
エナンチオマー混合物を脱ラセミ化する方法
本発明は、立体選択性アルコール脱水素酵素およびその補助因子を用いて酸化反応と還元反応との組合せによって第2アルコールのエナンチオマー混合物を酵素的に脱ラセミ化するための方法であって、この場合には、形式的に光学活性の第2アルコールのエナンチオマーは、選択的に相応するケトンに酸化され、引続きケトンは、選択的に光学的対掌体に還元され、他方、補助因子の還元された形は、添加酵素により還元反応に使用される、上記に脱ラセミ化法に関し、この方法は、
相反する立体選択性および異なる補助因子選択性を有する2つのアルコール脱水素酵素ならびに2つの属する異なる補助因子が、酸化反応または還元反応に使用され、酸化された補助因子および還元された補助因子が、同時に進行する、添加酵素を用いる酵素反応で互いに変換され、この場合2つのエナンチオマーの1つへの脱ラセミ化の方向は、2つのアルコール脱水素酵素の選択によって制御可能であるか、または2つの補助因子のための添加酵素の選択性の差につき制御可能であることによって特徴付けられる。

相反する立体選択性および異なる補助因子選択性を有する2つのアルコール脱水素酵素ならびに2つの属する異なる補助因子が、酸化反応または還元反応に使用され、酸化された補助因子および還元された補助因子が、同時に進行する、添加酵素を用いる酵素反応で互いに変換され、この場合2つのエナンチオマーの1つへの脱ラセミ化の方向は、2つのアルコール脱水素酵素の選択によって制御可能であるか、または2つの補助因子のための添加酵素の選択性の差につき制御可能であることによって特徴付けられる。
【発明の詳細な説明】
【技術分野】
【0001】
発明の技術分野
本発明は、酵素系を使用しながらエナンチオマー混合物を脱ラセミ化する方法に関する。
【0002】
先行技術
最近、立体異性体の分野でラセミ化、即ちラセミ混合物を得るために光学異性体の鏡像異性体への変換および脱ラセミ化の場合ならびに前記プロセスの正確な逆のプロセスの場合にめざましい進歩が達成された。ラセミ化は、立体的に不安定な化合物、例えばシアンヒドリン、半(チオ)アセタールα−置換カルボニル化合物およびα−置換ヒダントインの場合に、簡単な保護的な酸触媒反応または塩基触媒反応によって達成可能であり、一方で、立体的に安定な化合物、例えば第2アルコールおよびキラールアミンは、ラセミ化がはるかに困難である。
【0003】
最後のキラールアミンは、例えば遷移金属錯体触媒を用いて反応されるレドックス法により達成され、この場合キラリティー中心で必然的にsp3−ハイブリダイズされているエナンチオマーは、プロキラールなsp2ハイブリダイズされた中間生成物を経て別のエナンチオマーに変換される。例えば、O. PamiesおよびJ.- E. Baeckvall, Trends Biotechnol. 22, 130-135 (2004)およびChem. Rev. 103. 3247-3261 (2003); H. Pellissier, Tetrahedron 59, 8291-8327 (2003); M. J. Kim, Y. AhnおよびJ. Park, Curr. Opin. Biotechnol. 13, 578-587 (2002); V. Zimmermann, M. BellerおよびU. Kragi, Org. Process Res. Dev. 10, 622-627 (2006); Y. AsanoおよびS. Yamaguchi, J. Am. Chem. Soc. 127, 7696-7697 (2005)の論文参照。
【0004】
固有の高特異的な生合成の分野で、僅かな「真性」ラセミ体だけが公知であり、それというのも、工業界とは異なり、天然ではラセミ化は、殆んど必要とされていないからである。即ち、例えばα−ヒドロキシカルボン酸(例えば、マンデル酸誘導体)、α−アミノ酸およびヒダントインのラセミ化を促進するための特殊な酵素は、ほんの二三が公知である(例えば、B. Schnell, K. FaberおよびW. Kroutil, Adv. Synth. Catal. 345, 653-666 (2003))。第2アルコールおよび第1アミンをラセミ化するために、実際に定義された酵素は、久しく公知ではなかった。
【0005】
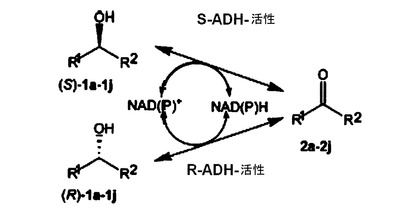
Chem.Eur.J.13,8271−8276に開示された、新しいラセミ化戦略の本発明者らの論文類は、動力学的考察形式の代わりに進行する反応の熱力学的考察形式を基礎とするものである。2つのエナンチオマーRおよびSならびにプロキラールな中間生成物Pからなる反応系においては、2つの光学対掌体のそれぞれは、中間生成物と一緒に化学的平衡および熱力学的平衡、即ちP⇔SおよびR⇔Pが存在する。同一の補助因子、NADまたはNADP(ニコチンアミド−アデニン−ジヌクレオチドまたは−ホスフェート)を利用する、2つの相反するエナンチオ選択性のアルコール脱水素酵素(以下、略してADHsと呼称される)からなる多数の組合せを使用しながら、アシロインを含めて種々の光学活性の第2アルコールのラセミ化が達成される。アルコール/ケトンの平衡は、補助因子の酸化された形と還元された形の量および比、即ちNAD+:NADHまたはNADP+:NADPHをアルコール側で適当に選択することによって維持される。詳細は図1参照。
【0006】
NAD(P)+の割合を最小に設定した場合には、純粋な(S)異性体から出発して数時間の反応時間後に望ましいラセミ体が生じた。ケトン中間生成物の量は、10%未満に減少させることができ、一部分は、1%未満に減少させることができた。これとは異なり、高い選択性のADHだけを用いる比較試験は、多くの場合に試験されたAHDsにとって不成功に終わった。単に、1つの場合には、14日の反応時間後、82%eeの収率("enantiomeric excess"、エナンチオマーの過剰量、即ち光学的収率)は、達成される。
【0007】
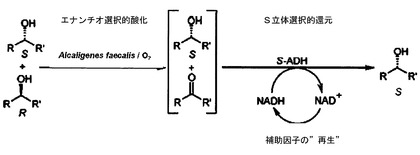
本発明者らの最初に短時間で発行された論文(C.V. Voss, C.C. GruberおよびW. Kroutil, Angew. Chem. Int. Ed. 47, 741-745 (2008))には、第2アルコールのラセミ化合物を、中間生成物としてのプロキラールなケトンを経てエナンチオ選択性の細菌酵素に由来するタンデム系を使用しながらアルコール酸化のために補助因子としての立体選択性ADHおよびNADの形で脱ラセミ化することが開示されている。補助因子は、いわば「再生され」、即ち同時に「添加酵素」または「補助酵素」としてのグルコース脱水素酵素(以下、GHDと略記する)を用いて促進される、グルコースからグルコノラクトンまたはグルコン酸への酸化を進行させることにより、酸化された形から還元された形へ戻された。
【0008】
親液性化されたアルカリ糞便菌細胞を使用しながらの最初の試験の場合には、意外なことに、脱ラセミ化が確認されるのではなく、むしろ、出発基質としてのエナンチオマー純粋のアルコールのラセミ化が確認され、このことは、親液性化による細胞透過性の上昇に帰因した。損傷を受けていない細胞膜を有する、新たに収穫された細胞を使用し、したがって酸化および還元は、空間的に互いに別々に進行し、種々の第2アルコールのラセミ化合物は、選択的に99%eeを上廻る収率で望ましいエナンチオマーに変換されることができた。
【0009】
しかし、この公知技術水準は、多数の欠点を有する。一方で、酵素混合物を酸化のために準備するアルカリ糞便菌系は、正確には定義不可能であり、したがって反応の進行の著しい変化をまねく可能性があり、ひいては再現可能性は、殆んど無い。他方、全ての従来法の場合、異性体化されたアルコール1モル当たり酸化のために酸素1モルが使用され、および補助因子の再生のためにグルコースが化学量論的に1モル使用され、その上、この場合には、グルコン酸またはグルコノラクトン1モルが副生成物として生じる。
【0010】
従って、本発明の課題は、上記の欠点を回避する、改善された脱ラセミ化法を準備することであった。
【0011】
発明の詳細な説明
意外なことに、立体選択性アルコール脱水素酵素およびその補助因子を用いて酸化反応と還元反応との組合せによって第2アルコールのエナンチオマー混合物を酵素的に脱ラセミ化するための改善された方法は、前記課題を解決することが見出され、この場合には、形式的に光学活性の第2アルコールのエナンチオマーは、選択的に相応するケトンに酸化され、引続きケトンは、選択的に光学的対掌体に還元され、他方、補助因子の還元された形は、添加酵素により還元反応に使用される。本発明による方法は、相反する立体選択性および異なる補助因子選択性を有する2つのアルコール脱水素酵素ならびに2つの属する異なる補助因子が、酸化反応または還元反応に使用され、酸化された補助因子および還元された補助因子が、同時に進行する、添加酵素を用いる酵素反応で互いに変換され、この場合2つのエナンチオマーの1つへの脱ラセミ化の方向は、2つのアルコール脱水素酵素の選択によって制御可能であるか、または2つの補助因子のための添加酵素の選択性の差につき制御可能であることによって特徴付けられる。
【0012】
