.,Houston,TX)を使用して48時間透析した。透析後、生成物を、凍結乾燥機(Labconco,Kansas City,MO)を使用して凍結乾燥した。塩形態の生成物は、0.95gの重さであった。EC−GAPの合成スキームは、図16に示す。
【0113】
(99mTc−EC−GAPの安定性アッセイ)
99mTcでのEC−GAPの放射性標識を、以前に記載されたのと同じ手順を使用して達成した。放射化学純度は、100%であった。99mTc−EC−GAPの安定性を、血清サンプル中で試験した。手短にいうと、740KBqの1mg99mTc−EC−GAPを、37℃で4時間、イヌ血清(200μl)中でインキュベートした。血清サンプルを、水中50%メタノールで希釈し、そして放射性TLCを、上記のように、0.5時間、2時間、および4時間で繰り返した。
【0114】
(シンチグラフィー画像化研究)
低エネルギーの、平行な穴のコリメータを備えたγカメラを使用して、シンチグラフィー画像を、18.5MBqの各放射性トレーサーの静脈内注射の0.5時間、2時間および4時間後に得た。
【0115】
(結果)
(99mTc−EC−GAPの安定性アッセイ)
99mTc−EC−GAPは、イヌ血清サンプル中で、0.5時間、2時間および4時間で安定であることが見出された。分解産物は、観察されなかった。
【0116】
(シンチグラフィー画像化研究)
異なる時点で得られたシンチグラフィー画像は、99mTc−EC−GAP群において腫瘍の可視化を示した。最適な取り込みは、投与後30分〜1時間である(図17)。
【0117】
(実施例4:腫瘍アポトーシス細胞の画像化)
(エチレンジシステイン−アネキシンV(EC−ANNEX)の合成)
炭酸水素ナトリウム(1N、1ml)を、EC(5mg、0.019mmol)の撹拌溶液に添加した。この無色溶液に、スルホNHS(4mg、0.019mmol)およびEDC(4mg、0.019mmol)を添加した。次いでアネキシンV(M.W.33kD、ヒト、Sigma Chemical Company)(0.3mg)を添加した。混合物を24時間室温で撹拌した。混合物を、10,000のカットオフを有するSpectra/POR分子多孔膜(Spectrum Medical Industries Inc.,Houston,TX)を使用して48時間透析した。透析後、生成物を、凍結乾燥機(Labconco,Kansas City,MO)を使用して凍結乾燥した。塩形態の生成物は、12mgの重さであった。
【0118】
(99mTc−EC−ANNEXの安定性アッセイ)
99mTcでのEC−ANNEXの放射性標識を、EC−GAPにおいて記載されたのと同じ手順を使用して達成した。放射化学純度は、100%であった。標識した99mTc−EC−ANNEXの安定性を、血清サンプル中で試験した。手短にいうと、740KBqの1mg99mTc−EC−ANNEXを、37℃で4時間、イヌ血清(200μl)中でインキュベートした。血清サンプルを、水中50%メタノールで希釈し、そして放射性TLCを、上記のように、0.5時間、2時間、および4時間で繰り返した。
【0119】
(シンチグラフィー画像化研究)
低エネルギーの、平行な穴のコリメータを備えたγカメラを使用して、シンチグラフィー画像を、18.5MBqの各放射性トレーサーの静脈内注射の0.5時間、2時間および4時間後に得た。使用した動物モデルは、乳房、卵巣および肉腫であった。乳房腫瘍および卵巣腫瘍を有する両方のラットは、高いアポトーシス性の細胞を過剰発現することが知られている。画像化研究を、腫瘍細胞接種後14日で行なった。腫瘍処置応答を確かめるために、画像化前のマウスを、パクリタキセル投与し(80mg/Kg、静脈内注射、14日目)、そして画像を18日目に取った。
【0120】
(結果)
(99mTc−EC−ANNEXの安定性アッセイ)
99mTc−EC−ANNEXは、イヌ血清サンプル中で、0.5時間、2時間および4時間で安定であることが見出された。分解産物は、観察されなかった。
【0121】
(シンチグラフィー画像化研究)
異なる時点で得られたシンチグラフィー画像は、99mTc−EC−ANNEX群において腫瘍の可視化を示した(図18〜20)。画像は、高度にアポトーシス性の細胞が、より多くの99mTc−EC−ANNEXの取り込みを有することを示した。高いアポトーシス(卵巣腫瘍保有)群(図19Aおよび図19B)および低アポトーシス(肉腫腫瘍保有)群(図20Aおよび図20B)におけるパクリタキセル処理前とパクリタキセル処理後との間に、腫瘍取り込みの顕著な差異は、存在しなかった。
【0122】
(実施例5:腫瘍新脈管形成の画像化)
((コルヒチンのアミノアナログ、COL−NH2)の合成)
コルヒチンの脱メチル化アミノアナログおよび脱メチル化ヒドロキシアナログを以前に記載された方法(Orrら、1995)に従って合成した。手短にいうと、コルヒチン(4g)を、25%硫酸を含む100mlの水中に溶解した。反応混合物を、100℃で5時間加熱した。混合物を、炭酸ナトリウムを用いて中和した。生成物をろ過し、そして、凍結乾燥機で乾燥し、2.4g(70%)の所望のアミノアナログ(融点153〜155℃、155〜157℃と報告されている)を生じた。ニンヒドリン(メタノール中2%)噴霧は、COL−NH2のアミノ基の陽性を示した。構造を、1H−NMRおよび質量分析法(FAB−MS)によって確かめた。
