
エトリコキシブの中間体、1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノンの調製方法の改良
【課題】COX−2阻害薬の医薬原体である、エトリコキシブ合成の中間体、1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノンを調製する改良された方法を提供する。
【解決手段】1−(6−メチルピリジン−3−イル)エタノンと、4−置換−フェニルメチルスルフィド、スルフォキシド、又はスルホンとを反応させるステップを含む方法により、上記中間体を調製する。
【解決手段】1−(6−メチルピリジン−3−イル)エタノンと、4−置換−フェニルメチルスルフィド、スルフォキシド、又はスルホンとを反応させるステップを含む方法により、上記中間体を調製する。
【発明の詳細な説明】
【技術分野】
【0001】
(発明の技術分野)
本発明の目的は、COX−2阻害薬の医薬原体(pharmaceutical active ingredient)である、エトリコキシブ(Etoricoxib)合成の中間体、1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノンを調製する方法である。
【背景技術】
【0002】
(従来技術)
CAS登録番号(RN)221615−75−4を有する、式(I):
【0003】
【化1】

の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノンは、COX−2阻害薬の分類に属する医薬原体であり、かつ登録商標アルコキシア(Arcoxia)で2002年から市販されている、エトリコキシブ合成用の重要な中間体である。
【0004】
【化2】
【0005】
COX−2阻害薬の合成に、このような化合物を用いる例は、WO99/55830、WO99/15503、及びDavies、Lan WらのJournal of Organic Chemistry(2000)、65(25)、8415〜8420に報告されている。
【0006】
このような重要な構成要素の種々の合成法は、前記特許文献に記載されたもの以外にも知られているが、LonzaとMerck&CoのWO2001/007410に記載された方法は、経済的観点から最も有利な方法であると思われる。このような方法の欠点は、最終ステップにおいて、タングステン酸ナトリウムで触発させた過酸化水素による酸化処理を含むことである。Zambon Group S.p.Aによる後続の出願WO2001/029004は、触媒(タングステン酸ナトリウム)及び酸(例えば、メタンスルホン酸)の存在下で、酸化剤の組み合わせ(例えば、過酢酸と過酸化水素の混合物)を含む酸化処理を行う改良された方法を報告している。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】WO99/55830
【特許文献2】WO99/15503
【特許文献3】WO2001/007410
【特許文献4】WO2001/029004
【非特許文献】
【0008】
【非特許文献1】Davies、Lan WらのJournal of Organic Chemistry(2000)、65(25)、8415〜8420
【発明の概要】
【発明が解決しようとする課題】
【0009】
(発明の概要)
本発明が取り上げる問題は、1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノンを調製する代替方法の提供である。
【課題を解決するための手段】
【0010】
このような問題は、付属の特許請求の範囲に記載したような、1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノンを調製する方法により解決される。そこでなされた定義は、本発明の開示の不可欠な要素である。
【0011】
本発明に関する方法の更なる特徴及び利点は、非限定的な実施例により与えられる、その好ましい実施形態に関する以下の記載から得られるであろう。
【図面の簡単な説明】
【0012】
(図の簡単な説明)
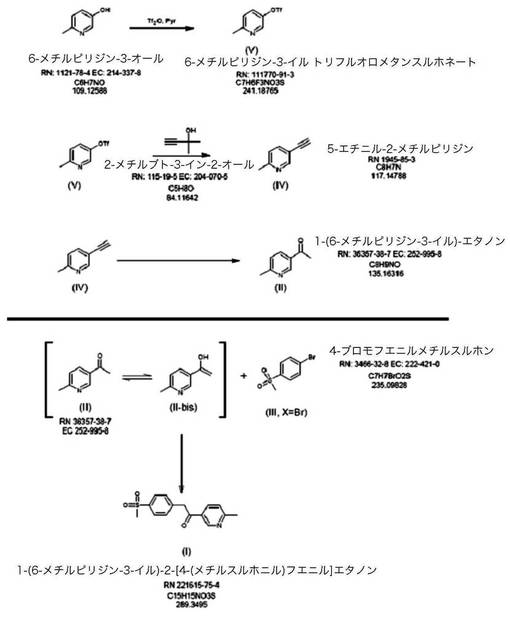
【図1】本発明の好ましい態様に従った、1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノンの合成の略図である。
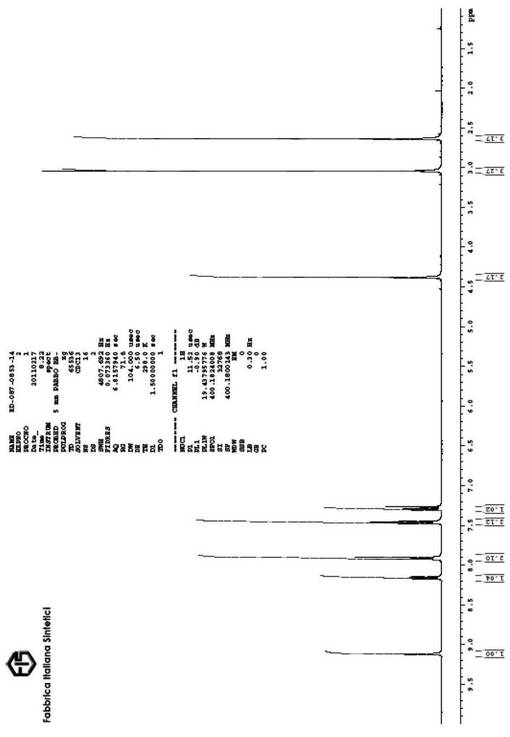
【図2】本発明の方法に従って得られた、式(I)の生成物の1H−NMRスペクトルを示す図である。
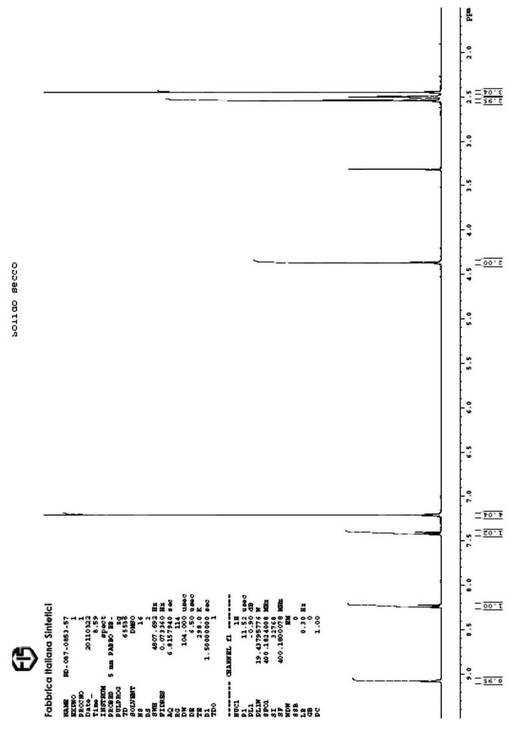
【図3】本発明の方法に従って得られた、式(VI)の生成物の1H−NMRスペクトルを示す図である。
【発明を実施するための形態】
【0013】
(発明の詳細な説明)
本発明は、式(I):
【0014】
【化3】
の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノン又はその塩を調製する方法であって、式(II):
【0015】
【化4】
の1−(6−メチルピリジン−3−イル)エタノンと、式(III):
【0016】
【化5】
の4−置換−フェニルメチルスルホンとを反応させることを含む方法に関する。
【0017】
ここで、Xは、F、Br、Cl、I、OTs、OTf、OMs、ONf、及びO(C=O)NR2からなる群から選ばれ、ここでRは、メチル、エチル、n−プロピル、iso−プロピル、n−ブチル、iso−ブチル、sec−ブチル、若しくはtert−ブチルの中から選ばれた、直鎖の、若しくは分枝したC1〜C4のアルキル置換基、又はフェニル若しくはベンジルである。TsOは脱離基トシラート(Tosylate)、TfOは脱離基トリフラート(Triflate)、MsOは脱離基メシラート(Mesylate)、NfOは脱離基ノナフラート(Nonaflate)である。
【0018】
式(III)の化合物で、Rはエチルであることが好ましい。
【0019】
本発明の好ましい態様に従えば、この方法は、4−ブロモフェニルメチルスルホンを用いて行われる。
【0020】
また、α−アリーレーション型カップリングに用いられるその他の脱離基は、この方法を実施のためにXに置換して使用することができ、従って、本発明の欠かせない要素として考えるべきである。
【0021】
また、式(II)の1−(6−メチルピリジン−3−イル)エタノンは、式(II−ビス):
【0022】
【化6】
の1−(6−メチルピリジン−3−イル)エテノールであるそれらの「エノール」型でも存在することができる。
【0023】
本発明の方法は、以下に説明する反応条件に従って行う、式(II)又は(II−ビス)と、式(III)の2つの試薬間のカップリングを考慮に入れている。
【0024】
この反応は、トルエン、キシレン、アルコール類などの有機溶剤中で、又はジオキサン、THF(テトラヒドロフラン)及びMe−TFHなどのエーテル溶剤中で、又はDMF(ジメチルホルムアミド)、DMSO(ジメチルスルホキシド)及びNMP(N−メチルピロリドン)中で行われ、エーテル溶剤が好ましく、かつNMP及びDMFが更により好ましい。
【0025】
好ましくは、反応は、6、8又は10容積(volume)の溶剤中で行われ、6容積が好ましい。
【0026】
反応は、tert−ブトキシカリウム、炭酸カリウム、又はリン酸カリウムなどの塩基の存在下で、好ましくはリン酸カリウムの存在下で行われる。
【0027】
留意すべき観点は、塩基の粉砕度合である。反応開始直前に、湿式粉砕機により行うリン酸カリウムの粉砕は、迅速な反応を促進し、かつ高収率で所望する生成物が得られる。
【0028】
1〜3モル塩基当量が使用され、3モル塩基当量が好ましい。3モル塩基当量の使用は、反応速度を増大し、かつ反応生成物中で試薬のより高い転化を促進するので好ましい。
【0029】
反応に用いる触媒前駆体は、Pd(OAc)2(酢酸パラジウム(II))、Pd(F6−acac)2(パラジウムビス(ヘキサフルオロ)アセチルアセトナト)、Pd(acac)2(酢酸パラジウム(II)アセチルアセトナト)、Pd2(dba)3(トリス(ジベンジリデンアセトン)ジパラジウム(0))、(PdallylCl)2(アリルパラジウム(II)クロリド(ダイマー))、又はPdCl2(塩化パラジウム(II))であり、Pd(OAc)2が好ましい。
【0030】
触媒前駆体の量は、式(II)の1−(6−メチルピリジン−3−イル)エタノンを基準に、0.05モル%と2モル%の間に包含される触媒量が通常使用される。好ましくは、0.15〜0.5モル%である。
【0031】
反応に用いられる配位子は、PPh3(トリフェニルホスフィン)、P(Cy)3(トリシクロヘキシルホスフィン)、Xantphos(キサントホス)、dppe(1,2−ビス(ジフェニルホスフィノ)エタン)、dppp(1,3−ビス(ジフェニルホスフィノ)プロパン)、dppf(1,1−ビス(ジフェニルホスフィノ)フェロセン)、Josiphos(ジョシホス)からなる群から選ばれる。キレート性ホスフィンのXantphosが好ましい。
【0032】
これは、約0.075モル%と1モル%の間に含まれる量での使用が好ましい。
【0033】