本発明による方法によって、脱ラセミ化は、実際に定量的な光学的収率、即ち99%ee超で達成可能であり、この場合には、後になお詳説されているように、前記系が安定した平衡を達成すると直ちに、試薬が同時の反応の経過中に化学量論的に使用されることはない。更に、正確に定義された純粋な酵素(2つのADHsおよび添加酵素)は、触媒反応に使用され、このことから専ら可逆的な反応が前記方法で生じ、ならびに顕著な再生可能性が生じる。最終的に、前記方法は、簡単な一槽反応法で実施されることができ、この場合個々の部分反応間での時間的または空間的な分離は、不要である。
【0013】
アルコール脱水素酵素として、特に市場で入手可能または簡単に入手可能なアルコール脱水素酵素、例えばバシラス属(Bacillus)、シュードモナス属(Pseudomonas)、コリネバクテリウム属(Corynebacterium)、ロドコッカス属(Rhodococcus)、乳酸菌(Lactobacillus)および/またはサーモアナエロビウム(Thermoanaerobium)に由来する細菌酵素、例えばロドコッカス・ルベル(Rhodococcus ruber)、ラクトバシラス・ケフィル(Lactobacillus kefir)またはサーモアナエロビウム・ブロキイ(Thermoanaerobium brockii)に由来する細菌酵素または酵母、例えばアスペルギルス(Aspergillus)、カンジダ(Candida)、ピキア(Pichia)またはサッカロミケス(Saccharomyces)に由来する酵素が使用され、それというのも、前記酵素は、エナンチオマー過剰量および反応速度に関連して特に良好な結果を生じるからである。しかし、適したAHD対の選択にとって決定的なことは、なかんずく2つのADHsが相反する立体選択性および異なる補助因子選択性を有しなければならないという要件にある。補助因子は、ADHsのそれぞれの選択から相応してもたらされ、通常は、NADおよびNADPであり、有利には、単に触媒量で使用される。
【0014】
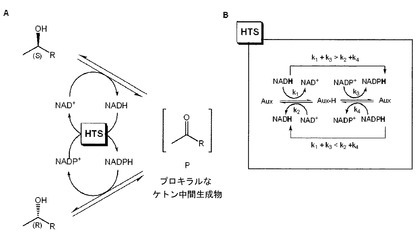
図3は、本発明に方法における反応経過を説明し、この場合HTSは、「Hydrid-Transfer-System水素化物移送系」を表わし、この中で「Aux」で示された添加酵素によって促進される副反応は、補助因子の「再生」、即ち酸化された形と還元された形の互いの変換のために行なわれる。k1〜k4は、水素化物移送系の第1の順序の反応の形式的な速度定数を表わす。
【0015】
図3Aに図示されているように、第2のアルコールエナンチオマーの酸化反応および還元反応は、2つの相反する立体選択性ADHs(図示されていない)によって促進される。(S)エナンチオマーから(R)エナンチオマーへの反応が進行する場合には、NADに対して補助因子の選択を有する、(S)選択性ADHの(S)異性体の酸化の際に、水素化物イオンは、アルコールから分離され、補助因子、NAD+、の酸化物の形に移行され、それによってこの補助因子は、還元された形、NADH、に移行される。本質的に同時に添加酵素Auxは、NADHの前記水素化物イオンを受け取り(このことは、「Aux−H」を生じる)、引続き酸化された形、NADP+の形で第2の補助因子に移行され、それによって第2の補助因子の還元された形のNADPHは、準備される。前記の第2の補助因子から再び水素化物は、NADPの選択性を有する第2の(R)選択性ADHによってケトン中間生成物Pに移行され、それによってこのケトン中間生成物Pは、(R)エナンチオマーに還元される。逆の方向に、即ち(R)異性体が(S)形に変換される際に、逆反応は、当然に同様に行なわれる。
【0016】
平衡で存在する酵素/補助因子系から出発する場合には、同一の水素化物イオンは、上記に説明した反応を経て、最終的に再び今や立体化学が逆転されたアルコール分子上に到達する。
【0017】
添加酵素がヌクレオチドトランスヒドロゲナーゼである場合には、水素化物イオンは、添加酵素によって1つの補助因子から別の補助因子へ、上記の簡単な形で移行する。この場合、図3B中のAux−Hは、酵素と水素化物イオンとの複合体を表わす。しかし、試験されたヌクレオチドトランスヒドロゲナーゼは、満足させる結果を生じないので、本発明者らは、試験の際に他の選択可能な方法により、水素化物を直接に移行するヌクレオチドトランスヒドロゲナーゼの代わりにもう1つの脱水素酵素/基質系もその役を引き受けうることが見い出された。この場合、Aux−Hは、添加酵素に相当する基質の還元された形を表わす。
【0018】
この種の添加酵素として、原理的に全ての補助因子に依存するオキシド還元酵素がこれに該当し、このオキシド還元酵素は、脱ラセミ化する第2アルコールの酸化反応および還元反応を損なわない。脱水素酵素、有利にグルコース脱水素酵素(GDH)、グルコース−6−ホスフェート脱水素酵素(G6PDH)およびホルミエート脱水素酵素(FDH)は、極めて良好な結果をもたらし、したがって好ましい添加酵素である。
【0019】
最初の2つの場合には、基質への水素化物の移行によって、グルコン酸またはグルコノラクトン、またはこれらの−6−ホスフェートは、グルコースまたは−6−ホスフェートへ還元され(このことは、"Aux−H"を生じる)、直ちに再び酸化される。同様のことは、第3の場合にもCO2で行なわれ、このCO2は、Aux−Hとしてのホルミエートで平衡状態になる。実際にホルミエートからCO2への酸化の反応平衡は、二酸化炭素側に遙かに広がるが、しかし、反応の原理的な可逆性は、正しいことが確認された。本発明においては、添加酵素は、全く使用されず(または殆んど使用されず)、したがって、微少量だけが必要とされるので、ホルミエート脱水素酵素/ホルミエート/CO2系は、本発明の目的に十分に好適であり、例えばこのことは、下記の実施例に示されている。
【0020】
上記したように、本発明による方法において、平衡状態が達成されると直ちに試薬の化学量論的使用は、全く生じない。この平衡状態は、特殊な酵素/基質の組合せおよびその選択性に依存するので、予想または予めの調整は、不可能である。それによって、実際には、この平衡は、脱ラセミ化法の開始時に生じる。前記の通常数分間継続する段階において、微少量の添加基質、即ちグルコースまたはグルコン酸、ホルミエートまたはCO2は、実際に使用される。
【0021】
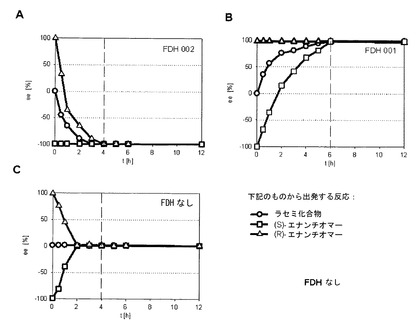
第2アルコールの異性体化がどの方向に進行するかにより、実際には最初に2つのADHsの立体選択性および補助因子選択性に依存するが、しかし、他の結果においては、2つの補助因子のための添加酵素の異なる選択性にも依存する。NADPの選択性を有する(S)選択性ADHおよびNADPの選択性を有する(R)選択性ADHから出発する、上記に示された例には、実際に添加酵素の補助因子選択性による脱ラセミ化の方向を規定することができ、そのために、これは、十分に「制御系」とも呼称されてよい。添加系の酸化法または還元法における補助因子選択性は、NADHおよびNADP+がNAD+およびNADPH(図3Bで:k1+k3>k2+k4)へ変換されることを導くか、またはNAD+およびNADPHがNADHおよびNADP+(図3Bで:k1+k3>k2+k4)へ変換されることを導き、このことは、第1の場合には、(R)エナンチオマーの形成を導き、別の場合には、(S)エナンチオマーの形成を導く。制御酵素またはその基質が省略されるか、または制御酵素が2つの補助因子の1つに対する選択性を有する(勿論、このことは、全くありそうもないことである)場合には、全く脱ラセミ化だけを観察することができるのではなく、光学的に純粋なアルコールから出発して、むしろ、逆反応、即ちラセミ化を観察することができ、例えば、これは、図4からも明らかである:図4Aおよび4Bは、それぞれ逆の補助因子選択性を有するホルミエート脱水素酵素に対して第2アルコールとしての2−オクタノールを用いた反応経過を示し、図4cは、FDHなしの系のための反応経過を示す。
【0022】
しかし、脱ラセミ化の方向は、逆に反対される立体選択性または補助因子選択性を有するADH対を選択することによって逆転される可能性もある。例えば、上記の記載において、同じ添加酵素の際にNADPの選択性を有する(S)選択性ADHおよびNADPの選択性を有する(R)選択性ADHを使用する場合には、ラセミ化合物から選択的に(R)エナンチオマーの代わりに光学的に純粋な(S)エナンチオマーが形成される。
【0023】
本発明による方法は、通常、水、水と1つ以上の有機溶剤とからなる単相または多相の混合物およびイオン性液体を含む群から選択された溶剤中で実施され、この場合費用および安定性の理由から特に従来の水性緩衝系が使用される。
【0024】
水性緩衝系は、溶剤をpH値に対して非敏感性にする物質、例えば塩を含有する水性溶剤である。公知の水性緩衝系は、例えば炭酸/重炭酸塩系、炭酸−珪酸塩緩衝液、酢酸/酢酸塩緩衝液、燐酸塩緩衝液、ミハエリス(Michaelis)によるベロナール/酢酸塩緩衝液、アンモニア緩衝液、HEPES(4−(2−ヒドロキシエチル)−1−ピペラジンエタンスルホン酸)およびMES(2−(N−モルホリノ)エタンスルホン酸)である。
【図面の簡単な説明】
【0025】
【図1】図1は、プロキラールなケトンを経て2つの特殊なADHsを用いて酵素触媒反応によりラセミ化を行なう原理を示す略図である。