【0123】
【数1】
(エチレンジシステイン−コルヒチン(EC−COL)の合成)
水酸化ナトリウム(2N,0.2ml)を、水(5ml)中のEC(134mg、0.50mmol)の撹拌溶液に添加した。この無色溶液に、スルホNHS(217mg、1.0mmol)およびEDC(192mg、1.0mmol)を添加した。次いでCOL−NH2(340mg、2.0mmol)を添加した。混合物を24時間室温で撹拌した。混合物を、500のカットオフを有するSpectra/POR分子多孔膜(Spectrum Medical Industries Inc.,Houston,TX)を使用して48時間透析した。透析後、生成物を、凍結乾燥機(Labconco,Kansas City,MO)を使用して凍結乾燥した。塩形態の生成物は、315mgの重さであった(収率55%)。
【0124】
【数2】
EC−COLの合成スキームを、図21に示す。
【0125】
(EC−COLおよびECの99mTcを用いる放射性標識)
99mTc−EC−COLの放射性合成を、EC−COLの凍結乾燥残渣(5mg)、SnCl2(100μg)、Na2HPO4(13.5mg)、アスコルビン酸(0.5mg)およびNaEDTA(0.5mg)を含む自家製キットへ必要な量の99mTc−過テクネチウム酸を添加することによって達成した。調製物の最終pHは、7.4であった。99mTc−ECはまた、pH10で、ECの凍結乾燥残渣(5mg)、SnCl2(100μg)、Na2HPO4(13.5mg)、アスコルビン酸(0.5mg)およびNaEDTA(0.5mg)を含む自家製キットを使用することによって得た。次いで、調製物の最終pHを7.4に調整した。放射化学純度を、酢酸アンモニウム(水中1M):メタノール(4:1)を用いて溶出するTLC(ITLC SG,Gelman Sciences,Ann Arbor,MI)によって決定した。放射線−薄層クロマトグラフィー(TLC,Bioscan,Washington,DC)を使用して、両方の放射線トレーサについての放射化学純度を分析した。
【0126】
(99mTc−EC−COLの安定性アッセイ)
標識された99mTc−EC−COLの安定性を、血清サンプル中で試験した。手短にいうと、740KBqの5mg99mTc−EC−COLを、37℃で4時間、ウサギ(rabbinate)血清(500μl)中でインキュベートした。血清サンプルを、水中50%メタノールで希釈し、そして放射性TLCを、上記のように、0.5時間、2時間、および4時間で繰り返した。
【0127】
(組織分布研究)
雌性Fischer344ラット(150±25g)(Harlan Sprague−Dawley,Indianapolis,IN)を、25ゲージ針を使用して後肢に、13762腫瘍細胞株懸濁液(10細胞/ラット、Fischerラットに特異的な腫瘍細胞株)由来の0.1mlの乳房腫瘍細胞で皮下接種した。研究を、腫瘍が直径約1cmに達した、移植後14〜17日で行なった。ラットを、各手順の前に、ケタミン(10〜15mg/ラット、腹腔内)で麻酔した。
【0128】
組織分布研究において、各動物を、370〜550KBqの99mTc−EC−COLまたは99mTc−ECで静脈内注射した(n=3/時点)。99mTc−EC−COLの注射された質量は、1匹のラット当り10μgであった。放射線トレーサの投与0.5時間、2時間および4時間後、ラットを屠殺し、そして選択した組織を切り出し、秤量し、そして放射能についてカウントした。各サンプルにおけるトレーサーの体内分布を、1gの組織湿重量当りの注射された用量の割合として計算した(%ID/g)。腫瘍/非標的組織カウント密度比を、対応する%ID/g値から計算した。スチューデントt検定を使用して、群の間の差異の有意性を評価した。
【0129】
(シンチグラフィー画像化研究)
低エネルギーの、平行な穴のコリメータを備えたγカメラ(Siemens Medical Systems,Inc.,Hoffman Estates,IL)を使用して、シンチグラフィー画像を、300μCiの99mTc−EC−COLおよび99mTc−ECの静脈内注射の0.5時間、2時間および4時間後に得た。コンピューターで概略した目的の領域(ROI)を使用して、腫瘍取り込み対正常筋肉取り込みを定量する(1画素当りのカウント)。
【0130】
(結果)
(99mTc−EC−COLの放射性合成および安定性)
99mTcでのEC−COLの放射性合成を、高い(>95%)放射化学的純度で達成した(図21)。99mTc−EC−COLは、は、ウサギ血清サンプル中で、0.5時間、2時間および4時間で安定であることが見出された。分解産物は、観察されなかった(図22)。
【0131】
(インビボ体内分布)