配位子のモル量は、通常パラジウム触媒前駆体の量の0.5〜2倍を使用し、好ましくは2倍である(式(II)の化合物を常に基準にして、パラジウム触媒前駆体が0.5モル%である時、配位子1モル%に等しい)。
【0034】
例えば、酢酸パラジウム(II)の場合、最も好ましい配位子/金属のモル比は、0.5であるが、2の比率も使用できる。
【0035】
反応は、60℃と140℃の間、好ましくは80℃と120℃の間の温度、より好ましくは約85℃〜約100℃で行われる。
【0036】
反応は、16〜30時間、好ましくは約18〜20時間で通常行われる。
【0037】
この反応は、少量のフェニルメチルスルホンが、4−ブロモフェニルメチルスルホンのプロト−脱臭素反応で発生することを除けば、その他の副生物がないことから、良好な選択性で進行する。
【0038】
更なる好ましい態様に従えば、この反応は無水環境下で行われる。
【0039】
次に、生成物は、抽出及び結晶化を含む従来の有機合成技術により分離される。
【0040】
本発明の実施形態に従えば、本発明の化合物を調製するために有用な、式(II):
【0041】
【化7】
の中間体1−(6−メチルピリジン−3−イル)エタノンは、式(IV):
【0042】
【化8】
の5−エチニル−2−メチルピリジンの水和反応により調製できることが好都合である。
【0043】
このような水和反応は、硫酸とトルエンの混合物、それぞれ2:1から4:1、好ましくはそれぞれ4:1の混合物中で行うことができるので都合がよい。反応は、50℃で16時間行い、又は80℃で3時間、又は70℃で2時間行うことができる。生成物を、酢酸エチルを用いて水相から抽出し、脱水の後に濃縮して残留物とする。
【0044】
この方法の通常のモル収率は、90%超である。
【0045】
本発明の実施形態に従えば、本発明の化合物を調製するために有用な式(IV):
【0046】
【化9】
の中間体5−エチニル−2−メチルピリジンの合成は、式(V):
【0047】
【化10】
の中間体6−メチルピリジン−3−イルトリフルオロメタンスルホネートと2−メチル−3−ブチン−2−オールとの反応を含む方法により行うことができる。
【0048】
この反応は、酢酸パラジウム(II)とホスフィンの存在下で行われる。また、PPh3、P(p−FPh)3(トリス(4−フルオロフェニル)ホスフィン)、P(p−tol)3(トリ−p−トリルホスフィン)、dppe、dppfなどの種々のホスフィンが使用できるが、高収率を与えるので、P(p−tol)3が好ましい。有機溶剤として、NMP/トルエン混合物(1:1)又はトルエン単独を使用できることが好都合である。この反応は、ピリジン、DBU(ジアザビシクロウンデセン)、ピペリジン、HNiPr2、TEA、DABCO(1,4−ジアザビシクロ[2,2,2]オクタン)、DIPEA(N,N−ジイソプロピルエチルアミン)などの塩基の存在下で行われる。
【0049】
その好ましい態様では、反応は、高収率になる理由から、ピペリジンの存在下で行われる。この反応は、通常、40℃で16時間行われる。化合物2−メチル−4−(6−メチルピリジン−3−イル)ブト−3−イン−2−オールが、本反応の中間体として形成される。この化合物は、式(IV−ビス):
【0050】
【化11】
で表され、所望により容易に分離される。
【0051】
式中、アセチレン官能基は、さらにアセトンアダクトの型で保護されている。このような保護は、トルエン中でソーダ処理して除去し、式(IV)の中間体を形成するために環流させる。
【0052】
また、このように式(IV)の化合物を分離しないので、式(V)の中間体から出発して式(II)の中間体を調製する方法は、同一容器内で行うことができる。
【0053】
本発明に従う1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノンを調製する方法は、タングステン系触媒の使用を避けるという利点がある。更なる利点は、本発明の方法が、市場から入手できる式(II)及び式(III)の中間体を使用することであり、かつ(III)は既に商標登録されたEINECS(少なくともX=Brの場合)である。
【0054】
更に、式(III)のスルホネート中間体は、既に、最終生成物に要求される酸化状態の硫黄を示しており、それ故に、このことは、既知技術の方法に比べると、最終的な酸化処理を回避することができ、潜在的には爆発性で危険物質である過酸化水素及び有機過酸化物の使用を回避できている。更に、このような酸化試薬は、Journal of Organic Chemistry(2000)、65(25)、8415〜8420に記載されたように、通常、反応副産物のピリジンN−オキシドを発生させる。
【0055】
最後に、本発明に関する方法の更なる利点は、記載の合成経路が、既知技術に比べて収斂することである。既知技術は、所望する製品の構造的核心部に関して、一連の2つの化学的変換を含んでいる。産業上の利用の観点から、この方法の収斂は、最終製品の品質から危ない工程要因の影響を分離可能にする。
【0056】
本発明に関する方法の変型は、式(II)の化合物が、式(III−ビス)の4−置換−フェニルメチルスルフィド又はスルホキシド:
【0057】
【化12】
と反応させることができる。
【0058】
式中、nは、それぞれ、0及び1であり、かつXは、それぞれ、式(VI):
【0059】
【化13】
の化合物1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルファニル)フェニル]エタノン、又は式(VI−ビス):
【0060】
【化14】
の化合物1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルフィニル)フェニル]エタノンを得るために、前記したとおりである。
【0061】
また、好ましい態様について云えば、この反応を行うための、溶剤、触媒前駆体、配位子、量などの条件は、式(III)の4−置換−フェニルメチルスルホンから出発する、既に上に記載した反応に類似する。
【0062】
これらの中間体化合物は、スルフィド基又はスルホキシド基をスルホン基に酸化することにより、式(I)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノン又はそれらの塩に転化できることが都合がよい。このような酸化は、先行技術、例えばJ.O.C.2000、65、8415〜8420に、又はWO2001/029004に、又はLonzaとMerck&CoによるWO2001/007410に既に記載されている。
【0063】
最後に述べた文献の検討は、式(VI)の、得られた中間体が同一であるから、本発明の方法が特に有利であり、かつシアン基の転化とそれに続く除去が不必要であるから、本発明の方法がより一層原子効率の良いことを示している。このことは、シアン化反応を行わないこと、及び工業的な量のシアン化物を取り扱わないことと、シアン化物を多く含む産業廃棄物の廃棄処理を阻止することに結び付く。
【0064】
更に、2ステップだけが必要であり、幾つかの合成ステップが回避される。それ故、4−置換−アニソール又は4−置換−フェニルメチルスルホオキシドを用い、かつ式(VI)又は式(VI−ビス)の生成物を酸化して式(I)の生成物を得るという本発明の方法を用いる場合であっても、この方法は、既存技術の方法と比較して、とにかく大変に有利である。
【0065】
式(III)の4−置換−フェニルメチルスルホンを用いるという本発明の方法の最善の場合で、式(I)の化合物が、単一の合成ステップで得られる。従って、この方法は、既知の方法に比較して、少なくとも3つの合成ステップをなくし、かつシアン化物の使用及び廃棄処理をなくしている。それ故、本発明の方法は、経済的な観点から大変に有利である。
【実施例】
【0066】
(実験的部分)
(例1)−式(V)の中間体6−メチルピリジン−3−イルトリフルオロメタンスルホネートの合成−本発明の典型例
【0067】
【化15】
【0068】
10.0gの5−ヒドロキシ−2−メチルピリジン(91.7mmol)、11.0mLのピリジン、及び100mLのジクロロエタンを、機械的攪拌器、温度計、及びサーモスタットを備えた250mLのフラスコに入れた。この溶液に、18.5mLの無水トリフルオロメタンスルホン酸(110.0mmol)を液滴状に加え、温度を0℃に維持した。1.5時間攪拌した後、2mLのMeOH及びNaHCO3で飽和させた水溶液を加えた。有機層を水及び塩水で洗浄した後、MgSO4上で乾燥させた。この溶液を真空下で濃縮して残留物を得た。この残留物を、溶離液として9:1から4:1のn−ヘキサン:酢酸エチルを用いてクロマトグラフのカラム上で精製した。
【0069】
20gの6−メチルピリジン−3−イルトリフルオロメタンスルホネートを得た。無色のオイルとして、90.1%のモル収率であった。
1H−NMR(CDCl3)δ:2.61(s,3H)、7.26(d、J=8.4Hz、1H)、7.52(dd、J=9.0、3.0Hz、1H)、8.47(d、J=3.0Hz、1H)。
【0070】
(例2)−式(IV)の中間体5−エチニル−2−メチルピリジンの合成−本発明の典型例
【0071】
【化16】
【0072】
176mgのP(p−tol)3(0.58mmol)、65.0mgのPd(OAc)2(0.29mmol)、及び5mLの式(V)の6−メチルピリジン−3−イルトリフルオロメタンスルフォネート(29.0mmol)を、磁気固定装置を備えた乾燥した反応容器に入れた。アルゴン及び真空のサイクルで脱ガスした後、NMP/トルエン(1:1,40mL)中の脱ガスしたピペリジン(11.0mL,117.4mmol)の溶液を加えた。