【図2】図2は、定義された菌株の背景を用いるラセミ化合物の分解および引続く補助因子の再生を示す略図である。
【図3】図3のAは、補助因子の再生のために2つの特異性ADHsおよび特異性補助酵素(水素化物移行系:HTS)を用いて本発明による酵素触媒反応により脱ラセミ化を行なう原理を示す略図であり、図3のBは、水素化物移行系の原理を示す略図である。
【図4】図4のAは、ホルミエート脱水素酵素(FDH002)を用いる1−フェニルエタノールのラセミ混合物の移動を示す略図であり、図4のBは、ホルミエート脱水素酵素(FDH001)を用いる1−フェニルエタノールのラセミ混合物の移動を示す略図であり、図4のCは、ホルミエート脱水素酵素(FDH)なしの1−フェニルエタノールのラセミ混合物の移動を示す略図である。
【図5】図5は、NADまたはNADP特異性ホルミエート脱水素酵素(FDH)を用いるラセミ混合物の移動を示す略図である。
【図6】図6は、反応平衡に対する種々の反応パラメーターの変更の影響を示す略図である。
【図7】図7は、ラセミ化平衡の調節に対する添加基質のグルコースの種々の濃度の影響を示し、図7のAは、種々のグルコース濃度についての時間的経過を示す略図であり、図7のBは、6時間の反応時間後のそれぞれのグルコース濃度への相関関係ee[%]を示す略図である。
【実施例】
【0026】
例:
次に、本発明を代表的な制限されていない実施例につき詳細に記載する。
【0027】
材料、関連資料および方法
酵素
ADH−A:ロドコッカス・ルベル(Rhodococcus ruber)のアルコール脱水素酵素(BioCatalytics Inc.,社から市場で入手可能、今やCodexis社, Pasadens在, USAから市場で入手可能)。
LK−ADH:乳酸桿菌属ケフィル(Lactobacillus kefir)のアルコール脱水素酵素(Sigma-Aldrich社, Wien在、から市場で入手可能、#05643、0.4 IE/mg)。
RE−ADH:ロドコッカス・エリトロポリス(Rhodococcus erythropolis)のアルコール脱水素酵素(Sigma-Aldrich社から市場で入手可能、#68482、20 IE/ml)。
LB−ADH:アルコール脱水素酵素002(Juelich Chiral Solutions社から市場で入手可能、今やCodexis社から市場で入手可能、#05.11)。
ADH−T:アルコール脱水素酵素005(Juelich Chiral Solutions社から市場で入手可能、今やCodexis社から市場で入手可能、#26.10)。
ADH−PR2:アルコール脱水素酵素007(Juelich Chiral Solutions社から市場で入手可能、今やCodexis社から市場で入手可能、#42.10)。
TB−ADH:テルモアナエロビウム・ブロキイ(Thermoanaerobium brockii)のアルコール脱水素酵素(Sigma-Aldrich社から市場で入手可能、#A9287,30−90 IE/mg)。
G6PDH:パン酵母のグルコース−6−ホスフェート脱水素酵素(Sigma-Aldrich社から市場で入手可能、#49271,240 IE/mg)。
GLY−DH:ゲオトリクム・カンディドゥム(Geotrichum candidum)のグリセリン脱水素酵素(Sigma-Aldrich社から市場で入手可能、#49860,30 IE/mg)。
LDH−SC:スラフィロコッキ(Sraphylococci)のD−ラクテート脱水素酵素(Sigma-Aldrich社から市場で入手可能、#17847,120 IE/mg)。
LDH−LS:ラクトバシラス・スプ(Lactobacillus sp.)のD−ラクテート脱水素酵素(Sigma-Aldrich社から市場で入手可能、#59023,400 IE/mg)。
LDH−RM:イエウサギの筋肉に由来するL−ラクテート脱水素酵素(Sigma-Aldrich社から市場で入手可能、#61311,500 IE/mg)。
FDHI:NADP特異性ホルミエート脱水素酵素001(Juelich Chiral Solutions社から市場で入手可能、今やCodexis社, Pasadena在, USAから市場で入手可能、#25.10,47 IE/ml)。
FDHI:NADP特異性ホルミエート脱水素酵素001(Juelich Chiral Solutions社から市場で入手可能、今やCodexis社, Pasadena在, USAから市場で入手可能、#25.10,47 IE/ml)。
FDH3:NAD特異性ホルミエート脱水素酵素001(Juelich Chiral Solutions社から市場で入手可能、今やCodexis社から市場で入手可能、#09.11,200 IE/ml)。
FDH4:酵母のホルミエート脱水素酵素(Boehringer Mannheim GmbH社から市場で入手可能、#204226,0.5 IE/mg)。
FDH5:カンディダ・ボイジニイ(Candida boidinii)のホルマール脱水素酵素(Martina Pohl、デュッセルドルフ大学、ドイツ連邦共和国、からの寄贈品)。
GDH−BM:D−グルコース脱水素酵素001(Juelich Chiral Solutions社から市場で入手可能、今やCodexis社から市場で入手可能、#22.10,30 IE/mg)。
GDH−BS:D−グルコース脱水素酵素002(Juelich Chiral Solutions社から市場で入手可能、今やCodexis社から市場で入手可能、#29.10,500 IE/ml)。
【0028】
酵素の性質
【表1】
【0029】
【表2】
【0030】
化学薬品
rac−2−オクタノール(#04504、MG 130.23g/mol)、(R)−2−オクタノール(#74864、MG 130.23g/mol)、(S)−2−オクタノール(#74863、MG 130.23g/mol)、2−オクタノン(#53220、MG 128.21g/mol)、アンモニウムホルミエート(#09739、63.96g/mol)、ナトリウムホルミエート(#3996−15−4、69.02g/mol)およびカリウム塩としての蟻酸(#57444−81−2、MG 85.13g/mol)は、Sigma−Aldrich社、Wien在、から購入した。抽出および後処理のための化学薬品:
抽出のためのエチルアセテート(#441977)は、Brenntag CEE GmbH社、Ort在、から購入され、新たに蒸留に使用された。アセチル化のためのDMAP(#29224、MG 122.17g/mol)および無水酢酸(#45830、MG 102.09g/mol)は、Sigma−Aldrich社、Wien在、から購入した。
【0031】
一般的なプロセス
光学的組成物のシフトのためのモデル的方法:市販の酵素の活性は、一般に国際単位(IE)で記載されている。しかし、この単位は、全体でここで使用されたものとは別の基質に対するそれぞれの酵素の活性を示す。従って、使用された酵素の活性は、適した"再生"系での2−オクタノンの還元の際に測定された(一般に、アンモニウムホルミエートを有するFDH、5当量)。全ての試験のためには、例えばADHsの1 IE2-オクタノールが使用された。
【0032】
系1:ADH−A、LK−ADH、NADP特異性FDH(2 IE)、アンモニウムホルムエート(基質濃度に対して3当量)、NAD+およびNADP+(基質3モル%)は、TRIS−HCl(50mM、pH7.5、全容量0.5ml)中に懸濁された。反応は、ラセミ体の2−オクタノールの添加によって開始された(0.5μl、8mmol/ml、ee<3%)。30℃で3時間の振盪(130rpm)後、混合物は、EtOAc(500μl)で抽出され、遠心分離され、相分離が生じた。
【0033】
系2:NAD特異性FDH(2 IE)の使用まで系1と同様。
【0034】
系3:GDH−BS(2 IE)およびα−D−グルコース(1当量、8mmol/L)の使用まで系1と同様。
【0035】
系4:ADH−TおよびADH−PR2の使用まで系1と同様。
【0036】
系5:ADH−T、ADH−PR2およびNAD特異性FDHの使用まで系1と同様。
【0037】
系6:RE−ADHおよびテルモアナエロビウム・ブロキイ(Thermoanaerobium brockii)のAORの使用まで系1と同様。
【0038】
分析法
キラルなGC−FID分析:
アルコールは、無水酢酸(100ml)およびDMAP(0.5mg)の添加によって30℃で2時間でアセチル化された。後処理後、生成物は、GC−FIDおよびGC−MSDを用いてキラルな固定相で分析された。
【0039】
キラルなGC−FID分析は、バリアン(Varian)−3900−ガスクロマトグラフィーでFID検出器を用いてクロムパック−キラシル−DEX−CB−カラム(Chrompack-Chirasil-DEX-CB-Saeule)(Varian社、25m×0.32mm×0.25mm、1.0バール H2)を使用して、検出器温度250℃、分配比90:1で行なわれた。
【0040】
キラルなGC−MSD分析:
キラルなGC−MSD分析は、アギレント(Agilent)−7890A−GCシステムでFID検出器を用いてクロムパック−キラシル−DEX−CB−カラム(Chrompack-Chirasil-DEX-CB-Saeule)(Varian社、25m×0.32mm×0.25mm、1.0バール H2)を使用して、検出器温度250℃、分配比90:1で行なわれた。
【0041】
キラルなHPLC分析:
HPLC分析は、DGU−20A5脱ガス装置、LC−20AD液体クロマトグラフ、SIL−20AC自動サンプラー、CBM−20Aコミュニケーションバスモジュール(Kommunikationsbusmodul)、SPD−M20Aダイオードアレー検出器およびCTO−20ACカラム炉を備えたShimadzu−HPLCシステムでn−ヘプタン/イソプロパノール=90:10、0.5ml/分、18℃を有するChiralpak−ADカラム(Daicel, 0.46×25cm)を使用して行なわれた。
【0042】
実施例1〜13、比較例1〜4
脱ラセミ化を、第2アルコールとしての2−オクタノールとの種々のAHD/添加酵素組合せ物を使用して次の条件下で実施した:基質濃度8mmol/l、反応時間3〜12時間、TRIS−HCl(pH7.5、50mM)または燐酸塩緩衝液(pH7.5、50mM)中で30℃、130rpmでの振盪。ADHsからそれぞれ約1 IE(基質としての2−オクタノールのため);触媒量(約3モル%)中のNAD+およびNADP+。添加酵素:2 IE(そのために、製造業者によって記載されたような天然基質)。添加基質(ホルミエート、グルコース、グルコース−6−ホスフェート、ラクテートおよびグリセリン):16mmol/L。この組成および結果は、次の第3表中に記載されている。
【0043】
【表3】
【0044】
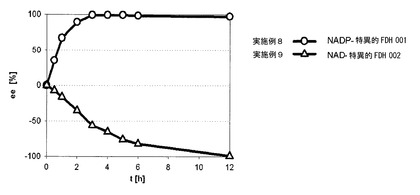
2−オクタノールラセミ化合物からなる本発明の実施例1〜13の試験された酵素組合せ物は、主に良好な一部分が実際に定量的なエナンチオマー過剰量およびエナンチオマーの1つの殆んど全て定量的な収量で供給されたことを確認することができる。同一のADH対を使用した場合、添加酵素の補助因子特異性を逆にすることによって、脱ラセミ化の方向を制御することができた。実施例1/2+3、8/9、10/11、12/13参照。また、実施例8および9の結果は、図5にグラフで図示されている。
【0045】
これに対して、比較例1〜4においてラクテート脱水素酵素またはグリセリン脱水素酵素を使用した場合には、脱ラセミ化は、全く生じなかった。
【0046】
実施例14〜22
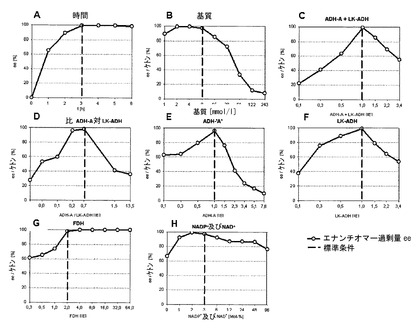
これらの実施例において、実施例1からの酵素系を使用して、種々の反応パラメーターが変更され、反応経過に対して前記反応パラメーターの作用が試験された。実施例14〜21の結果は、図6A〜Hにグラフで図示されており、実施例22の結果は、図7A〜Bに図示されている。
【0047】
実施例14
ここでは、反応時間を1〜6時間の間で変動させ、3時間後に既に定量的な変換が達成されることが見い出された。従って、前記群の他の実施例は、3時間に亘って実施された(図6A)。
【0048】
実施例15
アルコール濃度を1〜243m2/lの間で変動させ、この最適な結果は、6時間の所定の反応時間で2〜8mmol/lを生じた。よりいっそう高い濃度の場合には、よりいっそう長い反応時間またはよりいっそう大量の酵素が必要不可欠であり、完全な変換が達成される(図6B)。
【0049】
実施例16
2つのADHsの全体量を0.1〜3.4 IEの間で変動させ、この場合には、1 IEが最適な活性量であることが明らかになった(図6C)。
【0050】
実施例17
互いに2つのADHsの活性比(IEで)を0.01〜13.5の間で変動させ、約0.2〜約0.7の比は、最も効果的であることが判明したが、しかし、その際に、1:1の比の値は、欠いた(図6D)。
【0051】
実施例18および19
2つのADHsのそれぞれ一方の活性を0.1〜7.6 IEの間で、または3.4 IEで変動させ、その際に第2のADHの活性は、1 IEであり、また、第2の酵素のための最適な活性量は1 IEであり、したがって、2つのADHsの最適な活性比は1:1であることが見い出された(図6E、6F)。
【0052】
実施例20
FDHの量を0.3〜64.0 IEの間で変動させ、2 IEの量から既に定量的な変換が達成されることが判明した(図6G)。
【0053】
実施例21
補助因子NADおよびNADPの共通の濃度を0〜96モル%の間で変動させ、この場合約2〜3モル%の濃度は、最も効果的であった(図6H)。
【0054】
実施例22
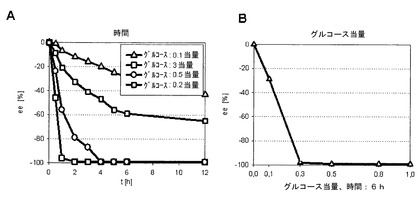
実施例5を繰り返したが、この場合添加基質、即ちグルコースの濃度は、図7Aに示されているように、0.5〜12時間の反応時間に亘ってアルコール濃度に対して0.1〜3当量の間で変動した。グルコース当量が0.1〜1の間で変動する際の6時間後のエナンチオマー過剰量eeは、図7Bに示されている。この場合には、>99%が既に0.3当量から達成され、このことは、明らかに化学量論的不足量の添加基質でも十分であることを示す。
【0055】
実施例23〜32
これらの実施例において、実施例1からの酵素系を使用して試験し、10個の別の第2アルコールのラセミ化合物を脱ラセミ化した。前記アルコールを、2つの関連したADHsのための専門書に記載された基質スペクトルを考慮して選択した。こうして、原理的には、本発明による方法により全ての基質が脱ラセミ化可能であり、この全ての基質は、2つのADHsの基質スペクトル中に含まれている。前記実施例中で使用された第2アルコールの構造は、次の第4表中に記載されている。
【0056】
【表4】
【0057】
ラセミ化の結果は、次の表中に記載されている。
【0058】
【表5】
【0059】
この表から、全ての試験されたラセミ化合物は、本発明による方法により顕著な選択性で短時間で殆んど定量的に脱ラセミ化されうることが判明し、その際に他の官能基の存在は、本発明による方法の作用を削減しなかった。
【0060】
本発明は、立体異性体化の分野の価値のある利益をもたらし、その理由から、本発明の産業上の利用可能性は、確実なことである。
【技術分野】
【0001】
発明の技術分野
本発明は、酵素系を使用しながらエナンチオマー混合物を脱ラセミ化する方法に関する。
【0002】
先行技術
最近、立体異性体の分野でラセミ化、即ちラセミ混合物を得るために光学異性体の鏡像異性体への変換および脱ラセミ化の場合ならびに前記プロセスの正確な逆のプロセスの場合にめざましい進歩が達成された。ラセミ化は、立体的に不安定な化合物、例えばシアンヒドリン、半(チオ)アセタールα−置換カルボニル化合物およびα−置換ヒダントインの場合に、簡単な保護的な酸触媒反応または塩基触媒反応によって達成可能であり、一方で、立体的に安定な化合物、例えば第2アルコールおよびキラールアミンは、ラセミ化がはるかに困難である。
【0003】
最後のキラールアミンは、例えば遷移金属錯体触媒を用いて反応されるレドックス法により達成され、この場合キラリティー中心で必然的にsp3−ハイブリダイズされているエナンチオマーは、プロキラールなsp2ハイブリダイズされた中間生成物を経て別のエナンチオマーに変換される。例えば、O. PamiesおよびJ.- E. Baeckvall, Trends Biotechnol. 22, 130-135 (2004)およびChem. Rev. 103. 3247-3261 (2003); H. Pellissier, Tetrahedron 59, 8291-8327 (2003); M. J. Kim, Y. AhnおよびJ. Park, Curr. Opin. Biotechnol. 13, 578-587 (2002); V. Zimmermann, M. BellerおよびU. Kragi, Org. Process Res. Dev. 10, 622-627 (2006); Y. AsanoおよびS. Yamaguchi, J. Am. Chem. Soc. 127, 7696-7697 (2005)の論文参照。
【0004】
固有の高特異的な生合成の分野で、僅かな「真性」ラセミ体だけが公知であり、それというのも、工業界とは異なり、天然ではラセミ化は、殆んど必要とされていないからである。即ち、例えばα−ヒドロキシカルボン酸(例えば、マンデル酸誘導体)、α−アミノ酸およびヒダントインのラセミ化を促進するための特殊な酵素は、ほんの二三が公知である(例えば、B. Schnell, K. FaberおよびW. Kroutil, Adv. Synth. Catal. 345, 653-666 (2003))。第2アルコールおよび第1アミンをラセミ化するために、実際に定義された酵素は、久しく公知ではなかった。
【0005】