乳房腫瘍保有ラットにおける99mTc−EC−COLおよび99mTc−ECのインビボ体内分布を、表4および6に示す。0.5時間、2時間および4時間における99mTc−EC−COLの腫瘍取り込み値(%ID/g)は、0.436±0.089、0.395±0.154および0.221±0.006であったが(表6)、99mTc−ECについての体内分布は、それぞれ0.342±0.163、0.115±0.002および0.097±0.005であった(表4)。時間の関数としての増加した腫瘍対血液比(0.52±0.12から0.72±0.07)および腫瘍対筋肉比(3.47±0.40から7.97±0.93)は、99mTc−EC−COL群において観察された(図23)。逆に、腫瘍対血液値および腫瘍対筋肉値は、同じ期間における99mTc−EC−COL群と比較した場合、99mTc−ECでの時間依存性の減少を示した(図24)。
【0132】
【表6】
*各ラットは、99mTc−EC−コルヒチン(10μCi、静脈内)を受けた。各値は、時間間隔あたり1グラム組織重量(n=3)当りの注射された用量のパーセントである。各データは、標準偏差を有する3つの測定値の平均値を示す。
【0133】
【表7】
(乳房腫瘍保有ラットにおける99mTc−EC−COLのγシンチグラフィー画像化)
投与後1時間での3匹の乳房腫瘍保有ラットにおけるインビトロ画像化研究は、腫瘍が99mTc−EC−COL群で十分に可視化され得ることを示したが(図25)、99mTc−EC群においてより少ない腫瘍取り込みが観察された(図26)。コンピューターで概略した目的の領域(ROI)は、99mTc−EC−COL群における腫瘍/バックグラウンド比が、99mTc−EC群よりも有意に高いことを示した(図27)。
【0134】
(腫瘍解糖標的化)
(実施例6:99mTc−EC−ネオマイシンの開発)
(ECの合成)
ECを、以前に記載された方法(RatnerおよびClarke,1937;Blondeauら、1967)に従って、2工程合成で調製した。前駆体(L−チアゾリジン−4−カルボン酸)を合成した(融点195°、196〜197°と報告されている)。次いでECを調製した(融点237°、251〜253°と報告されている)。構造を、1H−NMRおよび高速原子衝撃質量分析(FAB−MS)によって確認した。
【0135】
(エチレンジシステイン−ネオマイシン(EC−ネオマイシン)の合成)
水酸化ナトリウム(2N、0.2ml)を、水中(5ml)のEC(134mg、0.50mmol)の攪拌した溶液に添加した。この無色の溶液に、スルホ−NHS(217mg、1.0mmol)およびEDC(192mg、1.0mmol)を添加した。次いで、ネオマイシントリスルフェート塩(909mg、1.0mmol)を添加した。この混合物を、室温にて24時間攪拌した。この混合物を、500のカットオフ値を有するSpectra/POR分子多孔性膜(Spectrum Medical Industries Inc.,Houston,TX)を使用して48時間透析した。透析後、生成物を、凍結乾燥器(Labconco,Kansas City,MO)を使用して凍結乾燥した。生成物の重量は、720mg(収率83%)であった。EC−ネオマイシンの合成スキームを図36に示す。この構造を、1H−NMR(図38A〜B)、質量分析計(図39A〜B)および元素分析(Galbraith Laboratories,Inc,Knoxville,TN)によって確かめる。元素分析C39H75N10S4O19・15H2O(C、H、N、S)、計算値 C:33.77、H:7.58、N:10.11、S:9.23;実測値 C:32.44、H:5.90、N:10.47、S:10.58.EC−ネオマイシンのUV波長は、ECおよびネオマイシンと比較した場合に、270.5nmシフトした(図40A〜C)。
【0136】
(99mTcでのEC−MNおよびEC−ネオマイシンの放射標識)
99mTc−ECおよび99mTc−EC−ネオマイシンの放射合成(radiosynthesis)を、ECまたはEC−ネオマイシンの凍結乾燥化残渣(10mg)、SnCl2(100μg)、Na2HPO4(13.5mg)およびアスコルビン酸(0.5mg)を含む手製のキットに、必要量の99mTc−パラテクネテート(pertechnetate)を添加することによって達成した。次いで、0.1ml水中のNaEDTA(0.5mg)を添加した。調製物の最終pHは、7.4であった。放射化学的純度を、酢酸アンモニウム(水中1M):メタノール(4:1)で溶出されるTLC(ITLC SG,Gelman Sciences,Ann Arbor,MI)によって決定した。放射TLC(Bioscan,Washington,DC)分析(図41)およびHPLC分析(図42〜45)から、放射化学的純度は、両方の放射性トレーサーについて95%を超えた。
【0137】
(99mTc−ECおよび99mTc−EC−ネオマイシンの安定性アッセイ)
標識した99mTc−ECおよび99mTc−EC−ネオマイシンの安定性を、イヌ血清サンプル中で試験した。手短に言うと、740KBqの1mgの99mTc−ECおよび99mTc−EC−ネオマイシンを、イヌ血清中(200μl)で37℃にて4時間インキュベートした。この血清サンプルを、水中の50%メタノールで希釈し、そして放射−TLCを、上記のように0.5時間、2時間および4時間で繰り返した。
【0138】
(99mTc−EC−ネオマイシンの組織分布研究)