【0073】
次に、4.2mLの2−メチル−3−ブチン−2−オ−ル(43mmol)を注射器で加え、この混合物を40℃で一夜攪拌した。室温に冷却した後、混合物をNaHCO3で飽和させた水溶液(80mL)を用いて希釈し、次にこれをエチルエーテル(3×30mL)で抽出した。一体にした有機抽出物を、水(50mL)で洗浄し、MgSO4上で乾燥し、減圧真空下で濃縮した。油状の残留物を無水トルエン(100mL)に溶解し、次に11.0gの微粉砕NaOH(256mmol)を加えた。この溶液を加熱し2時間環流し、次にこれを濾過し、NaHCO3(3×20mL)の飽和溶液で洗浄し、最後にMgSO4上で乾燥した。
【0074】
有機相を真空下で濃縮し、次に未精製の生成物を昇華精製し、2.1gの生成物を得た。白色固体として63%のモル収率でであった。
1H−NMR(CDCl3)δ:2.55(s,3H)、3.15(s、1H)、7.10(d、J=7.9Hz、1H)、7.64(dd、J=7.9、2.13Hz、1H)、8.60(d、J=3.0Hz、1H)。
【0075】
(例3)−式(II)の中間体1−(6−メチルピリジン−3−イル)エタノンの合成−本発明の典型例
【0076】
【化17】
【0077】
1.0gの5−エチニル−2−メチルピリジン(8.5mmol)を、磁気式固定装置を備えたフラスコに入れ、10mLの1:4のトルエン/硫酸混合物(0.29mL)に溶解した。得られた溶液を50℃にして一夜加熱し、次に室温に冷却後、溶液をNaHCO3の添加により塩基性にし、次にこれを酢酸エチル(3×20mL)で抽出して、最後に無水MgSO4上で乾燥した。次に、有機相を真空下で濃縮し、1.06gの生成物を得た。黄色のオイルとして、91%のモル収率であった。
【0078】
(例4)−式(I)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノンの合成−本発明の典型例
【0079】
【化18】
【0080】
磁気式固定装置を備え、予め乾燥させたシュレンク反応管に、トリ−tert−ブチル−ホスホニウムテトラフルオロ硼素酸塩(5.4mg、18.5μmol)、Pd(OAc)2(2.1mg、9.2μmol)、4−ブロモフェニルメチルスルホン(130.4mg、0.55mmol)、及び式(II)の1−(6−メチルピリジン−3−イル)エタノン(50mg、0.37mmol)を入れ、かつ隔膜(septum)で閉じた。この容器をアルゴン3サイクルで脱ガスし、次に注射器で2mLの無水トルエンを加えた。
【0081】
tert−ブトキシカリウム(165.7mg、1.48mmol)を分けて加え、溶液を80℃にして16時間加熱した。混合物をNaHCO3の飽和溶液(20mL)で希釈し、酢酸エチル(3×20mL)で抽出した。一体にした有機相を、NaHCO3で飽和した水溶液(20mL)で洗浄し、MgSO4上で乾燥し、かつ真空中で濃縮した。
【0082】
残留物を、溶離液として酢酸エチル/シクロヘキサンを用いて、5:5から10:0の勾配で、フラッシュクロマトグラフで精製した。32.1mgの生成物を得た。白色結晶固体として30%のモル収率であった。
1H−NMR(CDCl3)δ:2.64(s,3H)、3.04(s、3H)、
4.38(s、2H)、7.29(d、J=8.2Hz、1H)、7.46(d、J=8.3Hz、2H)、7.91(d、J=8.3Hz、2H)、8.16(dd、J=8.2、2.2Hz、1H)、9.11(d、J=2.2Hz、1H)、(図2を参照)。
【0083】
(例5)−式(I)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノンの合成−本発明の典型例
【0084】
【化19】
【0085】
【表1】
【0086】
(例6)−式(I)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノンの合成−本発明の典型例
【0087】
【化20】
【0088】
Pd(F6−acac)2(10.4mg、0.02mmol、0.5mol%)及びXantphos(23.2mg、0.04mmol、1mol%)を、冷却器を備えた、フレアフラスコに入れる。それらに、式(III、X=Br)の4−ブロモフェニルメチルスルホン(1.17g、5mmol)、式(II)のアセチルピコリン(541mg、4mmol)及びK3PO4(2.55g、12.0mmol、3当量)を加える。一度、アルゴン雰囲気を、真空−アルゴンのサイクルで安定化させ、注射器を用いて脱ガスした無水NMP(15mL)を加える。この混合物を、アルゴン中で攪拌下、100℃で18時間保持する。ガス−クロマトグラフ検定は、94%の収率を示す。
【0089】
反応混合物を、10%の塩化アンモニウム水溶液(15mL)で希釈し、ジクロロメタン(2×10mL)で抽出する。一体にした有機相を、ダイカライト上で濾過し、食塩水(10mL)で洗浄し、かつ減圧下で濃縮する。得られた未精製の反応生成物を、10mLのアセトンから再結晶させ、974mgの式(I)の生成物を得る。分離した生成物のモル収率は84%である。
【0090】
(例7)−式(VI)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルファニル)フェニル]エタノンの合成−本発明の典型的発展例
【0091】
【化21】
【0092】
4−ブロモフェニル−メチルスルホンの代わりに4−ブロモチオアニソールを用いて、例6に記載のように反応を行う。ガス−クロマトグラフ検定は、式(VI)の生成物で94%の収率を示す。分離生成物のH−NMRスペクトルを、図3に収める。
【0093】
(例8)−式(I)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノンの合成−本発明の典型的発展例
【0094】
【化22】
【0095】
100gの式(VI)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルファニル)フェニル]エタノン(1.0当量)、150mLの氷冷酢酸(1.5vol)、及び30mLのメタンスルフォン酸(1.2当量、0.3vol−96.11g/mol;1.481g/mL)を、高温度の温度計、冷却器、及び滴下漏斗を備えた3,000mLのフラスコに入れる。反応混合物を5〜10℃に冷却する。
【0096】
120mLの30w/w%過酸化水素(3当量、1.20vol;34.02g/mol;1.13g/mL)を、5〜10℃で、攪拌下で加える。反応混合物を、20〜25℃で、少なくとも6時間攪拌する。反応の終わりに、0〜5℃に冷却し、かつ300gのチオ硫酸ナトリウム(3wt)及び水(10vol)からなる溶液を、分けて加える。
【0097】
約180〜260mLの30%水酸化ナトリウム水溶液を、pHが約5.5〜6.5に達するまで加えた。
【0098】
これを、20〜25℃で2時間攪拌し、懸濁液を濾過する。固体を、2×400mLの水で洗浄し、次に固体を、40℃で、真空中で少なくとも12時間乾燥する。105.7gの生成物を得る。94%HPLC(高速液体クロマトグラフィー)純度のモル収率は97.5%(A%)である。
【0099】
(例9)−式(I)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノンの合成−本発明の典型例
【0100】
【化23】
【0101】
Pd(acac)2(6.1mg、0.02mmol、0.5mol%)及びXantphos(23.2mg、0.04mmol、1.0mol%)を、冷却器を備えたフレアフラスコに入れた。これに、式(III、X=Br)の4−ブロモフェニルメチルスルホン(1.17g、5mmol)、式(II)のアセチルピコリン(541mg、4mmol)、及びK3PO4(2.55g、12.0mmol、3当量)を加える。一度、アルゴン雰囲気を真空−アルゴンサイクルで安定化させ、注射器を用いて脱ガスした無水NMP(15mL)を加える。次に、混合物をアルゴン雰囲気中で、100℃で18時間攪拌し続ける。転化は定量的に起きる。
【0102】
反応混合物を、NaHCO3の飽和溶液(50mL)で希釈し、かつ酢酸エチル(4×50mL)で抽出する。一体にした有機相を、NaHCO3で飽和させた水溶液(30mL)で洗浄し、MgSO4上で乾燥し、かつ真空中で濃縮した。残留物を、溶離液として酢酸エチル/シクロヘキサンを用いて、5:5から10:0の勾配で、シリカゲルクロマトグラフで精製した。1.05gの生成物を得た。白色結晶固体として91%のモル収率であった。
【0103】
(例10)−式(I)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノンの合成−特に好ましい実施形態に従う本発明の典型例
【0104】
【化24】
【0105】