Chem.Eur.J.13,8271−8276に開示された、新しいラセミ化戦略の本発明者らの論文類は、動力学的考察形式の代わりに進行する反応の熱力学的考察形式を基礎とするものである。2つのエナンチオマーRおよびSならびにプロキラールな中間生成物Pからなる反応系においては、2つの光学対掌体のそれぞれは、中間生成物と一緒に化学的平衡および熱力学的平衡、即ちP⇔SおよびR⇔Pが存在する。同一の補助因子、NADまたはNADP(ニコチンアミド−アデニン−ジヌクレオチドまたは−ホスフェート)を利用する、2つの相反するエナンチオ選択性のアルコール脱水素酵素(以下、略してADHsと呼称される)からなる多数の組合せを使用しながら、アシロインを含めて種々の光学活性の第2アルコールのラセミ化が達成される。アルコール/ケトンの平衡は、補助因子の酸化された形と還元された形の量および比、即ちNAD+:NADHまたはNADP+:NADPHをアルコール側で適当に選択することによって維持される。詳細は図1参照。
【0006】
NAD(P)+の割合を最小に設定した場合には、純粋な(S)異性体から出発して数時間の反応時間後に望ましいラセミ体が生じた。ケトン中間生成物の量は、10%未満に減少させることができ、一部分は、1%未満に減少させることができた。これとは異なり、高い選択性のADHだけを用いる比較試験は、多くの場合に試験されたAHDsにとって不成功に終わった。単に、1つの場合には、14日の反応時間後、82%eeの収率("enantiomeric excess"、エナンチオマーの過剰量、即ち光学的収率)は、達成される。
【0007】
本発明者らの最初に短時間で発行された論文(C.V. Voss, C.C. GruberおよびW. Kroutil, Angew. Chem. Int. Ed. 47, 741-745 (2008))には、第2アルコールのラセミ化合物を、中間生成物としてのプロキラールなケトンを経てエナンチオ選択性の細菌酵素に由来するタンデム系を使用しながらアルコール酸化のために補助因子としての立体選択性ADHおよびNADの形で脱ラセミ化することが開示されている。補助因子は、いわば「再生され」、即ち同時に「添加酵素」または「補助酵素」としてのグルコース脱水素酵素(以下、GHDと略記する)を用いて促進される、グルコースからグルコノラクトンまたはグルコン酸への酸化を進行させることにより、酸化された形から還元された形へ戻された。
【0008】
親液性化されたアルカリ糞便菌細胞を使用しながらの最初の試験の場合には、意外なことに、脱ラセミ化が確認されるのではなく、むしろ、出発基質としてのエナンチオマー純粋のアルコールのラセミ化が確認され、このことは、親液性化による細胞透過性の上昇に帰因した。損傷を受けていない細胞膜を有する、新たに収穫された細胞を使用し、したがって酸化および還元は、空間的に互いに別々に進行し、種々の第2アルコールのラセミ化合物は、選択的に99%eeを上廻る収率で望ましいエナンチオマーに変換されることができた。
【0009】
しかし、この公知技術水準は、多数の欠点を有する。一方で、酵素混合物を酸化のために準備するアルカリ糞便菌系は、正確には定義不可能であり、したがって反応の進行の著しい変化をまねく可能性があり、ひいては再現可能性は、殆んど無い。他方、全ての従来法の場合、異性体化されたアルコール1モル当たり酸化のために酸素1モルが使用され、および補助因子の再生のためにグルコースが化学量論的に1モル使用され、その上、この場合には、グルコン酸またはグルコノラクトン1モルが副生成物として生じる。
【0010】
従って、本発明の課題は、上記の欠点を回避する、改善された脱ラセミ化法を準備することであった。
【0011】
発明の詳細な説明
意外なことに、立体選択性アルコール脱水素酵素およびその補助因子を用いて酸化反応と還元反応との組合せによって第2アルコールのエナンチオマー混合物を酵素的に脱ラセミ化するための改善された方法は、前記課題を解決することが見出され、この場合には、形式的に光学活性の第2アルコールのエナンチオマーは、選択的に相応するケトンに酸化され、引続きケトンは、選択的に光学的対掌体に還元され、他方、補助因子の還元された形は、添加酵素により還元反応に使用される。本発明による方法は、相反する立体選択性および異なる補助因子選択性を有する2つのアルコール脱水素酵素ならびに2つの属する異なる補助因子が、酸化反応または還元反応に使用され、酸化された補助因子および還元された補助因子が、同時に進行する、添加酵素を用いる酵素反応で互いに変換され、この場合2つのエナンチオマーの1つへの脱ラセミ化の方向は、2つのアルコール脱水素酵素の選択によって制御可能であるか、または2つの補助因子のための添加酵素の選択性の差につき制御可能であることによって特徴付けられる。
【0012】
本発明による方法によって、脱ラセミ化は、実際に定量的な光学的収率、即ち99%ee超で達成可能であり、この場合には、後になお詳説されているように、前記系が安定した平衡を達成すると直ちに、試薬が同時の反応の経過中に化学量論的に使用されることはない。更に、正確に定義された純粋な酵素(2つのADHsおよび添加酵素)は、触媒反応に使用され、このことから専ら可逆的な反応が前記方法で生じ、ならびに顕著な再生可能性が生じる。最終的に、前記方法は、簡単な一槽反応法で実施されることができ、この場合個々の部分反応間での時間的または空間的な分離は、不要である。
【0013】
アルコール脱水素酵素として、特に市場で入手可能または簡単に入手可能なアルコール脱水素酵素、例えばバシラス属(Bacillus)、シュードモナス属(Pseudomonas)、コリネバクテリウム属(Corynebacterium)、ロドコッカス属(Rhodococcus)、乳酸菌(Lactobacillus)および/またはサーモアナエロビウム(Thermoanaerobium)に由来する細菌酵素、例えばロドコッカス・ルベル(Rhodococcus ruber)、ラクトバシラス・ケフィル(Lactobacillus kefir)またはサーモアナエロビウム・ブロキイ(Thermoanaerobium brockii)に由来する細菌酵素または酵母、例えばアスペルギルス(Aspergillus)、カンジダ(Candida)、ピキア(Pichia)またはサッカロミケス(Saccharomyces)に由来する酵素が使用され、それというのも、前記酵素は、エナンチオマー過剰量および反応速度に関連して特に良好な結果を生じるからである。しかし、適したAHD対の選択にとって決定的なことは、なかんずく2つのADHsが相反する立体選択性および異なる補助因子選択性を有しなければならないという要件にある。補助因子は、ADHsのそれぞれの選択から相応してもたらされ、通常は、NADおよびNADPであり、有利には、単に触媒量で使用される。
【0014】
図3は、本発明に方法における反応経過を説明し、この場合HTSは、「Hydrid-Transfer-System水素化物移送系」を表わし、この中で「Aux」で示された添加酵素によって促進される副反応は、補助因子の「再生」、即ち酸化された形と還元された形の互いの変換のために行なわれる。k1〜k4は、水素化物移送系の第1の順序の反応の形式的な速度定数を表わす。
【0015】
図3Aに図示されているように、第2のアルコールエナンチオマーの酸化反応および還元反応は、2つの相反する立体選択性ADHs(図示されていない)によって促進される。(S)エナンチオマーから(R)エナンチオマーへの反応が進行する場合には、NADに対して補助因子の選択を有する、(S)選択性ADHの(S)異性体の酸化の際に、水素化物イオンは、アルコールから分離され、補助因子、NAD+、の酸化物の形に移行され、それによってこの補助因子は、還元された形、NADH、に移行される。本質的に同時に添加酵素Auxは、NADHの前記水素化物イオンを受け取り(このことは、「Aux−H」を生じる)、引続き酸化された形、NADP+の形で第2の補助因子に移行され、それによって第2の補助因子の還元された形のNADPHは、準備される。前記の第2の補助因子から再び水素化物は、NADPの選択性を有する第2の(R)選択性ADHによってケトン中間生成物Pに移行され、それによってこのケトン中間生成物Pは、(R)エナンチオマーに還元される。逆の方向に、即ち(R)異性体が(S)形に変換される際に、逆反応は、当然に同様に行なわれる。
【0016】
平衡で存在する酵素/補助因子系から出発する場合には、同一の水素化物イオンは、上記に説明した反応を経て、最終的に再び今や立体化学が逆転されたアルコール分子上に到達する。
【0017】
添加酵素がヌクレオチドトランスヒドロゲナーゼである場合には、水素化物イオンは、添加酵素によって1つの補助因子から別の補助因子へ、上記の簡単な形で移行する。この場合、図3B中のAux−Hは、酵素と水素化物イオンとの複合体を表わす。しかし、試験されたヌクレオチドトランスヒドロゲナーゼは、満足させる結果を生じないので、本発明者らは、試験の際に他の選択可能な方法により、水素化物を直接に移行するヌクレオチドトランスヒドロゲナーゼの代わりにもう1つの脱水素酵素/基質系もその役を引き受けうることが見い出された。この場合、Aux−Hは、添加酵素に相当する基質の還元された形を表わす。
【0018】
この種の添加酵素として、原理的に全ての補助因子に依存するオキシド還元酵素がこれに該当し、このオキシド還元酵素は、脱ラセミ化する第2アルコールの酸化反応および還元反応を損なわない。脱水素酵素、有利にグルコース脱水素酵素(GDH)、グルコース−6−ホスフェート脱水素酵素(G6PDH)およびホルミエート脱水素酵素(FDH)は、極めて良好な結果をもたらし、したがって好ましい添加酵素である。
【0019】