雌性Fishcer 344ラット(150±25g)(Harlan Sprague−Dawley,Indianapolis,IN)に、25ゲージ針を用いて、13762腫瘍細胞株懸濁液由来の0.1mlの乳房腫瘍細胞(106細胞/ラット、Fischerラットに特異的な腫瘍細胞株)を後脚に皮下接種した。腫瘍が約1cm直径に到達する移植後14〜17日に、研究を行った。ラットを、各手順の前にケタミン(10〜15mg/ラット、腹腔内)で麻酔した。
【0139】
組織分布研究において、各動物に、10〜20μCiの99mTc−ECまたは99mTc−EC−ネオマイシン(n=3/時点)を静脈内注射した。99mTc−EC−ネオマイシンの注射した量は、200μg/ラットであった。これらの放射トレーサーの投与後0.5時間、2時間および4時間で、ラットを屠殺し、そして選択した組織を切り出し、重さを量り、そして放射活性を計数した。各サンプル中のトレーサーの体内分布を、1g組織湿重量あたりの注射用量のパーセンテージ(%ID/g)として計算した。腫瘍/非標的組織の計数密度比を、対応する%ID/g値から計算した。99mTc−EC(表4)および遊離テクネチウム(表9)と比較した場合、腫瘍対組織比は、99mTc−EC−ネオマイシン群において時間の関数として増加した(表8)。
【0140】
(シンチグラフィー画像化研究)
低エネルギーの平行穴コリメーターを備えたγカメラ(Simens Medical
Systems,Inc.,Hoffman Estates,IL)を使用して、シンチグラフィー画像を、100μCiの各放射トレーサーの静脈内注射後0.5時間、2時間および4時間で得た。99mTc−ECと比較して、腫瘍中の高い取り込みが観察された(図37A)。予備的な臨床画像化研究を、乳癌を有する患者において行った。腫瘍は、99mTc−EC−ネオマイシンの投与後2時間で十分に可視化された(図37B)。
【0141】
(表8)
(乳房腫瘍保有ラットにおける99mTc−EC−ネオマイシンの体内分布)
【0142】
【表8】
示された値は、3匹の動物からのデータの平均±標準偏差を表す。
【0143】
(表9)
(乳房腫瘍保有ラットにおける99mTcパーテクネテートの体内分布)
【0144】
【表9】
示された値は、3匹の動物からのデータの平均±標準偏差を表す。
【0145】
(99mTc−EC−薬物結合体のインビトロ細胞取り込み)
99mTc−EC−薬物結合体の細胞取り込みを評価するために、80,000個の細胞(A549肺癌細胞株)を含む各ウェルに、2μCiの99mTc−EC−ネオマイシンおよび18F−FDGを添加した。インキュベーション後0.5〜4時間で、細胞を、リン酸緩衝化生理食塩水で3回洗浄し、次いで、トリプシンによって細胞を死なせた。次いで、細胞を、γカウンターによって計数した。99mTc−EC−ネオマイシンは、ヒト肺癌細胞株中で試験した薬剤中で最も高い取り込みを示した(図46)。
【0146】
(99mTc−EC−ネオマイシンおよび18F−FDGの細胞取り込みに対するグルコースの効果)
ネオマイシンは、グルコース吸収に影響を及ぼすことが知られている(Rogersら、1968;Fanciulliら、1994)。先の実験は、99mTc−EC−ネオマイシンが、ヒト肺癌細胞株(A549)中で18F−FDGよりも高い取り込みを有することを示した。99mTc−EC−ネオマイシンの取り込みがグルコース関連機構を介して媒介されるか否かを決定するために、グルコース(0.1mg〜2.0mg)を、50,000個の(乳房)細胞または80,000個の(肺)細胞のいずれかを含有する各ウェルに、2μCiの99mTc−EC−ネオマイシンおよび18F−FDGと共に添加した。インキュベーション後、細胞を、リン酸緩衝化生理食塩水で3回洗浄し、次いで、トリプシンによって細胞を死なせた。次いで、細胞を、γカウンターによって計数した。
【0147】
0.1〜2.0mg/ウェルの濃度でグルコースを添加することによって、2つの肺癌細胞株および1つの乳房細胞株における99mTc−EC−ネオマイシンの取り込みの減少が観察された。類似の結果が、18F−FDG群において観察された。99mTc−EC(コントロール)は、取り込みを全く示さなかった。これらの知見は、99mTc−EC−ネオマイシンの細胞取り込みが、グルコース関連機構を介して媒介され得ることを示唆する(図47、48Aおよび48B)。
【0148】
(実施例7:99mTc−EC−デオキシグルコースでの腫瘍の代謝画像化)
(EC−デオキシグルコース(EC−DC)の合成)
水酸化ナトリウム(1N、1ml)を、水中(5ml)のEC(110mg、0.41mmol)の攪拌した溶液に添加した。この無色の溶液に、スルホ−NHS(241.6mg、1.12mmol)およびEDC(218.8mg、1.15mmol)を添加した。次いで、D−グルコサミン塩酸塩(356.8mg、1.65mmol)を添加した。この混合物を、室温にて24時間攪拌した。この混合物を、500のカットオフ値を有するSpectra/POR分子多孔性膜(Spectrum Medical Industries Inc.,Houston,TX)を使用して48時間透析した。透析後、生成物を、凍結乾燥器(Labconco,Kansas City,MO)を使用して凍結乾燥した。塩形態の生成物の重量は、568.8mgであった。この合成スキームを図59に示す。この構造を、質量分析計(図60)およびプロトンNMR(図61および62)で確かめた。99mTc−EC−DCの放射化学的純度は、放射−TLC(図63)およびHPLC(図64および65)分析によって決定した場合100%であった。