式(II)の1−(6−メチルピリジン−3−イル)エタノン(50g、1当量、0.370mol)、式(III;X=Br)の4−ブロモフェニル−メチルスルホン(87g、1当量、0.370mol),リン酸カリウム(235.6g、3当量、1.110mol)、Pd(acac)2(169mg、0.15当量.mol.%)、Xantphos(482mg、0.225当量.mol.%)、及びN,N−ジメチルホルムアミド(300mL)を、25℃で、窒素雰囲気下で2,000mLのフラスコに入れた。真空及び窒素の3サイクルを実施後、反応混合物を、85℃に加熱し、このような温度で、窒素雰囲気下で20時間攪拌した。次に、反応混合物を、50℃に冷却し、水(800mL)で希釈した。
【0106】
得られた混合物を、50℃で15分間攪拌し、次に、下部の水相をサイフォンで吸い上げて分離した。残留物を水(600mL)で希釈し、0〜5℃に冷却し、かつこのような温度で2時間攪拌した。得られた混合物を濾過し、固体を水(4×200mL)で洗浄し、65℃で10時間乾燥した。95.3gの1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノンを得た(モル収率=89%)。
【0107】
この場合、反応は、パラジウム触媒を基準にして、モル当量に換算して同量の配位子を用いて、ほぼ同様の方法で進行する、即ち式(II)の基体を基準にして、0.15%モル当量を用いて、ほぼ同様の方法で進行することに留意すべきである。
【0108】
(例11)−式(I)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノンの合成−もう一つの極めて特別に好ましい実施形態に従う本発明の典型例
【0109】
【化25】
【0110】
式(1)の1−(6−メチルピリジン−3−イル)エタノン(50g、370mmol)、式(2)の1−ブロモ−4−(メチルスルホニル)ベンゼン(87g、370mmol),リン酸カリウム(235.6g、3当量、1.11mol)、酢酸パラジウム(II)(125mg、0.15%mol、557μmol)、Xantphos(161mg、0.075%mol、278μmol)、及びN,N−ジメチルホルムアミド(300mL、6vol)を、この順番で、25℃かつ窒素雰囲気下で、機械式攪拌器、温度計、及び冷却器を備えた2,000mLのフラスコに入れた。
【0111】
反応混合物を25℃で攪拌し、真空及び窒素の3サイクルにかけ、次に85℃に加熱し、かつこのような温度で27時間攪拌した。次に、この反応混合物を45℃に冷却し、かつ攪拌を停止して、真空の助けで吸引吸い上げにより除去した水相の分離を促進させた。その結果得られた、高度の混合物を水(600mL、12vol)で希釈し、かつ3℃に冷却した。このような温度で2時間攪拌後、得られた懸濁液を濾過し、この生成物を水(4×200mL、4×4vol)で洗浄し、真空中60℃で乾燥した。黄色固体として、未精製の式(I)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノン(89.1g、83.2%)を収穫した。
【0112】
このようにして得られた未精製の式(I)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノン(80g)を、ジクロロメタン(560mL、7vol)、及び0.5MのHCl(384mL、4.8vol)の混合物に溶解した。分離した有機相を、0.5MのHCl(2×192mL、2×2.4vol及び96mL、1.2vol)で繰り返し抽出した。一体にした水相を、NaOH15%(91mL)を添加して中和した(pH6.8)。得られた水性混合物を15℃に冷却し、このような温度で2時間攪拌した。
得られた懸濁液を濾過し、生成物を水(4×320mL、4×4vol)で洗浄し、真空中65℃で乾燥した。ストローイエローの固体として、精製した式(I)の化合物(71.7g;89.6%)を収穫した。
【0113】
上記のようにして得られた、精製した式(I)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノン(70g)を、120℃でN,N−ジメチルホルムアミド(84mL、1.2vol)に溶解した。得られた溶液を25℃に冷却した。冷却中に生成物の結晶化が観察された。得られた懸濁液を、25℃で2時間攪拌し、次にこれを濾過し、生成物をアセトン(2×56mL、2×0.8vol)で洗浄し、真空中50℃で乾燥した。白色固体として、式(I)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノン(61.6g;88%)を収穫した。
【技術分野】
【0001】
(発明の技術分野)
本発明の目的は、COX−2阻害薬の医薬原体(pharmaceutical active ingredient)である、エトリコキシブ(Etoricoxib)合成の中間体、1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノンを調製する方法である。
【背景技術】
【0002】
(従来技術)
CAS登録番号(RN)221615−75−4を有する、式(I):
【0003】
【化1】
の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノンは、COX−2阻害薬の分類に属する医薬原体であり、かつ登録商標アルコキシア(Arcoxia)で2002年から市販されている、エトリコキシブ合成用の重要な中間体である。
【0004】
【化2】
【0005】
COX−2阻害薬の合成に、このような化合物を用いる例は、WO99/55830、WO99/15503、及びDavies、Lan WらのJournal of Organic Chemistry(2000)、65(25)、8415〜8420に報告されている。
【0006】
このような重要な構成要素の種々の合成法は、前記特許文献に記載されたもの以外にも知られているが、LonzaとMerck&CoのWO2001/007410に記載された方法は、経済的観点から最も有利な方法であると思われる。このような方法の欠点は、最終ステップにおいて、タングステン酸ナトリウムで触発させた過酸化水素による酸化処理を含むことである。Zambon Group S.p.Aによる後続の出願WO2001/029004は、触媒(タングステン酸ナトリウム)及び酸(例えば、メタンスルホン酸)の存在下で、酸化剤の組み合わせ(例えば、過酢酸と過酸化水素の混合物)を含む酸化処理を行う改良された方法を報告している。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】WO99/55830
【特許文献2】WO99/15503
【特許文献3】WO2001/007410
【特許文献4】WO2001/029004
【非特許文献】
【0008】
【非特許文献1】Davies、Lan WらのJournal of Organic Chemistry(2000)、65(25)、8415〜8420
【発明の概要】
【発明が解決しようとする課題】
【0009】
(発明の概要)
本発明が取り上げる問題は、1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノンを調製する代替方法の提供である。
【課題を解決するための手段】
【0010】
このような問題は、付属の特許請求の範囲に記載したような、1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノンを調製する方法により解決される。そこでなされた定義は、本発明の開示の不可欠な要素である。
【0011】
本発明に関する方法の更なる特徴及び利点は、非限定的な実施例により与えられる、その好ましい実施形態に関する以下の記載から得られるであろう。
【図面の簡単な説明】
【0012】
(図の簡単な説明)
【図1】本発明の好ましい態様に従った、1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノンの合成の略図である。
【図2】本発明の方法に従って得られた、式(I)の生成物の1H−NMRスペクトルを示す図である。
【図3】本発明の方法に従って得られた、式(VI)の生成物の1H−NMRスペクトルを示す図である。
【発明を実施するための形態】
【0013】
(発明の詳細な説明)
本発明は、式(I):
【0014】
【化3】
の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノン又はその塩を調製する方法であって、式(II):
【0015】
【化4】
の1−(6−メチルピリジン−3−イル)エタノンと、式(III):
【0016】
【化5】
の4−置換−フェニルメチルスルホンとを反応させることを含む方法に関する。
【0017】
ここで、Xは、F、Br、Cl、I、OTs、OTf、OMs、ONf、及びO(C=O)NR2からなる群から選ばれ、ここでRは、メチル、エチル、n−プロピル、iso−プロピル、n−ブチル、iso−ブチル、sec−ブチル、若しくはtert−ブチルの中から選ばれた、直鎖の、若しくは分枝したC1〜C4のアルキル置換基、又はフェニル若しくはベンジルである。TsOは脱離基トシラート(Tosylate)、TfOは脱離基トリフラート(Triflate)、MsOは脱離基メシラート(Mesylate)、NfOは脱離基ノナフラート(Nonaflate)である。
【0018】
式(III)の化合物で、Rはエチルであることが好ましい。
【0019】