最初の2つの場合には、基質への水素化物の移行によって、グルコン酸またはグルコノラクトン、またはこれらの−6−ホスフェートは、グルコースまたは−6−ホスフェートへ還元され(このことは、"Aux−H"を生じる)、直ちに再び酸化される。同様のことは、第3の場合にもCO2で行なわれ、このCO2は、Aux−Hとしてのホルミエートで平衡状態になる。実際にホルミエートからCO2への酸化の反応平衡は、二酸化炭素側に遙かに広がるが、しかし、反応の原理的な可逆性は、正しいことが確認された。本発明においては、添加酵素は、全く使用されず(または殆んど使用されず)、したがって、微少量だけが必要とされるので、ホルミエート脱水素酵素/ホルミエート/CO2系は、本発明の目的に十分に好適であり、例えばこのことは、下記の実施例に示されている。
【0020】
上記したように、本発明による方法において、平衡状態が達成されると直ちに試薬の化学量論的使用は、全く生じない。この平衡状態は、特殊な酵素/基質の組合せおよびその選択性に依存するので、予想または予めの調整は、不可能である。それによって、実際には、この平衡は、脱ラセミ化法の開始時に生じる。前記の通常数分間継続する段階において、微少量の添加基質、即ちグルコースまたはグルコン酸、ホルミエートまたはCO2は、実際に使用される。
【0021】
第2アルコールの異性体化がどの方向に進行するかにより、実際には最初に2つのADHsの立体選択性および補助因子選択性に依存するが、しかし、他の結果においては、2つの補助因子のための添加酵素の異なる選択性にも依存する。NADPの選択性を有する(S)選択性ADHおよびNADPの選択性を有する(R)選択性ADHから出発する、上記に示された例には、実際に添加酵素の補助因子選択性による脱ラセミ化の方向を規定することができ、そのために、これは、十分に「制御系」とも呼称されてよい。添加系の酸化法または還元法における補助因子選択性は、NADHおよびNADP+がNAD+およびNADPH(図3Bで:k1+k3>k2+k4)へ変換されることを導くか、またはNAD+およびNADPHがNADHおよびNADP+(図3Bで:k1+k3>k2+k4)へ変換されることを導き、このことは、第1の場合には、(R)エナンチオマーの形成を導き、別の場合には、(S)エナンチオマーの形成を導く。制御酵素またはその基質が省略されるか、または制御酵素が2つの補助因子の1つに対する選択性を有する(勿論、このことは、全くありそうもないことである)場合には、全く脱ラセミ化だけを観察することができるのではなく、光学的に純粋なアルコールから出発して、むしろ、逆反応、即ちラセミ化を観察することができ、例えば、これは、図4からも明らかである:図4Aおよび4Bは、それぞれ逆の補助因子選択性を有するホルミエート脱水素酵素に対して第2アルコールとしての2−オクタノールを用いた反応経過を示し、図4cは、FDHなしの系のための反応経過を示す。
【0022】
しかし、脱ラセミ化の方向は、逆に反対される立体選択性または補助因子選択性を有するADH対を選択することによって逆転される可能性もある。例えば、上記の記載において、同じ添加酵素の際にNADPの選択性を有する(S)選択性ADHおよびNADPの選択性を有する(R)選択性ADHを使用する場合には、ラセミ化合物から選択的に(R)エナンチオマーの代わりに光学的に純粋な(S)エナンチオマーが形成される。
【0023】
本発明による方法は、通常、水、水と1つ以上の有機溶剤とからなる単相または多相の混合物およびイオン性液体を含む群から選択された溶剤中で実施され、この場合費用および安定性の理由から特に従来の水性緩衝系が使用される。
【0024】
水性緩衝系は、溶剤をpH値に対して非敏感性にする物質、例えば塩を含有する水性溶剤である。公知の水性緩衝系は、例えば炭酸/重炭酸塩系、炭酸−珪酸塩緩衝液、酢酸/酢酸塩緩衝液、燐酸塩緩衝液、ミハエリス(Michaelis)によるベロナール/酢酸塩緩衝液、アンモニア緩衝液、HEPES(4−(2−ヒドロキシエチル)−1−ピペラジンエタンスルホン酸)およびMES(2−(N−モルホリノ)エタンスルホン酸)である。
【図面の簡単な説明】
【0025】
【図1】図1は、プロキラールなケトンを経て2つの特殊なADHsを用いて酵素触媒反応によりラセミ化を行なう原理を示す略図である。
【図2】図2は、定義された菌株の背景を用いるラセミ化合物の分解および引続く補助因子の再生を示す略図である。
【図3】図3のAは、補助因子の再生のために2つの特異性ADHsおよび特異性補助酵素(水素化物移行系:HTS)を用いて本発明による酵素触媒反応により脱ラセミ化を行なう原理を示す略図であり、図3のBは、水素化物移行系の原理を示す略図である。
【図4】図4のAは、ホルミエート脱水素酵素(FDH002)を用いる1−フェニルエタノールのラセミ混合物の移動を示す略図であり、図4のBは、ホルミエート脱水素酵素(FDH001)を用いる1−フェニルエタノールのラセミ混合物の移動を示す略図であり、図4のCは、ホルミエート脱水素酵素(FDH)なしの1−フェニルエタノールのラセミ混合物の移動を示す略図である。
【図5】図5は、NADまたはNADP特異性ホルミエート脱水素酵素(FDH)を用いるラセミ混合物の移動を示す略図である。
【図6】図6は、反応平衡に対する種々の反応パラメーターの変更の影響を示す略図である。
【図7】図7は、ラセミ化平衡の調節に対する添加基質のグルコースの種々の濃度の影響を示し、図7のAは、種々のグルコース濃度についての時間的経過を示す略図であり、図7のBは、6時間の反応時間後のそれぞれのグルコース濃度への相関関係ee[%]を示す略図である。
【実施例】
【0026】
例:
次に、本発明を代表的な制限されていない実施例につき詳細に記載する。
【0027】
材料、関連資料および方法
酵素
ADH−A:ロドコッカス・ルベル(Rhodococcus ruber)のアルコール脱水素酵素(BioCatalytics Inc.,社から市場で入手可能、今やCodexis社, Pasadens在, USAから市場で入手可能)。
LK−ADH:乳酸桿菌属ケフィル(Lactobacillus kefir)のアルコール脱水素酵素(Sigma-Aldrich社, Wien在、から市場で入手可能、#05643、0.4 IE/mg)。
RE−ADH:ロドコッカス・エリトロポリス(Rhodococcus erythropolis)のアルコール脱水素酵素(Sigma-Aldrich社から市場で入手可能、#68482、20 IE/ml)。
LB−ADH:アルコール脱水素酵素002(Juelich Chiral Solutions社から市場で入手可能、今やCodexis社から市場で入手可能、#05.11)。
ADH−T:アルコール脱水素酵素005(Juelich Chiral Solutions社から市場で入手可能、今やCodexis社から市場で入手可能、#26.10)。
ADH−PR2:アルコール脱水素酵素007(Juelich Chiral Solutions社から市場で入手可能、今やCodexis社から市場で入手可能、#42.10)。
TB−ADH:テルモアナエロビウム・ブロキイ(Thermoanaerobium brockii)のアルコール脱水素酵素(Sigma-Aldrich社から市場で入手可能、#A9287,30−90 IE/mg)。
G6PDH:パン酵母のグルコース−6−ホスフェート脱水素酵素(Sigma-Aldrich社から市場で入手可能、#49271,240 IE/mg)。
GLY−DH:ゲオトリクム・カンディドゥム(Geotrichum candidum)のグリセリン脱水素酵素(Sigma-Aldrich社から市場で入手可能、#49860,30 IE/mg)。
LDH−SC:スラフィロコッキ(Sraphylococci)のD−ラクテート脱水素酵素(Sigma-Aldrich社から市場で入手可能、#17847,120 IE/mg)。
LDH−LS:ラクトバシラス・スプ(Lactobacillus sp.)のD−ラクテート脱水素酵素(Sigma-Aldrich社から市場で入手可能、#59023,400 IE/mg)。
LDH−RM:イエウサギの筋肉に由来するL−ラクテート脱水素酵素(Sigma-Aldrich社から市場で入手可能、#61311,500 IE/mg)。
FDHI:NADP特異性ホルミエート脱水素酵素001(Juelich Chiral Solutions社から市場で入手可能、今やCodexis社, Pasadena在, USAから市場で入手可能、#25.10,47 IE/ml)。
FDHI:NADP特異性ホルミエート脱水素酵素001(Juelich Chiral Solutions社から市場で入手可能、今やCodexis社, Pasadena在, USAから市場で入手可能、#25.10,47 IE/ml)。
FDH3:NAD特異性ホルミエート脱水素酵素001(Juelich Chiral Solutions社から市場で入手可能、今やCodexis社から市場で入手可能、#09.11,200 IE/ml)。
FDH4:酵母のホルミエート脱水素酵素(Boehringer Mannheim GmbH社から市場で入手可能、#204226,0.5 IE/mg)。
FDH5:カンディダ・ボイジニイ(Candida boidinii)のホルマール脱水素酵素(Martina Pohl、デュッセルドルフ大学、ドイツ連邦共和国、からの寄贈品)。