【0149】
(ヘキソキナーゼアッセイ)
EC−DGがグルコースリン酸化を模倣するか否かを決定するために、ヘキソキナーゼアッセイを行った。既製のキット(Sigma Chemical Company)を使用して、EC−DG、グルコサミンおよびグルコース(標準)を、UV波長340nmでアッセイした。グルコース、EC−DGおよびグルコサミンは、陽性ヘキソキナーゼアッセイを示した(図66〜68)。
【0150】
(インビトロ細胞取り込みアッセイ)
インビトロ細胞取り込みアッセイを、ヒト肺癌細胞株(A549)を使用することによって行った。2μCiの99mTc−EC−DGおよび18F−FDGを、各80,000個の細胞を含有するウェルに添加した。インキュベーション後0.5〜4時間で、細胞を、リン酸緩衝化生理食塩水で3回洗浄し、次いで、トリプシンによって細胞を死なせた。次いで、細胞を、γカウンターによって計数した。99mTc−EC−DGの取り込みは、FDGに匹敵した(図69)。
【0151】
(99mTc−EC−デオキシグルコースおよび18F−FDGの細胞取り込みに対するd−グルコースおよびl−グルコースの効果)
99mTc−EC−デオキシグルコースの取り込みがd−グルコース関連機構を介して媒介されるか否かを評価するために、d−グルコースおよびl−グルコース(1mgおよび2.0mg)を、乳癌細胞または肺癌細胞(50,000個/0.5ml/ウェル)のいずれかを含有する各ウェルに、2μCiの99mTc−EC−デオキシグルコースおよび18F−FDGと共に添加した。2時間のインキュベーション後、細胞を、リン酸緩衝化生理食塩水で3回洗浄し、次いで、トリプシンによって細胞を死なせた。細胞を、γカウンターによって計数した。
【0152】
1〜2.0mg/ウェルの濃度でグルコースを添加することによって、乳癌細胞および肺癌細胞における99mTc−EC−デオキシグルコースおよび18F−FDGの取り込みの減少が観察された。しかし、l−グルコースによる両薬剤に対する影響は存在しなかった(図70〜73)。これらの知見は、99mTc−EC−デオキシグルコースの細胞取り込みが、d−グルコース機構を介して媒介されることを示唆する。
【0153】
(正常ラットにおける血中グルコースレベルに対するEC−デオキシグルコース負荷の効果)
先の実験は、99mTc−EC−デオキシグルコースの細胞取り込みが、FDGに類似することを示した。例えば、ヘキソキナーゼアッセイ(グルコースリン酸化)は、陽性であった。99mTc−EC−デオキシグルコースの取り込みは、d−グルコース機構を介して媒介される。この研究は、血中グルコースレベルがFDGまたはEC−デオキシグルコースのいずれかによって誘導され得、そしてインスリンによって抑制され得るか否かを決定することである。
【0154】
正常な健常Fischer 344ラット(重量145〜155g)を、実験前に一晩絶食させた。調製した塩酸グルコサミン、FDGおよびEC−デオキシグルコースの濃度は、60%および164%(mg/ml)であった。血中グルコースレベル(mg/dl)を、グルコースメーター(Glucometer DEX、Bayer Corporation,Elkhart,IN)によって測定した。この研究の前に、血中グルコースレベルのベースラインを得た。各ラット(n=3/群)に、1.2mmol/kgのグルコサミン、FDGおよびEC−デオキシグルコースを投与した。別の実験において、1群のラットに、EC−デオキシグルコースおよびFDGを投与した。インスリン(5ユニット)を、30分後に投与した。血液サンプルを、尾静脈から30分毎に投与後6時間まで収集した。
【0155】
血中グルコースレベルは、グルコサミン、FDGおよびEC−デオキシグルコースのボーラス静脈内投与によって誘導された。この血中グルコースレベルの増加は、EC−デオキシグルコースまたはFDGおよびインスリンの投与によって抑制され得た(図74および75)。
【0156】
(99mTc−EC−DGの組織分布研究)
乳房腫瘍保有動物モデルについて、雌性Fishcer 344ラット(150±25g)(Harlan Sprague−Dawley,Indianapolis,IN)に、25ゲージ針を用いて、13762腫瘍細胞株懸濁液由来の0.1mlの乳房腫瘍細胞(106細胞/ラット、Fischerラットに特異的な腫瘍細胞株)を後脚に皮下接種した。腫瘍が約1cm直径に到達する移植後14〜17日に、研究を行った。ラットを、各手順の前にケタミン(10〜15mg/ラット、腹腔内)で麻酔した。
【0157】
肺腫瘍保有動物モデルについて、各胸腺欠損ヌードマウス(20〜25g)に、25ゲージ針を用いて、A549腫瘍細胞株懸濁液由来の0.1mlのヒト肺腫瘍細胞(106細胞/マウス)を後脚に皮下接種した。腫瘍が約0.6cm直径に到達する移植後17〜21日に、研究を行った。