本発明の好ましい態様に従えば、この方法は、4−ブロモフェニルメチルスルホンを用いて行われる。
【0020】
また、α−アリーレーション型カップリングに用いられるその他の脱離基は、この方法を実施のためにXに置換して使用することができ、従って、本発明の欠かせない要素として考えるべきである。
【0021】
また、式(II)の1−(6−メチルピリジン−3−イル)エタノンは、式(II−ビス):
【0022】
【化6】
の1−(6−メチルピリジン−3−イル)エテノールであるそれらの「エノール」型でも存在することができる。
【0023】
本発明の方法は、以下に説明する反応条件に従って行う、式(II)又は(II−ビス)と、式(III)の2つの試薬間のカップリングを考慮に入れている。
【0024】
この反応は、トルエン、キシレン、アルコール類などの有機溶剤中で、又はジオキサン、THF(テトラヒドロフラン)及びMe−TFHなどのエーテル溶剤中で、又はDMF(ジメチルホルムアミド)、DMSO(ジメチルスルホキシド)及びNMP(N−メチルピロリドン)中で行われ、エーテル溶剤が好ましく、かつNMP及びDMFが更により好ましい。
【0025】
好ましくは、反応は、6、8又は10容積(volume)の溶剤中で行われ、6容積が好ましい。
【0026】
反応は、tert−ブトキシカリウム、炭酸カリウム、又はリン酸カリウムなどの塩基の存在下で、好ましくはリン酸カリウムの存在下で行われる。
【0027】
留意すべき観点は、塩基の粉砕度合である。反応開始直前に、湿式粉砕機により行うリン酸カリウムの粉砕は、迅速な反応を促進し、かつ高収率で所望する生成物が得られる。
【0028】
1〜3モル塩基当量が使用され、3モル塩基当量が好ましい。3モル塩基当量の使用は、反応速度を増大し、かつ反応生成物中で試薬のより高い転化を促進するので好ましい。
【0029】
反応に用いる触媒前駆体は、Pd(OAc)2(酢酸パラジウム(II))、Pd(F6−acac)2(パラジウムビス(ヘキサフルオロ)アセチルアセトナト)、Pd(acac)2(酢酸パラジウム(II)アセチルアセトナト)、Pd2(dba)3(トリス(ジベンジリデンアセトン)ジパラジウム(0))、(PdallylCl)2(アリルパラジウム(II)クロリド(ダイマー))、又はPdCl2(塩化パラジウム(II))であり、Pd(OAc)2が好ましい。
【0030】
触媒前駆体の量は、式(II)の1−(6−メチルピリジン−3−イル)エタノンを基準に、0.05モル%と2モル%の間に包含される触媒量が通常使用される。好ましくは、0.15〜0.5モル%である。
【0031】
反応に用いられる配位子は、PPh3(トリフェニルホスフィン)、P(Cy)3(トリシクロヘキシルホスフィン)、Xantphos(キサントホス)、dppe(1,2−ビス(ジフェニルホスフィノ)エタン)、dppp(1,3−ビス(ジフェニルホスフィノ)プロパン)、dppf(1,1−ビス(ジフェニルホスフィノ)フェロセン)、Josiphos(ジョシホス)からなる群から選ばれる。キレート性ホスフィンのXantphosが好ましい。
【0032】
これは、約0.075モル%と1モル%の間に含まれる量での使用が好ましい。
【0033】
配位子のモル量は、通常パラジウム触媒前駆体の量の0.5〜2倍を使用し、好ましくは2倍である(式(II)の化合物を常に基準にして、パラジウム触媒前駆体が0.5モル%である時、配位子1モル%に等しい)。
【0034】
例えば、酢酸パラジウム(II)の場合、最も好ましい配位子/金属のモル比は、0.5であるが、2の比率も使用できる。
【0035】
反応は、60℃と140℃の間、好ましくは80℃と120℃の間の温度、より好ましくは約85℃〜約100℃で行われる。
【0036】
反応は、16〜30時間、好ましくは約18〜20時間で通常行われる。
【0037】
この反応は、少量のフェニルメチルスルホンが、4−ブロモフェニルメチルスルホンのプロト−脱臭素反応で発生することを除けば、その他の副生物がないことから、良好な選択性で進行する。
【0038】
更なる好ましい態様に従えば、この反応は無水環境下で行われる。
【0039】
次に、生成物は、抽出及び結晶化を含む従来の有機合成技術により分離される。
【0040】
本発明の実施形態に従えば、本発明の化合物を調製するために有用な、式(II):
【0041】
【化7】
の中間体1−(6−メチルピリジン−3−イル)エタノンは、式(IV):
【0042】
【化8】
の5−エチニル−2−メチルピリジンの水和反応により調製できることが好都合である。
【0043】
このような水和反応は、硫酸とトルエンの混合物、それぞれ2:1から4:1、好ましくはそれぞれ4:1の混合物中で行うことができるので都合がよい。反応は、50℃で16時間行い、又は80℃で3時間、又は70℃で2時間行うことができる。生成物を、酢酸エチルを用いて水相から抽出し、脱水の後に濃縮して残留物とする。
【0044】
この方法の通常のモル収率は、90%超である。
【0045】
本発明の実施形態に従えば、本発明の化合物を調製するために有用な式(IV):
【0046】
【化9】
の中間体5−エチニル−2−メチルピリジンの合成は、式(V):
【0047】
【化10】
の中間体6−メチルピリジン−3−イルトリフルオロメタンスルホネートと2−メチル−3−ブチン−2−オールとの反応を含む方法により行うことができる。
【0048】
この反応は、酢酸パラジウム(II)とホスフィンの存在下で行われる。また、PPh3、P(p−FPh)3(トリス(4−フルオロフェニル)ホスフィン)、P(p−tol)3(トリ−p−トリルホスフィン)、dppe、dppfなどの種々のホスフィンが使用できるが、高収率を与えるので、P(p−tol)3が好ましい。有機溶剤として、NMP/トルエン混合物(1:1)又はトルエン単独を使用できることが好都合である。この反応は、ピリジン、DBU(ジアザビシクロウンデセン)、ピペリジン、HNiPr2、TEA、DABCO(1,4−ジアザビシクロ[2,2,2]オクタン)、DIPEA(N,N−ジイソプロピルエチルアミン)などの塩基の存在下で行われる。
【0049】
その好ましい態様では、反応は、高収率になる理由から、ピペリジンの存在下で行われる。この反応は、通常、40℃で16時間行われる。化合物2−メチル−4−(6−メチルピリジン−3−イル)ブト−3−イン−2−オールが、本反応の中間体として形成される。この化合物は、式(IV−ビス):
【0050】
【化11】
で表され、所望により容易に分離される。
【0051】
式中、アセチレン官能基は、さらにアセトンアダクトの型で保護されている。このような保護は、トルエン中でソーダ処理して除去し、式(IV)の中間体を形成するために環流させる。
【0052】
また、このように式(IV)の化合物を分離しないので、式(V)の中間体から出発して式(II)の中間体を調製する方法は、同一容器内で行うことができる。
【0053】
本発明に従う1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノンを調製する方法は、タングステン系触媒の使用を避けるという利点がある。更なる利点は、本発明の方法が、市場から入手できる式(II)及び式(III)の中間体を使用することであり、かつ(III)は既に商標登録されたEINECS(少なくともX=Brの場合)である。
【0054】
更に、式(III)のスルホネート中間体は、既に、最終生成物に要求される酸化状態の硫黄を示しており、それ故に、このことは、既知技術の方法に比べると、最終的な酸化処理を回避することができ、潜在的には爆発性で危険物質である過酸化水素及び有機過酸化物の使用を回避できている。更に、このような酸化試薬は、Journal of Organic Chemistry(2000)、65(25)、8415〜8420に記載されたように、通常、反応副産物のピリジンN−オキシドを発生させる。
【0055】
最後に、本発明に関する方法の更なる利点は、記載の合成経路が、既知技術に比べて収斂することである。既知技術は、所望する製品の構造的核心部に関して、一連の2つの化学的変換を含んでいる。産業上の利用の観点から、この方法の収斂は、最終製品の品質から危ない工程要因の影響を分離可能にする。
【0056】
本発明に関する方法の変型は、式(II)の化合物が、式(III−ビス)の4−置換−フェニルメチルスルフィド又はスルホキシド:
【0057】
【化12】
と反応させることができる。
【0058】
式中、nは、それぞれ、0及び1であり、かつXは、それぞれ、式(VI):
【0059】
【化13】
の化合物1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルファニル)フェニル]エタノン、又は式(VI−ビス):
【0060】
【化14】
の化合物1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルフィニル)フェニル]エタノンを得るために、前記したとおりである。
【0061】
また、好ましい態様について云えば、この反応を行うための、溶剤、触媒前駆体、配位子、量などの条件は、式(III)の4−置換−フェニルメチルスルホンから出発する、既に上に記載した反応に類似する。
【0062】