GDH−BM:D−グルコース脱水素酵素001(Juelich Chiral Solutions社から市場で入手可能、今やCodexis社から市場で入手可能、#22.10,30 IE/mg)。
GDH−BS:D−グルコース脱水素酵素002(Juelich Chiral Solutions社から市場で入手可能、今やCodexis社から市場で入手可能、#29.10,500 IE/ml)。
【0028】
酵素の性質
【表1】
【0029】
【表2】
【0030】
化学薬品
rac−2−オクタノール(#04504、MG 130.23g/mol)、(R)−2−オクタノール(#74864、MG 130.23g/mol)、(S)−2−オクタノール(#74863、MG 130.23g/mol)、2−オクタノン(#53220、MG 128.21g/mol)、アンモニウムホルミエート(#09739、63.96g/mol)、ナトリウムホルミエート(#3996−15−4、69.02g/mol)およびカリウム塩としての蟻酸(#57444−81−2、MG 85.13g/mol)は、Sigma−Aldrich社、Wien在、から購入した。抽出および後処理のための化学薬品:
抽出のためのエチルアセテート(#441977)は、Brenntag CEE GmbH社、Ort在、から購入され、新たに蒸留に使用された。アセチル化のためのDMAP(#29224、MG 122.17g/mol)および無水酢酸(#45830、MG 102.09g/mol)は、Sigma−Aldrich社、Wien在、から購入した。
【0031】
一般的なプロセス
光学的組成物のシフトのためのモデル的方法:市販の酵素の活性は、一般に国際単位(IE)で記載されている。しかし、この単位は、全体でここで使用されたものとは別の基質に対するそれぞれの酵素の活性を示す。従って、使用された酵素の活性は、適した"再生"系での2−オクタノンの還元の際に測定された(一般に、アンモニウムホルミエートを有するFDH、5当量)。全ての試験のためには、例えばADHsの1 IE2-オクタノールが使用された。
【0032】
系1:ADH−A、LK−ADH、NADP特異性FDH(2 IE)、アンモニウムホルムエート(基質濃度に対して3当量)、NAD+およびNADP+(基質3モル%)は、TRIS−HCl(50mM、pH7.5、全容量0.5ml)中に懸濁された。反応は、ラセミ体の2−オクタノールの添加によって開始された(0.5μl、8mmol/ml、ee<3%)。30℃で3時間の振盪(130rpm)後、混合物は、EtOAc(500μl)で抽出され、遠心分離され、相分離が生じた。
【0033】
系2:NAD特異性FDH(2 IE)の使用まで系1と同様。
【0034】
系3:GDH−BS(2 IE)およびα−D−グルコース(1当量、8mmol/L)の使用まで系1と同様。
【0035】
系4:ADH−TおよびADH−PR2の使用まで系1と同様。
【0036】
系5:ADH−T、ADH−PR2およびNAD特異性FDHの使用まで系1と同様。
【0037】
系6:RE−ADHおよびテルモアナエロビウム・ブロキイ(Thermoanaerobium brockii)のAORの使用まで系1と同様。
【0038】
分析法
キラルなGC−FID分析:
アルコールは、無水酢酸(100ml)およびDMAP(0.5mg)の添加によって30℃で2時間でアセチル化された。後処理後、生成物は、GC−FIDおよびGC−MSDを用いてキラルな固定相で分析された。
【0039】
キラルなGC−FID分析は、バリアン(Varian)−3900−ガスクロマトグラフィーでFID検出器を用いてクロムパック−キラシル−DEX−CB−カラム(Chrompack-Chirasil-DEX-CB-Saeule)(Varian社、25m×0.32mm×0.25mm、1.0バール H2)を使用して、検出器温度250℃、分配比90:1で行なわれた。
【0040】
キラルなGC−MSD分析:
キラルなGC−MSD分析は、アギレント(Agilent)−7890A−GCシステムでFID検出器を用いてクロムパック−キラシル−DEX−CB−カラム(Chrompack-Chirasil-DEX-CB-Saeule)(Varian社、25m×0.32mm×0.25mm、1.0バール H2)を使用して、検出器温度250℃、分配比90:1で行なわれた。
【0041】
キラルなHPLC分析:
HPLC分析は、DGU−20A5脱ガス装置、LC−20AD液体クロマトグラフ、SIL−20AC自動サンプラー、CBM−20Aコミュニケーションバスモジュール(Kommunikationsbusmodul)、SPD−M20Aダイオードアレー検出器およびCTO−20ACカラム炉を備えたShimadzu−HPLCシステムでn−ヘプタン/イソプロパノール=90:10、0.5ml/分、18℃を有するChiralpak−ADカラム(Daicel, 0.46×25cm)を使用して行なわれた。
【0042】
実施例1〜13、比較例1〜4
脱ラセミ化を、第2アルコールとしての2−オクタノールとの種々のAHD/添加酵素組合せ物を使用して次の条件下で実施した:基質濃度8mmol/l、反応時間3〜12時間、TRIS−HCl(pH7.5、50mM)または燐酸塩緩衝液(pH7.5、50mM)中で30℃、130rpmでの振盪。ADHsからそれぞれ約1 IE(基質としての2−オクタノールのため);触媒量(約3モル%)中のNAD+およびNADP+。添加酵素:2 IE(そのために、製造業者によって記載されたような天然基質)。添加基質(ホルミエート、グルコース、グルコース−6−ホスフェート、ラクテートおよびグリセリン):16mmol/L。この組成および結果は、次の第3表中に記載されている。
【0043】
【表3】
【0044】
2−オクタノールラセミ化合物からなる本発明の実施例1〜13の試験された酵素組合せ物は、主に良好な一部分が実際に定量的なエナンチオマー過剰量およびエナンチオマーの1つの殆んど全て定量的な収量で供給されたことを確認することができる。同一のADH対を使用した場合、添加酵素の補助因子特異性を逆にすることによって、脱ラセミ化の方向を制御することができた。実施例1/2+3、8/9、10/11、12/13参照。また、実施例8および9の結果は、図5にグラフで図示されている。
【0045】
これに対して、比較例1〜4においてラクテート脱水素酵素またはグリセリン脱水素酵素を使用した場合には、脱ラセミ化は、全く生じなかった。
【0046】
実施例14〜22
これらの実施例において、実施例1からの酵素系を使用して、種々の反応パラメーターが変更され、反応経過に対して前記反応パラメーターの作用が試験された。実施例14〜21の結果は、図6A〜Hにグラフで図示されており、実施例22の結果は、図7A〜Bに図示されている。
【0047】
実施例14
ここでは、反応時間を1〜6時間の間で変動させ、3時間後に既に定量的な変換が達成されることが見い出された。従って、前記群の他の実施例は、3時間に亘って実施された(図6A)。
【0048】
実施例15
アルコール濃度を1〜243m2/lの間で変動させ、この最適な結果は、6時間の所定の反応時間で2〜8mmol/lを生じた。よりいっそう高い濃度の場合には、よりいっそう長い反応時間またはよりいっそう大量の酵素が必要不可欠であり、完全な変換が達成される(図6B)。
【0049】
実施例16
2つのADHsの全体量を0.1〜3.4 IEの間で変動させ、この場合には、1 IEが最適な活性量であることが明らかになった(図6C)。
【0050】
実施例17
互いに2つのADHsの活性比(IEで)を0.01〜13.5の間で変動させ、約0.2〜約0.7の比は、最も効果的であることが判明したが、しかし、その際に、1:1の比の値は、欠いた(図6D)。
【0051】
実施例18および19
2つのADHsのそれぞれ一方の活性を0.1〜7.6 IEの間で、または3.4 IEで変動させ、その際に第2のADHの活性は、1 IEであり、また、第2の酵素のための最適な活性量は1 IEであり、したがって、2つのADHsの最適な活性比は1:1であることが見い出された(図6E、6F)。
【0052】
実施例20
FDHの量を0.3〜64.0 IEの間で変動させ、2 IEの量から既に定量的な変換が達成されることが判明した(図6G)。
【0053】
実施例21
補助因子NADおよびNADPの共通の濃度を0〜96モル%の間で変動させ、この場合約2〜3モル%の濃度は、最も効果的であった(図6H)。
【0054】
実施例22
実施例5を繰り返したが、この場合添加基質、即ちグルコースの濃度は、図7Aに示されているように、0.5〜12時間の反応時間に亘ってアルコール濃度に対して0.1〜3当量の間で変動した。グルコース当量が0.1〜1の間で変動する際の6時間後のエナンチオマー過剰量eeは、図7Bに示されている。この場合には、>99%が既に0.3当量から達成され、このことは、明らかに化学量論的不足量の添加基質でも十分であることを示す。
【0055】
実施例23〜32