【0158】
組織分布研究において、各動物に、10〜20μCi(1匹のラットあたり)または1〜2μCi(1匹のマウスあたり)の99mTc−ECまたは99mTc−EC−DG(n=3/時点)を静脈内注射した。99mTc−EC−DGの注射した量は、1mg/ラットであった。これらの放射トレーサーの投与後0.5時間、2時間および4時間で、これらの齧歯動物を屠殺し、そして選択した組織を切り出し、重さを量り、そして放射活性を計数した。各サンプル中のトレーサーの体内分布を、1g組織湿重量あたりの注射用量のパーセンテージ(%ID/g)として計算した。腫瘍/非標的組織の計数密度比を、対応する%ID/g値から計算した。99mTc−EC(表4)および遊離テクネチウム(表9)と比較した場合、腫瘍対組織比は、99mTc−EC−DG群において、時間の関数として増加した(図76〜80)。
【0159】
(シンチグラフィー画像化研究)
低エネルギーの平行穴コリメーターを備えたγカメラを使用して、シンチグラフィー画像を、100μCiの放射トレーサーの静脈内注射後0.5時間、2時間および4時間で得た。使用した動物モデルは、乳房腫瘍保有ラットであった。99mTc−EC(コントロール群)と比較した場合、腫瘍は十分に可視化され得た(図81)。予備的な臨床画像化研究を、5人の患者(3人の脳腫瘍および2人肺疾患患者)において行った。画像を、投与後1〜2時間で得た。99mTc−EC−DGは、良性腫瘍を悪性腫瘍に対して区別し得た。例えば、悪性星状細胞腫は、高い取り込みを示した(図82A,82B、83Aおよび83B)。良性髄膜腫は、悪性髄膜腫と比較して乏しい取り込みを示した(図84AおよびB)。乏しい取り込みは、TBを有する患者において観察されたが(図85Aおよび図85B)、高い取り込みが、肺腫瘍において観察された(図86A,図86B、および図86C)。
【0160】
本明細書中に開示されそして特許請求される全ての組成物および/または方法は、本開示を考慮して過度の実験を伴うことなく作製および実行される。本発明の組成物および方法は、好ましい実施形態について記載されたが、これらの組成物および/または方法に対して、および本明細書中に記載される方法の工程または工程の順序において、本発明の概念、精神および範囲を逸脱することなく改変が適用され得ることは、当業者に明らかである。より詳細には、化学的および生理学的の両方で関連する特定の薬剤が、本明細書中に記載される薬剤と置換され得、同じかまたは類似の結果が達成されることが、明らかである。当業者に明らかな全てのこのような類似の置換物および改変物は、添付の特許請求の範囲に規定されるように、本発明の精神、範囲および概念内にあるとみなされる。
【0161】
(参考文献)
以下の参考文献は、これらが本明細書中に記載されるものに対して補助的な、例示的手順または他の詳細を提供する程度まで、本明細書中に参考として詳細に援用される。
【0162】
【表10】
以下の図面は、本明細書の一部を形成し、そして本発明の特定の局面をさらに示すために含まれる。本発明は、本明細書中に示される特定の実施形態の詳細な説明と組み合わせて、これらの図面のうちの1つ以上を参照することによって良好に理解され得る。
【特許請求の範囲】
【請求項1】
本明細書中に記載される、画像化のための組成物、方法およびキット。
【請求項1】
本明細書中に記載される、画像化のための組成物、方法およびキット。
【図3】

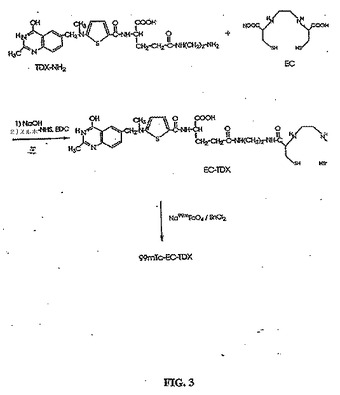
【図4】
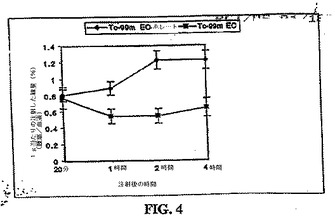
【図5】
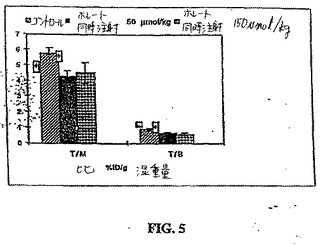
【図6】
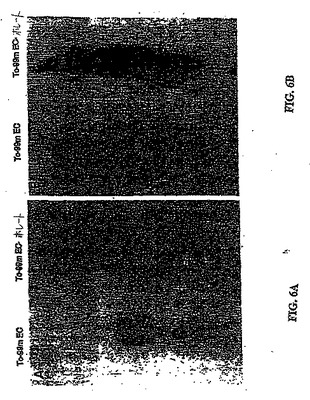
【図7】
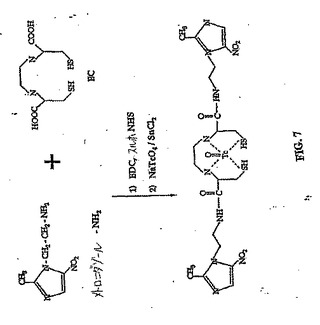
【図8A】
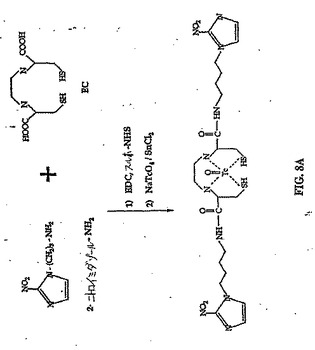
【図8B】
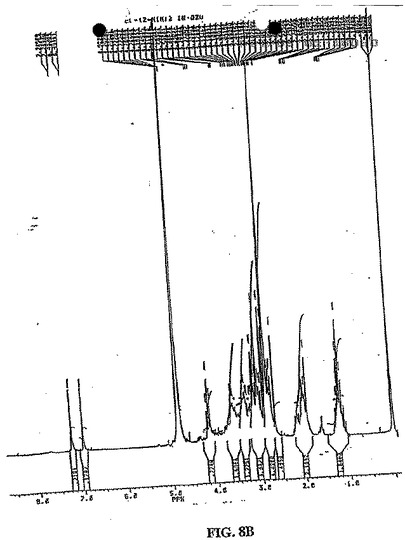
【図9】
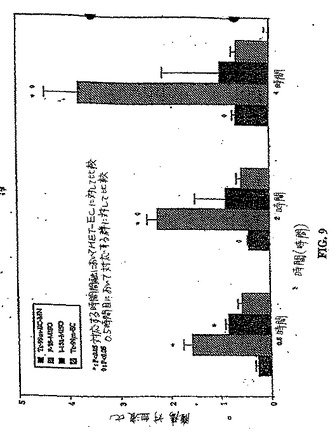
【図10】
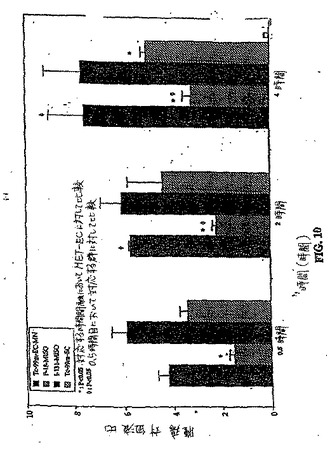
【図11】
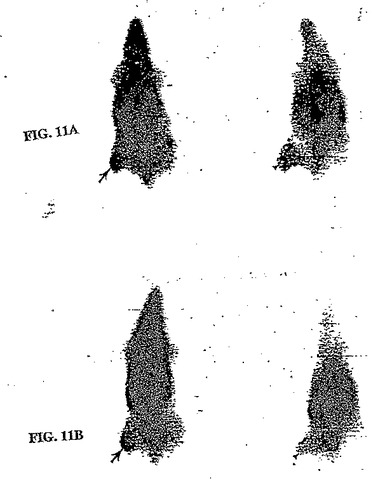
【図12】
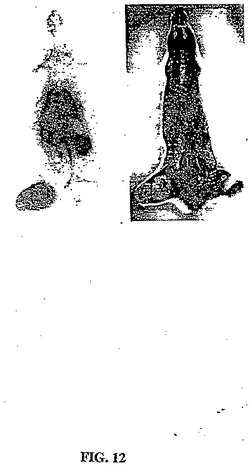
【図13】
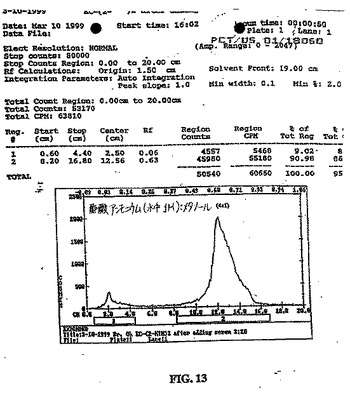
【図14A】
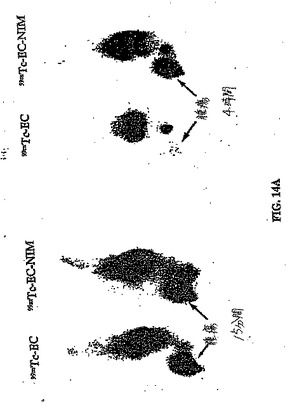
【図14B】
【図15A】
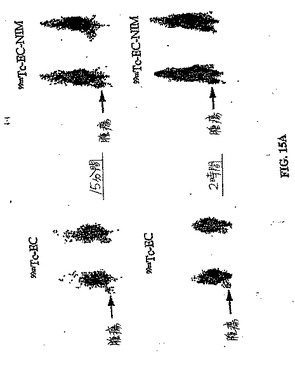
【図15B】
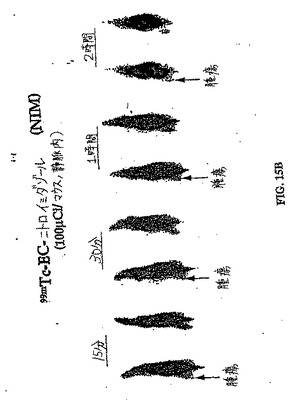
【図15C】

【図15D】
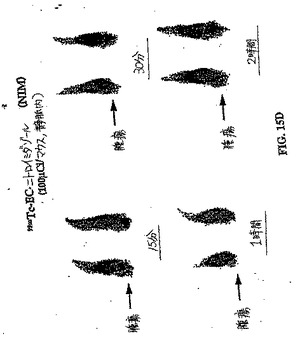
【図16】