これらの中間体化合物は、スルフィド基又はスルホキシド基をスルホン基に酸化することにより、式(I)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノン又はそれらの塩に転化できることが都合がよい。このような酸化は、先行技術、例えばJ.O.C.2000、65、8415〜8420に、又はWO2001/029004に、又はLonzaとMerck&CoによるWO2001/007410に既に記載されている。
【0063】
最後に述べた文献の検討は、式(VI)の、得られた中間体が同一であるから、本発明の方法が特に有利であり、かつシアン基の転化とそれに続く除去が不必要であるから、本発明の方法がより一層原子効率の良いことを示している。このことは、シアン化反応を行わないこと、及び工業的な量のシアン化物を取り扱わないことと、シアン化物を多く含む産業廃棄物の廃棄処理を阻止することに結び付く。
【0064】
更に、2ステップだけが必要であり、幾つかの合成ステップが回避される。それ故、4−置換−アニソール又は4−置換−フェニルメチルスルホオキシドを用い、かつ式(VI)又は式(VI−ビス)の生成物を酸化して式(I)の生成物を得るという本発明の方法を用いる場合であっても、この方法は、既存技術の方法と比較して、とにかく大変に有利である。
【0065】
式(III)の4−置換−フェニルメチルスルホンを用いるという本発明の方法の最善の場合で、式(I)の化合物が、単一の合成ステップで得られる。従って、この方法は、既知の方法に比較して、少なくとも3つの合成ステップをなくし、かつシアン化物の使用及び廃棄処理をなくしている。それ故、本発明の方法は、経済的な観点から大変に有利である。
【実施例】
【0066】
(実験的部分)
(例1)−式(V)の中間体6−メチルピリジン−3−イルトリフルオロメタンスルホネートの合成−本発明の典型例
【0067】
【化15】
【0068】
10.0gの5−ヒドロキシ−2−メチルピリジン(91.7mmol)、11.0mLのピリジン、及び100mLのジクロロエタンを、機械的攪拌器、温度計、及びサーモスタットを備えた250mLのフラスコに入れた。この溶液に、18.5mLの無水トリフルオロメタンスルホン酸(110.0mmol)を液滴状に加え、温度を0℃に維持した。1.5時間攪拌した後、2mLのMeOH及びNaHCO3で飽和させた水溶液を加えた。有機層を水及び塩水で洗浄した後、MgSO4上で乾燥させた。この溶液を真空下で濃縮して残留物を得た。この残留物を、溶離液として9:1から4:1のn−ヘキサン:酢酸エチルを用いてクロマトグラフのカラム上で精製した。
【0069】
20gの6−メチルピリジン−3−イルトリフルオロメタンスルホネートを得た。無色のオイルとして、90.1%のモル収率であった。
1H−NMR(CDCl3)δ:2.61(s,3H)、7.26(d、J=8.4Hz、1H)、7.52(dd、J=9.0、3.0Hz、1H)、8.47(d、J=3.0Hz、1H)。
【0070】
(例2)−式(IV)の中間体5−エチニル−2−メチルピリジンの合成−本発明の典型例
【0071】
【化16】
【0072】
176mgのP(p−tol)3(0.58mmol)、65.0mgのPd(OAc)2(0.29mmol)、及び5mLの式(V)の6−メチルピリジン−3−イルトリフルオロメタンスルフォネート(29.0mmol)を、磁気固定装置を備えた乾燥した反応容器に入れた。アルゴン及び真空のサイクルで脱ガスした後、NMP/トルエン(1:1,40mL)中の脱ガスしたピペリジン(11.0mL,117.4mmol)の溶液を加えた。
【0073】
次に、4.2mLの2−メチル−3−ブチン−2−オ−ル(43mmol)を注射器で加え、この混合物を40℃で一夜攪拌した。室温に冷却した後、混合物をNaHCO3で飽和させた水溶液(80mL)を用いて希釈し、次にこれをエチルエーテル(3×30mL)で抽出した。一体にした有機抽出物を、水(50mL)で洗浄し、MgSO4上で乾燥し、減圧真空下で濃縮した。油状の残留物を無水トルエン(100mL)に溶解し、次に11.0gの微粉砕NaOH(256mmol)を加えた。この溶液を加熱し2時間環流し、次にこれを濾過し、NaHCO3(3×20mL)の飽和溶液で洗浄し、最後にMgSO4上で乾燥した。
【0074】
有機相を真空下で濃縮し、次に未精製の生成物を昇華精製し、2.1gの生成物を得た。白色固体として63%のモル収率でであった。
1H−NMR(CDCl3)δ:2.55(s,3H)、3.15(s、1H)、7.10(d、J=7.9Hz、1H)、7.64(dd、J=7.9、2.13Hz、1H)、8.60(d、J=3.0Hz、1H)。
【0075】
(例3)−式(II)の中間体1−(6−メチルピリジン−3−イル)エタノンの合成−本発明の典型例
【0076】
【化17】
【0077】
1.0gの5−エチニル−2−メチルピリジン(8.5mmol)を、磁気式固定装置を備えたフラスコに入れ、10mLの1:4のトルエン/硫酸混合物(0.29mL)に溶解した。得られた溶液を50℃にして一夜加熱し、次に室温に冷却後、溶液をNaHCO3の添加により塩基性にし、次にこれを酢酸エチル(3×20mL)で抽出して、最後に無水MgSO4上で乾燥した。次に、有機相を真空下で濃縮し、1.06gの生成物を得た。黄色のオイルとして、91%のモル収率であった。
【0078】
(例4)−式(I)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノンの合成−本発明の典型例
【0079】
【化18】
【0080】
磁気式固定装置を備え、予め乾燥させたシュレンク反応管に、トリ−tert−ブチル−ホスホニウムテトラフルオロ硼素酸塩(5.4mg、18.5μmol)、Pd(OAc)2(2.1mg、9.2μmol)、4−ブロモフェニルメチルスルホン(130.4mg、0.55mmol)、及び式(II)の1−(6−メチルピリジン−3−イル)エタノン(50mg、0.37mmol)を入れ、かつ隔膜(septum)で閉じた。この容器をアルゴン3サイクルで脱ガスし、次に注射器で2mLの無水トルエンを加えた。
【0081】
tert−ブトキシカリウム(165.7mg、1.48mmol)を分けて加え、溶液を80℃にして16時間加熱した。混合物をNaHCO3の飽和溶液(20mL)で希釈し、酢酸エチル(3×20mL)で抽出した。一体にした有機相を、NaHCO3で飽和した水溶液(20mL)で洗浄し、MgSO4上で乾燥し、かつ真空中で濃縮した。
【0082】
残留物を、溶離液として酢酸エチル/シクロヘキサンを用いて、5:5から10:0の勾配で、フラッシュクロマトグラフで精製した。32.1mgの生成物を得た。白色結晶固体として30%のモル収率であった。
1H−NMR(CDCl3)δ:2.64(s,3H)、3.04(s、3H)、
4.38(s、2H)、7.29(d、J=8.2Hz、1H)、7.46(d、J=8.3Hz、2H)、7.91(d、J=8.3Hz、2H)、8.16(dd、J=8.2、2.2Hz、1H)、9.11(d、J=2.2Hz、1H)、(図2を参照)。
【0083】
(例5)−式(I)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノンの合成−本発明の典型例
【0084】
【化19】
【0085】
【表1】
【0086】
(例6)−式(I)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノンの合成−本発明の典型例
【0087】
【化20】
【0088】
Pd(F6−acac)2(10.4mg、0.02mmol、0.5mol%)及びXantphos(23.2mg、0.04mmol、1mol%)を、冷却器を備えた、フレアフラスコに入れる。それらに、式(III、X=Br)の4−ブロモフェニルメチルスルホン(1.17g、5mmol)、式(II)のアセチルピコリン(541mg、4mmol)及びK3PO4(2.55g、12.0mmol、3当量)を加える。一度、アルゴン雰囲気を、真空−アルゴンのサイクルで安定化させ、注射器を用いて脱ガスした無水NMP(15mL)を加える。この混合物を、アルゴン中で攪拌下、100℃で18時間保持する。ガス−クロマトグラフ検定は、94%の収率を示す。
【0089】
反応混合物を、10%の塩化アンモニウム水溶液(15mL)で希釈し、ジクロロメタン(2×10mL)で抽出する。一体にした有機相を、ダイカライト上で濾過し、食塩水(10mL)で洗浄し、かつ減圧下で濃縮する。得られた未精製の反応生成物を、10mLのアセトンから再結晶させ、974mgの式(I)の生成物を得る。分離した生成物のモル収率は84%である。
【0090】
(例7)−式(VI)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルファニル)フェニル]エタノンの合成−本発明の典型的発展例
【0091】
【化21】
【0092】
4−ブロモフェニル−メチルスルホンの代わりに4−ブロモチオアニソールを用いて、例6に記載のように反応を行う。ガス−クロマトグラフ検定は、式(VI)の生成物で94%の収率を示す。分離生成物のH−NMRスペクトルを、図3に収める。
【0093】
(例8)−式(I)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノンの合成−本発明の典型的発展例
【0094】
【化22】
【0095】