これらの実施例において、実施例1からの酵素系を使用して試験し、10個の別の第2アルコールのラセミ化合物を脱ラセミ化した。前記アルコールを、2つの関連したADHsのための専門書に記載された基質スペクトルを考慮して選択した。こうして、原理的には、本発明による方法により全ての基質が脱ラセミ化可能であり、この全ての基質は、2つのADHsの基質スペクトル中に含まれている。前記実施例中で使用された第2アルコールの構造は、次の第4表中に記載されている。
【0056】
【表4】
【0057】
ラセミ化の結果は、次の表中に記載されている。
【0058】
【表5】
【0059】
この表から、全ての試験されたラセミ化合物は、本発明による方法により顕著な選択性で短時間で殆んど定量的に脱ラセミ化されうることが判明し、その際に他の官能基の存在は、本発明による方法の作用を削減しなかった。
【0060】
本発明は、立体異性体化の分野の価値のある利益をもたらし、その理由から、本発明の産業上の利用可能性は、確実なことである。
【特許請求の範囲】
【請求項1】
立体選択性アルコール脱水素酵素およびその補助因子を用いて酸化反応と還元反応との組合せによって第2アルコールのエナンチオマー混合物を酵素的に脱ラセミ化するための方法であって、この場合には、形式的に光学活性の第2アルコールのエナンチオマーは、選択的に相応するケトンに酸化され、引続きケトンは、選択的に光学的対掌体に還元され、他方、補助因子の還元された形は、添加酵素により還元反応に使用される、上記の脱ラセミ化法において、相反する立体選択性および異なる補助因子選択性を有する2つのアルコール脱水素酵素ならびに2つの属する異なる補助因子が、酸化反応または還元反応に使用され、酸化された補助因子および還元された補助因子が、同時に進行する、添加酵素を用いる酵素反応で互いに変換され、この場合2つのエナンチオマーの1つへの脱ラセミ化の方向は、2つのアルコール脱水素酵素の選択によって制御可能であるか、または2つの補助因子のための添加酵素の選択性の差につき制御可能であることを特徴とする、上記の脱ラセミ化法。
【請求項2】
アルコール脱水素酵素として細菌由来のアルコール脱水素酵素を使用する、請求項1記載の方法。
【請求項3】
アルコール脱水素酵素としてバシラス属(Bacillus)、シュードモナス属(Pseudomonas)、コリネバクテリウム属(Corynebacterium)、ロドコッカス属(Rhodococcus)、乳酸菌(Lactobacillus)および/またはサーモアナエロビウム(Thermoanaerobium)に由来するアルコール脱水素酵素を使用する、請求項1または2記載の方法。
【請求項4】
アルコール脱水素酵素として酵母菌株に由来する酵素を使用する、請求項1記載の方法。
【請求項5】
アルコール脱水素酵素としてアスペルギルス(Aspergillus)、カンジダ(Candida)、ピキア(Pichia)またはサッカロミケス(Saccharomyces)に由来するアルコール脱水素酵素を使用する、請求項4記載の方法。
【請求項6】
2つのアルコール脱水素酵素を1:1の活性比で使用する、請求項1から5までのいずれか1項に記載の方法。
【請求項7】
2つのアルコール脱水素酵素を1 IEの全体量で使用する、請求項1から6までのいずれか1項に記載の方法。
【請求項8】
ラセミ化合物の第2アルコールを少なくとも2mmol/lの濃度で使用する、請求項1から7までのいずれか1項に記載の方法。
【請求項9】
添加酵素としてグルコース脱水素酵素、グルコース−6−ホスフェート脱水素酵素、ホルミエート脱水素酵素またはヌクレオチドトランスヒドロゲナーゼを使用する、請求項1から8までのいずれか1項に記載の方法。
【請求項10】
添加酵素を2 IEの量で使用する、請求項1から9までのいずれか1項に記載の方法。
【請求項11】
添加酵素の基質を第2アルコール1モル当たり少なくとも0.3モルの量で使用する、請求項1から10までのいずれか1項に記載の方法。
【請求項12】
補助因子を触媒量で使用する、請求項1から11までのいずれか1項に記載の方法。
【請求項13】
補助因子を第2アルコールに対して2〜3モル%の量で使用する、請求項9記載の方法。
【請求項14】
水、水と1つ以上の有機溶剤とからなる単相または多相の混合物ならびにイオン性液体から選択された溶剤を使用する、請求項1から13までのいずれか1項に記載の方法。
【請求項15】
溶剤として水性緩衝系を使用する、請求項14記載の方法。
【請求項1】
立体選択性アルコール脱水素酵素およびその補助因子を用いて酸化反応と還元反応との組合せによって第2アルコールのエナンチオマー混合物を酵素的に脱ラセミ化するための方法であって、この場合には、形式的に光学活性の第2アルコールのエナンチオマーは、選択的に相応するケトンに酸化され、引続きケトンは、選択的に光学的対掌体に還元され、他方、補助因子の還元された形は、添加酵素により還元反応に使用される、上記の脱ラセミ化法において、相反する立体選択性および異なる補助因子選択性を有する2つのアルコール脱水素酵素ならびに2つの属する異なる補助因子が、酸化反応または還元反応に使用され、酸化された補助因子および還元された補助因子が、同時に進行する、添加酵素を用いる酵素反応で互いに変換され、この場合2つのエナンチオマーの1つへの脱ラセミ化の方向は、2つのアルコール脱水素酵素の選択によって制御可能であるか、または2つの補助因子のための添加酵素の選択性の差につき制御可能であることを特徴とする、上記の脱ラセミ化法。
【請求項2】
アルコール脱水素酵素として細菌由来のアルコール脱水素酵素を使用する、請求項1記載の方法。
【請求項3】
アルコール脱水素酵素としてバシラス属(Bacillus)、シュードモナス属(Pseudomonas)、コリネバクテリウム属(Corynebacterium)、ロドコッカス属(Rhodococcus)、乳酸菌(Lactobacillus)および/またはサーモアナエロビウム(Thermoanaerobium)に由来するアルコール脱水素酵素を使用する、請求項1または2記載の方法。
【請求項4】
アルコール脱水素酵素として酵母菌株に由来する酵素を使用する、請求項1記載の方法。
【請求項5】
アルコール脱水素酵素としてアスペルギルス(Aspergillus)、カンジダ(Candida)、ピキア(Pichia)またはサッカロミケス(Saccharomyces)に由来するアルコール脱水素酵素を使用する、請求項4記載の方法。
【請求項6】
2つのアルコール脱水素酵素を1:1の活性比で使用する、請求項1から5までのいずれか1項に記載の方法。
【請求項7】
2つのアルコール脱水素酵素を1 IEの全体量で使用する、請求項1から6までのいずれか1項に記載の方法。
【請求項8】
ラセミ化合物の第2アルコールを少なくとも2mmol/lの濃度で使用する、請求項1から7までのいずれか1項に記載の方法。
【請求項9】
添加酵素としてグルコース脱水素酵素、グルコース−6−ホスフェート脱水素酵素、ホルミエート脱水素酵素またはヌクレオチドトランスヒドロゲナーゼを使用する、請求項1から8までのいずれか1項に記載の方法。
【請求項10】
添加酵素を2 IEの量で使用する、請求項1から9までのいずれか1項に記載の方法。
【請求項11】
添加酵素の基質を第2アルコール1モル当たり少なくとも0.3モルの量で使用する、請求項1から10までのいずれか1項に記載の方法。
【請求項12】
補助因子を触媒量で使用する、請求項1から11までのいずれか1項に記載の方法。
【請求項13】
補助因子を第2アルコールに対して2〜3モル%の量で使用する、請求項9記載の方法。
【請求項14】
水、水と1つ以上の有機溶剤とからなる単相または多相の混合物ならびにイオン性液体から選択された溶剤を使用する、請求項1から13までのいずれか1項に記載の方法。
【請求項15】
溶剤として水性緩衝系を使用する、請求項14記載の方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公表番号】特表2011−516053(P2011−516053A)
【公表日】平成23年5月26日(2011.5.26)
【国際特許分類】
【出願番号】特願2011−502345(P2011−502345)
【出願日】平成21年3月26日(2009.3.26)
【国際出願番号】PCT/EP2009/053576
【国際公開番号】WO2009/121785
【国際公開日】平成21年10月8日(2009.10.8)
【出願人】(501073862)エボニック デグサ ゲーエムベーハー (837)
【氏名又は名称原語表記】Evonik Degussa GmbH
【住所又は居所原語表記】Rellinghauser Strasse 1−11, D−45128 Essen, Germany
【Fターム(参考)】
【公表日】平成23年5月26日(2011.5.26)
【国際特許分類】
【出願日】平成21年3月26日(2009.3.26)
【国際出願番号】PCT/EP2009/053576
【国際公開番号】WO2009/121785
【国際公開日】平成21年10月8日(2009.10.8)
【出願人】(501073862)エボニック デグサ ゲーエムベーハー (837)
【氏名又は名称原語表記】Evonik Degussa GmbH
【住所又は居所原語表記】Rellinghauser Strasse 1−11, D−45128 Essen, Germany
【Fターム(参考)】
[ Back to top ]