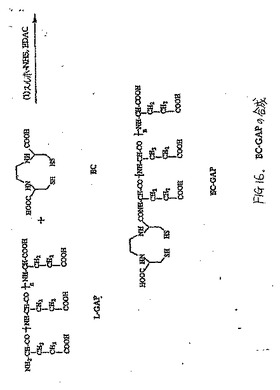
【図17】
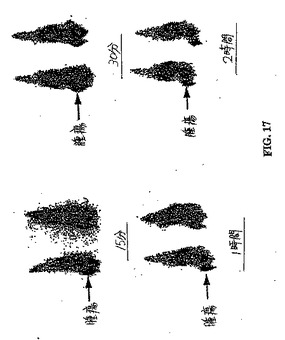
【図18】
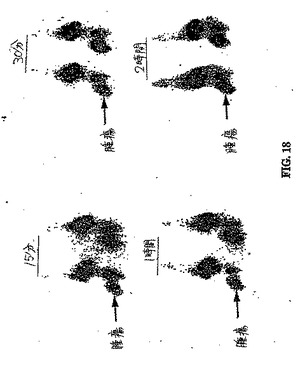
【図19A】
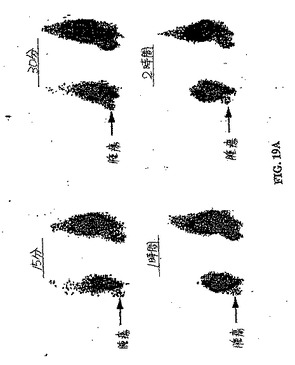
【図19B】
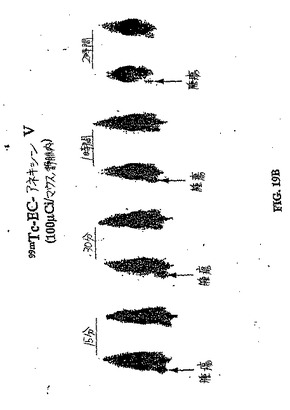
【図20A】

【図20B】
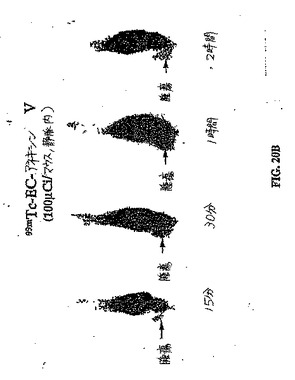
【図21】
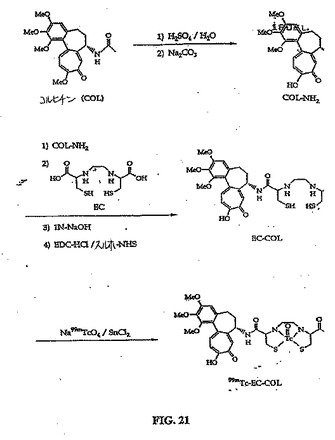
【図22】

【図23】
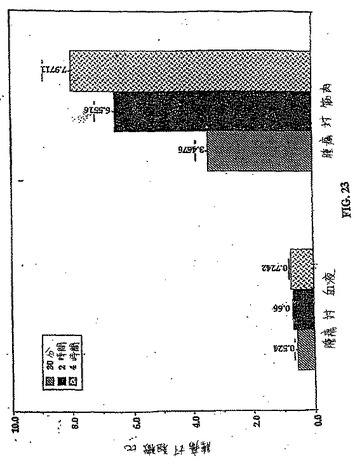
【図24】
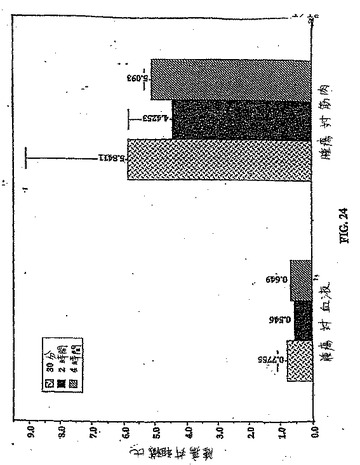
【図25】

【図26】

【図27】
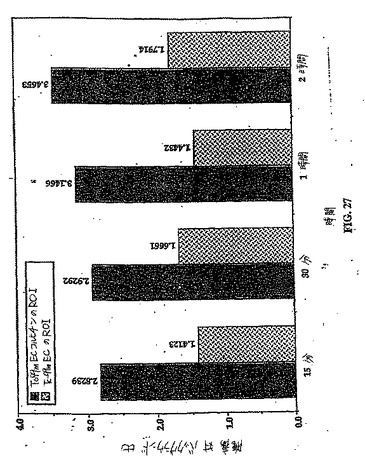
【図28】
【図29】
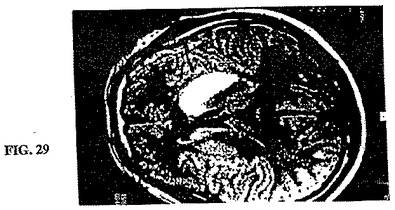
【図30】
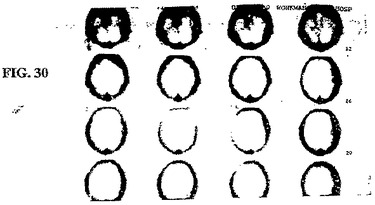
【図31】
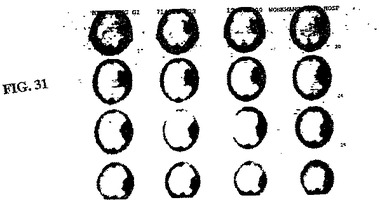
【図32】
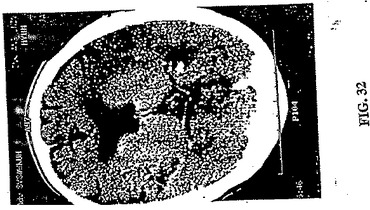
【図33】
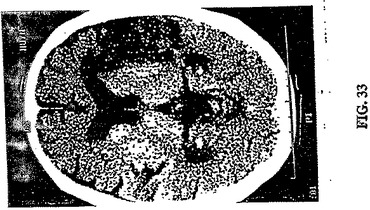
【図34】
【図35】
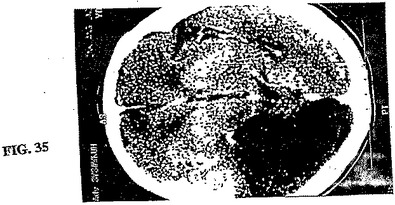
【図36】
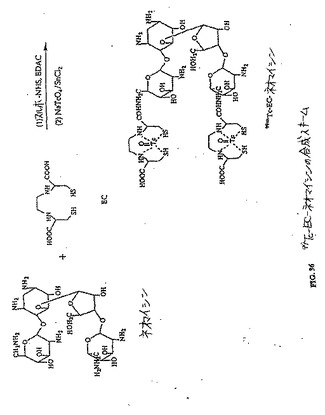
【図37A】
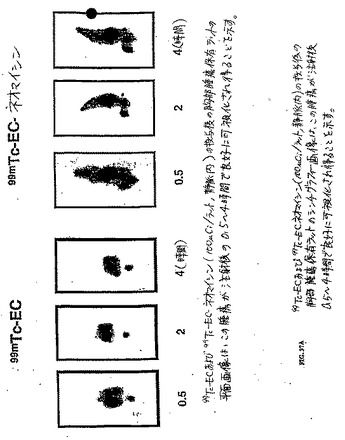
【図37B】
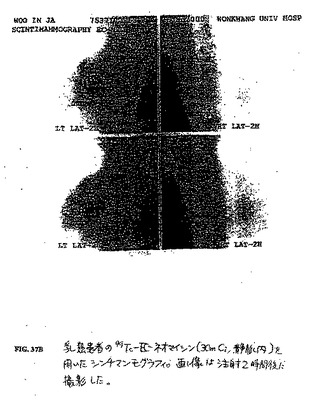
【図38A】
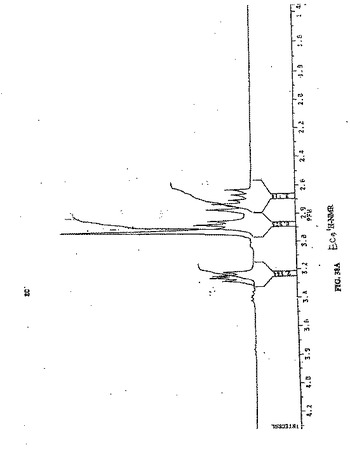
【図38B】
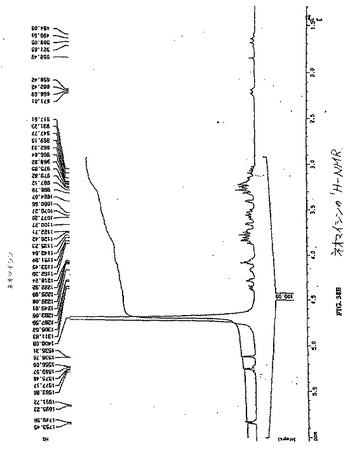
【図38C】
【図39】
【図40A】
【図40B】
【図40C】
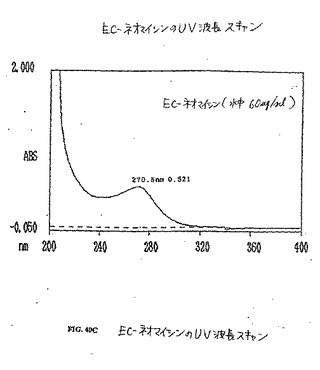
【図41】
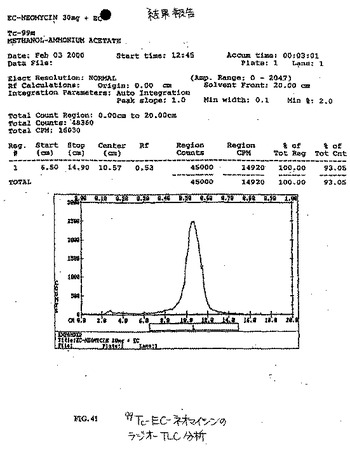
【図42】
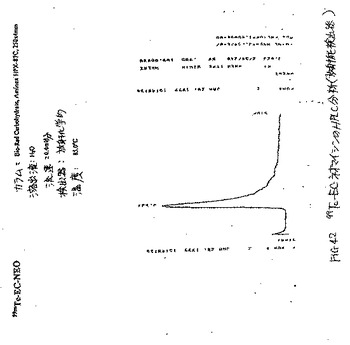
【図43】
【図44】
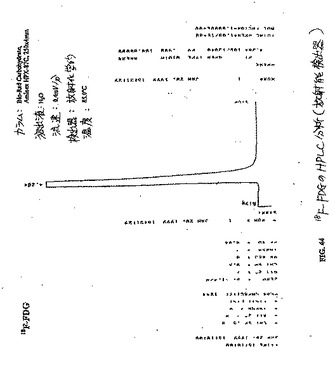
【図45】
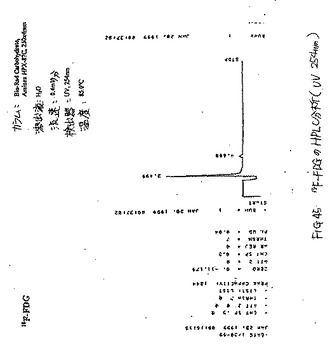
【図46】
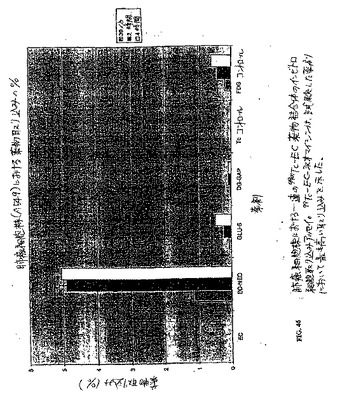
【図47】
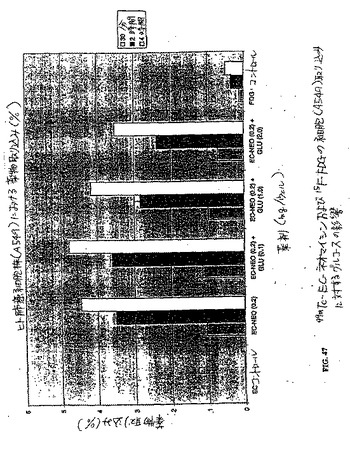
【図48A】
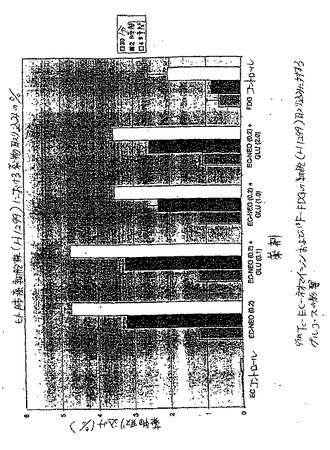
【図48B】
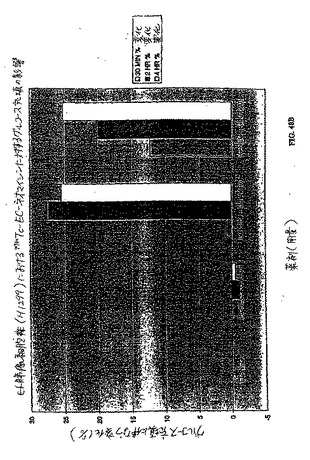
【図49】
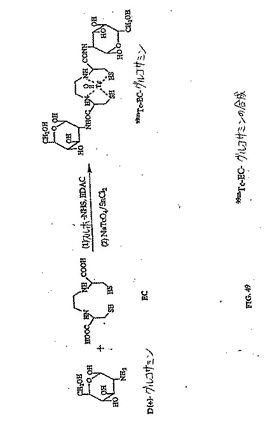
【図50】
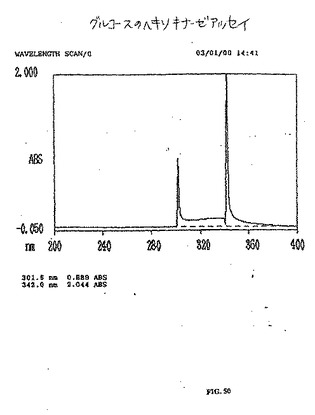
【図51】
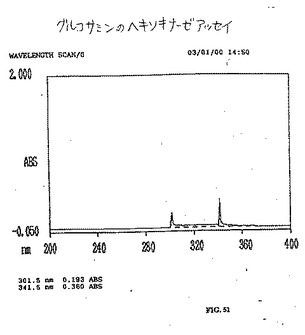
【図52】
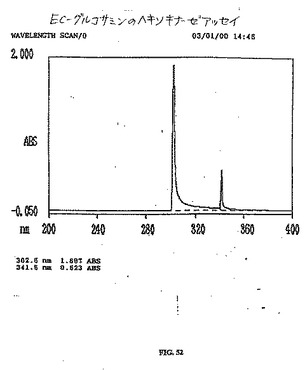
【図53】
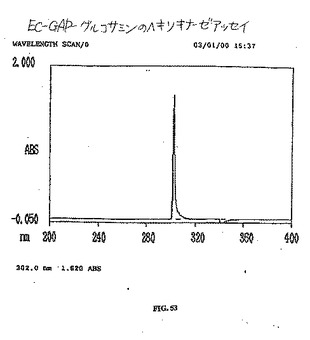
【図54】
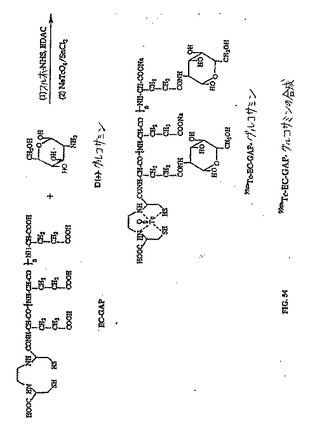
【図55A】
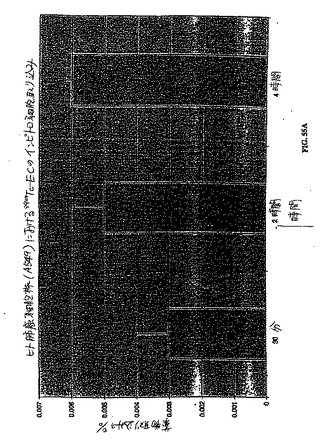
【図55B】

【図55C】

【図56】

【図57】
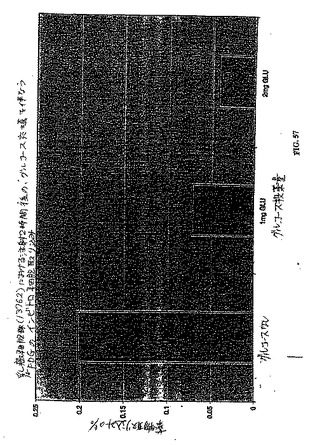
【図58】
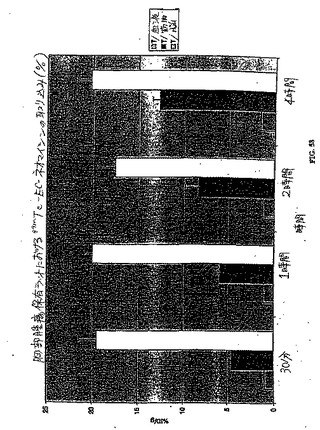
【図59】
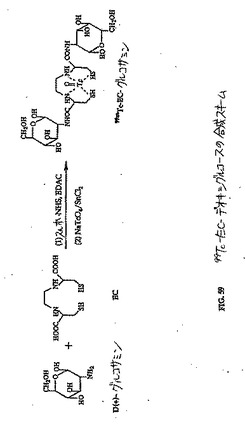
【図60】
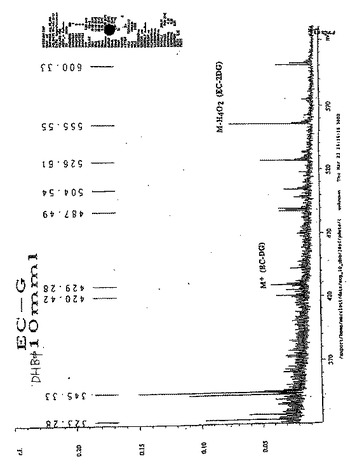
【図61】

【図62】
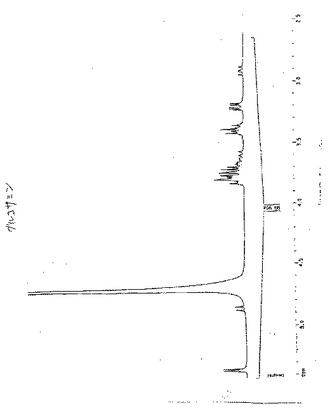
【図63】
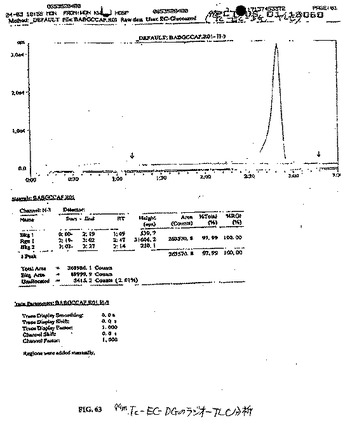
【図64】

【図65】
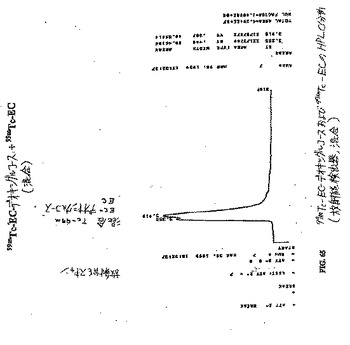
【図66】
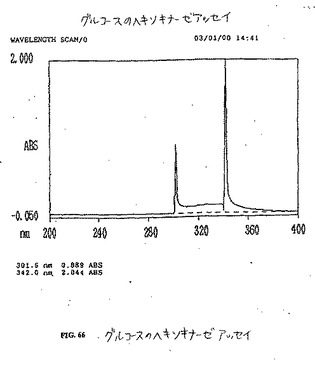
【図67】
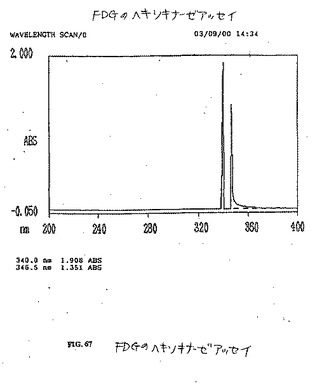
【図68】
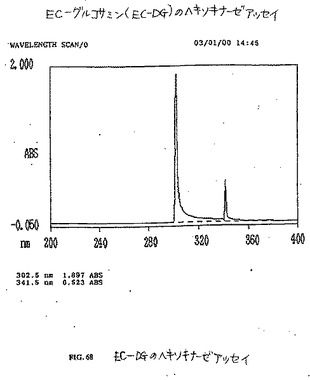