100gの式(VI)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルファニル)フェニル]エタノン(1.0当量)、150mLの氷冷酢酸(1.5vol)、及び30mLのメタンスルフォン酸(1.2当量、0.3vol−96.11g/mol;1.481g/mL)を、高温度の温度計、冷却器、及び滴下漏斗を備えた3,000mLのフラスコに入れる。反応混合物を5〜10℃に冷却する。
【0096】
120mLの30w/w%過酸化水素(3当量、1.20vol;34.02g/mol;1.13g/mL)を、5〜10℃で、攪拌下で加える。反応混合物を、20〜25℃で、少なくとも6時間攪拌する。反応の終わりに、0〜5℃に冷却し、かつ300gのチオ硫酸ナトリウム(3wt)及び水(10vol)からなる溶液を、分けて加える。
【0097】
約180〜260mLの30%水酸化ナトリウム水溶液を、pHが約5.5〜6.5に達するまで加えた。
【0098】
これを、20〜25℃で2時間攪拌し、懸濁液を濾過する。固体を、2×400mLの水で洗浄し、次に固体を、40℃で、真空中で少なくとも12時間乾燥する。105.7gの生成物を得る。94%HPLC(高速液体クロマトグラフィー)純度のモル収率は97.5%(A%)である。
【0099】
(例9)−式(I)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノンの合成−本発明の典型例
【0100】
【化23】
【0101】
Pd(acac)2(6.1mg、0.02mmol、0.5mol%)及びXantphos(23.2mg、0.04mmol、1.0mol%)を、冷却器を備えたフレアフラスコに入れた。これに、式(III、X=Br)の4−ブロモフェニルメチルスルホン(1.17g、5mmol)、式(II)のアセチルピコリン(541mg、4mmol)、及びK3PO4(2.55g、12.0mmol、3当量)を加える。一度、アルゴン雰囲気を真空−アルゴンサイクルで安定化させ、注射器を用いて脱ガスした無水NMP(15mL)を加える。次に、混合物をアルゴン雰囲気中で、100℃で18時間攪拌し続ける。転化は定量的に起きる。
【0102】
反応混合物を、NaHCO3の飽和溶液(50mL)で希釈し、かつ酢酸エチル(4×50mL)で抽出する。一体にした有機相を、NaHCO3で飽和させた水溶液(30mL)で洗浄し、MgSO4上で乾燥し、かつ真空中で濃縮した。残留物を、溶離液として酢酸エチル/シクロヘキサンを用いて、5:5から10:0の勾配で、シリカゲルクロマトグラフで精製した。1.05gの生成物を得た。白色結晶固体として91%のモル収率であった。
【0103】
(例10)−式(I)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノンの合成−特に好ましい実施形態に従う本発明の典型例
【0104】
【化24】
【0105】
式(II)の1−(6−メチルピリジン−3−イル)エタノン(50g、1当量、0.370mol)、式(III;X=Br)の4−ブロモフェニル−メチルスルホン(87g、1当量、0.370mol),リン酸カリウム(235.6g、3当量、1.110mol)、Pd(acac)2(169mg、0.15当量.mol.%)、Xantphos(482mg、0.225当量.mol.%)、及びN,N−ジメチルホルムアミド(300mL)を、25℃で、窒素雰囲気下で2,000mLのフラスコに入れた。真空及び窒素の3サイクルを実施後、反応混合物を、85℃に加熱し、このような温度で、窒素雰囲気下で20時間攪拌した。次に、反応混合物を、50℃に冷却し、水(800mL)で希釈した。
【0106】
得られた混合物を、50℃で15分間攪拌し、次に、下部の水相をサイフォンで吸い上げて分離した。残留物を水(600mL)で希釈し、0〜5℃に冷却し、かつこのような温度で2時間攪拌した。得られた混合物を濾過し、固体を水(4×200mL)で洗浄し、65℃で10時間乾燥した。95.3gの1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノンを得た(モル収率=89%)。
【0107】
この場合、反応は、パラジウム触媒を基準にして、モル当量に換算して同量の配位子を用いて、ほぼ同様の方法で進行する、即ち式(II)の基体を基準にして、0.15%モル当量を用いて、ほぼ同様の方法で進行することに留意すべきである。
【0108】
(例11)−式(I)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノンの合成−もう一つの極めて特別に好ましい実施形態に従う本発明の典型例
【0109】
【化25】
【0110】
式(1)の1−(6−メチルピリジン−3−イル)エタノン(50g、370mmol)、式(2)の1−ブロモ−4−(メチルスルホニル)ベンゼン(87g、370mmol),リン酸カリウム(235.6g、3当量、1.11mol)、酢酸パラジウム(II)(125mg、0.15%mol、557μmol)、Xantphos(161mg、0.075%mol、278μmol)、及びN,N−ジメチルホルムアミド(300mL、6vol)を、この順番で、25℃かつ窒素雰囲気下で、機械式攪拌器、温度計、及び冷却器を備えた2,000mLのフラスコに入れた。
【0111】
反応混合物を25℃で攪拌し、真空及び窒素の3サイクルにかけ、次に85℃に加熱し、かつこのような温度で27時間攪拌した。次に、この反応混合物を45℃に冷却し、かつ攪拌を停止して、真空の助けで吸引吸い上げにより除去した水相の分離を促進させた。その結果得られた、高度の混合物を水(600mL、12vol)で希釈し、かつ3℃に冷却した。このような温度で2時間攪拌後、得られた懸濁液を濾過し、この生成物を水(4×200mL、4×4vol)で洗浄し、真空中60℃で乾燥した。黄色固体として、未精製の式(I)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノン(89.1g、83.2%)を収穫した。
【0112】
このようにして得られた未精製の式(I)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノン(80g)を、ジクロロメタン(560mL、7vol)、及び0.5MのHCl(384mL、4.8vol)の混合物に溶解した。分離した有機相を、0.5MのHCl(2×192mL、2×2.4vol及び96mL、1.2vol)で繰り返し抽出した。一体にした水相を、NaOH15%(91mL)を添加して中和した(pH6.8)。得られた水性混合物を15℃に冷却し、このような温度で2時間攪拌した。
得られた懸濁液を濾過し、生成物を水(4×320mL、4×4vol)で洗浄し、真空中65℃で乾燥した。ストローイエローの固体として、精製した式(I)の化合物(71.7g;89.6%)を収穫した。
【0113】
上記のようにして得られた、精製した式(I)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノン(70g)を、120℃でN,N−ジメチルホルムアミド(84mL、1.2vol)に溶解した。得られた溶液を25℃に冷却した。冷却中に生成物の結晶化が観察された。得られた懸濁液を、25℃で2時間攪拌し、次にこれを濾過し、生成物をアセトン(2×56mL、2×0.8vol)で洗浄し、真空中50℃で乾燥した。白色固体として、式(I)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノン(61.6g;88%)を収穫した。
【特許請求の範囲】
【請求項1】
式(I):
【化1】
の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノン又はその塩を調製する方法であって、下記のステップ:(a)式(II):
【化2】
の1−(6−メチルピリジン−3−イル)エタノンと、式(III−ビス):
【化3】
の4−置換−フェニルメチルスルフィド、スルホキシド、又はスルホン(式中、nは0と2の間に含まれる整数であり、Xは、F、Br、Cl、I、OTs、OTf、OMs、ONf、及びO(C=O)NR2からなる群から選ばれ、ここでRは直鎖の、若しくは分枝したC1−C4アルキル置換基、又はフェニル、若しくはベンジルである)とを反応させるステップと;(b)nが0又は1である時、式(VI):
【化4】
又は式(VI−ビス):
【化5】
の関連中間体を更に酸化して式(I)の生成物を得るステップとを含む方法。
【請求項2】
式(III−ビス)の化合物が、2に等しいnを有する、即ち式(III):
【化6】
(式中、Xは請求項1に記載のとおりである)である、請求項1に記載の方法。
【請求項3】
Xが臭素である、請求項1〜2のいずれか一項に記載の方法。
【請求項4】
反応が、トルエン、キシレン、アルコール類、ジオキサン及びTHFなどのエーテル溶剤、Me−THF、DMF、DMSO、並びにNMPからなる群から選ばれた有機溶剤中で行われる、又はTHF、NMP、若しくはDMF中で行われる、請求項1〜3のいずれか一項に記載の方法。
【請求項5】
反応が、tert−ブトキシドカリウム、及びリン酸カリウムの中から選ばれた塩基の存在下で行われる、請求項1〜4のいずれか一項に記載の方法。
【請求項6】
1〜3モル塩基当量が使用される、又は3モル塩基当量が使用される、請求項1〜5のいずれか一項に記載の方法。
【請求項7】
反応が、Pd(OAc)2、Pd(F6−acac)2、及びPd(acac)2の中から選ばれた触媒前駆体の存在下で行われる、又はPd(OAc)2触媒前駆体の存在下で行われる、請求項1〜6のいずれか一項に記載の方法。
【請求項8】
式(II)の基体を基準にして、0.05〜2モル%の量の触媒が、反応で使用される、請求項1〜7のいずれか一項に記載の方法。
【請求項9】
式(II)の基体を基準にして、0.15〜0.5モル%の量の触媒が、反応で使用される、請求項1〜8のいずれか一項に記載の方法。
【請求項10】
反応が、PPh3、P(Cy)3、Josiphos、Xantphos、dppe、dppp、及びdppfからなる群から選ばれた反応用結合剤の存在下で行われる、又はXantphos反応用結合剤の存在下で行われる、請求項1〜9のいずれか一項に記載の方法。
【請求項11】
パラジウム触媒前駆体のモル量の0.5〜2倍の配位子のモル量が、使用される、請求項1〜10のいずれか一項に記載の方法。
【請求項12】
酢酸パラジウム触媒前駆体のモル量の約0.5倍の配位子のモル量が、使用される、請求項1〜11のいずれか一項に記載の方法。
【請求項13】
反応が、60℃と140℃の間で行われる、又は80℃と120℃の間で行われる、請求項1〜12のいずれか一項に記載の方法。
【請求項14】
反応が、85℃と100℃の間で行われる、請求項1〜13のいずれか一項に記載の方法。
【請求項15】
反応が、16〜30時間行われる、請求項1〜14のいずれか一項に記載の方法。
【請求項16】
反応が、18〜20時間行われる、請求項1〜15のいずれか一項に記載の方法。
【請求項17】
反応が、無水環境下で行われる、請求項1〜16のいずれか一項に記載の方法。
【請求項18】
式(II):
【化7】
の1−(6−メチルピリジン−3−イル)エタノンが、式(IV):
【化8】
の中間体5−エチニル−2−メチルピリジンの転化により得られる、請求項1〜17のいずれか一項に記載の方法。
【請求項19】
式(IV):
【化9】
の中間体5−エチニル−2−メチルピリジンが、式(V):
【化10】
の中間体6−メチルピリジン−3−イル トリフルオロメタンスルホネートの転化により得られる、請求項18に記載の方法。
【請求項20】
ステップ(a)が、DMF中で、塩基としてリン酸カリウム、触媒前駆体として酢酸パラジウム(II)、及び配位子としてXantphosの存在下で行われる、請求項1〜19のいずれか一項に記載の方法。
【請求項21】
このようにして得られた式(I):
【化11】
の化合物1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノン又はその塩を精製するステップを更に含む、請求項1〜20のいずれか一項に記載の方法。
【請求項22】
式(I)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノン又はその塩を調製するためのパラジウム触媒の使用。
【請求項23】
式(I)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノン又はその塩を調製するためのXantphos配位子の使用。
【請求項1】
式(I):
【化1】
の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノン又はその塩を調製する方法であって、下記のステップ:(a)式(II):
【化2】
の1−(6−メチルピリジン−3−イル)エタノンと、式(III−ビス):
【化3】
の4−置換−フェニルメチルスルフィド、スルホキシド、又はスルホン(式中、nは0と2の間に含まれる整数であり、Xは、F、Br、Cl、I、OTs、OTf、OMs、ONf、及びO(C=O)NR2からなる群から選ばれ、ここでRは直鎖の、若しくは分枝したC1−C4アルキル置換基、又はフェニル、若しくはベンジルである)とを反応させるステップと;(b)nが0又は1である時、式(VI):
【化4】
又は式(VI−ビス):
【化5】
の関連中間体を更に酸化して式(I)の生成物を得るステップとを含む方法。
【請求項2】
式(III−ビス)の化合物が、2に等しいnを有する、即ち式(III):
【化6】
(式中、Xは請求項1に記載のとおりである)である、請求項1に記載の方法。
【請求項3】
Xが臭素である、請求項1〜2のいずれか一項に記載の方法。
【請求項4】
反応が、トルエン、キシレン、アルコール類、ジオキサン及びTHFなどのエーテル溶剤、Me−THF、DMF、DMSO、並びにNMPからなる群から選ばれた有機溶剤中で行われる、又はTHF、NMP、若しくはDMF中で行われる、請求項1〜3のいずれか一項に記載の方法。
【請求項5】
反応が、tert−ブトキシドカリウム、及びリン酸カリウムの中から選ばれた塩基の存在下で行われる、請求項1〜4のいずれか一項に記載の方法。
【請求項6】
1〜3モル塩基当量が使用される、又は3モル塩基当量が使用される、請求項1〜5のいずれか一項に記載の方法。
【請求項7】
反応が、Pd(OAc)2、Pd(F6−acac)2、及びPd(acac)2の中から選ばれた触媒前駆体の存在下で行われる、又はPd(OAc)2触媒前駆体の存在下で行われる、請求項1〜6のいずれか一項に記載の方法。
【請求項8】
式(II)の基体を基準にして、0.05〜2モル%の量の触媒が、反応で使用される、請求項1〜7のいずれか一項に記載の方法。
【請求項9】
式(II)の基体を基準にして、0.15〜0.5モル%の量の触媒が、反応で使用される、請求項1〜8のいずれか一項に記載の方法。
【請求項10】
反応が、PPh3、P(Cy)3、Josiphos、Xantphos、dppe、dppp、及びdppfからなる群から選ばれた反応用結合剤の存在下で行われる、又はXantphos反応用結合剤の存在下で行われる、請求項1〜9のいずれか一項に記載の方法。
【請求項11】
パラジウム触媒前駆体のモル量の0.5〜2倍の配位子のモル量が、使用される、請求項1〜10のいずれか一項に記載の方法。
【請求項12】
酢酸パラジウム触媒前駆体のモル量の約0.5倍の配位子のモル量が、使用される、請求項1〜11のいずれか一項に記載の方法。
【請求項13】
反応が、60℃と140℃の間で行われる、又は80℃と120℃の間で行われる、請求項1〜12のいずれか一項に記載の方法。
【請求項14】
反応が、85℃と100℃の間で行われる、請求項1〜13のいずれか一項に記載の方法。
【請求項15】
反応が、16〜30時間行われる、請求項1〜14のいずれか一項に記載の方法。
【請求項16】
反応が、18〜20時間行われる、請求項1〜15のいずれか一項に記載の方法。
【請求項17】
反応が、無水環境下で行われる、請求項1〜16のいずれか一項に記載の方法。
【請求項18】
式(II):
【化7】
の1−(6−メチルピリジン−3−イル)エタノンが、式(IV):
【化8】
の中間体5−エチニル−2−メチルピリジンの転化により得られる、請求項1〜17のいずれか一項に記載の方法。
【請求項19】
式(IV):
【化9】
の中間体5−エチニル−2−メチルピリジンが、式(V):
【化10】
の中間体6−メチルピリジン−3−イル トリフルオロメタンスルホネートの転化により得られる、請求項18に記載の方法。
【請求項20】
ステップ(a)が、DMF中で、塩基としてリン酸カリウム、触媒前駆体として酢酸パラジウム(II)、及び配位子としてXantphosの存在下で行われる、請求項1〜19のいずれか一項に記載の方法。
【請求項21】
このようにして得られた式(I):
【化11】
の化合物1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノン又はその塩を精製するステップを更に含む、請求項1〜20のいずれか一項に記載の方法。
【請求項22】
式(I)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノン又はその塩を調製するためのパラジウム触媒の使用。
【請求項23】
式(I)の1−(6−メチルピリジン−3−イル)−2−[4−(メチルスルホニル)フェニル]エタノン又はその塩を調製するためのXantphos配位子の使用。
【図1】

【図2】
【図3】
【図2】
【図3】
【公開番号】特開2012−188414(P2012−188414A)
【公開日】平成24年10月4日(2012.10.4)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−2845(P2012−2845)
【出願日】平成24年1月11日(2012.1.11)
【出願人】(512008417)エッフェ.イ.エッセ.ファッブリカ イタリアーナ スィンテティチ ソシエタ ペル アチオニ (2)
【Fターム(参考)】
【公開日】平成24年10月4日(2012.10.4)
【国際特許分類】
【出願番号】特願2012−2845(P2012−2845)
【出願日】平成24年1月11日(2012.1.11)
【出願人】(512008417)エッフェ.イ.エッセ.ファッブリカ イタリアーナ スィンテティチ ソシエタ ペル アチオニ (2)
【Fターム(参考)】
[ Back to top ]