【図69】
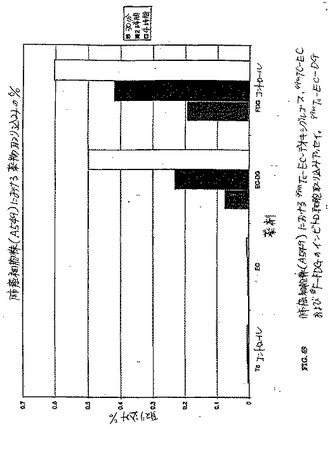
【図70】
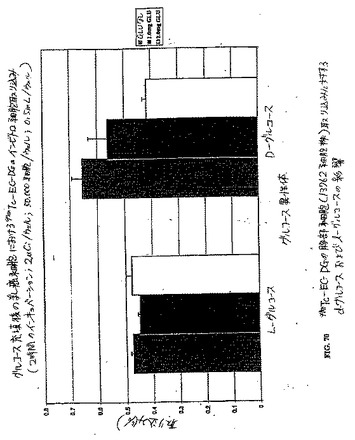
【図71】
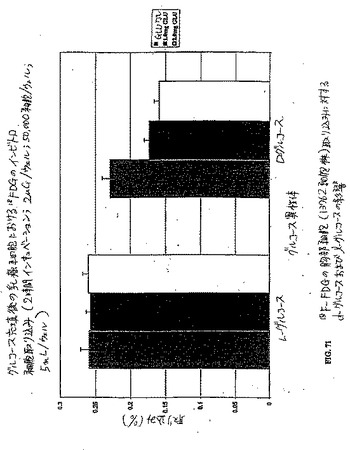
【図72】
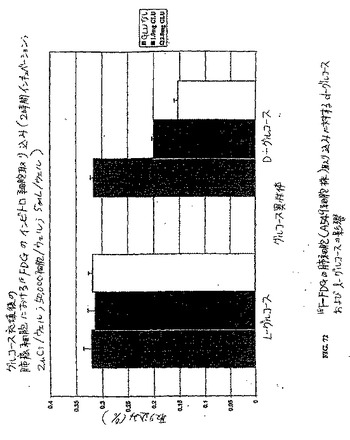
【図73】
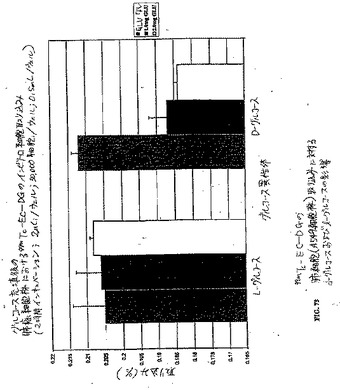
【図74】
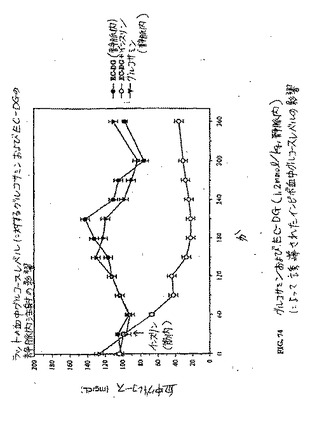
【図75】
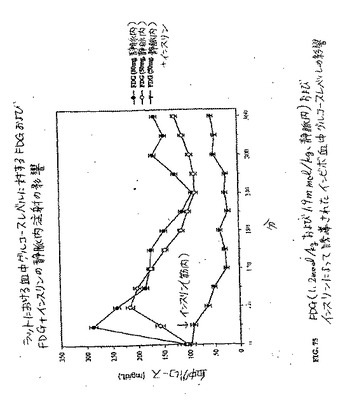
【図76】
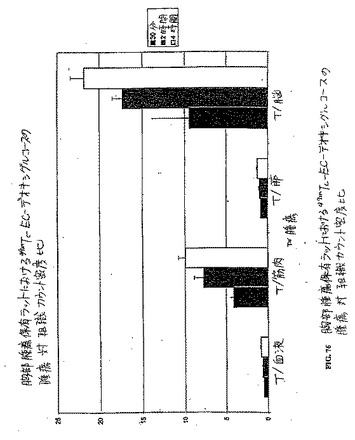
【図77】

【図78】
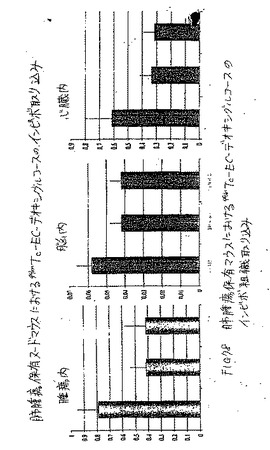
【図79】
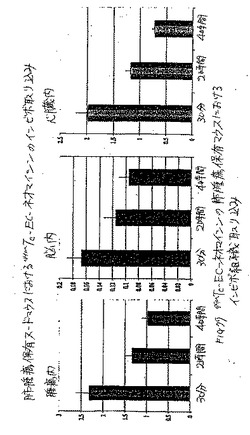
【図80】
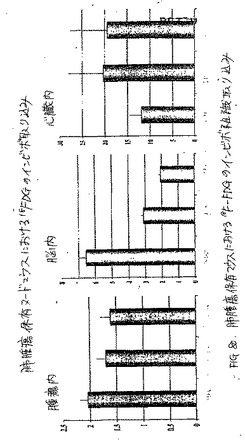
【図81】
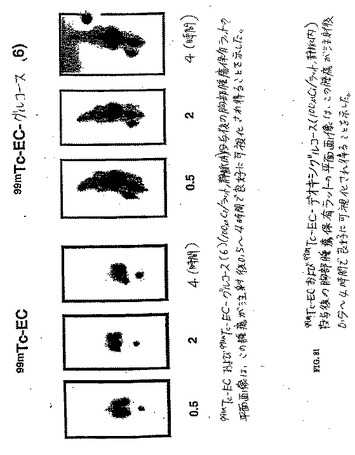
【図82A】
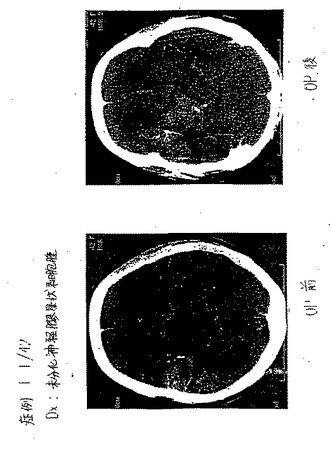
【図82B】
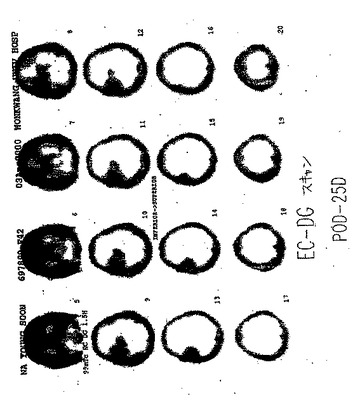
【図83A】
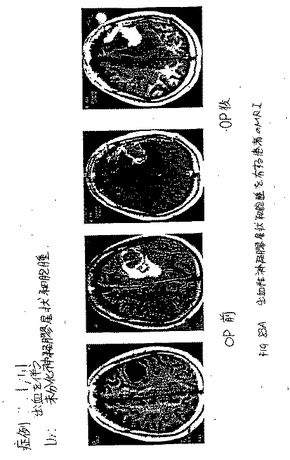
【図83B】
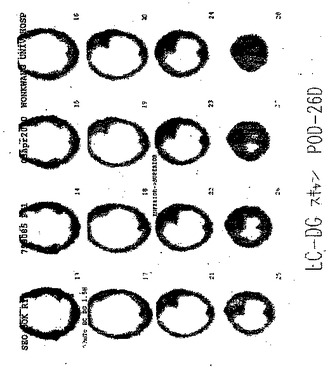
【図84A】
【図84B】
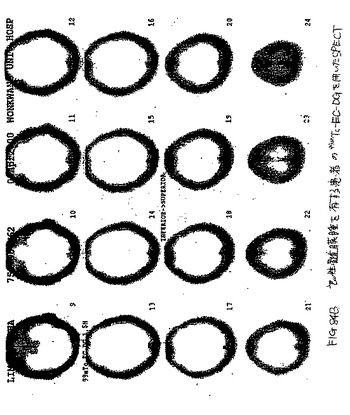
【図85A】
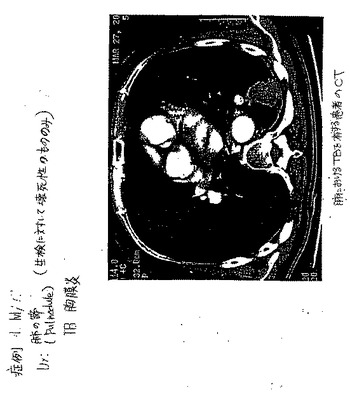
【図85B】

【図86A】
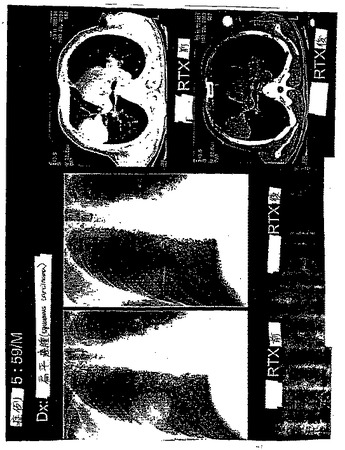
【図86B】
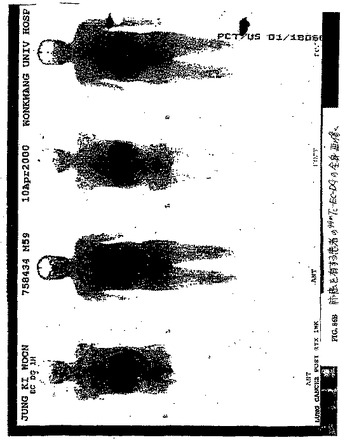
【図86C】
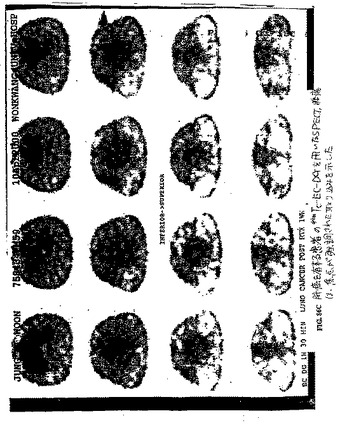
【図1】
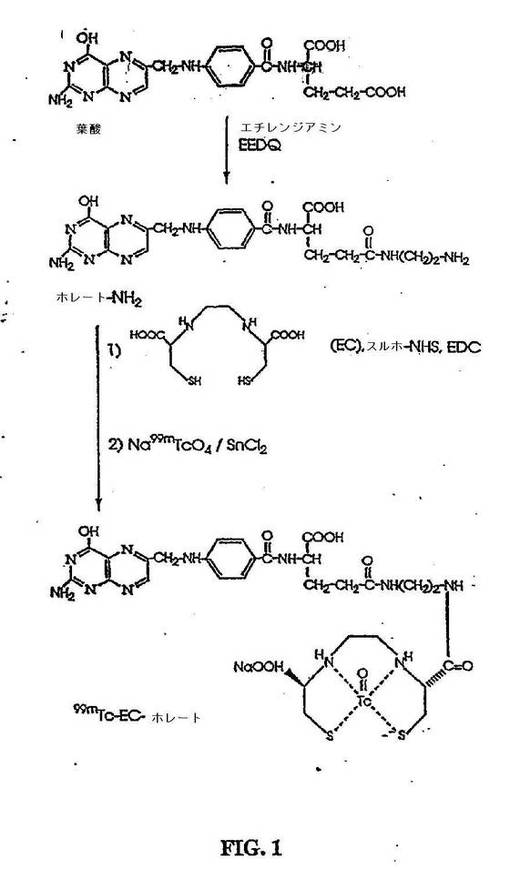
【図2】
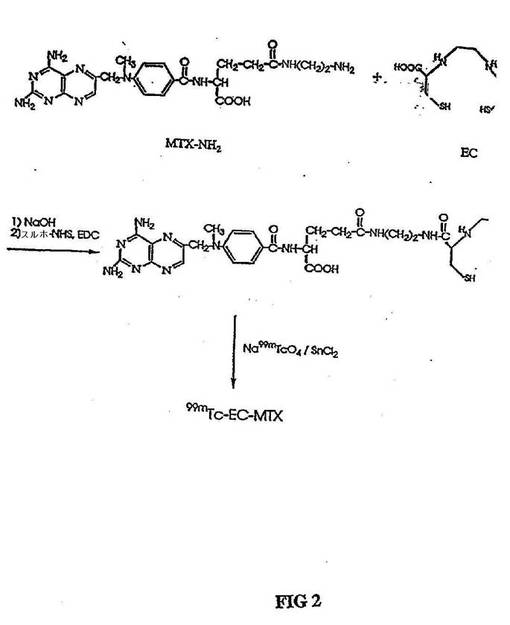
【図4】
【図5】
【図6】
【図7】
【図8A】
【図8B】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14A】
【図14B】
【図15A】
【図15B】
【図15C】
【図15D】
【図16】
【図17】
【図18】
【図19A】
【図19B】
【図20A】
【図20B】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30】
【図31】
【図32】
【図33】
【図34】
【図35】
【図36】
【図37A】
【図37B】
【図38A】
【図38B】
【図38C】
【図39】
【図40A】
【図40B】
【図40C】
【図41】
【図42】
【図43】
【図44】
【図45】
【図46】
【図47】
【図48A】
【図48B】
【図49】
【図50】
【図51】
【図52】
【図53】
【図54】
【図55A】
【図55B】
【図55C】
【図56】
【図57】
【図58】
【図59】
【図60】
【図61】
【図62】
【図63】
【図64】
【図65】
【図66】
【図67】
【図68】
【図69】
【図70】
【図71】
【図72】
【図73】
【図74】
【図75】
【図76】
【図77】
【図78】
【図79】
【図80】
【図81】
【図82A】
【図82B】
【図83A】
【図83B】
【図84A】
【図84B】
【図85A】
【図85B】
【図86A】
【図86B】
【図86C】
【図1】
【図2】
【公開番号】特開2012−193199(P2012−193199A)
【公開日】平成24年10月11日(2012.10.11)
【国際特許分類】
【出願番号】特願2012−155112(P2012−155112)
【出願日】平成24年7月11日(2012.7.11)
【分割の表示】特願2008−123946(P2008−123946)の分割
【原出願日】平成13年6月1日(2001.6.1)
【出願人】(500039463)ボード・オブ・リージエンツ,ザ・ユニバーシテイ・オブ・テキサス・システム (115)
【Fターム(参考)】
【公開日】平成24年10月11日(2012.10.11)
【国際特許分類】
【出願日】平成24年7月11日(2012.7.11)
【分割の表示】特願2008−123946(P2008−123946)の分割
【原出願日】平成13年6月1日(2001.6.1)
【出願人】(500039463)ボード・オブ・リージエンツ,ザ・ユニバーシテイ・オブ・テキサス・システム (115)
【Fターム(参考)】
[ Back to top ]